
“ANÁLISE DE HIDROCARBONETOS POLICÍCLICOS …
95
“ANÁLISE DE HIDROCARBONETOS POLICÍCLICOS AROMÁTICOS – HPAs – EM SEDIMENTOS DO RIBEIRÃO DO FUNIL NA REGIÃO DE OURO PRETO – MG” WESLEY ROBERT DE SOUZA Ouro Preto, MG 2007 UNIVERSIDADE FEDERAL DE OURO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM RECURSOS HÍDRICOS MESTRADO EM ENGENHARIA AMBIENTAL
Transcript of “ANÁLISE DE HIDROCARBONETOS POLICÍCLICOS …

“ANÁLISE DE HIDROCARBONETOS POLICÍCLICOS
AROMÁTICOS – HPAs – EM SEDIMENTOS DO RIBEIRÃO DO
FUNIL NA REGIÃO DE OURO PRETO – MG”
WESLEY ROBERT DE SOUZA
Ouro Preto, MG 2007
UNIVERSIDADE FEDERAL DE OURO PRETO
PROGRAMA DE PÓS-GRADUAÇÃO EM RECURSOS HÍDRICOS
MESTRADO EM ENGENHARIA AMBIENTAL

II
Catalogação: [email protected]
S729a Souza, Wesley Robert de.
Análise de hidrocarbonetos policíclicos aromáticos – HPAs – em sedimentos do Ribeirão do Funil na região de Ouro Preto – MG [manuscrito] / Wesley Robert de Souza – 2007. xiv, 79 f. : il. color., graf., tabs., mapas. Orientador: Prof. Dr. Robson José de Cássia Franco Afonso. Co-orientador: Prof. Dr. Cornélio de Freitas Carvalho. Dissertação (Mestrado) - Universidade Federal de Ouro Preto. Instituto de Ciências Exatas e Biológicas. Programa de Mestrado em Engenharia Ambiental. Área de concentração: Recursos hídricos.
1. Hidrocarbonetos policíclicos aromáticos - Teses. 2. Sedimentos fluviais - Ouro Preto (MG) - Teses. 3. Metais - Teses. I. Universidade Federal de Ouro Preto. II. Título. CDU: 547.53(815.1)

III
WESLEY ROBERT DE SOUZA
“ANÁLISE DE HIDROCARBONETOS POLICÍCLICOS AROMÁTICOS –
HPAs – EM SEDIMENTOS DO RIBEIRÃO DO FUNIL NA REGIÃO DE OURO
PRETO – MG”
Ouro Preto, MG
2007
Dissertação apresentada ao Programa de Pós-Graduação
em Recursos Hídricos, Universidade Federal de Ouro
Preto, como parte dos requisitos necessários para a
obtenção do título: “Mestre em Engenharia Ambiental –
Área de Concentração: Recursos Hídricos”
ORIENTADOR: Prof. Dr. Robson José de Cássia Franco Afonso
CO-ORIENTADOR: Prof. Dr. Cornélio de Freitas Carvalho

IV

V
"O único homem que está isento de erros, é aquele que não arrisca acertar"
(Einstein)

VI
Dedicatória
Aos meus pais, Valdir e Lourdes,
sempre presentes em tudo: Obrigado! Ao tio
Pedro pelas grandes lições de vida deixadas a todos.

VII
Agradecimentos
A Deus, pelo grande presente: a vida.
Ao Prof. Dr. Robson J.C.F. Afonso, pela orientação.
Ao Prof. Dr. Cornélio de F. Carvalho, pela co-orientação e pelo seu grande empenho
como coordenador do curso de mestrado.
Aos Professores Doutores: Jorge C. de Lena, Kátia M. Novack, Jorge L. Humberto,
Vagner R. Botaro, Cláudio G. dos Santos, Laurent F. Gil pelo apoio.
Ao Prof. Dr. Maurício Xavier Coutrim pelas orientações e colaborações.
Aos colegas do Laboratório de Análise de Águas, em especial ao Carlúcio e a Su pelo
apoio nos dias de coleta.
Aos professores responsáveis, técnicos e alunos dos seguintes laboratórios da UFOP:
LBCM, LGqA, LPM, LAMP e LABQOC, pelos equipamentos, análises, reagentes e
vidrarias disponibilizados para execução deste trabalho.
Aos meus pais, que sempre me apoiaram na luta pelos meus sonhos, meu porto seguro.
Aos meus irmãos, sobrinhos e toda família, por me acompanhar ao longo desta
trajetória.
À minha “boneca” Ana Luíza, que na graça de sua ingenuidade me proporciona
momentos de muita alegria e gosto pela vida. Papai te ama!
À minha “Linda Pequenina” pelo companheirismo, amor, carinho, amizade, dedicação,
compreensão, cumplicidade, paciência e pelos momentos felizes juntos. Amo Você!
Ao meu grande amigo Tio Pedro, que pelos inexplicáveis acontecimentos da vida nos
deixou e não vai poder estar presente neste momento. Saudades eternas!
Ao amigo-irmão Msc. Fausto Junior, pelos anos de convivência e eterna amizade.
Ao inesquecível amigo Leandro Martins “menino do mal”, que me apoiou bastante
neste trabalho e pela sua sincera amizade. Valeu mesmo! “Eu quero!”
À família Saideirana, que mais uma vez me acolheu com muito carinho.
Aos amigos: Fernanda Fernandes, Aniel Lima, Prof. Fred, Prof.ª Beth, Dani,
Hassegawa, Liana, “Feroz”, Ivan e Luciano pela amizade e momentos de descontração.
A CAPES pelos meses de bolsa de estudo e a todos que de alguma forma colaboraram
para a concretização deste sonho.
Muitíssimo obrigado!

VIII
Resumo
As águas de rios, córregos e lagos da região de Ouro Preto têm sido alvo de vários
estudos por diversos pesquisadores. O sistema hídrico desta região está sujeito à
contaminação por atividades mineradoras, industriais e outras formas antrópicas. Além
disso, sabe-se que ainda não se encontra na cidade um sistema eficiente de tratamento
da água antes de torná-la disponível ao consumo humano.
Neste trabalho, foram determinados e quantificados 11 Hidrocarbonetos Policíclicos
Aromáticos (HPAs) em amostras de sedimentos e foi feita uma avaliação dos
parâmetros físico-químicos nas amostras aquosas do Ribeirão do Funil. Foram definidos
4 pontos de amostragem sendo dois deles (pontos 1 e 2) à montante e dois ( pontos 3 e
4) à jusante de uma fábrica de alumínio localizada na cidade de Ouro Preto. Foram
realizadas 3 campanhas de amostragem durante o estudo sendo duas em época de seca e
uma em período chuvoso.
As amostras de sedimento foram submetidas à extração soxhlet e os extratos obtidos
concentrados em um sistema Kuderna-Danish (US-EPA METHOD 3540C).
Previamente à análise, foi feito o “clean-up” das amostras em uma coluna de sílica gel
ativada, conforme (US-EPA METHOD 3630 C). As amostras foram analisadas por
Cromatografia Líquida de Alta Eficiência – CLAE, com uma coluna C-18 de fase
reversa e detector de fluorescência. Nas amostras aquosas, foi feito um estudo da
qualidade das águas do ribeirão através da avaliação físico-química e metais.
Foram quantificados 11 HPAs, sendo que a maior concentração nas 3 coletas foi de
benzo(b)fluoranteno (3679,05 ng/g). Em algumas amostras encontrou-se benzo(a)pireno
em concentrações de até 712 ng/g de sedimento e sabe-se que este HPA apresenta alto
potencial carcinogênico. O ponto 3 foi o mais crítico na contaminação por HPAs e os
elevados teores destes contaminantes podem estar relacionados com a proximidade da
fábrica de alumínio, pois algum material contaminado com HPAs pode estar sendo
lançado nas águas do ribeirão por arraste das águas da chuva na lavagem do pátio e dos
telhados da fábrica ou até mesmo por lançamento de efluentes contaminados, que
posteriormente vão contaminar os sedimentos. Os resultados do estudo físico-químico
apontaram a baixa qualidade das águas quando comparados com os limites
estabelecidos pela CONAMA 357 (Março 2005) para águas de classe 1 e identificou-se

IX
a contaminação por metais de alta toxicidade, como arsênio em concentrações de até
39,08 µg/L e chumbo (160,6 µg/L).
A avaliação de HPAs em sedimentos desta região é inédita e pode ser considerada
de grande importância, pois estes contaminantes são conhecidos por serem
carcinogênicos e a presença do benzo(a)pireno em algumas amostras confere sérios
riscos de toxicidade que comprometem a qualidade ambiental da área estudada.

X
Abstract
The waters from rivers, streams and lakes from Ouro Preto region had been studied
by various researches. The hydric system of this region is subjected to contamination
from mining activities, industries and other anthropic sources. In spite of this, Ouro
Preto thus not have a proper water treatment plant.
In this work, were determined and quantified eleven Polycyclic Aromatic
Hydrocarbons (PAHs) in sediment samples and were evaluated the physico-chemical
proprieties of aqueous samples from the Ribeirão do Funil. The sampling points were
defined in four places along the stream. Two of them, were located upstream to the
aluminum smelting plant and two downstream to it. Those last two points are also
located after the urban center of Ouro Preto. The samples were collect in three different
sampling campaigns.
The sediment samples were subjected to soxhlet extraction and the extracted
produced was concentrated using Kuderna_Danish (US-EPA METHOD 3540C). The
extracts were clean-uped trough an activated silica gel column in accordance to the US-
EPA METHOD 3630 C. The samples were analyzed by High Performance Liquid
Chromatography (HPLC), using a C-18 reverse phase column and detected by
fluorescence emission spectrometry. For the aqueous samples were evaluated their
quality trough the physico-chemical parameters and metal contents.
For the quantified 11 PAHs, the highest concentration for the three campaigns
were found for the benzo(b)fluoranthene. In some samples was found benzo(a)pyrene,
knew for the high carcinogenic power. The sampling point just below the aluminum
smelting plant presented the highest total PAHs content, indicating the possible source
of contamination, since these kind of industries rely on fossil fuels in their process. The
physico-chemical analyses showed the pour quality for the aqueous samples which
presented high content of toxic metals such as arsenic and lead.
The evaluation of PAHs contents into sediments of this region is unknown, and
the results obtained should be considered for its importance, since these compounds are
well know for their high carcinogenesis. The presence of benzo(a)pyrene in some
samples indicate the risk for the human health exposed to this environment.

XI
Índice
RESUMO............................................................................................................... VIII
ABSTRACT................................................................................................................X
LISTA DE FIGURAS............................................................................................ XIII
LISTA DE TABELAS.............................................................................................XV
LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS ..................................... XVI
1. INTRODUÇÃO .................................................................................................1
2. OBJETIVOS ......................................................................................................3
3. REVISÃO BIBLIOGRÁFICA ..........................................................................4
3.1 - Hidrocarbonetos Policíclicos Aromáticos...........................................................4 3.1.1 – Técnicas de Extração dos HPAs .................................................................................................. 9 3.1.2 – Extração Soxhlet .........................................................................................................................12
3.2 - Avaliação Química das Águas...........................................................................12 3.2.1 – Parâmetros Físico-Químicos .......................................................................................................12 3.2.2 – Coleta das Amostras ...................................................................................................................12 3.2.3 – Metais .........................................................................................................................................13
4. CARACTERIZAÇÃO DA ÁREA DE ESTUDO ...........................................14
5. PARTE EXPERIMENTAL.............................................................................16
5.1 – Avaliação dos Hidrocarbonetos Policíclicos Aromáticos em Sedimentos.......16 5.1.1– HPAs Analisados .........................................................................................................................16 5.1.2 – Coleta das Amostras de Sedimento.............................................................................................17 5.1.3 – Secagem das Amostras de Sedimento.........................................................................................17 5.1.4 – Preparo das Amostras para Extração...........................................................................................18 5.1.5 – Extração Soxhlet .........................................................................................................................20 5.1.6 – Concentração no Kuderna-Danish (KD) .....................................................................................21 5.1.7 – Purificação dos extratos - Clean-up ............................................................................................22 5.1.8 – Condições Cromatográficas ........................................................................................................24 5.1.9 – Preparo da Solução-Padrão de HPAs..........................................................................................27 5.1.10 – Curvas Analíticas ......................................................................................................................27 5.1.11 - Cálculo do Índice de Recuperação.............................................................................................29
5.2 – Avaliação dos Parâmetros Físico-Químicos da Àgua......................................31 5.2.1 – Preparo dos Frascos de Coleta ....................................................................................................31 5.2.2 – Coleta das Amostras de Água .....................................................................................................31

XII
5.2.3 – Parâmetros Analisados................................................................................................................31 5.2.4 – Medidas de Vazão.......................................................................................................................32
5.3 – Avaliação dos Metais e Outros Elementos Químicos em Água.......................34 5.3.1 – Preparo dos Frascos de Coleta ....................................................................................................34 5.3.2 – Coleta das Amostras de Água .....................................................................................................34
6. RESULTADOS E DISCUSSÃO .....................................................................36
6.1 – Amostras Aquosas ............................................................................................36 6.1.1- Análises Físico-Químicas .............................................................................................................36 6.1.2- Análises de Metais e Outros Elementos Químicos .......................................................................38
6.2 – Avaliação dos Hidrocarbonetos Policíclicos Aromáticos ................................41 6.2.1 – Curvas Analíticas ........................................................................................................................41 6.2.2 – Cálculo do Índice de Recuperação..............................................................................................45 6.2.3 – HPAs nas Amostras Ambientais .................................................................................................48
7. CONCLUSÕES................................................................................................65
8. SUGESTÕES PARA TRABALHOS FUTUROS:..........................................67
9. REFERÊNCIAS BIBLIOGRÁFICAS............................................................69
10. ANEXOS ...........................................................................................................77

XIII
LISTA DE FIGURAS
Figura 1 - Estrutura dos 16 HPAs incluídos na lista de poluentes prioritários da EPA ....8
Figura 2 - Imagem de satélite mostrando a localização dos 4 pontos de Coleta no
Ribeirão do Funil .................................................................................................15
Figura 3 - Secagem dos sedimentos em capela fechada com aquecimento por lâmpadas
de infravermelho 250 watts ...................................................................................18
Figura 4 - Sedimento seco (4 pontos de coleta) ............................................................19
Figura 5 - Sedimento triturado e peneirado (4 pontos de coleta)...................................19
Figura 6 - Sedimento Quarteado (pronto para extração soxhlet) ...................................20
Figura 7 - (1) Esquema de um aparelho de soxhlet convencional e (2) experimento
realizado nas amostras de sedimento .....................................................................21
Figura 8 - Aparelho de Concentração das Amostras (KD)............................................22
Figura 9 - Fluxograma das etapas de preparação das amostras .....................................24
Figura 10 - Diagrama da Programação da Fase Móvel .................................................26
Figura 11 - Cálculo da Vazão (flutuador).....................................................................34
Figura 12 - Identificação dos 11 Padrões e tempos de retenção dos HPAs....................43
Figura 13 - Curvas Analíticas: Acenafteno+fluoreno, Fenantreno, Fluoranteno e
Benzo(a)pireno .....................................................................................................43
Figura 14 - Curvas Analíticas: Antraceno, Naftaleno e Benzo(b)fluoranteno ...............44
Figura 15 - Curvas Analíticas: Pireno e Criseno...........................................................44
Figura 16 - Curva Analítica do Benzo(k)fluoranteno ...................................................45
Figura 17 - Concentração dos HPAs na primeira coleta ...............................................52
Figura 18 - Concentração dos HPAs na segunda coleta................................................53
Figura 19 – Concentração dos HPAs na terceira coleta ................................................54
Figura 20 - Cromatograma de uma amostra de sedimento da 2º coleta (Ponto 2)
analisada por CLAE, detector de fluorescência......................................................55
Figura 21 - Concentração total dos HPAs nos pontos amostrados ................................59
Figura 22 - Concentração total dos HPAs nas 3 coletas................................................60
Figura 23 – Perfil da variação de concentração dos HPAs nos 4 pontos de coleta ........61
Figura 24 - Valores da razão BMM/AMM para HPAs nos sedimentos do Ribeirão do
Funil .....................................................................................................................63

XIV
Figura 26 - Cromatograma de uma amostra do ponto 2 (2º Coleta) ..............................77
Figura 27 - Cromatograma da mistura de Padrões HPAs para Curvas Analíticas..........77
Figura 28 – Cromatograma de uma amostra de sedimento do ponto 2 (1º Coleta) ........78

XV
LISTA DE TABELAS
Tabela 1 - Distribuição dos 4 pontos de coleta .............................................................15
Tabela 2 - Características dos 11 HPAs estudados .......................................................16
Tabela 3 - Dados das 3 Coletas de Sedimentos ............................................................17
Tabela 4 - Parâmetros das Análises Cromatográficas ...................................................25
Tabela 5 - Comprimentos de onda de excitação e emissão para os 11 compostos
avaliados...............................................................................................................26
Tabela 6 - Concentração, em µg/mL, dos HPAs nas doze soluções preparadas para as
curvas analíticas....................................................................................................28
Tabela 7 - Parâmetros, tempos de análise, preservação e quantidades das amostras .....32
Tabela 8 - Parâmetros das análises por ICP-OES .........................................................35
Tabela 9 - Resultados das análises físico-químicas de água..........................................37
Tabela 10 - Concentração dos Metais nas Amostras Aquosas do Ribeirão do Funil .....40
Tabela 11 - Concentrações dos HPAs na solução inicial ..............................................41
Tabela 12 - Coeficientes gerados nas curvas de calibração...........................................42
Tabela 13 - Concentração dos padrões de HPAs adicionados ao sedimento nas duas
contaminações ......................................................................................................46
Tabela 14 - Recuperação das extrações soxhlet para o spike 1 .....................................46
Tabela 15 - Recuperação das extrações soxhlet para o spike 2 .....................................47
Tabela 16 - Resultados da primeira coleta....................................................................49
Tabela 17 - Resultados das replicatas para a segunda coleta.........................................50
Tabela 18 - Resultados das replicatas para a terceira coleta..........................................51
Tabela 19 - Principais HPAs emitidos por algumas fontes . .........................................56
Tabela 20 - Concentrações totais dos 11 HPAs ............................................................58
Tabela 21 - Concentração total (ng/g) dos HPAs isolados............................................59
Tabela 22- Valores das concentrações (ng/g) dos HPAs para relação BMM/AMM......62
Tabela 23 - Valores das razões entre fenantreno/antraceno e fluoranteno/pireno ..........63
Tabela 24 - Concentrações das replicatas para o branco (teste de validação) ................78
Tabela 25 - Concentrações das replicatas para o Spike 1 ..............................................79
Tabela 26 - Concentrações das replicatas para o Spike 2 ..............................................79

XVI
LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS
ACN – Acetonitrila
AMM - Alta Massa Molecular
BMM - Baixa Massa Molecular
CLAE – Cromatografia líquida de alta eficiência
CONAMA – Conselho Nacional de Meio Ambiente
Em – emissão
Ex – excitação
HPAs – Hidrocarbonetos Policíclicos Aromáticos
ICP-OES – Inductively Coupled Plasma - Optical Emission Spectrometric
INMETRO – Instituto Nacional de Metrologia, Normalização e Qualidade Industrial
KD – Kuderna-Danish
ld – limite de detecção
ng/g – nanograma por grama
NTU – Unidade Nefelométrica de Turbidez
OD – oxigênio dissolvido
PCBs – Bifenilas policloradas
PET – poli-tereftalato de etileno
ppb – partes por bilhão
ppm – partes por milhão
US-EPA – United States Environmental Protection Agency
UV – Ultravioleta
λ – comprimento de onda

1
1. INTRODUÇÃO
Neste trabalho foram desenvolvidos e utilizados métodos analíticos para
avaliação de Hidrocarbonetos Policíclicos Aromáticos - HPAs - em amostras de
sedimentos do Ribeirão do Funil, pertencente ao sistema hídrico da região de Ouro
Preto – MG. As metodologias envolveram extração soxhlet, concentração no Kuderna-
Danish, clean-up com sílica gel, identificação e quantificação destes compostos por
Cromatografia Líquida de Alta Eficiência – CLAE, com coluna C-18 de fase reversa e
detecção por fluorescência. Foi feita ainda uma avaliação da qualidade das águas do
ribeirão, através de um estudo físico-químico das amostras aquosas.
A água, devido às suas propriedades de solvente e à sua capacidade de
transportar partículas, incorpora a si diversas impurezas, as quais definem a sua
qualidade.
A qualidade da água é resultante de fenômenos naturais e da atuação do homem.
A interferência do homem quer de uma forma concentrada, como na geração de
despejos domésticos ou industriais, quer de uma forma dispersa, como na aplicação de
defensivos agrícolas no solo, contribui na introdução de compostos na água, afetando
sua qualidade.
A água é o constituinte inorgânico mais abundante na matéria viva: no homem,
mais de 60% do seu peso é constituído por água, em certos animais aquáticos esta
porcentagem chega a 98%. A água é fundamental para a manutenção da vida.
No planeta, a água se distribui da seguinte forma: 97% água do mar, 2,2%
geleiras e 0,8% água doce. Desses 0,8%, 97% da água doce se encontra subterrânea e
apenas 3% superficial, ou seja, disponível para uso imediato e que pode ser mais
facilmente utilizada no abastecimento publico, pois sua extração é mais fácil. Daí, a
grande importância de se preservar os recursos hídricos na Terra (VON SPERLING,
1996). A qualidade da água requer a satisfação de diversos critérios para garantir a
preservação da saúde dos seres que a consome.
O propósito primário para a exigência de qualidade da água é a proteção à saúde
pública. Quase invariavelmente, o melhor método de assegurar água adequada para
consumo consiste em formas de proteção, evitando-se contaminações de dejetos animais

2
e humanos, os quais podem conter grande variedade de bactérias, vírus, protozoários e
helmintos (d’AGUILA et al., 2000).
A maior parte da água utilizada nos mais diversos processos retorna ao corpo de
água com algum tipo de poluente incorporado, quando não se faz um tratamento antes
do seu lançamento. Essa descarga de água contaminada altera todas as características de
qualidade do corpo receptor após receber o efluente. Os poluentes são responsáveis
diretos pela alteração na qualidade química, física e biológica do meio.
A contaminação que vem ocorrendo ao longo dos anos causada pelo
desenvolvimento industrial, crescimento demográfico e ocupação do solo de forma
intensa e acelerada tem provocado o comprometimento dos recursos hídricos
disponíveis para o consumo humano, aumentando consideravelmente o risco de doenças
de transmissão ou de origem hídrica (NEVES, 2003).
Os ecossistemas aquáticos acabam, de uma forma ou de outra, servindo como
reservatórios temporários ou finais de uma grande variedade e quantidade de poluentes
lançados no ar, no solo ou diretamente nos corpos de água. Desta forma, a poluição do
ambiente aquático, provocada pelo homem, de uma forma direta ou indireta, através da
introdução de substâncias inorgânicas ou orgânicas, produz efeitos deletérios tais como:
I) prejuízo aos seres vivos, II) perigo à saúde humana, III) efeitos negativos às
atividades aquáticas e IV) prejuízo à qualidade da água com respeito ao uso na
agricultura, indústria e outras atividade econômicas. A qualidade do ambiente aquático
pode ser determinada através de medidas quantitativas, como determinações físicas e
químicas ou através de medidas semiquantitativas e qualitativas, realizadas no campo ou
no laboratório e produzem vários tipos de informações, fornecendo diferentes
interpretações técnicas (RODRÍGUEZ, 2001).
Atualmente, grandes volumes de água estão sendo utilizados pelas indústrias,
pelo poder público e para irrigação de culturas. O resultado final destas atividades é um
impacto na estrutura física, química e biológica dos rios, lagos e represas, reduzindo o
potencial desses sistemas em oferecer condições para usos múltiplos. No Brasil, os rios
ainda são um sinônimo de degradação. Esse quadro de degradação teve inicio a partir
dos anos 50, com a intensificação do crescimento econômico, vinculado à
industrialização rápida e irrestrita, não levando em consideração os desequilíbrios de
ordem social, econômica e ambiental (SILVA, 2002).

3
Avaliação da presença de contaminantes orgânicos e inorgânicos em sedimentos
depositados em corpos d´água, juntamente com a avaliação de parâmetros físico
químicos da qualidade da água propicia informações importantes sobre o nível, bem
como a origem desta de contaminação.
2. OBJETIVOS
– Objetivo Geral:
O objetivo do presente trabalho foi avaliar a presença de hidrocarbonetos
policíclicos aromáticos em sedimentos, em quatro pontos de coleta, do Ribeirão do
Funil, na cidade de Ouro Preto – MG e identificar possíveis fontes de contaminação
destes compostos.
– Objetivos Específicos:
� Desenvolver uma metodologia analítica para extração, concentração,
identificação e quantificação de HPAs em amostras de sedimentos;
� Avaliar a qualidade das águas do Ribeirão do Funil, através da avaliação de
alguns parâmetros físico-químicos como: alcalinidade, cloreto, condutividade,
cor, dureza, nitrato, nitrito, resíduos sedimentáveis, sulfato, amônia, turbidez,
OD e pH sendo feita também a análise de metais;
� Desenvolvimento de uma metodologia para as análises cromatográficas, capaz
de quantificar os compostos de interesse (HPAs) através da cromatografia
líquida de alta eficiência (CLAE);
� Buscar identificar possíveis fontes de contaminação por HPAs no Ribeirão do
Funil.

4
3. REVISÃO BIBLIOGRÁFICA
3.1 - Hidrocarbonetos Policíclicos Aromáticos
Hidrocarbonetos Policíclicos Aromáticos (HPAs) são uma classe complexa de
compostos orgânicos contendo dois ou mais anéis aromáticos juntos e contendo apenas
carbono e hidrogênio como constituintes. As propriedades físicas e químicas dos HPAs
são determinadas pelo seu sistemas de elétrons Π, os quais dependem do número de
anéis aromáticos e da massa molar. Dentre eles, os naftalenos são os compostos de
menor massa molar contendo apenas dois anéis e são encontrados na atmosfera
normalmente na fase vapor.
Os HPAs são contaminantes difundidos no ambiente que surgem principalmente
de fontes antropogênicas como a combustão de produtos fósseis e liberação direta de
óleos e produtos oleosos. A difusão ambiental destes contaminantes tem sido
intensivamente estudada devido as suas propriedades mutagênicas e carcinogênicas
(BAUMARD etal, 1998). Eles são liberados na atmosfera como uma mistura complexa
de compostos durante a combustão incompleta de matérias orgânicas. Eles podem ser
emitidos pela queima de madeiras em fogões e lareiras, fotocopiadoras, queima de
carvão, fuligem de chaminé, exaustão de incineração de plantas e rejeitos, queima de
rejeitos na agricultura, descarga de veículos automotores, fumaça de cigarro, aplicações
de asfalto, etc. (POPPI, 2000; RIBEIRO, 2001) . Os HPAs são poluentes comuns
conhecidos por promover a formação de moléculas carcinogênicas em organismos
vivos, e se bioacumulam na cadeia alimentar. São formados quando materiais contendo
carbono são queimados incompletamente, na produção de derivados de alcatrão do
carvão, e também entram no ambiente como resultado do derramamento de óleos de
tanques, refinarias e de abertura de sítios de exploração de óleos. Estão presentes em
todos os meios: atmosfera, solos e água (CAPELO et al, 2005)
Embora no ambiente existam centenas de HPAs a US-EPA recomenda a
determinação e quantificação de 16 hidrocarbonetos policíclicos aromáticos, sendo
prioritários e devem ser rotineiramente monitorados (SUN et al., 1998, AZEVEDO et
al., 2004). O benzo(a)pireno, um HPA de cinco anéis, foi identificado como sendo
altamente carcinogênico (COMPENDIUM METHOD TO-13A, 1999). Apesar de

5
existirem centenas de HPAs identificados em materiais particulados na atmosfera, estão
disponíveis apenas os dados sobre toxidade a exposição de cerca de 33 compostos.
Os HPAs são os carcinógenos químicos mais potentes e os mais estudados.
Todos são cancerígenos indiretos dependendo da ativação pelo sistema enzimático.
Provocam tumores variados, conforme o local de introdução e as células presentes. Os
hidrocarbonetos aromáticos derivam dos núcleos do antraceno e do fenantreno.
Dependendo do tipo e local das modificações químicas das moléculas, a potência do
produto resultante é diferente. Um mecanismo de ação mutagênica desse grupo de
compostos cíclicos pode ser explicado por suas propriedades hidrofóbicas e planar,
através da qual se intercalam entre as bases do DNA; distorções provocadas na estrutura
dupla hélice do DNA facilitam a ocorrência de inserções/deleções no momento da
replicação (BOGLIOLO, 1998).
Substâncias químicas não seguras ou presumivelmente cancerígenas encontram-
se amplamente distribuídas na natureza e compreendem desde alimentos naturais até
compostos altamente modificados pelo homem. Muitos carcinógenos químicos têm
interesse apenas na carcinogênese experimental, outros são causa importante de
cânceres humanos. Eles são divididos em carcinógenos diretos e indiretos. Os diretos
são agentes alquilantes que possuem atividade eletrofilica intrínseca, podendo provocar
câncer diretamente. A maioria das substâncias cancerígenas precisa sofrer modificações
químicas no organismo para se tornarem eletrofilicas e ativas, que é o caso dos
carcinógenos indiretos. Os carcinógenos químicos diretos ou indiretos se ligam ao DNA
e causam mutações. O principal mecanismo de ação é a formação de compostos
covalentes com o DNA, que aumentam a probabilidade de ocorrerem erros durante a
replicação. Como o organismo dispõe de sistemas eficazes de reparação do DNA, nem
sempre uma mutação leva a formação de tumores. Existe grande variação entre os
indivíduos e entre os diferentes tecidos na reparação do DNA. Alguns cancerígenos
químicos (certos aldeídos, agentes alquilantes), além de sua ação mutagênica, podem
inibir a atividade de enzimas reparadoras. Então, pode-se dizer que substâncias
químicas provocam tumores na dependência de vários fatores do indivíduo e do
ambiente.
O processo de mutagênese está intimamente relacionado com a carcinogênese.
Quando uma substância química interage com o material genético, aparecem as

