
avaliação de diferentes tipos de fases estacionárias para ...
189
UNIVERSIDADE FEDERAL DE MINAS GERAIS FACULDADE DE FARMÁCIA PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS FARMACÊUTICAS LAURA MARIA FONTES PRADO AVALIAÇÃO DE DIFERENTES TIPOS DE FASES ESTACIONÁRIAS PARA SEPARAÇÕES RÁPIDAS EM CROMATOGRAFIA LÍQUIDA UTILIZANDO ANTIDIABÉTICOS ORAIS COMO MODELO Belo Horizonte - MG 2013
Transcript of avaliação de diferentes tipos de fases estacionárias para ...

UNIVERSIDADE FEDERAL DE MINAS GERAIS
FACULDADE DE FARMÁCIA
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS FARMACÊUTICAS
LAURA MARIA FONTES PRADO
AVALIAÇÃO DE DIFERENTES TIPOS DE
FASES ESTACIONÁRIAS PARA SEPARAÇÕES
RÁPIDAS EM CROMATOGRAFIA LÍQUIDA
UTILIZANDO ANTIDIABÉTICOS ORAIS COMO
MODELO
Belo Horizonte - MG 2013

LAURA MARIA FONTES PRADO
AVALIAÇÃO DE DIFERENTES TIPOS DE FASES
ESTACIONÁRIAS PARA SEPARAÇÕES RÁPIDAS EM
CROMATOGRAFIA LÍQUIDA UTILIZANDO
ANTIDIABÉTICOS ORAIS COMO MODELO
Dissertação apresentada ao Programa de Pós-
Graduação em Ciências Farmacêuticas da Faculdade
de Farmácia da Universidade Federal de Minas Gerais,
como requisito parcial à obtenção do grau de Mestre
em Ciências Farmacêuticas.
Orientador: Prof. Dr. Gerson Antônio Pianetti - UFMG
Co-orientador: Prof. Dr. Christian Fernandes - UFMG
Belo Horizonte - MG 2013

Prado, Laura Maria Fontes.
P896a
Avaliação de diferentes tipos de fases estacionárias para separações
rápidas em cromatografia líquida utilizando antidiabéticos orais como
modelo / Laura Maria Fontes Prado. – 2013.
188 f. : il.
Orientador: Dr. Gerson Antônio Pianetti. Co-orientador: Dr. Christian Fernandes. Dissertação (mestrado) - Universidade Federal de Minas Gerais,
Faculdade de Farmácia, Programa de Pós-Graduação em Ciências Farmacêuticas.
1. Cromatografia líquida de alta eficiência - Teses. 2. Medicamentos – Teses. 3. Validação de método – Teses. I. Pianetti, Gerson Antônio. II. Fernandes, Christian. III. Universidade Federal de Minas Gerais. Faculdade de Farmácia. IV.Título.
CDD: 615.4


Dedico este trabalho aos meus pais, Guilherme e Dôra, pelo amor e dedicação
incondicionais à ciência. Obrigada por me apresentarem à pesquisa científica e por
serem exemplos de pesquisadores!

AGRADECIMENTOS
Ao Prof. Dr. Gerson Antônio Pianetti, por fornecer todas as condições necessárias
para o desenvolvimento dessa dissertação e por confiar no meu trabalho.
Ao professor Dr. Christian Fernandes, pela co-orientação, dedicação, contribuições
técnicas e pelas valiosas discussões.
Ao Ricardo, pela ajuda na operação do UHPLC, pela amizade e disponibilidade em
ajudar durante toda a execução desse trabalho.
Aos amigos do LCQ, Betânia, Fernando, Geovani, Iara, Isabela, Juliana, Léo,
Luciano, Mariana, Paula Enéas, Paula Chelini, Ricardo, Taízia, Tiago e Vinícius,
pelos ensinamentos e amizade.
Aos professores e funcionários da Faculdade de Farmácia da UFMG, em especial
ao Eduardo, pela agradável convivência.
À CIFARMA e à FUNED pelo fornecimento das matérias primas utilizadas.
À minha mãe, minha orientadora desde sempre! Pela preocupação e incentivo
durante toda a minha formação acadêmica. Em especial pela colaboração na
correção desse trabalho, pelas intermináveis discussões e pelo entusiasmo
contagiante, capaz de transformar a correção de uma simples frase em algo
extremamente recompensante.
Ao meu pai, pelo apoio, incentivo e terno cuidado dispensados a mim.
Aos meus irmãos, Renata, Lívia, Paula, Fábio e Andréia, pelo companheirismo e
por, literalmente, “encherem” a minha vida.
Aos meus sobrinhos (Lalá e Biel), avós, tios, primos, cunhados e eternas amigas do
CP e da faculdade, pelos momentos de descontração compartilhados.

Ao Guilherme, que sonha comigo os meus sonhos, que me permite sonhar os seus
e, assim, juntos, sonhamos os nossos! Pela dedicação, carinho, companheirismo e
por encher a minha vida de amor e alegrias.
À Deus, pela oportunidade de concluir mais uma etapa de minha formação.

“Gostaria de te desejar tantas coisas.
Mas nada seria suficiente.
Então, desejo apenas que você tenha muitos desejos.
Desejos grandes.
E que eles possam te mover a cada minuto.”
(Carlos Drummond de Andrade)
“Ambição é o caminho para o sucesso. Persistência é o veículo no qual se chega lá”.
(Bill Eardley)

RESUMO
A Cromatografia Líquida de Alta Eficiência (CLAE) convencional, que emprega fase estacionária com
partículas totalmente porosas de diâmetro entre 3-5 µm (CV), apresenta limitações no que concerne à
possibilidade de gerar análises rápidas mantendo elevada eficiência cromatográfica. Diante da
demanda por métodos rápidos e eficientes, diferentes fases estacionárias foram desenvolvidas: fases
com partículas de núcleo fundido (NF), com partículas totalmente porosas com diâmetro inferior a
2 µm (Sub 2 μm) e monolíticas (MN). Como ainda há poucos estudos que aplicam e comparam essas
novas fases, o objetivo do presente trabalho foi comparar esses novos materiais, utilizando como
modelo métodos analíticos para determinação de antidiabéticos orais (clorpropamida, glibenclamida,
glimepirida e gliclazida). Inicialmente foi desenvolvido e otimizado método em coluna CV C18 100 x
2,1 mm, 5 μm, acetonitrila:Solução acetato de amônio 5 mM pH 3,0 (50:50) como fase móvel,
volume de injeção 2 μl, detecção em 230 nm, 30 oC. Esse método foi transferido para as colunas MN
(C18 100 x 2,0 mm), NF (C18 100 x 2,1 mm, 2,7 μm) e Sub 2 μm (C18 100 x 2,1 mm, 1,8 μm), após
ajustes nas proporções de acetonitrila e solução de acetato de amônio. Todas as colunas foram
caracterizadas quanto à porosidade total, capacidade de retenção, seletividade, permeabilidade e
volume morto. Com o objetivo de reduzir o efeito extracoluna os parâmetros instrumentais volume do
loop, diâmetro interno do tubo de PEEK e frequência de aquisição dos dados/constante de tempo do
detector foram otimizados. Aplicando-se os parâmetros otimizados e os métodos para determinação
de antidiabéticos, as colunas foram comparadas em termos de eficiência (análise das curvas de Van
Deemter, de Knox e gráficos de performance cinética), velocidade de análise, resolução, simetria do
pico e pressão. A coluna monolítica, apesar de compatível com equipamentos de HPLC e de propiciar
análises mais eficientes do que a convencional em vazões elevadas, apresentou eficiência inferior a
gerada pelas colunas de núcleo fundido e Sub 2 μm, além de dar origem a picos de pior simetria.
Considerando a determinação da glimepirida como exemplo, verificou-se que coluna de núcleo
fundido gerou análises com 90% da eficiência máxima e da velocidade ótima propiciadas pela coluna
Sub 2 μm, com a vantagem de não gerar pressões acima de 400 bar, o que permite o uso de
equipamento de HPLC. Além disso, assim como a coluna Sub 2 μm, a de núcleo fundido foi capaz de
manter elevada eficiência em altas velocidades de análise e de gerar picos de boa simetria. Com o
objetivo de comparar as colunas em uma situação limite de pressão e resolução, métodos
extremamente rápidos para determinação de antidiabéticos foram desenvolvidos. Verificou-se que o
método extremo com a coluna Sub 2 μm foi o mais rápido, mas levando-se em conta velocidade de
análise, eficiência e pressão, o método extremo com a coluna de núcleo fundido foi o ideal. Assim, o
método rápido com esta coluna foi validado, utilizando-se a glibenclamida como modelo. Demonstrou-
se a capacidade da coluna de núcleo fundido em gerar resultados quantitativos confiáveis.
Palavras-chave: coluna monolítica, coluna de núcleo fundido, partículas sub 2 μm, UHPLC, curva de
Van Deemter, gráficos de performance cinética, antidiabéticos orais

ABSTRACT
Conventional High Performance Liquid Chromatography (HPLC), which uses columns packed with
totally porous particles with diameter between 3-5 µm (CV), has some limitations regarding the
possibility to generate fast analysis maintaining high efficiency. Recently new stationary phases, such
as those packed with fused core particles (FC), packed with fully porous particles with diameter less
than 2 μm (Sub 2 μm) and monolithic phases (MN) have been introduced in the market in order to
reduce analysis time maintaining high resolution and efficiency. However, there are few studies
comparing and applying these new approaches. With this in view this study aimed to compare these
new stationary phases, using analytical methods for oral antidiabetics (chlorpropamide, glibenclamide,
gliclazide and glimepiride) determination as model. Initially the followed analytical method for
antidiabetics determination was developed and optimized in CV column (C18 column 100 x 2.1 mm, 5
μm): mobile phase composed of acetonitrile and ammonium acetate buffer 5 mM pH 3.0 (50:50),
volume of injection 2 μl, detection at 230 nm, 30 oC. This method has been transferred to MN (C18
100 x 2.0 mm), NF (C18 100 x 2.1 mm, 2.7 μm) and Sub 2 μm columns (C18 100 x 2.1 mm, 1.8 μm),
after adjustments in acetonitrile and buffer proportion. The columns (CV, MN, FC and Sub 2 μm) were
characterized in terms of dead volume, porosity, retention factor, selectivity and permeability. In order
to reduce extra-column volume the followed instrumental chromatographic parameters were
optimized: loop volume, internal diameter (id) of PEEK tube and data acquisition frequency/detector
time constant. Applying the optimized parameters and the methods for antidiabetics determination, the
columns were compared in terms of performance (analysis of Van Deemter, Knox and kinetic plots),
analysis speed, resolution, peak symmetry and pressure. According to the results fused core column
is the best alternative to conventional one. Although monolithic column conducted to more efficient
analyzes at higher flow rates than conventional one, it was less efficient than fused core and Sub 2 μm
columns. Besides, peaks from monolithic column presented worse symmetry. Considering the
determination of glimepiride as example, fused core column provided analysis with 90% of maximum
efficiency and optimal speed achieved by sub 2 μm column with the advantage of generate pressure
bellow 400 bar, which allows the use of conventional HPLC instrument. Furthermore, as Sub 2 μm
column, fused core was able to maintain high efficiency at high analysis speed and generate peak with
suitable symmetry. Aiming to compare the columns in a extreme situation of pressure and resolution,
fast methods for antidiabetics determination were developed. Extremely method developed in Sub 2
μm column was the fastest, but taking into account analysis speed, efficiency and pressure, the
extreme method developed in fused core column was the ideal. Considering that fused core column
showed the best relation between speed, efficiency and pressure, a quantitative evaluation of its
performance was carried out. The extreme method in fused core column was validated for
glibenclamide and the ability of this column to provide reliable quantitative results was demonstrated.
Key words: monolithic column, fused core column, sub 2- μm particles, UHPLC, Van Deemter curve,
kinetic plot, oral antidiabetics

LISTA DE FIGURAS
INTRODUÇÃO GERAL
1 Histórico do desenvolvimento de partículas para fases estacionárias..................... 27
CAPÍTULO 1
1.1 Estrutura geral das sulfoniluréias................................................................................. 35
1.2 Estruturas químicas da CL, GB, GM e GZ..................................................................... 36
1.3 Cromatograma de rosiglitazona (1), pioglitazona (2), glipizida (3), gliclazida (4),
glibenclamida (5), celecoxibe - padrão interno (6) e repaglinida (7). (a) branco, (b)
plasma humano contaminado com os padrões dos antidiabéticos e padrão
interno (c) mistura sintética contendo antidiabéticos e o padrão interno................
40
1.4 Curva de Van Deemter.................................................................................................... 44
1.5 Espectros de varredura dos antidiabéticos.................................................................. 51
1.6 Curvas de Van Deemter da CL em diferentes fases móveis....................................... 52
1.7 Curvas de Van Deemter da GZ em diferentes fases móveis....................................... 52
1.8 Curvas de Van Deemter da GB em diferentes fases móveis...................................... 52
1.9 Curvas de Van Deemter da GM em diferentes fases móveis...................................... 53
1.10 Relação entre percentual de ACN na fase móvel e k dos antidiabéticos.................. 53
1.11 Relação entre vazão e Rs (GM/GB) nas diferentes fases móveis............................... 54
1.12 Relação entre vazão e pressão do sistema nas diferentes fases móveis................. 55
1.13 Curvas de Van Deemter da GM nas concentrações de 100 e 250 μg/ml.................... 57
1.14 Curvas de Van Deemter da CL nos diferentes volumes de injeção........................... 59
1.15 Curvas de Van Deemter da GM nos diferentes volumes de injeção.......................... 59
1.16 Cromatograma obtido durante análise de antidiabéticos em coluna convencional 61
CAPÍTULO 2
2.1 Micrografia da fase monolítica de sílica....................................................................... 69
2.2 Macroporos de 2 μm (a) e mesoporos de 13 nm (b) da fase monolítica.................... 70
2.3 Micrografia de partícula de núcleo fundido.................................................................. 74
2.4 Esquema de partícula de núcleo fundido..................................................................... 74
2.5 Relação entre a porcentagem de acetonitrila presente na fase móvel e retenção
(k) da gliclazida nas diferentes colunas........................................................................
85
2.6 Relação entre a porcentagem de acetonitrila presente na fase móvel e a
seletividade (α) do par GZ e GB nas diferentes colunas.............................................
86

2.7 Relação entre u0 e ∆Pc nas diferentes colunas........................................................... 87
CAPÍTULO 3
3.1 Esquema de equipamento de HPLC - em vermelho, os fatores que contribuem
para o efeito extracoluna -.............................................................................................. 95
3.2 Medida da largura na linha de base (4 σ) e da largura na meia altura (2,354 σ)....... 97
3.3 Relação entre vazão e σ2
extracoluna em análises com loops de 2, 5 e 10 μl................. 103
3.4 Relação entre vazão e σ2
extracoluna em análises com PEEK de 0,0025 e 0,004
polegada de di.................................................................................................................
104
3.5 Relação entre vazão e pressão em análises com PEEK de 0,0025 e 0,004
polegada de di.................................................................................................................
105
3.6 Relação entre vazão e σ2
extracoluna em análises com diferentes pares de
F/t......................................................................................................................................
106
3.7 Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna CV..... 108
3.8 Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna MN..... 108
3.9 Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna NF...... 109
3.10 Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna Sub 2
μm.....................................................................................................................................
109
3.11 Relação entre F/t e Er dos antidiabéticos - vazão de 0,6 ml/min................................ 112
3.12 Relação entre F/t e Er dos antidiabéticos - vazão de 0,15 ml/min.............................. 112
CAPÍTULO 4
4.1 Efeito da difusão de Eddy (A) no alargamento do pico............................................... 119
4.2 Efeito da difusão longitudinal (B) no alargamento do pico........................................ 119
4.3 Efeito da resistência à transferência de massa (C) no alargamento do pico............ 120
4.4 Contribuição dos termos A, B e C na obtenção da curva de Van Deemter............... 120
4.5 Curvas de Van Deemter em quatro diferentes colunas obtidas para o
etilbenzeno....................................................................................................................... 122
4.6 Gráfico de performance cinética (N x t0) ...................................................................... 128
4.7 Transformações matemáticas para o cálculo de u0 otm................................................ 132
4.8 Curvas de Van Deemter da CL nas diferentes colunas............................................... 134
4.9 Curvas de Van Deemter da GZ nas diferentes colunas............................................... 134
4.10 Curvas de Van Deemter da GB nas diferentes colunas............................................... 135
4.11 Curvas de Van Deemter da GM nas diferentes colunas.............................................. 135
4.12 Curvas de Knox da GM nas diferentes colunas........................................................... 140

4.13 Gráfico de performance cinética N x t0/N2 da GM........................................................ 141
4.14 Gráfico de performance cinética N x L da GM.............................................................. 142
4.15 Gráfico de performance cinética np x tr da GM............................................................. 143
4.16 Cromatogramas relativos às análises dos antidiabéticos nas colunas (a) CV, (b)
MN, (c) NF e (d) Sub 2 μm - ordem de eluição: CL, GZ, GB, e GM.............................. 146
4.17 Relação entre vazão e pressão nas diferentes colunas.............................................. 148
CAPÍTULO 5
5.1 Cromatogramas referentes à formulação de lidocaína - (1) metilparabeno, (2) 2,6-
dimetilanilina, (3) propilparabeno e (4) lidocaína......................................................... 157
5.2 Cromatograma das substâncias relacionadas da glimepirida a (1) e b (2),
glibenclamida (3) e glimepirida (4)................................................................................ 158
5.3 Cromatogramas referentes às análises dos antidiabéticos utilizando os métodos
extremos desenvolvidos em coluna (a) CV (b) MN (c) NF (d) Sub 2 μm - ordem de
eluição: CL, GZ, GB e GM............................................................................................... 171
5.4 Cromatogramas da GB (a) padrão, (b) amostra, (c) diluente (d) placebo - coluna
CV..................................................................................................................................... 172
5.5 Cromatogramas da GB (a) padrão, (b) amostra, (c) diluente (d) placebo - coluna
NF...................................................................................................................................... 173
5.6 Curva analítica para a determinação de GB em comprimidos - coluna CV............... 176
5.7 Curva analítica para a determinação de GB em comprimidos - coluna NF............... 176
5.8 Gráfico de distribuição de resíduos - coluna CV......................................................... 176
5.9 Gráfico de distribuição de resíduos - coluna NF......................................................... 177

LISTA DE QUADROS
CAPÍTULO 1
1.1 Concentração de glicose plasmática (mg/dl) para diagnóstico de Diabetes
Mellitus e seus estágios pré-clínicos............................................................................ 33
1.2 Medicamentos utilizados no tratamento do DM 2........................................................ 34
1.3 Propriedades físico-químicas dos fármacos CL, GB, GM e GZ.................................. 37
1.4 Mé Métodos farmacopeicos de análise de CL, GB, GM e GZ............................................ 41
1.5 Parâmetros analíticos dos métodos cromatográficos farmacopeicos para
determinação de CL, GB, GM e GZ................................................................................ 42
CAPÍTULO 3
3.1 Volumes dos componentes típicos de equipamentos de HPLC e UHPLC................ 95

LISTA DE TABELAS
CAPÍTULO 1
1.1 Fabricante, número de lote, pureza e validade dos insumos farmacêuticos
ativos dos fármacos CL, GB, GM e GZ....................................................................... 46
1.2 Condições analíticas preliminares.............................................................................. 48
1.3 Otimização das condições cromatográficas em coluna convencional................... 49
1.4 Fatores de assimetria dos picos dos antidiabéticos nas diferentes fases
móveis............................................................................................................................ 56
1.5 Fatores de assimetria dos picos dos antidiabéticos nas concentrações de 100 e
250 μg/ml....................................................................................................................... 58
1.6 Condições de análise para determinação de antidiabéticos nas diferentes
colunas e valores de k.................................................................................................. 62
CAPÍTULO 2
2.1 Condições de análise para determinação de antidiabéticos nas diferentes
colunas.......................................................................................................................... 82
2.2 Pressões máximas suportadas pelas colunas e valores de vazão máximos
empregados em cada coluna para determinação de Kv0.......................................... 82
2.3 Volume morto e porosidade total das colunas.......................................................... 83
2.4 Inclinação do gráfico das retas u0 x ∆Pc, viscosidade da fase móvel,
comprimento e permeabilidade das colunas............................................................. 87
CAPÍTULO 3
3.1 Experimentos de otimização dos parâmetros instrumentais cromatográficos...... 100
3.2 Condições de análise para determinação de antidiabéticos nas diferentes
colunas.......................................................................................................................... 101
3.3 Er para a glibenclamida nas quatro colunas - vazão de 0,3 ml/min......................... 110
3.4 Er dos antidiabéticos na coluna sub 2 μm - vazão de 0,3 ml/min............................ 111
3.5 Er da clorpropamida em diferentes vazões e relações F/t........................................ 113
3.6 Er dos antidiabéticos em diferentes relações F/t - vazão de 0,6 ml/min.................. 113
3.7 Volumes dos componentes do equipamento de UHPLC (Acquity®) e volume
extracoluna ................................................................................................................... 114
CAPÍTULO 4
4.1 Condições de análise para determinação de antidiabéticos nas quatro
colunas..........................................................................................................................
131

4.2 Equações referentes às curvas de Van Deemter da CL e GZ - Figuras 4.8 e
4.9................................................................................................................................... 136
4.3 Equações referentes às curvas de Van Deemter da GB e GM - Figuras 4.10 e
4.11.................................................................................................................................
136
4.4 Valores de Hmin e u0 opt relativos às curvas de Van Deemter das figuras 4.8 a
4.11................................................................................................................................. 138
4.5 Equações e valores de hmin e v0 opt referentes às curvas de Knox da
GM.................................................................................................................................. 140
4.6 Valores de resolução entre GZ/CL, GB/GZ e GM/GB, nas diferentes colunas........ 144
4.7 Fatores de assimetria dos picos dos antidiabéticos orais nas diferentes
colunas.......................................................................................................................... 147
CAPÍTULO 5
5.1 Condições de análise para determinação de antidiabéticos nas diferentes
colunas.......................................................................................................................... 160
5.2 Pressão máxima suportada pelas diferentes colunas.............................................. 161
5.3 Função farmacotécnica, percentuais usuais e percentuais adicionados dos
excipientes presentes no comprimido referência de glibenclamida (Daonil®)....... 164
5.4 Preparo das soluções de glibenclamida para avaliação da linearidade................. 165
5.5 Preparo das soluções para o ensaio de exatidão...................................................... 167
5.6 Distribuição das variáveis nos experimentos de robustez...................................... 168
5.7 Condições analíticas para determinação extremamente rápida de antidiabéticos
nas diferentes colunas................................................................................................. 169
5.8 Parâmetros cromatográficos obtidos em análises de antidiabéticos utilizando
os métodos extremos desenvolvidos......................................................................... 170
5.9 Valores médios e DPR dos parâmetros de adequabilidade do sistema.................. 173
5.10 Áreas das soluções padrão de trabalho e amostra por período de 4 horas........... 174
5.11 Testes estatísticos aplicados aos dados de regressão linear................................. 174
5.12 Concentrações de glibenclamida e valores de área para a construção das
curvas analíticas........................................................................................................... 175
5.13 Valores de área e teor de glibenclamida em comprimidos para avaliação da
precisão dos métodos extremos relativos às colunas CV e NF............................... 177
5.14 Percentual de recuperação de glibenclamida adicionada ao placebo para
avaliação da exatidão dos métodos extremos nas colunas CV e NF...................... 178
5.15 Limites de detecção e quantificação.......................................................................... 179
5.16 Avaliação do efeito das variáveis na determinação de teor de glibenclamida
utilizando os métodos extremos nas colunas CV e NF............................................ 180

LISTA DE ABREVIATURAS, SIGLAS E SÍMBOLOS
A Coeficiente de difusão axial ou difusão de Eddy
ACN Acetonitrila
As Fator de assimetria
AU Absorbância
B Coeficiente de difusão longitudinal
BP Farmacopeia britânica
C Coeficiente de resistência à transferência de massa
CA Chemical Abstract
CL Clorpropamida
CLAE Cromatografia Líquida de Alta Eficiência
cp Centipoise
CV Coluna convencional
Conc. Concentração
Dm Coeficiente de difusão do analito
DM 1 Diabetes Mellitus tipo 1
DM 2 Diabetes Mellitus tipo 2
di Diâmetro interno
dp Diâmetro da partícula
DPR Desvio Padrão Relativo
Porosidade total
Er Eficiência remanescente
Eur. Farmacopeia europeia
ECV Extra-column volume
exp. Experimento
F Frequência de aquisição dos dados
F. Bras Farmacopeia Brasileira
FM Fase Móvel
GB Glibenclamida
GM Glimepirida
GZ Gliclazida
H Altura do prato teórico

h Altura do prato teórico reduzido
Hmin Altura mínima do prato teórico
hmin Altura mínima do prato teórico reduzido
HPLC High Performance Liquid Chromatography
HTLC High Temperature Liquid Chromatography
IBGE Instituto Brasileiro de Geografia e Estatística
K Constante característica da qualidade da injeção
k Fator de retenção
K+ATP Canais de potássio ATP dependentes
Kv0 Permeabilidade da coluna
L Comprimento da coluna
min. Minutos ou Mínima
max. Máxima
MN Coluna monolítica
N Número de pratos teóricos
Viscosidade da fase móvel
np Fator de capacidade do pico
NF Coluna com partículas de núcleo fundido
OMS Organização Mundial de Saúde
∆Pmax Pressão máxima
∆Pc Pressão da coluna
∆Pec Pressão extracoluna
∆Pt Pressão total
P Pressão do sistema cromatográfico
PEEK Poly(ether-ether-ketone)
rc Raio da coluna
r Coeficiente de correlação linear
Rs Resolução
RENAME Relação Nacional de Medicamentos Essenciais
Sub 2 μm Coluna com partículas porosas de diâmetro menor que 2 μm
t Constante de tempo do detector
T Temperatura
tr Tempo de retenção

t0 Tempo morto
u0 Velocidade linear da fase móvel
u0 otm Velocidade linear ótima da fase móvel
UHPLC Ultra High Performance Liquid Chromatography
USP United State Pharmacopeia
V Vazão
v0 Velocidade linear da fase móvel reduzida
v0 otm Velocidade linear ótima da fase móvel reduzida
Vm Volume morto
Vinj Volume de injeção
w0,5 Largura do pico na meia altura
w Largura do pico na linha de base
Comprimento de onda
α Seletividade
2
coluna/2
c Variância da coluna
2
extracoluna /2
ec Variância extracoluna
2
total/2
t Variância total
2
ec max Variância extracoluna máxima aceitável
Desvio padrão
Fator de resistência da coluna
Fração do alargamento do pico

SUMÁRIO
INTRODUÇÃO GERAL.................................................................................. 25
1 ESTADO DA ARTE DA CLAE.................................................................... 26
2 JUSTIFICATIVA.......................................................................................... 29
3 OBJETIVO GERAL..................................................................................... 29
REFERÊNCIAS BIBLIOGRAFICAS..............................................................
30
CAPÍTULO 1 DESENVOLVIMENTO DE MÉTODOS ANALÍTICOS POR
CROMATOGRAFIA LÍQUIDA EM DIFERENTES COLUNAS PARA
DETERMINAÇÃO DE ANTIDIABÉTICOS ORAIS........................................
31
1 REVISÃO BIBLIOGRÁFICA....................................................................... 32
1.1 Diabetes Mellitus...................................................................................... 32
1.2 Tratamento............................................................................................... 33
1.3 Sulfoniluréias............................................................................................ 35
1.4 Curva de Van Deemter............................................................................. 44
2 OBJETIVOS ESPECÍFICOS....................................................................... 45
3 MATERIAIS E MÉTODOS.......................................................................... 46
3.1 Materiais................................................................................................... 46
3.1.1 Insumos farmacêuticos ativos......................................................... 46
3.1.2 Solventes, reagentes e vidrarias..................................................... 46
3.1.3 Equipamentos e colunas cromatográficas....................................... 46
3.2 Métodos.................................................................................................... 47
3.2.1 Desenvolvimento de método analítico por cromatografia líquida
para a determinação de CL, GB, GM e GZ em coluna CV............................ 47
3.2.2 Otimização de método analítico por cromatografia líquida para a
determinação CL, GB, GM e GZ em coluna CV............................................. 48
3.2.3 Transferência do método desenvolvido e otimizado em coluna CV
para as colunas MN, NF e Sub 2 μm............................................................. 49
4 RESULTADOS E DISCUSSÃO.................................................................. 50
4.1 Desenvolvimento de método analítico por cromatografia líquida para a
determinação de CL, GB, GM e GZ em coluna CV........................................ 50

4.2 Otimização de método analítico por cromatografia líquida para a
determinação de CL, GB, GM e GZ em coluna CV........................................
51
4.2.1 Fase móvel...................................................................................... 51
4.2.2 Concentração.................................................................................. 57
4.2.3 Volume de injeção........................................................................... 58
4.2.4 Temperatura.................................................................................... 61
4.3 Transferência do método desenvolvido e otimizado em coluna CV para
as colunas MN, NF e Sub 2 μm..................................................................... 62
5 CONCLUSÃO............................................................................................. 63
REFERÊNCIAS BIBLIOGRÁFICAS..............................................................
64
CAPÍTULO 2 CARACTERIZAÇÃO FÍSICA DAS COLUNAS
CROMATOGRÁFICAS..................................................................................
67
1 REVISÃO BIBLIOGRÁFICA....................................................................... 68
1.1 Coluna monolítica..................................................................................... 68
1.2 Coluna empacotada com partículas de núcleo fundido............................ 72
1.3 Coluna empacotada com partículas porosas sub 2 µm........................... 75
2 OBJETIVO ESPECÍFICO............................................................................ 78
3 MATERIAIS E MÉTODOS.......................................................................... 78
3.1 Materiais................................................................................................... 78
3.1.1 Insumos farmacêuticos ativos......................................................... 78
3.1.2 Solventes, reagentes e vidrarias..................................................... 78
3.1.3 Equipamentos e colunas cromatográficas....................................... 78
3.2 Métodos.................................................................................................... 78
3.2.1 Estimativa do volume morto e porosidade total das colunas.......... 78
3.2.2 Avaliação da retenção e seletividade das colunas.......................... 79
3.2.3 Estimativa da permeabilidade das colunas..................................... 80
4 RESULTADOS E DISCUSSÃO.................................................................. 83
4.1 Estimativa do volume morto e porosidade total das colunas................... 83
4.2 Estimativa da retenção e seletividade das colunas.................................. 84
4.2.1 Retenção......................................................................................... 84
4.2.2 Seletividade..................................................................................... 86
4.3 Estimativa da permeabilidade das colunas.............................................. 87

5 CONCLUSÃO............................................................................................. 88
REFERÊNCIAS BIBLIOGRÁFICAS.............................................................. 90
CAPÍTULO 3 OTIMIZAÇÃO DE PARÂMETROS INSTRUMENTAIS
CROMATOGRÁFICOS..................................................................................
93
1 REVISÃO BIBLIOGRÁFICA....................................................................... 94
1.1 Efeitos extracoluna e a eficiência cromatográfica.................................... 94
2 OBJETIVOS ESPECÍFICOS....................................................................... 98
3 MATERIAIS E MÉTODOS.......................................................................... 99
3.1 Materiais................................................................................................... 99
3.1.1 Insumos farmacêuticos ativos......................................................... 99
3.1.2 Solventes, reagentes e vidrarias..................................................... 99
3.1.3 Equipamentos e colunas cromatográficas....................................... 99
3.1.4 Outros.............................................................................................. 99
3.2 Métodos.................................................................................................... 99
3.2.1 Otimização dos parâmetros instrumentais cromatográficos parte
I: avaliação do efeito extracoluna................................................................... 99
3.2.2 Otimização dos parâmetros instrumentais cromatográficos parte
II: avaliação da eficiência cromatográfica remanescente............................... 101
3.2.3 Determinação do volume extracoluna real (ECV)........................... 102
4 RESULTADOS E DISCUSSÃO.................................................................. 103
4.1 Otimização dos parâmetros instrumentais cromatográficos parte I:
avaliação do efeito extracoluna...................................................................... 103
4.1.1 Volume do loop................................................................................ 103
4.1.2 Diâmetro interno da tubulação de PEEK no trajeto da coluna ao
detector........................................................................................................... 104
4.1.3 Frequência de aquisição dos dados (F) e constante de tempo do
detector (t) ..................................................................................................... 106
4.2 Otimização dos parâmetros instrumentais cromatográficos parte II:
avaliação da eficiência cromatográfica remanescente................................... 107
4.2.1 Volume do loop................................................................................ 108
4.2.2 Frequência de aquisição dos dados (F) e constante de tempo do
detector (t) ..................................................................................................... 111

4.3 Cálculo do volume extracoluna (ECV) real............................................... 114
5 CONCLUSÃO............................................................................................. 114
REFERÊNCIAS BIBLIOGRÁFICAS..............................................................
116
CAPÍTULO 4 AVALIAÇÃO E COMPARAÇÃO QUALITATIVA DA
EFICIÊNCIA DE COLUNAS CROMATOGRÁFICAS.................................... 117
1 REVISÃO BIBLIOGRÁFICA....................................................................... 118
1.1 Curvas de Van Deemter e de Knox.......................................................... 118
1.2 Características das curvas de Van Deemter e de Knox das colunas
MN, NF e Sub 2 μm........................................................................................ 122
1.2.1 Coluna monolítica........................................................................... 122
1.2.2 Coluna empacotada com partículas de núcleo fundido.................. 123
1.2.3 Coluna empacotada com partículas sub 2 μm............................... 125
1.3 Gráficos de performance cinética............................................................. 126
2 OBJETIVO ESPECÍFICO............................................................................ 129
3 MATERIAIS E MÉTODOS.......................................................................... 130
3.1 Materiais................................................................................................... 130
3.1.1 Insumos farmacêuticos ativos......................................................... 130
3.1.2 Solventes, reagentes e vidrarias..................................................... 130
3.1.3 Equipamentos e colunas cromatográficas....................................... 130
3.2 Métodos.................................................................................................... 130
3.2.1 Avaliação da eficiência e velocidade de análise das colunas....... 130
3.2.2 Avaliação da resolução, fator de assimetria e pressão................... 133
4 RESULTADOS E DISCUSSÃO.................................................................. 134
4.1 Avaliação da eficiência e velocidade de análise das colunas.................. 134
4.1.1 Curvas de Van Deemter.................................................................. 134
4.1.2 Curvas de Knox............................................................................... 140
4.1.3 Gráficos de performance cinética.................................................... 141
4.2 Avaliação da resolução, fator de assimetria e pressão............................ 143
4.2.1 Resolução........................................................................................ 143
4.2.2 Fator de assimetria dos picos.......................................................... 146
4.2.3 Pressão do sistema cromatográfico................................................ 148
5 CONCLUSÃO............................................................................................. 149

REFERÊNCIAS BIBLIOGRÁFICAS.............................................................. 152
CAPÍTULO 5 DESENVOLVIMENTO DE MÉTODOS ANALÍTICOS
EXTREMAMENTE RÁPIDOS........................................................................
155
1 REVISÃO BIBLIOGRÁFICA....................................................................... 156
1.1 Métodos analíticos extremamente rápidos............................................... 156
1.2 Métodos analíticos para determinação de sulfoniluréias.......................... 158
2 OBJETIVOS ESPECÍFICOS....................................................................... 159
3 MATERIAIS E MÉTODOS.......................................................................... 159
3.1 Materiais................................................................................................... 159
3.1.1 Insumos farmacêuticos ativos......................................................... 159
3.1.2 Solventes, reagentes e vidrarias..................................................... 159
3.1.3 Equipamentos e colunas cromatográficas....................................... 160
3.1.4 Substâncias químicas de referência e amostras............................. 160
3.2 Métodos.................................................................................................... 160
3.2.1 Desenvolvimento e comparação de métodos analíticos
extremamente rápidos para determinação de antidiabéticos nas colunas
CV, MN, NF e Sub 2 μm.................................................................................
160
3.2.2 Comparação das colunas CV e NF em termos quantitativos.......... 162
4 RESULTADOS E DISCUSSÃO.................................................................. 169
4.1 Desenvolvimento e comparação de métodos analíticos extremamente
rápidos para determinação de antidiabéticos nas colunas CV, MN, NF e
Sub 2 μm........................................................................................................ 169
4.2 Comparação das colunas CV e NF em termos quantitativos................... 172
4.2.1 Seletividade e adequabilidade do sistema (system suitability)........ 172
4.2.2 Estabilidade das soluções padrão de trabalho e amostra............... 174
4.2.3 Linearidade...................................................................................... 174
4.2.4 Precisão intra-corrida e precisão inter-corridas............................... 177
4.2.5 Exatidão........................................................................................... 178
4.2.6 Limites de detecção e quantificação............................................... 179
4.2.7 Robustez......................................................................................... 179
5 CONCLUSÃO............................................................................................. 181
REFERÊNCIAS BIBLIOGRÁFICAS.............................................................. 182

CONCLUSÃO FINAL..................................................................................... 184
APÊNDICE - Trabalhos apresentados em congresso................................... 187

25
INTRODUÇÃO GERAL

26
1 ESTADO DA ARTE DA CLAE
A Cromatografia Líquida de Alta Eficiência (CLAE), em inglês High Peformance
Liquid Chromatography (HPLC), é considerada a técnica mais desenvolvida,
difundida e empregada em laboratórios analíticos de indústrias farmacêuticas,
químicas e de pesquisa. Isso se justifica, pois esta técnica é adequada para a
separação eficiente de substâncias, o que se aplica, por exemplo, às análises de
controle de qualidade, nas quais são realizadas determinações quantitativas de
fármacos, de impurezas de síntese e de produtos de degradação, análises
farmacocinéticas e estudos pré-clínicos de moléculas candidatas a fármacos [1, 2,
3].
Desde o início do desenvolvimento da Cromatografia a Líquido, na década de 1950,
até os dias atuais, muitos avanços, impulsionados pelo desenvolvimento de novas
partículas de fases estacionárias e da instrumentação, foram alcançados. A
necessidade de execução de análises mais rápidas, sem que houvesse o
comprometimento do desempenho cromatográfico, foi o que impulsionou as
pesquisas com o objetivo de tornar a técnica mais rápida e eficiente. Em 1950 eram
utilizadas colunas empacotadas com partículas irregulares de 100-200 μm, que
alcançavam eficiência de apenas 200 pratos/15 cm. Por volta dos anos 60 foram
introduzidas as colunas contendo partículas peliculares rígidas de 40-50 μm,
mecanicamente resistentes, que ofereciam maior eficiência (1000 pratos/15 cm). A
transição para partículas porosas menores com diâmetro em torno de 10 μm ocorreu
por volta dos anos 70, propiciando eficiência de 6000 pratos/15 cm. A partir dos
anos 80, com a introdução das partículas esféricas, o desenvolvimento voltou-se
basicamente para a redução do diâmetro das partículas da fase estacionária e
surgiram as primeiras partículas esféricas de diâmetro de 5 μm. Hoje já há
disponíveis colunas com partículas esféricas porosas de 1,7 μm, que permitem
melhor resolução e alta eficiência, que pode chegar a 30000 pratos/15 cm [3]. Na
Figura 1 há um resumo do histórico do desenvolvimento de partículas para fases
estacionárias de HPLC.
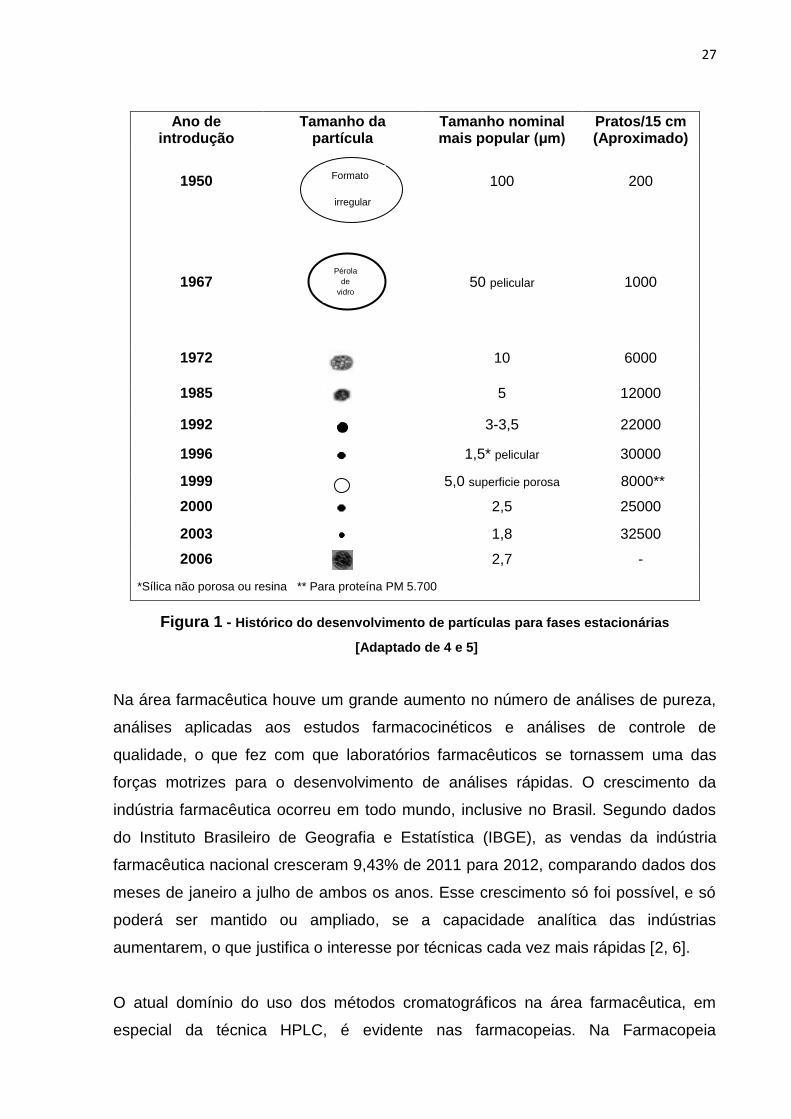
27
Ano de introdução
Tamanho da partícula
Tamanho nominal mais popular (μm)
Pratos/15 cm (Aproximado)
1950
100
200
1967
50 pelicular
1000
1972
10 6000
1985
5 12000
1992
3-3,5 22000
1996 1,5* pelicular 30000
1999 5,0 superficie porosa 8000**
2000 2,5 25000
2003 1,8 32500
2006 2,7 -
*Sílica não porosa ou resina ** Para proteína PM 5.700
Figura 1 - Histórico do desenvolvimento de partículas para fases estacionárias
[Adaptado de 4 e 5]
Na área farmacêutica houve um grande aumento no número de análises de pureza,
análises aplicadas aos estudos farmacocinéticos e análises de controle de
qualidade, o que fez com que laboratórios farmacêuticos se tornassem uma das
forças motrizes para o desenvolvimento de análises rápidas. O crescimento da
indústria farmacêutica ocorreu em todo mundo, inclusive no Brasil. Segundo dados
do Instituto Brasileiro de Geografia e Estatística (IBGE), as vendas da indústria
farmacêutica nacional cresceram 9,43% de 2011 para 2012, comparando dados dos
meses de janeiro a julho de ambos os anos. Esse crescimento só foi possível, e só
poderá ser mantido ou ampliado, se a capacidade analítica das indústrias
aumentarem, o que justifica o interesse por técnicas cada vez mais rápidas [2, 6].
O atual domínio do uso dos métodos cromatográficos na área farmacêutica, em
especial da técnica HPLC, é evidente nas farmacopeias. Na Farmacopeia
Formato
irregular
Pérola
de
vidro

28
Americana número 16, publicada em 1960, os métodos cromatográficos eram
empregados em aproximadamente 3% das monografias. Já na Farmacopeia
Americana número 34, de 2011, 77% das monografias continham ao menos um
método cromatográfico e 54% eram por HPLC. Esse aumento expressivo na
aplicação da cromatografia em análises farmacêuticas em substituição aos métodos
tradicionais (exemplos: espectrometria no visível e ultravioleta; volumetrias) resultou
em um controle mais efetivo da identidade, potência e pureza dos fármacos [7].
Atualmente a técnica HPLC que emprega fase estacionária convencional,
empacotada com partículas totalmente porosas de diâmetro entre 3 e 5 µm (CV),
ainda é a mais comum nos laboratórios. Cromatógrafos destinados a esse tipo de
cromatografia operam com valores de pressão máxima de 400 bar. Apesar de o
desempenho cromatográfico ser adequado, uma análise corrida analítica por HPLC
pode levar, em média, de 10 a 45 minutos [2, 8].
Alternativas estão sendo desenvolvidas para diminuir o tempo e melhorar a
eficiência de análises realizadas por HPLC. Uma delas refere-se à substituição das
colunas com partículas totalmente porosas tradicionais por colunas monolíticas (MN)
ou de núcleo fundido (NF). Outras opções são a Cromatografia Líquida de Ultra
Eficiência (UHPLC, do inglês Ultra High Performance Liquid Chromatography), em
que o diâmetro médio das partículas porosas constituintes da fase estacionária é
inferior a 2 µm (Sub 2 µm), e a Cromatografia Líquida de Alta Temperatura (HTLC,
do inglês High Temperature Liquid Chromatography) [9].
Nesse trabalho foi realizado um estudo dessas novas alternativas para reduzir o
tempo e melhorar a eficiência de análise por HPLC, excetuando-se a HTLC.
Portanto, colunas MN, NF e Sub 2 µm foram os objetos de estudo desse trabalho,
assim como a coluna CV, utilizada como referência. O presente estudo foi dividido
nos seguintes capítulos:
Capítulo I: Desenvolvimento de métodos analíticos por cromatografia líquida em
diferentes colunas para determinação de antidiabéticos orais
Capítulo II: Caracterização física das colunas cromatográficas

29
Capítulo III: Otimização de parâmetros instrumentais cromatográficos
Capítulo IV: Avaliação e comparação qualitativa da eficiência de colunas
cromatográficas
Capítulo V: Desenvolvimento de métodos analíticos extremamente rápidos
2 JUSTIFICATIVA
O atual crescimento das indústrias farmacêuticas vem acompanhado de uma
demanda por análises cada vez mais rápidas e, ao mesmo tempo, eficientes. As
análises rápidas por Cromatografia a Líquido de Alta Eficiência foram desenvolvidas
a partir do ano 2000, e ainda há poucos estudos em que se aplicam e comparam as
novas abordagens. Dentre essas abordagens destacam-se a utilização de colunas
cromatográficas monolíticas (MN), de partículas de núcleo fundido (NF) e de
partículas porosas sub 2 μm (Sub 2 μm). A importância da utilização dessas colunas
modernas em análises farmacêuticas justifica as pesquisas sobre o assunto, tendo
em vista a demanda por análises rápidas e eficientes.
Antidiabéticos orais (clorpropamida-CL, glibenclamida-GB, glimepirida-GM e
gliclazida-GZ) foram escolhidos como modelo para o estudo de diferentes tipos de
fases estacionárias para separações rápidas em cromatografia líquida. A escolha
desses fármacos se justifica pela sua ampla utilização, que é consequência da
elevada prevalência de Diabetes Mellitus tipo 2 na população. Faz-se necessário,
portanto, métodos rápidos e eficientes para o controle de qualidade desses
fármacos. Outra razão para a escolha desses fármacos é o fato de que eles, por
apresentarem polaridades diferentes, apresentam também retenções bem distintas,
o que possibilita que as colunas sejam comparadas de uma maneira mais completa.
3 OBJETIVO GERAL
Avaliar e comparar diferentes tipos de colunas cromatográficas (CV, MN, NF e Sub 2
μm) para separações rápidas utilizando fármacos antidiabéticos orais (CL, GB, GM e
GZ) como modelo.

30
REFERÊNCIAS BIBLIOGRÁFICAS
[1] AL-SAYAH, M. A.; RIZOS, P.; ANTONUCCI, V.; WU, N. High throughput screening of active pharmaceutical ingredients by UPLC. Journal of Separation Science. v. 31, p. 2167-2172, 2008. [2] MALDANER, L.; JARDIM, I. C. S. F. O estado da arte da cromatografia líquida de ultra eficiência. Química Nova. v. 32, n. 32, p. 214-222, 2009. [3] WREN, S. A. C.; TCHELITCHEFF, P. Use of ultra performance liquid chromatography in pharmaceutical development. Journal of Chromatography A. v. 1119, p. 140-146, 2006. [4] MAJORS, R. E. Fast and ultrafast HPLC on sub-2-µm porous particles - where do we go from here? LCGC North America. v. 23, n. 12, p. 1248-1255, 2005. [5] GUIOCHON, G.; GRITTI, F. Shell particles, trials, tribulations and triumphs. Journal of Chromatography A, v. 1218, p. 1915-1938, 2011. [6] INDICADORES ECONÔMICOS, 2012. Disponível em: <http://www. sindusfarma comunica.org.br/indicadores-economicos>. Acesso em: 30 de Agosto de 2012. [7] GÖRÖG, S. The paradigm shifting role of chromatographic methods in pharmaceutical analysis. Journal of Pharmaceutical and Biomedical Analysis. v. 69, p. 2-8, 2012. [8] GUILLARME, D.; NGUYEN, D.; RUDAZ, S.; VEUTHEY, J. Method transfer for fast liquid chromatography in pharmaceutical analysis: Application to short columns packed with small particle. Part I: Isocratic separation. European Journal of Pharmaceutics and Biopharmaceutics. v. 66, p. 475-482, 2007. [9] GUILLARME, D.; RUTA J.; RUDAZ, J.; VEUTHEY, J. New trends in fast and high-resolution liquid chromatography: a critical comparison of existing approaches. Analytical and Bioanalytical Chemistry. v. 397, p. 1069-1082, 2010.

31
CAPÍTULO 1
DESENVOLVIMENTO DE MÉTODOS ANALÍTICOS POR
CROMATOGRAFIA LÍQUIDA EM DIFERENTES COLUNAS
PARA DETERMINAÇÃO DE ANTIDIABÉTICOS ORAIS

32
1 REVISÃO BIBLIOGRÁFICA
1.1 Diabetes Mellitus
De acordo com dados da Organização Mundial de Saúde (OMS), aproximadamente
346 milhões de pessoas sofrem de Diabetes Mellitus (DM) no mundo. Em 2004,
cerca de 3,4 milhões de pessoas morreram em consequência de DM e de acordo
com projeções da OMS, entre 2005 e 2030, o número de mortes irá dobrar [1].
O DM não é uma única doença, mas um grupo heterogêneo de distúrbios
metabólicos que apresenta em comum a hiperglicemia, a qual é resultado de
defeitos na ação ou na secreção de insulina ou ambos. Atualmente é classificado
como DM tipo 1 (DM 1), DM tipo 2 (DM 2), DM gestacional e outros tipos específicos
[2].
O DM 1, presente em 5% a 10% dos casos, é o resultado da destruição de células
beta pancreáticas, com consequente deficiência da produção de insulina. Na maioria
das vezes essa destruição de células beta é mediada por autoimunidade, porém
existem casos em que não há evidência do processo autoimune, sendo, portanto,
referida como forma idiopática. O tratamento envolve a administração diária de
insulina. Sintomas incluem poliúria, polidipsia, fome constante, perda de peso,
alterações na visão e fadiga [1, 2].
Já o DM 2, presente em 90% a 95% dos casos, caracteriza-se por defeitos na ação
e secreção da insulina. A maioria dos pacientes apresenta sobrepeso ou obesidade
e a doença geralmente se manifesta após os 40 anos. Os pacientes não necessitam
de insulina exógena para sobreviver, entretanto podem necessitar da insulina para
controle metabólico adequado. Os sintomas são similares aos do DM 1, entretanto,
menos evidentes, o que faz com que a doença muitas vezes seja diagnosticada de
forma tardia. O tratamento atual visa diminuir a resistência à insulina e melhorar a
função da célula beta pancreática com dieta, exercícios, antidiabéticos orais, drogas
antiobesidade e insulina [1, 3].
33
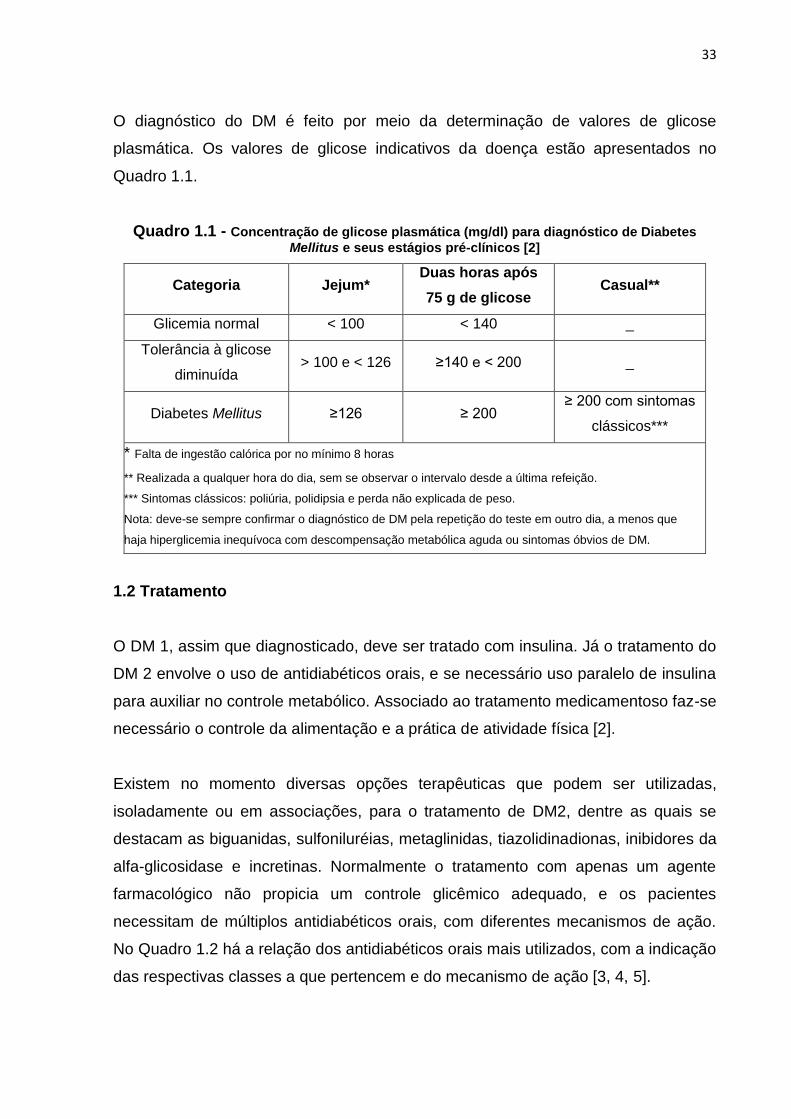
O diagnóstico do DM é feito por meio da determinação de valores de glicose
plasmática. Os valores de glicose indicativos da doença estão apresentados no
Quadro 1.1.
Quadro 1.1 - Concentração de glicose plasmática (mg/dl) para diagnóstico de Diabetes
Mellitus e seus estágios pré-clínicos [2]
Categoria Jejum* Duas horas após
75 g de glicose Casual**
Glicemia normal < 100 < 140 _
Tolerância à glicose
diminuída > 100 e < 126 ≥140 e < 200 _
Diabetes Mellitus ≥126 ≥ 200 ≥ 200 com sintomas
clássicos***
* Falta de ingestão calórica por no mínimo 8 horas
** Realizada a qualquer hora do dia, sem se observar o intervalo desde a última refeição.
*** Sintomas clássicos: poliúria, polidipsia e perda não explicada de peso.
Nota: deve-se sempre confirmar o diagnóstico de DM pela repetição do teste em outro dia, a menos que
haja hiperglicemia inequívoca com descompensação metabólica aguda ou sintomas óbvios de DM.
1.2 Tratamento
O DM 1, assim que diagnosticado, deve ser tratado com insulina. Já o tratamento do
DM 2 envolve o uso de antidiabéticos orais, e se necessário uso paralelo de insulina
para auxiliar no controle metabólico. Associado ao tratamento medicamentoso faz-se
necessário o controle da alimentação e a prática de atividade física [2].
Existem no momento diversas opções terapêuticas que podem ser utilizadas,
isoladamente ou em associações, para o tratamento de DM2, dentre as quais se
destacam as biguanidas, sulfoniluréias, metaglinidas, tiazolidinadionas, inibidores da
alfa-glicosidase e incretinas. Normalmente o tratamento com apenas um agente
farmacológico não propicia um controle glicêmico adequado, e os pacientes
necessitam de múltiplos antidiabéticos orais, com diferentes mecanismos de ação.
No Quadro 1.2 há a relação dos antidiabéticos orais mais utilizados, com a indicação
das respectivas classes a que pertencem e do mecanismo de ação [3, 4, 5].
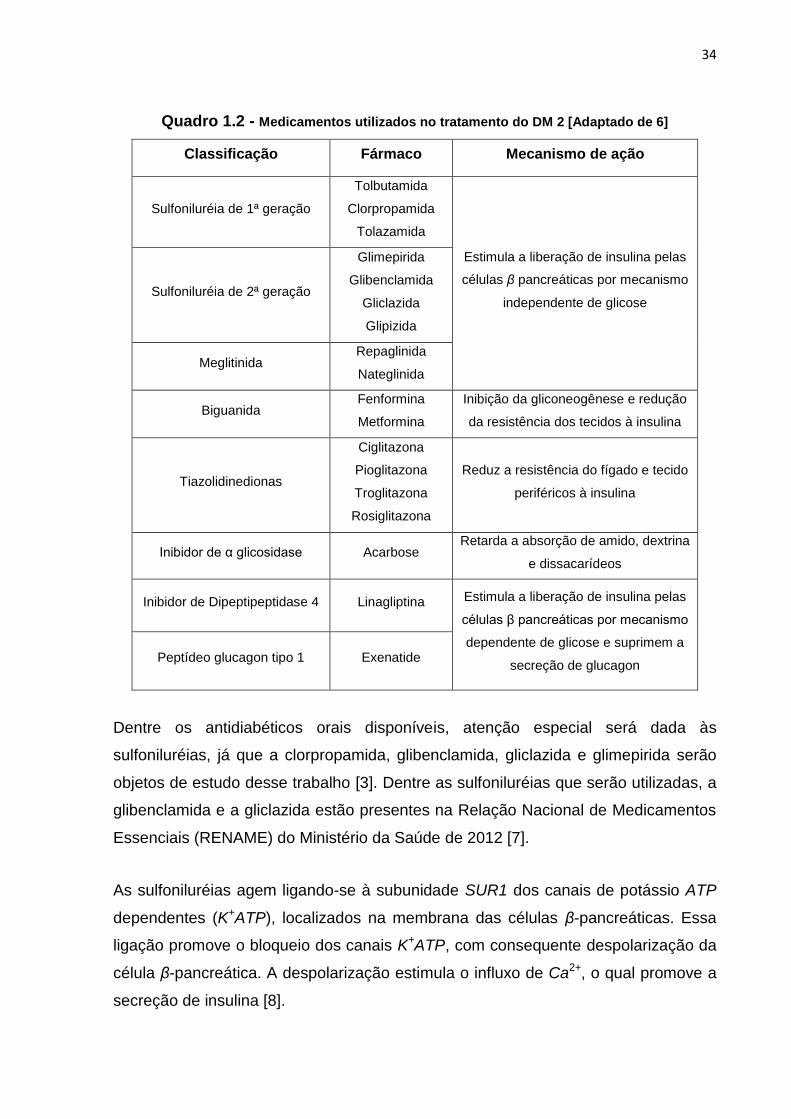
34
Quadro 1.2 - Medicamentos utilizados no tratamento do DM 2 [Adaptado de 6]
Classificação Fármaco Mecanismo de ação
Sulfoniluréia de 1ª geração
Tolbutamida
Clorpropamida
Tolazamida
Estimula a liberação de insulina pelas
células β pancreáticas por mecanismo
independente de glicose Sulfoniluréia de 2ª geração
Glimepirida
Glibenclamida
Gliclazida
Glipizida
Meglitinida Repaglinida
Nateglinida
Biguanida Fenformina
Metformina
Inibição da gliconeogênese e redução
da resistência dos tecidos à insulina
Tiazolidinedionas
Ciglitazona
Pioglitazona
Troglitazona
Rosiglitazona
Reduz a resistência do fígado e tecido
periféricos à insulina
Inibidor de α glicosidase Acarbose Retarda a absorção de amido, dextrina
e dissacarídeos
Inibidor de Dipeptipeptidase 4 Linagliptina Estimula a liberação de insulina pelas
células β pancreáticas por mecanismo
dependente de glicose e suprimem a
secreção de glucagon Peptídeo glucagon tipo 1 Exenatide
Dentre os antidiabéticos orais disponíveis, atenção especial será dada às
sulfoniluréias, já que a clorpropamida, glibenclamida, gliclazida e glimepirida serão
objetos de estudo desse trabalho [3]. Dentre as sulfoniluréias que serão utilizadas, a
glibenclamida e a gliclazida estão presentes na Relação Nacional de Medicamentos
Essenciais (RENAME) do Ministério da Saúde de 2012 [7].
As sulfoniluréias agem ligando-se à subunidade SUR1 dos canais de potássio ATP
dependentes (K+ATP), localizados na membrana das células β-pancreáticas. Essa
ligação promove o bloqueio dos canais K+ATP, com consequente despolarização da
célula β-pancreática. A despolarização estimula o influxo de Ca2+, o qual promove a
secreção de insulina [8].

