
AVALIAÇÃO DA PREVALÊNCIA DE INDIVÍDUOS … · cromossomo 16 estão inclusos genes que...
56
Pró- Reitoria de Graduação Biomedicina AVALIAÇÃO DA PREVALÊNCIA DE INDIVÍDUOS PORTADORES DE BETA TALASSEMIA NO HOSPITAL DAS FORÇAS ARMADAS DE BRASÍLIA- DF Camilla Eliza Wolf Sonza Orientadora: Msc.Cintia do Couto Mascarenhas Co-orientador: Msc. Paulo Roberto Sabino Jr. Brasília - DF 2010 Co-orientador : Esp. Fábio de França Martins
Transcript of AVALIAÇÃO DA PREVALÊNCIA DE INDIVÍDUOS … · cromossomo 16 estão inclusos genes que...

Pró- Reitoria de Graduação
Biomedicina
AVALIAÇÃO DA PREVALÊNCIA DE INDIVÍDUOS PORTADORES DE BETA TALASSEMIA NO HOSPITAL DAS
FORÇAS ARMADAS DE BRASÍLIA- DF
Camilla Eliza Wolf Sonza
Orientadora: Msc.Cintia do Couto Mascarenhas
Co-orientador: Msc. Paulo Roberto Sabino Jr.
Brasília - DF
2010
Co-orientador : Esp. Fábio de França Martins

CAMILLA ELIZA WOLF SONZA
AVALIAÇÃO DA PREVALÊNCIA DE PORTADORES DE BETA TALA SSEMIA NO HOSPITAL DAS FORÇAS ARMADAS DE BRASÍLIA – DF.
Projeto de TCC II apresentado ao curso de
graduação em Biomedicina da Universidade
Católica de Brasília como requisito para obtenção
do título de Bacharel em Biomedicina.
Orientadora: Msc. Cíntia do Couto Mascarenhas
Co-orientadores: Msc. Paulo Roberto Sabino Jr.
Esp. Fábio de França Martins
Brasília
2010

Projeto de TCC II, de autoria de Camilla Eliza Wolf Sonza, intitulado “Avaliação da
prevalência de indivíduos portadores da Beta Talassemia no Hospital das Forças Armadas
de Brasília- DF”, apresentado como requisito para a obtenção do grau de Bacharel em
Biomedicina da Universidade Católica de Brasília, em 04 de novembro de 2010, aprovada
pela banca examinadora abaixo assinada:
____________________________________________________
Prof. Msc. Cíntia do Couto Macarenhas
Orientadora
Doutoranda pela faculdade de ciências médicas da UNICAMP – Pesquisadora voluntária da UCB
_____________________________________________________
Prof. Dr. Anderson Ferreira da Cunha
Professor Adjunto da Universidade Federal de São Carlos
____________________________________________________
Prof. Dra. Rosângela Vieira de Andrade
Professor da Universidade Católica de Brasília
Curso de Biomedicina – UCB
Brasília
2010

RESUMO
SONZA, Camilla. Avaliação da prevalência de portadores da Beta Talassemia no
Hospital das Forças Armadas de Brasília- DF. Brasília. 2010 (57). Trabalho de conclusão
de curso de Biomedicina. Universidade Católica de Brasília.
As hemoglobinopatias são alterações genéticas relacionadas aos genes responsáveis pela síntese da hemoglobina. Com extensa distribuição mundial, principalmente na população do Mediterrâneo, consideradas problemas de saúde pública em muitos países, inclusive no Brasil, as hemoglobinopatias são divididas de acordo com suas alterações estruturais ou funcionais da hemoglobina, sendo identificadas como hemoglobinas variantes e talassemia, respectivamente. As talassemias são qualificadas pela pequena síntese ou ausência da síntese de cadeias globínicas que compõem a hemoglobina, e classificadas conforme a cadeia globínica afetada. Quando se tem uma alteração na cadeia beta da globina, caracteriza-se como beta talassemia, podendo esta apresentar-se de forma assintomática ou sintomática, o que conduz à necessidade de seu conhecimento e diagnóstico. Até o presente momento não há estudos publicados referenciando a prevalência de beta talassemia em Brasília- DF, o que evidencia a importância desta pesquisa. A escolha da referida localidade para ser a capital federal gerou correntes migratórias de várias regiões do nosso país, tornando-a uma área de grande miscigenação, indicando uma possível frequência desta patologia. O objetivo do trabalho foi avaliar entre o período de maio a agosto de 2010, a presença de indivíduos portadores de beta talassemia, em todos os níveis incluindo traço talassêmico. Para isso consideraram-se no estudo 129 pacientes e seis controles com alterações hematológicas ou que apresentaram requisição médica para o exame de eletroforese de hemoglobina por capilaridade, atendidos no laboratório de análises clínicas do Hospital das Forças Armadas de Brasília. O critério de inclusão foi baseado em alterações hematológicas sugestivas de beta talassemia observadas no hemograma, na eletroforese de hemoglobina em pH ácido e alcalino e pela quantificação de hemoglobinas através da eletroforese de hemoglobina por capilaridade. A prevalência encontrada de indivíduos sugestivos de portar traço beta talassêmico neste grupo de estudo, foi de 2,32%. Entretanto, não foram identificados pacientes portadores de beta talassemia maior ou intermediária. Conclui-se que não há grande transferência de genes alterados na população, reforçando a grande variabilidade étnica que há no Brasil em relação à hemoglobinopatias. Entretanto, é necessário realizar novos estudos com um universo amostral maior para determinar a prevalência desta doença na população de Brasília.
Palavras-chave: Hemoglobinopatias. Beta talassemia. Prevalência.

ABSTRACT
Hemoglobinopathies are genetic alterations relate on genes responsible for synthesis of hemoglobin. With an extensive worldwide distribution, mainly in the Mediterranean population, considered public healthy matter in several countries, including Brazil, hemoglobinopathies are divided according to their structural or functional changes of hemoglobin, identified as hemoglobin variants and thalassemia, respectively. Thalassemias are classified by absence or short synthesis of globin chains that compound hemoglobin, and classified according to the affect globin chain. The change in beta globin chain is characterized as beta thalassemia, which can be symptomatic or asymptomatic, what leads to the necessity of its perception and diagnosis. To date, there are no published studies referencing this disease prevalence in Brasília – DF, what glares the importance of this research. The choice of the mentioned location to be the capital of Brazil gave rise to a lot of migratory waves, making it an area of great miscegenation, suggesting a possible frequency of this pathology. The purpose of this study was evaluate between May - August 2010, the presence of individuals with beta thalassemia, in all levels including thalassemic traits. For this study were considered 129 patients and six controls with hematologic changes or had a medical requisition for the exam hemoglobin by capillary electrophoresis, that were attended at the clinical laboratory of Hospital das Forças Armadas de Brasília. The reckon criteria was based upon hematologic changes that suggested beta thalasemia observed in blood count, hemoglobin electrophoresis in acid and alkaline pH and hemoglobin electrophoresis capillary. The prevalence of individuals suggestive to be porting beta thalassemia traits in this population was 2.32%. However, there were no patients with beta thalassemia major or intermedia. We conclude there is no transfer of modified genes in this population, reinforcing the ethnic variability that exists in Brazil regarding hemoglobinopathies. However, it is necessary to perform new studies with a larger sampling universe, in order to define this disease prevalence in Brasília’s population
Keyword: Hemoglobinopathies. Beta thalassemia. Prevalence.

SUMÁRIO
1. INTRODUÇÃO 9
2. REFERENCIAL TEÓRICO 12
2.1. FISIOLOGIA E MANIFESTAÇÕES CLÍNICAS DA BETA TALASSEMIA 12
2.2. FORMAS CLÍNICAS DA BETA TALASSEMIA 16
2.2.1. Talassemia maior 16
2.2.2. Talassemia intermediária 17
2.2.3. Talassemia menor (traço talassêmico) 18
2.3. PREVALÊNCIA 18
2.4. DIAGNÓSTICO 20
2.4.1. Diagnóstico do indivíduo homozigoto 21
2.4.2. Diagnóstico do indivíduo heterozigoto 22
2.5. TRATAMENTO 22
2.5.1. Tratamento para beta-talassemia maior 22
2.5.2. Tratamento da beta-talassemia intermediária 24
2.5.1.3. Tratamento da beta-talassemia menor 24
3. JUSTIFICATIVAS 25
4. OBJETIVOS 26
4.1. OBJETIVO GERAL 26
4.2. OBJETIVOS ESPECÍFICOS 26
5. MATERIAS E MÉTODOS 27
5.1. AMOSTRAS 27
5.2. AVALIAÇÃO DAS AMOSTRAS DOS PACIENTES 28

5.2.1. Contagem automatizada e análise microscópica 28
5.2.2. Eletroforese de hemoglobina em pH ácido e alcalino (gel de agarose CELM-150). 29
5.2.3. Eletroforese de hemoglobina por capilaridade (MINICAP®) 30
6. ASPECTOS ÉTICOS 33
7. RESULTADOS 34
7.1. RESULTADOS DA SÉRIE VERMELHA DO HEMOGRAMA DOS PACIENTES SELECIONADOS PARA O ESTUDO A PARTIR DA ANÁLISE DO HEMOGRAMA 35
7.2. RESULTADO DOS 129 PACIENTES ANALISADOS ATRAVÉS DA ELETROFORESE DE HEMOGLOBINA POR CAPILARIDADE. 38
8. DISCUSSÃO 41
REFERÊNCIAS BIBLIOGRÁFICAS 45
ANEXOS 53
ANEXO A – TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO 53
ANEXO B - ELETROFORESE DE Hb EM pH ALCALINO 54
ANEXO C - ELETROFORESE DE Hb EM pH ÁCIDO 56

9
1. INTRODUÇÃO
As hemoglobinopatias são alterações hereditárias relacionadas aos genes responsáveis
pela síntese de globinas, formando assim hemoglobinas (Hb) anormais que levam ao
comprometimento funcional dos eritrócitos (SAKAMOTO et. al., 2008; VARGAS;
YAMAGUSHI, 2008).
Estas doenças têm ampla distribuição mundial, inclusive no Brasil, devido à grande
miscigenação da população. Podemos observar uma incidência variável (MELO et.al., 2008),
sobretudo nas regiões de Minas Gerais, Rio de Janeiro, São Paulo, região litorânea do
Nordeste, Mato Grosso, Mato Grosso do Sul e Goiás, devido à migração dos europeus e
asiáticos no Brasil, principalmente portugueses, italianos e espanhóis (SOUZA, 2006).
As hemoglobinopatias são divididas de acordo com suas alterações estruturais e
funcionais, sendo caracterizadas como hemoglobinas variantes e talassemias, respectivamente
(PATRINOS et. al., 2004; SAKAMOTO et al., 2008; PENMAN et al., 2009; SACHDEV et
al., 2010).
A hemoglobina é uma proteína constituída por quatro cadeias globínicas conexas a
grupos heme. Estes grupos são formados por um átomo de ferro em uma estrutura porfírica
(ZAMARO et al., 2002). Cada cadeia da globina é formada por uma sequência de
aminoácidos, sendo a cadeia alfa (α) composta por 141 aminoácidos e a cadeia beta (β) por
146 aminoácidos. As formas pelas quais as cadeias de proteínas se combinam geram
diferentes hemoglobinas presentes nos eritrócitos desde o período intra-uterino até a fase
adulta (GALIZA; PITOMBEIRA, 2003).
As mudanças que ocorrem na expressão dos genes estão relacionadas com a organização
destes grupos em dois clusters: no cromossomo 11 e no cromossomo 16 (ZAMARO, et.al.,
2002). Estão inclusos no cromossomo 16 os genes que sintetizam as cadeias zeta (ζ), alfa (α) e
teta (θ), já no cromossomo 11 são sintetizadas as cadeias épsilon (ε), gama (γA e γG), delta (δ)
e beta (β) (Figura 1.). Estes genes são ativados em ordem linear, da mesma forma em que são
distribuídos no cromossomo (SOUZA, 2006) (Figura 1.).

