
Bioinformática: Genômica e Transcriptômicabg515/seminario_genomica_transcriptomica... · - A...
79
Bioinformática: Genômica e Transcriptômica [email protected] Marcelo Falsarella Carazzolle Laboratório de Genômica e Proteômica Unicamp
Transcript of Bioinformática: Genômica e Transcriptômicabg515/seminario_genomica_transcriptomica... · - A...

Bioinformática: Genômica
e Transcriptômica
Marcelo Falsarella Carazzolle
Laboratório de Genômica e Proteômica
Unicamp

Resumo
- Introdução à genômica
- Estratégias de sequenciamento
- DNA
- mRNA
- Tecnologias de sequenciamento
- Sanger sequencing
- Illumina
- Anotação e bancos de dados biológicos
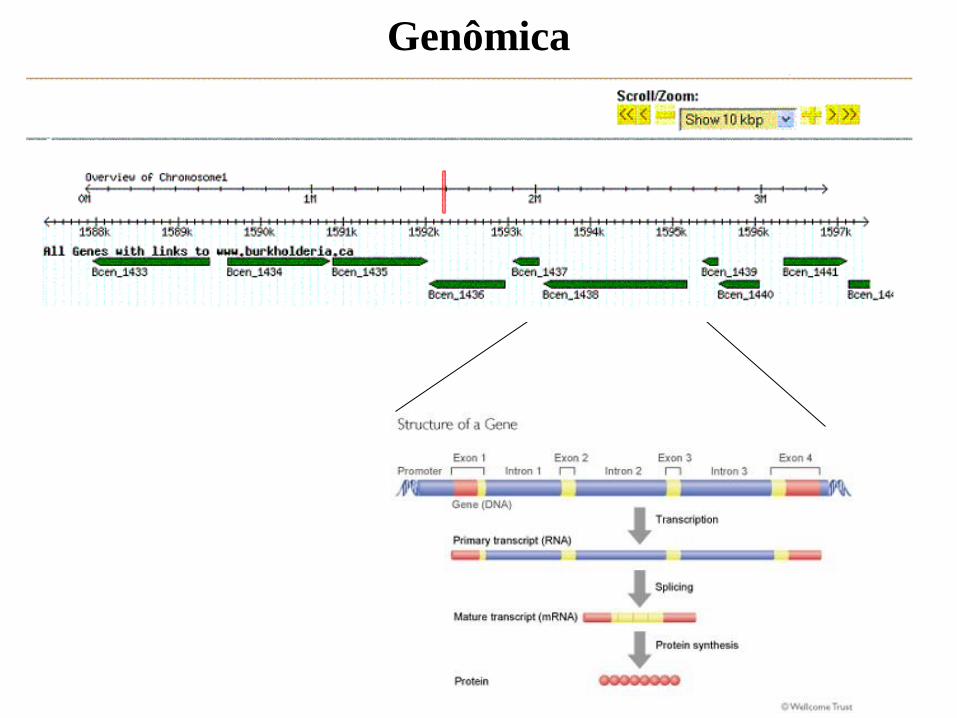
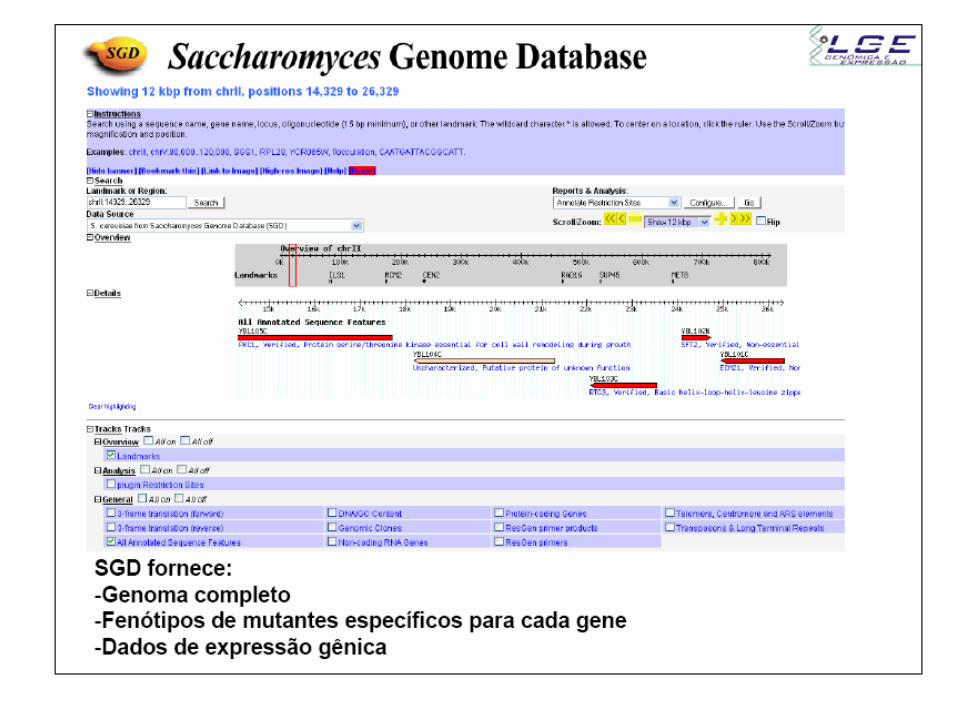
Genômica
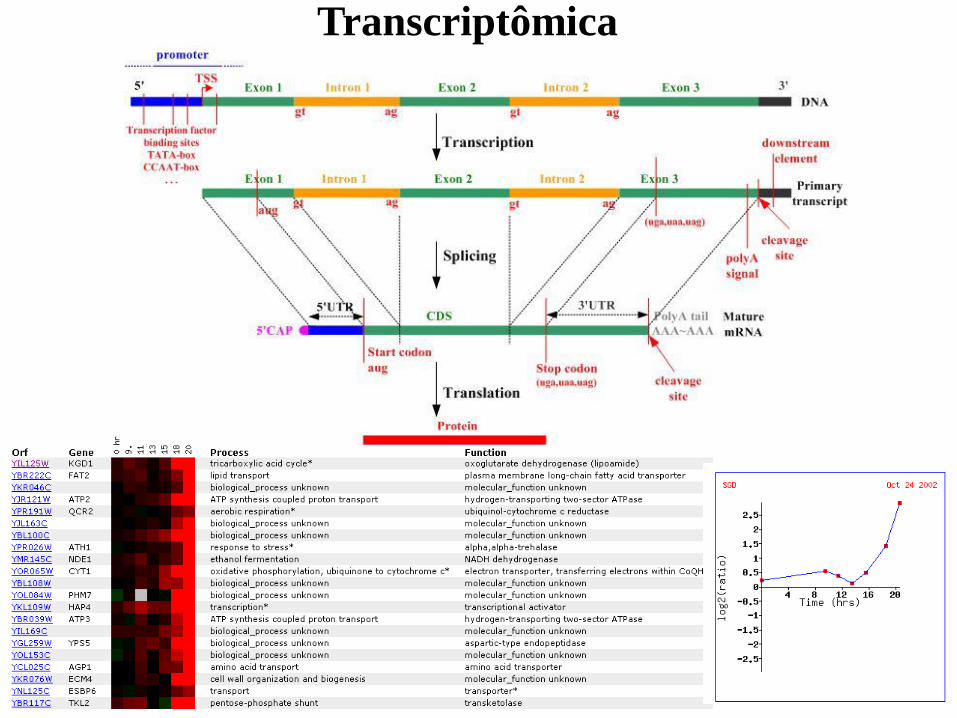
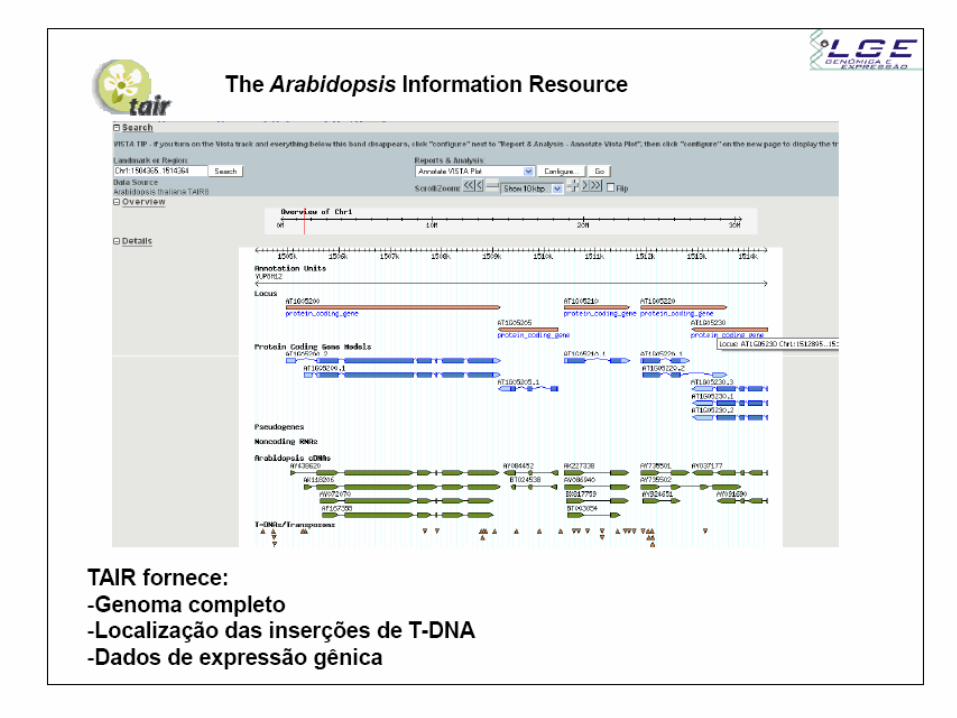
Transcriptômica

Diferenças entre as metodologias
- Sequenciamento de DNA, feito de forma aleatória, fornece :
- Informações sobre regiões codantes (genes) e promotores.
- Mas gera sequências em regiões inter-gênicas (a princípio
sem nenhuma função)
- Sequenciamento de mRNA fornece :
- Informação direta sobre os genes e também sobre a
expressão gênica.
- Mas genes pouco expressos são mais raros de serem
sequenciados por essa técnica
- A situação ideal para um projeto genoma é sequenciar ambos
DNA e mRNA