35
Por aumentarem a secreção de insulina, as sulfoniluréias são chamadas de
“secretagogos de insulina”. Em curto prazo promovem o aumento da secreção de
insulina, mas a longo prazo essa secreção pode estar igual ou até diminuída em
relação aos níveis iniciais. No entanto o efeito hipoglicemiante persiste devido às
ações extra-pancreáticas. Alguns estudos sugerem que as sulfoniluréias aumentam
o número de receptores de insulina e/ou possuem efeito pós-receptor, facilitando as
ações desse hormônio, além de atuarem reduzindo a sua depuração hepática [3, 9].
1.3 Sulfoniluréias
Sulfoniluréias são arilsulfoniluréias que diferem quanto aos substituintes na posição
para do anel benzênico e no nitrogênio terminal do grupo ureia (Figura 1.1) [9].
S
O
O
N
R2
O
N
R1
H H
Figura 1.1 - Estrutura geral das sulfoniluréias [10]
As sulfoniluréias são classificadas como de 1a geração (clorpropamida, tolazamida e
tolbutamida) e de 2a geração (glibenclamida, gliclazida, glimepirida e glipizida) [9]. As
estruturas químicas da clorpropamida (CL), glibenclamida (GB), glimepirida (GM) e
gliclazida (GZ) estão representadas na Figura 1.2.
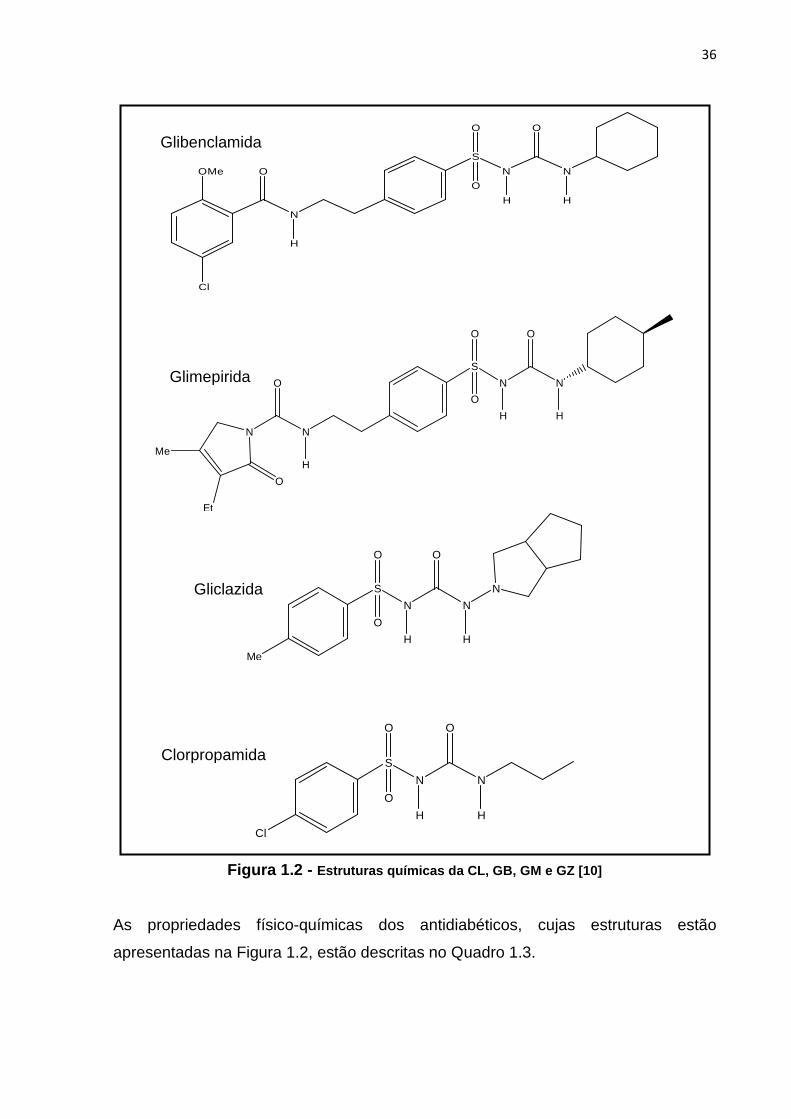
36
S
O
O
N
O
N
H H
N
OOMe
Cl
H
S
O
O
N
O
N
H H
N
O
N
H
O
Et
Me
S
O
O
N
Me
O
N
N
H H
S
O
O
N
Cl
O
N
H H Figura 1.2 - Estruturas químicas da CL, GB, GM e GZ [10]
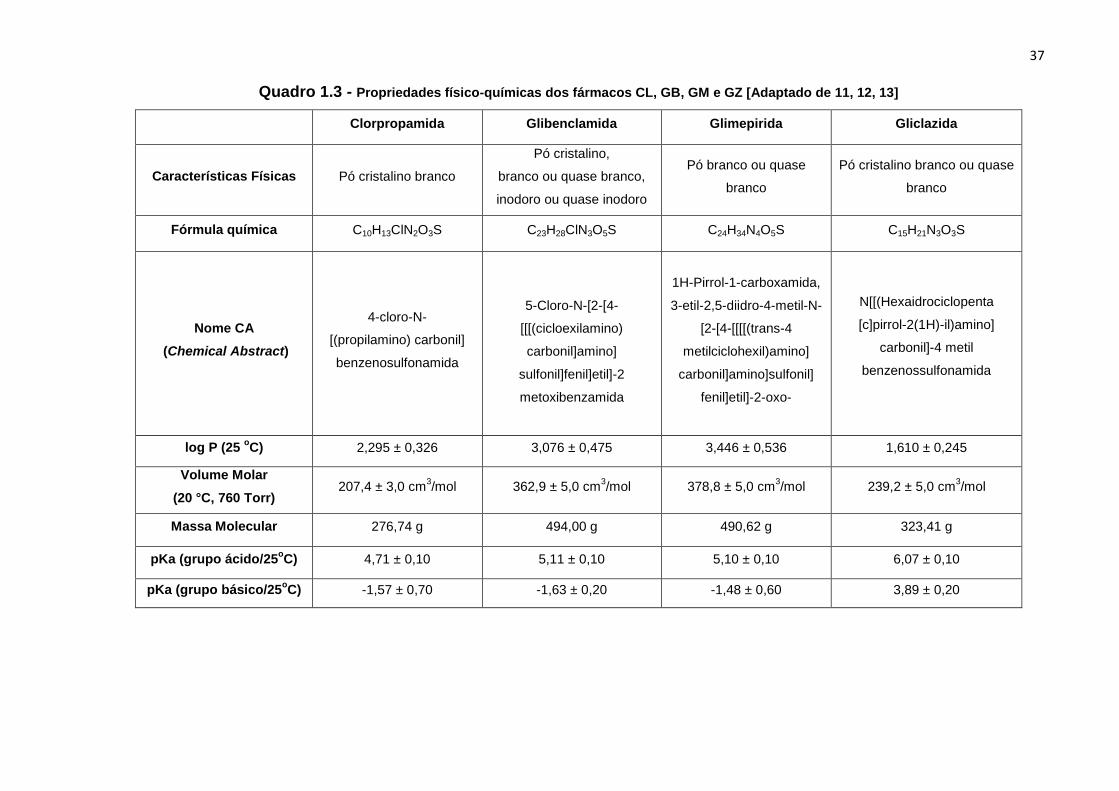
As propriedades físico-químicas dos antidiabéticos, cujas estruturas estão
apresentadas na Figura 1.2, estão descritas no Quadro 1.3.
Glibenclamida
Clorpropamida
Gliclazida
Glimepirida
37
Quadro 1.3 - Propriedades físico-químicas dos fármacos CL, GB, GM e GZ [Adaptado de 11, 12, 13]
Clorpropamida Glibenclamida Glimepirida Gliclazida
Características Físicas Pó cristalino branco
Pó cristalino,
branco ou quase branco,
inodoro ou quase inodoro
Pó branco ou quase
branco
Pó cristalino branco ou quase
branco
Fórmula química C10H13ClN2O3S C23H28ClN3O5S C24H34N4O5S C15H21N3O3S
Nome CA
(Chemical Abstract)
4-cloro-N-
[(propilamino) carbonil]
benzenosulfonamida
5-Cloro-N-[2-[4-
[[[(cicloexilamino)
carbonil]amino]
sulfonil]fenil]etil]-2
metoxibenzamida
1H-Pirrol-1-carboxamida,
3-etil-2,5-diidro-4-metil-N-
[2-[4-[[[[(trans-4
metilciclohexil)amino]
carbonil]amino]sulfonil]
fenil]etil]-2-oxo-
N[[(Hexaidrociclopenta
[c]pirrol-2(1H)-il)amino]
carbonil]-4 metil
benzenossulfonamida
log P (25 oC) 2,295 ± 0,326 3,076 ± 0,475 3,446 ± 0,536 1,610 ± 0,245
Volume Molar
(20 °C, 760 Torr) 207,4 ± 3,0 cm
3/mol 362,9 ± 5,0 cm
3/mol 378,8 ± 5,0 cm
3/mol 239,2 ± 5,0 cm
3/mol
Massa Molecular 276,74 g 494,00 g 490,62 g 323,41 g
pKa (grupo ácido/25oC) 4,71 ± 0,10 5,11 ± 0,10 5,10 ± 0,10 6,07 ± 0,10
pKa (grupo básico/25oC) -1,57 ± 0,70 -1,63 ± 0,20 -1,48 ± 0,60 3,89 ± 0,20
38
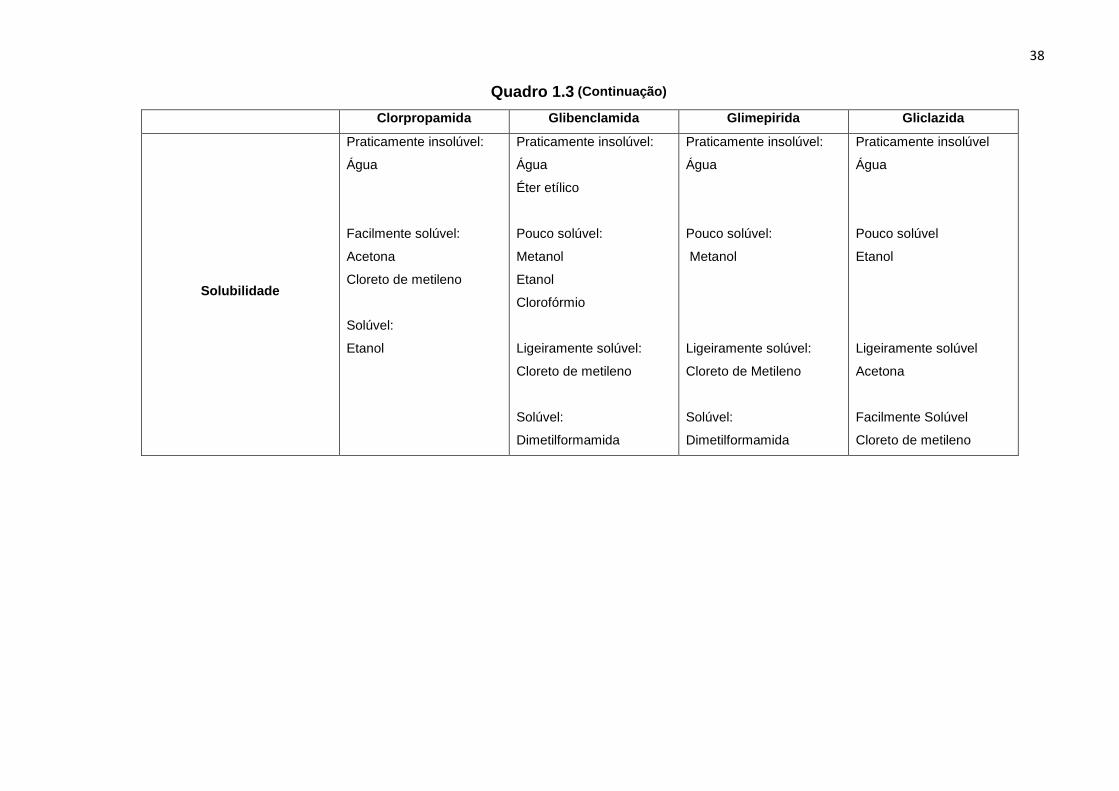
Quadro 1.3 (Continuação)
Clorpropamida Glibenclamida Glimepirida Gliclazida
Solubilidade
Praticamente insolúvel:
Água
Facilmente solúvel:
Acetona
Cloreto de metileno
Solúvel:
Etanol
Praticamente insolúvel:
Água
Éter etílico
Pouco solúvel:
Metanol
Etanol
Clorofórmio
Ligeiramente solúvel:
Cloreto de metileno
Solúvel:
Dimetilformamida
Praticamente insolúvel:
Água
Pouco solúvel:
Metanol
Ligeiramente solúvel:
Cloreto de Metileno
Solúvel:
Dimetilformamida
Praticamente insolúvel
Água
Pouco solúvel
Etanol
Ligeiramente solúvel
Acetona
Facilmente Solúvel
Cloreto de metileno

39
Encontram-se descritos na literatura inúmeros métodos para quantificação de
sulfoniluréias isoladamente ou associadas a fármacos de outras classes de
antidiabéticos orais.
Em trabalho desenvolvido para quantificar glimepirida em formulações
farmacêuticas, empregaram-se coluna C18 250 x 4,6 mm, 5 μm, detecção em 228
nm, vazão de 0,5 ml/min e fase móvel composta por acetonitrila e solução de ácido
fórmico 2%, pH 3,5 (80:20). O tempo de retenção foi de 7 minutos [14].
Em outro trabalho, com o intuito de determinar gliclazida, utilizaram-se coluna C18,
detecção em 230 nm, fase móvel composta de solução aquosa de fosfato de sódio
monobásico 0,1% p/v pH 2,1 ajustado com ácido fosfórico e acetonitrila (34:66). O
tempo de retenção foi de 5,4 minutos [15].
Em estudo realizado em 2011 empregou-se método por HPLC, modo isocrático,
para análise de comprimidos contendo glibenclamida. Para esta análise foram
utilizados coluna C18 250 x 4,6 mm, 5 µm, vazão de 1,0 ml/min e volume de injeção
20 µl. A análise foi realizada a temperatura ambiente e leitura em 210 nm. O tempo
de retenção da glibenclamida foi de 3,0 minutos [16].
Em estudo publicado em 2010, para determinação de clorpropamida e substâncias
relacionadas, empregou-se coluna C18 250 x 4,6 mm, 5 µm, vazão de 1,0 ml/min,
fase móvel composta de acetonitrila e diidrogenofosfato de amônia pH 3,5 ajustado
com ácido fosfórico (62,5:37,5) [17].
Há também na literatura a descrição de métodos para determinação simultânea de
antidiabéticos. Foi desenvolvido e validado método para determinação simultânea de
glibenclamida, gliclazida, glipizida, pioglitazona, repaglinida e rosiglitazona em
formulações farmacêuticas e em plasma. Empregaram-se coluna C18 250 x 4,6 mm,
5 μm, vazão de 1,0 ml/min, detecção em 260 nm e programa de gradiente de fase
móvel, composto por A (solução aquosa de ácido fórmico 0,05 M pH 3.0), B
(Água:Acetonitrila 5:95) e C (Água:Metanol 10:90). Na Figura 1.3 há um
cromatograma referente à análise [18].
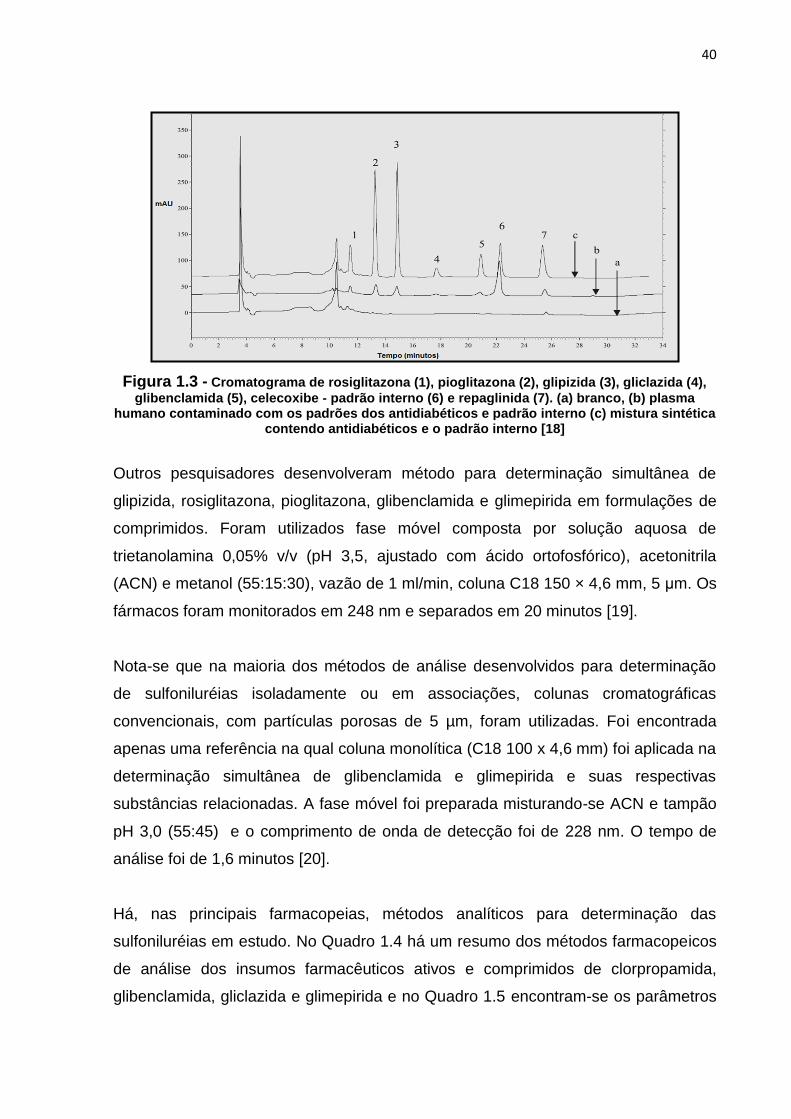
40
Figura 1.3 - Cromatograma de rosiglitazona (1), pioglitazona (2), glipizida (3), gliclazida (4),
glibenclamida (5), celecoxibe - padrão interno (6) e repaglinida (7). (a) branco, (b) plasma humano contaminado com os padrões dos antidiabéticos e padrão interno (c) mistura sintética
contendo antidiabéticos e o padrão interno [18]
Outros pesquisadores desenvolveram método para determinação simultânea de
glipizida, rosiglitazona, pioglitazona, glibenclamida e glimepirida em formulações de
comprimidos. Foram utilizados fase móvel composta por solução aquosa de
trietanolamina 0,05% v/v (pH 3,5, ajustado com ácido ortofosfórico), acetonitrila
(ACN) e metanol (55:15:30), vazão de 1 ml/min, coluna C18 150 × 4,6 mm, 5 μm. Os
fármacos foram monitorados em 248 nm e separados em 20 minutos [19].
Nota-se que na maioria dos métodos de análise desenvolvidos para determinação
de sulfoniluréias isoladamente ou em associações, colunas cromatográficas
convencionais, com partículas porosas de 5 µm, foram utilizadas. Foi encontrada
apenas uma referência na qual coluna monolítica (C18 100 x 4,6 mm) foi aplicada na
determinação simultânea de glibenclamida e glimepirida e suas respectivas
substâncias relacionadas. A fase móvel foi preparada misturando-se ACN e tampão
pH 3,0 (55:45) e o comprimento de onda de detecção foi de 228 nm. O tempo de
análise foi de 1,6 minutos [20].
Há, nas principais farmacopeias, métodos analíticos para determinação das
sulfoniluréias em estudo. No Quadro 1.4 há um resumo dos métodos farmacopeicos
de análise dos insumos farmacêuticos ativos e comprimidos de clorpropamida,
glibenclamida, gliclazida e glimepirida e no Quadro 1.5 encontram-se os parâmetros
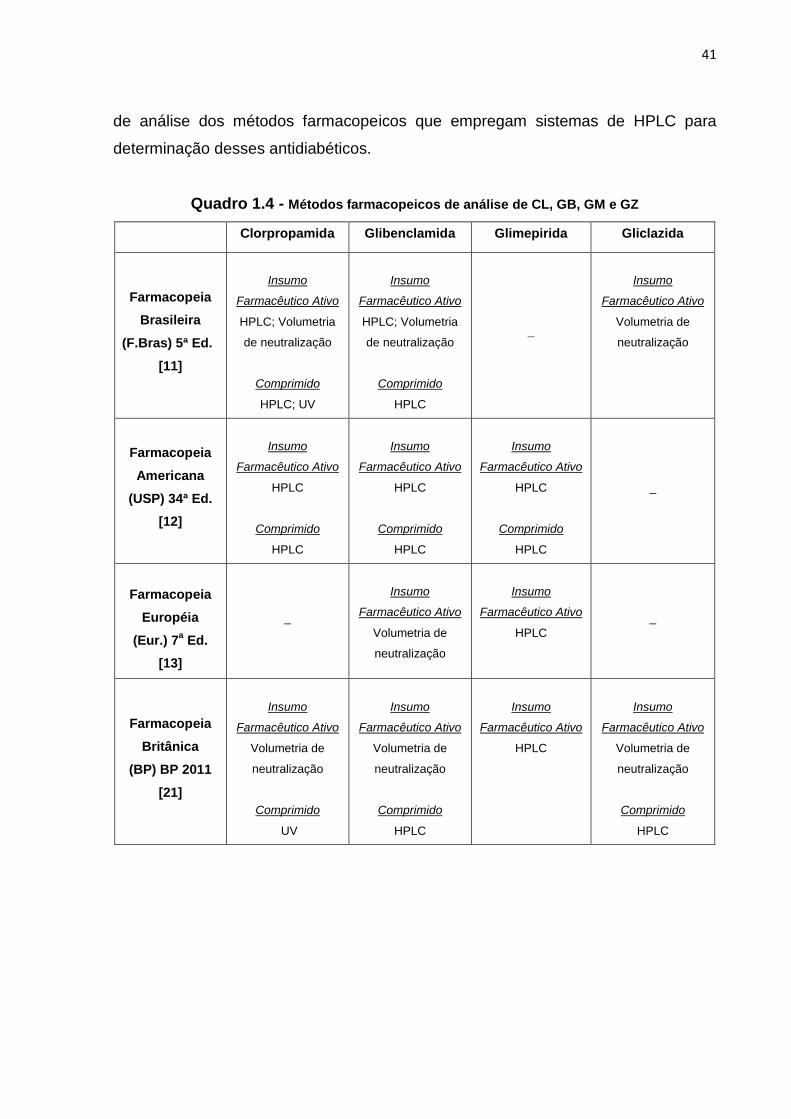
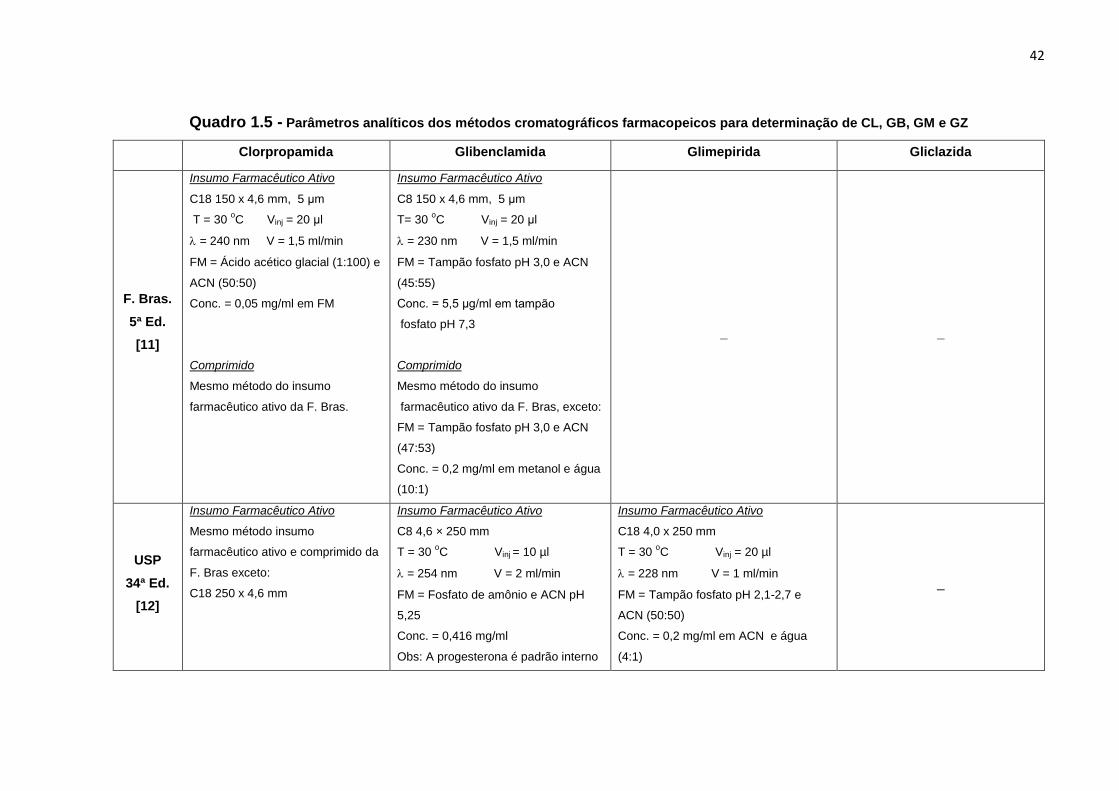
41
de análise dos métodos farmacopeicos que empregam sistemas de HPLC para
determinação desses antidiabéticos.
Quadro 1.4 - Métodos farmacopeicos de análise de CL, GB, GM e GZ
Clorpropamida Glibenclamida Glimepirida Gliclazida
Farmacopeia
Brasileira
(F.Bras) 5ª Ed.
[11]
Insumo
Farmacêutico Ativo
HPLC; Volumetria
de neutralização
Comprimido
HPLC; UV
Insumo
Farmacêutico Ativo
HPLC; Volumetria
de neutralização
Comprimido
HPLC
_
Insumo
Farmacêutico Ativo
Volumetria de
neutralização
Farmacopeia
Americana
(USP) 34ª Ed.
[12]
Insumo
Farmacêutico Ativo
HPLC
Comprimido
HPLC
Insumo
Farmacêutico Ativo
HPLC
Comprimido
HPLC
Insumo
Farmacêutico Ativo
HPLC
Comprimido
HPLC
_
Farmacopeia
Européia
(Eur.) 7a Ed.
[13]
_
Insumo
Farmacêutico Ativo
Volumetria de
neutralização
Insumo
Farmacêutico Ativo
HPLC
_
Farmacopeia
Britânica
(BP) BP 2011
[21]
Insumo
Farmacêutico Ativo
Volumetria de
neutralização
Comprimido
UV
Insumo
Farmacêutico Ativo
Volumetria de
neutralização
Comprimido
HPLC
Insumo
Farmacêutico Ativo
HPLC
Insumo
Farmacêutico Ativo
Volumetria de
neutralização
Comprimido
HPLC
42
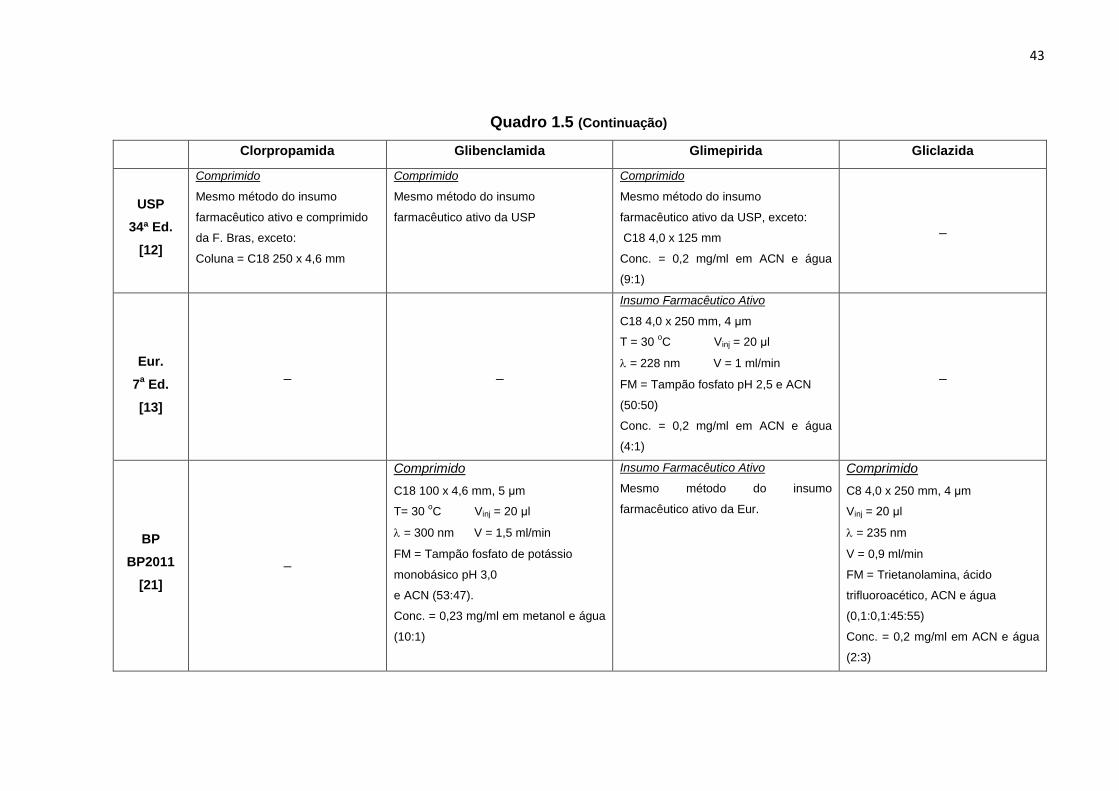
Quadro 1.5 - Parâmetros analíticos dos métodos cromatográficos farmacopeicos para determinação de CL, GB, GM e GZ
Clorpropamida Glibenclamida Glimepirida Gliclazida
F. Bras.
5ª Ed.
[11]
Insumo Farmacêutico Ativo
C18 150 x 4,6 mm, 5 μm
T = 30 oC Vinj = 20 μl
= 240 nm V = 1,5 ml/min
FM = Ácido acético glacial (1:100) e
ACN (50:50)
Conc. = 0,05 mg/ml em FM
Comprimido
Mesmo método do insumo
farmacêutico ativo da F. Bras.
Insumo Farmacêutico Ativo
C8 150 x 4,6 mm, 5 μm
T= 30 oC Vinj = 20 μl
= 230 nm V = 1,5 ml/min
FM = Tampão fosfato pH 3,0 e ACN
(45:55)
Conc. = 5,5 μg/ml em tampão
fosfato pH 7,3
Comprimido
Mesmo método do insumo
farmacêutico ativo da F. Bras, exceto:
FM = Tampão fosfato pH 3,0 e ACN
(47:53)
Conc. = 0,2 mg/ml em metanol e água
(10:1)
_ _
USP
34ª Ed.
[12]
Insumo Farmacêutico Ativo
Mesmo método insumo
farmacêutico ativo e comprimido da
F. Bras exceto:
C18 250 x 4,6 mm
Insumo Farmacêutico Ativo
C8 4,6 × 250 mm
T = 30 oC Vinj = 10 µl
= 254 nm V = 2 ml/min
FM = Fosfato de amônio e ACN pH
5,25
Conc. = 0,416 mg/ml
Obs: A progesterona é padrão interno
Insumo Farmacêutico Ativo
C18 4,0 x 250 mm
T = 30 oC Vinj = 20 µl
= 228 nm V = 1 ml/min
FM = Tampão fosfato pH 2,1-2,7 e
ACN (50:50)
Conc. = 0,2 mg/ml em ACN e água
(4:1)
_
43
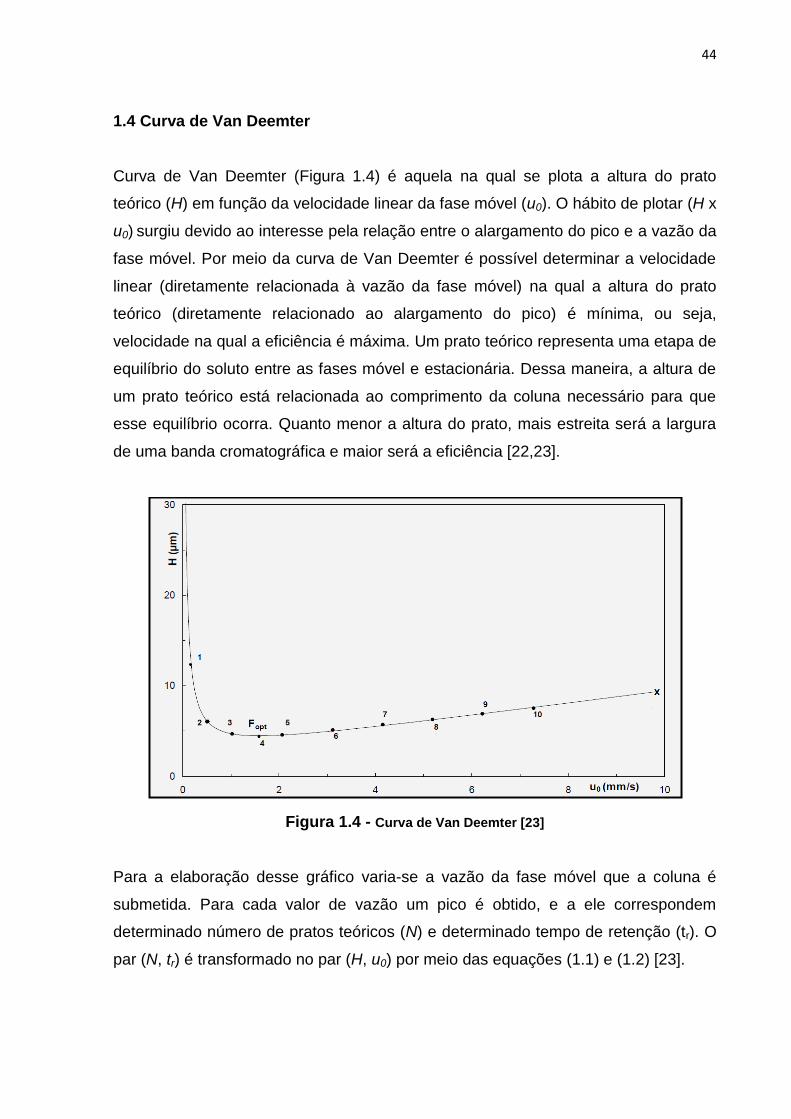
Quadro 1.5 (Continuação)
Clorpropamida Glibenclamida Glimepirida Gliclazida
USP
34ª Ed.
[12]
Comprimido
Mesmo método do insumo
farmacêutico ativo e comprimido
da F. Bras, exceto:
Coluna = C18 250 x 4,6 mm
Comprimido
Mesmo método do insumo
farmacêutico ativo da USP
Comprimido
Mesmo método do insumo
farmacêutico ativo da USP, exceto:
C18 4,0 x 125 mm
Conc. = 0,2 mg/ml em ACN e água
(9:1)
_
Eur.
7a Ed.
[13]
_ _
Insumo Farmacêutico Ativo
C18 4,0 x 250 mm, 4 μm
T = 30 oC Vinj = 20 μl
= 228 nm V = 1 ml/min
FM = Tampão fosfato pH 2,5 e ACN
(50:50)
Conc. = 0,2 mg/ml em ACN e água
(4:1)
_
BP
BP2011
[21]
_
Comprimido
C18 100 x 4,6 mm, 5 μm
T= 30 oC Vinj = 20 μl
= 300 nm V = 1,5 ml/min
FM = Tampão fosfato de potássio
monobásico pH 3,0
e ACN (53:47).
Conc. = 0,23 mg/ml em metanol e água
(10:1)
Insumo Farmacêutico Ativo
Mesmo método do insumo
farmacêutico ativo da Eur.
Comprimido
C8 4,0 x 250 mm, 4 μm
Vinj = 20 μl
= 235 nm
V = 0,9 ml/min
FM = Trietanolamina, ácido
trifluoroacético, ACN e água
(0,1:0,1:45:55)
Conc. = 0,2 mg/ml em ACN e água
(2:3)
44
1.4 Curva de Van Deemter
Curva de Van Deemter (Figura 1.4) é aquela na qual se plota a altura do prato
teórico (H) em função da velocidade linear da fase móvel (u0). O hábito de plotar (H x
u0) surgiu devido ao interesse pela relação entre o alargamento do pico e a vazão da
fase móvel. Por meio da curva de Van Deemter é possível determinar a velocidade
linear (diretamente relacionada à vazão da fase móvel) na qual a altura do prato
teórico (diretamente relacionado ao alargamento do pico) é mínima, ou seja,
velocidade na qual a eficiência é máxima. Um prato teórico representa uma etapa de
equilíbrio do soluto entre as fases móvel e estacionária. Dessa maneira, a altura de
um prato teórico está relacionada ao comprimento da coluna necessário para que
esse equilíbrio ocorra. Quanto menor a altura do prato, mais estreita será a largura
de uma banda cromatográfica e maior será a eficiência [22,23].
Figura 1.4 - Curva de Van Deemter [23]
Para a elaboração desse gráfico varia-se a vazão da fase móvel que a coluna é
submetida. Para cada valor de vazão um pico é obtido, e a ele correspondem
determinado número de pratos teóricos (N) e determinado tempo de retenção (tr). O
par (N, tr) é transformado no par (H, u0) por meio das equações (1.1) e (1.2) [23].

45
N
LH (1.1)
10 kt
Lu
r
(1.2)
H = altura do prato teórico, L = comprimento da coluna, N = número de pratos
teóricos, u0 = velocidade linear da fase móvel, tr = tempo de retenção, k = fator de
retenção.
O cálculo de k, necessário para estimar u0, é realizado utilizando-se a equação (1.3)
[24].
0
0
t
ttk r (1.3)
k = fator de retenção, tr = tempo de retenção e t0 = tempo morto.
2 OBJETIVOS ESPECÍFICOS
Foram objetivos dessa etapa do trabalho:
1- Desenvolver e otimizar método por cromatografia líquida utilizando coluna
convencional com partículas porosas de 5 µm de diâmetro (CV), para a
determinação simultânea dos antidiabéticos orais clorpropamida (CL), glibenclamida
(GB), glimepirida (GM) e gliclazida (GZ).
2- Transferir o método desenvolvido em coluna convencional para método de
separação em coluna monolítica (MN), coluna com partículas de núcleo fundido (NF)
e coluna com partículas porosas de diâmetro inferior a 2 μm (Sub 2 μm).

46
3 MATERIAIS E MÉTODOS
3.1 Materiais
3.1.1 Insumos farmacêuticos ativos
A identificação do fabricante, número de lote, pureza e validade dos insumos
farmacêuticos ativos clorpropamida, adquirido na farmácia artesanal, glibenclamida,
gentilmente cedido pela Fundação Ezequiel Dias, e glimepirida e gliclazida,
gentilmente cedidos pela CIFARMA, estão descritos na Tabela 1.1.
Tabela 1.1 - Fabricante, número de lote, pureza e validade dos insumos farmacêuticos ativos
dos fármacos CL, GB, GM e GZ
Insumo Fabricante Lote Pureza Validade
Clorpropamida - CP1007 100,12% 06/2015
Glibenclamida Cadila Pharmaceuticals 0GLL009 99,50% 12/2014
Glimepirida Mantena lab GD/021/11/2011 99,96% 10/2016
Gliclazida Shandong Keyuan
Pharmaceutical 0809130 99,10% 09/2012
3.1.2 Solventes, reagentes e vidrarias
Acetato de amônio Synth (Diadema, Brasil), ácido acético glacial Vetec (Duque de
Caxias, Brasil), acetonitrila grau HPLC Sigma Aldrich (St Louis, Estados Unidos da
America), uracila (Sigma Aldrich, St. Louis, Estados Unidos da America) membrana
de celulose regenerada com porosidade de 0,22 μm Sartorius (Goettingen,
Alemanha), dispositivos filtrantes de celulose regenerada Minisart 15 mm x 0,45 μm
(Goettingen, Alemanha), água ultra pura, pipetas e balões volumétricos calibrados,
béqueres, erlenmeyers, kit de filtração.
3.1.3 Equipamentos e colunas cromatográficas
Ultrapurificador de água (Millipore modelo Direct Q3), ultrassom (Max clean modelo
1400), balança analítica (Sartorius com precisão de 0,01 mg modelo BP210D),

47
UHPLC (Waters Acquity® UPLC), potenciômetro (Metrohm modelo 827 pH lab),
coluna com partículas porosas (ACE, C18, 100 x 2,1 mm, 5 µm - Convencional),
coluna com partículas de núcleo fundido (Poroshell®, Agilent, C18, 100 x 2,1 mm, 2,7
µm - Núcleo Fundido), coluna com partículas porosas (Zorbax®, Agilent, C18, 100 x
2,1 mm, 1,8 µm - Sub 2 μm), coluna monolítica (Onyx®, Phenomenex, C18, 100 x 2,0
mm - Monolítica)
3.2 Métodos
3.2.1 Desenvolvimento de método analítico por cromatografia líquida para a
determinação de CL, GB, GM e GZ em coluna CV
A seleção da fase móvel e diluente do método analítico para a determinação de CL,
GB, GM e GZ foi realizada com base nos métodos para quantificação de
sulfoniluréias descritos na literatura e nas características físico químicas dos
fármacos. O objetivo era que o diluente selecionado tivesse composição similar à
fase móvel, de maneira a evitar o alargamento dos picos cromatográficos.
Após seleção preliminar da fase móvel e do diluente foi realizada uma corrida
exploratória de varredura na região do ultravioleta (200-400 nm) de uma solução
contendo 250 μg/ml de cada um dos antidiabéticos, utilizando as condições
analíticas preliminares descritas na Tabela 1.2. Para obtenção dessa solução partiu-
se de soluções estoques de cada um dos antidiabéticos, a 1 mg/ml, preparadas em
ACN:água (4:1) e submetidas a banho de ultra som por 5 minutos. Posteriormente
realizaram-se diluições dessas soluções estoques de modo a obter uma solução de
trabalho com os quatro antidiabéticos na concentração de 250 μg/ml. O volume da
solução de trabalho foi completado com ACN:solução de acetato de amônio 5 mM
pH 3,0 (50:50). O objetivo dessa corrida exploratória de varredura foi determinar o
comprimento de onda de análise ideal, ou seja, aquele capaz de detectar
seletivamente os quatro antidiabéticos e propiciar a maior absorção possível.

48
Tabela 1.2 - Condições analíticas preliminares*
* Foram utilizados loop de 10 μl, tubo de poli éter cetona (poly ether-ether-ketone - PEEK) de 0,004 polegada e frequência de aquisição dos dados/constante de tempo do detector iguais a 20 Hz/0,1 s.
3.2.2 Otimização de método analítico por cromatografia líquida para a determinação
de CL, GB, GM e GZ em coluna CV
Após seleção das condições cromatográficas preliminares e do comprimento de
onda da análise, algumas condições cromatográficas foram otimizadas. Inicialmente
foram testadas três fases móveis com diferentes proporções de ACN e solução de
acetato de amônio 5 mM pH 3,0. Utilizando a fase móvel selecionada, foram
testadas três concentrações de trabalho. Em seguida, utilizando a fase móvel e a
concentração de trabalho selecionadas, testaram-se três volumes de injeção. Por
último, fazendo-se uso da fase móvel, concentração de trabalho e volume de injeção
selecionados, três temperaturas foram avaliadas. O esquema de otimização está
sumarizado na Tabela 1.3.
Em cada condição teste apresentada na Tabela 1.3, dez diferentes vazões de fase
móvel, variando de 0,02 a 0,6 ml/min, foram experimentadas. Determinaram-se o
número de pratos teóricos e o tempo de retenção dos picos referentes aos quatro
fármacos, em cada vazão avaliada, em cada condição teste. Outro parâmetro
determinado em cada vazão avaliada e em cada condição teste foi o tempo morto da
coluna. Para estimativa do tempo morto, injetaram-se 2 μl de uracila (10 μg/ml).
Parâmetros Condições
Fase móvel ACN:Solução de acetato de amônio 5mM pH 3,0
(50:50)
Vazão da fase móvel 0,2 ml/min
Volume de injeção 2 μl
Coluna C18 100 x 2,1 mm, 5 μm
Temperatura da coluna 30 oC
Solução de CL, GB, GM e GZ 250 μg/ml em fase móvel
Comprimento de onda 200-400 nm

49
Tabela 1.3 - Otimização das condições cromatográficas em coluna convencional
Experimento 1 Experimento 2 Experimento 3 Experimento 4
Fase Móvel Concentração Volume de injeção Temperatura
ACN:Solução (55:45)* 250 μg/ml 5 μl 30 oC
ACN :Solução (50:50)* 100 μg/ml 2 μl 35 oC
ACN:Solução(45:55)* 20 μg/ml 1 μl 40 oC
Condições
cromatográficas
Conc. = 250 μg/ml
Vinj = 2 μl
T = 30 oC
= 230 nm
V = 0,02 a 0,6 ml/min
Condições
cromatográficas
FM = Escolhida no
experimento 1
Vinj = 2 μl
T = 30 oC
= 230 nm
V = 0,02 a 0,6 ml/min
Condições
cromatográficas
FM = Escolhida no
experimento 1
Conc . = Escolhida no
experimento 2
T = 30 oC
= 230 nm
V = 0,02 a 0,6 ml/min
Condições
cromatográficas
FM = Escolhida no
experimento 1
Conc. = Escolhida no
experimento 2
Vinj = Escolhido no
experimento 3
= 230 nm
V = 0,02 a 0,6 ml/min
*Solução de acetato de amônio 5 mM pH 3,0
A partir dos dados de número de pratos teóricos, tempo de retenção e tempo morto,
foi construída uma curva de Van Deemter para cada antidiabético em cada condição
testada. Para os cálculos foram utilizadas as equações (1.1), (1.2) e (1.3).
A seleção da fase móvel, concentração, volume de injeção e temperatura ideais
foram realizadas com base na análise das curvas de Van Deemter e também com
base na análise dos parâmetros fator de retenção (k), resolução (Rs), fator de
assimetria dos picos (As) e pressão do sistema cromatográfico (P).
3.2.3 Transferência do método desenvolvido em coluna CV para as colunas MN, NF
e Sub 2 μm
O método desenvolvido e otimizado para análise de antidiabéticos orais em coluna
convencional foi aplicado às colunas cromatográficas empacotadas com partículas
sub 2 μm, com partículas de núcleo fundido e coluna monolítica. Todos os
parâmetros cromatográficos estabelecidos no método desenvolvido e otimizado na
coluna convencional foram mantidos, exceto a proporção de acetonitrila e solução
de acetato de amônio na fase móvel. A constituição da fase móvel foi ajustada para

50
cada coluna cromatográfica em estudo, de forma que os fatores de retenção de
todos os antidiabéticos fossem próximos aos verificados na coluna convencional. O
cálculo de k foi realizado utilizando-se a equação (1.3).
Para ajustar o percentual de acetonitrila e solução de acetato de amônio realizou-se
uma corrida, em cada coluna, aplicando-se o método desenvolvido e otimizado na
coluna convencional. Naquelas colunas em que a retenção foi superior à observada
na coluna convencional, aumentou-se o percentual de acetonitrila progressivamente
até que valores de k próximos aos objetivados fossem alcançados. Já nas colunas
nas quais a retenção foi inferior à observada na coluna convencional, reduziu-se o
percentual de acetonitrila até que valores de k próximos aos de interesse fossem
atingidos.
4 RESULTADOS E DISCUSSÃO
4.1 Desenvolvimento de método analítico por cromatografia líquida para a
determinação de CL, GB, GM e GZ em coluna CV
Verificou-se que nos métodos descritos na literatura para determinação simultânea
de sulfoniluréias foram utilizadas fases móveis compostas por uma mistura de
solvente orgânico e tampão com pH em torno de 3,0. Por isso tentou-se,
inicialmente, a utilização de solução de acetato de amônio pH 3,0 e acetonitrila, na
proporção de 50:50. Com base no pKa dos fármacos (CL - pKa 4,71 e -1,57;
GB - pKa 5,11 e -1,63; GM - pKa 5,10 e -1,48; GZ - pKa 6,07 e 3,89), espera-se que
em pH 3,0 aproximadamente 98% das moléculas de clorpropamida e 100% das
moléculas de glibenclamida e glimepirida estariam na forma não ionizada e que 90%
das moléculas de gliclazida estariam na forma ionizada. A seleção de um valor de
pH que propicie que a totalidade ou a quase totalidade das moléculas de
determinado analito estejam em sua forma ionizada ou não ionizada é importante
para evitar o alargamento dos picos [24].
Tentou-se inicialmente, sem sucesso, dissolver os insumos dos antidiabéticos em
acetonitrila. Posteriormente tentou-se a mistura ACN:água (4:1), sugerida como
diluente do padrão de glimepirida pelas farmacopeias européia 7ª edição e

