
Anemia de Fanconi -...
103
i CLAUDIA ESTELA GONÇALVES “ESTUDO MOLECULAR DO GENE FANCA EM PACIENTES COM QUADRO CLÍNICO DE ANEMIA DE FANCONI.” CAMPINAS 2014
Transcript of Anemia de Fanconi -...

i
CLAUDIA ESTELA GONÇALVES
“ESTUDO MOLECULAR DO GENE FANCA EM PACIENTES COM
QUADRO CLÍNICO DE ANEMIA DE FANCONI.”
CAMPINAS
2014

ii

iii
UNIVERSIDADE ESTADUAL DE CAMPINAS
FACULDADE DE CIÊNCIAS MÉDICAS
CLAUDIA ESTELA GONÇALVES
“ESTUDO MOLECULAR DO GENE FANCA EM PACIENTES COM
QUADRO CLÍNICO DE ANEMIA DE FANCONI.”
Orientadora: Profa. Dra. Carmen Sílvia Bertuzzo
Tese de Doutorado apresentada ao Programa de Pós-Graduação em Clínica
Médica da Faculdade de Ciências Médicas da Universidade de Campinas para obtenção de
título de Doutora em Clínica Médica, área de concentração Clínica Médica.
ESTE EXEMPLAR CORRESPONDE À VERSÃO FINAL DA
DISSERTAÇÃO TESE DEFENDIDA PELA ALUNA CLAUDIA ESTELA
GONÇALVES E ORIENTADA PELA PROFA. DRA. CARMEN SÍLVIA
BERTUZZO.
Assinatura da Orientadora
-------------------------------------------
\
CAMPINAS
2014

iv

v

vi

vii
Dedico este trabalho aos meus pais (Clélia e José),
aos meus irmãos (José Claudio, Leda Cristina e Maria Cristiani),
à minha filha (Amanda Bárbara)
e ao meu esposo Armando.

viii

ix
AGRADECIMENTOS
Em primeiro lugar à Deus pelo dom da vida.
À professora Carmen Sílvia Bertuzzo pela oportunidade, paciência na orientação e
incentivos que tornaram possível a conclusão desta monografia.
Ao meu esposo, minha filha, meus pais, meus irmãos, e a toda minha família que, com
muito carinho e apoio, não mediram esforços para que eu chegasse até esta etapa de minha
vida.
À amiga Lidiane Camila Rueda Rosada. Dizer obrigada não é o suficiente para agradecer
tudo o que você me ajudou.
À minha amiga Daniela Tenório Furgeri Buffalo pela amizade, colaboração, correção e por
toda ajuda.
E a todos os meus colegas do laboratório pelo respeito, colaboração e agradável
convivência.
Agradeço, portanto, àqueles que ajudaram a tornar este trabalho possível, das mais variadas
formas.

x

xi
“Conhecimento real é saber a extensão da própria ignorância.”
Confúcio

xii

xiii
RESUMO
A Anemia de Fanconi (AF) é uma alteração genética caracterizada por múltiplas
anomalias congênitas, anormalidades hematológicas e predisposição a uma variedade de
tumores. A incidência mundial da AF em todo o mundo é de aproximadamente três por
milhão e a frequência de heterozigotos é estimada em um para 300 na Europa e Estados
Unidos. É uma doença causada por mutações em genes relacionados ao sistema de reparo.
Até o momento foram descritos 16 genes que podem estar multados. São eles: FANCA,
FANCB, FANCC, FNCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ,
FANCL, FANCM, FANCN, FANCO, FANCP E PANCQ. Os grupos mais frequentes são
o FANCA e FANCC. De qualquer modo devido a essa heterogeneidade gênica, o
diagnóstico molecular dessa alteração é complexo. Com o intuito de testar uma estratégia
diagnóstica, o presente trabalho se propôs a identificar as mutações mais frequentes no
gene FANC por PCR e digestão enzimática e investigar mutações no gene FANCA, por
meio da Reação em Cadeia da Polimerase seguida de digestão enzimática da mutação
Brasileira e posterior sequenciamento dos 43 éxons em 60 pacientes portadores de Anemia
de Fanconi DEB positivos. Foram detectados 19 pacientes (27,94%), como sendo do
grupo C e 16 pacientes como grupo A (23,53%). A mutação Δ3788-3790 do gene FANCA
teve uma frequência alélica de 15,4%. Foram encontradas 3 mutações intrônicas, 1
mutação sinônima e 1 mutação de sentido trocado no gene FANCA. Não foram
encontradas correlações com as manifestações hematológicas, renais, baixo peso,
malformações congênitas de membros, machas e pigmentação de pele, sexo e idade.

xiv

xv
ABSTRACT
The Fanconi Anemia (FA) is a genetic disorder characterized by multiple congenital and
hematological abnormalities and predisposition to a variety of tumors. The worldwide
incidence of AF is approximately three per million and the frequency of heterozygotes is
estimated at one in 300 in Europe and the United States. It is a disease caused by mutations
in genes involved in the repair system. So far have been described 16 genes that may be
mutated. They are: FANCA , FANCB , FANCC , FNCD1 , FANCD2 , FANCE , FANCF ,
FANCG , FANCI , FANCJ , FANCL , FANCM , FANCN , FANCO , FANCP And
PANCQ . The most common groups are the FANCA and FANCC. However due to this
genetic heterogeneity, molecular diagnosis of this change is complex. In order to test a
diagnostic strategy, the present study aimed to identify the most frequent mutations in the
FANC gene by PCR and restriction enzyme digestion and investigate mutations in the
FANCA gene, using the polymerase chain reaction followed by enzymatic digestion of the
mutation Brazilian and subsequent sequencing of the 43 exons in 60 patients with Fanconi
Anemia positive DEB. 19 patients (27.94%) were detected as group C and 16 patients as
group A (23.53%). The Δ3788 - 3790 mutation in the FANCA gene had an allelic
frequency of 15.4%. Three intronic mutations, one synonymous mutation and one mutation
changed direction in FANCA gene were found. No correlation with hematologic, renal, low
weight manifestations of congenital malformations members, butches and skin
pigmentation, age and sex were found.

xvi

xvii
Lista de Abreviaturas e Siglas
AF Anemia de Fanconi
AT Ataxia Telangiectasia
ATM Ataxia Telangiectasia Mutated
ATR Ataxia Telangiectasia Related
BLM Gene da Síndrome de Bloom
BRCA1 Breast cancer gene 1
BRCA2 Breast cancer gene 2
cDNA Ácido desoxirribonucleico complementar
DEB Diepoxibutano
DNA Ácido desoxirribonucleico
dNTP Desoxinucleotideo tri-fosfato
EFARP European Fanconi Anaemia Research Programme
FAAP Fanconi Anemia-associated polypeptide
FANCD Proteína
FANCD2-S Isoforma: produto primário de tradução
FANCD2-L Isoforma: monoubiquitilada
HLA Human Leucocytes Antigens
IFAR International Fanconi Anemia Registry
Kda Kilodálton
LiCl Cloreto de lítio
LMA Leucemia Mielóide Aguda
mg Miligrama
mL Mililitro
MDS Síndrome Mielodisplásica
MMC Mitomicina C
MO Medula óssea
NBS Niejmegen Syndrome
OMIM Online Mendelian Inheritance in Man
pb Pares de bases
PCR Polymerase Chain Reaction
P53/TP53 Tumor protein p53
SMD Síndrome Mieloplásica
SSCP Single Strand Conformation Polymorphism
TCH Transplante de Células-tronco Hematopoiéticas

xviii

xix
Lista de Tabelas
pág.
Tabela 1: Os 16 genes envolvidos na AF e identificados até o momento (adaptado
de Wang e D’Andrea, 2004).
25
Tabela 2: Relação de mutações com respectivas enzimas utilizadas para a
digestão enzimática e fragmento para análise.
35
Tabela 3: Sequência dos Primers dos 43 éxons do gene FANCA que foram
utilizados no sequenciamento, com temperatura de anelamento (Tm) e
tamanho dos fragmentos.
41
Tabela 4: Sumário dos Dados Clínicos e Moleculares dos pacientes estudados. 45
Tabela 5: Frequência alélica das mutações deletérias encontradas. 49
Tabela 6: Frequência genotípica das mutações deletérias encontradas. 49
Tabela 7: Média de idade dos pacientes por grupo FANC. 50
Tabela 8: Correlação entre as variáveis clínicas e o Grupo FANC. 51
Tabela 9: Correlação entre as variáveis clínicas e o Grupo FANC, com grupo
FANCA e FANCC agrupados.
52
Tabela 10: Algoritmos de Predição testados com relação à mutação c.3982 A>G. 56

xx

xxi
Lista de Figuras
pág.
Figura 1: Paciente com alteração na implantação dos polegares e hipoplasia radial
(A) e manchas café-au-lait na região do tronco (B).
03
Figura 2: Indivíduo com AF apresentando dismorfismos faciais (A) e Indivíduo
com alterações físicas clássicas de AF, como baixa estatura, microcefalia,
microftalmia, hipoplasia de radio etc. (B). Fonte: Auerbach et al. (2002).
04
Figura 3: Metáfase de cultura de células tratadas com substâncias clastogênicas,
mostrando aberrações cromossômicas estruturais em indivíduos
portadores de AF. As setas indicam cromossomos quadrirradiais e
trirradiais formados após as quebras cromossômicas. Fonte: Adaptado de
Lyakhovich A, and Surrallés J (2007).
10
Figura 4: A figura representa um esquema onde quebras cromatídicas entre
cromossomos não homólogos leva a consequente formação de
cromossomos anômalos quadrirradiais em células de indivíduos afetados.
11
Figura 5: Teste de Complementação utilizando células de três indivíduos com AF
(P1, P2 e P3). A fusão de P1 com P2 não corrigiu o fenótipo celular
(sensibilidade à MMC), portanto são pertencentes ao mesmo grupo de
complementação. Já no segundo caso (entre P2 e P3) ocorre correção, o
produto proteico de uma célula substitui o ausente na outra após a
formação do híbrido e vice-versa, indicando que os indivíduos são
pertencentes ao mesmo grupo. S - sensível; R – resistente; MMC –
Mitomicina C. Fonte: Adaptado de Joenje e Patel, 2001.
15
Figura 6: Resultados baseados na análise de 241 famílias classificadas pelo EFARP
(European Fanconi Anaemia Research Programme – 1994 a 2003). O
número absoluto por grupo é: A-159, B-2, C-23, D1-8, D2-8, E-6, F-5,
G-21, I-4, J-4 e L-1. Fonte: Adaptado de Levitus et al., 2004.
17
Figura 7: Representação das três (1,2 e 3) vias relacionadas às proteínas da AF. A
proteína FANCD2 é fundamental no processo de reparo do DNA. Fonte:
Nakanishi et al., 2004; Tischkowitz, 2004.
21
Figura 8: Representação Gráfica do Conceito Atual da via FA/BRCA na
recombinação homóloga. O complexo de remodelação-DNA é formado
pelas proteínas FANCM (M), FAAP24, e pelas histonas-fold
heterodímeras MHF1 and MHF2 (I.; tons de laranja) que atuma na
duplicação do DNA.
22
Figura 9: Representação do gene FANCA com seus 43 éxons e a localização da
principal mutação: 3788-3790del no éxon 38.
28

xxii
Figura 10: Distribuição das mutações descritas no gene FANCA por categoria.
(Database statistics-Fanconi anemia database in:
HTTP://chromiun.liacs.nl/LOVD2/FANC/variants_statistics.php
29
Figura 11: Distribuição das mutações descritas no gene FANCA por éxon e íntron.
(Database statistics-Fanconi anemia database in:
HTTP://chromiun.liacs.nl/LOVD2/FANC/variants_statistics.php
30
Figura 12: Ciclo utilizado na reação de sequenciamento. 39
Figura 13: Classificação dos pacientes por gene mutado. 48
Figura 14: Eletroferograma da mutação Missense c.3982A>G, em A em
heterozigose e em B sequencia normal.
53
Figura 15: Eletroferograma da mutação T/C synonymous variant (1800358). 53
Figura 16: Eletroferograma da mutação C/T intron variant (rs34420680). 54
Figura 17: Eletroferograma da mutação C/A intron variant (rs373592545). 54
Figura 18: Eletroferograma da mutação C/T intron variant (rs1061646). 55

xxiii
SUMÁRIO
pág.
RESUMO xiii
ABSTRACT xiv
Lista de Abreviaturas e Siglas xv
Lista de Tabelas xvi
Lista de Figuras xvii
1.INTRODUÇÃO 1
1.1. ASPECTOS GERAIS 1
1.2 CARACTERÍSTICAS CLÍNICAS 2
1.2.1 Manifestações Físicas 2
1.2.2 Manifestações Não-Hematológicas 4
1.2.3 Manifestações Hematológicas 6
1.2.4 Desenvolvimento de Cânceres 8
1.3 FENÓTIPO CELULAR 9
1.4 TESTES DIAGNÓSTICOS E TESTE DE COMPLEMENTAÇÃO 12
1.5 AS PROTEÍNAS FANC 17
1.6 O GENE FANCC 23
1.7 O GENE FANCA 27
2. OBJETIVOS 31
2.1 Objetivo Geral 31
2.2 Objetivos Específicos 31
3.CASUÍSTICA E MÉTODOS 32
3.1 Casuística 32
3.2 Critérios de Inclusão 32
3.3 Métodos 33
3.3.1 Extração de DNA de Leucócitos Periféricos Utilizando-se LiCl (Cloreto
de Lítio) e Proteinase K.
33
3.3.2 Amplificação Gênica (PCR) e Digestão Enzimática. 33

xxiv
3.3.3 Sequenciamento 39
3.3.4 Análise dos Dados 43
3.3.5 Análise Estatística 43
4.RESULTADOS 44
5.DISCUSSÃO 57
6.CONCLUSÃO 63
7.REFERÊNCIAS BIBLIOGRÁFICAS 64
8.ANEXOS 75
8.1 Parecer do Comitê de Ética em Pesquisa. 76
8.2 Termo de Consentimento Livre e Esclarecido 77

1
1. INTRODUÇÃO
1.1 ASPECTOS GERAIS
A Anemia de Fanconi (AF) é uma alteração genética caracterizada por múltiplas
anomalias congênitas, anormalidades hematológicas e predisposição a uma variedade de
tumores. Foi descrita primeiramente pelo pediatra suíço Guido Fanconi em 1927 como uma
forma familial de anemia aplástica observada em três irmãos, com idades entre cinco e sete
anos, que apresentavam estatura baixa, malformações esqueléticas, hipogonadismo,
hipopigmentação na pele e pancitopenia (Alter, 1996; Alter, 2003; Fanconi, 1927;
Giampietro et al., 1997).
A incidência mundial da AF em todo o mundo é de aproximadamente três por
milhão e a frequência de heterozigotos é estimada em um para 300 na Europa e Estados
Unidos. Esse doença tem sido relatada em vários grupos étnicos, e mutações têm sido
descritas em judeus Ashkenazi, com frequência de 1 para 89, e em africanos da África do
Sul em 1 para 83(Tischkowitz e Dokal, 2004). No Brasil, não há dados sobre a prevalência
da AF.
A expectativa de vida dos pacientes deste distúrbio é em média 23 anos (0-50 anos)
(Joenje and Patel, 2001).
Entre as doenças associadas a aberrações cromossômicas estruturais e que possuem
um espectro de características semelhantes à AF estão: ataxia-telangiectasia, o xeroderma
pigmentoso, a síndrome de Cockayne, o câncer hereditário sem polipose, a síndrome de
Bloom, a neoplasia endócrina múltipla tipo 1 e a síndrome de Nijmegen (Jorde et al., 2004;
Tischkowitz e Hodgson, 2003).

