
ASPECTOS CLÍNICO- DEMOGRÁFICOS DA SÍNDROME DE DOWN …nico... · 2017. 9. 14. · de J. L. Down...
104
UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA HOSPITAL UNIVERSITÁRIO PROFESSOR EDGARD SANTOS CURSO DE PÓS-GRADUAÇÃO EM MEDICINA E SAÚDE ASPECTOS CLÍNICO- DEMOGRÁFICOS DA SÍNDROME DE DOWN EM SERVIÇO DE REFERÊNCIA NA BAHIA Tatiana Amorim DISSERTAÇÃO DE MESTRADO SALVADOR-BA, 2002
Transcript of ASPECTOS CLÍNICO- DEMOGRÁFICOS DA SÍNDROME DE DOWN …nico... · 2017. 9. 14. · de J. L. Down...

UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA HOSPITAL UNIVERSITÁRIO PROFESSOR EDGARD SANTOS CURSO DE PÓS-GRADUAÇÃO EM MEDICINA E SAÚDE
ASPECTOS CLÍNICO-DEMOGRÁFICOS DA SÍNDROME
DE DOWN EM SERVIÇO DE REFERÊNCIA NA BAHIA
Tatiana Amorim
DISSERTAÇÃO DE MESTRADO
SALVADOR-BA, 2002

UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA HOSPITAL UNIVERSITÁRIO PROFESSOR EDGARD SANTOS CURSO DE PÓS-GRADUAÇÃO EM MEDICINA E SAÚDE
ASPECTOS CLÍNICO-DEMOGRÁFICOS DA SÍNDROME
DE DOWN EM SERVIÇO DE REFERÊNCIA NA BAHIA
Projeto de Dissertação apresentado ao Colegiado do Curso de Pós-Graduação em Medicina e Saúde, da faculdade de Medicina da Universidade Federal da Bahia, como pré-requisito obrigatório para a obtenção do Grau de Mestre em Medicina, na área de Concentração em Medicina Interna.
AUTORA: Tatiana Regia Suzana Mattos de Amorim PROFESSORA-ORIENTADORA: Lícia Maria Oliveira Moreira
SALVADOR-BA, 2002.

UNIVERSIDADE FEDERAL DA BAHIA FACULDADE DE MEDICINA HOSPITAL UNIVERSITÁRIO PROFESSOR EDGARD SANTOS CURSO DE PÓS-GRADUAÇÃO EM MEDICINA E SAÚDE
Dissertação de Mestrado
ASPECTOS CLÍNICO- DEMOGRÁFICOS DA SÍNDROME DE
DOWN EM SERVIÇO DE REFERÊNCIA NA BAHIA
Membros Titulares
• Lícia Maria Oliveira Moreira (presidente), Professora Titular de Neonatologia do Departamento de Pediatria da Faculdade de Medicina da Universidade Federal da Bahia.
• Maria Betânia Pereira Toralles, Professora Adjunta de Genética do Departamento de Pediatria da Faculdade de Medicina da Universidade Federal da Bahia.
• Nelson de Assis Barros, Professor Titular de Pediatria do Departamento de Pediatria da Faculdade de Medicina da Universidade Federal da Bahia.
Membro Suplente:
• Argemiro d’Oliveira, Professor Adjunto do Departamento de Medicina da Faculdade de Medicina da Universidade Federal da Bahia.
SALVADOR-BA, 2002.

Por tanto amor, por tanta emoção, A vida me fez assim...
(Sérgio Magrão/Luis Carlos Sá)


DEDICATÓRIA

A Alana, Ana Paula, Allisson, Ailton, Aline, Ana
Beatriz, Aída Beatriz, Adriano, Adriele B., Adriele R., Ângelo, Ângelo Gabriel, Alexandre
Jorge, Alexandre S., Alessandra, Adriano, André, Bruna, Bruno, Carina, Carine, Caroline, Cleuto,
Carlos Eduardo, Deise, Daylan, Danielton, Elane, Evelyn, Eroilton, Elson, Eduarda, Eduardo, Francisco, Franciele, Gracielle, Gean, Geovana, Gabriel, Herbert, Henrique,
Jacob, Jadson, John Lennon, Jefferson, Jeniffer, João Victor, Leandro, Lucas, Lucas J., Lucas Luis,
Laís, Lucas N., Lucas C., Luiz Eduardo, Letícia, Luana, Milena R., Milena S., Mariana, Máximo, Maxuel, Michel, Matheus M., Maria Rita, Mateus A., Matheus S., Matheus C., Natália, Paulo Jr., Rute, Railane, Rodrigo, Rodrigo F., Rodrigo C.,
Sara, Suellen, Stefhanie, Ticiana, Tauana, Tailane, Tamares, Vitor Hugo, Victor, Vitor, William, Wesley e
Wilton –
VII

vocês foram a motivação, a inspiração e a alegria deste trabalho.

FONTES DE FINANCIAMENTO

1. Bolsa de Estudos da CAPES entre Março de 1999 e Fevereiro
de 2000
2. Bolsa de Pesquisa da FAPEX, entre Junho de 2001 e Junho de 2002.
X

AGRADECIMENTOS

• A minha mãe, pelo seu amor e apoio incondicionais, em todos os momentos da minha vida;
• A meu pai, pelo exemplo de homem da ciência;
• Ao meu marido, pela ajuda constante durante a realização deste trabalho;
• À equipe do Laboratório de Genética Médica, que viabilizou a realização
deste trabalho, em especial à bióloga Rita Alves, pela realização dos cariótipos e interesse constante nos pacientes;
• À Dra Cristina Maria Mascarenhas Fortuna, que guiou meus primeiros
passos na Genética Clínica;
• À Dra Isabella Queiróz, que prestou assistência psicológica fundamental às nossas crianças e suas famílias;
• À Dra Isabel Guimarães, que realizou o estudo ecocardiográfico dos
pacientes;
• À Dra Cláudia Dias, que nos apoiou com a avaliação oftalmológica de alguns pacientes;
• À acadêmica de Medicina Grace Milene Tavares, que participou ativamente
do atendimento ambulatorial nos seus primeiros momentos;
• À Dra Mônica Torres, pelo seu apoio na realização de exames de imagem, sempre que necessário;
• Ao Professor Argemiro D’Oliveira, pela sua orientação, sugestões, apoio, e
que me mostrou o caminho certo a seguir;
• À amiga Sônia Celino, secretária do Curso de Pós-graduação em Medicina e Saúde, pelo apoio e carinho durante a nossa trajetória;
• Ao Prof. José Tavares-Neto, pelo estímulo inicial e orientação dos
primeiros passos;
• À SER DOWN, Associação Baiana de Síndrome de Down, pelo acompanhamento constante;
• À Sra Jucélia de Oliveira Santos, bibliotecária do Hupes, pela realização da
ficha catalográfica;
• E principalmente, a estas crianças e seus maravilhosos pais, aos quais tentamos dar o melhor de nós.
XII

ÍNDICE

1.
Resumo
01
2.
Introdução
04
3.
Revisão da Literatura
07
4..
Objetivos
23
5.
Justificativas
25
6.
Casuística, Material e Métodos
27
7.
Resultados
32
8.
Discussão
39
9.
Propostas de Estudos
50
10.
Conclusões
54
11.
Summary
57
12.
Referências Bibliográficas
59
13.
Anexos
66
XIV

RESUMO

ASPECTOS CLÍNICO-DEMOGRÁFICOS DA SÍNDROME DE
DOWN EM SERVIÇO DE REFERÊNCIA NA BAHIA. A Síndrome de
Down é a cromossomopatia mais comum em humanos, e a principal
causa de Retardo Mental na população. Trata-se de entidade bastante
conhecida e estudada, e cujos portadores podem ter um grande
incremento na qualidade de vida, através de programas de
acompanhamento e estimulação, sendo inclusive possível a sua
inserção na sociedade e no mercado de trabalho. OBJETIVO:
descrever os aspectos clínico-demográficos de um grupo de pacientes
atendidos no Ambulatório de Genética de um Hospital de cuidados
terciários, visando o melhor conhecimento das alterações
relacionadas a esta doença no estado da Bahia. DESENHO E
ESTUDO: Corte Transversal. MATERIAL E MÉTODOS: A amostra foi
constituída de 87 pacientes portadores de Síndrome de Down de 0 a
18 anos acompanhados no Ambulatório de Genética do Hospital
Universitário Professor Edgard Santos, sendo excluídos os pacientes
com quadro clínico duvidoso que não pôde ser confirmado
citogeneticamente. A Análise Estatística realizada foi descritiva.
RESULTADOS: foi observado discreto predomínio de indivíduos do
sexo masculino (58,6%). A média de idade materna encontrada foi de
30,1 anos. Com respeito ao grupo racial, 69% das crianças foram
classificadas como brancas ou mulatas claras. Na avaliação
fenotípica, 100% das crianças apresentaram fenda palpebral oblíqua,
enquanto que apenas 32% apresentavam o achado de prega de
flexão palmar única (linha simiesca). A cardiopatia congênita ocorreu
em 44.9% dos pacientes avaliados. O hipotireoidismo foi detectado
em 6% dos pacientes. CONCLUSÕES: as características clínico-
demográficas do grupo de pacientes estudados assemelham-se ao
2

descrito na literatura, excetuando-se os achados referentes à idade
materna (a média da idade materna foi significantemente menor do
que o encontrado na literatura internacional, porém compatível com o
descrito para sociedades sul-americanas) e ao grupo racial. Tais
aspectos deverão ser mais estudados no futuro. O seguimento dos
pacientes ao longo do seu desenvolvimento poderá evidenciar outras
diferenças.
PALAVRAS-CHAVES: Síndrome de Down, Deficiência mental,
Fenótipo, Malformações
3

INTRODUÇÃO

A primeira referência a um grupo específico de deficientes
mentais data do século XIX. John Langdon Down, um médico pediatra
inglês, realizou um estudo descritivo dos pacientes institucionalizados
em um Serviço para deficientes mentais, enfatizando a questão
étnica. Seu artigo, intitulado “Observations on an ethnic classification
of idiots”, foi publicado em 1866, no Clinical Lectures and Reports, do
London Hospital, e chama à atenção para um grande número de
deficientes mentais que apresentam uma aparência asiática, apesar
de pertencerem comprovadamente a famílias européias. Down
descreve este grupo de indivíduos como portando características
fenotípicas semelhantes, o que permitiria agrupá-los como um "tipo
mongólico de idiotia"[1].
"(...) The great Mongolian family has numerous representatives,
and it is to this division, I wish, in this paper, to call special attention. A
very large number of congenital idiots are typical Mongols. So marked
is this, that when placed side by side, it is difficult to believe that the
specimens compared are not children of the same parents. (…)
The hair is not black, as in the real Mongol, but of a brownish
colour, straight and scanty. The face is flat and broad, and destitute of
proeminence. The checks are roundish, and extended laterally. The
eyes are obliquely placed, and the internal canthi more then normally
distant from one another. The palpebral fissure is very narrow (…).
The tongue is long, thick, and is much roughened. The nose is small.
The skin has a slight dirty yellowish tinge, and is deficient in elasticity,
giving the appearance of being too large for the body.
The boy's aspect is such that it is difficult to realize that he is a
child of Europeans, but so frequently are these characters presented,
5

that there can be no doubt that these ethnic features are the result of
degeneration.
The Mongolian type of idiocy occurs in more than ten per cent of
the cases which are presented to me. They are always congenital
idiots, and never result from accidents after uterine life. (…) They are
humorous, and a lively sense of the ridiculous often colours their
mimicry. (…). They are usually able to speak; the speech is thick and
indistinct, but may be improved very greatly by a well-directed scheme
of tongue gymnastics. (…) By systematic training, considerable
manipulative power may be obtained.
(…) The improvement which training effects in them is greatly in
excess of what would be predicated if one did not know the
characterists of the type. The life expectancy, however, is far below
the average, and the tendency is to the tuberculosis, which I believe to
be the hereditary origin of the degeneracy (...)"[2].
Apesar de algumas noções errôneas, como atribuir à
tuberculose parental a ocorrência de Síndrome de Down, a descrição
de J. L. Down é extremamente clara e concisa, servindo de base até
hoje para muitos estudos, além de delinear, ainda no século XIX, o
papel da estimulação na melhora do quadro clínico.
A Síndrome de Down é considerada a Cromossomopatia mais
comum em humanos, e é certamente a doença genética mais
conhecida, tanto no meio médico como leigo[1, 3]. É a causa genética
mais comum de deficiência mental, não apresentando diferenças
significantes entre os grupos raciais e sócio-econômicos, e
apresentando ema prevalência global estimada em 1:600 nascidos
vivos[1]
6

REVISÃO DA LITERATURA

Os primeiros registros sugestivos da Síndrome de Down são
ainda mais antigos do que o artigo original de JL Down. Juan Esquirol
(1838), Duncan (1866) e Seguin (1866) relataram casos clínicos que
sugerem Síndrome de Down, porém nenhum deles ateve-se a uma
descrição mais detalhada. Fora da esfera científica, temos um quadro
de Sir Joshua Reynolds, de 1773, intitulado Lady Cockburn y sus
hijos, que mostra uma criança com características faciais da SD[1].
O termo “Anomalia de Down” foi postulado em 1966, por
Penrose & Smith, em substituição a Mongolismo, e posteriormente a
doença passou a denominar-se Síndrome de Down[4].
Quase um século depois da publicação de JL Down, já em 1959,
Lejeune e colaboradores identificaram a presença de 3 cromossomos
21 (trissomia do 21) como a etiologia da Síndrome. Richards, em
1965, descreveu 225 exames citogenéticos de pacientes portadores
da doença[1].
Embora o defeito cromossômico na Síndrome de Down tenha
sido inicialmente descrito como a trissomia do cromossomo 21,
estudos posteriores mostraram que este excesso pode ser do
cromossomo inteiro ou de apenas uma parte dele. A trissomia livre
(causada por não-disjunção cromossômica, habitualmente de origem
meiótica) ocorre em cerca de 94% dos casos, seguida pelas
translocações Robertsonianas (rearranjos cromossômicos com ganho
de material genético) em 3,3% dos casos (envolvendo principalmente
os cromossomos 14 e 21) e pelo mosaicismo (linhagens celulares
normais e trissômicas em um mesmo indivíduo) em 2,4% dos casos[1,
5, 6].
8

Embora a trissomia livre seja responsável pela grande maioria
dos casos em qualquer faixa etária materna, em mães jovens, o risco
de recorrência está associado à presença de translocações
balanceadas em um dos pais (ou ambos). O risco de recorrência na
população geral encontra-se em torno de 1%[7].
A evolução das técnicas de análise de DNA permitiu o
desenvolvimento de marcadores altamente informativos para definição
da origem parental do cromossomo 21 extra, assim como a origem
meiótica ou mitótica do erro[8]. Com respeito à trissomia livre, sabe-se
hoje que a maioria dos erros na meiose levando a trissomia do
cromossomo 21 é de origem materna, a maioria destes ocorre na
meiose I e a idade materna média associada é de 32 anos. Em cerca
de 5% dos indivíduos, o cromossomo supranumerário parece resultar
de um erro na mitose. Nestes casos, não há relação com idade
materna elevada e o cromossomo é duplicado aleatoriamente[8, 9].
Em relação às translocações, a maioria das translocações de
novo (não herdadas) entre os cromossomos 14 e 21 [t(14:21)] tem sua
origem em células germinativas maternas, enquanto que nas
translocações entre dois cromossomos 21 [t(21:21)], a maioria dos
casos se deve à presença de um isocromossomo 21, sendo
semelhante a proporção de origem materna e paterna para este
erro[3].
O mapeamento do cromossomo 21 permitiu a identificação de
uma “região crítica” para Síndrome de Down, que em triplicata leva às
características da doença. Por ser o menor entre os cromossomos, na
espécie humana, o 21 já foi quase completamente mapeado, o que
gerou melhor compreensão das características da Síndrome[10].
Entretanto, em alguns casos, não foi encontrada nenhuma região
9

triplicada. É possível que estes pacientes não tenham trissomia do 21,
e sim sejam fenocópias de Síndrome de Down (indivíduos com o
quadro clínico típico da doença, sem que no entanto seja possível
identificar o defeito genético conhecidamente implicado na
etiologia)[1].
O aumento do risco de trissomia com a elevação da idade
materna foi inicialmente descrito por Penrose, em 1933[9] , sendo a
incidência de 1 em 1 500 dos 15 aos 19 anos, chegando em 1 em 50
após os 45 anos[3]. Beiguelman e colaboradores (1996) realizaram
estudo com pacientes portadores da Síndrome de Down
acompanhados da Universidade de Campinas (UNICAMP) e que
haviam realizado cariótipo neste serviço, objetivando estudar esta
relação, na ausência de dados na literatura brasileira. Encontraram
uma maior freqüência de mães jovens (menos de 34 anos), o que,
através de análise epidemiológica, foi atribuído a menor idade
materna na população geral, refletindo uma tendência sul-
americana[9, 11-14]. Em média, na população global, a prevalência da
Síndrome de Down atinge cerca de 1 para 600 nascidos vivos, sendo,
portanto, como já dito, considerado o padrão malformativo mais
comum em humanos.
Atualmente, a idade materna é considerada o único fator de
risco comprovadamente associado à maior ocorrência de Síndrome
de Down[7, 9, 13, 15]. Entretanto, estudos recentes têm mostrado um
possível papel de alterações na atividade da tetrahidrofolato-redutase
(enzima envolvida no metabolismo do ácido fólico) como implicadas
em um maior risco para a ocorrência de não-disjunção meiótica e ,
conseqüentemente, Síndrome de Down por trissomia livre[16, 17].
Tais estudos, após a devida confirmação, poderão vir a revolucionar o
10

aconselhamento genético para a Síndrome de Down, e possibilitar
estratégias de prevenção primária, não disponíveis atualmente.
Classicamente, não há relato de diferença na ocorrência da
Síndrome de Down entre os grupos raciais. Entretanto, até 1982, a
Síndrome de Down era considerada rara na África. Dados recentes
mostram que a ocorrência da Síndrome na África é tão alta quanto
entre os outros grupos populacionais, e que a aparente menor
prevalência se deve à falta de reconhecimento do quadro, tanto pelas
mães como pelos profissionais de saúde envolvidos com a criança,
levando à falta de diagnóstico ou diagnóstico tardio. A maioria das
mães não era capaz de identificar seu filho afetado pela Síndrome de
Down como fisicamente diferente dos outros filhos. Os profissionais
de saúde também apresentavam dificuldade em reconhecer a
Síndrome de Down, em parte devido ao fato das características
craniofaciais dos bebês negros normais se assemelharem àquelas
apresentadas pelos recém-nascidos portadores da Síndrome de
Down, e em parte devido ao próprio desconhecimento da doença,
suas características e implicações. É provável também que a falta de
acesso aos serviços de saúde na África seja responsável por um
grande número de óbitos de crianças com Síndrome de Down antes
que estas obtenham e diagnóstico[18].
Até o momento, não dispomos de dados clínico-demográficos
prévios sobre a Síndrome de Down na Bahia.
Atualmente, define-se como Síndrome de Down o conjunto de
sinais clínicos, incluindo fácies característico, retardo mental e
hipotonia, associado ao achado citogenético de um cromossomo 21
adicional (trissomia 21)[3].
11

