
CONTROLE ANALÍTICO DOS PRODUTOS DE FISSÃO EM …pelicano.ipen.br/PosG30/TextoCompleto/Maria...
396
INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES SECRETARIA DA INDÚSTRIA. COMÉRCIO, CIÊNCIA E TECNOLOGIA AUTARQUIA ASSOCIADA À UNIVERSIDADE DE SÃO PAULO CONTROLE ANALÍTICO DOS PRODUTOS DE FISSÃO EM SOLUÇÕES DO PROCESSO PUREX POR ESPECTROMETRIA GAMA Maria Augusta Gonçalves Dissertação apreservtada ao Instituto de Pesquisas Energéticas e Nucleares como parte dos requisitos para a obtenção do Grau de "Mestre - Área de Reatores Nucleares de Potência e Tecnologia do Coministfvel Nuclear". Orientador: Dra. Harico Tamura Matsuda São Paulo 19B2
Transcript of CONTROLE ANALÍTICO DOS PRODUTOS DE FISSÃO EM …pelicano.ipen.br/PosG30/TextoCompleto/Maria...

INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES SECRETARIA DA INDÚSTRIA. COMÉRCIO, CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA À UNIVERSIDADE DE SÃO PAULO
CONTROLE ANALÍTICO DOS PRODUTOS DE FISSÃO EM SOLUÇÕES DO PROCESSO PUREX POR ESPECTROMETRIA GAMA
Maria Augusta Gonçalves
Dissertação apreservtada ao Instituto de Pesquisas Energéticas e Nucleares como parte dos requisitos para a obtenção do Grau de "Mestre - Área de Reatores Nucleares de Potência e Tecnologia do Coministfvel Nuclear".
Orientador: Dra. Harico Tamura Matsuda
São Paulo 19B2

'1
INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES
SECRETARIA DA INDÚSTRIA, COMÉRCIO, CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA À UNIVERSIDADE DE SAO PAULO
J
CONTROLE ANALÍTICO DOS PRODUTOS DE FISSAO EM SOLUÇÕES
DO PROCESSO PUREX POR ESPECTROMETRIA GAMA
Maria Augusta Gonçalves
Dissertação apresentada ao Instituto de Pesquisas Energáticas e Nucleares como parte dos requisitos para a obtenção do grau de "IMestre — Área de Reatores Nucleares de Potência e Tecnologia do Combustível Nuclear".
Orientadora: Dra. Harko Tamura Matsuda
SÃO PAULO
1982

INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES
SECRETARIA DA INDÚSTRIA, COMÉRCIO, CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA À UNIVERSIDADE DE SAO PAULO
NTROLE ANALÍTICO DOS PRODUTOS DE FiSSAO EM SOLUÇÕES
DO PROCESSO PUREX POR ESPECTROMETRIA GAMA
Maria Augusta Gonçalves
L ! V R O \ Dissertação apresentada ao Instituto de Pesquisas Energéticas e Nucleares como parte dos requisitos para a obtenção do grau de "Mestre — Área de Reatores Nucleares de Potência e Tecnologia do Combustível Nuclear".
Orientadora: Dra Harko Tamura Matsuda
SÃO PAULO
1982

AGRADECIMENTOS
Harko Tamura Matsuda
Bertha Floh de Araújo
José Adroaldo de Araújo
Alcídio Abrão
Todos os colegas da Area de Reprocessamen
to e do Centro de Engenharia Química.
Pela orientação, colaboração e
incentivo na execução deste
trabalho.
rr»,,crRn Ki^rrmí r.F FKtRGiA NUCLEAR/SP - IPEH

SUMÁRIO
Página
RESUMO i
ABSTRACT i ü
CAPITULO I
INTRODUÇÃO
I.l - Objetivo.
CAPITULO II
A N A L I S E DE P R O D U T O S D E FISSAO POR E S P E C T R O M E T R I A GAMA 9
II. 1 - Produtos de Fissão 9
II. 2 - Espectrometria Gama 11
II.2.1 - Interação da Radiação com a Materia 12
II. 2. 2 - Detectores 13
II. 2. 2.1 - Detectores Semicondutores 13
II. 2. 2. 2 - Aplicações 15
II. 3 - Análise de Espectros 17
II. 3.1 - Programas "GELIGAM" 19

CAPITULO III
PARTE EXPERIMENTAL 22
111.1 - Equipamentos 22
111.2 - Reagentes 24
III. 3 - Amostras para Análise. 25
III.3.1 - Pontos de Retirada de Amostras para Con
trole de Processo e Estabelecimento da
Geometria de Contagem 25
III. 3.1.1 - Frasco de Amostra 2 7
III. 3.1.2 - Volume de Amostra 28
III. 3. 2 - Preparação das Soluções-Padrão 28
III.3.3 - Preparação das Amostras de Uranio Irra
diado 29
III. 4 - Análise Espectrométricas 33
III. 4.1 - Tempo de Contagem 33
III. 4. 2 - Análises Qualitativas 34
III. 4. 3 - Análises Quantitativas 34
111.4.3.1 - Calibração do Sistema Detec
tor 34
111.4.3.2 - Elaboração de Bibliotecas pa
ra Identificação de Radionu
elídeos 37
111.4.3.3 - Análise de Produtos de Fis
sao em Soluções de Uranio Ir
radiado 38

III. 5 - Dados Experimentais 38
III. 5.1 - Análises Qualitativas 39
III. 5. 2 - Análises Quantitativas 42
CAPITULO IV
DISCUSSÃO E CONCLUSÕES
APÊNDICE I 56
APÊNDICE II 58
APÊNDICE III 60
REFERÊNCIAS BIBLIOGRÁFICAS 64

.i.
"CONTROLE ANALÍTICO DOS PRODUTOS DE FISSÃO EM
SOLUÇÕES DO PROCESSO PUREX POR ESPECTROMETRIA GAMA"
MARIA AUGUSTA GONÇALVES
RESUMO
Apresenta-se neste trabalho o desenvolvimento de um mé
todo radioraetrico para o controle de produtos de fissão por espec
trometria gama em soluções de processo Purex. O estudo visa a apli
cação ao controle desses radionuclídeos na instalação de tratamen
to químico de uranio irradiado, em fase de implantação no Centro de
Engenharia Química do Instituto de Pesquisas Energéticas e Nuclea
res.
A principio, desenvolveram-se estudos para a definição
da geometria de contagem, levando-se em consideração as ativida
des encontradas nas soluções de processo, o sistema de preparação
de amostras nas células analíticas e o sistema de detecção gama u
tilizado. Da mesma forma, prepararam-se padrões de atividades co
nhecidas, seguindo a mesma geometria das amostras de análise.
Com a finalidade de se obter soluções com composições
semelhantes àquelas do processo Purex, irradiaram-se pequenas amos
tras de urânio natural e \irânio com 19,91% de enriquecimento em
U. Essas amostras foram dissolvidas com ácido nítrico, após um
curto período de resfriamento e, em seguida, preparadas para con
tagem em frascos padronizados, segundo a geometria definida.
Os espectros foram registrados utilizando-se um detec
tor semicondutor de Ge(Li) e analisados por meio do sistema de
programas "GELIGAM", em um computador PDP-11/05. Para a determina
ção das atividades dos produtos de fissão, prepararam-se bibliote
cas e realizaram-se calibrações, de modo a tornar esses programas

J cesso Purex.
.ii.
Mediante as análises dos dados fornecidos pelo siste
ma "GELIGAM", escolheu-se um programa para uso de rotina, levan
do-se em conta,não só a precisão dos resultados, mas também o
tempo total gasto no processamento.
adequados ãs análises de produtos de fissão em soluções de pro-

.iii.
"FISSION PRODUCTS CONTROL BY GAMMA SPECTROMETRY
IN PUREX PROCESS SOLUTIONS"
MARIA AUGUSTA GONÇALVES
ABSTRACT
This paper deals with a radiometric method for fission
products analysis by gamma spectrometry. This method will be afçlied
for fission products control at the irradiated material processing
facility, londer construction in Instituto de Pesquisas Energéti
cas e Nucleares.
Counting geometry was defined taking account the acti^
vities of process solutions to be analysed, the remotely operated
aliquotation device of analytical cell and the available detection
system.
Natural and 19,91% enriched uranium sairples vere irradiated
at IEAR-1 reactor in order to simulate the composition of Purex
process solutions. After a short decay time, the sartples were dissolved
with HNO^ and then, conditioned in standard flasks with defined
geometry.
The spectra were obtained by a Ge(Li) semiconductor
detector and analysed by the GELIGAM software system, losing a floppy
-disk connected to a PDP-11/05 computer. Libraries were prepared
and calibrations were made with standard sources to fit the programs
to the analysis of fission products in irradiated uranium solutions.
It was possible to choose the best program to be used
in routine analysis with the obtained data.
COMISCAC KAC:CN/L CE Lí.LF.GiA NUCLEAR/SP - iPEfi

.1.
CAPITULO I
INTRODUÇÃO
Uma fase importante do ciclo do combustível é o repro
cessamento do combustível nuclear. Este, apôs sua utilização no
reator, possui ainda um valor econômico, havendo interesse em re-
processá-lo a fim de se recuperar os elementos férteis e fisseis
nele ainda contidos, bem como aqueles que se formaram durante a
irradiação, separando-os dos produtos de fissão.
Os combustíveis nucleares devem ser reciclados perio
dicamente, porque os produtos de fissão que se formam durante a
irradiação limitam o uso do combustível no reator, alterando as
suas propriedades físicas. Além disso, alguns produtos de fissão
possuem alta secção de choque de captura de neutrons, prejudican
do a economia neutrônica no reator.
Um combustível irradiado pode ser considerado como una 38
mistura de cinco tipos de componentes :
o . • -, - T 235„ 239„ 241„ - o material fissil, como U, Pu, Pu
- o material fértil
236 237 242
- os isótopos pesados, tais como U, Np, Pu e
outros transurânicos
- os metais (magnesio, alumínio, molibdênio, zircônio,
aço inoxidável, etc.) que formam ligas com o combus
tível ou que constituem o revestimento
- os produtos de fissão

.2.
A unidade de reprocessamento recebe o uranio irradia
do, resfriado, cabendo-lhe separar físseis e férteis dos produtos
de fissão, permitindo, dessa forma, o seu reaproveitamento econô
mico para a reconstituição do elemento combustível.
Deve-se obter com o reprocessamento uma descontamina
ção total, isto é, diminuição das atividades B , Y ao nível de ati-3
vidade do urânio natural (0,67 Ci/t), a fim de se permitir o ma
nuseio direto do material recuperado.
A escolha do processo para o tratamento químico depen
de do tipo de elemento combustível, da queima do combustível, do
grau de descontaminação necessário, do grau de recuperação deseja
vel e das tecnologias disponíveis.
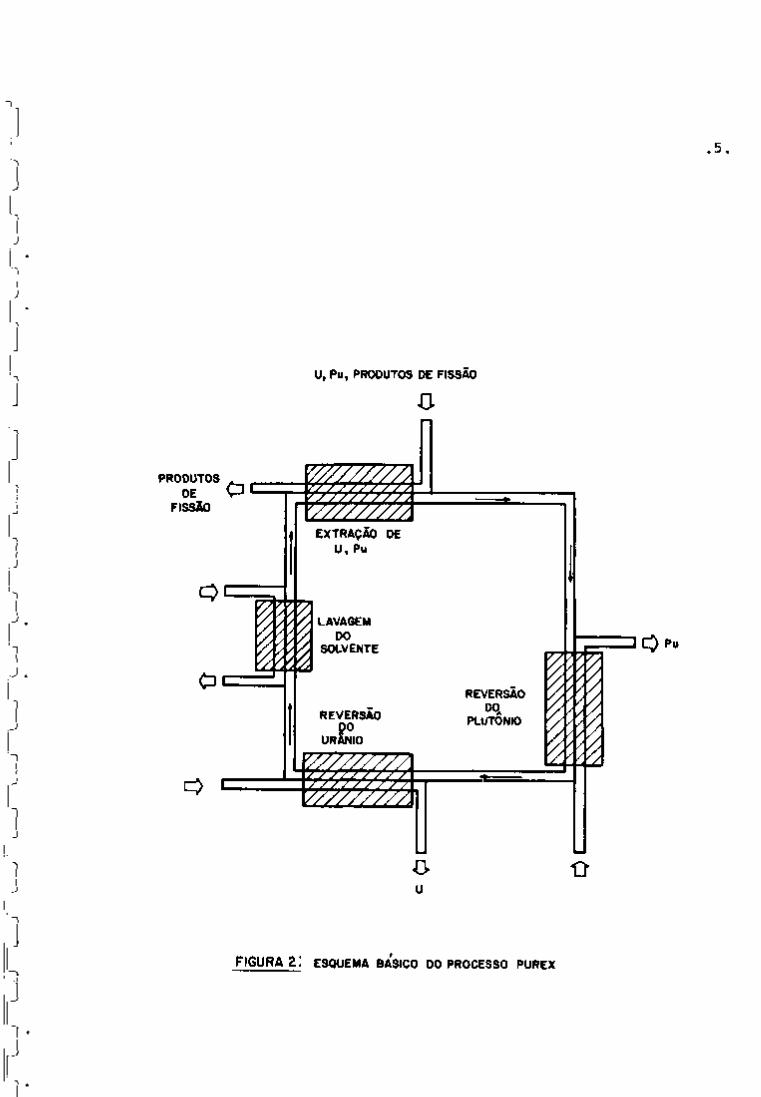
O processo Purex (Plutoni\im Uranium Refining by Ex
traction) é o mais usado dentre os processos utilizados para o ^ 1 8 2 3 3 8
tratamento químico do combustível do uranio ' ' .Ê um processo
líquido-líquido que utiliza o fosfato de tri-n-butila (TBP) como
agente extrator. Em relação aos outros processos que também empre
gam a técnica de extração por solventes orgânicos, apresenta as 1 8
seguintes vantagens :
- é um processo totalmente realizado em meio nítrico
e com recuperação total do ácido
- pode ser realizado em presença de radiação
- todas as operações são realizadas â temperatura am
biente e sem riscos de inflamabilidade
- o volume de efluentes é mínimo
O processo Purex básico apresenta, resumidamente, as
seguintes fases:
1) Operações preliminares ou de "head-end"
Os processos preliminares do tratamento incluem des-

.3.
mantelamento químico e/ou mecânico, dissolução em ácido nítrico e,
usualmente, pré-tratamentos químicos adicionais, terminando com o
ajuste da solução de alimentação do primeiro ciclo de extração.
2) Separação de urânio e plutónio dos produtos de fis^
são com TBP/diluente
Essa fase compreende:
-, extração conjunta dos nitratos de uranilo e plutó
nio, separando-os dos produtos de fissão,
- partição urânio-plutônio, baseada na redução de
Pu-IV a Pu-III,
- ciclos de purificação das soluções aquosas de urâ
nio e plutónio: são ciclos adicionais de extração
com TBP/diluente, para aumentar os fatores de des
contaminação em produtos de fissão.
3) Purificação final ou operações de "tail-end"
A purificação final do urânio é feita, geralmente, u-
tilizando-se uma coluna de sílica-gel. Para a purificação do plu
tónio, empregam-se operações de troca iónica ou extração com ami
nas terciárias.
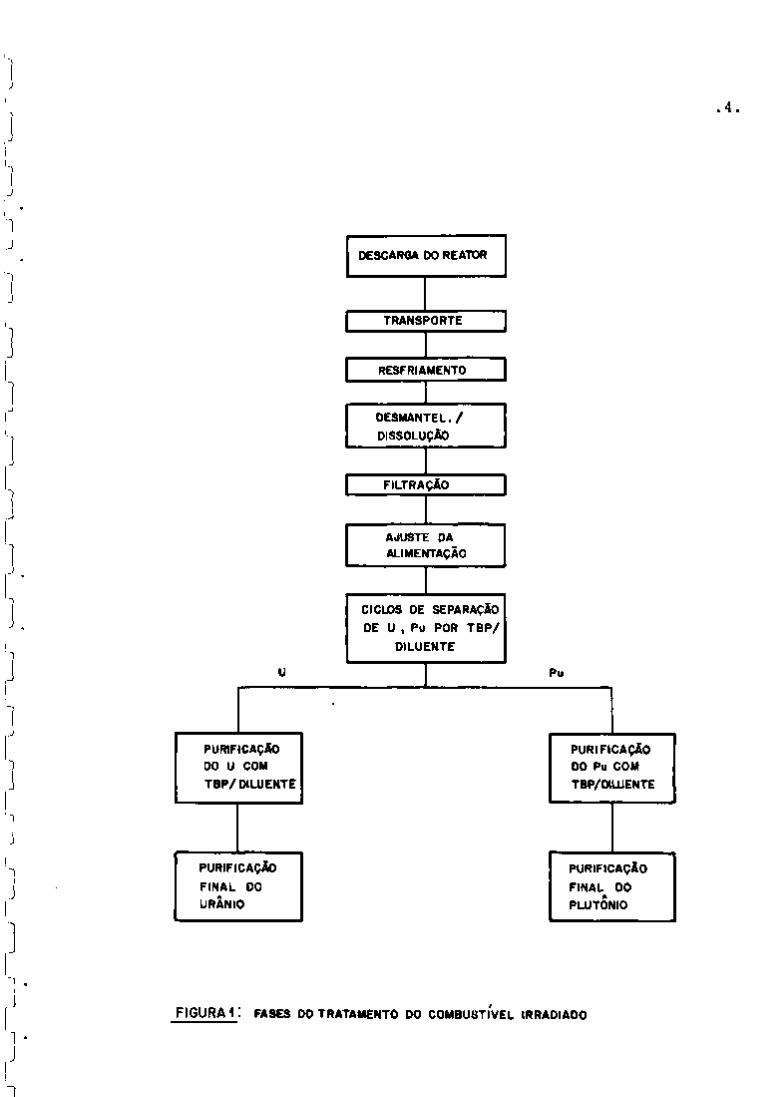
Na Figura 1 pode-se observar as principais fases do
tratamento do combustível irradiado e, na Figura 2, um esquema do
processo Purex.
Nos processos de separação e purificação dos actiní-
deos são exigidos fatores de descontaminação em produtos de fis-
~ 8 são da ordem de 10 . Para atingir tais fatores de descontaminaçãcv
torna-se necessário um controle rigoroso dos produtos de fissão em
diversas fases do tratamento químico.
.4,
DESCARGA 00 REATOR
D E S M A N T E L . / DISSOLUÇÃO
FILTRAÇÃO
AJUSTE DA ALIMENTAÇÃO
I TRANSPORTE "|
I RESFRIAMENTO "]
CICLOS OE SEPARAÇÃO DE U , Pu POR T B P /
DILUENTE
Pu
•
PURIFICAÇÃO DO U COM T B P / D I L U E N T E
PURIFICAÇÃO DO Pu COM TBP/DtUJENTE
PURIFICAÇÃO FINAL DO URÂNIO
PURIFICAÇÃO FINAL DO PLUTONIO
F I G U R A I : FASES DO TRATAMENTO DO COMBUSTÍVEL IRRADIADO
.5,
U, Pu, PRODUTOS DE FISSAO
PRODUTOS . DE O
FISSSO
EXTRAÇÃO DE U , Pu
LAVAGEM DO
SOLVENTE
REVERSÃO
UR^^IO
REVERSÃO
PLUTONIO
vyyy7///A
3 0 P "
u
FIGURA 2: ESQUEMA BÁSICO DO PROCESSO PUREX

.6.
A operação de vima instalação de reprocessamento re
quer um trabalho analítico organizado e árduo, pois são necessâ-4 , 1 7 , 18
rios diversos tipos de controle que podem, de uma forma geral,ser 17
agrupados em três categorias :
- Análises de processo: referem-se ã monitoração da
instalação por meio de amostras das soluções do pro
cesso. Os resultados apresentados devem ser rápidos,
sendo preferíveis os métodos analíticos instrumen
tais que não exijam separações químicas prévias.
- Análises para o balanço de materiais: incluem a con
tabilidade de materiais férteis e físseis e, também,
as análises de salvaguarda que previnem os riscos de
proliferação. O principal requisito neste tipo de a
nálise é a alta precisão do método empregado.
- Análises de segurança: servem para manter a seguran
ça interna (criticalidade, corrosão) e a segurança
externa (emissões radioativas) de uma instalação de
reprocessamento.
O problema principal na aplicação de métodos analíti
cos convencionais é o alto nível de atividade das amostras, que e
xige o emprego de técnicas por controle remoto, em células com pro
teção biológica, para o desenvolvimento das análises.
As análises mais frequentes são: as determinações de
urânio, de plutónio, de ácido nítrico, dos produtos de fissão, dos
estados de oxidação dos âctinídeos e dos produtos de degradação do
TBP, tanto em soluções aquosas quanto orgânicas do processo.
Os métodos analíticos básicos mais usados são a espec
trometria de fluorescência de raios-X, para a determinação de ele
mentos pesados, e a espectrometria de radiação nuclear de alta re

.7.
solução com detectores de estado sólido, para a análise de emisso
res alfa a nivel de traços e de produtos de fissão emissores gama.
Estes métodos são complementados por outros métodos analíticos,
tais como a potenciometria, a espectrofotometria, a fluorimetria,
a polarografia, a cromatografia gasosa, a espectrografía de emis
são e a espectrometria de massa.
I.l - Objetivo
A química analítica do combustível irradiado é muito
complexa dada a natureza e especificações do combustível. Cada e-
lemento combustível sofre após a irradiação um tratamento químico
diferente para a recuperação dos elementos férteis e fisseis, en
volvendo em cada processo \m programa analítico distinto.
Um dos problemas associados â análise do combustível
gasto é, naturalmente, o alto nível de radioatividade devido aos
produtos de fissão. Ê importante, durante o processo de separação
dos âctinídeos dos produtos de fissão, a análise rigorosa desses
nuclideos, a fim de se conhecer os fatores de descontaminação em
diversas fases do tratamento químico. No processo Purex, onde se
utiliza o TBP como agente extrator, a maioria dos produtos de fís
são não são extraídos juntamente com os âctinídeos. Os nuclídeos
que não apresentam uma distribuição desprezível neste solvente são
" z r - " H b , 1"-106^^.103-106^_ _ ^ ^ ^ ^ proporcio, l " - " « c e -
141-144 -r Pr. Todos esses nuclideos sao emissores gama.
Este trabalho é uma contribuição aos métodos analíti
cos radiometricos para o controle de produtos de fissão no trata
mento de materiais irradiados pelo processo Purex. O método será
aplicado na instalação em fase de implantação no Centro de En-
nharia Química (CEQ) do Instituto de Pesquisas Energéticas e Nu-

.8.
oleares (IPEN).
Apresenta-se um procedimento para a análise qualitati
va e quantitativa dos produtos de fissão emissores gama. Os espec
tros são obtidos por meio de um detector semicondutor de Ge(Li) e
as análises são efetuadas utilizando-se um sistema de programas de
nominado"GELIGAM". Uma das finalidades deste trabalho é o ajuste
dos dados fornecidos a esses programas para adaptá-los âs condi
ções do processo utilizado.

.9.
CAPITULO II
ANÁLISE DE PRODUTOS DE FISSÃO POR ESPECTROMETRIA GAMA
II.1 - Produtos de Fissão
38 A reação de fissão pode ser escrita como:
P ^1 P ^2 ^ " 92 O ^1 * 2 O
onde:
P-, + Pj = 92, A3^+ A 2 = 236 e V = 2,5
Chamam-se produtos de fissão aos novos nuclídeos F^
e resultantes da partição do elemento flssil. Os principais pro
~ 235 •« -dutos de fissão formados por irradiação de U com niutrons tér-
38 micos encontram-se na Tabela I.
~ 36
Pode-se dividir os produtos de fissao em tres tipos :
o primeiro tipo inclui os produtos de fissão que são produzidos di
retamente no processo de fissão ou são resultantes de precursores
de meias-vidas muito mais curtas que as suas próprias meias-vidas.
O segundo tipo abrange aqueles radionuclídeos que são descenden
tes dos produtos de fissão do primeiro tipo e que não estão em e-95
quillbrio secular com os pais, tal como o Nb. No terceiro tipo encontram-se os nuclídeos radioativos que se formam por reações
^ ~ - 134 neutronicas dos produtos de fissao, como e o caso de Cs.
Determinam-se teoricamente as atividades dos produtos
de fissão do primeiro tipo, como o •'•" Cs, ^^Sr, "'• Ce, •'" •'•Ce, ''•Y,
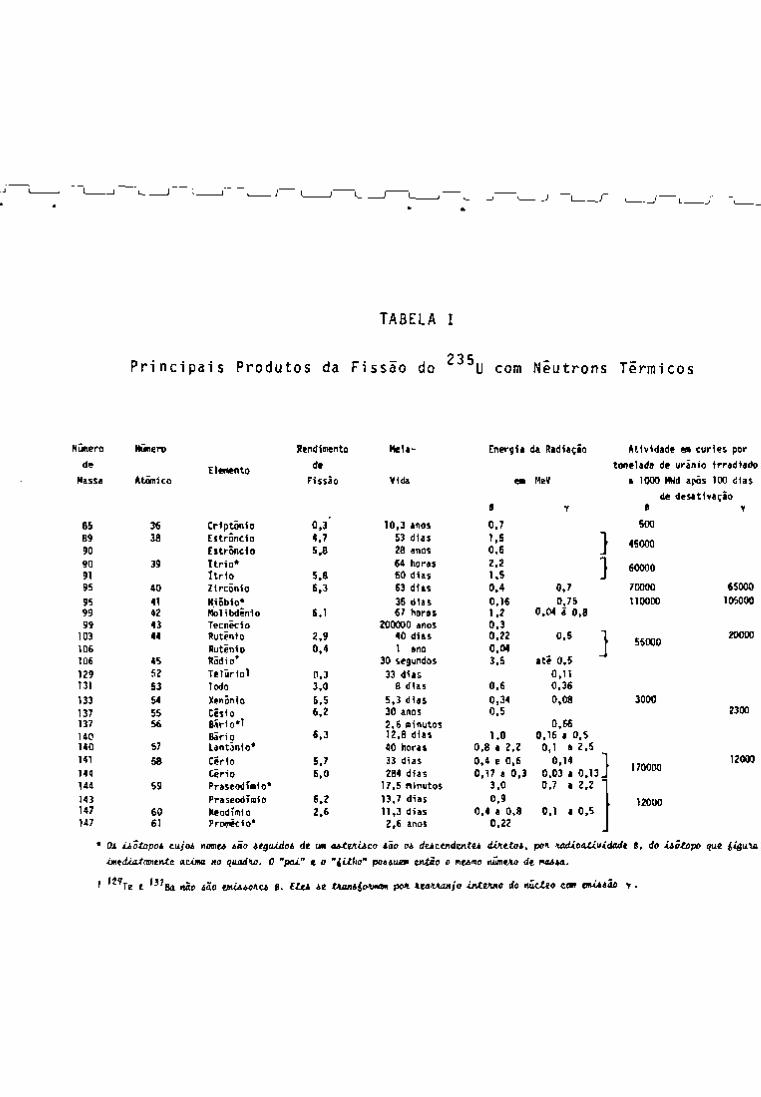
TABELA I
2 3 Principais Produtos da Fissão do U com Neutrons Térmicos
Número Número Rendimento Mela- Energía da Radiação Atividade em curies por
de Elemento
de tonelada de uranio irradiado
Massa Atômico Elemento
Fissão Vida em
e
MeV
Y
a 1000 MHd após 100 días
de desativação B Y
85 36 Crlptonio 0.3' 10,3 anos 0,7 500
89 38 Estroncio 4.7 53 días 1.5 • 45000
90 Estroncio 5.8 28 anos 0,6 _ • 45000
90 39 Itrio* 64 horas 2.2 60000 91 Itrio 5.8 60 días 1.5 _
60000
95 40 Zircônio 6.3 63 días 0,4 0,7 70000 65000
95 41 Niobio* 35 días 0,16 0,75 110000 105000 99 42 Molibdênio 6.1 67 horas 1.2 0,04 ã 0.8
99 43 Tecnicio 200000 anos 0,3 103 44 Ruteni0 2.9 40 días 0.22 O.S L crn«n 20000
55000 106 Rutinio 0.4 1 ano 0.04 L crn«n 20000
55000
106 45 Ródio* 30 segundos 3,5 até 0.5
129 52 Telúrio! 0,3 33 dias 0.11 131 53 lodo 3,0 8 dias 0.6 0.36
133 54 Xenóni0 6.5 5.3 dias 0.34 0,08 3000
137 55 . Césio 6,2 30 anos 0,5 2300
137 56 Bário*! 2.6 minutos 0.66
140 Bário 6.3 12,8 dias 1.0 0.16 a 0.5 140 57 Lantãnio* 40 horas 0,8 a 2.2 0,1 a 2,5 141 58 Cirio 5,7 33 dias 0.4 e 0.6 0.14
170000 144 Cirio 6,0 284 dias 0,17 a 0.3 0.03 a 0.13^
170000
144 59 PraseodTmio* 17,5 minutos 3.0 0.7 a 2,2 "
143 PraseodTmio 6.2 13.7 dias 0.9 12000 147 60 Neodimio 2,6 11,3 dias 0.4 a 0,8 0,1 a 0.5
12000
147 61 Promécio* 2,6 anos 0,22
* Oi ¿¿ótopoi cujoi nmu ião itgiUdoi di um oiteAiico ião oi ducendtntu dVieXoi, pon. ncdioatividadt B, do ¿iotopo quí
¿i«td¿at<me.ntc CLCMM no quadno. O "pai" e o "iilho" poauem então o mumo númeno de nana.
' '^'te e '^^8d vão ião emiaoiu B. Etu ie tAamiomm pol KMVutnjo ¿tvteAno do núcleo com enUiiâo y .
gwia

.11.
•^^^Pm, ^^Zr, •'• " Ru e '^^^Ru formados no processo de irradiação con
tínua do urânio (sob a forma de U^Og), pela expressão:
onde:
A. = - - — <t) o N Y d - e" ^^ir) ^ 3,7 X 10^
N = N — — o
sendo.
A^ = atividade do radioisótopo i em mCi
(t) = fluxo de niutrons
235
a = secçao de choque de fissao do U para neutrons
térmicos multiplicada pela abundância isotópica
N = número de átomos de urânio contido em 1,0 g de "3°8
Y = rendimento de fissão de cada isótopo
A = constante de desintegração de cada isótopo (h'"'')
t^^= tempo de irradiação (hora)
= número de Avogadro
m^ = massa de urânio contida no U^Og
My = peso atómico do urânio
II.2 - Espectrometria Gama
O decaimento de um radioisótopo é muitas vezes acompa
nhado pela emissão de um ou mais raios gama. Portanto, as medidas
das energias dos raios gama emitidos por um dado isótopo servem
para identificar o isótopo. Além disso, a taxa de emissão de raios
gama de uma amostra permite determinar a quantidade do material ra
dioativo na amostra.

.12,
1 1 2 7 3 0 3it ítO II.2.1 - Interação da Radiação com a Matéria ' ' ' '
Os principais mecanismos por meio dos quais a radia
ção eletromagnética interage com a matéria são o efeito fotoelé-
trico, o efeito Compton e a produção de pares.
No efeito fotoelétrico, a energia da radiação eletro
magnética (Ey) é totalmente absorvida por um átomo e é consiomida
para arrancar um elétron orbital e fornecer a este energia cinéti
ca (Eg), onde:
sendo, Ej^, a energia de ligação do elétron. O raio gama original
desaparece nesse processo, mas o átomo excitado emitirá em segui
da um ou mais raios X com energia total Ej^.
O processo de espalhamento Compton pode ser•considera
do como uma colisão elástica entre um fóton e um elétron livre ou
um elétron atômico, cuja energia de ligação seja desprezível com
parada ã energia da radiação incidente. Essa energia é dividida en
tre o fóton espalhado e o elétron de recuo.
Na produção de pares, há interação da radiação oom um
elétron ou núcleo atômico. O fóton desaparece e são criados um e-
létron e um positron, que terão a mesma energia cinética. A ener
gia cinética total é igual â energia do fóton menos a energia de
repouso das duas partículas. O positron pode sofrer aniquilação ao
se encontrar com um elétron do material e dar origem a dois raios
gama de 0,511 MeV cada um.
Para que ocorra a produção de pares, a energia do raio
gama incidente deve exceder a energia de repouso do elétron e do
positron, isto é, 1,02 MeV.

.13.
II.2.2 - Detectores
Os principais tipos de detectores utilizados para a
espectrometria gama são os detectores de cintilação e os detecto
res semicondutores. Estes são, atualmente, mais empregados para
a espectrometria gama devido ao seu alto poder de resolução com
parados aos cintiladores.
9 1 1 2 7 3 0 'tO II.2.2.1 - Detectores Semicondutores ' ' ' '
Os semicondutores são sólidos que, teoricamente, ã
temperatura de O K, são isolantes perfeitos, mas que, com a ele-2 7
vação da temperatura, tornam-se maus condutores
Os materiais mais utilizados para a fabricação de de
tectores semicondutores são o germânio e o silicio. Para a utili
zação desses materiais como detectores, torna-se necessário au
mentar sua resistividade especifica a fim de impedir a fuga ex
cessiva de corrente, quando da aplicação de um campo elétrico.Po
de-se evitar esta fuga por introdução de certas impurezas que
conferem ao cristal maior resistividade ã baixa temperatura.Quan
do tais impurezas são pentavalentes, o silicio ou o germânio são
denominados doadores de elétrons, tipo n(negativo). Por outro la
do, se as impurezas são trivalentes, são chamados receptores, ti
30 po p (positivo)
O comportamento dos semicondutores pode ser explica
do a partir das bandas de energia. No cristal, devido ã proximi
dade dos núcleos, os elétrons se misturam e os níveis de energia
onde estes se encontram, agrupam-se em bandas de energia denomi
nadas bandas permitidas. Estas são separadas por bandas chamadas
proibidas. Os elétrons das camadas mais externas são considera-
CCMÍSCÂÜ KACm-L LE LKH.GiA l\!UCLFAR/SP .

.14.
dos livres dentro da estrutura do cristal. A banda onde se loca
lizam estes elétrons é chamada banda de valencia. Os elétrons des
ta podem passar para a banda de condução, quando acelerados por
um campo elétrico, formando-se uma lacuna na banda de valencia que
é preenchida por elétrons vizinhos.
Quando uma partícula carregada, resultante da intera
ção da radiação eletromagnética com a matéria, passa através de
\im meio semicondutor, ela produz pares elétron-lacuna e, portanto,
cria uma capacidade de carga dentro do meio.
O número total de pares produzidos dentro do meio ê
E/e, onde E é a energia dissipada, e E é a energia média requeri
da para a produção do par elétron-lacuna.
Quando a partícula incidente produz pares elétron-la
cuna num melo semicondutor, este faz com que os portadores de car
ga se movam na direção dos eletrodos apropriados, induzindo carga
no circuito externo ao detector e, assim, pode-se detectar a pas
sagem da radiação incidente.
Existem três tipos principais de detector semicondu
tor: detector de união difusa, detector de barreira de superfície
30 è detector compensado com litio
Comparando-se o germânio e o silício em termos de sec
ção de choque para o efeito fotoelétrico, pode-se observar que o
91+0
germanio apresenta uma secçao de choque superior ao silicio ' . O
mesmo ocorre em relação à secção de choque para a produção de pa
res, enquanto para o efeito Compton, as secções de choque são a-
proximadamente iguais..0 germânio é mais eficiente que o silício
para espectrometría gama, embora apresente a dificuldade de opê-9 itO
rar a baixas temperaturas para evitar a precipitação do litio ' .

.15.
Os primeiros detectores de germânio compensados com
11
litio foram fabricados em 1962 e, desde entao, houve um pro
gresso rápido na aplicação e manufatura desses detectores, bem
como desenvolveu-se a eletrônica a eles associada.
Já na última década, houve vim grande desenvolvimento
dos detectores de germânio de alta pureza, que não necessitam de
baixas temperaturas e apresentam uma alta resolução no intervalo
das baixas energias. Em plantas de processamento de combustível
irradiado, esse tipo de detector é empregado principalmente para - . 1 5
a determinação da composição isotópica do plutonio '
II.2.2.2 - Aplicações
Desde os meados da década de sessenta, os detectores
de Ge(Li), devido ã sua excelente resolução, já se tornaram fer
ramentas poderosas em estudos de decaimentos nucleares e em aná-
« . 1 1 2 0 lise por ativação '
O uso de detectores em análise de produtos de fissão
permitiu o desenvolvimento de uma série de trabalhos visando a
determinação da queima do elemento combustível, por meio de rela
_ 7 l i t 2 2 2 9 36 çoes entre as atividades dos produtos de fissao. ' ' ' ' .
2 0
GORDON e colaboradores realizaram uma investigação
detalhada dos espectros gama de produtos de fissão obtidos com os
detectores de Ge(Li) e desenvolveram métodos para a determinação
dos alcances em alumínio e do rendimento de cerca de vinte des
ses radionuclídeos. 12
Por sua vez, CONTENSON e colaboradores verificaram
por espectrometria gama, utilizando detectores semicondutores, a
distribuição espacial dos produtos de fissão na estrutura do ele

.16.
mento combustível, durante e apôs a irradiação.
Da mesma forma, os detectores semicondutores ocupam
uma posição importante no controle analítico dos produtos de fis
são em soluções do processo de tratamento do combustível nuclear.
As razões são o alto conteúdo de informações do método, a nature
za não destrutiva das análises, a possibilidade de automatização
do método e a eliminação de procedimentos radioquímicos de sepa
ração, que são inconvenientes devido ãs altas atividades envolvi
das.
15
Segundo DENARD ,na instalação de Savannah River,EUA,
até 1966, a baixa resolução dos detectores cintiladores limitava
muito o uso da técnica de espectrometria gama. Atualmente, de
pois do advento dos detectores semicondutores, existem três sis
temas de espectrometria gama ligados a computadores, além de um
sistema portátil, que permitem a análise de rotina de 1200 amos
tras por mês, com uma precisão de + 10%. Ainda em Savannah River,
pode-se destacar o uso de um detector de Ge(Li) acoplado ao sis
tema de tratamento de "off-gases" da dissolução do combustível.
Esse detector mede a taxa de liberação do produto de fissão gaso
85 so Kr e, por meio desta, realiza-se o controle da dissolução do
10 elemento combustível
Na União Soviética, no V. G. Khlopin Institute, usa-
-se um sistema automático de espectrometria gama para o controle
dos produtos do processo de extração por solventes. Esse sistema 19
possui uma capacidade maxima de 150 analises por dia 8
Na Tchecoslováquia, BüLOVIC e colaboradores descre
veram um método de determinação de produtos de fissão em amostras
de combustível irradiado da Estação de Energia Atômica Al, basea
da em espectros gama obtidos com detectores semicondutores.
COMISCAC KAC;CK/l LZ llUmt^ NUCLEAR/SP - íiPES

.17.
1 7
Em Karlsruhe, Alemanha, ERTEL desenvolveu métodos por
espectrometria gama para o controle de produtos de fissão no tra
tamento do combustível nuclear irradiado usando detectores de es
tado sólido.
Na India, no Bhabha Atomic Research Centre, utiliza-
-se uma combinação de detectores cintiladores e detectores semi
condutores para as análises qualitativa e quantitativa dos produ
tos de fissão nas várias fases do processo de tratamento do coiji-2 8
bustivel nuclear
II.3 - Análise dos Espectros
A importância em se obter informações sobre radionu
clideos, levou vários pesquisadores a desenvolverem métodos para
o cálculo das taxas de desintegração a partir de espectros gama,
desde a época em que estes eram obtidos com detectores de cintila
ção.
1 3
Em 1959, COVELL apresentou um método simples de cál
culo da área do fotopico, que se baseava na soma das contagens nos
canais correspondentes ao fotopico e siibtração da área correspon
dente âs contagens de fundo. 2 5 ^ ,
LEE ,também em 1959, propôs o método de subtração de
espectros, no qual o espectro de um padrão de atividade conheci
da era subtraído do espectro referente ã amostra armazenado no a-
nalisador. Este método i similar ao "stripping" de espectros, que 6
foi usado por BONNEVIE-SVENDSEN para análise manual de espectros
gama de produtos de fissão, nas instalações de reprocessamento de
Kjeller, na Noruega. OLSON^^ utilizou xim método baseado ¡no mesmo
principio para análise dos produtos de fissão no Atomic Energy Ins
titute, de Idaho Falls.

. 18.
Porém, com o crescente uso dos detectores semicondu
tores e a necessidade de métodos mais rápidos para a interpreta
ção dos espectros, ampliou-se o uso de computadores para a avalia
ção dos dados, e, conseqüentemente, o desenvolvimento de ; progra
mas para equipamentos de grande e pequeno porte, bem como para caj.
35 37 culadoras programáveis '
If 2
Dessa forma, em 1968, YULE estudou os métodos de com
putação já existentes baseados no cálculo da área do fotopico.
Com esses estudos, verificou a possibilidade de utilização do mé-
todo de COVELL /até entao aplicado para espectros obtidos com de
tectores de cintilação, para os espectros obtidos com os detecto
res de Ge(Li). Concluiu que,para a obtenção de resultados precl-
sos, era necessária a combinação do método de COVELL a um método
derivativo para a localização dos fotopicos e de suas fronteiras. 21
GüNNINCK e NIDAY desenvolveram o programa denominado
"GAMANAL", para a realização de análises espectrométricas "in-li
ne", no Lawrence Livermore Laboratory, nos Estados Unidos. 39
Mais recentemente, SCHUBIGER e colaboradores desen
volveram o programa "JANE", para uso em grandes computadores. O
programa é composto de nove versões e executa funções de suaviza-
ção do espectro, determinação da posição dos picos, cálculos de
FWHM ("Full Width at Half Maximum") e da área dos picos, determi
nação das energias, bem como análises qualitativas e quantitati
vas dos radionuclideos presentes no espectro. 19 26 33 kl
Quanto aos programas para minicomputadores ' ' ' ,
19 ,
GOFMAN e colaboradores descreveram um programa, baseado no méto
do de soma de canais, que identifica e analisa quantitativamente
mais de dezesseis radionuclideos.
r/Minc!-,»:r • • « • r T » ' " r r r i.-rr r.;A wnr i FAR /SP . tPFIff

.19.
2
32 II.3.1 - Programas "GELIGAM"
Chama-se "GELIGAM" o conjunto de programas desenvolvi
do pela Ortec, para a análise de espectros gama obtidos com detec
tores de Ge(Li). Os programas "GELIGAM" operam sob o controle da
linguagem "ORACL", elaborada para o computador PDP-11/05, da Digi
tal. A linguagem "ORACL" é interpretativa e permite vmia interação
contínua entre o operador e o computador.
O conjunto "GELIGAM" é constituído por um sistema de
programas modulares autônomos. Esses programas permitem o cálculo
da resolução de um pico, preparação de bibliotecas, calibração do
sistema em energia e eficiência, verificação do conteúdo gravado
em um disco e outras funções, tais como o início da aquisição de
dados pelo analisador multicanal e gravação em disco do espectro
obtido. Os principais programas que realizam a análise qualitati
va e quantitativa dos radionuclideos emissores gama presentes em
uma amostra são o "GAMMAl", o "GAMMA2" e o "GAMMA3".
Estes três programas analisam um espectro a partir de
bibliotecas previamente elaboradas pelo operador. As bibliotecas
devem conter os radionuclideos de interesse com suas propriedades
BANASIK e colaboradores apresentaram um programa pa
ra um computador PDP-11/45, com xma estrutura em três níveis, que
além das análises espectrométricas alfa e gama, determina também
algumas propriedades fisico-quimicas dos nuclídeos.
No presente trabalho, utilizou-se o sistema de progra
32
mas "GELIGAM" ,elaborado pela Ortec Inc. Co., para a avaliação
qualitativa e quantitativa dos espectros obtidos com os detecto
res de Ge(Li), para o controle de produtos de fissão em soluções
de processo Pvirex.

. 20.
nucleares (meia-vida, energias e abundância). Outro requisito pa
ra a realização das análises é que o sistema esteja calibrado em
energia e eficiência. Faz-se essa calibração, utilizando-se pa
drões de atividades conhecidas.
O programa "GAMMAl" é o mais simples e analisa somen
te os fotopicos daqueles radionuclídeos contidos na biblioteca es
pecifiçada, sem utilizar uma rotina de pesquisa de picos.
O programa calcula a FWHM* (Full Width at Half Máxi
mum) de um determinado pico e a compara com a FWHM da calibração.
O pico i considerado válido apenas se a FWHM calculada estiver no
intervalo entre 0,8 e 1,2 vezes a FWHM da calibração. Se houver um
outro pico nas proximidades, o programa possui meios para analisa
-los separadamente, desde que os dois picos estejam registrados
na biblioteca.
Em seguida, calcula a energia do centroide (em KeV),
as contagens de fundo, as contagens de área do fotopico descontan
do as contagens de fundo (em contagens/segundo), a porcentagem de
incerteza nas contagens e a FWHM em KeV.
O centroide calculado deve estar dentro do intervalo
dos canais requerido pelo operador e deve concordar com a posição
do centroide estabelecida pela biblioteca, ou não é considerado ao
mo pertencente a um isótopo da biblioteca.
A incerteza em porcentagem calculada deve ser igual ou
menor que a sensitividade requerida pelo operador.
*FWtíM; HZÁoZução em eneAg-ca (em Kel/]

como:
.21.
Para os picos que são válidos, calcula-se a atividade
^ _ (Contagens do fotopico-contagens de fundo)xlOO
Eficiência x Intensidade em porcentagem (%/desint.)
A atividade (A) é calculada em microcuries.
Se existe um fotopico pertencente a um nuclldeo que,
apesar de se encontrar no intervalo de canais requerido, não ê vá
lido, este pico será usado apenas para o cálculo da atividade mí
nima detectável (MDA),
O programa "GAMMA2" difere pouco do programa "GAMMAl".
Este utiliza uma rotina de localização de picos, enquanto o "GPM/Sk2"
usa uma rotina de pesquisa de picos. Portanto, é possível o cálcu
lo de atividades diferentes para o mesmo isótopo analisado pelos
dois programas. O programa "GAMMA2" não calcula a atividade míni
ma detectável, mas apresenta a possibilidade de impressão dos da
dos intermediários.
O programa" "GAMMA3" difere dos programas anteriores
por utilizar duas bibliotecas. Coloca-se na primeira as energias
dos fotopicos livres de interferencia. Esses picos são analisados
e a atividade é registrada como na análise normal do "GAí»fl*íAl". U-
sa-se a segunda biblioteca para determinar a atividade dos isóto
pos que não possuem quaisquer linhas livres de interferência no
espectro, porém não se limita obrigatoriamente a estes.
A análise dos espectros utilizando-se os programas
"GAMMAl", "GAMMA2" e "GAMMAS" não destrói os dados armazenados no
analisador multicanal ou nos discos, possibilitando a repetição
das análises.

.22.
CAPITULO III
P A R T E E X P E R I M E N T A L
III.l - Equipamentos
- um espectrómetro de raios gama constituido de detec
tor Ge (Li) de 52,5 c m d e volume ativo, modelo 8001-0820
(resolução de 2,2 KeV para fotons de 1,33 MeV), pré
-amplificador modelo 120, amplificador modelo 450,
filtro de alta voltagem modelo 119, fonte de polari
zação 459, analisador multicanal de 4096 canais mo
delo 6240, da Ortec Incorporated Company, USA. A es
se sistema está acoplado uma unidade de processamen
to de dados PDP-11/05 com 24 K de memoria (Digital
Equipament Company, USA) com uma unidade de discos
flexivel ("floppy-disk") modelo 6200P da Ortec In
corporated Company, U.S.A., um teletipo (Teletype,
USA) e um registrador gráfico modelo 7004B-XY (Hew
lett Packard, USA). Na Figura 3 pode-se observar o
sistema utilizado.
- Balança analítica modelo H64, da Mettler, Suiça.
- Placa agitadora-aquecedora modelo PC-357,marca Cor
ning, Brasil.

.23.
WHÇ •S?,v;Br«sîsr.wst;it.i\ -
FIGURA 3 : SISTEMA USADO PARA ESPECTROMETRIA DE RAIOS GAMA
\ M U C l FAR/SP - IPEft

.24.
III.2 - Reagentes
- Soluções radioativas-padrão
- Rutênio-10 6, sob a forma de complexos de nitrosil
-rutênio, em meio HNO^ IM, com concentração radio
ativa original de 4,1 mCi/mL (01/02/76).
Procedência: Amersham International Limited.
- Cério-144, sob a forma de cloreto de cério-III, em
meio HCl IM, com concentração radioativa original
de 2,38 mCi/mL (12/06/79). Procedência: Amersham
International Limited.
- Manganês-54, sob a forma de cloreto de manganês,
em meio HCl 0,1M, com concentração radioativa ori
ginal de 0,09 2 mCi/mL (01/07/79). Procedência: A-
mersham International Limited.
- Americio-241, em meio HNO^ 3M, com atividade espe
cifica de 78,82 yCi/g em 06/04/81. Procedência: A
mersham International Limited.
- Cobalto-57, em meio HCl 0,2N com atividade espec¿
fica de 3,22 yCi/g em 22/07/80.
- Bário-133, com atividade especifica de 127,85 yCi/g
em 24/10/80. Procedência: New England Nuclear.
- Cobalto-60, com atividade especifica de 181,83 yCi/g
em 30/10/80. Procedência: Phillips Electronic Ins
truments.

.25.
Césio-137, em meio HNO^ 3M. Procedência: Amersham
International Limited.
As prooriedades nucleares desses radionuclideos 2 1 * 't 3
encontram-se na Tabela II .
- Urânio natural nuclearmente puro, sob a forma de
U^Og. Procedência: Instalação-piloto de purificação
de urânio do Centro de Engenharia Química do Insti
tuto de Pesquisas Energéticas e Nucleares.
235
- Urânio com 19,91% de enriquecimento em U, sob a
forma de U - O q . Procedência: United Nuclear Corpora-
tion, Chemicals División, Missouri, USA.
- Outros reagentes: grau analítico.
III.3 - Amostras para análise
III.3.1 - Pontos de Retirada de Amostras para Controle de Processo
e Estabelecimento da Geometria de Contagem
O presente trabalho é dirigido ao controle dos produ
tos de fissão por espectrometria gama das amostras provenientes do
processo de tratamento de materiais irradiados.
Nessa instalação, denominada CELESTE (Células para
Estudos e Testes em Extração), utiliza-se o processo Purex para a
recuperação e purificação dos âctinídeos.
As amostras para o controle de processo são coletadas
na fase de dissolução e durante todo o processo de separação e pu
rificação por extração com TBP/dodecano. Nesta fase, origem. da
maior parte das amostras, faz-se o controle das soluções aquosas e
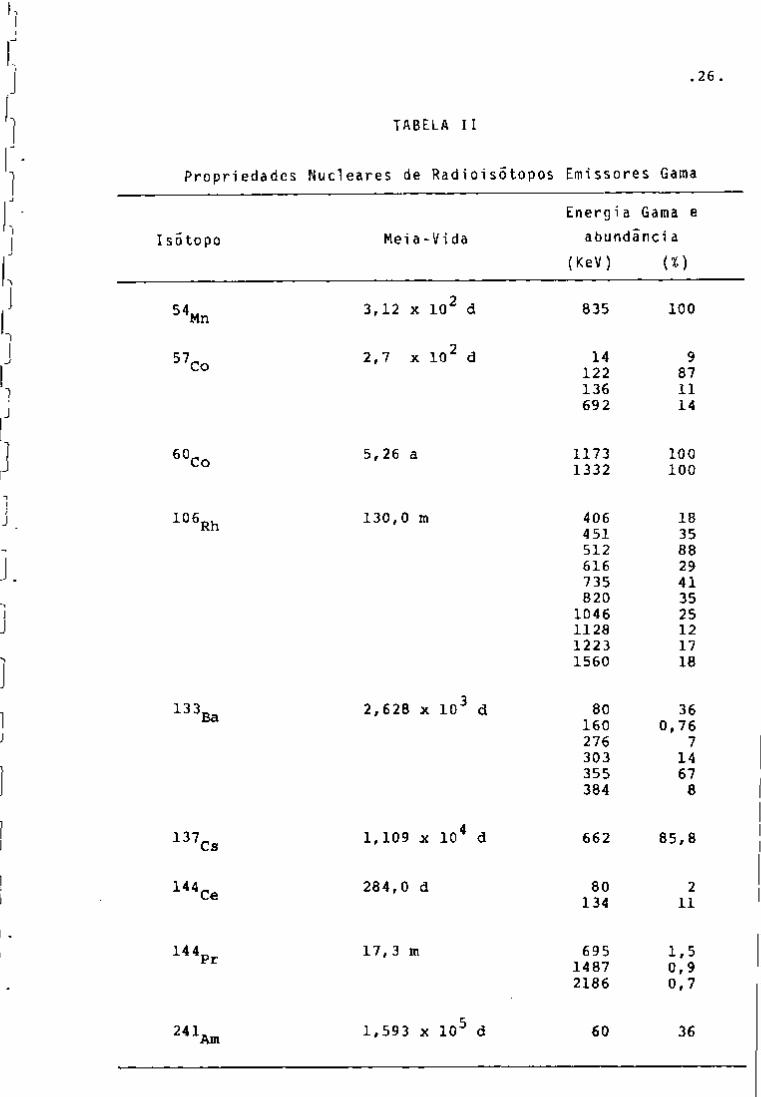
.26
(KeV) (%)
3,12 X 10^ d 835 100
2,7 X 10^ d 14 122 136 692
9 87 11 14
5,26 a 1173 1332
100 100
130,0 m 406 451 512 616 735 820
1046 1128 1223 1560
18 35 88 29 41 35 25 12 17 18
2,628 X 10^ d 80 160 276 303 355 384
36 0,76
7 14 67 8
1,109 X 10^ d 662 85,8
284,0 d 80 134
2 11
17,3 m 695 1487 2186
1,5 0,9 0,7
1,593 X 10^ d 60 36
TABELA II
Propriedades Nucleares de Radioisótopos Emissores Gama
Energia Gama e Isótopo Meia-Vida abundancia

.27.
orgânicas de alimentação e dos resíduos a fim de se conhecer os
fatores de descontaminação, em relação aos produtos de fissão, ao
longo do processo de separação.
Levando-se em conta que as atividades envolvidas na
instalação são da ordem de 10 Ci/L e, considerando-se a grande va
riedade de amostras com origens diferentes durante as diversas fa
ses do tratamento do material irradiado, definiu-se, para o início
dos trabalhos experimentais, a geometria de contagem para a análi
se. Estabeleceu-se, dessa forma, o tipo de frasco bem como o volu
me de amostra, a fim de manter sempre a mesma geometria.
III.3.1.1 - Frasco de Amostra
O tipo e a forma do frasco de amostra foram escolhi
dos levando-se em consideração que as amostras, devido ã sua ativi^
dade, devem ser preparadas em células analíticas com proteção bio
lógica e operações por controle remoto. Nessas condições, escolheu
-se um frasco que permite operações telemanipuláveis de abertura e
fechamento, bem como que facilite as operações de pipetagem.
Utilizou-se um frasco de vidro com capacidade de 5 mL,
de fundo chato, boca larga, com tampa rosqueada, compatível com o
sistema de preparação de amostras (pipetagem, diluição, separação)
por controle remoto em fase de instalação na CELESTE e, com geome
tria favorável ao sistema de detecção gama.

.28.
III.3.1.2 - Volume de Amostra
Considerando-se uma atividade 6 - Y de 10 Ci/L e saben
do-se que os limites mínimo e máximo de detecção e análise do sis
tema utilizado são, respectivamente, lo"" yCi e 10 uCi, estabele -
ceu-se um volume fixo de 1 mL, a fim de se manter a mesma geometri
a para todas as amostras.
Na escolha deste volume, considerou-se uma diluição
prévia de 1:50 v/v para as amostras mais ativas, provenientes da
solução nítrica do combustível, da solução de alimentação do pri
meiro ciclo de extração e do rafinado de alta atividade. Em segui
da, estas amostras, bem como aquelas originárias dos vários ciclos
de descontaminação podem sofrer diluições variáveis, dependendo da
atividade de cada uma, de tal forma a se obter sempre o volume fi
nal de 1 mL, com uma atividade máxima de lOyCi.
Por outro lado, prevé-se uma atividade da ordem de
10 "^yCi/mL para as correntes finais do processo. Dessa forma, o vo
lume de 1 mL ainda satisfará os limites estabelecidos.
Outro aspecto levado em conta, na escolha do volume
de 1 mL para análise, foi a minimização do volume de resíduos lí
quidos.
III.3.2 - Preparação das Soluções-Padrão
As fontes-padrão utilizadas foram preparadas no Labo
ratório de Metrologia Nuclear da Área de Física Nuclear do Centro
de Operação e Utilização do Reator de Pesquisa do IPEN. O método
consiste em pesar, em uma ampola, uma massa de aproximadamente 3,5g
de uma solução contendo o radionuclídeo de interesse e determinar
a atividade específica da solução por meio de contagem gama em

.29.
uma câmara de ionização tipo poço^^As fontes-padrão foram prepara
das colocando-se 1 mL dessas soluções, separadamente, em frascos
padronizados para controle gama.
As atividades dos padrões utilizados podem ser obser-2i4,ít3
vadas na Tabela III. Procurou-se obter padrões com atividades pró
ximas de 5 yCi, que está no intervalo de atividade estabelecido pa
ra o trabalho. Além disso, escolheu-se como padrões, radionuclí
deos que apresentassem raios gama característicos no intervalo de
100 a 1500 KeV, pois, é nesse intervalo que se encontram as energi
as dos produtos de fissão de interesse para o controle.
III.3.3 - Preparação das Amostras de Urânio Irradiado
Irradiaram-se quatro amostras de U^Og, sendo duas de
235
uranio natural e duas de uranio enriquecido (19,91% em ü ) . Es
sas irradiações foram realizadas com a finalidade de simular as
composições das soluções do processo Purex.
- Amostra 1
Irradiou-se 0,10006 g de U^Og (em pó) durante 8 horas
13 2
no reator lEA-Rl, sob um fluxo neutrônico de 10 n/cm s. A embala
gem interna utilizada foi de papel de al\imínio e a embalagem exter
na de polietileno. Após 15 horas de resfriamento, fez-se a dissolu
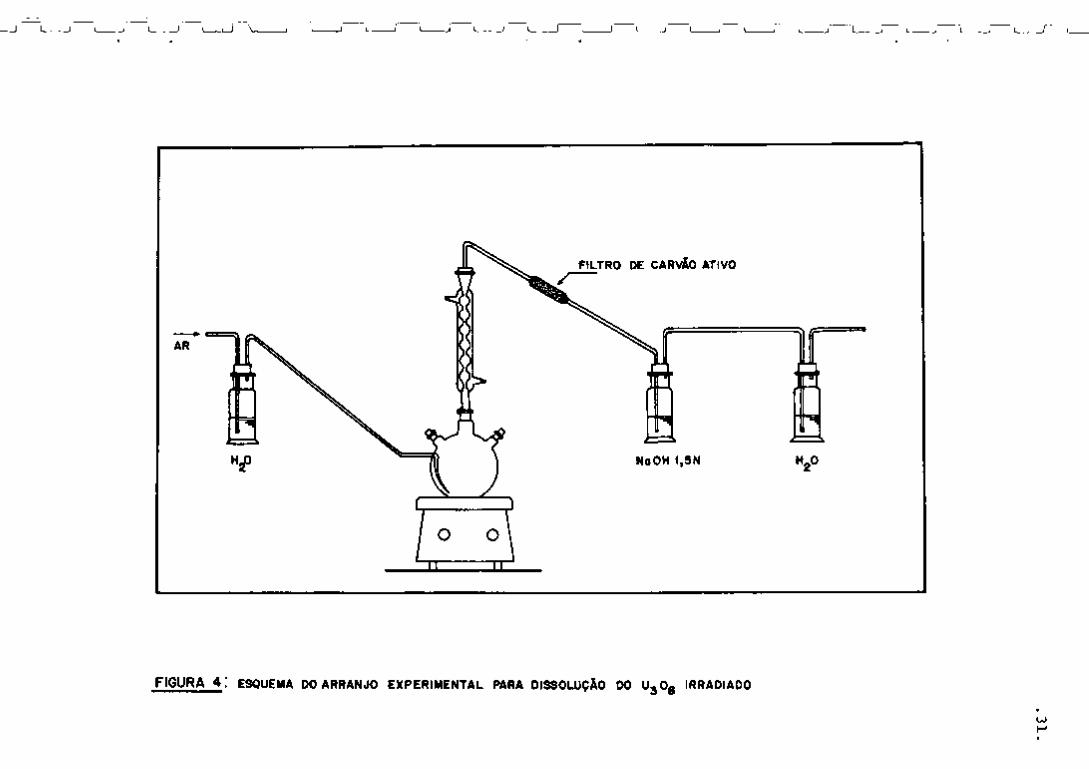
ção do U^Og irradiado com 12 mL de H N O 3 empregando-se o arran
jo experimental esquematizado na Figura 4. A solução resultante foi
diluída a 25 mL num balão volumétrico. Para a análise, colocou- se
uma alíquota de 100 vL da solução final no frasco padronizado para
controle gama e adicionou-se 900 uL de água destilada de modo a
completar um volume final de 1 mL.

i r { l' l _ J l i' l í L _ _ J L í i i k j 1 ; t I k r j í i i i i r
T A B E L A I I I
Atividades dos Padrões Utilizados para Calibração
em Energia e Eficiência
Radionuclídeos Energia Gama
(KeV)
Meia Vida Atividade
(PCi)
60 1,593x10^ d 2,00
" c o 122 2,7x10^ d 2,86
136
" 3 B a 80 2,628x10"^ d 5,20
160
220
276
30 3
355
384
662 1,109x10^ d
5^Mn * 835 3,12x10^ d
1173 5,2 a 1,64
1332
•Utilizado apenas para calibração em energia o
J L J \ I I i J i_ J \ i J I f k J I I i 1 I \ 1 -J 'l i I I L
AR
i 1
FILTRO DE CARVÃO ATIVO
NoOH 1,5N «2°
FIGURA 4 : ESQUEMA DO ARRANJO E X P E R I M E N T A L PARA DISSOLUÇÃO DO U j O g IRRADIADO
00

.32.
- Amostra 2
A segunda amostra (0,10000 g de U^Og em pó) foi irradiada nas mes
mas condições da Amostra 1, aumentando-se o tempo de irradiação pa
ra 43 horas. Utilizou-se como embalagem interna,papel de alumínio,
e como embalagem externa uma cápsula de alumínio. Após 2 dias de
resfriamento, efetuou-se a dissolução do material irradiado com
HNO^ 6M e a solução resultante foi diluída a 50 mL num balão volu
métrico. Retirou-se uma alíquota de 100 yL de solução final e adi
cionou-se 900 yL de água destilada de modo a completar um voliime
de 1 mL de solução.
- Amostra 3
Irradiou-se 1,27 mg de U^Og (com 19,91% de enriquecimen
235
to em u) durante 3 horas sob um fluxo de neutrons térmicos de
13 2 10 n/cm s. As embalagens utilizadas foram as mesmas da Amostra 2.
Após 18 horas de resfriamento, dissolveu-se o ü^Og ir
radiado com HNO^ 6M, juntamente com o papel de alumínio para evi
tar perdas de massa durante a transferência do material para o rea
tor de dissolução. Este procedimento pode ser adotado porque a ati
vidade do aluminio irradiado é desprezível comparada ^ atividade
dos produtos de irradiação do urânio. Completada a dissolução,
transferiu-se a solução para um balão volumétrico de 25 mL, comple
tando-se o volume com água destilada. Em seguida, retiraram-se ali
quotas de 100 yL e adicionaram-se 900 yL de água destilada, de tal
forma a se obter o volvmie final de 1 mL.

.33.
- Amostra 4
Esta amostra com uma massa de 1,28 mg de U^Og em pó
235
(com 19,91% de enriquecimento em U) foi irradiada durante 5 ho
ras nas mesmas condições da Amostra 3. Apôs 24 horas de resfriamen
to realizou-se a dissolução com HNO^ 6M; as amostras para contagem
foram obtidas de modo análogo ã Amostra 3.
III.4 - Análises Espectrométricas
3h II1.4.1 - Tempo de Contagem
Se n é o número total de contagens num intervalo de
tempo t, a taxa de contagem r será:
do como:
Este valor com seu desvio padrão pode ser estabeleci
nl/2 ^ 1 / 2 r ± - r = ^ ± ^ = r i -f-
Escrevendo-se em termos de erro percentual;
r -f i^O % = r i 100
(tr ,V2 „1/2
Da última equação, conclui-se que o erro percentual é
determinado pelo número total de contagens acumulado.

.34.
Levando-se em conta os aspectos descritos e a baixa e
ficiência do detector Ge(Li), escolheram-se temóos de contagem que
variaram entre 100 e 4000 segundos, de modo que o número de conta
gens totais acumuladas nunca fosse inferior a 100000 impulsos.
III.4.2 - Análises Qualitativas
A primeira fase do trabalho consistiu da análise qua
litativa de amostras de urânio natural irrar^iado (ver item III. 3.3)
com a finalidade de verificar,se os espectros obtidos naquelas con
diçõs de irradiação poderiam ser utilizados para simular os espec
tros correspondentes ãs soluções do processo Purex.
Os nuclídeos de interesse para o controle de processo
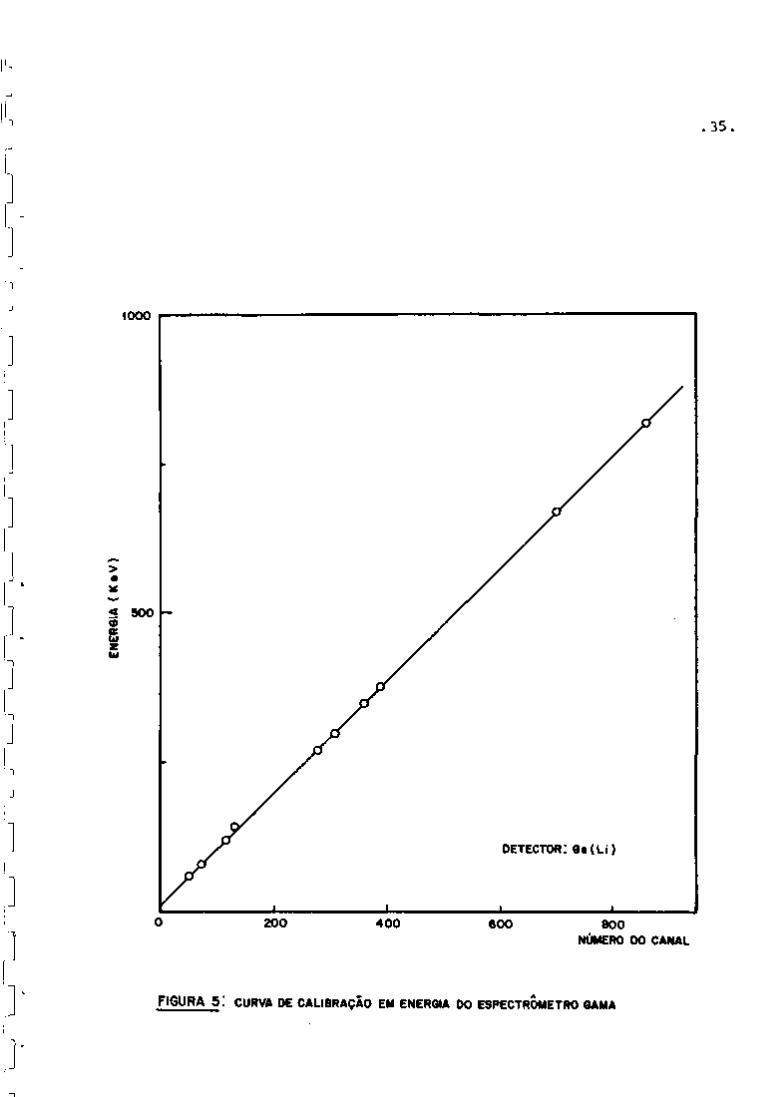
foram identificados mediante uma curva de calibração (Figura 5) ob
tida utilizando-se as fontes-padrão mencionadas no item III.3.2.
_ 32 III.4.3 - Analises Quantitativas
Realizaram-se as análises quantitativas usando-se o
sistema de programas "GELIGAM", descrito no item II.3.1.
III.4.3.1 - Calibração do Sistema Detector
O sistema "GELIGAM" apresenta o programa "CALIBR",
que opera, especialmente, para calibrações em energia e eficiência.
Fez-se a calibração a partir de um espectro de radionu
clídeos-padrão, de atividades conhecidas (item III.3.2). Nos en
saios preliminares, efetuou-se a calibração do sistema detector,
registrando-se separadamente o espectro de cada fonte-padrão. Com
COMISCAC r:Ãc;cN/L CE EM;;RGIA. N U C L E A R / S P - ÍPFI^
.35
1000
] 200 400 600 800
NÚMERO DO CANAL
FIGURA 5 : CURVA DE CALIBRAÇÃO EM ENERGIA DO ESPECTRÓMETRO GAMA

.36.
esse procedimento, não se obtiverem resultados satisfatórios, prin
cipalmente em relação ã calibração em eficiência.
Procurou-se, então, realizar a contagem simultaneados
padrões, colocando-se todos os frascos das fontes-padrão ao mesmo
tempo sobre o detector. Esse foi o procedimento adotado para a ca
libração do sistema para análise de produtos de fissão em soluções
de urânio irradiado.
Obtido o espectro dos padrões, o programa "CALIBR" re
aliza as calibrações em energia e eíiciência. Para a calibração em
energia, introduz-se o número do canal correspondente ao fotopico e
sua energia (em KeV).
Pode-se calibrar o sistema em eficiência de duas ma
neiras. Na primeira, utiliza-se uma biblioteca que contenha os ra-
dionuclídeos-padrão presentes no espectro e introduz-se o nome do
isótopo e sua atividade em microcuries. Nesse caso, o programa cor
rige as atividades para a data de preparação dos padrões (ativida
de original). Na segunda, não se utiliza uma biblioteca e faz-se a
relação das energias dos fotopicos com as correspondentes desinte
grações gama por segundo, no momento do registro do espectro. Nes
te trabalho, adotou-se o segundo procedimento.
A listagem do programa de calibração encontra-se no
Apêndice I. Observa-se, em seguida, os resultados da calibração
obtida com o uso desse programa, para a realização das análises dós
produtos de fissão.
a) Calibração em Energia
Energia (KeV) = 2,725 + 0,536 x (Número do canal) +
0,248x10"^ X (Número do canal)^

.37.
b) Calibração em Eficiencia
- Abaixo de 150 KeV:
log(Eficiência) = -6,9 7 + 0,531 x log(Energia em KeV)
- Acima de 150 KeV:
log(Eficiência) = 1,30 - 1,168 x log(Energia em KeV)
III.4.3.2 - Elaboração da biblioteca para identificação de radionu
elídeos
Uma vez calibrado o sistema em relação a energia e
eficiência, a fase seguinte é a elaboração de uma lista de radionu
elídeos de interesse que permita a identificação de nuclídeos em
uma amostra desconhecida.
Oomo o objetivo deste trabalho é a análise de produtos
de fissão emissores gama em soluções de urânio irradiado , elabo
rou-se uma lista contendo os principais nuclídeos emissores gama
presentes nas soluções de um combustível irradiado, segundo os da-
2 0 , 3 8
dos da literatura .Para o presente trabalho, incluiu-se tãrobem o
239 Np, proveniente da reação:
238„ , . 239„ g" 239„
uma vez que as amostras analisadas tiveram um tempo de desativação
curto, conforme descrito no item III.3.3.
A introdução dessa lista de nuclídeos com suas propri 24,1*3
edades nucleares ê feita mediante o programa denominado "USERLI",
do sistema "GELIGAM".
COMÍ SSAC U-QU'Ui le. BvERGiA N U C L E A R / S F - IPEK

.38.
Apresenta-se, no Apêndice II, vama biblioteca elabora
da para o desenvolvimento desse trabalho.
III.4.3.3 - Análise de Produtos de Fissão em Soluções de Urânio
Irradiado
Com o sistema calibrado em energia e eficiência e oom
a elaboração da biblioteca de radionuclídeos, o sistema GELIGAM es
tá apto a realizar as análises.
As amostras a serem analisadas (volvune de 1 mL) , colo
cadas em frascos padronizados, foram levadas ao detector de Ge(Li)
e contadas por um tempo que variou de 100 a 4000 segundos. A aqui
sição dos espectros pode ser iniciada por meio de programa "AGQUIRd'
do sistema "GELIGAM".
Realizaram-se as medidas diariamente durante o primei
ro mês após a irradiação, tuna vez por semana nos três meses seguin
tes e uma vez por mês daí por diante.
Os espectros obtidos foram analisados utilizando-se os
programas "GAMMAl", "GAMMA2" e "GAMMA3".
III.5 - Dados Experimentais
Os resultados apresentados referem-se aos dados obti
dos na análise qualitativa de amostras de urânio irradiado e aos
estudos realizados com os programas "GELIGAM", para a identifica
ção e determinação ce atividades de radionuclídeos para o controle
do processo de tratamento de urânio irradiado em fase de implanta
ção no CEQ/IPEN.

.39.
III.5.1 - Análises Qualitativas
Os primeiros ensaios para a análise qualitativa dos
produtos de fissão de interesse para o controle do processo foram
realizadas com as soluções resultantes da dissolução do urânio na
tural irradiado no reator lEA-Rl durante oito horas e com resfria
mento de quinze horas.
Como essas amostras apresentaram atividades baixas di
ficultando as análises, passou-se a trabalhar irradiando-se o urâ
nio por um período mais longo (40 horas).
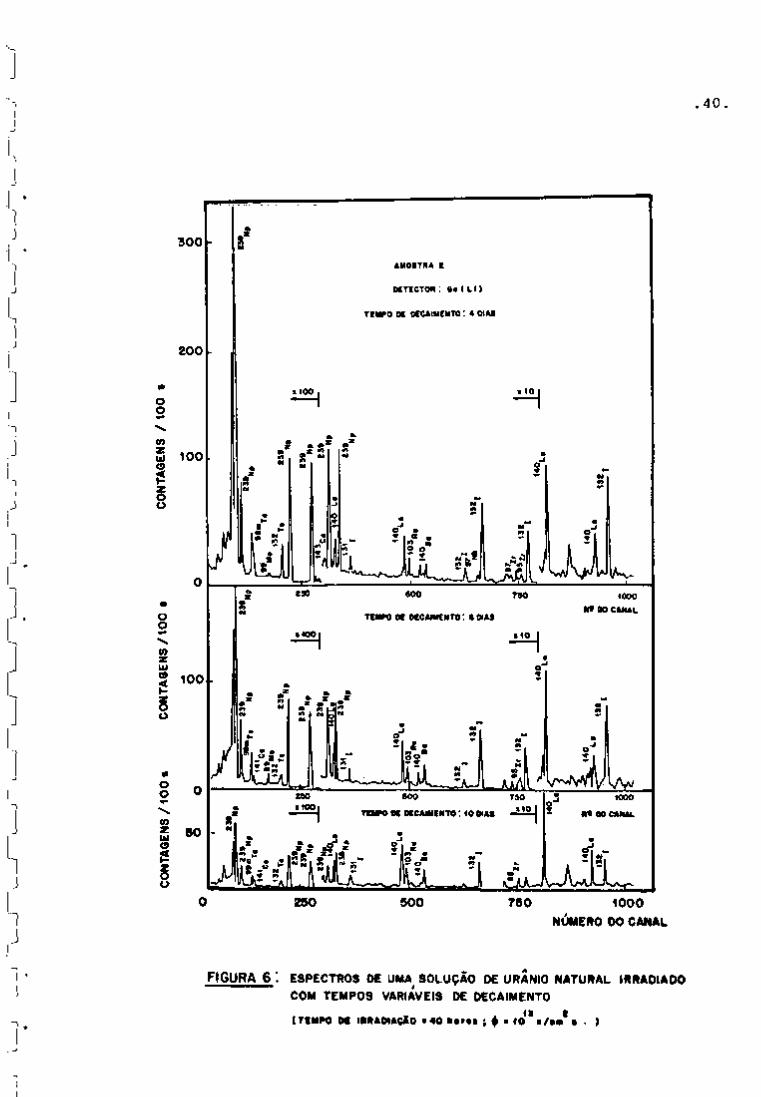
Registraram-se os espectros e, mediante a curva de ca
libração da Figura 5, obtiveram-se as energias correspondentes aos
fotopicos. Em seguida, identificaram-se os nuclídeos presentes com
a ajuda de uma tabela de radioisótopos ' e também pela determina
ção de meias-vidas.
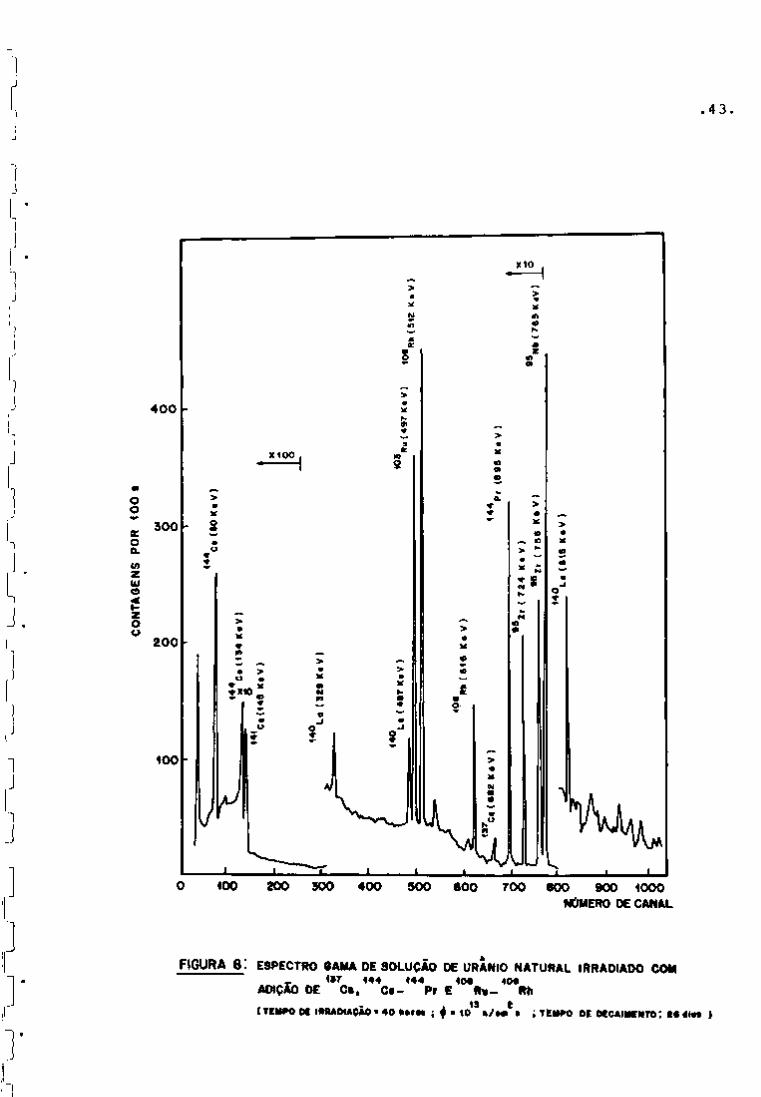
As Figuras 6 e 7 apresentam os espectros obtidos nes
sa fase de trabalho. Como pode se observar na Figura 6, nos primei
ros dez dias de resfriamento, há um predomínio dos fotopicos refe-
239
rentes ao Np (meia-vida de 2,34 dias) que decai quase completa
mente após vim período de aproximadamente 25 dias.
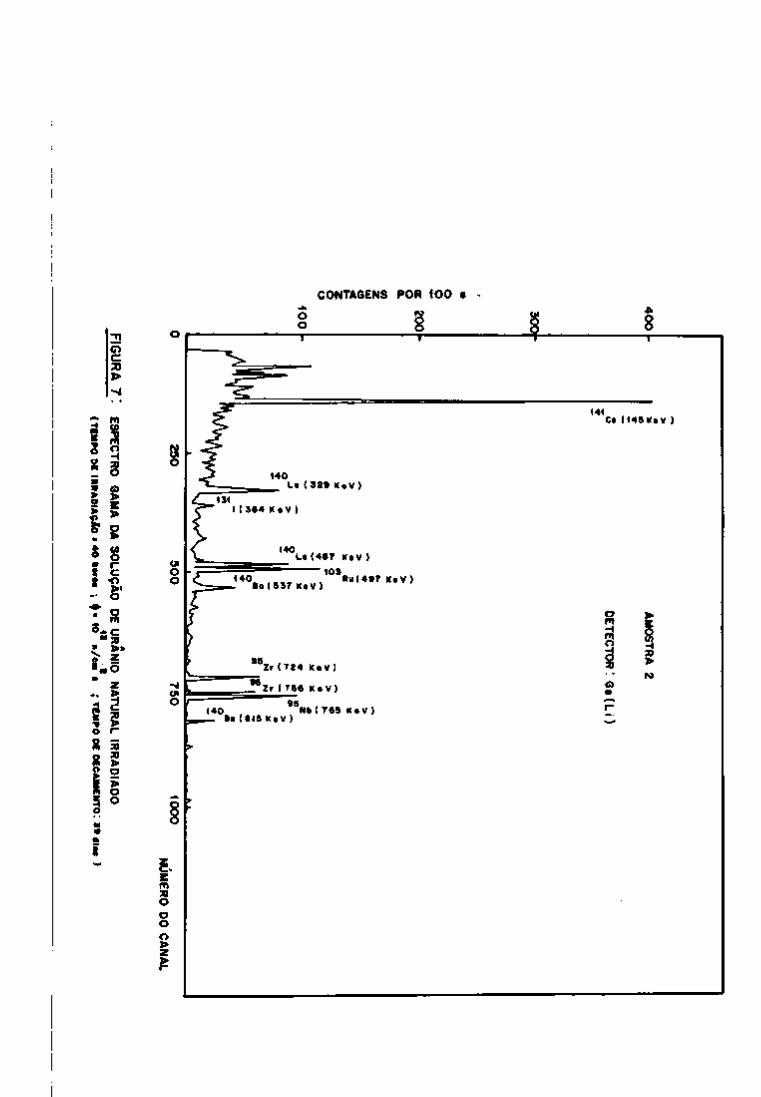
95 O Zr pode ser detectado por meio de seus fotopicos
de 724 e 756 KeV, como se vê na Figura 7. O ^^Nb, descendente do
95
Zr por decaimento g , tem seu fotopico também presente no espec
tro (765 KeV) .
Quanto ao rutênio, o isótopo detectado foi o " ' Ru
(497 KeV), sendo que não apareceram os fotopicos do ''" Rh, descen
dente do "^^^Ru.
O cério pode ser analisado por meio da linha de
145 KeV do • •''Ce, que se faz notar no espectro após aproximadamen-
.40
300
200
o o
m z U l
o O
100
§
( O z U i (9 100
o o
co z U l
s o
o
50
« M O S T R A 2
O C T E C T O I t : « • ( L l )
T E M P O DE D E C A I M E N T O : 4 DIAS
• 100 , l i o
u i
290
14001
a O O T 9 0
DE D C C A I M C N T O : 6 0 IA3
• 10
«000
Mt 0 0 C A N A L
S J
2S0 • rao
soo K d e c a i m e n t o : « o d i a s
790
_»«0 I 1000
n * 0 0 c a n a l
290 500 750 1000
NÚMERO DO CANAL
FIGURA 6 : ESPECTROS DE UMA SOLUÇÃO DE URANIO NATURAL IRRADIADO COM TEMPOS VARIÁVEIS DE DECAIMENTO
( T C H P O O C I R R â D I A Ç X O « 4 0 k e r o s ; f • 1 0 * ' . . )
o c >
l i
l i > I
i 8 I i - ê-
j r •«•o
5 c
S! »
il
a
UL O
m o o o o >
CONTAGENS POR 100 t >
O O
T -8
141
L a ( 9 2 9 K a V )
( 9 6 4 K • V )
140 L o ( 4 8 7 K * V )
103
5 9 7 K « V ) R u ( 4 « 7 K * V )
9B Zr ( 7 2 4 K a V )
M Z r ( 7 5 6 K « V )
» 5 . 140 ' N b ( 7 6 9 K . V )
Bo ( 8 4 5 K a V )
C * ( 1 4 S K t V )
1
IO

.42.
te dez dias de resfriamento.
O •'•" Cs, apesar de seu alto rendimento, não foi detec
tado na amostra analisada.
Além dos radioisótopos de interesse já citados, foram
também detectados os seguintes nuclídeos: o par ^^^Ba-'^^^La (537 e
329; 487 e 815 KeV); o par " - Te- I (230; 668 e 773 KeV) e o
•'•••'•I (364 KeV).
Como alguns radionuclídeos de interesse, tais como o
•••• Cs, -^^^Ce- -^^^Pr e •'•° Ru--'-° Rh, não foram obtidos nesta irradia
ção, adicionaram-se alíquotas de soluções-padrão desses nuclídeos
â amostra irradiada, para que seus fotopicos pudessem ser visuali
zados. O espectro resultante encontra-se na Figura 8.
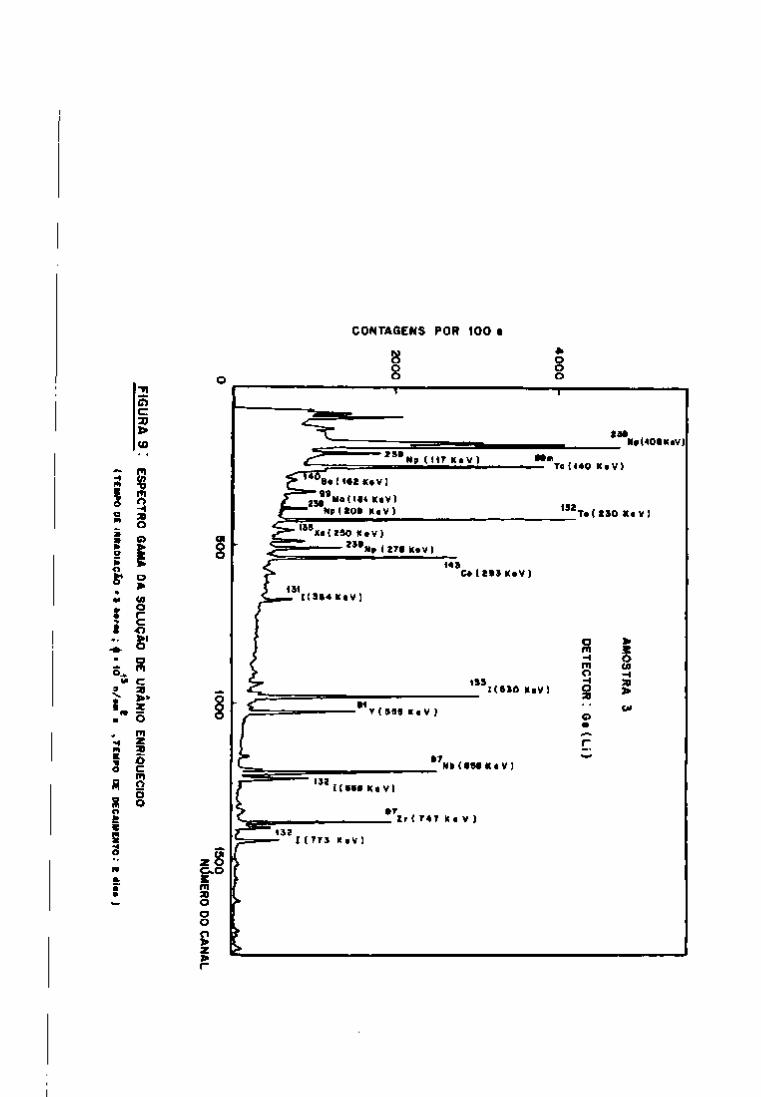
III.5.2 - Análises Quantitativas
As análises quantitativas dos principais produtos de
fissão de interesse para o controle das soluções do processo Purex
foram realizadas irradiando-se pequenas amostras de urânio enri
quecido (19,91% em ^ ^ ^ U ) .
Preparam-se as amostras para análise, conforme o pro
cedimento já descrito, e registraram-se os espectros para a iden
tificação dos radionuclídeos. Nas Figuras 9 e 10, observam-se os
espectros gama, com 2 dias e 101 dias de resfriamento, respectiva
mente.
A avaliação quantitativa foi realizada, utilizando-se
os programas "GAMMAl", "GAMMA2" e "GAMMA3", a fim de verificar o
comportamento de cada um dos programas em relação ao cálculo de a
tividade dos produtos de fissão.
.43,
4 0 0 -
O o K
O a. (O z UJ e 4 O U
300
200 -
100 •
100 200 300 400 500 600 700 800 900 1000 NÚMERO OE CANAL
FIGURA 8. ESPECTRO GAMA OE SOLUÇÃO DE URANIO NATURAL IRRADIADO COM 157 444 444 406 406
ADIÇÃO DE 08, C e - Pr E R u - Rh (TCMPO K IRRADIAÇÃO • 40 hera* ; ^ • 1o" » / o » ' • ; TEWPO DE DECAIMENTO: B6 610« )
CONTAGENS POR 100 S
3 G> C
> <0
1 S s > ? £ w (O
f I
w C
!• §> ^~ 5 • o
i i
o c
5
CONTAGENS POR 1000 •
Ci C >
i ^
? 3
1 IS ? s
«a s S, i .
O
S i S g 2 m m o S i » z 3
y : r
o 8
144 C * ( S O K * V )
C« (134 K « V )
L a ( 487 K * V )
Z a-
i l
103 Ru (497 K a V )
99 Z r ( 7 2 4 K a V )
99 Z r ( 7 5 e K a V ) 99
0> O O
141 C a ( 1 4 9 K a V )
I ra
- I > (M
N b { 7 8 9 K a V )