10
Durante o desenvolvimento ontogenético normal ocorrem mudanças na expressão dos
genes responsáveis pelos diferentes tipos de hemoglobinas humanas. No período embrionário
encontram-se as Hb Gower I (ζ2ε2), Gower II (α2ε2) e Portland (ε2γ2), no período fetal a
HbF (α2γ2), e na vida adulta a HbA (α2β2) e a HbA2 (α2δ2) (GALIZA; PITOMBEIRA,
2003; SOUZA, 2006) (Tabela 1.).
Figura 1. Organização dos genes, que compõem o grupo globina e que ao
se combinarem com a parte heme, formam a hemoglobina. No
cromossomo 16 estão inclusos genes que sintetizam cadeias zeta, alfa e
teta, enquanto no cromossomo 11 estão inclusos genes que sintetizam
cadeias épsilon, gama, delta e beta. FONTE: Naoum, 1990, adaptado.
Tabela 1. Representação dos diferentes tipos de hemoglobina relacionando
o principal período de produção e composição da cadeia globínica. FONTE:
GALIZA; PITOMBEIRA, 2003.
Tipo de
hemoglobina
Período preponderante
de síntese
Cadeias
globínicas
Gower-1 Embrião – até 3 meses ζ2ε2
Portland Embrião – até 3 meses ζ2γ2
Gower-2 Embrião – até 3 meses α2ε2
HbF Feto – até 6 meses α2γ2
HbA 2 Feto / Vida adulta α2δ2
HbA Vida adulta α2β2

11
As talassemias estão relacionadas a uma diminuição da síntese ou ausência da síntese
de cadeias globínicas, e são classificadas de acordo com a cadeia afetada, podendo apresentar-
se como alfa talassemia, beta talassemia, delta talassemia, delta-beta-talassemia ou gama-
delta-beta-talassemia (ZAGO, 2004). Quando a alteração ocorre na cadeia beta, caracteriza-se
uma beta talassemia, que faz parte de um grupo de doenças genéticas autossômicas recessivas
e que pode ser fatal, quando em homozigose (HAZIROLAM et al., 2009; AMINI et al., 2007;
DWIVEDI; KUMAR, 2007; ASSARI et.al., 2009; DSOUKY et al., 2009).
Na beta talassemia existem cerca de duzentas diferentes alterações moleculares
definidas, e podemos agrupá-las em: grandes deleções, pequenas deleções ou inserções e
mutações, sendo as mutações pontuais as mais freqüentes.
As mutações pontuais afetam a qualidade e a quantidade do mRNA produzido,
gerando uma tradução ineficaz (KIMURA et al., 2003; LONKAR et al., 2009;
CUNNINGHAM, 2010), tendo consequências diferentes de acordo com o local onde ocorre.
Estas mutações são classificadas de acordo com o comprometimento causado a regulação da
expressão do gene, podendo ser na transcrição, processamento do RNA ou tradução (THEIN,
2005).
O comprometimento da transcrição por alguma destas mutações envolvem sequências
de DNA que constituem o promotor da beta globina, resultando geralmente em uma
deficiência mínima na produção de β globina, podendo ser silenciosa em portadores de β
talassemia (THEIN, 2005).
O processamento alterado do DNA leva a formação de hemoglobinas com
funcionalidade prejudicada e assim originam a β talassemia, sendo esta maior, intemediária ou
menor (THEIN, 2005). Metade dos alelos de β talassemia comprometem diferentes estágios
da tradução do RNA, determinando uma talassemia β0. Isto ocorre devido ausência da
produção de cadeias β globínicas pela introdução prematura de códons de terminação
(THEIN, 2005).
Portanto, devido as alterações moleculares que ocorrem na beta talassemia, o
indivíduo pode ser heterozigoto (portador de um gene β talassêmico) ou homozigoto (portador
de dois genes β talassêmicos). No caso de homozigose as alterações moleculares podem

12
originar ausência completa ou apenas supressão parcial da síntese das cadeias β da globina,
denominando-se β0 e β+, respectivamente (ZAGO, 2004; THEIN, 2005).
A maior parte dos indivíduos, heterozigotos, é assintomática, tendo expectativa de
vida semelhante a dos indivíduos que não são portadores de hemoglobinas anormais. As
alterações são observadas através de exames laboratoriais (ESTEVÃO, 2009). Os indivíduos
homozigotos e os heterozigotos compostos (aqueles que carregam a herança de β talassemia
associada a outro defeito, funcional ou estrutural, na hemoglobina) têm manifestações clínicas
variando desde assintomáticos até as mais graves da doença (ZAGO, 2004).
2. REFERENCIAL TEÓRICO
2.1.FISIOLOGIA E MANIFESTAÇÕES CLÍNICAS DA BETA TALASSEMIA
Sollaino e colaboradores (2009) descrevem que as manifestações clínicas da beta
talassemia são variáveis e estão associadas à condição dependente de transfusão da beta
talassemia maior e intermediária, assim como o estado assintomático do indivíduo que possui
apenas o traço talassêmico.
Na beta talassemia maior as manifestações estão relacionadas diretamente a fatores
como: a intensidade da hemólise, os riscos relacionados às transfusões de hemácias
(CANÇADO, 2008), defeito da hemoglobinização, distúrbios proliferativos e metabólicos dos
eritroblastos, lesão da membrana eritrocitária e desequilíbrio da síntese de globina. (NAOUM,
1999; ZAGO, 2004).
Os principais sintomas dos pacientes com beta talassemia maior são astenia, palidez,
fraqueza muscular, hipodesenvolvimento físico e sexual, sopros no precórdio, taquicardia,
insuficiência cardíaca e maior susceptibilidade a infecções (NAOUM, 1990; ZAGO, 2004).

13
Como resultado da alteração molecular que ocorre na beta talassemia, há síntese
anormal de hemoglobina que leva ao excesso de cadeias α, tanto nos precursores eritróides,
quanto nos eritrócitos periféricos (SCOTT et, al., 1992). Estas cadeias α excedentes não são
viáveis para formar tetrâmeros e precipitam dentro do eritrócito gerando lesões na membrana,
além de causar comprometimento da mitose e do metabolismo celular desses eritrócitos
(ZAGO, 2004; THEIN, 2005; LITHANATUDOM et al., 2009).
Existem várias modificações celulares associadas aos eritrócitos dos indivíduos
talassêmicos, como hipocromia microcitose, anisopoiquilocitose e dano oxidativo da
membrana. A hipocromia e a microcitose são características encontradas tanto na anemia
ferropriva como nas talassemias (MATOS et. al., 2008). Já o dano oxidativo da membrana
pode ser potencializado devido ao ferro liberado pela degradação de cadeias alfa da
hemoglobina (VAN DENBERG et.al., 1993).
Também podemos observar nesta doença a diseritropoese que é caracterizada por
alterações morfológicas variadas nos eritroblastos, como núcleos com contornos irregulares,
segmentados ou em forma de trevo, células bi ou multinucleadas, micronúcleos e
fragmentação nuclear, pontes citoplasmáticas ou internucleares e vacuolização citoplasmática.
Observa-se ainda invaginação da membrana plasmática, acúmulo de ferro, agregados de
glicogênio, corpos de inclusão e alterações de membrana (ZAGO, 2004).
Os agregados da cadeia α podem ser visualizados nos citoplasmas dos eritroblastos pela
coloração com metilvioleta e com coloração panóptica, na qual as inclusões aparecem como
massas claras e homogêneas. Além disso, estas inclusões contribuem para a poiquilocitose por
meio de três mecanismos: lesão mecânica direta e formação de hemicromos aderidos à
membrana, alteração da estrutura de proteínas e peroxidação de lipídios e lesão da membrana
por fagocitose parcial quando os corpos de inclusão são retirados das células circulantes pelo
baço (NAOUM, P; NAOUM, F, 2004; ZAGO, 2004).
As anormalidades dos eritrócitos levam a uma anemia e eritropoese ineficaz, pois
ocorre a indução da apoptose na fase de eritroblasto policromático, devido à precipitação das
cadeias α e pelo acúmulo de ferro que levam à hemólise. A eritropoese ineficaz caracteriza-se
pela hiperplasia eritróide da medula óssea (CUNNINGHAM, 2010; LITHANATUDOM et
al., 2009, GALANELLO; ORIGA 2010)

14
A anemia é uma das manifestações clínicas mais importantes. Alguns pacientes
sintomáticos podem ter nível de hemoglobina mais elevado (ou próximo a normalidade),
quando comparados com pacientes com beta talassemia maior, (7 a 10 g/dL), sendo
classificados como talassêmicos intermediários. Já os pacientes com beta talassemia menor,
têm discreta redução dos níveis de hemoglobina e geralmente são assintomáticos.
Pacientes com beta talassemia maior apresentam uma produção eritróide aumentada
cerca de 7 a 30 vezes quando comparada ao indivíduo normal, na tentativa de compensar o
organismo da eritropoese ineficaz. As conseqüências são: aumento da absorção gastrintestinal
de ferro, alterações ósseas e desvio de nutrientes e energia alimentar para a MO
(LITHANATUDOM et al., 2009).
Entretanto, esta hiperplasia é ineficaz, pois a maioria das células produzidas são
destruídas ainda na MO (LITHANATUDOM et al., 2009; MELLOULI et.al., 2010). A
destruição celular contínua tem como conseqüência a liberação de enzimas intracelulares,
como a desidrogenase lática – DHL, e elevada produção de catabólitos derivados da
destruição de ácidos nucléicos e da hemoglobina, ácido úrico e bilirrubina, respectivamente
(LITHANATUDOM et.al., 2009; ZAGO, 2004).
Os eritrócitos que conseguem alcançar a circulação periférica são captados pelo baço,
que é sensível a qualquer tipo de alteração na membrana das hemácias, levando ao aumento
do baço. Então, a esplenomegalia ocorre devido à hiperplasia do sistema fagocítico pela
destruição de eritrócitos anormais, com ingurgitação e depósitos de ferro. O hiperesplenismo
ocorre devido à esplenomegalia, retendo e armazenando todos os tipos de células sangúineas,
reduzindo o número de eritrócitos, leucócitos e plaquetas. (SOUZA, 2006; ZAGO, 2004).
O aumento do ferro além de ser ocasionado pela hiperplasia da medula óssea, também
ocorre devido às transfusões sanguíneas, já que 2/3 do ferro do nosso organismo encontram-se
dentro das hemácias (ZAGO, 2004).
A sobrecarga do ferro, na ausência do tratamento, faz com que este se deposite na
forma de ferritina e hemossiderina em vários órgãos ou tecidos, como fígado, baço, medula
óssea, miocárdio e glândulas endócrinas, levando à hemocromatose (CANÇADO, 2008;
ESTEVÃO, 2009; ZAGO, 2004). A hemocromatose ocasiona alterações como o retardo no
crescimento e na maturidade sexual, anormalidades endocrinológicas, alterações cardíacas,