6
mutações, ou seja, alterações transmissíveis no material genético. A carcinogênese é
mais complexa e envolve duas etapas principais: a iniciação e a promoção. A iniciação
caracteriza-se pela ocorrência de uma mutação e a promoção pelo crescimento celular
intenso e desordenado das células iniciadas ou mutantes originando focos de
proliferação dos quais alguns se desenvolvem em tumores. A atividade carcinogênica de
um composto particular é dependente de vários fatores estruturais da molécula, como
modelo (forma), tamanho e fatores estéricos, e pode estar relacionada com o potencial
de ionização. A carcinogenicidade dos HPAs tem sido observada principalmente nos
compostos tri-, tetra-, penta- e hexacíclicos (POPPI, 2000). Além das propriedades
mutagênicas e carcinogênicas, os HPAs apresentam elevada hidrofobicidade e
resistência a biodegradação (BUSSETI et al., 2005).
A contaminação humana se dá geralmente através da contaminação ambiental,
pela inalação ou absorção pela pele, ou ingestão de alimentos contaminados tanto pelo
processo de cozimento, que pode gerar HPAs, quanto pela deposição atmosférica. Os
compostos aderidos às partículas são parcialmente dissolvidos e metabolizados nos
pulmões. Os metabólitos podem atuar sobre as células, ou entrar na corrente sangüínea e
chegar ao fígado, ou pode ser excretado sem passar por qualquer modificação, pela bílis.
Apenas uma pequena parte dos compostos é eliminada com a urina na forma
metabolizada ou na forma original. Os efeitos carcinogênicos dos HPAs são muito
conhecidos, principalmente na pele e pulmão, e têm sido observados em pessoas
expostas às inúmeras fontes emissoras existentes. O mecanismo de ação carcinogênica
dos HPAs é complexo, e envolve uma série de enzimas que catalisam reações de
oxidação, redução e hidrólise, pois a substância em si não é ativa e sim seus metabólitos
finais (RIBEIRO, 2001).
Os HPAs encontram-se muito difundidos no ambiente e sua importância prática
como causa de câncer é grande. Hoje eles apresentam o risco elevado devido à
multiplicidade das fontes de produção desses compostos, o consumo crescente de
alimentos industrializados e a generalização do hábito de fumar, um número cada vez
maior de indivíduos fica exposto a esse tipo de substância (BOGLIOLO, 1998).
Os hidrocarbonetos policíclicos aromáticos ocorrem largamente no meio
ambiente e podem ser formados durante a defumação de alguns alimentos. Substâncias
orgânicas apresentam aspecto cumulativo na cadeia alimentar e as aromáticas

7
complexas possuem baixa taxa de excreção corporal. Os HPAs são agentes
carcinogênicos humanos muito potentes. Eles ocorrem no solo, na água e nos alimentos,
naturais ou cozidos. Inicialmente foram encontrados no carvão e no óleo de xisto. Os
mais conhecidos são benzo(a)pireno, benzo(a)antraceno, benzo(b)fluoranteno e o
benzo(k)fluoranteno. O benzo(a)pireno ocorre em vegetais frescos, óleo de coco, chá,
presunto defumado, carnes cozidas em carvão, bifes e costelas de churrasco, em
concentrações superiores a 3,0 ppb (MONTORO & NOGUEIRA, 1983).
As maiores fontes de emissão de HPAs são: geradores de calor e energia,
incineradores, produção de coque, produção de carvão vegetal, motores de veículos e
incêndios de matas. Os HPAs podem ser encontrados na fase vapor e/ou partícula cuja
distribuição depende da pressão de vapor do HPA e da temperatura ambiente. Em
termos de estrutura, a 25 ºC, HPAs com 2 e 3 anéis condensados encontram-se na fase
vapor, com 4 e 5 anéis em ambas as fases e 6 ou mais praticamente na fase partícula. O
longo tempo de residência das partículas possibilita o transporte do material particulado
à longa distância juntamente com compostos químicos associados (POPPI, 2000).
No ambiente aquático, os HPAs podem contaminar tanto a água quanto o
sedimento, dependendo da sua hidrofobicidade, isto é, de sua afinidade com compostos
polares ou apolares. Os HPAs mais hidrofóbicos tendem a aderir nas partículas do
sedimento, ou nos tecidos biológicos, enquanto aqueles menos hidrofóbicos tendem a
ficar menos retidos. A contaminação do solo por esta classe de compostos se dá
principalmente por deposição atmosférica e despejo de águas e sedimentos
contaminados, e o transporte ocorre por difusão. Portanto, a permanência no solo está
intimamente relacionada à solubilidade de cada composto em água. De um modo geral,
os compostos contendo um número de anéis maior que 4 têm maior relevância do ponto
de vista ambiental, pois a maioria deles apresenta propriedades carcinogênicas e são
extremamente resistentes à degradação enzimática. Os HPAs maiores e mais
hidrofóbicos tendem a ser mais carcinogênicos, enquanto os HPAs menores (2 e 3
anéis) não são carcinogênicos. Apesar disto, estudos mostram que sob o efeito da luz
alguns deles podem se tornar extremamente tóxicos (RIBEIRO, 2001).
A Agência de Proteção Ambiental dos Estados Unidos – EPA - recomenda a
determinação e quantificação de 16 HPAs como poluentes prioritários (POPPI, 2000), e
são eles: acenafteno, acenaftileno, antraceno, benzo(a)antraceno, benzo(a)pireno,

8
benzo(b)fluoranteno, benzo(ghi)perileno, benzo(k)fluoranteno, criseno,
dibenzo(a,h)antraceno, fluoranteno, fluoreno, indeno(1,2,3-cd)pireno, naftaleno,
fenantreno e pireno. Estes compostos encontram-se listados no método 8310 US-EPA
(cromatografia líquida de alta eficiência com detecção por UV ou fluorescência),
enquanto que o método 8100 sugere o uso da cromatografia gasosa com detecção no
espectrômetro de massas (CAPELO et al., 2005). Os HPAs atingem as águas
superficiais através da deposição direta da atmosfera ou a partir de efluentes municipais
e industriais, poeira atmosférica, precipitação de cinzas do ar, enxurradas, lixiviação de
solos contaminados, etc. Por causa da sua baixa solubilidade em água os HPA´s são
sorvidos pelo material particulado, indo para as águas naturais e acumulando no
sedimento do fundo em altas concentrações.
A seguir, são mostradas as fórmulas estruturais dos 16 HPAs recomendados
como poluentes prioritários pela EPA:
A presença de alguns desses HPAs é indicativo de suas origens, como por
exemplo, o criseno e benzo(k)fluoranteno podem ser indicadores de emissões pela
Naftaleno Acenafteno Acenaftileno Fuoreno
Fenantreno Antraceno Fluoranteno Pireno
Benzo[a]Antraceno Criseno
Benzo[g,h,i]Perileno Dibenzo[a,h]Antraceno Indeno[1,2,3-cd]Pireno
Benzo[b]Fluoranteno Benzo[k]Fluoranteno
Benzo[a]Pireno
Figura 1 - Estrutura dos 16 HPAs incluídos na lista de poluentes prioritários da EPA

9
combustão de carvão mineral. Da mesma forma, benzo(g,h,i)perileno, coroneno e
fenantreno são indicadores de emissões por veículos a combustão; fenantreno,
fluoranteno e pireno são indicadores de material particulado de estradas; pireno e
fluoranteno estão associados com a incineração; fluoreno, fluoranteno e pireno estão
associados com a combustão de óleos. Sabe-se que próximo ao centro urbano de Ouro
Preto está sediada uma grande indústria de alumínio, e é sabido que as indústrias de
alumínio são importantes fontes de emissão de HPAs (KHALILI et al., 1995).
As amostras contendo policíclicos aromáticos são de difícil armazenagem, sua
preparação é demorada, sendo o procedimento de extração, a maior fonte de erro
durante o ciclo de análise. É necessário o maior cuidado possível nas etapas de extração
e purificação para se obter resultados quantitativos precisos. Recentemente, o
procedimento clássico de extração por Soxhlet para HPA´s em amostras sólidas como
solos e sedimentos, que incluam um ou mais ciclos de extração, tem sido substituído por
procedimentos mais rápidos, que utilizem menos ou nenhum solvente orgânico e que
possam ser automatizados (SUN et al., 1998, GFRERER et al., 2002, HUBERT et al.,
2000 e LUNDSTEDT et al., 2000).
3.1.1 – Técnicas de Extração dos HPAs
Os sedimentos constituem um fator muito importante do sistema aquático, por
sua participação no equilíbrio dos poluentes solúveis/insolúveis e por sua maior
permanência no corpo de água, sendo, em geral, integradores das cargas poluentes
recebidas pelas águas. Entende-se por sedimentos os materiais insolúveis que se
depositam no fundo dos corpos de água. Pode ser considerado também o material
insolúvel suspenso na água (AGUDO, 1987). A utilização dos sedimentos para estudos
de caracterização ambiental apresenta algumas vantagens com relação ao meio hídrico
circulante. Os sedimentos correspondem a um dos segmentos ambientais mais estáveis
em termos físico e químico. Eles podem apresentar um importante papel na fixação de
substâncias, atuando como depósitos de elementos e substâncias contaminantes
(BAISCH et al.)
A literatura atual reúne uma vasta coleção de dados acerca das técnicas de
extração de compostos orgânicos em matrizes sólidas. Alguns desses procedimentos

10
são extração por ultrasom (SUN et al, 1998), extração por líquidos pressurizados (PLE)
(BAVEL et al, 2000) , extração em fluido supercrítico (SFE) (SAIM, 1997; MAZEAS,
1997; BRUCE, 1998), extração assistida por microondas (MAE) (LOPEZ, 1996;
MARTENS, 1997; TOMANIOVA, 1998), extração em leito fluidizado (FBE)
(GFRERER et al, 2002), extração por solvente acelerada (ASE) (HUBERT et al, 2000).
Outra alternativa é a extração subcrítica usando água quente (KUBÁTOVÁ, 2002;
RAMOSA, 2002; HAWTHORNE, 2000; PÉREZ, 2000) como solvente de HPAs em
amostras sólidas, os extratos produzidos desta forma podem ser concentrados por
extração em fase sólida (SPE) (TREMBLEY, 2000; YANG, 1999). A literatura mostra
que as análises por cromatografia líquida de alta eficiência (CLAE) usando detectores
de fluorescência ou ultravioleta-visível, e cromatografia gasosa (CG) com detectores
por espectrometria de massas ou ionização em chama (DIC) são os métodos mais
comuns para a separação, identificação e quantificação dos hidrocarbonetos policíclicos
aromáticos em extratos de amostras ambientais (GORSHKOV et al., 2003,
HAWTHORNE et al., 2000 e AZEVEDO et al., 2004).
Recentemente tem sido desenvolvidas técnicas de extração para reduzir tempo e
uso de solventes orgânicos. Algumas técnicas propostas são: extração em líquido
pressurizado (PLE), extração em fluido supercrítico (SFE) e extração assistida por
microondas (MAE) (HAWTHORNE et al., 2000, TOMANIOVÁ et al. 1998 e BRUCE
et al., 1998). Mais recentemente foi desenvolvida a extração subcrítica com água
(SWE), onde é empregada a água quente sobre pressão para manter o estado líquido. É
usada para extrair uma variedade de componentes orgânicos polares e apolares de
amostras ambientais, baseado no fato de que a solubilidade de compostos orgânicos
aumenta bastante com o aumento da temperatura da água mantida no estado líquido
(KUBATOVÁ et al., 2002, YANG et al., 1999). O desenvolvimento de métodos
simplificados proporciona redução de tempo e de resíduos. A SWE seguida de
microextração em fase sólida (SPME) é usada para determinar HPAs e PCBs
(HAWTHORNE et al., 2000). A SPME apresenta vantagens como por exemplo a não
utilização de solventes e a reutilização das fibras (NETO et al., 2005). Geralmente são
métodos simples de desenvolver e requerem pouco ou nenhum solvente orgânico, e
também necessitam aparatos de baixo custo. Discos de extração (SDB-XC) estireno-
divinilbenzeno são utilizados para extração de HPAs (HAWTHORNE, et al., 2000)

11
Segundo Yang (1999), a extração subcrítica com água tem sido usada com
sucesso em extrações ambientais de muitas classes de compostos orgânicos. O princípio
da extração subcrítica é que a alteração da constante dielétrica, tensão superficial e
viscosidade da água são obtidos com o controle da pressão mantendo a água no estado
liquido a altas temperaturas. Como exemplo, a constante dielétrica da água é diminuída
de 80 para aproximadamente 30 numa temperatura de 250ºC. Os valores de tensão
superficial, viscosidade e constante dielétrica da água a 250ºC são similares ao etanol,
metanol ou acetonitrila em temperatura ambiente. Estudos recentes mostram que a água
a elevadas temperaturas e pressão é um excelente fluido de extração para muitos
compostos orgânicos de matrizes sólidas.
Compostos orgânicos apolares como HPAs e PCBs requerem temperatura da
água acima de 250ºC para uma melhor eficiência na extração. A extração de orgânicos
apolares e moderadamente polares é baseada na redução da polaridade, tensão
superficial e viscosidade da água a altas temperaturas (acima de 250ºC). Sob essas
condições , compostos orgânicos geralmente considerados insolúveis em água mostram
consideráveis aumentos de solubilidade. A troca das propriedades da água tem sido
usada para extrair uma variedade de poluentes orgânicos de sorbentes, solos
contaminados, lodos, material particulado e outras amostras ambientais. A eficiência da
extração subcrítica com água depende da temperatura e pressão suficiente para manter o
seu estado líquido (GRABANSKI, et al., 1998).
De acordo com SAIM, (1997), a escolha da técnica de extração é feita com base
no capital disponível, custos de operação, simplicidade de operação, quantidade de
solvente orgânico requerida e preparação da amostra. O artigo compara as técnicas de
extração de HPAs de solos altamente contaminados. As técnicas investigadas foram
extração soxhlet, assistida por microondas (MAE), extração em fluido supercrítico
(SFE) e extração acelerada por solvente (ASE). A extração soxhlet requer uma etapa de
clean-up (“limpeza”) antes da análise. Comparando os tempos de extração, temos a SFE
na faixa de 36 a 50 minutos, enquanto que a soxhlet temos de 2 a 24 horas. Se
compararmos os volumes de solvente orgânico gastos temos para a SFE (10 mL),
soxhlet (entre 60 e 500 mL).

12
3.1.2 – Extração Soxhlet
A técnica de extração soxhlet é usada para extrair compostos não-voláteis e
semivoláteis de matrizes sólidas como solos, lodos e resíduos. Neste processo garante-
se que a amostra mantém o contato permanente com o solvente de extração (SOXHLET
EXTRACTION, METHOD 3540C US-EPA). A extração por soxhlet apresenta uma
alta eficiência de extração e por muitos anos foi um método padrão para extração de
HPAs em matrizes sólidas, entretanto pode levar longos tempos de extração, cerca de 16
horas ou mais (SUN, et al, 1998).
3.2 - Avaliação Química das Águas
3.2.1 – Parâmetros Físico-Químicos
A água é essencial para a existência e o bem-estar do ser humano, devendo estar
disponível em quantidade suficiente e apresentar boa qualidade como garantia da
manutenção da vida. A água sendo um excelente solvente, através do seu ciclo
hidrológico permanece em contato com os constituintes do meio ambiente (ar e solo),
dissolvendo muitos elementos e carreando outros em suspensão. A qualidade da água é
vulnerável as condições ambientais a qual está exposta e, portanto, na maioria das
vezes, é necessário um tratamento para torná-la potável. O controle físico-químico das
águas tem uma importância fundamental na sua utilização (FREITAS et al., 2002).
3.2.2 – Coleta das Amostras
Uma grande parte do sucesso das análises de laboratório em estudos ambientais
depende da adoção de procedimentos adequados durante a etapa de coleta. Um erro
amostral repercutirá inevitavelmente sobre a qualidade dos resultados, mesmo que se
disponha de um avançado aparato analítico. As técnicas de coleta são determinadas em
função dos objetivos do estudo e das características do sistema ambiental.
A coleta de amostras é, provavelmente, o passo mais importante para a
caracterização de microconstituintes nas águas e é essencial que a coleta seja realizada

13
com precauções para evitar todas as fontes possíveis de contaminação e perdas. É
necessário obter uma amostra representativa e estabilizada do corpo de água amostrado
e das condições locais que podem interferir, tanto na interpretação dos dados quanto nas
próprias determinações laboratoriais (AGUDO, 1987).
3.2.3 – Metais
Os metais pesados atingem o ambiente através de efluentes industriais, podendo
contaminar a água e os alimentos, assim expondo o homem e animais à intoxicação
crônica. Também são contaminantes comuns em ambientes industriais e de trabalho de
alguns profissionais especializados, tendo grande importância em saúde ocupacional.
Dos metais pesados de interesse na contaminação ambiental e na saúde ocupacional, os
mais importantes são chumbo, mercúrio, cádmio e o arsênio (BOGLIOLO, 1998).
Metal pesado não implica necessariamente em “metal tóxico”. Muitos deles são
considerados nutrientes indispensáveis às plantas e seres vivos, desde que em baixas
concentrações. Vários íons metálicos são essenciais ao bom funcionamento dos
organismos vivos, e outros como sódio, cálcio, potássio, manganês, ferro, cobalto,
molibdênio, cobre e zinco têm relevante importância para o homem. Os metais são
essenciais aos organismos em pequenas quantidades, mas, quando em
excesso,desencadeiam diversos problemas. Uma vez que atingem o corpo d’água, esses
metais depositam nos sedimentos e sofrem processos complexos de adsorção, que ainda
são desconhecidos, especialmente em ambientes tropicais. O termo metal pesado é
amplamente utilizado para um grupo de metais que estão associados à poluição e
toxicidade, tais como chumbo, cádmio, mercúrio, arsênio e urânio. São também
incluídos cobalto, cobre, manganês, selênio e zinco, considerados biologicamente
essenciais em baixas concentrações (SILVA, 2002).
Os metais exercem um papel importante na manutenção da vida em nosso
planeta. Alguns elementos podem ser altamente tóxicos a várias formas de vida, outros
são considerados essenciais, mas podem ser tóxicos em doses mais elevadas
(TEMPLETON et al, 2000).
O Plasma Indutivamente Acoplado (ICP) é usado para determinar elementos
traços, incluindo metais, em solução (METHOD 6010 B EPA). O ICP acoplado ao

14
espectrômetro de massas (ICP-MS) é usado na determinação de concentrações
inferiores a µg/L de vários elementos em amostras aquosas. O ICP-MS pode ser
aplicado na determinação de cerca de 60 elementos em várias matrizes (METHOD 6020
EPA).
4. CARACTERIZAÇÃO DA ÁREA DE ESTUDO A área de estudo localiza-se próxima ao perímetro urbano da cidade de Ouro
Preto – MG, e têm seus quatro pontos de coleta distribuídos ao longo do Ribeirão do
Funil, que parte do seu percurso passa dentro de uma fábrica de alumínio seguindo pela
cidade onde tem trechos de contato com algumas residências.
A cidade de Ouro Preto está localizada na porção central do Estado, ocupa uma
área de 1246,23 Km2 tendo altitude máxima na divisa com o município de Santa
Bárbara (1891 m) e altitude mínima de 989 m, na foz do Rio Maracujá. O Ponto central
da cidade tem 1160 m de altitude. O índice pluviométrico anual é de cerca de 1600 mm,
e seu relevo é em maior parte montanhoso (55%), ondulado (40%) e plano (5%). A
temperatura média anual é de 18,5 ºC. Os principais rios são: Rio das Velhas e Ribeirão
do Funil (INDI, 2007).
O Ribeirão do Funil se destaca na região. Sua nascente está na proximidade de
Venda Nova-MG. Depois de tomar, sucessivamente, as denominações de Ribeirão do
Carmo e Rio do Carmo, deságua no Rio Piranga e divide a serra de Ouro Preto das
montanhas em que se destaca o Pico de Itacolomi. O sistema de drenagem é denso, e
fortemente declivoso, conseqüência do relevo movimentado da região.
A cidade de Ouro Preto localiza-se entre a meia encosta e o sopé da serra do
Veloso ao nível dos mananciais, onde nascem ou se juntam às primeiras linhas de água
que formam os dois canais que drenam para o Ribeirão do Funil (GUERREIRO, 2000).
Ao longo deste estudo foram coletadas amostras de águas e sedimentos em 3
épocas diferentes e em 4 pontos distintos do Ribeirão do Funil. As amostras de ambas
as matrizes foram coletadas nos mesmos pontos e mesmas datas para as 3 coletas. Os
pontos de coleta das amostras de água e sedimentos foram distribuídos da seguinte
forma, tendo como referência a fábrica de alumínio citada:
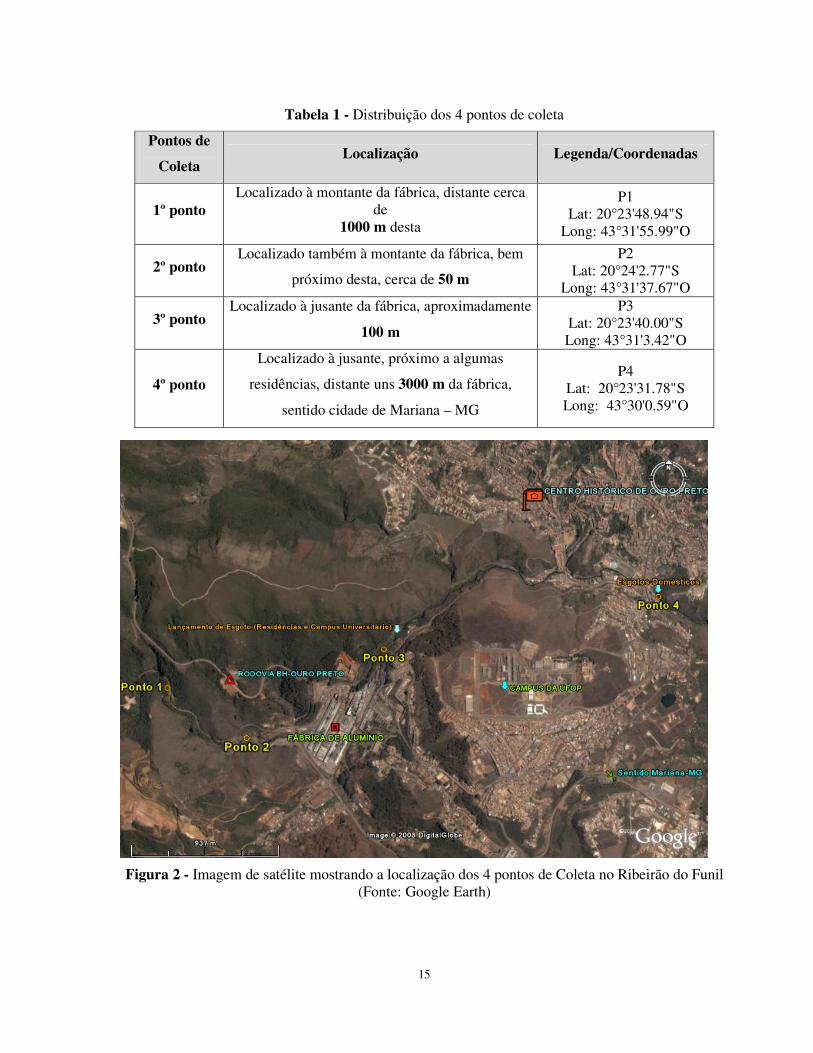
15
Tabela 1 - Distribuição dos 4 pontos de coleta
Pontos de
Coleta Localização Legenda/Coordenadas
1º ponto Localizado à montante da fábrica, distante cerca
de 1000 m desta
P1 Lat: 20°23'48.94"S
Long: 43°31'55.99"O
2º ponto Localizado também à montante da fábrica, bem
próximo desta, cerca de 50 m
P2 Lat: 20°24'2.77"S
Long: 43°31'37.67"O
3º ponto Localizado à jusante da fábrica, aproximadamente
100 m
P3 Lat: 20°23'40.00"S Long: 43°31'3.42"O
4º ponto
Localizado à jusante, próximo a algumas
residências, distante uns 3000 m da fábrica,
sentido cidade de Mariana – MG
P4 Lat: 20°23'31.78"S Long: 43°30'0.59"O
Figura 2 - Imagem de satélite mostrando a localização dos 4 pontos de Coleta no Ribeirão do Funil (Fonte: Google Earth)
16
5. PARTE EXPERIMENTAL
Os procedimentos adotados nas etapas de preparo das amostras, procedimento de
extração e limpeza dos extratos foram baseados em procedimentos adotados pela US-
EPA: (SOXHLET EXTRACTION, METHOD 3540C; SILICA GEL CLEAN-UP,
METHOD 3630 C). Na série de métodos de extração soxhlet também se encontra
disponível o procedimento para concentração das amostras usando o Kuderna-Danish.
5.1 – Avaliação dos Hidrocarbonetos Policíclicos Aromáticos em Sedimentos
5.1.1– HPAs Analisados
Foram avaliados 11 HPAs sendo: naftaleno (Naph), acenafteno (Ace), fluoreno
(Fluo), fenantreno (Phe), antraceno (Anth), fluoranteno (Flu), pireno (Pyr), criseno
(Chr), benzo(b)fluoranteno (BbFl), benzo(k)fluoranteno (BkFl) e benzo(a)pireno
(BaPy). Nos últimos anos os HPAs têm recebido uma maior atenção em vários estudos
devido ao conhecimento de sua alta capacidade carcinogênica e mutagênica.
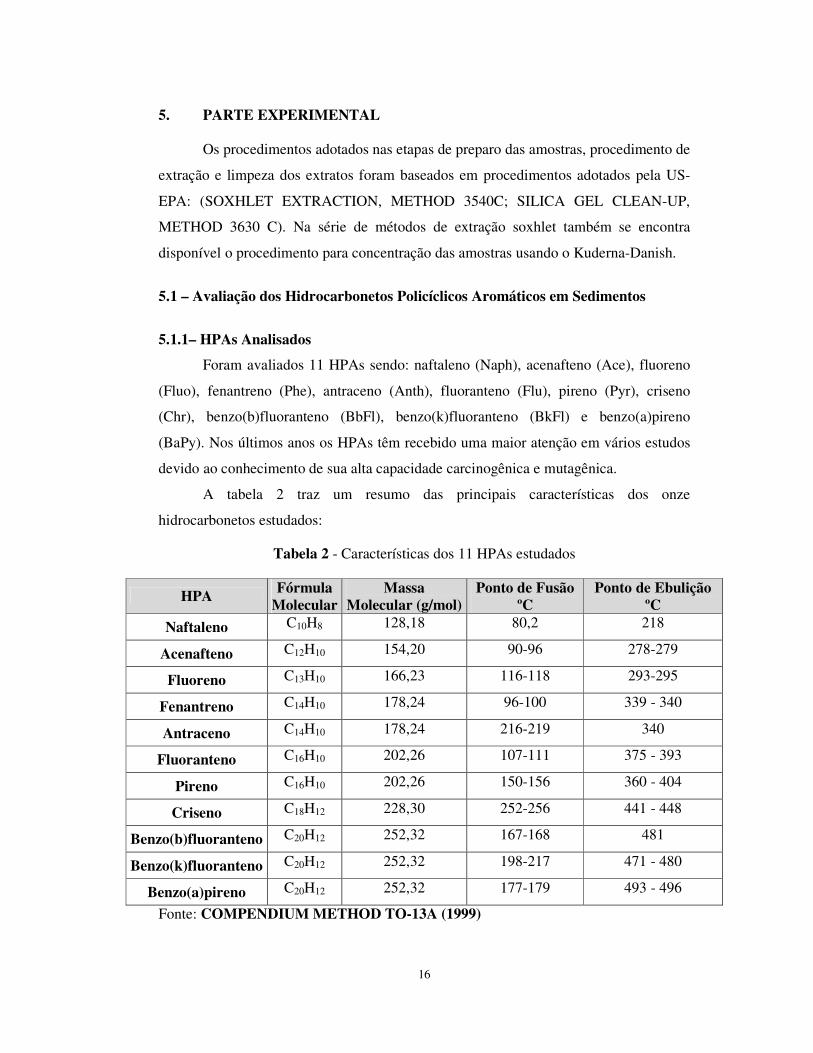
A tabela 2 traz um resumo das principais características dos onze
hidrocarbonetos estudados:
Tabela 2 - Características dos 11 HPAs estudados
HPA Fórmula Molecular
Massa Molecular (g/mol)
Ponto de Fusão ºC
Ponto de Ebulição ºC
Naftaleno C10H8 128,18 80,2 218
Acenafteno C12H10 154,20 90-96 278-279
Fluoreno C13H10 166,23 116-118 293-295
Fenantreno C14H10 178,24 96-100 339 - 340
Antraceno C14H10 178,24 216-219 340
Fluoranteno C16H10 202,26 107-111 375 - 393
Pireno C16H10 202,26 150-156 360 - 404
Criseno C18H12 228,30 252-256 441 - 448
Benzo(b)fluoranteno C20H12 252,32 167-168 481
Benzo(k)fluoranteno C20H12 252,32 198-217 471 - 480
Benzo(a)pireno C20H12 252,32 177-179 493 - 496
Fonte: COMPENDIUM METHOD TO-13A (1999)
17
5.1.2 – Coleta das Amostras de Sedimento
O Ribeirão do Funil apresenta, na maior parte do seu percurso, um leito
predominante de águas rasas, com trechos de sedimentação acumulada em alguns
pontos. Para cada um dos quatro pontos e nas 3 coletas realizadas, aproximadamente
500 g de sedimento foram retirados das margens do ribeirão com auxílio de espátulas de
polipropileno limpas. Os sedimentos coletados foram transferidos para sacos plásticos
limpos, levados ao laboratório até ser enviado para secagem. Cada saco plástico foi
vedado e etiquetado, identificando cada ponto de coleta.
Na tabela 3 são mostrados os dados das três coletas de sedimento realizadas ao
longo deste estudo:
Tabela 3 - Dados das 3 Coletas de Sedimentos
Coleta Data Quantidade de Amostras Características Climáticas
1a 30/03/2005 4 amostras (~ 500g cada) Seca
2a 18/11/2005 4 amostras (~ 500g cada) Chuva
3a 08/06/2006 4 amostras (~ 500g cada) Seca
5.1.3 – Secagem das Amostras de Sedimento
As amostras de sedimento foram secas em capelas fechadas com aquecimento
por lâmpadas de infravermelho (250 watts). A temperatura medida neste ambiente foi
na faixa de 30ºC a 40ºC. Em cada uma das 3 coletas, as massas (~ 500g) dos sedimentos
dos quatro pontos das foram acondicionadas em bandejas plásticas e colocadas na
capela. As amostras ficaram na capela até que fosse observada a completa secagem do
material e o tempo desta etapa foi de aproximadamente três semanas. Na figura 3
melhor se visualiza a etapa de secagem descrita.

