
Carlos Rangel Rodrigues - QNEscqnesc.sbq.org.br/online/cadernos/03/modelag.pdf · mento da...
7
43 Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001 Introdução U m dos mais importantes avanços no planejamento e descoberta de novos fármacos tem sido a utilização da modelagem molecular. Ela tem se firmado como uma ferra- menta indispensável não somente no processo de descoberta de novos fár- macos, mas também na otimização de um protótipo já existente ou obtido pelo próprio estudo de modelagem molecular. O grande desenvolvi- mento da modelagem mole- cular deveu-se em grande parte ao avanço dos recur- sos computacionais em ter- mos de hardware (velocida- de de cálculo) e software (programas de modelagem molecular). No passado, a utilização desta técnica era restrita a um seleto grupo de pessoas que desenvolviam os seus próprios programas de mo- delagem molecular. Atualmente, não é mais necessário ao usuário (modelista) compor o seu próprio programa em vir- tude deles poderem ser obtidos atra- vés de grandes companhias e de labo- ratórios acadêmicos. A modelagem molecular fornece in- formações importantes para o proces- so de descoberta de fármacos. Ela per- mite a obtenção de propriedades es- pecíficas de uma molécula que podem Carlos Rangel Rodrigues A modelagem molecular é uma ferramenta importante no desenvolvimento de fármacos. Neste trabalho, descrevemos os dois principais métodos empregados na elaboração de programas de modelagem molecu- lar: métodos de mecânica molecular e métodos semi-empíricos. A enzima HIV protease (HIVPR) foi selecionada como alvo terapêutico para mostrar como a modelagem molecular pode ser utilizada no planejamento racional de novos inibidores de HIVPR modelagem molecular, mecânica molecular, semi-empírico, HIVPR influenciar na interação com o recep- tor. Como exemplos, podemos citar o mapa de potencial eletrostático, o con- torno da densidade eletrônica e a energia e os coeficientes dos orbitais de fronteira HOMO (Highest Occupied Molecular Orbital) e do LUMO (Lowest Unoccupied Molecular Orbital ) etc. Outras informações importantes tam- bém podem ser obtidas a partir da comparação estrutural entre diferentes mo- léculas, o que pode permitir a geração de um índice de simila- ridade que po- de ser correla- cionado com a atividade farmacológica. A modelagem molecular também permite a visualiza- ção tridimensional (3D) do complexo fármaco-receptor e fornece informa- ções sobre os requisitos estruturais es- senciais que permitem uma interação adequada do fármaco no seu sítio re- ceptor. Esta ferramenta também tem o potencial de planejar teoricamente no- vas moléculas que satisfaçam as pro- priedades eletrônicas e estruturais para um perfeito encaixe no sítio receptor. A maioria dos programas de mode- lagem molecular é capaz de desenhar a estrutura molecular e realizar os cál- culos de otimização geométrica e es- tudos de análise conformacional. Os arquivos de saída destes cálculos podem ser utilizados como arquivos de entrada para outros programas. Desta forma, a primeira etapa em estudos de modelagem molecular é desenhar a estrutura da molécula. Em seguida, a molécula é otimizada objetivando encontrar parâmetros geométricos tais como comprimentos e ângulos de ligação (Figura 1) que estejam pró- ximos aos valores determinados expe- rimentalmente. Desta forma, pode-se avaliar a qualidade do programa de modelagem molecular selecionado pa- ra efetuar os cálculos considerando que ele deve ser capaz de representar corretamente a estrutura molecular sem que os parâmetros estruturais da referida molécula tenham sido usados para elaborá-lo. Assim, um programa de modela- gem molecular deve ser capaz de ado- tar o princípio da transferibilidade, ou seja, reconhecer e transferir os parâ- A maioria dos programas de modelagem molecular é capaz de desenhar a estrutura molecular e realizar os cálculos de otimização geométrica e estudos de análise conformacional Figura 1: Representação do comprimento de ligação (d21 e d32) e ângulo de ligação (a321) definidos pelos átomos At1, At2 e At3. Modelagem molecular
Transcript of Carlos Rangel Rodrigues - QNEscqnesc.sbq.org.br/online/cadernos/03/modelag.pdf · mento da...

43
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001
Introdução
Um dos mais importantes avançosno planejamento e descobertade novos fármacos tem sido a
utilização da modelagem molecular.Ela tem se firmado como uma ferra-menta indispensável não somente noprocesso de descoberta de novos fár-macos, mas também na otimização deum protótipo já existente ouobtido pelo próprio estudode modelagem molecular.