Entre as características da síndrome, destaca-se o retardo
mental de gravidade variável, associado a hipotonia, com tendência a
boca entreaberta e língua protrusa, assim como diátese de retos
abdominais e hipermobilidade articular[1, 3].
O fácies é típico, com face aplanada, fendas palpebrais oblíquas
para cima, nariz pequeno com ponte nasal baixa e epicanto. O
pescoço é curto[1]. Ao nascimento, os lábios da criança com
Síndrome de Down parecem ser de tamanho normal, entretanto,
sofrem um aumento progressivo, característico do fácies na doença. A
língua normalmente é aumentada, podendo a macroglossia se
absoluta ou relativa, conseqüente à micrognatia e à cavidade oral
pequena. Mais de 90% das crianças desenvolvem fissuras (de causa
desconhecida) na língua por volta dos cinco anos de idade. A
cavidade oral apresenta palato alto, estreito, e pode haver úvula
bífida, fenda palatina submucosa, ou mesmo fenda lábio-palatina[19].
Na pele, observa-se excesso de pele na nuca na infância (dado
útil para o diagnóstico pré-natal, através do estudo de translucência
nucal), cutis marmorata, pele seca e hiperceratótica com o tempo. Os
cabelos habitualmente são finos, macios e esparsos[3].
O crânio caracteriza-se por braquicefalia com occipital plano e
microcefalia leve, assim como implantação baixa de cabelos. As
fontanelas fecham-se tardiamente e há uma tendência a hipoplasia ou
aplasia de seios paranasais frontais e palato duro curto. O terço médio
da face é normalmente hipoplásico, levando a um prognatismo
mandibular relativo, com a aparência característica de face aplanada.
Os pacientes apresentam alterações significativas na face média,
mandíbula e endocrânio[19].
12

Cerca de 12% apresentam instabilidade atlanto-axial com risco
de compressão medular[19, 20]. Este fato reveste-se de especial
interesse quando se torna necessária a entubação oro-traqueal,
especialmente para a realização de anestesia geral. Assim, a coluna
cervical deve ser avaliada no pré-operatório, através de estudo
radiológico em perfil, extensão e flexão[19].
Nos olhos, além da obliqüidade das fendas palpebrais, pode-se
notar manchas (Brushfield spots) e hipoplasia da íris, opacificação
congênita fina do cristalino (visível ao exame com lâmpada de fenda),
erros de refração (principalmente miopia), nistagmo, estrabismo,
obstrução do canal lacrimal e catarata na vida adulta[21, 22].
As orelhas são habitualmente pequenas, proeminentes, com a
curva superior da hélice excessivamente dobrada. Os lóbulos podem
ser pequenos ou estar ausentes. Surdez ocorre por defeito de
condução, neurossensorial ou ambos. O diâmetro do canal auditivo
externo é significantemente diminuído, o que predispõe à otite externa
e impactação de cerume. A perda auditiva condutiva é também
causada por doença do ouvido médio, principalmente a otite média
crônica. O diâmetro das Trompas de Eustáquio apresenta-se
diminuído. Hipotonia e defeitos musculares do tensor do véu palatino
também já foram descritos como efeitos causais. A incidência de
perda auditiva neurossensorial também está aumentada, e tem com
causas principais as anomalias dos ossos temporais, como a cóclea
de Mondini, canais cocleares apicais curtos, órgão de Corti curto,
canais semicirculares e vestíbulo grandes, mesênquima residual no
ouvido médio, entre outros [19].
Crianças portadoras de Síndrome de Down tendem a ter uma
incidência de cáries menor que a média, porém a ocorrência de
13

doença periodontal é alta, e atribuível principalmente à higiene oral
precária[19]. A dentição geralmente é hipoplásica[23], havendo
erupção dentária irregular ou atrasada, tanto dos dentes decíduos
como permanentes em cerca de 75% dos casos [19].
Anomalias cardíacas incluem, em ordem decrescente de
freqüência, defeitos do coxim endocárdico, defeito septal ventricular,
persistência do canal arterial, defeito septal atrial e artéria subclávia
aberrante. A partir dos 18 anos de vida podem surgir regurgitações
valvulares. A cardiopatia congênita constitui-se na principal causa de
óbito nestes pacientes, a despeito da evolução das técnicas
cirúrgicas[1, 24, 25], especialmente em regiões onde o acesso aos
serviços de cirurgia cardíaca é difícil[18]. No entanto, trabalhos
recentes têm mostrado que a correção cirúrgica em idade precoce,
objetivando principalmente prevenir a ocorrência de alterações
vasculares pulmonares permanentes e o desenvolvimento de
Insuficiência Cardíaca Congestiva (mais comum em crianças com
Síndrome de Down do que naquelas sem a patologia), tem melhorado
significativamente a sobrevida destes pacientes[26]. Outros trabalhos
mostram também que a evolução pós-operatória das crianças com
Síndrome de Down submetidas à cirurgia para correção de
cardiopatias congênitas pode ser bastante favorável[27].
As vias aéreas da criança com Síndrome de Down têm sido
descritas como “tênues”, especialmente durante procedimentos
cirúrgicos. Devido à hipoplasia do andar médio da face, as regiões de
naso e orofaringe têm sido descritas como mais estreitas e menores.
As adenóides tendem a ser hipoplásicas, motivo pelo qual sua
remoção cirúrgica não parece muito eficaz no tratamento da obstrução
das vias aéreas, embora possa funcionar como um tratamento
14

coadjuvante às cirurgias específicas do palato e da faringe. Trabalhos
recentes sugerem que eventual entubação oro-traqueal seja realizada
com cânulas de diâmetro cerca de 0,5 milímetros menor do que o
preconizado para a idade, devido ao risco de estenose subglótica [19].
Apnéia obstrutiva do sono é mais comum do que na população
geral, e se caracteriza por movimentos paradoxais da parede
abdominal e torácica durante o sono, apnéia, hipopnéia, cianose;
quando acordados, os pacientes exibem com maior freqüência
obstrução nasal, respiração bucal, sonolência diurna excessiva e
problemas comportamentais. Atualmente, atribui-se a gênese do
distúrbio ao tônus muscular inadequado da faringe e ao lúmen
diminuído das vias aéreas superiores[19].
Ocasionalmente, os portadores de Síndrome de Down podem
apresentar convulsões, pectus excavatum ou carinatum, estenose
traqueal, atresia duodenal, onfalocele, doença de Hirchsprung e ânus
imperfurado[3].
As mãos do paciente portador de Síndrome de Down
apresentam metacarpianos e falanges curtas, 5o quirodáctilo curto
com hipoplasia da falange média. A prega única de flexão palmar
(linha simiesca) ocorre em 45% dos casos, segundo a literatura (vale
lembrar que esta característica, por muitos considerada clássica e
quase diagnóstica da Síndrome, ocorre em cerca de 5% da população
normal). O padrão dermatoglífico de arco ulnar está presente em
todos os dígitos em 35% dos casos. Ao nível dos pés observa-se
aumento do espaço entre 1o e 2o pododáctilos, linha plantar entre
estes dígitos e padrão dermatoglífico de campo aberto na área halucal
da planta [28].
15

Alterações genitais são descritas, como pênis relativamente
pequeno e volume testicular diminuído. Ocorre deficiência gonadal
primária e, embora mulheres possam reproduzir, não há relatos de
fertilidade masculina[5, 8].
A prevalência de doença celíaca é também maior que na
população geral, tendo os estudos encontrado uma freqüência de até
17% entre crianças com Síndrome de Down[29]. Estes números são
significantemente mais altos do que os 0,5% descritos para a
população geral[30]. Estudo norte-americano recente, objetivando
estimar a prevalência desta doença entre os portadores da Síndrome
de Down, utilizando pesquisa de anticorpos antigliadina e
antiendomísio, e confirmando os casos suspeitos com biópsia
intestinal, encontrou freqüência de cerca de 7% da doença. Tais
dados recomendam a utilização de testes de rastreamento para
doença celíaca entre os portadores da Síndrome de Down [31].
Alguns trabalhos vêm sendo desenvolvidos abordando a
operacionalização da triagem para doença celíaca em portadores da
Síndrome de Down. Csizmadia e colaboradores propõem uma
estratégia em dois passos, o primeiro identificando pacientes
predispostos através da ligação com determinado haplótipo de HLA
reconhecidamente relacionado à doença celíaca, e em seguida
pesquisando os anticorpos específicos. Os casos positivos foram
confirmados por biópsia. Os autores concluíram que a triagem não é
efetiva se realizada apenas uma vez na vida do indivíduo, uma vez
que foram identificados casos nos quais um teste positivo foi
identificado dois anos após uma triagem inicial negativa. A definição
de qual o intervalo ideal entre as avaliações ainda está em estudo. A
doença celíaca pode apresentar-se de maneira subclínica no indivíduo
16

com Síndrome de Down, e ser de difícil suspeição clínica. O
diagnóstico através e estratégias e rastreamento permite o tratamento
precoce, prevenindo complicações potenciais da Doença Celíaca não
tratada, a exemplo de anemia, osteoporose e possíveis neoplasias
intestinais. Deste modo a triagem para doença celíaca está indicada
entre os portadores da Síndrome de Down[30].
A hipotonia tende a melhorar com a idade, ao passo que o
desenvolvimento mental desacelera. Por outro lado, a performance
social é em média 3,3 anos superior à esperada para a idade mental.
São geralmente “bons bebês” e crianças felizes, amigáveis, têm bom
sentido de ritmo e adoram música. Caracterizam-se também por
serem travessos e obstinados, ocasionalmente há sérios problemas
emocionais. A coordenação motora é deficitária e a voz, rouca. O
crescimento é relativamente lento, com atraso da idade óssea. O
desenvolvimento sexual na adolescência é menos completo que o
normal[8].
Alguns trabalhos têm relatado que indivíduos com Síndrome de
Down têm uma percepção diminuída para a dor e para localização de
estímulos dolorosos. Hennequin e colaboradores, em 2000,
sugeriram que tal diferença na percepção dolorosa poderia ser
atribuída ao déficit cognitivo apresentado pelos pacientes, mais do
que a alguma alteração na transmissão do estímulo doloroso[32].
Entretanto, tem sido descrito um aumento de peptídeos opióides no
córtex cerebral destes indivíduos, e algumas evidências mostram que
a Síndrome de Down pode estar associada a uma função
neurossensorial periférica prejudicada, e isto poderia incluir o estímulo
doloroso, sendo o papel da deficiência mental menos relevante[33,
34].
17

Os pacientes apresentam relativa baixa estatura e tendem à
obesidade na adolescência[3]. O uso do hormônio do crescimento,
uma vez que é descrito um déficit de sua produção na Síndrome de
Down, já foi estudado. Os resultados mostraram que, embora o
tratamento possa resultar em velocidade de crescimento normal (que
declina quando o tratamento é descontinuado), não houve melhora no
que diz respeito ao perímetro cefálico ou ao desenvolvimento mental e
motor, motivo pelo qual os autores não recomendam o seu uso[35].
A doença tireoidiana é comum (sendo descrita na literatura uma
prevalência variando de 2 a 63%) especialmente o hipotireoidismo
que, quando não tratado, piora consideravelmente o prognóstico da
Síndrome de Down. O hipotireoidismo adquirido é devido, na maioria
dos casos, à tireoidite autoimune. Supõe-se que estes indivíduos
possam apresentar glândulas tireóide hipoplásicas, que manteriam
níveis normais de hormônios tireoidianos por diversos anos, porém
com o aumento da idade sua função se tornaria subnormal[36]. Assim,
a função tireoidiana deve ser aferida com regularidade[37-39].
Estudos científicos têm mostrado também que crianças portadoras da
Síndrome de Down podem apresentar elevação do THS (Hormônio
Tireoestimulante) basal, na presença de níveis normais de hormônios
tireoidianos e anticorpos antitireóide negativos, podendo não
apresentar quadro clínico de hipotireoidismo. Tal anormalidade pode
ser atribuída a algumas hipóteses, incluindo casos subclínicos em
estágio inicial (devem ser acompanhados), resistência tireoidiana ao
nível de receptores (hipótese ainda não comprovada), ou secreção
inapropriada de TSH, de origem hipotalâmica (o que parece mais
provável, embora a causa para esta disfunção permaneça
18

desconhecida). Tais casos necessitam de acompanhamento regular e
rigoroso, no intuito de evitar tratamentos inadequados[38].
A mortalidade por doença respiratória, principalmente
pneumonia, (assim como outras doenças infecciosas), é muito mais
alta que na população geral, em decorrência de disfunção
imunológica[40-45].
Ao longo dos anos, desde a sua descrição, inúmeros estudos
têm sido feitos sobre o assunto, abordando os mais variados
aspectos. A melhoria da qualidade de vida destes pacientes, com o
adequado controle das infecções, a correção precoce da cardiopatia e
as diversas modalidades de estimulação disponíveis, tornaram
imperioso o conhecimento cada vez mais preciso da doença. Assim,
desde a importância da aceitação da criança no âmbito da família[25]
até sua inserção no mercado de trabalho e na sociedade, passando
pela abordagem adequada dos problemas clínicos associados à
doença, a informação científica sobre a síndrome vem crescendo ao
longo dos anos.
O papel da estimulação e do ambiente enriquecido no
desenvolvimento das crianças com Síndrome de Down estão bem
estabelecidos. Durante algum tempo, o uso da droga Piracetam foi
postulado como tendo a capacidade de melhorar a performance
cognitiva destes indivíduos. O piracetam é habitualmente usado em
alguns casos de disfunção cerebral, a exemplo do Mal de Alzheimer e
da Dislexia comportamental. Entretanto, estudos abordando o uso
desta droga em crianças portadoras de Síndrome de Down, não
mostraram diferenças cognitivas ou comportamentais entre os grupos
(droga e placebo), além de ter evidenciado efeitos colaterais sobre o
19

sistema nervoso central. Deste modo, tal terapia não está indicada
para portadores de Síndrome de Down[46].
Por fim, e não menos importante, devemos considerar os
aspectos psicológicos relacionados a esta patologia. Tal abordagem
mostra-se difícil, dada a amplitude do tema. No entanto, e
especialmente ao longo das três últimas décadas, várias abordagens
deste aspecto têm sido realizadas. Vários autores de diversos campos
do conhecimento têm estudado as reações emocionais dos pais ao
diagnóstico de patologias específicas da infância, inclusive a
Síndrome de Down; entretanto, pouco é conhecido sobre o impacto de
características particulares de cada condição, como por exemplo tipo
de deficiência, curso, prognóstico, visibilidade, etc. Em trabalho de
1984, Cunningham e colaboradores abordaram a questão do
diagnóstico inicial, e relataram experiências negativas dos pais quanto
à maneira como a “notícia” de que seu bebê era portador da Síndrome
de Down. Os pesquisadores, então, desenvolveram um protocolo
padrão e o testaram, tendo como resultado um aumento significativo
na satisfação dos pais[47]. Garwick e colaboradores, em 1995,
desenvolveram um projeto de pesquisa de análise qualitativa,
abordando o mesmo tema e objetivando desenvolver recomendações
para que os profissionais de saúde possam informar as famílias, de
maneira efetiva, sobre a condição de sua criança. Este trabalho incluiu
crianças portadoras de Síndrome de Down e/ou cardiopatia congênita,
e seus familiares. Os estudiosos procuraram fatores que
influenciariam as reações familiares a respeito da condição crônica ou
deficiência da criança. Entrevistaram as famílias em suas próprias
residências, questionando-as sobre a ocasião em que inicialmente
foram informadas sobre a Síndrome de Down. Foram avaliados o
20

contexto no qual a família foi informada, as estratégias usadas pelo
profissional de saúde que informou o diagnóstico, e as reações
familiares – reações emocionais à notícia (choque, medo, tristeza,
culpa, otimismo), e reações à maneira como os profissionais de saúde
inicialmente deu a informação sobre a condição da criança. A maioria
das famílias obteve o diagnóstico logo após o nascimento, por um
médico da maternidade, e na maior parte dos casos os dois pais
estavam presentes. O choque foi a principal reação relatada. O estudo
mostra claramente como recomendado pelos próprios pais que a
notícia seja dada a ambos (pai e mãe) conjuntamente, e em ambiente
dotado de privacidade. Também chama a atenção para a importância
do fornecimento de informações atualizadas. Mas, principalmente,
aborda a necessidade do profissional de saúde apresentar-se, além
de bem informado, sensível aos sentimentos dos pais e capaz de
prestar-lhes apoio, considerando a criança portadora da Síndrome de
Down como um todo, ao invés de valorizar os aspectos negativos da
condição. Reações negativas ocorreram em situações que parecem
absurdas, porém podem ser verificadas, mesmo em nosso meio:
receber a notícia por telefone, em presença de estranhos, receber
material didático de décadas atrás, com títulos como “A criança
mongolóide e você”, “Mongolismo, você e seu bebê”, e com
informações como: “a maioria das crianças deve ser
institucionalizada”. Os autores recomendam que os médicos saibam
ouvir as famílias, sejam sensíveis aos seus sentimentos e evitem
expressões negativas, como “más-notícias”, e junto com informações
sobre as deficiências, forneçam também conhecimento sobre as
possibilidades da criança, sempre adaptando as informações Às
21

condições de compreensão e ansiedade da família, e levando em
consideração as questões por ela levantadas[48].
22

