
de Qualidade da Técnica de PCR -...
103
Faculdade de Farmácia da Universidade do Porto Mestrado em Controlo de Qualidade Controlo e Garantia de Qualidade da Técnica de PCR Ana Elisabete Pereira Correia de Oliveira Dezembro 2008
Transcript of de Qualidade da Técnica de PCR -...

Faculdade de Farmácia da Universidade do Porto
Mestrado em Controlo de Qualidade
Controlo e Garantia de Qualidade da Técnica
de PCR
Ana Elisabete Pereira Correia de Oliveira
Dezembro 2008

Faculdade de Farmácia da Universidade do Porto
Mestrado em Controlo de Qualidade
Controlo e Garantia de Qualidade da Técnica de PCR
Área: Medicamentos e Plantas Medicinais
Orientador:
Professor Doutor Rui Manuel de Medeiros Melo Silva
Co‐orientadora:
Professora Doutora Maria de São José Garcia Alexandre Nascimento da Fonseca
Dissertação de Candidatura ao Grau de Mestre
apresentado à Faculdade de Farmácia da
Universidade do Porto.
Trabalho realizado no laboratório de
Oncologia Molecular do IPO‐Porto
e financiado pela Fundação Astrazeneca.
Ana Elisabete Pereira Correia de Oliveira
Dezembro 2008

ii
Declaração relativa à reprodução:
É autorizada a reprodução integral desta dissertação apenas para efeitos de investigação
mediante declaração escrita do interessado, que a tal se compromete.

iii
Agradecimentos
O presente trabalho não seria possível sem a ajuda e o apoio de determinadas pessoas que
demonstraram o seu apoio e carinho incondicional. Tentando não esquecer ninguém, quero
agradecer:
Em primeiro lugar, à Fundação Astrazeneca, que financiou este projecto.
À Professora Doutora Maria Beatriz Prior Pinto Oliveira, coordenadora do Mestrado em
Controlo de Qualidade, pela sua preocupação, atenção e apoio prestados, pela rápida resposta e
bons conselhos sempre que solicitada, aproveitando para felicitá‐la pela excelente coordenação
deste mestrado.
Ao Professor Doutor Rui Medeiros, meu orientador neste trabalho, pela simpatia e prontidão
com que me recebeu, pelo apoio, pela ginástica temporal para me incluir na sua agenda, por si só
cheia, pela paciência e entendimento, e principalmente pela orientação neste projecto inesperado.
À Professora Doutora Maria de São José Nascimento, minha co‐orientadora que
prontamente aceitou co‐orientar este projecto, pelas palavras reconfortantes de força e
disponibilidade, que sempre me foi transmitindo nos últimos anos e pelo seu optimismo contagiante,
que se revelou muito útil nos últimos meses.
À Doutora Albina Marinho, do laboratório de Virologia do IPO‐Porto, pela simpatia e atenção
prestada e pela partilha de conhecimentos que em muito enriqueceram este trabalho.
A todos os meus colegas e recentes amigos do laboratório de Oncologia Molecular do IPO‐
‐Porto, pela simpatia com que me receberam e se deixaram observar nas suas rotinas diárias.
A todos os meus antigos colegas e amigos do departamento de Química Orgânica da
Faculdade de Farmácia da Universidade do Porto, especialmente à Professora Doutora Maria José
Gonzalez e ao Carlos, pela grande amizade, alegria e apoio que sempre me demonstraram.
A todos os meus familiares e amigos, pela presença e apoio constante.
À Cátia, amiga e antiga companheira de laboratório e mestrado, pela amizade incondicional.
Aos meus pais, pelo enorme carinho e apoio com que acompanharam as diversas fases deste
mestrado, desculpando todos os pequenos momentos de má disposição e falta de disponibilidade.
Ao Hélder, meu irmão, a pessoa que mais acredita em mim, por todas as horas infindáveis de
conversa e carinho.
Ao Pedro, que me acompanhou desde sempre neste mestrado, pela confiança, incentivo e
carinho nas horas certas.

iv
Resumo
Esta dissertação pretende apresentar uma proposta de avaliação interna da qualidade das
técnicas de PCR‐RFLP e Real‐Time PCR, utilizadas diariamente no laboratório de Oncologia Molecular
do IPO‐Porto, com fins de diagnóstico e investigação epidemiológica e farmacogenómica. Este
trabalho baseou‐se apenas na sua utilização qualitativa em projectos de investigação.
O controlo e a garantia de qualidade têm um papel vital hoje em dia, ajudando a garantir a
fiabilidade de resultados e produtos. Os critérios de qualidade tornaram‐se essenciais para assegurar
a correcta utilização de tecnologias genéticas que se desenvolveram nas últimas décadas, como é o
caso da amplificação dos ácidos nucleicos.
Desde a sua descoberta, a amplificação dos ácidos nucleicos in vitro, através da técnica de
PCR, tornou‐se uma ferramenta muito poderosa em laboratórios de diagnóstico. No entanto, uma
das desvantagens apontadas tem sido a falta de requisitos e regulamentos padronizados para esta
técnica, assim como a dificuldade de controlar todas as variáveis intervenientes, podendo
comprometer a qualidade dos resultados. Sendo assim, e face à nova tendência universal de garantia
de qualidade, é necessário implementar protocolos de controlo e sistemas de gestão de qualidade.
O HACCP é uma metodologia preventiva que assenta numa abordagem científica e
sistemática de identificação e avaliação de perigos específicos em todas as etapas da produção.
Apesar do uso do HACCP não se ter ainda expandido ao laboratório de técnicas genéticas, a base
deste sistema pode ser adaptada a diferentes sectores, com alta probabilidade de sucesso.
Através de uma cuidada observação do laboratório de Oncologia Molecular e seus
colaboradores e utilizando as bases do sistema de gestão de qualidade HACCP, foram elaborados os
fluxogramas das técnicas de PCR‐RFLP e Real‐Time PCR, e preenchidas tabelas de identificação de
perigos, pontos críticos de controlo, medidas de controlo e correctivas, reunindo todos os cuidados e
acções a realizar durante a técnica e na resolução de problemas. A partir daqui, foram adaptados
critérios de avaliação dos vários sectores do laboratório, utilizados na observação diária e que
poderão ser implementados futuramente como medidas de avaliação interna do local, sua
organização e colaboradores.
Com a utilização dos fundamentos do HACCP, os critérios e a observação, concluiu‐se que os
principais requisitos de trabalho eram, em geral, cumpridos, mas que faltava um protocolo que
avaliasse internamente a qualidade dos vários passos, para além dos resultados. Tendo em conta os
pontos críticos identificados e a necessária adaptação ao local, elaborou‐se um plano de controlo de
qualidade interno, a experimentar, melhorar e implementar num futuro próximo.

v
Abstract
This thesis aims to present a proposal for assessing internal quality of the technique of PCR‐
‐RFLP and Real‐Time PCR, used daily in the laboratory of Molecular Oncology of IPO‐Porto. These
techniques are used for purposes of diagnosis and epidemiological and pharmacogenomics research.
This work was based only on their qualitative use in research projects.
Quality control and assurance play a vital role nowadays helping to ensure the reliability of
tests results and industrial products. The criteria of quality have become essential to ensure proper
use of genetic technologies that have been developed in recent decades, such as the amplification of
nucleic acids.
In vitro amplification of nucleic acids using PCR has become, since its discovery, a powerful
diagnostic tool. However, the lack of officially approved, standardized regulations and instructions
has been pointed as one of the disadvantages, as well as the difficulty to control all the variables.
Both can compromise the quality of the results. So, it is necessary to introduce a quality control plan
and management system, in order to achieve the new universal trend of quality assurance.
HACCP is a preventive approach based on a scientific and logical system designed to identify
and evaluate specific hazards at all stages of the process. Despite the use of HACCP has not yet
expanded to the genetic laboratory, the foundation of this system can be adapted and implemented
in different sectors, with high probability of success.
Through a careful observation of the laboratory of Molecular Oncology and its collaborators,
and using the fundamentals of quality management system HACCP, the flowcharts were developed
for PCR‐RFLP and Real‐Time PCR techniques, as well as tables for the identification of hazards, critical
control points, control and corrective measures, bringing together all the care and actions to be
undertaken during the technique and problem‐solving. From here, some criteria were also adjusted
for evaluating the various sectors of the laboratory, and used for daily observation. These criteria
may be implemented as future measures of internal evaluation of the site, its organization and
employees.
Using the fundamentals of HACCP, the criteria and observation, it was concluded that the
main requirements of work were generally satisfied, but we needed a protocol to evaluate the
internal quality of the various steps, in addition to the results. Given the critical points identified and
the necessary adaptation to local, a plan of internal quality control was prepared, to experiment,
improve and implement in the near future.

vi
Objectivos e Organização da Dissertação
Face à importância actual da qualidade e à falta de documentos, registos e alguns
procedimentos que controlem e comprovem a qualidade dos resultados e ensaios em geral
praticados no laboratório de Oncologia Molecular do IPO‐Porto, foi objectivo desta dissertação iniciar
um capítulo de integração do controlo e garantia da qualidade no laboratório, começando assim por
exercer um controlo interno dos seus próprios ensaios.
Alguns termos relativos a material, reagentes ou técnicas foram mantidos em inglês ao longo
da dissertação, pela sua utilização habitual no laboratório.
A dissertação está organizada em 4 partes.
Na primeira parte, denominada Introdução, efectua‐se uma abordagem geral ao termo
Qualidade, à sua história e evolução até ao tempo actual.
A segunda parte, Controlo da Qualidade de Técnicas Genéticas, pretende rever o sector da
qualidade nos ensaios genéticos e está subdividida em três pontos. No primeiro descreve‐se, de uma
forma geral, a importância do controlo de qualidade nas técnicas genéticas; no segundo, faz‐se uma
abordagem do controlo interno utilizado em ensaios genéticos, das variáveis que podem afectar os
ensaios e que é necessário controlar; no terceiro, referem‐se brevemente os esquemas de controlo
externo e de ensaios interlaboratoriais já existentes para as técnicas genéticas.
Na terceira parte, intitulada HACCP, faz‐se uma descrição deste sistema de gestão da
qualidade, a sua história, normas, utilização presente e futura.
A quarta parte, Proposta de Modelo de Controlo e Garantia de Qualidade, constitui a parte
principal deste trabalho, em que são apresentados os fluxogramas, as tabelas HACCP, os critérios e a
proposta de plano de avaliação.
Na quinta parte, denominada Resultados e Discussão, apresentam‐se os resultados da
observação diária dos colaboradores do laboratório e da aplicação dos critérios referidos na quarta
parte. Segue‐se uma breve discussão desses resultados e do próprio modelo proposto.
A sexta parte, Conclusões e Perspectivas, resume as principais conclusões que foram
retiradas a partir deste trabalho e como se pretende evoluir a partir daqui.
Por último, a sétima parte compreende a Bibliografia utilizada na realização deste trabalho.

vii
Índice Geral
Agradecimentos iii
Resumo iv
Abstract v
Objectivos e Organização da Dissertação vi
Índice de Figuras ix
Índice de Tabelas x
Abreviaturas e Símbolos xii
I. Introdução 1
II. Controlo de Qualidade de Técnicas Genéticas 7
1. Considerações Gerais 8
2. Controlo de Qualidade Interno de Técnicas de Amplificação de Ácidos Nucleicos ‐ PCR 13
3. Esquemas de Controlo Externo 26
III. HACCP 31
IV. Proposta de Modelo de Controlo e Garantia de Qualidade 37
1. Adaptação de um Plano HACCP 38
2. Critérios de Avaliação 54
3. Plano de Avaliação de Qualidade 63
V. Resultados e Discussão 66
1. Adaptação de um Plano HACCP 67
2. Análise dos Resultados da Observação dos Colaboradores e Utilização dos Critérios 67
3. Plano de Avaliação de Qualidade 79

viii
VI. Conclusões e Perspectivas 80
VII. Bibliografia 83

ix
Índice de Figuras
Figura 1 ‐ Ciclo de Shewart/Deming 4
Figura 2 ‐ Evolução histórica da qualidade em Portugal 5
Figura 3 ‐ Variáveis pré‐analíticas em testes de laboratório 22
Figura 4 ‐ Erros presentes nas três fases do processo de um teste de laboratório 23
Figura 5 ‐ Anatomia de um esquema de controlo externo 27
Figura 6 – Fluxograma de PCR‐RFLP 40
Figura 7 – Fluxograma de Real‐Time PCR 41
Figura 8 – Árvore de decisão de pontos críticos de controlo 50

x
Índice de Tabelas
Tabela 1 ‐ Princípios de controlo de qualidade interno para métodos de genotipagem
baseados em PCR
14
Tabela 2 ‐ Operações realizadas em técnicas de ácidos nucleicos e potenciais fontes de erro 16
Tabela 3 ‐ Falsos positivos ‐ controlo e melhorias 17
Tabela 4 ‐ Falsos negativos ‐ causas, prevenção e detecção 19
Tabela 5 ‐ Fases do processo de um laboratório médico e possíveis erros 21
Tabela 6 ‐ Indicadores de qualidade e especificações (limites de aceitabilidade) da fase
pré‐analítica
28
Tabela 7 ‐ Indicadores de qualidade e especificações (limites de aceitabilidade) da fase
analítica
28
Tabela 8 ‐ Indicadores de qualidade e especificações (limites de aceitabilidade) da fase
pós‐analítica
29
Tabela 9 – Descrição do produto final 39
Tabela 10 – Índice de risco para diferentes valores de severidade e probabilidade 42
Tabela 11 – Descrição dos perigos, respectivas causas e medidas de controlo de cada
etapa pré‐analítica
43
Tabela 12 – Descrição dos perigos, respectivas causas e medidas de controlo de cada
etapa analítica
47
Tabela 13 – Descrição dos perigos, respectivas causas e medidas de controlo de cada
etapa pós‐analítica
49

xi
Tabela 14 – Identificação de pontos críticos de controlo 51
Tabela 15 – Monitorização e medidas correctivas de cada ponto crítico de controlo 52
Tabela 16 ‐ Cumprimento de critérios pré‐analíticos 68
Tabela 17 – Cumprimento de critérios analíticos 69
Tabela 18 – Cumprimento de critérios pós‐analíticos 72

xii
Abreviaturas e Símbolos
A260 Absorvância a 260nm
A280 Absorvância a 280nm
ACMG American College of Medical Genetics
ASAE Autoridade de Segurança Alimentar e Económica
ASCO American Society of Clinical Oncology
AZF Factor de Azoospermia
BRCA Cancro da Mama Hereditário
CAC Codex Alimentarius Commission
CAP College of American Pathologists
CE Comissão Europeia
CEE Comunidade Económica Europeia
CF Fibrose Cística
CLIAC Clinical Laboratory Improvement Advisory Committee
CLIA Clinical Laboratory Improvement Amendments
CMGS Clinical Molecular Genetics Society
CMQ Ciclo de Melhoria da Qualidade
CMT Doença de Charcot‐Marie‐Tooth
DMD Distrofia Muscular de Duchenne
DNA Ácido Desoxirribonucleico
DNAse Desoxirribonuclease
ε Coeficiente de Extinção Molar

xiii
EDTA Ácido Etilenodiaminotetracético
EMQN European Molecular Genetics Quality Network
EQA External Quality Assessment
EQUAL Multinational External Quality Assay programmes in Clinical Molecular
Diagnostics based on Performance and Interpretation of PCR assay methods
EU‐QCCA European Union Quality Control Concerted Action
FA Anemia de Fanconi
FAO Food and Agriculture Organization
FDA Food and Drug Administration
FDCA Federal Food, Drug and Cosmetic Act
FMIA Federal Meat Inspection Act
GMP Good Manufacturing Practices (Boas Práticas de Fabrico)
HACCP Hazard Analysis Critical Control Point
HCV Hepatitis C Virus (Vírus da Hepatite C)
HD Doença de Huntington
IEC International Electrotechnical Commission
IPO Instituto Português de Oncologia
IR Índice de Risco
ISO International Standard Organization
NASA National Aeronautics and Space Administration
NCCLS National Committee for Clinical Laboratory Standards
NIBSC National Institute for Biological Standards and Control
OMS Organização Mundial de Saúde

xiv
PCC Ponto Crítico de Controlo
PCR Polymerase Chain Reaction (Reacção de Polimerização em Cadeia)
PDCA Plan‐Do‐Check‐Act (Planear‐Fazer‐Verificar‐Agir)
QA Quality Assurance (Garantia de Qualidade)
QMS Quality Management System (Sistema de Gestão da Qualidade)
RNA Ácido Ribonucleico
RNAse Ribonuclease
RFLP Restriction Fragment Length Polymorphism
SACGT Secretaries Advisory Committee on Genetic Testing
SDS Dodecil Sulfato de Sódio
SoGAT Standardization of Gene Amplification Techniques
SOP Standard Operating Procedures
TAN Técnicas de Amplificação de Ácidos Nucleicos
TQM Total Quality Management (Gestão da Qualidade Total)
UNG Uracil‐N‐glicosilase
UV Ultravioleta

I. Introdução

Introdução
2
A Qualidade é um termo muito actual e de difícil definição. De uma forma geral, pode‐se
dizer que qualidade é antecipar os requisitos dos clientes e fornecê‐los com precisão todas as vezes.
Por outro lado, a qualidade total, termo mais actual, é vista como uma melhoria contínua, tendo
como objectivo satisfazer os requisitos dos clientes, a um custo óptimo, através do empenho de
todos [1]. No entanto, o termo “qualidade” é utilizado em muitas situações diferentes, variando a
sua definição.
Hoje em dia, a qualidade tem sido vista cada vez mais como uma estratégia efectiva para
redução de custos, assegurando a vitalidade de um negócio e surgindo como o elemento chave para
um aumento de produtividade e confiança, face às exigências cada vez maiores da população. Um
pequeno grupo de “Gurus da Qualidade” tem vindo a defender o caminho da qualidade como um
processo de melhoria, um ciclo interminável e indispensável, um caminho que pode ser percorrido
através de diferentes meios. Quatro dos mais conhecidos são William Deming, Joseph Juran, Philip
Crosby e William Conway, que defendem diferentes abordagens para um mesmo fim e diferentes
definições do termo qualidade [2].
Segundo Deming, boa qualidade não é necessariamente um sinónimo de alta qualidade, mas
sim “um grau previsível de uniformidade e de confiança ao mais baixo custo e adaptado às
necessidades do mercado”. É um forte defensor dos métodos estatísticos e cartas de controlo, de
modo a que não haja somente uma inspecção da matéria inicial e final, mas sim todo um controlo de
produção, um acompanhamento do processo total [2].
Juran, por sua vez, menciona qualidade como “adequação ao uso” e “conformidade com as
especificações”, interessa‐se pela gestão global da qualidade e defende o conceito de círculos de
qualidade, que melhoram a comunicação entre os órgãos de gestão e os trabalhadores [2].
Crosby definiu qualidade como a conformidade com as exigências dos clientes, só podendo
ser avaliada pelo custo da não conformidade. A sua abordagem defende que o único padrão de
performance aceitável é o de zero defeitos e que a prevenção é o único sistema a utilizar, querendo
com isso dizer a “perfeição”, não admitindo a existência de níveis estatísticos aceitáveis de qualidade
[2].
Por último, Conway não fala de uma definição específica de qualidade, mas engloba‐a numa
definição mais ampla de “gestão pela qualidade”, como sendo “o desenvolvimento, fabrico, gestão e
um fornecimento consistente de produtos e serviços a baixo custo que os clientes desejam e/ou
necessitam”. Defende a necessidade de criação de um novo sistema de gestão, cuja primeira tarefa
deve ser a melhoria contínua em todas as áreas. É também um grande defensor dos métodos
estatísticos [2].
Muitas outras definições de qualidade se podem encontrar, como sendo o “grau de
satisfação de requisitos dado por um conjunto de características intrínsecas” [3], ou “the right result

