
ERROS INATOS DO METABOLISMO -EIM- conceitos- MED UNIR
74
Núcleo de Saúde Departamento de Medicina Disciplina de Genética Médica • Discentes: - Carina A. Cabral da Costa - Gabriela de O. Toledo - Rafael S. Bortolato • Docente: - Profª. Drª. Vera Engracia Gama de Oliveira ERROS INATOS DO METABOLISMO - CONCEITOS
-
Upload
gabriela-toledo -
Category
Documents
-
view
8.905 -
download
9
Transcript of ERROS INATOS DO METABOLISMO -EIM- conceitos- MED UNIR

Núcleo de Saúde
Departamento de Medicina
Disciplina de Genética Médica
• Discentes:
- Carina A. Cabral da Costa
- Gabriela de O. Toledo
- Rafael S. Bortolato
• Docente:
- Profª. Drª. Vera EngraciaGama de Oliveira
ERROS INATOS DO
METABOLISMO - CONCEITOS

INTRODUÇÃO• Segundo, Archibald Garrod- o primeiro a empregar o termo EIM-
“ os EIM são manifestações de individualidade bioquímica”.
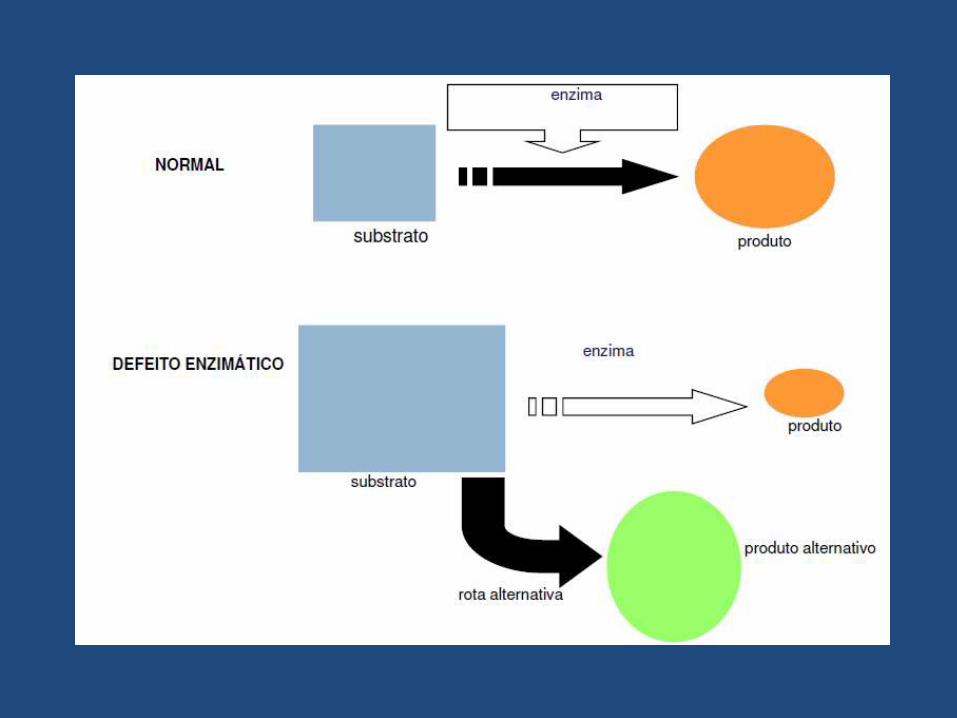
• Distúrbios de natureza genética devido a falta da atividade de umaou mais enzimas especificas ou defeitos nos transportes dasproteínas acarretando a interrupção de uma via metabólica.
• Podem ocasionar o acúmulo de substâncias, a deficiência deprodutos intermediários críticos, a deficiência de produtos finaisespecíficos ou ainda o excesso nocivo de produtos de viasmetabólicas acessórias.
• Doenças Metabólicas Hereditárias ausência de um produtoesperado, acúmulo de substrato da etapa anterior interrompida ou osurgimento de uma rota metabólica alternativa podem levar aocomprometimento dos processos celulares.