Estratégias de sequenciamento
- DNA
– Shotgun de genoma inteiro
– Shotgun em pedaços do genoma clonados em BACs
– Primer walking
- mRNA
– mRNA oriundos de diferentes tecidos ou condições
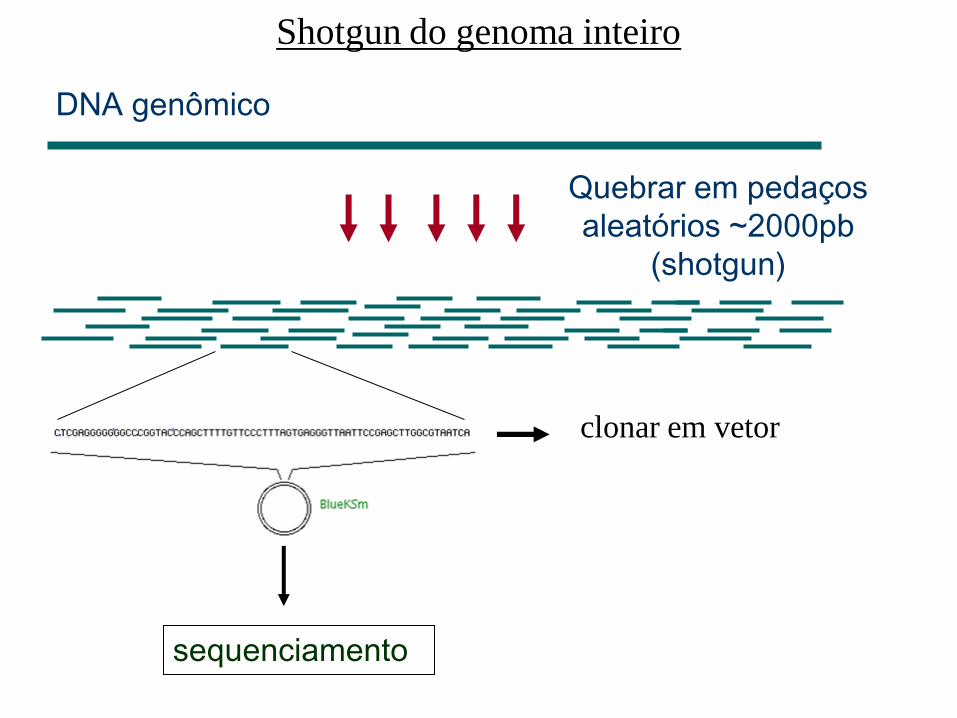
Quebrar em pedaços
aleatórios ~2000pb
(shotgun)
DNA genômico
clonar em vetor
sequenciamento
Shotgun do genoma inteiro
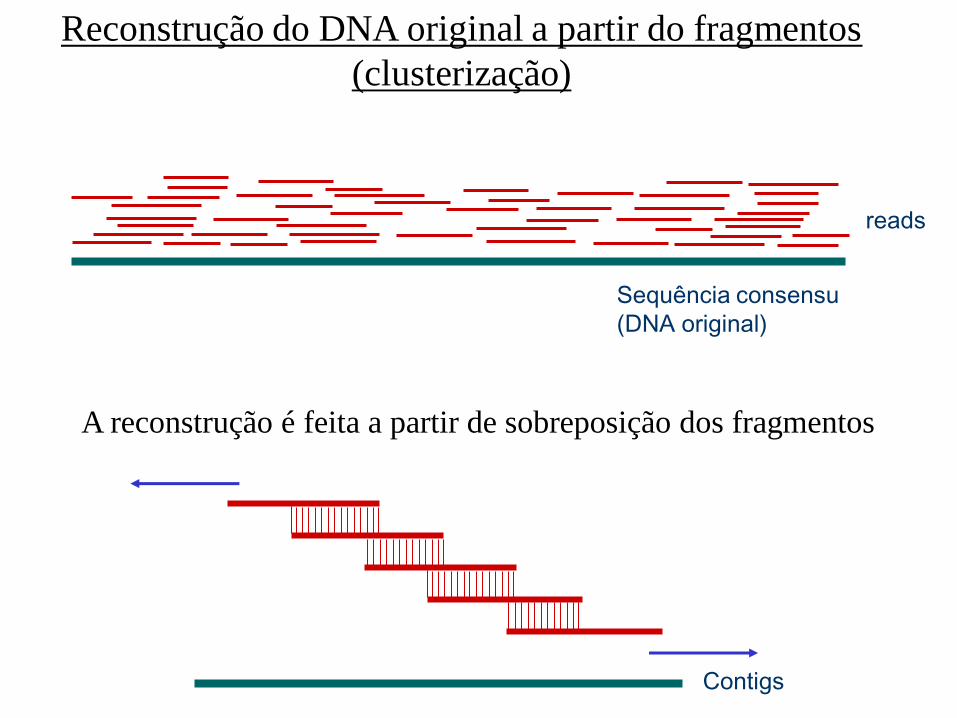
Reconstrução do DNA original a partir do fragmentos
(clusterização)
reads
Sequência consensu
(DNA original)
A reconstrução é feita a partir de sobreposição dos fragmentos
Contigs
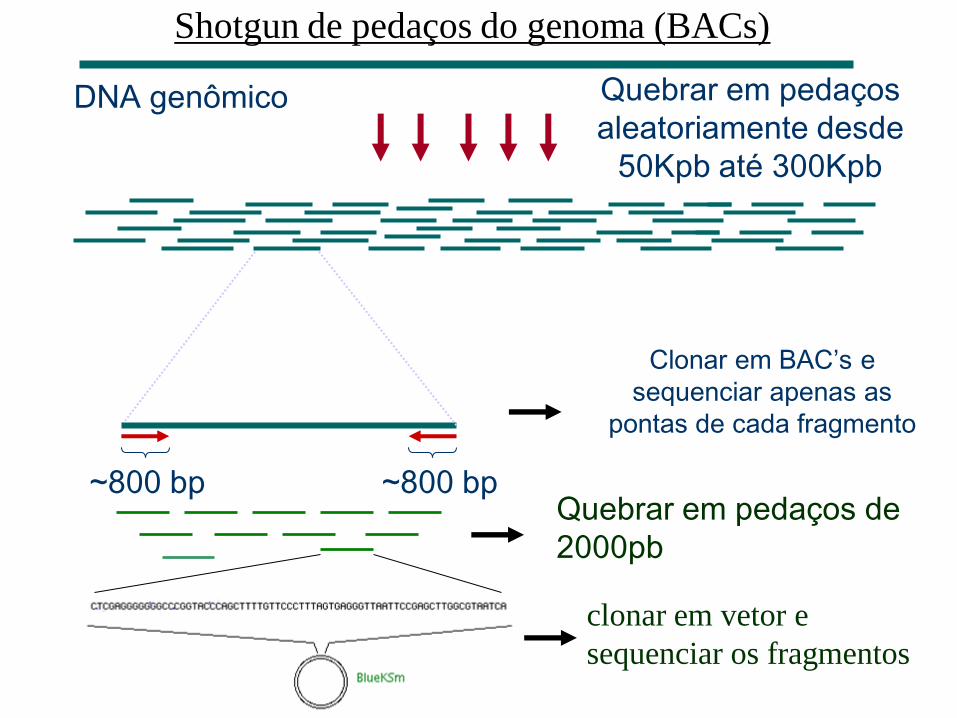
Quebrar em pedaços
aleatoriamente desde
50Kpb até 300Kpb
DNA genômico
~800 bp ~800 bp Quebrar em pedaços de
2000pb
Clonar em BAC’s e
sequenciar apenas as
pontas de cada fragmento
clonar em vetor e
sequenciar os fragmentos
Shotgun de pedaços do genoma (BACs)
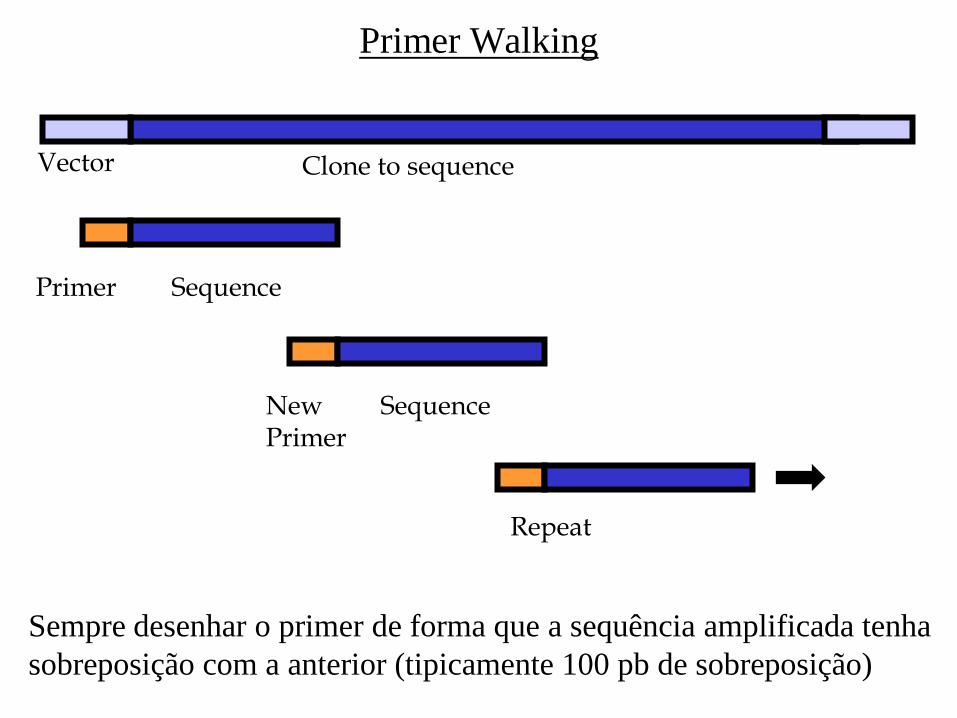
Primer Walking
Clone to sequence Vector
Primer Sequence
New Primer
Sequence
Repeat
Sempre desenhar o primer de forma que a sequência amplificada tenha
sobreposição com a anterior (tipicamente 100 pb de sobreposição)
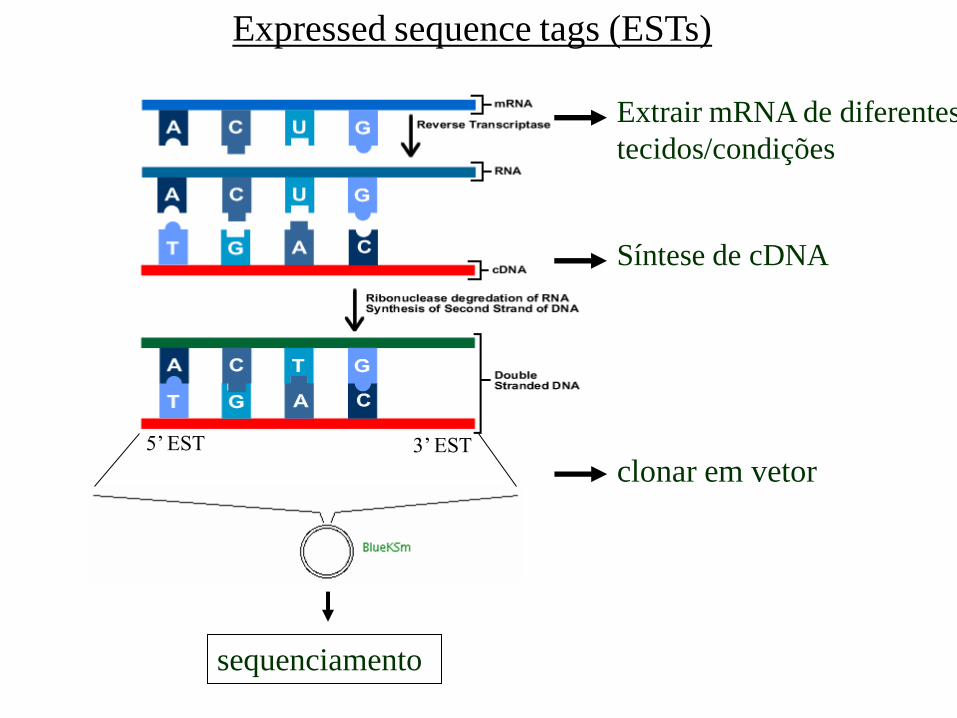
Expressed sequence tags (ESTs)
clonar em vetor
sequenciamento
3’ EST 5’ EST
Extrair mRNA de diferentes
tecidos/condições
Síntese de cDNA
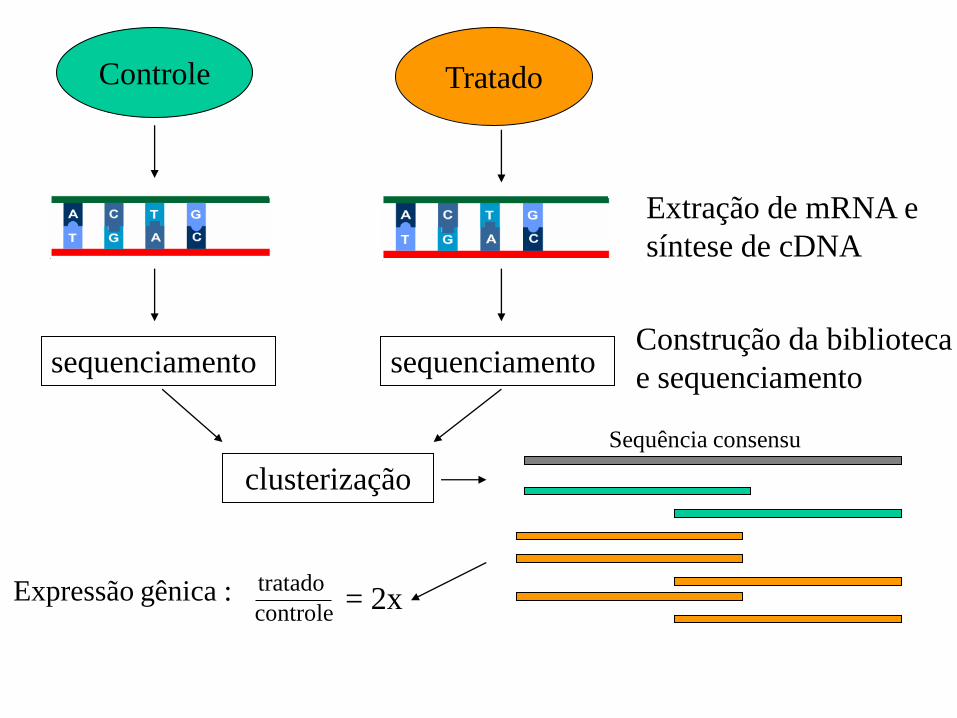
Controle Tratado
Extração de mRNA e
síntese de cDNA
sequenciamento sequenciamento
clusterização
Construção da biblioteca
e sequenciamento
Sequência consensu
Expressão gênica : controle
tratado = 2x

