
Implementação e validação de uma ... - Estudo Geral: Home · PDF...
81
Implementação e validação de uma metodologia para análise de fibra alimentar Ana Rita Mendes Mestrado em Química Forense Departamento de Química FCTUC Julho 2011
-
Upload
nguyenhuong -
Category
Documents
-
view
219 -
download
0
Transcript of Implementação e validação de uma ... - Estudo Geral: Home · PDF...

Implementação e validação de
uma metodologia para análise de
fibra alimentar
Ana Rita Mendes
Mestrado em Química Forense Departamento de Química
FCTUC
Julho 2011

1

2
Implementação e validação de uma
metodologia para determinação
de fibra alimentar
Ana Rita Mendes
Dissertação apresentada para provas de Mestrado em
Química Forense
Orientador: Prof. Rui Fausto
Co-orientador: Dr.ª Elsa Cancela
Julho 2011
Universidade de Coimbra

3
Índice
Índice 2
Lista de Abreviaturas 5
Resumo 6
Agradecimentos 8
I-Introdução 9
As Fibras Alimentares 12
1-Definição 12
2- Origem, características e composição da fibra alimentar 15
2.1- Origem: Parede Celular 15
2.2- Características 17
2.2.1- Classificação 18
2.3- Composição Química 21
2.3.1- Celulose 21
2.3.2- Hemicelulose 22
2.3.3- Pectinas 22
2.3.4- β-Glicanos 23
2.3.5- Amido Resistente 23
2.3.6- Oligossacarídeos não-digeríveis 24
2.3.7- Outros carbohidratos sintéticos 25
2.3.8- Gomas e mucilagens 26
2.3.9- Lignina 26
2.3.10- Outros componentes 27
II- A Química Forense na Segurança Alimentar 27
1- Aditivos Alimentares 27
2-Rotulagem 31

4
3-Polidextrose e Amido Modificado 33
3.1-Polidextrose 33
3.2-Amido Modificado 33
III- Métodos e Materiais 34
3.1- Cromatografia Líquida de Alta Eficiência (HPLC) 34
3.1.1. Introdução ao método 34
3.1.2. Princípios Teóricos e Instrumentais 35
3.1.3- Materiais usados em HPLC 41
3.2- Espectroscopia de Infravermelho, FTIR 41
3.2.1- Materiais usados em espectroscopia de infravermelho 43
3.3-Espectroscopia de Raman 43
3.3.1- Introdução ao método 43
3.3.2- Princípios teóricos 44
3.3.3- Instrumentação 46
3.3.4- Materiais usados em espectroscopia de Raman 48
IV- Procedimento Experimental Implementação do método AOAC 2009.01
na linha de análise de alimentos da ControlVet 49
4.1- Objectivo e domínio de aplicação 49
4.2- Referências 49
4.3- Resumo do processo 49
4.4- Reagentes 50
4.5- Aparelhos e Utensílios 51
4.6- Técnica 52
4.6.1 - Extracção da matéria gorda para amostras com teor de
gordura superior a 10%. 52
4.6.2 - Toma para análise 53
4.6.3 - Digestão enzimática e filtração 53
4.6.4 – Determinação de fibras de baixo peso molecular 54
4.6.4.1- Filtração 54

5
4.6.4.2- Desionização da amostra 54
4.6.4.2.1- Preparação da coluna de vidro 54
4.6.4.2.2- Desionização da amostra 54
4.7- Cálculos 55
V- Resultados 56
5.1- Resultados obtidos por HPLC 56
5.2- Resultados obtidos por espectroscopia de Infravermelho 70
5.3-Resultados obtidos por espectroscopia de Raman 74
VI- Conclusão 76
VII- Bibliografia 77

6
Lista de Abreviaturas
AOAC - Association of Official Analytical Chemists International
HPLC- Cromatografia Líquida de Alta Eficiência
GC- Cromatografia Gasosa
HPAEC- High-Performance Anion-Exchange Chromatography
HPAEC-PAD- High Performance Anion Exchange Chromatography with Pulsed Amperometric Detection
FAT- Fibra alimentar total
FAI- Fibra alimentar insolúvel
FAZ- Fibra alimentar solúvel
AGCC- Ácidos gordos de cadeia curta

7
Resumo
A determinação e a quantificação do teor de aditivos em alimentos é
fundamental na segurança alimentar, uma vez que estes podem ser
prejudiciais à saúde do consumidor. Sendo assim, todos os produtos
alimentares devem vir rotulados com as percentagens de aditivos que incluem,
sendo a cromatografia líquida de alta eficiência (HPLC) um método adequado
para esse fim. As fibras alimentares, em particular, são também um elemento
de caracterização geral de alimentos, cuja importância forense pode ser
relevante.
Neste trabalho, foi efectuada uma caracterização de oito amostras de
gelado da marca Pingo Doce por HPLC, de forma a quantificar-se a fibra
alimentar sintética (fibra alimentar de baixo peso molecular) e determinar se a
percentagem presente estava de acordo com a rotulada.
O método de análise usado (AOAC 2009.01) foi implementado pela
primeira vez no laboratório da empresa ControlVet durante este trabalho,
expandindo desta forma asa capacidades da empresa nesta área, visto que até
agora estava, este apenas habilitado a efectuar determinação de fibra de alto
peso molecular. O método foi validado obtendo-se o limite de quantificação
igual a 0.2g/ ml.
Nas amostras estudadas, observa-se que as percentagens de fibra de
baixo peso molecular obtidas pelo método aqui implementado se encontravam
dentro dos valores rotulados. Foram ainda realizados estudos por
espectroscopia de infravermelho para quatro das amostras, tendo-se concluído
que 50% das amostras injectadas no cromatógrafo corresponde a fibra de
baixo peso molecular, glucose e maltose. Os restantes 50% correspondem a
outro tipo de substâncias, cuja composição detalhada não foi possível
efectuar, deixando-se aqui campo aberto para futuros estudos.

8
Por último, iniciaram-se ensaios por espectroscopia de Raman das
amostras e padrões, para futura aplicação na caracterização e eventual
quantificação de fibras alimentares.

9
Agradecimentos
Desde já queria agradecer ao Professor Doutor Rui Fausto, por todos
os conhecimentos e ensinamentos transmitidos, por todas as suas palavras
sábias, e por toda a paciência e dedicação demonstrados ao longo deste ano de
trabalho que não é possível descrever num pequeno parágrafo. Desde já o meu
muito obrigado!
Quero agradecer também a todo o grupo de trabalho do Laboratório
de Crioscopia e Bio-espectroscopia Molecular, em especial à Susana.
Obrigado!
Agradeço também ao laboratório de química da ControlVet, em
particular à Dr.ª Ana e à Dr.ª Elsa pelas condições que me facultaram para
desenvolver o meu trabalho na empresa. O meu muito obrigado!
Agradeço ainda a todos os meus amigos, em especial à Tânia…pois
foram eles que me confortaram em momentos de desalento, foi com eles que
me diverti e são eles o meu porto de abrigo. Obrigado!
Como não poderia deixar de ser, quero agradecer à minha mãe, pois
sem ela nada disto era possível. Agradeço-te tudo o que fizeste e continuas a
fazer por mim. Sem Ti nada disto era possível. Muito obrigado!
Agradeço também à minha família, em especial aos meus sobrinhos,
Bernardo, Leonor e Filipa, por todos os sorrisos que me deram em momentos
de tristeza, e que me fizeram continuar a lutar, e a todas as pessoas que
acreditaram sempre em mim e me deram muita força para que nunca
desistisse de lutar pelos meus sonhos!
Agradeço ao meu namorado Ricardo, por todos os momentos
fantásticos que me proporcionaste, mas, acima de tudo, pela paciência que
tiveste comigo, especialmente este ano que não foi nada fácil, pelo teu
carinho, amor e dedicação, mesmo nos momentos mais complicados da minha
vida. Sem Ti nada disto era possível. Muito Obrigado!
A ti Coimbra!!!!

10
I- Introdução
A Química, em todas as suas modalidades, é instrumento de interesse
à área forense, sendo utilizada neste domínio de forma isolada ou associada a
outras ciências, já que uma única investigação num laboratório forense pode
envolver vários tipos de cientistas. A Química Forense é uma subdivisão da
grande área do conhecimento que é a Ciência Forense que se encarrega da
análise, classificação e determinação de elementos ou substâncias encontradas
nos locais de averiguação ou ocorrência de um delito, ou que podem estar
relacionadas a este.
A Química Forense tem vindo largamente a expandir-se, sendo cada
vez mais necessária diante da criminalidade que, ao longo dos anos, tem
evidenciado uma face sofisticada e procedimentos complexos de actuação. É
um meio seguro e eficaz na elucidação de diversos tipos de crimes, que
recorre ao uso de técnicas de diferentes leituras destinadas a esse fim.
Uma das áreas abrangidas pela Química Forense é a segurança
alimentar, na medida em que o uso indevido de aditivos alimentares ou com
rotulação deficiente de composição dos alimentos por exemplo, podem
afectar, por vezes gravemente, a saúde do consumidor. Assim, a Segurança
Alimentar surge actualmente como uma das principais preocupações da
Indústria Alimentar.
Existem hoje em dia regulamentos, leis e directivas múltiplas que
estabelecem critérios no uso de aditivos alimentares, bem como
especificações de rotulagem.
Neste trabalho, foi desenvolvido e implementado, de acordo com as
necessidades do laboratório de química da empresa ControlVet, um método
que permite quantificar o teor de fibra alimentar total (fibras de alto e baixo
peso molecular) presente nos alimentos. Este método (AOAC 2009.01)
assenta na determinação quantitativa das fibras alimentares de alto peso

11
molecular por técnicas enzimáticas e graviméticas e na quantificação das
fibras de baixo peso molecular por cromatografia líquida de alta eficiência
O conceito de fibra alimentar mudou consideravelmente nos últimos
anos. Actualmente, sabe-se que fibra alimentar abrange uma gama muito mais
ampla de substâncias do que se acreditava anteriormente, e que a fibra tem
maior significado fisiológico do que se pensava. Não existe, contudo, uma
definição universalmente aceite de fibra alimentar na Europa, ou no resto do
mundo. Existe, no entanto, consenso de que há necessidade de uma definição
com bases fisiológicas.
A não-digestibilidade no intestino delgado é uma característica
fisiológica de fundamental importância das fibras alimentares. Por isso,
definições recentes de fibra alimentar englobam, além dos polissacarídeos
não-amiláceos, outros carbohidratos não-digeríveis, como o amido resistente e
oligossacarídeos não-digeríveis. Por outro lado, estudos realizados ao longo
das últimas décadas identificaram diversos efeitos fisiológicos da fibra
alimentar, sendo os principais a melhoria no funcionamento do intestino
grosso, a redução do colesterol sanguíneo e a atenuação dos níveis
plasmáticos pós-prandiais de insulina e de glicose. Essas características
fisiológicas foram também incorporadas nas recentes definições de fibra
alimentar.
De acordo com as definições mais recentes, a fibra alimentar consiste
então em polímeros de carbohidratos e em polissacarídeos não-amiláceos que
são, basicamente, componentes da parede celular das plantas. Estes incluem a
celulose, hemiceluloses, hemiglicanos e pectinas, além de outros
polissacarídeos de origem vegetal e de algas, como as gomas e mucilagens.
Outros componentes que se podem incluir neste tipo de substâncias são os
oligossacarídeos não-digeríveis, como a inulina e o amido resistente. As
definições recentes consideram também como constituintes das fibras
alimentares outros carbohidratos que atravessam o intestino delgado sem
sofrer qualquer tipo de alteração, tais como maltodextrinas resistentes,
frutooligossacarídeos e galactooligossacarídeos, e ainda as celuloses

12
modificadas e polímeros de carbohidratos sintetizados, como a polidextrose.
Incluem-se ainda substâncias como a lignina e outras extraídas juntamente
com os polissacarídeos e oligossacarídeos quando da aplicação dos métodos
analíticos de fibra alimentar (por exemplo, ceras, cutina, polifenóis e
fitosteróis). Essas substâncias podem estar naturalmente presentes ou podem
ser sintetizadas.
Nesta tese, adopta-se o conceito de fibra alimentar proposto pelo
Codex Alimentarius [2], e o método para sua determinação proposto pela
AOAC 2009.01 (Association of Official Analytical Chemists International),
que permite determinar a fibra alimentar total de baixo e alto peso molecular
presente nos alimentos.
Os métodos anteriormente existentes apenas quantificavam fibra de
alto peso molecular, deixando a fibra de baixo peso molecular por quantificar.
O método aqui descrito não se encontrava implementado em Portugal, apesar
de já ser utilizado noutros países.
Nesta tese, começa-se por fazer uma breve referência às definições de
fibra alimentar propostas ao longo dos anos. Seguidamente são apresentadas
as origens principais e a composição das fibras alimentares, as suas
características e os seus efeitos fisiológicos. É feita depois referência ao papel
e relevância da química forense na segurança alimentar, onde são
especificados os cuidados a ter no uso de aditivos alimentares, de acordo com
a legislação em uso. Descrevem-se também sucintamente os principais
cuidados a ter na rotulagem de alimentos para que não se induza o
consumidor em erro, evitando assim fraudes.
Finalmente, são apresentados os resultados obtidos neste trabalho, em
particular os relativos à implementação do método de quantificação das fibras
alimentares, bem como o relatório de validação do mesmo.