HISTÓRICO
• Archibald Garrod é considerado primeiro geneticista
molecular.
Na primeira década do século XX livro sobre aalcaptonúria,albinismo,porfiria e pentosúria .
Garrod deu início à genética bioquímica ao tratar a “individualidadequímica”.
• Em meados do século XX: Beadle,Tatum e outros estudiosos:
“Todos os processos bioquímicos de organismo ocorrem sobcontrole gênico e propuseram a hipótese de “um gene- uma enzima”,portanto, mutações gênicas levam a rotas bioquímicas diferentes.
• Após o desenvolvimento de novas técnicas, como a cromatografia eeletroforese de proteínas e a tecnologia do DNA, atualmente,admite-se a existência de mais de 500 doenças metabólicashereditárias.

INCIDÊNCIA
• Representam 10% de todas as doenças genéticas.
• Baixa incidência,isoladamente Herança autossômica recessiva.
• Incidência acumulativa é de 1:5.000 nascidos vivos
• Aproximadamente 500 distúrbios conhecidos.
Padrões de herança
Autossômico recessivo
Autossômico dominante
Ligados ao cromossomo X
Herança mitocondrial
• Maioria, de herança autossômica recessiva, com risco de recorrência de25% a cada gestação de pais heterozigotos.
• Doenças em que a herança é ligada ao X, o risco de recorrência é de 50%para o sexo masculino e de 50% do sexo feminino serem portadoras epassarem aos seus filhos.
• Nas doenças de herança mitocondrial, o risco de recorrência é depraticamente 100% de comprimento dos filhos de ambos os sexos, quandoa mãe é portadora das mutações.

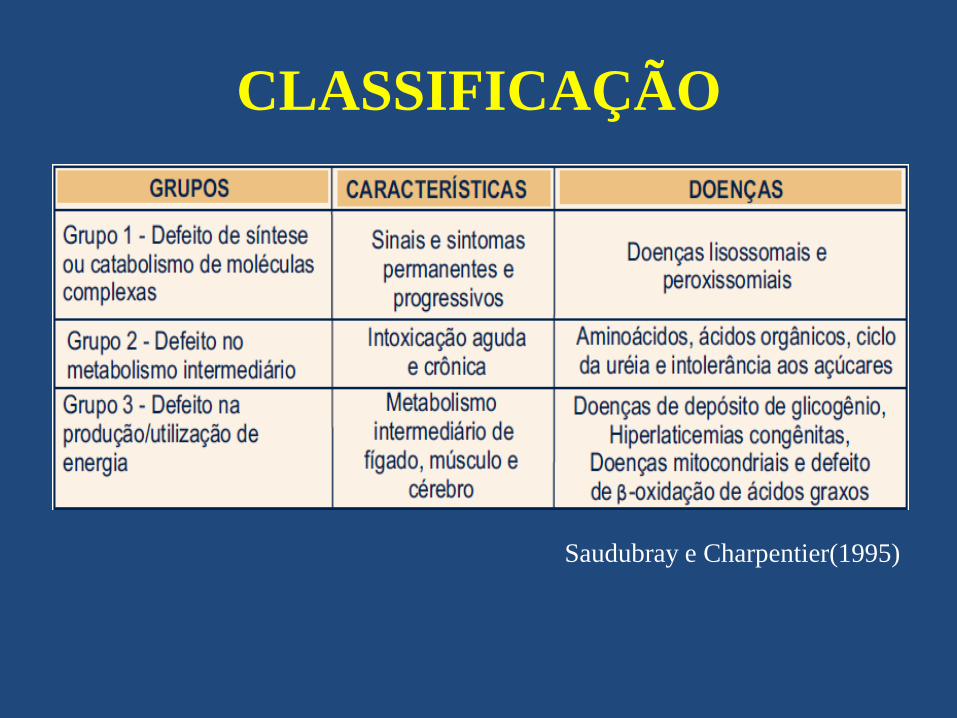
CLASSIFICAÇÃO
• Saudubray e Charpentier, estabeleceram a
classificação didática de aplicação clínica
utilizanado o fenótipo clinico das doenças.
Defeitos na síntese ou catabolismos de
moléculas complexas
Defeitos no metabolismo intermediário
Defeitos na produção e
utilização de energia
CLASSIFICAÇÃO
Saudubray e Charpentier(1995)