Tecnologias de sequenciamento
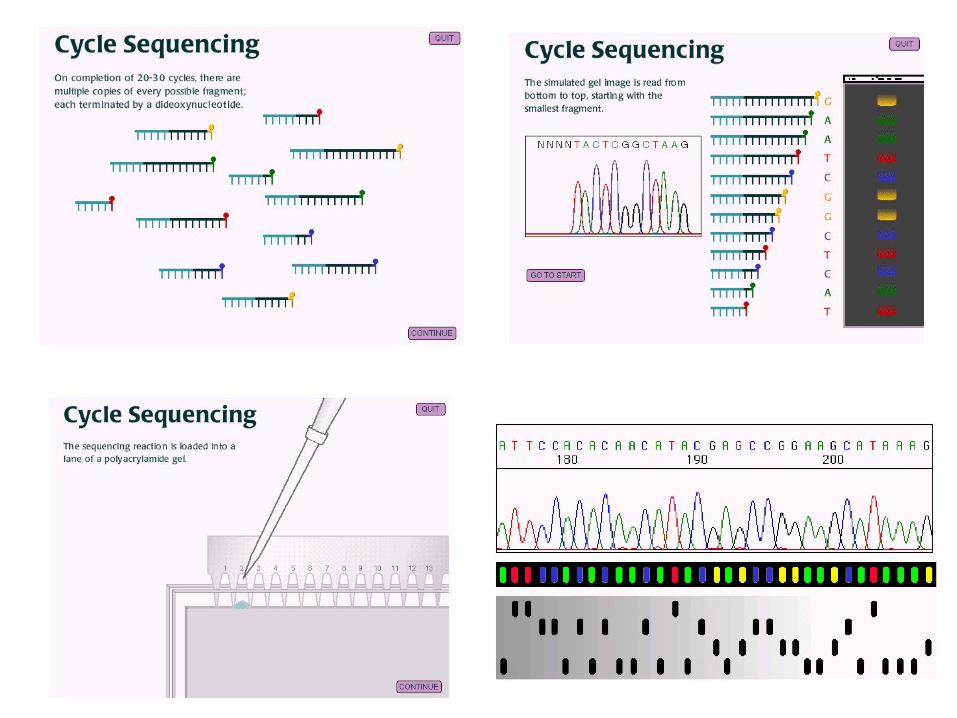
- Sanger sequencing
- PNAS 74 (1977), n. 12, 5463-5467
- Sequenciador MegaBACE (1Mpb/24 horas)
- Sequências em torno de 800 bp
- Illumina sequencing
- 70 Gbp/24 horas (a corrida dura 8 dias)
- Sequências de 150 bp
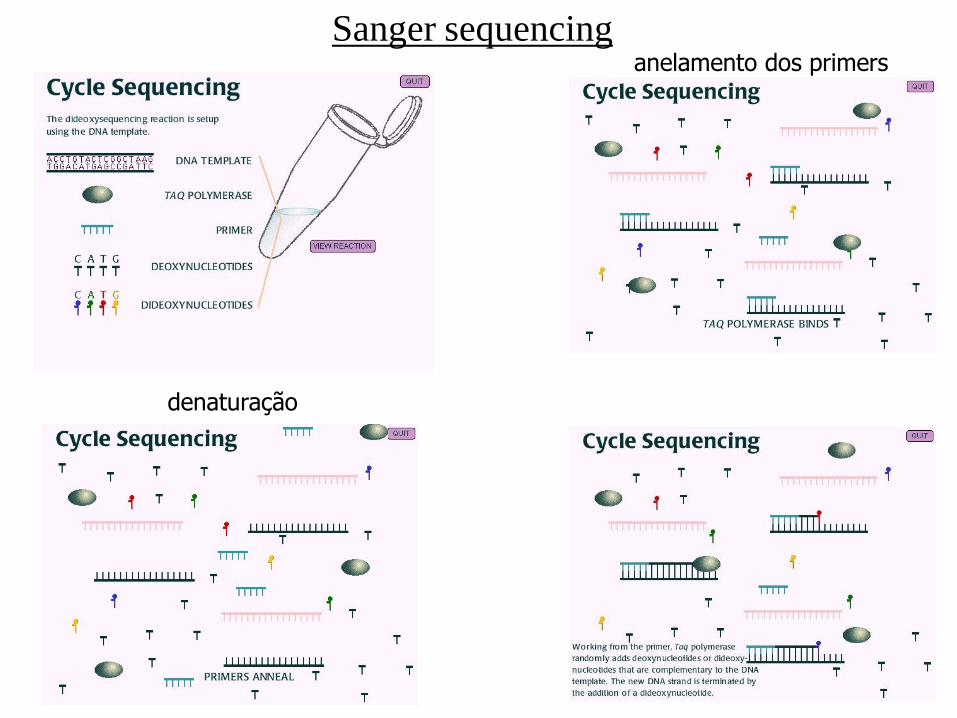
Sanger sequencing
denaturação
anelamento dos primers
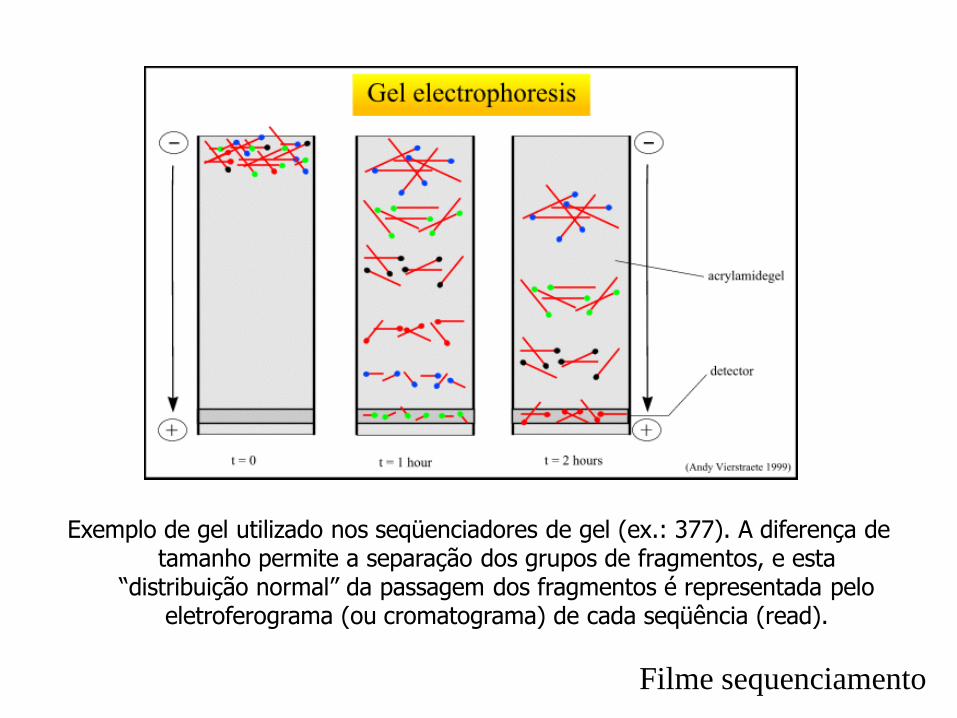
Exemplo de gel utilizado nos seqüenciadores de gel (ex.: 377). A diferença de tamanho permite a separação dos grupos de fragmentos, e esta
“distribuição normal” da passagem dos fragmentos é representada pelo eletroferograma (ou cromatograma) de cada seqüência (read).
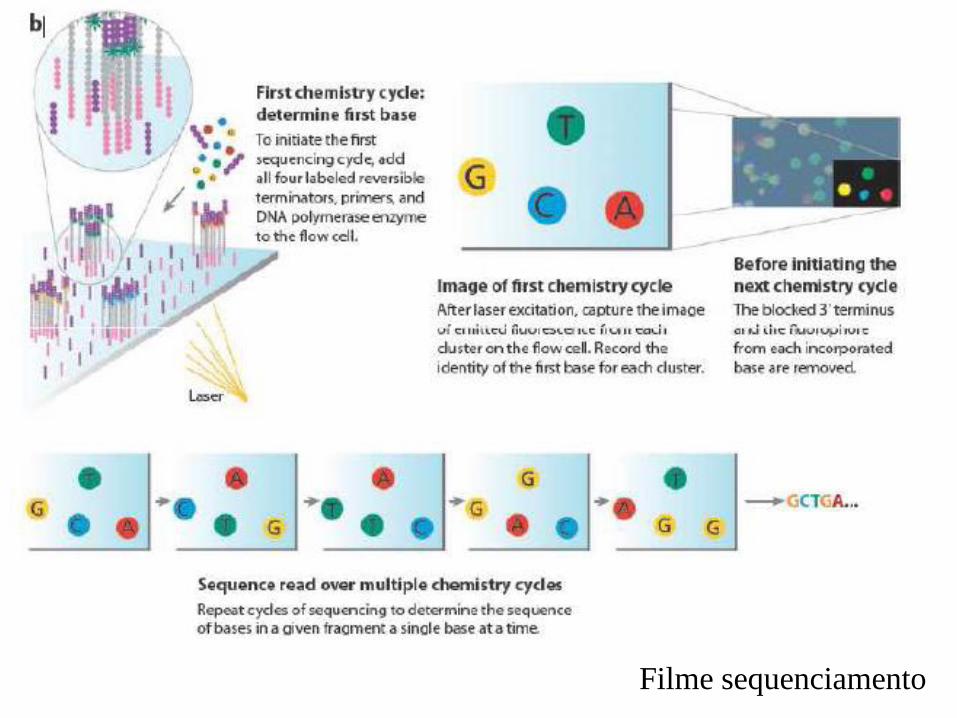
Filme sequenciamento
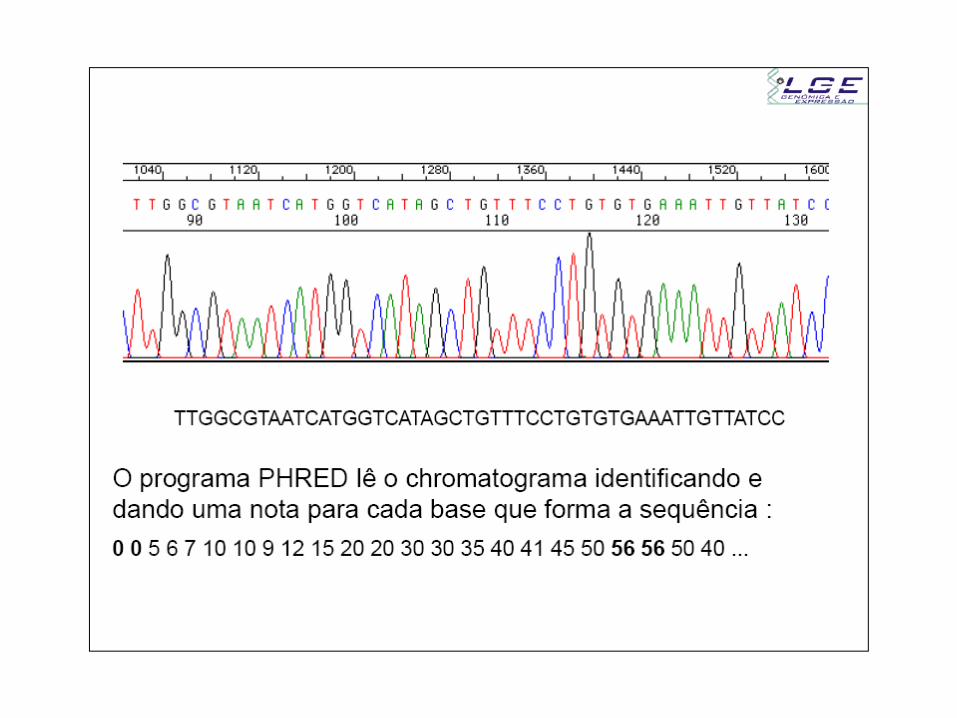
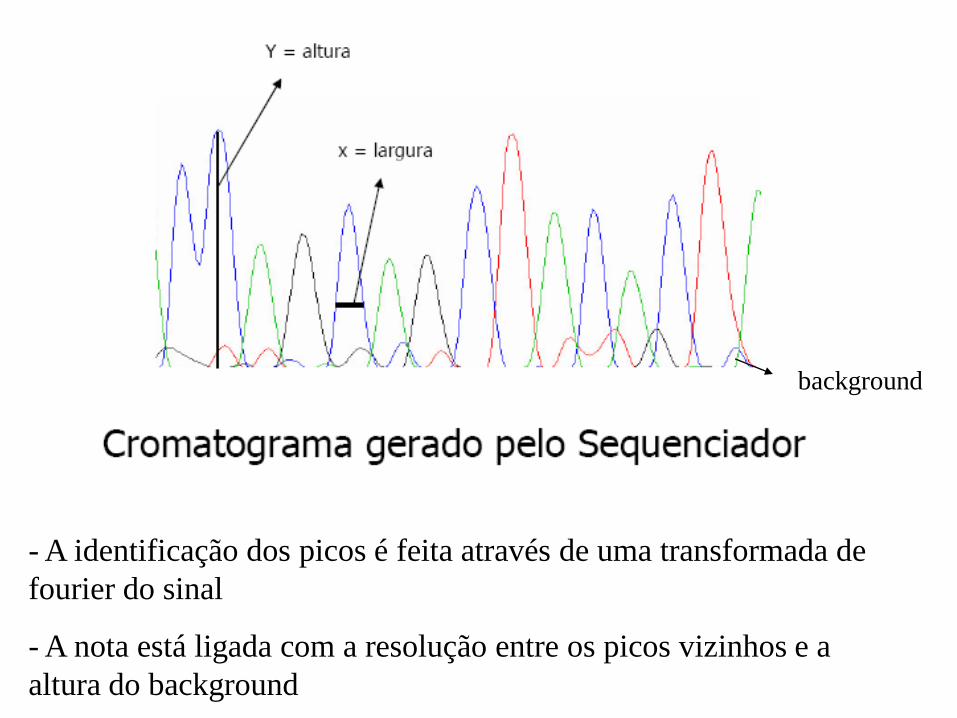
- A identificação dos picos é feita através de uma transformada de
fourier do sinal
- A nota está ligada com a resolução entre os picos vizinhos e a
altura do background
background
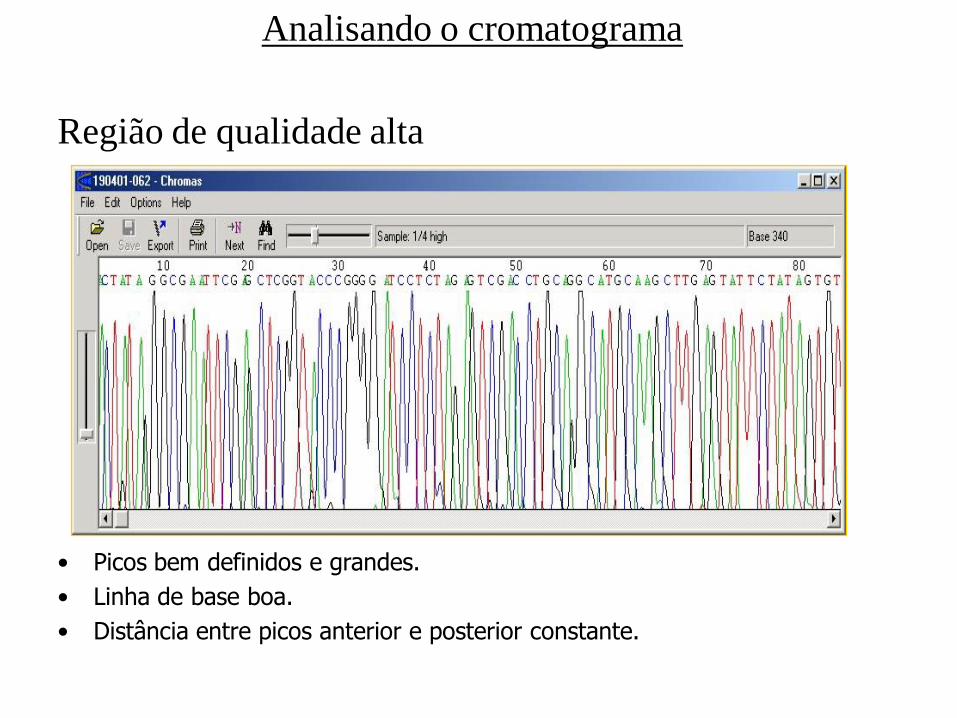
Região de qualidade alta
• Picos bem definidos e grandes.
• Linha de base boa.
• Distância entre picos anterior e posterior constante.
Analisando o cromatograma
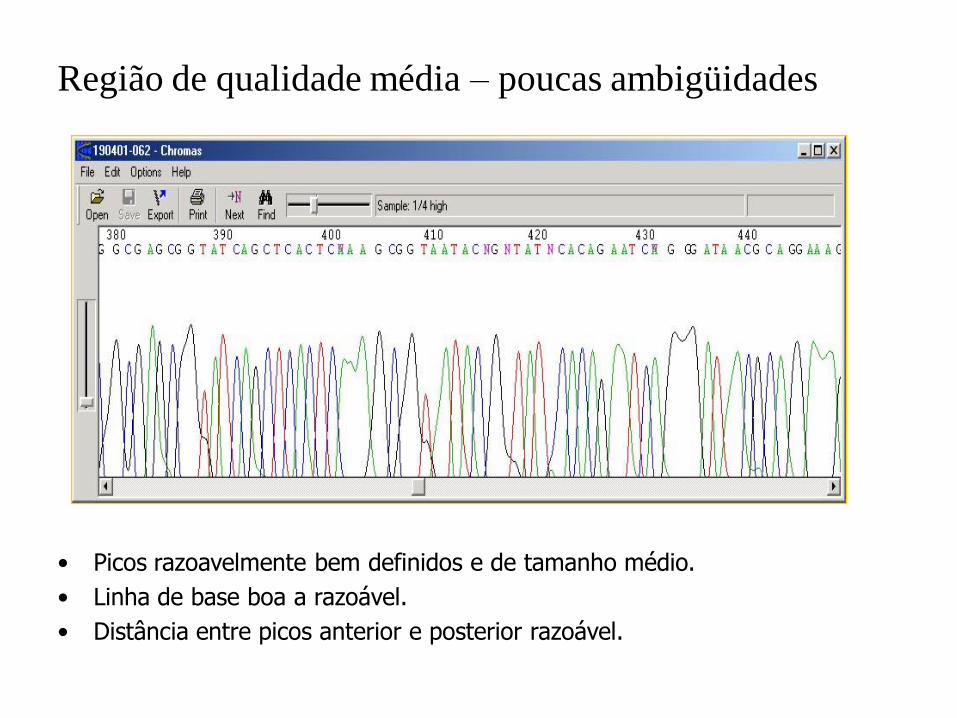
Região de qualidade média – poucas ambigüidades
• Picos razoavelmente bem definidos e de tamanho médio.
• Linha de base boa a razoável.
• Distância entre picos anterior e posterior razoável.

Região de qualidade baixa – baixa confiabilidade
• Picos mal definidos e de tamanho pequeno.
• Linha de base confusa.
• Distância entre picos anterior e posterior inconstante.
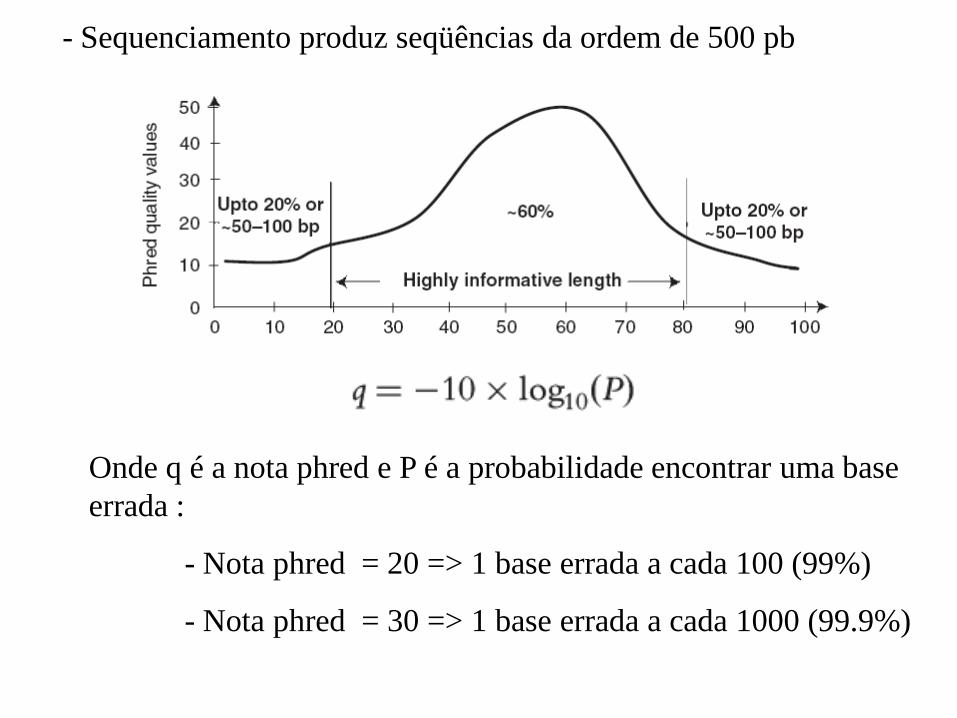
Onde q é a nota phred e P é a probabilidade encontrar uma base
errada :
- Nota phred = 20 => 1 base errada a cada 100 (99%)
- Nota phred = 30 => 1 base errada a cada 1000 (99.9%)
- Sequenciamento produz seqüências da ordem de 500 pb

Fevereiro 2001
Colins et al. Venter et al.