13
As Fibras Alimentares
1- Definição
A compreensão do significado fisiológico de substâncias definidas
como fibra alimentar e, portanto, do conceito de fibra alimentar, progrediu
consideravelmente ao longo dos últimos dez anos. Tal definição é vital para
os fabricantes de alimentos, para que possam fornecer informação válida e
precisa na rotulagem dos produtos. É também fundamental para a actividade
das entidades reguladoras das alegações de propriedades nutricionais e de
saúde. Essa informação é ainda necessária para os consumidores, que utilizam
as informações nutricionais declaradas nos rótulos dos alimentos e em
materiais associados.
A denominação fibra alimentar foi originalmente adoptada, em 1953,
por Hipsley, com o objectivo de descrever os componentes alimentares
provenientes da parede celular de vegetais [1-6].
Em 1970, Trowell definiu fibra alimentar [1-6] como “os
remanescentes da parede celular vegetal que não são hidrolisados pelas
enzimas digestivas do homem, que inclui celuloses, hemiceluloses, lignina,
gomas, celuloses modificadas, mucilagens, oligossacarídeos, pectinas e
substâncias menores associados, como ceras, cutina e suberina.”
Em 2006, a Comissão do Codex Alimentarius avançou com uma
definição para fibra que estabelece que: “O termo fibra dietética indica
polímeros de carbohidratos com um grau de polimerização (DP) não menor
do que três, os quais não são digeridos nem são absorvidos no intestino
delgado. O grau de polimerização não inferior a três pretende excluir os
mono e dissacarídeos; não pretende reflectir o grau de polimerização médio
de uma mistura. A fibra alimentar consiste em um ou mais:
- polímeros de carbohidratos comestíveis presentes naturalmente nos
alimentos na forma em que estes são consumidos;

14
- polímeros de carbohidratos que foram obtidos de material
alimentício cru por métodos físicos, enzimáticos ou químicos, ou de polímeros
de carbohidratos sintéticos”.
Na maioria dos países, a necessidade de definir precisamente fibra
alimentar resulta da necessidade de fornecer informação rigorosa ao
consumidor na rotulagem. As definições utilizadas baseiam-se em diversos
métodos analíticos adoptados pela Association of Official Analytical Chemists
International, inicialmente o método AOAC 985.29 (tabela 1). Este método é
baseado no conceito de resistência à digestão. O método utiliza a digestão
enzimática para eliminar componentes não fibrosos e a quantificação dos
resíduos por pesagem (portanto, o termo ‘gravimétrico’). Os procedimentos
analíticos e enzimas utilizados seguem critérios rígidos de desempenho e
pureza. O seu uso foi defendido em parte por causa da sua suposta
reprodutibilidade. No entanto, esta característica não foi confirmada por um
estudo de certificação da Comissão Europeia realizado em 1996 [3-6].
Actualmente, para finalidade de rotulagem, é utilizada a definição
proposta pelo Codex Alimentarius [7,8]. Este facto fez surgir a necessidade de
se criar um novo método de quantificação de fibras alimentares que
quantificasse todos os componentes da fibra alimentar, incluindo os
oligossacarídeos não-digeríveis, como a inulina e a polidextrose, e ainda o
amido resistente.
Assim, em 2009 foi desenvolvido um método enzimático-
gravimétrico e de cromatografia liquida de alta eficiência (AOAC 2009.01). A
implementação e validação deste método constituíram a componente principal
do trabalho apresentado nesta tese.

15
Tabela 1- Principais métodos de análise de fibra alimentar.
Nome Tipo Componentes
Fibra alimentar
total
Enzimático-gravimétrico
AOAC 985.29
AOAC 991.43
- Polissacarídeos solúveis
e insolúveis
- Lignina
Método de
Englyst
Enzimático-químico ou
GC ou HPLC
- Polissacarídeos não
amiláceos
Método Upsala
AOAC 994.13
Enzimático-químico -Polissacarídeos solúveis e
insolúveis
- Lignina
AOAC 995.16
AACC 32-33
Enzimático - β-Glicanos
Método de
Englyst para
amido resistente
Enzimático - Amido resistente
AOAC 2002-02
AACC 37.42
Enzimático - Amido resistente
- Fibra de algas
AOAC 999.03 Enzimático e
colorimétrico
- Frutanas
AOAC 997.08 Enzimático e
cromatografia de troca
iónica
- Frutanas
AOAC 2000.11 HPAEC - Polidextrose
AOAC 2001.02 HPAEC-PAD - Galactooligossacarídeos
AOAC 2001.03 Enzimático, gravimétrico
e HPLC
- Fibra Alimentar total em
alimentos contendo
maltodextrinas resistentes
AOAC 2009.01 Enzimático, gravimétrico
e HPLC
-Fibra alimentar total
- Oligissacarideos não
digeriveís
-Amido resistente

16
2- Origem, Características e Composição da
Fibra Alimentar
A principal fonte de fibra dietética é a parede celular vegetal, que
compreende uma série de polissacarídeos frequentemente associados a
proteínas e compostos fenólicos, juntamente com polímeros de lignina.
Figura 2.1- Esquema geral de uma parede celular, origem principal da fibra alimentar
2.1- Origem: Parede Celular
A parede da célula vegetal apresenta uma série de funções essenciais,
tais como conferir rigidez às células vegetais, proteger do ambiente, evitar a
desidratação e permitir a comunicação entre as células [9].
A célula viva segrega a sua parede celular, que compreende três partes
[10] (Figura 2.2):
• Lamela Mediana: é a camada mais externa. Consiste principalmente
de substâncias pécticas e tem uma estrutura coloidal, amorfa e
opticamente inactiva.

17
• Parede Primária: composta principalmente de celulose e algumas
hemiceluloses.
• Parede Secundária: Aparece quando as células envelhecem, de modo
que, neste ponto, termina o seu crescimento. É constituída por
celulose, hemicelulose, substâncias pécticas e lignina.
De acordo com os modelos mais recentes, a parede das células dos
vegetais é composta uma rede de celuloses / xiloglucanos (Figura 2.2) e uma
rede de pectinas, que interagem entre si e, em alguns casos, com uma terceira
rede composta por proteínas estruturais [10]:
• Rede de celuloses / xiloglucanos: a celulose é o principal componente
desta rede; as cadeias de celulose formam microfibrilas, que estão
embutidas dentro de uma matriz de hemiceluloses, principalmente
xiloglucanos e arabinoxilanos.
• Rede de pectinas: as substâncias pécticas constituem uma família de
polímeros e ácidos como o ramnogalacturonano e
homogalacturonanos; unidos a estes polímeros existem outros que são
neutros, como outras arabinanos, galactanos e arabinogalactanos.
• Proteínas estruturais: as proteínas são também parte importante da
parede celular.
Figura 2.2- Modelo da parede celular das dicotiledóneas

18
Embora a natureza da celulose varie muito pouco de uma planta para
outra, a composição da matriz que incorpora a celulose apresenta uma
variação considerável de um tecido para o outro dentro da mesma planta e
entre plantas. Portanto, a composição da parede celular depende não só da
espécie vegetal, mas também do tipo de tecido e da maturidade da planta na
época da colheita [11].
2.2- Características
Os constituintes das fibras alimentares são, na sua maior parte,
substâncias de origem vegetal, predominantemente originadas da parede
celular e, em menor parte, alguns polissacarídeos extraídos de outras partes
das plantas ou sintetizados por microorganismos. Sendo assim, a fibra
alimentar está presente na dieta humana através do consumo de alimentos de
origem vegetal, nos quais está presente normalmente como constituinte
natural, em alimentos em geral onde tenha sido adicionada e na forma de
suplementos alimentares ricos em fibra.
As substâncias constituintes da fibra, conforme a definição adoptada
nesta Tese, estão listadas na Tabela 2. A composição da fibra presente nos
vegetais é bastante variável, dependendo de muitos factores, podendo-se
considerar que esta variação provém principalmente de diferenças [12]:
a) entre diferentes espécies vegetais;
b) entre variedades, dentro da mesma espécie vegetal;
c) entre os diferentes órgãos ou tecidos vegetais, inclusive dentro da
mesma espécie;
d) num mesmo órgão ou tecido, na dependência da fase de
desenvolvimento e/ou maturação vegetal;
e) ocasionadas por condições de armazenamento;
f) ocasionadas por processamentos industriais ou culinários.

19
Tabela 2- Constituintes das Fibras de acordo com a definição do Codex
Alimentarius
2.2.1- Classificação
As fibras podem ser classificadas de acordo com a sua estrutura, a sua
solubilidade em água, e em relação ao seu grau de fermentação [12].
- Solubilidade em água
Segundo a grande maioria dos autores, a fibra alimentar total (FAT) é
classificada de acordo com a sua solubilidade em água em duas fracções: fibra
alimentar solúvel (FAS) e fibra alimentar insolúvel (FAI).
A FAI é a principal fracção (75%) de fibra da grande maioria dos
alimentos, sendo que em alguns alimentos é a única ou quase a única.
Grupos Constituintes
Polissacarídeos não-
amiláceos e
oligossacarídeos
resistentes à digestão
Celulose Hemicelulose Polifrutoses (inulinas e oligofrutanas)- solúveis Galactooligossacarídeos Gomas Mucilagens Pectinas
Carbohidratos análogos
ao primeiro grupo
Dextrinas indigerivéis Carbohidratos sintetizáveis (polidextrose e metilcelulose) Amidos resistentes à digestão
Lignina Lignina Substâncias associadas
com polissacarídeos e
lignina formadores de
fibra nas plantas
Fitatos Cutina Saponinas Taninos

20
Consiste principalmente de constituintes da parede celular das células
vegetais, que incluem a celulose, hemiceluloses e lignina. Apresenta efeito
mecânico no tracto gastrointestinal, é pouco fermentável e acelera o tempo de
trânsito intestinal devido à absorção de água. São encontradas principalmente
em verduras, farelo de trigo e grãos integrais [12-15].
Os seus principais efeitos metabólicos são:
• Aumentam o peso e a maciez das fezes;
• Aumentam a frequência da evacuação e diminuem o tempo de
trânsito no cólon;
• Reduzem a obstipação;
• Retêm água;
• São pouco fermentáveis;
• Aumentam a protecção contra infecção bacteriana.
A FAS consiste de polissacarídeos não-celulósicos, tais como
pectinas, gomas, mucilagens, inulina, FOS (frutooligossacarídeos) e
polifrutoses, grande parte das quais sofrem degradação/fermentação no
intestino grosso. Estas fibras têm a capacidade de se ligar à água e formar
géis. No tracto gastrointestinal, retardam o esvaziamento gástrico e o tempo
de trânsito intestinal, e diminuem o ritmo de absorção de glicose e colesterol.
São substratos para fermentação bacteriana que resultam em gases
(hidrogénio, metano e dióxido de carbono) e ácidos gordos de cadeia curta
(AGCC), importantes para o metabolismo intestinal. São encontradas
principalmente em frutas e verduras, mas também em cereais (aveia e cevada)
e leguminosas (feijão, grão de bico, lentilha e ervilha). Os principais efeitos
metabólicos das fibras solúveis são [12-15]:
• Retardam o esvaziamento gástrico e o trânsito intestinal;
• Alteram o metabolismo intestinal através da produção dos
AGCC;
• Modulam a mobilidade gastrointestinal
• Reduzem a diarreia (aumento na absorção de água);

