
Apresentação do PowerPoint -...
74
ERROS INATOS DO METABOLISMO Professor Isaías Soares de Paiva MUCOPOLISSACARIDOSES
Transcript of Apresentação do PowerPoint -...

ERROS INATOS DO METABOLISMO
Professor Isaías Soares de Paiva
MUCOPOLISSACARIDOSES

DOENÇAS DE DEPÓSITO LISOSSÔMICO
O LISOSSOMO

DOENÇAS DE DEPÓSITO LISOSSÔMICO

LipasesProteasesGlicosidasesNucleasesFosfolipasesArilsulfatases e Fosfatase ácida
Cerca de 50 enzimas
(Altamente glicosilada)
Membrana de parede simples
Estrutura do Lisossomo
Complexo de enzimas
O LISOSSOMO
Apresentador
Notas de apresentação
Lisossomo: estrutura que apresenta enzimas digestivas capazes de digerir um grande número de produtos orgânicos. Realiza a digestão intracelular. É importante nos glóbulos brancos e de modo geral para a célula já que digere as partes desta (autofagia) que serão substituídas por outras mais novas, o que ocorre com frequência em nossas células. Cerca de 50 enzimas estão localizadas nos lisossomas, incluindo lipases, proteases, glucosidases, nucleases, arilsulfatases e fosfatase ácida, que possibilitam decompor praticamente todo o tipo de macromoléculas nos seus constituintes fundamentais. Do ponto de vista da origem dos materiais degradados no seu interior, os lisossomas podem ser autolisossomas e heterolisossomas. Os lisossomos são caracterizados, não só por seu conteúdo enzimático, como por sua membrana envoltória única dentre as organelas: proteínas transportadoras contidas nessa membrana, permitem que os produtos finais da digestão de macromoléculas (tais como aminoácidos, açúcares, nucleotídeos e até mesmo pequenos peptídeos) transitem para o citosol onde serão excretados ou reutilizados pela célula. A membrana do lisossomo possui também bombas de H+, que, através da hidrólise de ATP, bombeiam íons H+ para o lúmen, mantendo assim o pH ácido, ideal para a ação enzimática. A maioria das membranas lisossomais é altamente glicosilada, de modo que lhe é conferida proteção das enzimas contidas no lúmen.

A secreção celular de proteínas (enzimas) pelo RE e complexo de Golgi - para o meio extracular
O LISOSSOMO
Ribossomo e Golgi
Gene (DNA)
mRNA
Lisossomo

Estrutura da manose-6-fosfato numa enzima
lisossomal.
As hidrolases lisossómi-cassão glicoproteinas que
possuem, na sua estrutura, uma manose-6-fosfato.
O LISOSSOMO
Apresentador
Notas de apresentação
As bolsas originadas pela fusão de lisossomos com fagossomos ou pinossomos são denominadas vacúolos digestivos; em seu interior, as substâncias originalmente presentes nos fagossomos ou pinossomos são digeridas pelas enzimas lisossômicas. À medida que a digestão intracelular vai ocorrendo, as partículas capturadas pelas células são quebradas em pequenas moléculas que atravessam a membrana do vacúolo digestivo, passando para o citosol. Essas moléculas serão utilizadas na fabricação de novas substâncias e no fornecimento de energia à célula. Eventuais restos do processo digestivo, constituídos por material que não foi digerido, permanecem dentro do vacúolo, que passa a ser chamado vacúolo residual. Muitas célula eliminam o conteúdo do vacúolo residual para o meio exterior. Nesse processo, denominado clasmocitose, o vacúolo residual encosta na membrana plasmática e fundem-se com ela, lançando seu conteúdo para o meio externo.

EIM – Grupo1 - deficiência enzimática no catabolismo de moléculas complexas.
Substrato A Degradação
Bloqueio
Enzima lisossômica
acumula
Acúmulo (substância A) nos tecidosExcreção Urinária Patogênese relacionada ao acúmulo (excesso)
DOENÇAS DE DEPÓSITO LISOSSÔMICO

DOENÇAS DE DEPÓSITO LISOSSÔMICO

Glicosaminoglicanos – GAGs.
A função dos GAGs A função das enzimas
Enzimas são proteínas especiais responsáveis pelas reações de
transformação que ocorrem no nosso corpo.
MUCOPOLISSACARIDOSES
Apresentador
Notas de apresentação
O que causa as MPS ? Nosso corpo é formado por muitas e muitas células. As células formam os tecidos. O espaço entre as células nos tecidos é preenchido pelos GAGs. Eles formam estruturas parecidas com uma “escovinha de mamadeira”, que junta água, como uma esponja. Assim ajudam a manter a forma e a resistência dos tecidos. Para entender como os GAGs acumulam e causam as MPS, é importante entender que no curso de uma vida normal existe um processo contínuo de construir novos mucopolissacarídeos e de quebrar os velhos – um processo de reciclagem. O andamento deste processo é necessário para manter o corpo saudável. Em uma célula saudável os GAGs se soltam da membrana e entram na célula para serem quebrados dentro de compartimentos chamados lisossomos. O processo de quebra e reciclagem precisa de uma série de ferramentas bioquímicas chamadas de enzimas. Para quebrar os GAGs, uma série de enzimas trabalha em sequência, uma após a outra, dentro do lisossomo. A cadeia de GAG é quebrada com a remoção de uma molécula de açúcar de cada vez começando em uma das pontas da cadeia. Enzimas são proteínas especiais responsáveis pelas reações de transformação que ocorrem no nosso corpo. Cada enzima no organismo possui uma tarefa especial e tem uma ação muito específica, da mesma maneira que uma chave e uma fechadura.

Normal Doença de depósito lisossômico
Acúmulo lisossômico de GAGsMUCOPOLISSACARIDOSES

Classificação
MPS IMPS IIMPS IIIMPS IVMPS VIMPS VIIMPS IX
Hurler ou ScheieHunterSanfilippoMórquioMaroteaux-LamySlyNatowicz
MUCOPOLISSACARIDOSES
Apresentador
Notas de apresentação
MPS I is part of a larger family of lysosomal storage disorders characterized by a deficiency in any one of 11 enzymes involved in the sequential degradation of GAGs (McKusick and Neufeld, 1983; Hopwood and Morris, 1990). The enzyme deficiency results in the accumulation of non-degraded or partially degraded GAGs, which are stored in lysosomes and excreted in urine. The seven MPS disorders share many clinical features, but each individual disorder has specific characteristics (Hopwood and Morris, 1990; Neufeld and Muenzer, 2001). All MPS disorders have a spectrum of clinical severity.