.46.
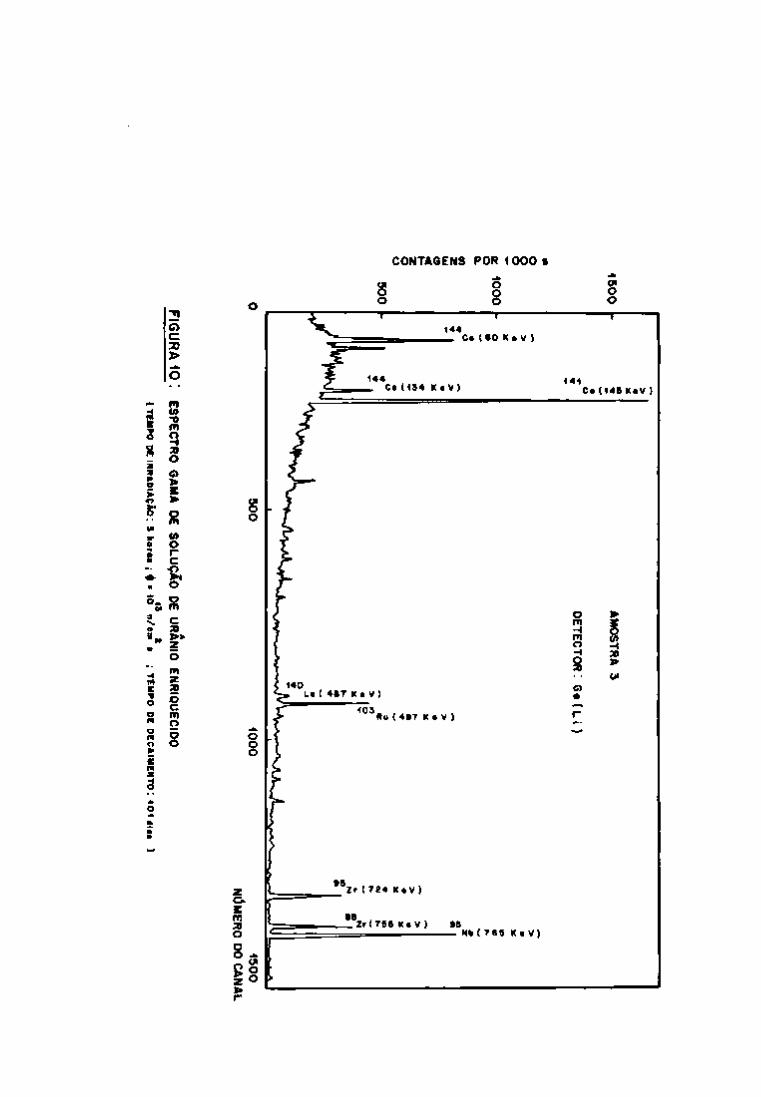
As listagens dos programas enoontram-se no Apêndice III.
Os resultados apresentados referem-se ãs análises de uma amostra
de 100 yL da solução resultante da dissolução do urânio irradiado
e diluída a 1 mL com água destilada. Essa alíquota é proveniente
da Amostra 3, que foi irradiada durante 3 horas, como um tempo de
resfriamento de 115 dias e o tempo de contagem de 2000 segundos.
Examinando-se os dados, verifica-se que após 115 dias
de resfriamento os radionuclideos identificados pelos programas são
aqueles previstos teoricamente, com exceção do e do Ce.
O ^ "Vc (energia gama de 140 KeV e meia-vida 0,25 dias)
141 foi identificado por influencia do fotopico do Ce (145 KeV)e o
• ' • Ce (energias gama de 293 e 725 KeV, meia-vida de 1,38 dias) de
95 vido a presença do fotopico de 724 KeV do Zr.
A amostra foi analisada durante aproximadamente 4 me
ses, registrando-se as atividades individuais dos produtos de fis
são, utilizando-se os programas "GAMMAl", "GAMMA2" e "GAMMA3".
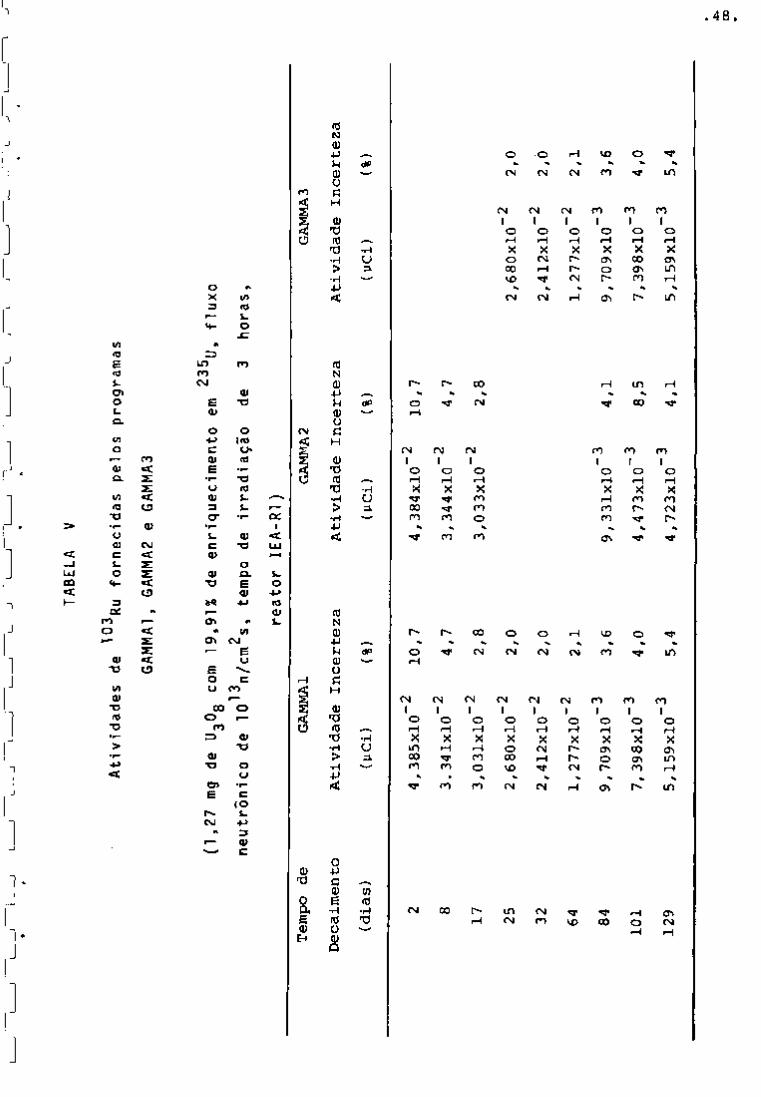
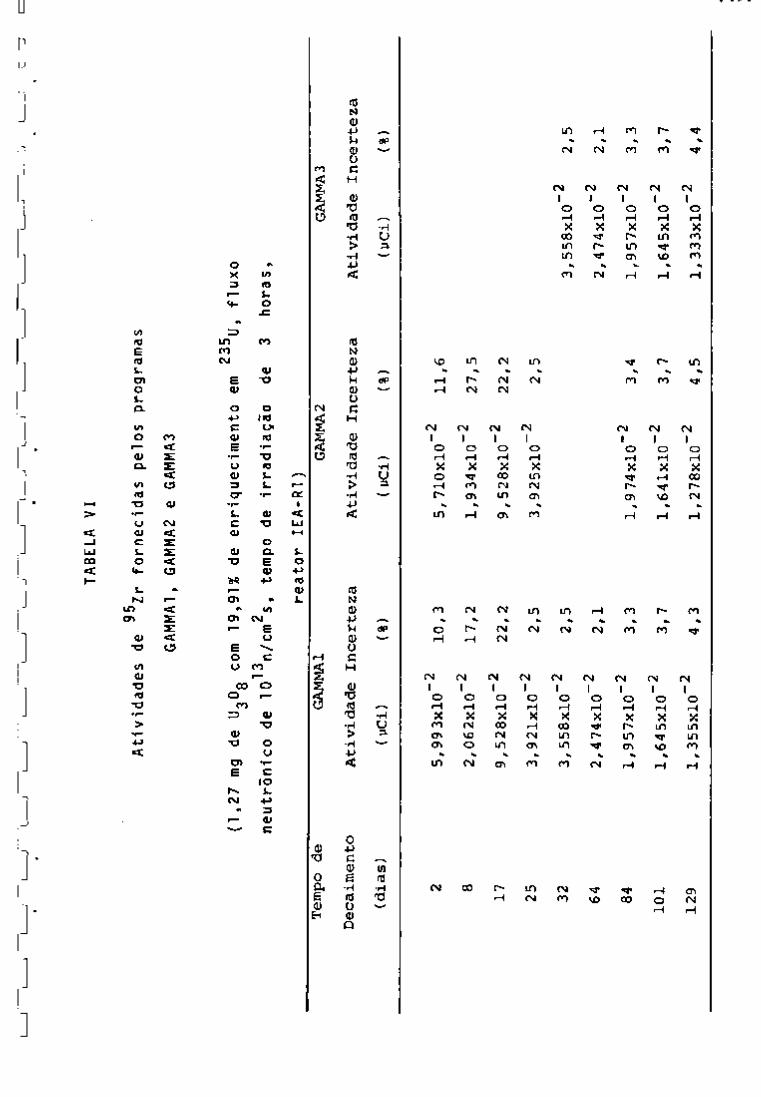
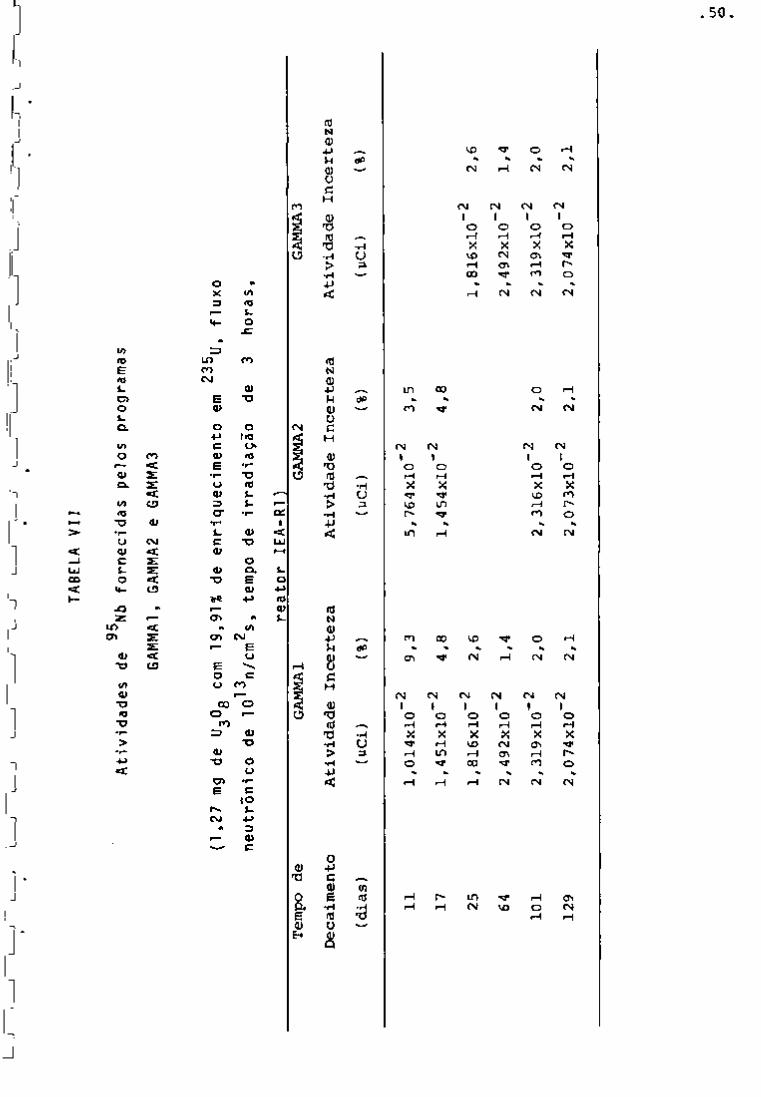
Nas Tabelas IV, V, VI e VII encontram-se as ativida-
95
des fornecidas pelos três programas para os radionuclideos Zr,
^^Nb, ^^"^Ce e ^^^Ru com vários tempos de desativação. Nessas tabe
Ias observa-se que as atividades fornecidas pelos três programas
"GAMMAl", "GAMMA2" e "GAMMAS" apresentam valores aproximadamente
iguais. Verifica-se ainda que as porcentagens de incerteza mais al
tas encontram-se nos primeiros dias de resfriamento. Isso se ex
plica porque, nesse período, a presença de produtos de fissão de 239
meia-vida curta, bem como do Np, faz com que os fotopicos so
fram interferências entre si.
Por outro lado, as atividades calculadas pelos progra
mas obedecem â lei exponencial de decaimento, como pode ser obser
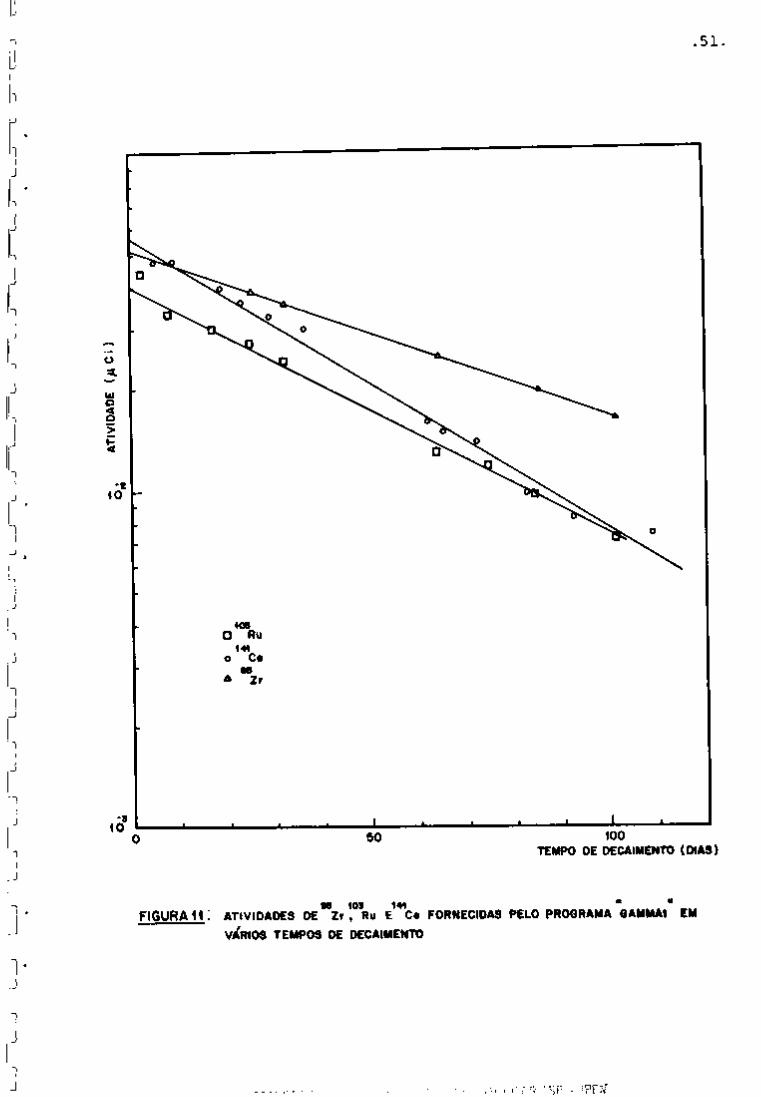
vado na Figxira 11.
J
L
I I
I J i_
J 1 I i r
J
L
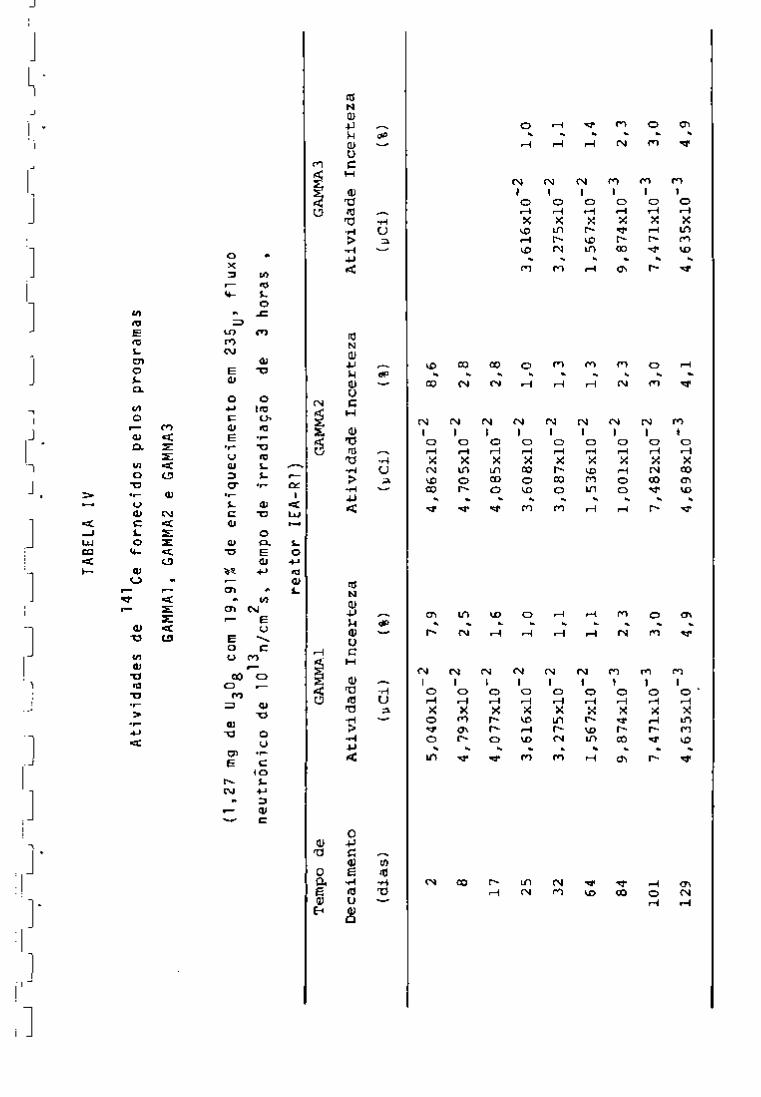
TABELA IV
141
Atividades de
Ce fornecidos pelos programas
GAMMAl, GAMMA2 e GAMMAS
(1,27 mg de UgOg com 19,91% de
13
2
neutrônico de 10
n/cm s, tempo enri quecimento
de irradiação
em 235^j
de
3
, fluxo
horas ,
reator lEA-Rl )
Tempo de
GAMMAl
GAMMA2
GAM-MA3
Decaimento
Atividade : Incerteza
Atividade Incerteza
Atividade Incerteza
(dias)
(yCi)
(%)
(yCi)
(%)
(yCi)
(%)
2
5,040x10"^
7,9
4,862x10"^
8,6
8
4,79 3x10"^
2,5
4,70 5x10"^
2,8
17
4,077x10"^
1,6
4,085x10"^
2,8
25
3,616x10"^
1,0
3,608x10"^
1,0
3,616x10"^
1,0
32
3,275x10"^
1,1
3,087x10"^
1,3
3,275x10"^
1,1
64
1,56 7x10"^
1,1
1,536x10"^
1,3
1,567x10"^
1,4
84
9,874x10'-^
2,3
1,001x10"^
2,3
9,874x10"-^
2,3
101
7,471xl0~3
3,0
7,482x10"^
3,0
7,471xl0~^
3,0
129
4,635x10"-^
4,9
4,698x10"^
4,1
4,635x10"^
4,9
1
i J i J
J
1
I í-
I i
"1
I L
1
I
_j
I I
I
r
TABELA V
Atividades de
fornecidas pelos programas
GAMMAl,
GAM
MA2 e GAMMAS
(1 ,
27 mg de
U
gOg com
1
9,9
1%
de enriquecimento em
23
5u
. fluxo
13
neutrônico de
10
2
n/cm s, tempo de irradiação
de
3
horas,
reator
lEA-Rl)
Tempo de
GAMMAl
GAMMA2
GAMMAS
Decaimento
Atividade Incerteza
Atividade Incerteza
Atividade Incerteza
(dias)
(uCi)
(%)
(wci)
(%)
(uci)
(%)
2
4,385x10"^
10,7
4,384x10"^
10,7
8
3.341x10"^
4,7
3,344x10"^
4,7
17
3,031x10"^
2,8
3,033x10"^
2,8
25
2,680x10"^
2,0
2,680x10"^
2,0
32
2,412x10"^
2,0
2,412x10"^
2,0
64
1,277x10"^
2,1
1,277x10"^
2,1
84
9,709x10"-^
3,6
9,331x10"^
4,1
9,709x10"^
3,6
101
7,398x10"^
4,0
4,473x10"^
8,5
7,398x10""^
4,0
129
5,159x10"^
5,4
4,723x10"^
4,1
5,159x10"^
5,4
00
I I
L
J L.
J I
T
TABELA VI
Atividades de
'hr
fornecidas pelos programas
GAMMAl,
GA
MM
A2 e
G
AM
MA
3
(1,2
7 mg de U
^Og com
19,
91% de enriquecimento em
23
5u , fluxo
neutrônico de
10
^^
2 n/cm s. tempo de irradiação
de
3 horas.
reator
lEA-Rl)
Tempo de
GAMMAl
GAMMA
2 GAMÍ4A3
Decaimento
Atividade Incerteza
Atividade Incerteza
Atividade Incerteza
(dias)
(yCi)
(%)
( yCi)
(%)
(uCi)
(%)
2 5
,99
3x
10
"^
1
0,3
5
,71
0x
10
"^
1
1,6
8 2
,06
2x
10
"^
1
7,2
1
,93
4x
10
"^
2
7,5
17
9,5
28
x1
0"
^
22
,2
9,5
28
x1
0"
^
22
,2
25
3,9
21
x1
0"
^
2,5
3
,92
5x
10
"^
2
,5
32
3,5
58
x1
0"
^
2,5
3
,55
8x
10
"^
2
,5
64
2,4
74
x1
0"
^
2,1
2
,47
4x
10
"^
2
,1
84
1,9
57
x1
0"
^
3,3
1
,97
4x
10
"^
3
,4
1,9
57
x1
0"
^
3,3
10
1 1
,64
5x
10
"^
3
,7
1,6
41
x1
0"
^
3,7
1
,64
5x
10
"^
3
,7
12
9 1
,35
5x
10
"^
4
,3
1,2
78
x1
0"
^
4,5
1
,33
3x
10
"^
4
,4
J
\ I
L
~\
1
I i
\ r
I r
-I L
J
I_
TABELA VII
95.
Atividades de
Nb fornecidas pelos programas
GAMMAl , GAMMA2 e GAMMAS
2S5.
(1,27 mg de U^Og com 19,9U de enriquecimento em
U, fluxo
tempo de
im
reator lEA-RU
13
2
neutrônico de 10
n/cm s, tempo de irradiação
de
S
horas.
Tempo de
Decaimento
GAMMAl
Atividade Incerteza
GAMMA2
Atividade Incerteza
GAMMAS
Atividade Incerteza
(dias)
(uCi)
(%)
(uci)
(%)
(yCi)
(%)
11
1,014x10"^
9,3
5,764x10"^
3,5
17
1,451x10"^
4,8
1,454x10'^
4,8
25
1,816x10"^
2,6
1,816x10"^
2,6
64
2,492x10"^
1,4
2,492x10"^
1,4
101
2,319x10"^
2,0
2,316x10"^
2,0
2,319x10"^
2,0
129
2,074x10"^
2,1
2,073x10"^
2,1
2,074x10"^
2,1
OI
O
.51,
TEMPO OE DECAIMENTO (DIAS)
00 109 141 " " FIGURA H : ATIVIDADES DE Zr , Ru E Ce FORNECIDAS PELO PROGRAMA GAMMAl EM
y/dmos T E M P O S DE DECAIMENTO
•st'" -

.52.
CAPÍTULO IV
DISCUSSÃO E CONCLUSÕES
O tratamento químico do combustível nuclear irradiado
para a recuperação dos elementos férteis e físseis processa-se, ñor
malmente, após um periodo de desativação, isto é, um periodo que
permita o decaimento dos produtos de fissão de meia-vida curta,di
minuindo-se, dessa forma, a atividade do material.
Portanto, permanecem os produtos de fissão de meias-
-vidas mais longas e, durante o processamento químico, apenas al
guns deles causam problemas, na fase de extração com TBP/diluente.
Dentre esses nuclídeos, destacam-se o Zr/Nb, o Ru/Rh e, em menor
proporção, o Ce/Pr, que não apresentam distribuição desprezível no
solvente usado.
Desde a dissolução do combustível, seguida de ciclos
de extração para a separação e purificação de urânio e plutónio,é
exigido um controle rigoroso desses produtos de fissão para a ve
rificação dos fatores de descontaminação ao longo de todo o proces
so.
Por outro lado, para o controle do processo, é impor
tante que se apliquem métodos analíticos de respostas rápidas. Sob
esse aspecto, o uso de um processador acoplado diretamente ao equi
pamento de medida e operado pelo analista é de grande \itilidade, pois
permite a obtenção imediata dos resultados das análises.
O número elevado de análises normalmente solicitadas
para o controle dos produtos de fissão em uma instalação de repro

.53.
cessamento e o tempo necessário para o cálculo da atividade de ca
da produto de fissão, requer o uso de um computador para a avalla
ção dos dados e diminuir, dessa forma, o tempo de resposta.
Com essa finalidade, desenvolveram-se estudos visando
o uso e adaptação do programa "GELIGAM" para o controle dos produ
tos de fissão do tratamento de urânio irradiado da instalação CE
LESTE, em fase de implantação no CEQ/IPEN.
A linguagem ORACL, empregada pelo sistema "GELIGAM" ,
por ser interpretativa e interativa, requer um tempo mais longo de
processamento, mas apresenta a possibilidade de correções e adap
tações dos dados de entrada, ã medida que o programa vai se desen
volvendo.
Os estudos foram realizados irradiando-se pequenas quan
» 2 35 tidades de uranio enriquecido a 19,91% em U, seguindo-se, apos
um curto período de resfriamento, ã dissolução com HNO^. A solução
resultante foi aquela utilizada para análise quantitativa, usando
-se os programas "GELIGAM".
Antes, porém, o sistema foi devidamente calibrado em
energia e eficiência, bem como foi definida a geometria de conta
gem.
A seleção do frasco de amostra foi feita baseando-se,
principalmente, numa geometria que facilitasse as operações poroon
trole remoto, visto que, devido â atividade das amostras, estas só
podem ser manipuladas ã distância, em células com proteção bioló
gica. Por outro lado, procurou-se trabalhar com uma geometria fa
vorável ao sistema de preparações de amostra, em fase de instala
ção na célula analítica, bem como ãs condições de processo e do sis
tema de detecção usado.

.54.
Levando-se em conta essas restrições elegeu-se, final
mente, um frasco de vidro de 5 ml, tampa rosqueada, contendo uma
borracha de silicone para vedação. O volume da amostra para análi
se de 1 mL foi limitado, considerando-se as atividades das soluções
de processo.
Quanto aos programas usados para a realização das a-
nálises, "GAMMAl", "GAMMA2" e "GAMMA3", verifica-se, pelos dados
das Tabelas IV, V, VI e VII, que estes apresentam resultados pró
ximos nos cálculos de atividades dos produtos de fissão emissores
gama. No entanto, o programa "GAMMAl" requer um tempo menor de pro
cessamento em relação ao "GAMMA2" e ao "GAMMAS". Este último, pos
sui a vantagem de analisar corretamente os picos muito próximos.
Entretanto, no controle de produtos de fissão em soluções proveni
entes do tratamento químico do urânio irradiado, os produtos de fis
são de interesse não apresentam outros picos interferentes.
Portanto, para as análises de rotina, o programa do sis
tema "GELIGAM" mais adequado é o "GAMMAl". Com os dados de ativi
dade obtidos por meio deste, procurou-se seguir o decaimento dos
principais nuclideos para o controle (Figura 11). Observou-se que
a diminuição de atividade segue a lei exponencial de decaimento.
Diante das dificuldades encontradas na seleção e na ãis
ponibilidade de nuclideos-padrão, na determinação de suas ativida
des em uma geometria compatível com a das soluções de análise, de
senvolveu-se esse estudo com um número limitado de padrões para a
calibração.
Dos dados obtidos pelos três programas, verificou- se
que o procedimento seguido para a calibração do sistema para a de
terminação das atividades introduziu alguns erros de geometria,bem
como um aumento do tempo morto. Esses fatores afetam a precisão da

.55.
calibração e, conseqüentemente, a determinação das atividades dos
nuclídeos.
Os resultados obtidos podem ser melhorados diante da
possibilidade da obtenção de uma única solução-padrão contendo nu
clideos-padrão com atividades conhecidas. Esse seria o procedimen
to mais correto para a calibração.
Verificou-se ainda que o tempo de processamento é me
nor, quanto menor for o número de radionuclídeos introduzidos nas
bibliotecas. Portanto, nas amostras retiradas das fases iniciais
de purificação pelo processo Purex, é interessante a utilização de
uma biblioteca completa, isto é, contendo todos os produtos de fis
são que podem estar presentes no combustível irradiado. Após este
conhecimento dos radionuclídeos presentes, é conveniente que se u
tilize uma biblioteca reduzida, que contenha apenas os produtos de
fissão de interesse para o controle de processo, de modo a diminu
ir o tempo de processamento.

.56,
APÊNDICE I
Programa para calibração em energia e eficiência
+ P U N C A L I B R
G E L I - G A M V 6 ( ^ 2 - J U M - 8 1 1 3 : 4 0 : 4 7
* * * G E L I - G A Í ' 1 S Y S T E M C A L I B P A T I O M ( V E P S I O N 7 )
DO Y O U W A N T T O ( 1 ) - P E R F O P M N C A L I B P A T I O N
( 2 ) - P E C A L L A P P E V I O U S C A L I B p A T I O M F I L E
( 3 ) - S T O P E C U P P E M T C A I . I B P A T I O M I N F O R M A T I O N
( 4 ) - P P I N T C U P R E N T C A L I E P A T I O t J I N F O R M A T I O N
O R ( 5 ) - E X I T T H I S R O U T I N E
? : 2
E N T E R D I S K U N I T A M D C A L I B P A T I O N F I L E M A M E : 0 ^ C A L 0 Í ? 5
C A J ^ I B R A T I O N I N F O R M A T I O N F O P C A L C ^ 0 5
C A L I B R A T I O N M A D E 1 3 - A P R - 3 1 1 5 : 0 0 : 4 3
> C A J ^ I B R A C A O 2 M D P O S I T I O N
> T E S T E S 1 3 - A P R - 3 1
>
>
9 E F F I C I E N C Y C A L I B P A T I O N P O I N T S S T O R E D
D O Y O U W A N T T O ( 1 ) - P E R F O R M N E W C A J ^ I B P A T I O M
( 2 ) - R E C A L L A P R E V I O U S C A L I B P A T I O M F I L E
( 3 ) - S T O R E C U R p e x l T C A L I B P A T I O N I N F O R M A T I O N
( 4 ) - P R I N T C U p P E M T C A L I B R A T I O N I N F O R M A T I O N
O R ( 5 ) - E X I T T H I S R O U T I N E
? : 4
C A L I B R A T I O N F I L E N A M E I S C A L 0 0 5 G E L I G A M S Y S T E M D I S K
E N E R G Y R A M G E A P P R O X . 0 T O 2 0 0 0 K E V
E N E R G Y . V S . C H A N N E L N I M B E R C A L I B p A T I O M
" E N E R G Y ( K E V ) = A + B * C H A N N E L + C * C H A M M E L T 2
A = 2 . 7 2 4 9 1 3 7 4
B = 0 . 5 3 6 4 1 9 4 3
C = 0 . 2 4 3 1 5 2 4 6 E - 0 7
P E A K S H A P E . V S . C H A M M E L M U ^ I B E P C A L I B P A T I O M
F W H M ( C H A M N E L S ) = A + B * C H A M M E L + C * C ! i A N M E L t 2
A = 0 . 3 3 7 1 4 4 5 4 E + 0 1
B = - 0 . 6 0 9 7 9 3 3 7 E - 0 5

.57.
E F F I C I E N C Y . V S . E M E P G Y C A L I B P A T I O M
D E T E C T O R ' K N E E ' E N E R G Y = 15!?. P K E V
B E L O W D E T E C T O R ' K N E E ' . . . . .
L O G C E F F ) = A + B * L O G ( K E V )
A = - 0 . 6 9 6 9 1 3 5 6 E + 0 1 B = 0. 5 3 0 3 6 6 7 2 E + 0 0
A B O V E D E T E C T O R ' K N E E '
L O G ( E F F ) = A + B * L O G ( K E V )
A = 0.1 3 0 4 5 6 4 9 E + 0 1 B = - 0 . 1 163 47 6 4 E + 0 1
N U M B E R O F E N E R G Y ^ E F F I CI EN CY P O I N T S S T O R E D =
I N D E X N O .
1 2 3 4 5 6 7 8 9
E N E R G Y ( K E V )
3 0 . 0 1 2 2 . 0 1 3 6 . 0 27 6. 0 3 0 3 . 0 3 5 5 . 0 33 4. 0
1 17 3. 0 1 3 3 3 . 0
E F F I CI E M C Y F A C T O R
0. 0 0 9 0 2 5 8 2 0 . 0 1 3 3 0 3 0 3 0. 0 1 5 6 9 5 7 3 0. 0 0 4 7 0 4 3 3 0. 0 0 5 4 5 8 6 1 0. 0 0 3 2 6 6 3 4
,0. 0 0 3 8 3 6 1 0 0. 0 0 1 0 4 3 4 5 0. 0 0 0 7 5 2 7 3
DO Y O U W A N T T O
O R
( 1 ) - P E R F O P M N E J C A L I B P A T I O M ( 2 ) - P E C A L L A P R E V I O U S C A L I B R A T I O N F I L E ( 3 ) - S T O R E C U R R E N T C A L I B R A T I O N I M F O R M A T I O N ( 4 ) - P R I N T C U R R E N T C A L I B R A T I O N I N F O R M A T I O N ( 5 ) - E X I T T H I S R O U T I N E
E N D O F C A L I B R A T I O N

.58,
APÊNDICE II
Listagem da biblioteca utilizada para as análises
* P U f J U S E R L I
O P T I O N ? L I S T
E M T E R D I S K U N I T A N D N A M E O F L I B P A P Y : Pl^ o F G A M 2
T H E , L I B R A R Y P F G A M 2 W A S C R E A T E D ON 2 S - J U L - S 1
A N D L A S T M O D I F I E D ON 2'3-JLn.-8 1.
T H E R E A R E 2 6 I S O T O P E S L I S T E D I N T H E L I B P A P Y WIT:!
6 G A M M A E N E R G I E S P E R I S O T O P E
M P - 2 39
1 0 6 . 0 0
H A L F L I F E
( 2 3 . 0 0 )
2 . 3¿i D A Y S
2 2 3 . 0 0 ( 12. 0 0 ) 2 7 8 . 0 0 ( 1 4 . 0 0 )
M O - 9 9
1 8 1 . 0 0
H A L F L I F E
( 7 . 0 0 0 )
2 . 7 8 D A Y S
7 4 0 . 0 0 ( 1 2. 0 0 ) 7 8 0. 0 0 ( 4. 0 0 0 )
T C - 9 9 M
1 4 0 . 0 0
H A L F L I F E
( 9 0 . 0 0 )
0 . 2 5 D A Y S
T E - 1 3 2
2 3 0 . 0 0
H A L F L I F E
( 9 0 . 0 0 )
3 . 2 4 D A Y S
I- 1 3 2
7 7 3 . 0 0
H A L F L I F E
( 8 9 . 0 0 )
0. 09 DAY S
9 5 5 . 0 0 ( 2 2 . 0 0 )
X E - 1 3 5
2 5 0 . 0 0
H A L F L I F E
( 9 1. 0 0 )
0. 38 DAY S
B A - 1 4 0
3 0 5 . 0 0
H A L F L I F E
( 6 . 0 0 0 )
1 2 . 8 0 D A Y S
4 3 8 . 0 0 ( 5. 0 0 0 ) 5 3 7 . 0 0 ( 3 4 . 0 0 )
L A - 1 4 0
3 2 9 . 0 0
1 59 6. 0 0
H A L F L I F E
( 2 0 . 0 0 )
( 9 6 . 0 0 )
1 . 6 8 DAY S
4 3 7 . 0 0 ( 4 0 . 0 0 ) 8 1 5. 0 0 ( 1 9 . 0 0 )
1 - 1 3 1
3 6 4 . 0 0
H A L F L I F E
C 3 2 . 0 0 )
3 . 0 5 D A Y S
R U - 1 0 3
4 9 7 . 0 0
H A L F L I F E
( 3 8 . 0 0 )
39 . 5 0 DAY S
Z R - 9 5
7 2 4 . 0 0
H A L F L I F E
( 4 9 . 0 0 )
6 5 . 5 0 D A Y S
7 5 6 . 0 0 ( 49 . 0 0 )
N B - 9 5
7 6 5 . 0 0
H A L F L I F E
C 1 0 0 . 0 )
3 5 . 0 0 D A Y S
C E - 1 4 1
1 4 5 . 0 0
H A L F L I F E
( 4 8 . 0 0 )
3 2 . 5 0 D A Y S
R H - 1 0 6
4 5 1 . 0 0
7 3 5 . 0 0
H A L F L I F E
( 3 5 . 0 0 )
( 4 1 . 0 0 )
0. 0 9 DAY S
5 1 2 . 0 0 ( 3 3 . 0 0 )
8 2 0 . 0 0 ( 3 5 . 0 0 )
6 1 6 . 0 0 ( 2 9 . 0 0 )
1 0 4 6 . 0 0 ( 2 5 . 0 0 )

.59.
CE- 1 4 4 H A L F LT F E = 1 3 4 . 0 0 ( 1 1. 0 0 )
PR- 1 4 4 HAI.F LI F E = 6 9 5 . 0 0 ( 1 . 5 0 0 )
CS- 1 37 H A L F LI F E = 6 6 2 . 0 0 ( 8 5 . 0 0 )
28 4. 0 0 D A Y S
0 . 0 1 DAY S 1 4 8 7 . 0 0 ( 0. 29 0 )
3 0 . 0 0 Y E A ^ S
K R - 8 5M 1 5 0 . 0 0
N B - 9 7 6 5 8 . 0 0
I- 1 3 3 5 3 0 . 0 0
BA- 1 39 1 6 6 . 0 0
CE- 1 4 3 29 3. 0 0
N D- 1 4 7 9 1. 0 0
7;R-97 7 4 3 . 5 0
Y - 9 IM 5 5 1 . 0 0
X E - 1 3 3 8 1 . 0 0
H A L F L I F E ( 7 4 . 0 0 )
H A L F L I F E ( 9 9 . 0 0 )
H A L F L I F E ( 9 0 . 0 0 )
H A L F L I F E ( 2 3 . 0 0 )
H A L F L I F E ( 4 6 . 0 0 )
H A L F L I F E ( 2 8 . 0 0 )
H A L F L I F E { 9 2 . 0 0 )
H A L F L I F E ( 9 5 . 0 0 )
H A L F L I F E ( 3 6 . 6 0 )
0 . 1 8 DAY S
0 . 0 5 DAY S
0.8 5 D A Y S
0 . 0 6 D A Y S
1 . 38 DAY S 7 2 5 . 0 0 C 8 . 0 0 0 )
1 1 . 0 6 D A Y S 3 1 9 . 0 0 ( 3. 0 0 0 )
17 . 0 0 H O U R ?
5 0 . 3 0 M I N U T E S
5 . 6 5 D A Y S
5 3 3 . 0 0 ( 1 3 . 0 0 )

.60
APÊNDICE III
Programas GAMMAl, GAMMA2 e GAMMA3
R U N G A M M A l
G E L I - G A M V 6 S P - D E C - S l 0 9 : 5 8 : 1 6
*^<c4c*>lc G A M M A I ( V 3 1 ) * * * * *
W H E R E I S D A T A ( M C A O P D I S K ) ? : M C A
I S I N P U T : N E i í ( N ) , S A M E ( S ) ^ O P T O B E M O D I F I E D ( M ) ? S
S T A R T C H A N N E L
S T O P C H A N N E L
S E N S I T I V I T Y
U N I T S
F A C T O R
D E C A Y C O R R E C T ?
1 5 0
3 0 0 0
1 0
M I C R O C U P I E S
1 . 0 0 0 0
N
L I B R A R Y U N I T N U M B E R A N D F I L E N A M E ? : 0 > p F G A M 2
S A M P L E D E S C R I P T I O N . . .
Q 0 0 0 1 9 2 7 - A U G - 8 1 1 4 : 1 1 : 4 3
A M O S T R A D E U R A N I O E N R I Q U E C I D O I R R A D I A D A D I A 0 4 - M A I 0 - 8 1
E S P E C T R O O B T I D O D I A 2 7 - A G 0 S T 0 - 8 1
D E T E C T O R S Y S T E M D E S C R I P T I O N . . .
C A L I B P A T I O N F I L E N A M E I S C A L 0 0 5 G E L I G A M
C A L I E R A C A O 2 N D P O S I T I O N
T E S T E S 1 3 - A P R - 8 1
— S P E C T P U M L I V E C O U N T T I M E = 2 0 0 0 S E C O N D S !
* * * * * S U M M A R Y O F N U C L I D E S I N S A M P L E * * * * *
T I M E O F C O U N T P E R C E N T
N U C L I D E A C T I V I T Y U N C E R T A I N T Y
( M I C R O C U P I E S ) ( 1 S I G M A )
N P - 2 3 9 < 3 . 7 * E - 3
• M O - 9 9 < 1 . 2 * E - 2
T C - 9 9 M 2 . 9 4 6 * E - 3 3 . 9
T E - 1 3 2 < 1 . 0 * E - 3 « I - 1 3 2 < 1 . 7 * E - 3
X E - 1 3 5 < 1 . 0 * E - 3
B A - 1 4 0 < 1 . 7 * E - 2
L A - 1 4 0 N O T P R E S E N T
I - 1 3 1 < 1 . 5 * E - 3
R U - 1 0 3 6. 1 3 8 * E - 3 4 . 8
Z R - 9 5 1 . 508* E - 2 4 . 2

.61,
N E - 9 5 2. 1 3 5 * E - 2 2. 1
C E - M l 5 . 5 7 7 + E - 3 3 . 3
P H - 1 0 6 < 4 . 1 * E - 3
C E - 1 4 4 < 6 . 3 * E - 3
p p - 1 Z i 4 < 5 . 0 * E - 1
C S - 1 3 7 < 1 . 7 * E - 3
K P - 8 5 M < 1 . 0 * E - 3
N B - 9 7 < 1 . 5 * E - 3
1 - 1 3 3 < 1 . 9 * E - 3
B A - 1 3 9 < 3 . 6 * E - 3
C E - 1 4 3 2 . 2 3 8 * E - 3 1 6 . 9
N D - 1 4 7 < 3 . 7 * E - 2
Z n - 9 7 < 1 . 7 * E - 3
Y - 9 I M < 2 . 0 * E - 3
X E - 1 3 3 < 0 . 0 * E 0
* * * U N U S E D P E A K S ( K E V ) * * *
+ 7 5 6 . 0 0 + 7 2 5 . 0 0
A N A L Y S I S F I N I S H E D A T 1 0 : 0 9 : 0 2
* R U N G A M M A 2
G E L I - G A M V 6 3 0 - D E C - 8 1 1 0 : 1 0 : 3 7
* * * * * G A M M A I I ( V 1 6 ) * * * * *
W H E P E I S D A T A ( M C A O R D I S K ) ? : M C A
I S I N P U T : N F W ( N ) , S A M E ( S ) ^ O R T O B E M O D I F I E D ( M ) ? S
S T A R T C H A N N E L
S T O P C H A N N E L
S E N S I T I V I T Y
U N I T S
F A C T O R
D E C A Y C O R R E C T ?
1 5 0
3 0 0 0
1 0
M I C R O C U R I E S
1 . 0 0 0 0
N
L I B R A R Y U N I T N U M B E R A N D F I L E N A M E ? : 0 , P F G A M 2
S A M P L E D E S C R I P T I O N . . .
Q 0 0 0 1 9 2 7 - A U G - 8 1 1 4 : 1 1 : 4 3
A M O S T R A D E U R A N I O E N R I Q U E C I D O I R R A D I A D A D I A 0 4 - M A I 0 - 8 1
E S P E C T R O O B T I D O D I A 2 7 - A G 0 S T 0 - 8 1
D E T E C T O R S Y S T E M D E S C R I P T I O N . . .

. 6 2 .
C A L I B R A T I O N F I L E N A M E I S C A L 0 P S 5 G E L I G A M S Y S T E M D I S K
C A L I B R A C A O 2 N D P O S I T I O N
T E S T E S 1 3 - A P R - 3 I
S P E C T R U M L I V E C O U N T T I M E = 2 0 0 0 S E C O N D S
* * * * * S U M M A R Y O F N U C L I D E S I N S A M p L E * * * * *
T I M E O F C O U N T P E R C E N T
N U C L I D E A C T I V I T Y U N C E R T A I N T Y
( M I C R O C U R I E S ) ( 1 S I G M A )
C E - M l 5 . 5 3 1 * E - 3 3 . 3
R U - 1 0 3 6 . . 1 2 3 * E - 3 4 . 3
Z R - 9 5 1 . 5 0 7 * E - 2 4 . 2
N B - 9 5 2 . 1 3 5 * E - 2 2 . 1
* * * U N U S E D P E A K S ( K E V ) : * * *
2 3 3 . 2 4 + 7 5 6 . 0 2 1 4 5 9 . 9 2
A N A L Y S I S F I N I S H E D A T 1 0 : 1 3 : 0 5
* R U N G A M M A 3
G E L I - G A M V 6 3 0 - D E C - 3 1 1 0 : 2 2 : 2 7
* * * * * G A M M A I I I ( V 2 3 ) * * * * *
W H E R E I S D A T A ( M C A O R D I S K ) ? : M C A
I S I N P U T : N E W ( N ) , S A M E ( S ) ^ O R T O B E M O D I F I E D ( M ) ? N
S T A R T C H A N N E L : 1 5 0
S T O P C H A N N E L : 3 0 0 0
S E N S I T I V I T Y : 1 0
U N I T S : M I C R O C U R I E S
F A C T O R ; 1 . 0 0 0 0
D E C A Y C O R R E C T ? : N
E N T E R T W O L I B R A R Y U N I T N L ' M B E R S A N D F I L E N A M E S
( 1 ) : 0 ^ P R F I S 7
( 2 ) : 0 , P R F I S 3
S A M P L E D E S C R I P T I O N . . .
Q 0 0 0 1 9 2 7 - A U G - 3 1 1 4 : 1 1 : 4 3
A M O S T R A D E U R A N I O E N R I Q U E C I D O I R R A D I A D A D I A 0 4 - M A I 0 - 3 1
E S P E C T R O O B T I D O D I A 2 7 - A G 0 S T 0 - 3 1

.63,
D E 1 £ C T 0 R S Y S T E M D E S C R I P T I O M . . .
C A L I B R A T I O N F I L E N A M E I S C A L 0 0 5
C A L I B R A C A O 2 N D P O S I T I O N
T E S T E S 1 3 - A P R - 8 1
G E L I G A M S Y S T E M D I S K
S P E C T R U M L I V E C O U N T T I M E = 2 0 0 0 S E C O N D S
* * * * * S U M M A R Y O F N U C L I D E S I N S A M P L E * * * * *
P E R C E N T
U N C E R T A I N T Y
C 1 S I G M A )
T I M E O F C O U N T
N U C L I D E A C T I V I T Y
( M I C R O C U R I E S )
N P - 2 3 9 < 3 . 7 * E - 3
M O - 9 9 < 1 . 2 * E - 2
1 - 1 3 2 < 1 . 7 * E - 3
X E - 1 3 5 < 1 . 0 * E - 3
B A - 1 4 0 < 0 . 0 * E 0
L A - 1 4 0 N O T P R E S E N T
1 - 1 3 1 < 1 . 5 * E - 3
N B - 9 5 2 . 1 3 5 * E - 2 2 . 1
C E - 1 4 1 5 . 5 7 7 * E - 3 3 . 8
R H - 1 0 6 < 4 . l * E - 3
C E - 1 4 4 < 6 . 3 * E - 3
P R - 1 4 4 < 5 . 0 * E - 1
K R - 3 5 M < 1 . 0 * E - 3
C E - 1 4 3 2 . 2 3 8 * E - 3 1 6 . 9
N D - 1 4 7 < 3 . 7 * E - 2
Z R - 9 7 < l . 7 * E - 3
Y - 9 1 M < 2 . 0 * E - 3
* * * E N D P A S S 1 * * *
T C - 9 9 M
T E - 1 3 2
2 . 9 4 6 * E - 3
< 1 . 0 * E - 3
3 . 9
R U - 1 0 3
Z R - 9 5
I - 1 3 3
X E - 1 3 3
6 . 1 3 3 * E - 3 4 . 3
4 . 3 1 . 4 7 2 * E - 2
< 2 . 2 * E - 3
< 0 . 0 * E 0
* * * E N D P A S S 2 * * *
A N A L Y S I S F I N I S H E D A T 1 0 : 3 3 : 5 3

. 64,
REFERÊNCIAS BIBLIOGRÁFICAS
1. AVERY'YANOVA, V, P. Nuclear radiation detectors based on high
purity germanivun. Sov. At. Energy, 34 (6):591-3, Dec. 1973.
2. BANASIK, Z.; KIERZEK, J.; PARUS, J.; ZOLTOWSKI, T.; ZALEWSKI ,
J. On-line computer system applied in a nuclear chemistry
laboratory. In: MEYER, H., ed. Real-time data handling and
process control: 1. European symposium on ... Berlin, Germa
ny, F. R., 23-25 October 1979. Amsterdam, North Holland,
1980. p.675-8.
3. BAUMGARTNER, F. The chemistry of nuclear reprocessing (content
of lectures) . Karlsruhe, Kernforschungszentrvim, 1977.
(KFK-2494).
4. BAUMGARTNER, F. & ERTEL, D. The modern purex process and its
analytical requirements. J. Radioanal. Chem., 58:11-28,
1980.
5. BELEN'KII, M. I.; BERDIKOV, V.V.; lOKHIN, B. S.; SUVOROV, M.
A. Gamma-spectrometric method for determining the isotopic
composition of plutonium. Sov. Radiochem., 21(1):127-31,
Jan./Feb. 1979.
6. BONNEVIE-SVENDSEN, M. Reprocessing analysis selected problems
and methods. In: INSTITUTT FOR ATOMENERGI. KJELLER RESEARCH
ESTABLISHMENT. Reprocessing of fuel from present and future
power reactors. Kjeller, Norway, Sep. 1967. (KR-126).
7. BULOVIC, V. F. Non-destructive determination of nuclear fuel
burn-up by measuring the gamma radiation of fission products
in irradiated uranium oxide. J. Radioanal. Chem., 4:99-107,
1970.

.65.
8. BULOVIC, V. P.; KRTIL, J.; SUS, F.; MAKSIMOVIC, Z.; KLOSOVA
E. Gamma-spectrometric determination of ''" Ru, ^"^^Cs,
^37^5 g ^^^ce in the fuel from the Czechoslovak atomic po
wer station Al. J. Radioanal. Chem., 51. (1) :153-60, 1979.
9. CAMP, D. C. Applications and optimization of the Ge(Li) detec
tor system. Livermore, Calif., California Univ., Lawrence
Radiation Lab., Mar. 1976. (UCRL-50156).
10. CANUETTE, R. P. Computer-controlled on-line gamma analysis for
Krypton-85. Aiken, SC, Du Pont de Nemours (E. I.) & Co.,
Savannah River Lab., 1980. (DP-MS-79-99; CONF-800303-15).
11. CAPPELLANI, F. & RESTELLI, G. X-ray and gamma-ray spectrosco
py. In: BERTOLINI, G. & COCHE, A.,ed. Semiconductor detec
tors. Amsterdam, North-Holland, 1968. p.365-407.
12. CONTENSON, G.; MONIER, J.; VIGNESOULT, N. Distribution des
produits de fission et localization par spectrometrie gamma
en cours d'irradiation et apres irradiation. In: INTERNA
TIONAL ATOMIC ENERGY AGENCY. Behaviour and chemical state
of irradiated ceramic fuels: proceedings of a panel on ...
held in Vienna, 7-11 August 1972. Vienna, 1974. p.191-200.
13. COVELL, D. F. Determination of gamma-ray abundance directly
from the total absorption peak. Anal. Chem., 31 (11):1785-
90, Nov. 1959.
14. CUNHA, I. I. L. Aplicação dos métodos radioquímico e de espec
trometria de raios gama direta para determinação da queima
do oxido de urânio irradiado. São Paulo, Instituto de Ener
gia Atômica, jun. 1979. (Dissertação de mestrado).
(IEA-DT-133).
15. DENARD, C. D. Applications of gamma spectrometry in the labo
ratories department at the Savannah River Plant. Aiken, SC,
. . . . . r r,0

.66.
20. GORDON, G. E.; HARVEY, J. W.; NAKAHARA, H. Measuring fission
spectra with semicondutor detectors. Nucleonics, 24:62-6,
Dec. 1966.
21. GUNNINCK, R.; NIDAY, J. B. Computerized quantitative analysis
by gamma-ray spectrometry. Livermore, Calif., California
Univ., Lawrence Livermore Lab., Mar. 1972. (UCRL-51061).
22. HARDWICK, W. H. Reprocessing analyses - general considerations
In: INSTITUTT FOR ATOMENERGI. KJELLER RESEARCH ESTABLISHMENT
Reprocessing of fuel from present and future power reactors,
K-ieller. Norwav. Sep. 1967. (KR-126). o. 143-8.
Du Pont de Nemours (E. I.) and Co., Sep. 1979.
{DPSPÜ-79-30-13).
16. DIAS, M. S. Calibração de um sistema de câmara de ionização
de poço 4TT -"y para medidas de atividades de radionuclideos.
São Paulo, Instituto de Energia Atômica, nov. 1978. (Dis
sertação de mestrado). (IEA-DT-087).
17. ERTEL, D. Equipment and instrvimentation of a laboratory for
purex process analytical chemistry. In: INTERNATIONAL ATO
MIC ENERGY AGENCY. Design and equipment for hot laborato
ries: proceedings of a symposium on ... Otaniemi, Finland,
2-6 August 1976. Vienna, 1976. p.159-64.
18. FLOH, B.; A R A O J O , J. A.; MATSUDA, H. T. Introdução aO estudo
do tratamento de combustíveis nucleares irradiados. São
Paulo, Instituto de Energia Atômica, jan. 1975.
(IEA-Inf-41).
19. GOFMAN, F. E.; ELISEEV, V. I.; ZAVOROTKIN, A. A.; KALENTAROV,
N. S.; LUZYANIN, E. N.; LYUBTSEV, R. I.; MOSYAZH, V. M.;
ORLOV, V. I. Automated laboratory gamma-spectrometric mo
nitoring of the solvent extraction reprocessing of a spent
fuel elements. Sov. Radiochem., 20(6):785-9, Jul. 1979.

.67.
26. LIMA, F. W.; ATALLA, L. T. A program in BASIC language for a-
nalyses of gamma spectra using On-line minicomputers. São
Paulo, Instituto de Energia Atômica, nov. 1973.
(IEA-Pub-317).
27. MAFRA, O. Y. Introdução ã técnica de medidas nucleares. São
Paulo, Instituto de Energia Atômica, jan. 1973.
(IEA-Inf^25).
28. MARATHE, S. G.; RAO, V. K.; BHARGAVA, V. K.; IYER, R. E.; RA-
MANIAH, M. V. A gamma monitoring assembly for the analysis
of fission products in reprocessing streams. Nucl. Instrum.
Meth., 127 (1):99-103, Jul, 1965 .
29. MATSUURA, S.; TSURÜTA, H.; SUZAKI, T.; OKASHITA, H.; UMEZAWA,
H. Non-destructive gamma-ray spectrometry on spent fuels
on a boiling water reactor. J. Nucl. Sci. Technol., 12 (1):
24-34, Jan. 1975.
30. MERTZIG , W. Estudo da técnica de eletrodeposição na prepara-
~ ~ 233 cao de amostras para determinação de U por espectrometria
alfa. São Paulo, Instituto de Pesquisas Energéticas e Nu
cleares, dez. 1979. (Dissertação de mestrado). (IPEN-DT-1),
23. KOCK, G. Recent developments in chemical processing of power
reactor fuels. In: COMPANHIA BRASILEIRA DE TECNOLOGIA NU
CLEAR. INSTITUTO DE ENERGIA NUCLEAR. Anais da primeira reu
nião de reprocessamento do combustível nuclear. Rio de Ja
neiro, 19-21 agosto 1974. Rio de Janeiro, 1974. p. 77-88.
24. LEDERER, C. M.; HOLLANDER, J. M.; PERLMAN, I. Table of isoto
pes. 6.ed. New York, N. Y., Wiley, 1967.
25. LEE, W. Direct estimation of gamma-ray abundances in radionu
clide mixtures. Complement subtraction method. Anal. Chem.,
31 (5):800-6, May 1959.

4
.68.
31. OLSON, D. G. Quantitative garama-ray spectrometrie analysis
of nuclides mixtures. Consecutive standard sources nullifica
tion. Idaho Falls, Idaho, Atomic Energy Institute, Nov.
1959. (IDO-14495).
32. OPERATORS Manual for GELIGAM (ORACL): Analytical Software. En
vironmental package. Model 6523. Oak Ridge, Tenn., Ortec
Inc., Dec. 1977.
33. PIERCE, T. B.; WEBSTER, R. K.; HALLETT, R.; MAPPER, D. Deve-
lopments in the use of small digital computers in activa
tion analysis systems. In: DeVOE, J. R.,ed. Modern trends
in activation analysis: proceedings of the 1968 internatio
nal conference held at Gaithersburg, Maryland, 7-11 October
1968. Washington, D. C , Jun. 1969, Vol. II, p.1116-20.
(NBS Special Publication 312, Vol. II).
34. PRICE, W. J. Nuclear radiation detectors. New York, N. Y.,
McGraw-Hill, 1958.
35. QUITTNER, P. & WAINERDI, R. E. Computer evaluation of Nal(Tl),
and Ge(Li) gamma-ray spectra. At. Energy Rev., 8 (2):361-
415, 1970.
36. RASMUSSEN, N- C.; SOVKA, J. A.; MAYMAN, S. A. The non-destruc-^
tive measurement of burn-up by gamma-ray spectroscopy. In:
INTERNATIONAL ATOMIC ENERGY AGENCY. Nuclear materials mana
gement: proceedings of a symposium on ... held by the IAEA j
at Vienna, 30 AugUst-3 September 1965. Vienna, 1966. p.829-
49.
37. ROCCA, H. C. Análise de espectros gama com calculadora progra
mável. São Paulo, Instituto de Energia Atômica, jul. 1978.
(IEA-Pub-514).

. 69.
38. SAUTERON, J. Les coinbustibles nucléaires. Paris, Hermann,
^ 1965.
J • 39. SCHUBIGER, P. A.; CHAKRABORTY, S.; WYTTENBACH, A.; BLASER, W.
Jane-an easy to handle computer program for different levels
of qualitative and quantitative gamma-ray spectra analysis.
J. Radioanal. Chem., 25:141-54, 1975.
40. SIFFERT, P. & COCHE, A. General characteristics of the inter
actions of nuclear radiations with matter and their conse
quences. In: BERTOLINI, G. & COCHE, A., ed'. Semiconduc
tor detectors. Amsterdam, North Holland, 1968. p.279-300.
41. THOMPSON, C. J. On-line activation analysis with a PDP-9 com
puter. In: DeVOE, J. R., ed. Modern Trends in activation
analysis: proceedings of the 1968 international conference
held at Gaithersburg, Maryland, 7-11 October 1968. Washing
ton, D. C , Jun. 1969, Vol. II, p.1116-20. (NBS Special
Publication 312, Vol. II).
42. YULE, H. P. Computation of Ge(Li) detectors peak areas for
activation analysis and gamma-ray spectrometry. Anal. Chem.,
40 (10):1480-6, Aug. 1968.
43. ZADDACH, G. Katalog von Ge(Li)>- Gamma S p e k t r e n . Jülich, Kern
forschungsanlage, 1973. {JÜ1-914-DE).

INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES SECRETARIA DA INDÚSTRIA, COMÉRCIO, CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA À UNIVERSIDADE DE SAO PAULO
ESTUDO DE DÍMEROS IONIZADOS DE GASES NOBRES PELO MÉTODO CELULAR VARIACIONAL
Renata M. M. Wentzcovitch
Dissertação apresentada ao Instituto de Pesquisas Energéticas e Nucleares como parte dos requisitos para obtenção do Grau de "Mestre na Area de Concentração: Tecnologia Nuclear".
Orientador: Dr. Josó Roberto Leite
São Paulo 1982

1
INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES SECRETARIA DA INDÚSTRIA. COMÉRCIO. CIÊNCIA E TECNOLOGÍA
AUTARQUÍA ASSOCIADA À UNIVERSIDADE DE SÂO PAULO
ESTUDO DE DÍMEROS IONIZADOS DE (¡ASES NOBRES
PELO MÉTODO CELULAR VARIACIONAL
R e n a t a M . M . W e n t z c o v i t c h
Orientador :
Dr. Josó Roberto Leite
Dissertação apresentada ao Instituto de Pesquisas Energéticas e Nucleares como parte dos requisitos para obtenção do grau de «Mestre" na Área de Concentração: Tecnologia Nuclear
S Ã O P A U L O
1 9 8 2 to O E
S t
N.
^OS MEUS PAIS
INSTITUTO D E P E S Q U I S A S È^fc-RÔètieAS E NUCLEARES I. P . E, N :

A G R A P E C I M E W T O S
Sinc&n.o.ò aQfiadtcimzntoò òão dzvldoò aò òzgalntzò
pa&oa.ò e ¿ni>t¿tu¿Q.oz¿, i
Ao PfLO^.Vn.. JoÁZ RobzAto Izltz, pzta. oA.<zntaçào
òZQafia e pzla. con^Zança dzpoòZtada du.fia.ntz a execução dz^
tz trabalho.
Ao VfLo{^.VK. Spzio Pznha Mofiato, pzlo apoio na f^a
òz inicial dzòtz tHabalko.
Ao Gzlòon T. Otanl z Tzizza C. Landgfta^, pzla co_
labofiação na pafitz computacional.
Aoò pAo{^zòòofLZ¿> z amlgoò quz dlHzta oa Indlfizta-
mzntz COntHlbalHam com zòclafizclmzntoò z òugzòtõzi.
 Sfia. Vayòz VaaHtz Calló pzlo capHlchoao traba
lho datllogfiallco.
Ao Instituto dz Pz¿qul¿a¿ Enzfigztlcaò z Naclza-
fizò (IPEW) pzla bol¿a dz mzòtfiado concedida.
Ao labofiatÓKlo dz E&tadoò Avançados [LEA] pzla
utilização dz òzu czntKo dz pKoczòòamznto dz dadoò na (¡a-
òz {^Inal dzòtz trabalho.

r Ñ V J c E
RESUMO i
ABSTRACT i i
1MTR0VUÇA0 1
I . MOLÉCULAS VO TIPO EXCIMER
I - l . I n t r o d u ç ã o 5
1 - 2 . H i s t ó r i c o 6
1 - 3 . Os H a l e t o s de G a s e s N o b r e s 8
1 -4 . M e c a n i s m o s de P e r d a nos H a l e t o s de G a s e s Nobres 13
I I . S I S T E M A S VE MUITAS P A R T Í C U L A S
I I - l . A p r o x i m a ç ã o de H a r t r e e - F o c k 18
I I - 2 . A p r o x i m a ç ã o Xa p a r a o P o t e n c i a l de T r o c a . . . . 24
I I I . O MiTOVO CELULAR VARIACIONAL (MCI/) ,
I I I - l . A F o r m u l a ç ã o O r i g i n a l 30
I I I - 2 . O MCV com P o l a r i z a ç ã o de S p i n 45
I V . RESULTADOS
I V - 1 . A M o l é c u l a de 49
I V - 2 . A M o l é c u l a de A r * 60
CONCLUSÃO 65
A P É N D I C E A - MOLtCULAS VlATOUlCAS 67
R E F E R E N C I A S 76

- 1 -
R H S II M O
ESTUDO DE DÍMEROS I O N I Z A D O S DE G A S E S NOBRES
PELO MÉTODO CL'LULAR V A R I A C I O N A L
R e n a t a M . M . W e n t z c o v i t c h
N e s t e t r a b a l h o f o i t e s t a d a a p o s s i b i l i d a d e de s e
u s a r o Método C e l u l a r V a r i a c i o n a l (MCV) p a r a o e s t u d o de mo
l é c u l a s i o n i z a d a s em s e u s e s t a d o s f u n d a m e n t a i s e e x c i t a
d o s . Os í o n s e s t u d a d o s foram Ne^ e A r ^ , s endo que o
Ar2 ê o s i s t e m a de m a i o r número de e l é t r o n s e n t r e os a t é
a g o r a c o n s i d e r a d o s a t r a v é s do MCV.
As t r a n s i ç õ e s e l e t r ô n i c a s e n t r e os e s t a d o s d e s
t e s s i s t e m a s s ã o i m p o r t a n t e s mecani smos de p e r d a de e f i c i ê n
c i a nos l a s e r s de h a l e t o s de g a s e s n o b r e s ("excimers l a ser s" ) .

- 1 1 -
A B S T R A C T
STUDY OF RARE-GAS DIMER IONS BY THE VARIATIONAL CELLULAR METHOD
Renata M.M. Wentzcovitch
We have used the Variational Cellular Method to
study ionized molecules in their ground and excited states
with the scope of testing the validity of such method in
these cases. The ions studied are Ne^ , Ar2 , where the
latter is the system with the largest number of electrons
tested by VCM so far.
The electronic transitions in these systems are
important mechanisms of efficiency decay for the noble gas
halide lasers ("excimer lasers").
p . E . N .

I N T R Õ V U Ç A O
A c l a s s e de l a s e r s de t r a n s i ç ã o e l e t r ô n i c a em m o l é c u
I a s c o n h e c i d a s como " e x c i m e r s " , a t u a l m e n t e ê um t ó p i c o de g r a n
de i n t e r e s s e na c l a s s e de l a s e r s a g â s . E s t e s l a s e r s s ã o pode
r o s a s f o n t e s de r a d i a ç ã o c o e r e n t e n a s r e g i õ e s v i s í v e l , u l t r a
v i o l e t a (UV) e v á c u o - u l t r a - v i o l e t a (VUV) do e s p e c t r o .
Em p a r t i c u l a r , os h a l e t o s de g a s e s n o b r e s , que tem os
menores c o m p r i m e n t o s de onda f o r a da p r o b l e m á t i c a r e g i ã o do v ã
c u o - u l t r a - v i o l e t a , tem r e c e b i d o a t e n ç ã o , t a n t o p o r p a r t e dos
t e ó r i c o s q u a n t o dos e x p e r i m e n t a i s . E n t r e e s t e s s i s t e m a s l a -
12 15
s e r s , o KrF ' e c o n s i d e r a d o como o m a i s i m p o r t a n t e d e v i d o
a s u a e f i c i ê n c i a e s e u compr imento de o n d a , que é o menor de t o
dos os que s e c o n h e c e f o r a do V U V .
Toda a a t e n ç ã o que e s t e s s i s t e m a s tem r e c e b i d o é j u s
t i f i ç a d a p e l a s u a o t i m i z a ç ã o , p o i s s u a s a p l i c a ç õ e s ^ ^ abrem n o
v a s p e r s p e c t i v a s a f o t o q u í m i c a ^ ^ , s e p a r a ç ã o i s o t ó p i c a e f u s ã o
t e r m o n u c l e a r c o n t r o l a d a ^ ^ .
A n í v e l t e ó r i c o , m u i t o s c á l c u l o s de e s t r u t u r a e l e t r ô
n i c a ^ ^ e de c i n é t i c a química^'* s ã o f e i t o s t e n t a n d o - s e a v a l i a r
os e v e n t u a i s p r o c e s s o s que venham a o c o r r e r na c a v i d a d e e s u a s
i m p o r t â n c i a s r e l a t i v a s . Cabe d e s t a c a r a q u i que a m a i o r i a d e s
t e s p r o c e s s o s e s t u d a d o s d i f i c i l m e n t e podem s e r i d e n t i f i c a d o s
e x p e r i m e n t a l m e n t e .
N e s t e t r a b a l h o n o s propomos a e s t u d a r a e s t r u t u r a e -
l e t r ô n i c a de d o i s d í m e r o s i o n i z a d o s de g a s e s n o b r e s : ^ 6 2 * e
A r 2 * . E s t a s e s p é c i e s s e formam na c a v i d a d e do l a s e r quando um
dos g a s e s n o b r e s e s t ã p r e s e n t e , s e j a como g â s " b u f f e r " ou como
um dos e l e m e n t o s do q u a l o meio a t i v o é c o m p o s t o . A e x i s t ê n c i a 6 7
d e s t a s e s p é c i e s i n f l u i d e c i s i v a m e n t e ' na e f i c i ê n c i a dos l a -

sers, pois suas fotodissociações requerem comprimentos de onda
que caem exatamente na região do espectro em que ocorre a ação
laser dos haletos de gases nobres, ou seja, o ultra-violeta.
Para este estudo, usamos o Método Celular Variacio -
nal (MCV)^^'^"^ como técnica de química quântica. Este é um me
todo que tem sua origem na física de estado sólido e se baseia
no método celular de Wigner-Seitz-Slater proposto em 1934^"^.
Neste método o potencial é tratado como um funcional de densi
dade local além de ser tratado por diferentes expressões, em
diferentes regiões do espaço.
Basicamente o MCV é um método intermediário entre os
métodos "ab initio" que se baseiam exclusivamente no formalis
mo de Hartree-Fock e na técnica de interação de configurações,
e os métodos semi-empíricos, nos quais as dificuldades ineren
tes ao formalismo de Hartree-Fock são eliminadas introduzindo-
-se parâmetros obtidos experimentalmente. Dizemos que o MCV é
um método intermediário pois chega a ser 100 vezes mais rápido
48
do ponto de vista computacional que os precisos métodos "ab
initio" e, embora mais lento que os métodos semi-empíricos, in
depende de qualquer resultado experimental para se parametri
zar .
No que se refere ã sua aplicação a moléculas, o MCV
já foi empregado em sistemas diatómicos covalentes tais como à âQ f \ à ftQ
H ^ , C ^ , N2 e CO , F2 e Ne2 , LÍ2 e HLi""^. Das informações
obtidas destes estudos, conclue-se que o MCV determina muito
bem posiçõeS' de equilíbrio Re e energias de dissociação De ,
entretan1;o as constantes elásticas Ke , que descrevem o compor
tamento das curvas de energia próximo da posição de equilíbrio
já diferem sensivelmente dos valores experimentais^^. Isto o-
corre porque o valor de Ke está estritamente relacionado com

a boa descrição das curvas de energia para todos os valores de
R e não apenas para os pontos em torno da posição de equilí
brio.
O MCV também já foi aplicado para o estudo de siste
mas iónicos como os próprios excimers: NeF, NeC£, ArF e ArCJi^.
Para estas moléculas, ele forneceu constantes espectroscópicas
e características de emissão bastante precisas, isto porque as
características das curvas de potencial destas moléculas são
bastante acentuadas.
Entretanto, moléculas ionizadas nunca foram submeti
das ao cálculo por este método. Assim a descrição de suas pro
priedades também pode servir como um teste para a exploração
deste método de cálculo aproximativo. Para isso, podemos compa
rar nossos resultados com cálculos precisos efetuados por meto
dos "ab initio" ou mesmo com resultados obtidos pelo método do
59
Espalhamento Múltiplo (MS-Xa) que utiliza o mesmo tipo de a-
proximação de densidade local para a descrição do potencial,em
bora este seja feito "muffin-tin".
É importante acentuar que este método, no que se re
fere a sua aplicação a moléculas, desempenhará importante pa
pel no cálculo de moléculas grandes, ou mesmo de "clusters" mo
leculares, que são sistemas para os quais os métodos"ab initio"
são impraticáveis, além do que sua aplicação a sistemas aber
tos ê perfeitamente viável, o que já não ocorre com o método
MS-Xa, cuja aplicação a estes sistemas é discutível.
Na seção I deste trabalho apresentamos um breve his
tórico da descoberta e desenvolvimento dos lasers de excimers
além de um resumo dos principais mecanismos de perda nos hale
tos degases nobres e do processo por nós estudado. Na seção II
mostramos como o problema de cálculo de estrutura eletrônica

- 4 -
pode s e r r e d u z i d o ao f o r m a l i s m o de H a r t r e e - F o c k , c h e g a n d o ã e -
q u a ç ã o de uma p a r t í c u l a num campo a u t o - c o n s i s t e n t e ( S C F ) e i n
t r o d u z i n d o na mesma a a p r o x i m a ç ã o de d e n s i d a d e l o c a l Xa p a r a
o p o t e n c i a l de t r o c a . Em s e g u i d a , na s e ç ã o I I I , a p r e s e n t a m o s
o Método C e l u l a r V a r i a c i o n a l na sua f o r m u l a ç ã o o r i g i n a l e a
s u a f o r m u l a ç ã o c o n s i d e r a n d o - s e a p o l a r i z a ç ã o de s p i n . Na s e ç ã o
I V a p r e s e n t a m o s n o s s o s r e s u l t a d o s c o m p a r a d o s e na s e ç ã o V , n o s
s a s c o n c l u s õ e s . E s t e t r a b a l h o contêm a i n d a um A p ê n d i c e que
d e s c r e v e a s i m e t r i a d o s o r b i t a i s e dos e s t a d o s das m o l é c u l a s
d i a t ó m i c a s , c u j o c o n h e c i m e n t o o t i m i z a a e s c o l h a d a s f u n ç õ e s s e
gundo a s q u a i s o s o r b i t a i s m o l e c u l a r e s s ã o e x p a n d i d o s . Com ba
se também n e s t a s s i m e t r i a s a p r e s e n t a m o s a s r e g r a s de s e l e ç ã o
p a r a as t r a n s i ç õ e s e n t r e os e s t a d o s m o l e c u l a r e s de s i s t e m a s di^
a t ô m i c o s .
I N S I I T U ÍO D E P Ê Ê Õ U feAS E N E R G É t | G > - S E N U C L S A R E S
I. P . E . N .
- C A P I T U L O I -
MOLÉCULAS VO TIPO EXCIMERS
I - l . INTRODUÇÃO
E x c i m e r s e e x c i p l e x s ã o f o r m a d o s p e l a i n t e r a ç ã o e n
t r e d o i s á tomos ou m o l é c u l a s , s endo que um d e l e s e n c o n t r a - s e
num e s t a d o e l e t r o n i c a m e n t e e x c i t a d o .
A + B* -»• ( A B ) *
E s t a m o l é c u l a pode d e c a i r r a d i o a t i v a m e n t e p a r a o e s
t a d o f u n d a m e n t a l e s e d i s s o c i a r
( A B ) * -> A + B + hv
P a r a i s t o , o e s t a d o f u n d a m e n t a l deve s e r r e p u l s i v o
ou l i g e i r a m e n t e l i g a d o , de forma a s e r i n s t á v e l a t e m p e r a t u r a s
n o r m a i s . Um d i a g r a m a de e n e r g i a p o t e n c i a l d e s t e s s i s t e m a s é
m o s t r a d o na F i g u r a I - l .
FIGURA 1 -7 .
Vlagrama do. znzrg¿a po-
tzncZat para um zxcimzr
( A B ) * .
DISTANCIA INTERNUCLEAR
P a r a s i s t e m a s c u j o e s t a d o f u n d a m e n t a l é r e p u l s i v o , a
e m i s s ã o e x c i m e r é c a r a c t e r i z a d a p o r uma l a r g a banda c o n t i n u a .
No c a s o d o s s i s t e m a s com e s t a d o f u n d a m e n t a l l i g e i r a m e n t e l i g a
do é p o s s í v e l se o b s e r v a r uma e s t r u t u r a v i b r a c i o n a l n o r m a l .
1 - 2 . H I S T Ó R I C O
A p r i m e i r a i d e n t i f i c a ç ã o de um e x c i m e r f o i f e i t a no
52
f i n a l da d e c a d a de 20 p o r L o r d R a y l e i g h , quando e s t u d a v a o
e s p e c t r o de e m i s s ã o de v a p o r de m e r c u r i o na r e g i ã o do u l t r a - v i o
l e t a . O b s e r v o u um e s p e c t r o c o n t i n u o com um máximo de e m i s s ã o
em 330 nm. E s t e e s p e c t r o e l e a t r i b u i u a uma t r a n s i ç ã o e n t r e
um e s t a d o do d í m e r o Hg^ e o e s t a d o f u n d a m e n t a l .
Na d é c a d a de 30 a p a r e c e m m a i s d o i s t r a b a l h o s v a l e n
d o - s e d e s t a mesma i d é i a . H o p f i e l d ^ ^ d e s c o b r i u a e m i s s ã o c o n t ^ *
nua do He2 na r e g i ã o do v a c u o - u l t r a - v i o l e t a , e em 1938 F i n -
23 k e l n b u r g d e s c r e v e a d e s c o b e r t a de l a r g a s b a n d a s de e m i s s ã o
* *
no u l t r a - v i o l e t a dos v a p o r e s de m e t a i s como Hg2 e C d 2 . N e s t e
t r a b a l h o e l e r e l a c i o n a e s t a s e m i s s õ e s c o n t í n u a s com t r a n s i ç õ e s
a t ô m i c a s d e s t e s m e t a i s .
- 80
J á na d é c a d a de 5 0 , T a n a k a e c o l a b o r a d o r e s d e s c o
brem a s b a n d a s de e m i s s ã o e x c i m e r dos g a s e s n o b r e s . E s t e s e s
p e c t r o s s ã o m o s t r a d o s na F i g u r a 1 - 2 .
500 1000 ^ 1500 > ( A )
2000
FIGURA 1 - 2 . EmlÁòão continua doò cxclmzfiò dz gãò nobfiz [dz
A s t r a n s i ç õ e s e x c i m e r s c o r r e s p o n d e m ã s b a n d a s l a r g a s
s i t u a d a s ã d i r e i t a dos p i c o s , que p o r sua v e z c o r r e s p o n d e m ã s
t r a n s i ç õ e s a t ô m i c a s e n t r e o s e s t a d o s e x c i t a d o s ou '''P e o

-/-
e s t a d o f u n d a m e n t a l . Além d e s t e e s t u d o d e s e n v o l v e r a m lampa
79
das p a r a a p r o d u ç ã o dos e s p e c t r o s e x c i m e r s , que d e s d e e n t ã o
tem s i d o u s a d a s como f o n t e de r a d i a ç ã o c o n t í n u a p a r a e s p e c t r o ^
c o p i a na r e g i ã o do v a c u o - u l t r a - v i o l e t a .
A i d é i a de s e u t i l i z a r e s t a s t r a n s i ç õ e s dos e x c i m e r s ,
ou s e j a , uma t r a n s i ç ã o " b o u n d - f r e e " p a r a s e o b t e r uma i n v e r s ã o
de p o p u l a ç ã o e a s s i m p o s s i b i l i t a r uma e m i s s ã o e s t i m u l a d a , f o i
s u g e r i d a p o r H o u r t e r m a n s ^ ^ em 1960. E n t r e t a n t o o p r i m e i r o r e
s u l t a d o c o n c r e t o de um l a s e r de e x c i m e r só v e i o a o c o r r e r em
1970 , quando Basov^ d e m o n s t r o u e m i s s ã o e s t i m u l a d a no Xe2 l í
q u i d o em t o r n o de 170 nm . Em 1972 , s u r g i u uma n o v a v e r s ã o d e £ 42
t e l a s e r , d e s t a v e z na f a s e g a s o s a a a l t a s p r e s s õ e s no q u a l
o bombeamento e r a f e i t o a t r a v é s de e l é t r o n s r e l a t i v í s t i c o s . Por
e s t e mesmo p r o c e s s o c o n s e g u i u - s e , nos anos s u b s e q u e n t e s , lasers • • -• j • *33 A *37 s i m i l a r e s dos e x c i m e r s Kr2 e Ar2
Os g a s e s n o b r e s também podem f o r m a r e x c i m e r s h e t e r o -
n u c l e a r e s ou e x c i p l e x com v á r i o s t i p o s de á t o m o s , i n c l u i n d o hi^
d r o g ê n i o ^ ^ , h a l o g ê n i o s , o x i g ê n i o e m e r c ú r i o . Em 1974 e 1975 a
p a r e c e m os t r a b a l h o s de P o w e l l ^ ^ e H u g h e s ^ ^ no q u a l e l e s c o n s e
guem a ç ã o l a s e r de uma m i s t u r a de o x i g ê n i o (©2) e g â s n o b r e ,
e x c i t a d a p o r um f e i x e de e l é t r o n s . A b a n d a de t r a n s i ç ã o e x c i
mer a p a r e c i a p e r t o de 558 nm , c o m p r i m e n t o de onda da r a d i a ç ã o
c o r r e s p o n d e n t e ã t r a n s i ç ã o a t ô m i c a 0(''"S) -»• O('^D) , c o n h e c i d a
como t r a n s i ç ã o " a u r o r e a l " na q u a l o e s t a d o ê m e t a e s t â v e l .
E n t r e t a n t o o m a i o r a v a n ç o no campo dos l a s e r s de e x
c i m e r s , começou com a d e s c o b e r t a de uma c l a s s e i n t e i r a m e n t e no
v a de e x c i m e r : os m o n o h a l e t o s de g a s e s n o b r e s .
A p o t e n c i a l i d a d e d e s t a s m o l é c u l a s como s i s t e m a s ú t e i s 27 81
a a ç ã o l a s e r f o i i n d i c a d a p o r G o l d e e V e l a z c o em 1974 , quan
do e s t u d a r a m a d e s a t i v a ç ã o do e s t a d o m e t a - e s t â v e l ^P2 dos g a

- 0 -
s e s Ar e Xe p o r h a l o g ê n i o s e o x i g ê n i o e o b s e r v a r a m e m i s s õ e s
e x c i m e r s de h a l e t o s e ó x i d o s de g a s e s n o b r e s . V e l a z c o c o n c l u i u
que a f o r m a ç ã o de h a l e t o s de Xe , X e X * , e r a p e l o menos dez ve
z e s m a i o r que a f o r m a ç ã o de XeO* , s e n d o p o r t a n t o m u i t o m a i s
e f i c i e n t e s p a r a a a ç ã o l a s e r que os ó x i d o s de g a s e s n o b r e s .
A a ç ã o l a s e r nos h a l e t o s de g a s e s n o b r e s f o i r a p i d a
mente d e m o n s t r a d a e n t r e 1975 e 1976 . E n t r e e s t e s c o m p o s t o s , o
K r F ^ ^ f o i c o n s i d e r a d o como o m e l h o r s i s t e m a d e v i d o ã a l t a e f i
c i ê n c i a do m e i o .
Os mono h a l e t o s de g a s e s n o b r e s p o s s i b i l i t a r a m a o b
t e n ç ã o de l a s e r s na r e g i ã o do v â c u o - u l t r a - v i o l e t a e do u l t r a
v i o l e t a , r e g i ã o a t e e n t ã o n u n c a a t i n g i d a p o r um l a s e r . I s t o
tem e s t i m u l a d o a p e s q u i s a t e ó r i c a da e s t r u t u r a e l e t r ô n i c a d e s
t e s c o m p o s t o s e dos mecan i smos de r e a ç õ e s a t é o s d i a s de h o -
j e 3 1 ' 3 9 _
1 - 3 . OS HALETOS DE G A S E S NOBRES
A e m i s s ã o e x c i m e r dos m o n o h a l e t o s de g a s e s n o b r e s ê
d e v i d a ã uma t r a n s i ç ã o de um e s t a d o i ó n i c o f o r t e m e n t e l i g a d o pa
r a um e s t a d o c o v a l e n t e r e s p u l s i v o ou l i g e i r a m e n t e l i g a d o . Os
p r i m e i r o s e s t u d o s t e ó r i c o s s o b r e e s t a s m o l é c u l a s f o r a m f e i t o s
em a n a l o g i a a s i s t e m a s de h a l e t o s a l c a l i n o s . A c o n f i g u r a ç ã o e -
l e t r ô n i c a de um átomo de g â s n o b r e e x c i t a d o ê b a s t a n t e s i m i l a r
ã de um m e t a l a l c a l i n o , i s t o é , um ú n i c o e l é t r o n s o r b i t a n d o
em t o r n o de um c a r o ç o de c a r g a p o s i t i v a u n i t á r i a . I s t o r e s u l t a
numa g r a n d e s e m e l h a n ç a e n t r e o s p o t e n c i a i s de i o n i z a ç ã o dos e £
t a d o s m e t a e s t ã v e i s "^P^ 2 de Ne , A r * , K r * e X e * e os e s
t a d o s f u n d a m e n t a i s de Na , K , Rb e Cs r e s p e c t i v a m e n t e . Em
p a r t i c u l a r , os g a s e s n o b r e s e x c i t a d o s formam l i g a ç õ e s i ó n i c a s

m u i t o f o r t e s p o r t r a n s f e r ê n c i a de c a r g a a á tomos e l e t r o n e g a t i -
v o s , t a i s como os h a l o g ê n i o s . Com b a s e n e s t a s e m e l h a n ç a e n t r e
17
os h a l e t o s de gases nobrese os h a l e t o s a l c a l i n o s , Ewing p r e v i u
o c o m p r i m e n t o de onda de e m i s s ã o p a r a m u i t a s d e s t a s m o l é c u l a s .
P a r a a c o m p r e e n s ã o a n í v e l t e ó r i c o da e s t r u t u r a e l e
t r ô n i c a e das p r o p r i e d a d e s r e l a c i o n a d a s d e s t a s m o l é c u l a s , f o
ram a p l i c a d o s p o d e r o s o s m é t o d o s "ab i n i t i o " i n c l u i n d o i n t e r a -29 13 30
ção de c o n f i g u r a ç ã o * ' . Também f o r a m a p l i c a d o s m o d e l o s u - 9
t i l i z a n d o a d e n s i d a d e de c a r g a do g a s de e l é t r o n s e a a p r o x i
mação do f u n c i o n a l de d e n s i d a d e Xa de S l a t e r . D e n t r o do e s
p í r i t o d e s t a ú l t i m a a p r o x i m a ç ã o , f o r a m f e i t o s c á l c u l o s a u t o -
c o n s i s t e n t e s a t r a v é s do método do e s p a l h a m e n t o m ú l t i p l o (MS-Xa), com 58 e sem^^ " o v e r l a p " de e s f e r a s , p a r a a m o l é c u l a de A r F e
a t r a v é s do Método C e l u l a r V a r i a c i o n a l CMCV) p a r a as m o l é c u l a s
A r F , A r C i . , NeF e N e C £ ^ . Todos e s t e s c á l c u l o s a j u d a r a m no en
t e n d i m e n t o das c a r a c t e r í s t i c a s de e m i s s ã o e d a s c o n s t a n t e s e s
p e c t r o s c ó p i c a s d e s t a s m o l é c u l a s .
O mecanismo de f o r m a ç ã o m a i s i m p o r t a n t e do e s t a d o ex
c i t a d o i ó n i c o ê d e v i d o a uma r e a ç ã o e n t r e o e s t a d o e x c i t a d o do
gâs n o b r e com m o l é c u l a s de h a l o g ê n e o . A a l t a a f i n i d a d e e l e t r ô
n i c a d a s m o l é c u l a s de h a l o g ê n e o e o b a i x o p o t e n c i a l de i o n i z a
ção d o s átomos de g â s n o b r e e x c i t a d o s p r o v o c a m um p r o c e s s o c h a
mado " h a r p o o n i n g " ^ ^ , p r o p o s t o a m u i t o tempo p a r a e x p l i c a r a
g r a n d e s e ç ã o de c h o q u e da r e a ç ã o de átomos a l c a l i n o s com m o l é
c u l a s de h a l o g ê n e o . E s t e p r o c e s s o e n v o l v e um e s t a d o t r i a t ó m i
c o i ó n i c o i n t e r m e d i á r i o e o " h a r p o o n " ( a r p ã o ) d e s t e mecan i smo
é o e l é t r o n f a c i l m e n t e i o n i z á v e l . O e l é t r o n p a s s a do átomo de
g á s n o b r e e x c i t a d o ou do átomo a l c a l i n o p a r a a m o l é c u l a de h a
l o g ê n e o e o f o r t e campo c o u l o m b i a n o a s s i m p r o d u z i d o a t r a i o í o n
d i a t ó m i c o n e g a t i v o e i s t o f a z com que o e l é t r o n " a r p ã o " r e t o r -

ne ao I o n p o s i t i v o t r a z e n d o c o n s i g o a m o l é c u l a de h a l o g ê n e o .
Rg* + X2 ^ 8 * ^ 2 ' * ^
F i n a l m e n t e o p a r i ó n i c o é formado p e l o r á p i d o d e c a i
mento da e s p é c i e t r i a t ó m i c a , que é um e s t a d o e l e t r o n i c a m e n t e
e x c i t a d o .
Um o u t r o mecan i smo de f o r m a ç ã o de g r a n d e importância^^
o c o r r e quando os e l é t r o n s u s a d o s p a r a o bombeamento ("pumping")
produzem Ions i s o l a d o s . A s e q u ê n c i a c i n é t i c a que p r o d u z o p a r
i ó n i c o é :
e + Rg ^ Rg"*" + e + e
e + X2 X " + X
X" + Rg"" + (M) ^ Rg*X~ + (M)
onde M é q u a l q u e r t e r c e i r o c o r p o , g e r a l m e n t e um átomo de g â s
n o b r e m a i s l e v e u s a d o como " b u f f e r " .
Na F i g u r a 1-3 s ã o a p r e s e n t a d a s c u r v a s t í p i c a s de e -
n e r g i a p o t e n c i a l p a r a os h a l e t o s de gase s n o b r e s .
As c u r v a s i n f e r i o r e s s ã o e s s e n c i a l m e n t e r e p u l s i v a s e
d e r i v a m da a p r o x i m a ç ã o de um átomo de g â s n o b r e e um de h a l o g ê
neo no e s t a d o f u n d a m e n t a l . A s s i m e t r i a s a t ô m i c a s s ã o : R g : ^ S
1 2 e X : P - , - 1 / o • Os e s t a d o s m o l e c u l a r e s que surgem s a o I E e
• j / ^ > 1/ 2
1 N dependendo da o r i e n t a ç ã o do o r b i t a l 2p d e s o c u p a d o do ha
l o g ê n e o , e n t r e t a n t o , d e v i d o ao a c o p l a m e n t o s p i n - ó r b i t a n e s t e
mesmo á t o m o , a d e g e n e r e s c e n c i a do e s t a d o N e q u e b r a d a e o s
e s t a d o s passam a s e r i n d i c a d o s p o r X ( l / 2 ) , A ( l / 2 ) e A ( 3 / 2 )
(em n o t a ç ã o e s p e c t r o s c ó p i c a ) . O b s e r v a - s e que a d e g e n e r e s c ê n
c i a d i m i n u i a m e d i d a que s e c a m i n h a na d i r e ç ã o dos h a l o g é n e o s
m a i s p e s a d o s .
<
õ
TRANSIÇÃO LASER
A ( l / 2 , 3/2)
X (1/2)
Rg-»- X
X + R g
-í f-
FIGURA 1-3.
DISTÂNCIA INTERNUCLEAR
CufLvaò tZpicaò de potencial para oò haletoò dz gáò
nobrz.
De f o r m a s i m i l a r e x i s t e m os e s t a d o s de n a t u r e z a i ô n i
c a 2 1* e 2 n formados a p a r t i r dos í o n s X" , e s f é r i c a m e n
t e s i m é t r i c o s num e s t a d o S e do í o n
2
Rg* com s i m e t r i a
^3 /2 1/2 * ^ e f e i t o do a c o p l a m e n t o s p i n - ó r b i t a nos í o n s de ga
s e s n o b r e s p e s a d o s é b a s t a n t e p r o n u n c i a d o , i s t o f a z com que a
d e g e n e r e s c ê n c i a do e s t a d o 2 II s e j a q u e b r a d a e os e s t a d o s s e
jam i n d i c a d o s p o r B ( l / 2 ) , C ( l / 2 ) e D ( 3 / 2 ) .
A banda de e m i s s ã o no u l t r a - v i o l e t a m a i s i n t e n s a c o r
r e s p o n d e ã t r a n s i ç ã o B — X e as m a i s f r a c a s â s t r a n s i ç õ e s
D — X e C — A . Todos os l a s e r s operam na t r a n s i ç ã o B — X ,
p o i s o e s t a d o f u n d a m e n t a l é a p e n a s f r a c a m e n t e r e p u l s i v o na r e -
18 _ -g i ã o d e s t a t r a n s i ç ã o . I s t o i m p l i c a que a b a n d a de e m i s s ã o e
m a i s e s t r e i t a {' 2 nm) e p o r t a n t o a s e ç ã o de choque p a r a a e m i s
s ã o e s t i m u l a d a é m a i o r .

