
CAPÍTULO 1A CLASSIFICAÇÃO DOS MÉTODOS 1 ANALÍTICOS
24
Ao longo deste livro, este ícone indica uma opor- tunidade de auto-aprendizado (em inglês) via in- ternet. Visite o endereço www.thomsonedu.com/chemistry/skoog para acessar tutoriais interativos, simulações orientadas e exercícios. 1A CLASSIFICAÇÃO DOS MÉTODOS ANALÍTICOS Os métodos analíticos são freqüentemente classificados como clássicos ou instrumentais. Os métodos clássicos, algumas vezes denominados métodos de via úmida, pre- cederam os métodos instrumentais por um século ou mais. 1A-1 Métodos clássicos Nos primeiros anos da química, muitas análises eram feitas após a separação dos componentes de interesse (os analitos) de uma amostra por precipitação, extração ou destilação. Em análises qualitativas, os componentes isolados eram tratados com reagentes, gerando produtos que poderiam ser reconhecidos por meio de suas cores, seus pontos de ebulição ou de fusão, suas solubilidades em uma série de solventes, seus cheiros, suas atividades ópticas ou seus índices de refração. Em análises quan- titativas, a quantidade do analito era determinada por medidas gravimétricas ou volumétricas. Nas medidas gravimétricas, a massa de um analito ou de algum composto produzido a partir do analito era determinada. Em procedimentos volumétricos, também denominados titulométricos, media-se o volume ou mas- sa de um regente padrão necessário para reagir comple- tamente com o analito. Esses métodos clássicos de separação e determi- nação de analitos ainda são empregados em muitos la- boratórios. A extensão de sua aplicação está, contudo, diminuindo com o passar dos anos e com o advento dos métodos instrumentais, que os têm suplantado. 1A-2 Métodos instrumentais Logo no início do século XX, os cientistas iniciaram a exploração de outros fenômenos alternativos àqueles empregados pelos métodos clássicos para a resolução de problemas analíticos. Assim, as medidas de propriedades físicas dos analitos, como condutividade, potencial de eletrodo, absorção ou emissão de luz, razão massa/carga e fluorescência começaram a ser utilizadas em análises quantitativas. Além disso, técnicas altamente eficientes de cromatografia a eletroforese começaram a substituir a destilação, a extração e a precipitação na separação dos componentes de misturas complexas antes da sua deter- minação qualitativa ou quantitativa. Esses novos méto- dos de separação e determinação de espécies químicas são coletivamente conhecidos como métodos instrumen- tais de análise. Introdução A química analítica lida com os métodos de determinação da composição química de amostras de materiais. Um método quali- tativo fornece informações sobre a identidade das espécies atômicas ou moleculares ou dos grupos funcionais presentes em uma amostra. Um método quantitativo, por outro lado, provê informações numéricas, como a quantidade relativa de um ou mais desses componentes. CAPÍTULO 1
Transcript of CAPÍTULO 1A CLASSIFICAÇÃO DOS MÉTODOS 1 ANALÍTICOS

Ao longo deste livro, este ícone indica uma opor-tunidade de auto-aprendizado (em inglês) via in-
ternet. Visite o endereço www.thomsonedu.com/chemistry/skoog para acessar tutoriais interativos, simulações orientadas e exercícios.
1A CLASSIFICAÇÃO DOS MÉTODOS ANALÍTICOS
Os métodos analíticos são freqüentemente classificados como clássicos ou instrumentais. Os métodos clássicos, algumas vezes denominados métodos de via úmida, pre-cederam os métodos instrumentais por um século ou mais.
1A-1 Métodos clássicos
Nos primeiros anos da química, muitas análises eram feitas após a separação dos componentes de interesse (os analitos) de uma amostra por precipitação, extração ou destilação. Em análises qualitativas, os componentes isolados eram tratados com reagentes, gerando produtos que poderiam ser reconhecidos por meio de suas cores, seus pontos de ebulição ou de fusão, suas solubilidades em uma série de solventes, seus cheiros, suas atividades ópticas ou seus índices de refração. Em análises quan-titativas, a quantidade do analito era determinada por medidas gravimétricas ou volumétricas.
Nas medidas gravimétricas, a massa de um analito ou de algum composto produzido a partir do analito era determinada. Em procedimentos volumétricos, também denominados titulométricos, media-se o volume ou mas-sa de um regente padrão necessário para reagir comple-tamente com o analito.
Esses métodos clássicos de separação e determi-nação de analitos ainda são empregados em muitos la-boratórios. A extensão de sua aplicação está, contudo, diminuindo com o passar dos anos e com o advento dos métodos instrumentais, que os têm suplantado.
1A-2 Métodos instrumentais
Logo no início do século XX, os cientistas iniciaram a exploração de outros fenômenos alternativos àqueles empregados pelos métodos clássicos para a resolução de problemas analíticos. Assim, as medidas de propriedades físicas dos analitos, como condutividade, potencial de eletrodo, absorção ou emissão de luz, razão massa/carga e fluorescência começaram a ser utilizadas em análises quantitativas. Além disso, técnicas altamente eficientes de cromatografia a eletroforese começaram a substituir a destilação, a extração e a precipitação na separação dos componentes de misturas complexas antes da sua deter-minação qualitativa ou quantitativa. Esses novos méto-dos de separação e determinação de espécies químicas são coletivamente conhecidos como métodos instrumen-tais de análise.
Introdução
A química analítica lida com os métodos de determinação da composição química de amostras de materiais. Um método quali-
tativo fornece informações sobre a identidade das espécies atômicas ou moleculares ou dos grupos funcionais presentes em uma amostra. Um método quantitativo, por outro lado, provê informações numéricas, como a quantidade relativa de um ou mais desses componentes.
CAPÍTULO
1

16 Princípios de Análise Instrumental
Muitos dos fenômenos por trás dos métodos ins-trumentais são conhecidos há um século ou mais. Sua aplicação por muitos cientistas, contudo, foi adiada pela falta de instrumentação simples e confiável. De fato, o crescimento dos métodos instrumentais de análise mo-dernos tem ocorrido paralelamente ao desenvolvimento das indústrias eletrônicas e de computadores.
1B TIPOS DE MÉTODOS INSTRUMENTAIS
Vamos primeiramente considerar algumas das caracte-rísticas químicas e físicas que são úteis em análises qua-litativas ou quantitativas. A Tabela 1-1 lista a maioria das propriedades características que são correntemente empregadas em análise instrumental. A maior parte das características mostradas na tabela necessita de uma fon-te de energia para estimular uma resposta mensurável do analito. Por exemplo, em uma emissão atômica, um aumento de temperatura do analito é inicialmente ne-cessário para produzir átomos gasosos do analito e, pos-teriormente, excitá-los a estados de energia mais altos. Os átomos excitados, então, emitem radiação eletromag-nética característica, que é a quantidade medida pelo instrumento. Fontes de energia podem tomar a forma de uma mudança térmica brusca, como no exemplo ante-rior; da radiação eletromagnética de uma região selecio-nada do espectro; da aplicação de uma grandeza elétrica,
como voltagem, corrente ou carga; ou talvez formas mais sutis, intrínsecas ao próprio analito.
Observe que as seis primeiras propriedades carac-terísticas listadas na Tabela 1-1 envolvem a interação do analito com a radiação eletromagnética. Na primei-ra propriedade, a radiação é produzida pelo analito; as próximas cinco propriedades envolvem alterações na ra-diação eletromagnética causadas pela sua interação com a amostra. Seguem quatro propriedades elétricas. Final-mente, outras cinco propriedades são agrupadas: massa, razão massa/carga, velocidade de reação, características térmicas e radioatividade.
A segunda coluna da Tabela 1-1 lista os métodos instrumentais baseados nas várias propriedades físicas e químicas. Fique certo de que não é sempre fácil selecio-nar o método mais adequado dentre as técnicas instru-mentais disponíveis e suas complementares clássicas. Al-gumas técnicas instrumentais são mais sensíveis do que as técnicas clássicas, mas outras não o são. Por meio de certas combinações de elementos e compostos, um méto-do instrumental pode ser mais seletivo, mas com outras, um método gravimétrico ou volumétrico pode sofrer menor interferência. Generalizações em termos de exa-tidão, conveniência ou rapidez são igualmente difíceis de serem estabelecidas. Também não é necessariamente verdadeiro afirmar que os procedimentos instrumentais empregam dispositivos mais caros ou sofisticados.
TABELA 1-1 Propriedades físicas e químicas empregadas pelos métodos instrumentais
Propriedades características Métodos instrumentais
Emissão de radiação Espectroscopia de emissão (raios X, UV, visível, eletrônica, Auger); fluorescência, fosfo-rescência e luminescência (raios X, UV e visível)
Absorção de radiação Espectrofotometria e fotometria (raios X, UV, visível, IR); espectroscopia fotoacústica, ressonância nuclear magnética e espectroscopia de ressonância de spin eletrônico
Espalhamento de radiação Turbidimetria; nefelometria; espectroscopia Raman
Refração de radiação Refratometria; interferometria
Difração de radiação Métodos de difração de raios X e de elétrons
Rotação de radiação Polarimetria; dispersão óptica rotatória; dicroísmo circular
Potencial elétrico Potenciometria; cronopotenciometria
Carga elétrica Coulometria
Corrente elétrica Amperometria. Polarografia
Resistência elétrica Condutometria
Massa Gravimetria (microbalança de quartzo)
Razão massa/carga Espectrometria de massas
Velocidade de reação Métodos cinéticos
Características térmicas Gravimetria e titulometria térmicas; calorimetria diferencial de varredura, análise térmi-ca diferencial, métodos condutométricos térmicos
Radioatividade Métodos de ativação e de diluição isotópica

