
Fluidos supercriticos
45
FLUIDOS SUPERCRÍTICOS Introdução e Questionamento Geral Rafael Carvalho Barreto Orientador: Sylvio Canuto USP 2007
-
Upload
valdecir-valdecir -
Category
Documents
-
view
66 -
download
3
description
Fluidos supercriticos
Transcript of Fluidos supercriticos

FLUIDOS SUPERCRÍTICOS
Introdução e Questionamento Geral
Rafael Carvalho Barreto
Orientador: Sylvio Canuto
USP 2007

1
Sumário
Capítulo 1 : Estados da matéria e o ponto crítico ................................................................... 2
Capítulo 2 : Caracterização geral do sistema fluido e fluido supercrítico.............................. 7
2.1 Densidade ............................................................................................................... 9
2.1.1 Equação de van der Waals................................................................................ 11
2.2 Energia e entropia................................................................................................. 20
Capítulo 3 : Características dos fluidos supercríticos........................................................... 26
Capítulo 4 : Algumas aplicações notáveis............................................................................ 29
4.1 Reciclagem química de embalagens PET (Poli Tereftalato de Etila / Polyethylene
Terephthalate)................................................................................................................... 29
4.2 Transformação da celulose em glicose................................................................. 31
4.3 Recuperação dos resíduos de TDI (Toluene diisocyanate) para reuso em sua
própria produção............................................................................................................... 33
Capítulo 5 : Extração e purificação via dióxido de carbono supercrítico [8] ....................... 35
Capítulo 6 : Empresas e grupos envolvidos com pesquisa na área de fluidos supercríticos
(alguns). ................................................................................................................................ 37
Capítulo 7 : Livros e periódicos ........................................................................................... 39
The Journal Of Supercritical Fluids (Elsevier)................................................................. 39
Chemical Reviews: 1999.................................................................................................. 41
2
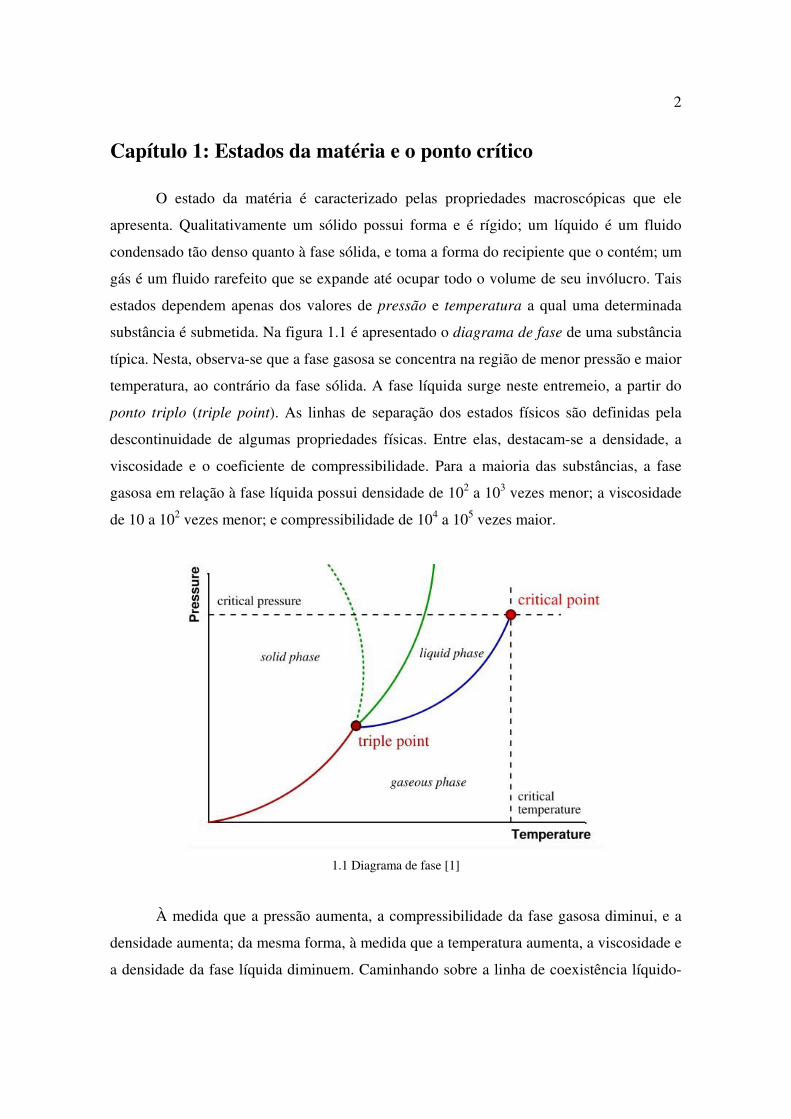
Capítulo 1: Estados da matéria e o ponto crítico
O estado da matéria é caracterizado pelas propriedades macroscópicas que ele
apresenta. Qualitativamente um sólido possui forma e é rígido; um líquido é um fluido
condensado tão denso quanto à fase sólida, e toma a forma do recipiente que o contém; um
gás é um fluido rarefeito que se expande até ocupar todo o volume de seu invólucro. Tais
estados dependem apenas dos valores de pressão e temperatura a qual uma determinada
substância é submetida. Na figura 1.1 é apresentado o diagrama de fase de uma substância
típica. Nesta, observa-se que a fase gasosa se concentra na região de menor pressão e maior
temperatura, ao contrário da fase sólida. A fase líquida surge neste entremeio, a partir do
ponto triplo (triple point). As linhas de separação dos estados físicos são definidas pela
descontinuidade de algumas propriedades físicas. Entre elas, destacam-se a densidade, a
viscosidade e o coeficiente de compressibilidade. Para a maioria das substâncias, a fase
gasosa em relação à fase líquida possui densidade de 102 a 103 vezes menor; a viscosidade
de 10 a 102 vezes menor; e compressibilidade de 104 a 105 vezes maior.
1.1 Diagrama de fase [1]
À medida que a pressão aumenta, a compressibilidade da fase gasosa diminui, e a
densidade aumenta; da mesma forma, à medida que a temperatura aumenta, a viscosidade e
a densidade da fase líquida diminuem. Caminhando sobre a linha de coexistência líquido-
3
gás, as diferenças entre as fases líquida e gasosa vão diminuindo até o chamado ponto
crítico (critical point): a partir deste ponto, conhecida como região supercrítica, a
transição entre a fase líquida e gasosa não é mais visível através da descontinuidade das
propriedades macroscópicas.
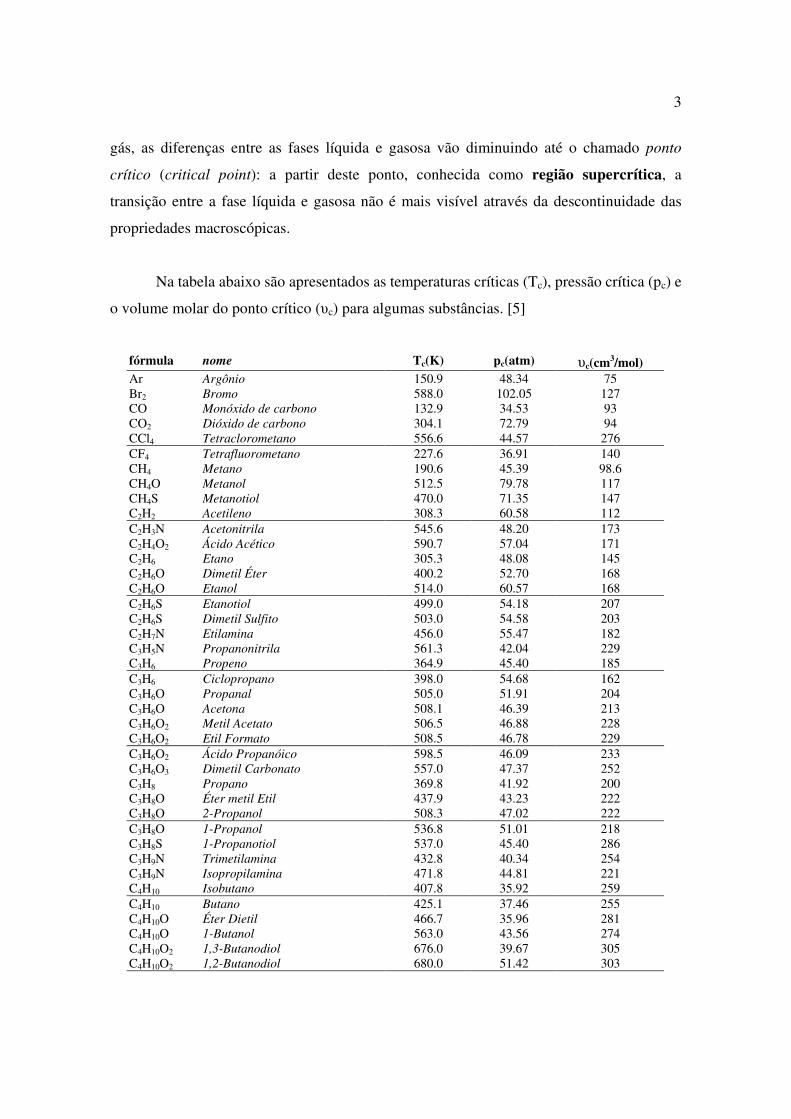
Na tabela abaixo são apresentados as temperaturas críticas (Tc), pressão crítica (pc) e
o volume molar do ponto crítico (υc) para algumas substâncias. [5]
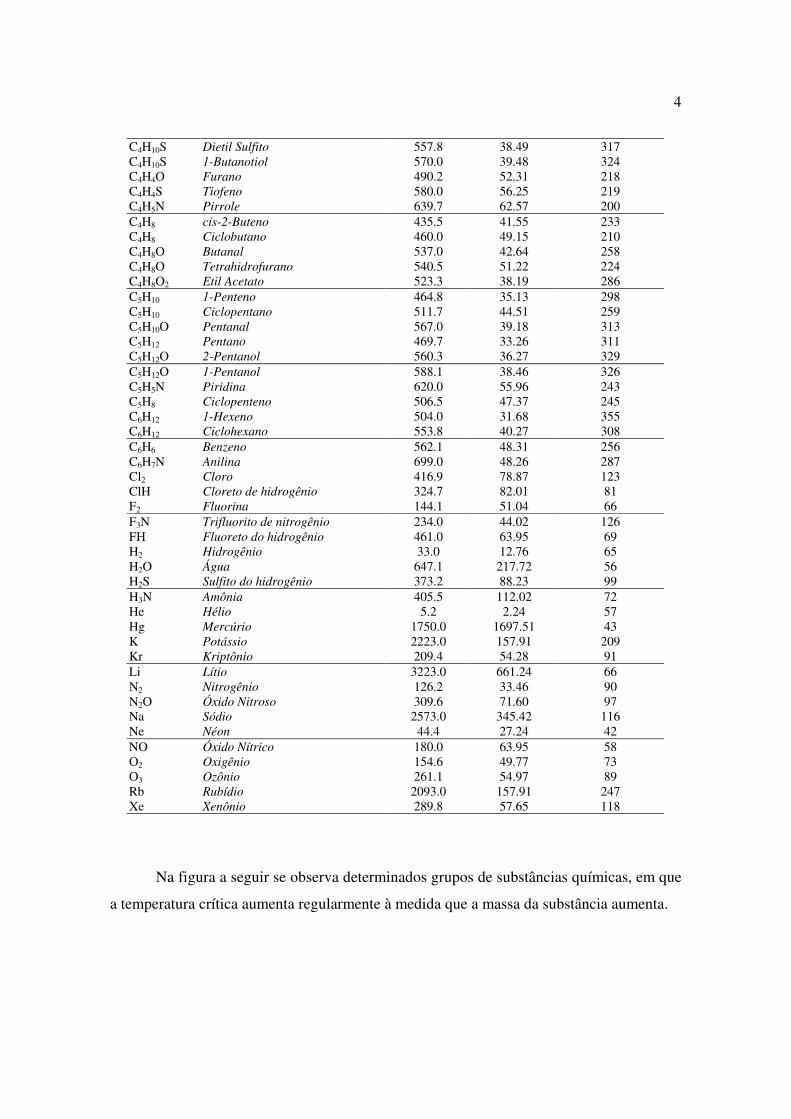