21
• Promovem o desenvolvimento da mucosa intestinal (íleo e
cólon);
• Proporcionam energia (devido à fermentação) para a mucosa
intestinal;
• Diminuem o pH do cólon;
• Melhoram a protecção contra a infecção (função de barreira,
imunidade);
• Aumentam tolerância à glicose;
• Diminuem os níveis de colesterol total.
- Estrutura
Grande parte das fibras pertence ao grupo dos polissacarídeos, os
quais são muito variáveis física e quimicamente. Normalmente, o que difere
nas fibras em termos de estrutura é essencialmente [12]:
- A quantidade de monossacarídeos;
- O tipo de monossacarídeos na cadeia polimérica;
- A sequência dos monossacarídeos na cadeia;
- A existência ou não de cadeias secundárias;
- O tipo de ligação, alfa ou beta, entre os monossacarídeos.
Assim, a classificação das fibras de acordo com a sua estrutura
subdivide-se em duas categorias: polissacarídeos não amiláceos (definidos
como polissacarídeos com grau de polimerização ≥10) e polissacarídeos
amiláceos.
- Fermentação bacteriana
A fermentação das fibras ocorre nos intestinos por acção das bactérias
anaeróbicas. O tracto gastrointestinal possui mais de 500 espécies diferentes
de bactérias. No cólon, a flora bacteriana consiste quase que totalmente de
bactérias anaeróbias estritas, como Bacteróides, Bifidobacterium, Clostridium

22
e Lactobacillus. O grau de fermentação no cólon é função da composição da
flora intestinal e das características químicas e físicas, ou seja, o tipo de fibra,
a solubilidade, a fonte, e a forma e o tamanho das partículas [13,16].
Os produtos da fermentação bacteriana das fibras são:
• Ácidos gordos de cadeia curta (AGCC): os mais
importantes obtidos por fermentação das hemiceluloses e
pectinas são os ácidos acético, butírico e propiónico; são
removidos do lúmen intestinal por difusão iónica e facilitam a
absorção do sódio e potássio.
• Gases: hidrogénio, metano e dióxido de carbono, que são
excretados por via rectal.
• Energia: utilizada para crescimento e manutenção das
bactérias.
A fermentação das fibras varia de 0% a 90%, e a fibra, só é
considerada fermentável se for no mínimo 60% fermentada. Quanto mais
solúvel for a fibra, maior o seu grau de fermentação (lignina, 0%; celulose,
15% a 60%; hemiceluloses, 56% a 85%; e pectinas, 90% a 95%) [13].
2.3- Composição Química
De acordo com a definição de fibra alimentar adoptada no âmbito
desta tese são descritos seguidamente os seus componentes principais.
2.3.1- Celulose
A celulose (Figura 2.3) é um polissacarídeo linear, não-ramificado,
consistindo apenas em unidades de glicose, com até 10.000 unidades de
glicose por molécula. As moléculas estão intimamente compactadas em forma
de fibras longas, muito insolúveis e resistente à digestão pelas enzimas
humanas. A celulose é o principal componente da parede celular da maioria
dos vegetais estando presente em frutas, hortaliças e cereais. Grande parte da
fibra no farelo de cereais é constituída por celulose. A celulose constitui cerca

23
de um quarto da fibra alimentar nos grãos e frutas e um terço nas hortaliças e
castanhas/nozes [17].
Figura 2.3- Estrutura química da celulose
2.3.2- Hemiceluloses
As hemiceluloses são polissacarídeos que contêm outros açúcares
além da glicose, estando associadas à celulose na parede celular dos vegetais.
As hemiceluloses tanto podem ser polímeros lineares como ramificados,
menores que a celulose; tipicamente, contêm 50-200 unidades de pentoses
(xilose e arabinose) e hexoses (glicose, galactose, manose, ramnose, ácidos
glucurónico e galacturónico). O nome hemicelulose descreve assim um grupo
heterogéneo de substâncias que estão presentes nos alimentos de origem
vegetal nas formas solúvel e insolúvel. Aproximadamente um terço da fibra
alimentar nas hortaliças, frutas, leguminosas e castanhas/nozes consiste de
hemiceluloses [17].
2.3.3- Pectinas
As pectinas são polissacarídeos que são solúveis em água quente e
formam géis depois de frias. São compostas principalmente de cadeias de
ácido galacturónico intercalado com unidades de ramnose, exibindo
ramificações com cadeias de unidades de pentose e hexose. Estão presentes

24
nas paredes celulares e tecidos intracelulares de frutas e hortaliças, sendo
utilizados como agentes gelificantes e espessantes em diversos produtos
alimentares.
Embora as frutas sejam a principal fonte de pectinas, estas substâncias
também representam 15% a 20% da fibra alimentar nas hortaliças,
leguminosas e castanhas/nozes [17].
2.3.4- β-Glicanos
Os β-glicanos são polímeros da glicose. Ao contrário da celulose, as
ligações entre as moléculas de glicose são variáveis. Possuem estrutura
ramificada e são menores que a celulose. Estas propriedades influenciam a sua
solubilidade, favorecendo a formação de soluções viscosas. Os β-glicanos são
importantes componentes do material da parede celular dos grãos de aveia e
cevada, mas estão presentes apenas em quantidades reduzidas no trigo. Os β-
glicanos têm despertado interesse enquanto fibras solúveis [17].
2.3.5- Amido Resistente
O amido e os seus produtos de degradação que não são absorvidos no
intestino delgado dos seres humanos, são conhecidos como amido resistente.
O amido resistente está presente numa ampla variedade de alimentos, em
proporções variáveis. Foram identificadas quatro classes de amido resistente:
amido fisicamente inacessível (RS1), grãos de amido nativo (RS2), amido
cristalizado (RS3) e amido quimicamente modificado (RS4). As leguminosas
constituem uma das principais fontes de RS1, por conterem uma espessa
parede celular que torna o amido inacessível às enzimas. Certos tipos de
amido, como o existente na batata crua e na banana verde, são muito
resistentes à hidrólise enzimática (RS2). Contudo, ao contrário da banana, a
batata é consumida depois de cozida e quase todos os processos de cozimento
promovem a gelatinização do amido. Portanto, a banana verde é a principal
fonte de RS2 na dieta humana. A quantidade de RS2 na banana depende do

25
seu grau de maturação. Outra categoria de RS2 é constituída pelos amidos
ricos em amilose, fontes frequentes de amido resistente industrial. O
cozimento, o arrefecimento e o armazenamento de alimentos sem prévia
desidratação provocam retrogradação (recristalinização) do amido
gelatinizado: RS3. O reaquecimento de, por exemplo, batata fria, pode reduzir
o conteúdo de RS3. Contudo, ciclos repetidos de aquecimento e arrefecimento
aumentam os níveis de RS3 nas batatas [17].
O amido quimicamente modificado (RS4) inclui éteres e ésteres do
amido, amidos com ligações cruzadas e amidos pirodextrinados. As
modificações químicas são a razão para a redução da digestibilidade do amido
no intestino delgado e, portanto, para a formação de RS4. Alguns amidos
quimicamente modificados nos quais a digestibilidade do amido não foi
modificada são utilizados como ingredientes, por exemplo, em alimentos
infantis.
O conteúdo de amido resistente num alimento pode mudar durante o
armazenamento, dependendo da temperatura e do conteúdo de água, e durante
a preparação do alimento. Consequentemente, é impossível uma quantificação
exacta da concentração de amido resistente num alimento por ocasião do seu
consumo.
2.3.6- Oligossacarídeos não-digeríveis
Os oligossacarídeos não-digeríveis com grau de polimerização entre
3-12 ocorrem naturalmente em alimentos de origem vegetal, principalmente
verduras, cereais e frutas. Estas substâncias também podem ser sintetizadas
por processos químicos ou enzimáticos a partir de monossacarídeos e
dissacarídeos, ou por hidrólise enzimática de polissacarídeos. Os
oligossacarídeos não-digeríveis estão incluídos na definição de fibra alimentar
porque, como resultado da sua não-digestibilidade, exibem efeitos fisiológicos
similares aos dos polissacarídeos maiores e seus congéneres. Em geral, os
oligossacarídeos não-digeríveis são altamente fermentáveis, e alguns possuem

26
as propriedades prebióticas. Alguns dos prebióticos mais conhecidos são as
frutanas, que incluem a inulina, e os frutooligossacarídeos ou oligofrutoses,
obtidos pela hidrólise enzimática da inulina de ocorrência natural (com grau
de polimerização 3-60), e seus análogos sintéticos obtidos por síntese
enzimática a partir da sacarose. A cebola, a chicória e a alcachofra são as
principais fontes alimentares de frutanas de ocorrência natural, a partir das
quais são obtidos a inulina e os frutooligossacarídeos [17,18].
2.3.7- Outros carbohidratos sintéticos
Como a própria celulose, os derivados sintéticos da celulose, e.g. a
metilcelulose e a hidroxipropilmetilcelulose, não são digeridos no intestino
delgado. Ao contrário da sua molécula-mãe, essas substâncias são solúveis,
mas dificilmente fermentáveis pela colónia macrobiótica.
A polidextrose (Figura 2.4) é um polissacarídeo ou oligossacarídeo
resistente e é também uma fibra solúvel resistente. É um polímero de
carbohidrato não-digerível, com um grau médio de polimerização de 12,
sintetizado a partir da glicose e do sorbitol, utilizando como catalisador um
ácido orgânico (por exemplo, ácido cítrico). O resultado é uma estrutura
complexa e resistente à hidrólise pelas enzimas digestivas humanas. A
polidextrose é parcialmente fermentada no colo – cerca de 50% em seres
humanos – e tem propriedades prebióticas e de formação de volume [17,18].
Figura 2.4- Estrutura química da polidextrose

27
As dextrinas resistentes são produzidas por calor em pH alcalino e por
tratamento enzimático de amidos, como o do milho e o da batata, resultando
num material com grau de polimerização aproximadamente igual a 15. As
dextrinas resistentes são parcialmente digeridas pelas enzimas digestivas
humanas e parcialmente fermentadas no cólon. Consequentemente, estas
substâncias comportam-se fisiologicamente como fibra alimentar. Os efeitos
prebióticos das dextrinas não foram ainda confirmados [17,18].
2.3.8- Gomas e mucilagens
Os hidrocolóides compreendem uma ampla gama de polissacarídeos
viscosos. Estas substâncias são derivadas de exsudatos vegetais (goma
arábica), sementes (gomas de guar e locust) e de extractos de algas marinhas
(agar, carragena e alginatos). As mucilagens estão presentes em células das
camadas externas de sementes da família “plantain”, por exemplo, isphagula
(Psyllium). Estes hidrocolóides são utilizados em certos alimentos, em
pequenas quantidades, como agentes gelificantes, espessantes, estabilizantes e
emulsificantes [17,18].
2.3.9- Lignina
A lignina não é um polissacarídeo, mas está quimicamente ligada à
hemicelulose na parede celular dos vegetais e, portanto, mantém uma íntima
associação com polissacarídeos da parede celular dos vegetais. A lignina
também influencia a fisiologia gastrointestinal. Está presente em alimentos
possuidores de componente “lenhoso”, como o aipo, e nas camadas externas
dos grãos de cereais [17-19].
Figura 2.5- Estrutura química da lignina