Capítulo 1 • Introdução 17
Como observado anteriormente, além dos métodos listados na segunda coluna da Tabela 1-1 há um grupo de procedimentos instrumentais que são empregados para separação e resolução de compostos muito semelhantes entre si. A maioria desses procedimentos é baseada em cromatografia, extração por solventes ou eletroforese. Uma das características listadas na Tabela 1-1 é empre-gada para completar a análise após se realizar as sepa-rações cromatográficas. Por exemplo, a condutividade térmica, a absorção no ultravioleta e no infravermelho, o índice de refração e a condutividade elétrica são empre-gados com esse propósito.
Este livro dedica-se aos princípios, às aplicações e às características de desempenho dos métodos instru-mentais listados na Tabela 1-1, bem como aos proce-dimentos de separação cromatográficos e eletroforéti-cos. Os métodos clássicos não serão abordados porque consideramos que o leitor já estudou essas técnicas previamente.
1C INSTRUMENTOS PARA ANÁLISES
Um instrumento para análise química converte infor-mação sobre as características físicas ou químicas de um analito em informação que pode ser manipulada e interpretada por um ser humano. Assim, um instrumen-to analítico pode ser visto como um dispositivo de co-municação entre o sistema em estudo e o analista. Para obter a informação desejada do analito, é necessário prover um estímulo, o qual usualmente se apresenta na forma de energia eletromagnética, elétrica, mecânica ou nuclear, como exemplificado na Figura 1-1. O estímu-lo evidencia uma resposta do sistema em estudo, cujas natureza e magnitude são governadas pelas leis funda-mentais da física e da química. A informação resultante está contida no fenômeno que resulta da interação do estímulo com o analito. Um exemplo familiar consiste em passar uma banda estreita de comprimentos de onda de luz visível através de uma amostra para medir a ex-tensão de sua absorção pelo analito. A intensidade da luz é determinada antes e depois da sua interação com a
amostra, e a razão dessas intensidades produz uma me-dida da concentração do analito.
Geralmente, os instrumentos para análises químicas são constituídos por alguns poucos componentes bási-cos, alguns dos quais estão listados na Tabela 1-2. Para compreender as relações entre os componentes dos ins-trumentos e o fluxo de informação das características do analito, as quais passam pelos componentes até a saída numérica ou gráfica produzidas pelo instrumento, é ins-trutivo que se explore como a informação de interesse pode ser representada e transformada.
1C-1 Domínios de dados
O processo de medida é auxiliado por uma variedade de dispositivos que convertem informação de uma forma a outra. Antes de investigar como os instrumentos fun-cionam, é importante compreender como a informação pode ser codificada (representada) por meio de caracte-rísticas físicas e químicas e, particularmente, por sinais elétricos, tais como a corrente, a diferença de potencial e a carga. As várias formas de codificação são denomi-nadas domínios de dados. Um esquema de classificação foi desenvolvido, com base nesse conceito, para simpli-ficar bastante a análise de sistemas instrumentais e para permitir a compreensão do processo de medida.1 Como mostrado no mapa de domínios de dados da Figura 1-2, os domínios de dados podem ser classificados primeira-mente em não-elétricos e elétricos.
1C-2 Domínios não-elétricos
O processo de medida inicia e termina em domínios não-elétricos. A informação química e física de interesse em um experimento particular reside nestes domínios de dados. Dentre essas características estão o comprimen-to, a densidade, a composição química, a intensidade de luz, a pressão e outras, listadas na primeira coluna da Tabela 1-1.
É possível realizar uma medida obtendo-se a infor-mação que reside inteiramente em domínios não-elétri-cos. Por exemplo, a determinação da massa de um objeto empregando-se uma balança mecânica de dois pratos en-volve a comparação da massa do objeto, que é colocado em um dos pratos, com massas padrão, que são colocadas no segundo prato. A informação que representa a massa do objeto em unidades padrão é codificada diretamente pelo interessado, que providencia o processamento da informação por meio da soma das massas padrão para obter um número. Em outras balanças mecânicas, a for-
1 C.G. Enke, Anal. Chem., 1971, 43, 69A.
Estímulo Resposta
Fonte deenergia
Sistemaem
estudo
Informaçãoanalítica
FIGURA 1-1 Diagrama de blocos mostrando o processo ge-
ral de uma medida instrumental.

18 Princípios de Análise Instrumental
ça da gravidade sobre uma massa é amplificada mecani-camente empregando-se um braço mais longo do que o outro, aumentando, desta forma, a resolução da medida.
A determinação das dimensões lineares de um objeto com uma régua e a medida de volume de uma amostra líquida com um cilindro graduado são outros exemplos de medidas realizadas exclusivamente em domínios não-elé-tricos. Tais medidas são sempre associadas aos métodos clássicos de análise. O advento dos processadores de si-nais eletrônicos de baixo custo, dos transdutores sensíveis e dos dispositivos de leitura tem levado ao desenvolvi-mento de um grande número de instrumentos eletrônicos, os quais adquirem a informação de um domínio não-elé-trico, a processam em domínios elétricos e, finalmente, a apresentam em uma forma não-elétrica. Dispositivos ele-trônicos processam a informação e a transformam de um domínio a outro de forma análoga à multiplicação da mas-sa em balanças de braços assimétricos. Uma vez que esses dispositivos estão disponíveis e são capazes de processar informação de forma rápida e sofisticada, os instrumentos que se atêm exclusivamente à transferência de informa-ção no domínio não-elétrico estão se tornando obsoletos. Contudo, a informação que buscamos inicia nas proprie-
TABELA 1-2 Alguns exemplos de componentes de instrumentos
Instrumento
Fonte de energia (estímulo)
Informação analítica
Seleção da informação
Trasdutor de entrada
Domínio de dados da informação transduzida
Processador de sinal/leitura
Fotômetro Lâmpada de tungstênio
Feixe de luz ate-nuado
Filtro Fotodiodo Correnteelétrica
Amplificador, di-gitalizador, mos-trador de LEDs
Espectrômetro de emissão atô-mica
Plasma induzido por acoplamento
Radiação UV ou visível
Monocromador Tubo fotomulti-plicador
Correnteelétrica
Amplifcador, di-gitalizador, mos-trador digital
Coulômetro Fonte de corren-te contínua
Carga requerida para reduzir ou oxidar um analito
Potencial da célula
Eletrodos Tempo Amplificador, relógio digital
pHmetro Amostra/eletrodo de vidro
Atividade do íon hidrogênio
Eletrodo de vidro
Eletrodos de vidro ecalomelano
Voltagemelétrica
Amplificador, di-gitalizador, mos-trador digital
Espectrômetro de massas
Fonte de íons Razão massa/carga
Analisador de massas
Multiplicador de elétrons
Correnteelétrica
Amplificador, digitalizador, sistema computa-cional
Cromatografia gasosa com ioni-zação em chama
Chama Concentração de íons vs. tempo
Cromatografia em coluna
Eletrodospolarizados
Correnteelétrica
Eletrômetro, digitalizador, sistema computa-cional
Analógica
Tempo
Dig
ital
Domíniofísico-químico
Domínio físico-químico
Domínios não-elétricos
Largura de pulso
FaseContagem
Serial
Paralelo
Número
Posição
do ponteiro
Corrente
Voltagem
Carga
Freq
üênc
ia
FIGURA 1-2 Mapa de domínios de dados. A metade superior
(sombreada) do mapa consiste dos domínios não-elétricos. A
metade inferior é constituída pelos domínios elétricos. Observe
que o domínio digital abrange ambos os domínios, elétrico e
não-elétrico.Tutorial: Aprenda mais sobre os domínios de dados.

Capítulo 1 • Introdução 19
dades do analito e termina em um número, e ambos cons-tituem representações não-elétricas. O objetivo final em uma medida analítica é obter um resultado numérico que seja, de alguma maneira, proporcional à característica físi-ca ou química que se busca do analito.
1C-3 Domínios elétricos
As formas de codificar informação como quantidades elétricas podem ser subdivididas em domínio analógico, domínio do tempo e domínio digital, como ilustrado na metade inferior do mapa circular da Figura 1-2. Obser-ve que o domínio digital não é constituído somente de sinais elétricos, mas também inclui uma representação não-elétrica devido ao fato de que os números, apresen-tados em um mostrador de qualquer tipo, constituem uma informação digital.
Qualquer processo de medida pode ser representado como uma série de conversões interdomínios. Por exem-plo, a Figura 1-3 ilustra a medida da intensidade de fluo-rescência de uma amostra de água tônica contendo traços de quinino e, de uma forma geral, algumas das conversões de domínio de dados que são necessárias para se chegar a um número relacionado à intensidade. A intensidade da fluorescência é significativa nesse contexto porque é proporcional à concentração de quinino na água tônica, a qual representa, em última instância a informação dese-jada. A informação inicia na solução da água tônica como
concentração de quinino. Essa informação é manifesta-da na amostra aplicando-se um estímulo sob a forma de energia eletromagnética do laser mostrado na Figura 1-3. A radiação interage com as moléculas de quinino presen-tes na água tônica e produz emissão por fluorescência em uma região espectral característica do quinino e de mag-nitude proporcional à sua concentração. A radiação que não se relaciona com a concentração do quinino é remo-vida do feixe de luz por um filtro óptico, como mostrado na Figura 1-3. A intensidade de emissão fluorescente, que é uma informação não-elétrica, é codificada em um sinal elétrico por um dispositivo especial denominado transdu-tor de entrada. O tipo particular de transdutor empregado nesse experimento é um fototransdutor, do qual há di-versos tipos; muitos deles serão discutidos nos Capítulos 6 e 7. Nesse exemplo, o transdutor de entrada converte a fluorescência da água tônica em uma corrente elétrica I, proporcional à intensidade da radiação. A relação ma-temática entre a saída elétrica e a potência radiante de entrada na sua superfície é denominada função de trans-ferência do transdutor.
A corrente do fototransdutor passa através de um resistor R que, de acordo com a lei de Ohm, produz uma diferença de potencial V proporcional a I que, por sua vez, é proporcional à intensidade de fluorescência. Final-mente, V é medido por um voltímetro digital para forne-cer uma leitura proporcional à concentração de quinino na amostra.
Intensidadede fluorescência
do analito
Intensidadeda fonte
Leis daquímica
e da física
Função detransferênciado transdutor
Lei deOhm V = IR
Função detransferênciado medidor
Fluxo deinformação
Fonte de energia
Laser
(b)
(a)
Governado por
(c)
Correnteelétrica I
Filtroóptico
Emissão porfluorescência
Resistor
Fototransdutor
Voltímetro digital
Água tônica(analito)
I R V
Voltagem V Número
FIGURA 1-3 Diagrama de blocos de um fluorímetro mostrando (a) um diagrama geral do instru-
mento, (b) um diagrama representando o fluxo de informação através de vários domínios de dados
no instrumento e (c) as regras que governam as transformações de domínio de dados durante o
processo de medida.
20 Princípios de Análise Instrumental
Voltímetros, mostradores alfa-numéricos, motores elétricos, telas de monitores de computadores e muitos outros dispositivos, que servem para converter dados dos domínios elétricos para os não-elétricos, são denomina-dos transdutores de saída. O voltímetro digital do fluorí-metro da Figura 1-3 é um dispositivo um tanto complexo que converte a voltagem V em um número num mostra-dor de cristal líquido de forma que ela pode ser lida e interpretada pelo usuário do instrumento. Iremos consi-derar detalhadamente a natureza do voltímetro digital e de vários outros circuitos elétricos e sinais nos Capítulos de 2 a 4.
Sinais no domínio analógico
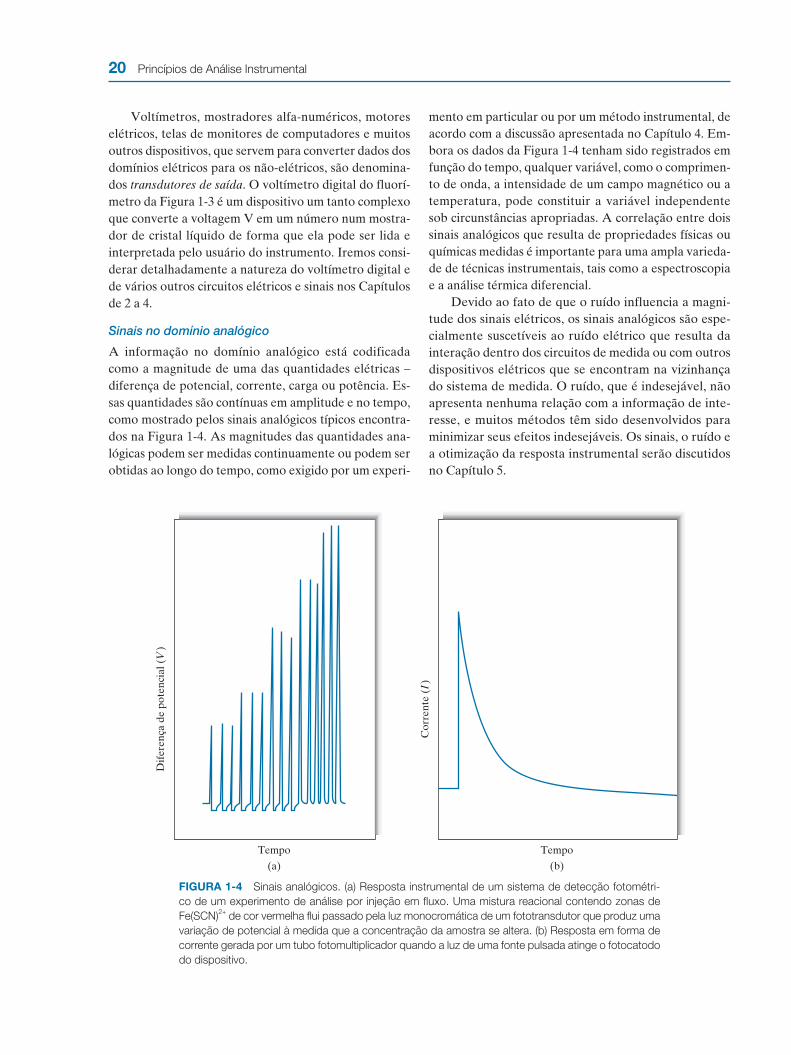
A informação no domínio analógico está codificada como a magnitude de uma das quantidades elétricas – diferença de potencial, corrente, carga ou potência. Es-sas quantidades são contínuas em amplitude e no tempo, como mostrado pelos sinais analógicos típicos encontra-dos na Figura 1-4. As magnitudes das quantidades ana-lógicas podem ser medidas continuamente ou podem ser obtidas ao longo do tempo, como exigido por um experi-
mento em particular ou por um método instrumental, de acordo com a discussão apresentada no Capítulo 4. Em-bora os dados da Figura 1-4 tenham sido registrados em função do tempo, qualquer variável, como o comprimen-to de onda, a intensidade de um campo magnético ou a temperatura, pode constituir a variável independente sob circunstâncias apropriadas. A correlação entre dois sinais analógicos que resulta de propriedades físicas ou químicas medidas é importante para uma ampla varieda-de de técnicas instrumentais, tais como a espectroscopia e a análise térmica diferencial.
Devido ao fato de que o ruído influencia a magni-tude dos sinais elétricos, os sinais analógicos são espe-cialmente suscetíveis ao ruído elétrico que resulta da interação dentro dos circuitos de medida ou com outros dispositivos elétricos que se encontram na vizinhança do sistema de medida. O ruído, que é indesejável, não apresenta nenhuma relação com a informação de inte-resse, e muitos métodos têm sido desenvolvidos para minimizar seus efeitos indesejáveis. Os sinais, o ruído e a otimização da resposta instrumental serão discutidos no Capítulo 5.
Dif
eren
ça d
e po
tenc
ial (
V)
Cor
rent
e (I
)
Tempo
(a)
Tempo
(b)
FIGURA 1-4 Sinais analógicos. (a) Resposta instrumental de um sistema de detecção fotométri-
co de um experimento de análise por injeção em fluxo. Uma mistura reacional contendo zonas de
Fe(SCN)2+
de cor vermelha flui passado pela luz monocromática de um fototransdutor que produz uma
variação de potencial à medida que a concentração da amostra se altera. (b) Resposta em forma de
corrente gerada por um tubo fotomultiplicador quando a luz de uma fonte pulsada atinge o fotocatodo
do dispositivo.