15
escurecimento da pele, lesões celulares e teciduais e o comprometimento hepático
(CANÇADO, 2008; LO; SINGER, 2002).
O ferro pode lesar vários tecidos, pois catalisa a reação que converte peróxidos de
oxigênio em íons de radicais livres, destruindo a membrana celular, proteínas e DNA,
causando hipertrofia cardíaca, dilatação e degeneração das fibras do miocárdio, provocando
injúria dos miócitos (HAZIROLAN et.al., 2009). Portanto, é importante que haja um
tratamento com quelante de ferro dos pacientes que apresentam níveis elevados de ferro
sérico, ferritina e transferrina (ZAGO, 2004).
Em um estudo realizado por Olivieri e colaboradores foi observada a importância da
determinação da ferritina sérica em pacientes beta-talassêmicos, já que estes autores
demonstraram que quanto maior o tempo que a ferritina for mantida menor que 2.500ng/mL,
menor a chance de desenvolver doença cardíaca e maior a sobrevida deste paciente
(OLIVIERI, 1999). Além disso, a sobrecarga de ferro pode contribuir para a osteopatia
talassêmica, pois provoca o hipoparatiroidismo e diminuição de vitamina C, que também pode
ocasionar osteoporose, escorbuto e hiperatividade dos osteoclastos (CANÇADO, 2008)
Além do aumento do ferro no organismo, a hiperplasia da medula óssea pode levar a
alterações endócrinas e o hipodesenvolvimento somático e sexual, pois esta consome muitos
nutrientes e energia, comprometendo o aporte para outros tecidos. O hipodesenvolvimento
somático e sexual se caracteriza por um menor crescimento pôndero-estatural na infância,
redução da massa muscular e ausência ou retardo da maturidade sexual na adolescência (LO;
SINGER, 2002). As alterações ósseas e articulares, observadas no rosto e crânio de indivíduos
portadores de beta talassemia maior, também são ocasionados devido a hiperplasia da medula
óssea (AMINI et al., 2007).
.

16
2.2. FORMAS CLÍNICAS DA BETA TALASSEMIA
2.2.1. Talassemia maior
Também conhecida como anemia do Mediterrâneo, anemia eritroblástica ou anemia de
Cooley, é a forma mais grave, sintomática e dependente de transfusão sanguínea. Com
manifestações clínicas presentes desde o primeiro ano de vida. A caracterização do indivíduos
como sendo portador de beta talassemia maior e intermediária é realizada pela frequência de
transfusões que esses pacientes recebem, bem como pela associação das manifestações
clinicas, relacionando-as aos exames laboratoriais que caracterizam o genótipo do indivíduo
(ZAGO, 2004).
As manifestações clínicas da beta talassemia maior são: anemia severa, episódios
recorrentes de febre, diarréia, irritabilidade, apatia, palidez e aumento progressivo do
abdômen devido à esplenomegalia, retardo no crescimento, icterícia, musculatura fraca,
úlceras nas pernas, desenvolvimento de massa extramedular e alterações ósseas decorrentes
da expansão da MO (CAO; GALANELLO, 2010; LIMA, 2001; LO; SINGER, 2002; ZAGO,
2004; THEIN, 2005). Estas alterações em decorrência da expansão da MO são: deformidades
nos ossos longos das pernas e alterações craniofaciais típicas – relevo do crânio, eminência
malar proeminente, inclinação mongolóide do olho, depressão da ponte do nariz, hipertrofia
maxilar que expõe os dentes superiores (GALANELLO; ORIGA, 2010).
O indivíduo acometido pode não apresentar anemia nos seus primeiros seis meses de
vida, devido ao nível de hemoglobina fetal presente. Porém com a diminuição dessa
hemoglobina instala-se uma anemia severa (CAO; GALANELLO, 2010), com hemoglobina
abaixo de 6,5g/dL (ZAGO, 2004). As células vermelhas apresentam alterações como
microcitose, hipocromia, anisiocitose, poiquilocitose e presença de eritroblastos no sangue
periférico (CAO; GALANELLO, 2010; LIMA, 2001).

17
2.2.2 Talassemia intermediária
É uma forma sintomática, porém de menor gravidade que a talassemia maior.
Apresentando níveis de hemoglobina entre 7 a 11 g/dL, e não dependem de transfusões
regulares (ZAGO, 2004; SOLLAINO et al., 2009).
As manifestações clínicas mais freqüentes da beta talassemia intermediária são:
palidez, icterícia, colestase, hepatoesplenomegalia moderada a severa, alterações faciais,
diminuição da massa muscular, úlceras crônicas nas pernas e anemia crônica (ELDOR;
RACHMILEWITZ, 2002; CAO; GALANELLO, 2010). Podem apresentar também cálculos
biliares, hipertrofia da medula eritróide com hematopoese medular e extramedular e suas
complicações – osteoporose, massas do tecido eritropoietico que afetam o baço, fígado,
linfonodos, tórax e coluna, e deformidades ósseas e alterações faciais (CAO; GALANELLO
2010).
Esta patologia resulta de combinações genéticas, como (THEIN, 2005):
- β+/β+ (homozigoto, com produção moderada de cadeia β, apresentando quantidade
moderada de HbA).
- β0/β0 (homozigoto, com produção moderada de cadeia - β, apresentando quantidade
moderada de HbA).
- β0/β+ (heterozigoto, com produção moderada de cadeia β, apresentando quantidade
moderada de HbA).
- Associação entre β+/δβ – talassemia;
- Presença de uma modificação adicional que reduz o excesso de cadeias α (co-herança
de α-talassemia) ou que aumenta a produção de cadeias γ (Persistência de HbF – PHHF);
- Heterozigose para o gene β- talassêmico particularmente grave.

18
2.2.3. Talassemia menor (traço talassêmico)
Normalmente estes pacientes são assintomáticos, apenas carregam o gene beta
talassêmico (heterozigotos) apresentando nível de hemoglobina ligeiramente diminuído em
algumas situações como na infância, na gravidez e na presença de infecções ou inflamação
crônica. (LO; SINGER, 2002; ZAGO, 2004).
Apesar de serem assintomáticos podem apresentar anemia, microcítica e hipocrômica,
além de aumento de HbA2 ( >4% ) (THEIN, 2005; YANG et.al., 2009). Ainda podem ter
redução do volume corpuscular médio (VCM) e hemoglobina corpuscular média (HCM),
entretanto não se observa eritroblastos presentes na circulação periférica (CAO;
GALANELLO, 2010).
2.3. PREVALÊNCIA
As hemoglobinopatias são doenças muito frequentes mundialmente, atingindo 7,0% da
população, sendo que a cada ano 300 a 400 mil recém-nascidos são acometidos por uma
forma mais severa (SOUZA, 2006).
De acordo com a Organização Mundial da Saúde 270 milhões de pessoas têm genes
que determinam hemoglobinopatias (SOUZA, 2006), e cerca de 90 milhões de pessoas têm
genes defeituosos de talassemia (DSOUKY et al., 2009). Hajibeiji (2009) cita que a cada ano
aproximadamente 60 mil bebês nascem no mundo com talassemia e Amini e colaboradores
(2007) relatam que 150 milhões de pessoas no mundo carregam genes beta-talassêmicos
(Figura 2.).

19
Figura 2. Distribuição geográfica das hemoglobinopatias como: talassemias, anemia falciforme,
hemoglobina E, hemoglobina C e hemoglobina D. FONTE: MANUAL DO EQUIPAMENTO SEBIA, 2010.
A beta talassemia ocorre mais frequentemente no Mediterrâneo, Norte e Oeste da
África, Índia, Oriente Médio e Ásia (AMINI et al., 2007; DSOUKY et al., 2009). No Egito
tem-se a beta-talassemia como causa mais comum de anemia hemolítica (DSOUKY et al.,
2009).
No Brasil temos um alto grau de miscigenação, devido à colonização por europeus e o
grande trabalho escravo vindo da África (VINCI DE MORAES, 2000; MELO et al., 2008),
principalmente para o Nordeste do Brasil, influenciando na prevalência de
hemoglobinopatias, e consequentemente da β-talassemia no país (DSOUKY et al., 2009).
Também contribuiu para o aumento da prevalência desta doença no Brasil, assim como todas
as outras hemoglobinopatias, a grande imigração de italianos, principalmente para o Sul do
Brasil, no final do século XIX. (ROSATELLI et al., 1992; LISOT; SILLA, 2004; ROBBINS
et al., 2005). Souza (2006) relata que no Brasil 10 milhões de indivíduos são heterozigotos
para hemoglobinopatias.

20
2.4. DIAGNÓSTICO
Como método de screening, de acordo com Protocolos de Metodologias Laboratoriais
Clássicas Para o Diagnóstico de Hemoglobinopatias da UNESP (2003) e Vargas e Yamagushi
(2008), tem-se basicamente a análise do eritrograma, da morfologia eritrocitária e o teste de
resistência ao NaCl.
A partir do eritrograma é possível avaliar parâmetros como índices de VCM, CHCM,
HCM, RDW, hematócrito e hemoglobina. Estas técnicas são de baixo custo, rápidas e
confiáveis (DSOUKY et al., 2009). Thein (2005) e colaboradores demonstraram em um
estudo que indivíduos que possuíam genes β0 talassêmicos apresentavam valores aproximados
de VCM = 63,1fl e HCM = 19,7 pg, enquanto que indivíduos que possuíam genes β+
talassêmicos apresentavam VCM de aproximadamente; 69,3 fl e HCM de 21,8 pg (THEIN,
2005).
Pela análise da morfologia eritrocitária observamos informações importantes sobre
qual é o tipo de anemia presente (como no caso de talassemias: anemia microcítica e
hipocrômica) e alterações como anisiopoiquilocitose, hemácias em alvo, pontos basofílicos
entre outras características da talassemia (LIMA, 2001).
O teste de resistência ao NaCl (0,36%) pode ser utilizado para auxiliar outros métodos
de diagnósticos para a beta talassemia. Ele é utilizado principalmente quando há suspeita de
beta talassemia heterozigótica. Nestes casos, os eritrócitos são mais resistentes à hemólise,
então quando os eritrócitos de um indivíduo beta talassêmico heterozigoto são submetidos a
essa concentração de NaCl não ocorre hemólise, já nos eritrócitos de um individuo controle
ocorre hemólise (UNESP, 2003).
Existem técnicas avançadas que podem revelar mais precisamente informações sobre
as Hb variantes e os tipos de talassemias, porém são técnicas de maior custo em relação às
usadas para o screening (DSOUKY et al., 2009). Dentre elas podemos citar a eletroforese de
Hb (ácida ou alcalina), a cromatografia líquida de alta pressão (HPLC), a eletroforese de
hemoglobina por capilaridade, a análise por espectrometria de massa e o sequenciamento do
DNA (BHAT et al., 2010).

