
DE BIOQUÍMICA E FISIOLOGIA MICROBIANA - Autenticação · BIOQUÍMICA E FISIOLOGIA MICROBIANA...
21
0 GUIA DE TRABALHOS LABORATORIAIS DE BIOQUÍMICA E FISIOLOGIA MICROBIANA Mestrado Integrado em Engenharia Biológica Ano Lectivo 2016/2017 Jorge H. Leitão (Área de Ciências Biológicas, DBE, IST)
Transcript of DE BIOQUÍMICA E FISIOLOGIA MICROBIANA - Autenticação · BIOQUÍMICA E FISIOLOGIA MICROBIANA...

0
GUIA DE TRABALHOS LABORATORIAIS
DE
BIOQUÍMICA E FISIOLOGIA MICROBIANA
Mestrado Integrado em Engenharia Biológica
Ano Lectivo 2016/2017
Jorge H. Leitão (Área de Ciências Biológicas, DBE, IST)
1
ACTIVIDADE DE SUPERÓXIDO DISMUTASE E DE GLUCOSE-6-FOSFATO DESIDROGENASE EM Pseudomonas aeruginosa I – Introdução
A exposição celular a factores ambientais tais como valores extremos de
temperatura ou do pH, radiação ionizante, agentes químicos tóxicos, níveis
elevados de metais pesados e outras agressões, podem levar ao aparecimento
intracelular de espécies moleculares altamente reactivas contendo oxigénio,
capazes de causar danos à célula, fenómeno conhecido por stress oxidativo
(Scandalios, 1993). Algumas espécies moleculares altamente reactivas (radicais
livres) podem ainda resultar de processos metabólicos como por exemplo a
respiração aeróbia.
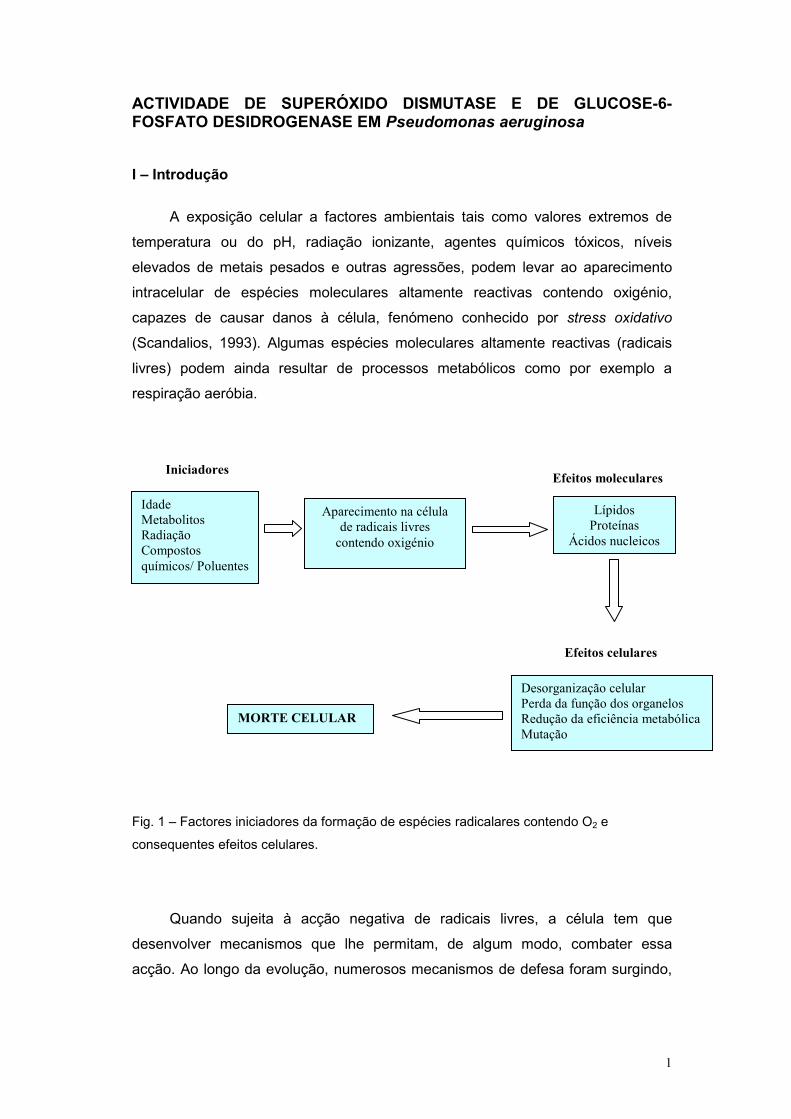
Fig. 1 – Factores iniciadores da formação de espécies radicalares contendo O2 e
consequentes efeitos celulares.
Quando sujeita à acção negativa de radicais livres, a célula tem que
desenvolver mecanismos que lhe permitam, de algum modo, combater essa
acção. Ao longo da evolução, numerosos mecanismos de defesa foram surgindo,
Idade Metabolitos Radiação Compostos químicos/ Poluentes
Aparecimento na célula de radicais livres
contendo oxigénio
Lípidos Proteínas
Ácidos nucleicos
Desorganização celular Perda da função dos organelos Redução da eficiência metabólica Mutação
MORTE CELULAR
Iniciadores Efeitos moleculares
Efeitos celulares