Capítulo 1 • Introdução 21
Informação no domínio do tempo
A informação é armazenada no domínio do tempo como variações do sinal em relação ao tempo em vez de suas amplitudes. A Figura 1-5 ilustra três diferentes sinais no domínio do tempo registrados como uma quantidade ana-lógica em função do tempo. As linhas interrompidas hori-zontais representam um limite arbitrário do sinal analógi-co que é empregado para se decidir se o sinal é HI (acima de um limite) ou LO (abaixo de um limite)*. As relações de tempo entre as transições do sinal de HI para LO ou de LO para HI contêm a informação de interesse. Para os instrumentos que produzem sinais periódicos, o número de ciclos do sinal por unidade de tempo é a freqüência, e o tempo necessário para cada ciclo é seu período. Dois exemplos de sistemas instrumentais que produzem infor-mação codificada no domínio da freqüência são a espec-troscopia Raman (Capítulo 18) e a análise instrumental por ativação de nêutrons (Capítulo 32). Nesses métodos, a freqüência dos fótons que atingem um detector é dire-tamente relacionada à intensidade de emissão do analito, a qual é proporcional à sua concentração.
O tempo entre sucessivas transições HI para LO é denominado período, e o tempo entre uma transição LO para HI e HI para LO é denominado largura de pulso.
Dispositivos como os conversores voltagem-freqüência e freqüência-voltagem podem ser empregados para se con-verter sinais no domínio do tempo em sinais no domínio analógico e vice versa. Estes e outros conversores de do-mínio de dados são discutidos nos Capítulos 2 a 4, como parte da nossa abordagem sobre os dispositivos eletrôni-cos, e serão revistos em outros contextos neste livro.
Informação digital
Os dados são codificados no domínio digital por dois ní-veis. A informação pode ser representada por meio do estado de uma lâmpada de filamento, um diodo emis-sor de luz, uma chave liga-desliga ou um sinal de nível lógico, para citar apenas poucos exemplos. A caracte-rística compartilhada por esses dispositivos é que cada um deles deve estar em um de somente dois estados possíveis. Por exemplo, lâmpadas e chaves podem es-tar somente LIGADOS ou DESLIGADOS, e os sinais lógicos podem ser somente HI ou LO. A definição do que representa LIGADO e DESLIGADO para chaves e lâmpadas é fácil de ser entendida, porém no caso dos sinais elétricos, como os sinais no domínio do tempo, um nível arbitrário deve ser definido de forma a distin-guir entre HI e LO. Tal informação pode ser dependen-te das condições do experimento ou pode depender das características dos dispositivos eletrônicos em uso. Por exemplo, o sinal representado na Figura 1-5c consiste em uma seqüência de pulsos produzida por um detec-tor nuclear. A função da medida é contar o número de pulsos durante um período de tempo fixo para se obter a grandeza da intensidade da radiação. A linha inter-rompida representa um nível de sinal que, além de ser baixo o suficiente para assegurar que nenhum pulso seja perdido, é também suficientemente alto para rejei-tar flutuações aleatórias no sinal que não se relacionam com o fenômeno nuclear de interesse. Se o sinal cruza o limite quatorze vezes, como no caso do sinal mostrado na Figura 1-5c, então podemos assumir que quatorze eventos nucleares ocorreram. Depois que os eventos foram contados, os dados são codificados no domínio digital por sinais HI-LO representando o número 14. No Capítulo 4 exploraremos as formas de decidirmos pelas transições eletrônicas HI-LO e de se codificar a informação no domínio digital.
Como mostrado pelo mapa de domínios de dados da Figura 1-2, o domínio digital abrange ambos os mé-todos de codificação, elétrico e não-elétrico. No exem-plo citado acima, os eventos nucleares são acumulados empregando-se um contador eletrônico e mostrados em um dispositivo de leitura digital. Quando se lê e interpre-ta o mostrador, o número que representa a quantidade medida está novamente em um domínio não-elétrico.
(a)
(b)
(c)
HI
LO
HI
LO
HI
Tempo
Sina
l
LO
FIGURA 1-5 Sinais no domínio do tempo. As linhas horizon-
tais interrompidas representam os limites dos sinais. Quando
cada sinal está acima do limite, o sinal é HI, e quando ele está
abaixo do limite, o sinal é LO.
* N. de T.: Os termos HI e LO foram mantidos para designar os sinais lógicos de nível alto e nível baixo, respectivamente, devido ao seu uso comum na língua portuguesa falada e escrita no Brasil. Os termos são abreviaturas de HIGH (HI) e LOW (LO), alto e baixo, em inglês.

22 Princípios de Análise Instrumental
Cada parte HI-LO dos dados que representa um evento nuclear constitui um bit (do inglês, binary digit) de infor-mação, que é a unidade fundamental de informação no domínio digital. Bits de informação que são transmitidos por um canal eletrônico único podem ser contados por um observador ou por um dispositivo eletrônico que es-teja monitorando esse canal; esses dados acumulados são denominados contagem digital de dados, a qual aparece no mapa de domínios de dados da Figura 1-2. Por exem-plo, o sinal da Figura 1-5a pode representar o número n � 8 porque existem oito pulsos completos no sinal. Da mesma forma, o sinal da Figura 1-5b pode corresponder a n � 5, e aquele da Figura 1-5c poderia representar n � 14. Embora efetiva, essa forma de transmissão de infor-mação não é muito eficiente.
Uma forma muito mais eficiente de se codificar a informação consiste no uso de números binários para re-presentar dados numéricos e alfa-numéricos. Para cons-tatar como esse tipo de codificação pode ser realizada, vamos considerar os sinais da Figura 1-6. O dado de con-tagem digital do sinal da Figura 1-6a representa o núme-ro n � 5, como anteriormente. Monitoramos o sinal e contamos o número de oscilações completas. O processo requer um período de tempo proporcional ao número de ciclos do sinal ou, nesse caso, cinco vezes a largura de um único intervalo de tempo, como indicado na Figura 1-6. Observe que os intervalos de tempo são numerados consecutivamente, iniciando-se com zero. Em um esque-ma de codificação binária, como mostrado para o sinal da Figura 1-6b, atribuímos um valor numérico para cada intervalo de tempo sucessivo. Por exemplo, o intervalo identificado pelo número zero representa 20 � 1, o in-tervalo identificado pelo número 1 representa 21 � 2, o
intervalo de tempo de número 2 representa 22 � 4, e as-sim por diante, como mostrado na Figura 1-6. Durante cada intervalo de tempo, necessitamos decidir somente se o sinal é HI ou LO. Se o sinal for HI durante um dado intervalo de tempo, então o valor correspondente àquele intervalo é somado ao total. Todos os intervalos que são LO contribuem com zero para o total.
Na Figura 1-6b, o sinal é HI somente nos intervalos zero e 2; portanto, o valor total representado é (1 � 20) � (0 � 21) � (1 � 22) � 5. Assim, no espaço de somente três intervalos de tempo, o número n � 5 foi representado. No exemplo de contagem digital da Figura 1-6a, cinco inter-valos de tempo são necessários para representar o mesmo número. Neste exemplo extremo, a codificação binária se-rial é aproximadamente duas vezes mais eficiente do que a contagem serial de dados. Um exemplo mais expressivo pode ser visto na contagem de n � 10 oscilações, de for-ma similar à da Figura 1-6a. Nos mesmos dez intervalos de tempo, dez bits HI-LO de informação no esquema de codi-ficação binária serial permitem a representação de núme-ros binários de 0 a 210 � 1 � 1024 números ou 0000000000 a 1111111111. A melhora na eficiência é 1024/10 ou cerca de 100 vezes. Em outras palavras, o esquema de contagem serial necessita de 1024 intervalos de tempo para represen-tar o número 1024, mas o esquema de codificação binário necessita de somente dez intervalos de tempo. Em conse-qüência da eficiência dos esquemas de codificação binária, a maior parte das informações digitais é codificada, trans-ferida, processada e decodificada na forma binária.
Dados representados por códigos binários em uma única linha de transmissão são denominados dados bi-nários codificados serialmente ou simplesmente dados seriais. Um exemplo comum de transmissão de dados
(a)
(b)
(c)
HI
LO
HI
Binário(serial)
Binário(paralelo)
Contagem
n = 4 + 1 = 5
n = 4 + 1 = 5
n = 5
Intervalo de tempo 4 3 2
22 21 20
1 0
LO
Tempo
FIGURA 1-6 Diagrama ilustrando três tipos de dados digitais: (a) contagem serial de dados, (b)
dados codificados de forma binária serial e (c) dados binários paralelos. Nos três casos, os dados
representam o número n = 5.