51
americana 34a edição. A mistura ACN:água (4:1) foi capaz de solubilizar todos os
quatro antidiabéticos orais, após banho de ultra som de 5 minutos, produzindo
soluções estoque de 1 mg/ml de cada um deles.
Os espectros de varredura dos antidiabéticos utilizando as condições analíticas
descritas na Tabela 1.2 estão apresentados na Figura 1.5.
Figura 1.5 - Espectros de varredura dos antidiabéticos
A partir da análise dos espectros foi selecionado 230 nm como o comprimento de
onda ideal, capaz de propiciar absortividades próximas às máximas de todos os
quatro fármacos.
4.2 Otimização de método analítico por cromatografia líquida para a
determinação de CL, GB, GM e GZ em coluna CV
Quatro parâmetros do método analítico desenvolvido em coluna convencional foram
otimizados: fase móvel, concentração de trabalho, volume de injeção e temperatura.
4.2.1 Fase móvel
O primeiro parâmetro otimizado foi a proporção de ACN e da solução de acetato de
amônio na fase móvel. As curvas de Van Deemter referentes aos quatro
antidiabéticos (Figuras 1.6, 1.7, 1.8 e 1.9), em cada uma das três fases móveis
testadas, são mostradas a seguir.
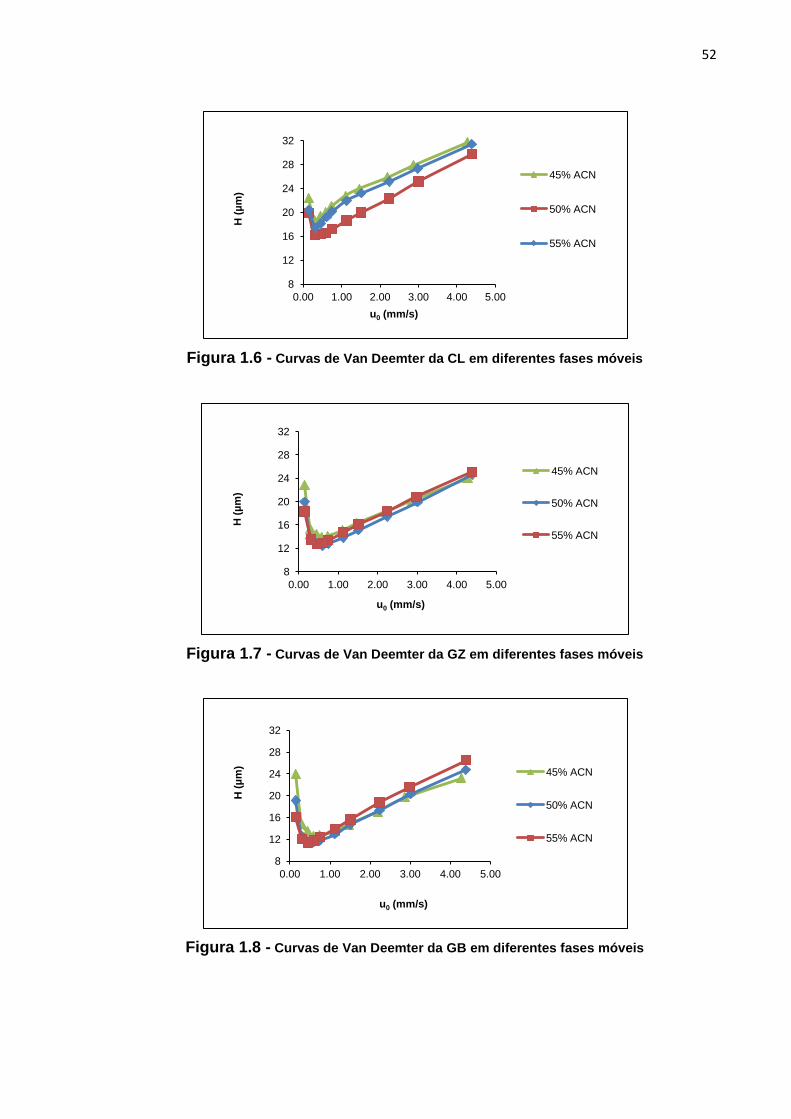
52
Figura 1.6 - Curvas de Van Deemter da CL em diferentes fases móveis
Figura 1.7 - Curvas de Van Deemter da GZ em diferentes fases móveis
Figura 1.8 - Curvas de Van Deemter da GB em diferentes fases móveis
8
12
16
20
24
28
32
0.00 1.00 2.00 3.00 4.00 5.00
H (
µm
)
u0 (mm/s)
45% ACN
50% ACN
55% ACN
8
12
16
20
24
28
32
0.00 1.00 2.00 3.00 4.00 5.00
H (
µm
)
u0 (mm/s)
45% ACN
50% ACN
55% ACN
8
12
16
20
24
28
32
0.00 1.00 2.00 3.00 4.00 5.00
H (
µm
)
u0 (mm/s)
45% ACN
50% ACN
55% ACN
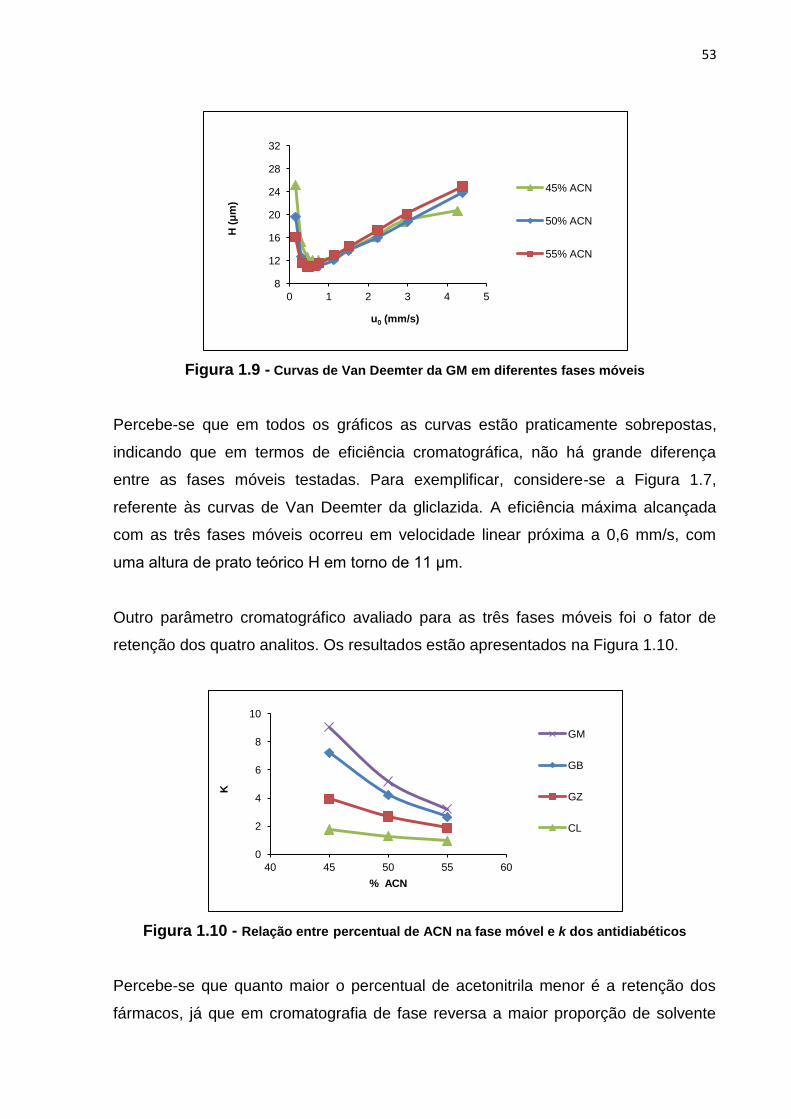
53
Figura 1.9 - Curvas de Van Deemter da GM em diferentes fases móveis
Percebe-se que em todos os gráficos as curvas estão praticamente sobrepostas,
indicando que em termos de eficiência cromatográfica, não há grande diferença
entre as fases móveis testadas. Para exemplificar, considere-se a Figura 1.7,
referente às curvas de Van Deemter da gliclazida. A eficiência máxima alcançada
com as três fases móveis ocorreu em velocidade linear próxima a 0,6 mm/s, com
uma altura de prato teórico H em torno de 11 μm.
Outro parâmetro cromatográfico avaliado para as três fases móveis foi o fator de
retenção dos quatro analitos. Os resultados estão apresentados na Figura 1.10.
Figura 1.10 - Relação entre percentual de ACN na fase móvel e k dos antidiabéticos
Percebe-se que quanto maior o percentual de acetonitrila menor é a retenção dos
fármacos, já que em cromatografia de fase reversa a maior proporção de solvente
8
12
16
20
24
28
32
0 1 2 3 4 5
H (
μm
)
u0 (mm/s)
45% ACN
50% ACN
55% ACN
0
2
4
6
8
10
40 45 50 55 60
K
% ACN
GM
GB
GZ
CL
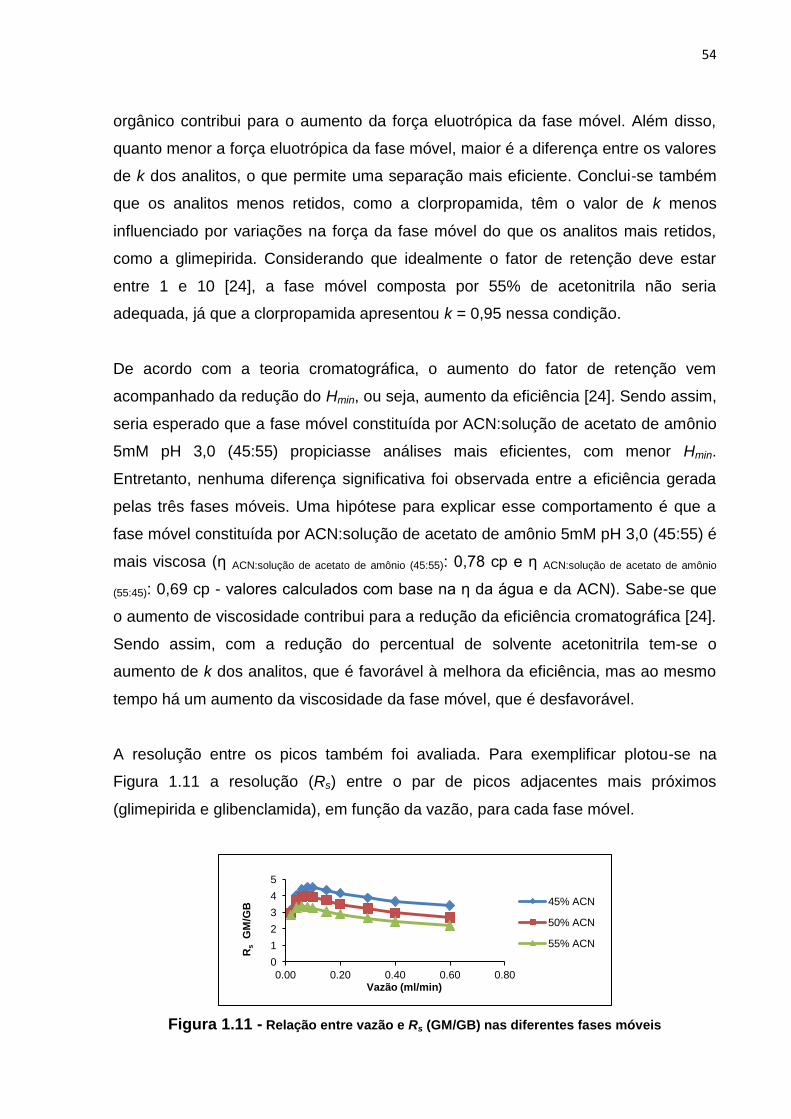
54
orgânico contribui para o aumento da força eluotrópica da fase móvel. Além disso,
quanto menor a força eluotrópica da fase móvel, maior é a diferença entre os valores
de k dos analitos, o que permite uma separação mais eficiente. Conclui-se também
que os analitos menos retidos, como a clorpropamida, têm o valor de k menos
influenciado por variações na força da fase móvel do que os analitos mais retidos,
como a glimepirida. Considerando que idealmente o fator de retenção deve estar
entre 1 e 10 [24], a fase móvel composta por 55% de acetonitrila não seria
adequada, já que a clorpropamida apresentou k = 0,95 nessa condição.
De acordo com a teoria cromatográfica, o aumento do fator de retenção vem
acompanhado da redução do Hmin, ou seja, aumento da eficiência [24]. Sendo assim,
seria esperado que a fase móvel constituída por ACN:solução de acetato de amônio
5mM pH 3,0 (45:55) propiciasse análises mais eficientes, com menor Hmin.
Entretanto, nenhuma diferença significativa foi observada entre a eficiência gerada
pelas três fases móveis. Uma hipótese para explicar esse comportamento é que a
fase móvel constituída por ACN:solução de acetato de amônio 5mM pH 3,0 (45:55) é
mais viscosa (η ACN:solução de acetato de amônio (45:55): 0,78 cp e η ACN:solução de acetato de amônio
(55:45): 0,69 cp - valores calculados com base na η da água e da ACN). Sabe-se que
o aumento de viscosidade contribui para a redução da eficiência cromatográfica [24].
Sendo assim, com a redução do percentual de solvente acetonitrila tem-se o
aumento de k dos analitos, que é favorável à melhora da eficiência, mas ao mesmo
tempo há um aumento da viscosidade da fase móvel, que é desfavorável.
A resolução entre os picos também foi avaliada. Para exemplificar plotou-se na
Figura 1.11 a resolução (Rs) entre o par de picos adjacentes mais próximos
(glimepirida e glibenclamida), em função da vazão, para cada fase móvel.
Figura 1.11 - Relação entre vazão e Rs (GM/GB) nas diferentes fases móveis
0
1
2
3
4
5
0.00 0.20 0.40 0.60 0.80
Rs G
M/G
B
Vazão (ml/min)
45% ACN
50% ACN
55% ACN
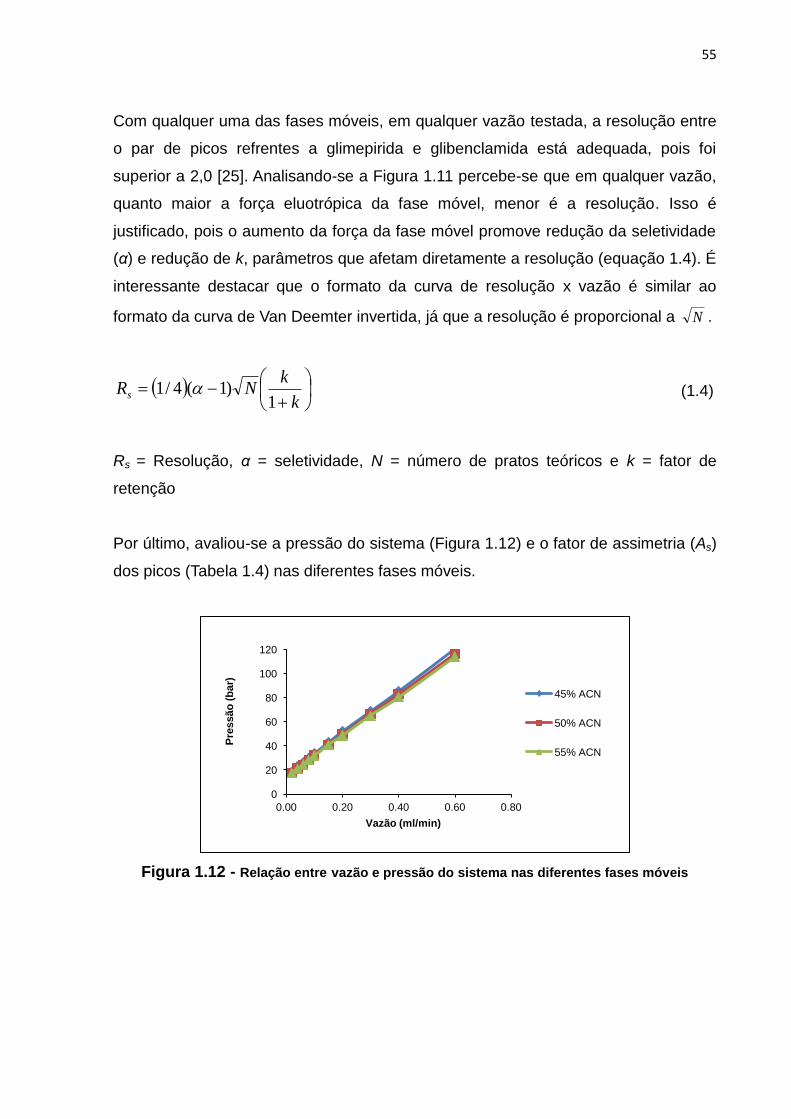
55
Com qualquer uma das fases móveis, em qualquer vazão testada, a resolução entre
o par de picos refrentes a glimepirida e glibenclamida está adequada, pois foi
superior a 2,0 [25]. Analisando-se a Figura 1.11 percebe-se que em qualquer vazão,
quanto maior a força eluotrópica da fase móvel, menor é a resolução. Isso é
justificado, pois o aumento da força da fase móvel promove redução da seletividade
(α) e redução de k, parâmetros que afetam diretamente a resolução (equação 1.4). É
interessante destacar que o formato da curva de resolução x vazão é similar ao
formato da curva de Van Deemter invertida, já que a resolução é proporcional a N .
k
kNRs
1)1(4/1 (1.4)
Rs = Resolução, α = seletividade, N = número de pratos teóricos e k = fator de
retenção
Por último, avaliou-se a pressão do sistema (Figura 1.12) e o fator de assimetria (As)
dos picos (Tabela 1.4) nas diferentes fases móveis.
Figura 1.12 - Relação entre vazão e pressão do sistema nas diferentes fases móveis
0
20
40
60
80
100
120
0.00 0.20 0.40 0.60 0.80
Pre
ssão
(b
ar)
Vazão (ml/min)
45% ACN
50% ACN
55% ACN
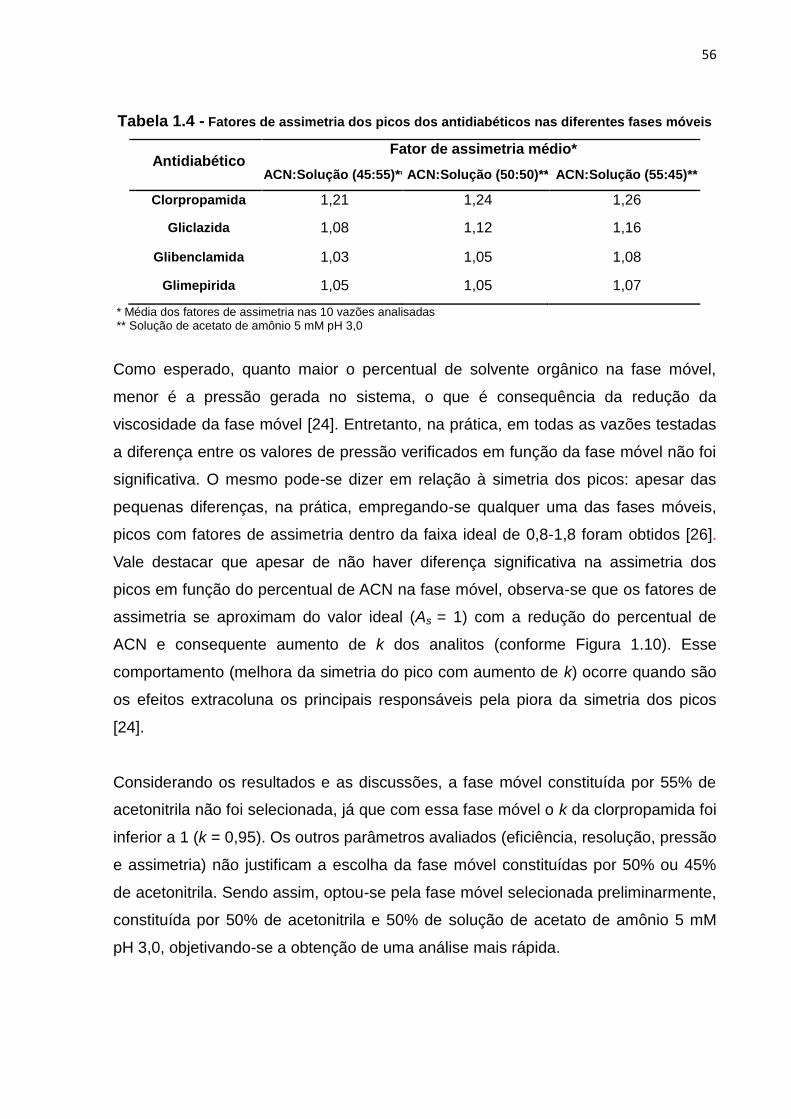
56
Tabela 1.4 - Fatores de assimetria dos picos dos antidiabéticos nas diferentes fases móveis
Antidiabético Fator de assimetria médio*
ACN:Solução (45:55)** ACN:Solução (50:50)** ACN:Solução (55:45)**
Clorpropamida 1,21 1,24 1,26
Gliclazida 1,08 1,12 1,16
Glibenclamida 1,03 1,05 1,08
Glimepirida 1,05 1,05 1,07
* Média dos fatores de assimetria nas 10 vazões analisadas ** Solução de acetato de amônio 5 mM pH 3,0
Como esperado, quanto maior o percentual de solvente orgânico na fase móvel,
menor é a pressão gerada no sistema, o que é consequência da redução da
viscosidade da fase móvel [24]. Entretanto, na prática, em todas as vazões testadas
a diferença entre os valores de pressão verificados em função da fase móvel não foi
significativa. O mesmo pode-se dizer em relação à simetria dos picos: apesar das
pequenas diferenças, na prática, empregando-se qualquer uma das fases móveis,
picos com fatores de assimetria dentro da faixa ideal de 0,8-1,8 foram obtidos [26].
Vale destacar que apesar de não haver diferença significativa na assimetria dos
picos em função do percentual de ACN na fase móvel, observa-se que os fatores de
assimetria se aproximam do valor ideal (As = 1) com a redução do percentual de
ACN e consequente aumento de k dos analitos (conforme Figura 1.10). Esse
comportamento (melhora da simetria do pico com aumento de k) ocorre quando são
os efeitos extracoluna os principais responsáveis pela piora da simetria dos picos
[24].
Considerando os resultados e as discussões, a fase móvel constituída por 55% de
acetonitrila não foi selecionada, já que com essa fase móvel o k da clorpropamida foi
inferior a 1 (k = 0,95). Os outros parâmetros avaliados (eficiência, resolução, pressão
e assimetria) não justificam a escolha da fase móvel constituídas por 50% ou 45%
de acetonitrila. Sendo assim, optou-se pela fase móvel selecionada preliminarmente,
constituída por 50% de acetonitrila e 50% de solução de acetato de amônio 5 mM
pH 3,0, objetivando-se a obtenção de uma análise mais rápida.
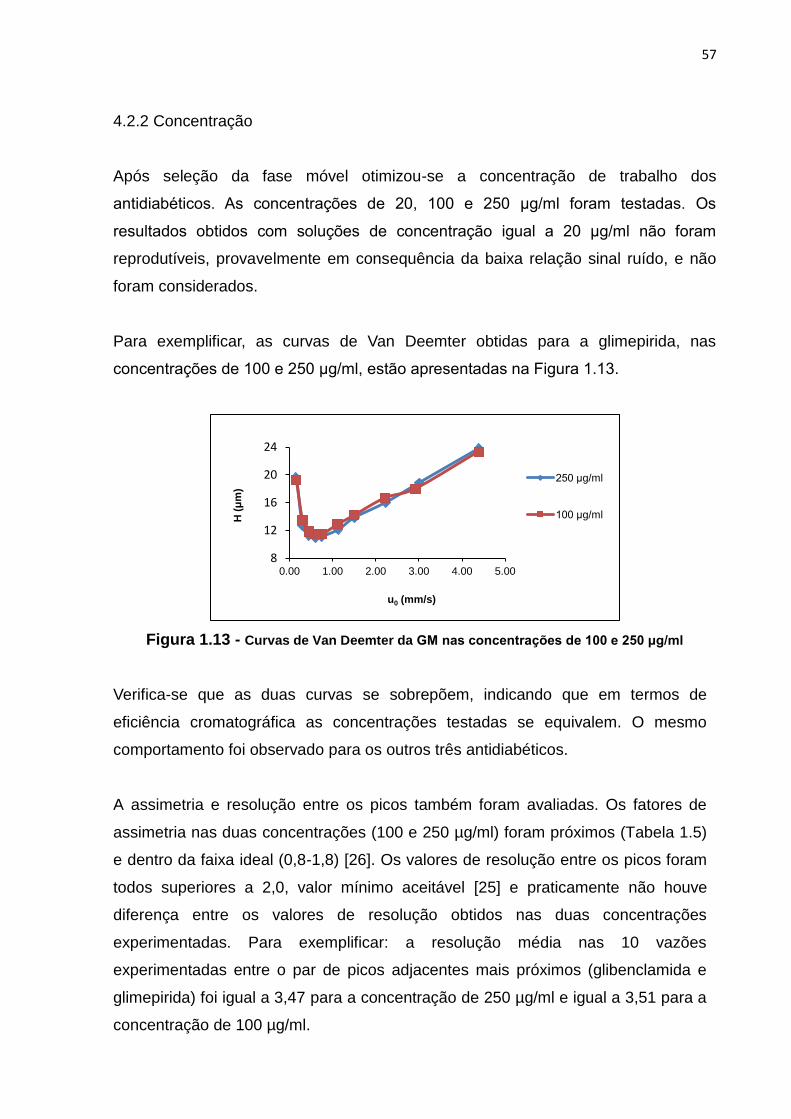
57
4.2.2 Concentração
Após seleção da fase móvel otimizou-se a concentração de trabalho dos
antidiabéticos. As concentrações de 20, 100 e 250 μg/ml foram testadas. Os
resultados obtidos com soluções de concentração igual a 20 μg/ml não foram
reprodutíveis, provavelmente em consequência da baixa relação sinal ruído, e não
foram considerados.
Para exemplificar, as curvas de Van Deemter obtidas para a glimepirida, nas
concentrações de 100 e 250 μg/ml, estão apresentadas na Figura 1.13.
Figura 1.13 - Curvas de Van Deemter da GM nas concentrações de 100 e 250 μg/ml
Verifica-se que as duas curvas se sobrepõem, indicando que em termos de
eficiência cromatográfica as concentrações testadas se equivalem. O mesmo
comportamento foi observado para os outros três antidiabéticos.
A assimetria e resolução entre os picos também foram avaliadas. Os fatores de
assimetria nas duas concentrações (100 e 250 µg/ml) foram próximos (Tabela 1.5)
e dentro da faixa ideal (0,8-1,8) [26]. Os valores de resolução entre os picos foram
todos superiores a 2,0, valor mínimo aceitável [25] e praticamente não houve
diferença entre os valores de resolução obtidos nas duas concentrações
experimentadas. Para exemplificar: a resolução média nas 10 vazões
experimentadas entre o par de picos adjacentes mais próximos (glibenclamida e
glimepirida) foi igual a 3,47 para a concentração de 250 µg/ml e igual a 3,51 para a
concentração de 100 µg/ml.
8
12
16
20
24
0.00 1.00 2.00 3.00 4.00 5.00
H (
μm
)
u0 (mm/s)
250 μg/ml
100 μg/ml

58
Tabela 1.5 - Fatores de assimetria dos picos dos antidiabéticos nas concentrações de 100 e
250 μg/ml
Antidiabético Fator de assimetria médio*
250 μg/ml 100 μg/ml
Clorpropamida 1,24 1,23
Glibenclamida 1,05 1,07
Glimepirida 1,05 1,05
Gliclazida 1,12 1,12
* Média da dos fatores de assimetria nas 10 vazões experimentadas
Como não houve diferença em termos de eficiência, resolução e fator de assimetria
entre as concentrações de 100 e 250 μg/ml, optou-se pela seleção da concentração
de 100 μg/ml, objetivando a simplificação dos cálculos durante a validação dos
métodos analíticos no capítulo 5 desse trabalho.
4.2.3 Volume de injeção
Após seleção da fase móvel e concentração ideais, avaliaram-se os seguintes
volumes de injeção: 1, 2 e 5 μl. Com a finalidade de evitar a dispersão da amostra e
consequente redução da eficiência cromatográfica, o volume de injeção deve ser
reduzido, embora volumes de injeção muito pequenos possam conduzir a uma baixa
detectabilidade do método [24,27]. Faz-se necessário, portanto, otimizar o volume
de injeção objetivando eficiência cromatográfica e detectabilidade satisfatórias.
A seguir encontram-se apresentadas as curvas de Van Deemter da clorpropamida
(Figura 1.14) e glimepirida (Figura 1.15), nos três volumes de injeção testados.
Os resultados da clorpropamida e glimepirida foram selecionados como exemplos
porque eles representam os analitos de menor (k = 1,3) e maior retenção (k = 5,2),
respectivamente.
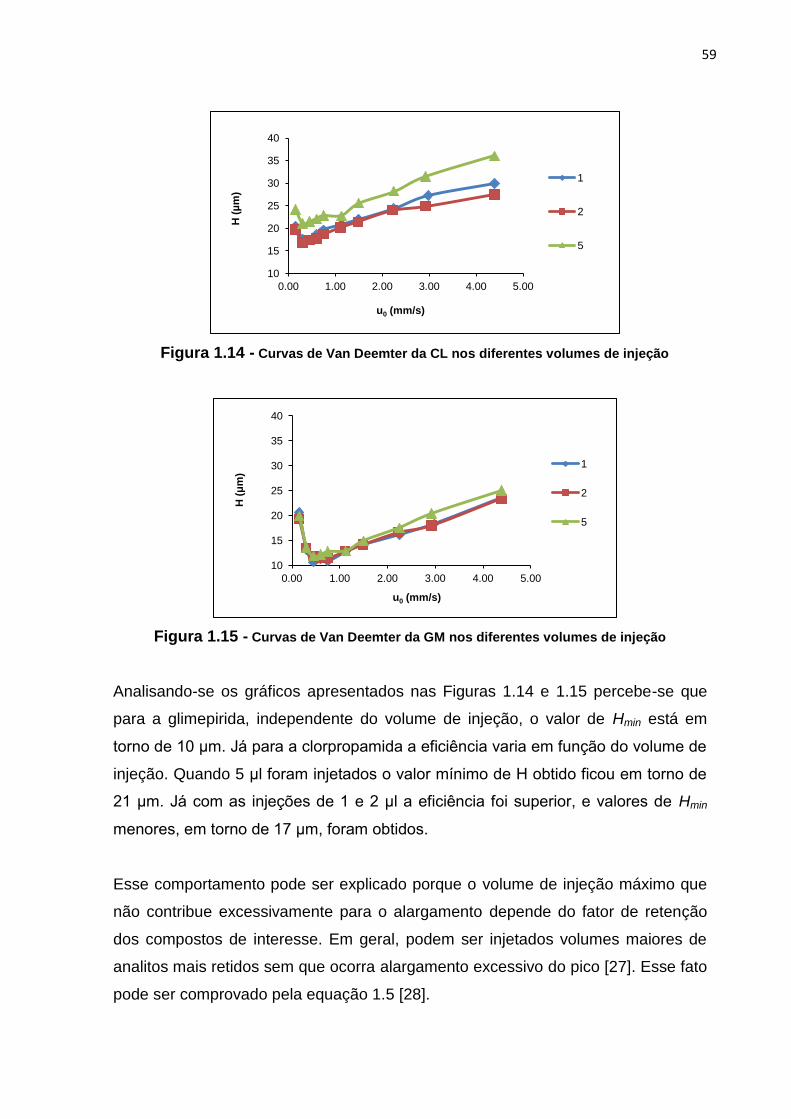
59
Figura 1.14 - Curvas de Van Deemter da CL nos diferentes volumes de injeção
Figura 1.15 - Curvas de Van Deemter da GM nos diferentes volumes de injeção
Analisando-se os gráficos apresentados nas Figuras 1.14 e 1.15 percebe-se que
para a glimepirida, independente do volume de injeção, o valor de Hmin está em
torno de 10 μm. Já para a clorpropamida a eficiência varia em função do volume de
injeção. Quando 5 μl foram injetados o valor mínimo de H obtido ficou em torno de
21 μm. Já com as injeções de 1 e 2 μl a eficiência foi superior, e valores de Hmin
menores, em torno de 17 μm, foram obtidos.
Esse comportamento pode ser explicado porque o volume de injeção máximo que
não contribue excessivamente para o alargamento depende do fator de retenção
dos compostos de interesse. Em geral, podem ser injetados volumes maiores de
analitos mais retidos sem que ocorra alargamento excessivo do pico [27]. Esse fato
pode ser comprovado pela equação 1.5 [28].
10
15
20
25
30
35
40
0.00 1.00 2.00 3.00 4.00 5.00
H (
µm
)
u0 (mm/s)
1
2
5
10
15
20
25
30
35
40
0.00 1.00 2.00 3.00 4.00 5.00
H (
μm
)
u0 (mm/s)
1
2
5

60
N
KVV r
inj
(1.5)
Vinj = volume de injeção, = fração do alargamento do pico, Vr = volume de
retenção, K = constante característica da qualidade da injeção normalmente igual a
2, N = número de pratos teóricos
Analisando-se a equação 1.5 verifica-se que considerando um mesmo volume de
injeção, analitos mais retidos (maior Vr e, portanto, maior fator de retenção k)
sofrerão menor alargamento (menor ).
Sendo assim, como a glimepirida possui maior fator de retenção (k = 5,2), o pico
relativo a ela sofreu menor alargamento quando o volume de injeção de 5 μl foi
injetado, tendo apresentado eficiência semelhante às verificadas quando volumes
de 1 e 2 μl foram utilizados. Já o pico da clorpropamida (k = 1,3) sofreu maior
alargamento quando 5 μl foram injetados, o que explica o fato de a eficiência
cromatográfica com esse volume de injeção ter sido inferior a eficiência observada
quando 1 e 2 μl foram injetados. O comportamento da gliclazida (k = 2,7) e da
glibenclamida (k = 4,2) foi similar ao da clorpropamida, ou seja, houve redução da
eficiência (aumento de Hmin) quando 5 μl foram injetados.
Considerando o exposto, os volumes de injeção de 1 e 2 μl poderiam ser utilizados,
já que garantem uma maior eficiência cromatográfica dos quatro antidiabéticos
quando comparados ao volume de injeção de 5 μl. Analisando-se os parâmetros
assimetria e resolução entre os picos, percebe-se que os volumes de 1 e 2 μl se
mostraram novamente equivalentes: valores de fator de assimetria e resolução
similares e satisfatórios (0,8<As<1,8 e Rs>2,0) [25, 26] foram obtidos. Para
exemplificar: a resolução média nas 10 vazões experimentadas, entre o par de
picos mais crítico (glibenclamida e glimepirida), foi igual a 3,49 para o volume de
injeção de 1 μl e igual a 3,51 para o volume de injeção de 2 μl; o fator de assimetria
médio do pico de glibenclamida nas 10 vazões testadas foi igual a 1,09 para o
volume de injeção de 1 μl e igual a 1,07 para o volume de injeção de 2 μl.
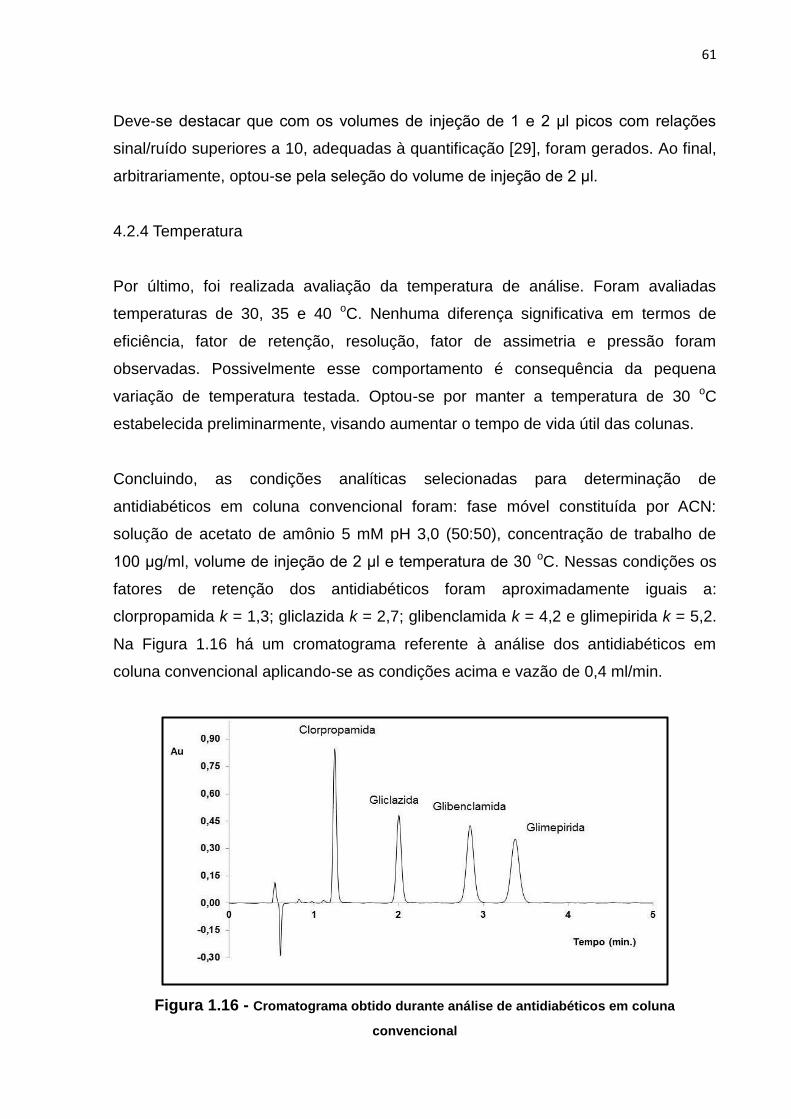
61
Deve-se destacar que com os volumes de injeção de 1 e 2 μl picos com relações
sinal/ruído superiores a 10, adequadas à quantificação [29], foram gerados. Ao final,
arbitrariamente, optou-se pela seleção do volume de injeção de 2 μl.
4.2.4 Temperatura
Por último, foi realizada avaliação da temperatura de análise. Foram avaliadas
temperaturas de 30, 35 e 40 oC. Nenhuma diferença significativa em termos de
eficiência, fator de retenção, resolução, fator de assimetria e pressão foram
observadas. Possivelmente esse comportamento é consequência da pequena
variação de temperatura testada. Optou-se por manter a temperatura de 30 oC
estabelecida preliminarmente, visando aumentar o tempo de vida útil das colunas.
Concluindo, as condições analíticas selecionadas para determinação de
antidiabéticos em coluna convencional foram: fase móvel constituída por ACN:
solução de acetato de amônio 5 mM pH 3,0 (50:50), concentração de trabalho de
100 μg/ml, volume de injeção de 2 μl e temperatura de 30 oC. Nessas condições os
fatores de retenção dos antidiabéticos foram aproximadamente iguais a:
clorpropamida k = 1,3; gliclazida k = 2,7; glibenclamida k = 4,2 e glimepirida k = 5,2.
Na Figura 1.16 há um cromatograma referente à análise dos antidiabéticos em
coluna convencional aplicando-se as condições acima e vazão de 0,4 ml/min.
Figura 1.16 - Cromatograma obtido durante análise de antidiabéticos em coluna
convencional

62
4.3 Transferência do método desenvolvido e otimizado em coluna CV para as
colunas MN, NF e Sub 2 μm
A proporção de acetonitrila e Solução de acetato de amônio 5 mM, pH 3,0 na fase
móvel foi ajustada para cada coluna cromatográfica (MN, NF e Sub 2 μm), de
maneira que os fatores de retenção (k) de todos os antidiabéticos ficassem próximos
aos verificados na coluna convencional. Esse procedimento foi realizado para
permitir a comparação do desempenho das colunas cromatográficas, que será
descrita no capítulo 4. Como os valores de k influenciam na eficiência e formato das
curvas de Van Deemter, devem ser ajustados em todas as colunas [30].
No caso da coluna monolítica, para que a retenção observada na coluna
convencional fosse mantida, foi necessário reduzir a força eluotrópica da fase móvel.
Já com a coluna de partículas sub 2 μm ocorreu o contrário e portanto a manutenção
dos fatores de retenção ocorreu mediante aumento da força eluotrópica da fase
móvel. Apesar de a coluna de núcleo fundido apresentar capacidade de retenção um
pouco menor do que a coluna convencional, na prática nenhum ajuste na força da
fase móvel foi necessário. Na Tabela 1.6 há a descrição das condições selecionadas
para a análise e os valores de k dos antidiabéticos em cada uma das colunas.
Tabela 1.6 - Condições de análise para determinação de antidiabéticos nas diferentes colunas
e valores de k
Colunas
Fase Móvel
Vinj
(μl)
(nm)
T
(oC)
Conc.
(μg/ml) K
Convencional ACN: Solução
(50:50)** 2 230 30 100
CL 1,3 - GB 2,7
GZ 4,2 - GM 5,2
Monolítica ACN: Solução
(43:57)** 2 230 30 100
CL 0,9 - GB 2,2
GZ 4,3 - GM 5,5
Núcleo
Fundido
ACN: Solução
(50:50)** 2 230 30 100
CL 1,3 - GB 2,7
GZ 4,5 - GM 5,6
Sub 2 μm ACN: Solução
(53:47)** 2 230 30 100
CL 1,2 - GB 2,7
GZ 4,2 - GM 5,2
* Foram utilizados loop de 10 μl, tubo de PEEK de 0,004 polegada e frequência de aquisição dos dados e constante de tempo do detector iguais a 20 Hz/0,1 s * *Solução de acetato de amônio 5 mM pH 3,0

63
5 CONCLUSÃO
Método analítico para quantificação de clorpropamida, glibenclamida, glimepirida e
gliclazida foi desenvolvido e otimizado em coluna convencional com partículas
totalmente porosas de 5 μm de diâmetro. Posteriormente, as condições analíticas
selecionadas foram transferidas e otimizadas para a análise dos antidiabéticos
utilizando colunas monolítica, empacotada com partícula de núcleo fundido e
empacotada com partículas totalmente porosas sub 2 μm. O único ajuste realizado
foi na proporção de acetonitrila e solução de acetato de amônio 5mM pH 3,0 na fase
móvel. Esses métodos, descritos na Tabela 1.6, foram utilizados nas etapas
subsequentes desse trabalho.

64
REFERÊNCIAS BIBLIOGRÁFICAS
[1] WORLD HEALTH ORGANIZATION. Disponível em: <http://www.who.int/ mediacentre/factsheets/fs312/en/index.html>. Acesso em: 29 de agosto de 2012. [2] DIRETRIZES DA SOCIEDADE BRASILEIRA DE DIABETES 2009. Disponível em: <http://www.diabetes.org.br/attachments/diretrizes09_final.pdf>. Acesso em: 28 de agosto de 2012. [3] ARAÚJO, L. M.B.; BRITTO, M. M.S; CRUZ, T. R. P. Tratamento do Diabetes Mellitus do Tipo 2: Novas Opções. Arquivos Brasileiros de Endocrinologia e Metabologia. v. 44, n. 6, p. 509-518, 2000. [4] LEBOVITZ, H. E. Type 2 diabetes mellitus-current therapies and the emergence of surgical options. Nature Reviews Endocrinology. v. 7, p. 408-419, 2011. [5] OVER, R. K.; RATNER, R. E. Combination Pharmacotherapy with Incretins: What Works Best and When? Current Diabetes Reports. v. 8, p. 361-367, 2008. [6] WEISSMAN, P. N. Reappraisal of the Pharmacologic Approach to Treatment of Type 2 Diabetes Mellitus. American Journal of Cardiology. v. 90 (supllement), p. 42-50, 2002. [7] RELAÇÃO NACIONAL DE MEDICAMENTOS – RENAME, 2012. Disponível em: <http://portal.saude.gov.br/portal/arquivos/pdf/anexos_rename_2012_pt_533_30_03_
12.pdf>. Acesso em: 28 de Agosto de 2012.
[8] SEINO, S. Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia. v. 55, p. 2096-2108, 2012. [9] BRUNTON, L.L.; LAZO, J. S.; PARKER, K. L (ED). Goodman & Gilman As bases farmacológicas da terapêutica. 11 ed. Rio de Janeiro: Mc Graw Hill, 2006. 1821 p. [10] CHEMICAL ABSTRACTS REACTION SEARCH SERVICE. Disponível em: <http://www.chem.ox.ac.uk/cheminfo/scifinder.html>. Acesso em: 20 de agosto de 2011. [11] FARMACOPEIA Brasileira. 5 ed. Rio de Janeiro: Editora Fiocruz, 2010. 1448 p. [12] THE UNITED States Pharmacopeia. 34 ed. Rockville: The United States Pharmacopeial Convention, 2011. 4638 p. [13] EUROPEAN Pharmacopeia. 7 ed. Convention on the Elaboration of a European Pharmacopeia, 2010. 3309 p. [14] KHAN, U.; ASLAM, F.; ASHFAQ, M.; ASGHAR, M. N. Determination of Glimepiride in Pharmaceutical Formulations Using HPLC and First-Derivative Spectrophotometric Methods. Journal of Analytical Chemistry, v. 64, n. 2, p. 171-175, 2009.

65
[15] CHOWDARY, K. P. R.; SUNDARI, P. T. HPLC estimation of gliclazide in formulations and in pharmacokinetic studies. Asian Journal of Chemistry. v. 21, n. 7, p. 5221-5227, 2009. [16] RAYANM, I. V.; RAO A. L.; RAMANA, M.V. Validated RP - HPLC Method for the Estimation of Glibenclamide in Formulation and Serum. International Journal of Research in Pharmaceutical and Biomedical Sciences. v. 2, n. 2, p. 856-862, 2011. [17] JIAN-YU, Z.; SU-FANG, T. RP-HPLC determination of the related substances in chlorpropamide. Chinese Journal of Pharmaceutical Analysis. 2010-09. [18] VENKATESH, P.; HARISUDHAN,T.; CHOUDHURY, H.; MULLANGI, R.; SRINIVAS, N. R. Simultaneous estimation of six anti-diabetic drugs - glibenclamide, gliclazide, glipizide, pioglitazone, repaglinide and rosiglitazone: development of a novel HPLC method for use in the analysis of pharmaceutical formulations and its application to human plasma assay. Biomedical Chromatography, v. 20, p. 1043-1048, 2006. [19] LAKSHMI, K. S.; RAJESH, T. Development and Validation of RP-HPLC Method for Simultaneous Determination of Glipizide, Rosiglitazone, Pioglitazone, Glibenclamide and Glimepiride in Pharmaceutical Dosage Forms and Human Plasma. Journal of The Iranian Chemical Society, v. 8, n. 1, p. 31-37, 2011. [20] DEEB, S. E.; SCHEPERS, U. Fast HPLC method for the determination of glimepiride, glibenclamide, and related substances using monolithic column and flow program. Journal of separation science. v. 29, p. 1571-1577, 2006. [21] BRITISH Pharmacopeia 2011. London: The Stationary Office, 2011. 4019 p. [22] HARRIS, D. C. Análise Química Quantitativa. 7 ed. Rio de Janeiro: LTC, 2008. 886 p. [23] THE BASICS OF THE KINETIC PLOT METHOD. Disponível em: <http://www.vub.ac.be/CHIS/Research/kp.html>. Acesso em: 01 de Abril de 2011
[24] SNYDER, L. R., KIRKLAND, J. J., GLAJCH, J. L. Practical HPLC method development. 2 ed. New York: John Wiley & Sons, 1997. 765 p. [25] RIBANI, M.; BOTTOLI, C. B. G.; COLLINS, C. H.; JARDIM, I. C. S. F.; MELO, L. F. C. Validação em métodos cromatográficos e eletroforéticos. Química Nova. v. 27, n. 5, p. 771-780, 2004. [26] PATNAIK, P. Handbook of environmental analysis. 2 ed. New York: CRC press, 1997. [27] AMPARO, M. R.; FRANZINI, C. D.; SCHIAVON, B.; CARVALHO, L. S.; de, NETO, A. J. S. Influência dos parâmetros instrumentais sobre o desempenho de coluna de HPLC com partículas superficialmente porosas sub-3 μm. Scientia Chromatographica. v. 3, n. 2, p.51-66, 2011.

66
[28] MEYER, V. R. High-performance liquid chromatographic theory for the practitioner. Journal of Chromatography,v. 334, p.197-209, 1985. [29] BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RE no. 899, de 29 de Maio de 2003. Guia para validação de métodos analíticos e bioanalíticos. Diário Oficial da União, Brasília, DF, Poder Executivo, de 02 de Junho de 2003c. [30] OLAH, E.; FEKETE, S.; FEKETE, J.; GANZLER, K. Comparative study of new shell-type, sub-2μm fully porous and monolith stationary phases, focusing on mass-transfer resistance. Journal of Chromatography A. v.1217, p. 3642-3653, 2010.

67
CAPÍTULO 2
CARACTERIZAÇÃO FÍSICA DAS COLUNAS
CROMATOGRÁFICAS

68
1 REVISÃO BIBLIOGRÁFICA
Nesta revisão serão apresentadas características físicas, vantagens e desvantagens
das colunas cromatográficas: monolítica (MN), empacotada com partículas de núcleo
fundido (NF) e empacotada com partículas porosas de diâmetro inferior a 2 m (Sub
2 m). Além disso, serão realizadas comparações dessas colunas com a coluna
convencional (CV), empacotada com partículas porosas de diâmetro entre 3-5 µm.
1.1 Coluna monolítica
As fases estacionárias monolíticas surgiram no final da década de 80 e início da
década de 90. A popularização dessas fases, entretanto, só aconteceu a partir do
ano 2000, quando monolitos de sílica se tornaram disponíveis comercialmente. Ao
longo dos últimos cinco anos os monolitos, que podem ser feitos de material
orgânico sintético (ex: resina acrílica), natural (ex: celulose) ou material inorgânico
(ex: sílica), foram amplamente difundidos [1, 2].
A fase monolítica de sílica é preparada no formato de um cilindro, pelo processo
“sol-gel”. Esse preparo não ocorre dentro das colunas cromatográficas
convencionais de aço inoxidável, pois após a etapa de secagem o cilindro monolítico
encolhe e perde o contato com a parede interna da coluna cromatográfica. Sendo
assim, materiais especiais, como o PEEK, devem ser utilizado, pois se ajustam mais
facilmente ao diâmetro do monolito de sílica [1].
Em um procedimento típico de preparo de monolitos de sílica pelo processo “sol-
gel”, tetrametoxissilano ou tetraetoxissilano é adicionado a uma solução aquosa de
poli(oxietileno) (agente porogênico) contendo ácido ou base (catalisador). A mistura
é agitada a 0 oC e a solução resultante é colocada dentro de um molde de
policarbonato para que ocorram as reações de hidrólise e policondensação. A
primeira etapa é a formação de um “sol”, suspensão coloidal de espécies sólidas em
um líquido. Em seguida, a suspensão é convertida em gel, por meio de um processo
de policondensação. A amostra na forma de gel é então envelhecida por alguns
dias. O cilindro de sílica úmido formado é colocado em uma solução aquosa para
formar as estruturas mesoporosas. Sucessivas evaporações, secagens e

69
tratamentos térmicos são realizados para decompor os constituintes orgânicos e
estabilizar a superfície do gel hidrofílico de sílica. Após as etapas de envelhecimento
e secagem, o monolito de sílica encolhe dentro do molde. O cilindro monolítico é
retirado do molde e encaixado em um tubo de PEEK, e por meio de aquecimento e
aplicação de pressão externa o tubo de PEEK se ajusta ao monolito, assegurando a
inexistência de espaços vazios. O monolito é posteriormente funcionalizado com o
reagente de interesse [3].
A fase monolítica é um meio contínuo de separação, contendo uma "partícula única".
Possui uma estrutura sólida e altamente porosa, de pequenos domínios, que fornece
elevada eficiência cromatográfica (Figura 2.1) [1].
Figura 2.1 - Micrografia da fase monolítica de sílica [4]
Os domínios do monolito podem ser micro, macro ou mesoporosos. Segundo a
IUPAC, os microporos são poros com diâmetros que não excedem 2 nm (20 Å), os
mesoporos são poros de tamanhos intermediários entre 2 e 50 nm e os macroporos
são definidos como poros com diâmetros superiores a 50 nm (500 Å). Nas colunas
monolíticas de sílica de primeira geração os macroporos, em inglês chamados de
through-pore, são de 2 µm. É principalmente através deles que a fase móvel passa
durante o processo de eluição cromatográfica. Já os mesoporos, chamados em
inglês de skeleton-pore, possuem poros de aproximadamente 13 nm nos monolitos
de primeira geração. São os mesoporos que geram a área superficial, normalmente
entre 50 a 300 m2/g, necessária à separação cromatográfica [1,5]. Na figura 2.2 há
um esquema de uma coluna monolítica e os respectivos macroporos e mesoporos.

70
Figura 2.2 - Macroporos de 2 μm (a) e mesoporos de 13 nm (b) da fase monolítica [4]
Característica física importante das colunas monolíticas é a elevada porosidade (ε),
situada em torno de 80% [5]. A alta porosidade justifica os elevados valores de
permeabilidade (Kv0) característicos desse tipo de coluna. Valores de permeabilidade
iguais a 5,00 x 10-14 e 8,00 x 10-14 m2 foram descritos na literatura para a coluna
monolítica da marca Onyx® [6, 7]. Verifica-se que, em consequência da elevada
porosidade e da baixa densidade características das colunas monolíticas, o fator de
retenção (k) dos analitos nesse tipo de coluna é inferior ao observada nas colunas
com partículas porosas convencionais de diâmetro entre 3 e 5 µm. Observa-se
também que colunas monolíticas e convencionais que possuem o mesmo grupo
químico ligante apresentam valores de seletividade (α) próximos, indicando que os
mecanismos que governam a separação em ambas são similares [8].
Como já foi relatado, a geometria dos canais dos monolitos apresentam estrutura
altamente permeável e, portanto, de menor resistência à vazão da fase móvel do
que a gerada pelas partículas porosas presentes nas colunas cromatográficas
convencionais. Sendo assim, é possível realizar análises rápidas, em vazões
elevadas (> 5 ml/min), utilizando instrumentos convencionais de cromatografia
líquida (HPLC), sem que haja aumento da pressão para valores acima do máximo
suportado por eles (400 bar) [1, 2].
Embora as colunas monolíticas apresentem as vantagens mencionadas, menos de
1% dos profissionais que trabalham com cromatografia as utilizam. Os motivos são o
pequeno número de fornecedores (apenas Merck e Phenomenex) e de grupos
químicos ligantes (sílica, C8, C18), inexistência de colunas com comprimento
superior a 100 mm (por limitações durante o processo produtivo), estabilidade

71
química limitada a uma estreita faixa de pH (2-8) e elevado consumo de solvente [9,
10]. Além disso, a limitação do número de fornecedores faz com que o custo das
colunas monolíticas seja elevado. Em cotação realizada no primeiro semetre de
2011, a coluna monolítica da marca Phenomenex (Onyx® C18 100 x 2,0 mm) foi
cotada em R$ 1700,00, enqunato que a coluna convencional da marca ACE (C18
100 x 2,1 mm) foi cotada em R$ 960,00.
Outro ponto negativo é que as fases monolíticas dão origem a picos de pior simetria
[11]. Além disso, a eficiência cromatográfica das colunas monolíticas, apesar de ser
comparável à apresentada por colunas convencionais com partículas porosas de
diâmetro entre 3,5 e 5,0 μm, é inferior à alcançada por colunas empacotadas com
partículas de núcleo fundido e partículas porosas sub 2 μm [1,10,12].
Com o objetivo de melhorar o desempenho das colunas monolíticas, a empresa
Merck introduziu no mercado, no fim de 2011, a fase monolítica de sílica de segunda
geração. Essa nova fase é chamada de Chromolithic High Resolution® e possui duas
vantagens em relação à fase de primeira geração: gera uma maior eficiência
cromatográfica (H fase monolítica 2a geração < 7 μm; H fase monolítica 1
a geração < 12,5 μm) e
propicia a obtenção de picos mais simétricos. Essas características foram
alcançadas mediante o desenvolvimento de um processo de fabricação mais
reprodutível, garantindo que poros de tamanhos homogêneos fossem formados,
além da redução do tamanho dos macroporos. Na fase de primeira geração o
macroporo possui tamanho entre 1,8-2,0 μm, já na de segunda geração esse
tamanho encontra-se situado entre 1,1-1,2 μm. É óbvio que a redução do tamanho
do macroporo conduziu a um aumento da pressão, entretanto as colunas
Chromolithic High Resolution® ainda podem ser utilizadas em cromatógrafos
convencionais, pois a pressão gerada é inferior a 400 bar [12].
Apesar de não serem utilizadas com elevada frequência, muitos trabalhos científicos
vem sendo realizados com os monolitos. Separações de compostos orgânicos sob
condições de fase reversa, dos quais se destacam fármacos, peptídeos e proteínas,
representam a quase totalidade dos trabalhos publicados [1]. Exemplo de aplicação
das colunas monolíticas é a determinação de tadalafil em coluna C18, 100 x 4,6 mm,
empregando-se vazão de 5 ml/min, em aproximadamente 10 minutos [13].

72
1.2 Coluna empacotada com partículas de núcleo fundido
O conceito de partículas peliculares ou superficialmente porosas, constituídas por
um núcleo sólido e uma fina camada porosa, foi inicialmente sugerido por Hórvath e
Lipsky, na década de 60, e foi aplicado a colunas de cromatografia de troca iônica,
um tipo de cromatografia líquido-sólido. O desenvolvimento dessas partículas foi
realizado considerando o princípio de que o menor caminho de difusão presente nas
partículas peliculares, comparado ao presente nas partículas totalmente porosas,
poderia propiciar uma difusão mais rápida do analito, aumentando a eficiência
cromatográfica. Apesar da excelente capacidade de separação apresentada por
essas partículas, elas não foram adotadas pela comunidade científica pelo fato de a
cromatografia de troca iônica apresentar aplicação limitada. Na época, o interesse
maior era pela cromatografia líquido-líquido [14].
Também na década de 60 e pelo mesmo motivo, Kirkland e colaboradores
desenvolveram as chamadas shell particles, constituídas por um núcleo sólido e por
uma camada porosa mais espessa do que a sugerida por Hórvath e Lipsky. Kirkland
foi o primeiro pesquisador a desenvolver partículas desse tipo aplicadas à
cromatografia líquido-líquido. Na década de 70, colunas como Corasil® I e II, da
Waters, empacotadas com shell particles, foram comercializadas. Entre 5 e 10% do
volume dessas partículas era ocupado pela camada porosa. Apesar do sucesso
inicial, com o passar do tempo percebeu-se que essas colunas eram instáveis:
rapidamente perdiam fase estacionária, arrastada pela fase móvel, gerando análises
de baixa reprodutibilidade. Além disso, constatou-se a baixa capacidade de amostra
em consequência da menor superfície porosa desse tipo de partícula [14].
Ao mesmo tempo em que as limitações das shell particles foram evidenciadas, eram
produzidas partículas totalmente porosas com diâmetros cada vez menores,
capazes de gerar análises extremamente eficientes. A redução do tamanho das
partículas totalmente porosas passou a ser o foco das pesquisas e os estudos
envolvendo as shell particles foram temporariamente interrompidos [14].
Recentemente, a necessidade de desenvolver colunas eficientes que pudessem
operar em equipamentos de HPLC levou ao desenvolvimento de novas gerações de

73
colunas empacotadas com shell particles. As colunas empacotadas com partículas
totalmente porosas de tamanho muito reduzido, apesar de eficientes, geram
pressões acima de 400 bar, sendo compatíveis com equipamentos de UHPLC.
Pensando nisso, em 1992 a Agilent desenvolveu a coluna Poroshell 300®,
constituída por partículas de 7 μm de diâmetro, envolvidas por camada porosa de
1 μm. Entretanto, o real sucesso dessas partículas ocorreu em 2006, com a
introdução da coluna Halo core-shell® pela Advanced Material Technologies. Essa
coluna é constituída por shell particles, de 2,7 μm de diâmetro, envolvida por 0,5 μm
de camada porosa. A camada porosa, agora estável quimicamente, ocupa 75% do
volume da partícula, e não apenas 10% como nas shell particles desenvolvidas na
década de 60, solucionando o problema da baixa capacidade de amostra [14].
Procedimento típico de preparo das shell particles envolve a suspensão de
partículas de sílica não porosas em uma mistura de etanol e água. Uma solução
concentrada de hidróxido de amônio deve ser vertida sobre a suspensão, fazendo
com que os grupos silanóis presentes na superfície da sílica fiquem carregados
negativamente. Em seguida adiciona-se uma solução de surfactante orgânico
catiônico (C18TABr+) e as moléculas desse surfactante, carregadas positivamente,
são adsorvidas às partículas de sílica, que estão carregadas negativamente. Por
último adiciona-se tetraetilortosilicato, que tem como função construir a estrutura de
sílica porosa em torno das moléculas de surfactante adsorvidas. Finalmente, as
novas partículas formadas são centrifugadas e o mesmo processo é repetido até
que a espessura da camada porosa desejada seja alcançada [14].
As shell particles também têm sido chamadas fused core, core shell e porous shell.
Em português, o termo mais empregado é partículas de núcleo fundido. As colunas
com partículas de núcleo fundido (Figura 2.3), ao contrário das monolíticas, não são
um sistema contínuo. São constituídas por um núcleo sólido de sílica fundida não
poroso (core), normalmente de 1,6 ou 1,7 µm de diâmetro, revestido por uma parte
externa porosa (shell) com espessura geralmente de 0,5 µm. Sendo assim, essas
partículas possuem um diâmetro total de 2,6 ou 2,7 µm (Figura 2.4) [15]. Já há no
mercado, entretanto, partículas de núcleo fundido com diâmetro total de 1,7 µm
(núcleo de 1,25 µm de diâmetro e a parte externa de 0,23 µm de espessura),

74
1,3 µm, assim como partículas de 2,6 µm de diâmetro total com parte externa porosa
de 0,35 µm e núcleo de 1,9 µm.
Figura 2.3 - Micrografia de partícula de núcleo fundido [16]
Figura 2.4 - Esquema de partícula de núcleo fundido [17]
As colunas de núcleo fundido possuem porosidade (ε) entre 50% e 60%. Na
literatura encontram-se descritos valores de ε iguais a 56% (Poroshell® 2,7 μm), 55%
(Kinetex® 2,6 μm) e 51% (Halo® 2,7 μm) [18, 19]. A permeabilidade (Kv0) desse tipo
de coluna é inferior à observada em colunas monolíticas. Valores de Kv0 iguais a
1,18 x 10-14 m2 (Kinetex® 2,6 μm), 1,02 x 10-14 m2 (Halo® 2,7 μm) e 0,99 x 10-14 m2
(Poroshell® 2,7 μm) foram descritos na literatura [20]. Observa-se que a retenção
dos analitos em colunas com partículas de núcleo fundido é inferior à observada em
colunas convencionais empacotadas com partíclas totalmente porosas. A explicação
está no fato de o volume de fase estacionária presente em partículas de núcleo
fundido ser inferior ao presente em partículas totalmente porosas [14].
O maior benefício de colunas com partículas de núcleo fundido é a possibilidade de
redução do tempo de análise, com eficiência cromatográfica comparável à propiciada
por colunas com partículas porosas sub 2 µm e manutenção da pressão em níveis
compatíveis com os requeridos por um instrumento de HPLC (400 bar) [21].

75
Apesar das vantagens, de modo similar às colunas monolíticas há um número
limitado de fabricantes (exemplos: Phenomenex, Agilent, Advanced Materials
Technologies, Supelco) e de grupos químicos ligantes disponíveis, o que é um fator
que contribui para a baixa utilização desse tipo de coluna [10]. Em cotação realizada
no primeiro semestre de 2011, a coluna de núcleo fundido da marca Phenomenex
(Kinetex® C18, 100 x 2,1mm, 2,6 µm) foi cotada em R$ 1490,00. Já a coluna da
marca Agilent (Poroshell® C18, 100 x 2,1mm, 2.7 µm) foi cotada em R$ 975,00,
preço próximo ao cobrado pelas tradicionais colunas convencionais.
Apesar de ainda pouco utilizada, há várias publicações empregando coluna de
núcleo fundido. Exemplo é o trabalho publicado em 2011 por Kirkland e
colaboradores, no qual é descrita a separação de nove proteínas, em menos de dois
minutos, com resolução mínima de 1,5, utilizando-se coluna de núcleo fundido [22].
1.3 Coluna empacotada com partículas porosas sub 2 µm
O desafio de desenvolver partículas com diâmetro inferior a 2 µm, resistentes à
pressão gerada no sistema cromatográfico em consequência da redução do
tamanho das partículas, foi vencido, inicialmente, pela obtenção de partículas não
porosas de 1,5 µm de diâmetro. Embora essas partículas não porosas sub 2 µm
conduzissem a análises de alta eficiência, apresentavam baixa capacidade de
amostra e baixa retenção devido à pequena superfície de contato com o analito.
Para que não houvesse redução da retenção e da capacidade de amostra era
preciso desenvolver partículas porosas sub 2 µm que fossem capazes de resistir a
altas pressões. Pensando nisso pesquisadores da Waters, em 2004, desenvolveram
a primeira coluna empacotada com partículas porosas de diâmetro inferior a 2 µm
resistentes mecanicamente. Nessas colunas, denominadas Acquity®, a resistência
das partículas porosas sub 2 µm é alcançada por meio de pontes de etano inseridas
na estrutura da sílica [23].
As colunas empacotadas com partículas porosas com diâmetro inferior a 2 µm, ao
contrário das monolíticas e das empacotadas com partículas de núcleo fundido,
aplicam-se a análises realizadas em instrumentos que operam sob pressão de até

76
1000 bar (UHPLC) [24]. Instrumentos de UHPLC ainda não são comuns devido ao
custo 25% maior quando comparado aos instrumentos de HPLC [15].
O primeiro cromatógrafo a suportar altas pressões foi desenvolvido pela Waters e
lançado em 2004 com o nome de Acquity UPLC®. As modificações requeridas em
um sistema desse tipo em relação ao HPLC são: resistência a altas pressões
(tipicamente até 1000 bar), volumes internos menores, melhoramento nos sistema
de controle de dados, injetores com precisão na faixa de pequenos volumes de
injeção, sistema de injeção rápido e capaz de suportar pressões elevadas, detector
com elevada frequência de aquisição de dados, filtros de colunas com porosidade
menor que 2 µm. Depois de 2004, outros sistemas de UHPLC, que suportam
pressões de até 1300 bar, foram desenvolvidos [25,26].
As colunas com partículas porosas sub 2 μm possuem valores de permeabilidade
(Kv0) da ordem de 10-15 e, portanto são menos permeáveis que as monolíticas. Na
literatura encontram-se relatados valores de Kv0 iguais a 6,28 x 10-15 m2 para a
coluna Zorbax SB® e 4,50 x 10-15 m2 para a coluna Acquity® [27]. Quanto à
porosidade, valor de 63% foi descrito para a coluna Acquity® [19]. Em relação à
retenção verifica-se que colunas com partículas porosas sub 2 μm apresentam maior
capacidade de retenção do que colunas com partículas de núcleo fundido. Isso se
deve ao fato de essas partículas serem menores e totalmente porosas, o que
aumenta a superfície de contato entre analito e fase estacionária [21].
A principal vantagem das colunas com partículas porosas sub 2 μm é possibilitar
análises rápidas, o que contribui para a redução do consumo de solventes, com
elevada eficiência de separação. A eficiência de uma análise, determinada pelo
número de pratos teóricos (N), é inversamente proporcional ao diâmetro das
partículas, o que explica o fato de colunas empacotadas com partículas tão
pequenas, como as de diâmetro inferior a 2 μm, propiciarem análises de elevada
eficiência [28]. Ao benefício da alta eficiência se contrapõe a elevação da pressão do
sistema cromatográfico, já que a pressão gerada é inversamente proporcional ao
quadrado do diâmetro das partículas (equação 2.1), requerendo a utilização de um
sistema de UHPLC, mais caros e de menor disponibilidade nos laboratórios [5, 23].