fórmula nome Tc(K) pc(atm) υc(cm3/mol) Ar Argônio 150.9 48.34 75 Br2 Bromo 588.0 102.05 127 CO Monóxido de carbono 132.9 34.53 93 CO2 Dióxido de carbono 304.1 72.79 94 CCl4 Tetraclorometano 556.6 44.57 276 CF4 Tetrafluorometano 227.6 36.91 140 CH4 Metano 190.6 45.39 98.6 CH4O Metanol 512.5 79.78 117 CH4S Metanotiol 470.0 71.35 147 C2H2 Acetileno 308.3 60.58 112 C2H3N Acetonitrila 545.6 48.20 173 C2H4O2 Ácido Acético 590.7 57.04 171 C2H6 Etano 305.3 48.08 145 C2H6O Dimetil Éter 400.2 52.70 168 C2H6O Etanol 514.0 60.57 168 C2H6S Etanotiol 499.0 54.18 207 C2H6S Dimetil Sulfito 503.0 54.58 203 C2H7N Etilamina 456.0 55.47 182 C3H5N Propanonitrila 561.3 42.04 229 C3H6 Propeno 364.9 45.40 185 C3H6 Ciclopropano 398.0 54.68 162 C3H6O Propanal 505.0 51.91 204 C3H6O Acetona 508.1 46.39 213 C3H6O2 Metil Acetato 506.5 46.88 228 C3H6O2 Etil Formato 508.5 46.78 229 C3H6O2 Ácido Propanóico 598.5 46.09 233 C3H6O3 Dimetil Carbonato 557.0 47.37 252 C3H8 Propano 369.8 41.92 200 C3H8O Éter metil Etil 437.9 43.23 222 C3H8O 2-Propanol 508.3 47.02 222 C3H8O 1-Propanol 536.8 51.01 218 C3H8S 1-Propanotiol 537.0 45.40 286 C3H9N Trimetilamina 432.8 40.34 254 C3H9N Isopropilamina 471.8 44.81 221 C4H10 Isobutano 407.8 35.92 259 C4H10 Butano 425.1 37.46 255 C4H10O Éter Dietil 466.7 35.96 281 C4H10O 1-Butanol 563.0 43.56 274 C4H10O2 1,3-Butanodiol 676.0 39.67 305 C4H10O2 1,2-Butanodiol 680.0 51.42 303
4
C4H10S Dietil Sulfito 557.8 38.49 317 C4H10S 1-Butanotiol 570.0 39.48 324 C4H4O Furano 490.2 52.31 218 C4H4S Tiofeno 580.0 56.25 219 C4H5N Pirrole 639.7 62.57 200 C4H8 cis-2-Buteno 435.5 41.55 233 C4H8 Ciclobutano 460.0 49.15 210 C4H8O Butanal 537.0 42.64 258 C4H8O Tetrahidrofurano 540.5 51.22 224 C4H8O2 Etil Acetato 523.3 38.19 286 C5H10 1-Penteno 464.8 35.13 298 C5H10 Ciclopentano 511.7 44.51 259 C5H10O Pentanal 567.0 39.18 313 C5H12 Pentano 469.7 33.26 311 C5H12O 2-Pentanol 560.3 36.27 329 C5H12O 1-Pentanol 588.1 38.46 326 C5H5N Piridina 620.0 55.96 243 C5H8 Ciclopenteno 506.5 47.37 245 C6H12 1-Hexeno 504.0 31.68 355 C6H12 Ciclohexano 553.8 40.27 308 C6H6 Benzeno 562.1 48.31 256 C6H7N Anilina 699.0 48.26 287 Cl2 Cloro 416.9 78.87 123 ClH Cloreto de hidrogênio 324.7 82.01 81 F2 Fluorina 144.1 51.04 66 F3N Trifluorito de nitrogênio 234.0 44.02 126 FH Fluoreto do hidrogênio 461.0 63.95 69 H2 Hidrogênio 33.0 12.76 65 H2O Água 647.1 217.72 56 H2S Sulfito do hidrogênio 373.2 88.23 99 H3N Amônia 405.5 112.02 72 He Hélio 5.2 2.24 57 Hg Mercúrio 1750.0 1697.51 43 K Potássio 2223.0 157.91 209 Kr Kriptônio 209.4 54.28 91 Li Lítio 3223.0 661.24 66 N2 Nitrogênio 126.2 33.46 90 N2O Óxido Nitroso 309.6 71.60 97 Na Sódio 2573.0 345.42 116 Ne Néon 44.4 27.24 42 NO Óxido Nítrico 180.0 63.95 58 O2 Oxigênio 154.6 49.77 73 O3 Ozônio 261.1 54.97 89 Rb Rubídio 2093.0 157.91 247 Xe Xenônio 289.8 57.65 118
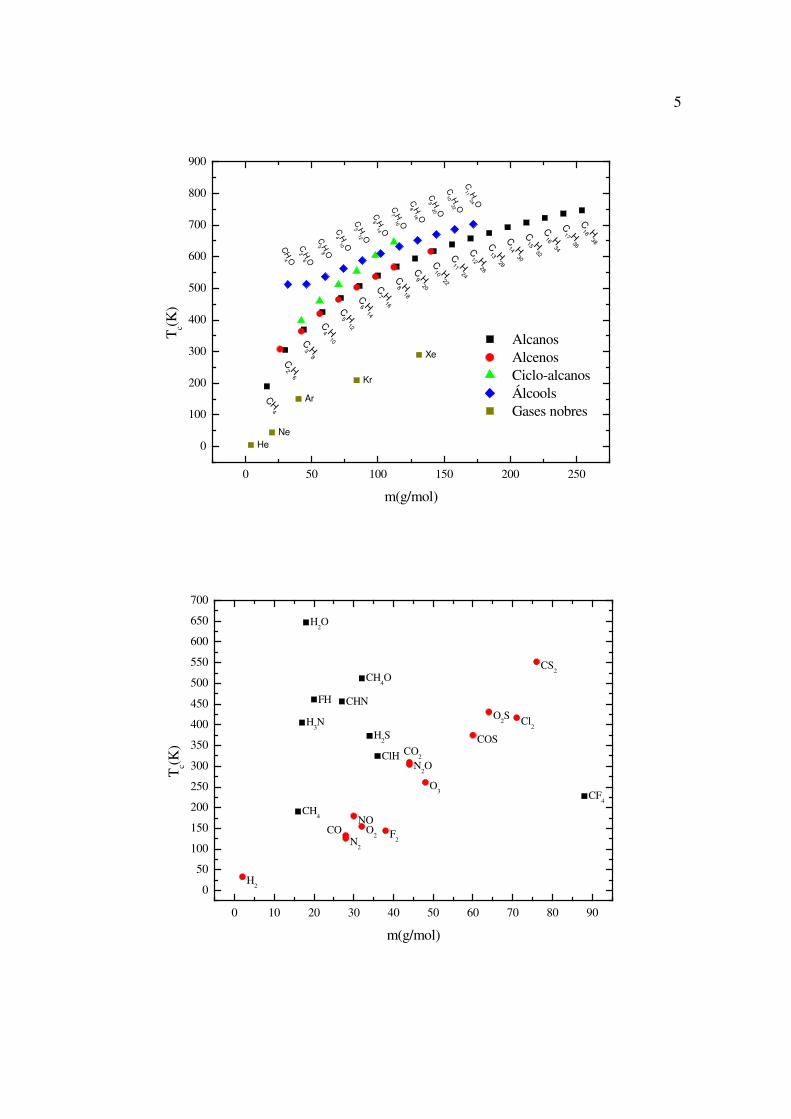
Na figura a seguir se observa determinados grupos de substâncias químicas, em que
a temperatura crítica aumenta regularmente à medida que a massa da substância aumenta.
5
0 50 100 150 200 250
0
100
200
300
400
500
600
700
800
900
CH4
C2 H
6
C3 H
8
C4 H
10
C5 H
12
C6 H
14
C7 H
16
C8 H
18
C9 H
20
C10 H
22
C11 H
24
C12 H
26
C13 H
28
C14 H
30
C15 H
32
C16 H
34
C17 H
36
C18 H
38
CH
4 O
C2 H
6 O
C3 H
8 O
C4 H
10 O
C5 H
12 O
C6 H
14 O
C7 H
16 O
C8 H
18 O
C9 H
20 O
C10 H
22 O
C11 H
24 O
He
Ne
Ar
Kr
Xe
Alcanos Alcenos Ciclo-alcanos Álcools Gases nobres
Tc(K
)
m(g/mol)
0 10 20 30 40 50 60 70 80 90
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
FH
H2O
H3N
CH4
H2S
CF4
CHN
ClH
CH4O
H2
F2
O2
N2
Cl2
O3
O2S
NO
N2O
COS
CS2
CO2
CO
Tc(K
)
m(g/mol)
6
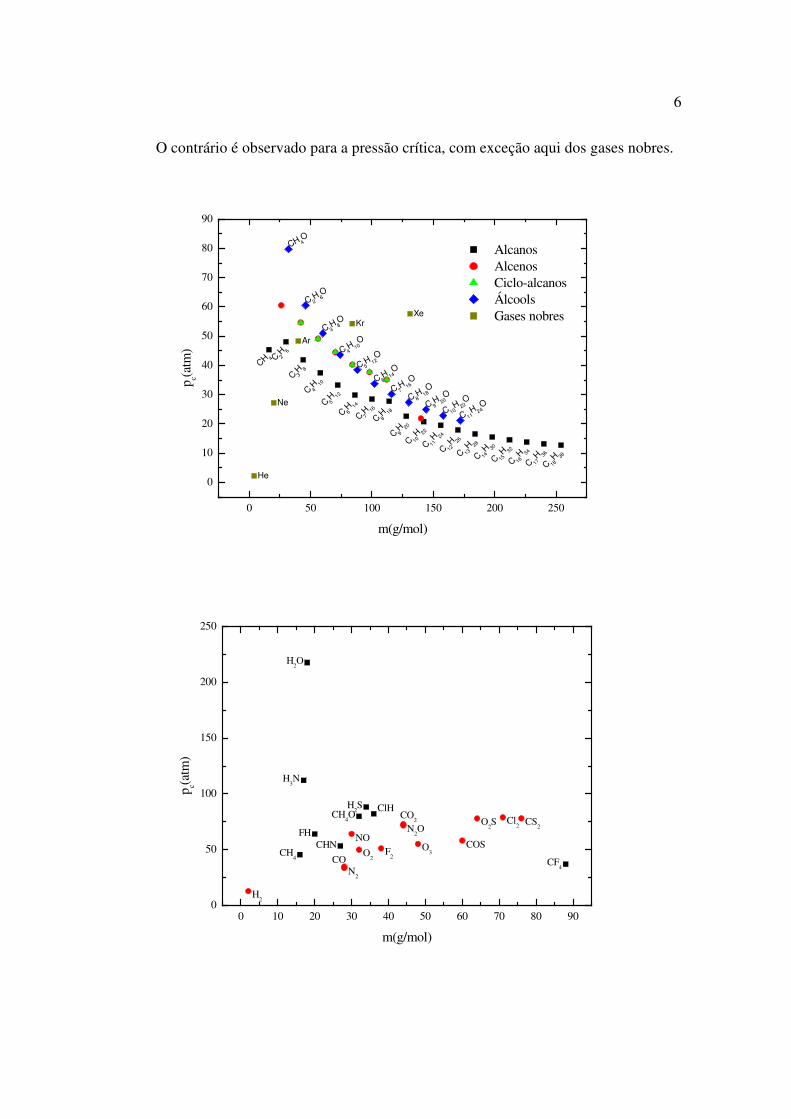
O contrário é observado para a pressão crítica, com exceção aqui dos gases nobres.
0 50 100 150 200 250
0
10
20
30
40
50
60
70
80
90
CH 4 C 2
H 6
C 3H 8
C 4H 1
0
C 5H 1
2
C 6H 1
4
C 7H 1
6
C 8H 1
8
C 9H 2
0
C 10H 2
2
C 11H 2
4
C 12H 2
6
C 13H 2
8
C 14H 3
0
C 15H 3
2
C 16H 3
4
C 17H 3
6
C 18H 3
8
CH 4O
C 2H 6
O
C 3H 8
O
C 4H 10
O
C 5H 12
O
C 6H 14
O
C 7H 16
O
C 8H 18
O
C 9H 20
O
C 10H 22
O
C 11H 24
O
He
Ne
Ar
Kr
Xe
Alcanos Alcenos Ciclo-alcanos Álcools Gases nobres
p c(atm
)
m(g/mol)
0 10 20 30 40 50 60 70 80 900
50
100
150
200
250
FH
H2O
H3N
CH4
H2S
CF4
CHN
CH4O
H2
F2O
2
N2
Cl2
O3
O2S
NON
2O
COS
CS2
ClHCO
2
CO
p c(atm
)
m(g/mol)
7
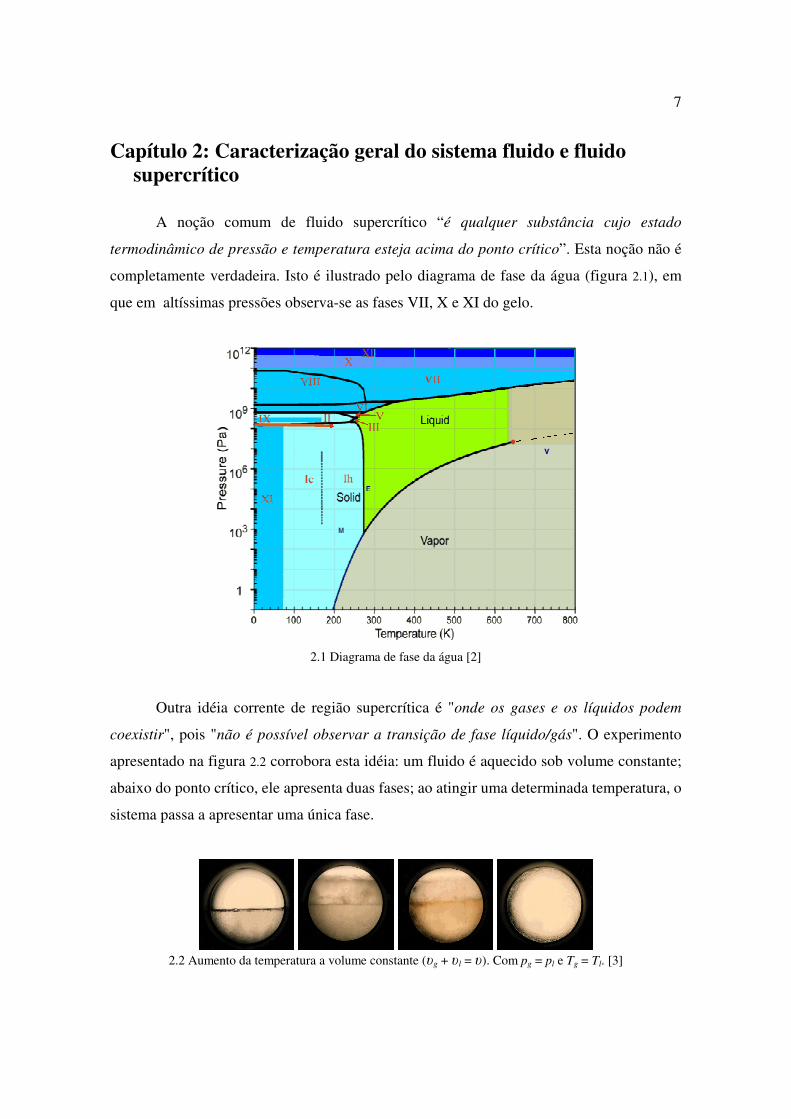
Capítulo 2: Caracterização geral do sistema fluido e fluido supercrítico
A noção comum de fluido supercrítico “é qualquer substância cujo estado
termodinâmico de pressão e temperatura esteja acima do ponto crítico”. Esta noção não é
completamente verdadeira. Isto é ilustrado pelo diagrama de fase da água (figura 2.1), em
que em altíssimas pressões observa-se as fases VII, X e XI do gelo.
2.1 Diagrama de fase da água [2]
Outra idéia corrente de região supercrítica é "onde os gases e os líquidos podem
coexistir", pois "não é possível observar a transição de fase líquido/gás". O experimento
apresentado na figura 2.2 corrobora esta idéia: um fluido é aquecido sob volume constante;
abaixo do ponto crítico, ele apresenta duas fases; ao atingir uma determinada temperatura, o
sistema passa a apresentar uma única fase.
2.2 Aumento da temperatura a volume constante (υg + υl = υ). Com pg = pl e Tg = Tl. [3]

