
A conexina 26 e sua relação com outras proteínas no órgão ... · energia e a Ori, por todos os...
188
Ana Carla Batissoco A conexina 26 e sua relação com outras proteínas no órgão de Corti São Paulo 2011
Transcript of A conexina 26 e sua relação com outras proteínas no órgão ... · energia e a Ori, por todos os...

Ana Carla Batissoco
A conexina 26 e sua relação com outras proteínas no órgão de Corti
São Paulo 2011

2
Ana Carla Batissoco
A conexina 26 e sua relação com outras proteínas no órgão de Corti
Tese apresentada ao Instituto de Biociências da Universidade de São Paulo, para a obtenção de Título de Doutor em Ciências, na Área de Biologia/Genética.
Orientadora: Profa. Dra. Regina Célia Mingroni Netto
Co-orientadora: Profa. Dra. Luciana Amaral Haddad
São Paulo 2011

3
Ficha Catalográfica
Batissoco, Ana Carla
A Conexina 26 e sua relação com outras proteínas no órgão de Corti
181 pp.
Tese (Doutorado) - Instituto de Biociências da Universidade de São Paulo. Departamento de Genética e Biologia Evolutiva.
1. GJB2
2.Conexina 26
3. Surdez hereditária
4. órgão de Corti
5. precipitação por afinidade de proteínas
6. células progenitoras
7. células de suporte
Comissão Julgadora:
Prof(a). Dr(a). Prof(a).Dr(a).
Prof(a). Dr(a). Prof(a).Dr(a).
Profa. Dra. Regina Célia Mingroni Netto Orientadora

4
Aos amores da minha vida;
Alê e Urko.

5
Agradecimentos
À minha orientadora Profa. Dra. Regina Célia Mingroni Netto, por sua
dedicação, confiança e conselhos valiosos.
À minha co-orientadora Profa. Dra. Luciana Amaral Haddad, por toda
orientação técnica e científica e pela grande disposição em me ajudar em todos os
momentos desse estudo.
Aos meus pais, Luiz e Zulmira, e meus irmãos, Miguel e Graça, aos quais
devo tudo o que sou.
Ao Alê pelo amor e apoio integral, ao Urko por ser a fonte de toda minha
energia e a Ori, por todos os momentos de descontração.
Ao Prof. Dr. Paulo Otto, por ter me apresentado ao Laboratório de Genética
Humana.
À Profa. Dra. Angela Vianna Morgante, pelo uso das dependências do
laboratório de Genética Humana.
Ao Dr. Ignácio del Castillo pela amostra controle cedida para a triagem da
deleção de 200Kb localizada nas proximidades do gene GJB6 e pelas análises de
MLPA.
Ao diretor da DERDIC (Divisão de Reabilitação dos Distúrbios da
Comunicação, PUC-SP), Dr. Alfredo Tabith Jr. e ao diretor clínico Dr. Mauro
Spinelli (in memoriam) e todos os demais membros do corpo profissional pelas
inúmeras avaliações clínicas e audiológicas realizadas nos pacientes e seus familiares.
Aos amigos Faculdade de Medicina da USP, Jeanne, Luiz e Karina, pela
amizade, pelo companheirismo e parceria na pesquisa e pelas ótimas discussões
científicas e não científicas.
Aos funcionários e alunos do Laboratório de Genética Humana, de
Genômica Funcional e de Oxidações Biológicas em Leveduras, que contribuíram de
alguma forma para que eu realizasse este projeto. Em especial, agradeço à Maraísa
pela amizade.
Aos meus colegas do laboratório de Genética Humana, Dayane, Magnólia,
Renata e Vítor. Agradeço em especial a Lilian e a Maria Teresa pelo apoio técnico,
científico e pela amizade. E também ao “agregado” Gustavo. E as ex-alunas Daniela
e Renata Thiele.

6
Aos meus amigos mais do queridos Deborah e Fernando, e também Crys,
Larissa e Juliana por todo o apoio durante as aventuras e “desventuras” da vida
acadêmica.
Aos funcionários do Departamento de Genética e Biologia Evolutiva e do
Centro de Estudos do Genoma Humano pelo apoio técnico.
Ao Prof. Dr. Alberto A. G. F. C. Ribeiro, por ter permitido o acesso ao seu
laboratório de Biologia Celular e Microscopia Eletrônica e em especial ao técnico
Waldir Caldeira, por todos os longos períodos de captura de imagens.
À Ana Lucia Garippo, do núcleo de microscopia confocal/fluorescência
Rede Multiusuários do Sistema FMUSP/HC pela captura de imagens e pelas
valiosas dicas de imunofluorescência.
Aos nosso colaboradores internacionais, Prof. Stefan Heller (Department of
Otolaryngology Head and Neck Surgery and Department of Molecular and Cellular
Physiology, Stanford University School of Medicine, Stanford, Califórnia, USA) e
Prof. Azel Zine (Institute for Neurosciences of Montpellier and Institute for
Research in Biotherapy, University of Montpellier I, Montpellier, France) por todo o
treinamento oferecido a nossa equipe de pesquisa em células-tronco/progenitoras
do órgão de Corti.
Ao Prof. Dr. Ricardo Bento, pela coordenação dos projetos INCT-CNPq
(Instituto Nacional de Ciência e Tecnologia de Células-Tronco) e RNTC-FAPESP
(Rede Nacional de Terapia Celular), cujos recursos financieros permitiram parte das
pesquisas aqui apresentadas.
À Profa. Dra. Mayana Zatz, pela coordenação dos projetos PRONEX-CNPq,
CEPID-FAPESP, Centro de Estudos do Genoma Humano e INCT-CNPq, cujos
recursos financeiros permitiram a execução da maioria das pesquisas aqui
apresentadas
Ao Instituto de Biociências e ao Departamento de Genética e Biologia
Evolutiva, pela infra-estrutura.
À FAPESP e CNPq pelos auxílio financeiro.
Aos nossos pacientes com deficiência auditiva e às suas famílias, pela enorme
paciência em colaborar com nossas pesquisas.
A todos os animais sacrificados que contribuíram para esse estudo.

7
SUMÁRIO
Apresentação 1
Resumo 4
Abstract 6
Capítulo 1 9
1. Revisão bibliográfica 9
1.1.Introdução 9
1.2.Fisiologia da audição 10
1.2.1.O órgão de Corti 11
1.2.1.1.Desenvolvimento, proliferação e diferenciação celular no órgão de Corti
13
1.2.1.2.A regeneração das células ciliadas como indicativo da presença de células-tronco na orelha interna
17
1.3.Surdez de etiologia genética 19
1.4.Surdez não-sindrômica 20
1.5.Surdez não-sindrômica de herança autossômica recessiva e o lócus DFNB1 21
1.6.As proteínas conexinas 26
1.6.1.Expressão e estrutura das conexinas 26
1.6.2.Funções das conexinas 28
1.6.3.Biossíntese das conexinas 30
1.6.4.Interação entre conexinas e outras proteínas 33
Capítulo 2. Objetivos 35
2.1. Objetivo geral 35
2.2. Objetivos específicos 35
Capítulo 3. Pesquisa de novos alelos patogênicos no lócus DFNB1 relacionados à surdez não-sindrômica de herança autossômica recessiva
36
3.1.Introdução 36
3.2.Objetivo 37
3.3.Casuística e Métodos 38
3.3.1.Pacientes 38
3.3.2.Amostra controle 39

8
3.3.3.Métodos 39
3.3.3.1.Sequenciamento das regiões de código, promotora e de splicing do gene GJB2
40
3.3.3.2.Triagem da deleção de 200kb localizada nas proximidades do gene GJB2
43
3.3.3.3.Pesquisa de variações no número de cópias nos exons dos genes GJB2, GJB6, GJB3 e WFS1 por MLPA
45
3.4.Resultados 52
3.4.1.Sequenciamento das regiões de código, promotora e doadora de splicing do intron 1 do gene GJB2
52
3.4.2.Triagem da deleção de 200kb a a 130kb da região 5’ do gene GJB6 53
3.4.3.Pesquisa de variações no número de cópias por MLPA e por PCR em tempo real quantitativa
56
3.5.Discussão 59
3.5.1.Sequenciamento das regiões de código, promotora e doadora de splicing do intron 1 do gene GJB2
59
3.5.2.Triagem da deleção de 200kb localizada a 130kb da região 5’ do gene GJB6
61
3.5.3.Pesquisa de variações no número de cópias por MLPA (Multiplex Ligation-dependent Probe Amplification) e por PCR em tempo real quantitativa
62
3.6.Conclusão 65
Capítulo 4. Estabelecimento de culturas primárias de células neuro-epiteliais do órgão de Corti de cobaias e camundongos
66
4.1.Introdução 66
4.2.Objetivo 68
4.3.Animais e Métodos 68
4.3.1.Animais 68
4.3.2.Métodos 69
4.3.2.1.Dissecção do órgão de Corti de cobaias e camundongos neonatos 70
4.3.2.2.Cultura em suspensão de células progenitoras do órgão de Corti 70
4.3.2.3.Caracterização fenotípica das células em cultura 73
4.3.2.4.Contagem celular 75
4.3.2.5.Análises estatísticas 75
4.4.Resultados 75
4.5.Discussão 85
4.6.Conclusão 90

9
Capítulo 5. Pesquisa e identificação de proteínas que interagem com a Cx26 91
5.1.Introdução 91 5.2.Objetivo 93 5.3.Animais e Métodos 94 5.3.1.Animais 94 5.3.2.Métodos 95 5.3.2.1.Clonagem e expressão de uma proteína com a sequência codificadora para a região C-terminal da Cx26 em fusão com GST
95
5.3.2.2.Obtenção de proteínas recombinantes em condições solúveis ou insolúveis 98 5.3.2.3. Ensaios de precipitação por afinidade entre a proteína de fusão recombinante (GST-Cx26) e proteínas do lisado celular
100
5.3.2.4.Identificação proteica por espectrometria de massas 102 5.3.2.5.Análises in silico 104 5.4. Resultados 105 5.4.1. Clonagem e expressão de uma proteína com a sequência codificadora para a região C-terminal da Cx26 em fusão com GST
105
5.4.2.Ensaios de precipitação por afinidade entre as proteínas recombinantes e proteínas do lisado celular
111
5.4.3.Análises das proteínas identificadas por espectrometria de massas 112 5.5.Discussão 127 5.6. Conclusão 142
Referências Bibliográficas 144 Anexos 165

10
Índice de Figuras
Figura 1.1. Esquema representando o aparelho auditivo humano: orelha externa (E), orelha média (M) e orelha interna (I).
12
Figura 1.2. Secção transversal de um dos giros da cóclea, mostrando sua divisão em três compartimentos longitudinais: as escalas vestibular, timpânica e média ou duto coclear.
12
Figura 1.3. Esquema ampliado do órgão de Corti, mostrando as células ciliadas internas e externas e as células de suporte.
13
Figura 1.4. Diferenciação das células progenitoras em células ciliadas e de suporte.
16
Figura 1.5. Representação esquemática dos genes GJB2 e GJB6.
23
Figura 1.6. Representação esquemática da região do lócus DFNB1, 13q12-11, onde estão presentes os genes GJB2 e GJB6 e das deleções descritas nessa região e associadas a surdez não-sindrômica de herança autossômica recessiva.
25
Figura 1.7. Representação esquemática da estrutura das conexinas, dos conexons e dos canais de junção do tipo fenda.
29
Figura 1.8. Representação esquemática da reciclagem dos íons potássio na cóclea.
31
Figura 1.9. Representação esquemática da síntese, montagem e degradação dos canais comunicantes do tipo fenda.
32
Figura 3.1. Representação esquemática do gene GJB2 e das suas regiões de código, promotora e doadora de splicing do intron 1 e da extensão das regiões sequenciadas nesse trabalho.
42
Figura 3.2. Exemplo de curva de dissociação específica mostrando um único pico máximo entre 80-84oC.
49
Figura 3.3. Exemplo das curvas de amplificação das diluições seriadas utilizadas para a construção das curvas padrão e cálculo da eficiência dos pares de iniciadores quanto a sequência alvo.
52
Figura 3.4. Nova mutação p.L76P(c.C227T) no gene GJB2.
53
Figura 3.5. Resultados da genotipagem de SNPs com o MegaBace SNuPe Genotyping Kit da nova mutação p.L76P (c.C227T).
54
Figura 3.6. Triagem da deleção de 200kb localizada a 130kb da região 5’ do gene GJB6.
54

11
Figura 3.7. Perfil das sondas de MLPA do kit SALSA P163-C1 Hearing loss no paciente em que foi detectada a duplicação do gene GJB2 associada à mutação 167delT.
57
Figura 3.8. Gráficos mostrando a quantificação relativa (QR) realizada a partir dos resultados da PCR em tempo real quantitativa no paciente controle sem duplicação e no paciente com a duplicação.
57
Figura 3.9. Representação esquemática do gene GJB2 e da região duplicada detectada no DNA genômico do paciente portador da mutação c.167delT.
59
Figura 3.10. Heredograma da família da paciente portadora da mutação c.167delT no gene GJB2 em heterozigose e na qual foi detectada a duplicação na região de código do gene GJB2 .
59
Figura 3.11. Análise do alinhamento do resíduo 76 na conexina 26, do gene GJB2, em diferentes espécies.
60
Figura 4.1. Dissecção da orelha interna de cobaias.
76
Figura 4.2. Morfologia dos diferentes tipos de esferas obtidas de culturas de células em suspensão do órgão de Corti de camundongos.
77
Figura 4.3. Análise do potencial de proliferação das células do órgão de Corti de cobaias e camundongos em suspensão.
79
Figura 4.4. Resultado do ensaio de auto-renovação das células do órgão de Corti em suspensão.
80
Figura 4.5. Análise do número de células presentes nas otoesferas obtidas com o cultivo em suspensão das células do órgão de Corti de cobaias e camundongos em meio enriquecido com TGFα ou bFGF.
82
Figura 4.6 Imunofluorescência indireta com os anticorpos anti-nestina e anti-Sox2 nas otoesferas de cobaias e camundongos, após uma ou duas passagens, cultivadas na presença de bFGF or TGFα.
83
Figura 4.7.: Imunofluorescência indireta com os anticorpos p27kip1 e Jagged 1 nas otoesferas de camundongos, após duas passagens, cultivadas na presença de TGF e submetidas a diferenciação celular.
84
Figura 4.8: Imunofluorescência indireta com os anticorpos miosina VIIa e Jagged 2, nas otoesferas de camundongos, após duas passagens, cultivadas na presença de TGF e submetidas a diferenciação celular.
84
Figura 5.1. Gráfico mostrando a variação da probabilidade de identificação correta de uma proteína pelo Scaffold 3 proteomic software, em função ao número de peptídeos identificados e probabilidade de identificação correta que reflete a porcentagem de similaridade.
104

12
Figura 5.2. Triagem das colônias recombinantes com o vetor pGEX-4T-1.
108
Figura 5.3. Padronização das condições de expressão das proteínas GST-Cx26 e GST.
109
Figura 5.4. Western blotting para confirmação da expressão das proteínas GST-Cx26 e GST.
109
Figura 5.5. Solubilidade das proteínas GST-Cx26 e GST.
110
Figura 5.6. Quantificação e purificação das proteínas GST-Cx26 e GST.
111
Figura 5.7. Eletroforese em SDS-PAGE a 10% dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26, tendo sido usada a extração de cérebro com o tampão PHEM.
114
Figura 5.8. Eletroforese em SDS-PAGE a 12% dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26 tendo sido usada a extração de cérebro com os tampões EGTA e EDTA.
114
Figura 5.9. Eletroforese em SDS-PAGE a 8 % dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26 tendo sido usada a extração de fígado com o tampão EGTA.
115
Figura 5.10. Eletroforese em SDS-PAGE a 8% dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26 tendo sido usada a extração de fígado com o tampão EGTA.
115
Figura 5.11. Diagrama de Venn mostrando as 23 proteínas candidatas a interagirem com a Cx-26 classificadas em cinco grupos distintos de acordo com identidade de ontologia gênica relacionada a processo biológico, compartimento celular e função molecular.
121

13
Índice de Tabelas
Tabela 1.1. Distribuição tecidual das conexinas (RNAm) em humanos (Cx hs) e camundongos (Cx mus).
27
Tabela 3.1. Relação das sondas que compõe o Kit SALSA MLPA P163 C1 (MRC - Holland) e as regiões cromossômicas abrangidas por elas.
46
Tabela 3.2. Programa de amplificação utilizado nos experimentos de PCR em tempo real.
49
Tabela 3.3. Sequência dos iniciadores de regiões diferentes do gene GJB2 utilizados nos experimentos de PCR em tempo real.
49
Tabela 3.4. Caracterização dos dezesseis pacientes com mutações monoalélicas nos genes GJB2 ou GJB6 e resultados das análises moleculares.
55
Tabela 3.5. Resultados das quantificações relativas (QR) das sequências alvo do gene GJB2, que indicam o seu número de cópias, e seus valores de P adotando-se intervalo de confiança de 95%.
58
Tabela 4.1. Relação dos anticorpos primários utilizados na caracterização fenotípica das culturas de células progenitoras do órgão de Corti de cobaias e camundongos.
74
Tabela 4.2. Análise dos parâmetros obtidos quanto ao número de otoesferas e o número de células por otoesferas presentes no cultivo em suspensão de células do órgão de Corti de cobaias e camundongos, cujo meio de cultura foi enriquecido com TGFα ou bFGF.
81
Tabela 5.1. Relação das conexinas descritas por interagir com os domínios PDZs das proteínas ZO-1 e a sequência dos últimos dez resíduos da região C-terminal de cada para a espécie Mus musculus.
93
Tabela 5.2. Composição dos três diferentes tampões de lise utilizados para o ensaio de precipitação por afinidade.
101
Tabela 5.3. Sequência dos dez aminoácidos que compõem a região C-terminal da Cx26 nas espécies Mus musculus e Homo sapiens.
106
Tabela 5.4. Relação dos 11 pares de fatias recortadas em geis de SDS-PAGE de concentrações diferentes referentes às bandas presentes no ensaio de interação por afinidade com a proteína de fusão GST-Cx26 e ausentes no ensaio com a GST.
113
Tabela 5.5. Relação do número de proteínas identificadas por meio da espectrometria de massas das 11 bandas isoladas em SDS-PAGE presentes apenas no ensaio de precipitação por afinidade com a proteína GST-Cx26 e das outras 11 bandas localizadas na mesma posição da canaleta vizinha correspondentes ao ensaio com a GST.
117

14
Tabela 5.6. Relação total das 49 proteínas identificadas por meio da espectrometria de massa após análises com o Scaffold 3 proteomic software das 11 bandas recortadas de géis SDS-PAGE presentes no ensaio de precipitação por afinidade com a proteína GST-Cx26.
118
Tabela 5.7. Relação das 23 proteínas candidatas a interagirem com a região C-terminal da Cx26.
122
Tabela 5.8.: Classificação funcional baseada na literatura científica das 22 proteínas candidatas em potencial a participarem do complexo molecular da Cx26.
140

15
Sites utilizados para Análises in silico
- DECIPHER: http://decipher.sanger.ac.uk/
- DGV - Database of Genomic Variants: http://projects.tcag.ca/variation/
- EMBL-EBI: http://www.ebi.ac.uk/
- Ensembl Genome Browser: http://www.ensembl.org/index.html
- ExPASy Proteomics server : http://expasy.org/
- Hereditary Hearing Loss Homepage-: http://hereditaryhearingloss.org/
- Kyte Doolittle Hydropathy:http://gcat.davidson.edu/rakarnik/KD.html
- MGI- Mouse Genome Informatic: http://www.informatics.jax.org/
- NCBI :http://www.ncbi.nlm.nih.gov/
- POLYPHEM-2: http://genetics.bwh.harvard.edu/pph2/
- PSORT: http://psort.hgc.jp/
- The Connexin-Deafness Homepage : http://davinci.crg.es/deafness/
- SWISSPROT: http://www.ebi.ac.uk/uniprot/
- UCSC Genome Bioinformatics: http://genome.ucsc.edu/

1
APRESENTAÇÃO
Estimou-se que 16% dos casos de surdez no Brasil, assim como 60% nos
países desenvolvidos, tenham causas genéticas. Desses, em 70% dos casos a surdez
é não-sindrômica. Dos casos hereditários, 80% têm herança autossômica recessiva.
A causa mais comum de surdez não-sindrômica de herança autossômica recessiva
são as mutações nos genes GJB2 e GJB6. Dentre os indivíduos com deficiência
auditiva recessiva associada ao gene GJB2, 10% a 50% apresentam uma única
mutação recessiva em heterozigose detectável por testes convencionais, frequência
muito superior à esperada com base na frequência de heterozigotos na população
geral.
Durante o projeto de mestrado que teve o objetivo de caracterizar os tipos e
estimar a frequência das mutações dos genes GJB2 e GJB6 (lócus DFNB1), que
codificam para as proteínas conexinas 26 e 30, respectivamente, estudamos uma
amostra de 300 indivíduos com perda auditiva e sem diagnóstico de síndrome
conhecida associada à surdez. Foram rastreadas as mutações c.35delG e c.167delT
no gene GJB2, del(GJB6-D13S1830) e del(GJB6-D13S1854) no gene GJB6, e
polimorfismos conformacionais de fita simples (SSCP) no gene GJB2, para posterior
identificação de mutações por sequenciamento. A mutação c.35delG no gene GJB2
foi a mais frequentemente detectada, presente em 37 indivíduos (12,4%), sendo 22
homozigotos e 15 heterozigotos. Entre os heterozigotos, em oito (53,3%) não foi
identificada uma segunda mutação no gene GJB2 ou no GJB6. Desde então, mais 8
indivíduos com situação semelhante foram identificados em nosso serviço de
aconselhamento genético, totalizando 16 casos.
A maioria dos trabalhos sobre mutações no gene GJB2 investigam sua região
codificadora e a região não codificadora foi pouco estudada. Alguns estudos já
revelaram mutações fora da região de código do gene que afetam o padrão de splicing
do RNA mensageiro ou o seu nível de expressão. O alto número de surdos
portadores de uma única mutação recessiva (sem a segunda mutação detectada)
sugere que a haplo-insuficiência no gene GJB2 possa interagir com outras mutações
no mesmo gene, no gene GJB6 vizinho ou até em outros genes.

2
A triagem de variantes patogênicas no gene GJB2 fora de sua região código,
bem como o estudo de grandes deleções, a exemplo das que ocorrem com o seu
gene vizinho, o GJB6, pode nos auxiliar a revelar os mecanismos moleculares que
relacionam os produtos desses dois genes, GJB2 e GJB6, além de poder explicar os
casos de pacientes surdos com uma única mutação detectada no gene GJB2. Para
dar continuidade aos estudos iniciados no mestrado e também contribuir para o
esclarecimento da patogênese da surdez de herança autossômica recessiva, o nosso
primeiro objetivo no doutorado foi a identificação de novos alelos patogênicos no
lócus DFNB1 (onde estão contidos os genes GJB2 e GJB6) que possam ser
responsáveis por surdez quando presentes com outros alelos patogênicos nos genes
GJB2 e GJB6, no material genético de pacientes surdos, previamente caracterizados
como portadores de uma única mutação recessiva patogênica.
Na orelha interna, mais especificamente na cóclea, o órgão de Corti, estrutura
responsável pela transdução do som em impulsos nervosos, é formado por dois
tipos celulares altamente diferenciados, as células ciliadas (internas e externas) e as
células de suporte. Uma característica marcante destas células é que, durante o
desenvolvimento embrionário, elas entram em diferenciação terminal e são mantidas
em quiescência mitótica ao longo da vida. Em aves, peixes e anfíbios, as células
ciliadas auditivas são capazes de se regenerarem após lesão celular como parte do
processo fisiológico de crescimento e manutenção tecidual, devido à proliferação e
transdiferenciação das células de suporte. Já em mamíferos, a perda das células
ciliadas auditivas é definitiva, representando uma das principais causas de surdez em
seres humanos. Recentemente, foi demonstrado que células ciliadas do órgão de
Corti podem ser obtidas a partir de células-tronco embrionárias e de células-tronco
adultas da orelha interna, da medula óssea e neurais dos camundongos. Essas células
são multipotentes e, em teoria, capazes de originar todos os tipos celulares da orelha
interna. Dada à extrema relevância do tema, o cultivo das células do órgão de Corti
em laboratório representa um passo fundamental, não somente para o
desenvolvimento de futuras terapias gênicas e celulares, mas também para
desvendar os mecanismos moleculares e celulares relacionados à fisiologia da
audição e, consequentemente, à sua perda. Assim, buscando realizar pesquisas

3
básicas sobre audição e alternativas ao tratamento da surdez foi formado, em 2007,
um grupo de pesquisa com integrantes do Instituto de Biociências da USP e do
Departamento de Oftalmologia e Otorrinolaringologia da Faculdade de Medicina da
USP, com o objetivo de estabelecer no Brasil condições para pesquisa básica e
clínica sobre audição, com ênfase no cultivo e no estudo de células do órgão de
Corti. Uma vez que a conexina 26, proteína codificada pelo GJB2, é expressa nas
células de suporte do órgão de Corti, o cultivo dessas células em laboratório
representa passo de fundamental importância em estudos básicos de identificação e
localização de proteínas, relacionadas à função auditiva. Desse modo, o segundo
objetivo do projeto de doutorado foi padronizar protocolos para cultivo e
diferenciação de células epiteliais do órgão de Corti de cobaias e camundongos, para
se criar, a médio prazo, condições experimentais fundamentais aos estudos
funcionais relacionados às conexinas e suas “parceiras” e, a longo prazo, ao
desenvolvimento de estratégias futuras de terapia celular e terapia gênica na
deficiência auditiva.
Diversos estudos têm sido realizados com o objetivo de demonstrar a
interação entre conexinas e outras proteínas, como as proteínas de zônula de
oclusão-1 (ZO-1 ou TJP1), as calmodulinas, as caderinas, algumas cinases e
proteínas do citoesqueleto. No entanto, até o momento, são poucas as proteínas
identificadas por interagirem com a Cx26. A identificação de proteínas que se
associam com a Cx26 amplia a base de nosso conhecimento funcional das conexinas
e indica novos genes candidatos como causa de surdez sindrômica e não-
sindrômica, que poderiam vir também a explicar os casos de pacientes surdos com
uma única mutação no gene GJB2. Estes casos poderiam ser explicados por herança
digênica, ou seja, interação gênica entre o loco da Cx26 e outro que codifique para
outra proteína, que se associa a ela física e funcionalmente. Desse modo, o terceiro
objetivo do nosso estudo foi identificar proteínas que interagem com a Cx26, em
diferentes tecidos que a expressam e também nas células de suporte do órgão de
Corti. Desta forma, pretende-se aprofundar o conhecimento sobre as vias
funcionais relacionadas à audição.

4
RESUMO
A causa mais frequente de surdez de herança autossômica recessiva são as
mutações no lócus DFNB1, onde estão os genes GJB2 e GJB6. Dentre os
indivíduos com deficiência auditiva associada a esse lócus, 10% a 50% apresentam
uma única mutação recessiva no gene GJB2, frequência muito superior à esperada
em função da frequência de heterozigotos na população geral. Apesar de alguns
desses casos terem sido elucidados após a identificação de grandes deleções no gene
GJB6 ou nas suas proximidades, a existência de muitos indivíduos com uma única
mutação patogênica no gene GJB2 sugere que a haplo-insuficiência nesse gene possa
interagir com outras mutações no mesmo gene, no gene GJB6 vizinho, ou até em
outros genes.
O objetivo desse estudo foi identificar novos alelos patogênicos, novas
proteínas e novos genes que interagem com o lócus DFNB1, do ponto de vista
molecular e celular, e que possam ser responsáveis por surdez de herança
autossômica recessiva. Desse modo, pretendemos contribuir para o esclarecimento
da patogênese da surdez de herança autossômica recessiva. Nesse trabalho, três
tipos de estudos foram realizados, com metodologias próprias.
Na primeira parte, buscamos identificar novos alelos patogênicos no lócus
DFNB1 que poderiam ser responsáveis por surdez quando presentes em
heterozigose composta com outros alelos patogênicos nos genes GJB2 e GJB6. Foi
realizada a análise do DNA de 16 pacientes surdos portadores de uma única
mutação patogênica em um desses dois genes por meio: (i) do sequenciamento das
regiões codificadora, promotora e doadora de splicing (intron 1) do gene GJB2, (ii) da
triagem de uma deleção de 200 kb localizada a 130 kb da proximidade distal da
região 5’ do gene GJB6 e (iii) da pesquisa de variações no número de cópias de um
ou mais exons dos genes GJB2, GJB6, GJB3 e WFS1 por MLPA (Multiplex Ligation-
dependent Probe Amplification). Detectamos uma segunda mutação provavelmente
patogênica em dois dos 16 pacientes heterozigotos: em um deles, a mutação p.L76P
(c.C227T) foi identificada na região de código do gene GJB2 e foi por nós descrita

5
pela primeira vez; no segundo caso, uma duplicação (0,4-1,2Kb) que inclui a região
de código do gene GJB2 foi detectada, também inédita na literatura.
Na segunda parte, tivemos como objetivo obter um modelo experimental
para estudos funcionais in vitro da proteína codificada pelo gene GJB2, a conexina
26, em seu local de expressão que são as células de suporte do órgão de Corti.
Padronizamos o cultivo in vitro de células progenitoras do órgão de Corti de
camundongos e de cobaias e conseguimos obter a diferenciação in vitro das
otoesferas dos camundongos em células que expressam marcadores de células
ciliadas (Miosina VIIa e Jagged2) e de células de suporte (p27kip e Jagged1).
Por fim, na terceira parte, buscamos por proteínas que interagem com a
conexina 26 por meio de ensaios de precipitação por afinidade. Para isso,
produzimos clones recombinantes de uma proteína de fusão GST-Cx26 e de uma
proteína controle (GST), e realizamos sua expressão in vitro em bactérias E.coli B21.
Ensaios de precipitação por afinidade entre a proteína de fusão GST-Cx26 ou GST
sozinha e proteínas extraídas de cérebro ou fígado de camundongos foram
realizados em diferentes condições. A identificação e a análise das proteínas
presentes em bandas de SDS-PAGE, obtidas no ensaio de precipitação com a
proteína de fusão GST-Cx26 e ausentes no ensaio com a GST, foi realizada por
espectrometria de massas. Identificamos um total de 49 proteínas candidatas a
interagirem com a região C-terminal da Cx26. Realizamos diversas análises in silico e
em literatura específica e após exclusão de candidatas por: (i) redundância de
representação no ensaio GST-Cx26, (ii) diferença entre a massa molecular esperada
e a obtida, (iii) precipitação inespecífica e (iv) localização subcelular incompatível
com a conexina 26, selecionamos um total de 22 proteínas candidatas a interagirem
com a região C-terminal da conexina 26, para estudos futuros. A confimação da
interação entre essas 22 proteínas e a conexina 26 é desejável por meio de estudos
de co-localização e imuno-coprecipitação.

6
ABSTRACT
The most frequent causes of nonsyndromic recessive hearing loss are
mutations in locus DFNB1, in the GJB2 and GJB6 genes. Among the individuals
with hearing loss with mutations in this locus, 10% to 50% present a single recessive
mutation in the GJB2 gene, frequency much higher than expected taking into
account the frequency of heterozygotes in the general population. Although some
of these cases have been elucidated after the identification of large deletions in GJB6
or its surrounding regions, the existence of many individuals with a single
pathogenic mutation in the GJB2 gene suggests that haplo-insufficiency of this gene
may interact with other types of mutations in the same gene, in the neighbor gene
GJB6, or even in other genes.
The aim of this study was to identify new pathogenic alleles, proteins and
genes that interact with the locus DFNB1, from the molecular and cellular
perspective, and that may be responsible for autosomal recessive deafness. Thus, we
aimed to contribute to the understanding of the pathogenesis of autosomal recessive
deafness. In this work, three different types of studies were performed, each one
with a particular methodology.
In the first part, we searched for new pathogenic alleles in the locus DFNB1
that could be responsible for deafness, when present in compound heterozygosis
with other pathogenic alleles in GJB2 and GJB6 genes. We performed DNA analysis
in samples from 16 deaf patients, carriers of a single pathogenic mutation in one of
these two genes by: (i) sequencing the coding, promoter and splice donor (intron 1)
regions of the GJB2 gene, (ii) screening for a deletion of 200 kb located 130 kb
upstream from GJB6 gene and (iii) investigating copy number variations in of one or
more exons of the genes GJB2, GJB6, GJB3 and WFS1 by MLPA (Multiplex
Ligation-dependent Probe Amplification). We detected a second mutation, probably
pathogenic, in two of the 16 heterozygous patients: in one case, the p.L76P
(c.C227T) mutation was identified in the coding region of the GJB2 gene and was

7
firstly described by us; in the second case, a novel duplication (0.4 - 1.2 Mb) that
includes the coding region of the GJB2 gene was detected.
In the second part, our objective was to obtain an experimental model for in
vitro functional studies of the protein encoded by the GJB2 gene, connexin 26, in its
site of expression, that is, in the supporting cells of the organ of Corti. We
standardized the culturing of guinea pigs and mice progenitor cells of organ of
Corti. We were also able to induce differentiation of mice’s otospheres into cells
that express markers of hair (myosin VIIa and Jagged2) and supporting cells (p27kip
and Jagged1).
Finally, we searched for connexin 26 interacting proteins by pull-down
assays. Recombinant clones expressing a fusion protein GST-Cx26 and a control
protein (GST) were produced, so that in vitro expression in E. coli B21 could be
performed. Pull-down experiments, perfomed with fusion protein GST-Cx26 or
GST alone, and with proteins from mice brain or liver extracts were done under
several different conditions. The identification and analysis of proteins present in
SDS-PAGE bands in experiments performed with the fusion protein GST-Cx26,
and absent in the GST assay, were performed by mass spectrometry. We identified a
total of 49 candidate proteins for interaction with the C-terminal region of Cx26. In
silico analyses performed in several databases and search in the literature allowed
exclusion of candidates by: (i) redundancy of representation in the GST-Cx26
experiments; (ii) discrepancy between the expected and the obtained molecular
weight; (iii) nonspecific precipitation and (iv) subcellular localization incompatible
with connexin 26 localization. Summing up, we selected a total of 22 candidate
proteins to interact with the C-terminal region of connexin 26. Confirmation of the
interaction between these proteins and connexin 26 is planned to be performed by
co-localization studies and by immuno-coprecipitation.

8
CAPÍTULO 1

9
CAPÍTULO 1 Revisão Bibliográfica
1.1. Introdução
Os termos “perda” ou “deficiência auditiva” são comumente utilizados para
se referir a qualquer déficit ou comprometimento auditivo. Já o termo “surdez”
implica em um comprometimento auditivo geralmente grave ou profundo (Smith e
col., 2011). A OMS (Organização Mundial da Saúde) considera a surdez leve como
capacitante e as demais incapacitantes (In: http://www.who.int/en/, 2011).
Utilizaremos neste trabalho, porém, os três termos como sinônimos, do mesmo
modo que esses termos têm sido tratados na maioria dos trabalhos de genética, pois
muitas vezes perdas auditivas com a mesma etiologia genética exibem gravidade de
manifestação muito variável.
Em países desenvolvidos, a taxa de perda auditiva congênita permanente
entre recém-nascidos varia de 1 a 3 por 1000 nascidos vivos (Nelson, Bougatsos e
Nygren, 2008). No Brasil, foi estimado que a cada mil nascimentos, cerca de quatro
crianças apresentam perda auditiva de grave a profunda, frequência maior que nos
países desenvolvidos, provavelmente devido a uma maior contribuição dos fatores
ambientais (Braga e col., 1999, Piatto e Maniglia, 2001). Segundo estimativa da
OMS, 10% das pessoas de qualquer população têm algum tipo de deficiência, sendo
que desses, 1,5% são portadores de deficiência auditiva. Resultados do Censo 2000
(IBGE - Fundação Instituto Brasileiro de Geografia e Estatística), em uma
população de aproximadamente 170 milhões, mostraram que 14,5% da população
total (24,5 milhões de brasileiros) apresentam algum tipo de incapacidade. Dentre
estes, os portadores de deficiência auditiva representam 16,7%, sendo essa a terceira
deficiência mais frequente e acometendo 3,4% da população total (5,7 milhões de
brasileiros) (In: http://www.ibge.gov.br/2011).
A surdez é clinicamente muito heterogênea. Sua apresentação difere quanto à
idade de manifestação, gravidade e lado do acometimento. A perda auditiva de

10
origem sensorioneural é o tipo mais frequente e pode decorrer da disfunção do
epitélio sensorial do órgão de Corti, dos neurônios do gânglio espiral da orelha
interna, do nervo auditivo, das vias centrais no tronco encefálico ou encéfalo
(Raphael e Altschuler 2003; Atar e Avraham 2005). Ruído excessivo, infecções,
doenças auto-imunes, fármacos e a própria idade podem levar à degeneração de
células ciliadas do órgão de Corti, assim como mutações em diversos genes (Zippora
e col., 2006), o que leva à deficiência auditiva.
1.2. Fisiologia da audição
A audição é a capacidade de detectar e interpretar as vibrações mecânicas que
se propagam pelo ar ou pela água por meio de ondas, que são chamadas de som, e
que resultam de variações na pressão do ar. Ela depende de um sistema complexo e
altamente desenvolvido capaz de assimilar e responder aos mais diversos estímulos
ambientais. Viabiliza nossa interação com o meio, sendo responsável por uma das
mais importantes, únicas e complexas características da espécie humana, a
comunicação. Este sistema depende de estruturas sensoriais altamente
especializadas, com características similares a neurônios, capazes de converter a
onda sonora mecânica em estímulo elétrico, transmitindo-o aos circuitos neurais
centrais e interpretando-o do ponto de vista cognitivo como estímulo auditivo
específico (Smith e col., 2011).
A orelha humana é capaz de detectar variações na pressão do ar caso estas
estejam dentro da faixa de frequências entre 20Hz e 20kHz. Ela está dividida em
orelha externa, média e interna. A orelha externa coleta as ondas sonoras
provenientes de uma grande área, concentrando-as no tímpano, que constitui a
fronteira entre a orelha externa e a orelha média. A orelha média é a cavidade cheia
de ar no osso temporal na qual residem os ossículos martelo, bigorna e estribo.
Devido ao acoplamento mecânico do tímpano e dos ossículos, a energia de vibração
do tímpano em resposta às ondas sonoras é conduzida por eles até a orelha interna.
A orelha interna é a estrutura onde estão o vestíbulo, que capta os sinais de
aceleração e velocidade da cabeça no espaço contribuindo para o equilíbrio, e a

11
cóclea, que converte os estímulos sonoros em sinais nervosos ou elétricos que serão
interpretados gerando a percepção auditiva (Figura 1.1). A cóclea é um canal ósseo-
membranoso em forma de caracol, dividido em três dutos preenchidos por fluidos:
a escala vestibular, a escala timpânica e a escala média ou duto coclear (Figuras 1.1 e
1.2). A diferença de composição iônica entre a endolinfa, fluido que preenche o
compartimento central da cóclea onde está o órgão de Corti, e a perilinfa, fluido dos
outros dois compartimentos, gera o potencial endococlear, que é de fundamental
importância para a transdução do estímulo sonoro. A perilinfa é caracterizada por
uma alta concentração de Na+ e baixa concentração de K+, semelhante a outros
fluidos extracelulares. Já a endolinfa é um fluido com alta concentração de K+ e
baixa de Na+ ou seja, com composição iônica distinta da perilinfa e semelhante à do
citosol. A estria vascular, no duto coclear, tem fundamental importância no
potencial endococlear, pois ela participa da reciclagem de potássio no órgão de Corti
e secreta a endolinfa (Figura 1.2) (Junqueira & Carneiro, 1999).
1.2.1. O órgão de Corti
O órgão de Corti, situado sobre a membrana basilar, é constituído por células
de suporte não sensoriais e por células ciliadas sensoriais. A porção apical das células
ciliadas sensoriais contém projeções especializadas ricas em actina denominadas
estereocílios. Existem dois tipos de células ciliadas, as externas e as internas. As
externas ocorrem em maior número, cerca de três vezes maior que as internas. Os
dois grupos de células diferem tanto na forma quanto na função. Enquanto as
células ciliadas internas desempenham a função da transdução do som em sinais
nervosos, as externas, que apresentam tanto elementos sensoriais quanto motores,
contribuem para uma maior sensibilidade e discriminação das frequências por meio
da amplificação da recepção do som. A membrana tectória apresenta-se conectada
às células ciliadas externas por meio dos estereocílios mais longos e permanece em
estreita proximidade em relação aos estereocílios das células ciliadas internas (Figura
1.2) (Junqueira & Carneiro, 1999).

12
Figura 1.1. Esquema representando o aparelho auditivo humano: orelha externa (E), orelha média (M) e orelha interna (I). Figura modificada da página Promenade round the cochlea Home Page (Pujol e col., 2011).
Figura 1.2. Secção transversal de um dos giros da cóclea, mostrando sua divisão em três compartimentos longitudinais: as escalas vestibular, timpânica e média ou duto coclear. O circulo em vermelho delimita o órgão de Corti no duto coclear (Figura modificada da página http://ccrma.stanford.edu, 2011).
Quando o som é captado, os fluidos se movem pela escala média e fazem
vibrar a membrana basilar e a membrana tectória. As vibrações são o gatilho para a
deflexão dos estereocílios, permitindo o influxo de íons potássio por meio da
abertura de canais que irão despolarizar a membrana das células ciliadas internas.
Essa despolarização provoca a liberação de neurotransmissores que irão

13
desencadear o impulso nervoso e assim transmitir a informação ao cérebro via
nervo vestíbulo-coclear (oitavo par craniano) (Dror e Avraham, 2009).
1.2.1.1. Desenvolvimento, proliferação e diferenciação celular no órgão
de Corti
O desenvolvimento do órgão de Corti resulta na diferenciação de dois tipos
de células ciliadas (internas e externas) e quatro tipos de células de suporte (Deiters,
Hensen, Claudius e pilares). Alguns autores classificam as células de suporte em até
doze subtipos. Essa classificação esta baseada no posicionamento dessas células no
órgão de Corti. Para esse estudo, dada a sua proximidade com as células ciliadas,
iremos incluir apenas dois subtipos; borders e phalengeals (Figura 1.3) (Smeti e col.,
2011). Cada um destes tipos celulares possui morfologia distinta e contribui de
modo único para as complexas propriedades estruturais e funcionais do órgão de
Corti.
Figura 1.3. Esquema ampliado do órgão de Corti, mostrando as células ciliadas internas e externas e as células de suporte (os quatro tipos: Deiters, Hensen, Claudius e pilares e os dois subtipos: borders e phalangeals (Figura modificada de Rio e col., 2002).
Uma característica marcante das células ciliadas é que, durante o
desenvolvimento embrionário, elas entram em diferenciação terminal e são mantidas
ativamente em quiescência mitótica pelo aumento da expressão de inibidores da
progressão do ciclo celular (Baumgartner e Harper, 2003). Estas células
permanecem pós-mitóticas ao longo da vida e, em mamíferos, a perda de células

14
ciliadas auditivas é definitiva, representando uma das principais causas de surdez em
seres humanos (Chen e Segil, 1999).
Além das células ciliadas sensoriais, o órgão de Corti também contém células
não-sensoriais denominadas células de suporte. Essas células, como o próprio nome
diz, servem de apoio às células ciliadas. Em aves, peixes e anfíbios, após a morte das
células ciliadas, as células de suporte não-sensoriais recebem sinais moleculares que
desencadeiam proliferação e/ou transdiferenciação em células ciliadas imaturas. Há
posteriormente, reinervação dessas novas células ciliadas, ocorrendo a aferência
sensorial e consequentemente, a manutenção da função auditiva (Kelley, 2006).
Durante o desenvolvimento da orelha interna, o primórdio sensorial e as
células que darão origem ao órgão de Corti saem do ciclo celular (mitose) em uma
onda relativamente sincrônica, tornando-se pós-mitóticas e constituindo a zona de
células não proliferantes (ZNPC, zone of nonproliferating cells), localizada na parede
dorsal do duto coclear do embrião. A mitose terminal coincide temporalmente com
a formação da ZNPC e se propaga mediada pelo aumento progressivo do gradiente
de expressão do p27Kip1, um inibidor de cinase dependente de ciclina (CKI, cyclin
dependent kinase inhibitors). Portanto, o p27Kip1 é um marcador molecular do órgão de
Corti rudimentar, o que permite sua identificação antes de qualquer outra alteração
bioquímica ou molecular e sinaliza precocemente a região do otocisto que dará
origem ao órgão sensorial maduro (Kelley, 2006).
Estudos de expressão gênica usando cócleas de camundongos evidenciaram
que inúmeros genes reguladores negativos da proliferação celular, além do p27kip1,
sofrem aumento de sua expressão nos estágios mais tardios do desenvolvimento
embrionário. Esses incluem outros CIK, como p19ink4d, p21 e genes supressores
tumorais, membros da família das pocket protein, como por exemplo o produto do
gene de suscetibilidade ao retinoblastoma (pRb, product of the retinoblastoma susceptibility
gene) (Kelley, 2006). Esse aumento de expressão coincide com a saída do ciclo celular,
o estabelecimento e a manutenção do estado de quiescência do epitélio sensorial
coclear (Figura 1.4) (Chen, 2003 e 2006).

15
É na ZNPC que o padrão de diferenciação celular dará origem ao mosaico
de células ciliadas e de suporte do órgão de Corti (Chen, 2003). Os sinais
moleculares indutores do aumento da expressão do p27Kip1 nas células progenitoras,
que darão origem tanto às células ciliadas quanto às células de suporte, ainda não
são bem conhecidos. A expressão do pRb inicia-se após a formação da ZNPC e
coincide com a onda de diferenciação celular, seguindo gradiente que se propaga da
base para o ápice da cóclea (Kelley, 2006). Na ZNPC, a determinação do fenótipo
celular ocorre após a saída das células-tronco sensoriais do ciclo celular de
proliferação. A diferenciação em células ciliadas é determinada por influência do
fator de transcrição do tipo hélice-alça-hélice básico (bHLH, basic helix-loop-helix),
também conhecido como Atoh-1 ou Math-1. O fenótipo de células de suporte é
sinalizado pela via do fator denominado Notch (Figura 1.4) (Chen, 2006).
À medida que a diferenciação progride no órgão de Corti, as expressões do
p27Kip1 e do pRb se tornam segregadas, pois o pRb passa a ser expresso apenas nas
células ciliadas e o p27Kip1 fica restrito às células de suporte. Esta segregação resulta
da queda na expressão do p27Kip1 nas células progenitoras, que darão origem às
células ciliadas e coincide com o aumento da expressão da miosina VIIa, marcador
molecular precoce e exclusivo destas células, sinalizando o início do gradiente de
diferenciação e um padrão de expressão mutuamente excludente desses dois
marcadores (Chen 2003, Kelley, 2006). O CIK que mantém a quiescência das células
ciliadas após a diferenciação é o p19Ink4d (Chen e col., 2003). Por outro lado, o
p27Kip1 continua a ser expresso nas células progenitoras que darão origem às células
de suporte, o que determina o fenótipo destas no órgão de Corti maduro, além de
manter sua quiescência após diferenciação (Baumgartner e Harper, 2003).
Apesar do Atho1/Math-1 ser fundamental na determinação do número e na
disposição das células ciliadas internas e externas na cóclea de mamíferos, os fatores
que regulam sua expressão espacial e temporal ainda são desconhecidos. Outros
fatores de transcrição bHLH podem influenciar a determinação do fenótipo celular,
agindo como reguladores negativos.