2
1.2 CARACTERÍSTICAS CLÍNICAS
A AF é um distúrbio clinicamente heterogêneo, caracterizado por múltiplas
anomalias congênitas, progressiva falha da medula óssea e alto risco de desenvolvimento
de câncer (Wang e D`Andrea, 2004).
1.2.1 Manifestações Físicas
As anomalias congênitas mais comuns em pacientes com AF são as malformações
do esqueleto como as observadas nos polegares que ocorrem em 39-55% dos casos. Entre
elas temos: hipoplasia, ausência dos polegares ou ainda polegares supranumerários.
Ausência ou hipoplasia radial estão envolvidas em 13-16% dos casos. Menos comumente
afetados estão a espinha (espinha bífida), costelas (hipoplasia) e quadris (deslocamento e
displasia) (de Kerviler et al., 2000). No entanto, de 25 a 40% dos pacientes são fisicamente
normais (Kee e D`Andrea, 2012).
A AF é também associada ao retardo de crescimento pré e pós-natal, em 54-77%
dos casos, e baixo peso ao nascimento. Isso pode estar relacionado à deficiência de
hormônio de crescimento ou hipotireoidismo, pois em estudo prospectivo com 54
indivíduos, 44% destes tiveram uma resposta subnormal à estimulação com hormônio de
crescimento e 36% apresentavam hipotireoidismo (Wajnrajch et al., 2001).
Outra característica muito comum da AF é a presença de anomalias cutâneas que se
devem à deposição de melanina em determinados locais da pele. Consistem principalmente
de manchas café-au-lait encontradas em 63-79% dos pacientes. Esse aumento da

3
pigmentação ocorre principalmente na região do tronco, ao redor do pescoço, na virilha e
axilas como mosqueado ou manchas largas de bordas difusas. Além disso, outra anomalia
cutânea frequente é a hipopigmentação, encontrada em 31% dos indivíduos que consiste de
manchas bem pequenas próximas umas das outras (de Kerviler et al., 2000) (Figuras 1A e
1B).
A baixa estatura aparece como uma das principais características físicas observadas
nessa doença, com mais de 60% dos indivíduos segundo Giampietro et al. (1993).
As malformações faciais observadas nos pacientes são principalmente: face
dismórfica, micrognatia e microcefalia, base nasal ampla, fendas palpebrais estreitas e
pregas epicantais (Tischkowitz e Hodgson, 2003) (Figura 2).
Figura 1: Paciente com alteração na implantação dos polegares e hipoplasia radial (A) e manchas
café-au-lait na região do tronco (B)
A B

4
Figura 2: Indivíduo com AF apresentando dismorfismos faciais (A) e Indivíduo com alterações
físicas clássicas de AF, como baixa estatura, microcefalia, microftalmia, hipoplasia de radio etc.
(B). Fonte: Auerbach et al. (2002).
1.2.2 Manifestações Não-Hematológicas
Na AF mais de um terço dos casos não apresentam anormalidades congênitas
clássicas, somente sendo diagnosticados quando outro indivíduo da família é afetado ou
quando desenvolvem alterações hematológicas. O diagnóstico estabelecido previamente ao
desenvolvimento de aplasia medular, antes dos oito anos de idade, ocorre em menos de
50% dos casos. Em estudo realizado por Giampietro et al. (1997), 144 dos 419 pacientes
analisados (34%) não apresentaram anomalias congênitas principais, sendo diagnosticados
somente via análise citogenética e/ou molecular (Berger et al., 1993).
B
A
B

5
Anomalias renais estão presentes em aproximadamente um terço dos pacientes e
incluem aplasia renal unilateral, hipoplasia, rins em forma de ferradura ou ureteres duplos.
Em homens há uma alta incidência de anomalias genitais como hipogenitália, que afeta
mais de 51% dos casos, hipospádia, sendo a infertilidade muito frequente (Liu et al., 1991).
Alterações nos níveis de glicose/insulina são comuns e os autores concluíram que
somado a baixa estatura, uma característica observada na maioria dos indivíduos
portadores, as endocrinopatias adquiridas devem prejudicar ainda mais o crescimento
(Tischkowitz e Dokal, 2004).
Surdez condutiva é relativamente comum, em geral moderada, podendo ou não
estar associada a malformações do ouvido externo (de Kerviler et al., 2000).
Anomalias menos comuns são as gastrintestinais, cardíacas e do sistema nervoso e
central (Kaplan et al., 1985).
Apesar dos pacientes apresentarem características em comum, existe um amplo
grau de variabilidade fenotípica entre famílias, como exemplificado por Koc et al. (1999)
que descreveram quatro indivíduos afetados, de duas famílias consanguíneas não
relacionadas, que apresentavam uma grande variação no peso ao nascimento, pigmentação
da pele e gravidade das anomalias esqueléticas, renais e genitais. De acordo com Lo Ten
Foe et al. (1997), é provável que mosaicismo somático seja a principal explicação para a
variação fenotípica nas famílias na AF. O mosaicismo é caracterizado pela presença, em
um mesmo indivíduo, de duas ou mais linhagens celulares, sendo a causa comum de
mosaicismo, a não disjunção dos cromossomos em uma mitose pós-zigótica inicial
(Nussbaum et al., 2002).

6
Como é de se esperar os mosaicos somáticos apresentam fenótipo hematológico
leve (Lo Ten Foe et al., 1997).
1.2.3 Manifestações Hematológicas
A mais importante das características clínicas na AF é a hematológica, responsável
pelo grande número de morbidade e mortalidade. A incidência de anemia aplástica da
síndrome mieloplásica (SMD) e da leucemia mielóide aguda (LMA) é alta em portadores
de AF. Ao nascimento, a contagem de células do sangue é geralmente normal e
macrocitose é geralmente a primeira anomalia detectada, seguida de trombocitopenia
(diminuição no número de plaquetas) e neutropenia (diminuição do número de neutrófilos).
Pancitopenia está presente tipicamente em crianças com idades entre cinco e 10 anos, em
média inicia-se aos sete anos. A pancitopenia é um quadro anômalo caracterizado pela
alteração de duas a três linhagens celulares sanguíneas. Progressiva, torna-se fatal devido a
infecções e hemorragias (Rosenberg et al., 2004).
A síndrome mielodisplásica (termo descrito por Bennet et al., (1982) ou
mielodisplasia são desordens hematológicas de cronicidade variável e pouco definidas que
evoluem para leucemias agudas. Pré-leucemia, termo introduzido por Block et al. (1953)
para também caracterizar mielodisplasia, foi descrito por Linman et al., (1970) que
encontrou pacientes com anemia que não tinham evidência de deficiência nutricional e que
se apresentavam refratários ao tratamento com ferro.
De acordo com Butturini (1994), os indivíduos que apresentam mielodisplasia são
caracterizados pela presença de 5% a 30% de blastos mielóides na medula óssea e de 5% a

7
10% de blastos no sangue, enquanto indivíduos com LMA apresentam mais de 30% dos
blastos na medula e mais de 20% no sangue.
Em estudo realizado por Alter et al. (2000), em pacientes com AF. 32% dos
indivíduos analisados apresentavam SMD com quadro clínico caracterizado por
pancitopenia e aumento no número de células da medula óssea.
De acordo com Stites et al. (2000) o termo leucemia é descrito como sendo uma
neoplasia hematológica caracterizada pela presença de células malignas na medula óssea e
no sangue. As leucemias mielogênicas iniciam-se pela produção cancerosa de células
mielogênicas jovens na medula óssea as quais são disseminadas por todo o corpo (Guyton e
Hall, 1998). Em pacientes com AF, a incidência cumulativa de leucemia está em torno de
10% em indivíduos portadores com até 25 anos de idade, de acordo com dados de Alter
(2003) e de mais dois estudos (Kutler et al., 2003 e Rosenberg et al., 2003).
Schaison et al. (1983) acompanharam a evolução de 44 pacientes com AF entre
1962 e 1980, na França. A idade média de diagnóstico para AF foi de oito anos (mas
podendo ser diagnosticada desde os sete meses a 29 anos) e a incidência de leucemia foi de
30% durante o período de acompanhamento dos pacientes (Rodriguez, 2003).
Auerbach e Allen (1991) analisaram todos os pacientes com AF do Registro
Internacional da Anemia de Fanconi (IFAR) e constataram uma incidência de leucemia
15.000 vezes maior que a observada em crianças da população em geral. Outro estudo com
388 pacientes com AF realizado por Butturini (1994) permitiu calcular o estimado risco
para o desenvolvimento de anormalidades hematopoiéticas e morte devido a causas
hematológicas até a idade de 40, sendo de 98% e 81%, respectivamente. Dos 388 pacientes,
85% desenvolveram anomalias hematológicas, sendo que as mais comuns foram a

8
trombocitopenia isolada e a pancitopenia, as quais estavam associadas à diminuição no
número de células da medula óssea em 75% dos casos estudados. O risco para pancitopenia
foi de 84% até os 20 anos, seguido de anomalias citogenéticas clonais (risco de 67% até os
30 anos) (Rodriguez, 2003).
1.2.4 Desenvolvimento de Cânceres
Para os pacientes que atingem a idade adulta, existe um alto risco para o
desenvolvimento de tumores sólidos, principalmente tumores hepáticos e carcinomas de
células escamosas de esôfago, orofaríngeo e da vulva (Alter, 1993; Alter, 1996; Lustig,
1995).
Outros tipos de tumores menos encontrados são os adenocarcinomas do estômago,
os tumores de mama, os meduloblastomas, os tumores de Wilms, os linfomas, os
retinoblastomas e os osteossarcomas (de Chadarévian et al., 1985; Gibbons et al., 1995;
Hilgigliol et al., 1981; Jacobs e Karabus, 1984; Levinson e Vicente, 1977; Van Niekerk et
al., 1987).
Um estudo com indivíduos do IFAR sugere a ocorrência de anemia aplástica
precocemente em indivíduos portadores (risco de 84% até 20 anos), seguido de anomalias
citogenéticas clonais (risco de 67% até 30 anos) e mais tarde leucemia (risco de 52% para
SMD e LMA até 40 anos de idade). Desenvolvimento de tumores hepáticos ocorre em 5%
dos pacientes descritos na literatura (Rosenberg et al., 2003).
Alguns estudos tiveram como objetivo a análise do risco de desenvolvimento de
cânceres em parentes de pacientes com AF. Um desses trabalhos foi realizado por Swift et
al. (1971), que analisaram 102 óbitos de parentes de casos de AF de oito famílias e

9
encontraram uma alta proporção de leucemia e cânceres de língua e estômago. Entretanto,
isto não foi estatisticamente significativo quando o estudo, realizado pelo mesmo grupo, foi
ampliado para 25 famílias. Não encontraram um aumento significativo de cânceres, como
também, casos de leucemia como esperado (Swift et al., 1980).
Da mesma forma, Potter et al. (1983) estudaram 125 indivíduos de nove famílias e
também apresentaram dados que comprova ser estatisticamente insignificante o risco de
câncer para os parentes de indivíduos com AF.
1.3 FENÓTIPO CELULAR
A AF também é denominada de síndrome da instabilidade cromossômica. As
células de pacientes com AF são caracterizadas por hipersensibilidade a agentes
clastogênicos, como mitomicina C (MMC), diepoxibutano (DEB), ciclofosfamida e
cisplatina, dando origem a quebras cromossômicas. Essas quebras cromossômicas,
descritas por Schroeder et al. (1964), são visíveis microscopicamente através da análise
citogenética de metáfases, tornando, esta característica, base para um teste diagnóstico
eficaz (Figura 3). Esse dano espontâneo do cromossomo está relacionado ao atraso no
término da fase S ou G2 (pós-replicação) do ciclo celular. Quando as células desses
pacientes são expostas, in vitro, a uma baixa dose de um agente clastogênico, como MMC,
elas respondem notadamente por meio de quebras cromatídicas e intercâmbios
cromatídicos, como quadrirradial, o que as diferenciam de células controles que respondem
a este tratamento, mas em uma porcentagem muito menor (Magdalena, 1999) (Figura 4).
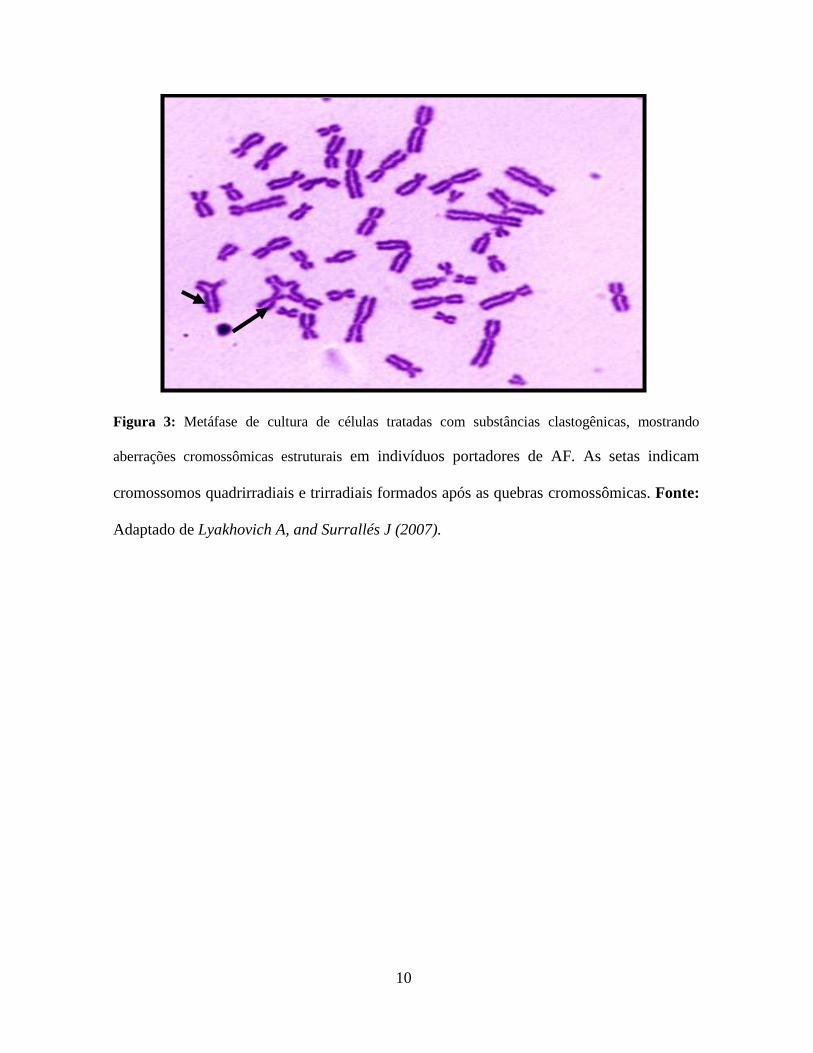
10
Figura 3: Metáfase de cultura de células tratadas com substâncias clastogênicas, mostrando
aberrações cromossômicas estruturais em indivíduos portadores de AF. As setas indicam
cromossomos quadrirradiais e trirradiais formados após as quebras cromossômicas. Fonte:
Adaptado de Lyakhovich A, and Surrallés J (2007).

11
Figura 4: A figura representa um esquema onde quebras cromatídicas entre cromossomos
não homólogos leva a consequente formação de cromossomos anômalos quadrirradiais em
células de indivíduos afetados.
No entanto, na AF, o intercâmbio cromatídico parece envolver pontos de ruptura
entre cromossomos não homólogos. Já, na Síndrome de Bloom, o intercâmbio envolve
preferencialmente cromossomos homólogos (Joenje e Patel, 2001; Tischkowitz et al.,
2004). Até 50% das células de portadores de AF, quando estimuladas, apresentam de uma a
três aberrações por célula. A instabilidade cromossômica espontânea, observada em células
de indivíduos com AF, é diferente da observada em outras síndromes, embora se assemelhe
àquelas presentes em células de portadores da Síndrome de Bloom (Joenje e Patel, 2001;
Rodriguez, 2003).