MPS Nome GAG Enzima Gene
MPS I Hurler DS e HS α-L-iduronidase 4p 16.3
MPS II Hunter DS e HS Iduronato Sulfatase Xq28
MPS III A Sanfilippo A HS Heparan-N-sulfatase 17q25.3
MPS III B Sanfilippo B HS α-N-acetil-glicosami-nidase 17q21
MPS III C Sanfilippo C HS Acetil CoA α-glicosa-minídeo acetiltransf. Desconh.
MPS III D Sanfilippo D HS N-acetilglicosamina-6-sulfatase 12q14
MPS IV A Mórquio A QS Galactose-6-sulfatase 16q24.3
MPS IV B Mórquio B QS β-galactosidase 3p21.33
MPS VI Marateaux-Lamy DS Arilsulfatase A 5q13-q14
MPS VII Sly DS e HS β-glicuronidase 7q21.11
MPS IX Natowicz CS Hialuronidase 3p21.1-p21.3
MUCOPOLISSACARIDOSES

MUCOPOLISSACARIDOSESDoença progressiva – MPS I forma severa
10 meses 12 meses
22 meses 34 meses
39 meses

Progressão da doença: evolução rápida
1 ano 6 anos 10 anos 16 anos 19 anos3 meses
Fotos: cortesia da The National MPS Society Inc.
Evolução rápida é visível nos primeiros anos de vida. Aos 12 meses de idade:
os traços grosseiros ainda não são marcantes. macrocefalia, anormalidades esqueléticas, mãos em garra, abdome
protuberante (hepatomegalia) e hérnia umbilical são evidentes.
MUCOPOLISSACARIDOSES

MUCOPOLISSACARIDOSESDoença progressiva – MPS I forma severa
0 3 6 9 12 30 35 40 45 50 55 60 65
Normal ao nascimento
Normal nos primeiros meses Diagnóstico
meses
Macrocefalia?

MUCOPOLISSACARIDOSES
Giordano Bruno Souza
Solicito:
Pesquisa de Glicosaminoglicanos(GAGs)
Material: urina
Pesquisa GAGs
Cromatografia GAGs
Ensaio Enzimático
Análise molecular
Diagnóstico

Reconheça o fenótipo
MPS
MUCOPOLISSACARIDOSES

Reconheça o fenótipoAtraso crescimentoAtraso DNPMRM progressivoMacrocefaliaAlterações osteoarticularesHepatoesplenomegaliaDismorfias faciais
MUCOPOLISSACARIDOSES
Fenótipo característico
Apresentador
Notas de apresentação
Developmental delay is usually apparent in patients with the severe form of MPS I by 12 to 24 months of age, with a maximum functional age of 2 to 4 years. This is followed by slowed development and progressive regression in developmental skills until death (Neufeld and Muenzer, 2001). Most of these patients develop only limited language skills likely related to a triad of developmental delay, chronic hearing loss, and enlarged tongue (Neufeld and Muenzer, 2001). In addition to the direct CNS effects, communicating high pressure hydrocephalus is also common in these patients. The obstruction to resorption of cerebrospinal fluid causes an associated increase in intracranial pressure, leading to brain compression. Rapidly increasing pressure can be the cause of rapid developmental decline in some patients. Symptoms may be difficult to assess, and progression can be insidious and is often underappreciated. Lumbar puncture with opening pressure is a preferred method for assessing the degree of pressure elevation.

MUCOPOLISSACARIDOSES
Macrocefalia
Baixa Estatura
Fenótipo facial
Abdome volumosoHepatoesplenomegalia
Mão “em garra”
Deformidades articulares
Pectus carinatum
Apresentador
Notas de apresentação
Patients at the severe end of the spectrum demonstrate skeletal manifestations of MPS I, early in life, by about 6 months of age. At that time, it is common to observe mild bone abnormalities (detected by radiological methods), particularly within the hip, ovoid vertebrae, as well as widening of the ribs. At the clinical level, skeletal involvement does not become obvious until the age of 10-14 months when a gibbus deformity of the back, or dorsolumbar kyphosis, is observed in severe patients. Eventually, progressive skeletal dysplasia involving all bones is seen in all types of MPS I. The vertebrae may become progressively flattened and beaked, often leading to spinal deformity. Typically the pelvis is poorly formed, with small femoral heads and coxa valga. Involvement of the femoral head leads to progressive and debilitating hip deformity. Clavicles may be short, thickened, and irregular (“oar-shaped”). By 3 years of age there is little progression of physical (linear) growth; children may not grow taller than 4 feet. The joints may become stiffened by the age of 2 years and progressive arthropathy affects all joints. The hands take on a characteristic claw deformity resulting from both phalangeal dysostosis and synovial thickening.

MUCOPOLISSACARIDOSES
Macrocefalia
Baixa Estatura
Fenótipo facial
Abdome volumosoHepatoesplenomegalia
Comprometimento osteoarticular

MUCOPOLISSACARIDOSES
Abdome globosoHepatoesplenomegalia
Fenótipo facial
Macrocefalia
Hérnia umbilical
Apresentador
Notas de apresentação
Progressive hepatosplenomegaly is common in MPS I (Neufeld and Muenzer, 2001). GAGs storage in the liver and spleen does not lead to organ dysfunction; however, organ size may be massive. �Loose stools and diarrhea are episodic problems for some patients.

MUCOPOLISSACARIDOSES
Hérnia umbilical
Apresentador
Notas de apresentação
The enlarged liver and spleen, in addition to abnormal connective tissue, may cause hernias to develop. Inguinal and umbilical hernias are common in MPS I. They are occasionally present at birth or develop within the first several months of life, and are often one of the first clinical signs noted.

MPS I
MUCOPOLISSACARIDOSESFenótipo MPS
Macrocefalia
Cabelos espessos
Sobrancelhas espessas
Base nasal plana
Macroglossia
Aspecto facial “coarse”
Lábios grossos

Macrocefalia
Nariz alargado com narinas antervertidas
Base nasal plana
Fronte proeminente
Aspecto facial “coarse”
Cabelos espessos
Macroglossia
Lábios grossos
MUCOPOLISSACARIDOSES

Fenótipo MPS
Nariz alargado com narinas antervertidas
Base nasal plana
Macroglossia
Aspecto facial “coarse”
MUCOPOLISSACARIDOSES

Opacificação da córnea. MPS I – 18 meses
Fenótipo MPSMUCOPOLISSACARIDOSES

Gibosidade. MPS I
MUCOPOLISSACARIDOSES
Apresentador
Notas de apresentação
These photos show two young children with a gibbus, characteristic in more severe cases of MPS I. Also note hepatosplenomegaly present in the child on the left.

Síndrome do túnel do Carpo. A maioria de pacientes faltam sintomas
típicos até que a compressão severa ocorra.
Fenótipo MPSMUCOPOLISSACARIDOSES
Apresentador
Notas de apresentação
Carpal tunnel syndrome and other nerve entrapments or nerve compressions are common in many patients with MPS I. Most patients lack typical symptoms until severe compression occurs (Neufeld and Muenzer, 2001), making early diagnosis of carpal tunnel syndrome challenging.

O comprometimento articular pode ser o primeiro sinal visível.Fique atento a “artrites” sem características de doença auto-imune.
MUCOPOLISSACARIDOSES

Diagnóstico clínico precoce
10 meses 12 meses
MUCOPOLISSACARIDOSES

MUCOPOLISSACARIDOSES
Diagnostico mais precoce !!!
Apresentador
Notas de apresentação
"I am very happy to be able to get in touch with your society... because in this way I realise there are other cases like mine in different parts of the world who share the same experiences. I have two boys - Jakeline Alves de Menezes (16 years old) and Valtercie Camilio de Menezes (15 years old), MPS sufferers with Morquio Syndrome and another yet undefined. ... Both are wonderful boys, united and loving and also much loved in the community where we live in Goiania Brazil."