GRUPO I
DEFEITOS NA SÍNTESE DE
MOLÉCULAS COMPLEXAS OU
MACROMOLÉCULAS

Doenças com sintomas permanentes e
progressivos, independentes de eventos
intercorrentes, como infecções.
I.1) Doenças Relacionadas as Organelas
I.2)Defeito de Metabolismo das Vitaminas
I.3)Doenças do Metabolismos de Lipídios e Acido Biliar
I.4) Doenças do Metabolismos do Ácido Nucléico e do Heme
I.5) Doenças do Transporte dos Metais
I.6) Doenças dos Neurotransmissores e Pequenos Peptídeos

DOENÇAS PEROXISSOMAIS

Os peroxissomos
• Organela presente em quase todas as células
eucarióticas
• Apresenta mais de 40 enzimas oxidativas e
catalisa várias reações essenciais em diferentes
rotas metabólicas
• Papel extremamente importante no
metabolismo
Figura 1

Processos metabólicos
• Beta-oxidação de ácidos graxos de cadeia muito longa
• Biossíntese de fosfolipídios, ácidos biliares, colesterol e intermediários de colesterol
• Papel importante na produção e eliminação de espécies reativas de oxigênio
– Catalase
– Glutationa peroxidase
– Superóxido dismutase

Doenças peroxissomais
As doenças peroxissomais estão distribuídas em
dois grandes grupos:
• Defeito de uma única enzima
– Estrutura peroxissomal intacta
– Defeito em uma única proteína
• Doenças da biogênese do peroxissomo
– Organela formada anormalmente
– Comprometimento de todas as vias metabólicas

Doenças
Defeito em uma
enzima
• X-ALD
• Hiperoxaliúria tipo I
• Doença de Refsum
Doenças da biogênese do
peroxissomo
• Síndrome de Zellweger
• Adrenoleucodistrofia
neonatal
• Condrodisplasia
Rizomélica tipo I

DOENÇA DO
ARMAZENAMENTO
LISOSSOMAL

Os lisossomos
• Organelas de formato redondo ou ovalado
• Catabolizam macromoléculas celulares e
extracelulares, gerando aminoácidos, ácidos
nucleicos, ácidos graxos e açúcares para
reutilização na síntese celular
• Principal centro de eliminação e reciclagem
das células
Figura 2

Doenças lisossomais
Desenvolvem-se em decorrência de mutações
que resultam em redução da síntese de enzimas
lisossomais

Figura 3

Figura 3 (continuação)

DOENÇAS DO METABOLISMO
DE LIPÍDIOS E ÁCIDO BILIAR

DEFEITOS DE PROTEÍNAS
RECEPTORAS
1974 - Goldstein e Brown;
Defeito do receptor de lipoproteínas de baixadensidade (LDL);
Hiperlipoproteinemia genética:
Níveis elevados de lipídios plasmáticos (colesterol, triglicerídios, ou ambos) e de lipoproteínas plasmáticas específicas;
IAM;
Hipercolesterolemia familiar.

Dislipidemias
Doenças do metabolismo endógeno das
lipoproteínas
• Aterosclerose

Defeito da síntese de ácido biliarXantoma

Classes de mutações no receptor de
lipoproteína de baixa densidade

Classes de mutações no receptor de
lipoproteína de baixa densidade
Mutações classe I
Alelos nulos que evitam a síntese de qualquerreceptor detectável;
Alguns alelos classe I são decorrentes dedeleções, enquanto outros produzemquantidades normais de mRNA para receptorde LDL e supostamente têm defeitos queimpedem a formação ou a estabilidade dopolipeptídio.

Classes de mutações no receptor de
lipoproteína de baixa densidade
Mutações classe II
Deficientes de transporte;
Os receptores de LDL se acumulam no sítio de
sua síntese, o RE, em vez de serem
transportados para o CG;
Supõe-se que impeçam o dobramento
apropriado da proteína, aparentemente um
requisito para a saída do RE.

Classes de mutações no receptor de
lipoproteína de baixa densidade
Mutações classe III
Superfície celular
Incapazes de ligar LDL
Um crossing desigual deletou parte do domínio deligação de LDL;
A recombinação homóloga desigual entre duascópias de uma sequência repetitiva de DNA foivista como uma causa frequente de deleções tantoneste gene quanto nos outros.