2
entre eles mecanismos enzimáticos capazes de remover, neutralizar ou quelatar
radicais livres e intermediários oxidantes (Scandalios, 1993).
Uma das espécies radicalares contendo oxigénio é o radical anião
superóxido, capaz de inactivar muitas enzimas essenciais à célula. Esta espécie
pode ser rapidamente convertida em peróxido de hidrogénio1 e oxigénio molecular
por acção da enzima superóxido dismutase (SOD) (Fridovich, 1986; Hassett et al.,
1995) (equação 1). A enzima SOD, pela primeira vez isolada a partir de sangue de
bovino, pode ser encontrada em organismos aeróbios, anaeróbios facultativos e
anaeróbios obrigatórios (Fridovich, 1986). Todas as SODs conhecidas são
metaloenzimas multiméricas, distinguindo-se 3 tipos com base no ião metálico que
apresentam no seu centro activo: Mn-SOD, Fe-SOD e Cu/Zn-SOD.
O2.- + O2
.- + 2H+ H2O2 + O2 (1)
Outra actividade enzimática envolvida na resposta celular ao stress oxidativo
é a actividade de glucose-6-fosfato desidrogenase, importante para a manutenção
de níveis intracelulares adequados de NADH (equação 2).
G-6-P + NADP+ NADPH + 6-Fosfogluconato (2)
No presente trabalho serão determinadas as actividades específicas de SOD e de
glucose-6-fosfato desidrogenase, presentes em extractos não purificados
preparados a partir de células de variantes mucosas de Pseudomonas aeruginosa
8821M, crescidas na ausência ou na presença de 2,5 mM de CuCl2. As variantes
mucosas de P. aeruginosa apenas ocorrem em estreita associação com infecções
respiratórias crónicas de doentes com Fibrose Quística, a doença genética mais
comum na raça caucasiana (Govan e Deretic, 1996), sendo razoavelmente
conhecidos os mecanismos moleculares subjacentes à conversão da forma não
mucosa (a habitualmente encontrada na natureza) à forma mucosa
(exclusivamente isolada de secreções de doentes com FQ cronicamente
infectados), produtora de grandes quantidades do exopolissacárido aniónico
alginato (Govan e Deretic, 1996).
1 O peróxido de hidrogénio é decomposto em água e O2, por acção das actividades enzimáticas catalase ou peroxidase, também estas envolvidas na defesa ao stress oxidativo.

3
II - Parte Experimental
- Introdução
Para a realização do presente trabalho experimental serão fornecidas
suspensões celulares congeladas de P. aeruginosa 8821M (variante altamente
mucosa produtora do exopolisaccárido alginato) (Alves et al., 1991). Estas
suspensões foram previamente preparadas a partir de culturas efectuadas em
meio LBS1 contendo ou não 2,5 mM CuCl2. As células de P. aeruginosa 8821M
foram recolhidas na fase exponencial de crescimento (DO640 = 1,0).
As suspensões celulares serão sujeitas a sonicação para libertação de
enzimas intracelulares. Os sobrenadantes obtidos após centrifugação das
suspensões sonicadas serão utilizados para determinação espectrofotométrica das
actividades enzimáticas de superóxido dismutase (SOD) e de glucose-6-fosfato
desidrogenase.
A actividade total de SOD será determinada com base na inibição da auto-
oxidação do pirogalol pela SOD. O pirogalol sofre auto-oxidação rápida
especialmente em soluções alcalinas, para valores de pH próximos de 8. A
reacção pode ser inibida em 99% pela enzima SOD, o que indica uma
dependência quase total da participação do radical superóxido O2 na reacção
(Marklund e Marklund, 1974). A reacção de auto-oxidação do pirogalol poderá ser
seguida espectrofotometricamente a 420 nm. As medições serão efectuadas
seguindo a evolução da Abs420 durante 3 minutos. Uma unidade de actividade
enzimática (1U) de SOD é definida como a quantidade de enzima capaz de inibir
em 50% a velocidade de auto-oxidação do pirogalol nas condições usadas.
A actividade de glucose-6-fosfato desidrogenase será determinada segundo
o método descrito por Lee et al. (1993), baseado na seguinte reacção:
NADP+ + G-6-P NADPH + 6-fosfogluconato (1)
Neste método o NADP é reduzido pela glucose-6-fosfato desidrogenase (G6P dh),
formando-se NADPH que pode ser quantificado espectrofotometricamente a 340
G6P dh

4
nm. Dado que a enzima também é capaz de usar como co-fator o NAD+, este será
o usado no presente trabalho.
A actividade total de glucose-6-fosfato desidrogenase é calculada definindo uma
unidade de actividade enzimática (1 U) como a quantidade de enzima capaz de
formar 1 µmol de NADPH por minuto, nas condições usadas. A conversão do
aumento de absorvância a 340 nm para moles de NADPH formadas é efectuada
com base no coeficiente de extinção molar (ε340 = 6220 M-1 cm-1).
Os extractos celulares serão ainda sujeitos a fraccionamento electroforético
em gel nativo de poliacrilamida e os géis obtidos serão tratados de modo a
visualizar no gel nativo as actividades de SOD dependentes de Fe e/ou de Mn
(Clare et al., 1984), indistinguíveis espectrofotometricamente.
1 O meio LBS consiste em meio LB suplementado com 10 g/l de glucose, 0,5 mM MgCl2 e 0,5 mM MnCl2.