OBJETIVOS

• Descrever os aspectos clínico-demográficos de indivíduos de
zero a dezoito anos de idade, com diagnóstico de Síndrome de Down,
acompanhados no ambulatório de Neurogenética do Hospital
Universitário Professor Edgard Santos.
24

JUSTIFICATIVAS

A Síndrome de Down é considerada a Cromossomopatia mais
comum em humanos, e é certamente a doença genética mais
conhecida, tanto no meio médico como leigo [1, 3, 5, 8, 25, 43-45, 49-
51] . Reveste-se de especial importância pelo seu caráter não letal,
com um retardo mental treinável e a possibilidade de investimento
nestes pacientes, tanto no que diz respeito ao tratamento das
condições clínicas patológicas, como pelos avanços da fisioterapia e
estimulação precoce, assim como também pela possibilidade de
inclusão destes pacientes na sociedade e no mercado de trabalho.
Em nosso meio, e em especial nos atendimentos ambulatoriais
do Hospital Universitário Professor Edgard Santos, observa-se a
constante presença destas crianças, porém até recentemente não
havia uma sistematização para o seu atendimento e condução clínica,
assim como para o suporte destas famílias, tanto no que diz respeito
ao Aconselhamento Genético como ao Acompanhamento Psicológico.
As crianças portadoras de Síndrome de Down obtinham diagnóstico e
orientações iniciais, porém permaneciam sem acompanhamento.
Estes fatos constituíram-se no estímulo inicial para a criação de um
serviço especializado no acompanhamento de crianças com Síndrome
de Down, somando-se a isto a total ausência de dados na literatura
nacional e internacional sobre a Síndrome de Down na Bahia.
26

CASUÍSTICA, MATERIAL E MÉTODOS

Para a realização do Projeto, foi criado, com o apoio do
Laboratório de Genética Médica e como parte integrante do
Ambulatório de Neurogenética, o Ambulatório de Assistência à criança
portadora de Síndrome de Down. Este Serviço foi iniciado em
Dezembro de 1999, no Ambulatório Professor Magalhães Netto, às
terças-feiras pela manhã. O atendimento de crianças com Síndrome
de Down encaminhadas à consulta genética, que era feito de maneira
rotineira no ambulatório de genética geral, onde se prestava uma
consultoria com emissão de laudo e encaminhamento para
acompanhamento em Postos de Saúde, a partir deste momento,
passou a ser por nós realizado, de maneira diferenciada, através de
sistematização e protocolo de atendimento (v. anexo I). O objetivo
deste empreendimento foi prestar uma assistência integrada e
qualificada às crianças com Síndrome de Down, ao tempo que nos
permitiria estudar e avaliar esta população.
O protocolo de atendimento usado foi desenvolvido a partir do
conhecimento sobre as características da doença e da literatura
disponível sobre diretrizes para acompanhamento e orientação
antecipatória para crianças portadoras da Síndrome de Down[37, 39,
52, 53] .
As entrevistas iniciais foram realizadas em primeira consulta, em
ambiente tranqüilo e com duração média de 90 (noventa) minutos, por
profissional médico, médico residente ou acadêmico de medicina
treinado, com a presença preferencial de ambos os pais. Na
impossibilidade da presença do genitor, as entrevistas foram
conduzidas com a mãe. Dados de nascimento e imunização foram
confirmados com o Cartão da Criança, sempre que disponível. Dados
de caráter psicológico foram questionados, seguindo uma
28

normatização. Assim, ao questionarmos a reação inicial da mãe ao
diagnóstico e sua principal preocupação inicial com a criança foram
oferecidas opções (v. anexo1). O item referente a como a mãe avalia
a maneira como foi informada do diagnóstico de seu filho constituiu-se
numa avaliação mais difícil. Optamos por classificar a informação em
três tipos, assim definidos: Clara, quando foi possível observar na
mãe o conhecimento sobre aspectos básicos da Síndrome de Down,
incluindo o diagnóstico, sua relação com retardo do desenvolvimento
neuropsicomotor e a necessidade de cuidados especiais; Pouco Informativa, quando a mãe não havia registrado nenhuma informação
relevante, afirmando que havia sido informada apenas de que a
criança teria “um problema”, ou apenas era capaz de relatar o nome
da doença; e Pejorativa, quando a mãe relatava ter percebido uma
atitude de preconceito ou repulsa por parte do profissional de saúde
em relação à criança, ou quando foram usadas palavras como
“mongol”, “retardado”, “vegetal”, e ainda quando foram emitidos pelo
citado profissional prognósticos sombrios, a exemplo de “nunca vai
andar ou falar”, “vai morrer cedo”.
Contamos no início do projeto com a participação voluntária de
uma psicóloga, que freqüentou ativamente o ambulatório, realizando
atendimento dos pacientes e suas famílias e participando de
discussão de casos, tendo inclusive desenvolvido um trabalho de
pesquisa relacionado.
Foi também firmada parceria com Cardiologista Pediátrica, no
intuito de fornecer a estas crianças esta avaliação, imprescindível
para o estudo da patologia em questão.
29

O Laboratório de Genética Médica do HUPES realizou o estudo
citogenético (cariótipo) dos pacientes. Foi também firmada parceria
em pesquisa com o Serviço de Pneumologia Pediátrica.
A demanda de pacientes para o "Ambulatório de Down" teve
crescimento rápido e espontâneo, sendo o veículo principal de
informação aquela propagada pelas famílias de pacientes e pela
classe médica que teve informação sobre a iniciativa, tendo tido
também papel importante a divulgação realizada pela "Ser Down"
(Associação Bahiana de Síndrome de Down). Atualmente, o
ambulatório recebe pacientes de várias Maternidades de Salvador e
de ambulatórios de outros Hospitais da capital e do interior do Estado.
No momento, estão sendo acompanhadas no serviço cerca de 180
crianças com Síndrome de Down, com idades variando de 0 a 18
anos.
A obtenção das referências bibliográficas teve como
metodologia pesquisa nos bancos de dados MEDLINE (usando as
palavras chaves “trisomy 21” ou “Down Syndrome”) e LILACS (usando
as palavras chaves “trissomia 21” ou “Down” e “Sindrome”), além de
busca por artigos relacionados nas bibliografias dos artigos
selecionados e consulta a livros-texto de Genética Médica.
O Desenho do estudo definido como corte transversal foi o
passo inicial para o conhecimento da coorte composta por indivíduos
portadores de Síndrome de Down.
A Amostra consistiu em 87 pacientes portadores de Síndrome
de Down de 0 a 18 anos acompanhados no referido ambulatório,
sendo:
30

A. Critérios de inclusão:
• Pacientes na faixa etária pediátrica (0 a 19 anos
incompletos);
• Pacientes que apresentem quadro clínico e/ou citogenético
compatível com Síndrome de Down.
B. Critérios de exclusão:
• Pacientes com quadro clínico duvidoso que não pôde ser
confirmado citogeneticamente;
A Análise Estatística realizada foi Descritiva.
31

RESULTADOS

Entre as 87 crianças estudadas, 51 (58,6%) são do gênero
masculino, enquanto que 36 (41,4%) pertencem ao gênero feminino.
A idade dos pacientes à época do início do acompanhamento variava
de 0 a 18 anos, com 32% abaixo de dois meses e 82,8% abaixo de
dois anos, com média de 16,74 meses e mediana de sete meses.
Quarenta e seis pacientes (52,9%) procediam da capital (Salvador),
15 (17,2%) da região metropolitana e cidades vizinhas (Camaçari,
Candeias, Lauro de Freitas e Simões Filho) e 26 (29,9%) do interior
do Estado. Quanto às instituições encaminhadoras, 20 crianças (23%)
foram encaminhadas pelo Campus Universitário (Hospital universitário
Professor Edgard Santos, Centro Pediátrico Professor Hosannah de
Oliveira, Maternidade Climério de Oliveira); igual número, pela APAE-
Salvador (Associação de Pais e Amigos de Excepcionais); 15 (17%)
foram encaminhados por maternidades da região metropolitana e 32
(36,8%) foram referenciados por outros (na maioria, pediatras).
Quanto ao grupo racial, 34 (39,1%) foram classificados como
brancos; 26 (29,9%), como mulatos claros; 25 (28,7%), como mulatos
médios; dois (2,3%) como mulatos escuros e nenhum como preto. No
que diz respeito às mães, 14 (17,3%) pertenciam ao grupo branco, 20
(24,7%) ao mulato claro, 36 (44,4%) ao mulato médio, nove (11,1%)
ao mulato escuro e duas (2,5%) ao preto. Em nove casos (6.9%) esta
informação não estava disponível (ausência da mãe à consulta ou
falha no preenchimento do protocolo). O grupo racial dos pais não foi
incluído, uma vez que estes raramente acompanhavam as crianças à
consulta, não sendo possível então sua classificação racial.
33

A idade materna, à época do nascimento da criança com
Síndrome de Down, variou de 15 a 50 anos, com média de 30,11 anos
e mediana de 31 anos. 58,6% das mães tinham menos de 34 anos
quando seus filhos nasceram. Apenas 3,3% tinham mais de 45 anos.
Cerca de 40% das mães tinham idade inferior a 28 anos à época do
parto.
No item escolaridade, três (3,5%) mães e sete (8,8%) pais eram
analfabetos, 47 (54,7%) mães e 40 (50%) pais foram alfabetizados,
mas não concluíram o ensino fundamental, sete (8,1%) mães e 10
(12,5%) pais concluíram o ensino fundamental, 14 (16,3%) mães e
seis (7,5%) pais iniciaram o ensino médio e uma (1,2%) mãe e 15
(18,8%) pais concluíram o ensino médio. Apenas uma mãe e dois pais
possuíam nível superior.
Quanto à ocupação, 55 (63,2%) mães eram donas-de-casa, 15
(17,2%) possuíam emprego formal, sete (8%) exerciam atividades de
maneira autônoma, cinco (5,7%) eram estudantes, duas (2,3%) eram
empregadas domésticas, uma (1,1%) exercia "biscates" e duas,
outros afazeres. Quanto aos pais, 38 (45,2%) possuíam algum
emprego formal, 13 (15,5%) exerciam atividades de prestação de
serviço, sem vínculo empregatício, 13 exerciam atividades
autônomas, oito (9,5%) exerciam "biscates", dois (2,4%) eram
estudantes, sete (8%) encontravam-se desempregados. Três (3,6%)
exerciam outras atividades e em três casos a informação não foi
disponível.
34

A renda familiar média das famílias esteve em 1,36 salários-
mínimos, com mediana de um salário-mínimo, e desvio-padrão de
1,46. O saneamento básico estava presente em 60 (71,4%) e ausente
em 24 (28,6%) residências.
Os dados referentes ao nascimento incluíram o tipo de parto
(normal em 65,1% dos casos, sendo pélvico em apenas um caso;
fórceps em 2 casos; cesárea em 25 casos, sendo dois pélvicos). A
antropometria ao nascimento revelou peso médio de 2.988g (mediana
2,910g), estatura média de 47,2cm (esta informação só esteve
disponível – através de informação da mãe ou documentos da
maternidade – em 65,5% dos casos), perímetro cefálico (informação
disponível em apenas 29,9% dos casos) médio 32,5 cm. O índice de
APGAR no 50 minuto de vida (disponível em apenas 25,3% dos
casos) foi de 6 ou 7 em 4,5% dos casos, 8 em 27,3% dos casos, 9 em
45,5% dos casos e 10 em 18,2% dos casos. Questionadas sobre a
idade gestacional, as genitoras (o percentual de informação médica
neste sentido foi insignificante) informaram parto a termo em 64,4%
dos casos e pré-termo em 17,2% dos casos.
Na ocasião da primeira consulta, 44 (50,6%) crianças já haviam
iniciado a estimulação precoce.
As alterações fenotípicas avaliadas encontram-se descritas na
tabela 1. O ecocardiograma foi obtido em 89,3% (resultados
disponíveis até o momento – o exame foi oferecido à totalidade dos
pacientes), o cariótipo em 20% dos casos, a avaliação hematológica
em 69% dos casos, avaliação tireoidiana em 82% dos casos e ultra-
sonografia abdominal em 59.8% dos pacientes.
35

TABELA 1– Alterações Fenotípicas em 87 indivíduos com diagnóstico de Síndrome de Down
ALTERAÇÃO FENOTÍPICA N0 % Fenda palpebral oblíqua para cima 87 100 Cabelos finos, lisos e esparsos 81 93,1 Epicanto 52 59,8 Orelhas dismórficas 48 55,2 Linha simiesca 28 32,2 Unilateral 5 5,7 Bilateral 23 26,4 Cardiopatia 39 44,9
A distribuição das alterações morfológicas cardíacas,
diagnosticadas através de Ecocardiograma bidimensional com
Doppler, encontra-se descrita na Tabela 2.
TABELA 2 – Alterações Ecocardiográficas em 73 indivíduos com diagnóstico de Síndrome de
Down
DIAGNÓSTICO ECOCARDIOGRÁFICO N0 % Comunicação Interatrial (CIA) 11 14,1 Comunicação Interventricular (CIV) 3 3,8 Defeito Átrio-ventricular total (DAVT) 2 2,6 Tetralogia de Fallot 2 2,6 Persistência do Canal Arterial (PCA) 3 3,8 CIA+CIV 3 3,8 CIA+PCA 4 5,1 CIV+PCA 3 3,8 Defeito Atrioventricular intermediário 3 3,8 Ecocardiograma normal 39 50
A avaliação da função tireoidiana foi realizada em 82 (94,7%)
pacientes, e hipotireoidismo foi detectado em 5 (6,1%) casos.
As avaliações especiais incluíram ultra-sonografia abdominal
(realizada em 59,8% dos casos), e a avaliação hematológica
(disponível no momento da análise em 69% dos casos). Estes dados
encontram-se descritos nas Tabelas 3 e 4.
36
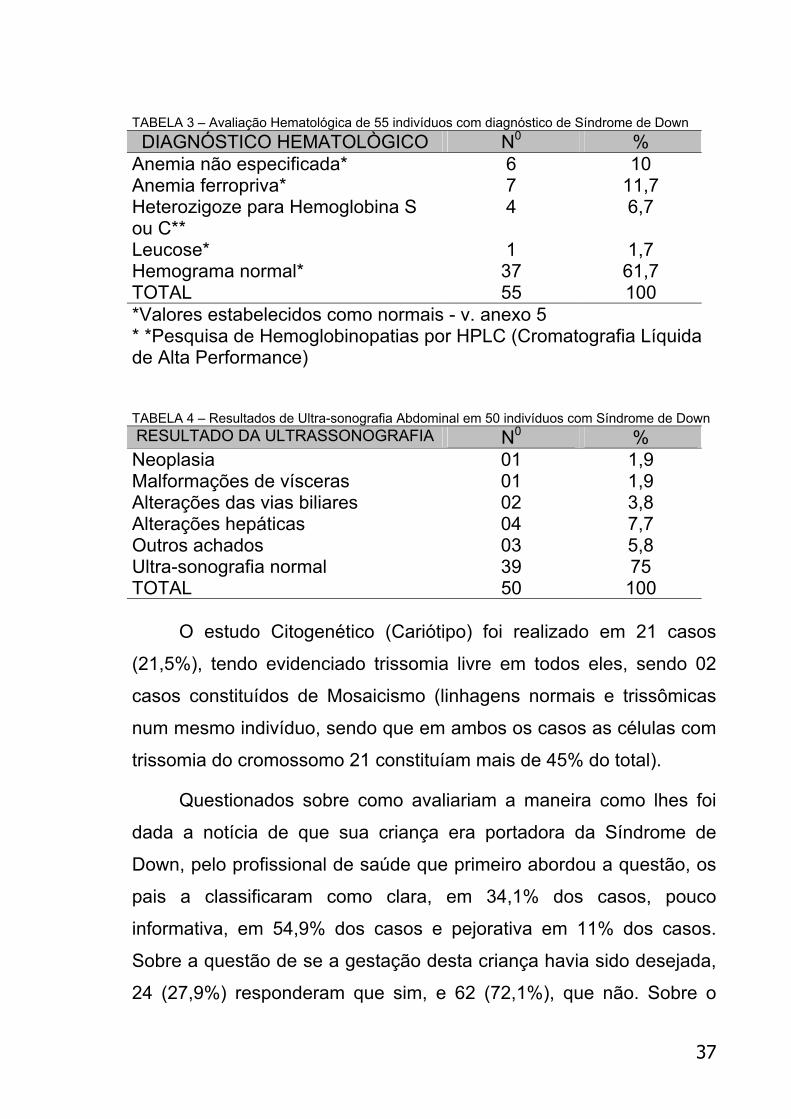
TABELA 3 – Avaliação Hematológica de 55 indivíduos com diagnóstico de Síndrome de Down DIAGNÓSTICO HEMATOLÒGICO N0 %
Anemia não especificada* 6 10 Anemia ferropriva* 7 11,7 Heterozigoze para Hemoglobina S ou C**
4 6,7
Leucose* 1 1,7 Hemograma normal* 37 61,7 TOTAL 55 100 *Valores estabelecidos como normais - v. anexo 5 * *Pesquisa de Hemoglobinopatias por HPLC (Cromatografia Líquida de Alta Performance) TABELA 4 – Resultados de Ultra-sonografia Abdominal em 50 indivíduos com Síndrome de Down RESULTADO DA ULTRASSONOGRAFIA N0 % Neoplasia 01 1,9 Malformações de vísceras 01 1,9 Alterações das vias biliares 02 3,8 Alterações hepáticas 04 7,7 Outros achados 03 5,8 Ultra-sonografia normal 39 75 TOTAL 50 100
O estudo Citogenético (Cariótipo) foi realizado em 21 casos
(21,5%), tendo evidenciado trissomia livre em todos eles, sendo 02
casos constituídos de Mosaicismo (linhagens normais e trissômicas
num mesmo indivíduo, sendo que em ambos os casos as células com
trissomia do cromossomo 21 constituíam mais de 45% do total).
Questionados sobre como avaliariam a maneira como lhes foi
dada a notícia de que sua criança era portadora da Síndrome de
Down, pelo profissional de saúde que primeiro abordou a questão, os
pais a classificaram como clara, em 34,1% dos casos, pouco
informativa, em 54,9% dos casos e pejorativa em 11% dos casos.
Sobre a questão de se a gestação desta criança havia sido desejada,
24 (27,9%) responderam que sim, e 62 (72,1%), que não. Sobre o
37

desejo de prole subseqüente, sete (8%) afirmaram desejar ter outros
filhos, enquanto que 75 (86,2%) relataram não desejar outros filhos, e
cinco (5,7%) não tinham opinião formada.
Vinte e quatro por cento das 87 mães entrevistadas (tivemos a
presença do pai em apenas um pequeno número de casos) colocaram
como principal preocupação inicial em relação `a sua criança o atraso
na (ou ausência de) aquisição de habilidades motoras, em especial a
deambulação. A preocupação com o preconceito da sociedade
esteve presente em 14,6% das entrevistas. Múltiplas preocupações,
não sendo as mães capazes de defini-la, foram relatadas por 19,5%
das genitoras. A preocupação com a habilidade intelectual a ser
alcançada pela criança foi relatada por 6,1% das mães; a capacidade
da criança viver de forma independente no futuro, 4,9%; a presença e
evolução da cardiopatia congênita, por 4,9%; a possibilidade de
comportamento social inadequado, por 1,2% e a freqüência
aumentada de infecções, por 1,2% das mães entrevistadas. Oito e
meio por cento das mães afirmaram não terem preocupações em
relação à criança e 13,8% relataram outras preocupações.
A maioria dos casais permanecia unida após o nascimento da
criança com Síndrome de Down (81,6%), enquanto que oito (9,2%) já
haviam se separado anteriormente ao nascimento desta e igual
número se separou após o nascimento da criança.
38