10 meses 12 meses
Caso Clínico 1: Menina, 10 meses. Observado aumento do PC e diminuição da velocidade de crescimento. Pais jovens, consanguíneos. Exame: hepatoesplenomegalia.
MUCOPOLISSACARIDOSES
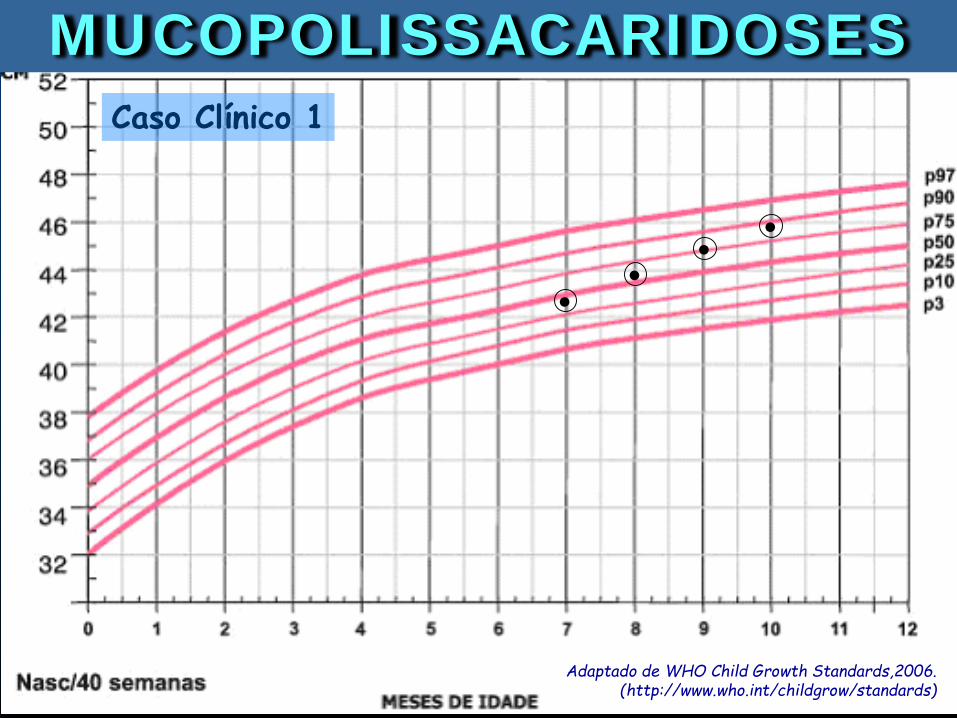
Adaptado de WHO Child Growth Standards,2006. (http://www.who.int/childgrow/standards)
. . . .Caso Clínico 1
MUCOPOLISSACARIDOSES

. . . .
Caso Clínico 1

Terapia de Reposição Enzimática (TRE)
MPS I, II, VIMPS III, IV
MPS IMPS IIMPS VI
MUCOPOLISSACARIDOSES

Professor Isaías Soares de Paiva
ADRENOLEUDISTROFIA

PEROXISSOMONúcleo
Citoplasma

PEROXISSOMO
Peroxissomos: organelas membranosas no citoplasma. Enzimas (oxidases) que catalisam o peróxido de hidrogênio (H2O2). Oxidam os AG
para a síntese de colesterol e como matériapara a Beta-oxidação dos AG. Rins e fígado.
2 H2O2 → Catalase → 2 H2O + O2
Apresentador
Notas de apresentação
Os peroxissomos são organelas membranosas presentes no citoplasma das células vegetais e animais, formando vesículas arredondadas, cuja função está relacionada ao armazenamento de enzimas que catalisam o peróxido de hidrogênio (água oxigenada - H2O2), uma substância tóxica que necessita ser degradada. Essas enzimas oxidam os ácidos graxos para a síntese de colesterol. E também são usados como matéria-prima na respiração celular com o intuito de obter energia. No corpo humano, os peroxissomos são encontrados nas células que formam os rins (células renais) e o fígado (células hepáticas). Peroxissomos: pequenas vesículas semelhantes aos lisossomos, porém sua enzima principal é a Peroxidase (Catalase), que degrada as moléculas do Peróxido de Hidrogênio (água oxigenada) que se formam como resultado do metabolismo celular. O Peróxido de Hidrogênio pode ser muito tóxico para a célula porque leva a produção de radicais livres, que danificam as células.

PEROXISSOMO
Os peroxissomas β-oxidam: AGCM, AGCL, AGCML. Ácidos dicarboxílicos de
cadeia média e longa. Prostanóides (prostaglandinas,
tromboxanos, leucotrie-nos) Cadeias laterais carboxílicas
de certos xenobióticos e dos intermediários dos ácidosbiliares, ácidos di- e trihidroxicoprostanóicos (emperoxissomas hepáticos).
Lazarow PB. Peroxisome structure, function, and biogenesis - Human patients and yeast mutants show strikingly similar defects in peroxisome biogenesis. Journal of Neuropathology and Experimental Neurology. 54, 720-25, 1995.
Sistema de β-oxidação
Apresentador
Notas de apresentação
Peroxissomo: O sistema de oxidacao de acidos gordos peroxissomal foi identificado em 1976 (Lazarow e De Duve, 1976). Os peroxissomas B-oxidam uma serie de substratos: acidos gordos de cadeia media, longa e muito longa, acidos dicarboxilicos de cadeia media e longa, acidos gordos com ramificacao 2-metil derivados do isoprenoido (exemplo: acido pristanico), prostanoides (prostaglandinas, tromboxanos, leucotrienos), as cadeias laterais carboxilicas de certos xenobioticos e dos intermediarios dos acidos biliares, acidos di- e trihidroxicoprostanoicos (em peroxissomas hepaticos), outros substratos sao o acido dolicoico e as cadeias carboxilicas laterais de vitaminas lipossoluveis (Cerdan et ai, 1988; Diczfalusy e Alexon, 1990; Jedlitschky et ai, 1991; K(|)lvraa e Gregersen, 1986; Mortensen, 1992; Singh et ai, 1993; Vamecq e Drayer, 1989; Mannaerts e van Veldhoven, 1993a).

ADRENOLEUDISTROFIAGenética Gene ABCD1
ATP Binding Casset, Sbfamily D, Member 1
Xq28
Mosser J, Douar A-M, Sarde C-O, Kioschis P, Feil R, Moser H, Poustka A-M, Mandel J-L, Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361: 726-730, 1993.
Apresentador
Notas de apresentação
Genética ALD-X: O gene da X-ALD, recentemente denominado ABCD1, foi identificado por tecnicas de clonagem posicionai (Mosser et ai, 1993), apos o estudo de um doente de X-ALD que, na regiao vizinha dos genes R/GCP (Red/ Green colour pigment genes) (Sack e Morrei, 1993) apresentava um complexo rearranjo que incluia duas deleccoes separadas por uma inversao de 140 kb (Feil et ai, 1991, Aubourg et dl, 1990b). Este gene de 21kb, compreende 10 exoes e juncoes intrao-exao com sequencias consenso para os dadores/aceitadores de splicing. O gene da X-ALD codifica para um mRNA com um quadro de leitura (open reading frame) de 2,235 bases codificando uma proteina (ALDP) de 745 aminoacidos (Sarde et ai, 1994a)(apendice 3). O exao 1 de 1286 bp, contem 386 bp de sequencia nao-traduzida- 5TJTR. O exao 10 contem 246bp de regiao codificadora e aproximadamente 1570 bp de sequencia nao-traduzida ate ao sinal putativo de poliadenilacao (figura 2).