21
2.4.1. Diagnóstico do indivíduo homozigoto
As principais alterações hematológicas presentes nos indivíduos homozigotos são a
anemia microcitica e hipocromica (Hb menor que 9g/dl), anisopoiquilocitose intensa,
hemácias e eritroblastos com granulações basofílicas, anel de Cabot, policromasia e
hemograma com desvio a esquerda nos granulócitos (ZAGO, 2004).
Os micrócitos encontrados são mais delgados que os eritrócitos normais, causa pela
qual a fragilidade osmótica está diminuída. A contagem de reticulócitos está aumentada e há
presença de inclusões intracitoplásmicas, que são os precipitados das cadeias alfa (LIMA,
2001). Também podemos observar os níveis de ferritina, ferro sérico e transferrina elevados
(LIMA, 2001; NAOUM, P.; NAOUM, F., 2004; ZAGO, 2004) além do aumento da
bilirrubina indireta (CAO, 2010) e do urobilinogênio (LIMA, 2001; ZAGO, 2004).
Nem todo indivíduo que tem beta talassemia apresenta aumento de HbF, o que ocorre
é que na ausência de HbA, a HbF é mais significativa (LIMA, 2001). A HbF é formada por
duas cadeias alfa e duas gama, com expressão dos genes γG e γA, pertencentes à família beta
globina (figura 1.). O gene da cadeia beta globina atuam em diferentes fases do
desenvolvimento, com pouca intensidade nas primeiras semanas de vida fetal, e após esse
período a síntese de cadeia γ é substituída pela síntese de cadeia β, formando a HbA
(GALIZA; PITOMBEIRA, 2002; ZAMARO et.al., 2003), e consequentemente reduzindo a
síntese de HbF. Portanto, quando há redução ou ausência da síntese de cadeia β é possível que
a HbF continue sendo expressa (ZAMARO, et.al., 2003).
Em indivíduos homozigotos pode-se encontrar na eletroforese presença de HbF (20 a
100%), aumento de HbA2 (cerca de 1 a 6% em relação ao normal) e presença de HbA (0 a
20%) (LIMA, 2001).

22
2.4.2. Diagnóstico do indivíduo heterozigoto
Em casos clássicos, é comum encontrar nos indivíduos heterozigotos níveis de Hb
ligeiramente inferiores ao normal (10,5 a 13g/dL), microcitose e hipocromia (nível de HCM
em torno de 20 pg), ferro sérico normal, aumento da HbA2 (3,5 a 6%) e níveis de HbF
normais (até 1%) ou ligeiramente aumentados (>5%) em relação ao normal (LIMA, 2001). As
alterações eritrocíticas podem ser as mesmas encontradas na talassemia maior, porém de
forma menos intensa (LIMA, 2001; ZAGO, 2004).
2.5. TRATAMENTO
O tratamento da beta talassemia é feito de acordo com o quadro clínico que o paciente
se encontra. Porém, mais frequentemente realiza-se um regime de transfusões sanguíneas e
terapia quelante de ferro com o objetivo de aumentar a sobrevida e reduzir a morbidade do
paciente (ORIGA et al., 2009 ; SUMBOONNANONDA et.al., 2009). O medicamento mais
comumente utilizado para o tratamento destes indivíduos além da hidroxiureia é o Deferasirox
(Exjade®), que é um quelante de ferro que se mostra eficaz na remoção do ferro hepático
além de oferecer uma proteção cardíaca (GAMARRA et al., 2009; ZAGO, 2204).
2.5.1. Tratamento para beta-talassemia maior
Alguns dos tratamentos utilizados para diminuir as manifestações clinicas destes
pacientes são transfusões regulares, supressão da eritropoese e inibição da absorção
gastrointestinal de ferro, utilizando terapia quelante de ferro, entretanto, a única forma de cura
é o transplante de MO por doador HLA idêntico (CAO; GALANELLO, 2010).

23
O transplante alogênico com doador familiar HLA - idêntico proporciona uma
sobrevida superior a 85% em relação aos indivíduos não transplantados, mas em pacientes
que têm complicações mais avançadas a sobrevida pode ser menor (LO; SINGER, 2002;
ZAGO, 2004).
O tratamento transfusional deste paciente é iniciado logo que ocorre diagnóstico (nos
primeiros anos de vida), quando a hemoglobina permanecer abaixo ou entre 6,5 a 7g/dL. O
paciente deve continuar sendo examinado semanalmente, para que consiga manter o nível de
Hb em torno de 10g/dL (LO; SINGER, 2002; ZAGO, 2004). Com este tratamento, percebe-se
a diminuição das manifestações e o crescimento do indivíduo ocorre normalmente até os 10
ou 11 anos. Após esta idade a criança apresenta o risco de desenvolver complicações graves
relacionadas à sobrecarga de ferro (CAO; GALANELLO, 2010).
Se não for possível alcançar este nível de hemoglobina, significa que pode haver
hiperesplenismo e consequentemente o consumo transfusional não diminui, então considera-
se a hipótese de esplenectomia (ZAGO, 2004). A esplenectomia é uma terapia auxiliar,
realizada quando as complicações excedem os benefícios da permanência do baço no
organismo. Uma das indicações é quando há elevado consumo de sangue transfundido (LO;
SINGER, 2002)
Após 10 a 12 meses de transfusões deve-se iniciar a terapia quelante de ferro (ZAGO,
2004), sendo esta a única forma para suprimir o ferro dos pacientes talassêmicos. Atualmente
o quelante mais usado é Desferioxamine ® (DFO) (VOGIATZY et al., 2009). Também há
terapia combinada (TC), que permite a redução das doses de cada droga, e age sobre
compartimentos distintos de ferro no organismo, somando para a eficiência da quelação
(PAULA et.al., 2003).
Um estudo recente mostrou que o Deferiprone ® por ser uma pequena molécula, tem
capacidade maior de penetrar em vários tecidos aumentando a eficiência na remoção do ferro
presente no coração, melhorando assim a função cardíaca (AESSOPOS et al., 2009).
Outro tratamento utilizado é a indução da produção de HbF utilizando a hidroxiuréia,
que atua principalmente reduzindo a massa extramedular e aumentando a síntese de HbF
(CAO; GALANELLO, 2010).

24
2.5.2. Tratamento da beta-talassemia intermediária
Apesar destes pacientes serem sintomáticos, não necessitam de transfusões regulares
pois conseguem manter níveis de Hb acima de 7g/dl. Entretanto, estes indivíduos devem
receber suplementação com ácido fólico (5mg/dia), pois a carência de folato pode acentuar a
anemia. Outra causa que agrava a anemia é a esplenomegalia, neste caso, a partir da avaliação
do quadro clínico, o paciente pode ser submetido à esplenectomia (CAO; GALANELLO,
2010).
2.5.1.3. Tratamento da beta-talassemia menor
Estes indivíduos são heterozigotos e assintomáticos, não sendo necessário tratamento
habitual apesar de que pode ser observada uma leve hipocromia, microcitose e níveis de Hb
ligeiramente inferiores ao normal (ZAGO, 2004).

25
3. JUSTIFICATIVAS
Não há estudos publicados até o presente momento descrevendo a prevalência da beta
talassemia em Brasília- DF, por isso a importância desta pesquisa, sendo, portanto a primeira
a descrever esta patologia em Brasília. Pois estamos nos referindo a uma região em que o
fluxo migratório foi intenso durante alguns anos devido às circunstâncias da época da
construção de Brasília, influenciando a possível presença de hemoglobinopatias na região.
Assim, nosso objetivo foi pesquisar a presença e prevalência de beta talassemia,
inclusive traço talassêmico, na população que apresentou alteração hematológica, atendida no
laboratório do Hospital das Forças Armadas de Brasília – DF (HFA – DF). Após este estudo,
o HFA poderá ser um hospital referência para estudos de hemoglobinopatias, devido às
técnicas já utilizadas na rotina laboratorial, como a eletroforese de hemoglobinas por
capilaridade.

26
4. OBJETIVOS
4.1. OBJETIVO GERAL
Identificar a prevalência da Beta Talassemia nos pacientes que apresentaram alteração
hematologia, atendidos no laboratório de análises clínicas do Hospital das Forças Armadas
de Brasília- DF, entre o período de maio a agosto de 2010.
4.2. OBJETIVOS ESPECÍFICOS
- Diagnosticar indivíduos com Beta Talassemia maior, intermediária e menor (traço
beta talassêmico).
- Identificar a prevalência de Beta Talassemia na população de estudo.

27
5. MATERIAS E MÉTODOS
5.1. AMOSTRAS
Após a assinatura do termo de consentimento livre e esclarecido, aprovado pelo
comitê de ética em pesquisa do HFA (Anexo A), 129 amostras e seis controles fizeram parte
do estudo, sendo estas coletadas, em tubo contendo anticoagulante EDTA, durante a rotina
laboratorial do Hospital das Forças Armadas de Brasília, entre o período de maio a agosto de
2010, não sendo identificados os pacientes, mantendo-se sigilo sobre seus dados.
A inclusão de pacientes para o grupo de estudo foi realizada por indivíduos que
apresentaram alguma alteração hematológica no hemograma ou de indivíduos que possuíam
requisição médica para realização de eletroforese de hemoglobina por capilaridade.
Os pacientes selecionados a partir do hemograma foram aqueles que apresentaram
diagnóstico de anemia e, para tal, foram analisadas as variáveis de Hb, VCM, HCM, CHCM e
alterações morfológicas das hemácias. A eletroforese de hemoglobina em pH ácido e alcalino,
foi realizada em seguida, demonstrando a mobilidade eletroforética de bandas normais e
anormais presentes no gel. Após a identificação de hemoglobinas em regiões não específicas
para HbA e HbA2 , foi realizada a eletroforese de hemoglobina por capilaridade (Figura 3.).
Os pacientes selecionados a partir da eletroforese de hemoglobina por capilaridade
foram aqueles que deram entrada no laboratório com pedido médico para a realização deste
exame. Portanto, esta seleção baseou-se na identificação e quantificação das hemoglobinas
presentes (Figura 3.).
Um grupo controle, de doadores de sangue sadios, foi utilizado como padrão para
todas as técnicas realizadas neste estudo.

28
Figura 3. Fluxograma das análises realizadas para rastreamento da beta talassemia na
população de estudo.
5.2. AVALIAÇÃO DAS AMOSTRAS DOS PACIENTES
5.2.1. Contagem automatizada e análise microscópica
As amostras de sangue periférico foram coletadas em tubos contendo anticoagulante
EDTA, seguindo para a realização do esfregaço sanguíneo utilizando 5µL de sangue e
coloração com corante Wright. Para a análise do eritrograma foi utilizado os valores de
referência padronizados pelo laboratório de acordo com a idade dos pacientes, apresentados
na tabela 2. A leitura dos índices hematimétricos foi realizada por contador eletrônico de
células (Beckman-Coulter®), sendo selecionados pacientes que apresentassem nível de

29
hemoglobina (Hb) inferior ao valor de referência, valores diminuídos de VCM, e/ou HCM
e/ou alterações morfológicas das hemácias que indicassem a existência de beta talassemia.
Tabela 2. Valores de referência do hemograma,
padronizado pelo laboratório de análises clínicas do
HFA - DF. Foi calculado uma média pra os valores de
eritrócitos, hemoglobina e hematócrito que variam de
acordo com a idade e sexo do paciente.
HEMOGRAMA Parâmetros Valores de referência Eritrócitos 4,1 – 5,1(x 106µL) Hemoglobina 11,7 a 15,7g/dL Hematócrito 35 a 47%
VCM 80 a 100fL HCM 24 a 34 pg
CHCM 31 a 37g/dL RDW 11,5 a 15,4%
5.2.2. Eletroforese de hemoglobina em pH ácido e alcalino (gel de agarose CELM-150).
Depois de realizado o hemograma e caracterizada uma anemia ou morfologia das
hemácias compatível com beta talassemia, foi realizada a seleção dos pacientes para nosso
estudo. Estas amostras foram submetidas à eletroforese de hemoglobina em pH ácido e
alcalino.
A eletroforese de hemoglobina é capaz de identificar anormalidades hemoglobínicas
qualitativas, através de suas diferentes mobilidades eletroforéticas devido ao peso molecular,
observando um aumento ou diminuição de frações e presença de frações anormais. Protocolos
– Anexo B e C.