O grande desenvolvi-mento da modelagem mole-cular deveu-se em grandeparte ao avanço dos recur-sos computacionais em ter-mos de hardware (velocida-de de cálculo) e software(programas de modelagemmolecular). No passado, a utilizaçãodesta técnica era restrita a um seletogrupo de pessoas que desenvolviamos seus próprios programas de mo-delagem molecular. Atualmente, não émais necessário ao usuário (modelista)compor o seu próprio programa em vir-tude deles poderem ser obtidos atra-vés de grandes companhias e de labo-ratórios acadêmicos.
A modelagem molecular fornece in-formações importantes para o proces-so de descoberta de fármacos. Ela per-mite a obtenção de propriedades es-pecíficas de uma molécula que podem
Carlos Rangel Rodrigues
A modelagem molecular é uma ferramenta importante no desenvolvimento de fármacos. Neste trabalho,descrevemos os dois principais métodos empregados na elaboração de programas de modelagem molecu-lar: métodos de mecânica molecular e métodos semi-empíricos. A enzima HIV protease (HIVPR) foi selecionadacomo alvo terapêutico para mostrar como a modelagem molecular pode ser utilizada no planejamento racionalde novos inibidores de HIVPR
modelagem molecular, mecânica molecular, semi-empírico, HIVPR
influenciar na interação com o recep-tor. Como exemplos, podemos citar omapa de potencial eletrostático, o con-torno da densidade eletrônica e aenergia e os coeficientes dos orbitaisde fronteira HOMO (Highest OccupiedMolecular Orbital) e do LUMO (LowestUnoccupied Molecular Orbital) etc.Outras informações importantes tam-bém podem ser obtidas a partir da
c o m p a r a ç ã oestrutural entrediferentes mo-léculas, o quepode permitir ageração de umíndice de simila-ridade que po-de ser correla-cionado com a
atividade farmacológica. A modelagemmolecular também permite a visualiza-ção tridimensional (3D) do complexofármaco-receptor e fornece informa-ções sobre os requisitos estruturais es-senciais que permitem uma interaçãoadequada do fármaco no seu sítio re-ceptor. Esta ferramenta também tem opotencial de planejar teoricamente no-vas moléculas que satisfaçam as pro-priedades eletrônicas e estruturais paraum perfeito encaixe no sítio receptor.
A maioria dos programas de mode-lagem molecular é capaz de desenhara estrutura molecular e realizar os cál-culos de otimização geométrica e es-
tudos de análise conformacional. Osarquivos de saída destes cálculospodem ser utilizados como arquivos deentrada para outros programas. Destaforma, a primeira etapa em estudos demodelagem molecular é desenhar aestrutura da molécula. Em seguida, amolécula é otimizada objetivandoencontrar parâmetros geométricos taiscomo comprimentos e ângulos deligação (Figura 1) que estejam pró-ximos aos valores determinados expe-rimentalmente. Desta forma, pode-seavaliar a qualidade do programa demodelagem molecular selecionado pa-ra efetuar os cálculos considerandoque ele deve ser capaz de representarcorretamente a estrutura molecularsem que os parâmetros estruturais dareferida molécula tenham sido usadospara elaborá-lo.
Assim, um programa de modela-gem molecular deve ser capaz de ado-tar o princípio da transferibilidade, ouseja, reconhecer e transferir os parâ-
A maioria dos programasde modelagem molecular é
capaz de desenhar aestrutura molecular erealizar os cálculos de
otimização geométrica eestudos de análise
conformacional
Figura 1: Representação do comprimentode ligação (d21 e d32) e ângulo de ligação(a321) definidos pelos átomos At1, At2 eAt3.
Modelagem molecular

44
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001
metros embutidos no programa parauma nova molécula que apresente asmesmas características estruturais eeletrônicas das moléculas usadas paraconfeccionar o programa (mesmo tipode átomos, funções químicas, hibridi-zação molecular etc).
A rotação entre ligações saturadas,como entre átomos de carbono, permi-te que uma única molécula adote diver-sas conformações. O estudo de aná-lise conformacional permite determinaras conformações de mínimo de ener-gia (confôrmeros). Estes confôrmerosindicam como os grupamentos funcio-nais estão orientados e, portanto, reve-lam aspectos relevantes de como amolécula pode interagir com um recep-tor específico, considerando que a con-formação mais estável deve estar emmaior número durante o processo deinteração com o receptor. Entretanto,devemos ressaltar que não existe umarelação entre a conformação maisestável e a conformação bioativa, poisa primeira pode sofrer mudanças nasua conformação original no momentoem que se aproxima do sítio receptor.Este estudo conformacional é realizadoconsiderando as rotações livres entredois átomos consecutivos que, por suavez, determinam um ângulo diedro,assinalando como devem estar orienta-dos os grupamentos adjacentes a
estes dois átomos (Figura 2).