18
Figura 3 - Secagem dos sedimentos em capela fechada com aquecimento por lâmpadas
de infravermelho 250 watts 5.1.4 – Preparo das Amostras para Extração
Os sedimentos secos foram colocados em tabuleiros de alumínio e triturados
com auxílio de um pistilo de porcelana. O sedimento desfragmentado foi peneirado
(peneira 8 mesh) eliminando folhas, paus e pedras. Em seguida, os sedimentos
peneirados passaram pelo processo de quarteamento e foram obtidas as amostras para
extração soxhlet Para a extração soxhlet foram separadas cerca de 50 g de amostra de
sedimento quarteado para cada ponto de coleta e nas 3 coletas realizadas. Na figura 4 é
mostrado o sedimento que já se encontrava completamente seco depois de
aproximadamente 20 dias de exposição ao calor de lâmpadas em capela.

19
Figura 4 - Sedimento seco (4 pontos de coleta)
Na figura 5 são mostrados, para uma das 3 coletas, os sedimentos dos 4 pontos
de coleta após secagem e peneiramento. A figura 6 mostra os sedimentos quarteados dos
4 pontos (~ 50 g cada) e prontos para a separação das amostras para extração soxhlet
Figura 5 - Sedimento triturado e peneirado (4 pontos de coleta)

20
Figura 6 - Sedimento Quarteado (pronto para extração soxhlet)
5.1.5 – Extração Soxhlet
A técnica de extração via soxhlet apresenta algumas desvantagens, como o uso
de grande quantidade de solvente e tempo prolongado, no entanto foi a técnica escolhida
por ser a que mais se adequou à estrutura disponível para realização deste estudo e por
ser considerado um procedimento de referência. O procedimento soxhlet é descrito no
método 3540C da US-EPA (SOXHLET EXTRACTION, METHOD 3540C).
Para cada ponto de coleta e nas 3 coletas realizadas, foi feita a extração em
triplicata. A massa pesada foi de 10 g de sedimento para cada extração. O solvente de
extração foi o diclorometano e o período de extração foi de 24 horas para cada
seqüência. A temperatura da manta aquecedora foi ajustada para que o sistema sifonasse
a cada 15 minutos.

21
5.1.6 – Concentração no Kuderna-Danish (KD)
Os extratos foram concentrados em um concentrador do tipo Kuderna-Danish
segundo metodologia proposta em um dos tópicos do procedimento de extração soxhlet
(SOXHLET EXTRACTION, METHOD 3540C). Toda a aparelhagem foi acoplada a
um sistema de destilação para recuperação do diclorometano. O sistema foi aquecido
por banho de água quente (banho-maria) com um aquecedor/agitador com controle de
temperatura baseado na evaporação do diclorometano. A temperatura de ebulição do
diclorometano está entre 39 – 40 ºC. A temperatura utilizada foi ligeiramente maior para
o processo de concentração. Nesta etapa o volume do extrato que era de
aproximadamente 150 mL foi reduzido para 10 mL (TABLE 1, SOXHLET
EXTRACTION, METHOD 3540C).
(1)
(2)
Figura 7 - (1) Esquema de um aparelho de soxhlet convencional e (2) experimento
realizado nas amostras de sedimento

22
Figura 8 - Aparelho de Concentração das Amostras (KD)
O extrato concentrado (10,0 mL) foi passado primeiramente por uma coluna de
sulfato de sódio anidro (Na2SO4 anidro), previamente purificado (eliminação de
orgânicos) em uma mufla (T=400ºC) por um período de 4 horas. Depois do tratamento
térmico o sulfato de sódio anidro foi mantido em estufa ou dessecador durante sua
utilização. Em uma coluna de vidro de 50 cm e diâmetro interno de 1 cm colocou-se
um chumaço de lã de vidro na extremidade inferior e adicionou-se o Na2SO4 até que a
altura deste dentro da coluna fosse de aproximadamente 10 cm. Adicionou-se os extrato
concentrado para a retirada da umidade, seguida da adição de 20 ml de diclorometano.
O eluído foi reservado para a etapa de purificação (clean-up).
5.1.7 – Purificação dos extratos - Clean-up
A limpeza ou “clean-up” dos extratos, com intuito de eliminar interferentes
orgânicos da etapa cromatográfica, foi realizado usando coluna com sílica (SiO2) gel
ativada (Sílica Gel 60/ 0,063-0,200 mm, para cromatografia em coluna/ VETEC)
(SILICA GEL CLEANUP, 1996). Foi preparada uma solução de 10 g de sílica em
diclorometano e transferida para a coluna cromatográfica (10 mm diâmetro interno e 50

23
cm de comprimento). Adicionou-se 1 a 2 cm de Na2SO4 anidro ao topo da coluna de
sílica para eliminar umidade na hora de transferir os extratos. A ativação da sílica foi
feita sob aquecimento em estufa (130ºC) por 16 horas. Antes do clean-up com sílica, o
solvente de extração foi trocado por cicloexano. A troca foi realizada adicionando 4 mL
de cicloexano seguindo a redução do extrato para 1-2 mL usando o Kuderna-Danish. Se
o volume do extrato for reduzido para menos de 1 mL, analitos semivoláteis podem ser
perdidos. Se o extrato secar, a extração deve ser repetida. Primeiramente, pré-eluiu a
coluna com 40 mL de pentano. O fluxo de todas a eluições foi de 2 mL/min. Descartou-
se o eluato e, antes do Na2SO4 entrar em contato com o ar, transferiu-se 2 mL de
cicloexano junto com o extrato na coluna usando 2 mL de cicloexano para completar a
transferência. Antes do Na2SO4 entrar em contato com o ar, adicionou-se 25 mL de
pentano e continuou a eluição na coluna. Descartou-se o eluato com pentano. Na
próxima etapa, eluiu-se a coluna com 25 mL de diclorometano/pentano 2:3 v/v para um
frasco de concentração. Concentrou-se a fração coletada para o volume recomendado
(1-10 mL) (SILICA GEL CLEANUP, METHOD 3630 C). No caso as amostras foram
concentradas para 1,0 mL. Esta amostra concentrada foi utilizada nas análises
cromatográficas.
24
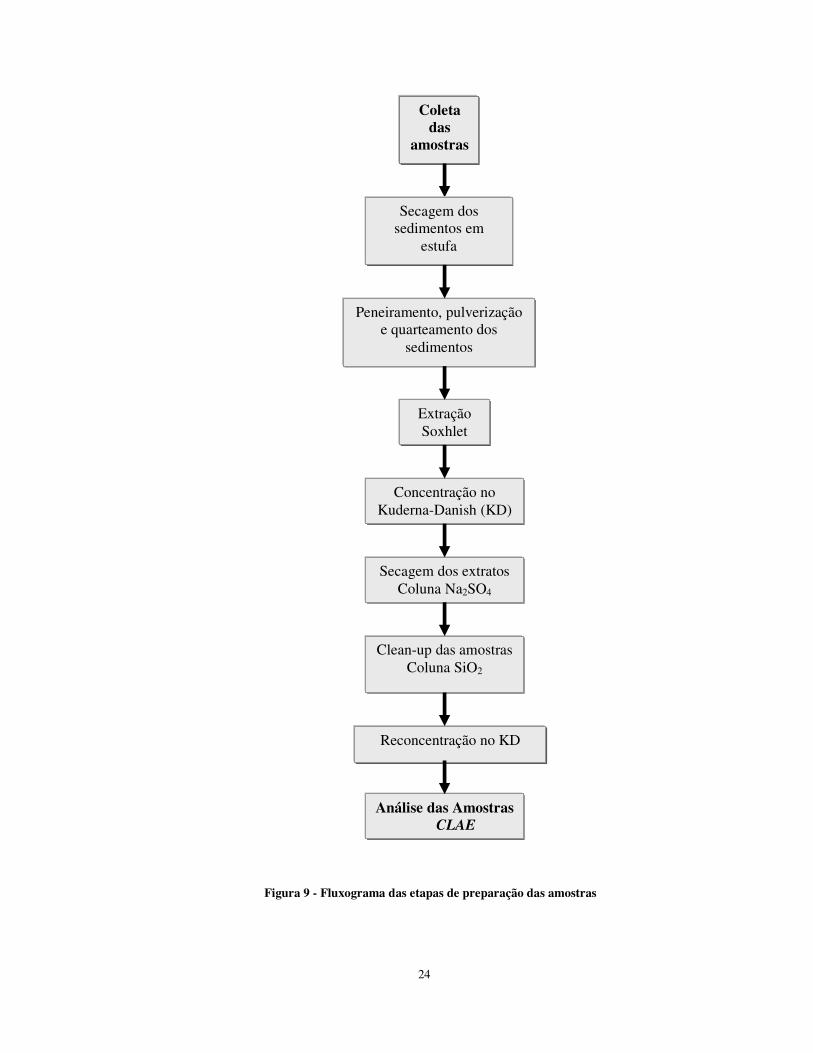
Figura 9 - Fluxograma das etapas de preparação das amostras
Coleta das
amostras
Secagem dos sedimentos em
estufa
Peneiramento, pulverização e quarteamento dos
sedimentos
Extração Soxhlet
Concentração no Kuderna-Danish (KD)
Secagem dos extratos Coluna Na2SO4
Clean-up das amostras Coluna SiO2
Reconcentração no KD
Análise das Amostras CLAE
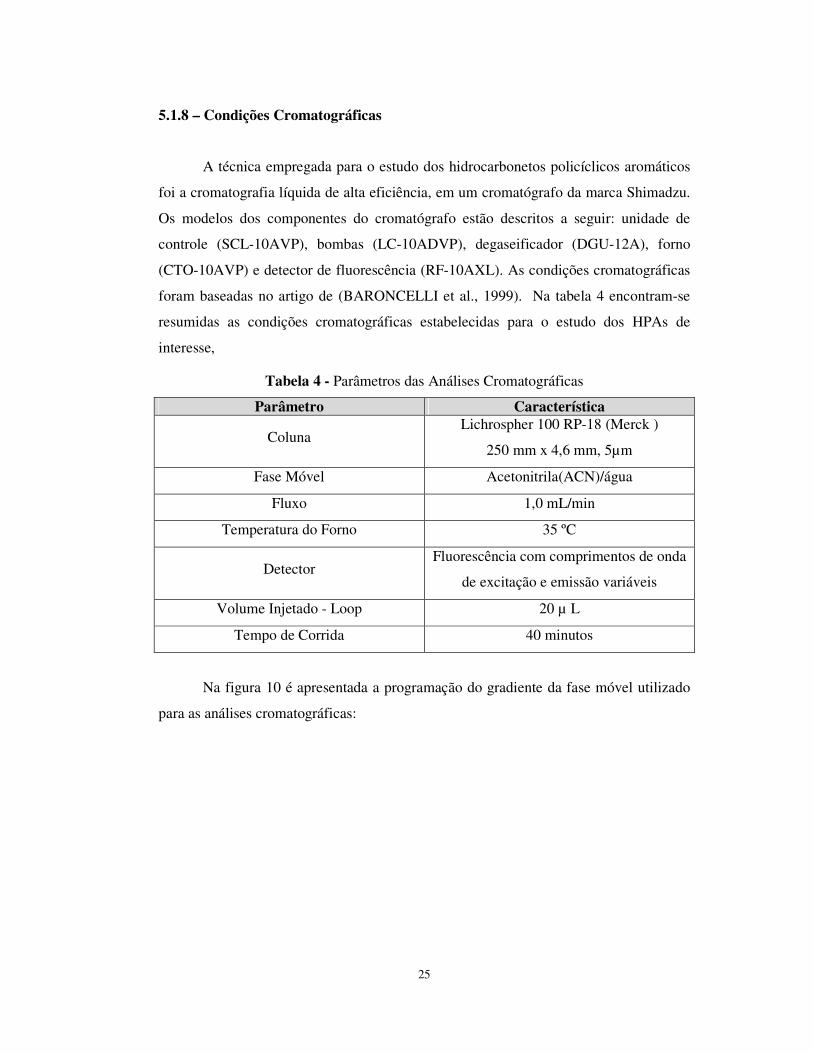
25
5.1.8 – Condições Cromatográficas
A técnica empregada para o estudo dos hidrocarbonetos policíclicos aromáticos
foi a cromatografia líquida de alta eficiência, em um cromatógrafo da marca Shimadzu.
Os modelos dos componentes do cromatógrafo estão descritos a seguir: unidade de
controle (SCL-10AVP), bombas (LC-10ADVP), degaseificador (DGU-12A), forno
(CTO-10AVP) e detector de fluorescência (RF-10AXL). As condições cromatográficas
foram baseadas no artigo de (BARONCELLI et al., 1999). Na tabela 4 encontram-se
resumidas as condições cromatográficas estabelecidas para o estudo dos HPAs de
interesse,
Tabela 4 - Parâmetros das Análises Cromatográficas
Parâmetro Característica
Coluna Lichrospher 100 RP-18 (Merck )
250 mm x 4,6 mm, 5µm
Fase Móvel Acetonitrila(ACN)/água
Fluxo 1,0 mL/min
Temperatura do Forno 35 ºC
Detector Fluorescência com comprimentos de onda
de excitação e emissão variáveis
Volume Injetado - Loop 20 µ L
Tempo de Corrida 40 minutos
Na figura 10 é apresentada a programação do gradiente da fase móvel utilizado
para as análises cromatográficas:
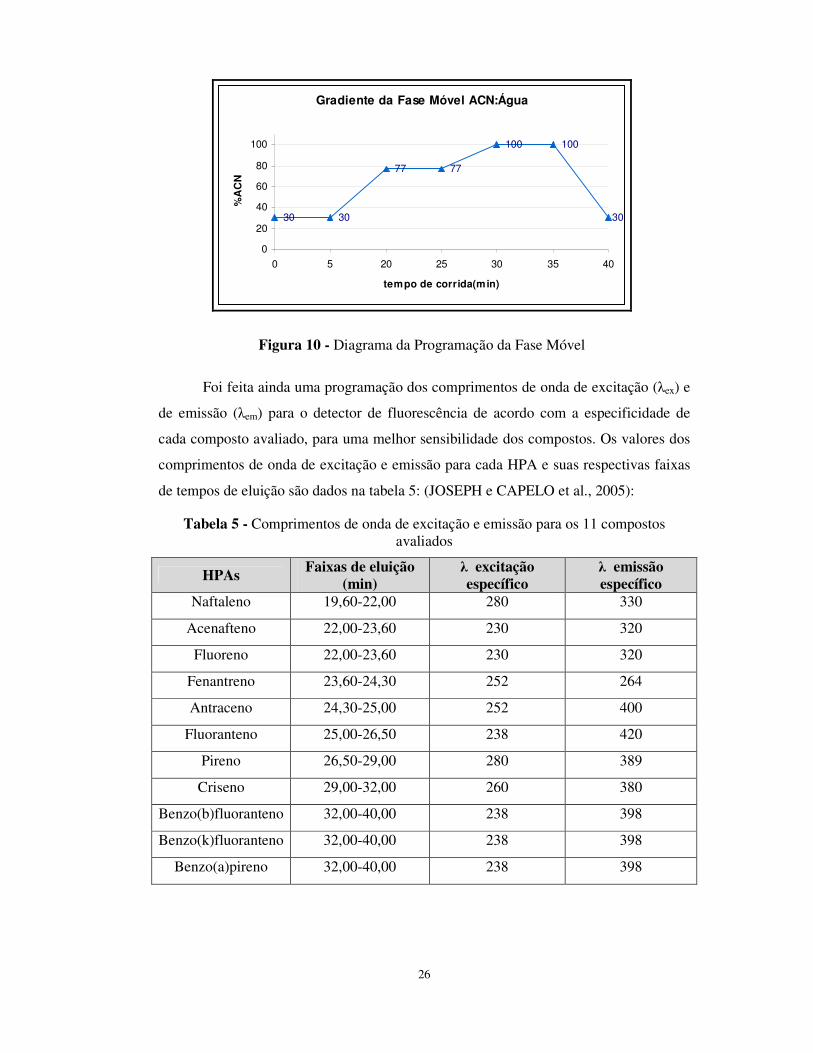
26
Foi feita ainda uma programação dos comprimentos de onda de excitação (λex) e
de emissão (λem) para o detector de fluorescência de acordo com a especificidade de
cada composto avaliado, para uma melhor sensibilidade dos compostos. Os valores dos
comprimentos de onda de excitação e emissão para cada HPA e suas respectivas faixas
de tempos de eluição são dados na tabela 5: (JOSEPH e CAPELO et al., 2005):
Tabela 5 - Comprimentos de onda de excitação e emissão para os 11 compostos avaliados
HPAs Faixas de eluição
(min) λ excitação específico
λ emissão específico
Naftaleno 19,60-22,00 280 330
Acenafteno 22,00-23,60 230 320
Fluoreno 22,00-23,60 230 320
Fenantreno 23,60-24,30 252 264
Antraceno 24,30-25,00 252 400
Fluoranteno 25,00-26,50 238 420
Pireno 26,50-29,00 280 389
Criseno 29,00-32,00 260 380
Benzo(b)fluoranteno 32,00-40,00 238 398
Benzo(k)fluoranteno 32,00-40,00 238 398
Benzo(a)pireno 32,00-40,00 238 398
Gradiente da Fase Móvel ACN:Água
30 30
77 77
100 100
30
0
20
40
60
80
100
0 5 20 25 30 35 40
tempo de corrida(min)
%A
CN
Figura 10 - Diagrama da Programação da Fase Móvel

27
A especificidade pode ser confirmada pelo estabelecimento dos tempos de
retenção e espectros de fluorescência excitação e emissão máxima, através da injeção
individual dos 11 HPAs padrões.
5.1.9 – Preparo da Solução-Padrão de HPAs
Uma solução estoque contendo os 11 padrões de HPAs foi preparada de acordo
com concentrações conhecidas de cada analito estabelecidas em um catálogo que
mostrava suas concentrações em uma mistura dos 16 HPAs prioritários da EPA e a
partir dela foram feitas todas as diluições necessárias para o estabelecimento das curvas
analíticas. As concentrações dos 11 padrões na solução estoque foram (em µg/mL):
Naftaleno (498,0), Acenafteno (218,0), Fluoreno (102,0), Fenantreno (50,0), Antraceno
(25,0), Fluoranteno (49,4), Pireno (111,4), Benzo(b)fluoranteno (26,0),
Benzo(k)fluoranteno (23,0), Benzo(a)pireno (56,8) e Criseno (51,4). Foram preparados
50,0 mL da solução estoque diluindo-se os padrões em uma mistura de
acetonitrila/metanol (90:10). Desta solução foram preparadas soluções para as curvas
analíticas.
5.1.10 – Curvas Analíticas
Depois de estabelecidas as condições cromatográficas de análise, com a
separação de sete dos onze padrões de HPAs, identificou-se cada hidrocarboneto
policíclico aromático nos cromatogramas da mistura de padrões de acordo com
BARONCELLI (1999). As curvas de analíticas foram estabelecidas pela regressão
linear de no mínimo 6 pontos para diferentes concentrações dos analitos e as respectivas
respostas em área dos picos cromatográficos. As concentrações utilizadas são descritas
na tabela 6. Foram preparadas 12 diluições no total, sendo as curvas analíticas ajustadas
de acordo com os pontos que garantiram melhor linearidade.
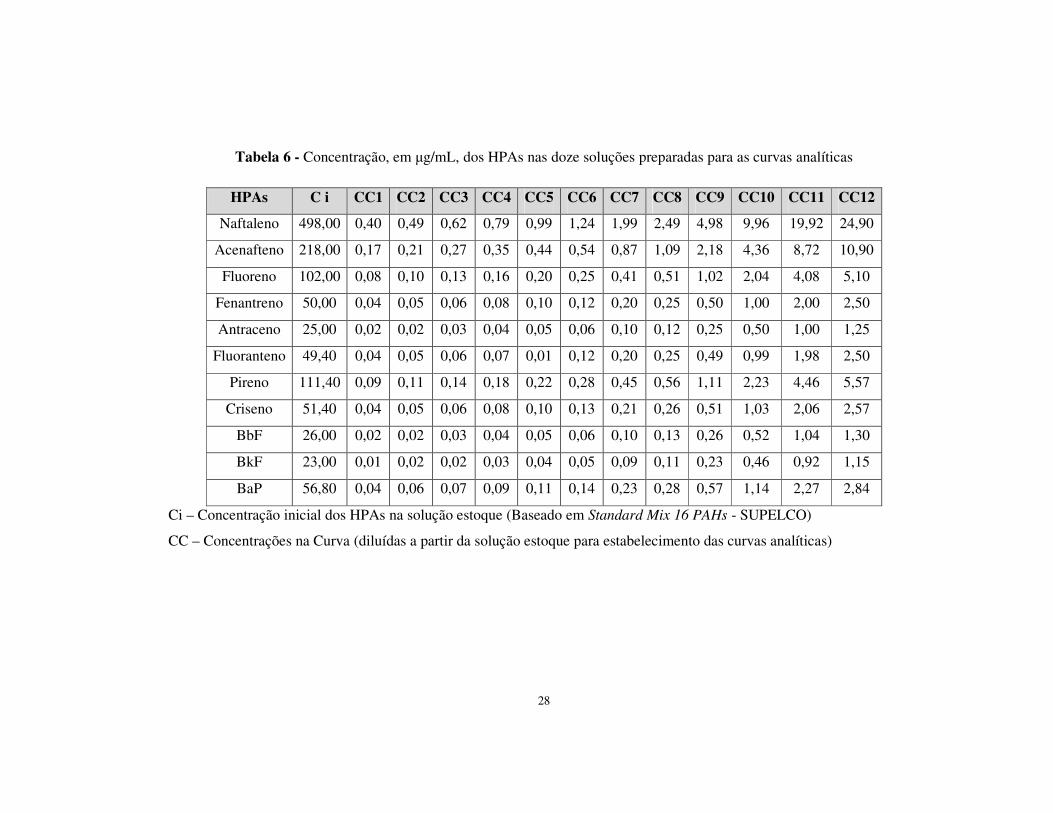
28
Tabela 6 - Concentração, em µg/mL, dos HPAs nas doze soluções preparadas para as curvas analíticas
HPAs C i CC1 CC2 CC3 CC4 CC5 CC6 CC7 CC8 CC9 CC10 CC11 CC12
Naftaleno 498,00 0,40 0,49 0,62 0,79 0,99 1,24 1,99 2,49 4,98 9,96 19,92 24,90
Acenafteno 218,00 0,17 0,21 0,27 0,35 0,44 0,54 0,87 1,09 2,18 4,36 8,72 10,90
Fluoreno 102,00 0,08 0,10 0,13 0,16 0,20 0,25 0,41 0,51 1,02 2,04 4,08 5,10
Fenantreno 50,00 0,04 0,05 0,06 0,08 0,10 0,12 0,20 0,25 0,50 1,00 2,00 2,50
Antraceno 25,00 0,02 0,02 0,03 0,04 0,05 0,06 0,10 0,12 0,25 0,50 1,00 1,25
Fluoranteno 49,40 0,04 0,05 0,06 0,07 0,01 0,12 0,20 0,25 0,49 0,99 1,98 2,50
Pireno 111,40 0,09 0,11 0,14 0,18 0,22 0,28 0,45 0,56 1,11 2,23 4,46 5,57
Criseno 51,40 0,04 0,05 0,06 0,08 0,10 0,13 0,21 0,26 0,51 1,03 2,06 2,57
BbF 26,00 0,02 0,02 0,03 0,04 0,05 0,06 0,10 0,13 0,26 0,52 1,04 1,30
BkF 23,00 0,01 0,02 0,02 0,03 0,04 0,05 0,09 0,11 0,23 0,46 0,92 1,15
BaP 56,80 0,04 0,06 0,07 0,09 0,11 0,14 0,23 0,28 0,57 1,14 2,27 2,84
Ci – Concentração inicial dos HPAs na solução estoque (Baseado em Standard Mix 16 PAHs - SUPELCO)
CC – Concentrações na Curva (diluídas a partir da solução estoque para estabelecimento das curvas analíticas)

29
As equações das regressões lineares produzidas foram utilizadas para a
determinação das concentrações nos extratos e cálculos da concentração nas
amostras.
5.1.11 - Cálculo do Índice de Recuperação
A exatidão de um método é definida pelo INMETRO como sendo a
concordância entre o resultado de um ensaio e o valor de referência aceito como
convencionalmente verdadeiro. A exatidão, quando aplicada a uma série de
resultados de ensaio, implica numa combinação de componentes de erros aleatórios e
sistemáticos (tendência). A determinação da tendência total com relação aos valores
de referência apropriados é importante no estabelecimento da rastreabilidade aos
padrões reconhecidos. A tendência pode ser expressa como recuperação analítica
(valor observado / valor esperado). A tendência deve ser corrigida ou demonstrada
ser desprezível, mas em ambos os casos, a incerteza associada com a determinação
da tendência permanece como um componente essencial da incerteza global. Os
processos normalmente utilizados para avaliar a exatidão de um método são, entre
outros: uso de materiais de referência, participação em comparações
interlaboratoriais e realização de ensaios de recuperação (INMETRO, 2003).
A necessidade de se mostrar a exatidão de medições químicas, através de sua
comparabilidade, rastreabilidade e confiabilidade, está sendo cada vez mais
reconhecida e exigida. A validação de um método analítico deve garantir, através de
estudos experimentais, que o método atenda às exigências das aplicações analíticas,
assegurando a confiabilidade dos resultados. É essencial que os estudos de validação
sejam representativos e conduzidos de modo que a variação da faixa de concentração
e os tipos de amostras sejam adequados (COLLINS et al., 2004).
Não estava disponível um material de referência certificado que se
assemelhasse às condições das amostras de sedimentos usadas neste trabalho.
Portanto, para o estabelecimento da exatidão do método fez-se necessário o estudo
para determinação dos índices de recuperação dos analitos. Segundo o INMETRO
(2003) a recuperação do analito pode ser estimada pela análise de amostras
adicionadas com quantidades conhecidas do mesmo (spike ou surrogate). As
amostras podem ser adicionadas com o analito em pelo menos três diferentes

30
concentrações, por exemplo, próximo ao limite de detecção, próximo à concentração
máxima permissível e em uma concentração próxima à média da faixa de uso do
método. A limitação deste procedimento é a de que o analito adicionado não está
necessariamente na mesma forma que a presente na amostra. A presença de analitos
adicionados em uma forma mais facilmente detectável pode ocasionar avaliações
excessivamente otimistas da recuperação.
Neste trabalho utilizou-se a técnica de adição de uma quantidade conhecida
dos analitos (spike) em duas concentrações nas amostras de sedimento seco coletadas
no ponto 4 da segunda coleta, que apresentou melhores resultados para as
concentrações no sedimento não contaminado (branco). Os índices de recuperação
deveriam ser estabelecidos pelas diferenças entre as concentrações nas amostras
puras e o percentual de recuperação dos valores adicionados.
Antes do preparo das duas soluções de contaminação, fez-se uma estimativa
dos valores de concentração esperados no sedimento sem adição dos padrões de
HPAs. Através dos resultados encontrados, calculou-se as razões que deveriam ser
adicionadas. Sendo uma com adição de cerca de 50% (spike 1) e 75% (spike 2) em
relação às concentrações estimadas do sedimento sem contaminação.
Preparou-se duas soluções (50 mL cada) em acetonitrila com concentrações
conhecidas dos HPAs. Devido à falta de quantidade suficiente do padrão do criseno
este não foi avaliado. Estas soluções foram adicionadas a 40g da amostra de
sedimento preparada em Beckers de 250 ml. As amostras foram homogeneizadas e o
de solvente evaporado em uma estufa à temperatura constante de 40ºC e por 12 horas
aproximadamente. Nestas condições houve a completa evaporação da acetonitrila.
Após a secagem, o material foi homogeneizado com o auxilio de um pistilo
de vidro e deste foram separadas quatro alíquotas, sendo que três delas foram
utilizadas no procedimento de extração.
Realizou-se as extrações soxhlet do sedimento preparado, do sedimento
preparado adicionado com a solução dos padrões 1 (spike 1) e do sedimento
preparado adicionado com a solução dos padrões 2 (spike 2).