12
1.4 TESTES DIAGNÓSTICOS E TESTE DE COMPLEMENTAÇÃO
Auerbach et al., (1981) desenvolveram um método citogenético para o diagnóstico
dos pacientes com AF, o qual utiliza preparações de metáfases obtidas a partir de cultura de
linfócitos de sangue periférico estimulados com fitohemaglutinina e tratados com DEB.
Esta técnica foi utilizada pela primeira vez em 1977.
Em 1984 foi fundado um laboratório de referência para a realização do teste
citogenético com o DEB teste na Universidade Rockfeller, Nova York, EUA. De acordo
com o IFAR, os homozigotos mutantes apresentam, em média, 8,96% de quebras
cromossômicas por células (variação de 1,3 a 23,9%) após cultura dos linfócitos do sangue
periférico com DEB, comparados com uma média de 0,06% (variação de 0 a 0,36%) em
controles normais (Auerbach et al., 1989; Auerbach, 1995; Rodriguez, 2003).
Os portadores heterozigotos são assintomáticos e suas células não apresentam
sensibilidade a fatores clastogênicos, dificultando assim o diagnóstico (Kutler et al., 2003;
Magdalena, 1999).
Apesar do DEB teste não detectar heterozigotos e de gerar resultados falso-
negativos, ele ainda permanece como o principal método utilizado no diagnóstico da AF
(Shimamura, 2002).
Ainda em diagnóstico citogenético, para distinção entre portadores de AF e o grupo
de doenças classificadas como anemia aplástica idiopática (características muito
semelhantes à AF), Cervenka et al. (1982) utilizaram a MMC como substância
clastogênica. Na concentração de 80 ng/mL de MMC no meio de cultura dos linfócitos, foi
observado um aumento de 50 vezes nas quebras cromossômicas e de 200 vezes nas figuras
radiais dos linfócitos de pacientes com AF, quando comparado com os valores médios para

13
as células de pacientes com anemia aplástica ou células de indivíduos saudáveis do grupo
controle.
Um segundo teste diagnóstico para o distúrbio é a citometria de fluxo baseado na
mensuração do aumento da porcentagem de células na fase G2/M em culturas de células
sanguíneas periféricas após sensibilização com mostarda de nitrogênio. Este teste foi
desenvolvido por Berger et al. (1993) e não deve ser utilizado em indivíduos com
complicações hematológicas como mielodisplasia ou leucemia.
Um terceiro teste relacionado à AF é o teste para análise do fenótipo celular
denominado Teste de Complementação.
Complementação é definida como a habilidade das células de pacientes com
fenótipos similares, devido a mutações em genes diferentes, de restabelecerem o fenótipo
selvagem quando hibridizadas (Thompson et al., 2002). No caso da avaliação da AF, o
fenótipo celular avaliado e o da instabilidade à cultura com substâncias como DEB e
MMC.
Yoshida (1980) fusionou fibroblastos de indivíduos normais e de pacientes afetados
com AF e observou uma taxa elevada na correção das aberrações cromossômicas, tanto
espontâneas quanto induzidas por MMC, ocorrendo, portanto, a correção do defeito celular.
Alguns pesquisadores utilizaram esta linha de raciocínio na realização da fusão de
células de dois pacientes com AF e observaram que, quando linhagens celulares de
linfoblastos de indivíduos não-consanguíneos são fundidas, são obtidas células híbridas que
apresentam resistência ou não a substâncias clastogênicas, tais como MMC. Ocorrendo
complementação do fenótipo, ou seja, surgimento de um híbrido 4n resistente a MMC, os
indivíduos são pertencentes a grupos de complementação distintos. As células de

14
indivíduos portadores que não se complementarem pertencem a um mesmo grupo de
complementação. (Duckworth-Rysiecki et al., 1985; Joenje et al., 1995; Joenje et al., 1997;
Zakrzewski e Sperling, 1980) (Figura 5).
Por meio do teste de complementação e consequente correção da hipersensibilidade
aos agentes clastogênicos utilizados, foram descobertos 16 grupos de complementação
distintos (A, MIM 607139; B, MIM 300515; C, MIM 227645; D1, MIM 605724; D2, MIM
227646; E, MIM 600901; F, MIM 603467; G, MIM 602956; I, MIM 609053; J, MIM
605882; L, MIM 608111; M, MIM 609644; N, MIM 610355; O, MIM 613390; P, MIM
613951 e Q, MIM 615272. (Kee e D’Andrea, 2012; Bogliolo et al., 2013). O grupo
FANCH foi posteriormente verificado ser na verdade FANCA (Joenje et al., 2000).

15
Figura 5: Teste de Complementação utilizando células de três indivíduos com AF (P1, P2
e P3). A fusão de P1 com P2 não corrigiu o fenótipo celular (sensibilidade à MMC),
portanto são pertencentes ao mesmo grupo de complementação. Já no segundo caso (entre
P2 e P3) ocorre correção, o produto proteico de uma célula substitui o ausente na outra
após a formação do híbrido e vice-versa, indicando que os indivíduos não pertencentes ao
mesmo grupo. S - sensível; R – resistente; MMC – Mitomicina C. Fonte: Adaptado de
Joenje e Patel, 2001.
Em se tratando ainda de pesquisas relacionadas aos genes de AF, o IFAR,
localizado na Universidade Rockefeller - Nova York - Estados Unidos, surgiu com a
finalidade de reunir informações genéticas e clínicas de pacientes com AF,
permitindo o estudo amplo da história natural desta rara doença em um grande
número de pacientes. Um total de 754 indivíduos apresentando DEB teste positivo
foram registrados entre 1982 e 2004, não somente da América do Norte como

16
também de países como Brasil, Índia e Turquia. Resultados de estudos a respeito das
mutações no FANCC e FANCA foram anteriormente relatados em pacientes do
IFAR. (Auerbach et al., 2003; Gillio et al., 1997; Kutler et al., 2003; Verlander et
al., 1995).
De acordo com o IFAR, 65% dos pacientes no mundo pertencem ao grupo de
complementação A, 15% ao grupo C e 8-10% ao grupo G (Meetei et al., 2003a;
Rutler et al., 2003). A análise de 241 famílias classificadas pelo EFARP (European
Fanconi Anaemia Research Programme), mostrou que a maioria de pacientes, cerca
de 66%, pertenciam ao grupo de complementação A, seguidos por 9,5% e 8,7% dos
grupos C e G, respectivamente (Figura 6).

17
Figura 6: Resultados baseados na análise de 241 famílias classificadas pelo EFARP
(European Fanconi Anaemia Research Programme – 1994 a 2003). O número absoluto por
grupo é: A-159, B-2, C-23, D1-8, D2-8, E-6, F-5, G-21, I-4, J-4 e L-1. Fonte: Adaptado de
Levitus et al., 2004.
1.5 AS PROTEÍNAS FANC
Em relação às proteínas codificadas pelos genes relatados, a ampla similaridade
fenotípica entre os indivíduos dos diferentes grupos de complementação da AF nos leva a
crer que, provavelmente, os seus produtos proteicos interajam em uma função comum
(D’Andrea e Grompe, 2003).
Segundo Van de Vrugt et al. (2002), a AF é classificada como um distúrbio de gene
de manutenção por causa de duas características da doença: as células em cultura de
indivíduos com AF apresentam um aumento de aberrações cromossômicas espontâneas e

18
são hipersensíveis a agentes clastogênicos como MMC. Os genes de manutenção estão
envolvidos em reparar danos ao DNA e manter a integridade genômica.
De acordo com Reuter et al. (2003), a hipótese funcional que surgiu por meio da
análise do fenótipo clínico dos pacientes sugere que esses produtos proteicos estejam
associados a cinco classes funcionais de proteínas: 1) responsáveis pela regulação da
transcrição, 2) sinalização, 3) metabolismo oxidativo, 4) transporte intracelular e 5)
apoptose. Por meio de um recente estudo realizado pelo mesmo grupo, cada uma das
proteínas codificadas pelos respectivos genes interage com, no mínimo, uma classe.
Evidências recentes indicam que um complexo central e nuclear composto por
FANCA, B, C, E, F, G, I, M e, juntamente com FANCL, convertam FANCD2-S em
FANCD2-L monoubiquitilado. Isso é desencadeado pelo dano ao DNA. Recentes
evidências sugerem que FANCL seja a principal E3 ubiquitina ligase que monoubiquitina
FANCD2 antes de BRCA1/BARD1, como se acreditava anteriormente (Meetei et al.,
2003a).
Embora as proteínas da AF formem um complexo, a sua função permanece
desconhecida. Pesquisas são necessárias para determinar se o complexo atua diretamente
detectando o dano do DNA, na reparação do mesmo ou na estabilização de estruturas
cromossômicas (Tischkowitz e Dokal, 2004).
A proteína FANCD2 é peça chave na ligação entre o complexo e as proteínas
diretamente relacionadas ao reparo de DNA. As proteínas que atuam antes da
monoubiquitinação de FANCD2 estão upstream e as que estão abaixo, dowstream. (Garcia-
Higuera, 2001).

19
Estudos iniciais indicam que FANCD2 existe em duas isoformas: FANCD2-S
(produto primário de tradução) e FANCD2-L (isoforma monoubiquilada). Importante dizer
que a isoforma FANCD2-L é observada somente em células de indivíduos normais ou
células de indivíduos com AF que são resistentes a MMC. Este achado indica que o
complexo nuclear e as outras subunidades proteicas são necessários para a conversão de
FANCD2-S para FANCD2-L (D’Andrea e Grompe, 2003; Timmers et al., 2001)
Análise de espectrometria de massa mostrou que a monoubiquitinação de FANCD2
se dá na lisina 561 (lis561) (K61). Esta lisina é conservada em muitas proteínas FANCD2
eucarióticas, por exemplo, em A. thaliana, C. elegans e Drosophila, o que indica que este
aminoácido apresenta função crucial para o funcionamento apropriado da proteína
(Timmers et al., 2001).
Mutações de lisina para arginina neste resíduo abalam a monoubiquitinação,
indicando que outros resíduos de lisina na proteína não podem servir como substitutos para
ligação da ubiquitina (D’Andrea e Grompe, 2003; Timmers et al., 2001).
A proteína FANCD2 está presente em três vias (Figura 7): na primeira delas, o
complexo nuclear, juntamente com FANCL, provoca a conversão de FANCD2S em
FANCD2L. Essa, por sua vez, ativa as proteínas que diretamente atuam no reparo de DNA
por recombinação homóloga. São elas: BRCA1, com FANCJ, BRCA2/FANCD1 e RAD51.
Na segunda via, a proteína FANCD2 interage independentemente com ATM/ATR.
Mutações na proteína ATM, codificada pelo gene Ataxia Telangiectasia Mutated, causa o
distúrbio autossômico recessivo denominado Ataxia Telangiectasia que, como na AF
resulta em quebras cromossômicas espontâneas e cânceres hematológicos. Diferentemente
da AF, os pacientes AT apresentam imunodeficiência e degeneração neural cerebelar. A

20
proteína ATM é responsável pela fosforilação de FANCD2S, a qual atua diretamente no
ponto de checagem da fase S do ciclo celular. Na terceira e última via, a proteína ATM,
ativada em resposta ao dano do DNA induzido por radiação ionizante, ativa a proteína NBS
responsável pela fosforilação de FANCD2S, a qual atua sobre o complexo MRE11 e
RAD50, proteínas responsáveis pelo ponto de checagem da fase S do ciclo celular (Couzin,
2003; D’Andrea e Grompe, 2003; Meetei et al., 2004; Nakanish et al., 2002; Taniguch et
al., 2002; Wang e D’Andrea, 2004; Zhang et al., 2004). Uma hipótese para a atuação das
proteínas FANC no período S está representado na Figura 8.
A síndrome de Nijmegen (NBS) é outro distúrbio autossômico recessivo. Similar a
AT é caracterizada por imunodeficiência e predisposição a linfoma. Uma quantidade
significativa de pacientes apresentam fenótipo semelhante à AF, como exemplo,
progressiva falha da medula óssea (MO) e LMA (D’Andrea e Grompe, 2003).
Em resumo, FANCD2 está relacionada a três vias: 1) as proteínas da AF formadoras
do complexo nuclear são responsáveis pela monoubiquitinação de FANCD2 que atuam
sobre proteínas do reparo; 2) em resposta à radiação ionizante, ATM/ATR fosforila NBS1
levando a fosforilação de FANCD2 que por sua vez promove a ativação do ponto de
checagem da fase S do ciclo celular, enquanto 3) em resposta a MMC, a NSB1 reúne-se em
um mesmo foci nuclear com MRE11/RAD50 e FANCD2 (Venkitaraman, 2003).
A descoberta de que FANCD1 é, de fato, BRCA2 indica que os passos envolvidos
na susceptibilidade ao câncer de mama e AF são interconectados em mais de um nível
(D’Andrea e Grompe, 2003; Witt e Ashworth, 2002).
Estudos mostraram que o complexo AF interage também com o complexo
BLM/BRAFT; o gene BLM é mutado na síndrome de Bloom, a qual apresenta

21
similaridades com AF e é possível que estes complexos atuem em conjunto no processo de
correção do dano do DNA (Meetei et al., 2003b).
Embora existam várias hipóteses em relação aos passos bioquímicos nos quais
atuam as proteínas envolvidas com AF, provavelmente estes modelos são muito
simplificados. Novas hipóteses alternativas têm sido propostas, onde AF e as proteínas
relacionadas trabalhem em uma rede de processos e não como simplesmente uma
sequência linear de eventos na proteção contra aberrações cromossômicas estruturais
(Venkitaraman, 2004).
Figura 7: Representação das três (1,2 e 3) vias relacionadas às proteínas da AF. A proteína
FANCD2 é fundamental no processo de reparo do DNA. Fonte: Nakanishi et al., 2004;
Tischkowitz, 2004.

22
Figura 8: Representação Gráfica do Conceito Atual da via FA/BRCA na recombinação
homóloga.
O complexo de remodelação-DNA é formado pelas proteínas FANCM (M),
FAAP24, e pelas histonas-fold heterodímeras MHF1 and MHF2 (I; tons de laranja) que
atuma na duplicação do DNA. Uma vez que é encontrada uma lesão na forquilha de
replicação (um cross-link entre as cadeias representadas por uma linha em zig-zag
vermelha), inicia-se a reunião das proteínas que formarão o complexo Core FA (II;
proteínas FANCA (A), -B, -C, -E, -F, -G, -L, e FAAP100; tons de vermelho). Este por sua
vez, monoubiquitila (U) FANCD2 e FANCI e os recruta a cromatina (III; Complexo: D2 e
I; verde). Múltiplos eventos de fosforilação (P-branco em esfera vermelha) também
contribuem para a ativação da via. A região da lesão do DNA é cortada (setas) por ação de
endonucleases específicas e desenrolada (IV; Coordenadores de reparo no DNA: FANCJ
(J) e FANCP (P); azul). A lacuna é preenchida por uma polimerase de baixa especificidade
e ocorre síntese translesão. Para a correção do nucleotídeo na posição exata da fita de
DNA, é necessário o envolvimento do FANCD1, FANCN, e FANCO (V; Componentes da
Recombinação Homóloga: D1, N, e O; lilás). Adaptado de

23
http://link.springer.com/referenceworkentry/10.1007%2F978-3-642-16483-
5_2116/fulltext.html - Fanconi Anemia
1.6 O GENE FANCC
Schroeder et al. (1976), analisando a segregação da AF em 90 famílias com
membros portadores do distúrbio, estabeleceram o padrão de herança autossômico
recessivo para esta doença e sugeriram a existência de uma heterogeneidade genética.
Com a descoberta de grupos de complementação distintos a próxima etapa seria a
procura pelos genes envolvidos na doença.
Foram identificados, até o momento, 16 genes (FANCA, B, C, D1, D2, E, F, G, I, J,
L, M, N, O, P e Q) nos grupos de complementação existentes, comprovando a grande
heterogeneidade genética na AF (Savino et al., 2003; Soulier et al., 2005) (Tabela 1).
Análises de mutações em genes responsáveis pela AF têm sido realizadas por países
ocidentais, de acordo com The Rockefeller University - Fanconi Anemia Database, sendo
que a prevalência por um subgrupo, o tipo de mutação e até mesmo a gravidade do fenótipo
dependem do grupo étnico a que os pacientes pertencem. Como citado anteriormente, a
mutação IVS4+4A>T no gene FANCC, por exemplo, é altamente prevalente em judeus da
população Ashkenazi. Interessantemente, essa mutação é a mais prevalente na população
japonesa, mas o fenótipo é menos grave comparado aos Ashkenazis, sugerindo que genes
modificadores de uma raça específica ou fatores ambientais influem na gravidade do
fenótipo (Tischkowitz e Hodgson, 2003).
No Brasil, em trabalho recente intitulado: “Estudo Molecular da Anemia de
Fanconi” (RODRIGUEZ, 2003), pacientes foram triados através da análise clínica e

24
posteriormente triados para a presença de mutações nos genes FANCA e FANCC. No
entanto, a maioria dos pacientes (64%), apesar da clínica compatível, não apresentou
mutações nestes genes.