8
2.3 Evolução do sistema bifásico sobre a linha de coexistência. [4]
Dois aspectos devem ser levantados neste ponto do texto para que não se caia em
armadilhas conceituais. Em primeiro, o experimento apresentado na figura 2.2 leva a
entender que o sistema apresenta uma única fase somente quando a temperatura atinge o
ponto crítico. Isto não é sempre verdade, como será discutido a seguir. Em segundo, a idéia
que na região supercrítica o estado não é nem líquido e nem gasoso será debatida; as
diferenças entre líquido e gás não são apenas qualitativas. Tais diferenças serão ressaltadas
nesta região, analisando principalmente a densidade e a energia do sistema.
9
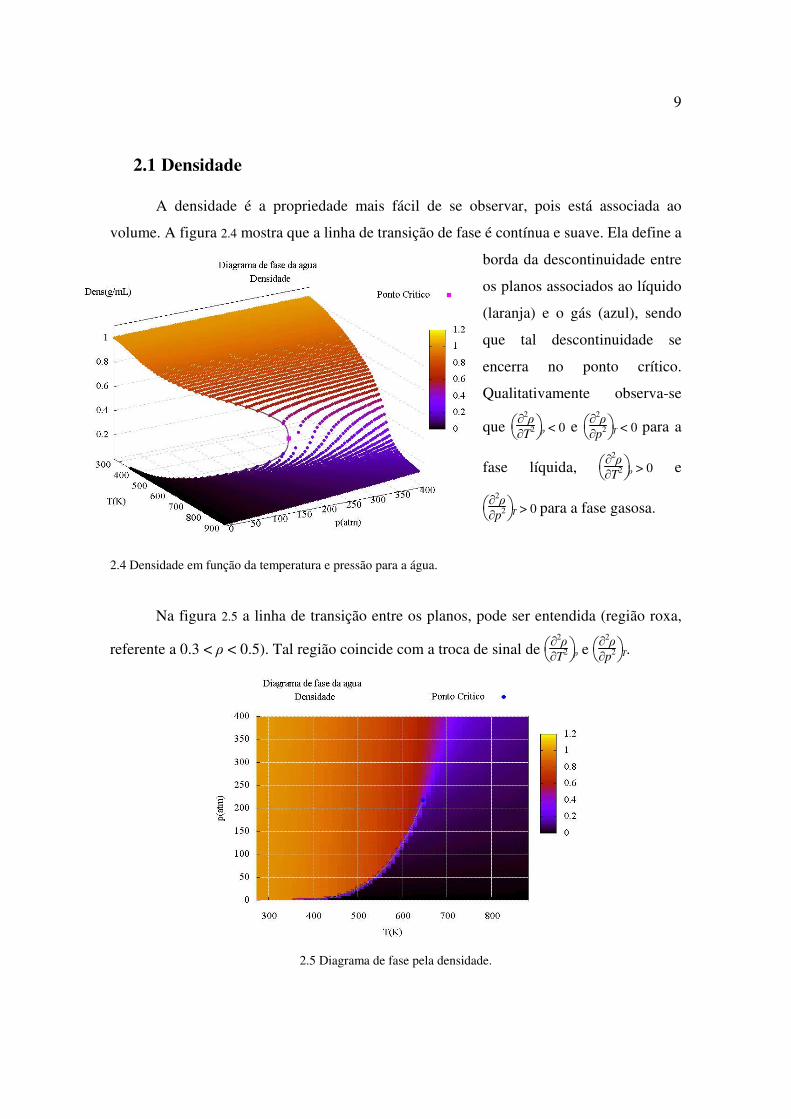
2.1 Densidade A densidade é a propriedade mais fácil de se observar, pois está associada ao
volume. A figura 2.4 mostra que a linha de transição de fase é contínua e suave. Ela define a
borda da descontinuidade entre
os planos associados ao líquido
(laranja) e o gás (azul), sendo
que tal descontinuidade se
encerra no ponto crítico.
Qualitativamente observa-se
que
∂
2ρ
∂T2 p < 0 e
∂
2ρ
∂p2 T < 0 para a
fase líquida,
∂
2ρ
∂T2 p > 0 e
∂
2ρ
∂p2 T > 0 para a fase gasosa.
2.4 Densidade em função da temperatura e pressão para a água.
Na figura 2.5 a linha de transição entre os planos, pode ser entendida (região roxa,
referente a 0.3 < ρ < 0.5). Tal região coincide com a troca de sinal de
∂
2ρ
∂T2 p e
∂
2ρ
∂p2 T.
2.5 Diagrama de fase pela densidade.
10
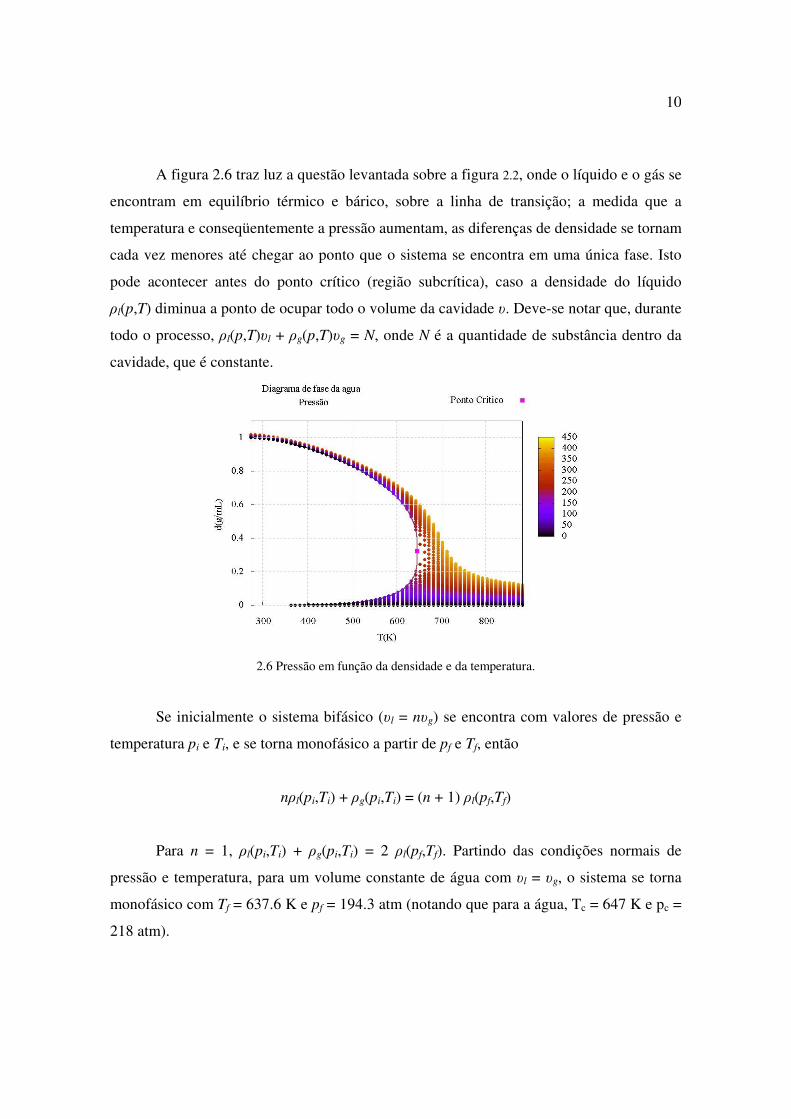
A figura 2.6 traz luz a questão levantada sobre a figura 2.2, onde o líquido e o gás se
encontram em equilíbrio térmico e bárico, sobre a linha de transição; a medida que a
temperatura e conseqüentemente a pressão aumentam, as diferenças de densidade se tornam
cada vez menores até chegar ao ponto que o sistema se encontra em uma única fase. Isto
pode acontecer antes do ponto crítico (região subcrítica), caso a densidade do líquido
ρl(p,T) diminua a ponto de ocupar todo o volume da cavidade υ. Deve-se notar que, durante
todo o processo, ρl(p,T)υl + ρg(p,T)υg = N, onde N é a quantidade de substância dentro da
cavidade, que é constante.
2.6 Pressão em função da densidade e da temperatura.
Se inicialmente o sistema bifásico (υl = nυg) se encontra com valores de pressão e
temperatura pi e Ti, e se torna monofásico a partir de pf e Tf, então
nρl(pi,Ti) + ρg(pi,Ti) = (n + 1) ρl(pf,Tf)
Para n = 1, ρl(pi,Ti) + ρg(pi,Ti) = 2 ρl(pf,Tf). Partindo das condições normais de
pressão e temperatura, para um volume constante de água com υl = υg, o sistema se torna
monofásico com Tf = 637.6 K e pf = 194.3 atm (notando que para a água, Tc = 647 K e pc =
218 atm).
11
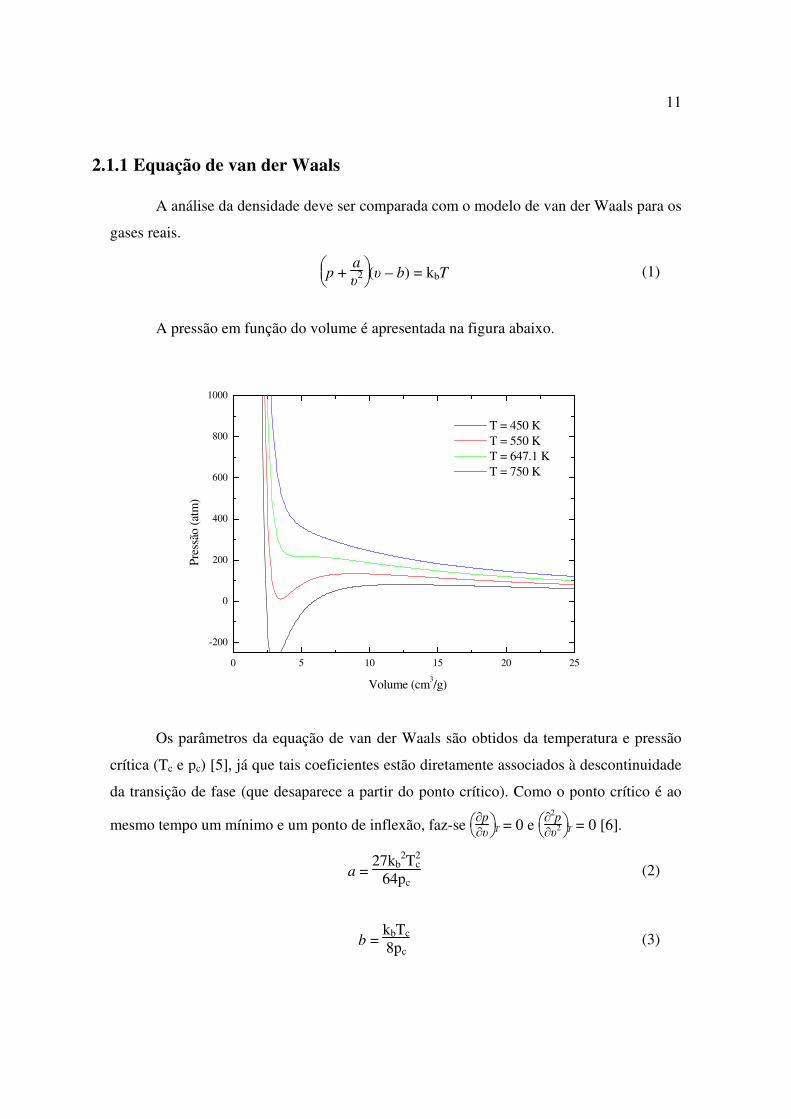
2.1.1 Equação de van der Waals A análise da densidade deve ser comparada com o modelo de van der Waals para os
gases reais.
p +
aυ
2 (υ – b) = kbT (1)
A pressão em função do volume é apresentada na figura abaixo.
0 5 10 15 20 25
-200
0
200
400
600
800
1000
Pres
são
(atm
)
Volume (cm3/g)
T = 450 K T = 550 K T = 647.1 K T = 750 K
Os parâmetros da equação de van der Waals são obtidos da temperatura e pressão
crítica (Tc e pc) [5], já que tais coeficientes estão diretamente associados à descontinuidade
da transição de fase (que desaparece a partir do ponto crítico). Como o ponto crítico é ao
mesmo tempo um mínimo e um ponto de inflexão, faz-se
∂p
∂υ T = 0 e
∂
2p∂υ
2 T = 0 [6].
a = 27kb
2Tc2
64pc (2)
b = kbTc
8pc (3)