31
5.2 – Avaliação dos Parâmetros Físico-Químicos da Àgua 5.2.1 – Preparo dos Frascos de Coleta
Os frascos utilizados para coleta das amostras de água foram as garrafas de
plástico tipo PET com capacidade volumétrica de 2000 mL. Os recipientes tiveram
seus rótulos retirados, foi feita escovação com detergente e enxágüe dos mesmos em
água corrente até retirar todo o excesso de detergente. Posteriormente realizou-se a
lavagem com água destilada repetindo o processo por 5 vezes.
5.2.2 – Coleta das Amostras de Água
As amostras de água foram coletadas nos mesmos pontos e dias das coletas de
sedimento. A coleta foi realizada através da imersão do frasco coletor na parte
central do leito do ribeirão e em seguida transferidas para os frascos de coleta,
através de um funil de plástico. O volume coletado das amostras de água em cada
ponto e coleta foi de 2L, armazenados em frascos PET deste volume.
A água foi recolhida da superfície do fluxo, em contra corrente. Antes de
recolher a amostra para análise, o frasco coletor foi homogeneizado com água do
ribeirão por três vezes. O frasco receptor da amostra (PET 2L) também foi
enxaguado por três vezes antes da coleta definitiva. Durante a coleta, os frascos
foram mantidos refrigerados pelo acondicionamento em caixas de isopor contendo
gelo para a preservação das amostras para a avaliação de vários parâmetros. No
laboratório, foram mantidas sob refrigeração em geladeira, dentro do prazo de
validade dos ensaios, antes de serem submetidas às análises.
5.2.3 – Parâmetros Analisados
A qualidade da água pode ser representada através de diversos parâmetros,
que traduzem as suas principais características físicas, químicas e biológicas. Foram
avaliados os seguintes parâmetros: alcalinidade, cloreto, condutividade, cor, dureza,
metais, nitrato, nitrito, resíduos sedimentáveis, sulfato, amônia, turbidez, OD e pH.
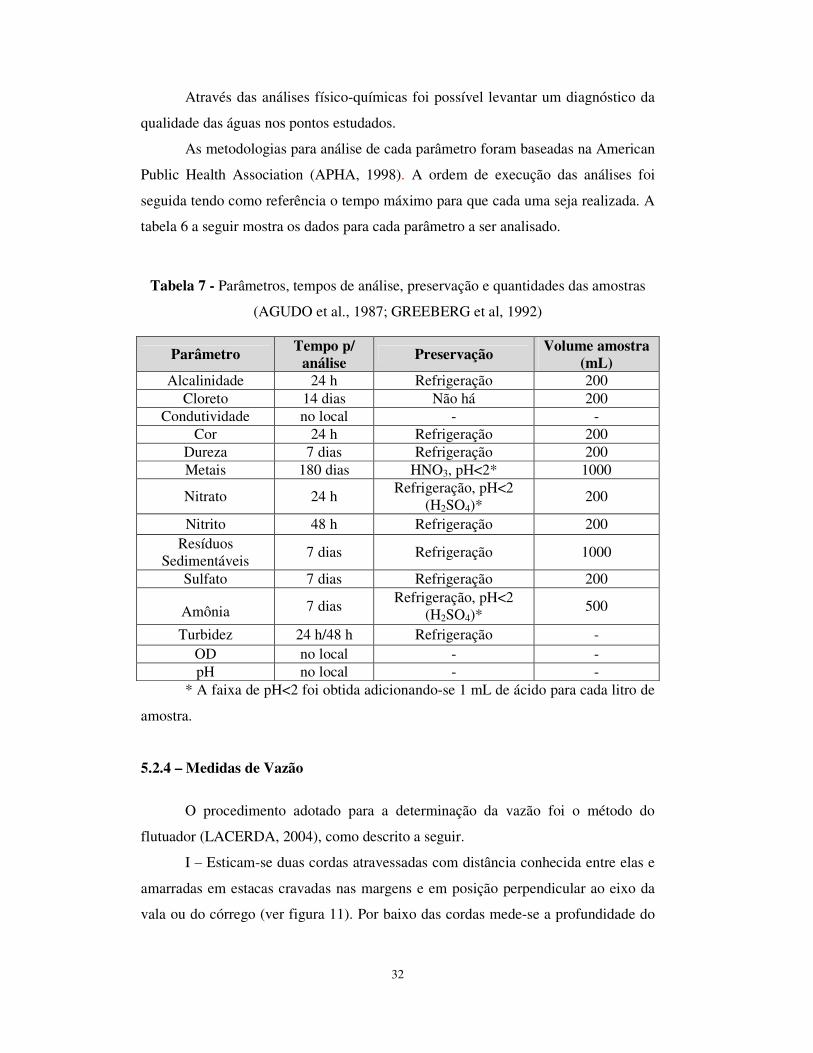
32
Através das análises físico-químicas foi possível levantar um diagnóstico da
qualidade das águas nos pontos estudados.
As metodologias para análise de cada parâmetro foram baseadas na American
Public Health Association (APHA, 1998). A ordem de execução das análises foi
seguida tendo como referência o tempo máximo para que cada uma seja realizada. A
tabela 6 a seguir mostra os dados para cada parâmetro a ser analisado.
Tabela 7 - Parâmetros, tempos de análise, preservação e quantidades das amostras
(AGUDO et al., 1987; GREEBERG et al, 1992)
Parâmetro Tempo p/ análise
Preservação Volume amostra (mL)
Alcalinidade 24 h Refrigeração 200 Cloreto 14 dias Não há 200
Condutividade no local - - Cor 24 h Refrigeração 200
Dureza 7 dias Refrigeração 200 Metais 180 dias HNO3, pH<2* 1000
Nitrato 24 h Refrigeração, pH<2
(H2SO4)* 200
Nitrito 48 h Refrigeração 200 Resíduos
Sedimentáveis 7 dias Refrigeração 1000
Sulfato 7 dias Refrigeração 200
Amônia 7 dias Refrigeração, pH<2
(H2SO4)* 500
Turbidez 24 h/48 h Refrigeração - OD no local - - pH no local - -
* A faixa de pH<2 foi obtida adicionando-se 1 mL de ácido para cada litro de
amostra.
5.2.4 – Medidas de Vazão
O procedimento adotado para a determinação da vazão foi o método do
flutuador (LACERDA, 2004), como descrito a seguir.
I – Esticam-se duas cordas atravessadas com distância conhecida entre elas e
amarradas em estacas cravadas nas margens e em posição perpendicular ao eixo da
vala ou do córrego (ver figura 11). Por baixo das cordas mede-se a profundidade do

33
rio em dez locais diferentes (cinco em baixo de cada corda) e calcula-se a
profundidade média.
II – Mede-se a largura do rio em metros (m).
III – Calcula-se a área (A) da seção do rio:
LhA ×=
Onde:
A = área média da seção transversal do rio, em metros quadrados (m²)
h = profundidade média do rio, em metros (m)
L = largura do rio, em metros (m)
VI – Mede-se a velocidade média (V ) do rio.
Para isto, utiliza-se um flutuador (por exemplo, uma garrafa com água pela
metade). Joga-se o flutuador no rio antes da primeira corda e mede-se o tempo (em
segundos) gasto para o flutuador percorrer a distância entre as duas cordas esticadas.
A velocidade do flutuador é convertida em velocidade média do rio usando-se um
coeficiente de redução, que comumente é 0,85 para flutuador superficial.
tCVFlutuador = FlutuadorRio VV ×= 85,0
Onde:
FlutuadorV = velocidade do flutuador, em m/s
C = distância entre as cordas, em metros (de 4 a 5 vezes a largura do rio)
t = tempo gasto pelo flutuador para ir de uma corda a outra, em segundos (s)
RioV = velocidade média do rio, em m/s
V – Calcula-se a vazão (Q) em m³/s pela fórmula:
RioVAQ ×=

34
Figura 11 - Cálculo da Vazão (flutuador)
5.3 – Avaliação dos Metais e Outros Elementos Químicos em Água 5.3.1 – Preparo dos Frascos de Coleta
A limpeza dos frascos de vidro foi realizada utilizando água e detergente. Os
frascos, as tampas e os batoques foram colocados em um recipiente de grande
volume, mergulhados numa solução água+detergente e deixados de “molho” por
cerca de três semanas. Foi feita a troca periódica da solução durante o tempo de
limpeza. Posteriormente, os frascos foram esvaziados e enxaguados em água de
torneira até eliminar todos os resíduos de detergente. Em seguida, foram enxaguados
com água destilada para garantir a completa descontaminação.
Para avaliação de metais, os frascos coletores receberam tratamento
diferenciado. Após a lavagem com água e detergente, colocou-se ácido clorídrico 1:1
até a metade do frasco, tampou-se e agitou-se energicamente; esvaziou-se o frasco e
enxaguou-se cinco vezes com água destilada. Procedeu-se da mesma maneira com as
tampas (AGUDO, 1987).
5.3.2 – Coleta das Amostras de Água
Para cada um dos quatro pontos amostrados, foram coletados 1000 mL de
água para análise de metais nos mesmos pontos e dias das coletas de sedimento. A
coleta foi realizada como descrito em (5.2.2) para as análises físico-químicas. Com o
auxílio do funil, transferiram-se as amostras de água para os frascos âmbar de 1000
mL. Os frascos foram estocados em caixas de isopor contendo gelo até a chegada ao
laboratório, assegurando a preservação das propriedades físico e químicas das
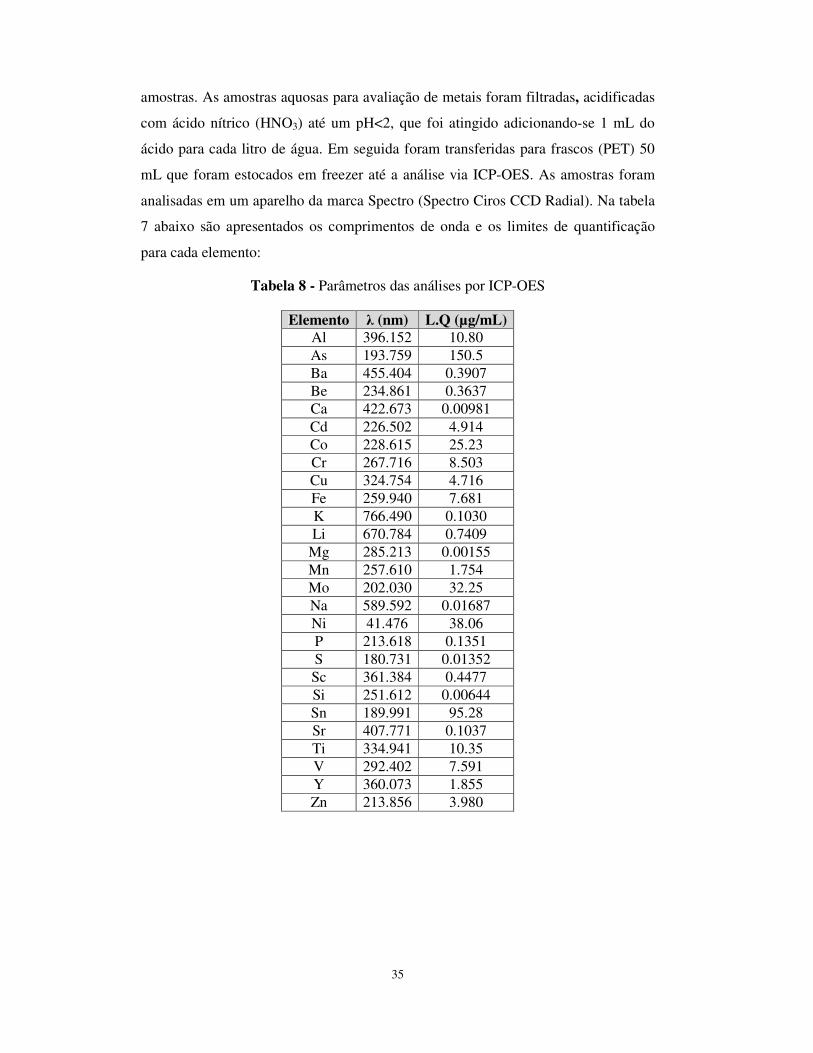
35
amostras. As amostras aquosas para avaliação de metais foram filtradas, acidificadas
com ácido nítrico (HNO3) até um pH<2, que foi atingido adicionando-se 1 mL do
ácido para cada litro de água. Em seguida foram transferidas para frascos (PET) 50
mL que foram estocados em freezer até a análise via ICP-OES. As amostras foram
analisadas em um aparelho da marca Spectro (Spectro Ciros CCD Radial). Na tabela
7 abaixo são apresentados os comprimentos de onda e os limites de quantificação
para cada elemento:
Tabela 8 - Parâmetros das análises por ICP-OES
Elemento λ (nm) L.Q (µg/mL) Al 396.152 10.80 As 193.759 150.5 Ba 455.404 0.3907 Be 234.861 0.3637 Ca 422.673 0.00981 Cd 226.502 4.914 Co 228.615 25.23 Cr 267.716 8.503 Cu 324.754 4.716 Fe 259.940 7.681 K 766.490 0.1030 Li 670.784 0.7409
Mg 285.213 0.00155 Mn 257.610 1.754 Mo 202.030 32.25 Na 589.592 0.01687 Ni 41.476 38.06 P 213.618 0.1351 S 180.731 0.01352 Sc 361.384 0.4477 Si 251.612 0.00644 Sn 189.991 95.28 Sr 407.771 0.1037 Ti 334.941 10.35 V 292.402 7.591 Y 360.073 1.855 Zn 213.856 3.980

36
6. RESULTADOS E DISCUSSÃO
6.1 – Amostras Aquosas
6.1.1- Análises Físico-Químicas
Na tabela 9, são apresentados os dados obtidos e os limites máximos
permitidos para cada parâmetro de acordo com a Resolução CONAMA 357, de 17 de
Março de 2005, que dispõe sobre a classificação dos corpos de água e diretrizes
ambientais para seu enquadramento. Foram considerados os limites estabelecidos por
esta resolução para as águas doces de Classe 1. De acordo com os dados obtidos
pode-se afirmar que, de um modo geral, os parâmetros físico-químicos se
encontraram dentro dos limites estabelecidos por esta resolução.
Para alguns casos específicos, verificou-se o não atendimento aos limites do
CONAMA, como foi o caso do oxigênio dissolvido. Para os pontos 1 (2º coleta) e
para os pontos 3 e 4 (3º coleta) os valores encontrados para OD estão fora dos
padrões estabelecidos para estas amostras. O maior consumo de oxigênio nos pontos
citados pode estar relacionado com o despejo de algum efluente de alto teor de
matéria orgânica que esteja contaminando as águas. Foram encontrados valores de
turbidez acima do limite para as amostras da segunda coleta, fato que pode estar
relacionado com o período de chuvas. Também foram verificadas amostras com
elevados valores no parâmetro cor e uma maior vazão do leito do rio para a segunda
coleta. Apesar de estarem dentro do limite da resolução 357, os teores de nitrito e
nitrato aumentaram no sentido a jusante do rio, pontos 3 e 4, confirmando a
contribuição dos lançamentos de esgotos nas águas do ribeirão. Elevados valores de
alcalinidade foram encontrados nas amostras da terceira coleta, principalmente nos
pontos 1 e 2. Os valores de cloreto são altos, em geral, nos dois primeiros pontos
amostrados. A dureza apresentou valores altos principalmente para os dois primeiros
pontos na segunda coleta. Os valores de sulfato variaram na primeira e terceira
coletas em um patamar mais elevado que na segunda. Os maiores valores foram
identificados nos pontos 1 e 2 na primeira campanha de amostragem. O
comportamento do pH foi de ligeiramente ácido, passando pela neutralidade e a
tendência ao pH alcalino nas 3 coletas.
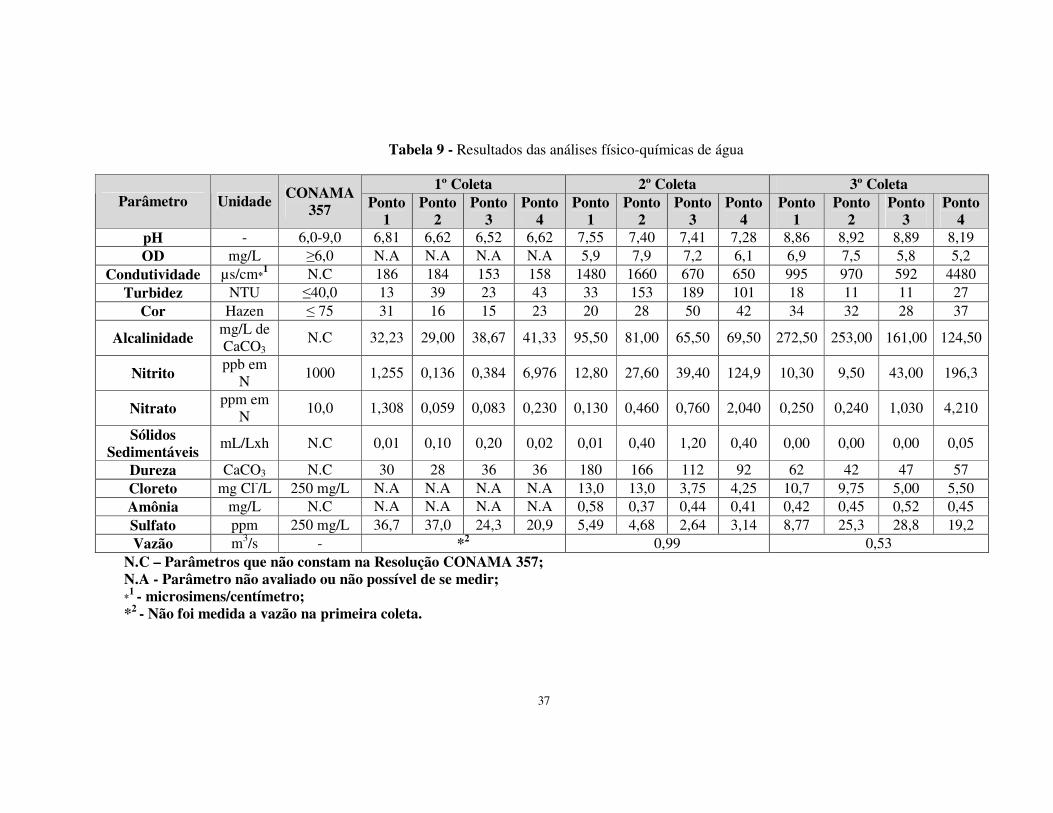
37
Tabela 9 - Resultados das análises físico-químicas de água
N.C – Parâmetros que não constam na Resolução CONAMA 357; N.A - Parâmetro não avaliado ou não possível de se medir; *
1 - microsimens/centímetro; *2 - Não foi medida a vazão na primeira coleta.
1º Coleta 2º Coleta 3º Coleta Parâmetro Unidade CONAMA
357 Ponto 1
Ponto 2
Ponto 3
Ponto 4
Ponto 1
Ponto 2
Ponto 3
Ponto 4
Ponto 1
Ponto 2
Ponto 3
Ponto 4
pH - 6,0-9,0 6,81 6,62 6,52 6,62 7,55 7,40 7,41 7,28 8,86 8,92 8,89 8,19 OD mg/L ≥6,0 N.A N.A N.A N.A 5,9 7,9 7,2 6,1 6,9 7,5 5,8 5,2
Condutividade µs/cm*1 N.C 186 184 153 158 1480 1660 670 650 995 970 592 4480
Turbidez NTU ≤40,0 13 39 23 43 33 153 189 101 18 11 11 27 Cor Hazen ≤ 75 31 16 15 23 20 28 50 42 34 32 28 37
Alcalinidade mg/L de CaCO3
N.C 32,23 29,00 38,67 41,33 95,50 81,00 65,50 69,50 272,50 253,00 161,00 124,50
Nitrito ppb em
N 1000 1,255 0,136 0,384 6,976 12,80 27,60 39,40 124,9 10,30 9,50 43,00 196,3
Nitrato ppm em
N 10,0 1,308 0,059 0,083 0,230 0,130 0,460 0,760 2,040 0,250 0,240 1,030 4,210
Sólidos Sedimentáveis
mL/Lxh N.C 0,01 0,10 0,20 0,02 0,01 0,40 1,20 0,40 0,00 0,00 0,00 0,05
Dureza CaCO3 N.C 30 28 36 36 180 166 112 92 62 42 47 57 Cloreto mg Cl-/L 250 mg/L N.A N.A N.A N.A 13,0 13,0 3,75 4,25 10,7 9,75 5,00 5,50 Amônia mg/L N.C N.A N.A N.A N.A 0,58 0,37 0,44 0,41 0,42 0,45 0,52 0,45 Sulfato ppm 250 mg/L 36,7 37,0 24,3 20,9 5,49 4,68 2,64 3,14 8,77 25,3 28,8 19,2 Vazão m3/s - *2 0,99 0,53

38
6.1.2- Análises de Metais e Outros Elementos Químicos
Os valores de concentração dos metais analisados por ICP-OES são
apresentados na Tabela 9. Os limites máximos permitidos, segundo a Resolução
CONAMA nº. 357 de 17 de Março de 2005, para a concentração de metais em águas
doces de Classe 1, foram comparados com os resultados encontrados para o presente
trabalho.
Teores elevados de Alumínio foram observados na maioria dos pontos
amostrados e em todas as campanhas de coleta, sendo que no ponto 3 da 2º coleta o
valor encontrado foi superior em cerca de 10 vezes o valor máximo permitido para este
metal. Estes valores podem ser explicados pela concentração natural de alumínio em
minerais da região, como a bauxita ou pela presença da fábrica de alumínio.
Arsênio foi encontrado acima do limite estabelecido para os pontos 2 e 4 da 1º
coleta sendo que neste último ponto o valor estava cerca de 4 vezes acima do máximo
estabelecido pela resolução no 357. Estes valores são preocupantes, porém podem ser
explicados pela formação geológica da região, com presença de arseno-piritas dentre
outros minerais.
As concentrações de cobre foram elevadas para todos os pontos da 1º coleta e
para o ponto 2 da 2º coleta que pode estar relacionado com lançamento de efluentes
contendo metais nas águas do ribeirão.
Algumas amostras apresentaram concentrações elevadas no teor de ferro. Para
todos os pontos amostrados e em todas as coletas, os níveis de manganês presentes nas
amostras foram superiores aos limites estabelecidos pelo órgão ambiental. Estes valores
podem ser explicados pela formação de complexos minerais ferríferos presentes na
região de Ouro Preto.
Os valores de concentração de fósforo aumentam nos pontos 3 e 4 o que
confirma a presença de esgotos lançados no ribeirão. Estes pontos se encontram a
jusante das várias emissões de esgoto na cidade de Ouro Preto.
Foi avaliada a presença de chumbo nas amostras aquosas apenas na primeira
coleta. Os valores encontrados indicam valores bastante superiores aos limites do
CONAMA. Isto pode indicar que o Ribeirão do Funil vem sendo contaminado por
efluentes que contêm metais tóxicos.