O genoma humano é praticamente o mesmo (99.9%) em todas as pessoas.
Apenas 2% do genoma contém genes
Humanos tem cerca de 25 mil genes; a função de muitos deles é desconhecida
Maioria das proteínas humanas tem similaridade com outros organismos
Pequenas diferenças genéticas entre indivíduos podem causar grandes diferenças nos efeitos de fármacos

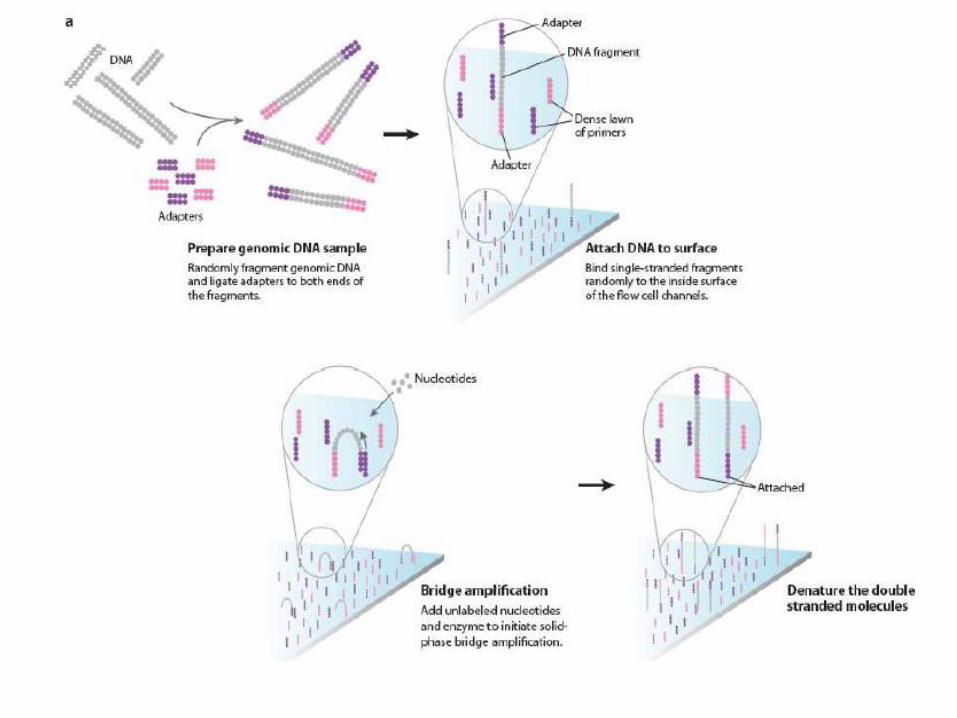
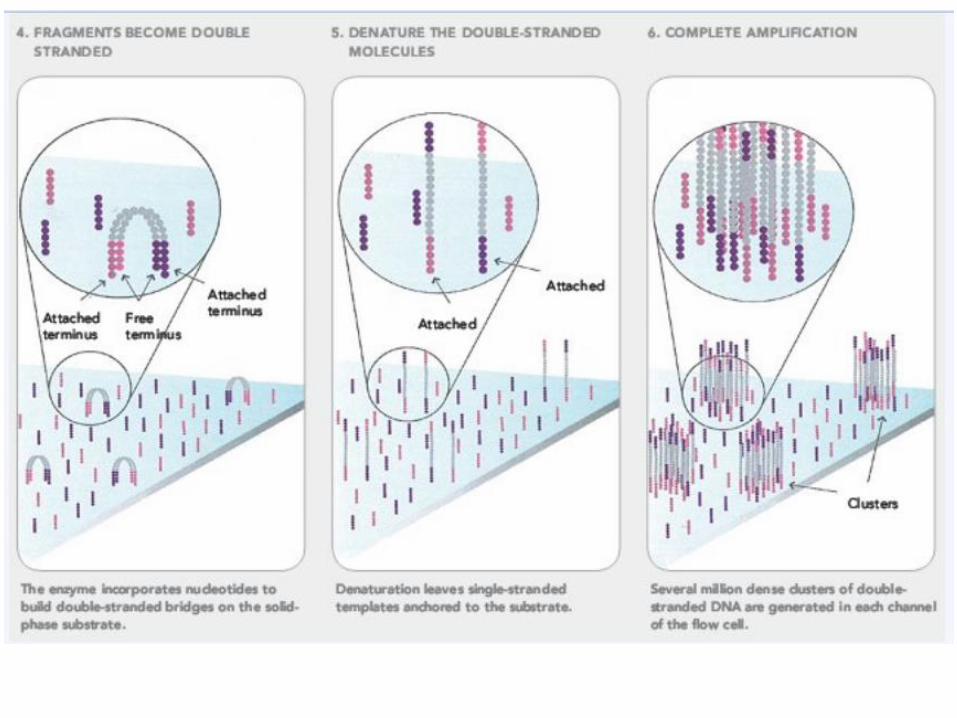
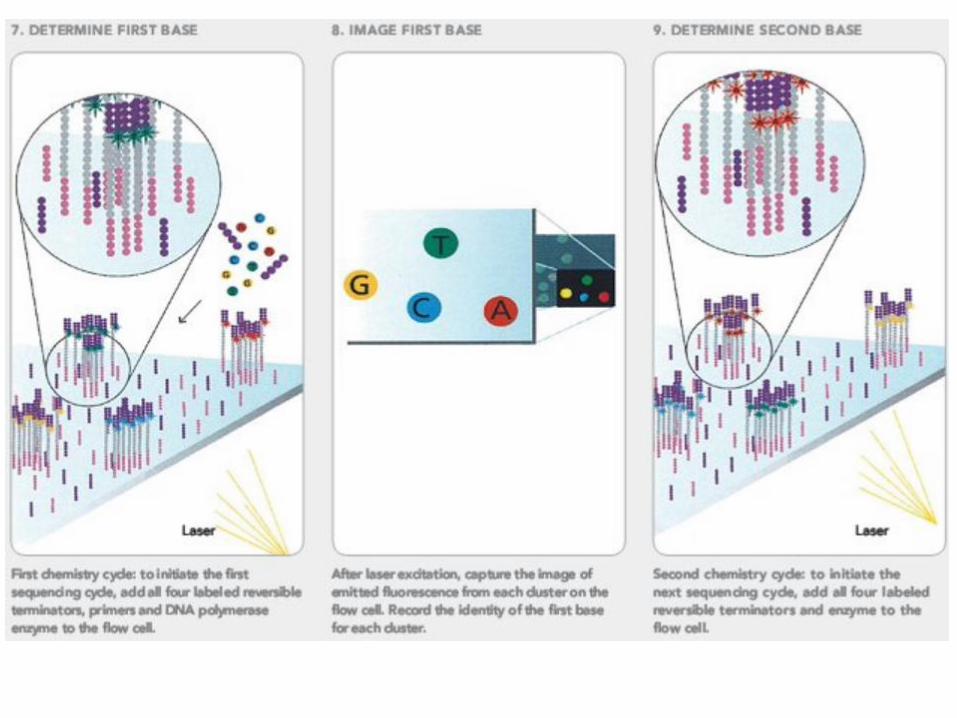
Solexa sequencing
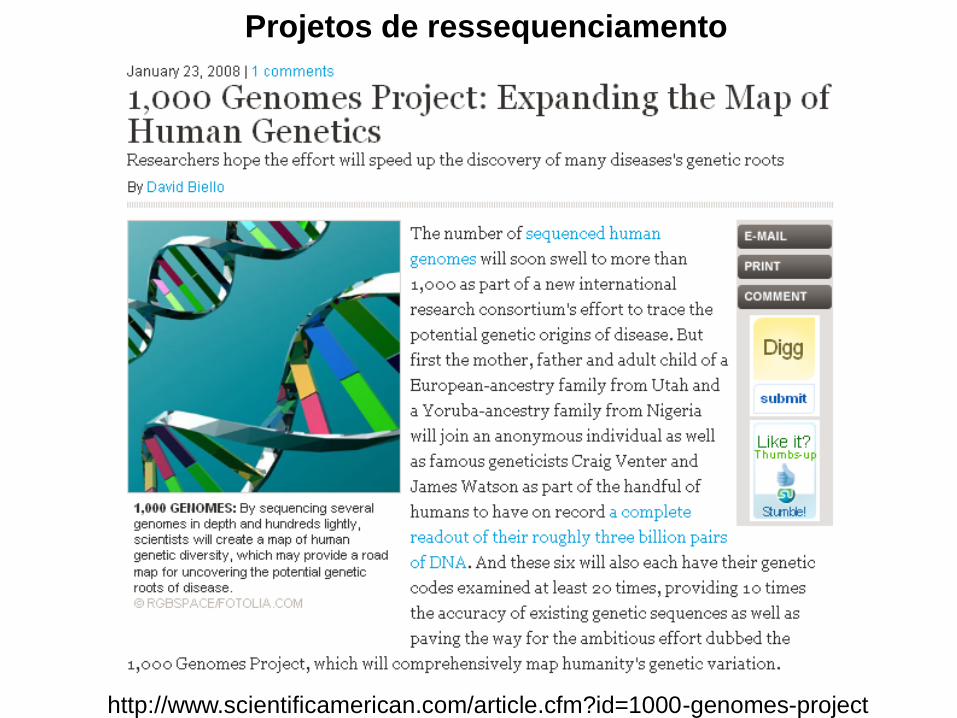
http://www.scientificamerican.com/article.cfm?id=1000-genomes-project
Projetos de ressequenciamento

3ª geração de sequenciadores
Sequenciador pessoal
Nanopore technology
$1000 mil dólares o aparelho + $900 do chip
1 chip = 25Gb
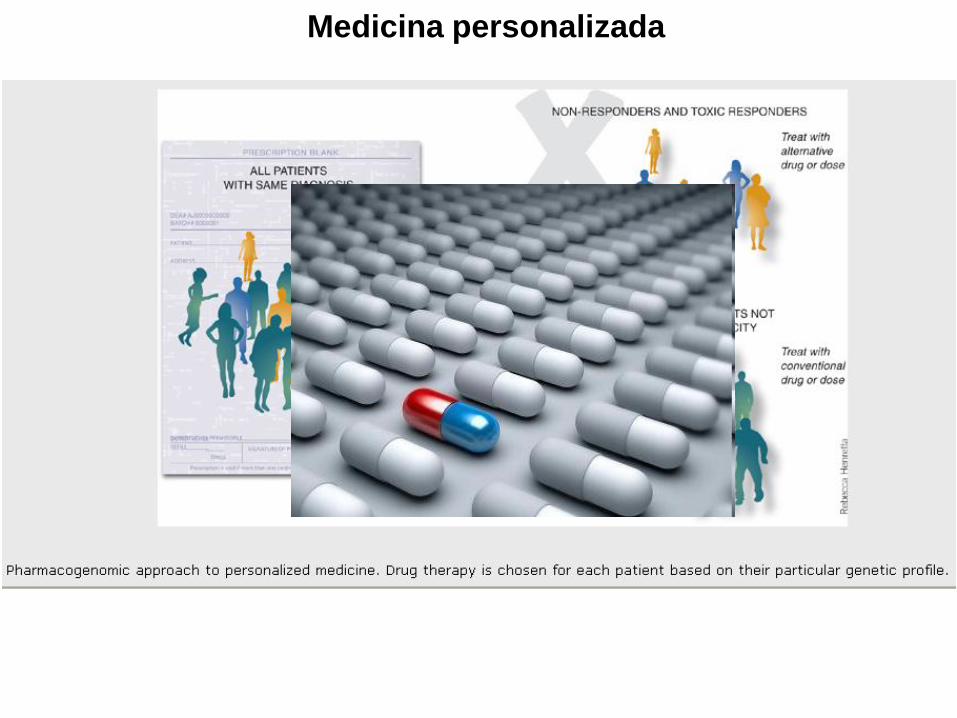
Medicina personalizada

Processamento de sequências brutas
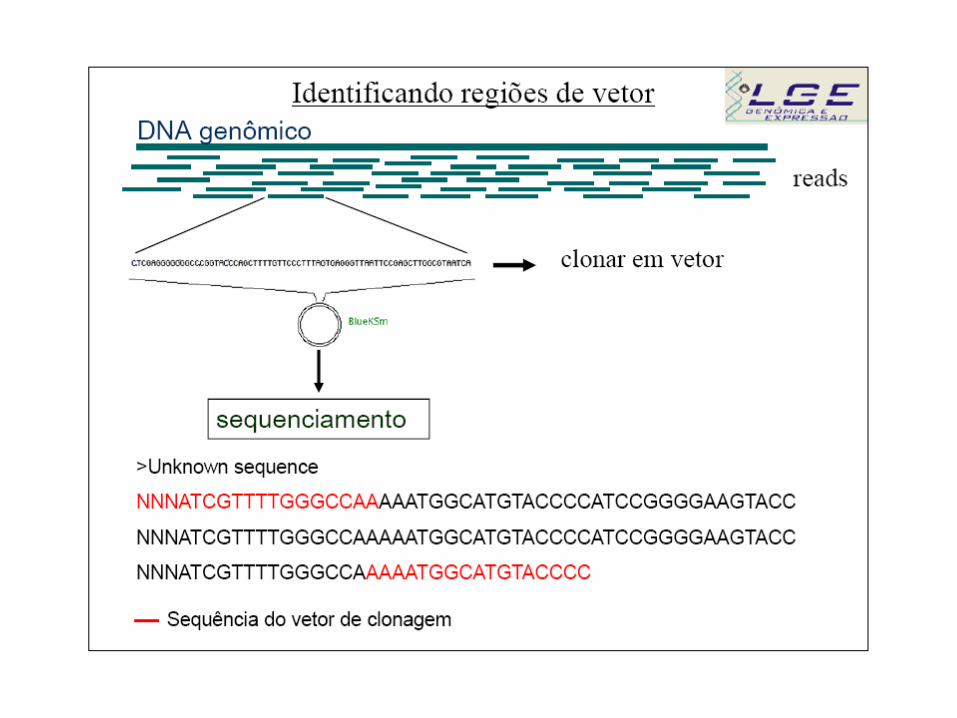

A identificação da região de vetor é feita através da comparação, via
BLAST (bl2seq), da sequência fasta com a sequência do vetor. A sequência
do vetor utilizado na clonagem pode ser obtida no site do
fabricante/distribuidor :
http://www.invitrogen.com/content.cfm?pageid=94
Outra possibilidade é utilizando o banco de sequências de vetores do NCBI
(BLASTn): http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html

Para análises em larga escala o programa cross_match (linux) faz a
identificação da região do vetor através da comparação entre as
sequências fasta e o banco de vetores mascarando a região do vetor
na sequência fasta. Isto é, substitui os nucleotídeos identificados com
vetor pela letra X :
>Unknown sequence
XXXXXXXXXXXXXXXXXXXAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAAAAATGGCATGTACCCCATCCGGGGAAGTACC
NNNATCGTTTTGGGCCAXXXXXXXXXXXXXXXXXX
X => Sequência do vetor de clonagem
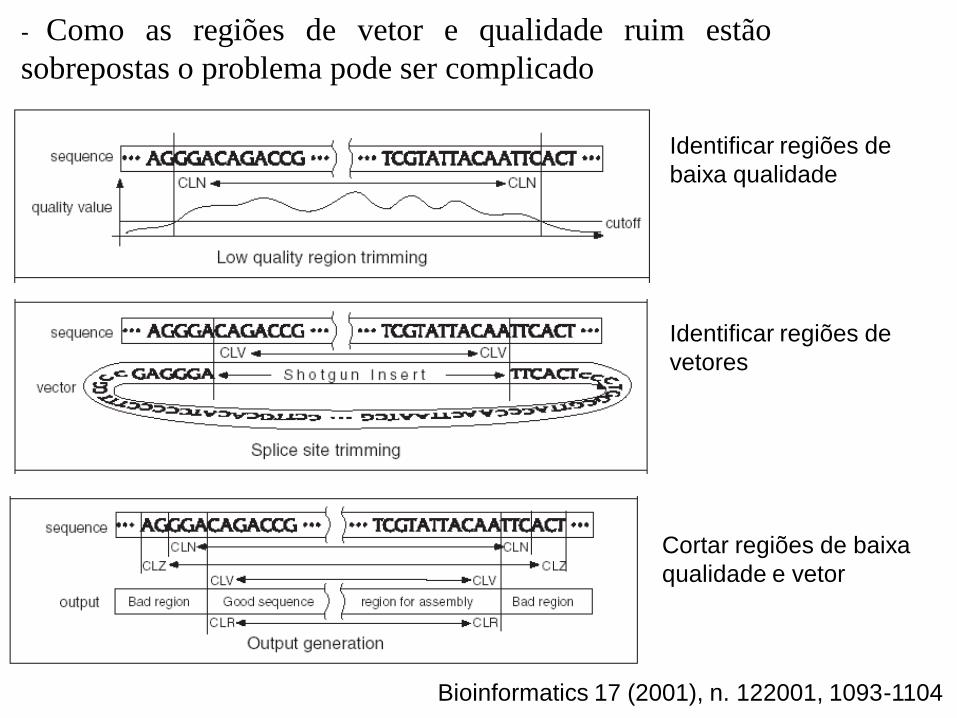
Identificar regiões de
baixa qualidade
Identificar regiões de
vetores
Cortar regiões de baixa
qualidade e vetor
- Como as regiões de vetor e qualidade ruim estão
sobrepostas o problema pode ser complicado
Bioinformatics 17 (2001), n. 122001, 1093-1104