5
- Procedimento experimental
A. Preparação dos extractos celulares
1. Descongelar as suspensões celulares fornecidas de P. aeruginosa 8821M.
2. Proceder à desintegração celular por sonicação, aplicando a cada amostra 3
ciclos de sonicação de 30 segundos esperando 1 minuto entre cada ciclo.
Manter sempre a amostra no gelo, mesmo durante a sonicação.
3. Centrifugar as amostras 45 minutos a 15000 rpm (centrífuga refrigerada 2K 15,
rotor 12141).
4. Recolher cuidadosamente os sobrenadantes para tubos limpos, tendo o
cuidado de não perturbar os sedimentos.
5. Manter as amostras no gelo.
B. Determinação da concentração proteíca nos extractos celulares (Bradford,
1976)
1. Diluir os extractos de forma a obter soluções contendo entre 100 e 500 µg/ml
de proteína total.
2. Em tubos de ensaio de vidro adicionar 2,5 ml de reagente de Coomassie a 50
µl de extracto diluido. Efetuar ensaios em triplicado.
3. Agitar vigorosamente em vórtex e esperar cerca de 15 min.
4. Proceder à leitura da absorvância das soluções a 595 nm, das soluções
preparadas em 3., tendo o cuidado de calibrar previamente o
espectrofotómetro preparando um branco em que o volume de amostra a
analisar é substituido por água destilada.
5. Com base na recta de calibração fornecida, que relaciona a absorvância a 595
nm com a concentração de proteína, calcular a concentração de proteína nos
extractos.

6
C. Determinação espectrofotométrica das actividades de SOD e glucose-6-fosfato
desidrogenase
A actividade de SOD será determinada segundo o método de Marklund e Marklund
(1974). Neste método é determinada a redução da velocidade de auto-oxidação do
pirogalol a 25°C, devida à actividade de SOD, que se traduz numa redução da
variação de absorvância a 420 nm. A actividade de glucose-6-fosfato
desidrogenase será determinada usando o método descrito por Lee et al. (1993).
C1 - Determinação da velocidade de auto-oxidação do pirogalol
Em cuvettes de 1 ml adicionar:
- 990 µl de tampão Tris.Cl 50 mM, EDTA 1 mM, pH 8,2
- 10 µl de solução de pirogalol em HCl 10 mM
Registar o aumento da absorvância a 420 nm durante três minutos.
Obter 3 registos independentes de modo a calcular a velocidade média de
auto-oxidação do pirogalol nas condições experimentais usadas.
C2 - Determinação da actividade total de SOD presente nos extractos
Em cuvettes de 1 ml adicionar:
- 970 µl de tampão Tris Cl 50 mM, EDTA 1 mM, pH 8,2
- 20 µl de extrato celular
- 10 µl de pirogalol
Registar a absorvância a 420 nm durante 3 min.
Repetir o processo de modo a obter resultados em triplicado para cada
extracto a analisar.