39
A presença de alguns metais e outros elementos químicos nas amostras
analisadas em concentrações superiores aos máximos estabelecidos pela resolução
CONAMA 357 confirma a baixa qualidade das águas do ribeirão no trecho avaliado e
indica fontes que contribuem para a degradação ambiental desta região.
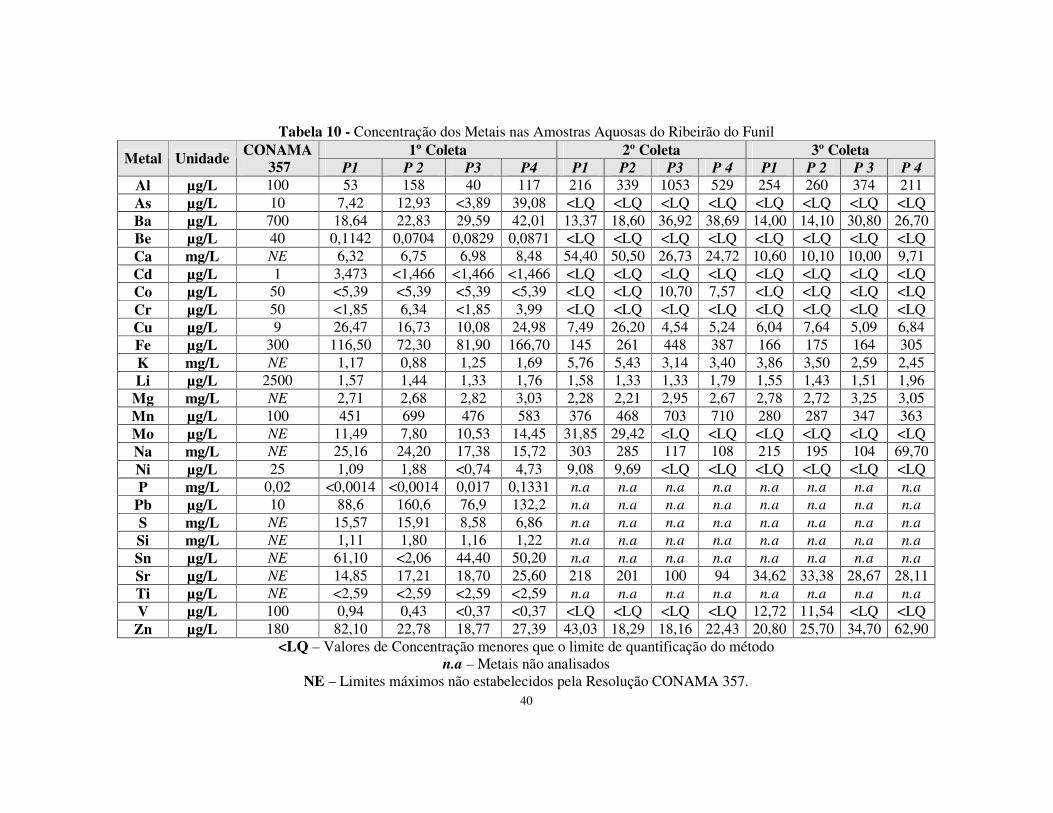
40
Tabela 10 - Concentração dos Metais nas Amostras Aquosas do Ribeirão do Funil 1º Coleta 2º Coleta 3º Coleta Metal Unidade CONAMA
357 P1 P 2 P3 P4 P1 P2 P3 P 4 P1 P 2 P 3 P 4
Al µg/L 100 53 158 40 117 216 339 1053 529 254 260 374 211 As µg/L 10 7,42 12,93 <3,89 39,08 <LQ <LQ <LQ <LQ <LQ <LQ <LQ <LQ Ba µg/L 700 18,64 22,83 29,59 42,01 13,37 18,60 36,92 38,69 14,00 14,10 30,80 26,70 Be µg/L 40 0,1142 0,0704 0,0829 0,0871 <LQ <LQ <LQ <LQ <LQ <LQ <LQ <LQ Ca mg/L NE 6,32 6,75 6,98 8,48 54,40 50,50 26,73 24,72 10,60 10,10 10,00 9,71 Cd µg/L 1 3,473 <1,466 <1,466 <1,466 <LQ <LQ <LQ <LQ <LQ <LQ <LQ <LQ Co µg/L 50 <5,39 <5,39 <5,39 <5,39 <LQ <LQ 10,70 7,57 <LQ <LQ <LQ <LQ Cr µg/L 50 <1,85 6,34 <1,85 3,99 <LQ <LQ <LQ <LQ <LQ <LQ <LQ <LQ Cu µg/L 9 26,47 16,73 10,08 24,98 7,49 26,20 4,54 5,24 6,04 7,64 5,09 6,84 Fe µg/L 300 116,50 72,30 81,90 166,70 145 261 448 387 166 175 164 305 K mg/L NE 1,17 0,88 1,25 1,69 5,76 5,43 3,14 3,40 3,86 3,50 2,59 2,45 Li µg/L 2500 1,57 1,44 1,33 1,76 1,58 1,33 1,33 1,79 1,55 1,43 1,51 1,96
Mg mg/L NE 2,71 2,68 2,82 3,03 2,28 2,21 2,95 2,67 2,78 2,72 3,25 3,05 Mn µg/L 100 451 699 476 583 376 468 703 710 280 287 347 363 Mo µg/L NE 11,49 7,80 10,53 14,45 31,85 29,42 <LQ <LQ <LQ <LQ <LQ <LQ Na mg/L NE 25,16 24,20 17,38 15,72 303 285 117 108 215 195 104 69,70 Ni µg/L 25 1,09 1,88 <0,74 4,73 9,08 9,69 <LQ <LQ <LQ <LQ <LQ <LQ P mg/L 0,02 <0,0014 <0,0014 0,017 0,1331 n.a n.a n.a n.a n.a n.a n.a n.a
Pb µg/L 10 88,6 160,6 76,9 132,2 n.a n.a n.a n.a n.a n.a n.a n.a S mg/L NE 15,57 15,91 8,58 6,86 n.a n.a n.a n.a n.a n.a n.a n.a Si mg/L NE 1,11 1,80 1,16 1,22 n.a n.a n.a n.a n.a n.a n.a n.a Sn µg/L NE 61,10 <2,06 44,40 50,20 n.a n.a n.a n.a n.a n.a n.a n.a Sr µg/L NE 14,85 17,21 18,70 25,60 218 201 100 94 34,62 33,38 28,67 28,11 Ti µg/L NE <2,59 <2,59 <2,59 <2,59 n.a n.a n.a n.a n.a n.a n.a n.a V µg/L 100 0,94 0,43 <0,37 <0,37 <LQ <LQ <LQ <LQ 12,72 11,54 <LQ <LQ Zn µg/L 180 82,10 22,78 18,77 27,39 43,03 18,29 18,16 22,43 20,80 25,70 34,70 62,90
<LQ – Valores de Concentração menores que o limite de quantificação do método n.a – Metais não analisados
NE – Limites máximos não estabelecidos pela Resolução CONAMA 357.
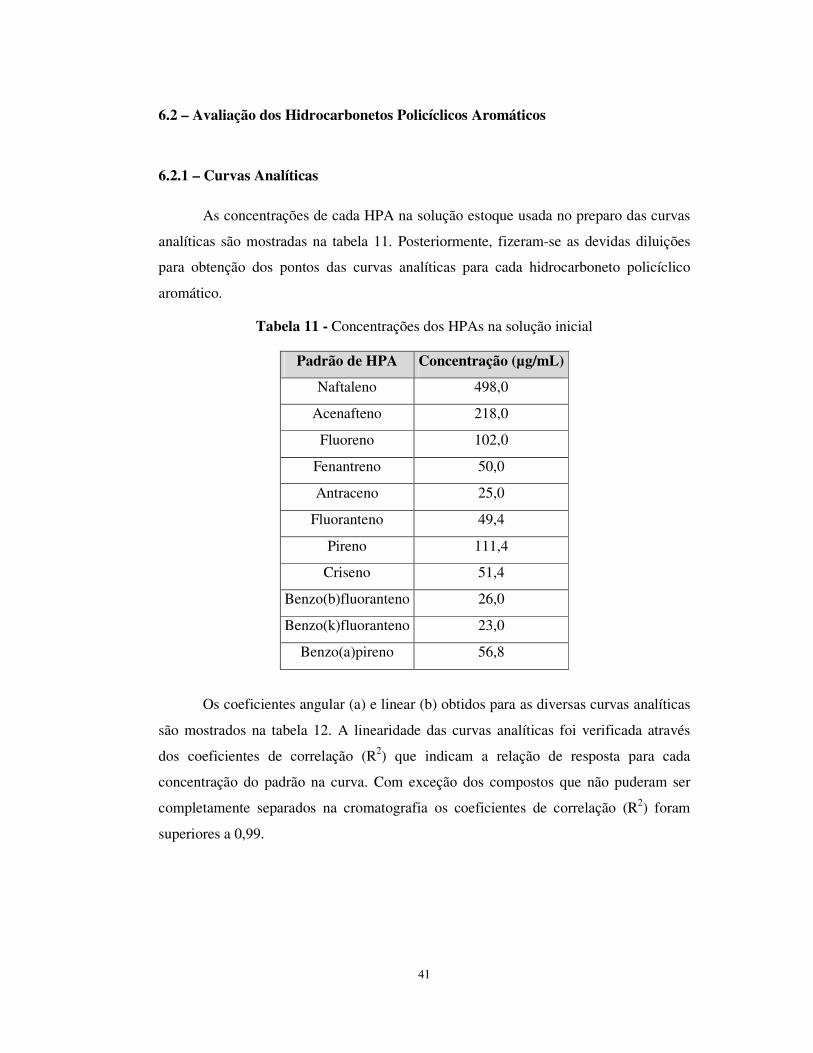
41
6.2 – Avaliação dos Hidrocarbonetos Policíclicos Aromáticos
6.2.1 – Curvas Analíticas
As concentrações de cada HPA na solução estoque usada no preparo das curvas
analíticas são mostradas na tabela 11. Posteriormente, fizeram-se as devidas diluições
para obtenção dos pontos das curvas analíticas para cada hidrocarboneto policíclico
aromático.
Tabela 11 - Concentrações dos HPAs na solução inicial
Padrão de HPA Concentração (µg/mL)
Naftaleno 498,0
Acenafteno 218,0
Fluoreno 102,0
Fenantreno 50,0
Antraceno 25,0
Fluoranteno 49,4
Pireno 111,4
Criseno 51,4
Benzo(b)fluoranteno 26,0
Benzo(k)fluoranteno 23,0
Benzo(a)pireno 56,8
Os coeficientes angular (a) e linear (b) obtidos para as diversas curvas analíticas
são mostrados na tabela 12. A linearidade das curvas analíticas foi verificada através
dos coeficientes de correlação (R2) que indicam a relação de resposta para cada
concentração do padrão na curva. Com exceção dos compostos que não puderam ser
completamente separados na cromatografia os coeficientes de correlação (R2) foram
superiores a 0,99.
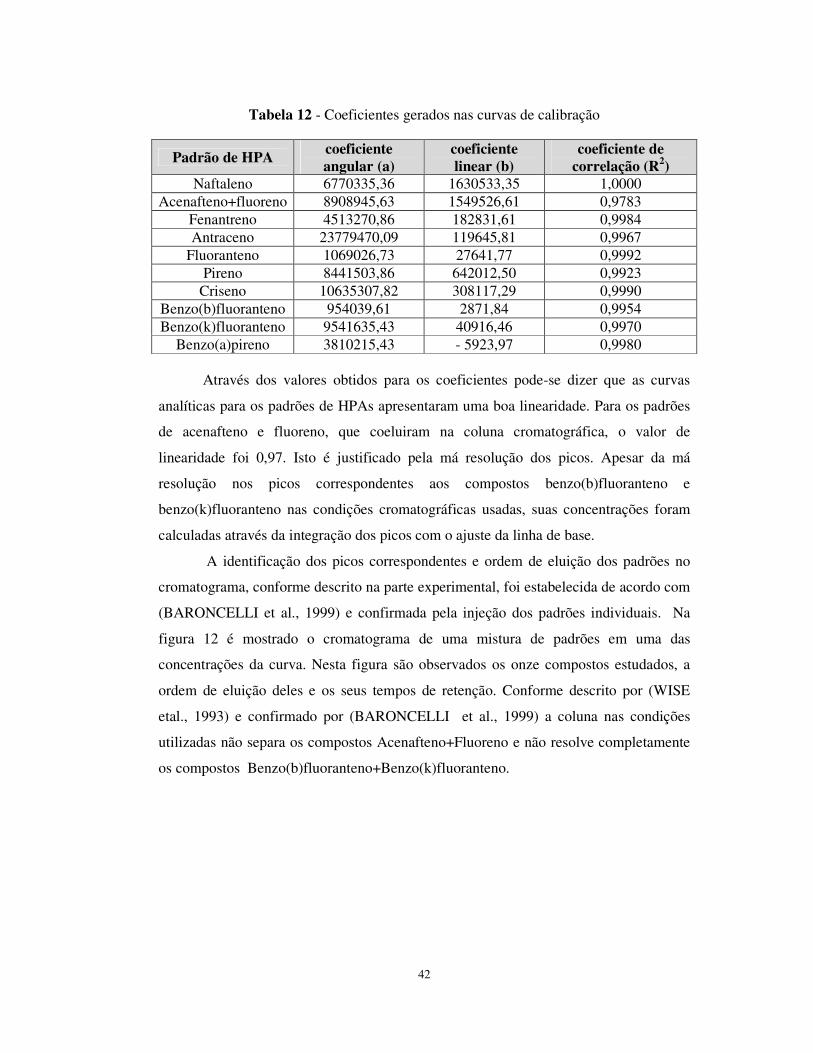
42
Tabela 12 - Coeficientes gerados nas curvas de calibração
Padrão de HPA coeficiente angular (a)
coeficiente linear (b)
coeficiente de correlação (R2)
Naftaleno 6770335,36 1630533,35 1,0000 Acenafteno+fluoreno 8908945,63 1549526,61 0,9783
Fenantreno 4513270,86 182831,61 0,9984 Antraceno 23779470,09 119645,81 0,9967
Fluoranteno 1069026,73 27641,77 0,9992 Pireno 8441503,86 642012,50 0,9923
Criseno 10635307,82 308117,29 0,9990 Benzo(b)fluoranteno 954039,61 2871,84 0,9954 Benzo(k)fluoranteno 9541635,43 40916,46 0,9970
Benzo(a)pireno 3810215,43 - 5923,97 0,9980
Através dos valores obtidos para os coeficientes pode-se dizer que as curvas
analíticas para os padrões de HPAs apresentaram uma boa linearidade. Para os padrões
de acenafteno e fluoreno, que coeluiram na coluna cromatográfica, o valor de
linearidade foi 0,97. Isto é justificado pela má resolução dos picos. Apesar da má
resolução nos picos correspondentes aos compostos benzo(b)fluoranteno e
benzo(k)fluoranteno nas condições cromatográficas usadas, suas concentrações foram
calculadas através da integração dos picos com o ajuste da linha de base.
A identificação dos picos correspondentes e ordem de eluição dos padrões no
cromatograma, conforme descrito na parte experimental, foi estabelecida de acordo com
(BARONCELLI et al., 1999) e confirmada pela injeção dos padrões individuais. Na
figura 12 é mostrado o cromatograma de uma mistura de padrões em uma das
concentrações da curva. Nesta figura são observados os onze compostos estudados, a
ordem de eluição deles e os seus tempos de retenção. Conforme descrito por (WISE
etal., 1993) e confirmado por (BARONCELLI et al., 1999) a coluna nas condições
utilizadas não separa os compostos Acenafteno+Fluoreno e não resolve completamente
os compostos Benzo(b)fluoranteno+Benzo(k)fluoranteno.
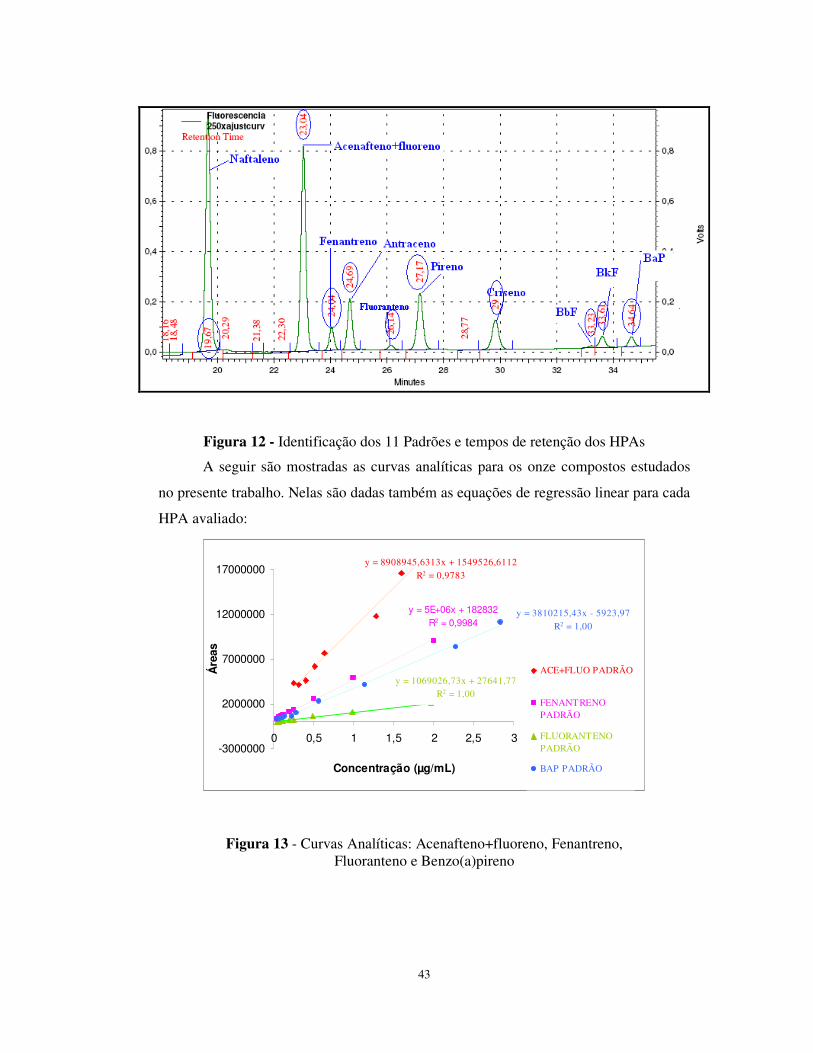
43
Figura 12 - Identificação dos 11 Padrões e tempos de retenção dos HPAs
A seguir são mostradas as curvas analíticas para os onze compostos estudados
no presente trabalho. Nelas são dadas também as equações de regressão linear para cada
HPA avaliado:
y = 8908945,6313x + 1549526,6112R2 = 0,9783
y = 5E+06x + 182832
R2 = 0,9984
y = 1069026,73x + 27641,77R2 = 1,00
y = 3810215,43x - 5923,97R2 = 1,00
-3000000
2000000
7000000
12000000
17000000
0 0,5 1 1,5 2 2,5 3
Concentração (µg/mL)
Áre
as
ACE+FLUO PADRÃO
FENANTRENOPADRÃO
FLUORANTENOPADRÃO
BAP PADRÃO
Figura 13 - Curvas Analíticas: Acenafteno+fluoreno, Fenantreno, Fluoranteno e Benzo(a)pireno
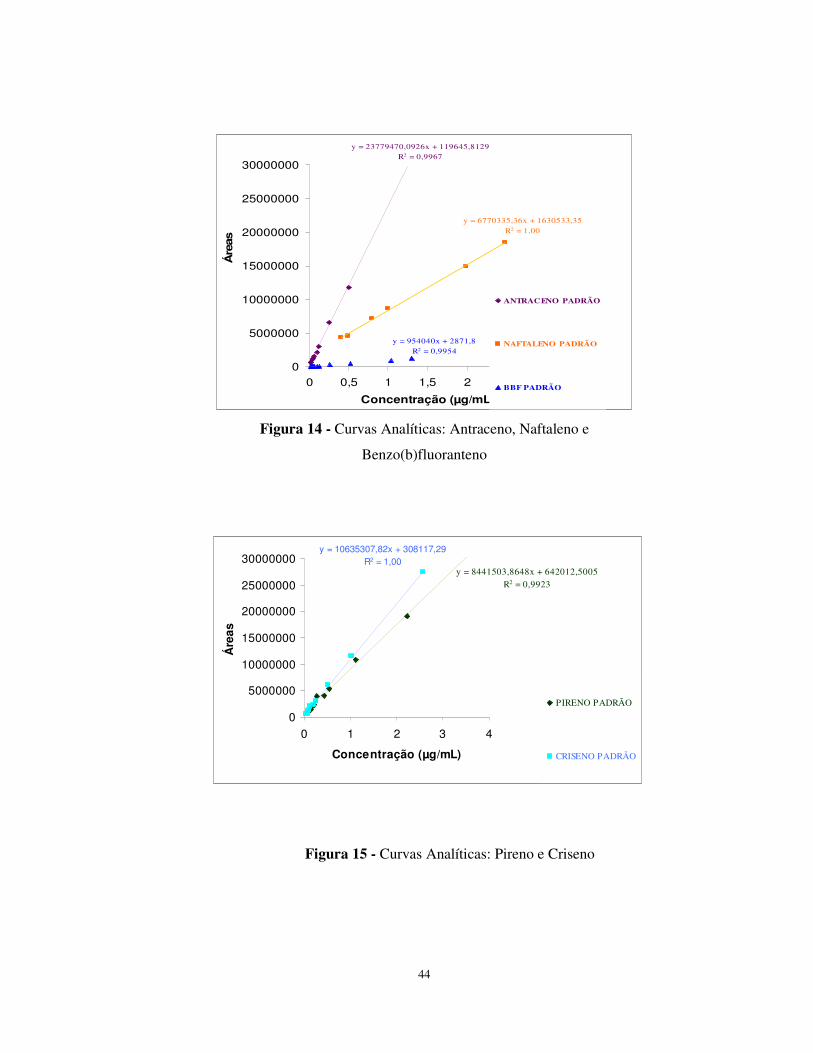
44
y = 23779470,0926x + 119645,8129R2 = 0,9967
y = 6770335,36x + 1630533,35R2 = 1,00
y = 954040x + 2871,8R2 = 0,9954
0
5000000
10000000
15000000
20000000
25000000
30000000
0 0,5 1 1,5 2 2,5 3
Concentração (µg/mL)
Áre
as
ANTRACENO PADRÃO
NAFTALENO PADRÃO
BBF PADRÃO
Figura 14 - Curvas Analíticas: Antraceno, Naftaleno e
Benzo(b)fluoranteno
y = 8441503,8648x + 642012,5005R2 = 0,9923
y = 10635307,82x + 308117,29
R2 = 1,00
0
5000000
10000000
15000000
20000000
25000000
30000000
0 1 2 3 4
Concentração (µg/mL)
Áre
as
PIRENO PADRÃO
CRISENO PADRÃO
Figura 15 - Curvas Analíticas: Pireno e Criseno
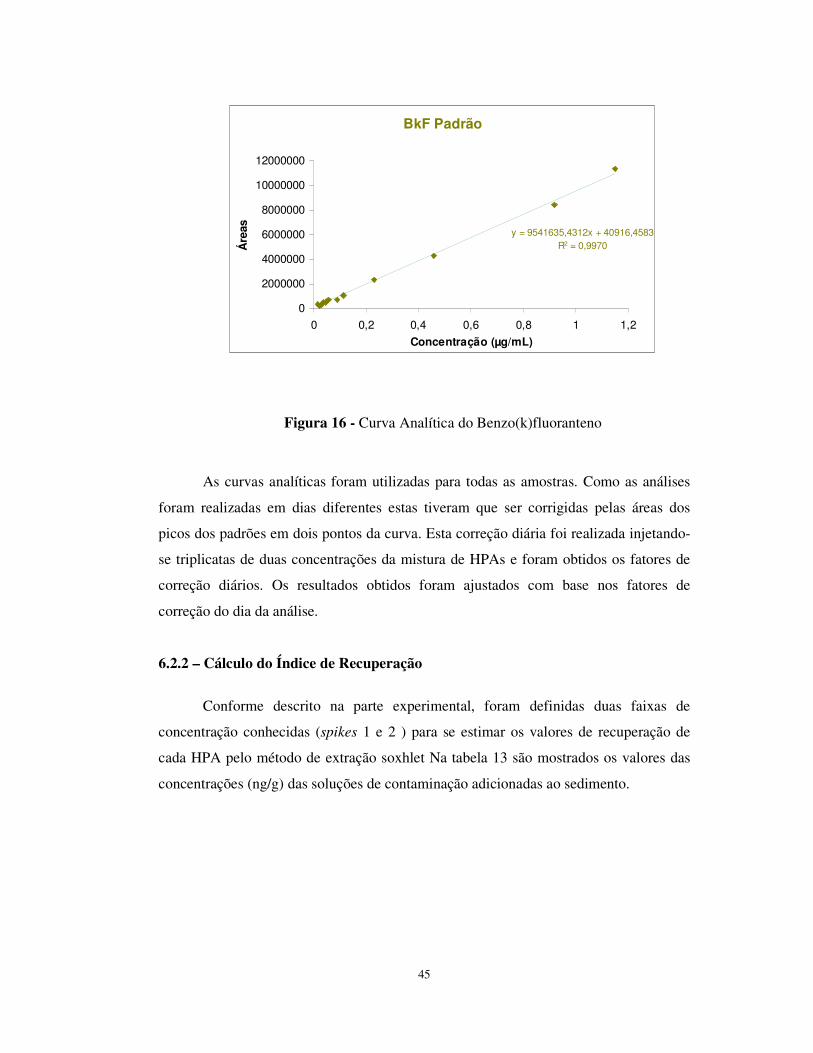
45
As curvas analíticas foram utilizadas para todas as amostras. Como as análises
foram realizadas em dias diferentes estas tiveram que ser corrigidas pelas áreas dos
picos dos padrões em dois pontos da curva. Esta correção diária foi realizada injetando-
se triplicatas de duas concentrações da mistura de HPAs e foram obtidos os fatores de
correção diários. Os resultados obtidos foram ajustados com base nos fatores de
correção do dia da análise.
6.2.2 – Cálculo do Índice de Recuperação
Conforme descrito na parte experimental, foram definidas duas faixas de
concentração conhecidas (spikes 1 e 2 ) para se estimar os valores de recuperação de
cada HPA pelo método de extração soxhlet Na tabela 13 são mostrados os valores das
concentrações (ng/g) das soluções de contaminação adicionadas ao sedimento.
BkF Padrão
y = 9541635,4312x + 40916,4583
R2 = 0,9970
0
2000000
4000000
6000000
8000000
10000000
12000000
0 0,2 0,4 0,6 0,8 1 1,2
Concentração (µg/mL)
Áre
as
Figura 16 - Curva Analítica do Benzo(k)fluoranteno
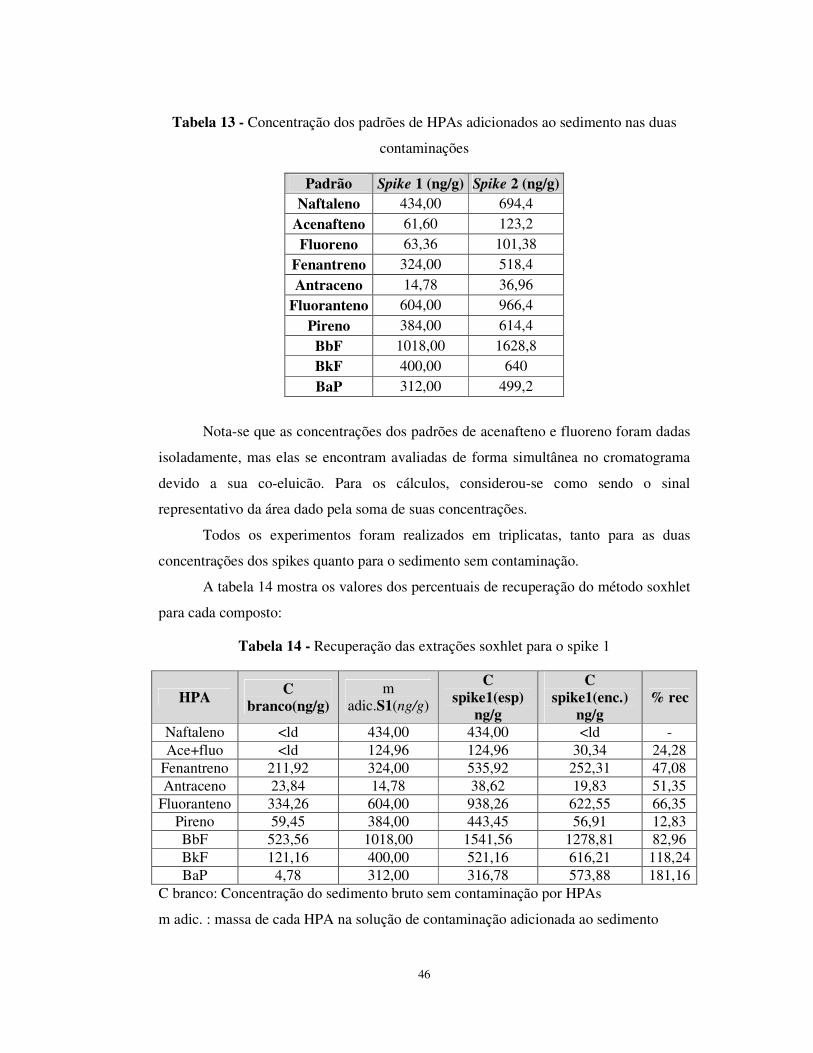
46
Tabela 13 - Concentração dos padrões de HPAs adicionados ao sedimento nas duas
contaminações
Padrão Spike 1 (ng/g) Spike 2 (ng/g) Naftaleno 434,00 694,4
Acenafteno 61,60 123,2 Fluoreno 63,36 101,38
Fenantreno 324,00 518,4
Antraceno 14,78 36,96 Fluoranteno 604,00 966,4
Pireno 384,00 614,4 BbF 1018,00 1628,8 BkF 400,00 640 BaP 312,00 499,2
Nota-se que as concentrações dos padrões de acenafteno e fluoreno foram dadas
isoladamente, mas elas se encontram avaliadas de forma simultânea no cromatograma
devido a sua co-eluicão. Para os cálculos, considerou-se como sendo o sinal
representativo da área dado pela soma de suas concentrações.
Todos os experimentos foram realizados em triplicatas, tanto para as duas
concentrações dos spikes quanto para o sedimento sem contaminação.
A tabela 14 mostra os valores dos percentuais de recuperação do método soxhlet
para cada composto:
Tabela 14 - Recuperação das extrações soxhlet para o spike 1
HPA C
branco(ng/g) m
adic.S1(ng/g)
C spike1(esp)
ng/g
C spike1(enc.)
ng/g % rec
Naftaleno <ld 434,00 434,00 <ld - Ace+fluo <ld 124,96 124,96 30,34 24,28
Fenantreno 211,92 324,00 535,92 252,31 47,08 Antraceno 23,84 14,78 38,62 19,83 51,35
Fluoranteno 334,26 604,00 938,26 622,55 66,35 Pireno 59,45 384,00 443,45 56,91 12,83 BbF 523,56 1018,00 1541,56 1278,81 82,96 BkF 121,16 400,00 521,16 616,21 118,24 BaP 4,78 312,00 316,78 573,88 181,16
C branco: Concentração do sedimento bruto sem contaminação por HPAs
m adic. : massa de cada HPA na solução de contaminação adicionada ao sedimento
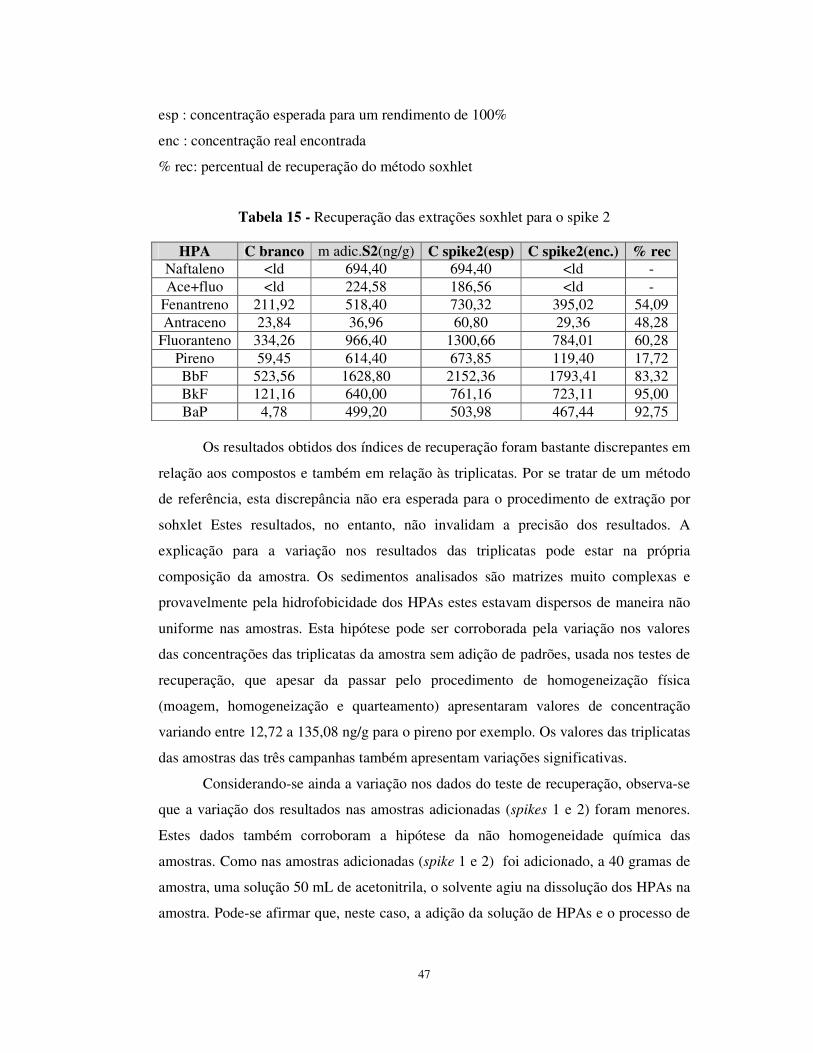
47
esp : concentração esperada para um rendimento de 100%
enc : concentração real encontrada
% rec: percentual de recuperação do método soxhlet
Tabela 15 - Recuperação das extrações soxhlet para o spike 2
HPA C branco m adic.S2(ng/g) C spike2(esp) C spike2(enc.) % rec Naftaleno <ld 694,40 694,40 <ld - Ace+fluo <ld 224,58 186,56 <ld -
Fenantreno 211,92 518,40 730,32 395,02 54,09 Antraceno 23,84 36,96 60,80 29,36 48,28
Fluoranteno 334,26 966,40 1300,66 784,01 60,28 Pireno 59,45 614,40 673,85 119,40 17,72 BbF 523,56 1628,80 2152,36 1793,41 83,32 BkF 121,16 640,00 761,16 723,11 95,00 BaP 4,78 499,20 503,98 467,44 92,75
Os resultados obtidos dos índices de recuperação foram bastante discrepantes em
relação aos compostos e também em relação às triplicatas. Por se tratar de um método
de referência, esta discrepância não era esperada para o procedimento de extração por
sohxlet Estes resultados, no entanto, não invalidam a precisão dos resultados. A
explicação para a variação nos resultados das triplicatas pode estar na própria
composição da amostra. Os sedimentos analisados são matrizes muito complexas e
provavelmente pela hidrofobicidade dos HPAs estes estavam dispersos de maneira não
uniforme nas amostras. Esta hipótese pode ser corroborada pela variação nos valores
das concentrações das triplicatas da amostra sem adição de padrões, usada nos testes de
recuperação, que apesar da passar pelo procedimento de homogeneização física
(moagem, homogeneização e quarteamento) apresentaram valores de concentração
variando entre 12,72 a 135,08 ng/g para o pireno por exemplo. Os valores das triplicatas
das amostras das três campanhas também apresentam variações significativas.
Considerando-se ainda a variação nos dados do teste de recuperação, observa-se
que a variação dos resultados nas amostras adicionadas (spikes 1 e 2) foram menores.
Estes dados também corroboram a hipótese da não homogeneidade química das
amostras. Como nas amostras adicionadas (spike 1 e 2) foi adicionado, a 40 gramas de
amostra, uma solução 50 mL de acetonitrila, o solvente agiu na dissolução dos HPAs na
amostra. Pode-se afirmar que, neste caso, a adição da solução de HPAs e o processo de