ADRENOLEUDISTROFIAPatogenia
Apresentador
Notas de apresentação
Genética:

ADRENOLEUDISTROFIAGenética – Gene ABCD1
A mutação descrita é uma deleção da adenina que ocorre no 477º aminoácido (Glutamato) → ALDP mutadonão possui sítio funcional de ligação ao ATP.
Valadares ER, Trindade ALC, Oliveira LR, Arantes RR, Daker MV, Viana BM, Haase VG, Jardim LB, Lopes GC, Godard ALB. Novel exon nucleotide deletion causes adrenoleukodystrophy in a Brazilian family. Genetics and molecular research : GMR. 10. 65-74, 10.4238/vol10-1gmr975. 2011.
Apresentador
Notas de apresentação
Genética – Gene ABCD1: A) gene ABCD1, localizado no cromossomo X, consistindo de 10 exons, codifica a proteína adrenoleucodistrofia (ALDP) com 745 aminoácidos. Esta proteína possui dois domínios: a superfamília da membrana ABC, um domínio transmembrana com seis segmentos helicoidais e a superfamília da NTPase P-loop, um domínio de ligação a ATP. Observa-se que este último começa pouco antes do 500º aminoácido da proteína. Como a mutação descrita neste estudo é uma deleção da adenina que ocorre no 477º aminoácido (glutamato), podemos concluir que o ALDP mutado não possui sítio funcional de ligação ao ATP. B) Alinhamento múltiplo de proteínas da sequência parcial de aminoácidos do ALDP. A comparação da sequência da proteína normal obtida do NCBI (NP_000024.2) com a proteína mutada resultante da deleção da adenina, obtida da ferramenta Translate do ExPASy Proteomics Server, mostra uma identidade de sequência de 64%. Pode-se observar a partir desse alinhamento, realizado pelo programa Blastp 2.2.21+, utilizando como referência o ALDP do tipo selvagem e o banco de dados de seqüências de proteínas não redundantes (nr), que existe uma conservação do aminoácido glutamato (E ) em várias espécies, incluindo mamíferos, anfíbios, peixes, invertebrados e fungos. Vermelho indica o aminoácido preservado e azul indica o aminoácido que diferencia as seqüências das espécies comparadas.

ADRENOLEUDISTROFIAGenética
Mosser J, Douar A-M, Sarde C-O, Kioschis P, Feil R, Moser H, Poustka A-M, Mandel J-L, Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361: 726-730, 1993.
Gene ABCD1ATP Binding Casset, Sbfamily D, Member 1
21 Kb. 10 éxonsALDP = 745 aminoácidosDomínio Transmembrana (Aa 75-352)Domínio de Ligação a Nucleotíceos(ATP) - (Aa 474-700)
Apresentador
Notas de apresentação
Genética – Gene ABCD1: O gene consiste em 10 exons (caixas) com o códon de início no exon 1. A proteína ALDP contém um DTM (aminoácidos 75-352) e um domínio de ligação a nucleotídeos (NBD, também chamado de região de ligação a ATP, aminoácidos 474-700). A marca X vermelha mostra a posição da mutação S149R no éxon 1 do gene ABCD1. A proteína ALDP possui dois domínios: a superfamília da membrana ABC, um domínio transmembrana com seis segmentos helicoidais e a superfamília da NTPase P-loop, um domínio de ligação a ATP.

ADRENOLEUDISTROFIAGenética – Gene ABCD1
Domínio Transmembrana Domínio ligação a neucleotídeo - ATP
Wang YF, Ying W, Li H, Chen H, Xu JC, Chao. S149R, a novel mutation in the ABCD1 gene Causing X-linked adrenoleukodystrophy. Oncotarget. 8. 10.18632/, 2017.
Apresentador
Notas de apresentação
Genética – Gene ABCD1: Análise de mutação do gene ABCD1. (A) Proteína (ALDP) e gene (ABCD1): Estrutura genômica do gene ABCD1 humano. O gene consiste em 10 exons (caixas) com o códon de início no exon 1. As letras vermelhas no gene indicam a posição da mutação missense, que está localizada no exon 1 do gene ABCD1. (B) Diagrama de seqüenciamento de parte do exon 1 no gene ABCD1. A seta indica a alteração do nucleotídeo envolvido. O painel superior é a sequência normal, enquanto o painel inferior é a sequência mutada. (C) Esquema da estrutura molecular do ALDP. ALDP contém um DTM (aminoácidos 75-352) e um domínio de ligação a nucleotídeos (NBD, também chamado de região de ligação a ATP, aminoácidos 474-700). A marca X vermelha mostra a posição da mutação S149R no éxon 1 do gene ABCD1. A proteína ALDP possui dois domínios: a superfamília da membrana ABC, um domínio transmembrana com seis segmentos helicoidais e a superfamília da NTPase P-loop, um domínio de ligação a ATP.

ADRENOLEUDISTROFIAGenética – Gene ABCD1
Soardi FC, Esquiaveto-Aun AM, Guerra-Júnior G, Lemos-Marini SHV, Mello MP. Phenotypic variability in a family with x-linked adrenoleukodystrophy caused by the p.Trp132Ter mutation. Arquivos Brasileiros de Endocrinologia e Metabologia. 54. 738-43, 2010.
> 750 mutações descritas
Apresentador
Notas de apresentação
Genética – Gene ABCD1: Representação esquemática dos 10 exons do gene ABCD1. Resíduos de aminoácidos nos quais mais de dez casos foram relatados com mutações diferentes são indicados. Os números de casos relatados estão entre parênteses. A mutação descrita neste relatório é representada em negrito.

ADRENOLEUDISTROFIA
Dubois-Dalcq M, Feigenbaum V, Aubourg P. The Neurobiology of X- linked Adrenoleukodystrophy (ALD). A demyelinating peroxisomal disorder. Trends in Neurose. 22(1):4-12, 1999.
Mutação Gene ABCD1
Função da proteínaALDP
AGCML não sãotransportados parao Perixossomo
Acúmulo de AGCML
Lesão da bainha de mielina, córtexsupra-renal e células testiculares
Apresentador
Notas de apresentação
Ald-X – Patogenia: As inclusoes lipidicas trilamelares visiveis em microscopia electronica, no cerebro e glandula supra-renal dos doentes com X-ALD, sao o resultado da acumulacao de acidos gordos de cadeia muito longa saturados (AGCML) (Powers e Schaumburg, 1974). A medida que a doenca avanca, todo o cortex da glandula supra-renal atrofia. Em fetos com X-ALD sao visiveis alteracoes na zona da glandula supra-renal fetal (Powers et ai, 1982). Individuos do sexo masculino em idade pos-pubertaria desenvolvem estruturas lipidicas lamelares no citoplasma das celulas de Leydig, semelhantes as encontradas nas celulas adrenocorticotroficas (Powers e Schaumburg, 1981).

ADRENOLEUDISTROFIA
Dubois-Dalcq M, Feigenbaum V, Aubourg P. The Neurobiology of X- linked Adrenoleukodystrophy (ALD). A demyelinating peroxisomal disorder. Trends in Neurose. 22(1):4-12, 1999.
Ácumulo de AGCML:
nas membranas das células do SNC → alterações estruturais→ função dos receptoresmembranares.
induz uma resposta inflama-tória → citoquinas → aumen-tam efeitos acúmulo AGCML → morte dos oligodendrócitose desmielinização.
instabilidade na mielina e morte dos oligodendrócitos.
Patogenia
Apresentador
Notas de apresentação
ALD-X – Patogenia: Varias hipoteses surgiram para explicar os mecanismos atraves dos quais a acumulacao de AGCML originam alteracoes no sistema nervoso central (SNC) em doentes com X-ALD: (i) a acumulacao das longas cadeias de AGCML nas membranas das celulas do SNC provoca alteracoes estruturais perturbando a funcao dos receptores membranares (Ho et ai, 1995); (ii) a acumulacao de AGCML induz uma resposta inflamatoria e as citoquinas, por sua vez, aumentam os efeitos causados pela acumulacao de AGCML, originando morte dos oligodendrocitos e desmielinizacao; (iii) a acumulacao de AGCML causa instabilidade na mielina e morte dos oligodendrocitos (Dubois-Dalcq et ai, 1999).