7
C3 - Determinação da actividade de glucose-6-fosfato desidrogenase
Às duas células espectrofotométricas (reaccional e de referência) adicionar:
- 500 µl de tampão Tris 100 mM pH 7,5
- 200 µl MgCl2 25 mM
- 100 µl de NADP 5 mM
- 100 µl de extrato celular
- À célula reaccional adicionar ainda 100 µl de uma solução de glucose-
6-fosfato 10 mM que na célula de referência são substituídos por água
destilada, de forma a descontar a actividade endógena dos extractos.
Registar os valores de absorvância a 340 nm durante 2,5 minutos num
espectrofotómetro de feixe duplo. Efectuar pelo menos, 3 determinações
independentes.
8
D. Visualização das actividades enzimáticas de SOD em gel nativo de
poliacrilamida
Tendo como objectivo esclarecer o efeito da presença no meio de cultura do ião
Cu2+ na actividade das enzimas Mn-SOD e Fe-SOD, os extractos celulares
preparados como descrito em A. serão sujeitas a electroforese em gel nativo de
poliacrilamida, procedendo-se de seguida à detecção, no gel, das actividades
enzimáticas das duas enzimas. A distinção entre as formas de SOD dependentes
de Mn e Fe será efectuada com base na inibição da Fe-SOD pelo peróxido de
hidrogénio (Clare et al., 1984).
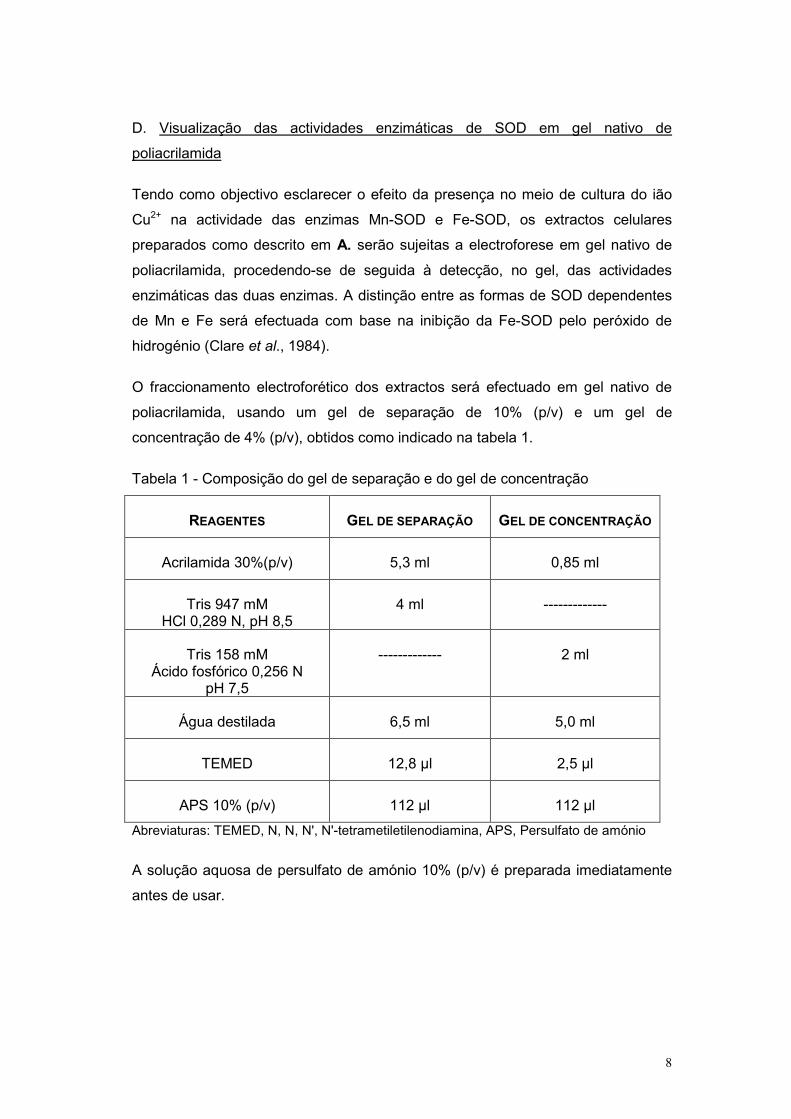
O fraccionamento electroforético dos extractos será efectuado em gel nativo de
poliacrilamida, usando um gel de separação de 10% (p/v) e um gel de
concentração de 4% (p/v), obtidos como indicado na tabela 1.
Tabela 1 - Composição do gel de separação e do gel de concentração
REAGENTES GEL DE SEPARAÇÃO GEL DE CONCENTRAÇÃO
Acrilamida 30%(p/v) 5,3 ml 0,85 ml
Tris 947 mM HCl 0,289 N, pH 8,5
4 ml -------------
Tris 158 mM Ácido fosfórico 0,256 N
pH 7,5
------------- 2 ml
Água destilada 6,5 ml 5,0 ml
TEMED 12,8 µl 2,5 µl
APS 10% (p/v) 112 µl 112 µl
Abreviaturas: TEMED, N, N, N', N'-tetrametiletilenodiamina, APS, Persulfato de amónio
A solução aquosa de persulfato de amónio 10% (p/v) é preparada imediatamente
antes de usar.

9
D1 – Preparação do gel
Após montagem do aparelho de electroforese a usar (Hoeffer), verter a mistura
para o gel de separação no molde. Adicionar à parte superior da mistura cerca de
1 mm de altura de isopropanol (o oxigénio inibe a reacção de polimerização). Após
polimerização da acrilamida (cerca de 15 minutos) remover a água e verter a
mistura para o gel de concentração no molde.
Colocar o molde na tina de electroforese que contém o tampão de corrida para a
parte inferior, "lower tank buffer" (Tris 63 mM, HCl 50 mM, pH 7,5). Colocar na
parte superior o "upper tank buffer" (Tris 37,5 mM, glicina 40 mM, pH 8,9).
Colocar nos poços do gel alíquotas dos extractos celulares a analisar contendo 50
µg de proteína total [determinados com base no método de Bradford (1976), ponto
B.], às quais se adicionou 5 µl de corante de aplicação (50% p/v de sacarose,
0,1% p/v de azul de bromofenol). Carregar 2 géis em duplicado. Submeter os géis
a electroforese durante cerca de 1 hora a 100 V.
D2 – Visualização das actividades de Mn- e Fe-SOD
Mergulhar um dos géis em solução aquosa de NBT (Nitro Blue Tetrazolium,
Sigma) 2,5 mM durante 20 minutos, à temperatura ambiente e no escuro e o outro
em idênticas condições adicionando peróxido de hidrogénio à solução de NBT de
modo a obter uma concentração final de H2O2 3 mM.
Substituir a solução de NBT por solução de revelação (fosfato de potássio 36 mM,
TEMED 2,8 mM, riboflavina 2,8x10-5 M, pH 7,8). Manter o gel no escuro durante
mais 20 minutos.
Remover a solução de revelação e irradiar com luz visível (lâmpada de 60 W) até
ao aparecimento de bandas acromáticas nos géis que entretanto se tornaram
roxos (Clare et al., 1984). Durante o tempo em que o gel está exposto à luz ocorre
redução fotoquímica da riboflavina que por sua vez reduz o dioxigénio (O2) ao
anião superóxido (O2-) o qual reduz o NBT originando um composto de cor roxa.
Nos locais do gel onde se encontra a actividade de SOD esta inactiva os radicais
O2- levando ao aparecimento de bandas acromáticas num gel roxo. Guardar o gel
em ácido acético 7,5% (v/v) até ser fotografado.