48
homogeneização em fase líquida garantiram amostras com maior homogeneidade e
portanto, maior repetibilidade de resultados.
Baixos índices de recuperação eram esperados em compostos mais voláteis, de
menor número de anéis aromáticos, como o naftaleno. As várias etapas necessárias ao
preparo dos extratos: secagem da amostra; extração soxhlet; concentração e evaporação
de solventes; e “clean up” podem levar a perdas significativas dos compostos com
maior pressão de vapor.
Por outro lado, os dados obtidos na etapa de cromatografia (CLAE) foram
bastante repetitivos, em termos de variabilidade para as áreas dos picos, mesmo quando
realizados em dias diferentes. Isto pode ser observado nos dados das curvas analíticas,
que apresentam boa linearidade e dados repetitivos nos valores de áreas dos padrões de
ajuste das curvas em diferentes dias.
Diante do apresentado acima, os resultados analíticos para as amostras das três
campanhas devem ser considerados como a média das determinações, pois mesmo após
as etapas de homogeneização física, não foi possível garantir a homogeneidade química.
A amostragem, neste caso, deve ser considerada como amostragem composta e
os teores de HPAs nas amostras dados pelas médias das determinações.
6.2.3 – HPAs nas Amostras Ambientais
Através dos cromatogramas dos padrões com os tempos de retenção de cada
composto, foi possível identificar cada HPA presente na amostra e os valores das suas
respectivas áreas foram usados como dados para sua quantificação. Nas tabelas 16, 17 e
18, são mostradas as concentrações dos HPAs nas três coletas. Não foi realizada
triplicata para a extração soxhlet das amostras na primeira coleta. Não foram analisados
os HPAs nos pontos 3 e 4 por perdas nas amostras antes da etapa final de preparação.
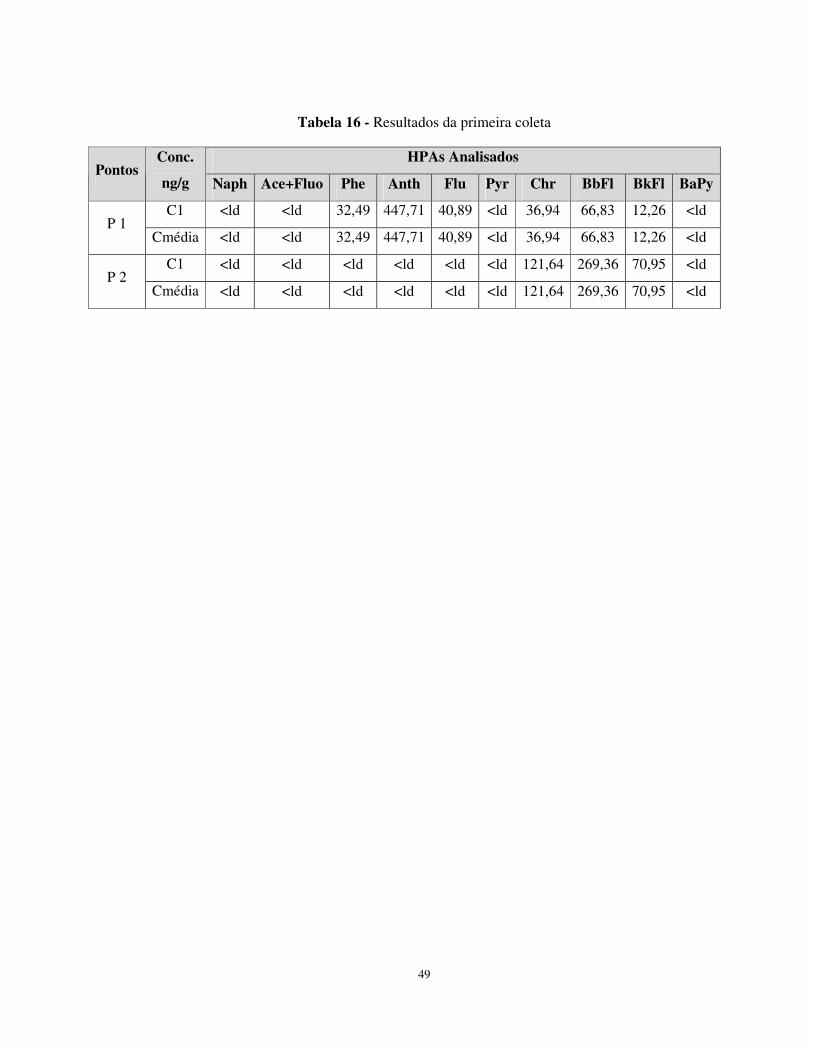
49
Tabela 16 - Resultados da primeira coleta
HPAs Analisados Pontos
Conc.
ng/g Naph Ace+Fluo Phe Anth Flu Pyr Chr BbFl BkFl BaPy
C1 <ld <ld 32,49 447,71 40,89 <ld 36,94 66,83 12,26 <ld P 1
Cmédia <ld <ld 32,49 447,71 40,89 <ld 36,94 66,83 12,26 <ld
C1 <ld <ld <ld <ld <ld <ld 121,64 269,36 70,95 <ld P 2
Cmédia <ld <ld <ld <ld <ld <ld 121,64 269,36 70,95 <ld
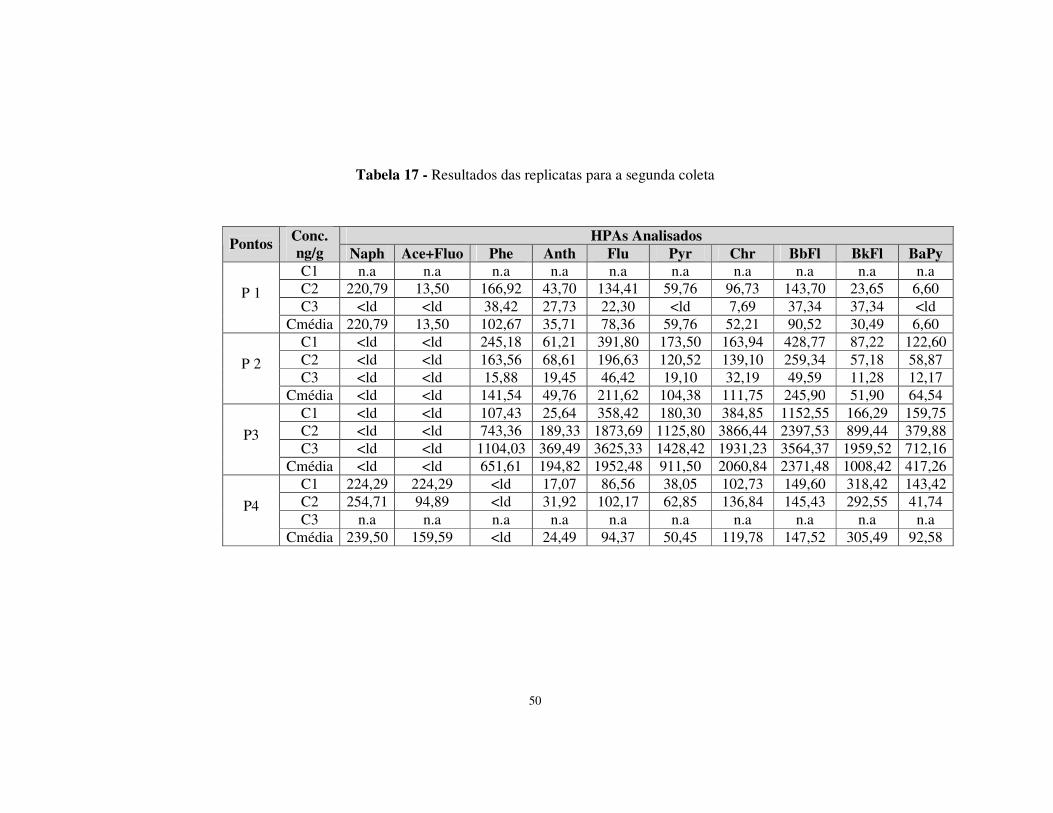
50
Tabela 17 - Resultados das replicatas para a segunda coleta
HPAs Analisados Pontos Conc. ng/g Naph Ace+Fluo Phe Anth Flu Pyr Chr BbFl BkFl BaPy C1 n.a n.a n.a n.a n.a n.a n.a n.a n.a n.a C2 220,79 13,50 166,92 43,70 134,41 59,76 96,73 143,70 23,65 6,60 C3 <ld <ld 38,42 27,73 22,30 <ld 7,69 37,34 37,34 <ld
P 1
Cmédia 220,79 13,50 102,67 35,71 78,36 59,76 52,21 90,52 30,49 6,60 C1 <ld <ld 245,18 61,21 391,80 173,50 163,94 428,77 87,22 122,60 C2 <ld <ld 163,56 68,61 196,63 120,52 139,10 259,34 57,18 58,87 C3 <ld <ld 15,88 19,45 46,42 19,10 32,19 49,59 11,28 12,17
P 2
Cmédia <ld <ld 141,54 49,76 211,62 104,38 111,75 245,90 51,90 64,54 C1 <ld <ld 107,43 25,64 358,42 180,30 384,85 1152,55 166,29 159,75 C2 <ld <ld 743,36 189,33 1873,69 1125,80 3866,44 2397,53 899,44 379,88 C3 <ld <ld 1104,03 369,49 3625,33 1428,42 1931,23 3564,37 1959,52 712,16
P3
Cmédia <ld <ld 651,61 194,82 1952,48 911,50 2060,84 2371,48 1008,42 417,26 C1 224,29 224,29 <ld 17,07 86,56 38,05 102,73 149,60 318,42 143,42 C2 254,71 94,89 <ld 31,92 102,17 62,85 136,84 145,43 292,55 41,74 C3 n.a n.a n.a n.a n.a n.a n.a n.a n.a n.a
P4
Cmédia 239,50 159,59 <ld 24,49 94,37 50,45 119,78 147,52 305,49 92,58
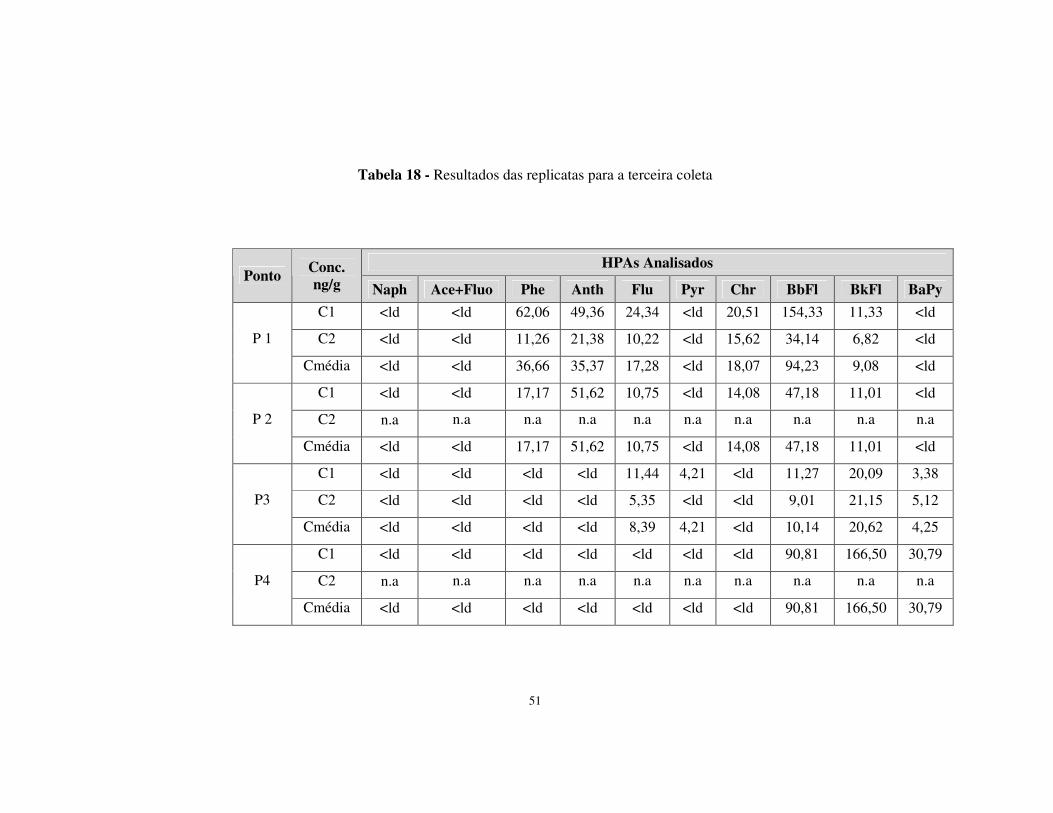
51
Tabela 18 - Resultados das replicatas para a terceira coleta
HPAs Analisados Ponto
Conc. ng/g Naph Ace+Fluo Phe Anth Flu Pyr Chr BbFl BkFl BaPy
C1 <ld <ld 62,06 49,36 24,34 <ld 20,51 154,33 11,33 <ld
C2 <ld <ld 11,26 21,38 10,22 <ld 15,62 34,14 6,82 <ld P 1
Cmédia <ld <ld 36,66 35,37 17,28 <ld 18,07 94,23 9,08 <ld
C1 <ld <ld 17,17 51,62 10,75 <ld 14,08 47,18 11,01 <ld
C2 n.a n.a n.a n.a n.a n.a n.a n.a n.a n.a P 2
Cmédia <ld <ld 17,17 51,62 10,75 <ld 14,08 47,18 11,01 <ld
C1 <ld <ld <ld <ld 11,44 4,21 <ld 11,27 20,09 3,38
C2 <ld <ld <ld <ld 5,35 <ld <ld 9,01 21,15 5,12 P3
Cmédia <ld <ld <ld <ld 8,39 4,21 <ld 10,14 20,62 4,25
C1 <ld <ld <ld <ld <ld <ld <ld 90,81 166,50 30,79
C2 n.a n.a n.a n.a n.a n.a n.a n.a n.a n.a P4
Cmédia <ld <ld <ld <ld <ld <ld <ld 90,81 166,50 30,79
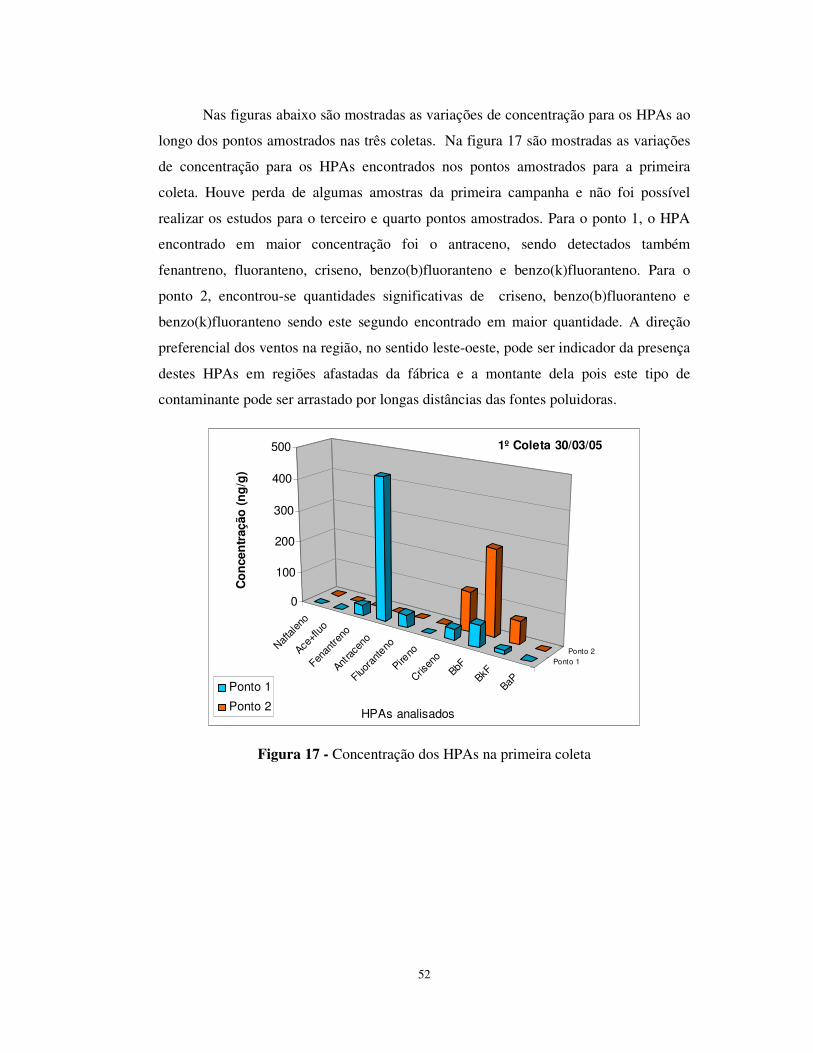
52
Nas figuras abaixo são mostradas as variações de concentração para os HPAs ao
longo dos pontos amostrados nas três coletas. Na figura 17 são mostradas as variações
de concentração para os HPAs encontrados nos pontos amostrados para a primeira
coleta. Houve perda de algumas amostras da primeira campanha e não foi possível
realizar os estudos para o terceiro e quarto pontos amostrados. Para o ponto 1, o HPA
encontrado em maior concentração foi o antraceno, sendo detectados também
fenantreno, fluoranteno, criseno, benzo(b)fluoranteno e benzo(k)fluoranteno. Para o
ponto 2, encontrou-se quantidades significativas de criseno, benzo(b)fluoranteno e
benzo(k)fluoranteno sendo este segundo encontrado em maior quantidade. A direção
preferencial dos ventos na região, no sentido leste-oeste, pode ser indicador da presença
destes HPAs em regiões afastadas da fábrica e a montante dela pois este tipo de
contaminante pode ser arrastado por longas distâncias das fontes poluidoras.
Naf
tale
no
Ace+f
luo
Fenan
treno
Antra
ceno
Fluor
ante
no
Pireno
Cris
eno
BbF
BkF
BaP
Ponto 1
Ponto 2
0
100
200
300
400
500
Co
nce
ntr
ação
(n
g/g
)
HPAs analisados
1º Coleta 30/03/05
Ponto 1
Ponto 2
Figura 17 - Concentração dos HPAs na primeira coleta
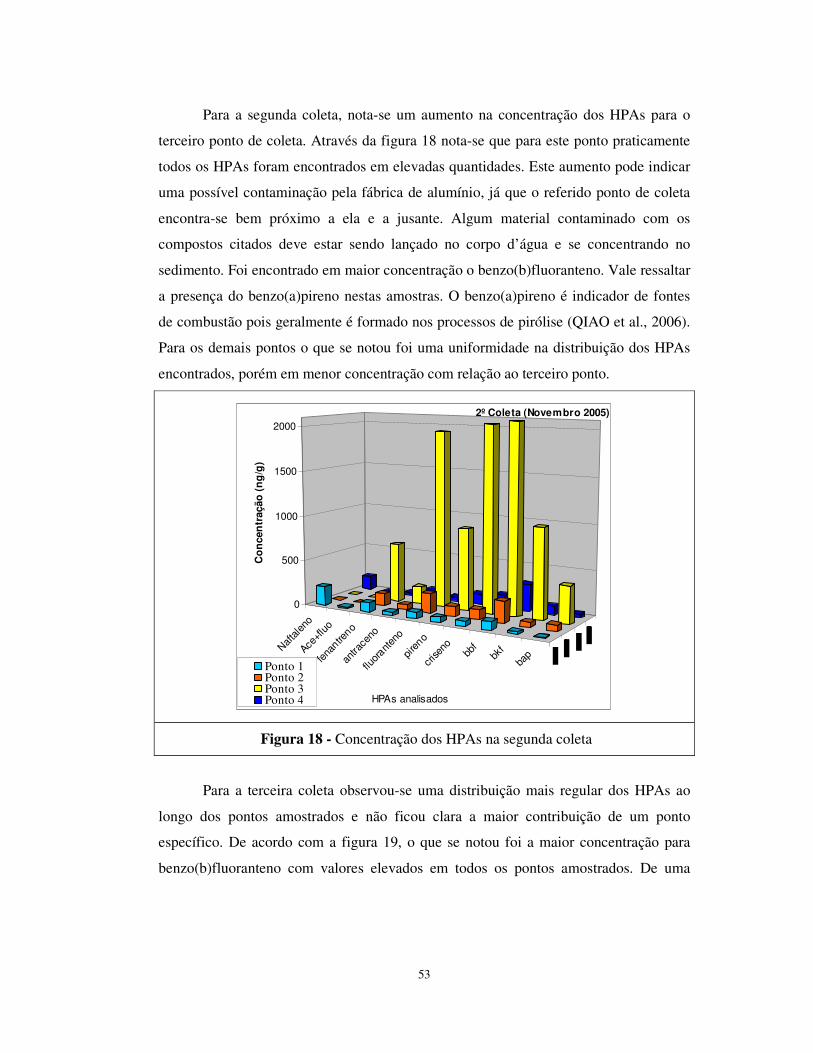
53
Para a segunda coleta, nota-se um aumento na concentração dos HPAs para o
terceiro ponto de coleta. Através da figura 18 nota-se que para este ponto praticamente
todos os HPAs foram encontrados em elevadas quantidades. Este aumento pode indicar
uma possível contaminação pela fábrica de alumínio, já que o referido ponto de coleta
encontra-se bem próximo a ela e a jusante. Algum material contaminado com os
compostos citados deve estar sendo lançado no corpo d’água e se concentrando no
sedimento. Foi encontrado em maior concentração o benzo(b)fluoranteno. Vale ressaltar
a presença do benzo(a)pireno nestas amostras. O benzo(a)pireno é indicador de fontes
de combustão pois geralmente é formado nos processos de pirólise (QIAO et al., 2006).
Para os demais pontos o que se notou foi uma uniformidade na distribuição dos HPAs
encontrados, porém em menor concentração com relação ao terceiro ponto.
Naf
tale
no
Ace+f
luo
fena
ntreno
antra
ceno
fluor
anten
o
pire
no
crisen
obb
fbk
f
bap
0
500
1000
1500
2000
Co
nce
ntr
ação
(n
g/g
)
HPAs analisados
2º Coleta (Novembro 2005)
Ponto 1Ponto 2Ponto 3Ponto 4
Figura 18 - Concentração dos HPAs na segunda coleta
Para a terceira coleta observou-se uma distribuição mais regular dos HPAs ao
longo dos pontos amostrados e não ficou clara a maior contribuição de um ponto
específico. De acordo com a figura 19, o que se notou foi a maior concentração para
benzo(b)fluoranteno com valores elevados em todos os pontos amostrados. De uma
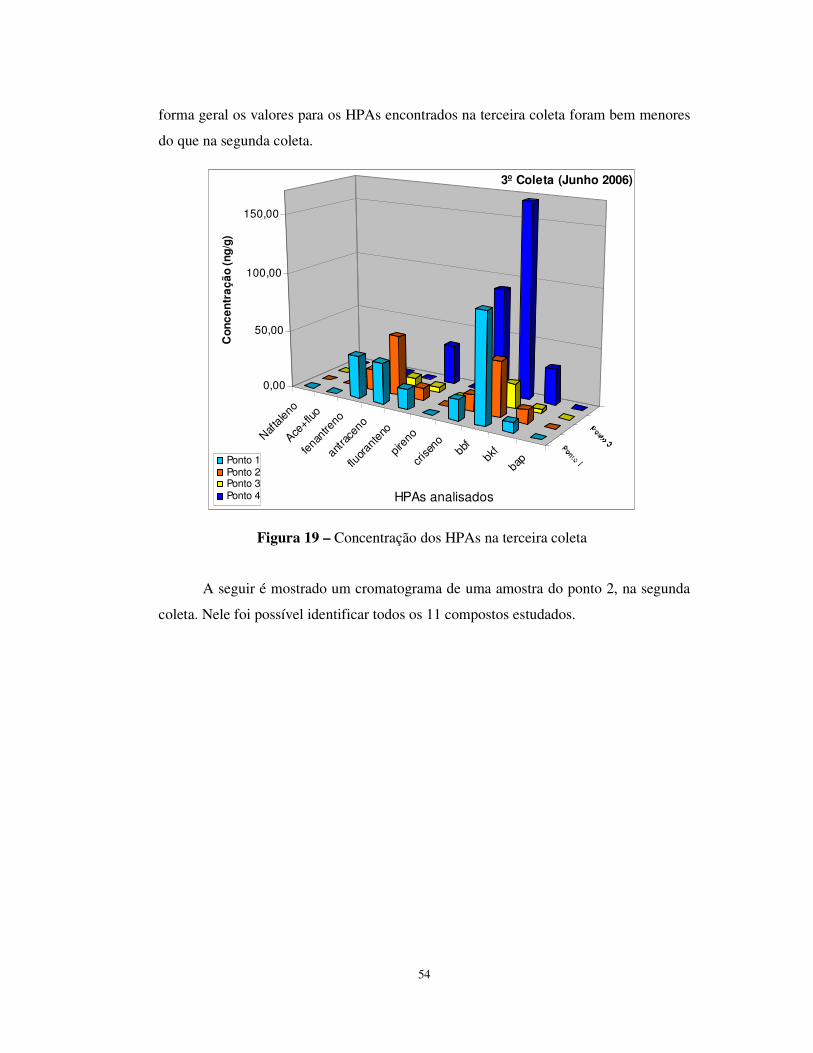
54
forma geral os valores para os HPAs encontrados na terceira coleta foram bem menores
do que na segunda coleta.
Naf
taleno
Ace
+fluo
fena
ntre
no
antra
ceno
fluor
ante
no
pire
no
crisen
obb
f
bkf
bap
0,00
50,00
100,00
150,00C
on
ce
ntr
aç
ão
(n
g/g
)
HPAs analisados
3º Coleta (Junho 2006)
Ponto 1Ponto 2Ponto 3Ponto 4
Figura 19 – Concentração dos HPAs na terceira coleta
A seguir é mostrado um cromatograma de uma amostra do ponto 2, na segunda
coleta. Nele foi possível identificar todos os 11 compostos estudados.
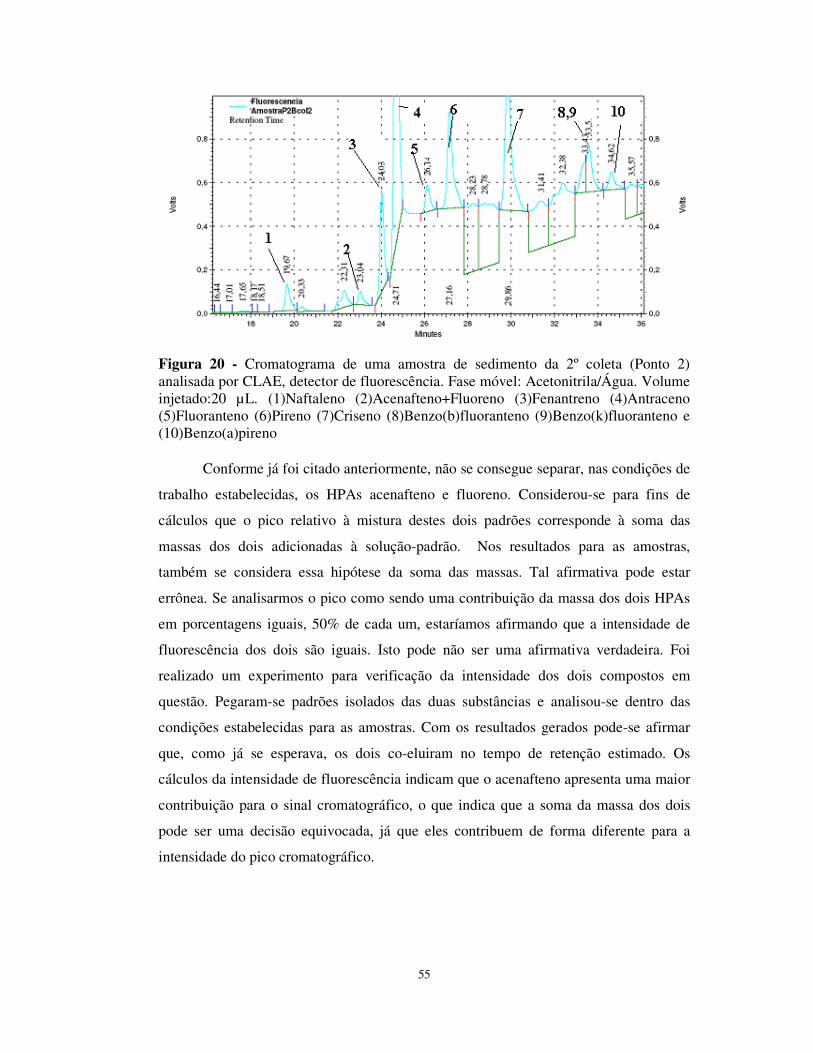
55
Conforme já foi citado anteriormente, não se consegue separar, nas condições de
trabalho estabelecidas, os HPAs acenafteno e fluoreno. Considerou-se para fins de
cálculos que o pico relativo à mistura destes dois padrões corresponde à soma das
massas dos dois adicionadas à solução-padrão. Nos resultados para as amostras,
também se considera essa hipótese da soma das massas. Tal afirmativa pode estar
errônea. Se analisarmos o pico como sendo uma contribuição da massa dos dois HPAs
em porcentagens iguais, 50% de cada um, estaríamos afirmando que a intensidade de
fluorescência dos dois são iguais. Isto pode não ser uma afirmativa verdadeira. Foi
realizado um experimento para verificação da intensidade dos dois compostos em
questão. Pegaram-se padrões isolados das duas substâncias e analisou-se dentro das
condições estabelecidas para as amostras. Com os resultados gerados pode-se afirmar
que, como já se esperava, os dois co-eluiram no tempo de retenção estimado. Os
cálculos da intensidade de fluorescência indicam que o acenafteno apresenta uma maior
contribuição para o sinal cromatográfico, o que indica que a soma da massa dos dois
pode ser uma decisão equivocada, já que eles contribuem de forma diferente para a
intensidade do pico cromatográfico.
Figura 20 - Cromatograma de uma amostra de sedimento da 2º coleta (Ponto 2) analisada por CLAE, detector de fluorescência. Fase móvel: Acetonitrila/Água. Volume injetado:20 µL. (1)Naftaleno (2)Acenafteno+Fluoreno (3)Fenantreno (4)Antraceno (5)Fluoranteno (6)Pireno (7)Criseno (8)Benzo(b)fluoranteno (9)Benzo(k)fluoranteno e (10)Benzo(a)pireno
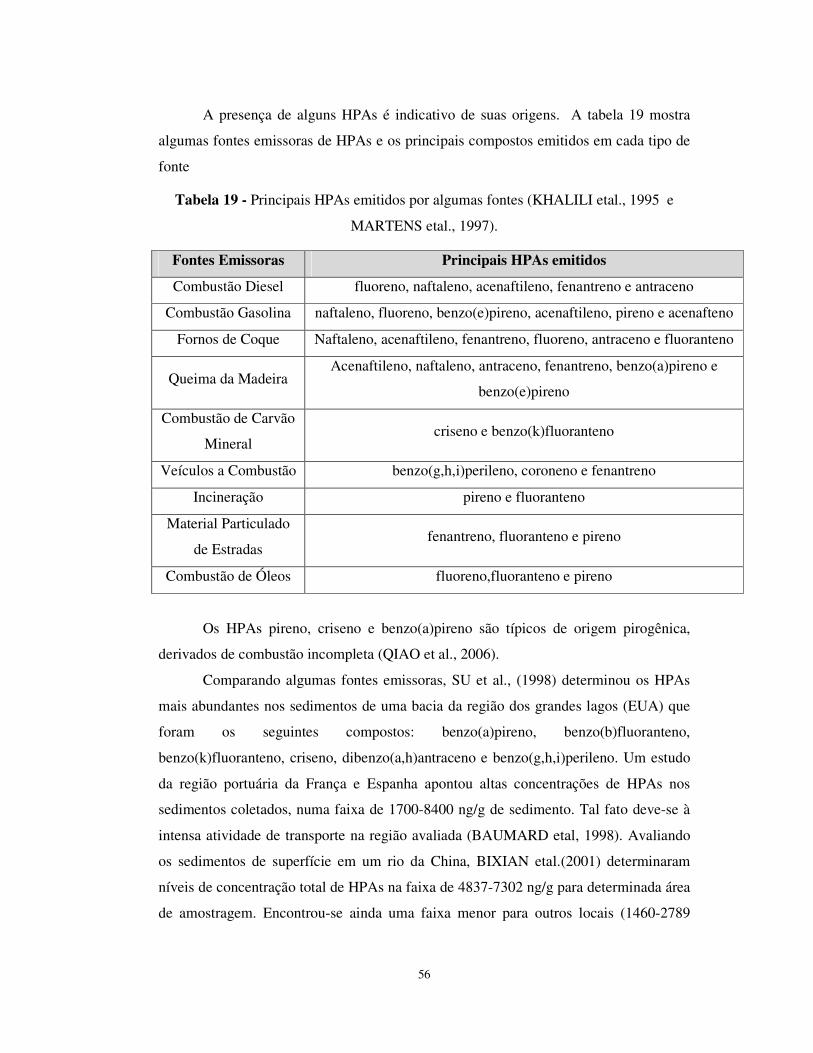
56
A presença de alguns HPAs é indicativo de suas origens. A tabela 19 mostra
algumas fontes emissoras de HPAs e os principais compostos emitidos em cada tipo de
fonte
Tabela 19 - Principais HPAs emitidos por algumas fontes (KHALILI etal., 1995 e
MARTENS etal., 1997).
Fontes Emissoras Principais HPAs emitidos
Combustão Diesel fluoreno, naftaleno, acenaftileno, fenantreno e antraceno
Combustão Gasolina naftaleno, fluoreno, benzo(e)pireno, acenaftileno, pireno e acenafteno
Fornos de Coque Naftaleno, acenaftileno, fenantreno, fluoreno, antraceno e fluoranteno
Queima da Madeira Acenaftileno, naftaleno, antraceno, fenantreno, benzo(a)pireno e
benzo(e)pireno
Combustão de Carvão
Mineral criseno e benzo(k)fluoranteno
Veículos a Combustão benzo(g,h,i)perileno, coroneno e fenantreno
Incineração pireno e fluoranteno
Material Particulado
de Estradas fenantreno, fluoranteno e pireno
Combustão de Óleos fluoreno,fluoranteno e pireno
Os HPAs pireno, criseno e benzo(a)pireno são típicos de origem pirogênica,
derivados de combustão incompleta (QIAO et al., 2006).
Comparando algumas fontes emissoras, SU et al., (1998) determinou os HPAs
mais abundantes nos sedimentos de uma bacia da região dos grandes lagos (EUA) que
foram os seguintes compostos: benzo(a)pireno, benzo(b)fluoranteno,
benzo(k)fluoranteno, criseno, dibenzo(a,h)antraceno e benzo(g,h,i)perileno. Um estudo
da região portuária da França e Espanha apontou altas concentrações de HPAs nos
sedimentos coletados, numa faixa de 1700-8400 ng/g de sedimento. Tal fato deve-se à
intensa atividade de transporte na região avaliada (BAUMARD etal, 1998). Avaliando
os sedimentos de superfície em um rio da China, BIXIAN etal.(2001) determinaram
níveis de concentração total de HPAs na faixa de 4837-7302 ng/g para determinada área
de amostragem. Encontrou-se ainda uma faixa menor para outros locais (1460-2789