30
5.2.3. Eletroforese de hemoglobina por capilaridade (MINICAP®)
Conforme o Colégio Americano de Patologistas (CAP) recomenda, o diagnóstico para
portadores de traço β talassêmico deve ser realizado usando a técnica de cromatografia líquida
de alta pressão (HPLC). Entretanto, foi relatada também, para diagnóstico, a técnica de
eletroforese por capilaridade, que mostrou ter um bom desempenho para este diagnóstico
(HIGGINS et. al., 2009; Yang et.al., 2009).
As amostras submetidas a este procedimento foram aquelas cadastradas no sistema do
laboratório para realização de eletroforese de hemoglobina por capilaridade, bem como as que
apresentaram alterações na eletroforese de hemoglobina em pH ácido e/ou alcalino como:
- presença de banda mais espessa de Hb A2;
- presença de HbF;
- bandas presentes no gel em região não específica para HbA e HbA2 (UNESP, 2003).
O MINICAP realiza vários testes simultaneamente e é totalmente automatizado,
possuindo, por exemplo, um bom coeficiente de variância para HbA2 (MANUAL DO
EQUIPAMENTO SEBIA, 2010; YANG et.al., 2009). Na tabela 3. mostramos outras
vantagens deste método em relação ao HPLC.
O aparelho MINICAP® realiza eletroforese de hemoglobina por capilaridade,
separando as hemoglobinas de acordo com ponto isoelétrico em um campo elétrico em meio
líquido. Por conseguinte quantifica todas as hemoglobinas presentes na amostra (MANUAL
DO EQUIPAMENTO SEBIA, 2010).
O sistema é composto por um carrossel de amostras, uma unidade de análise, um
compartimento de reagentes, uma unidade de controle e o software de processamento de
dados. As diluições das amostras são feitas pelo próprio aparelho
A unidade de análise é composta por: dois cartuchos termo condutores, uma unidade
de controle de temperatura para manter os capilares a uma temperatura estável, um sistema

31
de sensores, um sistema hidráulico que faz circular os reagentes dos capilares e uma fonte
de alta voltagem ligada a um conector de platina
Tabela 3. Comparação entre o método de HPLC e MINICAP, em relação à identificação e quantificação das hemoglobinas.
MINICAP HPLC Capacidade 34 testes por hora 10 testes por hora
Reprodutibilidade Muito boa Muito boa Tubo primário Não pode ser realizado com o sangue
total, apenas após remoção do plasma Realizado com o sangue total
Intereferência das proteínas plasmáticas
Sem interferência Possíveis interferências
Principais variantes Todas detectadas (mais de 50 variantes identificadas)
Todas detectadas (em torno de 45 identificadas)
Variantes raras Possível superposição com principais picos
Possível superposição com principais picos
Hb Bart’s e HbH Detecta e quantifica A fração glicada da HbA interfere na quantificação
Formas glicadas Não são detectadas. As frações são incluídas na hemoglobina
correspondente
Todas as frações glicadas são separadas.
Interferências das formas glicadas Sem interferência na quantificação Interferência da HbS glicada nos valores da HbA2
Quantificação dos picos Maior exatidão Valores de HbF em amostras
normais Sempre são detectados concentrações
menores que 0,5% Menor sensibilidade, devido a
forma glicada da HbA
HbA2 em amostras normais Igual Igual HbE Separada da HbA2 Não é separada da fração A2,
sendo impossível o diagnóstico de beta talassemia com presença de
HbE.
Para realização do exame o plasma é separado das hemácias, e em seguida a amostra á
colocada dentro do MINICAP. Após isso, o equipamento aspira a amostra e coloca-as em
cubetas, para que o hemolisante seja adicionado. Depois da lise das hemácias, esta é
transferida para os capilares, onde está presente o tampão de corrida (pH 9,4). A corrida de
eletroforese ocorre nesse capilar em meio líquido, e as hemoglobinas presentes na amostra são
detectadas e lidas por um programa chamado phoresis.
As hemoglobinas lidas pelo programa phoresis são apresentadas em um gráfico
(eletroferograma), representado por zonas que variam de Z1 a Z15, encontradas no eixo x do
gráfico, de acordo com a densidade óptica da hemoglobina (figura 4.). As hemoglobinas A e
A2, que são encontradas normalmente, são apresentadas sempre nas zonas Z9 e Z3 do gráfico,
respectivamente.

32
A validação do método é realizada utilizando amostras controles do próprio aparelho.
Para calibração e controle do equipamento, a amostra controle é passada no aparelho três
vezes, apresentando como referência, a densidade óptica das hemoglobinas A e A2 de 150 e
240, respectivamente. Assim, a quantificação das Hb A e A2, seguindo suas densidades
ópticas são 96,8 a 97,8% de HbA e 2,2 a 3,2% de HbA2.
Vários estudos descrevem que dosagens de HbA2 aumentadas fazem parte da triagem
para beta talassemia (LISOT; SILLA, 2004). Portanto, por ser indicativo desta patologia,
utilizamos um cutoff de 3,2%, para esta hemoglobina, conforme os valores de referência
(figura 4.).
Figura 4. Amostra controle do equipamento MINICAP. No
método de eletroforese de hemoglobina por capilaridade, as
hemoglobinas são apresentadas em zonas, que variam de Z1 a
Z15. As hemoglobinas encontradas normalmente, HbA e HbA2,
são apresentadas sempre nas zonas Z9 e Z3, respectivamente.

33
6. ASPECTOS ÉTICOS
O estudo foi realizado após aprovação do Comitê de Ética em pesquisa do Hospital
das Forças Armadas de Brasília. Nenhum material adicional foi coletado. Os procedimentos
foram realizados somente após assinatura do termo de consentimento livre e esclarecido pelo
paciente.

34
7. RESULTADOS
Foram incluídos neste estudo pacientes que apresentaram alteração hematológica
sugestiva de beta talassemia e pacientes que apresentaram requisição médica para realização
do exame de eletroforese de hemoglobina por capilaridade. Portanto foram 129 pacientes e
seis controles, entre o período de maio a agosto de 2010, sendo 78 mulheres e 51 homens
(tabela 4.).
Tabela 4. População de estudo avaliada a partir de dois métodos: o hemograma e a eletroforese de
hemoglobina por capilaridade, e os valores de referência do laboratório de análises clínicas do Hospital das
Forças Armadas de Brasilia- DF. As siglas “N”, “D” e “A”, significam normal, diminuído e aumentado,
respectivamente, em relação ao valor de referência.
Pacientes selecionados a partir do hemograma
Hemograma Total de pacientes 65 Sexo feminino Sexo masculino 39 26 Parâmetros Valores de referência Seleção dos pacientes Eritrócitos 4,1 – 5,1(x 106µL) N ou D Hemoglobina 11,7 a 15,7g/dL D Hematócrito 35 a 47% N ou D VCM 80 a 100fL D HCM 24 a 34 pg N ou D CHCM 31 a 37g/dL N ou D RDW 11,5 a 15,4% N ou A Análise microscópica
Alterações morfológicas das hemácias
Microcitose e/ou hipocromia ou alterações compatíveis com beta
talassemia Eletroforese de hemoglobina em pH ácido e alcalino
Mobilidade eletroforética das hemoglobinas
Bandas: HbA e HbA2 Bandas de HbA2 mais espessa, ou presença de HbF, ou bandas em
região não específicas para HbA e HbA2
Eletroforese de hemoglobina por capilaridade
Confirmação das hemoglobinas presentes pela eletroforese ácida e alcalina e quantificação de todas as hemoglobinas presentes
HbA: 96,8 a 97,8% HbA: < valor de referência
HbA2: 2,2 a 3,2% HbA2: > valor de referência
Pacientes selecionados a partir da eletroforese de hemoglobina por capilaridade Eletroforese de hemoglobina por capilaridade Total de pacientes 64 Sexo feminino Sexo masculino 39 25 Parâmetros Valores de referência Seleção dos pacientes Identificação e quantificação de
todas as hemoglobinas presentes HbA: 96,8 a 97,8% HbA: < valor de referência
HbA2: 2,2 a 3,2% HbA2: > valor de referência

35
Um exemplo de resultado de eletroforese em pH ácido e alcalino, que foi realizado
para os sessenta e cinco pacientes selecionados pelo hemograma, é demostrado pelo gel da
figura 5. Todos estes pacientes foram submetidos à eletroforese em pH ácido e alcalino,
portanto, confirmando a presença das hemoglobinas presentes.
Figura 5. Gel de eletroforese em pH ácido e alcalino, respectivamente. O controle utilizado para os dois géis
está demonstrado pela amostra número 10. O gel ácido apresenta as hemoglobinas F, A, S e Hb variante
(var.). O gel alcalino apresenta as hemoglobinas A, S e A2.
7.1. RESULTADOS DA SÉRIE VERMELHA DO HEMOGRAMA DOS PACIENTES SELECIONADOS PARA O ESTUDO A PARTIR DA ANÁLISE DO HEMOGRAMA
Sessenta e cinco pacientes foram selecionados a partir da avaliação do nível de
hemoglobina (Hb) compatível com anemia, dos valores diminuídos de VCM, e/ou HCM e/ou
alterações morfológicas das hemácias que indicassem a existência de beta talassemia.
A maioria desta população 87,69% (57/65) apresentou hemoglobina abaixo dos seus
valores de referência (gráfico 1.). A relação dos índices hematimétricos de VCM e HCM
estão expostos nos gráficos 2 e 3, indicando que o VCM da população de estudo foi menor em
relação aos valores de referência, enquanto o HCM manteve-se em seu nível normal em
grande parte da população. Dentre os 65 pacientes, ao realizarmos uma média 34,61% desta
população apresentou anemia microcítica e hipocrômica. Para o laboratório do HFA – DF a
microcitose e hipocromia são determinadas por “+”, indicando assim o grau de microcitose e
hipocromia do paciente (gráfico 4.).

36
Gráfico 1. Demonstração dos níveis de hemoglobina encontrados nos 65
pacientes. Todos os pacientes apresentaram hemoglobina abaixo do valor de
referência, utilizado pelo laboratório de análises clínicas do HFA- DF.

37
Gráfico 2. Análise do VCM na população de 65 pacientes.
Neste gráfico, os níveis de VCM estão separados em relação ao
valor de referência, ou seja, ≤ 80fL, entre 80 a 100 fL e ≥ 100fL.
Gráfico 3. Análise do HCM na população de 65 pacientes. Os níveis
de HCM estão separados conforme seus valores de referência, ou
seja, <26pg, entre 26 a 34pg e >34pg.