Capítulo 1 • Introdução 23
seriais é o modem de um computador, que é um disposi-tivo para transmissão de dados entre computadores via telefone por meio de um único fio condutor (e de uma conexão comum).
Um método ainda mais eficiente de codificar da-dos no domínio digital é mostrado na Figura 1-6c. Neste caso, usamos três lâmpadas para representar os três dí-gitos binários: 20 � 1, 21 � 2 e 22 � 4. No entanto, pode-ríamos empregar chaves, fios, diodos emissores de luz ou qualquer outro dispositivo eletrônico, dentre os muitos existentes, para codificar a informação. Nesse esquema, LIGADO � 1 e DESLIGADO � 0, de forma que nosso número é codificado como mostrado na Figura 1-6, com a primeira e a terceira lâmpadas LIGADAS e a do meio DESLIGADA, o que representa 4 � 0 � 1 � 5. Esse esquema é altamente eficiente porque toda a informação desejada é apresentada para nós simultaneamente, assim como todos os dígitos em um mostrador do voltímetro digital da Figura 1-3 aparecem simultaneamente. Os da-dos apresentados desta maneira são designados como dados digitais paralelos. Os dados são transmitidos den-tro dos instrumentos analíticos e dos computadores por meio de transmissão paralela de dados. Uma vez que os dados são transmitidos a distâncias relativamente curtas dentro desses dispositivos, é barato e eficiente empregar a transferência paralela de informação. Essa economia encontrada para transmissão a curtas distâncias contras-ta com a situação na qual dados devem ser transportados a longas distâncias de um instrumento a outro ou de um computador a outro. Nessas instâncias, a comunicação é realizada serialmente por meio do uso de modems ou de outros esquemas mais rápidos e sofisticados de trans-missão de dados serial. Consideraremos estas idéias com mais detalhes no Capítulo 4.
1C-4 Detectores, transdutores e sensores
Os termos detector, transdutor e sensor são freqüente-mente empregados como sinônimos, mas, de fato, eles têm significados diferentes. O termo mais geral dos três, detector, refere-se a um dispositivo mecânico, elétrico ou químico que identifica, registra ou indica uma mudança em uma das variáveis em seu ambiente, como pressão, temperatura, carga elétrica, radiação eletromagnética, radiação nuclear, presença de partículas ou moléculas. Esse termo generalizou-se de tal forma que instrumen-tos completos são freqüentemente referidos como detec-tores. No contexto da análise instrumental, deveríamos empregar o termo detector no sentido geral que defini-mos e o termo sistema de detecção para nos referirmos aos conjuntos completos que indicam ou registram quan-
tidades físicas ou químicas. Um exemplo é o detector UV (ultravioleta), comumente empregado para indicar e registrar a presença dos analitos eluídos em cromato-grafia líquida.
O termo transdutor refere-se especificamente aos dispositivos que convertem a informação em um domí-nio não-elétrico em informação nos domínios elétricos e vice-versa. Exemplos incluem fotodiodos, fotomultipli-cadoras e outros fotodetectores eletrônicos que produ-zem uma corrente ou voltagem proporcional à potência da radiação eletromagnética que atinge suas superfícies. Outros exemplos incluem termistores, sensores de ten-são e transdutores de efeito Hall (intensidade de cam-po magnético). Como sugerido anteriormente, a relação matemática entre a saída elétrica e o aporte de potência radiante, temperatura, força ou campo magnético é cha-mada de função de transferência do transdutor.
O termo sensor também se tornou de uso amplo, mas neste livro reservaremos esse termo para designar a classe de dispositivos analíticos que são capazes de mo-nitorar espécies químicas de forma específica, continua e reversível. Existem numerosos exemplos de sensores neste livro, incluindo o eletrodo de vidro e outros eletro-dos íon-seletivos, que são tratados no Capítulo 23; o ele-trodo de oxigênio de Clark, que é descrito no Capítulo 25; e os sensores de fibra óptica (optodos) que aparecem no Capítulo 14. Os sensores consistem de um transdutor acoplado a uma fase de reconhecimento quimicamen-te seletiva, como mostrado na Figura 1-7. Desta forma, por exemplo, os optodos consistem de um fototransdu-tor acoplado a uma fibra óptica que é recoberta na ter-minação oposta ao transdutor por uma substância que responde especificamente a uma característica física ou química em particular de um analito.
Um sensor que é especialmente interessante e ins-trutivo é aquele feito de uma microbalança de cristal de quartzo, ou MCQ. Esse dispositivo é baseado nas ca-racterísticas piezelétrico do quartzo. Quando o quartzo é deformado mecanicamente, uma diferença de poten-cial elétrico desenvolve-se na sua superfície. Além disso, quando uma voltagem é aplicada através das faces de um cristal de quartzo, o cristal se deforma. Um cristal conectado a um circuito elétrico adequado oscila a uma freqüência que é característica da massa e da forma do cristal, e esta é surpreendentemente constante enquanto a massa do cristal permanece constante. Essa proprieda-de de alguns materiais cristalinos é denominada efeito piezelétrico e constitui a base de uma MCQ. Além dis-so, a freqüência característica do cristal de quartzo é a base dos modernos e altamente precisos relógios, bases de tempo, contadores, temporizadores e medidores de

24 Princípios de Análise Instrumental
freqüência os quais, por sua vez, têm permitido o desen-volvimento de muitos sistemas analíticos instrumentais altamente precisos e exatos.
Se um cristal de quartzo é recoberto com um políme-ro que adsorve seletivamente certas moléculas, a massa do cristal aumenta se as moléculas estão presentes, cau-sando assim um decréscimo da freqüência ressonante do cristal. Quando as moléculas são dessorvidas da superfí-cie, o cristal retorna à sua freqüência original. A relação entre a mudança da freqüência de oscilação do cristal �f e a alteração de massa do cristal �M é dada por
onde M é a massa do cristal, A é sua área superficial, f é a freqüência de oscilação do cristal e C é uma constan-te de proporcionalidade. Essa relação indica que é pos-sível medir pequenas alterações na massa do cristal se a freqüência do cristal puder ser medida com precisão. Como esperado, é possível medir facilmente mudanças na freqüência da ordem de 1 parte em 107 com o uso de instrumentação de baixo custo. O limite de detecção de um sensor piezelétrico desse tipo é estimado como sendo 1 pg, ou 10�12 g. Esses sensores têm sido empre-gados para detectar uma grande variedade de analitos presentes em fase gasosa, incluindo formaldeído, clore-to de hidrogênio, sulfeto de hidrogênio e benzeno. Eles têm sido também propostos como sensores de agentes químicos de guerra, como o gás mostarda e o fosgênio.
O sensor piezelétrico de massa representa um exem-plo excelente de transdutor que converte uma proprie-dade do analito, sua massa, nesse caso, em uma mudança de uma propriedade elétrica, a freqüência de ressonân-
cia do cristal de quartzo. Esse exemplo também ilustra a distinção entre um transdutor e um sensor. Na MCQ, o transdutor é o cristal de quartzo, e a segunda fase seleti-va é a cobertura polimérica. A combinação do transdu-tor e da fase seletiva constitui o sensor.
1C-5 Dispositivos de leitura
Um dispositivo de leitura é um transdutor que conver-te a informação de um domínio elétrico em uma forma que possa ser compreendida por um observador hu-mano. Usualmente, o sinal transduzido toma a forma de uma saída alfa-numérica ou gráfica em um tubo de raios catódicos, uma série de números em um mostra-dor digital, a posição de um ponteiro em uma escala ou, ocasionalmente, causa o escurecimento de uma pla-ca fotográfica ou gera um traço sobre um papel de um registrador. Em algumas instâncias, o dispositivo de leitura pode ser manipulado para fornecer diretamente a concentração do analito.
1C-6 Computadores em instrumentos
A maioria dos instrumentos analíticos modernos con-tém ou está ligada a um ou mais dispositivos eletrônicos mais sofisticados e a conversores de domínio de dados, como amplificadores operacionais, circuitos integra-dos, conversores analógico-digital e digital-analógico, microprocessadores e computadores. Para poder ava-liar o potencial e as limitações desses instrumentos, os investigadores necessitam desenvolver pelo menos um conhecimento qualitativo sobre como esses dispositivos funcionam e o que eles podem realizar. Os Capítulos 3 e 4 tratam de forma resumida desses tópicos.
EnzimasAnticorposReceptoresPolímerosOrganelasMicróbiosCélulasTecidos
EletrodoSemicondutorDispositivo MCQFototransdutorTransdutor de somTermistor
Sinal químico,massa, luz, calor,som, pressão, elétrico
Saídaelétrica
Fase de reconhecimentomolecular Transdutor
FIGURA 1-7 Um sensor químico. O sensor consiste de um elemento de reconhecimento molecular
e de um transdutor. Uma ampla variedade de elementos de reconhecimento é possível. Estão mos-
trados aqui alguns elementos de reconhecimento particularmente úteis aos biosensores. A fase de
reconhecimento converte a informação de interesse em uma característica detectável, como outra
espécie química, massa, luz ou calor. O transdutor converte a característica em um sinal elétrico que
pode ser medido.

Capítulo 1 • Introdução 25
1D CALIBRAÇÃO DOS MÉTODOS INSTRUMENTAIS
Uma etapa muito importante de todos os procedimentos analíticos consiste no processo de calibração e de padro-nização. A calibração determina a relação entre a res-posta analítica e a concentração do analito. Geralmente ela é determinada com o uso de padrões químicos.
Quase todos os métodos analíticos requerem al-gum tipo de calibração com padrões químicos. Os mé-todos gravimétricos e alguns métodos coulométricos estão entre os poucos métodos ditos absolutos, que não necessitam de calibração com padrões químicos. Diver-sos tipos de procedimentos de calibração são descritos nessa seção.
1D-1 Comparação com padrões
Dois tipos de métodos de comparação são descritos aqui, a técnica de comparação direta e o procedimento titulo-métrico.
Comparação direta
Alguns procedimentos analíticos envolvem a compara-ção de uma propriedade do analito (ou do produto de uma reação com o analito) com padrões, de forma que a propriedade que está sendo avaliada se iguala ou quase se iguala àquela do padrão. Por exemplo, nos coloríme-tros antigos, a cor resultante de uma reação química do analito era comparada com a cor produzida pela reação de padrões. Se a concentração do padrão era variada por diluição, por exemplo, era possível se obter uma igualda-de de cor bastante exata. Então, a concentração do ana-lito era igual à concentração do padrão após a diluição. Tal procedimento é denominado comparação de nulo ou método de equiparação.2
Titulações
As tilulações estão entre os procedimentos analíticos mais exatos. Em uma titulação, o analito reage com um reagente padronizado (o titulante) em uma reação de estequiometria conhecida. Geralmente a quantidade de titulante é variada até que a equivalência química seja encontrada, sendo detectada por meio da mudança de cor de um indicador químico ou por meio da alteração de uma resposta instrumental. A quantidade de reagente padronizado necessário para se obter a equivalência quí-mica pode então ser relacionada à quantidade de analito
2 Veja, por exemplo, H.V. Malmstad and J.D. Winefordner, Anal. Chim. Acta, 1960, 20, 283; Ramaley and C.G. Enke, Anal. Chem., 1965, 37, 1073.
presente. A titulação é, desta forma, um tipo de equipa-ração química.3
1D-2 Calibração com padrões externos
Um padrão externo é preparado separadamente da amostra. Por outro lado, um padrão interno é adicionado à amostra. Padrões externos são empregados para se ca-librar instrumentos e procedimentos quando não há efei-tos interferentes advindos dos componentes da matriz presentes na solução do analito. Uma série de padrões externos contendo o analito em concentrações conheci-das é preparada. De forma ideal, três ou mais soluções são utilizadas em um processo de calibração. Contudo, em algumas análises de rotina as calibrações com dois pontos podem ser confiáveis.
A calibração é realizada pela obtenção do sinal de resposta (absorbância, altura de pico, área de pico) como uma função da concentração conhecida do analito. Uma curva de calibração (ou curva analítica) é preparada cons-truindo-se um gráfico a partir dos dados ou ajustando-os a uma equação matemática adequada, como aquela em-pregada no método dos quadrados mínimos, utilizando o coeficiente angular e linear. O próximo passo é a etapa de previsão, na qual o sinal de resposta é obtido para a amostra e empregado para prever a concentração desco-nhecida do analito, cx, a partir de uma curva de calibração ou da equação que obteve o melhor ajuste. A concentra-ção do analito na amostra original é calculada a partir de cx, aplicando-se os fatores de diluição apropriados gera-dos pelas etapas de preparação da amostra.
O método dos quadrados mínimos
Uma curva de calibração típica é mostrada na Figura 1-8 para a determinação de isoctano em uma amostra de hi-drocarbonetos. Neste caso, uma série de padrões de isoc-tano foi injetada em um cromatógrafo a gás, e a área do pico do isoctano foi obtida em função da concentração. A ordenada é a variável dependente e corresponde à área do pico. A abscissa é a variável independente e corresponde à porcentagem molar de isoctano (mol %). Como é comum e usualmente desejável, o gráfico aproxima-se de uma li-nha reta. Note, no entanto, que devido aos erros indeter-minados no processo de medida, nem todos os pontos se ajustam exatamente à linha. Assim, o investigador deve
3 Veja D.A. Skoog, D.M. West, F.J. Holler and S.R. Crouch, Fun-damentals of Analytical Chemistry, 8th ed. Belmont, CA: Brooks/Cole, 2004, Chps. 13-17.
Tutorial: Aprenda mais sobre calibração.