Na T a b e l a I - l m o s t r a m o s os X ' s de e m i s s ã o dos h a l e
t o s de gases nobres, o n d e , a a ç ã o l a s e r ê o b s e r v a d a nas t r a n s i ç õ e s
c u j o s c o m p r i m e n t o s de onda e s t ã o s u b l i n h a d o s . Os o u t r o s compri^
mentos de onda c i t a d o s c o r r e s p o n d e m â o b s e r v a ç ã o de f l u o r e s c ê n
c i a .
T A B E L A I - ]
Comprimento do, onda de eml&&õe& "excimerÁ" doò ha
leto& de QOLeò nobreò {dadoò retirados da rei. [3 9 ] )
F Cl Br I
X (nm) X (nm) X (nm) X (nm)
B - - X 351 308 282 253
Xe C -- A 450 350 302 263
B - - X 249 222 206
Kr C -- A 275
B - X 193 175
A r C -- A 203 199
B - X 108
Ne C - - A 117
Alem d e s t e s e s t a d o s e l e t r ô n i c o s o u t r o s e s t a d o s m a i s
e x c i t a d o s s ã o p o s s í v e i s e , dependendo de s u a s c a r a c t e r í s t i c a s ,
assumem i m p o r t a n t e p a p e l no que se r e f e r e ao desempenho do l a
s e r .

1-4 . MECANISMOS DE PERDA NOS HALETOS DE G A S E S NOBRES
P a r a a c o m p r e e n s ã o do desempenho d e s t e s l a s e r s é p r e
c i s o que s e c o n h e ç a os p r o c e s s o s c i n é t i c o s e n v o l v i d o s . No í t e m
a n t e r i o r c i t a m o s d o i s p r o c e s s o s de f o r m a ç ã o dos e s t a d o s e x c i t a
d o s i ó n i c o s , e n t r e t a n t o também e x i s t e m p r o c e s s o s de d e s t r u i ç ã o
d e s t e s mesmos e s t a d o s , a l ém da p o s s í v e l t r a n s i ç ã o p a r a o e s t a
do f u n d a m e n t a l . A l é m d i s s o , é p o s s í v e l que o c o r r a a b s o r ç ã o da
r a d i a ç ã o da t r a n s i ç ã o " e x c i m e r " p o r o u t r a s e s p é c i e s que e v e n
t u a l m e n t e venham a se f o r m a r na c a v i d a d e do l a s e r . É j u s t a m e n
t e n e s t e p o n t o que os c á l c u l o s t e ó r i c o s a d q u i r e m i m p o r t â n c i a ,
p o i s o s p r o c e s s o s que o c o r r e m na c a v i d a d e d i f i c i l m e n t e podem
s e r c a r a c t e r i z a d o s de forma e x p e r i m e n t a l .
A d e s c r i ç ã o dos p r o c e s s o s c i n é t i c o s nos l a s e r s de ha
l e t o s de g a s e s n o b r e s f o i p r i m e i r a m e n t e a p r e s e n t a d a p o r R o k n i
e t a l ^ ^ que c o n s i d e r o u em d e t a l h e s a f o r m a ç ã o e os p r o c e s s o s
de p e r d a nos l a s e r s de X e F e K r F e x c i t a d o s p o r f e i x e de e l ê -
19 -
t r o n s e d e s c a r g a e l é t r i c a . Ewing também a p r e s e n t a uma c o l e
t â n e a de d a d o s t e ó r i c o s e e x p e r i m e n t a i s que tem s i d o ú t i l no
e n t e n d i m e n t o e na o t i m i z a ç ã o do desempenho d e s t e s l a s e r s . A q u i
a p r e s e n t a m o s a p e n a s uma s í n t e s e dos p r o c e s s o s m a i s r e l e v a n t e s
que i n f l u e n c i a m no desempenho d e s t e s l a s e r s .
E x i s t e um mode lo b a s t a n t e s i m p l e s que e x p l i c a p o r q u e
em a l g u n s h a l e t o s de gases nobres a a ç ã o l a s e r é e f i c i e n t e e em
o u t r o s n ã o . Embora t o d a s e s t a s m o l é c u l a s tenham a s mesmas c u r
v a s de e n e r g i a p o t e n c i a l c a r a c t e r í s t i c a s e a s mesmas p r o p r i e d a
des de e m i s s ã o do e s t a d o i ó n i c o p a r a o e s t a d o r e p u l s i v o , somen
t e s e i s ( A r F , K r F , X e F , K r C í , , XeCil e X e B r ) d o s p o s s í v e i s d e z e s
s e i s c o m p o s t o s s e mostram ú t e i s . O u t r o s q u a t r o ( N e F , A r C £ ,
K r B r e X e l ) e x i b e m a p e n a s f l u o r e s c ê n c i a e o s r e s t a n t e s não emi

tem p r a t i c a m e n t e n a d a .
Uma e x p l i c a ç ã o p a r a e s t e f a t o é que o u t r o s e s t a d o s
e x c i t a d o s , c u j o l i m i t e de á tomos s e p a r a d o s ê dado p o r (Rg + X * ) ,
onde X se r e f e r e ã p r i m e i r a e x c i t a ç ã o do h a l o g ê n e o , tem c u r
v a s de e n e r g i a p o t e n c i a l que cruzam a c u r v a do e s t a d o i ó n i c o .
A i m p o r t â n c i a do c r u z a m e n t o d e s t a s c u r v a s f o i a p o n t a d a p o r E -
wing e Brau"*^^ ao f a z e r e m a a n a l o g i a d e s t a s m o l é c u l a s com os ha
l e t o s a l c a l i n o s . E s t a s c u r v a s podem s e r v i s t a s na F i g u r a 1-3
l o g o a c i m a da c u r v a c a r a c t e r í s t i c a do e s t a d o i ó n i c o . A i d é i a
e que se a p o s i ç ã o d a s c u r v a s (Rg + X ) e t a l que e l a s c r u
zam a s c u r v a s i ó n i c a s , e n t ã o a e f i c i ê n c i a de p r o d u ç ã o do e s t a
do i ó n i c o s e r á r e d u z i d a havendo também a f o r m a ç ã o d e s t e s e s t a
dos c o v a l e n t e s que costumam s e r chamados de e s t a d o s de R y d b e r g .
C o n h e c e n d o - s e a e n e r g i a dos s i s t e m a s (Rg + X * ) e
(Rg* + X ~ ) no l i m i t e de á tomos s e p a r a d o s e a s s u m i n d o - s e e s s e n
c i a l m e n t e a f o r m a p l a n a p a r a a c u r v a c o v a l e n t e e a forma - —i—
p a r a a c u r v a i ó n i c a , p o d e - s e v e r i f i c a r s e e s t a s c u r v a s s e c r u
zam. P a r a a s m o l é c u l a s A r F , K r F , X e F e XeCí , e s t a s c u r v a s não
s e c r u z a m e p o r t a n t o é de s e e s p e r a r que o e s t a d o c o v a l e n t e não
i n t e r f i r a na t r a n s i ç ã o l a s e r , como r e a l m e n t e a c o n t e c e . No c a
so d a s m o l é c u l a s N e C í , , N e B r , N e l e A r i a d i s t â n c i a Re na q u a l
e s t a s c u r v a s s e c r u z a m é menor do que a d i s t â n c i a de e q u i l í b r i o
Re da m o l é c u l a R g * X ~ . I s t o s i g n i f i c a que a c u r v a do e s t a d o
c o v a l e n t e e s t á a b a i x o da c u r v a i ó n i c a e p o r t a n t o o e s t a d o iôni
co deve s e r f a c i l m e n t e p r e d i s s o c i a d o . De f a t o , nenhuma d e s s a s
m o l é c u l a s a p r e s e n t a a ç ã o l a s e r ou mesmo f l u o r e s c e n c i a . A s o i t o
m o l é c u l a s r e s t a n t e s s e c l a s s i f i c a m e n t r e e s t e s d o i s l i m i t e s .
X e l , K r B r , ArCfi, e NeF a p e n a s f l u o r e s c e m , A r B r e K r l não a p r e
sen tam nem mesmo f l u o r e s c e n c i a e i s t o pode s e r e n t e n d i d o p o i s
Rc e Re p a r a e s t a s m o l é c u l a s d i f e r e m a p e n a s de 3I"^"^ o que s i £

- l e
n i f i c a que deve h a v e r uma f o r t e p r e d i s s o c i a ç a o . A s m o l é c u l a s
r e s t a n t e s X e B r e KrCí , a p r e s e n t a m a ç ã o l a s e r e n e s t a s m o l é c u
l a s a p r e d i s s o c i a ç a o do e s t a d o i ó n i c o não e x i s t e , p o i s Rc e Re
d i f e r e m m a i s de 2001"^" .
A e f i c i ê n c i a da p r o d u ç ã o de e x c i m e r s também pode s e r
r e d u z i d a a t r a v é s da d e s a t i v a ç ã o do e s t a d o e x c i t a d o i ó n i c o p o r
á tomos d e g a s e s nobres, formando a s s i m e s p é c i e s t r i a t ó m i c a s . A e
x i s t ê n c i a de e s p é c i e s Ar2F e Kr2F f o i p o s t u l a d a p o r L o -
r e n t z ^ ^ p a r a e x p l i c a r a e x i s t ê n c i a de duas b a n d a s de e m i s s ã o a
d i c i o n a i s no s i s t e m a A r - K r - F 2 • E s t a s m o l é c u l a s s e r i a m f o r
madas p e l a r e a ç ã o :
K r F * + Kr + [Rg] ^^2^* ^
ou A r F * + A r + [Rg] -> Ar2F* + [Rg'
E s t a s m o l é c u l a s também só e x i s t e m no e s t a d o e x c i t a -
do ' i ó n i c o , formado p o r Rg2 e F na c o n f i g u r a ç ã o de um
t r i â n g u l o i s ó s c e l e s , e a e m i s s ã o n a s b a n d a s o b s e r v a d a s c o r r e s
pondem ã t r a n s i ç ã o d e s t e e s t a d o p a r a o e s t a d o f u n d a m e n t a l r e
p u l s i v o no q u a l e s t a s m o l é c u l a s s e quebram.
A s s i m no s i s t e m a l a s e r K r F , a p r e s s ã o p a r c i a l do Kr
deve s e r t a l que a f o r m a ç ã o do e s t a d o i ó n i c o s e j a f a v o r e c i d a e
a d e s a t i v a ç ã o do mesmo, a t r a v é s da r e a ç ã o a c i m a , s e j a e v i t a d a .
M i s t u r a s t í p i c a s d e s t e s g a s e s c o n t é m : 0 , 2 - 0,S% de F2. 5 - 1 0 %
de Kr e 90% de A r , onde e s t e u l t i m o é u s a d o como g á s " b u f -
f e r " " » .
A e f i c i ê n c i a dos l a s e r s de m o n o h a l e t o s de gases nobres
também é r e d u z i d a p o r a b s o r ç õ e s p a r a s í t i c a s d a s e s p é c i e s c r i a
d a s d u r a n t e a e x c i t a ç ã o da m i s t u r a de g a s e s . Uma e s p é c i e que
se forma na c a v i d a d e é o í o n n e g a t i v o X do h a l o g ê n e o e n v o l
v i d o . A m u i t o tempo se s a b e ^ ^ que e s t a e s p é c i e tem a l t a s e ç ã o

de c h o q u e no u l t r a - v i o l e t a p a r a a f o t o - n e u t r a l i z a ç a o do i o n .
Na v e r d a d e , q u a n t o m a i s p e s a d o o í o n , m a i o r ê a s e ç ã o de c h o
que e i s t o c o n t r i b u i p a r a que os s i s t e m a s A r F e K r F s e j a m os
m a i s e f i c i e n t e s , s endo os menos a f e t a d o s p o r e s t e p r o c e s s o .
De i g u a l i m p o r t â n c i a p a r a a a b s o r ç ã o da e m i s s ã o l a
s e r é a f o r m a ç ã o dos d í m e r o s Rg2 . E s t a s m o l é c u l a s s e formam
p e l a c o l i s ã o de t r ê s c o r p o s :
Rg" + Rg + [ R g ' ] ^ R g ¡ + [Rg'J
Do l i m i t e de á tomos s e p a r a d o s R g * ( ^ P ) + Rg('^S) , na
a u s ê n c i a de a c o p l a m e n t o s p i n - o r b i t a , surgem q u a t r o e s t a d o s mo
l e c u l a r e s : ^ Z * , , e ^I* , sendo que os d o i s p r i m e i -
u g u g ^ ^
r o s e s t a d o s s ã o l i g a d o s d e v i d o a urna l i g e i r a c o v a l ê n c i a e os
o u t r o s d o i s s ã o e s s e n c i a l m e n t e r e p u l s i v o s .
P a r a c o m p r e e n d e r o compor tamento d e s s a s m o l é c u l a s ,
m u i t o s c á l c u l o s foram f e i t o s , d e s d e c á l c u l o s H a r t r e e - F o c k 26 78
(HF) ' , p a s s a n d o p o r c á l c u l o s e n v o l v e n d o i n t e r a ç ã o de conf i_
g u r a ç ã o (Cl) ' '^^ ' ' , a t é c á l c u l o s u t i l i z a n d o a a p r o x i m a ç ã o de
58
d e n s i d a d e l o c a l p a r a o p o t e n c i a l de t r o c a . O r e s u l t a d o m a i s
i m p o r t a n t e , no que s e r e f e r e a o s l a s e r s de h a l e t o s de g a s e s no
b r e s é que a f o r t e t r a n s i ç ã o ^ Z * ^ T * , que c o r r e s p o n d e a u -
ma f o t o - d i s s o c i a ç ã o , o c o r r e no u l t r a - v i o l e t a ( 2 0 0 - 4 0 0 n m ) , e -
x a t a m e n t e na r e g i ã o onde o c o r r e a t r a n s i ç ã o l a s e r d o s h a l e t o s
de g a s e s n o b r e s . 83
P a r a a a v a l i a ç ã o d i r e t a do p r o b l e m a da absorção, Wadt
c a l c u l o u a s e ç ã o de c h o q u e de a b s o r ç ã o d e s t a s m o l é c u l a s p a r a
a l g u m a s t e m p e r a t u r a s e c o n c l u i u que os p i c o s d a s b a n d a s de a b
s o r ç ã o s e e n c o n t r a m em: 260 nm, 330 nm, 350 nm e 390 nm p a r a
N e ^ , A r 2 , Kr2 e Xe2 r e s p e c t i v a m e n t e . E s t a s c o n c l u s õ e s s e r v e m
de c r i t é r i o p a r a a e s c o l h a do g á s n o b r e que é u t i l i z a d o como

"buffer", de tal forma que o mínimo de absorção ocorra no com
primento de onda do laser.
Mais adiante, daremos maiores detalhes acerca das con
figurações eletrônicas destas moléculas.

- C A P Í T U L O I I -
SISTEMAS VE MUITAS PARTÍCULAS
I I - l . APROXIMAÇÃO DE HARTREE-FOCK
A d e s c r i ç ã o e x a t a do c o m p o r t a m e n t o de s i s t e m a s de mui
t a s p a r t í c u l a s ê um p r o b l e m a i n s o l ú v e l , mesmo c l a s s i c a m e n t e .
Quando p a s s a m o s p a r a a r e g i ã o de d o m í n i o da m e c â n i c a q u â n t i c a ,
não podemos e s p e r a r i r m u i t o l o n g e s e p o d e r o s a s a p r o x i m a ç õ e s
não forem f e i t a s .
No c a s o dos f enômenos r e l a t i v o s ã e s t r u t u r a e l e t r ô n i .
c a , p a r a os q u a i s os e f e i t o s r e l a t i v í s t i c o s s ã o d e s p r e z í v e i s ,
o p r o b l e m a c e n t r a l ê d e t e r m i n a r a s s o l u ç õ e s da e q u a ç ã o de S c h r o
d i n g e r i n d e p e n d e n t e do tempo p a r a um s i s t e m a de n ú c l e o s e e l é
t r o n s , dada p o r :
H ^(rS) = ^e ^"^^'^^ (.r,t) = E Y C r . ^ ) ( I I . 1 )
onde Tj^ e T^ s ã o os o p e r a d o r e s de e n e r g i a c i n é t i c a t o t a l d o s
n ú c l e o s e dos e l é t r o n s r e s p e c t i v a m e n t e , e V ( r , ] ^ ) é o o p e r a -
dor de e n e r g i a p o t e n c i a l de i n t e r a ç ã o e n t r e t o d a s a s p a r t í c u
l a s , s e n d o que r r e p r e s e n t a a s c o o r d e n a d a s dos e l é t r o n s e
a s c o o r d e n a d a s d o s n ú c l e o s .
L e v a n d o - s e em c o n t a que a m a s s a dos n ú c l e o s é aproxi^
madamente 10^ v e z e s m a i o r que a massa dos e l é t r o n s , e a s s i m tem
uma v e l o c i d a d e m u i t o menor que e s t e s , é p o s s í v e l d e s a c o p l a r o
movimento d e s t e s d o i s t i p o s de p a r t í c u l a s e p a r a c a d a d i s p o s i
ç ã o dos n ú c l e o s p r o c u r a r - s e " i n s t a n t a n e a m e n t e " uma s o l u ç ã o p a
r a a f u n ç ã o de onda t o t a l dos e l é t r o n s , d a í e s t e t r a t a m e n t o ser
c o n h e c i d o como a p r o x i m a ç ã o a d i a b á t i c a ^ .
E s t a i d é i a f o r m a l i z a d a nos l e v a a r e s o l v e r s e p a r a d a -

mente d u a s h a m i l t o n i a n a s , uma s o p a r a os e l é t r o n s , na q u a l a
p o s i ç ã o d o s n ú c l e o s é f i x a , e o u t r a so p a r a os n ú c l e o s , na q u a l
a e n e r g i a e l e t r ô n i c a t o t a l e n t r a como um p o t e n c i a l e f e t i v o . As
s im a s o l u ç ã o de ( I I . 1 ) é o p r o d u t o das s o l u ç õ e s d e s t a s d u a s
e q u a ç õ e s d e s a c o p l a d a s .
D e n t r o do e s p í r i t o d e s t a a p r o x i m a ç ã o , o h a m i l t o n i a n o
de um s i s t e m a de N e l é t r o n s no campo de M n ú c l e o s f i x o s é
o s e g u i n t e
H = - I V . 2 - ^ l 2 . l l -J— ( I I . 2 ) i = l i = l y = l r . ^ 1=1 j = l r . .
em u n i d a d e s a t ô m i c a s . A q u i r . = | r . - ^ | . onde ^ é o
v e t o r p o s i ç ã o do y - é s i m o n ú c l e o de c a r g a , r^ é a c o o r d e
n a d a do i - é s i m o e l é t r o n e r ^ j = | r ^ - r ^ | .
^ - 28 Em 1928 H a r t r e e p r o p ô s uma s o l u ç ã o d e s t e p r o b l e m a
p a r a o c a s o a t ô m i c o , e c o n s i s t i a em c o n s i d e r a r c a d a e l é t r o n s u
j e i t o a um p o t e n c i a l médio e s f é r i c o d e v i d o ao n ú c l e o e a o s d e
m a i s e l é t r o n s . N e s t a é p o c a j á e x i s t i a m s u f i c i e n t e s e v i d ê n c i a s
e x p e r i m e n t a i s de que t o d o s o s e l é t r o n s podiam s e r c o n s i d e r a d o s
74
s u j e i t o s a um campo c e n t r a l . A s s i m p o d i a - s e r e s o l v e r uma "e
q u a ç ã o de S c h r o d i n g e r " p a r a c a d a e l é t r o n e a a u t o - f u n ç ã o do ha
m i l t o n i a n o ( I I . 2 ) s e r c o n s i d e r a d a como p r o d u t o das f u n ç õ e s de
onda de t o d o s o s e l é t r o n s .
A s e q u a ç õ e s m o n t a d a s p o r H a r t r e e de m a n e i r a i n t u i t i
v a , também podem s e r d e r i v a d a s de um p r i n c í p i o v a r i a c i o n a l , no
q u a l o v a l o r e s p e r a d o do h a m i l t o n i a n o ( I I . 2 ) ê m i n i m i z a d o v a -
r i a n d o - s e a s f u n ç õ e s de onda de um e l é t r o n . E n t r e t a n t o e s t a ma
n e i r a de t r a t a r o p r o b l e m a não dâ c o n t a do p r i n c í p i o de e x c l u
s ã o de P a u l i . P a r a que e s t e p r i n c í p i o s e j a o b e d e c i d o é n e c e s
s á r i o que os e l é t r o n s s e j a m t r a t a d o s como f é r m i o n s de a c o r d o
com o p o s t u l a d o de a n t i s s i m e t r i a de D i r a c .

Slater em 1929''^ propõe que a função de onda de um
sistema de fêrmions seja descrita por urna combinação linear an
tissimetrizada de produtos de funções que também dependam do
spin. Tal função antissimétrica pode ser escrita na forma de
um determinante.
1
N 1*
N
conhecido como determinante de Slater, onde
N
4» N
N N
C U . 3 )
= * (r ) n (a ) a a y ex y
(II.4)
sendo que a designa os diferentes spin-orbitais e r ^ e
as coordenadas espaciais e de spin do y-esimo elétron. Além
disto estes spin-orbitais são linearmente independentes e po
dem ser ortonormalizados.
Em 1930^^ Fock propôs a aplicação do principio varia
cional ã função antissimétrica í> , da mesma forma que se po
dia fazer para deduzir as equações de Hartree. Definindo:
< $ H $ > ^HF
(II.5)
e impondo a ortonormalização dos spin-orbitais
= 6 a6
(II.6)
podemos escrever a energia de Hartree-Fock como:
^HF I "a a
<t^i^ "i K^h^
I N S T I T U T O D E P E S Q U I S A S E N E R G É T I C A S E N U C L E A R E S
1. P . E . N .

-¿1-
+ — I — y N N .
2 a 3 " ^ 12
- 6
" 12
( I I . 7 )
onde M
l -M = l r
( I I . 8 )
l y
e n^ é a p o p u l a ç ã o do a - e s i m o e s t a d o (n^ = O ou 1 ) .
O p r i n c i p i o v a r i a c i o n a l impõe que o m e l h o r p r o d u t o
a n t i s s i m é t r i c o t o r n a Ej^p e x t r e m o , a s s i m , f a z e n d o v a r i a ç õ e s
a r b i t r a r i a s e i n d e p e n d e n t e s n o s 'í'jj's da e x p r e s s ã o ( I I . 7) e im
pondo que ^^^ij: = O o b t e m o s :
«1 ^ l "3 * 6 ( Í 2 ^ — • E ' ^ 2 Í ^^^2
- l 6
12
^12 ( I I . 9 )
onde é o m u l t i p l i c a d o r de L a g r a n g e c o r r e s p o n d e n t e ao v í n
c u l o ( I I . 6 ) .
E s t a e q u a ç ã o é c o n h e c i d a como e q u a ç ã o de H a r t r e e - F o c k
e d i f e r e da e q u a ç ã o de H a r t r e e a p e n a s p e l a e x i s t e n c i a do ú l t i
mo termo do l a d o e s q u e r d o .
Ê p o s s í v e l e s c r e v e r e s t a e q u a ç ã o na forma de urna e -
q u a ç ã o de S c h r o d i n g e r p a r a um e l é t r o n como:
E „ •A(I'J) (11.10)
se d e f i n i r m o s :

M
^12 CU.11)
^ X H F ^ ^ l ^ = <í>„Cr,) < Cr,) ( 1 1 . 1 2 )
A e x p r e s s ã o ( 1 1 . 1 1 ) é f a c i l m e n t e i n t e r p r e t a d a como o
p o t e n c i a l c o u l o m b i a n o g e r a d o p e l o s n ú c l e o s e e l é t r o n s do s i s t e
ma, e f o i e s t a s i m p l e s i n t e r p r e t a ç ã o que l e v o u H a r t r e e a e s c r e
v e r e s t a mesma e q u a ç ã o , e x c e t o p e l o ú l t i m o t e r m o . E n t r e t a n t o a
e x p r e s s ã o ( 1 1 . 1 2 ) é um termo que não p o s s u i a n á l o g o c l á s s i c o e
sua o r i g e m é a i m p o s i ç ã o de a n t i s s i m e t r i a da f u n ç ã o de onda t o
t a l . E s t e termo é c o n h e c i d o como p o t e n c i a l de t r o c a ( e x c h a n
g e ) , e sô o c o r r e e n t r e e l é t r o n s de mesmo s p i n .
Na d e d u ç ã o d a s e q u a ç õ e s de H a r t r e e - F o c k , a l g u m a s a s
s e r ç õ e s f o r a m f e i t a s i m p l i c i t a m e n t e . A p r i m e i r a ê que o hamil_
t o n i a n o H não contém o p e r a d o r e s de s p i n , podendo a f u n ç ã o de
onda s e r e s c r i t a como em ( I I . 4 ) . A s e g u n d a é que a p a r t e o r b i
t a l da f u n ç ã o ( I I . 4 ) i n d e p e n d e do s p i n , e n t r e t a n t o podemos o b
s e r v a r n a e x p r e s s ã o ( 1 1 . 1 2 ) que a i n t e r a ç ã o e x p r e s s a p o r e s t e
termo s5 e x i s t e e n t r e e l é t r o n s de mesmo s p i n . A s s i m , quando
t r a t a m o s s i s t e m a s de camadas a b e r t a s , onde o número de e lé trons
com s p i n "up" é d i f e r e n t e do número de e l é t r o n s com spin "down",
o p o t e n c i a l V^j^p depende do s p i n da p a r t í c u l a e p o r t a n t o a
s o l u ç ã o <t>^ também. A e q u a ç ã o ( 1 1 . 1 0 ) deve p o r t a n t o s e r e s
c r i t a da s e g u i n t e f o r m a :
(II.13a)

( I I . 1 3 b )
sendo que a g o r a f i c a e v i d e n c i a d o que o p o t e n c i a l de t r o c a , a
p a r t e o r b i t a l da f u n ç ã o de onda e o a u t o - v a l o r de e n e r g i a d e
pendem do s p i n .
Quando o numero de e l é t r o n s com s p i n "up" ê i g u a l ao
número de e l é t r o n s com s p i n "down", o p o t e n c i a l V^ j p assume
um ú n i c o v a l o r p o n t u a l m e n t e , e p a r a c a d a s o l u ç ã o 4) de (11.10),
c o r r e s p o n d e n t e ao a u t o - v a l o r , e x i s t e m d o i s e l é t r o n s : um
com s p i n "up" e o u t r o com "down".
O e f e i t o d e s t e p o t e n c i a l de t r o c a f r e q u e n t e m e n t e ê
chamado de c o r r e l a ç ã o e s t a t í s t i c a d e v i d o ao f a t o que os e l é
t r o n s obedecem ã e s t a t í s t i c a de F e r m i - D i r a c .
Na e q u a ç ã o de H a r t r e e - F o c k o a u t o - v a l o r é i g u a l
ã d i f e r e n ç a de e n e r g i a t o t a l e n t r e s i s t e m a s de N e l é t r o n s ,
com <() o c u p a d o , e N - 1 e l é t r o n s , com ()) d e s o c u p a d o , sem
que h a j a um r e a r r a n j o dos o r b i t a i s . E s t e ê o t e o r e m a do K o o p -
46 mans , que e e x p r e s s o p o r :
O l h a n d o p a r a as e q u a ç õ e s ( 1 1 . 1 0 ) e ( 1 1 . 1 3 ) vemos que
p r e c i s a m o s c o n h e c e r e V^j^p p a r a r e s o l v ê - l a s e o b t e r
e , e n t r e t a n t o a s e q u a ç õ e s ( 1 1 . 1 1 ) e ( 1 1 . 1 2 ) mostram que
p a r a d e t e r m i n a r e V^^^p p r e c i s a m o s c o n h e c e r t o d a s a s s o l u
ç õ e s < l )o ' s . E s t e p r o b l e m a é r e s o l v i d o a t r a v é s de um p r o c e s s o
i t e r a t i v o a p a r t i r de um p o t e n c i a l t e n t a t i v a i n t r o d u z i d o na e -
q u a ç ã o ( 1 1 . 1 0 ) . Com i s t o d e t e r m i n a m - s e as s o l u ç õ e s a p r o x i m a -
das <í>Qf's , que p o r s u a v e z v ã o g e r a r n o v o s p o t e n c i a i s V^, e
^ X H F novamente i n t r o d u z i d o s em ( 1 1 . 1 0 ) . E s t e p r o c e s s o

ê r e p e t i d o a t e que um c r i t é r i o de c o n v e r g ê n c i a p r é - e s t a b e l e c i -
do s e j a o b e d e c i d o . E s t e p r o c e d i m e n t o é c o n h e c i d o como o m é t o
do do campo a u t o - c o n s i s t e n t e .
Cabe a i n d a d e s t a c a r que o d e t e r m i n a n t e de S l a t e r uti_
l i z a d o p a r a a d e t e r m i n a ç ã o d a s e q u a ç õ e s de H a r t r e e - F o c k não é
a s o l u ç ã o m a i s g e r a l p a r a um s i s t e m a de m u i t o s e l é t r o n s . O u s o
de um ú n i c o d e t e r m i n a n t e g e r a e q u a ç õ e s n a s quais nem todas as cor
r e l a ç õ e s e n t r e os m o v i m e n t o s e l e t r ô n i c o s i n d i v i d u a i s e s t ã o i n
c l u i d o s . A p e n a s a c o r r e l a ç ã o e s t a t í s t i c a , que t r a d u z o p r i n c í
p i o de e x c l u s ã o de P a u l i , e s t á i n c l u í d a , e n t r e t a n t o nenhuma cor
r e l a ç ã o e n t r e e l é t r o n s de s p i n s o p o s t o s a p a r e c e . E s t e t i p o de
e f e i t o pode s e r l e v a d o em c o n t a quando a s o l u ç ã o t e n t a t i v a p a
r a o método v a r i a c i o n a l de H a r t r e e - F o c k é uma c o m b i n a ç ã o l i
n e a r de d e t e r m i n a n t e s de S l a t e r ' ' ^ . E s t e t r a t a m e n t o é d e s c r i t o
como i n t e r a ç ã o de c o n f i g u r a ç ã o ( C l ) e f o r n e c e o v a l o r e x a t o da
e n e r g i a , a menos de e f e i t o s r e l a t i v í s t i c o s , no c a s o de se com
b i n a r i n f i n i t o s d e t e r m i n a n t e s .
I I - 2 . APROXIMAÇÃO Xg PARA O P O T E N C I A L DE TROCA
Embora e s t e esquema s i m p l i f i q u e c o n s i d e r a v e l m e n t e o
t r a t a m e n t o de s i s t e m a s de m u i t a s p a r t í c u l a s , o c á l c u l o do p o
t e n c i a l de t r o c a no método de H a r t r e e - F o c k é m u i t o t r a b a l h o s o ,
j u s t a m e n t e p o r s e r um termo não l o c a l . Além d i s t o , os e f e i t o s
de c o r r e l a ç ã o não s ã o i n c l u í d o s n e s t e f o r m a l i s m o . E s t e s m o t i
v o s tem l e v a d o m u i t o s a u t o r e s a proporem a u t i l i z a ç ã o de um po
t e n c i a l e f e t i v o l o c a l , ao i n v é s do p o t e n c i a l de t r o c a d a s e q u a
ç õ e s de H a r t r e e - F o c k . Não d e s c r e v e r e m o s a q u i t o d a s as a p r o x i
m a ç õ e s , mas a p e n a s a q u e l a s que deram o r i g e m ã a p r o x i m a ç ã o X a .
Em 1930 , Dirac''^''" c a l c u l o u o termo de e x c h a n g e p a r a um

g á s de e l é t r o n s l i v r e s no e s t a d o f u n d a m e n t a l , ou s e j a , no z e r o
a b s o l u t o . N e s t a s i t u a ç ã o , a p a r t e o r b i t a l da f u n ç ã o de onda
dos e l é t r o n s é dada p o r :
1 ^K'^ Ã7T ^
( 1 1 . 1 5 )
onde V é o v o l u m e o c u p a d o p e l o g á s e p a r a c a d a 4) temos um
e l é t r o n com s p i n "up" e o u t r o com s p i n "down". Os ^ q j ' s p o s
s í v e i s s ã o t o d o s a q u e l e s m e n o r e s , em m ó d u l o , que o momento de
F e r m i , que pode s e r d e f i n i d o em t e r m o s da d e n s i d a d e do g á s c o
mo :
37T 2 N
V J
Sir" e c a d a um d e s t e s v e t o r e s k ^ ' s o c u p a um v o l u m e i g u a l a ^
no e s p a ç o r e c í p r o c o .
S u b s t i t u i n d o a e x p r e s s ã o ( 1 1 . 1 5 ) em ( 1 1 . 1 2 ) o b t e m o s :
12 ( 1 1 . 1 6 )
N S u b s t i t u i n d o a \ p o r
3=1
do ambas a s i n t e g r a i s o b t e m o s :
Xp = - — kp F(TI) b ir
V
onde
n = a
F(n) 4 - . J L ^ . n ^ 4n
1 n
d KO e r e a l i z a n p —
( 1 1 . 1 7 )
( 1 1 . 1 8 a )
( I I . 1 8 b )
Em 1951 S l a t e r p r o p ô s uma a p r o x i m a ç ã o p a r a o p o t e n
c i a l de t r o c a ' ' ^ que s e t o r n o u c l á s s i c a p e l a s s i m p l i f i c a ç õ e s i n
t r o d u z i d a s . B a s e i a - s e n a s s e g u i n t e s h i p ó t e s e s .
1) A d e n s i d a d e e l e t r ô n i c a de s i s t e m a s não h o m o g ê n e o s , t a i s c o -

mo á t o m o s , s o l i d o s , e t c . . . , pode s e r a p r o x i m a d a p e l a d e n s i
dade e l e t r ô n i c a de um g á s de e l é t r o n s l i v r e s .
2) Todos os e l é t r o n s do s i s t e m a e s t ã o s u b m e t i d o s a um mesmo po
t e n c i a l de t r o c a , ou s e j a , a um p o t e n c i a l de t r o c a m é d i o .
O p o t e n c i a l de t r o c a médio de um g á s homogêneo é:
d^k
( 1 1 . 1 9 )
a
e p o r t a n t o , p e l a p r i m e i r a h i p ó t e s e t e m o s :
= - 6 L 8TT
P ( r ) ( 1 1 . 2 0 )
onde P ( r ) é a d e n s i d a d e l o c a l do s i s t e m a não homogêneo .
E s t a a p r o x i m a ç ã o pode s e r a p l i c a d a a s i s t e m a s e l e t r ô
n i c o s com número de e l é t r o n s de s p i n "up" i g u a l ao número de e
l é t r o n s com s p i n "down". Quando e s t e s números s ã o d i f e r e n t e s ,
podemos a p r o x i m a r o p o t e n c i a l de t r o c a do s i s t e m a ao de d o i s
g a s e s homogêneos com s p i n s d i f e r e n t e s , d e f i n i n d o d o i s momentos
de F e r m i . r o . -.1/3
( I I . 2 1 a ) 'F+ Ó T T P ^ ( r )
Ó T T ^ P ^ ( r )
- | l / 3
n l / 3 ( I I . 2 1 b )
e a s s i m obtemos
- 6
- 6
47R ^
4TT
n l / 3
1/3
Quando h o u v e r e q u i l í b r i o de s p i n s t e r e m o s :
P ^ ( r ) = P ^ ( r ) P ( r )
( I I . 2 2 a )
( I I . 2 2 b )
( 1 1 . 2 3 )

' • ^ F 3Tr^ p(r)
1/3 ( 1 1 . 2 4 )
I n t r o d u z i n d o a a p r o x i m a ç ã o de S l a t e r n a s e q u a ç õ e s
( 1 1 . 1 3 ) , obtemos a s e q u a ç õ e s de H a r t r e e - F o c k - S l a t e r com p o l a r ^
z a ç ã o 4 E s p i n ,
- n l / 3 1
- + Y^ir^) - 6 4Tr
( I I . 2 5 a )
- V^" + V ^ ( r p - 6
n l / 3
4Tr P , ( r , )
( I I . 2 5 b )
E s t a e x p r e s s ã o f a c i l i t a m u i t o o c a l c u l o das f u n ç õ e s
de onda e d a s e n e r g i a s , e n t r e t a n t o v e r i f i c o u - s e que os v a l o r e s
de e n e r g i a de s i s t e m a s a t ô m i c o s c a l c u l a d o s d e s t a m a n e i r a eram
m a i o r e s que os v a l o r e s f o r n e c i d o s p e l a e q u a ç ã o de H a r t r e e - F o c k .
O u t r a a p r o x i m a ç ã o l o c a l , que s u r g i u l o g o a p ó s ã de
25 44
S l a t e r , e a de G a s p a r - K o h n - S h a m ' , b a s e a d a também em g a s e s
h o m o g ê n e o s , mas com um p r o c e d i m e n t o d i f e r e n t e . A e x p r e s s ã o ob
t i d a p o r e l e s p a r a o p o t e n c i a l de t r o c a , também pode s e r o b t i
da s e a a p r o x i m a ç ã o l o c a l do g â s de e l é t r o n s l i v r e s é f e i t a na
e x p r e s s ã o da e n e r g i a t o t a l de H a r t r e e - F o c k ( I I . 7) e d e p o i s é
f e i t a a v a r i a ç ã o das a u t o - f u n ç õ e s ' ^ ^ p a r a a o b t e n ç ã o da e -
q u a ç ã o de a u t o - v a l o r e s . O p o t e n c i a l de t r o c a de G a s p a r - K o h n -
Sham é:
GKS 3 ^ S ( 1 1 . 2 6 )
E n t r e t a n t o o s c á l c u l o s e f e t u a d o s com e s t a a p r o x i m a -
ç ã o também d i s c o r d a m d o s v a l o r e s e x p e r i m e n t a i s e d o s c á l c u l o s
H a r t r e e - F o c k , so que e s t a a p r o x i m a ç ã o f o r n e c e v a l o r e s de e n e r -

g i a menores que os v a l o r e s de H a r t r e e - F o c k .
O p r o x i m o p a s s o e n t ã o , f o i t e n t a r u s a r um termo de
t r o c a i n t e r m e d i á r i o e n t r e Xg e ^gj^g do t i p o :
Xa = a Xg ( 1 1 . 2 7 )
onde a é um p a r â m e t r o podendo a s s u m i r v a l o r e s e n t r e - y - e
7 7
1. Snow em 1964, num c a l c u l o de b a n d a s de c o b r e , mostrou que
u s a n d o - s e um a = p o d i a o b t e r r e s u l t a d o s m u i t o m a i s p r o x i
mos dos v a l o r e s e x p e r i m e n t a i s . A s s i m s u r g i r a m v á r i o s c r i t é
r i o s p a r a d e t e r m i n a r o v a l o r de a a s e r u t i l i z a d o e o s p r i n
c i p a i s s ã o os s e g u i n t e s : 2
1) B e r r e n d o e G o c i n s k y . E s t e método c o n s i s t e em d e s c o b r i r o
a , p a r a c a d a e l e m e n t o , que s a t i s f a ç a o t e o r e m a do V i r i a l
p a r a a e n e r g i a t o t a l de H a r t r e e - F o c k . E s t e c r i t é r i o s u r g i u
a p a r t i r da o b s e r v a ç ã o que a s f u n ç õ e s c a l c u l a d a s p e
l a s e q u a ç õ e s de H a r t r e e - F o c k s a t i s f a z e m o t eorema do v i r i a l ,
o que não a c o n t e c e n e c e s s a r i a m e n t e quando s e u s a a s a p r o x i
mações de S l a t e r ou G a s p a r - K o h n - S h a m . A s s i m , d e f i n i r a m a
de modo que a s f u n ç õ e s de onda o b t i d a s p o r e s t a a p r o x i m a ç ã o
s a t i s f i z e s s e m a r e l a ç ã o : < V ( X a ) > = - 2 < T(Xa) > ( 1 1 . 2 8 )
41 -
2) Kmetko . O b t e v e o p a r â m e t r o a m i n i m i z a n d o a e n e r g i a t o
t a l de H a r t r e e - F o c k em f u n ç ã o de a . I s t o é f e i t o i n t r o d u -
z i n d o - s e o s o r b i t a i s 'l' Cot) , o b t i d o s p e l o método Xo , na
e x p r e s s ã o ( I I . 7) e a s s i m a c h a r o v a l o r de a que t o r n a E^^p
m í n i m o .
6 7
3) S c h w a r z . E s t e c r i t é r i o d e t e r m i n a o v a l o r de a impondo
que a e n e r g i a t o t a l o b t i d a p e l a a p r o x i m a ç ã o Xa s e j a i g u a l
ã e n e r g i a t o t a l de H a r t r e e - F o c k . Convém n o t a r que e s t e p r o

- Z Y -
C E D I M E N T O D E P E N D E D A E X I S T Ê N C I A D E U M C A L C U L O A T Ô M I C O D O TI^
P O H A R T R E E - F O C K . S C H W A R Z B A S E O U - S E E M C Á L C U L O S ATÔMICOS £EI^
T O S P O R M A N N ^ ^ P A R A D E T E R M I N A R E S T E P A R Â M E T R O P A R A U M A G R A N
D E Q U A N T I D A D E D E Á T O M O S .
O B S E R V A - S E Q U E O V A L O R D E A V A R I A N Ã O S 5 D E E L E M E N
T O P A R A E L E M E N T O , M A S T A M B É M C O M A I O N I C I D A D E E A C O N F I G U R A Ç Ã O
D O S Á T O M O S . I S T O T R Á S U M S É R I O P R O B L E M A Q U A N D O Q U E R E M O S A P L I
C A R A A P R O X I M A Ç Ã O X A P A R A M O L É C U L A S O U S Ó L I D O S , P O I S É IMPOS^
S Í V E L S E D E T E R M I N A R " A P R I O R I " Q U A L S E R Á A I O N I C I D A D E D E U M E -
L E M E N T O AO P A R T I C I P A R D E U M A L I G A Ç Ã O Q U Í M I C A .
E S T E A S P E C T O T A M B É M F O I I N V E S T I G A D O P O R S C H W A R Z ^ ^ E
C O N C L U I U Q U E P A R A D I F E R E N T E S C O N F I G U R A Ç Õ E S A T Ô M I C A S A S V A R I A
Ç Õ E S N O V A L O R D E A N Ã O U L T R A P A S S A M A T E R C E I R A C A S A D E C I M A L .
A S S I M P O D E S E R J U S T I F I C A D O Q U E O Á T O M O C A R R E G U E O S E U A P A R A
O S Ó L I D O O U M O L É C U L A .
C O M O V E R E M O S M A I S A D I A N T E , E S T A A P R O X I M A Ç Ã O T E M U M A
P R O F U N D A I N F L U Ê N C I A E M N O S S O S C Á L C U L O S .

- C A P Í T U L O I I I -
O UtTOVO CELULAR I /ARIÂCIONAL (MCI/)
III-l. A FORMULAÇÃO ORIGINAL
No capítulo anterior mostramos como os problemas de
cálculo de estrutura eletrônica podem ser reduzidos ã solução
da equação de Hartree-Fock. No caso atômico,os potenciais que
entram nas equações de auto-valores são esférico simétricos.
Isto traz uma certa simplicidade no tratamento deste problema,
podendo a parte orbital da função de onda de uma partícula ser
escrita como o produto de uma função radial por uma harmônica
esférica.
Quando passamos a estudar moléculas e solidos, nos
defrontamos com alguns problemas adicionais, por exemplo, como
descrever os potenciais médios aos quais os elétrons estão su
jeitos ou como descrever as funções de onda. As diferentes a-
proximações para contornar estas questões, deram origem aos mê
todos de cálculo hoje existentes.
O Método Celular Variacional (MCV) proposto por Fer-
20 21 - -reira e Leite ' constitui-se numa versão do método celular
4 7 88 de Wigner-Seitz-Slater . Assim como sua formulação original ,
baseia-se na decomposição do espaço molecular ou cristalino em
células, cada uma circundando um átomo ou uma região intersti
cial. Dentro de cada uma destas células o verdadeiro poten
cial é aproximado por sua média esférica em relação ao centro
da célula, o que simplifica a solução da equação de onda de um
elétron. A diferença em relação ã formulação original é que a
gora o problema da condição de contorno das funções de onda so
bre as células é reformulado de forma variacional.

A flexibilidade da escolha das células no MCV contri^
bui para urna representação mais realista do espaço molecular
ou cristalino, podendo superar as dificuldades que a aprocima-
ção muffin-tin para o potencial, utilizada por outros méto-
4 0 4 3
dos ' , acarreta em sistemas cristalinos nao compactos ou em
sistemas moleculares onde a região de potencial constante é mui
to grande.
No que se refere ã sua aplicação a moléculas, atual
mente a auto-consistência implantada no método S 5 se aplica a
sistemas diatómicos, entretanto não existe, em princípio, ne
nhuma limitação quanto ã sua extensão para sistemas poliatómi
cos .
Nas moléculas diatómicas as células são construídas
de tal forma que nos possibilite escrever o potencial de um e-
létron como sendo esférico simétrico dentro de cada uma delas,
sem que nos afastemos muito do potencial verdadeiro.
Em princípio, o MCV admite formas arbitrarias para
as células, entretanto os cálculos podem ser bastante simplifi_
cados se as superfícies que limitam as células forem planas ou
esféricas.
Uma análise do comportamento do potencial em diferen
tes regiões do espaço pode servir de guia para a construção des
tas células. Em regiões próximas dos núcleos atómicos, pode-
-se considerar que o elétron está sujeito a um potencial cen
tral com origem nos respectivos núcleos. Já nas regiões dis
tantes dos núcleos atômicos, o potencial sentido por um elé
tron não é muito diferente do potencial devido a uma carga efe
tiva situada no centro de massa da molécula.
Este raciocínio conduz ã construção de três células,
duas em torno dos átomos e uma terceira abrangendo o resto do
" • 1. P . E . N .

e s p a ç o . A f o r m a d e s t a s c é l u l a s é a p r e s e n t a d a na F i g u r a I I I - l
FIGURA JÍJ-1. Viviòao do e s p a ç o molzcLilafi para mol2.cu.la0 dia
tómicas .
I n t e r n a m e n t e à e s t a s c é l u l a s s ã o d e f i n i d a s a l g u m a s
r e g i õ e s l i m i t a d a s p o r e s f e r a s que podem ou não t a n g e n c i a r a s
c a l o t a s , onde a l é m do p o t e n c i a l a d e n s i d a d e de c a r g a também é
f e i t a e s f é r i c o s i m é t r i c a . Na r e g i ã o e x t e r n a a e s t a s e s f e r a s
i n s c r i t a s a d e n s i d a d e e l e t r ô n i c a ê f e i t a c o n s t a n t e .
Na d e f i n i ç ã o das d i m e n s õ e s da c é l u l a , os p a r â m e t r o s
a-j , a2 , b^ , b2 e p podem v a r i a r l i v r e m e n t e , e n t r e t a n t o e s
t e e x c e s s o de l i b e r d a d e t o r n a d i f í c i l um e s t u d o s o b r e a c o n v e r
g ê n c i a da e n e r g i a . Como s u g e r i d o p e l o s a u t o r e s ^ ^ , o p l a n o
se p o s i c i o n a de t a l forma que a d i s t â n c i a i n t e r a t ô m i c a s e j a di^
v i d i d a p r o p o r c i o n a l m e n t e ao r a i o c o v a l e n t e dos á tomos e a d i s
t â n c i a p pode s e r e s c o l h i d a de modo a t o r n a r mínima a e n e r
g i a .
F e i t a a a p r o x i m a ç ã o p a r a a d e s c r i ç ã o do p o t e n c i a l , o
p r o b l e m a se resume em r e s o l v e r a e q u a ç ã o de S c h r o d i n g e r d e n t r o
de c a d a uma d e s t a s r e g i õ e s e impor a c o n t i n u i d a d e da f u n ç ã o de
(D

- .5 O -
onda nas f r o n t e i r a s d a s c é l u l a s , o que no MCV se resume num
s5 p r o b l e m a . A d o t a - s e a s e g u i n t e e x p r e s s ã o v a r i a c i o n a l p a r a o
a u t o - v a l o r de e n e r g i a de urna p a r t í c u l a :
I i
3 * d r . d). (j).
1 ^1 ^1 = 1 i
d ^ r . (})! C - + V) <)). +
l
d S . . ((j). - <|).) O <i>* - 9 4>*) i j ^^1 ^ n^j n^i^
ds.. (C^. - * J ) 0 ^ * J - , ( I I I . l )
onde as i n t e g r a i s em d r^ s ã o i n t e g r a i s de vo lume d e n t r o de
c a d a uma d a s c é l u l a s e s p e c i f i c a d a s p e l o í n d i c e i , e a s i n t e
g r a i s em d S ^ j , s ã o i n t e g r a i s n a s s u p e r f í c i e s de s e p a r a ç ã o en
t r e a s c é l u l a s i e j . Os t e r m o s de s u p e r f í c i e tem no i n t e
g r a n d o e x p r e s s õ e s como ^.^^^ ' Q^e s ã o d e r i v a d a s n o r m a i s da
f u n ç ã o de onda s o b r e a s u p e r f í c i e S ^ j , d i r i g i d a p a r a f o r a da
c é l u l a i . N o t a - s e também que e s t e f u n c i o n a l é e s c r i t o em t e r
mos de u n i d a d e s a t ô m i c a s , onde -íí = 1 , m = - y - e e = /2 . _ *
Quando f a z e m o s v a r i a ç õ e s 5^.^ na f u n ç ã o de onda den
t r o de uma c é l u l a k , obtemos que o f u n c i o n a l e da e q u a ç ã o
( I I I . 1 ) é e x t r e m o s e :
(- + V) <j>. = e (j>. ( I I I . 2 )
* i S . .
( I I I . 3 )
n ^ i - 8 ( | ) .
n ^ j S . . ' 1 )
ou s e j a , a e q u a ç ã o de S c h r o d i n g e r deve s e r o b e d e c i d a d e n t r o de
c a d a uma d a s c é l u l a s e a f u n ç ã o de onda deve s e r c o n t í n u a s o
b r e a f r o n t e i r a das c é l u l a s . A s s i m r e s o l v e r a e q u a ç ã o de S c h r o
d i n g e r é e q u i v a l e n t e a e n c o n t r a r a f u n ç ã o de onda que t o r n a o

f u n c i o n a l e um e x t r e m o .
É um p r o c e d i m e n t o b a s t a n t e g e n é r i c o e x p a n d i r a f u n
ç ã o de onda <t>^ d e n t r o de c a d a c é l u l a em t e r m o s de um c o n j u n
t o c o m p l e t o de f u n ç õ e s e impor que a f u n ç ã o de onda a d e q u a d a é
a q u e l a c u j o s c o e f i c i e n t e s de e x p a n s ã o fazem e da equação ( I I I . l )
um e x t r e m o . No MCV, d e n t r o de c a d a c é l u l a o p o t e n c i a l é e s f é
r i c o s i m é t r i c o , p o r t a n t o p o d e - s e f a z e r uma e x p a n s ã o em t e r m o s
de h a r m ô n i c a s e s f é r i c a s c e n t r a d a s em c a d a uma d a s c é l u l a s , a s
s im
com. f ( r - ) R , ° ( r , ) Y , ( í , ) ( I I I . 5 )
onde o í n d i c e X i n d i c a o p a r de í n d i c e s S, e m d a s h a r m ô n i
c a s e s f é r i c a s ^ ¿ ( ^ i ^ ^ ^ ^ s o l u ç ã o da p a r t e r a d i a l da
e q u a ç ã o de S c h r o d i n g e r
AL . V ( r ) . dr r
e o
r ( r )
( I I I . 6 )
p a r a a e n e r g i a e^ q u e , a n t e s de se e s p e c i f i c a r a s c o n d i ç õ e s
de c o n t o r n o , pode a s s u m i r q u a l q u e r v a l o r , d e s d e que R^^ s e j a
r e g u l a r na o r i g e m e no i n f i n i t o .
P a r a e n c o n t r a r o s c o e f i c i e n t e s A ^ ^ , devemos subs t i^
t u i r a e x p r e s s ã o ( I I I . 4 ) na e q u a ç ã o ( I I I . 1 ) , f a z e r v a r i a ç õ e s * _
a r b i t r a r i a s n o s c o e f i c i e n t e s A ^ ^ e impor que a v a r i a ç ã o na e
n e r g i a e s e j a n u l a . Com i s t o chegamos ao s e g u i n t e s i s t e m a
de e q u a ç õ e s l i n e a r e s :
I n' L
(III.7)

o n d e ,
(1 - S . . ) d S . . f . , ,8 £ . * , + f * , 3 „ f , j X ' n i X ' i X n j X
( I I I . 8 )
E s t e s i s t e m a so t e r á s o l u ç ã o s e a m a t r i z dos c o e f i c i e n t e s A . , ,
t i v e r d e t e r m i n a n t e n u l o , ou s e j a .
d e t - V 2 . V ( r ) - el f .^ .dV . H . , ^ . , . = O ( I I I . 9 )
N a e x p r e s s ã o a c i m a as f u n ç õ e s f^^ dependem i m p l i c i
t a m e n t e do p a r â m e t r o , e i s t o n o s p e r m i t e f i x a r e s t e v a l o r
como s e n d o o v a l o r p r o c u r a d o e e x p l i c i t a d o na mesma e q u a ç ã o ,
p o r t a n t o a m a t r i z s e c u l a r pode s e r s i m p l i f i c a d a p a r a :
y H - , , A . - , = O ( I I I . 1 0 )
Temos p o r t a n t o uma m a t r i z c u j o s e l e m e n t o s s ã o i n t e
g r a i s de s u p e r f í c i e de f u n ç õ e s que dependem i m p l i c i t a m e n t e de
e . Com i s t o , m u d a n d o - s e o v a l o r de e , p o d e - s e l e v a n t a r uma
c u r v a d e t ] H(e) | e d e t e r m i n a r os v a l o r e s de e p a r a os quais
a c u r v a c o r t a o e i x o da e n e r g i a . Os v a l o r e s de e p a r a os
q u a i s o d e t e r m i n a n t e s e a n u l a s ã o os a u t o - v a l o r e s p r o c u r a d o s
de e n e r g i a .
Convém n o t a r que no MCV a b u s c a dos a u t o - v a l o r e s é
f e i t a b u s c a n d o - s e não os z e r o s de d e t | H(e) | , mas s im os ze
-1
t r H Ve) r o s de , ou s e j a , os z e r o s do i n v e r s o do t r a ç o
da m a t r i z i n v e r s a de H , p o i s s e d e t | H | = O , e n t ã o
-1 t r H
-1 O .
O b s e r v a m o s que uma r e d u ç ã o d a s d i m e n s õ e s da m a t r i z H
a t r a v é s de uma e s c o l h a a d e q u a d a do c o n j u n t o de f u n ç õ e s de e x
p a n s ã o o t i m i z a os c á l c u l o s t o r n a n d o - o s m a i s r á p i d o s . Uma e s c o

lha eficiente das funções da expansão pode ser feita tendo-se
em mente a simetria do orbital molecular para o qual se estã
procurando o auto-valor. A indicação de como isto pode ser fei
to encontra-se no Apêndice A.
Alem disso, no MCV, ê possível obter uma expressão
para um critério de precisão que permite controlar a qualidade
21
da base escolhida. Este fato e citado como uma grande vanta
gem do MCV sobre outros métodos para cálculos de estrutura ele
tronica.
Vemos também que, para a construção da matriz secu
lar, precisamos integrar a parte radial da equação de Schrodin
ger e obter as funções radiais . Para isto é preciso que
o potencial de um elétron esteja definido. No MCV a definição
do potencial eletrostatico está intimamente relacionada com o
cálculo da energia total da molécula, visto que a energia ele-
trostática de interação depende da densidade de carga assumida
e do potencial gerado por esta mesma distribuição de carga. 2 2 - IV
Segundo os autores , a energia total da molécula
deve ser calculada através de uma expressão variacional, cujo
extremo define o potencial. A expressão adotada é a seguinte:
E = I K a ^
(¡) , (j) a a + E ^n U[n-p,c] - S[p_ V p-n^
(III.11)
cujos termos tem o seguinte significado:
p(r) = l Pc((r) = I "a*-- "a -* ^ verdadeira densida
de de carga eletrônica total do sistema, enquanto que
n(r) . é a densidade de carga assumida como sendo "muffin-tin".
Ao longo do processo de auto-consistência, n(r) deve tender
a p (r) e quando este processo estiver convergido, em princí^
pio, estas duas densidades de carga devem ser iguais.

p(i") ó ( r - r ) é a densidade de carga dos nú-n
I K a
cieos, considerados como pontuais.
ê o funcional de energia cinética ele
trônica que, segundo a expressão (III.1), inclui inte
grais de superfície com a finalidade de tornar a função de on
da contínua em todo o espaço. Aqui a soma em a é feita so
bre todos os estados eletrônicos.
n X - a
33 n(r) d r (III.12)
é a energia de interação de troca segundo a aproximação
Xa explicitada no capítulo anterior. Nesta expressão 3 é a
constante - 6 (3/8TT) "''' , n(r) é a densidade de carga assumi
da e x^ é um parâmetro que pode assumir valores entre 1, ca
so em que a aproximação local para o potencial de exchange é
feita segundo Slater^^, e 2/3, caso em que a aproximação é fei
44 ta segundo Gaspar-Kohn-Sham
u[n-p,cj é o funcional de energia eletrostâtica de in
teração entre as cargas, que depende da densidade de car
ga dos elétrons n(r) e dos núcleos p(r) e do potencial cou
lombiano gerado por estas mesmas cargas. No MCV este funcio -
nal é expresso por:
U[n-p ,c] (n-p) 16ír
c
16lT i ds: (c!- c.) 3„c: - c.(9 c. + 3 C l )
^ 1 1' n 1 1 n 1 n 1
16ir i j dS.. (c.3 c. + C . 3 c.) (III.13)
onde S^ são as superfícies das esferas inscritas ãs células
i , cj (r) é o potencial coulombiano dentro das esferas inseri^

tas e c^(r) é esta função entre a esfera inscrita e a super
ficie delimitadora da célula i. A segunda soma é feita sobre
as superficies de separação entre as células i e j .
A expressão deste funcional é tomada em analogia ao
funcional de energia cinética da expressão (III.l) e nos diz
que U é extremo com relação a c(r) quando é solução da e-
quação de Poisson
V e = - 8IT (n-p) (III.14)
e é continuo com derivadas continuas sobre todas as superficies.
Quando c(r) satisfaz a equação (III.14) o funcio
nal U pode ser escrito como:
U (n-p) c(r) d r + termos de superficie (III.15)
onde os termos de superficie são os termos debaixo das somató
rias da equação (III.13).
Sendo solução da equação de Poisson, c(r) pode ser
escrito como:
C ( Í )
n(r') - p(r') d\
r - r (III.16)
onde a integral de volume é feita sobre todo o espaço.
A integral da equação (III.16) nos conduz a diferen
tes expressões para o potencial coulombiano dentro de cada uma
das regiões especificadas. Dentro das esferas inscritas obte
mos :
c:(r) = A: 2 Z .
1 8TT r n^(r) dr - Sir r n^(r) dr
(III.17)
onde é a carga elétrica do núcleo em torno do qual é cons
truída a i-ésima célula e n^(r) é a densidade de carga feita
INBIITUiO D E PÈSQU 'SAS fe".'h R Í E UC^vS 6 NUCLEARES
1. P. E. N.

esférico simétrica nesta região. Para a região interna ã esfe
ra inscrita na terceira célula obtemos:
c¿(r) 8TT r^ n^(r) dr + BIT r n^(r) dr (III.18)
Fora das esferas inscritas obtemos:
c.(r) = A. + -ÍJ-J_ _ 4 ^ — ^2 (III.19)
1 1 5 o Y J 1
onde n^ e a densidade de carga feita constante nesta região.
Nas equações acima as constantes Al , A. e A. ,
podem assumir quaisquer valores sem que com isto a equação
(III. 14) deixe de ser obedecida, entretanto para que U e con
sequentemente E sejam extremos é preciso que as continuida
des de c(r) e de sua derivada sejam garantidas.
Estas constantes são ajustadas substituindo-se as e-
quações (III.17), (III.18) e (III.19) na expressão (III.13),
com isto a dependência de U com a função passa a ser uma de
pendência com relação ãs constantes A. e Al . Para U ser
um extremo com relação ã escolha da função c , é preciso que
9 U 9 U 9 U = o o que quer dizer que —ãrr = O e —R-R = O . Pode U W V^V.. H - - ^ " - - - ^ M " - 9A: " " 9A.
3U -se mostrar que „.< = O independentemente dos valores de
dA^
A| , A^ ^ ^ • i 1 • Isto significa que o potencial dentro das
esferas inscritas pode ser deslocado sem com isto alterar o va
lor de U, o que permite forçar a continuidade de c(r) sobre
as esferas inscritas. Neste caso restam 6 valores de A para
serem determinados, ou seja, A. onde n = 0 , l e n = l , X) n
2, 3. Pode-se mostrar que das 6 equações lineares que se ob-
tém a partir de = O , somente 3 delas são independen-
tes podendo-se eliminar as equações correspondentes a —ç-R =
= O e fixar â vontade os parâmetros A ^ . Uma maneira de fi.

xar estes parâmetros, ê impondo a continuidade do potencial da
do pela equação (III.19) com o potencial da célula externa da
do pela equação (III.18). Apos a determinação das constantes
^ , impõem-se a continuidade sobre as esferas inscritas e
assim as constantes A.' são determinadas. 1
S[p] ê a auto-energia dos núcleos, interação estaque
estã incluída indevidamente na energia eletrostâtica U
e portanto deve ser descontada.
V(p-n) dr é um termo em que V é um multiplicador
de Lagrange e é introduzido na expressão da energia glo
bal para que se possa trabalhar com a densidade de carga n ao
invés da densidade verdadeira p.
A energia global calculada desta maneira é portanto
função de <{) , 0^ , n , V e c e, de acordo com o principio
variacional, deve-se exigir que ele seja estacionario para va
riações arbitrárias destas mesmas funções.
a) Variações em 4) : o vinculo que normaliza a função de onda
do a-ésimo estado
r * 3 (j) (() d r (III.20)
pode ser incluído na expressão da energia global usando a téc
nica dos multiplicadores de Lagrange. Assim,
E- = E + 5: e a
a 1 - l i
4) -4) . d r. a, 1 a , 1 1
(III.21)
Isto é feito apenas para garantir a normalização das funções
de onda. Para uma variação - , as equações (III.1), (III.13) Oi , 1
e (III.21) permitem escrever que 6E' = 0 se as condições
(- + V) 4> a,i
= e 4) . a , 1
(III.22)

d) . = (t) . sobre S. .
8 (j) = - 3 (f) - sobre S. - , (III.22)
que são as condições jã impostas pelo funcional de energia ci
nética.
b) Variações em V: para uma variação em V, a energia global
ê estacionaria quando n = p .
c) Variações em n: A energia global é estacionária para uma
variação em n quando
V = + , (III.23) ôn Ón
onde E e U são os integrandos da energia de troca e da e-X
nergia eletrostâtica. A equação (III.22) indica que V ê opo
tencial de um elétron, o que permite concluir que deve
ser identificado como o potencial coulombiano c(r) .
c) Como já foi frisado anteriormente, a energia global será ex
trema se âE ÔU
ôc ôc = O (III.24)
O processo de determinação da energia global E da
equação (III.11) é feito de forma auto-consistente. Assume-se
primeiramente uma densidade de carga esférico simétrica paraca
da um dos átomos, do tipo^'^
N ô 1 N Ó2 2 n(r) = -^-^ - - + - - (III.25)
47T r 4Tr r
Nesta equação com quatro parâmetros N-j , N^ , 6.^ e
¿2 , dois deles, N ^ e N2 , podem ser relacionados resolvendo-
-se a equação de Poisson com as seguintes condições de contor
no :

c(r) se r -> O
c(r) 2Z
se
Assim, obtém-se que
c(r) = -2N^ -6^r
e 2N2 -¿2^
e (III.26)
onde N- + N2 = Z .
As outras relações que determinam estes parâmetros,
são encontradas impondo-se que este funcional obedeça ao mode
lo atômico estatístico de Thomas-Fermi tornando seu funcional
um extremo, e também torne a energia total do átomo um extremo.
A partir destas densidades de cargas atômicas, gera-
-se uma densidade de carga esférico simétrica dentro das esfe
ras inscritas, tomando-se a média esférica da soma das densida
des atômicas assumidas. Fora das esferas inscritas a densida
de de carga ê feita constante de acordo com:
n. P. 3
(III.27)
onde P. é o volume da célula i fora da esfera inscrita e Q.
é definido como:
Z. - 4TT R. 2 -' r n^ (r) dr , para as células atômicas
(III.28)
- 471 r Uj(r) dr , para a célula externa
onde R. é o raio da esfera inscrita ã célula i .
Com esta densidade de carga assumida, calcula-se a
Contribuição do termo de troca para a energia global de acordo
com a equação (III.12) e em seguida gera-se o potencial coulom
biano de acordo com as equações (III.17), (III.18) e (III.19).

auto-energia dos núcleos e determina-se a diferença -j-
Tendo-se a densidade de carga e o valor do potencial
coulombiano em todo o espaço, calcula-se a energia eletrostáti^
ca conforme a equação (III.13), da qual se subtrai o termo de
qc -
- U[q,c] , entre as equações (III. 13) e o primeiro termo de
(III.15), que ê um controle da convergencia do processo.
Feito isto, o potencial de um elétron V pode ser de
terminado somando-se as contribuições do potencial coulombiano
e do potencial de exchange de acordo com a equação (III .23). Es
te é considerado o potencial inicial da primeira iteração.
A partir deste potencial, resolve-se a parte radial
da equação de Schrodinger para cada função f^^ da expansão
de um orbital, em seguida monta-se a equação secular de acordo
-1 -1
com a equação (III. 8) e busca-se o valor de e que faz(tr H )
igual a zero. Feito isto, seria natural voltar ao sistema de
equações (III.10), onde o valor de e encontrado no estagio
anterior é um parâmetro implícito, e determinar os coeficien
tes da expansão da função de onda e assim gerar uma distribui
ção devida a este orbital, entretanto, o MCV é capaz de forne
cer uma expressão para a densidade de carga onde ê necessário
apenas o conhecimento dos elementos da matriz H ''" . Assim a
densidade de cargas devida a função de onda de um orbital mole
cular é gerada. Este procedimento deve-se repetir para todos
os orbitais moleculares da configuração eletrônica do estado
molecular para o qual se está procurando uma solução.
Somando-se todas estas densidades de carga, obtemos
uma nova função n(r) e a partir dela um novo potencial cou
lombiano. Este potencial ê entendido como o potencial final da
primeira iteração e, nesta altura, calcula-se a máxima diferen
ça entre os potenciais inicial e final, sendo este valor um ou

tro controle da convergencia.
Finalmente calcula-se a energia global da molécula a
través da expressão (III. 11). Convém notar que, os auto-valo
res de energia corretos de cada orbital, ou seja, os valores
-1 -1
que tornam (det H ) igual a zero, fazem as funções de on
da continuas, o que quer dizer que os termos de superficie do
funcional de energia cinética de (III.11) são nulos. Isto per mite reagrupar as integrais de volume (t>* (-V ) <i> d^r
V P d^r nesta mesma expressão, sendo que assim a energia glo
bal pode ser calculada por
V(r) n(r) d^r.
(III.29)
onde a soma sobre e a soma dos auto-valores dos orbitais
moleculares (MO's) já determinados.
A partir daqui inicia-se realmente o processo de au
to-consistência, misturando-se os potenciais final e inicial da
- j . . ^ 22-IV
primeira iteração de maneira conveniente para que a con
vergência do processo seja acelerada, gerando-se assim um po
tencial inicial para a segunda iteração. Com este potencial
resolve-se novamente a parte radial da equação de Schrodinger
para todas as funções f^^ da expansão dos MO's, monta-se a
matriz H e recalcula-se uma densidade de cargas para cada MO
ocupado. Assim gera-se um novo potencial que é misturado de
forma conveniente com potenciais anteriores para dar inicio a
uma nova iteração.
Este ciclo é repetido até que um dos dois critérios
de convergência citados seja aceitável.

III-2. O MCV COM POLARIZAÇÃO DE SPIN^^
Como vimos no Capítulo II, na aproximação de Hartree
-Fock o potencial médio sentido por um elétron depende de seu
spin, caso estejamos tratando um sistema de camadas abertas.
Este efeito aumenta ã medida em que o número de elétrons de
spin "up" se afasta do número de elétrons de spin "down".
Esta dependencia é bastante visível nos auto-valores
das equações de Hartree-Fock, entretanto pode ser considerado
relativamente pequeno no valor de energia total, caso o número
de elétrons com spin "up" não seja muito diferente do número
de elétrons com spin "down". Além disso, para que este efeito
seja levado em consideração o volume de cálculo a ser efetuado
deve ser duplicado. Por estas razões a formulação original do
MCV não leva em consideração a dependência das funções de onda
e dos auto-valores com o spin.
A polarização de spin surge naturalmente no MCV se
definirmos dois potenciais de um elétron
V(r) — «
V para elétrons de spin "up"
V^ para elétrons de spin "down"
esféricamente simétricos em todas as regiões do espaço molecu
lar.
Assim as funções de onda de elétrons do mesmo orbi
tal devem ser diferentes na sua parte radial,
<í).(a) = l A.^(a) R^°(r-,a) Y^(í.) (III.30)
\
onde a ê a coordenada de spin, e desta forma as energias or
bitais que definem as funções radiais também dependem do
spin. Estes valores de energia podem ser determinados por uma
expressão variacional análoga ã expressão (III.l), onde agora

deve ser inserido o potencial correto
l i
d^r. (í)!(a) (í).(a) 1 1 ^ ^1
i d\. ^l(a-) - V + V(a)
S. .
[ dS. . (j). (a) - 4). (a)
dS. .
4,^(0) +
9^4)j (a) + 3j^*iCa) (III.31)
onde e(a) assume os valores e correspondente aos po
tenciais e . Observamos assim que o número de orbi
tais e de auto-valores é duplicado.
Este tratamento gera duas distribuições de carga:
a a
a'
(III.32)
Evidentemente a expressão variacional (III.11) para
a energia molecular total não pode ser usada. O novo funcio -
nal deve ser definido em função de densidades, potenciais e fim
ções de onda que dependam da polarização de spin.
Podemos adotar a seguinte expressão variacional:
E = I K a '- a'
+ E [ n . 1 + E [ n , ' ] + u r n . + n , - p , c l +
S[p] (III.33)
Nesta expressão n^ e n^ sao as densidades de car-

ga assumidas pelo método celular, como sendo muffin-tin. E [n ]
e E [n ] são os funcionais de troca que nos obrigaram a con-
siderar o spin e são dados por:
E
X
= - 6 a
- 6 a
L 4TT
3
n,
n
nl/3
nl/3
(III.34a)
L 4Tr (III.34b)
segundo a aproximação Xa citada no segundo capítulo.
Ë interessante notar que no limite n. = n n
onde n é a densidade total, a soma das equações (III.34a) e
(III.34b) se reduz ao funcional da expressão (III.12).
Os funcionais de energia eletrostâtica U[n_ ^4. ~ P'*íl
e de auto-energia dos núcleos S[p] não sofrem alterações,
já que dependem apenas da densidade total de elétrons, do po
tencial coulombiano c e da densidade de protons.
As funções e funcionam como multiplicado -
res de Lagrange e, como vimos no item anterior, são os poten
ciais de um elétron, que agora são escritos como:
V = c + Xa^ para elétrons de spin "up"
V(ö) = <
V, c + Xa, para elétrons de spin "down'
Da mesma forma que o funcional da expressão (III.11)
este novo funcional deve ser estacionário para variações arbi-^ * *
trarias em x , <j>„ > <¡>„. n. , n, , V. , V, e a , T a,t a , + a,y t + t +
c . Assim, variações nas funções '^ç^ ® -i- tornam o fun
cional estacionário quando as mesmas são contínuas com deriva
das normais contínuas sobre as superfícies das células, e as e
quações

são obedecidas.
Estas são as considerações básicas para se realizar
cálculos auto-consistentes de estrutura eletrônica de molécu
las com camadas abertas, restando apenas definir as densidades
de carga iniciais n^ e • Estas podem ser definidas como
sendo iguais ã metade da densidade total inicial definida no i
tem anterior. Após a primeira iteração do cálculo, as densida
des iteradas se tornam diferentes para cada spin, e assim pros^
seguem ate atingir a auto-consistência.
Na próxima seção, mostramos também quanto a polariza
ção de spin influencia nos valores da energia total e das ener
gias orbitais.
. S T N U C L E A R E S

- CAPÍTULO IV -
R E S U L T A V O S
IV -1 . A MOLÉCULA DE Ne2
O primeiro sistema estudado por nós foi o Ne2• No
limite de átomos separados temos um átomo de neônio no estado
fundamental "S e um íon positivo de neônio também no estado
2 - <•
fundamental P . A medida que o átomo e o íon se aproximam e
interagem, as degenerescências atômicas são quebradas e os or
bitais atômicos se distorcem e se misturam dando origem aos or
bitais moleculares (MO's).
Quando esta molécula é formada, podemos dizer que es
tamos diante de um sistema homonuclear com simetria de inver
são. Assim, como está indicado no Apêndice A, os estados mole
culares gerados a partir deste limite de átomos separados são: ^Z* , , e ^E* . Em princípio não podemos saber em u ' g u g y y y
que ordem de energia estes estados aparecem. Entretanto se os
orbitais moleculares aparecem segundo a estrutura de níveis in
dicada na Figura A-l, então os estados moleculares aparecem se
gundo a ordem citada acima. As configurações moleculares des-
tes estados aparecem na Tabela IV - 1 , onde os orbitais molécula
res indicados são apenas os que surgem a partir dos orbitais a
tómicos de valencia, ou seja
^ 2 6 Rg (caroço) ns np
r, + r ^ 2 5 Rg (caroço) ns np
orbitais atômicos de valencia

T A B E L A II/-J
Conilgaraçào doò ofibitaiò rnoZículamò de vaZtncZa do&
dZmíKoò ionizadoò de gaò&ò nobizò.
ESTADO ORBITAIS MOLECULARES DE VALENCIA
\ \
(ICg^) (la^2) ( 2 a g V (ITT^^ (iTTgV (2a^2)
No caso do Ne^ os orbitais moleculares de va lencia
são os provenientes dos orbitais atômicos 2s e 2p . Os orbi
tais moleculares e provenientes dos orbitais atômicos
Is , considerados como orbitais de caroço, são tratados de for
ma especial pelo MCV. Por continuarem com um caráter essencial
mente atômico, seus auto-valores de energia são determinados
impondo-se que suas funções de onda se anulem em pontos próxi
mos ãs superfícies esféricas inscritas nas duas células atômi-
22-IX
cas
Nossos cálculos auto-consistentes foram feitos, a
princípio, sem se levar em conta a polarização de spin. Adota
mos a divisão em células conforme a Figura IV -1 , e sobre estas
superfícies utilizamos 10 pontos para o cálculos das integrais
de superfície.
A expansão das funções dos orbitais de valencia, foi
feita de acordo com (III.4) e as harmônicas esféricas utiliza
das são indicadas na Tabela IV -2.

- ox -
FIGURA II/-?.
Vivióão do zòpaço motzcutaA.
em céíulaò. Na {¡igura.
'2 ' C- 2a
1 '
T A B E L A II/-2
Expanòão em harmónicas e.&{¡zr¿ca& em cada célala, pa
ra oò orbitais de va£enc-¿a da mo£écu-£a de We2 •
ORBITAIS DE
VALÊNCIA
CÉLULAS ATÔMICAS
CÉLULA EXTERNA
il = 0, 1, 2 £ = 0, 2
a u
£ = 0, 1, 2 £ = 1,3
g £ = 1, 2, 3 £ = 2,4
TT U
£ = 1, 2 , 3 £ = 1,3
Os orbitais cr e a originados a partir dos orbi-
tais atômicos Is foram descritos por um único orbital com o-
cupação 4 e usamos apenas uma função do tipo s na expansão da
função de onda. Com isso tínhamos 7 orbitais a serem calcula
dos .
A densidade de carga foi feita "muffin-tin" e igual
nas células atômicas pois estamos estudando um sistema homonu
clear .

o parâmetro a utilizado para descrever o potencial
de troca foi a = 0.7300 retirado dos cálculos de Schwarz^^
e, segundo este criterio de determinação de a, a energia total
atômica obtida deve ser igual ã energia de Hartree-Fock.
Os resultados obtidos para as energias orbitais e e-
nergias totais deste sistema no estado fundamental , são
apresentados na Tabela IV-3. Estes resultados são apresentados
em função da distância internuclear.
O criterio de convergencia adotado para a obtenção
destes resultados foi que a maior diferença, ponto a ponto, en
tre os potenciais final e inicial de uma iteração deveriam ser
-4
menores que 10 a.u.. Isto acontecia geralmente apos vinte i
terações. A busca dos auto-valores de energias orbitais na pri
meira iteração foi feita em intervalos de energias em tomo dos ^ 8
auto-valores correspondentes aos orbitais atômicos do Ne . Es
te ê um procedimento simples e facilita esta procura quando es
tamos tratando sistemas em que as ligações não são fortes.
Esta tabela foi apresentada para que se possa obser
var que os auto-valores das energias orbitais, assim como as e
nergias totais, variam de forma continua ã medida que a distan
cia internuclear aumenta. Esta observação também facilita a
busca dos auto-valores nas primeiras iterações.
Nossos resultados sobre este estado e sobre os ou
tros três encontram-se sintetizados na Figura IV-2.
2„ +
TABELA ÍV-S.
Energias orbitais e energias totais para o estado fundamental
do
We.
^.m fun
ção da distância
internuclear.
DISTÂNCIA
E N ERGIAS
ORBI TAIS
(Ry)
ENERGIA
TOTAL
ENERGIA
TOTAL
a
o
g
u
%
"g
(Ry)
2.2
-61.9670
-3.9540
-3.5035
-2.1938
-2.1754
-1.9086
-1.4899
-512,3176
2.3
-61.9825
-3.9031
-3.5363
-2.1780
-2.1606
-1.9243
-1 . 5860
-512.4299
2.5
-62.0029
-3.8207
-3.5803
-2.1365
-2.1049
-1.9423
-1.7174
-512.5765
2.6
-62.0103
-3.7887
-3.5950
-2.1168
-2.0827
-1.9048
-1.7629
-512 .6357
2.7
-62.0120
-3.7582
-3.6026
-2.0946
-2.0601
-1.9480
-1.7954
-512.6641
2.8
-62.0110
-3.7321
-3.6061
-2.0727
-2.0394
-1.9452
-1.8197
-512.6830
3.0
-62.0028
-3.6835
-3.6039
-2.0300
-2.0008
-1.9362
-1.8486
-512.7057
3.3
-61.9818
-3.6277
-3.5876
-1.9729
-1.9522
-1.9149
-1.8636
-512.7209
3.6
-61.9571
-3.5849
-3.5649
-1.9261
-1.9126
-1.8910
-1.8607
-512.7283
4.0
-61.9256
-3.5420
-3.5343
-1.8782
-1.8713
-1.8608
-1.8457
-512.7360
4.6
-61.8885
-3.4988
-3.4910
-1.8306
-1.8285
-1.8249
-1.8197
-512.7479
5.0
-61.8698
-3.4786
-3.4778
-1.8090
-1.8082
-1.8064
-1.8039
-512.7549
5.5
-61.8524
-3.4602
-3.4600
-1.7900
-1.7897
-1.7890
-1.7880
-512.7630
6.0
-61.8380
-3.4455
-3.4454
-1.7750
-1.7750
-1.7748
-1.7744
-512.7685
6.5
-61.8268
-3.4341
-3.4340
-1.7636
-1.7637
-1.7635
-1.7633
-512.7745
7.0
-61.8176
-3.4245
-3.4245
-1.7540
-1.7540
-1.7540
-1.7539
-512.7791
8.0
-61.8072
-3.4083
-3.4083
-1.7381
-1.7381
-1.7381
-1.7381
-512.7866
- 511.5
-512.0
>»
CU
< (S DC UJ z Ui
-512.5
-513.0 I—'—I
1—I—r T — r 1—I—I—I—I—I—1—I—I—r 1—r
I . , I J—I I I—I—I I.
Ne+Ne*
I I I
2.5 3.0 3.5 4.0 4.5 5.0
FIGURA H/-2. CuiA\ja¿ do. energía potencial para o¿> estados ele
trônicos , , e do We^ calculadas peto MCI/
desprezando o efeito do alinhamento de spins.
Como observamos, estas curvas são repulsivas e ten
dem todas ao mesmo limite de átomos separados. Neste limite,
temos também outro valor de energias assinalado. Este outro va
lor foi obtido da seguinte maneira:
E(~) = 2 E^p(Ne) + P.I. (Ne) - 512.6034 Ry (IV.1)
O valor de Ej^p(Ne) foi retirado dos cálculos de o
Clementi e P.I.(Ne) é o potencial de ionização experimental

do Ne, 21.559 eV^^.
Em princípio, o valor de a adotado por nos deve re
produzir o mesmo valor de E^p(Ne) para o átomo de Ne e um va
lor proximo de E^p(Ne) + P.I.(Ne) para o íon Ne . Apenas
não deve ser o mesmo para o íon pois este sistema deve ter ou
tro valor de a. Assim, não ë de se estranhar que o valor de
energia fornecido pelo MCV, para grandes distâncias interat5m¿
cas, não tenda a este limite assinalado, entretanto poderíamos
esperar que estas curvas fossem representativas em torno da po
sição de equilíbrio destas moléculas.
Antes de fazermos qualquer comentário sobre estas cur
vas, vamos apresentar uma síntese dos resultados de outros cál
culos sobre estes estados.
T A B E L A lV-4
2.+ "n Valorzò de fe e Re para o¿ <L¿tado¿ Z , u ,
2 + + - 9 e E do We^ obtldoò por outroò métodos.
'n u
ESTADOS MOLECULARES
h* u \ h
u
1.31^ 0.17^
0.07^
0.06^
0.003^
repulsiva
De(eV) 1.20^
1.30^
1.65'
0.17^
0.07^
0.06^
0.003^ repulsiva
3.19^ 4.05^ 4.76^
4.80^
repulsiva
Re(a^) 3.30^
3.31^
3.20^
4.10^
4.76^
4.80^ repulsiva
MSOS-Xa (referência 59)
Cálculo "ab initio" com interação de configuração
(referência 10)
Experimental (referência 32)
Cálculo "ab initio" MO-SCF (referência 26)

• o cálculo que melhor reproduz os resultados experi -
mentais ê o cálculo "ab initio" com interação de configurações
da referência [lOj. Neste cálculo, cada estado da molécula ê
descrito por uma combinação linear de 30 configurações, incluin
do configurações iónicas. Daí podemos imaginar a complexidade
deste cálculo, embora não seja auto-consistente.
O cálculo com o Espalhamento Múltiplo (MS), adota a-
proximações similares ãs nossas, ou seja, aproximação Xa pa
ra o potencial de troca, e diferentes expressões para o poten
cial total em regiões previamente delimitadas no espaço molecu
lar. Entretanto, adota também um procedimento que tem sido al_
vo de muitas críticas: a sobreposição das esferas atômicas
(overlaping spheres - OS). Isto implica numa falta de critério
para a delimitação das regiões do espaço molecular.
A mais importante característica que estes resulta -
dos mostram é que o estado fundamental é ligado, com uma ener
gia de dissociação De com valor aproximado de 1.30 eV e a
distância interatômica nesta ligação é de 3.30 a^ . Este ê um
resultado que não se obtêm a partir de nossos cálculos. Entre
tanto, se nos detivermos apenas em torno da região de equilí^
brio desta molécula e compararmos nosso valor de energia do e^
tado fundamental com o limite de átomos separados, vamos obter:
De = E(oo) - Ej^^^(3.30 a^) = 1.60 eV
Este valor é bastante proximo dos valores obtidos em
outros trabalhos, entretanto o poço de potencial típico de es
tados ligados não é observado.
Uma primeira causa para este efeito poderia ser a po
larização de spins, que até o momento tinha sido desprezada. O
Ne2 ê um sistema de camada aberta com 10 elétrons de spin "up"
e 9 elétrons de spin "down". O efeito da polarização de spins
sobre os auto-valores de uma partícula pode ser visto na Tabe
la IV-5.
T A B E L A II/-6
Efeito da polarização de ¿pin no¿ auto-valoreé de ama par
tZeula e na energia total para o We^ 5 di&tâneia de 3.3 a^
SIMETRIAS
DOS
OM's
ENERGIAS (Ry) SIMETRIAS
DOS
OM's Orbitais
Polari zados Orbitais nao Polarizados
(caroço)
-62.0038(+)
-61 . 9552 (4-) -61.9818
a g
-3.6707(+)
-3.5811 (4-) -3.6277
0
u
-3.6316(f)
-3. 5397 (4-) -3.5876
-2.0152(+)
-1.9270 (4-) -1.9729
IT
u
-1.9951 (f)
-1.9058 (> ) -1.9522
g
-1.9594(+)
-1 . 8667 (4-) -1.9149
a u
-1.9100(+) -1.8636
Energia total
-512.7432 -512.7209
Observamos que os auto-valores dos orbitais não pola
rizados encontram-se entre os auto-valores dos orbitais polari_
zados. Quando o número de elétrons com spin "up" for igual ao
número de elétrons com spin "down", os auto-valores não depen-
derão do spin e serão iguais aos auto-valores dos orbitais não
polarizados.
O efeito da polarização de spin na energia total po
de ser visto na Figura IV-3.
-512,50
Q:
< -512,75 < O OC u
UJ
- 513,00
T—I—^—I—n—R- - |—I—I—I—I—I—r "I—I—r T — I — r
oi. = 0. 7300
Ne 4-Ne*
^ d." 0.7384
J L J L J L J \ L J — I L _ L
2.0 3.0 3.3 4.0 5.0
RCa^)
FIGURA II / - 3 . CufLvas dz potencial para o estado T.^ do He^;
sem polarização de spin; com polarl
zação.
Observamos que a influência da polarização de spin
na energia total é praticamente constante ao longo da curva des
te estado e não contribui significativamente para a mudança da
forma da curva.
Para estimarmos o efeito da constante a, calculamos
o valor da energia total do estado fundamental na posição de e
quilíbrio usando um a = 0.7334 . Este a ê igual a -j- (a ^ +
59
+ a|yjg + ) • Vemos que a mudança na energia total e pequena com
parada com o valor desta mesma energia total, entretanto a in-

fluência desta constante ê suficientemente grande para deslo
car as curvas de potencial em relação ao limite de átomos sepa
rados. Podemos ir mais alem e supor que, no caso desta molecu
la, o valor de a varie ao longo da curva de potencial, pois
a ionicidade dos componentes desta molécula varia.
Este problema da ionicidade ê bastante delicado. No
limite de átomos separados temos Ne e Ne^ , ao passo que na
posição de equilíbrio temos Ne^ , onde o ultimo elétron é i-
gualmente compartilhado numa covalência. Para obtermos corre
tamente as energias dos estados em função da separação internu
clear, deveríamos poder prever a transferência de carga para o
íon ã medida que afastamos os núcleos. Este fenômeno seria na
tural caso todas as correlações eletrônicas estivessem sendo
tratadas, e é isto o que a interação de configurações descreve
bem.
Para a análise dos estados excitados devemos obter
as energias das transições permitidas. As regras de seleção
para as transições por dipolo encontram-se no Apêndice A.
Na Tabela IV-7 temos as energias das excitações ver
ticais calculadas pelo MCV sem considerar a polarização de spin
e as energias previstas por outros métodos.
Segundo nossos cálculos estas transições tem ener
gias bem menores do que as previstas pelos métodos "ab initio".
Acreditamos que isto seja devido ã má descrição dos estados ex
citados, pois neste caso a aproximação do potencial de troca
por um funcional de densidade local, no caso a aproximação Xa,
se torna duplamente questionável.

T A B E L A I I / - 7
1 + l Z •> lí pa/í.a a. a g ' Emrgiaò das tn.ansZc.diS
molécula do. We^ adotando-se. como distância de equilí
brio 3.3a
TRTiiNSIÇAO AE (eV) X (nm)
4 . 4 3 ' ' 280
u g
4.45^
4.77^
278
260^
1.57^ 788^
2.01^ 617^
h* ^ h u g
1.97^ 630^ h* ^ h u g
1.36^ 1680^
^ cálculo "ab initio" com interação de configurações
(referência 10)
^ MSOS-Xa (referência 59)
Cálculo "ab initio" (referência 80)
Este trabalho
IV-2. A MOLÉCULA DE Ar2
Quando passamos a tratar um sistema ionizado com um
número de elétrons maior, os erros que a constante a do poten
ciai de troca acarreta devem ser relativamente menores.
Neste caso, os orbitais moleculares de valencia espe
cifiçados na Tabela IV-1, são os provenientes dos orbitais ato
micos 3s e 3p , e para descrevê-los usamos a mesma expansão
em harmônicas esféricas indicada na Tabela IV-2. Os orbitais
originados a partir dos orbitais atômicos Is e 2s foram des
critos como dois orbitais o de caroço com ocupação 4. Os or
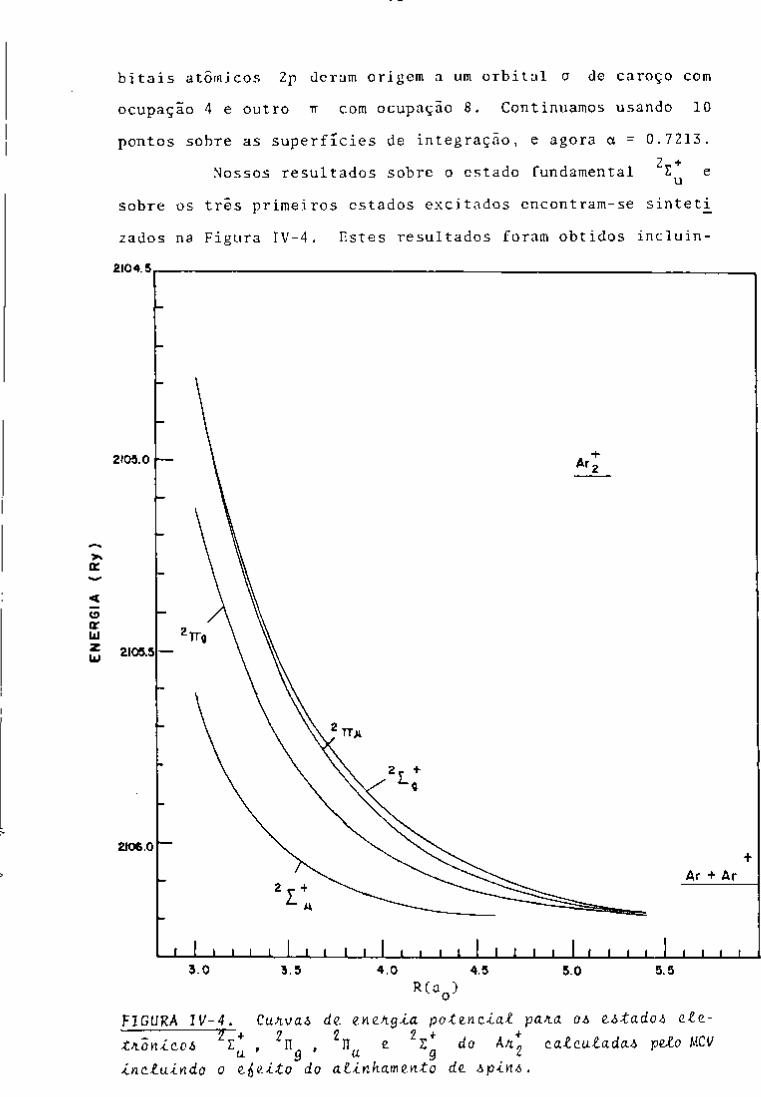
bitais atômicos 2p deram origem a um orbital o de caroço com
ocupação 4 e outro ir com ocupação 8. Continuamos usando 10
pontos sobre as superfícies de integração, e agora a = 0.7213.
Nossos resultados sobre o estado fundamental ^
sobre os três primeiros estados excitados encontram-se sinteti^
zados na Figura IV-4. Estes resultados foram obtidos incluin-
2104.5
2105.0
O
UJ
2 2105.5
2106.0
Ar + A r
J L_J L I I I I 1 I I I I I I I ' » • ' J I I I 3.0 3.5 4.0 4.5 5.0 5.5
R(a^)
FIGURA JV-4. Curvas dz znzrgZa potznzlal para o& Zòtadoò zlz-
trónlcos "^^^ , , z do kr*^ calculadas pzlo MCI/
Incluindo o zfzito do alinhamznto dz spins.

do-se a polarização de spins, através de um calculo auto-consis
tente envolvendo 19 orbitais.
O limite de átomos separados assinalado nesta figura
foi obtido da mesma maneira anterior:
= 2 E^p(Ne)° + P.I. (Ne) 87 = - 2106.1112 Ry .
Como observamos, este valor limite de energia é mais
próximo do valor para o qual tendem os nossos resultados. Na
Tabela IV-8 apresentamos uma síntese dos resultados obtidos por
outros cálculos.
T A B E L A JV~S
2„ + •n Valores de Ve e Re para os estados 1 , xi ,
2 + + ^ ^ Q 1 do Ne„ obttdos por outros métodos.
9 2
MSOS-Xa (referência 59)
^POL-CI (referência 85)
^Cálculo "ab initio" (referência 78)
^MO-SCF (referência 26)
Experimental (referencia 60)
ESTADOS MOLECULARES
u h g
\ h* g
1.30^ 0.10^ 0.05^ repulsiva^
1.24^ 1.24^ 0.12^ repulsiva^
De (eV) 1.20*
1.25<
1.33^
4.59^ 5.71^
6.33'^
6.42^ repulsiva
4.69^
5.71^
6.33'^ repul. repulsiva
Re (a^) 4.65^
4.60^
5.22^
T l C - S f e N U C L E A R E S

Excetuando os cálculos pelo método do Espalhamento
Múltiplo da referência [59], todos os demais são extremamente
longos e trabalhosos. Estes resultados indicam que há uma li
gação de aproximadamente 1.30 eV , e que a distancia de equi
librio desta molécula é 4.60 a . S e adotamos o mesmo procedi o ^ —
mentó anterior e comparamos o valor da energia do estado funda
mental com o limite de átomos separados vamos obter:
De = E(<») - Ej (. (4.60 a^) = 1.10 eV
Este valor difere do valor esperimental de 151, ao
passo que a estimativa similar para a molécula de Ne2 aponta
va uma diferença de 23%. Isto corrobora nossas espectativas de
que o erro provocado pela aproximação Xa neste sistema seja
menor, embora seja evidente o aspecto repulsivo da curva do es
tado fundamental.
No que se refere ãs transições eletrônicas, nossos
resultados para as transições verticais e os obtidos por ou
tros métodos encontram-se na Tabela IV-9.
Embora tenhamos melhorado a descrição do estado fun
damental em torno da posição de equilíbrio para esta molécula
com um maior número de elétrons, vemos que as transições ele
trônicas continuam sendo mau descritas pelo MCV. O efeito da a
proximação Xa para o potencial de troca no cálculo dos esta
dos excitados parece não ser abrandado com o aumento do número
de elétrons.