38
Gráfico 4. Avaliação de pacientes que apresentaram microcitose e hipocromia,
entre o total de 65 pacientes analisados pelo resultado do hemograma. Nenhum
dos pacientes apresentou alteração morfológica das hemácias.
7.2. RESULTADO DOS 129 PACIENTES ANALISADOS ATRAVÉS DA ELETROFORESE DE HEMOGLOBINA POR CAPILARIDADE.
A eletroforese de hemoglobina por capilaridade foi realizada para os 65 pacientes
selecionados por apresentarem alteração hematológica observada pelo hemograma, bem como
para os 64 pacientes incluídos no estudo a partir da requisição médica para a realização da
eletroforese de hemoglobina por capilaridade, e que, portanto não apresentaram dados do
hemograma (figura 3. e tabela 4.).
Assim, o equipamento MINICAP indicou 40,31% (52/129) da população analisada
com presença de hemoglobinas anormais ou aumento de HbA2. Desta população (n=129), a
freqüência seguiu em 14,72% (19/129) para HbS, 12,40% (16/129) para HbF, 5,42% (7/129)
para HbC e 12,40% (16/129) para aumento de HbA2 (gráfico 5.).
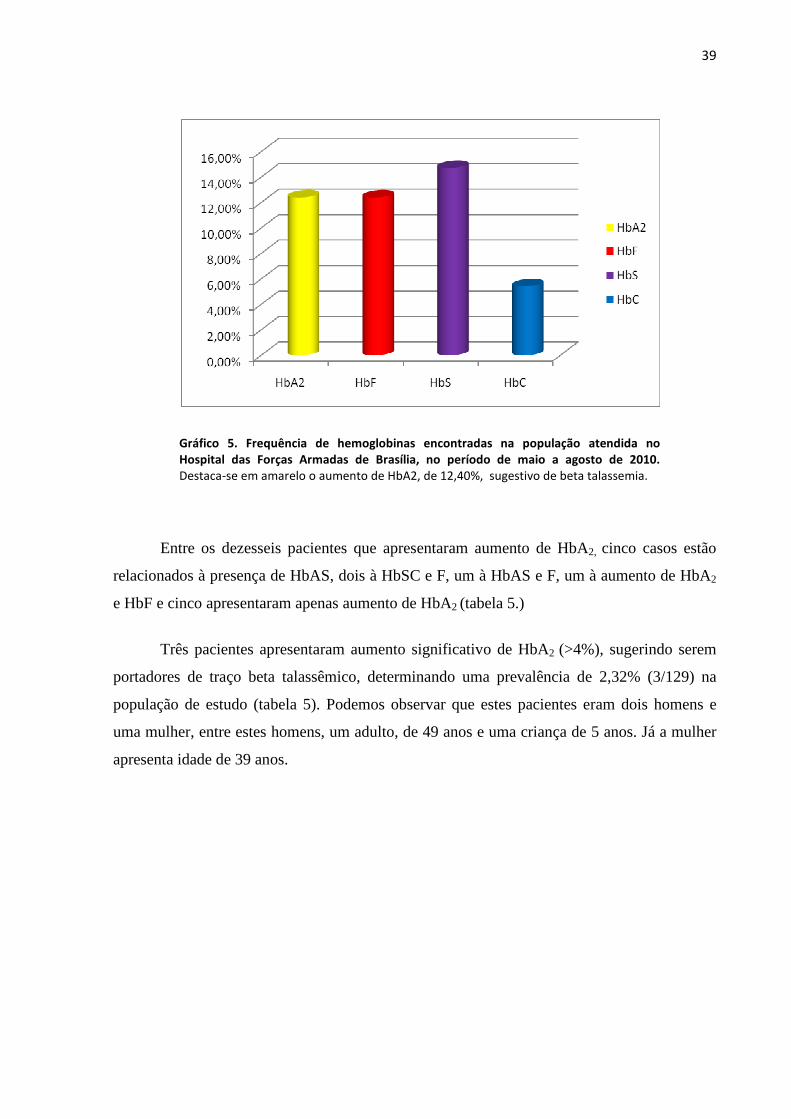
39
Gráfico 5. Frequência de hemoglobinas encontradas na população atendida no
Hospital das Forças Armadas de Brasília, no período de maio a agosto de 2010.
Destaca-se em amarelo o aumento de HbA2, de 12,40%, sugestivo de beta talassemia.
Entre os dezesseis pacientes que apresentaram aumento de HbA2, cinco casos estão
relacionados à presença de HbAS, dois à HbSC e F, um à HbAS e F, um à aumento de HbA2
e HbF e cinco apresentaram apenas aumento de HbA2 (tabela 5.)
Três pacientes apresentaram aumento significativo de HbA2 (>4%), sugerindo serem
portadores de traço beta talassêmico, determinando uma prevalência de 2,32% (3/129) na
população de estudo (tabela 5). Podemos observar que estes pacientes eram dois homens e
uma mulher, entre estes homens, um adulto, de 49 anos e uma criança de 5 anos. Já a mulher
apresenta idade de 39 anos.

40
Tabela 5. Pacientes que apresentaram aumento de HbA2 quando realizada a eletroforese de hemoglobina
por capilaridade. São indicados, em azul, três pacientes sugestivos de serem portadores de beta talassemia.
Treze pacientes sem indicativo de beta talassemia apresentaram aumento de HbA2. Os pacientes em negrito
são os pacientes selecionados através do hemograma.
Paciente Sexo Idade HbA HbA2 HbF HbS HbC β talassemia
2 F 49 55,40% 3,30% -- 41,30% -- Não
1 M 1 96,40% 3,30% 0,40% -- -- Não
3 F 16 -- 3,50% 1,80% 50,30% 44,50% Não
4 M 19 61% 3,60% -- 35,50% -- Não
5 M 14 -- 4% 0,90% 51% 44,10% Não
6 M 22 17,60% 4,50% 5,30% 72,50% -- Não
7 M 34 55,4 3,4 -- 41,2 -- Não
8 M 48 55 3,4 -- 41,6 -- Não
9 M 7 96 3,5 0,5 -- -- Não
11 M 19 61 3,6 -- 35,5 -- Não
10 F 52 96,4 3,6 -- -- -- Não
12 F 59 97,3 3,7 -- -- -- Não
13 M 38 97,3 3,7 -- -- -- Não
14 M 5 95,6 4,4 -- -- -- Sim
15 F 39 94,5 5,2 0,3 -- -- Sim
16 M 49 94,2 5,8 -- -- -- Sim
A seguir estão expostos os eletroferogramas plotados pelo aparelho MINICAP®,
exemplificando os pacientes que apresentaram aumento de HbA2 mais significativo, acima de
4%, de acordo com Yang et. al. (2009), sugerindo traço beta talassêmico.
Figura 6. Pacientes sugestivos de traço beta talassêmico por apresentar aumento de HbA2, detectada quando realizada a
eletroforese de hemoglobina por capilaridade. As hemoglobinas presentes foram HbA HbA2 e um paciente apresentou HbF.

41
8. DISCUSSÃO
Em âmbito de diagnóstico, os métodos de escolha para o rastreamento da beta
talassemia são análise da série vermelha do hemograma e a eletroforese de hemoglobina em
pH alcalino. Sendo que, a eletroforese ácida é utilizada para confirmação ou quando há
resultados inconclusivos. Para confirmação do diagnóstico, a história clínica do paciente deve
ser avaliada e se necessário utiliza-se a quantificação de hemoglobinas através de métodos
como eluição de hemoglobina específica, HPLC e a eletroforese de hemoglobina por
capilaridade (UNESP, 2003; ZAMARO et.al., 2002; YANG et.al., 2009).
Os pacientes selecionados a partir do hemograma apresentaram com maior freqüência
níveis de hemoglobina menor do que os valores de referência e, portanto seria um dado inicial
para o rastreamento de provável beta talassemia (THEIN, 2005; SOLLAINO et al., 2009;
ZAGO, 2004).
Entretanto, para complementar o diagnóstico, além da anemia seria necessário que
esta se caracterizasse como microcítica e hipocrômica (TIWARI et.al., 2009), anisocitose,
poiquilocitose e presença de eritroblastos para ser qualificada como beta talassemia maior. Os
pacientes portadores de beta talassemia apresentam anemia com diminuição nos níveis de
VCM e HCM, e com ausência de eritroblastos (CAO, 2010). Assim, pode-se sugerir que os
pacientes apresentam características de portadores de traço beta talassêmico.
Observa-se que, 34,61% dos pacientes que apresentaram anemia caracterizaram-se
com microcitose e hipocromia. Contudo, como a microcitose e hipocromia são comumente
encontradas no hemograma de pacientes que possuem carência de ferro e em pacientes beta
talassêmicos (HAZIROLAN et.al., 2009; TIWARI et.al., 2009), não podemos concluir que
estes pacientes sejam portadores de beta talassemia, com base apenas neste dado.
A distinção entre anemia ferropriva e a beta talassemia é realizada através das
dosagens de ferro, ferritina e transferrina (TIWARI et.al., 2009). No entanto, não foi possível

42
realizar a verificação destas dosagens, pois muitos pacientes não tinham amostras colhidas em
tubo sem anticoagulante.
Com isso, para todas as amostras que apresentaram índices sugestivos de anemia
realizamos a eletroforese de hemoglobina em pH ácido e alcalino, de forma paralela,
identificando as hemoglobinas presentes. O gel apresentado pela figura 5. mostra por
exemplo, que a amostra 80 apresentou presença de Hb variante que indica, quando em pH
ácido, uma possível HbC ou HbA2, porém quando realizada a eletroforese em pH alcalino,
confirmou-se a presença de HbA2 nesta amostra. A amostra número 10 foi utilizada como
nosso controle na eletroforese de pH alcalino e ácido, por apresentar apenas HbA e HbA2.
A quantificação de todas as hemoglobinas encontradas, dos 129 pacientes analisados
no estudo, foi realizada através da eletroforese de hemoglobina por capilaridade. As técnicas
de eletroforese têm grande importância para o nosso estudo devido à quantificação das
hemoglobinas, principalmente em relação à detecção e quantificação de HbF e de HbA2, que
são sugestivas de beta talassemia (CAO; GALANELLO, 2010; LISOT; SILLA, 2004; YANG
et.al., 2009). De acordo com Cao e Galanello (2010), portadores de beta talassemia
apresentam níveis de HbA2 >3,5% , HbF 0,5 a 4% e HbA 92 a 95%. Enquanto Yang e
colaboradores (2010) e Omar e colaboradores (2010) descrevem que o diagnóstico de traço
beta talassêmico é determinado pelo aumento de HbA2 >4%, independente dos dados do
hemograma do paciente.
Deste modo, após a análise da quantificação das hemoglobinas 40,31% (52/129) da
população apresentou hemoglobinas anormais ou aumento da HbA2. Em relação ao aumento
de HbA2, a frequência apresentada pela população foi de 12,40% (16/129) (Gráfico 5.).
O aumento de HbA2, mostrou-se associado também com outras hemoglobinas, como
Hb S, C e F. Embora nestes casos não possamos sugerir um diagnóstico de beta talassemia, já
que o aumento de HbA2 também ocorre devido à presença de HbS, sabe-se que a anemia
falciforme pode existir junto com a beta talassemia, contribuindo para o aumento da HbA2
destes pacientes (YANG et.al., 2009).
Considerando que, o diagnóstico de traço beta talassêmico pode ser baseado apenas no
aumento de HbA2 >4% (OMAR et.al., 2010; YANG. et.al., 2009; LISTOT; SILLA, 2004), a
prevalência de traço beta talassêmico foi de 2,32% (3/129) para pacientes que apresentaram