Métodos empregados na elaboraçãodos programas de modelagemmolecular
Um programa de modelagem mo-lecular permite a representação,visualização, manipulação e determi-nação de parâmetros geométricos(comprimento e ângulo de ligação) eeletrônicos (energia dos orbitais defronteira, momento de dipolo, potencialde ionização etc) de umamolécula isolada, além derealizar estudos em macro-moléculas (proteínas) e com-plexos droga–receptor.
A grande maioria dosprogramas de modelagemmolecular é capaz de retratarentidades químicas com um alto graude precisão. Esta afirmação é oriundade estudos comparativos de pa-râmetros eletrônicos e geométricosobtidos experimentalmente. Os méto-dos mais empregados para a obten-ção de propriedades moleculares são:a mecânica molecular e os métodossemi-empíricos.
Mecânica molecularMecânica molecular (MM) é um mé-
todo que calcula a estrutura e a ener-gia das moléculas com base nos
movimentos dos núcleos. Os elétronsnão são considerados explicitamentemas, ao contrário, é assumido que elesencontrarão uma distribuição ótima,uma vez que as posições dos núcleossão conhecidas. Esta idéia é baseadana aproximação de Born-Oppenhei-mer. Esta aproximação estabelece queos núcleos são mais pesados e, por-tanto, movem-se mais lentamente doque os elétrons. Desta forma, os movi-mentos nucleares, as vibrações e as
rotações po-dem ser estu-dadas separa-d a m e n t e ,admitindo queos elétrons mo-vem-se rapida-mente e ajus-
tam-se aos movimentos do núcleo.Assim, pode-se admitir que a mecâ-
nica molecular trata a molécula comouma coleção de esferas conectadaspor molas, onde as esferas represen-tam os núcleos e as molas represen-tam as ligações (Figura 3).
O campo de força é usado para cal-cular a energia e a geometria de umamolécula. Ele é elaborado de formaque contenha uma coleção de diferen-tes tipos de átomos, parâmetros (paracomprimento e ângulos de ligação eetc.) e equações para calcular a ener-gia de uma molécula. Em um determi-nado campo de força, um dado ele-mento pode ter diferentes tipos de áto-mos. Por exemplo, o etilbenzeno con-tém átomos de carbono com hibri-dização sp3 e átomos de carbono aro-mático (sp2). Os átomos de carbonocom hibridização sp3 apresentam umageometria de ligação tetraédrica, en-quanto átomos de carbono aromáticotêm uma geometria trigonal (planar). O
Modelagem molecular
Figura 2: Representação do ângulo de diedro delimitados por quatro átomos consecu-tivos, assinalando diferentes valores de ângulo de diedro.
Figura 3: Representação de uma moléculautilizando princípios de modelagem mole-cular onde as esferas são os átomos e amola representa a ligação entre eles.
Uma das dificuldades doestudo da modelagem
molecular é que não existeuma relação entre a
conformação mais estável ea conformação bioativa

45
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001
comprimento de ligação entre osátomos de carbono (C-C) no grupo etildifere do comprimento C-C no grupofenila e o comprimento da ligação en-tre o átomo de carbono etila e o átomode carbono da fenila ligados direta-mente é diferente de todos os compri-mentos de ligação no etilbenzeno. Oscampos de força contêm parâmetrospara estes diferentes tipos de ligação.A energia total de uma molécula podeser dividida em várias partes denomi-nadas forças potenciais ou equaçõesde energia potencial. Estas forçaspotenciais são calculadas independen-temente e somadas para obter a ener-gia total da molécula. Exemplos deforças potenciais são as equações deenergias associadas com a deforma-ção do comprimento (s), ângulo deligação (a), ângulo de torsão (t), inte-rações de van der Waals (vdW) etc. Es-tas equações definem a superfície deenergia potencial de uma molécula.
ETOTAL = Es + Ea + Et + EvdW (1)
Deformação no comprimento deligação
Se uma determinada ligação écomprimida ou estirada a energia sobe(Figura 4). Desta forma, o campo deforça é parametrizado com valores dedistância, por exemplo, para uma liga-ção C-C com hibridização sp3 e odesvio destes valores acarreta um au-mento na energia da molécula
Deformação ângularA função potencial para a deforma-
ção angular (Figura 5) deve consideraros diferentes tipos de átomos e hibri-dização molecular. Assim, o campo deforça deve ser capaz de contemplarcasos especiais, como por exemplo ociclobutano.