Introdução
3
on the right specimen from the right patient that is accurate, timely and properly interpreted” [4,5],
uma definição que talvez se aproxime mais do campo de diagnóstico, ou uma definição mais
corrente, em que a palavra qualidade é usada para significar “excelência”.
De qualquer modo, seja qual for a definição usada, a qualidade é um conceito horizontal, que
abrange todas as actividades e todos os sectores. É um factor competitivo que pode diferenciar os
produtos e as competências e que abrange muitos conceitos, como controlo, garantia e gestão de
qualidade.
O controlo de qualidade é utilizado para garantir que os produtos ou serviços são realizados
ou produzidos de modo a satisfazer ou exceder os requisitos do consumidor. A garantia de qualidade
(QA, Quality Assurance) é a parte da gestão da qualidade orientada no sentido de gerar confiança
quanto à satisfação dos requisitos da qualidade [3] e inclui todas as actividades desde o design,
desenvolvimento, produção, instalação, manutenção e documentação. No fundo, a QA refere‐se a
processos de produção planeados e sistemáticos que providenciam confiança na adequação do
produto aos seus requisitos iniciais. A QA não consegue garantir completamente a produção de
produtos de qualidade, mas torna‐a mais provável. Pode‐se dizer que enquanto o controlo de
qualidade enfatiza o teste e o bloqueio da libertação de produtos defeituosos, a QA preocupa‐se com
a melhoria e estabilização da produção de modo a evitar ou, pelo menos, minimizar questões que
possam levar a produtos defeituosos. Mas a QA não elimina a necessidade de controlo de qualidade,
pois alguns parâmetros são de tal maneira importantes que o seu teste é necessário, como
prevenção. Dois documentos internacionais importantes onde a garantia de qualidade é mencionada
e onde se especificam requisitos e meios para a conseguir são a ISO 17025 e a ISO 9000.
A gestão da qualidade é o conjunto de actividades coordenadas para dirigir e controlar uma
organização no que respeita à qualidade [3]. Resumidamente, assegura que todas as actividades
necessárias para desenhar, desenvolver e implementar um produto ou serviço são eficientes, no que
respeita à sua performance. A gestão de qualidade pode ter três componentes: controlo, garantia e
melhoria de qualidade, focando‐se não só na qualidade do produto mas também nos meios para a
alcançar. Existem muitos métodos para gerir e melhorar a qualidade, melhorando o produto, o
processo e a formação das pessoas. Uma das estratégias mais usadas é o Ciclo de Melhoria da
Qualidade (CMQ), uma abordagem sistemática e disciplinada para identificar oportunidades de
melhoria da qualidade e implementar soluções duradouras [1]. A implementação baseia‐se num
processo em quatro passos, muito utilizado na melhoria de empresas e negócios: Plan‐Do‐Check‐Act
(PDCA) ou Planear, Fazer, Verificar e Agir, muitas vezes conhecido como ciclo de Deming ou de
Shewhart. As diferentes fases deste processo estão esquematizadas na figura seguinte:

Introdução
4
Figura 1 ‐ Ciclo de Shewhart/Deming (adaptado de [1]).
O CMQ ajuda a resolver problemas e a identificar/implementar soluções. Quando todos os
gestores utilizam o mesmo processo, o trabalho em grupo pode ser melhorado de forma significativa
[1]. Resumidamente, ao planear, estabelecem‐se os objectivos e processos necessários para atingir
os resultados esperados, ou seja, para o output ser o esperado; ao fazer, implementa‐se um novo
processo, uma nova solução; ao avaliar, controla‐se o novo processo, comparando os resultados
obtidos com os esperados, avaliando os desvios e estudando as razões; por último, agir consiste em
determinar as causas dos desvios, implementar o processo e estudar ou reflectir sobre o
desenvolvimento.
Outra estratégia designa‐se por gestão da qualidade total (TQM, Total Quality Management).
Este método é uma estratégia de gestão que visa incorporar a consciência de qualidade em todos os
processos organizacionais. Há uma focalização nos clientes, a participação de todos os sectores,
inclusive clientes e fornecedores, de modo a que se possa avaliar o desempenho de todos os
processos e assim conseguir uma melhoria contínua [5]. A TQM é vista como uma gestão de futuro
em Portugal, como se pode ver na esquematização da evolução histórica da qualidade em Portugal:

Introdução
5
Figura 2 ‐ Evolução histórica da qualidade em Portugal (adaptado de [6]).
Muitas outras estratégias são conhecidas e utilizadas, como os círculos de qualidade, em que
existe uma abordagem de grupo para a melhoria, ou as normas da qualidade, mundialmente aceites.
A International Organization for Standardization (ISO) criou as normas de Sistema de Gestão da
Qualidade (QMS, Quality Management System), a série ISO 9000 (9001, 9002 e 9003), aplicáveis a
diferentes indústrias e que são revistas regularmente. As normas ISO 9000 são um ponto de partida
que não definem técnicas de gestão da qualidade, mas definem critérios uniformes pelos quais pode
ser avaliado um sistema de qualidade e, por consequência, melhorado. A revisão do ano 2000 (série
ISO 9000:2000), por necessidade de alargar o seu âmbito a outros processos, levou à integração da
ISO 9002 e 9003 numa única norma, a ISO 9001:2000. Sendo assim, a ISO 9000:2000 passa a referir‐
‐se aos fundamentos e vocabulário de sistemas de gestão da qualidade e a ISO 9001:2000 aos seus
requisitos, exigências e processos necessários para que o produto alcance as expectativas de
qualidade. Nesta série 9000:2000, formulou‐se também a ISO 9004:2000, que descreve as guidelines
para uma melhoria da qualidade acima do padrão básico da ISO 9001:2000, ou seja, as linhas de
orientação para a melhoria do desempenho de um sistema de gestão da qualidade. A nova estrutura
das ISO 9000:2000 baseia‐se no PDCA já mencionado, na existência de um ciclo de qualidade,
aproximando‐se muito da TQM, numa abordagem por processos na gestão da qualidade [7].
Recentemente a ISO publicou uma nova edição das normas ISO 9001, a ISO 9001:2008. Nesta nova
revisão não foram introduzidos novos requisitos, apenas algumas alterações e principalmente
clarificações do texto da norma, resultando numa transição fácil para aqueles que já têm um sistema
de gestão de qualidade correctamente implementado e de acordo com a ISO 9001:2000.
Estas normas do QMS permitem a certificação dos processos e da organização e não do
produto ou serviço, não certificando a sua qualidade. Recentemente a ISO lançou a ISO 22000:2005,
para a indústria alimentar, que cobre os princípios da ISO 9000 e do Hazard Analysis Critical Control

Introdução
6
Point (HACCP), originando uma única norma integrada para esta indústria. Além disso, existem
algumas normas que suportam a gestão da qualidade, descrevendo processos, como a ISO 12207 e
ISO 15288, e outras que tratam da avaliação e melhoria, como a ISO 15504.
Face à cada vez maior preocupação com a qualidade e a vasta oferta de programas e
soluções, cada empresa ou local deve adequar o seu sistema de qualidade às suas próprias
possibilidades e necessidades, passando muitas vezes pelo próprio design do modelo de qualidade a
utilizar, que pode integrar diferentes partes das várias estratégias faladas.
O objectivo deste trabalho, a proposta de um modelo de controlo de qualidade interno com
base no HACCP, passa por isso mesmo. Pretende‐se iniciar um processo, tendo como base um
sistema de gestão de qualidade, o HACCP, aproveitando‐se certas características de outros métodos,
como o PDCA, cujos objectivos e fundamentos nunca devem ser esquecidos, assim como as suas
ferramentas, como a esquematização, ou a preocupação da gestão por processos, retirada do
método TQM. Com o HACCP, o planeamento tem de ser muito bem feito, avalia‐se todo o processo,
todas as possíveis fontes e causas de erro, as suas medidas preventivas e correctivas e respectiva
avaliação. A implementação futura de controlo, garantia e gestão de qualidade deve ser feita
segundo esta abordagem, assim como melhorias após implementação. No fundo, trata‐se de evoluir
o laboratório na qualidade da mesma forma que o próprio conceito foi evoluindo, como está
representado na Figura 2, permitindo assim um maior grau de confiança, credibilidade e
reconhecimento.

II. Controlo de Qualidade de Técnicas Genéticas

Controlo de Qualidade de Técnicas Genéticas
8
1. Considerações Gerais
O percurso dos laboratórios de diagnóstico de doenças humanas tem sido permanentemente
alterado pelo rápido aparecimento e desenvolvimento da tecnologia molecular e pela sequenciação
do genoma humano [8]. Poucas décadas passaram desde que Kan e Dozy reportaram o primeiro
teste de DNA com uma enzima de restrição para testar anemia falsiforme [9], assinalando o
nascimento da clínica genética e molecular moderna. O que começou como um pequeno conjunto de
laboratórios académicos, que realizavam ensaios genéticos para doenças raras e muitas vezes
debilitantes, tornou‐se essencial hoje em dia, levando a que as técnicas genéticas e moleculares
entrassem em muitas áreas de diagnóstico e clínica. O mundo da biologia molecular está em
constante mudança e desenvolvimento, em que cada novo e pequeno passo representa uma
expansão do impacto e da utilidade que esta tecnologia pode ter na clínica, nos pacientes e
familiares.
Os testes genéticos desenvolveram‐se a meio da década de 80, em laboratórios que
tentavam identificar genes relacionados com determinadas doenças. Nos últimos 15 anos, os testes
genéticos passaram do “nada” para se tornarem parte principal de um laboratório de medicina [10].
Os ensaios genéticos de DNA ou RNA, para diagnóstico de doenças infecciosas, neoplásicas ou
genéticas, têm sido amplamente utilizados devido ao recente e enorme progresso da biologia
molecular e da biotecnologia e são essenciais para o tratamento e qualidade de vida do doente,
providenciando um diagnóstico precoce e mais preciso [11].
Os dados sobre o número de testes genéticos que são realizados são escassos, mas sabe‐se
que só no Reino Unido são feitos mais de 50.000 testes por ano, o que indica que talvez sejam
milhões em todo o mundo. A tendência é para que estes números aumentem, principalmente com o
conhecimento da sequência genómica humana. Para além da sua utilização como método de
diagnóstico ou de reconhecimento de uma mutação, os testes genéticos estão a expandir este papel
mais tradicional para a previsão, prevenção e tratamento de doenças crónicas, ou seja, para
determinar a susceptibilidade a determinadas patologias, como a diabetes, doenças cardíacas,
cancro [12,13], infecções, e também para uma aplicação farmacogenómica [14], de modo a prever a
resposta a um fármaco [8,10]. Esta expansão só foi possível através do conhecimento que o indivíduo
pode ser mais ou menos susceptível a uma doença, dependendo da sua base genética. A combinação
das variações genéticas e da exposição ambiental influencia a probabilidade de um indivíduo
desenvolver determinada patologia [8], e é isso que cada vez mais se tenta determinar.
Com a necessidade de usar estas poderosas novas tecnologias no mundo médico, pouca
atenção se tem prestado aos critérios de qualidade. O grande desenvolvimento dos ensaios
genéticos tornou mais visível a necessidade de regulamentação e padronização e nos últimos anos

Controlo de Qualidade de Técnicas Genéticas
9
começaram a surgir esforços para implementar e testar a qualidade dos resultados dos ensaios
genéticos. Esta necessidade de certificação e qualidade é realçada pela alta expectativa que está
aliada a esta tecnologia, compreensivelmente, sendo necessário manter um determinado grau de
confiança nestas técnicas. Agências regulamentares como a Food and Drug Administration (FDA), a
Secretaries Advisory Committee on Genetic Testing (SACGT) e a Clinical Laboratory Improvement
Advisory Committee (CLIAC) discutem este assunto e principalmente as duas últimas agências têm
deliberado sobre como os testes genéticos devem ser ordenados, realizados, comunicados e
supervisionados nos Estados Unidos.
Os ensaios genéticos são diferentes dos outros testes laboratoriais de diagnóstico em
diversos aspectos, acentuando a necessidade de assegurar a qualidade dos resultados:
A. A “composição” genética de um indivíduo não se altera com o tempo, os pacientes
normalmente são testados uma só vez e um resultado incorrecto pode ficar para sempre
como certo [8,10];
B. Os resultados de um teste genético podem resultar em consequências para outros membros
da família [10];
C. As pessoas em geral têm um alto grau de confiança nos resultados genéticos e poucos
questionam a sua validade [10];
D. No que se refere à avaliação de susceptibilidade, o teste é muitas vezes realizado em pessoas
assintomáticas e os resultados podem ou não ser suportados por outros dados, como a
história familiar [8].
Sendo assim, tudo o que se referiu contribui para a necessidade de estabelecimento de
medidas de controlo de qualidade. Já foram mencionadas algumas definições de qualidade, seu
controlo e garantia, mas dentro do universo dos laboratórios de técnicas genéticas, o controlo de
qualidade pode ser definido como o controlo de um processo para garantir que os resultados
tenham a qualidade exigida [15,16]. A aplicação do controlo de qualidade nos laboratórios foi
introduzida por Levey e Jennings em 1950, como uma ferramenta para melhorar o desempenho de
um laboratório [16,17]. Desde aí, os fundamentos práticos não sofreram grandes alterações,
podendo‐se enumerar algumas modificações na frequência de análise de amostras controlo, no
número de controlos e nas regras de aceitação ou rejeição de um teste, baseando‐se nos resultados
dos controlos [16].
Face à importância que o conceito de controlo de qualidade tem hoje em dia, a garantia que
é necessário dar e comprovar ao público‐alvo, os laboratórios procuram implementar esquemas de
qualidade e todas as medidas recomendadas, que podem ser indiscutivelmente importantes, como a

Controlo de Qualidade de Técnicas Genéticas
10
inclusão de um controlo positivo e negativo em cada ensaio, ou de importância dúbia, como o registo
diário da temperatura do frigorífico [18]. Independentemente dessa importância, um modelo de
controlo de qualidade é concebido com todas essas ferramentas, para que se consiga o resultado
considerado como “certo”. Pode‐se definir um resultado “certo” pela negativa, ou seja, não é um
falso positivo ou um falso negativo. Um resultado “certo” deve ser sim reprodutível, independente
do genótipo, principalmente na quantificação, relevante clinicamente e reembolsável [18]. É tendo
em conta todos os conceitos e a importância relativa de cada um que se vai construindo um modelo
de controlo de qualidade, passo por passo.
Um programa de controlo de qualidade genérico de diagnóstico deve ser constituído por,
pelo menos, três componentes essenciais: controlos, procedimentos e regras. Os controlos contêm
um analito em concentração conhecida, usados para se conseguir precisão e reprodutibilidade. Os
procedimentos referem‐se essencialmente ao número de vezes que os controlos devem ser testados
e onde são colocados. As regras definem como os resultados dos controlos vão ser interpretados
para aceitar, solucionar ou rejeitar um ensaio. Na grande maioria das vezes aplicam‐se aqui as regras
de Westgard. No entanto, se os controlos forem fornecidos como parte de um kit, normalmente
também são fornecidas sugestões ou requerimentos para o uso dos controlos [19]. Estes três pontos
essenciais cobrem principalmente a fase analítica do processo, mas não o processo total, que se foi
descobrindo ser essencial num programa de controlo e garantia de qualidade.
Actualmente, a implementação de esquemas de controlo de qualidade e a padronização de
protocolos nos laboratórios são cada vez mais necessários, englobando a preparação da amostra,
laboratório, equipamento, amostras de referência, acreditação do laboratório, treino do pessoal e
validação de protocolos. Além disso, os estudos de controlo de qualidade externo e estudos
interlaboratoriais são essenciais [10,20]. É com o desenvolvimento de métodos de controlo de
qualidade para os testes genéticos humanos que vai continuar o crescimento destas técnicas e a sua
integração na prática clínica [8].
Já se têm desenvolvido determinadas guidelines e surgem cada vez mais propostas e
recomendações, como é o caso das desenvolvidas pelo National Committee for Clinical Laboratory
Standards (NCCLS), agora conhecido por Clinical and Laboratory Standards Institute, pelo American
College of Medical Genetics (ACMG), pelo Clinical Laboratory Improvement Amendments (CLIA) ou
pelo College of American Pathologists (CAP), em que alguns surgiram como resposta às deliberações
do SACGT e estabeleceram relações de trabalho com a FDA através de discussões com a FDA‐
‐Professional Organization Roundtable, com o objectivo de desenvolver um plano prático de
supervisionamento [8,21‐25].
O CLIA define muitos dos sistemas básicos de qualidade que um laboratório requer mas
faltam‐lhe guidelines específicas para ensaios genéticos e moleculares. No entanto há pelo menos

Controlo de Qualidade de Técnicas Genéticas
11
três guidelines com este objectivo que podem ser aplicadas a muitas áreas dos testes moleculares,
independentemente do campo de estudo [26]. O ACMG contém guidelines que remetem para a
citogenética, genética bioquímica e genética molecular. Para além disso, desenvolveu guidelines
específicas para determinadas doenças, que remetem para problemas específicos que aparecem
frequentemente em ensaios mais complexos. Todas estas guidelines conseguem cobrir as regras de
boas práticas num laboratório, validação de ensaios e assuntos técnicos específicos para métodos ou
doenças [26]. O CAP é a organização por excelência para acreditação dos laboratórios [26].
Uma outra organização, a Clinical and Laboratory Standards Institute utiliza peritos em
determinados campos para desenvolver guidelines para diagnósticos moleculares, incluindo testes
de genética molecular, hematopatologia molecular, sequenciação de DNA, diagnóstico por
microarray e testes de proficiência [26].
Falando mais especificamente da área de oncologia, destaca‐se uma organização, a American
Society of Clinical Oncology (ASCO), que representa os especialistas da área envolvidos nos cuidados
dos pacientes e pesquisa clínica. Esta organização reconheceu a necessidade de recomendações
específicas para esta área, principalmente na pesquisa de predisposição genética para cancro e
medicina preventiva. Os testes genéticos envolvidos têm de sofrer um controlo apertado, para
garantir resultados de qualidade elevada. Segundo eles, existem elementos críticos que têm de ser
controlados, tem de existir supervisão dos reagentes, ensaios, pessoal que realiza os ensaios,
controlo de qualidade e um formato padrão de comunicação dos resultados. Além disso, defendem a
participação dos laboratórios em programas de controlo externo e de acreditação, como os do CAP e
ACMG [27].
Em todas as organizações se assume que os programas de controlo interno e externo são
estabelecidos para garantir que os laboratórios consigam produzir e reproduzir resultados de alta
qualidade. Os testes de proficiência ou controlo externo identificam fraquezas e funcionam como um
guia para o desenvolvimento [24]. No entanto, um programa de QA engloba mais do que um
controlo interno e externo. Os programas podem variar entre laboratórios mas em geral
compreendem um manual de garantia de qualidade, os objectivos do programa, a qualidade dos
recursos, os procedimentos padronizados (SOP, Standard Operating Procedures), controlo de
qualidade interno e externo [28]. É o conjunto de todas estas acções e de todos os dados que vai
conseguir minimizar os erros e garantir uma maior confiança nos resultados dos testes do
laboratório.
Apesar do imenso crescimento que a área tem sofrido e do estabelecimento de todas estas
organizações, ainda existem muitos problemas no controlo e garantia de qualidade das técnicas
genéticas. Entre os factores que mais impedem o desenvolvimento de programas que assegurem e
avaliem a qualidade, foi eleito como um dos mais preponderantes, no controlo interno e externo, a

Controlo de Qualidade de Técnicas Genéticas
12
falta de amostras positivas ou amostras com mutações bem definidas associadas com doenças de
saúde pública. Além disso, a falta de métodos padronizados e os diferentes métodos analíticos de
cada laboratório também complicam a criação de um modelo de controlo de qualidade global,
principalmente de controlo externo [8].

Controlo de Qualidade de Técnicas Genéticas
13
2. Controlo de Qualidade Interno de Técnicas de Amplificação de Ácidos Nucleicos ‐ PCR
O controlo de qualidade tem um papel cada vez mais importante na implementação das
técnicas de amplificação de ácidos nucleicos (TAN) no diagnóstico. Nos últimos 10 anos estas técnicas
revolucionaram a medicina, mas para serem aceites têm de demonstrar continuamente a sua
precisão, fiabilidade e relevância clínica para o paciente. Sendo assim, os responsáveis do laboratório
e os fabricantes de reagentes e kits de diagnóstico têm de estabelecer e implementar sistemas
efectivos de controlo de qualidade [18]. Nos primeiros anos de utilização destas técnicas a
percentagem de resultados errados era muito superior, demonstrando a importância e a diferença
que a automatização e o controlo de qualidade podem fazer [18]. Um controlo de qualidade
optimizado vai permitir a redução de erros e da comunicação de resultados errados, a comunicação
de bons resultados de modo mais rápido e seguro e a poupança de dinheiro pela prevenção do
aparecimento de erros e por se implementar apenas o controlo necessário e não o excedente [19]. A
avaliação da qualidade pode ser interna ou externa e, recentemente, foram introduzidos alguns
programas de avaliação externa para as técnicas genéticas, mas uma menor atenção tem sido
prestada à avaliação interna [29].
Uma definição de controlo interno foi estabelecida pela Organização Mundial de Saúde
(OMS), através do departamento de External Assessment of Health Laboratories, como sendo “um
conjunto de procedimentos realizados pelos colaboradores do laboratório para avaliar o trabalho e
os seus resultados, de modo a decidir se são suficientemente fiáveis ou não para serem
comunicados” [30]. Assim, os procedimentos que se vão executar têm um efeito imediato na
actividade do laboratório, com o dever de controlar e não apenas de examinar os resultados [30].
Alguns autores, tendo em conta a necessidade deste género de controlo, já começaram a
investir um pouco mais nesta área. Um exemplo é o grupo de trabalho de Bladbjerg et al. (2002), que
apresentou um exemplo de esquema de controlo de qualidade interno para análise de polimorfismos
através da reacção de polimerização em cadeia (PCR, Polymerase Chain Reaction), que pode ser visto
na Tabela 1.