16
À medida que o desenvolvimento progride, ocorre inibição da expressão de
ID (ID - inhibitors of differentiation and DNA binding) nas células progenitoras que irão
se diferenciar em células ciliadas, o que permite a atividade do Math-1 nestas células.
Naquelas em que a expressão do ID é mantida, a atividade do Math-1 é inibida,
assim como o surgimento de células ciliadas também é inibido e surgem assim as
células de suporte. Isto sugere um papel indireto dos ID na regulação da expressão
do Math-1 e no fenótipo das células ciliadas (Kelley, 2006).
Figura 1.4. Diferenciação das células progenitoras em células ciliadas e de suporte. Na orelha interna de camundongos, as células progenitoras que passam a expressar p27kip1 podem diferenciar-se em células ciliadas ou de suporte, dependendo se ocorrer expressão de pRb ou p27kip1. A expressão do Math-1 determina o desenvolvimento das células ciliadas e é controlado pela expressão da proteína ID, cuja presença determina o desenvolvimento das células de suporte. Genes expressos na via Notch contribuem também ao fenótipo de células de suporte: os ligantes de Notch, Jag2 e Delta1 (Dlt1) ativam Notch, que aumenta os níveis de transcrição dos genes Hes1 e Hes5 (Figura modificada de Kelley, 2006).
As células ciliadas em diferenciação geram sinais inibitórios laterais, por meio
da expressão de dois ligantes de Notch, Jagged2 e Delta1, que ativam Notch1 e
aumentam a transcrição de dois genes alvos de Notch, Hes1 e Hes5, nas células
vizinhas que irão se desenvolver como células de suporte (Figura 1.4). A expressão
simultânea de ID, com Hes1 ou Hes5, nas células progenitoras inibe a transcrição do
Math-1 e induz o fenótipo de células de suporte. Portanto, a diferenciação das
células de suporte é dependente de sinais indutores gerados pelas células ciliadas

17
adjacentes (Jones e col., 2006). Hes1 passa a ser expresso durante o período de
diferenciação das células ciliadas, sua expressão se eleva ao nascimento e se mantém
na vida adulta, em regiões adjacentes a estas células (Zheng e col., 2000).
1.2.1.2. A regeneração das células ciliadas como indicativo da presença
de células-tronco na orelha interna
A maioria das perdas auditivas sensorioneurais, adquiridas ou congênitas,
decorre da perda irreversível das células ciliadas cocleares ou de seus neurônios. Até
pouco tempo acreditava-se que os vertebrados nasciam com um estoque limitado de
células ciliadas, e, por isto, sua lesão era sempre permanente. Este conceito sofreu
uma reviravolta na década de 1980, com a descoberta de que a maioria dos
vertebrados, incluindo aves, peixes e anfíbios, é capaz de regenerar as células ciliadas
do órgão de Corti e do vestíbulo, tanto após lesão celular ou como parte do
processo fisiológico de crescimento e manutenção tecidual. Nestas espécies, a lesão
das células ciliadas não é definitiva, e sim seguida por reparo espontâneo e funcional.
Esse fato é oposto ao observado usualmente em mamíferos, incluindo a espécie
humana (Corwin e Cotanche, 1988; Ryals e Rubel, 1988). No entanto, já foi
demonstrado que a regeneração das células ciliadas em resposta a ototoxicidade
induzida por aminoglicosídeos ocorre no epitélio sensorial vestibular de mamíferos
adultos (cobaias e camundongos), embora de forma muito menos impressionante
do que a vista em aves (Forge e col., 1993; Warchol e col., 1993).
Estudos preliminares em aves têm demonstrado que a regeneração das
células ciliadas ocorre por meio da re-entrada das células de suporte adjacentes no
ciclo celular e subsequente divisão assimétrica, gerando novas células ciliadas e
novas células de suporte. Essa regeneração parece ocorrer também pela conversão
fenotípica, também conhecida como transdiferenciação, na qual células de suporte
são convertidas em células ciliadas sem que haja mitose (Walchol e Corwin, 1996;
Roberson e col., 2004; Brigande e Heller, 2009). Ao que parece, as células de
suporte ou, pelo menos, uma subpopulação de células de suporte atuam, na verdade,

18
como células-tronco específicas do órgão de Corti, atuando como precursoras para
as células ciliadas (Edge e Chen, 2008; Groves, 2010).
A existência de outros sistemas sensoriais no corpo humano com capacidade
regenerativa e contendo população de células-tronco adultas quiescentes, capazes de
reparo após lesão aguda, como por exemplo, o neuroepitélio olfativo, somado ao
fato de que a divisão assimétrica das células de suporte para gerar novas células
ciliadas e cópias idênticas de si mesma é uma das características que definem as
células-tronco em geral, levou os pesquisadores a testarem o próprio órgão de Corti
da orelha interna como fonte de células precursoras (progenitoras e/ou células-
tronco) tanto para reparo biológico como para estudos relacionados à fisiologia
auditiva (Leung e col., 2007).
Em estudos relativamente recentes foi possível isolar e demonstrar a
presença de células-tronco na cóclea (órgão de Corti, gânglio em espiral e estria
vascular) de camundongos neonatos e no vestíbulo de camundongos adultos.
Nesses estudos, foi demonstrado que essas células são pluripotentes e com
capacidade de originar in vitro e in vivo diferentes tipos celulares, sendo que no caso
do vestíbulo estão incluídas células das linhagens ectodérmica, mesodérmica e
endodérmica (Li e col., 2003, Senn e col., 2007, Oshima e col., 2007). No entanto, as
células-tronco da cóclea estão presentes somente até a terceira semana de vida
enquanto que as do vestíbulo persistem no camundongo adulto (Li e col., 2003,
Senn e col., 2007, Oshima e col., 2007). Em humanos, a presença de células
progenitoras neurais já foi demonstrada no gânglio espiral de onde emerge o nervo
vestíbulo-coclear. Entretanto, ainda faltam evidências da existência de células-tronco
na cóclea humana (Rask-Andersen e col., 2005).
O cultivo em suspensão de células dissociadas do órgão de Corti em
condições não aderentes leva à formação de colônias de células flutuantes,
denominadas “otoesferas”, que, quando dissociadas, são capazes de se auto-renovar,
proliferar e expressar marcadores prováveis de células progenitoras como nestina,
Sox2 (células progenitoras) e Abcg2, considerado um marcador universal de células-
tronco. Além disso, as células provenientes dessas otoesferas são capazes de se

19
diferenciar in vitro em células que expressam marcadores para células ciliadas, células
de suporte e neurônios (Li e col., 2003; Zhai e col., 2005; White e col., 2006;
Oshima e col., 2007; Savary e col., 2007; Senn e col., 2007; Zhang e col., 2007;
Savary e col., 2008).
Dada a extrema relevância do tema, diversos estudos têm sido realizados
com o objetivo de descobrir quais os principais mecanismos reguladores do reparo
das células da audição e porque este fenômeno é diferente em mamíferos quando se
compara aos outros vertebrados. Entre os fatores aparentemente responsáveis,
destacam-se o grau de desenvolvimento do sistema responsável pela audição e os
fatores relacionados ao número de células-tronco e a sua capacidade de proliferação
(Li e col., 2003, Oshima e col, 2007).
1.3. Surdez de etiologia genética
O sistema auditivo é complexo e especializado e requer interação entre a
função de diversos genes e proteínas para funcionar normalmente. Existem
evidências da ação de mecanismos gênicos em cascata no desenvolvimento do
sistema auditivo humano. O estudo da surdez genética tem auxiliado a compreensão
de como a orelha interna funciona em nível molecular, mostrando que muitos genes
codificam para bombas ou canais iônicos, fatores de transcrição, componentes da
matriz extracelular, componentes do citoesqueleto celular, moléculas
transportadoras (por exemplo, proteínas essenciais para o tráfego de vesículas
sinápticas), moléculas sinalizadoras, receptores, reguladores da proliferação celular,
enzimas ou junções do tipo fenda para comunicação intercelular (Eisena e Ryugoa,
2007).
Além do enorme número de genes relacionados com a surdez, outros fatores
contribuem para aumentar a complexidade da genética da audição: em um mesmo
gene, podem existir mutações que estão associadas a diferentes padrões de herança.
Por exemplo, mutações nos genes GJB2, GJB6, MYO7A, TECTA e TMC1 podem
ter efeito dominante ou recessivo. Estudos moleculares também têm demonstrado
que diferentes mutações no mesmo gene podem causar surdez sindrômica ou não-

20
sindrômica, como é o caso dos genes SLC26A4, USH1C e WFS1 (Smith e col.,
2010). Dessa maneira, fica evidente a grande heterogeneidade genética da surdez.
Além disso, a surdez também pode resultar de mecanismo multifatorial, ou
seja, resultar da interação de vários fatores genéticos e ambientais. Um exemplo é a
presbiacusia, que se caracteriza pela diminuição da capacidade auditiva relacionada
ao envelhecimento. Até o momento, poucos genes foram associados a esse tipo de
surdez. Não se sabe ao certo se genes relacionados a formas monogênicas de surdez
também podem estar associados à surdez de mecanismo multifatorial (Hilgert e col.,
2009).
No Brasil, foi estimado, na década de 1990, que as causas hereditárias
contribuem com cerca de 16% dos casos de deficiência auditiva (Braga e col., 1999).
1.4. Surdez não-sindrômica
Apesar de existirem mais de 400 doenças ou síndromes que incluem a surdez
como um de seus sinais clínicos, 70% dos casos de surdez hereditária são não-
sindrômicos (Keats e Berlin, 1999). Algumas das principais síndromes que
apresentam surdez como um de seus sinais mais característicos são: Waardenburg
(com defeitos de pigmentação e telecanto), Treacher Collins (com anormalidades
craniofaciais), Usher (com retinose pigmentar), Pendred (com bócio) e Alport (com
defeitos renais).
Dentre os casos de surdez não-sindrômica, o padrão de herança autossômico
recessivo é o mais frequentemente encontrado (75-80% dos casos genéticos), sendo
seguido pelos casos de herança autossômica dominante (20%) e apenas 1 a 1,5%
com herança ligada ao cromossomo X. As mutações mitocondriais contribuem em
pelo menos 1% dos casos de surdez não sindrômica (Smith e col., 2011) e
alcançaram 2% em um estudo realizado na população brasileira (Abreu-Silva e col.,
2006).
Os diferentes loci ou regiões candidatas a conterem um ou mais genes
responsáveis por surdez não-sindrômica são designados DFN (do inglês DeaFNess)

21
e numerados seguindo a ordem de descoberta. Os loci que supostamente contêm
genes de surdez de herança autossômica recessiva são denominados DFNB, aqueles
com padrão autossômico dominante de DFNA e os loci que estão no cromossomo
X são designados DFNX. Segundo Van Camp e Smith (2011), até o momento, 61
loci de surdez não-sindrômica de herança autossômica recessiva foram mapeados
com a identificação de 36 genes; 49 loci foram mapeados com 24 genes
identificados para a herança autossômica dominante e, para a herança ligada ao X,
cinco loci foram mapeados com a identificação de três genes. Adicionalmente, dois
loci identificados como genes modificadores e um lócus no cromossomo Y também
já foram mapeados. Em relação à herança mitocondrial, dois principais genes foram
identificados e muitas mutações diferentes nesses genes foram relacionadas à surdez
hereditária não-sindrômica.
1.5. Surdez não-sindrômica de herança autossômica recessiva
e o lócus DFNB1
Cerca de 80% dos casos de surdez não-sindrômica apresentam herança
autossômica recessiva. As perdas auditivas com esse padrão de herança geralmente
são sensorioneurais, de manifestação pré-lingual, de graves a profundas,
estacionárias e geralmente atingem todas as frequências (Keats e Berlin, 1999).
Apesar do grande número de loci mapeados, o DFNB1 (13q11-12), que
contém o gene da conexina 26 (Cx26, gene GJB2), é o mais importante, já que mais
de 50% dos casos de surdez não-sindrômica autossômica recessiva são devidos às
mutações nesse gene. Mais de 100 mutações diferentes já foram descritas na região
codificadora do gene GJB2, sendo a c.35delG a mutação predominante, presente em
até 75% dos casos (Denoyelle e col., 1997; Sundstrom e col., 1999; Connexin deafness
homepage: http://davinci.crg.es/deafness/, 2011). A c.35delG se caracteriza pela
deleção de uma base guanina (G) em uma sequência de seis guaninas, que resulta em
mutação de quadro de leitura e possivelmente leva à tradução de uma proteína
truncada com apenas treze aminoácidos.

22
A frequência de heterozigotos com a mutação c.35delG na população
brasileira foi estimada em 1% (Sartorato e col., 2000); todavia essa frequência pode
atingir até 3,5% em algumas populações de origem mediterrânea, como a população
grega (Gasparini e col., 2000).
A alta frequência da mutação c.35delG na população e a variabilidade do
quadro clínico a ela associado tornam mandatória sua triagem em todos os
indivíduos com deficiência auditiva sensorioneural de causa desconhecida. Essa
indicação é válida tanto para os casos isolados como para os familiais, ainda que o
padrão de herança aparente não seja o recessivo. Várias mutações detectadas no
gene GJB2 da conexina 26 parecem ser características de certos grupos
populacionais, como a c.35delG, comumentemente encontrada na população
caucasiana, principalmente da Europa e dos EUA, a c.167delT nos judeus askhenazi,
a c.235delC nos asiáticos e a c.T427C (p.R143W) nos africanos (Lerer e col., 2000;
Abe e col., 2000; Brobby e col., 1998).
O gene GJB2 é um gene pequeno, possui dois exons, mas o exon 1 não é
codificador. Apresenta uma região promotora altamente conservada entre as
diferentes espécies (Figura 1.5A) (Kiang e col., 1997). A conexina 26, proteína
codificada por esse gene, possui 226 aminoácidos e massa molecular aproximada de
26kDa. A maioria dos alelos mutados até agora descritos no gene GJB2 se
comportam como recessivos e acarretam surdez pré-lingual (Kenneson e col., 2002).
No entanto, algumas mutações no gene da conexina 26 têm sido descritas causando
surdez de herança dominante, caracterizando o lócus DFNA3.
Dentre os indivíduos com deficiência auditiva associada ao lócus DFNB1,
10% a 50% apresentam uma única mutação recessiva no gene GJB2, frequência esta
muito superior à esperada em função da frequência de heterozigotos na população
geral. O alto número de surdos portadores da mutação c.35delG (ou de outra
mutação patogênica) em heterozigose (sem a segunda mutação detectada) sugere
que a heterozigose no gene GJB2 possa interagir com outras mutações no mesmo
gene ou em outros genes, resultando em surdez. Diversos estudos têm sido
realizados com o objetivo de identificar uma segunda mutação patogênica nesse

23
gene ou em um gene diferente, que em associação à mutação no gene GJB2 levaria à
perda auditiva.
O gene vizinho ao GJB2 é o GJB6, que codifica a conexina 30, uma proteína
com 261 aminoácidos e massa molecular aproximada de 30Kda. Os genes GJB2 e
GJB6 estão na mesma região cromossômica, 13q11-12, a cerca de 35 kb um do
outro e expressam proteínas com 77% de identidade de sequência de aminoácidos
entre elas (Pallares-Ruiz e col., 2002).
Figura 1.5. Representação esquemática dos genes GJB2 e GJB6. Em A) representação do gene GJB2 (Genbank BC017048). Na figura podemos observar os dois exons, a região 5’ UTR que inclui o exon 1, a região codificadora, o intron 1 e a região 3’ UTR. Em B), representação esquemática do gene GJB6 (Genbank BC038934). Na figura podemos observar a região 5’UTR que contém os dois exons não codificantes, a região codificadora e a região 3’UTR.
O gene GJB6 apresenta uma organização mais complexa, com 6 exons,
sendo apenas o último exon codificador. Esse gene apresenta diferentes transcritos
com diferentes números de exons incluídos. A inclusão dos outros 5 exons não
codificadores irá depender do tipo de transcrito (Essenfelder e col., 2005). De
acordo os bancos de dados (Genbank e Ensembl), o transcrito de maior prevalência
do gene GJB6, nos diferentes tecidos incluindo a orelha interna, tem 3 exons, sendo
o último exon codificador (Figura 1.5.B). Ao contrário do gene GJB2, até o

24
momento, nenhuma mutação de ponto no gene GJB6 foi associada à surdez
recessiva não-sindrômica, apenas a formas de surdez sindrômica (In: Connexin
deafness homepage; OMIM 604418).
Em um estudo realizado com 33 indivíduos surdos previamente
caracterizados como portadores de uma única mutação patogênica no gene GJB2,
del Castillo e col. (2002) identificaram em 22 deles uma deleção de 309Kb no gene
GJB6, a 5’ do gene GJB2. Esta deleção, denominada del(GJB6-D13S1830), revelou-
se, depois da mutação c.35delG, como a mutação mais frequente entre afetados por
surdez pré-lingual na população espanhola. Posteriormente, del Castillo e col. (2005)
caracterizaram outra grande deleção de 232kb no gene GJB6 que foi nomeada
del(GJB6-D13S1854). Foi verificado que essas deleções removem todo ou parte do
gene GJB6 e acarretam surdez quando presentes em homozigose ou em
heterozigose composta com uma mutação recessiva no gene GJB2 (del Castillo e
col., 2002; del Castillo e col., 2005). Existe ainda um caso descrito de um paciente
com surdez portador de uma mutação no gene GJB2 (p.V84M) e de uma deleção de
pelo menos 920 kb que remove os genes GJB2 e GJB6 no cromossomo homólogo
(Feldmann e col., 2009). Mais recentemente, Wilch e col. (2010) descreveram uma
nova deleção no lócus DFNB1 que evidencia a existência de uma região regulatória
que controla a expressão dos genes GJB2 e GJB6. Essa deleção de 131,4 kb, cujo
ponto de quebra proximal dista mais de 100kb de GJB2 e GJB6, foi encontrada em
quatro indivíduos não aparentados heterozigotos com a mutação c.35delG e que
apresentavam níveis reduzidos de expressão do RNAm em ambos os alelos. Outra
deleção com tamanho estimado em 200kb em DFNB1, que não inclui as regiões de
código dos genes GJB2 e GJB6, foi encontrada em indivíduos heterozigotos com a
mutação c.35delG em GJB2, sugerindo que essa deleção deve eliminar um elemento
regulatório essencial à expressão de GJB2 na orelha interna (Del Castillo e col.,
2009). Na figura 1.6, está a representação esquemática das cinco deleções
identificadas no lócus DFNB1, responsáveis por surdez recessiva não-sindrômica
descritas na literatura até o momento.

25
Figura 1.6. Representação esquemática da região do lócus DFNB1, 13q12-11, onde estão
presentes os genes GJB2 e GJB6 e das deleções descritas nessa região e associadas a surdez não-sindrômica de herança autossômica recessiva. Sendo (1) del(GJB6-D13S1830), (2) del(GJB6-D13S1854), (3) deleção de 131,4 kb descrita por Wilch e col. (2010), (4) deleção de 200 kb descrita por del Castillo e col. (2009) e (5) deleção de >920kb descrita por Feldman e col. (2009). As extensões das deleções estão identificadas pelas barras horizontais em vermelho. A barra horizontal verde indica a região em comum as cinco deleções (Figura modificada de Feldmann e col., 2009).
Esses resultados mostram que o DFNB1 é um lócus complexo, já que
contêm dois genes, o GJB2 e o GJB6, ambos associados à deficiência auditiva. A
presença de duas mutações em genes diferentes leva à evidência de que a surdez
nesses indivíduos pode estar associada a uma mutação em um segmento regulatório
ou em uma região de controle de lócus regulando a co-expressão desses dois genes,
GJB2 e GJB6 (Common e col., 2005; Wilch e col., 2006 e 2010).
A pesquisa por mutações em outras regiões do gene GJB2, GJB6 ou fora da
sua região de código levou à identificação de duas mutações diferentes associadas à
surdez não-sindrômica de herança autossômica recessiva, a mutação IVS 1 +
1G>A, localizada no primeiro intron do gene GJB2, entre o exon 1 não codificador
e o exon 2, no sítio doador de splicing e a mutação -3438C>T na região promotora
do gene GJB2 (Denoyelle e col., 1999; Mattos e col, 2007).

26
1.6. As proteínas conexinas
1.6.1. Expressão e estrutura das conexinas
No genoma humano estão presentes os genes para 20 tipos de conexinas
diferentes e 21 tipos diferentes ocorrem no genoma dos camundongos
(Krutovskikh e Yamasaki, 2000; Marziano e col., 2003). O mesmo tipo de conexina
pode ser expresso em diferentes tipos celulares e um mesmo tipo celular pode
expressar conexinas diferentes (Tabela 1.1). Diversos estudos evidenciaram a
presença das proteínas Cx26 (gene GJB2) e Cx30 (gene GJB6) na orelha interna,
mais especificamente na cóclea (Kelsell e col., 1997; Grifa e col., 1999, Lautermann
e col., 1999). Na cóclea, as conexinas 26 e 30 se co-localizam nas células de suporte
do órgão de Corti, na região basal da estria vascular e nos fibrócitos do tipo 1 do
ligamento em espiral, sendo que nenhuma outra conexina foi detectada nesses tipos
celulares (Forge e col., 2003).
Além das conexinas 26 (gene GJB2) e 30 (gene GJB6), outras conexinas que
também são expressas na cóclea são as conexinas 31 (gene GJB3) e a conexina 43
(gene GJA1). Entretanto, somente os genes correspondentes às conexinas 26, 30 e
31 estão associados à surdez de herança autossômica recessiva e/ou dominante
(Van Camp e Smith, 2011).
As proteínas conexinas são divididas em três subfamílias α, β e γ, de acordo
com a similaridade da sequência de seus aminoácidos, sendo nomeadas pela sigla
“Cx”, seguida de sua massa molecular em kilodaltons (kDa), como por exemplo,
Cx26 e Cx30.
Todas as conexinas possuem uma estrutura em comum: quatro domínios
transmembrânicos ligados entre si por duas alças extracelulares e uma alça
citoplasmática, com os grupos amino (N-terminal) e carbóxi-terminais (C-terminal)
também citoplasmáticos. Variações nas sequências entre as diferentes proteínas
conexinas ocorrem principalmente no grupo carbóxi-terminal e na alça
citoplasmática, sendo os domínios transmembrânicos e as alças extracelulares
altamente conservados (Figura 1.7) (Bruzzone e col., 1996; Liu e col. 2000).

27
Tabela 1.1. Distribuição tecidual das conexinas (RNAm) em humanos (Cx hs) e camundongos (Cx mus) . (Modificado de Rackauskas, 2010)
Cx hs Cx mus Local de expressão
Cx23 Cx23 -
Cx25 -
Cx26 Cx26 cóclea, pele, glândula mamária, glândula salivar, fígado, útero, testículo, células da glia, pâncreas, pulmão, estômago, tireóide,
paratireóide, cérebro
Cx30 Cx30 cóclea, pele, cérebro, pulmão, útero, cristalino
Cx31.3 Cx29 oligodentrócitos, músculo esquelético, fígado, pâncreas, rins
Cx30.3 Cx30.3 pele, rins, blastócito
Cx31 Cx31 pele, cóclea, epitélio das vias aéreas, placenta, blastócito, rins, testículos, olhos
Cx31.1 Cx31.1 pele, testículos
Cx31.9 Cx30.2 coração e cérebro
Cx32 Cx32 Fígado, pele, células de Schwann, rins, baço, oligodentrócitos, pulmão, cérebro, glândula mamária, pâncreas, glândula salivar, testículos
Cx33 testículos
Cx36 Cx36 retina, pâncreas, cérebro
Cx37 Cx37 rins, pulmão, pele, endotélio, células musculares cardíacas, ovários,
Cx40 Cx40 endotélio, blastócitos, ovários, útero, pulmão, coração, fibras de Purkinje
Cx40.1 Cx39 -
Cx43 Cx43 34 tecidos e 86 tipos celulares (Laird, 2006)
Cx45 Cx45 células musculares cardíacas, blastócitos, pulmão, pele, coração, cérebro
Cx46 Cx46 pulmão, cristalino, células musculares cardíacas, células de Schwann
Cx47 Cx47 cérebro, oligodentrócitos
Cx50 Cx50 cristalino e células epiteliais da córnea
Cx59 -
Cx62 Cx57 retina, oócitos de camundongos
As conexinas são proteínas integrantes da membrana plasmática que se
hexamerizam para formar canais chamados conexons. Cada conexon pode conter

28
um único tipo de conexina se for homomérico, ou vários tipos de conexinas, se for
heteromérico. A união de dois conexons idênticos, um de cada célula, forma um
canal de junção celular homotípico. Um canal de junção celular heterotípico é
formado por conexons diferentes (Figura 1.7) (Spray e col., 2006). Cada conexon de
uma célula se alinha no espaço extracelular ao da célula adjacente, formando um
canal de junção do tipo fenda (gap junction). Esta é uma região especializada da
membrana plasmática de duas células vizinhas que as aproximam, sem se fundirem,
delimitando um espaço entre as membranas ou um intevalo, “gap”, de 2 - 3 nm. As
junções do tipo fenda são responsáveis pela regulação da passagem de íons
orgânicos e pequenas moléculas entre células epiteliais, o que facilita o fluxo de
sinais intercelulares específicos e consequentemente a cooperação metabólica entre
as células vizinhas de um mesmo tecido (Figura 1.7). Os diferentes tipos de
conexinas que irão formar o canal de junção comunicante proporcionam
características de permeabilidade únicas, o que é refletido nos tipos de metabólitos e
na sinalização celular que fluem através dele, assim como nas propriedades de
abertura e fechamento desses canais (gating) (Cottrell e Burt, 2005).
1.6.2. Funções das conexinas
Muito do que conhecemos sobre as conexinas na homeostasia dos diferentes
órgãos foi revelado por estudos de mutações em genes para as conexinas humanas
causando perda de função em uma variedade de doenças genéticas. Esse tipo de
estudo para o gene GJB2 nas diferentes formas de surdez tem associado à Cx26 a
vários aspectos da transmissão do som (Rabionet e col., 2000; Bruzzone e col, 2003).
Recentemente, por meio de análises de cristalografia da estrutura da Cx26, dois
trabalhos diferentes demonstraram pela primeira vez que os resíduos de
aminoácidos mais cruciais ao desenvolvimento da surdez hereditária sindrômica e
não-sindrômica estão localizados em regiões envolvidas em interações intra ou
intermoleculares ou em sítios de modificações pós-traducionais (Locke e col., 2009 e
Maeda e col., 2009).

29
Figura 1.7. Representação esquemática da estrutura das conexinas, dos conexons e dos canais de junção do tipo fenda. Em A) temos a representação da estrutura da conexina com seus quatro domínios transmembrânicos, duas alças extracelulares, uma alça citoplasmática, um domínio N-terminal e um C-terminal. Em B) organização dos conexons pela hexamerização das conexinas, que podem ser homoméricos quando formado pelo mesmo tipo de conexina ou heteroméricos se formado por conexinas diferentes. E em C) organização do canais de junção do tipo fenda, que quando for formado por dois conexons idênticos é chamado homotípico e quando for formado por dois conexons diferentes é heterotípico (Figura modificada das páginas http://www.nature.com, 2011 e http://commons.wikimedia.org , 2011).
Uma das funções dos canais de junção do tipo fenda na cóclea é regular o
transporte dos íons, principalmente os íons potássio na endolinfa. Os canais de

30
junção do tipo fenda fornecem um sistema único de separação, transferência e
reciclagem dos íons, sendo responsáveis pela manutenção dos altos níveis de
potássio na endolinfa, e, por conseguinte, do seu potencial endococlear, necessário
para a transdução de impulsos sonoros em impulsos elétricos. Hipóteses mais
antigas que relacionam as mutações do gene GJB2 às disfunções do seu produto, a
Cx26, incluem a perda de capacidade de formação dos conexons, com interrupção
da reciclagem do potássio nas células ciliadas e sua concentração elevada na
endolinfa dos dutos cocleares, o que resultaria na intoxicação do órgão de Corti pelo
potássio e consequente morte celular e perda da audição (Figura 1.8) (Lefebvre e
Van de Water, 2000; Rabionet e col., 2000; Bruzzone e col, 2003). No entanto,
algumas mutações na Cx26 que causam surdez em humanos (p.V84L, p.V95M e
p.A88S) alteram apenas a permeabilidade a moléculas grandes in vitro, como IP3
(inositol trifosfato), mas mantêm a permeabilidade iônica normal do potássio nos
canais de junção do tipo fenda (Beltramello e col., 2005; Zhang e col., 2005). Além
disso, trabalhos com camundongos nocautes nulos e condicionais para o gene da
Cx26 têm demonstrado que a surdez nesses animais ocorre antes do
estabelecimento do potencial endococlear e que outras estruturas como o túnel de
Corti (Figuras 1.2., 1.3. e 1.8), já estariam comprometidas mesmo antes do início da
morte das células de suporte e células ciliadas (Cohen-Salmon e col., 2002; Kudo e
col., 2003; Sun e col., 2009; Wang e col., 2009). Esses trabalhos sugerem que a Cx26
possa ter outras funções no órgão de Corti, além da reciclagem dos íons potássio.
De qualquer modo, apesar da teoria da reciclagem dos íons potássio ser a mais
aceita entre os pesquisadores como causa de surdez, o papel exato dos canais de
junção do tipo fenda na reciclagem dos íons potássio e na geração do potencial
endococlear ainda não foi demonstrado diretamente.
1.6.3. Biossíntese das conexinas
Assim como a maioria das proteínas de membrana, as conexinas são
sintetizadas pela via clássica das proteínas secretoras. Desse modo, as conexinas são
sintetizadas no retículo endoplasmático rugoso (RER) e transportadas à membrana
plasmática por meio do complexo de Golgi e vesículas secretoras. Durante sua

31
tradução, as cadeias polipeptídicas das conexinas em alongamento adquirem sua
estrutura secundária e são integradas as membranas do RE co-traducionalmente,
onde adotam sua topologia final. Algumas conexinas, no entanto, passam pelo
compartimento intermediário e atingem sua topologia final na porção distal no
complexo de Golgi (Laird, 2005, 2006 e 2009).
Figura 1.8. Representação esquemática da reciclagem dos íons potássio na cóclea. O movimento da membrana basilar promove a abertura dos canais de junção do tipo fenda. Os íons de potássio saem das células ciliadas em direção ao espaço extracelular no interior do órgão de Corti, circulando pelas células de suporte e da estria vascular, para serem liberados nos espaços onde estejam com seus níveis diminuídos (Figura modificada da página http://www.ncbi.nlm.nih.gov, 2011).
Segundo Koval (2006), as conexinas não devem seguir a via clássica de
oligomerização das proteínas integrantes ao formarem os hexâmeros e o local de
oligomerização das conexinas em conexons não é precisamente definido. Embora,
aparentemente, seja no complexo de Golgi que a maioria das conexinas se
oligomerizem para formar os conexons, alguns estudos têm revelado, sobretudo
para as Cx26 e Cx32, que esse processo pode ser realizado no interior do RE (Falk e
col., 1997; Falk e Gilula, 1998; Laird, 2005).

32
Como os conexons alcançam à membrana plasmática e como eles se
reagrupam para formar as placas juncionais ainda não está totalmente esclarecido.
Acredita-se que os conexons sejam inseridos na membrana plasmática, onde
permanecem como hemicanais, por meio de vesículas transportadoras ou através de
extensões tubulares e reencontram ao acaso os conexons das células vizinhas,
formando as junções do tipo fenda, para então se deslocarem até o local da placa
juncional por meio de movimentos laterais da bicamada lipídica da membrana
(Koval 2006; Laird, 2006 e 2009). Moléculas de adesão, como as caderinas E e N,
poderiam facilitar a interação entre os hemicanais (Fujimoto e col., 1997) (Figura
1.9).
Figura 1.9. Representação esquemática da síntese, montagem e degradação dos canais comunicantes do tipo fenda. As conexinas são sintetizadas no RE onde adquirem sua estrutura secundária ou passam pelo compartimento intermediário e atingem sua topologia final no complexo de Golgi. Os conexons são formados no complexo de Golgi e inseridos na membrana plasmática por vesículas transportadoras ou por extensões tubulares e reencontram ao acaso os conexons das células vizinhas para formar as junções do tipo fenda e se deslocarem até o local da placa juncional. Toda a placa juncional ou parte dela é internalizada em uma das células adjacentes para formar a junção anular, que será degradada pela via lisossômica ou proteassômica. (Figura modificada de Laird, 2006).

33
As conexinas são proteínas que apresentam uma meia vida curta, de apenas
poucas horas (Fallon e col., 1981, Laird e col., 1991; Laing e col., 1995; Beardslee e
col., 1998). Desse modo, elas são proteínas programadas para serem biossintetizadas
e degradadas constantemente. Devido a forte interação conexon-conexon no espaço
intercelular que os impede de se dissociar, toda a placa juncional ou parte dela é
internalizada em uma das células adjacentes, formando uma estrutura de dupla-
membrana chamada de junção anular. Essas junções anulares são degradadas pela
via lisossômica ou proteassômica dependente de ubiquitina (Laird, 2006 e 2009).
Estudos com inibidores de proteassomas e linhagens celulares que não apresentam a
enzima E1, necessária para a ubiquitinação, têm demonstrado que os proteassomas
possuem papel crítico na degradação tanto das conexinas tipo-selvagem quanto das
mutantes (Berthoud e col., 2004).
1.6.4.Interação entre conexinas e outras proteínas
O transporte intracelular das conexinas à membrana plasmática, a formação
dos canais de junção do tipo fenda entre as células adjacentes, a estabilidade, a
modulação da atividade e a degradação dos canais de junção do tipo fenda são
propriedades determinadas pelas proteínas conexinas constituintes dos conexons e
pela interação dessas proteínas conexinas com outras proteínas (Cascio, 2005; Laird ,
2005, 2006 e 2009).
Diversos estudos têm demonstrado interação entre as conexinas e outras
proteínas, como as da zônula de oclusão-1 (ZO-1), as calmodulinas, as caderinas,
algumas quinases e proteínas do citoesqueleto (Hervé e col., 2004). No entanto, são
poucas as proteínas identificadas por interagirem física e funcionalmente a conexina
26.
Henzl e col. (2004) verificaram que a Cx26 interage com as proteínas Fbx2 e
Skp1 na cóclea. A Fbx2 é uma ubiquitina ligase que atua como sinalizadora das
proteínas que serão degradadas via ERAD (degradação associada ao RE), sendo a
Skp1 uma proteína ligante a Fbx2 (Meusser e col., 2005). Esses estudos sugerem que
essas proteínas são substrato para a ubiquitinação e degradação em um conjunto que

34
inclui a Cx26, por meio do proteassomo 26S. A Fbx2 e Skp1 foram identificadas
pela primeira vez por Thalmann e col., (1997) no órgão de Corti de cobaias (Cavea
porcellus) e foram denominadas de OCP1 e OCP2, respectivamente. Entretanto, em
um trabalho recente de Nelson e col., (2007), a co-imuoprecipitação in vitro direta da
Cx26 com essas duas proteínas não foi observada. Nesse mesmo trabalho, foi
observado que células de suporte do órgão de Corti de camundongos com o gene
Fbx2 deletado apresentavam uma concentração elevada de Cx26, associada com
degeneração dessas células e com alteração da integridade da membrana celular. Em
resumo, a OCP1/Fbx2 deve estar relacionada à degradação da Cx26.
Recentemente, del Castillo e col. (2010) identificaram uma proteína ligante
comum às conexinas 26 e 30, por meio da técnica do duplo-híbrido da levedura. De
acordo com os autores, essa proteína, denominada consortina, atua como receptora
das conexinas 26 e 30 na porção distal do complexo do Golgi e atua no
direcionamento das conexinas para a membrana plasmática e na sua reciclagem na
superfície celular.
A identificação de novas proteínas que interagem com a Cx26 pode ampliar
o conhecimento funcional das conexinas na audição, indicar novos genes candidatos
à surdez sindrômica e não-sindrômica, além de poder explicar os casos de surdez
decorrentes de mutação isolada no gene GJB2.

35
CAPÍTULO 2

35
CAPÍTULO 2 Objetivos
2.1. Objetivo Geral
Identificar novos alelos patogênicos, novas proteínas e novos genes
relacionados com o lócus DFNB1 (genes das conexinas 26 e 30), do ponto de vista
molecular e celular, e que possam ser responsáveis por surdez de herança
autossômica recessiva.
2.2. Objetivos Específicos
� Identificar novos alelos patogênicos no lócus DFNB1 que possam ser
responsáveis por surdez, quando presentes em heterozigose composta com outros
alelos patogênicos no gene da Cx26 ou Cx30.
� Padronizar protocolos de cultivo e diferenciação in vitro de células epiteliais
do órgão de Corti, para desenvolver condições experimentais fundamentais a
estudos funcionais relacionados às proteínas conexinas e suas “parceiras” a médio
prazo e, a longo prazo, ao desenvolvimento de estratégias de terapia celular e gênica
na deficiência auditiva.
� Identificar proteínas que interagem com a Cx26 para contribuir com a
identificação de novas vias funcionais ou mecanismos moleculares que possam
resultar na descoberta de novos genes de surdez.
Pretendemos contribuir, com esse estudo, para o esclarecimento da
patogênese da surdez de herança autossômica recessiva.
Os métodos, os resultados e as discussões relacionados aos três objetivos
descritos acima serão apresentados, a seguir, sob a forma de três capítulos,
numerados de 3, 4 e 5, respectivamente.

36
CAPÍTULO 3

36
CAPÍTULO 3 Pesquisa de novos alelos patogênicos no lócus DFNB1 relacionados
à surdez não-sindrômica de herança autossômica recessiva
3.1. Introdução
Dentre os indivíduos com deficiência auditiva associada ao lócus DFNB1,
10% a 50% apresentam uma única mutação recessiva no gene GJB2, frequência
muito superior à esperada em função da frequência de heterozigotos na população
geral. Apesar de alguns casos de indivíduos com distúrbios de audição que
apresentam uma única mutação recessiva no gene GJB2 terem sido elucidados após
a identificação de cinco diferentes deleções no gene GJB6 ou nas suas proximidades
(del Castillo e col., 2003, 2005 e 2009; Feldmann e col., 2009; Wilsh e col., 2010) a
existência de muitos indivíduos com uma única mutação patogênica nesse gene
sugere que mutações em outras regiões do lócus DFNB1, como por exemplo em
outras regiões do gene GJB2, GJB6, fora da sua região de código, possam estar
presentes de forma a causar a surdez desses indivíduos. Diversos estudos têm
procurado identificar mutações nas regiões 5´UTR (no exon 1 e no sítio doador de
splicing) e nas regiões promotoras do gene GJB2. Um exemplo é a mutação IVS 1 +
1G>A, localizada no primeiro intron do gene GJB2, entre o exon 1 não codificador
e o exon 2, no sítio doador de splicing. A troca de uma guanina por uma adenina
nessa posição afeta o splicing e leva, aparentemente, à falta do RNA mensageiro
(Denoyelle e col., 1999; Shahin, 2002). O estudo dessa mutação, por Seeman e col.
(2006), possibilitou o diagnóstico molecular de 45% de uma amostra de pacientes
tchecos com uma única mutação patogênica na região codificadora do gene GJB2. Já
na Holanda, onde ela também foi estudada, a mutação IVS 1 + 1G>A foi apontada
como a terceira mutação mais frequente em indivíduos surdos, após a c.35delG e a
del(GJB6-D13S1830) (Santos e col., 2005). Outros estudos descreveram essa
mutação em diversos países (Áustria, China, EUA, Egito, Itália, Irã, Hungria e
Turquia) (Najmabadi e col., 2005; Snoeckx e col., 2005; Sirmaci e col., 2006;
Chaleshtori e col., 2007; Putcha e col, 2007; Ramsebner e col., 2007; Tóth e col.,
2007; Cama e col., 2009; Yuan e col., 2010). No Brasil, Silva-Costa e col. (2009)

37
identificaram a mutação IVS 1 + 1G>A em 2 dos 43 (4,6%) pacientes surdos já
previamente caracterizados como portadores de uma única mutação recessiva na
região codificadora do gene GJB2.
Mattos e col. (2007) identificaram pela primeira vez uma mutação patogênica
na região promotora do gene GJB2 associada à surdez não-sindrômica de herança
autossômica recessiva em uma família portuguesa. Essa mutação, -3438C>T, foi
identificada em trans com a mutação patogênica p.V84M.
A existência do alto número de indivíduos com uma única mutação
patogênica recessiva nos genes GJB2 e GJB6 sugere que outras mutações não
identificadas no lócus DFNB1 possam estar presentes de forma a causar surdez
nesses indivíduos. A triagem de variantes patogênicas no gene GJB2, fora de sua
região codificadora, bem como o estudo de grandes deleções ou inserções, a
exemplo das que ocorrem com o gene GJB6, podem nos auxiliar a revelar os
mecanismos moleculares que relacionam os produtos dos genes GJB2 e GJB6, além
de poder explicar os casos de pacientes surdos com uma única mutação detectada
no gene GJB2.
3.2. Objetivo
Identificar novos alelos patogênicos no lócus DFNB1 que possam ser
responsáveis por surdez, quando presentes em heterozigose composta com outros
alelos patogênicos nos genes da Cx26 ou Cx30.
Para atingir esse objetivo, foram executadas as seguintes etapas:
� Pesquisa de mutações por meio do sequenciamento das regiões
codificadora, promotora e doadora de splicing (intron 1) do gene GJB2 nos pacientes
previamente caracterizados como portadores de uma única mutação patogênica e
recessiva nos genes GJB2 ou GJB6.
� Pesquisa da deleção de 200kb descrita por del Castillo e col., (2009),
localizada a 130kb da proximidade distal da região 5’ do gene GJB6 em pacientes

38
previamente caracterizados como portadores de uma única mutação patogênica e
recessiva nos genes GJB2 ou GJB6, por meio da PCR específica para amplificar essa
deleção.
� Pesquisa de variações no número de cópias de um ou mais exons dos
genes GJB2, GJB6, GJB3 e WFS1 nos pacientes previamente caracterizados como
portadores de uma única mutação patogênica e recessiva no gene GJB2, por meio da
técnica MLPA (Multiplex Ligation-dependent Probe Amplification).
3.3. Casuística e Métodos
3.3.1. Pacientes
Nossa casuística é formada por portadores de distúrbios auditivos que
procuram aconselhamento genético no Laboratório de Genética Humana do
Departamento de Genética e Biologia Evolutiva do Instituto de Biociências da
Universidade de São Paulo (LGH – IBUSP). Os pacientes foram encaminhados
pelas instituições DERDIC – Divisão de Educação e Reabilitação de Distúrbios da
Comunicação da Pontifica Universidade Católica de São Paulo, pelo Hospital das
Clínicas da Faculdade de Medicina da Universidade de São Paulo, pelo CEPRO -
Centro de Ensino Profissionalizante Rotary do Colégio Rio Branco, pelo
Departamento de Biologia Celular e Genética da Universidade Estadual de Maringá,
pelo Departamento de Genética da Universidade Federal do Paraná e outras
instituições.
Todas as pessoas portadoras de distúrbios de audição que comparecem ao
serviço de aconselhamento genético são entrevistadas e assinam o termo de
consentimento para o exame e a utilização dos dados para pesquisa. O protocolo
para trabalho com esses pacientes foi submetido à Comissão de Ética em Pesquisa -
Seres Humanos do Instituto de Biociências da Universidade de São Paulo e
aprovado (Protocolo de pesquisa Nº062/2007).
Na casuística de cerca de 700 propósitos com distúrbios de audição, não-
sindrômicos e sindrômicos, foram triadas como rotina do nosso laboratório as

39
mutações c.35delG e c.167delT no gene GJB2 e del(GJB6-D13S1830) e del(GJB6-
D13S1854) no gene GJB6 por meio de técnicas específicas. Em alguns propósitos já
havia sido realizado previamente o sequenciamento da região de código do gene
GJB2 (Batissoco, 2006).
Foram selecionados para o nosso estudo 16 indivíduos com uma única
mutação, sem que fosse identificada uma segunda mutação nos genes GJB2 ou GJB6,
ou seja, portadores de uma única mutação recessiva em um desses dois genes. Para a
identificação de novos alelos patogênicos no lócus DFNB1, utilizamos o DNA
purificado a partir de sangue periférico desses 16 indivíduos com deficiência auditiva.
3.3.2. Amostra Controle
Estudamos também uma amostra controle formada por indivíduos ouvintes
que trabalham no Instituto de Biociências, moradores do Conjunto Residencial da
Universidade de São Paulo – CRUSP, doadores de sangue da Fundação Pró-Sangue
Hemocentro de São Paulo e por trabalhadores de empresas metalúrgicas e
condutores de ônibus da cidade de São Paulo-SP. Todos os que concordaram em
participar do estudo foram entrevistados quanto à existência de deficientes auditivos
na família e também assinaram o termo de esclarecimento livre e esclarecido. Os
indivíduos foram classificados como brancos, pardos ou negros em função da
origem dos seus ancestrais declarada em entrevista, o que permitiu a divisão
posterior da amostra em um grupo constituído por 50 indivíduos classificados como
brancos e um grupo constituído por 50 indivíduos classificados como pardos ou
negros.
3.3.3. Métodos
As amostras de sangue periférico de cada indivíduo tiveram o DNA extraído
com fenol e clorofórmio, segundo protocolo de rotina do Laboratório de Genética
Humana. Parte das amostras foi extraída no aparelho Autopure LS (Gentra Systems,
Minneapolis, Minnesota, USA). Posteriormente, cada amostra de DNA foi

40
quantificada pelo espectrofotômetro NanoDrop ND-1000 (NanoDrop Technologies,
Rockland, Delaware, USA).
O DNA extraído dos indivíduos surdos, identificados como heterozigotos
com única mutação recessiva nos genes GJB2 ou GJB6, foi estudado por meio das
seguintes técnicas:
i) Sequenciamento das regiões de código, promotora e doadora de splicing do
intron 1 do gene GJB2.
ii) Triagem da deleção de 200kb localizada nas proximidades do gene GJB6.
iii) Pesquisa de variações do número de cópias por MLPA (Multiplex Ligation-
dependent Probe Amplification).
3.3.3.1. Sequenciamento das regiões de código, promotora e de
splicing do gene GJB2
Sequenciamento da região de código do gene GJB2
Para o sequenciamento, a região de código do gene GJB2 (exon2) foi dividida
em dois fragmentos. Desse modo, foram utilizados dois pares de iniciadores: um
deles 1F (5’ GTG TTG TGT GCA TTC GTC TTT TC- 3’) e 3R (5’ ACC TTC
TGG GTT TTG ATC TCC TC 3’) para amplificar um fragmento de 425pb e o par
de iniciadores 4F (5’ GGA AGT TCA TCA AGG GGG AGA TA 3’) e 2R (5’ CCT
CAT CCC TCT CAT GCT GTC TA 3’) para amplificar um segundo fragmento de
422pb.
Para a realização da PCR, em um volume final de reação de 50uL, contendo
100ng de DNA total, foram utilizados 10 pmoles de cada iniciador, 1U de Taq
polimerase, 1,5mM de MgCl2, 20mM TRIS pH = 8.4, 50mM KCl e 0,2mM de cada
dNTP As condições da amplificação para os dois fragmentos foram as seguintes:
um ciclo de desnaturação a 95ºC por 5 min, 9 ciclos touchdown de desnaturação a
96ºC por 15s, hibridação a 69ºC por 15s para o primeiro ciclo com redução de 1ºC
por ciclo e extensão a 72ºC por 30s, 25 ciclos de desnaturação a 96ºC por 15s,

41
hibridação a 60ºC por 15s, extensão a 72ºC por 30s e extensão final a 72ºC por 10
min.
Os produtos da amplificação foram purificados pelo Illustra GFX PCR DNA
and gel purification Kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK),
segundo instruções do fabricante. Os produtos purificados foram então submetidos
à reação de sequenciamento, na qual a cada produto foi adicionada uma solução de
reação contendo um dos respectivos primers e o BigDye Terminator v3.1 Cycle Sequencing
(Applied Biosystems, Foster City, California, USA). Por fim, o sequenciamento
automático foi realizado pelo aparelho ABI 3730xl DNA Analyzer (Applied
Biosystems, Foster City, California, USA).
Sequenciamento das regiões promotora e de splicing do gene GJB2
Para o sequenciamento das regiões promotora e de splicing do gene GJB2
foram utilizados os iniciadores PromCX26F 5’ – GGG GCT CAA AGG AAC
TAG GA – 3’ e PromCX26R 5’ AAG GAC GTG TGT TGG TCC AG - 3’ para
amplificar um fragmento de 499pb. Esses iniciadores amplificam a região
promotora, parte da região 5’ UTR que inclui o exon 1 e o sítio doador de splicing do
intron 1. Essa reação permite identificar as mutações IVS 1 + 1G>A (-3170G>A)
no exon 1 não codificador que afeta o splicing e a mutação -3438C>T, localizada na
região promotora do gene (Figura 3.1).
Para a realização da PCR, em um volume final de reação de 25µL, contendo
100ng de DNA total, foram utilizados 10 pmoles de cada iniciador, 1U de Taq
polimerase, 1,5mM de MgCl2, 20mM TRIS pH = 8.4, 50mM KCl e 0,2mM de cada
dNTP. As condições da amplificação foram as seguintes: um ciclo de desnaturação a
95ºC por 5 min, 5 ciclos touchdown de desnaturação a 96ºC por 15s, hibridação a
67ºC por 15s para o primeiro ciclo com redução de 1ºC por ciclo e extensão a 72ºC
por 30s, 25 ciclos de desnaturação a 96ºC por 15s, hibridação a 57ºC por 15s,
extensão a 72ºC por 30s e extensão final a 72ºC por 10 min.
Os produtos da amplificação foram purificados pelo Illustra GFX PCR DNA
and gel purification Kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK),

42
segundo instruções do fabricante. Os produtos purificados foram então submetidos
à reação de sequenciamento, na qual a cada produto foi adicionada uma solução de
reação contendo um dos respectivos primers e o BigDye Terminator v3.1 Cycle Sequencing
(Applied Biosystems, Foster City, California, USA). Por fim, o sequenciamento
automático foi realizado pelo aparelho ABI 3730xl DNA Analyzer (Applied
Biosystems, Foster City, California, USA).
Na figura 3.1 apresentamos a representação esquemática das regiões sequenciadas no gene GJB2.
Figura 3.1. Representação esquemática do gene GJB2 e das suas regiões de código, promotora e doadora de splicing do intron 1 e da extensão das regiões sequenciadas nesse trabalho.
Triagem da nova mutação detectada no sequenciamento da região de código
do gene GJB2 na amostra controle
A nova mutação p.L76P (c.227C<T), detectada por meio de sequenciamento
em uma família com dois pacientes surdos heterozigotos quanto a c.35delG, foi
rastreada por meio da técnica de genotipagem automatizada de SNPs, com a
utilização do MegaBace SNuPe Genotyping Kit (Amersham Biosciences,
Buckinghamshire, UK) e do analisador genético MegaBACETM1000 (Amersham
Biosciences, Buckinghamshire, UK), disponível no Centro de Estudos do Genoma
Humano, na amostra controle constituída por 100 indivíduos ouvintes.