26 Princípios de Análise Instrumental
tentar traçar a “melhor” linha reta entre os pontos dos da-dos. A análise de regressão permite a obtenção objetiva desta linha reta e especifica as incertezas associadas a seu uso subseqüente. As incertezas estão relacionadas com os resíduos mostrados na Figura 1-8, os quais representam uma medida da distância entre os pontos dos dados e a melhor linha reta. O método dos quadrados mínimos (veja Apêndice 1, Seção a1D) é frequentemente aplicado a fim de se obter a equação para a melhor linha reta.4
O método dos quadrados mínimos é baseado em duas hipóteses. A primeira é que realmente existe uma relação linear entre a resposta medida y e a concentração padrão do analito, x. A relação matemática que descre-
4 Para uma discussão sobre o uso de planilhas eletrônicas em aná-lise linear de regressão, veja S. R. Crouch and F. J. Holler, Appli-cations of Microsoft® Excel in Analytical Chemistry, Belmont, CA: Brooks/Cole, 2004, Chap. 4.
ve essa hipótese é chamada de modelo de regressão, que pode ser representada por
y � mx � b
onde b é o intercepto (valor de y quando x é igual a zero) e m é a inclinação da linha (veja Figura 1-8). Assumimos também que qualquer desvio dos pontos individuais de uma linha reta provém de um erro na medida. Isto é, as-sumimos que não há erro nos valores dos pontos x (con-centrações). Ambas as considerações são apropriadas para muitos métodos analíticos, mas tenha em mente que sempre que houver uma incerteza significativa nos dados x, a análise básica linear por quadrados mínimos pode não encontrar a melhor linha reta. Neste caso, uma análise de correlação pode ser necessária. Além disso, a análise básica de quadrados mínimos pode não ser apro-priada quando as incertezas nos valores y variam signifi-cativamente com x. Nesse caso, pode ser necessário apli-car diferentes fatores de peso aos pontos e realizar uma análise de quadrados mínimos ponderada.5
Nos casos em que os dados não se ajustam a um mo-delo linear, métodos de regressão não-lineares podem ser empregados.6 Alguns desses métodos empregam mo-delos polinomiais ou procedimentos de regressão múlti-pla. Existem até mesmo programas computacionais que irão encontrar um modelo que descreve um conjunto de dados experimentais a partir de um conjunto de equa-ções internas ou definidas pelo usuário.7
A inclinação m e o intercepto (coeficiente linear) b de uma linha de regressão linear por quadrados mínimos são determinados com o uso das equações a1-34 e a1-35 do Apêndice 1. Para se determinar uma concentração desconhecida cx a partir da reta obtida por quadrados mí-nimos, o valor da resposta instrumental yc é obtido para a amostra, e a inclinação e o intercepto são empregados para se calcular a concentração desconhecida, como mostrado pela Equação 1-1.
(1-1)
O desvio padrão na concentração sc pode ser encon-trado a partir do erro padrão da estimativa sy, também denominado desvio padrão da regressão, como dado pela Equação 1-2:
5 Veja P. R. Bevington and D. K. Robinson, Data Reduction and Error Analysis for the Physical Sciences, 3rd ed., New York: Mc-Graw-Hill, 2002.6 J. L. Devore, Probability and Statistics for Engineering and the Sciences, 6th ed., Pacific Grove, CA: Duxbury Press at Brooks/Cole, 2004.7 Veja, por exemplo, TableCurve, Systat Software, Point Rich-mond, CA.
y, Á
rea
do p
ico,
uni
dade
s ar
bitr
ária
s
x, Concentração de isoctano, mol %0,0 0,5 1,0 1,5 2,0
5,0
4,0
3,0
2,0
1,0
0
Resíduo =yi – (mxi + b)
FIGURA 1-8 Curva de calibração para a determinação de
isoctano em uma mistura de hidrocarbonetos. O resíduo é a di-
ferença entre o ponto do dado experimental yi e aquele calcula-
do por meio do modelo de regressão, mxi + b, como mostrado
no detalhe acima.

Capítulo 1 • Introdução 27
(1-2)
onde M é o número de resultados de replicatas, N é o número de pontos da curva de calibração (número de pa-drões), c é a resposta média para a amostra de concen-tração desconhecida e é o valor médio dos valores y dos resultados de calibração. A quantidade Sxx é a soma dos quadrados dos desvios dos valores de x em relação à mé-dia, como fornecido pela Equação a1-31 do Apêndice 1.
Erros na calibração por padrões externos
Quando empregamos padrões externos, assumimos que as mesmas respostas serão obtidas quando a mesma con-centração do analito estiver presente na amostra e no padrão. Assim, a relação funcional (equação) de calibra-ção entre a resposta e a concentração do analito deve também se aplicar à amostra. Usualmente, em uma de-terminação, não se emprega a resposta bruta (original) do instrumento. Ao invés disso, a resposta bruta é corri-gida pela medida do branco. Um branco ideal é idêntico à amostra, mas não contém o analito. Na prática, em se tratando de amostras complexas, é muito lento ou mes-mo impossível preparar um branco ideal, e uma simplifi-cação precisa ser feita. Freqüentemente, um branco real é somente um branco do solvente, contendo o mesmo solvente no qual a amostra está dissolvida, ou um branco de reagente, contendo o solvente e todos os reagentes empregados na preparação da amostra.
Mesmo com as correções do branco, muitos outros fatores podem invalidar a suposição básica do método de padrões externos. Efeitos de matriz, devido às espé-cies estranhas presentes na amostras, mas que não estão presentes no branco ou nos padrões, podem levar a res-postas distintas para uma amostra e para um padrão con-tendo a mesma concentração do analito.8 Diferenças nas variáveis experimentais nos momentos em que o branco, a amostra ou o padrão são medidos podem invalidar a função de calibração determinada. Mesmo quando a su-posição básica é válida, erros ainda podem ocorrer devi-do à contaminação durante a amostragem ou as etapas de preparação da amostra.
Além disso, erros sistemáticos podem ocorrer durante o processo de calibração. Por exemplo, se os padrões são preparados de forma incorreta, certamente um erro irá ocorrer. A exatidão com a qual os padrões são preparados depende da exatidão das técnicas e dos equipamentos gra-vimétricos e volumétricos utilizados. A forma química dos padrões deve ser idêntica à do analito na amostra; o estado
8 A matriz inclui o analito e os outros constituintes, os quais são denominados de concomitantes.
de oxidação, isomerização ou complexação do analito pode alterar a resposta. Uma vez preparado, a concentração dos padrões pode mudar devido à sua decomposição, volatili-zação ou adsorção nas paredes do recipiente onde estão armazenados. A contaminação de padrões pode resultar também em uma concentração do analito maior do que a esperada. Um erro sistemático pode ocorrer se houver al-gum erro sistemático ou viés no modelo de calibração. Por exemplo, erros podem ocorrer se a função de calibração for obtida sem o uso de padrões suficientes para se obter uma boa estimativa estatística dos parâmetros.
Erros aleatórios também podem influenciar a exa-tidão dos resultados obtidos por meio de curvas de ca-libração, como ilustrado na Figura 1-9. A incerteza na concentração do analito S’c obtida de uma curva de ca-libração é mínima quando a resposta está próxima do valor médio . O ponto , representa o centróide da linha de regressão. Note que as medidas feitas próximas ao centro da curva geram uma incerteza menor na con-centração do analito do que aquelas feitas nos extremos.
Calibração multivariada
O procedimento de quadrados mínimos descrito é um exemplo de procedimento de calibração univariada por-que somente uma resposta é empregada por amostra. O processo de relacionar múltiplas respostas instrumentais a um analito ou a uma mistura de analitos é conhecido como calibração multivariada. Os métodos de calibração multivariados9 tornaram-se bastante populares atual-mente à medida que os novos instrumentos disponibili-zados tornaram-se capazes de produzir respostas multidi-mensionais (absorbância de muitas amostras a múltiplos comprimentos de onda, espectros de massas de compo-nentes separados por cromatografia, etc.). Os métodos de calibração multivariada são muito poderosos. Eles po-dem ser empregados para determinar simultaneamente diversos componentes em misturas e causam redundân-cia nas medidas de forma a melhorar a precisão porque a repetição de uma medida N vezes implica uma melhoria de na precisão do valor médio (veja o Apêndice 1, Seção a1 B-1). Eles também podem ser empregados para detectar a presença de interferentes que não poderiam ser identificados em uma calibração univariada.
1D-3 Métodos de adição de padrão
Os métodos de adição de padrão são particularmente úteis na análise de amostras complexas nas quais os efei-
9 Para uma discussão mais abrangente, veja K. R. Beebe, R. J. Pell, and M. B. Seasholtz, Chemometrics: A Practical Guide, New York: Wiley, 1998, Chap. 5; H. Martens and T. Naes, Multivariate Calibra-tion, New York: Wiley, 1989.
28 Princípios de Análise Instrumental
tos de matriz são significativos. Um método de adição de padrão pode tomar diversas formas.10 Uma das mais comuns envolve a adição de um ou mais incrementos de uma solução padrão às alíquotas de iguais volumes da amostra. Esse processo é denominado fortificação (em inglês spinking). Cada solução é, então, diluída a um volume fixo antes da medida. Observe que quando a quantidade de amostra é limitada, as adições de padrão podem ser realizadas por meio da introdução sucessiva de incrementos do padrão em um único volume medido da amostra. As medidas são feitas na amostra original e na amostra mais o padrão após cada adição. Na maio-ria das versões do método de adição de padrão, a matriz da amostra permanece praticamente idêntica após cada adição, sendo que a única diferença é a concentração do analito ou, nos casos envolvendo a adição de um excesso de reagente analítico, a concentração do reagente. Todos os outros constituintes da mistura reacional devem ser idênticos porque os padrões são preparados em alíquo-tas da amostra.
10 Veja M. Bader, J. Chem. Educ., 1980, 57, 703.
Consideremos que várias alíquotas Vx da solução da amostra com concentração cx são transferidas para balões volumétricos de volume Vt. Em cada um desses balões é adicionado um volume variável Vp de uma solução padrão do analito de concentração conhecida cp. Os reagentes apropriados são, então, adicionados, e cada solução é di-luída ao volume. As medidas instrumentais são realizadas para cada uma dessas soluções e corrigidas para a respos-ta do branco a fim de se obter a resposta líquida do ins-trumento S. Se a resposta do instrumento corrigida pelo branco é proporcional à concentração, como considerada no método de adição de padrão, podemos escrever
(1-3)
onde k é uma constante de proporcionalidade. Um gráfi-co de S em função de Vp é uma linha reta da forma
S � mVp � b
onde a inclinação m e o intercepto b são dados por
12,5
10
7,5
5
2,5
0
Res
post
a
1 3 5 7 9Concentração
Centróide, x, y
sc'
sr
sc
FIGURA 1-9 Efeito da incerteza da curva de calibração. As linhas interrompidas mostram os limites
de confiança para concentrações determinadas a partir da linha de regressão. Note que as incertezas
aumentam nas extremidades do gráfico. Usualmente, estimamos a incerteza na concentração do
analito somente com o desvio padrão da resposta. A incerteza da curva de calibração pode aumentar
significativamente a incerteza na concentração do analito de sc para s´c.