Controlo de Qualidade de Técnicas Genéticas
14
Tabela 1 ‐ Princípios de controlo de qualidade interno para métodos de genotipagem baseados em PCR (adaptado de [29]).
Fase Processo Passos para assegurar qualidade
Variação pré‐analítica Manuseamento da amostra ‐ Quantificação do DNA
Isolamento do DNA ‐ Cálculo do ratio DNA/proteína
‐ Teste de DNAses
‐ Diluição das amostras para
concentração de DNA semelhante
‐ Utilização de tubos e pontas livres de
DNAse/RNAse
Variação analítica Amplificação do DNA (PCR) ‐ Inclusão de amostras com genótipo
conhecido
Digestão com enzima de
restrição
‐ Inclusão de branco de reagentes
Separação de fragmentos por
electroforese
‐ Inclusão de marcador de pares de
bases
Confirmação de resultados ‐ Em populações: reanálise de 5‐10%
‐ Em pacientes: reanálise de portadores
do alelo da doença e de 5‐10% de não
portadores
‐ Reanálise com método diferente, se
possível
‐ Associação com outra variação
‐ Se discordam mais de 5% dos
resultados, reanalisar toda a série
Variação pós‐analítica Leitura dos resultados ‐ Duas leituras independentes
Entrada na base de dados ‐ Dupla entrada na base de dados
A maior parte das técnicas moleculares, senão todas, usa a técnica de PCR. A obtenção
rápida de resultados, o bom limite de detecção, selectividade, especificidade, sensibilidade e
potencial para automatização compõem as suas principais vantagens [31] e fazem com que esta
técnica seja utilizada em laboratórios de todo o Mundo, com diversas finalidades. Cada vez mais
protocolos específicos e automatizados para determinado diagnóstico estão a ser criados. São
exemplos os protocolos para o RNA do vírus de hepatite C (HCV, Hepatitis C Virus) [11] e para a
infecção meningocócica [20]. A importância desta técnica para o diagnóstico da infecção viral ou da
infecção bacteriana é muito grande, principalmente quando se trata do rastreio de doenças
potencialmente transmissíveis por transfusão. Na década de 90 essa importância foi descoberta e
assim, em 1995, no Reino Unido, o Standardization of Gene Amplification Techniques (SoGAT), foi
estabelecido no National Institute for Biological Standards and Control (NIBSC). O SoGAT é um grupo
de discussão, o único fórum internacional para troca de informação de aspectos científicos da técnica
de PCR, com o intuito de promover a padronização de reagentes, da amplificação e validação,

Controlo de Qualidade de Técnicas Genéticas
15
ajudando na definição de protocolos como o do HCV [32]. Face a isto, é notória a importância do PCR
e, por isso mesmo, os seus aspectos técnicos e principais cuidados devem constituir a primeira
camada de um projecto completo de QA [20].
No entanto, apesar das grandes vantagens da PCR relativamente a outros métodos, alguns
problemas têm sido discutidos. A novidade tecnológica, o investimento económico e a falta de SOP
têm sido as desvantagens mais apontadas [31]. A falta de padronização é sem dúvida o problema que
mais preocupa os laboratórios e tal deve‐se à complexidade dos métodos moleculares. As técnicas de
amplificação são baseadas em reacções enzimáticas cíclicas muito sensíveis a qualquer
contaminação, gerando facilmente falsos positivos. A técnica de PCR é particularmente sensível às
inúmeras variáveis, previsíveis ou não, que podem influenciar negativamente a amplificação [33].
Além disso, tal como todos os testes de biologia molecular, é tecnologicamente mais exigente e exige
maior experiência do que a maioria dos testes convencionais [34]. Na Tabela 2 estão resumidas as
principais operações efectuadas numa amplificação e alguns dos potenciais problemas que podem
aparecer.
A maior parte destes aspectos negativos refere‐se a protocolos de amplificação
inapropriados, má preparação ou quantificação dos ácidos nucleicos alvo, baixa recuperação,
presença de inibidores, uso de reagentes ou termocicladores ineficientes e má interpretação dos
resultados [33]. A importância destes pontos é visível em certos estudos como o de Grundmann et al.
(1997), que concluíram que utilizando reagentes padrão, de qualidade controlada, protocolos de
extracção de DNA e condições de amplificação padronizadas, é possível ultrapassar os problemas de
reprodutibilidade em diferentes laboratórios [35].

Controlo de Qualidade de Técnicas Genéticas
16
Tabela 2 ‐ Operações realizadas em técnicas de ácidos nucleicos e potenciais fontes de erro (adaptado de [19]).
Extracção/Isolamento do ácido nucleico
RNA ou DNA degradado
Introdução de inibidores ou contaminantes
Extracção incompleta, erro de amostragem
Ligação do analito a um anticorpo ou coluna
Transformação (ex: RNA a cDNA)
Erros de transcrição
Falha da enzima
Amplificação
Insensibilidade
Não especificidade
Inibição
Contaminação
Degradação enzimática
Detecção
Inespecificidade
Falha da enzima
Falha de calibração
Uma técnica de amplificação por si só apresenta, então, inúmeras variáveis que necessitam
de ser controladas. As principais preocupações concentram‐se normalmente nos falsos positivos e
nos falsos negativos. Os falsos positivos são conhecidos desde os anos 90, constituindo uma das mais
graves preocupações, e devem‐se principalmente à contaminação por produtos que foram
anteriormente amplificados [18,36,37]. Embora seja um problema que possa estar presente em
diversos ensaios, neste caso assume um carácter especial, devido à extrema sensibilidade destas
técnicas que, teoricamente, são capazes de detectar um único microorganismo, por exemplo, numa
amostra de um paciente. As primeiras recomendações para redução do risco de contaminação foram
descritas por Kwok e Higuchi (1989), que defenderam a importância de um protocolo bem delineado
como instrumento para evitar um mau desfecho da experiência [38]. Hoje em dia, essas
recomendações continuam actuais, foram incorporadas em programas de controlo de qualidade e
melhoradas com a introdução de novas medidas, como os sistemas de controlo de contaminação
enzimáticos ou sistemas automatizados [18], como se pode ver na Tabela 3.

Controlo de Qualidade de Técnicas Genéticas
17
Tabela 3 ‐ Falsos positivos ‐ controlo e melhorias (adaptado de [18]).
Controlo de contaminação (Kwok e Higuchi, 1989)
Separação física de pré e pós‐PCR
Material próprio de cada área (micropipetas, pontas, centrífugas, tubos, luvas,
batas, canetas, pessoal)
Reagentes em alíquotas
Manuseamento semi‐estéril
Controlos positivos (pequeno número, baixa concentração)
Controlos negativos (em maior número)
Melhorias recentes
Pontas resistentes a aerossóis
Semi‐automatização
Uracil‐N‐glicosilase
Sistema fechado (Real‐Time PCR)
Automatização completa (processamento da amostra + amplificação + detecção)
Para ultrapassar os falsos positivos e garantir a integridade de um resultado positivo, foram
formuladas algumas regras (A‐F):
A. Um teste de PCR deve ser realizado, pelo menos, em três áreas diferentes: uma de
preparação dos reagentes, uma de preparação da amostra e uma terceira de amplificação e
consequente detecção [34,39]. Embora os cuidados básicos e essenciais dos pontos B‐F não
variem de autor para autor, existem diferentes opiniões quanto à divisão de espaço. Por
exemplo, a Farmacopeia Europeia estabelece guidelines para a técnica de PCR, aconselhando
a sub‐divisão em quatro áreas: área de master‐mix, pré‐PCR, amplificação e detecção pós‐
‐PCR [40]; Neumaier et al. (1998) concordam também com esta divisão em quatro
compartimentos ou espaços: preparação e armazenamento de reagentes ‐ onde se recebem,
se dividem em alíquotas e se preparam reagentes e master‐mix, preparação da amostra,
amplificação e, por último, análise dos produtos de PCR [41], espaços que vão de encontro
aos designados pela Farmacopeia Europeia;
B. Estes espaços não devem ser adjacentes, mas sim em diferentes pisos ou edifícios e o
sentido de trabalho deve ser unidireccional [34,39‐42];
C. A organização do laboratório é essencial para evitar a contaminação. Cada zona deve ter
batas, luvas e material próprio, não havendo trocas entre áreas. Os reagentes devem ser
armazenados em alíquotas, ou seja, guardados em pequenos volumes para diminuir a
contaminação, e as bancadas devem ser descontaminadas através da utilização de radiação

Controlo de Qualidade de Técnicas Genéticas
18
ultravioleta (UV) ou de químicos como o hipoclorito de sódio. A técnica de pipetagem é
também muito importante, devendo‐se evitar a formação de aerossóis, o que se pode
conseguir utilizando condições de pressão positiva no espaço dedicado ao pré‐PCR ou de
pressão reduzida no espaço dedicado à amplificação [34,39‐42];
D. Cuidado extra com o material possível de contaminar o sistema, principalmente com as
pontas utilizadas, utilizando‐se pipetas com filtro [34]. É necessário também ter alguns
cuidados com o equipamento, como as verificações e calibrações das câmaras,
termocicladores e micropipetas;
E. Utilização de controlos, em que neste caso é extremamente importante o controlo negativo.
Se este controlo apresentar um resultado positivo, o ensaio deve ser repetido; se o resultado
positivo se mantiver, é um indicador de uma contaminação geral do laboratório e todos os
reagentes devem ser novamente preparados e todo o material, assim como os espaços,
devem ser devidamente limpo [34]. As amostras devem ser analisadas em duplicado [41];
F. Para detectar uma possível contaminação do ambiente do laboratório, um controlo
ambiental, ou seja um tubo que contém a master‐mix e foi deixado aberto no espaço
dedicado ao PCR, deve ser analisado, em intervalos regulares [42].
Ao analisar estas regras pode parecer que esta técnica não é de fácil implementação num
laboratório. No entanto, é uma das mais utilizadas hoje em dia, por isso muitas das dificuldades
foram ultrapassadas. Um dos métodos que ajudou foi um método de controlo bioquímico designado
uracil‐N‐glicosilase (UNG), um controlo anti‐contaminação. A ideia básica é marcar os produtos
amplificados produzidos durante a PCR para que sejam discriminados do alvo natural de DNA, o que
vai ser crucial para reconhecer a contaminação com produtos amplificados de um PCR anterior. Este
método pode permitir que toda a reacção se passe num só espaço, dividido nas três zonas já faladas,
desde que todas as outras precauções se mantenham [34,36,39,41,42]; pode também ser combinado
com uma amplificação hot‐start para reduzir a ligação não específica dos primers [42]. Uma
alternativa pós‐PCR é a geração fotoquímica de aductos de DNA através de compostos de
isopsoraleno, na presença de luz UV de longo comprimento de onda. Este método previne a
contaminação, pois os aductos de DNA são refractários à amplificação mas não interferem com os
procedimentos de hibridização pós‐PCR [41]. No entanto, é necessário não esquecer que o ideal deve
ser a prevenção da contaminação e não a utilização de métodos de remoção de contaminação. Estes
devem ser utilizados com precaução, para não criarem a sensação de falsa segurança [41].

Controlo de Qualidade de Técnicas Genéticas
19
Um outro problema grave, que se tem verificado na amplificação, tem sido o número de
resultados falsos negativos, como já foi referido. As principais causas, medidas de precaução e
detecção estão resumidas na Tabela 4.
Tabela 4 ‐ Falsos negativos ‐ causas, prevenção e detecção (adaptado de [18]).
Causas
Variações na sequência dos primers
Perda do ácido nucleico da amostra
Digestão do ácido nucleico da amostra (DNAses, RNAses)
Inibição da Taq polimerase
Prevenção
Evitar heparina, hemoglobina, etanol, fenol, SDS
Uso de técnicas de extracção apropriadas
Detecção
Controlos positivos
Controlo interno
Os resultados falsos negativos podem então gerar‐se devido a imensas possibilidades, como
os erros técnicos, baixa concentração da amostra, erro humano ou presença de inibidores [34]. Dois
controlos são essenciais para eliminar/detectar falsos negativos:
A. O uso de um controlo positivo vai ajudar a resolver principalmente os problemas técnicos e
no caso de dar negativo, todo o ensaio é considerado inválido e deve ser repetido com novos
reagentes e instrumentos calibrados [34];
B. O uso de um controlo interno, que se destina a identificar a presença de inibidores,
monitorizando a amplificação em cada teste PCR. Um controlo interno é um fragmento de
DNA adicionado a uma amplificação e que origina sempre um resultado positivo. Para isso
tem de ser o mais semelhante possível ao DNA alvo mas com uma característica que o
distinga deste. Caso haja inibição o ensaio deve ser repetido; se não resultar opta‐se por
diluir a amostra (1:5‐1:20), de modo a diluir o inibidor a uma concentração que não afecte a
amplificação. O único cuidado é não diluir o DNA alvo demasiado, para que não deixe de ser
amplificado. Outras resoluções já foram apresentadas, como aquecimento ou arrefecimento
da amostra, que ajudará se o inibidor for termolábil, ou a purificação do ácido nucleico [34].
Para além do uso de controlos, os primers são também uma fonte de preocupação, cuja
qualidade, design, pureza e validação devem ser controladas. Assim, cada novo lote de primers deve

Controlo de Qualidade de Técnicas Genéticas
20
ser testado para especificidade, eficiência de amplificação e ausência de inibidores antes de serem
aceites e utilizados [40].
As causas destes falsos positivos e negativos centram‐se mais na fase analítica do processo,
mas como se pode ver pelas tabelas anteriores e pelo que já foi referido, estes resultados podem
gerar‐se noutras fases do processo. Sendo assim, o primeiro passo ideal ao analisar um processo de
PCR será ter em conta a sua divisão em três fases, onde se inserem os erros falados anteriormente e
identificam‐se as suas causas e respectivas medidas de controlo [29]:
A. Pré‐analítica (recolha padronizada da amostra, verificação dos dados do paciente,
manuseamento da amostra, isolamento do DNA);
B. Analítica (amplificação, digestão com enzimas de restrição, electroforese);
C. Pós‐analítica (leitura, processamento e comunicação dos resultados).
Há mesmo autores que consideram a divisão de qualquer laboratório em cinco fases [43,44]:
A. Pré‐pré‐analítica (estudo do caso e decisão do teste a realizar);
B. Pré‐analítica;
C. Analítica;
D. Pós‐analítica;
E. Pós‐pós‐analítica (interpretação dos resultados pelo médico e tomada de decisões).
Estas duas fases extra têm outras causas de erros e pontos críticos de controlo e a análise de
todas as cinco fases vai permitir controlar todo o processo. A Tabela 5 mostra os principais passos
dessa análise e exemplos de erros comuns, num teste laboratorial de diagnóstico.
A importância desta separação de fases é indiscutível e todos os conceitos de diferentes
fases, medidas de controlo e regras estão incluídos na ISO/IEC 15189:2003 Medical laboratories ‐
particular requirements for quality and competence, desenvolvida especificamente para os
laboratórios de diagnóstico, onde se incluem também as TAN. Até ao desenvolvimento desta ISO,
não havia nenhuma que se referisse à gestão de qualidade e competência técnica nos laboratórios,
recorrendo‐se somente à ISO 9001:2000, com os requerimentos de gestão de qualidade aplicáveis a
qualquer empresa, ou à ISO 17025:1999, que se refere à acreditação e calibração. Assim, o
aparecimento desta ISO foi bem visto pela comunidade científica e médica, cobrindo muitos dos
campos que estavam incompletos [45,46].

Controlo de Qualidade de Técnicas Genéticas
21
Tabela 5 ‐ Fases do processo de um laboratório médico e possíveis erros (adaptado de [43]).
Pré‐pré‐analíticaClínico necessita de informação Clínico não interpreta bem a informação do
paciente Clínico tem uma hipótese Hipótese erradaClínico decide o teste a realizar Teste errado para a hipótese; Erro de
comunicação Clínico preenche um formulário ou
encarrega alguém de o fazer
Formulário errado
Pré‐analítica Identificação do paciente Troca de informaçãoEscolha do tubo (tamanho, anticoagulante) Informação errada do paciente no sistemaRecolha de sangue Tubo errado; técnica de recolha errada Formulário e amostra (sangue) são enviados
para o laboratório (condições de transporte) Amostra a temperatura errada
Amostras preparadas para análise Amostra é abanada, tubo contaminado, tubo partido, troca entre amostras
Analítica Aparelhos são verificados Amostra não analisadaRealizam‐se as reacções Amostra colocada no local errado Resultados são enviados para o sistema Diluição errada, falha no controlo de
qualidade, valor errado, falha no instrumento, protocolo não foi seguido, análise errada
Pós‐analítica Resultados são verificados Resultados improváveis não são vistos Resultados são comunicados Falha do computadorResultados são escritos Falha na impressora, erro de cálculo, erro de
escrita, teste errado comunicado, destino da comunicação errado
Pós‐pós‐analíticaClínico lê os resultados Problema de comunicação Clínico interpreta os resultados Má interpretaçãoClínico toma uma decisão Conclusão errada
Embora a fase analítica seja aquela que parece necessitar de uma maior atenção e controlo,
muitas das precauções têm de ser tomadas já na fase pré‐analítica. Apesar de ainda haver muito a
desenvolver, as técnicas estão cada vez mais evoluídas e automatizadas e por isso os erros analíticos
são cada vez menos prováveis. Os erros que ocorrem nas fases extra‐analítica continuam a ser uma
grande fonte de preocupação [43,44,47,48]. Lippi et al. (2006) reportaram que a falta de SOP para a
recolha, qualidade, manuseamento e armazenamento da amostra constituem mais de 93% dos erros
encontrados num processo de diagnóstico [47]. A Figura 3 pretende representar a importância de
variáveis “escondidas”, como as variáveis pré‐analíticas:

Controlo de Qualidade de Técnicas Genéticas
22
De facto, a qualidade dos resultados obtidos em biologia molecular depende muito do
controlo dos erros pré‐analíticos e analíticos associados à técnica. Erros pré‐analíticos ocorrem
muitas vezes no isolamento do DNA/RNA, em que o detergente usado para lise celular pode afectar a
amplificação do DNA posteriormente, assim como o anticoagulante escolhido [49]. As variáveis pré‐
‐analíticas, tal como a amostra, métodos de recolha e condições de armazenamento não conseguem
ser monitorizadas por métodos tradicionais de controlo de qualidade, apesar da inclusão dos
controlos para detectar a presença de inibidores ou a degradação da amostra possa ser muito útil
para preencher esse vazio [19]. Um ponto essencial parece ser o controlo da contaminação no
ambiente de trabalho, minimizando os erros analíticos e extra‐analíticos [49].
A validade dos resultados está dependente das medidas adoptadas em cada uma das três
fases do processo, sendo essencial padronizar todo o processo desde a solicitação do teste até à
saída dos resultados, atingindo‐se então o padrão de qualidade desejado. A importância dos erros
nas três fases do processo está esquematizada na Figura 4:
Teste de laboratório
Variabilidade biológica
Variáveis ambientais
Identificação do paciente
Colheita da amostra
Manuseamento da amostra
Figura 3 ‐ Variáveis pré‐analíticas em testes de laboratório (adaptado de [47]).