43
A reação de PCR foi feita sob as seguintes condições: 50µL, contendo 100ng
de DNA total, foram utilizados 10 pmoles de cada iniciador (1F - 5’ GTG TTG
TGT GCA TTC GTC TTT TC- 3’ e 3R - 5’ ACC TTC TGG GTT TTG ATC TCC
TC 3’), 1U de Taq polimerase, 1,5mM de MgCl2, 20mM TRIS pH = 8.4, 50mM KCl
e 0,2mM de cada dNTP. As condições da amplificação foram as seguintes: um ciclo
de desnaturação a 95ºC por 5 min, 9 ciclos touchdown de desnaturação a 96ºC por
15s, hibridação a 69ºC por 15s para o primeiro ciclo com redução de 1ºC por ciclo e
extensão a 72ºC por 30s, 25 ciclos de desnaturação a 96ºC por 15s, hibridação a
60ºC por 15s, extensão a 72ºC por 30s e extensão final a 72ºC por 10 min.
O produto da PCR (8µL) foi purificado com 2,5U da enzima Exonuclease I
(Amersham Biosciences, Buckinghamshire, UK) e 0,5U da enzima Shrimp Alkaline
Phosphatase (Amersham Biosciences, Buckinghamshire, UK), incubados a 37ºC por
1h e posteriormente submetidos a uma temperatura de 80ºC por 15 min para a
inativação das enzimas. Para a reação de SNuPe (volume final de 10µL) foram
utilizados 4µL de SNuPe premix (Amersham Biosciences, Buckinghamshire, UK ),
0,2µM do iniciador SNuPe, 3µL do produto da PCR já purificado e 2µL de água
estéril. Após a reação de SNuPe, os produtos foram novamente purificados com
AutoSeq96 Dye Terminator Clean-up, segundo instruções do fabricante 5µL do produto
da reação de SNuPe purificado foram misturados a 0,02µL de Multi Injection Marker e
4,98µL de Loading Solution (Amersham Biosciences, Buckinghamshire, UK), sendo
então carregados no analisador genético MegaBACETM1000 (Amersham
Biosciences, Buckinghamshire, UK).
3.3.3.2. Triagem da deleção de 200kb localizada nas proximidades do
gene GJB6
Para a detecção da deleção de 200kb próxima ao gene GJB6 foi utilizado um
protocolo sugerido pelo Dr. Ignácio del Castillo, que coordena a equipe que
descreveu esta deleção (del Castillo e col., 2009).
A pesquisa da deleção de 200kb foi feita por meio de uma PCR multiplex,
que permite o estudo de outras duas deleções, del(GJB6-D13S1830) de 309kb e

44
del(GJB6-D13S1854) de 232kb. Para a PCR, foram utilizados quatro pares de
iniciadores, um par que amplifica a região que corresponde ao exon 4 do gene GJB6
(como banda controle para verificar se a PCR está funcionando) e que origina um
fragmento de 333pb (sense 5’ – CGT CTT TGG GGG TGT TGC TT – 3’ e anti-
sense 5’ – CAT GAA GAG GGC GTA CAA GTT AGA A - 3’) e três pares de
iniciadores que amplificam o DNA na região de junção dos pontos de quebra de
cada uma das deleções. Del(GJB6-D13S1830) iniciadores: sense 5’ – TTT AGG GCA
TGA TTG GGG TGA TTT – 3’ e anti-sense 5’ – CAC CAT GCG TAG GCT TAA
CCA TTT T– 3’) que amplificam um fragmento de 460pb no caso de deleção.
Del(GJB6-D13S1854) iniciadores: sense 5’ – TCA TAG TGA AGA ACT CGA
TGC TGT TT– 3’ e anti-sense 5’ –CAG CGG CTA CCC TAG TTG TGG T– 3’)
que amplificam um fragmento de 564pb no caso de deleção. Deleção de 200kb
iniciadores: sense 5’ – GCC AGG GCT TGA GCA TAG AAC – 3’ e anti-sense 5’ –
GAA TTC TGG CAC TCT GAA TTA GAA AAC – 3’) que irá amplificar um
fragmento de 857pb se houver deleção. Para um volume final de reação de 20µL,
contendo 75ng de DNA total, foram utilizados 0,5 pmol dos pares de iniciadores
referentes às deleções del(GJB6-D13S1830) e del(GJB6-D13S1854), 0,375 pmol do
par de iniciadores referente ao exon 4 do gene GJB6 (controle) e 1,25 pmol do par
de iniciadores referente à deleção de 200kb nas proximidades do gene GJB6, 0,5U
de Taq polimerase platinum Pfx (Invitrogen, Carlsbad, CA, USA), 1,5mM de MgSO4,
20mM TRIS pH = 8.4, 50mM KCl e 0,2mM de cada dNTP. As condições da
amplificação foram as seguintes: um ciclo de desnaturação a 94ºC por 6 min, 5
ciclos touchdown de desnaturação a 94ºC por 40s, hibridação a 62ºC por 40s para o
primeiro ciclo com redução de 1ºC por ciclo e extensão a 72ºC por 30s, 25 ciclos de
desnaturação a 94ºC por 40s, hibridação a 58ºC por 40s, extensão a 72ºC por 60s e
extensão final a 72ºC por 7 min. Os produtos da PCR obtidos foram visualizados
em gel de agarose a 2% após coloração com brometo de etídeo. Foi utilizada como
controle uma amostra de DNA de um indivíduo já previamente identificado com a
deleção de 200kb em heterozigose. Essa amostra foi fornecida pelo Dr. Ignácio del
Castillo (Hospital Ramón Y Cajal, Madri, Espanha). Heterozigotos quanto à deleção
de 200kb terão duas bandas amplificadas, uma delas é a controle (333pb) e a outra é
referente à amplificação da região de junção dos pontos de quebra da deleção

45
(857pb); indivíduos homozigotos quanto à deleção terão apenas a banda de 857pb e
indivíduos homozigotos sem a deleção apresentam apenas a banda de 333pb. Os
indivíduos com a del(GJB6-D13S1830) em heterozigose terão as bandas de 333pb
(controle) e de 460pb (ponto de quebra) e em homozigose somente a banda de
460pb. Já os indivíduos com a del(GJB6-D13S1854) em heterozigose terão as
bandas de 333pb (controle) e de 564pb (ponto de quebra) e em homozigose
somente a banda de 564pb.
3.3.3.3. Pesquisa de variações no número de cópias nos exons dos
genes GJB2, GJB6, GJB3 e WFS1 por MLPA (Multiplex Ligation-dependent
Probe Amplification)
A técnica de MLPA permite detectar alterações no número de cópias em até
50 sequências diferentes de DNA em uma única reação de amplificação. Em outras
palavras, essa técnica possibilita a identificação de duplicações e deleções em um
grande número de sequências simultaneamente. A identificação do número de
cópias de uma dada sequência de DNA é efetuada pela hibridação de pares de
sondas específicas a essa sequência. Essas sondas apresentam sequências nas
extremidades que hibridam a um único par de iniciadores permitindo a amplificação
de todas as sequências, se houver hibridação das sondas. A peculiaridade desta
técnica está no fato de que não é o DNA genômico que é alvo da amplificação por
PCR, e sim as sondas que se hibridam ao DNA genômico. Quando existem
patologias onde o gene ou parte dele encontra-se duplicado ou deletado, as técnicas
laboratoriais de PCR e sequenciamento apresentam limitações na detecção devido à
amplificação exclusiva do alelo normal na amostra. Nesse caso, a análise pela técnica
de MLPA é uma alternativa apropriada para contornar as limitações dessas técnicas.
A empresa que desenvolveu a técnica de MLPA, MRC-Holland
(http://www.mrc-holland.com), disponibiliza centenas de kits de sondas para o
estudo de diversas doenças genéticas ou síndromes conhecidas, entre eles um kit
contendo sondas para os genes GJB2, GJB6, WFS1 e POU3F4 (kit SALSA MLPA
KIT P163-C1 Hearing loss).
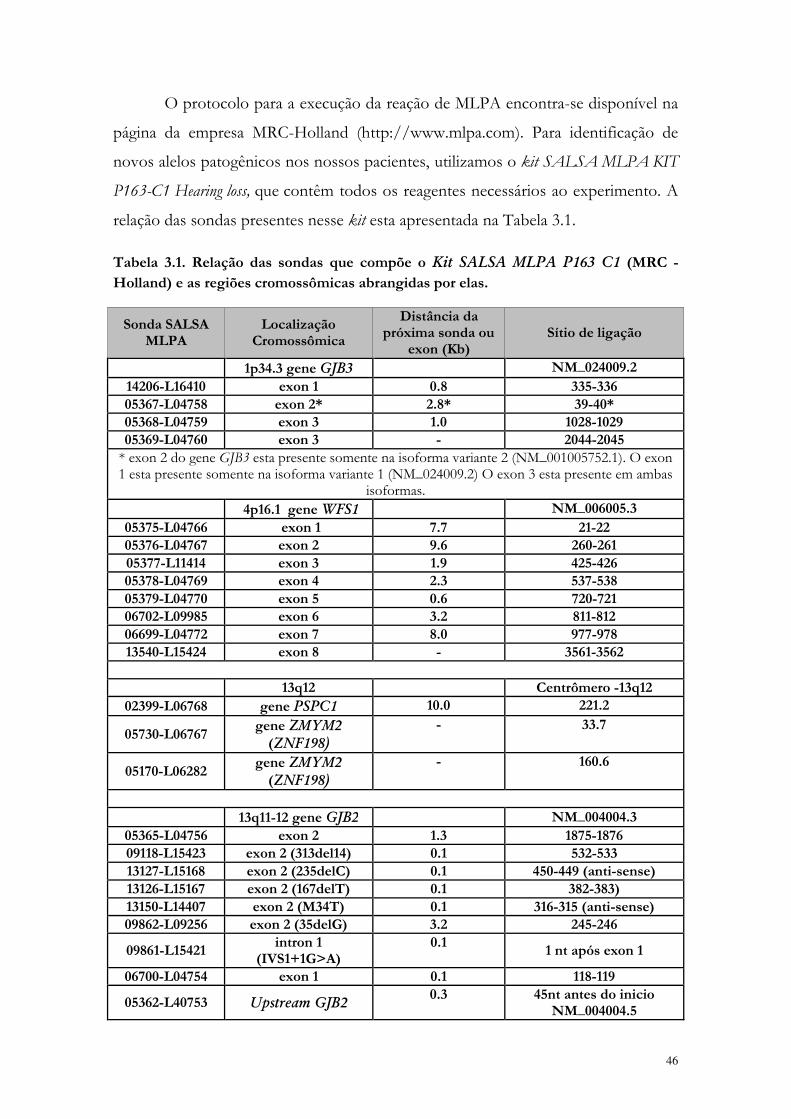
46
O protocolo para a execução da reação de MLPA encontra-se disponível na
página da empresa MRC-Holland (http://www.mlpa.com). Para identificação de
novos alelos patogênicos nos nossos pacientes, utilizamos o kit SALSA MLPA KIT
P163-C1 Hearing loss, que contêm todos os reagentes necessários ao experimento. A
relação das sondas presentes nesse kit esta apresentada na Tabela 3.1.
Tabela 3.1. Relação das sondas que compõe o Kit SALSA MLPA P163 C1 (MRC - Holland) e as regiões cromossômicas abrangidas por elas.
Sonda SALSA MLPA
Localização Cromossômica
Distância da próxima sonda ou
exon (Kb) Sítio de ligação
1p34.3 gene GJB3 NM_024009.2 14206-L16410 exon 1 0.8 335-336 05367-L04758 exon 2* 2.8* 39-40* 05368-L04759 exon 3 1.0 1028-1029 05369-L04760 exon 3 - 2044-2045
* exon 2 do gene GJB3 esta presente somente na isoforma variante 2 (NM_001005752.1). O exon 1 esta presente somente na isoforma variante 1 (NM_024009.2) O exon 3 esta presente em ambas
isoformas. 4p16.1 gene WFS1 NM_006005.3
05375-L04766 exon 1 7.7 21-22 05376-L04767 exon 2 9.6 260-261 05377-L11414 exon 3 1.9 425-426 05378-L04769 exon 4 2.3 537-538 05379-L04770 exon 5 0.6 720-721 06702-L09985 exon 6 3.2 811-812 06699-L04772 exon 7 8.0 977-978 13540-L15424 exon 8 - 3561-3562
13q12 Centrômero -13q12
02399-L06768 gene PSPC1 10.0 221.2
05730-L06767 gene ZMYM2 (ZNF198)
- 33.7
05170-L06282 gene ZMYM2 (ZNF198)
- 160.6
13q11-12 gene GJB2 NM_004004.3
05365-L04756 exon 2 1.3 1875-1876 09118-L15423 exon 2 (313del14) 0.1 532-533 13127-L15168 exon 2 (235delC) 0.1 450-449 (anti-sense) 13126-L15167 exon 2 (167delT) 0.1 382-383) 13150-L14407 exon 2 (M34T) 0.1 316-315 (anti-sense) 09862-L09256 exon 2 (35delG) 3.2 245-246
09861-L15421 intron 1
(IVS1+1G>A) 0.1
1 nt após exon 1
06700-L04754 exon 1 0.1 118-119
05362-L40753 Upstream GJB2 0.3 45nt antes do inicio
NM_004004.5

47
Sonda SALSA MLPA
Localização Cromossômica
Distância da próxima sonda ou
exon (Kb) Sítio de ligação
06698-L04752 Upstream GJB2 28.7 377nt antes do inicio
NM_004004.5
13q11-12 gene GJB6 NM_006783.4 05374-L04765 exon 3 1.5 2029-2030 05373-L15422 exon 3 6.2 578-579 06701-L04763 exon 2 1.2 424-425 05371-L04762 exon 1 0.3 313-314
05370-L04761 Upstream GJB6 743.8 332nt antes do inicio
NM_006783.4
13q12 13q12 06704-L03639 gene LATS2 - -
Xq21.1 gene POU3F4 NM_000307.3
13141-L14398 upstream 0.1 - 13143-L14400 upstream 52 - 13140-L14397 upstream 0.3 - 13139-L14396 upstream 137.3 - 13142-L14399 upstream 0.2 - 13144-L14401 upstream 783.7 - 12771-L13887 exon 1 1 92-93 12773-L13889 exon 1 - 1141-1142
sonda controle 11q23.3 - 11q23.3 sonda controle Cromossomo X - Cromossomo X sonda controle Cromossomo Y - Cromossomo Y sonda controle 18q23 - 18q23 sonda controle 2q24 - 2q24 sonda controle 13q14.3 - 13q14.3 sonda controle 3q21 - 3q21
A reação de MLPA foi iniciada pela desnaturação de 8µL de DNA (200ng) a
98°C por 5 min. Em seguida, foram adicionados a cada amostra 3µL de mix
contendo as sondas e realizou-se a desnaturação por 1 min a 95°C e incubação para
hibridação a 60°C por 16h. Após incubação, a ligação das sondas proximais e distais
foi feita com a adição da enzima ligase. Somente os pares de sondas que são ligados
pela ligase serão amplificados pela PCR. Para ligação, as amostras foram incubadas
por 15 min a 54°C, seguidos de 5 min a 98°C para inativação térmica da enzima.
Posteriormente, as amostras foram submetidas a amplificação por PCR, com as
seguintes condições: 35 ciclos de 30s a 95°C, 30s a 60°C e 60s a 72°C, e por fim 20
min de incubação a 72°C.

48
Após a amplificação pela PCR, os produtos obtidos foram separados
juntamente com um padrão de peso molecular por eletroforese em capilar no
equipamento ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City,
California, USA). A análise da quantidade das sondas fluorescentes amplificadas foi
realizada pelo software GeneMarker (SoftGenetics, State College, Pennsylvania, USA),
que lê os picos de fluorescência e já fornece os resultados analisados. Na análise dos
resultados, as alturas dos picos de fluorescência de cada sonda e de cada indivíduo
são normalizadas entre si, de modo a indicar um valor que permite estimar o
número de cópias de cada sequência de um indivíduo. Valores abaixo de 0,7 indicam
deleção e valores acima de 1,3 indicam duplicação da região. Espera-se que
indivíduos com número normal de cópias para uma determinada sequência tenham
o valor correspondente próximo de 1,0.
Uma vez que é o conjunto da altura dos picos que irá ao final fornecer
informações acerca do número de cópias das sequências, são necessários cuidados
para a obtenção de resultados confiáveis. A comparação dos dados (isto é, das
alturas dos picos de fluorescência) deve ser feita sempre entre as amostras da mesma
corrida, ou seja, amostras que foram submetidas simultaneamente à mesma reação
de MLPA, o que significa que uma única amostra de DNA de baixa qualidade
compromete os resultados. A intensidade da fluorescência de cada sonda também
deve se aproximar das condições ideais. Além disso, cada corrida de MLPA requer a
análise de pelo menos três amostras de indivíduos normais como controle.
Confirmação da presença de duplicação por PCR em tempo real quantitativa
As reações de PCR quantitativo em tempo real foram feitas no sistema de
detecção Light Cycler 480 (Roche Applied Science, Indianapolis, IN, USA). A reação
de PCR em tempo real teve volume final de 15µL, cerca de 2ng de DNA, 0,1µM de
cada iniciador e 7,5µL de SYBR®Green PCR Master Mix 2X (Applied Biosystems
Foster City, CA, USA). As condições de amplificação estão descritas na Tabela 3.2.
Todas as reações de amplificação das amostras foram feitas em triplicata, utilizando-
se a média do três resultados entre elas para os cálculos das quantificações relativas.

49
As sequências dos iniciadores foram desenhadas usando o programa Primer-
Blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) ou Primer3
(www.frodo.wi.mit.edu) e estão descritas na Tabela 3.3. Os iniciadores foram
testados por meio de uma análise de curva de dissociação, realizada após uma reação
de PCR em tempo real, para avaliar a formação de dímeros de iniciadores ou a
amplificação de produtos inespecíficos, o que interfere na quantificação real da
sequência alvo. Dessa forma, só foram utilizados iniciadores cuja curva de
dissociação indicava especificidade (Figura 3.2).
Tabela 3.2. Programa de amplificação utilizado nos experimentos de PCR em tempo real.
Tempo Temperatura Rampa Etapa 2min 50ºC 4,4ºC/s Ativação
10min 95ºC 4,4ºC/s Ativação 15s 95ºC 2,2ºC/s PCR - 60 ciclos - modo de análise: quantificação
1min 60ºC 2,2ºC/s PCR - 60 ciclos - modo de análise: quantificação 15s 95ºC 4,4ºC/s Dissociação - curvas de desnaturação
1min 60ºC 2,2ºC/s Dissociação - curvas de desnaturação aquisição 95ºC 0,11ºC/s Dissociação - curvas de desnaturação
Tabela 3.3. Sequência dos iniciadores de regiões diferentes do gene GJB2 utilizados nos experimentos de PCR em tempo real.
Sequências-alvo Iniciador sense Iniciador anti-sense GJB2_35 (c.-35 a c.77) CGTCTTTTCCAGAGCAAACCG GTGAGCCAGATCTTTCCAATGC
GJB2_167 (c.52 a c.199) ACCAGCATTGGAAAGATCTGG GATCGTAGCACACGTTCTTGC
GJB2_235 (c.177 a c.306) CTGCAAGAACGTGTGCTACGA TCTTCTCATGTCTCCGGTAGG
GJB2_UTR (c.746 a c.845) GCTGTCAAGGCTCAGTCGCTA GCATCTGGAGTTTCACCTGAG
GJB2_intron (c.-519 a c.-622) CACTGCTACATGCCACGTCTG GGCCAAGTACAGGAGAACCGT
Figura 3.2. Exemplo de curva de dissociação específica mostrando um único pico máximo entre 80-84oC.
Três amostras controles de ouvintes que apresentaram resultados normais
quanto ao gene GJB2 na técnica de MLPA foram utilizadas como amostras de
referência nas reações de PCR em tempo real. Uma dessas amostras foi também

50
utilizada para a construção de curvas padrão da amplificação das sequências alvo do
gene GJB2, com o objetivo de calcular as eficiências de amplificação de cada
iniciador. Foram feitas quatro diluições seriadas a partir da amostra controle original
(20ng, 2ng, 0,2ng e 0,02ng), para avaliar se existia amplificação proporcional à
quantidade inicial do DNA genômico na amostra. Dessa forma, quanto maior a
diluição, mais tardiamente a amplificação é detectada pelo termociclador e maior é o
Ct (Figura 3.3.). Os gráficos de dispersão log da quantidade X Ct das amostras
geraram curvas padrão lineares para todas as sequências alvo (valores de R2 entre
0,98 e 1), mostrando uma amplificação proporcional dessas sequências. O
coeficiente da reta (R) foi utilizado para calcular o valor de eficiência do iniciador,
segundo a fórmula: E=-1/coeficiente da reta. Um valor de E=2 representa uma
amplificação de 100% da quantidade inicial de DNA a cada ciclo do PCR. Assim,
para considerarmos que o iniciador tem uma boa eficiência, este valor deve estar
entre 1,7 e 2,3. Os valores de eficiência de cada iniciador são utilizados nos cálculos
da quantificação relativa das sequências alvo conforme descrito a seguir. Em relação
ao experimento para realizar a quantificação relativa do número de cópias das
sequências alvo do gene GJB2 na paciente com a duplicação, amplificamos três
amostras controle ou “referência” e a média dos valores de Ct (vide definição
abaixo) das três foi utilizada como o valor de referência. Também realizamos a
quantificação relativa de amostra escolhida para ser controle, selecionada, pois
pertencia a um indivíduo surdo com resultados normais quanto ao número de
cópias do GJB2 após MLPA. Também como controle, foi estudada a amplificação
por PCR em tempo real de uma sequência exônica do gene CCDC88A, localizado
na região cromossômica 2q16.1.
O modelo matemático utilizado para determinar a quantificação relativa de
cada sequência alvo (sequências possivelmente duplicadas do gene GJB2) em
comparação a uma sequência de referência (sequência exônica do gene CCDC88A,
localizado na região cromossômica 2q16.1) foi o descrito por Pfaffl (2001). Esse
modelo está apresentado na fórmula abaixo.

51
� QR: Quantificação relativa ou razão que corresponde ao número de cópias
da sequência alvo por genoma. Foi calculado um valor de QR para cada sequência
alvo em cada um dos três experimentos realizados. Dessa forma, quando estamos
estudando sequências ou genes de cópia única, são esperados valores ao redor de 1
para sequências não duplicadas e valores ao redor de 1,5 para sequências duplicadas.
O experimento de amplificação em tempo real com o objetivo de realizar a
quantificação relativa das sequências alvo do gene GJB2 foi realizado três vezes.
Dessa forma, obtivemos médias e desvios padrão do valor estimado para o número
de cópias de cada sequência alvo.
� Ct (ou Cp): O Ct (cycle threshold) ou Cp (crossing point) é o número decimal
de ciclos da PCR no qual a fluorescência cruza a linha do limiar, o que ocorre
quando a reação de PCR entra em sua fase exponencial. A linha do limiar (threshold)
é o ponto ou nível no qual a reação alcança uma intensidade de fluorescência acima
da fluorescência basal.
� Cálculo do ∆Ct: Este valor é obtido subtraindo-se o valor de Ct obtido
para a amostra experimental do valor do Ct de uma amostra referência. Utilizamos
como amostra de referência as três amostras controle cujo resultado foi normal
quanto ao número de cópias do gene GJB2 pela técnica de MLPA e obtivemos a
média entre elas.
� Cálculo do numerador e do denominador da equação: No numerador,
teremos o valor resultante da eficiência dos iniciadores da sequência alvo (calculada
por meio da curva padrão) elevada ao valor do seu ∆Ct, calculado conforme descrito
no parágrafo acima. No denominador, teremos o valor resultante da eficiência dos
iniciadores da sequência de referência elevada ao valor do seu ∆Ct.
As análises por meio da técnica de PCR em tempo real quantitativa foram
realizadas com a ajuda da Dra. Karina Lezirovitz, do Laboratório de

52
Otorrinolaringologia/LIM32 do Hospital das Clínicas da Faculdade de Medicina da
Universidade de São Paulo.
Figura 3.3. Exemplo das curvas de amplificação das diluições seriadas utilizadas para a construção das curvas padrão e cálculo da eficiência dos pares de iniciadores quanto a sequência alvo.
3.4. Resultados
Selecionamos para esse estudo 16 pacientes não aparentados que foram
previamente caracterizados como portadores de uma única mutação recessiva e
patogênica na região de código do gene GJB2 ou como portadores de uma das duas
deleções do gene GJB6 em heterozigose. Dez apresentavam a mutação c.35delG,
dois a mutação c.167delT, dois a mutação p.R127H e em dois deles estava presente
a deleção del(GJB6-D13S1854) no gene GJB6, todas em heterozigose. A Tabela 3.4.
relaciona todos os pacientes estudados, assim como todos os testes realizados e o
resultados encontrados.
3.4.1. Sequenciamento das regiões de código, promotora e doadora de
splicing do intron 1 do gene GJB2
Em nenhum dos 16 pacientes analisados foi detectada mutação nas regiões
promotora ou doadora de splicing do intron 1 do gene GJB2. Essa região sequenciada

53
inclui as mutações -3438C>T na região promotora e a IVS 1 + 1G>A na região
doadora de splicing do intron 1. No entanto, por meio do sequenciamento da região
de código do gene GJB2 do DNA dos indivíduos de uma família com surdez,
identificamos uma nova mutação, a p.L76P ou c.C227T associada à mutação
c.35delG (paciente 1, Tabela 3.4).
A nova mutação p.L76P foi identificada nos dois indivíduos afetados por
surdez na irmandade, associada à mutação c.35delG (Figura 3.4). A análise
molecular dos pais normais evidenciou que essas mutações estão em trans. A
mutação não foi identificada em nenhum de 100 indivíduos ouvintes analisados por
meio da técnica de genotipagem automatizada de SNPs (MegaBace SNuPe Genotyping
Kit) (Figura 3.5).
Figura 3.4. Nova mutação p.L76P no gene GJB2. a) Heredograma da família onde foi identificada a mutação p.L76P (c.C227T). O pai (I-1) é portador da mutação p.L76P em heterozigose e a mãe (I-2) é portadora da mutação c.35delG em heterozigose. Os irmãos (II-1 e II-2) apresentam ambas as mutações p.L76P e c.35delG. Sequenciamento da região codificadora do gene GJB2, que corresponde aos nucleotídeos 222 – 231, do pai heterozigoto (b) e do controle (c).
3.4.2. Triagem da deleção de 200kb localizada a 130kb da região 5’ do
gene GJB6
Em nenhum dos 16 pacientes analisados foi detectada a deleção de 200kb
próxima ao gene GJB6. Na figura 3.6. podem ser observadas as 2 bandas, uma de
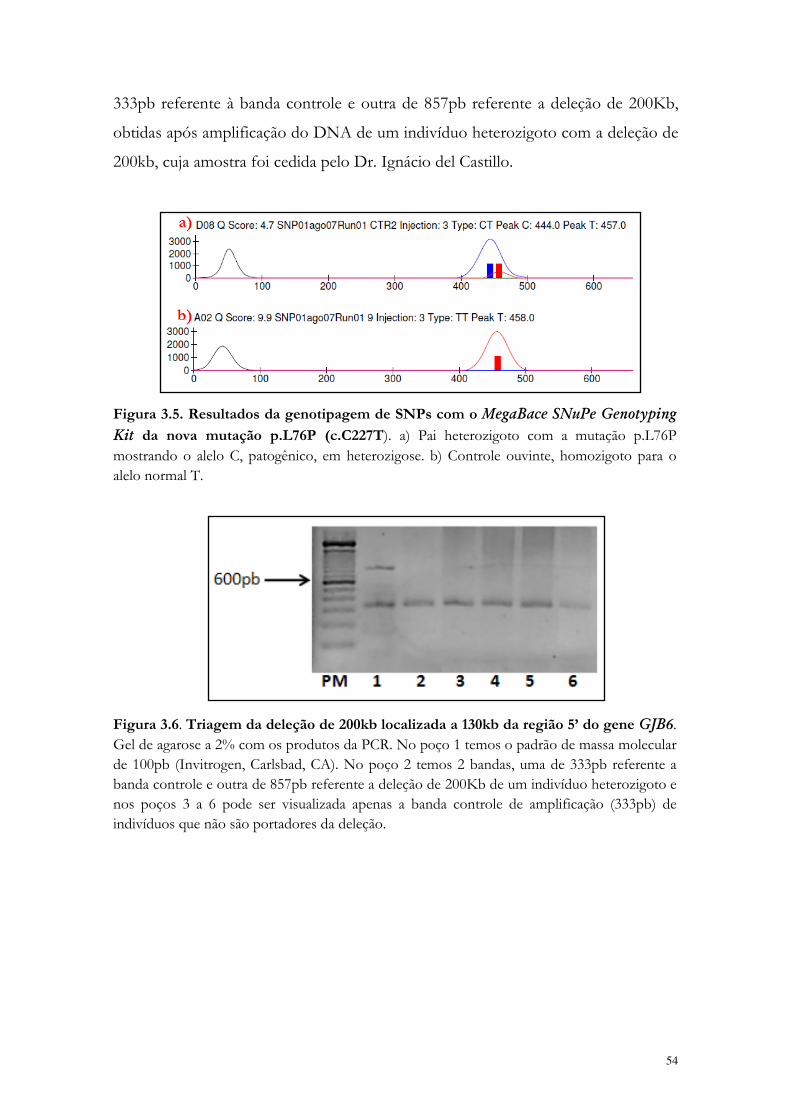
54
333pb referente à banda controle e outra de 857pb referente a deleção de 200Kb,
obtidas após amplificação do DNA de um indivíduo heterozigoto com a deleção de
200kb, cuja amostra foi cedida pelo Dr. Ignácio del Castillo.
Figura 3.5. Resultados da genotipagem de SNPs com o MegaBace SNuPe Genotyping
Kit da nova mutação p.L76P (c.C227T). a) Pai heterozigoto com a mutação p.L76P mostrando o alelo C, patogênico, em heterozigose. b) Controle ouvinte, homozigoto para o alelo normal T.
Figura 3.6. Triagem da deleção de 200kb localizada a 130kb da região 5’ do gene GJB6. Gel de agarose a 2% com os produtos da PCR. No poço 1 temos o padrão de massa molecular de 100pb (Invitrogen, Carlsbad, CA). No poço 2 temos 2 bandas, uma de 333pb referente a banda controle e outra de 857pb referente a deleção de 200Kb de um indivíduo heterozigoto e nos poços 3 a 6 pode ser visualizada apenas a banda controle de amplificação (333pb) de indivíduos que não são portadores da deleção.

55
Tabela 3.4. Caracterização dos dezesseis pacientes com mutações monoalélicas nos genes GJB2 ou GJB6 e resultados das análises moleculares. Abreviações: Pac: paciente, nl: normal, dup: duplicação, del200Kb: deleção de 200kb localizada a 130kb da região 5’ do gene GJB6 e A: ausente. Cód: região de código, P: região promotora, S: região doadora de splicing. Seq: sequenciamento.
Pac Mutações em heterozigose
MLPA
Seq GJB2 del 200kb
Classificação Clínica de Surdez
Observações e/ou outros sinais clínicos
Cód P/S
1 c.35delG (GJB2) nl c.35delG/ p.L76P
nl A sensórioneural, familial, pré-lingual, moderada e estacionária
História familial compatível com herança autossômica recessiva
2 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, isolada, pré-lingual, profunda e estacionária
História familial compatível com herança autossômica recessiva
3 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, isolada, pós-lingual, e profunda História familial compatível com herança autossômica recessiva
4 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, familial, pré-lingual, grave e estacionária História familial compatível com herança autossômica recessiva
5 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, familial, pré-lingual, profunda e estacionária
História familial compatível com herança autossômica recessiva
6 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, familial, pós-lingual, grave e estacionária História familial compatível com herança autossômica recessiva
7 c.35delG (GJB2) nl c.35delG/nl nl A isolada (paciente não apresentou exames audiológicos) Face triangular, clinodactilia e osso frontal amplo: sindrômico
8 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, isolada, moderada e progressiva Retinose pigmentar e deficiência visual: Síndrome de Usher
9 c.35delG (GJB2) nl c.35delG/nl nl A pré-lingual e familial (paciente não apresentou exames audiológicos)
Consanguinidade parental. Hérnia inguinal e coarctação da aorta: sindrômico
10 c.35delG (GJB2) nl c.35delG/nl nl A sensórioneural, familial, pós-lingual, moderada e estacionária
Surdez familial compatível com herança autossômica dominante
11 c.167delT(GJB2) Dup GJB2
c.167delT/nl nl A sensórioneural, isolada, pré-lingual, grave e estacionária Consanguinidade parental. Hérnia inguinal e estrabismo.
12 c.167delT(GJB2) nl c.167delT/nl nl A sensórioneural, familial, pós-lingual, grave e progressiva Surdez familial compatível com herança autossômica dominante
13 p.R127H(GJB2) nl p.R127H/nl nl A sensórioneural, isolada, pré-lingual, grave e progressiva -
14 p.R127H(GJB2) nl p.R127H/nl nl A sensórioneural, isolada, pós-lingual, profunda e progressiva
-
15 del(GJB6-D13S1854) (GJB6)
nl nl/nl nl A familial e pré-lingual (paciente não apresentou exames audiológicos)
História familial compatível com Síndrome de Waardenburg
16 del(GJB6-D13S1854) (GJB6)
nl nl/nl nl A sensórioneural, isolada, pré-lingual, profunda e progressiva
Atraso neuropsicomotor: sindrômico Retardo mental?

56
3.4.3. Pesquisa de variações no número de cópias por MLPA (Multiplex
Ligation-dependent Probe Amplification) e por PCR em tempo real quantitativa
Por meio da análise com o kit SALSA MLPA KIT P163-C1 Hearing loss (MRC
Holland), identificamos em um dos nossos pacientes (paciente 11, Tabela 3.4.), portador
da mutação c.167delT (gene GJB2) em heterozigose, uma duplicação de uma região do
gene GJB2. A análise dos resultados obtidos pelo MLPA com esse kit revelou que cinco
dentre um total de dez sondas para o gene GJB2 estavam duplicadas, sendo que todas se
encontram na região codificadora do gene GJB2, ou seja no exon 2, 09862-L09256
(GJB2_ex2_35delG), 13150-L14407 (GJB2_ex2_M34T), 13126-L15167
(GJB2_ex2_167delT), 13127-L15168 (GJB2_ex2_ 235delC) e 09118-L15423
(GJB2_ex2_313del14) (Tabela 3.1 e Figura 3.7.). Para confirmar a variação no número
de cópias e mapear melhor a duplicação detectada por MLPA no DNA genômico desse
paciente, realizamos a técnica de PCR em tempo real quantitativa. Na Figura 3.8
observa-se que três segmentos estão duplicados. O cálculo do número de cópias por
meio da quantificação relativa (QR) revelou valores ao redor de 1,5 quanto as três
sequências alvo da região codificadora do gene GJB2 nas amostras do paciente 11,
indicando que ele possuía duplicação dessas três sequências (Tabela 3.5.). Como
esperado, foram obtidos valores ao redor de 1,0 para o número de cópias das sequências
alvo da região codificadora do gene GJB2 nas amostras do paciente controle, cujos
resultados haviam sido normais quanto ao gene GJB2 na técnica de MLPA, indicando o
número normal de 1 cópia da sequência por genoma. Em relação às sequências alvo do
gene GJB2 localizadas nas regiões intrônica e 3’ UTR, tanto as amostras do paciente 11
quanto a do paciente controle revelaram valores ao redor de 1,0, portanto, número
normal de cópias (Tabela 3.5. e Figura 3.8.). Dessa forma os dados obtidos por meio da
técnica de PCR em tempo real corroboraram os achados da técnica de MLPA.
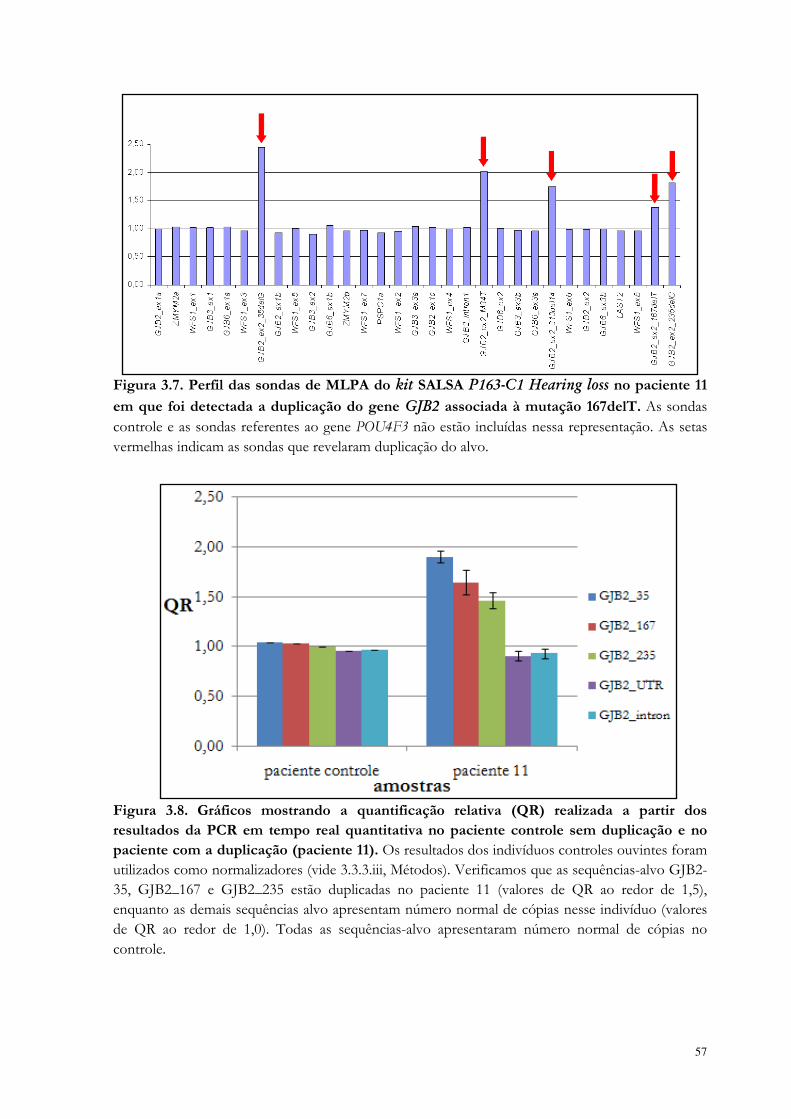
57
Figura 3.7. Perfil das sondas de MLPA do kit SALSA P163-C1 Hearing loss no paciente 11
em que foi detectada a duplicação do gene GJB2 associada à mutação 167delT. As sondas controle e as sondas referentes ao gene POU4F3 não estão incluídas nessa representação. As setas vermelhas indicam as sondas que revelaram duplicação do alvo.
Figura 3.8. Gráficos mostrando a quantificação relativa (QR) realizada a partir dos resultados da PCR em tempo real quantitativa no paciente controle sem duplicação e no paciente com a duplicação (paciente 11). Os resultados dos indivíduos controles ouvintes foram utilizados como normalizadores (vide 3.3.3.iii, Métodos). Verificamos que as sequências-alvo GJB2-35, GJB2_167 e GJB2_235 estão duplicadas no paciente 11 (valores de QR ao redor de 1,5), enquanto as demais sequências alvo apresentam número normal de cópias nesse indivíduo (valores de QR ao redor de 1,0). Todas as sequências-alvo apresentaram número normal de cópias no controle.

58
Tabela 3.5. Resultados das quantificações relativas (QR) das sequências alvo do gene GJB2, que indicam o seu número de cópias, e seus valores de P adotando-se intervalo de confiança de 95%. São consideradas normais as QR iguais a 1 correspondendo a duas cópias da sequência alvo. Valores de razão ao redor de 1.5 correspondem a três cópias da sequência alvo.
Lócus Amostras Média ± dp (QR)
GJB2_35 Paciente 11 1.90 ± 0.19 Paciente Controle 1.04 ± 0.06
Referência 1.00 ± 0.00 Comparação das médias Paciente 11/ Paciente Controle Paciente 11 / Referência Paciente Controle / Referência
GJB2_167 Paciente 11 1.64 ± 0.24 Paciente Controle 1.03 ± 0.12
Referência 1.00 ± 0.00 Comparação das médias Paciente 11/ Paciente Controle Paciente 11 / Referência Paciente Controle / Referência
GJB2_235 Paciente 11 1.46 ± 0.15 Paciente Controle 1.00 ± 0.08
Referência 1.00 ± 0.00 Comparação das médias Paciente 11/ Paciente Controle Paciente 11 / Referência Paciente Controle / Referência
GJB2_intron Paciente 11 0.93 ± 0.12 Paciente Controle 0.97 ± 0.05
Referência 1.00 ± 0.00 Comparação das médias Paciente 11/ Paciente Controle Paciente 11 / Referência Paciente Controle / Referência
GJB2_3’UTR Paciente 11 0.90 ± 0.04 Paciente Controle 0.96 ± 0.05
Referência 1.00 ± 0.00 Comparação das médias Paciente 11/ Paciente Controle Paciente 11 / Referência
Paciente Controle / Referência
Após a realização da PCR em Tempo Real concluímos que a região duplicada no
DNA genômico desse paciente pode apresentar uma extensão mínima de 427pb e
máxima de 1,2Kb (figura 3.9.).
A paciente 11 é filha de consanguíneos (Figura 3.10.) e a sua surdez se caracteriza
por ser sensórioneural, grave, pré-lingual e isolada. Os únicos sinais clínicos adicionais
são hérnia inguinal corrigida logo após o nascimento e estrabismo, o que sugere um
quadro de surdez não-sindrômica de herança autossômica recessiva. Além disso, a
paciente tem origem judaica, como esperado para portadores da mutação c.167delT.

59
Figura 3.9. Representação esquemática do gene GJB2 e da região duplicada detectada no DNA genômico do paciente portador da mutação c.167delT (paciente 11, Tabela 3.4.).
Figura 3.10. Heredograma da família da paciente portadora da mutação c.167delT no gene
GJB2 em heterozigose e na qual foi detectada a duplicação na região de código do gene
GJB2 (paciente 11, Tabela 3.4.).
3.5. Discussão
3.5.1. Sequenciamento das regiões de código, promotora e doadora de
splicing do intron 1 do gene GJB2
A investigação de outras mutações patogênicas que possam estar associadas com
as mutações já caracterizadas em nossos pacientes por meio do sequenciamento do

60
DNA genômico das regiões de código, promotora e doadora de splicing do gene GJB2
identificou em um dos pacientes a nova mutação p.L76P (c.C227T), na região de código,
que até a ocasião não havia sido descrita (Batissoco e col. , 2009, Anexo A).
Análises de bioinformática (world wide web PolyPhen - Polymorphism Phenotyping
(Ramensky e col., 2002; http://genetics.bwh.harvard.edu/pph/, 2011) que levam em
consideração a estrutura da proteína e o tipo dos aminoácidos substituídos indicaram
essa mutação como provavelmente patogênica. Além disso, foi observado que o
aminoácido códon 76, na conexina 26, é conservado tanto entre as diferentes espécies
animais (Figura 3.11) quanto em relação aos vários tipos de conexinas humanas
expressas (dados não mostrados).
Figura 3.11. Análise do alinhamento do resíduo 76 na conexina 26, do gene GJB2, em diferentes espécies. O resíduo do aminoácido 76 está identificado em vermelho.
As conexinas possuem uma estrutura em comum: quatro domínios
transmembrânicos, ligados entre si por duas alças extracelulares e uma alça
citoplasmática, com os grupos amino e carboxi-terminais citoplasmáticos. A mutação
p.L76P ocorre no segundo domínio transmembrânico da proteína e leva à substituição
de uma leucina por uma prolina no códon 76. Algumas mutações patogênicas, tanto
dominantes que levam à surdez sindrômica e não-sindrômica (p.R75W e p.R75Q),
quanto recessivas que levam à surdez não sindrômica (p.W77X, p.W77R) já foram
descritas nessa região. Essas mutações foram associadas com alterações de abertura e
fechamento dos canais de junção do tipo fenda formados pelas conexinas e também
com modificações no ancoramento entre os conexons. Esses fatos indicam ser essa uma
região crítica da proteína conexina 26.

61
A segregação da nova mutação c.C227T (p.L76P) em trans com a c.35delG nessa
família indica o modo de herança provavelmente autossômico recessivo, uma vez que a
mãe é heterozigota com a c.35delG e o pai heterozigoto com a p.L76P e ambos
apresentam audição normal (dados não mostrados). Além disso, a associação entre a
mutação p.L76P e a deficiência auditiva é sustentada pela ausência dessa mutação na
amostra controle de 100 indivíduos e pela análise com o programa Polyphen que indica
essa mutação como provavelmente patogênica.
Por meio do sequenciamento do DNA genômico das regiões promotora e
doadora de splicing do gene GJB2 não identificamos as mutações IVS 1 + 1G>A (-
3170G>A) no exon 1 não codificador que afeta o splicing e a mutação -3438C>T,
localizada na região promotora do gene. A mutação -3438C>T, localizada na região
promotora do gene GJB2 nunca foi descrita na nossa população e apesar da mutação
IVS 1 + 1G>A já ter sido descrita no Brasil, com uma frequência de 4,6% entre os
pacientes surdos já previamente caracterizados como portadores de uma mutação
recessiva na região codificadora do gene GJB2 (Silva-Costa e col., 2009), os nossos
resultados indicam que essas duas mutações devem ser raras em nossa população.
3.5.2 Triagem da deleção de 200kb localizada a 130kb da região 5’ do gene
GJB6
A deleção de 200kb localizada a 130kb da região 5’ do gene GJB6 não foi
identificada em nenhum dos 16 pacientes que foram previamente caracterizados como
portadores de uma única mutação recessiva e patogênica na região de código do gene
GJB2 ou como portadores de uma das duas deleções do gene GJB6 em heterozigose.
Nossos resultados sugerem que essa deleção é rara no Brasil.
Essa deleção de 200kb foi identificada nos indivíduos surdos de uma família
espanhola, associada à mutação c.35delG (del Castillo e col.,2009). Posteriormente, em
um estudo realizado por meio da técnica de MLPA para triar regiões consideradas
“quentes” para conter as deleções no gene GJB6 ou nas suas proximidades, em 236
pacientes surdos e portadores de uma única mutação patogênica no gene GJB2 da
Espanha, do Brasil, da Polônia e da Holanda, a deleção de 200kb localizada a 130kb da

62
região 5’ do gene GJB6 não foi identificada em nenhum dos pacientes analisados (del
Castillo e col., 2011). Essa amostra inclui seis pacientes da amostra aqui descrita. Uma
vez que nenhum outro caso foi descrito, esses resultados demonstram que de fato a
deleção de 200kb localizada nas proximidades do gene GJB6 é muito rara.
É importante lembrar que a prevalência das mutações no gene GJB2 e das
deleções localizadas no gene GJB6 ou nas suas proximidades estão relacionadas com a
origem da população estudada e que os propósitos aqui avaliados resultam de uma
amostra heterogênea etnicamente, fato comum na população brasileira. Porém, há
predomínio de indivíduos com origem européia na nossa casuística.
3.5.3. Pesquisa de variações no número de cópias por MLPA (Multiplex
Ligation-dependent Probe Amplification) e por PCR em tempo real quantitativa.
Por meio da análise de MLPA com o kit SALSA MLPA P163 GJB-WFS1 (MRC-
Holland) identificamos uma região duplicada no gene GJB2, associada à mutação
c.167delT, em um dos nossos pacientes (paciente 11, Tabela 3.4). Para confirmar a
variação no número de cópias no DNA genômico e mapear melhor a região do gene
GJB2 que contém a duplicação detectada realizamos a técnica de PCR em tempo real
quantitativa. A análise dos dados obtidos por meio dessas duas técnicas revelou que a
duplicação presente no DNA genômico desse paciente tem entre 0,4kb e 1,2Kb e
abrange principalmente a região codificadora do gene GJB2. No entanto, não foi ainda
possível localizar o ponto de quebra dessa duplicação e, aparentemente, parte da regiões
5’UTR do exon 2 e 3’UTR podem estar incluídas na região duplicada (Figura 3.9). A
porção da 5’ UTR contida no exon 1 não esta duplicada de acordo com os resultados de
MLPA e PCR em tempo real.
Por meio de análises in silico em bancos de dados de CNVs (copy number variants)
verificamos que essa duplicação não esta catalogada no DGV (Database of Genomic
Variants) que compila as CNVs presentes na população geral e nem há variantes
catalogadas que incluam a região de código do GJB2 (DECIPHER (Database of
Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources ).