12
Para obter analiticamente a densidade, parte-se da equação de van der Waals, que
pode ser re-escrita como (4) (onde ρ = 1/υ é a densidade)
ρ3 –
1b ρ2 +
pb + kbT
ab ρ – p
ab = 0 (4)
sendo reescrita
ρ3 + 3A ρ2 + B ρ + C = 0 (5)
e fazendo uma translação (ρ = y - A)
y3 + ( )B – 3A2 y + ( )2A3 – AB + C = 0 (6)
recai na forma
y3 + 3D y + E = 0 (7)
se ∆ = E2 + 4D3 > 0, a única solução real é dada por
y = –
E – ∆
2
1/3
+
E + ∆
2
1/3
(8)
caso contrário [7] há 3 soluções reais, dadas por
y = 2 r1/3 cos(t/3 + 2kπ/3) (9)
onde
r = |D3| t = acos(-E/2r) k = {0,1,2}
sendo que destas 3 soluções, as únicas duas que são fisicamente aceitáveis, isto é
∂p
∂υ T ≤ 0,
são as soluções com k = 1 e 0. A solução com k = 1 por sua vez fornece a possibilidade de
"adicionar" aos coeficientes de van der Waals outros tipos de força de coesão que devem
aparecer a baixas temperaturas.
13
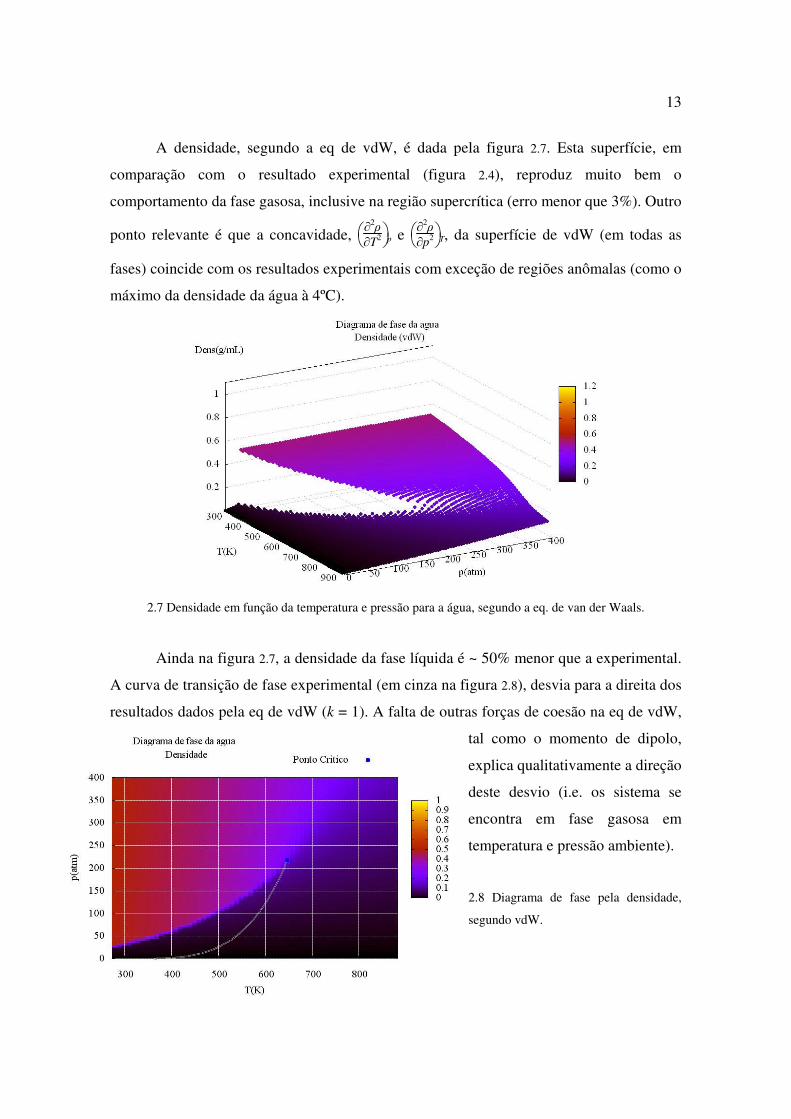
A densidade, segundo a eq de vdW, é dada pela figura 2.7. Esta superfície, em
comparação com o resultado experimental (figura 2.4), reproduz muito bem o
comportamento da fase gasosa, inclusive na região supercrítica (erro menor que 3%). Outro
ponto relevante é que a concavidade,
∂
2ρ
∂T2 p e
∂
2ρ
∂p2 T, da superfície de vdW (em todas as
fases) coincide com os resultados experimentais com exceção de regiões anômalas (como o
máximo da densidade da água à 4ºC).
2.7 Densidade em função da temperatura e pressão para a água, segundo a eq. de van der Waals.
Ainda na figura 2.7, a densidade da fase líquida é ~ 50% menor que a experimental.
A curva de transição de fase experimental (em cinza na figura 2.8), desvia para a direita dos
resultados dados pela eq de vdW (k = 1). A falta de outras forças de coesão na eq de vdW,
tal como o momento de dipolo,
explica qualitativamente a direção
deste desvio (i.e. os sistema se
encontra em fase gasosa em
temperatura e pressão ambiente).
2.8 Diagrama de fase pela densidade,
segundo vdW.
14
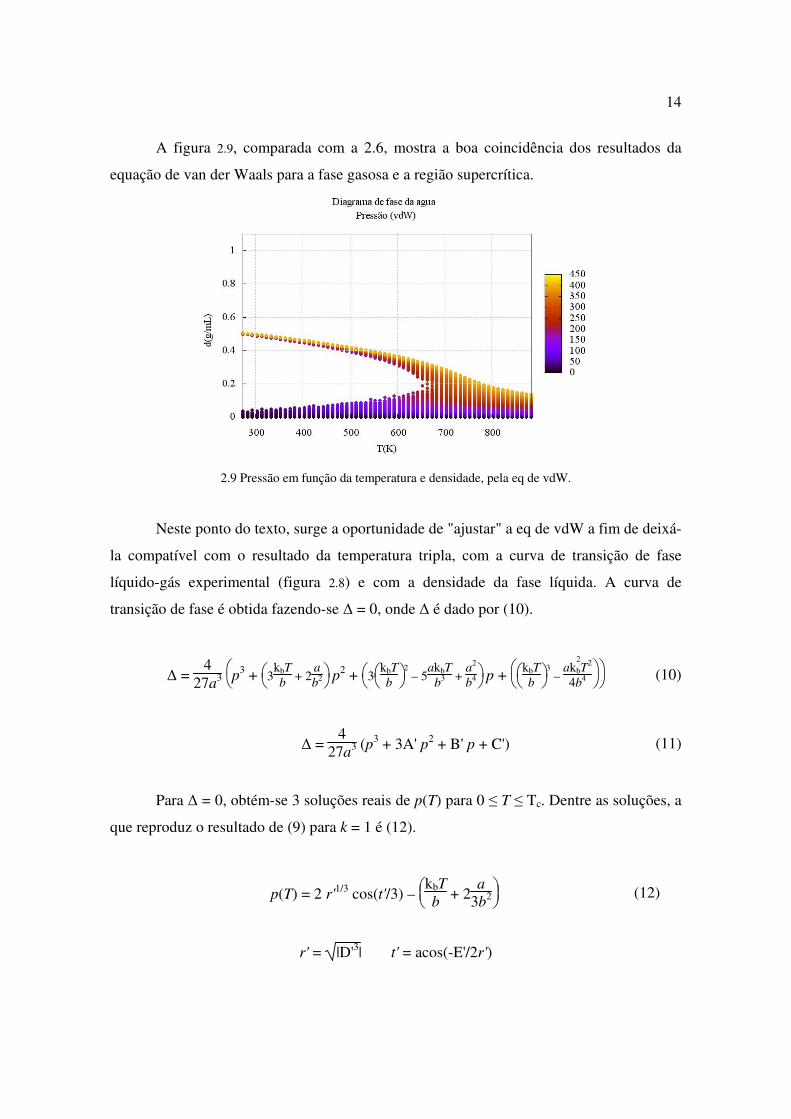
A figura 2.9, comparada com a 2.6, mostra a boa coincidência dos resultados da
equação de van der Waals para a fase gasosa e a região supercrítica.
2.9 Pressão em função da temperatura e densidade, pela eq de vdW.
Neste ponto do texto, surge a oportunidade de "ajustar" a eq de vdW a fim de deixá-
la compatível com o resultado da temperatura tripla, com a curva de transição de fase
líquido-gás experimental (figura 2.8) e com a densidade da fase líquida. A curva de
transição de fase é obtida fazendo-se ∆ = 0, onde ∆ é dado por (10).
∆ = 4
27a3
p3 +
3
kbTb + 2
ab2 p2 +
3
kbT
b2 – 5
akbTb3 +
a2
b4 p +
kbT
b3 –
ak2
bT2
4b4 (10)
∆ = 4
27a3 (p3 + 3A' p2 + B' p + C') (11)
Para ∆ = 0, obtém-se 3 soluções reais de p(T) para 0 ≤ T ≤ Tc. Dentre as soluções, a
que reproduz o resultado de (9) para k = 1 é (12).
p(T) = 2 r'1/3 cos(t'/3) –
kbT
b + 2a
3b2 (12)
r' = |D'3| t' = acos(-E'/2r')
15
D' = –
3
a(kbT)b3 +
a2
9b4 (13)
E' = 274
a(kbT)2
b4 + 5a2(kbT)
b5 – 2
27a3
b6 (14)
r' =
a
b23/2
27
(kbT)3
b3 + 3a(kbT)2
b4 + a2(kbT)
9b5 + a3
36b61/2
(15)
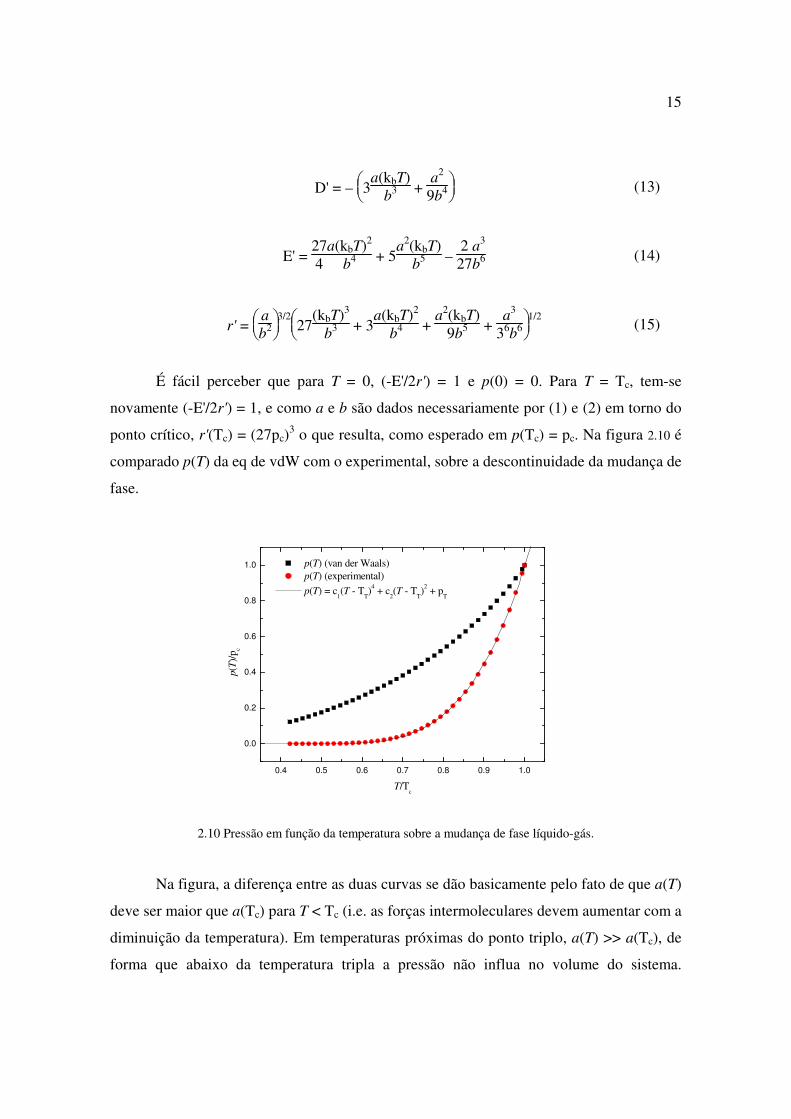
É fácil perceber que para T = 0, (-E'/2r') = 1 e p(0) = 0. Para T = Tc, tem-se
novamente (-E'/2r') = 1, e como a e b são dados necessariamente por (1) e (2) em torno do
ponto crítico, r'(Tc) = (27pc)3 o que resulta, como esperado em p(Tc) = pc. Na figura 2.10 é
comparado p(T) da eq de vdW com o experimental, sobre a descontinuidade da mudança de
fase.
0.4 0.5 0.6 0.7 0.8 0.9 1.0
0.0
0.2
0.4
0.6
0.8
1.0 p(T) (van der Waals) p(T) (experimental)
p(T) = c1(T - T
T)4 + c
2(T - T
T)2 + p
T
p(T)
/pc
T/Tc
2.10 Pressão em função da temperatura sobre a mudança de fase líquido-gás.
Na figura, a diferença entre as duas curvas se dão basicamente pelo fato de que a(T)
deve ser maior que a(Tc) para T < Tc (i.e. as forças intermoleculares devem aumentar com a
diminuição da temperatura). Em temperaturas próximas do ponto triplo, a(T) >> a(Tc), de
forma que abaixo da temperatura tripla a pressão não influa no volume do sistema.
16
Tomando este raciocínio como válido, e assumindo a(TT) deva ser tão grande que a pressão
possa ser desprezada na eq de vdW, é possível obter b(TT) em função de a(TT) e υT
(líquido).
b(TT) = υT – υT2 kbTT
a(TT) (16)
Tomando kbT/a(T) → 0 nas vizinhanças do ponto triplo, obtém-se (-E'/2r') = 1. Isto
implica que t' ~ 0, de tal forma que a pressão possa ser expandida em torno do ponto triplo
em potências de T2 (como feito na figura 2.10). Este raciocínio se justifica pela forma da
pressão ser co-seno, sendo a variação em torno do ponto triplo igual a zero.
0.4 0.6 0.8 1.0 1.2 1.4
0
1
2
3
4
5
a(Tc) [(T
c - T
T)/(T - T
T)]2
27(kbT)2/(64p
c)
a(Tc)
a(T)
/a(T
c)
T/Tc
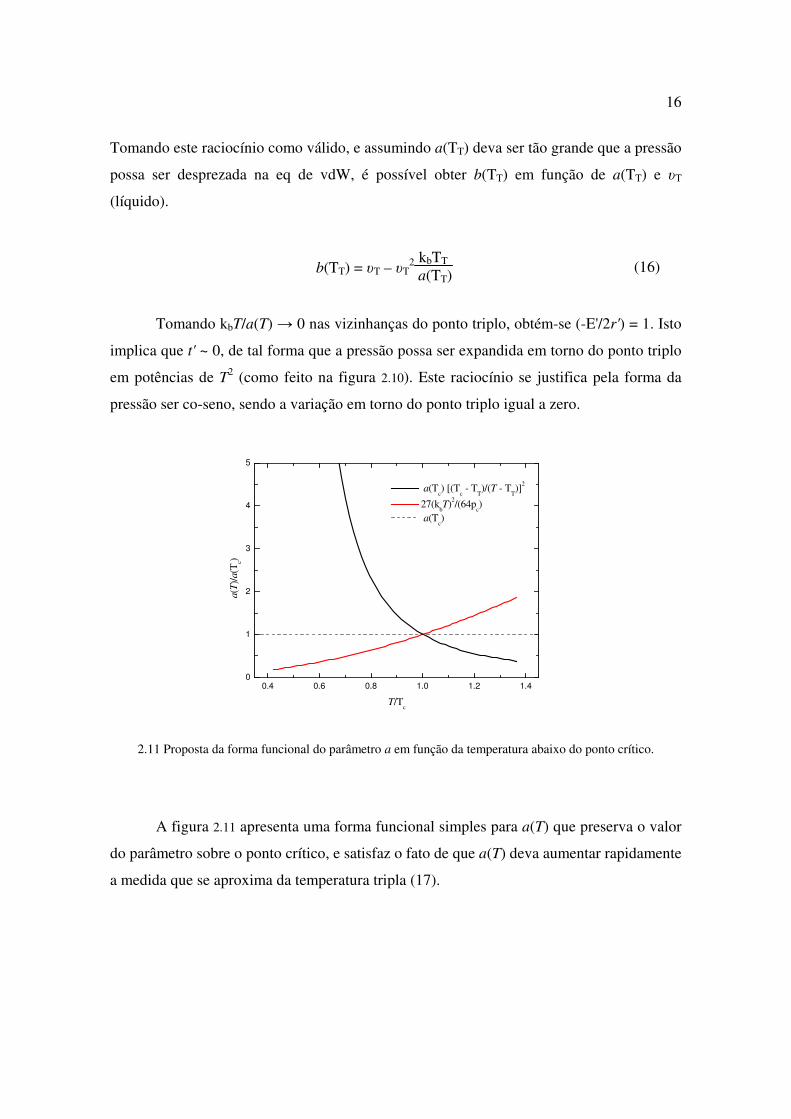
2.11 Proposta da forma funcional do parâmetro a em função da temperatura abaixo do ponto crítico.
A figura 2.11 apresenta uma forma funcional simples para a(T) que preserva o valor
do parâmetro sobre o ponto crítico, e satisfaz o fato de que a(T) deva aumentar rapidamente
a medida que se aproxima da temperatura tripla (17).
17
a(T) = a(Tc)
Tc – T'T
T – T'T2 (17)
Se T'T = TT, então p(TT) = – kbTT/b em (12), o que não é fisicamente aceitável.
Desta forma, escolheu-se um valor de T'T <~ TT, que fornecesse p(TT) = pT. Os resultados
para a densidade, utilizando (17) são apresentados na figura abaixo.
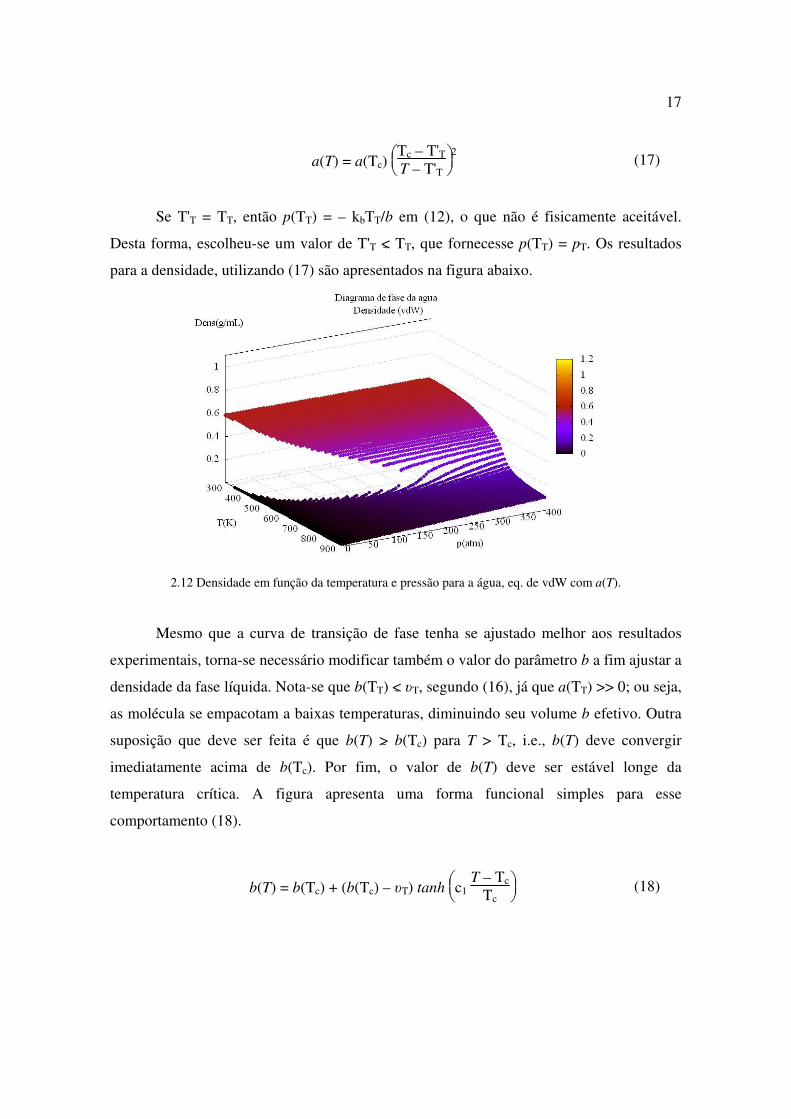
2.12 Densidade em função da temperatura e pressão para a água, eq. de vdW com a(T).
Mesmo que a curva de transição de fase tenha se ajustado melhor aos resultados
experimentais, torna-se necessário modificar também o valor do parâmetro b a fim ajustar a
densidade da fase líquida. Nota-se que b(TT) <~ υT, segundo (16), já que a(TT) >> 0; ou seja,
as molécula se empacotam a baixas temperaturas, diminuindo seu volume b efetivo. Outra
suposição que deve ser feita é que b(T) >~ b(Tc) para T > Tc, i.e., b(T) deve convergir
imediatamente acima de b(Tc). Por fim, o valor de b(T) deve ser estável longe da
temperatura crítica. A figura apresenta uma forma funcional simples para esse
comportamento (18).
b(T) = b(Tc) + (b(Tc) – υT) tanh
c1
T – Tc
Tc (18)
18
0.4 0.6 0.8 1.0 1.2 1.4
0.4
0.6
0.8
1.0
1.2
1.4 kbT/(8p
c)
b(Tc) + (b(T
c) - v
T) tanh(c
1(T - T
c)/T
c)
b(Tc)
b(T)
/b(T
c)
T/Tc
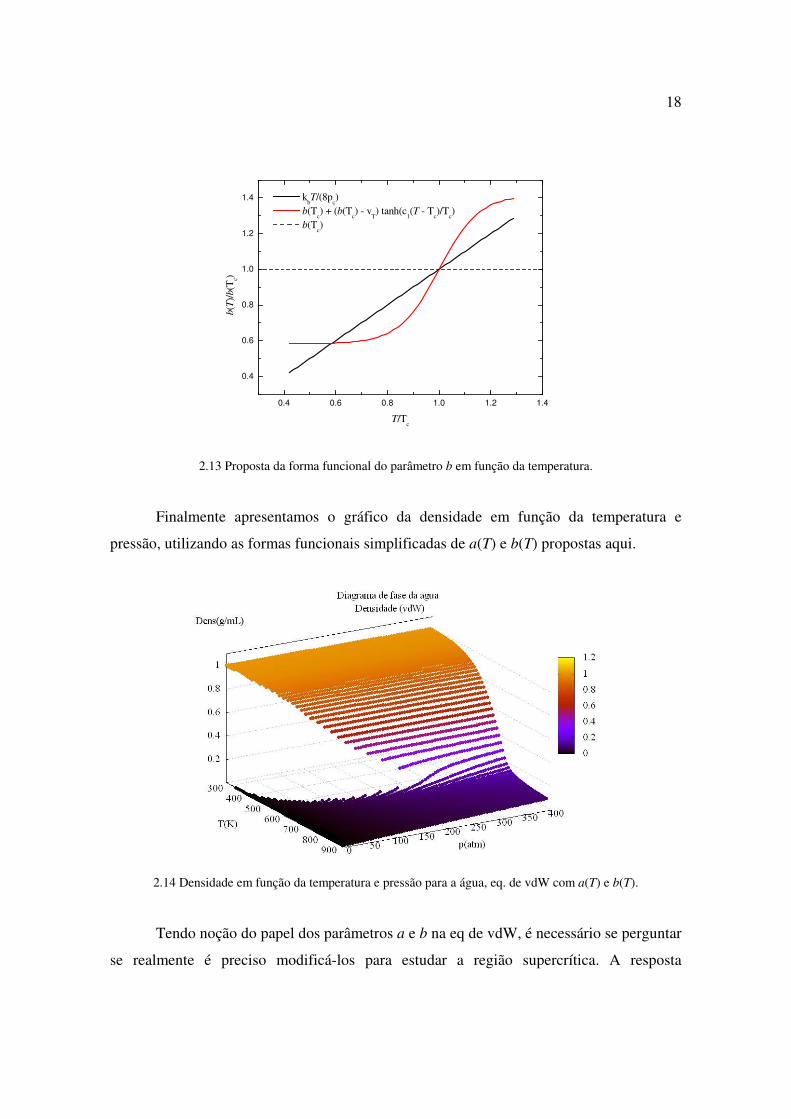
2.13 Proposta da forma funcional do parâmetro b em função da temperatura.
Finalmente apresentamos o gráfico da densidade em função da temperatura e
pressão, utilizando as formas funcionais simplificadas de a(T) e b(T) propostas aqui.
2.14 Densidade em função da temperatura e pressão para a água, eq. de vdW com a(T) e b(T).
Tendo noção do papel dos parâmetros a e b na eq de vdW, é necessário se perguntar
se realmente é preciso modificá-los para estudar a região supercrítica. A resposta