Controlo de Qualidade de Técnicas Genéticas
23
Figura 4 ‐ Erros presentes nas três fases do processo de um teste de laboratório (adaptado de [44]).
Como a total eliminação de erros não é possível, especialmente os que se relacionam com
fases extra‐analíticas, demonstra‐se mais uma vez a grande importância da existência de SOP, boas
práticas de laboratórios, sistemas de qualidade e acreditação do laboratório, de modo a não só
verificar a existência de um erro mas sim de o prevenir e reduzir. Só reduzindo a elevada frequência
destes erros se consegue melhorar a qualidade do laboratório e evitar o impacto negativo na
população que estes erros podem causar.
No entanto, toda esta ênfase na melhoria da qualidade e do seu controlo nas fases extra‐
‐analíticas traz alguns efeitos negativos. Um exemplo ocorreu nos Estados Unidos, em que a CLIA
propôs uma redução no controlo de qualidade para os processos analíticos como consequência à
crescente necessidade de aumentar e melhor assegurar a qualidade nas outras fases [48]. O facto é
que, apesar do número de erros analíticos ter diminuído e do número de erros extra‐analíticos ser
mais provável, existem diversas evidências que demonstram que a qualidade analítica ainda é uma
fonte de preocupação e não pode ser esquecida, tendo sido descritos procedimentos analíticos
menos satisfatórios em diferentes campos, inclusive no da biologia molecular [48]. É necessário
saber encontrar o equilíbrio que permite tentar controlar ao máximo os erros possíveis nas três
principais fases do processo.
Quando se pensa num plano de controlo de qualidade relacionado com uma técnica de
amplificação como a PCR, uma outra questão essencial e primária, que afecta o modelo de controlo
de qualidade escolhido e implementado refere‐se ao género de ensaio que se está a realizar, se é
qualitativo ou quantitativo. Ambos podem beneficiar de um sistema de controlo de qualidade
implementado, mas cada um tem os seus requerimentos.
Os métodos qualitativos têm somente um ponto de decisão: positivo/negativo ou
presença/ausência. Neste caso, parece que o essencial é ter um controlo de cada posição, ou seja,

Controlo de Qualidade de Técnicas Genéticas
24
um controlo positivo/presença e um negativo/ausência, e ambos ajudariam a detectar o erro, o que
torna estes controlos bastante úteis e essenciais para a eliminação de resultados errados [16,19].
Garrett (2001) fala também da hipótese de um único controlo, em que a concentração de analito
estaria exactamente no ponto de decisão [19]. Embora possa também ajudar na detecção de erro,
esta hipótese ainda não conseguiu ser validada e parece ter algumas falhas.
Os métodos quantitativos validados devem ser testados pela sua linearidade num
determinado intervalo. Aqueles que exibem essa linearidade requerem normalmente controlos em
menos pontos desse intervalo que aqueles que não exibem linearidade. Normalmente utilizam‐se
nestes casos as regras de Westgard, para identificar um erro ou decidir a rejeição de um ensaio. É
importante, tanto nos métodos qualitativos como nos quantitativos, o conhecimento do limite de
detecção, do viés do teste e da reprodutibilidade [16,19].
Resumidamente, após a referência a tantas variáveis e possíveis medidas de controlo, torna‐
‐se necessário concluir sobre os controlos que realmente devem ser implementados para a técnica
de PCR. A nível de controlo de qualidade interno são essenciais os seguintes passos:
A. Controlos relacionados com a preparação da amostra: no que diz respeito ao DNA, o mais
habitual é a electroforese em gel de agarose precedida ou não de uma digestão com uma
endonuclease, como a EcoRI, podendo‐se assim controlar a presença de inibidores da
actividade enzimática. A presença de potenciais inibidores é normalmente controlada
usando um espectrofotómetro, fazendo a leitura da absorvância a 260 (A260) e 280nm (A280);
o ratio A260/A280 deve estar entre 1,75‐2,0. Relativamente ao RNA, o método mais rápido
baseia‐se na electroforese em gel de agarose, em condições não desnaturantes, tal como no
DNA. Em caso de dúvida pode‐se utilizar um gel com condições desnaturantes, para verificar
a sua integridade [41];
B. Controlos para a síntese de cDNA e amplificação: a nível de amplificação são importantes o
controlo positivo e interno, para evitar falsos negativos, e controlo negativo e de reagentes,
para evitar falsos positivos [41,42]. A nível da síntese de cDNA, o controlo crucial é a
utilização de um controlo interno [41];
C. Controlos para a avaliação dos resultados: inicialmente tem de existir um controlo da
digestão com enzimas de restrição. A digestão do DNA genómico pode ser diminuída pela
presença de inibidores enzimáticos, condições inapropriadas ou actividade enzimática
reduzida. Através de uma electroforese em gel de agarose consegue‐se ver os resultados da

Controlo de Qualidade de Técnicas Genéticas
25
digestão e podem‐se aplicar determinados critérios que irão ajudar a decidir se a digestão foi
bem‐feita ou não. Por exemplo, verificar que há um intervalo de bandas com pesos
moleculares diferentes, de alta massa molecular até à mais baixa; adicionar uma quantidade
conhecida de um marcador molecular de elevado peso molecular, cuja digestibilidade é
conhecida; bandas satélites distintas que aparecem devido às sequências repetitivas do DNA
genómico, indicando uma electroforese bem sucedida. No entanto, nem todas as enzimas as
conseguem gerar [41]. Relativamente à electroforese, o controlo envolve a presença de
marcadores para o tamanho dos fragmentos (marcadores de pares de bases) e concentração
[41].
O objectivo deste trabalho consistiu na proposta de um modelo de controlo e garantia de
qualidade interno para PCR‐RFLP e Real‐Time PCR, ambas as técnicas qualitativas. O que já foi
referido aplica‐se essencialmente ao PCR‐RFLP, embora também se possa adaptar ao Real‐Time PCR.
Na parte IV deste trabalho, ao estabelecer a proposta de controlo e garantia de qualidade, os dois
métodos são mencionados.

Controlo de Qualidade de Técnicas Genéticas
26
3. Esquemas de Controlo Externo
Muitos passos de um teste podem ser controlados, mas apenas com o estudo dos resultados
dos testes se consegue chegar a uma conclusão sobre a fiabilidade destes [10]. Na Europa é usual
utilizarem‐se processos de avaliação externa de qualidade, conhecidos nos Estados Unidos como
testes de proficiência, em que amostras idênticas são enviadas para cada laboratório participante e
os resultados são comparados entre eles e/ou com um valor padrão ou certo [10]. A necessidade dos
laboratórios, incluindo aqueles que utilizam TAN, usarem este meio para assegurar a qualidade é
cada vez mais reconhecida, mas o seu uso é ainda baixo, especialmente nas técnicas baseadas em
PCR [50].
Os esquemas de controlo externo mais recentes incluem o processo analítico completo:
componentes pré‐analíticas, analíticas e pós‐analíticas. Esta era uma abordagem ainda pouco usual
em controlo externo mas que revela a importância nas técnicas genéticas de interpretar cada
resultado num contexto particular [10] e que se vai afirmando cada vez mais. A estrutura adoptada
pelo European Molecular Genetics Quality Network (EMQN) é um exemplo típico de um modelo
moderno de avaliação externa (EQA, External Quality Assessment) em técnicas genéticas e está
representado na Figura 5.

Controlo de Qualidade de Técnicas Genéticas
27
Figura 5 ‐ Anatomia de um esquema de controlo externo. AZF, Factor de Azoospermia; BRCA, Cancro da Mama Hereditário; CF, Fibrose Cística; CMT, doença de Charcot‐Marie‐Tooth; DMD, Distrofia Muscular de Duchenne; EMQN, European Molecular Genetics Quality Network; FA, Anemia de Fanconi; HD, Doença de Huntington (adaptado de [10]).
Outros exemplos de organizações/programas para controlo externo são: a Clinical Molecular
Genetics Society (CMGS), fundada em 1988 no Reino Unido, para promover a qualidade através do
treino, educação, pesquisa e recolha de dados e estabelecendo também esquemas de controlo
externo, e dois programas desenhados pelo CAP, o Q‐Probes, fundado em 1989, e o Q‐Tracks,
fundado em 1998. Estes dois últimos programas lidam com as três fases do processo (pré‐analítica,
analítica e pós‐analítica) e com as medidas a tomar em cada uma delas, com o objectivo de melhorar
a qualidade [51]. São identificados indicadores de qualidade, para os processos extra‐analíticos,
necessários para os laboratórios clínicos detectarem um fraco desempenho existente numa parte do
processo e estabelecerem padronização e especificações de qualidade (limites de aceitabilidade). Os
indicadores de qualidade e especificações para as fases pré‐analítica, analítica e pós‐analítica estão
reunidos nas Tabelas 6, 7 e 8, respectivamente. Quando os indicadores não estão dentro dos limites
de aceitabilidade são necessárias medidas correctivas na área em questão [51].

Controlo de Qualidade de Técnicas Genéticas
28
Tabela 6 ‐ Indicadores de qualidade e especificações (limites de aceitabilidade) da fase pré‐analítica (adaptado de [51]).
Indicador de qualidade Relacionado com Especificação (%)
Pedidos
Número de pedidos
Erros na identificação do paciente 0,08 Falta da identificação do médico 0,50 Especificação errada da unidade hospitalar 0,60 Pedido não perceptível 0,10 Correcção de erros nos testes requisitados
0,30
Amostragem
Número de pedidos
Pedido de flebotomia de pacientes internados não
recolhido 7,00
Pedido de flebotomia de pacientes ambulatoriais não recolhido
0,30
Torniquetes contaminados com sangue 2,50 Acidentes com agulhas por cada 100.000
venopunções 0,01
Recolha de sangue para monitorização da terapêutica no tempo errado
24,0
Erros nas pulseiras de identificação
3,00
Transporte e recepção das amostras
Número de amostras
Recolha e transporte inadequado da amostra 0,004Rejeição da amostra (todo o hemograma) 0,45 Rejeição da amostra (química) 0,35 Amostra perdida/não recebida 0,12 Rotulagem não apropriada do tubo 0,002Tubo de recolha não adequado 0,015Amostra danificada no transporte 0,002Amostra coagulada (hematologia) 0,20 Amostra coagulada (química) 0,006Amostra hemolisada (hematologia) 0,009Amostra hemolisada (química) 0,20 Acidente de laboratório 0,004Volume de amostra insuficiente 0,05 Ratio inadequado de volume de
amostra/anticoagulante 0,02
Tabela 7 ‐ Indicadores de qualidade e especificações (limites de aceitabilidade) da fase analítica (adaptado de [51]).
Indicador de qualidade Relacionado com Especificação (%)
Resultados inaceitáveis em controlos internos Número de resultados 0,07Resultados inaceitáveis em testes de proficiência do laboratório
Número de resultados 1,40

Controlo de Qualidade de Técnicas Genéticas
29
Tabela 8 ‐ Indicadores de qualidade e especificações (limites de aceitabilidade) da fase pós‐analítica (adaptado de [51])
Indicador de qualidade Relacionado com Especificação
Validação do relatório Número de relatórios
Relatório com o pedido de teste mas não completo 1,4% Relatório com o teste completo mas não pedido 1,1% Relatório com disparidades no nome do médico
1,9%
Relatórios intra‐laboratoriais Número de relatórios
Erros de comunicação do laboratório 0,05%Entrega fora do tempo
11,0%
Serviço de consulta Tempo médio de comunicação de valores críticos
aos pacientes internados 6min.
Tempo médio de comunicação de valores críticosaos pacientes de ambulatório
14min.
Inquéritos telefónicos não concluídos
Número de inquéritos telefónicos
21,3
Disponibilidade do computador do laboratório Número de episódios de inactividade 30 dias 3 episódiosMédia cumulativa de inactividade
4h
Competência do pessoal Taxa de erros de pessoal não técnico Número de
empregados 0,9‐2,9%
Taxa de erros de pessoal técnico 0,9‐6,4%
O objectivo de um esquema de EQA é educar e não punir, de modo a que os participantes
consigam atingir um padrão superior de qualidade [10] e controlar o seu próprio modelo de
qualidade interno. A organização de modelos de EQA é útil por diversas razões [10]:
A. Erros de genotipagem permitem aos participantes descobrir lacunas no seu controlo interno
e detectar a fonte dos seus erros;
B. Erros de genotipagem feitos por vários laboratórios apontam para a existência de lacunas no
método aplicado;
C. Os dados dos laboratórios participantes permitem uma análise geral do progresso feito na
qualidade das técnicas genéticas que estão a ser alvo de estudo;
D. Podem ser uma boa ferramenta para verificar o desenvolvimento de estratégias/testes
usados nos laboratórios de diagnóstico.
Embora muito úteis, alguns destes programas têm algumas desvantagens: apenas algumas
amostras são incluídas no painel (2 a 4), as amostras não são muitas vezes bem caracterizadas e os
dados geralmente não são publicados [18]. Uma extraordinária excepção é a European Union Quality

Controlo de Qualidade de Técnicas Genéticas
30
Control Concerted Action (EU‐QCCA), criada em 1998 com o objectivo de estabelecer um programa
de controlo de qualidade externo para a avaliação de TAN em diagnóstico viral, já existentes e em
desenvolvimento. Este programa foi continuado e expandido através de uma nova organização, a
Quality Control for Molecular Diagnostics, em colaboração com a European Society for Clinical
Diseases e a European Society for Clinical Virology [18].
Existem programas de controlo externo específicos para doenças, tanto nos Estados Unidos
como na Europa, mas estão descritos, por um grupo italiano, os resultados de um programa mais
genérico que compara a qualidade dos produtos de amplificação de DNA genómico por PCR, obtidos
por diversos laboratórios. Programas de controlo externo para testes de diagnóstico baseados na
amplificação de ácidos nucleicos não têm sido muito implementados e ainda existem muitas
limitações. Neste estudo, eles monitorizaram a extracção de DNA, de forma quantitativa e
qualitativa, a PCR (especificidade e eficiência), resultados da electroforese e interpretação,
atribuindo uma pontuação a cada laboratório [24,33]. Este é um modelo que tenta seguir as
tendências mais recentes do controlo externo de qualidade e que reflecte a qualidade geral dos
ensaios genéticos na Europa. No entanto, as guidelines do CLIA requerem que os laboratórios dos
Estados Unidos participem em programas de controlo externo para cada analito/doença e não para
cada método. Ambas as opções, a mais expansiva com os desafios de doenças específicas, ou a mais
redutora com desafios baseados nos métodos gerais têm as suas vantagens e desvantagens [24].
Um programa em evidência nos últimos anos é o programa Multinational External Quality
Assay programmes in Clinical Molecular Diagnostics based on Performance and Interpretation of PCR
assay methods (programa EQUAL), que define três programas de controlo externo diferentes, para
técnicas baseadas em PCR, independentemente do analito: EQUAL‐qual, para PCR qualitativo,
EQUAL‐quant, para PCR quantitativo, e EQUAL‐seq, para sequenciação com PCR e ensaios
relacionados [50,52].
Este programa tem em conta o uso extenso da técnica de PCR em todo o mundo e as
limitações existentes a nível de programas de controlo externo nesta técnica, permitindo uma
avaliação dos aspectos analíticos mais importantes e que são comuns à maioria dos testes
moleculares que utilizam a PCR [50,52].
O programa EQUAL‐qual foi desenvolvido para avaliar os ensaios da PCR a três níveis:
extracção de DNA (qualitativa e quantitativamente); PCR (especificidade e eficiência); interpretação
dos resultados após electroforese. Com tudo isto consegue actuar a nível pré‐analítico, analítico e
pós‐analítico, que, como já foi referido, é a forma ideal de controlar a qualidade [52].

III. HACCP

HACCP
32
A segurança é um conceito que acompanha e que é fonte de preocupação em diferentes
áreas. Desde os laboratórios de diagnóstico à indústria alimentar, é visível a necessidade de
assegurar qualidade, de a controlar e de demonstrar segurança nos resultados ou produtos.
A nível alimentar as preocupações surgiram mais cedo, o que levou a uma melhor
organização dos programas de controlo de qualidade e da legislação. Já desde o início do século XX
que a preocupação a nível alimentar resultou na formulação de regulamentos e formação de
entidades reguladoras, como é o caso da Federal Meat Inspection Act (FMIA) em 1906 ou a Federal
Food, Drug and Cosmetic Act (FDCA), que surgiu na mesma época [53]. No século XXI, a ênfase na
segurança alimentar continuou, virando‐se agora para um novo conceito, o HACCP. Este sistema de
gestão da qualidade foi desenvolvido pela Pillsbury Company em associação com a National
Aeronautics and Space Administration (NASA) e os laboratórios do exército americano. Devido ao
programa espacial dos anos 60, surgiu a necessidade de manter a qualidade dos alimentos dos
astronautas, principalmente a qualidade microbiológica [53]. No entanto, só nos anos 70 foi então
aplicado à indústria conserveira americana e em 1980 a OMS e a Food and Agriculture Organization
(FAO) recomendaram a sua aplicação às pequenas e médias empresas. Em 1993, através da Directiva
93/43/CEE, o HACCP entrou para a regulamentação europeia, tendo por base de aplicação os
Princípios expressos no Codex Alimentarius. Em 2006, o Regulamento (CE) nº 852/2006 do
Parlamento Europeu e do Conselho, de 29 de Abril de 2004, relativo à higiene dos géneros
alimentícios e que revoga a Directiva 93/43/CEE, estipula que todos os operadores do sector
alimentar devem criar, aplicar e manter um processo ou processos permanentes baseados nos sete
Princípios do HACCP [54]. A ISO 22000:2005 providencia um sistema de gestão de segurança
completo, fundindo os requisitos da ISO 9001 com o plano HACCP, sendo um passo à frente do
sistema de gestão HACCP inicial.
O sistema HACCP consiste numa abordagem científica e sistemática de identificação e
avaliação de perigos específicos em todas as etapas da produção de alimentos, definindo medidas
para o seu controlo, com o objectivo de garantir alimentos seguros [55]. Trata‐se de uma
metodologia preventiva e que assenta numa forte componente de autocontrolo, com o objectivo de
evitar potenciais riscos que podem causar danos aos consumidores, através da eliminação ou
redução de perigos [54]. Uma outra definição é a de Schmidt, 1996: sistema lógico desenhado para
identificar perigos e/ou situações críticas e produzir um plano estruturado para controlar essas
situações [53]. Genericamente, o HACCP baseia‐se na aplicação de princípios técnicos e científicos na
produção e manipulação dos géneros alimentícios desde “o prado até ao prato” [54]. Foram
definidos sete Princípios pela FAO/OMS/Codex Alimentarius Commission (CAC) e que o CAC
incorporou num documento que pode ser utilizado em todo o mundo para estabelecer e manter este
sistema de gestão de qualidade [53].

HACCP
33
Os sete Princípios em que o sistema HACCP se baseia são, de acordo com o Codex
Alimentarius [53‐55]:
1. Conduzir a análise de perigos – identificar quaisquer perigos que devam ser evitados,
eliminados ou reduzidos para níveis aceitáveis;
2. Determinar os pontos críticos de controlo (PCC) – identificar os PCC na fase ou fases em que
o controlo é essencial para evitar ou eliminar um risco ou para o reduzir a níveis aceitáveis;
3. Estabelecer os limites críticos para cada medida associada a cada PCC – estabelecer os limites
em PCC, que separem a aceitabilidade da não aceitabilidade com vista à prevenção,
eliminação ou redução dos riscos identificados;
4. Estabelecer um sistema para monitorizar/controlar cada PCC – estabelecer e aplicar
processos eficazes de vigilância em PCC;
5. Estabelecer a acção correctiva a empreender quando a monitorização indica que um
determinado PCC não está sob controlo;
6. Estabelecer procedimentos de verificação para confirmar se o sistema HACCP está a
funcionar eficazmente – estabelecer processos a efectuar regularmente, para verificar que as
medidas referidas nos Princípios 1 a 5 funcionam eficazmente;
7. Estabelecer documentação acerca de todos os procedimentos e registos apropriados a estes
Princípios e sua aplicação – elaboração de documentos e registos adequados à natureza e
dimensão das empresas, a fim de demonstrar a aplicação eficaz das medidas referidas nos
Princípios 1 a 6.
Nestes Princípios utiliza‐se os conceitos “perigo” e “ponto crítico de controlo”. Um perigo é
representado por um agente biológico, químico ou físico presente ou a condição em que este ocorre,
que pode causar um efeito adverso à saúde [55,56]. Um PCC é uma etapa na qual devem ser
aplicadas medidas de controlo, sendo estas essenciais para prevenir, eliminar ou reduzir um perigo a
níveis aceitáveis [56].
Uma análise de perigos permite avaliar os perigos potenciais de um processo e determinar se
são significativos para a segurança do produto em causa e se devem ou não ser introduzidos num
plano HACCP. A equipa HACCP tem de identificar o perigo e avaliar o seu risco e significância. O risco
é a probabilidade do perigo ocorrer e a significância transmite a severidade desse perigo, caso
ocorra. A relação entre esses dois conceitos é avaliada e determina se um perigo é ou não
significativo. Os perigos considerados significativos são avaliados com o uso de uma “árvore de
decisão”, ou seja, uma sequência de perguntas que vai determinar se esse ponto do processo é um
PCC ou se consegue ser controlado pelos pré‐requisitos de segurança [53].