77
2
0
p
td
uLP
(2.1)
Pt = pressão no sistema cromatográficos, = fator de resistência da coluna, L =
comprimento da coluna, u0 = velocidade linear da fase móvel, dp = diâmetro da
partícula
Vale destacar que, ao contrário do que ocorre com as colunas monolíticas e de
partículas de núcleo fundido, há uma grande variedade de grupos químicos ligantes
e de fabricantes das colunas com partículas porosas sub 2 μm disponíveis no
mercado [10]. Em cotação realizada no primeiro semestre de 2011, a coluna da
marca Waters (Acquity UPLC BEH C18, 100 x 2,1mm, 1,7 µm) foi cotada em
R$ 3310,18. Já a coluna da marca Agilent (Zorbax C18, 100 x 2,1mm, 1,8 µm) foi
cotada em R$ 975,00.
Além das vantagens já relatadas, com partículas porosas sub 2 μm são obtidos
picos com larguras na meia altura (W0,5) inferiores a 1 segundo, propiciando um
aumento de 2 a 3 vezes na detectabilidade se comparada à propiciada por colunas
convencionais [21].
Característica importante das colunas com partículas sub 2 µm é a limitação do
diâmetro interno, que deve estar compreendido na faixa de 1,0 a 2,1 mm, a fim de
minimizar o efeito frictional heating. Esse efeito é caracterizado pelo aquecimento
dos solventes da fase móvel quando submetidos a altos valores de pressão,
promovendo gradiente de temperatura ao longo da coluna e consequente redução
da eficiência [9, 10, 23]. Outros problemas a serem destacados nesse tipo de coluna
é a menor reprodutibilidade de empacotamento, o que é consequência da redução
do tamanho das partículas, e o menor tempo de vida útil, que é consequência dos
altos valores de pressão a que essas colunas são submetidas [9, 29].
A determinação de triancinolona e compostos relacionados em 1,5 minuto é um
exemplo da aplicação das colunas porosas com partículas sub 2 μm em análises
farmacêuticas. A análise foi conduzida em um UHPLC (Acquity UPLC®), coluna C18

78
2,1 x 50 mm, 1,7 μm, a 25 oC, utilizando fase móvel composta de ACN e água
(40:60) e vazão variando de 0,5 a 0,6 ml/min [29].
2 OBJETIVO ESPECÍFICO
O objetivo do trabalho descrito nesse capítulo foi caracterizar fisicamente as colunas
CV, MN, NF e Sub 2 μm utilizando os parâmetros volume morto, porosidade total,
fator de retenção, seletividade e permeabilidade.
3 MATERIAIS E MÉTODOS
3.1 Materiais
3.1.1 Insumos farmacêuticos ativos
Os mesmos descritos no capítulo 1, item 3.1.1.
3.1.2 Solventes, reagentes e vidrarias
Os mesmos descritos no capítulo 1, item 3.1.2.
3.1.3 Equipamentos e colunas cromatográficas
Os mesmos descritos no capítulo 1, item 3.1.3.
3.2 Métodos
3.2.1 Estimativa do volume morto e porosidade total das colunas
Volume morto de uma coluna cromatográfica representa o volume total de líquido
que é capaz de ocupá-la, e está diretamente relacionado à porosidade [28]. Para
estimá-lo injetaram-se, em triplicata, em todas as quatro colunas, solução de uracila
(10 µg/ml em fase móvel). A fase móvel foi composta por ACN:solução de acetato de
amônio 5 mM, pH 3,0 (50:50). A análise foi conduzida sob temperatura de 30 ºC,

79
vazão de 0,1 ml/min, volume de injeção de 1 μl e comprimento de onda de 254 nm.
Utilizando o valor médio do tempo de detecção da uracila (tempo morto), o volume
morto de cada coluna foi calculado por meio da equação (2.2) [30].
0tVVm (2.2)
Vm = volume morto, V = vazão da fase móvel, t0 = tempo morto
A partir dos valores de volume morto foi possível estimar a porosidade total de cada
coluna utilizando-se a equação (2.3) [28]. A porosidade representa o espaço livre
total de uma coluna, e é composta pela porosidade externa (espaços vazios entre as
partículas) e pela porosidade interna (espaços vazios dentro das partículas) [19].
Lr
V
c
m
214,3
(2.3)
ε = porosidade total Vm = volume morto, rc = raio da coluna, L = comprimento da
coluna
3.2.2 Avaliação da retenção e seletividade das colunas
O fator de retenção (k) é o parâmetro utilizado para avaliação da retenção, que está
relacionada à capacidade da coluna em manter as moléculas dos analitos retidas na
fase estacionária. Já a seletividade (α) está relacionada à capacidade de separação
de duas substâncias que eluem lado a lado [28].
A seletividade e o fator de retenção das colunas foram estimados em três fases
móveis de forças eluotrópicas diferentes, utilizando como modelo os fármacos
clorpropamida (CL), glibenclamida (GB), glimepirida (GM) e gliclazida (GZ). Para a
execução do experimento foram utilizadas condições previamente estabelecidas:
fase móvel composta por ACN e solução de acetato de amônio 5 mM pH 3,0;
volume de injeção 2 µl; temperatura 30 oC; vazão de 0,2 ml/min e comprimento de
onda 230 nm.

80
As três fases móveis utilizadas foram ACN: solução de acetato de amônio nas
proporções 55:45, 50:50 e 45:55. Utilizando cada uma delas, injetou-se, em
triplicata, solução contendo 100 μg/ml de cada um dos antidiabéticos e solução de
uracila 10 μg/ml para determinação do tempo morto. O procedimento acima descrito
foi executado nas quatro colunas cromatográficas.
Para o cálculo do fator de retenção determinou-se o tempo de retenção médio de
cada analito e o tempo morto médio, em cada uma das colunas, empregando-se
cada uma das fases móveis. O cálculo foi realizado por meio da equação (2.4) [28].
0
0
t
ttk r
(2.4)
k = fator de retenção, tr = tempo de retenção, t0 = tempo morto
A seletividade (α) também foi determinada para cada uma das colunas e fases
móveis experimentadas para os pares de analitos adjacentes. A equação (2.5) foi
utilizada para realização dos cálculos [28].
1
2
k
k
(2.5)
α = seletividade, k2 = fator de retenção do analito mais retido e k1 = fator de retenção
do analito menos retido.
3.2.3 Estimativa da permeabilidade das colunas
A permeabilidade (Kv0) das colunas foi determinada com o auxílio do gráfico de
velocidade linear (u0) em função da pressão na coluna (∆Pc). A permeabilidade pode
ser calculada por meio da equação (2.6), e a pressão da coluna e a velocidade linear
são determinadas com auxílio das equações (2.7) e (2.8), respectivamente [6, 7].

81
c
vP
LuK
00 (2.6)
ectc PPP (2.7)
00 / tLu (2.8)
Kv0 = permeabilidade, u0 = velocidade linear da fase móvel, η = viscosidade da fase
móvel, L = comprimento da coluna, ∆Pc = pressão na coluna, ∆Pt = pressão total do
sistema com a coluna, ∆Pec = pressão extracoluna (pressão do sistema na ausência
da coluna), t0 = tempo morto
Considerando-se u0 como variável independente (x) e ∆Pc como variável dependente
(y), a seguinte transformação pode ser realizada na equação (2.6):
c
vP
LuK
00
y
LxKv
0
0vK
Lxy
A partir da relação matemática apresentada, conclui-se que a inclinação do gráfico
u0 x ∆Pc pode ser escrita como:
0vK
LInclinação
Portanto, Kv0 pode ser calculado com o auxílio da equação (2.9).
Inclinação
LKv
0
(2.9)
Para a execução dos experimentos de determinação de permeabilidade, método
analítico desenvolvido para determinação dos antidiabéticos orais em coluna CV, e
aqueles obtidos após transferência para as colunas MN, NF e sub 2 μm, foram
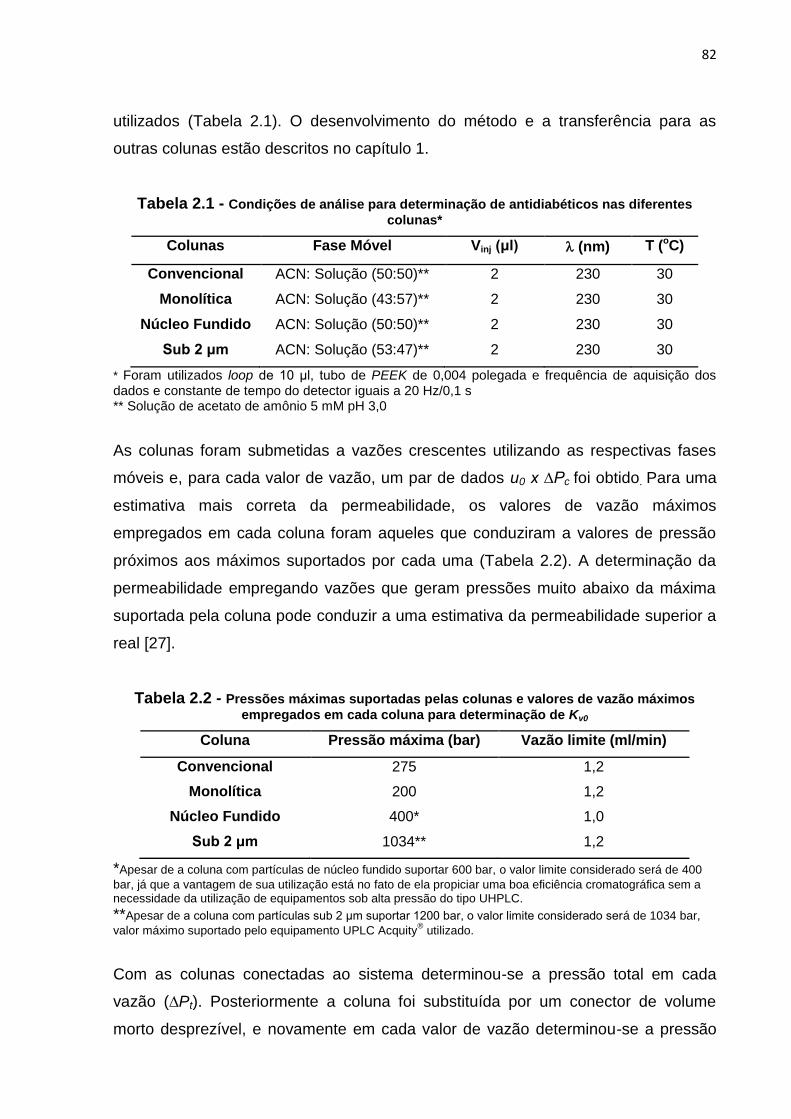
82
utilizados (Tabela 2.1). O desenvolvimento do método e a transferência para as
outras colunas estão descritos no capítulo 1.
Tabela 2.1 - Condições de análise para determinação de antidiabéticos nas diferentes
colunas*
Colunas Fase Móvel Vinj (μl) (nm) T (oC)
Convencional ACN: Solução (50:50)** 2 230 30
Monolítica ACN: Solução (43:57)** 2 230 30
Núcleo Fundido ACN: Solução (50:50)** 2 230 30
Sub 2 μm ACN: Solução (53:47)** 2 230 30
* Foram utilizados loop de 10 μl, tubo de PEEK de 0,004 polegada e frequência de aquisição dos dados e constante de tempo do detector iguais a 20 Hz/0,1 s ** Solução de acetato de amônio 5 mM pH 3,0
As colunas foram submetidas a vazões crescentes utilizando as respectivas fases
móveis e, para cada valor de vazão, um par de dados u0 x ∆Pc foi obtido. Para uma
estimativa mais correta da permeabilidade, os valores de vazão máximos
empregados em cada coluna foram aqueles que conduziram a valores de pressão
próximos aos máximos suportados por cada uma (Tabela 2.2). A determinação da
permeabilidade empregando vazões que geram pressões muito abaixo da máxima
suportada pela coluna pode conduzir a uma estimativa da permeabilidade superior a
real [27].
Tabela 2.2 - Pressões máximas suportadas pelas colunas e valores de vazão máximos
empregados em cada coluna para determinação de Kv0
Coluna Pressão máxima (bar) Vazão limite (ml/min)
Convencional 275 1,2
Monolítica 200 1,2
Núcleo Fundido 400* 1,0
Sub 2 μm 1034** 1,2
*Apesar de a coluna com partículas de núcleo fundido suportar 600 bar, o valor limite considerado será de 400
bar, já que a vantagem de sua utilização está no fato de ela propiciar uma boa eficiência cromatográfica sem a necessidade da utilização de equipamentos sob alta pressão do tipo UHPLC.
**Apesar de a coluna com partículas sub 2 μm suportar 1200 bar, o valor limite considerado será de 1034 bar,
valor máximo suportado pelo equipamento UPLC Acquity® utilizado.
Com as colunas conectadas ao sistema determinou-se a pressão total em cada
vazão (∆Pt). Posteriormente a coluna foi substituída por um conector de volume
morto desprezível, e novamente em cada valor de vazão determinou-se a pressão

83
extracoluna (∆Pec). Subtraindo-se a pressão total da extracoluna, conforme equação
2.7, determinou-se a pressão em cada coluna (∆Pc).
Para cálculo de u0 foi necessária a determinação do tempo morto (t0) em cada uma
das vazões, em cada uma das colunas, por meio da injeção de solução de uracila a
10 μg/ml. Dividindo-se o comprimento de cada coluna (L = 100 mm), pelo tempo
morto (equação 2.8), foi possível calcular u0 em cada coluna e vazão.
Os pares de valores u0 x ∆Pc relativos a cada coluna foram plotados. Fazendo-se
uso do método dos mínimos quadrados realizou-se análise de regressão linear e os
valores de inclinação dos gráficos foram determinados. Utilizando-se os valores de
inclinação determinados, os valores tabelados de viscosidade da fase móvel de
acordo com a proporção dos componentes orgânico e aquoso [30] e empregando-se
o comprimento das colunas (L = 100 mm), os valores de permeabilidade das colunas
foram calculados por meio da equação (2.9).
4 RESULTADOS E DISCUSSÃO
4.1 Estimativa do volume morto e porosidade total das colunas
Na Tabela 2.3 estão apresentados os valores estimados de volume morto e
porosidade total das colunas.
Tabela 2.3 - Volume morto e porosidade total das colunas
Coluna Volume morto (μL) Porosidade (%)
Convencional 221 64
Monolítica 261 83
Núcleo Fundido 208 60
Sub 2 μm 196 57
Encontra-se descrito na literatura que a porosidade de coluna monolítica é de cerca
de 80% [1], portanto o valor encontrado, 83%, é coerente. Observa-se ainda que a
coluna monolítica é aquela que apresenta maior volume morto (261 μl). Isso é

84
explicado pelo fato de a coluna mais porosa ser a que apresenta maior volume de
espaços vazios que podem ser percorridos pela fase móvel.
Na literatura foram descritos valores de porosidade em torno de 66% para coluna
convencional [2] e de 63% para coluna com partículas sub 2 μm (Acquity®) [19].
Portanto, os valores encontrados (ε CV= 64% e ε sub 2 μm= 57%) estão próximos aos
descritos.
Considerando-se que nas partículas de núcleo fundido apenas a parte externa é
porosa [13], pode-se afirma que a porosidade interna (espaços vazios dentro das
partículas) é menor do que a presente nas partículas totalmente porosas de 5 e 2
μm. Entretanto, para se avaliar a porosidade total deve-se considerar também a
porosidade externa (espaços vazios entre as partículas). Sabe-se que o aumento do
diâmetro das partículas contribui para o aumento da porosidade externa [19]. Sendo
assim, a maior porosidade total da coluna convencional em relação à coluna de
núcleo fundido ( CV = 64% > ε NF = 60%), verificada experimentalmente, era
esperada, já que as colunas com partículas totalmente porosas de 5 μm
apresentam, além da maior porosidade externa, maior porosidade interna.
Verificou-se também que a porosidade total da coluna com partículas de núcleo
fundido foi superior àquela da coluna com partículas sub 2 μm ( NF = 60% >
sub2 = 57%), o que não está de acordo com os resultados apresentados na
literatura. Na literatura valores de ε = 56% (Poroshell® 2,7 μm) [18] e 51% (Halo®
2,7 μm) [19] foram descritos para colunas de núcleo fundido, e valores superiores
(ε = 63%) foram relatados para colunas sub 2 μm.
4.2 Estimativa da retenção e seletividade das colunas
4.2.1 Retenção
Para demonstrar o comportamento das colunas quanto à capacidade de retenção,
está apresentado a seguir, na Figura 2.5, o gráfico que relaciona o percentual de
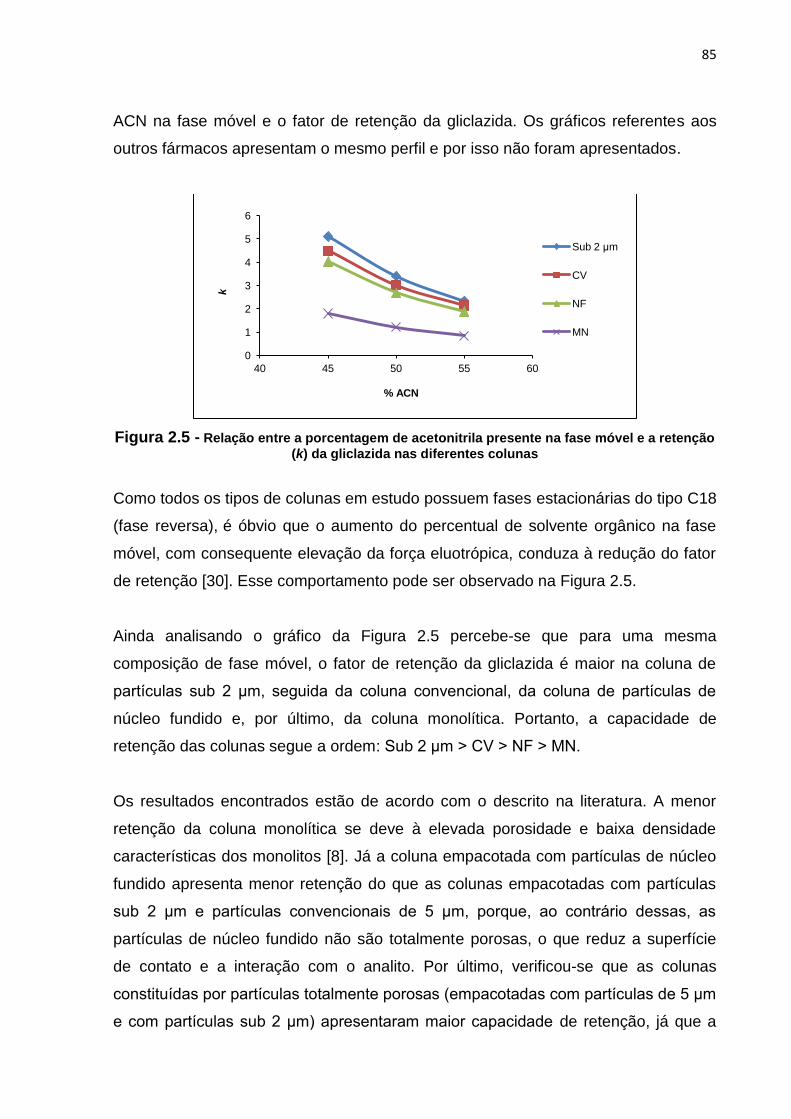
85
ACN na fase móvel e o fator de retenção da gliclazida. Os gráficos referentes aos
outros fármacos apresentam o mesmo perfil e por isso não foram apresentados.
Figura 2.5 - Relação entre a porcentagem de acetonitrila presente na fase móvel e a retenção
(k) da gliclazida nas diferentes colunas
Como todos os tipos de colunas em estudo possuem fases estacionárias do tipo C18
(fase reversa), é óbvio que o aumento do percentual de solvente orgânico na fase
móvel, com consequente elevação da força eluotrópica, conduza à redução do fator
de retenção [30]. Esse comportamento pode ser observado na Figura 2.5.
Ainda analisando o gráfico da Figura 2.5 percebe-se que para uma mesma
composição de fase móvel, o fator de retenção da gliclazida é maior na coluna de
partículas sub 2 μm, seguida da coluna convencional, da coluna de partículas de
núcleo fundido e, por último, da coluna monolítica. Portanto, a capacidade de
retenção das colunas segue a ordem: Sub 2 μm > CV > NF > MN.
Os resultados encontrados estão de acordo com o descrito na literatura. A menor
retenção da coluna monolítica se deve à elevada porosidade e baixa densidade
características dos monolitos [8]. Já a coluna empacotada com partículas de núcleo
fundido apresenta menor retenção do que as colunas empacotadas com partículas
sub 2 μm e partículas convencionais de 5 μm, porque, ao contrário dessas, as
partículas de núcleo fundido não são totalmente porosas, o que reduz a superfície
de contato e a interação com o analito. Por último, verificou-se que as colunas
constituídas por partículas totalmente porosas (empacotadas com partículas de 5 μm
e com partículas sub 2 μm) apresentaram maior capacidade de retenção, já que a
0
1
2
3
4
5
6
40 45 50 55 60
k
% ACN
Sub 2 μm
CV
NF
MN
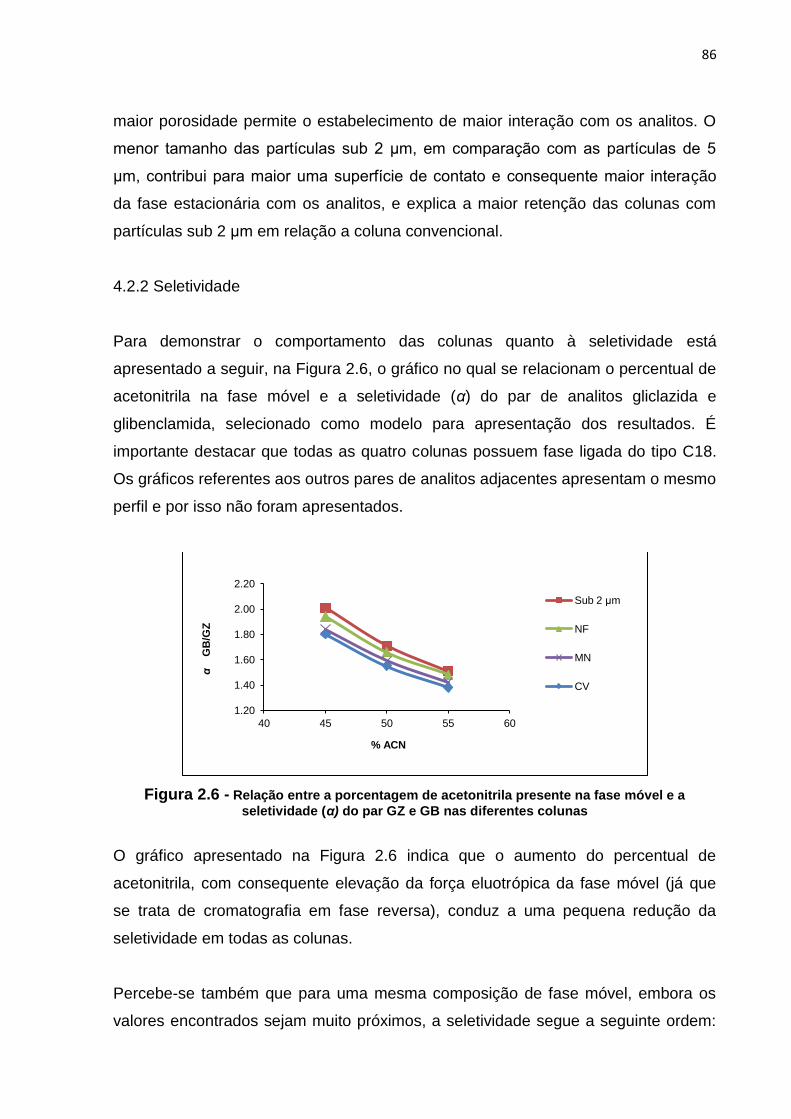
86
maior porosidade permite o estabelecimento de maior interação com os analitos. O
menor tamanho das partículas sub 2 μm, em comparação com as partículas de 5
μm, contribui para maior uma superfície de contato e consequente maior interação
da fase estacionária com os analitos, e explica a maior retenção das colunas com
partículas sub 2 μm em relação a coluna convencional.
4.2.2 Seletividade
Para demonstrar o comportamento das colunas quanto à seletividade está
apresentado a seguir, na Figura 2.6, o gráfico no qual se relacionam o percentual de
acetonitrila na fase móvel e a seletividade (α) do par de analitos gliclazida e
glibenclamida, selecionado como modelo para apresentação dos resultados. É
importante destacar que todas as quatro colunas possuem fase ligada do tipo C18.
Os gráficos referentes aos outros pares de analitos adjacentes apresentam o mesmo
perfil e por isso não foram apresentados.
Figura 2.6 - Relação entre a porcentagem de acetonitrila presente na fase móvel e a
seletividade (α) do par GZ e GB nas diferentes colunas
O gráfico apresentado na Figura 2.6 indica que o aumento do percentual de
acetonitrila, com consequente elevação da força eluotrópica da fase móvel (já que
se trata de cromatografia em fase reversa), conduz a uma pequena redução da
seletividade em todas as colunas.
Percebe-se também que para uma mesma composição de fase móvel, embora os
valores encontrados sejam muito próximos, a seletividade segue a seguinte ordem:
1.20
1.40
1.60
1.80
2.00
2.20
40 45 50 55 60
α
GB
/GZ
% ACN
Sub 2 μm
NF
MN
CV
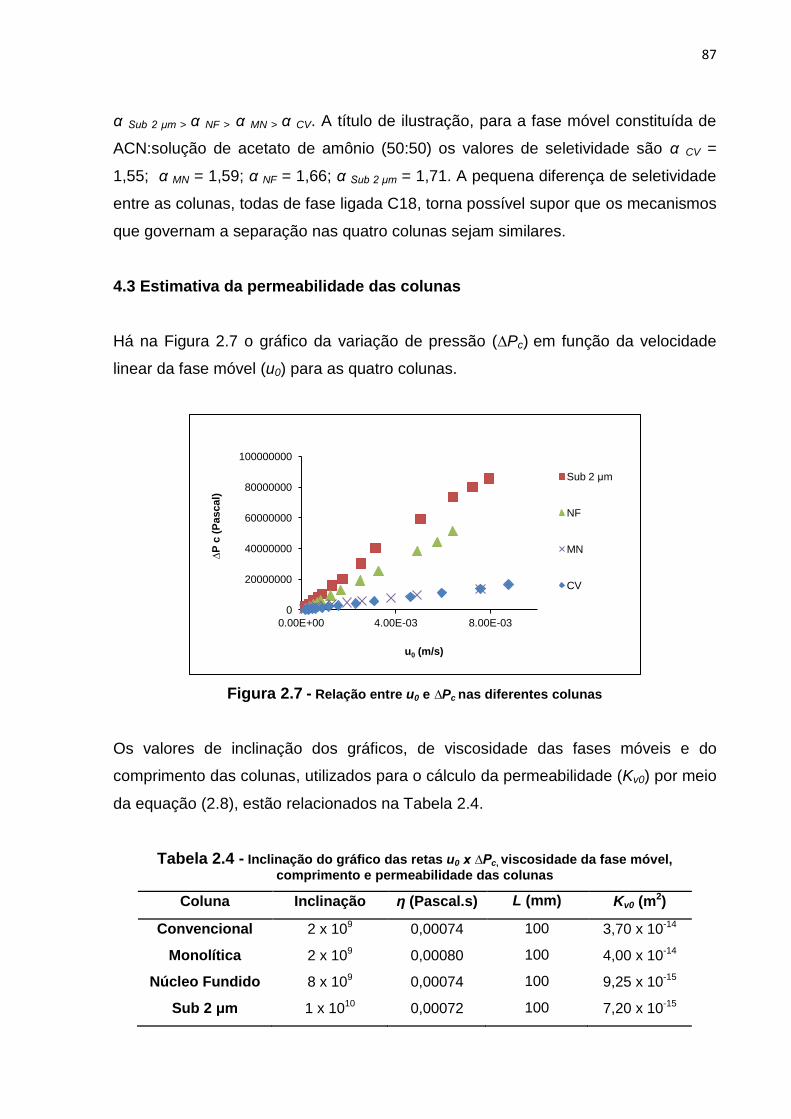
87
α Sub 2 μm > α NF > α MN > α CV. A título de ilustração, para a fase móvel constituída de
ACN:solução de acetato de amônio (50:50) os valores de seletividade são α CV =
1,55; α MN = 1,59; α NF = 1,66; α Sub 2 μm = 1,71. A pequena diferença de seletividade
entre as colunas, todas de fase ligada C18, torna possível supor que os mecanismos
que governam a separação nas quatro colunas sejam similares.
4.3 Estimativa da permeabilidade das colunas
Há na Figura 2.7 o gráfico da variação de pressão (∆Pc) em função da velocidade
linear da fase móvel (u0) para as quatro colunas.
Figura 2.7 - Relação entre u0 e ∆Pc nas diferentes colunas
Os valores de inclinação dos gráficos, de viscosidade das fases móveis e do
comprimento das colunas, utilizados para o cálculo da permeabilidade (Kv0) por meio
da equação (2.8), estão relacionados na Tabela 2.4.
Tabela 2.4 - Inclinação do gráfico das retas u0 x ∆Pc, viscosidade da fase móvel,
comprimento e permeabilidade das colunas
Coluna Inclinação η (Pascal.s) L (mm) Kv0 (m2)
Convencional 2 x 109 0,00074 100 3,70 x 10-14
Monolítica 2 x 109 0,00080 100 4,00 x 10-14
Núcleo Fundido 8 x 109 0,00074 100 9,25 x 10-15
Sub 2 μm 1 x 1010 0,00072 100 7,20 x 10-15
0
20000000
40000000
60000000
80000000
100000000
0.00E+00 4.00E-03 8.00E-03
∆P
c (
Pascal)
u0 (m/s)
Sub 2 μm
NF
MN
CV

88
Analisando-se os resultados da Tabela 2.4 verifica-se que a permeabilidade das
colunas segue a seguinte ordem: Kv0 MN > Kv0 CV > Kv0 NF > Kv0 Sub2. A ordem
encontrada e os valores de Kv0 são similares aos descritos na literatura e podem ser
explicados com base nas características de cada coluna [6, 7, 27].
A maior permeabilidade apresentada pela coluna monolítica (Kv0 = 4,00 x 10-14 m2)
deve-se à estrutura de macroporos, pelos quais a fase móvel passa sem grande
resistência [1]. Na literatura, Kv0 = 6,00 x 10-14 m2 [6] e Kv0 = 8,00 x 10-14 m2 [7] foram
descritos para coluna a monolítica Onyx®.
Já a coluna empacotada com partícula totalmente porosa sub 2 µm foi a de menor
permeabilidade (Kv0 = 7,20 x 10-15 m2). A despeito de a coluna ser totalmente porosa,
o que contribuiria para aumentar permeabilidade, as partículas são muito pequenas
(1,8 μm) e o que dificulta a passagem da fase móvel entre as partículas. Isso
contribui para a menor permeabilidade desse tipo de coluna e explica a alta pressão
que ela gera no sistema. Na literatura foram descritos valores de Kv0 iguais a 6,28 x
10-15 m2 para a coluna Zorbax SB® e 4,50 x 10-15 m2 para a coluna Acquity® [27].
Por outro lado, a coluna empacotada com partículas de núcleo fundido de 2,7 μm de
diâmetro apresentou permeabilidade igual a 9,25 x 10-15 m2, valor intermediário entre
as permeabilidades das colunas convencional (partícula totalmente porosa de 5 μm)
e de partículas sub 2 μm (partícula totalmente porosa de 1,8 μm). A menor
permeabilidade comparada à coluna convencional pode ser explicada pela estrutura
parcialmente porosa e por possuir tamanho de partícula inferior (2,7 μm < 5 μm). Já
a maior permeabilidade comparada à coluna de partículas sub 2 μm é explicada pelo
maior tamanho de partícula (2,7 μm > 1,8 μm). Na literatura foram descritos valores
de Kv0 para colunas de núcleo fundido iguais a 1,18 x 10-14 m2 (Kinetex® 2,6 μm),
1,02 x 10-14 m2 (Halo® 2,7 μm) e 0,99 x 10-14 m2 (Poroshell® 2,7 μm) [20].
5 CONCLUSÃO
As colunas foram caracterizadas segundo os seguintes parâmetros: volume morto,
porosidade, retenção, seletividade e permeabilidade. Verificou-se que a coluna
monolítica apresentou maior porosidade (83%), e consequentemente maior volume

89
morto, o que é justificável pela presença de uma estrutura de meso e macroporos.
Já as outras três colunas apresentaram porosidades na faixa de 57% a 64%.
Constatou-se que as colunas, todas de fase ligada C18, apresentaram seletividades
similares, o que permite supor que os mecanismos que governam a separação nas
quatro colunas sejam similares.
A coluna monolítica apresentou a menor capacidade de retenção, em função da alta
porosidade e menor interação dos analitos com a fase estacionária. Analisando-se
as outras três colunas verifica-se que os fatores de retenção, apesar de maiores na
coluna de partículas sub 2 μm, seguida da convencional e da empacotada com
partículas de núcleo fundido, não foram muito diferentes.
O último parâmetro avaliado foi a permeabilidade (Kv0). Os valores de
permeabilidade das colunas em estudo estão de acordo com os descritos na
literatura e seguem a seguinte ordem: Kv0 MN > Kv0 CV > Kv0 NF > Kv0 Sub2. É importante
destacar que a permeabilidade da coluna está diretamente relacionada com a
pressão que ela gera no sistema cromatográfico. Sendo assim, a menor
permeabilidade da coluna empacotada com partículas sub 2 μm explica o fato de
esse tipo de partícula gerar pressões na ordem de 1000 bar, justificando a
necessidade do emprego de um equipamento de UHPLC. Já a maior permeabilidade
das colunas monolíticas explica o fato de elas poderem ser submetidas a altos
valores de vazão de fase móvel e mesmo assim poderem ser utilizadas em
equipamento de HPLC que operam sob pressão máxima de 400 bar.

90
REFERÊNCIAS BIBLIOGRÁFICAS
[1] FARIA, A. M.; BOTTOLI, C. B. G.; JARDIM, C. S. F.; COLLINS, C. H. Fases estacionárias monolíticas para separações cromatográficas. Química Nova. v. 29, n.2, p. 300-309, 2006. [2] MISTRY, K.; GRINBERG, N. Application of Monolithic Columns in High Performance Liquid Chromatography. Journal of Liquid Chromatography and Related Technologies. v. 28, p. 1055-1074, 2005. [3] SIOUFFI, A. M. Silica gel-based monoliths prepared by the sol–gel method: facts and figures. Journal of Chromatography A. v. 1000, p. 801-818, 2003. [4] MONOLITHIC SILICA COLUMNS: BENEFITS AND APPLICATIONS OF CHROMOLITH®HPLC COLUMNSHTTP. Disponível em: <www.docstoc.com/docs/ 24844699/Monolithic-silica-columns-benefits-and-applications-of-Chromolith>. Acesso em: 05 de Dezembro de 2012. [5] UNGER, K. K.; SKUDAS, R.; SCHULTE, M. M. Particle packed columns and monolithic columns in high-performance liquid chromatography comparison and critical appraisal. Journal of Chromatography A. v. 1184, p. 393-415, 2008. [6] DESMET, G.; CLICQ, D.; NGUYEN, D. T.T.; GUILLARME, D.; RUDAZ, S.; VEUTHEY, J.L.; VERVOORT, N.; TOROK, G.; CABOOTER, D.; GZIL, P. Practical constraints in the kinetic plot representation of chromatographic performance data: theory and application to experimental data. Analytical Chemistry. v. 78, p. 2150-2162, 2006. [7] CABOOTER, D.; VILLIERS, A.; CLICQ, D. SZUCS, R.; SANDRA, P.; DESMET,G. Method to predict and compare the influence of the particle size on the isocratic peak capacity of high-performance liquid chromatography columns. Journal of Chromatography A. v. 1147, p. 183-191, 2007. [8] WUA, N.; DEMPSEYB, J.; YEHLA, P. M.; DOVLETOGLOUA, A.; ELLISONA, D.; WYVRATTA, J. Practical aspects of fast HPLC separations for pharmaceutical process development using monolithic columns. Analytica Chimica Acta. v. 523, p. 149-156, 2004. [9] MALDANER, L.; JARDIM, I. C. S. F. O estado da arte da cromatografia líquida de ultra-eficiência. Química Nova. v. 32, n. 32, p. 214-222, 2009. [10] GUILLERME, D.; RUTA J.; RUDAZ, J.; VEUTHEY, J. New trends in fast and high-resolution liquid chromatography: a critical comparison of existing approaches. Analytical and Bioanalytical Chemistry. v. 397, p. 1069-1082, 2010. [11] MCCALLEY, D. V. Comparison of conventional microparticulate and a monolithic reversed-phase column for high-efficiency fast liquid chromatography of basic
compounds. Journal of Chromatography A. v. 965, p. 51–64, 2002

91
[12] CABRERA, K. A new generation of silica-based monolithic HPLC columns with improved performance. LC-GC North America. Supplement, p. 56-60, 2012. [13] ABOUL-ENEIN, H. Y.; ALI, I. Determination of tadalafil in pharmaceutical preparation by HPLC using monolithic silica column. Talanta. v. 65, p. 276-280, 2005. [14] GUIOCHON, G.; GRITTI, F. Shell particles, trials, tribulations and triumphs. Journal of Chromatography A, v. 1218, p. 1915-1938, 2011. [15] SALISBURY, J. J. Fused-Core Particles: A Pratical Alternative to Sub-2 Micron Particles. Journal of Chromatographic. v. 46, p. 883-886, 2008. [16] NEW FUSED-CORE™ PARTICLES FOR VERY FAST HPLC SEPARATIONS. Disponível em: <www.hplc.hu/PDFs/HALOFusedCore.pdf> Acesso em: 11 de Dezembro de 2012. [17] HALO FUSED-CORE™ PARTICLE TECHNOLOGY FOR HIPPER-FAST AND SUPER-RUGGED TECHNOLOGY Disponível em: <http://www.advanced-materials-tech.com/halo.html>. Acesso em: 15 de Dezembro de 2012. [18] LIEKENS, A.; DENAYER, J.; DESMET, G. Experimental investigation of the difference in B-term dominated band broadening between fully porous and porous-shell particles for liquid chromatography using the effective medium theory. Journal of Chromatography A. v. 1218, p. 4406-4416, 2011. [19] BAKER, J. S.; VINCI, J. C.; MOORE, A. D.; COLON, L. A. Physical characterization and evaluation of HPLC columns packed with superficially porous particles. Journal of Separation Science. v. 33, p. 2547-2557, 2010. [20] CABOOTER, D.; FANIGLIULO, A.; BELLAZZI, G., ALLIERI, B. ROTTIGNIB, A.; DESMET, G. Relationship between the particle size distribution of commercial fully porous and superficially porous high-performance liquid chromatography column packings and their chromatographic performance. Journal of Chromatography A. v. 1217, p. 7074-7081, 2010. [21] ABRAHIM, A. A.; MOHAMMAD A.; SKRDLA, P.; BEREZNITSKI, Y.; CHEN, Y.; WU, N. Pratical comparasion of 2,7 µm fused-core silica particles and porous sub-2 µm particles for fast separation in pharmaceutical process development. Journal of Pharmaceutical and Biomedical Analysis, v. 51, p. 131-137, 2010. [22] KIRKLAND, J. J.; TRUSZKOWSKI, F. A., DILKS, C.H.; ENGEL, G. S. Superficially porous silica microspheres for fast high-performance liquid chromatography of macromolecules. Journal of Chromatography A, v. 890, p. 3-13, 2000. [23] SWARTZ. M. E. UPLC: An Introduction and Review. Journal of Liquid Chromatography and Related Technologies. v. 28, p. 1253-1263, 2005.

92
[24] AL-SAYAH, M. A.; RIZOS, P.; ANTONUCCI, V.; WU, N. High throughput of active pharmaceutical ingredients by UPLC. Journal of Separation Science. v.31, p. 2167-2172, 2008. [25] FEKETE, S.; OLAH, E.; FEKETE, J. Fast liquid chromatography: The domination of core–shell and very fine particles. Journal of Chromatography A. v. 1228, p. 57-71, 2012. [26] WREN, S. A. C.; TCHELITCHEFF, P. Use of ultra-performance liquid chromatography in pharmaceutical development. Journal of Chromatography A. v. 1119, p. 140-146, 2006. [27] CABOOTER, D.; BILLEN, J.; TERRYN, H.; LYNEN, F.; SANDRA, P.; DESMET, G. Kinetic plot and particle size distribution analysis to discuss the performance limits of sub-2 μm and supra-2 μm particle columns. Journal of Chromatography A. v. 1204, p.1-10, 2008. [28] LOUGH, W. J.; WAINER, I. W. High performance liquid chromatography: fundamental principles and practice. London: Black Academic & Professional, 1996. 276 p. [29] NOVÁKOVÁ, L.; MATYSOVÁ, L.; SOLICH, P. Advantages of application of UPLC in pharmaceutical analysis. Talanta. v. 68, p. 908-918, 2006. [30] SNYDER, L. R.; KIRKLAND, J. J.; GLAJCH, J. L. Practical HPLC method development. 2 ed. New York: John Wiley & Sons, 1997. 765 p.

93
CAPÍTULO 3
OTIMIZAÇÃO DE PARÂMETROS INSTRUMENTAIS
CROMATOGRÁFICOS

94
1 REVISÃO BIBLIOGRÁFICA
1.1 Efeitos extracoluna e a eficiência cromatográfica
Equipamentos de HPLC não são capazes de operar sob pressão superior a 400 bar,
e, portanto, são inadequados para análises com colunas empacotadas com
partículas com diâmetro sub 2 μm. Essa limitação levou ao desenvolvimento de
sistemas de UHPLC, que suportam pressão de aproximadamente 1000 bar. Além de
bombas capazes de trabalhar em pressões superiores, sistemas de injeção,
detecção e tubulação foram adaptados para que a eficiência das partículas sub 2 μm
fosse aproveitada e para que houvesse redução do efeito extracoluna [1].
Atualmente, além de UHPLC, outros sistemas cromatográficos são empregados para
separações rápidas (em inglês Rapid Resolution Liquid Chromatography, Ultra-Fast
Liquid Chromatography, Rapid Separation Liquid Chromatography). Os
equipamentos para execução da cromatografia rápida assemelham-se aos de
HPLC, que suportam até 400 bar, porém apresentam redução dos efeitos
extracoluna. As técnicas de cromatografia rápida são as mais adequadas para que
colunas de alta eficiência, como a empacotada com partículas de núcleo fundido e a
monolítica, que não geram altas pressões, possam ser usadas com sucesso, sendo
desnecessária a utilização de cromatógrafos como UHPLC [2].
Do ponto de vista estatístico, a largura do pico cromatográfico é definida por meio da
variância total (2 total), que corresponde à soma da variância da coluna (2
coluna) e da
variância extracoluna (2 extracoluna). Portanto, a eficiência total de uma separação
decorre da coluna (eficiência intrínseca) e de aspectos extracoluna [2].
A eficiência intrínseca da coluna é matematicamente definida pela equação de Van
Deemter e envolve os termos A (difusão de Eddy), B (difusão longitudinal) e C
(resistência à transferência de massa) [2]. Já os aspectos extracoluna podem ser
divididos em dois tipos. O primeiro é de natureza volumétrica, chamado de volume
extracoluna (ECV, do inglês extra-column volume) e está relacionado aos volumes
de injeção, do detector e dos tubos de conexão. O segundo refere-se a eventos de
tempo, como frequência de aquisição dos dados e constante de tempo do detector.

95
É importante destacar que a variância extracoluna também depende da vazão da
fase móvel. O aumento da vazão, até determinado limite, contribui para o aumento
da variância extracoluna. A partir da vazão na qual um fluxo turbulento é
estabelecido, a variância extracoluna deixa de aumentar com a vazão. Os fatores
responsáveis pelos efeitos extracoluna estão descritos na equação (3.1) e
representados na Figura 3.1 [1, 2].
22
sec
2
det
22
sdadosquisiçãodoeventosdeaonexõestuboectorinjetoraextracolun (3.1)
Figura 3.1- Esquema de equipamento de HPLC - em vermelho, os fatores que contribuem
para o efeito extracoluna – [2]
Exemplos de volumes dos componentes típicos de equipamentos de HPLC e
UHPLC estão apresentados no Quadro 3.1 [3,4].
Quadro 3.1 - Volumes dos componentes típicos de equipamentos de HPLC e UHPLC
[Adaptado de 3,4]
Descrição UHPLC Acquity® UPLC HPLC Agilent® 1100
Tubo de entrada 6,3 μl 17,7 μl
Tubo de saída 2,3 μl 10,1 μl
Volume do loop 2,0 μl 10,0 μl
Assento capilar da agulha NA 3,7 μl
Célula de fluxo do detector 0,5 μl 8,0 μl
Volume extracoluna total (ECV)* 11,1μl 49,5 μl
* Normalmente o ECV de UHPLC está próximo a 10 μL e de HPLC entre 30-50 μL [4].

96
Em um sistema bem configurado a contribuição dos processos ocorridos fora da
coluna deve ser desprezível, evitando-se a redução da eficiência de separação. É
desejável que a variância extracoluna seja minimizada de forma que a redução do
número de pratos teóricos em consequência do efeito extracoluna não exceda 10%,
ou seja, a eficiência total deve ser no mínimo 90% da capacidade da coluna. A
variância extracoluna máxima aceitável é definida pela equação (3.2) [3]. O valor,
em porcentagem, que a eficiência total de uma análise (estimada na presença dos
efeitos extracoluna) representa em relação à eficiência da coluna, é denominado
eficiência remanescente (Er) e é calculada com o auxílio da equação 3.3 [5].
N
kLrccolunaec
224222
max
)1(10,010,0
(3.2)
2
2
.100total
colunarE
(3.3)
2ec max = variância extracoluna máxima aceitável, 2
coluna = variância da coluna,
L = comprimento da coluna, rc = raio da coluna, ε = porosidade da coluna, k = fator
de retenção do analito, N = número de pratos teóricos, Er = eficiência remanescente,
2total = variância total
Analisando a equação (3.2), percebe-se que para colunas com menor raio (menor rc)
e mais curtas (menor L) a variância máxima aceitável é menor e, portanto, essas
colunas são mais susceptíveis aos efeitos extracoluna. O mesmo raciocínio se aplica
àquelas colunas empacotadas com partículas porosas sub 2 μm e de núcleo
fundido, que apresentam maior eficiência (maior N). Além disso, a equação (3.2)
indica que para analitos menos retidos (menor k) a variância extracoluna máxima
aceitável é menor do que para os analitos mais retidos.
A estimativa da 2extracoluna é feita a partir de pico obtido em um sistema
cromatográfico sem coluna. As condições de análise devem ser similares às que
seriam utilizadas para a análise cromatográfica (mesma fase móvel, vazão, volume
de injeção). Todavia, no local da coluna, deve ser utilizado um conector com volume

97
morto desprezível. Deve-se injetar solução contendo um dos analitos de interesse
ou, alternativamente, uracila ou acetona. A partir do pico registrado calcula-se o
valor de tangenciando o pico nos pontos de inflexão até a linha de base (largura
entre as tangentes na linha de base = 4) ou medindo-se a largura do pico na meia
altura (largura do pico na meia altura = 2,354), como demonstrado na Figura 3.2
[2,4].
Figura 3.2 - Medida da largura na linha de base (4σ) e da largura na meia altura (2,354σ) [6]
O valor de , calculado em unidade de tempo, deve ser multiplicado pela vazão, de
maneira a se encontrar o valor de em unidade volumétrica. O quadrado do valor
encontrado corresponde a 2extracoluna [2,4].
Para estimar o ECV do sistema cromatográfico deve-se multiplicar 4, medido em
unidade de tempo, pela vazão empregada. Esse cálculo é apenas uma estimativa, já
que parte-se do princípio de que toda 2extracoluna é advinda do ECV. De fato há
também a contribuição dos eventos relacionados ao tempo, como frequência de
aquisição dos dados e constante de tempo do detector. O valor real de ECV consiste
na adição de todos os volumes do sistema: volumes de injeção, do tubo de PEEK do
injetor a coluna, do tubo de PEEK da coluna ao detector, da célula de detecção,
filtros de linha e demais conectores [4].
Há estratégias para reduzir os efeitos extracoluna de um sistema cromatográfico. A
primeira delas é ajustar a condição de detecção: a constante de tempo do detector
(t) deve ser suficientemente reduzida pelo fato de as larguras de base dos picos

98
serem estreitas (apenas poucos segundos); deve haver alta frequência de aquisição
dos dados (F) para que sejam adquiridos dados suficientes em pouco tempo [7]. Em
sistemas de UHPLC a frequência deve ser de no mínimo 20 Hz e a constante de
tempo do detector 0,1 s [8]; em sistemas de HPLC a frequência deve ser de no
mínimo 10 Hz e a constante de tempo do detector 0,3 s [7]. Outras estratégias são
substituir a tubulação do equipamento por tubos de menor volume e reduzir os
volumes de injeção da amostra e da cela do detector [7].
Normalmente, sistemas de HPLC apresentam variância extracoluna entre 40 e
60 μl2, enquanto que em sistemas de UHPLC a variância extracoluna encontra-se
entre 4 a 10 μl2 [4]. Embora em sistemas de UHPLC a variância extracoluna seja
reduzida, em se tratando de análise com colunas muito eficientes, pode haver perda
de eficiência superior aos 10% aceitáveis [2]. Como exemplo cita-se estudo
publicado em 2011, no qual foi verificada perda de 35% da eficiência em análise
realizada utilizando sistema de UHPLC da Waters e coluna C18 - 50 × 2,1 mm, 1,7
μm [9]. Os resultados desse estudo confirmam que mesmo utilizando sistemas de
reduzido volume extracoluna, como os de UHPLC, a perda de eficiência
cromatográfica devido à variância extracoluna pode ser elevada quando colunas de
alta eficiência são utilizadas.
2 OBJETIVOS ESPECÍFICOS
Foram objetivos dessa etapa do trabalho:
1- Otimizar parâmetros instrumentais cromatográficos do equipamento de UHPLC
visando à redução da variância extracoluna.
2- Investigar a influência da variância extracoluna na eficiência remanescente das
colunas em estudo (convencional, sub 2 µm, núcleo fundido e monolítica), durante
análise dos quatro antidiabéticos orais (clorpropamida, glibenclamida, glimepirida e
gliclazida), procurando compreender como o tipo de coluna e os fatores de retenção
dos analitos interferem no grau de influência da variância extracoluna na eficiência
remanescente.

99
3- Verificar se os níveis dos parâmetros instrumentais otimizados com base na
variância extracoluna (objetivo 1) são os ideais para garantir a maior eficiência
remanescente durante análise dos antidiabéticos (objetivo 2).
3 MATERIAIS E MÉTODOS
3.1 Materiais
3.1.1 Insumos farmacêuticos ativos
Os mesmos descritos no capítulo 1, item 3.1.1.
3.1.2 Solventes, reagentes e vidrarias
Os mesmos descritos no capítulo 1, item 3.1.2.
3.1.3 Equipamentos e colunas cromatográficas
Os mesmos descritos no capítulo1, item 3.1.3.
3.1.4 Outros materiais
Loops de 10 μl, 5 μL e 2 μl.
Tubos de PEEK de diâmetro interno iguais a 0,004 polegada (0,1016 mm) e 0,0025
polegada (0,0635 mm) e 270 mm de comprimento.
3.2 Métodos
3.2.1 Otimização dos parâmetros instrumentais cromatográficos parte I: avaliação do
efeito extracoluna
Os parâmetros instrumentais cromatográficos submetidos à otimização, os níveis em
que foram testados, bem como o desenho experimental univariado empregado para
a realização da otimização, encontram-se descritos na Tabela 3.1.

100
Tabela 3.1 - Experimentos de otimização dos parâmetros instrumentais cromatográficos
Experimento 1 Experimento 2 Experimento 3
Loop PEEK F/t
2 μl 0,004 polegada 10 Hz/0,4 s = 25
5 μl 0,0025 polegada 20 Hz/0,2 s = 100
10 μl 20 Hz/0,1 s = 200
40 Hz/0,1 s = 400
40 Hz/0,025 s = 1600
Parâmetros fixos
PEEK: 0,004 polegada
F/t: 20 Hz/0,1 s
Parâmetros fixos
Loop: Escolhido no exp. 1
F/t: 20 Hz/0,1 s
Parâmetros fixos
Loop: Escolhido no exp. 1
PEEK: Escolhido no exp. 2
Para decidir sobre o melhor nível de cada um dos parâmetros, a variância
extracoluna (2extracoluna) e o volume extracoluna (ECV) foram estimados. No local da
coluna foi inserido um conector com volume morto desprezível e injetou-se solução
de uracila a 10 μg/ml, em vazões crescentes (de 0,02 a 0,6 ml/min). As condições de
análise desenvolvidas para determinação simultânea de antidiabéticos orais em
coluna convencional, descritas no Capítulo 1, foram utilizadas (fase móvel composta
de ACN:solução de acetato de amônio 5 mM pH 3,0 (50:50), temperatura 30 oC e
volume de injeção 2 μl).
Para cada valor de vazão, a solução de uracila foi injetada em triplicata e a largura
na meia altura média (w0,5) dos picos registrada. Para as estimativas da σ2extracoluna e
do ECV as equações (3.4), (3.5), (3.6) e (3.7) foram utilizadas [4].
354,25,0w (3.4)
354,2/5,0w (3.5)
22 )( vazãoaextracolun (3.6)
vazãoECV 4 (3.7)
101
w0,5= largura na meia altura, σ = desvio padrão, σ2extracoluna = variância extracoluna,
ECV = volume extracoluna
3.2.2 Otimização dos parâmetros instrumentais cromatográficos parte II: avaliação
da eficiência cromatográfica remanescente
Nessa segunda etapa loops de diferentes volumes e diferentes pares de frequência
de aquisição dos dados (F) e constante de tempo do detector (t) foram avaliados. O
diâmetro interno do tubo de PEEK que conecta a coluna ao detector não foi avaliado
porque resultados descritos no item 4.1.2 deste capítulo demonstraram que os
PEEKs testados (0,004 e 0,0025 polegada) pouco se diferenciaram em termos de
σ2 extracoluna.
Os volumes de loop foram avaliados nas quatro colunas. Foram realizadas três
injeções de solução contendo 100 μg/ml dos antidiabéticos, conforme os métodos
analíticos desenvolvidos no capítulo 1 (Tabela 3.2), empregando-se loops de 2 e
10 μl. O experimento foi realizado em vazões de 0,02 a 0,6 ml/min.
Tabela 3.2 - Condições de análise para determinação de antidiabéticos nas diferentes
colunas*
Colunas Fase Móvel Vinj (μl) (nm) T (oC)
Convencional ACN: Solução (50:50)** 2 230 30
Monolítica ACN: Solução (43:57)** 2 230 30
Núcleo Fundido ACN: Solução (50:50)** 2 230 30
Sub 2 μm ACN: Solução (53:47)** 2 230 30
*Utilizou-se PEEK de 0,004 polegada e F/t = 40 Hz/0,0025 s **Solução de acetato de amônio 5 mM pH 3,0
Para cada valor de vazão, em cada um dos loops e colunas, determinou-se, para os
quatro antidiabéticos, a média da largura na meia altura (w0,5). Esse valor foi
utilizado para a determinação da σ2total (equação 3.8). A σ2
extracoluna, necessária para
o cálculo da σ2coluna, foi estimada em cada valor de vazão, com loops de 2 e 10 μl,
utilizando-se os métodos descritos na Tabela 3.2, substituindo-se a coluna por tubo
de volume morto desprezível e injetando-se uracila (conforme descrito no item
3.2.1). Com os valores de σ2total e σ2
extracoluna foi possível determinar σ2coluna.