T A B E L A îl/-g
1 + 2 + Enefigloiò da¿ transições I (
2 + 1 Z n para a a 3
molécula de Ar^ adotando-se como distancia de equilZ -
brio 4.60 a .
TRANSIÇÃO AE (eV) X (nm)
3.89^ 319^
4.20^ 295^
u g
4.13^
4.13'
300^
300^
3.75^ 330®
1.39^ 893^
1.66^ 745^
2.03^ 610^
h* . h U g
1.73^ 716^ h* . h U g
1.65^ 753®
0.68^ 1822^
POL-CI (referência 85)
^ MSOS-Xa (referência 59)
"ab initio" Cl (referência 80)
^ "ab initio" (referência 78)
Experimental (referencia 60)
Este trabalho

C O N C L U S Ã O
Os resultados obtidos nos levam a crer que a mã des
crição dos estados destas moléculas tenha duas razões fundamen
tais. A primeira é uma questão de simetria; para distâncias
internucleares relativamente maiores que a distância de equilí^
brio torna-se questionável a simetria de inversão, pois o ülti
mo elétron passa a se localizar em uma das células de tal for
ma que no limite de átomos separados tenhamos Rg + Rg^ . Esta
localização de carga surgiria naturalmente se todas as correia
ções eletrônicas estivessem sendo tratadas de forma adequada.
A segunda causa é a aproximação Xa para o poten
cial de troca. Nesta aproximação utilizamos o parâmetro a que
tinha sido ajustado para reproduzir as energias dos átomos de
gás nobre. Entretanto este parâmetro deve mudar com a ionici
dade do átomo na molécula e, portanto, deve depender da distan
cia internuclear. O ajuste deste parâmetro é uma tentativa de
introduzir os efeitos de correlação de forma empírica no áto
mo, entretanto estes mesmos efeitos mudam quando passamos para
a molécula.
Na verdade estas duas razões abordadas são reflexos
de uma causa primeira: a má descrição dos efeitos de correla
ção. No caso das ligações dos estados fundamentais destas mo
léculas, estamos tentando descrever um efeito de 1 eV em
7 xlO" eV e 29 xio^ eV respectivamente para Ne^ e Ar^ •
Este é um efeito muito pequeno e para descrevê-lo parece inevi.
tável a utilização de uma aproximação mais elaborada para o po
tencial de troca que inclua os efeitos de correlação.
Dentro do mesmo espírito das aproximações locais pa-

ra o potencial de troca, apresenta-se a aproximação do operador
de massa • principal vantagem desta aproximação sobre a
aproximação Xa , ê que o funcional do operador de massa ê o
mesmo para todos os sistemas, ou seja, átomos, moléculas e s5li
dos. Além disso, inclui os efeitos de correlação explicitamen
te .
Acreditamos que a partir desta aproximação poderemos
obter bons resultados para os estados fundamentais destas molé
culas. Com relação aos estados excitados a questão é mais deli
cada, pois neste caso o tratamento dos efeitos de correlação e
troca por um funcional de densidade local não é rigoroso^^. Po
de-se provar que este procedimento é válido para a descrição dos
34
estados fundamentais , entretanto, nao se provou ainda que tal
procedimento seja válido para os estados excitados. Isto toda
via não tem impedido o largo uso destas aproximações locais pa
ra o estudo dos estados excitados de átomos e moléculas, devido
ã grande simplificação que esta aproximação acarreta.

- A P Ê N D I C E A -
M O L É C U L A S P I A T 0 M I C A S -"-
As moléculas diatómicas são sistemas que possuem si
metria cilíndrica e podem ser homonucleares, com simetria do
grupo pontual D j , ou heteronucleares, com simetria C^^ . Es
tes dois grupos são formados por operações de simetria segundo
as quais tanto o hamiltoniano total quanto o hamiltoniano de u
ma partícula são invariantes.
O grupo C^^ contêm as seguintes operações: rota
ções de um ângulo qualquer (¡) sobre o eixo que passa pelos
dois núcleos (eixo z), reflexões sobre os planos que contém o
eixo z e a operação de identidade. O grupo D j contém to
das as operações do grupo C^^ e mais a operação de inversão,
além das operações impróprias que surgem do produto direto en
tre os grupos C^^ e i .
O objetivo de se estudar estes grupos de simetria ê
a caracterização das simetrias dos estados eletrônicos molecu
lares e dos orbitais moleculares. Com isto pode-se, por exem
plo, otimizar a escolha dos orbitais atômicos que vão descre
ver um certo orbital molecular, ou então obter-se regras de se
leção para transições eletrônicas induzidas por uma perturba -
ção externa H' .
A-l. GRUPO C^^
As operações de simetria deste grupo estão divididas
nas seguintes classes:
a) E , operação identidade;
b) Todas as operações de reflexão sobre os planos a

- 0 0 -
pertencem a outra classe;
c) As rotações de um ângulo nos dois sentidos per
tencem ã classe (2C^) . Como o ângulo (p pode ser qualquer, e-
xistem infinitas classes 2C, .
Devido ã simetria em torno do eixo z sabemos que
-. , H = O . Isto indica que < L > e uma constante de _ d <p I Z
movimento e as funções que se transformam segundo as represen
tações irredutíveis deste grupo, assim como no caso atômico,
são funções do tipo e~^l^'"^ , onde X é um número inteiro.
Este número identificámos como sendo o modulo da projeção do mo
mento angular no eixo z e ê o número que serve de base para a
construção da tabela de caracteres.
A notação convencional dos estados eletrônicos das
moléculas diatómicas, costuma indicar o número X com letra
grega minúscula na descrição dos orbitais moleculares e maiús
cula para a descrição dos estados eletrônicos totais, em anal£
gia ã notação atômica.
X = O orbital o ou estado E
X = 1 orbital -rr ou estado n , etc...
As representações caracterizadas por X ^ O são du
plamente degeneradas e sob uma operação C^ o traço destas re
presentações é:
9
O
iX(|)
X^^hc^) = Tr = 2 C O S X<J) para X ^ O
(A.l)
Para descobrir o traço destas representações sob a
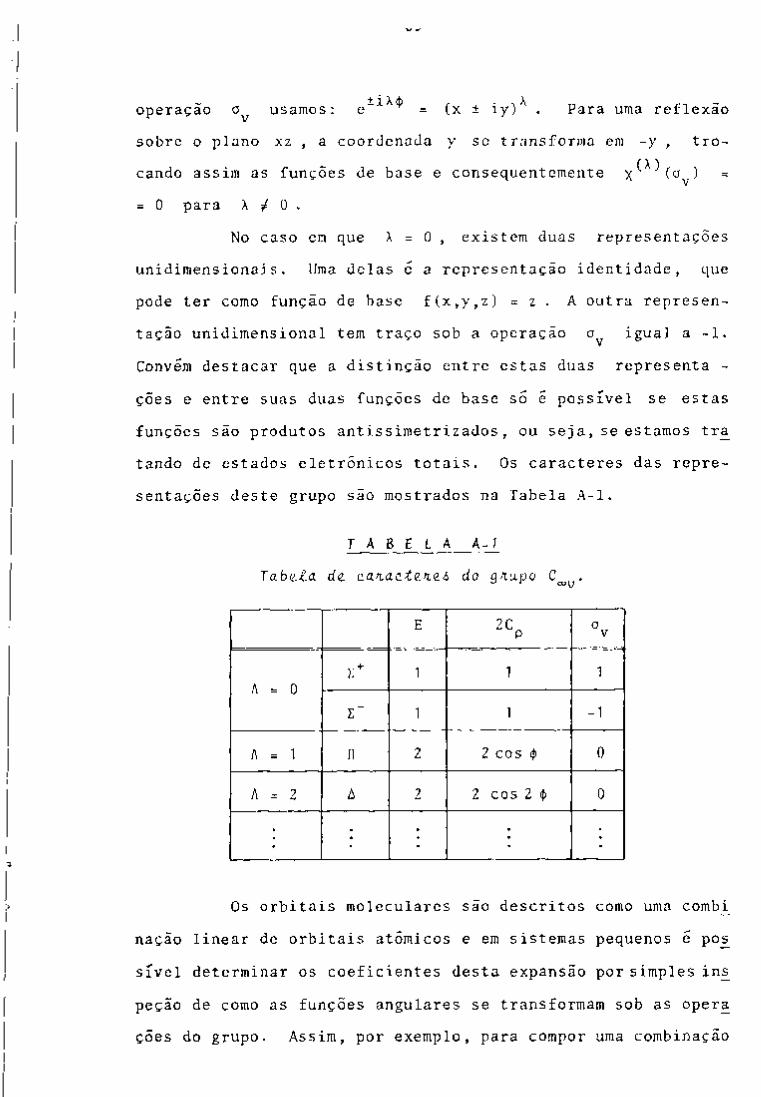
operação usamos: e*''" ' = (x ± iy)^ . Para uma reflexão
sobre o plano xz , a coordenada y se transforma em -y , tro
cando assim as funções de base e consequentemente x^^^ (^^^ =
= O para X 5^ O .
No caso em que A = O , existem duas representações
unidimensionais. Uma delas ê a representação identidade, que
pode ter como função de base f(x,y,z) = z . A outra represen
tação unidimensional tem traço sob a operação igual a -1.
Convêm destacar que a distinção entre estas duas representa -
ções e entre suas duas funções de base sõ ê possível se estas
funções são produtos antissimetrizados, ou seja, se estamos tra
tando de estados eletrônicos totais. Os caracteres das repre
sentações deste grupo são mostrados na Tabela A-l.
T A B E L A A-í
Tabela de ean.aeten.eo do gnapo C^^.
E 2 ^ \
A = 0 l* 1 1 1
A = 0
1 1 -1
A = 1 n 2 2 cos (|) 0
A = 2 A 2 2 cos 2 (J) 0
• • •
« •
• •
•
Os orbitais moleculares são descritos como uma combi^
nação linear de orbitais atômicos e em sistemas pequenos é pos
sível determinar os coeficientes desta expansão por simples in£
peção de como as funções angulares se transformam sob as opera
ções do grupo. Assim, por exemplo, para compor uma combinação

que descreva um orbital com |m| = A devemos tomar apenas fun
ções atômicas cuja dependência angular seja do tipo Y*"* .
Este grupo C^^ ê menor que o grupo da esfera, por
tanto as degenerescências das funções atômicas são quebradas.
O caracter das representações do grupo da esfera, que tem como
funções débase as harmônicas esféricas, sob uma rotação é
igual a
, fí,) . sin(£+l/2) ())
sin<})/2
Isto pode ser reescrito como:
x ' - ^ - ' c c ) = 1 + l IzosM (A.2) ^ m=l
Reduzindo estas representações ãs representações do grupo C^^,
concluimos que os orbitais atômicos geram os seguintes or
bitais moleculares:
£ = O -> a
£ = 1 ^ o + TT
£ = 2 ^ a + T T + õ , etc .. .
A-2. GRUPO D .
<»h
Como este grupo pode ser obtido pelo produto direto
entre os grupos C . e i , então ele terá o dobro do numero
de classes e consequentemente o dobro do niímero de representa
ções. Neste caso as representações podem ser pares ou ímpares,
segundo seus caracteres mudem ou não de sinal diante da opera
ção de inversão. Assim A = O correspondera a representações par e ímpar, A = 1 terá representações ir e ''y >
As funções de base das representações deste grupo de

vem ser pares ou ímpares, portanto a expansão do orbital mole
cular em termos de orbitais atômicos deve conter obrigatoria
mente orbitais centrados em ambos os átomos que agora se encon
tram em situação equivalente diante do centro de inversão. Com
isto o número de funções harmônicas esféricas a ser combinado
é duplicado e o caracter final do grupo da esfera frente ãs o-
perações deste grupo acabam sendo iguais aos caracteres origi
nais vezes o número de átomos que não trocam de posição quando
uma certa operação é efetuada. Desta maneira:
x ' - ^ - ' cC . ) = 2 + ^ 4 COS m i l * (A.3) m = 1
e X^^^(iC^) = O
Fazendo a redução destas representações ãs represen
tações do grupo D j , obtemos que combinações de orbitais
geram os seguintes orbitais moleculares:
1 = 0 ^ °g ^
£ = 2 - O g + ^g ^ % * 'g * "^U
etc. . .
Embora possamos saber como as degenerescencias atômi^
cas são quebradas, a ordenação destes orbitais em termos de e-
nergia não pode ser especificada, entretanto o esquema de ní
veis seguido pela maioria das moléculas diatómicas homonuclea
res é o indicado na Figura A-l.
Nesta figura está indicado entre paréntesis o número
de elétrons que cada orbital comporta, sendo que os orbitais
não degenerados podem ter dois elétrons de spins contrários e
os orbitais degenerados podem ter até quatro elétrons.
O estado global da molécula pode ser determinado sub
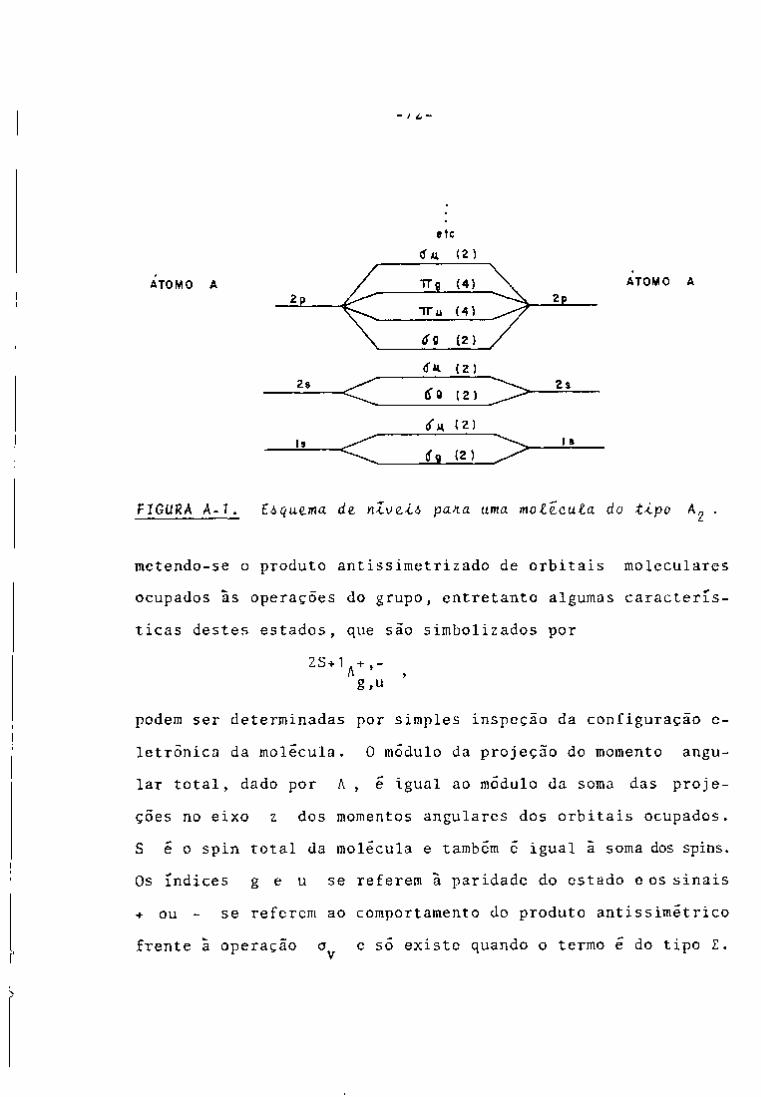
- I L -
A T O M O A 2p
28
Is <
etc
itt (2)
T T 9 (4 )
-TTu (41
(To (2)
(fU (2)
(Í0 (2;
(Tu (2)
(fa (2) >
A T O M O A
2p
2s
I s
FIGURA A- 7 . Eòqatma dz nlvzls pa.na ama molécula do tipo A^ •
metendo-se o produto antissimetrizado de orbitais moleculares
ocupados às operações do grupo, entretanto algumas caracterís
ticas destes estados, que são simbolizados por
2S+1,+ ,-
g,u
podem ser determinadas por simples inspeção da configuração e-
letrônica da molécula. O módulo da projeção do momento angu
lar total, dado por A , é igual ao módulo da soma das proje
ções no eixo z dos momentos angulares dos orbitais ocupados.
S é o spin total da molécula e também é igual ã soma dos spins.
Os índices g e u se referem ã paridade do estado e os sinais
+ ou - se referem ao comportamento do produto antissimétrico
frente ã operação e só existe quando o termo é do tipo Z.

A - 3 . REGRAS DE SELEÇÃO
kk' X
kk' z
,k* ^ ,k' , kk'
E ^ z D X
k* ij; Ey y 1 D T ,
Para obter as regras de seleção, primeiramente deter
minamos as representações irredutíveis de acordo com as quais
as partes de H' se transformam. As funções x e y se tran¿
formam, sob as operações do grupo C^^ , de acordo com a repre
sentação irredutível TT . A função z é invariante sob as ope
rações deste grupo e portanto se transforma de acordo com a
representação . Conhecendo-se R^^, e as representações se
k k ' gundo as quais as funções ip e ii se transformam, nota-se
que H^}¡) pertence a alguma representação incluída no produ-
f k ' )
to direto R^j, x . Como todas as funções de base sao or f k)
togonais, segue que se a representação T , segundo a qual
\¡) se transforma, não estiver incluída no produto direto
As regras de seleção para as transições entre esta
dos eletrônicos são determinadas pelas auto-£unções dos esta
dos envolvidos, ou mais especificamente, pela simetria dos es
tados envolvidos.
Qualquer que seja a perturbação a induzir uma transi^
ção, sabe-se que a probabilidade de transição entre dois esta
dos é proporcional ao quadrado do modulo do operador de pertur
bação conectando estes estados (Golden Rule). Para transições
induzidas por dipolo elétrico, o operador de perturbação é
H' = er.^ e, nestas circunstâncias, a probabilidade de transi^
ção é proporcional ao quadrado do módulo da quantidade veto-
nal r , onde

fk M f k* k' ^H' ^ ^ ' ^ integral ^ H' i|; dx sera nula.
O produto direto de qualquer representação r com
a representação E" reproduz a representação r*- - , assim as
regras de seleção induzidas pela componente z do momento de
dipolo são:
E" E" , E" ^ E" ,
n lí , A A , etc. . .
O produto direto de qualquer representação r - com
a representação f - gera uma representação que tem caracter
( A X T T ) ^ ^ ^ ^ = 4 C O S Acj) coscj) quando suas funções de base são
submetidas ã operação . Este caracter pode ser expresso co
mo:
C O S (A + 1) (f) + C O S (A - 1) ({)
portanto, r » " " ' , r » * " . r""!' (A.6)
assim, polarizações (x,y) geram transições entre estados tais
que AA = ± 1 . Combinando estes resultados, temos as seguin
tes regras de seleção:
i* i* , n
E" ^ E" , n
n -> E" , E~ , n , A
A -> n , A , $
No caso de moléculas diatómicas homonucleares, o gru
po de simetria é o D^j^ , sendo que a componente z do dipolo
se transforma de acordo com a representação E^ e a componen
te (x,y) de acordo com 11 . Como este grupo surge do produto
direto do grupo C^^ com o grupo da inversão, as regras de se

leção são as mesmas a menos do fato de que a paridade das fun
ções deve ser mudada pois x , y e z são funções ímpares. Por
tanto as transições são as seguintes:
E" ->- z" , n g u ' u E" z" , n u g ' g
g ^u' ^ü' \' ^
- ^¡ ' "g
"u - ^¡ ' ^¡ ' "g ' ^g
etc. . .

R E F E R É W C I A S
1. BASOV, N.G.; DANILYCHEV, V.A.; POPOV, Y.M.; KHODKEVITCH,
P.N. Laser operating in the vacuum region o£ the spectrum
by excitation o£ liquid xenon with an electron beam. JETP
Lett. , U : 329-31 , 1970.
2. BERRONDO, M.; GOSCINSKY, 0. Local exchange approximation
and the virial theorem. Phys. Rev., j J ^ C I ) : 10-16, 1969.
3. BORN, M.; OPENHEIMER, J.R. Ann. Physik, ¿7: 457, 1927
apud Tinkham M. Group Theory and Quantum Mechanics. New
York, McGraw-Hill Book Company, 1964.
4. BRESCANSIN, L.M.; LEITE, J.R.; FERREIRA, L.G. A study o£
the ground states of ionization energies of H2, C2, N2, F2
and CO molecules by the variational cellular method. J.
Chem. Phys. , ¿ 1 0 2 ) : 4923-30 , 1979 .
5. ; FERREIRA, L.G.; LEITE, J.R.; PEREIRA,
J.R.N. Variational cellular model of the electronic struc
ture of the rare gas halide excimers NeF, NeCí-, ArF and
ArC£. Rev. Brasil, de Física, 1982 - in press.
6. CHAMPAGNE, L.F.; HARRIS, N.W. The influence of diluent
gas on the XeF laser. App. Phys. Lett., 31 (8): 513-15,
1 977 .
7. Efficient operation of the electron-beam-
pumped XeC£ laser. App. Phys. Lett., 33(6): 523-25, 1978.
8. CLEMENTI, E. Tables of atomic functions. IBM J. Res.
Devel., 9: 2, 1965. ,
9. CLUGSTON, M.J.; GORDON, R.G. Electron-gas model for open
shell-closed shell interactions. I. Application to the
emission spectra of the diatomic noble-gas halides. J.
Chem. Phys. , ¿¿ü) : 239-43, 1977.

10. COHEN, J.S.; SCHNEIDER, B. Ground and excited states of
Ne2 and Ne2- I- Potential curves with and without spin-
orbit coupling. J. Chem. Phys., 6^(8^^ 3230-39, 1974.
11. DIRAC, P.A.M. Note on exchange phenomena in the Thomas
atom. Proc. Camb. Phyl. Soc. , 26 : 376-83, 1930.
12. DUNNING, T.H.J.; HAY, P.J. Electronic states of KrF.
App. Phys. Lett., 28(11): 649-51, 1976.
13. ; The covalent and ionic states
of the rare gas monofluorides. J. Chem. Phys., 69(1):
134-49, 1978.
14. EDEN, J.G.; WAYNANT, R.W.; SEARLES, S.K.; BURNHAM, R. New
quenching rates applicable to the KrF laser. App. Phys.
Lett., ¿¿(11): 733-35, 1978.
15. EWING, J.J.; BRAU, C A . Emission spectrum of Xel in elec
tron-beam-excited Xe/l2 mixtures. Phys. Rev. A, 12: 129-
32, 1975.
2 + 2 + 16. ; Laser action on the ^^/2 ^1/2
bands of KrF and XeCÄ. App. Phys. Lett. , 2^(6): 350-2 ,
1975.
17. ; High efficiency UV lasers. In:
Moradian, A. Tunable lasers and Applications. Springer-
Verlag, Berlin.
18. Rare gas halide lasers. Phys. Today., maio:
32-9, 1978.
19. Excimer lasers. In: Stitch, M.L. Laser
Handbook. North-Holland Publishing Company, New York,
1979, p. 135-196.
20. FERREIRA, L.G.; LEITE, J.R. Variational cellular model of
the molecular and crystal electronic structure. Phys. Rev.
Lett., 40(1): 49-52, 1978.

- / ti
ll. ; General formulation of the
variational cellular method for molecules and crystals.
Phys. Rev. A, ¿8(2): 335-43, 1978.
22. Método celular variacional auto-consisten
te para moléculas diatómicas - Manual e Programas. São Pau
lo, 1979.
23. FINKELNBURG, W. Kontinuiuliche Spektren, Springer-Verlag,
1938 apud Stitch, M.L. Laser Handbook, North-Holland Publ.
Co., New York, 1979, p. 138.
24. FOCK, V. Naherungsmethode zur hosung des quantenmecha-
nischen Mehrkorperproblems. Z. Phys.. 61: 126-31, 1930.
25. GASPAR, R. Acta Phys. Acad. Sci. Hung., 3: 263, 1954 apud
Kohn, W.; Sham, L.G. Self consistent equations including
exchange and correlation effects. Phys. Rev. A, 140(4):
1133-8, 1965.
26. GILBERT, T.L.; WAHL, A.C. Single-configuration wave func
tions and potential curves for low-lying states of He2,
+ + - - * Ne2, Ar2, F2, C5,2 and the ground state of Ci,2' J. Chem.
Phys., ¿5(11): 5247-61, 1971.
27. GOLDE, M.F.; THRUSH, B.A. Vaccum UV emission from reactions
of metastable inert gas atoms: chemiluminescence of ArO and
ArCA. Chem. Phys. Lett., ¿9(4): 486-90, 1974.
28. HARTREE, D.R. The wave mechanics of an atom with a non-
coulomb central field. Proc. Camb. Phyl. Soc, 24, 111-19,
1928.
29. HAY, P.J.; DUNNING, T.H. Jr. The electronic states of KrF.
J. Chem. Phys., 66(3): 1306-16, 1976.
30. ; The covalent and ionic states
of the xenon halides. J. Chem. Phys., 69(5): 2209-20, 1978.

31. ; WADT, W.R.; DUNNING, T.H. Jr. Theoretical
studies of molecular electronic transition lasers. Ann.
Rev. Phys. Chem., ¿0: 311-46, 1978.
32. HERZBERG, G. Molecular Spectra and Molecular Structure -
IV - Constants of Diatomic Molecules. D. Van Nostrand
Reinhold Company, New York, 1979.
33. HOFF, P.W.; SWINGLE, J.C. ; RHODES, C.K. Observations of
stimulated emission from high-pressure krypton and argon/
xenon mixtures. App. Phys. Lett., ¿¿(5): 245-6, 1973.
34. HOHEMBERG, P.; KOHN, W. Inhomogeneous electron gas. Phys.
Rev. B, 136(2): 864-71, 1964.
35. HOPFIELD, J.J. Absorption and Emission Spectra in the
region X600- 1100. Phys. Rev., 35: 1133-4, 1930.
36. HOUTERMANS, F.G. Über Maser-Wirkung im optischem spektral
gebiet und die möglichkeit absolut negativer absorption für
einige Fälle von molekulspektren (Licht-Lawine). Helv.
Phys. Acta, ¿3: 933-40, 1960.
37. HUGHES, W.M.; SHANNON, J.; HUNTER, R. 126.1 nm molecular
argon laser. App. Phys. Lett., Z^{10): 488-90, 1974.
38. ; OLSON, N.T.; HUNTER, R. Experiments on 558 nm
argon oxide laser system. App. Phys. Lett., ¿8(2): 81-3,
1975.
39. ATCHINSON, M.H. Excimers and Excimer Lasers. App. Phys.,
21: 95-114, 1980.
40. JOHNSON, K.H. Quantum Chemistry. Ann. Rev. Phys. Chem.,
¿6: 39-57, 1975.
41. KMETKO, E.A. Single parameter free-electron exchange ap
proximation in free atoms. Phys. Rev. A, ¿(1): 37-8, 1970.
42. KOEHLER, H.A.; FERBERDER, L. J. ; REDHEAD, P.L.; EBERT, P.J.
Stimulated VUV emission in high-pressure xenon excited by

- i S U -
high-current relativistic electron beams. App. Phys. Lett.,
21(5): 198-200, 1972.
43. KOHN, W.; ROSTOKER, N. Solution of the Schrödinger Equation
in periodic lattices with an application to Metallic Lithium.
Phys. Rev., 94(5): 1111-20, 1954.
44. ; SHAM, L.G. Self-consistent equations including
exchange and correlations effects. Phys. Rev. A, 140 (4):
1133-8, 1965.
45. KOMPA, K.L.; WALTHER, H. High-power lasers and applications.
Springer-Verlag, Berlin. (Springer Series in Optical Sciences,
9).
46. KOOPMANS, J.H. Über die Z u o r d n u n g von wellenfunktionen und
e i g e n w e r t e n zu deu eizelnen e l e k t r o n e n eines atoms. Physica
1: 104-9, 1933.
47. LEITE, J.R.; BENNETT, B.I.; HERMAN, F. Electronic structure
of the diamond crystal based on an improved cellular calcu
lation. Phys. Rev. B, ¿2(4): 1466-81, 1975.
48. 0 método celular moderno. São Paulo, 1974 (Te
se de Livre-Docência, Instituto de Física, Universidade de
São Paulo).
49. ; FAZZIO, A.; LIMA, M.A.P.; DIAS, A.M.; ROSATO,
A.; SEGRE, E. The variational cellular method for quantum
applications: Calculations of the ground and excited states
of F2 and Ne2 molecules. Int. J. Quant. Chem., 15: 401-8,
1981.
50. LETOKHOV, V.S. Laser-induced chemical processes. Phys.
Today, novembro: 34-41, 1980.
51. LIMA, M.A.; FERREIRA, L.G. One parameter electronic densi
ties in atoms. Phys. Rev. A, 2¿: 343-7, 1980.

-o j . -
52. LORD RAYLEIGH, F.R.S. Series.of emission and absorption
bands in the mercury spectrum. Proc. R. Soc. London,
A116: 702-19, 1927.
53. LORENTS, D.C.; HUESTIS, D.L.; McCUSKER, M.V. NAKANO, H.H.;
HILL, R.M. Optical emission of triatomic rare gas halides.
J. Chem. Phys., 68(10), 1978.
54. MAGEE, J.L. The mechanism of reaction involving excited
electronic states. J. Chem. Phys., 8: 687-92, 1940.
55. MANDL, A. Ionization cross sections of F 2 and by elec
tron impact. J. Chem. Phys., ¿7(10): 4104-6, 1972.
56. MANN, J.B. Atomic structure calculations. Los Alamos
Scientific Lab. Rep. 1967 (LA-3690).
57. MICHELS, H.H.; HOBBS, R.H.; WRIGHT, L.A. The electronic
structure of ArF and Ar^F. Chem. Phys. Lett., £8(1):
158-61, 1977.
58. ; ; ; CONNOLLY, J.W.D.
Electronic structure of excimer molecular lasers. Int. J.
Quant. Chem., 13: 169-87, 1978.
59. ; ; Electronic struc
ture of noble gas dimer ions. I. Potential energy curves
and spectroscopic constants. J. Chem. Phys., 69(11):
5151-62, 1978.
60. MOSELEY, J.T.; SAXON, R.P.; HUBER, B.A.; COSBY, P.C.;
ABONAF, R.; TADJEDDINE, M. Photofragment spectroscopy and
potential curver of Ar2. J. Chem. Phys. , 67: 1659-68, 1977.
61. PAVÃO, A . C ; LEITE, J.R. An application of the exchange-
correlation mass operator approximation to the study of
s-doublet structure of some 3d-transition metal ions. J.
Phys. Chem. Sol., ¿1(9): 953-8, 1980.

62. POWELL, H.T.; MURRAY, J.R.; RHODES, C.K. Laser oscillation
on the Green bands of XeO and KrO. App. Phys. Lett., 2 5 (12)
730-2, 1974.
63. ROKNI, M.; MANGANO, J.A.; JACOB, J.H.; HSIA, J.C. Rare gas
fluoride lasers. IEEE J. Quant. Chem., 14(7): 464-81, 1978.
64. SANTOS, M.C. Polarização de spin no método celular varia
cional . São Paulo, 1981 (Tese de mestrado. Instituto de
Física, Universidade de São Paulo).
65. SCHNEIDER, B.I.; COHEN, J.S. Ground and excited states of
Ne2 and Ne^. II. Spectroscopic properties and radiative
lifetimes. J. Chem. Phys., ( 8 ) : 3240-3, 1974.
66. SCHLÜTER, M.; SHAM, L.J. Density funtional theory. Phys.
Today, Fevereiro: 36-43, 1982.
67. SCHWARZ, K. Optimization of the statistical exchange
parameter a for the free atoms H through Nb. Phys. Rev. B,
5(7): 2466-68, 1972.
68. SEARS, V.F. Laser for inertial confinement fusion research
Chalk River, Ontario, Chalk River Nuclear Lab., Aug. 1980,
(AECL-7058).
69. SILVA, E.G.F. Propriedades elásticas e momento de dipolo
em moléculas diatômicas. São Paulo, 1982 (Tese de mestra
do. Instituto de Física, Universidade de São Paulo).
70. SLATER, J.C. The theory of complex spectra. Phys. Rev.,
¿4: 1293-9, 1929.
71. Electronic energy bands in metals. Phys.
Rev., £5: 794-9, 1934.
72. Wave function in a periodic potential. Phys.
Rev., 5^: 846-51, 1937.
73. A simplification of the Hartree-Fock method.
Phys. Rev., 81: 385-92, 1951.

74. Quantum theory of atomic structure. Vol. I,
Cap. 8: 188-209, New York, McGraw-Hill Book Comp., 1960.
75. Quantum theory of atomic structure, Vo1. II,
Cap. 18: 31-47, New York, McGraw-Hill Book Comp., 1960.
76. SLOCOMB, C.A.; MILLER, W.H.; SCHAEFER, H.F. Collisional
quenching of metastable hydrogen atoms. J. Chem. Phys.,
¿5(2): 926-32, 1971.
77. SNOW, E.G. CANFIELD, J.M.; WABER, J.T. Total energies from
numerical self-consistent field calculations. Phys. Rev. A,
¿35(4): 969-73, 1964.
78. STEVENS, W.J.; GARDNER, M.; JULIENE, P.; KARO, A.
Theoretical determination of bound-free absorption cross
sections inAr^. J. Chem. Phys., 67(6): 2860-7, 1977.
79. TANAKA, Y. Continuous emission spectra of rare gases in
the vacuum ultraviolet region. J. Opt. Soc. Am., £5(9):
710-3, 1955.
80. ; JURSA, A.S.; LE BLANC, F.J. Continuous emission
spectra of rare gases in the vacuum ultraviolet region. II.
Neon and Helium. J. Opt. Soc. Am., 4^(5): 304-8, 1958.
81. TINKHAN, M. Group theory and quantum mechanics. Cap. 7,
New York, McGraw-Hill Book Comp., 1974.
82. VELAZCO, J.E.; SETZER, P.W. Bound-free emission spectra of
diatomic xenon halides. J. Chem. Phys. , 62^(5): 1990-1,
1975 .
83. WADT, W.R.; CARTWRIGHT, D.C.; COHEN, J.C. Theoretical
absorption spectra for Ne2, Ar^, Kr^ and Xe2 in the near
ultraviolet. App. Phys. Lett., ¿1(10): 672-4, 1977.
84. ; HAY, P.J. The low-lying electronic states of
Ar2F. App. Phys. Lett., jj(ll): 573-5, 1977.

- 5 4 -
85. The electronic states of Ar2, ^ "2 ' ®2' *
tential curves with and without spin-orbit coupling. J.
Chem. Phys., 68(2): 402-14, 1978.
86. Electronic states of Ar2F and Kr2F. J. Chem.
Phys., ¿8(8): 3850-63, 1978.
87. WEAST, R.C. Handbook of Chemical and Physics - 53- ed.
Cleveland, Ohio, Chemical Rubber Co., 1972-73, E 56.
88. WIGNER, E.; SEITZ, F. On the constitution of metallic
sodium. Phys. Rev., 43: 804-9, 1933.

INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES SECRETARIA DA INDÚSTRIA. COMÉRCIO. CIÊNCIA E TECNOLOGIA
AUTARQUIA ASSOCIADA A UNIVERSIDADE DE SÃO PAULO
ESTUDO E CARACTERIZAÇÃO DE DIURANATO DE AMÓNIO E TRIÓXIDO DE URÂNIO POR TERMOGRAVIMETRIA E
CALORIMETRIA EXPLORATÓRIA DIFERENCIAL
JOSÉ MAIA DANTAS
Dissertação apresentada como parte dos requisitos para a olitençao do Grau de "Mestre na Área de Concentração em Reatores Nucleares de Potdnda e Tecnologia do Combustível Nuclear".
Orientador: Dr. Alcídio Abrão
Sao Paulo 1983

INSTITUTO DE PESQUISAS ENERGÉTICAS E NUCLEARES
AUTARQUIA ASSOCIADA À UNIVERSIDADE DE SÂO PAULO
V
ESTUDO E CARACTERIZAÇÃO DE DIURANATO DE AMÓNIO E
TRIÓXIDO DE URÂNIO POR TERMOGRAVIMETRIA E
CALORIMETRIA EXPLORATÓRIA DIFERENCIAL
José Mala Dantas
Dissertação apresentada como parte dos
requisitos para obtenção do Grau de "Mestre
na Área de Concentração em Reatores
Nucleares de Potência e Tecnologia do.
Combustível Nuclear". '
Orientador: Dr. Alcídio Abrão
1 SAO PAULO
1983
INST I T U T O O E P H G Q U ' S * ' s e N U C Í F A R E s J

Ä Beth
Ao Joòé. Gullke.rrm
Ao¿ rma¿ pais e Zrmãoò

AGRAVECJMENTOS
O auton. deszja. zxpà.zssafL ¿ua gratidão a
todos aqueles íue, diretamente ou Indiretamente, eo¿abo_
raram na realização deste trabalho, Em especial, agra
deço:
- Dr. AlaZdlo Abrão, orientador desta dis
sertação, pela orientação segura, pelos eselareclmen -
tos, pelos ensinamentos, pelo apolo, pela dedicação e
pela amizade demonstrada durante a execução deste traba
Ihoí
- Instituto de Pesquisas Energéticas e Nu
cleares e, em particular, ao Departamento de Engenha
ria duZmlca, em cajos laboratorios me fol dada a o porta
nldade de realizar o trabalho experimental;
- Vra. Ludmlla federgrun e sua equipe do
laboratório Analítico, ptla contribuição concedida;
- Vra. laura Tognoll Atalla, responsãvel
pelo Laboratorio de Análises Instrumentais, pela colabo_
ração e compreensão;
- Integrantes da ComA,ssão de Pos - Graduação,
da Vlvlsão de Informação e Vocumentação Cientifica;
- Colegas do Vepartamento de Engenharia
Química, pela cooperação e companheirismo,

CSTUDO E CARACTERIZAÇÃO DE DIURANATO DE AMÕNIO E TRIÕXIDO DE
URÂNIO POR TERMOGRAVIMETRIA E CALORIMETRIA EXPLORATÓRIA DIFE-
RENCIAL
JOSÉ MAIA DANTAS
R E S U M O
O comportamento térmico de diuranatos de amónio
e trióxido de urânio, produzidos no Departamento de Engenharia
Química do Instituto de Pesquisas Energéticas e Nucleares, foi
estudado por termogravimetria (TG), termogravimetria derivada
(DTG) e calorimetria exploratória diferencial (DSC). Os resul
tados iniciais confirmaram os dados da literatura, constatando
se a influência da variação dos parâmetros de precipitação e
secagem sobre o comportamento térmico destes compostos.
Em estudos posteriores verificou-se que o tempo
de envelhecimento dos diuranatos de amónio i muito importante
e foi considerado na avaliação da influSncia dos parâmetros de
precipitação e secagem sobre o comportamento térmico destes
compostos.

A caracterização dos compostos foi realizada
por meio das razões molares entre seus constituintes e respec
tivos õxidos resultantes da termodecomposição.
A interpretação das curvas TG e DTG permitiu i.
dentificar, para cada amostra, as temperaturas de eliminação
de água (livre e de cristalização), evolução e oxidação de a-
raônla, decomposição do nitrato de amonio ocluido e perda de o
xiginio, bem como as fases intermediárias e a estabilidade
térmica, fatos estes confirmados pelas curvas DSC. Os proce£
SOS endotérmico e exotérmico correspondentes foram assinala -
dos nas curvas DSC.
Deste estudo resultou um número considerável
de informações as quais poderão ser coordenadas e interpreta
das para uso e orientação nas operações das usinas piloto de
obtenção de DUA, UO3 e UF^.

STUDY AND CHARACTERIZATION OF AMMONIUM DIURANATE AND URANIUM
TRIOXIDE BY THERMOGRAVIMETRY AND DIFFERENTIAL SCANNING
CALORIMETRY.
JOSÉ MAIA DANTAS
A B S T R A C T
; Thermogravimetry (TG), Differential Ther
mogravimetry (DTG) and Differential Scanning Calorimetry (DSC)
were used to characterize the thermal behavior of ammonium dinrana
te (ADU) and uranium trioxide (UOg) produced at IPEN'S Chemical
Engineering Department.
Preliminary studies showed that t h e r e is
an influence of the precipitation and drying parameters on the
thermal behavior of these compounds in agreement with the r e s u l t s
available from the literature. Further studies showed that, for
t h e ADU powder, the precipitation and drying parameters are
strongly influenced by ageing time.
Compounds characterization was done using
t h e m o l a r r a t i o s among t h e compounds and t h e o x i d e s r e s u l t i n g f r o m
t h e r m a l d e c o m p c s i t i o n .
The TG and DTG c u r v e s r e g i s t e r e d f o r e a c h

sample were used for the determination of the following tempera tures :
- temperature of water evolution (free and crystallized water);
- ammonia evolution and oxidation temperature; - ocluded ammonium nitrate decomposition temperature and - oxygen release temperature.
The intermediate phases and'their ther mal stabilities were also identified by TG and DTG and con firmed by DSC curves, DSC curves showed also the exothermic and endothermic behavior of the processes involved.
Finally, the great amount of data collected in this study can be handed as a guide by the profes_ sionals responsible for the operation of ADU, UOg and UF^ pilot plants.

S U M A R I O
Pág.
RESUMO
ABSTRACT
I.-* INTRODUÇÃO 1
1.2 - Objetivo 2
1.3 - Estudos realizados 3
¥ 1.4 - Conceitos e considerações gerais sobre
as técnicas termoanalíticas 4
•I.U.l - Introdução 4
1.4.2 - Revisão bibliográfica 8
< 1.4.3 - Características das curvas ter
moanalíticas 10
1.4.4 - Medida de temperatura em análi
se térmica 13
1.4.4.1 - Termopares 14
1.4.5 - Fontes de erros em termogravi
metria e análise térmica dife
rencial 15
»1.4.6 - Comparação entre termogravime
tria e termogravimetria deriva
da ' 18
»t 1.4.7 - Comparação entre analise têrmi
ca diferencial e calorimetria
exploratoria diferencial 20
I

1.4.8 - Aplicação da termogravimetria,
análise térmica diferencial e :.
calorimetría exploratoria dife
rencial • 26
II. REVISÃO BIBLIOGRÁFICA 29
II. 1 - Preparação, composição e estrutura de
diuranato de amonio 29
II. 2 - Preparação de UO3 37
11.2.1 - Decomposição térmica de nitra
to de uranilo 38
11.2.2 - Decomposição térmica do peróxido
de urânio 39
11.2.3 - Decomposição térmica do tricar-
bonato de uranilo e amonio 39
11.2.4 - Decomposição térmica do diurana
to de amonio 40
11.3 - Decomposição de compostos de urânio por
técnicas termoanalíticas 40
II.3.1 - Decomposição térmica de diurana
tos de amonio 41
11.3.1.1 - Decomposição térmica do DUA
ao ar 44
11.3.1.2 - Decomposição térmica do DUA
em atmosfera inerte 46
11.4 - Influência das propriedades do DUA e UOg
sobre as características do UOj 47

Pig
III. PARTE EXPERIMENTAL 50
III.l - Equipamentos 50
III.1.1 - Especificações dos módulos de
analise térmica 5 0
111.1.1.1 - Modulo central 50
111.1.1.2 - Célula base 53
111.1.1.3 - célula de calorimetria expio
ratoria diferencial 54
111.1.1.4 - célula de analise térmica di
fereficial (500°C) 56
111.1.1.5 - Analisador termogravimetrico. 57
III. 2 - Materiais 6 2
III. 3 - Comportamento térmico 63
111.3.1 - Variação de cor dos diuranatos
de amónio e trióxido de urânio
durante o aquecimento 6 3
111.3.2 - Influência da etapa de secagem so
bre o comportamento térmico do
diuranato de amonio 6 5
111.3.3 - Decomposição térmica de diurana
tos de amonio envelhecidos 6 6
III.4 - Caracterização dos diuranatos de amo
nio e trióxidos de urânio 6 7
111.4.1 - Curvas termogravimétricas e ter-
mograviraétricas derivadas 67
111.4.2 - Curvas calorimétricas explorató
rias diferenciais 68

Pâg.
III.5 - Procedimentos analíticos 6 9
1 1 1 . 5 . 1 - Determinação de urânio 6 9
1 1 1 . 5 . 2 - Determinação do teor de amonia.. 7 0
III. 5 . 3 - Determinação de lons nitrato- ... 7 0
1 1 1 . 5 . 4 - Determinação do teor de água ... 7 0
1 1 1 . 5 . 5 - Determinação das razões molares
N H J / U , NO3 / U e H2O / U 7 1
1 1 1 . 5 . 6 - Determinação da razão O / U no re
síduo de calcinação dos compos
tos 7 1
1 1 1 . 5 . 7 - Determinação da densidade 7 2
1 1 1 . 5 . 8 - Determinação da superfácie es
pecífica 7 2
IV - RESULTADOS 7 3
IV.1 - Experimentos preliminares 7 3
IV.1 . 1 - Quantidade de amostra 7 3
IV. 1 . 2 - Tamanho do grão 7 4
IV.1 . 3 - Variação de cor durante o aqueci
mento dos diuranatos de amonio ao
ar 7 4
IV.1 . 4 - Estudo compar^ativo do comportamen
to térmico de quatro tipos de diu
ranato- de amonio envelhecidos... 7 6
IV. 2 - Estudo da influência dos parâmetros de
secagem sobre o comportamento térmico
dos diuranatos de amonio 8 3

Pág.
IV.2.1 - Secagem no módulo de termogravi
metria e decomposição dos diura
natos maciços 83
IV.2.2 - Secagem em estufa e decomposição
térmica dos compostos pulveriza
dos 9 2
IV.3 - Estudo da influência do tempo de enve
lhecimento dos diuranatos de amónio
sobre o comportamento térmico 102
IV.4 - Estudo da influência da atmosfera na
decomposição dos diuranatos de amónio. 110
IV.5 - Influência do teor de nitrato de amó
nio sobre o comportamento térmico dos
diuranatos 118
IV.6 - Caracterização dos diuranatos de amó
nio 126
IV.6.1 - Curvas termogravimétricas e ter
mogravimitricas derivadas 126
IV.6.2 - Curvas de calorimetria explórate
ria diferencial 127
IV.7 - Caracterização do trióxido de urânio.. 140
IV.7.1 - Curvas termogravimétricas e ter
mo gravimêtricas derivadas 140
IV. 7.2 - Curvas de calorimetria explórate
ria diferencial 140
IV. 8 - Dados analíticos 147

Pãg.
V - Discussão e Conclusões 155
V.l - Efeito da secagem sobre o comportamento
térmico e composição do diuranato de amo
nio 155
V.2 - Efeito da calcinação sobre o comportamen
to térmico e composição do trióxido de
urânio 158
V.3 - Efeito do tempo de envelhecimento sobre
o comportamento térmico do diuranato de
amonio 160
V.4 - Efeito do nitrato na decomposição térmi
ca do diuranato de amonio 160
V.5 - Mecanismo geral da decomposição térmica
dos diuranatos de amonio 163
V.6 - Conclusão geral 172
Apêndice 1 - Calculo do teor de urânio em óxidos
de urânio por termogravimetria 17 6
Apêndice 2 - Resultados da decomposição térmica
dos diuranatos 2-C, 3-C e 12-C, seca
dos a 75°C e 150°C por II4I4Ü, 2880 e
. 4320 minutos 182
Apêndice 3 - Resultados da decomposição térmica
dos diuranatos 2-C, 3-C e 12-C, seca
dos a 75°C por 2880 minutos . e a
150°C por 4320 minutos (2-C) e 2880
minutos (3-C e 12-C) 197
REFERÊNCIAS BIBLIOGRÁFICAS 204

I. INTRODUÇÃO
O interesse no estudo do diuranato de amonio
(DUA) e trióxido de uranio (UO^) se prende, principalmente,ao
fato de estes compostos serem utilizados na produção seqüenci
al de dióxido de uranio (UO2), tetrafluoreto de uranio • (UF )
e hexafluoreto de uranio (UF-), essenciais ao ciclo do uranio
como combustível nuclear.
A produção destes compostos, no Departamento
de Engenharia Química do Instituto de Pesquisas Energéticas e
Nucleares, é controlada em termos do teor de agua livre to-
tal, teor de uranio e óxidos totais, teor de impurezas metáli
cas, densidade, superfície específica e tamanho médio de par
tículas . O tempo consumido na execução e interpretação dos da
dos obtidos destes procedimentos analíticos, tendo em vista a
avaliação das propriedades e características de cada composto,
é grande e constitui-se num problema para o processamento do
uranio, uma vez que, o bom andamento das fases subseqüentes ã
obtenção do DUA refletem, de certo modo, as condições de ori-
- (33) gem de sua preparação
Indiferente ao sistema de produção (contínuo
ou descontínuo), do ponto de vista tecnológico, o interesse
principal concentra-se na reprodutibilidade durante a produ
ção do DUA, com o objetivo de obter-se UG2 ou UF ^ com caracte
risticas inalteradas. Assim, é necessário caracterizá-lo

(DUA) por um método simples e rápido.
As técnicas termoanalíticas, além da simplici
dade e rapidez de caracterização, apresentam a vantagem de in
dicar com exatidão as tempreaturas de eliminação de água (li
vre e de cristalização), da decomposição do nitrato de amonio
ocluido, de decomposição térmica, de transformação do UOg em
UgOg, de determinar o teor de óxidos de urânio no DUA, bem co
mo mostrar os correspondentes processos endotêrmidos e exotér
micos ocorridos. São uteis, também, para se determinar a tem
peratura ótima de secagem do DUA e conhecer a reação de óxido-
redução da amonia retida pelo óxido intermediário. Desse mo
do, estas técnicas proporcionam um meio valioso para o estudo
das reações, caracterização e estabelecimento do mecanismo e
estequiometria da decomposição térmica do DUA e UO^, tendo em
vista o uso destes compostos na produção do UO2 e UF^.
1.2. OBJETIVO
O objetivo principal do presente trabalho con-
Sistiuem conhecer a natureza geral da decomposição termina do
diuranato de amónio e trióxido de urânio, produzidos nas uni
dades piloto do DEQ-IPEN, até a fase U^Og, por termogravime
tria e calorimetria exploratória diferencial. Paralelamente,
estabelecer uma correlação entre os dados da termodecomposi -
ção com os valores dos parâmetros de precipitação e secagem
do diuranato de amónio.
I N S T I T U T O D E P J S Q U í ^ a S E ' . ^ P : É " I C S E N U C L E A R E S
i. P . t. N.

1.3. ESTUDOS REALIZADOS
Os estudos descritos neste trabalho se referem
âs características da decomposição térmica de pós de diurana
to de amonio e trióxido de urânio, produzidos nas unidades pi.
loto do DEQ-IPEN. Foi estudada a decomposição térmica desses
compostos em atmosfera comum (ar) e nitrogênio seco.
Os resultados de experimentos iniciais, com
compostos envelhecidos em ambiente de laboratório, são apre
sentados no Capítulo IV. Nestes experimentos verificou-se al
gumas duvidas acerca da influência dos parâmetros de precipi
tação e secagem, sobre as características térmicas presentes
nas curvas termoanalíticas desses diuranatos de amonio. Com -
postos novos foram então utilizados no estudo da influência
dos parâmetros de preparação e do tempo de envelhecimento do
DUA sobre o seu comportamento térmico. Em experimentos poste
riores, apresentados também no Capítulo.IV, foram feitas ob
servações a respeito da variação da cor dos compostos em fun
ção do aquecimento.
De acordo com os objetivos, foram estudados
preferencialmente, os compostos produzidos diretamente nas u-
nidades piloto do DEQ-IPEN, nas condições usuais, 'descritas
no Capitulo III. Na sua maioria, as amostras selecionadas fo
ram precipitadas em pH acima de 7,0, portanto, produzidas em
excesso de amonia.
O conhecimento do comportamento térmico de vâ-

rios diuranatos produzidos sob condições determinadas possibi^
litou a obtenção de importantes informações acerca da composi
ção e características térmicas desses compostos. Foi dada ên
fase especial aos parâmetros de precipitação e secagem e a
sua influência sobre os teores de amonia e nitrato ocluidos
nos sólidos, bem como sobre o comportamento térmico do trióxi.
do de urânio.
A caracterização dos coiiipostos foi realizada
pela associação,dos resultados termoanaliticos com os dados a
naliticos, referentes aos teores de urânio, amonia e nitrato,
obtidos por técnicas volumétricas e pela espectrofotometria
de absorção molecular.
Faz parte desse trabalho uma revisão do grande
número de publicações que tratam da quimica dos compostos de
urânio, em especial, diuranato de amonio e trióxido de urâ
nio, pelo uso das técnicas termoanalíticas.
1.4. CONCEITOS E CONSIDERAÇÜES GERAIS SOBRE AS TÉCNICAS TERMO
ANALÍTICAS
1.4.1. Introdução
Algumas considerações teóricas serão feitas ,

5.
relativas somente às técnicas termoanalíticas que, no presen
te trabalho, foram posteriormente utilizadas na pratica. De£
se modo, as técnicas consideradas serão: a termogravimetria,
a termogravimetria derivada, analise térmica diferencial e a
calorimetría exploratoria diferencial.
As informações descritas a seguir estão de a-
cordo com a revisão da nomenclatura em análise térmica, con-
densada por lONASHIRO e GIOLITO em 1980.
^ As técnicas termo ana1ít i cas são aquelas que
envolvem a medição de uma propriedade física de Jsubstâncias
ou materiais capazes de sofrer variações em função da tempe
ratura. Em princípio têm ym procedimento comum de operação
que consiste em submeter a amostra a um aquecimento ou res
friamento segundo um programa pré-determinado, enquanto se
registra alguma propriedade da amostra como uma função da
temperatura. O registro obtido é a curva termoanalítica.
Entre as técnicas termoanalíticas destacam-se,
atualmente, pelas numerosas aplicações nos mais variados cam
pos científicos e tecnológicos, a termogravimetria (TG), a
análise térmica diferencial (DTA) e a calorimetría explórate
ria diferencial (DSC). A Tabela I.l relaciona estas técni
cas, o parâmetro registrado, a instrumentação usada e a cur
va termoanalítica típica.
A termogravimetria é uma técnica na qual a va
riação de massa de uma substância e/ou de seu(s) produto (s)
de reação é medida em função da temperatura, enquanto a subs_

6 .
tância e submetida a uma programação controlada de tempera
tura. A curva correspondente, de perda ou ganho de massa
em função da temperatura, é denominada curva termogravime -
trica ou simplesmente curva TG.
A analise térmica diferencial é uma técnica
que consiste em registrar a diferença de temperatura entre
uma substancia e/ou de seu(s) produto(s) de reação e um ma
terial de referência em função da temperatura, enquanto as
duas substâncias são.submetidas a uma programação controla
da de temperatura. O gráfico obtido é denominado curva tér
mica diferencial ou curva DTA.
A calorimetria exploratória diferencial é u-
ma técnica na qual se mede a diferença de energia fornecida
a uma substância e/ou seu(s) produto(s) de reação e a um
material de referência em função da temperatura, :. enquanto
os dois materiais são submetidos a uma programação controla
da de temperatura. O registro é a curva calorimétrica ex
ploratória diferencial ou curva DSC.
A caracterização térmica de uma substância,
aquecida a temperaturas elevadas, por meio dessas técnicas
pode fornecer informações a respeito da cinética e varia
ções de entalpia de reação de decomposição, composição quí
mica de produtos intermediários e resíduos, estabilidade
térmica, temperaturas de transições de fase e calores de re
ação.
As informações obtidas por estas ¿técnicas.

TABE
LA
I.l
- T
ÉC
NIC
AS
T
ER
MO
AN
AL
ITIC
AS
U
SA
DA
S
NE
ST
E
TR
AB
AL
HO
E
S
UA
S
CA
RA
CT
ER
ÍST
ICA
S
TÉ
CN
ICA
P
AR
ÁM
ET
RO
M
ED
IDO
IN
ST
RU
ME
NT
O
RE
GIS
TR
O
TÍP
ICO
TE
RM
OG
RA
VIM
ET
RIA
(T
G)
TE
RM
OG
RA
VIM
ET
RIA
DE
RIV
AD
A (
DT
G)
MA
SS
A
dm
/dt
AT
(T
a-T
r)
FL
UX
O D
E C
ALO
R
• dQ
dt
TE
RM
OB
AL
AN
ÇA
TE
RM
OB
AL
AN
ÇA
SIS
TE
MA
D
TA
CA
LO
RÍM
ET
RO
AN
AL
ISE
T
ÉR
MIC
A
DIF
ER
EN
CIA
L (D
TA
)
CA
LO
RIM
ET
RIA
E
XP
LO
RA
TO
RIA
D
IFE
RE
NC
IAL
(DS
C)
T
= T
EM
PE
RA
TU
RA
Ta
s T
EM
PE
RA
TU
RA
D
A
AM
OS
TR
A
Tr=
T
EM
PE
RA
TU
RA
D
O
MA
TE
RIA
L IN
ER
TE
U
SA
DO
C
OM
O
RE
FE
RE
NC
IA
Q
= C
ALO
R
MA
SS
A
dm
/dt
(+)
AT
(-)
dQ
/dt

8.
quando associadas com aquelas resultantes da difração de
raios-X,análise óptica e química dos resíduos e gases produ
zidos, fornecem uma avaliação quantitativa das reações no
estado sólido.
1.4.2. Revisão Bibliográfica
A representação gráfica da influência de va
riações de temperatura sobre as propriedades térmicas dos
materiais tem sido estudada por vários métodos desde o sécu
(22)
Io XIX . Entretanto, somente alguns desses métodos se
desenvolveram e obtiveram ampla aplicação nos campos cientí
fieos e tecnológicos nos últimos trinta anos.
A análise térmica diferencial, DTA, é sem dú
vida o método termoanalítico mais utilizado. Seu princípio
(135
foi estabelecido por Le CHATELIER , no século passado,
por meio de um trabalho sobre a caracterização de argilas e
minerais.
A termogravimetria, TG, foi introduzida no i
nício deste século, quando foi possível a obtenção do regi£
tro contínuo da variação de massa em função da .temperatura
com os trabalhos de NERNST e RIESENFELD e com a
( 23)
construção da primeira termobalança. por HONDA , . Da mesma
maneira como ocorreu com a análise térmica diferencial, por
muito tempo ignorada pelos químicos, mas indispensável aos
trabalhos dos mineralogistas e ceramistas na identificação

9.
A termogravimetria derivada, DTG, teve seu
início em 19 34 . ,, por meio de medidas e registro manuais,
(27)
posteriormente, aperfeiçoada por ERDEY em 19 54, apresen
tando curvas TG, muito semelhantes âs curvas DTA, com picos
seqüenciais ou sobrepostos correspondentes ã velocidade de
variação de massa da amostra em função de tempo ou da tempe
ratura.
Dos métodos termoanaliticos, a calorimetria
exploratoria diferencial, DSC, é o mais recente, desenvolvi^
do pela Perkin Elmer Corporation e descrito pela primeira f o -1 >
vez em 1964 por WATSON e colab. .
Os métodos de análise térmica têm sido ampla
mente aceitos na quimica analítica, inorgânica, orgânica e
outras áreas de pesquisa nos últimos 2 5 anos. Por meio de
encontros e simpósios, as três Sociedades de interesse co
mum neste campo de trabalho, a North American Thermal
Analysis Society (NATAS), a International Confederation of
Thermal Analysis (ICTA) e a Society of Calorimetry Analysis,
promovem a discussão e as aplicações das técnicas de análi^
se térmica.
de minerais e argilas, houve um longo intervalo de tempo até
que a termogravimetria fosse aplicada na resolução de
problemas analíticos. Apesar dos esforços e trabalhos produ
zidos por pesquisadores, das escolas japonesa e francesa de
( 3 6 )
termogravimetria , no estudo da decomposição térmica de
compostos inorgânicos naturais e de inúmeros precipitados ,
(23) . - . somente em 1947, DUVAL introduziu, de modo sistemático,
suas aplicações no campo da análise gravimétrica inorgânica.

10.
o progresso nessa area de pesquisa recebeu
contribuição preponderante pela centralização de publica
ções em três periódicos especializados e reconhecidos, o
Journal of Thermal Analysis (1969), Thermochimica Acta
(1970) e Thermal Analysis Abstracts (1972).
1.4.3. Características das curvas termoanalíticas
* As seguintes características podem ser iden
tificadas na Figura I.l., que mostra uma curva termogravime
trica (a), uma curva termogravimétrica derivada (a) e uma
curva de analise térmica diferencial (b) representativas de
um processo que ocorre em uma única etapa, de acordo com
e RE:
,(54.)
COATS e REDFERN - ^ - , lONASHIRO e GIOLITO^^^, NEWKIRK e
SIMONS
Figura I.l.a: O patamar AB é a parte da curva TG onde a
massa é essencialmente constante. O segmen
to A'B', equivalente ao patamar AB da curva TG, é a parte
da curva DTG onde a velocidade de variação de massa é igual
a zero.
A temperatura inicial, T ^ - B na curva TG, é
a temperatura (nas escalas Celsius ou Kelvin) na qual as va
riações acumuladas de massa totalizam o valor que a balança
é capaz de detectar.
B' na curva DTG, é a temperatura (nas esca-

11,
A temperatura do pico, T^ na curva DTG, é
a temperatura (nas escalas Celsius ou Kelvin) na qual a ve
locidade de variação de massa atinge valor máximo. Na cur
va TG, Tp pode ser caracterizada como sendo a temperatu
ra de inclinação máxima da inflexão BC.
O intervalo de reação é a diferença de tem
peratura entre T^ (C ou C ) e T^ (B ou B'), definidos aci
ma .
Figura I.l.b: A linha base, AB e DE, corresponde ã por
ção ou porções da curva DTA, nas quais AT é aproximadamen
te zero.
Um pico, BCD, é a porção.da curva DTA - que
se afasta da linha base, e, posteriormente, retorna ã mes
ma .
•¡"iTpESGU'ÑASE'.ÍRRJÉTLC.'SE N U C L E A R E S .| Í M 3 T I T U T O D
i. P . 'd. N .
las Celsius ou Kelvin) na qual a velocidade de variação, de
massa apresenta um valor diferente de zero, que a balança
é capaz de detectar.
A temperatura final, '^f ~ Ç curva TG,
e a temperatura (nas escalas Celsius ou Kelvin) na qual as
variações acumuladas de massa atingem valor máximo.
C' na curva DTG, i a temperatura (nas esca
las Celsius ou Kelvin) na qual a velocidade de variação de
massa atinge valor zero.
12.
Tou t
(a)
(b)
Tout
FIGURA r . l CURVAS TG e DTG (a) e D T A ( b )

13.
1.4.4. Medida de temperatura em análise térmica
A caracterização térmica de um material por
meio de técnicas termoanalíticas, de algum modo, envolve a
miedida de temperatura.
Um pico endotérmico ou endoterma é um pico
no qual a temperatura da amostra torna-se menor que a do
material de referência, isto é, AT é negativo.
Um pico exotérmico ou exoterma é um pico no
qual a temperatura da amostra torna-se maior que a do mate
rial de referencia, isto é, AT é positivo.
Largura do pico, B'D', é o intervalo de tem
po ou de temperatura entre seus pontos de saída e de retor
no ã linha base.
Altura do pico, €F, é a distancia, perpendi
cular em relação ao eixo dos tempos ou temperaturas, - entre
a linha base interpolada, e a extremidade do pico (C).
Area do pico, BCDB, é a área compreendida pe
lo pico e a linha base interpolada.
O inicio extrapolado, G, é o ponto de inter
secção da tangente ao ponto de máxima inclinação, no lado
principal do pico, BC, com a linha base extrapolada, BG. ^

Em análise térmica, de modo geral, a medida
de temperatura e de variação de temperatura é feita por
meio de termopares.
1.4.4.1. Termopares
Os termopares são formados por fios de dois
metais de natureza diferente soldados ou apenas mantidos em
contacto entre si em dois pontos. Sempre que as duas extre
midades estejam a temperaturas diferentes, o circuito origi^
nará uma força eletromotriz (fem). Este fenômeno é conheci-
( ) do como efeito SEEBECK ou termoeleétrico .
A fem de um par termoelétrico resulta das a ^
terações que a feraperatuf a provoca na densidade de elétrons
do metal no qual estejam mais fracamante ligados para o ou
tro metal. A junção funciona, portanto, como um gerador de
fem e, como por meio desse fenômeno, há passagem de corren
te de um metal para o outro, hã, também, libertação ou ab
sorção de energia que se traduz por um escoamento térmico
V, • (35 ) entre as junções e o ambiente - .-.
Muitos termopares tem sido usados na m.edida
de temperatura em análise térmica. Os mais utilizados na
prática estão relacionados na Tabela 1.4.1, juntamente com
o intervalo de temperatura e temperaturas máximas de opera-
~ (34) çao
A escolha de termopares depende da temperatu

15.
TABELA 1.2. Termopares comuns, intervalo de .temperatura e
temperatura máxima de operação ,
Termopar
Intervalo de
Prático
Temperatura
Máxima
Platina x Platina-10% RÓ.dio 0 a 1450 1700
Chromel-Alumel -190 a 1100 1350
Ferro-Constantán -190 a 760 .1000
Cobre-Constantán -190 a 300 600
Chromel-Constantan abaixo de 8 00 1000
I.U.5. Fontes de erros em termogravimetria e análise
térmica diferencial
As principais fontes de erro em TG e DTA, em
INSTITUTO oe PESO UIPA-SENERGÉTiCSF NUCLEARES
p C. N J
ra operacional máxima do instrumento, da resposta térmica
esperada para o termopar e da reatividade química dos com -
postos em estudo. Em geral os termopares formados por me
tais nobres (Pt/Pt-Rh, Au/Pt-PtRh, W-Mo) operam em tempera
turas altas e são mais resistentes ao ataque químico. Os
termopares formados por metais com.uns -í (Cu-Constantan,
Ni-NiCr) apresentam uma fem muito maior, em resposta ã exci
tação térmica do que aqueles com metais nobres, são mais ba
ratos, porém, operam em temperaturas baixas

Ib.
geral, derivam de dois grupos de fatores que afetam, as cur
vas termoanalíticas. Esses fatores, descritos e discutidos
detalhadamente por COATS e REDFERN^ "'•'^ GARN^^'^\ MURPHY
(51,52) ^ G I O L I T O ^ f o r a m classificados em dois grupos:
- Fatores instrumentais: Velocidade de aquecimento do
forno; velocidade do registrador ou do papel; atmosfera do
forno; geometria do suporte da amostra e do forno; profundi
dade e raio do orifício do suporte no qual a amostra e colo
cada (DTA); localização, natureza e dimensões dos termopa
res diferenciais (DTA).
- Fatores ligados às •características da amostra: Quan
tidade da amostra; solubilidade dos gases libertados na prõ
pria amostra; tamanho das partículas; calor da reação; com
pactação da amostra; natureza da amostra e condutividade
térmica da amostra; natureza da substância inerte, utiliza
da como referé"ncia.
A instrumentação moderna é fabricada de modo
a tornar praticamente negligenciáveis certas causas de erro
tais como: flutuações erráticas do sistema registrador e da
balança, efeitos de indução provocados pelo forno, efeitos
eletrostáticos sobre as peças que compõem a suspensão da ba
lança, ajuste do papel no mecanismo registrador e reações
(36)
da amostra com o cadinho- •. Entretanto, algumas causas de
erro podem influenciar a precisão dos resultados e devem
ser conhecidas pelo pesquisador:
a) Termogravimetria.

17.
b) Análise Térmica Diferencial
- O efeito da impulsão do ar sobre o
cadinho e seu suporte
Provoca um aumento aparente crescente da mas_
sa do cadinho, ã medida que a .temperatura vai sendo eleva
da; pode ser determinado realizando-se a curva termogravime
trica do cadinho vazio.
- Correntes de convecgão e turbulencia
no forno
A perda de massa aparente causada pelo fluxo
ascendente de ar quente sob o cadinho e o ganho de massa de
vido a turbulencia do ar dependem, diretamente, do tamanho
e da forma do cadinho. O ganho de massa, numa atmosfera di
námica, i proporcional ao fluxo do gás e amassa molecular.
- Medições de temperatura e calibração
A temperatura da amostra pode ser ligeiramen
te maior ou menor que a temperatura indicada pelo termopar,
dependendo do tipo de reação de decomposição (endotérmica ou
exotérmica), da geometria do cadinho e suporte, da velocida
de de aquecimento e da condutividade térmica da amostra. Des_
se modo, é necessário observar os cuidados práticos relacio
nados com a calibração de termopares e as correções a serem
' . ^ (36) aplicadas •

18.
1.4.6. Comparação entre termogravimetria e termogravimetria
derivada (14.26,36J,__
- Posicionamento dos termopares
Quando não estão colocados no centro da amo£
tra ou referência, há um aumento gradual na diferença de
temperatura entre os dois termopares, provocando um desvio
da linha base e uma variação na forma do pico^
- Compactação da amostra e da referência
A maior ou menor compactação da amostra e da
referência pode -afetar o aspecto da curva DTA, pois modifi
ca não apenas a transferência de calor-da parede dos recipi
entes para as junções dos termopares mas também, no caso de
reações de decomposição, interfere sobre o fluxo de gases
de dentro para fora da amostra e vice-versa. Em geral, pre
fere-se fazer umia compactação firme porque, de um lado dimi
nui o gradiente térmico, pois aumenta a condutividade térmd^
^ ^' ,(34,36) ca, e, de outro, por ser mais reprodutível '
- Tamanho das partículas da amostra
Amostras com partículas menores são melhor
compactadas , gerando umi aumento na condutividade térmica do
material. Assim, as temperaturas nas quais a reação inicia
e termina diminuem com a diminuição do tamanho das partícu-
(51) las

19.
Na termogravimetria sao obtidas curvas de
massa da amostra, m, em função da temperatura (T) ou do tem
po (t). Assim,
m = f (T ou t)
Portanto, nestas curvas os desníveis em rela
ção ao eixo das ordenadas correspondem ãs variações de massa
(perda ou ganho) sofridas pela amostra e permitem obter da
dos que podem ser utilizados com finalidades quantitativas
(35)
Na termogravimetria derivada são obtidas cur
vas da derivada da massa em relação ao tempo, dm/dt, em fun
ção da temperatura ou do tempo. Desse modo,
dm ^ , „ , X • : 3 q r = f (T ou t) dt
Logo, por esta técnica, as curvas obtidas
correspondem ã derivada primeira da curva termogravimétrica
e nas quais as inflexões são substituídas por picos que de-
limiitam áreas proporcionais ãs alterações de massa sofridas
n ( 3 6) pela amostra • .
A curva termoanalítica derivada miatematica -
mente ou registrada diretamente, não contém mais infoririação
do que uma curva termoanalítica. integral, obtida sob as
mesmas condições experimentais; simplesmente é registrada
de modo diferente^. Picos sobrepostos sobre a curva DTG,
geralmente, são melhores indicadores de reações sucessivas

20.
- As curvas DTG indicam, com exatidão, as tem
peraturas correspondentes ao inicio, ao instante em que a
velocidade e máxima e ao instante em que a reação chegou ao
seu término;
- Os picos agudos das curvas DTG permitem dis
tinguir, claramente, uma sucessão de reações que, pelo fato
de apresentarem estágios coincidentes, não podem ser clara
mente distinguidas por meio das curvas termogravimêtricas;
- As áreas dos picos correspondem, exatamiente,
a perda ou ganho de niassa e podem, ser u t i l Í 2.adas em determi
nações quantitativas.
1.4,7. Comparação ent:-e analise térmica diferencial e calo-
T • • T (34,49 ,51, 52 ,81,82) rimetria explopatoria diferencial 5 5 5 5 5
Quando uma substância sob aquecimiento sofre
uma transição com liberação de calor (exotérmiica) ou absor
ção de calor (endotérmica), a temperatura, no momento da
transição, aumenta ou diminui. Essa variação de temiperatura
pode ser medida e registrada facilm.ente num instrumento de
DTA, em relação a um material de referência, aquecido nas
mesmas condições, em função da temperatura ou do tempo. Num
instrumento de DTA registra-se, portanto, a diferença de
I N S T I T U T O D E P E S Q U ' S A S E'•J P R £ É T I C ^ S E N U C L E A R E S
I. P . E . N .
sobrepostas do que inflexões na curva TG. Assim, podem ser
^ (36) atribuidas certas vantagens a termogravimetria derivada- .:

21.
temperatura: AT = - Tj (T^ = temperatura da amostra;
Tj = temperatura da referencia) .
De modo geral, os sistemas de analise térmi-
(82) ca diferencial podem ser classificados em tres tipos ' :
(1) DTA convencional ou clássica
(2) DTA de "BOERSMA" e
(3) DSC (Figura I. .2)
No caso da DTA clássica, as temperaturas são
medidas por termopares colocados em contacto direto com a a
mostra e o material usado como referencia (Figura 1.2.1).
A área do pico DTA não depende somente do ca
lor da reação e da massa dã amostra mas, também, do calor
específico e da condutividade térmica da amostra, que podem
variar durante a decomposição térmica da substancia. Assim,
do ponto de vista quantitativo, a obtenção de dados por DTA
convencional ou clássica é dificultada, principalmente, pe
lo fato de o termopar esta posicionado diretamente na amo£
tra, que atua tanto como fonte de calor como consumidor de
calor.
Na DTA de "BOERSMA", pelo fato de os termopa
res estarem posicionados juntos aos suportes da amostra e
referencia porém, sem contactar os miateriais (Figura 1. 2.2) .,
a variação na condutividade•térmica da amostra não exerce
influencia na medida da diferença de temperatura (AT) en
tre a amostra e a referencia. Portanto, nesse caso, o fluxo

22.
de calor entre a amostra e a referencia em função da tempe
ratura ou do tempo i proporcional ã diferença de temperatu
ra (AT). Assim, pode-se dizer que a DTA de "BOERSMA" ê
uma técnica que se baseia na indicação indireta da diferen
ça do fluxo de calor espontâneo entre a amostra e a substân
cia usada como referência em função da temperatura ou do
tempo.
A calorimetria exploratória diferencial (DSC)
é uma técnica que produz dados semelhantes â DTA, onde a â-
rea do pico DSC é diretamente pr'oporcional ã quantidade de
energia total liberada ou absorvida pela amostra. A diferen
ça principal entre os sistemas de DTA e DSC e que, enquanto
na DTA tanto a amostra como a referência são aquecidos por
uma única fonte de calor, na DSC os dois miateriais (amostra
e referência) possuem fontes individuais de aquecimento (Fi
gura 1. 2. 3) .
(49)
De acordo com MACKENZIE existem dois ti
pos de DSC: - DSC com fluxo de calor (descrito em ill.1.1.3);
- DSC com compensação de energia.
O sistema de DSC com compensação de energia
representado esquematicamente na Figura I. 2.3, do mesmo mo
do que a DTA, submete a amostra e a referência a um aumento
ou diminuição de temperatura uniforme. Quando ocorre uma
transição (endotérmica ou exotérmica), um sistema adicional
de controle detecta a diferença de temperatura (AT) entre
a amostra (A) e a referência (R) e, fornece a energia nece£
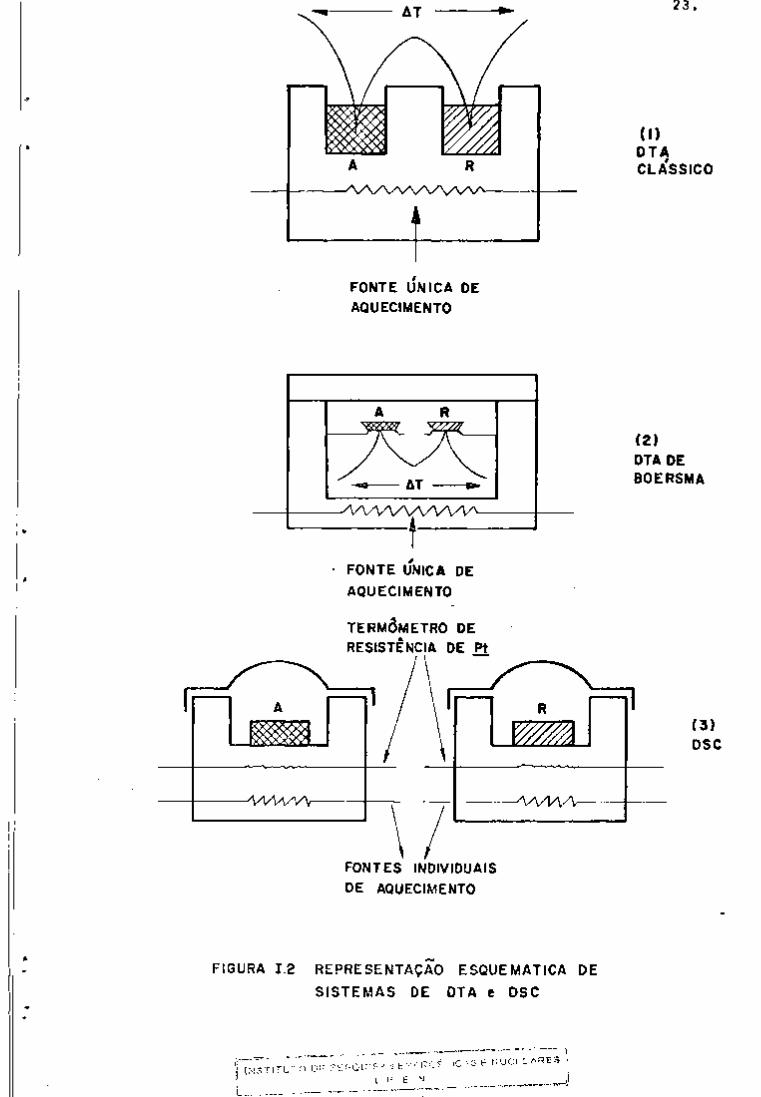
23.
A R
F O N T E Ú N I C A D E
A Q U E C I M E N T O
(I)
D T A
CLA'SSICO
F O N T E ÚNICA D E
A Q U E C I M E N T O
T E R M Ó M E T R O D E
RESISTENCIA D E Pt
F O N T E S INDIVIDUAIS
D E AQUECIMENTO
(2 )
OTA D E
B O E R S M A
( 3 )
D S C
FIGURA 1.2 R E P R E S E N T A Ç Ã O E S Q U E M Á T I C A D E
S I S T E M A S D E D T A e D S C
1, P . Ê . N . _ _ J

sâria para manter as duas substâncias (A e R) sob a mesma
temperatura (isto é, mantém AT = 0). Como a energia forne
cida é diretamente proporcional ã quantidade de energia en
volvida na transição 'da amostra^ um registro desta compensa
çáo de energia resulta numa medida calorimétrica direta da
energia de transição.
A grande vantagem do instrumento de DSC so
bre o de DTA convencional é a capacidade de medir diretamen
te a quantidade de energia envolvida numa transição, inde -
pendente do calor específico da amostra e da razão de aque
cimento, onde a amplitude do pico registrado é medida como
uma; razão da energia liberada ou absorvida em mcals./s e a
área. do pico é igual â energia total da transição ém mcals.
(81) •
A figura 1.3,. mostra de forma esquemática as
possibilidades para os sistemas de DTA e DSC comercialmente
disponíveis, onde os equipamentos representados em 1.3.1 e
1-.3.-2 são produzidos por muitos fabricantes; os tipos 1.3.3,
1.3.4 e 1.3.5 são produzidos, respectivamente, pela Du Pont,
Mettler e Setaram e o tipo 1.3.6 é produzido pela Perkin
Elmer e Rigaku^^'^\ Conforme a Figura 1.3, existem dois sis
temas típicos de DTA ( 1 . 3 . 1 e 1.3.2), três sistemas caracte
rísiticos de DSC ( 1.3.4, 1.3.5 e 1.3.6) e um sistema classi
ficado tanto de DTA (BOERSMA) como de DSC (com fluxo de ca
lor) ( 1.3.3).
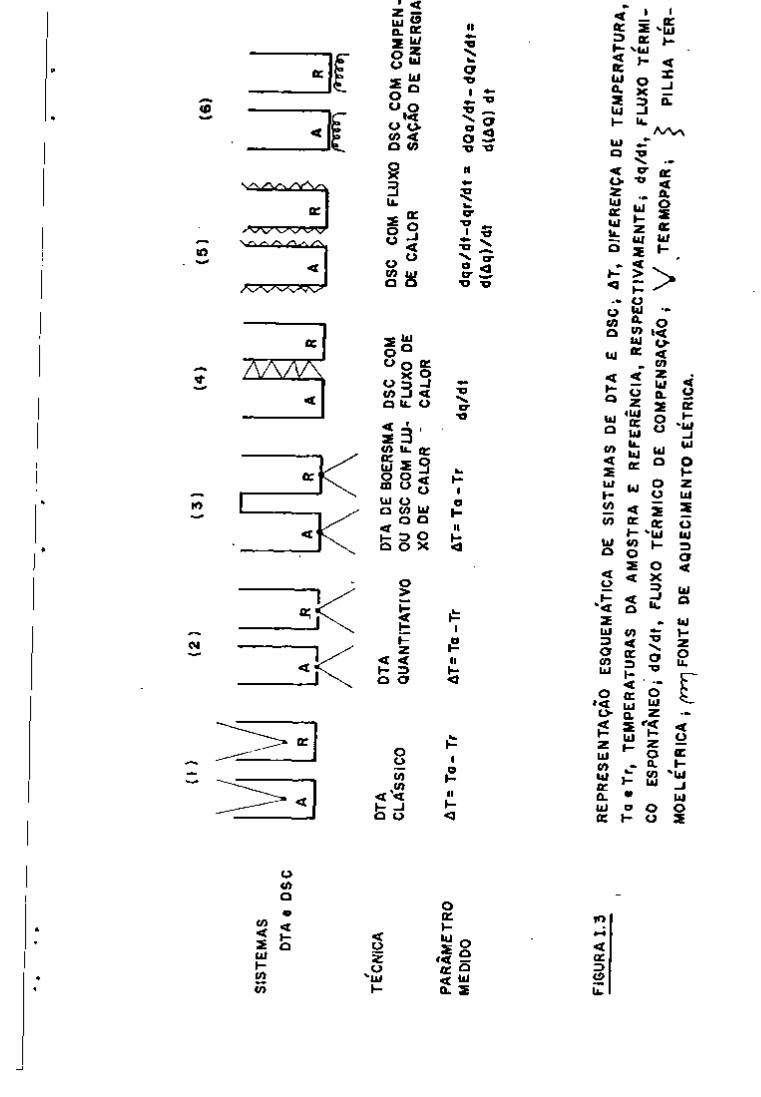
SIS
TE
MA
S
DT
A e
DS
C
TÉ
CN
ICA
PA
RÁ
ME
TR
O
ME
DID
O
(I )
OTA
CLA'SSICO
AT
=
To
-
Tr
(2)
DT
A
QU
AN
TIT
AT
IVO
AT
=T
a-T
r
(3
) (4) >
>
DT
A
DE
BO
ER
SM
A
DS
C C
OM
OU
D
SC
CO
M F
UJ-
FL
UX
O
DE
XO
D
E
CA
LO
R
" C
AL
OR
AT
=
Ta
-T
r d
q/d
t
(5)
1
< <
(6)
6
DS
C
CO
M F
LU
XO
D
SC
C
OM
CO
MP
EN
-
DE
C
AL
OR
S
AÇ
ÃO
D
E
EN
ER
GIA
dq
o/d
t-d
qr/
dt
=
dQ
o/d
t-d
Qr/d
t =
d(A
q)/
dt
d(A
Q)
dt
FIG
UR
A
1.3
REPRESENTAÇÃO
ESQUEMÁTICA
DE SISTEMAS DE DTA E DSC-. ÍT. D
IFERENÇA DE
TEMPERATURA
To
.T
r,
TEMPERATURAS
DA AMOSTRA
E REFERÊNCIA. RESPECTIVAMENTE
i d
,/d
t, FLUXO
TÉRMI
CO
ESPONTANEO,
dO
/d..
FLUXO
TÉRMICO DE COMPENSAÇÃO,
N/
TERMOPAR,
?
P.LHA TÉR-
MOELETRICA ,
FONTE DE AQUECIMENTO ELÉTRICA.
^
K3

2b.
1.4.8. Aplicação da termogravimetria, análise térmica dife
rencial e calorimetria exploratória diferencial
60)
O número de fenômenos que pode ser detectado
por uma operação DTA ou DSC é muito maior do que numa opera
ção TG pois, reações do tipo fusão, transição cristalina,
transição vitrea, cristalização e reação sólido-sólido sem
a liberação de produtos voláteis, não podem ser detectadas
pela termogravimetria, uma vez que não ocorre variação de
massa na amostra. Entretanto, o uso da termogravimetria per
mite estabelecer a estequiometria exata das reações químicas,
enquanto por DTA ou DSC estas mudanças são indicadas apenas
por um pico endotérmico ou exotérmico.
Na TG, quando há decomposição com reações so
brepostas, os componentes não podem ser identificados devido
ã baixa resolução da técnica e ã baixa seletividade da deter
minação. Por outro lado, na DTG, DTA ou DSC, a situação é o
inverso, com relação ã resolução do método. As transforma
ções são registradas sob a forma de picos de fácil observa -
ção, que ocorrem em temperaturas bem definidas. Assim, TG,
DTG, DTA ou DSC são técnicas complementares onde a informa
ção obtida pela aplicação de uma é, freqüentemente, comple
tada pela aplicação da outra.
A aplicação dos métodos termoanaliticos, iso
lados ou simultâneos, na área da química inorgânica, analíti
ca e orgânica é muito vasta. Por exemplo, no estudo da compo

. 2 7 .
sição, estrutura cristalina, temperatura de secagem ou de de
composição de precipitados, com formação de produtos interme
diários de composição estequiomëtrica e massa constante; na
cinética de decomposição térmica de complexos metálicos; na
detecção e determinação de constituintes minerais; na quími
ca e tecnologia de plásticos e polímeros: acompanhar proces
sos de condensação e polimerização, verificar a estabilidade
térmica e a eficiência de estabilizadores e ativadores, de
terminar a porcentagem de cristalinidade durante o aquecimen
to.
A Figura 1.4 representa um diagrama que mos
tra como as técnicas termoanalíticas podem ser associadas a
outros métodos no estudo de compostos, segundo ASSIS e GIOL^
T0<".
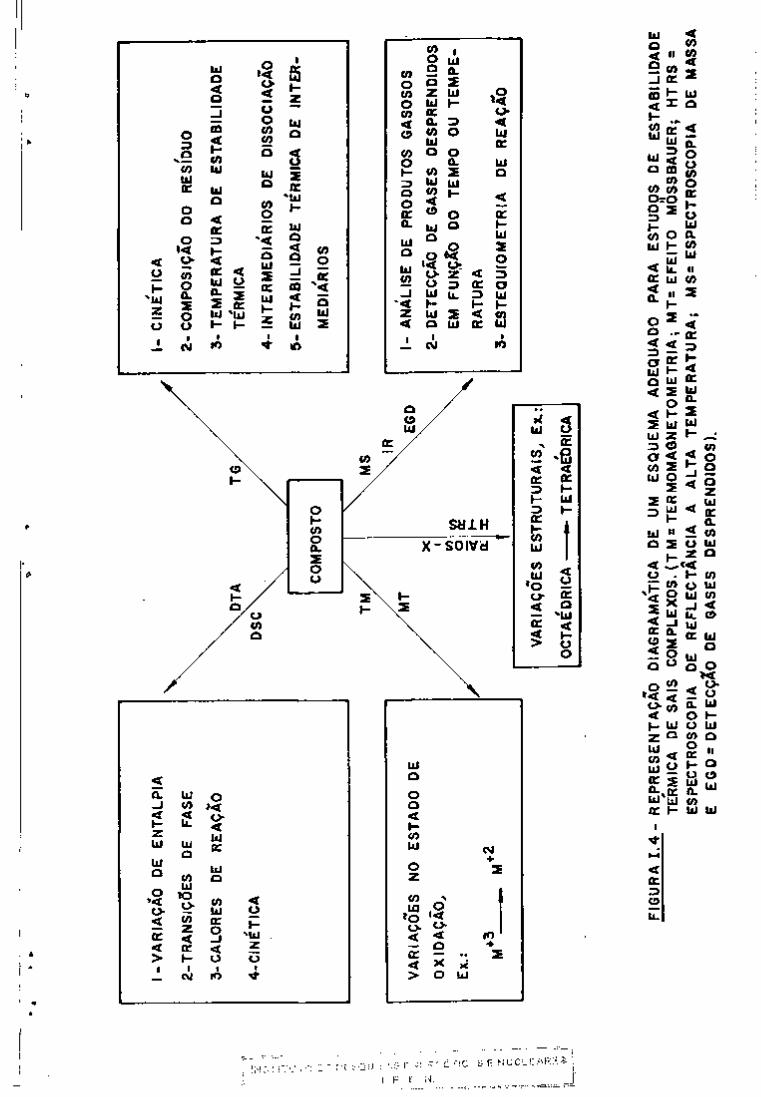
1 i;
yi
i' --1
;
(...
a:
O C m.
•
0
'
Cl
1
m 1
i ^
:
I-V
AR
IAÇ
ÃO
D
E
EN
TA
LP
IA
2-T
RA
NS
lÇÕ
ES
D
E
FA
SE
3-
CA
LO
RE
S
DE
R
EA
ÇÃ
O
4-C
INÉ
TIC
A
VA
RIA
ÇÕ
ES
N
O
ES
TA
DO
D
E
OX
IDA
ÇÃ
O^
Ex.:
M •3
+2
COMPOSTO
TM
MS
\|R
/MT
\E6D
X 1
to
o
(O
< h- z
VARIAÇÕES ESTRUTURAIS^
Ex.:
OCTAÉDRICA
^
TETRAE'DRICA
I- CINÉTICA
2-COMPOSIÇÃO
00
RESÍDUO
3-TEMPERATURA
DE
ESTABILIDADE
TE'RMICA
4-INTERMEDIARIOS
DE
DISSOCIAÇÃO
5-E
STABILIDADE
TÉRMICA DE INTER
MEDIARIOS
1- ANALISE OE PRODUTOS
GASOSOS
2- DETECÇÃO DE GASES
DESPRENDIDOS
EM FUNÇÃO
DO TEMPO OU TEMPE
RATURA
3-ESTEQUIOMETRIA
DE
REAÇÃO
FIGURA 1.4 - REPRESENTAÇÃO
DIAGRAMATICA
DE UM
ESQUEMA
ADEQUADO
PARA
ESTUDOS DE
ESTABILIDADE
TÉRMICA DE SAIS COMPLEXOS. (
T M -
TERMOMAGNETOMETRIA , M T
-EFEITO
MOSSBAUER;
HTRS»
ESPECTROSCOPIA
OE REFLECTÃNCIA A
ALTA
TEMPERATURA;
MS= ESPECTROSCOPIA
DE MASSA
E
EGO» DETECÇÃO DE GASES
DESPRENDIDOS).
IS3
00

29
II. REVISÃO BIBLIOGRAnCA
O levantamento bibliográfico apresentado nes_
te estudo faz referência à produção, composição e caracterÍ£
ticas estruturais de diuranato de amonio (DUA) e trióxido de
urânio (UOg), bem. como uma análise breve da influência dos
parâmetros de preparação desses compostos sobre as proprieda
des do dióxido de urânio (UO2).
II.1. PREPARAÇÃO, COMPOSIÇÃO E ESTRUTURA DE DIURANATO
DE AMONIO
A preparação de diuranato de amonio pode ser
feita pelo processo de precipitação, em descontínuo ou cont^
nio, a partir de solução de nitrato ou sulfato de ' uranilo,
tratada em geral, com hidróxido de amonio ou amonia gasosa .
Ê obtido sob a forma de um sal hidratado, de consistência
( 3 3 ' 65 ) pastosa e de composição estequiomëtrica mal definida .
No estudo da produção de UO2, WATSON^.^^^ em
1957, verificou que o processo de precipitação em batelada
(descontínuo) produz um DUA mais sensível aos parâmetros de
precipitação, do que pelo processo de precipitação contínuo
e, independente do tipo de processo, o produto da precipita
ção com gás NHg é mais reprodutível.