43
alguma alteração hematológica ou como nesses três casos possuíam apenas a requisição
médica para a realização da eletroforese de hemoglobina por capilaridade. Ou seja, três
pacientes são sugestivos de serem portadores de traço beta talassêmico (tabela 5).
Entre os três pacientes, dois apresentaram aumento de 1,2% e de 2,6% de HbA2, em
relação ao seu cutoff de 3,2%. Enquanto, o outro paciente apresentou 0,5% de HbF na zona 7,
juntamente com 2% de HbA2 acima de seu cutoff de 3,2% (figura 6.). Este dado corrobora
para os casos em que Thein (2005) e Mosca et.al. (2009) citam que a HbF pode ser encontrada
na beta talassemia homozigótica e heterozigótica, porém com aumento variável. Nos casos de
heterozigose, como o traço beta talassêmico, a HbF pode estar presente de forma ligeiramente
aumentada (CAO; GALANELLO, 2010; MOSCA et.al., 2009; ZAMARO et.al., 2003).
Assim, a prevalência encontrada como sugestiva de traço beta talassêmico nesta
população, é um fator importante por dois motivos: para evidenciar que não há grande
transferência de genes alterados para beta talassemia na população estudada, e que mesmo
essa prevalência sendo pequena, pode revelar uma provável descendência de povos italianos,
africanos, do sudeste da Ásia ou do Mediterrêneo, de onde mais se têm prevalência de beta
talassemia no mundo (ESTEVÃO, 2009; MELO et.al., 2008; SOUZA, 2006).
Outro fator importante ao encontrar este resultado para esta população, é que estes
pacientes bem como suas famílias devem ter um acompanhamento médico, a fim de ter um
aconselhamento genético correto. Pois, um dos pacientes sugestivo de portar traço beta
talassêmico tem apenas cinco anos de idade, portanto é importante que se faça o diagnóstico
dos pais para prevenção de transferência deste gene para outro filho. Até mesmo para que
futuramente este paciente seja consciente que é portador de um gene beta talassêmico e saiba
quais são as possíveis complicações se combinado a outro gene alterado, ou mesmo quais são
as possibilidades de transferir este gene para um filho.
No Brasil esta prevalência é variável, devido à variabilidade étnica da população
(DUCATTI et.al., 2001; ESTEVÃO, 2009; MELO et. al., 2008; SOUZA, 2006), sendo
encontrada mais frequentemente nas regiões do Sul e Sudeste, devido à imigração italiana
(SOUZA, 2006). Este fato explica a diferença dos resultados em vários estudos brasileiros
(DUCATTI et.al., 2001).

44
Um estudo realizado por Lisot e Silla (2004) com 9.000 doadores de sangue no Rio
Grande do Sul, apresentou 12% da população com alguma hemoglobinopatia, sendo 9,87%
portadores de beta talassemia. Melo et.al. (2000) determinaram em uma população de 23.981
doadores de sangue em Minas Gerais, 820 pacientes com hemoglobinopatias, sendo 0,13%
portadores de beta talassemia menor. Orlando e colaboradores. (2000) em São José do Rio
Preto – SP demonstraram em 265 amostras de doadores de sangue, 13 pacientes com
hemoglobinopatias, sendo 0,38% indivíduos portadores de beta talassemia. Já Ducatti e
colaboradores (2001) em uma pesquisa realizada em cordão umbilical de 913 recém-nascidos
(RN) em São José do Rio Preto – SP encontraram 100 RN portadores de alguma
hemoglobinopatia, sendo 23 portadores de beta talassemia.
Assim, quando comparado com outros estudos, podemos concluir que nossa pesquisa,
embora realizada com um pequeno universo amostral, reforça que há no Brasil uma
prevalência variável de beta talassemia, assim como de outras hemoglobinopatias. Entretanto,
novos estudos englobando uma população maior precisam ser realizados.

45
REFERÊNCIAS BIBLIOGRÁFICAS
AESSOPOS, A et.al. “Prevention of cardiomyopathy in transfusion-dependent homozygous thalassemia today and the role of cardiac magnetic resonance imaging”. Advances in Hematology, 2009.
AMINI, F. et. al. “A cephalometric study on craniofacial morphology of Iranian children with beta-thalassemia major” Orthodontics and Craniofacial Research, v. 10, 2007. p. 36 – 44.
ARMELI, C. et. al. “Comparing knowledge of beta-thalasemia in samples of Italians, Italian-Americans, and non-Italian-Americans”. Journal of Genetic Counseling, v.14, n. 15, 2005. p. 365-76.
ASSARI, B.H et. al. “Anxiety and depression affects life and sleep quality in adults with beta-thalassemia”. Indian Journal of Hematology and Blood Transfusion, v. 25, n.2, 2009. p. 59 – 65.
BHAT, V.S et al. “Characterization of hemoglobin variant: HbQ-India/IVS 1-1 [G >T] – beta-thalassemia”. Indian Hournal of clinical Biochemistry , v. 25, n.2, 2010. p. 99 – 104.
CANÇADO, R.D. “Talassemia beta maior: uma nova era”. Revista Brasileira de Hematologia e Hemoterapia, v.30, n.6, 2008 p. 433 – 436.
CAO, A.; GALANELLO, R. “Beta-thalassemia”, In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2000 Sep 28 [updated 2010 Jun 17]. Disponível em: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=b-thal

46
CASTRO, F.S. et.al. “Prevalência de talassemias e hemoglobinas variantes em pacientes portadores de lúpus eritematoso sistêmico”. Revista Brasileira de Hematologia e Hemoterapia, v. 30, n.1, 2008.
CUNNINGHAM, M.J. “Update on Thalasemia: clinical care and complications”. Hematol. Oncol. Clin., v. 24, 2010. p. 215-227.
DSOUKY, O.S. et.al.“Biophysical characterization of beta-thalassemic red blood cells”. Cell Biochemistry and Biophysics, v.55, n.1, 2009. p. 45-53.
DUCATTI, R.P. et al. “Investigação de hemoglobinopatias em sangue de cordão umbilical de recém-nascidos do Hospital de Base de São José do Rio Preto”. Revista Brasileira de Hematologia e Hemoterapia, v. 23, n.1, 2001. p. 23 – 29.
DWIVEDI, S.; KUMAR, V. “Beta-thalassemia, hiperlipoproteinemia and metabolic syndrome: its low-cost holistic therapy”. Journal of Alternative and Complamentary Medicine, v. 13, n.2, 2007. p. 287-9.
ELDOR, A; RACHMILEWITZ, E.A. “The hypercoagulable state in thalassemia” Blood, v. 99, n.1, 2002. p. 36-43.
ESTEVÃO, I.F. “Frequência dos mutantes C282Y e H63D do gene HFE e sua influência no metabolismo do ferro e na expressão da beta talassemia heterozigota” Revista Brasileira de hematologia e hemoterapia, v. 31, n.1, 2009. p.55-56.
GALANELLO, R.; ORIGA, R. “Beta thalassemia”. Orphanate Journal of rare diseases, v.5, 11, 2005.
GALIZA, G.C.de N.; PITOMBEIRA, M. da S. “Aspectos moleculares da anemia falciforme”. Jornal Brasileiro de Patologia e Medicina Laboratorial, v.39, n.1, 2003. p.51-56.

47
GAMARRA, S. et al. “Beta-thalassemia major in a Spanish patient due to a compound heterozygosity for CD39 C --> T/-28 A --> C”. Advances in Hematology, 2009.
HAZIROLAN, T. et al. “Dual-echo TFE MRI for the assessment of myocardial iron overload in beta-thalassemia major patients”. Diagnostic and Interventional Radiology, v. 16, n.1, 2009. p.59-62.
HIGGINS, T.N. et.al. “ Quantification of HbA2 in Patients With and Without β-Thalassemia
and in the Presence of HbS, HbC, HbE, and HbD Punjab Hemoglobin Variant- comparison of two systems”. American society for Clinical Pathology, 2009. p. 357-62.
KIMURA, E.M. et.al. “Thalassemia intermedia as a result of heterozygosis for beta 0 – thalassemia and alpha anti 3,7 genotype in a Brazilian patient”. Brazilian Journal of Medical and Biological Research, v. 36, n.6, 2003. p.699 – 701.
LIMA, A. O. Métodos de laboratóio aplicados à cínica: técnicas e interpretação. 8. Ed. Rio de Janeiro, RJ: Guanabara Koogan, 2001.
LISOT, C.L.A.; SILLA, L.M.R. “Screening for hemoglobinopathies in blood dornors from Caias do Sul, Rio Grande do Sul, Brazil: prevalence in na Italian colony”. Cadernos de Saúde Pública, Rio de Janeiro, v. 20, n.6, 2004, p. 1595- 601.
LITHANATUDOM, P. et. al. “A mechanism of ineffective erythropoiesis in beta-thalassemia/HbE disease” Haematologica, v. 95, n. 5, 2009. p. 716 – 23.
LO, L.; SINGER, S.T. “Thalassemia: current approach to an old disease”. Pediatric clinics of North American , v. 49, n.6, 2002, p. 1165-91.
LONKAR, P. et.al. “Target correction of a thalassemia – associated beta-globin mutation induced by pseudo-complementary peptide nucleic acids”. Nucleic Acids Research, v. 37, n.11, 2009, p. 3635 – 44.

48
MANUAL DO EQUIPAMENTO SEBIA, MiniCap. Eletroforese de hemoglobina por capilaridade, 2010
MATOS, J.F. et. al. “O papel do RDW, da morfologia eritrocitária e de parâmetros plaquetários na diferenciação entre anemias microcíticas e hipocrômicas”. Revista Brasileira de Hematologia e Hemoterapia, v. 30, n.6, 2008. p. 463-469.
MELO, L.M.S. et.al. “Rastreamento de hemoglobinas variantes e talassemias com associação de métodos de diagnóstico”. Revista Brasileira de Hematologia e Hemoterapia, v. 30, n.1, 2008. p.12-17.
MELO, S.M.A. et. al. “Prevalência de hemoglobinpatias em doadores de sangue do hemocentro regional de Uberlândia – MG”. Revista Brasileira de Hematologia e Hemoterapia, v.22, 2000.
MELLOULI, F. et.al. “Switch from Beta-thalassemia major to Beta—thalassemia intermedia after secondary graft failure- Case report”. Experimental and clinical transplantation, v.8, n.3, 2010.
MINISTÉRIO DA SAÚDE. Portaria nº 822, Brasília, Brasil, 2001.
MORAWAKAGE, R.L. et.al. “Huperhemolysis in a patient with beta-thalassemia major”. Asian Journal of Transfusion Science, v.3, n.1, 2009. p. 26-7.
MOSCA, A. et.al. “The relevance of hemoglobin F measurement in the diagnosis of thalassemias and related hemoglobinopathies”. Clinical Biochemistry, v. 42, n. 18, 2009. p. 1797-801.
NAOUM, P.C.; NAOUM, F.A. “Síntese genética da hemoglobina”. Disponível em: http://www.hemoglobinopatias.com.br/hb-normais/intro.htm. Acesso em: 3 mar. 2010.