25
Tabela 1: Os 16 genes envolvidos na AF e identificados até o momento. (Adaptado
de Wang e D’Andrea, 2004)
Grupo de
Complementação
Gene localização Tamanho
da
Proteína
(kD)
Função proteica
A FANCA 16q24.3 163 Complexo central FA
B FANCB Xp22.31 95 Complexo central FA
C FANCC 9q22.3 63 Complexo central FA
D1 FANCD1/BRCA2 13q12.13 380 Recruta RAD51 e promove
Recombinação Homóloga
D2 FANCD2 3p25.3 155/162 Monoubiquitilado recruta FAN1,
FANCP para a cromatina
E FANCE 6p21.22 60 Complexo central FA. Provável
adaptador de FANCD2
F FANCF 11p15 42 Complexo central FA
G FANCG/XRCC9 9p13 68 Complexo central FA
I FANCI/KIAA1794 15q25-26 146 Monoubiquitilado forma
heterodímero com FANCD2
J FANCJ/BRIP1/BACH1 17q22-24 150 Interage com BRCA1, DNA

26
helicase e ATPase
L FANCL/PHF9 2p16.1 43 Recruta UBE2T via domínio
PHD3. Monoubiquitila FANCD2
e FNCI
M FANCM 14q21.3 250 Complexo central FA. Interage
com BLM
N FANCN/PALB2 16p12 130 Media a interação entre BRCA1
e BRCA2 durante a
Recombinação Homóloga
O FANCO/RAD51C 17q23 37 Promove Recombinação
Homóloga. Interage com RAD52
e suas homólogas.
P FANCP/SLX4 16p13.3 268 Junção da Resolvase com a
SLX1. Interação com as
nucleases XPF-ERCC1 e EME1-
MUS81.
Q FANCQ/XPF4/ERCC4 16p13.12 100 Interage com ERCC1 and SLX4,
atuando no reparo de excisão
Mutações nos genes FANCA, FANCC e FANCG acometem cerca de 85% dos
casos diagnosticados, por isso a grande importância em estudá-los (Kutler et al., 2003).

27
1.7 O GENE FANCA
O gene FANCA foi o segundo gene clonado. Foi clonado por dois grupos de
pesquisadores independentes (Apostolou et al., 1996 e Lo Ten Foe et al., 1997). Localiza-
se no braço longo do cromossomo 16 região 24.3.
O gene FANCA tem 5.5 Kb e codifica uma proteína com 1455 aminoácidos, com
uma massa molecular de 163 kD, que também não tem homologia significativa com outras
proteínas. Consiste de 43 éxons que variam de 34 a 188 pb (Rodriguez, 2003) (Figura 9).
Ianzano et al. (1997) descreveram 3 splicing alternativos que resultavam em perda do éxon
37, uma deleção de 23 pb na porção 5’ final do éxon 41, e uma inserção GCAG na porção
3’ do éxon 41. Verificou também que na região promotora tem uma região rica em GC,
típica de genes housekeeping.
A mutação brasileira, “the common Brazilian mutation”, para FANCA é a Δ3788-
3790. Em um estudo com 350 indivíduos, realizado por Levran et al. (1997), 10% deles
apresentaram essa mutação.
No Brasil a mutação 3788-3790del foi encontrada em 30% dos pacientes
analisados, ou seja, vinte quatro em uma amostra de 80 pacientes não consanguíneos de
todas as regiões do Brasil. O trabalho confirmou que dos 24 pacientes, treze (16,25%) eram
homozigotos mutantes e 11(13,75%) eram heterozigotos compostos. Assim confirmando
uma alta frequência da mutação 3788-3790del do gene FANCA em brasileiros (Magdalena
et al., 2005).
Estudo de mutação relacionada à etnia é que ocorre com a população da África do
Sul. Caracterizada pela alta incidência da doença, quase todos os pacientes apresentam
mutações no gene FANCA, e esta alta frequência de mutações encontradas foi atribuída a

28
um casal que imigrou para o sul da África no final do século XVII (efeito fundador)
(Demuth et al., 2000; Joenje e Patel, 2001; Tipping et al., 2001).
Figura 9: Representação do gene FANCA com seus 43 éxons e a localização da principal
mutação: 3788-3790del no éxon 38.
Há um alto número de mutações deletérias no gene FANCA, cerca de 1.555, sendo
extremamente heterogêneas, mutações de ponto, de splicing, inserções e grande deleções
intragênicas, provavelmente mediadas por sequências Alu (Morgan et al., 1999; Levran et
al., 2005) (Figura 10).
1 2 3 4 5 6 7 8 910 1112 13 1415 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38
Δ3788_3790

29
Figura 10: Distribuição das mutações descritas no gene FANCA por categoria. (Database
statistics- fanconi anemia database in:
HTTP://chromiun.liacs.nl/LOVD2/FANC/variants_statistics.php)
Série1; Substituição; 596
Série1; Deleção; 851
Série1; Duplicação; 83
Série1; Inserção; 15
Série1; Inserção/Deleção; 2
Série1; 2variantes; 1
Série1; Desconhecida; 7
Série1; Total; 1555

30
Figura 11: Distribuição das mutações descritas no gene FANCA por éxon e íntron.
(Database statistics- fanconi anemia database in:
HTTP://chromiun.liacs.nl/LOVD2/FANC/variants_statistics.php

31
2. OBJETIVO
2.1 Objetivo Geral
Identificar as mutações do gene FANCA, em pacientes com quadro clínico de
Anemia de Fanconi.
2.2 Objetivos Específicos
Verificar a prevalência da mutação Δ3788-3790, em pacientes que apresentam
quadro clínico sugestivo de Anemia de Fanconi e com DEB teste positivo.
Verificar a presença de outras alterações no gene FANCA
Correlacionar os achados laboratoriais com as variáveis clínicas dos paciente.

32
3. CASUÍSTICA E MÉTODOS
3.1 Casuística
O presente estudo foi aprovado pelo Comitê de Ética em Pesquisa – Unicamp,
(anexo 1).
Foram analisados 68 indivíduos com suspeita clínica de AF, escolhidos através de
uma triagem realizada nos Ambulatórios de Genética Médica do Hospital das
Clínicas/UNICAMP, nos ambulatórios de Hematologia e casos que foram enviados de
diversos hematologistas da região de Campinas. A análise citogenética por meio da cultura
de linfócitos com o agente clastogênico DEB, foi realizada pelo Laboratório de
Citogenética do Hemocentro, coordenado pela Prof.ª. Dar. Carmen Sílvia Passos Lima ou
em laboratórios particulares. Foi preenchido um formulário com os dados clínicos pelo
médico responsável pelo paciente.
3.2 Critérios de Inclusão
Os indivíduos que compuseram a amostra apresentaram quadro clínico compatível
com o distúrbio e DEB teste positivo, bem como concordaram em participar da pesquisa
via assinatura de um termo de consentimento. Os sujeitos que apresentaram DEB teste
negativo e/ou se recusaram a participar foram excluídos da pesquisa.

33
3.3 Métodos
As análises efetuadas foram:
1- Análise molecular das mutações mais frequentes no gene FANCC para exclusão
de pacientes
2- Análise molecular da mutação Brasileira no FANCA
3- Nos casos nos quais foi encontrado apenas um alelo mutante no gene FANCA e
nenhum alelo das mutações investigadas, foi realizado o sequenciamento do
gene FANCA.
3.3.1 Extração de DNA de Leucócitos Periféricos Utilizando-se LiCl (Cloreto De Lítio)
e Proteinase K.
Foram retirados assepticamente 5mL de sangue venoso de cada um dos pacientes,
preparadas as alíquotas e a extração de DNA realizada de acordo com protocolo descrito
por WOODHEAD et al., 1986 .
3.3.2 Amplificação Gênica (PCR) e Digestão Enzimática.
A técnica de reação em cadeia da polimerase (denominada de PCR, abreviatura do
nome em inglês, “Polymerase Chain Reaction” descrita por SAIKI et al., 1989), possibilita
a amplificação de uma pequena sequência de DNA através do uso de dois iniciadores
(“primer”) que flanqueiam a região do DNA a ser amplificada, onde se hibridizam devido à
complementaridade de bases de suas sequências. A amostra de DNA é misturada em uma
solução com os iniciadores e, logo em seguida, são realizados, em um aparelho
termociclador, repetidos ciclos de desnaturação térmica do DNA, anelamento dos

34
iniciadores em temperaturas adequadas e a extensão dos iniciadores promovida pela enzima
Taq DNA polimerase, levando ao acúmulo exponencial da sequência de DNA alvo.
A reação da PCR para análise das mutações foi realizada em volume total de 50 L,
com a seguinte concentração de reagentes: 36,5 L água de injeção; 5,0 L tampão 10x;
2,0 L MgCl2 50 mM; 2,5 L dNTP 1,25 mM; 1,0 L primer forward 5 pmol e 1,0 L
primer reverse 5 pmol, 3,0 L de Taq DNA polimerase e 1,0 L de DNA genômico. A
reação de amplificação para as mutações foi realizada em aparelho termociclador e
consistiu em uma desnaturação inicial de 94oC por 5 min, seguida de 30 ciclos de 94
oC por
1 minuto, 56-61oC por 1 minuto, dependendo do primer utilizado, 72°C durante 1 minuto e,
no final, 72oC por 5 minutos de extensão.
As mutações e as temperaturas de anelamento utilizadas para análise estão descritas
na tabela 3.

35
Tabela 2: Relação de mutações com respectivas enzimas utilizadas para a digestão
enzimática e fragmento para análise.
Genes
de AF
Mutações
Estudadas
Enzima de
Restrição
T(ºC)
Incubação
Fragmentos
Normais (pb)
Fragmentos
Mutantes (pb)
FANCC Q13X Bcl I 50 227 + 23 250
FANCC W22X Fok I 37 204 + 17 187 + 17 + 17
FANCC G322 Bsp1286I 37 128 + 22 151
FANCC IVS4+4 A > T ScaI 37 108 + 23 131
FANCC R185X NlaIII 37 105 + 17 74 + 31 + 17
FANCC L496R HhaI 37 303 218 + 85
FANCC R548X AvaI 37 231 + 133 364
FANCC L554P BbvI 37 260 + 104 364
FANCA 3788-3790 MboII 37 130 + 30 160
Fonte: Adaptado de GIBSON et al., 1996.
Para detecção da mutação IVS4+4A>T utilizamos análise dos fragmentos de
restrição, em gel, obtidos pela enzima ScaI (Tabela 3). Houve a amplificação de um
fragmento de 131 pb. A enzima Sca I reconhece o sítio (AGTACT) no íntron quatro. A
mutação abole um sítio de restrição. Depois da digestão, um indivíduo normal apresentará
uma banda visível de 108 pb e outra de 23pb. Um homozigoto mutante terá uma única
banda de 131 pb e, um heterozigoto, o qual apresenta um alelo normal e o outro mutante, as
três bandas.
A reação de digestão consistiu em adicionar 10 u de enzima Sca I, diretamente ao
tubo de PCR. Após incubação a 37°C por 16 horas o resultado da digestão foi avaliado em
eletroforese em gel de poliacrilamida 12% corado com brometo de etídio.

36
Para análise da mutação G322 utilizamos somente a enzima Bsp1286 I que
reconhece um sítio de restrição na sequência normal. O amplificado após a PCR apresenta
151 pb. Após a digestão um indivíduo normal apresentará bandas de 128 e 22 pb visíveis
no gel. Um indivíduo afetado apresenta uma banda única de 151pb e um heterozigoto as
bandas de 22, 128 e 151 pb. Importante ressaltar que o fragmento de 22 pb, por ser muito
pequeno, não aparece no gel após a eletroforese. A reação de digestão consistiu em
adicionar 10 u de enzima Bsp1286 I, diretamente ao tubo de PCR. Após incubação a 37°C
por 16 horas o resultado da digestão foi avaliado em eletroforese em gel de poliacrilamida
12% corado com brometo de etídio.
Para análise da mutação Q13X utilizamos a mesma técnica descrita anteriormente,
porém utilizando a enzima Bcl I para clivagem do DNA (Tabela 3). A enzima reconhece
um sítio de restrição na sequência normal. O amplificado após a PCR apresenta 250 pb.
Após a digestão um indivíduo normal apresentará bandas de 227 e 23 pb visíveis no gel.
Um indivíduo afetado apresenta uma banda única de 250 pb e um heterozigoto as bandas
de 23, 227 e 250 pb. Importante ressaltar que o fragmento de 23 pb, por ser muito pequeno,
não aparece no gel após a eletroforese.
Na reação de digestão foi adicionado 10 u da enzima Bcl I diretamente ao tubo de
PCR. Após incubação por 16 horas a 37°C o resultado foi avaliado em gel de
poliacrilamida 12% corado com brometo de etídio.
Utilizamos a enzima Fok I para detecção da mutação W22X (Tabela3). A reação
de digestão consistiu em adicionar 10 u de enzima Fok I, diretamente ao tubo de PCR.
Após incubação a 37°C por 16 horas o resultado da digestão foi avaliado em eletroforese
em gel de poliacrilamida 12% corado com brometo de etídio. Houve a amplificação de um

37
fragmento de 221 pb. O indivíduo que apresenta os dois alelos com a mutação apresentará
duas bandas visíveis no gel, 187, 17 e17 pb. O heterozigoto três bandas, 204, 187 e 17. O
indivíduo normal uma banda de 204 e outra 17 pb.
Utilizamos a enzima Ava I para detecção da mutação R548X (Tabela3). A reação
de digestão consistiu em adicionar 10u de enzima Ava I, diretamente ao tubo de PCR. Após
incubação a 37°C por 16 horas o resultado da digestão foi avaliado em eletroforese em gel
de poliacrilamida 12% corado com brometo de etídio. Houve a amplificação de um
fragmento de 364 pb. O indivíduo que apresenta os dois alelos com a mutação apresentará
uma banda visível no gel, 364pb. O heterozigoto apresentará três bandas, 364, 231 e 133. O
indivíduo normal duas bandas de 231 e outra 133 pb.
Para análise da mutação L554P utilizamos a mesma técnica descrita anteriormente,
porém utilizando a enzima Bbv I para clivagem do DNA (Tabela 3). A enzima reconhece
um sítio de restrição na sequência normal. O amplificado após a PCR apresenta 364 pb.
Após a digestão um indivíduo normal apresentará bandas de 260pb e 104pb visíveis no gel.
Um indivíduo afetado apresenta uma banda única de 364 pb e um heterozigoto as bandas
de364, 260 e 104 pb.
Para análise da mutação L496R utilizamos somente a enzima Hha I que adiciona
um sítio de restrição na sequência normal. O amplificado após a PCR apresenta 303 pb.
Após a digestão um indivíduo normal apresentará uma banda de 303pb visível no gel. Um
indivíduo afetado apresenta duas bandas uma de 218 e 85 pb e um heterozigoto as bandas
de 303, 218 e 85 pb. A reação de digestão consistiu em adicionar 10u de enzima Bsp1286 I,

38
diretamente ao tubo de PCR. Após incubação a 37°C por 16 horas o resultado da digestão
foi avaliado em eletroforese em gel de poliacrilamida 12% corado com brometo de etídio.
Para detecção da mutação R185X utilizamos análise dos fragmentos de restrição,
em gel, obtidos pela enzima NlaIII (Tabela 3). Houve a amplificação de um fragmento de
122 pb. A mutação cria um sítio de restrição. Depois da digestão, um indivíduo normal
apresentará duas bandas uma de 105 e outra de 17pb, mas apenas uma é visível de 105 pb.
Um homozigoto para a mutação apresentará três bandas uma de 74, uma de 31 e outra de
17pb e, um heterozigoto, o qual apresenta um alelo normal e o outro mutante, quatro
bandas.
A reação de digestão consistiu em adicionar 10u de enzima Nla I, diretamente ao tubo de
PCR. Após incubação a 37°C por 16 horas o resultado da digestão foi avaliado em
eletroforese em gel de poliacrilamida 12% corado com brometo de etídio.
Para detecção da mutação 3788-3790 utilizamos análise dos fragmentos de
restrição, em gel, obtidos pela enzima MboII (Tabela 3). Houve a amplificação de um
fragmento de 160 pb. A mutação elimina o sítio de restrição. Depois da digestão, um
indivíduo normal apresentará duas bandas uma de 130 e outra de 30 pb. Um homozigoto
para a mutação apresentará apenas a banda de 160 bp e, um heterozigoto, o qual apresenta
um alelo normal e o outro mutante, as bandas de 160, 130 e 30 pb.
A reação de digestão consistiu em adicionar 10 u de enzima MboII, diretamente ao
tubo de PCR. Após incubação a 37°C por 16 horas o resultado da digestão foi avaliado em
eletroforese em gel de poliacrilamida 12% corado com brometo de etídio.