Barreira de energia de rotaçõesintramoleculares (ângulos de torsão)
A análise conformacional envolve arotação do ângulo de torsão θ que éformado por quatro átomos (A1, A2, A3e A4). Estas rotações intramolecularesexigem energia. Na Figura 6, o valorde ângulo de torsão θ é de 180° e posi-ciona os átomos A1 e A4 o mais dis-tante possível (conformação mais es-tável). Mudanças no ângulo de torsãoθ ocasionam uma aproximação dosreferidos átomos provocando um au-mento de energia do sistema.
Interações de van der WaalsO raio de van der Waals de um áto-
mo é o seu tamanho efetivo. Quandodois átomos não ligados são aproxi-mados, a atração de van der Waalsentre eles aumenta (decréscimo naenergia). Quando a distância entre elesé igual à soma dos raios de van derWaals, a atração é máxima. Se os áto-mos são aproximados ainda mais,ocorre uma forte repulsão de van derWaals (Figura 7).
Métodos semi-empíricosOs métodos semi-empíricos são
baseados no mesmo formalismo dosmétodos ab initio, mas parte de seusparâmetros são ajustados a dadosexperimentais. A parametrização dosmétodos semi-empíricoscom dados experimentaisaumentou significativa-mente a acuracidade quí-mica e a velocidade dosmétodos de orbitais mo-leculares. O sucesso des-ta abordagem é indicadopor inúmeros estudos,cujos resultados de cál-culos de energia produzem variaçõesna faixa de 1,0 kcal.mol-1 em relaçãoaos dados experimentais. Os métodossemi-empíricos mais recentes são AM1(Austin Model 1) e PM3 (ParametricMethod 3) contidos em diversos paco-tes de cálculos teóricos. Do ponto de
vista da estrutura das ligações hidro-gênio, importantes em sistemas bioló-gicos, o método PM3 tem apresentadoresultados mais próximos aos obtidosexperimentalmente e por cálculos abinitio.
As diversas aproximações semi-empíricas permitem evitar o cálculo deum grande número de integrais, o quepossibilita a aplicação destes métodosem sistemas com um número maior deátomos. Nestes métodos, os núcleossão assumidos em sucessivas posi-ções estacionárias, sobre as quais adistribuição espacial ótima dos elétronsé calculada pela resolução da equa-ção de Schrödinger. O processo érepetido até que a energia não maisvarie dentro de um limite escolhido, ouseja, até se alcançar um ponto esta-cionário da superfície de energia. Emum sistema no estado fundamental,isto significa que a geometria é tal queo calor de formação (∆Hf) é um mínimoirredutível (na verdade um mínimo irre-dutível local), ou seja, todas as suasconstantes de força são positivas; paraestados de transição, o sistema deveter exatamente uma constante de forçanegativa. Deste modo, tem-se tornadoprática comum nos trabalhos teóricosde qualidade a avaliação de todas assegundas derivadas (constantes deforça) da energia molecular em funçãodos parâmetros moleculares, para sedeterminar inequivocamente a nature-za dos pontos estacionários encontra-dos no processo de otimização dageometria molecular.
Modelagem molecular
Figura 4: Deformação no comprimento deligação.
Figura 5: Deformação no ângulo de ligação.
Figura 6: Ângulo de torsâo (θ) com um valor de 180°.
Figura 7: Interações de van der Waals.

46
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001
Planejamento de novos inibidorescom base na estrutura do complexoda enzima HIV protease co-cristalizado com um ligante
Inibidores de HIV proteaseOs estudos experimentais de crista-
lografia de raios-X do complexo droga-receptor permitem identificar a confor-mação bioativa da droga que interagecom o bioreceptor. O conhecimentodesta bioconformação é de grandeutilidade para planejar novos ligantesque apresentem os grupamentos far-macofóricos na mesma disposiçãoestereoespacial, permitindo uma per-feita interação com a biomacromo-lécula. Assim, a primeira etapa noprocesso de planejamento de novosfármacos consiste na análise confor-macional das moléculas-alvo. O co-nhecimento das conformações maisestáveis permite a sobreposição dasmesmas com a conformação doinibidor complexado no sítio, anteci-pando possíveis modificações estru-turais que atendam aos requisitos con-formacionais essenciais para interaçãocom o receptor.
Baseando-se na importância dosgrupos químicos presentes no seus sí-tios catalíticos, as proteases são se-paradas em quatro grandes classes –serinil (como a tripsina), metalo ( comoa carboxipeptidase), cisteinil (cruzipaí-na) e aspartil ( como a HIV-1 protease).