28
2.3.10- Outros componentes
O ácido fítico (hexafosfato de inositol) está associado à fibra em
alguns alimentos, sobretudo nos grãos de cereais. Os seus grupos fosfatos
ligam-se muito fortemente com iões positivamente carregados como o ferro, o
zinco, o cálcio e o magnésio, podendo interferir na absorção dos minerais.
Outros constituintes dos vegetais associados à fibra alimentar são, por
exemplo, polifenóis (taninos), cutinas e fitosteróis [17-19].
II- A Química Forense na Segurança Alimentar
No contexto desta Tese seguiu-se o Regulamento (CE) nº 1333/2008,
de 16.12.08 referente ao uso de aditivos alimentares e, em questões de
rotulagem, a Directiva 2000/12/CE de 20 de Março de 2000, uma vez que
uma das fibras quantificadas neste trabalho (polidextrose) é considerada um
aditivo alimentar.
1- Aditivos Alimentares
De acordo com o regulamento atrás referido, são considerados
aditivos alimentares as substâncias que não são consumidas habitualmente
como géneros alimentícios em si mesmas, mas que são intencionalmente
adicionadas aos géneros alimentícios para atingir determinado objectivo
tecnológico, como, por exemplo, a conservação dos géneros alimentícios.
Todos os aditivos alimentares deverão ser abrangidos pelo regulamento citado
acima e, por conseguinte, a lista das classes funcionais deverá ser actualizada
à luz do progresso científico e dos avanços tecnológicos. Contudo, não
deverão ser consideradas aditivos alimentares as substâncias cuja utilização

29
tenha por objectivo conferir determinado aroma e/ou sabor ou tenha fins
nutricionais, tais como os sucedâneos do sal, as vitaminas e os minerais. Além
disso, as substâncias consideradas géneros alimentícios que podem ser
utilizadas com um objectivo tecnológico, tais como o cloreto de sódio ou o
açafrão — utilizado para conferir cor — assim como as enzimas alimentares,
também não deverão ser abrangidas pelo âmbito do presente regulamento. No
entanto, deverão ser consideradas aditivos, as preparações obtidas a partir de
géneros alimentícios ou de outros materiais de base naturais e que se destinem
a ter um efeito tecnológico a nível do produto final e sejam obtidas por
extracção selectiva de componentes (por exemplo, pigmentos) em relação aos
componentes nutritivos ou aromáticos.
Os aditivos alimentares só podem ser autorizados e utilizados se
preencherem os critérios definidos no regulamento. A utilização dos aditivos
alimentares deve ser segura, deve decorrer de uma necessidade tecnológica,
não deve induzir o consumidor em erro e deve ser vantajosa para o
consumidor. Induzir o consumidor em erro inclui, por exemplo, as alegações
relacionadas com a qualidade dos ingredientes utilizados, com o carácter
natural de um produto ou do modo de produção, com o valor nutricional do
produto, incluindo o seu teor de frutos e legumes. A aprovação de aditivos
alimentares requer igualmente a consideração de outros factores pertinentes
para a matéria em apreço, incluindo os factores societais, económicos,
tradicionais, éticos e ambientais, o princípio da precaução e a viabilidade dos
controlos. O uso e os níveis máximos de um aditivo alimentar têm de ter em
conta a dose do aditivo alimentar proveniente de outras fontes e a exposição
ao mesmo a que estão sujeitos grupos especiais de consumidores (por
exemplo, consumidores alérgicos).
Os aditivos alimentares estão classificados da seguinte forma:
1. Edulcorantes: substâncias utilizadas para conferir um sabor doce
aos géneros alimentícios ou utilizadas nos edulcorantes de mesa.
2. Corantes: substâncias que conferem ou restituem cor a um género
alimentício; incluem componentes naturais de géneros alimentícios e

30
substâncias naturais que normalmente não são consumidos como géneros
alimentícios em si mesmos nem utilizados como ingredientes característicos
dos géneros alimentícios. São consideradas corantes as preparações obtidas a
partir de géneros alimentícios ou de outros materiais de base naturais
comestíveis obtidas por extracção física e/ou química de modo a provocar a
extracção selectiva dos pigmentos em relação aos componentes nutritivos ou
aromáticos.
3. Conservantes: substâncias que prolongam o prazo de conservação
dos géneros alimentícios, protegendo-os contra a deterioração causada por
microrganismos e/ou contra o desenvolvimento de microrganismos
patogénicos.
4. Antioxidantes: substâncias que prolongam o prazo de conservação
dos géneros alimentícios, protegendo-os contra a deterioração causada pela
oxidação, tal como a rancidez das gorduras e as alterações de cor.
5. Agentes de transporte: substâncias utilizadas para dissolver,
diluir, dispersar ou de outro modo modificar fisicamente um aditivo alimentar,
um aroma alimentar, uma enzima alimentar, um nutriente e/ou outra
substância adicionada a um género alimentício para efeitos nutricionais ou
fisiológicos sem alterar a sua função (e sem que elas próprias exerçam
quaisquer efeitos tecnológicos), a fim de facilitar o respectivo manuseamento,
aplicação ou utilização.
6. Acidificantes: substâncias que aumentam a acidez dos géneros
alimentícios e/ou lhes conferem um sabor acre.
7. Reguladores de acidez: substâncias que alteram ou controlam a
acidez ou a alcalinidade dos géneros alimentícios.
8. Antiaglomerantes: substâncias que reduzem a tendência das
partículas isoladas dos géneros alimentícios para aderirem umas às outras.
9. Antiespumas: substâncias que impedem ou reduzem a formação de
espuma.

31
10. Agentes de volume: substâncias que contribuem para dar volume
aos géneros alimentícios sem contribuírem significativamente para o seu valor
energético disponível.
11. Emulsionantes: substâncias que tornam possível a formação ou a
manutenção de uma mistura homogénea de duas ou mais fases imiscíveis,
como óleo e água, nos géneros alimentícios.
12. Sais de fusão: substâncias que convertem as proteínas contidas no
queijo numa forma dispersa, daí resultando uma distribuição homogénea das
gorduras e outros componentes.
13. Agentes de endurecimento: substâncias que tornam ou mantêm
firmes ou estaladiços os tecidos dos frutos ou dos produtos hortícolas, ou
actuam em conjunto com gelificantes para produzir ou reforçar um gel.
14. Intensificadores de sabor: substâncias que intensificam o sabor
e/ou o cheiro dos géneros alimentícios.
15. Espumantes: substâncias que tornam possível a dispersão
homogénea de uma fase gasosa nos géneros alimentícios líquidos ou sólidos.
16. Gelificantes: substâncias que dão textura aos géneros alimentícios
através da formação de um gel.
17. Agentes de revestimento (incluindo lubrificantes): substâncias
que, quando aplicadas na superfície externa dos géneros alimentícios, lhes
conferem uma aparência brilhante ou um revestimento protector.
18. Humidificantes: substâncias que impedem os géneros
alimentícios de secar por contrabalançarem o efeito de uma atmosfera com
baixo grau de humidade, ou promovem a dissolução de um pó num meio
aquoso.
19. Amidos modificados: substâncias obtidas através de um ou mais
tratamentos químicos de amidos comestíveis, que podem ter sofrido um
tratamento físico ou enzimático e podem ser fluidificadas por via ácida ou
alcalina ou branqueadas.

32
20. Gases de embalagem: gases, com excepção do ar, introduzidos
em recipientes antes, durante ou após a colocação dos géneros alimentícios
nesses recipientes.
21. Propulsores: gases, com excepção do ar, que expelem os géneros
alimentícios dos recipientes.
22. Levedantes químicos: substâncias ou combinações de
substâncias que libertam gás, aumentando assim o volume das massas ou
polmes de farinha.
23. Sequestrantes: substâncias que formam complexos químicos com
iões metálicos.
24. Estabilizadores: substâncias que tornam possível a manutenção
do estado físico-químico dos géneros alimentícios. Os estabilizadores incluem
as substâncias que permitem a manutenção de uma dispersão homogénea de
duas ou mais substâncias imiscíveis nos géneros alimentícios, as substâncias
que estabilizam, retêm ou intensificam a cor natural dos géneros alimentícios
e as substâncias que aumentam a capacidade de aglomeração do género
alimentício, incluindo a formação de ligações cruzadas entre proteínas que
permitem a aglomeração dos elementos alimentares para a formação de um
género alimentício reconstituído.
25. Espessantes: substâncias que aumentam a viscosidade dos
géneros alimentícios.
26. Agentes de tratamento da farinha: substâncias, com excepção
dos emulsionantes, adicionadas à farinha ou à massa para melhorar a
qualidade da cozedura.
2- Rotulagem
De acordo com a directiva mencionada anteriormente entende-se por
rotulagem as menções, indicações, marcas de fabrico ou de comércio,
imagens ou símbolos referentes a um género alimentício e que figurem em

33
qualquer embalagem, documento, aviso, rótulo, anel ou gargantilha que
acompanhe ou seja referente a este género alimentício.
Entenda-se por género alimentício a unidade de venda destinada a ser
apresentada como tal ao consumidor final e às colectividades, constituída por
um género alimentício e pela embalagem em que foi acondicionado, antes de
ser apresentado para venda, quer a embalagem o cubra na totalidade ou
parcialmente, mas de tal modo que o conteúdo não possa ser alterado sem que
a embalagem seja aberta ou alterada.
Segundo esta directiva todos os ingredientes presentes no alimento
devem vir especificados no rótulo nas proporções presentes no alimento. Esta
regra aplica-se tanto aos componentes naturais dos alimentos como aos
aditivos. Assim, e com uma legislação mais rígida, uma vez que os aditivos
alimentares quando consumidos em demasia são prejudiciais para a saúde
humana, estes têm regras de rotulagem, sendo a mais relevante a de nele ter de
vir indicado o nome. Por outro lado, os aditivos têm de ser fabricados em
conformidade com as boas práticas e as suas dosagens indicadas nos rótulos
para que o consumidor não seja induzido em erro.
Figura 3.1- Rotulagem de alimentos

34
3- Polidextrose e Amido Modificado
3.1- Polidextrose
A polidextrose, a que já me referi anteriormente, além de ser uma
fibra sintética de baixo peso molecular, é classificada nos termos referidos
como sendo um aditivo alimentar, uma vez que pode ser usada como agente
de volume e de textura, humidificante, estabilizador e espessante. Assim, tem
de obedecer tanto aos critérios de rotulagem bem como aos dos aditivos
alimentares. Logo, o seu nome tem de vir especificado no rótulo bem como a
sua quantidade.
3.2- Amido Modificado
O mesmo acontece com o amido modificado, uma vez que este
também é obtido por tratamentos químicos de amidos comestíveis, que podem
ter sofrido um tratamento físico ou enzimático e podem ser fluidificados por
via ácida ou alcalina ou branqueados. Pode mesmo não ser de origem vegetal
e conter glúten, acabando por poder ser prejudicial à saúde do consumidor
afectado por alergia a esta ultima substância.

35
III- Materiais e Métodos
3.1- Cromatografia Líquida de Alta Eficiência
(HPLC)
3.1.1. Introdução ao método
A cromatografia, em todas as suas variantes, é um método de
separação de componentes de uma mistura em que a separação depende da
distribuição das diferentes moléculas entre duas fases: uma fase estacionária e
uma fase móvel. Os métodos cromatográficos classificam-se de acordo com a
natureza das fases estacionária e móvel, os seus estados físicos e os
mecanismos de separação [20].
Em cromatografia líquida, a fase móvel é um líquido que escoa ao
longo da fase estacionária numa direcção definida. Os compostos que são
fracamente retidos pelo sorbente da fase estacionária escoam mais
rapidamente ao longo do enchimento, enquanto os que estabelecem
interacções mais fortes com a fase estacionária saem mais lentamente, até à
separação completa dos componentes da mistura.
O processo de escoamento dos compostos, que são arrastados pela
fase móvel ao longo da coluna até à saída, designa-se eluição [21]. O detector
regista o resultado na forma de um cromatograma, tal como o mostrado na
Figura 3.1.
Assim, um cromatograma é a resposta do detector na forma de
gráfico, representando a concentração de analito em função do tempo ou do
volume de eluição.