39
3.3.3 Sequenciamento
Os pacientes que não tiveram mutações detectadas pela técnica de PCR e digestão
enzimática específica ou que foi detectado um alelo da mutação 3788-3790 foram
sequenciadas, totalizando 41 amostras. Utilizou-se o kit BigDye® Terminator v3.1 Cycle
Sequencing (Applied Biosystems®) para realização da reação de sequenciamento. O
protocolo incluiu tampão de sequenciamento 5 X, big dye termination, PCR, primer (5
pmoles) e H2O.
O procedimento foi realizado em um aparelho termociclador (Mastercycler
eppendorf) com programa de ciclagem específico (Figura 10).
Figura 12: Ciclo utilizado na reação de sequenciamento
Posteriormente, as reações foram purificadas utilizando-se isopropanol e etanol,
também para remover as impurezas que poderiam afetar a qualidade da reação de
sequenciamento.

40
Por fim, os fragmentos foram submetidos a um sistema de eletroforese capilar no
equipamento Applied Biosystems 3500xl Genetic Analyzer® de 24 capilares para
determinação da sequência linear.
Todas as amostras de sequenciamento e da PCR foram realizadas em duplicatas e
repetidas para confirmação, quando encontrada qualquer tipo de alteração.

41
Tabela 3: Sequência dos Primers dos 43 éxons do gene FANCA que foram utilizados no
sequenciamento, com temperatura de anelamento (Tm) e tamanho dos fragmentos.
EXON Sequência dos Primers (5´ => 3´) Tm Produto da PCR (pb)
1 F: GGCCGCAGCCAATAGGAAGG 60 222
R: GATCGGGAACCGGGCGAAAC
2 F:GATGGTGGGTTTCTCCGCCGG 59 355
R:GAACTCCCGGGCTCAGGCGAC
3 F:CCCATCTCGGCCTCCCAAAGT 56 286
R:GGAAACCCATCGCCTGAGAAA
4 F: GTGTTGCATCTTAAAATAAGGC 60 315
R: GCTATGTCCTATTTTCCCAAC
5 F:ACCTGCCCGTTGTTACTTTTA 55 248
R:AGAACATTGCCTGGAACACTG
6 F: GCCCAAAGGCCAGGGAGTTT 58 384
R: AGCCAGAAATCAAACCCGTCTGA
7 F: GGGACATATGGCTCAACTCAATC 60 291
R: GAGACAGGCTGTTCTGCCTCGC
8 F: CTGAAGTGGATGGTCTGTGCC 59 278
R: CCCGTAAATAGGTACAAACAGC
9 F:GCAGGTATCACACAAATTACAG 60 189
R:GCACAGTGAAACATACAGAGG
10 F: GTAGAAGTCTTGATGGATGTG 60 297
R: CTGCTGAAGCTCTGGAAGTG
11 F: GATTGGTGGGTTGCGTGCAAGGC 60 272
R: AGAATTCCTGGCATCTCCAGTC
12 F:CCCACAACTTTTTGATCTCTG 55 221
R:CGTCCACGGCAGGCAGCATG
13 F: GAGCTGTCACAGCTCCATGTG 60 398
R: GTGGGAAGGGCTTCACTGAG
14 F: GGAATACTTGATCACCCAGC 59 337
R: GCTGACAGCAAGGTTGCTCAC
15 F: AGGAGGCCGACTACAGC 58 268
R: CTTGGGGAGGCCAAGGCAGTC
16 F: CAGCACTGTGGATGTTGGAAG 58 202
R: GAAAACAAAGCAGTTTCTGCTGG
17 F:CCATGCCCACTCCTCACACC 62 156
R:AAAAGAAACTGGACCTTTGCAT
18 F: CGCACAGCATGTGGGCCTTTACC 59 277
R: GAATAGAGTGGCCAACCTG
19 F: GAAACACCGGTCACCGTCTGTG 55 353
R: AGATCCACGATTCTTCGCATTGTC
21 F:CAGGCTCATACTGTACACAG 58 331
R:CACCGGCTTGAGCTGGCACAG
22 F: GTTCACAGCTCTGTATTTGAC 58 330

42
R: CTACTGGACTAGGAAGAC
23 F: AGCAGGATCCGTGGAATCGTAC 55 328
R:GAGAAGGCTCCATGCGTCTAATG
24 F:GCTGGCCGGCTTCCTTC 55 230
R:CGGAGACGAGCTCATGAGTC
25 F:TTCTTCGGCCGCTGGTGGTTG 56 324
R:CGAGAATGAGGGTGGCAGAGCAGA
26 F: GAGGGCCAGGCTGCTACTT 60 424
R: GACAGATAAAATTCTGGAAGG
27 F: CAGGCCATCCAGTTCGGAATG 60 284
R: CCTTCCGGTCCGAAAGCTGC
28 F: GTGTGGGCTGTTGATGGTCTGTT 55 361
R: CTGTTCTTGCCCGAGGAGCACA
29 F: GTGTGTTCTGTCCTCATTG 58 214
R:GATTCAAGAGATCTCCTGCC
30 F:GTCCCGAGCCGCCAGTC 56 380
R:AAGGCAGACCCACCCTAAG
31 F:GGAGAACTGGGAACTTCAGCACTA 56 302
R:GTTCTGGGATTACAGGCGTGAG
32 F: CTTGCCCTGTCCACTGTGGAG 58 369
R:CTCACTACAAAGAACCTCTAGG
33 F:GACACAGGCCAAGGCTCTG 58 390
R:GGCATTCCAGACACTGTTCC
34 F: GCCAAGCCTTAGCGAGTGTTT 56 300
R: AGCAGGAGGTCAGCGGTTTGT
35 F:GGATCCTCCTGTCAGCTTCCTG 60 313
R:TTTCCCTGAGATGGTAACACC
36 F: GTCATGGCTGGGGCAGCGGAG 62 260
R: GCATCTGGGCGGGCACAC
37 F: GGTGACAGGTGGGAATAAGGAC 59 347
R: CTTGCTCCAAGCCACATATTTG
38 F: AGGATTTATGGCCTAGATGTAAAAA 50 240
R: CTGGTGCCCCTGCCTGG
39 F: GCAAAGAGGAAATGCCCTGTT 55 288
R: GGGCTCGTTCTTAACCATTTG
40 F: CCAGCTGCTGACAGGTACC 55 310
R: GGACCCAGAAGTGCTGAGATG
41 F: CCCCATCTCAGCACTTCTGGGTCC 60 382
R: CCATAGTCTCCATGCTGTGC
42 F: GCACAGCATGCAGACTATGG 58 271
R: GTCGAGTTGTATTGCCAGCC
43 F:CCTGGCTGGCAATACAACTC 56 219
R:GGCAGGTCCCGTCAGAAGAGAGAT
Fonte: Adaptado de GIBSON, et al., 1996 e MAGDALENA, 1999.

43
3.4 Análise dos Dados
A análise dos sequenciamentos foi realizada com o auxílio do programa Chromas
LiteTM
e DNA Baser (v. 3.5) para a visualização da sequência linear dos eletroferogramas e
com o auxilio do programa Gene Runner® para comparar as sequências obtidas com as
postadas como referência em base de dados. As alterações encontradas foram pesquisadas
nos seguintes bancos de dados: NCBI, Ensembl Genome Browser (GRch 37.p10), HGMD
(Human Genome Mutation Database) , dbSNP (short genetic variations_build 37.3 ) e
Exome Variant Server.
3.5 Análise Estatística
Para análise da associação entre as variáveis clínicas e a presença de mutação foram
utilizados os testes: teste Exato de Fisher e o teste de Mann-Whitney, com valor de p
positivo para valores inferiores a 0,05.

44
4. RESULTADOS
Na amostra de 152 pacientes enviados para o laboratório com quadro clínico de AF na
região de Campinas no gene FANCC, foram excluídos 84 pacientes, pois não tinham
realizado o DEB teste, ou tinham resultado negativo. Nesses pacientes foi realizada apenas
a investigação das mutações mais frequentes e os resultados não foram considerados para
este estudo. Com isso, a nossa amostra totalizou 68 pacientes com uma média de idade de
16,62 anos (DP=8,72). Do total da amostra 40 pacientes eram do sexo feminino e 15 eram
negroides (Tabela 4).
Os resultados dos testes moleculares encontram-se sumarizados na tabela 4. Com o
método utilizado foi possível classificar como sendo do grupo C, 19 pacientes (27,94%),
como sendo do grupo A, 16 casos (23,53%) e 33 pacientes que não foram classificados
como grupo A ou C (48,53) (Figura 14). No caso do gene FANCC as mutações mais
frequentes foram a Δ3788-3790 no gene FANCA e IVS4+4 A>T no gene FANCC (Tabela
5). Com relação a distribuição genotípica, o genótipo mais frequente foi o heterozigoto
composto Δ3788-3790 com uma segunda mutação não identificada (16,17%) no gene
FANCA e o heterozigoto composto com a mutação IVS4+4 A>T e uma segunda mutação
não identificada (10,29%) (Tabela 6).
Na tabela 7 temos a média das idades dos pacientes por grupo FANC.

45
Tabela 4: Sumário dos Dados Clínicos e Moleculares dos pacientes estudados
Caso Sexo Idade COR Mutação Encontrada Aspectos clínicos
1 F 10 C Δ3788-3790 Estatura baixa, pancitopenia
2 F 8 C IVS4+4 A>T Formação de esquimoses (sangramento
cutâneo) esporádico, estrabismo, microftalmia e pancitopenia.
3 M 18 C IVS4+4 A>T/ΔG322 Sangramento, esquimoses,
hiperpigmentação da pele, ausência do rim esquerdo e pancitopenia.
4 F 10 C IVS4+4 A>T/ΔG322 Sangramento, esquimoses,
hiperpigmentação da pele, microftalmia e pancitopenia.
5 M 12 C IVS4+4 A>T/ΔG322 Sangramento, esquimoses,
hiperpigmentação da pele, deformidade de polegar e pancitopenia.
6 F 18 N IVS4+4 A>T Sangramento, esquimoses,
hiperpigmentação da, microcefalia manchas café com leite e pancitopenia
7 F 15 N IVS4+4 A>T Sangramento, esquimoses,
hiperpigmentação da, microcefalia e pancitopenia
8 F 20 N R548X Sangramento, esquimoses,
hiperpigmentação da, microcefalia manchas café com leite e pancitopenia
9 M 14 C R548X Anemia Aplástica, SMD, sangramento.
10 F 33 C Q13X Pancitopenia, SMD,
11 M 17 N Δ3788-3790/C/T hetero intron
variant (rs1061646)
Mancha café com leite criptorquidia, baixa estatura, polegares com implantação
anormal e pancitopenia.
12 F 15 c Δ3788-3790/C/T hetero intron
variant (rs1061646) Estatura baixa, pancitopenia.
13 F 14 C W22X/W22X Sangramento, anemia, leucopenia,
hipoplasia de medula.
14 F 21 N Missense c.3982A>G Polegar duplicado, anemia,
hiperpigmentaçao, manchas café com leite, microcefalia, microftalmia.
15 F 19 N L554P Anemia, leucopenia hipoplasia de medula,
baixa estatura, manchas hipercromicas.
16 M 28 C Δ3788-3790/Δ3788-3790 Mancha café com leite criptorquidia, baixa
estatura, polegares com implantação anormal, pancitopenia e LMA.
17 F 10 N C/T homo intron variant
(rs1061646) Baixa estatura, pancitopenia,
hiperpigmentação, microcefalia.
18 F 7 C C/T hetero intron variant
(rs1061646)
Leucopenia, manchas café com leitepancitopenia, hiperpigmentação,
microcefalia.

46
19 F 8 c Δ3788-3790/C/T hetero intron
variant (rs1061646) Polidactilia, pancitopenia, hiperpigmentação
da, microcefalia
20 F 6 c Δ3788-3790/C/T hetero intron
variant (rs1061646) Polidactilia, pancitopenia, hiperpigmentação,
microcefalia.
21 F 8 C C/T hetero intron variant
(rs1061646) Baixa estatura, hipoplasia de polegar,
ausência rim esquerdo.
22 F 1 C IVS4+4 A>T Pancitopenia, aplasia de medula nódulos no
pâncreas.
23 M 2 C W22X Pancitopenia, aplasia de medula, baixa
estatura, manchas hircrômicas, manchas café com leite.
24 F 10 C C/T hetero intron variant
(rs1061646) Pancitopenia, aplasia de medula.
25 F 23 N Δ3788-3790 SMD, micrognatia, orelha esquerda com
alteração na cartilagem devido à retirada de tumor.
26 M 8 C Δ3788-3790 Sangramento, esquimoses,
hiperpigmentação da e pancitopenia
27 M 9 C Δ3788-3790/Missense c.3982A>G Mancha hipercromicas grande em dorso,
prega palpebral, hipertelorismo
28 F 25 C Q13X Neutropenia crônica idiopática, e hipoplasia
de medula e manchas hipercromicas.
29 F 35 C W22X hiperpigmentação da , microcefalia e
pancitopenia, nódulo axilar, leucopenia, plaquetopenia e SMD
30 M 29 C Δ3788-3790/Δ3788-3790 Neutropenia crônica idiopática, e hipoplasia
de medula e manchas hipercromicas.
31 F 30 C Δ3788-3790/Δ3788-3790 Neutropenia crônica idiopática, e hipoplasia
de medula e manchas hipercromicas.
32 M 11 C Δ3788-3790/C/T intron variant
(rs34420680) Polidactilia, pancitopenia, hiperpigmentação
da microcefalia.
33 M 9 C T/C synonymous variant
(1800358)/C/T intron variant (rs34420680)
Pancitopenia, aplasia de medula.
34 M 12 N T/C synonymous variant
(1800358)/C/T intron variant (rs34420680)
Síndrome Mielodisplásica (SMD)
35 F 12 C T/C synonymous variant
(1800358)/C/T intron variant (rs34420680)
Sangramento, esquimoses, hiperpigmentação da e pancitopenia
36 F 23 N Δ3788-3790/Δ3788-3790 Mancha hipercromicas grande em dorso,
prega palpebral, hipertelorismo
37 F 2 C Δ3788-3790/Missense c.3982A>G Neutropenia crônica idiopática, hipoplasia de medula e manchas hipercromicas em MMSS.
38 M 21 C T/C synonymous variant (1800358) SMD