DISCUSSÃO

O presente trabalho apresenta-se como relevante pelo fato de
não dispormos de dados locais, e ter sido este o primeiro Serviço
montado especificamente com este objetivo, no Estado da Bahia. Tal
iniciativa gerou parcerias importantes, que tiveram como resultado,
além do melhor nível de assistência aos pacientes atendidos, o
estímulo à pesquisa em vários aspectos relacionados à Síndrome de
Down.
Neste estudo houve um predomínio de pacientes do gênero
masculino (58,6%), numa proporção de 1,4:1. Embora seja aceito que
não exista diferença na prevalência de SD de acordo com o gênero,
diversos estudos têm encontrado uma maior proporção do sexo
masculino [6, 24, 54-56], sendo que a maioria deles obteve amostras
em ambulatórios de acompanhamento, fato que pode explicar este
achado de acordo com a sugestão de Boy, baseado em Carnevale e
colaboradores, de que nas sociedades latino-americanas há uma
maior preocupação com a saúde do filho masculino[56].
Houve uma grande variação na idade do início de
acompanhamento dos pacientes (1o mês de vida até os 18 anos), com
grande predomínio da faixa etária menor de dois anos (82,8%) e com
cerca de 1/3 dos pacientes iniciando o acompanhamento até o
segundo mês de vida. A freqüência de quase 20% de crianças
iniciando o acompanhamento após os 24 meses de idade pode ser
explicada pela ausência de um serviço especializado em atender
crianças com Síndrome de Down no estado da Bahia, e da
inexistência de um programa de triagem e orientação bem definido
nas maternidades do estado, sendo encaminhadas ao ambulatório,
crianças já com idade mais avançada. Entretanto, a idade de início do
40

acompanhamento vem diminuído, à medida que o ambulatório se
estabelece como serviço de referência no estado da Bahia.
A ocorrência de uma maior demanda de pacientes provenientes
da capital do estado, região metropolitana e cidades vizinhas (70%
dos casos) pode ser explicada pelas melhores condições das
unidades de saúde destas regiões e pela proximidade geográfica.
Este fato é reforçado pela maior participação de Organizações Não-
governamentais ligadas à criança com deficiência (Associação de
Pais e Amigos dos Excepcionais - APAE, Ser Down) e Universitárias
(Hospital Universitário Professor Edgard Santos, Maternidade Climério
de Oliveira) no encaminhamento de pacientes ao Ambulatório
(praticamente metade dos pacientes atendidos), todas elas situadas
na cidade de Salvador. O pouco conhecimento dos médicos
estabelecidos no interior do estado, sobre a existência de um
Ambulatório de Referência para crianças com Síndrome de Down,
possivelmente também influenciou na menor participação de unidades
do interior.
A taxa de mortalidade encontrada no presente estudo (2,3%) é
semelhante a de outros estudos de mortalidade em Síndrome de
Down, confirmando uma tendência de queda do obituário,
principalmente associada ao melhor controle das doenças infecciosas
e correção cirúrgica das doenças congênitas, sobretudo as
cardiopatias[27, 42, 50]. Um estudo realizado no Rio de Janeiro
encontrou mortalidade semelhante (2,4%) cuja etiologia foi atribuída a
sepse e insuficiência cardíaca. Infecções, principalmente do sistema
respiratório, e doenças cardíacas, foram as principais causas de óbito
(75%) no estudo conduzido por Thase em 1982[42, 56]. A existência
de protocolos de acompanhamento tem colaborado para reduzir a
41

mortalidade registrada neste trabalho e em outros realizados no Brasil
e em outros países[37, 53]. Na presente amostra, os óbitos ocorreram
no primeiro ano de vida. Alguns estudos demonstram que as mais
altas taxas de mortalidade ocorrem no primeiro ano de vida, com
índices de 28 a 54%, atingindo proporções até 30 vezes maiores que
na população geral. Indivíduos com Síndrome de Down com mais de
40 anos de idade também experimentam maior mortalidade que a
população em geral[42, 50].
É conhecida a predominância de mulatos e negros no estado da
Bahia, principalmente na cidade do Salvador, o que justifica a
predominância de genitoras mulatas (80,2%) no presente estudo.
Outros estudos realizados no Brasil, mais especificamente no Rio de
Janeiro e Rio Grande do Sul, não descrevem o perfil racial
materno[24, 56]. Granzotti, em estudo realizado em Ribeirão
Preto/SP, relata 85% de pacientes com Síndrome de Down da cor
branca, porém não faz citação a cor materna[55]. Estudos em
crianças negras da África mostram prevalência semelhante aos outros
grupos raciais[18]. Na presente amostragem, praticamente 40% das
crianças com Síndrome de Down foram classificadas como brancas,
de acordo com a classificação proposta por Krieger e
colaboradores[57]. A ausência de dados sobre a cor paterna, o
tamanho amostral e o desenho do estudo não permitem maiores
explicações sobre o alto percentual de crianças com Síndrome de
Down de cor branca, mesmo com o maior predomínio de genitoras
mulatas. Como não existem estudos em sociedades com
miscigenação racial semelhante à encontrada na Bahia, estudos
direcionados e construídos com esse intuito devem ser realizados.
42

Existe consenso na literatura sobre a influência da idade
materna no risco de prole com Síndrome de Down por trissomia livre
(uma vez que a ocorrência de trissomia por translocação não sofre
influência da idade materna), sendo até de 20,5 vezes em genitoras
com mais de 40 anos[1, 9, 22]. Estudo realizado no Chile encontrou
risco de um para nove entre mulheres com mais de 44 anos de idade
a época do nascimento da prole[58]. No presente estudo não se
analisou a taxa de incidência de nascimentos de crianças com
Síndrome de Down da população, nem sua relação com a idade
materna, pois não houve uma abrangência de todas as maternidades
existentes no estado da Bahia. Em relação à idade materna à época
da gestação dos pacientes acompanhados nesta amostra, o achado
de idade média de 30,11 anos, com uma mediana de 31 anos,
corresponde ao descrito em outros estudos. Mutton encontrou idade
materna média de 32,7 anos numa análise de 5.537 casos de
Síndrome de Down na Inglaterra[6]. Nazer, em estudo com 53
pacientes com SD, encontrou idade materna média de 34,91 anos.
Deve-se ressaltar que, no citado estudo, mulheres com idade acima
de 34 anos foram responsáveis por 58% de todos os nascidos com
Síndrome de Down, enquanto no presente trabalho apenas 42% das
genitoras apresentavam idade superior a 34 anos. Estudo realizado
no Rio Grande do Sul[24], Brasil, com uma amostra de 50 casos
encontrou idade materna média de 31 anos e 8 meses (mediana de
32,5 anos). Resultados semelhantes foram encontrados por Dipierri
na Argentina, em 206 genitoras de crianças com Síndrome de Down,
com idade média de 32,4 anos[54].
Na análise desta amostra de 87 pacientes com Síndrome de
Down, a existência de cerca de 60% de mães com idade inferior a 34
43

anos e apenas 3,3% de genitoras com mais de 45 anos, pode ser
explicada pela alta taxa de fecundidade e nascimentos entre mulheres
jovens na população latino-americana[7, 9, 11, 13, 14]. Alguns
estudos já demonstram que no final da década de 90, partos entre
mulheres de 10 a 20 anos já representavam 1/3 de todos os
procedimentos obstétricos realizados na rede SUS do estado da
Bahia[12]. Mutton, analisando 207 casos de pacientes com Síndrome
de Down e translocação robertsoniana encontrou idade materna
média de 27,1 anos, e 84,7% das translocações ocorreram em
mulheres com menos de 30 anos[6]. Até o momento foram realizados
cariótipos em 24,1% dos pacientes da amostra e, para fins de
aconselhamento genético, uma vez que estes casais poderiam
programar outro filho, foi dada prioridade à realização do exame nas
crianças cujas mães tinham menos de 34 anos. Entre estes exames,
não houve registro de translocação. Este fato, embora de forma não
definitiva, acentua a importância da alta taxa de nascimentos em
mulheres jovens, na população geral, como fator mais plausível para
explicar a menor média de idade encontrada entre as genitoras desta
amostra, embora este achado não seja discordante da literatura em
geral, principalmente dos estudos realizados na América Latina.
Os resultados obtidos em relação à escolaridade dos genitores,
com predominância de escolaridade até o ensino fundamental (66,3%
das mães e 71,3% dos pais), associados à renda familiar média de
1,36 salários-mínimos, demonstram a predominância de indivíduos de
classe média baixa e classe baixa, dentre as famílias atendidas no
ambulatório, característica esperada por se tratar de serviço ligado à
rede SUS do estado da Bahia. O perfil de inserção no mercado de
44

trabalho dos genitores das crianças estudadas nesta amostragem
reforça o exposto acima.
Alterações fenotípicas encontradas não diferiram do descrito na
literatura[1, 3, 8, 28, 59].
A freqüência de cardiopatias associadas à Síndrome de Down
foi de 50%, o que é concordante com a maioria dos estudos.
Entretanto, a cardiopatia mais freqüente nesta amostra foi a
Comunicação Interatrial (CIA), e não a comunicação Interventricular,
como é descrito na literatura[1, 26, 55, 60]. Tais achados podem estar
relacionados ao tamanho amostral. Outro fato que pode ser levado em
consideração é a possibilidade da freqüência de CIA estar sendo
superestimada pela presença dos forames, cardiopatias congênitas
benignas e de resolução espontânea, presentes da população em
geral, e que não estariam relacionadas à trissomia do 21[61].
O estudo hematológico, através de triagem (hemograma)
semestral, visou a detecção e o tratamento precoce das alterações, e
foi realizado em quase 70% dos pacientes. O achado mais freqüente
foi a anemia, sendo cerca da metade dos casos de etiologia
ferropriva. Heterozigoze para hemoglobinopatia foi detectada em
apenas quatro casos, e leucose (leucemia linfóide aguda) em apenas
um. O achado de policitemia em dois casos coincidiu com a presença
de cardiopatia congênita cianótica. Estima-se que a falta desta
triagem em 30% da amostra deva-se ao estágio precoce do
acompanhamento (primeiras consultas), e à baixa idade das crianças
(o primeiro hemograma de rotina, segundo o protocolo, foi realizado
aos seis meses de idade). O pequeno número de alteração
neoplásicas, não condizente com a literatura[1, 3, 5, 25, 40], pode ser
atribuído ao tamanho da amostra e ao tempo de estudo utilizado.
45

A avaliação tireoidiana, realizada ao nascer e repetida de 6/6
meses, e disponível em cerca de 95% dos casos, mostrou
hipotireoidismo em 6%, sendo apenas dois congênitos, e os demais
detectados através da triagem de rotina. Todos estes pacientes foram
encaminhados para Serviço especializado no HUPES, tiveram seu
diagnóstico confirmado através da avaliação dos hormônios
tireoidianos[62], e vêm evoluindo sem seqüelas neurológicas
atribuíveis ao hipotireoidismo, exceto uma criança, que iniciou o
acompanhamento no ambulatório aos oito anos, tinha diagnóstico de
hipotireoidismo congênito, porém só havia iniciado tratamento após o
50 mês de vida. A literatura descreve hipotireoidismo em cerca de 10%
dos casos, o que se aproxima dos resultados obtidos[1, 36]. Em três
casos (3,5%) houve o achado de aumento transitório do TSH, sem
aumento concomitante dos hormônios tireoidianos. Estes pacientes
não foram diagnosticados como portadores do hipotireoidismo, e
mantém-se em acompanhamento regular.
O estudo ultrassonográfico do abdome, visando principalmente a
detecção de calculose biliar (complicação potencialmente deletéria na
Síndrome de Down, pela associação com colangite e sepse),
realizado em cerca de 60% dos casos, com exame inicial aos três
meses, detectou dois casos de litíase biliar, quatro casos de alteração
parenquimatosa hepática transitória, um caso de neoplasia (teratoma)
e um caso de malformação (baço supranumerário). Estes achados,
em número inferior ao encontrado na literatura, podem ser explicados
pelo tamanho reduzido da amostra, assim como pela faixa etária das
crianças – é possível que, com o seguimento destes pacientes, surjam
novos achados referentes a estes aspectos[56].
46

Durante a avaliação inicial (primeira consulta), obteve-se dados
sobre o impacto do nascimento da criança com Síndrome de Down
sobre os familiares: 11% dos responsáveis classificaram como
pejorativo o modo como o primeiro profissional de saúde atendeu a
criança (habitualmente nas maternidades). As mães descreveram tal
atendimento com palavras como "grosseira" e "brutal", com o uso de
expressões como "mongol", "retardado", e informações errôneas, do
tipo "nunca vai andar e falar", "irá morrer logo". Em uma minoria dos
casos houve o cuidado em informar ao casal conjuntamente sobre a
condição da criança. Aproximadamente metade dos pais referiu que o
profissional foi gentil, porém pouco claro e não capacitado ou
disponível para tirar dúvidas e encaminhar adequadamente. A
informação foi classificada pelos pais como clara e elucidativa em 1/3
dos casos. Tal achado chama a atenção para a falta de treinamento
de nossas maternidades em como lidar com o nascimento de crianças
portadoras de doenças genéticas, mesmo tão conhecidas como a
Síndrome de Down.
Em apenas dois casos, o Ecocardiograma foi solicitado na
Maternidade, procedimento que deveria ser de rotina adotado por todo
serviço de Neonatologia, antes mesmo da avaliação genética e
independente de evidências clínicas de cardiopatia[53, 55].
A gestação da criança afetada pela Síndrome de Down não
havia sido desejada em cerca de 70% dos casos. Este dado reflete
uma tendência nacional, especialmente do Nordeste do país, onde
ainda não é rotina a realização do planejamento familiar.
Os anseios dos pais, com relação à criança portadora de
deficiência, concentraram-se prioritariamente na questão da
motricidade (especialmente deambulação), ocupando segundo lugar a
47

questão do preconceito. Uma boa parte não conseguia definir sua
preocupação principal, num momento inicial. É interessante observar
que, à medida que o acompanhamento prossegue, este foco pode
tender a mudar. Tal fato, associado à variação nas idades do início do
acompanhamento, torna este dado pouco fidedigno, uma vez que,
com o crescimento da criança, e a constatação dos pais de suas
capacidades motoras, a preocupação provavelmente tenderá a
desviar-se para as questões cognitivas e de inserção na sociedade e
no mercado de trabalho.
Menos de 10% dos pais manifestaram desejo de ter outros
filhos; em todos estes casos, a criança afetada era o primeiro filho, o
que poderia ser explicado pelo desejo de ter um filho não afetado.
Entre os pais que não desejavam prole subseqüente, parte referia
como motivo a presença de uma prole já numerosa, e parte a
incapacidade de cuidar de outra criança, tendo um filho portador de
deficiência. Nesta última categoria, as mães eram na maioria
primíparas. Este dado, aparentemente, também poderá vir a sofrer a
influência do fato de ter sido obtido num primeiro momento, uma vez
que, com o superar das primeiras dificuldades (especialmente após o
primeiro ano), alguns casais poderão passar a considerar a hipótese
de um outro filho. Uma pequena parcela dos pais não tinha opinião
formada a este respeito, durante a avaliação inicial.
Finalmente, sobre o impacto na unidade do casal, foi observado
que quase 10% dos casais separaram-se após o nascimento da
criança com Síndrome de Down, por motivos atribuíveis a ela. Na
maioria dos casos, a iniciativa partiu do genitor. Em um caso, houve
reconstituição da unidade familiar alguns meses depois, com retorno
48

do pai à família, após rejeição inicial à criança, e permanência da
unidade familiar até o momento (dois anos de seguimento).
Estes dados também reforçam a necessidade da estruturação
de uma rede hospitalar de apoio as crianças com SD que envolva
equipes multidisciplinares, incluindo psicólogos[63], fonoaudiólogos,
terapeutas ocupacionais, fisioterapeutas, oftalmologistas,
otorrinolaringologistas, neuropediatras, cardiologistas, além de
disponibilidade de recursos laboratoriais e de imagem
complementares para a adoção efetiva de um protocolo de
acompanhamento adequado, que reduza riscos e possibilite a
realização de intervenções clínico-cirúrgicas em tempo hábil[4, 25, 39,
52, 53, 63, 64].
Entretanto,. visto tratar-se de Hospital Escola, com atendimento
a nível terciário, estima-se que a amostra possa não ter sido
representativa da população. Assim, estudos populacionais se farão
necessários, com triagem para recém-nascidos com Síndrome de
Down nas maternidades. Tal trabalho coloca-se agora como um
objetivo a ser alcançado a curto e médio prazo.
Os principais obstáculos encontrados na operacionalização
deste trabalho foram de ordem técnica – recursos humanos e
materiais, dificuldade em coordenar ações de estimulação de maneira
multidisciplinar (por falta de profissionais disponíveis na comunidade
universitária) e a falta de programas visando à integração futura
destes pacientes na Sociedade e no mercado de trabalho.
49