63
Resultados preliminares obtidos por meio de tentativa do sequenciamento do
DNA genômico desse paciente utilizando-se diferentes combinações de iniciadores para
amplificar a região duplicada, sugerem que essa duplicação não está em tandem (dados
não mostrados). Trabalhos adicionais serão em breve conduzidos para tentar localizar o
exato ponto de quebra.
Mais de 100 mutações patogênicas (recessivas e dominantes) no gene GJB2 e
cinco diferentes deleções localizadas no gene GJB6 ou nas suas proximidades já foram
descritas e associadas à surdez (del Castillo e col., 2003, 2005 e 2009; Feldmann e col.,
2009; Wilsh e col., 2010; In: Connexin deafness homepage). No entanto, essa é a primeira
duplicação descrita no gene GJB2 associada à surdez de herança autossômica recessiva
(Mingroni-Netto e col., 2011a). Esses resultados foram apresentados no 8th Molecular
Biology of Hearing and Deafness Conference.
Na literatura, são poucas as duplicações descritas associadas à surdez. A maioria
dos casos de variações de número de cópias são deleções de grandes extensões
localizadas em outras regiões cromossômicas, distintas da região 13q11-12 (locús
DFNB1), com exceção das cinco deleções localizadas no gene GJB6 ou nas suas
proximidades. Além disso, nesses casos a surdez é frequentemente acompanhada de
sinais clínicos adicionais (Aller e col., 2010; Bernardini e col., 2008 e 2009; Chen e col.,
2008; Lehman e e col., 2009; Lundin e col., 2010; Mathijssen e col., 2005). Somente três
casos de duplicação genômica são descritos na literatura onde a surdez aparece como
sinal clínico isolado: um caso no gene POU3F4 (Xq21.1.), outro no gene TPJ2 (6q22) e
um terceiro que engloba parte de dois genes, IMMP2L e DOCK4 (7q31.1) (de Kok e col.,
1995; Walsh e col., 2010; Uehara, 2010; Mingroni-Netto e col., 2011b). O primeiro caso
é uma duplicação de 150kb, localizada a 170kb da região proximal ao gene POU3F4
(lócus DFN2) (de Kok e col., 1995). A surdez associada a mutações nesse gene é de
herança ligada ao X, do tipo mista e associada à fixação do estribo. Já a duplicação
descrita no gene TJP2 (lócus DFNA51) possui 270kb e foi identificada em uma família
israelense, cuja surdez se caracterizava por ser sensórioneural, pós-lingual, progressiva e
de herança autossômica dominante (Walsh e col., 2010). Nesses dois casos as
duplicações foram identificadas em tandem. Já o terceiro caso é uma duplicação de no
mínimo 438kb e que engloba os exons 1, 2 e 3 do gene IMMP2L e pelos menos os

64
exons 45 a 52 do gene DOCK4. Essa duplicação foi identificada em uma família com
surdez não-sindrômica de herança autossômica dominante. O gene DOCK4 possui uma
isoforma que se localiza nos estereocílios das células ciliadas e interage com uma
importante proteína relacionada à audição, a harmonina. Essa informação é suficiente
para fazer suspeitar que DOCK4 seja um novo gene responsável por surdez. A
associação de IMMP2L com surdez é menos provável, devido ao grande número de
CNVs (Copy Number Variations) não patogênicas que incluem partes desse gene (Uehara,
2010; Mingroni-Netto e col., 2011b).
Por meio da técnica de MLPA, além da análise por nós realizada com o kit
SALSA MLPA KIT P163-C1 Hearing loss, foram feitas análises adicionais em amostras
de DNA de seis dos nossos 16 pacientes (pacientes 2 a 6 e 11, Tabela 3.4.). Essas
análises foram realizadas no laboratório do Dr. Ignácio del Castillo (Hospital Ramón Y
Cajal, Madri, Espanha) em estudo colaborativo com um kit especificamente desenhado
pela sua equipe para estudar as regiões consideradas “quentes” para conter as deleções
no gene GJB6 ou em regiões vizinhas a ele. Em nenhum desses seis pacientes foi
identificada qualquer alteração (del Castillo e col., 2011). Esse kit de MLPA, desenhado
pela equipe do Dr. Del Castillo, contém sondas que estão localizadas nas regiões
abrangidas pelas deleções de 131,4Kb descrita por Wilch e col., (2010) e de <920Kb
descrita por Feldmann e col., (2009). Desse modo, podemos excluir também a presença
dessas duas deleções nesses seis pacientes (pacientes 2 a 6 e 11, Tabela 3.4.). Quanto aos
outros 10 pacientes restantes, por meio do uso do kit SALSA MLPA KIT P163-C1
Hearing loss, foi possível excluir apenas a presença da deleção de <920Kb.
Entre os 16 pacientes selecionados para o nosso estudo por serem portadores de
uma única mutação recessiva e patogênica na região de código do gene GJB2 ou como
portadores de uma das duas deleções do gene GJB6, delGJB6(D13S1830) e
delGJB6(D13S1854), em heterozigose, cinco (pacientes 7, 8, 9, 15 e 16, Tabela 3.4.)
apresentam outros sinais clínicos além de surdez que os caracterizam como sindrômicos.
Em dois desses pacientes considerados sindrômicos, 8 e 15 (Tabela 3.4.), foram
confirmados os diagnósticos de Síndrome de Usher e de Síndrome de Waardenburg,
respectivamente. Já em dois dos 16 pacientes (pacientes 10 e 12, Tabela 3.4.), o quadro
clínico de surdez não-sindrômica familial apresentado é compatível com o de herança

65
autossômica dominante. Em um desses pacientes (paciente 10, Tabela 3.4.) foi
posteriormente detectada uma mutação patogênica não descrita no gene TECTA,
responsável por surdez não-sindrômica de herança autossômica dominante
(DFNA8/12) que segrega na família (Lezirovitz e col., 2011). Desse modo, concluímos
que as mutações c.35delG e c.167delT no gene GJB2 e a delGJB6(D13S1854) no gene
GJB6 nesses sete pacientes (7 a 10, 12, 15 e 16) ocorreram ao acaso e não explicam o
quadro clínico apresentado por esses indivíduos. Entre esses sete pacientes, quatro
(pacientes 7 a 10) apresentam a mutação c.35delG em heterozigose. Dado que a
frequência de heterozigotos para a mutação c.35delG na nossa população foi estimada
em 1/100 (Sartorato e col., 2000), e que o tamanho da casuística do laboratório é de
cerca de 700 pacientes, em teoria sua ocorrência está de acordo com o esperado para
nossa população.
3.6. Conclusão
Detectamos a segunda mutação provavelmente patogênica apenas em dois dos
nossos 16 pacientes heterozigotos quanto a mutações patogênicas e recessivas nos genes
GJB2 ou GJB6: a mutação p.L76P, localizada na região de código do gene GJB2 descrita
por nós pela primeira vez e uma duplicação que inclui a região de código do gene GJB2,
também inédita na literatura. Em sete dos 16 pacientes, concluímos que as mutações
únicas nos genes GJB2 ou GJB6 provavelmente não estão relacionadas diretamente à
causa de surdez, permanecendo sete casos com etiologia a esclarecer.
.

66
CAPÍTULO 4

66
CAPÍTULO 4 Estabelecimento de culturas primárias de células neuro-epiteliais do
órgão de Corti de cobaias e camundongos
4.1. Introdução
Diversas mutações que causam surdez em humanos já foram identificadas
tanto em genes expressos nas células ciliadas (MYO7A, KCNQ4) quanto em genes
expressos nas células de suporte (GJB2 e GJB6) do órgão de Corti (Van Camp e
Smith, 2011).
Embora a função das células ciliadas sensoriais e de seus neurônios
associados já estejam estabelecidas, as funções das células de suporte emergiram de
estudos mais recentes. Diversos estudos têm demonstrado que essas células não
sensoriais apresentam papel importante no desenvolvimento, na função e na
manutenção da homeostasia da orelha interna (Cotanche e Kaiser, 2010, Gomes-
Casati e col., 2010; Warchol e col., 2010). Além disso, as células de suporte parecem
ser importantes reguladores da atividade sináptica no órgão de Corti (Stankovic e
col., 2004; Sugawara e col., 2005; Glowatzki e col., 2006; Tritsch e col., 2007).
A expressão do inibidor do ciclo celular p27kip1 nas células de suporte do
órgão de Corti maduro está relacionado com o seu estado de quiescência mitótica.
Estudos com camundongos nocautes para o p27kip1 têm demonstrado que a sua
deleção resulta na proliferação contínua das células de suporte do órgão de Corti
(Chen e Segil,1999; Lowenheim e col., 1999, Ono e col., 2009). White e col., (2006),
após dissociação e purificação das células de suporte de camundongos por FACS
(Fluorescence-activated cell sorting) verificaram que essas células são capazes de se
proliferarem e de se transdiferenciarem em células ciliadas e que esse fenômeno é
acompanhado pela redução da expressão do p27kip1. Esses estudos sugerem que há
uma correlação entre os níveis de expressão do p27kip1 e a capacidade das células de
suporte dos mamíferos de re-entrarem no ciclo celular, e que embora as células de
suporte dos mamíferos apresentem in vitro potencial de regeneração semelhante ao
observado em aves, esse potencial deve ser suprimido quando no interior tecido.

67
As células menos diferenciadas encontradas na cóclea dos mamíferos são
progenitoras das células ciliadas e das de suporte. No órgão de Corti, as células com
características de progenitoras localizam-se em regiões onde residem as células de
suporte mais próximas às células ciliadas, como por exemplo, as células Deiters
vizinhas das células ciliadas externas e as inner phalengeals junto às células ciliadas
internas (Figura 1.3, pág. 13, Capítulo 1) (Smeti e col., 2010). Com base nos ensaios
para o isolamento de células-tronco do sistema nervoso central de mamíferos
(Reynolds e Rieze, 2005), em diversos estudos foi possível purificar e isolar uma
população de células progenitoras na cóclea de camundongos recém-nascidos
(Malgrange e col., 2002; Li e col., 2003; Oshima e col., 2007; Savary e col., 2007;
Senn e col., 2007). Essas células, quando mantidas em condições de cultivo não
aderentes, são capazes de formar colônias de células flutuantes, denominadas
“otoesferas”. As células das otoesferas, quando dissociadas, são capazes de formar
novas esferas, ou seja, são capazes de se auto-renovar, expressam marcadores que
são produtos de genes importantes para o desenvolvimento tanto da orelha interna
(ex.: Pax2, BMP-4, BMP-7) quanto do sistema nervoso (ex.: nestina) e podem se
diferenciar nos seguintes tipos celulares: células que expressam marcadores de
células de suporte (ex.: p27kip1, Jagged 1) ou marcadores de células ciliadas do
órgão de Corti (Miosina VIIa, Jagged 2) (Li e col., 2003; Oshima e col., 2007; Savary
e col., 2007; Senn e col., 2007). No entanto, os fatores moleculares responsáveis pela
promoção da auto-renovação e da formação dessas esferas celulares ainda não
foram completamente caracterizados (Doetzlhofer e col., 2004; Zhai e col., 2005;
White e col., 2006; Savary e col., 2008).
Em virtude da irreversibilidade da maior parte dos casos de surdez, pesquisas
recentes buscam desvendar os mecanismos moleculares e celulares relacionados à
fisiologia da audição e, consequentemente, à sua perda, por meio de diversas
estratégias. Modelos animais, culturas de células e estudos funcionais sobre
mutações que causam surdez têm auxiliado a revelar o papel de várias moléculas na
fisiologia da audição. Dado que as proteínas conexinas 26 e 30 se co-localizam nas
células de suporte do órgão de Corti, onde possuem papel importante em vários
processos de transdução auditiva, o cultivo de células epiteliais do órgão de Corti em

68
laboratório e o estudo da sua diferenciação in vitro representam passo fundamental
para estudos básicos de identificação e localização de proteínas relacionadas à
função auditiva.
4.2. Objetivo
Padronizar protocolos para cultivo e diferenciação in vitro de células epiteliais
do órgão de Corti, para criarmos, a médio prazo, condições experimentais
fundamentais para estudos relacionados ao papel das conexinas e suas “parceiras”
na audição e, a longo prazo, para o desenvolvimento de estratégias de terapia celular
e gênica na deficiência auditiva.
4.3. Animais e Métodos
4.3.1. Animais
Cobaias (Cavia porcellus)
As cobaias foram fornecidas pelo CEDEME - UNIFESP (Centro de
Desenvolvimento de Modelos Experimentais para Medicina e Biologia da
Universidade Federal de São Paulo, São Paulo, Brasil), que foi responsável pela sua
manutenção e manejo. O protocolo para trabalho com esses animais foi submetido
à Comissão de Ética para Análise de Projetos de Pesquisa – CAPPesq da Diretoria
Clínica do Hospital das Clínicas e da Faculdade de Medicina da Universidade de São
Paulo e aprovado (Protocolo de pesquisa No 1221/06).
Na fase de pré-experimento, os animais foram mantidos em gaiolas grandes
de polipropileno, com maravalha esterilizada no forro do piso, ração específica e
água acrescida de vitamina C (20 gotas – 200 mg/litro), renovadas diariamente. Os
animais foram transportados em gaiolas do biotério ao Instituto de Biociências da
USP, onde foram realizados os experimentos. Foram utilizados somente animais
recém-nascidos (3 – 4 dias), machos e/ou fêmeas, saudáveis, pesando entre 100 -
300 gramas. Os animais foram tratados de acordo com os princípios éticos e

69
práticos do uso de animais de experimentação. Este estudo experimental envolveu
sacrifício de todos os animais para remoção do material de interesse, sendo realizado
em câmara de dióxido de carbono por cinco minutos. Esse método, recomendado
para pequenos roedores, é de baixo custo, seguro e tem rápida ação letal ao
provocar depressão do sistema nervoso central, minimizando o sofrimento do
animal.
Foi utilizado um total de 92 animais, sendo quatro animais por experimento,
totalizando 23 experimentos.
Camundongos Balb/c (Mus musculus)
Os animais foram fornecidos pelo Instituto de Ciências Biomédicas (ICB) da
USP - Biotério de Criação de Camundongos Isogênicos (São Paulo, Brasil), que foi
responsável pela sua manutenção e manejo. O protocolo para trabalho com
camundongos foi submetido à Comissão de Ética para Análise de Projetos de
Pesquisa – CAPPesq da Diretoria Clínica do Hospital das Clínicas e da Faculdade de
Medicina da Universidade de São Paulo e aprovado (Protocolo de pesquisa No
0466/08).
Foram utilizados somente animais recém-nascidos (1-3 dias), machos e/ou
fêmeas, saudáveis. O procedimento para a sua manipulação foram os mesmos
utilizados para as cobaias.
Foi utilizado um total de 256 animais, sendo que cada cultura foi realizada
com órgão de Corti de oito camundongos, totalizando 32 experimentos.
4.3.2. Métodos
Os procedimentos foram realizados no Laboratório de Genômica Funcional
(IB-USP), sob co-orientação da Profa. Dra. Luciana Amaral Haddad. Experimentos
iniciais também foram realizados no laboratório de Genética Humana, da Profa. Dra.
Angela Vianna Morgante.

70
4.3.2.1. Dissecção do órgão de Corti de cobaias e camundongos
neonatos
A antissepsia do animal sacrificado era feita em banho de álcool absoluto,
seguida da decapitação com tesoura Metzenbaum e incisão longitudinal do arco
mandibular bilateral. Esta abordagem permitiu acesso inferior à bula timpânica e
retirada integral do osso temporal após o afastamento das fáscias e músculos
adjacentes. O material era colocado em placa de Petri contendo meio Leibovitz
(Sigma-Aldrich, St Louis, MO, EUA), aberto para exposição da cóclea e submetido
à dissecção micro-cirúrgica sob visão de um estéreo-microscópio (lupa Tecnival,
SQF-F), com magnificação de 20 vezes. A cápsula coclear era removida usando
micro-pinças e micro-estiletes para exposição dos giros apical, médio e basal. Os
tecidos, contendo o órgão de Corti eram coletados em placa de Petri contendo meio
Dulbecco's Phosphate Buffered Saline (DPBS) (Sigma-Aldrich, St Louis, MO, EUA)
acrescido de glicose 6g/L (Sigma-Aldrich, St Louis, MO, EUA) a 4ºC.
Todo o processo de dissecção foi realizado pelos médicos
otorrinolaringologistas Dra. Jeanne Oiticica e doutorando Luiz Carlos de Melo
Barbosa Júnior do Departamento de Otorrinolaringologia da Faculdade de Medicina
da Universidade de São Paulo, nossos colaboradores nessa pesquisa.
4.3.2.2. Cultura em suspensão de células progenitoras do órgão de
Corti
Cultura primária
A cada experimento, o tecido obtido de todas as cócleas dissecadas à lupa
era submetido à dissociação enzimática e mecânica para obtenção de células
isoladas. Na dissociação enzimática, o tecido era incubado a 37ºC, por 15 min, em
meio HBSS (Hank’s Balanced Salt Solution, NaCl a 137mM, KCl a 5,4mM, Na2HPO4 a
0,3mM, KH2PO4 a 0,4mM, NaHCO3 a 4,2mM, glicose a 5,6mM, HEPES a 300
mM, pH 7,4) contendo elastase (Sigma-Aldrich, St Louis, MO, EUA) a 0,05 U/mL.
Após incubação, era adicionado CaCl2 para uma concentração final de 3mM e
colagenase tipo II (Invitrogen, Carslbad, CA, EUA) com concentração entre 600 a

71
1200 U/mL. Após 15 min de incubação a 37ºC, era adicionado Tryple Express
(Invitrogen, Carslbad, CA, EUA) para uma concentração final de 0,05% e o tecido
era incubado por 15 min, a 37ºC. Após esperar o tecido depositar no fundo do tubo
devido à ação da gravidade e sob gelo, o sobrenadante era descartado e o tecido
lavado duas vezes com HBSS, sendo o sobrenadante novamente descartado. O
tecido obtido era então ressuspendido em 1mL de HBSS, dissociado mecanicamente
com três pipetas Pasteur de calibres decrescentes para obtenção das células e
posteriormente filtrado para remover debris celulares (filtro de 100 µm, BD
FalconTM). Eram utilizados 20 µL do sobrenadante para observar a morfologia e
realizar a contagem das células no microscópio Axiovert 40C (Zeiss, Alemanha). As
células eram plaqueadas em placas de culturas de 24 poços, previamente revestidas
com poli-HEME (Poli -2-hidroxietil metacrilato, Sigma-Aldrich, St Louis, MO,
EUA) para evitar adesão celular (Savary e col, 2007). Para o revestimento, poli-
HEME foi solubilizado em metanol à concentração de 50mg/mL e diluído em
etanol para concentração final de 10 mg/mL. Para preparar o revestimento, eram
adicionados 100µL da solução de poli-HEME para cada poço utilizado da placa de
24 poços, deixado secar ao fluxo laminar, seguido de três lavagens em HBSS. Em
cada poço da placa de 24 poços, eram semeadas 104 células em DMEM:F12 (1:1,
Invitrogen, Carslbad, CA, EUA), glutamina a 2mM (Invitrogen, Carslbad, CA,
EUA), N2 1X (Invitrogen, Carslbad, CA, EUA), 50 µL/mL de ampicilina (TEUTO,
Brasil), suplementado com 20ng/mL do fator de crescimento epidérmico (EGF,
Invitrogen, Carslbad, CA, EUA). Os fatores de crescimento transformante de
crescimento alfa (TGFα, Invitrogen, Carslbad, CA, EUA) (Savary e col., 2007) ou
básico de fibroblastos (bFGF, Invitrogen, Carslbad, CA, EUA) foram adicionados
respectivamente para as concentrações de 20ng/mL e 10ng/ml. O número de
células viáveis era determinado pela exclusão do corante azul de tripan em câmara
de Neubauer. As placas eram mantidas a 37ºC, em incubadora com CO2 a 5% e a
troca de 50% do meio de cultura era realizada a cada dois dias (Lou e col., 2007).
Expansão
As culturas primárias eram mantidas por sete dias in vitro (7DIV), enquanto
que os repiques referentes às passagens P1 e P2 eram realizados entre o terceiro e o

72
quarto DIV. Para os repiques, otoesferas e meio de cultura eram transferidos para
um microtubo de capacidade de 2mL e centrifugados a 200 x g, por cinco min, a
4ºC. O sobrenadante era removido e Tryple Express (Invitrogen, Carslbad, CA, EUA)
era adicionado na proporção 1:1 para dissociação das otoesferas. Esta suspensão era
incubada por 10 min, a 37ºC, seguida de nova centrifugação a 400 x g, por cinco
minutos a 4ºC. O sobrenadante era removido e as células eram ressuspendidas em
500µL de meio de cultura completo e dissociadas com pipeta Pasteur, repetidas vezes.
Como alternativa, a dissociação foi feita também com o kit NeuroCult Chemical
Dissociation (Stem Cell Technologies, Vancouver, Canadá), de acordo as instruções
do fabricante.
O número de células viáveis era determinado em câmara de Neubauer e as
células dissociadas eram plaqueadas (104 células/poço) nas mesmas condições
descritas acima. Este protocolo permitiu a análise da auto-renovação de células
provenientes de otoesferas primárias, secundárias e terciárias.
Diferenciação das otoesferas
Para o ensaio de diferenciação, as otoesferas e todo o meio de cultura eram
transferidos para câmaras (BD FalconTM) de oito poços previamente revestidas por
16 horas, com poli-D-ornitina (Sigma-Aldrich, St Louis, MO, EUA) a 0.1mg/mL e
fibronectina (Sigma-Aldrich, St Louis, MO, EUA) a 5 µg/mL por duas a quatro
horas, a 37ºC e mantidas em condições para adesão celular, usando o mesmo meio
de cultura em suspensão, embora desprovido dos fatores tróficos. A cultura era
mantida a 37ºC, em incubadora com CO2 a 5% e após a adesão celular, era realizada
a troca de 80% do meio, sendo que esse procedimento era repetido no terceiro ou
quarto DIV. Após sete dias de cultivo, as células diferenciadas eram fixadas e
analisadas por imunofluorescência indireta.
Fixação
Antes de realizar a imunofluorescência indireta, tanto as otoesferas quanto as
células diferenciadas eram fixadas com PFA (paraformaldeído).

73
Para fixação das otoesferas presentes na cultura em suspensão, todas as
otoesferas formadas e todo o meio de cultura eram transferidos para câmaras de
oito poços previamente revestidas por 16 horas, com poli-D-ornitina (Sigma-
Aldrich, St Louis, MO, EUA) a 0,1 µg/mL e fibronectina (Sigma-Aldrich, St Louis,
MO, EUA) a 5 µg/mL por duas a quatro horas, a 37ºC. As câmaras eram mantidas
por duas horas 37ºC e 5% de CO2 e eram centrifugadas a 4ºC, por dois minutos a
200 x g para depósito das células (Widera e col., 2006). Todo o meio de cultura era
removido gentilmente e as células eram fixadas em paraformaldeído a 4%, por 60
minutos, a 37ºC e 5% de CO2. O paraformaldeído era retirado e as otoesferas eram
lavadas duas vezes com HBSS.
Para fixação das células resultantes do ensaio de diferenciação todo o meio
de cultura era removido e as células eram fixadas em paraformaldeído a 4%, por 60
minutos, a 37ºC. O paraformaldeído era retirado e as células eram lavadas duas
vezes com HBSS.
4.3.2.3. Caracterização fenotípica das células em cultura
A caracterização fenotípica das células fixadas (em suspensão ou
diferenciadas) era feita por imunofluorescência indireta, com anticorpos específicos
para diversos tipos de marcadores celulares, esperados nesse tipo de cultura. O
protocolo de imunofluorescência foi adaptado de Pranchevicius e col., 2008. Na
tabela 4.1. temos a relação de todos os anticorpos primários utilizados e suas
respectivas diluições de uso. Todos esses anticorpos primários foram utilizados
diluídos em HBSS contendo BSA (Bovine Serum Albumin, Invitrogen, Carslbad, CA,
EUA) a 0,3%.
Após a fixação, as células eram permeabilizadas em triton X-100 a 0,3%, por
20 minutos, à temperatura ambiente. A seguir, as células eram bloqueadas por uma
hora, à temperatura ambiente, com IgG não imune (Santa Cruz Biothnologies,
Santa Cruz, CA, EUA), específica para a espécie que da origem ao anticorpo
secundário a 5% em HBSS, e BSA (Bovine Serum Albumine, Invitrogen, Carslbad, CA,
EUA) a 0,3%, e incubadas com o anticorpo primário diluído em HBSS e BSA a

74
0,3% por 16 horas à temperatura ambiente. Após incubação, as células eram lavadas
em HBSS e incubadas novamente, por uma hora, à temperatura ambiente com os
anticorpos secundários conjugados aos fluorocromos e diluídos em HBSS na
proporção 1:500 (Cy3, ALEXA 546 ou ALEXA 488 IgG de anti-camundongo,
FITC, ALEXA 546 ou ALEXA 488 IgG de anti-coelho e ALEXA 546 ou
ALEXA 488 IgG de anti-cabra) (Invitrogen, Carslbad, CA, EUA). As lamínulas
eram montadas em lâminas contendo ProLong Gold Antifade (Invitrogen,
Carslbad, CA, EUA) contendo DAPI (4’, 4-diamidina-fenil indol) para identificação
do núcleo e seladas com esmalte. As imagens eram observadas em microscópio de
imunofluorescência Axioplan (Carl Zeiss, Alemanha) e capturadas com software Isis
Fish Imaging Meta System ou em microscópio confocal (LSM 410 ou LSM 510,Carl
Zeiss, Alemanha) e capturadas com o software Zeiss LSM Image Browser.
Tabela 4.1. Relação dos anticorpos primários utilizados na caracterização fenotípica das culturas de células progenitoras do órgão de Corti de cobaias e camundongos (cultura de células em suspensão e diferenciadas).
Anticorpo primário
Tipo/Animal de origem
Empresa Tipo celular/localização Diluição de uso
Anti-nestina (clone rat-401)
Monoclonal/ camundongo
Chemicon Filamento intermediário expresso em de células-tronco
neuroepiteliais durante o desenvolvimento embrionário.
Marcador de neurônios imaturos e neuroblastos(Lopez e col., 2004; Wiese e col., 2004)
1:200
Anti-Sox2 (clone S-15)
Policlonal/ cabra
Santa Cruz Fator de transcrição que participa do desenvolvimento sensorial da orelha interna, da
determinação do fenótipo celular e da manutenção do
estado de células-tronco. (Yerukhimovich e col., 2007).
1:100
Anti-Miosina VIIa (clone PA1-
936)
Policlonal/ coelho
Affinity Bioreagentes
Proteína motora específica das células ciliadas do órgão de
Corti
1:100
Anti-p27kip1 (clone Y236)
Monoclonal/ coelho
Abcan Inibidor de cinase dependente de ciclina. Marcador molecular do órgão de corti rudimentar e das células de suporte maduras.
1:100
Anti- Jagged1 (clone C-20)
Policlonal/ cabra
Santa Cruz Proteína de membrana tipo I das células de suporte do órgão
de Corti
1:100
Anti-Jagged2 (clone C-17)
Policlonal/ cabra
Santa Cruz Proteína de membrana tipo I das células ciliadas do órgão de
Corti
1:100

75
4.3.2.4. Contagem celular
A contagem direta do número de esferas ou células foi realizada em 20
campos consecutivos do microscópio com objetiva de 40x para cada lâmina. Em
cada campo era contado o número de núcleos marcados com DAPI refletindo o
número total de células e/ou o número de células marcadas com o(s) anticorpo(s)
primário(s) utilizado(s). Os seguintes parâmetros foram analisados: (i) número total
de células ou de otoesferas para cada campo; (ii) número total de células marcadas
com o anticorpo primário; (iii) número de células em otoesferas presentes e (iv)
número de células marcadas com o anti-corpo primário em cada esfera.
4.3.2.5. Análises Estatísticas
Os resultados foram expressos como média ± desvio padrão da porcentagem
de células marcadas, para cada condição analisada. As variáveis contínuas foram
comparadas pelo teste t de Student. O nível de significância estatística foi
estabelecido em p<0,05. As análises estatísticas foram realizadas com o sofware
GraphPad Instant.
4.4. Resultados
Para a padronização das culturas de células em suspensão do órgão de Corti,
utilizamos cobaias e camundongos. Iniciamos os trabalhos com cobaias porque a
técnica de dissecção já estava padronizada pelo grupo de pesquisa do Departamento
de Otorrinolaringologia da Faculdade de Medicina da Universidade de São Paulo.
Durante o curso do trabalho, a escolha de se utilizar também camundongos foi
reforçada pela disponibilidade de protocolos para o cultivo bem estabelecidos na
literatura. Além disso, o genoma do camundongo é mais conhecido e há maior
número de anticorpos específicos a esta espécie disponíveis comercialmente.
Realizamos um total de 55 experimentos (23 com cobaias e 32 com
camundongos) para padronização da cultura de células do órgão de Corti de cobaias
e camundongos. Nesse período, padronizamos: (i) a técnica de dissecção (Figura
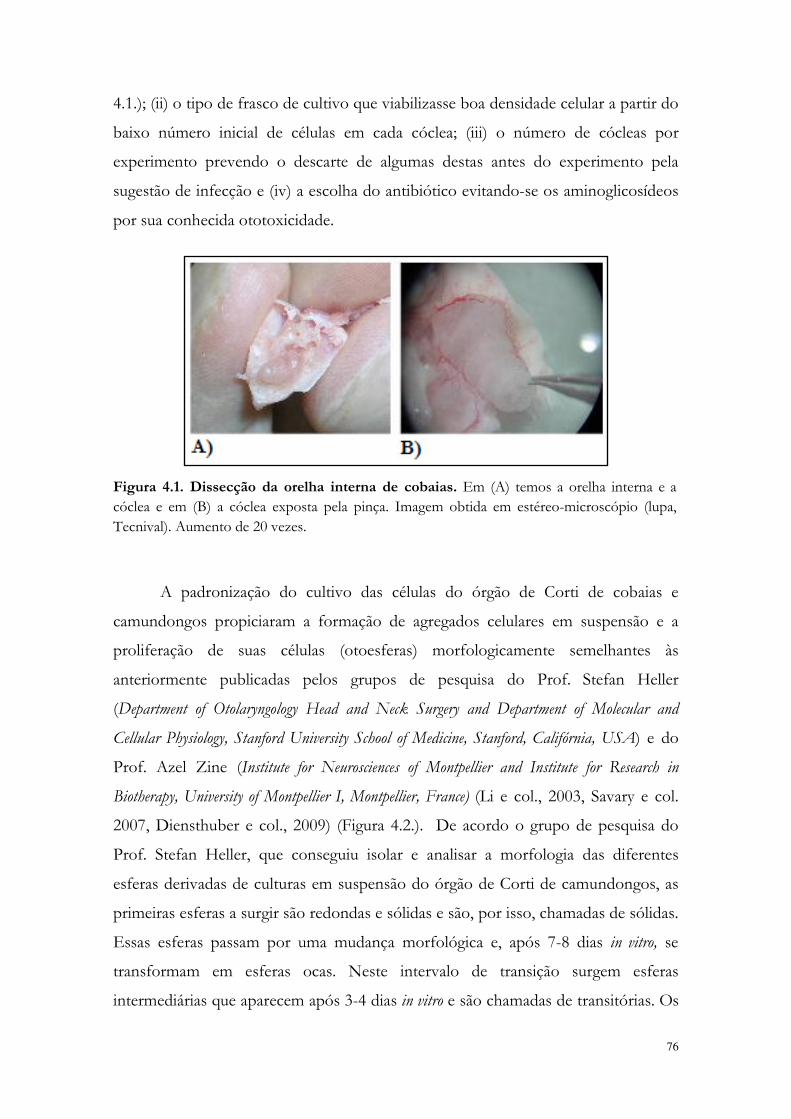
76
4.1.); (ii) o tipo de frasco de cultivo que viabilizasse boa densidade celular a partir do
baixo número inicial de células em cada cóclea; (iii) o número de cócleas por
experimento prevendo o descarte de algumas destas antes do experimento pela
sugestão de infecção e (iv) a escolha do antibiótico evitando-se os aminoglicosídeos
por sua conhecida ototoxicidade.
Figura 4.1. Dissecção da orelha interna de cobaias. Em (A) temos a orelha interna e a cóclea e em (B) a cóclea exposta pela pinça. Imagem obtida em estéreo-microscópio (lupa, Tecnival). Aumento de 20 vezes.
A padronização do cultivo das células do órgão de Corti de cobaias e
camundongos propiciaram a formação de agregados celulares em suspensão e a
proliferação de suas células (otoesferas) morfologicamente semelhantes às
anteriormente publicadas pelos grupos de pesquisa do Prof. Stefan Heller
(Department of Otolaryngology Head and Neck Surgery and Department of Molecular and
Cellular Physiology, Stanford University School of Medicine, Stanford, Califórnia, USA) e do
Prof. Azel Zine (Institute for Neurosciences of Montpellier and Institute for Research in
Biotherapy, University of Montpellier I, Montpellier, France) (Li e col., 2003, Savary e col.
2007, Diensthuber e col., 2009) (Figura 4.2.). De acordo o grupo de pesquisa do
Prof. Stefan Heller, que conseguiu isolar e analisar a morfologia das diferentes
esferas derivadas de culturas em suspensão do órgão de Corti de camundongos, as
primeiras esferas a surgir são redondas e sólidas e são, por isso, chamadas de sólidas.
Essas esferas passam por uma mudança morfológica e, após 7-8 dias in vitro, se
transformam em esferas ocas. Neste intervalo de transição surgem esferas
intermediárias que aparecem após 3-4 dias in vitro e são chamadas de transitórias. Os
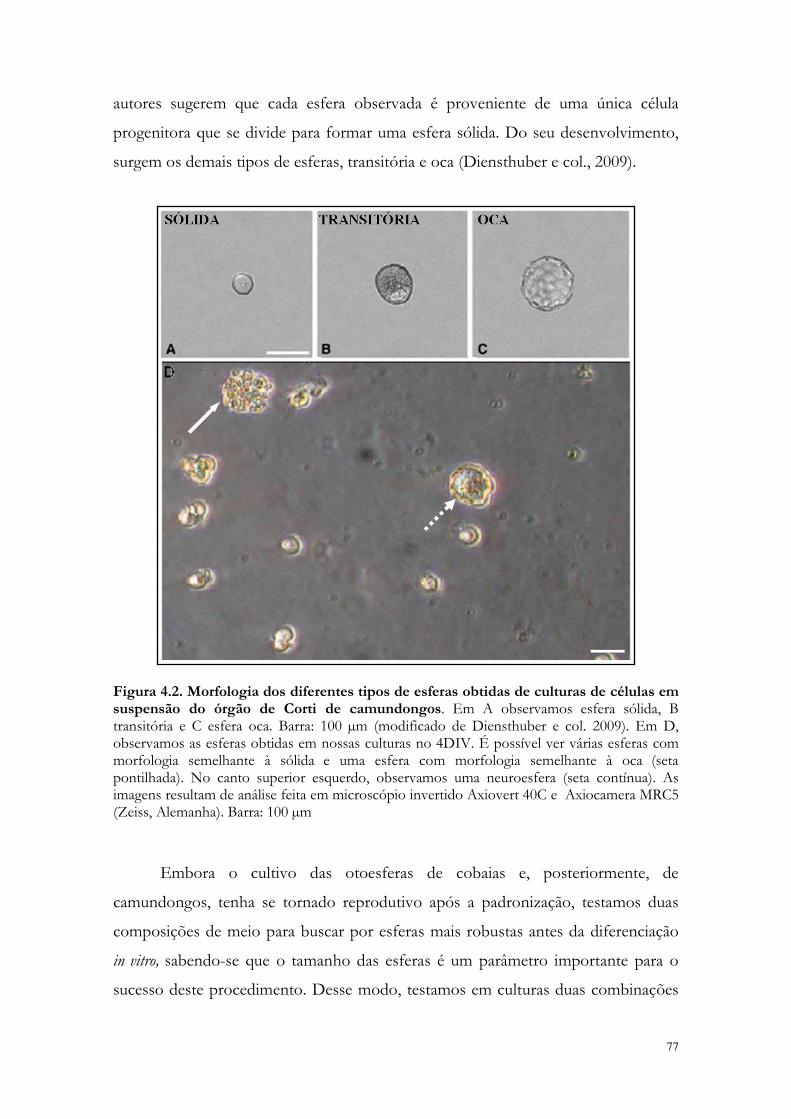
77
autores sugerem que cada esfera observada é proveniente de uma única célula
progenitora que se divide para formar uma esfera sólida. Do seu desenvolvimento,
surgem os demais tipos de esferas, transitória e oca (Diensthuber e col., 2009).
Figura 4.2. Morfologia dos diferentes tipos de esferas obtidas de culturas de células em suspensão do órgão de Corti de camundongos. Em A observamos esfera sólida, B transitória e C esfera oca. Barra: 100 µm (modificado de Diensthuber e col. 2009). Em D, observamos as esferas obtidas em nossas culturas no 4DIV. É possível ver várias esferas com morfologia semelhante à sólida e uma esfera com morfologia semelhante à oca (seta pontilhada). No canto superior esquerdo, observamos uma neuroesfera (seta contínua). As imagens resultam de análise feita em microscópio invertido Axiovert 40C e Axiocamera MRC5 (Zeiss, Alemanha). Barra: 100 µm
Embora o cultivo das otoesferas de cobaias e, posteriormente, de
camundongos, tenha se tornado reprodutivo após a padronização, testamos duas
composições de meio para buscar por esferas mais robustas antes da diferenciação
in vitro, sabendo-se que o tamanho das esferas é um parâmetro importante para o
sucesso deste procedimento. Desse modo, testamos em culturas duas combinações

78
diferentes de fatores de crescimento para enriquecer o meio de cultura e produção
de esferas, baseando-se em resultados do grupo de pesquisa do Prof. Azel Zine
(University of Montpellier, France) (Savary e col., 2008) com o fator de crescimento
epidérmico (EGF) associado ao transformador de crescimento alfa (TGFα).
Denominamos esse primeiro grupo de cultivo de TGFα e comparamos os
resultados obtidos em cultura com esse grupo com os resultados obtidos das
culturas tratadas com o fator de crescimento epidérmico (EGF) associado ao fator
de crescimento básico de fibroblastos (bFGF) denominado grupo bFGF. Os
resultados obtidos com as culturas de segunda passagem (P2) com esses dois grupos
de fatores de crescimento e sua comparação foram publicados no periódico Journal
of Translation Medicine (Oiticica e col., 2010 - Anexo B) e serão resumidamente
descritos a seguir.
A partir do cultivo em suspensão de células do órgão de Corti de cobaias e
camundongos, verificamos que as otoesferas obtidas em ambas as culturas
apresentaram potencial de proliferação. Não padronizamos um ensaio mensurável
da proliferação celular para esse sistema. Mesmo assim, evidenciamos facilmente
aumento do número das células e tamanho das esferas na cultura primária após
alguns dias in vitro (Figura 4.3). A auto-renovação foi evidenciada pelo surgimento de
novas esferas após repique da cultura primária, ou seja, subcultivo de esferas
dissociadas (Figura 4.4). No primeiro dia de subcultivo as células apareceram
isoladas ou formando colônias de duas a três células. Após três dias in vitro, elas
formaram colônias multicelulares flutuantes, ou seja, otoesferas (Figura 4.3.).
Observamos ainda o aumento no tamanho das esferas em cultura, ao longo do
tempo, sugerindo que, após o repique, as células obtidas a partir de esferas
dissociadas são capazes de proliferar e formar novas esferas (Figura 4.4.). Esses
resultados foram observados utilizando-se os dois grupos de fatores de crescimento.
Estas são evidências da capacidade de proliferação e auto-renovação de células do
órgão de Corti de camundongos e cobaias, mantidas em cultura de suspensão.
Comparando os dois grupos de fatores de crescimento (TGFα e bFGF)
utilizados no meio de cultura para o cultivo em suspensão de células do órgão de
Corti de cobaias e camundongos, verificamos que houve uma diferença significativa

79
em relação ao número de otoesferas obtidas em ambas as espécies, com maior
número de otoesferas observadas no grupo TGFα (23.3 ± 8.5) em relação ao grupo
bFGF (9 ± 1, p=0.044, teste-t de Student) (Tabela 4.2 e Figura 4.5.).
Figura 4.3. Análise do potencial de proliferação das células do órgão de Corti de cobaias e camundongos em suspensão. As imagens representam a análise feita em microscópio invertido Axiovert 40C e Axiocamera MRC5 (Zeiss, Alemanha) de otoesferas observadas em contraste de fase durante cultura em suspensão do órgão de Corti de cócleas de
cobaias e de camundongos, cujos meios foram enriquecidos com TGFα. Do mesmo modo, as otoesferas cultivadas em meio enriquecido com bFGF apresentaram o mesmo padrão de proliferação celular (dados não mostrados). Todas as imagens são de células cultivadas após primeira passagem no primeiro (1DIV) ou quarto (4DIV) dias in vitro . As setas indicam as
otoesferas secundárias. Barra 50µm.
Quando analisamos o número de otoesferas obtidas, comparando cobaias
(11.5 ± 4.9) e camundongos (18.5 ± 11), não foi observada nenhuma diferença
significativa (p=0.458, teste-t de Student). Por outro lado, verificamos que o número
de células presentes em otoesferas das culturas de camundongos (32.6 ± 30.5) foi
maior quando comparado com o número de células em otoesferas de cobaias (12.5
± 5.8, p=0.041, test-t de Student). Deste modo, concluímos que; (i) TGFα em
presença do EGF resulta em maior número de otoesferas a partir do cultivo em
suspensão de células do órgão de Corti de cobaias e camundongos, quando
comparado ao número de otoesferas obtidas no grupo bFGF; e que (ii) cócleas de
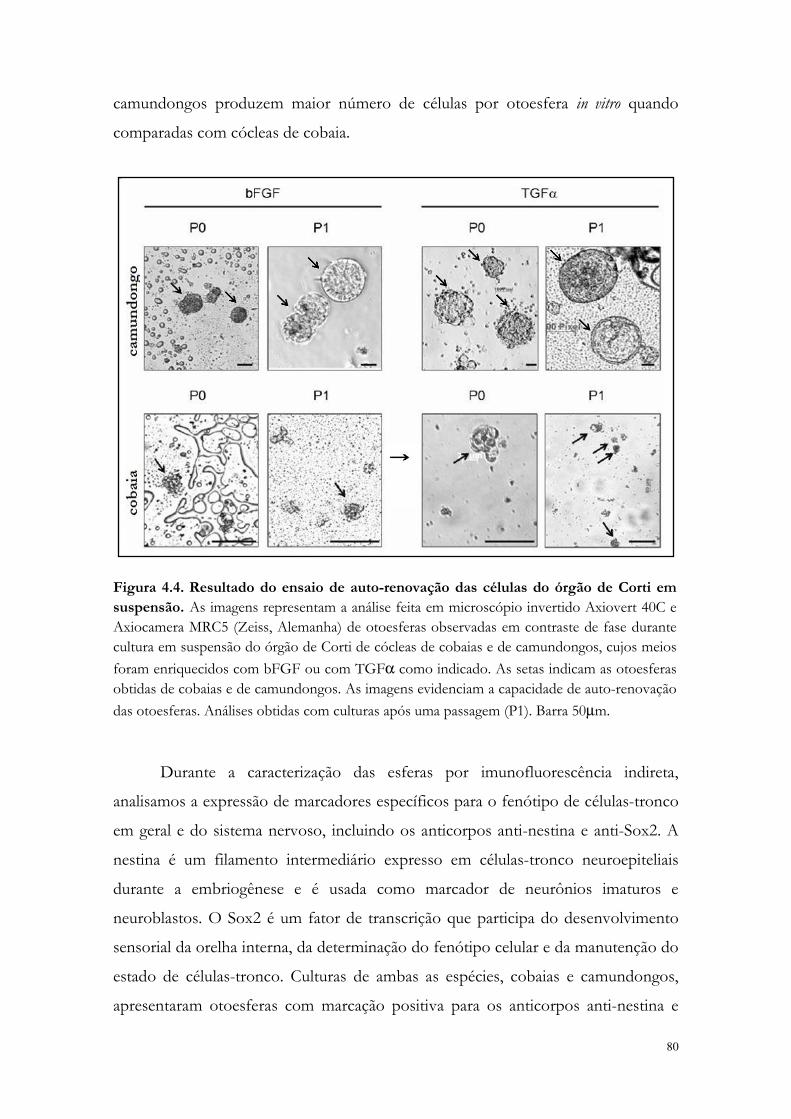
80
camundongos produzem maior número de células por otoesfera in vitro quando
comparadas com cócleas de cobaia.
Figura 4.4. Resultado do ensaio de auto-renovação das células do órgão de Corti em suspensão. As imagens representam a análise feita em microscópio invertido Axiovert 40C e Axiocamera MRC5 (Zeiss, Alemanha) de otoesferas observadas em contraste de fase durante cultura em suspensão do órgão de Corti de cócleas de cobaias e de camundongos, cujos meios
foram enriquecidos com bFGF ou com TGFα como indicado. As setas indicam as otoesferas obtidas de cobaias e de camundongos. As imagens evidenciam a capacidade de auto-renovação
das otoesferas. Análises obtidas com culturas após uma passagem (P1). Barra 50µm.
Durante a caracterização das esferas por imunofluorescência indireta,
analisamos a expressão de marcadores específicos para o fenótipo de células-tronco
em geral e do sistema nervoso, incluindo os anticorpos anti-nestina e anti-Sox2. A
nestina é um filamento intermediário expresso em células-tronco neuroepiteliais
durante a embriogênese e é usada como marcador de neurônios imaturos e
neuroblastos. O Sox2 é um fator de transcrição que participa do desenvolvimento
sensorial da orelha interna, da determinação do fenótipo celular e da manutenção do
estado de células-tronco. Culturas de ambas as espécies, cobaias e camundongos,
apresentaram otoesferas com marcação positiva para os anticorpos anti-nestina e

81
anti-Sox2, com distribuição citoplasmática em aproximadamente 40% das células
(Figura 4.6.). Portanto, o material da cóclea de ambos os animais, camundongos e
cobaias, originou esferas que expressam marcadores de pluripotência. Por outro
lado, não observamos marcação nuclear para Sox2, o que poderia ser esperado uma
vez que esse fator transcricional transita entre o núcleo e o citoplasma (Hume e col.,
2007).
Tabela 4.2. Análise dos parâmetros obtidos quanto ao número de otoesferas e o número de células por otoesferas presentes no cultivo em suspensão de células do órgão de Corti de cobaias e camundongos, cujo meio de cultura foi enriquecido com TGFα ou bFGF.
Grupos bFGF TGFα p camundongo* cobaia* p
nº de otoesferas
9±1 23.3±8.5 0.044 18.5±11 11.5±4.9 0.458
nº de células por otoesfera
16.3±4.1 37.6±23.5 0.098 32.6±30.5 12.5±5.8 0.041
Esses valores representam o desvio padrão da média ±1, onde p foi obtido por meio do test- t de Student. * foram considerados ambos os grupos de fatores de crescimento. As análises foram realizadas em culturas após duas passagens (P2).
As condições para a diferenciação in vitro das otoesferas em células ciliadas e
células de suporte já foram descritas por Li e col. em 2003. Com base nesse estudo,
cultivamos otoesferas de camundongos após uma ou duas passagens (P1 ou P2) sob
condições aderentes e em meio de cultura que favorece a diferenciação em células
de suporte e células ciliadas. Nesse caso, foi utilizado o mesmo meio de cultura para
o cultivo em suspensão, mas desprovido dos fatores de crescimento. Após
diferenciação, observamos nas culturas de camundongos otoesferas que
apresentaram células que expressam marcadores para células de suporte (p27kip1 e
jagged1) e otoesferas que apresentaram células que expressam marcadores para
células ciliadas (miosina VIIa e jagged2), indicando o direcionamento destas células
progenitoras do órgão de Corti à expressão de fenótipos de células de suporte ou
células ciliadas (Figuras 4.7. e 4.8.). Esta conclusão é reforçada pela observação da
distribuição subcelular esperada para esses marcadores. Anticorpo anti-miosina VIIa
marcou finos processos da membrana plasmática (Figuras 4.8A e 4.8C). Jagged 1 foi
observado na membrana plasmática (Figura 4.7B), assim como Jagged 2 (Figura

82
4.8B). Marcação nuclear foi visualizada em células incubadas com o anticorpo
p27kip1 (Figura 4.7A). Quanto às otoesferas obtidas de cobaias, não conseguimos
mantê-las sob condições de cultivo aderente, pois as células morreram. Desse modo,
não foi possível conseguir a diferenciação celular. Este resultado pode ser devido ao
baixo número de células observado nas otoesferas de cobaias, quando comparado
com as otoesferas de camundongos. Estas evidências, em conjunto, confirmaram
que a espécie Mus musculus é útil como modelo experimental para o cultivo de
otoesferas e sua diferenciação
Figura 4.5. Análise do número de células presentes nas otoesferas obtidas com o cultivo em suspensão das células do órgão de Corti de cobaias e camundongos em meio enriquecido com TGFα ou bFGF. As imagens resultam de análise feita em microscópio invertido Axiovert 40C e Axiocamera MRC5 (Zeiss, Alemanha) de otoesferas observadas em contraste de fase durante cultura em suspensão do órgão de Corti de cócleas de cobaias e de
camundongos, cujos meios foram enriquecidos tanto com bFGF como com TGFα. Barra
50µm.

83
Figura 4.6. Imunofluorescência indireta com os anticorpos anti-nestina e anti-Sox2 nas otoesferas de cobaias e camundongos, após uma
ou duas passagens, cultivadas na presença de bFGF or TGFαααα. Os marcadores de células-tronco sox2 e nestina, foram testados para
caracterização fenotípica das otoesferas e o DAPI foi usado na marcação do núcleo celular. Barra 10µm. Análise realizada em microscópio confocal (LSM 410, Carl Zeiss, Alemanha).
83

84
Figura 4.7.: Imunofluorescência indireta com os anticorpos p27kip1 e Jagged 1 nas otoesferas de camundongos, após duas passagens, cultivadas na presença de TGF e submetidas a diferenciação celular. Em A) C observamos o p27kip1 e sua marcação vermelha (Alexa 546) no núcleo das células de suporte. Em B) Jagged 1, outro marcador para células de suporte e sua marcação peculiar em verde (Alexa 488) corando a membrana plasmática. O DAPI marca em azul o DNA nuclear. Barra 10µm (LSM 410 Carl Zeiss, Germany).
Figura 4.8: Imunofluorescência indireta com os anticorpos miosina VIIa e Jagged 2, nas otoesferas de camundongos, após duas passagens, cultivadas na presença de TGF e submetidas a diferenciação celular. Em A e C podemos observar miosina VIIa (MyoVIIA), marcador de células ciliadas, corado por Alexa 488. As setas indicam os processos da membrana plasmática, onde há um acúmulo maior de miosina VIIa. Em B) podemos observar a marcação com Jagged2, outro marcador de células ciliadas, corado por Alexa 546. As setas indicam maiores concentrações da marcação próxima à membrana plasmática. O DAPI identifica os núcleos celulares marcados em azul Barra 10µm (LSM 410 Carl Zeiss, Alemanha).