O vírus da imunodeficiência huma-na (HIV) foi identificado como o prová-vel agente causador da síndrome daimunodeficiência adquirida (AIDS).Vários processos biológicos têm sidoalvos para uma intervenção terapêu-tica. Um dos alvos mais promissoresfoi a identificação de uma aspartil pro-tease essencial para o processo dereplicação viral (HIV-PR). Esta proteasetambém está relacionada ao alto graude infectividade viral. Isto foi compro-vado através da mutação direcionadaem determinados resíduos de aminoá-cidos aspárticos do sítio catalítico.
O conhecimento da estrutura tridi-mensional da HIV protease co-cristali-zada com um ligante permite o planeja-mento de novos inibidores que apre-sentem grupamentos químicos queaumentam a interação pela enzima
provocando um maior potencial inibi-tório. Esta nova abordagem é conhe-cida como “planejamento de novosfármacos baseando-se na estrutura 3Dda enzima co-cristalizada com o li-gante” e exige o desenvolvimento deprogramas de modelagem molecularque sejam capazes de responder adeterminadas questões, como selecio-nar e priorizar novos ligantes no sítioreceptor. A utilização desta nova meto-dologia exige a estrutura 3D da proteí-na, enzima ou receptor cristalizada ouum modelo teórico obtido por estudosde homologia baseando-se na seqüên-cia primária de enzimas da mesmafamília complexada com um ligante.Este complexo age como ponto departida para identificar o modo de liga-ção, a conformação do ligante eminvestigação e aspectos essenciaisque determinam a sua afinidade pelaenzima alvo. Este conhecimento é usa-do para conceber novas idéias decomo otimizar um ligante existente oucomo desenvolver novos análogosalternativos. As discussões com espe-cialistas em química orgânica sintética,nessa fase, são importantes para eli-minar propostas sintéticas sem um pro-pósito prático. Este novo compostodeve ser sintetizado e avaliado farma-cologicamente. Em seguida, o seu mo-do de ligação pode ser determinadopor métodos bioquímicos, cristalo-gráficos e espetroscópicos. A vanta-gem importante desta abordagem é ainformação fornecida pela nova estru-tura 3D do complexo proteína-ligante.Se um ligante não demonstra a afinida-de esperada ou o perfil inibitório dese-jado, as análises citadas anteriormentefornecem, freqüentemente, informa-
ções que podem explicar a ausênciade atividade. Isto pode fornecer novasidéias de como é a interação com aenzima e, desta forma, propor novosinibidores.
A estrutura cristalina da HIV aspartilprotease foi determinada por cristalo-grafia de raios-X e um grande númerode inibidores foram propostos empre-gando-se técnicas de modelagemmolecular, que visam a proposição oua modificação estrutural de inibidoresconhecidos utilizando as informações3D do cristal, de modo a obter-se umaperfeita interação com o sítio catalíticoda enzima. Uma nomenclatura padrãoé usada para designar os resíduos ousub-unidades do substrato/inibidor(P3, P2, P1, P1’, P2’, P3’) que se ligamaos sub-sítios correspondentes naenzima (S3, S2, S1, S1’, S2’, S3’ )(Figura 8).
Um grupo de pesquisadores daDupont Merck (De Lucca et al., 1997;De Lucca et. al., 1999) descreveu o pla-nejamento racional de uma série dederivados, com a sub-unidade uréiainserida em um anel de sete membros,como potentes inibidores da HIVPRprotease. Esta nova classe de inibido-res foi descoberta com base em estu-dos preliminares da relação entre aestrutura molecular dos derivados dia-mino dióis com suas atividades bioló-gicas, o que permitiu a construção deum modelo farmacofórico 3D. Estemodelo foi utilizado para a pesquisa denovos candidatos a inibidores em ban-cos de dados de estruturas molecu-lares, onde os derivados cíclicos dauréia foram identificados. Estes deriva-dos são uma versão cíclica dos inibi-dores lineares diamino diol. A estrutura
Modelagem molecular
Figura 8: A letra P indica as regiões do substrato que interagem com os respectivos resíduosde amino ácidos do sítio catalítico da enzima (denotados pela letra S).

47
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001
de raios-X da HIVPR protease foi utili-zada e através do emprego de técnicascomputacionais, modificações estrutu-rais nos derivados da uréia foramidealizadas com o objetivo de promo-ver interações com os principais sítiosda aspartil protease. O átomo de oxigê-nio da carbonila da uréia foi planejadopara deslocar a única molécula deágua estrutural presente em comple-xos com inibidores lineares (Figura 9).