10
REFERÊNCIAS
Alves, MJ, et al. Temperature profiles and alginate synthesis in mucoid and non-mucoid variants of Pseudomonas aeruginosa. Letters in Applied Microbiology 12, 244-248, 1991.
Bradford, MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein dye binding. Analytical Biochemistry 72, 248-254, 1976.
Clare, DA et al. Effects of molecular oxygen on the detection of superoxide radical with nitroblue tetrazolium and an activity stain for catalase. Analytical Biochemistry 140, 532-537, 1984.
Fridovich, I. Superoxide dismutases. Advances in Enzymology 58, 62-97, 1986.
Govan, JRW, and Deretic, V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiological Reviews 60, 539-574, 1996.
Hassett, DJ et al. Pseudomonas aeruginosa sodA and sodB mutants defective in manganese- and iron-cofactored superoxide dismutase activity demonstrate the importance of the iron-cofactored form in aerobic metabolism. Journal of Bacteriology 177, 6330-6337, 1995.
Lee, J.-S., Y.-C. Hah, J.-H. Roe. The induction of oxidative stress in Streptomyces coelicolor upon hydrogen peroxide treatment. Journal of General Microbiology 139, 1013-1018, 1993.
Marklund, S, and Marklund G. Involvement of the superoxide anion radical in the autooxidation of pyrogallol and a convenient assay for superoxide dismutase. European Journal of Biochemistry 47, 469-474, 1974.
Perry, ACF, et al. Molecular characterization of the gor gene encoding glutathione reductase from Pseudomonas aeruginosa. Determinants of substrate specificity among pyridine nucleotide-disulphide oxidoreductases. Molecular Microbiology 5, 163-171, 1991.
Scandalios, JG. Oxygen stress and superoxide dismutases. Plant Physiology 101, 7-12, 1993.

11
TP2. PRODUÇÃO E PURIFICAÇÃO DA PROTEÍNA TRANSPORTADORA DE
GRUPOS ACILO, Acp, DE Burkholderia cepacia J2315
1 – Introdução
A biossíntese de ácidos gordos tem sido intensivamente estudada em eucariontes
e procariontes. Em eucariontes e em micobactérias, as actividades enzimáticas
envolvidas nos vários passos da biossíntese de ácidos gordos encontram-se todas
juntas num complexo multienzimático, dizendo-se que a síntese é do tipo I. Em
procariontes, a biossíntese é dita do tipo II ou dissociada, pois é conduzida de
forma dissociada por cada um dos enzimas intervenientes.
O acetil-CoA é o precursor dos ácidos gordos. Em procariotas e eucariotas, a
proteína Acp (acyl carrier protein) desempenha um papel fundamental na
biossíntese de ácidos gordos, encontrando-se os intermediários da via
normalmente ligados a esta proteína.
No presente trabalho irá ser usada a proteína Acp de Burkholderia cenocepacia.
Esta espécie bacteriana pertence ao complexo Burkholderia cepacia, um conjunto
de 17 espécies muito próximas, contendo patogénios oportunistas capazes de
causar infecções respiratórias graves, particularmente em doentes com fibrose
quística (Mahenthiralingam et al., 2005; Richau et al., 2000; Leitão et al., 2010).
O gene que codifica a proteína Acp foi identificado usando uma estratégia que
envolveu a preparação de uma colecção de mutantes de B. cenocepacia J2315
por mutagénese aleatória com o plasposão pTnModOCm (Dennis and Zylstra,
1989), seguida da selecção de mutantes atenuados na sua virulência para o
nemátodo Caenorhabditis elegans (Sousa et al., 2008). Uma estratégia similar
permitiu anteriormente identificar um agrupamento de genes envolvidos na
biossíntese de exopolissacárido por bactérias do complexo Burkholderia cepacia
(Moreira et al., 2003). A digestão do DNA total do mutante B. cenocepacia SJ1
com a enzima de restrição BamHI, seguida de auto-ligação e transformação em
Escherichia coli, permitiu obter um plasmídeo recombinante contendo DNA de B.
cenocepacia J2315, cuja sequenciação revelou que o plasposão tinha
interrompido o gene acp pertencente à organização genética representada na Fig.
1. Os genes fabF, fabG, fabD e fabH codificam actividades enzimáticas de β-
cetoacil-Acp sintase II, β-cetoacil-ACP redutase, malonil-CoA-Acp transacilase, e
β-cetoacil Acp sintase III, respectivamente. Embora não seja totalmente conhecido