30.
2U02(N03)2 + BNHj OH - -(NH ) 2U2O.7 + 3H2O + NH NOg
Verificaram também, que durante esta reação formava-se nitra
to básico de uranilo, hidróxido de uranilo e monouranato de
amônio.
( R7)
NOTZ e col. •, em 1960, observaram que o
diuranato de amônio formado pela polimerização dos compostos
HO-UO2-NO3 ; HO-UO2-OH ; H0-U02-0-NHi^ e NH^^-0-U02-0-NH^^
(DUA), com eliminação de H2O, HNO3, NH^OH ou NH NOg, é, prova
velmentejU'a mistura de poliuranatos, cuja estrutura média
pode ser representada por
O H O II / V II
HO - U - O O - U - OH II -v / II O N O
Nos pós de DUA, o tamanho da partícula e sua
estrutura podem ser controladas, durante a precipitação, pe
la concentração de uranio, razão molar NH^iU e tempo de resi
dincia. Para baixa razão molar NH^-'U, por exemplo, pode-se
encontrar três tipos diferentes de partículas: cristalitos e
lementares, agrupamento de cristalitos e agrupamento de agio (59)
merados . .
( 72 78) — Alguns autores • ' - atribuíram a formula
XWí^),p^^ (DUA) ao precipitado formado pela reação do nitra
to ou sulfato de uranilo com hidróxido de amonio ou amonia
gasosa, cuja reação global poderia ser representada por:

3J. ,
í 18 )
(NHi^)2U0^.
O heptauranato ( (NH^) 2Uy022 e tetrauranato ( ( NH ^ ) 2U O-j 3 fo
ram indicados como os compostos mais importantes no estudo
da formação dos uranatos de amônio. Desse modo mostraram
que outros sais de urânio e amônio também se formam juntamen
te com o DUA, durante essa reação, onde a razão NH^/U nesses
compostos aumenta com o aumento de pH de precipitação.
Em 19 61, DEANE estudou varias amostras
de DUA por espectroscopia no infra-vermelho e verificou que
apresentavam um máximo de 0,60 mol e 1,30 mol, respectivamen
te, de NHg e H^O por mol de urânio. Observou também, que uma
estrutura do tipo UO2(OH)2.xNHg.yH20 representa melhor esses
compostos demonstrando que mesmo com grande excesso de a m o
nia não podia ser obtido um composto com a formula
(NHi^)2U20^ (DUA).
IPPOLITOVA e 001.^*^°.^ em 1961, determinaram
a composição dos uranatos de amônio formados durante a rea
ção de nitrato de uranilo e hidróxido de amônio, em diferen
tes valores de pH, por meio de métodos potenciometricos e
condutométricos. No intervalo de pH de 3,70 a 9,30 (razão mo
lar NH OH":U = 1:1 a 4:1), encontraram os seguintes compostos:
U02(OH)2 ; U02(OH)N03 ; iU02(N03)2. U02(OH)2 ;
(NH )2U.7022 i (NHi^)2U^0^3 ; (NHi^)2U207 (DUA) e

32.
A reação entre nitrato de uranilo e hidróxi
do de amonio, estudada por DEPTULA '''- ' em 1952 , revelou que
os uranatos de amonio são formados pela polimerização do íon
Uo'^'^ até o tipo U02( (OH)2U02)^'^ , onde os grupos UO2.H2O são
combinados em cadeia por celas do tipo:
O I
- U -
HO OH
Verificou, também, que a estrutura do precipitado depende da
forma de adição da amonia. Uma adição lenta resulta ém urana
to relativamente grosso e uma adição rápida resulta em preci
pitado amorfo facilmente hidrolizável. O precipitado obtido
numa razão molar NHj'j/U = 1:2 apresentou composição que foi
expressa pela fórmula ((NH^)2O)^.(UO^.H2O)2q.7H2O.
CORDFUNKE^.^^\ em 1952 , identificou quatro
fases sólidas para o sistema ternario NH2-UO2-H2O, em solu
ções aquosas:
I. UO3.2H2O (pH = 3,5) ; II. 3UO3.NH3.5H2O (pH = U,0)
III. 2UO3.NH3.3H2O (pH > 7,0) ; IV. 3UO3.2NH3.UH2O (pH ' 7,0) ,
com composições intermediárias do tipo I+II, II+III, etc. O
composto II ao ser preparado apresentou excesso de água, que
foi liberado em contato com o ar, diminuindo a razão molar
H2O/UO3, porém, mantendo constante a razão molar NH3/UO3.Foi
identificado como o mais estável dos três uranatos. Ao con
trário, os compostos III e IV são higroscópicos e instáveis

ao ar úmido, pois apresentaram diminuição gradual da razão
molar NH^/UO^ com o tempo. Este fenômeno foi atribuído ã
substituição de parte da NH^, do retículo cristalino, pela ã
gua do ar, por um mecanismo de troca iónica.
Em 1966, FODOR e col. , estudaram a forma
ção de uranatos de amônio e encontraram uma estrutura do ti
po trimérica no produto inicial (UO2.2H2O), onde a amonia é
incorporada (máximo de 0,66 mol de NHg por mol de U) não por
uma simples troca com a água mas, envolvendo a quebra de pon
tes de hidrogênio formadas por grupos OH e moléculas de água.
Assim, observaram que o aumento no teor de amonia leva a uma
diminuição no teor de água, provocando transformações estru
turais no sistema que pode ser representado, aproximadamente,
por
UO3.(2-x)H20.xNH2.
Os trabalhos realizados por STUART e
(71) -
WHATELEY em 1969, por sua vez, nao revelaram quatro com
postos diferentes para representar os uranatos de amônio. Ve
rificaram que estes precipitados constituem-se numa fase sim
pies na qual a razão NH^/U pode variar de modo contínuo. En
contraram a seguinte fórmula para representar o sistema de
uranatos de amônio:
UO 2(OH)2_^(ONHi;^)^yH20,
onde X varia continuamente nos limites d e x = 0 a x = 0 , 7

34 .
O mesmo com.portamento foi observado por SAPA
((UO^)„(OH)., (ONH^)„ (H„0). ^.;p-n.H_0. ¿ m /(m-nJ 4 ¿n ¿ (.m-n;. ¿
STUART e MILLER^^^^ em 1973, estudaram os
produtos da reação, de UO2.2H2O e UO^ anidro com NH OH ou NH^
gasoso. Os uranatos de amionio formados apresentaram caracte
rísticas de um sistema continuo, não estequiométrico, com
propriedades zeolíticas. Estas propriedades foram associadas
ao tamanho da partícula. Os compostos formados por partícu -
las grandes tinham uma hidratação, lenta, absorvendo preferen
cialmente NH *!, enquanto a competição entre os processos de
hidratação e amoniação levava a uma razão NH^rU menor para
os compostos formados por partículas menores.
Um mecanismo para exr^licar a formação do DUA
(73)
dura?ite a precipitação foi proposto j.or TURCANU e DEJU em
1979:
U02(N03)2.xH^O (U02(N03).(x+l)H^0) + NO3 (1)
(UO^CNOg) . (x + DH^O)^ + OH U02(N03) . (OH) .XH2O (2)
2U02(N03) .(OH) .XH2O (NO 3 )U02 C Q|]^ UO2 (NO 3) (3)
(x-l)H20 (x-l)H20
CU e CISMARU em 1972, quando atribuíram a fórmula seguin
te, como sendo a mais provável para representar o sistema de
uranatos de amonio:
(66)

35.
U02(N03) .OH.XH2O + 0 H " ^ = U02(OH)2.(X-1)H20 (4)
"2° "2° H_0 H_D • / OH ^ i i / OH ^ OH f
HO-UO_^ ^ UO^-OH e HO - UO,^ / UO^ UO^-OH : ^ OH : 2 i 2^ OH'^ ^OH : 2
H2O H2O H2O H2O
De acordo com estas fórmulas somente duas moléculas, do tipo
NH^, podem ser adicionadas em substituição aos', hidrogenios
ionizáveis nas extremidades da cadeia. Apesar do mecanismo a
cima, uma fórmula real para representar o DUA não pode ser
estabelecida pois, durante as etapas de levagem e secagem,há
degradação do precipitado. Entretanto, uma fórmula geral do
tipo nUO2.mNH3.pH2O pode ser aceita.
Os resultados apresentados acima são diferen
tes, em alguns aspectos contraditórios, porém, no geral, os
trabalhos encontrados na literatura apresentam evidências de
que:
_ ^. , D - • ' • ^ I C - S E NUCLEARES I
OH^ ^ OH ^ ^ OH 2U02(0H)..(x-l)H„0 UO. ^ UO^ ...
2 2 2 .. 2 Q '2...
(x-l)H20 (x-l)H20
^ OH ^ 2+
Onde a cela (UO. ^ UO^) pode tomar a forma triméri OH ^
ca abaixo:
^ O H ^ ^ O H ^ 2 + (U0„. ^ U0„ UO^) , que compõe os diuranatos com
^ OH ^ OH ^
estruturas mais estáveis, respectivamente^

36.
a) E importante representar o DUA por um sistema em equili
brio multifásico e pode-se aceitar as duas hipóteses a-. (12,14,15,19,40,44,5Q,59 ,71,
baixo, como as mais prováveis
73,76,85):
a-|) O DUA e um sistema homogêneo monofásico no qual as
razões molares NH^iU e H^OrU podem variar dentro de
limites ampios;
3-2) O DUA ó um sistema de composição variável, onde a
forma física e propriedades químicas dependem das
condições de precipitação. -
(Vi
il''' l\
b) A formula CNE^^) ^'^^ inadequada para representar o DUA
obtido em soluções aquosas mesmo em grande excesso de a- n''
mônia, pois se trata de um composto de natureza comple-
'(7 ,15 ,20 , 3 5 ,43 , 57 ,63) xa
c) 0 DUA por ser um composto não estequiométrico, cuja com
posição depende das condições de preparação não pode
ser representado por uma fórmula definida; porém, pode
ser aceita uma fórmula geral do tipo nUO2.mNH2.pH2O ou
do tipo UO2(OH)2_^(ONH^) .ZH2O, onde x varia de modo con
. . (3 5,63,70 tmuo no intervalo limitado por x = 0 e x = 0,7 73) ..
d) A amonia está presente na forma do íon NH^. Em ambiente
úmido, seu teor varia por meio de ura mecanismo de troca
iónica, conferindo propriedades zeolíticas ao DUA, onde
tanto o íon NH^ como a água podem ocupar "sítios" na ce-
• • ^ - - ' • (15,71,74,7Sy Ia unitaria básica do oxido de uranio

37.
e) A precipitação de DUA em solução de nitrato ou sulfato
)~ ou SO^' «• — 2—
de uranilo acarreta a oclusão de íons N0_ ou SO ^ em quan
tidades variáveis "^^^ A absorção de íos nitrato é maior
em altos valores de pH e em velocidades de precipitação
rápidas. A absorçã(
lores de pH baixos
rápidas. A absorção de íons sulfates é favorecida em va-
( 2 , 6 ,"114,43,70 ,73 , 7 4 , 8 4 , 85 ,86 ) .
f) A razão N H l | / U na fase sólida depende do pH final de pre
. ~ í 1 5 ,2 9 , 4 0 , 4 3 , 71,'8 4 , 85;) cipitaçao »,-.»..-».»___» s
g) As etapas de lavagem e secagem dos precipitados têm gran
de influência sobre a estrutura e composição dos diurana
tos. A lavagem do material provoca mudança , na razão
NH^/U e no teor de nitrato. A secagem em altas temperatu
ras (maior que 1 5 0 ° C ) pode mudar a composição ou mesmo
^ ^ ' • j T ^ T T A - (29v43v44,57,63 ,7 3 , 74 ) alterar a estrutura química do DUA.. * ' ' » '
h) O aumento do pH favorece o aumento da área superficial
do pó., mas provoca a diminuição do tamanho dos cristali-
-, j ^ T M t A (43, 58,8 5,86) tos e aglomerados do DUA ' ' • '
II.2. PREPARAÇÃO DE UO3
~~ A preparação de trióxido de urânio ê feita,
normalmente, pela decomposição térmica dos seguintes compos_
tos de urânio :

38.
Nitrato de uranilo
11.2.1. Decomposição térmica de nitrato de uranilo
Dependendo das condições experimentais, a
decomposição térmica do nitrato de uranilo e seus- hidratos
- G 8 apresenta diferentes fases cristalográficas para o UO^ ; '
41, 4 8,58)^ geral, o produto da decomposição térmica do
nitrato de uranilo hexahidratado é o gama-UOg. Entretanto,
os possíveis produtos para sua decomposição térmica são in
dicados a seguir: UOg amorfo, g, alfa-UO^, beta- UO3,
eta-UOg, gama-UOg, teta-UO^ e zeta-UO^. O mecanismo de des-
nitração ocorre em dois estádios, por um processo endotérmi
C O , segundo as equações:
U02(N03)2.6H20 - UO2(NO3)2.H2O + 5H2O
U02(N03)2.H2'C)' - UO3 + H2O + 2NO2 + I/2O2
O nitrato de uranilo anidro, sob vácuo, no intervalo de tem
peratura de 250°C a 450°C, apresenta UO3 amorfo como produ
to dar\ reação de decomposição, enquanto, a 500°C, forma-se a
mistura UO3 amorfo + U30g. Os produtos da decomposição do
~ (58) nitrato de uranilo dihidratado ao ar ou sob vacuo sao. i
Peróxido de uranio
Tricarbonato de uranilo e amonio
Diuranato de amonio

39
UOg amorfo + alfa-UO^ vácuo (250OC)
vácuo (300 - UOO°C)
ar (250 - 450OC)
ar 1500OC)
ar (550OC)
II.2.2. Decomposição térmica do peróxido de urânio
O processo de decomposição do peróxi
do de urânio, acima de 400°C, ocorre com perda de oxi
gênio e água, produzindo trióxido de urânio, cuja es
tru
-7 7)
trutura depende das condições de preparação. •• ~ -
, Em geral, obtem-se UO^ amorfo e alfa-UOg,
II.2.3. Decomposição térmica do tricarbonato de
uranilo. e.amonio
Este sal decompõe-se facilmente quan-
^ ^ ~ (37, 65) do aquecido, de acordo com a reação :
(NH^)^^ U02(C03)3^ UNHg - 3CO2 * ^"2°
Em atmosfera inerte, ar ou hidrogênio, a 3 80°C, o mes
mo composto, HO^^ia^O^^ 25' obtido, cuja formação se
' (37) da por meio das seguintes etapas • ' :
UOg amorfo
gama-UOg
gama-UOg + beta-UOg
•beta-UOo

40.
UO '3^"2O^l,0^ "03^20)^^33-. U03(H20)o^3o 003^20)^^25
II. 2. 4. De compos igão térm:ica do diuranato de amônio
A reação de decomposição térmica do diurana
to de amônio sera analisada com mais detalhes em II.3.1. O
trióxido de urânio obtido nesta reação, a 500°C, depende,en
tre outros fatores, da velocidade de aquecimento. Em veloci
dade de aquecimento muito baixa (l°C/min) produz-se UO3 a-
morfo puro. Em velocidade de aquecimento rápida (10°C/min)
obtém-se misturas de beta-UOg + UO3 amorfo em qüantidaçíes
não reprodutíveis^.
II. 3. DECOMPOSIÇÃO DE COMPOSTOS DE URÂNIO POR TÉCNICAS TERMOAMALTTTCAS
O estudo da caracterização térmica de com -
postos de urânio pelo uso dos métodos termoanaliticos é : um
assunto que vem sendo explorado há mais de vinte anos, ini
ciado com o comportamento termogravimetrico de vários preci-
( 23)
pitados de uranio e documentado no livro de DUVAL r . DUVAL
caracteriza esses precipitados e alguns de seus óxidos e dis_
cute, num estudo comparativo, a estabilidade térmica destes
compostos e de seus intermediários,
A aplicação, de modo complementar ou simul-

41.
tâneo, dos métodos termoanaliticos (TG, DTG, DTA e DSC)
ao estudo de compostos de urânio já é bastante extensa
e constituída por trabalhos publicados nos últimos vante
anos.
No geral, os trabalhos encontrados na
literatura usam os métodos termoanaliticos associados as
técnicas complementares, tais como a espectroscopia no.
infravermelho, a difração de raios-X e a análise de ga
ses por espectrometria de massa e cromatografia gasosa,
nos quais os autores procuram não somente conhecer a
composição, estrutura e estabilidade térmica, processos
endotérmicos e exotérmicos, como também correlacionar
os dados termoanaliticos com outras propriedades dos com
postos.
II.3.1 - Decomposição térmica de diuranatos de amônio
O diuranato de amônio por ser um composto
não estequiométrico apresenta um comportamento térmico va
riável. O mecanismo do processo da decomposição téraica do
DUA tem sido estudado, principalmente, pelos métodos ana
líticos. Os resultados são contraditórios quanto à compo
sição e estrutura,porem, em sua i}pioria,são de inteira concordai

42,
cia em relação as etapas envolvidas durante sua decomposi-
- (21,24,57,68,70,83) çao 9 » Í . 9 >
As curvas TG e DTA mostram quatro efeitos
térmicos em temperaturas correspondentes, indicando que a
decomposição térmica do DUA se processa em quatro estádios
(.21,57,66,70 ,83).
a) Estádio I (20 a 120OC) e Estádio II (120 a 200OC)
Foram eles caracterizados por um processo endotérmico de
desidratação do DUA ^^1. 24, > 0 , 4^, ..57,- 68, • 70, 73. 74,
75,83)
. A curva TG mostra perdas de massa relacionadas
com a liberação de água livre (absorvida fisicamente), mo
léculas de água coordenadas (cristalização) e alguma de-
sidroxilação (composição). Dois picos endotérmicos cor
respondentes, com mínimos em torno de 60°C ou 80°C e
135°C ou 160°C ou 193°C, respectivamente, aparecem na
curva DTA^24, 45 , 68 , 70 ) j;m alguns casos foi observada
a liberação de xm excesso de amonia nos intervalos de
temperatura 115 a 145°C ^^6 > j^^O a 135°C " ^ ou em tor
no de 150°C A área superficial varia nesses dois
- (63) -estádios : até lOO^C (secagem) ela diminui e acima de 100^0, após secagem, ela aumenta (57)
b) Estádio III: 200 a 350°C
Nesse intervalo de temperatura ocorre a decomposição tér
mica da estrutura do DUA e do nitrato de amônio ocluido,
com perda simultânea de H2O, NH^ e óxidos de nitrogênio,
e formação de trióxido dè.¡uranio amorfo e beta-UOg. No

43.
final desse estadio, a curva TG mostra um excesso de apro
ximadamente 2 % de massa em relação ã esperada para o UOg.
Esse excesso de massa foi atribuído ã amonia retida na e£
trutura do UO^, sob a forma de amoniato (UOg.xNHg). A li
beração máxima de amonia ocorre em 330°C e a ruptura com
pleta do retículo cristalino do DUA se da em 350°C. Um
pico exotérmico na curva DTA foi associado ã decomposição
do nitrato de amônio. Para DUA precipitado de solução de
sulfato de uranilo, a curva DTA mostra um pico exotérmico
somente em torno de 415°C. Nesse estádio o aumento da su
perfície específica foi associada ã decomposição do nitra
to ocluido ÍIO.24,40,44, 57,^€8,71,73:,7:4V83,8.4;
c) Estádio IV: 350°C a 450°C
Esse estádio foi relacionado com a reação de oxidação ou
redução da amonia retida no UOg, de acordo com a atmosfe
ra de decomposição. Nesse intervalo de tempreatura obser
va-se um aumento significativo na superfície específica
do material, acompanhado de mudanças no tamanho do crista
lito. Esse aumento na superfície específica foi associado
ã reação de auto-redução do oxido de urânio pelo craqueio
da amonia e foi maior para compostos com mais alto teor
de NHg, independente do teor de nitrato ocluido (24,44,57,
83,84,86)
Acima de 450'~'C observa-se a ocorrência do
UO3 anidro e sua decomposição térmica para formação de U^Og,
que varia segundo a atmosfera de decomposição. Observa-^se uma

44.
diminuição rápida. na superficie específica, associada ao
^ , • 4- -.- (10,24,40,44, 57,68 ,70,73,74,83,84) crescimento do cristalito. ' * ' ' * * ' ' ' '
O estudo da decomposição térmica do DUA,de£
crito em varios trabalhos, mostra que as reações envolvidas
são sensíveis ãs mudanças das condições externas, onde o fa
tor dominante, que determina os produtos finais das rea
ções, é o tipo de atmosfera, de acordo com o diagrama apre-
, , T (11,21,40,42,44,57,63,66,58,70, -73, sentado na Figura II. 1 ' - ' * • • » ' .L
74,75,83,84). ' "V • '
II.3.1.1. Decomposição térmica do DUA ao ar
Quando a decomposição térmica é feita na
presença de ar, um efeito térmico bem característico (pioo
exotérmico) aparece na curva DTA, em torno de 390°C, resul
tante da reação da amonia retida com o oxigênio da atmosfe-
^ 3 (24 , 40 , .57 , 63, 68, 830._ ^.sse efeito estaré presen
te, nessa temperatura, em menor ou maior extensão de acordo
com a velocidade de aquecimento. Em baixas velocidades de
aquecimento, 5.°C/min por exemplo, essa reação consome toda
a amonia retida antes que a reação de auto-redução possa o-
correr. O produto não contém U-IV e apresenta a cor alaran
jada característica do m^^^^' -^h-
Os õxidos intermediários formados nesta at
mosfera são o UOg amorfo, que se decompõe entre 430 e 450°C,
o beta-UOg, que se decompõe entre 525 e 550°C, e o UO2 g ,
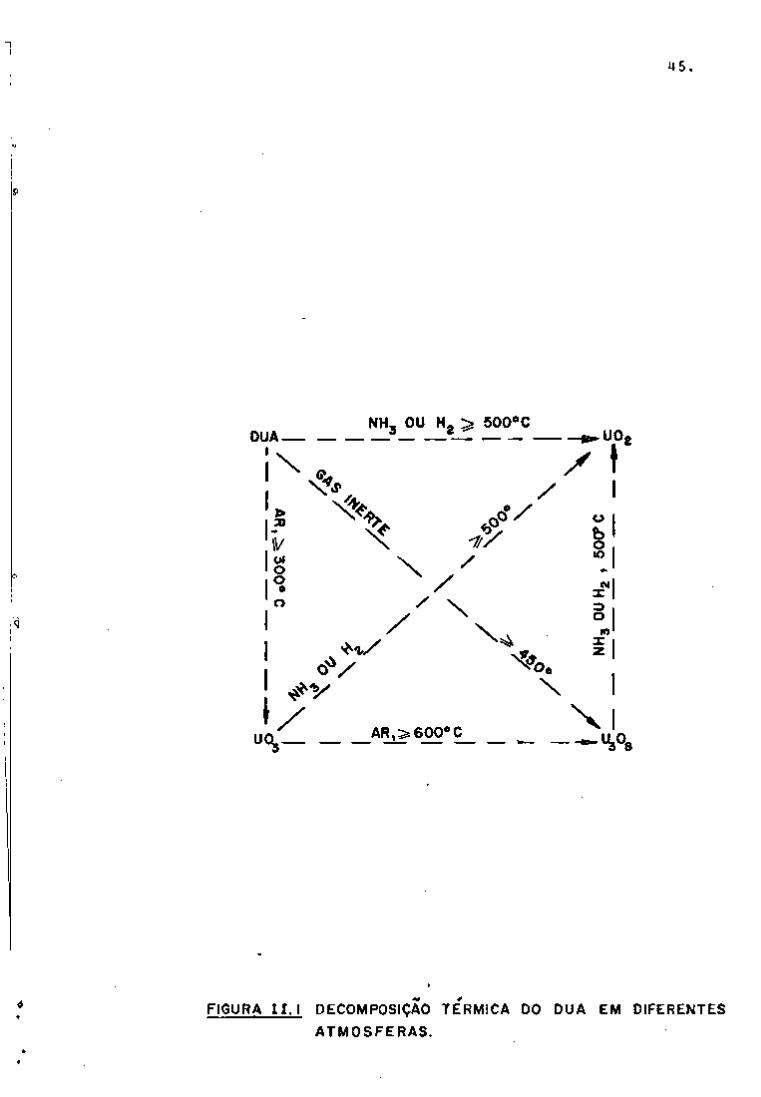
45.
DUA. NHg OU Hg ^ 500»C
UO
>
V Oi O
O
o O
/ 1
/ I
2
^7/ Oi
Oi
IO X
FIGURA II. I DECOMPOSIÇÃO T É R M I C A DO D U A E M D I F E R E N T E S
A T M O S F E R A S .

46
ncnOr. ( 0, 44, 55, 84)
que se decompõe em 550 C • '
Os resultados das curvas DTA e TG indicam
que o UgOg êa fase predominante em torno de 600°C, embora a
difração de raios-X mostre que ele também se forma em torno
o (11, 21, 24 , 40, 57, 73,: :74, 75, 84) de 500°C » ' ' ' ' '
II.3.1.2. Decomposição térmica do DUA em atmosfera inerte
As curvas termoanalíticas obtidas em atmos
fera inerte apresentam características semelhantes aquelas
resultantes da decomposição sob vácuo. Em sua maioria, os
trabalhos apresentam concordância de resultados em relação
ãs temperaturas e aos produtos da decomposição térmica do
DUA em atmosfera inerte.
Nessa atmosfera, a mistura UO^-U^Og tem si
do detectada, em torno de 400^0, por espectroscopia no in
fravermelho e difração de raios-X. A presença de U^Og nesta
temperatura foi explicada como função da reação de auto-re
dução que se processa no interior do solido. O beta-UO^ e o
UO2 que se formaram mantêm-se estáveis em temperaturas mais
altas, devido ã ausência de agente redutor na atmosfera de
(57, 61, 63, 68, 83, 84, 86) reação ' ' ' .
Quando a decomposição do DUA é feita em pre
sença de hidrogênio, uma perda de massa na curva TG e uma
exoterma, correspondente, na curva DTA, em 400°C, foram as
sociadas â formação de UgOg. No intervalo de temperatura de

U50OC a 510OC foi observada a formação de
U7.
(70,83,84)
Ao lado da grande influSncia do tipo de at
mosfera empregada sobre os dados das curvas termoanalíticas
durante o processo de decomposição, outros fatores também
podem ter efeito, como por exemplo, a velocidade de aqueci
mento, o volume da amostra, o material e a forma do cadinho.
A decomposição térmica do DUA depende ainda de sua composi
ção, especialmente do teor de amonia e de nitrato ou sulfa
to de amônio ocluidos durante a precipitação ^ ' '
63, 70, 86) . A reação de aüto-redução, por exemplo, e afeta
da pela presença de nitrato: maior razão NHj^NOg:U leva a
(63 70 ) uma menor porcentagem de U(IV) no oxido ' .
11.4. INFLUÊNCIA DAS PROPRIEDADES DO ^UA E UO3 SOBRE AS CA
RACTERÍSTICAS DO UO2
As propriedades físicas que caracterizam o
UO25 como densidade, superfície específica, poDosidade, ta
manho e distribuição de cristais, são determinantes na rea
tividade química do óxido para produção de UF^ ou nas carac
terísticas de compactabilidade e de sintetização exigidas
na produção de óxidos cerâmicos. Em sua maioria, os traba
lhos na área de fabricação do combustível nuclear concordam
que as propiredades do UO2 dependem das propriedades do pó . (1,59,
original (DUA, ATCU, nitrato de uranilo, UO3 ou UgOg)
64, 85)

48 .
O prçcesso de produção, o tipo de reagente
e alguns parámetros, como por exemplo, ordem de adição e
concentração dos reagentes, agitação, velocidade, pH e tem
peratura de precipitação, podem ter influencia sobre as pro
priedades do DUA (composição e morfologia), bem como, sobre
as propriedades das fases subseqüentes ã sua preparação (6 ,12, 19 , 2 5, 28 ,3 3 ,40,59 ,64,79 ,80 ,86 ,
(UO3, UgOg, UO2 e UFj ) • ' ' •' ' '
87).
O dióxido de urânio obtido a partir de poli^
uranatos de amonio tem densidade e reatividade mais alta do
(.3 2 59) que o UO2 preparado a partir de outros sais ' . Se o
DUA de partida foi precipitado em pH acima de 6,5, o dióxi-
— ( 6 12 79) do de urânio apresenta alta sinterabilidade " . Co
mo conseqüência, ò UF^ produzido a partir de UO^-DUA sinte-
riza mais facilmente em temperaturas mais baixas, isto é, e
le e indicado para fluoridretações em baixas temperaturas (65),
(450-500OC) .
Durante a decomposição térmica do DUA ocor
rem mudanças rio tamanho da partícula e na superfície especí
fica, até a formação de UO^ ou U^Og. Na redução posterior
desses õxidos, para formação de UO2, não hâ variação no ta
manho da partícula. Portanto, é a preparação do UOgOuU^Og ,
mais do que as propriedades do DUA, que, normalmente, deter
(li 8 6) mina as propriedades do UO2 '
A superfície específica aumenta durante a
decomposição térmica do DUA,, apresentante um valor máximo,
na fase UO^, em torno de 450°C, e uma queda brusca acima de •3

49 .
5 5 0 O C , para a fase UgOg. Embora RIBAS ^^^^ ( 1 9 7 3 ) não tenha
encontrado relação alguma entre as superfícies específicas
do DUA de partida e do UO^ produzido, em 1 9 7 8 , WOOLFREY
( 8 6 ) - ^
verificou que, para produzir dioxido de uranio com su
perficie específica alta deve-se usar DUA com superficie es
pecifica alta, ter alto teor de amonia na estrutura e baixo
teor de nitrato ocluido.
4

50
III. PARTE EXPERIMENTAL
III.1. EQUIPAMENTOS
- Sistema Modular de Análise Térmica, Du Pont:
Analisador Térmico, modelo 9 90; Analisador
Termogravimetrico, modelo 9 51; Célula Base;
Célula de Calorimetria Exploratoria Diferen
cial e célula de Análise Térmica Diferenci
al (500^0.
- Espectrofotometro de feixe duplo com regis
trador, Perkin Elmer, modelo 3 56.
- Estufa Fanen, até 200°C.
- Mufla Heraus, até 1000°C.
- Balança Analítica, modelo H 16, Mettler.
III.1.1. Especificações dos mõdulos de análise térmica
III.1.1.1. Modulo Central
O modulo central, modelo 990, tem duas uni-

dades funcionais distintas, comuns a todas as técnicas ter
moanalíticas, que são completamente independentes uma da ou
tra:
a) Programador-Controlador de Temperatura
b) Registrador.
Independente da técnica de análise térmica
empregada, a temperatura do ambiente da amostra, sob aqueci
mento, é controlada pelo Programador (a), enquanto a varia
ção na propriedade a ser estudada (isto é, absorção ou libe
ração de energia, mudança de massa, etc.) é registrada nos
eixos Y e y em função da temperatura da amostra sobre o ei
xo X do Registrador (b). As Figuras III.l e III.2 mostram,
respectivamente, em diagrama de bloco, a termobalança e a
célula DTA conectadas ao módulo central (990).
Características e especificaçóes do móàulo
central (990):
- Razão de aquecimento e resfriamento linear
em todo intervalo de temperatura, para todos os módulos.
- Seletor de razão de aquecimento, escala fi
xa (0,5; 1; 2; 5; 10; 20; 50 e 100°C/min.) e escalas variá
veis C (O a lOOC/min.) A e B (O a lOO^C).
- Exatidão da razão de aquecimento: Í 2 % ou
0,02OC/min.
- Precisão da razão de aquecimento: 1 % ou
íU.:_.,.^_. , . I. P . e. N . " .

52.
0,05OC/min.
- Corrente máxima do sistema de aquecimento :
amperes.
-Intervalo de temperatura programavel: -190
a 1 6 0 0 O C .
Características e especificações do regis -
trader:
- Marca Honeywell, Modelo Y540D2, X-Y-Y'.
- Escalas do eixo X: 5-, 10; 20; 50; 100°C/po-
legada (0,2; 0,4; 0,8; 2; 4 mV/polegada).
- Escalas dos eixos Y e Y': 0,05; 0,1; 0,2;
0,5; 1; 2; 5; 10; 20; 50/polegada (TG = mg/pol.; DTG=
(mg/min)/pol; DTA = <^C/pol. e DSC = (mcal./seg)/pol.) (1;
- 2; 4; 10; 20; 40; 100; 200; 400; 1000 mV/pol.).
- Linearidade da razão de aquecimento (termo
pares Platinei II e Pt/Pt/Rh 13 % ) : + 1 % ou 0,01°C/min.
- Exatidão da temperatura em operação isotér
mica: ± 1 % (± lOC).
- Exatidão da temperatura final: ± 2 % (í 1°C).

- Exatidão de registro: 0,25 % de toda esca
la, para todos os eixos.
- Precisão de registro: 0,20 % de toda,esca
la, para todos os eixos.
- Linearidade de registro: 0,10 % de toda es_
cala, para todos os eixos.
- Escalas da base de tempo: 0,05; 0,1; 0,2;
0,5; 1; 2; 5 e 10 min./pol.
- Exatidão da base de tempo: ± 1 % da razão
indicada, para todas as escalas.
- Linearidade da base de tempo: ± 1 % de to
da escala, para todas as razões.
III.1.1.2. Célula Base
O Módulo Célula Base opera conectado ao Mó
dulo Central (990) e é usado para suporte das Células
de DTA (Padrão-500°C, P200OC e leOO^C) e Célula DSC.
Durante a operação possui as funções básicas seguintes:
a) Amplificador AT. O ganbo do amplificador AT é ajus^
tado automaticamente para cada célula, permitindo re

gistrar diretamente nas unidades de medida dos eixos
Y e Y' .
b) Circuitos da junção de referência eletrônica para a-
mostra e termopares de controle (compensação eletrô
nica que substitui o banho de gelo externo).
c) Contrôle da linha de base. Permite minimizar o des
vio da linha de base para cada Célula.
d) Sistema para controle de atmosfera e gases para res
friamento .
III.1.1.3. Célula de CaTorimetria Exploratória
Diferencial
A Célula de DSC emprega um disco de Cons -
tantán como fonte principal de fluxo de calor para amo£
tra e referência, o qual atua também, como um dos ele -
mentos de um termopar. Dois termopares diferenciais
(Chromel-Constantan) é fornecido pela solda de dois
fios - termoeletricamente complementares ao material do
disco - ãs bases dos locais que suportam os recipientes
de amostra e de referência. Desde que a resistência tér
mica da amostra e referência seja mantida constante, as
<j,¡ temperaturas diferenciais são diretamente proporcionais
aos fluxos diferenciais de calor. Um par termoelétrico

bb .
(Chromel-Alumel) adicional é fixado no locai da amostra,
a fim de medir a tempreatura da amostra. As variáveis re
gistradas a partir desse sistema são AQ (mcal) no eixo Y
e temperatura .da amostra ou tempo no eixo X. A Figura
III. 3 mostra detalhes da Célula DSC^'^'^\
Características e especificações da Célula
de DSC:
- Intervalo, de temperatura: da ambiente ate
700OC (atmosfera inerte ou redutora) ou até 600°C (atmo£
fera oxidante).
- Quantidade de amostra: 0,5 a 10 0 mg.
- Volume de amostra: 0,0 5 mL.
- Precisão da temperatura: í 1°C.
- Sensibilidade calorimétrica:
0,01 (mcal)seg.)/pol.
- Precisão calorimétrica: ± 1 %
- Sensibilidade da derivada:
0,01'((mcal/seg.)min.)/pol.
- Termopar diferencial: Chromel-Constantan
- Termopar da amostra: Chromel-Alumel

56
- Termopar de controle: Platinei II
- Volume da Célula: 2 mL.
III.IJ.1.4. célula de Análise Térmica Diferencial
(500°C)
Características e especificações da Célula
DTA:
- Bloco de prata para aquecimento, com quatro
cavidades: cavidade central (aquecedor), cavidades fron
tais (recipientes e termopares de amostra e referencia )
e cavidade posterior (termopar de controle).
- Intervalo de temperatura: da ambiente até
500°C.
- Sensibilidade: 0,05OC/pol.
Sensibilidade da derivada: (O,O5°C/min)/pol.
- TermoDar de controle: Platinei II
- Termopar de amostra: Chromel-Alumel
1>? - Termopar de referencia: Chromel-Alumel

A Figura 111,2 mostra, em diagrama de bloco,
a Célula DTA conectada ao Módulo Central.
III. 1.1. 5 . Analisador Terinogravimetrico (951)
O funcionamento desse módulo tem como prin
cípio básico o balanço nulo. Opera de modo continuo em
equilibrio, pois os eventuais deslocamentos do braço de
amostra são detectados por um feixe luminoso - anteparo-
fotocélula e o equilibrio é restabelecido por raeio da
força de um motor de torque magnético. O transdutor, nés_
te caso,é um detector fotossensivel nulo, colocado numa
das extremidades da balança. Consiste de uma lampada, u-
ma placa com uma ranhura conectada ao braço da balança e
um par de diodos fotossensíveis. A placa na posição ñor
mal é tal que a ranhura permite passar igual radiação lu
miñosa para cada fotocélula, produzindo um sinal de sal
da zero, que corresponde ao equilibrio estabelecido no
inicio da operação. Quaisquer mudanças na massa da amos_
tra, resultantes de transições térmicas, provocam um des_
vio desse equilibrio. Tal desvio faz com que a placa se
mova, de modo que a luz incida desigualmente sobre as fo
tocélulas. A força eletromotriz. resultante é amplificada
e volta alimentada como uma corrente para o motor de tor
que restabelecer o equilibrio da balança. Esta corrente
é proporcional ã variação de massa e é registrada no ei-
xo Y ou Y'

58 .
Características e especificações do Analisa
dor Termogravimetrico (951):
- Capacidade: Ig
- Intervalo de temperatura: da ambiente até
1200°C.
- Supressão de massa: até 110 mg.
- Exatidão da supressão de massa: í 0,04 %
- Sensibilidade da medida de massa: 0,2 % de
toda escala.
- Precisão da medida de massa: 0,4 % de toda
escala .
- Exatidão da medida de massa: t 1,0 % de to
da escala .
- Pressão: da atmosférica até 1 Torr.
- Fluxo de gás: até 1 L/min.
- Sensibilidade da derivada:
'0,05 até 50 (mg/min. )/pol.
- Termopar da amostra: Chromel-Alumel
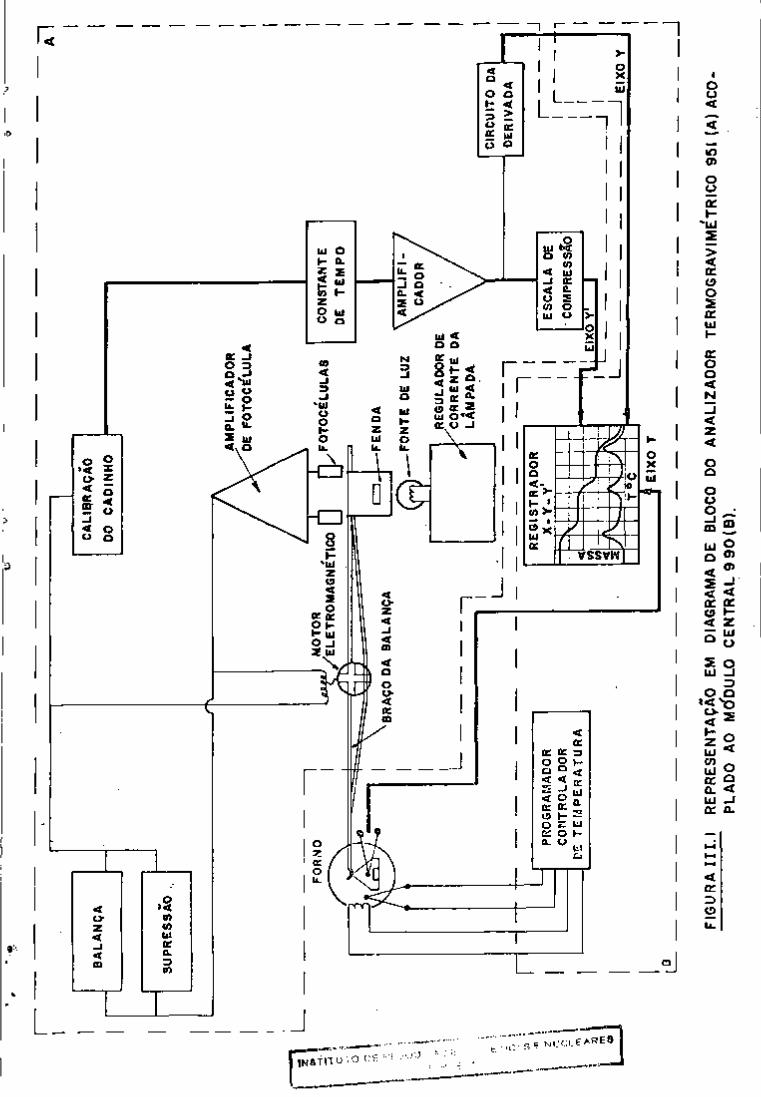
CA
LI9R
AÇ
A0
00
CA
DIN
HO
AM
PLI
FIC
AD
OR
DE
FO
TO
CÉ
LU
LA
FOTO
CÉ
LULA
S
FE
ND
A
CO
NS
TAN
TE
DE
TE
MP
O
FO
NT
E D
E L
UZ
\AM
PL
IFI-
^CA
bO
R
RE
GU
LAD
OR
DE
^
CO
RR
EN
TE
DA
LA
MP
AD
A.
RE
GIS
TR
AD
OR
X-Y
-Y'
\ V)
O
T i L
\ V)
O
T i J
T
I E
SC
AL
A D
E
CO
MPR
ESSÃ
O
Ij^E
IXO
Y'
I
¿1
CIR
CU
ITO
DA
D
ER
IVA
DA
._l
J E
IXO
Y
EIX
O T
1 en
03
FIGURA III
.l REPRESENTAÇÃO
EM
DIAGRAMA DE BLOCO DO ANALIZADOR
TERMOGRAVIMETRICO
951 (A) ACO
PLADO AO
MCTDULO CENTRAL 990(B).
60 .
AMPLIFICADOR A T
R E G I S T R A D O R
<- Y-•Y
r V
UJ L. 1 \ ta
g \ ( ta
g V
f T E M P D A A M O S T R A
I L
SINAL D E T E M P E R A T
DIFERENCIAL
C É L U L A DTA
P R O G R A M A D O R
C O N T R O L A D O R
D E T E M P E R A T U R A
FIGURA III.2 R E P R E S E N T A Ç Ã O E M D I A G R A M A D E B L O C O D A
CE'LULA D T A (500**C) A C O P L A D A A O M O D U L O
C E N T R A L 9 9 0 .
61.
A N E L DE PRATA
PRATO DE AMOSTRA
JUNÇÃO DO TERMOPAR
BLOCO DE AQUECIMENTO
ENTRADA DE G^S
TAMPA
PRATO DE REFERENCIA
DISCO T E R M O E L E -TRICO (CONSTANTAN) CHROMEL
FIO DE ALUMEL
FIOS DE CHROMEL
F I G U R A I I I . 3 R E P R E S E N T A Ç Ã O E M D E T A L H E S DA C É L U L A
D E D S C .

- Termopar de controle: Platinei II.
III.2. MATERIAIS
O diuranato de amonio usado nos experimen
tos provem de lotes diferentes, retirado diretamente da
linha de produção, úmido ou seco, das unidades piloto
do Departamento de Engenharia Química (MQ) do Instituto
de Pesquisas Energéticas e Nucleares (IPEN).
Um tipo de diuranato identificado por
DUAN-B e outro tipo identificado por DUAN-C, foram pro
duzidos, respectivamente, por precipitação em batelada
e continua, em solução de nitrato de uranilo com amónia
gasosa, filtração contínua e secagem estacionaria em es
tufa ou secagem contínua em forno elétrico do tipo tú-
(9,33) nel ' , com cinta rolante de aço.
A amostragem dos compostos foi feita levan
do em conta não só o tipo de precipitação (batelada ou
contínua) mas também, os valores dos parâmetros de pre
cipitação (pH e temperatura) e de secagem (temperatura
e tempo). Na Tabela III.l estão relacionados vários ti
pos de diuranato de amônio, utilizados neste trabalho.
A Figura III.l mostra, em digrama de blo
co, a termobalança conectada ao Módulo Central (990).

com os respectivos valores dos parâmetros de precipita
ção e secagem.
O trióxido de urânio (UOg) utilizado
neste trabalho, como no caso do DUA, foi retirado dire
tamente da linha de produção. Foi obtido a partir de
DUAN-C, por aquecimento em forno do tipo túnel, cujas
zonas de aquecimento estavam, respectivamente, a 200°C, ( o)
300OC e 400OC ' . A amostragem do material foi feita
de acordo com as condições de preparação do diuranato
de amonio (composto de partida) e com a cor do - produto
obtido (UOg - fração amarela, alaranjada e verde oliva).
Após homogeneização, em almofariz de á-
gata, os compostos secos.foram transferidos para peque
nos frascos claros com tampa e guardados.
III.3. COMPORTAMENTO TERMICO
III. 3.1. Variação de cor dos diuranatos de amor) i o e
trióxido de urânio durante o aquecimento
Quantidades pequenas de amostras, colo
cadas em suportes de porcelana, foram submetidas ao a-
quecimento no intervalo de temperatura de 100' C a 650°C ,
em mufla comum de laboratório. No decorrer dessa opera
ção observou-se o comportamento dos materiais em rela -
TABELA „111.1« Dados da produção dos diuranatos de amonio
PRECIPITAÇÃO LAVAGEM SECAGEM
DUA pH T (OC)
t (min.)
NH^NOg
2 ,5 %
T (OC)
t (min.)
1-B 9,0 85 3 5 não 90 2160
2-C 9,2 60 120'. não
3-C 9,2 20 120 não Ä
4-B 9,1 85 35 não 115 5760 no»0.5
5-B 9,0- 70 50 sim 90 2160
6-B 8,7 75 40 não 180 240
7-B 8,7- 90 30 não 280 720
8-B 8,3 60 35 sim 115 5760,
9-B 8,2 70 40 sim 90 2160
10-B 7,6 65 35 sim 90 2160
11-B 7,6 70 40 • sim 130 4320 1 2 \\oí¿
12-C 7,6 60 12 0 não
13-B 7,5 60 4 0 sim 115 5760
14-B 7,3 65 40 não 180 ' 240
15-B 6,3 60 25 não 180 240
16-B 6,0 70 35 não 90 2160
17-B 6,0 65 35 não 130 4320
18-B 6,0 65 35 sim 220 a440
19-B 5,8 70 •30 sim. 130 4320
20-C 4,0 65 180 não 280 720
21-B 3,9 65 35 não 130 4320
* Secadas a 50°C tivãmente, por
por 120 minutos 120, 1440, 2880
e a 75°C e 150°C, e 4320 minutos.
respec-
H NUC'-EARE3
obtidos pelos processos de precipitação em
batelada (B) e continuo (C) utilizados nes
te trabalho.

6 5 .
ção ãs mudanças de cor, em intervalos de 50°C, apôs 20
minutos em cada temperatura.
III.3.2. Influência da etapa de secagem sobre o com -
portamento térmico do diuranato de amonio
Algumas amostras de diuranatos, do ti
po DUAN-C, foram retiradas da linha de produção, ainda
úmidas, com a finalidade de se verificar a influência
dos parâmetros de secagem (T e t) sobre o comportamen
to termoanalítico destes compostos. Segundo os valores
de pH e temperatura de precipitação, estes compostos
foram separados em 3 tipos diferentes (Tabela III.l) :
2-C, 3-C e 12-C.
A secagem desses materiais foi realiza
da de dois modos, em estufa comum de laboratorio e no
módulo de termogravimetria.
Placas úmidas de DUAN-C foram coloca
das em vidro de relógio e submetidas ao aquecimento em
estufa, a 75°C e 150°C, respectivamente, por 120, 1.H40
e 2880 minutos. Em alguns casos, o processo foi reali
zado por tempos maiores (4320 minutos), a fim de simu
lar melhor as condições operacionais de secagem utili
zadas nas unidades piloto.
Do mesmo modo, por termogravimetria, es

66.
III.3.3. Decompôsição térmica de diuranatos de amónio
envelhecidos
ses compostos foram secados a 50°C, 75°C e 150°C, res
pectivamente por 120 minutos, com purga de ar comprimi^
do num fluxo de 100 mL/minuto.
As amostras secadas em estufa foram
trituradas em almofariz de ágata e conservadas em des-
secador. Nesta operação, foi possível classificar es
tes diuranatos segundo um grau de dur^eza relativo, on
de a variação nos valores dos parâmetros de secagem
não apresentou influência:
DUA tipo 2-:C: material mole;
DUA tipo 3-C: material duro;
DUA tipo 12-C: material muito duro.
Uma alíquota de cada composto foi usa
da para o tratamento térmico (TG, DTG e DSC) e outra
parte foi estocada em condições de laboratório, para
estudos posteriores sobre o tempo de envelhecimento dos
diuranatos.
Os compostos secados no módulo de ter
mogravimetria foram utilizados imediatamente no estudo
da decomposição térmica.

57.
Compostos envelhecidos, selecionados se
gundo os parâmetros de precipitação (pH e T) e secagem
(T e t ) , foram caracterizados por TG e DTG como descri
to em III.U.l.
III.U. CARÁCTER!ZAÇÃO DOS DIURANATOS DE AMÕNIO E TRIÕXI
DOS DE URÂNIO
III.4.1. Curvas termogravimêtricas é termogravimétricas
derivadas
As curvas TG e DTG foram obtidas ao ar
e em atmosfera de N^, com um fluxo de 100 mL por minu
to, por meio do módulo Analisador Termogravimetrico 951,
com capacidade de operação da temperatura ambiente ati
1200^0. O controle e ,a medição de temperatura foram re
alizados por meio de termopares de Platinei II e Chro
mel-Alumel.
A massa das amostras, colocadas em cadi
nho de platina, oscilou em torno de 2 7 mg para a maio
ria dos diuranatos e óxidos (UO^). Em alguns casos, u-
saram-se amostras com cerca de 18 mg.
O fundo de escala de 10 polegadas do
sistema balança-registrador foi ajustado de modo que um
deslocamento de uma polegada no traço do registrador

6 8 .
(eixo Y) correspondesse a uma perda de massa de 0,5 mg.
III.4.2, Curvas calorimétricas exploratórias
diferenciais
As curvas DSC foram obtidas também em
atmosfera de nitrogênio e ar, num fluxo de 100 mL por
minuto, com a Célula de DSC acoplada aos módulos Célula
Base e Central (990), descritos em III.l.
A massa das amostras, colocadas em cadi
nho de alumínio, sem diluição foi da ordem de 22 mg. Um
cadinho de alumínio vazio foi usado como material de re
ferência. Com intuito de melhorar a reprobutibilidade
durante o enchimento do cadinho, foi usada sempre a me£
ma técnica para compactar as amostras.
D registro das curvas foi feito numa
O sistema de registro para o eixo das
ordenadas (temperatura) foi fixado numa ordem de uma po
legada por 100°C e a razão de aquecimento foi da ordem
de 5°C por minuto.
O intervalo de temperatura estudado foi
de 25^0 a 650°C ou, em alguns casos, ate 800OC.
O oxalato de calcio monoíhidratado foi u
sado como composto padrão para verificar o .desempenho
do modulo de termogravimetria.

69.
III. 5. PROCEDIMENTOS ANALÍTICOS
III. 5.1. Determinação de urânio
A análise quantitativa de urânio
nesses compostos foi feita por dois métodos: o método
termogravimetrico, que consiste no aquecimento do mate
rial em termobalança, até 650°C ou 900^0, obtendo-se
UgOg, descrito no Apêndice 1; e, o método volumétrico,
que consiste na titulação com dicromato de potássio, u-
sensibilidade de 1,0 mcal/seg. por polegada, razão de a
quecimento de 5°C por minuto e variação de uma polegada
por 100°C no eixo das ordenadas.
O intervalo de temperatura estudado foi
de 25 a OOO^C para todas as amostras.
A medida das temperaturas de fusão do
índio metálico e do zinco metálico foram usadas para ca
libração do modulo de DSC.
Neste trabalho, o uso da Célula de DSC
envolveu apenas o aspecto qualitativo da caracterização
dos materiais (DUA e UO^), em substituição a Célula de
DTA que tem sua operação limitada ã temperatura de
500°C (III.1.4).

70.
sando difenilamina como indicador, conforme técnica des
crita por FEDERGRÜN e ABRAO o segundo método foi
usado apenas para verificação de alguns resultados obti
dos pelo método termogravimetrico.
III.5.2. Determinação do teor de amónia
A determinação de amónia nos diuranatos
foi feita em células de microdifusão, do tipo CONWAY, u
sando o método de titulação por retorno (back-titration )
(3)
A verificação dos resultados foi feita pela deter
minação da amónia por meio do método espectrofométrico,
usando o reagente de Nessier ^ \
111. 5 . 3 . Determiriaçâo de íons nitrato
Os íons nitrato, ocluidos nesses compo£
tos, foram determinados por espectrofotometria de absor
ção molecular, após separação do urânio por troca ióni
ca. O reagente colorimétrico usado para sensibilizar a
presença de nitrato foi o ácido 1-2-4 fenoldissulfônico
(62) ,fazendo-se a leitura em 405 nm.
III.5.4. Determiriaçao do teor de água
Foi feita pelo método termogravimétri -
C O , a partir de cálculos de perda de massa (curvas TG e

71.
DTG), no intervalo de temperatura de 20°C a 200°C
(215°C ou 2.250c, dependendo do tipo de material; água
livre e água de cristalização), pelo uso das temperatu
ras inicial e final dos picos DTG, cOmo indicadores do
inicio e término da perda de água. A água de composi
ção foi calculada do mesmo modo, no intervalo de tempe
ratura de .200^0 a 4 50°C, por diferença de massa após co
nhecer as porcentagens de NH3, NOg e 1103:% combinada
quimicamente = 100 - (% UOg + % NHg + % NO3 + % H2O de
cristalização).
III. 5.5. Determinação das razões molares NK^^./\¿ ,
NO f / \ l e Ef/U
Foram calculadas a partir da relação en
tre os dados obtidos nos itens III.5.2, III.5.3 e III.
5.4.com os dados do item III.5.1.
III.5.6. Determinação da razão O/U no resíduo de calei
nação dos compostos
A composição da fase U30g_^ foi calcula
da a partir da perda de massa do material acima de
5700c, considerando-se que a fase UO3 é estequiométrica
(método termogravimetrico). A verificação dos resulta -.
dos foi feita pelo método volumétrico citado em III.5.1.

72
III.5.7. Determinação da densidade
A densidade media ou aparente, chamada
comumente "densidade solta", foi obtida de acordo com o ml
(65)
todo padrão que consiste em encher, sob condições de
terminadas, um recipiente de dimensões específicas (25Í
i 0,05 cm ) com o p5 e pesar essa quantidade, calculando -
se em seguida a relação massa/volume.
A densidade batida foi determinada de
acordo com o método clássico, que consiste em colocar uma
quantidade conhecida de põ numa proveta graduada, deixando
a cair livremente 20 vezes de uma altura de 15 cm sobre u-
ma base de borracha semi-dura, calculando-se a seguir a
densidade do põ a partir da massa da amostra e do volume
(65) que esta ocupa na proveta . .
III.5.8. Determinação da superfície específica
Foi determinada pela adsorção de nitro
gênio, usando o princípio B.E.T. (Brunauer, Emmett e
Teller) . ^ ^v
!. P - t . N.
é-,,cíSÊ N U C L E A R E S ,

73.
IV. RESULTADOS
IV.1. EXPERIMENTOS PRELIMINARES
Algumas operações iniciais foram realiza
das, por TG e.DTGy a fim de avaliar a melhor quantidade de
amostra e a influincia do tamanho do grão , nã -decompçsição
térmica dos diuranatos de amonio.
IV.1.1. Quantidade de amostra
Em geral, a decomposição térmica do DUA,
no intervalo de temperatura de 20°C a 650°C, apresenta^, apro
ximadamente, de 12 a 20 % de perda de massa. As reações en
volvidas apresentam variações de massa entre 0,10 % a 8,00 %
da massa original.
Apesar da alta sensibilidade da termoba
lança (Item III. 1.1.5), verificou-se que quantidades de amos_
tra abaixo de 5 mg resultavam em curvas com baixa resolução
para determinadas reações. No entanto, devido ao pequeno ta
manho do cadinho, massas acima de 35 mg resultavam em perdas
por transbordamento. Assim, resultados reprodutíveis foram
obtidos empregando-se cargas de amostras com massas entre 10
e 30 mg.

74.
A escolha de cargas de amostra em torno
de 27 mg pode ser atribuida ã melhor resolução dos eventos
sobre as curvas TG e DTG, que facilitou a interpretação dos
resultados, principalmente, para as reações com baixa por -
centagem de perda de massa (menor 0,20 % ) .
IV.1.2. Tamanho do grão
Pos de DUA, em geral, apresentam-se sob
a forma de aglomerados de pequenas partículas. Por peneira-
mento, separaram-se os grãos em duas frações:
- Fração grossa, formada de partículas e
aglomerados de partículas, > O,10 mm (~ 10 % ) ;
- Fração fina, composta por partículas e
aglomerados de partículas, ^ 0,08 mm (- 90 % ) .
Curvas TG e DTG foram obtidas para as
frações grossa, fina e amostra original. Os resultados reve
laram o mesmo comportamento térmico para as três frações. Por
tanto, não foi necessário fazer separação prévia de grãos pa
ra estudo da decomposição térmica desses compostos.
IV.1.3. Variação de gor -durante o aquecimento dos diurana -
tos de amônio ao ar
Os diuranatos de amônio são .compostos de

75.
cor amarelada, que apresentam variações de cores quando sub
metidos ao aquecimento. Neste trabalho, utilizando três di
ferentes lotes de cada tipo.^de diuranato, verificou-se que a
intensidade da cor amarela nos pôs de DUA (envelhecidos) va
riou em função dos parâmetros de secagem:
- Amarela: foi observada nos compostos se
cados em temperatura e tempo, respectivamente, de ate 10 5°C
e 5800 minutos.
- Amarela muito.clara: nos compostos seca
dos no intervalo de temperatura de 130 a 180°C, por tempo su
perior a 240 minutos.
- Alaranjada: nos compostos secados : em
temperatura e tempo, respectivamente, acima de 200°C e 360
minutos.
A cor dos compostos começa a mudar em
temperaturas acima de 150°C, passando, gradualmente, de uma
cor amarelada a alaranjado no intervalo de temperatura de
150OC a 2500c.
De 250OC a 450°Cos compostos apresentam
uma mudança gradual de cor, do alaranjado a uma mistura de
cores do alaranjado com o.marrón e o verde oliva, ou do ala
ranjado ao marrón ou verde oliva, ou do alaranjado ao verme
lho tijolo, dependendo do diuranato. Esse intervalo de tem
peratura coincidiu com as temperaturas de liberação de amó
nia e decomposição de nitrato.
De acordo com a descrição em II.3.1 Cde-

76.
composição térmica do DUA), entre 450°C e 550°C, o UO^ é a
fase predominante para os compostos -que apresentaram as co -
res alaranjado e vermelho tijolo e uma possível mistura de _
UOg e UgOg para os compostos com as cores marron e verde o-
liva. \ i/o
Acima de 55 0°C, a cor dos compostos pas
sa a ser o verde oliva ou o preto. A diferença de cor no re
síduo, a 650°C, pode ser atribuída ã formação da fase ^3^3
com diferente razão O/U.
Na Tabela IV.1 estão as variações de cor
observadas ao se aquecer os diuranatos de amônio relaciona -
dos na Tabela III.l.
IV.1.4. Estudo comparativo do comportamento térmico de qua
tro tipos de diuranato de amônio envelhecidos
De acordo com as informações em II.1, o
pH de precipitação foi considerado, entre outros fatores, o
parâmetro mais importante na produção de DUA. Portanto, com
o objetivo de avaliar a eficiência dos métodos termoanaliti
cos no estudo da decomposição térmica do DUA, algumas amos
tras, 1-B, 4-B, 6-B e 7-B (Tabela III.l), produzidas, aproxi
madamente, no mesmo pH (9,0) foram caracterizadas por TG e
DTG.
As Figuras IV.1 e IV.2 ilustram, respec
tivamente, os resultados da termogravimetria (curvas TG) e
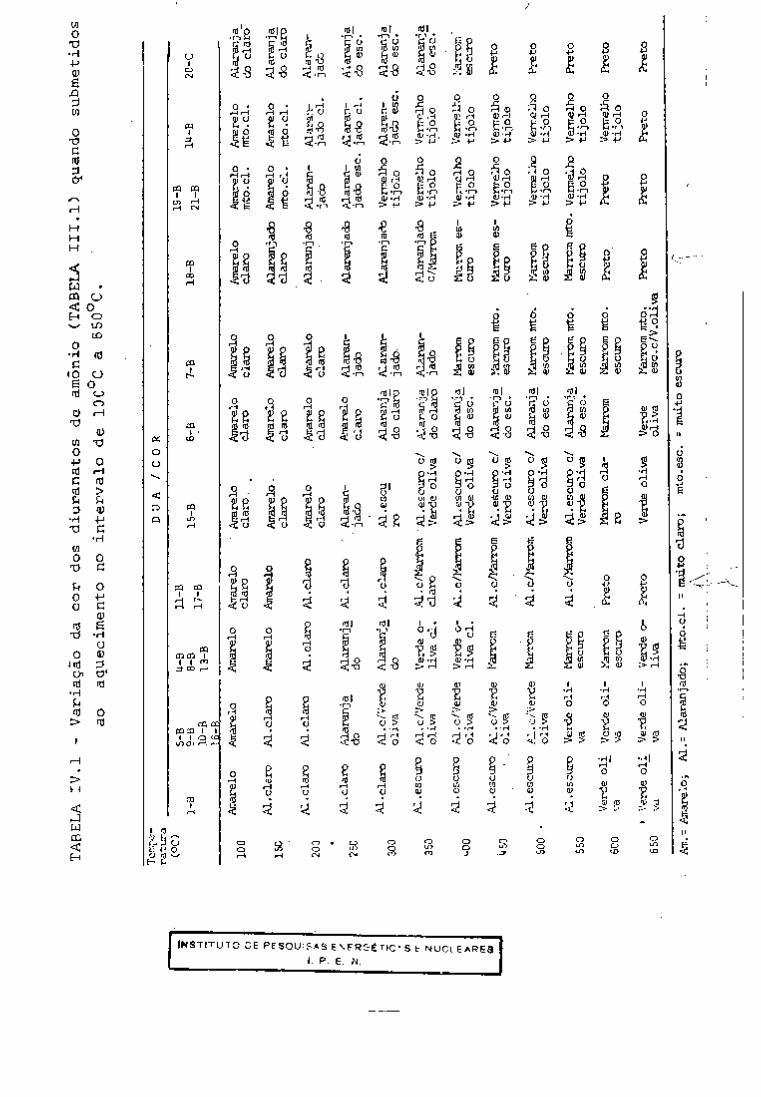
TABELA IV.1 - Variação da cor dos diuranatos de amonio (TABELA III.l) quando submetidos
ao
aquecimento no intervalo de 10 0°C a 6 50°C,
ratijra
DU
A /
CO
R
2 m
H H C H
O o
M T3
Tl
O)
o c
—
''f>
y CO
_ F
m ^
. Tl
:D
Q
m.
H O > CO
M 2 C O (—
M > (11
OB
(OC)
1-B
5-B
9-B
10-B
1B-R
U-B
8-B
13-B
11-B
17-B
15-B
6-B
7-B
18-B
19-B
21-B
14-B
20-C
100
Airarelo
Amarelo
Anarelo
Aitarelo
claro
AiiHrelo
el aro,
Anarelo
claro
Amarelo
claro
Anarelo
claro
Amarelo
mto.cl.
Anarelo
mto.cl.
Alaran ja_
do claro
150
Al.clcxo
Al.claro
Airarelo
Anarelo
Amarelo.
claro
Anarelo
claro
Anarelo
claro
Alaran jacto
claro
Amarelo
rato.cl. Anarelo
ntto.cl.
Alaranja
do claro
200
. claro Al.claro
Al.claro /U..claro
Anarelo
claro
Anarelo
claro
Anarelo
claro
Alaranjado Alaran-
jacto
Alaran
jado cl.
Alaran
jado
250
Al.claro
Alaiwija
do
Al aranja
do
Al.cü.aro
Alaran
jado '
Anarelo
claro
jado
Álaian jacto Alaran
jado esc. Alaran
jado cl.
Alaranja
do esc.
300
Al.claro
Al.c/Vercüe
oliva
Alaranja
do
Al.claro
.Al. escu
ro
~
Alaranja
do claro Alaran
jado/
Alaranjado Vermelho
tijolo.
Alaran-
jacto esc. Alaranja
do esc.
350
AJ. escuro Al.c/Verde
oliva
Verde o-
liva d. .Al.c/
faríom
ciLaro .
"Al.escuro c/
Verde oliva
Alaranja
do claro Alaran
jado
Alaran jacto
c/Marrom
Vermelho
tijolo
Vemielho
tijolo
/laranja
do esc.
400
Al.escuro Al.c/Verde
oliva
Verde o-
liva el. íú..o/Vsrcam.
Al.escuro c/
Verde oliva
Alaranja
do esci.
Marrom
escuro
MniTom es
curo
VerTirelho
tijolo
Vermelho
tijolo
Farrom'
escuro
it50
Al.escuro Al.c/Verde
oliva
ítorcín
Al.c/íferrom Al.escuro c/
Verde oliva
Alaranja
do esc.
Marrom ntto.
escuro
Marrom es
curo
Vermelho
tijolo
Vermelho
tijolo
Preto
500 • Al.escuro /JL.c/Verde
oliva
Marrom
Al.c/tterom Al.escuro c/
Verde oliva
Alaranja
do esc.
Marrom mto-.
escuro
l^rrom •
esciuro
Vermelho
tijolo
Verr.eliio
tijolo
Preto
550
Al.escuro Verde oli
va
Marrcm
escuro
Al.c/Itorom Al.escuro c/
Verde oliva
Alaranja
do esc.
>FEI
TOM mto.
escuro
hferrom rato,
escuro
Vermelho
tijolo
Vermelho
tijolo
Preto
600
Vercie dlx
va
Verde oli
va
>örrom
escuro
Preto
Marrom cla
ro
Marrom
Marrom mto.
escuro
Preto
Preto
Vermelho
tijolo
Preto
650 ' Vertie oli
va
Verde oli
va
Verde o-
liva
Preto
Verde oliva
Verde
oliva
Itorom rato.
esc.c/V.oliva Preto
Preto
Preto
Preto
Am.= Airarelo; Al. = Alaranjado; rttto.cl. = muito claro;
ntto.esc. = muito escuro
A h

78.
da termogravimetria derivada (curvas DTG) para a decomposi
ção térmica desses diuranatos. Os dados relativos ãs cur
vas TG e DTG estão apresentados na Tabela IV.2.
O número de picos nas curvas DTG (Figu
ra IV.2)5 por exemplo, até a formação do composto intermedi
a r i o , que inicia acima de 400^0, mostram 5 (cinco) reações
para o DUA tipo 1-B, 4 (quatro) reações para os tipos 4-B e
6-B e 3 (três) reações para o tipo 7-B.. Com exceção da rea
ção em 19 8°C, observada nas curvas DTG dos diuranatos 1-B e
6-B e em 585°C, observada nas curvas DTG dos compostos 2-B
e 6-B, todas as outras reações ocorreram em temperaturas e
intervalos de temperatura diferentes (Tabela IV.2). Associ
adas a essas observações, pode-se assinalar a grande varia
ção nos teores de voláteis liberados em cada reação (Tabela
IV.2), revelando composições químicas diferentes para esses
compostos.
A estabilidade térmica dos compostos in
termediarios (UO^) formados a partir das amostras 4-B e 7-B
foi menor do que aqueles formados a partir das amostras 1-B
e 6-B, cujas reações de decomposição se iniciaram, respec
tivamente, em torno de 520°C e 557°C. No primeiro caso, a
reação de decomposição térmica se deu por meio de duas eta
pas, com perda de massa acima de 2 %, enquanto, no segundo
caso essa reação ocorreu por meio de uma sõ etapa, apresen
tando perda de massa em torno de 1,6 % (Figura IV.2 e Tabe
la IV.2).
As diferenças, acima apontadas, para os

79.
valores de temperatura e perda de massa (%) verificadas nas
curvas TG e DTG, correspondentes ã decomposição térmica de£
ses diuranatos, mostram que os parâmetros de precipitação,
por si so, não estabelecem um comportamento térmico defini
do para esses compostos. Assim, pode-se concluir que o com
portamento térmico dos diuranatos de amônio varia em fun
ção,' também, dos parâmetros de s.-cagem (temperatura e tem -
po). Esses resultados estão de acordo com os dados da lite
ratura descritos em II.1.
Para verificar a influência da variação
dos parâmetros de secagem, isolada ou associada ãs condi
ções de precipitação, sobre.o comportamento térmico dos diu
ranatos de amônio, foram executados experimentos com amos
tras de DUA produzido pelo processo de precipitação contí
nua, conforme descrito em III.3.2.
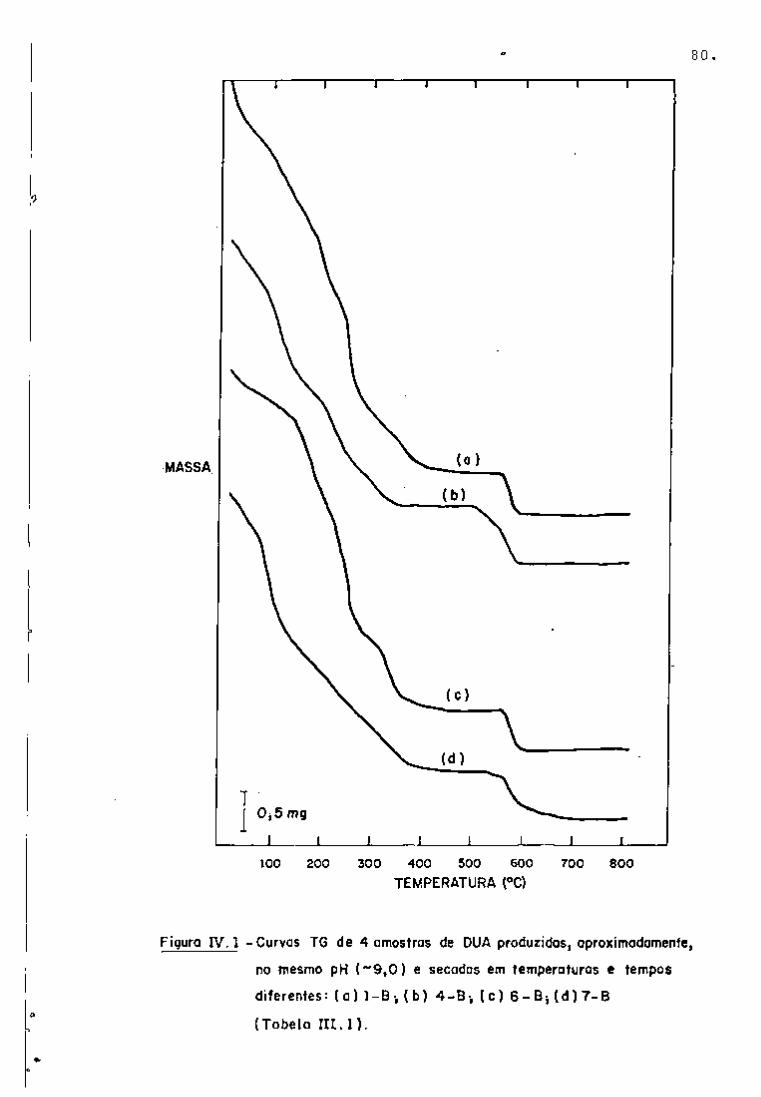
80
MASSA
100 200 300 400 500 600 TEMPERATURA (°C)
700 800
Figuro IV. 1 - Curvas TG de 4 amostras de DUA produzidas, aproximadamente,
no mesmo pH ( ~ 9 , 0 ) e secadas em temperaturas e tempos
diferentes: ( a ) 1-B-, ( b ) 4 - B - , ( c ) 6 - B i ( d ) 7 - B
(Tabe la I I I . l ) .
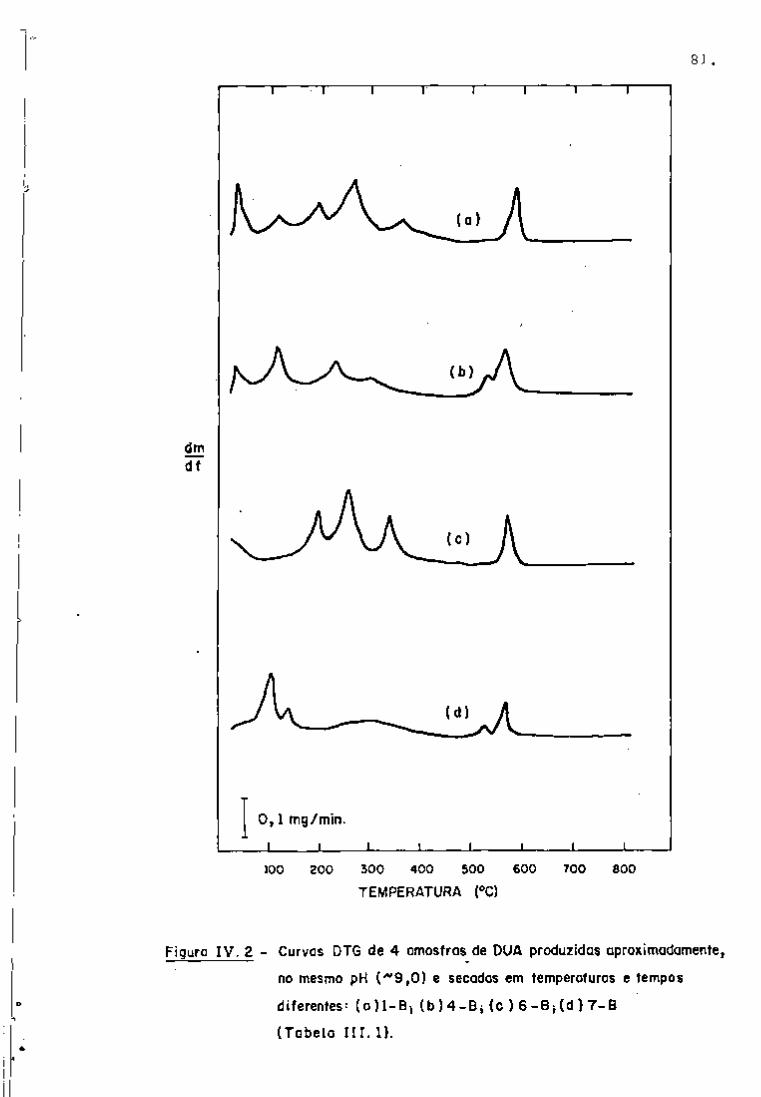
81
dm
d t
100 200 300 400 500 600
TEMPERATURA (''C)
700 800
Figura I V . 2 - Curvas DTG de 4 amostras de DUA produzidas aproximadamente,
no mesmo pH ( ' ^ 9 , 0 ) e secadas em temperaturas e tempos
diferentes: ( o J l - B - , (b ) 4 - 8 , (o ) 6 - B » ( d ) 7 - B
( T a b e l o I I I . l ) .
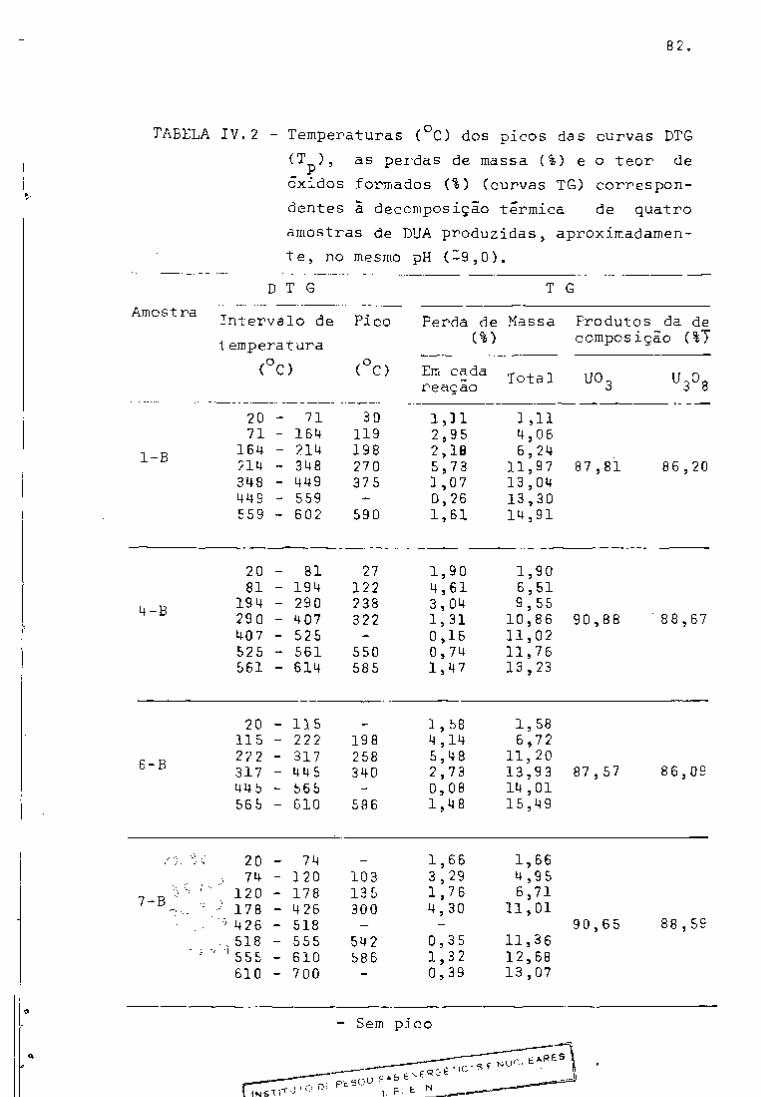
82.
TABELA IV.2 - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes ã decomposição térmica de quatro
amostras de DUA produzidas, aproximadamen
te, no mesmo pH (-9,0).
D T G T G
Amostra Intervalo de
temperatura
Pico Perda de Massa (%)
Produtos da de composição (%T
^C) (°C) Em cada reação
Total UO3 U3°8
20 - 71 30 1,11 1,11 71 - 164 119 2,95 4,06
^"^ 214 - 214 - 348
198 270
2,18 5,73
6,24 11,97 87 ,81 86,20
348 - 449 375 1,07 13,04 449 - 559 - 0,26 13 ,30 559 - 602 590 1,61 14,91
20 - 81 27 1,90 1,90 81 - 194 122 4,61 6,51
4-B ^ ^ 290
- 290 - 407
238 322
3,04 1,31
9,55 10,86 90,88 • 88,67
407 - 525 - 0,16 11,02 525 - 561 550 0,74 11,76 561 - 614 585 . 1,47 13 ,23
20 - 115 1, 58 1, 58 115 - 222 198 4,14 6,72
K B 222 ^ ^ 317
- 317 258 5,48 11,20 K B 222 ^ ^ 317 - 44 5 340 2,73 13,93 87, 57 86,09
445 - 565 - 0,08 14,01 565 - 610 586 1,48 15,49
70.. C 2 0 - 74 1,66 1,66 - 120 103 3 ,29 4,95
7 R ^ ^ • • 120 - 178 - 426
135 300
1,76 4,30
6,71 11,01
-^426 - 518 - - 90,65 88 , 59 . .„ 518 - 555 542 0,3 5 11,36 "' "555 - 610 586 1,32 12,68
610 - 7 00 0,39 13 ,07
- Sem pico