ADRENOLEUDISTROFIAPatogenia
Moser HW, Moser AB, et al. Adrenoleukodystrophy: survey of 303 cases: biochemistry, diagnosis and theraphy. Ann Neurol. 16, 628-41, 1984.
Deminielização
Apresentador
Notas de apresentação
ALD-X – Patogenia:

ADRENOLEUDISTROFIA
Frequência
1.1/100.000 nascidos vivos(Moser et ai, 1995c).
1.6/100.000 (Kirk et ai, 1998 e Ruiz et ai, 1998).
1/200.000 indivíduos do sexo masculino(van Geel et ai, 1994).
2/3 das portadoras desenvolvem sintomas. Frequência: 1/10.000. (Moser et al, 1995c).
Geral: 1/100.000.
Apresentador
Notas de apresentação
Frequência: A X-ALD e uma doenca genetica, que apresenta um padrao de hereditariedade recessivo ligado ao cromossoma X (Moser et al, 1984b) e uma frequencia que pode variar entre 1.1 por 100 000 nados vivos nos EUA (Moser et ai, 1995c), 1.6 por 100 000 na Australia e Espanha (Kirk et ai, 1998 e Ruiz et ai, 1998) e 1 por 200 000 individuos do sexo masculino na Holanda (van Geel et ai, 1994). A tabela 1 lista a frequencia dos diferentes fenotipos clinicos observados em doentes com X-ALD de diversos paises. Dois tercos das portadoras desenvolvem sintomas de X-ALD, aumentando a frequencia desta patologia para 1:10 000 (Moser et al, 1995c).

ADRENOLEUDISTROFIA
Adrenoleucodistrofia Autossômica Recessiva
Adrenoleucodistrofia ligada ao cromossomo X
Forma Cerebral InfantilForma Cerebral AdolescenteForma Cerebral do AdultoAdrenomieloneuropatia (AMN)Doença de Addison (isolada)
Fenótipos Clínicos
Defeito intrinseco dos Peroxissomos –(ausentes ou estruturalmente anormais).
Apresentador
Notas de apresentação
ALD ligada ao cromossoma X versus ALD neonatal: O termo adrenoleucodistrofia e usado para descrever duas entidades geneticas completamente distintas com alteracoes no cortex das glandulas supra-renais e na mielina do sistema nervoso. Na forma ligada ao cromossoma X, a alteracao bioquimica presente e a acumulacao de acidos gordos de cadeia muito longa nos tecidos e no plasma, encontrando-se a estrutura do peroxissoma normal. Na forma neonatal (Ulrich et al, 1978) o padrao de hereditariedade e autossomico recessivo e verifica-se uma diminuicao do numero e tamanho dos peroxissomas (Aubourg et ai, 1986). A acumulacao de acidos gordos de cadeia muito longa, e uma caracteristica comum as duas formas de adrenoleucodistrofia que, no entanto, resulta de diferentes mecanismos: na forma neonatal, existe uma alteracao em todo o processo de p-oxidacao daqueles substratos; na forma ligada ao cromossoma X, o defeito envolve apenas activacao dos acidos gordos (Goldfischer et ai, 1985). Existem outras diferencas fundamentais entre a forma ligada ao X (X- ALD) e a neonatal: (i) na forma neonatal, grande parte dos peroxissomas estao ausentes e os que estao presentes sao morfologicamente distintos dos existentes em celulas de doentes com a forma de adrenoleucodistrofia ligada ao cromossoma X que apresentam uma estrutura normal (Vamecq et ai, 1986); (ii) os doentes com a forma neonatal apresentam uma incapacidade de sintetizar plasmalogeneos e oxidar acido fitanico (Wanders et ai, 1987a), tem niveis aumentados de acido pipecolico (Kelley e Moser, 1984) e dos intermediarios dos acidos biliares (Noetzel et ai, 1983), enquanto que os doentes com X-ALD nao apresentam nenhuma destas alteracoes; (iii) as duas formas de adrenoleucodistrofia nunca ocorreram na mesma familia (Lazarow e Moser 1995).

ADRENOLEUDISTROFIAForma cerebral infantil
Assintomático: 3 a 10 anos (geralmente 4 – 5).
Sintomas de dificitsmotores e sensoriais –
hemiparésia, alteraçõesvisuais e auditivas.
Evolução até um estadograve, vegetativo.
Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15:1761-6.
Nascimento 5 10
Moser HW, Moser AB, et al. Adrenoleukodystrophy: survey of 303 cases: biochemistry, diagnosis and theraphy. Ann Neurol. 16, 628-41, 1984.
1512
Idade (anos)
Óbito 2 ou 3 anos apósinício dos sintomas).

ADRENOLEUDISTROFIAForma Cerebral Infantil Sintomas neurológicos precoces:
distúrbios comportamentais, baixodesempenho escolar, deterioração da visão e hipoacusia.
Curso progressivo com convulsões, tetraplegia espástica e demência se manifestam em meses.
Óbito em 2 a 3 anos após o início dos sintomas neurológicos na maioriados casos.
Menino de 7 anos com CCALD. Até os 6 anos de idade DNPM era normal. Aos 6 anos: hipoacusia, seguido diminuição da visão; surgiram convulsões e uma tetraparesiaespástica. Óbito 2 anos após o início dos sinto-mas.
van Geel BM, Assies J, Wanders RJA, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. J. Neurol, Neurosurg, and Psych; 63:4–14, 1997.
Apresentador
Notas de apresentação
ALD Cerebral Infantil: A ALD cerebral na infantil (CCALD) é caracterizada por desmielinização cerebral rapidamente progressiva. O início ocorre entre 3 e 10 anos de idade. Sintomas neurológicos precoces freqüentes são distúrbios comportamentais, baixo desempenho escolar, deterioração da visão e discriminação auditiva prejudicada. O curso é implacavelmente progressivo, e convulsões, tetraplegia espástica e demência se manifestam em meses. A figura 2 mostra um paciente típico com CCALD. A maioria dos pacientes morre dentro de 2 a 3 anos após o início dos sintomas neurológicos. Alguns pacientes sobrevivem por mais tempo, embora em estado vegetativo persistente. Em 80% dos pacientes com CCALD, a RNM cerebral tipicamente apresenta desmielinização extensa na substância branca periventricular occipital. Os primeiros sintomas da CCALD são frequentemente atribuídos a um distúrbio de déficit de atenção ou hiperatividade. Antes do advento de testes diagnósticos confiáveis e RNM, leucodistrofia metacromática, lipofuscinose ceróide, leucodistrofia de células globóides (doença de Krabbe) e panencefalite esclerosante subaguda foram às vezes diagnosticados em vez de CCALD.