Classes de mutações no receptor de
lipoproteína de baixa densidade
Mutações classe IV
Prejudicam a localização do receptor nadepressão revestida e, consequentemente, oLDL ligado não é internalizado;
Alteram ou retiram o domínio citoplasmáticono terminal carboxila do receptor;
Uma substituição de tirosina por cisteína noéxon 17, altere a conformação do domíniocitoplasmático do receptor.

Classes de mutações no receptor de
lipoproteína de baixa densidade
Mutações classe V
Alelos com defeito de reciclagem;
Requer a dissociação do receptor e do LDL ligadono endossomo e é mediada pelo domínio dehomologia do precursor do fator de crescimentoepidérmico;
As mutações neste domínio, ambas as deleções desegmentos dele, bem como algumas substituiçõesde sentido trocado, impedem a liberação doligante;
Degradação do receptor.

DEFEITOS DE TRANSPORTE
Fibrose cística 1960;
Distúrbio genético autossômico recessivo fatal maiscomum de crianças nas populações caucasianas;
Gene da FC: Proteína integrante da membrana;
Gene CFTR: canal de Cl¯ situado na membrana apicaldas células epiteliais afetadas pela doença;
Proteína Cftr (regulador de condutânciatransmembranar de CF);
A perda de função de Cftr significa que o Cl¯ no dutoda glândula não pode fluir pela luz, e através dascélulas do duto para a corrente sanguínea.

A Genética da Fibrose Cística
Mutações no polipeptídio Cftr
Deleção de fenilalanina na posição 508
(∆F508) na primeira dobra de ligação de ATO
(NBD1);
70% de todos os alelos CF nas populações
caucasianas;
Mutações de substituição de sentido trocado.

A Genética da Fibrose Cística
Mutações de classe I: defeito na produção daproteína -associadas a códons finalizadoresprematuros ou mutações que geram RNAsinstáveis. Ex.: proteína Cftr;
Mutações de classe II: processamento proteicodefeituoso decorrente de mau dobramento daproteína. Ex.: ∆F508;
Mutações de classe III: perturbam a regulação daproteína. Situam-se em NBDs e no domínio R;
Mutações classe IV: condução defeituosa decloreto. Situam-se em MSDs.

Deficiência de MCAD
Erro inato do metabolismo de ácido graxo
mais comum, o qual resulta de uma deficiência
da cadeia média acil-coenzima A
desidrogenase (MCAD);
90% dos alelos possuem uma mutação de
sentido trocado de A para, que resulta na
substituição da lisina por glutamato.

Deficiência de LCHAD
É um dos distúrbios mais severos de FAO;
Os ácidos graxos de cadeia longa acil-CoA sãometabolizados em uma sequência de etapascatalisadas por várias enzimas;
O segundo passo do metabolismo dos ácidosgraxos é catalisado por enzimas que são parte deum complexo enzimático, a proteína trifuncionalmitocondrial (TFP);
Uma das enzimas de TFP é uma L-3-hidroxiacil-CoA desidrogenase de cadeia longa (LCHAD).

DOENÇAS DO METABOLISMO
DO ÁCIDO NUCLEICO E DO
HEME

Doenças do metabolismo das purinas
• Síntese aumentada de purina, resulta no aumento
da síntese e degradação de ácido úrico,
ocasionando a gota, a síndrome de Lesh-Nyan e a
Imunodeficiência.

Porfirias hepáticas
Porfiria intermitente aguda (tipo sueco)
Doença autossômica dominante à disfunçãoneurológica intermitente;
Grande número de medicações, hormônios esteróides einanição;
Deficiência de porfobilinogênio (PBG) desaminase –enzima na via biossintética do heme;
Aumento da síntese de citocromos hepáticos P450;
A deficiência de heme devido à redução de PBGdesaminase e a consequente diminuição nosreservatórios de heme, culminam no aumentosecundário na sintase em níveis maiores que o normal.

Doença da Hemoglobina M
Ametemoglobina se forma quando a hemoglobina é oxidada àforma férrica;
A hemoglobina é de novo reduzida pela enzima DNPHmetemoglobina-redutase (diaforase);
A substituição de um aminoácido modifica a relação com o grupoheme, de modo que o ferro está permanentemente no estadoférrico;
Metemoglobinemia com cianose e hipóxia;
Herança dominante em heterozigotos;
Hb M-Boston: tirosina substitui a histidina na porção 58 dacadeia alfa (componente de metemoglobinas fetal e adulta);
Hb M-Iwate: anormalidade da cadeia alfa;
Hb M-Saskatoon: tirosina substitui a histidina homóloga naposição 63;
Hb M-Milwaukee: ácido glutâmico substitui a valina na posição67 da cadeia beta.