19
provavelmente é não. Os parâmetros a e b devem se manter aproximadamente constantes
acima da temperatura crítica. Neste regime, as velocidades moleculares devem ser
suficientemente grandes para que as interações de multipolo influenciem pouco na coesão
ou na forma de compactação das moléculas.
20
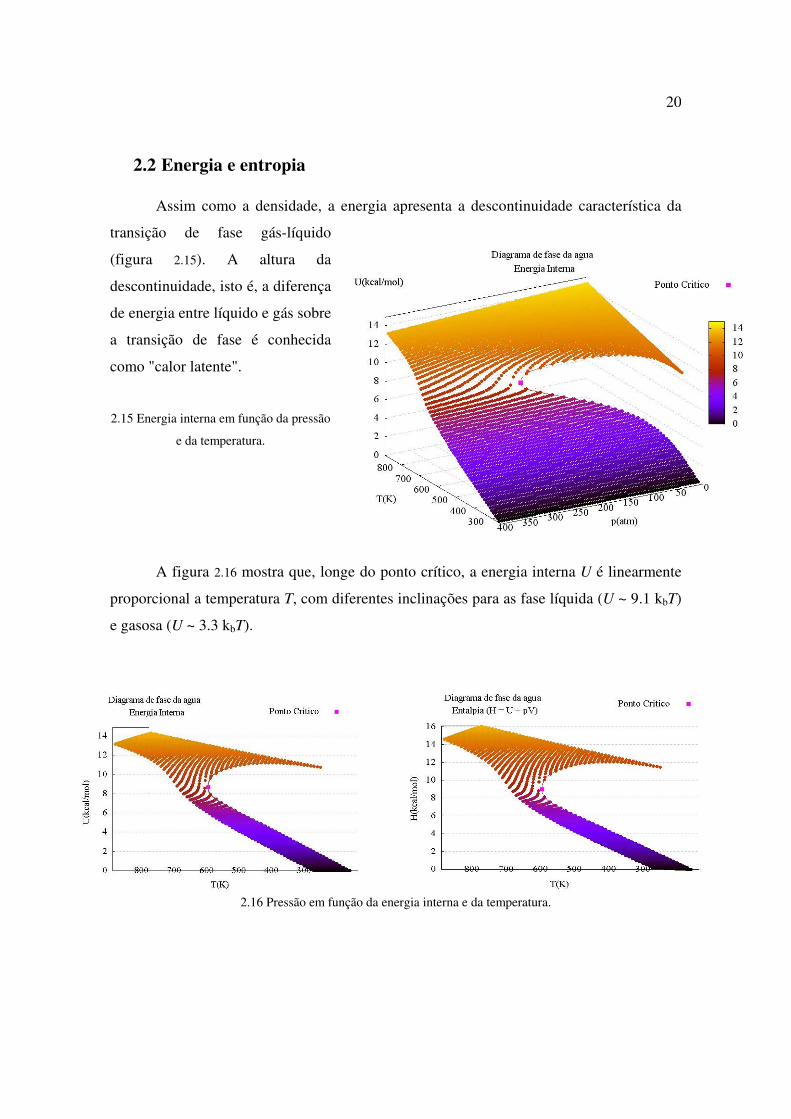
2.2 Energia e entropia Assim como a densidade, a energia apresenta a descontinuidade característica da
transição de fase gás-líquido
(figura 2.15). A altura da
descontinuidade, isto é, a diferença
de energia entre líquido e gás sobre
a transição de fase é conhecida
como "calor latente".
2.15 Energia interna em função da pressão
e da temperatura.
A figura 2.16 mostra que, longe do ponto crítico, a energia interna U é linearmente
proporcional a temperatura T, com diferentes inclinações para as fase líquida (U ~ 9.1 kbT)
e gasosa (U ~ 3.3 kbT).
2.16 Pressão em função da energia interna e da temperatura.