12
o papel do gene plsX, pensa-se que tem um papel no metabolismo do 1-
acilglicerol-3-fosfato e, muito possivelmente, na regulação da concentração
intracelular de acil-Acp.
Fig 1 –Organização genética do agrupamento de genes fab (fatty acid biosynthesis) em B.
cenocepacia J2315. A bandeira colocada por cima do gene acp (a negro) indica a posição do
plasposão a interromper o gene acp. Adaptado de Sousa et al. (2008).
Em bactérias gram-negativas, as proteínas Acp e os seus intermediários acilados
participam ainda na síntese de ácidos gordos β-hidroxilados e na síntese das
moléculas-sinal usadas em sistemas de quorum-sensing, as acilhomosserina
lactonas. A proteína Acp de B. cenocepacia é 73% identica à proteína Acp de E.
coli, mostrando-se na Fig. 2 a similaridade entre a estrutura prevista para a
proteína Acp de B. cenocepacia e a estrutura, determinada experimentalmente, da
proteína Acp de E. coli.
Fig. 2 – Estrutura das proteínas Acp de E. coli (A) e de B. cenocepacia J2315 (B). Adaptado de
Sousa et al. (2008).
No presente trabalho experimental, pretende-se purificar a proteína Acp
recombinada, contendo 6 histidinas adicionais (cauda de histidinas) na região C-
terminal. Para tal, a proteína será sobre-expressa em E. coli e purificada por
cromatografia de afinidade. Será posteriormente comprovada a sua identidade por
AA
α1
C
N
α3
α2
α4 BB
α1
C N
α3
α2
α4
α3
plsX fabH fabF acp fabG fabD

13
recurso a hibridação de western usando um anticorpo específico para a cauda de
histidinas.
2- Procedimento experimental:
a) Sobre-expressão da proteína Acp em E. coli
1- Inocular a estirpe E. coli BL21-DE3(pSAS6) em 20 ml de meio LB
suplementado com o antibiótico ampicilina (150 mg/ml).
2- Incubar 2 a 3 horas a 37ºC com agitação de 250 rpm.
3- Medir a D.O. (640 nm) dos pré-inóculos.
4- A partir dessas culturas inocular 100 ml de meio SB (Anexo), contendo
ampicilina, de forma a que a D.O. inicial seja de 0,1.
5- Deixar crescer a cultura a 37ºC até que a D.O. seja de 0,5
(aproximadamente 2-3h). Retirar uma amostra de cada cultura (volume
equivalente a 1,2/D.O. da cultura), centrifugar e ressuspender as células em
80 µl de tampão de aplicação em SDS-PAGE. Colocar as amostras no gelo.
6- Induzir adicionando 0,4 mM de IPTG (isopropil-β-D-tiogalactósido).
7- Deixar crescer 2 h nas mesmas condições e findo este tempo retirar
igualmente amostras das culturas induzidas (volume equivalente a 1,2/D.O.),
centrifugar e ressuspender as células em 80 µl de tampão de aplicação em
SDS-PAGE. Ferver todas as amostras durante 5 min e guardar a –20ºC até
posterior utilização.
8- Recolher a cultura de E. coli BL21-DE3(pSAS6), centrifugando-a 5 min a
7000 rpm (4ºC). Ressuspender o sedimento celular em 4 ml de tampão I
(tabela I), transferir para um tubo apropriado e congelar a –80ºC.

14
b) Lise das células por recurso a ultrassons e purificação da proteína
recombinante por cromatografia de afinidade
1- A lise celular é efectuada por uso de ultrassons (sonicador Branson Sonifier
250). Após descongelação, o tubo contendo as células é mantido em gelo e
estas são sujeitas a 3 sonicações a 70Watts de 30s cada, intervalando cerca
de 1 min entre cada sonicação.
2- A amostra proteica é de seguida centrifugada durante 45 min a 15000 rpm e
a 4ºC. O sobrenadante é transferido para novo tubo e guardado no gelo até
se iniciar a purificação.
3- Preparar a coluna HisTrap (Amersham). Usando uma seringa, lavar a coluna
com 5 ml de água destilada, tendo o cuidado de não formar bolhas de ar.
4- Aplicar 0,5 ml de solução de NiCl2 0,1M e equilibrar de seguida a coluna com
10 ml de tampão I.
5- Aplicar a amostra proteica na coluna com a seringa e recolher o eluído.
6- Lavar a coluna com 5 ml de tampão I.
7- Aplicar 3 ml de tampão II e recolher o eluído em três fracções de 1 ml.
8- Repetir o passo 7, mas utilizando os tampões III e IV (Tabela I).
Tabela I- Soluções tampão usadas durante a purificação da proteína His-Acp.
Tampão Concentração de
Imidazole (mM)
Tampão fosfato
8x (pH 7.4)
Imidazole 2M
(pH7.4)
Água destilada
I 10 3,0 ml 0,12 ml 20,88 ml
II 50 1,0 ml 0,20 ml 6,8 ml
III 100 1,0 ml 0,40 ml 6,6 ml
IV 200 1,0 ml 0,80 ml 6,2 ml
Tampão fosfato 8x (8mM tampão fosfato; 0,4M NaCl, pH 7,4)