DOENÇA DE TRANSPORTE
DE METAIS

Doença de Menkes
Deficiência de cobre;
Distúrbio recessivo ligado ao X descrito em1962 por John Menkes;
A proteína ATP7A é o gene causador;
O cobre pode ser absorvido pelo epitéliogastrintestinal, mas não pode ser exportadopara a corrente sanguínea;
Retardo mental, convulsões e morte naprimeira infância.

Doença de Wilson
Distúrbio autossômico recessivo, descrito por
Kinnear Wilson em 1912, chamado degeneração
hepatolenticular;
Mutações no gene altamente homólogos de MND,
ATP7B.
Resultante do excesso de cobre causado pela
excreção defeituosa do mesmo para o trato biliar;
Causa doença hepática progressiva e anomalias
neurológicas.

Acrodermatite enteropática
Defeito na absorção de zinco do trato
intestinal;
Mutações em SLC39A4, que codifica uma
proteína transportadora de zinco expressa na
membrana apical das células epiteliais do
intestino delgado.

DOENÇAS DOS
NEUROTRANSMISSORES E
PEQUENOS PEPTÍDIOS

Deficiência de ácido gama amino
butírico (GABA)
Principal neurotransmissor inibitório do
encéfalo;
Quando se liga ao receptor, permite a entrada
de Cl¯ na célula;
É responsável pela sintonia fina e coordenação
dos movimentos;
Sua deficiência leva a algumas formas de
Esquizofrenia;.

Deficiência de peptídios
Endorfinas ou encefalinas: opiáceos endógenos
capazes de modular a dor e o estresse;
Sistema límbico, mesencéfalo e produzidos por
glândulas pituitárias e liberados como
hormônios e envolvidos na redução da dor
(substância P), pressão (aumentam a produção
de dopamina) e hibernação;

GRUPO II
DEFEITOS DECORRENTES DE
ERROS INATOS DO
METABOLISMO
INTERMEDIÁRIO

DEFEITOS DECORRENTES DE
ERROS INATOS DO METABOLISMO
INTERMEDIÁRIO
Esse grupo apresenta doenças com intoxicação
aguda e recorrente ou crônica e progressiva. O
grupo é dividido em dois subtipos:
II.1)Aminoacidopatias
II.2)Doenças do Metabolismo do Carboidratos

AMINOACIDOPATIAS

AMINOACIDOPATIAS
• Hiperfenilalaninemias aumento no nívelsanguíneo de fenilalanina.
Causas
Mutações de perda de função no gene quecodifica a fenilalanina hidroxilase (PAH).
Mutação dos genes necessários para a síntese ou areutilização de seu co-fator, a tetraidrobiopterina(BH4).

FENILCETONURIA (PKU)
PhenylKetonUria
• Distúrbio autossômico recessivo
• Resultante da deficiência da enzima fenilalanina hidroxilase
(FAL-OH) hepática, que converte o aminoácido fenilalanina
em tirosina.
• A reação acontece corretamente na presença do co-fator
tetrahidrobiopterina (BH4)
Erros Inatos do Metabolismo - Martins,A.M.

SÍNTESE DE NEUROTRANSMISSORES NA PRESENÇA DAS ENZIMAS TIROSINA (TIR-OH) E
TRIPTOFANO HIDROXILASE (TRIP-OH) E DO CO-FATOR TETRAHIDROBIOPTERINA (BH4).
• Distúrbio ocorre no cromossoma par 12
Genótipo FF fenótipo saudável;
Genótipo Ff fenótipo saudável (portador do alelo
que provoca doença) ;
Genótipo ff fenótipo doente.

PKU-ClássicaSinais Clínicos
• Quadro clinico mais grave ,não tem boa resposta aotratamento com dieta pobre de fenilalanina, devidodeficiência na síntese dos neurotransmissores.
• A fenilalanina acumulada no sangue, é desviada parafenilpiruvato, fenilacetato e fenilactato, produtos tóxicospara o organismo.
• Prejudica o desenvolvimento do sistema nervoso centralno início da lactância.
• Interfere no funcionamento do cérebro maduro.