21
A escala mostrada na figura tem seu início sobre o ponto triplo para a fase líquida,
de tal forma que U = 0 a T = 273.16 K. Notando que à pressão nula (ausência de forças
externas) são acessíveis apenas os estados sólido e gasoso, U = 0 implica na transição entre
estes dois estados abaixo do ponto triplo. Entendendo a fase líquida como um condensado
de moléculas, sua energia total é menor ou igual a zero. O trabalho exercido pelas forças
externas para condensar um gás de volume infinito até um volume υ é igual a (19); dada a
temperatura da linha de transição sólido-fluido, os deslocamentos moleculares devem
tender a zero para qualquer υ', de tal forma que p(υ') → 0 com ∆W ~ 0. Note-se que estas
condições se referem a energia E constante, p(υ',E) = p(υ'), sendo que T → 0 para υ → ∞.
∆W = ⌡⌠∞
υ
dυ' p(υ',E) (19)
Dada a boa concordância da eq. de vdW com os resultados da fase gasosa, podemos
partir desta relação a fim de calcular ∆W. Tem-se que a pressão é dada por (20).
p(υ') = kbT'
(υ' – b) – aυ'2 (20)
No gás ideal, o trabalho exercido pelas forças externas se converte tão somente em
energia cinética (21). Sendo v a velocidade associada à temperatura e n o número de graus
de liberdade, de tal forma que nkbT' = mv2. Desta, deriva-se que T'υ'2/n é constante.
p(υ')dυ' + mvdv = 0 (21)
Assumindo que no gás de vdW, T'υ'2/n ~ constante, é possível derivar (22), onde T' =
T'(υ,E).
p(υ') ~ kbT υ
2/n
υ'2/n(υ' – b) – aυ'2 (22)
Substituindo (22) em (19), expandindo υ >> b, obtém-se (23).
∆W ~ aυ – kbT
n
2 + nb
(n + 2)υ (23)
Tomando ∆W + U = 0, a energia interna é dada aproximadamente por (24).
22
U ~ kbT
n
2 + nb
(n + 2)υ – aυ (24)
Este resultado pode ser apreciado na figura 2.17, onde se utilizou n = 6.
2.17 Energia interna obtida aproximadamente via equação de van der Waals.
Assim como a densidade, a energia interna apresenta inversão da concavidade na
região supercrítica. Esta inversão é ilustrada pela figura 2.18; a região vermelha (7 < U < 8)
representa o limite do estado líquido e a laranja (10 < U < 11), o limite do estado gasoso
dentro da região supercrítica.
2.18 Diagrama de fase pela energia.
23
Esta mudança de estado dentro da região supercritica é reforçada pela figura 2.19
(área roxa, em extensão a transição de fase). A figura fornece uma visão superior da
superfície pV (figura 2.20) em função pra pressão e da temperatura.
2.19 Diagrama de fase em pV.
2.20 pV em função da pressão e da temperatura.
Deve-se enfatizar neste ponto do texto, que nesta região em que a concavidade
sobre inversão, pequenas variações da pressão e temperatura causam grandes variações na
densidade, solubilidade, viscosidade, e propriedades afins. Sugere-se aqui a necessidade de
se definir, dentro da região supercrítica, uma região líquida e uma gasosa.

24
2.21 Entropia em função da pressão e da temperatura.
A figura 2.21 apresenta o comportamento da entropia. A entropia diverge para
pressão nula – fase gasosa (o que é esperado, já que infinitos estados são acessíveis). Assim
como a energia e a densidade, expandindo a linha de transição de fase para dentro da região
supercrítica, observa-se a inversão da concavidade.
Sobre o ponto crítico, a primeira derivada diverge, e assim o calor específico, cp =
T
∂S
∂T , vai a infinito (assim como outras primeiras variações, como a compressibilidade).
25
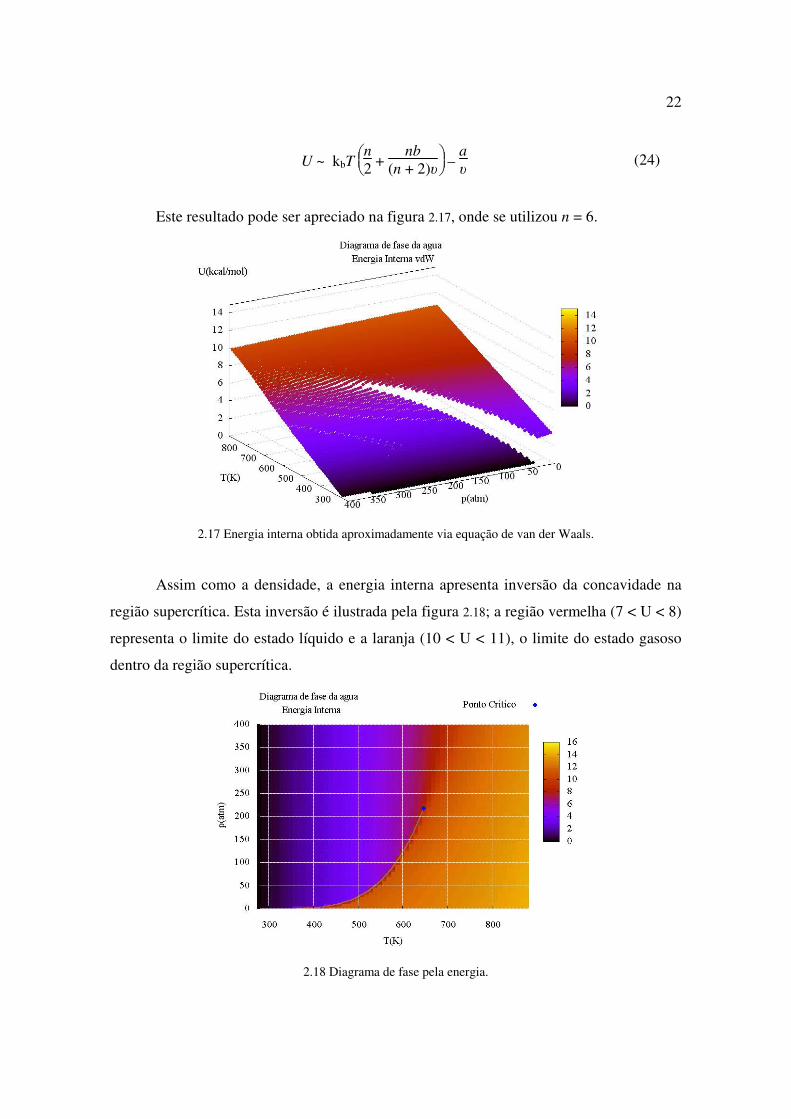
Observa-se que o máximo do calor específico, marcando a transição de fase entre a
fase líquida e a fase gasosa se estende dentro da região supercrítica. Ou seja,
∂cp
∂T p < 0 para
a gás supercrítico, e
∂cp
∂T p > 0 para líquido supercrítico.
2.22 Viscosidade em função da pressão e temperatura.
Por fim, a viscosidade µ é definida pela relação F = Aµ∂v
∂y , onde F é a força para
arrastar uma lâmina de área A, a um gradiente de velocidades ∂v
∂y . Observa-se na figura
2.22 que a viscosidade cai exponencialmente com a temperatura a temperaturas próximas do
ponto de fusão, crescendo linearmente na fase gasosa. Observa-se que a pressão não
influencia no valor da viscosidade, com exceção da mudança de fase líquido-gás. O mínimo
da viscosidade se dá sobre a transição entre a fase líquida e a fase gasosa, se entendendo
dentro da região supercrítica. Isto implica que
∂µ
∂T p < 0 para líquido supercrítico, e
∂µ
∂T p > 0
para gás supercrítico.
26
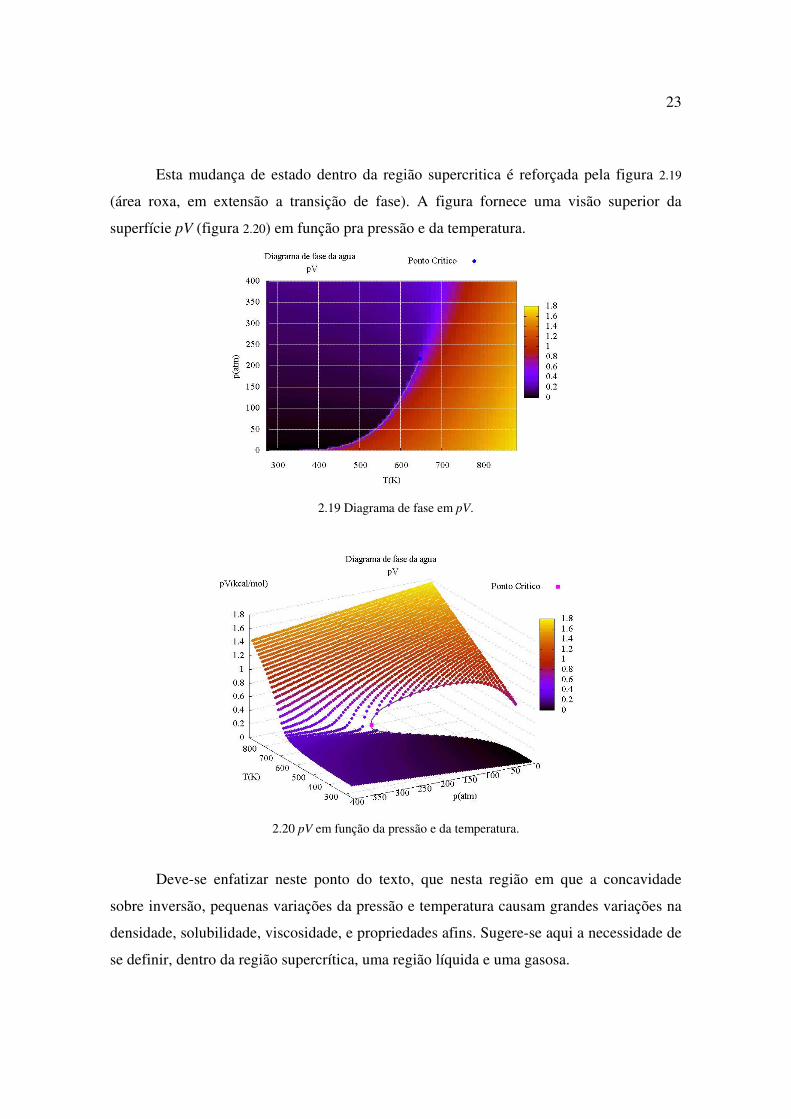
Capítulo 3: Características dos fluidos supercríticos
3.1) Muda sua densidade de forma contínua; a constante dielétrica é reduzida com
o aumento da temperatura e a diminuição da densidade, tornando substâncias polares bons
solventes de compostos orgânicos; [8]
3.2) Aumento da capacidade de transporte (extração) com a diminuição da
viscosidade (de 10 a 100 vezes menor) e o aumento do coeficiente de difusão (de 10 a 100
vezes maior); também associado com a alteração da concentração de íons livres (produto
iônico, Kw=[H+][OH-] em água, e.g.);

27
3.3) Pode substituir os solventes orgânicos na indústria alimentícia, farmacológica e
de polímeros; nos processos de extração (SFE), cromatografia (SFC), concentração,
purificação, infusão, e reações químicas (hidrogenação e polimerização); solvente
ecológico;
Exemplo de cromatografia de coluna.
Fase imóvel: gel de sílica; Fase móvel:
solventes orgânicos + folhas frescas.
É uma alternativa aos métodos GC (gas chromatography) e HPLC (High
Performance Liquid Chromatography). SCF oferece maior sensibilidade de suas
propriedades químicas em função das propriedades termodinâmicas, além de ser mais
rápido que HPLC e pode ser feito em temperaturas menores que GC;

28
3.4) É aplicado no processamento de materiais: granulação fina, deposição de filmes
finos, refinamento de fibras usando o método RESS (Rapid Expansion of the Supercritical
Solutions); como exemplo, produção de micro partículas ou micro fibras de sílica (SiO2),
na qual sílica monocristalina é dissolvida em água supercrítica. [8]
(823K,100MPa) (823K, 63MPa)

29
Capítulo 4: Algumas aplicações notáveis
4.1 Reciclagem química de embalagens PET (Poli Tereftalato de Etila / Polyethylene Terephthalate).
PET é um poliéster, polímero termoplástico ou plástico, desenvolvido por dois
químicos britânicos Whinfield e Dickson em 1941, formado pela reação entre o ácido
tereftálico (dihidroxi benzeno) e o etileno glicol (etanodiol), formando um poliéster, utiliza-
se principalmente na forma de fibras para tecelagem e de embalagens para bebidas. [1]
(PET)
(dihidroxi benzeno) (etanodiol)
Possui propriedade termoplástica, isto é, pode ser reprocessado diversas vezes pelo
mesmo ou por outro processo de transformação. Quando aquecidos a temperaturas
adequadas, esses plásticos amolecem, fundem e podem ser novamente moldados.
As garrafas produzidas com este polímero só começaram a ser fabricadas na década
de 70, após cuidadosa revisão dos aspectos de segurança e meio ambiente.
No começo dos anos 80, os Estados Unidos e o Canadá iniciaram a coleta dessas
garrafas, reciclando-as inicialmente para fazer enchimento de almofadas. Com a melhoria
da qualidade do PET reciclado, surgiram aplicações importantes, como tecidos, lâminas e
garrafas para produtos não alimentícios.
Mais tarde na década de 90, o governo americano autorizou o uso destes material
reciclado em embalagens de alimentos.