36
Figura 3.1- Esquema geral de um cromatograma, mostrando dois picos. Vêr texto
para significado dos símbolos apresentados na figura.
De um cromatograma podem obter-se vários parâmetros que
caracterizam a separação (ver Figura 3.1):
� tM (tempo morto da coluna): também conhecido como tempo
extra da coluna, é o tempo que demora o soluto que não foi
retido na coluna para ser eluído, isto é, um composto que é
insolúvel ou não adsorvido na fase estacionária.
� tR (tempo de retenção): é o tempo que demora um soluto a ser
totalmente eluído da coluna. Na Figura 3.1 estão
representadas por tR1 e tR2 as respostas para dois solutos
diferentes.
� w50 : largura a meia altura do pico de eluição
� wb (largura do pico): resultante da intersecção das duas
tangentes aos pontos de inflexão do pico
3.1.2. Princípios Teóricos e Instrumentais
A cromatografia líquida de alta eficiência (HPLC, High Perfomance
Liquid Chromatography) foi inventada na década de 60 do século XX. É uma
extensão da cromatografia líquida clássica e é caracterizada pelo uso de
colunas em aço inox muito mais estreitas, com diâmetro interno de 2-5 mm,
empacotadas com partículas de tamanho muito pequeno, 3-10 µm, que
constituem a fase estacionária. A fase móvel circula sob alta pressão ao longo

37
da coluna, com um fluxo controlado. A alta pressão permite análises rápidas,
e o uso de fases estacionárias constituídas por micro-partículas permite uma
elevada eficiência na separação [20].
A técnica de HPLC revelou-se um dos mais eficientes métodos
cromatográficos, também em virtude do desenvolvimento de instrumentação
automatizada. Permitiu a injecção de volumes de amostra cada vez mais
pequenos e reprodutíveis, e a detecção de quantidades de analito cada vez
menores, em sistemas de detecção em fluxo, que indicam quando os
componentes sofrem eluição da coluna.
Esta técnica veio complementar a cromatografia de gases (GC) no
tipo de compostos separados. Apresenta também a vantagem de conduzir a
tempos de retenção mais curtos e permitir assim, um maior número de
análises por unidade de tempo. A introdução de colunas capilares, ofereceu
um maior número de pratos teóricos e uma melhor resolução do que as
colunas padrão. Outra vantagem que esta técnica apresenta é a menor
quantidade de fase móvel, o que permite utilizar solventes tóxicos, raros, ou
caros, bem como o uso de fases estacionárias dispendiosas [20].
Um sistema de HPLC consiste em quatro componentes principais:
uma bomba, um sistema de injecção, uma coluna de separação e um detector,
todos conectados numa instalação resistente a altas pressões (Figura 3.2), que
podem ir até 300 atm.
Figura 3.2- Esquema representativo de Cromatografia líquida

38
Em geral, a técnica de HPLC é um processo dinâmico onde as
moléculas dos analitos se movem através de um enchimento poroso, por acção
da fase móvel bombeada continuamente, e interagem com diferentes
afinidades com o material da fase estacionária.
Consoante as características específicas do sistema experimental, a
cromatografia líquida de alta eficiência pode ser classificada como
cromatografia de fase normal (HPLC-NP) ou cromatografia de fase reversa
(HPLC-RP).
Na cromatografia em fase normal, é usada uma fase estacionária polar
(normalmente sílica) para a retenção de analitos polares. As separações em
fase reversa baseiam-se em forças atractivas entre solutos não polares e
grupos funcionais não polares ligados a um suporte de sílica.
A maioria das separações são levadas a cabo por HPLC de fase
reversa, pois, ao contrário de HPLC de fase normal, existem muitos poucos
compostos que sejam retidos permanentemente na coluna. As colunas tendem
também a ser mais robustas [22].
Para um soluto ser retido por um sistema cromatográfico, deve ser
transferido da fase móvel para a fase estacionária. A associação do soluto com
a fase estacionária pode envolver partição, adsorção ou a combinação de
ambas.
Num processo por partição, o soluto está praticamente todo embebido
na fase estacionária, enquanto num processo por adsorção o soluto apenas está
em contacto superficial com a fase estacionária e não totalmente embebido
nela. Esses dois processos foram usados para desenvolver as duas teorias
principais que descrevem o mecanismo de retenção molecular em HPLC-RP
[23].
Na fase reversa existe uma relação exponencial entre o factor de
retenção k e a fracção do volume do solvente orgânico na fase móvel. Um erro
de 1% na quantidade de solvente orgânico pode variar o tempo de retenção
(tR) entre 5% e 15% [24].

39
Quando a composição da fase móvel é alterada gradualmente ao longo
da eluição cromatográfica, por aumento e posterior diminuição de
percentagem de solvente orgânico, fala-se de uma eluição em gradiente. Nos
casos em que a composição da fase móvel se mantém constante durante toda a
análise, o tipo de eluição é conhecido como isocrático.
Pode dizer-se que, nas cromatografias de fase reversa, os compostos
apolares ou fracamente polares são mais fortemente retidos na fase
estacionária do que na fase móvel. A retenção de um composto na fase
estacionária é tanto maior quanto menor é a sua solubilidade na fase móvel.
Um detector de HPLC deve seguir os seguintes requisitos [25]:
• Suficiente sensibilidade
• Limite de detecção suficientemente baixo
• Amplo intervalo dinâmico linear
• Suficiente estabilidade e reprodutibilidade do sinal
• Resposta do sensor rápida
• Ligação entre a coluna e o detector curta e directa
• Detector com volume tão pequeno quanto possível
Existem outras características que afectam a aplicabilidade do
detector, por exemplo, se é necessário que o detector mostre a mesma
sensibilidade para todos os solutos detectados, e que o sinal seja o menos
possível influenciado pela temperatura, a velocidade do fluxo e a composição
da fase móvel.
O detector refractivo é um dos menos sensíveis em HPLC. É muito
sensível às mudanças de temperatura, às mudanças de pressão, às alterações
na taxa de fluxo e não pode ser usado para a eluição de gradiente. Apesar
destas desvantagens, este detector é extremamente útil para detectar
compostos que são não iónicos, que não absorvem no UV, e que não
fluorescem. Existem muitos sistemas ópticos utilizados em detectores
refractivos, mas um dos mais comuns é o detector de índice de refracção
diferencial, representado esquematicamente na Figura 3.3.

40
Figura 3.3- Esquema representativo do detector de índice de refracção diferencial
O refractómetro apresentado na Figura 3.3 mostra a deflexão de um
feixe de luz causada pela diferença do índice de refracção entre o conteúdo da
célula da amostra e da célula de referência. Um feixe de luz (obtido
geralmente a partir de uma lâmpada incandescente) passa através de uma
máscara óptica que limita a iluminação à região da célula. Uma lente colima o
feixe de luz que passa através de ambas as células (da amostra e referência) e
o fazem incidir num espelho plano. O espelho reflecte o feixe de volta através
das células da amostra e referência, após o que o feixe é focado por uma lente
numa célula fotoeléctrica. A localização do feixe, ao invés da sua intensidade,
é determinada pelo desvio angular do feixe resultante da diferença do índice
de refracção entre o conteúdo das duas células. O sinal é então
electronicamente modificado para fornecer um sinal proporcional à
concentração de solutos na célula da amostra [25].
O detector de índice de refracção é quase sempre uma "escolha de
último recurso" e está marcado para as aplicações onde, por uma razão ou
outra, todos os outros detectores são inadequados ou impraticáveis. No
entanto, o detector possui uma área de aplicação específica onde é utilizado
correntemente: a separação e análise de polímeros. Para os polímeros que

41
contêm mais de dez unidades de monómero, o índice de refracção é
directamente proporcional à concentração do polímero e é praticamente
independente da massa molecular. Uma análise quantitativa de uma mistura
de polímero pode, portanto, ser obtida pela normalização simples das áreas
dos picos no cromatograma. Algumas especificações típicas para o detector de
índice de refracção são as seguintes:
Sensibilidade 1x10-6 g/ml
Intervalo dinâmico linear 1x10-6 a 1x10
-4 g/ml
Factor resposta 0.97 – 1.03
Uma aplicação típica do detector de índice de refracção é a análise de
carbohidratos. Os carbohidratos não absorvem no UV, não se ionizam e,
apesar de poderem ser produzidos derivados fluorescentes, o procedimento é
complexo e demorado. Um exemplo de aplicação deste tipo de sistema de
detecção está representado na Figura 3.4, onde se apresenta a separação dos
produtos da hidrólise da β-ciclodextrina.
Figura 3.4- Cromatograma de separação dos produtos de hidrólise da β-ciclodextrina utilizando
um detector de índice de refracção [25]

42
3.1.3- Materiais usados em HPLC
Para a realização deste trabalhado, foi usado um equipamento de
HPLC da marca Shimadzu, acoplado a um detector refractivo da mesma
marca, com as seguintes características: temperatura de 60 ºC, fluxo 0.5
mL/min, coluna da marca Macherey- Nagel Nucleogel Sugar 810 CA Va
300/7.8 e fase móvel constituída por uma solução de água destilada com
Na2CaEDTA (50 mg/L). Foi ainda usada, numa fase inicial de
experimentação, uma coluna da marca Waters Carbohydrate Analyses de
3.9x300 mm destinada à análise de açúcares.
Foram obtidos os cromatogramas de oito amostras de gelados da
marca Pingo Doce de três sabores, morango, baunilha e chocolate.
3.2- Espectroscopia de Infravermelho, FTIR
A espectroscopia estuda a interacção da radiação electromagnética
com a matéria, sendo um dos seus principais objectivos o estudo dos níveis de
energia de átomos ou moléculas.
A espectroscopia de absorção na região do infravermelho permite a
identificação de uma substância orgânica pelos grupos funcionais presentes no
material em análise. Baseia-se na medida da energia absorvida para excitação
vibracional dos diferentes modos vibracionais de uma molécula. A amostra é
submetida a uma radiação de comprimento de onda na região do
infravermelho e a faixa de radiação utilizada situa-se, em geral, entre os 4000
os 400 cm-1.
Uma técnica importante no estudo de sistemas moleculares complexos
é a espectroscopia de infravermelho por transformada de Fourier. Esta técnica
baseia-se no método interferométrico de Michelson. (Figura 3.5) A energia
da fonte de radiação é dirigida para o divisor do feixe, que gera dois percursos
ópticos ortogonais, em que um colide num espelho fixo e outro num espelho

43
móvel. Tanto o espelho móvel como o espelho fixo reflectem novamente a
respectiva radiação para o divisor do feixe onde se recombinam.
Figura.3.5.Instrumentação típica para espectroscopia IR usando transformada de Fourier.
Este feixe de luz recombinado gera uma interferência que, consoante
o valor do deslocamento do espelho móvel, se manifesta de forma
opticamente construtiva ou destrutiva, seguindo depois em direcção do
detector.
Quando o espelho móvel do interferómetro se desloca num certo
espaço de tempo faz surgir diferente padrão e interferência que são
observadas no detector como bandas ou espaços negros. É, assim, registado
um interferograma, que, matematicamente, é uma soma de funções cosseno
que descrevem a intensidade da radiação em função do tempo ou do espaço
percorrido,
I(δ)=∫ I(ν[1+cos(2πδν)]δν (3.2.1)
onde I(δ) é a intensidade do sinal no detector; I(ν) é a intensidade da radiação
como função do tempo da frequência (número de onda, (ν)) e δ é o valor da
retardação óptica. Para determinar a partir do interferograma a função
intensidade da radiação vs o número de onda, calcula-se a Transformada de
Fourier do interferograma, a partir da qual se pode obter o espectro;

44
I(ν)=4∫ [I(δ)-1/2I(δ =0)](cos(2πνδ)dδ (3.2.1)
Quando o composto não está presente, o espectro obtido é o
característico do material óptico do espectrómetro e das condições ambientais.
Quando o composto está presente, por subtracção do espectro anterior, obtêm-
se o espectro de infravermelho característico do composto [26].
3.2.1- Materiais usados em espectroscopia de
Infravermelho
Os espectros de Infravermelho foram obtidos num espectrofotómetro
da marca Bomem (MB104), com resolução de 2 cm-1 e óptica de ZnSe.
Obtiveram-se os espectros de quatro amostras de gelado de diferentes
sabores, bem como o de glucose, maltose e de uma mistura padrão de
oligossacarídeos (LCRT), usando o método da pastilha em brometo de
potássio, tendo-se registado as bandas características dos referidos compostos,
à temperatura ambiente, entre 4000 e 550 cm-1.
3.3-Espectroscopia de Raman
3.3.1 Introdução ao método
Quando um feixe de luz monocromática é difundido pelas moléculas
de um composto, verifica-se que uma pequena parte da luz difundida tem uma
frequência que difere da radiação incidente. Tal fenómeno é chamado efeito
Raman.
Desde a sua descoberta, em 1928, o efeito Raman tornou-se a base de
um método importante de elucidação de estruturas moleculares, de localização
de diversos grupos funcionais ou ligações químicas na molécula, e de análise
quantitativa de misturas complexas, em particular no que respeita aos seus
componentes maiores. Embora os espectros de Raman estejam relacionados