VARIANTES
• Heterogenicidade: três fenótipos clínicos com
mutações no gene PAH.
• Há alto grau de heterogeneidade alélica no lócus.
Dois alelos diferentes causadores da doença
(maioria dos casos).
Hiperfenilalaninemia não-PKU e PKU variante.
Enzima PAH mutante tem alguma atividade
residual.

A Hiperfenilalaninemia não-PKU
Concentração plasmática de fenilalanina
menor que 1mM, em dieta normal.
Identificados apenas por triagem neonatal;
PKU Variante
Categoria que inclui pacientes com tolerância
intermediaria entre a PKU clássica e a
Hiperfenilalaninemia não-PKU.

DEFEITO NO METABOLISMO DO TETRADROBIOPTERINA (BH4)
• Cerca de 1% a 3% apresentam o gene PAH normal.
• Defeito genéticos na formação ou na reciclagem do co-fator PAH,tetraidobiopterina (BH4) . Distúrbio autossômico recessivo.
• Proteínas codificadas pelos genes que manifestam heterogeneidadeatuam em etapas diferentes em uma única via bioquímica.
• Os pacientes com deficiência em BH4, têm defeitos em uma das etapasda biossíntese de BH4 a partir de guanosina trifosfato (GTP) ou naregeneração de BH4.
• Tratamento difere da PKU clássica Crianças comhiperfenilalaninemia devem ser testadas quanto a deficiência de BH4.
Controle do níveis de fenilalanina do sangue,
Normalizar os neurotransmissores no cérebro administrando os produtosda tirosina hidroxilase e do triptofano hidroxilase, L-dopa e 5-hidroxitriptofano.

Fenilcetonúria Materna
• Filhos heterozigotos de mães com PKU:
Retardo mental,
Microcefalia,
Prejuízo de crescimento e malformações
• Causas:
Efeito altamente teratogênico dos níveis elevados de
fenilalanina na circulação materna.
• Mulheres PKU com pretensão de engravidar,
necessitam impreterivelmente de uma dieta pobre em
fenilalanina antes de engravidar.

DOENÇAS DO METABOLISMO
DO CARBOIDRATOS

DOENÇAS DO METABOLISMO
DO CARBOIDRATOS
• Os Carboidratos são as substancias orgânicas maisabundantes da Terra.
• Funcionam como substratos para a produção earmazenamento de energia,como intermediários de viasmetabólicas e arcabouço estrutural do DNA e do RNA.
• São metabolizados em três monossacarídeos principais:glicose,galactose e frutose.

Galactosemia
• Afeta 1 a cada 55.000 neonatos
• GALACTOSEMIA CLÁSSICA: Mutação no gene
que codifica a galactose-1-fosfatouridil transferase
(GAL-1-P uridil transferase)

• O gene possui 11 éxons distribuídos ao longo de 4kb deDNA.
• Única mutação de sentido trocado no éxon 6, nos aleloscausadores da galactosemia (70% dos casos).
• Resultam em atividade diminuída da GAL-1-P uridiltransferase.
Conseqüências:
Indivíduos não podem converter efetivamente agalactose em glicose.
Glicose é metabolizada em Galactitol e Galactonato .

Sinais Clínicos
• Falha no desenvolvimento, insuficiência hepática,
cataratas e atraso no crescimento (habilidades motoras ou
retardo mental).
• Diagnosticada através da medida da atividade da GAL-1-
P uridil transferase no plasma de uma gota de sangue.
• Eliminação da galactose na dieta (precocemente),reduz
substancialmente a morbidade associada com efeitos
agudos do altos níveis de metabolitos da galactose.

Outra causa para a Galactosemia:
• Mutação no gene que codifica Galactocinase ouUridina Difosfato Galactose-4-Epimerase(UDP-Galactose-4-Epimerase).
• Limitada às hemácias e leucócitos Não causaefeitos prejudiciais.
• Sistêmica Sintomas similares a GalactosemiaClássica, formação de catarata.
• Tratamento: restrição dietética da galactose.