15
c) Separação electroforética de proteínas por SDS-PAGE
1- Preparar o sistema para fazer o gel.
2- Preparar o gel de resolução (12,5% de acrilamida), juntando:
-1,25 ml de solução I (Tabela II)
-2,08 ml de solução de acrilamida
-1,64 ml de água destilada
-8 µl de TEMED
-33 µl de persulfato de amónia 10%
Misturar cuidadosamente e pipetar imediatamente a solução entre as placas
de vidro e silica até uma altura de aproximadamente 6 cm. Colocar uma
camada de 1 ml de isopropanol de forma a obter uma superfície plana.
Deixar polimerizar.
3- Após polimerização remover o isopropanol com papel absorvente.
4- Preparar o gel de concentração (6% de acrilamida) juntando:
-0,4 ml de solução II (Tabela II)
-0,4 ml de solução de acrilamida
-1,2 ml de água destilada
-4 µl de TEMED
-12 µl de persulfato de amónia 10%
Misturar cuidadosamente, encher as placas com esta solução e colocar o
pente. Deixar polimerizar.
5- Montar o sistema de electroforese e preencher os tanques com tampão de
electroforese 1x (Tabela II)
6- Preparar as amostras para aplicação no gel, juntando 10 µg (20 µl) de cada
uma das fracções de purificação com 5 µl de tampão da amostra. Ferver
durante 5 min.
7- Proceder à electroforese aplicando uma voltagem constante de 150 volts,
durante 1h.
8- Retirar o gel e colocá-lo numa solução corante (Tabela II). Levar ao micro-
ondas 30 s e agitar 10 min.
9- Colocar o gel numa solução descorante (Tabela II). Levar ao micro-ondas 30
s e agitar 10 min. Findo este tempo, as bandas de proteínas devem ser
visíveis.
16
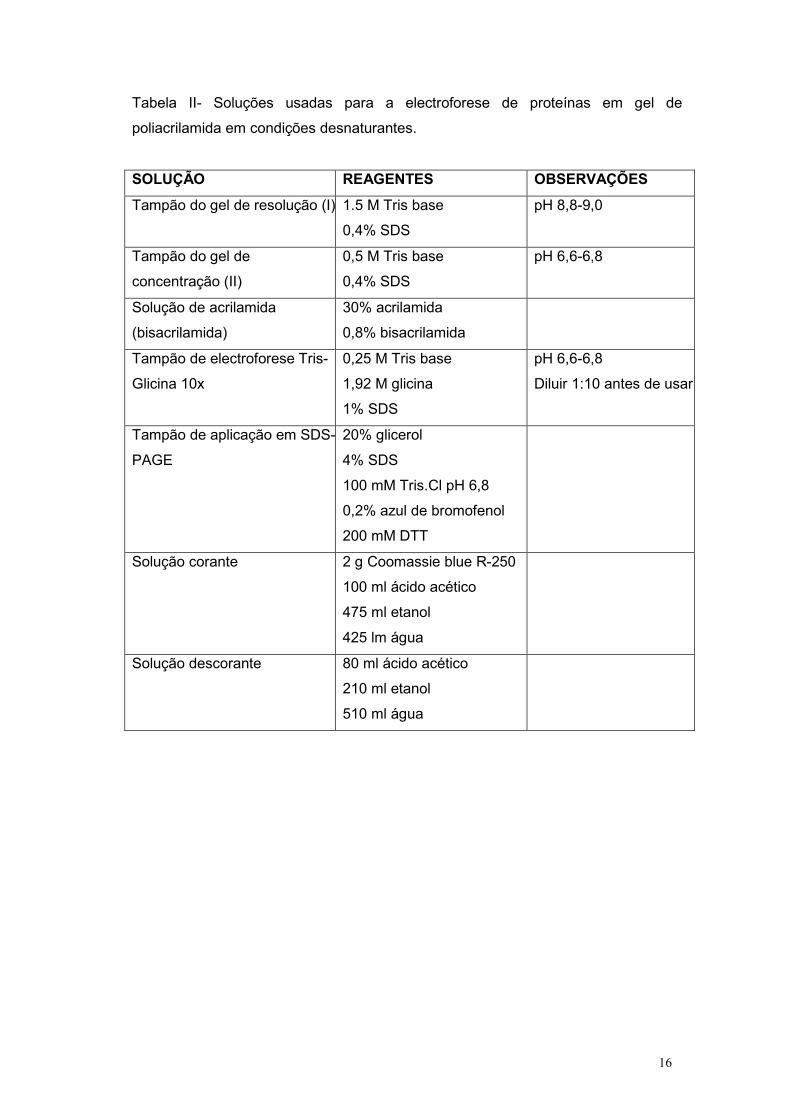
Tabela II- Soluções usadas para a electroforese de proteínas em gel de
poliacrilamida em condições desnaturantes.
SOLUÇÃO REAGENTES OBSERVAÇÕES
Tampão do gel de resolução (I) 1.5 M Tris base
0,4% SDS
pH 8,8-9,0
Tampão do gel de
concentração (II)
0,5 M Tris base
0,4% SDS
pH 6,6-6,8
Solução de acrilamida
(bisacrilamida)
30% acrilamida
0,8% bisacrilamida
Tampão de electroforese Tris-
Glicina 10x
0,25 M Tris base
1,92 M glicina
1% SDS
pH 6,6-6,8
Diluir 1:10 antes de usar
Tampão de aplicação em SDS-
PAGE
20% glicerol
4% SDS
100 mM Tris.Cl pH 6,8
0,2% azul de bromofenol
200 mM DTT
Solução corante 2 g Coomassie blue R-250
100 ml ácido acético
475 ml etanol
425 lm água
Solução descorante 80 ml ácido acético
210 ml etanol
510 ml água

17
d) Transferência de proteínas para uma membrana (western blot)
1 – Após separação electroforética, mergulhar cuidadosamente o gel em tampão
Towbin durante 15 minutos à temperatura ambiente com agitação suave.
2- Preparar uma membrana de nitrocelulose (55 mm X 85 mm) e 6 folhas de papel
Whatman 3MM de igual dimensão e mergulhar em tampão Towbin durante 5
minutos.
3- Remover o gel de concentração e colocar o gel invertido sobre uma placa de
vidro, tendo o cuidado de o manter molhado com solução tampão. Colocar a
membrana por cima do gel e sobre aquela colocar 3 folhas de papel Whatman.
Remover as bolhas de ar.
4- Voltar o o conjunto, remover a placa de vidro e colocar por cima do gel 3 folhas
de papel Whatman, tendo o cuidado de remover eventuais bolhas de ar.
5 – Colocar o conjunto no aparelho Trans-Blot SD Semi-Dry Transfer Cell (BIO-
RAD). Remover o excesso de líquido. Efectuar a transferência durante 45 minutos a
15 volts, 250 mA.