HACCP
34
Os 7 Princípios podem ser implementados num processo industrial ou empresa em 12 Passos
sequenciais [55]:
1. Designar a equipa HACCP (deve incluir representantes das diferentes actividades do
processo);
2. Descrever detalhadamente o produto (designação, características, condições de
armazenagem, etc.);
3. Identificar o uso pretendido do produto (descrição detalhada do produto final);
4. Elaborar o diagrama de fluxo e o esquema da área de fabrico (layout);
5. Confirmar in loco os diagramas de fluxo e layout;
6. Listar todos os perigos potenciais, conduzir uma análise de perigos e considerar as medidas
de controlo (Princípio 1);
7. Determinar os PCC (Princípio 2);
8. Estabelecer os limites críticos para cada PCC (Princípio 3);
9. Estabelecer um sistema de monitorização para cada PCC (Princípio 4);
10. Estabelecer medidas correctivas (Princípio 5);
11. Estabelecer os procedimentos de validação/verificação (Princípio 6);
12. Estabelecer a documentação e conservar os registos (Princípio 7 – plano HACCP).
Para que a implementação de um plano HACCP seja feita correctamente é necessário
esforço, empenho e compromisso por parte da administração, para seleccionar as pessoas, o devido
cargo de cada pessoa, disponibilizar os recursos financeiros e humanos, agendar reuniões, avaliações
do pessoal e do próprio plano HACCP, de modo a que possa ser revisto e alterado sempre que
necessário; a administração está também encarregue de motivar todas as pessoas que fazem parte
do sistema [53]. Tudo isto pertence então a um primeiro requerimento fundamental da
administração, para que o plano HACCP funcione.
Um segundo requerimento para a implementação será a formação sobre a metodologia
HACCP [53]. Essa formação é indispensável e deve ter diferentes níveis de aprofundamento dos
conhecimentos, ou seja, devem‐se realizar diferentes acções de formação para a administração, a
equipa HACCP, o pessoal de monitorização de qualidade, os operadores envolvidos e outros
colaboradores da empresa. Todos deverão conhecer o plano HACCP existente mas, conforme o seu
cargo e envolvimento na empresa e no próprio plano, os conhecimentos que têm de adquirir vão ser
diferentes.
Um terceiro requerimento será a existência de pré‐requisitos, que serão a base de um plano
HACCP e vão permitir a sua aplicação efectiva [53,54]. Pode‐se dizer que os pré‐requisitos controlam

HACCP
35
os perigos associados ao meio envolvente do processo e ao cumprimento das boas práticas em geral,
assim como os perigos considerados não significativos após avaliação, enquanto o sistema HACCP
controla os perigos associados ao processo [54]. Para ser operacional, é fundamental que o
funcionamento do sistema seja paralelo com determinados procedimentos previamente
implementados, como as Boas Práticas de Fabrico (GMP, Good Manufacturing Practices) e as Boas
Práticas de Higiene, utilizados a nível industrial.
Após a implementação do plano HACCP é necessário uma gestão do sistema, ou seja,
assegurar que o plano é monitorizado, o que inclui a verificação e validação do Princípio 6. A
verificação pode ser feita diariamente pelo próprio departamento que desempenha correctamente
as funções, mas pode ser acompanhada por programas de auditoria interna, que têm de ocorrer com
determinada e suficiente frequência. Se são identificados erros ou potenciais erros durante a rotina
diária ou nas auditorias, devem ser documentados, incluindo a sua causa e medidas que foram
tomadas [53].
A nível de laboratórios de diagnóstico e técnicas genéticas, o sistema HACCP não é utilizado.
No entanto é possível de adaptar e nos últimos anos já surgiram referências a esse cruzamento
HACCP/genética. Os sete Princípios e os 12 Passos de implementação são possíveis de adaptar a um
laboratório ou a uma técnica de amplificação e existem pré‐requisitos para diversas técnicas, como
as guidelines existentes para a amplificação de ácidos nucleicos da Farmacopeia Europeia [40],
parecendo então que todos os conceitos teóricos do HACCP encaixam na nova tendência de controlo
de qualidade das técnicas genéticas. Além disso, faz parte das etapas da validação de um método,
como por exemplo qualquer técnica de biologia molecular, o controlo de qualidade e a determinação
dos PCC [42], o que encaixaria perfeitamente num sistema de HACCP. Foi já referido que o ideal para
um plano de qualidade será controlar todo o processo e não só a fase analítica e de um ponto de
vista prático e multidisciplinar, o controlo de qualidade de um processo inteiro, com identificação,
redução e prevenção do erro, pode ser conseguido através de um modelo como o HACCP [43,47].
Como se pode concluir com tudo o que já foi referido, o objectivo do sistema HACCP é
prevenir os erros, levando a uma reforma eficiente do processo, com vista a um melhor
desempenho, eficiência e qualidade. Assim, com todos os passos críticos identificados pode‐se
reajustar o processo nos pontos críticos, com controlos adicionais intermédios. Pode‐se dizer que o
HACCP representa uma abordagem integral e sequencial de todo o processo e não apenas parcial,
como o são outras formas de controlo já existente a nível dos laboratórios [43].
Com a implementação deste sistema nas técnicas genéticas muitos dos objectivos citados
por alguns autores conseguiriam ser cumpridos. Plebani (2006) escreve sobre a importância da
identificação de erros e não conformidades, segundo as ISO, de uma acção correctiva e preventiva,

HACCP
36
da revisão do processo total em intervalos regulares, de modo a conseguir identificar os erros que
vão acontecendo e verificar a acção correctiva e preventiva, e da introdução de indicadores de
avaliação de qualidade nas diferentes fases [44]. Tudo isto vai melhorar a qualidade do laboratório e
pode ser conseguido por uma análise completa dos PCC e pela implementação total do HACCP.

IV. Proposta de Modelo de Controlo e Garantia da
Qualidade

Proposta de Modelo de Controlo e Garantia da Qualidade
38
O objectivo deste trabalho consistiu em sistematizar as condições ideais e necessárias para
realizar a técnica de PCR com qualidade e avaliar a qualidade dos resultados. A proposta de um
modelo de controlo e garantia de qualidade para a execução da técnica de PCR‐RFLP e Real‐Time PCR
no Instituto Português de Oncologia (IPO) do Porto dividiu‐se em três fases: adaptação de um plano
HACCP, proposta de guidelines internas com verificação do seu cumprimento e proposta de um
plano de avaliação de qualidade interna, apropriado ao local e com potencial implementação.
1. Adaptação de um Plano HACCP
Na elaboração desta proposta recorreu‐se, inicialmente, ao sistema de gestão de qualidade
HACCP. Tal como já foi referido, este sistema multidisciplinar é uma abordagem científica e
sistemática, um sistema lógico de identificação e avaliação de perigos e/ou situações críticas em
todas as etapas de um processo, com o objectivo de produzir um plano estruturado para controlar
essas situações [53,54]. Aparentemente, as linhas do HACCP aplicam‐se mais à gestão de empresas
do que ao trabalho de rotina de um laboratório. No entanto, a base destas orientações é que todos
são pessoalmente responsáveis por garantir que a empresa/laboratório onde trabalham produza um
produto ou serviço de qualidade.
Nas Tabelas e Figuras seguintes estão esquematizadas as medidas de adaptação do plano
HACCP às técnicas de PCR‐RFLP e Real‐Time PCR.

Proposta de Modelo de Controlo e Garantia da Qualidade
39
A. Descrição do produto final – Produto genético
Tabela 9 – Descrição do produto final.
Denominação do produto Produto genético amplificado
Características biológicas Isento de contaminantes biológicosCaracterísticas químicas Isento de contaminantes químicosCaracterísticas físicas Isento de contaminantes físicosCondições de armazenagem Não aplicável Método de distribuição Não aplicável Prazo de validade Não aplicável Material de embalagem Não aplicável Rotulagem Não aplicável Requisitos regulamentares Codex Alimentarius
ISO 22000:2005 Guidelines EMQN
B. Identificação do uso pretendido para o produto
Diagnóstico/projecto de investigação.
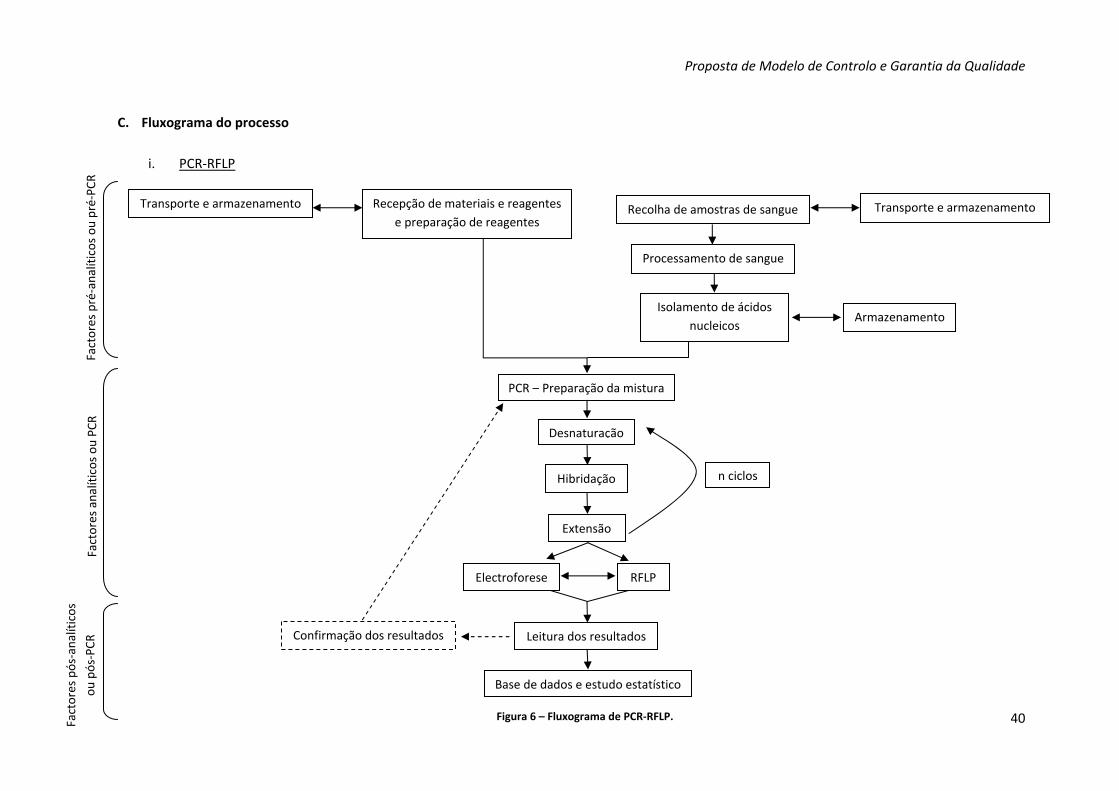
Proposta de Modelo de Controlo e Garantia da Qualidade
40
C. Fluxograma do processo
i. PCR‐RFLP
Figura 6 – Fluxograma de PCR‐RFLP.
Factores pós‐analíticos
ou pós‐PCR
Factores analíticos ou PCR
Factores pré‐analíticos ou pré‐PCR
Transporte e armazenamento Recepção de materiais e reagentes
e preparação de reagentes Recolha de amostras de sangue Transporte e armazenamento
Isolamento de ácidos
nucleicos
PCR – Preparação da mistura
Desnaturação
Hibridação
Extensão
RFLPElectroforese
Base de dados e estudo estatístico
Confirmação dos resultados
n ciclos
Processamento de sangue
Armazenamento
Leitura dos resultados

Proposta de Modelo de Controlo e Garantia da Qualidade
41
ii. Real‐Time PCR
Figura 7 – Fluxograma de Real‐Time PCR.
Factores pós‐analíticos
ou pós‐PCR
Factores analíticos ou PCR
Factores pré‐analíticos ou pré‐PCR
Transporte e armazenamento Recepção de materiais e reagentes
e preparação de reagentes Recolha de amostras de sangue Transporte e armazenamento
Isolamento de ácidos
nucleicos
Real‐Time PCR – Preparação
Desnaturação
Hibridação
Extensão
Base de dados e estudo estatístico
Confirmação dos resultados
n ciclos
Processamento de sangue
Armazenamento
Leitura dos resultados

Proposta de Modelo de Controlo e Garantia da Qualidade
42
D. Análise de perigos
O índice de risco (IR) de cada perigo/etapa é calculado multiplicando o valor atribuído à severidade (S) pelo valor atribuído à probabilidade (P) do
respectivo perigo. Os perigos classificados com IR≥3 foram considerados significativos e seguiram para análise através da árvore de decisão. Os outros foram
controlados pelos pré‐requisitos.
Tabela 10 – Índice de risco para diferentes valores de severidade e probabilidade (IR = S x P) (adaptado de [55,56]).
Severidad
e
(S)
Probabilidade (P)
Baixa (1) Média (2) Alta (3)Baixa (1) 1 2 3Média (2) 2 4 6Alta (3) 3 6 9

Proposta de Modelo de Controlo e Garantia da Qualidade
43
i. Factores pré‐analíticos ou pré‐PCR
Tabela 11 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa pré‐analítica.
Etapa Descrição dos perigos Causas P S IR Medidas de controloRecepção de materiais e reagentes/ Preparação e
armazenamento de reagentes
‐ Contaminantes biológicos, químicos e físicos
‐ Deficiente manipulação‐ Práticas incorrectas ‐ Fornecedor sem certificado de qualidade ‐ Quando os reagentes são armazenados em grandes frascos, as constantes utilizações podem levar a contaminações ‐ Temperatura de armazenamento inadequada
1 3 3 ‐ Área separada para preparação e armazenamento de reagentes ‐ Todas as áreas com equipamento/material próprio, não sendo permitidas quaisquer trocas ‐ Sistema unidireccional ‐ Tubos descartáveis livres de DNAse e RNAse ‐ Manutenção do local limpo ‐ Selecção de fornecedores com certificado de qualidade ‐ Verificação da integridade da embalagem, prazo de validade, rotulagem e certificação de qualidade ‐ Etiquetagem (conteúdo e data) ‐ Armazenamento e congelamento em alíquotas
Transporte de reagentes e materiais
‐ Contaminantes biológicos, químicos ou físicos
‐ Temperatura inadequada‐ Transporte inadequado
1 2 2 ‐ Utilização de métodos de transporte e verificação adequados

Proposta de Modelo de Controlo e Garantia da Qualidade
44
Tabela 11 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa pré‐analítica (cont.).
Etapa Descrição dos perigos Causas P S IR Medidas de controloRecolha da amostra
de sangue
‐ Contaminantes biológicos, físicos e químicos (tubo) ‐ Amostra hemolisada, insuficiente, incorrecta ou coagulada ‐ Alteração da concentração de metabolitos e de volume plasmático ‐ Hemoconcentração (aumento da concentração de moléculas grandes) ‐ Diluição de analitos extracelulares
‐ Falta de procedimentos padronizados ‐ Exercício físico regular influencia as variações de volume plasmático e metabolitos, influenciando muitas variáveis bioquímicas e hematológicas ‐ Dano vascular e/ou celular durante flebotomia ‐ Estase venosa por prolongamento do tempo de utilização do torniquete ‐ Hemólise in vitro
1 2 2 ‐ Procedimento padronizado para a recolha de amostra ‐ Antes da recolha da amostra, parâmetros individuais de estilo de vida e ritmos biológicos devem ser tidos em conta, como o exercício físico regular, dieta, stress ‐ Adopção de medidas preventivas para minimizar a influência da estase venosa ‐ Certificação de flebotomistas e treino de toda a equipa de recolha de amostra ‐ Material estéril/autoclavado ‐ Precauções com o anticoagulante escolhido: heparina é um inibidor da amplificação
Manuseamento da amostra
(processamento de sangue e extracção de ácidos nucleicos)
‐ Contaminantes biológicos, físicos e químicos introduzidos ou de amostras anteriores ‐ Degradação do DNA/RNA ‐ Presença de inibidores ‐ Extracção incompleta
‐ Tubos contaminados‐ Contaminação cruzada (amostras, material, bancadas) ‐ Ácido nucleico contaminado com inibidores
2 3 6 ‐ Área própria, sem troca de material com as outras áreas – sistema unidireccional ‐ Tubos descartáveis ‐ Etiquetagem ‐ Luvas mudadas com frequência ‐ Pontas descartáveis com filtro ‐ Lavagens periódicas das bancadas com lixívia 10% e etanol 70%, podendo‐se recorrer à presença de luz UV (bancadas, microcentrífuga, luvas) para descontaminação ‐ Tubos e pipetas livres de DNase/RNase ‐ Autoclavagem do material de vidro ‐ Separação/processamento da amostra efectuada dentro de 72h após colheita ‐ Reagentes próprios em pequenas alíquotas ‐ Não produzir aerossóis quando se está a trabalhar/pipetar DNA

Proposta de Modelo de Controlo e Garantia da Qualidade
45
Tabela 11 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa pré‐analítica (cont.).
Etapa Descrição dos perigos Causas P S IR Medidas de controloManuseamento da
amostra (processamento de sangue e extracção de ácidos nucleicos)
(cont.)
‐ Controlo do DNA extraído:a) Controlo do DNA através de electroforese em gel de agarose b) Digestão do DNA com endonuclease, por exemplo EcoRI, seguida de separação electroforética – controlo da presença de inibidores enzimáticos c) Quantificação de DNA d) Diluição das amostras para concentração de DNA semelhante (10ng/µL) ‐ sucesso de amplificação depende da quantidade e da qualidade de DNA e) Avaliação da pureza de DNA (A260/A280) ‐ Controlo do RNA extraído: a) Electroforese em gel de agarose em condições não desnaturantes (tal como o DNA) e, em caso de dúvida, em condições desnaturantes, para avaliar integridade do RNA

Proposta de Modelo de Controlo e Garantia da Qualidade
46
Tabela 11 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa pré‐analítica (cont.).
Etapa Descrição dos perigos Causas P S IR Medidas de controloTransporte e
armazenamento da amostra/ácidos
nucleicos
‐ Degradação do DNA/RNA‐ Alteração da amostra ‐ Contaminantes biológicos, químicos e físicos
‐ Temperatura inadequada‐ Enzimas que degradam DNA ‐ Práticas incorrectas
1 3 3 ‐ Conservação do tubo de colheita: até 8h à temperatura ambiente (≈ 22°C) ou até 72h quando acondicionado entre 2‐10°C ‐ Armazenamento do plasma: até 4h à temperatura ambiente, até 7 dias entre 2‐10°C e sem limite a ‐70°C ou inferior ‐ Amostras para análise de DNA devem ser guardadas em tampão de 10mmol/l Tris, 1mmol/l EDTA (pH 7,5‐8,0), a 4°C ‐ Amostras para análise de RNA devem ser guardadas em soluções tamponadas de preferência a ‐80°C ou em azoto líquido; também pode ser como um precipitado de etanol a ‐20°C ‐ Teste DNAse para verificar se houve degradação do DNA durante armazenamento ‐ Armazenamento de amostras deve ser feito na mesma área da sua preparação ‐ Controlo e registo da temperatura ‐ Transporte segundo as normas (quando necessário)
IR, Índice de Risco; P, Probabilidade; S, Severidade.
Tabela elaborada com base nas seguintes referências: [19,29,39,41,47,55‐58].

Proposta de Modelo de Controlo e Garantia da Qualidade
47
ii. Factores analíticos ou PCR
Tabela 12 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa analítica.
Etapa Descrição dos perigos Causas P S IR Medidas de controlo
Preparação da mistura de
PCR/Master‐mix
‐ Contaminantes biológicos, químicos e físicos ‐ Contaminação devido a PCR anterior
‐ Técnicas incorrectas‐ Contaminação
1 3 3 ‐ Reagentes em alíquotas‐ Área própria, sem troca de material com as outras áreas – sistema unidireccional; ‐ Limpeza (químicos/UV) ‐ Todos os reagentes, excepto o DNA, são misturados num único tubo – Master‐mix
PCR e Real Time PCR+
RFLP +
Electroforese
‐ PCR e Real‐Time PCR:a) Contaminantes biológicos, químicos ou físicos – falsos positivos e negativos b) Produtos não definidos ou que não eram esperados (falta de especificidade) c) Inibição da reacção d) Contaminação por PCR anterior e) Degradação enzimática f) Protocolos não optimizados (primers longos, alta concentração de sal, etc.)
‐ Contaminação cruzada‐ Erros (analíticos, humanos, etc.) ‐ Contaminação dos reagentes ‐ Má purificação ‐ Restos de determinados reagentes inibidores
1 3 3 ‐ PCR e Real‐Time PCR:a) Áreas separadas, sem troca de material com as outras áreas – sistema unidireccional b) Limpeza (químicos/UV) c) Reagentes próprios em alíquotas d) Termociclador regulado e calibrado, micropipetas calibradas e) Optimização das condições f) Uso de controlo positivo, negativo, controlo interno e controlo ambiental g) Dividir a master‐mix em tubos individuais e só depois o DNA é adicionado (aumenta a eficiência e diminui a contaminação) h) Bom uso das câmaras

Proposta de Modelo de Controlo e Garantia da Qualidade
48
Tabela 12 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa analítica (cont.).
Etapa Descrição dos perigos Causas P S IR Medidas de controlo
PCR e Real Time PCR+
RFLP +
Electroforese (cont.)
‐ RFLP: a) Os sais podem inibir as enzimas, a actividade enzimática pode estar diminuída devido à temperatura ou as condições podem não ser as mais apropriadas ‐ Electroforese: a) Concentração errada do gel, força iónica ou pH e alta concentração de sais na amostra podem distorcer o resultado
‐ RFLP:a) Quando a digestão não funciona, permanece uma grande fracção de elevado peso molecular (mas nem sempre é um indicador) b) Adicionar uma quantidade conhecida de um marcador de alto peso molecular e monitorizar a sua digestibilidade c) Utilizar sequências repetitivas de genoma humano, como DNA mitocondrial, que originam bandas satélites, indicando sucesso de digestão (no entanto, nem todas as enzimas originam essas bandas satélites) ‐ Electroforese: a) Marcador de pares de bases ‐ Os resultados devem ser confirmados: a) Em populações: reanalisar 5‐10% b) Em pacientes: reanalisar os portadores do alelo responsável pela doença e 5‐10% dos não portadores c) Reanalisar com um método diferente, se possível d) Associar com outra variação
IR, Índice de Risco; P, Probabilidade; S, Severidade.
Tabela elaborada com base nas seguintes referências: [18,19,29,34,39,41,42,55,56,58].