GRUPO III
DEFEITOS DECORRENTES NA PRODUÇÃO
OU UTILIZAÇÃO DE ENERGIA POR
ERROS INATOS DO METABOLISMO

DEFEITOS DECORRENTES NA PRODUÇÃO
OU UTILIZAÇÃO DE ENERGIA POR ERROS
INATOS DO METABOLISMO
Esse erros decorrem de erros do metabolismo
intermediário no fígado, miocárdio ou cérebro.
O grupo é dividido em dois subtipos:
III. 1)Doenças de Deposito de Glicogênio
III. 2)Doenças do Metabolismo Energético das Mitocôndrias

DOENÇA DO METABOLISMO
DAS MITOCÔNDRIAS

Distúrbios mitocondriais
Os sintomas surgem de tecidos cujas células
possuem muitas mitocôndrias, como músculos
esqueléticos;
Miopatias mitocondriais: músculos flácidos e
intolerância ao exercício; fibras dos músculos
esqueléticos apresentam-se vermelhas e
anfractuosas, com muitas mitocôndrias
anormais próximas à membrana celular.

Distúrbios mitocondriais
Neuropatia óptica hereditária de Leber
(LHON): prejudica a visão;
Mutação em gene mitocondrial que codifica
tRNA ou rRNA pode ser devastadora, pois
prejudica a capacidade da célula produzir
proteínas;
MELAS: síndrome da acidose lática com
miopatia e encefalopatia.

DISTÚRBIO DO DEPÓSITO DE
GLICOGÊNIO

Doenças do armazenamento de
glicogênio
Distúrbio na síntese ou degradação de glicogênio;
15 defeitos enzimáticos, com quadros que ainda
não possuem classificação;
Doença de von Gierke: primeiro erro inato do
metabolismo no qual a deficiência tecidual
conhecida foi demonstrada;
Desordens do fígado ou músculo, isolado ou em
combinação com o coração, rim e sistema
nervoso;

Doenças do armazenamento de
glicogênioGlicogenoses:
Hepáticas: armazenamento de glicogênio no fígado ehipoglicemia;
Doença de von Gierke ou glicogenose tipo I:deficiência da enzima glicose-6-fosfatase;
Doença de Cori ou glicogenose tipo III: deficiência daenzima desramificadora;
Doença de Hers ou glicogenose tipo VI: deficiência defosforilase hepática;
Glicogenoses tipos VIII ou IX: deficiência defosfoquinase b hepática.

Doenças do armazenamento de
glicogênio Miopáticas: fraqueza músculos estriados;
Doença de McArdle ou glicogenose tipo V: deficiências dafosforilase muscular;
Glicogenose tipo VII: deficiência da fosfofrutoquinasemuscular
Específicas: deficiência de a -glicosidases e falta de enzimaramificadora não se adaptam nas categorias
Doença de Pompe ou glicogenose tipo II: deficiência daenzima alfa-1,4-glicosidase lisossômica (maltase ácida);
Doença de Andersen ou glicogenose tipo IV ouAmilopectinose: deficiência da enzima alfa-1,4-glucan-6-glicosiltransferase (ramificadora).

Referências • DEON, MARION. Avaliação de stress oxidativo em adrenoleucodistrofia
ligada ao cromossomo x e doenças do espectro Zellweger. UniversidadeFederal do Rio Grande do Sul. Porto Alegre, 2009
• PEDROSO, PEDRO MIGUEL. Doença do armazenamento lisossomalinduzida pelo consumo de Sida carpinifolia (MALVACEAE) em herbívorosno Rio Grande do Sul. Universidade Federal do Rio Grande do Sul. PortoAlegre, 2009.
• JORDE,L. B. et al . Genética Médica. Rio de Janeiro : Elsevier,2004.
• MARTINS, A. M. Erros Inatos do Metabolismo, Abordagem Clinica .Disponível em :http://www.supportnet.com.br/artigos/pdf/monografia.pdf.Acessado em: 01/08/2013.
• Centro de Referência de Erros Inatos do Metabolismo- Centro de GenéticaMédica UNIFESP. Disponível em: http://www.unifesp.br/centros/creim/ .Acessado em: 01/08/2013.
• NUSSBAUM, RL. Thompson & Thompson – Genética Médica. RJ.Guanabara Koogan, 2002.
• LEWIS, Ricki. Genética Humana: conceitos e aplicações. Rio de Janeiro:Guanabara Koogan, 2004.
• NORA, J.J; FRASER, F.C. Genética Médica. 3.ed. Rio de Janeiro.Guanabara-Koogan.


