Bioinformatics 17 (2001), n. 122001, 1093-1104
- Possíveis combinações de regiões com qualidade ruim
e vetores
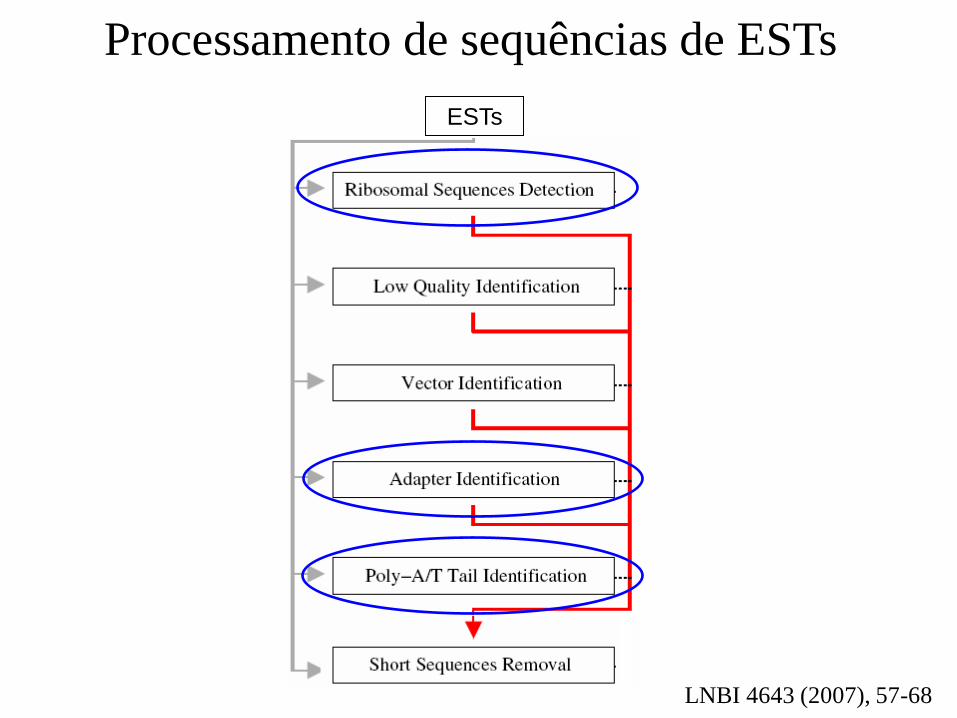
Processamento de sequências de ESTs
LNBI 4643 (2007), 57-68
ESTs

Montagem de sequências de DNA ou ESTs
- Utilizado para :
- montagem de genomas completos
- melhoria da qualidade de uma sequência de
interesse
- agrupar ESTs para análises de padrões de
expressões gênicas
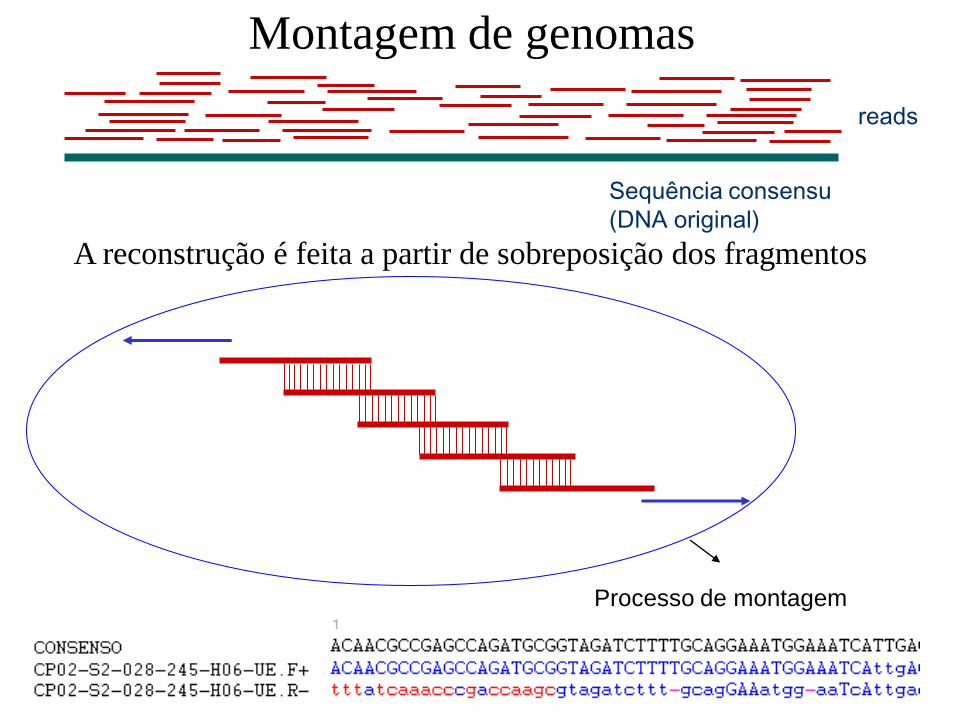
reads
Sequência consensu
(DNA original)
A reconstrução é feita a partir de sobreposição dos fragmentos
Processo de montagem
Montagem de genomas
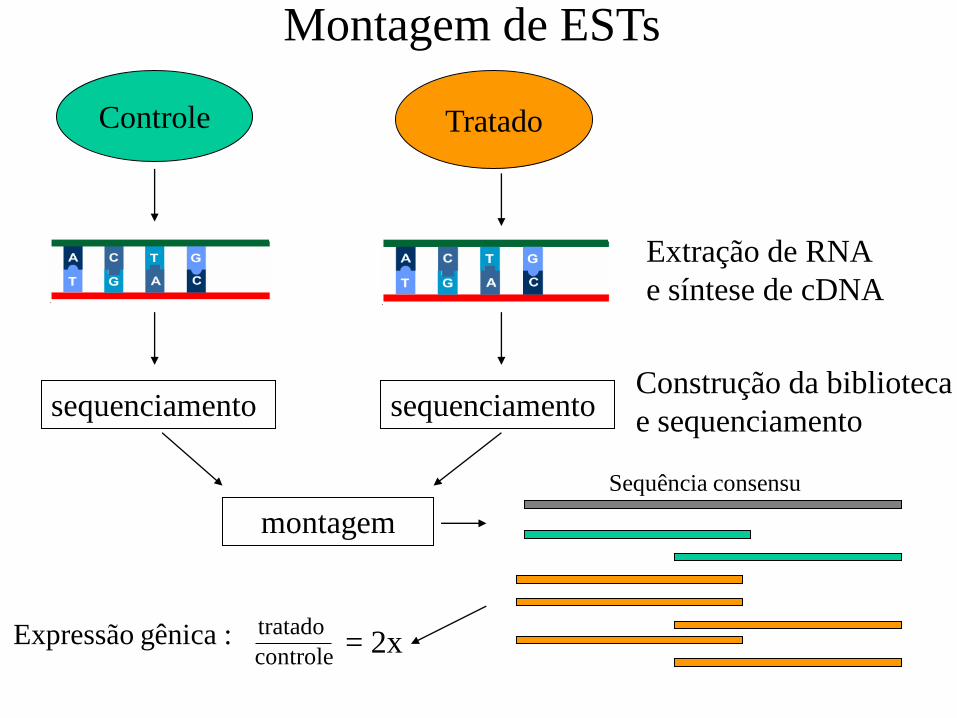
Controle Tratado
Extração de RNA
e síntese de cDNA
sequenciamento sequenciamento
montagem
Construção da biblioteca
e sequenciamento
Sequência consensu
Expressão gênica : controle
tratado = 2x
Montagem de ESTs

- Uma forma rápida de agregar alguma informação sobre uma
sequência desconhecida é compará-la com um banco de dados de
sequências com funções conhecidas
- Esta comparação é feita através de alinhamentos par a par entre
as sequências. Isto é, se o banco de dados possuir 1000 sequências
conhecidas serão realizados 1000 alinhamentos
?
Alinhamento de sequências
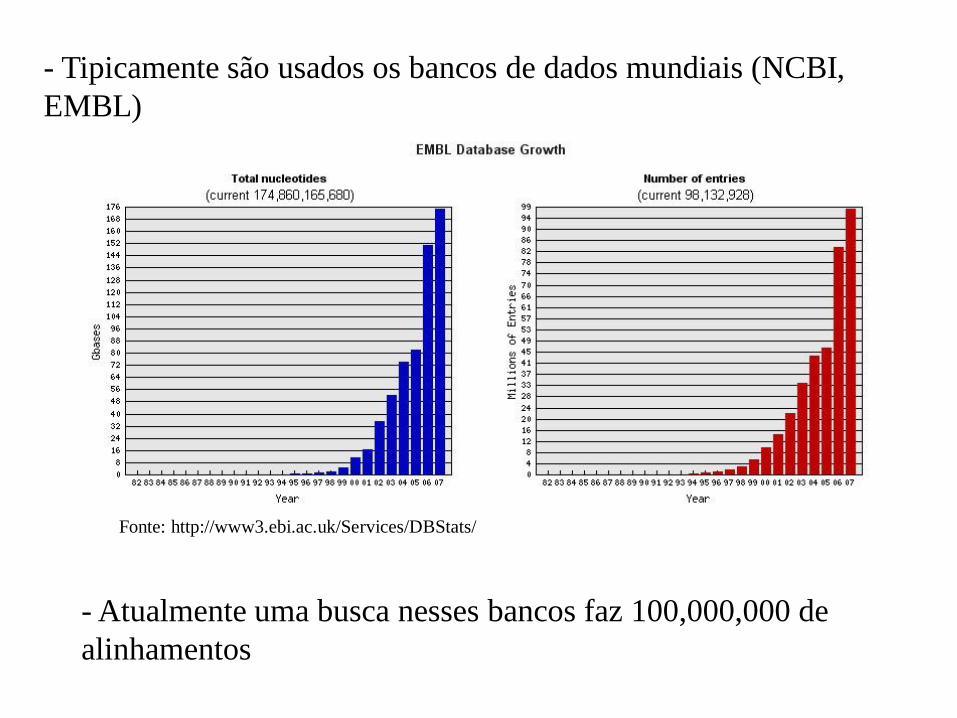
- Tipicamente são usados os bancos de dados mundiais (NCBI,
EMBL)
Fonte: http://www3.ebi.ac.uk/Services/DBStats/
- Atualmente uma busca nesses bancos faz 100,000,000 de
alinhamentos
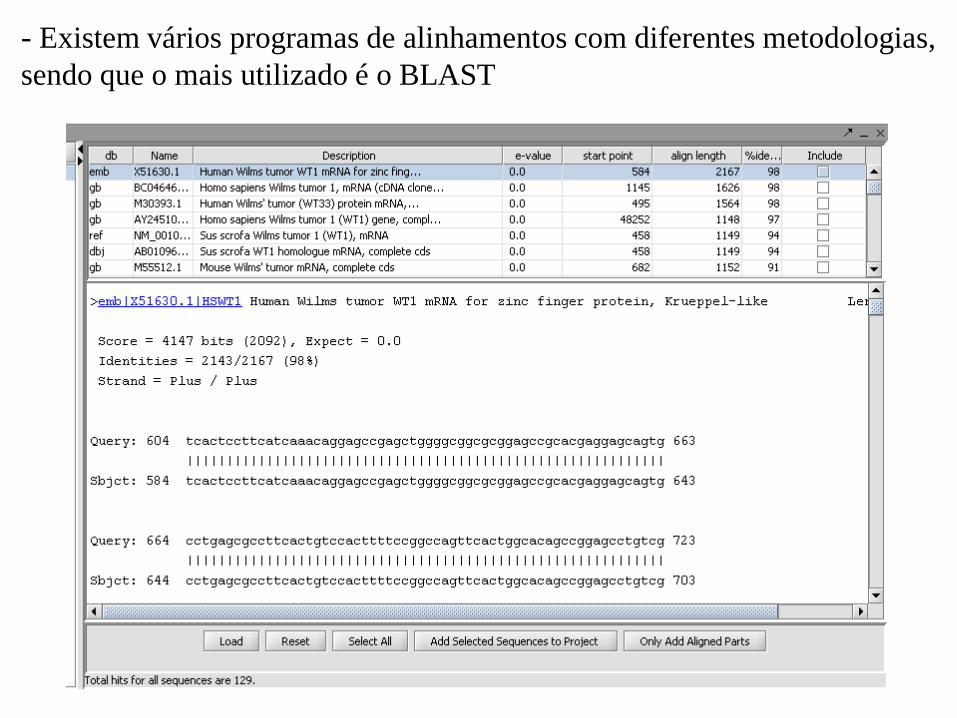
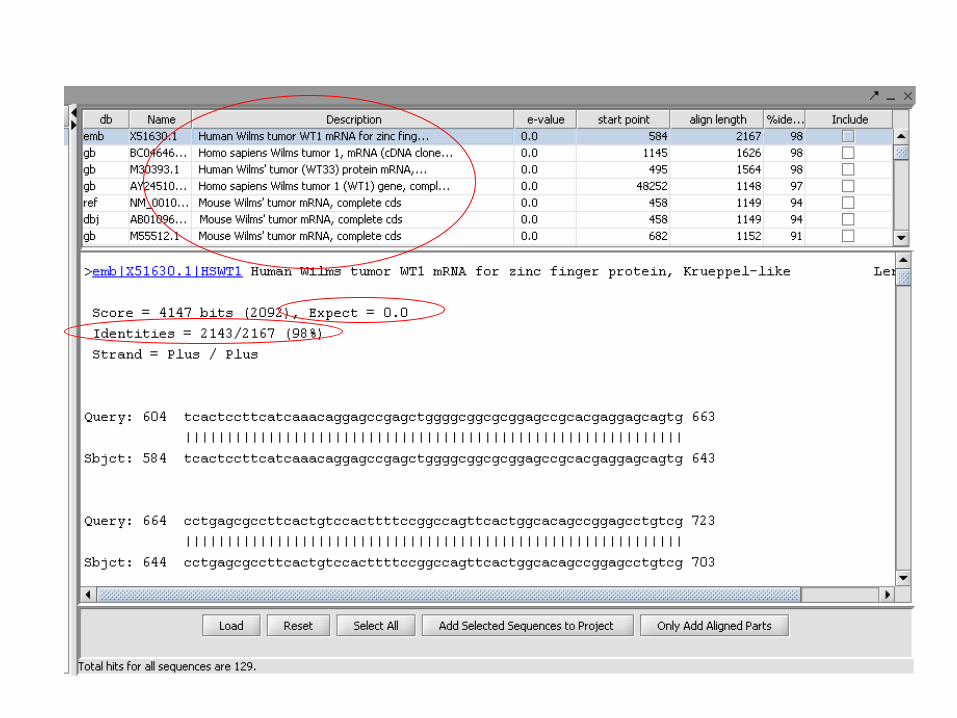
- Existem vários programas de alinhamentos com diferentes metodologias,
sendo que o mais utilizado é o BLAST

Outras aplicações de alinhamento de sequências
- Montagem de genomas e transcriptomas
- Comparação entre genomas
- ...