102
2
5,02
354,2
vazão
wtotal (3.8)
222
aextracoluntotalcoluna (3.9)
Em seguida, com os valores de σ2total e σ
2coluna, calculou-se a eficiência remanescente
(Er) por meio da equação 3.3 [5].
Já os parâmetros frequência de aquisição e constante de tempo do detector foram
avaliados utilizando-se apenas a coluna de partículas sub 2 μm. Essa coluna foi
escolhida porque, dentre as quatro em estudo, é a que, de acordo com dados da
literatura, possui maior eficiência cromatográfica. Isso significa que a performance
da coluna de partículas sub 2 μm é influenciada de forma mais significativa por
aumentos da σ2extracoluna advindas de mudanças em F/t. O desenho experimental
incluiu a injeção em triplicata de solução de antidiabéticos orais (100 μg/ml),
seguindo método analítico desenvolvido para a coluna de partículas sub 2 μm
descrito na Tabela 3.2, utilizando-se loops de 2 μl e PEEK de 0,004 di. Os seguintes
pares de F/t foram avaliados: 10 Hz/0,4 s , 20 Hz/0,2 s, 20 Hz/0,1 s, 40 Hz/0,1 s e 40
Hz/0,025 s. O experimento foi realizado nas vazões de 0,15 ml/min e 0,6 ml/min.
Para cada valor de vazão e par de F/t determinou-se a média da largura na meia
altura (w0,5), a σ2total (equação 3.8) e a σ2
coluna (equação 3.9), para os quatro
antidiabéticos. A σ2extracoluna, necessária para o cálculo da σ2
coluna, foi estimada
conforme procedimento descrito no item 3.2.1, nas vazões de 0,15 e 0,6 ml/min, em
todos os pares F/t. Em seguida, com os valores de σ2total e σ
2extracoluna, calculou-se a
eficiência remanescente (Er) por meio da equação 3.3 [5].
3.2.3 Determinação do volume extracoluna real (ECV)
O valor real do volume extracoluna, determinado após otimização dos parâmetros
intrumentais cromatográficos, foi calculado somando-se os volumes dos seguintes
componentes do equipamento Acquity® UPLC: tubo de PEEK de entrada, tubo de
PEEK de saída, volume do loop e célula de fluxo do detector.
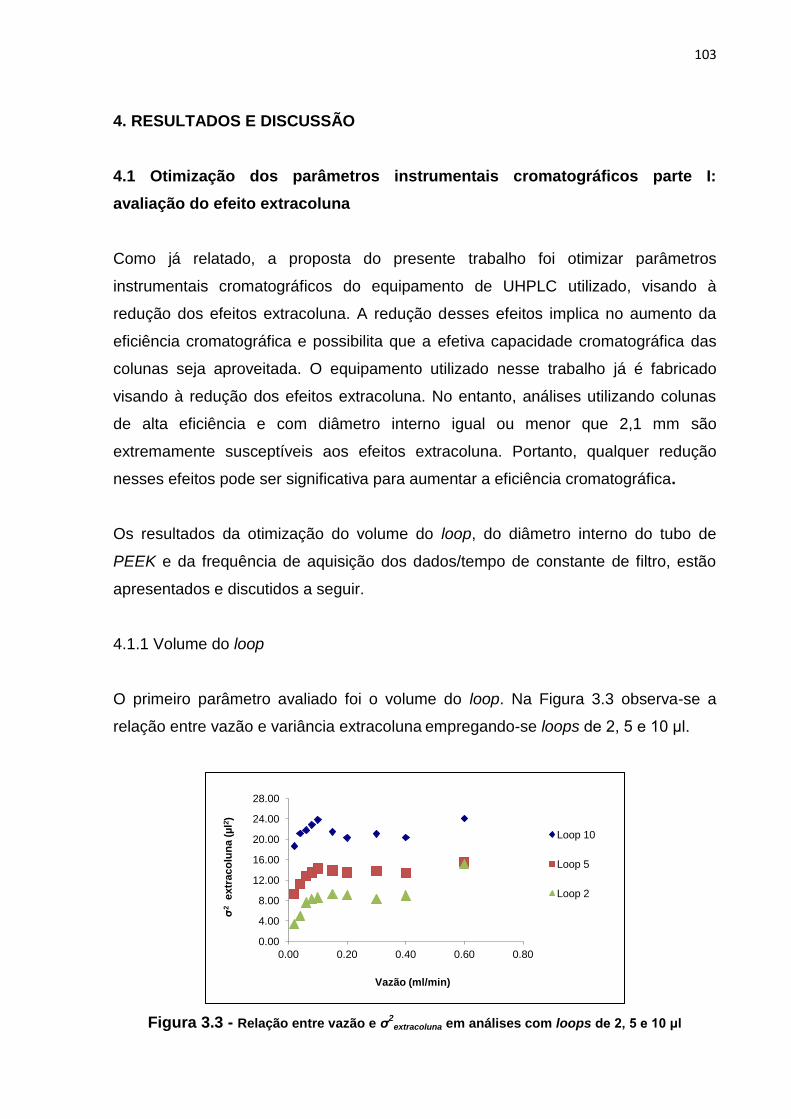
103
4. RESULTADOS E DISCUSSÃO
4.1 Otimização dos parâmetros instrumentais cromatográficos parte I:
avaliação do efeito extracoluna
Como já relatado, a proposta do presente trabalho foi otimizar parâmetros
instrumentais cromatográficos do equipamento de UHPLC utilizado, visando à
redução dos efeitos extracoluna. A redução desses efeitos implica no aumento da
eficiência cromatográfica e possibilita que a efetiva capacidade cromatográfica das
colunas seja aproveitada. O equipamento utilizado nesse trabalho já é fabricado
visando à redução dos efeitos extracoluna. No entanto, análises utilizando colunas
de alta eficiência e com diâmetro interno igual ou menor que 2,1 mm são
extremamente susceptíveis aos efeitos extracoluna. Portanto, qualquer redução
nesses efeitos pode ser significativa para aumentar a eficiência cromatográfica.
Os resultados da otimização do volume do loop, do diâmetro interno do tubo de
PEEK e da frequência de aquisição dos dados/tempo de constante de filtro, estão
apresentados e discutidos a seguir.
4.1.1 Volume do loop
O primeiro parâmetro avaliado foi o volume do loop. Na Figura 3.3 observa-se a
relação entre vazão e variância extracoluna empregando-se loops de 2, 5 e 10 μl.
Figura 3.3 - Relação entre vazão e σ2
extracoluna em análises com loops de 2, 5 e 10 μl
0.00
4.00
8.00
12.00
16.00
20.00
24.00
28.00
0.00 0.20 0.40 0.60 0.80
σ2 extr
aco
lun
a (
µl2
)
Vazão (ml/min)
Loop 10
Loop 5
Loop 2
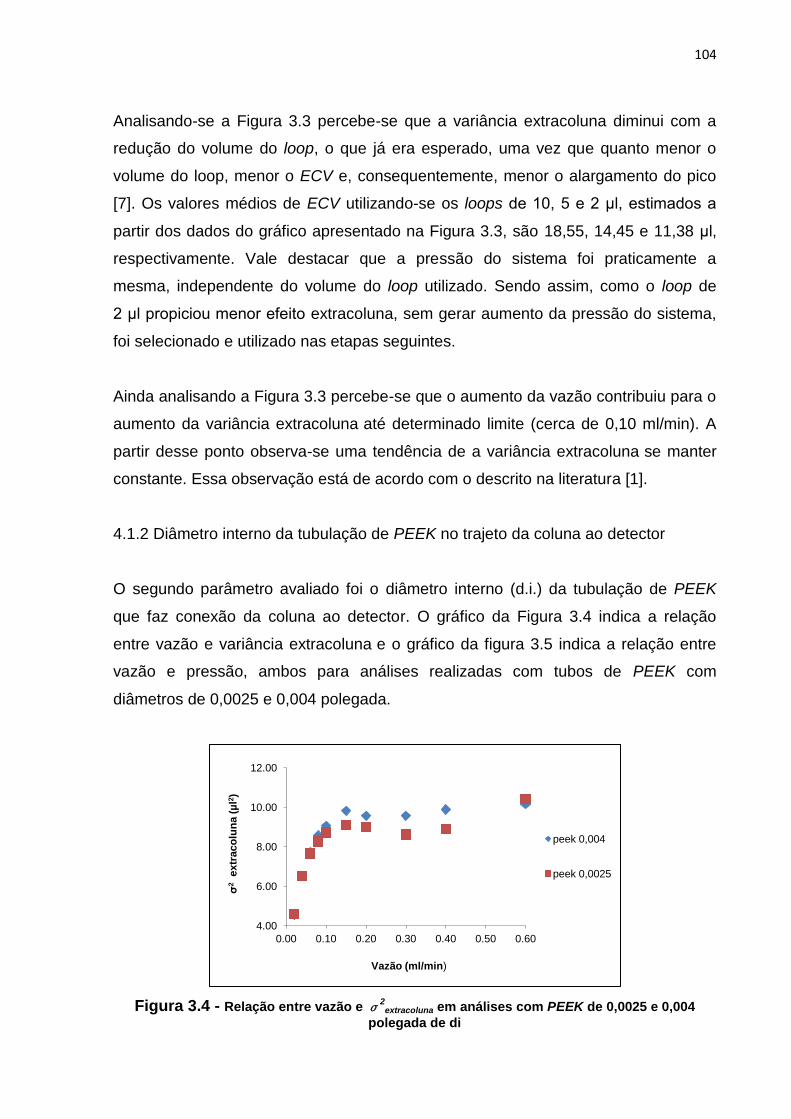
104
Analisando-se a Figura 3.3 percebe-se que a variância extracoluna diminui com a
redução do volume do loop, o que já era esperado, uma vez que quanto menor o
volume do loop, menor o ECV e, consequentemente, menor o alargamento do pico
[7]. Os valores médios de ECV utilizando-se os loops de 10, 5 e 2 μl, estimados a
partir dos dados do gráfico apresentado na Figura 3.3, são 18,55, 14,45 e 11,38 μl,
respectivamente. Vale destacar que a pressão do sistema foi praticamente a
mesma, independente do volume do loop utilizado. Sendo assim, como o loop de
2 μl propiciou menor efeito extracoluna, sem gerar aumento da pressão do sistema,
foi selecionado e utilizado nas etapas seguintes.
Ainda analisando a Figura 3.3 percebe-se que o aumento da vazão contribuiu para o
aumento da variância extracoluna até determinado limite (cerca de 0,10 ml/min). A
partir desse ponto observa-se uma tendência de a variância extracoluna se manter
constante. Essa observação está de acordo com o descrito na literatura [1].
4.1.2 Diâmetro interno da tubulação de PEEK no trajeto da coluna ao detector
O segundo parâmetro avaliado foi o diâmetro interno (d.i.) da tubulação de PEEK
que faz conexão da coluna ao detector. O gráfico da Figura 3.4 indica a relação
entre vazão e variância extracoluna e o gráfico da figura 3.5 indica a relação entre
vazão e pressão, ambos para análises realizadas com tubos de PEEK com
diâmetros de 0,0025 e 0,004 polegada.
Figura 3.4 - Relação entre vazão e σ2extracoluna em análises com PEEK de 0,0025 e 0,004
polegada de di
4.00
6.00
8.00
10.00
12.00
0.00 0.10 0.20 0.30 0.40 0.50 0.60
σ2 extr
aco
lun
a (
µl2
)
Vazão (ml/min)
peek 0,004
peek 0,0025
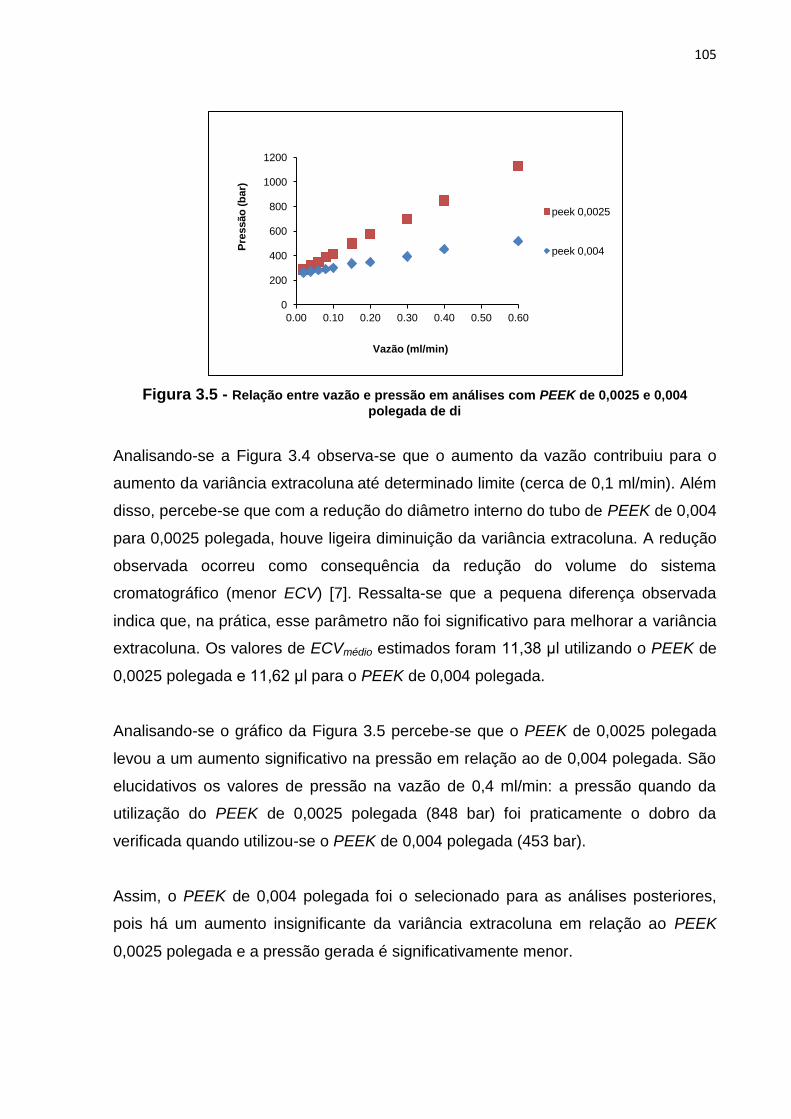
105
Figura 3.5 - Relação entre vazão e pressão em análises com PEEK de 0,0025 e 0,004
polegada de di
Analisando-se a Figura 3.4 observa-se que o aumento da vazão contribuiu para o
aumento da variância extracoluna até determinado limite (cerca de 0,1 ml/min). Além
disso, percebe-se que com a redução do diâmetro interno do tubo de PEEK de 0,004
para 0,0025 polegada, houve ligeira diminuição da variância extracoluna. A redução
observada ocorreu como consequência da redução do volume do sistema
cromatográfico (menor ECV) [7]. Ressalta-se que a pequena diferença observada
indica que, na prática, esse parâmetro não foi significativo para melhorar a variância
extracoluna. Os valores de ECVmédio estimados foram 11,38 μl utilizando o PEEK de
0,0025 polegada e 11,62 μl para o PEEK de 0,004 polegada.
Analisando-se o gráfico da Figura 3.5 percebe-se que o PEEK de 0,0025 polegada
levou a um aumento significativo na pressão em relação ao de 0,004 polegada. São
elucidativos os valores de pressão na vazão de 0,4 ml/min: a pressão quando da
utilização do PEEK de 0,0025 polegada (848 bar) foi praticamente o dobro da
verificada quando utilizou-se o PEEK de 0,004 polegada (453 bar).
Assim, o PEEK de 0,004 polegada foi o selecionado para as análises posteriores,
pois há um aumento insignificante da variância extracoluna em relação ao PEEK
0,0025 polegada e a pressão gerada é significativamente menor.
0
200
400
600
800
1000
1200
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Pre
ssão
(b
ar)
Vazão (ml/min)
peek 0,0025
peek 0,004
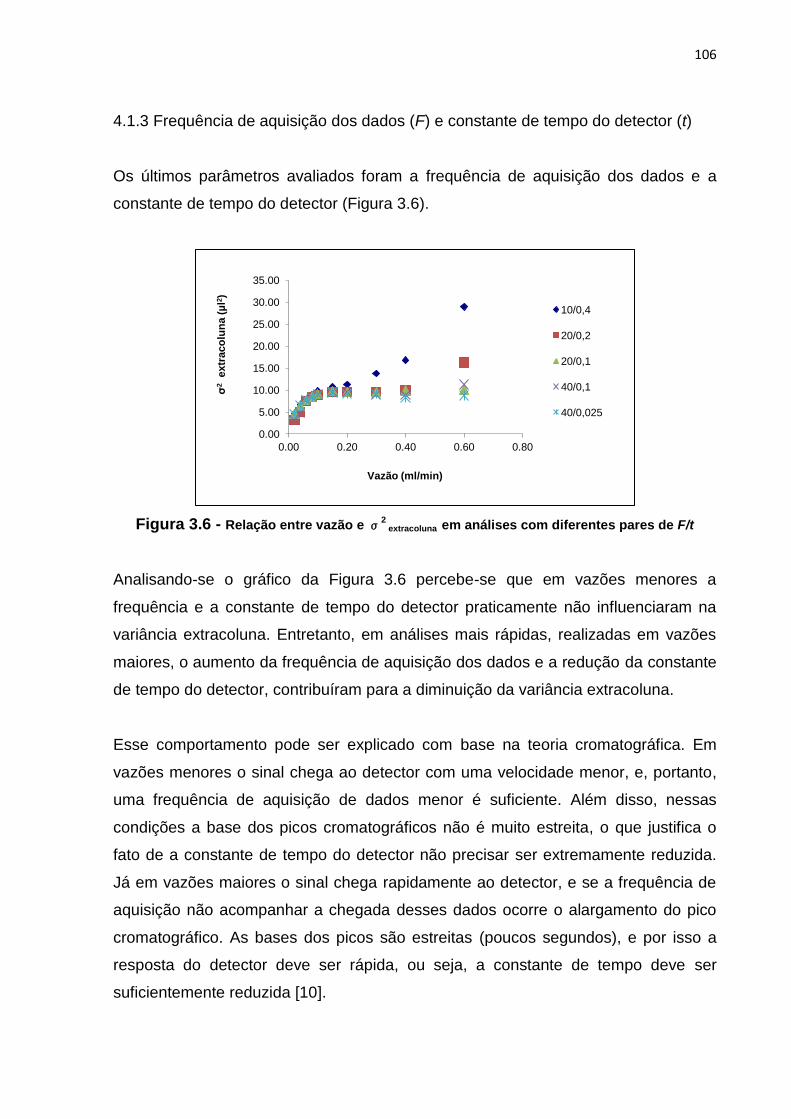
106
4.1.3 Frequência de aquisição dos dados (F) e constante de tempo do detector (t)
Os últimos parâmetros avaliados foram a frequência de aquisição dos dados e a
constante de tempo do detector (Figura 3.6).
Figura 3.6 - Relação entre vazão e σ2
extracoluna em análises com diferentes pares de F/t
Analisando-se o gráfico da Figura 3.6 percebe-se que em vazões menores a
frequência e a constante de tempo do detector praticamente não influenciaram na
variância extracoluna. Entretanto, em análises mais rápidas, realizadas em vazões
maiores, o aumento da frequência de aquisição dos dados e a redução da constante
de tempo do detector, contribuíram para a diminuição da variância extracoluna.
Esse comportamento pode ser explicado com base na teoria cromatográfica. Em
vazões menores o sinal chega ao detector com uma velocidade menor, e, portanto,
uma frequência de aquisição de dados menor é suficiente. Além disso, nessas
condições a base dos picos cromatográficos não é muito estreita, o que justifica o
fato de a constante de tempo do detector não precisar ser extremamente reduzida.
Já em vazões maiores o sinal chega rapidamente ao detector, e se a frequência de
aquisição não acompanhar a chegada desses dados ocorre o alargamento do pico
cromatográfico. As bases dos picos são estreitas (poucos segundos), e por isso a
resposta do detector deve ser rápida, ou seja, a constante de tempo deve ser
suficientemente reduzida [10].
0.00
5.00
10.00
15.00
20.00
25.00
30.00
35.00
0.00 0.20 0.40 0.60 0.80
σ2 extr
aco
lun
a (
µl2
)
Vazão (ml/min)
10/0,4
20/0,2
20/0,1
40/0,1
40/0,025

107
Conforme verificado no gráfico da Figura 3.6, a variância extracoluna para os valores
F/t de 10 Hz/0,4 s e 20 Hz/0,2 s são maiores que os valores de variância verificados
para os outros três pares de F/t investigados, sobretudo em vazões maiores. A
despeito de as diferenças dos valores de variância extracoluna serem pequenas nas
análises realizadas com F/t de 20 Hz/0,1 s, 40 Hz/0,1 s e 40 Hz/0,025 s e serem
menos influenciadas pela vazão, a maior frequência (40 Hz) e a menor constante de
tempo do detector (0,025 s) levaram à menor variância extracoluna
(σ2extracoluna média = 8,10 μl). Dessa forma, objetivando-se a máxima redução do efeito
extracoluna, o par 40 Hz/0,025 s foi selecionado como o ideal.
Concluindo, os seguintes níveis dos parâmetros instrumentais foram selecionados:
loop de 2 μl, PEEK com d.i de 0,004 polegada, F de 40 Hz e t de 0,025 s. A variância
extracoluna média, calculada a partir dos valores de variância extracoluna obtidos
em cada vazão testada aplicando-se os parâmetros instrumentais cromatográficos
otimizados foi igual a 8,10 μl2 e o ECVmédio igual a 11,33 μl. Esses valores estão
próximos aos descritos na literatura para equipamentos de UHPLC: a variância
extracoluna deve estar entre 4 e 10 μl2 e o ECV próximo a 10 μl [2,4].
4.2 Otimização dos parâmetros instrumentais cromatográficos parte II:
avaliação da eficiência cromatográfica remanescente
A avaliação do efeito extracoluna como critério para seleção dos níveis ideais dos
parâmetros instrumentais cromatográficos, descrita no item 4.1, é uma maneira
indireta de otimizar a separação cromatográfica, uma vez que há uma relação
inversa entre a variância extracoluna e a eficiência cromatográfica. A intensidade
dessa relação pode variar em função do tipo de coluna e do fator de retenção dos
analitos [6]. Por outro lado, a eficiência cromatográfica remanescente (Er) relaciona-
se diretamente com a eficiência cromatográfica e também pode ser usada para a
seleção dos níveis ideais dos parâmetros instrumentais cromatográficos.
Os resultados da otimização do volume do loop e da frequência de aquisição (F) e
tempo da constante de filtro (t) utilizando Er como critério, e discussões sobre como
o tipo de coluna e o fator de retenção dos analitos interferem na intensidade da
relação entre efeito extracoluna e a Er, estão apresentados a seguir.
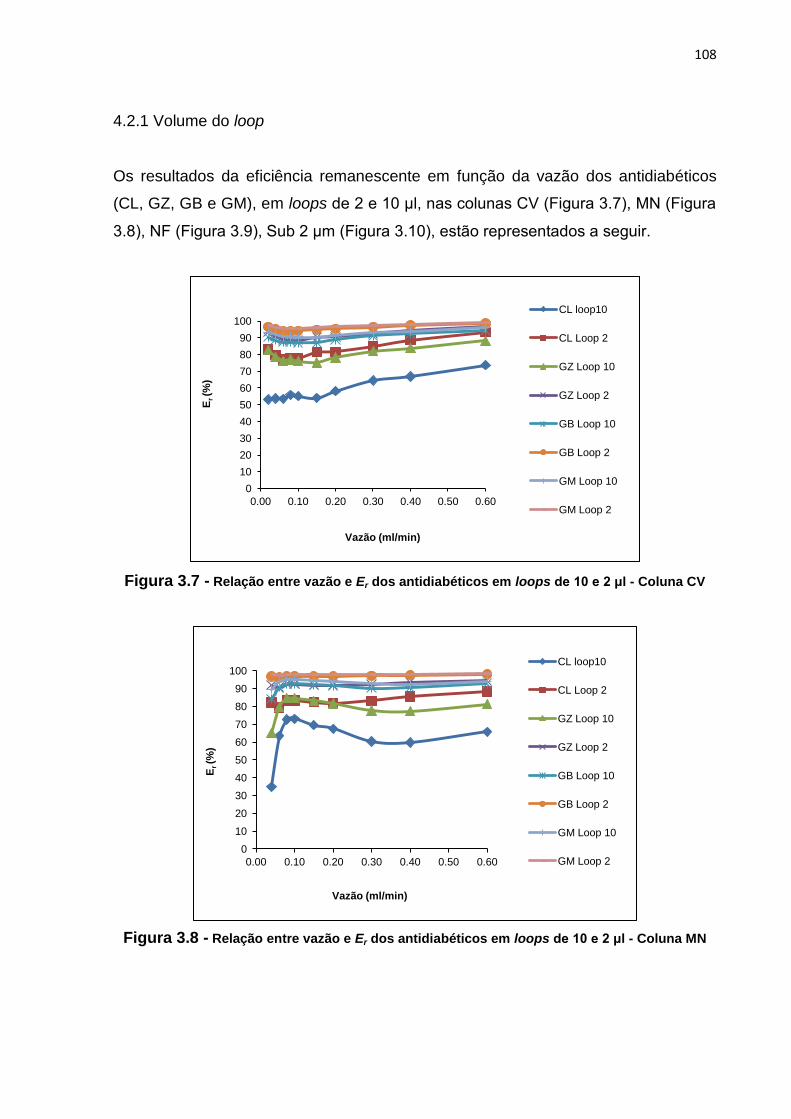
108
4.2.1 Volume do loop
Os resultados da eficiência remanescente em função da vazão dos antidiabéticos
(CL, GZ, GB e GM), em loops de 2 e 10 μl, nas colunas CV (Figura 3.7), MN (Figura
3.8), NF (Figura 3.9), Sub 2 μm (Figura 3.10), estão representados a seguir.
Figura 3.7 - Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna CV
Figura 3.8 - Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna MN
0
10
20
30
40
50
60
70
80
90
100
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Er (%
)
Vazão (ml/min)
CL loop10
CL Loop 2
GZ Loop 10
GZ Loop 2
GB Loop 10
GB Loop 2
GM Loop 10
GM Loop 2
0
10
20
30
40
50
60
70
80
90
100
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Er (%
)
Vazão (ml/min)
CL loop10
CL Loop 2
GZ Loop 10
GZ Loop 2
GB Loop 10
GB Loop 2
GM Loop 10
GM Loop 2
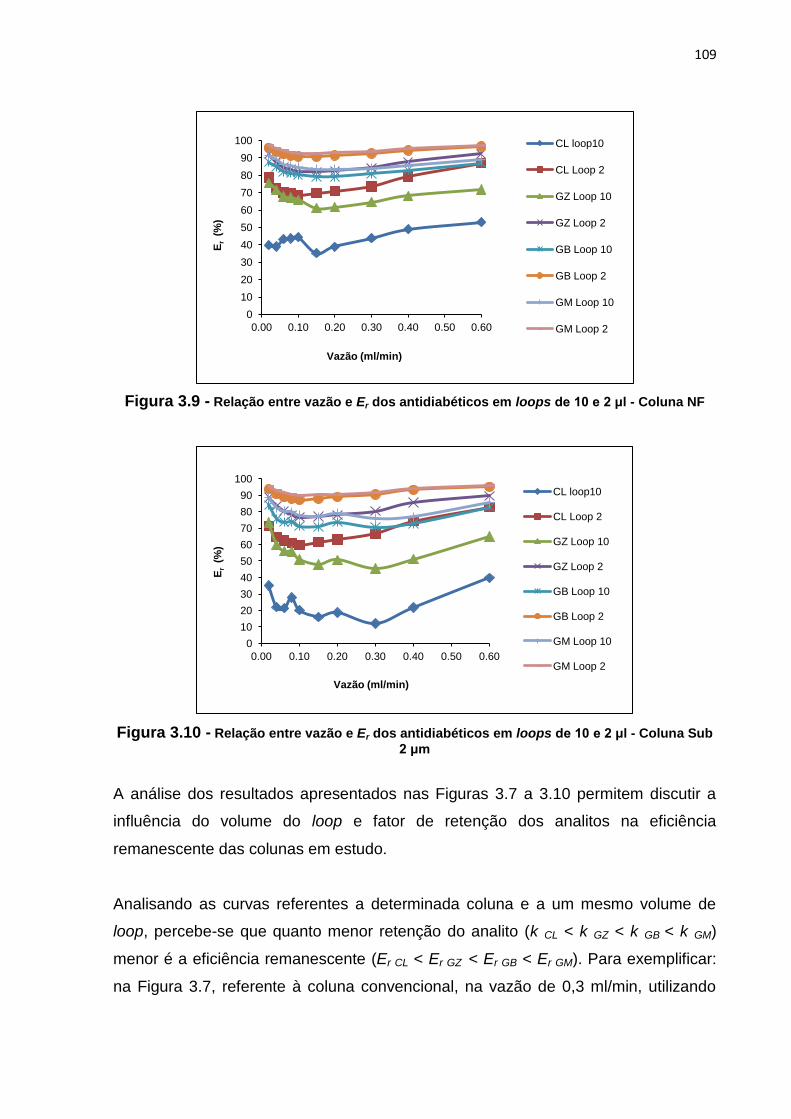
109
Figura 3.9 - Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna NF
Figura 3.10 - Relação entre vazão e Er dos antidiabéticos em loops de 10 e 2 μl - Coluna Sub
2 μm
A análise dos resultados apresentados nas Figuras 3.7 a 3.10 permitem discutir a
influência do volume do loop e fator de retenção dos analitos na eficiência
remanescente das colunas em estudo.
Analisando as curvas referentes a determinada coluna e a um mesmo volume de
loop, percebe-se que quanto menor retenção do analito (k CL < k GZ < k GB < k GM)
menor é a eficiência remanescente (Er CL < Er GZ < Er GB < Er GM). Para exemplificar:
na Figura 3.7, referente à coluna convencional, na vazão de 0,3 ml/min, utilizando
0
10
20
30
40
50
60
70
80
90
100
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Er (
%)
Vazão (ml/min)
CL loop10
CL Loop 2
GZ Loop 10
GZ Loop 2
GB Loop 10
GB Loop 2
GM Loop 10
GM Loop 2
0
10
20
30
40
50
60
70
80
90
100
0.00 0.10 0.20 0.30 0.40 0.50 0.60
Er (
%)
Vazão (ml/min)
CL loop10
CL Loop 2
GZ Loop 10
GZ Loop 2
GB Loop 10
GB Loop 2
GM Loop 10
GM Loop 2

110
loop de 10 μl, as eficiências remanescentes da CL, GZ, GB e GM foram de 67%,
82%, 91% e 93% respectivamente.
Analisando as curvas de todas as colunas relativas a um mesmo volume de loop e
mesmo analito verifica-se que a Er é menor na coluna sub 2 μm e aumenta
sucessivamente nas colunas de núcleo fundido, monolítica e convencional.
Exemplificando: utilizando o loop de 10 μl, na vazão de 0,3 ml/min, as eficiências
remanescentes para a clorpropamida nas colunas sub 2 μm, NF, MN e CV foram de
12%, 44%, 60% e 67% respectivamente.
A análise das curvas referentes a determinada coluna e a um mesmo analito permite
observar que o aumento do volume do loop, o qual promove aumento da variância
extracoluna (conforme demonstrado na Figura 3.3 - item 4.1.1), vem acompanhado
da redução da Er. Por exemplo, na figura 3.9, observa-se que para a gliclazida,
utilizando coluna de NF e vazão de 0,3 ml/min, as eficiências remanescentes foram
de 92,51% quando se usou loop de 2 μl e de 64,58% no caso de loop de 10 μl.
Analisando-se as curvas de determinada coluna e determinado analito, percebe-se
que a influência do aumento do volume do loop, com consequente aumento da
variância extracoluna (conforme demonstrado na Figura 3.3 - item 4.1.1), na redução
da Er, é mais significativa na coluna de sub 2 μm, seguida das colunas NF, MN e CV.
A título de exemplo, encontram-se relacionados a seguir, na Tabela 3.3, os valores
de eficiência remanescente obtidos para a glibenclamida nas análises realizadas
com as quatro colunas, vazão de 0,3 ml/min, loops de 2 e 10 μl, bem como a
diferença entre os valores de Er quando os diferentes volumes de loop foram
empregados.
Tabela 3.3 - Er para a glibenclamida nas quatro colunas - vazão de 0,3 ml/min
loop 2 μl loop 10 μl Er loop 2 μl - Er loop 10 μl
Er Sub2 90% 71% 19%
Er NF 93% 81% 12%
Er MN 97% 90% 7%
Er CV 91% 96% 5%

111
Nas curvas referentes a determinada coluna, percebe-se que a influência do
aumento do volume do loop, com consequente aumento da variância extracoluna, na
redução da Er, é mais significativa para analitos menos retidos. Nos dados
apresentados a seguir referentes à Figura 3.10, para coluna sub 2 μm, loops de 2 e
10 μl, vazão de 0,3 ml/min, torna-se evidente esta observação (Tabela 3.4)
Tabela 3.4 - Er dos antidiabéticos na coluna sub 2 μm - vazão de 0,3 ml/min
loop 2 μl loop 10 μl Er loop 2 μl - Er loop 10 μl
Er CL 67% 12% 55%
Er GZ 80% 46% 34%
Er GB 90% 71% 19%
Er GM 92% 76% 16%
Destaca-se ainda que os gráficos de vazão versus Er apresentam aspecto inverso
aos gráficos de vazão versus σ2extracoluna. O aumento da vazão contribui para o
aumento da σ2extracoluna (até determinado limite) e esse aumento σ2
extracoluna resulta
em redução da eficiência cromatográfica remanescente.
Os resultados apresentados indicam que a influência negativa do aumento da
variância extracoluna na eficiência cromatográfica, como resultado do aumento do
volume do loop, foi maior para os analitos menos retidos (clorpropamida e gliclazida)
e para as colunas mais eficientes (Sub 2 μm, NF). Os resultados obtidos estão de
acordo com os da literatura [1, 6] e baseado neles confirma-se a escolha do loop de
2 μl descrito no item 4.1.1 como o ideal para garantir uma maior Er, ou seja, garantir
uma melhor eficiência cromatográfica.
4.2.2 Frequência de aquisição dos dados (F) e constante de tempo do detector (t)
Os resultados da eficiência remanescente (Er) em função de F/t, dos quatro
antidiabéticos, na coluna de partículas sub 2 μm, estão apresentados a seguir. Nas
Figuras 3.11 e 3.12 encontram-se os resultados referentes às análises realizadas
nas vazões de 0,6 e 0,15 ml/min, respectivamente. Ë importante destacar que na
abscissa dos gráficos não estão plotados os valores nominais de F/t (10 Hz/0,4 s,
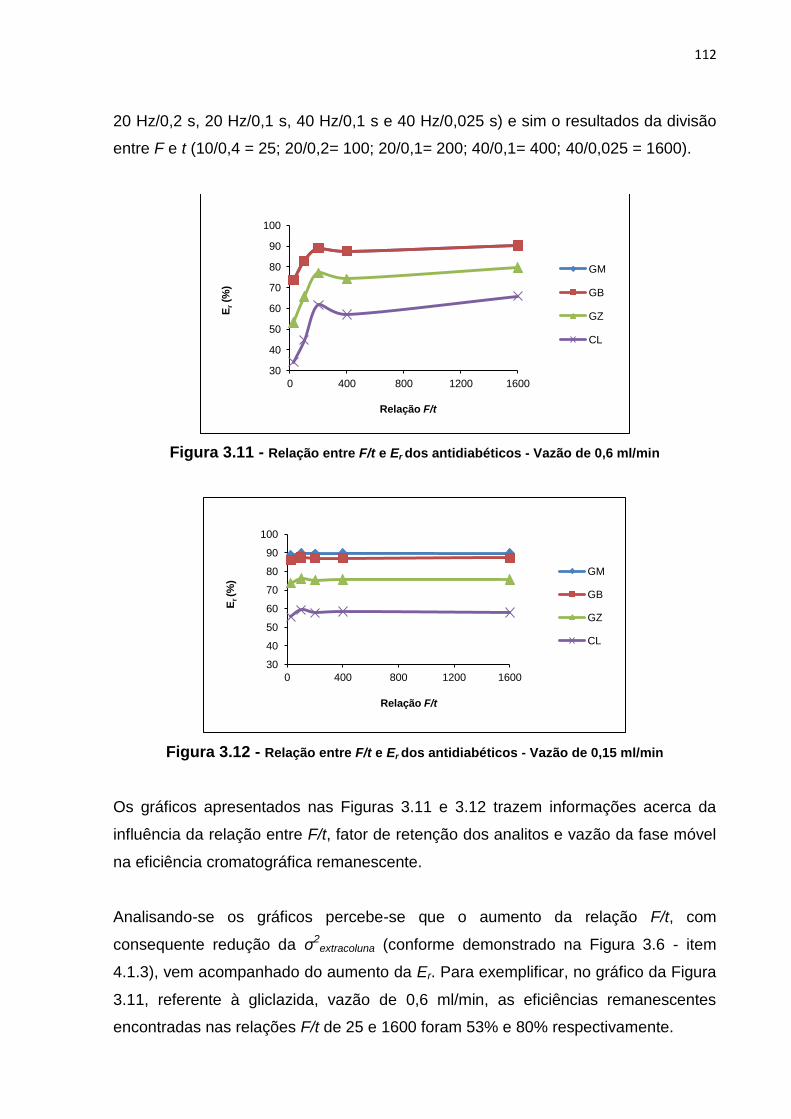
112
20 Hz/0,2 s, 20 Hz/0,1 s, 40 Hz/0,1 s e 40 Hz/0,025 s) e sim o resultados da divisão
entre F e t (10/0,4 = 25; 20/0,2= 100; 20/0,1= 200; 40/0,1= 400; 40/0,025 = 1600).
Figura 3.11 - Relação entre F/t e Er dos antidiabéticos - Vazão de 0,6 ml/min
Figura 3.12 - Relação entre F/t e Er dos antidiabéticos - Vazão de 0,15 ml/min
Os gráficos apresentados nas Figuras 3.11 e 3.12 trazem informações acerca da
influência da relação entre F/t, fator de retenção dos analitos e vazão da fase móvel
na eficiência cromatográfica remanescente.
Analisando-se os gráficos percebe-se que o aumento da relação F/t, com
consequente redução da σ2extracoluna (conforme demonstrado na Figura 3.6 - item
4.1.3), vem acompanhado do aumento da Er. Para exemplificar, no gráfico da Figura
3.11, referente à gliclazida, vazão de 0,6 ml/min, as eficiências remanescentes
encontradas nas relações F/t de 25 e 1600 foram 53% e 80% respectivamente.
30
40
50
60
70
80
90
100
0 400 800 1200 1600
Er (%
)
Relação F/t
GM
GB
GZ
CL
30
40
50
60
70
80
90
100
0 400 800 1200 1600
Er (%
)
Relação F/t
GM
GB
GZ
CL
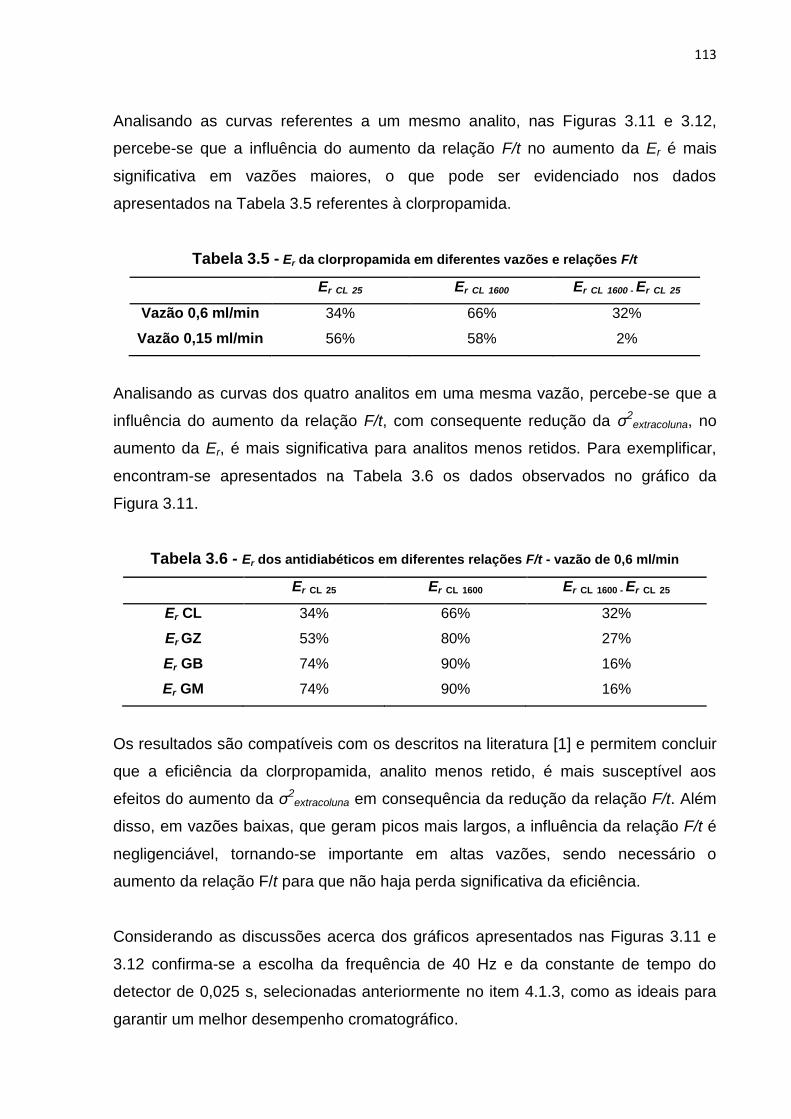
113
Analisando as curvas referentes a um mesmo analito, nas Figuras 3.11 e 3.12,
percebe-se que a influência do aumento da relação F/t no aumento da Er é mais
significativa em vazões maiores, o que pode ser evidenciado nos dados
apresentados na Tabela 3.5 referentes à clorpropamida.
Tabela 3.5 - Er da clorpropamida em diferentes vazões e relações F/t
Er CL 25 Er CL 1600 Er CL 1600 - Er CL 25
Vazão 0,6 ml/min 34% 66% 32%
Vazão 0,15 ml/min 56% 58% 2%
Analisando as curvas dos quatro analitos em uma mesma vazão, percebe-se que a
influência do aumento da relação F/t, com consequente redução da σ2extracoluna, no
aumento da Er, é mais significativa para analitos menos retidos. Para exemplificar,
encontram-se apresentados na Tabela 3.6 os dados observados no gráfico da
Figura 3.11.
Tabela 3.6 - Er dos antidiabéticos em diferentes relações F/t - vazão de 0,6 ml/min
Er CL 25 Er CL 1600 Er CL 1600 - Er CL 25
Er CL 34% 66% 32%
Er GZ 53% 80% 27%
Er GB 74% 90% 16%
Er GM 74% 90% 16%
Os resultados são compatíveis com os descritos na literatura [1] e permitem concluir
que a eficiência da clorpropamida, analito menos retido, é mais susceptível aos
efeitos do aumento da σ2extracoluna em consequência da redução da relação F/t. Além
disso, em vazões baixas, que geram picos mais largos, a influência da relação F/t é
negligenciável, tornando-se importante em altas vazões, sendo necessário o
aumento da relação F/t para que não haja perda significativa da eficiência.
Considerando as discussões acerca dos gráficos apresentados nas Figuras 3.11 e
3.12 confirma-se a escolha da frequência de 40 Hz e da constante de tempo do
detector de 0,025 s, selecionadas anteriormente no item 4.1.3, como as ideais para
garantir um melhor desempenho cromatográfico.

114
Vale destacar que o aumento da relação F/t, apesar de contribuir para diminuir a
variância extracoluna, leva também à redução da relação sinal/ruído (S/R) [6].
Entretanto, analisando a relação S/R dos picos dos antidiabéticos, verificou-se que
mesmo com uma frequência elevada (40 Hz) e uma constante de tempo do detector
reduzida (0,025 s), as relações S/R, em todas as vazões testadas, foram superiores
a 10, relação mínima recomendada para uma análise quantitativa exata e precisa
segundo a RDC 899 [11].
4.3 Cálculo do volume extracoluna (ECV) real
Os volumes dos componentes do equipamento de UHPLC (desde a injeção até a
detecção), que dão origem ao volume extracoluna, estão descritos na Tabela 3.7.
Tabela 3.7 - Volumes estimados dos componentes do equipamento de UHPLC (Acquity®) e
volume extracoluna
Fonte Volume (μl)
Tubo de entrada 5,0
Tubo de saída 2,2
Volume do loop 2
Célula de fluxo do detector 0,5
Volume extracoluna 9,7
5 CONCLUSÃO
Conclui-se que os melhores níveis de parâmetros instrumentais cromatográficos,
selecionados por meio da análise dos menores valores de variância extracoluna, e
confirmados por meio de análises da eficiência cromatográfica remanescente, foram:
loop de 2 μl, PEEK de 0,004 polegada de diâmetro interno e frequência de aquisição
dos dados/constante de tempo do detector de 40 Hz/0,025 s.
A influência negativa da 2extracoluna na eficiência cromatográfica foi maior para a
clorpropamida e gliclazida (analitos menos retidos) e quando colunas mais eficientes
(sub 2 μm e núcleo fundido) foram empregadas. Em condições de elevada 2extracoluna
(baixa relação F/t, elevado volume de loop), os valores de Er da clorpropamida e

115
gliclazida nas colunas de núcleo fundido e de partículas sub 2 μm variaram entre 20
e 60%, o que significa que foram bem inferiores aos 90% recomendados. Entretanto,
após a otimização e consequente redução da 2extracoluna (alta relação F/t, reduzido
volume de loop), houve uma melhora significativa: os valores de Er da clorpropamida
e gliclazida nas colunas de núcleo fundido e de partículas sub 2 μm passaram a ficar
entre 70 e 80% e portanto mais próximos ao ideal valor de 90%.
Apesar de menos significativa, a melhora da 2extracoluna também contribuiu para o
aumento da eficiência remanescente durante a análise dos analitos mais retidos
(glibenclamida e glimepirida) nas colunas convencional e monolítica. Após redução
da 2extracoluna, os valores de Er da glibenclamida e glimepirida nas colunas
convencional e monolítica passaram de aproximadamente 90% para 95%.
Portanto, a otimização do sistema cromatográfico foi essencial para permitir que a
performance das colunas cromatográficas, em especial das colunas empacotadas
com partículas sub 2 µm e com partículas de núcleo fundido, fosse bem aproveitada,
apesar das limitações relacionadas à instrumentação e às próprias colunas. Por
último, vale destacar que o volume extracoluna estimado, calculado por meio da
soma dos volumes dos componentes do equipamento de UHPLC Acquity UPLC®, foi
igual a 9,7 μL e está de acordo com o valor próximo a 10 μL previsto na literatura
[2,4].

116
REFERÊNCIAS BIBLIOGRÁFICAS
[1] FOUNTAIN, K. J.; NEUE, U. D.; GRUMBACH, E. S.; DIEHL, D. M. Effects of extra-column band spreading, liquid chromatography system operating pressure, and column temperature on the performance of sub-2μm porous particles. Journal of Chromatography A. v. 1216, p. 5979-5988, 2009. [2] NETO, A. J. S. Como obter maior eficiência com partículas superficialmente porosas em HPLC. Scientia Chromatographica, v. 3, n.1, p. 65-87, 2011. [3] BRADLEY, A. C.; WELCH, C. J.; ZHANG, L. Effect of extra-column volume on practical chromatographic parameters of sub-2-μm particle-packed columns in ultra-high pressure liquid chromatography. Journal of separation science. v. 35, p. 2018-2025, 2012. [4] HOW TO MEASURE AND REDUCE HPLC EQUIPMENT EXTRA COLUMN VOLUME. Disponível em: <http://www.mac-mod.com/pdf/MMA084-ReduceECV_ MM_Singles.pdf>. Acesso em: 20 de agosto de 2012. [5] FEKETE, S.; FEKETE, J. The impact of extra-column band broadening on the chromatographic efficiency of 5 cm long narrow-bore very efficient columns. Journal of Chromatography A. v. 1218, p. 5286-5291, 2011. [6] MAJORS, R. E. Are You Getting the Most Out of Your HPLC Column? LCGC North america. v. 21, n. 12, p.1124-1133, 2003. [7] AMPARO, M. R.; FRANZINI, C. D.; SCHIAVON, B.; CARVALHO, L. S.; de, NETO, A. J. S. Influência dos parâmetros instrumentais sobre o desempenho de coluna de HPLC com partículas superficialmente porosas sub-3 μm. Scientia Chromatographica. v. 3, n. 2, p.51-66, 2011. [8] GUIDELINES FOR THE USE OF UHPLC INSTRUMENTS. Disponível em: <http://www.unige.ch/sciences/pharm/fanal/lcap/Guidelines%20for%20the%20use%20of%20UHPLC%20instruments%20-%20site%20WEB%20labo.pdf>. Acesso em: 20 de Novembro de 2012. [9] FEKETEA, S.; GANZLER, K.; FEKETE,J. Efficiency of the new sub-2_m core–shell (KinetexTM) column in practice, applied for small and large molecule separation. Journal of Pharmaceutical and Biomedical Analysis. v. 54, p. 482-490, 2011. [10] MCCALLEY, D. V. Instrumental considerations for the effective operation of short, highly efficient fused-core columns. Investigation of performance at high flow rates and elevated temperatures. Journal of Chromatography A. v. 1217, p. 4561-4567, 2010. [11] BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RE no. 899, de 29 de Maio de 2003. Guia para validação de métodos analíticos e bioanalíticos. Diário Oficial da União, Brasília, DF, Poder Executivo, de 02 de Junho de 2003c.

117
CAPÍTULO 4
AVALIAÇÃO E COMPARAÇÃO QUALITATIVA DA
EFICIÊNCIA DE COLUNAS CROMATOGRÁFICAS

118
1 REVISÃO BIBLIOGRÁFICA
1.1 Curvas de Van Deemter e de Knox
Tradicionalmente, colunas cromatográficas são caracterizadas e comparadas por
meio de suas respectivas curvas de Van Deemter, nas quais se plota a altura do
prato teórico (H) em função da velocidade linear da fase móvel (u0). A obtenção da
curva (H x u0) permite relacionar o alargamento do pico cromatográfico e a vazão da
fase móvel e determinar a velocidade linear (diretamente relacionada à vazão da
fase móvel) na qual a altura do prato teórico (diretamente relacionada ao
alargamento do pico) é mínima, ou seja, velocidade na qual a eficiência da coluna é
máxima. A altura mínima do prato teórica é abreviada como Hmin e a velocidade
linear ótima é abreviada como u0 otm. Um prato teórico representa uma etapa de
equilíbrio do soluto entre as fases móvel e estacionária. Dessa maneira, a altura de
um prato teórico está relacionada ao comprimento da coluna necessário para que
ocorra o equilíbrio. Quanto menor a altura do prato, mais estreita será a largura de
uma banda cromatográfica e maior a eficiência [1,2, 3].
A curva de Van Deemter é regida pela equação (4.1):
0
0
uCu
BAH (4.1)
H = altura do prato teórico, A = coeficiente de difusão de Eddy, B = coeficiente de
difusão longitudinal, C = coeficiente de resistência à transferência de massa, u0 =
velocidade linear da fase móvel.
Altos valores do termo A (coeficiente de difusão de Eddy) ocorrem em colunas que
não são bem empacotadas, o que resulta na presença de caminhos múltiplos e,
consequentemente, na difusão axial do soluto e alargamento do pico (Figura 4.1).
Quanto menor o diâmetro das partículas, menor é a possibilidade de formação de
caminhos múltiplos e menor é o coeficiente A. O termo A independe da velocidade
linear da fase móvel [4].

119
Figura 4.1 - Efeito da difusão de Eddy (A) no alargamento do pico [3]
Altos valores do termo B (coeficiente de difusão longitudinal) ocorrem em vazões
baixas da fase móvel (baixa velocidade linear) e quando um soluto de alto
coeficiente de difusão é analisado. Nessas condições a difusão longitudinal do soluto
aumenta, o que resulta em alargamento do pico (Figura 4.2) [4].
Figura 4.2 - Efeito da difusão longitudinal (B) no alargamento do pico [3]
Já o termo C (coeficiente de resistência à transferência de massa) diz respeito à
resistência à transferência de massa do analito entre as fases estacionária e móvel
(Figura 4.3) e está relacionado à taxa de sorção/dessorção ou difusão do soluto
entre as fases. Fenômenos como a estagnação da fase móvel entre os poros da
fase estacionária, diferentes velocidades de transferência do analito ao longo da
coluna e baixa dessorção e/ou difusão do analito contribuem para o aumento do
termo C e consequente alargamento do pico. Fatores que colaboram para a
elevação do termo C são o aumento da velocidade linear, do tamanho das partículas
da fase estacionária e a diminuição do coeficiente de difusão do analito [4].
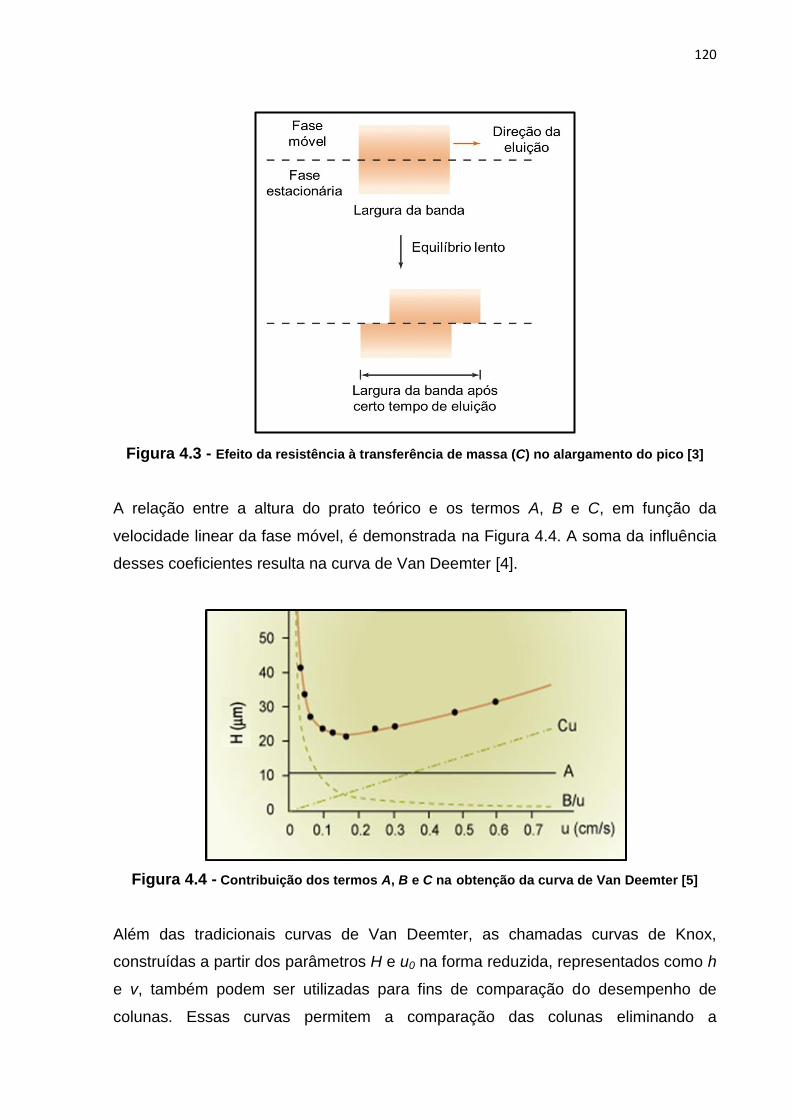
120
Figura 4.3 - Efeito da resistência à transferência de massa (C) no alargamento do pico [3]
A relação entre a altura do prato teórico e os termos A, B e C, em função da
velocidade linear da fase móvel, é demonstrada na Figura 4.4. A soma da influência
desses coeficientes resulta na curva de Van Deemter [4].
Figura 4.4 - Contribuição dos termos A, B e C na obtenção da curva de Van Deemter [5]
Além das tradicionais curvas de Van Deemter, as chamadas curvas de Knox,
construídas a partir dos parâmetros H e u0 na forma reduzida, representados como h
e v, também podem ser utilizadas para fins de comparação do desempenho de
colunas. Essas curvas permitem a comparação das colunas eliminando a

121
variabilidade proporcionada pelos diferentes tamanhos de partículas das colunas e
coeficientes de difusão dos analitos, e são regidas pela equação de Knox (equação
4.2). A maior eficiência cromatográfica ocorre no menor valor de h (hmin), e a
velocidade linear reduzida ótima na qual hmin ocorre é abreviada como v0 opt. Baixos
valores de hmin estão relacionados a um empacotamento de alta qualidade. Para o
cálculo dos parâmetros reduzidos h e v aplicam-se as equações (4.3) e (4.4) [6].
CvBvAvh 3
1
(4.2)
pd
Hh (4.3)
m
p
D
duv
0 (4.4)
A = coeficiente de difusão de Eddy, B = coeficiente de difusão longitudinal,
C = coeficiente de resistência à transferência de massa, h = altura do prato teórico
reduzido, v = velocidade linear da fase móvel reduzida, u0 = velocidade linear da
fase móvel, H = altura do prato teórico, dp = diâmetro da partícula, Dm = coeficiente
de difusão do analito
Os formatos das curvas de Van Deemter e de Knox são influenciados pelo fator de
retenção do analito (k), já que os termos B e C de ambas dependem de k. Assim,
para comparação de duas ou mais colunas por meio de suas respectivas curvas de
Van Deemter ou Knox, o analito deve possuir fatores de retenção similares nas
colunas em estudo. Para tanto se faz necessário ajustar a composição da fase
móvel em cada coluna para garantir valores de k similares. É importante destacar
que o ajuste na composição da fase móvel introduz alterações em termos de
viscosidade da fase móvel e difusão do analito, fatores que também interferem no
formato das curvas de Van Deemter [7].
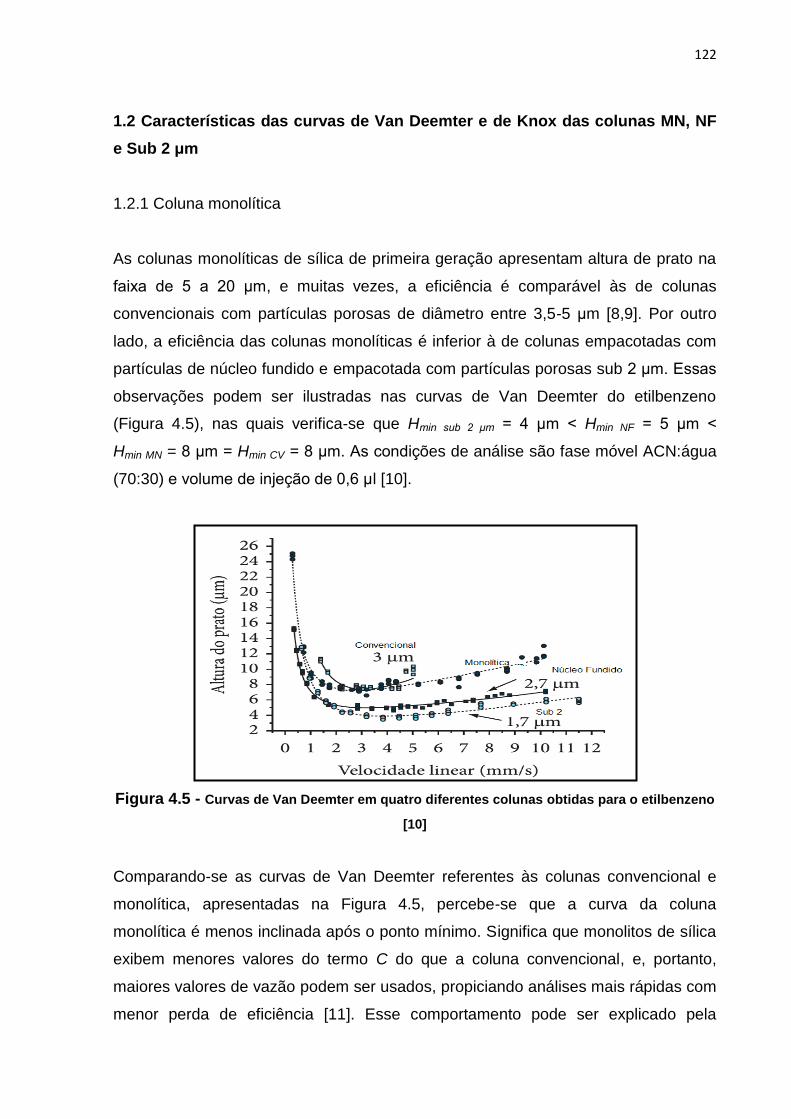
122
1.2 Características das curvas de Van Deemter e de Knox das colunas MN, NF
e Sub 2 μm
1.2.1 Coluna monolítica
As colunas monolíticas de sílica de primeira geração apresentam altura de prato na
faixa de 5 a 20 μm, e muitas vezes, a eficiência é comparável às de colunas
convencionais com partículas porosas de diâmetro entre 3,5-5 μm [8,9]. Por outro
lado, a eficiência das colunas monolíticas é inferior à de colunas empacotadas com
partículas de núcleo fundido e empacotada com partículas porosas sub 2 μm. Essas
observações podem ser ilustradas nas curvas de Van Deemter do etilbenzeno
(Figura 4.5), nas quais verifica-se que Hmin sub 2 μm = 4 μm < Hmin NF = 5 μm <
Hmin MN = 8 μm = Hmin CV = 8 μm. As condições de análise são fase móvel ACN:água
(70:30) e volume de injeção de 0,6 μl [10].
Figura 4.5 - Curvas de Van Deemter em quatro diferentes colunas obtidas para o etilbenzeno
[10]
Comparando-se as curvas de Van Deemter referentes às colunas convencional e
monolítica, apresentadas na Figura 4.5, percebe-se que a curva da coluna
monolítica é menos inclinada após o ponto mínimo. Significa que monolitos de sílica
exibem menores valores do termo C do que a coluna convencional, e, portanto,
maiores valores de vazão podem ser usados, propiciando análises mais rápidas com
menor perda de eficiência [11]. Esse comportamento pode ser explicado pela

123
natureza porosa do monolito de sílica, que propicia uma aprimorada transferência de
massa do analito entre as fases estacionária e móvel. Outra vantagem dessa
estrutura porosa e altamente permeável é a geração de valores de pressão baixos,
compatíveis com equipamento de HPLC [12].
Em trabalho publicado em 2004, verificou-se que coluna monolítica C18
(Chromolith®) foi comparável em termos de seletividade, reprodutibilidade e
eficiência à coluna particulada totalmente porosa de 3 μm, C18 (YMC®). A pressão
produzida em coluna monolítica foi 3,5 vezes menor que a produzida pela coluna
particulada, e a retenção foi 3,5 vezes menor. O fator de cauda obtido com a coluna
monolítica foi maior, entretanto aceitável [13]. Além desse, outro estudo também
destaca a tendência das colunas monolíticas (em especial as de primeira geração)
de gerar picos mais assimétricos, principalmente durante a análise de substâncias
básicas [14].
1.2.2 Coluna empacotada com partículas de núcleo fundido
As partículas de núcleo fundido são constituídas por um núcleo sólido, normalmente
de 1,7 µm de diâmetro, e uma camada porosa, normalmente de 0,5 µm, pela qual
ocorre o processo de difusão do analito. O menor caminho de difusão (0,5 µm),
quando comparado ao caminho de difusão presente nas partículas totalmente
porosas, facilita a transferência de massa e reduz o caminho intra partícula, o que
contribui para a redução do termo C (coeficiente de resistência à transferência de
massa) da equação de Van Deemter. A redução do coeficiente C minimiza o
alargamento dos picos, contribuindo para o aumento da eficiência, e é
especialmente importante em elevados valores de vazão da fase móvel, condição
que favorece o aumento do termo C [15].
Outra característica a ser destacada em colunas com partículas de núcleo fundido,
quando comparada às partículas totalmente porosas, é a distribuição mais
homogênea do tamanho das partículas no leito cromatográfico. Acredita-se que a
maior aspereza da superfície das partículas de núcleo fundido seja essencial para a
obtenção do leito cromatográfico mais homogêneo [16]. Como consequência há
menos caminhos múltiplos, o que diminui a difusão axial do analito. O resultado é a