30
RECICLAGEM - Contaminantes
Os principais contaminantes do PET reciclado de garrafas de refrigerantes são os
adesivos (polietireno e a cola usada em sua base) usados como rótulo e ("base cup") - A
famosa base de alguns refrigerantes (Polipropileno). A maioria dos processos de lavagens
não impede que traços destes produtos
indesejáveis permaneçam no floco de
PET.
A cola age como catalisador de
degradação hidrolítica quando o material
é submetido à alta temperatura no
processo de extrusão, além de escurecer
e endurecer o reciclado. O mesmo pode
ocorrer com o cloreto de polivinila
(PVC), que compõe outros tipos de
garrafas e não pode misturar-se com a
sucata de PET, pois o PVC reage com o
PET, transformando-o em outra
substância.
Exemplo de reação de hidrólise. (~ 30 min)
Antes da reação (esq)
grânulos de PET + água
Depois da decomposição (dir)
ácido tereftálico (em finas partículas) +
solução aquosa de etileno glicol

31
4.2 Transformação da celulose em glicose.
A celulose é um polímero de "cadeia longa" composto de um só monômero,
carboidratado, classificado como polissacarídeo. É o componente estrutural primário das
plantas e não é digerível pelo homem.
A celulose tem uma estrutura linear ou fibrosa, na qual se estabelecem múltiplas
pontes de hidrogênio entre os grupos hidroxilas das distintas cadeias justapostas de glicose,
fazendo-as impenetráveis a água, e originando fibras compactas que constituem a parede
celular dos vegetais.
Industrialmente, a celulose é extraída da madeira de árvores como o pinho, o
eucalipto ou o abeto ou de plantas herbáceas com grande quantidade de celulose no talo,
como a cana-de-açúcar, diversas gramíneas e juncos. O algodão puro é formado em 99,8%
de celulose. Outras fibras têxteis, como a juta, o cânhamo, o rami e o linho também
possuem grande proporção desse polissacarídeo. Conforme o tipo de árvore se obtém a
celulose de fibra curta ou de fibra longa. Esta característica torna o papel resultante mais
absorvente ou mais resistente, respectivamente.
A glicose ou dextrose é um carboidrato do tipo monossacarídeo. Cristal sólido de
sabor adocicado, de formula molecular C6H12O6, encontrado na natureza na forma livre ou
combinada. Juntamente com a frutose e a galactose, é o carboidrato fundamental de

32
carboidratos maiores, como sacarose e maltose. Amido e celulose são polímeros de glicose.
É encontrada nas uvas e em vários frutos. Industrialmente é obtida a partir do amido.
No metabolismo, a glicose é uma das principais fontes de energia e fornece 4
calorias de energia por grama. A glicose hidratada (como no soro glicosado) fornece 3,4
calorias por grama. Sua degradação química durante o processo de respiração celular dá
origem a energia química (armazenada em moléculas de ATP - entre 36 e 38 moléculas de
ATP por moléculas de glicose), gás carbônico e água.
A glicose pode ser extraída da celulose, via água supercrítica. As cadeias são
quebradas, segundo a reação:
(C6H10O5)n + n(H2O) → n(C6H12O6)
33
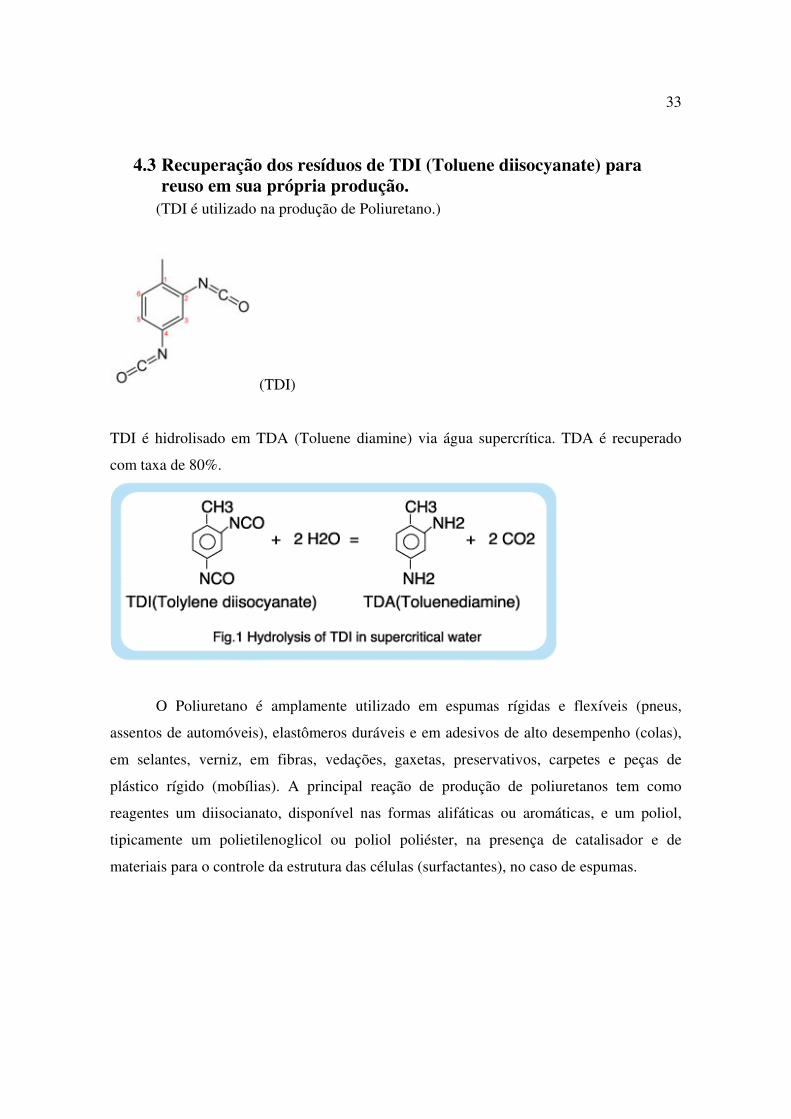
4.3 Recuperação dos resíduos de TDI (Toluene diisocyanate) para reuso em sua própria produção.
(TDI é utilizado na produção de Poliuretano.)
(TDI)
TDI é hidrolisado em TDA (Toluene diamine) via água supercrítica. TDA é recuperado
com taxa de 80%.
O Poliuretano é amplamente utilizado em espumas rígidas e flexíveis (pneus,
assentos de automóveis), elastômeros duráveis e em adesivos de alto desempenho (colas),
em selantes, verniz, em fibras, vedações, gaxetas, preservativos, carpetes e peças de
plástico rígido (mobílias). A principal reação de produção de poliuretanos tem como
reagentes um diisocianato, disponível nas formas alifáticas ou aromáticas, e um poliol,
tipicamente um polietilenoglicol ou poliol poliéster, na presença de catalisador e de
materiais para o controle da estrutura das células (surfactantes), no caso de espumas.
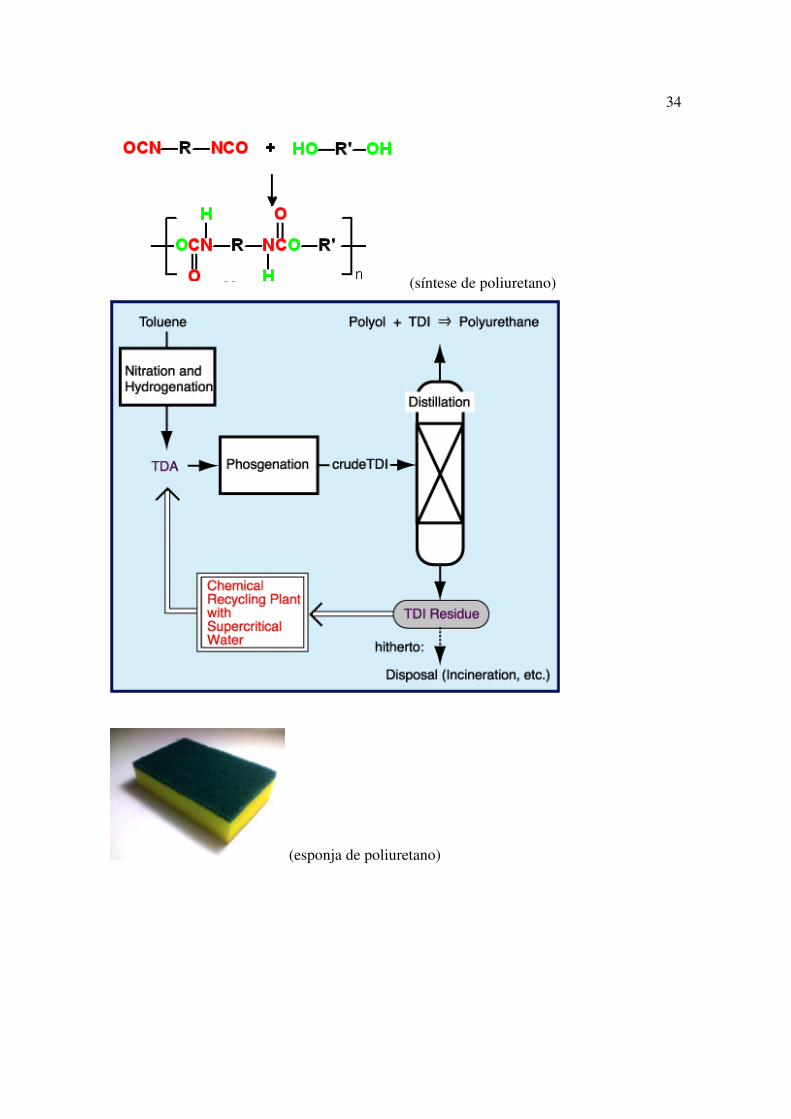
34
(síntese de poliuretano)
(esponja de poliuretano)
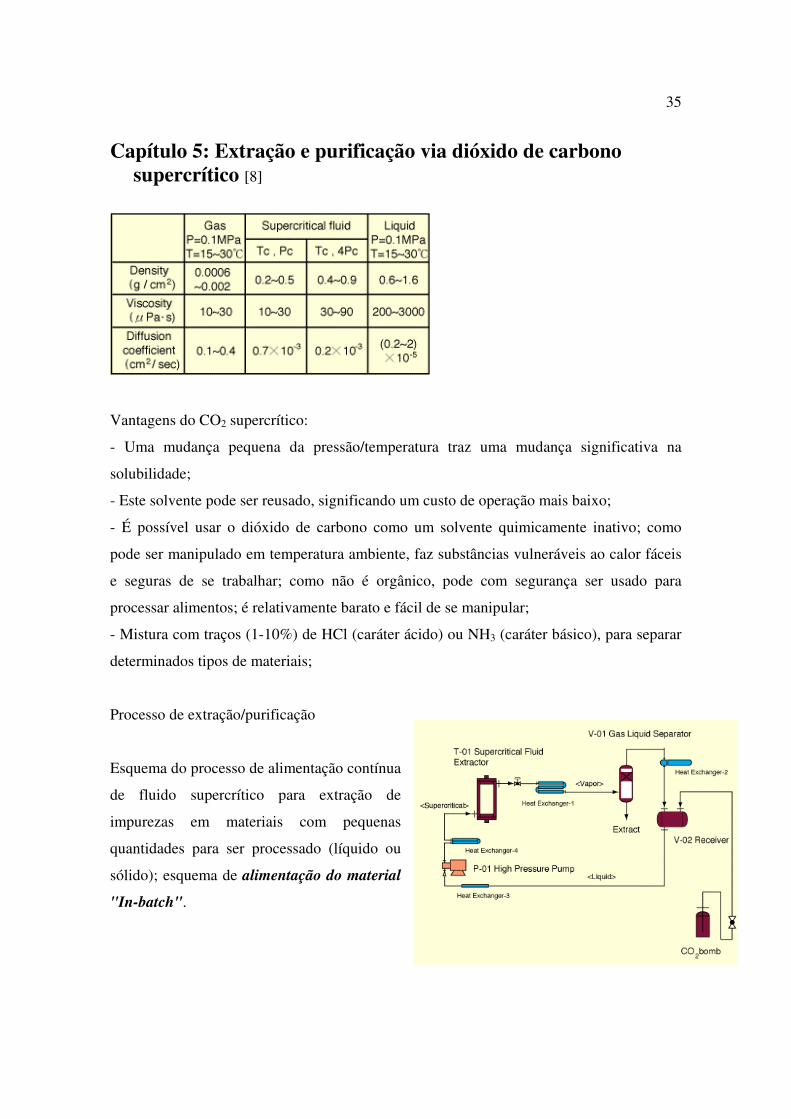
35
Capítulo 5: Extração e purificação via dióxido de carbono supercrítico [8]
Vantagens do CO2 supercrítico:
- Uma mudança pequena da pressão/temperatura traz uma mudança significativa na
solubilidade;
- Este solvente pode ser reusado, significando um custo de operação mais baixo;
- É possível usar o dióxido de carbono como um solvente quimicamente inativo; como
pode ser manipulado em temperatura ambiente, faz substâncias vulneráveis ao calor fáceis
e seguras de se trabalhar; como não é orgânico, pode com segurança ser usado para
processar alimentos; é relativamente barato e fácil de se manipular;
- Mistura com traços (1-10%) de HCl (caráter ácido) ou NH3 (caráter básico), para separar
determinados tipos de materiais;
Processo de extração/purificação
Esquema do processo de alimentação contínua
de fluido supercrítico para extração de
impurezas em materiais com pequenas
quantidades para ser processado (líquido ou
sólido); esquema de alimentação do material
"In-batch".

36
Aplica-se, por exemplo, na extração de odorantes e colorantes; remoção de pesticidas do
extrato ou pó de ginseng.
Projeto piloto para o processo de
concentração/purificação de solução crua de etanol,
através do esquema de alimentação contínua
(grandes quantidades de líquido).
Aplica-se na conversão de misturas azeotrópicas
como a solução aquosa de etanol em álcool absoluto.
Aumento da eficiência da extração de pesticida
(trichlorophenoxyacetic acid) de grãos em função
do aumento da pressão.