Desta forma, os derivados da uréiade sete membros mostraram um ali-nhamento adequado e apresentaramum alto grau de complementariedadedos seus substituintes com os respec-tivos resíduos de aminoácidos do sítiocatalítico da aspartil protease. Este tra-balho resultou em dois candidatos paraestudos clínicos: DMP323 e DMP450(Figura 10). É importante ressaltar que
estas uréias cíclicas foram identificadasa partir do inibidor linear diamino diol.
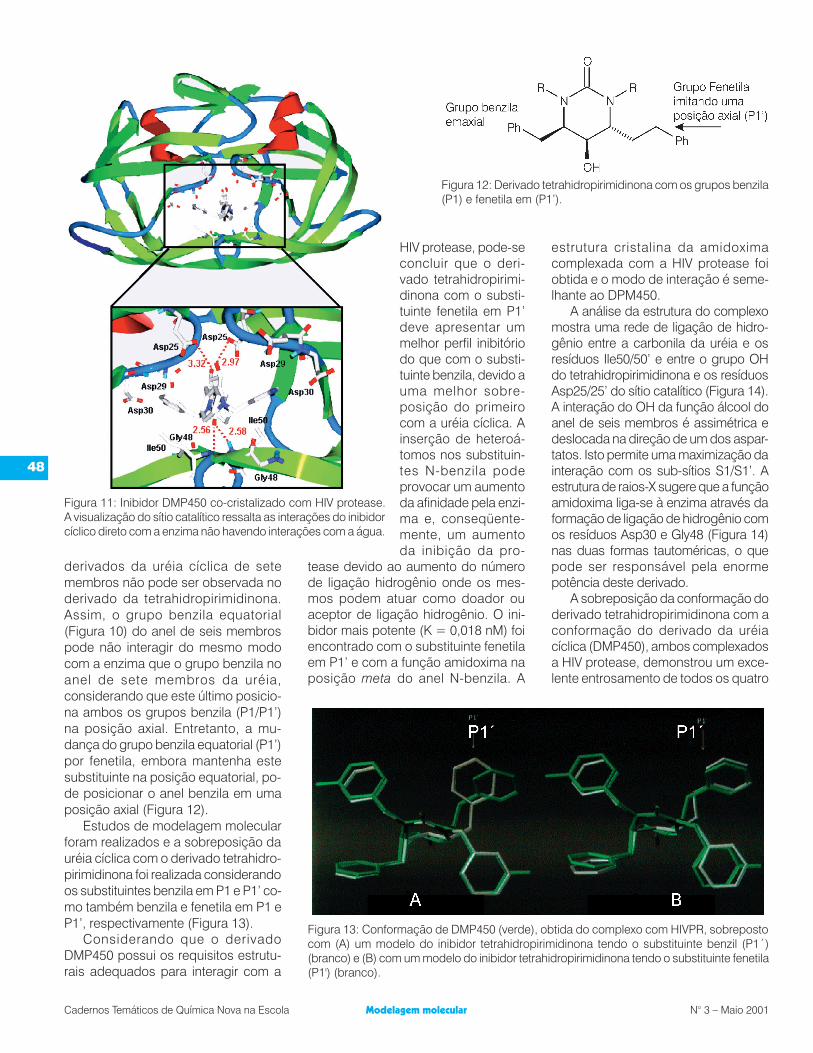
A análise da estrutura do inibidorDMP450 co-cristalizado com a HIVPRpermitiu compreender os requisitosestruturais que tornam as uréias cícli-cas potentes inibidores (Figura 11). A
característica mais evidente é uma re-de de ligações de hidrogênio entreIle50/50´ e a função carbonílica dauréia e entre os grupos dióis e os resí-duos dos aminoácidos aspárticos cata-líticos -Asp25 e 25’- (Figura 9). Umaoutra característica importante destesinibidores é a restrição conformacionalque arranja os grupos benzílicos transdiaxial em P1 em uma disposição quepermite uma perfeita interação com osresíduos de aminoácidos lipofílicos dosub-sítio S1 (Figura 8). Os grupos N-benzílicos desempenham duas fun-ções importantes: contribuem parauma interação com o sub-sítio lipofílicoS2 da protease como também orien-tam os substituintes nas posições metae para na direção dos sub-sítios S2/S3da enzima que possuem resíduos deaminoácidos polares tais como Asp29,Asp30 e Gly48.
Uma abordagem semelhante foiadotada para a descoberta de novosinibidores mais potentes de HIV pro-tease. Assim, foi explorada a possibi-lidade de se ciclizar outros inibidoreslineares de HIV protease. Entretanto,neste processo é importante manter aestereoquímica correta dos átomos decarbono como também observar aconformação do anel e a sua interaçãocom a enzima. O derivado diamino ál-cool, um dos primeiros inbidores linea-res desenvolvidos pela Abbott, foi sele-cionado para o processo de ciclização.