47
39 F 23 C T/C synonymous variant
(1800358)/C/T intron variant (rs34420680)
Sangramento, esquimoses, hiperpigmentação da e pancitopenia.
40 M 12 C IVS4+4 A>T Mancha hipercromica grande em dorso prega
palpebral, hipertelorismo.
41 M 24 N Δ3788-3790/Δ3788-3790 Polidactilia, pancitopenia, hiperpigmentação,
microcefalia.
42 M 21 C Missense c.3982A>G Polidactilia, pancitopenia, hiperpigmentação,
microcefalia.
43 F 21 C Δ3788-3790/C/T intron variant
(rs34420680) Baixo peso, baixa estatura, mancha café com
leite.
44 M 15 C IVS4+4 A>T Aplasia medula, Polidactilia, pancitopenia,
hiperpigmentação da, microcefalia
45 F 11 C T/C synonymous variant (1800358) Polidactilia, pancitopenia, hiperpigmentação,
microcefalia.
46 M 17 C T/C synonymous variant (1800358) Neutropenia crônica idiopática, hipoplasia de medula e manchas hipercromicas em MMSS
e dorso
47 M 21 C T/C synonymous variant (1800358) Neutropenia crônica idiopática, hipoplasia de
medula e manchas hipercromicas em membros e dorso.
48 F 33 C IVS4+4 A>T Aplasia radial bilateral
49 F 18 N ΔG322/W22X Neutropenia crônica idiopática, hipoplasia de
medula e manchas hipercromicas.
50 M 28 C C/T hetero intron variant
(rs1061646)
Mancha café com leite criptorquidia D, baixa estatura, polegares com implantação
anormal e pancitopenia.
51 F 30 c ΔG322/ Sangramento, anemia, leucopenia,
hipoplasia de medula.
52 M 32 C C/T hetero intron variant
(rs1061646)
Polegar duplicado, anemia, hiperpigmentaçao, manchas café com leite,
microcefalia, microftalmia.
53 F 10 C C/T intron variant (1061646) Linfonodo em cadeia submandibular, cianose nas falanges distal, AA, neutropenia, SMD.
54 F 31 N C/T hetero intron variant
(rs1061646) Polidactilia, pancitopenia, hiperpigmentação,
microcefalia.
55 M 5 C T/C synonymous variant
(1800358)/C/T hetero intron variant (rs1061646)
Mancha café com leite criptorquidia, baixa estatura, polegares com implantação
anormal e pancitopenia.
56 M 4 C Missense c.3982A>G Polegar duplicado, anemia,
hiperpigmentaçao, manchas café com leite, microcefalia, microftalmia.
57 F 10 C C/T homo intron variant
(rs1061646)
Polegar duplicado, anemia, hiperpigmentaçao, manchas café com leite,
microcefalia, microftalmia.
58 F 21 C C/T homo intron variant
(rs1061646) Estatura baixa, pancitopenia

48
59 F 5 N T/C synonymous variant (1800358) Sangramento, anemia, leucopenia,
hipoplasia de medula
60 M 34 N C/T homo intron variant
(rs1061646) Estatura baixa ,pancitopenia
61 F 12 C T/C synonymous variant
(1800358)/C/T intron variant (rs34420680)
Polegar duplicado, anemia, hiperpigmentaçao, manchas café com leite,
microcefalia, microftalmia
62 M 23 C C/T intron variant (rs34420680) sangramento, polidactilia, esquimoses, hiperpigmentação da , microcefalia e
pancitopenia
63 M 8 C T/C synonymous variant (1800358) sangramento, esquimoses, hiperpigmentação
da e pancitopenia
64 F 6 C C/A intron variant (rs 373592545) sangramento, esquimoses, hiperpigmentação
da e pancitopenia
65 F 11 C C/T hetero intron variant
(rs1061646)/C/T homo intron variant (rs1061646)
Mancha hipercromica grande em dorso, prega palpebral, hipertelorismo
66 M 23 C C/T homo intron variant
(rs1061646)
Mancha café com leite criptorquidia D, baixa estatura, polegares com implantação
anormal e pancitopenia
67 M 14 C C/T hetero intron variant
(rs1061646) estatura baixa ,pancitopenia, microftalmia
68 F 19 C T/C synonymous variant
(1800358)/C/T hetero intron variant (rs1061646)
sangramento, anemia, leucopenia, hipoplasia de medula
Figura 13: Classificação dos pacientes por gene mutado.
FANCA FANC FANX

49
Tabela 5: Frequência alélica das mutações deletérias encontradas.
MUTAÇÃO FREQUÊNCIA ALÉLICA
NA AMOSTRA (N)
Δ3788-3790 0,154 (21)
IVS4+4
A>T 0,073 (10)
ΔG322 0,036 (5)
W22X 0,029 (4)
R548X 0,014 (2)
Q13X 0,014 (2)
L554P 0,007 (1)
Tabela 6: Frequência genotípica das mutações deletérias encontradas (Des – mutação não
identificada).
GENÓTIPO
FREQUÊNCIA
GENOTÍPICA
NA AMOSTRA (N)
Δ3788-3790/Desc. 0,1617 (11)
IVS4+4 A>T/Desc. 0,1029 (7)
Δ3788-3790/ Δ3788-3790 0,0735 (5)
IVS4+4 A>T/ ΔG322 0,0441 (3)
R548X/ Desc. 0,0294 (2)
Q13X/ Desc. 0,0294 (2)
W22X/ Desc. 0,0294 (2)
W22X/ W22X 0,0147 (1)
L554P/ Desc. 0,0147 (1)
ΔG322/W22X 0,0147 (1)
ΔG322/ Desc. 0,0147 (1)
Desc./ Desc. 0,4705 (32)

50
Tabela 7: Média de idade dos pacientes por grupo FANC
Grupo Média (DP) da idade Comparação
entre os grupos
FANCA 16,50 (±9,048)
P=0,623 FANCC 17,84 (±9,907)
Desconhecido 15,48 (±8,345)
Nas Tabelas 8 e 9 estão os resultados dos testes de correlação para as variáveis
clínicas. Sendo que na Tabela 9, os dados de FANCA e FANC foram agrupados.

51
Tabela 8: Correlação entre as variáveis clínicas e o Grupo FANC
Variáveis Mutação no gene FANC Total p-value
A C Desconhecido
Sexo Feminino 9 14 17 40
0,286 Masculino 7 5 16 28
Total 16 19 33 68
Etnia Caucasoide 12 14 27 53
0,752 Não caucasoide 4 5 6 15
Total 16 19 33 68
Manif.
Hematológica
Ausente 2 1 4 7 0,696
Presente 14 18 29 61
Total 16 19 33 68
Alter. Membros Ausente 12 16 20 48
0,180 Presente 4 3 13 20
Total 16 19 33 68
Alt. Cutâneas Ausente 3 7 13 23
0,340 Presente 13 12 20 45
Total 16 19 33 68
Alt. Renais Ausente 16 18 32 66
0,656 Presente 0 1 1 2
Total 16 19 33 68
Baixa estatura Ausente 11 16 24 51
0,526 Presente 5 3 9 17
Total 16 19 33 68
Outras Ausente 7 12 18 37
0,517 Presente 9 7 15 31
Total 16 19 33 68
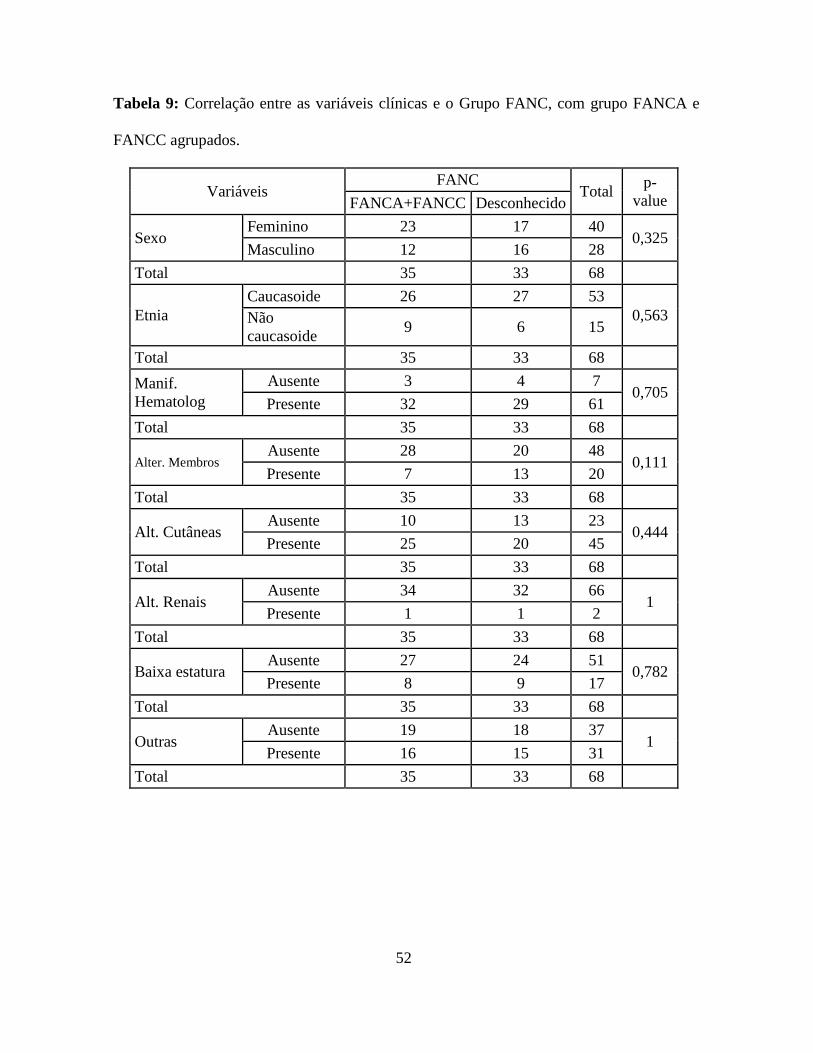
52
Tabela 9: Correlação entre as variáveis clínicas e o Grupo FANC, com grupo FANCA e
FANCC agrupados.
Variáveis FANC
Total p-
value FANCA+FANCC Desconhecido
Sexo Feminino 23 17 40
0,325 Masculino 12 16 28
Total 35 33 68
Etnia
Caucasoide 26 27 53
0,563 Não
caucasoide 9 6 15
Total 35 33 68
Manif.
Hematolog
Ausente 3 4 7 0,705
Presente 32 29 61
Total 35 33 68
Alter. Membros Ausente 28 20 48
0,111 Presente 7 13 20
Total 35 33 68
Alt. Cutâneas Ausente 10 13 23
0,444 Presente 25 20 45
Total 35 33 68
Alt. Renais Ausente 34 32 66
1 Presente 1 1 2
Total 35 33 68
Baixa estatura Ausente 27 24 51
0,782 Presente 8 9 17
Total 35 33 68
Outras Ausente 19 18 37
1 Presente 16 15 31
Total 35 33 68
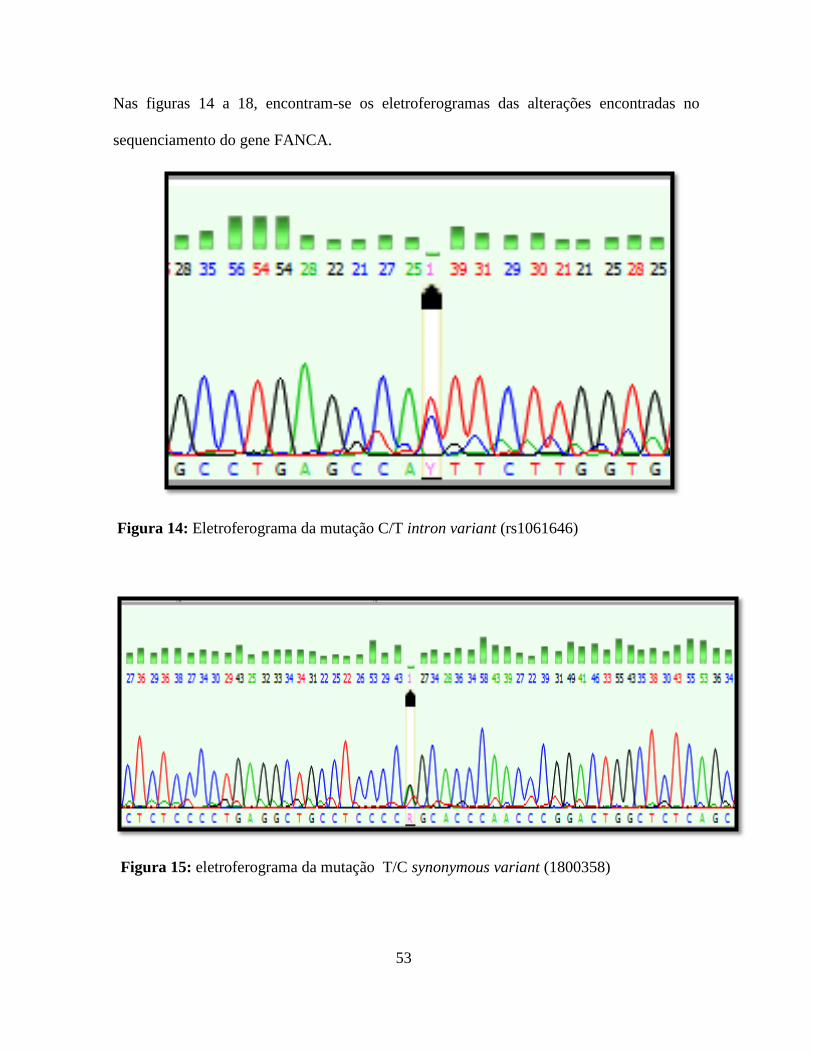
53
Nas figuras 14 a 18, encontram-se os eletroferogramas das alterações encontradas no
sequenciamento do gene FANCA.
Figura 14: Eletroferograma da mutação C/T intron variant (rs1061646)
Figura 15: eletroferograma da mutação T/C synonymous variant (1800358)

54
Figura 16: Eletroferograma da mutação C/T intron variant (rs34420680)
Figura 17: Eletroferograma da mutação C/A intron variant (rs 373592545)

55
Figura 18: eletroferograma da mutação Missense c.3982A>G , em A em heterozigose e
em B sequencia normal
Analisando a mutação c.3982A>G pelos algoritmos de predição obtivemos os resultados
que estão apresentados na tabela 5.
A
B
56
Tabela 10: Algoritmos de Predição testados com relação à mutação c.3982 A>G
Algoritmo Índice Probabilidade Efeito
PolyPhen 0.005 0.74 Benigna
Snps&GO 9 - Neutra
SNAP 2 70% Não-Neutra
PhD-SNP 7 - Neutra
MutPred - 0.116 Neutra

57
5. DISCUSSÃO
Desde a primeira descoberta de um gene FANC (FANCC por Strathdee et al.,
1992), tem havido um significativo avanço no conhecimento da patogênese e bases
moleculares da Anemia de Fanconi. Percebe-se que se trata de uma doença bem mais
complexa do que se imaginava no inicio, principalmente com relação às proteínas
envolvidas e seus genes.
Com relação ao diagnóstico da doença, o teste “ouro” é o DEB teste, onde é
verificado se as células em cultura, geralmente linfócitos, perderam sua capacidade de
corrigir danos provocados por agentes clastogênicos. No entanto, alguns estudos tem
demonstrado a ineficiência do método, onde pode haver artefatos de cultura e falsos
negativos, uma vez que temos o mosaicismo clonal dos linfócitos (Auerbach, 2009). Com
isso, há uma parcela de pacientes que não são adequadamente diagnosticados. Frente a esse
problema, com a descrição do primeiro gene FANC, surgiram esperanças que o teste
molecular poderia substituir o DEB teste com mais eficiência. No entanto, à medida que os
outros genes foram sendo descobertos, percebeu-se que isso não poderia ocorrer, levando-
se em consideração os métodos que atualmente estavam disponíveis na prática clínica.
Assim partiu-se para o estudo molecular desses genes a fim de efetivar o diagnóstico dos
pacientes, avaliar o núcleo familiar, selecionar potenciais candidatos a doadores nos casos
em que houvesse aplasia de medula e mesmo disponibilizar o diagnóstico pré-natal, pré-
implantação e pré-fecundação para os casais interessados.