49
NAOUM, P.C. Eletroforese: técnicas e diagnósticos. 2.ed., Santos, 1990.
NAOUM, P.C. “Hemoglobinopatias e talassemias. As tecnologias de identificação devem ser ampliadas em neonatos?” Revista Brasileira de Hematologia e Hemoterapia, v. 30, n.1, 2008.
NAOUM, P.C.; NAOUM, F.A. Doença das anemias das células falciformes. Sarvier, São Paulo, 2004.
OLIVIERI, N.F. “The ß-Thalassemias”. The new England Journal of Medicine, 1999. p. 99-109.
OMAR, S. et.al. “Une bêta thalassémie mineure masquée par um mutant de L’hémoglobine A2”. La Tunise Medicale, v. 88, n.9, 2010. p. 678-681.
ORIGA, R. et.al. “Pregnancy and beta-thalasemia: na Italian multicenter experience”. Haematologica, v. 95, n.3, 2009. p. 376-81.
ORLANDO, G. M. et.al. “Diagnóstico diferencial de hemoglobinopatias em populações diferenciadas”. Revista Brasileira de Hematologia e Hemoterapia, v.22, 2000. p. 111-21.
PATRINOS, G.P. et.al. “Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies”. Nucleics Acids Research, v.32, 2004.
PAULA, E.V. et.al. “Quelação oral de ferro na beta-talassemia”. Revista Brasileira de Hematologia e Hemoterapia, v. 25, n.1, 2003. p. 59-63
PENMAN, B.S. et.al. “Epistativ interactions between genetic disorders of hemoglobin can explain why the sickle-cell gene is uncommon in the Mediterranean”. Proceedings of the National Academy of Sciences, v. 150, n.50, 2009.

50
ROSATELLI, M.C. et.al. “Molecular screening and fetal diagnosis of beta-thalassemia in the Italian population”. Human Genetics, v.89, n.6, 1992. p. 585-9.
SACHDEV, R. et.al. “Detection of Hb variants and hemoglobinopathies in Indian population using HPLC: report of 2600 cases”. Indian Journal of Pathology and Microbiology, v. 53, n.1, 2010. p.57-62.
SADIQ, M.F. et.al. “Spectrum of b-thalassemia in Jordan: Identification of two novel mutations”. American Journal of Hematology, v. 68, 2001. p. 16-22.
SAKAMOTO, T.M. et.al. “Talassemia ß intermediária em gestante”. Revista Brasileira de Hematologia e Hemoterapia, v. 30, n.6, 2008. p. 498-500.
SCHRIER, S.L. et.al. “The role of oxidant injury in the pathophisiology of human thalassemias”. Redox Report, v.8, n.5, 2003. p. 241-5
SCOTT, M.D. et.al. “Effec of excess alpha-hemoglobin chains on cellular and membrane oxidation in model beta-thalassemic erythrocytes”. The Journal of Clinical Investigation, v. 91, n.4, 1993. p. 1706-12.
SOLLAINO, M.C. et.al. “Association of alpha globin gene quadruplication and heterozygous beta thalassemia in patients with thalassemia intermedia”. Haematologica, v. 90, n.10, 2009. p. 1445 – 8.
SOUZA, R.A.V. de. Análise crítica do programa de triagem de neonatal para detecção de hemoglobinopatias em Dourados – MS. Brasília. 2006. 149 f. Dissertação (Mestrado em ciências da saúde) – Universidade de Brasília e Centro Universitário da Grande Dourados, Brasília, 2006.
SUMBOONNANONDA, A. et.al. “Renal tubule function in beta-thalassemia after hematopoietic stem cell transplantation”. Pediatric Nephrology, v.24, n.1, 2009. p. 183-7.

51
THEIN, S.L. “Pathophysiology of beta thalassemia--a guide to molecular therapies”. Hematology American Society of Hematology Education Program, 2005. p.31-7.
THEIN, S.L. “Genetic modifiers of β-thalassemia” The Hematology Journal, v.90, n.5, 2005, p.649-660.
TIWARI,A.K. et.al. “Approach to blood donors with microcytosis. Transfusion Medicine, Oficial Journal of the British Blood Transfusion Society, 2009.
UNESP. Campus de São José do Rio Preto- Instituto de Biociências, Letras e Ciências Exatas. Protocolos de metodologias laboratoriais clássicas para o diagnóstico de hemoglobinopatias, 2003.
VARGAS, S.P.; YAMAGUSHI, M.U. “Diagnóstico laboratorial para talassemias”. Revista de Saúde e Pesquisa, v.1, n.1, 2008. p. 85-88.
VINCI DE MOARAES, J.G. Caminhos das civilizações: História integrada e geral do Brasil. São Paulo, SP: Atual, 2000.
VOGIATZI, M.G. et.al. “Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassemia syndromes in North America”. British Journal of Haematology, v. 146, n.5, 2009. p. 546-56.
YANG, Z. et.al. “ Prevalence of Elevated Hemoglobin A
2 Measured by the CAPILLARYS
System”. American Journal of Clinical Pathology, 2009. p. 42-48.
ZAGO, M.A.; FALCÃO, R.P.; PASQUINI, R. Hematologia: fundamentos e prática. São Paulo, SP: Atheneu, 2004.

52
ZAMARO, P.J.A. et.al. “Diagnóstico laboratorial de hemoglobinas semelhantes à HbS”. Jornal Brasileiro de Patologia e Medicina Laboratorial, v. 38, n. 4, 2002. p. 261-266.
ZAMARO, P.J.A. et.al. “Análise quantitativa e molecular de hemoglobina fetal em indivíduos da população brasileira”. Revista Brasileira de Hematologia e Hemoterapia, v.25, n.4, 2003. p.223-229.

53
ANEXOS
ANEXO A – TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
Eu, __________________________________________________________________ ( )(Concordo) ( )(Discordo) que o material coletado para realização dos exames constantes nesta guia, assim como os resultados destas análises, os quais estarão armazenados nas bases de dados do LAC, sejam utilizados na composição de pesquisas e outros trabalhos executados pelo laboratório com finalidades acadêmicas e científicas, inclusive para publicação, desde que seja preservado o sigilo da minha identidade.

54
ANEXO B - ELETROFORESE DE Hb EM pH ALCALINO (Protocolos de Metodologias Clássicas para o Diagnóstico de Hemoglobinopatias- UNESP, 2003, modificado).
Amostra: sangue total com EDTA
Material necessário:
Filme de agarose para separação eletroforética das Hb em pH alcalino (Hb alcalina)
Seringa micro-volume para pipetar amostras para o filme de agarose.
Tampão alcalino TRIS pH 9,5 ±0,2 CELMGEL
Hemolisante (do Kit)
Corante Ponceau S (5,0 g CELM + 50,0 g de ácido tricloroacético)
Descorante para eletroforese de Hb (100mL ácido acético glacial + 50 mL metanol
P.A, completando com água até 1000 mL)
Placas de Petri
Secador
Aparelho CELM FEA – 250
Procedimento
Preparação do hemolisado
Centrifugar parte do sangue total com EDTA e desprezar o plasma.
Lavar hemácias com solução salina 0,9% ( para cada 100µL de hemácias acrescentar
200µL de solução fisiológica) até obter sobrenadante límpido.
Misturar partes iguais das hemácias e hemolisante (para 100µL de hemácias 100µL de
hemolisante), em um tubo de ensaio.
Agitar vigorosamente em vortex por aproximadamente 30 segundos.
Determinar a concentração de hemoglobina no equipamento de hemograma e ajustar a
concentração de Hb em aproximadamente 1g/dL, diluindo com água destilada ou deionizada
até atingir concentração desejada.
O hemolisado obtido é estável por 24 horas quando armazenado entre 2 e 8º C.
Eletroforese
Com seringa de micro-volume, pipetar 1,0 µL de cada hemolisado em uma cavidade
numerada no gel alcalino.
Ligar o aparelho CELM FEA – 250 e ajustar para 30 minutos e 150 volts.

55
Colocar o gel alcalino no porta filme, coincidindo os pólos positivos e negativos do
filme e da cuba.
Colocar 80 mL de tampão alcalino gelado (2 a 8ºC), na cuba para cobrir o eletroldo
metálico.
Colocar o porta filme com o gel alcalino dentro da cuba ligá-la.
Após os 30 minutos deve-se desligar e retirar o porta-filme da cuba e deixar escorrer o
excesso de tampão em papel toalha.
Retirar o gel alcalino e colocá-lo a 60ºC (secador) até secar completamente, por
aproximadamente 15 minutos.
Mergulhar o gel em placa Petri, contendo o corante Ponceau S, por 10 minutos, sem
agitação.
Retira-lo do corante Ponceau e colocá-lo em outra placa contendo o descorante (ácido
acético / metanol), por 10 minutos.
Repetir o procedimento acima com descorante limpo por 10 minutos.
Completar a descoloração deixando o gel alcalino mergulhado por 10 minutos ou até
sua total descoloração em ácido acético 5,0%.
Secar completamente o gel alcalino à 60ºC (secador).

56
ANEXO C - ELETROFORESE DE Hb EM pH ÁCIDO (Protocolos de Metodologias Clássicas para o Diagnóstico de Hemoglobinopatias- UNESP, 2003, modificado).
Amostra: sangue total com EDTA
Material necessário:
Filme de agarose para diferenciação da Hb S, C e F(Hb ácida)
Seringa micro-volume para pipetar amostras para o filme de agarose
Tampão citrato pH 6,3 ± 0,2 CELMGEL
Hemolisante (do Kit)
Corante Amido-Black (Negro amido 0,1% em ácido acético a 5%)
Descorante para eletroforese de Hb (ácido acético a 5%)
Placas de Petri
Secador
Aparelho CELM FEA – 250
Procedimento
Preparação do hemolisado
Centrifugar parte do sangue total com EDTA e desprezar o plasma.
Lavar hemácias com solução salina 0,9% (para cada 100µL de hemácias acrescentar
200 µL de solução fisiológica) até obter sobrenadante límpido.
Misturar partes iguais das hemácias e hemolisante (para 100µL de hemácias 100µL de
hemolisante), em um tubo de ensaio.
Agitar vigorosamente em um vórtex por aproximadamente 30 segundos.
Determinar a concentração de hemoglobina no esquipamento de hemograma e ajustar
a concentração de Hb em aproximadamente 1g/dL, diluindo com água destilada ou deionizada
até atingir concentração desejada.
O hemolisado obtido é estável por 24 horas quando armazenado entre 2 e 8º C.
Eletroforese
Com seringa de micro-volume, pipetar 0,5µL de cada hemolisado em uma cavidade
numerada no gel ácido. Aguardar alguns segundos para que o hemolisado penetre no gel e
aplicar mais 0,5µL do hemolisado.

57
Ligar o aparelho CELM FEA – 250 e ajustar para 55 minutos e 50 volts.
Colocar o gel ácido no porta-filme, coincidindo o pólo positivo da cuba com o pólo
negativo do gel ácido.
Colocar 90 mL de tampão citrato pH 6,3 ± 0,2 gelado (2 a 8ºC), na cuba para cobrir o
eletroldo metálico.
Colocar o porta filme com o gel alcalino dentro da cuba ligá-la.
Após os 55 minutos deve-se desligar e retirar o porta-filme da cuba e deixar escorrer o
excesso de tampão em papel toalha.
Retirar o gel ácido e colocá-lo em uma placa de Petri com o corante Amido-Black por
5 minutos, sem agitação.
Retira-lo do corante Amido – Black e colocá-lo em outra placa contendo o descorante
(ácido acético 5%), por 5 minutos.
Retirar o excesso de descorante do gel e colocá-lo à 60º (secador) até secá-lo
completamente, por aproximadamente 15 minutos.
Completar descoloração deixando o gel ácido mergulhado em ácido acético 5% até
que o fundo fique transparente, se necessário trocar o descorante.
Colocar o gel ácido em uma placa de Petri com água destilada por 2 minutos.
Secar completamente o gell ácido a 60ºC (secador).


