57
ng/g) o que foi atribuído à biodegradação e desorção de HPAs de baixa massa
molecular.
A concentração total dos 11 HPAs analisados nas três coletas ao longo dos
quatro pontos de coleta é mostrada na figura 21. Para cada ponto, somaram-se as
concentrações encontradas para os onze compostos nas três campanhas de coleta e
assim foi distribuído para cada ponto de coleta. Nota-se que a concentração variou numa
escala regular para os pontos 1,2 e 4. O ponto três obteve um maior valor para o
somatório das concentrações. O intervalo variou entre o mínimo 1363,12 (ponto 4) até o
máximo de 9616,03 ng/g para o ponto 3. Os maiores valores totais para a soma dos 11
HPAs foram verificadas para os pontos 2 e 3, sendo extremamente elevada neste último
ponto. Uma explicação para tais valores mais elevados, em destaque para o ponto 3, foi
provavelmente devido a contribuição da fábrica de alumínio próxima a este pontos. O
ponto 3 talvez esteja recebendo, além da contribuição por deposição atmosférica, uma
carga de contaminantes levada ao córrego pelos efluentes contaminados e se
depositando sedimentos do ribeirão. No seu trabalho, YAMADA (2006), encontrou
valores variando entre 79,76 ng/g e 219,94 ng/g de HPAs totais nos sedimentos da
represa de Ibirité, na região metropolitana de Belo Horizonte. Os valores determinados
no presente trabalho para o Ribeirão do Funil, na região estudada, são bem mais
elevados do que os encontrados naquela represa.
Um estudo realizado nos sedimento da baía de Todos os Santos, localizada no
Recôncavo Baiano, apontou níveis bastante elevados nas concentrações de HPAs para
regiões portuárias variando entre 0,9 ng/g a 408629,1 ng/g. Em outros sítios do mesmo
estudo os níveis de concentração foram bastante reduzidos, sendo que nas áreas de
referência os valores de concentração dos HPAs foram de 10,2 ng/g. Os níveis de
poluição são apontados como produtos da pirólise do petróleo na região estudada,
devido as atividades da Petrobrás. Fontes antropogênicas como atividades marinhas
relacionadas à recreação também contribuem para o lançamento de HPAs no ambiente
avaliado (CELINO & QUEIROZ, 2006). As regiões portuárias são conhecidas por
apresentarem níveis extremamente altos na concentração de HPAs (>>100 000 ng/g) e a
fusão do alumínio pode afetar algumas regiões. Um estudo global das concentrações de
HPAs sugere que rios, lagos, estuários, portos e regiões costeiras apresentam diferentes
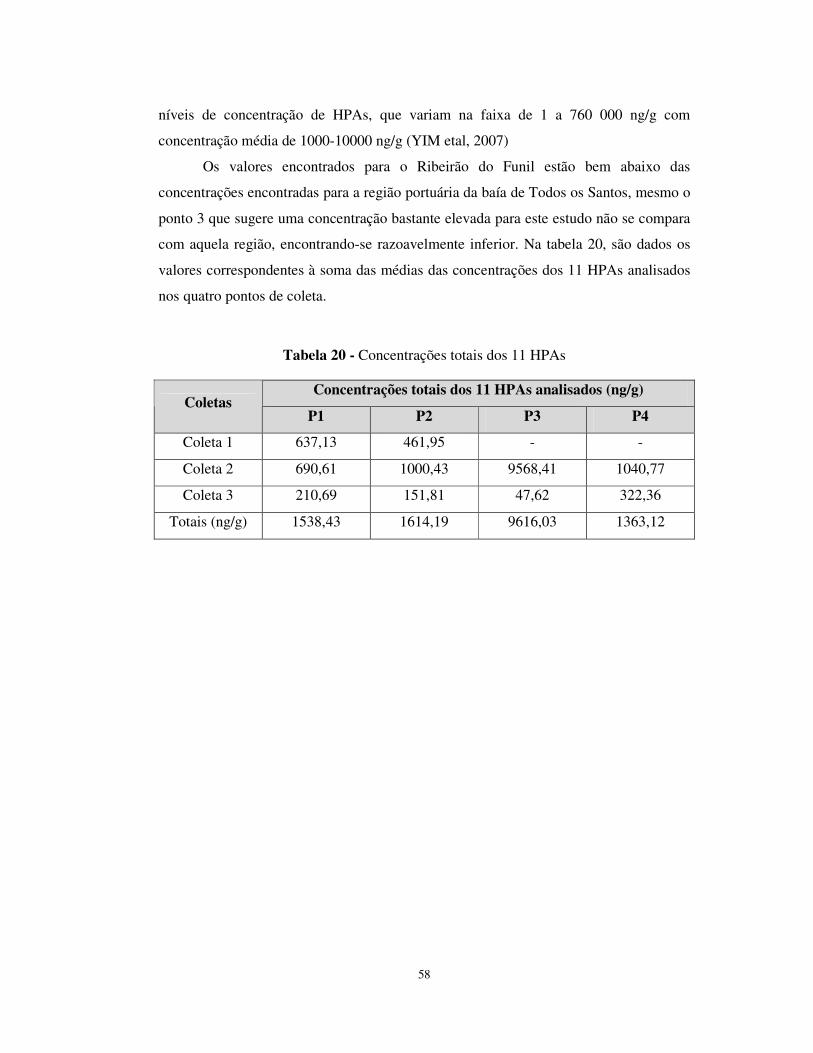
58
níveis de concentração de HPAs, que variam na faixa de 1 a 760 000 ng/g com
concentração média de 1000-10000 ng/g (YIM etal, 2007)
Os valores encontrados para o Ribeirão do Funil estão bem abaixo das
concentrações encontradas para a região portuária da baía de Todos os Santos, mesmo o
ponto 3 que sugere uma concentração bastante elevada para este estudo não se compara
com aquela região, encontrando-se razoavelmente inferior. Na tabela 20, são dados os
valores correspondentes à soma das médias das concentrações dos 11 HPAs analisados
nos quatro pontos de coleta.
Tabela 20 - Concentrações totais dos 11 HPAs
Concentrações totais dos 11 HPAs analisados (ng/g) Coletas
P1 P2 P3 P4
Coleta 1 637,13 461,95 - -
Coleta 2 690,61 1000,43 9568,41 1040,77
Coleta 3 210,69 151,81 47,62 322,36
Totais (ng/g) 1538,43 1614,19 9616,03 1363,12
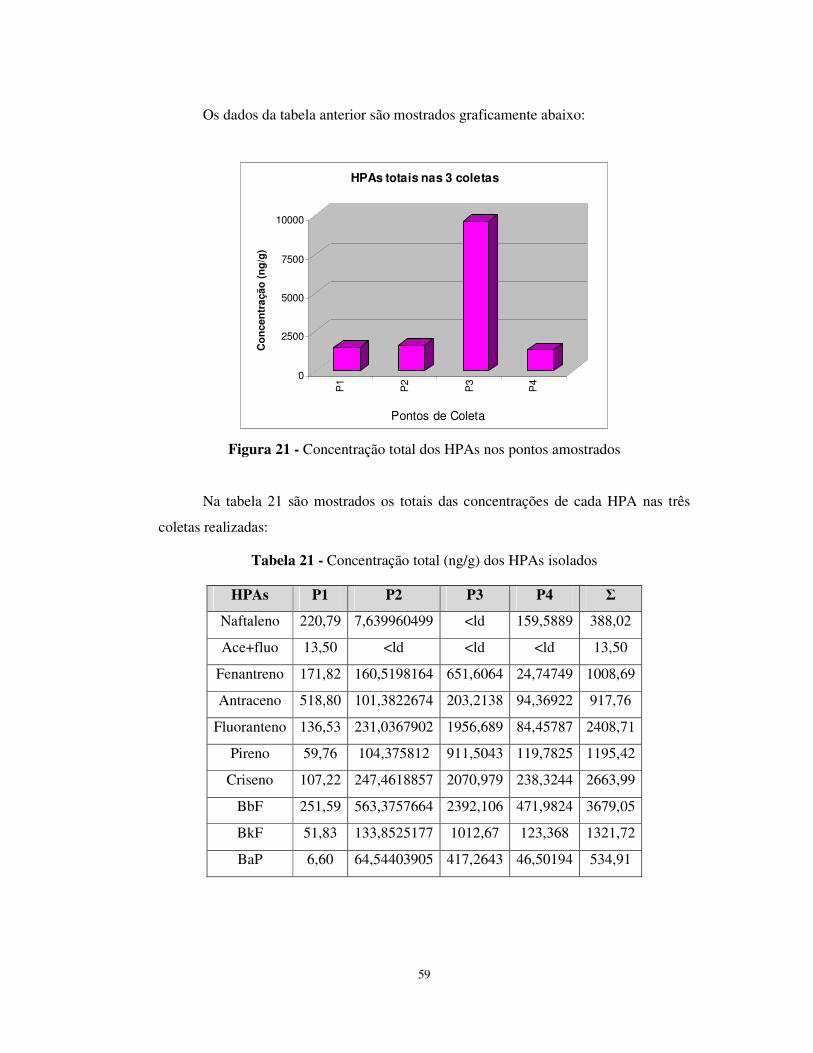
59
Os dados da tabela anterior são mostrados graficamente abaixo:
0
2500
5000
7500
10000
Co
nce
ntr
ação
(n
g/g
)
P1
P2
P3
P4
Pontos de Coleta
HPAs totais nas 3 coletas
Figura 21 - Concentração total dos HPAs nos pontos amostrados
Na tabela 21 são mostrados os totais das concentrações de cada HPA nas três
coletas realizadas:
Tabela 21 - Concentração total (ng/g) dos HPAs isolados
HPAs P1 P2 P3 P4 Σ
Naftaleno 220,79 7,639960499 <ld 159,5889 388,02
Ace+fluo 13,50 <ld <ld <ld 13,50
Fenantreno 171,82 160,5198164 651,6064 24,74749 1008,69
Antraceno 518,80 101,3822674 203,2138 94,36922 917,76
Fluoranteno 136,53 231,0367902 1956,689 84,45787 2408,71
Pireno 59,76 104,375812 911,5043 119,7825 1195,42
Criseno 107,22 247,4618857 2070,979 238,3244 2663,99
BbF 251,59 563,3757664 2392,106 471,9824 3679,05
BkF 51,83 133,8525177 1012,67 123,368 1321,72
BaP 6,60 64,54403905 417,2643 46,50194 534,91
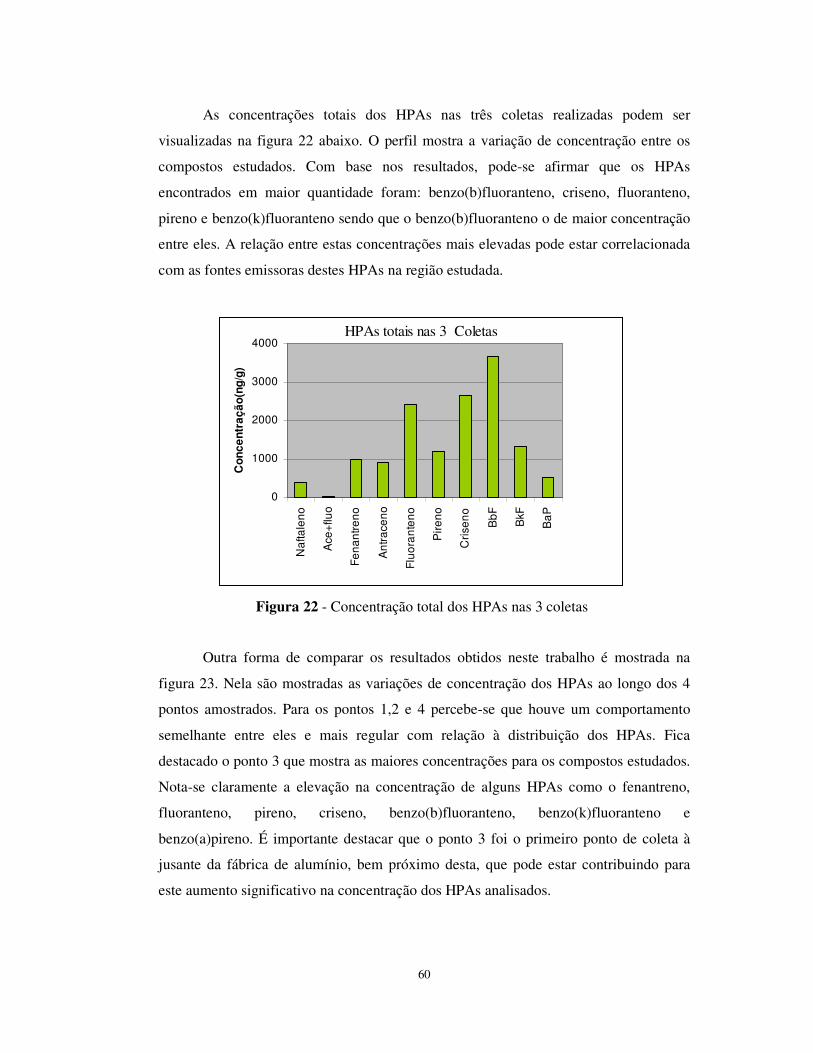
60
As concentrações totais dos HPAs nas três coletas realizadas podem ser
visualizadas na figura 22 abaixo. O perfil mostra a variação de concentração entre os
compostos estudados. Com base nos resultados, pode-se afirmar que os HPAs
encontrados em maior quantidade foram: benzo(b)fluoranteno, criseno, fluoranteno,
pireno e benzo(k)fluoranteno sendo que o benzo(b)fluoranteno o de maior concentração
entre eles. A relação entre estas concentrações mais elevadas pode estar correlacionada
com as fontes emissoras destes HPAs na região estudada.
HPAs totais nas 3 Coletas
0
1000
2000
3000
4000
Na
fta
len
o
Ace
+flu
o
Fe
na
ntr
en
o
An
tra
ce
no
Flu
ora
nte
no
Pir
en
o
Cri
se
no
Bb
F
BkF
Ba
P
Co
nc
en
tra
çã
o(n
g/g
)
Figura 22 - Concentração total dos HPAs nas 3 coletas
Outra forma de comparar os resultados obtidos neste trabalho é mostrada na
figura 23. Nela são mostradas as variações de concentração dos HPAs ao longo dos 4
pontos amostrados. Para os pontos 1,2 e 4 percebe-se que houve um comportamento
semelhante entre eles e mais regular com relação à distribuição dos HPAs. Fica
destacado o ponto 3 que mostra as maiores concentrações para os compostos estudados.
Nota-se claramente a elevação na concentração de alguns HPAs como o fenantreno,
fluoranteno, pireno, criseno, benzo(b)fluoranteno, benzo(k)fluoranteno e
benzo(a)pireno. É importante destacar que o ponto 3 foi o primeiro ponto de coleta à
jusante da fábrica de alumínio, bem próximo desta, que pode estar contribuindo para
este aumento significativo na concentração dos HPAs analisados.
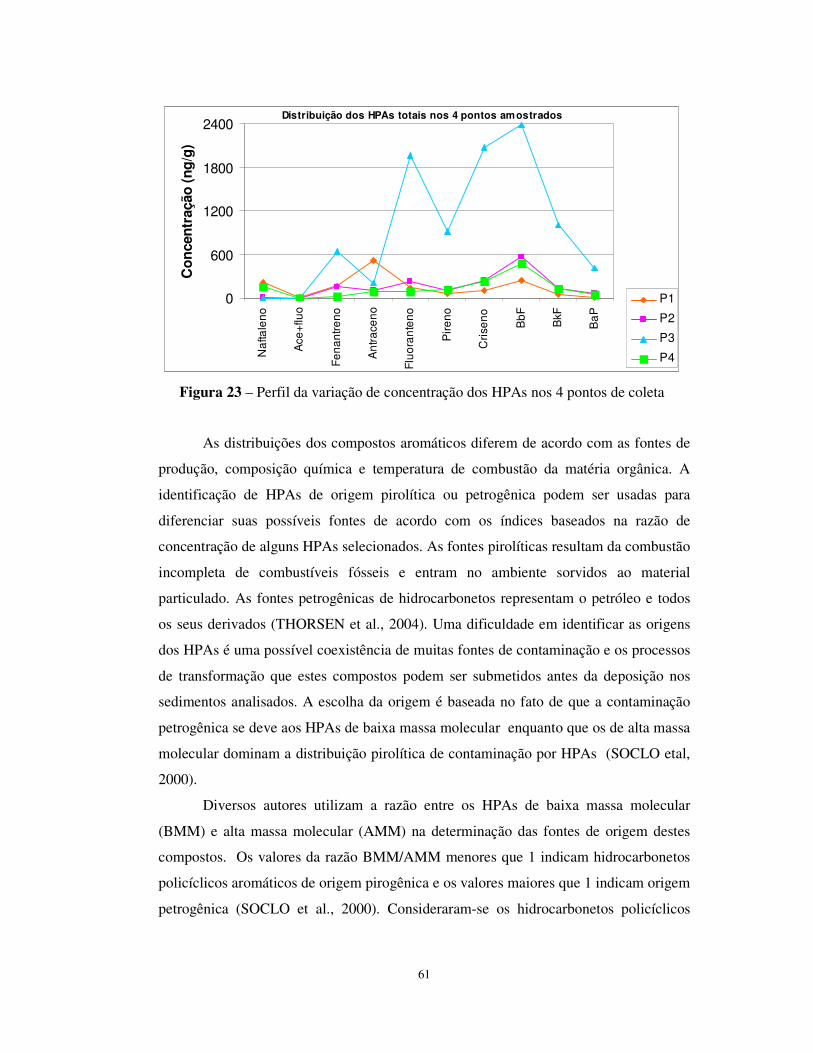
61
As distribuições dos compostos aromáticos diferem de acordo com as fontes de
produção, composição química e temperatura de combustão da matéria orgânica. A
identificação de HPAs de origem pirolítica ou petrogênica podem ser usadas para
diferenciar suas possíveis fontes de acordo com os índices baseados na razão de
concentração de alguns HPAs selecionados. As fontes pirolíticas resultam da combustão
incompleta de combustíveis fósseis e entram no ambiente sorvidos ao material
particulado. As fontes petrogênicas de hidrocarbonetos representam o petróleo e todos
os seus derivados (THORSEN et al., 2004). Uma dificuldade em identificar as origens
dos HPAs é uma possível coexistência de muitas fontes de contaminação e os processos
de transformação que estes compostos podem ser submetidos antes da deposição nos
sedimentos analisados. A escolha da origem é baseada no fato de que a contaminação
petrogênica se deve aos HPAs de baixa massa molecular enquanto que os de alta massa
molecular dominam a distribuição pirolítica de contaminação por HPAs (SOCLO etal,
2000).
Diversos autores utilizam a razão entre os HPAs de baixa massa molecular
(BMM) e alta massa molecular (AMM) na determinação das fontes de origem destes
compostos. Os valores da razão BMM/AMM menores que 1 indicam hidrocarbonetos
policíclicos aromáticos de origem pirogênica e os valores maiores que 1 indicam origem
petrogênica (SOCLO et al., 2000). Consideraram-se os hidrocarbonetos policíclicos
Distribuição dos HPAs totais nos 4 pontos amostrados
0
600
1200
1800
2400
Na
fta
len
o
Ace
+flu
o
Fe
na
ntr
en
o
An
tra
ce
no
Flu
ora
nte
no
Pir
en
o
Cri
se
no
Bb
F
BkF
Ba
P
Co
nce
ntr
ação
(n
g/g
)
P1
P2
P3
P4
Figura 23 – Perfil da variação de concentração dos HPAs nos 4 pontos de coleta
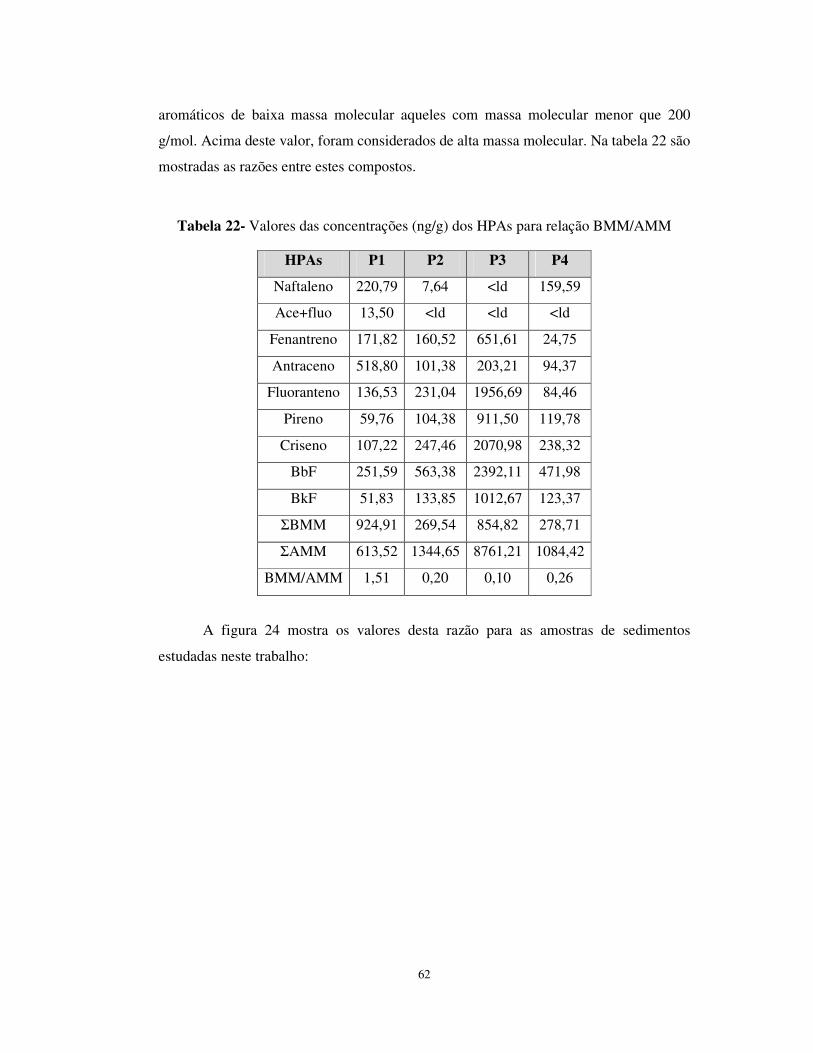
62
aromáticos de baixa massa molecular aqueles com massa molecular menor que 200
g/mol. Acima deste valor, foram considerados de alta massa molecular. Na tabela 22 são
mostradas as razões entre estes compostos.
Tabela 22- Valores das concentrações (ng/g) dos HPAs para relação BMM/AMM
HPAs P1 P2 P3 P4
Naftaleno 220,79 7,64 <ld 159,59
Ace+fluo 13,50 <ld <ld <ld
Fenantreno 171,82 160,52 651,61 24,75
Antraceno 518,80 101,38 203,21 94,37
Fluoranteno 136,53 231,04 1956,69 84,46
Pireno 59,76 104,38 911,50 119,78
Criseno 107,22 247,46 2070,98 238,32
BbF 251,59 563,38 2392,11 471,98
BkF 51,83 133,85 1012,67 123,37
ΣBMM 924,91 269,54 854,82 278,71
ΣAMM 613,52 1344,65 8761,21 1084,42
BMM/AMM 1,51 0,20 0,10 0,26
A figura 24 mostra os valores desta razão para as amostras de sedimentos
estudadas neste trabalho:
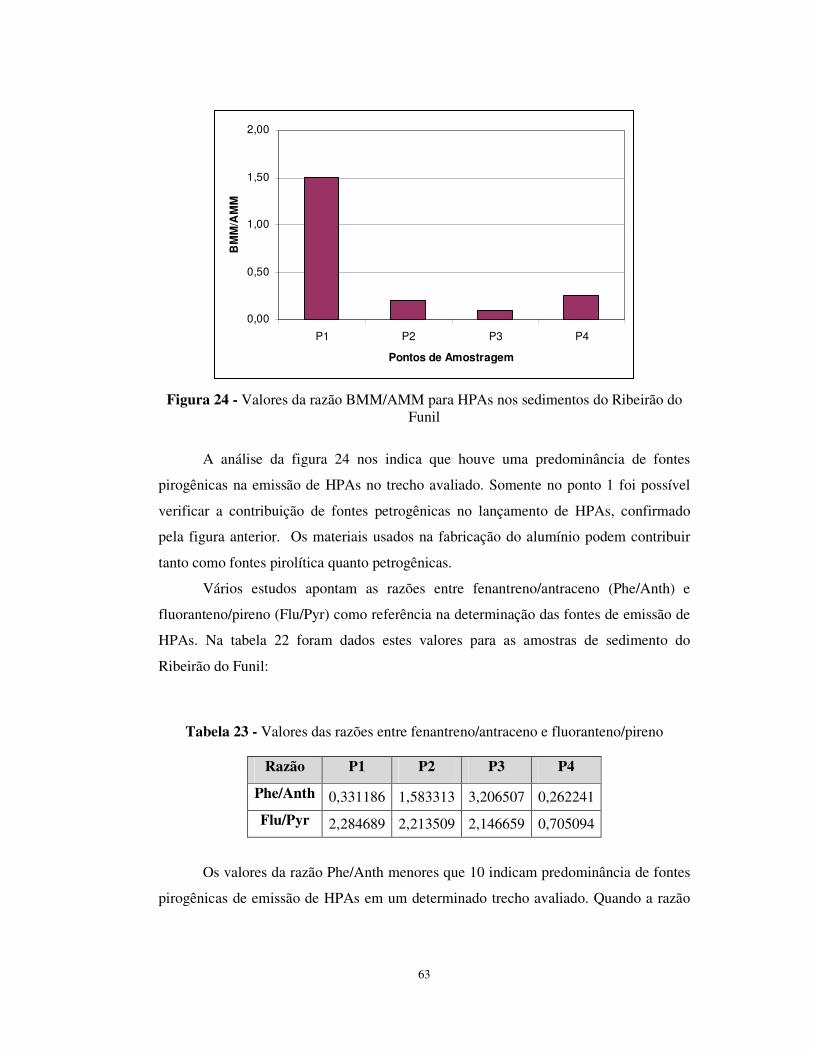
63
0,00
0,50
1,00
1,50
2,00
P1 P2 P3 P4
Pontos de Amostragem
BM
M/A
MM
Figura 24 - Valores da razão BMM/AMM para HPAs nos sedimentos do Ribeirão do Funil
A análise da figura 24 nos indica que houve uma predominância de fontes
pirogênicas na emissão de HPAs no trecho avaliado. Somente no ponto 1 foi possível
verificar a contribuição de fontes petrogênicas no lançamento de HPAs, confirmado
pela figura anterior. Os materiais usados na fabricação do alumínio podem contribuir
tanto como fontes pirolítica quanto petrogênicas.
Vários estudos apontam as razões entre fenantreno/antraceno (Phe/Anth) e
fluoranteno/pireno (Flu/Pyr) como referência na determinação das fontes de emissão de
HPAs. Na tabela 22 foram dados estes valores para as amostras de sedimento do
Ribeirão do Funil:
Tabela 23 - Valores das razões entre fenantreno/antraceno e fluoranteno/pireno
Razão P1 P2 P3 P4
Phe/Anth 0,331186 1,583313 3,206507 0,262241
Flu/Pyr 2,284689 2,213509 2,146659 0,705094
Os valores da razão Phe/Anth menores que 10 indicam predominância de fontes
pirogênicas de emissão de HPAs em um determinado trecho avaliado. Quando a razão
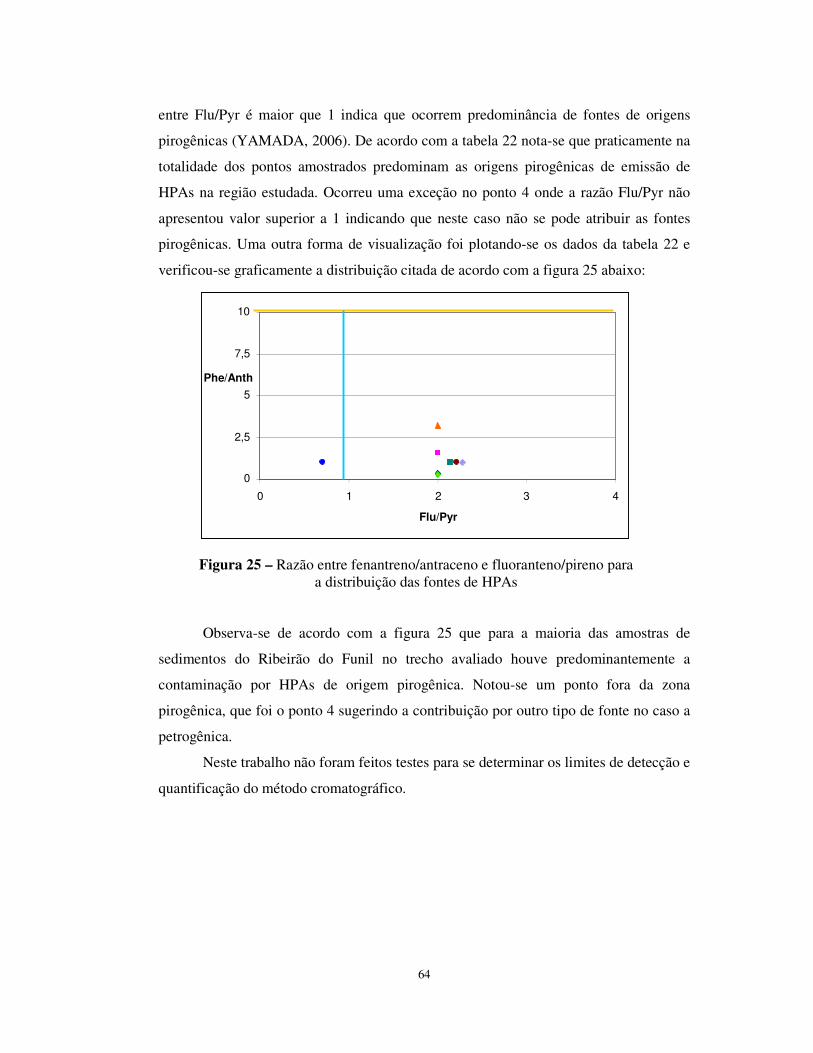
64
entre Flu/Pyr é maior que 1 indica que ocorrem predominância de fontes de origens
pirogênicas (YAMADA, 2006). De acordo com a tabela 22 nota-se que praticamente na
totalidade dos pontos amostrados predominam as origens pirogênicas de emissão de
HPAs na região estudada. Ocorreu uma exceção no ponto 4 onde a razão Flu/Pyr não
apresentou valor superior a 1 indicando que neste caso não se pode atribuir as fontes
pirogênicas. Uma outra forma de visualização foi plotando-se os dados da tabela 22 e
verificou-se graficamente a distribuição citada de acordo com a figura 25 abaixo:
Observa-se de acordo com a figura 25 que para a maioria das amostras de
sedimentos do Ribeirão do Funil no trecho avaliado houve predominantemente a
contaminação por HPAs de origem pirogênica. Notou-se um ponto fora da zona
pirogênica, que foi o ponto 4 sugerindo a contribuição por outro tipo de fonte no caso a
petrogênica.
Neste trabalho não foram feitos testes para se determinar os limites de detecção e
quantificação do método cromatográfico.
0
2,5
5
7,5
10
0 1 2 3 4
Flu/Pyr
Phe/Anth
Figura 25 – Razão entre fenantreno/antraceno e fluoranteno/pireno para a distribuição das fontes de HPAs

65
7. CONCLUSÕES As avaliações dos diversos parâmetros físico-químicos mostraram a baixa qualidade das
águas analisadas.
Elevados valores de alcalinidade foram encontrados nas amostras da terceira coleta,
principalmente nos pontos 1 e 2.
Os valores de cloreto são altos, em geral, nos dois primeiros pontos amostrados.
A cor das águas do Ribeirão do Funil apresentou, quase na totalidade das amostras,
valores baixos, com exceção dos pontos 3 e 4 na segunda coleta.
Outro parâmetro que apresentou valores altos foi a dureza, principalmente para os dois
primeiros pontos na segunda coleta. Isto atribui a característica de águas duras do
ribeirão estudado.
Os teores de nitrato e nitrito foram elevados nos pontos 3 e 4, o que confirma a presença
de esgotos nos locais amostrados, o que também é indicado pelo alto teor de fósforo
nestes pontos.
Os valores de sulfato variaram na primeira e terceira coletas em um patamar mais
elevado que na segunda. Os maiores valores foram identificados nos pontos 1 e 2 na
primeira campanha de amostragem.
A turbidez apresentou valores elevados para a segunda amostragem, o que pode ser
atribuído ao período chuvoso no dia de coleta.
O comportamento do pH foi de ligeiramente ácido, passando pela neutralidade e a
tendência ao pH alcalino nas 3 coletas.