O anel tetrahidropirimidinona deseis membros possui os substituintesaromáticos com uma orientação trans-1,3-di-benzila (substituintes P1/P1’)com um dos grupos benzilas na po-sição axial e o outro na posição equa-torial (Figura 10). Desta forma, a orien-tação di-axial observada para os
Modelagem molecular
Figura 9: Inibidor linear co-cristalizado com HIV protease. O sítio catalítico é mostradoevidenciando a interação do inibidor peptídico do tipo substrato com uma molécula deágua (ligação de hidrogênio em tracejado vermelho) que é substituída nos inibidores cíclicospela interação direta com a enzima.
Figura 10: Estrutura dos compostos DMP323 e DMP450.
48
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001Modelagem molecular
derivados da uréia cíclica de setemembros não pode ser observada noderivado da tetrahidropirimidinona.Assim, o grupo benzila equatorial(Figura 10) do anel de seis membrospode não interagir do mesmo modocom a enzima que o grupo benzila noanel de sete membros da uréia,considerando que este último posicio-na ambos os grupos benzila (P1/P1’)na posição axial. Entretanto, a mu-dança do grupo benzila equatorial (P1’)por fenetila, embora mantenha estesubstituinte na posição equatorial, po-de posicionar o anel benzila em umaposição axial (Figura 12).
Estudos de modelagem molecularforam realizados e a sobreposição dauréia cíclica com o derivado tetrahidro-pirimidinona foi realizada considerandoos substituintes benzila em P1 e P1’ co-mo também benzila e fenetila em P1 eP1’, respectivamente (Figura 13).
Considerando que o derivadoDMP450 possui os requisitos estrutu-rais adequados para interagir com a
Figura 11: Inibidor DMP450 co-cristalizado com HIV protease.A visualização do sítio catalítico ressalta as interações do inibidorcíclico direto com a enzima não havendo interações com a água.
Figura 12: Derivado tetrahidropirimidinona com os grupos benzila(P1) e fenetila em (P1’).
HIV protease, pode-seconcluir que o deri-vado tetrahidropirimi-dinona com o substi-tuinte fenetila em P1’deve apresentar ummelhor perfil inibitóriodo que com o substi-tuinte benzila, devido auma melhor sobre-posição do primeirocom a uréia cíclica. Ainserção de heteroá-tomos nos substituin-tes N-benzila podeprovocar um aumentoda afinidade pela enzi-ma e, conseqüente-mente, um aumentoda inibição da pro-
tease devido ao aumento do númerode ligação hidrogênio onde os mes-mos podem atuar como doador ouaceptor de ligação hidrogênio. O ini-bidor mais potente (K = 0,018 nM) foiencontrado com o substituinte fenetilaem P1’ e com a função amidoxima naposição meta do anel N-benzila. A
estrutura cristalina da amidoximacomplexada com a HIV protease foiobtida e o modo de interação é seme-lhante ao DPM450.
A análise da estrutura do complexomostra uma rede de ligação de hidro-gênio entre a carbonila da uréia e osresíduos Ile50/50’ e entre o grupo OHdo tetrahidropirimidinona e os resíduosAsp25/25’ do sítio catalítico (Figura 14).A interação do OH da função álcool doanel de seis membros é assimétrica edeslocada na direção de um dos aspar-tatos. Isto permite uma maximização dainteração com os sub-sítios S1/S1’. Aestrutura de raios-X sugere que a funçãoamidoxima liga-se à enzima através daformação de ligação de hidrogênio comos resíduos Asp30 e Gly48 (Figura 14)nas duas formas tautoméricas, o quepode ser responsável pela enormepotência deste derivado.
A sobreposição da conformação doderivado tetrahidropirimidinona com aconformação do derivado da uréiacíclica (DMP450), ambos complexadosa HIV protease, demonstrou um exce-lente entrosamento de todos os quatro
Figura 13: Conformação de DMP450 (verde), obtida do complexo com HIVPR, sobrepostocom (A) um modelo do inibidor tetrahidropirimidinona tendo o substituinte benzil (P1´)(branco) e (B) com um modelo do inibidor tetrahidropirimidinona tendo o substituinte fenetila(P1') (branco).

49
Cadernos Temáticos de Química Nova na Escola N° 3 – Maio 2001
Leitura complementarALLINGER, N.L. A Hydrocarbon force
field utilizing V1 and V2 torsional terms. J.Am. Chem. Soc., v. 99, p. 8127, 1977.