45
com os espectros de absorção no infravermelho, o seu mecanismo de
produção é totalmente diferente, o que permite obter informação
complementar. As vibrações activas em Raman podem ser inactivas no
infravermelho, e vice-versa. Um aspecto importante da difusão Raman é o
facto de cada sinal espectroscópico ter uma polarização característica. Esta
particularidade constitui um elemento adicional de análise, também
relacionado com a estrutura molecular do composto, em particular com a
simetria das vibrações e com a simetria global da molécula.
3.3.2- Princípios teóricos
Observa-se o efeito Raman quando um feixe intenso de radiação
monocromática, proveniente, por exemplo, de um laser, incide numa amostra
que contém moléculas susceptíveis de sofrerem uma alteração na sua
polarizabilidade molecular à medida que vibram.
A maior parte das colisões dos fotões incidentes com as moléculas da
amostra são elásticas (difusão de Rayleigh). Porém, cerca de uma em cada
milhão de colisões é inelástica, e envolve uma troca quantificada de energia
entre a molécula e o fotão incidente, o que dá origem a sinais
espectroscópicos distintos da banda correspondente à radiação excitadora. As
frequências destes sinais estão relacionadas com as frequências de vibração
específicas do material. De acordo com a interpretação do fenómeno Raman à
luz da Mecânica Quântica, o fotão incidente eleva a molécula difusora até a
um estado vibracional virtual, com um valor acima do nível inicial igual à
energia da radiação excitadora, Figura 3.5. No regresso a um nível de menor
energia, um quantum de energia vibracional pode permanecer associado à
molécula, de onde resulta um decréscimo da frequência da radiação re-
emitida. Se a molécula se encontra já num nível vibracional excitado, pode
ceder um quantum de energia vibracional, transitando, como resultado do
processo global, para um nível de energia inferior, enquanto aumenta a
frequência de radiação difundida. Em qualquer dos casos, o desvio da

46
frequência da radiação difundida de Raman varia proporcionalmente à energia
vibracional que intervêm na transição. Assim, o espectro de Raman ocorre sob
a forma de uma serie de bandas, situadas simetricamente acima e abaixo da
frequência da radiação incidente, segundo um modelo característico da
molécula. O desvio é independente da frequência da luz incidente, mas a
intensidade da radiação difractada varia inversamente à quarta potência dessa
frequência.
Figura 3.6- Transições vibracionais em Espectroscopia de Raman
As bandas habitualmente estudadas são as que se situam na zona de
menores frequências, as chamadas bandas de Stokes, as quais são mais
intensas do que as de frequências superiores à da radiação excitadora (bandas
de anti-Stokes). Por convenção, a posição das bandas de Raman são expressas
em números de onda (cm-1).
Observam-se certos inconvenientes na técnica de Raman. Assim, a
frequência da radiação excitadora tem de ser inferior às que correspondem às
transições electrónicas da molécula, para que não ocorra qualquer absorção da
luz incidente por alteração o estado electrónico. Por vezes, a amostra emite
fluorescência a uma destas frequências de excitação, podendo essa
fluorescência ser intrínseca à substância em estudo ou devida à presença na
amostra de uma impureza, ainda que em pequenas quantidades. A
fluorescência, com uma intensidade em geral muito maior à do efeito Raman,
pode obscurecer completamente o espectro de Raman. A frequência da

47
radiação excitadora é então escolhida de forma que se situe abaixo da maior
parte das transições electrónicas S→S*, e acima da maior parte das
frequências vibracionais fundamentais [27].
3.3.3- Instrumentação
Os espectrómetros de Raman combinam, em geral, uma fonte de
radiação, um dispositivo de amostragem, um sistema de dispersão ou selecção
de radiação e um detector de radiação interfaciado com equipamento para
registo e processamento do sinal.
Num espectrómetro dispersivo com rede de difracção, a radiação é
monocromatizada com base numa rede de difracção, Figura 3.6. Uma rede de
difracção é um dispositivo óptico que contém um grande número de estrias
paralelas, produzidas mecânica ou fotograficamente no material suporte, e que
dispersa a radiação por interferência.
Para além do elemento dispersivo, têm-se ainda outros elementos
necessários para gerar, transportar e detectar radiação: fonte de radiação
adequada, espelhos, colimadores e fibras ópticas, detectores de radiação e
conversores de radiação em sinais eléctricos, e ainda conversores analógico-
digitais e sistemas informáticos.
Os espectrómetros de Raman exigem fontes de radiação muito intensa
e monocromática. Sendo assim, utilizam-se lasers (de plasma, de estado
sólido ou de corantes - consoante a frequência de excitação desejada). O laser
de plasma mais vulgarmente utilizado é o de árgon, cujas frequências
dominantes de emissão correspondem ao comprimentos de onda λ= 488 e λ=
515 nm.
Quanto aos detectores, estes são dispositivos que têm como finalidade
essencial converter uma propriedade associada a uma radiação numa carga,
corrente ou potencial eléctrico. Uma vez o sinal gerado, este pode ser
utilizado directamente para traçar um espectro, ou digitalizado para
armazenamento e posterior processamento analítico em sistemas informáticos.

48
Em espectroscopia de Raman, porque os sinais originais são intrinsecamente
muito pouco intensos, a detecção convencional exige a utilização de
fotomultiplicadores (em geral arrefecidos termoelectricamente), dispositivos
que amplificam o número de fotões incidentes, através de uma reacção em
cadeia em que o elemento iniciador são os próprios fotões, de forma a
produzir um fluxo electrónico suficientemente intenso para gerar um sinal
eléctrico significativo. Uma alternativa ao uso de fotomultiplicadores consiste
na utilização de múltiplos detectores miniatura (rede de fotodíodos), dispostos
numa matriz. Este arranjo permite que o registo de um espectro se possa fazer
de uma só vez, após separação espacial das diferentes frequências que o
compõem, enquanto na detecção convencional se torna necessário recolher os
dados individualmente para cada valor de frequência analisada. O registo
sequencial de um grande número de espectros assim obtidos para a mesma
amostra, seguido da sua adição, traduz-se num aumento da relação sinal-
ruído, sem acarretar um aumento do tempo total de obtenção do espectro [28].
Figura 3.7- Esquema geral simplificado de um Espectrómetro de Raman dispersivo

49
3.3.4- Materiais usados em espectroscopia de Raman
Para a realização deste trabalhado, foi usado um espectrómetro de
Raman com as seguintes características: comprimento de onda do laser 780
nm, potencia do laser (máx 14 mW) 10.0 mW, abertura da fenda 50 µm, com
uma resolução estimada 10.1 – 18.6 cm-1.
Foram obtidos os espectros de glucose, de uma mistura padrão de
oligossacarídeos (LCRT) e de uma amostra de gelado da marca Pingo Doce
de sabor a morango.

50
IV- Procedimento Experimental
Implementação do método AOAC 2009.01 [2]
na linha de análise de alimentos da ControlVet
4.1- Objectivo e domínio de aplicação
O método quantifica fibras alimentares totais incluindo amido
resistente (RS) e oligossacarídeos não digeríveis com DP≥3.
4.2-Referências
Este método resulta da combinação de vários métodos da AOAC,
nomeadamente os 985.29, 991.43, 2001.03 e 2002.02.
4.3-Resumo do processo
As amostras são incubadas durante 16 h, em frascos de 100 mL, com
α-amilase pancreática e amiloglucosidase, a 37 ºC, em banho vibratório.
Durante este passo, o amido não resistente é solubilizado e hidrolisado a
glucose e maltose, por combinação da acção das duas enzimas.
A reacção termina com o ajuste do pH a 8.2 e aquecimento. A
proteína na amostra é digerida pela protease. Para a quantificação de fibras de
alto peso molecular, é adicionado etanol, e as fibras alimentares solúveis e
insolúveis são capturadas, lavadas com etanol e acetona, aquecidas e pesadas.
Um dos duplicados é analisado para proteína e o outro para cinza. As fibras
que não precipitaram encontram-se no filtrado (solução etanólica) e são
posteriormente concentradas por evaporador rotativo, dessalinizadas em
coluna de resinas, novamente concentradas, e quantificadas por HPLC.

51
4.4- Reagentes
• Solução tampão maleato de sódio 50 mM, pH = 6.0
Dissolver 11.6 g de ácido maleico em 1600 mL de água destilada e
ajustar o pH com solução tampão de hidróxido de sódio, 4 M.
• Solução tampão de hidróxido de sódio, 4 M
Dissolver 160 g/L de hidróxido de sódio em água destilada. Perfazer o
volume até 1 L. De seguida, adicionar 0.6 g de CaCl2 (cloreto de sódio) e 0.4
g de azida de sódio (NaN3)- Não adicionar NaN3 até o pH ser ajustado a pH
6.0. Usar máscara. A solução é estável durante um ano, em refrigeração.
• Solução de “trizma base” 0.75 M
Dissolver 90.8 g de trizma base em aproximadamente 800 mL de água
destilada. Perfazer o volume até 1 L.
• Solução de ácido acético, 2 M
Num balão de 1 L com aproximadamente 500 mL de água destilada,
adicionar 115 mL de ácido glacial acético e perfazer o volume.
• D-sorbitol (padrão interno para coluna SugarPaK), 100
mg/L, grau de pureza ≥≥≥≥ 99.5%
Pesar 10 g de D-sorbitol para um balão de 100 ml. Dissolver em 80
mL de azida de sódio a 0.02%. Perfazer até aos 100 mL.
• Solução de NaN3, 0.02% (m/v)
Dissolver 0.2 g de azida de sódio (NaN3) num balão de 1 L, com água
destilada. (Usar máscara)
• Solução para determinação do tempo de retenção padrão
(LCRT)
Dissolver 2.5 g da porção de mistura padrão de oligossacarídeos em
água destilada, e transferir para um balão de 100 mL. Pipetar 10 mL de D-
sorbitol. Perfazer o volume com a solução de azida de sódio a 0.02%.
Transferir as soluções para um frasco Duran de 100 mL.
• D-Glucose padrão para cromatografia líquida, 2, 5, 10, 15,
20 mg/L, com um grau de pureza ≥≥≥≥ 99.5%

52
Pesar 0.2, 0.5, 1.0, 1.5 e 2.0 g de D-glucose (Sigma) e adicionar em
separado as porções em balões de 100 mL. Pipetar 10 mL da solução de D-
sorbitol para cada balão. Perfazer o volume com solução de azida de sódio a
0.02%. Transferir as soluções para frascos Duran.
• Kit enzimático MEGAZYME
Contém enzimas α-amilase pancreática (3300 U/mL), protease (50
mg/mL) e amiloglucosidase (3.4 U/mL).
• Álcool etílico 78 %
Num balão de 1 L, diluir 800 mL de álcool etílico e perfazer o volume
com água destilada.
• Álcool etílico 96%
• Amberlite FPA53
• Ambersep 200
• Acetona
• Areia do mar lavada
4.5-Aparelhos e Utensílios
• Balança sensível à quarta casa decimal
• Cadinhos de vidro de placa filtrante de porosidade 2 (40 a
100 µm).
Colocam-se na mufla a 525 ºC, com uma colher de areia do mar,
durante duas horas. Arrefecem-se no excicador e regista-se a massa.
• Banho de água com temperatura regulável até 100 ºC
• Banho de água vibratório com temperatura regulável: T=
37 ± -1 ºC e 60±1ºC
• Banho de areia
• Mufla de temperatura regulável até 525 ± 5 ºC
• Bomba de vácuo
• Coluna de vidro
• Potenciómetro