124
redução do termo A (coeficiente de difusão de Eddy) da equação de Van Deemter, o
que minimiza o alargamento dos picos e contribui para o aumento da eficiência [15].
Os pequenos valores de hmin, (entre 1,5 e 4,0 μm) descritos na literatura para colunas
com partículas de núcleo fundido evidenciam a qualidade do leito cromatográfico
desse tipo de coluna [17].
Em trabalho de revisão publicado em 2011 foi relatado que em colunas com
partículas de núcleo fundido também há a redução da difusão longitudinal, ou seja,
redução do coeficiente B da equação de Van Deemter [16]. Esse comportamento é
decorrente do fato de aproximadamente 20% do volume da coluna ser ocupado por
uma sílica não porosa pela qual o analito não pode se difundir [17].
Resumindo, pode-se dizer que a melhora na eficiência cromatográfica verificada nas
colunas empacotadas com partículas de núcleo fundido, quando comparada às
colunas empacotadas com partículas totalmente porosas convencionais, é
consequência da redução dos termos A, B e C da equação de Van Deemter [16].
Analisando-se as curvas de Van Deemter referentes ao etilbenzeno (Figura 4.5),
observa-se o aumento da eficiência já que Hmin NF = 5 μm < Hmin CV = 8 μm [10]. Em
levantamento bibliográfico valores de Hmin entre 3 e 11 μm foram relatados para
análises em colunas de núcleo fundido de diferentes marcas [17].
Vale destacar que a redução do termo C justifica o fato de as curvas de Van
Deemter referentes às colunas de núcleo fundido serem menos inclinadas do que as
curvas das colunas com partículas porosas convencionais. Em colunas de partículas
de núcleo fundido, ao contrário do que acontece nas convencionais, verifica-se que
mesmo com o aumento da velocidade linear da fase móvel não há perda significativa
de eficiência, ou seja, não há aumento representativo de H. Sendo assim, utilizando-
se colunas com partículas de núcleo fundido, é possível a realização de análises
rápidas, com elevada velocidade linear da fase móvel, sem perda significativa de
eficiência. Além disso, deve-se lembrar de que as pressões geradas pelas colunas
com partícula de núcleo fundido são compatíveis com HPLC [16].
Estudos têm sido realizados atualmente com o objetivo de comparar as colunas de
partículas de núcleo fundido com aquelas empacotadas com partículas porosas sub

125
2 µm. Em 2008 um estudo foi realizado utilizando-se como modelo um método
analítico para separação de torcetrapibe e impurezas. Comparada com a coluna de
partícula porosa sub 2 µm, a de núcleo fundido atingiu 80% da eficiência e gerou
aproximadamente a metade da pressão [10].
1.2.3 Coluna empacotada com partículas sub 2 μm
As colunas com partículas totalmente porosas de diâmetro inferior a 2 μm
apresentam características que permitem análises rápidas e eficientes [18]. O
pequeno diâmetro das partículas (dp) contribui para a redução dos termos A e C da
curva de Van Deemter, o que minimiza o alargamento dos picos e aumenta a
eficiência (redução da altura do prato teórico H) [4].
A redução do termo A como resultado da redução do tamanho das partículas ocorre
porque o empacotamento de partículas menores gera menos espaços vazios dentro
do leito cromatográfico. Como consequência há menos caminhos, o que reduz a
difusão de Eddy das moléculas do soluto por entre a fase estacionária [4]. Já a
redução do termo C, também como consequência da redução do tamanho das
partículas, é explicada pelo fato de o menor diâmetro das partículas corresponder a
um menor caminho percorrido pelo analito na fase estacionária, facilitando a
transferência de massa do analito entre as fases estacionária e móvel [4].
Apesar das vantagens relatadas, a redução do diâmetro das partículas vem
acompanhada do aumento da pressão do sistema cromatográfico, já que a pressão
gerada é inversamente proporcional ao quadrado do diâmetro das partículas. Sendo
assim faz-se necessário a utilização de sistemas cromatográficos denominados
UHPLC, que suportam valores de pressões na ordem de 1000 bar [16,18].
Analisando-se o formato típico de uma curva de Van Deemter relativa à coluna de
partículas sub 2 μm (Figura 4.5), observa-se que de maneira similar ao que ocorre
com as colunas de núcleo fundido, o aumento da velocidade linear da fase móvel
não implica em perda significativa da eficiência, ou seja, não há aumento
significativo de H. É importante destacar que esse comportamento, que é
consequência da redução do termo C, é mais pronunciado nas colunas com

126
partículas sub 2 μm do que nas colunas com partículas de núcleo fundido. Além
disso, a eficiência cromatográfica gerada por colunas com partículas sub 2 μm é
superior à gerada por colunas com partículas de núcleo fundido, o que é evidenciado
comparando-se os valores de Hmin do etilbenzeno (Figura 4.5): Hmin NF = 5 μm > Hmin
Sub 2 μm = 4 μm [10]. Segundo dados da literatura, valores de Hmin entre 3 e 7 μm
foram encontrados para colunas com partículas sub 2 μm de diferentes marcas [17].
Encontram-se na literatura várias publicações relacionadas ao emprego de coluna
sub 2 µm e comparando-a com outras colunas. Exemplo é a análise de extrato de
raiz de gengibre por HPLC (coluna C18 - 2,1 x 100 mm - 5 µm) e por UHPLC (coluna
C18 - 2,1 x 100 mm - 1,7 µm). Foi verificado que o uso de UHPLC resultou em
aumento da resolução, da velocidade de análise, e melhora na detectabilidade [18].
1.3 Gráficos de performance cinética
Quando apenas colunas cromatográficas empacotadas eram utilizadas, elas podiam
ser comparadas a partir de seus respectivos valores de altura do prato teórico (H) ou
prato teórico reduzido (h). Entretanto, com a introdução de colunas monolíticas,
poliméricas e outras com diferentes formas e tamanhos, que apresentam maior
permeabilidade e menor resistência ao fluxo, a comparação em função dos valores
mínimos de H e h deixou de ser suficiente. Analisando as equações de Van Deemter
e de Knox verifica-se que a permeabilidade do leito cromatográfico não é um fator
considerado, e, portanto, informações sobre resistência ao fluxo e a possibilidade de
se utilizar altos valores de vazão e/ou colunas de maior comprimento não são
consideradas [19].
Para minimizar o problema que emergiu com a evolução das colunas
cromatográficas, pesquisadores passaram a avaliar a altura do prato teórico (H) e o
coeficiente de permeabilidade (Kv0) para comparação entre colunas. Entretanto,
tornou-se difícil avaliar simultaneamente os dois parâmetros. Além disso, nenhum
desses parâmetros trazia informação acerca do tempo de análise [19].
Na tentativa de correlacionar permeabilidade do leito cromatográfico, eficiência
cromatográfica e tempo de análise, Poppe e colaboradores propuseram a

127
elaboração de um gráfico de performance cinética (do inglês Kinetic Performance
Plot) do tipo (N x t0). Entretanto, a construção desse gráfico envolvia a utilização de
algoritmos e cálculos matemáticos, o que limitou a sua aplicabilidade [19,20].
Em 2005, Desmet e colaboradores desenvolveram fórmulas matemáticas simples
para a construção do gráfico de performance cinética (N x t0), anteriormente
proposto por Poppe. Para a construção desse gráfico faz-se necessário aplicar às
colunas cromatográficas valores crescentes de vazão da fase móvel. Para cada
valor de vazão testado um par de valores (u0 x H) é calculado (equações 4.5 e 4.6).
Posteriormente, fazendo-se uso dos pares de valores u0 e H e dos valores de
viscosidade da fase móvel, permeabilidade do leito cromatográfico e pressão
máxima suportada por cada coluna, os pares de valores (N x t0) são calculados
utilizando-se as equações (4.7) e (4.8) e, em seguida, plotados (Figura 4.6) [19].
N
LH (4.5)
10 kt
Lu
r
(4.6)
Hu
KvPN
0
0max
(4.7)
2
0
0max0
u
KvPt
(4.8)
H = altura do prato teórico, L = comprimento da coluna, N = número de pratos
teóricos, u0 = velocidade linear da fase móvel, tr = tempo de retenção, k = fator de
retenção, ΔPMax = pressão máxima suportada pelo sistema, η = viscosidade da fase
móvel, Kv0 = permeabilidade da coluna, u0 = velocidade linear da fase móvel, t0 =
tempo morto
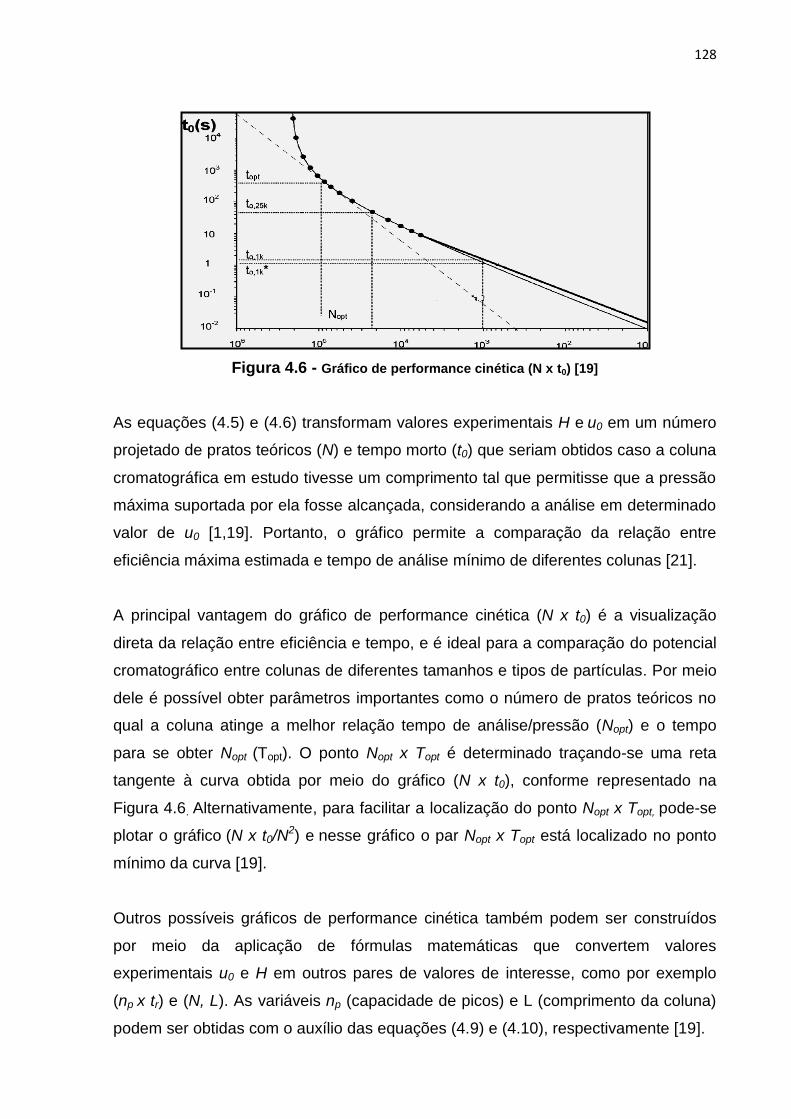
128
Figura 4.6 - Gráfico de performance cinética (N x t0) [19]
As equações (4.5) e (4.6) transformam valores experimentais H e u0 em um número
projetado de pratos teóricos (N) e tempo morto (t0) que seriam obtidos caso a coluna
cromatográfica em estudo tivesse um comprimento tal que permitisse que a pressão
máxima suportada por ela fosse alcançada, considerando a análise em determinado
valor de u0 [1,19]. Portanto, o gráfico permite a comparação da relação entre
eficiência máxima estimada e tempo de análise mínimo de diferentes colunas [21].
A principal vantagem do gráfico de performance cinética (N x t0) é a visualização
direta da relação entre eficiência e tempo, e é ideal para a comparação do potencial
cromatográfico entre colunas de diferentes tamanhos e tipos de partículas. Por meio
dele é possível obter parâmetros importantes como o número de pratos teóricos no
qual a coluna atinge a melhor relação tempo de análise/pressão (Nopt) e o tempo
para se obter Nopt (Topt). O ponto Nopt x Topt é determinado traçando-se uma reta
tangente à curva obtida por meio do gráfico (N x t0), conforme representado na
Figura 4.6. Alternativamente, para facilitar a localização do ponto Nopt x Topt, pode-se
plotar o gráfico (N x t0/N2) e nesse gráfico o par Nopt x Topt está localizado no ponto
mínimo da curva [19].
Outros possíveis gráficos de performance cinética também podem ser construídos
por meio da aplicação de fórmulas matemáticas que convertem valores
experimentais u0 e H em outros pares de valores de interesse, como por exemplo
(np x tr) e (N, L). As variáveis np (capacidade de picos) e L (comprimento da coluna)
podem ser obtidas com o auxílio das equações (4.9) e (4.10), respectivamente [19].

129
kN
np 1ln4
1 (4.9)
HNL (4.10)
np = capacidade de picos, N = número de pratos teóricos, k = fator de retenção, L =
comprimento da coluna, H = altura do prato teórico
O gráfico de performance (np x tr) possibilita determinar o tempo mínimo necessário
para que determinado fator de capacidade de picos seja alcançado ou determinar o
maior fator de capacidade de picos que pode ser alcançado em determinado tempo,
considerando que a coluna é longa o bastante para que a pressão máxima por ela
suportada seja alcançada [1]. A capacidade de picos representa o número máximo de
picos que podem ser separados entre o tempo morto e o tempo de eluição do último
analito [22]. Já o gráfico (N x L) possibilita determinar o comprimento da coluna
cromatográfica necessário para que determinado valor N projetado seja alcançado.
Dessa forma é possível verificar se os valores projetados de N correspondem a
colunas de comprimento impraticáveis (muito longas ou curtas) [1].
Embora haja vantagens, os gráficos de performance cinética apresentam limitações.
Assume-se, para fins de cálculos, que a viscosidade da fase móvel (η), o fator de
retenção (k) e a permeabilidade do leito cromatográfico (Kv0) são constantes, ou
seja, que independem da vazão da fase móvel. Isso é verdade em condições
cromatográficas de pressão inferior a 400 bar. Sabe-se, entretanto, que em pressões
mais elevadas o aquecimento da fase móvel promove alterações nesses
parâmetros, muitas vezes difíceis de serem previstas. Assim, a análise dos gráficos
de performance cinética pode conduzir a estimativas incorretas acerca dos
potenciais cromatográficos das colunas que estão sendo comparadas [20, 23].
2 OBJETIVO ESPECÍFICO
O objetivo do estudo descrito nesse capítulo foi comparar colunas cromatográficas
monolítica (MN) e empacotadas com partículas porosas de 5 μm (CV), com

130
partículas de núcleo fundido de 2,7 μm (NF) e com partículas porosas de 1,8 μm
(Sub 2 μm), utilizando como modelo método analítico para determinação de
antidiabéticos orais (CL, GB, GM e GZ) e os parâmetros eficiência, velocidade de
análise, pressão, resolução entre os picos e fator de assimetria dos picos.
3 MATERIAIS E MÉTODOS
3.1 Materiais
3.1.1 Insumos farmacêuticos ativos
Os mesmos descritos no capítulo 1, item 3.1.1.
3.1.2 Solventes, reagentes e vidrarias
Os mesmos descritos no capítulo 1, item 3.1.2.
3.1.3 Equipamentos e colunas cromatográficas
Os mesmos descritos no capítulo1, item 3.1.3.
3.2 Métodos
3.2.1 Avaliação da eficiência e velocidade de análise das colunas
Para comparar a eficiência e velocidade de análise das colunas cromatográficas
utilizou-se como modelo o método analítico para determinação de antidiabéticos
orais desenvolvido em coluna convencional e aplicado às outras colunas após os
necessários ajustes na composição da fase móvel, conforme foi descrito no capítulo
1. O ajuste na composição da fase móvel foi realizado com o objetivo de manter os
valores dos fatores de retenção (k) dos analitos próximos em todas as colunas, já
que as curvas de Van Deemter e de Knox, usadas para comparar a eficiência das
colunas e velocidade de análise, têm o formato influenciado por k [7].

131
Os parâmetros instrumentais cromatográficos otimizados, descritos no capítulo 3,
foram utilizados nessa etapa do trabalho. O volume do loop, o diâmetro interno do
tubo de PEEK e a frequência de aquisição dos dados/tempo da constante de filtro
que promoveram a redução do efeito da variância extracoluna foram essenciais para
uma comparação mais fidedigna do real desempenho das colunas cromatográficas.
Na Tabela 4.1 encontra-se a descrição das condições de análise utilizadas.
Tabela 4.1 - Condições de análise para determinação de antidiabéticos nas quatro colunas*
Colunas Fase Móvel Vinj (μl) (nm) T (oC) Conc. (μg/ml)
Convencional ACN: Solução (50:50)** 2 230 30 100
Monolítica ACN: Solução (43:57)** 2 230 30 100
Núcleo Fundido ACN: Solução (50:50)** 2 230 30 100
Sub 2 μm ACN: Solução (53:47)** 2 230 30 100
* Parâmetros instrumentais cromatográficos: loop de 2 μl, tubo de PEEK de 0,004 polegada e frequência de aquisição dos dados e tempo da constante de filtros iguais a 40 Hz/0,025 s ** Solução de acetato de amônio 5 mM pH 3,0
Além das curvas de Van Deemter e de Knox, gráficos de performance cinética
também foram usados para comparação da performance e velocidade de análise
obtidas com cada uma das colunas cromatográficas avaliadas.
3.2.1.1 Curvas de Van Deemter
Aplicando-se os respectivos métodos analíticos apresentados na Tabela 4.1, cada
coluna foi submetida a valores crescentes de vazão (0,02 a 0,6 ml/min para as
colunas CV, NF e Sub 2 μm e 0,02 a 1,0 ml/min para a coluna MN). Determinaram-
se o número de pratos teóricos e o tempo de retenção dos picos referentes aos
quatro fármacos, em cada vazão avaliada e em cada coluna. Outro parâmetro
determinado em cada vazão e em cada coluna foi o tempo morto. Para estimativa do
tempo morto injetaram-se 2 μl de uracila (10 μg/ml).
A partir do número de pratos teóricos, tempo de retenção, tempo morto e fatores de
retenção (k), as alturas dos pratos teóricos (H) e os valores de velocidade linear da
fase móvel (u0) para cada antidiabético, em cada uma das vazões e colunas, foram
calculados. Para a realização dos cálculos foram utilizadas as equações (4.5) e

132
(4.6). A partir dos pares de valores (u0 x H) calculados, foram construídas curvas de
Van Deemter para os quatro antidiabético em cada coluna.
As equações matemáticas que descrevem cada uma das curvas de Van Deemter
construídas foram determinadas com o auxílio de um macro do Excel. A significância
das equações foi comprovada por meio da análise de variância (α = 5%) e a
significância dos coeficientes A, B e C de cada equação foi comprovada a partir da
aplicação do teste t-student (α = 5%).
A partir das equações matemáticas referentes às curvas de Van Deemter, os valores
de u0 opt e Hmin de cada analito, em cada coluna, foram calculados. Considerando
que o par de valores u0 opt e Hmin ocorre no ponto mínimo da curva, no qual a
inclinação da reta tangente é nula, pode-se dizer que a derivada da equação nesse
ponto é igual a zero. Considerando os arranjos matemáticos (Figura 4.7) u0 opt foi
estimado a partir das constantes B e C da equação de Van Deemter.
0
0
0 )( Cuu
BAuH
2
0
0'u
BCuH 0'
2
0
0 otm
otmu
BCuH
C
Bu otm 0
Figura 4.7 - Transformações matemáticas para o cálculo de u0 otm
Sendo assim, calculando-se a raiz da razão entre as constantes B e C, os valores de
u0 opt de cada analito, em cada coluna, foram determinados. Os valores de u0 opt
foram aplicados às equações de Van Deemter determinadas para cálculo dos
respectivos valores de Hmin.
3.2.1.2 Curvas de Knox
A partir dos pares de valores de u0 e H, usados na construção das curvas de Van
Deemter, foram calculados os parâmetros reduzidos v0 e h (equações 4.3 e 4.4).

133
O coeficiente de difusão (Dm) de cada analito foi estimado por meio da aplicação da
equação de Wilke Chang [24] e o diâmetro das partículas fornecido pelo fabricante
de cada coluna foram utilizados para a realização dos cálculos dos parâmetros
reduzidos. Como a coluna monolítica não possui partículas, o tamanho médio dos
poros do monolito de sílica da marca Onyx®, descrito na literatura como sendo igual
a 3,5 μm, foi utilizado em substituição ao diâmetro das partículas [25].
3.2.1.3 Gráficos de performance cinética
A partir dos pares de valores de u0 e H, usados na construção das curvas de Van
Deemter, foram calculados os pares de valores (N x t0), (np x tr) e (N, L) por meio das
equações 4.7. 4.8, 4.9 e 4.10. Em seguida esses pares de valores foram plotados
em gráficos. Para a realização dos cálculos foram utilizadas planilhas do Excel
desenvolvidas por Desmet e colaboradores [26].
3.2.2 Avaliação da resolução, fator de assimetria e pressão
Aplicando-se os métodos analíticos da Tabela 4.1, cada coluna foi submetida a
vazões crescentes de fase móvel. Para cada valor de vazão a pressão do sistema
foi registrada e os valores de resolução entre os pares de picos adjacentes e o fator
de assimetria dos picos calculados utilizando-se as equações (4.11) e (4.12) [2].
21
122
ww
ttR rr
s
(4.11)
A
BAs (4.12)
Rs = Resolução, tr1 e tr2 = tempos de retenção, w1 e w2 = larguras dos picos na linha
de base, As = Fator de assimetria, A = largura da metade direita do pico dividido a
partir do ponto de altura máxima, a 10% da altura, B = largura da metade esquerda
do pico dividido a partir do ponto de altura máxima, a 10 % da altura.
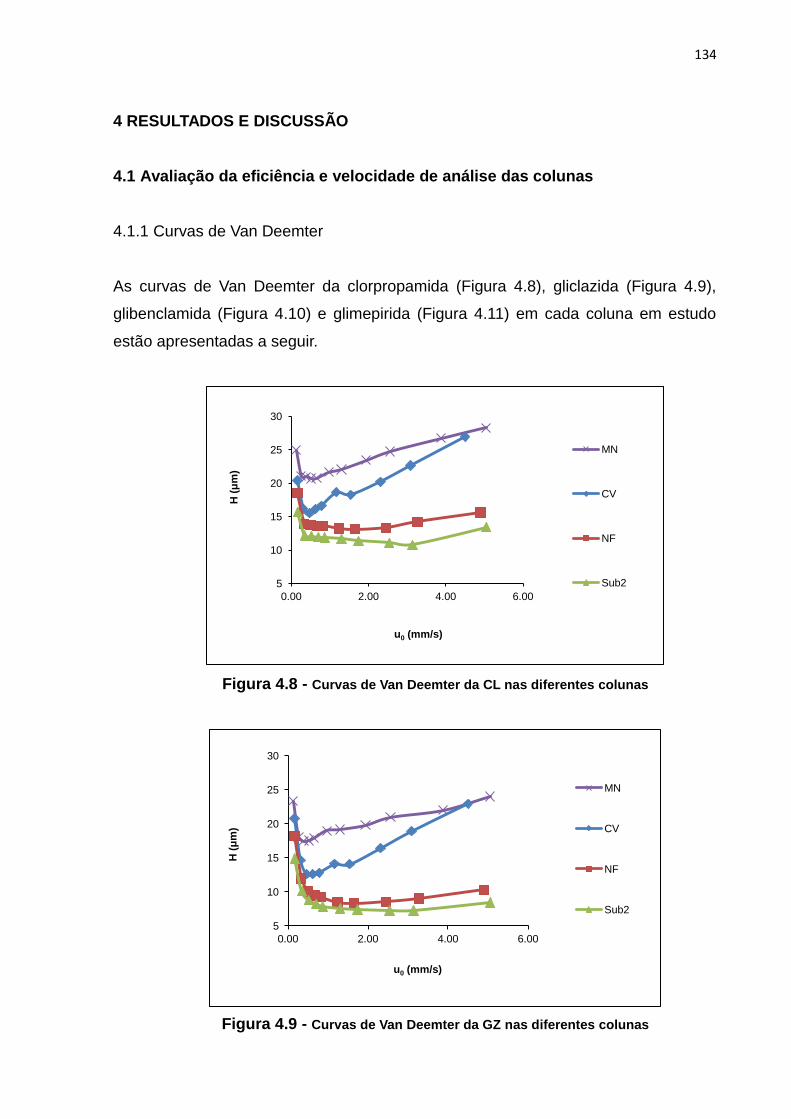
134
4 RESULTADOS E DISCUSSÃO
4.1 Avaliação da eficiência e velocidade de análise das colunas
4.1.1 Curvas de Van Deemter
As curvas de Van Deemter da clorpropamida (Figura 4.8), gliclazida (Figura 4.9),
glibenclamida (Figura 4.10) e glimepirida (Figura 4.11) em cada coluna em estudo
estão apresentadas a seguir.
Figura 4.8 - Curvas de Van Deemter da CL nas diferentes colunas
Figura 4.9 - Curvas de Van Deemter da GZ nas diferentes colunas
5
10
15
20
25
30
0.00 2.00 4.00 6.00
H (
μm
)
u0 (mm/s)
MN
CV
NF
Sub2
5
10
15
20
25
30
0.00 2.00 4.00 6.00
H (
μm
)
u0 (mm/s)
MN
CV
NF
Sub2
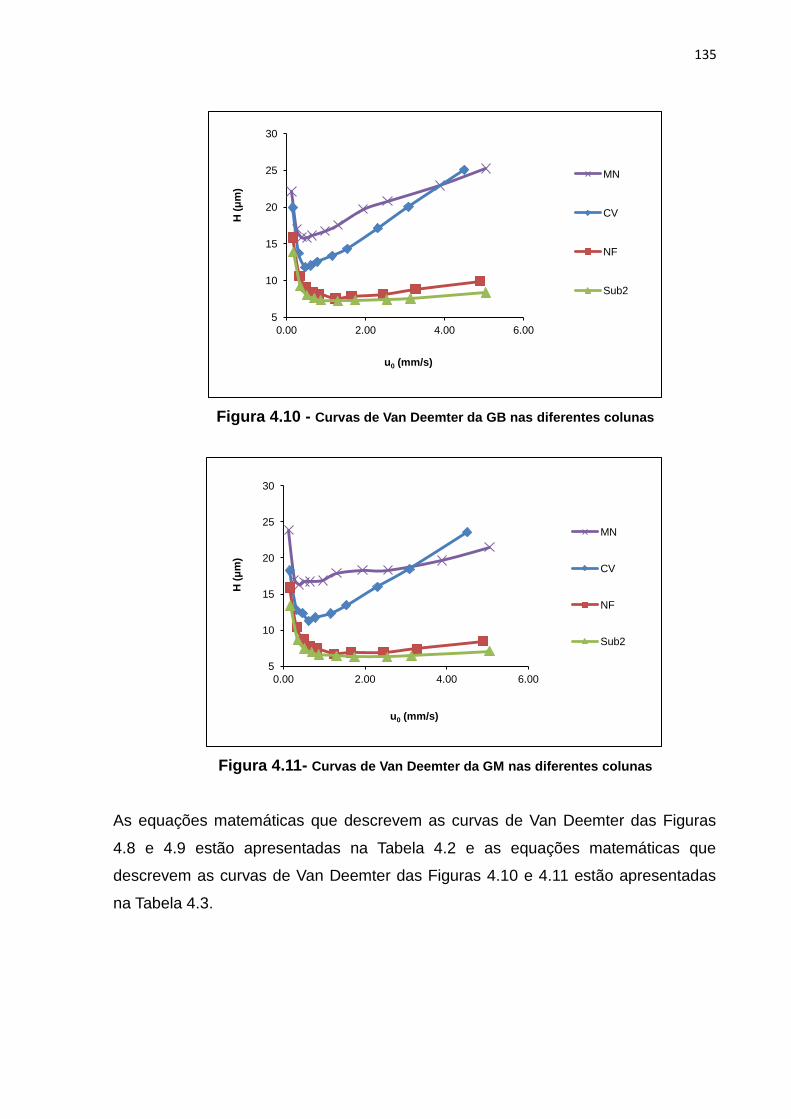
135
Figura 4.10 - Curvas de Van Deemter da GB nas diferentes colunas
Figura 4.11- Curvas de Van Deemter da GM nas diferentes colunas
As equações matemáticas que descrevem as curvas de Van Deemter das Figuras
4.8 e 4.9 estão apresentadas na Tabela 4.2 e as equações matemáticas que
descrevem as curvas de Van Deemter das Figuras 4.10 e 4.11 estão apresentadas
na Tabela 4.3.
5
10
15
20
25
30
0.00 2.00 4.00 6.00
H (
μm
)
u0 (mm/s)
MN
CV
NF
Sub2
5
10
15
20
25
30
0.00 2.00 4.00 6.00
H (
μm
)
u0 (mm/s)
MN
CV
NF
Sub2

136
Tabela 4.2 - Equações referentes as curvas de Van Deemter da CL e GZ - Figuras 4.8 e 4.9
Coluna Analito
Clorpropamida Gliclazida
CV H = 12,66 + 1,04/u0 + 3,14u0 H = 7,71 + 1,89/u0 + 3,36u0
MN H = 18,91 + 0,67/u0 + 1,84u0 H = 15,77 + 0,80/u0 + 1,58u0
NF H = 11,00 + 1,17/u0 + 0,88u0 H = 5,66 + 2,06/u0 + 0,85u0
Sub 2 μm H = 9,81 + 0,98/u0 + 0,54u0 H = 5,52 + 1,60/u0 + 0,48u0
Tabela 4.3 - Equações referentes as curvas de Van Deemter da GB e GM - Figuras 4.10 e 4.11
Coluna Analito
Glibenclamida Glimepirida
CV H = 6,63 + 1,90/u0 + 4,08u0 H = 6,59 + 1,69/u0 + 3,70u0
MN H = 13,57 + 0,97/u0 + 2,24u0 H = 13,91 + 1,14/u0 + 1,50u0
NF H = 5,24 + 1,72/u0 + 0,90u0 H = 4,55 + 1,85/u0 + 0,73u0
Sub 2 μm H = 5,14 + 1,46/u0 + 0,61u0 H = 4,44 + 1,51/u0 + 0,48u0
Analisando-se o perfil das curvas de Van Deemter apresentadas nas Figuras 4.8 a
4.11, verifica-se que aquelas referentes à coluna sub 2 μm, seguida da coluna com
partículas de núcleo fundido, são as menos inclinadas após o ponto de máxima
eficiência (Hmin). Significa que mesmo em altas velocidades lineares da fase móvel é
possível obter valores de eficiência próximos ao máximo quando essas colunas são
usadas. Por outro lado, as curvas de Van Deemter referentes à coluna convencional
são as mais inclinadas. Portanto, o emprego de altas velocidades lineares da fase
móvel na coluna convencional implica na obtenção de valores de eficiência distantes
do máximo que ela poderia propiciar. Já as curvas de Van Deemter relativas à
coluna monolítica apresentaram inclinações intermediárias. Curvas com aspectos
similares aos observados foram descritas na literatura, destacando-se trabalho
publicado em 2009 por Brice e colaboradores [10].
É sabido que quanto maior a influência do aumento da velocidade linear da fase
móvel na perda da eficiência cromatográfica, maior é a inclinação da curva de Van
Deemter após o ponto de máxima eficiência e maior é o coeficiente C [4]. Sendo
assim e considerando o exposto no parágrafo anterior, esperar-se-ia que para um

137
mesmo analito as equações relativas à coluna convencional apresentassem os
maiores valores do termo C, seguidos daqueles das colunas monolítica,
empacotadas com partículas de núcleo fundido e com partículas sub 2 μm. Os
resultados encontrados para o termo C, apresentadas na Tabela 4.2 e 4.3, são
compatíveis com o previsto. Para ilustrar, os termos C das equações relativas a
glimepirida em cada coluna são: C CV = 3,70; C MN =1,50; C NF = 0,73 e
C sub 2μm = 0,48.
Os dados apresentados na Tabela 4.2 e 4.3, evidenciam que o coeficiente A das
equações relativas a um mesmo analito foi maior nas análises realizadas em coluna
monolítica, seguida das análises em colunas convencional, empacotadas com
partículas de núcleo fundido e com partículas sub 2 μm. Para exemplificar os termos
A das equações relativas a glimepirida em cada coluna são: A MN = 13,91;
A CV = 6,59; A NF = 4,55 e A sub 2μm = 4,44. Para compreender os resultados é
importante considerar que o coeficiente A é tanto maior quanto maior for o número
de caminhos múltiplos e que o número de caminhos múltiplos é diretamente
proporcional ao diâmetro das partículas (dp) e inversamente proporcional à qualidade
do empacotamento/formação da coluna [4]. Sendo assim, como o diâmetro das
partículas na coluna sub 2 μm é 1,8 μm, menor que o presente na coluna de
partículas de núcleo fundido (dp = 2,7 μm), há menos caminhos múltiplos na coluna
sub 2 μm, o que explica o menor valor de A nas equações de Van Deemter relativas
à coluna sub 2 μm. O mesmo raciocínio se aplica para explicar a razão de o termo A
das equações relativas à coluna de núcleo fundido (dp = 2,7 μm) ser menor do que o
presente nas equações relativas à coluna convencional (dp = 5 μm). Para justificar os
maiores valores do termo A verificados nas equações relativas à coluna monolítica
pode-se supor a presença de um leito cromatográfico de baixa qualidade, com baixa
homogeneidade de tamanho dos poros, como já foi relatado em trabalhos da
literatura [10].
Na Tabela 4.4, apresentada a seguir, estão descritos os valores de Hmin e u0 opt das
curvas de Van Deemter apresentadas nas Figuras 4.8 a 4.11.
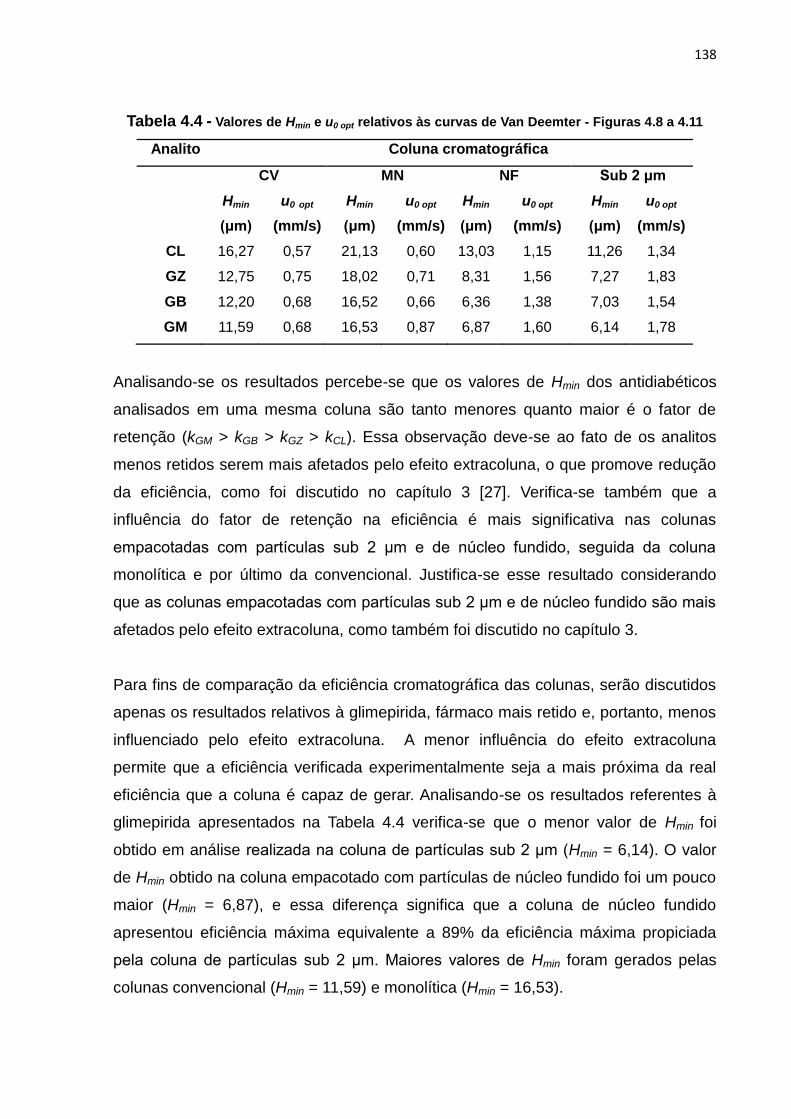
138
Tabela 4.4 - Valores de Hmin e u0 opt relativos às curvas de Van Deemter - Figuras 4.8 a 4.11
Analito Coluna cromatográfica
CV MN NF Sub 2 μm
Hmin
(μm)
u0 opt
(mm/s)
Hmin
(μm)
u0 opt
(mm/s)
Hmin
(μm)
u0 opt
(mm/s)
Hmin
(μm)
u0 opt
(mm/s)
CL 16,27 0,57 21,13 0,60 13,03 1,15 11,26 1,34
GZ 12,75 0,75 18,02 0,71 8,31 1,56 7,27 1,83
GB 12,20 0,68 16,52 0,66 6,36 1,38 7,03 1,54
GM 11,59 0,68 16,53 0,87 6,87 1,60 6,14 1,78
Analisando-se os resultados percebe-se que os valores de Hmin dos antidiabéticos
analisados em uma mesma coluna são tanto menores quanto maior é o fator de
retenção (kGM > kGB > kGZ > kCL). Essa observação deve-se ao fato de os analitos
menos retidos serem mais afetados pelo efeito extracoluna, o que promove redução
da eficiência, como foi discutido no capítulo 3 [27]. Verifica-se também que a
influência do fator de retenção na eficiência é mais significativa nas colunas
empacotadas com partículas sub 2 μm e de núcleo fundido, seguida da coluna
monolítica e por último da convencional. Justifica-se esse resultado considerando
que as colunas empacotadas com partículas sub 2 μm e de núcleo fundido são mais
afetados pelo efeito extracoluna, como também foi discutido no capítulo 3.
Para fins de comparação da eficiência cromatográfica das colunas, serão discutidos
apenas os resultados relativos à glimepirida, fármaco mais retido e, portanto, menos
influenciado pelo efeito extracoluna. A menor influência do efeito extracoluna
permite que a eficiência verificada experimentalmente seja a mais próxima da real
eficiência que a coluna é capaz de gerar. Analisando-se os resultados referentes à
glimepirida apresentados na Tabela 4.4 verifica-se que o menor valor de Hmin foi
obtido em análise realizada na coluna de partículas sub 2 μm (Hmin = 6,14). O valor
de Hmin obtido na coluna empacotado com partículas de núcleo fundido foi um pouco
maior (Hmin = 6,87), e essa diferença significa que a coluna de núcleo fundido
apresentou eficiência máxima equivalente a 89% da eficiência máxima propiciada
pela coluna de partículas sub 2 μm. Maiores valores de Hmin foram gerados pelas
colunas convencional (Hmin = 11,59) e monolítica (Hmin = 16,53).

139
De acordo com a literatura era de se esperar que a coluna com partículas sub 2 μm
fosse a mais eficiente (em função do menor tamanho de partícula) e que a eficiência
da coluna de partículas de núcleo fundido, também elevada (em função da estrutura
superficialmente porosa), fosse um pouco inferior [15,18]. Entretanto, a maior
eficiência da coluna convencional quando comparada à propiciada pela coluna
monolítica não está de acordo com a literatura. Esperar-se-ia que a coluna
monolítica apresentasse eficiência comparável à da coluna convencional [8, 9].
É importante também comparar os valores de velocidade ótima (u0 opt) das quatro
colunas cromatográficas, apresentados na Tabela 4.4. Como a velocidade linear
ótima da fase móvel é inversamente proporcional ao diâmetro das partículas (dp), era
de se esperar que a coluna convencional (dp = 5 μm) apresentasse a menor
velocidade (u0 opt = 0,68 mm/s - vazão de aproximadamente 0,08 ml/min) e que a
coluna com partículas sub 2 μm (dp = 1,8 μm) apresentasse a maior velocidade
(u0 opt = 1,78 mm/s - vazão de aproximadamente 0,2 ml/min) [28]. A velocidade linear
ótima de análise da glimepirida em coluna de núcleo fundido (u0 opt = 1,60 mm/s -
vazão um pouco abaixo de 0,2 ml/min), apesar de um pouco menor, foi próxima a
obtida em coluna com partículas sub 2 μm (u0 opt = 1,78 mm/s). Resultados similares
já foram descritos na literatura. Em experimento realizado com benzofenona
verificou-se que a velocidade linear ótima de análise em colunas de núcleo fundido e
de partículas sub 2 μm foram bem próximas, ficando em torno de 3 mm/s [10].
Quanto à velocidade linear ótima da coluna monolítica (u0 opt = 0,87 mm/s - vazão de
aproximadamente 0,15 ml/min), verificou-se que ela foi maior do que a apresentada
pela coluna convencional, mas inferior à das outras colunas.
Considerando as discussões sobre o formato das curvas de Van Deemter e sobre
valores de Hmin e u0 opt, conclui-se que as colunas com partículas sub 2 μm e com
partículas de núcleo fundido apresentaram maior eficiência máxima, ou seja,
menores valores de Hmin, os quais ocorreram em maiores velocidades. O formato
menos inclinado das curvas de Van Deemter referentes a essas colunas permite o
emprego de altas velocidades lineares da fase móvel com obtenção de eficiência
próxima ao máximo que elas poderiam propiciar, ou seja, há nessas colunas um
maior compromisso entre eficiência e velocidade de análise. As colunas monolítica e
convencional são menos eficientes (maiores valores de Hmin) e ao aumento da
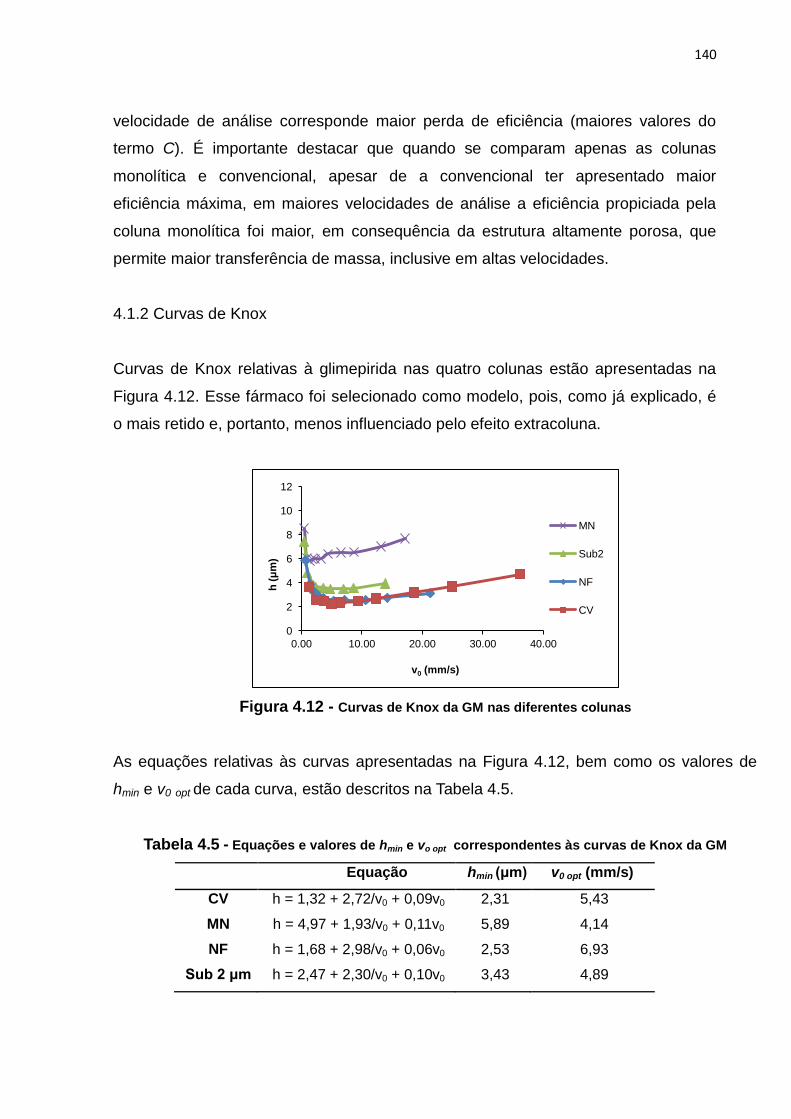
140
velocidade de análise corresponde maior perda de eficiência (maiores valores do
termo C). É importante destacar que quando se comparam apenas as colunas
monolítica e convencional, apesar de a convencional ter apresentado maior
eficiência máxima, em maiores velocidades de análise a eficiência propiciada pela
coluna monolítica foi maior, em consequência da estrutura altamente porosa, que
permite maior transferência de massa, inclusive em altas velocidades.
4.1.2 Curvas de Knox
Curvas de Knox relativas à glimepirida nas quatro colunas estão apresentadas na
Figura 4.12. Esse fármaco foi selecionado como modelo, pois, como já explicado, é
o mais retido e, portanto, menos influenciado pelo efeito extracoluna.
Figura 4.12 - Curvas de Knox da GM nas diferentes colunas
As equações relativas às curvas apresentadas na Figura 4.12, bem como os valores de
hmin e v0 opt de cada curva, estão descritos na Tabela 4.5.
Tabela 4.5 - Equações e valores de hmin e vo opt correspondentes às curvas de Knox da GM
Equação hmin (μm) v0 opt (mm/s)
CV h = 1,32 + 2,72/v0 + 0,09v0 2,31 5,43
MN h = 4,97 + 1,93/v0 + 0,11v0 5,89 4,14
NF h = 1,68 + 2,98/v0 + 0,06v0 2,53 6,93
Sub 2 μm h = 2,47 + 2,30/v0 + 0,10v0 3,43 4,89
0
2
4
6
8
10
12
0.00 10.00 20.00 30.00 40.00
h (
μm
)
v0 (mm/s)
MN
Sub2
NF
CV
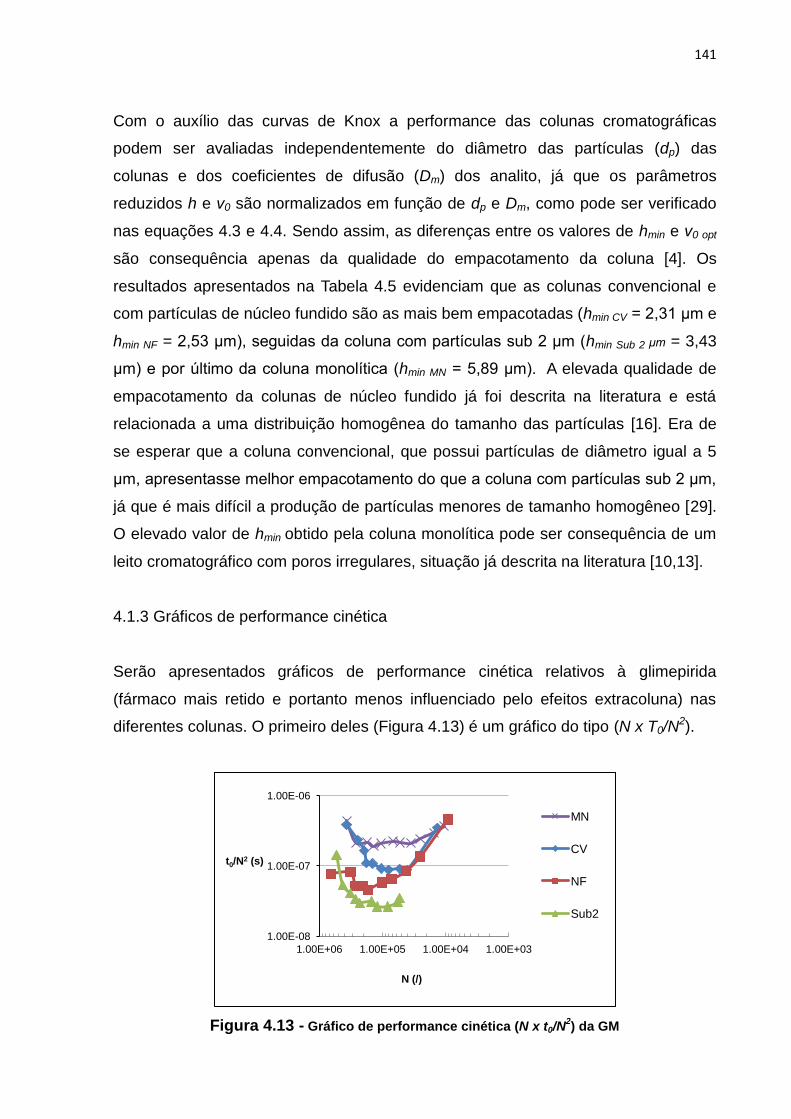
141
Com o auxílio das curvas de Knox a performance das colunas cromatográficas
podem ser avaliadas independentemente do diâmetro das partículas (dp) das
colunas e dos coeficientes de difusão (Dm) dos analito, já que os parâmetros
reduzidos h e v0 são normalizados em função de dp e Dm, como pode ser verificado
nas equações 4.3 e 4.4. Sendo assim, as diferenças entre os valores de hmin e v0 opt
são consequência apenas da qualidade do empacotamento da coluna [4]. Os
resultados apresentados na Tabela 4.5 evidenciam que as colunas convencional e
com partículas de núcleo fundido são as mais bem empacotadas (hmin CV = 2,31 μm e
hmin NF = 2,53 μm), seguidas da coluna com partículas sub 2 μm (hmin Sub 2 μm = 3,43
μm) e por último da coluna monolítica (hmin MN = 5,89 μm). A elevada qualidade de
empacotamento da colunas de núcleo fundido já foi descrita na literatura e está
relacionada a uma distribuição homogênea do tamanho das partículas [16]. Era de
se esperar que a coluna convencional, que possui partículas de diâmetro igual a 5
μm, apresentasse melhor empacotamento do que a coluna com partículas sub 2 μm,
já que é mais difícil a produção de partículas menores de tamanho homogêneo [29].
O elevado valor de hmin obtido pela coluna monolítica pode ser consequência de um
leito cromatográfico com poros irregulares, situação já descrita na literatura [10,13].
4.1.3 Gráficos de performance cinética
Serão apresentados gráficos de performance cinética relativos à glimepirida
(fármaco mais retido e portanto menos influenciado pelo efeitos extracoluna) nas
diferentes colunas. O primeiro deles (Figura 4.13) é um gráfico do tipo (N x T0/N2).
Figura 4.13 - Gráfico de performance cinética (N x t0/N2) da GM
1.00E-08
1.00E-07
1.00E-06
1.00E+03 1.00E+04 1.00E+05 1.00E+06
MN
CV
NF
Sub2
t0/N2 (s)
N (/)
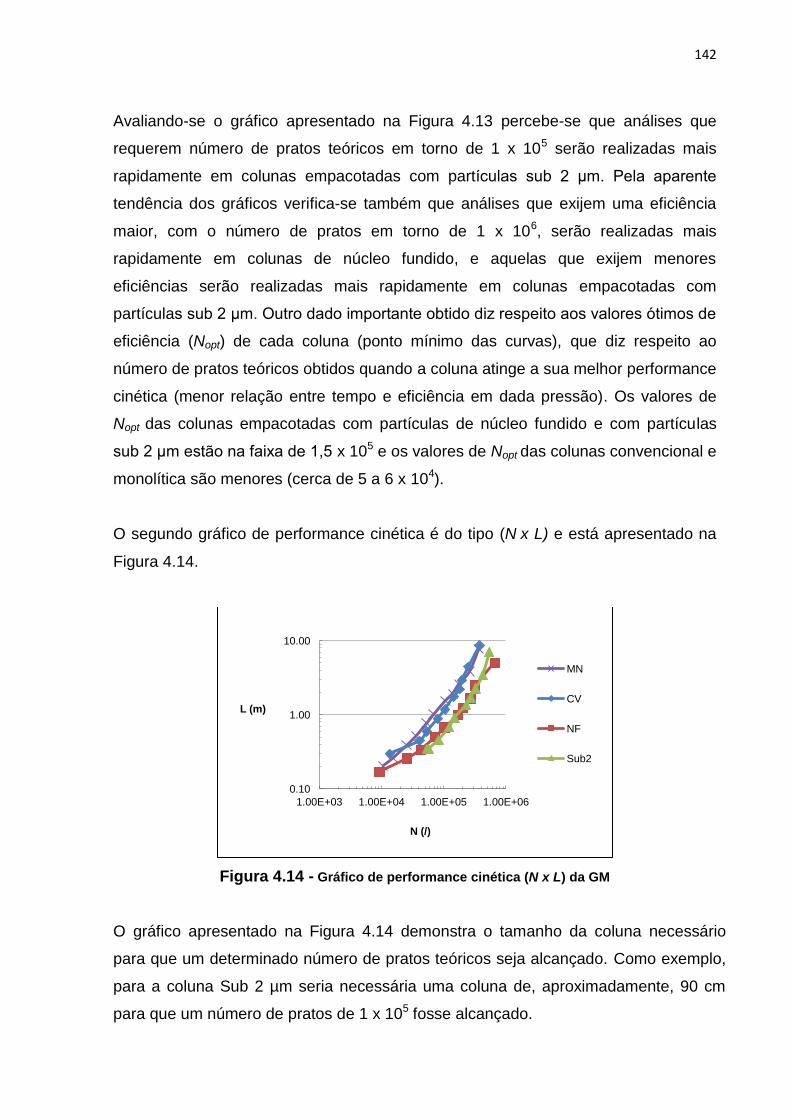
142
Avaliando-se o gráfico apresentado na Figura 4.13 percebe-se que análises que
requerem número de pratos teóricos em torno de 1 x 105 serão realizadas mais
rapidamente em colunas empacotadas com partículas sub 2 μm. Pela aparente
tendência dos gráficos verifica-se também que análises que exijem uma eficiência
maior, com o número de pratos em torno de 1 x 106, serão realizadas mais
rapidamente em colunas de núcleo fundido, e aquelas que exijem menores
eficiências serão realizadas mais rapidamente em colunas empacotadas com
partículas sub 2 μm. Outro dado importante obtido diz respeito aos valores ótimos de
eficiência (Nopt) de cada coluna (ponto mínimo das curvas), que diz respeito ao
número de pratos teóricos obtidos quando a coluna atinge a sua melhor performance
cinética (menor relação entre tempo e eficiência em dada pressão). Os valores de
Nopt das colunas empacotadas com partículas de núcleo fundido e com partículas
sub 2 μm estão na faixa de 1,5 x 105 e os valores de Nopt das colunas convencional e
monolítica são menores (cerca de 5 a 6 x 104).
O segundo gráfico de performance cinética é do tipo (N x L) e está apresentado na
Figura 4.14.
Figura 4.14 - Gráfico de performance cinética (N x L) da GM
O gráfico apresentado na Figura 4.14 demonstra o tamanho da coluna necessário
para que um determinado número de pratos teóricos seja alcançado. Como exemplo,
para a coluna Sub 2 µm seria necessária uma coluna de, aproximadamente, 90 cm
para que um número de pratos de 1 x 105 fosse alcançado.
0.10
1.00
10.00
1.00E+03 1.00E+04 1.00E+05 1.00E+06
MN
CV
NF
Sub2
L (m)
N (/)
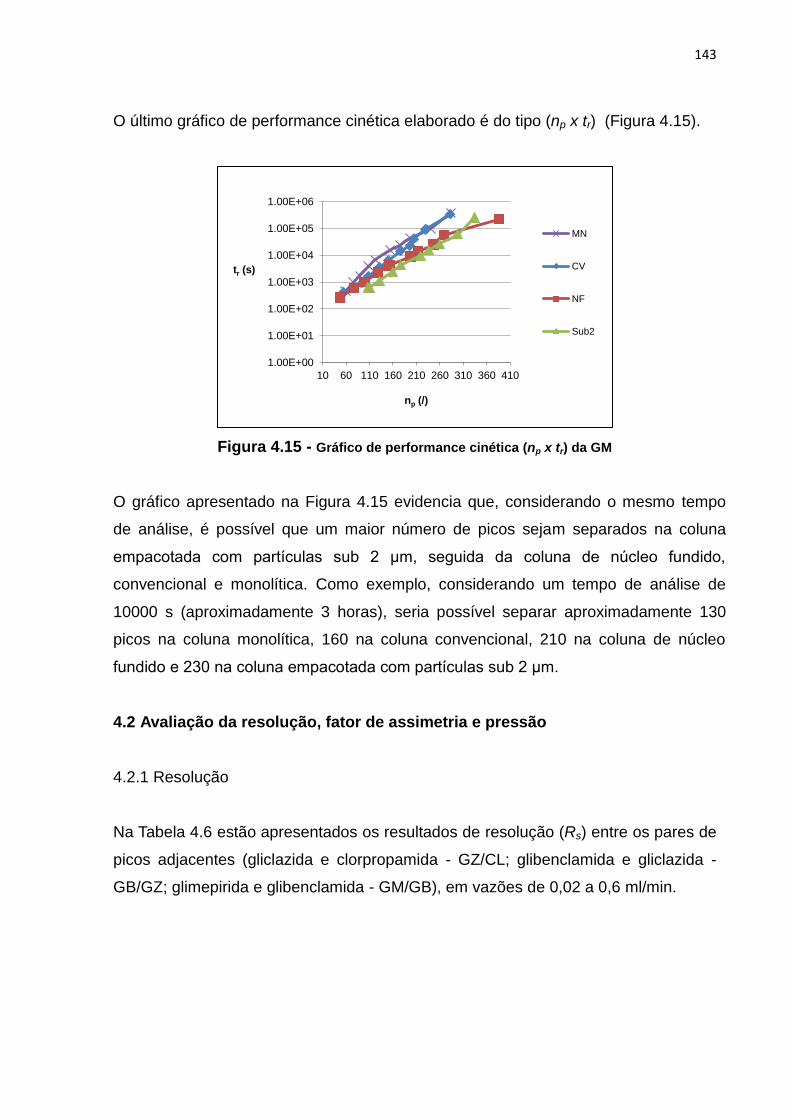
143
O último gráfico de performance cinética elaborado é do tipo (np x tr) (Figura 4.15).
Figura 4.15 - Gráfico de performance cinética (np x tr) da GM
O gráfico apresentado na Figura 4.15 evidencia que, considerando o mesmo tempo
de análise, é possível que um maior número de picos sejam separados na coluna
empacotada com partículas sub 2 μm, seguida da coluna de núcleo fundido,
convencional e monolítica. Como exemplo, considerando um tempo de análise de
10000 s (aproximadamente 3 horas), seria possível separar aproximadamente 130
picos na coluna monolítica, 160 na coluna convencional, 210 na coluna de núcleo
fundido e 230 na coluna empacotada com partículas sub 2 μm.
4.2 Avaliação da resolução, fator de assimetria e pressão
4.2.1 Resolução
Na Tabela 4.6 estão apresentados os resultados de resolução (Rs) entre os pares de
picos adjacentes (gliclazida e clorpropamida - GZ/CL; glibenclamida e gliclazida -
GB/GZ; glimepirida e glibenclamida - GM/GB), em vazões de 0,02 a 0,6 ml/min.
1.00E+00
1.00E+01
1.00E+02
1.00E+03
1.00E+04
1.00E+05
1.00E+06
10 60 110 160 210 260 310 360 410
MN
CV
NF
Sub2
tr (s)
np (/)
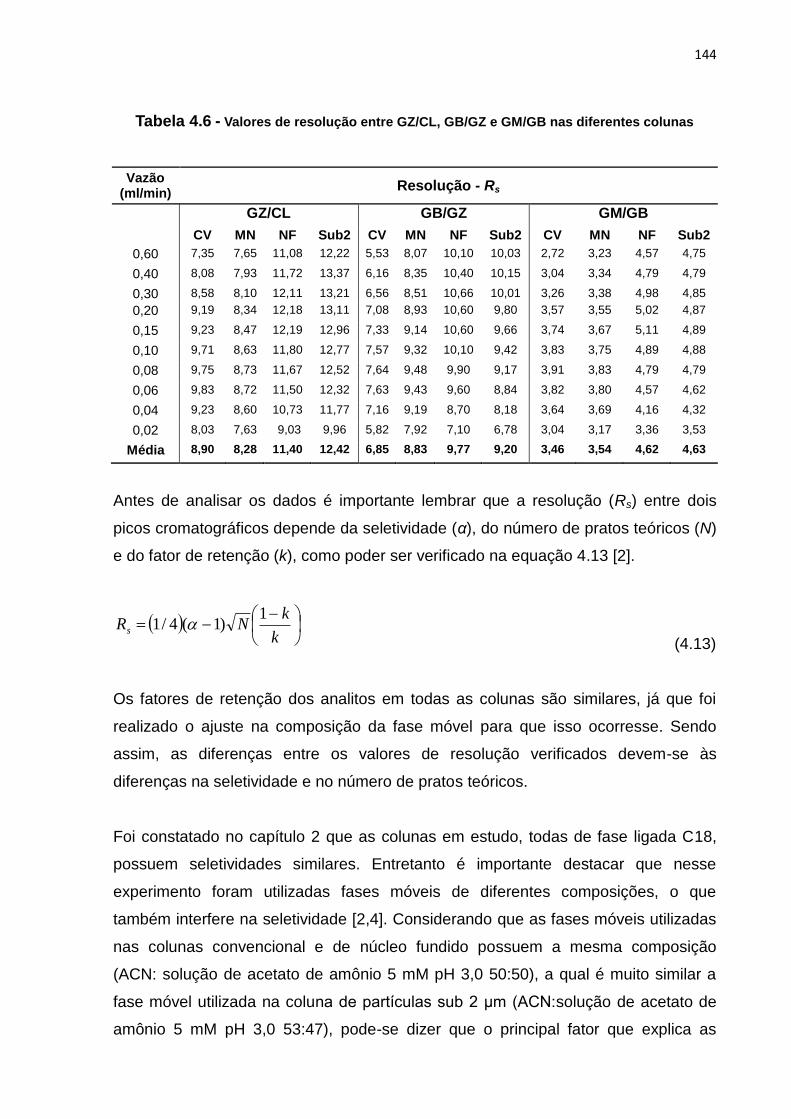
144
Tabela 4.6 - Valores de resolução entre GZ/CL, GB/GZ e GM/GB nas diferentes colunas
Vazão (ml/min)
Resolução - Rs
GZ/CL GB/GZ GM/GB
CV MN NF Sub2 CV MN NF Sub2 CV MN NF Sub2
0,60 7,35 7,65 11,08 12,22 5,53 8,07 10,10 10,03 2,72 3,23 4,57 4,75
0,40 8,08 7,93 11,72 13,37 6,16 8,35 10,40 10,15 3,04 3,34 4,79 4,79
0,30 8,58 8,10 12,11 13,21 6,56 8,51 10,66 10,01 3,26 3,38 4,98 4,85
0,20 9,19 8,34 12,18 13,11 7,08 8,93 10,60 9,80 3,57 3,55 5,02 4,87
0,15 9,23 8,47 12,19 12,96 7,33 9,14 10,60 9,66 3,74 3,67 5,11 4,89
0,10 9,71 8,63 11,80 12,77 7,57 9,32 10,10 9,42 3,83 3,75 4,89 4,88
0,08 9,75 8,73 11,67 12,52 7,64 9,48 9,90 9,17 3,91 3,83 4,79 4,79
0,06 9,83 8,72 11,50 12,32 7,63 9,43 9,60 8,84 3,82 3,80 4,57 4,62
0,04 9,23 8,60 10,73 11,77 7,16 9,19 8,70 8,18 3,64 3,69 4,16 4,32
0,02 8,03 7,63 9,03 9,96 5,82 7,92 7,10 6,78 3,04 3,17 3,36 3,53
Média 8,90 8,28 11,40 12,42 6,85 8,83 9,77 9,20 3,46 3,54 4,62 4,63
Antes de analisar os dados é importante lembrar que a resolução (Rs) entre dois
picos cromatográficos depende da seletividade (α), do número de pratos teóricos (N)
e do fator de retenção (k), como poder ser verificado na equação 4.13 [2].
k
kNRs
1)1(4/1
(4.13)
Os fatores de retenção dos analitos em todas as colunas são similares, já que foi
realizado o ajuste na composição da fase móvel para que isso ocorresse. Sendo
assim, as diferenças entre os valores de resolução verificados devem-se às
diferenças na seletividade e no número de pratos teóricos.
Foi constatado no capítulo 2 que as colunas em estudo, todas de fase ligada C18,
possuem seletividades similares. Entretanto é importante destacar que nesse
experimento foram utilizadas fases móveis de diferentes composições, o que
também interfere na seletividade [2,4]. Considerando que as fases móveis utilizadas
nas colunas convencional e de núcleo fundido possuem a mesma composição
(ACN: solução de acetato de amônio 5 mM pH 3,0 50:50), a qual é muito similar a
fase móvel utilizada na coluna de partículas sub 2 μm (ACN:solução de acetato de
amônio 5 mM pH 3,0 53:47), pode-se dizer que o principal fator que explica as