ADRENOLEUDISTROFIAForma Cerebral Infantil
RNM ponderada em T2 cerebral de um menino de 8 anos de idade com deficiência visual e convulsões devidoà CCALD. Primeiros sintomas neurológicos con-sistiram em estrabismo divergentepara o qual ele foi operado duas ve-zes. Óbito aos 10 anos.
van Geel BM, Assies J, Wanders RJA, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. J. Neurol, Neurosurg, and Psych; 63:4–14, 1997.
Apresentador
Notas de apresentação
Forma Cerebral Infantil: RNM ponderada em T2 cerebral de um menino de 8 anos de idade com deficiência visual e convulsões devido à CCALD. O dano extensivo infligido na substância branca periventricular occipital é evidente. Três de seus tios maternos morreram antes dos 10 anos de idade, mas sua mãe tinha VLCFAs normais em fibroblastos de plasma e pele e de alguma forma entendera que ela não podia ser um heterozigoto. Retrospectivamente, seus primeiros sintomas neurológicos consistiram em estrabismo divergente para o qual ele foi operado duas vezes. Ele morreu com a idade de 10 anos (cortesia de MRI Mojoart Dr. EL Hospital Universitário, Groningen, na Holanda.)

Em aproximadamente 85% a RNM mostra um padrão característico de sinal T-2 aumentado simétrico na região parieto-occipital com realce de contraste na margem avançada.
Moser HW, Moser AB, Steinberg SJ, Kemp S. Diagnosis of ALD. https://adrenoleukodystrophy.info/clinical-diagnosis/diagnosis-of-ald.
ADRENOLEUDISTROFIAForma Cerebral Infantil
Apresentador
Notas de apresentação
A RNM do cérebro é sempre anormal em homens neurologicamente sintomáticos e geralmente fornece a primeira pista diagnóstica. Em aproximadamente 85% dos indivíduos afetados, a RNM mostra um padrão característico de sinal T-2 aumentado simétrico na região parieto-occipital com realce de contraste na margem avançada.

ADRENOLEUDISTROFIA
1993 George Miller
Apresentador
Notas de apresentação
Filme Óleo de Lorenzo: Lorenzo viveu até os 30 anos, desmistificando todas as pesquisas que indicavam uma morte precoce para os portadores de ALD, e morreu em 2008, um dia após seu aniversário.

ADRENOLEUDISTROFIA
Lorenzo Odone viveuaté os 30 anos, faleceuem 2008. Michaela Odone faleceu em2000, vitima de cân-cer, e Augusto Odonemorreu, na Itália em2013 por PNM.
Apresentador
Notas de apresentação
Filme Óleo de Lorenzo: Lorenzo viveu até os 30 anos, desmistificando todas as pesquisas que indicavam uma morte precoce para os portadores de ALD, e morreu em 2008, um dia após seu aniversário. Michaela Odone faleceu em 2000, vitima de câncer, e Augusto Odone morreu, na Itália, no dia 25 de outubro de 2013 em decorrência de uma infecção pulmonar.

ADRENOLEUDISTROFIAForma Cerebral Adolescente
van Geel BM, Assies J, Wanders RJA, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. J. Neurol, Neurosurg, and Psych; 63:4–14, 1997.
Menor frequência. Os sintomas e progressão são
semelhantes aos do CCALD. Início é entre 10 e 21 anos de
idade.
Metamorfose dramática de um meninosaudável de 6 anos em um adolescentede 17 anos demente e espástico. O diagnóstico inicial foi de EpilepsiaMioclonia, mas retrospectivamenteAdolCALD foi diagnosticado após um primo dele ter morrido de CCALD.
Apresentador
Notas de apresentação
Forma Cerebral Adolescente: ALD cerebral adolescente (AdolCALD) ocorre com muito menos frequência. Os sintomas e progressão são semelhantes aos do CCALD, mas o início é entre 10 e 21 anos de idade.

ADRENOLEUDISTROFIAAdrenomieloneuropatia (AMN)
van Geel BM, Assies J, Wanders RJA, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. Journal of Neurology, Neurosurgery, and Psychiatry; 63:4–14, 1997.
Fenótipo mais comum na Holanda. Início na terceira e quarta décadas de vida. Sintomas neurológicos decorrentes da
mielopatia (principal) e neuropatia. Sintomas: paraparesia espástica, distúrbios
miccionais. Em 50% envolvimento cerebral na RNM,
(desmielinização semelhante CCALD). A medula espinhal é freqüentemente atrófica. EMG: polineuropatia sensorimotora axonal. A expectativa de vida é normal, se tratamento
adequado. Diagnósticado como outras doenças
neurológicas (esclerose múltipla progressiva e paraparesia espástica hereditária).
Apresentador
Notas de apresentação
AMN Pele menos na Holanda, o AMN é o fenótipo mais comum de X-ALD. O início dos sintomas neurológicos nesse fenótipo geralmente ocorre na terceira e quarta décadas. Os déficits neurológicos são devidos principalmente à mielopatia e em menor grau, a neuropatia. Os pacientes desenvolvem gradualmente uma paraparesia espástica, muitas vezes em combinação com o sentido de vibração perturbado nas pernas e distúrbios miccionais. Cerca de 50% dos homens com AMN apresentam envolvimento cerebral leve a moderado na RNM, e em alguns casos as anormalidades na substância branca podem se assemelhar à desmielinização observada na CCALD; a medula espinhal é freqüentemente atrófica. Estudos de condução nervosa e EMG são compatíveis com polineuropatia sensorimotora predominantemente axonal. A expectativa de vida é provavelmente normal, a menos que os pacientes desenvolvam adicionalmente desmielinização cerebral, ou quando a insuficiência adrenocortical não seja reconhecida e permaneça sem tratamento. Em muitos pacientes com doenças neurológicas AMN, como esclerose múltipla progressiva crônica e paraparesia espástica hereditária foram inicialmente diagnosticados.

ADRENOLEUDISTROFIA
van Geel BM, Assies J, Wanders RJA, Barth PG. X linked adrenoleukodystrophy: clinical presentation, diagnosis, and therapy. Journal of Neurology, Neurosurgery, and Psychiatry; 63:4–14, 1997.
A escassez de cabelo no
courocabeludo é
típico e podeser visto em
todos osfenótipos.
Este pacientecom AMN
desenvolveuo padrão de
cabelodurante a
adolescência.
Adrenomieloneuropatia (AMN)
Apresentador
Notas de apresentação
AMN A escassez de cabelo no couro cabeludo é típico e pode ser visto em todos os fenótipos. Este paciente com AMN desenvolveu o padrão de cabelo durante a adolescência

ADRENOLEUDISTROFIAInsuficiência Supra-renal
Powers JM, Moser HW, Moser AB, Schaumburg HH. Adrenoleukodystrophy: The significance of pathologic lesions in adrenal gland and testis. Hum Pathol. 13, 1013-15, 1982.
Moser HW, Bergin A, Naidu S, Ladenson PW. Adrenoleukodystrophy: new aspects of adrenal cortical disease. Endocrinol Metab Clin North Am. 20, 1-9, 1991.
Em 95% dos casos de ADL-X infantil e juvenil. Em 20% constitui o primeiro sintoma. A insuficiência periférica – funções glico e mineralo-corticóides sem alterações ao nível da medula.
Apresentador
Notas de apresentação
A insuficiencia da glandula supra-renal ocorre em 95% dos casos de X-ALD infantil e juvenil e constitui em cerca de 20% dos casos como o primeiro sintoma da patologia (Moser et ai, 1991b). A insuficiencia periferica das glandulas supra-renais, observada na X-ALD, caracteriza-se por uma perturbacao das funcoes gluco- e mineralo-corticoides (Powers et ai, 1982) que nao e acompanhada por alteracoes ao nivel da medula (Bakos et ai, 1995).