PROPOSTAS DE ESTUDOS

• Idade Materna: como já descrito anteriormente, em nossa amostra
a média de idade materna revelou-se mais baixa do que a descrita na
literatura mundial. Embora seja esta uma característica comum às
populações latino-americanas, estudos mais aprofundados envolvendo
taxa de fecundidade e estudos epidemiológicos populacionais podem
ser de grande ajuda para melhor entendimento destes achados[15].
• Infecções: Sabe-se que crianças com Síndrome de Down
apresentam maior morbidade por doenças infecciosas, especialmente
pneumonias. A existência de um grupo de pacientes sob
acompanhamento regular, tendo ainda como vantagem adicional o
suporte de um Hospital Universitário, permitirá analisar a incidência
destas doenças, itens predisponentes ou etiologias, evolução clínica,
opções de tratamento, em comparação com crianças sem Síndrome de
Down, estratégias preventivas e o uso de vacinas especiais (ainda não
disponíveis no Sistema Único de Saúde para crianças portadoras da
Síndrome)[49].
• Doença Celíaca: é descrita na literatura a maior ocorrência deste
distúrbio entre crianças com Síndrome de Down, sendo pertinente um
programa de rastreamento para tal doença, permitindo melhor
conhecimento e detecção e tratamento precoces da mesma[30, 31, 65].
• Alterações eletroencefalográficas: visando estabelecer um padrão
de atividade elétrica cerebral destas crianças, ao tempo que também
permitiria detecção precoce da Síndrome de West, o que gera
51

tratamento e melhor prognóstico. Este trabalho já se encontra em
andamento, em parceria com o Serviço de EEG do HUPES.
• Metabolismo do Folato: estudos recentes têm evidenciado
associação entre alterações enzimáticas (determinadas geneticamente,
com mutação já identificada no cromossomo 1) envolvidas no
metabolismo do ácido fólico e a maior predisposição à não-disjunção.
Tal abordagem poderia levar a uma revolução na questão do
Aconselhamento Genético e a possíveis estratégias de prevenção
primária, recurso até hoje não disponível para a esta
cromossomopatia[16, 17].
• Cardiologia: avaliando a evolução destas crianças, especialmente
no pós-operatório, além de comparar o perfil das cardiopatias em
relação à população sem Síndrome de Down[60].
• Endocrinologia: determinação das endocrinopatias associadas à
Síndrome de Down, além de estudos específicos dos distúrbios da
Tireóide[38].
• Psicologia: abordando os benefícios de um acompanhamento
precoce, o impacto da Síndrome de Down sobre as famílias, o
desenvolvimento neuropsicomotor e as estratégias de inserção da
criança na sociedade[47, 63].
• Oftalmologia: cerca de 40% dos pacientes foram avaliados por
oftalmologista até o momento. Entretanto, uma vez que alguns achados
52

não foram compatíveis com a literatura revisada, optamos por expandir
esta amostra, antes de expor tais dados.
• Nosso objetivo imediato, a partir deste trabalho inicial, de
caracterização global de uma amostra de pacientes com Síndrome de
Down, deverá incluir a manutenção de linhas de pesquisa sobre o
assunto, usando como ponto de partida as possibilidades acima, o que
resultará em benefício ao conhecimento científico, aprimoramento
profissional e, principalmente, em melhor assistência aos pacientes
portadores da Síndrome de Down.
53

CONCLUSÕES

O presente estudo procurou delinear um perfil clínico e demográfico
das crianças portadoras de Síndrome de Down no Estado da Bahia,
mediante uma análise descritiva de um grupo de pacientes atendidos em
ambulatório especializado do Hospital Universitário Professor Edgard
Santos.
• Dentre os dados demográficos, o perfil sócio-econômico observado
foi compatível com o encontrado entre os usuários do Sistema Único de
Saúde (SUS).
• Os pacientes aqui estudados foram predominantemente
classificados como da raça branca. Tal particularidade necessita de
avaliação mais detalhada, incluindo grupo racial paterno, para melhor
avaliação, uma vez que não existe descrição na literatura de prevalência
diferenciada por raças. Entretanto, na revisão bibliográfica, não se
encontraram relatos de estudos realizados em regiões com
miscigenação racial semelhante à observada na cidade de Salvador.
• Verificou-se que o perfil fenotípico assemelhou-se bastante ao
descrito na literatura, assim como a freqüência das anomalias maiores,
como as cardiopatias.
• Em relação à cardiopatia congênita, principal causa de morbidade
na Síndrome de Down, existe a possibilidade desta freqüência não estar
sendo corretamente estimada. A idade do início do acompanhamento
dos pacientes variou bastante, e a maioria não havia realizado
ecocardiograma anteriormente, sendo então o diagnóstico dado neste
primeiro momento. Entretanto, algumas alterações, a exemplo do
Forame Oval Patente ou da Persistência do Canal Arterial, tendem
55

normalmente a resolver-se espontaneamente. A realização do
ecocardiograma tardiamente (após o primeiro ano de vida) poderia estar
deixando de detectar alterações sem repercussões clínicas e que teriam
desaparecido espontaneamente, e que deste modo não
necessariamente estariam relacionadas a esta síndrome genética. A
estratificação da amostra por faixa etária poderia dar uma informação
mais fidedigna sobre este aspecto.
• A idade materna média, inferior à classicamente descrita para a
Síndrome de Down foi, no entanto, semelhante à encontrada em outros
estudos latino-americanos. Estudos com base populacional seriam
importantes para melhor entendimento deste aspecto.
• O presente trabalho, utilizando uma ferramenta de atendimento
(protocolo) para sistematização da informação, permitiu o conhecimento
das principais características de um grupo de pacientes, o que será de
utilidade para pesquisas futuras.
56

SUMMARY

CLINICAL AND DEMOGRAPHIC ASPECTS OF DOWN’S SYNDROME IN
AN OUTPATIENT REFERENCE MEDICAL CENTER IN BAHIA –
BRAZIL. Down’s Syndrome is the most frequent chromosomal anomaly
among humans, and the first cause of mental disability in the worldwide
population. Down’s syndrome is a well-known and studied pathology, and
the affected persons that are offered programs of stimulation and
prevention can experience a great improvement in their life quality, social
insertion and work. OBJECTIVE: to describe the clinical and
demographic aspects of a sample of outpatients in a Medical Genetic
Center, part of a Universitary Hospital, in order to achieve a better
knowledge about Down’s Syndrome in Bahia. STUDY DESIGN: Cross-
sectional. SUBJECTS AND METHODS: 87 children with Down’s
Syndrome (aged 0-18 years) seen between January 2000 and January
2002 at Genetics Clinic oh the Hospital Universitário Professor Edgard
Santos. Those children whose diagnosis were not clear, and could not de
confirmed by caryotype analysis, were not enrolled. Statistic analysis was
descriptive. RESULTS: there was an excess of male individuals (58,6%).
The mean maternal age was 30,1y. Racial group: 69% children were
white or light mulatto. All children showed slant palpebral fissures,
meanwhile, only 32% showed simian creases. Congenital Heart Disease
was seen in 44.9% of the children. Hypothyroidism was detected in 6% of
patients. CONCLUSIONS: the clinical and demographic characteristics of
the sample of patients are similar to that described worldwide, except
about maternal age (this aspect, however, showed results similar to
those seen in South America) and racial group. These points must be
better studied latter. The follow-up of these children will probably give us
more important assessments.
KEY WORDS: Down Syndrome, Mental Deficiency, Phenotype, Malformations.
58

REFERÊNCIAS BIBLIOGRÁFICAS

1. Mustacchi, Z., Síndrome de Down, in Genética Baseada em Evidências: Síndromes e Heranças, Z. Mustacchi, Editor. 2000, CID Editora: São Paulo. p. 817-94.
2. Observations on an ethnic classification of idiots. Disponível em: <http://home.vicnet.net.au/~dealccinc/Downs.htm>. Acesso em: 04/01/2002.
3. Jones, K.L., Smith´s Recognizable Patterns of Human Malformation. 5a ed. 1997, New York: Saunders. 8-13.
4. Trisomy 21: The Story of Down Syndrome. Disponível em: <http://www.ds-health.com/trisomy.htm>. Acesso em: 30/03/2002.
5. Hayes, A.& Batshaw, M.L., Down Syndrome. Pediatric Clinics of North America, 1993. 40(3): p. 523-35.
6. Mutton, D., Alberman, E.&Hook, E.B., Cytogenetic and epidemiological findings in Down syndrome, England and Wales 1989 to 1993. Journal of Medical Genetics, 1996. 33: p. 387-94.
7. Risk and Recurrence Risk of Down Syndrome. Disponível em: <http://www.nas.com/downsyn/benke.html>. Acesso em: 04/01/2002.
8. Trisomy 21. Disponível em: <http://www3.ncbi.nim.nih.gov/htbin-post/Omim/dispmin?190685>. Acesso em: 06/02/2001.
9. Penrose, L.S., The Relative Effects of Parental and Maternal age in Mongolism. Journal of Genetics, 1933. 27: p. 219.
10. The DNA sequence of human chromosome 21. Disponível em: <http://www.nature.com/cgitaf/DynaPage.taf?file=/nature/jour.../405311a0_r.html&filetype>. Acesso em: 04/01/2002.
11. Oteiza, M.E.F., Lotti, F.A., Muñiz, I.C., Ortiz, J.R., Valle, A.P.&Lloret, M.C.E., Tendencias del Síndrome de Down en Cuba. Su relación com edad materna y tasa de fecundidad. Revista Cubana de Pediatria, 1998. 70(3): p. 141-7.
12. Boa Sorte, N.C.A. Perfil dos Partos entre adolescentes na Rede SUS da Bahia entre 1998 e 2000. In: VII Congresso Brasileiro de Adolescência. Salvador, 2001
13. Cuckle, H., Maternal age-standardization of prevalence of Down’s Syndrome. Lancet, 1999. 354: p. 529-30.
14. Bieguelman, B., Krieger, H.&Silva, L.M., Maternal age and Down Syndrome in Southeastern Brazil. Brazilian Journal of Genetics, 1996. 19(4): p. 637-40.
60

15. Palomaki, G.E., Age-related prevalence of Down syndrome. American Journal of Obstetrics and Gynecology, 1999. 180(6): p. 102-10.
16. Da Silva, L.R.J. Genes Envolvidos do Metabolismo da Homocisteína e sua relação com risco aumentado para Síndrome de Down. In: XIV Congresso Brasileiro de Genética Clínica. Ribeirão Preto-SP, 21(p.), 2002
17. Palekar, A.G., Preconceptional intake of folate and vitamin B12 in the prevention of neural tube defects and Down syndrome. American Journal of Obstetrics and Gynecology, 2001.
18. Christianson, A.L., Down syndrome in black South African infants and children - clinical features and delayed diagnosis. South African Medical Journal, 1997. 87(8): p. 992-95.
19. Kanamori, G., Witter, M., Brown, J.&Williams-Smith, L., Syndromic and other congenital anomalies of the head and neck. Otolaryngologic Manifestations of Down Syndrome. Otolaryngologic Clinics of North America, 2000. 33(6): p. 227-38.
20. Barros Filho, T.E.P., Oliveira, R.P., Rodrigues, N.R., Galvão, P.E.C.&Souza, M.P., Instabilidade atlanto-axial na síndrome de Down. Relato de 10 casos tratados cirurgicamente. Revista Brasileira de Ortopedia, 1998. 33(2): p. 91-4.
21. Tamayo F, M.L., Villegas, J.E.B.&Frias, J.L., Anomalías Oculares y Auditivas en el Síndrome de Down. Pediatria (Bogota), 1994. 29(1): p. 53-7.
22. M Rosales, J.L.&Rojas, P.P.M., Estudio oftalmológico de los niños con síndrome de Down. Revista Oftalmologica Venezoelana, 1996. 52: p. 16-8.
23. Elias, C.L.L.F.&Elias, R., Síndrome de Down, in Odontologia de Alto Risco: pacientes especiais, R. Elias, Editor. 1995, Revinter: São Paulo. p. 15-52.
24. Lima, G.B., Capra, M.E.Z., Frantz, B.C., Leite, J.C.L.&Giugliani, R., Síndrome de Down: características clínicas, perfil epidemiológico e citogenético em recém-nascidos no Hospital de Clínicas de Porto Alegre. Revista Associação Médica do Rio Grande do Sul, 1996. 40(1): p. 8-13.
25. Jijón, M., Sindrome de Down. Pautas mínimas para su entendimiento y atención. 1a ed. Genética. 1997, Quito: Ed. Ecuaoffset Cía Ltda. 123.
61

26. Marino, B., Anaclerio, S.&DI Donato, R., Age at Operation for Children With Atrioventricular Canal. Pediatrics, 2001. 108(1): p. 85.
27. Reller, M.D.&Morris, C.D., Is Down syndrome a risk factor for poor outcome after repair of congenital heart defects? Journal of Pediatrics, 1998. 132(4): p. 738-41.
28. Rex, A.P.&Preus, M., A diagnostic index for Down syndrome. The journal of Pediatrics, 1982. 100(6): p. 903-6.
29. Walker-Smith, J.A., Celiac disease and Down syndrome. Journal of Pediatrics, 2000. 137(6): p. 85-90.
30. Csizmadia, C.G.D.S., Mearin, M.L., Oren, A., Kromhout, A., Crusius, J.B.A., Blomberg, B.M.E., Pena, A.S., Wiggers, M.N.L.&Vandenbroucke, J.P., Accuracy and cost-effectiveness of a new strategy to screen for celiac disease in children with Down syndrome. Journal of Pediatrics, 2000. 137(6): p. 756-60.
31. Zachor, D.A., Mroczek-Musulman, E.&Brown, P., Prevalence of Celiac Disease in Down Syndrome in the United States. Journal of Pediatric Gastroenterology and Nutrition, 2000. 31(3): p. 275-79.
32. Hannequin, M., Morin, C.&Feine, J.S., Pain expression and stimulus localization in individuals with Down's syndrome. Lancet, 2000. 356: p. 1882-87.
33. Jessop, D., Pain in Down's Syndrome. The lancet, 2001. 357(9261): p. 1041.
34. Brandt, P.R., Pain in Down's Syndrome. The lancet, 2001. 357: p. 1041-42.
35. Annerén, G., Tuvemo, T., Carlsson-Skwirut, C., Lönnerholm, T., Bang, P., Sara, V.R.&Gustafsson, J., Growth hormone treatment in young children with Down´s Syndrome: effect on growth and psychomotor development. Archives Disease in Childhood, 1999. 80(4): p. 334-38.
36. Ivarsson, S.A., Ericsson, U.B., Gustafsson, J., Forslund, M., Vegfors, P.&Annerén, G., The impact of thyroid autoimmunity in children and adolescents with Down syndrome. Acta Paediatrica, 1997. 86(10): p. 1065-67.
37. Seashore, M.R., Cho, S., Desposito, F., Sherman, J., Wappner, R.S.&Wilson, M.G., Health Supervision for Children With Down Syndrome. Pediatrics, 1994. 93(5): p. 855-559.
62

38. Oliveira, A.T.A., Longui, C.A., Calliari, L.E.P., Ferone, E.A.,
Kawaguti, F.S.&Monte, O., Avaliação do eixo hipotalâmico-hipofisário-tireoidiano em crianças com síndrome de Down. Jornal de Pediatria, 2002. 78(4): p. 295-300.
39. Foreman, P.J., Paediatric Management Practices in Down Syndrome: a follow-up survey. Journal of Paediatric Children Health, 1993. 29(1): p. 27-31.
40. Castilla, E.E., Rittler, M., Dutra, M.G., Lopez-Camelo, J.S., Campaña, H., Paz, J.E., Orioli, I.M.&Group, E.-D., Survival of Children With Down Syndrome in South America. American Journal of Medical Genetics, 1998. 79: p. 108-11.
41. Orlicek, S.L., Walker, M.S.&Kuhls, T.L., Severe Mycoplasma Pneumonia in Yong Children with Down Syndrome. Clinical Pediatrics, 1992. 31(7): p. 409-12.
42. Thase, M.E., Longevity and mortality in Down´s syndrome. Journal of Mental Deficiency Research, 1982. 26: p. 177-92.
43. Ugazio, A.G., Maccario, R., Notarangelo, L.D.&Burgio, R., Immunology of Down Syndrome: A Review. American Journal of Medical Genetics, 1990. Supplement 7: p. 204-12.
44. Nespoli, L., Burgio, G.R., Ugazio, A.G.&Maccario, R., Immunological features of Down´s Syndrome: a review. Journal of Intellectual Disability Research, 1993. 37(6): p. 543-51.
45. Ablin, R.J., Immunity in Down´s Syndrome. European Journal of Pediatrics, 1978. 127(2): p. 149-52.
46. Lobaugh, N.J., Karaskov, V., Rombough, V., Rovet, J., Bryson, S., Greenbaun, R., Haslam, R.H.&Koren, G., Piracetan Therapy Does Not Enhance Cognitive Functioning in Children With Down Syndrome. Archives of Pediatrics and Adolescent Medicine, 2001. 155(4): p. 442-48.
47. Cunningham, C.C., Morgan, P.A.&McGucken, R.B., Down's syndrome: is dissatisfaction with disclosure of diagnosis inevitable? Devices in Medical Children Neurology, 1984. 26(1): p. 33-39.
48. Garwick, A.W., Patterson, J., Bennett, F.C.&Blum, R.W., Breaking the News - How Families First Learn About Their Child's Chronic Condition. Archives of Pediatrics and Adolescent Medicine, 1995. 149(9): p. 991-97.
63