53
• Estufa com temperatura regulável
• HPLC (características referidas anteriormente na secção
dos materiais)
- T= 60 ºC
- fase móvel: solução de água destilada com Na2CaEDTA (50 mg/L)
- velocidade do fluxo: 0.5 mL/min
- coluna: Nucleogel Sugar 810 CA Va 300/7.8
- Detector refractivo
4.6- Técnica
De uma forma genérica, o processo para determinar o teor de fibra
alimentar de acordo com o método AOAC 2009.01 segue o seguinte
procedimento:
4.6.1 - Extracção da matéria gorda para amostras com teor de
gordura superior a 10%.
Produtos sólidos secos
A toma de amostra é feita para um cadinho de placa filtrante, onde é
posteriormente lavada com acetona. O cadinho é seco na estufa o tempo
necessário para evaporar o solvente ou deixar o cadinho submerso em acetona
durante a noite; no dia seguinte, filtrar e lavar com acetona. Depois de seca, a
amostra é transferida cuidadosamente para o copo onde decorre a digestão, de
modo a evitar perdas.
Produtos sólidos pastosos
A toma da amostra é feita para um cadinho de placa filtrante, tendo o
cuidado de esta não agarrar à placa do cadinho e, para isso, tentar fixá-la na
parte lateral do cadinho. O cadinho é seco na estufa até a amostra solidificar,

54
de modo a que possa ser desengordurada. O procedimento posterior é o
mesmo que se descreveu para produtos sólidos.
4.6.2 - Toma para análise
A toma de amostra varia consoante o teor de humidade e de matéria
gorda presente na amostra a analisar, tendo como massa de referência1.0 g.
4.6.3 - Digestão enzimática e filtração
a) A amostra é pesada em duplicado para a determinação de
cinza e proteína, não devendo a massa exceder 0.02 g uma da outra. A toma
da amostra é feita para garrafas de vidro de 100 mL.
b) Adicionar 40 mL da solução tampão de maleato de sódio (pH=
6) contendo a mistura de α-amilase pancreática e AMG, e verificar o pH.
Ajustar se não for igual a 6.0 ± 0.1.
c) Incubar em banho vibratório (159 r/min), à temperatura de 37
ºC, durante 16 h (17h00-9h00).
d) Adicionar 3.0 mL de trizma base (pH 7.9-8.4) e ajustar o pH a
8.2
e) Incubar em banho de água (95-100 ºC), durante 20 minutos.
f) Remover os frascos do banho de água, com luvas apropriadas,
e deixar arrefecer o banho até 60 ºC
g) Adicionar 0.1 mL de protease (solução viscosa).
h) Incubar a 60 ºC em banho de água durante 30 minutos. Deixar
arrefecer à temperatura ambiente.
i) Adicionar 4.0 mL de ácido acético (2 M) a cada frasco e ajustar
o pH a 4.3.
j) Adicionar 1 mL da solução D-sorbitol a cada frasco e,
posteriormente, à temperatura ambiente, adicionar 180 mL, de etanol a 95%,
pré aquecido a 60 ºC. A precipitação de fibra ocorre em 1 hora.

55
k) Usando um sistema de vácuo, filtrar a amostra digerida para o
cadinho já pesado e lavar o resíduo com 2x15 mL de etanol a 78%, 2x15 ml
de etanol a 95% e 2x15 mL de acetona. Reter os filtrados e as lavagens para a
determinação de fibra de baixo peso molecular.
l) Secar o resíduo na estufa a 105 ºC durante a noite.
m) Arrefecer em excicador, aproximadamente durante uma hora, e
registar o peso do cadinho com o resíduo
n) Num dos duplicados é determinado o teor de proteína a partir
da análise de nitrogénio e, no outro, o teor de cinza (5 horas a 525 ºC)
o) A massa do resíduo de cinza obtém-se por diferença do
cadinho + resíduo e do cadinho vazio, e a massa resíduo de proteína obtém-se
por diferença do cadinho + resíduo e do cadinho vazio x 6.25 (factor
resultante da conversão do teor de nitrogénio em proteína assumindo que em
média a percentagem de nitrogénio numa proteína é 16%).
4.6.4 – Determinação de fibras de baixo peso
molecular
4.6.4.1- Filtração
Transferir o filtrado para um balão e concentrar a amostra em banho
de areia.
4.6.4.2- Desionização da amostra
4.6.4.2.1- Preparação da coluna de vidro
Misturar 4 g de Amberlite e 4 g de Ambersep, e empacotar na coluna
para a análise de cada amostra teste.
4.6.4.2.2- Desionização da amostra
Dissolver a amostra em 5 mL de água desionizada, e transferir 2 ml
desta para a coluna.

56
Colectar o eluato (0.5 para 2 mL/min) para um balão de 100 mL.
Continuar a extracção na coluna com 20 mL de água desionizada.
Posteriormente, levar à secura em banho de areia. Redissolver em 2 mL de
água desionizada. Filtrar e injectar.
4.7- Cálculos
O teor de fibra, em percentagem, é obtido pela seguinte fórmula de
cálculo:
(((Mrp+Mrc/2)-P-C)/ (Mc+Mp/2)) + (((Mg*PAA)/(Mc*PAG)*100)*2.5)
Onde:
Mrp– massa do resíduo da proteína (g);
Mrc – massa do resíduo de cinza (g);
P – Massa de proteína da amostra (g);
C- Massa de cinza (g);
Mc – massa de amostra para cinza (g)
Mp – massa da amostra para proteína (g);
Mg – massa do padrão glucose (g)
PAA – área do pico da amostra;
PAG – área do pico da glucose

57
V- Resultados
5.1- Resultados obtidos por HPLC
Numa fase inicial de implementação do método da AOAC 2009.01 e
enquanto se aguardava a chegada da coluna destinada à análise de fibra de
baixo peso molecular, foram realizados ensaios com uma coluna da marca
Waters destinada à análise de açúcares. Sendo assim, e como primeiro passo,
foram realizados testes para a obtenção dos padrões, com diferentes
proporções de fase móvel. Os resultados encontram-se nas tabelas 5.1 e 5.2.
Tabela 5.1- Resultados para os padrões de glucose a diferentes concentrações e mistura padrão de oligossacarídeos (LCRT) com fase móvel nas seguintes proporções 80 acetonitrilo (ACN):20 água ( H2O) e fluxo a 2 mL/min
Tempo de retenção (min) Área do pico Glucose 0.5% 4.947 456704 Glucose 1 % 5.067 896565 Glucose 2 % 5.009 1496549 LCRT 5.455 2027845
Tabela 5.2- Resultado para o padrão de glucose com a fase móvel na proporção 75 ACN:25 H20
Tempo de retenção (min) Área do pico Glucose1% 2.107 1945334
Como a glucose saiu com um tempo de retenção de 2.107 com a fase
móvel a 75 ACN: 25 H2O, ou seja muito cedo, optou-se por usar a fase móvel
na proporção de 80 ACN:20 H2O.
Seguidamente, foi realizado um ensaio com três amostras do qual se
concluiu que o sorbitol (usado como padrão interno) adicionado durante o
processo não deveria ser utilizado, uma vez que o seu tempo de retenção era o
mesmo da glucose para esta coluna. Com isto, retirou-se o sorbitol do ensaio e

58
passou-se a realizá-lo pelo método de referência externo (sem uso do padrão
interno, apenas com o padrão de glucose).
Posteriormente foi repetido todo o processo, para três amostras tendo-
se obtido os seguintes resultados:
Figura 5.1- Resultados obtidos para amostras e padrões a fluxo 2 mL/min, fase móvel a 80 ACN:20 H20 e volume de injecção de 20 µL.
Aquando da análise destes resultados concluiu-se haver necessidade
de tarar balões, para que fosse possível saber a massa que é recuperada e
Tempo de retenção (min) Área do pico Glucose 0.5% 4.965 456704 Glucose 1% 5.070 896565 Glucose 2% 5.070 1730645 Branco C --------- -------------- Branco P --------- -------------- 11594 C 5.699 20047 11595 C 6.070 21365 11596 C 5.664 25000
5 10 15
Amostra 11594
Amostra 11595
Amostra 11596
Glucose 2%
tempo (min)

59
posteriormente diluída em água, e assim se quantificar a percentagem de fibra
presente nas amostras. Mais tarde, concluiu-se que a percentagem de fibra
seria a mesma tanto no resíduo como na amostra pesada inicialmente. Os
resultados para as amostras com balões tarados encontram-se na tabela e
cromatograma a seguir:
*Vinj = volume de solução injectada Figura 5.2- Resultados obtidos para amostras e padrões a fluxo 2 mL/min com a fase
móvel nas seguintes proporções 80ACN:20H2O.
Massas(g) Vinj*(µl) Tr (min) Área %Glucose
Glucose1% 20 4.771 863825
11595C 0.9836 20 3.724 123132 1.45
20 5.337 401461 4.73
11595P 0.3072 20 3.671 44134 1.66
20 5.350 167996 6.33
11596P 0.6506 20 3.776 197703 3.52
20 5.422 410424 7.3
11596C 0.7726 20 3.698 125704 1.88
20 5.323 641843 9.62
11594 P e C REPETIU-SE
5 10 15
Amostra 11595
Glucose1%
Amostra 11596
tempo (min)

60
Da análise destes resultados conclui-se que o segundo pico, observado
na Figura 5.2, tinha o mesmo tempo de retenção do padrão da glucose.
Pensou-se ser este o pico correspondente às porções de fibra de baixo peso
molecular, uma vez que aquando da contaminação directa em vial, da amostra
este veio evidenciado. No entanto, os valores obtidos para a percentagem de
fibra de baixo peso molecular não estavam de acordo com o esperado. Depois
da realização de vários ensaios com esta coluna, chegou-se à conclusão que
definitivamente esta não podia ser utilizada, uma vez que depois da realização
de ensaios de recuperação se pode verificar que os picos que se estavam a
quantificar não podiam corresponder à fibra de baixo peso molecular e a
interpretação dos picos correctos não ser trivial.
Procederam-se então, seguidamente a ensaios com a coluna de marca
Macherey-Nagel destinada para a análise de fibra, usando como fase móvel
água com Na2CaEDTA (50 mg/L) e um fluxo de 0.5 mL/min, podendo este
ser variável. Antes da realização do ensaio, foram testados os padrões, sendo
um deles uma mistura de oligossacarídeos (LCRT), que serviu apenas como
ponte de referência para as amostras, a maltose e o sorbitol, que servem como
marcadores (sendo o sorbitol usado como padrão interno) e, por fim, a
glucose, para quantificar as amostras. Seguidamente está reproduzido o
cromatograma dos padrões.
Tr (min) Área LCRT 2.5% 8.186 5351794 Maltose 2% 10.463 8398648 Sorbitol 0.1% 30.74 693636 Glucose1% 12.483 2595306

61
Figura 5.3 - Cromatogramas obtidos para os padrões a fluxo 0.5 mL/min e volume de injecção de 20 µL e na tabela os respectivos tempos de retenção (Tr) e áreas dos picos.
No seguimento da análise dos padrões, foi injectada uma amostra,
tendo-se procedido à sua quantificação. Os resultados apresentam-se a seguir.
Figura 5.4- Cromatograma de padrões e amostra a fluxo 0.5 mL/min e volume de injecção de
20 µL e tabela com respectivos tempos de retenção e áreas dos picos
Tr (min) Área LCRT 2.5% 8.186 5351794 Maltose 2% 10.463 8398648 Sorbitol 0.1% 30.74 693636 Glucose1% 12.483 2595306 11595 7.942 9093402
5 10 15 20 25 30 35
LCRT 2.5%
maltose 2%
glucose 1%
tempo(min)
5 10 15 20 25 30 35
m altose 2%
glucose 1%
11595
tem po(m in)
FB PM

62
Comparando o cromatograma da amostra com o padrão de maltose e
o LCRT, conclui-se que a fibra de baixo peso molecular é tudo o que se
encontra para trás do padrão de maltose, tendo-se obtido num primeiro ensaio
para esta amostra uma percentagem de fibra de baixo peso molecular igual a
3.73%. A percentagem de fibra de alto peso molecular obtida, pela fase
gravimétrica, foi de 0.33%, sendo o total de fibra igual a 4.05%. Comparando
este valor com o valor rotulado da amostra em estudo (gelado de baunilha)
que é de 4.80 %, este resultado encontra-se dentro do critério de aceitação da
rotulagem do produto (20%).
Com isto, e depois de se ter obtido este resultado, procedeu-se a
realização do ensaio para as oito amostras de gelados da marca Pingo Doce de
diferentes sabores (já referenciados anteriormente). Os resultados encontram-
se nos cromatogramas na Figura 5.5 e na Tabela 5.3,.
5 10 15 20 25 30 35 40 45
Tempo (min)
11594
11595
11596
12273
13514
15187
15188
15189
maltose 2%
FBPM
Figura 5.5- Cromatogramas obtidos para diferentes amostras a fluxo 0.4 mL/min e volume de
injecção de 20 µL