ADRENOLEUDISTROFIA
Apresentador
Notas de apresentação
ALD-X em mulheres: Estado sintomático e idade. As barras indicam a percentagem de portadoras de ADL-X considerada sintomática dentro de cada grupo etário (isto é, diagnosticada com uma mielopatia e / ou uma neuropatia periférica). Os pontos mostram cada portador da ADL-X na coorte, classificada como sintomática ou assintomática.

ADRENOLEUDISTROFIA
Espectro Clínico
ALD Info. https://adrenoleukodystrophy.info/clinical-diagnosis/facts-on-ald.
Apresentador
Notas de apresentação
Espectro Clínico ALD: espectro clínico de ALD em homens. Pacientes com ALD não apresentam nenhum sintoma ao nascimento. As barras coloridas indicam a faixa etária de início da insuficiência adrenal (barra azul), mielopatia (barra lilás) e ALD cerebral (barra verde). O início da insuficiência adrenal pode ser tão cedo quanto aos 5 meses de idade. Na idade adulta, os homens invariavelmente desenvolvem uma mielopatia progressiva crônica. A ALD cerebral pode ocorrer em qualquer idade, com o paciente mais jovem relatado aos 3 anos de idade. O defeito primário no gene ALD e o armazenamento de VLCFA nos tecidos resulta em insuficiência adrenal e mielopatia (em conjunto, denominada adrenomieloneuropatia ou AMN). O início da ALD cerebral é mais provavelmente definido pela interação do defeito do gene primário da ALD e uma combinação de fatores desencadeantes ambientais e / ou genéticos ainda desconhecidos. É importante reconhecer que pacientes com insuficiência adrenal e / ou mielopatia permanecem em risco de desenvolver ALD cerebral.

ADRENOLEUDISTROFIAGenética - Herança
Apresentador
Notas de apresentação
Genética:

Mensuração da concentração ds AGCML. Os níveis de AGCML estão elevados em 99,9% dos homens com ALD-X em todas
as idades, independentemente da presença ou ausência de sintomas clínicos. Os três parâmetros analisados são: a concentração de C26:0, a razão de
C24:0/C22:0 e a relação de C26:0/C22:0.
Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S. Analysis of very long-chain fatty acids usingelectrospray ionization mass spectrometry. Mol Genet Metab; 79(3):189-96, 2003.
ADRENOLEUDISTROFIADiagnóstico
Apresentador
Notas de apresentação
Diagnóstico: Masculino: O ensaio laboratorial mais importante é a mensuração da concentração de ácidos graxos de cadeia muito longa (VLCFA) no plasma. Os níveis de VLCFA estão elevados em 99,9% dos homens com ALD-X em todas as idades, independentemente da presença ou ausência de sintomas clínicos. Os três parâmetros analisados são: a concentração de C26: 0, a razão de C24: 0 / C22: 0 e a relação de C26: 0 / C22: 0. A Tabela 1 mostra os resultados médios para controles normais, homens afetados e fêmeas portadoras.

ADRENOLEUDISTROFIA
Tratamento dietético (ingestão ristrita de ácidos graxosde cadeia longa para reduzir o acúmulo). Óleo de Lorenzo. Quimioterapia. Transplante de células-tronco. Transplante de medula óssea. Terapia Gênica Terapia de Reposição Hormonal (para tratar a
insuficiência adrenal)
Tratamento
Apresentador
Notas de apresentação
Tratamento: Os outros tratamentos utilizados abordam os sintomas da doença e não a causa da doença. Tratamento dietético (ingestão ristrict de ácidos graxos de cadeia longa para reduzir o acúmulo) Quimioterapia Transplante de células-tronco Transplante de medula óssea (não usado como comum, devido aos riscos e desafios da utilização da medula óssea de um indivíduo) Terapia de Reposição Hormonal (para tratar a insuficiência adrenal)

ADRENOLEUDISTROFIAÓleo de Lorenzo
Odone A, Odone M. Lorenzo’s oil: a new treatment for adrenoleukodystrophy. J Pediatr Neurosci 5:55–61, 1989.
Ácido cerótico (C26:0) Ácido oleico (C18:l ω 9-
trioleato de glicerol-GTO) Ácido erucico (C22:l ω 9 -
trierucicato de glicerol-GTE).
Bloquear a síntese endógenados AGCML.
Apresentador
Notas de apresentação
Óleo de Lorenzo: Essencialmente do acido cerotico (C26:0), e por outro na ingestao de determinados oleos: acido oleico (C18:l xu 9-trioleato de glicerol-GTO) e acido erucico (C22:l w 9 - trierucicato de glicerol-GTE), capazes de bloquear a sintese endogena desses mesmos acidos gordos

ADRENOLEUDISTROFIATratamento –Terapia Gênica
Apresentador
Notas de apresentação
Terapia Genética: Embora não tenha sido amplamente utilizado, o uso da terapia genética foi testado em pacientes em que uma correspondência apropriada pode ser encontrada. O processo envolve a modificação de vetores lentivirais apropriados para expressar o tipo selvagem ABCD1, ao invés da versão mutante. Os vetores lentivirais são vetores que podem infectar células divididas e não divididas e criar uma expressão gênica mais duradoura do que outros retrovírus. O vetor lentiviral usado neste processo é derivado de versões deficientes do HIV. Este vetor é então transplantado para os pacientes usando um processo semelhante a um transplante de medula óssea ou de células-tronco. Este processo foi feito principalmente na França até agora. O raciocínio por trás do uso de terapia genética foi que não havia correspondência encontrada para um transplante tradicional. Na maioria dos casos, até agora, houve sucesso na resolução do processo de desmielinização e na diminuição / resolução dos sintomas neurológicos. Com isso dito, ainda há altos níveis de plasma VLCFA no paciente, mesmo após o tratamento de terapia gênica. Um grande risco do uso da terapia gênica é que a inserção desses vetores poderia interferir com a função biológica normal da célula e levar à leucemia. Este resultado não ocorreu em ensaios de tratamento de ALD, mas tem no tratamento de terapia gênica de outras doenças.

ADRENOLEUDISTROFIATratamento –Terapia Gênica
Apresentador
Notas de apresentação
Terapia Genética:

TMO é o único tratamento que pode interromper o processo de desmielinização de pacientes com ALD-X, se as ce lulas do dador atravessarem a barreira hematoencefa lica do receptor.
Necessário que seja feito no estágio inicial da doença1.
Sobrevida de 5 anos de 89% a 95% contra uma de sobrevida de 45% a 54% em pacientes não tratados com o TMO2 3 4.
Particularmente em pacientes do sexo masculino sem sintomasneurológicos, e a desmielinização cerebral em meninos pode serinterrompida ou revertida por TMO.
1 – Cartier N, Aubourg P. Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linkedadrenoleukodystrophy. Brain Pathol. 2010;20:857–62.
2 – Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experiencefrom 1982 to 1999. Blood. 2004;104:881–8.
3 – Mahmood A, Raymond GV, Dubey P, et al. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linkedadrenoleukodystrophy: a comparison study. Lancet Neurol. 2007; 6:687–92. 11.
4 – Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118:1971–8.
5 – Krivit W, Lockman LA, Watkins PA, Hirsch J, Shapiro EG. The future for treatment by bone marrow transplantation for adrenoleukodystrophy, metachromatic leukodystrophy, globoid cell leukodystrophy and Hurler syndrome. J Inherit Metab Dis 18:398–412, 1995.
ADRENOLEUDISTROFIATransplante de Medula Óssea
Apresentador
Notas de apresentação
TMO: TMO é o único tratamento que pode interromper o processo de desmielinização de pacientes com ALD-X e resultar em uma boa qualidade de vida em longo prazo, desde que o procedimento seja feito no estágio inicial da doença1, mostrando uma sobrevida de 5 anos de 89% a 95% contra uma probabilidade de sobrevida de 45% a 54% em pacientes não existe. O transplante de medula ossea (TMO) em doentes com X-ALD, permite restaurar, pelo menos em parte, os locais desmielinizados se as celulas do dador atravessarem a barreira hematoencefalica do receptor. A experiencia com este tratamento e de certa forma positiva, sendo que os transplantados apresentam uma regressao das lesoes desmielinizantes com melhoramento de algumas funcoes cerebrais nomeadamente as cognitivas (Aubourg et ai, 1990a). Esta aproximacao terapeutica nao e no entanto eficaz em doentes em que ja ocorreu o aparecimento de sinais neurologicos, sendo especialmente aplicada em doentes cujos sinais de envolvimento neurologico sao detectados no estado ainda precoce ou em doentes assintomaticos, com sinais de progressao da patologia (Moser et ai, 1984c).