Proposta de Modelo de Controlo e Garantia da Qualidade
49
iii. Factores pós‐analíticos ou pós‐PCR
Tabela 13 – Descrição dos perigos, respectivas causas e medidas de controlo de cada etapa pós‐analítica.
Etapa Descrição dos perigos Causas P S IR Medidas de controlo
Leitura dos resultados
‐ Leitura errada leva a conclusões erradas
‐ Leitura errada 1 3 3 ‐ Leitura feita independentemente por duas pessoas
Base de dados e estudo estatístico
‐ Resultados e conclusões erradas
‐ Erro na digitação 1 3 3 ‐ Entrada dupla dos dados
IR, Índice de Risco; P, Probabilidade; S, Severidade.
Tabela elaborada com base nas seguintes referências: [29,39,55,56].

Proposta de Modelo de Controlo e Garantia da Qualidade
50
E. Identificação dos PCC através da árvore de decisão
Modificar o passo, processo ou produto
Sim Não
Sim
Sim
Sim
Sim
Não
Não
Não
Não
Não é PCC
Não é PCC
Nesta etapa é necessário um controlo para garantir a segurança?
Questão 2 (Q2): Esta etapa elimina ou reduz o perigo a um nível aceitável?
Questão 3 (Q3): Pode ocorrer contaminação pelo perigo ou aumento deste para valores inaceitáveis?
Questão 4 (Q4): Existe alguma etapa seguinte que elimine ou reduza o perigo a níveis aceitáveis?
PCC
PCC Não é PCC
Figura 8 – Árvore de decisão de pontos críticos de controlo (adaptado de [55]).
Questão 1 (Q1): Existem medidas preventivas e de controlo para o perigo em questão?

Proposta de Modelo de Controlo e Garantia da Qualidade
51
Tabela 14 – Identificação de pontos críticos de controlo (adaptado de [56]).
Etapa Q1 Q2 Q3 Q4 PCC (Sim/Não) Nº PCC
Recepção de materiais e reagentes/Preparação e armazenamento de
reagentes
Sim Não Sim Não Sim 1
Manuseamento da amostra
(processamento de sangue e extracção de
ácidos nucleicos)
Sim Não Sim Não Sim 2
Transporte e armazenamento da
amostra/ácidos nucleicos
Sim Não Sim Não Sim 3
Preparação da mistura de PCR/Master‐mix
Sim Não Não ‐ Não ‐
PCR e Real‐Time PCR+RFLP + Electroforese
Sim Não Sim Não Sim 4
Leitura de resultados
Sim Não Sim Não Sim 5
Base de dados e estudo estatístico
Sim Não Sim Não Sim 6
PCC, Ponto Crítico de Controlo; Q1, Questão 1; Q2, Questão 2; Q3, Questão 3; Q4, Questão 4.

Proposta de Modelo de Controlo e Garantia da Qualidade
52
F. Plano HACCP
Tabela 15 – Monitorização e medidas correctivas de cada ponto crítico de controlo.
Nº PCC
Limite crítico Monitorização Medidas correctivas Documentos
Frequência Método Responsável
1 ‐ Limite de temperatura (aplica‐se a reagentes com temperatura controlada) ‐ Bula – certificação da qualidade
‐ No momento da recepção/elaboração
‐ Visual‐ Registo
‐ Funcionário destacado
‐ Rejeição do produto que não se encontra em boas condições, certificado ou à temperatura adequada
‐ Registo de recepção, verificação e preparação dos reagentes e materiais
2 ‐ Separação efectuada dentro de 72 horas após colheita ‐ Electroforese de DNA ‐ Quantidade de DNA adequada ‐ Concentração de DNA semelhante em todas as amostras ‐ 1,75< A260/A280 >2,0
‐ 0,5‐1% das amostras ‐ Absorvância‐ Electroforese
‐ Funcionário destacado
‐ Rejeição das amostras não convenientes ‐ Kits de purificação de DNA ‐ Acertar concentração de DNA ‐ Investigar a causa
‐ A cada novo controlo de qualidade preencher uma folha de registo com resultados e observações
3 ‐ Limite de temperatura ‐ Conservação do tubo de colheita: até 8h à temperatura ambiente (≈ 22°C) ou até 72h quando acondicionado entre 2‐10°C ‐ Armazenamento do plasma: até 4h à temperatura ambiente, entre 2‐10°C até 7 dias e a ‐70°C ou inferior sem limite ‐ Teste de degradação do DNA negativo
‐ Temperatura: todos os dias ‐ Teste de degradação DNA ‐ 0,5‐1% das amostras
‐ Temperatura‐ Teste de degradação (DNAses)
‐ Funcionário destacado
‐ Rejeição das amostras de DNA degradado ‐ Em caso de avaria do equipamento, mudar tudo para outro refrigerador rapidamente ‐ Rejeição se a temperatura limite tiver sido atingida ‐ Investigação da causa
‐ Registo da temperatura, avarias e manutenção do equipamento
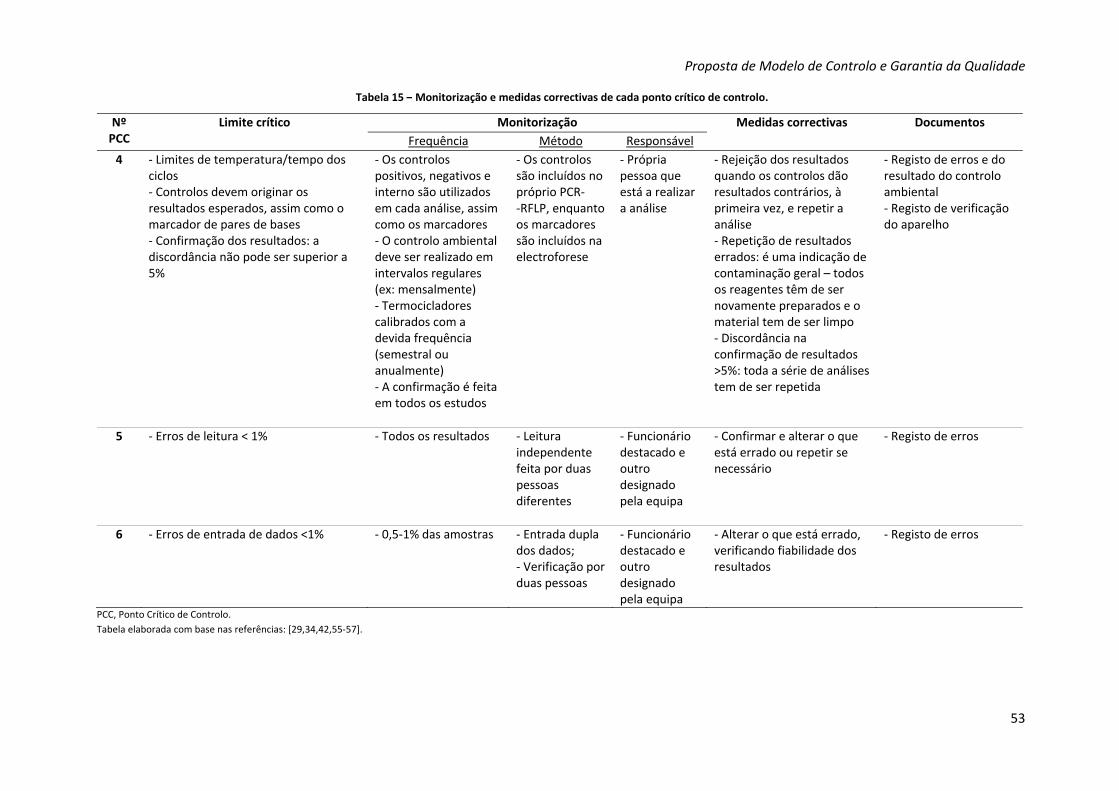
Proposta de Modelo de Controlo e Garantia da Qualidade
53
Tabela 15 – Monitorização e medidas correctivas de cada ponto crítico de controlo.
Nº PCC
Limite crítico Monitorização Medidas correctivas Documentos
Frequência Método Responsável
4 ‐ Limites de temperatura/tempo dos ciclos ‐ Controlos devem originar os resultados esperados, assim como o marcador de pares de bases ‐ Confirmação dos resultados: a discordância não pode ser superior a 5%
‐ Os controlos positivos, negativos e interno são utilizados em cada análise, assim como os marcadores ‐ O controlo ambiental deve ser realizado em intervalos regulares (ex: mensalmente) ‐ Termocicladores calibrados com a devida frequência (semestral ou anualmente) ‐ A confirmação é feita em todos os estudos
‐ Os controlos são incluídos no próprio PCR‐ ‐RFLP, enquanto os marcadores são incluídos na electroforese
‐ Própria pessoa que está a realizar a análise
‐ Rejeição dos resultados quando os controlos dão resultados contrários, à primeira vez, e repetir a análise ‐ Repetição de resultados errados: é uma indicação de contaminação geral – todos os reagentes têm de ser novamente preparados e o material tem de ser limpo ‐ Discordância na confirmação de resultados >5%: toda a série de análises tem de ser repetida
‐ Registo de erros e do resultado do controlo ambiental ‐ Registo de verificação do aparelho
5 ‐ Erros de leitura < 1% ‐ Todos os resultados ‐ Leitura independente feita por duas pessoas diferentes
‐ Funcionário destacado e outro designado pela equipa
‐ Confirmar e alterar o que está errado ou repetir se necessário
‐ Registo de erros
6 ‐ Erros de entrada de dados <1% ‐ 0,5‐1% das amostras ‐ Entrada dupla dos dados; ‐ Verificação por duas pessoas
‐ Funcionário destacado e outro designado pela equipa
‐ Alterar o que está errado, verificando fiabilidade dos resultados
‐ Registo de erros
PCC, Ponto Crítico de Controlo.
Tabela elaborada com base nas referências: [29,34,42,55‐57].

Proposta de Modelo de Controlo e Garantia da Qualidade
54
2. Critérios de Avaliação
Tendo em conta o que foi sistematizado nas Tabelas 11‐15 e tendo como base
principalmente as guidelines da EMQN [30], elaborou‐se um conjunto de critérios para as várias fases
do processo. Os critérios remetem tanto para a parte laboratorial (cuidados, material, etc.) como
para toda a organização de informação e do próprio laboratório.
Estes critérios foram utilizados unicamente como guia durante a observação das várias
pessoas que constituem a equipa de investigação do grupo de Oncologia Molecular do IPO‐Porto. O
objectivo era simplesmente verificar se estes critérios são diariamente cumpridos ou não.
Relativamente aos critérios mais relacionados com a organização do laboratório e de informação, o
seu cumprimento também foi verificado. No entanto, estes critérios podem ter outras utilidades
futuras, como servirem de base à elaboração de fichas de registo de cumprimento de funções ou
mesmo de auditorias internas. Segue‐se a discrição dos critérios adoptados neste trabalho:
A. Recepção da amostra
i. Informação do paciente armazenada em base de dados, protegida por palavra‐chave,
com acesso restricto;
Existência de uma cópia de segurança da informação;
Pessoal dedicado à manutenção e realização de cópia de segurança da base de dados;
ii. Procedimento para lidar com amostras com informação incompleta ou incorrecta;
iii. Tubos da amostra têm de ser etiquetados com pelo menos duas informações de
identificação que os unem;
iv. Transferência de informação manual? Etiquetas com informação são impressas?
v. Quando é alterada alguma informação, como se processa? Inclusão da razão dessa
alteração? Existe um procedimento padronizado?

Proposta de Modelo de Controlo e Garantia da Qualidade
55
B. Reagentes – recepção, transporte e armazenamento
i. Minimização da contaminação – limpeza de bancadas e material com álcool 70°;
ii. Área separada para recepção e preparação de reagentes e master‐mix;
iii. O sistema é unidireccional, não se voltando a esta sala;
iv. Material próprio na área, sem trocas com outro compartimento (luvas, batas, pontas,
micropipetas, etc.);
v. Pontas com filtro e tubos descartáveis livres de DNAse e RNAse;
vi. Utilização de lâmpadas UV colocadas sobre a bancada e material, ligadas pelo menos
15 minutos antes de se iniciar o trabalho;
vii. Fornecedores bem seleccionados, com garantia de qualidade (bula);
viii. Verificação da integridade da embalagem, prazo de validade, rotulagem;
ix. Os reagentes são armazenados em alíquotas, bem etiquetadas (conteúdo e data);
x. Quando se tem de congelar, congela‐se em alíquotas;
xi. Rejeição de reagentes contaminados.
C. Transporte e armazenamento da amostra
i. Conservação do tubo de colheita até 8h à temperatura ambiente (≈22°C) ou até 72h
quando armazenado entre 2‐10°C;
ii. Armazenamento do plasma: até 4h à temperatura ambiente; até 7 dias entre 2‐10°C;
sem limite a ‐70°C ou inferior;

Proposta de Modelo de Controlo e Garantia da Qualidade
56
iii. DNA armazenado a 4°, ‐20° ou ‐40°C;
iv. Controlo da temperatura;
v. Quando necessário transporte, as condições para a amostra/reagente em causa são
respeitadas;
vi. Amostras para análise de DNA guardadas em tampão de 10mmol/l Tris, 1mmol/l EDTA
(pH 7,5‐8,0), a 4°C;
vii. Amostras para análise de RNA guardadas em soluções tamponadas de preferência a
‐80°C ou em azoto líquido ou como um precipitado de etanol a ‐20°C;
viii. Minimização do processo congelar/descongelar da amostra;
Alíquota de trabalho armazenada a 4°C enquanto o teste se realiza;
ix. Duplicado da amostra de sangue/ácido nucleico armazenado antes e/ou depois da
extracção de ácidos nucleicos;
Duplicados são guardados por um período mínimo de um ano, num segundo
frigorífico, de preferência noutro departamento ou edifício ou realiza‐se
armazenamento de gotas de sangue em papel 3MM;
x. As etiquetas em amostras de DNA ou papéis com gotas de sangue incluem pelo menos
duas características de identificação;
Utilizam mais registos em números ou em letras? Registos em números são mais
susceptíveis a erros.
D. Processamento de sangue e extracção de ácidos nucleicos
i. Medidas para minimizar o risco de contaminação durante a extracção: lavagens
periódicas das bancadas com lixívia 10% e etanol 70%, pode‐se utilizar luz UV em cima
da bancada e material (centrífuga, luvas, pontas), limpeza da bancada e material com
álcool antes de iniciar o trabalho;

Proposta de Modelo de Controlo e Garantia da Qualidade
57
ii. Uso de espaços dedicados somente ao processo de processamento e extracção,
cabines de segurança e material próprio da área (luvas, batas, micropipetas e pontas);
iii. Sistema unidireccional, não se voltando a esta sala;
iv. Processamento da amostra (sangue) efectuado dentro de 72h após colheita;
v. Utilização de pontas com filtro e tubos descartáveis livres de DNAse e RNAse;
micropipetas bem calibradas;
vi. Material de vidro autoclavado;
vii. Todos os tubos, eppendorfs, etc. devem ser bem identificados;
viii. Controlo da quantidade e qualidade do DNA;
ix. Controlo da quantidade e qualidade do RNA;
x. Diluição das amostras para uma concentração de DNA semelhante (a concentração de
DNA tem de ser constante (10ng/µl));
xi. Reagentes e DNA em alíquotas;
xii. Se os reagentes não foram completamente removidos, o DNA é retomado num maior
volume de tampão e repete‐se a precipitação;
xiii. Produção de aerossóis mínima (pipetar devagar, com cuidado, evitar correntes de ar,
portas fechadas, para evitar contaminação).

Proposta de Modelo de Controlo e Garantia da Qualidade
58
E. Manuseamento da amostra
i. Todas as transferências de tubos durante a extracção de ácidos nucleicos, PCR e
digestão são independentemente verificadas pelo operador ou outro membro
qualificado, para minimizar o risco de misturar/contaminar a amostra;
Folhas realizadas pelo verificador para fornecer uma prova da verificação;
Alternativamente, duplicado da amostra é montado em paralelo e analisado
juntamente com o original;
ii. São guardados todos os números de lote de todos os reagentes e soluções do
laboratório, para rastreabilidade e resolução de problemas;
iii. Separação das áreas de pré e pós‐PCR. Material próprio em cada área.
F. Preparação da mistura PCR
i. Todos os reagentes misturados num único tubo, excepto o DNA – master‐mix;
ii. Reagentes armazenado em alíquotas;
iii. Área própria, com material próprio, sem trocas ‐ sistema unidireccional;
iv. Pontas com filtro e micropipetas calibradas;
v. Realizado em ambiente semi‐estéril;
vi. No caso de câmara de acrílico: material (caixa de pontas, micropipetas, suportes) é
limpo com álcool, introduzido na câmara, ligam‐se os UV durante 15 minutos e só
depois se inicia o trabalho. No final, retira‐se todo o material, limpa‐se e volta‐se a
ligar os UV por 15 minutos;
vii. No espaço dedicado ao pré‐PCR a pressão deve ser positiva e no espaço de
amplificação a pressão deve ser reduzida;

Proposta de Modelo de Controlo e Garantia da Qualidade
59
viii. Primers: qualidade, design, pureza e validação são controlados. Cada novo lote é
testado para especificidade, eficiência de amplificação e ausência de inibidores antes
de serem utilizados;
ix. Todas as soluções devem estar fora da sua temperatura habitual o menor tempo
possível – uso de gelo durante a sua utilização;
x. A enzima deve ser retirada do congelador imediatamente antes de ser adicionada e é a
última a ser introduzida na master‐mix;
G. PCR‐RFLP/Electroforese
i. Área de amplificação e detecção separada das áreas de armazenamento e
processamento. A amplificação e a detecção podem ser ou não na mesma área.
Sistema unidireccional;
ii. Material próprio, sem troca;
iii. Micropipetas bem calibradas;
iv. Limpeza das bancas (químicos/UV);
v. Utilização de DNA guardado em alíquotas, recorrendo‐se ao DNA original apenas
quando a quantidade da alíquota já não é suficiente;
vi. Identificação da amostra certa ‐ os eppendorfs devem estar todos numerados na
tampa e não pode haver erros;
vii. Todas as amostras que vão ser utilizadas têm de ser centrifugadas antes;
viii. Dividir a master‐mix em tubos de PCR e só depois o DNA de cada amostra é adicionado
(aumenta eficiência e diminui contaminação);

Proposta de Modelo de Controlo e Garantia da Qualidade
60
ix. PCR: uso de controlo positivo (mutações específicas) ou amostras de genótipo
conhecido, controlo negativo e controlo interno. Realizar um controlo ambiental, em
intervalos regulares, para verificar contaminação a partir do meio ambiente;
x. Procedimento padronizado a realizar quando os resultados dos controlos são
diferentes dos esperados;
xi. PCR: termociclador regulado e calibrado; optimização de protocolo;
xii. RFLP: tem de ser controlado – adicionar marcador de peso molecular, tal como na
electroforese. Também se pode controlar utilizando bandas satélite. Inclusão de
controlo negativo;
xiii. RFLP: overnight, mínimo de 16 horas. Controlo da temperatura da estufa (37°C).
H. Real‐Time PCR
i. Utilização de alíquotas de DNA, recorrendo‐se ao DNA original apenas quando a
quantidade da alíquota já não é suficiente;
ii. Identificação da amostra certa. Os eppendorfs devem estar todos numerados na tampa
e não pode haver erros;
iii. Centrifugação de cada amostra;
iv. Utilização de controlos negativos em maior número;
v. Utilização de controlo positivo (ou amostras de genótipo conhecido);
vi. A preparação da placa que vai ser lida é feita em câmara de fluxo laminar ‐ a câmara
tem de ser analisada periodicamente;

Proposta de Modelo de Controlo e Garantia da Qualidade
61
vii. As luzes UV devem estar ligadas durante 15 minutos. O material tem de ser limpo com
álcool antes de ser introduzido na câmara e quando se retira. No final voltar a ligar UV.
O tempo de purga deve ser respeitado;
viii. Micropipetas calibradas;
ix. Neste caso, a master‐mix já vem preparada. Verificação das condições de
armazenamento e a bula que a acompanha para certificar que cumpre todos os
requisitos;
x. Assay: verificação de qualidade pelo fornecedor;
xi. Não esquecer de tapar a placa com tira para não saltar entre poços ao centrifugar nem
evaporar durante a leitura.
I. Resultados
i. Quando não se consegue concluir ou existem dúvidas, tem de se repetir;
ii. Presença de um protocolo de confirmação dos resultados;
iii. Reanálise das amostras com outro método se for possível;
iv. Associação do estudo da variação em causa com outra;
v. Se na reanálise os resultados diferirem mais de 5%, reanalisa‐se toda a série;
vi. A leitura dos resultados é feita independentemente por duas pessoas;
vii. A entrada dos resultados em base de dados é dupla;
viii. Os resultados das experiências são guardados por um período mínimo de 5 anos, em
formato electrónico, em base de dados.