49. Burns, D.A.&Esterl, S.I., As alterações imunológicas na Síndrome
de Down, in Genética Baseada em Evidências - Síndromes e Heranças, Z. Mustacchi, Editor. 2000, CID Editora: São Paulo. p. 558-93.
50. Bertolini, D.L., Vitalle, M.S.S.&Fisberg, M., Morbimortalidade em Indivíduos Portadores da Síndrome de Down. Jornal Brasileiro de Medicina, 1991. 61(3): p. 13-25.
51. Ugazio, A.G., Down´s Syndrome: problems of immunodeficiency. Human Genetic Supplies, 1981. 2: p. 33-39.
52. Aracena, A.M., Supervisión de la salud en personas com síndrome de Down. Revista Chilena de Pediatria, 1993. 64(1,supl): p. 20-23.
53. Saenz, R.B., Primary Care of Infants and Young Children with Down Syndrome. American Family Physician, 1999. 59(2): p. 381-90.
54. Dipierri, J.E., Quinteros, R.C., Alfaro, E.&Romero, M., Epidemiología del síndrome de Down en la provincia de Jujuy. Archivos Argentinos de Pediatria, 1994. 92(4): p. 205-11.
55. Granzotti, J.A., Paneto, I.L.C., Amaral, F.T.V.&Nunes, M.A., Incidência de cardiopatias congênitas na Síndrome de Down. Jornal de Pediatria, 1995. 71(1): p. 28-30.
56. Boy, R., Neto, J.G.B., Vargas, F.R., Fontana, C., Almeida, J.C.C.&Lherena Jr, J., Síndrome de Down - análise clínica, citogenética e epidemiológica de 165 casos. Jornal de Pediatria, 1995. 71(2): p. 88-92.
57. Krieger, H., Morton, N.E., Mi, M.P., Azevêdo, E., Freire-Maia, A.&Yasuda, N., Racial admixture in northeastern Brazil. Annals of Human Genetics, 1965. 29: p. 113-25.
58. Nazer, J.H., Eaglin, M.A.&Cifuentes, L.O., Incidencia del sindrome de Down en la maternidad del Hospital Clínico de la Universidad de Chile. Un registro de 25 años: 1972-1997. Revista Médica de Chile, 1998. 126(4): p. 383-90.
59. Molina, J.J.P., Alfaro, N.A., Garcia, T.R.&Castellanos, E.A., Síndrome de Down, prevalencia en 18,509 nacimientos consecutivos y frecuencia de 11 características morfológicas externas. Revista Médica del Instituto Mexicano del Seguro Social, 1993. 31(4): p. 263-64.
60. Ferrín, L.M., Atik, E., Ikari, N.M., Martins, T.C., Marcial, M.B.&Ebaid, M., Defeito Total do Septo Atrioventricular. Correlação Anatomofuncional entre Pacientes com e sem Síndrome de Down. Arquivos Brasileiros de Cardiologia, 1997. 69(1): p. 19-23.
64

61. McConnell, M.E., Heart Murmurs in Pediatric Patients: When Do You Refer? American Family Physician, 1999. 60(2): p. 315-19.
62. Calliari, L.E., Hipotireoidismo, in Endocrinologia para o Pediatra, Atheneu, Editor. 1998, Monte, O: São Paulo. p. 114-18.
63. Sadir, M.A. Síndrome de Down:acompanhamento psicológico integrado à consulta do geneticista. In: XI Congresso Brasileiro de Genética Clínica. Salvador, 46(p.), 1999
64. Cronk, C., Crocker, A.C., Pueshel, S.M., Shea, A.M., Zackai, E., Pickens, G.&Reed, R.B., Growth Charts for Children With Down Syndrome: 1 Month to 18 Years of Age. Pediatrics, 1988. 81(1): p. 102-10.
65. Carlsson, A., Axelsson, I., Borulf, S., Bredberg, A., Forslund, M., Lindberg, B., Sjöberg, K.&Ivarsson, S.A., Prevalence of IgA - Antigliadin Antibodies and IgA - Antiendomisium Antibodies Related to Celiac Disease in Children With Down Syndrome. Pediatrics, 1998. 101(2): p. 272-75.
66. Rosenfeld, R., Alterações Eritrocitárias, in Diagnóstico Laboratorial em Pediatria, Sarvier, Editor. 2000, Carraza, FR: São Paulo. p. 26-38.
67. Rotondi, M.E.A., Alterações Leucocitárias, in Diagnóstico Laboratorial em Pediatria, Sarvier, Editor. 2000, Carraza, FR: São Paulo. p. 39-62.
65

ANEXOS

ANEXO 1 – PROTOCOLO DE ATENDIMENTO
Universidade Federal da Bahia HUPES - Laboratório de Genética Médica Protocolo de Atendimento Ambulatorial
Síndrome de Down
1. IDENTIFICAÇÃO
Data da primeira consulta: ______________________ Encaminhado por: HUPES/CPPHO/MCO MTB HGRS MAS IPERBA
HGCandeias HGCamaçari Outros: ______________ Motivo do encaminhamento: Diagnóstica Acompanhamento Registro HUPES: ______________ Consulta Genética: _____________ Informante: ______________ Precisão: Boa Regular Precária Paciente:________________________________________________ Nascimento: ___/___/___ Idade ____ Sexo ___ Grupo Racial ___________ Mãe _____________________________________________ Idade _____ Grupo Racial ____________ Naturalidade ___________________________ Estado Civil ____________ Ocupação ______________________________ Escolaridade: analfabeto 1o GI 1o GC 2o GI 2o GC 3o GI 3o GC NS Pai ______________________________________________ Idade _____ Grupo Racial ____________ Naturalidade ___________________________ Estado Civil ____________ Ocupação ______________________________ Escolaridade: analfabeto 1o GI 1o GC 2o GI 2o GC 3o GI 3o GC NS Adendos ___________________________________________________ Endereço ____________________________________________________ CEP _____________ Telefone (_____) ___________ Contato ___________ Adendos ____________________________________________________ Renda Familiar: ____ salários-mínimos Residência: Urbana Suburbana Rural Habitação: própria alugada cedida / instituição / outros Número de Cômodos: 1 2 3 4 5 6 + de 6 Saneamento: Sim Não Número de pessoas na família: 2 3 4 5 6 7 8 9 10 +10
67

2. ANAMNESE/ANTECEDENTES História Clínica
Notou alteração ao nascimento? Sim Não
Foi informada do diagnóstico ao nascimento? Sim Não
Notou alteração no DNPM? Sim Não
Notou alguma outra alteração? Sim Não
Qual? ___________________________________________________
Antecedentes Obstétricos Maternos: G ______ P ______ A ______ G Conc.* Nome Sexo Idade Observação
*NV= Nativivo / NM= Natimorto / AE= Aborto Espontâneo / AP= Aborto Provocado
Antecedentes Pré-natais:
Pré-Natal: Fez - Local: ________________ A partir do ____ mês Não Fez
Movimentos Fetais: _____ mês Intensidade ↓ Normal ↑ ________
Diferenças outras gestações ________________________________GS____
Exames ______________________________________________________
USG ________________________________________________________
Fumante: Sim Não Bebidas Alcoólicas: Sim Não
Engravidou usando anticoncepcional: Sim _______________________ Não
Antecedentes Paternos: ___________________________________ GS ___
68

Complicações e/ou Intervenções durante a gestação S/N Detalhes Mês/Sem. Doenças Agudas Doenças Crônicas Metrorragia Imunizações Fatores Físicos Medicamentos Drogas Procedimentos Radiação Cont. com animais
Antecedentes Peri e Neonatais:
Parto: PSNV PSNP PSAC PSAF _______________
Local________________________________________________________
Assistência _____ Sedação ______ Anestesia _____ Duração TP ________
Rotura Precoce de Membranas (> 24h antes do parto) Sim Não
Peso _____ g Estatura ____ cm PC ____ cm PT ___ cm Apgar 1 ___ 5 ____
Capurro _______ ( Termo Pré-termo) Grupo Sangüíneo ______
Perman. Mat. _______Eventos sugestivos de hipóxia: Sim Não
Intercorrências/Observações _____________________________________
____________________________________________________________
____________________________________________________________
TESTE DO PEZINHO:___________________________________________ Evolução Neuromotora: Evolução 2 3 4 5 6 7 8 9 10 11 12 13 14 15-24 >24 Sorriso Social Firmou cabeça Sentou c/ apoio Sentou s/ apoio Palavra Andou s/ auxilio
Esfíncteres: vesical - diurno _____ m noturno _____ m anal _____ m
Fontanelas: FA _____ m FP ______ m Primeiro dente _______ m
Observações: _________________________________________________
Terapias e Escolaridade Tipo Local Idade Início Fisioterapia Fonoaudiologia T. Ocupacional Psicologia Escola
69

Antecedentes Patológicos Imunizações Vacinas 1a Dose 2a Dose 3a Dose 1o Reforço 2o Reforço BCG Hepatite B Pólio DPT H. Influenzae Sarampo Febre Amarela Hepatite A Anti-pneumocócica Tétano Varicela Gripe (Influenza)
Doenças Comuns na Infância e Doenças Agudas
Viroses IVAS Pneumonias Infecções cutâneas Diarréia
Doenças Crônicas
Cardiopatia Rinite Obstipação
Cirurgias
Cardíacas Gastrointestinais Ortopédicas
Outros _________________________________________________________________________
_________________________________________________________________________________
_____________________________________________________________________________
Antecedentes Familiares
Casal consangüíneo: Não Sim Grau: ____________________________________
Defeitos Congênitos na família: Não Sim _____________________________
Deficiência Mental na família: Não Sim __________________________________________
Problema semelhante ao do propósito: Não Sim ____________________________________
Outros Problemas: Não Sim_________________________________________________
_______________________________________________________________________________
Casal adota planejamento familiar? Não Sim
Método Contraceptivo utilizado ______________________________________________________
Casal desejou este filho? Não Sim
Casal aceitou a gravidez do propósito? Não Sim
70

Como avalia a maneira como foi informado do diagnóstico
Clara Pouco informativa Pejorativa Preocupação inicial e dúvidas em relação ao problema
Habilidade Motora Habilidade Intelectual Independência Comportamento social Cardiopatia Infecções Discriminação social Outros _________________________________________________
Pais desejam ter mais filhos?
Sim Não Não sabem Pais separados?
Sim, antes do nascimento da criança Sim, logo após o nascimento da criança Sim, mais de 2 anos após o nascimento da criança Não Reação inicial da mãe ao ser informada do diagnóstico
Revolta Tristeza Negação Rejeição Culpa Outra____________________________________ Heredograma
71

3. EXAME FÍSICO
Sinais clínicos associados à Síndrome de Down Sinal Clínico Presente Ausente
Hipotonia muscular Baixa estatura relativa
RDNPM/Retardo Mental Braquicefalia
Cabelos finos, lisos e esparsos Perfil facial achatado
Hipoplasia malar Fendas palpebrais oblíquas para cima
Ptose palpebral Nistagmo Epicanto
Blefarite/conjuntivite Manchas de Brushfield
Ponte nasal baixa Boca permanentemente aberta
Língua protrusa Língua fissurada Dentes anormais
Palato estreito (Ogival ____) Micrognatia/retrognatia
Orelhas dismórficas Pescoço curto
Pele redundante no pescoço Defeito cardíaco
Diátese de retos/hérnia umbilical Frouxidão ligamentar
Mãos pequenas e largas 5o quirodáctilo curto (prega única ___)
Clinodactilia de 5o quirodáctilo Prega de flexão palmar única D ( ) E ( )
Intervalo aumentado entre 1o e 2o pododáctilos Cutis marmorata
Observações relativas ao exame físico: ____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
72

14. DIAGNÓSTICOS, CONDUTAS E INFORMAÇÕES ADICIONAIS
Diagnóstico Clínico Diagnóstico Etiológico: Conduta Inicial __________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________ Informações adicionais:
AVALAIÇÂO ESPECIALIZADA CARDIOLÓGICA
ao nascer 10 ano 20 ano 30 ano 40 ano
OFTALMOLÓGICA
Demais ao nascer 10 ano 20 ano 30 ano 40 ano
TIREOIDIANA
Demais ORTOPÉDICA ULTRASSONOGRÁFICA
Paciente
Mãe
CITOGENÉTICA
Pai
ao nascer 10 ano 20 ano 30 ano 40 ano 50 ano
OTORRINOLARINGOLÓGICA
Demais PSICOLÓGICA
HEMATOLÓGICA
73

5. EVOLUÇÃO E SEGUIMENTO Data: Idade: Compareceu:_________________________ Atendido por:_______ _________________ Marcado p/: _______ P: E: PC:
Data: Idade: Compareceu:_________________________ Atendido por:_______ _________________ Marcado p/: _______ P: E: PC:
_
Data: Idade: Compareceu:_________________________ Atendido por:_______ _________________ Marcado p/: _______ P: E: PC:
74

Hospital Universitário Prof. Edgard Santos – HUPES Laboratório de Genética Médica
Protocolo de Atendimento Ambulatorial – Síndrome de Down
RESUMO DO CASO
NOME: __________________________________________________________________________ GÊNERO: ____________ GRUPO RACIAL: ______________ DATA NASC: _____________________ REG:___________ CG:__________ ENCAMINHADO POR: __________________________________ MOTIVO DO ENCAMINHAMENTO:
Aconselhamento Genético _________________________________________________________
Confirmação Diagnóstica __________________________________________________________
Outro________________________________________________________________________ TÉCNICAS DE ESTUDO E AVALIAÇÕES ESPECIALIZADAS: Cariótipo
Cardiologia
F. Tireoidiana
Otorrino
Hemograma
Oftalmologia
Radiografias
Neurologia
USG
Psicologia
Imunização especial Fisioterapia T. Ocupacional
Outros
Outra
DIAGNÓSTICO CLÍNICO DIAGNÓSTICO ETIOLÓGICO
Trissomia livre Translocação ( ________ ) Outro ACONSELHAMENTO GENÉTICO Data: ___________ IM _____ IP ______ Consangüinidade: sim não Risco de recorrência: ____________________________________________ OBSERVAÇÕES: RESUMO PRELIMINAR REALIZADO EM: / / . Sujeito a atualizações. Ass:
75

ANEXO 2 -TERMO DE CONSENTIMENTO
ASPECTOS CLÍNICO-DEMOGRÁFICOS DA SÍNDROME DE DOWN EM SERVIÇO DE REFERÊNCIA NA BAHIA
Eu, ________________________________, portador do RG n0
____________, responsável pelo paciente
___________________________________, acompanhado no
ambulatório de Neurogenética do Hospital Universitário Professor Edgard
Santos, declaro que fui devidamente informada pela Dra Tatiana Amorim,
CRM 12367, sobre a inclusão do citado paciente em uma pesquisa por ela
coordenada, e sobre o objetivo da mesma. Estou ciente que o objetivo
principal desta pesquisa é descrever as características Clínicas dos
portadores de Síndrome de Down no nosso Estado, visando um melhor
estudo e condução destes pacientes. O paciente poderá ser fotografado e
as fotografias usadas apenas para fins científicos. A Dra Tatiana Amorim
deixou claro que, caso eu não concorde com a participação do paciente
sob minha responsabilidade, este não sofrerá nenhum tipo de prejuízo no
seu acompanhamento, e caso eu concorde, os dados servirão apenas para
pesquisa. Informo estar ciente de que todas as informações sobre o
paciente serão mantidas em sigilo e que este não poderá ser identificado
como participante da pesquisa. Após estas informações a mim prestadas,
autorizo a Dra Tatiana Amorim a utilizar os dados clínicos do paciente sob
minha responsabilidade, assim como resultado de exames laboratoriais e
complementares e fotografias do paciente, no presente projeto de
pesquisa.
Salvador, ______ / _______ / _______.
Assinatura: _____________________________________
Voluntário (a)
76

ANEXO 3 – MODELOS DE SOLICITAÇÃO DE AVALIAÇÕES
ESPECIALIZADAS FORA DA UFBA
UFBA – Universidade Federal da Bahia HUPES - Hospital universitário Professor Edgard Santos LGM – Laboratório de Genética Médica
SOLICITAÇÃO DE AVALIAÇÃO ___________________ Prezada Dra ________________________________________ Instituição: _________________________________________ Estamos encaminhando o (a) paciente ____________________________, portador (a) de Síndrome de Down, para sua avaliação. Atenciosamente,
_________________________ Ambulatório de Neurogenética
Resposta de Consulta (se necessário) _____________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________________
77

ANEXO 4 – RELATÓRIO PARA INSS UNIVERSIDADE FEDERAL DA BAHIA
HOSPITAL UNIVERSITÁRIO PROF EDGARD SANTOS LABORATÓRIO DE GENÉTICA MÉDICA
ATESTADO MÉDICO
Atesto para os devidos fins que o (a) paciente
__________________________________________é portador de CID Q90
+ F72.
Salvador, ______de ____________de ________.
_____________________________ Ambulatório de Neurogenética
78