85
4.5. Discussão
A alta prevalência de surdez em humanos combinada à perda da capacidade
de regeneração das células ciliadas em mamíferos tem alimentado o interesse na
identificação de células-tronco ou células progenitoras que possam vir a ser
utilizadas em terapias para regeneração celular da orelha interna e restauração da
audição (Parker e Cotanche, 2004; Edge e Chen, 2008). Em mamíferos, as células
ciliadas auditivas são produzidas apenas durante o desenvolvimento embrionário e
não são capazes de se regenerarem ao longo da vida (Stone e col., 1998). Desse
modo, a perda das células ciliadas é permanente e resulta em disfunção do órgão.
São várias as evidências da presença das células progenitoras no epitélio
sensorial da orelha interna. Em primeiro lugar, a formação de colônias de células
flutuantes ou otoesferas a partir do epitélio sensorial de aves (Corwin e Cotanche,
1988; Ryals e Rubel, 1988) e peixes (Hernandez e col., 2007), do vestíbulo e da
cóclea neonatal de camundongos (Li e col., 2003; Oshima e col., 2007) e do gânglio
espiral de humanos e cobaias (Rask-Andersen e col., 2005). O segundo ponto é que
as células dessas otoesferas são capazes de proliferar e se auto-renovar (Li e col.,
2003; Oshima e col., 2007). Além disso, as células presentes nas otoesferas
provenientes do vestíbulo e da cóclea são capazes de se diferenciar em células
ciliadas e células com fenótipo neuronal (Oshima e col., 2007; Martinez-Monedero e
col., 2008). Finalmente, já foi demonstrado que as otoesferas obtidas do vestíbulo
são capazes de se diferenciar em células das linhagens ectodérmica, mesodérmica e
endodérmica (Oshima e col., 2007).
O processo de regeneração das células ciliadas ocorre por meio da re-entrada
das células de suporte no ciclo celular e subsequente divisão assimétrica, gerando
novas células ciliadas e novas células de suporte. No entanto, em alguns casos se
observa a conversão fenotípica, também conhecida como transdiferenciação, na
qual células de suporte são convertidas em células ciliadas sem que haja mitose.
Apesar desse mecanismo ser potencialmente mais rápido na geração de novas
células ciliadas, ele também requer subsequentes divisões para reabastecimento das
células de suporte (Brigande e Heller, 2009).

86
Estudos in vivo em aves têm demonstrado que a capacidade de regeneração
das células ciliadas frente a lesões após exposição por aminoglicosídeos é muito
rápida. O início da regeneração das células ciliadas ocorre a partir da reentrada no
ciclo celular das células de suporte que as circundam e ocorre dentro de um período
de 16 horas, sendo que dentre dois a três dias já é possível observar que todas as
células ciliadas lesadas foram substituídas. Aparentemente o processo de
regeneração em aves envolve ambas as vias, transdiferenciação das células de
suporte em células ciliadas seguida da proliferação das células de suporte. No
entanto, a alteração fenotípica das células de suporte em células ciliadas ainda não
foi totalmente caracterizada (Walchol e Corwin, 1996; Roberson e col., 2004;
Duncan e col., 2006; Edge e Chen, 2008; Groves, 2010; Warchol, 2010).
Em mamíferos, apenas o epitélio vestibular apresenta potencial de
regeneração, ainda que limitado, sendo necessário quatro a doze semanas para total
regeneração das células ciliadas (Forge e col., 1998). Estudos com ratos
demonstraram que as células de suporte do vestíbulo mantêm a habilidade de se
proliferar até o quinto dia pós natal (P5) e que essa proliferação pode ser induzida
até P12. A partir desse ponto, a capacidade de proliferação dessas células é perdida
(Gu e col., 2007). No órgão de Corti, a capacidade de proliferação das células de
suporte é aparentemente perdida durante o desenvolvimento embrionário. No
entanto, já foi demonstrado in vivo que as células de suporte do órgão de Corti de
cobaias adultas têm capacidade de proliferação frente à lesão nas células ciliadas,
porém esse potencial é limitado (Yamasoba e Kondo, 2006). Aparentemente o
estabelecimento da quiescência nas células de suporte relaciona-se com o aumento
de expressão de inibidores de cinases dependentes de ciclinas e com alterações na
configuração das proteínas do citoesqueleto (Groves e col., 2010; Warchol, 2010).
Estudos sobre a regeneração das células ciliadas do vestíbulo e do órgão de
Corti de aves e mamíferos sugerem que as células de suporte atuam como
precursores das células ciliadas e que essas células apresentam características de
células-tronco. No entanto, essas propriedades variam entre os diferentes tipos de
células de suporte, que apresentam morfologias e perfis de expressão gênica
diferentes (Malgrange e col., 2002; Savary e col., 2007; Doetzlhofer e col., 2006;

87
White e col., 2006; Ismati e col., 2011). A expressão de diversos marcadores de
células-tronco como Sox2, Nestina, Musashi, Notch, Prox1 e Islet1 já foi
demonstrada nas células de suporte de camundongos pós-natal (P3)(Lanford e col.,
1999; Li e col., 2004; Lopez e col., 2004; Sakaguchi e col., 2004; Stone e col., 2004;
Hume e col., 2007).
Essas evidências têm despertado o interesse em estudos sobre as células de
suporte do órgão de Corti para utilização tanto em novas abordagens para
regeneração das células ciliadas quanto para identificação de genes e/ou vias de
sinalização responsáveis pela sua reentrada no ciclo de divisão celular e/ou pelo seu
fenótipo ou estado de quiescência. Além disso, o cultivo dessas células pode revelar
mais sobre sua biologia e auxiliar na identificação de moléculas na fisiologia auditiva.
Apesar de o cultivo in vitro de células do órgão de Corti permitir a
sobrevivência de células epiteliais e neurogliais em meio de cultura completo, as
células provenientes do órgão de Corti têm demonstrado baixa viabilidade in vitro e
metodologias diferentes têm sido testadas para aumentar o rendimento dessas
culturas (Zheng e col., 1997; Zhao e col., 2001;Malgrange e col., 2002; Li e col.,
2003a;Li e col., 2003b; Doetzlhofer e col., 2004.
Estudos anteriores do nosso grupo, em condições de cultivo aderentes com a
espécie Cavia porcellus, haviam mostrado que no 13DIV algumas células
apresentaram marcação positiva para nestina, marcador de células progenitoras
neurais. No entanto, esse tipo de cultivo leva a franca proliferação dos fibroblastos
(Oiticica e col., 2007). A partir desses resultados e com base em estudos prévios que
demonstraram que o cultivo de células dissociadas do órgão de Corti em condições
não aderentes forma colônias de células flutuantes (otoesferas) com características
de células progenitoras (Li e col., 2003a; Oshima e col., 2007, Savary e col., 2007 e
2008), iniciamos a padronização do cultivo de células progenitoras do órgão de
Corti de cobaias e camundongos em condições não aderentes.
O cultivo de células progenitoras do órgão de Corti em condições não
aderentes é um desafio uma vez que in vitro a densidade e a proliferação celular são
muito baixas. De acordo com a literatura, são vários os fatores de crescimento que

88
podem promover a proliferação in vivo do epitélio vestibular após lesão induzida por
aminoglicosídeos, incluindo os fatores EGF, bFGF, TGFα e IGF1 (insulin like growth
factor 1) entre outros (Li e col., 2003b; Kopke e col., 2001; Kuntz e col., 1998). Em
geral, a cultura em condições não aderentes de células dissociadas do órgão de Corti
a partir de 20 cócleas de camundongos (P3) e com cerca de 104 células semeadas irá
produzir 4 ± 2,08 otoesferas após 6DIV na ausência de qualquer fator de
crescimento (Zine e Ribaupierre, 1998). Na presença dos fatores de crescimento
EGF e TGFα, esse mesmo experimento apresenta um número de otoesferas
significativamente maior (41,25 ± 3,50, p < 0.05). Além disso, esses autores
observaram que 50% dessas otoesferas apresentam expressão do marcador universal
de células-tronco, Abcg2 (Savary e col., 2008). Os efeitos desses dois fatores de
crescimento sobre a formação das otoesferas são confirmados pelos resultados dos
nossos experimentos e de estudos prévios que relacionam a presença de EGF e de
TGFα com a proliferação in vitro em culturas organotípicas do vestíbulo e do órgão
de Corti (Yamashita e col., 1995; Kuntz e Oesterle, 1998; Zine e Ribaupierre, 1998).
Nossos experimentos demonstram que o TGFα tem um efeito adicional
sobre o número de otoesferas formadas, resultando em número 2,5 vezes maior
quando comparado com o grupo bFGF, o que está de acordo com a literatura.
Nenhuma diferença significativa foi observada no número de células presentes em
cada otoesfera. No entanto, houve uma tendência para valores mais elevados no
grupo TGFα. Não foi possível demonstrarmos a atividade proliferativa direta das
células das otoesferas, por meio do ensaio com BrdU (Bromodeoxiuridina) devido à
marcação inespecífica na imunofluorescência (dados não mostrados). Embora esse
protocolo precise ser padronizado para essas condições in vitro, conseguimos
registrar durante o período de cultivo a expansão do tamanho das otoesferas de
ambas as espécies (cobaias e camundongos), o que é indiretamente sugestivo de
proliferação celular (Figura 4.4.).
Observamos que as otoesferas de cobaias e camundongos apresentaram, por
meio de reação de imunofluorescência indireta, marcação positiva para os
anticorpos anti-Sox2 e anti-nestina. A presença de células que expressam esses dois
marcadores corrobora as evidências da presença de células progenitoras na orelha

89
interna de camundongos e cobaias no período pós-natal, que retêm um fenótipo
indiferenciado. É importante salientar que, em cobaias, essa foi a primeira
demonstração da presença de células com características de progenitoras no órgão
de Corti em condições de cultivo não aderentes (Oiticica e col., 2010 – Anexo B).
Nossos resultados mostram claramente marcação positiva com os anticorpos anti-
miosina VIIa e anti-jagged2, característica de células ciliadas, e anti-p27kip1 e anti-
jagged1, característica de células de suporte, evidenciando a diferenciação das
otoesferas em condições adequadas de cultivo (Figuras 4.7. e 4.8.). Em todos os
marcadores utilizados foi possível observar a distribuição subcelular esperada, ou
seja, miosina VIIa em processos celulares, jagged1 e 2 na membrana plasmática e
localização nuclear para p27kip1. Esses resultados confirmam o fenótipo
indiferenciado das otoesferas e seu comprometimento para a diferenciação dos
diferentes tipos celulares presentes na orelha interna.
Acreditamos que o fato de não conseguirmos demonstrar a diferenciação das
otoesferas de cobaias em células de suporte e ciliadas é devido, provavelmente, ao
número limitado de células obtidas durante o seu cultivo (Tabela 4.2. e Figura 4.5.)
Tal evento também pode estar relacionado à maturação relativamente precoce da
cóclea em cobaias. Nessa espécie, já foi demonstrado que o potencial endococlear é
estabelecido ainda no período pré-natal, cerca de 12 a 15 dias antes do nascimento
(período gestacional 69-72 dias), enquanto que em camundongos, isso ocorre entre
P12 e P14 (período gestacional 19-21 dias)(Pujol, 1985). O potencial endococlear é
estabelecido somente após maturação completa do órgão de Corti e sinaliza o início
do processo auditivo. Em um estudo realizado pelo grupo de pesquisa do Prof.
Stephan Heller (Stanford University School of Medicine, Stanford, USA) foi relatada a
formação de um número reduzido de otoesferas a partir do cultivo de células do
órgão de Corti de camundongos P21, que corresponde a aproximadamente nove
dias após a completa maturação do órgão de Corti (Oshima e col., 2007).
Considerando que utilizamos cobaias com três dias de vida e que a maturação do
órgão de Corti se dá cerca de 15 dias antes do nascimento, a obtenção de células
com características de progenitoras nessa espécie deve ser mesmo dificultada. O
limitado número de otoesferas produzidas in vitro e o restrito potencial de

90
diferenciação observados nas culturas do órgão de Corti de cobaias são também
evidências da maturação precoce da cóclea nessas espécies quando comparada a dos
camundongos.
4.6. Conclusão
Durante o período referente ao doutorado, conseguimos padronizar o cultivo
de células dissociadas do órgão de Corti de cobaias e camundongos, caracterizar as
células presentes nas otoesferas formadas nessas culturas por imunofluorescência
indireta e avaliar o seu potencial de renovação e de diferenciação em células ciliadas
e de suporte, atingindo o nosso objetivo. Nossos resultados indicam que o
enriquecimento do meio de cultura com os fatores de crescimento EGF e TGFα é
uma boa alternativa para obtenção de maior número otoesferas. A espécie Mus
musculus se mostrou ser a mais adequada ao estabelecimento de culturas em
suspensão de otoesferas quando analisadas entre P2 e P3.
Em experimentos futuros, pretendemos selecionar as células de suporte
obtidas após diferenciação e avaliarmos sua capacidade de cultivo in vitro, para
futuramente podermos utilizar essas células em nossos estudos sobre proteínas
parceiras da conexina 26.

91
CAPÍTULO 5

91
CAPÍTULO 5 Pesquisa e identificação de proteínas que interagem com a Cx26
5.1. Introdução
Regiões de interação entre células ou entre célula e substrato são
especializações da membrana plasmática formadas por complexos protéicos que se
associam à membrana plasmática, às moléculas do citoesqueleto e da matriz
extracelular. Tais complexos incluem as junções ocludentes (tight junction), as junções
de ancoramento (junções aderentes, desmossomas e hemidesmossomas) e as
junções do tipo fenda (Duffy e col., 2002).
Nos vertebrados, as conexinas são as proteínas estruturais constituintes dos
canais de junção do tipo fenda. No desenvolvimento, crescimento e diferenciação
celular, esses canais e desempenham importante função na regulação da homeostasia
tecidual, por meio da manutenção do pH e de concentrações iônicas. Além disso,
promovem rápido transporte de pequenas moléculas entre células respondendo pelo
fluxo direto de moléculas com massa molecular inferior a 2000 dáltons, como íons
(K+, Cl-), mensageiros secundários (IP3, AMPc e Ca+2), nucleotídeos (ADP, ATP e
CTP), compostos do metabolismo intermediário (aminoácidos) e RNAs curtos. Os
canais de junções do tipo fenda estão localizados em pontos específicos da
membrana plasmática de duas células adjacentes formando estruturas altamente
organizadas denominadas placas juncionais (Alberts e col., 2007, Dbouk e col.,
2009). Nesses locais, tem-se demonstrado que as conexinas interagem com diversos
outros componentes celulares que incluem elementos do citoesqueleto, enzimas
(cinases e fosfatases), moléculas de adesão e moléculas sinalizadoras (Duffy e col.,
2002; Hervé e col., 2007; Derangeon e col., 2008; Dbouk e col., 2009; Dror e
Avraham, 2009; Laird e col., 2009).
Na região apical das células polarizadas, tem se observado frequentemente
que as placas juncionais do tipo fenda mostram co-localização em relação às placas
juncionais ocludentes, formando um complexo juncional. Vários estudos mostraram
interação entre as conexinas e as proteínas das zônulas ocludentes ZO-1, ZO-2 e

92
ZO-3 (sinônimo atualmente das Tight Junction Protein: TJP1, TJP2 e TJP3,
respectivamente) (Hervé e col., 2004; Hervé e col., 2007; Derangeon e col., 2008;
Dbouk e col., 2009; Dror e Avraham, 2009; Laird e col., 2009). As proteínas ZO das
junções ocludentes de células epiteliais e endoteliais apresentam vários domínios
PDZ (densidade pós-sináptica 95, grandes discos, zônula-ocludentes-1),
responsáveis por ancorar proteínas integrantes de membrana às do citoesqueleto,
que apresentam o motivo de ligação a PDZ ou PDZbm (Hervé e col., 2004; Brône e
Eggermont, 2005). Esses estudos sugeriram que essas interações entre as proteínas
ZO e as conexinas podem ter um papel regulatório na organização das placas de
junções do tipo fenda (Kausalya e col., 2001; Hervé e col., 2004; Laird, 2005). O
motivo PDZbm já foi funcionalmente demonstrado na região C-terminal das
conexinas Cx30, Cx31.9, Cx32, Cx36, Cx40, Cx43, Cx45, Cx46, Cx47 e Cx50
(Tabela 5.1.). Interações entre domínios PDZ e PDZbm são um modelo recorrente
para explicar o ancoramento de proteínas integrantes da membrana plasmática.
Observa-se pela tabela 5.1. que, apesar da divergência em geral dos últimos dez
resíduos C-terminais dessas conexinas, há algumas posições conservadas, como a
última em que se observa serina (S), isoleucina (I), cisteína (C) ou valina (V) e três
resíduos anteriores a última com a presença de serina (S), aspartato (D), cisteína (C)
ou treonina (T) (marcados com asterisco).
Por outro lado, são poucas as proteínas conhecidas por interagirem física e
funcionalmente com a Cx26. Em linhagem celular do intestino humano, foi descrita
interação entre a Cx26 e um domínio específico da ocludina, que por sua vez
interage com o domínio PDZ da ZO-1 (Nusrat e col., 2000). São conhecidas as
proteínas OCP1/Fbox2 e OCP2/Skip1 que parecem relacionar-se à degradação da
Cx26 e a consortina, proteína relacionada com o trânsito das Cx26 e Cx30 do
complexo de Golgi à membrana plasmática (Nusrat e col., 2000; Nelson e col., 2007;
Del Castillo e col., 2010).
A identificação de novas proteínas que interagem com a Cx26 pode ampliar
o conhecimento funcional sobre as conexinas, indicar novos genes candidatos à
surdez sindrômica e não-sindrômica, além de poder explicar os casos de surdez
decorrentes de mutação isolada no gene GJB2. Estes casos poderiam ser atribuídos à

93
herança digênica, ou seja, interação gênica entre o lócus da Cx26 e outros
codificadores de proteínas distintas, porém associadas física e funcionalmente a ela.
Alterações na sequência de aminoácidos ou nos níveis de expressão dessas proteínas
poderiam perturbar sua interação com a Cx26 e levar à disfunção coclear e perda
auditiva. Se essas alterações forem determinadas por alterações nos genes que
codificam essas proteínas, novos genes e novas mutações potencialmente
causadoras de surdez poderão ser identificados.
Tabela 5.1. Relação das conexinas descritas por interagir com os domínios PDZs das proteínas ZO-1 e a sequência dos últimos dez resíduos da região C-terminal de cada para a espécie (Mus musculus).
Cx ID NCBI Ligante Domínio 10 últimos resíduos
do CT (mus) Referência
* * Cx30 NP_001010937.1
ZO-1 PDZ-2 G G N A I T S F P S Penes e col., 2005
Cx31.9CAC93843.1 ZO-1 PDZs L A T V R G D L A I Kojima e col., 2001
Cx32 NP_032150.2 ZO-1 PDZ-1 A E K S D R C S A C Flores e col., 2008 Cx36 AD13684.1 ZO-1 PDZ-1 R T Q S S D S A Y V Li e col., 2004ª
Cx40 AA43850.1 ZO-1 PDZ-1 S K A R S D D L S V Nagasawa e col, 2006
Cx43 NP_034418.1 ZO-1 PDZ-2 S R P R P D D L E I
Toyofuku e col., 1998 Barker e col., 2002 Duffy e col., 2004 Sorgen e col., 2004 Jin e col., 2004 Hunter e col., 2005 Girao e col., 2007
Cx45 NP_032148.1 ZO-1 PDZs S G D G K T S V W I Kausalya e col., 2001 Laing e col., 2005
Cx46 NP_058671.2 ZO-1 PDZ-2 G R A R P G D L A I Nielsen e col, 2001
Cx47 NP_780661.2 ZO-1 PDZ-2 S R D G K A T V W I Li e col., 2004b
Cx50 AAG42241.1 ZO-1 PDZ-2 S R A R S D D L T I Nielsen e col, 2003
5.2. Objetivo
O objetivo desta etapa do trabalho foi identificar proteínas que interagem
com a Cx26 e, desta forma, procurar compreender melhor as vias funcionais que
atuam nessas células. Esses estudos podem culminar com a descoberta de novos
genes candidatos para surdez, que possam interagir com o gene GJB2. Pretendemos

94
contribuir, com esse estudo, para o esclarecimento de aspectos da fisiologia da
audição e da patogênese da surdez de herança autossômica recessiva.
Para atingir esses objetivos, foram executadas as seguintes etapas:
� Construção de um clone recombinante contendo a sequência codificadora
da região C-terminal da Cx26 em fusão com a da proteína GST (glutationa-S-
transferase) e produção de uma proteína de fusão Cx26/C-term-GST ou GST-Cx26
em fração solúvel.
� Realização de ensaios de precipitação por afinidade entre as proteínas GST-
Cx26 ou GST (GST sozinha) e as proteínas extraídas de tecido de camundongo que
expressa a Cx26.
� Identificação e análise das proteínas que interagem com a região C-terminal
da Cx26, por meio de análise por espectrometria de massas das proteínas presentes
em bandas de SDS-PAGE, obtidas no ensaio de precipitação com a proteína de
fusão GST-Cx26, mas não obtidas no ensaio com a GST.
5.3. Animais e Métodos
5.3.1. Animais
Utilizamos animais da espécie Mus musculus (camundongos C57BL/6) para
obtenção de tecidos que expressam a Cx26. A escolha dos tecidos com expressão
para a Cx26 foi feita com base na análise in silico realizada no banco de dados
UniGene e em literatura específica (NCBI; Nagy e col., 2001; Rash e col., 2001;
Altevogt e Paul, 2004). Os tecidos, cérebro e fígado, foram utilizados para obtenção
de DNA genômico para a amplificação pela PCR do fragmento que contém a
sequência codificadora terminal do gene Gjb2, a partir da primeira base do primeiro
códon após o quarto domínio transmembrânico até o códon de parada (região C-
terminal da Cx26). Esse fragmento foi utilizado para construção de uma proteína de
fusão. Além disso, com esses tecidos foram preparados os lisados celulares
utilizados no ensaio de precipitação por afinidade de proteínas.

95
Os animais vieram de outro estudo do Laboratório de Genômica Funcional
(CEP 109/2010). Todos os protocolos de manipulação realizados com esses animais
foram os mesmos que estão descritos na página 70, item 4.3.1., do capítulo 4.
Amostras de fígado e cérebro, após dissecção, foram imediatamente congeladas a -
80ºC.
5.3.2. Métodos
5.3.2.1. Clonagem e expressão de uma proteína com a sequência
codificadora para a região C-terminal da Cx26 em fusão com a GST
O vetor pGEX-4T-1 (GE Healthcare, Little Chalfont, Buckinghamshire,
UK) contém a sequência de cDNA codificadora para a Glutationa-S-Transferase de
Shistosoma japonicum seguida pelo sítio múltiplo de clonagem e códons de parada da
tradução, ou seja, é um vetor de expressão.
Utilizamos DNA genômico de camundongos para a clonagem neste vetor de
um segmento codificador para a região C-terminal da Cx26, que está contido no
único exon codificador do gene Gjb2. O DNA genômico foi procedente de fígado
total de camundongos (machos e fêmeas). Para extração do DNA genômico foi
utilizado o primeiro sobrenadante obtido da lise celular em LiCl 3M e uréia 6M
(20mg/mL de tecido) que foi homogenizado com o auxílio de uma seringa (10ml) e
agulha 8 mm em jato. Em seguida, o tecido foi incubado por 16 horas a 4ºC e
centrifugado por uma hora a 10.000 x g 4ºC. O DNA foi precipitado em álcool
isopropílico, ressuspendido em 200µL de água ultrapura, quantificado por
espectrofotômetro NanoDrop ND-1000 (NanoDrop Technologies, Rockland,
Delaware, USA) e conservado a -20ºC.
Para amplificar o segmento que contém a sequência codificadora para a
região C-terminal da Cx26, foi utilizado o par de iniciadores: CX26MOUSE – 1S 5’
CGA GGC CCG GGT TAT TGC TCA GGA AAG TCC A 3’ e MOUSECX26 –
1AS 5’ CGA GGG CGG CCG CTG GGT TCC TCT CTC CTG TC 3’ que
definem um segmento de 414pb. Para a realização da PCR, em um volume final de

96
10μL, contendo 30ng de DNA genômico, foram utilizados 0,5 pmoles de cada
iniciador, 1U de Taq polimerase, MgCl2 1,5mM, Tris 20mM pH8.4, KCl 50mM e
30μM de cada dNTP. As condições de amplificação foram as seguintes:
desnaturação inicial 95ºC por cinco min, 30 ciclos de desnaturação a 95ºC por 45
seg, hibridação a 58ºC por 45 seg, extensão a 72ºC por cinco min.
Após purificação do produto de PCR em gel agarose com o Kit in Concert
(Invitrogen, Carlsbad, CA, USA), de acordo com os procedimentos recomendados
pelo fabricante, ele foi inicialmente clonado com o sistema TOPO TA no vetor
pCR2.1-TOPO (Invitrogen, Carlsbad, CA, USA) em bactérias E.coli DH5α
transformadas por choque térmico. Colônias isoladas, crescidas em placas com meio
LB (Luria Broth) (Bacto-Triptona 10g/L, Bacto-Yeast 5g/L e NaCl 10g/L, pH 7.0)
com ágar 1% e 50µg/mL de ampicilina (TEUTO, Brasil) foram triadas pela PCR
com os iniciadores M13F (5’ GTA AAA CGA CGG CCA G 3’ ) e M13R (5’ CAG
GAA ACA GCT ATG AC 3’ em volume final de 10µL. O produto obtido da PCR
de clones que se revelaram recombinantes (fragmento de 615pb) foi então
purificado pelo Illustra GFX PCR DNA and gel purification Kit (GE Healthcare, Little
Chalfont, Buckinghamshire, UK), segundo instruções do fabricante e submetido à
reação de sequenciamento automático no ABI 3730xl DNA Analyzer (Applied
Biosystems, Foster City, California, USA), para confirmação de que o produto
inserido no vetor correspondia à sequência correta do fragmento de 414pb clonado,
que incluía a região C-terminal da Cx26.
A extração dos plasmídios recombinantes foi realizada pelo método de lise
alcalina de acordo com procedimentos já bem estabelecidos (Sambrook e col., 1989).
A sequência dos iniciadores SMACX26-1S e NOTCX26-1A utilizados para
amplificação do DNA genômico de camundongo (414pb) incluem sítios para as
enzimas de restrição SmaI e NotI. Assim, após extração do DNA dos plasmídeos
recombinantes por lise alcalina foi realizada a digestão com as enzimas SmaI e NotI
(Invitrogen, Carlsbad, CA, USA), de acordo com as instruções do fabricante, para a
liberação do inserto e posterior subclonagem em DNA do vetor pGEX-4T-1, em
seu sítio de clonagem múltipla, digerido com as mesmas enzimas e desfosforilado

97
pela Calf Intestinal Alkaline Phosphatase (CIAP) (Invitrogen, Carlsbad, CA, USA),
seguindo as recomendações do fabricante.
O DNA plasmidial dos vetores pGEX-4T-1 contendo GST ou GST-Cx26
foi usado para transformar E. coli BL21, linhagem celular que apresenta alto
potencial de expressão de proteínas. Para expressão proteica, foram semeadas
células E.coli BL21 recombinantes, a partir do estoque a -80°C, em placa de Petri
contendo meio LB ágar com 50µg/mL ampicilina (TEUTO, Brasil). As placas
foram mantidas em estufa a 37°C por 16 horas. Colônias frescas foram transferidas
da placa para um tubo plástico de 15mL contendo 6 mL de meio LB líquido com
50µg/mL ampicilina (TEUTO, Brasil) e crescidas sob agitação a 37°C por 16 horas.
Um mililitro do meio foi utilizado para quantificação no espectrofotômetro à luz
visível (600nM) usando meio LB estéril como referência para medir a D.O.
(densidade óptica). Após verificação da D.O., as culturas contidas em meio LB
líquido foram diluídas para obtenção de solução com uma D.O. de 0,2 e crescidas
sob agitação a 37°C por duas horas para atingir D.O. entre 0,4 e 0,6. Quatro
alíquotas de 1mL de cada cultura, GST-Cx26 e GST, foram separadas e a três de
cada quatro foi adicionado IPTG (Isopropyl β-D-1-thiogalactopyranoside) (Invitrogen,
Carlsbad, CA, USA) para obtenção das concentrações finais de 0,05mM, 0,2mM e
0,5mM, respectivamente. Em uma das alíquotas não foi adicionado IPTG. As
amostras foram incubadas sob agitação a 37ºC por duas horas. Após indução com
IPTG, as suspensões de bactérias foram centrifugadas a 10.000 x g por cinco min
para obtenção de pellet que continha a proteína expressa. O pellet foi ressuspendido
em tampão de amostra, GSD 3X (glicerol 34%, SDS 20%, DTT 1M, azul de
bromofenol) e metade da amostra foi submetida a eletroforese em NuPAGE®
Novex® Bis-Tris 12% (Invitrogen, Carlsbad, CA, USA) ou em gel SDS-PAGE
(dodecil sultato de sódio) 10% e corados com azul de Coomassie (Sigma-Aldrich, St
Louis, MO, USA) para análise das proteínas. A outra metade da amostra foi utilizada
para realização do Western blotting.
Para confirmação da presença das bandas referentes às proteínas GST-Cx26
e GST após indução de expressão por IPTG foi realizado o Western blotting com o
anticorpo primário Anti-GST (Invitrogen, Carlsbad, CA, USA). As amostras

98
contendo as proteínas GST-Cx26 e GST foram submetidas à eletroforese em gel
SDS-PAGE (dodecil sultato de sódio) 12% e transferidas para membranas de
nitrocelulose (Nitrocellulose Membrane - BioRad, Hercules, CA, USA). Para o
bloqueio da membrana de nitrocelulose foram utilizados 1% de caseína (Sigma-
Aldrich, St Louis, MO, USA) em TBST (TBS: Tris 50mM, NaCl 250mM, pH 7,6
com 0,05% de Tween 20/Sigma-Aldrich, St Louis, MO, USA) e a membrana foi
incubada sob agitação por uma hora à temperatura ambiente. Após o bloqueio, a
membrana foi lavada em TBST três vezes e foi adicionado o anticorpo primário
anti-GST (Invitrogen, Carlsbad, CA, USA) na proporção de 1:5000, seguido de
incubação por uma hora à temperatura ambiente. O segundo anticorpo, conjugado a
anti-IgG (camundongo), foi adicionado na diluição de 1:20000 (Amersham
Biosciences, Buckinghamshire, UK) e incubado por uma hora, seguido de três
lavagens com TBS e em seguida incubado por uma hora com ECL plusTM para a
peroxidase (Amersham Biosciences, Buckinghamshire, UK). Após este
procedimento a membrana de nylon foi revelada em filme Kodak X-Omat K XK-1 13
X 18 cm.
5.3.2.2. Obtenção de proteínas recombinantes em condições solúveis
ou insolúveis
Uma vez determinada a melhor condição para expressão das proteínas GST-
Cx26 e GST, clones GST-Cx26 e GST foram crescidos em meio LB líquido com
ampicilina 50 µg/mL (TEUTO, Brasil) e IPTG (Invitrogen, Carlsbad, CA, USA)
com concentração final de 0.5mM para indução das culturas, até atingir densidade
óptica entre 0.4 e 0.6 a 600nm ao espectrofotômetro. Foram separadas duas
alíquotas de 1,5ml de cultura que foram centrifugadas por 1min a 10.000 x g. As
duas alíquotas foram submetidas a processos diferentes para que fosse verificada a
solubilidade da proteína de fusão.
Para isolamento de ambas as proteínas na fração solúvel, após centrifugação,
o pellet de uma das alíquotas foi ressuspendido em PBS (NaCl 137mM, KCl 2,7mM,
Na2HPO4 10.0mM e KH2PO4 1,76mM, pH 7.4) contendo inibidor de protease

99
(Pefabloc- Roche Applied Science, Indianapolis, IN, USA) 100ng/µL e lisozima
(Sigma-Aldrich, St Louis, MO, USA) a 10mg/mL, e incubado em gelo por 15
minutos. A amostra foi submetida a dois ciclos rápidos de congelamento e fusão,
seguido de centrifugação por 15 min, 10.000 x g a 4ºC para a separação do
sobrenadante que contém as proteínas solúveis. Ao sobrenadante foi adicionado
GSD 3X na proporção 1:3.
Para isolamento de cada proteína GST-Cx26 e GST na fração insolúvel, após
centrifugação, o pellet da outra alíquota foi ressuspendido em 100µL de TNE (NaCl
100mM, EDTA 1mM, Tris 50mM pH 8.0) contendo lisozima a 10mg/mL (Sigma-
Aldrich, St Louis, MO, EUA), incubado à temperatura ambiente por 20 minutos e
centrifugado por 10min, 10.000 x g a 4ºC. O sobrenadante foi descartado e o pellet
foi ressuspendido em TNE gelado contendo 0,1% de DOC (Ácido Desoxicólico -
Sigma-Aldrich, St Louis, MO, USA), seguido de nova incubação em gelo por dez
min com agitação ocasional. Foi adicionado MgCl2 para uma concentração final de
8mM e 10U/µL de DNase (Invitrogen, Carlsbad, CA, USA), a amostra foi
centrifugada por 15 min, 10.000 x g a 4ºC e o pellet foi lavado com TNE gelado
adicionado de 1% de Igepal 30 (Sigma-Aldrich, St Louis, MO, USA) e centrifugado
por dez min, 10.000 x g a 4ºC, seguido de 2 lavagens com TNE gelado. O pellet foi
então ressuspendido em 30µL de GSD 3X.
Ambas as alíquotas, fração solúvel e insolúvel, após adição de GSD 3X,
foram incubadas a 95ºc por 10 min e submetidas à eletroforese em mini-géis
NuPAGE® Novex® Bis-Tris 12% (Invitrogen, Carlsbad, CA, USA) ou em gel SDS-
PAGE (dodecil sultato de sódio) 10% e coradas com azul de Coomassie (Sigma-
Aldrich, St Louis, MO, USA) para identificação da proteína de fusão nas duas
frações (solúvel e/ou insolúvel).
Após confirmar a solubilidade da proteína de fusão, ensaios de expressão em
grande quantidade foram realizados para obtenção de grande volume de ambas
proteínas, GST-Cx26 e GST. Essas proteínas foram quantificadas e alíquotas de
600pmoles para o ensaio de precipitação por afinidade foram preparadas como
descrito abaixo.

100
Para quantificação, 100µL do sobrenadante da fração solúvel das proteínas
GST-Cx26 ou GST e 40 µL de sefarose GST Bind Resin ligada à glutationa (GSH)
em 50% de PBS (Novagen, Darmstadt, Germany) foram incubadas, sob agitação, a
temperatura ambiente por 30 min. Após incubação as amostras foram lavadas três
vezes com tampão PBS e foi adicionado 20µL de GSD 3X. As amostras foram
incubadas a 95ºc por 10 min e submetidas à eletroforese em mini-géis NuPAGE®
Novex® Bis-Tris 12% (Invitrogen, Carlsbad, CA, USA) ou em gel SDS-PAGE 10%
junto com BSA (Invitrogen, Carlsbad, CA, USA) em concentrações diferentes e
crescentes para quantificação das proteínas por comparação de intensidade de
bandas com o BSA e submetidas à eletroforese. Os geis foram corados com azul de
Coomassie (Sigma-Aldrich, St Louis, MO, USA).
Após quantificação, alíquotas de 600pmoles das proteínas GST-Cx26 e GST
foram preparadas e armazenadas a -80ºC. Essas alíquotas foram utilizadas para o
ensaio de precipitação por afinidade.
5.3.2.3. Ensaios de precipitação por afinidade entre a proteína de fusão
recombinante (GST-Cx26) e proteínas do lisado celular
Para o preparo do lisado celular foram utilizados três tampões diferentes de
lise para se tentar obter os melhores resultados da extração de proteínas do
complexo da Cx26, maximizando-se as chances de resultados reprodutíveis e
ligações específicas. As composições desses tampões estão descritas na Tabela 5.2.
Cerca de 100mg de tecidos obtidos de fígado e cérebro foram
homogeneizados em 5mL dos tampões de lise EGTA e/ou EDTA, lisados no gelo
por 30 min e centrifugados por 30 min, por 14.000 x g a 4°C. Os sobrenadantes
foram transferidos para tubo plástico de 15mL e foram utilizados para ensaio de
precipitação por afinidade.
Cerca de 50mg de tecidos obtidos de fígado e cérebro foram
homogeneizados em 10mL de tampão PHEM e incubados por dois min a 37ºC.
Procedeu-se a centrifugação por 5 min, 14.000 x g a 4ºC. O sobrenadante, que

101
contém as proteínas solúveis, foi separado para ensaio de precipitação por afinidade
e o pellet, que contém a fração proteica insolúvel, foi ressuspendido em 200µL de
tampão PHEM, desprovido de EGTA e MgCl2, mas adicionado incluindo EDTA
0,5mM. As amostras oriundas do pellet foram centrifugadas por 20 min, 14.000 x g a
4ºC e os sobrenadantes foram transferidos para tubo plástico de 15mL e utilizados
para ensaio de precipitação por afinidade.
Tabela 5.2. Composição dos três diferentes tampões de lise utilizados para o ensaio de precipitação por afinidade.
Tampão EGTA Tampão EDTA Tampão PHEM
Tris-HCl pH 7,4 50mM Tris-HCl pH 7,4 50mM PIPES pH 6,9 60mM
NaCl 150mM NaCl 150mM HEPES 25mM
EGTA 10mM EDTA 1 mM EGTA 10mM
MgCl2 2mM MgCl2 2mM
Inibidor de protease 1 1:50 Inibidor de protease1 1:50 Inibidor de protease 1 1:50
Na3VO4 2 2mM Na3VO4 2 2mM
NaF 3 10mM NaF 3 10mM
Falacidina 4 5µM
Triton X-100 0,75% Triton X-100 0,75% Triton X-100 0,75%
1 inibidor de protease de mamífero (Sigma-Aldrich, St Louis, MO, EUA). Na3VO42 : inibidor de fosfatase de resíduo de tirosina fosforilada: (Sigma-Aldrich, St Louis, MO, EUA ). NaF 3: inibidor de fosfatase de resíduo de serina ou treonina fosforilada: (Sigma-Aldrich, St Louis, MO, EUA). inibidor de fosfatase de resíduo de tirosina fosforilada: (Sigma-Aldrich, St Louis, MO, EUA ). 4 Falacidina (Sigma-Aldrich, St Louis, MO, EUA)
Para o ensaio de precipitação por afinidade, a proteína de fusão expressa
deve ter afinidade por outra molécula já conhecida, ligada de modo covalente a
contas de sefarose. O sistema utilizado neste estudo foi o da ligação da proteína de
fusão com glutationa-S-transferase (GST), a qual se liga com avidez à glutationa
(GSH) das contas de sefarose. Para ligação das proteínas GST-Cx26 ou GST, as
alíquotas de 600pmoles de ambas as proteínas e 200µL de sefarose GST Bind Resin
ligada à glutationa (GSH) em 50% de PBS (Novagen, Darmstadt, Germany) foram
incubadas, sob agitação, à temperatura ambiente por 30 min. Após incubação as
amostras foram lavadas três vezes com tampão PBS (Invitrogen, Carlsbad, CA,

102
USA) contendo inibidor de protease (Pefabloc, Roche Applied Science, Indianapolis,
IN, EUA) e utilizadas para ensaio de precipitação por afinidade.
Após a ligação das alíquotas de 600pmoles das proteínas GST-Cx26 ou GST
às contas de sefarose com GSH, foram adicionados 1mL dos lisados celulares
obtidos de fígado ou cérebro (tampões EGTA, EDTA e PHEM) e as amostras
foram mantidas sob agitação por 16h a 4ºC. Como controle negativo, foram
utilizadas alíquotas de 600pmoles da proteína de fusão recombinante ligadas
somente às contas de sefarose (sem lisado) e 200µL de contas de sefarose com GSH
ligadas aos lisados celulares (sem GST ou GST-Cx26). Alíquotas de 200µl contendo
somente as contas de sefarose também foram utilizadas como controle.
Após incubação, as amostras foram lavadas 4 vezes em seus respectivos
tampões de lise, desprovidos do detergente, e ressuspendidas em 100µL de PBS
(Invitrogen, Carlsbad, CA) e 30 µL de GSD 3X. As amostras foram incubadas a
95ºc por 10 min e submetidas à eletroforese em geis SDS-PAGE (dodecil sultato de
sódio) de diferentes concentrações (6%, 8%, 10%, 12% e 14%) e coradas com azul
de Coomassie.
5.3.2.4. Identificação proteica por espectrometria de massas
Após análise dos geis de SDS-PAGE para verificar as bandas presentes no
ensaio de precipitação com a proteína GST-Cx26 e ausentes no ensaio com a GST,
as bandas identificadas foram recortadas e enviadas para análise por espectrometria
de massa para o CHUQ, Centre Hospitalier Universitarie Quebéc, no Canadá. Para evitar
resultados falso-positivos, como experimento controle, também foram recortadas as
fatias dos geis da pista referente ao ensaio de precipitação com a proteína GST
(sozinha), cuja posição era correspondente às das fatias de geis recortadas que
continham as bandas presentes apenas no ensaio com a proteína GST-Cx26.
A identificação das proteínas precipitadas e presentes nas bandas isoladas de
geis para as canaletas GST ou GST-Cx26 foi realizada por prestação de serviço em
Quebéc, Canadá, no CHUQ Centre Hospitalier Universitarie Quebéc. Basicamente, as

103
fatias recortadas dos geis de SDS-PAGE foram digeridas com tripsina, como
descrito previamente por Jeronimo e col., (2004) e os peptídeos trípticos resultantes
foram purificados e identificados por espectrometria de massas (LC-MS/MS) com a
seguinte configuração: microcapilares com colunas de fase reversa e sob alta pressão,
acoplados a espectrômetro de massas com analisador do tipo íon quadrupolo
DecaXP LCQ (ThermoFinnigan) ou LTQ-LTQ Orbitrap (ThermoElectron) e ionizador do
tipo nanospray. Os resultados foram recebidos por correio eletrônico e analisados
com o Scaffold 3 proteomic software (Searle, 2010)
As análises das proteínas identificadas por meio do Scaffold 3 proteomic software
foram realizadas de acordo com as instruções recebidas pelo CHUQ Centre
Hospitalier Universitarie Quebéc. Esse software permitiu o acesso à lista de todas as
proteínas identificadas tanto nas bandas recortadas dos geis de SDS-PAGE do
ensaio de precipitação por afinidade com a proteína GST-Cx26, quanto nas bandas
correspondentes ao ensaio da proteína GST (grupo controle). Para facilitar as
análises, os resultados foram agrupados em duplas, listando as proteínas
identificadas e o número de peptídeos com similaridade para cada uma em cada
ensaio, GST ou GST-Cx26. O programa permite localizar os peptídeos na sequência
da proteína identificada (exemplo no Anexo D) e a porcentagem de resíduos da
proteína identificados por peptídeos trípticos.
A probabilidade de identificação correta de uma proteína pelo Scaffold 3
proteomic software é calculada separadamente para cada amostra, com base no número
de peptídeos identificados com similaridade à sua sequência de aminoácidos e no
percentual de similaridade entre a sequência do peptídeo identificado e da proteína,
de acordo com o banco de dados utilizado. Consideramos como resultado positivo
toda proteína com pelo menos duas sequências peptídicas identificadas com
similaridade de pelo menos 95%, o que resulta em uma probabilidade de
identificação da proteína maior que 95% (Figura 5.1.).
Para seleção das proteínas candidatas que interagem com a região C-terminal
da Cx26 para análise foram utilizados os seguintes critérios:
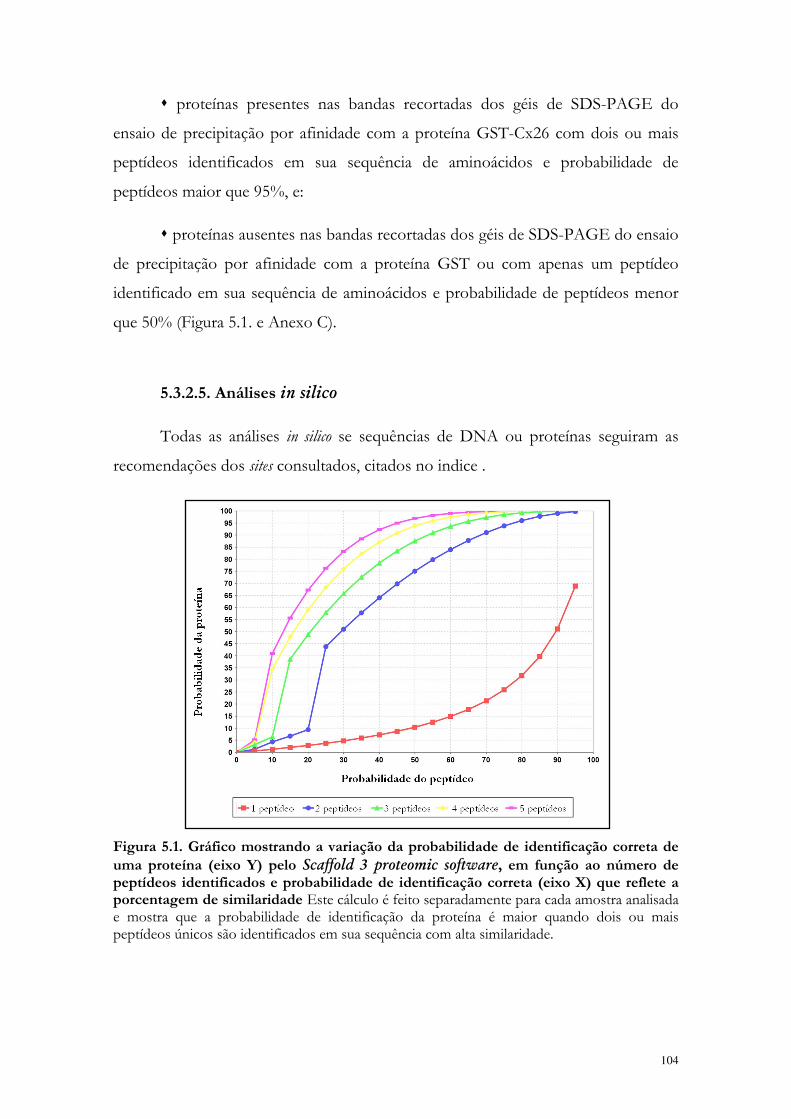
104
� proteínas presentes nas bandas recortadas dos géis de SDS-PAGE do
ensaio de precipitação por afinidade com a proteína GST-Cx26 com dois ou mais
peptídeos identificados em sua sequência de aminoácidos e probabilidade de
peptídeos maior que 95%, e:
� proteínas ausentes nas bandas recortadas dos géis de SDS-PAGE do ensaio
de precipitação por afinidade com a proteína GST ou com apenas um peptídeo
identificado em sua sequência de aminoácidos e probabilidade de peptídeos menor
que 50% (Figura 5.1. e Anexo C).
5.3.2.5. Análises in silico
Todas as análises in silico se sequências de DNA ou proteínas seguiram as
recomendações dos sites consultados, citados no indice .
Figura 5.1. Gráfico mostrando a variação da probabilidade de identificação correta de uma proteína (eixo Y) pelo Scaffold 3 proteomic software, em função ao número de peptídeos identificados e probabilidade de identificação correta (eixo X) que reflete a porcentagem de similaridade Este cálculo é feito separadamente para cada amostra analisada e mostra que a probabilidade de identificação da proteína é maior quando dois ou mais peptídeos únicos são identificados em sua sequência com alta similaridade.

105
5.4. Resultados
5.4.1. Clonagem e expressão de uma proteína com a sequência
codificadora para a região C-terminal da Cx26 em fusão com a GST
As análises in silico da sequência da Cx26 de Mus musculus nos bancos de
dados PSORT WWW server, ExPASy Proteomics server, KYTE DOOLITTLE
HYDROPATHY PLOT e NCBI mostraram que a extensão do domínio C-terminal
da conexina 26 após o último domínio transmembrânico corresponde a 10
aminoácidos, sendo que essa região difere em apenas um único aminoácido entre as
espécies Mus musculus e Homo sapiens (Tabela 5.3.). Observamos que esta região C-
terminal da Cx26 é divergente da de outras dez conexinas da Tabela 5.1. (página 93),
com similaridade apenas com a valina (V) C-terminal das conexinas 36 e 40.
A partir do conhecimento da extensão da região C-terminal citosólica da
Cx26, de Mus musculus um par de iniciadores foi desenhado que definia um
fragmento de 414pb a ser amplificado a partir de DNA genômico, uma vez que está
contido inteiramente no único exon codificador do gene Gjb2. O segmento inclui a
região codificadora terminal do gene Gjb2 de camundongos, desde a primeira base
do primeiro códon após o quarto domínio transmembrânico até o códon de parada
e parte da região 3’UTR. Sítios de restrição estavam presentes na extremidade 5’ de
cada iniciador, respectivamente, para as enzimas SmaI (iniciador sense) e NotI
(iniciador antisense) para permitir a clonagem direcional no vetor pGEX-4T-1. A
sequência do sítio de restrição para SmaI (CCCGGG) está contida nas trincas de
bases 225 (TTC), 226 (CCG) e 227 (GGT) que contêm os códons para os
aminoácidos fenilalanina (F), prolina (P) e glicina (G), respectivamente. Esses
códons fazem parte da sequência codificadora para uma região C-terminal
adicionada a Glutationa S-Transferase (GST) e se encontram no sítio múltiplo de
clonagem do vetor pGEX-4T-1 (Schistosoma japonicum). O vetor pGEX-4T-1 é
próprio para a expressão de proteínas em bactérias a partir da ativação do promotor
lac. As células portadoras do vetor de expressão contendo o gene de interesse devem
expressá-lo após a adição do indutor IPTG ao meio de cultura, o qual vai ativar a
região operadora (lac o) do promotor lac, permitindo que o gene alvo seja expresso
(http://www.gelifesciences.com; Handbook GST Gene Fusion System).