BARREIRO, EJ; RODRIGUES C.R., AL-BUQUERQUE M.G., SANT’ANNA C.M.R.e ALENCASTRO, R.B. Modelagem mo-lecular: Uma ferramenta para o planeja-mento racional de fármacos em químicamedicinal. Química Nova, v. 20 n. 3, p.300-310, 1997.
DE LUCCA, G.V.; LIANG, J.; ALDRICH,P.E.; CALABRESE, J.; CORDOVA, B;KLABE, R.M.; RAYNER, M.M. e CHANG,C-H. Design, synthesis, and evaluation of
Modelagem molecular
Figura 14: (A) Estrutura de raios-X do complexo da amidoximacom a HIV protease mostra as interações de ligação hidrogênio.O átomo de oxigênio da uréia atua como aceptor de ligaçãohidrogênio de ambos os resíduos de Ile50/50' sem a participaçãoda molécula de água estrutural. O grupo hidróxi está assimetri-camente ligado aos dois resíduos Asp catalíticos. (B) Esquemada ligação hidrogênio da amidoxima com a HIV protease com asdistâncias assinaladas em Å. Note que o substituinte amidoximaparece ligar-se nas duas diferentes formas tautoméricas.
Figura 15: Sobreposição de DMP450 (verde) com a amidoxima (bran-co) nas suas conformações obtidas a partir de análise de raios-X dosseus respectivos complexos com a HIVPR. Existe um bom ajuste en-tre os quatro anéis fenila, maximizando as interações com os resíduosde aminoácidos do sítio catalítico (ver Figura 14 B).
anéis fenilas e confirmou os estudos de planejamento iniciaisutilizando a modelagem molecular (Figura 15). A síntese doderivado benzila foi realizada e o resultado farmacolígicomostrou que o mesmo é cerca de 100 vezes menos ativo doque o derivado fenetila na posição P1’. Isto comprova outravez os dados obtidos por modelagem molecular.
Conclusões
Os estudos de modelagem molecular propiciaram a obteçãode novos inibidores de HIV protease mais potentes. Assim, amodelagem molecular tem se tornado uma ferramenta impor-tante no planejamento de novos fármacos. A sua importânciapode ser observada no grande interesse de companhiasvoltadas na elaboração de programas de modelagem (soft-ware) que buscam retratar com acurácia os parâmetroseletrônicos e estrutrais tanto de moléculas isoladas quantocomplexadas a uma biomacromolécula (enzima, proteína oureceptor) traduzindo com maior precisão o processo de inte-ração fármaco-receptor.
Carlos Rangel Rodrigues ([email protected]), graduado em ciências farmacêuticaspela Faculdade de Farmácia da UFRJ em 1991, mestre e doutor pelo DQO-IQ da UFRJ,é professor adjunto da Faculdade Farmácia – UFRJ e responsável pelo setor de QuímicaComputacional do LASSBio, desenvolvendo projetos na área de química medicinal.
tetrahydropyrimidinones as an example ofa general approach to nonpeptide HIV pro-tease inhibitors J. Med. Chem., v. 40 n. 11,p. 1707-1719, 1997.
DE LUCCA, G.V.; LIANG, J. E DE LUCCA,I. Stereospecific synthesis, structure-activ-ity relationship, and oral bioavailability oftetrahydropyrimidin-2-one HIV protease in-hibitors. J. Med. Chem., v. 42 n. 1, p. 135-152, 1999.
DEWAR, M.J.S.; ZOEBISCH, E.G.; HEALY,E.F. e STEWART, J.J.P. AM1: A new generalpurpose quantum mechanical molecularmodel. J. Am. Chem. Soc., v. 107, p. 3902-3909, 1985.
LEUNG, D.; ABBENANTE, G. e FAIRLIE,
D.P. Protease inhibitors: Current status andfuture prospects. J. Med. Chem., v. 43 n.3, p. 305-341, 2000.
MMX force field of PCMODEL“PCMODEL”, Gilbert, K. Serena Software:Bloomington, IN, 1993.
NUGIEL, D.A.; JACOBS, K.; WORLEY,T.; PATEL, M., KALTENBACH III, R.F.;MEYER, D.T.; JADHAV, P.K.; DE LUCCA,G.V, SMYSER, T.E.; KLABE, R.M,BACHELER, L.T.; RAYNER, M.M. e SEITZ,P.S. Preparation and structure-activity re-lationship of novel P1/P1'-substituted cy-clic urea-based human immunodeficiencyvirus type-1 protease inhibitors. J. Med.Chem., v. 39 p. 11, n. 2156-2169, 1996.


