Proposta de Modelo de Controlo e Garantia da Qualidade
62
J. Documentação
i. Conjunto de protocolos‐padrão para todas as técnicas usadas.
K. Validação
i. Testes de diagnóstico são validados para assegurar procedimentos‐padrão e que são
adequados para o objectivo proposto;
ii. Comparação com resultados obtidos noutros laboratórios (resultados devem ser
consistentes);
iii. Procedimentos de controlo de qualidade.
L. Treino da equipa
i. As pessoas são devidamente qualificadas e especificamente treinadas nos métodos
usados e também nas regras de segurança;
ii. A equipa é encorajada para fazer continuamente uma actualização do seu
conhecimento, através de leitura de comunicados recentes e presença em seminários
e conferências apropriadas;
Mantém‐se um registo dessa formação contínua de cada membro da equipa.

Proposta de Modelo de Controlo e Garantia da Qualidade
63
3. Plano de Avaliação Qualidade
De acordo com os PCC identificados no ponto 1, com os critérios estabelecidos, com as
observações feitas nas instalações e com o plano de controlo de qualidade interno descrito por
Bladbjerg et al. (2002) [29], tenta‐se estabelecer uma proposta para controlo e avaliação da
qualidade interna da técnica de PCR efectuada no laboratório de Oncologia Molecular do IPO‐Porto.
Cumprindo os vários pontos é possível avaliar o próprio processo, concluir sobre a qualidade do
processo e resultados, e instalar uma rotina de controlo.
O protocolo a experimentar e implementar será o seguinte para cada PCC:
A. PCC nº 1
i. Certificação da qualidade, prazo, embalagem, condições do produto;
ii. Elaboração de uma ficha de registo por cada reagente ou material
recebido/preparado, com lote e data;
iii. Frequência: sempre que haja recepção de novos materiais/reagentes ou preparação
de novos reagentes.
B. PCC nº 2
i. Controlo da qualidade do DNA extraído;
ii. Controlo de DNA através de electroforese em gel de agarose; pode ser utilizada uma
digestão do DNA com uma endonuclease para controlo da presença de inibidores
enzimáticos;
iii. Quantificação do DNA extraído ‐ leitura da absorvância a 260nm (A260) e cálculo da
concentração utilizando o coeficiente de extinção molar (ε = 20ml/(mg x cm)); fazer
média e desvio padrão dos valores das diferentes amostras;
iv. Avaliação da pureza de DNA – leitura da absorvância a 260 e 280nm, divisão dos
valores (A260/A280) e verificação de que se encontram dentro do intervalo
(1,75<A260/A280>2,0) e quantos estão fora dos parâmetros;
v. Possibilidade de elaboração de cartas de controlo e registos;
vi. Frequência: 0,5‐1% das amostras mensais;
vii. Elaboração de ficha de registo da calibração das micropipetas; estas devem ser
calibradas semestral ou anualmente.

Proposta de Modelo de Controlo e Garantia da Qualidade
64
C. PCC nº 3
i. Elaboração de fichas de registo da temperatura para cada frigorífico e arca – garantia
de bons processos de conservação; podem‐se elaborar cartas de controlo;
ii. Frequência de registo da temperatura: todos os dias, com a assinatura da pessoa
responsável;
iii. Se possível, implementar o teste de DNAses – garantia de DNA não degradado por
exclusão de enzimas que o degradem durante o seu armazenamento; resultado
positivo ou negativo;
iv. Frequência do teste de DNAses – em 0,5‐1% das amostras mensais.
D. PCC nº 4
i. Utilização de controlos negativos no PCR‐RFLP e Real‐Time PCR em todas as análises;
ii. Utilização de controlo positivo ou amostra de genótipo conhecido, quando possível, no
PCR‐RFLP e Real‐Time PCR;
iii. Não é viável a utilização de controlo interno;
iv. Inclusão de marcador de peso molecular na electroforese em todas as análises;
v. Utilização de um controlo ambiental no PCR‐RFLP e Real‐Time PCR, mensalmente;
vi. Confirmação dos resultados: repetição de 10% dos casos de estudo, aleatoriamente,
em todos os estudos;
vii. Registo de casos em que os controlos ou a confirmação não dão os resultados
esperados (repetição de toda a série);
viii. Estudo da percentagem de casos em que os controlos não dão o resultado esperado e
de reanálises;
ix. Registo das calibrações e verificações do termociclador (anualmente);
x. Elaboração de ficha de registo da calibração das micropipetas; estas devem ser
calibradas semestral ou anualmente;
xi. Elaboração da ficha de registo da temperatura da estufa (37°C) utilizada no RFLP – uma
vez por dia o operador regista a temperatura e assina.
E. PCC nº 5
i. Leitura feita por duas pessoas, de modo independente, em todas as análises;

Proposta de Modelo de Controlo e Garantia da Qualidade
65
ii. Registo de percentagem de erros ocorridos durante o controlo (<1%).
F. PCC nº 6
i. Entrada dupla dos dados, feita por duas pessoas de modo independente, em 0,5‐1%
das amostras;
ii. Registo de percentagem de erros ocorridos durante o controlo (<1%);

V. Resultados e Discussão

Resultados e Discussão
67
1. Adaptação de um Plano HACCP
O plano HACCP apresentado pretende resumir as etapas mais importantes da técnica de PCR
e os seus principais cuidados, identificar perigos, PCC, medidas preventivas, de controlo e
correctivas. Dos sete Princípios do HACCP, apenas foram cumpridos os Princípios 1‐5 e com a
implementação do plano de avaliação da qualidade interna tornar‐se‐á viável a realização dos
Princípios 6 e 7. Os Princípios 1‐5 foram sendo cumpridos à medida que se seguiram os 12 Passos
estipulados para a implementação do sistema HACCP. Os passos 2‐10 foram cumpridos através da
elaboração das Tabelas 9‐15 e Figuras 6 e 7. Também aqui, os passos em falta (1, 11 e 12) poderão
ser cumpridos ao implementar o plano da avaliação da qualidade estipulado. Com essa
implementação vai nascer a necessidade de designar uma equipa HACCP, responsável por cumprir o
protocolo, por verificar que todo o plano é cumprido e verificar e elaborar os registos, atingindo‐se
assim os pontos 1, 11 e 12. Não se estabeleceu um documento de requerimentos‐base e pré‐
‐requisitos do laboratório necessários ao estabelecimento deste plano, no entanto, muitas das
indicações descritas nas tabelas de cada etapa da técnica constituem as boas práticas necessárias
para o trabalho diário.
Através da adaptação de um plano HACCP, foram identificados seis PCC para a técnica de
PCR e estabelecida uma proposta para o seu controlo e avaliação, como já foi referido.
Nas tabelas foram referidos os cuidados e especificações necessárias durante a prática de
PCR, que foram também utilizadas nos critérios. O cumprimento desses critérios será discutido no
ponto seguinte.
2. Análise dos Resultados da Observação dos Colaboradores e Utilização dos Critérios
Após a elaboração dos critérios, os vários colaboradores do laboratório foram
acompanhados, na prática de PCR‐RFLP e Real‐Time PCR, com o objectivo de verificar se cumpriam as
condições necessárias. Essa observação está resumida nas Tabelas 16‐18.

Resultados e Discussão
68
A. Critérios pré‐analíticos
Tabela 16 ‐ Cumprimento de critérios pré‐analíticos.
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Processamento de sangue e extracção dos ácidos nucleicos
Medidas para minimizar risco de contaminação
‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ S ‐ S ‐
Uso de espaços dedicados somente ao processo de processamento e extracção
‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ S ‐ S ‐
Utilização de material próprio da área e do protocolo
‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ S ‐ S ‐
Processamento da amostra (sangue) efectuado dentro de 72h após colheita
‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ ‐ ‐ S ‐
Controlo da quantidade e qualidade do DNA
‐ ‐ ‐ ‐ ‐ ‐ ‐ ‐ ‐ ‐ ‐ ‐ N ‐ ‐ ‐
Reagentes e DNA em alíquotas
‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ S ‐ S ‐
Produção de aerossóis Mínima
‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ S ‐ S ‐
Etiquetagem ‐ ‐ ‐ ‐ ‐ S ‐ ‐ ‐ ‐ ‐ ‐ S ‐ S ‐

Resultados e Discussão
69
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Manuseamento da amostra
Verificação por outro operador
N N N N N N N N N N N N N N ‐ N
Etiquetagem S S S S S S S S S S S S S S ‐ S S, Sim; N, Não.
B. Critérios analíticos
Tabela 17 – Cumprimento de critérios analíticos.
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Preparação da master‐mix
Reagentes em alíquotas
S S S S S S S S S S S S S ‐ ‐ S
Área própria
N N N N N N N N N N N N N ‐ ‐ N
Material adequado existente na área
S S S S S S S S S S S S S ‐ ‐ S
Realizada em câmara
S S S S S S S S S S S S S ‐ ‐ S
Micropipetas calibradas
N N N N N N N N N N N N N N ‐ N
Funcionamento adequado dos UV da câmara
S S S S S S S S S S S S S S ‐ S
Teste dos primers N N N N N N N N N N N N N N ‐ N
Tabela 16 ‐ Cumprimento de critérios pré‐analíticos (cont.).

Resultados e Discussão
70
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Preparação da master‐mix (cont.)
Cuidados com a temperatura dos reagentes
S S S S S S S S S S S S S S ‐ S
Uso de gelo
N N N N N N N N N N N N N N ‐ N
Adição da enzima em último
S S ‐ S S S ‐ S S S S S S ‐ ‐ S
PCR‐RFLP
Áreas separadas
S S N S S S ‐ S S S S S S ‐ ‐ S
Sistema unidireccional
N N N N N N ‐ N N N N N N ‐ ‐ N
Material próprio da área
S S S S S S ‐ S S S S S S ‐ ‐ S
Pipetas calibradas
N N N N N N ‐ N N N N N N ‐ ‐ N
Utilização de alíquotas de DNA
S S ‐ S S S ‐ S S S S S S ‐ ‐ S
Correcta identificação dos casos/controlos
S S S S S S ‐ S S S S S S ‐ ‐ S
Centrifugação prévia dos casos
S S S S S S ‐ N N S S S N ‐ ‐ S
Primeiro adiciona‐se a master‐mix a cada tubo e só depois o DNA
S S S S S S ‐ S S S S S S ‐ ‐ S
Tabela 17 – Cumprimento de critérios analíticos (cont.).

Resultados e Discussão
71
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
PCR‐RFLP (cont.)
Controlo positivo/amostras com genótipo conhecido
N N S N N N ‐ N S N N N N ‐ ‐ S
Controlo negativo
S S S S S S ‐ S S S S S S ‐ ‐ S
Controlo interno
N N N N N N ‐ N N N N N N ‐ ‐ N
Controlo ambiental
N N N N N N ‐ N N N N N N ‐ ‐ N
Controlo negativo e marcador de peso molecular
S S S S S S ‐ S S S S S S ‐ ‐ S
Real‐time PCR
Utilização de alíquotas de DNA
S S ‐ S S S ‐ ‐ ‐ ‐ ‐ S S S ‐ S
Identificação correcta dos casos
S S ‐ S S S S ‐ ‐ ‐ ‐ S S S ‐ S
Centrifugação dos casos
S S ‐ S S S S ‐ ‐ ‐ ‐ S N S ‐ S
Controlos negativos
S S ‐ S S S S ‐ ‐ ‐ ‐ S S S ‐ S
Controlo positivo
N N ‐ N N N S ‐ ‐ ‐ ‐ N N N ‐ N
Controlo interno
N N ‐ N N N S ‐ ‐ ‐ ‐ N N N ‐ N
Pipetas calibradas
N N ‐ N N N N ‐ ‐ ‐ ‐ N N N ‐ N
Tabela 17 – Cumprimento de critérios analíticos (cont.).

Resultados e Discussão
72
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Real‐time PCR (cont.)
Utilização correcta da câmara: UV/Purga
S/N S/N ‐ S/N S/N S/N S/S ‐ ‐ ‐ ‐ S/N S/N S/N ‐ S/N
Cumprimento de todos os cuidados necessários durante a manipulação
S S ‐ S S S S ‐ ‐ ‐ ‐ S S S ‐ S
S, Sim; N, Não.
C. Critérios pós‐analíticos
Tabela 18 – Cumprimento de critérios pós‐analíticos.
Colaborador (nº)
Etapa/Critério 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Resultados
Confirmação dos resultados
S S ‐ S S S ‐ S S S S S S S ‐ S
Leitura dos resultados feita independentemente por duas pessoas
S S N S S S N S S S S S S S ‐ S
Entrada dupla na base de dados
N N N N N N N N N N N N N N ‐ N
S, Sim; N, Não.
Tabela 17 – Cumprimento de critérios analíticos (cont.).

Resultados e Discussão
73
Tendo em conta as Tabelas HACCP elaboradas para a técnica de PCR, os critérios que
remeteram para os colaboradores e a observação destes no laboratório, podem‐se retirar algumas
conclusões em cada etapa do processo. De um modo geral, todos os colaboradores cumpriram os
requisitos mínimos da prática de PCR.
A. Critérios pré‐analíticos
Rapidamente se verifica nas etapas de processamento de sangue, extracção de DNA e
manuseamento da amostra (Tabela 16) que os dois únicos pontos não cumpridos são o controlo da
qualidade e quantidade de DNA e a verificação por outro operador. O controlo do DNA foi incluído
no plano de avaliação da qualidade proposto, mas a verificação por outro operador de todo o
manuseamento da amostra pode ser complicado de implementar e por isso pode ser substituído por
folhas de registo em que o operador assinala se efectuou ou não todos os requisitos.
B. Critérios analíticos
Relativamente aos critérios analíticos (Tabela 17), é de salientar os seguintes aspectos:
i. Embora existam áreas separadas e muitas das guidelines para as áreas sejam
cumpridas, existem algumas excepções, como é o caso da preparação da master‐mix que é feita na
mesma área de amplificação e de algumas das salas se encontrarem no mesmo piso, de modo
contínuo; apesar disso, existe uma boa separação das diferentes fases do processo, que parece
garantir a obtenção de bons resultados;
ii. A calibração das micropipetas é muito importante e não existe nenhum registo da
sua frequência, tendo‐se verificado que grande parte se encontra descalibrada; por essa mesma
razão é proposta a elaboração de um registo e a calibração das micropipetas semestral ou
anualmente;
iii. Os primers não são testados antes de serem utilizados nos ensaios; no entanto a
certificação de qualidade que os acompanha é verificada. Apesar da importância que tem, o controlo
dos primers não foi incluído no plano de avaliação; futuramente, se necessário, este ponto será
também incluído;

Resultados e Discussão
74
iv. O uso de gelo é aconselhado pois, para além de manter a temperatura ideal para a
conservação das enzimas utilizadas durante o trabalho, evita que os colaboradores se levantem a
meio do PCR, o que pode levar a algumas contaminações (correntes de ar, esquecimento da troca de
luvas, etc.), assim como se cumpriria melhor o sistema unidireccional de salas;
v. Centrifugação das amostras nem sempre é feita, provavelmente por esquecimento;
em amostras com pouca quantidade, a centrifugação é essencial e na maioria das vezes é feita;
vi. A utilização de um controlo positivo de referência ou de uma amostra de genótipo
conhecido é muito comum em dois colaboradores, em testes de virologia, onde também se utiliza
controlo interno, mas cujos ensaios já obedecem a um plano de controlo de qualidade; no PCR‐RFLP
e Real‐Time que se avaliou, não é habitual usar, demonstrando a dificuldade de obtenção de um
controlo de referência em testes genéticos, principalmente quando se trabalha com
mutações/polimorfismos; o uso de uma amostra cujo genótipo é já conhecido pode representar um
maior gasto da amostra, que ainda pode ser útil noutros estudos, ficando a cargo do responsável o
seu uso ou não;
vii. Não se utiliza controlo interno, o que se justifica pelo alargado número de
polimorfismos que são estudados, o que iria introduzir um gasto superior ao normal; registou‐se a
utilização de controlo interno nos testes de virologia, onde se utilizam kits para identificar a presença
de alguns vírus e onde o controlo de qualidade está bem implementado, como já se referiu;
viii. O controlo ambiental não é utilizado, mas está proposto no plano de avaliação da
qualidade;
ix. A câmara de fluxo laminar exige um certo número de cuidados para o seu
funcionamento, como os UV durante 15 minutos (início e fim) e um tempo de purgação (5 minutos);
só depois destes dois passos se deve iniciar o trabalho na câmara, mas nem sempre é cumprido; no
entanto, pensa‐se que este pequeno esquecimento não tem sido determinante nos resultados;
x. A câmara de fluxo laminar é verificada todos os anos, já existindo um registo, que se
encontra actualizado;

Resultados e Discussão
75
xi. Um parâmetro de prevenção de contaminação muito importante é o uso de luvas.
Em geral, todos os colaboradores usam luvas nas mais diversas operações, com excepção de um
esquecimento que se deveu à inexperiência do operador. No entanto, a troca de luvas não é por
vezes respeitada.
C. Critérios pós‐analíticos
Em geral, todos os resultados são verificados por duas pessoas. A introdução na base de
dados é feita apenas por uma pessoa e não parece exequível que se faça sempre por duas pessoas.
Por esta razão estabeleceu‐se no plano de avaliação da qualidade que só para uma certa
percentagem de amostras ou ensaios se faria a entrada através de duas pessoas. Nos casos em que
se verifique que os erros são em grande escala, terão de ser tomadas as medidas adequadas.
D. Critérios gerais referentes ao laboratório
Para além dos critérios relacionados com a prática diária dos colaboradores nas técnicas de
PCR, também foram adaptados critérios gerais do laboratório em termos de organização, de
recursos, etc., que foram verificados diariamente, por visualização, ou por questões a alguns
colaboradores mais antigos do laboratório. Os critérios são os descritos na parte IV deste trabalho e
os resultados da visualização/questionário encontram‐se explicados seguidamente. De salientar que
existe uma fase do processo explicada nas tabelas HACCP que não se encontra nos critérios: a
recolha da amostra. Esta fase não é feita por nenhum dos colaboradores do laboratório e por isso é
de difícil controlo por parte do laboratório de Oncologia Molecular. No entanto é uma fase
determinante do processo, que deve ser controlada pelos pré‐requisitos, cumprindo‐se todos os
cuidados.
i. Recepção da amostra
A nível da recepção da amostra, os critérios são em geral cumpridos. A informação
está armazenada em base de dados, havendo mesmo um grupo de indivíduos responsável, mas estas
não possuem palavra‐chave. No entanto, essa é uma medida a implementar, assim como a criação
de uma cópia de segurança.
Quando uma amostra do IPO é recebida com uma informação incompleta ou
incorrecta, recorrem ao arquivo de registo clínico; se for do Hospital de S. João ou do Hospital

Resultados e Discussão
76
Militar, entra‐se em contacto com o médico responsável pela base de dados desse local. Quando há
alguma alteração a fazer, actualiza‐se simplesmente a base de dados.
Os tubos têm etiquetas impressas, com duas ou mais informações do doente.
ii. Reagentes – recepção, transporte e armazenamento
Neste ponto quase todos os critérios são cumpridos, desde as limpezas, ao material
utilizado, alíquotas e fornecedores. As únicas excepções, já referidas anteriormente, dizem respeito à
separação de áreas e sistema unidireccional, mas tal não parece comprometer a qualidade dos
resultados.
iii. Transporte e armazenamento da amostra
Em relação ao transporte e armazenamento da amostra, não se consegue ter um bom
controlo no transporte dos tubos de colheitas com origem noutro Hospital que não o IPO. No
entanto, está garantido o processamento do sangue no próprio dia em que a amostra é recolhida e
recebida.
Relativamente a todas as outras especificações de temperatura e cuidados da amostra,
estas são cumpridas, com excepção do controlo de temperatura das arcas e frigoríficos e por isso
mesmo foi sugerido o registo dessa mesma temperatura por um membro responsável. No que diz
respeito ao duplicado, existe uma reserva de sangue e pellet celular, mas não se armazena gotas de
sangue em papel 3MM como acontece em alguns laboratórios de diagnóstico. Na identificação dos
tubos e eppendorfs utilizam‐se mais dados em números, pois embora mais sujeitos a erros,
correspondem a números internos, o que facilita a procura de informação, quando necessário.
iv. Processamento de sangue e extracção de ácidos nucleicos
Todos os cuidados relativos à limpeza e material são cumpridos, assim como os
cuidados de pipetagem. Os restantes requisitos de separação de salas, sistema unidireccional e
cuidados dos colaboradores já foram referidos.
Existe um registo de todos os tubos de sangue recebidos, com as informações
necessárias, assinado pelo próprio operador.
É importante referir que, no decorrer deste trabalho, o laboratório esteve em processo
de automatização da etapa de extracção dos ácidos nucleicos e vai ser possível a utilização desse
sistema em breve.