Capítulo 1 • Introdução 29
e
O gráfico de adição de padrão é mostrado na Figura 1-10.
Uma análise de quadrados mínimos (Apêndice 1, Seção a1D) pode ser empregada para se determinar m e b; cx pode, então, ser obtida d0a razão dessas duas quan-tidades e dos valores conhecidos de cp, Vx e Vp. Assim,
ou
(1-4)
O desvio padrão na concentração pode, então, ser obtido primeiramente calculando-se o desvio padrão do volume sv e, depois, empregando-se a relação entre o volume e a concentração. O desvio padrão do volume é encontrado por meio da Equação 1-2 com pequenas al-terações. Devido ao fato de que extrapolamos a curva de calibração ao eixo x no método de adição de padrão, o valor de y para a amostra é 0, e o termo 1/M não aparece. Desta forma, a equação para sv torna-se
(1-5)
Conforme mostrado pela linha interrompida da Figura 1-10, a diferença entre o volume do padrão adicionado na origem (zero) e o valor do volume na interseção da linha reta com o eixo x, ou o intercepto em x para (Vp)0, é o volu-me de padrão equivalente à quantidade de analito na amos-tra. Além disso, o intercepto em x corresponde ao zero da resposta do instrumento, de forma que podemos escrever
(1-6)
Resolvendo a Equação 1-6 para cx, obtemos
(1-7)
O desvio padrão na concentração sc é, então
(1-8)
EXEMPLO 1-1
Alíquotas de 10 mililitros de uma amostra de água natural foram pipetadas para balões volumétricos de 50,00 mL. Volumes exatos de 0,00; 5,00; 10,00; 15,00 e 20,00 mL de uma solução padrão contendo 11,1 ppm de Fe3� foram adicionados em cada balão, seguidos de excesso de íon tiocianato, para formar o complexo de cor vermelha Fe(SCN)2�. Após diluição ao volume, as respostas do instrumento S para cada uma das cinco
1,2
1,0
0,8
0,6
0,4
0,2
0,0
P, a
bsor
bânc
ia
–10,0 0,0
(Vp)0 = –6,31 mL(calculado ou extrapolado)
b = 0,2412
m = 0,03820
10,0Vp, mL
20,0
FIGURA 1-10 Curva linear de calibração para o método de adição de padrão. A concentração da
amostra pode ser calculada a partir da inclinação m e do intercepto b ou pode ser determinada por
extrapolação, como explicado no texto.
30 Princípios de Análise Instrumental
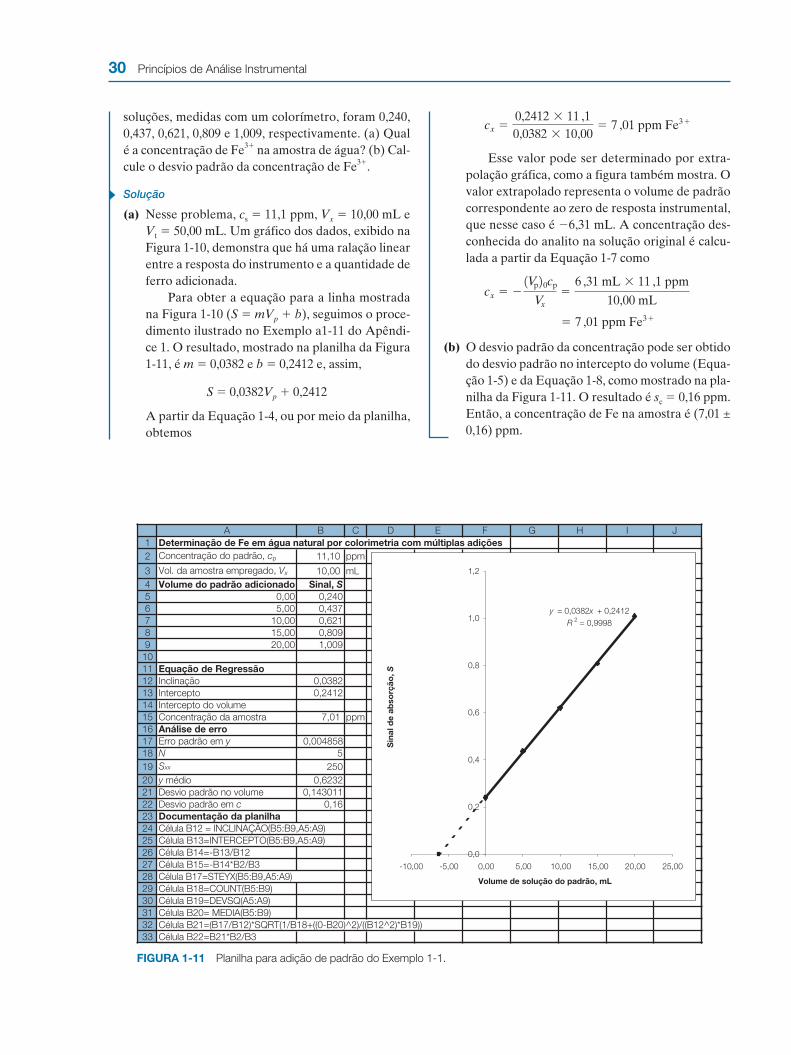
soluções, medidas com um colorímetro, foram 0,240, 0,437, 0,621, 0,809 e 1,009, respectivamente. (a) Qual é a concentração de Fe3� na amostra de água? (b) Cal-cule o desvio padrão da concentração de Fe3�.
Solução
(a) Nesse problema, cs � 11,1 ppm, Vx � 10,00 mL e Vt � 50,00 mL. Um gráfico dos dados, exibido na Figura 1-10, demonstra que há uma ralação linear entre a resposta do instrumento e a quantidade de ferro adicionada.
Para obter a equação para a linha mostrada na Figura 1-10 (S � mVp � b), seguimos o proce-dimento ilustrado no Exemplo a1-11 do Apêndi-ce 1. O resultado, mostrado na planilha da Figura 1-11, é m � 0,0382 e b � 0,2412 e, assim,
S � 0,0382Vp � 0,2412
A partir da Equação 1-4, ou por meio da planilha, obtemos
Esse valor pode ser determinado por extra-polação gráfica, como a figura também mostra. O valor extrapolado representa o volume de padrão correspondente ao zero de resposta instrumental, que nesse caso é �6,31 mL. A concentração des-conhecida do analito na solução original é calcu-lada a partir da Equação 1-7 como
(b) O desvio padrão da concentração pode ser obtido do desvio padrão no intercepto do volume (Equa-ção 1-5) e da Equação 1-8, como mostrado na pla-nilha da Figura 1-11. O resultado é sc � 0,16 ppm. Então, a concentração de Fe na amostra é (7,01 ± 0,16) ppm.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
A B C D E F G H I J
Determinação de Fe em água natural por colorimetria com múltiplas adiçõesConcentração do padrão, cp 11,10 ppm
7,01 ppm
Vol. da amostra empregado, Vx 10,00 mL
Volume do padrão adicionado Sinal, S0,00 0,240
5,00 0,437
10,00 0,621
15,00 0,809
20,00 1,009
Equação de Regressão0,0382Inclinação
0,2412Intercepto
Intercepto do volume
Concentração da amostra
Análise de erroErro padrão em y 0,004858
N 5
Sxx 250
y médio 0,6232
Desvio padrão no volume
Desvio padrão em c 0,16
Documentação da planilhaCélula B12 = INCLINAÇÃO(B5:B9,A5:A9)
Célula B13=INTERCEPTO(B5:B9,A5:A9)
Célula B14=-B13/B12
Célula B15=-B14*B2/B3
Célula B17=STEYX(B5:B9,A5:A9)
Célula B18=COUNT(B5:B9)
Célula B19=DEVSQ(A5:A9)
Célula B20= MEDIA(B5:B9)
Célula B21=(B17/B12)*SQRT(1/B18+((0-B20)^2)/((B12^2)*B19))
Célula B22=B21*B2/B3
y = 0,0382x + 0,2412
R2 = 0,9998
0,0
0,2
0,4
0,6
0,8
1,0
1,2
-10,00 -5,00 0,00 5,00 10,00 15,00 20,00 25,00
Volume de solução do padrão, mL
Sin
al d
e ab
sorç
ão, S
0,143011
FIGURA 1-11 Planilha para adição de padrão do Exemplo 1-1.

Capítulo 1 • Introdução 31
Com a finalidade de consumir menos tempo ou amostra, é possível realizar uma adição de padrão empregando-se somente dois incrementos da amostra. Neste caso, uma adição simples de Vp mL do padrão de-verá ser realizada em uma ou duas amostras, podendo-se escrever
onde S1 e S2 são os sinais resultantes da amostra diluída e da amostra diluída contendo o padrão, respectivamen-te. Dividindo a segunda equação pela primeira obtemos, após rearranjo,
O método de adição única de padrão é um pouco perigoso porque ele presume uma relação linear e não fornece nenhuma forma de verificar se essa relação real-mente existe. O método das adições múltiplas possibilita, pelo menos, a verificação da suposição da linearidade.
1D-4 O método do padrão interno
Um padrão interno é uma substância que é adicionada em uma quantidade constante na amostra, no branco e nos padrões de calibração em uma análise. Alternati-vamente, ele pode ser um dos constituintes principais das amostras e dos padrões que esteja presente em uma grande quantidade, de forma que sua concentra-ção possa ser considerada constante em todos os casos. A calibração envolve, então, a construção da curva da razão entre o sinal do analito e o sinal do padrão inter-no em função da concentração do analito nos padrões. Essa mesma razão calculada para as amostras é, então, empregada para se obter suas concentrações a partir da curva de calibração.
Um padrão interno, se escolhido e empregado adequa-damente, pode compensar diversos tipos de erros, quer se-jam estes aleatórios ou sistemáticos. Assim, se os sinais do analito e do padrão interno respondem proporcionalmente às flutuações aleatórias instrumentais e do método, a razão desses sinais é independente dessas flutuações. Se os dois sinais são influenciados da mesma forma pelos efeitos de matriz, a compensação desses efeitos também ocorre. Nos casos em que o padrão interno é um constituinte principal das amostras e dos padrões, uma compensação de erros causados pela preparação da amostra, pela diluição e pelo tratamento da amostra também pode ocorrer.
Uma dificuldade relevante que surge na aplicação do método de padrão interno está em encontrar uma substância adequada para servir como padrão interno e em introduzi-la nas amostras e padrões de forma repro-dutível. De maneira geral, o padrão interno deve prover um sinal que seja similar ao sinal do analito, mas sufi-cientemente diferente, de forma que os dois sinais são distinguíveis pelo instrumento. Se o padrão interno não for tomado como um constituinte principal da amostra, esse não deve estar presente na matriz da amostra, de forma que a única fonte de padrão seja a quantidade adi-cionada. Por exemplo, o lítio é um bom padrão interno para a determinação de sódio e potássio em soro sanguí-neo porque o comportamento químico do lítio é similar ao dos dois analitos, mas ele não ocorre no sangue natu-ralmente.
Um exemplo de determinação de sódio em sangue por espectrometria de chama usando lítio como padrão interno é mostrado na Figura 1-12. Ela mostra uma curva de calibração normal da intensidade emitida pelo sódio versus a sua concentração em ppm. Embora um gráfico bastante linear seja obtido, um espalhamento dos pontos é observado. O gráfico abaixo mostra a razão das inten-sidades para sódio e lítio versus a concentração de só-dio em ppm. Observe a melhoria na curva de calibração quando o padrão interno é utilizado.
No desenvolvimento de qualquer método de padrão interno, devemos verificar se as alterações na concentra-ção do analito não afetam a intensidade do sinal produ-zido pelo padrão interno e se o padrão interno não supri-me ou aumenta o sinal do analito.
1E SELEÇÃO DE UM MÉTODO ANALÍTICO
A coluna 2 da Tabela 1-1 mostra que atualmente temos uma enorme quantidade de ferramentas para realizar análises químicas. De fato, há tantas que a escolha entre elas é sempre difícil. Nesta seção, descreveremos breve-mente como essas escolhas são feitas.
1E-1 Definição do problema
Para selecionar um método analítico de forma inteligen-te, é essencial que se defina claramente a natureza do problema analítico. Tal definição requer a resposta às seguintes questões:
1. Qual é a exatidão necessária?2. Qual é a quantidade de amostra disponível?3. Qual é a faixa de concentração do analito?4. Quais componentes da amostra podem causar in-
terferências?