58
Frente a essas perspectivas, o presente estudo se propôs a estudar o gene FANCA
nessa amostra de pacientes com apresentação clínica compatível com Anemia de Fanconi e
com DEB teste positivo. Para facilitar esse estudo, uma vez que os Grupos mais
prevalentes são os FANCA e FANCC, iniciamos com uma análise das mutações mais
prevalentes do gene FANCC segundo Auerbach et al. 2003. Com isso, tínhamos uma
grande probabilidade de no restante da amostra detectar pacientes FANCA. Vale ressaltar
aqui que seria interessante se tivéssemos disponível o teste de complementação para
direcionarmos o gene a ser pesquisado (Joenje e Patel, 2001). Infelizmente em nosso País
esse teste não é realizado e são poucos os centros no restante do mundo que o realizam.
Em nossa amostra detectamos 19 pacientes (27,94%), como sendo do grupo C e 16
pacientes como grupo A (23,53% ). Diferente dos números do IFAR, onde 65% dos
pacientes no mundo pertencem ao grupo de complementação A e 15% ao grupo C. É
importante fazer aqui a ressalva que entre os pacientes que não foram identificadas
mutações podem existir pacientes FANCC que tenham outras mutações que não as
analisadas e pacientes FANCA que tenham mutação na região promotora ou em regiões
mais internas dos íntron e grandes deleções. E essa pode ser a causa da diferença de
proporção encontrada no resto do mundo.
No caso do gene FANCA a mutação mais frequentes foi a Δ3788-3790 e no
FANCC, a IVS4+4 A>T, o que vem de encontro com outros estudos realizados com
pacientes brasileiros (Magdalena et al. 2005 ; Dear et al., 2005).
Com relação aos dados clínicos houve uma predominância de manifestações
hematológicas (89,7%) e seguidos de alterações na pele, como manchas e
hiperpigmentação (66,2%). Em 20 (29,4%) pacientes ocorreram alterações de polegares e

59
17 (25%) pacientes tinham baixa estatura. Esses sinais clínicos estão entre os mais
frequentes na Anemia de Fanconi. Anormalidades Hematológicas ocorrem em algum
momento da vida dos pacientes (média de 7 anos, mas variando do nascimento a 41 anos),
segundo Butturini em 1994 e Kutler et al, 2003. No ínicio da doença as manifestações
hematológicas são variadas. A trombocitopenia é frequentemente associada a elevados
níveis de hemoglobina fetal e macrocitose e geralmente precede o aparecimento de anemia
ou neutropenia. Notavelmente, alguns pacientes apresentaram síndrome mielodisplásica
(SMD) ou leucemia mielóide aguda (LMA), sem diagnóstico prévio de anemia aplástica
(AA) (Auerbach, 2009).
Dos 754 pacientes do IFAR , 120 pacientes (16%) apresentaram SMD e / ou LMA.
A incidência cumulativa de SDM ou LMA por 40 anos foi de 33% (Kutler et al., 2003).
Com base em uma pesquisa com pacientes com FA realizados por Rosenberg et al. 2003, a
idade média de início da leucemia foi de 11,3 anos. Em nosso estudo encontramos 13,11%
dos pacientes apresentando anemia, 60,65% apresentando pancitopenia, 11,47% anemia
aplástica, 14,75% SMD e 1,63% LMA, sendo que o único paciente que apresentou LMA
tinha 28 anos e era homozigoto Δ3788-3790 e foi a óbito. Portanto, parece que em nosso
meio a evolução do quadro hematológico tem sido mais lenta, com uma proporção maior
de pacientes com SMD.
Com relação a baixa estatura, essa é uma característica bem reconhecida da FA e é
frequentemente secundária a deficiências hormonais, que incluem hipofunção da pituitária
com hipogonadismo, deficiência de hormônio de crescimento, disfunção da tireoide e
deficiência ou resistência de insulina com intolerância à glicose (Auerbach, 2009). Um
estudo prospectivo por Wajnrajch et al. (2001) de 54 pacientes de 47 famílias não

60
relacionadas, mostrou que endocrinopatias são uma característica comum das FA,
manifestando-se principalmente como anormalidades da glicose / insulina, insuficiência do
hormônio do crescimento e hipotireoidismo, sendo que em 46% dos pacientes a baixa
estatura estava presente. Em nosso estudo, ela esteve presente em 25% dos pacientes.
Como a via metabólica tem sido recentemente desvendada, a relação entre os genes
mutados e a presença de anormalidades congênitas, hematológicas, predisposição a câncer,
ou seja, ao fenótipo, ainda é incipiente e necessita de muitos estudos que avaliem essa
correlação fenótipo/genótipo para que algumas considerações possam ser feitas. No
presente estudo, não encontramos uma correlação entre o grupamento FANC e as variáveis
clínicas apresentadas pelos pacientes.
Com relação a análise molecular do gene FANCA, Magdalena et al. (2005) estudou
80 pacientes AF , não relacionados com relação a presença da mutação Δ3788-3790. Em
16% da amostra foi encontrada homozigose dessa mutação e em 13,75 % heterozigose.
Levran et al. (1997) analisando 97 pacientes, a maioria deles eram do grupo de
complementação A, encontrou em 67 pelo menos uma mutação no gene da FANCA. Dos
restantes 30 (31%) pacientes, nenhuma mutação no gene FANCA foi evidenciada. Foram
identificados dois tipos principais de mutações no presente estudo: substituições de base e
microdeleções / microinserções. Em nosso estudo, onde não tínhamos a informação com
relação ao grupo de complementação, 23,53% da amostra tinha pelo menos uma mutação
detectada no gene FANCA.
Com relação às mutações não deletérias detectadas, todas são descritas
anteriormente. Apenas a mutação rs1061646 foi detectada em homozigose (T/T) e em
heterozigose (C/T). As demais só correram em heterozigose. A mutação rs1061646 é uma

61
troca C/T no íntron 40, sendo que sua prevalência varia entre as populações. Em africanos
o alelo T tem uma prevalência de 58%, em Americanos 50%%, em Asiáticos 76% e em
Europeus 31% (Ensembl-1). Em nossa amostra o alelo T teve uma prevalência de 29%,
semelhante à encontrada em Europeus.
A mutação rs1800358T/C é uma variação sinônima ou mutação neutra, onde não
ocorre troca do aminoácido. Ela ocorre no códon 1218 no éxon 37 do FANCA e o códon
GGT muta para GGC e continua codificando Prolina. O alelo C tem uma prevalência de
19% em Africanos, em Americanos 5%%, em Asiáticos 79% e em Europeus 8% (Ensembl-
2). Em nossa amostra o alelo T teve uma prevalência de 16%, semelhante à encontrada em
Africanos.
A mutação rs34420680 é uma variação no íntron 37 onde ocorre uma troca C/T. O
alelo T tem uma prevalência de 1% em Africanos, em Americanos 3% , em Asiáticos 21%
e em Europeus 7% (Ensembl-3). Em nossa amostra o alelo T teve uma prevalência de
9,7%, semelhante à encontrada em Europeus.
A mutação rs 373592545 é uma variação no íntron 38 onde ocorre uma troca C/A .
É uma variação rara. O alelo A foi detectado em uma população de afro-americanos na
proporção de 1 entre 4.395 alelos analisados e em uma população de americanos de
ascendência europeia de 1 entre 8.599 alelos (Ensembl-4). Em nossa amostra o alelo A foi
detectado em um alelo (1,21%).
A mutação c.3982A>G é uma mutação de sentido trocado ou missense, onde ocorre
troca do aminoácido. Ela ocorre no códon 1218 no éxon 40 do FANCA e o códon ACC
muta para GCC e muda o aminoácido de treonina para Alanina. Essa mutação foi
depositada no Banco de dados por Arleen Auerbach e foi encontrada em um paciente com

62
Anemia de Fanconi que era um heterozigoto composto dessa mutação com
c.2316_2316+2delGGT. Foram analisados os pais, sendo que eram heterozigotos para cada
uma das mutações (Ensembl-5). Em nossa amostra encontramos dois paciente onde já
havia sido identificado a mutação Δ3788-3790 e em outros dois onde nenhuma outra
mutação foi identificada. Apesar dos algoritmos de predição em sua maioria apontarem
para uma mutação não deletéria (Tabela 5), acredito que essa mutação mereça um maior
cuidado.
Com o surgimento das novas tecnologias de sequenciamento, a dificuldade do
diagnóstico molecular poderia ser resolvido a um custo menor. Knies et al em 2012,
verificou a possibilidade da utilização do sequenciamento do Exoma (WES) no diagnóstico
molecular da Anemia de Fanconi. Foram analisados separadamente quatro pacientes
utilizando diferentes tecnologias de enriquecimento e sequenciamento. O custo de cada
uma das análises variou de $800 a $4.500,00, mas acredita-se que a tendência seria de
haver diminuição de custos. Com relação às limitações do método, é importante lembrar
que o WES sozinho não é capaz de detectar grandes deleções e rearranjos complexos,
sendo necessária a associação ao MLPA. Nos casos de mutações intrônicas que ocorrem
longe da junção éxon/íntron, o método WES não é capaz de realizar a detecção.
Já Chandrasekharappa et al. (2013) propõe o uso em conjunto do CGH array, o
WES e o sequenciamento do RNA, uma vez que utilizando esses três métodos em conjunto
conseguiu detectar os dois alelos mutantes em 27 pacientes com Anemia de Fanconi.
Portanto, apenas a nova geração de tecnologias, a custo baixo pode resolver o
problema do diagnóstico molecular de uma alteração tão heterogênea como a Anemia de
Fanconi.

63
6. CONCLUSÃO
-Em nossa amostra foram detectados 19 pacientes (27,94%), como sendo do grupo C e 16
pacientes como grupo A (23,53% ).
- A mutação Δ3788-3790 do gene FANCA teve uma frequência alélica de 15,4% na
amostra estudada.
- Não foram encontradas correlações com as manifestações hematológicas, renais, baixo
peso, malformações congênitas de membros, machas e pigmentação de pele, sexo e idade.
-Foram encontradas 3 mutações intrônicas, 1 mutação sinônima e 1 mutação de sentido
trocado no gene FANCA, com frequência alélica de 0.29 , 0.16, 0.09 e 0.01.

64
7. REFERÊNCIAS BIBLIOGRÁFICAS
Alter BP. Fanconi's anaemia and its variability. Br J Haematol. 1993; 85(1):9-14.
Alter BP. Fanconi’s anemia and malignancies. Am. J. Hematol 1996; 53: 99-110
Alter BP, Caruso JP, Drachtman RA, Uchida T, Velagaleti GV, Elghetany MT: Fanconi
anemia: myelodysplasia as a predictor of outcome. Cancer Genet Cytogenet 2000;
117(2):125-31.
Apostolous S, Whitmore AS, Crawford J. Positional cloning of the Fanconi anemia group
C gene. Nat Genet 1996; 14:324.
Auerbach AD, Adler B e Chaganti RS. Prenatal and postnatal diagnosis an carrier detection
of Fanconi anemia by a cytogenetic method. Pediatrics 1981; 67(1):128-135.
Auerbach AD, Rogatko A, Achroeder-Kurth TM. International Fanconi Anemia Registry:
relation of clinical symptoms to dyepoxibutane sensitivity. Blood 1989; 73(2):391-396.
Auerbach AD, Allen RG. Leukemia and preleukemia in Fanconi anemia patients: A review
of the literature and report of the International Fanconi Anemia Registry. Cancer Genet
Cytogenet 1991; 51:1-12.
Auerbach AD. Fanconi anemia. Dermatol Clinic 1995; 13(1):41-9.
Auerbach AD, Greenbaum J, Pujara K, Batish SD, Bitencourt MA, Kokemohr I, et al.
Spectrum of sequence variation in the FANCG gene: an International of Fanconi Anemia
Registry (IFAR) study. Hum Mutat 2003; 21(2):158-168.
Auerbach AD. Fanconi anemia and and its diagnosis.. Mutat Res. 2009 Jul 31;668(1-2):410

65
Block M, Jacobson Lo, Bethard Wf. Preleukemic acute human leukemia. Am J Med
1953; 152:1018-28.
Bogliolo, M., Schuster, B., Stoepker, C., Derkunt, B., Su, Y., Raams, A., Trujillo, J. P.,
Minguillon, J., Ramirez, M. J., Pujol, R., Casado, J. A., Banos, R., and 10 others. Mutations
in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J.
Hum. Genet. 92: 800-806, 2013.
Butturini A. Hematologic abnormalities in Fanconi anemia: an International Fanconi
Anemia Registy study. Blood 1994; 84:1650-55.
Cervenka J, Arthur D, Yasis C. Mitomycin C test for diagnostic differentiation of
idiopathic Aplastic anemia and Fanconi anemia. Pediatrics 1982; 67(1):119-27.
Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, et al. Massively
parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive
molecular diagnosis of Fanconi anemia. Blood. 2013 May 30;121(22):e138-48.
Couzin J. The twists and turns in BRCA`s path. Science 2003; 302:591-93.
DÀndrea AD e Grompe M. The Fanconi anemia/BRCA pathway. Nature Reviews Cancer
2003; 3:23-34.
De Chadarévian JP, Vekemans M, e Bernstein M. Fanconi’s anemia medulloblastoma,
Wilms’ tumor, horseshoe kidney and gonadal dysgenesis. Arch Pathol Lab Med 1985;
109(4):367-69.
De Kerviler E, Guermazi A, Zagdanski AM, Gluckman E, Frija J. The clinical and
radiological features of Fanconi’s anaemia. Clin Radiol 2000; 55:340-45.

66
Demuth I, Wlodarski M, Tipping AJ, Morgan NV, de Winter JP, Thiel M, et al. Spectrum
of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur J Hum Genet
2000; 8:861-68.
Dear; Lima CSP; Lourenco GJ; Figueiredo ME; Carneiro JDA; Tone LG; Llerena Junior
JC; Wpj; Costa Ff; Bertuzzo CS. Molecular Analysis Of The Most Prevalent Mutations of
The FANCA and FANCC Genes in Brazilian Patients with Fanconi Anemia. Genetics and
Molecular Biology, v. 28, n.2, p. 205-209, 2005.
Duckworth-Rysiecki G, Cornish K, Clarke CA, Buchwald M. Identification of two
complementation groups in Fanconi anemia. Somat Cell Mol Genet 1985; 11:35-41.
Ensembl-1:
http://www.ensembl.org/Homo_sapiens/Variation/Summary?db=core;g=ENSG000001877
41;r=16:8980395789883065;source=dbSNP;t=ENST00000389301;v=rs1061646;vdb=vari
ation;vf=849212
Ensembl-2:
http://www.ensembl.org/Homo_sapiens/Variation/Summary?db=core;g=ENSG000001877
41;r=16:8980395789883065;source=dbSNP;t=ENST00000389301;v=rs1800358;vdb=vari
ation;vf=1368960
Ensembl-3:
http://www.ensembl.org/Homo_sapiens/Variation/Summary?db=core;g=ENSG000001877
41;r=16:89803957-
89883065;source=dbSNP;t=ENST00000389301;v=rs34420680;vdb=variation;vf=1084527
Ensembl-4:
http://www.ensembl.org/Homo_sapiens/Variation/Summary?db=core;g=ENSG000001877
41;r=16:8980395789883065;source=dbSNP;t=ENST00000389301;v=rs373592545;vdb=va
riation;vf=61966826

67
Ensembl-5:
http://www.ensembl.org/Homo_sapiens/Variation/Summary?db=core;g=ENSG000001877
41;r=16:8980395789883065;source=PhenCode;t=ENST00000389301;v=FANCA:c.3982A
%3EG;vdb=variation;vf=68315735
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, et
al. Interaction of Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cel
2001; 7:249-262.
Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD.
The need for more accurate and timely diagnosis in Fanconi anemia: a report from the
International Fanconi Anemia Registry. Pediatrics. 1993; 91(6):1116-20.
Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in
patients without congenital malformations: an International Fanconi Anemia Registry
study. Am J Med Genet 1997; 68:58-61
.
Gibbons B, Scott D, Hungerford JL, Cheung KL, Harrison C, Attard-Montalto S, et al.
Retinoblastoma in association with the chromosome breakage syndromes Fanconi’s anemia
ad Bloom’s syndrome: clinical and cytogenetics findings. Clin Genet 1995; 47(6):311-17.
Gillio AP, Verlander PC, Batish SD, Giampietro PF, Auerbach AD. Phenotypic
consequences of mutation in the Fanconi anemia FAC gene: an International Fanconi
Anemia Registry study. Blood 1997; 90:105-10.
Jacobs P, Karabus C. Fanconi’s anemia. A family study with 20- year follow-up including
associated breast pathology. Cancer 1984; 54(9): 1850-53.