106
Tabela 5.3. Sequência dos dez aminoácidos que compõem a região C-terminal da Cx26 nas espécies Mus musculus e Homo sapiens.
Sequência de aminoácidos da região C-terminal da Cx 26
Organismo (ID NCBI)
YCSGKSKRPV Mus musculus (NM_008125.3 ) YCSGKSKKPV Homo sapiens (NP_003995.2 )
Para expressar em bactérias a proteína de fusão (ou proteína GST-Cx26) que
foi utilizada como “isca” para identificação de proteínas que interagem com a Cx26,
o amplicon de 414pb que contém a sequência codificadora para a região C-terminal
da Cx26 foi purificado e clonado primeiramente no sistema TOPO e,
posteriormente, foi feita a subclonagem no vetor pGEX-4T-1. A escolha da
clonagem por meio do sistema TOPO seguida da subclonagem no vetor pGEX-4T-
1 ocorreu pela melhor eficiência observada em laboratório da clonagem no TOPO.
Após clonagem do fragmento de 414pb no sistema TOPO, os clones
recombinantes contendo a sequência codificadora para a região C-terminal da Cx26
foram digeridos com as enzimas de restrição SmaI e NotI e subclonados no vetor
pGEX-4T-1. Sequenciamos os DNAs plasmidiais obtidos dos clones pGEX-4T-1
recombinantes com a sequência de DNA da GST em fusão à região C-terminal da
Cx26 (ou clones GST-Cx26) para confirmar a fase de leitura para tradução. Os
clones GST-Cx26 confirmados como apresentando a sequência inteiramente correta
e em fase foram utilizados para transformar linhagens de E. coli BL21 (Figura 5.2). A
linhagem de E. coli BL21 foi escolhida por apresentar características importantes
para o processo de expressão como a deleção de genes que codificam proteases (ton
e ompT) e uma mutação no gene rne131 (RnaseE) responsável pela degradação de
RNAm, o que aumenta a estabilidade do RNAm transcrito e consequentemente a
síntese protéica. Além destas deleções, esta linhagem apresenta no genoma o gene
lacI e uma cópia do gene T7 RNA polimerase, importantes para a indução da
expressão da proteína de interesse (http://www.promega.com; Technical Bulletin:
E. coli Competent Cells).
Para a indução de expressão das proteínas, foram testadas concentrações
crescentes de IPTG (0,01mM, 0,25mM e 0,5mM) e duas diferentes temperaturas,
30ºC e 37ºC. A melhor condição para expressão foi a que utilizou IPTG a 0,5mM e

107
a temperatura de 37ºC (Figura 5.3.). Como explicado, o sistema de expressão do
vetor é induzido por IPTG e se busca por uma expressão específica para ambos os
clones: um deles expressando a GST sozinha (GST) e o recombinante para a Cx26,
expressando a proteína de fusão (GST-Cx26) (Figuras 5.2. B e C). A proteína GST
possui massa molecular de 26kDa. Já a proteína GST-Cx26, ou seja, a proteína GST
em fusão com a região C-terminal da Cx26 tem massa molecular de 26,71kDa. O
amplicon de 414pb clonado no vetor pGEX-4T-1, contém a sequência
correspondente aos 10 códons para a região C-terminal da Cx26 e a seguir ocorre
um códon de parada. Ao ser inserido no vetor pGEX-4T-1 ele remove oito códons
da sequência da GST recombinante (GST-Cx26). Desse modo, a diferença de
massa molecular entre as duas proteínas expressas, GST e GST-Cx26, corresponde
apenas dois aminoácidos. Uma vez que ambas as proteínas apresentam massas
moleculares praticamente idênticas, não é possível discriminar em SDS-PAGE as
bandas referentes às duas proteínas. Para confirmar a expressão das proteínas GST-
Cx26 e GST e sua localização, realizamos Western blotting com o anticorpo primário
Anti-GST a partir de uma alíquota do lisado contendo as proteínas expressas (Figura
5.4.).
Uma vez determinadas as melhores condições para expressão da proteína de
fusão, fizemos extração das frações proteicas solúveis e insolúveis em detergente a
0,75% (Figura 5.5.). Saber se a proteína expressa está na fração solúvel e/ou
insolúvel é um passo importante, pois as análises por precipitação por afinidade são
de preferência realizadas com proteínas solúveis. Ensaios realizados com proteínas
insolúveis podem causar resultados falso-positivos. Observamos a GST-Cx26 em
ambas as frações, solúvel e insolúvel (Figura 5.5.), o que pode ser frequentemente
observado neste sistema de expressão em bactérias induzido por IPTG, dado o
excesso de proteína de fusão produzida. Na situação de excesso, uma proteína pode
perder sua forma nativa, ou seja, uma proteína solúvel pode se apresentar como
insolúvel em corpúsculos de inclusão. Uma vez verificada a solubilidade das
proteínas, adotamos o protocolo de extração de proteínas solúveis e obtivemos após
quantificação, alíquotas de 600pmoles de ambas as proteínas, GST-Cx26 e GST
(Figura 5.6.).

108
Figura 5.2. Triagem das colônias recombinantes com o vetor pGEX-4T-1. Em A) exemplo de sequenciamento parcial de um clone contendo o segmento genômico para expressão da região C-terminal da Cx26 ligado ao vetor pGEX-4T-1. O eletroferograma mostra o resultado da sequenciamento realizado com iniciador sense específico para o vetor. A sequência na linha superior é a esperada do vetor, em preto, a do inserto, em vermelho. O sítio de restrição para SmaI encontra-se sublinhado, demonstrando assim que o fragmento foi inserido na fase correta. Em B) sequência do vetor pGEX-4T-1 (ou GST) com seus códons em fase de leitura até o códon de parada. As setas vermelhas indicam a região de corte das enzimas de restrição SmaI e NotI, indicando o local onde o amplicon de 414pb contendo a sequência para a região C-terminal da Cx26 foi inserido. Em C) sequência da proteína de fusão recombinante GST-Cx26. Em azul está representada a sequência do segmento de nucleotídeos inserido, incluindo o sítio de restrição da enzima SmaI (CCCGGG), seguido de uma T, para não alterar a fase de leitura e os dez códons da região C-terminal da Cx26, incluindo seu códon de parada.
O ensaio de precipitação por afinidade prevê a interação de uma matriz
sólida e inerte (sefarose) modificada para apresentar um componente com alta
afinidade (GSH) a uma molécula que servirá como isca (GST-Cx26). No nosso caso,
a matriz sólida foi composta por sefarose ligada de forma covalente à glutationa
(GSH) (GST-Bind Resin - Novagen, Darmstadt, Germany), aqui denominada GSH-
beads. GSH tem alta afinidade por GST, a qual corresponde à maior parte da
proteína de fusão.
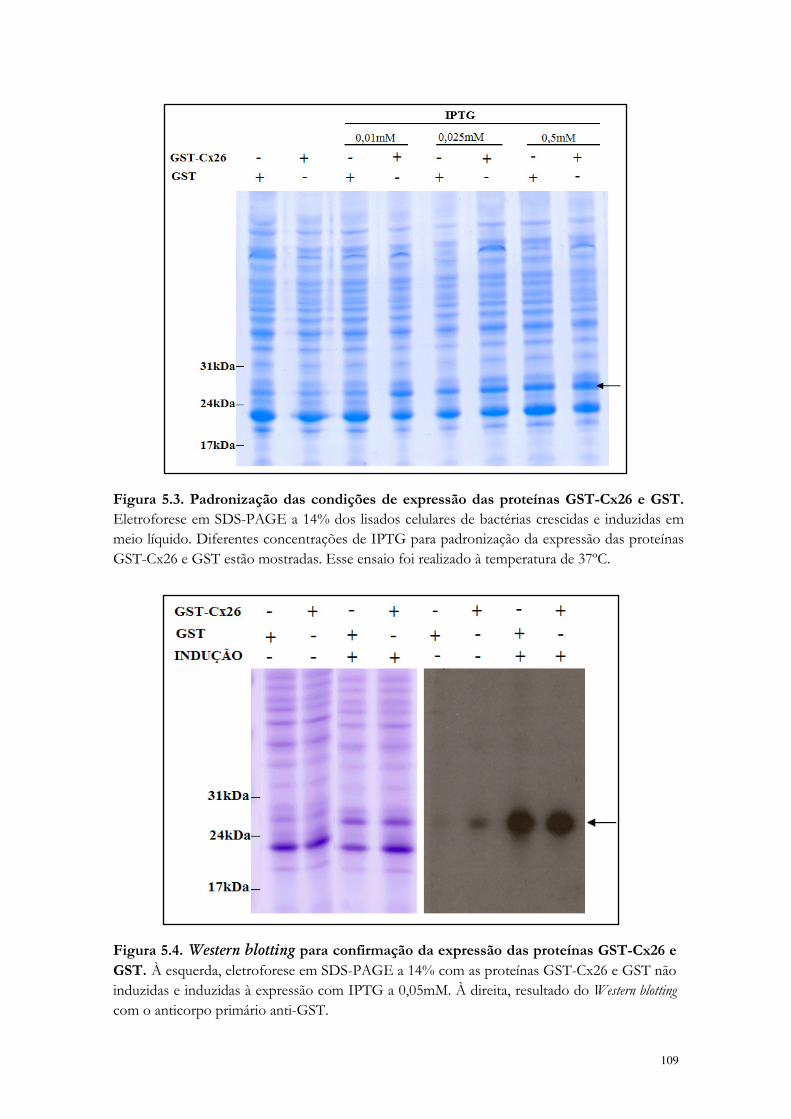
109
Figura 5.3. Padronização das condições de expressão das proteínas GST-Cx26 e GST. Eletroforese em SDS-PAGE a 14% dos lisados celulares de bactérias crescidas e induzidas em meio líquido. Diferentes concentrações de IPTG para padronização da expressão das proteínas GST-Cx26 e GST estão mostradas. Esse ensaio foi realizado à temperatura de 37ºC.
Figura 5.4. Western blotting para confirmação da expressão das proteínas GST-Cx26 e GST. À esquerda, eletroforese em SDS-PAGE a 14% com as proteínas GST-Cx26 e GST não induzidas e induzidas à expressão com IPTG a 0,05mM. À direita, resultado do Western blotting com o anticorpo primário anti-GST.

110
Para um ensaio de precipitação por afinidade a mistura deve conter além das
GSH-beads e da proteína de fusão, um lisado de tecido com relevância funcional
para o alvo, em razoável volume. Sabendo que o órgão de Corti possui um número
pequeno de células ciliadas (cerca de 20.000 ao nascimento) e um número de 3 a 5
vezes maior de células de suporte (Morris e col., 2005; Smeti e col., 2011), onde a
Cx26 é expressa, um número muito alto de animais seria necessário (pelo menos 20
animais para cada ensaio) para obtermos resultados reprodutíveis. Dessa forma,
decidimos escolher outros tecidos que expressam a Cx26 para nosso ensaio. Por isso,
desenvolvemos os ensaios com cérebro total e fígado de camundongos.
Figura 5.5. Solubilidade das proteínas GST-Cx26 e GST. Eletroforese em SDS-PAGE a 12% contendo as bandas das proteínas GST-Cx26 e GST expressas por indução com IPTG e isoladas da fração solúvel (S) e insolúvel (I). A seta indica a banda referente às proteínas GST-Cx26 e GST. T:lisado total.
Buscando possibilitar interações protéicas em condições iônicas distintas,
utilizamos nos ensaios de precipitação por afinidade três tampões diferentes para
lise de cérebro ou fígado (Tabela 5.2., página 101). Dois dos tampões de lise
utilizados apresentam praticamente a mesma composição, sendo a única diferença
entre eles, os quelantes, EDTA e EGTA, e no segundo caso houve a inclusão de
MgCl2 . O terceiro tampão de lise celular utilizado, “PHEM”, apresenta reagentes
específicos para estabilização dos elementos do citoesqueleto (falacidina) com alta

111
afinidade por actina filamentosa, além de triton X-100 que não desnatura esses
filamentos. Neste caso, nós temos duas frações, solúvel e insolúvel, sendo que a
fração insolúvel contém os componentes do citoesqueleto, enquanto que a fração
solúvel contêm as demais proteínas. Em todos os tampões utilizamos como
detergente o triton X-100 na concentração de 0,75%.
Figura 5.6. Quantificação e purificação das proteínas GST-Cx26 e GST. Eletroforese em SDS-PAGE a 12% em que houve eletroforese de lisados de bactérias após expressão por indução com IPTG isoladamente da fração solúvel que foi posteriormente ligada às contas de sefarose GSH. O BSA (Bovine Serun Albumin - Sigma-Aldrich, St Louis, MO, USA) foi utilizado para quantificação T: lisado total. S: lisado solúvel.
5.4.2. Ensaios de precipitação por afinidade entre as proteínas
recombinantes e proteínas do lisado celular
Os ensaios de precipitação por afinidade foram repetidos várias vezes com o
objetivo de se buscar reprodutibilidade. Os resultados foram analisados em geis de
poliacrilamida de concentrações diferentes para separar a banda de interesse de
outras vizinhas e para identificar de modo reprodutível bandas presentes
unicamente na amostra teste (GST-Cx26). Os experimentos controles constituíam:
(i) ensaio sem a proteína recombinante GST-Cx26, (ii) ensaio com o lisado
substituído por tampão, (iii) ensaios com os sobrenadantes restantes de cada ensaio,

112
(iv) ensaio com lisado total e/ou suas frações solúvel e insolúvel (sem GST-Cx26,
sem GSH-beads) e (v) ensaios com GSH-beads (sem GST-Cx26 e sem lisado).
Após o ensaio de precipitação por afinidade e análise dos géis de SDS-PAGE
contendo as amostras de experimentos com diferentes condições de reações
(tampões de lise celular: EGTA, EDTA e PHEM), onze fatias de gel contendo as
bandas presentes somente no ensaio de precipitação com a proteína de fusão GST-
Cx26 de forma reprodutível foram isoladas do restante do gel. Como controle,
isolamos as bandas obtidas no controle (GST) e localizadas na mesma posição em
canaleta vizinha. Na Tabela 5.4., temos a relação dos onze pares de fatias de gel dos
ensaios GST-Cx26 e GST que foram enviadas para o CHUQ Centre Hospitalier
Universitarie Quebéc e analisadas pela técnica de espectrotometria de massas. Entre as
11 bandas recortadas do ensaio com a proteína GST-Cx26, seis foram obtidas a
partir do ensaio de precipitação por afinidade com o tecido do cérebro, sendo três
identificadas na fração solúvel do lisado celular obtido com o tampão PHEM (com
massas moleculares observadas de 50kDa, 75kDa e 110kDa) (Figura 5.7.), duas com
o tampão EGTA (com massas moleculares observadas de 50kDa e 60kDa) (Figura
5.8.) e uma com o tampão de lise EDTA (com massa molecular observada de
40kDa (Figura 5.8).
As demais cinco bandas foram obtidas a partir do ensaio de precipitação por
afinidade com tecido do fígado, sendo todas identificadas a partir do lisado celular
obtido com o tampão de lise EGTA. Uma banda possui massa molecular
observada de 25 kDa e as outras quatro com massas moleculares entre 140-150kDa
(Figura 5.9). Essas quatro bandas foram cortadas a partir de géis de SDS-PAGE de
concentrações diferentes 8% e 14% (Figura 5.10.).
5.4.3. Análises das proteínas identificadas por espectrometria de
massas
As análises por espectrometria de massas dos produtos da digestão tríptica de
proteínas presentes nas 22 fatias de gel foram feitas aos pares, comparando a banda
do ensaio da GST-Cx26 e a equivalente do ensaio GST. Essas análises geraram uma

113
tabela para cada um dos onze pares de bandas, contendo as proteínas identificadas
(exemplo no Anexo C). Foi considerado como resultado positivo de detecção de
proteína para o ensaio da GST-Cx26 cada identificação no banco SWISSPROT que
apresentasse pelo menos dois peptídeos, com probabilidade maior que 95% de
identificação da proteína correta. Nas onze tabelas (exemplo no Anexo C), estão
listadas 447 proteínas. Dessas, identificamos 83 proteínas presentes no ensaio com
GST-Cx26 e não no GST correspondente, sendo 31 proteínas obtidas no ensaio
com o tecido cerebral e 52 no ensaio com fígado (Tabela 5.4.). No entanto, quando
comparamos as proteínas presentes no conjunto dos controles GST verificamos que
entre as 83 proteínas, 34 também estavam presentes em algum outro controle GST,
indicando interação com a porção GST ou precipitação inespecífica. Desse modo,
essas 34 proteínas foram excluídas (Tabela 5.5.)
Tabela 5.4. Relação dos 11 pares de fatias recortadas em geis de SDS-PAGE de concentrações diferentes referentes às bandas presentes no ensaio de interação por afinidade com a proteína de fusão GST-Cx26 (2, 4, 6, 8, 10, 12, 14, 16, 18, 20 e 22) e ausentes no ensaio com a GST (1, 3, 5, 7, 9, 11, 13, 15, 17, 19 e 21).
Nº da fatia/ensaio
Tecido Tampão de lise Massa observada (kDa)
Concentração do gel (SDS-
PAGE)
Figura
1 - GST cérebro fração solúvel PHEM
75 10% 5.7.
2 - GST-Cx26 3 - GST cérebro fração solúvel
PHEM 110 10% 5.7.
4 - GST-Cx26 5 - GST cérebro fração solúvel
PHEM 50 10% 5.7.
6 - GST-Cx26 7 - GST cérebro EGTA 50 12% 5.8.
8 - GST-Cx26 9 - GST cérebro EDTA 40 12% 5.8.
10 - GST-Cx26 11 - GST cérebro EGTA 60 12% 5.8.
12 - GST-Cx26 13 - GST fígado EGTA 140-150 8% 5.9.
14 - GST-Cx26 15 - GST fígado EGTA 140-150 14% 5.10.
16 - GST-Cx26 17 - GST fígado EGTA 140-150 14% 5.10.
18 - GST-Cx26 19 - GST fígado EGTA 25 14% 5.10.
20 - GST-Cx26 21 - GST fígado EGTA 140-150 8% 5.9.
22 - GST-Cx26
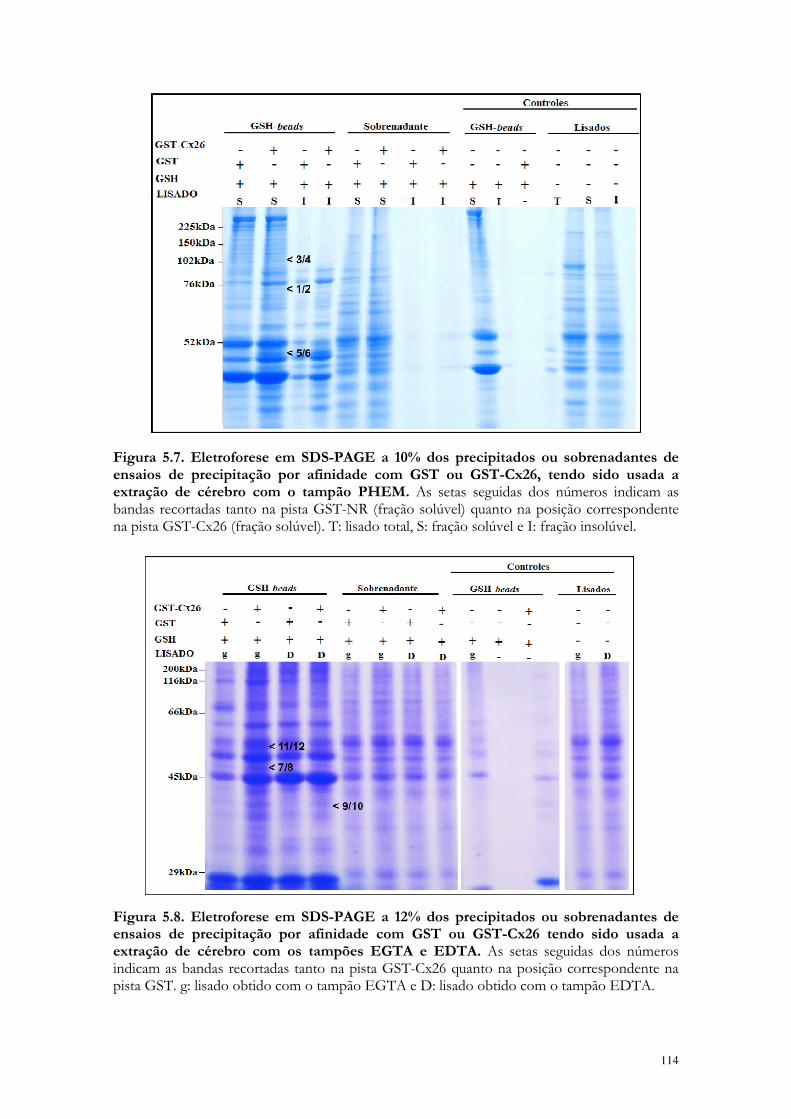
114
Figura 5.7. Eletroforese em SDS-PAGE a 10% dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26, tendo sido usada a extração de cérebro com o tampão PHEM. As setas seguidas dos números indicam as bandas recortadas tanto na pista GST-NR (fração solúvel) quanto na posição correspondente na pista GST-Cx26 (fração solúvel). T: lisado total, S: fração solúvel e I: fração insolúvel.
Figura 5.8. Eletroforese em SDS-PAGE a 12% dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26 tendo sido usada a extração de cérebro com os tampões EGTA e EDTA. As setas seguidas dos números indicam as bandas recortadas tanto na pista GST-Cx26 quanto na posição correspondente na pista GST. g: lisado obtido com o tampão EGTA e D: lisado obtido com o tampão EDTA.

115
Figura 5.9. Eletroforese em SDS-PAGE a 8 % dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26 tendo sido usada a extração de fígado com o tampão EGTA. As setas seguidas dos números indicam as bandas recortadas tanto na pista GST-Cx26 quanto na posição correspondente na pista GST. g: lisado obtido com o tampão EGTA.
Figura 5.10. Eletroforese em SDS-PAGE a 8 dos precipitados ou sobrenadantes de ensaios de precipitação por afinidade com GST ou GST-Cx26 tendo sido usada a extração de fígado com o tampão EGTA. As setas seguidas dos números em amarelo indicam as bandas recortadas tanto na pista GST-Cx26 quanto na posição correspondente na pista GST. g: lisado obtido com o tampão EGTA.

116
Dentre as 49 proteínas restantes, oito (O88568, P28653, Q02788, Q6S385,
B7ZNJ1, P59242, O08638 e UPI00001954ED) foram identificadas em mais de um
ensaio, sendo cinco identificadas duas vezes e três proteínas identificadas três vezes.
Desse modo, por meio da análise por espectrometria de massas das proteínas
contidas nas bandas dos geis SDS-PAGE presentes apenas no ensaio de
precipitação com a proteína de fusão GST-Cx26 e subtraída a redundância de
representação, identificamos um total de 38 proteínas diferentes candidatas a
interagirem com a região C-terminal da Cx26 (Tabela 5.6.).
Nove das 38 proteínas identificadas apresentavam massa molecular esperada
não correspondente à massa molecular observada no gel de SDS-PAGE do qual a
fatia foi retirada para análise (B2RQQ5, Q80WC3, Q80X90, Q69ZX3, P28653,
Q6S385, B7ZNJ1, O08638 e UPI00001954ED). Entre as nove proteínas, sete
apresentavam massa molecular observada menor do que a esperada, de acordo com
os bancos de dados (B2RQQ5, Q80WC3, Q80X90, Q69ZX3, Q6S385, B7ZNJ1 e
O08638). Grandes reduções de massa molecular entre o esperado e o observado
podem ocorrer por causa da clivagem da proteína ou splicing alternativo do transcrito
correspondente. No entanto, os peptídeos identificados por espectrometria de
massas para quatro dessas sete proteínas estavam distribuídos ao longo de toda a sua
sequência (exemplo no Anexo D). Esta observação invalida essas hipóteses de
clivagem ou splicing, levando-nos a excluir essas quatro proteínas por causa dessa
inconsistência (B2RQQ5, Q80WC3, Q6S385 e O08638, Tabela 5.6.). Duas
proteínas (P28653 e UPI00001954ED) tinham massa molecular observada na banda
recortada do gel de SDS-PAGE maior que a massa molecular esperada e isso
poderia ser justificado por modificações pós-traducionais que poderiam aumentar os
valores de massa em 2kDa, como no caso da fosforilação, ou em 9kDa, como no
caso da ubiquitinação, entre outras modificações. Mesmo na possibilidade de um
extenso conjunto de modificações pós traducionais, excluímos essas proteínas pela
grande diferença entre as massas moleculares observadas e as esperadas, inexplicável
somente por modificações pós-traducionais (Tabela 5.6.). No entanto, a proteína de
número de acesso UPI00001954ED não foi excluída, pois foi identificada em outra

117
banda e com massa molecular correspondente à observada em gel (Tabela 5.6.,
banda 20 - GST-Cx26).
Tabela 5.5. Relação do número de proteínas identificadas por meio da espectrometria de massas das 11 bandas isoladas em SDS-PAGE presentes apenas no ensaio de precipitação por afinidade com a proteína GST-Cx26 e das outras 11 bandas localizadas na mesma posição da canaleta vizinha correspondentes ao ensaio com a GST.
Fatias Ensaio nº total de proteínas
presentes por duplas1
nº proteínas apenas GST-
Cx262
nº proteínas final presentes apenas
GST-Cx263
1 - GST cérebro 59 7 4 2 - GST-Cx26 3 - GST cérebro 89 7 6 4 - GST-Cx26 5 - GST cérebro 42 1 0 6 - GST-Cx26 7 - GST cérebro 32 6 5 8 - GST-Cx26 9 - GST cérebro 41 3 1 10 - GST-Cx26 11 - GST cérebro 41 7 2 12 - GST-Cx26 Nº total de proteínas cérebro
304 31 18
13 - GST fígado 37 12 7 14 - GST-Cx26 15 - GST fígado 27 10 4 16 - GST-Cx26 17 - GST fígado 31 13 8 18 - GST-Cx26 19 - GST fígado 20 8 6 20 - GST-Cx26 21 - GST fígado 28 9 6 22 - GST-Cx26 Nº total de proteínas fígado 143 52 31 Nº total de protéinas 447 83 49
1 número total de proteínas identificadas nas onze duplas de fatias de gel isoladas do ensaio com a GST-Cx26 e GST. 2 número total de proteínas identificadas apenas no ensaio com a GST-Cx26. 3 número total final de proteínas identificadas no ensaio com a GST-Cx26, apos exclusão das proteínas presentes em outros ensaios GST.
Portanto, com base na incompatibilidade entre a massa molecular da proteína identificada e a massa molecular observada na banda de gel recortada, cinco proteínas foram excluídas, restando 33 proteínas candidatas a interagirem com a região C-terminal da Cx26 (Tabela 5.6.).

118
Tabela 5.6. Relação total das 49 proteínas identificadas por meio da espectrometria de massa após análises com o Scaffold 3 proteomic software das 11 bandas recortadas de géis SDS-PAGE presentes no ensaio de precipitação por afinidade com a proteína GST-Cx26.
Fatia/ ensaio
ID* Proteínas presentes no ensaio de
interação por afinidade com a proteína GST-Cx26 e ausente com a GST
MME (kDa)
MMO (kDa)
Ação
2 GST-Cx26
cérebro
Q99KI0 ACON aconitate hydratase mitochondrial 85 75 �
O88568 Heterogeneous nuclear ribonucleoprotein U
88 75 �
Q02257 PLAK_mouse Junction plakoglobin 82 75 �
Q3UHT7 Q3UHT7 Putative uncharacterized protein 113 75 �
4 GST-Cx26
cérebro
Q6ZQ74 MKIAA0666 protein fragment 125 110 � O88735 MAP7 ensconsin 82 110 � Q571M2 MKIAA4025 103 110 � Q3UDE9 Q3UDE9 Putative uncharacterized protein 115 110 � B2RQQ5 Microtubule associated protein 1B 270 110 exc1 Q3TXZ6 Q3TXZ6 Putative uncharacterized protein 106 110 �
8 GST-Cx26
cérebro
Q9QWW1
HOME homer protein homolog 2 41 50 �
Q8R2R9 AP3M2 AP-3 complex subunit mu-2 47 50 � O88712 CTBP1 C-terminal-binding protein 1 48 50 � 8R570 SNP47 Synaptosomal associated protein47 47 50 � Q9JJK2 LANC2 LanC-like protein 2 51 50 �
10 GST-Cx26
cérebro Q8R001
MARE2 microtubule-associated protein RP/ EB family member 2
37 40 �
12 GST-Cx26
cérebro
Q80WC3 TNC18 Trinucleotide repeat-containing gene 18 protein
308 60 exc1
Q9WV31 ARC Activity-regulated cytoskeleton associated protein
45 60 �
14 GST-Cx26 fígado
Q80X90 FLNB Filamin B 278 140-150 � Q69ZX3 MKIAA0866 228 140-150 � P28653 PGS1 Biglycan 42 140-150 exc1 Q02788 CO6A2 Collagen alpha-2(VI) chain 110 140-150 � Q6S385 Plectin 10 518 140-150 exc1 B7ZNJ1 Fn1 Fibronectin1 240 140-150 � P59242 CING Cingulin 136 140-150 �
16 GST-Cx26 fígado
O08638 MYH11 Myosin 11 227 140-150 exc1 Q02788 CO6A2 Collagen alpha-2 (VI) chain 110 140-150 exc2 UPI00001954ED
Preditcted: hypotetical protein isoform 1 36 140-150 exc1
P59242 CING Cingulin 136 140-150 exc2
ID*: número de acesso do banco SWISSPROT. MME: massa molecular esperada. MMO: massa molecular observada em gel de SDS-PAGE. Sombreado: proteínas que aparecem em mais de um ensaio. Em vermelho estão identificadas as massas moleculares das proteínas que não correspondem às massas moleculares observadas nos géis de SDS-PAGE. Exc1: proteínas excluídas por diferença entre a massa molecular esperada e a observada. Exc2: proteínas excluídas por redundância. � proteínas selecionadas.

119
Continuação da Tabela 5.5.
Fatia/ ensaio
ID*
Proteínas presentes no ensaio de interação por afinidade com a
proteína GST-Cx26 e ausente com a GST
MME (kDa)
MMO (kDa)
Ação
18 GST-CX26 fígado
B9EHJ3 Tpj1 Tight protein junction 1 189 140-150 �
O08638 MYH11 Myosin 11 227 140-150 exc1,2 O88568 Heterogeneous nuclear ribonuleoprotein
U 88 140-150 exc2
P59242 CING cingulin 136 140-150 exc2 Q02788 CO6A2 Collagen alpha-2 (VI) chain - 110 140-150 exc2 Q6S385 Q6S385 plectin 10 518 140-150 exc1,2 Q9EP71 RAIA4 Ankycorbin 109 140-150 �
P28653 PGS1 Biglycan 42 140-150 exc1,2
20GST-Cx26 fígado
UPI00001954ED
Preditcted: hypotetical protein isoform 1 36 25 �
Q561M8 Cps1 protein (fragment) 32 25 �
P16460 ASSY Argininosuccinate synhase 47 25 �
P62991 UBIQ ubiquitina 9 25 �
Q8VBW8 TTC36 Tetratricopeptide repeat protein 36
20 25 �
B2RY90 Isoc2a protein 22 25 �
22GST-Cx26 fígado
B7ZNJ Fibronectin1 protein 240 140-150 exc2 Q64727 VINC Vinculin 117 140-150 �
Q6S385 Plectin 10 518 140-150 exc1,2 Q01149 CO1A2 Collagen alpha2(I) chain 130 140-150 �
P08122 CO4A2 Canstatin 167 140-150 �
Q80X36 Myosin IE 127 140-150 �
ID*: numero de acesso do banco SWISSPROT. MME: massa molecular esperada. MMO: massa molecular observada em gel de SDS-PAGE. Sombreado: proteínas que aparecem em mais de um ensaio. Em vermelho estão identificadas as massas moleculares das proteínas que não correspondem às massas moleculares observadas nos géis de SDS-PAGE. Exc1: proteínas excluídas por diferença entre a massa molecular esperada e a observada. Exc2: proteínas excluídas por redundância. � proteínas selecionadas.
As proteínas miosina MKIAA0866 (Q69ZX3) e miosina IE (Q80X36)
tiveram parálogas (miosina 9/Q8VDD5, Miosina heavy/Q5SV64, miosinas
1B/Q9WTI7 e 1C/P46735) observadas em ambas às frações GST e GST-CX26. O
mesmo foi observado para os colágenos CO6A2 (Q02788), CO1A2 (Q01149) e
CO4A2 (P08122), com suas parálogas observadas também no controle negativo
(CO6A1/Q04857, Prócolageno VI/UPI0000F2). Por isso, decidimos excluí-las
também. Fortalecendo esta decisão está o fato de colágenos serem proteínas
secretadas, além de serem fibrosas, o que favorece sua precipitação inespecífica in
vitro. A orientação citosólica da porção C-terminal da Cx26 reduz a probabilidade de

120
ocorrer interação entre essa proteína e a via secretora. Pelo mesmo motivo, o
precursor de fibronectina foi excluído (B7ZNJ1).
Duas proteínas, a sintase de aconitato (Q99KI0), que participa do ciclo de
Krebs na matriz mitocondrial e a ribonucleoproteína U (O88568), concentrada no
nucleoplasma, foram excluídas pela baixa probabilidade de terem a mesma
distribuição subcelular que a Cx26 (PSORT). Duas proteínas identificadas por
espectrometria de massas fazem parte do ciclo da uréia. Uma delas, a sintase de
carbamoil-fosfato (Q561M8), está presente na matriz mitocondrial e foi excluída por
causa da sua localização (PSORT). A segunda, sintase de argininosuccinato (P16460),
é citosólica e já foi descrita por se associar à membrana plasmática, em cavéolas e,
portanto permaneceu como candidata em nosso estudo (Flam e col., 2001).
Prosseguimos as análises in silico das 24 proteínas restantes e observamos que
a proteína identificada P62991 corresponde à ubiquitina nesta proteína, sugerindo
que a interação pode ter ocorrido por causa da digestão tríptica de qualquer proteína
da fração GST-Cx26 que estivesse ubiquitinada. Assim, a proteína com número de
acesso (P62991) foi excluída, deixando a lista com 23 candidatos.
Estas 23 proteínas identificadas estão listadas na Tabela 5.7., onde se
observam os números de peptídeos únicos detectados com similaridade a cada
proteína e a porcentagem de resíduos identificados por espectrometria de massas.
As 22 proteínas foram analisadas por uma ferramenta de antologia gênica (Gene
Ontology), classificando-as de acordo com o que se conhece delas sobre sua função
molecular, os processos biológicos e os componentes celulares (Tabela 5.7.). Desta
análise pudemos discriminá-las em quatro grupos com algumas congruências como
se vê na Figura 5.11.: (i) proteínas do citoesqueleto, (ii) proteínas de junções
celulares, (iii) proteínas da via secretora (retículo endoplasmático e aparelho de
Golgi) e/ou endocítica, e (iv) proteínas envolvidas na sinalização celular. Quatro das
22 proteínas não foram incluídas em nenhum desses grupos baseando-se somente
nos dados de ontologia gênica (Tabela 5.7.) sendo agrupadas como “outros” (Figura
5.11.). Na Tabela 5.7. os termos utilizados para classificar essas 23 proteínas em
cinco grupos funcionais distintos encontram-se em negrito e sublinhados.

121
Figura 5.11. Diagrama de Venn mostrando as 23 proteínas candidatas a interagirem com a Cx-26 classificadas em cinco grupos distintos de acordo com identidade de ontologia genica relacionada a processo biológico, compartimento celular e função molecular (Gene Ontology).

12
2
Tabela 5.7. Relação das 23 proteínas candidatas a interagirem com a região C-terminal da Cx26. Estão mostrados o número de peptídeos únicos detectados com similaridade a cada proteína, a porcentagem de resíduos identificados na sequência protéica por espectrometria de massas e sua classificação de acordo sua identidade de Ontologia Gênica para processo biológico, compartimento celular e função molecular.
Nº Nome da proteína (ID Swissprot)
Nº peptídeos únicos1
% da sequência2
GO processo biológico* GO compartimento celular*
GO função molecular*
1 Anquicorbina (Q9EP71)
3 5% Não há termos GO associados
Citoplasma Citoesqueleto Células do Córtex Mitocôndria
Ligante de proteína
2 Subunidade µµµµ2 da proteína adaptadora 3 (AP3) (Q8R2R9)
2 12% Transporte intracelular de proteínas Transporte mediado por vesículas
Complexo de Golgi Complexo adaptador de clatrina Vesícula citoplasmática Membrana
Não há termos GO associados
3 Proteína associada ao citoesqueleto regulada por atividade (ARCAP) (Q9WV31)
2 5,1% Endocitose Junção celular Endossoma Densidade pós-sináptica Membrana pós-sináptica Espinha dentrítica
Ligante de actina
4 Sintase de argininossuccinato (P16460)
2 6,3% Biossíntese da arginina Ciclo de uréia
Mitocôndria Núcleo Citoplasma Reticulo Endoplasmático Lisossomo Fibra do corpo celular
Atividade de síntese do argininossuccinato Atividade de ligase Ligante de nucleotídeo Ligante de ATP
5 Fosfodiesterase dependente de GMP 3',5'-cíclico (Q3TXZ6)
4 6,7% Sinal de transdução Processo catabólico de nucleotídeo
Citosol Membrana raft
Hidrolase Ligante de íon Metal
Nº de peptídeos únicos1 : Número dede peptídeos únicos identificados . % da sequência 2: Porcentagem da sequência identificada por peptídeos. GO: Identidades de Ontologia Gênica. *Resultados obtidos através da ferramenta Gene Ontology

12
3
Continuação da Tabela 5.7.
Nº Nome da proteína (ID Swissprot)
Nº de peptídeos únicos1
% da sequência2
GO processo biológico* GO compartimento celular*
GO função molecular*
6 Cingulina (P59242)
7 2 4
9,8% 3,9% 5,8%
Não há termos GO associados
Junção celular apical Junções ocludentes Complexo de miosinas
Ligante de proteína Atividade motora Ligante de actina
7 Proteína ligante 1 de região C-terminal (CTB-bp1) (O88712)
2 5,4% Regulação negativa de transcrição dependente de DNA Diferenciação celular
Citosol Complexo repressor transcricional
Ligante de NAD Ligante de domínio Atividade de homodimerização Ligante de fator de Transcrição
8 Ativador da morfogênese associado a disheveled (DAAM1) (Q6ZQ74)
3 5% Organização do citoesqueleto de actina
Citoplasma Membrana plasmática
Ligante de actina Ligante de Rho GTPase
9 Ensconsina (O88735)
3 5,2% Morfogênese celular Proliferação celular Homeostasia do nº de células Formação dos feixes de microtúbulos Organização do núcleo Desenvolvimento de órgãos Localização da proteína na membrana plasmática Resposta ao estresse osmótico
Microtúbulos Membrana plasmática basolateral Região perinuclear do citoplasma
Ligante de receptor
Nº de peptídeos únicos1 : Número dede peptídeos únicos identificados . % da sequência 2: Porcentagem da sequência identificada por peptídeos. GO: Identidades de Ontologia Gênica. *Resultados obtidos através da ferramenta Gene Ontology

12
4
Continuação da Tabela 5.7.
Nº Nome da proteína (ID Swissprot)
Nº de peptídeos únicos1
% da sequência2
GO processo biológico* GO compartimento celular*
GO função molecular*
10
Membro 2 da família de proteínas estruturais associadas à zona ativa na citomatriz (CAST2) (Q3UHT7)
2 3,8% Não há termos GO associados
Junção celular Ligante de domínio PDZ Ligante de complexo de proteínas
11 Filamina B (Q80X90)
2 1,1% Processo de desenvolvimento
Citoplasma Citoesqueleto Núcleo Membrana plasmática Adesão focal Fibra estresse
Ligante de actina Ligante de proteína
12 Homer 2 (Q9QWW1)
3 15% Via de sinalização do receptor de glutamato
Junção celular Citoplasma Densidade pós-sináptica
Atividade de scaffold Ligante de actina
13 Proteína 4 de choque térmico (HSP4) (Q571M2)
2 3% Montagem do complexo proteico mediado por chaperona Ligante de proteína
Citoplasma Citosol
Ligante de ATP Ligante de Nucleotídeo
14 Proteína ligante de proteína de choque térmico (HSPBP21) (Q8VBW8)
2 13% Não há termos GO associados
Não há termos GO associados
Ligante
Nº de peptídeos únicos1 : Número dede peptídeos únicos identificados . % da sequência 2: Porcentagem da sequência identificada por peptídeos. GO: Identidades de Ontologia Gênica. *Resultados obtidos através da ferramenta Gene Ontology

12
5
Continuação da Tabela 5.7.
Nº Nome da proteína (ID Swissprot)
Nº de peptídeos únicos1
% da sequência 2
GO processo biológico* GO compartimento celular*
GO função molecular*
15 Precursor da proteína 2A contendo domínio de isocorismatase (ISOC2A) (B2RY90)
2 24% Processo metabólico Desestabilização de proteínas
Núcleo Mitocôndria Citoplasma
Atividade Catalítica
16 Proteína 2 semelhante ao componente C da sintase de lantionina (LANCL2) (Q9JJK2)
2 4,9% Regulação negativa de transcrição dependente de DNA
Citosol Núcleo Membrana plasmática
Atividade catalítica Ligante de fosfolipídio
17 Placoglobina (Q02257)
2 4,3% Adesão celular Montagem desmossomos Desenvolvimento da pele
Citoesqueleto de actina Junção celular Desmossomos
Ligante alfa catenina Proteína RPTP-like
18 Membro 2 da família RP/EB de proteínas associadas a microtúbulos (Q8R001)
2 11% Ciclo celular Divisão celular Mitose
Citoplasma Citoesqueleto Microtúbulos Microtúbulos
Ligante Microtúbulos Ligante de Proteína
19 Proteína 47 associada a sinaptossomo (SAP47) (8R570)
3 11% Não há termos GO associados Citoplasma Membrana Região perinuclear do citoplasma
Não há termos GO associados
Nº de peptídeos únicos1 : Número dede peptídeos únicos identificados . % da sequência 2: Porcentagem da sequência identificada por peptídeos. GO: Identidades de Ontologia Gênica. *Resultados obtidos através da ferramenta Gene Ontology

12
6
Continuação da Tabela 5.7.
Nº Nome da proteína (ID Swissprot)
Nº de peptídeos únicos1
% da sequência 2
GO processo biológico* GO compartimento celular*
GO função molecular*
20 Proteína de ligação a junção ocludente 1/ZO1 (B9EHJ3)
2 1,1% Formação blastocisto Junções ocludentes Junções aderentes Citoplasma Núcleo Membrana plasmática Aparelho de Golgi Complexos juncionais apicais
Ligante de domínio Ligante da região C-term
21 Proteína cinase de tirosina beta2 (TYKbeta2) (Q3UDE9)
2 2,9% Organização dos filamentos de actina Organização do complexo de sinalização
Citoesqueleto Adesão focal
Ligante de ATP Ligante de Proteína Atividade de tirosina cinase Atividade transdutora de sinal
22 Gliceraldeído 3-fosfato desidrogenase (GAPDH) (UPI00001954ED)
3 15% Desenvolvimento de organismo multicelular
Citoplasma Mitocôndria
Atividade de Gliceraldeído 3-fosfato desidrogenase
23 Vinculina (Q64727)
2 3,7% Adesão celular Adesão celular epitelial Formação de lamepódios Morfogênese de epitélio Contração muscular (6936) Localização da proteína na superfície celular Regulação na migração celular
Região extracelular Filamentos de actina Citoesqueleto Junções aderentes Citoplasma Complexo de proteínas Fibra de estresse Fascias aderentes Adesão focal
Ligante de proteína Ligante de Actina Atividade de molécula estrutural Ligante Alfa-catenina Ligante de RhoGTase
Nº de peptídeos únicos1 : Número dede peptídeos únicos identificados . % da sequência 2: Porcentagem da sequência identificada por peptídeos. GO: Identidades de Ontologia Gênica. *Resultados obtidos através da ferramenta Gene Ontology

127
5.5. Discussão
Em virtude da irreversibilidade da maior parte dos casos de surdez, pesquisas
exaustivas buscam desvendar os mecanismos moleculares e celulares relacionados à
fisiologia da audição e, consequentemente, à sua perda, por meio de diversas
estratégias. O mapeamento de genes de surdez em grandes famílias levou ao
conhecimento de mais de uma centena de locos gênicos relacionados à surdez
hereditária. Cerca de 50 produtos gênicos foram identificados desde 1990, o que
contribuiu de maneira fascinante à compreensão da fisiologia da audição. Modelos
animais e estudos funcionais sobre mutações que causam surdez também revelaram
o papel de várias moléculas. No entanto, um grande número de locos mapeados
relacionados à surdez permanece ainda sem ter o seu respectivo gene identificado
(Van Camp e Smith, 2011). Mais recentemente, diversos estudos funcionais têm
sido realizados com o objetivo de identificar proteínas expressas na orelha interna,
que interagem entre si física e funcionalmente, por meio de domínios específicos,
como por exemplo, proteínas das junções celulares, proteínas sinalizadoras, fatores
de transcrição, moléculas do citoesqueleto, enzimas, etc. (Duffy e col., 2002; Hervé e
col., 2007; Derangeon e col., 2008; Dbouk e col., 2009; Dror e Avraham, 2009;
Laird e col., 2009). Esses estudos são ferramentas importantes para a elucidação das
diferentes funções relacionadas a essas interações protéicas e para identificação de
novas via moleculares na orelha interna relacionadas à função auditiva, contribuindo
assim para a descoberta de novos genes candidatos à surdez.
A determinação de interações físicas entre proteínas depende de
metodologias extensas e de alta complexidade. A maioria das abordagens utilizadas é
composta por ensaios in vitro utilizando-se proteínas recombinantes com potencial
de gerar um número elevado de interações falso-positivas, levando à necessidade de
se usar metodogias diferentes para confirmar a interação proteica detectada (Hervé e
col., 2007; Derangeon e col., 2009). O ensaio de precipitação por afinidade é um
método utilizado in vitro para determinar a interação física entre duas proteínas ou
identificar proteínas de um mesmo complexo macromolecular. É útil como teste de
triagem para a primeira identificação de interações proteicas e para confirmar
associações previamente observadas entre proteínas por outros métodos, como por

128
exemplo, co-imunoprecipitação, ensaio do duplo-híbrido da levedura, centrifugação
em gradiente de densidade, etc. (Smith e Johnson, 1988). Tem a vantagem de não
depender de bibliotecas de expressão de cDNA, empregando o conjunto de
proteínas disponível no tecido e obtido nas condições do tampão. Por outro lado,
uma vez que a isca, em nosso caso, foi expressa em bactérias, esta proteína não
apresenta modificações pós-traducionais que podem ser relevantes às interações
moleculares in vivo.
Neste estudo, o nosso objetivo foi buscar por proteínas que interagem com a
região C-terminal da Cx26 citoplasmática que consiste de 10 aminoácidos após o
último domínio transmembrânico da proteína. Esta é uma das regiões mais variáveis
entre as diferentes conexinas na busca por ligantes específicos à Cx26 (Maeda e col.,
2009). Para isso, a porção do gene que contém a região codificadora deste segmento
de camundongo (Mus musculus) foi clonada em fase à sequência codificadora do
peptídeo marcador GST seguida da expressão em bactérias E. coli BL21. A principal
vantagem desse sistema é que o peptídeo GST possui diversos resíduos hidrofílicos,
o que aumenta a solubilidade das proteínas de fusão insolúveis expressas pela E. coli
BL21. No entanto, uma vez que as proteínas são expressas em um vetor
procariótico, interações que exigem dobramento ou modificações pós-traducionais
de uma ou ambas as proteínas não serão identificadas (Phillips-Mason e col., 2006;
Simader e col., 2006). É importante salientar que já foram descritos dois sítios de
metilação na região C-terminal da Cx26 nos resíduos -3 (K) e -7 (G) em humanos.
Porém, a espécie Mus musculus tem no resíduo -3 o aminoácido R (Locke e col.,
2009).
Para facilitar a extração das proteínas nativas e assim criar condições para
interações proteicas utilizamos três tampões de lise celular diferentes. É de
conhecimento que a força iônica (tipo e concentração de sal) e o pH do tampão de
lise utilizado podem afetar significativamente a interação entres proteínas e que a
escolha do detergente para o tampão, assim como sua concentração, deve ter como
parâmetro a quantidade de membrana que será permeabilizada para exposição das
proteínas intracelulares, sem chegar a desnaturá-la. Geralmente concentrações de
0,1% a 1% de detergentes não iônicos comprometem a integridade das membranas