83.
IV.2.. ESTUDO DA INFLUENCIA DOS PARÂMETROS DE SECAGEM SOBRE
O COMPORTAMENTO TÉRMICO DOS DIURANATOS DE AMÕNIO
IV,2.1. Secagem no módulo de termogravimetria e decomposigao
térmica dos diuranatos maciços.
Na Figura IV.3 estão representadas . as
curvas TG e DTG que mostram ai-variação do comportamento tér
mico em função da variação da temperatura e tempo de secagem,
respectivamente, para os diuranatos 2-C, 3-C e 12-C (Tabela
III.l) secados no modulo de termogravimetria. De acordo com
essas curvas, os compostos estão relativamente secos após 50,
25 e 10 minutos, respectivamente, a 50°C, 75°C e 15Ü°C.
Os teores de voláteis liberados durante
a secagem nessas temperaturas, por 120 minutos, apresentados
sob a forma de perda de massa (%) em função do tempo, ' estão
na Tabela IV.3. Apesar das curvas TG e DTG apresentarem o
mesmo formato para a secagem desses compostos, verificou -se
maior perda de massa (%) para DUA tipo 12-C secado a 75°C e
150°C (Tabela IV.3).
Para cada temperatura de secagem encon -
tram-se nas Figuras IV.U e IV.5 as curvas TG e DTG correspon
dentes ã decomposição térmica desses compostos, no estado ma
ciço, até a fase U^Og. Os dados relativos a essas curvas es
tão apresentados nas Tabelas IV.U, IV.5 e IV.6.
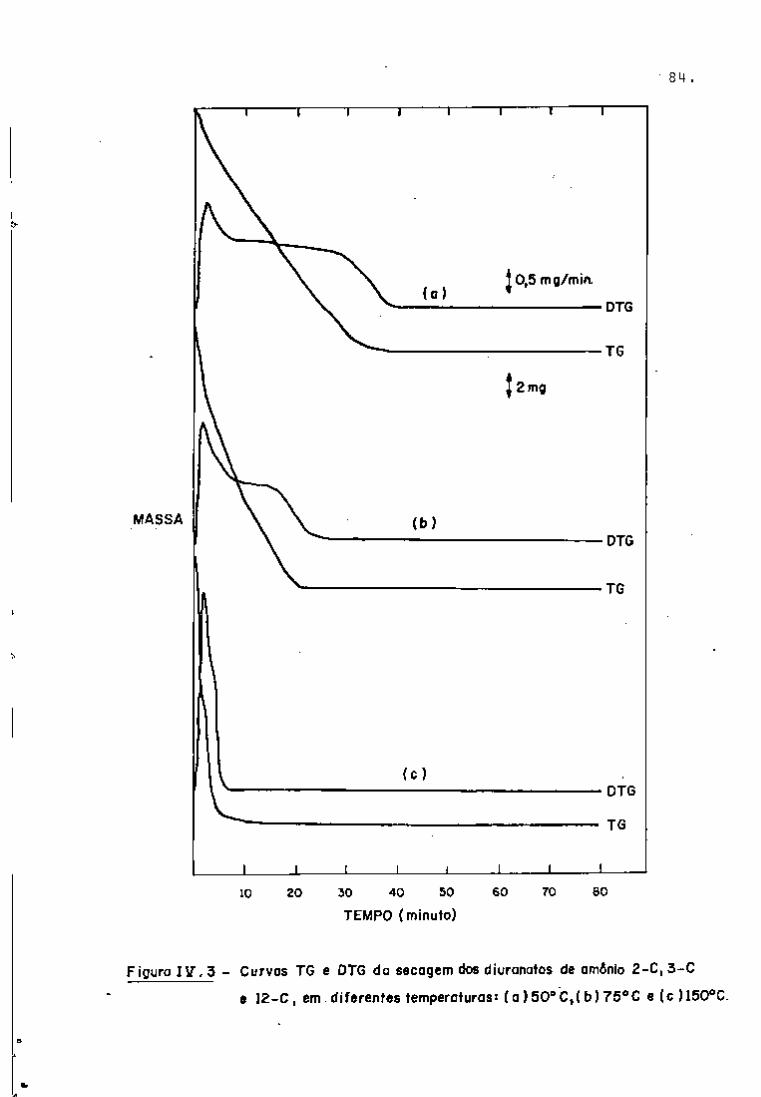
8 4
MASSA
10 20 30 40 50 60 70 80
TEMPO (minuto)
Figuro I V . S - Curvas TG e DTG da secagem dos diuranatos de amônio 2 - C , 3 - C
e 1 2 - C , em diferentes temperaturas--(a )50* 'c , (b) 7 5 * 0 e ( c ) 1 5 0 » C .
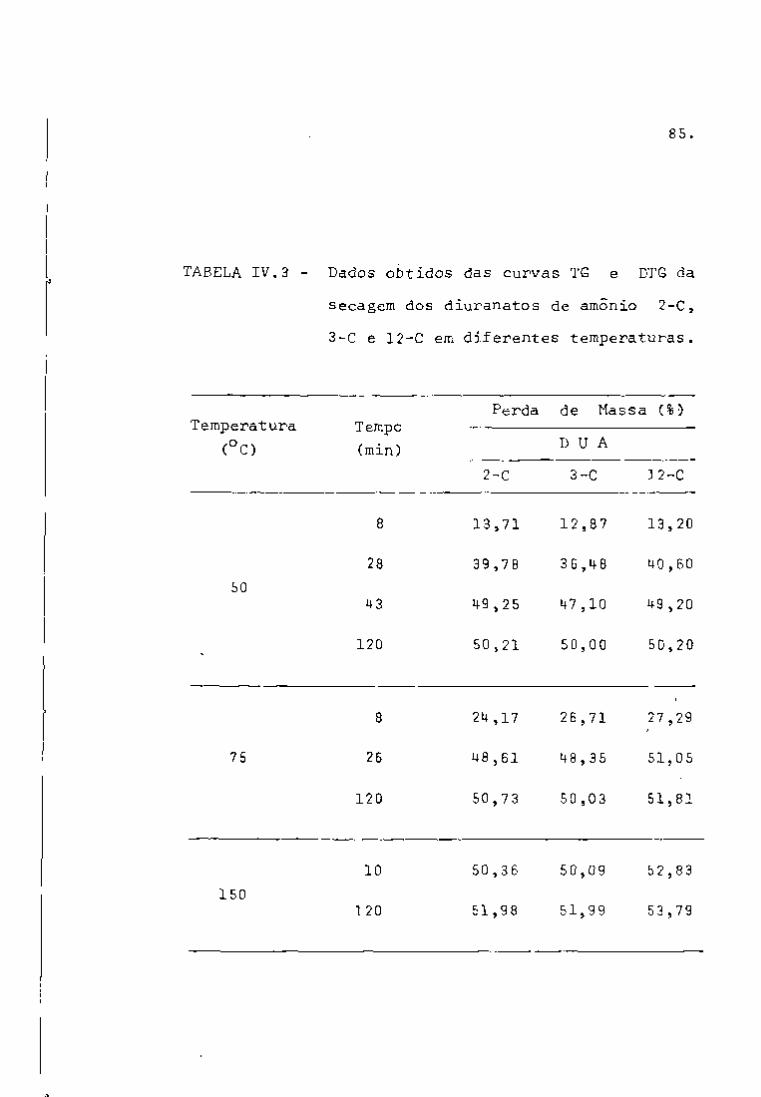
85.
TABELA IV.3 - Dados obtidos das curvas TG e DTG da secagem dos diuranatos de amonio 2-C, 3-C e 12-C em diferentes temperaturas.
Temperatura ( C)
Tempo (min)
Perda de Massa (%) Temperatura
( C) Tempo (min) D U A
2-C 3-C 12-C
8 13 ,71 12,87 13,20
28 39,78 36,48 40,60 50
43 49 ,25 47,10 49 ,20
120 50,21 50,00 50,20
8 24,17 26,71 27 ,29
75 26 48 ,61 48,35 51,05
120 50,73 50,03 51,81
10 50 ,36 50,09 52,83 150
120 51,98 51,99 53,79
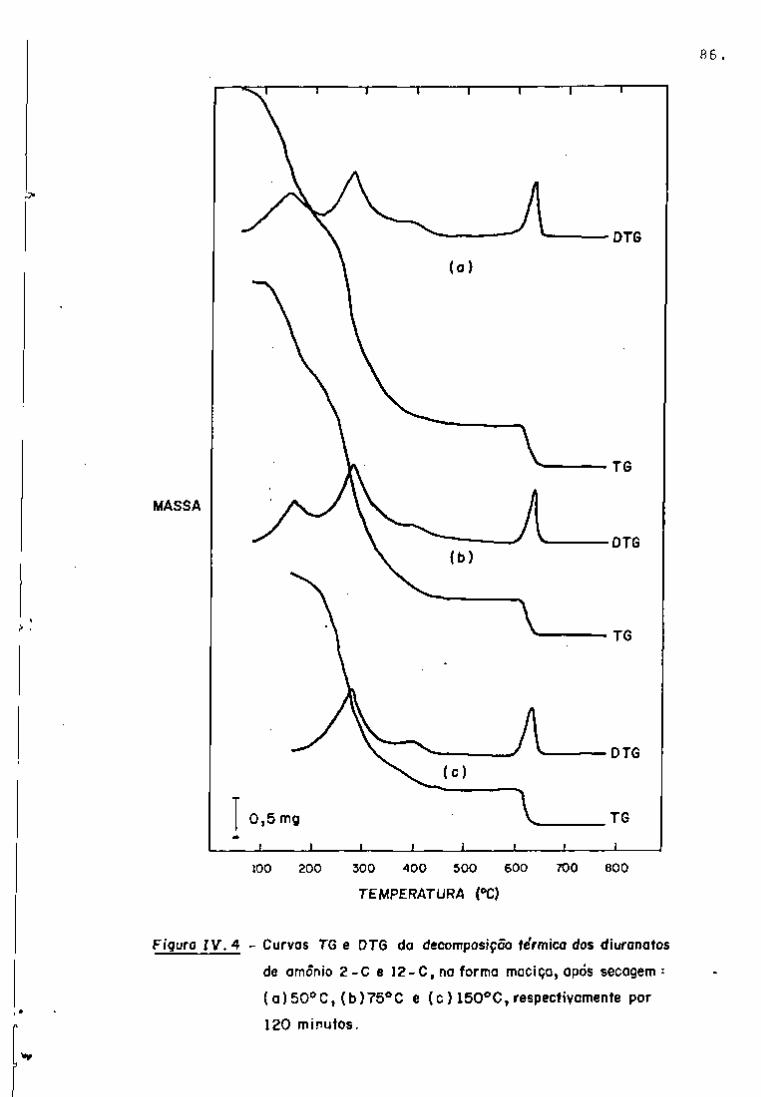
8 6
MASSA
100 200 300 400 500 600 700 800
TEMPERATURA ("O
Figuro I V . 4 - Curvas TG e DTG da decomposição te'rmica dos diuronatos
de amonio 2 - C e 1 2 - C , na forma maciça, após secagem =
( a ) 5 0 * ' C , ( b ) 7 5 * * C e ( c ) 1 5 0 * 0 , respectivamente por
120 minutos.
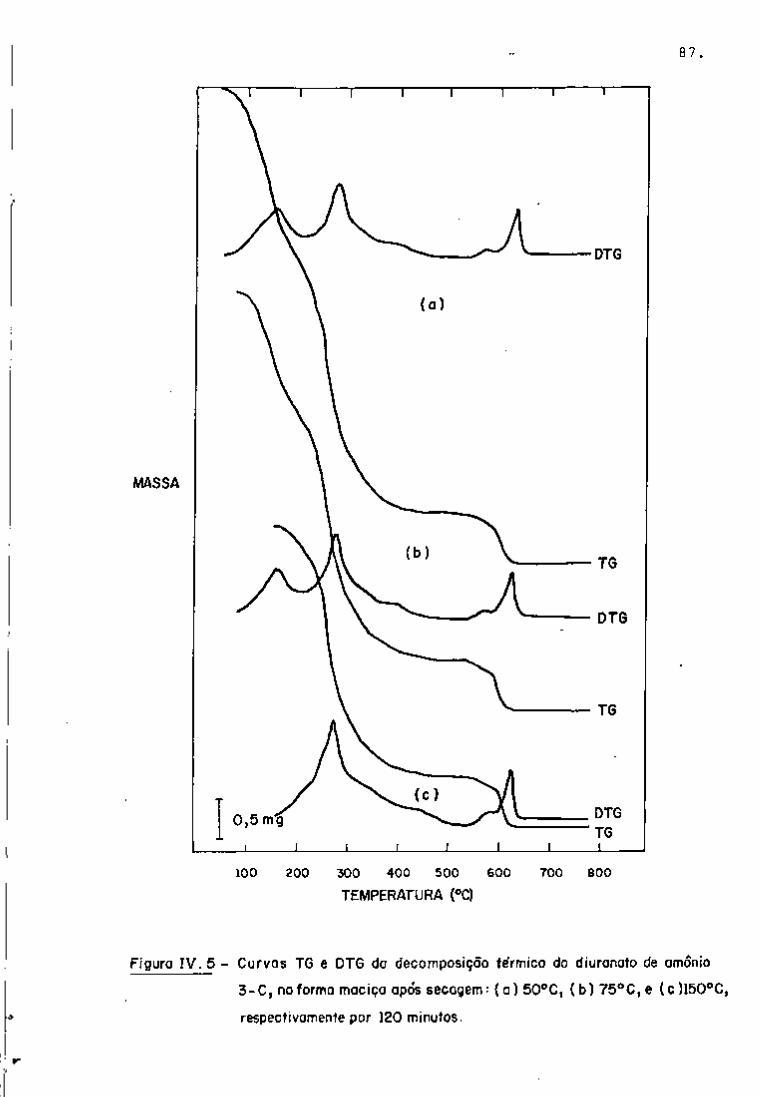
87.
MASSA
100 200 300 400 500 600
TEMPERATURA («>C)
700 800
Figuro I V . 5 - Curvas TG e DTG do decomposição te'rmica do diuronato de amonio
3 - C , na forma maciça após secagem: ( a ) 50* 'C, ( b ) 75**C,e (c)150'*C,
respectivamente por 120 minutos.
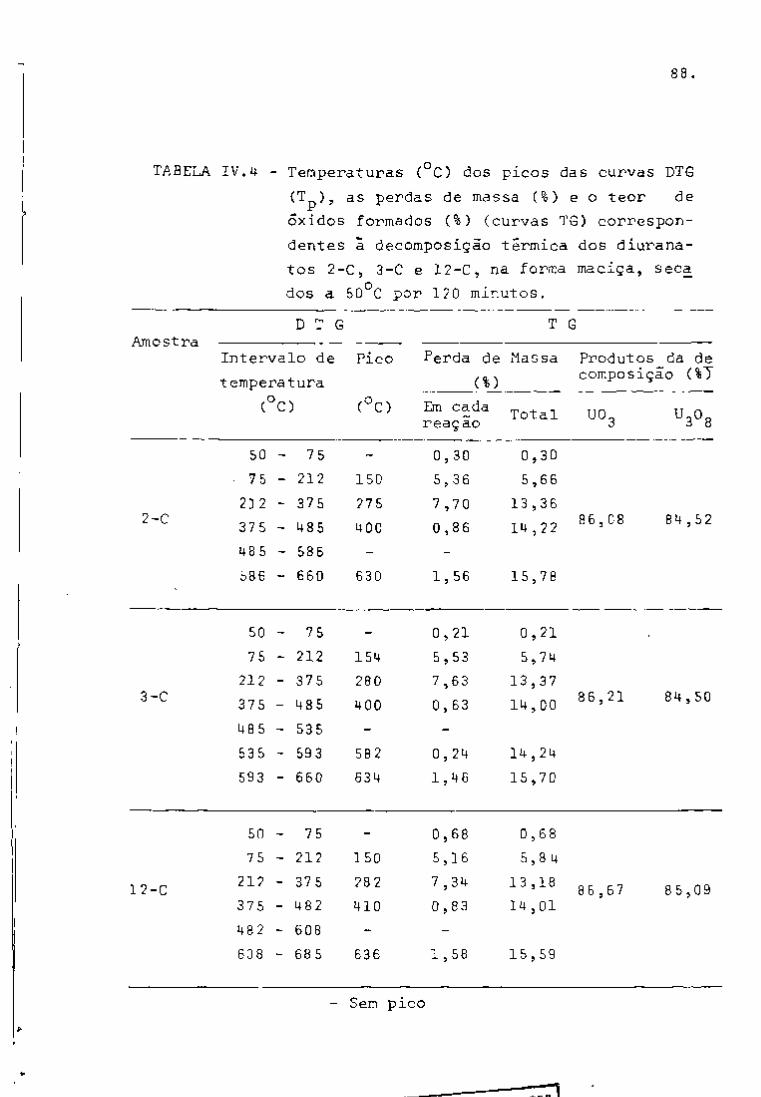
88.
TABELA IV.U - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, na forma maciça, seca
dos a 50°C por 120 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da de composição (%7
(° O (°C) Em cada reação
Total UO3 "3O8
50 - 75 - 0,30 0,30
• 75 - 212 150 5,36 5,6 6
2-C 212
375
1+85
- 375
- 485
~ 586
275
400
7,70
0,86
13,36
14 ,22 86,08 84,52
586 - 660 630 1,56 15,78
50 - 75 - 0,21 0,21
75 - 212 154 5,53 5,74
3-C 212
375
485
- 375
-485
- 535
280
400
7 ,63
0,63
13,37
14,00 86,21 84,50
535 - 5 9 3 582 0,24 14,24
593 - 660 634 1,46 15,70
50 - 75 - 0,68 0,68
75 - 212 150 5,16 5,8 4
12-C 212
375
482
- 375
- 482
- 608
282
410
7,34
0,83
13 ,18
14,01 86,67 85,09
608 - 685 636 1,58 15,59
- Sem pico
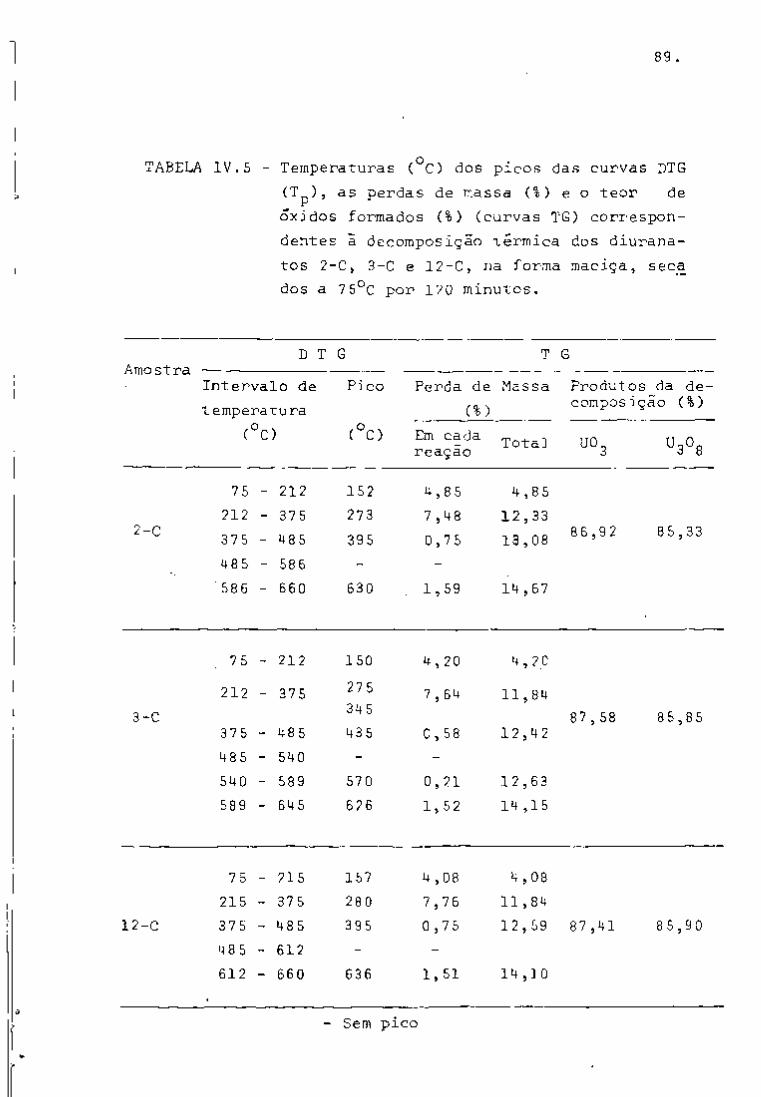
89 .
TABELA IV.5 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica dos diurana-
tos 2 -C, 3-C e 12-C, na forma maciça, seca
dos a 7 5°C por 120 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da decomposição (%)
(° C) (°C) Em cada reação
Total UO3 UgOg
75 - 212 152 4,85 4,85
2-C 212
375
485
- 375
- 485
- 586
273
395
7,48
0,75
12,33
13,08 86,92 85,33
586 - 660 630 1,59 14 ,67
75 • -212 150 4,20 4,2 0
212 • - 375 275 7,64 11,84
3-C 375 -
485 -
- 485
- 540
345
435 0-,58 12,42 87 , 58 85,85
540 -- 589 570 0,21 12,63
589 -- 645 626 1,52 14 ,15
75 -- 215 157 4 ,08 4,08
215 -- 375 280 7,76 11,84
i2-C 375 -
485 -
- 485
• 612
395 0,75 12, 59 87,41 85,90
612 -- 660 636 1,51 14 ,10
- Sem pico
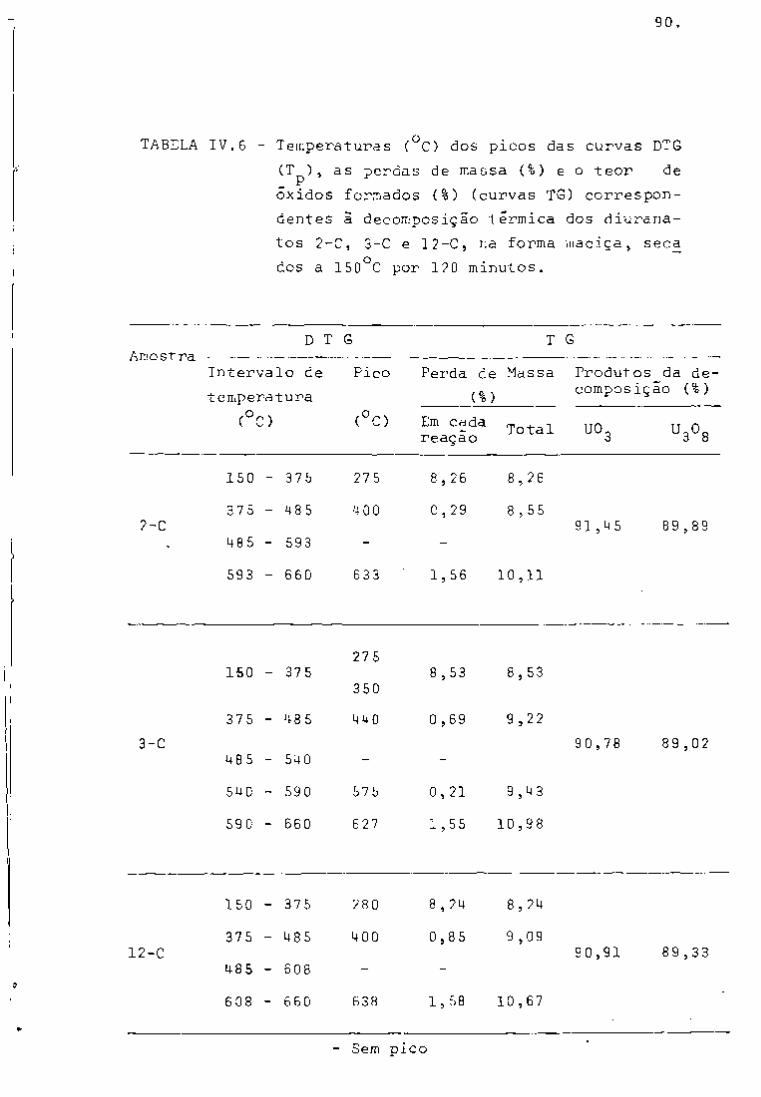
90.
TABELA IV.6 - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, na forma maciça, seca
dos a 150°C por 120 minutos.
Amostra — D T G T G
Intervalo de
temperatura
(°C)
Pico
(°C)
Perda de Massa . Produtos da de-ç, composição (%)
Em cada reação
Total UO, U3°8
2-C
150
375
485
593
375
485
593
660
275 8,26 8,26
400 0,29 8,55
633 1,56 10,11
91,45 89,89
3-C
15 0
375
485
540
590
375
485
540
590
660
275
350
440
575
627
8,53 8,53
0,69 9,22
0,21 9,43
1,55 10,98
90,78 89,02
150 - 375 280 8 , 24 8,24
12-C 375 -
485 -
485
508
400 0,85 9 ,09 90,91 89,33
608 - 660 638 1, 58 10,67
- Sem pico

91.
Os resultados mostraram que o número de"
reações envolvidas na decomposição'térmica dos diurana
tos de amonio depende da temperatura de secagem:
- 50°C: a agua livre não .foi totalmente eliminada,
observando-se 5 perdas de massa para os DUA 2-C e
12-C e 6 perdas de massa para o DUA 3-C (Tabela
IV.U);
- 75°C: ausencia de água livre, observam-se M- perdas
de massa para os DUA 2-C e 12-C e 5 perdas de massa
para o DUA 3-C (Tabela IV. 5);
- 150°C: ausencia de água livre e eliminação parcial
da água de cristalização, observam-se apenas 3 per-
das de massa para os DUA 2-C e 12-C e 4 perdas de
- massa para o DUA 3-C (Tabela IV.6).
Os compostos precipitados em pH acima
de 9,0 e a 60°C (tipo 2-C), apresentaram diminuição da
perda de massa na reação em torno de UOO°C, em função do
aumento da temperatura de secagem. Por outro lado, essa
perda de massa permaneceu aproximadamente constante na
decomposição térmica dos compostos precipitados em pH a-
cima de 9,0 e a 20°C (tipo 3-C) e, em pH acima de 7,0 e
a 60°C (tipo 12-C) ; (Tabelas IV.4 , IV.5 e IV.6).
Os dados das Tabelas IV.4 , IV.5 e IV.6
mostram que o oxido intermediario (UOg) obtido do DUA
3-C tem menor estabilidade térmica que os demais, com
reação de decomposição térmica se processando em duas e-
tapas sobrepostas (Figura IV.5).

92.
IV.2.2 - Secagem em estufa e decomposição térmica dos
compostos pulverizados
Os produtos secados em estufa, conforme
descrição em III.3.2, foram previamente pulverizados, an
tes do tratamento termoanalítico.
As curvas TG e DTG obtidas da decomposição
térmica desses produtos, secados a 75°C e 150°C por 120
minutos, estão nas Figuras de IV.6 a IV,9. As perdas de
massa (%) e a formação porcentual dos óxidos de urânio
correspondentes estão nas Tabelas IV.7 e IV.8.
Os dados apresentados nessas figuras e ta
belas, comparados com aqueles da decomposição dos produ
tos no estado maciço (IV.2.1), mostram um comportamento
térmico com as seguintes características:
- Independente do tipo de diuranato e da temperatura
de secagem, as temperaturas das reações são mais
altas e os intervalos de temperatura corresponden
tes são maiores para os produtos no estado maciço.
- Os produtos na forma de pó apresentaram duas rea
ções no intervalo de temperatura de 20°C a 205°C ;
com exceção dos diuranatos precipitados em pH aci
ma de 7,0 e a 60°C (tipo 12-C), secado a 75°C (Ta
bela IV.7), que apresentaram três reações. A per
da de massa (%) nesse nintervalo .de temperatura
foi menor para os produtos no estado maciço, mos
trando que o excesso de perda de massa (%) verifi
cado nas curvas TG dos produtos na forma de pó, po
de ser atribuído ã égua absorvida durante a pulve-

93.
rização. Nos produtos pulverizados a temperatura
do pico (Tp) na curva DTG para a reação acima de
75°C foi mais alta (169±U°C) quando secados a
150°C do que a 75°C (Tabelas IV,7 e IV.8).
Nos produtos pulverizados verificam-se duas rea
ções para o diuranato 12-C e apenas uma reação pa
ra os diuranatos 2-C e 3-C, independente da tempe
ratura de secagem, no intervalo de temperatura de
20 5°C a 37 0°C. Para temperatura de secagem mais
alta (150°C), a liberação de voláteis foi maior
na curva TG da decomposição do diuranato 2-C (põ)
do que naquelas dos demais produtos (põ), que ao
contrário, apresentaram .menor perda de massa (.%)
do que nas curvas TG da decomposição desses pro
dutos secados a 75°C. Nesse intervalo de tempera
tura a perda de m.assa (%) foi maior para os produ
tos no estado maciço.
Antes da formação de um patamar na curva TG há
uma reação comum a esses produtos, que na forma de
põ apresentou maior perda de massa (%) para o diu
ranato 3-C (-1,46%) e maior intervalo de tempera
tura para o diuranato 12-C (370-520°C), indepen -
dente da temperatura de secagem. As curvas TG
dos diuranatos 2-C e 3-C, na forma de põ, apresen
taram maior perda de massa (%) do que aquelas dos
mesmos produtos no estado maciço. Essa perda de
massa (%) foi aproximadamente a mesma nas curvas
TG do diuranato 12-C, secado a 150°C, nos estados
maciço e pulverizado, e menor na forma de po quan
do secado a 7 5°C,

94.
- Um patamar nas curvas TG dos diuranatos no estado
pulverizado, acima de 44 5°C, caracteriza a forma -
ção do composto intermediario, UO^, que apresentou
perda de massa lenta e gradual até sua reação de
decomposição. Para os diuranatos 2-C e 3-C essa
perda de massa (%) foi maior quando secados a
150°C. No caso do diuranato 12-C, secado a 75°C e
150°C, verificou-se, aproximadamente, a mesma per
da de massa (%) . Nas curvas:TG desses produtos
no estado maciço esse patamar não apresentou varia
ção de massa.
- A decomposição térmica do composto intermediário
(UOg),, na forma de pó, ocorreu por meio de uma
reação (~585°C) para os diuranatos 2-C e 3-C, e
duas reações sobrepostas (~540°C e 590°C) para o
produto 12-C, que apresentou menor estabilidade
térmica.
- Os teores de óxidos formados, com uma variação em
torno de 1%, foram de 87% em UO^ e 8 5% em UgOg pa
ra os produtos secados a 75°C, e de 91% em UO^ e
89l em U^Og para os produtos secados a 150°C, tan
to no estado maciço como na forma de po .
As demais curvas TG e DTG correspondentes
â decomposição térmica desses diuranatos de amonio, se
cados a 75°C e 150°C, respectivamente, por 1440 e 2880
ou 43 2 0 minutos estão, juntamente, com os dados dessas
curvas no Apêndice 2.

95.
A partir dessas informações observou-se
que, independentemente do diuranato e da temperatura
de secagem, ocorre diminuição no teor de voláteis li
berados na primeira reação (3 5°C) e aumento na perda de
massa (%) na reação em torno de 3 75°C, para tempos de
secagem maiores. Também,. independentemente do tipo
de diuranato, os teores de óxidos formados (%) foram
mais altos para os produtos,secados a 150°C e cresceu
com o aumento do tempo de secagem. No caso da secagem
a 7 5^C, a formação porcentual dos óxidos • de urânio
(UOg e UgOg) permaneceu, aproximadamente,igual.
O trióxido de urânio obtido do diurana
to 12-C, secado a 75°C por 120 e 1440 minutos e a
105^C por 120 minutos, apresentou maior estabilidade
térmica do que os demais UOg e a reação de decomposd^
ção ocorreu em duas etapas sobrepostas. O UO3 produzi
do a partir do diuranato 12-C, secado a 75°C por 2880
minutos ou a 150°C por 1440, 2880 e 4320 minutos, apre
sentou o mesmo comportamento térmico daqueles obtidos
dos produtos 2-C e 3-C.
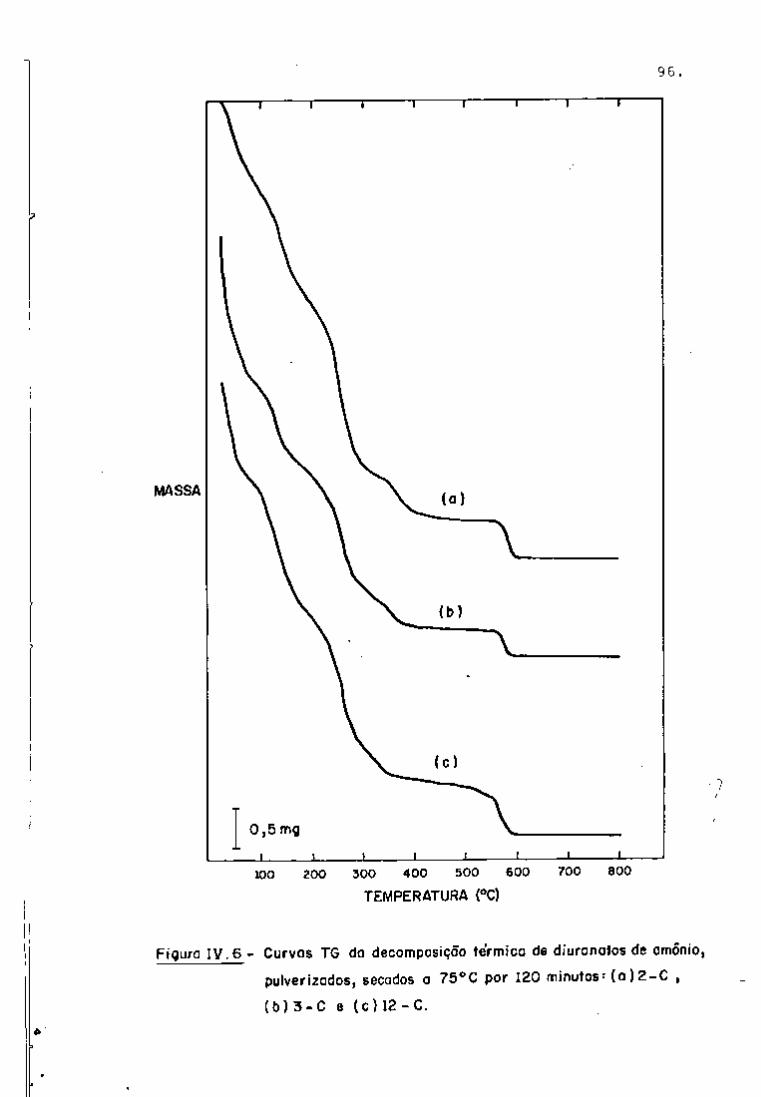
96,
MASSA
100 200 300 4 0 0 500 600
TEMPERATURA (*>C)
700 800
Figura I Y . 6 - Curvas TG da decomposição te'rmica de diuranatos de amonio,
pulverizados, secados a 75°C por 120 minutos: ( o ) 2 - C ,
( b ) 3 - C e ( c ) 1 2 - C .
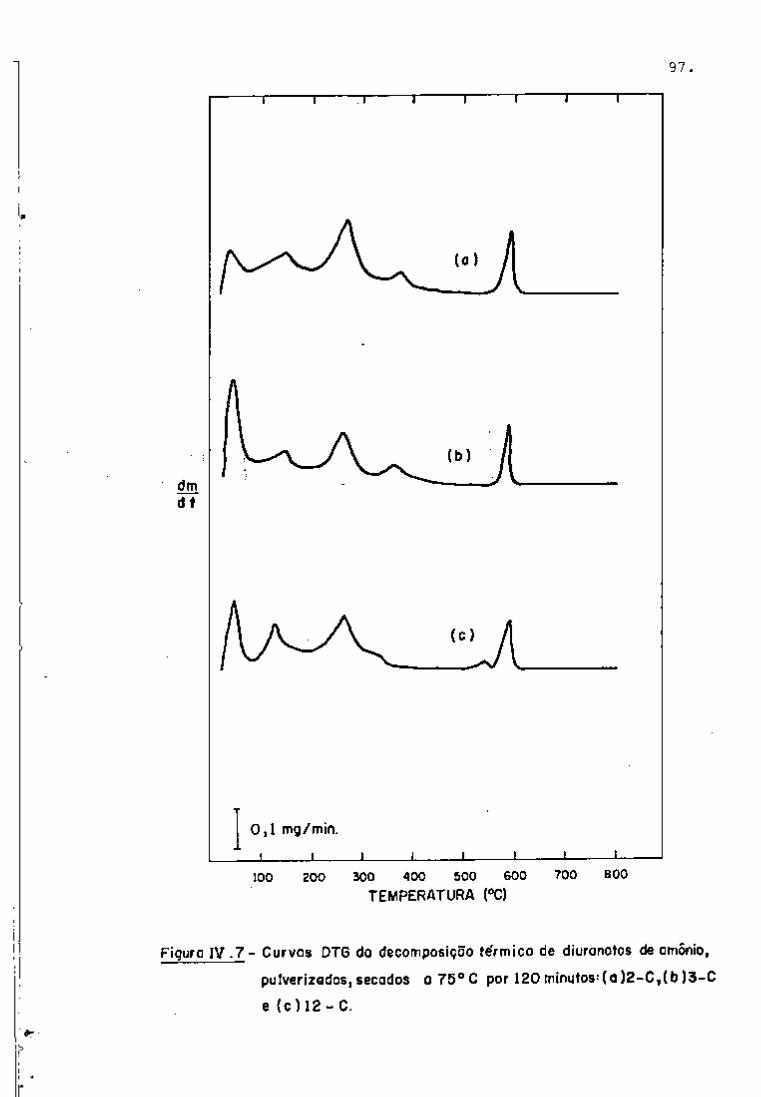
97.
dm d t
100 200 300 400 500 600
TEMPERATURA (»C)
700 800
Figuro IV . 7 - Curvas DTG da decomposição te'rmica de diuranatos de amônio,
pulverizados,secados a 7 5 * ' C por 1 2 0 m i n u t o s : ( a ) 2 - C , ( b ) 3 - C
e { c ) 1 2 - C .
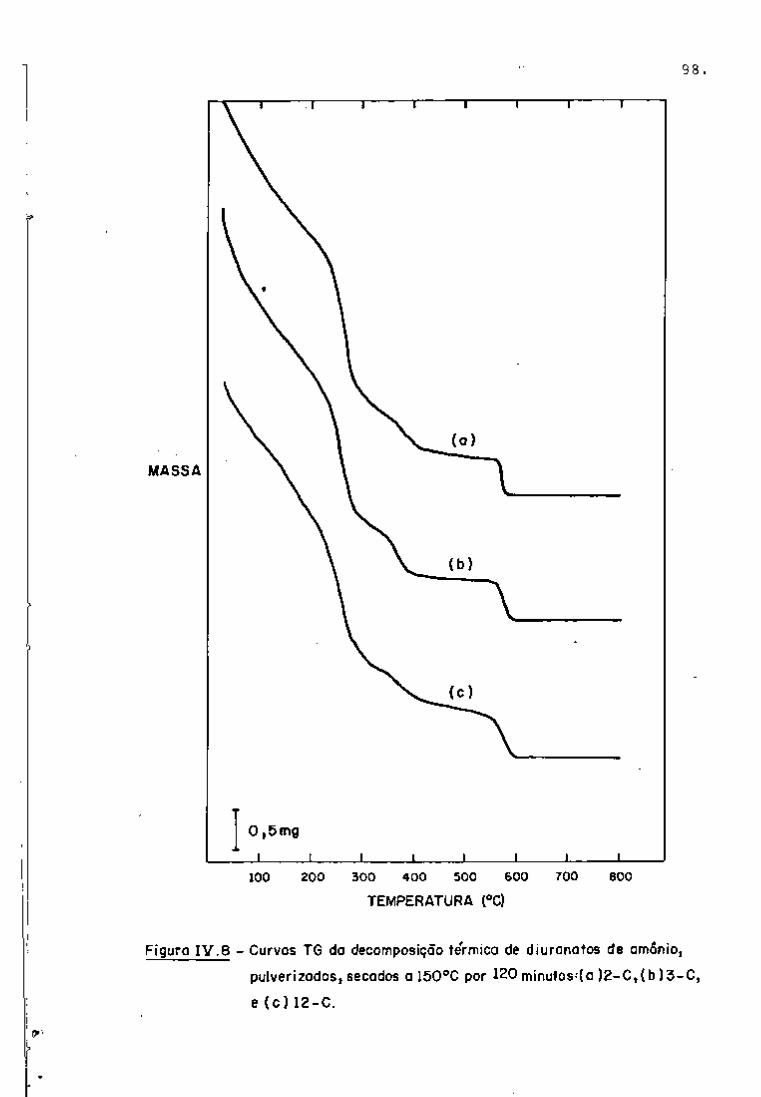
98.
M A S S A
100 200 300 4 0 0 500 600
TEMPERATURA {°C)
700 800
Figura I V . 8 - Curvas TG da decomposição térmica de diuranatos de amonio,
pulver izados, secados a ISO^C por 120 minutos^a ) 2 - C , ( b ) 3 - C ,
e { c ) 1 2 - C .
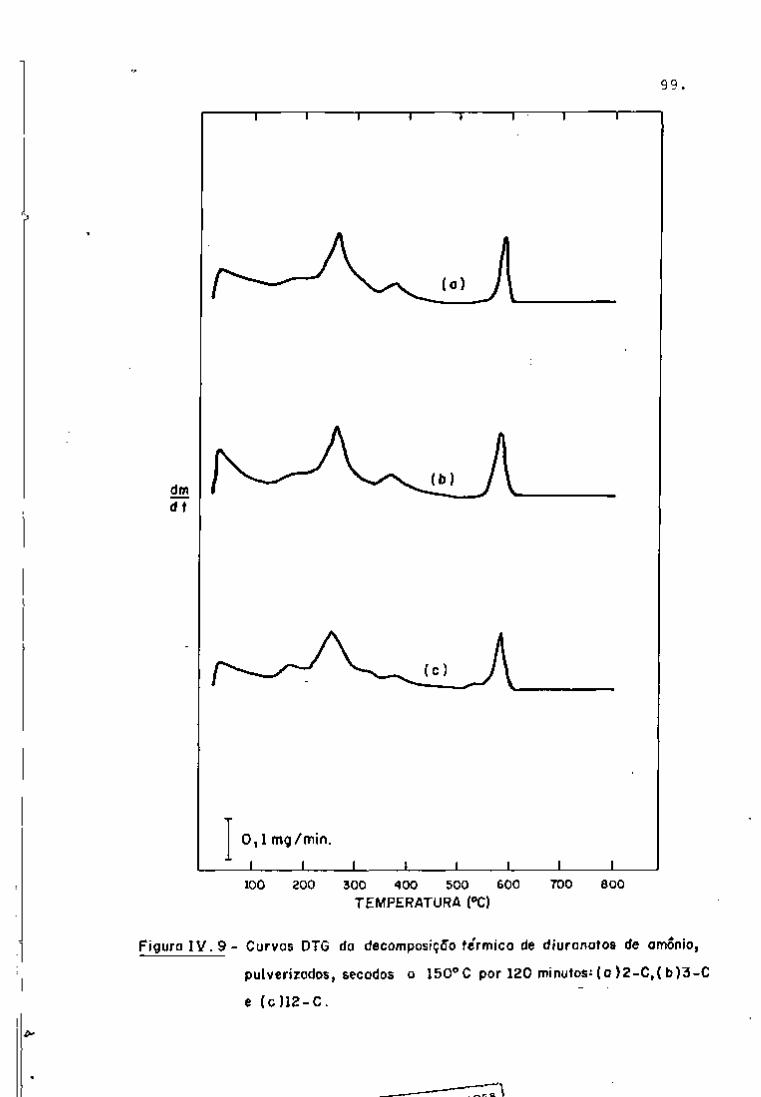
99
100 200 300 400 500 600 TEMPERATURA («C)
700 800
Figura I V . 9 - Curvas DTG da decomposição te'rmica de diuranatos de amonio,
pulverizados, secodos o 150*'C por 120 minutos: (a ) 2 - C , ( b ) 3 - C
e ( c ) 1 2 - C .
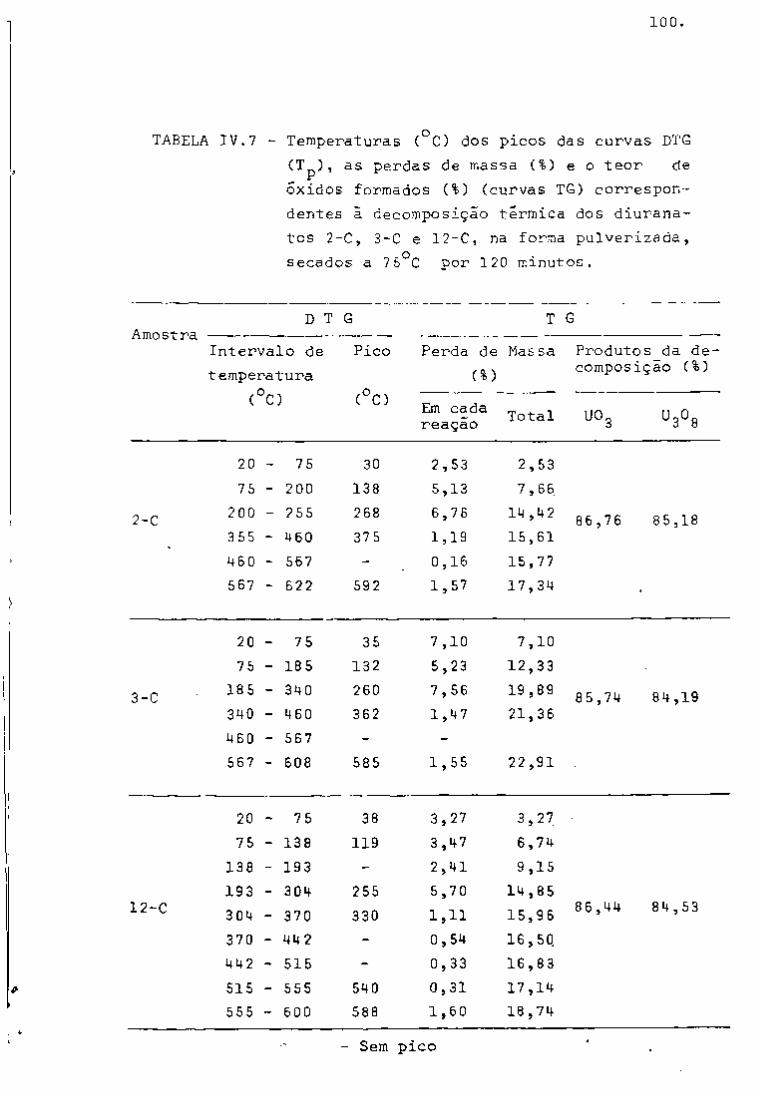
100.
TABELA IV.7 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes i. decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, na forma pulverizada,
secados a 75°C por 120 minutos.
D T G T G Amostra
Intervalo de
temperatura
Pico Perda de Massa Produtos da decomposição (%)
(%)
( 'o (°C) Em cada reação Total UO3 U3°8
20 - 75 30 2,53 2,53
75 - 200 138 5,13 7,66
2-C 200
355
- 255
- 460
268
375
6:y7 6
1,19
14,42
15,61 86,76 85 ,18
460 - 567 - 0,16 15,77
567 - 622 592 1,57 17,34 •
20 - 75 35 7,10 7,10
75 - 185 132 5,23 12,33
3-C 185
340
460
- 340
- 460
- 567
260
362
7,56
1,47
19,89
21,36 8 5,74 84,19
567 - 608 585 1,55 22,91
20 - 75 38 3,27 3,27
75 - 138 119 3,47 6,74
138 - 19 3 - 2,41 9,15
12-C 193
304
- 304
- 370
255
330
5,70
1,11
14,85
15,96 8 6,44 84,53
370 - 442 - 0,54. 16,50
442 - 515 - 0,33 16,8 3
515 - 555 540 0,31 17,14
555 - 600 588 1,60 18,74
- Sem pico
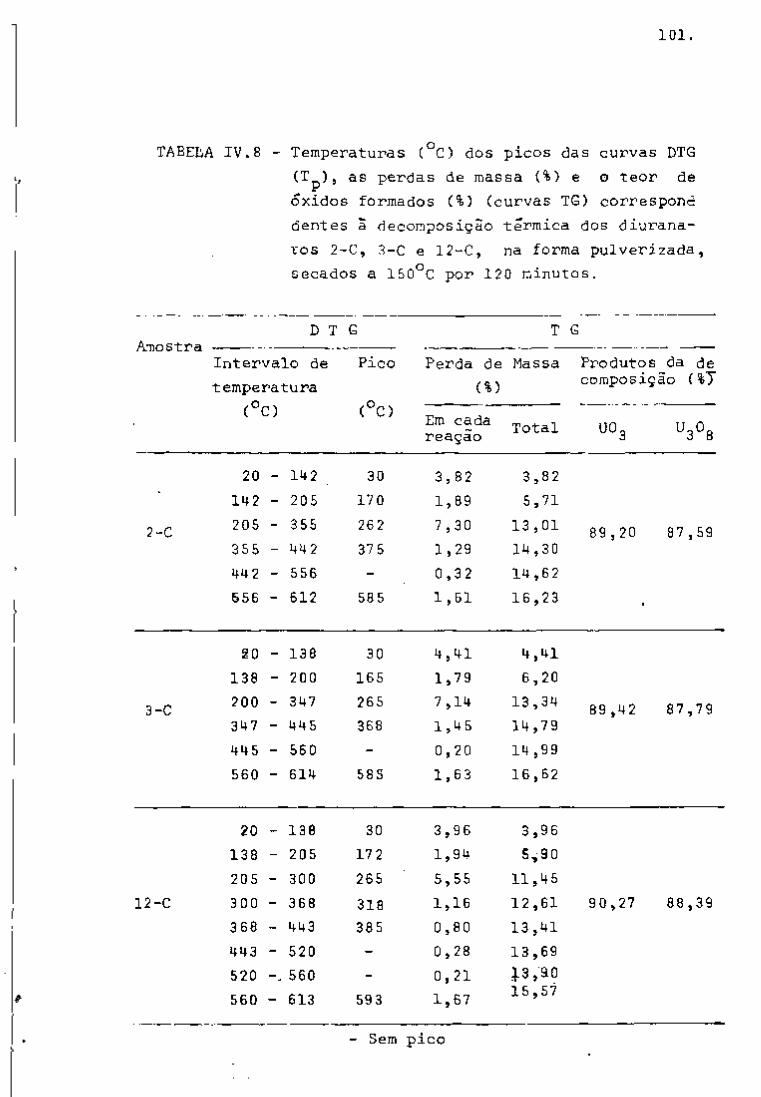
101.
TABELA IV.8 - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e ó teor de
óxidos formados (%) (curvas TG) corresponé
dentes a decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, na forma pulverizada,
secados a 150°C por 120 minutos.
D T G T G Amostra
Intervalo de
temperatura
Pico Perda de Massa Produtos da de composição (%y
(°C) (°C) Em cada reação
Total UO3 U3O8
20 - 142 30 3,82 3,82
142 - 205 170 1,89 5,71
2-C 205 -
355 -
442 -
355
442
556
262
375
7,30
1,29
0,32
13,01
14,30
14,62
89 ,20 87,59
556 - 612 585 1,61 16,23
20 - 138 30 4,41 4,41
138 - 200 165 1,79 6,20
3-C 200 -
347 -
347
445
265
368
7,14
1,45
13,34
14,79 89 ,42 87,79
445 - 560 - 0,20 14,99
560 - 614 585 1,63 16,62
20 - 138 30 3,96 3,96
138 - 205 17 2 1,94 5^90
205 - 300 265 5,55 11,45
12-C 300 - 368 318 . 1,16 12,61 90,27 88,39
368 - 443 38 5 0,80 13,41
443 - 520 - 0,28 13,69
520
560 -
560
613 593
0,21
1,67
|3,âO 15,57
- Sem pico

102.
IV.3., ESTUDO DA INFLUENCIA DO TEMPO DE ENVELHECIMENTO DOS
DIURANATOS DE AMONIO SOBRE O COMPORTAMENTO TÉRMICO
Com o objetivo de coletar dados adicio
nais que permitissem elucidar a termodecomposição dos diura
natos em estudo, com respeito ao envelhecimento, uma vez
que a literatura nada registra sobre o assunto, foram obti
das curvas termogravimêtricas e termogravimêtricas deriva -
das de alguns compostos.
Esse estudo foi feito, também, em fun
ção da variação dos parâmetros de secagem. Para tanto, pro
dutos secados em estufa, a 75°C por 2880 minutos e a ISO^C
por 12 0 e 2880 ou 4320 minutos, de acordo com III.3.2, arma
zenados durante um ano, foram novamente caracterizados por
TG e DTG.
As curvas termoanalíticas obtidas da de
composição térmica dos diuranatos 2-C, 3-C e 12-C (Tabela
III.l), secados a 150* 0 por 120 minutos, foram comparadas
com aquelas registradas para os mesmos compostos, logo apos
a secagem, e estão ilustradas nas Figuras de IV.10 a IV.12.
Os dados referentes a estas figuras estão apresentados nas
Tabelas IV.8 (operação de 1980 ) e IV. 9 (operação de 1981).
As demais curvas TG e DTG da decomposi
ção térmica desses diuranatos, secados a 75°C e 150°C, res
pectivamente, por 2880 ou 4320 minutos estão, juntamente
com os dados dessas curvas, no Apêndice 3.

103.
Uma analise desses resultados mostra
que o comportamento térmico desses compostos'^ variou em fun
ção do tempo de envelhecimento e foi influenciado, também ,
pela variação dos parâmetros de precipitação e secagem.
As curvas termoanalíticas da decomposi
ção desses diuranatos (2-C, 3-C e 12-C) envelhecidos • apre
sentaram algumas mudanças em comum,' independentemente dos
parâmetros de precipitação e secagem, quando comparadas com
aquelas obtidas em IV.2.2:
- Maior numero de reações, com picos DTG
bem definidos (Figuras IV.lO, IV.11 è IV.12);
- Menor perda de massa (%) e temperatura
mais alta para a reação, antes observada, em torno de 2 70°C
(Tabelas IV.8 e IV.9);
- Maior perda de massa (%) para a reação
em torno de 37 0°C (Tabelas IV.8 e IV.9).
Os resultados apresentados no Apêndice
3, quando comparados com aqueles em IV.2.2, mostram que es
sas observações também ocorrem nos compostos secados a 7 5°C
e 150OC por 2880 ou 4320 minutos.
Outras variações, observadas nas curvas
TG e DTG dos compostos envelhecidos, foram influenciadas pe
los parâmetros de secagem:
- As curvas termoanalíticas para a seca
gem a 75°C sõ apresentaram mudanças, em relação aquelas ob-

104.
tidas em IV.2.2, para tempos de secagem superiores a 1440 mi
nutos.* As curvas DTG obtidas a essa temperatura, por exem
plo, por 2 880 minutos, mostraram menor estabilidade térmica
para o trióxido de urânio (UO^) produzido a partir dos diura
natos envelhecidos (Tabela 2.3 - Apêndice 2; Tabelas 3.1,3.2
e 3.3 - Apêndice 3) .
- Independentemente do tipo de diuranato,
ou seja,; dos parâmetros de precipitação, os compostos enve
lhecidos, secados a 150°C por tempos superiores a 1440 minu
tos, produziram UO^ com estabilidade térmica menor do que a-
quela observada em IV.2.2, cuja decomposição ocorreu por
meio de duas reações. As curvas TG permitiram observar um
segundo intermediário (acima de 570°C) com composição aproxi
madamente equivalente ã do g (Tabelas 2.4 e 2.5 - Apên
dice 2; Tabelas 3.1, 3.2 e 3.3 - Apêndice 3). Ao contrário,
quando secados a 150°C por 120 minutos, esses diuranatos en
velhecidos produziram UOg com estabilidade térmica maior (Ta
belas IV.8 e IV.9).
- Os teores de óxidos de urânio C%) foram
aproximadamente os mesmos_ nas curvas TG desses compostos se
cados a 7 5°C e menores nas curvas TG dos diuranatos seca
dos a 150°C, quando comparados com aqueles obtidos logo após
a secagem, em IV.2.2.
- Com algumas exceções, os produtos enve
lhecidos e secados a 75°C apresentaram maior perda de massa
(%) no intervalo de temperatura entre 20 e 220 e' na reação
em torno de 2 7 0°C, e temperatura mais alta para a reação em
370°C, do que quando secados a 150°C.

105.
- As curvas DTG dos diuranatos envelhecidos,
secados a 150°C por tempos superiores a 144Ü minutos, a-
presentaram um pico bem definido em torno de 160°C.
Ao lado da influência dos parâmetros de
secagem sobre o comportamento térmico dos compostos enve
Ihecidos, observou-se também, alguma influência dos para
metros de precipitação. Os produtos secados a 7 5°C apre
sentaram as seguintes diferenças, no intervalo de tempe
ratura de 20 a 2 2 0°C, em relação ao comportamento térmi
co anterior (IV.2.2):
- Observou-se o mesmo número de reações para
os diuranatos 2-C e 12-C e duas novas reações, respecti
vamente, em 137°C e 198°C, substituindo aquela em 148°C,
para o diuranato 3-C. Não so as temperaturas, mas tam
bém, as perdas de massa (%) em cada reação variaram em
função do tipo de diuranato (Tabela 2.3 - Apêndice 2 e
Tabelas 3.1, 3.2 e 3.3 - Apêndice 3).
Do mesmo modo, quando secados a 150°C,os
compostos envelhecidos apresentaram duas novas reações
em substituição aquelas observadas anteriormente, no
intervalo de temperatura de 20°C a 2 20°C, cujas tempera
turas de reação e perdas de massa (%) correspondentes va
riaram em função dos parâmetros de precipitação (Tabelas
IV.8 e IV.IV.9; Tabela 2.4 - Apêndice 2 e Tabelas 3.1,
3.2 e 3.3 - Apêndice 3).
A decomposição do trióxido de urânio, com
formação de um segundo intermediário, nos compostos seca
dos a Í50°C por 28b0 ou 4320 minutos, apresentou varia
ções no intervalo de temperatura da reação e na perda de
massa (%) correspondente, em função do tipo de diuranato.
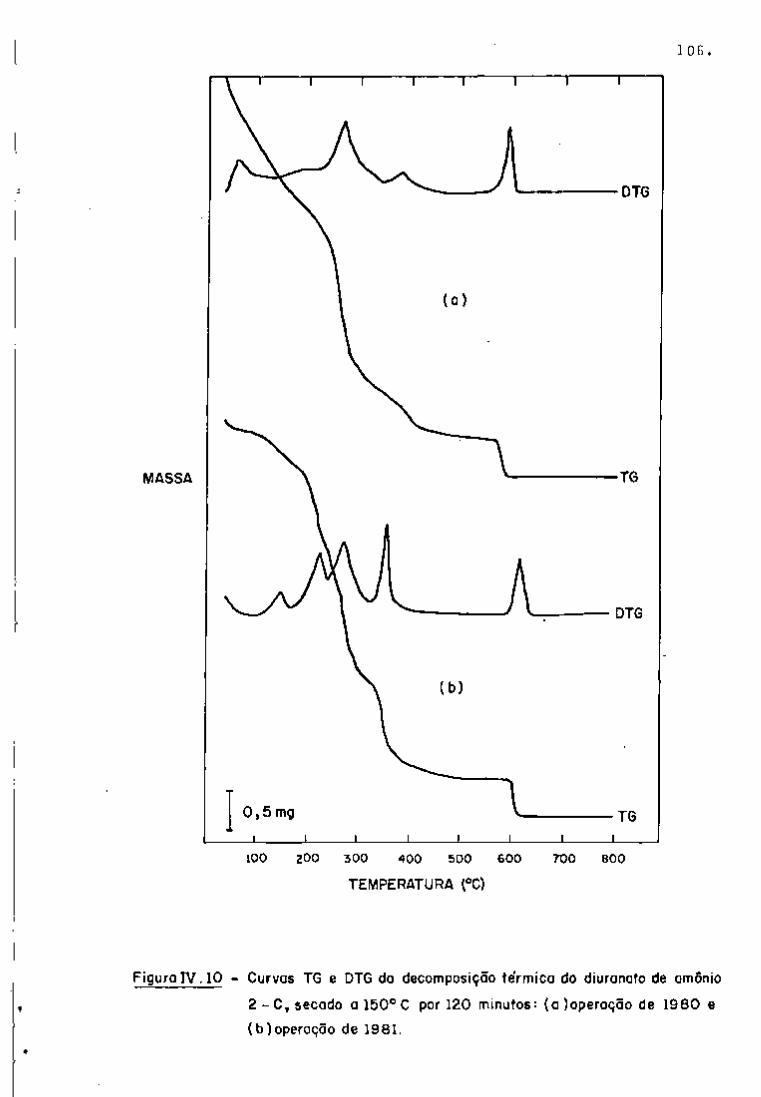
106,
MASSA
100 200 300 400 500 600
TEMPERATURA ( X )
700 800
Figuro I V . 10 - Curvas TG e DTG da decomposição térmica do diuranoto de amonio
2 - C , secado a l 5 0 ° C por 120 minutos: (a )operação de 1 9 8 0 e
(b )operação de 1981 .
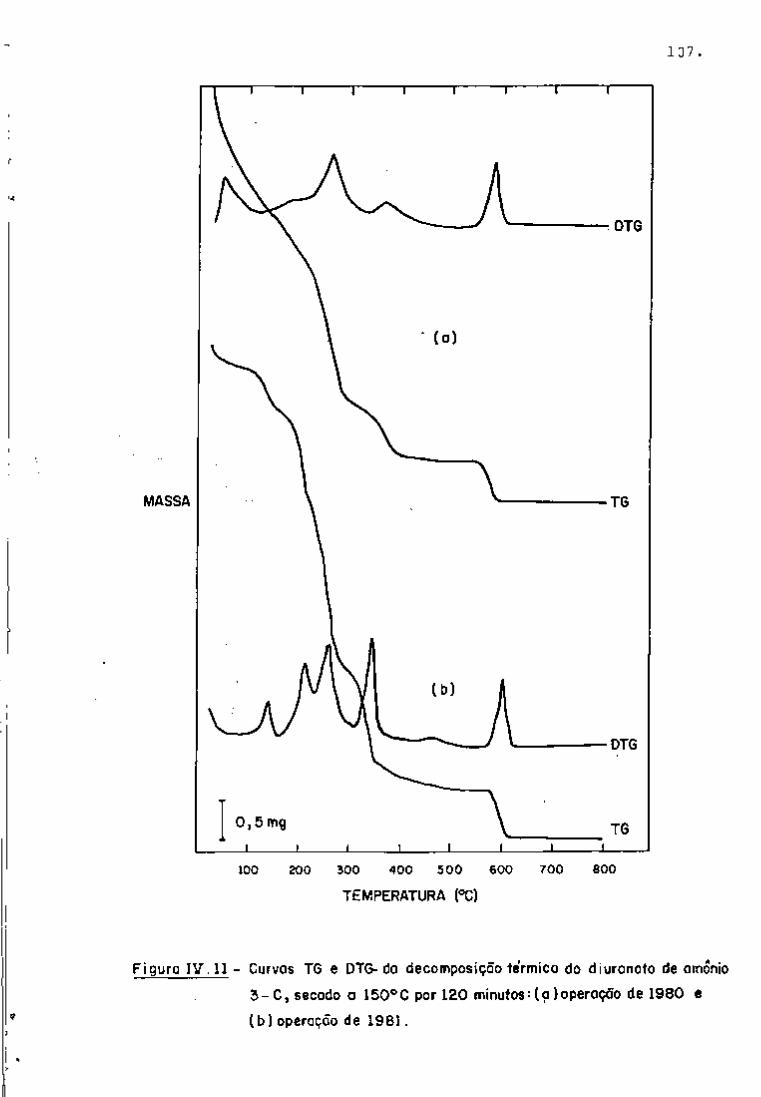
107 .
MASSA
100 200 300 400 5 0 0 600
TEMPERATURA {°C)
700 800
Figuro I V . 11 - Curvas TG e DTG-da decomposicoo térmica do diuronato de omonio
3 - C , secado o 1 5 0 * 0 por 120 minutos:(o)operação de 1980 e
( b ) operação de 1 9 8 1 .
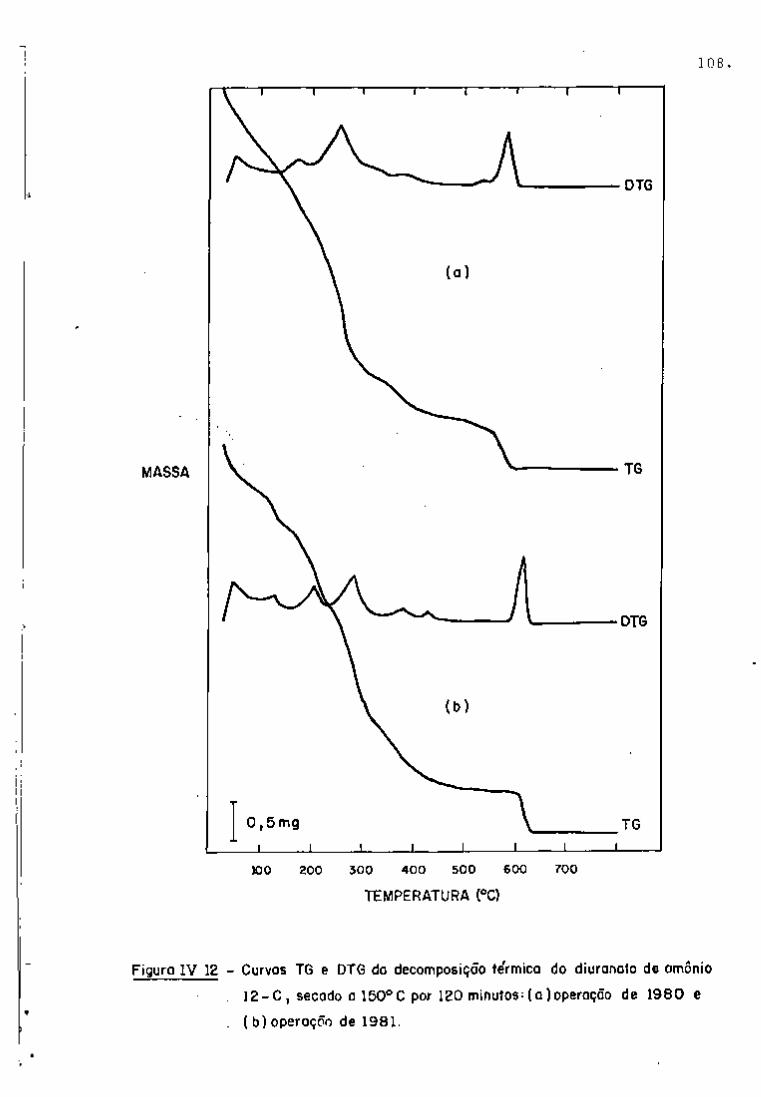
101
MASSA
100 200 300 400 500 600
TEMPERATURA («O
700
Figuro I V 12 - Curvas TG e DTG da decomposição te'rmica do diuranato de amônio
. 1 2 - C , secodo o 1 5 0 * 0 por 120 minutos:(o)operação de 1 9 8 0 e
. ( b) operaçõo de 1 9 8 1 .
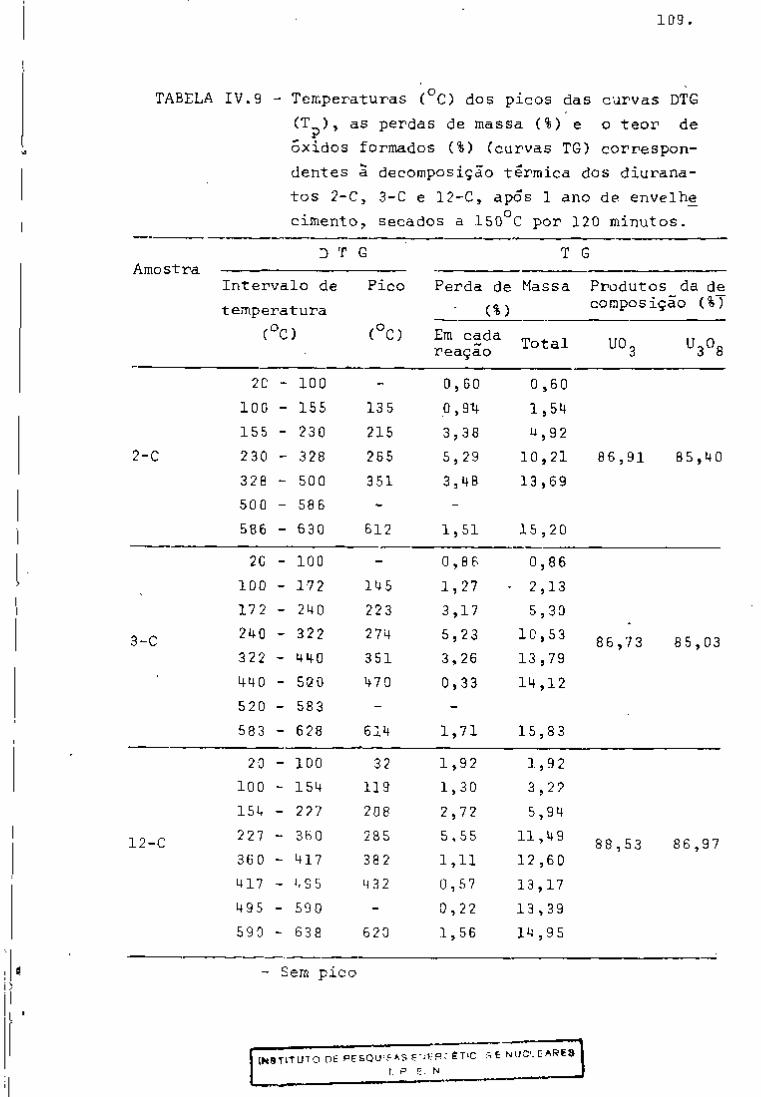
109.
TABELA IV.9 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes â decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, após 1 ano de envelhe
cimento, secados a 150 C por 120 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
• (%)
Massa Produtos da de composição (%)
( °C) (°C) Em cada reação Total UO3 UgOg
20 - 100 - 0,60 0,60
100 - 155 135 0,9-4 1,54
155 - 230 215 3,38 4,92
2-C 230 - 328 265 5,29 10,21 86,91 85,40
328 - 500 351 3,48 13,69
500 - 586 - -
586 - 630 612 1,51 15,20
20 - 100 - 0,86 0,86
100 - 172 145 1,27 2,13
17 2 - 240 223 3,17 5,30
3-C 240
322
- 322
- 440
274
351
5,23
3,26
10,53
13 ,79 86,73 85,03
440 - 520 470 0,33 14,12
520 - 583 - -583 - 628 614 1,71 15,83
20 - 100 32 1,92 1,92
100 - 154 119 1,30 3,22
154 - 227 208 2,72 5,94
12-C 227
360
- 360
- 417
285
382
5,55
1,11
11,49
12,60 88,53 86,97
417 - 435 432 0,57 13,17
495 - 590 - 0,22 13,39
590 - 638 620 1,56 14,95
- Sem pico
INSTITUTO D E P E S Q U I S A S E N f í R G É T l C - ' S E N U C L E A R E S
I, P . E . N .

110.
IV.4 ESTUDO DA INFLUÊNCIA DA ATMOSFERA NA RECOMPOSIÇÃO
DOS DIURANATOS DE AMÕNIO
Conforme mostrado em II.3.1, a decompo
sição térmica do DUA pode ser influenciada pelo tipo de
atmosfera, Foi assinalado também, que entre as rea
ções que ocorrem acima de 300°C, aquela entre a amonia
retida e o oxido de urânio (em atmosfera inerte ou redu
tora) ou com o oxigênio do- ar (fluxo de ar ou O2) é a
mais sensível. Os õxidos de urânio catalizam o cra
queio da molécula NH^, decompondo-se em nitrogênio e
hidrogênio. Este, em atmosfera inerte, reduz o oxido
de urânio a UO2. Em atmosfera oxidante, isto é, em flu
xo de ar ou oxigênio, o hidrogênio é consumido, não o-
correndo a redução do oxido de urânio.
A composição dos diuranatos de amônio u-
sados neste trabalho apresentou-se bastante variável,
principalmente em relação ao teor de amonia. Para me
lhorar ou complementar sua caracterização, foram obti
das curvas TG, DTG e DSC de diuranatos, em ar e nitro
gênio, as quais se encontram, respectivamente, nas Fi
guras IV.13, IV.14 e IV.15. Estes compostos foram obti
dos em condições diferentes de precipitação e secagem,
Nas Tabelas IV.10 e IV.11 estão as perdas de massa (%)
e a indicação dos õxidos de urânio formados pela decom
posição, bem como os intervalos de temperatura de cada
reação e seu tipo, nas duas atmosferas.
Os resultados apresentados nestas figu
ras e tabelas indicam que, de um modo geral, as reações

111.
no intervalo de 20 a 49 0°C se processaram em temperatu
ras mais elevadas para a decomposição em nitrogênio do
que na presença de ar.
Acima de U9 0°C, todos os compostos apre
sentaram UOg como oxido intermediario com maior estabi
lidade térmica para a decomposição em ar. .
Em relação â variação de massa durante a
decomposição térmica desses compostos, nas duas atmos
feras, pode-se assinalar as seguintes observações (Ta
bela 10):
- no intervalo de temperatura de 70 a 215°C, a perda
de massa (%) foi aproximadamente a mesma, nas
duas atmosferas, para os produtos do tipo 1-B e
maior, em atmosfera inerte (N2), para os produ
tos do tipo 21-B.
- Para as reações entre 215 e 350°C observou-se
maior perda de massa (%), on atmosfera inerte, para
ambos os produtos. As curvas DTG do diuranato 1-B
apresentaram duas reações na decomposição em'atmo£
fera inerte e apenas uma reação na decomposição
em atmosfera oxidante (ar) (Figura IV.14).
- Os dois produtos (1-B e 21-B) apresentaram maior
perda de massa (%) para a reação entre 350 e 450°C
em atmosfera oxidante (ar).
- Na reação de decomposição do oxido intermediário,
tanto para o produto que apresenta uma sõ etapa
(1-B), quanto para aquele que apresenta duas eta-

1 1 2 .
pas ( 2 1 - B ) , a perda de massa total foi maior em j
atmosfera inerte ( N 2 ) . Nas curvas T(3 do diurana
to 21-B verificou-se que, em atmosfera inerte, há
um aumento na perda de massa (%) na primeira etapa
desta reação e uma diminuição na segunda.
Quanto ao teor de óxidos (UO^ e U^Og), os diurana
tos do tipo 1-B apresentaram, aproximadamente, a
mesma porcentagem em ambas as atmosferas. Os pro
dutos do tipo 2 1-B, apresentaram uma porcentagem
de óxidos menor em atmosfera inerte.
113
MASSA
ÍOO 2 0 0 300 4 0 0 500 6 0 0 7 0 0 8 0 0
TEMPERATURA ("O
Figura I Y . 1 3 - C u r v a s TG da decomposição térmica dos diuranatos de amonio
1 - B ( a ) e 2 1 - B ( b ) , em diferentes atmosferas (ar e N g ) .
114.
I R 1 1 R
A ar
(O)
A
( b )
ar
0,1 m g / m i n .
—I L 1 I I I I I
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
TEMPERATURA (*»C)
7 0 0 8 0 0
Figuro I V . 14 - Curvas DTG da decomposição te'rmico dos diuranatos de amônio
1 - 8 ( o ) e 2 1 - B ( b ) , em diferentes otmosferos (or e N g ) .
I. P . E , N .
1 1 5 ,
ENDO
100 200 300 400 500 600
TEMPERATURA CO
700 800
Figuro rv. 15 - Curvas DSC da decomposição térmica dos diuronatos de omônio
1 -B ( d ) e 2 1 - B ( b ) , em diferentes atmosferas (or e N2)-
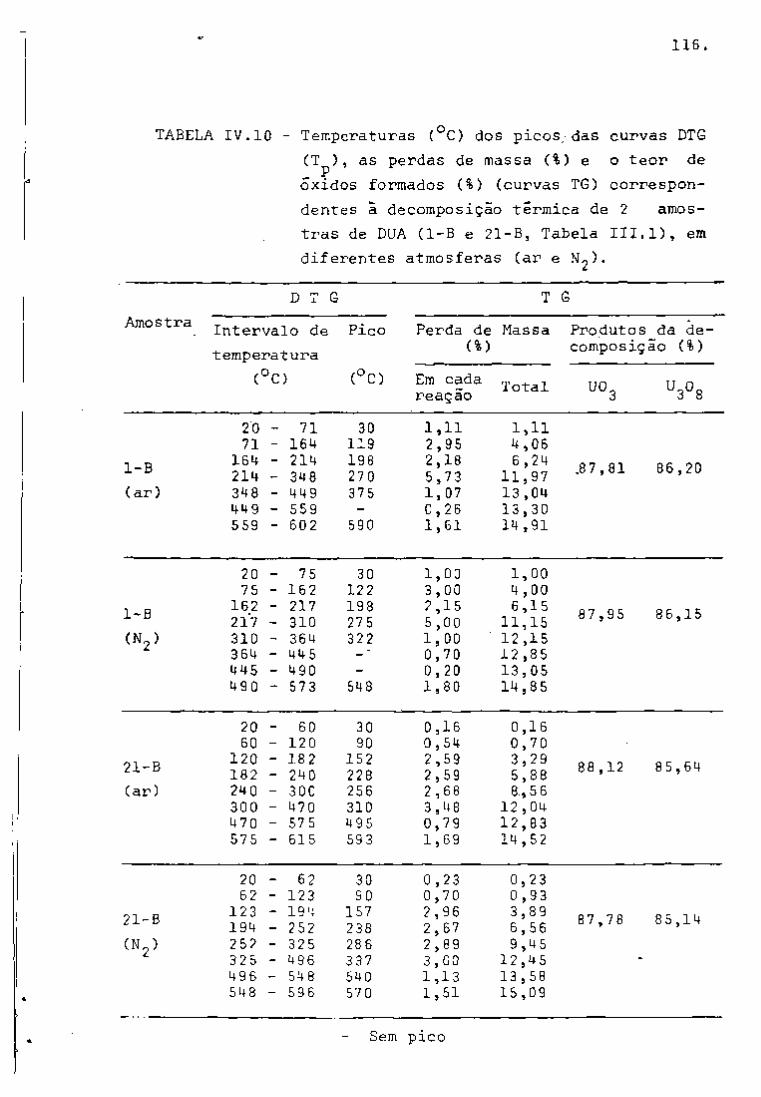
116.
TABELA IV.10 - Temperaturas ( C) dos picosydas curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes ã decomposição térmica de 2 amos
tras de DUA (1-B e 21-B, Tabela III.l), em
diferentes atmosferas (ar e N2).
D T G T G
Amostra Intervalo de Pico
temperatura
Perda de Massa Produtos da de-(%) composição (%)
(° 0 (°C) Em cada reação Total
2:0. -. 71 30 1,11 1,11 71 - 164 119 2,95 4,06
164 - 214 198 2,18 6,24 214 - 348 27 0 5,73 11,97 348 - 449 375 1,07 13,04 449 - 559 - 0,26 13,30 559 - 602 590 1,61 14,91
UO.
1-B
(ar)
.87 ,81 86 ,20
20 -- 75 30 1,00 1,00 75 • - 162 122 3,00 4,00
1-B 162 -217 -
- 217 - 310
198 275
2,15 5,00
6,15 11,15
87,95 86,15
(N.) 310 -- 364 322 1,00 • 12,15 364 -- 445 0,70 12,85 445 -- 490 - 0,20 13,05 490 -- 573 548 1,80 14,85
20 -- 60 30 0,16 0,16 60 -- 120 90 0,54 0,70
21-B 120 -182 -
- 182 - 240
152 228
2,59 2,59
3,29 5,88 88,12 85,64
(ar) 240 -- 300 256 2,68 8,5 6 300 -- 470 310 3,48 12 ,04 470 -- 575 495 0,79 12,83 575 -- 615 593 1,69 14,52
20 -- 62 30 0,23 0,23 62 -- 123 90 0,70 0,93
21-B 123 -194 -
- 194 - 252
157 238
2,96 2,67
3,89 6,56
87,78 85,14
(N.) 252 -- 325 286 2,89 9,45 ¿ 325 -- 496 337 3,00 12,45
496 -- 548 540 1,13 13,58 548 -- 596 570 1,51 15,09
Sem pico
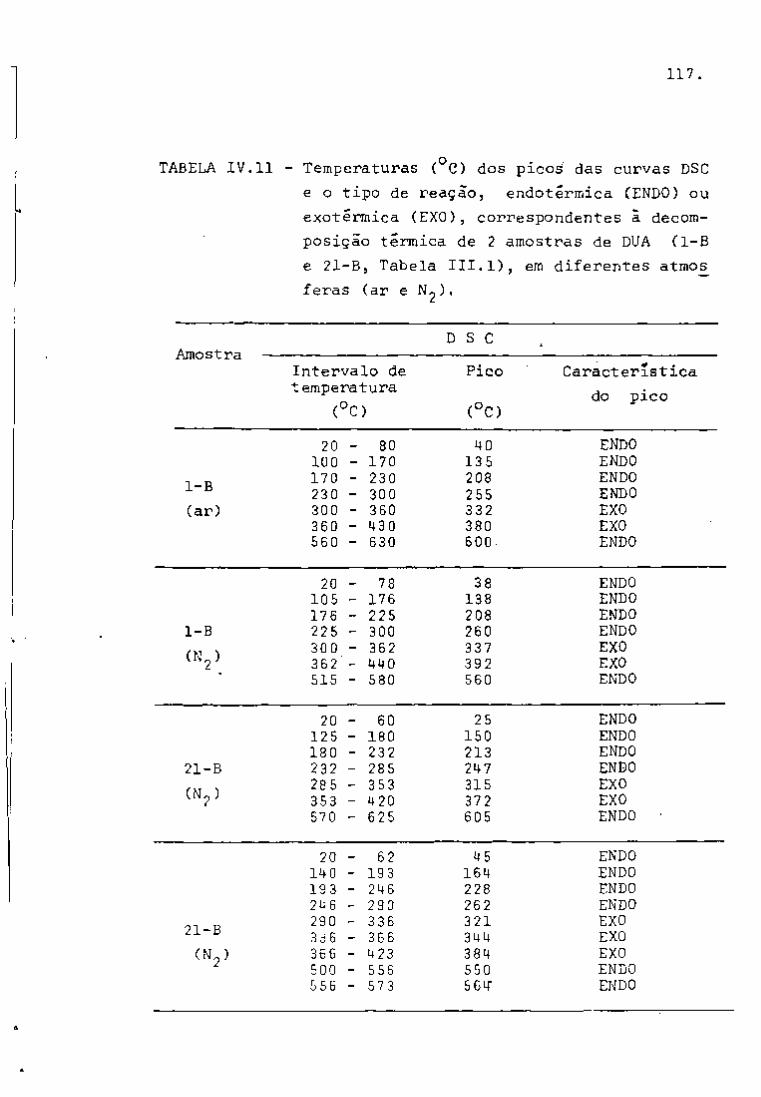
117 .
TABELA IV. 11 - Temperaturas ( C) dos picos' das curvas DSC
e o tipo de reação, endotérmica (ENDO) ou
exotérmica (EXO), correspondentes â decom
posição térmica de 2 amostras de DUA (1-B
e 21-B, Tabela III.l), em diferentes atmo£
feras (ar e N2).
D S C Amostra
Intervalo de temperatura
(°C)
Pico
(°C)
Característica
do pico
20 80 40 ENDO 100 - 170 135 ENDO
1-B 170 - 230 208 ENDO 1-B 230 - 300 255 ENDO (ar) 300 - 360 332 EXO
360 - 430 380 EXO 560. • — 630 600 ENDO
20 78 38 ENDO 105 - 176 138 ENDO 176 - 225 208 ENDO
1-B 225 - 300 260 ENDO
(N2) 300 - 362 337 EXO
(N2) 362' - 440 392 EXO 515 580 560 ENDO
20 60 25 ENDO 125 - 180 150 ENDO 180 - 232 213 ENDO
21-B 232 - 285 247 ENDO
(N2) 285 - 353 315 EXO
(N2) 353 - 420 372 EXO 570 — 625 605 ENDO •
20 _ 62 45 ENDO 140 - 193 164 ENDO 193 - 246 228 ENDO 246 - 290 262 ENDO
21-B 290 336
336 366
321 344
EXO EXO
(N„) 366 - 423 384 EXO ¿ 500 - 556 550 ENDO
556 — 573 564- ENDO

118.
IV.5 INFLUÊNCIA DO TEOR DE NITRATO DE AMONIO SOBRE O J
COMPORTAMENTO TÉRMICO DOS DIURANATOS
Um diuranato com baixo teor de amonia
(-1%) (20-C, Tabela III.l), foi empregado na prepara
ção de misturas solidas DUA-NH^^NOg, por homogeneização
em almofariz de ágata. As porcentagens de nitrato de
amonio adicionadas foram, respectivamente, 1, 5 e 10%
(m/m).
As curvas TG, DTG e DSC do diuranato ori
ginal e destas misturas sólidas estão representadas nas
Figuras de IV.16 a IV.18 e foram obtidas em atmosfera
inerte (N2).
As Tabelas IV.12 e IV.13 mostram a va
riação na liberação de voláteis, bem como o tipo de
reação (endotérmica ou exotérmica) envolvida durante a
decomposição térmica destas amostras.
Os efeitos térmicos observados nas cur
vas TG, DTG e DSC foram intensificados ã medida que au
mentou o teor de NH NOg nesse DUA e foram acompanha
dos por mudanças na temperatura e perda de massa.
O aumento no teor de NH^NO^ nesse DUA r£
sultou nas seguintes mudanças, de acordo com as curvas
TG, DTG e DSC (Figuras IV.16 a IV.18):
a) de 20 a U90°C foi maior a quantidade de voláteis
liberada.
b) A perda de massa na primeira reação, em torno de
¡ I N S T I T U T O D E PE í iQU ' f i , . - s s E N U c i e A R e ' S I, P- E, N .

119.
6 5°C, foi a mesma tanto para o DUA original como
para a amostra com 1% de NH^NO^, mas aumentou nas
amostras com 5 e 10% de nitrato de amônio (Tabela
IV.12).
c) Surgiu uma nova reação entre 160 e 220°C nas amos
tras com 5 e 10% de NH^^NOg.
d) Apareceu, também, uma reação bem definida em torno
de 270°C, para os compostos com 1 e 5% de NH^NOg
ou em 285°C par.a o- composto com 10% de NH^NO^, cur
vas DTG e DSC (Figuras IV.17 e IV.18), a qual ca
racterizou a decomposição do nitrato de amônio.
Esta reação envolveu uma perda de massa proporcio
nal ao aumento do teor de NH^NO^ adicionado ã amo£
tra.
e) O teor de voláteis liberado na reação em torno de
410°C diminuiu com o aumento da porcentagem de
NHj NOg no DUA.
f) A decomposição do oxido intermediário na amostra
original ocorreu por meio de uma perda de massa rá
pida em 5 25°C, acompanhada de liberação de volá
teis lenta e gradual até 7 8 5°C. A temperatura
final desta reação diminuiu na seguinte ordem: DUA
original (78 5°C) < DUA + 1% NH^NO^ (770°C) < DUA
+ 5% NH^NOg (650°C) < DUA + 10% NH^^NO^ (577°C). A
temperatura da primeira etapa (reação mais rápi
da), por outro lado, aumentou de 52 5 para 54 2°C,

120.
para as amostras com 5 e 10% de NHj^NO^, respecti-
vãmente.
g).A reação global de decomposição do intermediario
apresentou aumento na perda de massa.
As curvas DSC, Figura IV.18, mostram no
vas reações, â medida que aumenta o teor de nitrato de
amonio no DUA, as quais não apresentaram, exatamente,
os mesmos valores de temperaturas observados nas curvas
TG e DTG. Dentre elas convém destacar aquela em 208°C
e, principalmente, as reações exotérmicas em 325 e
372°C, que substituíram gradativamente a reação exotér
mica em torno de 410°C, observada na amostra original.
121
M A S S A
T E M P E R A T U R A («C)
F i g u r o I V . 16 - C u r v o s T G d a d e c o m p o s i ç ã o t é r m i c o d o d i u r a n o t o d e a m o n i o 2 0 - C
( T ó b e l o I I I . 1 ) ( o ) e d e suos m i s t u r a s s ó l i d o s c o m n i t r a t o d e amonio,
r e s p e c t i v a m e n t e , 1% NH4NO3 ( b ) , 5 % NH4NO3 ( c ) e 1 0 % NH4NO3
( d ) , e m a t m o s f e r a i n e r t e ( N g ) .
122,
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
T E M P E R A T U R A (°C)
7 0 0 8 0 0
F i g u r a l Y . 1 7 - C u r v a s D T G d a d e c o m p o s i ç ã o t é r m i c a d o d i u r a n o t o d e a m o n t o 2 0 - C
( T ó b e l o I I I . 1 ) ( a ) e de s u o s m i s t u r a s s ó l i d a s c o m n i t r a t o d e a m o n i o ,
r e s p e c t i v a m e n t e , 1% NH4NO3 ( b ) , 5 7 o NH4NO3 ( c ) e 1 0 % NH4NO3
( d ) , e m a t m o s f e r a i n e r t e ( N g ) -
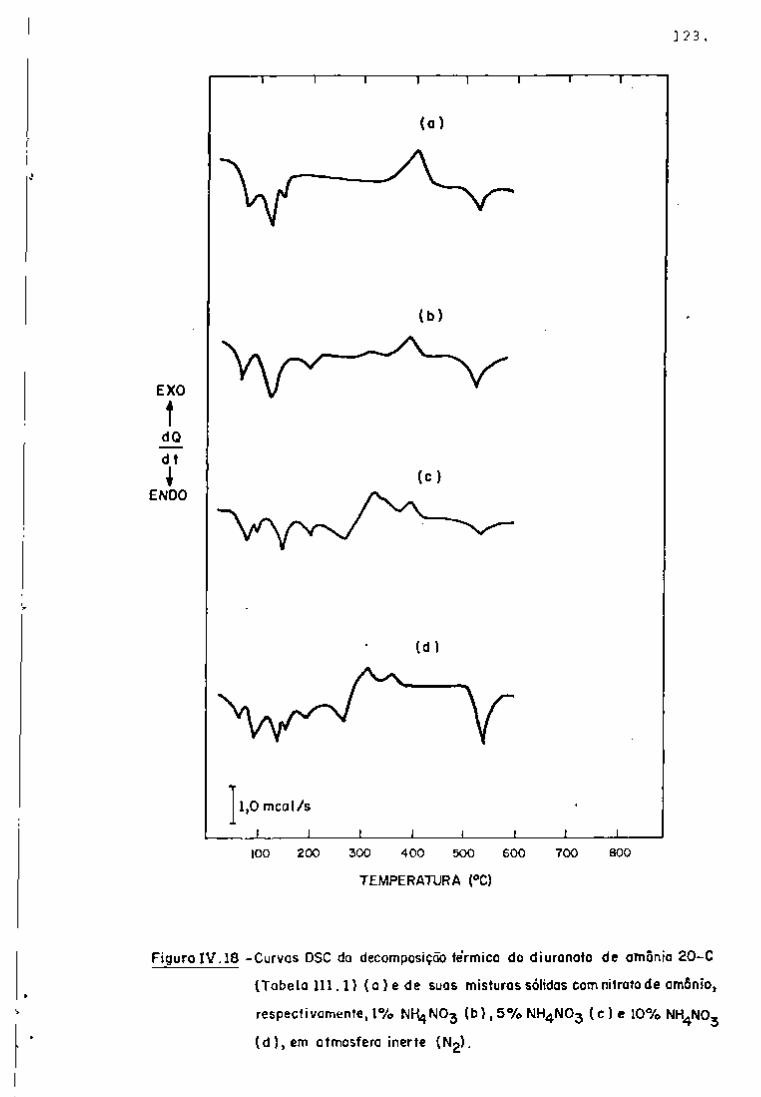
123
EXO
dO
d t
ENDO
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
TEMPERATURA (°C)
7 0 0 8 0 0
Figuro IV.18 -Curvas DSC da decomposição te'rmico do diuranoto de amônio 20 -C
(Tabelo 111.1) ( a ) e d e suas misturos sólidas com nitrato de omônio,
respectivamente, 17o NH4NO3 ( b ) , 5 % NH4NO3 (c ) e 10% NH^NOg
( d ) , em atmosfera inerte (Ng) .
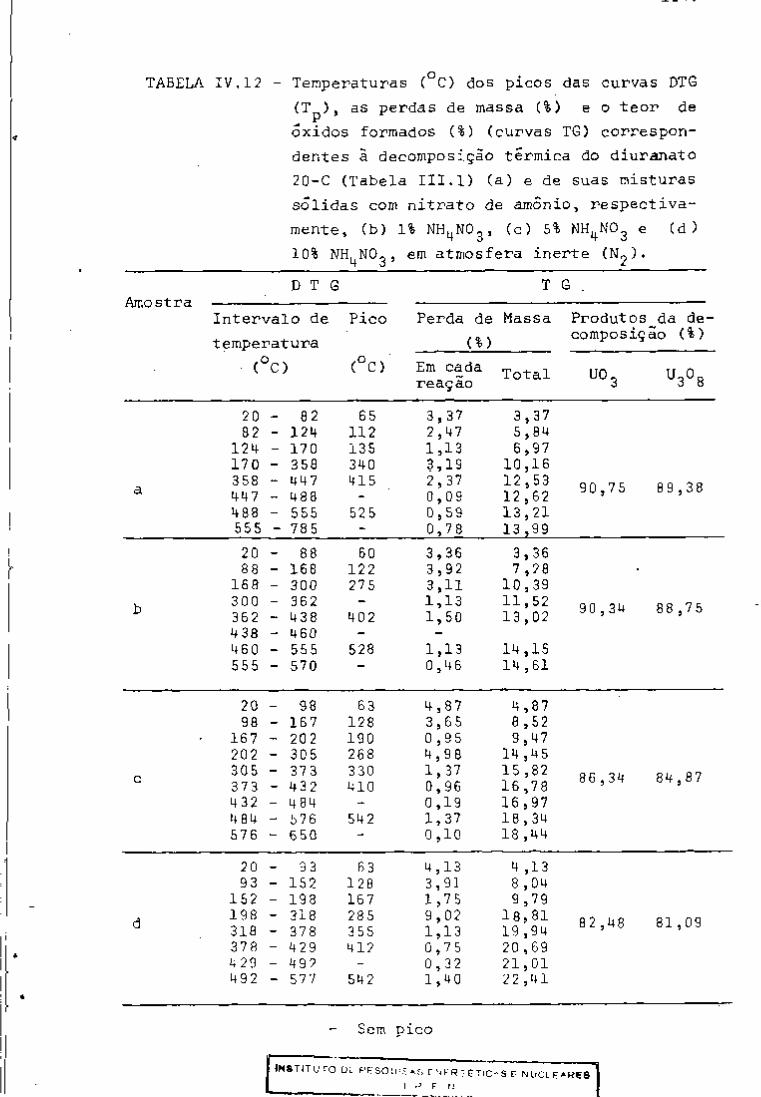
124.
TABELA IV.12 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes â decomposição térmica do diuranato
20-C (Tabela III.l) (a) e de suas misturas
solidas com nitrato de amonio, respectiva
mente, (b) 1% NHi^NOg, (c) 5% NHj NOg e (d )
10% NH^^NOg, em atmosfera inerte (N2).
D T G T G Amostra
Intervalo de Pico
temperatura
Perda de Massa
(%)
Produtos da decomposição (%)
(°C) (°C) Em cada reação
Total UO,
20 82 65 3,37 3,37 82 - 124 112 2,47 5,84
124 - 170 135 1,13 6,97 170 - 358 340 3,19 10,16
a 358 447
—. 447 488
415 2, 37 0,09
12,53 12,62 90,75 89,38
488 - 555 525 0,59 13,21 555 - 785 - 0,78 13 ,99
20 — 88 60 3,36 3,36 88 - 168 122 3,92 7 ,28 -
168 - 300 275 3,11 10,39
b 300 36 2 438
-362 438 460
402 1,13 1,50
11,52 13,02
90 ,34 88,75
460 - 555 528 1,13 14,15 555 — 570 0,46 14,61
20 — 98 63 4,87 4,87 98 - 167 128 3,65 8 ,52
167 - 202 190 0,95 9,47 202 - 305 26 8 4,98 14,45
c 305 373
— 373 432
330 410
1,37 0,96
15 ,82 16,78
86 ,34 84,87
432 - 484 - 0,19 16,97 484 - 576 542 1,37 18,34 576 - 650 - 0,10 18 ,44
20 — 93 63 4,13 4 ,13 93 - 152 128 3,91 8 ,04
152 - 198 167 1,75 9 ,79 18,81 19 ,94 d 198
318 — 318
378 285 355
9,02 1,13
9 ,79 18,81 19 ,94 82 ,48 81,09
378 - 429 412 0,75 20 ,69 429 - 497 - 0,32 21,01 492 577 542 1,40 22 ,41
Sem pico
Í N S T I T U T O DE P e S Q U : S A S E , ^ t - R C - e T l C - ' S E N U C L E A R E S
I. P. E . N,
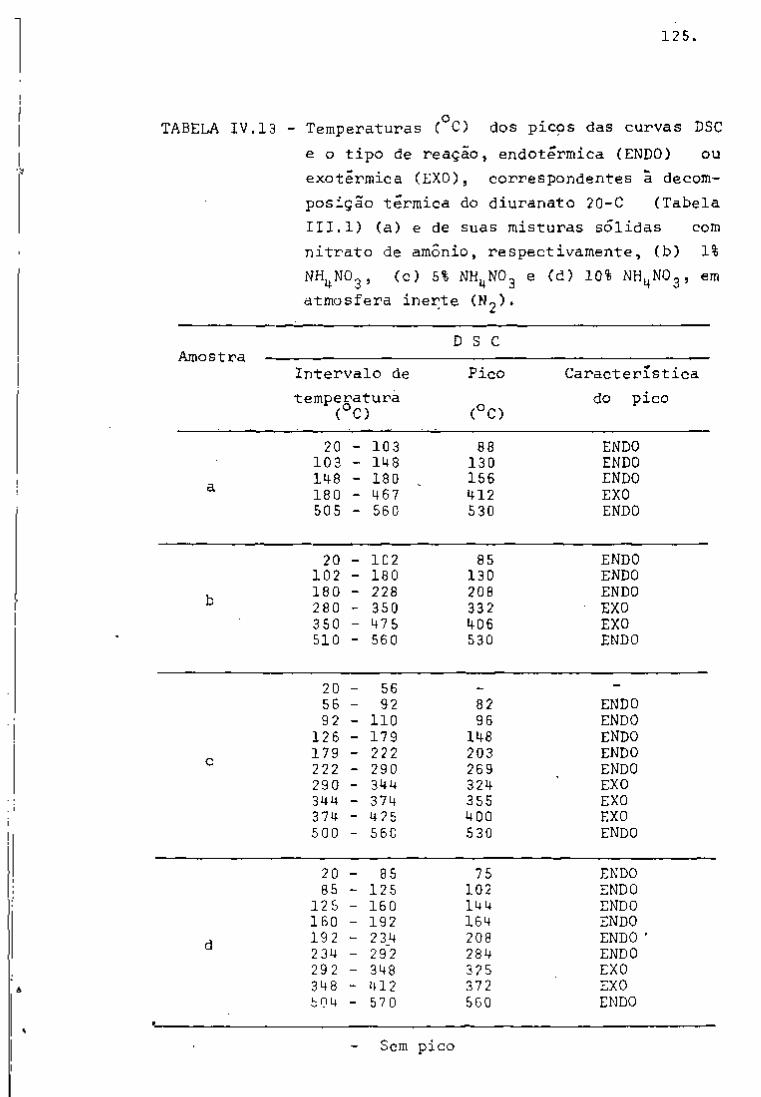
125.
• a
TABELA IV.13 - Temperaturas (°C) dos picos das curvas DSC
e o tipo de reação, endotérmica (ENDO) ou
exotérmica (EXO), correspondentes ã decom
posição térmica do diuranato 20-C (Tabela
III.l) (a) e de suas misturas sólidas com
nitrato de amonio, respectivamente, (b) 1%
NH^^NOg, (c) 5% NHj NOg e (d) 10% NH^^NO^, em
atmosfera inerte (N2).
D S C Amostra
Intervalo de
temperatura (°C)
Pico
(°C)
Característica
do pico
20 - 103 88 ENDO 103 - 14 8 130 ENDO 148 - 180 156 ENDO
a 180 - 467 412 EXO 505 - 560 530 ENDO
20 - 102 85 ENDO 102 - 180 130 ENDO 180 - 228 208 ENDO
D 280 - 350 332 EXO 350 - 475 406 EXO 510 - 560 530 ENDO
20 - 56 — -•
56 - 92 82 ENDO 92 - 110 96 ENDO
126 - 179 148 ENDO 179 - 222 203 ENDO
C 222 - 290 269 ENDO 290 - 344 324 EXO 344 - 374 355 EXO 374 - 425 400 EXO 500 - 560 530 ENDO
20 - 85 75 ENDO 85 - 125 102 ENDO
125 - 160 144 ENDO 160 - 192 164 ENDO
d 192 - 234 208 ENDO • 2 34 - 292 284 ENDO 292 - 348 325 EXO 348 - 412 372 EXO 504 - 570 560 ENDO
Sem pico

126.
IV. 6 CARACTERIZAÇÃO DOS DIURANATOS DE AMONIO y
IV.6.1 Curvas termogravimêtricas e termogravi
mêtricas derivadas
A comparação dos perfis das curvas TG e
DTG permitiu classificar os diuranatos de amonio (enve
Ihecidos) estudados em seis gruposdiferentes. . Essa
classificação foi influenciada diretamente pelo modo de
secagem:
Grupo I compostos secados a 90 C por 216 0 minu-
16-B
Grupo II - compostos secados a 115 C por 57 60 minu
tos.
Grupo III -
Grupo IV -
compostos secados a 13 0 C por 43 20 minu
tos.
compostos secados a 18 0 C por 240 minu-
tos. Í 4 - B
Grupo V
Grupo VI -
compostos secados a 220°C por 4320 mi-
ñutos. I'í-P)
compostos secados a 280°C por 720 minu-
2 0 ' c tos.
As curvas TG e DTG desses compostos são
apresentadas nas Figuras de IV. 19 a IV. 24 e foram cons_i
deradas as mais representativas para cada grupo.