32 Princípios de Análise Instrumental
5. Quais são as propriedades físicas e químicas da matriz da amostra?
6. Quantas amostras devem ser analisadas?
A resposta à questão 1 é de vital importância porque ela determina quanto tempo e cuidado serão necessários para se realizar a análise. As respostas às questões 2 e 3 determinam qual é a sensibilidade que o método deve apresentar e qual é a extensão da faixa de concentração que deve ser acomodada. A resposta à questão 4 deter-mina a seletividade requerida para o método. As res-postas à questão 5 são importantes, pois alguns métodos analíticos listados na Tabela 1-1 se aplicam a soluções (usualmente aquosas) do analito. Outros métodos são aplicados mais facilmente a amostras gasosas, e, ainda, outros são adequados à análise direta de sólidos.
O número de amostras a ser analisado (questão 6) também é uma consideração importante do ponto de vista econômico. Se esse número é alto, tempo e recur-sos consideráveis podem ser gastos na instrumentação, no desenvolvimento do método e na calibração. Além disso, se o número é alto, deve-se escolher um método que requeira o mínimo de tempo do operador por amos-
tra. Por outro lado, se somente poucas amostras deverão ser analisadas, um método simples, porém mais lento e que necessite de pouco ou nenhum trabalho preliminar, é sempre a escolha mais sábia.
Com as respostas a essas seis questões, um método pode ser escolhido, considerando que as características de desempenho dos vários instrumentos mostrados na Tabela 1-1 são conhecidas.
1E-2 Características de desempenho dos instrumentos
A Tabela 1-3 lista os critérios quantitativos de desem-penho dos instrumentos que podem ser empregados na decisão de que um dado método instrumental é adequado para se abordar um problema analítico. Es-sas características são expressas em termos numéricos, denominadas figuras de mérito. As figuras de mérito nos permitem selecionar relativamente poucos instru-mentos adequados para um dado problema analítico. A escolha entre esses poucos pode então ser feita com base nos critérios de desempenho qualitativos listados na Tabela 1-4.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
A B C D E F G H I J K
Método de padrão interno para espectrometria de chama1.000 ppm de Li adicionado como padrão interno
Conc. de Na ppm INa ILi INa /ILi
0,10 0,11 86 0,001279
0,50 0,52 80 0,0065
1,00 1,8 128 0,014063
5,00 5,9 91 0,064835
10,00 9,5 73 0,130137
613640,0594,4Amostra
Equação de regressão0,012975
0,000285
3,54759
0,000556
Inclinação
Intercepto
Concentração da amostra
Análise de erroErro padrão em Y
N 5
Sxx 71,148
0,043363
M 1
Desvio padrão de c 0,046925
DocumentaçãoCélula D4=B4/C4
Célula B11= INCLINAÇÃO(D4:D8,A4:A8)
Célula B12=INTERCEPTO(D4:D8,A4:A8)
Célula B13=(D9-B12)/B11
Célula B15=STEYX(D4:D8,A4:A8)
Célula B16=COUNT(A4:A8)
Célula B17=DEVSQ(A4:A8)
Célula B18=MÉDIA(D4:D8)
Célula B19=entre com o nº de replicatas
Célula B20=B15/B11*SQRT(1/B19+1/B16+((D9-B18)^2)/((B11^2)*B17))
y = 0,947x + 0,422
R2 = 0,9816
0
2
4
6
8
10
12
0,00 2,00 4,00 6,00 8,00 10,00
Conc. de Na, ppm
I Na
y = 0,013x + 0,0003
R2 = 0,9999
0
0,02
0,04
0,06
0,08
0,1
0.12
0,14
0,00 2,00 4,00 6,00 8,00 10,00
Conc. de Na, ppm
I Na/I L
i
y médio (y barra)
FIGURA 1-12 Planilha ilustrando o método de padrão interno para a determinação de sódio por espectrometria de chama.

Capítulo 1 • Introdução 33
Nessa seção definiremos cada uma das seis figuras de mérito listadas na Tabela 1-3. Essas figuras serão en-tão empregadas no restante do livro para a discussão dos vários instrumentos e métodos instrumentais.
Precisão
Como mostramos na Seção a1A-1, Apêndice 1, a preci-são dos dados analíticos é o grau de concordância mútua entre dados que foram obtidos da mesma forma. A pre-cisão fornece uma medida do erro aleatório ou indeter-minado de uma análise. As figuras de mérito de precisão incluem o desvio padrão absoluto, o desvio padrão relati-vo, o erro padrão da média, o coeficiente de variação e a variância. Esses termos são definidos na Tabela 1-5.
Viés
Como mostrado na Seção a1A-2, Apêndice 1, o viés pro-porciona uma medida do erro sistemático, ou determi-nado, de um método analítico. O viés � é definido pela equação
� � m � t (1-9)
onde m é a média da população para a concentração do analito em uma amostra e t é o valor verdadeiro.
A determinação do viés envolve a análise de um ou mais materiais de referência cuja concentração do ana-lito é conhecida. Fontes de tais materiais são discutidas na Seção a1A-2 do Apêndice 1. Os resultados de tais análises irão, contudo, conter ambos os erros, aleatório e sistemático, mas se repetirmos as medidas por um núme-ro suficiente de vezes, o valor médio pode ser determi-nado com um certo grau de confiança. Como mostrado na Seção a1B-1, Apêndice 1, a média de vinte ou trinta replicatas pode usualmente ser tomada como uma boa estimativa da média da população m na Equação 1-9. Qualquer diferença entre essa média e a concentração conhecida do analito no material de referência pode ser atribuída ao viés.
Se a realização de vinte replicatas de um padrão é impraticável, a presença ou ausência provável de um viés pode ser avaliada como mostrado no Exemplo a1-10 no Apêndice 1. Geralmente, ao se desenvolver um método analítico, procuramos identificar a fonte de erro sistemá-tico (viés), eliminá-la ou corrigir os dados pelo uso de brancos e pelo uso de calibração do instrumento.
Sensibilidade
Existe um consenso de que a sensibilidade de um ins-trumento ou método é uma medida da sua habilidade em discriminar pequenas diferenças na concentração do analito. Dois fatores limitam a sensibilidade: a inclinação da curva de calibração e a reprodutibilidade ou precisão do dispositivo de medida. Entre dois métodos que apre-
TABELA 1-3 Critérios numéricos para seleção de
métodos analíticos
Critério Figura de mérito
1. Precisão Desvio padrão absoluto, desvio padrão relativo, coeficiente de varia-ção, variância
2. Viés Erro absoluto sistemático, erro rela-tivo sistemático
3. Sensibilidade Sensibilidade da calibração, sensibili-dade analítica
4. Limite de detecção Branco mais três vezes o desvio pa-drão do branco
5. Faixa dinâmica Limite de quantificação expresso em concentração (LDQ) até a concen-tração limite de linearidade (LDL)
6. Seletividade Coeficiente de seletividade
TABELA 1-4 Outras características a serem
consideradas na escolha de um método
1. Velocidade2. Facilidade e conveniência3. Habilidade requerida do operador4. Custo e disponibilidade do equipamento5. Custo por amostra
TABELA 1-5 Figuras de mérito para a precisão dos
métodos analíticos
Termos Definição*
Desvio padrão absoluto, s
Desvio padrão relativo (DPR)
Erro padrão da média, sm
Coeficiente de variação (CV)
Variância
* xi = valor numérico da inésima medida
= média de N medidas =

34 Princípios de Análise Instrumental
sentam a mesma precisão, aquele que mostra uma curva de calibração mais inclinada (com maior coeficiente an-gular) será o mais sensível.
Um corolário dessa afirmação é que se dois métodos apresentam curvas de calibração com inclinações iguais, aquele que exibe a melhor precisão é o mais sensível.
A definição quantitativa da sensibilidade que é acei-ta pela IUPAC (International Union of Pure and Applied Chemistry) é a sensibilidade de calibração, que consiste na inclinação da curva de calibração na concentração de interesse. A maioria das curvas de calibração emprega-das em química analítica é linear e pode ser representa-das pela equação
S � mc � Sbr (1-10)
onde S é o sinal medido, c é a concentração do analito, Sbr é o sinal instrumental para o branco e m é a inclinação da linha reta. A quantidade Sbr é o intercepto em y da li-nha reta. Com essas curvas, a sensibilidade da calibração é independente da concentração c e é igual a m. A sensi-bilidade de calibração como uma figura de mérito falha em considerar a precisão individual das medidas.
Mandel e Stiehler11 reconheceram a necessidade da inclusão da precisão em uma definição matemática signi-ficativa da sensibilidade e propuseram a seguinte defini-ção para a sensibilidade analítica :
g � m/sS (1-11)
Nessa equação, m é novamente a inclinação da curva de calibração e ss é o desvio padrão da medida.
A sensibilidade analítica oferece a vantagem de ser relativamente insensível aos fatores de amplificação. Por exemplo, o aumento do ganho de um instrumento por um fator de cinco produzirá um aumento de cinco vezes em m. Contudo, normalmente esse aumento será acompanhado por um aumento correspondente em ss, mantendo assim a sensibilidade analítica mais ou menos constante. Uma segunda vantagem da sensibilidade ana-lítica é que ela independe das unidades de medida de S.
Uma desvantagem da sensibilidade analítica é que ela é freqüentemente dependente da concentração, por-que ss pode variar com a concentração.
Limite de detecção
A definição mais aceita para o limite de detecção é que ele é igual à concentração, ou massa mínima, do analito que pode ser detectada em um dado nível de confian-ça. Esse limite depende da razão da magnitude do sinal
11 J. Mandel and R. D. Stiehler, J. Res. Natl. Bur. Std., 1964, A53, 155.
analítico em relação à grandeza da flutuação estatística do sinal do branco. Isso é, a menos que o sinal analítico seja maior que o sinal do branco por algum múltiplo k da variação do branco devido aos erros aleatórios, é im-possível se detectar o sinal analítico com certeza. Assim, à medida que se aproxima do limite de detecção, o sinal analítico e seu desvio padrão se aproximam do sinal do branco Sbr e seu desvio padrão sbr. O menor sinal ana-lítico distinguível Sm é, então, tomado como a soma do sinal médio do branco Sbr mais um múltiplo k do desvio padrão do branco. Isso é
(1-12)
Experimentalmente, Sm pode ser determinado re-alizando-se entre vinte a trinta medidas do branco, de preferência durante um longo período de tempo. Os dados resultantes são tratados estatisticamente para se obter Sbr e sbr. Finalmente, a inclinação da Equação 1-10 é empregada para se converter Sm para cm, que é definida como o limite de detecção. O limite de detecção é, então, dado por
(1-13)
Como mostrado por Ingle,12 muitas alternativas, basea-das de forma correta ou incorreta na estatística de t ou z (Seção a1B-2, Apêndice 1), têm sido utilizadas para se determinar o valor de k na Equação 1-12. Kaiser13 argumenta que um valor razoável para a constante é k � 3. Ele mostra que não é correto assumir uma distri-buição normal dos resultados das medidas do branco, e que quando k � 3 o nível de confiança da detecção será de 95% na maioria dos casos. Além disso, ele argumen-ta que há um pequeno ganho em empregar um valor maior de k e, assim, obter um nível de confiança maior. Long e Winefordner,14 em uma discussão sobre limite de detecção, também recomendam o uso de k � 3.
EXEMPLO 1-2
Uma análise de quadrados mínimos dos dados de ca-libração para a determinação de chumbo, baseada em seu espectro de emissão em chama, forneceu a seguin-te equação
S � 1,12cPb � 0,312
12 J. D. Ingle Jr., J. Chem. Educ., 1974, 51, 100.13 H. Kaiser, Anal. Chem., 1987, 42, 53A.14 G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712A.