129
celulares, facilitando a lise das células e a extração das proteínas solúveis em
condições nativas. Porém, alguns detergentes não iônicos como o triton-X-100,
apesar de excelentes bloqueadores de interação inespecífica, podem causar grande
perda de ligações específicas e desse modo, levar a resultados falso-negativos.
Interações inespecíficas podem ser minimizadas por meio de pequenos ajustes na
concentração do sal inorgânico ou pela adição de baixas concentrações de
detergentes. Outros componentes críticos são os inibidores de proteases e os
agentes quelantes que evitam a atividade catalítica por meio da degradação de
enzimas ou pelo sequestro dos íons metálicos, respectivamente. Inibidores de
fosfatases permitirão manter interação dependente de resíduos de serina ou treonina
fosforiladas, com o uso de fluoreto de sódio (NaF) ou de tirosinas fosforiladas
como o ortovanadato de sódio (Na3VO4 )(Evans, 1987; Macnname, 1989).
Dentre as 11 fatias dos géis identificadas por conterem as bandas presentes
no ensaio de precipitação com a proteína de fusão GST-Cx26 e ausentes no ensaio
com a GST, sete foram identificadas a partir dos ensaios realizados com o tampão
de lise EGTA, uma com o tampão EDTA e três foram identificadas na fração
solúvel do tampão PHEM. Esses resultados indicam que a maior parte das
interações entre a proteína GST-Cx26 e as proteínas do lisado celular ocorrem na
presença de íons magnésio.
A escolha do tampão PHEM ocorreu porque ele é específico para o
isolamento dos elementos solúveis do citosol em relação aos componentes
insolúveis em triton X-100 do citoesqueleto. O tampão PHEM contém falacidina, a
qual se liga às subunidades dos filamentos de actina estabilizando-os e evitando sua
despolimerização durante a lise celular (Keller e Nigli, 1993). Porém, esse protocolo
não se mostrou eficaz uma vez que observamos que as proteínas das frações
insolúveis de ambos os lisados, cérebro (Figura 5.7.) e fígado (dados não mostrados)
precipitaram-se de forma inespecífica, diminuindo consideravelmente o número de
bandas detectadas à coloração pelo azul de Coomassie.
Devido ao número reduzido de células de suporte onde se expressa a Cx26
no órgão de Corti, utilizamos tecido cerebral e fígado para o ensaio de precipitação

130
por afinidade com a proteína GST-Cx26. No fígado, a expressão da Cx26 é alta,
uma vez que ela está presente nas junções do tipo fenda dos hepatócitos (Zhang e
Nicholson, 1989). Já no cérebro adulto a expressão da Cx26 é restrita a áreas
específicas. Estudos diferentes já demonstraram a presença da Cx26 nas junções do
tipo fenda dos astrócitos, das células da glia e das leptomeninges (Nagy e col., 2001;
Rash e col., 2001, Altevogt e Paul, 2004). A escolha do tecido cerebral também se
deve ao fato de que o proteoma do cérebro é mais diversificado do que o do fígado.
Além disso, um protocolo de lise para esse tecido já estava padronizado no
laboratório de Genômica Funcional.
Como mostrado na Tabela 5.5 (página 117) observamos um número maior
de proteínas identificadas por duplas (GST-Cx26 e GST) nos ensaios com o tecido
cerebral (304 proteínas) em relação ao fígado (143 proteínas), o que provavelmente
deve estar relacionado à maior diversidade de proteínas presentes no cérebro.
Porém, quando observamos as proteínas presentes apenas no ensaio com a GST-
Cx26, verificamos um número maior de proteínas identificadas no ensaio com
fígado (52) em relação ao ensaio com o cérebro (31). No ensaio de precipitação por
afinidade a proteína expressa (GST ou GST-Cx26) ao se prender às contas de
sefarose por afinidade, traz consigo ao precipitado as moléculas que se associam
com qualquer uma de suas partes, C-terminal Cx26, GST ou GSH, nas condições do
ensaio. Desse modo, esses resultados demonstram alta afinidade das proteínas dos
lisados com a GST e a GSH, ou seja, precipitação inespecífica. Isso deve justificar o
grande número de proteínas identificadas com o ensaio controle da GST e ratifica a
importância da utilização dos controles para evitar resultados falso-positivos. Dois
aspectos são importantes lembrar em relação ao fígado: em primeiro lugar, é um
órgão com uma diversidade de tipos celulares bem mais baixa do que o cérebro, por
causa do predomínio de hepatócitos em relação às demais células. A organização
dos hepatócitos em lóbulos e a comunicação entre essas células epiteliais através de
junções do tipo fenda demonstram que, apesar de apresentarem um proteoma
menos rico que neurônios e glia cerebrais, o fígado deve estar enriquecido com
proteínas relevantes para as junções onde ocorrem as conexinas; o segundo aspecto
importante em relação ao fígado é a sua rica matriz extracelular unindo grupos de

131
hepatócitos nos lóbulos hepáticos. Este fato deve justificar a grande representação
de proteínas da matriz extracelular, muitas delas fibrosas como o colágeno, que
devem, por isso, apresentar-se mais propensas à precipitação in vitro inespecífica.
Por meio da espectrometria de massas, identificamos 83 proteínas nos 11
pares de fatias de gel, presentes somente na fração GST-Cx26 nas comparações por
duplas. Porém dentre essas, 34 estavam presentes em outros controles negativos
(GST) e foram excluídas (Tabela 5.5, página 117). Na tabela 5.6 (página 118) temos
a relação das 49 proteínas restantes.
Prosseguimos com as análises para as 49 proteínas identificadas e após
exclusão por (i) redundância de representação no ensaio GST-Cx26, (ii) diferença
entre a massa molecular esperada e a observada, (iii) precipitação inespecífica em
potencial e (iv) localização subcelular incompatível com a Cx26, selecionamos um
total de 23 proteínas candidatas a interagirem com a região C-terminal da Cx26.
Entre essas 23 proteínas, em apenas uma delas (HOMER 2) foi verificado
que o gene que codifica a sua órtologa em humanos está localizado em região
cromossômica com gene de surdez mapeado, porém não identificado. O gene
HOMER2 em humanos está localizado na região cromossômica 15q24.3. Nessa
região cromossômica, está mapeado o lócus DFNB48 (15q23-q25.1) responsável
por surdez hereditária não sindrômica autossômica recessiva (Ahmad e col., 2005;
Van Camp e Smith; 2011). Seria uma interessante hipótese a investigar se o gene
HOMER2 não corresponde ao DFNB48.
Todas as 23 proteínas tiveram confirmação de sua expressão no tecido que
originou o ensaio, fígado ou cérebro, por meio de análises in silico. Porém, não
confirmamos a expressão na orelha interna em três delas (DAAM1, HSPBP21 e
ISOC2A). Para essas análises, levamos em consideração os dados obtidos de quatro
bancos diferentes, Unigene (NCBI), dois bancos específicos a genes expressos na
orelha interna, sendo um de camundongos e outro de humanos (Home e col., 2011
in: Hereditary Hearing Loss Homepage; Morton Cochlear ESTs in
http://brighamandwomens.org, 2011) e em um estudo de análise por microarray de
expressão de genes na orelha interna de camundongos, nas fases embrionária e

132
adulta (Sajan e col., 2007). Com base nessa pesquisa, confirmamos que 20 delas se
expressam na orelha interna. Não podemos afirmar com segurança que as outras
três não o sejam, porque esses bancos de dados são baseados em análises de RNA e
a expressão pode ocorrer em período de desenvolvimento não avaliado.
Por meio de análises in silico realizadas no MGI (Mouse Genome Informatics)
verificamos que dentre os genes dessas 23 proteínas, treze deles (genes que
codificam para as proteínas: CAST2, TYKbeta2, HSPA4, ensconsina, DAAM1,
HOMER2, AP3, CTB-p1, ARCAP, Filamina B, vinculina, placoglobina e
fosfodiesterase) foram explorados em modelos animais de camundongos. No MGI
estão listados os fenótipos que resultam de mutações espontâneas ou induzidas e de
animais geneticamente modificados. Nos modelos animais correspondentes para
esses doze genes, não foi verificado o fenótipo de surdez. Uma vez que esse banco
de dados inclui animais utilizados em pesquisas de consórcios internacionais
associados a diversos grupos de investigação em surdez, acreditamos que a presença
desse sinal clínico também tenha sido um fenótipo avaliado nesses animais.
Os genes de três dentre essas 23 proteínas estão associados a doenças
hereditárias humanas (filamina B, placoglobina e sintase de arginossuccinato).
Dentre as doenças humanas associadas a filamina B (gene FLNB), em duas a surdez
aparece como sinal clínico adicional. São elas a síndrome de Sinostose
Espondilocarpotársica (OMIM#272460), condição rara de herança autossômica
recessiva, cuja principal característica é a fusão dos ossos da coluna, das mãos e dos
pés e a Osteogênese Imperteita do tipo III (OMIM#108721) de herança
autossômica dominante, caracterizada pela fragilidade óssea que leva a fraturas
espontâneas e a articulações hiperflexíveis (Langer e col., 1994; Steiner e Col., 2008).
As doenças humanas associadas aos genes da placoglobina (JUP) e da sintase de
arginossuccinato (ASS1) não têm surdez como sinal clínico. Porém, a placoglobina
esta associada a Doença de Naxos (OMIM#601214), uma genodermatose de
herança autossômica recessiva que se caracteriza pela cardiomiopatia arritmogênica
do ventrículo direito e pela presença de queratoderma palmoplantar (Hammill e col,
1988). No gene Cx26 ja foram descritas mutações dominantes diferentes, associadas
a síndromes que apresentam como sinais clínicos surdez e queratoderma

133
palmoplantar, entre elas, síndrome de Bart-Pumphrey (OMIM#149200),
queratoderma palmoplantar associada a surdez (OMIM#148350), síndrome de
Vohwinkel (OMIM#124500) e a síndrome de KIDS (OMIM#148210) (Richard e
col., 1998, 2002 e 2004, Maestrini e col., 1999; Uyguner e col., 2002;Yotsumoto e
col., 2003).
Um trabalho recente demonstrou que a isoforma da fosfodiesterase
dependente de GMP (Q3TX26) identificada em nossas análises é detectada
exclusivamente na matriz mitocondrial (Acin-Perez e col., 2011). É possível que esta
e outras proteínas de localização na matriz mitocondrial que identificamos façam
parte de um ou mais complexos moleculares que apresentem alguma interação com
uma parceira da Cx26, mas não com a própria. Desta forma, consideramos essa
isoforma da fosfodiesterase dependente de GMP como um resultado falso-positivo
e a excluímos da lista de candidatos a associar com a Cx26, deixando-a com 22
proteínas.
Entre essas 22 proteínas, além da TJP1 que interage com dez diferentes
membros da família das conexinas (Tabela 5.1, página 93), a RB/EB e a vinculina
também já foram descritas por se associarem à Cx43 (Shaw e col., 2007;Xu e col.,
2006). Estudos diferentes demonstraram que interações entre conexinas e proteínas
adaptadoras citosólicas, como os domínios citoplasmáticos das proteínas das
junções oclusivas ZO-1, estão relacionadas à formação, extensão e internalização
das placas juncionais (Hunter e col, 2005; Gilleron e col., 2008; Kieken e col., 2009 ).
A proteína TJP1 contém três domínios PDZ e um domínio SH3 que interagem com
proteínas de membrana, componentes do citoesqueleto e moléculas sinalizadoras. A
TJP1 interage diretamente com a actina filamentosa (actina F) formando uma ponte
entre as proteínas claudinas e ocludinas e o citoesqueleto (Faning e col., 2002). A
tricelulina, codificada pelo gene TRIC, apresenta em sua região C-terminal um
domínio para a ocludina, que media sua interação com a TJP1. Na orelha interna
essa proteína é expressa nas junções ocludentes das células de suporte e ciliadas.
Mutações no gene TRIC que levam à formação de uma proteína truncada e
consequentemente à perda do domínio de interação com a ocludina já foram
identificadas associadas à surdez em humanos (Riazudin e col, 2006). Este cenário

134
reforça a hipótese de que TJP1 possa realmente interagir com a Cx26 e que essa
associação deva ser indireta.
A vinculina é uma proteína adaptadora entre proteínas integrantes da
membrana plasmática nas junções entre células e entre a célula e a matriz
extracelular, com função de ancoramento das suas proteínas ligantes ao
citoesqueleto de actina. Nas placas de adesão focal, essas proteínas são responsáveis
pela interação entre as móleculas de adesão integrinas e os microfilamentos de actina
(Ziegler e col., 2008). A interação da vinculina com a Cx43 já foi demonstrada por
meio de estudos in vitro que incluem co-imunoprecipitação e co-localização (Xu e
col., 2006).
A RB/EB é uma proteína relacionada com a organização e a dinâmica do
microtúbulos. Ela está associada à extremidade plus, citosólica dos microtúbulos,
sendo co-responsável pelo rápido crescimento dessas estruturas distanciando-se de
centros organizadores de microtúbulos como o centrossomo. Além disso, essas
proteínas são responsáveis pelo trânsito de suas proteínas ligantes entre os
compartimentos subcelulares e a membrana plasmática (Schuyler e Pellman, 2001).
A interação da RB/EB com a Cx43 promove o trânsito dos conexons do interior
celular à membrana plasmática até o local onde se encontram junções aderentes
(Shaw e col, 2007). Desta forma, a RB/EB pode ser considerada uma proteína
adaptadora entre o sistema vesicular da via secretora e o sistema de transporte de
organelas pelos microtúbulos.
A seguir temos uma breve revisão sobre a função de cada uma das 19 outras
proteínas selecionadas a interagir com a região C-terminal da Cx26. Onze proteínas
foram associadas ao citoesqueleto pela ferramenta GO. Duas delas interagem
diretamente com microtúbulos, sendo a RB/EB, já citada, e a ensconsina. A
ensconsina é expressa predominantemente nas células epiteliais e está envolvida na
dinâmica dos microtúbulos, tendo papel fundamental na polarização e na
diferenciação celular (Masson e Kreis, 1993; Bulinski e col., 2001). Sua expressão
aumenta no período de estabelecimento de junção intercelular in vitro em
queratinócitos (Fabre-Jonca e col., e col., 1999).

135
Nove proteínas classificadas com distribuição ao citoesqueleto estão direta
ou indiretamente associadas à actina F. Uma delas é a vinculina, já discutida acima.
A anquicorbina (proteína contendo estrutura de coiled-coil e repetições de
anquirina) é uma proteína adaptadora, com função de manutenção e organização do
citoesqueleto e se associa indiretamente aos filamentos de actina em fibras de
estresse e sítios de contato entre células (Peng e col., 2000).
Também conhecida como γ-catenina, a placoglobina é um dos principais
componentes dos desmossomos e das junções aderentes. Essa proteína funciona
como uma proteína intracelular adaptadora, responsável pela associação dos
filamentos intermediários ou microfilamentos de actina às moléculas de adesão,
como as caderinas (Delva e col., 2009). Foi demonstrado que pacientes com o
transtorno cardiocutâneo de Naxos possuem mutações no gene codificador para a
placoglobina e expressão reduzida da Cx43 nas junções intercelulares dos
ventrículos cardíacos, esquerdo e direito (Kaplan e col., 2004), sugerindo que o
remodelamento de junções do tipo fenda seja um evento precoce nessa doença e
decorrente de uma associação anormal entre o citoesqueleto e junções entre células.
A proteína associada ao citoesqueleto e regulada por atividade (ARCAP) é
uma proteína citosólica que interage aos microfilamentos de actina, atuando como
proteína efetora em diversas vias da sinalização neuronal. Essa proteína está
diretamente relacionada à regulação da sinapse e plasticidade neuronal (Shepherd e
Bear, 2011). Foi demonstrado que, nas sinapses, a ARCAP interage com a dinamina
e a endofilina, proteínas envolvidas na endocitose mediada por clatrina, facilitando a
remoção dos receptores de glutamato do tipo AMPA da membrana plasmática
(Chowdhury e col, 2006). Diante disso, pode-se inferir a ARCAP na regulação da
distribuição subcelular dessas proteínas integrantes da membrana plasmática à
atividade sináptica.
A cingulina é uma proteína que ocorre como homodímero em junções
ocludentes de células epiteliais. Essa proteína adaptadora faz intermediação entre as
proteínas ZO, por meio de um motivo conservado de ligação PDZ, e várias outras
proteínas das junções ocludentes, com os microfilamentos de actina e miosinas

136
(Cordenonsi e col., 1999; D'Atri e Citi, 2001). Diferente de outras proteínas
ocludentes, a cingulina não está aparentemente envolvida na organização e
manutenção das placas juncionais. Porém, estudos in vitro já evidenciaram que a
cingulina funciona como uma proteína adaptadora de sinalização que regula a
atividade da RhoA, uma GTPase conhecida por regular o citoesqueleto de actina na
formação das fibras de estresse (Aijaz e col., 2005; Guillemot e Citi, 2006).
A proteína associada a dishevelled e ativadora da morfogênese (DAAM1) é
uma proteína da família das forminas (FH) com papel importante na organização do
citoesqueleto de actina através da profilina e na polaridade celular por meio da
ativação da GTPase Rho. Ela é uma das integrantes da via não canônica de
sinalização Wnt, também conhecida como via de polaridade celular planar, que
regula os movimentos celulares por meio de modificações do citoesqueleto de actina
(Liu e col., 2008).
A CAST2 é uma integrante da família ERC (ELKS-Rab6-interacting protein-
CAST; CAST: Cytomatrix at the active zone-associated structural protein), cujos membros
são conhecidos por se ligarem a proteínas localizadas na zona ativa (ZA)
responsáveis pela regulação de neurotransmissores. A ZA é uma região especializada
da membrana plasmática pré-sináptica onde as vesículas sinápticas se ancoram e se
fundem. As ERCs estão relacionadas à organização e à localização de complexos
protéicos em domínios específicos do citoesqueleto (CZA) responsáveis pela
liberação de neurotransmissores (Ko e col., 2003; Hida e Ohtsuka, 2010). Essas
proteínas também podem ser localizadas no citosol, porém a CAST2 está localizada
exclusivamente nas ZA. Essa proteína apresenta em sua região C-terminal um
motivo de ligação PDZ (PDZbm) (Ko e col., 2003).
A filamina B ou filamina beta é uma proteína citoplasmática associada aos
microfilamentos de actina. Elas são responsáveis pela regulação do citoesqueleto,
pois ancoram os microfilamentos de actina à membrana plasmática, permitindo a
comunicação e a sinalização intracelular entre o citoesqueleto e a célula. Além disso,
as filaminas são responsáveis pela regulação das vias de sinalização responsáveis
pelo desenvolvimento esquelético (Stossel e col., 2001; Lu e col., 2007b)

137
A proteína 2 semelhante ao componente C da sintase de lantionina
(LANCL2) é uma proteína associada ao citoesqueleto de actina, embora também
possa ser encontrada no núcleo celular. A miristolação da região N-terminal da
LANCL2 foi demonstrada. Esta reação de transferência de uma cadeia do lípide
miristato pela enzima coA-miristoil parece promover a associação de proteínas
específicas entre si, como também a domínios na membrana plasmática, no caso da
LANCL2. Este tipo de modificação pós-traducional parece induzir uma variedade
de cascatas de transdução de sinal (Farazi e col., 2001; Landlinger e col., 2006).
Desta forma, além de se associar ao citoesqueleto, essa é uma proteína que deve
estar envolvida na sinalização celular. Da mesma forma, HOMER-2 é uma proteína
associada ao citoesqueleto de actina pela pequena GTPase CDC42 e drebrina, além
de ser uma efetora na sinalização por cálcio (Shiraishi-Yamaguchi e Furuichi, 2009).
Ainda, como será visto abaixo, a enzima gliceraldeído-3-fosfato desidrogenase
(GAPDH) está associada ao citoesqueleto (Trisdale c col., 2009; Tristan e col., 2011).
Pelas informações apresentadas acima, pudemos compor um grupo funcional
relacionado ao citoesqueleto, formado por 13 proteínas, candidatas em potencial a
participar de complexos macromoleculares contendo a Cx26, como listado na
Tabela 5.8. No entanto, esta lista não indica que todas elas façam parte de um ou
mais complexos funcionais com a Cx26. Neste aspecto, é importante discutir que,
mesmo dentro de um possível complexo funcional que inclui a Cx26, as proteínas a
ele pertencentes podem não apresentar interação física direta com a Cx26, mas
somente uma interação funcional. Por outro lado, uma vez que algumas dessas
proteínas, como a cingulina, interagem com outras desse grupo do citoesquelto e
também com proteínas que identificamos como resultados falso-positivos (ex.
miosinas), um cenário possível é que, neste exemplo, a cingulina interaja com a
Cx26 e tenha trazido consigo ao precipitado proteínas parceiras como a
placoglobina e TJP1, mas que podem não ser funcionalmente associados à Cx26.
A subunidade (µ2) da proteína AP3 é parte do complexo adaptador de
clatrina relacionada à endocitose de vesículas sinápticas e ao transporte intracelular
de proteínas mediado por vesículas. A formação de vesículas para o transporte entre
organelas na via secretora requer a montagem de um complexo de proteínas

138
envoltórias, recrutadas a partir do citosol. As proteínas da família AP são
responsáveis pelo sequestro e concentração das proteínas-cargo que serão
transportadas e pela invaginação da membrana para formação das vesículas
transportadoras (Stepp e col., 1997; Rous e col., 2002).
A proteína AP3, juntamente com a proteína RP/EB, agrupam-se em uma
segunda classe funcional, desta vez relacionada ao direcionamento de proteínas à
membrana plasmática, fazendo parte da via secretora celular que inclui a síntese
protéica no retículo endoplasmático rugoso, sua modificação pós-traducional no
complexo de Golgi e o transporte vesicular à membrana plasmática.
A proteína 4 de choque térmico (HSP4) e a proteína 21 ligante de chaperona
de choque térmico (HSPB21) são proteínas de choque térmico da família das
chaperonas, cuja função é auxiliar ao dobramento correto da proteína ou a sua
degradação quando não atinge sua conformação correta. A HSP4 é uma das
isoformas da HSP70. Ela auxilia no dobramento das proteínas que são sintetizadas
nos ribossomos que se encontram livres no citosol ou das proteínas que estão
presentes no retículo endoplasmático. Além disso, ela está relacionada com o
transporte de proteínas para o interior das mitocôndrias e dos cloroplastos (Mayer e
Bukau, 2007; Kriechbaumer e col., 2011). Já a HSPB21 faz parte de um grupo de
proteínas que apresentam um número elevado de repetições tetratricopeptídicas
(TPR), cuja interação com a HSP70 já foi demonstrada (Liu e col., 2008). Podemos
considerar ambas as proteínas, HSP4 e HSPBP21, como integrantes do sistema de
direcionamento de proteínas à membrana plasmática no segundo grupo funcional
nessa classificação (Tabela 5.8). O gene da proteína HSP70 já foi relacionado a
susceptibilidade a perda auditiva induzida por ruído (PAIR) e a proteção à
ototoxicidade por aminoglicosídeos (Konings e col., 2009)
A gliceraldeído-3-fosfato desidrogenase (GAPDH) converte gliceraldeído-3-
fosfato em 1,3 D-gliceroil bisfosfato na presença de NAD+ (dinucleotídeo de
nicotinamida e adenina) e de fosfato inorgânico e media a formação de NADH e
trifosfato de adenosina (ATP). É uma enzima da via glicolítica, conhecida por
interagir com a tubulina e a actina, facilitando a agregação dos microtúbulos e a
polimerização da actina, respectivamente. Ela também é conhecida como proteína

139
adaptadora, responsável por intermediar o trânsito vesicular de proteínas nos
compartimentos celulares como o aparelho de Golgi e o retículo endoplasmático
(Tisdale e col., 2009, Tristan e col., 2011).
A Proteína ligante 1 de região C-terminal (CTB-bp1) faz parte de uma família
de proteínas nucleares que atuam como co-repressores de transcrição. Essas
proteínas também apresentam funções citoplasmáticas diferentes, relacionadas à
manutenção do Complexo de Golgi, que inclui a cisão da membrana para o
transporte intracelular e processo de fragmentação durante a mitose (Corda e col.,
2006). Além disso, a CTB-bp1 possui homologia estrutural com a família das
desidrogenases e já foi demonstrado que ela possui capacidade de se ligar a NAD+
(Fjeld e col., 2003; Zhang e col., 2002). Dada a participação das proteínas GAPDH
e CTB-bp1 no transporte vesicular, elas foram agrupadas na classe das proteínas
envolvidas no direcionamento de proteínas à membrana plasmática (Tabela 5.8). A
CTB-bp1 também esta associada a com a organização das fitas sinápticas. A
otoferlina, produto do gene OTOF também está relacionada à organização das fitas
sinapticas (ribbon sinaptic) das células auditivas ciliadas internas. Em humanos,
mutações nesse gene já foram associadas a neuropatia auditiva (Roux e col., 2007;
Rodrigues-Ballesteros e col., 2008).
O precursor da proteína 2A contendo domínio de isocorismatase (ISOC2A)
é uma proteína citoplasmática cuja única função conhecida é a atividade catalítica na
conversão do isocorismato em 2,3 di-hidrobenzoato e glutamato. Essa é uma
proteína conservada entre as diferentes espécies e já foi vista por interagir com a
proteína inibidora de cinase dependente de ciclina p16INK4a, uma supressora de
tumor. Foi demonstrado que a super expressão da ISOC2A inibe a expressão da
p16INK4a, sugerindo que o gene que codifica a proteína ISOC2A possa ter um papel
importante no desenvolvimento de tumores (Huang e col., 2007).
A sintase de arginossuccinato é citosólica e participa do ciclo da uréia,
catalizando a reação a produção de succinato de arginina, a partir de citrulina e
aspartato. Além disso, a citrulina e o óxido nítrico são produtos da catálise pela
sintase de óxido nítrico a partir de arginina. O óxido nítrico (NO) é uma molécula

140
gasosa na sinalização celular, fundamental em diversos processos celulares (Zhao e
col., 2008). Já foi descrito que enzimas envolvidas na reciclagem de citrulina em
arginina, como as sintases de argininossuccinato e de óxido nitrico podem ser
redistribuídas a domínios específicos da membrana plasmática denominados
caveólas, o que leva à hipótese de que o processo de regeneração da citrulina em
arginina possa ocorrer nesses locais (Flam e col., 2001).
Tabela 5.8.: Classificação funcional baseada na literatura científica das 22 proteínas candidatas em potencial a participarem do complexo molecular da Cx26.
Grupo funcional Proteínas
Relacionado ao citoesqueleto, com ênfase na manutenção e ancoramento de proteínas integrantes da membrana plasmática
1. Anquicorbina 2. Ativador da morfogênese associado a disheveled (DAAM1) 3. Cingulina 4. Ensconsina 5. Filamina B 6. Gliceraldeído 3-fosfato desidrogenase (GAPDH)* 7. Homer 2* 8. Membro 2 da família de proteínas estruturais associadas à zona
ativa na citomatriz (CAST2) 9. Placoglobina 10. Proteína 2 semelhante ao componente C da sintase de lantionina
(LANCL2)* 11. Proteína associada ao citoesqueleto regulada por atividade
(ARCAP) 12. Proteína de ligação a junção ocludente 1/ZO1 (TJP1) 13. Vinculina
Transporte na via secretora
1. Gliceraldeído-3-fosfato desidrogenase (GAPDH)* 2. Membro 2 da família RP/EB de proteínas associadas a
microtúbulos 3. Proteína 4 de choque térmico (HSP4) 4. Proteína ligante 1 de região C-terminal (CTB-bp1) 5. Proteína ligante de proteína de choque térmico (HSPBP21) 6. Subunidade µ2 da proteína adaptadora 3 (AP3)
Sinalização celular 1. Homer2* 2. Precursor da proteína 2A contendo domínio de
isocorismatase (ISOC2A) 3. Proteína 2 semelhante ao componente C da sintase de
lantionina (LANCL2)* 4. Proteína 47 associada a sinaptossomo (SAP47) 5. Proteína cinase de tirosina beta2 (TYKbeta2) 6. Sintase de argininossuccinato
*Proteínas classificadas em mais de um grupo funcional.
A proteína cinase de tirosina beta2 (TYKbeta2) é uma proteína da família das
cinases de adesão focal, cuja atividade é induzida pela interação com as integrinas e

141
os fatores de crescimento. Sua função está associada com a regulação e a
organização do citoesqueleto de actina, com a migração celular e com a reabsorção
óssea. Além disso, essa cinase de tirosina citoplasmática está envolvida na regulação
induzida por Ca2+ dos canais iônicos e na ativação da via de sinalização MAPK. (Du
e col., 2001, Xiong e Feng, 2003).
A HOMER2, assim como outras isoformas, HOMER1 e HOMER3, são
proteínas adaptadoras, presentes nas junções celulares relacionadas à via de
sinalização do Ca2+. Essas proteínas estão envolvidas no direcionamento e na
localização de proteínas sinalizadoras de Ca2+ em domínios específicos da
membrana plasmática e na regulação da atividade dessas proteínas no interior do
complexo proteico de sinalização do Ca2+ (Worley e col., 2007). Como dito, a
LANCL2 também está envolvida na sinalização celular.
O gene que codifica para a proteína 47 associada ao sinaptossomo (SAP47) é
membro de uma família de genes filogeneticamente conservados cuja função é
desconhecida até o momento. Em Drosophila, a SAP47 é expressa em todas as
regiões sinápticas do cérebro larval do tipo selvagem. Drosofilas mutadas com
mutações em que o gene que codifica para essa proteína se encontra parcialmente
deletado, se mostraram viáveis e férteis. No entanto, a curto prazo, foi observada
alteração da plasticidade neuronal (Saumweber e col., 2011).
Basendo-nos nas análises da literatura, revimos a Figura 5.11. e propomos
aqui, uma nova classificação funcional para este grupo das 22 proteínas com
potencial de participar de complexos macromoleculares com a Cx26 (Tabela 5.7.). O
terceiro grupo funcional é composto por seis proteínas relacionadas diretamente às
vias de sinalização na célula.
É fundamental, em experimentos futuros, confirmar a interação desssas 22
proteínas por meio de ensaios in vivo como a co-imunoprecipitação e demonstrar a
co-imunolocalização da Cx26 e sua(s) parceira(s) no órgão de Corti de
camundongos, confirmando a relevância da associação. Esta relevância deve ser
demonstrada funcionalmente in vitro, por meio da análise de processos biológicos
que ocorrem quando a expressão de um dos dois genes é inibida, por exemplo, por

142
meio de interferência por RNA. Os processos biológicos que se mostraram mais
interessantes tendo em vista nossos objetivos são o transporte da Cx26 à membrana
plasmática pela via secretora, a regulação da sua distribuição subcelular, a
manutenção das junções do tipo fenda e o controle de sua atividade a partir da
sinalização inter e intracelular.
5.6. Conclusões
Por meio do ensaio de precipitação por afinidade com a proteína de fusão
GST-Cx26 e análise por espectrometria de massas das proteínas presentes em
bandas de SDS-PAGE, obtidas no ensaio com a GST-Cx26 e ausentes no ensaio
com a GST, identificamos um total de 49 proteínas candidatas a interagirem com a
região C-terminal da Cx26.
Dessas 49 proteínas, dezesseis foram excluídas por redundância de
representação no ensaio GST-Cx26 ou por diferença entre a massa molecular
esperada e a observada. Seis proteínas foram excluídas por precipitação inespecífica.
Das 27 proteínas restantes, após análises in silico em diferentes bancos de dados,
quatro foram exlcuídas por localização subcelular incompatível com a Cx26 e uma
protéina foi excluída por ter sido identificada por similaridade do peptídio à
ubiquitina, sugerindo se tratar de alguma proteína ubiquitinada.
Vinte duas proteínas candidatas em potencial a interagir com a Cx26 foram
analisadas e classificadas de acordo com a ferramenta de ontologia gênica, definindo
a priori quatro grupos funcionais e um quinto grupo misto. Análises da literatura
científica reagruparam essas 22 proteínas em três classes principais: proteínas
responsáveis pela manutenção e ancoramento de proteínas integrantes da membrana
plasmática ao citoesqueleto; proteínas envolvidas no transporte vesicular de
moléculas na via secretora; e proteínas envolvidas na sinalização celular.
Dezenove das 22 proteínas são produtos de genes, cujos transcritos já foram
demonstrados na orelha interna. A confimação da interação entre essas 22 proteínas

143
e a conexina 26, será realizada em experimentos futuros por meio de estudos de co-
localização e imuno-coprecipitação.

144
REFERÊNCIAS BIBLIOGRÁFICAS

144
REFERÊNCIAS BIBLIOGRÁFICAS Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2)
mutations in Japanese. J Med Genet. 2000;37:41–43. Abreu-Silva RS, Lezirovitz K, Braga MC, Spinelli M, Pirana S, Della-Rosa VA, Otto PA,
Mingroni-Netto RC. Prevalence of the A1555G (12S rRNA) and tRNA Ser(UCN) mitochondrial mutations in hearing-impaired Brazilian patients. Braz J Med Biol Res 2006; 39(2):219-26.
Acin-Perez R, Russwurm M, Günnewig K, Gertz M, Zoidl G, Ramos L, Buck J, Levin LR,
Rassow J, Manfredi G, Steegborn C. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J Biol Chem. 2011 [Epub ahead of print]
Ahmad J, Khan SN, Khan SY, Ramzan K, Riazuddin S, Ahmed ZM, Wilcox ER, Friedman
TB, Riazuddin S. DFNB48, a new nonsyndromic recessive deafness locus, maps to chromosome 15q23-q25.1. Hum Genet 2005; 116(5): 407-12.
Ahn KS, Jeon SJ, Jung JY, Kim YS, Kang JH, Shin S, Choi T, Choi SJ, Chung P, Shim H.
Isolation of embryonic stem cells from enhanced green fluorescent protein-transgenic mouse and their survival in the cochlea after allotransplantation. Cytotherapy. 2008;10(7):759-69.
Aijaz S, D'Atri F, Citi S, Balda MS, Matter K. Binding of GEF-H1 to the tight junction-
associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition.Dev Cell. 2005 May;8(5):777-86.
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 5a
Ed. Garland Science, New York, 2007. Aller E, Jaijo T, García-García G, Aparisi MJ, Blesa D, Díaz-Llopis M, Ayuso C, Millán JM.
Identification of large rearrangements of the PCDH15 gene by combined MLPA and a CGH: large duplications are responsible for Usher syndrome. Invest Ophthalmol Vis Sci. 2010 Nov;51(11):5480-5.
Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J
Neurosci. 2004; 24(18): 4313-23. Atar O, Avraham KB. Therapeutics of hearing loss: expectations vs reality. Drug Discov Today
2005;10(19):1323-30. Barker RJ, Price RL, Gourdie RG. Increased association of ZO-1 with connexin43 during
remodeling of cardiac gap junctions. Circ Res 2002; 90(3): 317-24. Batissoco AC. Mutações nos genes GJB2 e GJB6 em Indivíduos com Deficiência Auditiva.
Dissertação de Mestrado. Instituto de Biociências da USP. São Paulo, SP. (2006). Baumgartner B, Harper JW. Deafening cycle. Nat Cell Biol 2003;5(5):385-7. Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat
heart. Circ Res. 1998 Sep 21;83(6):629-35.

145
Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F.Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness.Nat Cell Biol. 2005 Jan;7(1):63-9.
Bernardini L, Palka C, Ceccarini C, Capalbo A, Bottillo I, Mingarelli R, Novelli A, Dallapiccola
BComplex rearrangement of chromosomes 7q21.13-q22.1 confirms the ectrodactyly-deafness locus and suggests new candidate genes. Am J Med Genet A. 2008 Jan 15;146A(2):238-44.
Bernardini L, Sinibaldi L, Capalbo A, Bottillo I, Mancuso B, Torres B, Novelli A, Digilio MC,
Dallapiccola B. HDR (Hypoparathyroidism, Deafness, Renal dysplasia) syndrome associated to GATA3 gene duplication. Clin Genet. 2009 Jul;76(1):117-9.
Berthoud VM, Minogue PJ, Laing JG, Beyer EC. Pathways for degradation of connexins and
gap junctions. Cardiovasc Res. 2004 May 1;62(2):256-67. Review. Braga MCC, Otto PA, Spinelli M. Recurrence risks in cases of non-syndromic deafness. Braz J
Dysmorph Speech Hearing Dis. 1999. 2:33-40. Brigande JV, Heller S. Quo vadis, hair cell regeneration? Nat Neurosci. 2009 Jun;12(6):679-85.
Review. Brobby et al 1998 Connexin 26 R143W mutation associated with recessive non-syndromic
sensorineural deafness in Africa. N Engl J Med 1998, 338:8, 548-50 Brône B, Eggermont J. PDZ proteins retain and regulate membrane transporters in polarized
epithelial cell membranes. Am J Physiol Cell Physiol 2005; 288(1): C20-9. Brownstein Z, Avraham KB. Future Trends and Potential for Treatment of Sensorineural
Hearing Loss. Seminars in hearing. 2006; 27(3). Bruzzone R, Veronesi V, Gomès D, Bicego M, Duval N, Marlin S, Petit C, D'Andrea P, White
TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003 Jan 2;533(1-3):79-88.
Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct
intercellular signaling. Eur J Biochem. 1996 May 15;238(1):1-27. Review. Bulinski JC, Odde DJ, Howell BJ, Salmon TD, Waterman-Storer CM. Rapid dynamics of the
microtubule binding of ensconsin in vivo. J Cell Sci 2001; 114(21): 3885-97. Cama E, Melchionda S, Palladino T, Carella M, Santarelli R, Genovese E, Benettazzo F,
Zelante L, Arslan E. Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int J Audiol. 2009 Jan;48(1):12-7.
Cascio M. Connexins and their environment: effects of lipids composition on ion channels.
Bioch and Biophy Acta 2005;1711:142-153. Chen E, Obolensky E, Rauen KA, Shaffer LG, Li X. Cytogenetic and array CGH
characterization of de novo 1p36 duplications and deletion in a patient with congenital cataracts, hearing loss, choanal atresia, and mental retardation. Am J Med Genet A. 2008 Nov 1;146A(21):2785-90.

146
Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development. 1999 Apr;126(8):1581-90.
Chen P, Zindy F, Abdala C, Liu F, Li X, Roussel MF, et al. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat Cell Biol 2003;5(5):422-6
Chen ZY. Applications of genomics in the inner ear. Pharmacogenomics 2003;4(6):735-45.
Chen ZY. Cell cycle, differentiation and regeneration: where to begin? Cell Cycle 2006;5(22):2609-12.
Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytotic machinery to regulate AMPA receptor trafficking. Neuron 2006; 52: 445-459.
Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002 Jul 9;12(13):1106-11.
Common JE, Bitner-Glindzicz M, O'Toole EA, Barnes MR, Jenkins L, Forge A, Kelsell DP. Specific loss of connexin 26 expression in ductal sweat gland epithelium associated with the deletion mutation del(GJB6-D13S1830). Clin Exp dermatol. 2005 Nov;30(6):688-93.
Corda D, Colanzi A, Luini A.The multiple activities of CtBP/BARS proteins: the Golgi view.
Trends Cell Biol. 2006 Mar;16(3):167-73. Epub 2006 Feb 17. Review.
Cordenonsi M, D'Atri F, Hammar E, Parry DA, Kendrick-Jones J, Shore D, Citi S. Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J Cell Biol 1999; 147(7): 1569–82.
Corwin JT, Cotanche DA. Regeneration of sensory hair cells after acoustic trauma. Science.1988;240:1772–1774.
Cotanche DA, Kaiser CL.Hair cell fate decisions in cochlear development and regeneration. Hear Res. 2010 Jul;266(1-2):18-25.
Cottrell GT, Burt JM. Functional consequences of heterogeneous gap junction channel
formation and its influence in health and disease. Biochim Biophys Acta. 2005 Jun 10;1711(2):126-41. Review.
D'Atri F, Citi S. Cingulin interacts with F-actin in vitro. FEBS Lett 2001; 507(1): 21–4. Dbouk HA, Mroue RM, El-Sabban ME, Talhouk RS. Connexins: a myriad of functions
extending beyond assembly of gap junction channels. Cell Commun Signal 2009; 12:7-4. de Kok YJ, Merkx GF, van der Maarel SM, Huber I, Malcolm S, Ropers HH, Cremers FP. A
duplication/paracentric inversion associated with familial X-linked deafness (DFN3) suggests the presence of a regulatory element more than 400 kb upstream of the POU3F4 gene. Hum Mol Genet. 1995 Nov;4(11):2145-50.
del Castillo F, Aguirre L, Rodriguez-Ballesteros M, Villamar M, Moreno-Pelayo M, Moreno F,
del Castillo I. A novel 200-kb deletion not involving the GBB6 (Connexin-30) gene at the DFNB1 locus is found in the compound heterozygous state with a GJB2 (Connexin-26) mutation in subjects with autosomal recessive hearing impairment. 7th Molecular Biology of Hearing and Deafness. Livro de resumos:85. 2009.

147
del Castillo FJ, Cohen-Salmon M, Charollais A, Caille D, Lampe PD, Chavrier P, Meda P, Petit C. Consortin, a trans-Golgi network cargo receptor for the plasma membrane targeting and recycling of connexins. Hum Mol Genet 2010; 19(2): 262-75.
del Castillo FJ, Gandía M, Pollak A, Hoefsloot L, Silva-Costa SM, Batissoco A, Sartorato EL,
Mingroni-Netto RC, Lechowicz L, Mueller-Malesinska M, Skarzynski H, Piotr H. Skarzynsk PS, Moreno-Pelayo MA, Villamar M, Moreno F, Kremer H, Ploski R, del Castillo I. A Multiplex Ligation-dependent Probe Amplification (MLPA) assay specifically developed to detect novel deletions causing nonsyndromic hearing impairment at the DFNB1 locus. In: 8th Molecular Biology of Hearing and Deafness Conference, 2011. Livro de resumos P51.
del Castillo FJ, Rodríguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA,
Azaiez H, Brownstein Z, Avenarius MR, Marlin S, Pandya A, Shahin H, Siemering KR, Weil D, Wuyts W, Aguirre LA, Martín Y, Moreno-Pelayo MA, Villamar M, Avraham KB, Dahl HH, Kanaan M, Nance WE, Petit C, Smith RJ, Van Camp G, Sartorato EL, Murgia A, Moreno F, del Castillo I. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005 Jul;42(7):588-94.
del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D,
Menéndez I, Moreno F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002 Jan 24;346(4):243-9.
Delva E, Tucker DK, Kowalczyk AP. The Desmosome. Cold Spring Harb Perspect Biol 2009;
1:a002543. Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabédian EN, Petit C. Clinical features
of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet. 1999 Apr 17;353(9161):1298-303.
Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, Osborn AH, Dahl
HH, Middleton A, Houseman MJ, Dodé C, Marlin S, Boulila-ElGaïed A, Grati M, Ayadi H, BenArab S, Bitoun P, Lina-Granade G, Godet J, Mustapha M, Loiselet J, El-Zir E, Aubois A, Joannard A, Petit C, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997 Nov;6(12):2173-7.
Derangeon M, Bourmeyster N, Plaisance I, Pinet-Charvet C, Chen Q, Duthe F, Popoff MR,
Sarrouilhe D, Hervé JC. RhoA GTPase and F-actin dynamically regulate the permeability of Cx43-made channels in rat cardiac myocytes. J Biol Chem 2008; 283(45): 30754-65.
Derangeon M, Spray DC, Bourmeyster N, Sarrouilhe D, Hervé JC. Reciprocal influence of
connexins and apical junction proteins on their expressions and functions. Biochim Biophys Acta. 2009 Apr;1788(4):768-78. Epub 2008 Nov 11. Review.
Diensthuber M, Oshima K, Heller S.Stem/progenitor cells derived from the cochlear sensory
epithelium give rise to spheres with distinct morphologies and features.J Assoc Res Otolaryngol. 2009 Jun;10(2):173-90.
Doetzlhofer A, White P, Lee YS, Groves A, Segil N. Prospective identification and purification
of hair cell and supporting cell progenitors from the embryonic cochlea. Brain Res. 2006;1091:282–288.

148
Doetzlhofer A, White PM, Johnson JE, Segil N, Groves AK. In vitro growth and differentiation of mammalian sensory hair cell progenitors: a requirement for EGF and periotic mesenchyme. Dev Biol 2004;272(2):432-47.
Dror AA, Avraham KB. Hearing loss: mechanisms revealed by genetics and cell biology. Annu
Rev Genet 2009; 43:411-37. Du QS, Ren XR, Xie Y, Wang Q, Mei L, Xiong WC. Inhibition of PYK2-induced actin
cytoskeleton reorganization, PYK2 autophosphorylation and focal adhesion targeting by FAK. J Cell Sci. 2001 Aug;114(Pt 16):2977-87.
Duffy HS, Ashton AW, O’Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC.
Regulation of connexin43 protein complexes by intracellular acidification. Circ Res 2004; 94(2): 215-22.
Duffy HS, Delmar M, Spray DC. Formation of the gap junction nexus: binding partners for
connexins. J Physiol Paris 2002; 96(3-4): 243-9. Duncan LJ, Mangiardi DA, Matsui JI, Anderson JK, McLaughlin-Williamson K, Cotanche DA.
Differential expression of unconventional myosins in apoptotic and regenerating chick hair cells confirms two regeneration mechanisms. J Comp Neurol. 2006 Dec 10;499(5):691-701.
Edge AS, Chen ZY. Hair cell regeneration. Curr Opin Neurobiol. 2008 Aug;18(4):377-82.
Review. Eisen MD, Ryugo DK. Hearing molecules: contributions from genetic deafness. Cell Mol Life
Sci. 2007 Mar;64(5):566-80. Review. Essenfelder GM, Larderet G, Waksman G, Lamartine J. Gene structure and promoter analysis
of the human GJB6 gene encoding connexin 30. Gene. 2005 Apr 25;350(1):33-40. Evans, WH. In J.B.C. Findlay and W.H. Evans (Eds.), Biological Membranes: A Practical
Approach. IRL Press, Oxford, England, 1987, p. 1-25. Fabre-Jonca N, Viard I, French LE, Masson D. Upregulation and redistribution of E-MAP 115
epithelial microtubule-associated protein of 115 kDa) terminally differentiating keratinocytes is coincident with the formation of intercellular contacts. J Invest Dermatol. 1999 Feb;112(2):216-25.
Falk MM, Buehler LK, Kumar Mn, Gilula NB. Cell-free synthesis and assembly of connexins
into funcional gap junction membrane channels. EMBO J 1997;16:2703-2716. Falk MM, Gilula NB. Connexin membrane protein biosynthesis is influenced by polypeptide
positioning within the translocon and signal peptidades access. J Biol Chem 1998;273:7856-7864.
Fallon RF, Goodenough DA. Five-hour half-life of mouse liver gap-junction protein. J Cell
Biol. 1981 Aug;90(2):521-6. Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin
binding region in the tight junction protein ZO-1. FASEB J. 2002 Nov;16(13):1835-7. Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation.
J Biol Chem 2001; 276(43): 39501-4.

149
Feldmann D, Le Maréchal C, Jonard L, Thierry P, Czajka C, Couderc R, Ferec C, Denoyelle F, Marlin S, Fellmann F. A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur J Med Genet. 2009 Jul-Aug;52(4):195-200.
Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the
transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci USA 2003; 100(16): 9202-7.
Flam BR, Hartmann PJ, Harrell-Booth M, Solomonson LP, Eichler DC. Caveolar localization
of arginine regeneration enzymes, argininosuccinate synthase, and lyase, with endothelial nitric oxide synthase.Nitric Oxide. 2001 Apr;5(2):187-97.
Flores CE, Li X, Bennett MV, Nagy JI, Pereda AE. Interaction between connexin35 and
zonula occludens-1 and its potential role in the regulation of electrical synapses. Proc Natl Acad Sci USA 2008; 105(34): 12545-50.
Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G. Gap junctions in the inner
ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J Comp Neurol. 2003 Dec 8;467(2):207-31.
Forge A, Li L, Nevill G. Hair cell recovery in the vestibular sensory epithelia of mature guinea
pigs. J Comp Neurol 1998;397(1):69-88. Fujimoto K, Nagafuchi A, Tsukita S, Kuraoka A, Ohokuma A, Shibata Y. Dynamics of
connexins, E-cadherin and alpha-catenin on cell membranes during gap junction formation. J Cell Sci. 1997 Feb;110 ( Pt 3):311-22.
Gasparini P, Rabionet R, Barbujani G, Melçhionda S, Petersen M, Brøndum-Nielsen K,
Metspalu A, Oitmaa E, Pisano M, Fortina P, Zelante L, Estivill X. High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG. Eur J Hum Genet. 2000 Jan;8(1):19-23.
Gilleron J, Fiorini C, Carette D, Avondet C, Falk MM, Segretain D, Pointis G. Molecular
reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci. 2008 Dec 15;121(Pt 24):4069-78.
Girao H, Pereira P. J Cell Biochem. 2007 Oct 15;102(3):719-28. The proteasome regulates the
interaction between Cx43 and ZO-1. J Cell Biochem 2007; 102(3): 719-28. Glowatzki E, Cheng N, Hiel H, Yi E, Tanaka K, Ellis-Davies GC, Rothstein JD, Bergles DE
The glutamate-aspartate transporter GLAST mediates glutamate uptake at inner hair cell afferent synapses in the mammalian cochlea. J Neurosci. 2006 Jul 19;26(29):7659-64.
Gómez-Casati ME, Murtie J, Taylor B, Corfas G.Cell-specific inducible gene recombination in
postnatal inner ear supporting cells and glia. J Assoc Res Otolaryngol. 2010 Mar;11(1):19-26. Groves AK. The challenge of hair cell regeneration. Exp Biol Med (Maywood). 2010
Apr;235(4):434-46. Review. Grifa A, Wagner CA, D'Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R,
Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet. 1999 Sep;23(1):16-8.