ANEXO 5 – VALORES DE REFERÊNCIA PARA AVALIAÇÃO HEMATOLÓGICA
1. TESTES DE TRIAGEM PARA ANEMIA (limites inferiores do valor de referência) Idade (anos) e sexo Hemoglobina (g/dl) Hematócrito(%) VCM (µ3) HCM
(pg) 0,5 – 4 11,0 32 72 24 5 – 10 11,0 33 75 25 11 – 14 feminino 11,5 34 78 26 11 – 14 masculino 12,0 35 78 26 15 – 19 feminino 12,0 35 79 27 15 – 19 masculino 13,0 39 79 27 VCM – volume corpuscular médio HCM – hemoglobina corpuscular média Fonte: [66] 2. VALORES NORMAIS DE FERRO SÉRICO
Idade Masculino Feminino µg/dl µmol/l µg/dl µmol/l 1 – 30 dias 32 – 112 5,7 – 20 29 – 127 5,2 – 22,7 1 – 12 meses 27 – 109 4,8 – 19,5 25 –126 4,5 – 22,6 1 – 3 anos 29 – 91 5,2 – 17,3 25 – 101 4,5 – 18,1 4 – 6 anos 25 – 115 4,5 – 20,6 28 – 93 5 – 16,7 7 – 9 anos 27 – 96 4,8 – 17,2 30 – 104 5,4 – 18,6 10 – 12 anos 28 – 112 5 – 20 32 – 104 5,7 – 18,6 13 – 15 anos 26 – 110 4,7 – 19,7 30 – 109 5,4 – 19,5 16 – 18 anos 27 - 138 4,8 – 24,7 33 – 102 5,9 – 18,3 Fonte: [66] 3. TESTES CONFIRMATÓRIOS PARA DEFICIÊNCIA DE FERRO (valor de corte) Idade (anos) Ferritina (ng/ml) Saturação da Transferrina (%) 0,5 – 4 <10 < 12 5 – 10 <10 < 14 11 – 14 <10 < 16 15 – 19 2 < 16 Fonte: [66]
79
4. VALORES DE REFERÊNCIA PARA LEUCÓCITOS CONFORME A IDADE (n0/mm3)
Idade Leucócitos totais neutrófilos Linfócitos 6 meses 6,0 – 17,5 1,0 – 8,5 4 – 13,5
1 ano 6,0 – 17,5 1,5 – 8,5 4,0 – 10,5 2 anos 6,0 – 17 1,5 – 8,5 3,0 – 9,5 4 anos 5,5 – 15,5 1,5 – 8,5 2,0 – 8,0 6 anos 5,0 – 14,5 1,5 – 8,0 1,5 – 7,0 8 anos 4,5 – 13,5 1,5 – 8,0 1,5 – 6,8 10 anos 4,5 – 13,5 1,8 – 8,0 1,5 – 6,5 16 anos 4,5 – 13 1,8 – 8,0 1,2 – 5,2 21 anos 4,5 - 11 1,8 – 7,7 1,0 – 4,8
Fonte: [67] 5. VALORES DE REFERÊNCIA PARA PLAQUETAS
Idade Masculino feminino X103/ml X103/ml 3 – 6 meses 275 – 566 288 - 598 7 meses – 2 anos 219 – 452 229 – 465 2 – 6 anos 204 – 405 204 – 402 7 – 12 anos 194 – 364 183 – 369 13 – 18 anos 165 – 332 185 - 335 Fonte: [66]
80

ANEXO 6– NORMAS PARA PUBLICAÇÃO O Jornal de Pediatria é a publicação científica da Sociedade Brasileira de Pediatria (SBP), com circulação regular desde 1934. Atualmente, sua versão impressa em língua portuguesa atinge mais de 14.000 leitores e instituições no Brasil e em toda a América Latina. Todo o conteúdo do Jornal de Pediatria está disponível em português e inglês no site http://www.jped.com.br, que é de livre acesso. O material publicado se destina a elevar o padrão da prática pediátrica e do atendimento médico de crianças e adolescentes em geral, bem como a promover o debate sobre a saúde.
O Jornal de Pediatria aceita a submissão de artigos originais, relatos de casos, artigos especiais e cartas ao editor em português, espanhol e inglês. Editoriais e comentários, que geralmente referem-se a artigos selecionados, são encomendados a autoridades em áreas específicas. O Conselho Editorial poderá eventualmente considerar a publicação de comentários submetidos espontaneamente. Da mesma forma, profissionais de reconhecida experiência em assuntos de interesse especial para os leitores são em geral convidados a escrever artigos de revisão, que são avaliações críticas e ordenadas da literatura em relação a temas de importância clínica, com ênfase em fatores como causas e prevenção de doenças, seu diagnóstico, tratamento e prognóstico. Metanálises se incluem nesta categoria. Autores não convidados podem também submeter previamente ao Conselho Editorial uma proposta de artigo de revisão, com um roteiro. Se aprovado, o autor pode desenvolver o roteiro e submetê-lo para publicação. Artigos de revisão devem limitar-se a 6.000 palavras, excluindo referências e tabelas. As referências bibliográficas deverão ser atuais e em número mínimo de 30. Os artigos em português e inglês são publicados, na versão impressa, em português e, no site, em português e inglês. Os artigos em espanhol são publicados, na versão impressa, na língua original e, no site, em português e em inglês, com uma versão em PDF na língua original.
Artigos originais incluem estudos controlados e randomizados, estudos de testes diagnósticos e de triagem e outros estudos descritivos e de intervenção, bem como pesquisa básica com animais de laboratório. O texto deve ter entre 2.000 e 3.000 palavras, excluindo tabelas e referências; o número de referências não deve exceder a 30.
Relatos de casos tratam de pacientes ou situações singulares, doenças especialmente raras ou nunca descritas, assim como formas inovadoras de diagnóstico ou tratamento. O texto é composto por uma introdução breve que situa o leitor em relação à importância do assunto e apresenta os objetivos da apresentação do(s) caso(s); por um relato resumido do caso; e por comentários que discutem aspectos relevantes e comparam o relato com a literatura. O número de palavras deve ser inferior a 2.000, excluindo referências e tabelas. O número máximo de referências é 15. Recomenda-se não incluir mais de duas figuras.
Artigos especiais são textos não classificáveis nas categorias acima, que o Conselho Editorial julgue de especial relevância para a saúde da criança. Sua revisão admite critérios próprios, não havendo limite de tamanho ou exigências prévias quanto à bibliografia.
Cartas ao editor são altamente estimuladas. Em princípio, devem comentar, discutir ou criticar artigos publicados no Jornal de Pediatria, mas também podem versar sobre outros temas médicos de interesse geral. Também são bem-vindos comunicados de investigação de assuntos relevantes, cujo conteúdo não seja suficientemente desenvolvido para ter sua publicação como artigo original. Recomenda-se tamanho máximo de 1.000 palavras, incluindo referências bibliográficas, que não devem exceder a seis. Sempre que possível, uma resposta dos autores será publicada junto com a carta.
O Jornal de Pediatria dá preferência ao envio de material submetido à publicação por correio eletrônico (e-mail), desde que não contenha desenhos ou fotografias digitalizados. Caso o artigo inclua figuras que necessitem ser digitalizadas, e sempre que for de preferência dos autores, o material pode ser enviado por correio comum.
81

ANEXO 7 – CARTA DE RECEBIMENTO DO ARTIGO PARA PUBLICAÇÃO Artigo : 2495 Aspectos clínicos-demográficos da Síndrome de Down na Bahia. Dr(a). Tatiana R. S. M. Amorim Recebemos os originais do seu artigo e o protocolamos com o número citado acima. Já o enviamos aos revisores e assim que tivermos uma posição sobre o mesmo, entraremos em contato. Desde já agradecemos sua colaboração. Atenciosamente, Maria de Fátima Machado Assistente do Jornal de Pediatria [email protected] (51) 3328.9520
82

ANEXO 8 - ARTIGO SUBMETIDO A PUBLICAÇÃO Aspectos clínico-demográficos da
Síndrome de Down na Bahia Clinical and demographic aspects of Down Syndrome in Bahia
Tatiana Amorim1, Ney Boa Sorte2, Grace Andrade3, Isabella Queiroz4,
Isabel Guimarães5, Lícia Moreira6. Resumo
OBJETIVO: delinear o perfil clínico-demográfico de um grupo de pacientes atendidos em um ambulatório de Genética de Hospital Universitário, visando o melhor conhecimento das alterações relacionadas a esta patologia no estado da Bahia.
DESENHO E ESTUDO: Observacional descritivo. MATERIAL E MÉTODOS: A amostra foi constituída de 87 pacientes portadores de Síndrome de Down (SD), com idade variando de sete dias a 18 anos. A análise estatística utilizou medidas descritivas.
RESULTADOS: não houve variação significante entre os gêneros. A média da idade materna foi significantemente menor do que o encontrado na literatura internacional, porém compatível com o descrito para sociedades sul-americanas. Houve um predomínio de crianças de grupo racial branco ou mulato-claro. As malformações maiores apresentaram prevalência semelhante à descrita na literatura, sendo a presença de fendas palpebrais oblíquas uma constante, o achado de linha de flexão palmar única (linha simiesca) presente em cerca da metade dos casos, e a cardiopatia mais freqüente a Comunicação Interatrial. O hipotireoidismo mostrou freqüência de 8,2%, semelhante ao descrito na literatura.
CONCLUSÕES: as características clínico-demográficas do grupo de pacientes estudados assemelham-se ao descrito na literatura, excetuando-se os achados referentes à idade materna e grupo racial. Tais aspectos deverão ser mais estudados no futuro. O seguimento dos pacientes ao longo do seu desenvolvimento poderá evidenciar outras diferenças.
PALAVRAS-CHAVES: Síndrome de Down, Deficiência mental, Fenótipo, Malformações
Abstract OBJECTIVE: to show the clinical and
demographic aspects of clinical follow-up of 87 children in a outpatient program at the Ambulatório de Genética (Hospital Universitário Professor Edgard Santos), looking for a better knowledge of Down Syndrome in Bahia.
METHODS: The sample constituted of 87 patients with Down Syndrome (SD) of age ranging 0 to 20 years. Statistical analysis performed included evaluation of central tendency and variability measures.
RESULTS: there was no significant variation among gender. Maternal age was lower than in international literature; meanwhile, it was similar to the results found in South America. We found a predominance of white children, not described in other publications. The major malformations showed a pattern similar to that described in literature (congenital cardiopathy, slant palpebral fissures, simiam crease). The hypothiroidism showed prevalence similar to those seen in literature.
CONCLUSIONS: the clinical and demographic patterns of the group of patients studied resembles to that seen in the international literature, except those related to maternal age, and racial group. These aspects should be better studied in future. The continuing follow-up of these children might show other developmental differences. KEY WORDS: Down Syndrome, Mental Deficiency, Phenotype, Malformations. 1Pediatra, Mestranda em Medicina Interna, UFBA 2Pediatra, Mestrando em Epidemiologia Clínica, UFBA 3Acadêmica de Medicina, UFBA 4Psicóloga, Psicopedagoga e Psicomotricista, APAE-Salvador 5Cardiopediatra do Hospital Santo Amaro, Mestre em Medicina pela UFBA 6Doutora, Professora Titular de Neonatologia da Faculdade de Medicina da Universidade Federal da Bahia (UFBA)
83

INTRODUÇÃO A Síndrome de Down (SD), caracterizada
citogeneticamente pela presença do cromossomo 21 em triplicata (trissomia do 21[1]), constitui-se na cromossomopatia mais comum na espécie humana, e na primeira causa genética de retardo mental.[2-4] Estima-se que sua prevalência global esteja em torno de um afetado para cada 700 nascidos vivos. Foi inicialmente reconhecida como uma entidade isolada em 1866 por John Langdon Down, um pediatra inglês, cuja descrição das características clínicas permanece atual, mais de um século depois[5].
Além do fenótipo clássico e bem conhecido, está bem estabelecida a morbi-mortalidade mais elevada destas crianças, quando comparadas à população geral, especialmente na primeira infância, o que se deve tanto às malformações (em especial a cardiopatia) como a outras complicações clínicas (pneumonias, doenças mieloproliferativas, etc)[6-8].
No presente estudo, o primeiro realizado com pacientes portadores de SD no estado da Bahia, no qual objetivou-se traçar um perfil clínico-demográfico destas crianças, avaliamos a freqüência das características fenotípicas principais, das malformações maiores (como a cardiopatia) e das complicações clínicas potencialmente incapacitantes (e, principalmente, tratáveis). Procuramos, também, avaliar os aspectos demográficos relacionados a estes pacientes e suas famílias, assim como tentar avaliar o impacto do nascimento da criança com SD sobre a estrutura familiar.
MATERIAL E MÉTODOS Trata-se de um estudo cujo desenho foi
observacional, descritivo, com uma amostra de 87 pacientes, com idades variando de 7 dias a 18 anos, que apresentam quadro clínico compatível com SD (foram excluídos pacientes cujo quadro clínico fosse duvidoso e nos quais não foi possível a confirmação da suspeita
através da citogenética), atendidos no Serviço de Genética do Hospital Universitário Professor Edgard Santos (HUPES) – Universidade Federal da Bahia (UFBA). O ambulatório conta com a atuação de pediatra, geneticista, citogeneticista, neuropediatra e psicólogo[9], havendo ainda a realização de avaliação cardiológica (rotineiramente).
O atendimento clínico foi realizado através da utilização de protocolo específico, de maneira prospectiva, e em seguida mantido o acompanhamento, de acordo com a necessidade, à medida que iam sendo realizadas as avaliações complementares[10-14]. O aconselhamento genético foi iniciado na primeira consulta, e confirmado quando da realização do cariótipo (especialmente no caso de mães jovens)[15, 16].
As variáveis de interesse estabelecidas foram agrupadas em três grupos - demográficas, incluindo idade da criança ao iniciar o acompanhamento, procedência, gênero, grupo racial da criança e da mãe, idade materna à época da gestação, renda familiar (em salários mínimos), condições de saneamento da habitação, dados relativos ao parto (tipo, idade gestacional e antropometria ao nascimento); clínicas, como fenda palpebral oblíqua, pregas epicânticas e linhas simiescas, avaliação citogenética e avaliações especiais (ecocardiográfica, tireoidiana, hematológica, e ultra-sonografia abdominal); e psicossociais, referindo-se ao desejo da gestação, à maneira como os pais avaliaram a maneira como foi exposto o diagnóstico pelo profissional que inicialmente avaliou a criança, à preocupação inicial dos pais quanto ao futuro da criança e à intenção do casal em ter outros filhos.
Todas as crianças foram submetidas à anamnese e ao exame clínico direcionados. Quanto ao grupo racial foram classificadas de acordo com as normas de Krieger e Azevedo[17]. Os controles hematológicos e tireoidianos foram realizados semestralmente.
A análise estatística incluiu medidas de tendência central e de dispersão, sendo os dados analisados através do software SPSS (Versão 7,0).
O presente trabalho foi aprovado pelo Comitê de Ética do Hospital Universitário Professor Edgard Santos.
84
Ney Cristian Amaral Boa Sorte
Ser mais informativa sobre a síndrome com parágrfao curtos e sem dar muitos detalhes, como que Down era pediatra inglês, que permanece atual o que ele descreveu, que delineou as bases da reabilitação – Isto é um artigo. Falar mais com menos.
Ney Cristian Amaral Boa Sorte
Parágrafo sem nexo que não expõe uma idéia clara.
Ney Cristian Amaral Boa Sorte
“procuramos traçar” – linguagem de sonorização ruim. Talvez obteve-se um perfil, procurou-se caracterizar estas crianças, avaliando..., ou outros...
Ney Cristian Amaral Boa Sorte
Talvez ser mais descritiva dos objetivos. “ avaliando a freqüência das características fenotípicas, alterações cardiológicas, oftalmológicas, ultrassonográficas, tireoidianas, ... e o impacto do nascimento da criança na estrutura familiar. \(talvez o rudimentar seje desnecessário aqui\).

RESULTADOS Entre as 87 crianças estudadas, 51 (58,6%)
são do gênero masculino, enquanto que 36 (41,4%) pertencem ao gênero feminino. A idade dos pacientes à época do início do acompanhamento variava de 0 a 18 anos, com 32% abaixo de dois meses e 82,8% abaixo de dois anos, com média de 16,74 meses e mediana de sete meses. Quarenta e seis pacientes (52,9%) procediam da capital (Salvador), 15 (17,2%) da região metropolitana e 26 (29,9%) do Interior do Estado.
Quanto ao grupo racial, 34 (39,1%) foram classificados como brancos; 26 (29,9%), como mulatos claros; 25 (28,7%), como mulatos médios; dois (2,3%) como mulatos escuros e nenhum como preto. No que diz respeito às mães, 14 (17,3%) pertenciam ao grupo branco, 20 (24,7%) ao mulato claro, 36 (44,4%) ao mulato médio, nove (11,1%) ao mulato escuro e duas (2,5%) ao preto. Em nove casos (6.9%) esta informação não estava disponível. O grupo racial dos pais não foi incluído, uma vez que estes raramente acompanhavam as crianças à consulta, não sendo possível então sua classificação racial.
A idade materna na época do nascimento da criança com SD variou de 15 a 50 anos, com média de 30,11 anos e mediana de 31 anos. 58,6% das mães tinham menos de 34 anos quando seus filhos nasceram. Apenas 3,3% tinham mais de 45 anos. 40,2% das mães tinham idade inferior a 28 anos à época do parto.
A renda familiar média das famílias esteve em 1,36 salário-mínimo, com mediana de um salário-mínimo, e desvio-padrão de 1,46. O saneamento básico estava presente em 60 (71,4%) e ausente em 24 (28,6%) residências.
Os dados referentes ao nascimento incluíram o tipo de parto (normal em 65,1% dos casos, sendo pélvico em apenas um caso; fórceps em 2 casos; cesárea em 25 casos, sendo dois pélvicos). A antropometria ao nascimento revelou peso médio de 2.988g (mediana 2,910g), estatura média de 47,2cm (esta informação só esteve disponível – através de informação da mãe ou documentos da maternidade – em 65,5% dos casos), perímetro cefálico (informação disponível em apenas 29,9% dos casos) médio 32,5 cm. Questionadas sobre a idade gestacional, as
genitoras (o percentual de informação médica neste sentido foi irrelevante) informaram parto a termo em 64,4% dos casos e pré-termo em 17,2% dos casos.
As alterações fenotípicas avaliadas encontram-se descritas na tabela 1. O ecocardiograma foi obtido em 89,3% (não realizado apenas nos pacientes que não mantiveram o acompanhamento), o cariótipo em 24,1% dos casos, a avaliação hematológica em 69% dos casos, avaliação tireoidiana em 82% dos casos, ultrassonografia abdominal em 59.8% dos pacientes e a avaliação oftalmológica em 44,8%.
TABELA 1 – Alterações Fenotípicas ALTERAÇÃO N0 % Fenda palpebral
oblíqua para cima 87 100
Epicanto 52 59,8Linha simiesca 28 32,2 Unilateral 5 5,7 Bilateral 23 26,4Cardiopatia 39 44,9 A distribuição das cardiopatias encontra-se
descrita na Tabela 2. TABELA 2 – Alterações
Ecocardiográficas CARDIOPATIA N0 % CIA 9 11,5 CIV 3 3,8 Defeito AV total 2 2,6 Tetralogia de Fallot 2 2,6 PCA 3 3,8 CIA+CIV 3 3,8 CIA+PCA 4 5,1 CIV+PCA 3 3,8 Defeito AV
intermediário 3 3,8
Forame Oval Patente 2 2,6 Outros 5 6,4 Ecocardiograma
normal 39 50
78 100
85
Ney Cristian Amaral Boa Sorte
Litoral Norte não é região metropolitana de Salvador
Ney Cristian Amaral Boa Sorte
Talvez seja interessante falar o percentual de mães jovens \(menos de 25 anos\).