127.
Fara facilitar a discussão dos resulta-
dos, foram calculadas as perdas de massa devidas â de
composição térmica dos compostos, considerando-se como
resíduo, o oxido U^Og, e as perdas de massa evidencia -
das pelas curvas TG, conforme descrição no Apêndice 1.
Os dados encontram-se nas Tabelas de IV. 14 a IV. 25. Nes_
tas tabelas estão reunidos também, os intervalos de tem
peraturas de cada estádio da decomposição dos compostos
e as temperaturas correspondentes a cada reação, eviden
ciadas pelas curvas DTG. Esses dados possibilitaram os
cálculos sobre o numero de moléculas de água de crista
lização' nos compostos e: a composição dos intermediários
evidenciados nas curvas TG.
IV,6.2 Curvas de calorimetria exploratoria
diferencial
As curvas DSC obtidas para os compostos
estudados, agrupados de modo semelhante ao das respecti^
vas curvas TG e DTG, são apresentadas, também, nas Fi
guras de IV,19 a IV.24.
As temperaturas e intervalos de tempera
turas correspondentes aos picos observados nas curvas
DSC foram reunidos, respectivamente, nas Tabelas IV.15,
IV.17, IV,19, IV.21, IV.23 e IV.25.
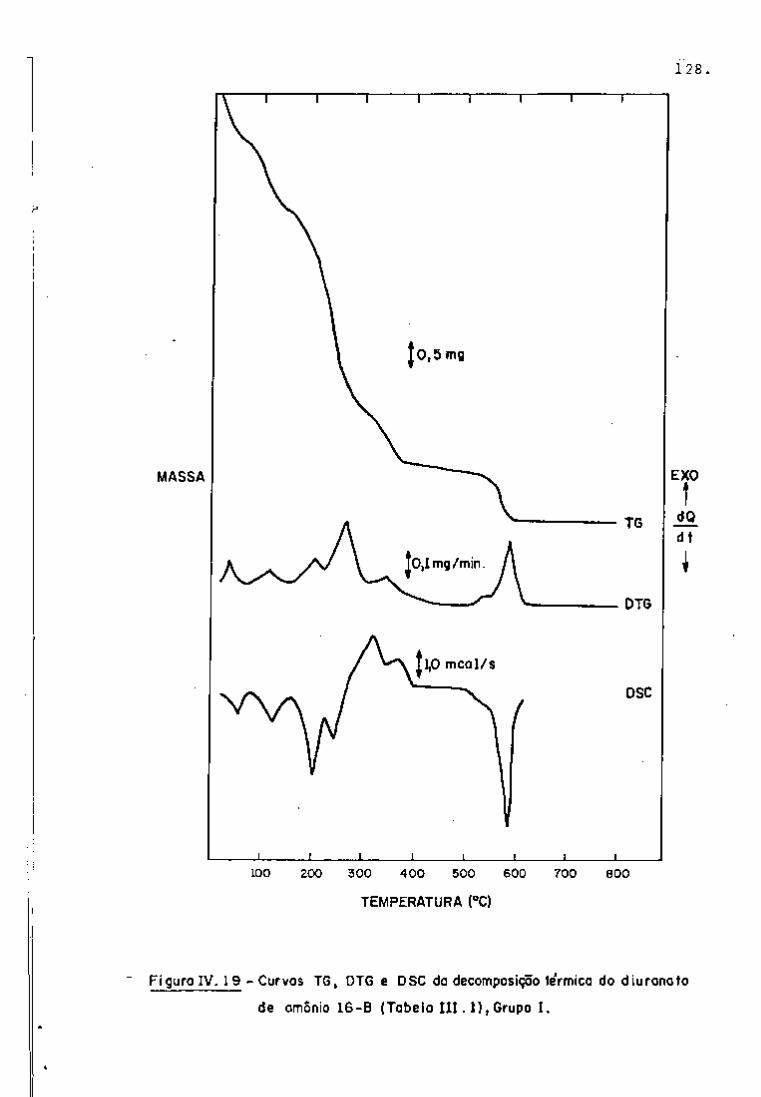
128
MASSA
T 1 T
0,1 mg/min.
mca l / s
TG
DTG
DSC
-J 1 I I I I 1 L
EXO
f dQ
d t
100 200 300 4 0 0 500 600 700 800
TEMPERATURA («O
Figuro IV. 19 - Curvas T G , DTG e DSC da decomposição te'rmico do diuronato
de amonio 1 6 - B (Tabela I I I . 1 ) , Grupo I.
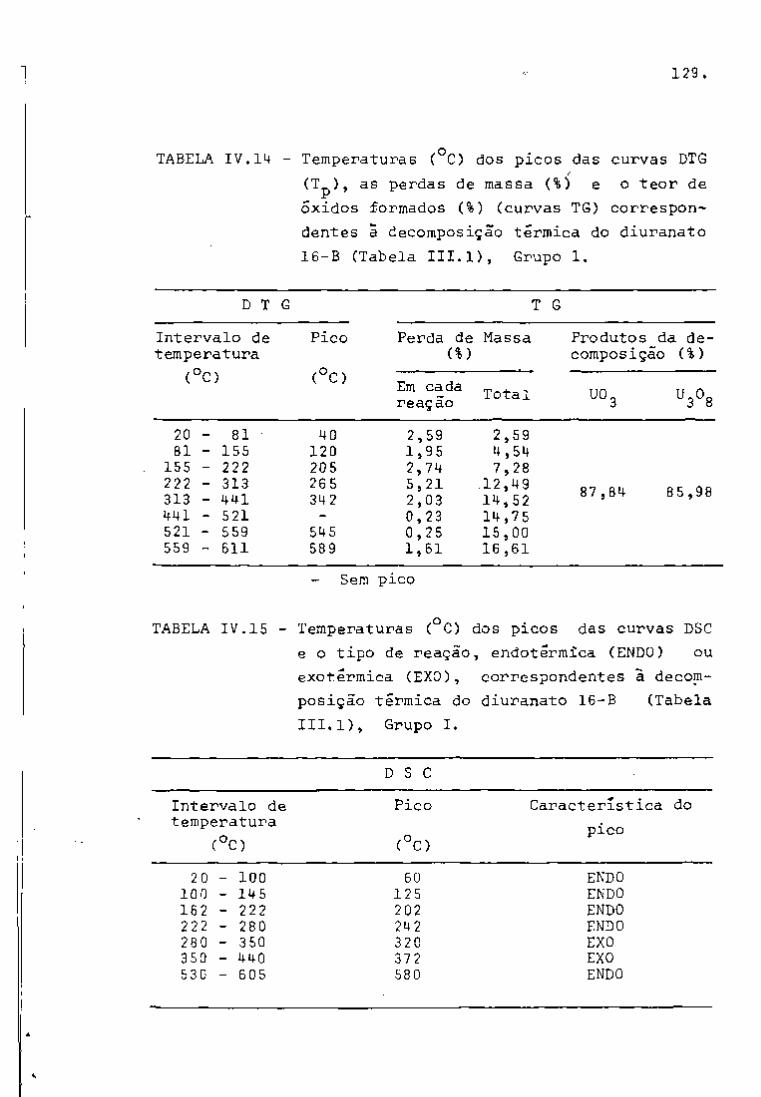
129.
TABELA IV.14 - Temperaturas ( C) dos picos das curvas DTG y
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica do diuranato
16-B (Tabela III.l), Grupo 1.
D T G T G
Intervalo de temperatura
Pico Perda de (%)
Massa Produtos da decomposição (%)
(°C) ( C) ^ •' Em cada
reação Total UO3 UgOg
20 - Bl 81 - 15.5
155 - 222 222 - 313 313 - 441 441 - 521 521 - 559 559 - 611
40 2,59 120 1,95 205 2,74 265 5,21 34 2 2,03
0,23 545 0,25 589 1,61
2,59 4,54 7,28
ll'll 87,84 85,98
14',75 15,00 16,61
Sem pico
TABELA IV.15 - Temperaturas (°C) dos picos das curvas DSC
e o tipo de reação. endotérmica (ENDO) ou
exotérmica (EXO), correspondentes a decom-
posição térmica do diuranato 16-B (Tabela
III.l), Grupo I.
D S C
Intervalo de temperatura
(°C)
Pico
(°C)
Característica do
pico
20 - 100 100 - 145 162 - 222 222 - 280 280 - 350 350 - 440 530 - 605
60 125 202 242 320 372 580
ENDO ENDO ENDO ENDO EXO EXO ENDO
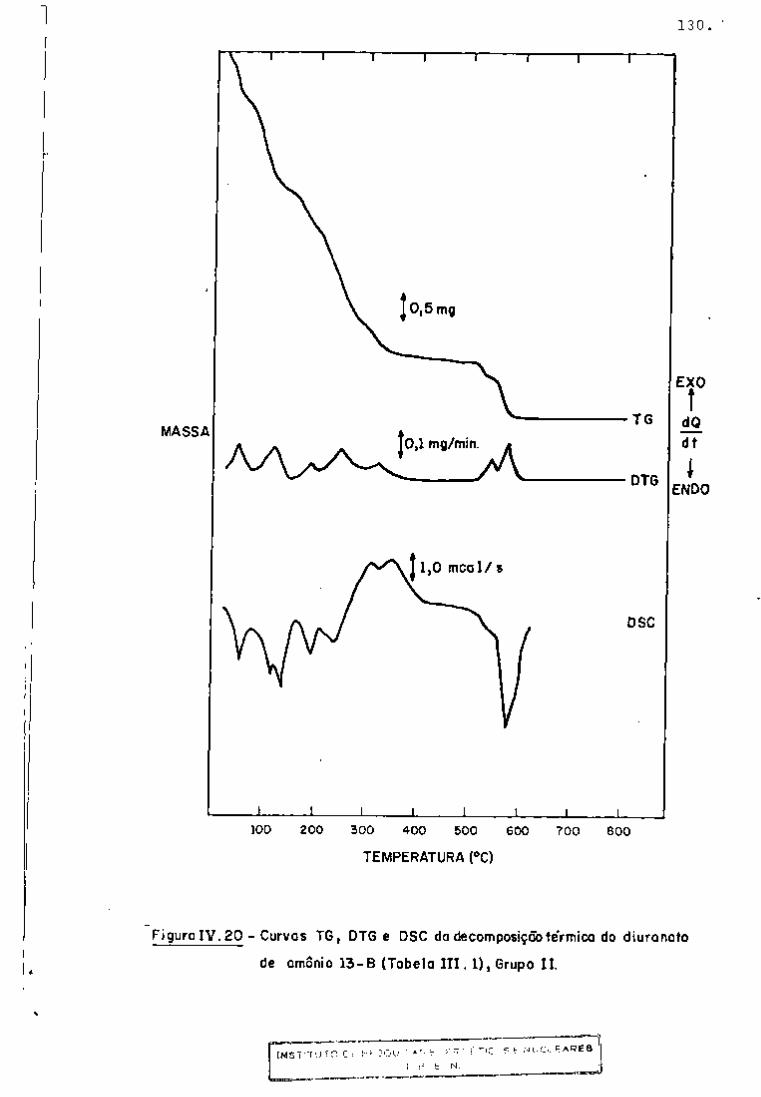
130.
M A S S A
T T
A /\ /V i O , l m g / m i n . M
1,0 m c a l / s
T G
D T G
D S C
J L J I \ I L
E ) | 0
dQ
dt
E N D O
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
T E M P E R A T U R A («C)
7 0 0 8 0 0
F i g u r o I V . 2 0 - C u r v a s T G , D T G e D S C d o d e c o m p o s i ç ã o t é r m i c o d o d i u r a n o t o
d e a m o n i o 1 3 - B ( T ó b e l o I I I . 1 ) , G r u p o I I .
INGTITU-ÍO DE PE S Q U ' A S E F P É - I C S E N U C L E A R E S
i. P. £ . N .
ni
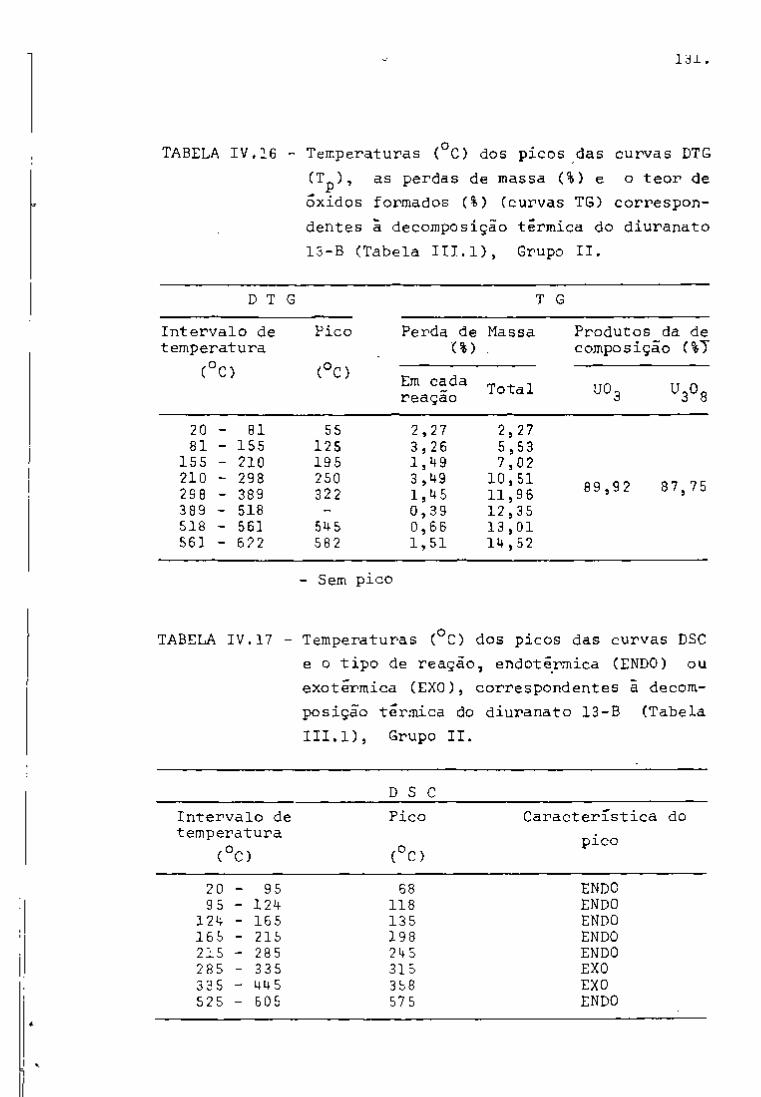
131.
TABELA IV.16 - Temperaturas ( C) dos picos .das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica do diuranato
13-B (Tabela III.l), Grupo II.
D T G T G
Intervalo de temperatura
(°C)
Pico
(°C)
Perda de Massa (%)
Em cada reação
Total
Produtos da de composição .(%T
UO,
20 - 81 55 81 - 155 125
155 - 210 195 210 - 298 250 298 - 389 322 389 - 518 -518 - 561 545 561 - 622 582
2,27 3,26 1,49 3,49 1,45 0,39 0,66 1,51
2,27 5,53 7,02
10,51 11,9 6 12,35 13,01 14,52
89,92 87,75
- Sem pico
TABELA IV.17 - Temperaturas (°C) dos picos das curvas DSC
e o tipo de reação , endotérmica (ENDO) ou
exot érmica (EXO), correspondentes ã decom-
posição térmica do diuranato 13-B (Tabela
III.l), Grupo II.
D S C
Intervalo de Pico Característica do temperatura pico
(°C) (°C) pico
20 - 95 68 ENDO 95 - 124 118 ENDO
124 - 165 135 ENDO 155 - 215 198 ENDO 215 - 285 245 ENDO 285 - 335 315 EXO 335 - 445 358 EXO 525 - 505 575 ENDO
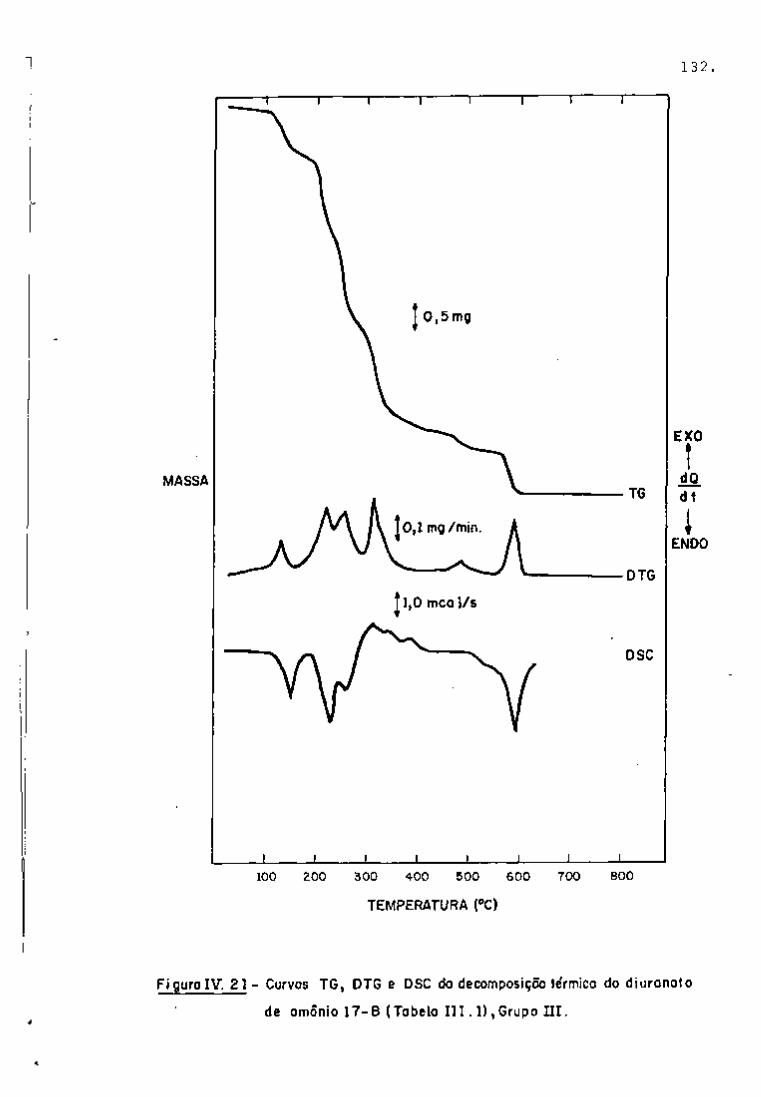
132
MASSA
ENDO
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0 8 0 0
TEMPERATURA ( " O
Fi guro IV. 2 1 - Curvas T G , DTG e DSC do decomposicoo térmico do diuronato
de omonio 1 7 - B (Tabelo I I I . 1) .Grupo I I I .
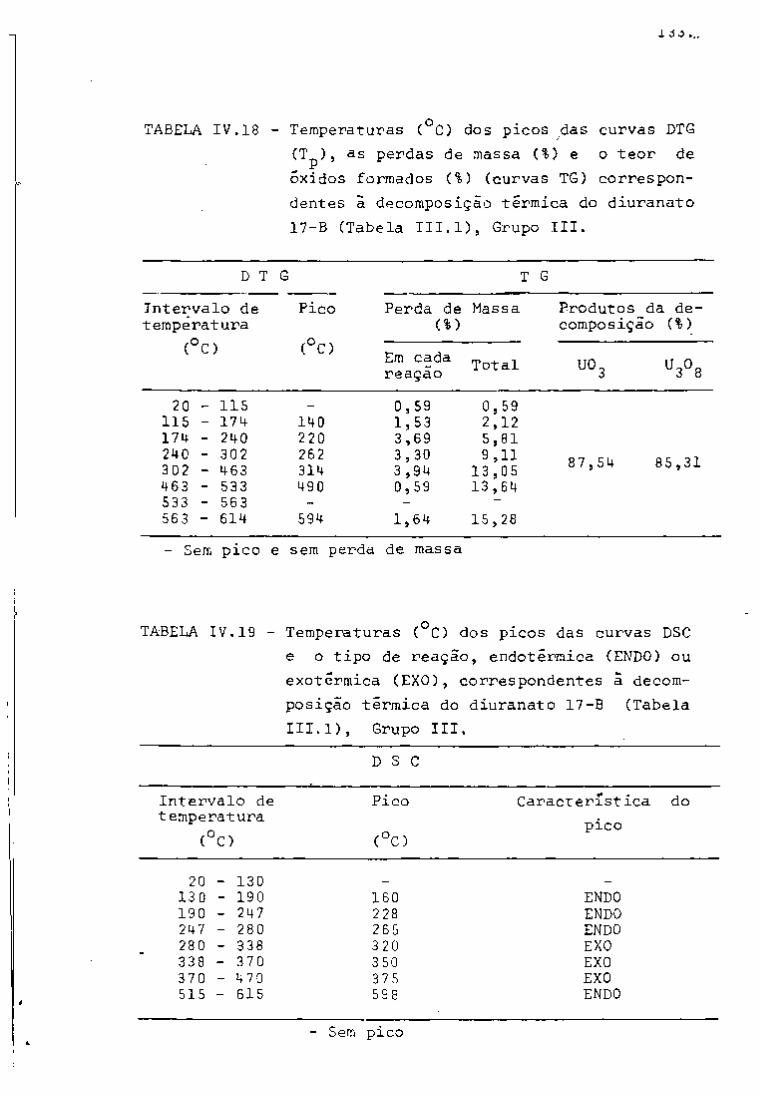
TABELA IV.18 - Temperaturas ( C) dos picos^das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes â decomposição térmica do diuranato
17-B (Tabela III.l), Grupo III.
D T G T G
Intervalo de temperatura
(°C)
Pico
(°C)
Perda de (%)
Massa Produtos da decomposição (%).
Intervalo de temperatura
(°C)
Pico
(°C) Em cada reação
Total UO3 U3O3
20 - 115 l i s - 174 174 - 240 240 - 302 302 - 463 463 - 533 533 - 563 563 - 614
140 220 262 314 490
594
0,59 1,53 3,69 3,30 3 ,94 0,59
1,64
0,59 2,12 5,81 9,11
13,05 13,64
15,28
87,54 85,31
- Sem pico e sem perda de massa
TABELA IV.19 - Temperaturas ( C) dos picos das curvas DSC
e o tipo de reação, endotérmica (ENDO) ou
exotérmica (EXO), correspondentes ã decom
posição térmica do diuranato 17-B (Tabela
III.l), Grupo III.
D S C
Intervalo de Pico Característica do temperatura pico
(° C) (°C)
20 - 130 _ _ 130 - 190 160 ENDO 190 - 247 228 ENDO 247 - 280 265 ENDO 280 - 338 320 EXO 338 - 370 350 EXO 370 - 470 375 EXO 515 - 615 598 ENDO
- Sem pico
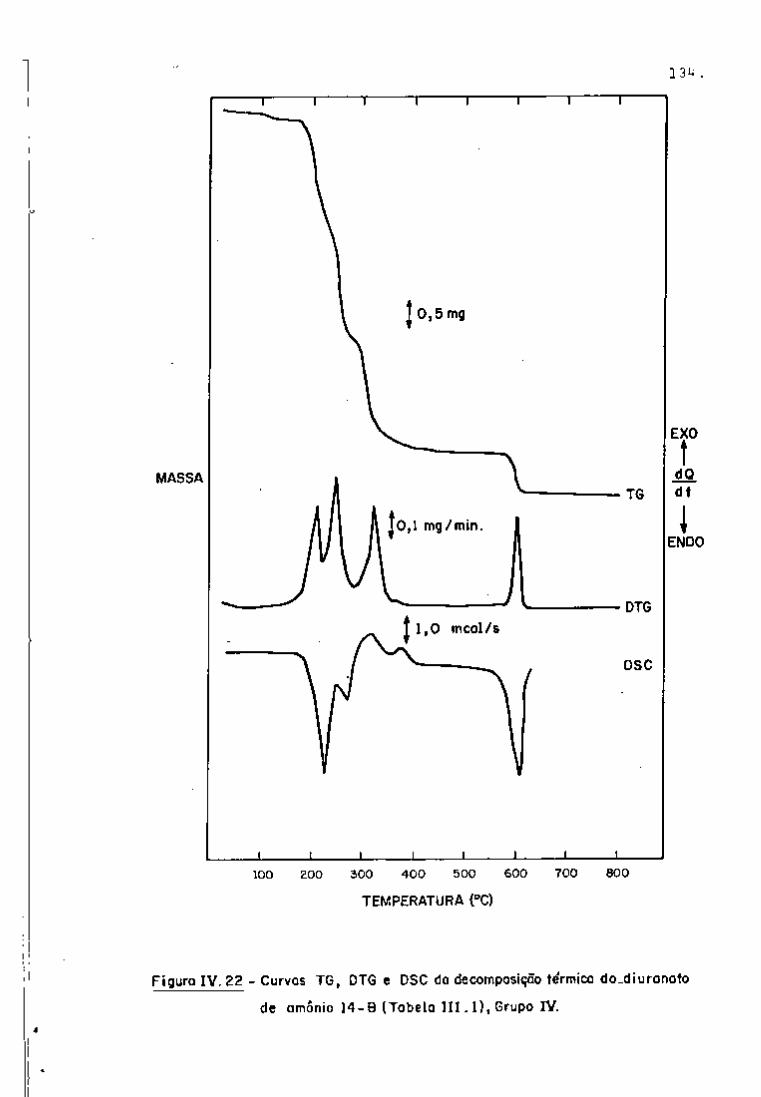
MASSA
0 , 5 mg
^0 ,1 m g / m i n .
1,0 mcal /s
134 .
1 1 1 1 T
TG
DTG
DSC
EXO
dQ
d t
1 ENDO
J I I \ I L
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0 8 0 0
TEMPERATURA ( "O
Figura lY . 22 - Curvas T G , DTG e DSC da decomposição te'rmico do.diuranoto
de amônio 1 4 - B (Tabelo I I I . 1 ) , Grupo lY.
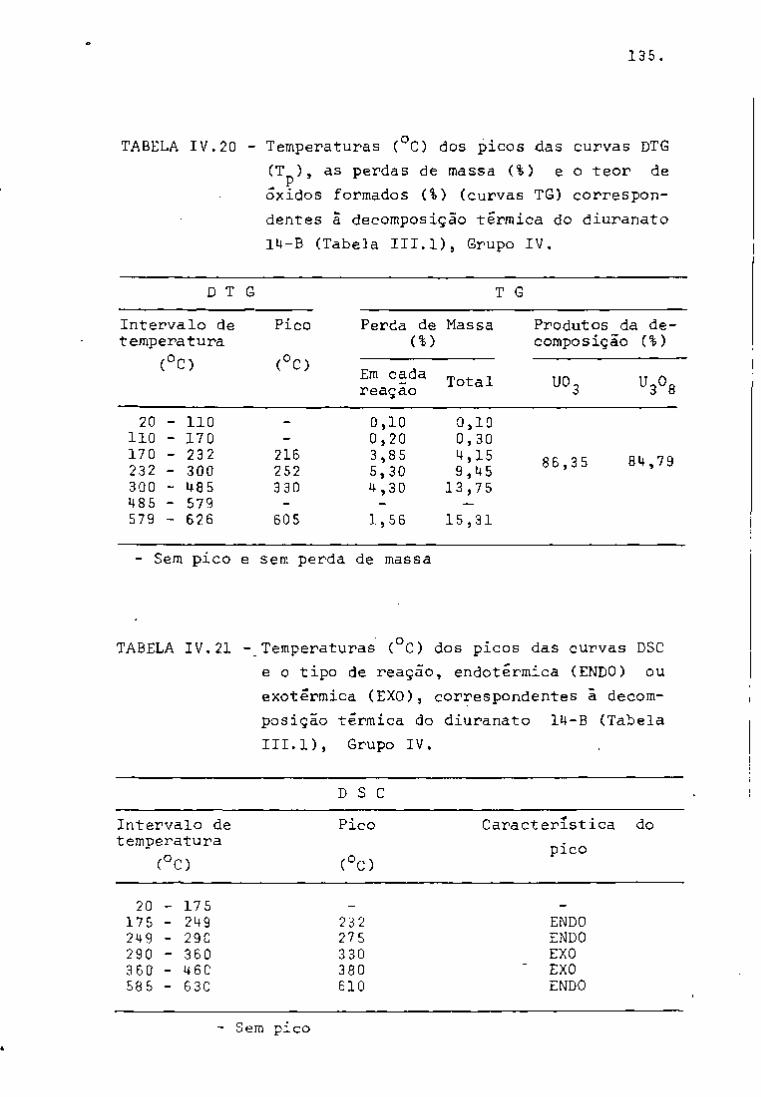
135.
TABELA IV.20 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes â decomposição térmica do diuranato
m-B (Tabela III.l), Grupo IV.
D T G T G
Intervalo de temperatura
Pico Perda de Massa (%)
Produtos da decomposição (%)
("C) ("C) Em cada reação Total
20 - 110 — 0,10 0,10 110 - 170 - 0,20 0,30 170 - 232 216 3,85 4,15 232 - 300 252 5,30 9,45 300 - 485 330 4,30 13,75 485 - 579 - - •. —
579 - 626 605 1,56 15,31
UO. "3^8
86,35 84,79
- Sem pico e sem perda de massa
TABELA IV.21 -Temperaturas ( C) dos picos das curvas DSC
e o tipo de reação, endotérmica (ENDO) ou
exotérmica (EXO), correspondentes ã decom
posição térmica do diuranato 14-B (Tabela
III.l), Grupo IV.
D S C .
Intervalo de Pico Característica do temperatura pico
(°C) (°C) pico
20 - 175 175 - 249 232 ENDO 249 - 290 275 ENDO 290 - 360 330 EXO 360 - 460 380 " EXO 585 - 630 610 ENDO
- Sem pico
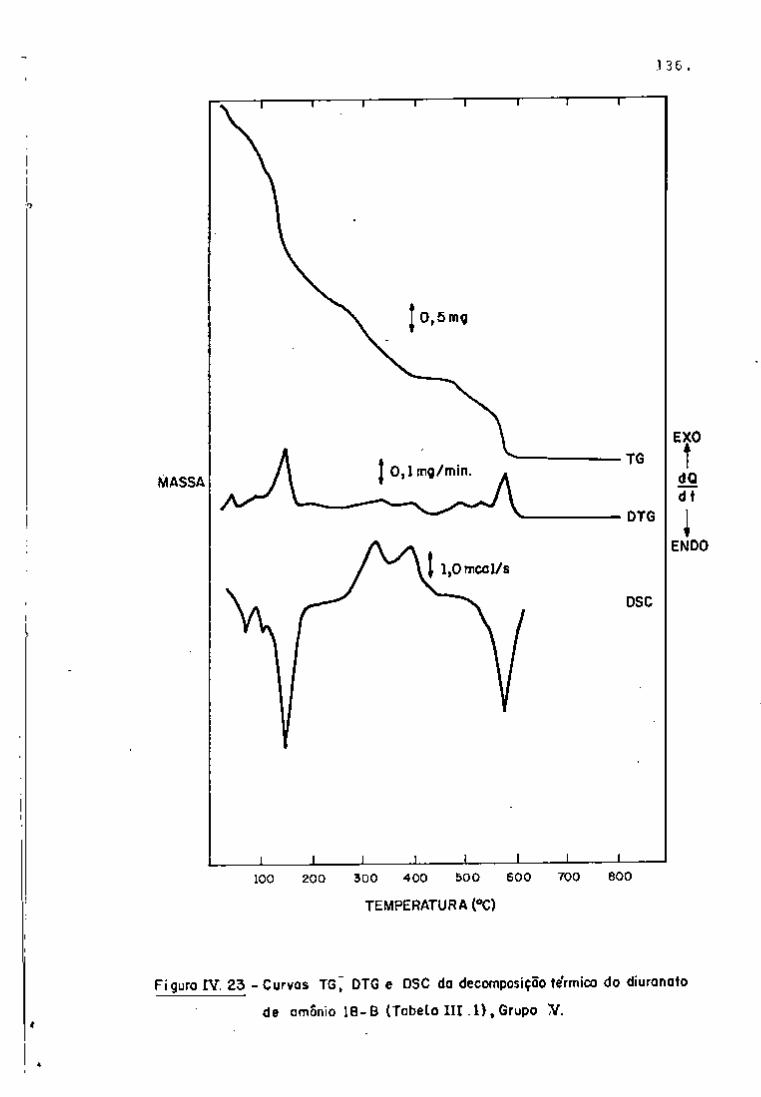
136 .
MASSA
ENDO
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
TEMPERATURA («O
7 0 0 8 0 0
Figura lY. 23 - Curvas TGJ DTG e DSC da decomposição térmica do diuranoto
de amonio 1 8 - B (Tóbelo I I I . 1 ) , Grupo V .

137.
TABELA IV.22 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes â decomposição térmica do diuranato
18-B (Tabela III.l), Grupo V.
D T G T G
Intervalo de temperatura
(°C)
Pico
(°C)
Perda de Massa Produtos da de-(%) • composição (%)
Em cada reação Total UOg UgOg
20 - 70 55 70 - 115 105
115 - 186 145 186 - 276 -276 - 370 340 370 - 437 395 437 - 470 -470 - 529 500 529 - 555 538 555 - 605 584
0,99 1,38 3,89 1,71 2,00 0,83
0,88 0,44 1,56
0,99 2,37 6,26 7,97 9,97
10,80
11,68 12,12 13,68
90,19 87,31
- Sem pico e sem perda de massa
TABELA IV.23 - Temperaturas (°C) dos picos das curvas DSC
e o tipo de reação, endotérmica (ENDO) ou
exotérmica (EXO), correspondentes ã decom
posição térmica do diuranato 18-B -(Tabela
III.l), Grupo V.
D S C ,
Intervalo de Pico Característica do temperatura pico
(°C) (°C)
20 - 85 70 ENDO 85 - 125 110 ENDO
125 - 190 152 ENDO 250 - 355 324 EXO 355 - 440 390 EXO 510 - 600 580 ENDO
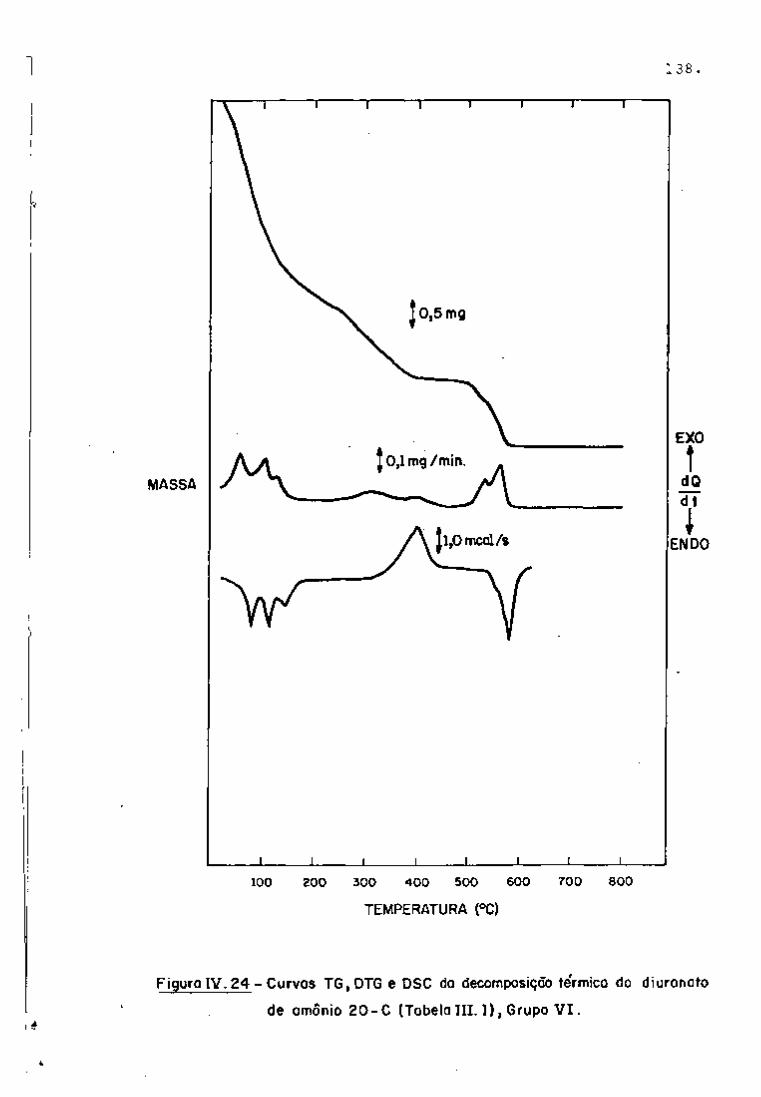
1 3 8 .
MASSA
100 200 300 400 500 600 700 800
TEMPERATURA (*C)
Figuro IV . 2 4 - Curvos T 6 , DTG e D S C do decomposição te'rmico do diuranoto
de omônio 2 0 - C ( T ó b e l a I I I . 1 ) , Grupo V I .
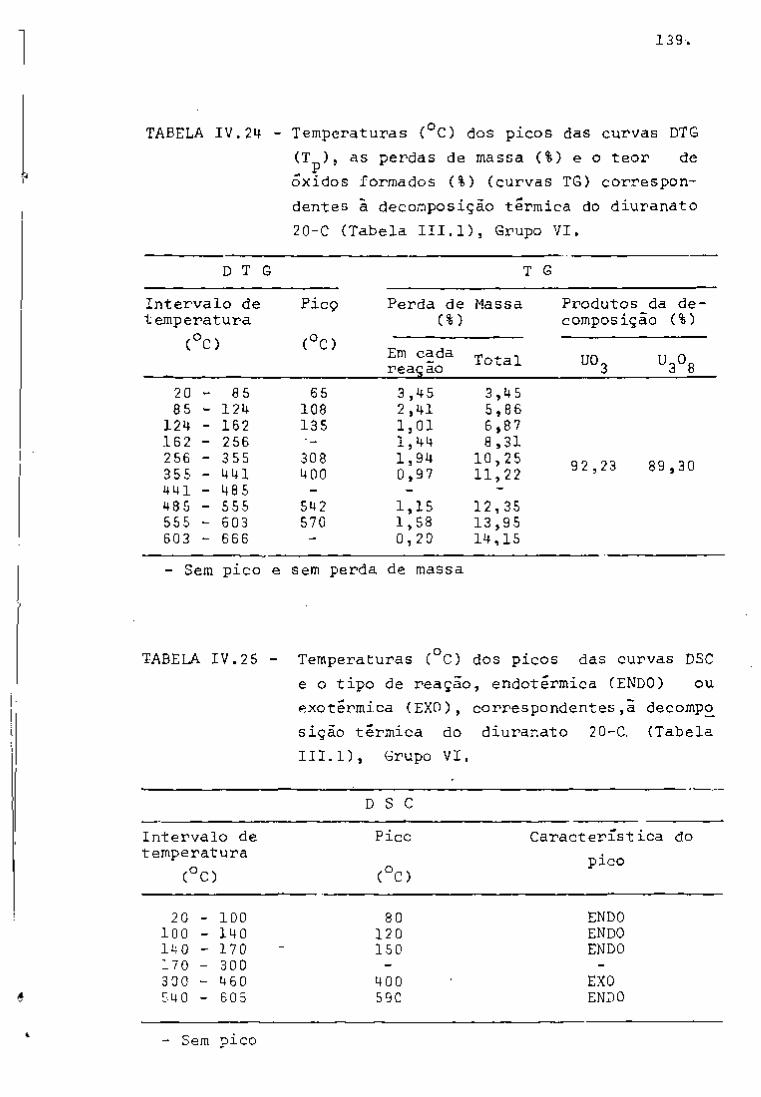
139v
TABELA IV.24 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon-
dentes a. decomposição térmica do diuranato
20-C (Tabela III.l), Grupo VI.
D T G T G
Intervalo de temperatura
Pico Perda de (%)
Massa Produtos da decomposição (%)
( C) (°C) ^ Em cada
reação Total UO3 U3O3
20 - 85 85 - 124
124 - 162 162 - 256 256 - 355 355 - 441 441 - 485 485 - 555 555 - 603 603 - 666
6 5 3,45 108 2,41 135 1,01
1,44 308 1,94 400 0,97
542 1,15 570 1,58
0,20
3,45 5,86 6,87 8,31
10,25 11,22
12,35 13,95 14,15
92,23 89,30
- Sem pico e sem perda de massa
TABELA IV.25 - Temperaturas (°C) dos picos das curvas DSC
e o tipo de reação , endotérmica (ENDO) ou
exotérmica (EXO), correspondentes,ã decompo
sição térmica do diuranato 20-C. (Tabela
III.l), Grupo VI.
D S C
Intervalo de temperatura
(°C)
Pico
(°C)
Característica do
pico
20 - 100 100 - 140 140 - 170 170 - 300 300 - 460 54 0 - 605
80 120 150
400 590
ENDO ENDO ENDO
EXO ENDO
- Sem pico

140.
IV.7 CARACTERIZAÇÃO DO TRIÕXIDO DE URÂNIO J
IV.7.1 Curvas termogravimêtricas e termogravi
mêtricas derivadas
Foram obtidas as curvas TG e DTG dos di
ferentes tipos de trióxidos de urânio, ou seja, aqueles
de cor amarelada 1, amarelada 2, alaranjada e "verde oli
va". -Na realidade, este óxido daqui para frente assinala
do como "verde oliva" é u'a mistura de UO2 e UO3.
As curvas TG e DTG desses compostos são
apresentadas nas Figuras IV.25 e IV.26.
Nas Tabelas IV.26 e IV.27 são apresenta
dos os dados de perdas de massa calculados consideran
do-se o óxido U„0_ correspondente, como produto final de
calcinação dos compostos e as perdas de massa para cada
reação da decomposição e totais verificadas nas curvas
TG.
As temperaturas inicial e final de cada
reação da decomposição dos diuranatos, bem como as tempe
raturas correspondentes a cada reação, indicadas' pelas
curvas DTG, estão reunidas nas Tabelas IV. 26 a IV. 27.
IV.7. 2 Curvas de calorimetria exploratória
diferencial
As curvas DSC obtidas para os quatro di
ferentes tipos de UOg, ou seja, o de cor amarelada 1,
amarelada 2, alaranjada e verde oliva, são apresenta-

lUl.
das na Figura IV.27. •
As temperaturas correspondentes aos pi-
eos observados nas curvas DSC, bem como o tipo de
reação, endotérmica ou exotérmica, foram reunidas nas
Tabelas IV. 26 e IV.27.
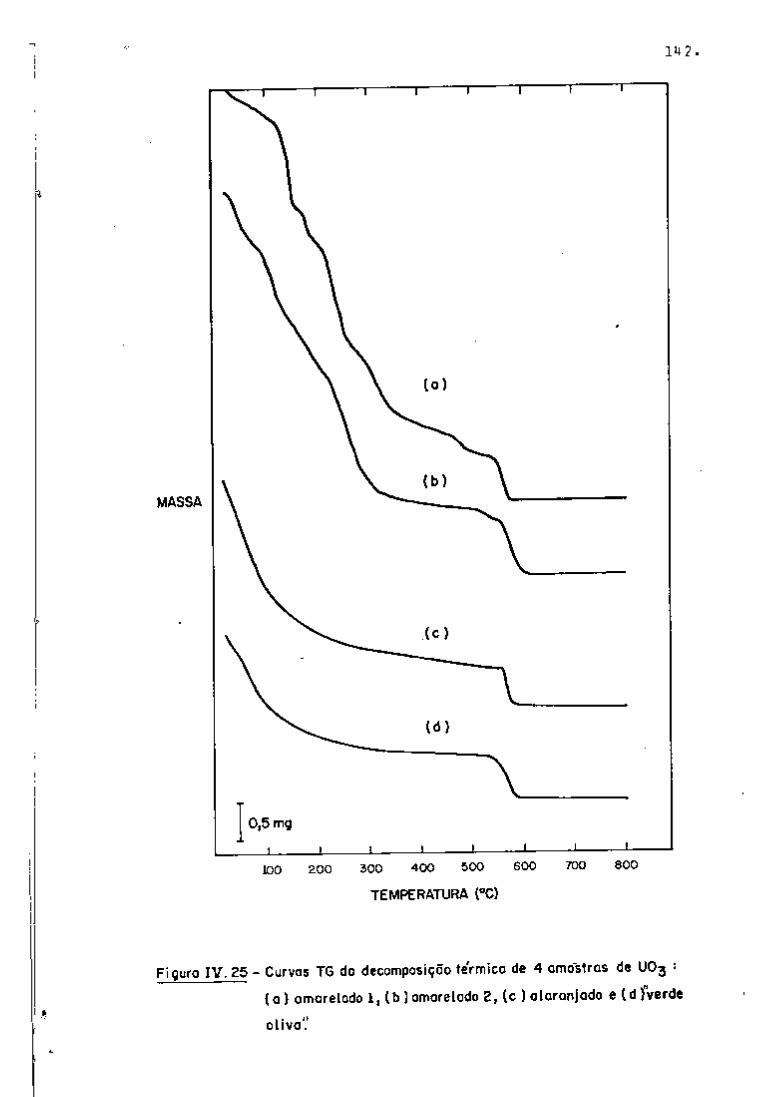
1U2.
MASSA
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
TEMPERATURA (°C)
7 0 0 8 0 0
Figuro I V . 25 - Curvas TG do decomposição te'rmica de 4 amostras de UO3 •
( o ) amarelado 1, ( b ) omarelodo 2 , (c ) alaranjado e ( d )"verde
oliva'.'
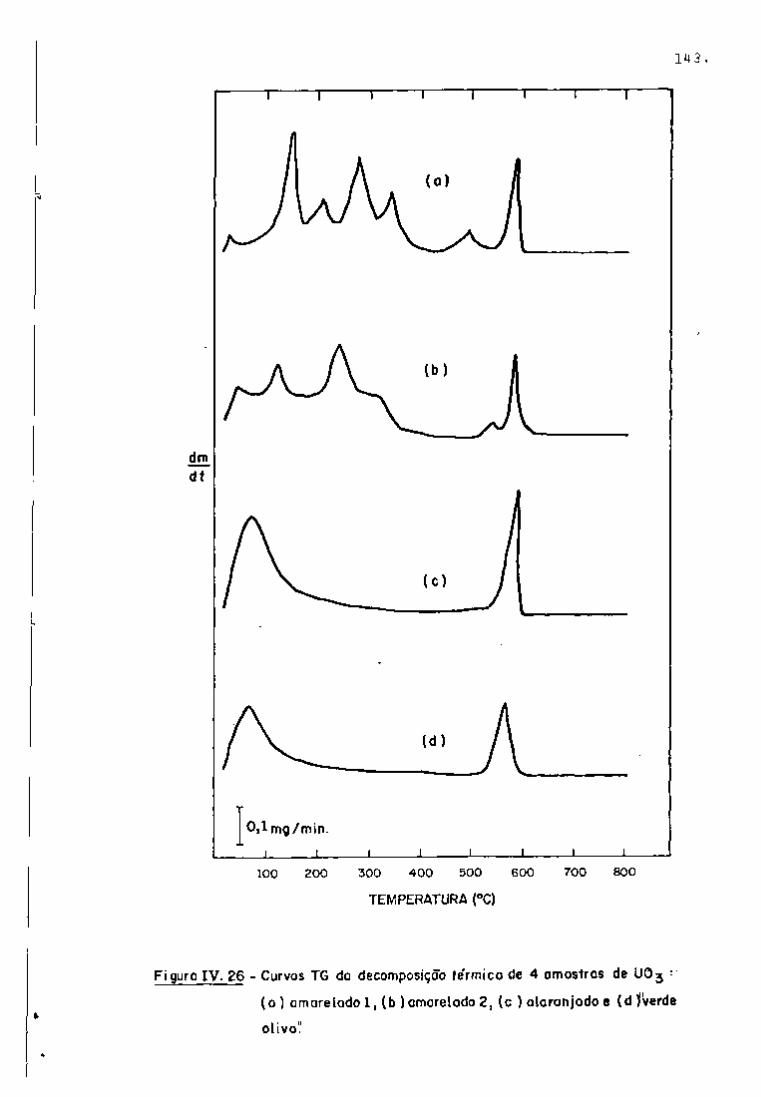
143 ,
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
TEMPERATURA ( "O
7 0 0 8 0 0
Figuro I V . 26 - Curvas T6 do decomposição te'rmico de 4 amostras de UO3 5
(a ) amarelado 1, (b )amarelado 2 , (c ) alaranjado e (d Jverde
olivo'.'
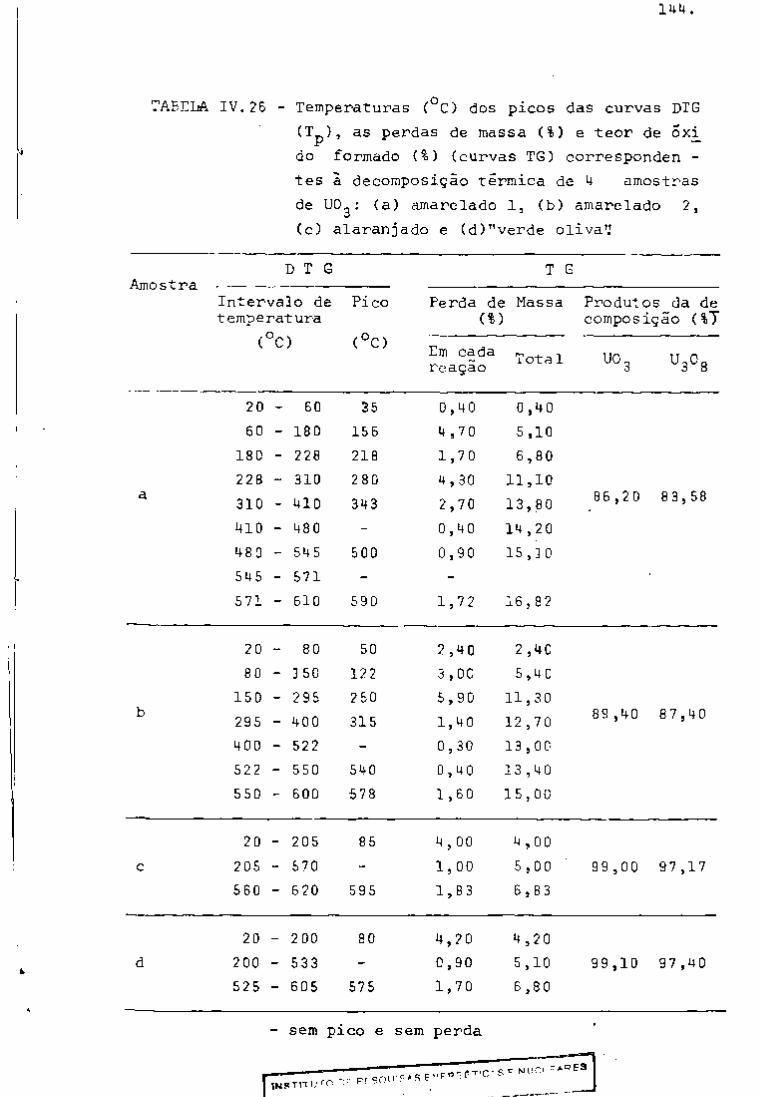
144.
TABELiA IV. 26 - Temperaturas ( ° C ) dos picos das curvas DTG
(Tp), as perdas de massa (%) e teor de oxi
do formado (%) (curvas TG) corresponden -
tes à decomposição térmica de 4 amostras
de UO^: (a) amarelado 1, (b) amarelado 2,
(c) alaranjado e (d)"verde oliva'.'
D T G T G Amostra
Intervalo de temperatura
Pico Perda de Massa Produtos da de (%) composição (%T
( ° C ) ( ° C ) Em cada Total UoOo reação
Total 3 3 8
20 - 60 35 0,40 0,40
60 - 180 156 4,70 5,10
180 - 228 218 1,70 6,80
228 - 310 280 4,30 11,10 a 310 - 410 343, 2,70 13,8 0 86,20 83,58
410 - 480 - 0,40 14,2 0
480 - 545 500 0,90 15,10
545 - 571 - -571 - 610 590 1,72 16,82
20 - 80 50 2,40 2 ,40
80 - 150 122 3 ,00 5,40
150 - 295 250 5,90 11,30 b 295 - 400 315, 1,40 12,70
89 ,40 87,40
400 - 522 - 0,30 13,00
522 - 550 540 0,40 13 ,40
550 - 600 678 1,60 15, 00
20 - 205 85 4, 00 4,00
c 205 - 570 - 1,00 5,00 99,00 97,17
560 - 620 595 1,83 6,83
20 - 200 80 4,20 4,2 0
d 200 - 533 - 0,90 5,10 99,10 97,40
525 - 605 575 1,70 6,80
- sem pico e sem perda
* P E 8
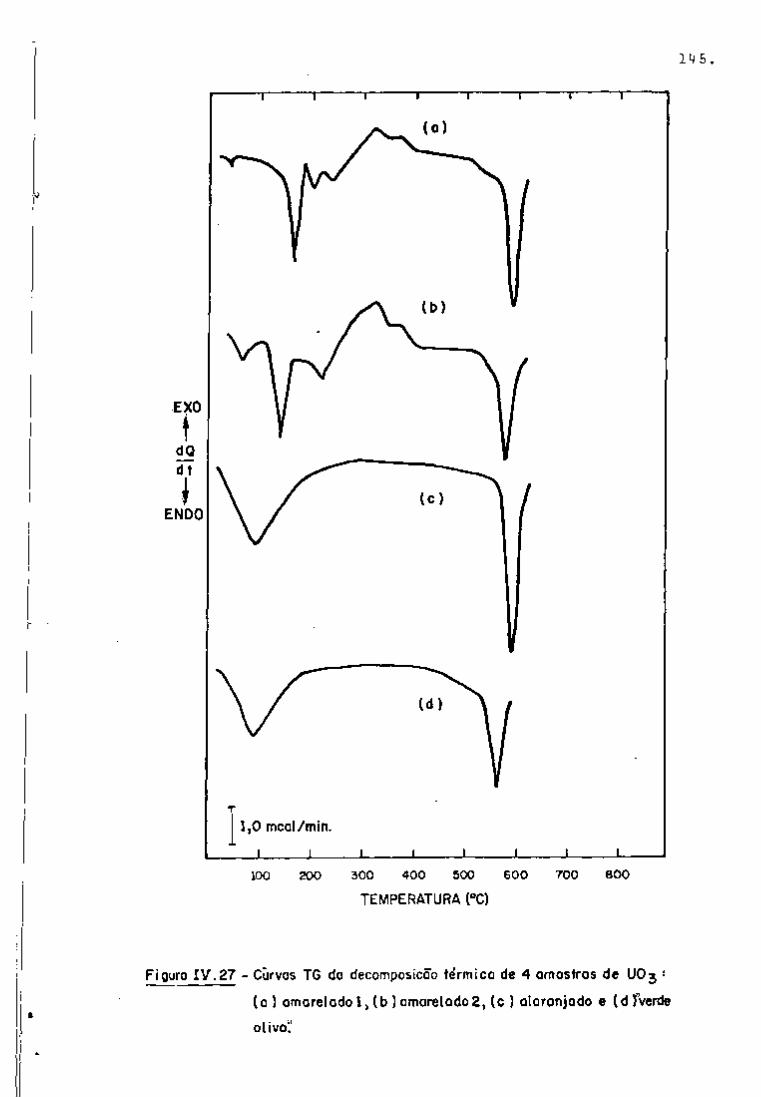
145.
100 200 300 400 500 6 0 0
TEMPERATURA ( "O
700 800
Figuro I V . 2 7 - Curvos TG do decomposicoo te'rmica de 4 amostras de UO3 ••
( o ) a m o r e l a d o l , ( b ) a m a r e l a d o 2 , (c ) olaronjodo e (díverde
olivo'.'
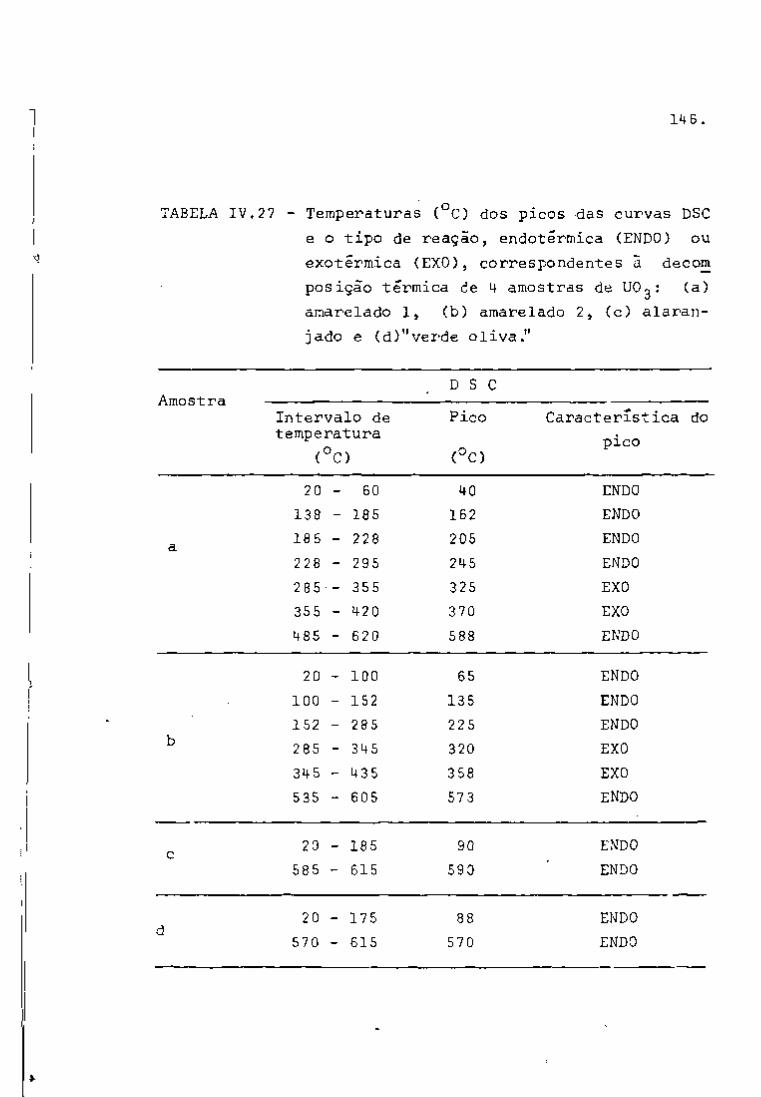
146.
y .
TABELA IV.27 - Temperaturas ( C) dos picos -das curvas DSC
e o tipo de reação, endotérmica (ENDO) ou
exotérmica (EXO), correspondentes a decom
posição térmica de 4 amostras de UO^: (a)
amarelado 1, (b) amarelado 2, (c) alaran
jado e (d)"verde oliva."
D S C Amostra
Intervalo de temperatura
(°C)
Pico
(°C)
Característica do
pico
20 - 60 40 ENDO
138 - 185 162 ENDO
a 185 - 228 205 ENDO
228 - 295 245 ENDO
285 - 355 325 EXO
355 - 420 370 EXO
485 - 620 588 ENDO
20 - 100 65 ENDO
100 - 152 135 ENDO
152 - 285 225 ENDO b 285 - 345 320 EXO
345 - 435 358 EXO
535 - 605 573 ENDO
c 20 - 185 90 ExNDO
585 - 615 590 ENDO
d 20
570
- 175
- 615
88
570
ENDO
ENDO

1U7.
I V . 8 DADOS ANALÍTICOS
Os dados analíticos de uranio, nitrato, a-
mônia e água, determinados de acordo com a descrição em
I I I . 4 , apenas para os diuranatos de amonio representati
vos de cada grupo, estão reunidos na Tabela I V . 28. Esses
dados associados aqueles obtidos a partir das curvas
TG e DTG, apresentados em I V .5.1., possibilitaram a de
terminação da composição e do mecanismo da decomposição
térmica dos diuranatos, apresentados em V .
Na Tabela I V .28 também são apresentados
os valores calculados para as razões molares NOg/U, NH^/U
e H2O/U nos diuranatos.
Os resultados da densidade (solta e bati
da) e superficie específica dos diuranatos, bem como os
valores calculados para a razão O/U nos produtos de cal
cinação desses compostos estão na Tabela I V .29,
Os diuranatos de amonio, 2 - C , 3-C e 12 - C ,
usados no estudo de secagem ( I V . 2 ) e envelhecidos por 1
ano, foram agrupados segundo a classificação em I V \ è tam
bém em função dos parâmetros de secagem:
Grupo I - Compostos secados a 7 5°C, respectivamente,
por 120, 1440 e 2880 minutos.
Grupo I I I - Compostos secados a 150°C, respectivamente,
por 1440, 2880 e 4320 minutos.
Grupo IV - Compostos secados a 150°C por.120 minutos.
Na Tabela I V . 30 estão os dados analíticos obtidos para u-
ránio, nitrato, amónia e água e os valores calculados pa

148.
tos. Os resultados da densidade (solta e batida) e su-
ra as razões molares NO-ZU, NH, /U e HO/U nos diurana-O H Z .1
perfície específica desses diuranatos estão na Tabela
IV.31, juntamente com os valores calculados para a ra
zão O/U nos produtos de calcinação desses compostos.
Os resultados apresentados nas Tabelas
IV. 3 0 e IV.31 correspondem aos diuranatos secados a TS^C
por 2880 minutos (Grupo I), secados a 150°C por 2880
minutos (Grupo III) e a 150°C por 120 minutos (Gru-
IV) .
Para os trióxidos de urânio também fo
ram determinados os teores de urânio, nitrato e amónia +
e foram calculados os valores das razões NO^/U, NHi /U
e H2O/U. Os resultados estão na Tabela IV.32. Os da
dos da densidade (solta e batida) e superfície especí
fica desses ©"xidos estão na Tabela IV. 33, juntamente
com os valores calculados para a razão O/U nos produtos
de calcinação desses compostos.
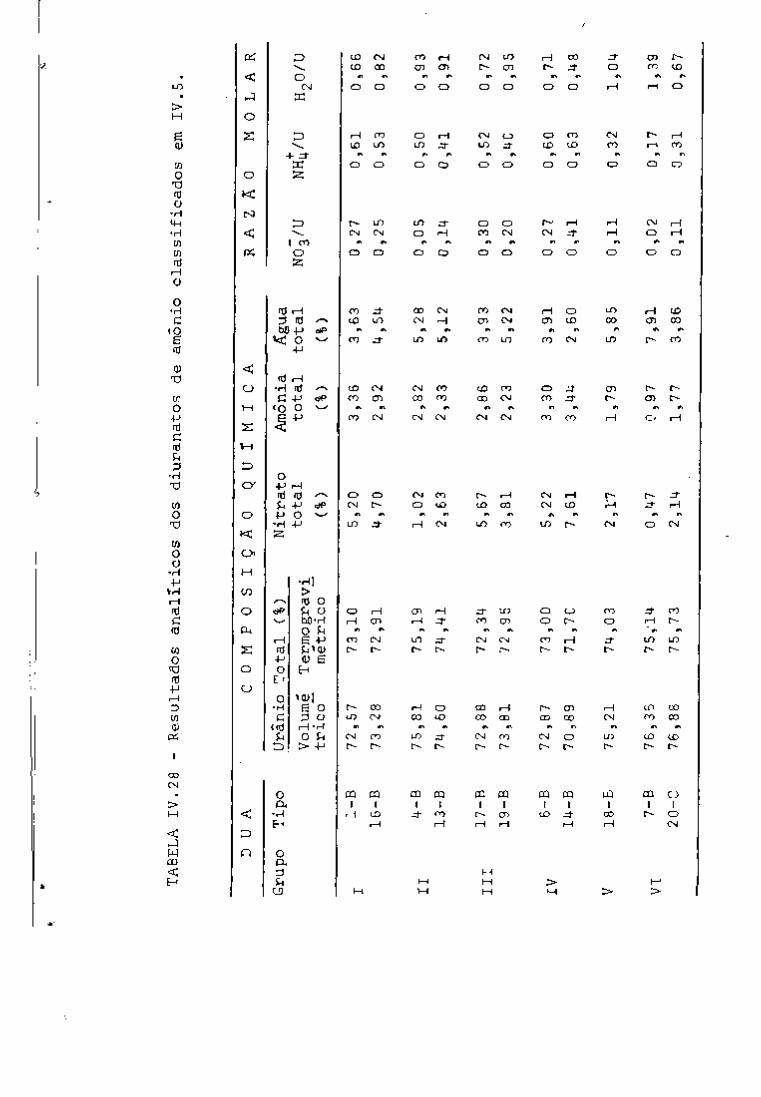
TABELA I
V.2 8 - Resultados a
nalíticos
dos diuranatos d
e amonio c
lass
ific
ados e
m IV
.5.
D U A
C 0 M
P 0 S I CAO
QU
I M
I C A
R A Z
Ä 0
M 0
LAR
Grupo
Tipo
Uranio T
otal (%)
Nitrato
Amón
ia
Agua
Volume
trico
Term
ogra
vi
métr
ico
total
(%)
total
(%)
tota
l (%)
NO3/
U
NH
J/U
H2
O/U
I 1-B
72,57
73 ,10
5,20
3 ,36
3,63
0 ,27
0 ,61
0 ,66
16-B
73,28
72,91
4,70
2,92
4,54
. 0,25
0,53
0,82
II
4-B
75,81
75,19
1,02
2,82
5,28
0,05
0,50
0,93
13-B
74,60
74,41
2,63
2,33
5,12
0 ,14
0,41
0 ,91
III
17-B
72,88
72,34
5,67
2,86
3,93
0 ,30
0 ,52
0,72
19-B
73,81
72,95
3,81
2,23
5,22
0 ,20
0,40
0,95
IV
6-B
72,87
73,00
5,22
3,30
3,91
0,27
0,60
0,71
14-B
70 ,89
71,70
7,61
3,44
2,60
0,41
0 ,63
0 ,48
V 18-E
75,21
74,03
2,17
1,79
5,85
0 ,11
0,32
1,04
VI
7-B
76,36
7 5,-14
0,47
0,97
7 ,91
0,02
0,17
1,39
20-C
76 ,88
75,73
2,14
1,77
3,86
Q, 1
1 0,31
0,67
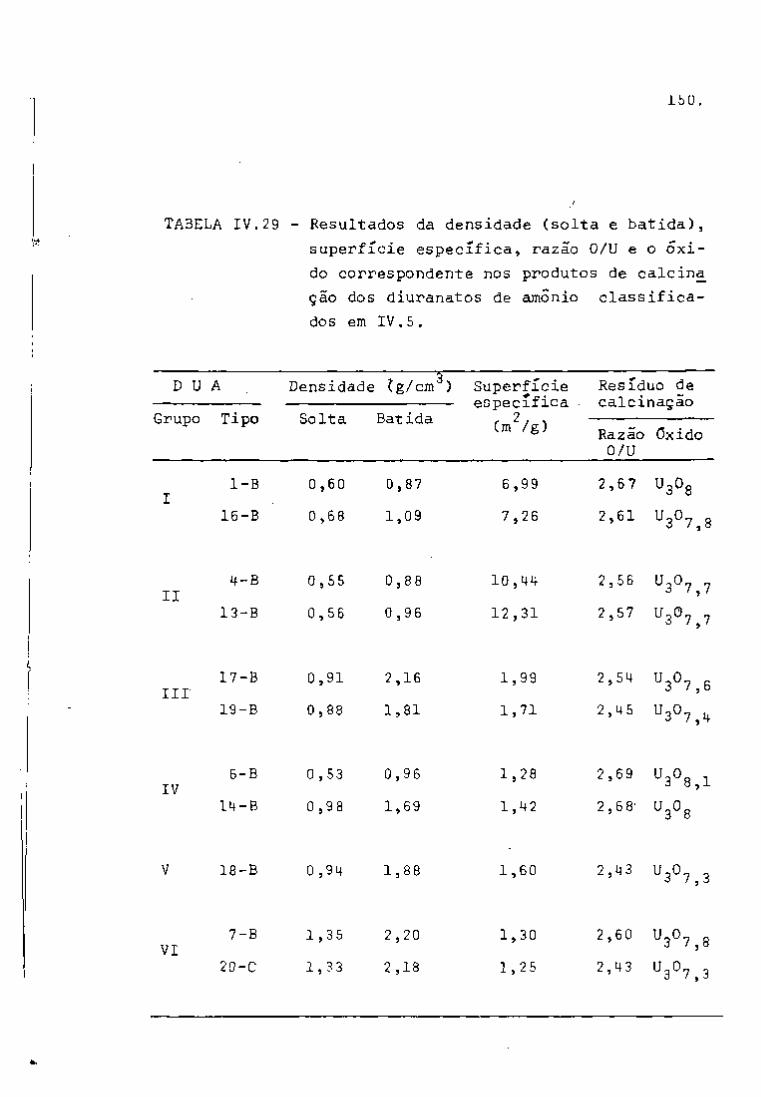
150.
TABELA IV.29 - Resultados da densidade (solta e batida),
superficie específica, razão O/U e o oxi
do correspondente nos produtos de calcina
ção dos diuranatos de amonio classifica
dos em IV.5 .
D U A Densidade tg/cm ) Superficie Residuo de especifica calcinação
Grupo Tipo Solfa Batida (m^/g) Razão Oxido O/U
1-B 0,60 0,87
16-B 0,68 1,09
6,99
7,26
2,67 UgOg
2,61 UgO,^
II 4-B
13-B
0,55
0,56
0,88
0,96
10,44
12,31
2.56 UgO^^^
2.57 UgO^^,
iir 17-B
19-B
0,91
0,88
2,16
1,81
1,99
1,71
UgO^^g
2,45 UgO^^^
IV 6-B
14-B
0,53
0,98
0,96
1,69
1,28
1,42
2'69 UgOg^,
2,68' U3O3
18-B 0,94 1,88 1,60 2,43 UgO^ 3
VI 7-B 1,35 2,20
20-C 1,33 2,18
1,30
1,25
2,60 UgO.^g
2,43 U30,^3
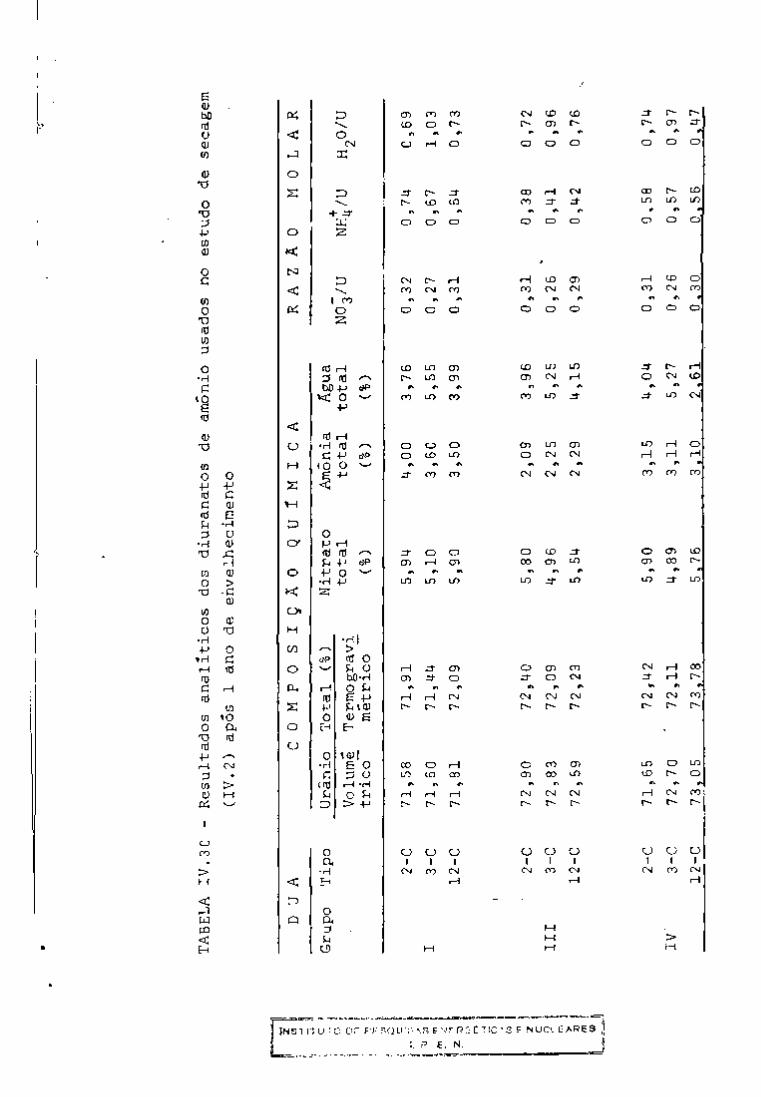
TABE
LA I
V.3
0 -
Resultados a
nalíticos
dos
diur
anat
os d
e amonio u
sado
s no e
studo
de s
ecag
em
(IV.
2) após
1 an
o de e
nvel
heci
ment
o
z o o S I
en
O
c
2 ? co
m Z c p > ííl
CO
D U A
C
0 M
P 0
S I CAO
Q U
I
M I
C A
R A
Z A
0 M
0 LAR
Grupo
Tipo
Uran
io
Tota
l (%)
Nitrato
Amón
ia
Agua
X
Volumé
tric
o Te
rmog
ravi
mé
tric
o to
tal
(%)
total
(%)
tota
l (%)
NO3/
U
NH
J/U
H
^O/U
2-C
7
1,5
8 7
1,9
1 5
,94
4,0
0 3
,76
0,3
2
0,7
4 0
,69
I 3-
C
71
,60
71
,44
5,1
0 3
,60
5,5
5 0
,27
0 ,6
7 1
,03
12
-C
71
,81
7 2
,09
5,9
0 3
,50
3,9
9 0
,31
0 ,5
4 0
,73
2-C
7
2,9
0 7
2,4
0 5
,80
2,0
9 3
,96
0,3
1 0
,38
0 ,7
2
III
3-C
7
2,8
3 7
2,0
9 4
,96
2,2
5 5
,25
0,2
6 0
,41
0 ,9
6
12
-C
72
, 59
7
2,2
3 5
,54
2,2
9 4
,15
0 ,2
9 0
,42
0 ,7
6
2-C
7
1,6
5 7
2,4
2 5
,90
3,1
5 4
,04
0 ,3
1 0
,58
0 ,7
4
IV"
3-C
7
2,7
0 7
2,1
1 4
,89
3,1
1 5
,27
0,2
6 0
,57
0 ,9
7
12
-C
7 3
,05
73
,78
5,7
6 3
,10
2,6
1 0
.30
0,5
6
0 ,4
7 EN
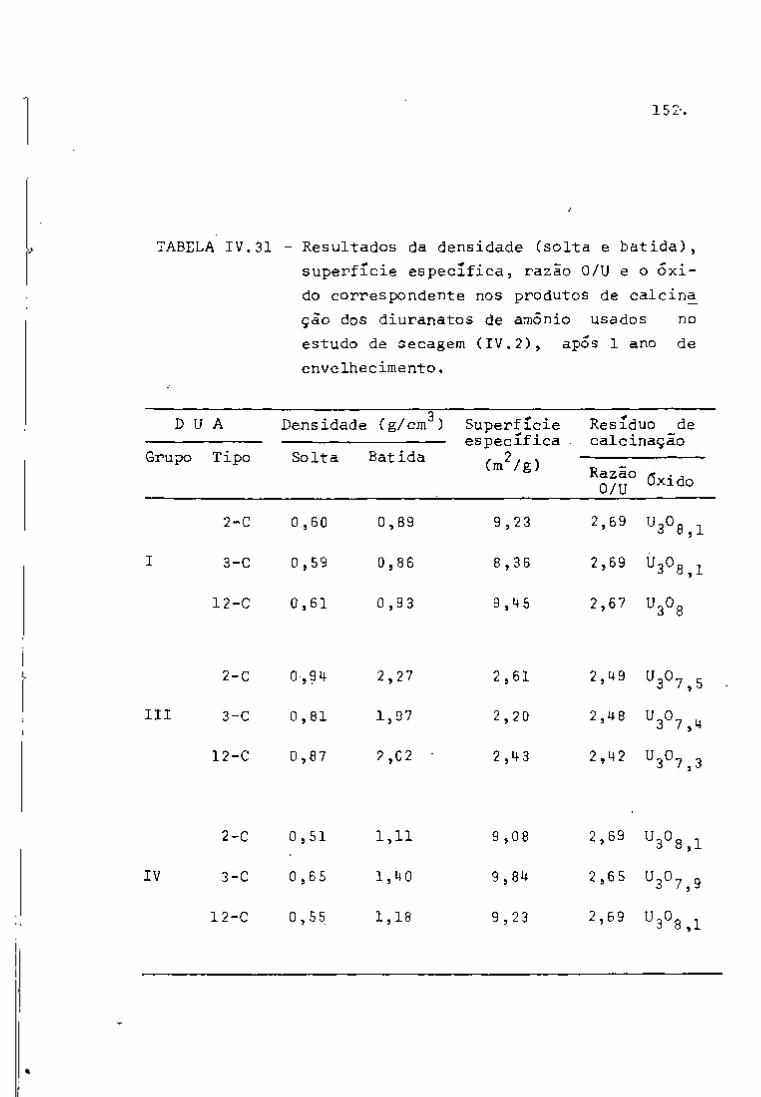
152'.
TABELA IV.31 - Resultados da densidade (solta e batida),
superficie especifica, razão O/U e o oxi
do correspondente nos produtos de calcina
ção dos diuranatos de amonio usados no
estudo de secagem (IV.2), apos 1 ano de
envelhecimento.
D U A Densidade (g/cm^) Superficie especifica
(mVg)
Residuo de calcinação
Grupo Tipo Solta Batida
Superficie especifica
(mVg)
Residuo de calcinação
Grupo Tipo Solta Batida
Superficie especifica
(mVg) Razão O/U óxido
2-C 0,60 0,89 9,23 2,69 "303,1
I 3-C 0,59 0,8 6 . 8,36 2,69 "303,1
12-C 0,61 • 0,93 9,45 2,67 ^ 8
2-C 0',?4 2,27 2,61 2,49 "307,5
III 3-C 0 ,81 1,97 2,20 2,48 "307,4
12-C 0,87 2 ,02 - 2,43 2,42 "307,3
2-C 0,51 1,11 9,08 2,69 "308,1
IV 3-C 0,65 1,40 9 ,84 2,65 "307,9
12-C 0,5 5 1,18 9 ,23 2,69 "308,1
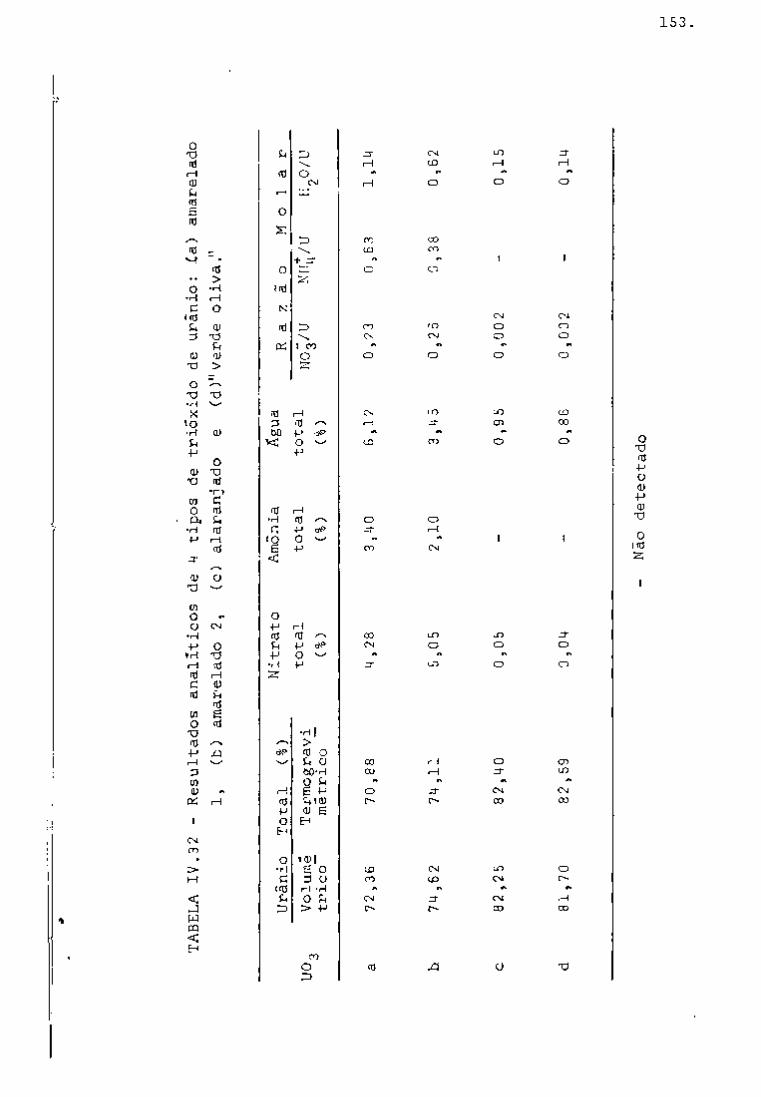
TABELA IV.
3 2 - Resultados analíticos de
4 tipos de trióxido de urânio: ,Ca) amarelado
1,
(b) amarelado
2
, (c) alaranjado
e
(d)"verde oliva."
U03
Urânio
Total
(%)
Nitrato
total
(%)
Amónia
total
(%)
Agua
total
(%.)
R a z ã 0
Mo lar
U03
Volume
trico
Tex^'mogravi
métrico
Nitrato
total
(%)
Amónia
total
(%)
Agua
total
(%.)
NO
¡/U
N
HJ/
U
H^O
/U
a
72
,36
70
,88
4,2
8 3
,40
6 ,1
2 0
,23
0 ,6
3 1
,14
b
7U
,62
74
,11
5,0
5 2
,10
3,4
5 0
,26
0,3
8 0
,62
c
82
,25
82
,40
0,0
5 -
0,9
5 0
,00
2 -
0,1
5
d
81
,70
82
,59
0,0
4 -
0,8
6 0
,00
2 -
•0 ,
14
- Não detectado
cn
CO
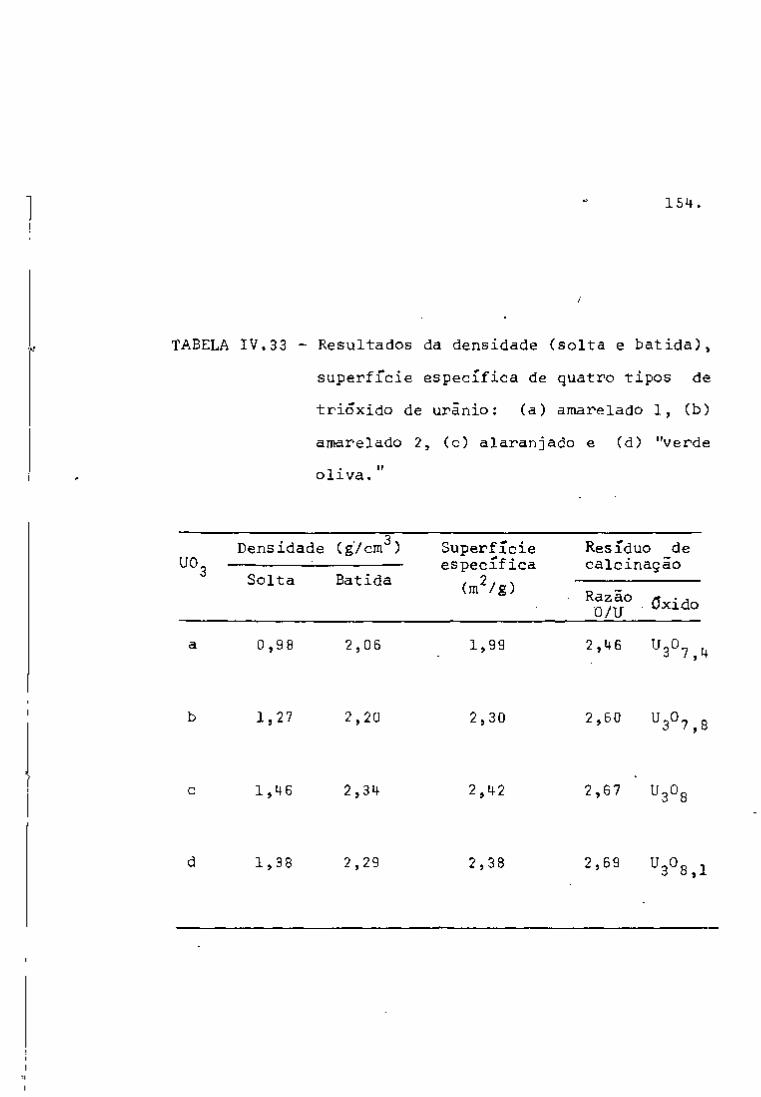
154.
TABELA IV.33 - Resultados da densidade (solta e batida),
superfície específica de quatro tipos de
trióxido de urânio: (a) amarelado 1, (b)
amarelado 2, (c) alaranjado e (d) "verde
olxva.
3 ^ *• Densidade (g/cm ) Superfície Resíduo de
U0„ • ' específica calcinação Solta Batida . 2 (m /g) , 5° Oxide
0,98 2,06 1,99 . 2,46 UgO^ ^
1,27 2,20 2,30 2,60 UgO^ g
1,46 2,34 2,42 2,67 UgOg
1,38 2,29 2,38 2,69 UgOg ^

155.
V. DISCUSSÃO E CONCLUSÕES
De acordo com a proposição inicial e pro
cedimento experimental realizado, este trabalho teve
basicamente duas finalidades:
a) conhecer de modo geral a decomposição térmica
do DUA e UOg, produzidos no IPENj por meio
de técnicas termoanalíticas (TG, DTG e DSC)
e,
b) correlacionar os dados da termodecomposição
com os parâmetros de precipitação e secagem,
no caso do DUA, e de calcinação no caso do
UO3.
O objetivo comum desses estudos foi o de estabelecer um
método eficiente, simples e rápido de caracterização de£
ses compostos por termogravimetria e calorimetria explo
ratoria diferencial.
V.l - EFEITO DA SECAGEM SOBRE O COMPORTAMENTO TÉRMICO
E COMPOSIÇÃO DO DIURANATO DE AMÕNIO
Como foi visto, no estudo da secagem em
IV.2, o procedimento experimental executado neste tra
balho consistiu de duas experiências: numa delas os com
postos foram aquecidos em termobalança sob fluxo de
ar, e na outra foram secados em estufa comum de laborato
rio.

156.
De acordo com consultas realizadas na li-
teratura, uma das técnicas comumente adotada para evi
tar a mudança na composição do DUA é a de proceder-se
sua secagem ao ar, apos lavagem com álcool etílico ou
acetona, ã temperatura ambiente e depois coloca-lo em
recipiente contendo ^ 2 ^ ^ ^ concentrado ou P2O5' ' ^ ' ^
(15 7 6) sua massa permaneça constante '
Traços de água livre geralmente não podem
ser removidos ã temperatura ambiente. Outra maneira de
se evitar a mudança na composição do DUA ê aquecê-lo a
uma temperatura .mais elevada, a fim de não diminuir o tem
po de secagem, pois ã temperatura ambiente, mesmo por
um tempo de 10200 minutos, a secagem não e eficien
te KRTiL e colaboradores concluíram que 50°C era
a temperatura õtima desse aquecimento, por um período de
tempo variável, até 480 minutos, dependendo da quantida
de de material.
Em outros trabalhos^^5 ,40,45,73,74,75)
autores usando temperaturas de secagem mais elevadas
(100°C,150°C e 200°C) verificaram que as razões molares
N H | ^ / U e H2O / U (água de cristalização e composição) di
minuíam. Portanto, secagem em altas temperaturas provoca
alterações na composição química do diuranato de amônio.
Os resultados apresentados em IV.2 mos
tram que o numero de reações envolvidas na decomposição
térmica dos diuranatos vcriou em função da temperatura e
do tempo de secagem, modificando o comportamento térmico
desses compostos. Do mesmo modo, os dados analíticos a-
presentados na Tabela IV.3O mostram que o teor de amô-

15?.
nia diminuiu com o aumento da temperatura e tempo de
secagem: Grupo I (75°C/2880 minutos) > Grupo IV (150°C/
/120 minutos) > Grupo III (150°C/2880 minutos). Em re
lação aos dados da Tabela IV.31 verificou-se apenas que
a razão O/U nos produtos de calcinação aumentou para va
lores mais altos da densidade e superfície especifica
dos diuranatos,
Alem das informações apresentadas em
IV.1.4, não se conheciamdados mais concretos para a e£
colha da temperatura e do tempo de secagem, bem como
as características dos diuranatos (usados neste traba
lho) e o seu comportamento térmico durante a secagem.
Assim, foi necessário conhecer bem a decomposição tér
mica desses diuranatos secados em diferentes condições,
a fim de que fosse escolhida a temperatura na qual es
ses compostos atingissem massa constante, sem altera
ção de sua estrutura.
Os dados apresentados em IV.2 permitem
concluir que 7 5°C é a melhor temperatura de secagem dos
diuranatos de amónio, num intervalo de tempo entre 40
e 24 0 minutos,• dependendo da quantidade e altura da ca
mada do precipitado.
As diferenças no comportamento térmico
assinaladas em IV.2.2, entre os diuranatos nas formas
maciça e pó, derivam principalmente, do fato de que
durante o aquecimento os gases liberados difundem
mais rapidamente através da amostra (de dentro para fo
ra), quando esta se encontra na forma de pó, do que
quando está na forma maciça.