150
Gu R, Montcouquiol M, Marchionni M, Corwin JT.Proliferative responses to growth factors decline rapidly during postnatal maturation of mammalian hair cell epithelia. Eur J Neurosci. 2007 Mar;25(5):1363-72.
Guillemot L, Citi S. Cingulin regulates claudin-2 expression and cell proliferation through the
small GTPase RhoA. Mol Biol Cell 2006; 17(8): 3569-77. Hammill, W. W., Fyfe, D. A., Gillette, P. C., Taylor, A., Dobson, R. L., Thompson, R. P.
Cardiomyopathy with arrhythmias and ectodermal dysplasia: a previously unreported association. Am Heart J 1988; 115: 373-377.
Henzl MT, Thalmann I, Larson JD, Ignatova EG, Thalmann R. The cochlear F-box protein
OCP1 associates with OCP2 and connexin 26. Hear Res. 2004 May;191(1-2):101-9. Hernandez PP, Olivari FA, Sarrazin AF, Sandoval PC, Allende ML. Regeneration in zebrafish
lateral line neuromasts: expression of the neural progenitor cell marker sox2 and proliferation-dependent and-independent mechanisms of hair cell renewal. Dev Neurobiol.2007;67:637–654.
Hervé JC, Bourmeyster N, Sarrouilhe D. Diversity in protein-protein interactions of connexins:
emerging roles. Biochim Biophys Acta. 2004 Mar 23;1662(1-2):22-41. Review. Hervé JC, Derangeon M, Bahbouhi B, Mesnil M, Sarrouilhe D. The connexin turnover, an
important modulating factor of the level of cell-to-cell junctional communication: comparison with other integral membrane proteins. J Memb Biol 2007; 217(1-3):21-33.
Hida Y, Ohtsuka T. CAST and ELKS proteins: structural and functional determinants of the
presynaptic active zone. J Biochem 2010; 148(2): 131-137. Hilgert N, Smith RJ, Van Camp G. Function and expression pattern of nonsyndromic deafness
genes. Curr Mol Med. 2009 Jun;9(5):546-64. Review. Holme, Ralph H., Bussoli, Tracy J. and Steel, Karen P. Table of gene expression in the
developing ear. World Wide Web URL: http://www.ihr.mrc.ac.uk/legacy/hereditary/ genetable/index.html
Huang X, Shi Z, Wang W, Bai J, Chen Z, Xu J, Zhang D, Fu S. Identification and
characterization of a novel protein ISOC2 that interacts with p16INK4a. Biochem Biophys Res Comm 2007; 361: 287–293.
Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear
development. Gene Expr Patterns. 2007;7:798–807. Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap
junction size and organization by influencing channel accretion. Mol Biol Cell 2005; 16(12): 5686-98.
Jeronimo C, Forget D, Bouchard A, Li Q, Chua G, Poitras C, Thérien C, Bergeron D, Bourassa S, Greenblatt J, Chabot B, Poirier GG, Hughes TR, Blanchette M, Price DH, Coulombe B.Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007 Jul 20;27(2):262-74.

151
Jin C, Martyn KD, Kurata WE, Warn-Cramer BJ, Lau AF. Connexin43 PDZ2 binding domain mutants create functional gap junctions and exhibit altered phosphorylation. Cell Commun Adhes 2004; 11(2-4): 67-87.
Jones JM, Montcouquiol M, Dabdoub A, Woods C, Kelley MW. Inhibitors of differentiation
and DNA binding (Ids) regulate Math1 and hair cell formation during the development of the organ of Corti. J Neurosci 2006;26(2):550-8.
Junqueira LC; Carneiro J. Histologia Básica. 9º edição – Editora Guanabara Koogan S.A., Rio
de Janeiro. 1999. Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A,
Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm 2004; 1(1): 3-11.
Kausalya PJ, Reichert M, Hunziker W. Connexin45 directly binds to ZO-1 and localizes to the
tight junction region in epithelial MDCK cells. FEBS Lett 2001; 505(1): 92-6. Kawamoto K, Yagi M, Stöver T, Kanzaki S, Raphael Y.Hearing and hair cells are protected by
adenoviral gene therapy with TGF-beta1 and GDNF. Mol Ther. 2003 Apr;7(4):484-92. Kazuya Ono , Takayuki Nakagawa, Ken Kojima, Masahiro Matsumoto, Takeshi Kawauchi,
Mikio Hoshino, Juichi Ito. Silencing p27 reverses post-mitotic state of supporting cells in neonatal mouse cochleae. Molecular and Cellular Neuroscience 42 (2009) 391–398
Keats BJ, Berlin CI. Genomics and hearing impairment. Genome Res. 1999 Jan;9(1):7-16.
Review. Keller HU, Niggli V. Colchicine-induced stimulation of PMN motility related to cytoskeletal
changes in actin, alpha-actinin, and myosin. Cell Motil Cytoskeleton 1993; 25(1): 10-8. Kelley MW. Regulation of cell fate in the sensory epithelia of the inner ear. Nat Rev Neurosci.
2006;7(11):837-49. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM.
Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997 May 1;387(6628):80-3.
Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic
sensorineural hearing loss: a HuGE review. Genet Med. 2002 Jul-Aug;4(4):258-74. Review. Kiang DT, Jin N, Tu ZJ, Lin HH. Upstream genomic sequence of the human connexin26 gene.
Gene. 1997 Oct 15;199(1-2):165-71. Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, Hirst-Jensen BJ, Duffy HS,
Sorgen PL. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009 May 8;104(9):1103-12.
Ko J, Na M, Kim S, Lee JR, Kim E. Interaction of the ERC family of RIM-binding proteins
with the liprin-alpha family of multidomain proteins. J Biol Chem. 2003 Oct 24;278(43):42377-85. Epub 2003 Aug 15.

152
Kojima T,Kokai Y, Chiba H, Yamamoto, M. Mochizuki Y, Sawada N., Cx32 but not Cx26 is associated with tight junctions in primary cultures of rat hepatocytes, Exp. Cell Res. 263 (2001) 193–201
Konings A, Van Laer L, Van Camp G. Genetic studies on noise-induced hearing loss: a review.
Ear Hear. 2009 Apr;30(2):151-9. Kopke RD, Jackson RL, Li G, Rasmussen MD, Hoffer ME, Frenz DA, Costello M, Schultheiss
P, Van De Water TR. Growth factor treatment enhances vestibular hair cell renewal and results in improved vestibular function. Proc Natl Acad Sci USA. 2001;98:5886–5891.
Koval M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006
Mar;16(3):159-66. Review. Kriechbaumer V, von Löffelholz O, Abell BM. Chaperone receptors: guiding proteins to
intracellular compartments. Protoplasma. 2011 Apr 3. [Epub ahead of print] PMID:21461941
Krutovskikh V, Yamasaki H. Connexin gene mutations in human genetic diseases. Mutat Res.
2000 Apr;462(2-3):197-207. Review. Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki
Y, Kobayashi T, Matsubara Y Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet. 2003 May 1;12(9):995-1004.
Kuntz AL, Oesterle EC. Transforming growth factor alpha with insulin stimulates cell
proliferation in vivo in adult rat vestibular sensory epithelium. J Comp Neurol. 1998;399:413–423.
Laing JG, Beyer EC. The gap junction protein connexin43 is degraded via the ubiquitin
proteasome pathway. J Biol Chem. 1995 Nov 3;270(44):26399-403. Laing JG, Koval M, Steinberg TH. Association with ZO-1 correlates with plasma membrane
partitioning in truncated connexin45 mutants. J Membr Biol 2005; 207(1): 45-53. Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap
junction protein in cultured cardiac myocytes. Biochem J. 1991 Jan 1;273(Pt 1):67-72. Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction
internalization and degradation. Biochim Biophys Acta. 2005 Jun 10;1711(2):172-82. Review. Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006 Mar 15;394(Pt
3):527-43. Review. Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010
Feb;20(2):92-101. Review. Landlinger C, Salzer U, Prohaska R. Myristoylation of human LanC-like protein 2 (LANCL2) is
essential for the interaction with the plasma membrane and theincrease in cellular sensitivity to adriamycin. Biochim Biophys Acta 2006; 1758(11):1759-67.

153
Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, Kelley MW. Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet. 1999;21:289–292.
Langer, L. O., Jr., Gorlin, R. J., Donnai, D., Hamel, B. C. J., Clericuzio, C. Spondylocarpotarsal
synostosis syndrome (with or without unilateral unsegmented bar). Am J Med Genet 1994; 51: 1-8.
Lautermann J, Frank HG, Jahnke K, Traub O, Winterhager E. Developmental expression
patterns of connexin26 and -30 in the rat cochlea. Dev Genet. 1999;25(4):306-11. Lefebvre PP, Van De Water TR. Connexins, hearing and deafness: clinical aspects of mutations
in the connexin 26 gene. Brain Res Brain Res Rev. 2000 Apr;32(1):159-62. Review. Lehman AM, Friedman JM, Chai D, Zahir FR, Marra MA, Prisman L, Tsang E, Eydoux P,
Armstrong L. A characteristic syndrome associated with microduplication of 8q12, inclusive of CHD7. Eur J Med Genet. 2009 Nov-Dec;52(6):436-9.
Lerer I, Sagi M, Malamud E, Levi H, Raas-Rothschild A, Abeliovich D. Contribution of
connexin 26 mutations to nonsyndromic deafness in Ashkenazi patients and the variable phenotypic effect of the mutation 167delT. Am J Med Genet. 2000 Nov 6;95(1):53-6.
Leung CT, Coulombe PA, Reed RR. Contribution of olfactory neural stem cells to tissue
maintenance and regeneration. Nat Neurosci. 2007 Jun;10(6):720-6. Lezirovitz K, Batissoco AC, Lima FT, Auricchio MTBM, Dantas VGL, Oiticica J, Mingroni-
Netto RC. Splice donor site deletion in the TECTA gene causing autosomal dominant deafness in a Brazilian family. In: 8th Molecular Biology of Hearing and Deafness Conference, 2011. Livro de resumos P15.
Li X, Olson C, Lu S, N. Kamasawa, T. Yasumura, J.E. Rash, J.I. Nagy, Neuronal connexin36
association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1, Eur. J. Neurosci. 19 (2004c)2132–2146.
Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita N, Davidson KG, Yasumura T, Rash
JE, Nagy JI, Connexin47, connexin29 and connexin32 coexpression in oligodendrocytes and Cx47 association with zonula occludens-1(ZO-1) in mouse brain, Neuroscience 126 (2004c) 611–630.
Li H, Corrales CE, Edge A, Heller S. Stem cells as therapy for hearing loss. Trends Mol
Med.2004;10:309–315. Li H, Liu H, Heller S. Pluripotent stem cells from the adult mouse inner ear. Nat Med
2003;9(10):1293-9 (b). Li H, Roblin G, Liu H, Heller S. Generation of hair cells by stepwise differentiation of
embryonic stem cells. Proc Natl Acad Sci USA. 2003;100(23):13495-500 (a). Liu Q, Gao J, Chen X, Chen Y, Chen J, Wang S, Liu J, Liu X, Li J. HBP21: a novel member of
tpr motif family, as a potential chaperone of heat shock protein 70 in proliferative vitreoretinopathy (PVR) and breast cancer. Mol Biotechnol 2008; 40: 231–40.
Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, Habas R. Mechanism of activation
of the Formin protein Daam1. Proc Natl Acad Sci USA 2008; 105(1): 210–5.

154
Liu XZ, Xia XJ, Xu LR, Pandya A, Liang CY, Blanton SH, Brown SD, Steel KP, Nance WE.
Mutations in connexin31 underlie recessive as well as dominant non-syndromic hearing loss. Hum Mol Genet. 2000 Jan 1;9(1):63-7.
Locke D, Bian S, Li H, Harris AL. Post-translational modifications of connexin26 revealed by mass spectrometry. Biochem J. 2009 Dec 10;424(3):385-98.
Lopez IA, Zhao PM, Yamaguchi M, de Vellis J, Espinosa-Jeffrey A.Stem/progenitor cells in
the postnatal inner ear of the GFP-nestin transgenic mouse. Int J Dev Neurosci. 2004 Jun;22(4):205-13
Lou X, Zhang Y, Yuan C. Multipotent stem cells from the young rat inner ear. Neurosci Lett
2007;416(1):28-33. Löwenheim H, Furness DN, Kil J, Zinn C, Gültig K, Fero ML, Frost D, Gummer AW,
Roberts JM, Rubel EW, Hackney CM, Zenner HP. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of corti. Proc Natl Acad Sci USA. 1999 Mar 30;96(7):4084-8.
Löwenheim, H., Furness, D.N., Kil, J., Zinn, C., Gültig, K., Fero, M.L., Frost, D., Gummer,
A.W., Roberts, J.M., Rubel, E.W., Hackney, C.M., Zenner, H.P., 1999. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of Corti. Proc. Natl. Acad. Sci. U. S. A. 96, 4084–4088.
Lu J, Lian G, Lenkinski R, De Grand A, Vaid RR, Bryce T, Stasenko M, Boskey A, Walsh C,
Sheen V. Filamin B mutations cause chondrocyte defects in skeletal development. Hum Mol Genet 2007; 16(14): 1661–75. (b)
Lu J, Meng W, Poy F, Maiti S, Goode BL, Eck MJ. Structure of the FH2 domain of Daam1:
implications for formin regulation of actin assembly. J Mol Biol 2007; 369(5): 1258–69.(a) Lundin J, Söderhäll C, Lundén L, Hammarsjö A, White I, Schoumans J, Läckgren G, Kockum
CC, Nordenskjöld A. 22q11.2 microduplication in two patients with bladder exstrophy and hearing impairment. Eur J Med Genet. 2010 Mar-Apr;53(2):61-5.
Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Structure of
the connexin 26 gap junction channel at 3.5 A resolution. Nature 2009; 458(7238): 597-602. Maestrini, E., Korge, B. P., Ocana-Sierra, J., Calzolari, E., Cambiaghi, S., Scudder, P. M.,
Hovnanian, A., Monaco, A. P., Munro, C. S. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Molec Genet 1999; 8: 1237-1243.
Martinez-Monedero R, Yi E, Oshima K, Glowatzki E, Edge AS. Differentiation of inner ear
stem cells to functional sensory neurons. Dev Neurobiol. 2008;68:669–684. Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for
connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003 Apr 15;12(8):805-12.
Masson D, Kreis TE. Identification and molecular characterization of E-MAP-115, a novel
microtubule-associated protein predominantly expressed in epithelial cells. J Cell Biol 1993; 123(2): 357-71.

155
Mathijssen IB, Hoovers JM, Mul AN, Man HY, Ket JL, Hennekam RC. Array comparative genomic hybridization analysis of a familial duplication of chromosome 13q: a recognizable syndrome. Am J Med Genet A. 2005 Jul 1;136(1):76-80.
Matos TD, Caria H, Simões-Teixeira H, Aasen T, Nickel R, Jagger DJ, O'Neill A, Kelsell DP,
Fialho G. A novel hearing-loss-related mutation occurring in the GJB2 basal promoter. J Med Genet. 2007 Nov;44(11):721-5.
Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol
Life Sci 2005; 62(6): 670-84. McNamee MG. Isolation and characterization of cell membranes. Biotechniques 1989; 7(5):
466-75.
Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005 Aug;7(8):766-72. Review.
Mingroni Netto RC, Uehara DT, Freitas EL, Mazzeu JF, Auricchio MTBM, Tabith Jr A, Rosenberg C. A Duplication in a Patient with Nonsyndromic Deafness Reveals a Novel Candidate Gene for Deafness, DOCK4. In: 8th Molecular Biology of Hearing and Deafness Conference, 2011. Livro de resumos T113.
Mingroni-Netto RC, Lezirovitz K, Uehara DT, Batissoco AC. Novel GJB2 duplication identified in a patient with nonsyndromic recessive hearing loss by MLPA (Multiplex Ligation-dependent Probe Amplification). In: 8th Molecular Biology of Hearing and Deafness Conference, 2011. Livro de resumos P52.
Morris KA, Snir E, Pompeia C, Koroleva IV, Kachar B, Hayashizaki Y, Carninci P, Soares MB, Beisel KW. Differential expression of genes within the cochlea as defined by a custom mouse inner ear microarray. J Assoc Res Otolaryngol 2005; 6(1):75-89.
Nagasawa K, Chiba H, Fujita H, Kojima T, Saito T, Endo T, Sawada N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and ling endothelial cells. J Cell Physiol 2006; 208(1): 123-32.
Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE. Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol 2001; 441(4): 302-23.
Najmabadi H, Nishimura C, Kahrizi K, Riazalhosseini Y, Malekpour M, Daneshi A, Farhadi M, Mohseni M, Mahdieh N, Ebrahimi A, Bazazzadegan N, Naghavi A, Avenarius M,Arzhangi S, Smith RJ. GJB2 mutations: passage through Iran.Am J Med Genet A. 2005 Mar 1;133A(2):132-7.
Nakagawa T, Lomb DJ, Haigis MC, Guarente L.SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009; 137(3): 560-70.
Nelson HD, Bougatsos C, Nygren P; 2001 US Preventive Services Task Force. Universal newborn hearing screening: systematic review to update the 2001 US Preventive Services Task Force Recommendation. Pediatrics. 2008 Jul;122(1):e266-76. Review. Erratum in: Pediatrics. 2008 Sep;122(3):689.
Nelson RF, Glenn KA, Zhang Y, Wen H, Knutson T, Gouvion CM, Robinson BK, Zhou Z, Yang B, Smith RJ, Paulson HL. Selective cochlear degeneration in mice lacking the F-box protein, Fbx2, a glycoprotein-specific ubiquitin ligase subunit. J Neurosci. 2007 May 9;27(19):5163-71.

156
Nielsen PA, Baruch A, Giepmans BN, Kumar NM. Characterization of the association of connexins and ZO-1 in the lens. Cell Commun Adhes 2001; 8(4-6): 213-7.
Nielsen PA, Baruch A, Shestopalov VI, Giepmans BN, Dunia I, Benedetti EL, Kumar NM.
Lens connexins alpha3Cx46 and alpha8Cx50 interact with zonula occludens protein-1 (ZO-1). Mol Biol Cell 2003; 14(6): 2470-81.
Nusrat A, Chen JA, Foley CS, Liang TW, Tom J, Cromwell M, Quan C, Mrsny RJ. The coiled-
coil domain of occludin can act to organize structural and functional elements of the epithelial tight junction. J Biol Chem 2000; 275(38): 29816-22.
Oiticica J, Batissoco AC, Junior LCMB, Netto RCM, Haddad LA, Bento RF. Organ of Corti
culture for functional analysis of precursor, support and hair cells. Arq Int Otorrinolaringol 2007;11(4):433-437.
Ono K, Nakagawa T, Kojima K, Matsumoto M, Kawauchi T, Hoshino M, Ito J. Silencing p27
reverses post-mitotic state of supporting cells in neonatal mouse cochleae. Mol Cell Neurosci. 2009 Dec;42(4):391-8.
Oshima K, Grimm CM, Corrales CE, Senn P, Martinez Monedero R, Geleoc GS, Edge A,
Holt JR, Heller S. Differential distribution of stem cells in the auditory and vestibular organs of the inner ear. J Assoc Res Otolaryngol. 2007;8:18–31.
Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including
most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet. 2002 Jan;10(1):72-6.
Parker MA, Cotanche DA. The potential use of stem cells for cochlear repair. Audiol
Neurootol 2004;9(2):72-80. Parker MA, Jiang K, Kempfle JS, Mizutari K, Simmons CL, Bieber R, Adams J, Edge
AS.TAK1 Expression in the Cochlea: A Specific Marker for Adult Supporting Cells.J Assoc Res Otolaryngol. 2011 Aug;12(4):471-83.
Penes MC, Li X, Nagy JI. Expression of zonula occludens-1 (ZO-1) and the transcription
factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur J Neurosci 2005; 22(2): 404-18.
Peng YF, Mandai K, Sakisaka T, Okabe N, Yamamoto Y, Yokoyama S, Mizoguchi A, Shiozaki
H, Monden M, Takai Y. Ankycorbin: a novel actin cytoskeleton-associated protein. Genes Cells 2000; 5(12): 1001-8.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR.Nucleic
Acids Res. 2001 May 1;29(9):e45. Phillips-Mason PJ, Gates TJ, Major DL, Sacks DB, Brady-Kalnay SM. The receptor protein-
tyrosine phosphatase PTPmu interacts with IQGAP1. J Biol Chem. 2006 Feb 24;281(8):4903-10.
Piatto VB, Maniglia JV. Importância do gene Conexina 26 na etiologia da deficiência
sensorioneural não sindrômica. Acta Awho. 2001. 20:106-112.

157
Pranchevicius MC, Baqui MM, Ishikawa-Ankerhold HC, Lourenço EV, Leão RM, Banzi SR, dos Santos CT, Roque-Barreira MC, Espreafico EM, Larson RE.Myosin Va phosphorylated on Ser1650 is found in nuclear speckles and redistributes to nucleoli upon inhibition of transcription. Cell Motil Cytoskeleton. 2008 Jun;65(6):441-56.
Pujol R, Réclar-Enjalbert V, Pujol T. Promenade round the cochlea. World Wide Web
URL:http://www.iurc.montp.inserm.fr/cric/audition/english/index.htm. Acesso em: Julho 2011
Pujol R. Morphology, synaptology and electrophysiology of the developing cochlea. Acta
Otolaryngol Suppl. 1985;421:5–9. Putcha GV, Bejjani BA, Bleoo S, Booker JK, Carey JC, Carson N, Das S, Dempsey MA,
Gastier-Foster JM, Greinwald JH Jr, Hoffmann ML, Jeng LJ, Kenna MA, Khababa I,Lilley M, Mao R, Muralidharan K, Otani IM, Rehm HL, Schaefer F, Seltzer WK, Spector EB, Springer MA, Weck KE, Wenstrup RJ, Withrow S, Wu BL, Zariwala MA, Schrijver I. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med. 2007 Jul;9(7):413-26.
Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations
in gap junction genes encoding beta connexins. Hum Mutat. 2000 Sep;16(3):190-202. Review.
Rackauskas M, Neverauskas V, Skeberdis VA. Diversity and properties of connexin gap
junction channels. Medicina (Kaunas). 2010;46(1):1-12. Review. Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey.Nucleic
Acids Res. 2002 Sep 1;30(17):3894-900. Ramsebner R, Volker R, Lucas T, Hamader G, Weipoltshammer K, Baumgartner WD,
Wachtler FJ, Kirschhofer K, Frei K. High incidence of GJB2 mutations during screening of newborns for hearing loss in Austria. Ear Hear. 2007 Jun;28(3):298-301.
Ramsebner R, Volker R, Lucas T, Hamader G, Weipoltshammer K, Baumgartner
WD, Wachtler FJ, Kirschhofer K, Frei K. High incidence of GJB2 mutations during screening of newborns for hearing loss in Austria. Ear Hear. 2007 Jun;28(3):298-301.
Raphael Y, Altschuler RA. Structure and innervation of the cochlea. Brain Res Bull 2003;60(5-
6):397-422. Rash JE, Yasumura T, Davidson KG, Furman CS, Dudek FE, Nagy JI. Identification of cells
expressing Cx43, Cx30, Cx26, Cx32 and Cx36 in gap junctions of rat brain and spinal cord. Cell Commun Adhes. 2001;8(4-6):315-20.
Rask-Andersen H, Bostrom M, Gerdin B, Kinnefors A, Nyberg G, Engstrand T, Miller JM,
Lindholm D. Regeneration of human auditory nerve. In vitro/in video demonstration of neural progenitor cells in adult human and guinea pig spiral ganglion. Hear Res. 2005;203:180–191.
Rask-Andersen H, Boström M, Gerdin B, Kinnefors A, Nyberg G, Engstrand T, Miller JM,
Lindholm D. Ryals BM, Rubel EW. Hair cell regeneration after acoustic trauma in adult Coturnix quail.Science. 1988;240:1774–1776.

158
Reynolds BA, Rietze RL.Neural stem cells and neurospheres--re-evaluating the relationship. Nat Methods. 2005 May;2(5):333-6.
Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, Ramzan K, Khan SN, Chattaraj P, Friedman PL, Anderson JM, Belyantseva IA, Forge A, Riazuddin S, Friedman TB.Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet. 2006 Dec;79(6):1040-51. Epub 2006 Oct 31.
Richard G, Brown N, Ishida-Yamamoto A, Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart-Pumphrey syndrome is caused by a novel missense mutation in GJB2.J Invest Dermatol 2004; 123(5): 856-63.
Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 1998; 103(4): 393-9.
Richard, G., Rouan, F., Willoughby, C. E., Brown, N., Chung, P., Ryynanen, M., Jabs, E. W., Bale, S. J., DiGiovanna, J. J., Uitto, J., Russell, L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet 2002; 70: 1341-1348.
Rio C, Dikkes P, Liberman MC, Corfas G. Glial fibrillary acidic protein expression and promoter activity in the inner ear of developing and adult mice. J Comp Neurol. 2002 Jan 7;442(2):156-62.
Roberson DW, Alosi JA, Cotanche DA. Direct transdifferentiation gives rise to the earliest new hair cells in regenerating avian auditory epithelium. J Neurosci Res. 2004 Nov 15;78(4):461-71.
Rodríguez-Ballesteros M, Reynoso R, Olarte M, Villamar M, Morera C, Santarelli R, Arslan E,
Medá C, Curet C, Völter C, Sainz-Quevedo M, Castorina P, Ambrosetti U, Berrettini S, Frei K, Tedín S, Smith J, Cruz Tapia M, Cavallé L, Gelvez N, Primignani P, Gómez-Rosas E, Martín M, Moreno-Pelayo MA, Tamayo M, Moreno-Barral J, Moreno F, del Castillo I. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy.Hum Mutat. 2008 Jun;29(6):823-31.
Rous BA, Reaves BJ, Ihrke G, Briggs JAG, Gray SR, Stephens DJ, Banting G, Luzio JP. Role
of Adaptor Complex AP-3 in Targeting Wild- Type and Mutated CD63 to Lysosomes. Mol Biol Cell 2002; 13: 1071–1082.
Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M,
Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C.Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2007 Oct 20;127(2):277-89.
Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang DS, Garcia-Anoveros J, Hinds
PW, Corwin JT, Corey DP, Chen ZY. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science. 2005 Feb 18;307(5712):1056-8.
Sajan SA, Warchol ME, Lovett M. Toward a systems biology of mouse inner ear
organogenesis: gene expression pathways, patterns and network analysis. Genetics 2007; 177(1): 631-53.

159
Sakaguchi H, Yaoi T, Suzuki T, Okano H, Hisa Y, Fushiki S. Spatiotemporal patterns of Musashi1 expression during inner ear development. Neuroreport. 2004;15:997–1001.
Sambrook J., Fritsch E.F. & Maniatis T. Molecular cloning: a laboratory manual. 2a Ed. Cold
Spring Harbor Laboratory Press, New York, 1989. Sartorato EL, Gottardi E, de Oliveira CA, Magna LA, Annichino-Bizzacchi JM, Seixas CA,
Maciel-Guerra AT. Determination of the frequency of the 35delG allele in Brazilian neonates. Clin Genet. 2000 Oct;58(4):339-40.
Santos RL, Aulchenko YS, Huygen PL, van der Donk KP, de Wijs IJ, Kemperman MH,
Admiraal RJ, Kremer H, Hoefsloot LH, Cremers CW. Hearing impairment in Dutch patients with connexin 26 (GJB2) and connexin 30 (GJB6) mutations. Int J Pediatr Otorhinolaryngol. 2005 Feb;69(2):165-74.
Saumweber T, Weyhersmüller A, Hallermann S, Diegelmann S, Michels B, Bucher D, Funk N, Reisch D, Krohne G, Wegener S, Buchner E, Gerber B. Behavioral and synaptic plasticity are impaired upon lack of the synaptic protein SAP47. J Neurosci 2011; 31(9): 3508-18.
Savary E, Hugnot JP, Chassigneux Y, Travo C, Duperray C, Van De Water T, et al. Distinct
population of hair cell progenitors can be isolated from the postnatal mouse cochlea using side population analysis. Stem Cells 2007;25(2):332-9.
Savary E, Sabourin JC, Santo J, Hugnot JP, Chabbert C, Van De Water T, et al. Cochlear
stem/progenitor cells from a postnatal cochlea respond to Jagged1 and demonstrate that notch signaling promotes sphere formation and sensory potential. Mech Dev 2008;125(8):674-86.
Schuyler SC, Pellman D.Search, capture and signal: games microtubules and centrosomes play.
J Cell Sci. 2001 Jan;114(Pt 2):247-55. Review. Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies.
Proteomics 2010; 10(6):1265-9. Seeman P, Sakmaryová I. High prevalence of the IVS 1 + 1 G to A/GJB2 mutation among
Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2. Clin Genet. 2006 May;69(5):410-3
Senn P, Oshima K, Teo D, Grimm C, Heller S. Robust postmortem survival of murine
vestibular and cochlear stem cells. J Assoc Res Otolaryngol. 2007 Jun;8(2):194-204. Shahin H, Walsh T, Sobe T, Lynch E, King MC, Avraham KB, Kanaan M.Genetics of
congenital deafness in the Palestinian population: multiple connexin 26 alleles with shared origins in the Middle East. Hum Genet. 2002 Mar;110(3):284-9.
Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY.Microtubule plus-end-
tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007 Feb 9;128(3):547-60. Erratum in: Cell. 2008 Apr 18;133(2):376.
Shepherd JD, Bear MF.New views of Arc, a master regulator of synaptic plasticity. Nat
Neurosci. 2011 Mar;14(3):279-84. Shiraishi-Yamaguchi Y, Furuichi T.The Homer family proteins. Genome Biol. 2007;8(2):206.
Review.

160
Silva-Costa SM, Coeli FB, Lincoln-de-Carvalho CR, Marques-de-Faria AP, Kurc M, Pereira T, Pomilio MC, Sartorato EL. Screening for the GJB2 c.-3170 G>A (IVS 1+1 G>A) mutation in Brazilian deaf individuals using multiplex ligation-dependent probe amplification. Genet Test Mol Biomarkers. 2009 Oct;13(5):701-4.
Simader H, Hothorn M, Köhler C, Basquin J, Simos G, Suck D. Structural basis of yeast
aminoacyl-tRNA synthetase complex formation revealed by crystal structures of two binary sub-complexes. Nucleic Acids Res 2006; 34(14): 3968-79.
Sirmaci A, Akcayoz-Duman D, Tekin M. The c.IVS1+1G>A mutation in the GJB2 gene is
prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J Genet. 2006 Dec;85(3):213-6.
Smeti I, Savary E, Capelle V, Hugnot JP, Uziel A, Zine A. Expression of candidate markers for
stem/progenitor cells in the inner ears of developing and adult GFAP and nestin promoter-GFP transgenic mice. Gene Expr Patterns 2011; 11(1-2):22-32.
Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli
as fusions with glutathione S-transferase. Gene. 1988 Jul 15;67(1):31-40. Smith RJH, Hildebrand MS, Van Camp G. Deafness and hereditary hearing loss overview. In:
Pagon RA (Ed.). Gene reviews. Seattle: University of Washington, Seattle, 1993-. <http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=deafness-overview>. Acesso em: Junho 2011.
Smith, D.B. & Johnson, K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions of glutathione-Stransferase. Gene 1988; 67: 31–40. Snoeckx RL, Hassan DM, Kamal NM, Van Den Bogaert K, Van Camp G. Mutation analysis of
the GJB2 (connexin 26) gene in Egypt. Hum Mutat. 2005 Jul;26(1):60-1. Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the
carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem 2004; 279(52): 54695-701.
Spray DC, Ye ZC, Ransom BR. Functional connexin "hemichannels": a critical appraisal. Glia.
2006 Nov 15;54(7):758-73. Review. Steiner, C. E., Torriani, M., Norato, D. Y. J., Marques-de-Faria, A. P. Spondylocarpotarsal
synostosis with ocular findings. Am J Med Genet 2000; 91: 131-134. Stepp JD, Huang K, Lemmon SK. The yeast adaptor protein complex, ap-3, is essential for the
efficient delivery of alkaline phosphatase by the alternate pathway to the vacuole. J Cell Biol 1997; 139(7): 1761–1774
Stern, H. J., Graham, J. M., Jr., Lachman, R. S., Horton, W., Bernini, P. M., Spiegel, P. K.,
Bodurtha, J., Ives, E. J., Bocian, M., Rimoin, D. L. Atelosteogenesis type III: a distinct skeletal dysplasia with features overlapping atelosteogenesis and oto-palato-digital syndrome type II. Am. J. Med. Genet. 36: 183-195, 1990
Stone JS, Cotanche DA. Hair cell regeneration in the avian auditory epithelium. Int J Dev Biol.
2007;51(6-7):633-47.

161
Stone JS, Oesterle EC, Rubel EW. Recent insights into regeneration of auditory and vestibular hair cells. Curr Opin Neurol. 1998 Feb;11(1):17-24. Review.
Stone JS, Rubel EW. Cellular studies of auditory hair cell regeneration in birds. Proc Natl Acad
Sci USA 2000;97(22):11714-21. Stone JS, Shang JL, Tomarev S. cProx1 immunoreactivity distinguishes progenitor cells and
predicts hair cell fate during avian hair cell regeneration. Dev Dyn. 2004 Aug;230(4):597-614.
Stossel, T.P., Condeelis, J., Cooley, L., Hartwig, J.H., Noegel, A.,Schleicher, M. and Shapiro,
S.S. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2001; 2: 138-145.
Sugawara M, Corfas G, Liberman MC.Influence of supporting cells on neuronal degeneration
after hair cell loss. J Assoc Res Otolaryngol. 2005 Jun;6(2):136-47. Sun Y, Tang W, Chang Q, Wang Y, Kong W, Lin X. Connexin30 null and conditional
connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J Comp Neurol. 2009 Oct 20;516(6):569-79.
Sundstrom RA, Van Laer L, Van Camp G, Smith RJ. Autosomal recessive nonsyndromic
hearing loss. Am J Med Genet. 1999 Sep 24;89(3):123-9. Review. Stankovic K, Rio C, Xia A, Sugawara M, Adams JC, Liberman MC, Corfas G.J Survival of adult
spiral ganglion neurons requires erbB receptor signaling in the inner ear. Neurosci. 2004 Oct 6;24(40):8651-61.
Thalmann R, Henzl MT, Thalmann I. Specific proteins of the organ of Corti. Acta Otolaryngol. 1997 Mar;117(2):265-8.
Tisdale EJ, Azizi F, Artalejo CR. Rab2 utilizes glyceraldehyde-3-phosphate dehydrogenase and protein kinase C{iota} to associate with microtubules and to recruit dynein. J Biol Chem. 2009 Feb 27;284(9):5876-84. Epub 2008 Dec 23.
Tóth T, Kupka S, Haack B, Fazakas F, Muszbek L, Blin N, Pfister M, Sziklai I. Coincidence of mutations in different connexin genes in Hungarian patients. Int J Mol Med. 2007 Sep;20(3):315-21.
Toyofuku T, Yabuki M, Otsu K, Kuzuva T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 1998; 273(21): 12725:31.
Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell Signal. 2011 Feb;23(2):317-23.
Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007 Nov 1;450(7166):50-5.
Uehara, DT. Pesquisa de microrrearranjos em genes candidatos a surdez sindrômica e não-sindrômica. Dissertação de Mestrado. Instituto de Biociências da USP. São Paulo. SP. 2010.
Uyguner, O., Tukel, T., Baykal, C., Eris, H., Emiroglu, M., Hafiz, G., Ghanbari, A., Baserer, N., Yuksel-Apak, M., Wollnik, B. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin Genet 2002; 62: 306-309.

162
Van Camp G, Smith RJH. Hereditary hearing loss homepage. URL:
<http://hereditaryhearingloss.org/>. Acesso em: Julho 2011. Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, Roeb W,
McCarthy S, Nord AS, Gordon CR, Ben-Neriah Z, Sebat J, Kanaan M, Lee MK, Frydman M,King MC, Avraham KB.Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet. 2010 Jul 9;87(1):101-9.
Wang Y, Chang Q, Tang W, Sun Y, Zhou B, Li H, Lin X. Targeted connexin26 ablation arrests
postnatal development of the organ of Corti. Biochem Biophys Res Commun. 2009 Jul 17;385(1):33-7.
Warchol ME, Corwin JT. Regenerative proliferation in organ cultures of the avian cochlea:
identification of the initial progenitors and determination of the latency of the proliferative response. J Neurosci. 1996 Sep 1;16(17):5466-77.
Warchol ME, Lambert PR, Goldstein BJ, Forge A, Corwin JT. Regenerative proliferation in
inner ear sensory epithelia from adult guinea pigs and humans. Science. 1993 Mar 12;259(5101):1619-22.
Warchol ME. Sensory regeneration in the vertebrate inner ear: differences at the levels of cells
and species. Hear Res. 2011 Mar;273(1-2):72-9. Review. Warchol,M.E.,Corwin,J.T.,1996. Regenerative proliferation in organ cultures of the avian
cochlea: identification of the initial progenitors and determination of the latency of the proliferative response.J.Neurosci.16,5466e5477.
White PM, Doetzlhofer A, Lee YS, Groves AK, Segil N. Mammalian cochlear supporting cells
can divide and trans-differentiate into hair cells. Nature 2006;441(7096):984-7. Widera D, Mikenberg I, Kaus A, Kaltschmidt C, Kaltschmidt B. Nuclear Factor-kappaB
controls the reaggregation of 3D neurosphere cultures in vitro. Eur Cell Mater 2006;11:76-84; discussion 85.
Wiese C, Rolletschek A, Kania G, Blyszczuk P, Tarasov KV, Tarasova Y, Wersto RP, Boheler
KR, Wobus AM. Nestin expression--a property of multi-lineage progenitor cells? Cell Mol Life Sci. 2004 Oct;61(19-20):2510-22. Review.
Wilch E, Azaiez H, Fisher RA, Elfenbein J, Murgia A, Birkenhäger R, Bolz H, Da Silva-Costa
SM, Del Castillo I, Haaf T, Hoefsloot L, Kremer H, Kubisch C, Le Marechal C, Pandya A, Sartorato EL, Schneider E, Van Camp G, Wuyts W, Smith RJ, Friderici KH. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin Genet. 2010 Sep;78(3):267-74.
Wilch E, Zhu M, Burkhart KB, Regier M, Elfenbein JL, Fisher RA, Friderici KH.Expression of
GJB2 and GJB6 is reduced in a novel DFNB1 allele. Am J Hum Genet. 2006 Jul;79(1):174-9.
Worley PF, Zeng W, Huang G, Kimd JY, Shin DM, Kimc MS, Yuan JP, Kiselyov K, Muallemc
S. Homer proteins in Ca2+ignaling by excitable and non-excitable cells. Cell Calcium 2007; 42: 363–371.

163
Xiong WC, Feng X. PYK2 and FAK in osteoclasts. Front Biosci. 2003 Sep 1;8:d1219-26. Xu X, Francis R, Wei CJ, Linask KL, Lo CW. Connexin 43-mediated modulation of polarized
cell movement and the directional migration of cardiac neural crest cells. Development. 2006 Sep;133(18):3629-39.
Yamashita H, Oesterle EC. Induction of cell proliferation in mammalian inner-ear sensory
epithelia by transforming growth factor alpha and epidermal growth factor. Proc Natl Acad Sci USA. 1995;92:3152–3155.
Yamasoba T, Kondo K. Supporting cell proliferation after hair cell injury in mature guinea pig
cochlea in vivo. Cell Tissue Res. 2006 Jul;325(1):23-31. Yamasoba,T., Kondo,K. Supporting cell proliferation after hair cell injury in mature guinea pig
cochlea in vivo. Cell Tissue Res. 2006 .325, 23e31. Yerukhimovich MV, Bai L, Chen DH, Miller RH, Alagramam KN. Identification and
characterization of mouse cochlear stem cells. Dev Neurosci. 2007;29:251–260. Yotsumoto S, Hashiguchi T, Chen X, Ohtake N, Tomitaka A, Akamatsu H, Matsunaga K,
Shiraishi S, Miura H, Adachi J, Kanzaki T. Novel mutations in GJB2 encoding connexin-26 in Japanese patients with keratitis-ichthyosis-deafness syndrome. Br J Dermatol 2003; 148(4): 649-53.
Yuan Y, Yu F, Wang G, Huang S, Yu R, Zhang X, Huang D, Han D, Dai P. Prevalence of
the GJB2 IVS1+1G >A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. J Transl Med. 2010 Dec 2;8:127.
Zhai S, Shi L, Wang BE, Zheng G, Song W, Hu Y, Gao WQ. Isolation and culture of hair cell
progenitors from postnatal rat cochleae. J Neurobiol. 2005 Dec;65(3):282-93. Zhang JT, Nicholson BJ. Sequence and tissue distribution of a second protein of hepatic gap
junctions, Cx26, as deduced from its cDNA. J Cell Biol 1989; 109(6 Pt 2): 3391-401. Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH.
Science. 2002 Mar 8;295(5561):1895-7 Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular
biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc Natl Acad Sci USA. 2005 Oct 18;102(42):15201-6.
Zhang Y, Zhai SQ, Shou J, Song W, Sun JH, Guo W, Zheng GL, Hu YY, Gao WQ. Isolation,
growth and differentiation of hair cell progenitors from the newborn rat cochlear greater epithelial ridge. J Neurosci Methods. 2007 Aug 30;164(2):271-9.
Zhao HB. Long-term natural culture of cochlear sensory epithelia of guinea pigs. Neurosci Lett
2001;315(1-2):73-6. Zhao Y, Zhang J, Li H, Li Y, Ren J, Luo M, Zheng X.An NADPH sensor protein (HSCARG)
down-regulates nitric oxide synthesis by association with argininosuccinate synthetase and is essential for epithelial cell viability. J Biol Chem. 2008 Apr 18;283(16):11004-13.
Zheng JL, Gao WQ. Overexpression of Math1 induces robust production of extra hair cells in
postnatal rat inner ears. Nat Neurosci 2000;3(6):580-6.

164
Zheng JL, Helbig C, Gao WQ. Induction of cell proliferation by fibroblast and insulin-like
growth factors in pure rat inner ear epithelial cell cultures. J Neurosci. 1997 Jan 1;17(1):216-26.
Ziegler WH, Gingras AR, Critchley DR, Emsley J. Integrin connections to the cytoskeleton
through talin and vinculin. Biochem Soc Trans. 2008 Apr;36(Pt 2):235-9. Review. Zine A, de Ribaupierre F. Replacement of mammalian auditory hair cells. Neuroreport.
1998;9:263–268.

ANEXOS

165
ANEXOS
Anexo A - Artigo publicado no periódico Brazilian Brazilian Journal of Medical and
Biological Research, 2009.
Anexo B - Artigo publicado no periódico Journal Translation Medicine , 2010.
Anexo C - Exemplo de uma tabela gerada pelo programa Scaffold com as
proteínas identificadas por espectrometria de massas nas fatias isoladas de géis
SDS-PAGE referente a canaleta da GST-Cx26 ou GST, contendo as bandas
identificadas apenas no ensaio com a GST-Cx26
Anexo D - Exemplo de uma análise realizada para exclusão ou inclusão das
proteínas identificadas somente no ensaio com a GST-Cx26 devido à
diferença entre a massa molecular observada e a esperada

180
Anexo C - Exemplo de uma tabela gerada pelo programa Scaffold com as proteínas identificadas por espectrometria de massas nas fatias isoladas em SDS-PAGE referente a canaleta da GST-Cx26 ou GST, contendo as bandas identificadas apenas no ensaio com a GST-Cx26.

181
Anexo D - Exemplo de uma análise realizada para exclusão ou inclusão das proteínas identificadas somente no ensaio com a GST-Cx26 devido à diferença entre a massa molecular observada e a esperada. Nesse exemplo, temos a sequência da proteína Q69ZX3, cuja massa molecular observada foi menor que a esperada de acordo com os bancos de dados. Uma vez que os peptídeos identificados por espectrometria de massas estavam distribuídos ao longo de toda sua sequncia, essa proteína foi excluída do nosso estudo.
Q69ZX3 (99%), 228.274,0 Da
Q69ZX3_MOUSE MKIAA0866 protein (Fragment) [Mus musculus (Mouse) [10090]]
2 unique peptides, 2 unique spectra, 2 total spectra, 114/1984 amino acids (6% coverage)
S T P G V D L G P P D I M A Q K G Q L S D D E K F L F V D K N F M N S P M A Q A D W V A K K L V W V P S E K Q G F E A A
S I K E E K G D E V V V E L V E N G K K V T V G K D D I Q K M N P P K F S K V E D M A E L T C L N E A S V L H N L R E R
Y F S G L I Y T Y S G L F C V V V N P Y K Y L P I Y S E K I V D M Y K G K K R H E M P P H I Y A I A D T A Y R S M L Q D
R E D Q S I L C T G E S G A G K T E N T K K V I Q Y L A V V A S S H K G K K D S S I T G E L E K Q L L Q A N P I L E A F
G N A K T V K N D N S S R F G K F I R I N F D V T G Y I V G A N I E T Y L L E K S R A I R Q A R D E R T F H I F Y Y L L
A G A K E K M K S D L L L E S F N S Y T F L S N G F V P I P A A Q D D E M F Q E T L E A M S I M G F N E E E Q L A I L K
V V S S V L Q L G N I V F K K E R N T D Q A S M P D N T A A Q K V C H L V G I N V T D F T R A I L T P R I K V G R D V V
Q K A Q T K E Q A D F A I E A L A K A T Y E R L F R W I L S R V N K A L D K T H R Q G A S F L G I L D I A G F E I F E V
N S F E Q L C I N Y T N E K L Q Q L F N H T M F I L E Q E E Y Q R E G I E W N F I D F G L D L Q P C I E L I E R P N N P
P G V L A L L D E E C W F P K A T D K S F V E K L C S E Q G N H P K F Q K P K Q L K D K T E F S I I H Y A G K V D Y N A
S A W L T K N M D P L N D N V T S L L N A S S D K F V A D L W K D V D R I V G L D Q M A K M T E S S L P S A S K T K K G
M F R T V G Q L Y K E Q L G K L M T T L R N T T P N F V R C I I P N H E K R S G K L D A F L V L E Q L R C N G V L E G I
R I C R Q G F P N R I V F Q E F R Q R Y E I L A A N A I P K G F M D G K Q A C I L M I K A L E L D P N L Y R I G Q S K I
F F R T G V L A H L E E E R D L K I T D V I M A F Q A M C R G Y L A R K A F T K R Q Q Q L T A M K V I Q R N C A A Y L K
L R N W Q W W R L F T K V K P L L Q V T R Q E E E M Q A K E E E M Q K I K E R Q Q K A E T E L K E L E Q K H T Q L A E E
K T L L Q E Q L Q A E T E L Y A E A E E M R V R L A A K K Q E L E E I L H E M E A R L E E E E D R G Q Q L Q A E R K K M
A Q Q M L D L E E Q L E E E E A A R Q K L Q L E K V T A E A K I K K L E D D I L V M D D Q N S K L S K E R K L L E E R V
S D L T T N L A E E E E K A K N L T K L K S K H E S M I S E L E V R L K K E E K S R Q E L E K L K R K L E G D A S D F H
E Q I A D L Q A Q I A E L K M Q L A K K E E E L Q A A L A R L D E E I A Q K N N A L K K I R E L E G H I S D L Q E D L D
S E R A A R N K A E K Q K R D L G E E L E A L K T E L E D T L D S T A T Q Q E L R A K R E Q E V T V L K K A L D E E T R
S H E A Q V Q E M R Q K H T Q A V E E L T E Q L E Q F K R A K A N L D K S K Q T L E K E N A D L A G E L R V L G Q A K Q
E V E H K K K K L E V Q L Q D L Q S K C S D G E R A R A E L S D K V H K L Q N E V E S V T G M L N E A E G K A I K L A K
D V A S L G S Q L Q D T Q E L L Q E E T R Q K L N V S T K L R Q L E D E R N S L Q D Q L D E E M E A K Q N L E R H V S T
L N I Q L S D S K K K L Q D F A S T I E V M E E G K K R L Q K E M E G L S Q Q Y E E K A A A Y D K L E K T K N R L Q Q E
L D D L V V D L D N Q R Q L V S N L E K K Q K K F D Q L L A E E K N I S S K Y A D E R D R A E A E A R E K E T K A L S L
A R A L E E A L E A K E E L E R T N K M L K A E M E D L V S S K D D V G K N V H E L E K S K R A L E T Q M E E M K T Q L
E E L E D E L Q A T E D A K L R L E V N M Q A L K G Q F E R D L Q A R D E Q N E E K R R Q L Q R Q L H E Y E T E L E D E
R K Q R A L A A A A K K K L E G D L K D L E L Q A D S A I K G R E E A I K Q L R K L Q A Q M K D F Q R E L D D A R A S R
D E I F A T S K E N E K K A K S L E A D L M Q L Q E D L A A A E R A R K Q A D L E K E E L A E E L A S S L S G R N T L Q
D E K R R L E A R I A Q L E E E L E E E Q G N M E A M S D R V R K A T L Q A E Q L S N E L A T E R S T A Q K N E S A R Q
Q L E R Q N K E L R S K L Q E V E G A V K A K L K S T V A A L E A K I A Q L E E Q V E Q E A R E K Q A A T K S L K Q K D
K K L K E V L L Q V E D E R K M A E Q Y K E Q A E K G N T K V K Q L K R Q L E E A E E E S Q R I N A N R R K L Q R E L D
E A T E S N E A M G R E V N A L K S K L R R G N E A S F V P S R R A G G R R V I E N T D G S E E E M D A R D S D F N G T
K A S E


