145
diferenças nos valores de resolução verificados entre as colunas convencional, de
partículas de núcleo fundido e de partículas sub 2 μm é o número de pratos teóricos.
Já a coluna monolítica foi submetida a uma fase móvel de composição bem distinta,
de menor força eluotrópica (ACN:solução de acetato de amônio 5 mM pH 3,0 43:57).
Sendo assim as diferenças nos valores de resolução verificados entre a coluna
monolítica e as outras é devido aos fatores seletividade e número de pratos teóricos.
Analisando-se os valores médios de resolução entre os picos (Tabela 4.6), constata-
se que os maiores valores foram alcançados com as colunas de partículas sub 2 μm
e de núcleo fundido e os menores valores correspondem às colunas monolítica e
convencional. Esse resultado pode ser atribuído ao fato de as colunas de partículas
sub 2 μm e de núcleo fundido gerarem maior eficiência (maior número de pratos
teóricos) do que as colunas monolítica e convencional. A eficiência cromatográfica
dessas colunas pode ser constatada nas curvas de Van Deemter apresentadas nas
Figuras 4.8 a 4.11. A vantagem, em termos de resolução, propiciada pelas colunas
de partículas de núcleo fundido e sub 2 μm em relação às colunas monolítica e
convencional já foram relatadas na literatura [10, 30, 31].
É importante destacar que apesar de a coluna monolítica ter sido menos eficiente do
que a coluna convencional, como verificado nas Figuras 4.8 a 4.11, as resoluções
propiciadas por ela foram próximos e até maiores do que as observadas entre os
picos gerados pela coluna convencional. Esse comportamento é devido à maior
seletividade da fase móvel utilizada na análise com coluna monolítica, a qual possui
menor força eluotrópica [2,4]. Avaliando-se a equação 4.13 percebe-se que a
resolução é mais influenciada pela seletividade (α) do que pelo número de pratos
teóricos (N) [32]. Isso explica o fato de os valores de resolução entre os picos
obtidos em coluna monolítica, apesar de apresentarem menor número de pratos
teóricos do que os obtidos em coluna convencional, serem próximos e até maiores
do que os valores de resolução entre os picos obtidos em coluna convencional.
Para ilustrar, estão apresentados na Figura 4.16 os cromatogramas referentes às
análises dos antidiabéticos em cada coluna, na vazão de 0,4 ml/min.
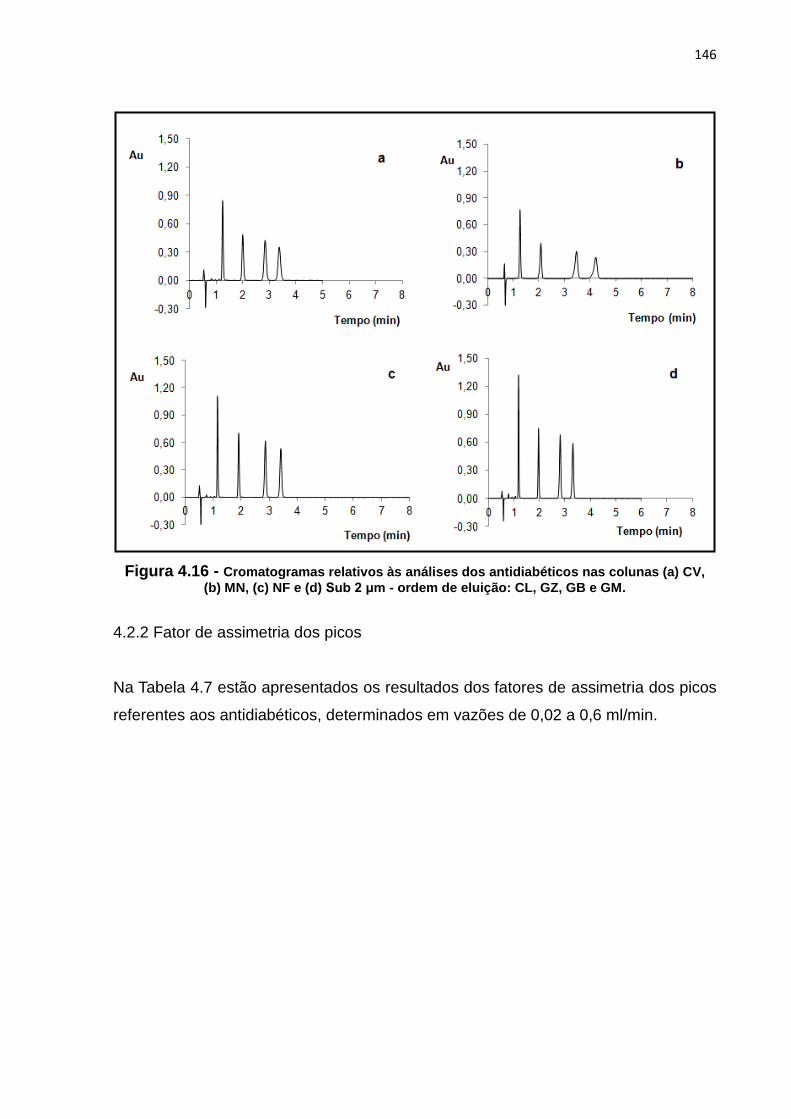
146
Figura 4.16 - Cromatogramas relativos às análises dos antidiabéticos nas colunas (a) CV,
(b) MN, (c) NF e (d) Sub 2 μm - ordem de eluição: CL, GZ, GB e GM.
4.2.2 Fator de assimetria dos picos
Na Tabela 4.7 estão apresentados os resultados dos fatores de assimetria dos picos
referentes aos antidiabéticos, determinados em vazões de 0,02 a 0,6 ml/min.
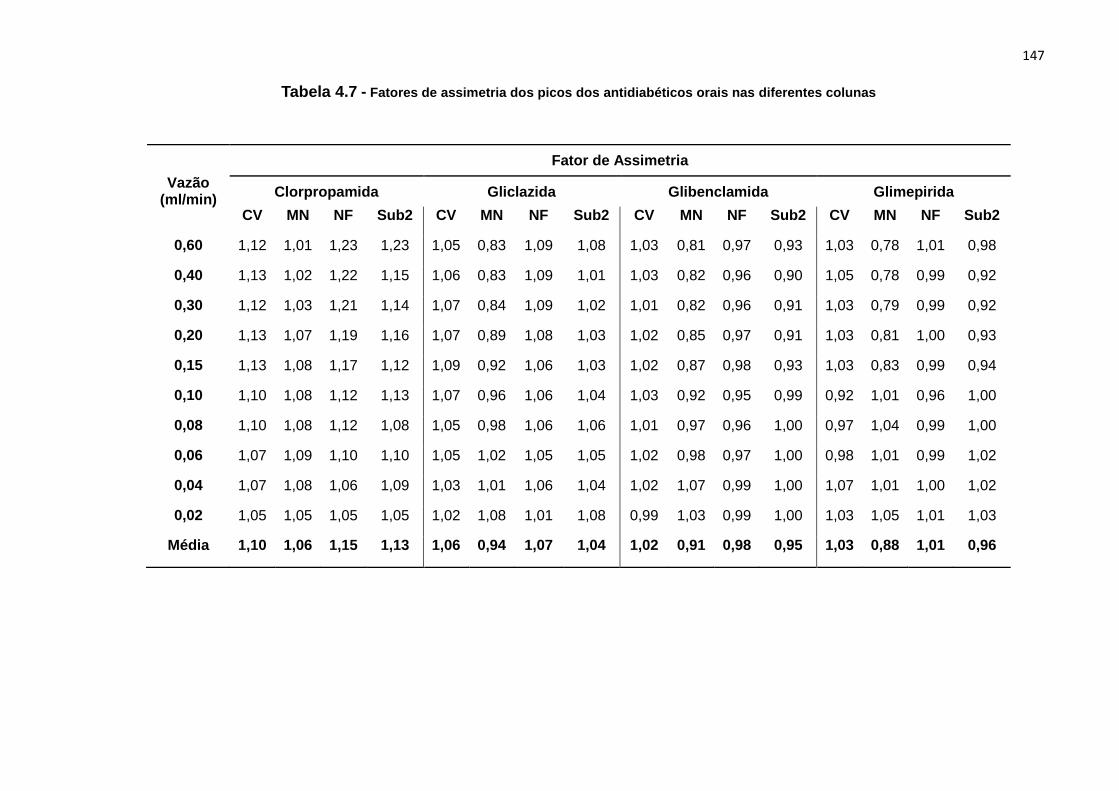
147
Tabela 4.7 - Fatores de assimetria dos picos dos antidiabéticos orais nas diferentes colunas
Vazão (ml/min)
Fator de Assimetria
Clorpropamida Gliclazida Glibenclamida Glimepirida
CV MN NF Sub2 CV MN NF Sub2 CV MN NF Sub2 CV MN NF Sub2
0,60 1,12 1,01 1,23 1,23 1,05 0,83 1,09 1,08 1,03 0,81 0,97 0,93 1,03 0,78 1,01 0,98
0,40 1,13 1,02 1,22 1,15 1,06 0,83 1,09 1,01 1,03 0,82 0,96 0,90 1,05 0,78 0,99 0,92
0,30 1,12 1,03 1,21 1,14 1,07 0,84 1,09 1,02 1,01 0,82 0,96 0,91 1,03 0,79 0,99 0,92
0,20 1,13 1,07 1,19 1,16 1,07 0,89 1,08 1,03 1,02 0,85 0,97 0,91 1,03 0,81 1,00 0,93
0,15 1,13 1,08 1,17 1,12 1,09 0,92 1,06 1,03 1,02 0,87 0,98 0,93 1,03 0,83 0,99 0,94
0,10 1,10 1,08 1,12 1,13 1,07 0,96 1,06 1,04 1,03 0,92 0,95 0,99 0,92 1,01 0,96 1,00
0,08 1,10 1,08 1,12 1,08 1,05 0,98 1,06 1,06 1,01 0,97 0,96 1,00 0,97 1,04 0,99 1,00
0,06 1,07 1,09 1,10 1,10 1,05 1,02 1,05 1,05 1,02 0,98 0,97 1,00 0,98 1,01 0,99 1,02
0,04 1,07 1,08 1,06 1,09 1,03 1,01 1,06 1,04 1,02 1,07 0,99 1,00 1,07 1,01 1,00 1,02
0,02 1,05 1,05 1,05 1,05 1,02 1,08 1,01 1,08 0,99 1,03 0,99 1,00 1,03 1,05 1,01 1,03
Média 1,10 1,06 1,15 1,13 1,06 0,94 1,07 1,04 1,02 0,91 0,98 0,95 1,03 0,88 1,01 0,96
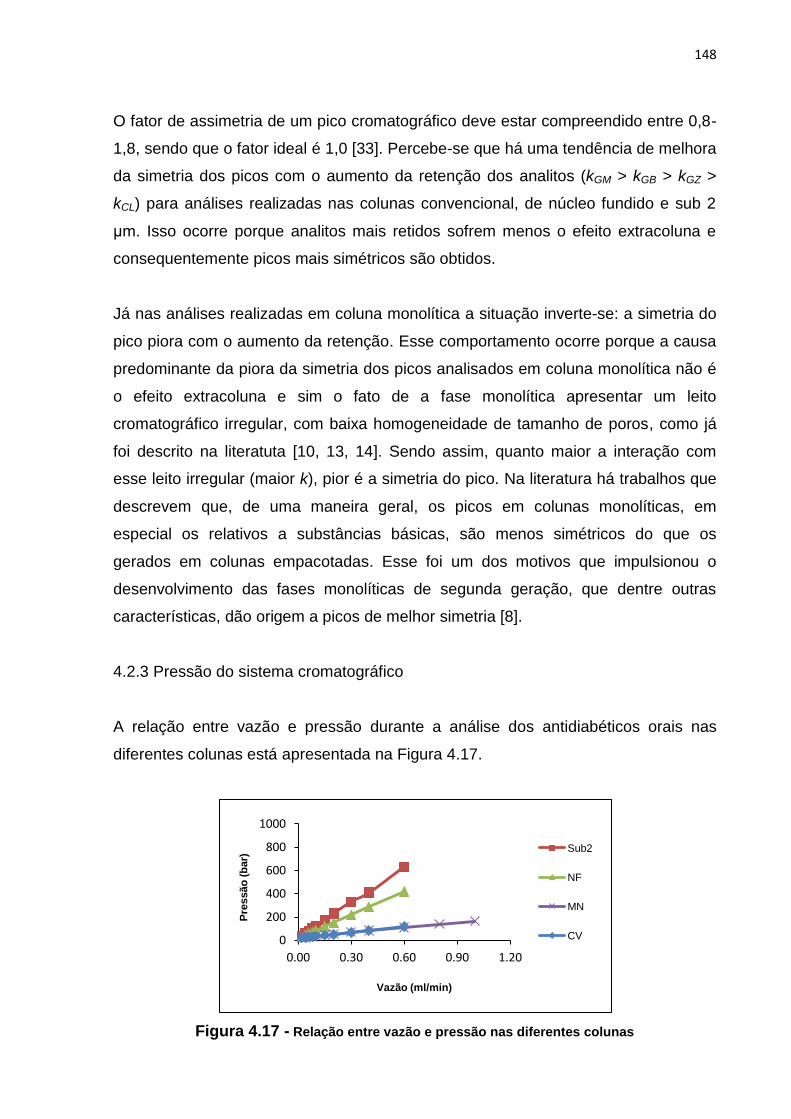
148
O fator de assimetria de um pico cromatográfico deve estar compreendido entre 0,8-
1,8, sendo que o fator ideal é 1,0 [33]. Percebe-se que há uma tendência de melhora
da simetria dos picos com o aumento da retenção dos analitos (kGM > kGB > kGZ >
kCL) para análises realizadas nas colunas convencional, de núcleo fundido e sub 2
μm. Isso ocorre porque analitos mais retidos sofrem menos o efeito extracoluna e
consequentemente picos mais simétricos são obtidos.
Já nas análises realizadas em coluna monolítica a situação inverte-se: a simetria do
pico piora com o aumento da retenção. Esse comportamento ocorre porque a causa
predominante da piora da simetria dos picos analisados em coluna monolítica não é
o efeito extracoluna e sim o fato de a fase monolítica apresentar um leito
cromatográfico irregular, com baixa homogeneidade de tamanho de poros, como já
foi descrito na literatuta [10, 13, 14]. Sendo assim, quanto maior a interação com
esse leito irregular (maior k), pior é a simetria do pico. Na literatura há trabalhos que
descrevem que, de uma maneira geral, os picos em colunas monolíticas, em
especial os relativos a substâncias básicas, são menos simétricos do que os
gerados em colunas empacotadas. Esse foi um dos motivos que impulsionou o
desenvolvimento das fases monolíticas de segunda geração, que dentre outras
características, dão origem a picos de melhor simetria [8].
4.2.3 Pressão do sistema cromatográfico
A relação entre vazão e pressão durante a análise dos antidiabéticos orais nas
diferentes colunas está apresentada na Figura 4.17.
Figura 4.17 - Relação entre vazão e pressão nas diferentes colunas
0
200
400
600
800
1000
0.00 0.30 0.60 0.90 1.20
Pre
ssão
(b
ar)
Vazão (ml/min)
Sub2
NF
MN
CV

149
Verifica-se que em um mesmo valor de vazão os maiores valores de pressão foram
gerados pela coluna com partículas totalmente porosas sub 2 μm. Para exemplificar:
na vazão de 0,6 ml/min a pressão gerada pela coluna sub 2 μm foi de 632 bar,
aproximadamente cinco vezes maior do que a pressão gerada pelas colunas
convencionais e monolítica, e 1,5 vezes maior do que a gerada pela coluna com
partículas de núcleo fundido. Considerando que a pressão é inversamente
proporcional ao quadrado do diâmetro das partículas da coluna, e que a coluna sub
2 μm utilizada possui partículas de apenas 1,8 μm de diâmetro, elevados valores de
pressão, já descritos na literatura, eram esperados. Esse aumento da pressão
justifica a necessidade da utilização de um equipamento de UHPLC [15,18].
Como já foi dito, quando se trabalhou com partículas de núcleo fundido a pressão foi
de aproximadamente 65% da gerada por partículas totalmente porosas sub 2 μm.
Na literatura há trabalhos que demonstram que essa relação está em torno de 50%
[10]. É importante destacar que a pressão gerada pela coluna de núcleo fundido foi
compatível com o sistema de HPLC (Pmax = 400 bar).
As colunas monolítica e convencional apresentaram valores de pressão mais baixos,
compatíveis com sistemas de HPLC, e próximos entre si.
5 CONCLUSÃO
Conclui-se que as colunas com partículas sub 2 μm e com partículas de núcleo
fundido apresentaram maior eficiência máxima (Ex.: Hmin Sub 2μm GM = 6,14 μm e
Hmin NF GM = 6,87 μm), os quais ocorreram em maiores velocidades lineares de fase
móvel (Ex.: u0 opt Sub 2μm GM = 1,78 mm/s e u0 opt NF GM = 1,60 mm/s). O formato menos
inclinado das curvas de Van Deemter referentes a essas colunas permite o emprego
de altas velocidades de fase móvel com obtenção de eficiência próxima a máxima.
Já as colunas monolítica e convencional foram menos eficientes
(Ex.: Hmin MN GM = 16,53 μm e Hmin CV = 11,59 μm) e com o aumento da velocidade de
análise houve uma maior perda de eficiência (maiores valores do termo C). Quando
se comparam apenas as colunas monolítica e convencional, apesar de a
convencional ter propiciado análises de maior eficiência máxima, em altas

150
velocidades a eficiência gerada pela monolítica foi maior, o que é consequência da
estrutura permeável, que permite melhor transferência de massa.
Analisando-se as curvas de Knox percebe-se que as colunas convencional e com
partículas de núcleo fundido foram melhor empacotadas (Ex.: hmin CV GM = 2,31 μm e
hmin NF GM = 2,53 μm), seguidas das colunas com partículas sub 2 μm
(Ex.: hmin Sub 2 μm GM = 3,43 μm). A coluna monolítica apresentou o pior processo
produtivo (Ex.: hmin MN GM = 5,89 μm).
Avaliando-se os gráficos de performance cinética verifica-se que a coluna
empacotada com partículas sub 2 μm, seguida da coluna de núcleo fundido, são
capazes de dar origem a análises com maior capacidade de picos e com maior
número de pratos teóricos em tempos de análise mais curtos.
Analisando-se a resolução constata-se que maiores valores foram alcançados com
as colunas de partículas sub 2 μm e de núcleo fundido. Quanto à simetria nota-se
que com exceção da monolítica, que gerou picos mais assimétricos, todas as outras
colunas deram origem a picos de boa simetria. Quanto à pressão nota-se que a
gerada pela coluna sub 2 μm foi aproximadamente 1,5 maior do que a gerada pela
coluna com partículas de núcleo fundido, sendo que em vazões acima de 0,4 ml/min
a coluna sub 2 μm gerou pressões que requerem sistemas de UHPLC, ao contrário
da coluna de núcleo fundido, que gerou pressão compatível com sistemas de HPLC.
As colunas monolítica e convencional geraram os valores mais baixos de pressão,
também compatíveis com sistemas de HPLC.
Considerando o exposto conclui-se que a coluna empacotada com partículas de
núcleo fundido é a melhor alternativa à coluna convencional. A coluna monolítica,
apesar de compatível com HPLC e de propiciar análises mais rápidas com maior
eficiência do que a coluna convencional, apresenta eficiência cromatográfica bem
inferior à gerada pelas colunas de núcleo fundido e sub 2 μm, além de dar origem a
picos de pior simetria e ser, dentre todas as colunas, a de maior custo.
Considerando a análise da glimepirida para exemplificar, verifica-se que coluna de
núcleo fundido gerou análises com 90% da eficiência máxima e da velocidade ótima
e aproximadamente a mesma resolução propiciada pela coluna sub 2 μm, com a

151
grande vantagem de não gerar no sistema pressões acima de 400 bar, e portanto
não requerer a utilização de sistemas de UHPLC, mais caros e menos disponíveis.
Da mesma maneira que a coluna sub 2 μm, a coluna de núcleo fundido foi capaz de
manter elevada eficiência em altas velocidades de análise e de gerar picos de boa
simetria. Além disso, já é possível encontrar no mercado colunas de núcleo fundido
com aproximadamente o mesmo preço que colunas convencionais.

152
REFERÊNCIAS BIBLIOGRÁFICAS
[1] THE KINETIC PLOT METHOD. Disponível em: <http://www.vub.ac.be/CHIS/ Research/ kp.html>. Acesso em: 01 de Abril de 2012. [2] SNYDER, L. R., KIRKLAND, J. J., GLAJCH, J. L. Practical HPLC method development. 2 ed. New York: John Wiley & Sons, 1997. 765 p. [3] HARRIS, D. C. Análise Química Quantitativa. 7 ed. Rio de Janeiro: LTC, 2008. 886 p.
[4] LOUGH, W. J.; WAINER, I. W. High performance liquid chromatography: fundamental principles and practice. London: Black Academic & Professional, 1996. 276 p. [5] THE VAN DEEMTER EQUATION <http://www.chromedia.org/chromedia? waxtrapp=wnjedDsHqnOxmOlIEcCbCmF&caller=pdoxfDsHqnOxmOlIEsGvG&zoekres=yes&scrv=subwnjedDsHqnOxmOlIEcCbCmF>. Acesso em: 11 de dezembro de 2012 [6] NGUYEN, D. T. T.; GUILLARME, D.; RUDAZ, S.; VEUTHEY,J. L. Chromatographic behaviour and comparison of column packed with sub-2μm stationary phases in liquid chromatography. Journal of Chromatography A, v.1128, p. 105-113, 2006. [7] OLAH, E.; FEKETE, S.; FEKETE, J.; GANZLER,K. Comparative study of new shell-type, sub-2μm fully porous and monolith stationary phases, focusing on mass-transfer resistance. Journal of Chromatography A. v.1217, p. 3642-3653, 2010. [8] CABRERA, K. A new generation of silica-based monolithic HPLC columns with improved performance. LC-GC North America. Supllement, p.56-60, 2012. [9] FARIA, A. M.; BOTTOLI, C. B. G.; JARDIM, C. S. F.; COLLINS, C. H. Fases estacionárias monolíticas para separações cromatográficas. Química Nova. v. 29, n. 2, p. 300-309, 2006. [10] BRICE, R. W.; ZHANG, X.; COLÓN, L. A. Fused-core, sub-2μm packings, and monolithic HPLC columns: a comparative evaluation. Journal of separation science. v. 32, p. 2723-2731, 2009. [11] SIOUFFI, A. M. About the C term in the van Deemter’s equation of plate height in monoliths. Journal of Chromatography A. v. 1126, p. 86-94, 2006.
[12] MISTRY, K. Application of Monolithic Columns in High Performance Liquid Chromatography. Journal of Liquid Chromatography & Related Technologies. v. 28, p. 1055-1074, 2005.

153
[13] WU, N.; DEMPSEY, J.; YEHL, P. M.; DOVLETOGLOU, A.; ELLISON, D.;
WYVRATT, J. Analytica Chimica Acta. Practical aspects of fast HPLC separations for
pharmaceutical process development using monolithic columns. v. 523, p. 149-156,
2004.
[14] MCCALLEY, D. V. Comparison of conventional microparticulate and a monolithic reversed-phase column for high-efficiency fast liquid chromatography of basic
compounds. Journal of Chromatography A. v. 965, p. 51–64, 2002.
[15] ABRAHIM, A. A.; MOHAMMAD A.; SKRDLA, P.; BEREZNITSKI, Y.; CHEN, Y.; WU, N. Pratical comparasion of 2,7 µm fused-core silica particles and porous sub-2 µm particles for fast separation in pharmaceutical process development. Journal of Pharmaceutical and Biomedical Analysis, v. 51, p. 131-137, 2010. [16] GUIOCHON, G.; GRITTI, F. Shell particles, trials, tribulations and triumphs. Journal of Chromatography A. v. 1218, p. 1915-1938, 2011. [17] FEKETE, S.; OLAH, E.; FEKETE, J. Fast liquid chromatography: The domination of core–shell and very fine particles. Journal of Chromatography A. v. 1228, p. 57-71, 2012. [18] SWARTZ. M. E. UPLC: An Introduction and Review. Journal of Liquid Chromatography and Related Technologies. v. 28, p. 1253-1263, 2005. [19] DESMET, G.; CLICQ, D.; GZIL, P. Geometry-Independent Plate Hight Representation Methods for the direct comparision of the kinetic performance of LC supports with a different size or morphology. Analytycal Chemistry, v. 77, n. 13, p. 4958-4070, 2005. [20] CABOOTER, D. C.; LESTREMAU, F.; LYNEN, F.; SANDRA, P.; DESMET, G. Kinetic plot as a tool to design coupled column systems producing 100000 theoretical plates in the shortest possible time. Journal of Chromatography A, v. 1212, p. 23-34, 2008. [21] FANIGLIULO, A.; CABOOTER,D.; BELLAZZI, G.; ALLIERI, B.; ROTTIGNIA, A.; DESMET,G. Kinetic performance of reversed-phase C18 high-performance liquid chromatography columns compared by means of the Kinetic Plot Method in pharmaceutically relevant applications. Journal of Chromatography A. v. 1218, p. 3351-3359, 2011. [22] GRUSHKA,E. Chromatographic peak capacity and the factors influencing it. Analytical chemistry, v. 42, n. 11, p. 1142-1147,1970. [23] CABOOTER, D. C.; LESTREMAU, F.; VILLIERS, A.; BROECKHOVEN, K.; LYNEN, F.; SANDRA, P.; DESMET, G. Investigation of the validity of the Kinetic Plot to predict the performance of coupled column systems operated at high pressures under different thermal conditions. Journal of Chromatography A, v. 1216, p. 3895-3903, 2009.

154
[24] LI, J.; CARR, P. W. Accuracy of Empirical Correlations for Estimating Diffusion Coefficients in Aqueous Organic Mixtures. Analytical Chemistry. v. 69, p. 2530-2536, 1997. [25] GRITTI, F.; GUIOCHON, G. Measurement of the eddy diffusion term in chromatographic columns. Application to the first generation of 4.6 mm I.D. monolithic columns. Journal of Chromatography A. v. 1218, p. 5216-5227, 2011. [26] THE KINETIC PLOT ANALYSER. Disponível em: <http://www.vub.ac.be/ CHIS/ Backup %20 Website/ Research/ kp/kp_analyser/KineticPlotAnalyser.html>. Acesso em: 01 de Abril de 2012 [27] BRADLEY, A. C.; WELCH, C. J.; ZHANG, L. Effect of extra-column volume on practical chromatographic parameters of sub-2-μm particle-packed columns in ultra-high pressure liquid chromatography. Journal of separation science. v. 35, p. 2018-2025, 2012. [28] WREN, S. A. C.; TCHELITCHEFF, P. Use of ultra-performance liquid chromatography in pharmaceutical development. Journal of Chromatography A, v. 1119, p. 140-146, 2006. [29] VILLIERS, A. F. L.; SZUCS, R.; GELEBART, S.; DAVID, F.; SANDRA,P. Evaluation of ultra performance liquid chromatography Part I. Possibilities and limitations. Journal of Chromatography A, v.1127, 60-69, 2006. [30] MANCHÓN, N.; D’ARRIGO, M.; GARCÍA-LAFUENTE, A.; GUILLAMÓN,E.; VILLARES, A.; MARTÍNEZ, J. A.; RAMOS, A.; ROSTAGNO, M. A. Comparison of different types of stationary phases for the analysis of soy isoflavones by HPLC. Analytical and Bioanalytical Chemistry. v. 400, p.1251-1261, 2011. [31] PIETROGRANDE, M. C., DONDI, F., CIOGLI, A.; GASPARRINI, F.; PICCINC, A.; SERAFINIC, M. Characterization of new types of stationary phases for fast and ultra-fast liquid chromatography by signal processing based on auto covariance Function: A case study of application to Passiflora incarnata L. extract separations. Journal of Chromatography A. v.1217, p. 4355-4364, 2010. [32] UNGER, K. K.; SKUDAS, R.; SCHULTE, M. M. Particle packed columns and monolithic columns in high-performance liquid chromatography-comparison and critical appraisal. Journal of Chromatography A. v. 1184, p. 393-415, 2008. [33] PATNAIK, P. Handbook of environmental analysis. 2 ed. New York: CRC press,1997.

155
CAPÍTULO 5
DESENVOLVIMENTO DE MÉTODOS ANALÍTICOS
EXTREMAMENTE RÁPIDOS

156
1 REVISÃO BIBLIOGRÁFICA
1.1 Métodos analíticos extremamente rápidos
Há uma crescente demanda para aumentar a produtividade e reduzir os custos em
análise farmacêutica. Recentemente, novas colunas cromatográficas (monolítica,
empacotada com partículas de núcleo fundido e empacotada com partículas porosas
de diâmetro inferior a 2 μm), novas geometrias (menores comprimento e diâmetro
interno), novas abordagens (como o uso de temperatura elevada - HTLC) e novos
instrumentos (UHPLC) foram introduzidos no mercado de modo a reduzir o tempo de
análise mantendo a eficiência cromatográfica [1].
Associado às novas alternativas, o aumento da vazão da fase móvel é uma maneira
simples de reduzir o tempo de análise. Entretanto, com o aumento da vazão tem-se
elevação da pressão e, portanto, para se estabelecer a vazão máxima à qual uma
coluna pode ser submetida deve-se considerar a pressão máxima que ela suporta.
[1, 2].
As colunas convencional com partículas totalmente porosas (CV), monolítica (MN),
empacotada com partículas de núcleo fundido (NF) e empacotada com partículas
porosas de diâmetro inferior a 2 μm (Sub 2 μm), podem ser submetidas a diferentes
valores máximos de pressão. Normalmente, as colunas monolíticas de sílica
suportam uma pressão máxima de 200 bar, as empacotadas com partículas de
núcleo fundido resistem a uma pressão de até 600 bar e as empacotadas com
partículas totalmente porosas de diâmetro inferior a 2 μm podem ser submetidas a
valores de pressão em torno de 1000 bar [3]. Colunas convencionais, empacotadas
com partículas totalmente porosas de diâmetro entre 3 e 5 μm suportam pressão
máxima de aproximadamente 275 bar [4].
Em função da demanda para aumentar a produtividade em análise farmacêutica, há
trabalhos na literatura que descrevem o desenvolvimento de análises extremamente
rápidas utilizando novas colunas cromatográficas. Em um deles métodos rápidos
para análise de formulação de lidocaína foram desenvolvidos e validados em coluna
monolítica e em coluna empacotada com partículas sub 2 μm. Em seguida,
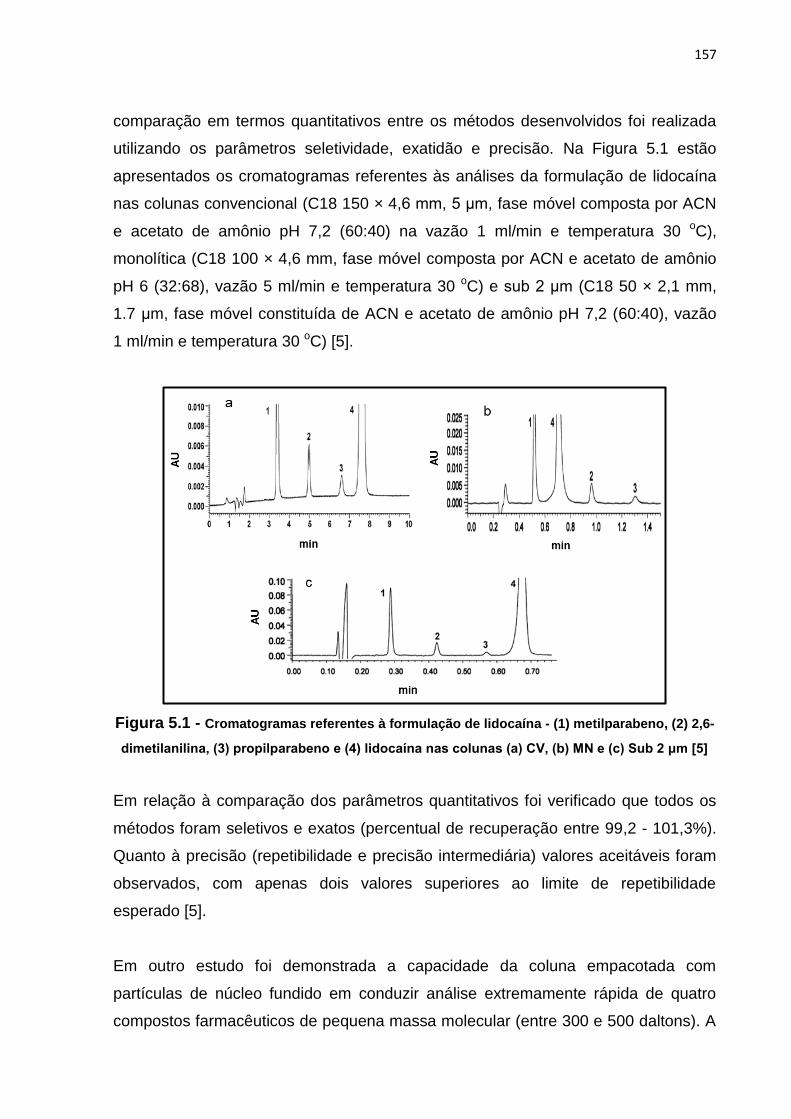
157
comparação em termos quantitativos entre os métodos desenvolvidos foi realizada
utilizando os parâmetros seletividade, exatidão e precisão. Na Figura 5.1 estão
apresentados os cromatogramas referentes às análises da formulação de lidocaína
nas colunas convencional (C18 150 × 4,6 mm, 5 μm, fase móvel composta por ACN
e acetato de amônio pH 7,2 (60:40) na vazão 1 ml/min e temperatura 30 oC),
monolítica (C18 100 × 4,6 mm, fase móvel composta por ACN e acetato de amônio
pH 6 (32:68), vazão 5 ml/min e temperatura 30 oC) e sub 2 μm (C18 50 × 2,1 mm,
1.7 μm, fase móvel constituída de ACN e acetato de amônio pH 7,2 (60:40), vazão
1 ml/min e temperatura 30 oC) [5].
Figura 5.1 - Cromatogramas referentes à formulação de lidocaína - (1) metilparabeno, (2) 2,6-
dimetilanilina, (3) propilparabeno e (4) lidocaína nas colunas (a) CV, (b) MN e (c) Sub 2 μm [5]
Em relação à comparação dos parâmetros quantitativos foi verificado que todos os
métodos foram seletivos e exatos (percentual de recuperação entre 99,2 - 101,3%).
Quanto à precisão (repetibilidade e precisão intermediária) valores aceitáveis foram
observados, com apenas dois valores superiores ao limite de repetibilidade
esperado [5].
Em outro estudo foi demonstrada a capacidade da coluna empacotada com
partículas de núcleo fundido em conduzir análise extremamente rápida de quatro
compostos farmacêuticos de pequena massa molecular (entre 300 e 500 daltons). A
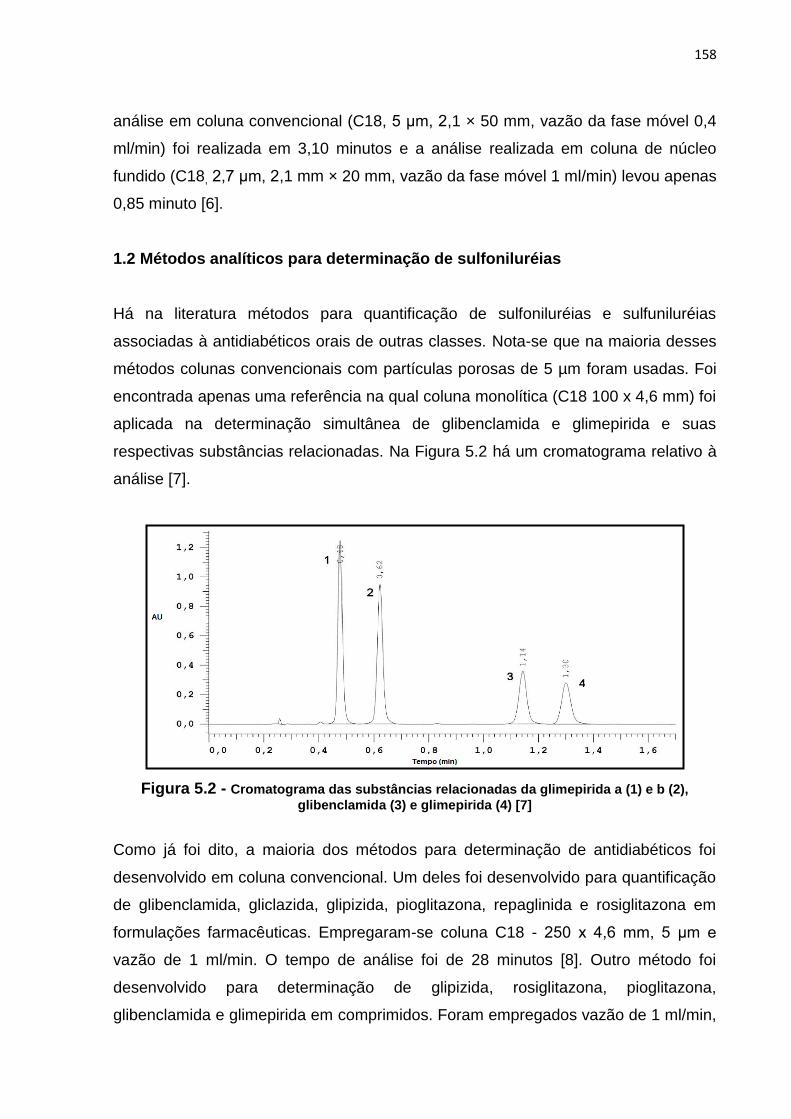
158
análise em coluna convencional (C18, 5 μm, 2,1 × 50 mm, vazão da fase móvel 0,4
ml/min) foi realizada em 3,10 minutos e a análise realizada em coluna de núcleo
fundido (C18, 2,7 μm, 2,1 mm × 20 mm, vazão da fase móvel 1 ml/min) levou apenas
0,85 minuto [6].
1.2 Métodos analíticos para determinação de sulfoniluréias
Há na literatura métodos para quantificação de sulfoniluréias e sulfuniluréias
associadas à antidiabéticos orais de outras classes. Nota-se que na maioria desses
métodos colunas convencionais com partículas porosas de 5 µm foram usadas. Foi
encontrada apenas uma referência na qual coluna monolítica (C18 100 x 4,6 mm) foi
aplicada na determinação simultânea de glibenclamida e glimepirida e suas
respectivas substâncias relacionadas. Na Figura 5.2 há um cromatograma relativo à
análise [7].
Figura 5.2 - Cromatograma das substâncias relacionadas da glimepirida a (1) e b (2),
glibenclamida (3) e glimepirida (4) [7]
Como já foi dito, a maioria dos métodos para determinação de antidiabéticos foi
desenvolvido em coluna convencional. Um deles foi desenvolvido para quantificação
de glibenclamida, gliclazida, glipizida, pioglitazona, repaglinida e rosiglitazona em
formulações farmacêuticas. Empregaram-se coluna C18 - 250 x 4,6 mm, 5 μm e
vazão de 1 ml/min. O tempo de análise foi de 28 minutos [8]. Outro método foi
desenvolvido para determinação de glipizida, rosiglitazona, pioglitazona,
glibenclamida e glimepirida em comprimidos. Foram empregados vazão de 1 ml/min,

159
fase móvel composta por solução de trietanolamina 0,05% v/v (pH 3,5, ajustado com
ácido ortofosfórico), acetonitrila e metanol, na proporção de 55:15:30, coluna C18 -
150 × 4,6 mm, 5 μm. Os fármacos foram separados em 20 minutos [9].
Não foi encontrado na literatura nenhum método para quantificação simultânea de
clorpropamida, glibenclamida, glimepirida e glicazida em coluna convencional ou em
colunas que permitem análises mais rápidas (monolítica, com partículas de núcleo
fundido ou com partículas totalmente porosas Sub 2 µm).
2 OBJETIVOS ESPECÍFICOS
Foram objetivos dessa etapa do trabalho:
1- Desenvolver e comparar métodos analíticos extremamente rápidos para
quantificação dos antidiabéticos orais clorpropamida (CL), glibenclamida (GB),
glimepirida (GM) e gliclazida (GZ), utilizando as colunas CV, MN, NF e Sub 2 μm.
2- Validar os métodos analíticos extremamente rápidos desenvolvidos em coluna CV
e de NF e comparar essas colunas cromatográficas em termos quantitativos,
verificando a capacidade de originarem métodos confiáveis.
3 MATERIAIS E MÉTODOS
3.1 Materiais
3.1.1 Insumos farmacêuticos ativos
Os mesmos descritos no capítulo 1, item 3.1.1.
3.1.2 Solventes, reagentes e vidrarias
Os mesmos descritos no capítulo 1, item 3.1.2.

160
3.1.3 Equipamentos e colunas cromatográficas
Os mesmos descritos no capítulo 1, item 3.1.3.
3.1.4 Substâncias químicas de referência e amostras
Glibenclamida SQR (Farmacopeia Brasileira, lote 1018) e Daonil® comprimidos
(5 mg de glibenclamida, lote 139672, validade 09/201, Sanofi Aventis – São Paulo,
Brasil)
3.2 Métodos
3.2.1 Desenvolvimento e comparação de métodos analíticos extremamente rápidos
para determinação de antidiabéticos nas colunas CV, MN, NF e Sub 2 μm
O desenvolvimento de métodos analíticos extremamente rápidos para quantificação
de antidiabéticos orais (métodos extremos) em cada coluna foi realizado a partir dos
métodos previamente desenvolvidos e descritos no capítulo 1, e utilizando-se os
parâmetros instrumentais cromatográficos otimizados no capítulo 3 (Tabela 5.1).
Tabela 5.1 - Condições de análise para determinação de antidiabéticos nas diferentes
colunas*
* Parâmetros instrumentais: loop de 2 μl, tubo de PEEK de 0,004 polegada e frequência de aquisição dos dados e constante de tempo do detector iguais a 40 Hz/0,025 s ** Solução de acetato de amônio 5 mM pH 3,0
Para desenvolver os métodos extremos a partir dos métodos apresentados na
Tabela 5.1, as etapas 1 e 2 descritas a seguir foram realizadas em cada coluna.
Colunas Fase Móvel Vinj (μl) (nm) T (oC) Conc. (μg/ml)
Convencional ACN: Solução (50:50)** 2 230 30 100
Monolítica ACN: Solução (43:57)** 2 230 30 100
Núcleo Fundido ACN: Solução (50:50)** 2 230 30 100
Sub 2 μm ACN: Solução (53:47)** 2 230 30 100

161
Etapa 1 - Determinação da vazão máxima
Análises de antidiabéticos foram conduzidas em cada coluna utilizando-se os
respectivos parâmetros analíticos descritos na Tabela 5.1. A vazão da fase móvel foi
aumentada de forma progressiva até que a pressão máxima suportada por cada
coluna (Tabela 5.2) ou que resolução mínima próxima a 2,5 entre os picos referentes
à glibenclamida e glimepirida (picos adjacentes mais próximos) fosse atingida. Se a
pressão máxima foi atingida, mas a resolução mínima não, prosseguiu-se com a
etapa 2. Se a resolução mínima ou ambas as condições foram alcançadas concluiu-
se que o método extremo relativo à coluna em teste apresentava as condições de
análise descritas na Tabela 5.1 e a maior vazão testada.
Tabela 5.2 - Pressão máxima suportada pelas diferentes colunas
Colunas Pressão máxima (bar)
Convencional 275
Monolítica 200
Núcleo Fundido 400 *
Sub 2 μm 1034**
* Apesar da coluna com partículas de núcleo fundido suportar 600 bar, o valor limite será de 400 bar, já que a
vantagem de sua utilização está no fato de ela propiciar uma boa eficiência cromatográfica sem a necessidade da utilização de UHPLC. ** Apesar da coluna com partículas sub 2 μm suportar 1200 bar, o valor limite considerado será de 1034 bar, valor máximo suportado pelo equipamento UPLC Acquity
® utilizado.
Etapa 2 - Determinação da composição da fase móvel
Análises de antidiabéticos foram conduzidas em cada coluna utilizando-se os
respectivos parâmetros analíticos descritos na Tabela 5.1 e a vazão selecionada na
etapa 1. Aumentou-se progressivamente o percentual de acetonitrila da fase móvel
até que resolução mínima próxima a 2,5 entre os picos da glibenclamida e
glimepirida fosse atingida. Concluiu-se que o método extremo relativo à coluna em
teste apresentava a vazão máxima determinada na etapa 1, a composição da fase
móvel determinada na etapa 2 e o restante das condições de análise conforme
descrito na Tabela 5.1.

162
Após serem desenvolvidos, os métodos analíticos extremos referentes a cada
coluna foram comparados em termos de velocidade de análise e eficiência. Para
tanto foram utilizados como parâmetros o tempo de retenção do analito mais retido
(glimepirida) e as alturas de prato teórico (H) dos picos relativos aos antidiabéticos.
3.2.2 Comparação das colunas CV e NF em termos quantitativos
Além de avaliar as colunas cromatográficas em termos qualitativos, faz-se
necessário também avaliar o impacto dessas novas colunas em termos
quantitativos. Avaliações qualitativas já foram abordadas nos capítulos 2 e 4, e
dizem respeito aos parâmetros seletividade, fator de retenção, pressão, eficiência,
resolução, simetria, tempo de análise. Já as avaliações quantitativas têm por
objetivo comparar a capacidade das novas colunas em dar origem a métodos
analíticos que assegurem a confiabilidade dos resultados.
Para realização da avaliação quantitativa foram validados os métodos em condições
extremas nas colunas convencional e de núcleo fundido, entretanto apenas a
glibenclamida foi utilizada como modelo nos procedimentos experimentais de
validação. Os resultados da validação do método extremo desenvolvido em coluna
convencional serviram de base para avaliação dos resultados da validação do
método extremo em coluna de núcleo fundido.
Para a realização das validações foi necessário determinar os parâmetros
seletividade, adequabilidade do sistema, estabilidade das soluções, linearidade,
precisão, exatidão e robustez, de acordo com as normas da RE 899/2003 da
ANVISA e outras referências complementares. Esses parâmetros foram
determinados nas colunas convencional e de núcleo fundido, utilizando os
respectivos métodos extremos desenvolvidos.

163
3.2.2.1 Seletividade e adequabilidade do sistema (system suitability)
As soluções a seguir foram preparadas e injetadas no cromatógrafo em triplicata,
exceto a solução padrão de trabalho, que foi injetada cinco vezes para avaliação da
adequabilidade do sistema.
- Diluente: ACN:solução de acetato de amônio 5 mM pH 3,0 (1:1)
- Solução padrão de trabalho (100 μg/ml): Pesaram-se exatamente cerca de 25,0 mg
de glibenclamida SQR em balão volumétrico de 25 ml. Adicionaram-se cerca de 20
ml de acetonitrila:solução de acetato de amônio (4:1) e o balão foi levado para
banho de ultrassom por 5 minutos. O volume foi completado com ACN:solução de
acetato de amônio (4:1). Pipetou-se 1 ml dessa solução para balão de 10 ml e o
volume foi completado com acetonitrila:solução de acetato de amônio (1:1).
- Solução amostra de comprimido de glibenclamida (100 μg/ml): O peso médio de 20
comprimidos foi determinado e em seguida os comprimidos foram triturados até
formarem pó. Pesou-se quantidade de pó correspondente a 1 peso médio,
equivalente a 5 mg de glibenclamida, e transferiu-se para balão volumétrico de 25
ml. Foram adicionados cerca de 20 ml de ACN:solução de acetato de amônio (4:1) e
o balão foi levado para banho de ultrassom por 30 minutos. Em seguida o volume do
balão foi completado com ACN:solução de acetato de amônio (4:1). A solução foi
filtrada, descartando-se os primeiros 5 ml. Pipetaram-se 5 ml do filtrado para balão
volumétrico de 10 ml e o volume foi completado com ACN:solução de acetato de
amônio (1:4).
- Solução placebo de comprimido de glibenclamida: Pesaram-se cerca de 157,0 mg
de placebo (Tabela 5.3) em balão volumétrico de 25 ml. Adicionaram-se cerca de 20
ml de ACN:solução de acetato de amônio (4:1) e o balão foi levado para banho de
ultrassom por 30 minutos. Em seguida o volume do balão foi completado com
ACN:solução de acetato de amônio (4:1). A solução foi filtrada e os primeiros 5 ml
descartados. Pipetaram-se 5 ml do filtrado para balão volumétrico de 10 ml e o
volume foi completado com acetonitrila:solução de acetato de amônio (1:4).
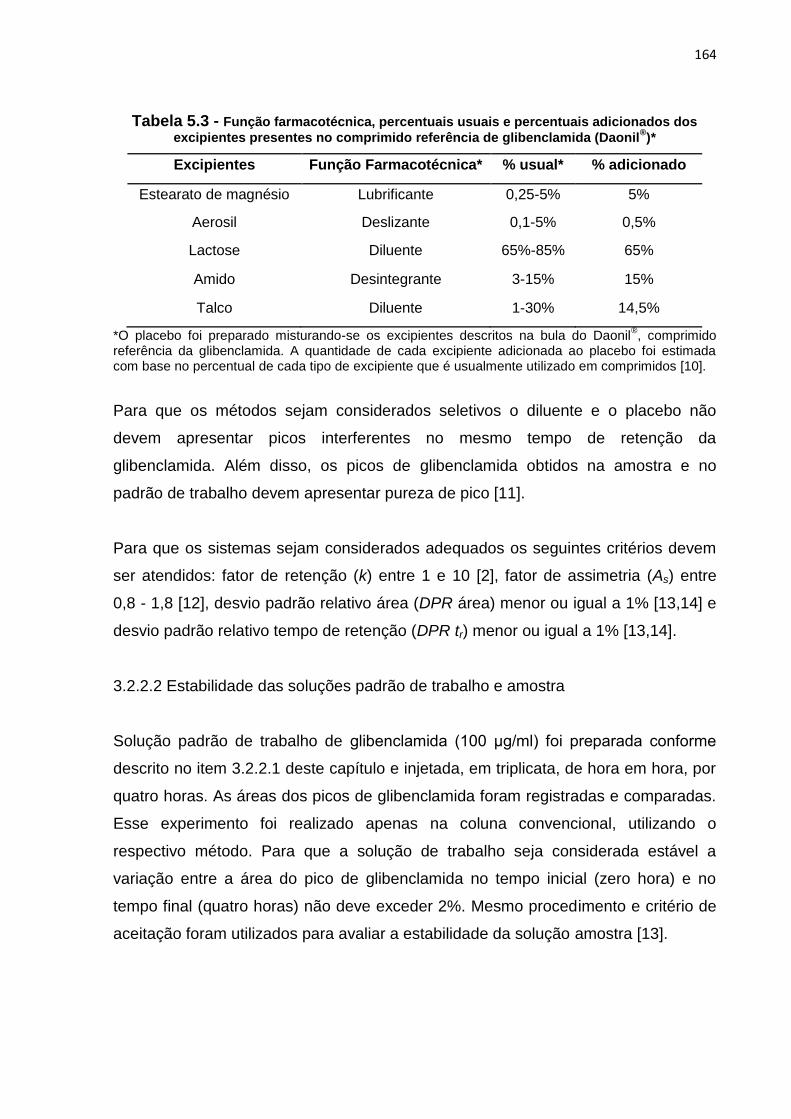
164
Tabela 5.3 - Função farmacotécnica, percentuais usuais e percentuais adicionados dos
excipientes presentes no comprimido referência de glibenclamida (Daonil®)*
Excipientes Função Farmacotécnica* % usual* % adicionado
Estearato de magnésio Lubrificante 0,25-5% 5%
Aerosil Deslizante 0,1-5% 0,5%
Lactose Diluente 65%-85% 65%
Amido Desintegrante 3-15% 15%
Talco Diluente 1-30% 14,5%
*O placebo foi preparado misturando-se os excipientes descritos na bula do Daonil®, comprimido
referência da glibenclamida. A quantidade de cada excipiente adicionada ao placebo foi estimada com base no percentual de cada tipo de excipiente que é usualmente utilizado em comprimidos [10].
Para que os métodos sejam considerados seletivos o diluente e o placebo não
devem apresentar picos interferentes no mesmo tempo de retenção da
glibenclamida. Além disso, os picos de glibenclamida obtidos na amostra e no
padrão de trabalho devem apresentar pureza de pico [11].
Para que os sistemas sejam considerados adequados os seguintes critérios devem
ser atendidos: fator de retenção (k) entre 1 e 10 [2], fator de assimetria (As) entre
0,8 - 1,8 [12], desvio padrão relativo área (DPR área) menor ou igual a 1% [13,14] e
desvio padrão relativo tempo de retenção (DPR tr) menor ou igual a 1% [13,14].
3.2.2.2 Estabilidade das soluções padrão de trabalho e amostra
Solução padrão de trabalho de glibenclamida (100 μg/ml) foi preparada conforme
descrito no item 3.2.2.1 deste capítulo e injetada, em triplicata, de hora em hora, por
quatro horas. As áreas dos picos de glibenclamida foram registradas e comparadas.
Esse experimento foi realizado apenas na coluna convencional, utilizando o
respectivo método. Para que a solução de trabalho seja considerada estável a
variação entre a área do pico de glibenclamida no tempo inicial (zero hora) e no
tempo final (quatro horas) não deve exceder 2%. Mesmo procedimento e critério de
aceitação foram utilizados para avaliar a estabilidade da solução amostra [13].