18
f) Detecção imunológica
1 – Após a transferência das proteinas para a membrana, recolher a membrana e
incubá-la durante 1 hora com 40 ml de solução de bloqueio (tampão PBS contendo
5% de leite magro em pó) à temperatura ambiente com agitação suave.
2 – Lavar a membrana 3X 5 minutos com tampão PBS contendo 0,05% Tween 20.
3 – Incubar durante 1 hora com agitação suave a membrana com solução de
anticorpo monoclonal de rato anti- poli-histidinas conjugado com peroxidase (His-1,
Sigma-Aldrich), diluido 1:1500 em PBS 1X contendo 0,05% de Tween 20 e 1% de
leite magro em pó. Alternativamente, a incubação pode ser efectuada a 4ºC durante
a noite.
4- Lavar 3 vezes (5 minutos cada) com PBS contendo 0,05% Tween 20, de forma a
remover o excesso de anticorpo.
5 - Deixar escorrer o líquido e colocar a membrana sobre uma película de filme
aderente, com a face contendo a proteína virada para cima. Colocar 3 ml de
substrato da peroxidase (3,3´,5,5´ tetrametilbenzidina, TMB, Sigma). Deixar actuar
5-15 minutos. Monitorizar visualmente a reacção.
6 - Remover o substrato quando as bandas de proteína estiverem visíveis e o sinal
de fundo for ainda baixo.
7 - Lavar a membrana com água destilada durante 1 hora e guardar a membrana
seca entre 2 folhas de papel 3MM, no escuro.

19
Referências
Mahenthiralingam, E., T.A. Urban, J. Goldberg (2005). The multifarious, multireplicon Burkholderia cepacia complex. Nature Reviews Microbiology 3 144-156. Moreira, L.M., P.A. Videira, S.A. Sousa, J.H. Leitão, M.V. Cunha, I. Sá-Correia (2003). Identification and physical organization of the gene cluster involved in the biosynthesis of Burkholderia cepacia complex exopolysaccharide. Biochemical and Biophysical Research Communications 312, 323-333. Leitão, J.H., S.A. Sousa, A.S. Ferreira, C.G. Ramos, I.N. Silva, L.M. Moreira (2010). Pathogenicity, virulence factors, and strategies to fight against Burkholderia cepacia complex pathogens and related species. Applied Microbiology and Biotechnology 87, 31-40. Richau, J. A., J.H. Leitão, M. Correia, L. Lito, M.J. Salgado, C. Barreto, P. Cescutti, I. Sá-Correia, (2000). Molecular typing and exopolysaccharide biosynthesis of Burkholderia cepacia isolates from a Portuguese cystic fibrosis center. Journal of Clinical Microbiology 38 1651-1655. Sousa, SA, C. G. Ramos, F. Almeida, L. Meirinhos-Soares, J. Wopperer, S. Schwager, L. Eberl, J. H. Leitão (2008). Burkholderia cenocepacia J2315 acyl carrier protein: a potential target for antimicrobials´ development?. Microbial Pathogenesis 45 331-336.

20
ANEXO
MEIO SB: 32g/l de triptona; 20 g/l de extracto de levedura; 5 g/l de NaCl. Adicionar
água e ajustar pH o 7,5. Esterilizar em autoclave.
MARCADOR DE PESOS MOLECULARES PARA PROTEÍNAS LMW (BioRad) 97,0; 66,0; 45,0; 30,0; 20,1; 14,4 kDa Tampão Towbin: 48 mM Tris-base; 39 mM glicina; 0,04% (p/v) SDS; 20% metanol,
pH 8,2 – 8,4.
Tampão PBS 10X : 1,4 M NaCl; 27 mM KCl; 10 mM Na2HPO4, 18 mM KH2PO4; pH
7,4.













![DE BIOQUÍMICA E FISIOLOGIA MICROBIANA · µg de proteína total [determinados com base no método de Bradford (1976), ponto B.], às quais se adicionou 5 µl de corante de aplicação](https://static.fdocumentos.com/doc/165x107/612e62cf1ecc51586942c7c4/de-bioqumica-e-fisiologia-microbiana-g-de-protena-total-determinados-com.jpg)
![[PPT]Bioquímica e Fisiologia Microbiana - Técnico … · Web viewBioquímica e Fisiologia Microbiana Ciclo celular bacteriano Sistema Arc (aerobic respiration control) A schematic](https://static.fdocumentos.com/doc/165x107/5bebae9109d3f2cb318c0a7e/pptbioquimica-e-fisiologia-microbiana-tecnico-web-viewbioquimica-e-fisiologia.jpg)