66
Para análise de metais foram encontrados elevados teores para: alumínio, arsênio, ferro,
chumbo, manganês, cálcio, sódio e estrôncio. A presença de metais pesados como:
arsênio, cádmio e chumbo nas amostras evidenciam a toxicidade das águas e sua baixa
qualidade, bem como a influência das fontes poluidoras na região.
Os HPAs encontrados nas diversas amostras de sedimentos do Ribeirão do Funil sugere
que ocorrem variadas fontes de contaminação. Os onze compostos analisados
encontraram-se distribuídos em concentrações variadas nas campanhas de amostragem e
nos 4 pontos coletados. Os resultados apontam para uma heterogeneidade de fontes de
emissão podendo haver contribuição de: combustão do carvão mineral, emissões por
veículos a combustão, material particulado de estrada, já que alguns pontos se localizam
próximo a uma das rodovias que ligam Ouro Preto a Belo Horizonte, combustão de
óleos, combustão de diesel, combustão de gasolina, fornos de coque, queima de madeira
entre outras.
A soma dos HPAs totais sugere um elevado valor para o terceiro ponto de amostragem.
Este ponto se localiza a jusante da fábrica de alumínio e bem próximo a ela o que pode
nos indicar uma suposta contaminação por algumas fontes especificas de emissão de
hidrocarbonetos policíclicos aromáticos por este tipo de indústria. Nessa fábrica ocorre
tanto a queima de materiais oleosos quanto despejos de resíduos contendo HPAs no seu
ambiente. Os produtos podem estar sendo lançados via atmosférica através da queima e
posterior deposição nas águas do Ribeirão ou por processos de limpeza, carregados pela
chuva ou até mesmo descartados na região onde ocorre uma maior concentração dos
compostos avaliados.
A presença do benzo(a)pireno em algumas amostras de sedimentos sugere uma
preocupação com a contaminação por HPAs no trecho avaliado pois este hidrocarboneto
é conhecido por ter propriedades altamente mutagênicas e carcinogênicas e serve de
alerta com relação aos sérios riscos de degradação ambiental do Ribeirão do Funil.
A avaliação da razão entre HPAs de baixa e alta massa molecular (BMM/AMM), entre
fenantreno e antraceno (Phe/Anth) e a razão entre fluoranteno e pireno(Flu/Pyr),

67
apontaram que a contaminação dos sedimentos para a área estudada se deve
principalmente e na grande maioria por fontes de origem pirogênicas.
Sendo Ouro preto uma cidade de destaque nacional e patrimônio da humanidade, uma
política de melhoria da qualidade de suas águas é indispensável para a manutenção da
qualidade de vida de seus habitantes e da preservação de sua imagem no cenário
turístico que é tão importante para sua economia.
Este primeiro estudo envolvendo HPAs pode contribuir para a avaliação da qualidade
ambiental na região e indicar as fontes de poluição mais significativas, que promovem a
degradação do sistema hídrico estudado afetando a qualidade de suas águas e podendo
comprometer a saúde da população.
8. SUGESTÕES PARA TRABALHOS FUTUROS:
⇒ Propor o desenvolvimento de um método que resulte em melhores rendimentos
dos índices de recuperação, com ensaios de validação mais confiáveis;
⇒ Desenvolver e até mesmo comparar métodos de homogeneização das amostras
que atendam melhor com relação aos resultados esperados;
⇒ Realizar as análises dos HPAs com uma coluna especifica para estes compostos
melhorando a resolução dos picos cromatográficos principalmente para os
isômeros que não se separam e analisar outros HPAs além dos onze estudados;
⇒ Aprofundar a investigação sobre as possíveis fontes de HPAs na região (ar,
água, sedimentos) podendo relacionar as contaminações de uma forma mais
especifica;

68
⇒ Testar outros métodos e solventes de extração de HPAs nas matrizes e até
mesmo compara-los com o método soxhlet, através dos rendimentos de cada
um;
⇒ Analisar os HPAs por outro método cromatográfico como, por exemplo, o
CG/MS.

69
9. REFERÊNCIAS BIBLIOGRÁFICAS
Agudo, E. G. (1987). Guia de Coleta e Preservação de Amostras de Água. CETESB,
23-28, 51-61, São Paulo.
American Public Health Association (APHA). (1998). Standard Methods for The
Examination of Water and Wastewater. Washington. 1193 p.
Azevedo, Débora de A.; Gerchon, Elaine., Reis, Ederson O. dos (2004),. Monitoring of
Pesticides and Polycyclic Aromatic Hydrocarbons in Water from Paraíba do Sul River,
Brazil. J. Braz. Chem. Soc., vol. 15, 292-299
Baisch, P. e Mirlean, N., Lima, G. Protocolos de Coleta e Análise dos Sedimentos e
Material em Suspensão. Projeto Millenium. FURG, Rio Grande do Sul- RS.
Baroncelli, F., Oliveira, L.S., Afonso, R.J.C.F.e Pataca, L.C.M.(1999) Avaliação da
potencialidade carcinogênica das partículas inaláveis presentes na atmosfera da região
central de Belo Horizonte. Rev. Automação e Instrumentação Aplicadas ao Meio
Ambiente. 48-57
Baumard, P.; Budzinski, H., Michon, Q., Garrigues, P., Burgeot, t. & Bellocq J. (1998)
Origin and bioavailability of PAHs in the Mediterranean Sea from Mussel and Sediment
Records. Estuarine, Coastal and Shelf Science. 47, 77-90
Bixian, M.; Jiamo, F., Gan, Z., Zheng, L., Yushun, M., Guoying, S. e Xingmin, W.
(2001) Polycyclic aromatic hydrocarbons in sediments from the Pearl river and estuary,
China: spatial and temporal distribution and sources. Applied Geochemistry.16, 1429-
1445.

70
Bøwadt, S.; Mazeas, L., Miller, D. J., Hawthorne, S. B. (1997). Field-portable
determination of polychlorinated biphenyls and polynuclear aromatic hydrocarbons in
soil using supercritical fluid extraction. J. Chromatogr. A, 785, 205–217.
Busetti, F.; Heitz, A., Cuomo, M., Badoer, S., Traverso, P. (2005). Determination of
Sixteen Polycyclic Aromatic Hydrocarbons in Aqueous and Solid Samples from an
Italian Wastewater Treatment Plant. J. Chromatogr. A. 1-12
Capelo, J.L.; Galesio, M.M., G.M, Felisberto., Vaz, C., Pessoa, J.C. (2005). Micro-
focused ultrasonic solid-liquid extraction (µFUSLE) combined with CLAE and
fluorescence detection for PAHs determination in sediments: optimization and linking
with the analytical minimalism concept. Talanta. 66, 1272-1280.
Celino, J.J & Queiroz, A.F.S. (2006) Fonte e grau da contaminação por hidrocarbonetos
policíclicos aromáticos (HPAs) de baixa massa molecular em sedimentos da baía de
Todos os Santos, Bahia. Revista Escola de Minas, REM. 59, 265-270
Collins, C.H.; Ribani, M., Bottoli, C.B.G., Jardim, I.C.S.F. & Melo, L.F.C. (2004)
Validação em Métodos Cromatográficos e Eletroforéticos. Química Nova. 27, 771-780
Compendium Method TO-13A. (1999). Determination of Polycyclic Aromatic
Hydrocarbons (PAHs) in Ambient Air Using Gas Chromatography/Mass Spectrometry
(GC/MS). Center for Environmental Research Information Office of Research and
Development U.S. Environmental Protection Agency-EPA.
CONAMA (2005), Conselho Nacional de Meio Ambiente. Resolução nº 357, de 17 de
março de 2005.
d’Aguila, P. S.; Cruz, O.C.R., Miranda, C.A.S. e Ferreira, A.P. (2000). Avaliação da
qualidade de água para abastecimento público do Município de Nova Iguaçu. Cad.
Saúde Pública. Departamento de Saneamento e Saúde Ambiental, Escola Nacional de
Saúde Pública. Fundação Oswaldo Cruz, 16,791-798.

71
Fernández-Pérez V. e Castro, M.D.L. (2000). Micelle formation for improvement of
continuous subcritical water extraction of polycyclic aromatic hydrocarbons in soil prior
to high-performance liquid chromatography–fluorescence detection. J. Chromatogr. A.
902, 357-367.
Filho, G.B. (1998). Patologia Geral. Ed. Guaranabara Koogan. 2a ed. p. 182-183, 262
Freitas, P.S.; Rígido, B.M., Badolato, M.I.C., Alaburda, J.F. (2002). Padrão Físico-
Químico da Água de Abastecimento Público da Região de Campinas. Rev. Inst. Adolfo
Lutz, Inst. Adolfo Lutz. 61, 51-58.
Gfrerer, M.; Serschen, M., Lankmayr, E.(2002). Optimized extraction of polycyclic
aromatic hydrocarbons from contaminated soil samples. J. Biochem. Biophys. Methods.
53, 203–216.
Greeberg, A. E.; Clesceri, L. S., Eaton, A. D. (1992). Standard Methods for the
Examination of Water and Wastewater, 18th Edition. 1-22.
Guerreiro, M.R. (2000). A Lógica Territorial na Gênese e Formação das Cidades
Brasileiras - O Caso de Ouro Preto - Comunicação apresentada no Colóquio "A
Construção do Brasil Urbano". Convento da Arrábida - Lisboa
http://urban.iscte.pt/revista/numero3/artigos/artigo_11.htm acessado em 25/02/2007
Hawthorne, S. B.; Grabanski, C. B., Martin, E., J. Miller, David. (2000) Comparisons of
Soxhlet extraction, pressurized liquid extraction, supercritical fluid extraction and
subcritical water extraction for environmental solids: recovery, selectivity and effects on
sample matrix. J. Chromatogr. A. 892, 421-433.
Hawthorne, S. B.; Trembley, S., Moniot, C.L., Grabanski C.B., Miller, D.J. (2000)
Static subcritical water extraction with simultaneous solid-phase extraction for

72
determining polycyclic aromatic hydrocarbons on environmental solids. J. Chromatogr.
A. 886, 237-244.
Hubert, A.; Wenzel, K.D., Manz, M., Weissflog, L., W. Engewald. (2000). High
Extraction Efficiency for POPs in Real Contaminated Soil Samples Using Accelerated
Solvent Extraction. Anal. Chem. 72, 1294-1300.
INDI- Instituto de Desenvolvimento Industrial de Minas Gerais. Perfil Municipal de
Ouro Preto. http://www.indi.mg.gov.br acessado em 25/02/2007.
Inductively Coupled Plasma-Atomic Emission Spectrometry. METHOD 6010B. EPA-
Environmental Protection Agency. USA.
INMETRO (Instituto Nacional de Metrologia, Normalização e Qualidade Industrial).
Orientações sobre validação de métodos de ensaios químicos. DOQ-CGCRE-008-
Revisão: 01- Março/2003.
JOSEPH, Maureen. CLAE Detector Options for the Determination of Polynuclear
Aromatic Hydrocarbons. Varian Application Note, n.7. Catálogo.
Benner., B.A.Jr. (1998). Summarizing the Effectiveness of Supercritical Fluid
Extraction of Polycyclic Aromatic Hydrocarbons from Natural Matrix Environmental
Samples, Anal. Chem. 70, 4594-4601.
Khalili, N. R.; Scheffn, P.A. , Holsen, T.M. (1995). PAH Source Fingerprints for Coke
Ovens, Diesel and Gasoline Engines, Highway Tunnels, and Wood Combustions
Emissions. Atmos. Environ. 29, 533-542.
Kubátová, A.; Jansen, B., Vaudoisot, J.F., Hawthorne, S. B. (2002). Thermodynamic
and kinetic models for the extraction of essential oil from savory and polycyclic
aromatic hydrocarbons from soil with hot (subcritical) water and supercritical CO2. J.
Chromatogr. A. 975,175-188.

73
Lacerda, J.P. (2004). Estudo do Impacto Ambiental nos Cursos d’Água Causado pelo
Lançamento de Efluentes de Indústrias do Município de Itabirito / MG. Dissertação de
Mestrado. Universidade Federal de Ouro Preto. PROÁGUA/UFOP, Ouro Preto, MG. 97
p.
Lopez-Avila, V.; Young, R., Teplitsky, N. (1996). Microwave-assisted extraction as an
alternative to Soxhlet, sonication and supercritical fluid extraction, J. A.O.A.C Int. 79,
142–156.
Lundstedt, S.; van Bavel, B., Haglund, P., Tysklind, M., Berg, L. (2000). Pressurised
liquid extraction of polycyclic aromatic hydrocarbons from contaminated soils. J.
Chromatogr. A. 883, 151– 162.
Martens, D., Maguhn, J., Spitzauer, P., Kettrup, A.(1997). Occurrence and distribution
of polycyclic aromatic hydrocarbons (PAHs) in an agricultural ecosystem. J. Anal.
Chem. 359, 546-554.
Neto, A.J.S. e Siqueira, M.E.P.B. (2005). Análise de Praguicidas Organofosforados em
Água por Extração em Fase Sólida (SPE) Utilizando Discos C-18 e Cromatografia em
Fase Gasosa: Avaliação da Contaminação do Reservatório de Furnas (MG-Brasil).
Química Nova. 28, 747-750.
Neves, K.O. (2003). Qualidade Microbiológica da Água de Abastecimento Público e
Alternativo no Município de Ouro Preto/MG. Dissertação de Mestrado. Universidade
Federal de Ouro Preto. PROÁGUA/UFOP, Ouro Preto, MG. 132 p.
Poppi, N.R. (2000). Hidrocarbonetos Policíclicos Aromáticos e Outras Substâncias
Orgânicas na Combustão de Madeira para Produção de Carvão e em Particulado
Atmosférico da Cidade de Campo Grande – MS. Tese de Doutorado. IQ/UNESP,
Araraquara-SP. 242 p.

74
Qiao, M.; Wang, C., Huang, S., Wang, G. e Wang, Z. (2006). Composition, sources and
potencial toxicological significance of PAHs in surface sediments of the Meiliang Bay,
Taihu Lake. China. Env. Intern. 32, 28-33.
Ramosa, L.; Kristensonb, E.M., Brinkmanb, U.A.Th. (2002). Current use of pressurised
liquid extraction and subcritical water extraction in environmental analysis. J.
Chromatogr. A. 975,3-29.
Ribeiro, F.A.L. (2001). Aplicação de Métodos de Análise Multivariada no Estudo de
Hidrocarbonetos Policíclicos Aromáticos. Dissertação de Mestrado. Instituto de
Química, IQ/UNICAMP, Campinas, SP. 174 p.
Rodríguez, M.P. (2001). Avaliação da Qualidade da Água da Bacia do Alto do
Jacareí/SP (Ribeirão do Feijão e Rio do Monjolinho) Através de Variáveis Físicas,
Químicas e Biológicas. Tese de Doutorado.. Universidade de São Paulo/USP, São
Carlos, SP. 175 p.
Saim, N.; Dean, J.R., Abdullah, M.P., Zakaria, Z. (1997). Extraction of polycyclic
aromatic hydrocarbons from contaminated soil using Soxhlet extraction, pressurised and
atmospheric microwave-assisted extraction, supercritical fluid extraction and
accelerated solvent extraction. J. Chromatogr. A.791,361–366.
Silica Gel Cleanup (1996). Method 3630 C, US-EPA - Environmental Protection
Agency. USA. Disponível em www.epa.gov acessado em 15/04/2005.
Soxhlet Extraction (1996). Method 3540 C, US-EPA - Environmental Protection
Agency. USA. Disponível em www.epa.gov acessado em 05/04/2005.
Silva, M.R.C. (2002). Estudo de Sedimento da Bacia Hidrográfica do Moji-Guaçu, com
Ênfase na Determinação de Metais. Dissertação de Mestrado. Universidade de São
Paulo/IQ-USP, São Carlos, SP. 175 p.

75
Soclo, H.H.; Garrigues, PH. & Ewald, M. (2000). Origin of Polycyclic Aromatic
Hydrocarbons (PAHs) in Coastal Marine Sediments: Case Studies in Cotonou (Benin)
and Aquitaine (France) Areas. Marine Pollution Bulletin. 40, 387-396.
Su, Ming-Chien.; Christensen, E.R., Karls, J.F. (1998). Determination of PAH sources
in dated sediments from Green Bay, Wisconsin, by a chemical mass balance model.
Env. Pollution. 98, 411-419.
Sun, F.; Littlejohn, D., Gibson, M.D. (1998). Ultrasonication extraction and solid phase
extraction clean-up for determination of US EPA 16 priority pollutant polycyclic
aromatic hydrocarbons in soils by reversed-phase liquid chromatography with
ultraviolet absorption detection. Anal. Chim. Acta. 364, 1–11.
Templeton, D. M.; Ariese, F., Cornelis, R., Danielsson, L. G., Muntau, H., van
Leeuwen, H. P., Łobiński, R.(2000). Guidelines for terms related to chemical speciation
and fractionation of elements. Definitions, structural aspects, and methodological
approaches. Pure Appl. Chem. 72, 1453-1470.
Tomaniová, M.; Hajsˇlova´ , J., Pavelka Jr, J., Kocourek, V., Holadova´, K., Klímova´,
L. (1998). Microwave-assisted solvent extraction—a new method for isolation of
polynuclear aromatic hydrocarbons from plants. J. Chromatogr. A. 827, 21– 29.
Von Sperling, M. (1996). Introdução à Qualidade das Águas e ao Tratamento de
Esgotos, 2º edição. Departamento de Engenharia Sanitária e Ambiental. Universidade
Federal de Minas Gerais - UFMG. Belo Horizonte, MG. 243 p.Yamada, T.M. (2006).
Determinação de Fontes de Hidrocarbonetos Policíclicos Aromáticos e Alifáticos em
Sedimentos de Ambientes Aquáticos. Dissertação de Mestrado. Universidade Federal de
São Carlos/UFSCAR. São Carlos, SP. 86 p.
Yang, Y. e B. Li. (1999). Subcritical Water Extraction Coupled to High-Performance
Liquid Chromatography. Anal.Chem. 71, 1491 – 1495.

76
Yim, U.H.; Hong, S.H. & Shim, W.J. (2007). Distribution and characteristics of PAHs
in sediments from the marine environment of Korea. Chemosphere. 68, 85–92.
Thorsen, W.A.; Cope, W.G.e Shean, D. (2004). Bioavailability of PAHs: effects of soot
and PAH source. Environm. Sci. Technol. 38, 2029.
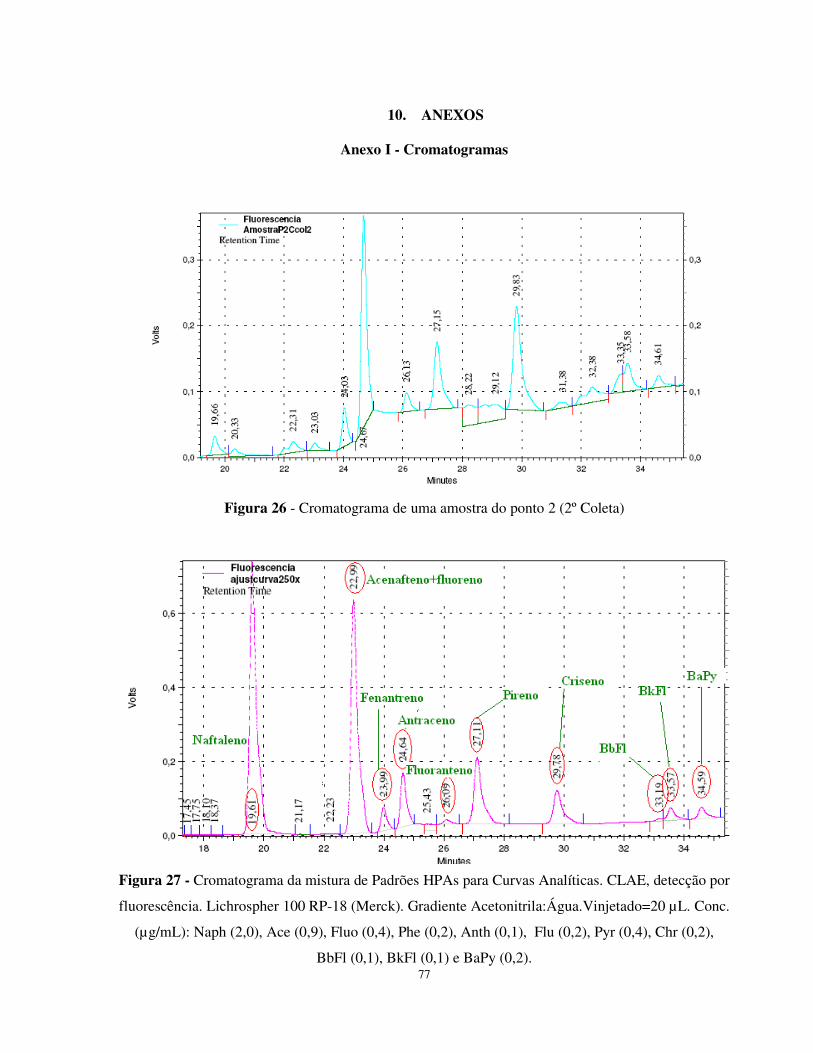
77
10. ANEXOS
Anexo I - Cromatogramas
Figura 26 - Cromatograma de uma amostra do ponto 2 (2º Coleta)
Figura 27 - Cromatograma da mistura de Padrões HPAs para Curvas Analíticas. CLAE, detecção por
fluorescência. Lichrospher 100 RP-18 (Merck). Gradiente Acetonitrila:Água.Vinjetado=20 µL. Conc.
(µg/mL): Naph (2,0), Ace (0,9), Fluo (0,4), Phe (0,2), Anth (0,1), Flu (0,2), Pyr (0,4), Chr (0,2),
BbFl (0,1), BkFl (0,1) e BaPy (0,2).
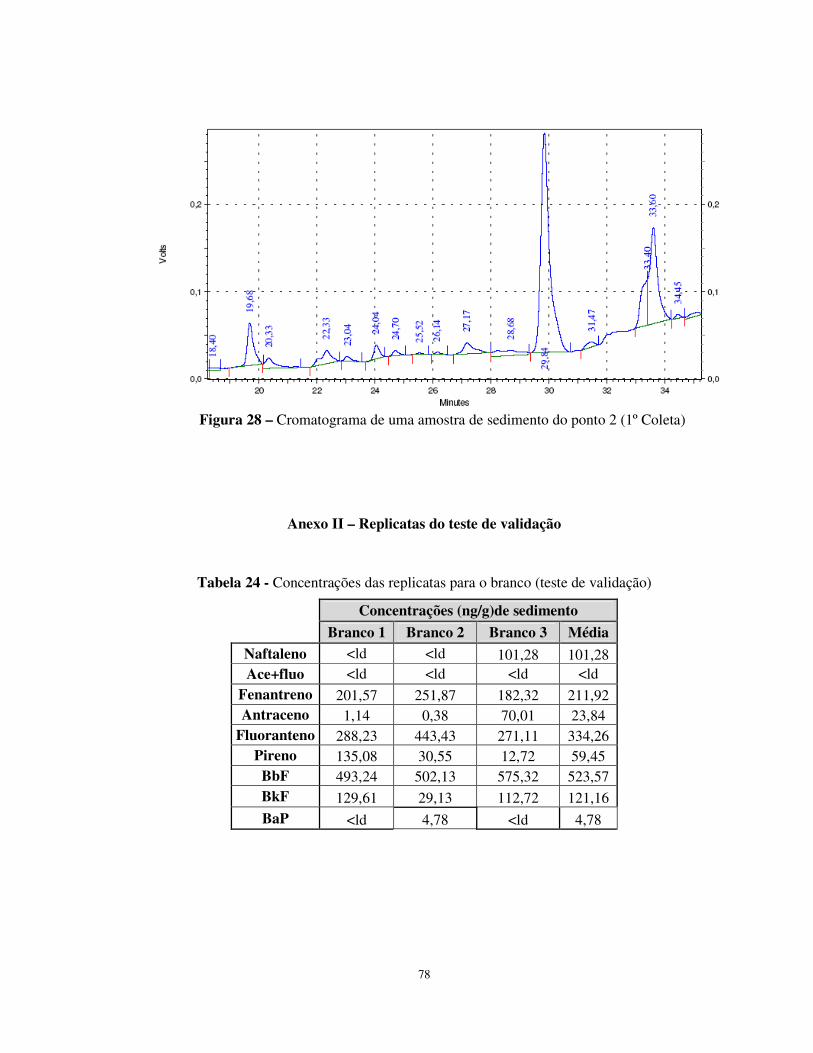
78
Anexo II – Replicatas do teste de validação
Tabela 24 - Concentrações das replicatas para o branco (teste de validação)
Concentrações (ng/g)de sedimento Branco 1 Branco 2 Branco 3 Média
Naftaleno <ld <ld 101,28 101,28 Ace+fluo <ld <ld <ld <ld
Fenantreno 201,57 251,87 182,32 211,92 Antraceno 1,14 0,38 70,01 23,84
Fluoranteno 288,23 443,43 271,11 334,26 Pireno 135,08 30,55 12,72 59,45 BbF 493,24 502,13 575,32 523,57 BkF 129,61 29,13 112,72 121,16 BaP <ld 4,78 <ld 4,78
Figura 28 – Cromatograma de uma amostra de sedimento do ponto 2 (1º Coleta)
79
Tabela 25 - Concentrações das replicatas para o Spike 1
Concentrações (ng/g)de sedimento Spike 1A Spike 1B Spike 1C Média
Naftaleno <ld <ld <ld <ld Ace+fluo <ld <ld 30,34 30,34
Fenantreno 284,05 26,70 220,56 252,31 Antraceno 19,41 <ld 20,25 19,83
Fluoranteno 552,60 87,34 692,50 622,55 Pireno 51,53 57,53 61,68 56,91 BbF 1786,67 320,07 1729,68 1278,81 BkF 868,11 107,84 872,69 616,21 BaP 841,35 55,04 825,24 573,88
Tabela 26 - Concentrações das replicatas para o Spike 2
Concentrações (ng/g)de sedimento
Spike 2A Spike 2B Spike 2C Média Naftaleno <ld <ld <ld <ld Ace+fluo <ld <ld <ld <ld
Fenantreno 335,32 454,72 64,00 395,02 Antraceno 31,11 27,61 4,05 29,36
Fluoranteno 512,59 1055,44 179,74 784,01 Pireno 101,92 140,94 115,33 119,40 BbF 1132,66 2454,16 613,72 1793,41 BkF 605,24 1368,56 195,54 723,11 BaP 277,37 1017,91 107,05 467,44


