Capítulo 1 • Introdução 35
onde cPb é a concentração de chumbo em partes por milhão e S é uma medida da intensidade relativa da linha de emissão do chumbo. Os seguintes dados de replicatas foram obtidos:
Conc.ppm Pb
Nº de replicatas
Valor médio de S s
10,0 10 11,62 0,15
1,00 10 1,12 0,025
0,000 24 0,0296 0,0082
Calcular (a) a sensibilidade de calibração, (b) a sensi-bilidade analítica a 1 e a 10 ppm de Pb e (c) o limite de detecção.
Solução
(a) Por definição, a sensibilidade de calibração é dada pela inclinação m � 1,12.
(b) Para 10 ppm de Pb, g � m/ss � 1,12/0,15 � 7,5.Para 1 ppm de Pb, g � 1,12/0,025 � 45.Observe que a sensibilidade analítica depende bastante da concentração. Por causa disso, ela não é mencionada tão freqüentemente como a sensibilidade de calibração.
(c) Aplicando a Equação 1-12,
S � 0,0296 � 3 × 0,0082 � 0,054
Substituindo na Equação 1-13, obtém-se
Faixa dinâmica
A Figura 1-13 ilustra a definição de faixa dinâmica de um método analítico, a qual se estende desde a menor concentração para a qual medidas quantitativas podem ser realizadas (limite de quantificação, ou LDQ) até a concentração na qual a curva de calibração se afasta da linearidade por uma quantia especificada (limite de linearidade, ou LDL). Usualmente, um desvio de 5% da linearidade é considerado o limite superior. Desvios da linearidade são comuns em altas concentrações por causa da resposta não-ideal dos detectores ou devido a efeitos químicos. O limite inferior das medidas quan-titativas é geralmente tomado como sendo igual a dez vezes o desvio padrão de medidas repetitivas de um branco, ou 10sbr. Nesse ponto, o desvio padrão relati-vo é aproximadamente 30% e decresce rapidamente à medida que a concentração torna-se maior. Para ser útil, um método analítico deve ter uma faixa dinâmica de pelo menos algumas ordens de magnitude. Algumas
técnicas analíticas, como a espectrometria de absor-ção, são lineares somente por uma ou duas ordens de magnitude. Outros métodos, como a espectrometria de massas e a fluorescência molecular, podem exibir uma linearidade que se estende por quatro ou cinco ordens de magnitude.
Seletividade
A seletividade de um método analítico refere-se a quan-to a resposta que ele fornece para o analito se mostra livre de interferência por outras espécies contidas na ma-triz da amostra. Infelizmente, nenhum método analítico está totalmente livre de interferências de outras espécies, e freqüentemente são necessárias diversas etapas para se minimizar os efeitos dessas interferências.
Considere, por exemplo, uma amostra contendo um analito A e também as espécies potencialmente interfe-rentes B e C. Se cA, cB e cC são as concentrações dessas espécies, e mA, mB e mC são suas sensibilidades de cali-bração, então o sinal total do instrumento será dado por uma versão modificada da Equação 1-10. Isso é,
S � mAcA � mBcB � mCcC � Sbr (1-14)
Vamos definir agora o coeficiente de seletividade para A com respeito a B, kB,A, como
kB,A � mB/mA (1-15)
O coeficiente de seletividade nos dá, então, a respos-ta do método para a espécie B em relação à espécie A. Um coeficiente similar para A com respeito a C é
kC,A � mC/mA (1-16)
Res
post
a do
inst
rum
ento
Concentração
cmLDQ
LDL
Faixa dinâmica
FIGURA 1-13 Faixa útil de um método analítico. LDQ = limite
de medida quantitativa; LDL = limite de resposta linear.

36 Princípios de Análise Instrumental
Substituindo essas relações na Equação 1-14 obtemos
S � mA(cA � kB,AcB � kC,AcC) � Sbr (1-17)
Os coeficientes de seletividade podem variar de zero (nenhuma interferência) até valores consideravel-mente maiores que a unidade. Observe que um coefi-ciente é negativo quando a interferência leva a uma re-dução da intensidade do sinal do analito. Por exemplo, se a presença do interferente B causa uma redução em S na Equação 1-14, mB terá um sinal negativo, assim como kB,A.
Os coeficientes de seletividade são figuras de mé-rito úteis para se descrever a seletividade dos métodos analíticos. Infelizmente, eles não são empregados com freqüência exceto para caracterizar o desempenho de eletrodos seletivos a íons (Capítulo 23). O Exemplo 1-3 ilustra o uso dos coeficientes de seletividade quando eles estão disponíveis.
EXEMPLO 1-3
O coeficiente de seletividade de um eletrodo íon-se-letivo para K� com respeito a Na� é 0,052. Calcule o erro relativo na determinação de K� em uma solução
na qual a concentração de K� é igual a 3,00 � 10�3 mol L�1 se a concentração de Na� for (a) 2,00 � 10�2 mol L�1; (b) 2,00 � 10�3 mol L�1; (c) 2,00 � 10�4 mol L�1. Assuma que Sbr para uma série de brancos é apro-ximadamente igual a zero.
Solução
(a) Substituindo na Equação 1-17, obtém-se
Se o Na� não estivesse presente,
S/mK� � 3,00 × 10�3
O erro relativo em será idêntico ao erro relativo em S/m (veja Seção a, Apêndice 1). Portanto,
Procedendo da mesma forma, encontraremos
(b) Erel � 3,5%(c) Erel � 0,35%
QUESTÕES E PROBLEMAS
* As respostas dos problemas marcados com asterisco estão no final do livro.
Os problemas com este ícone são resolvidos mais facilmente com o uso de planilhas eletrônicas.
1-1 O que é um transdutor em um instrumento analítico?
1-2 Qual é o processador de informação em um instrumento de medida visual da cor de uma solução?
1-3 Qual é o detector de um espectrógrafo no qual as linhas espectrais são registradas fotograficamente?
1-4 Qual é o transdutor em um detector de fumaça?
1-5 O que é um domínio de dados?
1-6 Indique alguns sinais elétricos que são considerados analógicos. Como a informação está codificada em um sinal analógico?
1-7 Liste quatro transdutores de saída e descreva como eles são empregados.
1-8 O que é uma figura de mérito?
*1-9 Uma amostra de 25,0 mL contendo Cu2� forneceu uma resposta instrumental de 23,6 unidades (corrigida pelo branco). Quando exatamente 0,500 mL de uma solução 0,0286 mol L�1 de Cu(NO3)2 foram adicionados à amostra, o sinal aumentou para 37,9

Capítulo 1 • Introdução 37
unidades. Calcule a concentração em mol L�1 considerando que o sinal é diretamente proporcional à concentração do analito.
*1-10 Os dados na tabela abaixo foram obtidos durante uma determinação colorimétrica de glicose em soro sanguíneo.
Concentração de glicose, mmol L�1 Absorbância, A
0,0 0,002
2,0 0,150
4,0 0,294
6,0 0,434
8,0 0,570
10,0 0,704
(a) Considerando uma relação linear, encontre as estimativas por quadrados míni-mos da inclinação e do intercepto.
(b) Empregue a função LINEST do Excel para encontrar os desvios padrão da incli-nação e do intercepto.15 Qual é o erro padrão da estimativa?
(c) Determine o intervalo de confiança em 95% para a inclinação e o intercepto.(d) Uma amostra de soro produziu uma absorbância de 0,350. Encontre a concen-
tração de glicose e seu desvio padrão.
1-11 Alíquotas exatas de 5,00 mL de uma solução contendo fenobarbital foram transferidas para balões volumétricos de 50,00 mL e alcalinizadas com KOH. Os seguintes volumes de uma solução padrão contendo 2,000 mg mL�1 de fenobarbital foram introduzidos em cada balão, e a mistura foi diluída ao volume: 0,000, 0,500, 1,00, 1,50 e 2,00 mL. A fluorescência destas soluções foram medidas com um fluorímetro, o qual forneceu valores de 3,26; 4,80; 6,41; 8,02 e 9,56, respectivamente.(a) Faça um gráfico dos dados.
*(b) Empregando o gráfico obtido em (a), calcule a concentração de fenobarbital na amostra.
*(c) Determine a equação para os dados por quadrados mínimos.*(d) Encontre a concentração de fenobarbital a partir da equação obtida em (c).
(e) Calcule o desvio padrão para a concentração obtida em (d).
Problema desafiador
1-12 (a) Use um programa de busca para encontrar o site da IUPAC na Web e localize o Compendium of Analytical Nomenclature. Em qual ano a última edição foi pu-blicada? Quem foram os autores?
(b) Encontre a definição de viés (bias) recomendada pelo Compendium. A definição e a simbologia diferem daquelas deste capítulo? Explique.
(c) Encontre a definição de limite de detecção recomendada no Compendium. A definição e a simbologia diferem daquelas desse capítulo? Explique.
(d) Quais são as diferenças entre sensibilidade de calibração e sensibilidade analítica?(e) Os seguintes dados de calibração foram obtidos por meio de um método instru-
mental para a determinação da espécie X em solução aquosa.
15 Veja S. R. Crouch and F. J. Holler, Applications of Microsoft® Excel in Analytical Chemistry, Belmont, CA:Brooks/Cole, 2004, Chap. 4.

38 Princípios de Análise Instrumental
Conc. X, ppm Nº replicatasSinal analítico
médio Desvio padrão
0,00 25 0,031 0,0079
2,00 5 0,173 0,0094
6,00 5 0,422 0,0084
10,00 5 0,702 0,0084
14,00 5 0,956 0,0085
18,00 5 1,248 0,0110
(i) Calcule a sensibilidade de calibração. (ii) Determine a sensibilidade analítica a cada concentração do analito. (iii) Encontre o coeficiente de variação para a média de cada grupo de replicatas. (iv) Qual é o limite de detecção do método?











![DIAGRAMAS ANALÍTICOS [Modo de Compatibilidade]](https://static.fdocumentos.com/doc/165x107/553c7fa04a7959d8258b4993/diagramas-analiticos-modo-de-compatibilidade.jpg)