Alinhamentos de DNA
- A comparação entre sequências de DNA de organismos
diferentes é baseada no conceito de que estes organismos
originaram-se de um ancestral comum.
- No contexto de evolução as sequências de DNA sofrem
mutações. Estas modificações locais entre os nucleotídeos podem
ser :
- Inserções : inserção de uma base ou várias bases na
sequência
- Deleções : deleção de uma base ou mais bases na sequência
- Substituições : substituição de uma base por outra
- Portanto um programa de alinhamento de sequências biológicas
tem que considerar essas mutações

Exemplo :
Match = 1 Mismatch = -1
Gap = -2
- Gaps representam as inserções e deleções entre as sequências
- O melhor alinhamento entre duas sequências é aquele que
maximiza o score :
- Score = #Matchs * (1) + #Mismatch * (-1) + #Gaps * (-2)
= 24 – 4 – 10 = 10
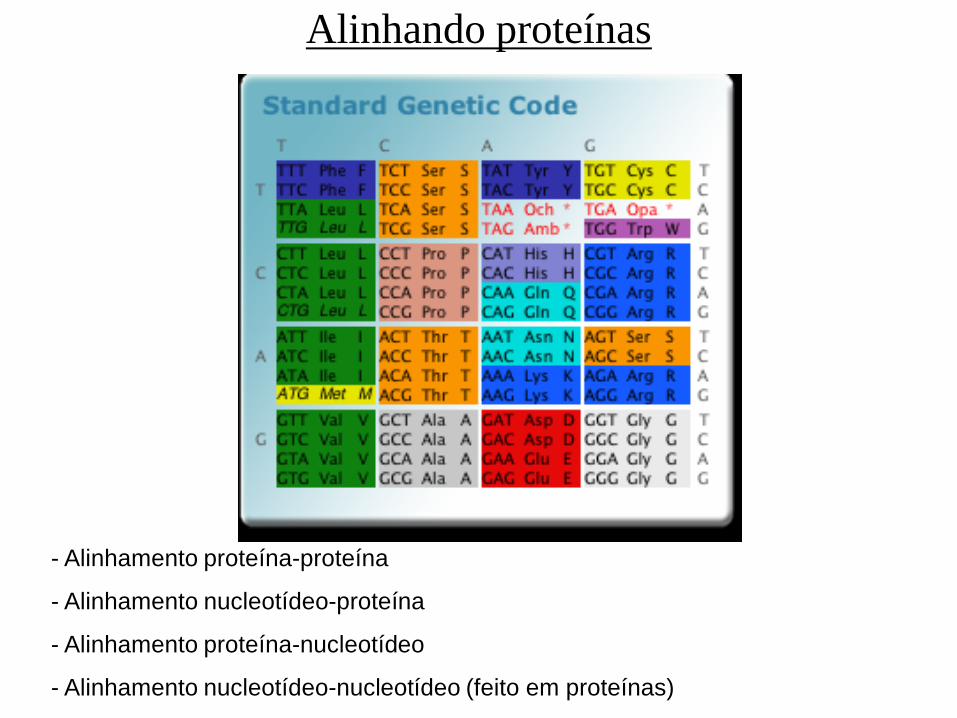
Alinhando proteínas
- Alinhamento proteína-proteína
- Alinhamento nucleotídeo-proteína
- Alinhamento proteína-nucleotídeo
- Alinhamento nucleotídeo-nucleotídeo (feito em proteínas)
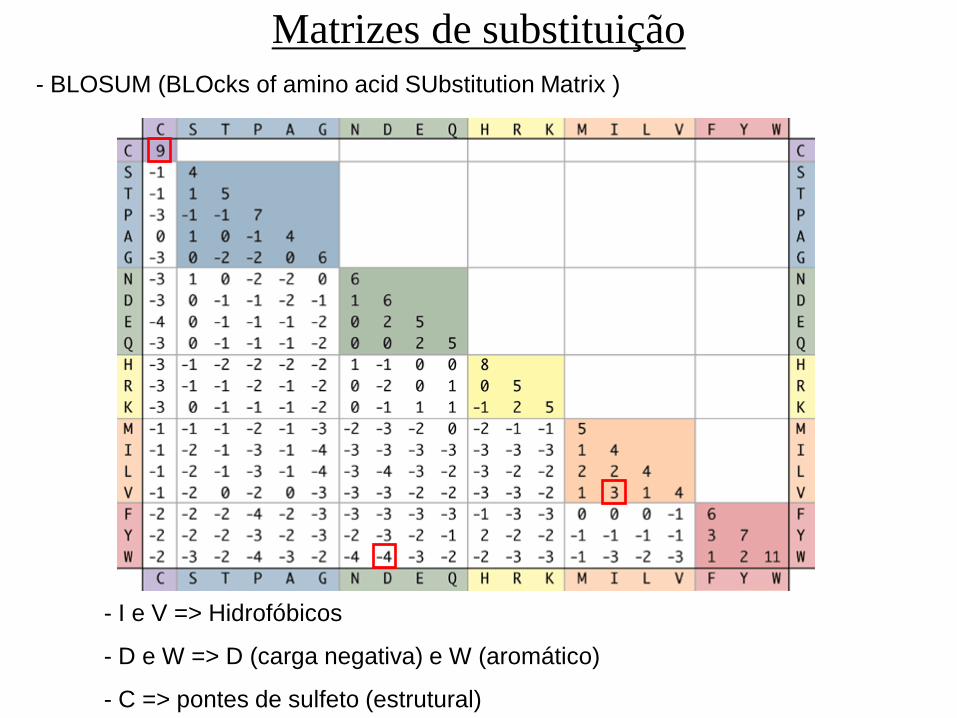
Matrizes de substituição
- BLOSUM (BLOcks of amino acid SUbstitution Matrix )
- I e V => Hidrofóbicos
- D e W => D (carga negativa) e W (aromático)
- C => pontes de sulfeto (estrutural)

- A matriz foi construída a partir de alinhamentos múltiplos globais
de 504 grupos de proteínas
- BLOSUM 62 : grupos com similaridade >62%
- BLOSUM 80 : grupos com similaridade >80%
- BLOSUM 45 : grupos com similaridade >45%
Query Length Substitution Matrix
<35 PAM-30
35-50 PAM-70
50-85 BLOSUM-80
>85 BLOSUM-62 PNAS 89 (1992), 10915-19919

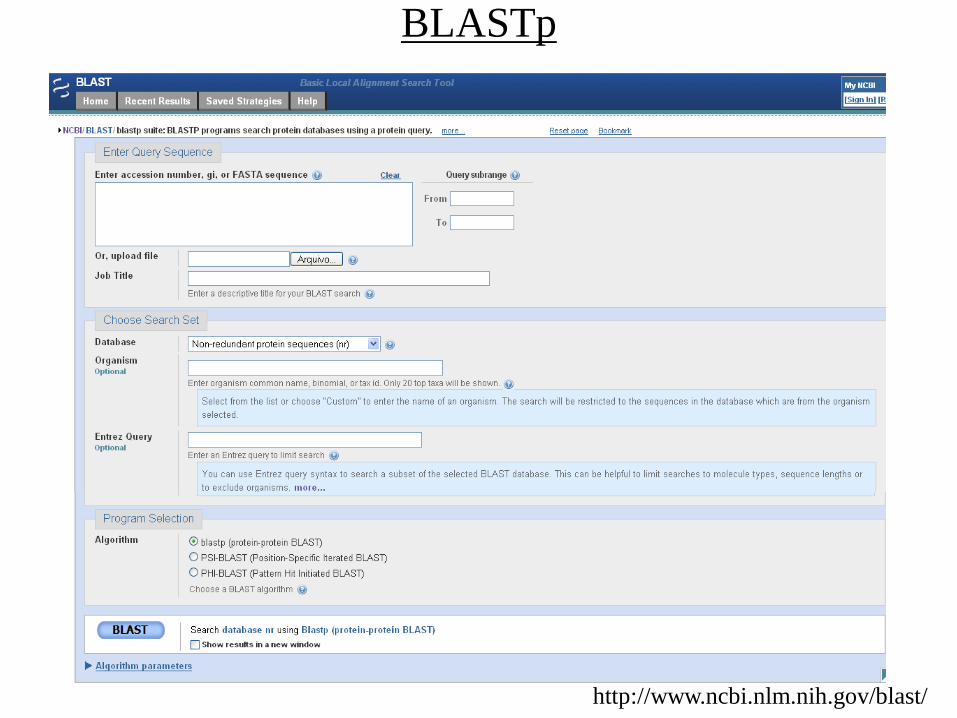
BLAST
• Basic Local Alignment Search Tool
• Algoritmo BLAST (Alstchul et al.; 1990 – J. Biol., 215, 403-410)
• Implementações: NCBI BLAST e WU-BLAST
• Acesso via web / local (linux)
• Consulta de seqüências em BDs biológicos (nt ou proteínas)
• Alinhamento – sobreposição de trechos semelhante de duas seqüências (seqs). BLAST traz pontuação e mostra alinhamentos.
• Similaridade – grau de semelhança de seqs num alinhamento.
• Homologia – genes com ancestral comum