37
Capítulo 6: Empresas e grupos envolvidos com pesquisa na área de fluidos supercríticos (alguns).
Empresas:
* Petrotech; detecção, processamento, extração, caracterização, tratamento químico de
hidrocarbonetos, reservas de óleo e gás, reservas de águas subterrâneas, e traços de
elementos tóxicos, corrosivos e poluentes; bases na Noruega, Reino Unido, Países Baixos,
Malásia, Austrália e Emirado Árabes Unidos;
* Petronas; empresa governamental da Malásia, controladora e dona de todos os recursos
petrolíferos (gás e óleo) da Malásia (é uma das 500 maiores corporações do planeta);
* Kobe Steel; maior empresa japonesa na produção de embalagens de alumínio; trabalha
com processamento de polímeros, plásticos, sílica, aço, titânio, cobre, deposição em
superfícies, reciclagem, extração, equipamentos...;
* Jasco; empresa britânica especialista em maquinário de caracterização e controle de
substâncias, tecnologia em espectroscopia e cromatografia (em SCF), microscópios,
bombas de pressão e controle de temperatura;
* Supercritical Fluid Technologies; empresa americana especializada em tecnologia e
equipamentos para extração e processamento de materiais utilizando fluidos supercríticos;
* Thar; empresa americana especializada na pesquisa de tecnologia em fluidos
supercríticos, desenvolvendo processos e equipamentos, em colaboração com pesquisa
governamental e acadêmica;
* Restek; empresa americana especializada em equipamentos de cromatografia (análise e
colunas de separação), e métodos de separação química em geral;

38
* Phasex Corporation; empresa americana especializada em produtos e processos com
fluidos supercríticos (extração, fracionamento de polímeros, purificação e esterilização de
componentes médicos, concentração de nutricêuticos...);
****************************************
Grupos:
Neil R Foster, Grupo de Fluidos Supercríticos, New South Wales, Sydney, Australia
http://www.supercritical.unsw.edu.au
Jerry King, Applied Chemical Technologies (C-ACT), Los Alamos National Lab
http://scrub.lanl.gov/2002/scf/pubs/scf_king.htm
Pacific Northwest National Laboratory (PNNL)
http://www.pnl.gov/supercriticalfluid/index.stm
*********
Fernando M. Lanças, Laboratório de Cromatografia, IQSC-USP
http://www.iqsc.usp.br/iqsc/grupos_pesquisa/dqfm/croma/index_arquivos/Page1353.htm
Reinaldo Camino Bazito, DQF, IQUSP
Linha de Pesquisa: Química Verde: (1) Fluidos supercríticos como solventes alternativos
(2) Degradação de poluentes persistentes e (3) Otimização de processos químicos
http://www2.iq.usp.br/docente/bazito/
Alessandra Lopes de Oliveira, laboratório de tecnologia a alta pressão em produtos
naturais, USP
Pesquisa na área de Extração com dióxido de carbono supercrítico de princípio ativo e
aroma de frutos tropicais

39
Capítulo 7: Livros e periódicos
Supercritical Fluid Extraction (2nd Edition) McHugh, Mark A.; Krukonis, Val J. 1994 Elsevier Fundamentos básicos + aplicações industriais Supercritical Fluid Science and Technology (Hardcover) Y. Arai, T. Sako, Y. Takebayashi 2001 Springer Estudo microscópico + simulação Supercritical Fluid Technology in Oil and Lipid Chemistry, J. W. King and G. R. List (eds.), AOCS Press, Champaign, IL (1996)
The Journal Of Supercritical Fluids (Elsevier) Editor-in-Chief: Erdogan Kiran De cobertura multidisciplinar, inclui tópicos básicos e aplicados. Termodinâmica, e
equilíbrio de fase, cinética e processos de reação, propriedades térmicas e de transporte, e
todos os tópicos relacionados ao processamento tais como separações (extração,
fracionamento, purificação, cromatografia), nucleação e impregnação estão dentro deste
foco. São incluídas considerações sobre aplicações de engenharia específicas tais como as
encontradas nas indústrias de alimento, combustível, produtos naturais, minerais,
farmacêuticos e de polímero. Os tópicos relacionaram-se ao projeto de equipamentos de
alta pressão, técnicas analíticas, sensores, e as metodologias de controle de processo estão
também dentro do enfoque do jornal. O jornal publica contribuições originais em todos os
aspectos teóricos e experimentais da ciência e da tecnologia de processos e fluidos
supercríticos. Artigos que descrevem instrumentação, metodologias e técnicas
experimentais novas, procedimentos preditivos e de revisão são também aceitáveis. [9]
(fev/07) 1. Editorial Board • EDITORIAL BOARD Thermodynamics, Solubility, Phase Equilibria 2. High-pressure phase behavior of CO2/acetone/ionic liquid system • ARTICLE Zhaofu Zhang, Weize Wu, Bo Wang, Jiawei Chen, Dong Shen and Buxing Han

40
3. Determination of vapor pressure and solubility correlation of phenolic compounds in supercritical CO2 • ARTICLE Marleny D.A. Saldaña, Bruno Tomberli, Selma E. Guigard, Saul Goldman, Chris G. Gray and Feral Temelli 4. Raman spectral shifts of CO2 as measure of CO2-philicity of solutes in supercritical carbon dioxide • ARTICLE Yoshihiro Kachi, Takehiko Tsukahara, Yoshihito Kayaki, Takao Ikariya, Jun Sato and Yasuhisa Ikeda 5. The application of a shear mode piezoelectric sensor to monitoring the high-pressure phase behaviour of asymmetric binary systems • ARTICLE Jie Ke, Katherine E. Reid and Martyn Poliakoff 6. Investigation of the enhanced solubility of fluorinated methanes in CO2 by Monte Carlo simulation: Absolute free energy of solvation and structural properties of solution • ARTICLE Mohsen Tafazzoli and Ali Khanlarkhani Extractions, Separations 7. Separation of natural tocopherols from soybean oil byproduct with supercritical carbon dioxide • ARTICLE Tao Fang, Motonobu Goto, Xianbao Wang, Xiaolin Ding, Jianguo Geng, Mitsuru Sasaki and Tsutomu Hirose 8. Recovery of squalene from vegetable oil sources using countercurrent supercritical carbon dioxide extraction • ARTICLE Luis Vázquez, Carlos F. Torres, Tiziana Fornari, F. Javier Señoráns and Guillermo Reglero 9. Separation of enantiomers by diastereomeric salt formation and precipitation in supercritical carbon dioxide: Application to the resolution of mandelic acid • ARTICLE A. Martín and M.J. Cocero Reactions, Reactors 10. Kinetic analysis on alcohol concentration and mixture effect in supercritical water oxidation of methanol and ethanol by elementary reaction model • ARTICLE Rumiko Hayashi, Masato Onishi, Masakazu Sugiyama, Seiichiro Koda and Yoshito Oshima 11. Study of natural convection in supercritical CO2 cold wall reactors: Simulations and experiments • ARTICLE Xiaoying Shan, David P. Schmidt and James J. Watkins Bioavailability, Bioreactivity 12. On the importance of the supporting material for activity of immobilized Candida antarctica lipase B in ionic liquid/hexane and ionic liquid/supercritical carbon dioxide biphasic media • ARTICLE Pedro Lozano, Teresa De Diego, Tanja Sauer, Michel Vaultier, Said Gmouh and José L. Iborra 13. Bioavailability enhancement of an active substance by supercritical antisolvent precipitation • ARTICLE Viktor Majerik, Gérard Charbit, Elisabeth Badens, Géza Horváth, László Szokonya, Nathalie Bosc and Eric Teillaud Particle Formation

41
14. A Plackett–Burman design for screening of the operation variables in the formation of salbutamol sulphate particles by supercritical antisolvent • ARTICLE A. Vatanara, A. Rouholamini Najafabadi, K. Gilani, R. Asgharian, M. Darabi and M. Rafiee-Tehrani 15. Formation of TiO2–polymer composite microparticles by rapid expansion of CO2 saturated polymer suspensions with high shear mixing • ARTICLE Kiyoshi Matsuyama and Kenji Mishima Polymers, Polymer Processing 16. High-pressure phase equilibria in the system linear low density polyethylene + isohexane: Experimental results and modelling • ARTICLE Istvan Nagy, Ryan A. Krenz, Robert A. Heidemann and Theo W. de Loos 17. Effect of material properties and processing conditions on RESS of poly(l-lactide) • ARTICLE Amporn Sane and Mark C. Thies 18. Production of controlled polymeric foams by supercritical CO2 • ARTICLE E. Reverchon and S. Cardea 19. The effect of supercritical CO2 as a reversible plasticizer and foaming agent on the hot stage extrusion of itraconazole with EC 20 cps • ARTICLE Geert Verreck, Annelies Decorte, Koen Heymans, Jef Adriaensen, Dehua Liu, David L. Tomasko, Albertina Arien, Jef Peeters, Patrick Rombaut, Guy Van den Mooter and Marcus E. Brewster
Chemical Reviews: 1999
Uma série de artigos envolvendo fluidos supercríticos foi publicada na edição 99 da
Chemical Reviews, em 1999.
Supercritical Fluids: Introduction (Ryoji Noyori – Nagoya University)
Fluidos supercríticos (SCF) apresentam uma grande oportunidade de descobrir
novos fenômenos químicos não vistos nas fases convencionais. Mesmo tendo sido matéria
de grande interesse desde o século XIX, seu potencial ainda não foi totalmente atingido.
Todo composto estável possui um ponto triplo e um ponto crítico. Todo gás se torna
supercrítico quando comprimido mais do que a pressão crítica (pc) e acima da temperatura
crítica (Tc). As propriedades dos SCF são diferentes das vistas para líquidos e gases
ordinários e são ajustáveis apenas mudando a temperatura e a pressão. Em particular a
densidade e a viscosidade variam drasticamente perto do ponto crítico. O poder de
solvente é muito menor que aqueles de solventes fluidos convencionais, mas isto pode ser

42
usado para gerar clusters moleculares únicos ou agregados em fase homogênea. Tais
fenômenos foram reconhecidos em estudos espectroscópicos, e os mesmos efeitos são
esperados para mudar seletividade e reatividade química.
SCFs oferecem uma série de vantagens técnicas. Suas características foram
exploradas em cromatografia e engenharia química, particularmente nos processos de
separação e extração, mas SCF pode ainda ser usado como meio de reação. Eles formam
uma mistura de única fase com vários reagentes, algumas vezes evitando o passo de limite
de taxa de transferência de massa, e portanto aumentando as taxas de reação. Atualmente,
processamento químico em larga escala enfrenta sério problema com solventes em
conexão com questões ambientais. A regulação do uso de solventes orgânicos perigosos,
tais como hidrocarbonetos clorinados, está se tornando cada vez mais severo e forçando o
desenvolvimento de meios de reação conscientes quanto ao ambiente e econômicos. Esta é
uma matéria de urgência. Dióxido de carbono supercrítico (scCO2), facilmente acessível a
31ºC e 73 atm, tem excelente potencial para atingir este objetivo. É abundante, barato, não
inflamável, não tóxico, e benigno ao ambiente. Tem alta solubilidade para compostos
orgânicos. Embora que sua habilidade de dissolver compostos polares, iônicos, e
poliméricos seja excessivamente limitada, pequenas quantidades de entrainer1 ou um
tensoativo (surfactant2) apropriado alteram dramaticamente o "micro-ambiente",
aumentando enormemente a solubilidade destas substâncias. Compostos perfluorados são
particularmente efetivos para este propósito, expandindo grandemente a aplicabilidade do
scCO2. Esta permutabilidade é ilimitada. Desde que o uso de scCO2 permita fácil
separação de reagentes, catalizadores, e produtos, pode ser eventualmente usado como um
substituto de solventes menos aceitáveis ao meio ambiente. Porém, apesar de uma
variedade de propriedades atrativas, o uso de SCFs para síntese orgânica e polimerização
permanece largamente inexplorado. Outros SCFs além de CO2 também apresentam
propriedades interessantes. Na perspectiva de altos potenciais científicos e tecnológicos
para este tema, o estado da arte está ilustrado por esta edição especial do ChemRev.
Esta edição trata tanto de aspectos fundamentais quanto aplicações. Três dos 11
artigos focam nos aspectos físicos básicos de SCFs que afetam a química molecular.
1 Substância difundida no meio, como pequenas bolhas de ar no concreto para fazer-lhe resistente às variações de temperatura. 2 Substância misturada no líquido a fim de lhe diminuir a tensão superficial.

43
Kajimoto descreve a solvatação em SCFs e seu efeito na transferência de energia e
reatividade química de diversos compostos. Uma perspectiva nas inomogeneidades da
densidade do solvente em SCFs é dado por Tucker, enquanto o comportamento de multi-
fase de fluido em sistemas próximos a CO2 crítico é analisado por Peters e Gauter.
Brennecke e Chateauneuf resumem várias reações orgânicas homogêneas como sondas
mecanicistas em SCF. O próximo tema a ser analisado é catálise. Baiker de forma
abrangente revê a catálise heterogênea em SCF, enquanto Jessop, Ikariya e Noyori
avaliam o progresso da catálise homogênea usando complexos organo-metálicos em SCF,
o qual é detalhado por Darr e Poliakoff. Mas a ciência intrigante em SCF não é limitada a
bem definidas pequenas moléculas, mas seu uso se estende a química de polímeros.
Kendall, Canelas, Young e DeSimone apresentam uma revisão extensiva em polimerização
em SCFs que inclui polimerização pode "chain-growth" e "step-growth", e Kirby e
McHugh tratam com diagramas de fase de misturas polímeros/SCF e com o
comportamento de homopolímeros, copolímeros e fluoropolímeros nestes fluidos não
ortodoxos. Em conexão com esta tecnologia verde, água é outro meio SCF ideal.
Surpreendentemente, diferente de água em condições ambiente, scH2O é relativamente não
polar mas altamente ácido, ainda que necessite de condições extremas (Tc = 374ºC e pc =
218atm). Esta fase é utilizada para decompor resíduos químicos. Um ensaio na síntese e
conversão química em scH2O é dada por Savage. Finalmente, Mesiano, Beckman, e Russel
cobrem biocatálise sobre condições SCF, incluindo parâmetros afetando reações
enzimáticas e aplicações.
Comparado com solventes líquidos convencionais, SCFs não são uma panacéia;
eles têm méritos e desvantagens. Muitas reações químicas feitas melhor em soluções
fluidas ordinárias. Porém, a utilização dos SCFs ainda é uma vertente jovem e
inexplorada. As inomogeneidades locais dos SCFs é um tema da maior importância, no
entanto conhecimento científico nesta área ainda é escassa. Um estudo sistemático do
comportamento molecular em SCF afetando propriedades do estado fundamental e de
transição em reações químicas irá aumentar em muito sua utilidade em ciência e
tecnologia. Eu espero que esta compilação presente provida uma previsão de futuras
direções.

44
Referências
Kobe Steel, LTD. Wikipedia Jasco NIST [1] Wikipedia
[2] http://www.martin.chaplin.btinternet.co.uk/images/phase.gif
[3] http://www.chem.leeds.ac.uk/People/CMR/criticalpics.html
[4] http://www.criticalprocesses.com/SCFmore.html
[5] CRC Handbook
[6] Livro do Mário
[7] http://pessoal.sercomtel.com.br/matematica/medio/polinom/tartaglia.htm
[8] http://www.kobelco.co.jp/eng/p14/sfe01.htm
[9] http://www.elsevier.com/wps/find/journaldescription.cws_home/600250/description


