68
Joenje H, Lo ten Foe JR, Oostra AB, Van Berkel CG, Rooimans MA, Schroeder-Kurth T,
et al. Classification of Fanconi anemia patients by complementation analysis: evidence for
fifth genetic subtype. Blood 1995; 86:2156-60.
Joenje H, Oostra AB, Wijker M, Di Summa FM, Van Berkel CG, Rooimans MA, et al.
Evidence for at least eight Fanconi anemia genes. Am J Hum Genet 1997; 61:940-44.
Joenje H, Patel KJ .The emerging genetic and molecular basis of Fanconi Anemia. Nat Rev
Genet 2001; 2:446-57.
Jorde Lynn B, Carey John C, Bamshad Michael J, White Raymond L. Genética Médica. 3ª
edição. Ed. Elsevier. 2004; 415p.
Kaplan MJ, Sabio H, Wanebo HJ, Cantrell RW. Squamous cell carcinoma in the
immunosuppressed patient: Fanconi’s anemia. Laryngoscope 1985; 95:771-75.
Koc A, Pronk JC, Alikasifoglu M, Joenje H, Altay C. Variable pathogenicity of éxon 43del
(FAA) in four Fanconi anaemia patients within a consanguineous family. Br J Haematol
1999; 104:127-130.
Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, et al. A 20-year
perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003;
101(4):1249-56.
Levinson S, Vincent KA. Multifocal osteosarcoma in a patient with Fanconi anemia. J
Pediatr Hematol Oncol 1977; 19(3):251-53.
Levitus M, Rooimans MA, Steltenpool J, Cool NF, Oostra AB, Mathew CG, et al.
Heterogeneity in Fanconi anemia: evidence for two new genetic subtypes. Blood 2004;
103:2498-2503.

69
Levran O, Erlich T, Magdalena N, Gregory JJ, Batish SD, Verlander PC, et al. Sequence
variation in the Fanconi anemia gene FAA. Proceedings of the National Academy of
Sciences. 1997; 94:13051-56.
Linman JW. Myelomonocytic leukemia and its preleukemic phase. J Chronic 1970;
22(11):713-6.
Liu JM, Auerbach AD, Young NS. Fanconi anemia presenting unexpectedly in an adult
kindred with no dysmorphic features. Am J Med 1991; 91:555-57.
Lo Ten Foe JR, Kwee ML, Rooimans MA, Oostra AB, Veerman AJ, van Weel M, et al.
Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur J
Hum Genet 1997; 5:137-148.
Lustig JP. Head and neck carcinoma in Fanconi’s anaemia: report of a case and review of
the literature. Eur J Cancer B Oral Oncol 1995; 31B(1):68-72.
Magdalena NIR. Estudo das variações da sequência do gene FANCA da Anemia de
Fanconi. 1999. Tese (Doutorado) – Faculdade de Ciências Biológicas da Universidade
Federal do Paraná, Curitiba.
Magdalena N, Pilonetto DV, Bitencourt MA, Pereira NF, Ribeiro RC, Jeng M, et al.
Frequency of Fanconi anemia in Braziland efficacy of screening for the FANCA 3788-
3790del mutation. Brazilian Journal of Medical and Biological Research 2005; 38: 669-673
Meetei AR, De Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, et al.
A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 2003a; 35(2):165-170.
Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, et al. A multiprotein
nuclear complex connects Fanconi anemia and Bloom syndrome. Molecular and Cellular
Biology 2003b; 23: 3417-26.

70
Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, et al. X-linked
inheritance of Fanconi anemia complementation group B. Nat Genet 2004; 36:1219-24.
Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, et al.
Interaction of FANCD2 and NBS1 in the DNA damage response. Nature Cell Biology
2002; 4:913-20.
Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, et al. Human
Fanconi anaemia monoubiquitination pathway promotes homologous DNA repair. PNAS
2005; 102(4).
Nussbaum RL, McInnes RR, Willard HF. Thompson & Thompson Genética Médica. In:
Fundamentos de Citogenética Clínica. Rio de Janeiro: Guanabara Koogan; 2002. p.118-37.
Potter NU, Sarmousakis C, Li FP. Cancer in relatives of patients with aplastic anemia.
Cancer Genet Cytogenet 1983; 9:61-5.
Reuter TY, Medhurst AL, Waisfisz Q, Zhi Y, Herterich S, Hoehn H, Gross HJ, Joenje
H, Hoatlin ME, Mathew CG, Huber PA.Yeast two-hybrid screens imply involvement of
Fanconi Anemia proteins in transcription regulation, cell signaling, oxidative metabolism
and cellular transport. Exp Cell Res. 2003 Oct 1;289(2):211-21.
Rodriguez DEA. Estudo molecular da Anemia de Fanconi. 2003. Tese (Doutorado) –
Faculdade de Ciências Biológicas da Universidade Estadual de Campinas.
Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia.
Blood 2003;101:822–826.
Rosenberg PS, Huang Y. Individualized risks of first adverse events in patients with
Fanconi anemia. Blood 2004; Prepublished online 2004.
http://www.ncbi.nlm.nih.gov/pubmed/?term=Medhurst%20AL%5BAuthor%5D&cauthor=true&cauthor_uid=14499622

71
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, et al. Primer-directed
enzymatic amplification of DNA with thermostable DNA polymerase. Science 1989;
239:487-91.
Savino M, Borriello A, D'Apolito M, Criscuolo M, Del Vecchio M, Bianco AM, et al.
Spectrum of FANCA mutations in Italian Fanconi anemia patients: identification of six
novel alleles and phenotypic characterization of the S858R variant. Hum Mut 2003; 6:1-8.
Schaison G. Fanconi’s anemia. Incidence of its development intro leukemia. Presse Med
1983; 12(20):1269-74.
Schroeder TM, Anschutz F, Knopp A. Spontane chromosomen aberrationne bei familiäerer
panmyelopathie. Humangenetik 1964; 1:194-6.
Schroeder TM, Tilgen D, Krüger J, Vogel F. Formal genetics of Fanconi’s anemia. Hum
Genet 1976; 32(3):257-88.
Shimamura A. A novel diagnostic screen for detects in the Fanconi anemia pathway. Blood
2002; 100(13):4649-54.
Soulier J, Leblanc T, Larghero J, Dastot H, Shimamura A, Guardiola P, et al. Detection of
somatic mosaicism and classification of Fanconi Anemia patients by analysis of FA/BRCA
pathway. Blood 2005; 105:1329-36.
Stites DP, Terr AI, Parslow TG. Imunologia Médica. In: Atwater SK. Neoplasias do
Sistema Imune. Rio de Janeiro: Guanabara Koogan; 2000. p.502-23.
Strathdee CA, Gavish H, Shannon WR, Buchwald M. Cloning of cDNAs for Fanconi`s
anaemia by functional complementation. Nature 1992; 356:763-67.

72
Swift M. Fanconi’s anemia in the genetics of the neoplasia. Nature 1971; 230:370-73.
Swift M, Caldwell RJ, Chase C. Reassessment of cancer predisposition of Fanconi anemia
heterozygotes. J Natl Cancer Inst 1980; 65:863-67.
Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, et al.
Convergence of the Fanconi anaemia and ataxia telangiectasia signaling pathways. Cell
2002; 109:459-72.
The Rockefeller University. Fanconi Anemia Mutation Database. Disponível em: <
http://www.rockefeller.edu/fanconi/mutate/jumpg.html>. Acesso em: 16 jun. 2003.
Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox
B, Olson S, D'Andrea AD, Moses R, Grompe M. Positional cloning of a novel Fanconi
Anemia Gene, FANCD2. Molecular Cell 2001; 7:241-48.
Tipping AJ, Pearson T, Morgan NV, Gibson RA, Kuyt LP, Havenga C, et al. Molecular
and genealogical evidence for a founder effect in Fanconi Anemia families of the Afrikaner
population of South Africa. Proc Natl Acad Sci 2001; 98:5734-39.
Tischkowitz MD, Dokal I. Fanconi anaemia and leukaemia -clinical and molecular aspects
2004; 126:176-191.
Tischkowitz MD, Morgan NV, Grimwade D, Eddy C, Ball S, Vorechovsky I, et al.
Deletion and reduced expression of the Fanconi anemia FANCA gene in sporadic acute
myeloid leukemia. Leukemia 2004; 18:420-25.
Van de Vrugt HJ, Koomen M, Berns MA, De Vries Y, Rooimans MA, Van der Weel L, et
al. Characterization, expression and complex formation of the murine Fanconi anaemia
gene product Fancg. Genes to Cells 2002; 7(3):333-42.
http://www.ncbi.nlm.nih.gov/pubmed/?term=Taniguchi%20T%5BAuthor%5D&cauthor=true&cauthor_uid=11239453

73
Van Niekerk CH, Jordaan C, Badenhorst PN. Pancytopenia secondary to primary
malignant lymphoma of bone marrow as the first hematologic manifestation of Estren-
Dameshek variant of fanconi’s anemia. Eur J Pediatr 1987; 152(8):691-93.
Venkitaraman AR. A growing network of cancer-susceptibility genes. New England
Journal of Medicine 2003; 348:1917-19.
Venkitaraman AR. Tracing the network connecting BRCA and Fanconi anaemia proteins.
Nature Reviews Cancer 2004; 4:266-76.
Verlander PC, Kaporis A, Liu Q, Zhang Q, Seligsohn U, Auerbach AD. Carrier frequency
of the IVS4 + 4A→T mutation of the Fanconi Anemia gene FAC in the Ashkenazi Jewish
population. Blood 1995; 86:4034-38.
Wajnrajch MP, Gertner JM, Huma Z, Popovic J, Lin K, Verlander PC, et al. Evaluation of
growth and hormonal status in patients referred to the International Fanconi Anemia
Registry (IFAR). Pediatrics 2001; 107:744-54.
Wang X, D’Andrea A.D. The interplay of Fanconi anemia proteins in the DNA damage
response. DNA Repair 2004; 3:1063-69.
Witt E, Ashworth A. Biomedicine. D-Day for BRCA2. Science. 2002; 26;297.
Woodhead JL, Fallon R, Figueredo H, Langdale J, Malcom ADB. Alternative methodology
of genes diagnosis. In: DAVIES, K. E. Human genetics disease – a practical approach.
Oxford: IRL Press Limited, 1986, p. 51-54.
Yoshida MC. Suppression of spontaneous and mitomycin C-induced chromosome
aberrations in Fanconi's anemia by cell fusion with normal human fibroblasts. Hum Genet.
1980; 55(2):223-6.

74
Zakrzewski S, Sperling K. Genetic heterogeneity of Fanconi’s anemia demonstrated by
somatic cell hybrids. Hum Genet 1980; 56(1)81-84.
Zhang X, Li J, Sejas DP, Rathbun KR, Bagby GC, Pang Q. The Fanconi anemia proteins
functionally interact with the protein kinase regulated by RNA (PKR). The journal of
Biological Chemistry 2004; 279:43910-19.

75
8 ANEXOS
8.1 Parecer do Comitê de Ética e Pesquisa

76

77
8.2. Termo de Consentimento Livre e Esclarecido
Termo de Consentimento
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
Título do projeto: Estudo molecular do gene FANCA em pacientes com quadro clínico
de Anemia de Fanconi
Eu entendo que fui convidado a participar em um projeto de pesquisa
envolvendo indivíduos com Anemia de Fanconi. O objetivo geral desse trabalho será
estudar as mutações do gene FANCA, em pacientes com quadro clínico de Anemia de
Fanconi.
Se concordar em participar desse estudo, os pesquisadores participantes farão
perguntas a respeito dos meus antecedentes médicos e familiares. Será coletada uma
amostra de sangue venoso (7 ml, o equivalente a 6 colheres de sopa) dos participantes. Os
riscos associados a esse procedimento são mínimos, podendo ocorrer dor e manchas roxas
(equimoses) no local da coleta do sangue. O desconforto será mínimo, pois se trata de uma
coleta de sangue geralmente da veia do braço que será realizado por profissional treinado e
devidamente habilitado para realizar esse procedimento.
A única vantagem que poderei ter é saber se há alguma alteração no gene FANCA
ou nos 43 éxons do mesmo gene. No entanto fui informado (a) que se for detectada alguma
alteração, serei imediatamente comunicado(a), sendo que todas as consequências serão
devidamente explicadas a meus parentes próximos, e se assim desejarem, poderão realizar
Universidade Estadual de Campinas
Departamento de Genética Médica

78
o exame. Qualquer dúvida ou informação poderei contatar a UNICAMP no tel. (019)
3521-8907 (Dra: Claudia Estela Gonçalves).
Eu entendo que toda informação médica, assim como os resultados dos testes
genéticos decorrentes desse projeto de pesquisa, serão sigilosos. O sigilo será mantido em
todo o estudo através da utilização de um número de código para a identificação dos
indivíduos participantes. Se os resultados ou informações fornecidas forem utilizados para
fins de publicação científica, nenhum nome será utilizado. Fui informado que poderei
autorizar que esse material genético seja guardado e que qualquer outro projeto que
pretenda utilizá-lo, deverá ser aprovado pelo Comitê de Ética em Pesquisa da FCM. Eu
estou ciente que a minha participação neste projeto de pesquisa não me acarretará nenhuma
despesa, já que a coleta de material será realizada em minhas consultas regulares à
Unicamp.
Eu entendo que a minha participação é voluntária e que eu posso me recusar a
participar ou retirar meu consentimento e interromper a minha participação no estudo a
qualquer momento (incluindo a retirada da amostra de sangue) sem comprometer os
cuidados médicos que minha família recebe atualmente ou receberei no futuro no HC-
UNICAMP.
Em caso de recurso ou reclamações contatar a secretaria do Comitê de Ética da
Faculdade de Ciências Médicas - UNICAMP, tel. (019) 3521-8936.
Eu confirmo que a Dra. Claudia Estela Gonçalves, explicou-me o objetivo do
estudo, os procedimentos aos quais serei submetido e os riscos ou desconfortos advindos
desse projeto de pesquisa. Eu li e/ou me foi explicado, assim como compreendi esse
formulário de consentimento e estou de pleno acordo em participar desse estudo.
__________________________________________________________________
Nome e RG do sujeito da pesquisa
__________________________________________________________________
Nome e RG do responsável legal

79
_______________________________________ ______________
Assinatura do sujeito da pesquisa Data
_______________________________________ ______________
Assinatura do responsável legal Data
AUTORIZAÇÃO PARA ARMAZENAMENTO DA AMOSTRA
Eu concordo que o DNA de minha família seja estocado
Eu não concordo que o DNA de minha família seja estocado
RESPONSABILIDADE DO PESQUISADOR:
Eu expliquei a _______________________________________________ o objetivo do estudo, os procedimentos requeridos e os possíveis riscos que poderão advir do estudo, usando o melhor do meu conhecimento. Eu
me comprometo a fornecer uma cópia desse formulário de consentimento ao participante ou responsável.
__________________________________________________________________
Nome e RG do pesquisador
________________________________________ ______________
Assinatura do pesquisador Data


