• BDs – nucleotídeos, proteínas, domínios,
genomas específicos, dados particulares
• Blastp – prot / prot (distantes)
• Blastn – nt / nt (próximos)
• Blastx – nt trad / prot (novas seqs)
• Tblastn – prot / nt trad (regiões não anotadas)
• Tblastx – nt trad / nt trad
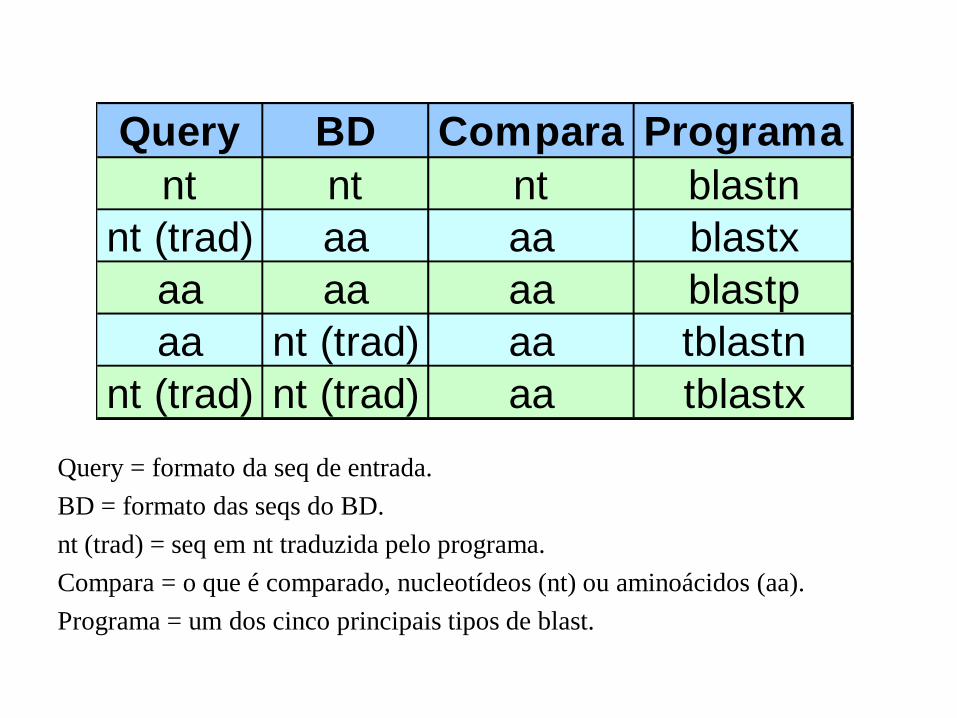
Query BD Compara Programa
nt nt nt blastn
nt (trad) aa aa blastx
aa aa aa blastp
aa nt (trad) aa tblastn
nt (trad) nt (trad) aa tblastx
Query = formato da seq de entrada.
BD = formato das seqs do BD.
nt (trad) = seq em nt traduzida pelo programa.
Compara = o que é comparado, nucleotídeos (nt) ou aminoácidos (aa).
Programa = um dos cinco principais tipos de blast.
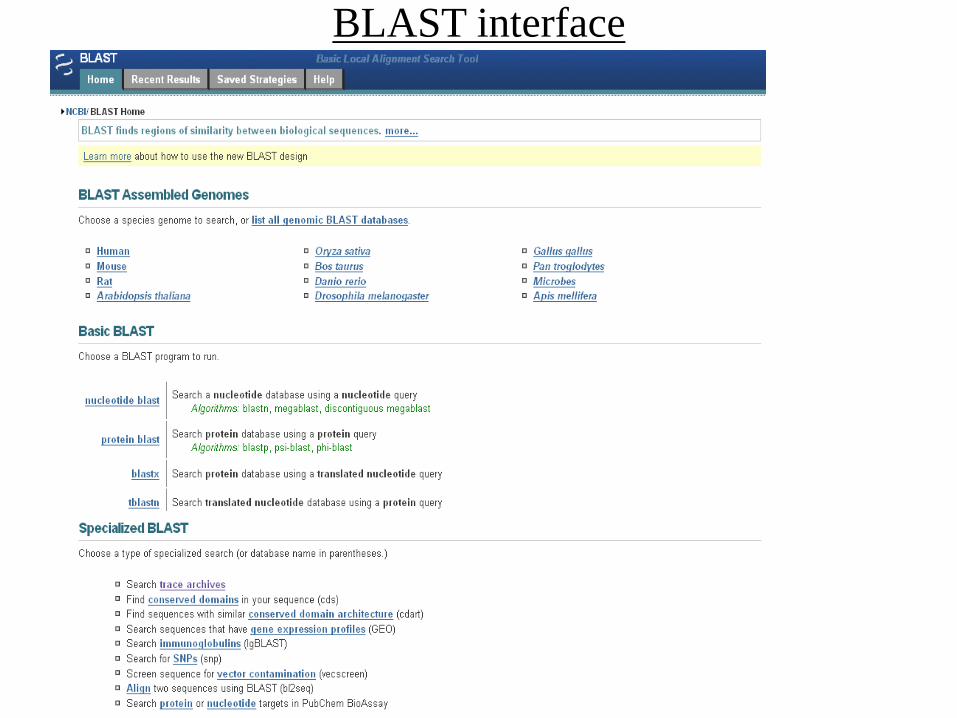
BLAST interface
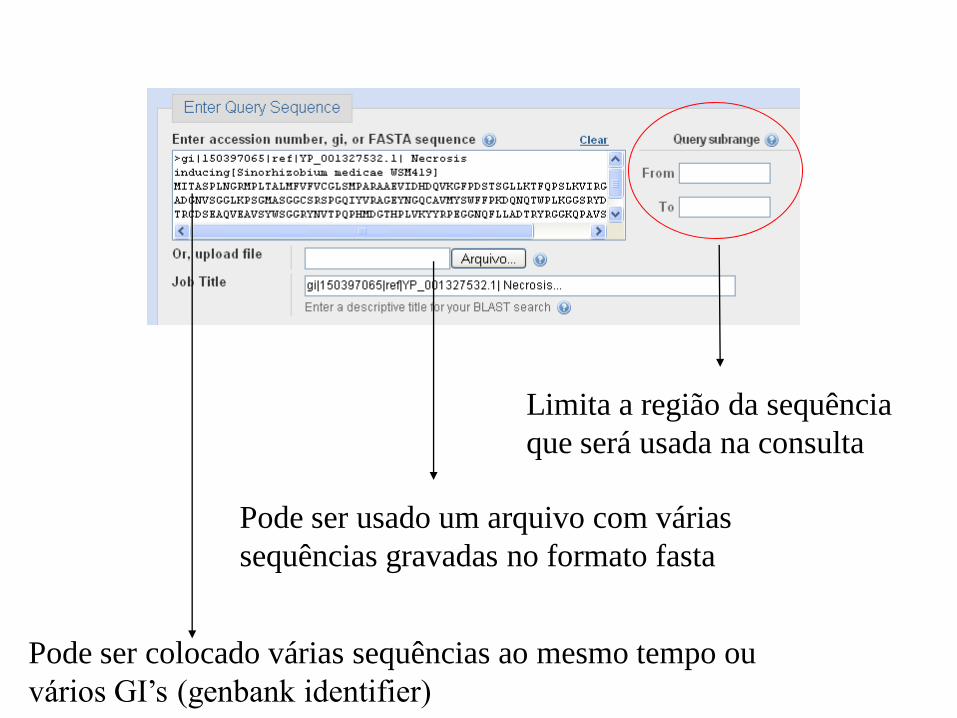
Limita a região da sequência
que será usada na consulta
Pode ser usado um arquivo com várias
sequências gravadas no formato fasta
Pode ser colocado várias sequências ao mesmo tempo ou
vários GI’s (genbank identifier)
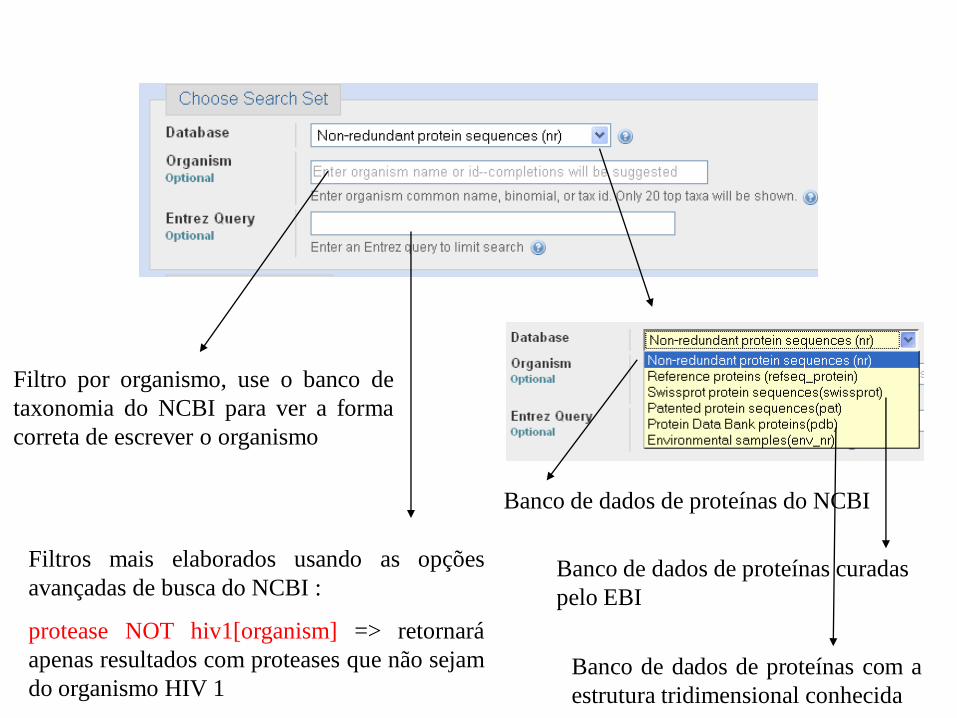
Banco de dados de proteínas do NCBI
Filtro por organismo, use o banco de
taxonomia do NCBI para ver a forma
correta de escrever o organismo
Filtros mais elaborados usando as opções
avançadas de busca do NCBI :
protease NOT hiv1[organism] => retornará