A avaliação da função tireoidiana foi
realizada em 82 (94,7%) pacientes, e hipotireoidismo foi detectado em sete (8,2%) casos. As avaliações especiais incluíram ultra-sonografia abdominal (realizada em 59,8% dos casos) e a avaliação hematológica (disponível no momento da análise em 69% dos casos). Estes dados encontram-se descritos nas Tabelas 3 e 4.
TABELA 3 – Ultra-sonografia Abdominal ALTERAÇÃO N0 % Neoplasia 1 1,9 Malformações de
vísceras 1 1,9
Alterações das vias biliares
2 3,8
Alterações hepáticas 4 7,7 Outros achados 3 5,8 Ultra-sonografia
normal 39 75
50 100 TABELA 4– Avaliação Hematológica ALTERAÇÃO N0 % Anemia não
especificada 6 10
Anemia ferropriva 7 11,7 Hemoglobinopatia 4 6,7 Leucose 1 1,7 Hemograma normal 37 61,7 55 100 O estudo Citogenético (Cariótipo) foi
realizado em 21 casos (24,1%), tendo evidenciado trissomia livre em 100%, sendo encontrados 02 casos de mosaicismo, ambos com mais de 40% de células trissômicas e sem diferenças fenotípicas em relação aos outros casos.
Questionados sobre como avaliariam a maneira como lhes foi dada a notícia de que
sua criança era portadora da SD, pelo profissional de saúde que primeiro atendeu a criança, os pais a classificaram como clara, em 34,1% dos casos, pouco informativa, em 54,9% dos casos e pejorativa em 11% dos casos. Sobre a questão de se a gestação desta criança havia sido desejada, 24 (27,9%) responderam que sim, e 62 (72,1%), que não. Sobre o desejo de prole subseqüente, sete (8%) afirmaram desejar ter outros filhos, enquanto que 75 (86,2%) relataram não desejar outros filhos, e 5 (5,7%) não tinham opinião formada.
Vinte e quatro por cento dos pais entrevistados colocaram como principal preocupação inicial com a criança a aquisição de habilidades motoras. Múltiplas preocupações, não sendo os pais capazes de defini-la, foram relatadas por 19,5% dos pais, preconceito da sociedade, por 14,6%, habilidade intelectual, por 6,1%, independência, por 4,9%, cardiopatia por 4,9%, comportamento social por 1,2% e infecções por 1,2% dos pais entrevistados. 8,5% dos pais afirmaram não terem preocupações em relação à criança e 13,8% relataram preocupações variadas.
DISCUSSÃO
Ao longo do estudo, observamos discreta
predominância do gênero masculino, o que é compatível com a literatura descrita[18-22]. A idade de início do acompanhamento foi variada, com mediana (mais fidedigna que a média, devido a influência que esta sofre de medidas extremas) de sete meses. Tal idade encontra-se acima do ideal, uma vez que seria desejável que estas crianças iniciassem o acompanhamento ainda no primeiro mês de vida[12]. A maioria das crianças procedia da capital do Estado; entretanto, um número significativo (29,9%) era residente no interior do estado, o que denota a falta de assistência especializada nestas regiões.
Quanto ao grupo racial, foi notado evidente predomínio de crianças brancas e mulatas claras (69%), e ausência de crianças negras. Tal achado não reflete a distribuição racial das crianças habitualmente atendidas na Instituição. Entretanto, visto não haver relatos de diferença na incidência de SD entre os
86

grupos raciais, a possibilidade mais verossímil é que tal distribuição seja devida a fatores socioeconômicos e culturais, que possibilitariam a grupos mais favorecidos um melhor acesso à Saúde. Entrementes, a renda familiar média foi de 1,6 salário mínimo.
Outro dado de interesse refere-se à idade materna, com média de 30,11 anos, sendo mais de 58% das mães com idade inferior a 34 anos (limite que classicamente marca o início do risco aumentado para trissomias). Este dado, embora não compatível com populações do primeiro mundo[20, 23], assemelha-se a outros estudos realizados no Brasil e América Latina, com possíveis motivos culturais (como sugerido por Beigelman e colaboradores no sudeste do Brasil em 1996, e Oteiza e colaboradores em Cuba, 1998), como a idade mais precoce do início da procriação e a ausência de programas efetivos de planejamento familiar[19, 24-27]. Entretanto, inferências mais exatas sobre este tópico não podem ser realizadas no momento, uma vez que trata-se de amostra direcionada. Para tal, seria necessária a realização de um estudo de prevalência com base populacional, para que fosse possível avaliar a real freqüência de gestações afetadas pela trissomia do 21 em cada faixa etária.
Os dados referentes ao nascimento incluíram peso, apresentação, estatura, e perímetro cefálico. O peso e a estatura encontrados foram compatíveis com a literatura; entretanto, a apresentação pélvica, descrita como mais comum na SD, não foi prevalente neste estudo (apenas 01 caso), e os dados de PC, assim como a idade gestacional em semanas (a maioria das mães referiu parto a termo, não havendo informação escrita), foram muito escassos, por falta de informação. Este último item aponta, mais uma vez, para a necessidade de abordagem mais adequada nas maternidades.
O Estudo Ecocardiográfico identificou algum tipo de cardiopatia em cerca de 45% dos pacientes, sendo a mais comum a Comunicação Interatrial (CIA), seguida da associação de CIA com Persistência do Canal Arterial (PCA), e em terceiro lugar, com a mesma freqüência, Comunicação Interventricular (CIV), CIA+CIV, CIV+PCA e Defeito atrioventricular intermediário. As outras cardiopatias tiveram freqüências
menores, incluindo o Defeito Atrioventricular total em 2 casos, PCA em 3 casos e Tetralogia de Fallot em 2 casos. Embora a freqüência de cardiopatia se assemelhe significantemente ao descrito na literatura disponível, chama a atenção a preponderância da Comunicação Interatrial (CIA), quando a literatura nacional e internacional mostra a Comunicação Interventricular (CIV) isolada como o defeito cardíaco mais comum na SD[2, 28, 29]. Foram encontrados dois casos de tetralogia de Fallot (2,6%), o que também não é comum na bibliografia disponível. Em ambos os casos, o aumento no tamanho da amostra poderá evidenciar novos padrões. No caso específico da CIA, seria necessário levar em consideração a possibilidade de defeitos septais comuns no período neonatal, que se resolvem espontaneamente e não estariam relacionados à SD. É possível que uma estratificação por faixa etária venha a mostrar resultados diferentes.
A avaliação tireoidiana detectou hipotireoidismo em 7 (8,2%) casos entre os submetidos a esta avaliação, sendo dois congênitos. Tal taxa é bastante significativa, sendo semelhante a descrita na literatura e reforça a necessidade de avaliações tireoidianas rotineiras, uma vez que o hipotireoidismo não tratado piora consideravelmente a performance, tanto motora quanto cognitiva, da criança com SD[2, 3, 21, 28].
A ultrassonografia abdominal mostrou alterações sem relevância clínica em 04 casos, porém funcionou como método diagnóstico de triagem para neoplasia em 01 caso e litíase biliar em outros dois, além de alterações hepáticas (transitórias) em quatro. Embora com uma freqüênia baixa de achados patológicos, estes foram de grande importância, uma vez que o tratamento (cirúrgico) de neoplasia foi curativo, e a detecção precoce de litíase biliar permitiu o seguimento e rastreamento de complicações, como a colangite, que é uma patoloia grave e potencialmente fatal no portador de SD. Embora não seja consenso, alguns autores já recomendam a realização rotineira da ultrassonografia abdominal total [22].
A avaliação hematológica mostrou apenas um caso de doença mieloproliferativa. Entretanto, uma vez que a grande maioria das
87

crianças apresentava baixa idade à época do estudo, é possível que tal freqüência venha a aumentar com o seguimento[8].
Os aspectos psicossociais relacionados às crianças e seus pais, e os dados indicativos de uma relação médico paciente inadequada (atitude pejorativa, pouco esclarecimento, orientação inadequada) enfatizam a necessidade da atuação de equipe multidisciplinar, incluindo e profissional da psicologia, assim como a educação continuada dos profissionais de saúde que lidam com a perinatologia[9, 30].
CONCLUSÕES A avaliação realizada incluiu a
investigação das principais características associadas à Síndrome de Down. Em nosso Serviço, incluiu também rotineiramente exame oftalmológico, avaliação otorrinolaringológica e avaliação ortopédica, cujos achados não foram incluídos neste trabalho por motivos estatísticos.
Chamamos a atenção para a virtual ausência de casos cuja cardiopatia tenha sido investigada no período neonatal, procedimento que deveria ser rotina em todas as maternidades.
A avaliação citogenética é fundamental para a confirmação diagnóstica e o aconselhamento genético dos casais, especialmente no caso de mães jovens[16]. Contudo, o diagnóstico pode ser clínico na maioria das vezes, estando o profissional médico adequadamente treinado e atento[31]. A baixa realização de cariótipo em nosso Serviço, em conseqüência de problemas não relacionados ao atendimento clínico, deve melhorar com o seguimento e o estabelecimento de linhas de pesquisa.
A utilização de um protocolo clínico específico para crianças portadoras de síndrome de Down revelou-se um instrumento valioso, tanto no conhecimento das características clínicas da população, como na possibilidade de realizar detecção precoce de complicações, que poderão ser tratadas, com impacto positivo na morbi-mortalidade, permitindo a estas crianças um crescimento saudável e a oportunidade de integração sócia,
com conseqüente melhora na qualidade de vida[6, 10-14, 18].
É importante salientar que os dados aqui descritos referem-se a uma amostra selecionada em um Serviço de Referência. Há a necessidade de ampliação destes estudos, com uma amostra mais ampla de base populacional.
Resta lembrar que ao profissional da genética clínica cabe a confirmação diagnóstica, o aconselhamento genético e o fornecimento da orientação antecipatória e de eventuais conhecimentos que se façam necessários. É o Pediatra o profissional responsável pelo cuidado destas crianças, seu acompanhamento e seu encaminhamento para uma entrada saudável na vida adulta.
AGRADECIMENTOS
Agradecemos a valiosa contribuição do Laboratório de Genética Médica do Hospital Universitário Professor Edgard Santos da Universidade Federal da Bahia, pelo seu papel da nossa formação e apoio na criação e manutenção de um ambulatório direcionado a portadores de Síndrome de Down. Agradecemos também à SER DOWN – Associação Baiana de Síndrome de Down, pela doação do equipamento de microscopia que permitiu a realização dos exames de cariótipo e pela participação constante de seus associados ao longo do nosso trabalho.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Hattori, M.F., A; Taylor, TD; Watanabe, H; Yada, T; Park, HS; Toyoda, A et al, The DNA sequence of human chromosome 21. 2000, Nature.
2. Mustacchi, Z., Síndrome de Down, in Genética Baseada em Evidências: Síndromes e Heranças, Z. Mustachi, Editor. 2000, CID Editora: São Paulo. p. 817-94.
3. McKusick, V., Trisomy 21. 2001. 4. Leshin, L., Trisomy 21: The Story of
Down Syndrome. 2001. 5. Down, J., Observations on an ethnic
classification os idiots. 1866. 6. Thase, M., Longevity and mortality in
Down´s syndrome. Journal of Mental
88

Deficiency Research, 1982. 26: p. 177-92.
7. Burns, D.E., SI, As alterações imunológicas na Síndrome de Down, in Genética Baseada em Evidências - Síndromes e Heranças, Z. Mustachi, Editor. 2000, CID Editora: São Paulo. p. 558-93.
8. Bertolini, D.V., MSS; Fisberg, M, Morbimortalidade em Indivíduos Portadortes da Síndrome de Down. Jornal Brasileiro de Medicina, 1991. 61(3): p. 13-25.
9. Sadir, M.A. Síndrome de Down:acompanhamento psicológico integrado à consulta do geneticista. in XI Congresso Brasileiro de Genética Clínica. 1999. Salvador: SBGC.
10. Aracena, A., Supervisíon de la salud en personas com síndrome de Down. Rev Chil Pediatr, 1993. 64(1,supl): p. 20-23.
11. Cronk, C.C., AC; Pueshel, SM; Shea, AM; Zackai, E; Pickens, G; Reed, RB, Growth Charts for Children With Down Syndrome: 1 Month to 18 Years of Age. Pediatrics, 1988. 81(1): p. 102-10.
12. Foreman, P., Paediatric Manegement Pratices in Down Syndrome: a follow-up survey. Journal of Paediatric Children Health, 1993. 29(1): p. 27-31.
13. Saenz, R., Primary Care of Infants and Young Children with Down Syndrome. American Family Physician, 1999. 59(2): p. 381-90.
14. Seashore, M.C., S; Desposito, F; Sherman, J; Wappner, RS; Wilson, MG, Health Supervision for Children With Down Syndrome. Pediatrics, 1994. 93(5): p. 855-59.
15. Penrose, L.S., The Relative Effects of Parental and Maternal age in Mongolism. Journal of Genetics, 1933. 27: p. 219.
16. Benke, P.C., V; Donahue, R, Risk and Recurrence Risk of Down Syndrome. 1995.
17. Krieger, H.M., NE; Mi, MP; Azevêdo, E; Freire-Maia, A; Yasuda, N, Racial admixture in north-eastern Brazil.
Annals of Human Genetics, 1965. 29: p. 113-25.
18. Castilla, E.R., M; Dutra, MG; Lopez-Camelo, JS; Campaña, H; Paz, JE; Orioli, IM and the ECLAMC_Downsurv Group, Survival of Children With Down Syndrome in South America. American Journal of Medical Genetics, 1998. 79: p. 108-11.
19. Molina, J.A., NA; Garcia, TR; Castellanos, EA, Síndrome de Down, prevalencia en 18,509 nacimientos consecutivos y frecuencia de 11 características morfológicas externas. Revista Médica del Instituto Mexicano del Seguro Social, 1993. 31(4): p. 263-4.
20. Mutton, D.A., E; Hook, EB, Cytogenetic and epidemiological findings in Down syndrome, England and Wales 1989 to 1993. Journal Medical Genetics, 1996. 33: p. 387-94.
21. Hayes, A.B., ML, Down Syndrome. Pediatric Clinics of North America, 1993. 40(3): p. 523-35.
22. Boy, R.N., JGB; Vargas, FR; Fontana, C; Almeida, JCC; Jr. Lherena, J, Síndrome de Down - análise clínica, citogenética e epidemiológica de 165 casos. Jornal de Pediatria, 1995. 71(2): p. 88-92.
23. Cuckle, H., Maternal age-standardisation of prevalence of Down´s Syndrome. The Lancet, 1999. 354: p. 529-30.
24. Bieguelman, B.K., H; Silva, LM, Maternal age and Down Syndrome in Southeastern Brazil. Brazilian Jornal of Genetics, 1996. 19(4): p. 637-40.
25. Boa Sorte, N. Perfil dos Partos entre adolescentes na Rede SUS da Bahia entre 1998 e 2000. in VII Congresso Brasileiro de Adolescência. 2001. Salvador.
26. Lima, G.C., MEZ; Frantz, BC; Leite, JCL; Giugliani, R, Síndrome de Down: características clínicas, perfil epidemiológico e citogenético em recém-nascidos no Hospital de Clínicas de Porto Alegre. Revista Associação Médica do Rio Grande do Sul, 1996. 40(1): p. 8-13.
89

27. Oteiza, M.L., FA; Muñiz, IC; Ortiz, JR; Valle, AP; Lloret, MCE, Tendencias del Síndrome de Down en Cuba. Su relación com edad materna y tasa de fecundidad. Revista Cubana de Pediatria, 1998. 70(3): p. 141-7.
28. Jones, K., Smith´s Reconizable Patterns of Human Malformation. 5a ed. 1997, New York: Saunders. 8-13.
29. Granzotti, J.P., ILC; Amaral, FTV; Nunes, MA, Incidência de cardiopatias congênitas na Síndrome de Down. Jornal de Pediatria, 1995. 71(1): p. 28-30.
30. Jijón, M., Sindrome de Down. Pautas mínimas para su entendimiento y
atencíon. 1a ed. Genética. 1997, Quito: Ed. Ecuaoffset Cía Ltda. 123.
31. Rex, A.P., M, A diagnostic index for Down syndrome. The journal of Pediatrics, 1982. 100(6): p. 903-6.
Endereço para correspondência:
Dra Tatiana Amorim Hospital Universitário Professor Edgard
Santos - Laboratório de Genética Médica – 6o andar
e-mail: tatiamorim @ig.com.Br Fone: (71) 339 6304/334 1105
90


