ADRENOLEUDISTROFIAEscore de Loes
Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15:1761-6.
Estágio bastante precoce = < 4 Estágio precoce = 4 e 8 Estágio tardio = 9 e 13 Estágio muito tardio = > 13.
Avaliar progressão (padrão ouro).
Escala de gravidade na RNM de 34 pontos.
Sistema de pontuação baseado na localização e extensão das lesões de desminielização.
Locais selecionadas: substâncias brancas parieto-occipital, temporal anterior e frontal, corpo caloso, vias visuais e auditivas, fibras de projeção, cerebelo e núcleos da base.
TMO se escore < 9. Melhor < 4. Se = 0 (não indica).
Apresentador
Notas de apresentação
Critérios de Loes: O padrão ouro para avaliar a progressão da doença na ALD-X foi proposto por Loes em 1994, em uma escala de gravidade na RM de 34 pontos usando um sistema de pontuação baseado na localização e extensão do envolvimento e avaliação de presença de atrofia local ou global. As localizações selecionadas para o escore de gravidade são as substâncias brancas parieto-occipital, temporal anterior e frontal, corpo caloso, vias visuais e auditivas, fibras de projeção, cerebelo e núcleos da base. O escore de gravidade auxilia na determinação da extensão do dano na mielina no encéfalo. Considera-se um estágio bastante precoce se os escores forem menores que 4, um estágio precoce é considerado com escores entre 4 e 8, um estágio tardio é entre 9 e 13 e um estágio muito tardio se os escores forem maiores que 13. O escore de Loes é um dos parâmetros usados no seguimento e orienta decisões terapêuticas para o transplante de células hematopoiéticas (TMO). A progressão das alterações na RM na ALD-X depende da idade do paciente, escore inicial ao diagnóstico e localização anatômica das lesões (padrão da RM). Quando estes dados são interpretados em conjunto, auxiliam a predizer o curso da doença e a seleção de candidatos para o TMO. . Peters et al. sugerem que o paciente com escore de Loes menor que 9 pode se beneficiar do TMO, mas opiniões mais recentes sinalizam que até mesmo escores menores (< 4) podem ser melhores na seleção de candidatos para o TMO. O TMO não pode ser justificado na ausência de evidências de desmielinização (i.e., escore de Loes igual a zero), porque 60% a 85% dos meninos não desenvolverão a doença cerebral durante a infância. O TMO é o único tratamento que pode interromper o processo de desmielinização de pacientes com ALD-X e resultar em uma boa qualidade de vida em longo prazo, desde que o procedimento seja feito no estágio inicial da doença, mostrando uma sobrevida de 5 anos de 89% a 95% contra uma probabi- lidade de sobrevida de 45% a 54% em pacientes não tratados com o TMO, mas uma cura definitiva ainda não existe. Considerando que o diagnóstico precoce e o tranplante no momento certo são essenciais para que os resultados sejam otimizados, nós esperamos que o uso das técnicas avançadas de RM possa auxiliar nas decisões terapêuticas.

ADRENOLEUDISTROFIA
Espectro Clínico
ALD Info. https://adrenoleukodystrophy.info/clinical-diagnosis/facts-on-ald.
Apresentador
Notas de apresentação
Espectro Clínico ALD: espectro clínico de ALD em homens. Pacientes com ALD não apresentam nenhum sintoma ao nascimento. As barras coloridas indicam a faixa etária de início da insuficiência adrenal (barra azul), mielopatia (barra lilás) e ALD cerebral (barra verde). O início da insuficiência adrenal pode ser tão cedo quanto aos 5 meses de idade. Na idade adulta, os homens invariavelmente desenvolvem uma mielopatia progressiva crônica. A ALD cerebral pode ocorrer em qualquer idade, com o paciente mais jovem relatado aos 3 anos de idade. O defeito primário no gene ALD e o armazenamento de VLCFA nos tecidos resulta em insuficiência adrenal e mielopatia (em conjunto, denominada adrenomieloneuropatia ou AMN). O início da ALD cerebral é mais provavelmente definido pela interação do defeito do gene primário da ALD e uma combinação de fatores desencadeantes ambientais e / ou genéticos ainda desconhecidos. É importante reconhecer que pacientes com insuficiência adrenal e / ou mielopatia permanecem em risco de desenvolver ALD cerebral.

ADRENOLEUDISTROFIA

O diagnóstico de ALD-X deve ser considerado em quatro situações clínicas distintas:Meninos com sintomas de TDAH, distúrbio comportamental
progressivo, perda de visão, dificuldade na linguagem falada, incoordenação, sinais de demência ou outros distúrbios neurológicos.
Homens jovens ou de meia idade com distúrbios progressivos da marcha, rigidez ou fraqueza em MMII, anormalidades do controle esfincteriano e disfunção sexual, com ou sem insuficiência adrenal ou déficits cognitivos ou comportamentais.
Todos os homens com insuficiência adrenocortical primária (adrenal), com ou sem evidência de anormalidade neurológica.
Mulheres de meia-idade ou mais velhas com paraparesia progressiva, anormalidades do controle esfincteriano e distúrbios sensoriais afetando principalmente os MMII.
Moser HW, Moser AB, Steinberg SJ, Kemp S. Diagnosis of ALD. https://adrenoleukodystrophy.info/clinical-diagnosis/diagnosis-of-ald.
ADRENOLEUDISTROFIA
Apresentador
Notas de apresentação
Diagnóstico clínico O diagnóstico de ALD-X deve ser considerado em quatro situações clínicas distintas: Meninos com sintomas de TDAH, que também apresentam distúrbio comportamental progressivo, perda de visão, dificuldade em compreender a linguagem falada, piora da caligrafia, incoordenação, sinais de demência ou outros distúrbios neurológicos. Homens jovens ou de meia idade com distúrbios progressivos da marcha, rigidez ou fraqueza nas pernas, anormalidades do controle esfincteriano e disfunção sexual, com ou sem insuficiência adrenal ou déficits cognitivos ou comportamentais. Todos os homens com insuficiência adrenocortical primária (adrenal), com ou sem evidência de anormalidade neurológica. Mulheres de meia-idade ou mais velhas com paraparesia progressiva, anormalidades do controle esfincteriano e distúrbios sensoriais afetando principalmente as pernas. Pode ser difícil estabelecer o diagnóstico de ALD-X em uma mulher com histórico familiar negativo. O diagnóstico baseia-se em características clínicas, mais comumente paraparesia espástica progressiva, e um painel de testes laboratoriais.

ADRENOLEUDISTROFIA


