apenas resultados com proteases que não sejam
do organismo HIV 1
Banco de dados de proteínas curadas
pelo EBI
Banco de dados de proteínas com a
estrutura tridimensional conhecida
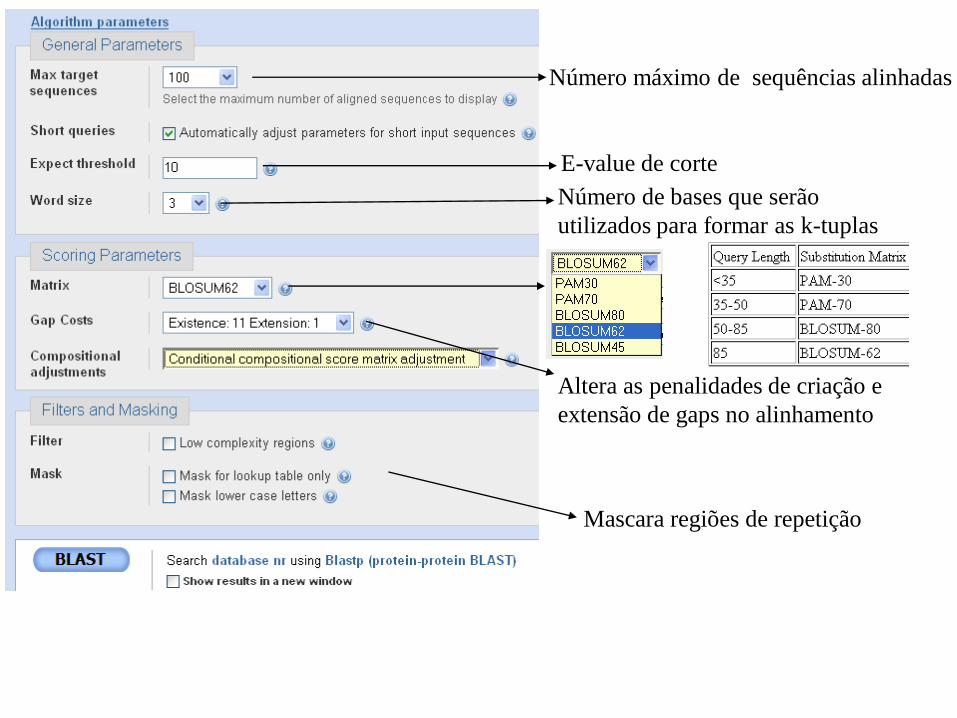
Número máximo de sequências alinhadas
E-value de corte
Número de bases que serão
utilizados para formar as k-tuplas
Altera as penalidades de criação e
extensão de gaps no alinhamento
Mascara regiões de repetição
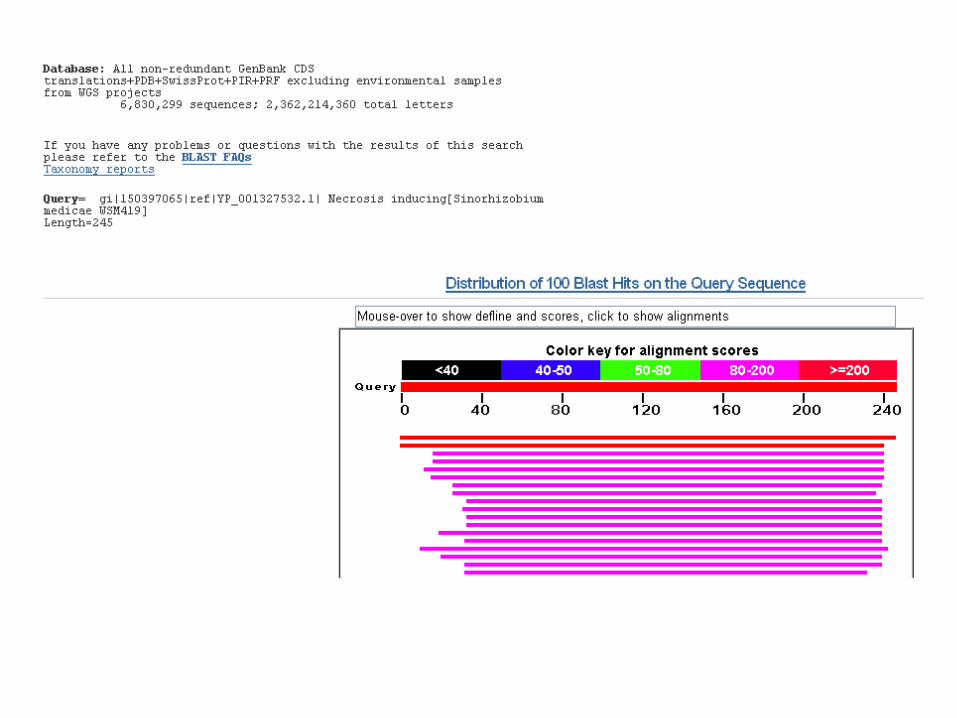
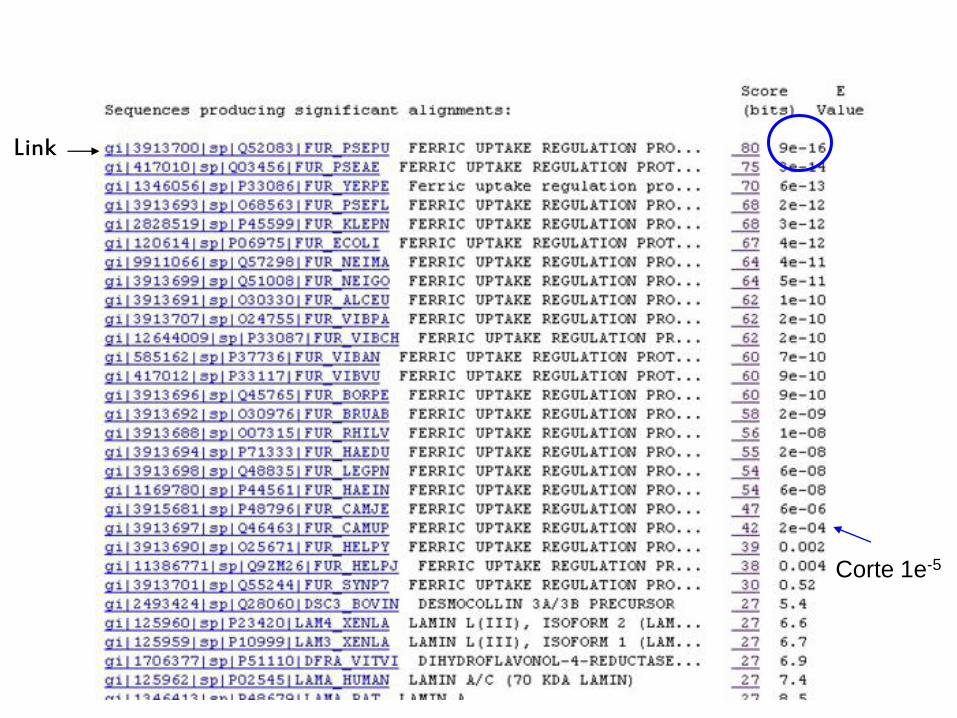
Link
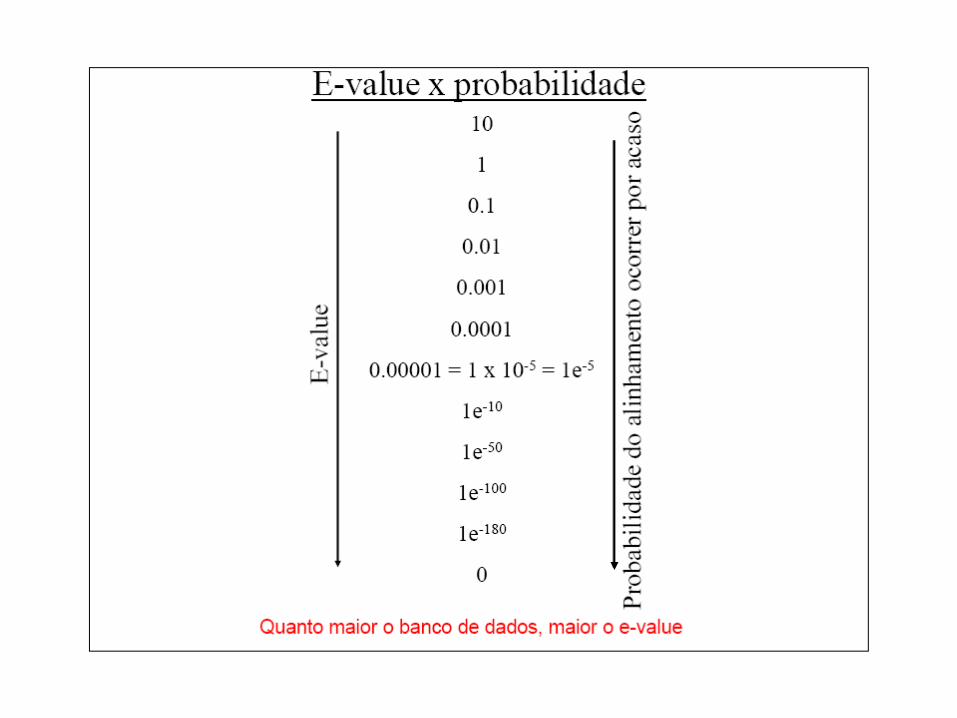
Corte 1e-5
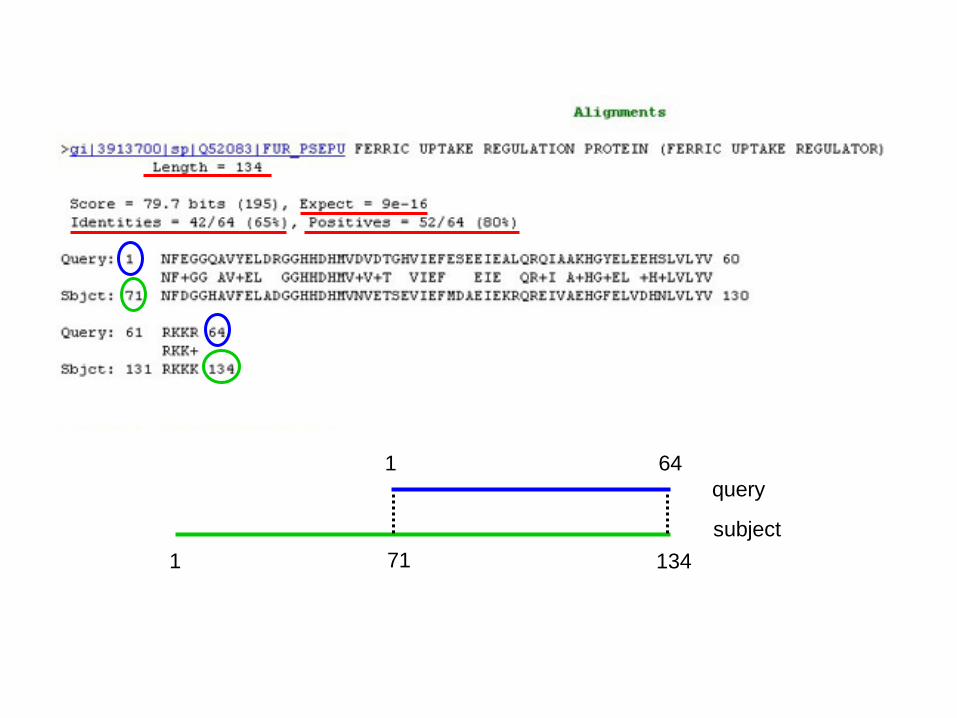
1
subject
query
71
1 64
134
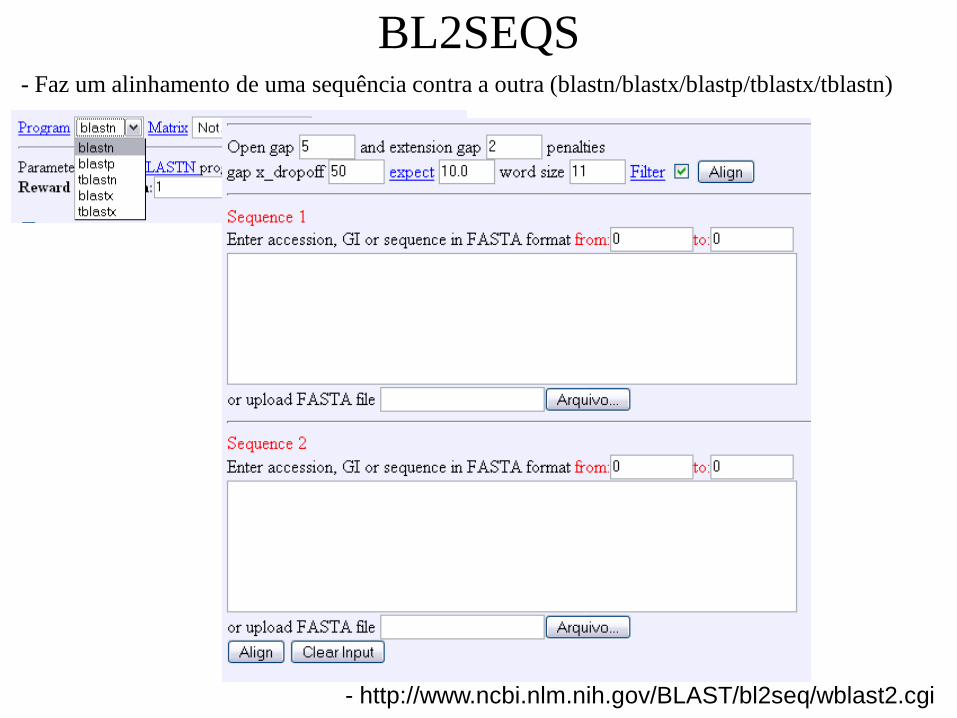
BL2SEQS
- http://www.ncbi.nlm.nih.gov/BLAST/bl2seq/wblast2.cgi
- Faz um alinhamento de uma sequência contra a outra (blastn/blastx/blastp/tblastx/tblastn)
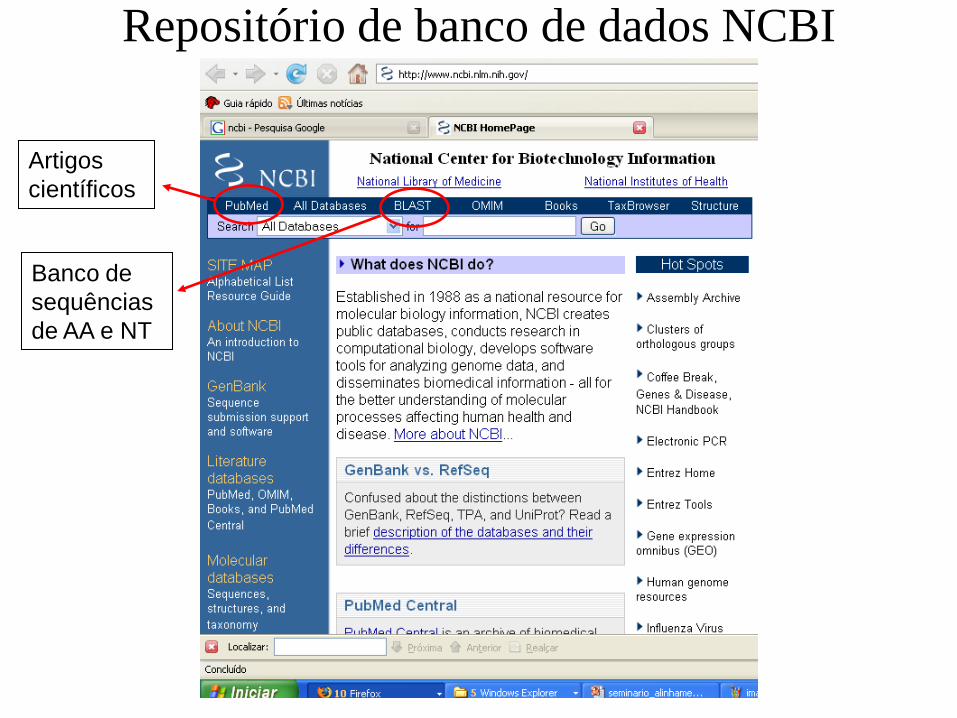
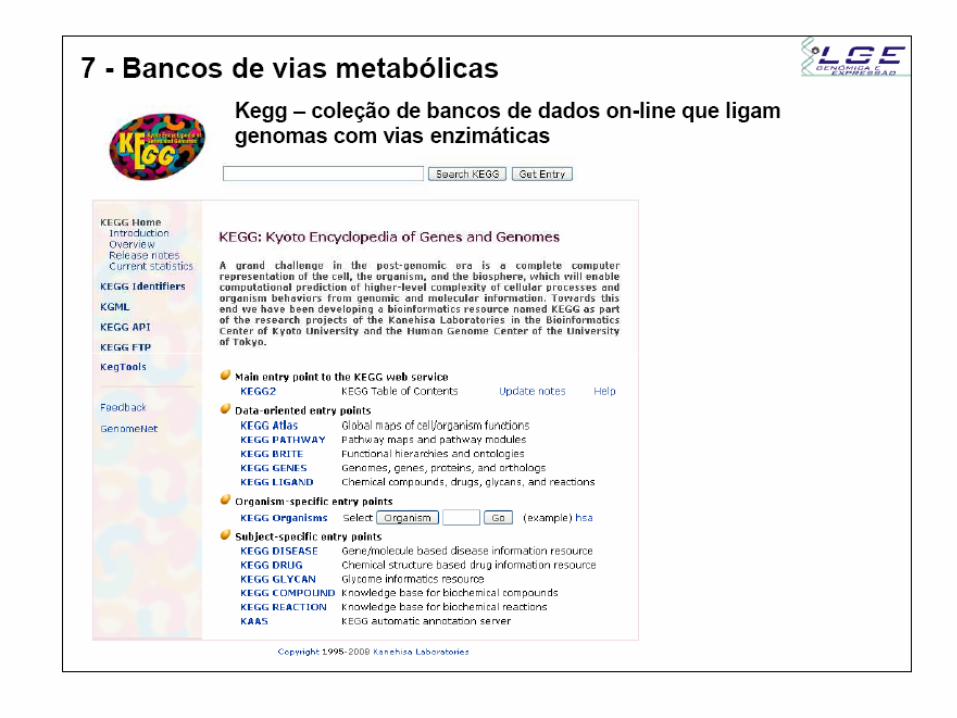
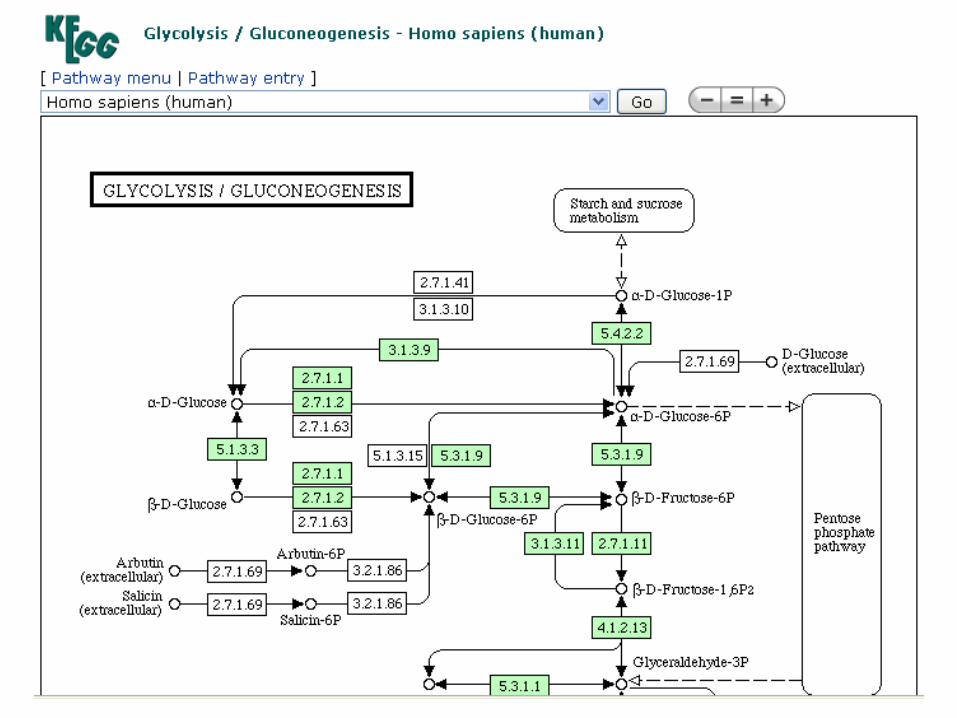
Repositório de banco de dados NCBI
Artigos
científicos
Banco de
sequências
de AA e NT
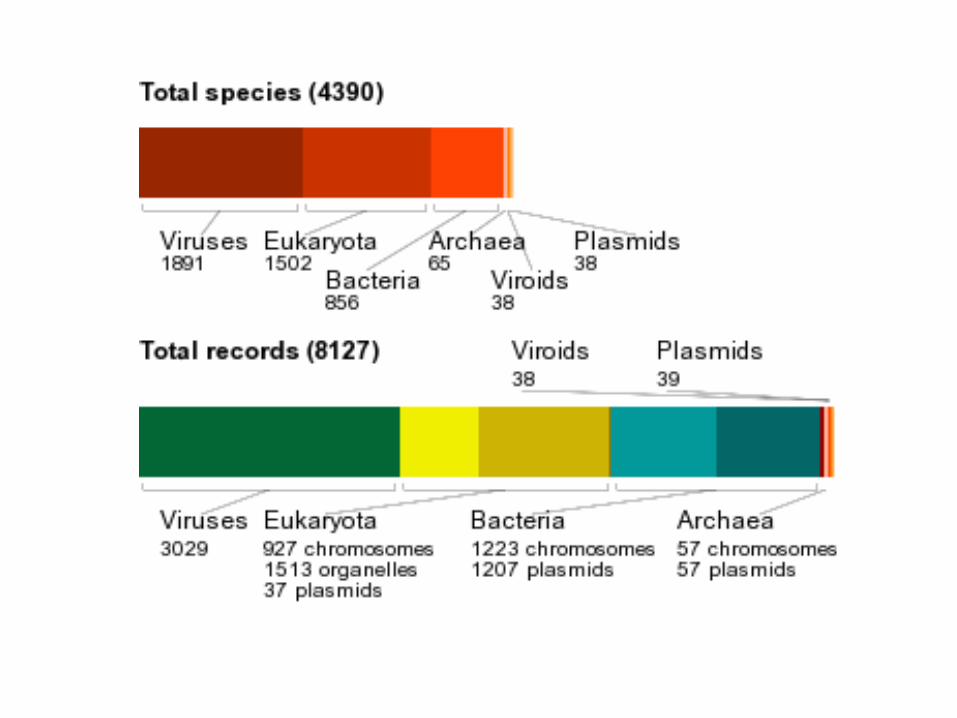
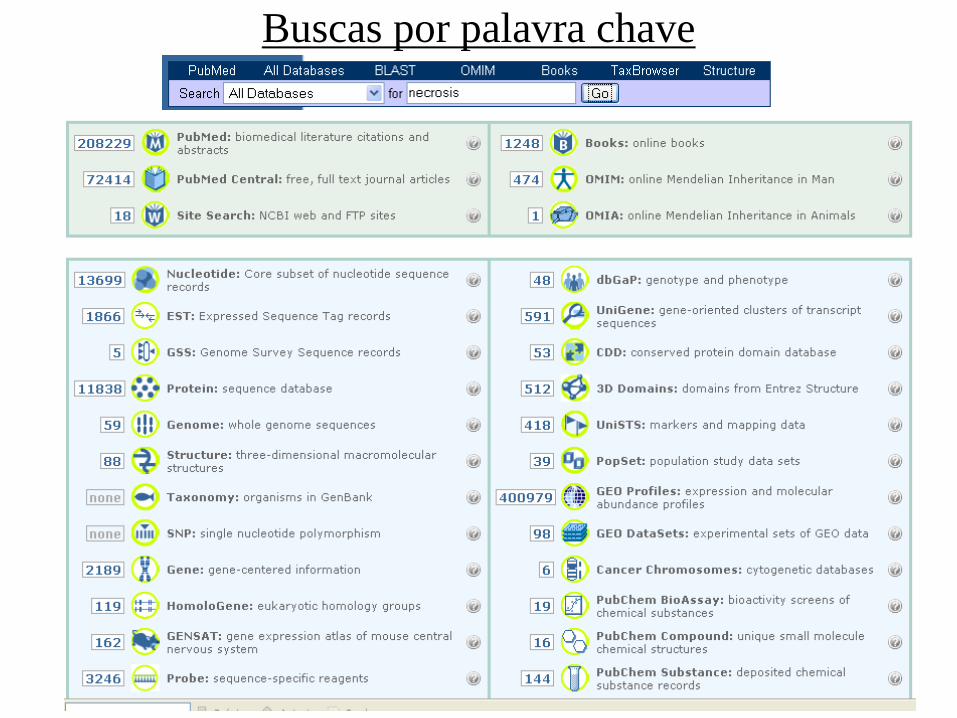
Buscas por palavra chave
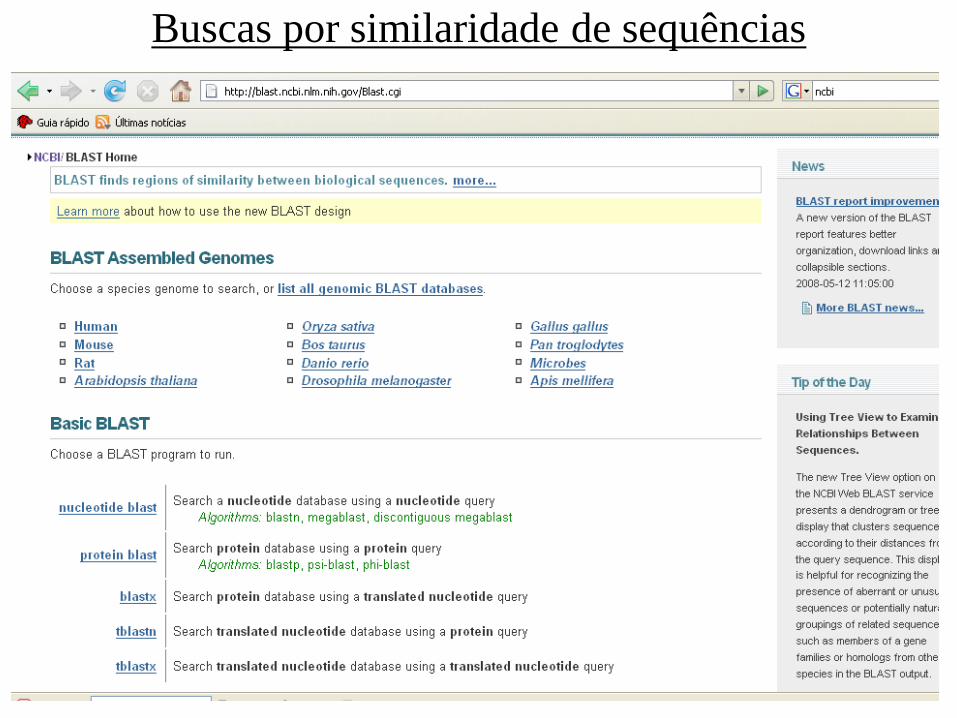
Buscas por similaridade de sequências





FIM


