165
3.2.2.3 Linearidade
Foram preparadas, em triplicata, soluções estoque de glibenclamida a 1 mg/ml, em
acetonitrila:solução de acetato de amônio (4:1). A partir de cada solução estoque,
foram feitas diluições para cinco diferentes concentrações, conforme Tabela 5.4,
totalizando 15 soluções. Cada solução foi injetada em triplicata.
Tabela 5.4 - Preparo das soluções de glibenclamida para avaliação da linearidade
*Em relação à concentração de trabalho de 100 μg/ml
As curvas analíticas foram construídas com os valores de área dos picos de
glibenclamida obtidos em cada nível de concentração. Os dados foram submetidos
aos seguintes testes estatísticos: outlier (teste do resíduo padronizado Jacknife),
normalidade dos resíduos (teste de Ryan-Joiner), independência dos resíduos (teste
de Durbin-Watson), homoscedasticidade dos resíduos (teste de Brown-Forsythe ou
Levene modificado). Após a aplicação dos testes, as regressões lineares foram
calculadas pelo método dos mínimos quadrados e foram construídos os gráficos de
distribuição de resíduos. Ao final, avaliação da significância das regressões foram
realizadas por meio da aplicação do teste de análise de variância (ANOVA). Todos
os testes estatísticos foram realizados com auxílio de planilha desenvolvida por
Souza e colaboradores [15].
Para que haja linearidade a regressão deve ser estatisticamente significativa, o
coeficiente de correlação (r) deve ser superior a 0,99 e o gráfico de resíduos não
deve apresentar nenhuma tendência [11, 14].
Nível de concentração*
Volume de solução estoque (ml)
ACN:Solução (1:1) q.s.p. (ml)
Concentração (μg/ml)
60% 1,5 25,0 60
80% 2,0 25,0 80
100% 2,5 25,0 100
120% 3,0 25,0 120
140% 3,5 25,0 140

166
3.2.2.4 Precisão intra-corrida e precisão inter-corridas
A precisão intra-corrida (repetibilidade) foi avaliada por meio de seis determinações
a 100% da concentração de trabalho (100 μg/ml de glibenclamida). As soluções
amostra para a análise foram preparadas conforme procedimento descrito no item
3.2.2.1 e injetadas três vezes. A precisão inter-corridas (precisão intermediária) foi
realizada pela repetição deste procedimento em outro dia. Os resultados foram
agrupados e calcularam-se os teores de glibenclamida e os desvios padrões
relativos (DPR) das seis determinações do primeiro dia e das 12 determinações
realizadas no primeiro e segundo dias. Para que os métodos apresentem precisão
intra e inter-corridas o DPR entre as seis e 12 determinações não deve exceder 2%
[11, 13, 14].
3.2.2.5 Exatidão
A determinação da exatidão foi realizada em três níveis de concentração, com a
adição de quantidades conhecidas de glibenclamida SQR ao placebo do comprimido
de referência de glibenclamida (Daonil®). O placebo foi preparado conforme Tabela
5.3.
Foram preparadas três soluções padrão estoque de glibenclamida, na concentração
de 1 mg/ml, em acetonitrila:solução de acetato de amônio 5 mM pH 3,0 (4:1). A partir
de cada solução padrão estoque, foram preparadas três soluções com
concentrações equivalentes a 80%, 100% e 120% da concentração de trabalho de
glibenclamida (100 μg/ml). Todas as soluções foram fortificadas com 157,0 mg de
placebo (massa correspondente à quantidade de excipientes na amostra na
concentração de trabalho do doseamento), e submetidas ao procedimento de
preparo da amostra de comprimido de glibenclamida, descrito no item 3.2.2.1. Cada
uma das nove soluções foi injetada três vezes. Na Tabela 5.5 há um resumo de
como foram preparadas as soluções para o ensaio de exatidão.

167
Tabela 5.5 - Preparo das soluções para o ensaio de exatidão
Níveis de concentração
Massa de placebo (mg)
Volume de solução estoque de padrão (ml)
Conc. Glibenclamida
(µg/ml)
Exatidão 80% 157,0 4 80
Exatidão 100% 157,0 5 100
Exatidão 120% 157,0 6 120
A exatidão foi calculada pela relação entre a concentração obtida experimentalmente
e a concentração teórica de glibenclamida nas soluções fortificadas com o placebo.
Para que os métodos sejam considerados exatos o percentual de recuperação deve
estar entre 98% - 102% [13].
3.2.2.6 Limites de detecção e quantificação
Para determinação dos limites de quantificação e detecção foram realizadas
diluições seriadas a partir de uma solução padrão de trabalho de glibenclamida (100
μg/ml), preparada de acordo com o item 3.2.2.1. Cada solução foi injetada três
vezes. Determinou-se o desvio padrão relativo (DPR) entre as áreas dos picos, bem
como a relação sinal/ruído (S/R).
O limite de detecção de cada método foi estimado como sendo igual a concentração
da solução que produziu S/R = 3. Já o limite de quantificação foi considerado como
sendo igual a concentração da solução que deu origem a um pico de S/R = 10 e a
áreas reprodutíveis (DPR ≤ 2% entre as áreas de três injeções) [11].
3.2.2.7 Robustez
A robustez dos métodos foi avaliada variando-se deliberadamente algumas
condições cromatográficas e verificando-se a influência das modificações nos
resultados do doseamento de glibenclamida em comprimido. Em cada condição
modificada foram realizadas injeções do padrão (cinco) e da amostra (três). Os
parâmetros foram variados de acordo com o método estatístico de Youden e Steiner
[16]. As variáveis modificadas, bem como a distribuição dessas variáveis nos
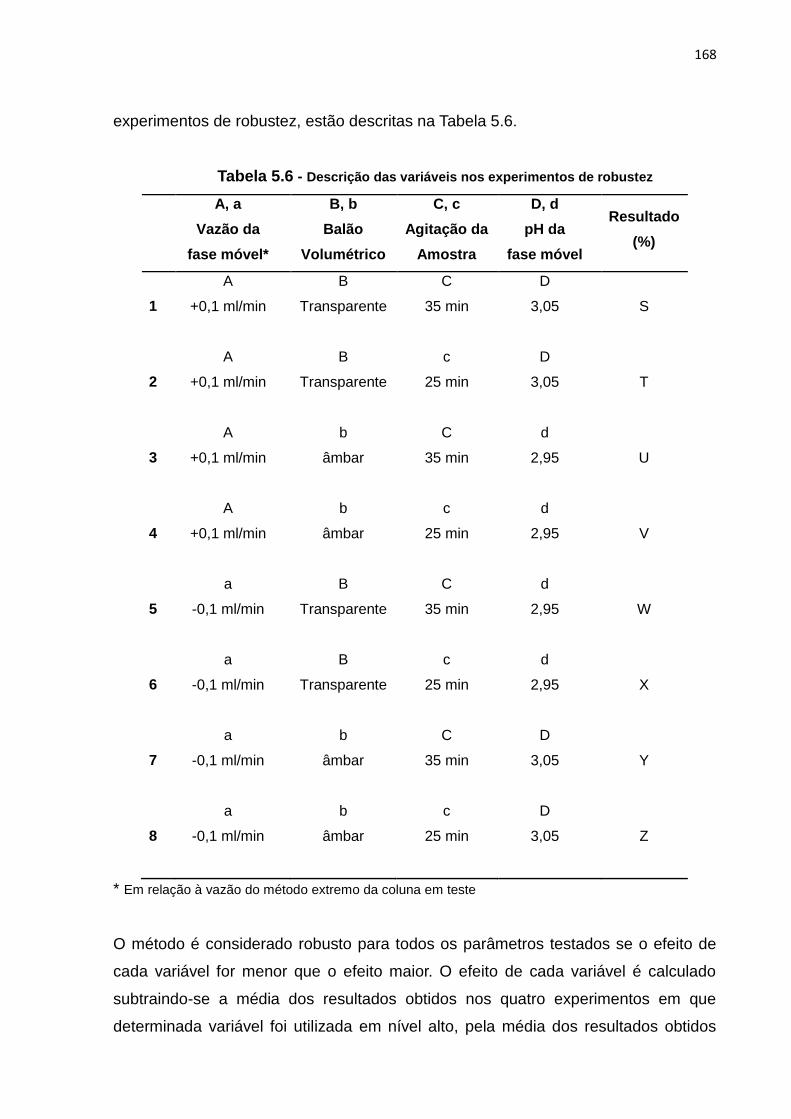
168
experimentos de robustez, estão descritas na Tabela 5.6.
Tabela 5.6 - Descrição das variáveis nos experimentos de robustez
A, a
Vazão da
fase móvel*
B, b
Balão
Volumétrico
C, c
Agitação da
Amostra
D, d
pH da
fase móvel
Resultado
(%)
1
A
+0,1 ml/min
B
Transparente
C
35 min
D
3,05 S
2
A
+0,1 ml/min
B
Transparente
c
25 min
D
3,05
T
3
A
+0,1 ml/min
b
âmbar
C
35 min
d
2,95
U
4
A
+0,1 ml/min
b
âmbar
c
25 min
d
2,95
V
5
a
-0,1 ml/min
B
Transparente
C
35 min
d
2,95
W
6
a
-0,1 ml/min
B
Transparente
c
25 min
d
2,95
X
7
a
-0,1 ml/min
b
âmbar
C
35 min
D
3,05
Y
8
a
-0,1 ml/min
b
âmbar
c
25 min
D
3,05
Z
* Em relação à vazão do método extremo da coluna em teste
O método é considerado robusto para todos os parâmetros testados se o efeito de
cada variável for menor que o efeito maior. O efeito de cada variável é calculado
subtraindo-se a média dos resultados obtidos nos quatro experimentos em que
determinada variável foi utilizada em nível alto, pela média dos resultados obtidos

169
nos quatro experimentos em que a mesma variável foi utilizada em nível baixo,
conforme demonstrado na equação 5.1 [16].
Efeito da variável A/a = (S + T + U + V)/4 – (W+X+Y+Z)/4 (5.1)
Já o cálculo do efeito maior é realizado multiplicando-se o desvio padrão relativo
entre os valores de teor obtidos nos oito experimentos por raiz de dois.
4 RESULTADOS E DISCUSSÃO
4.1 Desenvolvimento e comparação de métodos analíticos extremamente
rápidos para determinação de antidiabéticos nas colunas CV, MN, NF e Sub 2
μm
Na Tabela 5.7 estão descritas as condições analíticas dos métodos extremamente
rápidos desenvolvidos em cada coluna.
Tabela 5.7 - Condições analíticas para determinação extremamente rápida de antidiabéticos
nas diferentes colunas*
Colunas Fase Móvel Vazão
(ml/min)
Vinj
(μl)
(nm)
T
(o C)
Conc.
(μg/ml)
Convencional ACN: Solução (50:50)** 1,0 2 230 30 100
Monolítica ACN: Solução (50:50)** 1,2 2 230 30 100
Núcleo Fundido ACN: Solução (65:35)** 0,6 2 230 30 100
Sub 2 μm ACN: Solução (65:35)** 1,2 2 230 30 100
* Parâmetros instrumentais: loop de 2 μl, tubo de PEEK de 0,004 polegada e frequência de aquisição dos dados e constante de tempo do detector iguais a 40 Hz/0,025 s ** Solução de acetato de amônio 5 mM pH 3,0
Analisando-se a Tabela 5.7 verifica-se que foi possível desenvolver um método
extremo em coluna empacotada com partículas sub 2 μm que apresentou elevado
valor de vazão e alto percentual de acetonitrila na fase móvel. Isso provavelmente foi
possível porque a coluna com partículas sub 2 μm resiste mecanicamente ao
aumento de pressão provocado pelo aumento da vazão (suporta pressão de até
1200 bar) e possui elevado poder de resolução, o que permite que haja resolução
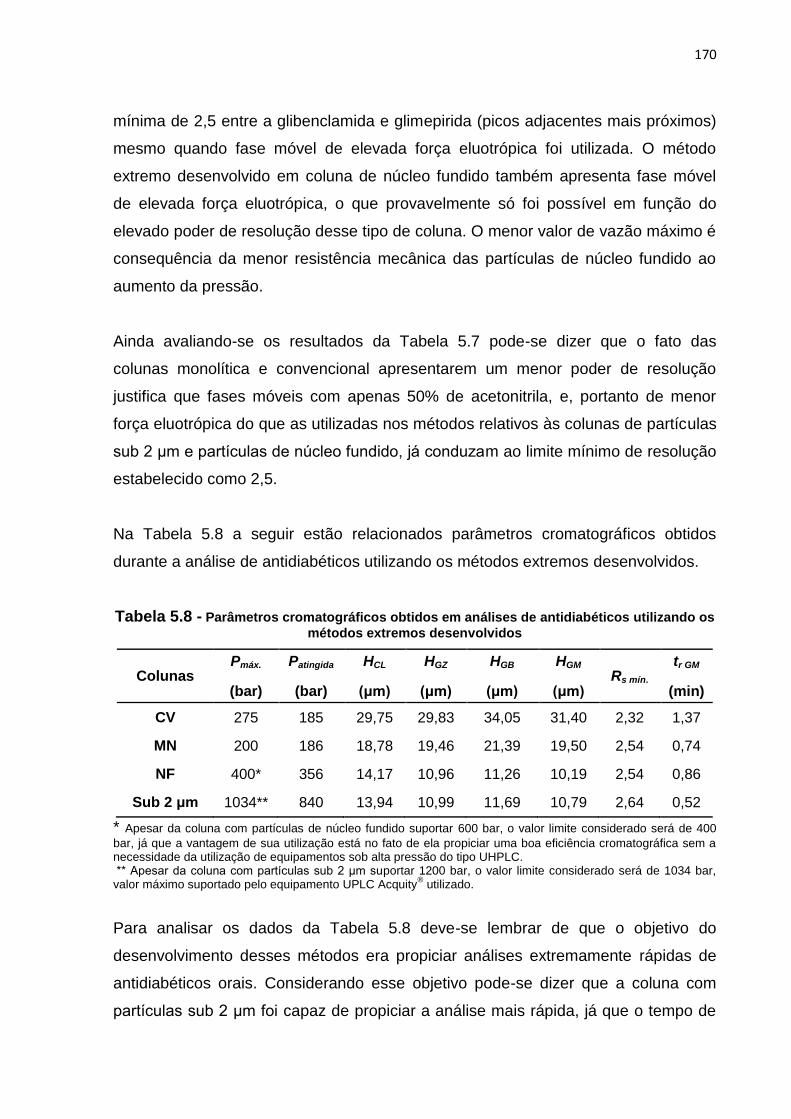
170
mínima de 2,5 entre a glibenclamida e glimepirida (picos adjacentes mais próximos)
mesmo quando fase móvel de elevada força eluotrópica foi utilizada. O método
extremo desenvolvido em coluna de núcleo fundido também apresenta fase móvel
de elevada força eluotrópica, o que provavelmente só foi possível em função do
elevado poder de resolução desse tipo de coluna. O menor valor de vazão máximo é
consequência da menor resistência mecânica das partículas de núcleo fundido ao
aumento da pressão.
Ainda avaliando-se os resultados da Tabela 5.7 pode-se dizer que o fato das
colunas monolítica e convencional apresentarem um menor poder de resolução
justifica que fases móveis com apenas 50% de acetonitrila, e, portanto de menor
força eluotrópica do que as utilizadas nos métodos relativos às colunas de partículas
sub 2 μm e partículas de núcleo fundido, já conduzam ao limite mínimo de resolução
estabelecido como 2,5.
Na Tabela 5.8 a seguir estão relacionados parâmetros cromatográficos obtidos
durante a análise de antidiabéticos utilizando os métodos extremos desenvolvidos.
Tabela 5.8 - Parâmetros cromatográficos obtidos em análises de antidiabéticos utilizando os
métodos extremos desenvolvidos
Colunas Pmáx.
(bar)
Patingida
(bar)
HCL
(μm)
HGZ
(μm)
HGB
(μm)
HGM
(μm) Rs mín.
tr GM
(min)
CV 275 185 29,75 29,83 34,05 31,40 2,32 1,37
MN 200 186 18,78 19,46 21,39 19,50 2,54 0,74
NF 400* 356 14,17 10,96 11,26 10,19 2,54 0,86
Sub 2 μm 1034** 840 13,94 10,99 11,69 10,79 2,64 0,52
* Apesar da coluna com partículas de núcleo fundido suportar 600 bar, o valor limite considerado será de 400
bar, já que a vantagem de sua utilização está no fato de ela propiciar uma boa eficiência cromatográfica sem a necessidade da utilização de equipamentos sob alta pressão do tipo UHPLC. ** Apesar da coluna com partículas sub 2 μm suportar 1200 bar, o valor limite considerado será de 1034 bar, valor máximo suportado pelo equipamento UPLC Acquity
® utilizado.
Para analisar os dados da Tabela 5.8 deve-se lembrar de que o objetivo do
desenvolvimento desses métodos era propiciar análises extremamente rápidas de
antidiabéticos orais. Considerando esse objetivo pode-se dizer que a coluna com
partículas sub 2 μm foi capaz de propiciar a análise mais rápida, já que o tempo de
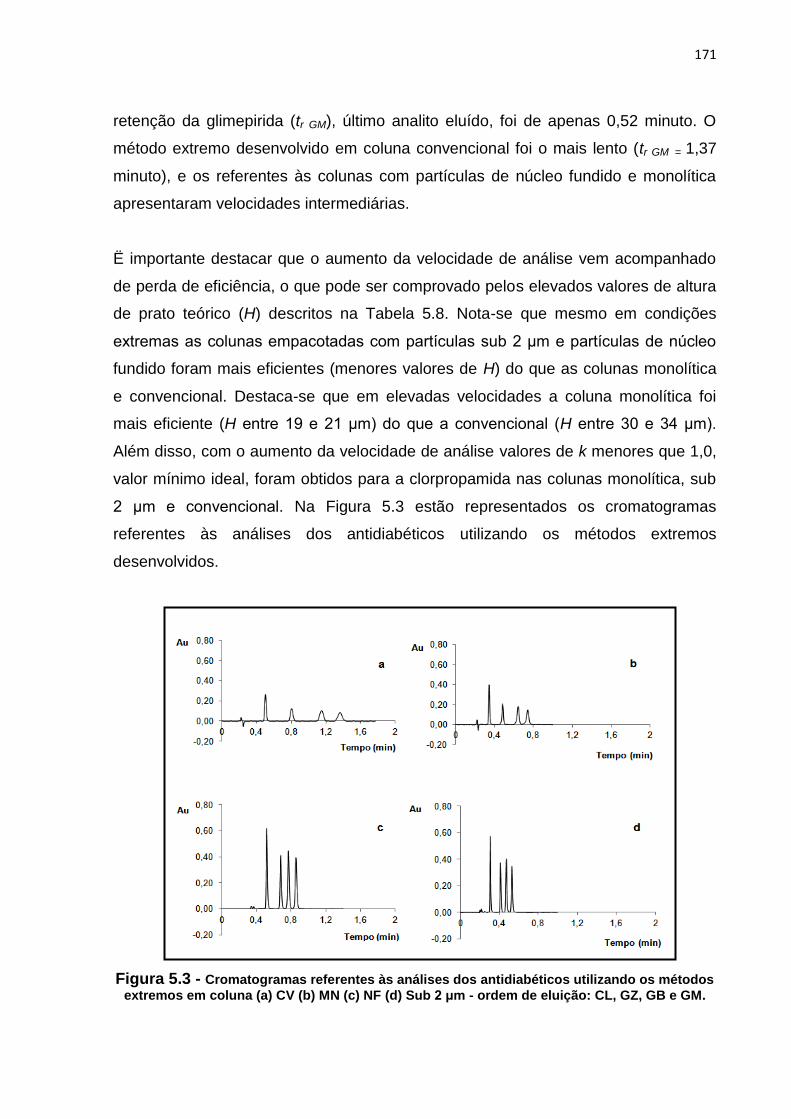
171
retenção da glimepirida (tr GM), último analito eluído, foi de apenas 0,52 minuto. O
método extremo desenvolvido em coluna convencional foi o mais lento (tr GM = 1,37
minuto), e os referentes às colunas com partículas de núcleo fundido e monolítica
apresentaram velocidades intermediárias.
Ë importante destacar que o aumento da velocidade de análise vem acompanhado
de perda de eficiência, o que pode ser comprovado pelos elevados valores de altura
de prato teórico (H) descritos na Tabela 5.8. Nota-se que mesmo em condições
extremas as colunas empacotadas com partículas sub 2 μm e partículas de núcleo
fundido foram mais eficientes (menores valores de H) do que as colunas monolítica
e convencional. Destaca-se que em elevadas velocidades a coluna monolítica foi
mais eficiente (H entre 19 e 21 μm) do que a convencional (H entre 30 e 34 μm).
Além disso, com o aumento da velocidade de análise valores de k menores que 1,0,
valor mínimo ideal, foram obtidos para a clorpropamida nas colunas monolítica, sub
2 μm e convencional. Na Figura 5.3 estão representados os cromatogramas
referentes às análises dos antidiabéticos utilizando os métodos extremos
desenvolvidos.
Figura 5.3 - Cromatogramas referentes às análises dos antidiabéticos utilizando os métodos
extremos em coluna (a) CV (b) MN (c) NF (d) Sub 2 μm - ordem de eluição: CL, GZ, GB e GM.

172
Analisando-se os cromatogramas nota-se que os picos gerados em análises
realizadas nas colunas de núcleo fundido e sub 2 μm (Figura 5.3 c e d) são mais
altos, indicando a capacidade dessas colunas em propiciar métodos com maior
detectabilidade do que as colunas convencional e monolítica (Figura 5.3 a e b).
4.2 Comparação das colunas CV e NF em termos quantitativos
A fim de facilitar a execução experimental, a validação foi realizada apenas para a
glibenclamida. O método extremo desenvolvido em coluna de núcleo fundido foi
selecionado para ser validado e em paralelo, para comparação, realizou-se
validação em coluna convencional.
4.2.1 Seletividade e adequabilidade do sistema (system suitability)
Os métodos desenvolvidos nas colunas convencional e de núcleo fundido foram
seletivos, o que significa que o diluente e o placebo não apresentaram picos
interferentes no mesmo tempo de retenção da glibenclamida e que foi verificada a
pureza dos picos referentes à glibenclamida nas soluções padrão de trabalho e
amostra. Para exemplificar, nas Figuras 5.4 e 5.5 estão apresentados os
cromatogramas referentes ao diluente e soluções placebo, padrão de trabalho e
amostra em coluna convencional e de núcleo fundido, respectivamente.
Figura 5.4 - Cromatogramas da GB - (a) padrão, (b) amostra, (c) diluente (d) placebo -
coluna CV
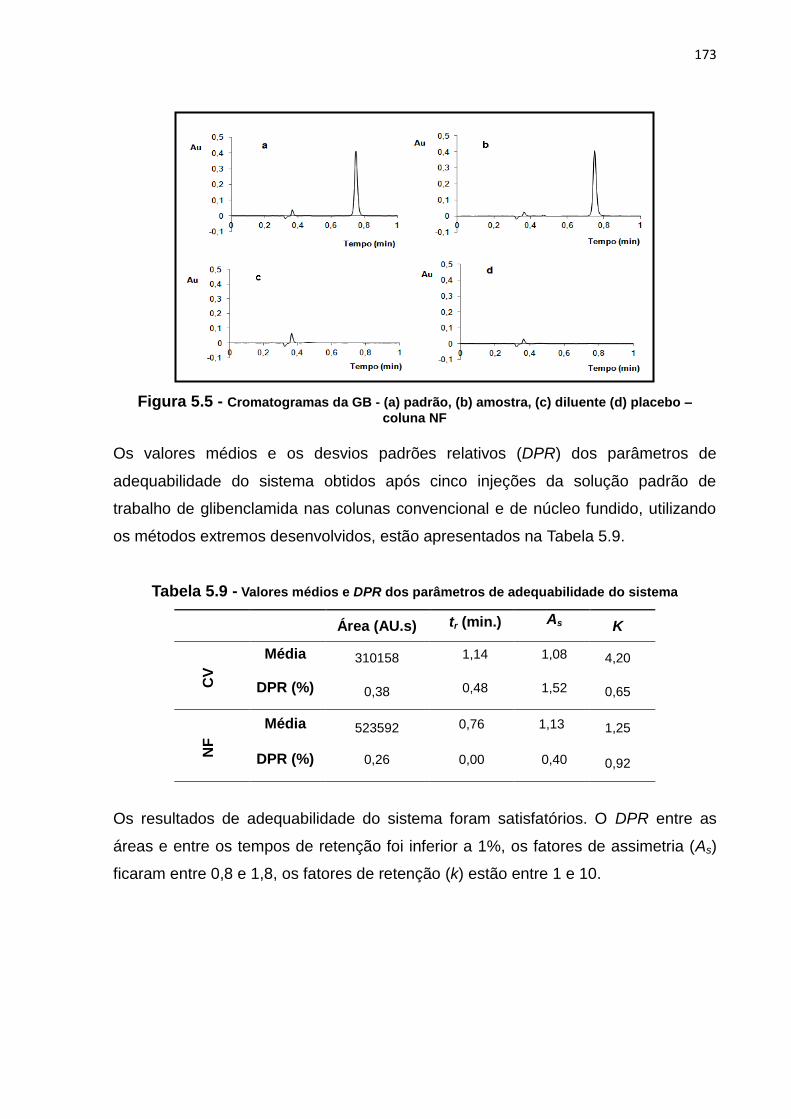
173
Figura 5.5 - Cromatogramas da GB - (a) padrão, (b) amostra, (c) diluente (d) placebo –
coluna NF
Os valores médios e os desvios padrões relativos (DPR) dos parâmetros de
adequabilidade do sistema obtidos após cinco injeções da solução padrão de
trabalho de glibenclamida nas colunas convencional e de núcleo fundido, utilizando
os métodos extremos desenvolvidos, estão apresentados na Tabela 5.9.
Tabela 5.9 - Valores médios e DPR dos parâmetros de adequabilidade do sistema
Área (AU.s) tr (min.) As K
CV
Média 310158 1,14 1,08 4,20
DPR (%) 0,38 0,48 1,52 0,65
NF
Média 523592 0,76 1,13 1,25
DPR (%) 0,26 0,00 0,40 0,92
Os resultados de adequabilidade do sistema foram satisfatórios. O DPR entre as
áreas e entre os tempos de retenção foi inferior a 1%, os fatores de assimetria (As)
ficaram entre 0,8 e 1,8, os fatores de retenção (k) estão entre 1 e 10.

174
4.2.2 Estabilidade das soluções padrão de trabalho e amostra
As áreas relativas ao pico de glibenclamida nas soluções padrão de trabalho e
amostra, determinadas de hora em hora por 4 horas, em coluna convencional, estão
descritas na Tabela 5.10.
Tabela 5.10 - Áreas das soluções padrão de trabalho e amostra em período de 4 horas
Hora Área (AU.s) da amostra
Área (AU.s) do padrão
0 317660 310158
1 316784 311415
2 316115 312496
3 317345 312647
4 318899 313485
∆ % (Hora 0 – Hora 4) 0,39% 1,07%
Como houve uma variação inferior a 2% entre as áreas registradas no início (hora
zero) e no final do experimento (hora quatro), conclui-se que as soluções padrão de
trabalho e amostra são estáveis por, pelo menos, 4 horas.
4.2.3 Linearidade
Para estimar as regressões lineares, as quais estabelecem a relação entre área e
concentração de glibenclamida, optou-se pela utilização do método dos mínimos
quadrados (MMQ). Antes da aplicação do MMQ testes estatísticos foram aplicados
aos dados de regressão linear (Tabela 5.11).
Tabela 5.11 - Testes estatísticos aplicados aos dados de regressão linear
Outliers
Resíduo Jacknife
Normalidade
Ryan-Joiner
Homocedasticidade
Levene modificado
Independência
Durbin-Watson
CV Não há outliers Dados seguem a
normal
Dados são
homocedásticos
Dados são
independentes
NF Há 1 outlier Dados seguem a
normal
Dados são
homocedásticos
Dados são
independentes
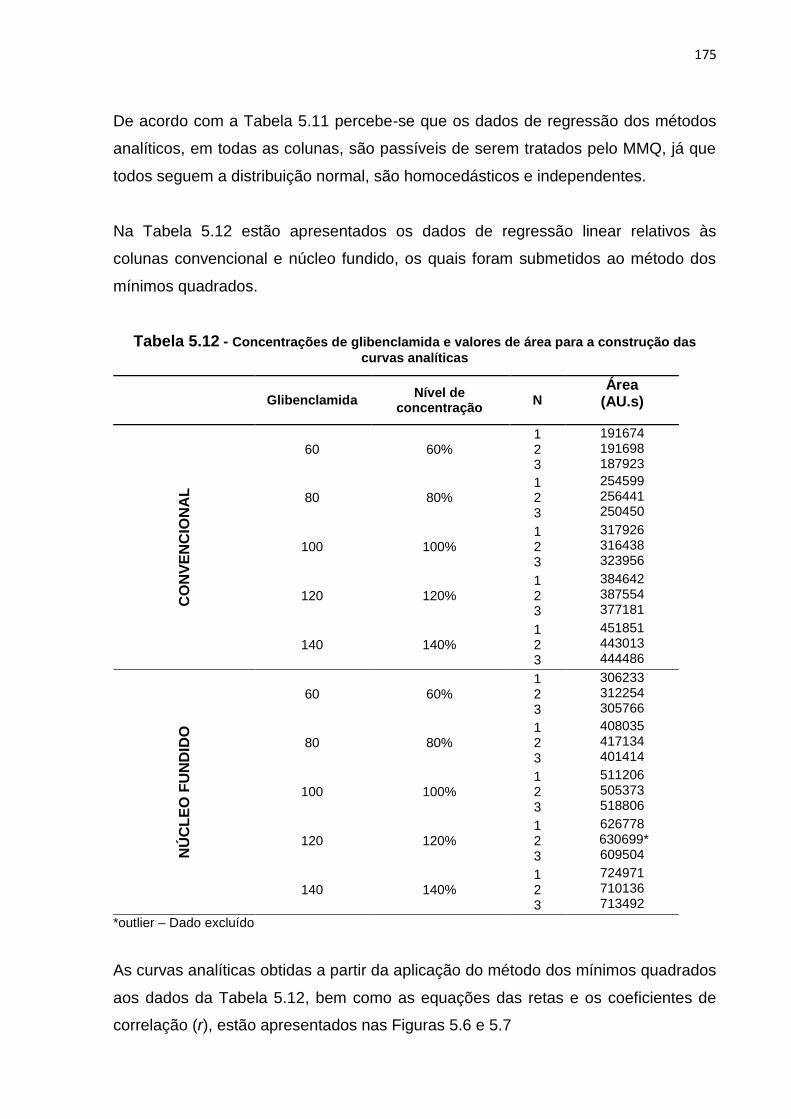
175
De acordo com a Tabela 5.11 percebe-se que os dados de regressão dos métodos
analíticos, em todas as colunas, são passíveis de serem tratados pelo MMQ, já que
todos seguem a distribuição normal, são homocedásticos e independentes.
Na Tabela 5.12 estão apresentados os dados de regressão linear relativos às
colunas convencional e núcleo fundido, os quais foram submetidos ao método dos
mínimos quadrados.
Tabela 5.12 - Concentrações de glibenclamida e valores de área para a construção das
curvas analíticas
Glibenclamida Nível de
concentração N
Área (AU.s)
CO
NV
EN
CIO
NA
L
60 60% 1 2 3
191674 191698 187923
80 80% 1 2 3
254599 256441 250450
100 100% 1 2 3
317926 316438 323956
120 120% 1 2 3
384642 387554 377181
140 140% 1 2 3
451851 443013 444486
NÚ
CL
EO
FU
ND
IDO
60 60% 1 2 3
306233 312254 305766
80 80% 1 2 3
408035 417134 401414
100 100% 1 2 3
511206 505373 518806
120 120% 1 2 3
626778 630699* 609504
140 140% 1 2 3
724971 710136 713492
*outlier – Dado excluído
As curvas analíticas obtidas a partir da aplicação do método dos mínimos quadrados
aos dados da Tabela 5.12, bem como as equações das retas e os coeficientes de
correlação (r), estão apresentados nas Figuras 5.6 e 5.7
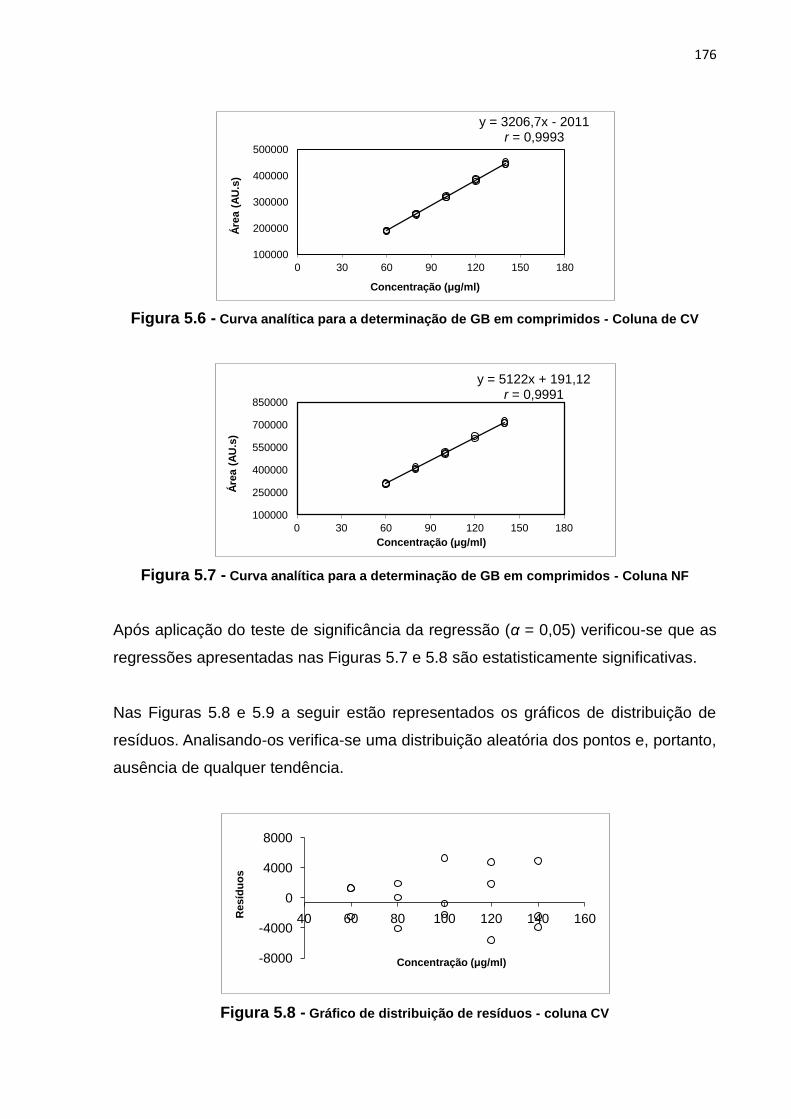
176
Figura 5.6 - Curva analítica para a determinação de GB em comprimidos - Coluna de CV
Figura 5.7 - Curva analítica para a determinação de GB em comprimidos - Coluna NF
Após aplicação do teste de significância da regressão (α = 0,05) verificou-se que as
regressões apresentadas nas Figuras 5.7 e 5.8 são estatisticamente significativas.
Nas Figuras 5.8 e 5.9 a seguir estão representados os gráficos de distribuição de
resíduos. Analisando-os verifica-se uma distribuição aleatória dos pontos e, portanto,
ausência de qualquer tendência.
Figura 5.8 - Gráfico de distribuição de resíduos - coluna CV
y = 3206,7x - 2011 r = 0,9993
100000
200000
300000
400000
500000
0 30 60 90 120 150 180
Áre
a (
AU
.s)
Concentração (μg/ml)
y = 5122x + 191,12 r = 0,9991
100000
250000
400000
550000
700000
850000
0 30 60 90 120 150 180
Áre
a (
AU
.s)
Concentração (μg/ml)
-8000
-4000
0
4000
8000
40 60 80 100 120 140 160 Resíd
uo
s
Concentração (μg/ml)

177
Figura 5.9 - Gráfico de distribuição de resíduos - coluna NF
Considerando as discussões apresentadas pode-se concluir que os dois métodos
apresentam linearidade. Os resultados das análises estatísticas indicaram ajustes
adequados ao modelo de regressão linear: o coeficiente de correlação r foi superior
a 0,99 e a ANOVA demonstrou que as regressões foram estatisticamente
significativas a um nível de significância de 5%. Além disso, analisando-se os
gráficos de resíduos, não se observou nenhuma tendência.
4.2.4 Precisão intra-corrida e precisão inter-corridas
Avaliou-se a precisão intra-corrida dos métodos por meio de seis determinações a
100% da concentração de trabalho (100 μg/ml) e a inter-corrida por meio de 12
determinações Os valores de teor e DPR estão apresentados na Tabela 5.13.
Tabela 5.13 - Valores de área e teor de glibenclamida em comprimidos para avaliação da
precisão dos métodos extremos relativos às colunas CV e NF
CV NF
1o d
ia
Intr
a-
co
rrid
a Área Média (AU. s) 311576 531507
Teor médio (AU. s) 100,40 100,46
DPR (%) 0,24 0,18
2o d
ia
Intr
a-
co
rrid
a Área Média (AU. s) 310241 526201
Teor médio (AU. s) 99,97 99,45
DPR (%) 0,22 0,34
Inte
r-
co
rrid
as Área Média (AU. s) 310908
528854
Teor médio (AU. s) 100,18 99,95
DPR (%) 0,33 0,58
-14000
-7000
0
7000
14000
40 60 80 100 120 140 160 Resíd
uo
s
Concentração (μg/ml)
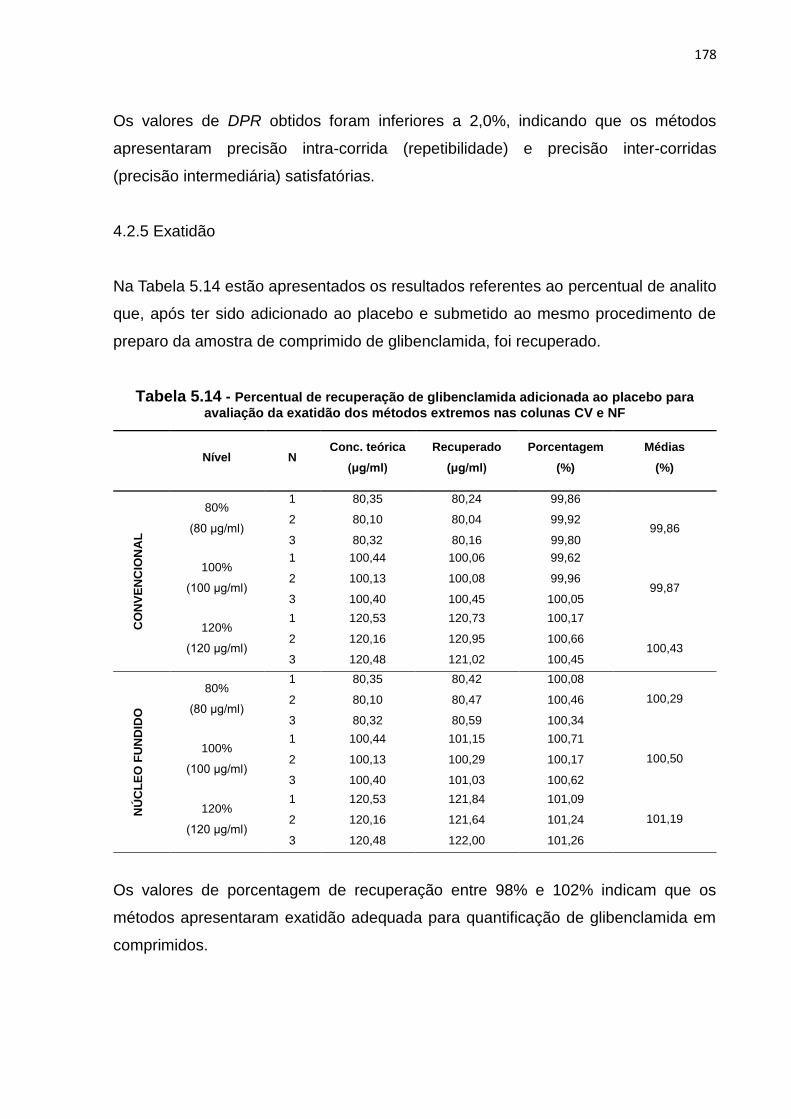
178
Os valores de DPR obtidos foram inferiores a 2,0%, indicando que os métodos
apresentaram precisão intra-corrida (repetibilidade) e precisão inter-corridas
(precisão intermediária) satisfatórias.
4.2.5 Exatidão
Na Tabela 5.14 estão apresentados os resultados referentes ao percentual de analito
que, após ter sido adicionado ao placebo e submetido ao mesmo procedimento de
preparo da amostra de comprimido de glibenclamida, foi recuperado.
Tabela 5.14 - Percentual de recuperação de glibenclamida adicionada ao placebo para
avaliação da exatidão dos métodos extremos nas colunas CV e NF
Nível N Conc. teórica
(μg/ml)
Recuperado
(μg/ml)
Porcentagem
(%)
Médias
(%)
CO
NV
EN
CIO
NA
L
80%
(80 μg/ml)
1
2
3
80,35
80,10
80,32
80,24
80,04
80,16
99,86
99,92
99,80
99,86
100%
(100 μg/ml)
1
2
3
100,44
100,13
100,40
100,06
100,08
100,45
99,62
99,96
100,05
99,87
120%
(120 μg/ml)
1
2
3
120,53
120,16
120,48
120,73
120,95
121,02
100,17
100,66
100,45
100,43
NÚ
CL
EO
FU
ND
IDO
80%
(80 μg/ml)
1
2
3
80,35
80,10
80,32
80,42
80,47
80,59
100,08
100,46
100,34
100,29
100%
(100 μg/ml)
1
2
3
100,44
100,13
100,40
101,15
100,29
101,03
100,71
100,17
100,62
100,50
120%
(120 μg/ml)
1
2
3
120,53
120,16
120,48
121,84
121,64
122,00
101,09
101,24
101,26
101,19
Os valores de porcentagem de recuperação entre 98% e 102% indicam que os
métodos apresentaram exatidão adequada para quantificação de glibenclamida em
comprimidos.

179
4.2.6 Limites de detecção e quantificação
Os limites de detecção e quantificação de glibenclamida referentes aos métodos
extremos desenvolvidos nas colunas CV e NF estão descritos na Tabela 5.15.
Tabela 5.15 - Limites de detecção e quantificação
Coluna LD (μg/ml) LQ (μg/ml)
CV 2,5 7,5
NF 0,5 2,5
Os resultados apresentados na Tabela 5.15 são apenas informativos já que os
métodos em questão são para determinação quantitativa de glibenclamida em
comprimidos, e, portanto, o limite de detecção não precisaria ser determinado. Além
disso, como a concentração de trabalho é alta (100 μg/ml) o limite de quantificação
também não precisaria ser estimado.
Analisando-se os dados nota-se que o método extremo desenvolvido em coluna de
núcleo fundido apresentou menor limite de detecção e de quantificação do que o
método extremo desenvolvido em coluna convencional. Isso é consequência da
redução do tamanho das partículas (de 5 μm na coluna CV para 2,7 μm na coluna
de NF) e do menor caminho de difusão presente nas partículas de núcleo fundido.
Ambos os fatores contribuem para um menor alargamento do pico cromatográfico,
dando origem a um pico de maior relação sinal/ruído [17,18].
4.2.7 Robustez
Os resultados de robustez estão apresentados na Tabela 5.16. Os efeitos de cada
variável e o efeito maior foram calculados para que a influência das modificações
propostas na quantificação da glibenclamida em comprimidos fosse avaliada.
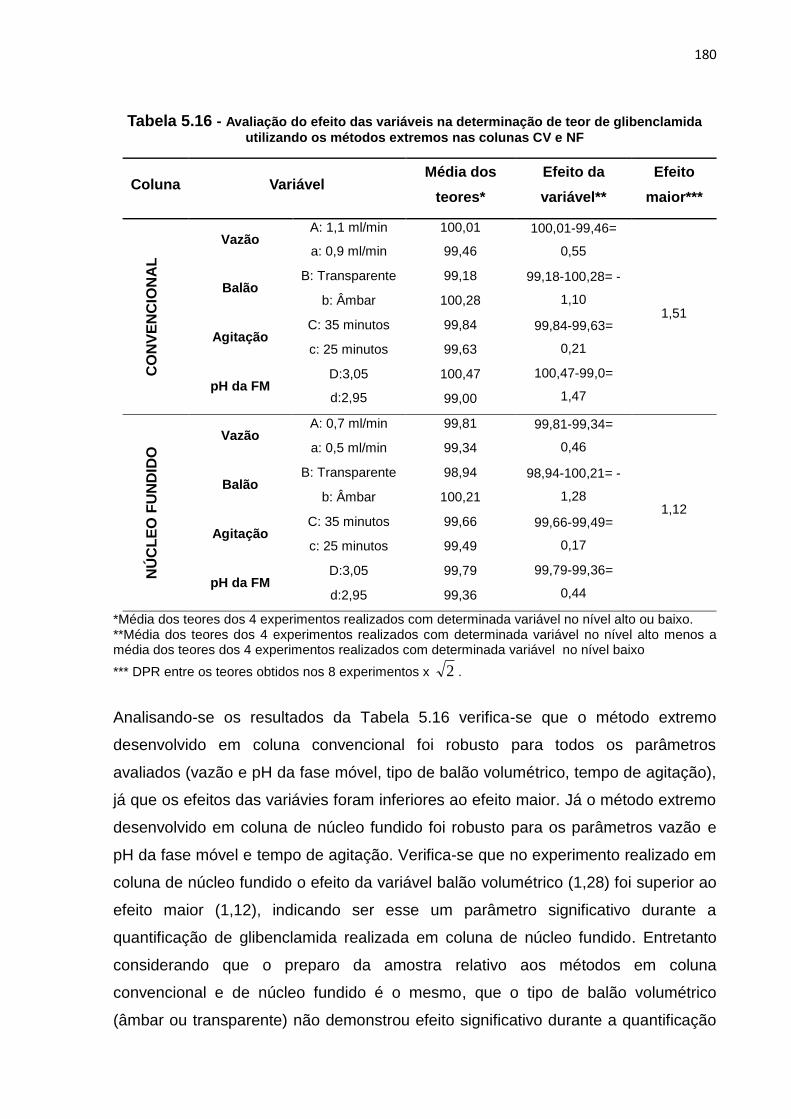
180
Tabela 5.16 - Avaliação do efeito das variáveis na determinação de teor de glibenclamida
utilizando os métodos extremos nas colunas CV e NF
Coluna Variável Média dos
teores*
Efeito da
variável**
Efeito
maior***
CO
NV
EN
CIO
NA
L
Vazão A: 1,1 ml/min 100,01 100,01-99,46=
0,55
1,51
a: 0,9 ml/min 99,46
Balão B: Transparente 99,18 99,18-100,28= -
1,10 b: Âmbar 100,28
Agitação C: 35 minutos 99,84 99,84-99,63=
0,21 c: 25 minutos 99,63
pH da FM D:3,05 100,47 100,47-99,0=
1,47
d:2,95
99,00
NÚ
CL
EO
FU
ND
IDO
Vazão A: 0,7 ml/min 99,81 99,81-99,34=
0,46
1,12
a: 0,5 ml/min 99,34
Balão B: Transparente 98,94 98,94-100,21= -
1,28 b: Âmbar 100,21
Agitação C: 35 minutos 99,66 99,66-99,49=
0,17 c: 25 minutos 99,49
pH da FM D:3,05 99,79 99,79-99,36=
0,44
d:2,95 99,36
*Média dos teores dos 4 experimentos realizados com determinada variável no nível alto ou baixo. **Média dos teores dos 4 experimentos realizados com determinada variável no nível alto menos a média dos teores dos 4 experimentos realizados com determinada variável no nível baixo
*** DPR entre os teores obtidos nos 8 experimentos x 2 .
Analisando-se os resultados da Tabela 5.16 verifica-se que o método extremo
desenvolvido em coluna convencional foi robusto para todos os parâmetros
avaliados (vazão e pH da fase móvel, tipo de balão volumétrico, tempo de agitação),
já que os efeitos das variávies foram inferiores ao efeito maior. Já o método extremo
desenvolvido em coluna de núcleo fundido foi robusto para os parâmetros vazão e
pH da fase móvel e tempo de agitação. Verifica-se que no experimento realizado em
coluna de núcleo fundido o efeito da variável balão volumétrico (1,28) foi superior ao
efeito maior (1,12), indicando ser esse um parâmetro significativo durante a
quantificação de glibenclamida realizada em coluna de núcleo fundido. Entretanto
considerando que o preparo da amostra relativo aos métodos em coluna
convencional e de núcleo fundido é o mesmo, que o tipo de balão volumétrico
(âmbar ou transparente) não demonstrou efeito significativo durante a quantificação

181
de glibenclamida realizada em coluna convencional e que referências da literatura
apontam para a fotoestabilidade da glibenclamida [19], não faz sentido que o tipo de
balão seja considerado parâmetro de efeito significativo na determinação de
glibenclamida em coluna de núcleo fundido.
5 CONCLUSÃO
O método extremo desenvolvido em coluna empacotada com partículas sub 2 μm foi
o mais rápido, já que o tempo de retenção da glimepirida, último analito a ser eluído,
foi de apenas 0,52 minutos. O método extremo desenvolvido em coluna
convencional foi o mais lento (tr GM = 1,37 minuto), e os referentes às colunas
empacotadas com partículas de núcleo fundido (tr GM = 0,86 minuto) e monolítica
(tr GM = 0,74 minuto) apresentaram velocidades intermediárias.
Verificou-se que mesmo em condições extremas as colunas empacotadas com
partículas sub 2 μm e partículas de núcleo fundido geraram análises mais eficientes
do que as colunas monolítica e convencional. É importante destacar que as
pressões atingidas pelas colunas convencional, monolítica e de núcleo fundido
foram compatíveis com HPLC (inferior a 400 bar). Já a pressão atingida pela coluna
de partículas sub 2 μm é compatível com UHPLC (próxima a 1000 bar).
Considerando apenas velocidade de análise o método extremo desenvolvido em
coluna Sub 2 μm foi o ideal, já que propiciou a análise mais rápida. Entretanto
considerando simultaneamente os fatores velocidade, eficiência e a limitação de
pressão, o método extremo relativo à coluna de núcleo fundido foi o ideal.
Os métodos extremos em coluna convencional e de núcleo fundido para
determinação de glibenclamida em comprimido foram adequadamente validados
quanto aos parâmetros seletividade, linearidade, precisão, exatidão, limites de
detecção e quantificação e robustez. Além disso, verificou-se a adequabilidade de
ambos os sistemas (system suitability). Conclui-se que o método desenvolvido em
coluna de núcleo fundido, assim como o método desenvolvido na coluna
convencional, é confiável para determinação quantitativa de glibenclamida em
comprimidos.

182
REFERÊNCIAS BIBLIOGRÁFICAS
[1] AL-SAYAH, M. A.; RIZOS, P.; ANTONUCCI, V.; WU, N. High throughput of active pharmaceutical ingredients by UPLC. Journal of Separation Science. v. 31, p. 2167-2172, 2008. [2] SNYDER, L. R., KIRKLAND, J. J., GLAJCH, J. L. Practical HPLC method development. 2 ed. New York: John Wiley & Sons, 1997. 765 p. [3] GUILLARME, D.; RUTA, J.; RUDAZ, J.; VEUTHEY, J. New trends in fast and high-resolution liquid chromatography: a critical comparison of existing approaches. Analytical and Bioanalytical Chemistry. v. 397, p. 1069-1082, 2010. [4] MCMASTER, M. C. HPLC A Practical User’s Guide. 2 ed. New York: John Wiley & Sons, 2007. 238 p. [5] GUILLARME, D.; NGUYEN, D. T.T.; RUDAZ, S.; VEUTHEY, J. L. Recent developments in liquid chromatography-Impact on qualitative and quantitative performance. Journal of Chromatography A. v. 1149, p. 20-29, 2007. [6] BADMAN, E. R.; BEARDSLEY, R. L., LIANG, Z.; BANSAL, S. Accelerating high quality bioanalytical LC/MS/MS assays using fused-core columns. Journal of Chromatography B. v. 878, p. 2307-2313, 2010. [7] KAMINSKI, L.; DEEB, S. E.; WATZIG, H. Repeatability of monolithic HPLC columns while using a flow program Journal of separation science. v. 31, p. 1745-1749, 2008. [8] VENKATESH, P.; HARISUDHAN,T.; CHOUDHURY, H.; MULLANGI, R.; SRINIVAS, N. R. Simultaneous estimation of six anti-diabetic drugs - glibenclamide, gliclazide, glipizide, pioglitazone, repaglinide and rosiglitazone: development of a novel HPLC method for use in the analysis of pharmaceutical formulations and its application to human plasma assay. Biomedical Chromatography, v. 20, p.1043-1048, 2006. [9] LAKSHMI, K. S.; RAJESH, T. Development and Validation of RP-HPLC Method for Simultaneous Determination of Glipizide, Rosiglitazone, Pioglitazone, Glibenclamide and Glimepiride in Pharmaceutical Dosage Forms and Human Plasma. Journal of The Iranian Chemical Society, v. 8, n. 1, p. 31-37, 2011. [10] FERREIRA, Anderson de Oliveira. Guia prático da farmácia magistral. 4. ed. São Paulo: Pharmabooks, 2010-2011. v.1. 736 p. [11] BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RE no. 899, de 29 de Maio de 2003. Guia para validação de métodos analíticos e bioanalíticos. Diário Oficial da União, Brasília, DF, Poder Executivo, de 02 de Junho de 2003c. [12] PATNAIK, P. Handbook of environmental analysis. 2 ed. New York: CRC press,1997.

183
[13] GREEN, J. M. A practical guide to analytical method validation. Analytical Chemistry. v. 68, p. 305-309, 1996. [14] RIBANI, M.; BOTTOLI, C. B. G.; COLLINS, C. H.; JARDIM, I. C. S. F.; MELO, L. F. C. Validação em métodos cromatográficos e eletroforéticos. Química Nova. v. 27, n. 5, p. 771-780, 2004. [15] SOUZA, S. V. C.; JUNQUEIRA, R. G. A procedure to assess linearity by ordinary least squares method. Analytica Chimica Acta. v. 552, p. 25-35, 2005. [16] YOUDEN, W. J.; STEINER E. H. Statistical Manual of the Association of Official Analytical Chemists. 48 ed. Arlington: AOAC, 1975. 88 p.
[17] FEKETE, S.; OLAH, E.; FEKETE, J. Fast liquid chromatography: The domination of core–shell and very fine particles. Journal of Chromatography A. v. 1228, p. 57-71, 2012. [18] LOUGH, W. J.; WAINER, I. W. High performance liquid chromatography: fundamental principles and practice. London: Black Academic & Professional, 1996. 276 p.
[19] BANNSAL, G.; SINGH, M.; JINDAL, K. C.; SINGH, S. Ultraviolet-Photodiode
Array an High-Performance Liquid Chromatographic/ Mass Spectrometric Studies on
forced degradation behaviorof glibenclamide and development on a validated
stability-indicating method. Journal of AOAC International. v. 91, n. 4, p. 709-719,
2008.

184
CONCLUSÃO FINAL

185
- Método analítico para quantificação de clorpropamida, glibenclamida, glimepirida e
gliclazida foi desenvolvido e otimizado em coluna convencional com partículas
porosas de 5 μm de diâmetro e transferido para as colunas monolítica, empacotada
com partícula de núcleo fundido e empacotada com partículas porosas sub 2 μm.
- As quatro colunas foram caracterizadas de acordo com os seguintes parâmetros:
volume morto, porosidade, retenção, seletividade e permeabilidade.
- Parâmetros instrumentais cromatográficos foram otimizados para permitir que a
performance das colunas cromatográficas fosse aproveitada ao máximo durante a
análise dos antidiabéticos orais. A otimização foi realizada baseada na análise dos
valores de variância extracoluna e eficiência cromatográfica remanescente.
- Verificou-se que a influência negativa da variância extracoluna na eficiência
cromatográfica foi mais intensa durante a análise da clorpropamida e gliclazida
(analitos menos retidos) e quando colunas mais eficientes (sub 2 μm e núcleo
fundido) foram empregadas. Portanto, a otimização do sistema cromatográfico foi
especialmente importante para que o potencial cromatográfico das colunas
empacotadas com partículas sub 2 µm e com partículas de núcleo fundido fosse
aproveitado. Dessa forma foi possível realizar uma comparação fidedigna do
desempenho cromatográfico das quatro colunas em estudo.
- Considerando as análises das curvas de Van Deemter e de Knox, gráficos de
performance cinética, resolução, simetria e pressão, conclui-se que a coluna
empacotada com partículas de núcleo fundido é a melhor alternativa à coluna
convencional quando o objetivo é a obtenção de análises rápidas e ao mesmo
tempo eficientes. A coluna monolítica, apesar de compatível com equipamento de
HPLC e de propiciar análises mais rápidas com maior eficiência do que a coluna
convencional, apresenta eficiência cromatográfica bem inferior à gerada pelas
colunas de núcleo fundido e sub 2 μm, além de dar origem a picos de pior simetria.
Para exemplificar, considerando a análise da glimepirida, verificou-se que coluna de
núcleo fundido gerou análises com 90% da eficiência máxima e da velocidade ótima
e aproximadamente a mesma resolução propiciada pela coluna sub 2 μm, com a
grande vantagem de não gerar no sistema pressões acima de 400 bar, e portanto

186
não requerer a utilização de sistemas de UHPLC, mais caros e menos disponíveis.
Além disso, assim como a coluna sub 2 μm, a coluna de núcleo fundido foi capaz de
manter elevada eficiência em altas velocidades de análise e de gerar picos de boa
simetria.
- O método extremo desenvolvido em coluna empacotada com partículas sub 2 μm
foi o mais rápido, o desenvolvido em coluna convencional o mais lento (tr GM = 1,37
minuto), e os referentes às colunas empacotadas com partículas de núcleo fundido
(tr GM = 0,86 minuto) e monolítica (tr GM = 0,74 minuto) apresentaram velocidades
intermediárias. Verificou-se que mesmo em condições extremas as colunas
empacotadas com partículas sub 2 μm e partículas de núcleo fundido foram mais
eficientes. É importante destacar que as pressões limites atingidas pelas colunas
convencional, monolítica e de núcleo fundido foram compatíveis com equipamentos
de HPLC (inferior a 400 bar). Já a pressão atingida pela coluna de partículas sub 2
μm é compatível com equipamento de UHPLC (próximo a 1000 bar).
- Considerando apenas velocidade de análise o método extremo desenvolvido em
coluna empacotada com partículas sub 2 μm é o ideal. Entretanto considerando a
velocidade de análise, eficiência e a limitação da pressão máxima, o método
extremo desenvolvido em coluna de núcleo fundido é o ideal.
- Os métodos extremos em coluna convencional e de núcleo fundido para
determinação de glibenclamida em comprimido foram adequadamente validados.
Conclui-se que o método desenvolvido em coluna de núcleo fundido, assim como o
método desenvolvido na coluna convencional, é confiável para determinação
quantitativa de glibenclamida em comprimidos.
- Por último destaca-se a importância desse tipo de estudo para a escolha,
compreensão das limitaçoes, vantagens e cuidados necessários ao aproveitamento
dessas novas fases estacionárias disponíveis no mercado.

187
APÊNDICE
TRABALHOS APRESENTADOS EM CONGRESSO

188


