158 .
As demais observações, em relação ao com
portamento térmico, foram atribuídas ao tipo de mate
rial, ou seja, aos parâmetros de precipitação.
y.2 - EFEITO DA CALCINAÇÃO SOBRE O COMPORTAMENTO
TÉRMICO E COMPOSIÇÃO DO TRIÕXIDO DE URÂNIO
Em II.2 apresentaram algumas possibili
dades para a produção de UO^, entre as quais está a
sua formação a partir da decomposição térmica do diura
nato de amônio, descrita com mais detalhes em II.3.1.
Os resultados apresentados em IV.7 (Figu
ras IV.25, IV.26 e IV.27 e Tabelas IV.26 e IV.27) e
os dados analíticos da Tabela IV.32 revelaram que os
trióxidos de urânio de cor amarelada tem composição e
comportamento térmico semelhantes aos dos diuranatos de
amónio. Os compostos de cores alaranjada e verde oliva,
entretanto, apresentaram composição e comportamento
- . . . ('+6 térmico iguais aos descritos na literatura para UOg '
48,58,64,65,75)
•
Uma vez que a temperatura e o tempo de
calcinação foram iguais para todos os óxidos, pode-se
concluir que as frações amareladas não sofreram decom
posição, talvez devido ao excesso de material (DUA) a-
cumulado em certos pontos do forno de calcinação. Por
tanto, para se ter uma transformação completa do DUA em
UOg, recomenda-se que o diuranato seja colocado no for
no de tal forma que a altura da camada e a quantida
de de amostra permitam um aquecimento uniforme em toda
a sua extensão.

1 5 9 .
Em relação ã decomposição térmica destes
óxidos, verificou-se que hã variação na 'estabilidade
térmica tanto dos trióxidos de urânio, retirados dire
tamente da linha de produção, como também, daqueles
observados nas curvas termoanalíticas dos diuranatos de
amonio.
Alguns trabalhos assinalam que a estabili
dade térmica do UO^ esta relacionada ãs diferentes fa
ses cristalográficas por ele assumidas e aumentam na
seguinte o r d e m ^ ^ ^ , 4 8 , 5 5 , 5 8 , 6 4 , 6 5 , 7 7 ) ^ ^ ^ ^ ^ ^^^^o^^ ,
( 4 6 0 ° C ) < amorfo ( 5 0 0 ° C ) < beta ( 5 3 0 ° C ) < U0„ q ( 5 8 0 ° C ) <
< delta ( 5 9 0 ° C ) < gama ( 6 3 0 ° C ) . As fases alfa e amorfa
tem o g como intermediário, enquanto a fase eta
tem o -j . Os demais UOg decompõem-se diretamente
para UO^^g^ (U3O3).
Ass-im., pelos resultados apresentados po
de-se observar as seguintes fases cristalográficas:
- U0„ amorfo ( 5 0 0 ° C ) e U0_ „ ( 5 9 0 ° C ) no óxido ama-
relado 1 .
- Beta-UOg ( 5 4 0 ° C ) e UO^ g ( 5 7 8 ° C ) no óxido amare
lado 2 .
- Delta-UOg ( 5 9 5 ° C ) no óxido alaranjado.
- UO^ g ( 5 7 5 ° C ) no óxido verde oliva.
Os valores para a razão O/U e o tipo de
óxido correspondente, apresentados na Tabela IV. 3 3 , pa
ra o resíduo de calcinação do trióxido de urânio de
cor verde oliva não estão corretos pois, foram calcula-
,N8TlTUTO D E P E SQU S AS E N t É - I C - S f. N U C L E A R E S
I- P - E . N. _ _ _ _ _ _

160.
dos a partir da massa de óxido encontrada na curva TG,
considerando-o estequiometricamente igual ao UO^ (O/U =
= 3). Refazendo os cálculos em relação ao óxido com a
composição do UO2 g, o residuo pode ser representado
pela fórmula U„0„ _ cuja razão O/U = 2,60. o / , o
v.3 - EFEITO DO TEMPO DE ENVELHECIMENTO SOBRE O
COMPORTAMENTO TÉRMICO DO DIURANATO DE A M O N I O
Conforme assinalado em IV.3, a decomposi
ção térmica do diuranato de amonio apresentou várias
mudanças em relação ao tempo de envelhecimento, algu
mas independentes e outras associadas aos parâmetros
de precipitação e secagem.
Entre outras observações já feitas, pode-
se destacar como. mais importante, a mudança na estrutu
ra e composição química do D U A , retratada nas curvas
termoanalíticas em termos do número de reações e teores
de gases liberados. Uma comparação dos dados apresen
tados no Apêndice 2 com aqueles do Apêndice 3, permite
concluir que o tempo de envelhecimento provoca altera
ções profundas na estequiometria da decomposição tér
mica do D U A .
V.4 - EFEITO DO NITRATO NA DECOMPOSIÇÃO TÉRMICA DO
DIURANATO D E .AMÕNIO
A presença de nitrato no D U A pode ser ex
plicada pela oclusão desse ânion, durante a reação de
precipitação. Esta oclusão é favorecida em altos valo

(215 res de pH e velocidades de precipitação rápidas » '
43 , 63, 69 , 74, 85, 86)^ \ ^ g^^^, g , ^¿ ^.^^^^^
sente no DUA pode ser eliminado com agua, porém, parece
ser impossível eliminá-lo totalmente na etapa de lava-
(15, 43) gem '
Os diuranatos usados neste trabalho apre
sentaram um teor de nitrato bas-tante variável, no ínter
valo de 0,47% (7-B) a 7,61% (14-B), de acordo com os
dados analíticos das Tabelas IV.28 e IV.30.
As informações da literatura, em relação
á decomposição térmica do DUA, apresentadas em II.3.1
permitem interpretar a curva DSC (a) da Figura IV. 18, on
de:
- os tres picos endotérmicos em 88°C, 130°C e 156°C
foram relacionados ã perda de água livre e de
cristalização;
- o pico exotérmico em 412°C caracteriza a reação
de auto-redução do UO3, em atmosfera inerte (N2),
provocada pelo craqueio da amonia retida no inte
rior do óxido;
- a decomposição térmica do óxido intermediário(UOg)
ocorre por meio de uma reação endotérmica em 530°C.
Nas demais curvas (DSC (b), (c) e (d)) da mesma figura
pode-se observar varias mudanças provocadas pelo aumen
to da porcentagem de nitrato de amonio no DUA.
Pelos resultados apresentados em IV. 5, po
de-se concluir que:

A perda de massa observada nas curvas TG, no ínter
valo de temperatura de 200°C a 3 20°C, pico DTG em
torno de 275°C, corresponde principalmente i rea
ção de decomposição do nitrato ocluido. Na curva
DSC esta reação ocorre em temperaturas mais altas,
pico DSC em torno de 330°C (curvas DSC (c) e (d)
da Figura IV,18).
A reação de auto-redução do UO3, pelo craqueio da
amónia retida (~410°C) pode ser inibida pelo au
mento do teor de nitrato ocluido ,pois observou-se
diminuição na porcentagem de amónia retida e na
temperatura desta reação (412°C para 372°C).
Em atmosfera inerte, o aum.ento no teor de nitrato
de amonio favorece a decomposição térmica do UO3
em temperaturas mais baixas.
O pico endotérmico em 20 8°C está associado ao au
mento da porcentagem de nitrato de amonio no • DUA
5%). EL-FEKEY ^24,25)^ ^ trabalhos recentes,
verificou que um pico endotérmico na curva DTA do
DUA, em 210°C, representava, principalmente, a
decomposição do nitrato de amonio.

X D O .
V.5 - MECANISMO GERAL DA DECOMPOSIÇÃO TÉRMICA DOS
DIURANATOS DE A M O N I O
Uma vez conhecidos os teores de todos os
constituintes dos diuranatos, foram calculadas as ra
zões molares NO^/U, N H | J ^ / U e H^O/U e os resultados se en
centram nas Tabelas IV.28 e IV.30.
Com o conhecimento do histórico e das
condições nas quais foram obtidos esses diuranatos, nos
resultados do estudo de sua decomposição térmica e nos
dados das Tabelas IV.28 e IV.30, que dão a composição
química desses materiais, procurou-se tirar algumas con
clusoes,
Os diuranatos de amonio, portanto, são
compostos de estequiometria variável, que podem ser re
presentados pela fórmula geral yUOg.xNH2.zH2O, onde as
relações NH^/UO^ e H2O/UO2variam, principalmente, em
função do tratamento de secagem e do envelhecimento.
A fórmula geral,sugerida acima para re
presentar esses diuranatos de amonio produzidos pelo
processo de precipitação em batelada ou contínuo, a par
tir de solução de nitrato de uranilo, com amónia gaso
sa, está de acordo com os resultados obtidos por BALL
DEANE FODOR ^^^^ e TURCANU ^^^'^^^ Estes
autores concluir'am que uma fórmula real não pode ser es_
tabelecida para esses compostos, pois a sua composição
varia durante as etapas de lavagem e secagem.
Os resultados termoanaliticos dos diura
natos de amonio usados neste trabalho são bastante se-

164.
melhantes àqueles encontrados na literatura. Portanto,
pela associação das informações jã publicadas e descri
tas em II.3.1 com esses resultados, pode-se estabele
cer um mecanismo geral para representar a decomposição
térmica desses compostos.
De um modo geral, as curvas TG dos diura
natos mostraram perda de massa contínua até a temperatu
ra de 4 50°C, O aquecimento desses compostos a tempera
turas mais elevadas, 500°C e 800°C, produzem óxidos de
urânio, UO^ ou misturas UO3 + U^Og e U^Og ou U20g_^,
respectivamente. A decomposição térmica dos diurana
tos de amônio é caracterizada então, pelas reações
observadas no intervalo de temperatura entre 20°C e
450°C.
Nesse intervalo de temperatura, as cur
vas DSC mostraram reações endotérmicas até 29 0°C e rea
ções exotérmicas de 290°C a 450°C.
A observação em experimentos na termoba
lança, com o aquecimento dos vários tipos de DUA até
80°C, 180°C, 220°C, 300°C, 350°C e 450°C, e, posterior
determinação dos teores de amónia e nitrato, revelou
que a perda de amonia inicia em temperaturas superiores
a 180°C e cessa em tomo de 400°C; entre 220°C e 350°C
observou-se a decomposição do nitrato.
As reações exotérmicas presentes nas cur
vas DSC desses compostos em torno de 330°C e 350°C ou
39 0°C foram atribuídas, respectivamente, ã decomposi
ção do nitrato e oxidação da amonia retida. Observações

165.
(17 18
semelhantes foram feitas por outros pesquisadores ' '
43,56,57,61,62,74,76) ^^.^ curvas^TG e DTA, pa ra as reações em 250°C a 350°C e 350°C a 450°C.
Com base nessas informações, nos resulta
dos analíticos e nos cálculos feitos a partir das cur
vas TG, a perda que ocorre na primeira reação (20°C a
8 0 ou 110°C) corresponde ã liberação de água livre, en
quanto, na segunda (80°C a .160 ou 180°C) resulta da
liberação da. água de cristalização e, na terceira
(160°C a 220: ou 240°C) e relacionada ã perda simul
tânea de água de cristalização e amonia.
Essas observações foram também feitas
(39 63 70 7 3)
por alguns pesquisadores ^ ' ' » ' a partir dos re
sultados termoanaliticos, associados aos de raios-X e
infravermelho, dos diuranatos de amonio por eles prepa
rados, que mo.stram a reação de desidratação ocorrendo
entre 20 e 2 20°C, em duas ou três etapas sem liberação ^ C 4 2 U 3 6 6 6 8 ) de amonia. Outros ^ ' » ' verificaram que esse
processo pode ocorrer em duas ou três etapas, porém com
liberação simultânea de amonia absorvida, que se dá
entre 12 0 e 16 0°C.
A decomposição térmica desses diuranatos
caracterizada pela perda de água, amonia e nitrato,o-
correu por meio de cinco ou seis reações sobrepostas,re
sultando na formação de trióxido de urânio. Entretanto,
pode-se considerar que a formação de UO^ a partir de
DUA se dá, basicamente, através de quatro estádios:

166.
(1) processo endotérmico de perda de água livre
(20°C a 80 ou 110°C);
(2) processo endotérmico de perda de água de cris_
talização e amonia (80°C a 180 ou 220°C) e
de água de composição, amonia e algum nitrato
(180°C a 270°C);
(3) processo exotérmico de decomposição de nitra
to, associada á perda de amonia (27 0°C a
350°C);
(U) processo exotérmico de perda de amonia, por
reação de oxidação (3 50°C a 4 5 0°C).
Em condições ambientes,a maioria dos diu
ranatos é higroscópica, o que é indicado pelo fato de
as curvas TG e DTG iniciarem,respectivamente, com uma
inflexão e um "pico que se estendem até 8 0 ou 110°C, mos_
trando perda de água em temperaturas abaixo daquela na
qual os diuranatos foram secados.
A interpretação das curvas TG e DTG,asso
ciada as informações já apresentadas, sugere que um
mecanismo da reação de decomposição desses diuranatos
pode ser representado pelas seguintes equações:

IbV .
- Diuranatos do Grupo I,
UO3 . OjSSNHg . 0,82H2O (0,25 NO^)^ estádio II (80 - 220°C)
UO 3 . 0,i+6NH3 . 0,04H2O (0,25 NO3) estádio III (220 - 313°C)
ÜO3 . 0,37NH3 estádio IV (313°C - 4U0°C)
+
UO3 ^ (545°C)
UO 2,9 0
^ (589°C) UO
2,67 Diuranatos do Grupo II,
UO3 . 0,41NH3 . 0,91H20 (0,14 NO") estádio II (80 - 210°C)
+ UOg . 0,34NH3 . 0,14H20 (0,14 NO")
estádio III (210 - 298°C) + .
UO3 . 0,26NH3 . 0,07 H2O estádio IV (298 - 389°C)
UO, 3 (5it5°C)
(682°C)

168.
Diuranatos do Grupo III,
UOg . 0,29NHg
estadio IV (302 - 463°C)
(490°C)
UO 2,8 0
^ (594°C) "°2,54
- Diuranatos do Grupo IV,
UO 3 . 0,63NH3 . 0,48H2O (0,41 NO")
estadio II " (110 - 232°C)
UOg . 0,36NH3 (0,41 NO3)
estadio III (232 - 300°C)
4-UOg . 0,36 NHg
estadio IV (300 - 485°C)
UO3 (605°C)
UO 2,68
UOg . 0,52NHg . 0,72H2O (0,30 NO^)'
estadio II (115 - 240°C) +
UOg . 0,29NH3 ^°'30 NOg)
estadio III (240 - 302°C)

169.
Diuranatos do Grupo V,
UO3 . 0,32NH3 . l,04H2O (0,11 N0~)/
estadio II (70 - 186°C)
4.
UO3 . 0,32NH3 . 0,10H2O (0,11 N0~)
estadio III (186 - 276°C)
4-
UO3 . 0,15NH3 (0,10 NO3)
estadio IV (276 - 437°C)
4-
UO3
(538°C)
"I' UO
2,80
UO
^ (574°C)
2,43
Diuranatos do Grüifo :VI,
UO3 . 0,31NH3 . 0,67H2O (0,11 N0¡)
estadio II (85 - 162°C)
•I-
UO3 . 0,31NH3 . 0,08H2O (0,11 NO3)
estadio III (162 - 256°C)
UO 3 . 0,17NH3 (0,11 NO3)
estádio IV (256 - 441°C)
4-UO 3
(542°C)
4-
UO 2,90
(570°C) 4-
UO 2,43

.17 0.
Um mecanismo semelhante poderá ser estabe
belecido também, para a reação de decomposição térmica
dos diuranatos de amônio usados no estudo da secagem
(2-C, 3-C e 12-C), cujos dados foram apresentados em
IV.2, Tabelas IV.30 e IV.31 e nos Apêndices 2 e 3.
A variação na estequiometria da decompôsi
ção térmica dos diuranatos de amônio, observada nos mo
delos acima, mostra que a composição desses compostos
muda em função das condições de preparação. A alteração
nos estádios da decomposição é um reflexo dos teores de
água, amónia e nitrato, que diferem de amostra para
mostra.
Considerando seu uso na produção seqüen
cial de óxidos de urânio, verificou-se que dependendo
do tipo de DUA obtém-se UO^, cuja decomposição pode o-
correr entre 49 0°C e 630°C. Os óxidos de urânio obtidos
nesse intervalo de temperatura apresentaram composição
variável, retratada pela razão O/U que variou de 2,90 a
2,4 2 e pode estar relacionada ã variação de cores apre
sentadas para o aquecimento destes compostos na Tabela
IV.1.
A não-estequiometria dos produtos de cal
cinação do DUA, observada neste ti"'abalho, pode estar re
lacionada ã reação de auto-redução do óxido intermediá
rio, provocada pelo craqueio da amónia retida na rede
cristalina do UO . -Apesar da atmosfera oxidante (ar),
de algum modo, esta reação pode ocorrer.
Alguns autores ^ ' ' ^ verificaram esta
possibilidade e afirmaram que a extensão dessa reação
iU I N S T I T U T O D E P E S Q U I P . A S I G N E R C É T i C 3 f NUCLSARSo'1
I. P . E . N .

171.
depende das características físico-químicas do DUA e j
das condições de aquecimento: em baixas velocidades de
aquecimento (5°C/min.) praticamente toda a amonia reti
da é consumida pela reação de oxidação com o oxigênio
do ar; em velocidades de aquecimento mais altas (50°C/
min.), 27% do uranio no DUA pode ser reduzido a U-IV;
a velocidade da reação de auto-redução ê da ordem de
10 vezes maior do que da reação de oxidação. Também, a
ausência de agua na atmosfera de decomposição favorece
a reação de auto-redução. Num ambiente úmido esta rea
ção é inibida pois, a agua promove a liberação da amo
nia na forma gasosa.
Os resultados e conclusões apresentadas,
respectivamente, em IV.5 e V.4 mostraram que a quanti
dade de amonia retida no oxido intermediario não era
proporcional ã sua concentração inicial no DUA e, dimi
nuía para teores mais altos de nitrato de amonio. Estes
(63)
resultados concordam com aqueles obtidos por PRICE
que explicou o efeito do nitrato de amonio na diminui
ção da porcentagem de amónia retida: nos gases da de
composição do nitrato de amonio tem água e oxigênio; a
água provoca maior liberação de a m o n i a e o oxigênio con
some amonia pela reação de oxidação.

172.
V.6 - CONCLUSÃO GERAL
As técnicas termoanalíticas quando empre
gadas na caracterização de compostos, sem a associação
com outros métodos analíticos, em geral, fornecem in
formações ambíguas para o mecanismo e estequiometria
do processo de decomposição térmica. Essa ambigüidade
é mais acentuada quando a decomposição é complexa e
composta por reações sucessivas e sobrepostas (libera
ção de dois ou mais produtos voláteis no mesmo interva
Io de temperatura), como é o caso do diuranato de amo
nio .
Embora as curvas TG e DTG tornem possí
vel a avaliação quantitativa da variação de massa, e
as curvas DSC o tipo de processo envolvido, endotérmi
co ou exotérmico, elas não podem fornecer informações
separadas das perdas de massa sobrepostas.
Na decomposição térmica do DNA, por exem
pio, ocorre perda simultanea de água, amonia e nitrato,
no intervalo de temperatura, de 200°C a 350°C; sem as
informações complementares obtidas pelas técnicas volu
métricas e espectrofotométrica, utilizadas neste traba
lho, seria impossível a determinação dessas substâncias.
Em relação aos resultados, obtidos por
termogravimetria, para o teor de urânio nos diuranatos
e trióxidos de urânio, apresentados nas tabelas IV.28,
IV.30 e IV.32, pode-se afirmar que são compatíveis com
aqueles obtidos pelo método volumétrico.
Por se tratar de um método gravimetrico,
a determinação de ur ãnio nesses compostos por termogra
vimetria poderá apresentar alguma imprecisão, quando

173.
ao erro da técnica se somarem erros provenientes de
contaminantes metálicos, não voláteis, presentes nas
amostras.
Mesmo com essa limitação, os resultados
mostraram que a termogravimetria pode ser usada na de
terminação da porcentagem de uranio no DUA e UO3 nu
clearmente puros, visto que os teores de impurezas me
tálicos nesses compostos são extremamente baixos, da
ordem de 0 , 0 0 01%.
Entretanto, o emprego da termogravime
tria com essa finalidade ficaria condicionada, quando
necessário,ã introdução de uma correção por um fator
gravimetrico, para as possíveis impurezas metálicas
presentes, de acordo com a descrição no Apêndice 1 . 1
Durante a execução deste trabalho ve
rificou-se com sucesso .que a termogravimetria e a ca
lorimetria exploratoria diferencial podem ser aplica
das também, na caracterização de outros compostos,
tais como o nitrato de uranilo, tricarbonato de urani
lo e amônio, peróxido de urânio, nitrato e sulfato de
zircônio.
A caracterização completa do DUA e UO^
poderá ser concluída em trabalhos futuros, de forma a
se conhecer o tipo de estrutura, grau de cristalinida
de e tamanho de grãos por meio de estudos com difra
ção de raios-X e microscopia eletrônica.
Em resumo, os resultados obtidos neste
trabalho demonstram que essas técnicas (TG, DTG, e

174.
DSC) podem ser aplicadas satisfatoriamente no acompanha
mento da produção de compostos de urânio.
Nota-se aqui a grande vahtagem de se tra
balhar com os métodos termoanaliticos pois, ao serem
usados simultaneamente com outras técnicas que possibili
tem a identificação de substâncias, por exemplo, a cro
matografia ga:sosa ou espectrometria de massa, além da
determinação simultânea de todos os constituintes, numa
única operação, pode-se obter informações sobre a esta
bilidade térmica dos compostos,
Desde que se mantenham condições pré-de-
terminadas para sua utilização, por exemplo, massa de
amostra, razão de aquecimento, fluxo e tipo de atmosfe
ra, o uso dessas técnicas é simples e rápido e o seu
estudo merece ser ampliado para possibilitar a sua a-
plicação em outros tipos de amostras consistindo uma
ferramenta analítica valiosa para acompanhar as diver
sas fases do processamento de urânio.

A P Ê N D I C E 1
C A L C U L O D O T E O R D E U R A N I O E M Õ X I D O S D E U R A N I O P O R
T E R M O G R A V I M E T R I A

APÊNDICE 1
Cálculo da perda de massa a partir das curvas TG " ^
A decomposição térmica dos diuranatos de
amônio envolve várias reações com liberação, de gases:
água, amonia, gases nitrosos e oxigênio. As curvas TG
desses compostos mostraram transformações térmicas com
plexas e superpostas em certos intervalos de temperatu
ra. A ausência de patamares definidos dificultou a me
dida das perdas de massa diretamente das curvas TG. En-
tratanto, a melhor resolução dos efeitos térmicos (pi
cos) das curvas DTG, permitiu determinar as temperatu
ras e intervalo de temperatura para cada reação. O cál
culo das perdas de massa e, posteriormente, da massa
do produto formado após cada reação foram executados oon
forme ilustração da Figura 1.1.
Determinou-se a massa do diuranato no i-
nicio da operação (M^^), no próprio modulo de termogra
vimetria, e, a massa do residuo (M^) foi obtida dire
tamente a partir do desnível observado entre o início
e o fim da curva.
Os diuranatos de amônio ou trióxido de
urânio quando aquecidos ao ar, em temperaturas superio
res a 600°C, produzem óxidos de fórmula U^Og; conhe
cendo-se a massa da tomada de ensaio do composto em
análise e a natureza do produto final (UgOg), pode-se
calcular a perda ocorrida durante a decomposição térmi
I N S T I T U T O D E P E S O U .S A S E E R O : É T IC • C E N U C L t A R E a
I. P. _ _ _ _

ca, bem como a porcentagem de óxido final obtido pela
decomposição térmica do diuranato.
Determinada a massa do produto final e
conhecida sua composição, pode-se calcular a massa to
tal de urânio nesse resíduo, que é a mesma no diurana
to ou trióxido de urânio, pois, durante a decomposição
térmica não há perdas de urânio. A Figura 1.1 ilustra
a forma de calcular as variaçóes de massa a partir das
curvas TG e DTG, . de acordo com a descrição acima.
cálcalo da razão O/U no resíduo de calcinação do diura
nato de amônio ou trióxido de urânio a partir das Curvas
TG ^ ' ^
A razão O/U pode ser calculada a partir
das perdas de massa das curvas TG de acordo com as in
formações descritas no item anterior, admitindo-se que
as fases UO^ e U - O Q O U U ^ O ^ são representadas pelos d O O O O —X
patamares em torno de 520°C e 650°C, respectivamente.
A decomposição do intermediário (UO^) o-
corre por meio da reação UO^ — UO + xO onde, para
composição estequiométrica, ou seja, razão O/U = 2,667,
28 6,03 mg de UO^ perdem 5,33 mg de oxigênio para dar
X = 0,333. Assim, por exemplo, numa amostra de UO^ cu
ja massa, 24,375 mg, perde 0,450 mg de oxigênio, tem-se
X = (_-L.AÍ2_) . ( 286,030 ) ^ (Q^333j ^ ^^33^^
5,330 24,375
Portanto, a composição do produto formado será:
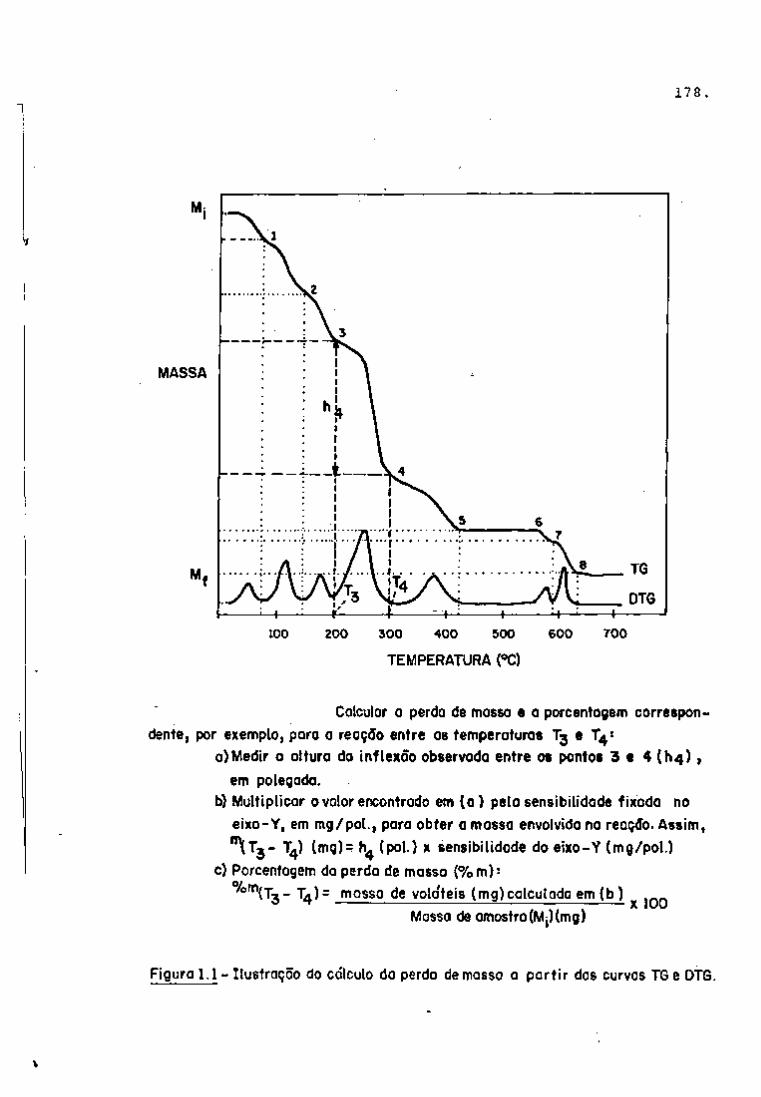
178.
M;
MASSA
M.
100 200 300 400 500 600
TEMPERATURA {°C)
700
Coicutor o perda de mosso e o porcentagem correspon
dente, por exemplo, poro o reação entre os temperaturas T3 e T4:
o)Medir o altura do inf lexão observodo entre os pontos 3 e 4 ( h 4 ) ,
em polegodo.
b) Multiplicar o volor encontrado em ( o ) pelo sensibilidade fixado no
e ixo -Y , em m g / p o l . , poro obter o mossa envolvida no reação. Ass im,
"XTj- T4) ( m g ) = (pol . ) x sensibilidode do e ixo-Y (mg /po l . )
c) Porcentagem da perda de mossa ( % m ) :
7 o m ^ - T4) = mosso de voláteis (mg) calculado em ( b ) ^
Mossa de amostro (M|)(mg)
Figura 1.1 - Ilustroçõo do cálculo do perda de mossa o part i r das curvas TG e DTG.

l/a .
cálculo para correção de impurezas metálicas na deter
minação gravimétrica de urânio em õxidos de alta pure-
za
Cxidos de urânio de alta pureza sâo calcina
dos a 900°C ±25°C em atmosfera de oxigênio até massa
constante. Apôs resfriamento, a massa do resíduo de
calcinação (UgOg) é determinada em balança analítica.
Visto que esse resíduo não é totalmente for
iriado por U^Og mas, contém impurezas não voláteis, é
necessário que se façamalgumas correções, admitindo-se
que estas impurezas estão presentes na forma de õxidos
metálicos. Se o teor de cada impureza metálica for me
nor do que 70 ppm, o efeito das impurezas pode ser de£
prezado, fazendo-se uma correção padrão de 0,01% no
teor total de urânio na amostra. Entretanto, se algum
elemento - impureza tiver teor superior a 7 0ppm
deve-se somar ã correção padrão, um fator de correção
calculado para a contribuição total de todos os elemen
tos-impureza, usando a formula abaixo:
(1) -fi h
0,01% + (% total de U não corrigida).10 Z X,F i=l
onde,
h = numero de impurezas não voláteis';
X = concentração de uma dada impureza metálica, em
ppm;
F = fator de conversão para calcular a massa da im
pureza (Tabela 1).
O cálculo do teor de urânio ê feito pelo uso
da fórmula:

Massa do Resíduo (U_0 ) % Total U = ( — — .f) - (1)
Massa da Amostra
onde.
f = fator para converter a massa de U^Og em f = 0,84803
TABELA 1 - Fatores de correção para impurezas metálicas
no UgOg
, Fórmula do Fator Gravimetrico Impureza x^aa^
oxido (pj^
Al A I 2 O 3 1,89
As As 2° 5 1,30
B 2,73
Ba BaO 1,12
Be BeO 2,78
Bi B Í 2 O 3 1,11
Ca -CaO 1,40
Cd CdO 1,14
Co C O 2 O 3 1,41
'Cr 1,46
Cu CuO 1,25
Fe ^^2^3 1,43
In In203 1,03
Li L Í 2 O 2,15
Mg MgO 1,66
Mn ^^2^3 1,58
Mo M 0 O 3 1,50
Na Na20 1,35
Ni Nio 1,27
P 2,29
Pb Pb02 1,15
Sb Sb20, 1,26
Si S Í O 2 2,14
Sn Sn0 2 1,27
Ta Ta205 1,22
Ti T Í O 2 1,67
Th Th02 0,96
V V 2 O 5
1,79
Zn ZnO 1,24
Zr ZrO^ 1,22
W WO 3 . - 1,26
P _ Massa molecular dõ óxido-impureza 3U
Massa, atômica ^ 3 ^ 3

18 2.
A P Ê N D I C E 2
RESULTADOS DA DECOMPOSIÇÃO TÉRMICA DOS DIURANATOS 2-C,
3-C E 12-C, SECADOS A 75°C E 150°C POR 1440, 2880 E
43 2 0 MINUTOS.
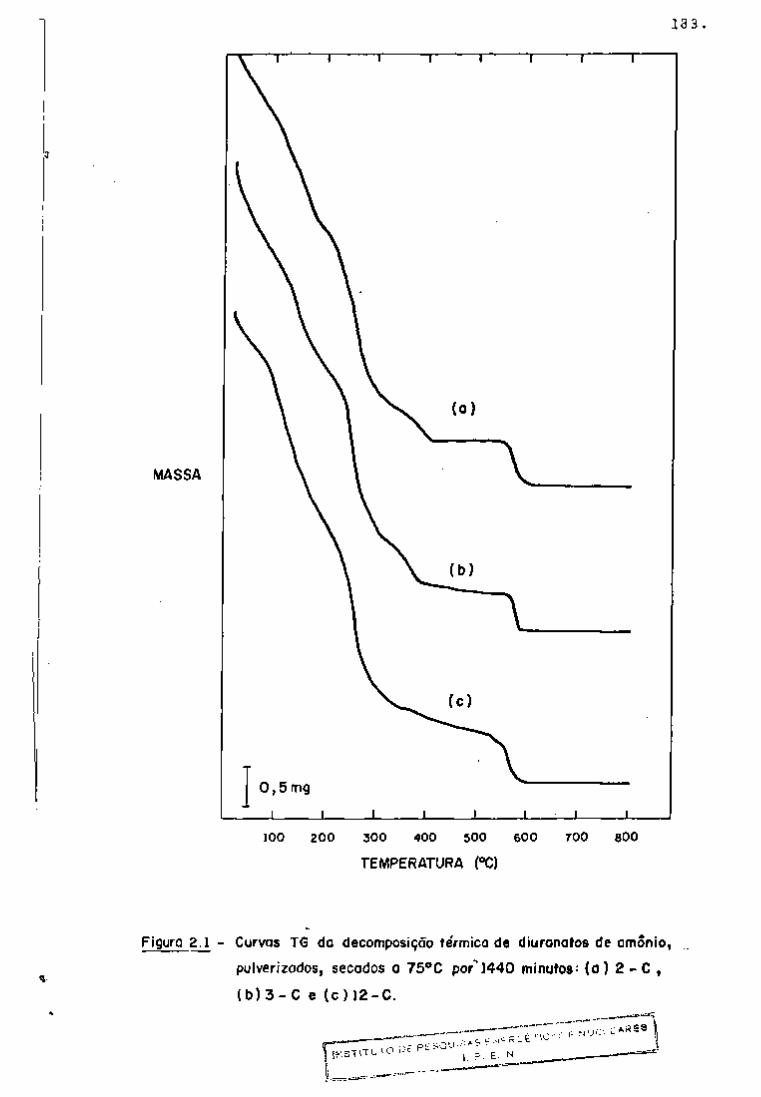
183
MASSA
100 200 300 400 500 600 700 800
TEMPERATURA CKi)
Figuro 2 1 - Curvas TG da decomposição te'rmico de diuranatos de amonio,
pulverizodos, secodos o 75**C por" 1440 minutos^ (o ) 2 - C ,
( b ) 3 - C e ( c ) l 2 - C .
. • r- N U C . f A R S 3
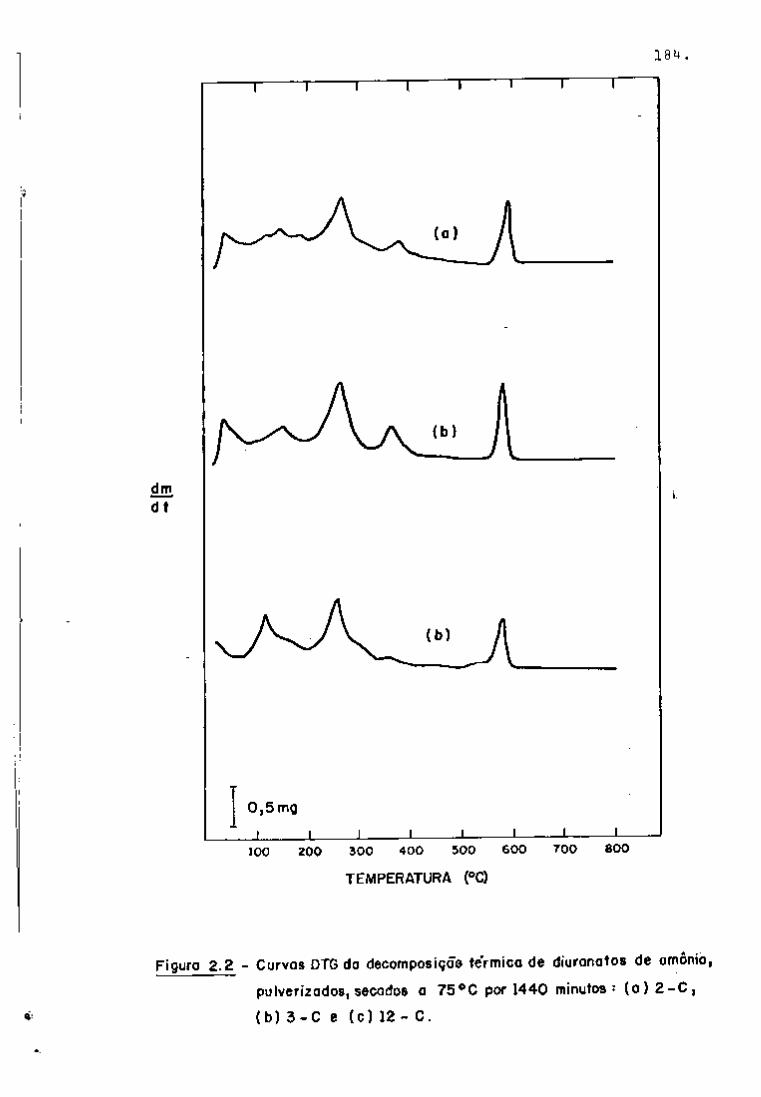
1814.
dm
d t
100 200 3 0 0 4 0 0 500 6 0 0
T E M P E R A T U R A ("O
700 800
Figura 2 .2 - Curvas DTG da decomposicoo te'rmica de diuronatos de amonio,
pulver izados, secados a 75**C por 1440 minutos: ( Q ) 2 - C ,
( b ) 3 - C e ( c ) 12 - C .
les.
M A S S A
100 200 300 400 500 6 0 0
TEMPERATURA (°C)
700 8 0 0
Figuro 2 . 3 - Curvas TG da decomposição te'rmica de diuranatos de amonio,
pulver izados, secados a 150**C por 1440 minutos-' ( a ) 2 - C ,
( b ) 3 - C , e ( c ) 1 2 - C .
186
dm
d t
100 200 300 400 500 600
TEMPERATURA («C)
700 8 0 0
Figuro 2 . 4 - Curvas DTG da decomposiqõo térmico de diuranatos de amônio,
pulverizados, secodos o 150**C por 1 4 4 0 minutos: ( o ) 2 - C ,
( b ) 3 - C e ( c ) 1 2 - C.
187
MASSA
100 200 300 400 500 600 700 800
TEMPERATURA («O
Figuro 2.5 - Curvas TG do decomposição térmico de diuranotos de omonio,
pulverizados, secados o 75** C por 2 8 8 0 minutos: (o ) 2 - C ,
( b ) 3 - C e ( c ) 1 2 - C .
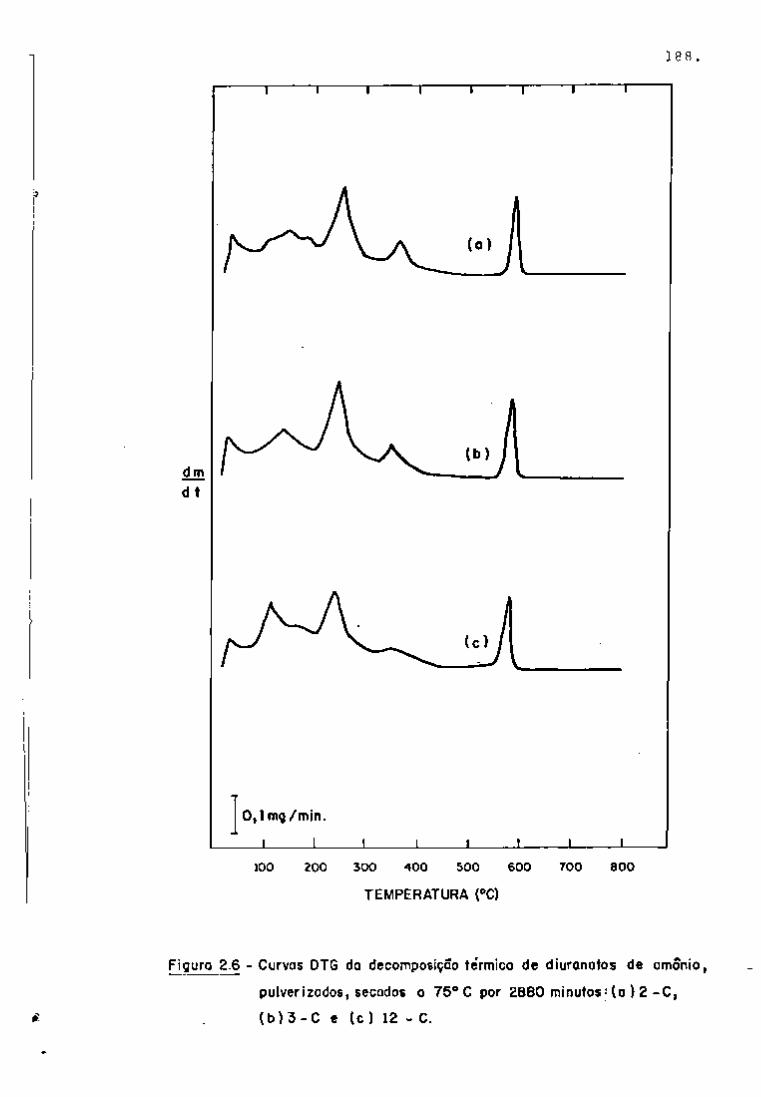
188
100 200 300 400 500 600
T E M P E R A T U R A ( " O
700 800
F i g u r o 2 .6 - C u r v a s D T G d o d e c o m p o s i ç ã o te ' rm ica d e d i u r o n a t o s d e a m o n i o ,
p u l v e r i z a d o s , s e c a d o s a 7 5 " C p o r 2 8 8 0 m i n u t o s : ( o ) 2 - C ,
( b ) 3 - C e ( c ) 12 - C .
18 9 .
MASSA
100 200 300 400 500 6 0 0
TEMPERATURA ( X )
700 800
Figuro 2 . 7 - Curvos TG do decomposição térmica de diuranotos de amonio,
pulverizados, secados o 150**C por 2 8 8 0 minutos = ( a ) 2 - C ,
( b ) 3 - C e ( c ) 1 2 - C .
190
d m
d t
100 zoo 300 400 500 6 0 0
TEMPERATURA {°Q
700 800
Figuro 2 .8 - Curvas DTG do decomposição te'rmico de diuranotos de omônio,
pulverizados, secados a 150" C por 2 8 8 0 minutos ¡ ( o ) 2 - C ,
( b ) 3 - C e ( c ) 1 2 - C .
INSTITUTO D E P E S Q U S A S E . \ f R C É T I C • S e N U C L E A R E S
I. P . E . N .
j 91
MASSA
100 200 300 400 500 600
TEMPERATURA ( "O
700 800
Figuro 2.9 - Curvas TG e DTG do decomposição térmica de diuranatos de amônio,
pulverizados, secodos o 150°C por 4 3 2 0 minutos-( o ) 2 - C e ( b ) 1 2 - C .
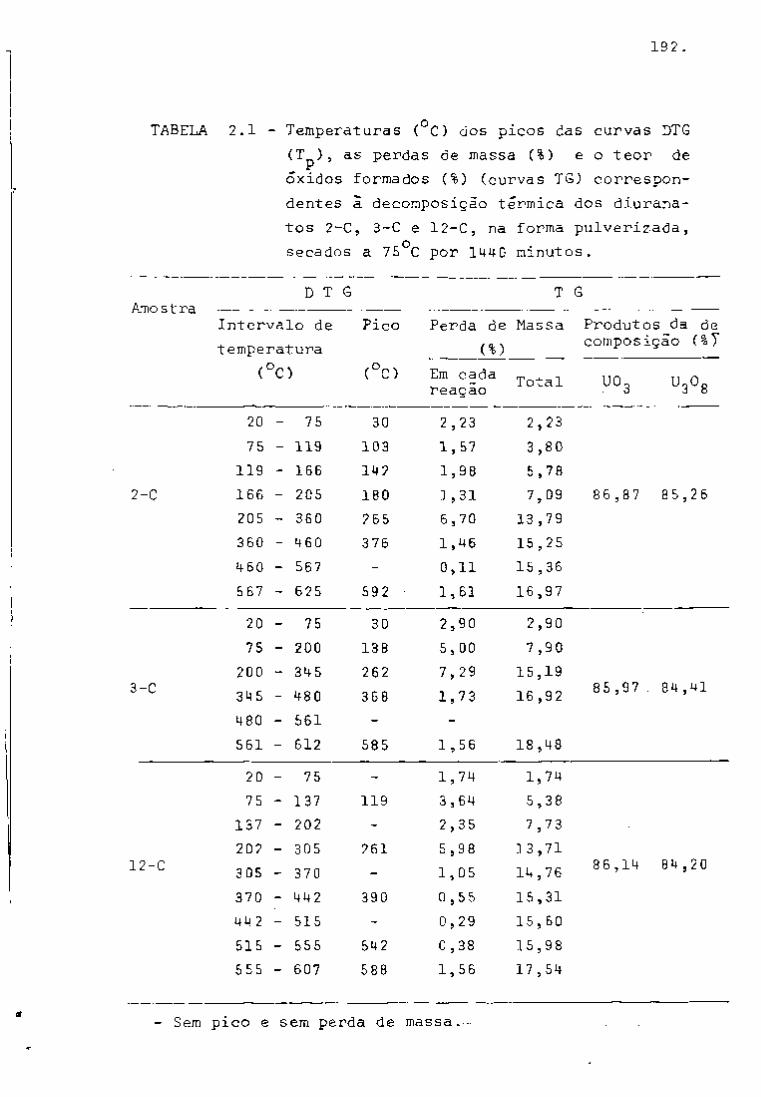
192 .
TABELA 2.1 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon-
dentes ã decomposição térmica dos diurana-
tos 2-C , 3-C e 12-C, na forma pulverizada,
secados a 75°C por 1440 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da de composição (%y
( °C) (°C) Em cada reação
Total "O3 UgOg
20 - 75 30 2,23 2,23
75 - 119 103 1,57 3,80
119 - 166 142 1,98 5,78
2-C 166 - 205 180 1,31 7,09 86,87 85,26
205 - 360 265 6,70 13 ,79
360 - 460 376 1,46 15,25
460 - 567 - 0,11 15 ,36
567 - 625 592 • 1,61 16,97
20 - 75 30 2,90 2,90
75 - 200 138 5,00 7,90
3-C 200
345
480
- 345
- 480
- 561
262
368
7,29
1,73
15,19
16,92 85,97 . 84,41
561 - 612 585 1,56 18,48
20 - 75 - 1,74 1,74
75 - 137 119 3,64 5,38
137 - 202 - 2,35 7,73
12-C 202
305
- 305
- 370
261 5,98
1,05
13,71
14,76 86,14 84,20
370 - 442 390 0,55 15,31
442 - 515 - 0,29 15,60
515 - 555 542 0,38 15,98
555 - 607 588 1,56 17,54
- Sem pico e sem perda de massa.-
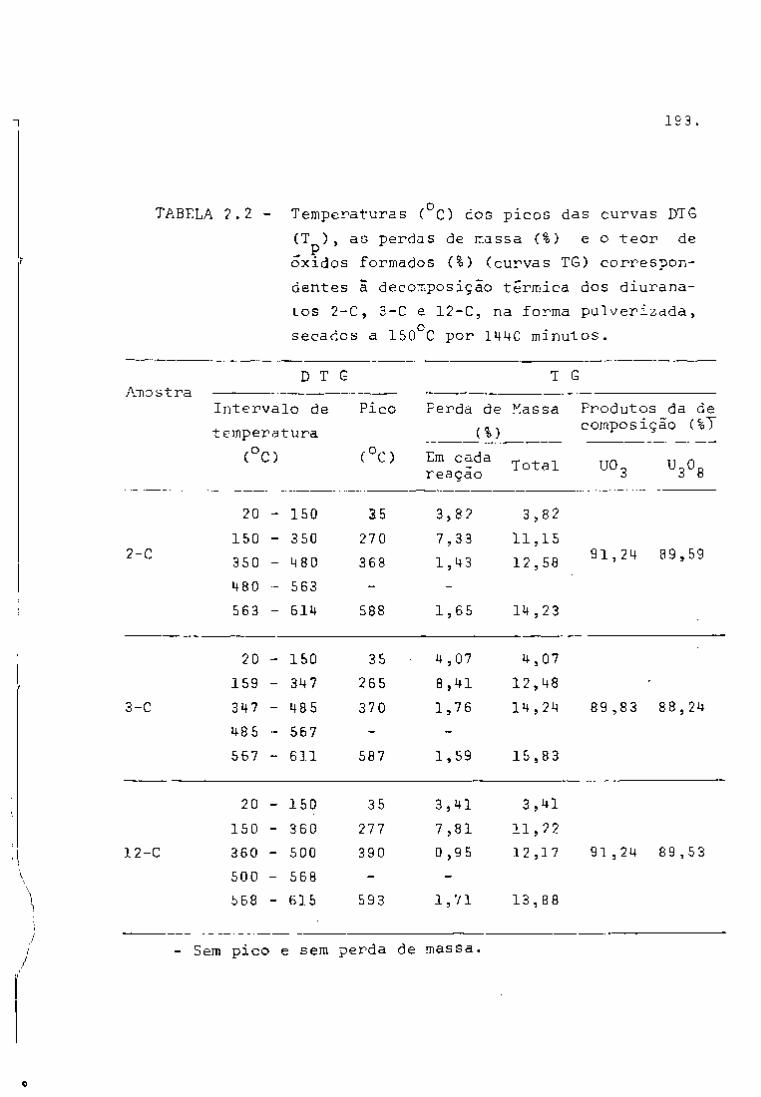
193.
TABELA 2.2- Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, na forma pulverizada,
secados a 150°C por .1440 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da de composição (%y
(°C) (°C) Em cada reação
Total UO3 U3O8
20 - 150 35 3,82 3,8 2
2-C 150 -
350 -
480 -
350
480
563
270
368
7,33
1,43
11,15
12,58 91,24 89,59
563 - 614 588 1,65 14,23
20 - 150 35 4,07 4,07
159 - 347 265 8,41 12,48 •
3-C 347 -
485 -
485
567
370 1,76 14,24 89,83 88,24
567 - 611 587 1,59 15,83
20 - 150 35 3,41 3,41
150 - 360 277 7 ,81 11,22
12-C 360 -
500 -
500
568
390 0,95 12 ,17 91,24 89,53
568 - 615 593 1,71 13,8 8
- Sem pico e sem perda de massa,
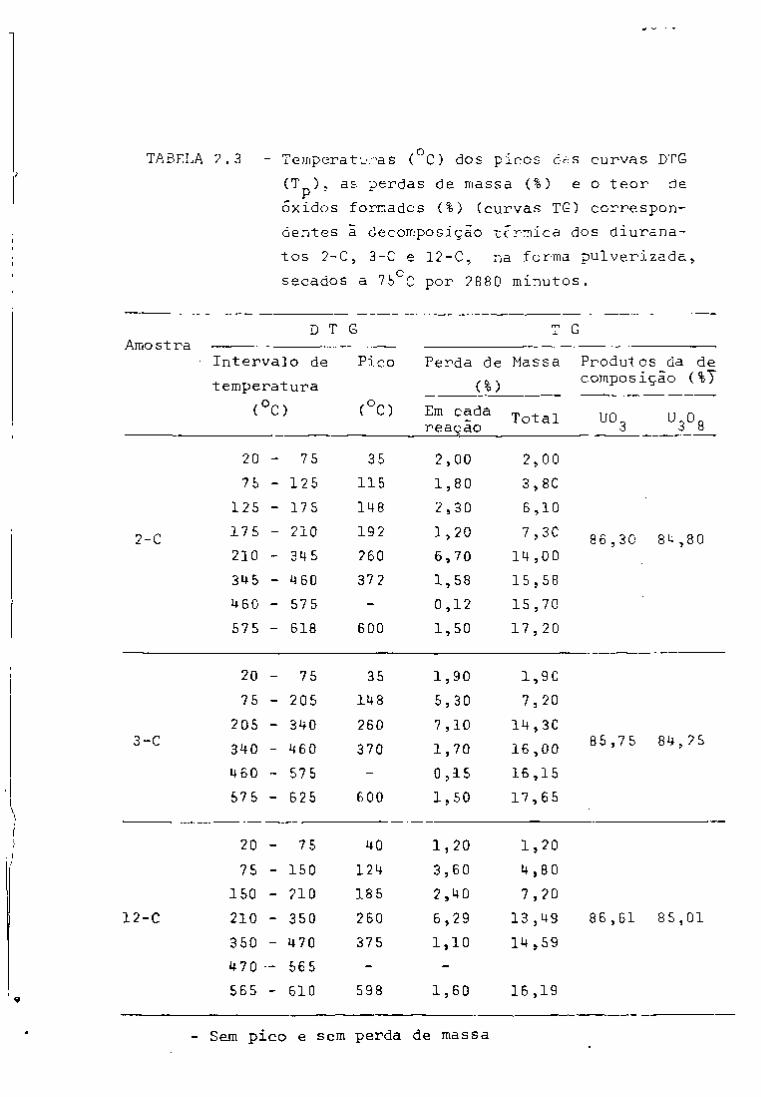
TABELA 2.3 - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon-
dentes ã decomposição térmica dos diurana-
tos 2-C , 3-C e 12-C, na forma pulverizada.
secados a 75°C por 28 8 0 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da de composição (%7
( °C) (°C) Em cada reação
Total "O3 "3O8
20 - 75 35 2, 00 2,00
75 - 125 115 1,80 3,80
125 - 175 148 2,30 6 ,10
2-C 175
210
- 210
- 345
192
260
1, 20
6,70
7,30
14,00 86,30 84,80
345 - 4 60 372 1,58 15,58
460 - 575 - 0,12 15,70
575 - 618 600 1,50 17, 20
20 - 75 3 5 1,90 1,90
75 - 205 148 5,30 7,20
3-C 205
340
- 340
- 460
260
370
7,10
1,70
14,30
16 ,00 85,75 84,25
460 - 575 - 0,15 16,15
575 - 625 600 1,50 17,65
20 - 75 40 1,20 1,20
75 - 150 124 3,60 4,80
150 - 210 185 2,40 7 ,20
12-C 210 - 350 260 6,29 13 ,49 86,61 85,01
350 - 470 37 5 1,10 14 ,59
470 — 565 - -
565 - 610 59 8 1,60 16,19
- Sem pico e sem perda de massa
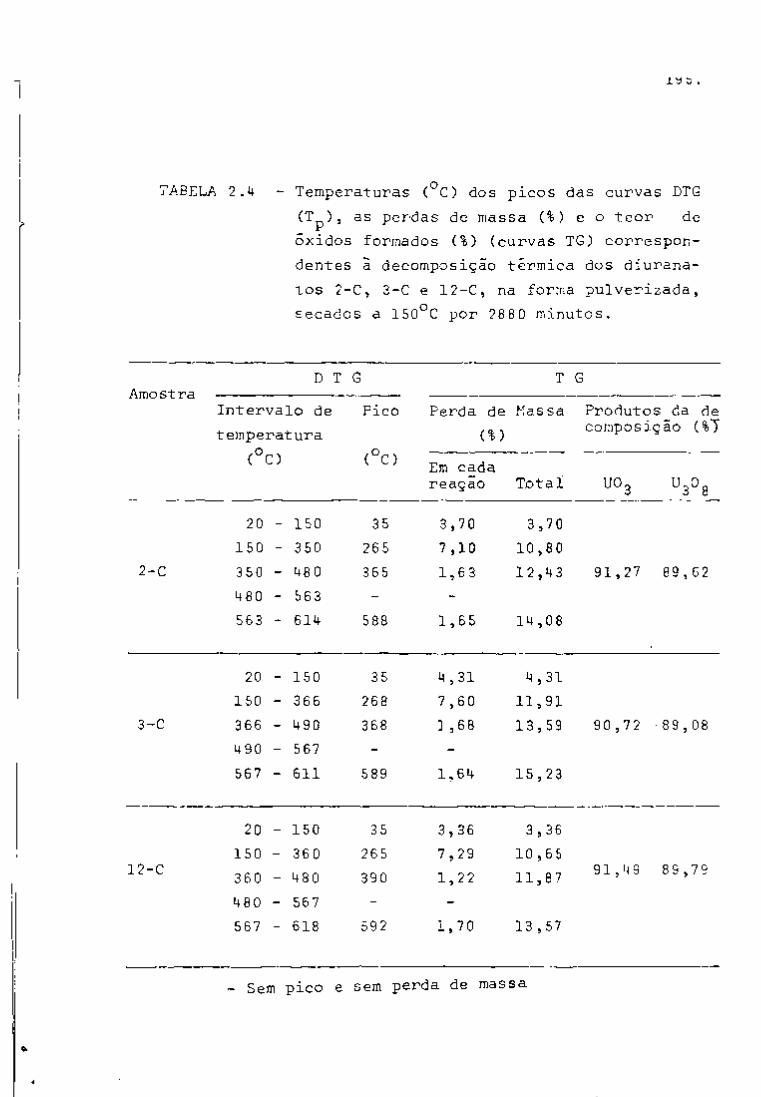
xa o .
TABELA 2.U - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes i decomposição térmica dos diurana
tos 2-C, 3-C e 12-C, na forma pulverizada,
secados a 150°C por 2880 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico
(°C)
Perda de
(%)
Mas sa Produtos da de composição (%T
(°C)
Pico
(°C) Em cada reação Total; U0,3 . UgO.g
20 - 150 35 3,70 3,70
150 - 350 - 265 7,10 10,80
2-C 350 -
480 -
480
563
365 1,63 12,43 91,27 89,62
563 - 614 588 1,65 14,08
20 - 150 35 4,31 4,31
150 - 366 268 7,60 11,91
3-C 366 -
490 -
490
567
368 1,68 13,59 90,72 89,08
567 - 611 589 1,64 15,23
20 - 150 35 3,36 3,36
12-C 150 -
360 -
480 -
360
480
567
265
390
7,29
1,22
10,65
11,87 91,4-9 89 ,79
567 - 518 592 1,70 13 ,57
- Sem pico e sem perda de massa
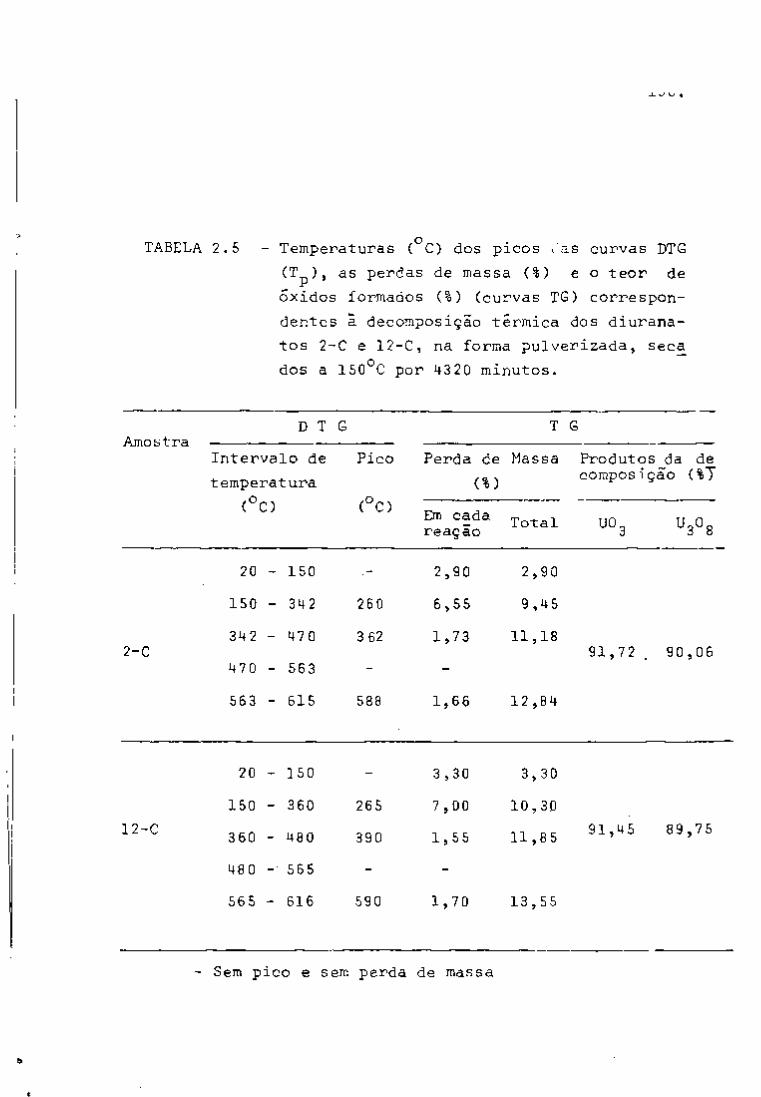
TABELA 2.5 - Temperaturas ( C) dos picos cas curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica dos diurana
tos 2-C e 12-C, na forma pulverizada, seca
dos a 150°C por 43 20 minutos.
Amostra D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da de composição (%7
(°C) (°C) Em cada reação
Total UO3 "3°8
20 - 150 .- 2,90 2,90
150 - 342 260 6,55 9,45
2-C 342 -
470 -
470
563
3 62 1,73 11,18 91,72 . 90,06
563 - 615 588 1,66 12 ,84
20 - 150 - 3,30 3,30
150 - 360 265 7,00 10,30
12-C 360 -
480 -•
480
565
390 1,55 11,85 91,45 89,75
565 - 616 590 1,70 13,55
- Sem pico e sem perda de massa

A P Ê N D I C E 3
RESULTADOS DA DECOMPOSIÇÃO TÉRMICA DOS DIURANATOS 2-C,
3-C e 12-C, SECADOS A 75°C POR 2880 MINUTOS E A 150°C
POR 4320 MINUTOS (2-C) E 2880 MINUTOS (3-C .. E 12-C).
198
MASSA
1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0
TEMPERATURA (*^)
7 0 0 8 0 0
Figura 3.1 - Curvas TG e DTG da decomposição te'rmica do diuranoto de amonio,
2 - C , opo's 1 ano de envelhecimento: ( o ) secado a 75**C por 2 8 8 0
minutos e { b) secado a ISO^C por 4 3 2 0 minutos.
I N S T I T U T O D G P E S Q U ÍÍAS F k R: .É ' .C
I- P- E . N .
]99 .
MASSA
100 200 3 0 0 400 5 0 0 600
TEMPERATURA {%)
700 800
Figuro 3 .2 - Curvas T6 e DTG do decomposição térmico do diuranato de amônio
3 - C , apos 1 ano de envelhecimento:(a) secodo a 75**C por 2 8 8 0
minutos e ( b ) secado o 1 5 0 * 0 por 4320minutos.
200 .
MASSA
100 200 300 400 500 600
TEMPERATURA ( "O
700 800
Figuro 3 .3 - Curvas TG e DTG da decomposição térmico do diuranoto de omonio
1 2 - C , opo's 1 ono de envelhecimento: ( o ) secodo o 75*»C por 2 8 8 0
minutos e ( b ) secado a 150**C por 2 8 8 0 minutos.
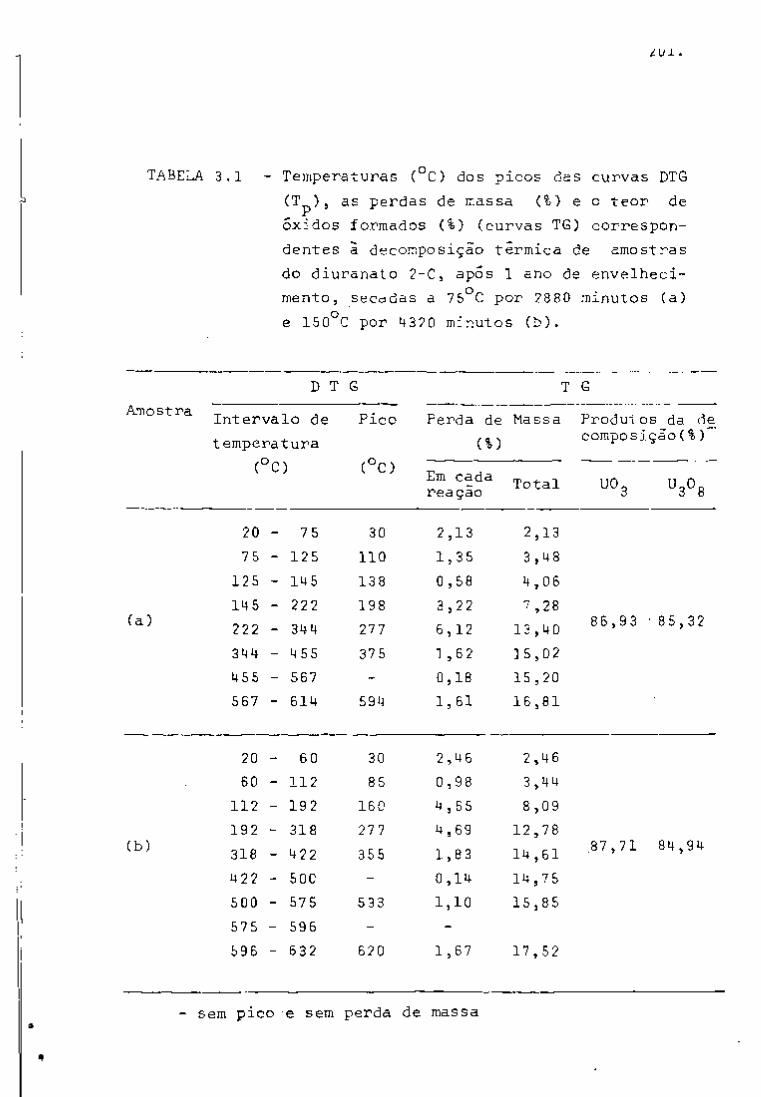
TABELA 3.1 - Temperaturas ( C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a. decomposição térmica de amostras
do diuranato 2-C, após 1 ano de envelheci
mento, secadas a 75°C por 2880 minutos (a)
e 15 0°C por 4 32 0 minutos (b).
D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da de composição(%)
(° O (°C) Em cada reação Total UO3 U3O8
20 - 75 30 2,13 2,13
75 - 125 110 1,35 3,48
125 - 145 138 0,58 4,06
(a) 14 5
222
- 222
- 344
198
277
3,22
6,12
7 ,28
13,40 86,93 •85,32
344 - 455 375 1,62 15,02
455 - 567 0,18 15,20
567 - 614 594 1,61 16,81
(b)
20 - 6 0 30 2,46 2,46
60 - 112 85 0,98 3,44
112 - 192 160 4 ,65 8,09
192 - 318 277 4,69 12,78
318 - 422 355 1,83 14,61
422 - 500 - 0,14 14,75
500 - 575 533 1,10 15,85
575 - 596 - -596 - 6 32 620 1,67 17,52
pico e sem perda de massa
,87 ,71 84 ,94
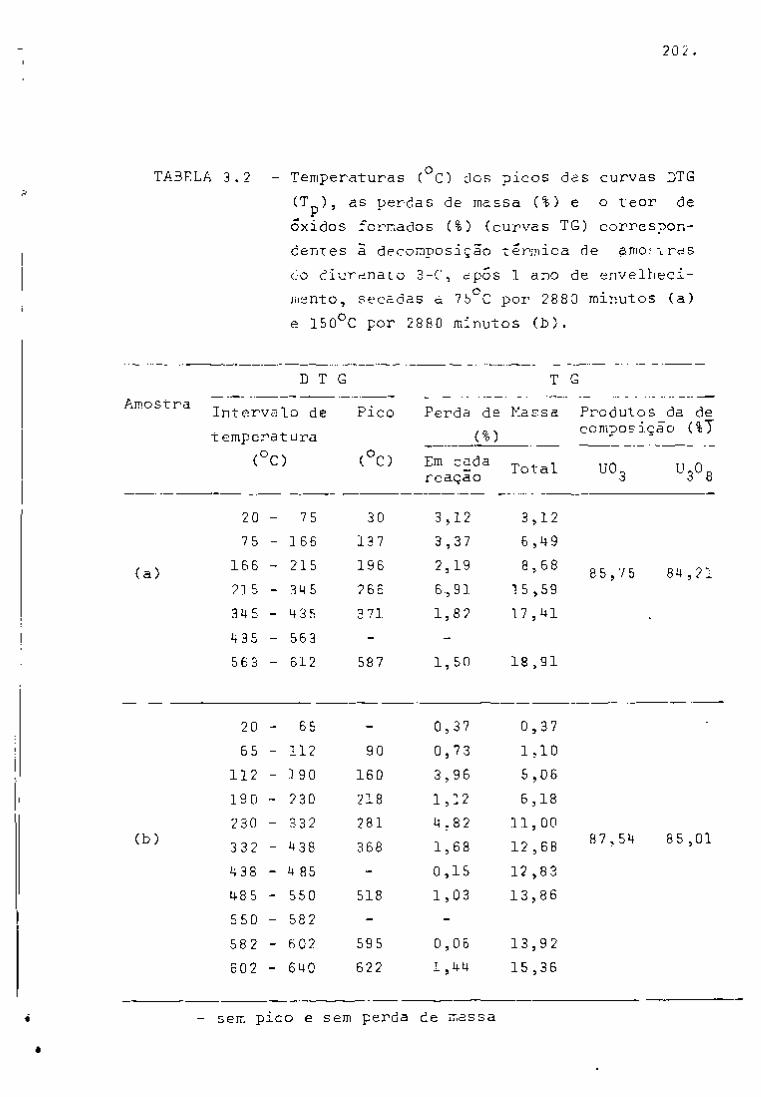
202 .
TABELA 3.2 - Temperaturas (°C) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes a decomposição térmica de amoí-"tras
do diuranato 3-C, apos 1 ano de envelheci
mento, secadas a 75* 0 por 2880 minutos (a)
e 150°C por 2880 minutos (b).
D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da composição
de (%T
( C) (°C) Em cada reação Total UO3 U 3° 8
20 - 75 30 3,12 3,12
75 - 166 137 3,37 5,49
(a) 165
215
— 215
345
196
266
2,19
6,91
8,68
15 ,59 85,75 84 ,21
345 - 435 371 1,82 17,41
435 - 563 - -
563 612 58 7 1,50 18,91
20 - 65 - 0,3 7 0,37
65 - 112 90 0,73 1,10
112 - 190 160 3,96 5,06
190 - 230 218 1,12 6,18
(b) 230
332
— 332
438
281
368
4,82
1,68
11,00
12 ,68 87,54 85 ,01
438 - 4 85 - 0,15 12 ,83
485 - 550 518 1,03 13,86
550 - 582 - -
582 - 602 595 0,06 13,92
602 — 640 622 1,44 15 ,36
- sem pioo e sem perda de massa
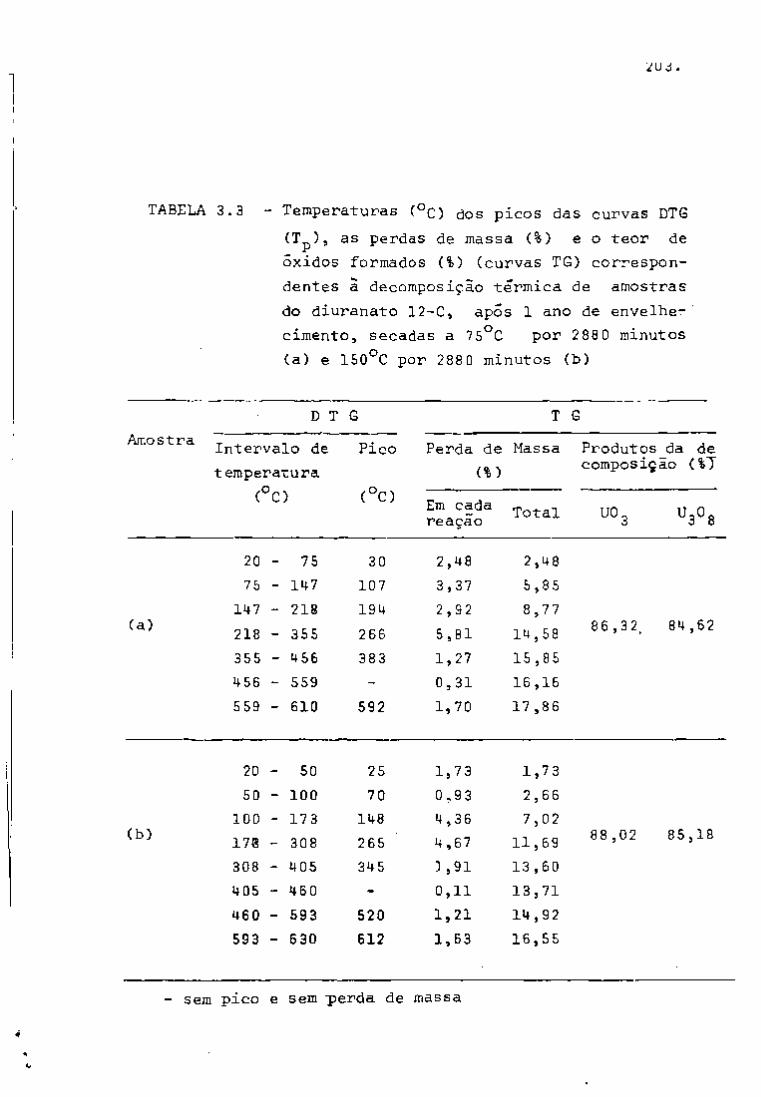
TABELA 3.3 - Temperaturas (°c) dos picos das curvas DTG
(Tp), as perdas de massa (%) e o teor de
óxidos formados (%) (curvas TG) correspon
dentes â decomposição térmica de amostras
do diuranato 12-C, após 1 ano de envelhe
cimento, secadas a 75°C por 2880 minutos
(a) e 150°C por 288 0 minutos (b)
D T G T G
Amostra Intervalo de
temperatura
Pico Perda de
(%)
Massa Produtos da composição
de (%T
(^ 'O (°C) Em cada reação
Total U O 3 U 3^8
20 - 75 30 2,48 2,48
75 - 147 107 3,37 5,85
(a) 147
218
- 218
- 355
194
266
2,92
5,81
8,77
14,58 86 ,32. 84 ,62
355 - 456 383 1,27 15,85
456 - 559 • - 0,31 16,16
559 - 610 592 1,70 17 ,86
20 - 50 25 1,73 1,73
50 - 100 70 0,93 2,66
(b) 100
17S
- 173
- 308
148
265
4,36
4,67
7,02
11,69 88,02 85 ,18
308 - 405 345 1,91 13 ,60
405 - 46 0 - 0,11 13,71
46 0 - 59 3 520 1,21 14,92
593 - 630 612 1,63 16,55
- sem pico e sem 3)erda de massa

204
1. A B R A O , A. Thermogravimetric behaviour of some uranium
compounds. Application to 0:U ratio determination.
São Paulo, Instituto de Energia Atômica, 1965.
(IEA-Pub-105).
2. ABRAO, A; ARAÚJO, J.A.; FRANÇA JR., J.M. Precipitação
reversa de diuranato de amônio a partir de soluções
de sulfato de uranilo: descontaminação do íon sulfa
to . São Paulo, Instituto de Energia Atômica, 1972.
(IEA-Pub-278).
3. ALTMANN, C.J.G.; DE HEER, B.H.J.; HERMANS, M.E.A. A
Modified Conway method for determining ammonia in
uranium compounds. Anal. Chem., 35:59 6-7, 1963.
4. AÍ1ERICAN SOCIETY FOR TESTING AND MATERIALS. Standard
methods for chemical, mass spectrometrie and spectro
chemical analysis of nuclear grade, uranium dioxide
powders and pellets: surface area by nitrogen
absorption method. May 30 1980. (ASTM-C-696-80).
In: 198 0 ANNUAL book of ASTM standards part 45:
nuclear standards. p.2 3 5-8.
5. AMERICAN SOCIETY FOR TESTING AND MATERIALS. Standard
test methods for ammonia nitrogen in water. (ASTM-
D-1426-79). In: 1980 ANNUAL book of ASTM standards
part-31. water. p.283-92.
B. ARAÚJO, J.A. Estudo da precipitação contínua de DUA
para 'implantação na iristalação piloto do CEQ-IEA.
São Paulo, Instituto de Energia Atômica, 1974. (lEA
Pub-339).
'7. ARMSON, .F.J.-, DIBBEN, H.E.-, MANSON, H.
An investigation into the formation and composition
of ammonium diuranate by potenciometric titration of
uranyl salts with ammonium hydroxide. Risley, UKAEA,
1949. (Declassified 1959). (SCS-R-30).
REFERÊNCIAS BIBLIOGRÁFICAS

205.
ASSIS, M.D. & GIOLITO, I. Estudo termoanalítico de
três séries de compostos de adição entre iodetos de
lantanídeos : (III) e itrio CIII) e o tetrametileno -
sulfõxido (TMSO) e oxido de tioxano CTSC). São Pau
lo, 19,81. (Dissertação c-; mestrado, Instituto de
Quimica, Universidade de . j.o Paulo).
' 9 . BABA, S . Operação de uma unidade s em i - ijldu sjtr i.aj. _ d e
produção de UO3. São Paulo, 1983. (Dissertação de
mestrado. Instituto de Pesquisas Energéticas e Nucle
ares).
10. BALL, M.C.-, BIRKETT, C.R.G; BROWN, D.S.; JAYCOCK, M.
J. The thermal .decomposition of ammonium diuranate.
J. Inorg. Nucl. Chem., 36:1527-29, 1974.
11. BARRCROFT, R. & WATSON, L.C. Preparationof uranium
dioxide for use in ceramic fuels. Pt.3 ammonium
diur-anate reduction studies. Chalk River, Ontario,
Atomic Energy of Canada 19 58. (AECL-7 57).
12. BOURNS, W.T. &.WATSON, L.C. Preparation of uranium
dioxide for use in ceramic fuels. . Pt. 1 -Batch
precipitation of ammonium diuranate. Chalk River,
Ontario, Atomic Energy of Canada, 1958. (AECL-757).
13. CHATELIER, H.L. Bull. Soc. Franc. Mineral., 10:204,
1877 apud opus cit ref. , 7.
14. COATS, A. W. & REDFERN, J.P. Thermogravimetric
analysis. A review. Analyst, 88:906-25, 1963.
15. CORKFUNKE, E.H.P. On the uranates of ammonium. 1.
The ternary system NH3-UO3-H2O. J. Inorg. Nucl. Chem.
24:303-07, 1962..
16. CORDFUNKE, E.H.P. VAN'DER.GIESSEN, A.A. Texture and
reactivity of uranium oxides. In: REACTIVITY ' of
solids, international symposium held in Munich, 1964.
Amsterdam, Elsevier, 1965 . p. 456-66 . (separata)..

17. DANTAS, J.M. & ABRAO, A. Estudo da caracterização
e da decomposição térmica de compostos -de urânio
por termogravimetria e calorimetría exploratória di
ferencial . São Paulo, Instituto de Pesquisas Ener
géticas e Nucleares, 1981. (IPEN-Pub-21).
18. DEANE, A.M. The infra-red spectra and structures of
some hydrated uranium trioxides and ammonium diura
nates. J. Inorg. Nucl. Chem,, 21:238-52, 1961.
19. DEPTULA, A. A study of composition of ammonium ura
nates. N^kleonilca, M_: 265-75 , 1962.
20. DRAGULESCU, C. & JULEAN, I. Study of the precipita
tion of ammmonium uranates. 1. Reaction between
uranyl nitrate and ammonia. apud NSA;14:21438,1960.
21. DURA, J. Description of ammonium diuranate produced
in the pilot plant at Kalna. apud NSA, 21:40681,
1967.
22. DUVAL, C. Historique de la thermogravimetrie. In:
REDFERN, J.P. ed. Thermal analysis,1965,proceedings
of the first international conference on held in
Aberdeen, Scotland, 6-9 Sept., 1965. London, Mac
Millan, 1965. p. 37.9.
23I DUVAL;, C. Inorganic_ Thermogravimetric analysis.
Amsterdam, Elsevier, 19 53.
24. EL-FEKEY, S.A.; EL-HAKIM, M.N.A.; ROFAIL, N. H.; ^
KHILLA, M.A. Solid phase decomposition of ammonium
uranate. ThermOchim. Acta, 54:327-36, 1982.
25. EL-FEKEY, S.A.; FARAH, M.Y. ; ROFAIL, N.H.
Stoichiometry of the U^O. phase form during cal-o o
cination of ammonium uranate.- Anal. Chim. Acta, 89 :
413-17, 1977.

2 0 7 .
2 6 . E R D E Y , L . The examination of materials by means of
derivation thermogravimetry. VJashington, D . C . ,
U S A E C , 1 9 6 0 . (AEC-tr - 3 9 U 0 ) .
2 7 . E R D E Y , L . ; PAULIK, F . ; PAULIK, J . Differential
thermogravimetry. Nature, 1 7 4 : 8 8 5 - 6 , 1 9 5 4 .
2 8 . F A R A H , M . Y . & E L - F E K E Y , S . A . Effect of tolerance in
mode of ammonium uranate preparation on the particle
size distribution and surface area of the inter
mediate U3O8 oxide. Arab . J . Nucl. Sci. Appl., 8 _ ( 2 ) :
6 7 - 7 3 , 1 9 7 5 .
2 9 . F A R A H , J . Y . & E L - F E K E Y , S . A . Thermal and chemical
analysis of ammonium uranates and intermediate oxides
Arab. J . Nucl. Sci. Appl., 1 0 . : 1 - 1 0 , 1 9 7 2 .
3 0 . F E D E R G R Ü N , L . & A B R A O , A . Determinação volumétrica
da relação O/U em pastilhas de õxidos cerâmicos U09.t.y
e U094.y.ThO?. São Paulo, Instituto de Energia Atômi
ca, 1 9 7 2 . (IEA-Pub -276 ) . i
3 1 . FODOR, M . & POKÕ, Z. On the formation, thermal
decomposition and structure of ammonium uranates.
Proceed. Anal. Chem. Conf., _ 3 ; 2 8 1 - 8 8 , 1 9 6 5 .
3 2 . F O G A Ç A FILHO, N.; G E N T I L E , E.F. ; M O U R A O , M . B . ; SANTOS,
T.D.S.; HAYDT, H . M . Contribuição ao estudo da fa -
bricação de pastilhas de U02.. São Paulo, Instituto
de Energia Atômica, 1 9 7 7 . (IEA-Pub-4 6 0 ) .
3 3 . F R A N Ç A , J . M : Usina piloto de purificação de urânio
pelo processo de colunas pulsadas em operação no Ins
titutò de Energia Atômica. São Paulo, Instituto de
Energia Atômica, 1 9 7 2 . C I E A - P u b - 2 7 7 ) .
3 4 . GARN, P . . D . Thermoanalytical methods of investigation.
New York, Academic, 1 9 6 5 .
INST ITUTO D E P E S Q U S A S E E R Ó É T ,0 ' S E N U C l . . R E S
I. P- E-

208 .
35. GENTILE, P. & COLLOPY, T.J. Ammonia precipitation
and filtration studies from uranyl nitrate solution.
Cincinnati, National Lead Company of Ohio, 19 61.
(NLCO-645).
36. GIOLITO, I. Análise termograv_imétrica e análise tér
mica diferencial. São Paulo, USP, 1974.
37. HALLDAHL, L. S^ SÖRENSEN, O.T. Thermal analysis of
the decomposition of amonium uranyl carbonate
(AUC) in different atmospheres. - Thermochim. Acta,
29:253-9, 1979.
38. HARTLAND, S. & NESBITT, R.J. Thermal decomposition
of uranyl nitrate hexahydrate. J. Apl. Chem.. , 14:
406-12, 1964.
39. lONASHIRO, M. & GIOLITO, I. Nomenclatura, padrões
e apresentação dos resultados em análise térmica.
Cerâmica, 26(121):17-24, 1980.
40. IPPOLITOVA, E.A.; PECHUROVA, N.l. ; GRIBENNIK., E. N.
. Investigation of the reaction process between uranyl
nitrate and ammonium hydroxide. Argonne, II. Ar-
gonne National Lab., 19 61. p.114 (ANL-Trans-33).
41. IWAMOTO, K. Thermal decomposition of uranyl nitrate
hexahydrate in the presence of graphite. J. Nucl.
Sci. Tech., 1(4):113-19, 1964.
42. JAKES, D. & LANDSPERSKY, H. Development of ceramic
nuclear fuel in Czechoslovakia. (A-CONF-28/8350)
apud NSA 1£:38368, 1964.
43. JANOV, J.; ALFREDSON, P.S.; VILKATTIS, V.K. The
influence of precipitation conditions on the proper
ties of ammonium diuranate and uranium dioxide
powders. J. Nucl. Mat., 44:161-74, 1972.

209
44. KOZHINA, I.I.; RADIONOVA, L.P.; TOLMACHEV, Y.M. Study of the thermal decomposition of ammonium uranates. Sov. Radiochem., 2 ( 2 : 206-9 , 1980 .
45. KRTIL, J.; VACHUSKA, J.; SPEYACKOVA, V.; KUVIK, V.; LUKAVSKY, J. The determination of uranium content in the real samples of ammonium diuranate. Radiochem. Radioanal. Lett., 30:7-12, 1977.
46. LANDSPERSKY, H.; SEDLAKOVA, L.; JAKES, D. Thermal decomposition of hydrated UO3. J. Appl. Chem., 14:
. 559-63, 1964.
47. LEVY, P.F. Thermal analysis. An overview. Amer. Lab. Jan. 1970 (separata).
48. LODDING, W. & OJAMAA, L. Dehydration and thermal decomposition of uranyl nitrates in the presence : of steam. J. Inorg. Nucl. Chem., _2J:1261-8 , 1965 .
49. MACKENZIE, R.C. Fundamental and quantitative applications of DTA and DSC. Anal. Proc., 17(6):215 -52, 1980.
50. MINTZ, M.H.; VAKNIN, S.; KREMENER, A.; HADARI, Z Nuclear fuel research and development. Influence of various manufacturing parameters on some characteris tics of UO2 powders and their sintering behaviour. Vienna, International Atomic Energy Agency, 19 77. (INIS-mf-4052).
51. MURPHY, C.B. Differential thermal analysis. Review. Anal. Chem., 30:867-72, 1958.
52. MURPHY, C.B. Thermal analysis review. Anal. Chem., ^:347 R-54R, 1964.
53. Natl. Bur. Stand. Tech. News Bull., 47:131, 1963 apud opus cit ref n9 51.

210 .
54. NEWKIRK, A.E. & SIMONS, L.L: Comment on the inter
pretation of dinamic derivarive thermogravimetric
curves. Talanta, l_3:1401-4, 1966.
55. NiceLAS-JARLETON, J. The role of alfa-UO^ in the
production of UOg and UF,, from beta-UOa . 0 ,5H70.
Saclay, Fr. CFA, Centre d'etuces nucleaires, 1970.
(CEA-R-M-074) .
56. NOTZ JR., K.J. Thermal decomposition of uranyl sulfa
te . Cincinnati, OH,. National Lead Company, of Ohio,
1960. (NLCO-Sll).
57. NOTZ, K.J.; MENDELL, M.G.; HUNTINGTON, C.W.; COLLOPY,
T.J. On the structure and thermal decomposition of
ammonium diuranate. Oak Ridge, Tn. USAEC Technical
Information Center, 1960. (TID-6228).
58. . ONDREJCIN, R.S. & GARRET JR., T.P. Thermal
decomposition of anhydrous uranyl nitrate and uranyl
nitrate dihydrate. J. Phys. Chem., 6^:470-73, 1961.
59. OOLMAN, T. Characterization of ammonium polyuranate
powders from a continuous precipitation.Ames, Iowa
state University of Science and Technology, Jan..
1979. (IS-T-846).
60. PAULIK, F.; PAULIK, P.; ERDEY,L. Derivatography. A
complex method in thermal analysis. Talanta, 13:
1405-30, 1966.
61. PEDREGAL, J.D. & SOLANO, R.R. Self-reduction of
ammonium diuranate to uranium dioxide. apud NSA 13.:
6526, 1959.
62.'^ PIRES, M.A.F.; ATALLA, L.T.; ABRAO , A. Separação de
nitrato em compostos de uranio por troca iónica, sua
determinação espectrofotométrica e por cromatografía
de lons. In: ASSOCIAÇÃO BRASILEIRA DE QUÍMICA. Quí
mica: '23? Congresso brasileiro realizado em Blumenau
S.C. 10-15 out. 19 82. (preprint).

211.
63. PRICE, G.H. SejI-reduction in ammonium uranates.
J. Inorg. Nucl. Chem., 3^:4085-92, 1971.
64. RAMAKRISHNAM, E.S. Determination oí the composition^
of the U3O8 phase from the decomposition of beta-UOs
Anal. Chim. Acta, 67:209-12, 1973.
65. RIBAS, A.G. & ABRAO; A. Preparação de UO9 apropria
do para obtenção de UF14. São Paulo, Instituto de E-
nergia Atômica, 1973. (IEA-Pub-318).
66. SAPACU, P. & CISMARU, G.D. Behaviour of uranium
compounds on heating: Thermal decomposition of
ammonium uraTiates. Rev.. Roum. Phys., 17(6):947--59,
1972 apud NSA 2^:55910, 1972.
67. SATO, T. Thermal decomposition of uranium peroxide
hydrates. J. Appl.' Chem. Biotech., 26 : 207-13 , 1976
apud I_NIS 2:2678 30 , 197 6 .
i 68. STUART, W.I. Thermal decomposition of ammonium • *<' —
' uranate an IR study. J. Inorg. Nucl. Chem., 38(7): - .
1378-9, 1976.
69. STUART, W.I. & MILLER, D.J. The nature of ammonium
uranates. J. Inorg. Nucl. Chem., 3^:2109-11, 1973.
y 70. STUART, W.I. & PRICE, G.H. Thermal decomposition of <
ammonium uranates. Lucas Heights, Australian Atomic
Energy Commission Research Establishment, 197 3.
(AAEC-E-276).
71. STUART., W.I. & WHATELEY, T.L. Composition • and
structure of ammonium uranates. J. Inorg. Nucl. Chem.
31:1639-47, 1959.
72, TRIDOT, G. Recherches sur les conditions de precipi
tation e t de stabilité en mileau aqueux des uranates
d'ammonium de sodium e t de potassium. Ann, Chim.,5 :
358-97, 1950.

212.
73. TURCANU, C.N. & DEJU, R. Thermal analysis of ammonium diuranate. Nucl. Tech. , 45.:18 8-92 , 1979.
74. TURCANU, C.N.; CORNESCU, M.; DEJU, R. Thermal analysis of ammonium diuranate. Romania, Institute Pentru Technologii Nucleare, Pitesti-Colibasi, 1976. (ITN-107).
75. UKAJI, R. On the reactivity of uranouranic oxide. 1. The thermal decomposition of uranyl nitrate (UNH) ammonium diuranate (ADU) and uranium peroxide (UPO). Washington, D.C. USAEC, 1959. (AEC-tr-4939).
76. URBANEK, V.; SARA, V. ; MORAVEC, J. Study of foniation and composition of ammonium uranates. J. Inorg. Nucl. Chem., 41(4): 537-40 , 1979.
77. VIDAVSKII, L.M.; BYAKHIVA, N.I.; KOVAILCHUK, V .-Y . IPPOLITOVA, E.A. Preparation of amorphous uranium trioxide by thermal decomposition of uranium peroxide dihydrate. Vestn. Mosk. Univ. Ser. II, Khim 4:33-34 1964 apud NSA 19:11121, 1965.
78. VIULLEMEY, R. Contribuition a 1'etude chimique et' technologique de la precipitation de diuranate • d'ammonium. Gif-sur-Ivëtte, Pr., CEA, Centre d'Etudes Nucleaires de Saclay, 1962. r:CEA-2204).
79. WARREN, H.S. Thermal dehydration of uranyl nitrate hydrates. J'.' Ihorg. Nuc 1. Chem. , 30:1761-8 , 1968.
80. WATSON, L.C. Preparation of UO2 for ceramic fuels. Oak Ridge, Tn., USAEC Technical Information Center , 1957. p.384. (TID-7546).
81. WATSON, E.S.; O'NEILL, M.J.; JUSTIN, J.; BRENNER, N . A differential scanning calorimeter for quantitative differential thermal analysis. Anal. Chem., 36(7) : 1233-8, 1964.


