Resultados e Discussão
77
v. Manuseamento da amostra
Como já foi referido, as operações não são habitualmente verificadas por outro
operador, não existe registo das várias fases do processo, nem nenhum duplicado é analisado em
paralelo, pois não se considera importante nesta fase. No plano de avaliação da qualidade sugere‐se
um registo que possa ser verificado ou preenchido pelo próprio operador.
Relativamente aos reagentes, alguns são preparados pelos próprios colaboradores do
laboratório, ficando isso registado, mas outros são adquiridos aos fornecedores. Quando há recepção
destes reagentes, para além de todos os cuidados de verificação de embalagem, prazo, certificação
de qualidade, etc., deve‐se criar uma folha de registo, com o lote, prazo e todas as informações
necessárias, de modo a facilitar a consulta de informação e a identificação de um lote que se conclua
mais tarde que não se encontra nas devidas condições. Este procedimento ainda não é realizado pelo
laboratório.
vi. Preparação da mistura PCR, PCR‐RFLP/Electroforese, Real‐Time PCR
Os requisitos referentes a estes três pontos já foram, na sua maioria, mencionados e
discutidos anteriormente, a partir da Tabela 17. É importante confirmar que não há controlo sobre as
condições de pressão atmosférica nos locais de pré‐PCR e de amplificação, mas parece não ter
importância o suficiente para ser colocado no plano de avaliação da qualidade. Além disso, já foi
referida a necessidade de registo de funcionamento do equipamento, e por isso se sugeriu a
elaboração de folhas de registo e de calibração dos termocicladores. O protocolo de PCR é sempre
optimizado no início da experiência.
Quando os controlos positivos ou negativos apresentam um resultado diferente do
esperado, a análise é repetida. No caso de se manter essa diferença, tenta‐se analisar em grupo as
razões que podem levar a esses resultados e como as corrigir (mudança de reagentes, primers,
melhoria das limpezas efectuadas, etc.).
vii. Resultados
Sempre que existe alguma dúvida no resultado, o ensaio é repetido e já existe no
laboratório um protocolo de confirmação de resultados que se manteve no plano de avaliação de
qualidade proposto: confirmação de 10% dos resultados, escolhidos ao acaso. Na Tabela 12
apresenta‐se um plano de confirmação proposto por Bladbjerg et al. (2002) [29], muito completo
mas que não é facilmente exequível numa primeira fase, parecendo a confirmação de 10% uma boa

Resultados e Discussão
78
escolha de momento. O cruzamento de resultados dos estudos de diferentes polimorfismos proposto
nesse mesmo plano de confirmação dos resultados já é habitualmente feito pelos colaboradores.
Limitou‐se o erro máximo possível na confirmação de resultados a 5% de discordância. Os dados são
armazenados sem período mínimo, ficando sempre disponíveis para consulta.
viii. Documentação
Relativamente à documentação, durante o decorrer deste trabalho estavam a ser
elaborados protocolos‐padrão para cada etapa da técnica de PCR.
ix. Validação
Ainda não existem procedimentos de validação para as técnicas de PCR utilizadas na
análise de polimorfismos no laboratório de Oncologia Molecular do IPO‐Porto. Existe um bom
sistema de garantia de qualidade para os testes de virologia, que poderá, no futuro, servir como
exemplo, mas que não é o âmbito deste trabalho. Com a proposta deste modelo de controlo de
qualidade e a sua futura implementação, espera‐se criar algumas bases que possam levar a
procedimentos de validação.
Na validação devem ser incluídos controlos de referência e a participação em
esquemas de controlo de qualidade externo, assegurando‐se regularmente o desempenho técnico
dos testes. Não esquecer, no entanto, que a validação pode ser particularmente difícil para testes
genéticos de doenças raras, onde pode ser mais complicado obter controlos positivos de mutações.
Existem também poucos guias nos requisitos mínimos de validação.
x. Treino da equipa
Cada colaborador é ensinado, treinado e supervisionado inicialmente, de modo a
adquirir todas as competências necessárias. Além disso, é continuamente motivado a adquirir mais
conhecimentos e melhorar os já existentes, em seminários e conferências, mas não se mantém no
laboratório um registo dessa actualização constante.

Resultados e Discussão
79
3. Plano de Avaliação de Qualidade
O plano de avaliação de qualidade resume a avaliação dos PCC da técnica de PCR, adaptada
ao laboratório de Oncologia Molecular do IPO‐Porto. Este plano introduz a elaboração de folhas de
registo de cada etapa e a sua avaliação, novos testes de controlo (ex: teste de DNAses, controlo de
qualidade do DNA através da leitura da absorvância) e a proposta de uma frequência de controlo
adequada a cada etapa e ao laboratório, de modo a possibilitar o estabelecimento de uma rotina de
controlo apropriada e de fácil execução.
O plano de avaliação de qualidade proposto ainda não foi testado ou implementado. O
objectivo é o de introduzir este modelo na rotina diária do laboratório e inserir modificações práticas
que sejam necessárias, para que possa ser aperfeiçoado de forma contínua, evoluir e ser
implementado futuramente.

VI. Conclusões e Perspectivas

Conclusões e Perspectivas
81
Nas últimas décadas, os grandes avanços na área da qualidade, nomeadamente no seu
controlo, garantia e gestão, fizeram‐se sentir nas mais diversas áreas. Iniciando‐se esta preocupação
principalmente com a revolução industrial, em que as técnicas de produção passaram a ser baseadas
no conceito de produção em série, rapidamente se notou a necessidade de uma inspecção final ao
produto. Gradualmente este género de controlo foi evoluindo, introduzindo‐se as cartas de controlo,
a inspecção por amostragem e a necessidade de controlar não só o produto final mas todas as
diferentes fases do processo. Surgiu posteriormente o conceito de TQM, já referido neste trabalho,
como uma metodologia de gestão, fonte inspiradora de modelos de qualidade, em que para além de
se ter em consideração os princípios da garantia da qualidade, incorpora‐se também a necessidade
de cumprir os requisitos implícitos e explícitos dos clientes, accionistas, fornecedores e da sociedade
em que se integra a organização.
O principal e primeiro sector onde todos estes conceitos evoluíram foi o sector industrial de
produção. No entanto, hoje em dia a qualidade alcançou todos os sectores, empresas, organizações
e, inclusive, laboratórios de análise e diagnóstico, onde a necessidade de padronização, controlo e
garantia de qualidade se sentiu, tornando‐se pontos muito importantes da rotina diária. O
laboratório clínico actual está rapidamente a transformar‐se num sistema altamente eficiente e
automatizado, ao qual os testes genéticos não podem escapar, necessitando de controlo e validação
como qualquer outro ensaio. A técnica de PCR é utilizada em grande parte dos testes genéticos de
biologia molecular, constituindo uma técnica muito sensível, onde o controlo de qualidade pode, por
vezes, tornar‐se complicado.
Neste trabalho pretendeu‐se propor um modelo de avaliação da qualidade interna para a
técnica de PCR‐RFLP e Real‐Time PCR, ambos qualitativos, no laboratório de Oncologia Molecular do
IPO‐Porto. Ambas são técnicas de rotina, praticadas diariamente neste laboratório para investigação
de polimorfismos e também nos testes de virologia, em que esta última parte não foi objecto de
estudo pois possui já uma forte componente de controlo de qualidade. Utilizou‐se como base para
este modelo o sistema de gestão de qualidade HACCP, cuja eficiência está já comprovada.
Como já foi referido, o projecto consistiu na observação dos vários colaboradores do
laboratório, de modo a poder desenhar‐se os fluxogramas das duas técnicas, sintetizar cuidados,
critérios, verificar o seu cumprimento e identificar os principais PCC. Posto isto, propor um plano de
avaliação adoptado ao laboratório em causa.
As observações feitas, juntamente com a devida revisão bibliográfica, permitiram a síntese
dos vários passos, medidas de prevenção, controlo e correcção, onde se utilizaram os Princípios e
tabelas HACCP como base do trabalho. Tendo em conta isto e as guidelines de Patton e Stenhouse
(2002) [30], elaboraram‐se folhas de critérios, facilmente utilizadas durante a observação de cada
colaborador. No final das observações, e como se pode verificar na parte VI deste trabalho, pode‐se

Conclusões e Perspectivas
82
concluir que, em geral, os cuidados básicos são cumpridos, o que por si só pode garantir a qualidade
dos resultados obtidos. No entanto, existe falta de registos de erros, falta de calibração de
instrumentos e, principalmente, não existe controlo da qualidade durante o processo, especialmente
durante o manuseamento da amostra e extracção dos ácidos nucleicos. Por isso mesmo, ao elaborar
o plano de controlo, introduziu‐se em todas as fases a elaboração de folhas de registos (temperatura,
verificação de procedimentos, erros, calibrações, etc.) e uma fase de controlo a realizar após a
extracção dos ácidos nucleicos.
Apesar da grande necessidade de controlo e garantia de qualidade, não se torna exequível a
realização de todos os passos do plano em todas as amostras. Existem aqueles em que isso é
necessário, por exemplo na utilização de controlo negativo, mas em muitos deles, para além de
trabalhoso, seria um gasto desnecessário. Por isso, futuramente, ao experimentar e implementar
este plano, convém que se faça apenas numa pequena percentagem de amostras (0,5‐1%), por um
grupo de pessoas destacadas para o caso.
Com o trabalho realizado e o modelo proposto levantaram‐se questões importantes neste
laboratório e talvez se possa iniciar uma nova etapa no futuro, com a implementação deste
protocolo na rotina do laboratório. Este trabalho não pretende apresentar um modelo final ou
estático, mas sim um começo, algo que tem de ser experimentado, praticado e continuamente
melhorado, de modo a integrar as tendências mais futuristas da qualidade, que têm vindo a surgir
nos últimos anos. Sem dúvida que entramos em anos de revolução de qualidade, de mudança, que
parecem trazer novas oportunidades para gerir e regular a qualidade de hoje e melhorar a qualidade
de amanhã, e que todos os laboratórios, de diagnóstico ou investigação, devem tentar acompanhar.
A forma mais tradicional de controlo de qualidade ainda se mantém, mas cada vez mais o caminho
certo parece ser o de compreensão de cada passo isolado, a função de cada passo no processo e o
processo total, de modo a identificar, corrigir e prevenir. Compreender também o que o sistema de
controlo de qualidade pode ou não fazer. No fundo, adoptar um conceito de design, de desenhar ou
adoptar um esquema de controlo e garantia de qualidade mais completo, que monitorize mais
pormenorizadamente os elos mais fracos do processo [19].
No entanto, não se pode esquecer uma questão fundamental: será que todo este movimento
de implementar qualidade irá realmente melhorar a qualidade dos ensaios genéticos e assim
proteger os pacientes? Não irá ele duplicar os mecanismos reguladores e acreditações, aumentar o
trabalho administrativo e os custos de laboratório e assim limitar o acesso público aos testes
genéticos [23]? As respostas a estas perguntas ainda não são conhecidas dos laboratórios. Mas
pensa‐se que uma das vantagens de adoptar um modelo de qualidade desenhado e adaptado ao
local será o de contornar essa possível desvantagem, permitindo o desenvolvimento de novas
abordagens para a gestão da qualidade total de um laboratório.

VII. Bibliografia

Bibliografia
84
1. Ferramentas da qualidade. Texto de apoio. Curso de Especialização em Auditorias Internas da
Qualidade. Empresa "Significado". 2003.
2. Gestão integrada da qualidade ambiente, higiene e segurança no trabalho. Texto de apoio.
Curso de Especialização em Auditorias Internas da Qualidade. Empresa "Significado". 2003.
3. NP EN ISO 9000:2000 2ª Edição. Sistemas de gestão da qualidade, fundamentos e vocabulário
(ISO 9000:2000).
4. Health Protection Agency. Quality assurance in the diagnostic virology and serology laboratory.
National Standard Method QSOP 27 (5.1). Disponível em http://www.hpa‐
standardmethods.org.uk/pdf_sops.asp [acedido em 22/04/2009].
5. Marinho, A. Qualidade no laboratório de virologia e serologia. Texto de apoio. Journal Club de
Virologia, IPO‐Porto; Dezembro 2008; Porto, Portugal.
6. Gomes MAP. Avaliação de laboratórios clínicos. Texto de apoio. Seminário do IPAC: Acreditação
dos Laboratórios Clínicos; 2 Julho 2008; Caparica, Portugal.
7. ISO 9000:2000, qualidade, certificação e auditorias. Texto de apoio. Curso de Especialização em
Auditorias Internas da Qualidade. Empresa "Significado". 2003.
8. Williams LO, Cole EC, Lubin IM, Iglesias NI, Jordan RL, Elliott LE. Quality assurance in human
molecular genetics testing. Arch Pathol Lab Med. 2003 Oct; 127 (10): 1353‐8.
9. Kan YW, Dozy AM. Polymorphism of DNA sequence adjacent to human β‐globin structural gene:
relationship to sickle mutation. Proc Natl Acad Sci USA. 1978 Nov; 75 (11): 5631‐5.
10. Dequeker E, Ramsden S, Grody WW, Stenzel TT, Barton DE. Quality control in molecular genetic
testing. Nat Rev Genet. 2001 Sep; 2 (9): 717‐23.
11. Miyachi H. The present status and future prospect of the molecular diagnostic tests. Rinsho
Byori. 2001 Feb; 49 (2): 139‐49.
12. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996 Oct; 87 (2): 159‐
70.

Bibliografia
85
13. Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al. The risk of cancer
associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med.
1997 May; 336 (20): 1401‐8.
14. Krynetski EY, Evans WE. Pharmacogenetics of cancer therapy: getting personal. Am J Hum
Genet. 1998 Jul; 63 (1): 11‐6.
15. Cembrowski GS. Thoughts on quality‐control systems: a laboratorian’s perspective. Clin Chem.
1997 May; 43 (5): 886‐92.
16. Kazmierczak SC. Laboratory quality control: using patient data to assess analytical performance.
Clin Chem Lab Med. 2003 May; 41 (5): 617‐27.
17. Levey S, Jennings ER. The use of control charts in the clinical laboratory. Am J Clin Pathol. 1950
Nov; 20 (11): 1059‐66.
18. Valentine‐Thon E. Quality control in nucleic acid testing – where do we stand?. J Clin Virol. 2002
Dec; 25 Suppl 3: S13‐21.
19. Garrett PE. Quality control for nucleic acid tests: common ground and special issues. J Clin Virol.
2001 Jan; 20 (1‐2): 15‐21.
20. Taha M‐K, Fox A. Quality assessed nonculture techniques for detection and typing of
meningococci. FEMS Microbiol Rev. 2007 Jan; 31 (1): 37‐42.
21. Clinical Laboratory Improvement Amendments of 1988. Fed Regist. 1992; 57: 7002‐7003.
22. American College of Medical Genetics. Standards and Guidelines: Clinical Genetic Laboratories,
Final Draft. 2nd ed. Bethesda, Md: American College of Medical Genetics Laboratory Practice
Committee. 1999.
23. Kaul KL, Leonard DGB, Gonzalez A, Garrett CT. Oversight of genetic testing: an update. J Mol
Diagn. 2001 Aug; 3 (3): 85‐91.
24. Richards CS, Grody WW. Alternative approaches to proficiency testing in molecular genetics. Clin
Chem. 2003 May; 49 (5): 717‐8.

Bibliografia
86
25. College of American Pathologists. Checklist for molecular pathology laboratories. Disponível em
http://www.cap.org/ [acedido em 5/09/2008].
26. Bellissimo DB. Practice guidelines and proficiency testing for molecular assays. Transfusion. 2007
Jul; 47 (Suppl 1): 79S‐84S.
27. American Society of Clinical Oncology. American Society of Clinical Oncology policy statement
update: genetic testing for cancer susceptibility. J Clin Oncol. 2003 Jun; 21 (12): 2397‐406.
28. Bellamy JEC. Quality assurance considerations for detection of waterborne zoonotic parasites
using Cryptosporidium oocyst detection as the main example. Vet Parasitol. 2004 Dec; 126 (1‐2):
235‐48.
29. Bladbjerg E‐M, Gram J, Jespersen J, Maat MPM. Internal quality control of PCR‐based
genotyping methods: practical experiences. Vascul Pharmacol. 2002 Aug; 39 (3): 127‐9.
30. Patton S, Stenhouse S, EMQN. Draft best practice guidelines for laboratory internal quality
control. Disponível em http://www.emqn.org/emqn/BestPractice.html [acedido a 8/10/2008].
31. Malorny B, Tassios PT, Radström P, Cook N, Wagner M, Hoorfar J. Standardization of diagnostic
PCR for the detection of foodborne pathogens. Int J Food Microbiol. 2003 May; 83 (1): 39‐48.
32. Robertson JS. International Standardization of Gene Amplification Technology. Biologicals. 1998
Jun; 26 (2): 111‐3.
33. Raggi CC, Pinzani P, Paradiso A, Pazzagli M, Orlando C. External quality assurance program for
PCR amplification of genomic DNA: an Italian experience. Clin Chem. 2003 May; 49 (5): 782‐91.
34. Burkardt H‐J. Standardization and quality control of PCR analyses. Clin Chem Lab Med. 2000 Feb;
38 (2): 87‐91.
35. Grundmann HJ, Towner KJ, Dijkshoorn L, Gerner‐Smidt P, Maher M, Seifert H, et al. Multicenter
study using standardized protocols and reagents for evaluation of reproducibility of PCR‐based
fingerprinting of Acinetobacter spp. J Clin Microbiol. 1997 Dec; 35 (12): 3071‐7.
36. Persing DH. Polymerase Chain Reaction: trenches to benches. J Clin Microbiol. 1991 Jul; 29 (7):
1281‐5.

Bibliografia
87
37. Niesters HGM. Clinical virology in real time. J ClinVirol. 2002 Dec; 25 Suppl 3: S3‐12.
38. Kowk S, Higuchi R. Avoiding false positives with PCR. Nature. 1989 May; 339 (6221): 237‐8.
39. Hübner P, Studer E, Häfliger D, Stadler M, Wolf C, Looser M. Detection of genetically modified
organisms in food: critical points for quality assurance. Accred Qual Assur. 1999; 4 (7): 292‐8.
40. Nucleic acid amplification techniques. In: European Pharmacopeia 4th Ed. Strasbourg: Council of
Europe; 2003. p. 153‐5.
41. Neumaier M, Braun A, Wagener C. Fundamentals of quality assessment of molecular
amplification methods in clinical diagnostics. Clin Chem. 1998 Jan; 44 (1): 12‐26.
42. Conraths FJ, Schares G. Validation of molecular‐diagnostic techniques in the parasitological
laboratory. Vet Parasitol. 2006 Mar; 136 (2): 91‐8.
43. Stroobants AK, Goldschmidt HMJ, Plebani M. Errorbudget calculations in laboratory medicine:
linking the concepts of biological variation and allowable medical errors. Clin Chim Acta. 2003
Jul; 333 (2): 169‐76.
44. Plebani M. Errors in clinical laboratories or errors in laboratory medicine?. Clin Chem Lab Med.
2006; 44 (6): 750‐9.
45. Plebani M. Appropriateness in programs for continuous quality improvement in clinical
laboratories. Clin Chim Acta. 2003 Jul; 333 (2): 131‐9.
46. Burnett D. ISO 15189:2003 – quality management, evaluation and continual improvement. Clin
Chem Lab Med. 2006; 44 (6): 733‐9.
47. Lippi G, Guidi GC, Mattiuzzi C, Plebani M. Preanalytical variability: the dark side of the moon in
laboratory testing. Clin Chem Lab Med. 2006; 44 (4): 358‐65.
48. Plebani M. Errors in laboratory medicine and patient safety: the road ahead. Clin Chem Lab
Med. 2007; 45 (6): 700‐7.
49. Hamasaki N, Iida H, Kinoshita S. Molecular biology techniques as clinical laboratory tests. Rinsho
Byori. 2001 Jan; 49 (1): 9‐18.

Bibliografia
88
50. Orlando C, Verderio P, Maatman R, Danneberg J, Ramsden S, Neumaier M, et al. Equal‐qual: a
european program for external quality assessment of genomic DNA extraction and PCR
amplification. Clin Chem. 2007 Jul; 53 (7): 1349‐57.
51. Ricós C, García‐Victoria M, Fuente B. Quality indicators and specifications for the extra‐analytical
phases in clinical laboratory management. Clin Chem Lab Med. 2004; 42 (6): 578‐82.
52. The EQUAL‐qual programme. Disponível em http://www.ec‐4.org/equal/home.htm [acedido em
6/10/2008].
53. Newslow D. Hazard Analysis Critical Control Poin (HACCP). In: Schmidt RH, Rodrick GE, editors.
Food safety handbook. New Jersey: John Wiley & Sons; 2003. p. 363‐79.
54. ASAE. O que é HACCP [homepage na internet]. Disponível em http://www.asae.pt [acedido em
7/10/2008].
55. Codex Alimentarius Comission. CAC/RCP 1‐1969 Rev. 4 – Recommended International Code of
Practice, General Principles of Food Hygiene. 2003: 1‐31.
56. Mota MVT. O essencial do HACCP. Texto de apoio. Mestrado em Controlo de Qualidade,
Faculdade de Farmácia do Porto; Fevereiro 2007; Porto, Portugal.
57. Camacho R, Araújo F, Sargento C, Queirós L, Pereira J. Utilização de técnicas de ácidos nucleicos
no rastreio das colheitas de sangues. ABO. 2000; 2: 9‐14.
58. Roux KH. Optimization and troubleshooting in PCR. PCR Methods Appl. 1995 Apr; 4 (5): S185‐94.


