63
Tabela 5.3- Resultados obtidos para as oito amostras de gelados a fluxo 0.4 mL/min e volume de injecção de 20 µL tanto para os padrões como para as amostras
Da análise destes resultados, e comparando estes com os valores
rotulados, chega-se à conclusão que a percentagem de recuperação de fibra de
baixo peso molecular é aceitável. Relativamente às amostras 11594 e 12273,
os valores obtidos foram os únicos dissemelhantes relativamente aos valores
rotulados, facto que se atribuiu à degradação da amostra, uma vez que esta já
evidenciava sinais de presença de bolor. Quanto às restantes amostras, os
resultados apresentados são semelhantes aos valores rotulados. A amostra
11595 apresenta como percentagem de fibra total 3.84%, sendo o valor
rotulado de 4.8%. A amostra 11596 possui 5.23% de fibra total e o valor
rotulado é 5.3%.Quanto à amostra 13514, o valor rotulado é de 4.3%, e o
QA da Amostra
Massa da
amostra (g)
Massa dos
resíduos (g)
Fibra de alto peso
molecular (g/100g)
Fibra de baixo peso molecular
Total de fibra alimentar (%)
Valores Rotulados
dos gelados
da marca PD (%)
11594 C 2.5470 0.0117 0.3127 2.5471 2.86 4.7%
11594 P 2.5547 0.0112 11595 C 2.5543 0.0083
0.1066 3.7380 3.84 4.8% 11595 P 2.7320 0.0061 11596 C 2.5507 0.0145
0.4672 4.7600 5.23 5.3% 11596 P 2.5190 0.0211 12273 C 2.5692 0.0033
0.0410 4.0680 4.11 3.0% 12273 P 2.7061 0.0101 13514 C 2.6493 0.0227
0.3386 3.8679 4.21 4.3% 13514 P 2.5978 0.0042 15187 C 2.4939 0.0474
1.0155 3.4688 4.48 4.8% 15187 P 2.6006 0.0458 15188 C 2.5505 0.0861
0.1041 5.5062 5.61 5.3% 15188 P 2.7971 0.0152 15189 C 2.8374 0.0091
0.1832 3.6200 3.80 3.7% 15189 P 2.5843 0.0056

64
obtido foi de 4.21%. Na amostra 15187, obteve-se 4.48% e o esperado era de
4.8%. A amostra 15188 teve como resultado 5.61% e o rotulado era 5.3%. Por
último, a amostra 15189 tem valor rotulado de 3.7% e obteve-se 3.80%.
Com isto, e visto que resultados serem bastante aceitáveis, procedeu-
se à validação do método, sendo que a amostra usada para tal foi a 15189.
Seguidamente são apresentados os resultados obtidos, bem como os seus
objectivos e critérios de validação.
O processo de validação tem por objectivo evidenciar que o método
permite obter resultados cuja qualidade é adequada aos fins com que são
usados. Para a validação deste método de ensaio o laboratório utiliza as
seguintes avaliações:
a) Avaliação dos fundamentos teóricos do método e adequabilidade
b) Calibração analítica
Estudo da linearidade da recta de calibração Usualmente o estudo da
linearidade de uma gama de trabalho em cromatografia é realizado de acordo
com o teste RIKILT. Este permite avaliar a linearidade através do estudo dos
resíduos. Contudo, para a validação deste método, utilizou-se a norma ISO
8466-01.
Prepararam-se os padrões necessários, de acordo com o definido no
método de ensaio, e efectuou-se a sua leitura. Introduziram-se os resultados
obtidos no Modelo 789 (modelo baseado em normas ISO) e avaliou-se a
linearidade.
Para avaliar a linearidade utilizaram-se, então, os padrões referidos no
método da AOAC 2009.01: 0.2 g/mL, 0.5 g/mL, 1.0 g/mL, 1.5 g/mL e 2.0
g/mL.

65
Gráfico 1: Curva de calibração para doseamento das fibras de baixo
peso molecular. Utilizou-se a fase móvel referida no método 2009.01, fluxo de 0.4 mL/min, temperatura de 60 ºC e detector RID.
A função de calibração é linear, fornecendo a equação da recta y =
4x106x+221519 e um coeficiente de correlação igual a 0.99227 para
concentrações entre 0.2 e 2.0 g/mL de glucose, obedecendo ao critério de
aceitação do laboratório (r ≥ 0.99). Procedendo de acordo com a norma ISO
referida anteriormente, a partir de um conjunto de pares ordenados (y=área;
x=concentração) calculam-se para a função de calibração linear e não linear
os respectivos desvios-padrão residuais, Sy/x, Sy2. A diferença das variâncias
(DS2) é calculada pela equação seguinte:
( ) ( ) 22/
2
232 yxy SNSNDS −−−=
em que N é o número de padrões de calibração.
Calcula-se o valor teste F, dado por.:
2
2
.
2y
CalcS
DSF =
e compara-se com o F tabelado. Como o valor de F obtido (1.99) é menor que
o F tabelado (15.98) a função é linear.

66
Estudo do limite de quantificação- Neste trabalho, o estudo do limite de
quantificação foi realizado através do primeiro padrão da curva de calibração
(padrão residual). Neste caso, efectuaram-se 10 leituras do primeiro padrão da
curva de calibração, em condições de precisão intermédia, e, posteriormente,
efectuou-se o cálculo da média, desvio padrão. Se o erro relativo e coeficiente
de variação dos valores obtidos foram inferiores a 10% o limite de
quantificação é igual ou inferior ao valor testado. Caso contrário, outro limite
de quantificação terá que ser estudado. O estudo do limite de quantificação do
método foi apurado no Modelo 759.
Tabela 5.4- Concentrações obtidos para o primeiro padrão da recta de calibração
Ensaio Data Resultado (g/ml) 1 16-02-2011 0.2020 2 23-02-2011 0.1840 3 25-02-2011 0.2093 4 28-02-2011 0.1970 5 28-02-2011 0.2000 6 28-02-2011 0.2000 7 1-03-2011 0.2001 8 1-03-2011 0.2020 9 1-03-2011 0.2002
10 1-03-2011 0.2004
O coeficiente de variação obtido é igual a 3.13% e o erro relativo é de
0.39% logo o limite de quantificação do método é 0.2 g/mL de glucose e
corresponde ao primeiro padrão da curva de calibração.
Estudo da repetibilidade- O ensaio de repetibilidade consiste em repetir o
mesmo ensaio, sobre a mesma amostra, pelo mesmo analista, mesmo
equipamento e mesmos reagentes, em curtos intervalos de tempo. Para se
determinar a repetibilidade do método efectuaram-se 10 repetições do ensaio
para a amostra 15189 nas condições anteriormente descritas.
Para um nível de confiança de 95%, o limite de repetibilidade (r) é
avaliado segundo:

67
riSr 8,2=
Sendo que,
riS é o desvio padrão de repetibilidade associada aos resultados considerados;
(NOTA: Avaliou-se a existência de valores aberrantes, através de teste de
“Outliers”-teste de Grubb’s.)
O coeficiente de variação de repetibilidade ( )rCV é expresso em
percentagem, e é dado por:
100x
SCV ri
r =
onde,
riS é o desvio padrão de repetibilidade
O erro relativo da repetibilidade (Er) é dado por:
100×−
=médio
médioobtido
x
xxEr
Considera-se a repetibilidade aceitável quando CVr ≤ 10%.
Tabela 5.5- Concentrações obtidas para a amostra 15189, repetida nas condições descritas no texto ( ver acima )
Ensaio Resultado
1 1.300
2 1.300
3 1.290
4 1.300
5 1.280
6 1.290
7 1.290
8 1.300
9 1.290
10 1.290

68
A repetibilidade obtida foi de 1.47% em valor relativo e com CVr
igual a 0.52%.
Estudo da especificidade, selectividade e exactidão do método (Ensaios de
recuperação)- Diz-se que um método é específico quando permite
discriminar um analito relativamente a outras substâncias, eventualmente
presentes na amostra a analisar, ou seja, quando oferece garantias que a
grandeza medida provém apenas do analito. A selectividade é a capacidade de
um método identificar e distinguir um analito numa mistura complexa, sem
interferência de outros componentes.
Para avaliar as interferências de matriz, efectuou-se um teste de
recuperação utilizando uma amostra real representativa do tipo de matriz, com
valor quantificável. Esta amostra é contaminada com analito a diferentes
níveis de concentração, incluindo os seus valores extremos.
O cálculo da recuperação é efectuado da seguinte forma:
η = [Cf–Ca) / Ct] *100
η = recuperação
Cf = concentração final (analito adicionado + amostra)
Ca = concentração da amostra
Ct = concentração esperada de analito
Considera-se a recuperação aceitável sempre que η=100±20% (métodos cromatográficos).

69
Tabela 5.6- Resultados obtidos a diferentes concentrações para os ensaios de recuperação
Conclui-se que o método é exacto, uma vez que cumpre o critério de
aceitação.
Estudo da precisão intermédia- Estudou-se a precisão intermédia
efectuando no mínimo 15 ensaios fazendo variar as condições susceptíveis de
variação tais como o tempo (variação de dias).
A precisão intermédia (S) é avaliada através da seguinte expressão:
( )1
,1
2
−
−
=∑
=
i
ni
iiji
n
yyS
Considera-se a precisão intermédia (S) aceitável se cumprir o valor de
referência CV ≤20%.
Concentração do analito na
amostra
Massa Amostra
(g)
Massa Analito
Adicionada (g)
Concentração Esperada
(%)
Concentração Obtida
(%)
Recuperação
(%)
0.08 1.0338 0.3750 0.20 0.29 103.50
0.16 1.0508 0.7000 0.50 0.57 81.50
0.33 1.0656 0.1000 1.00 1.44 111.90

70
Tabela 5.7- Resultados obtidos em g/100 g para a mesma amostra desfasada no tempo
Ensaio Resultado
1 1.300
2 1.300
3 1.290
4 1.300
5 1.280
6 1.290
7 1.290
8 1.300
9 1.290
10 1.290
11 1.130
12 1.280
13 1.300
14 1.100
15 1.100
Como o CV=6.06%, cumpre o critério de aceitação relativamente à
precisão intermédia.
Avaliação da incerteza- A incerteza do método foi calculada a partir
de ensaios de recuperação (exactidão) e da precisão intermédia.
A incerteza foi calculada a partir de ensaios de recuperação, sendo o
valor obtido de 8.88 % em valor relativo.
Atendendo aos resultados obtidos durante o processo de validação,
conclui-se que este método se encontra validado, podendo ser realizado em
trabalho de rotina do laboratório.

71
5.2- Resultados obtidos por espectroscopia de Infravermelho
Com estes estudos, pretendeu-se efectuar uma caracterização
espectroscópica das amostras a injectar no cromatógrafo, utilizando depois a
informação obtida para tirar ilações de natureza semi-quantitativa.
Numa fase inicial de experimentação, foram registados espectros de
infravermelho de glucose, de maltose e da amostra 13514 (gelado de morango
da marca Pingo Doce), com a finalidade de se tentar perceber onde se
encontravam as bandas características da fibra de baixo peso molecular. Na
Figura 5.7 encontram-se representados os mesmos:
Figura 5.7. Espectros de infravermelho de glucose, maltose, LCRT e da amostra 13514 obtidos à temperatura ambiente
A análise dos espectros obtidos revelou que estes apresentavam
grande grau de semelhança entre si, inviabilizando, desta forma, qualquer
análise quantitativa relativa ao teor de fibra por simples análise espectral.
Foi, então, desenvolvida uma metodologia de análise dos espectros
alternativa, baseada na simulação dos espectros esperados para cada amostra a
partir de espectros de referência da glucose, maltose e LCRT. Os teores de
4000 3500 3000 2500 2000 1500 1000 500
G lu cose
M a ltose
LC R T
nu m ero d e on d a / cm-1
1 35 1 4
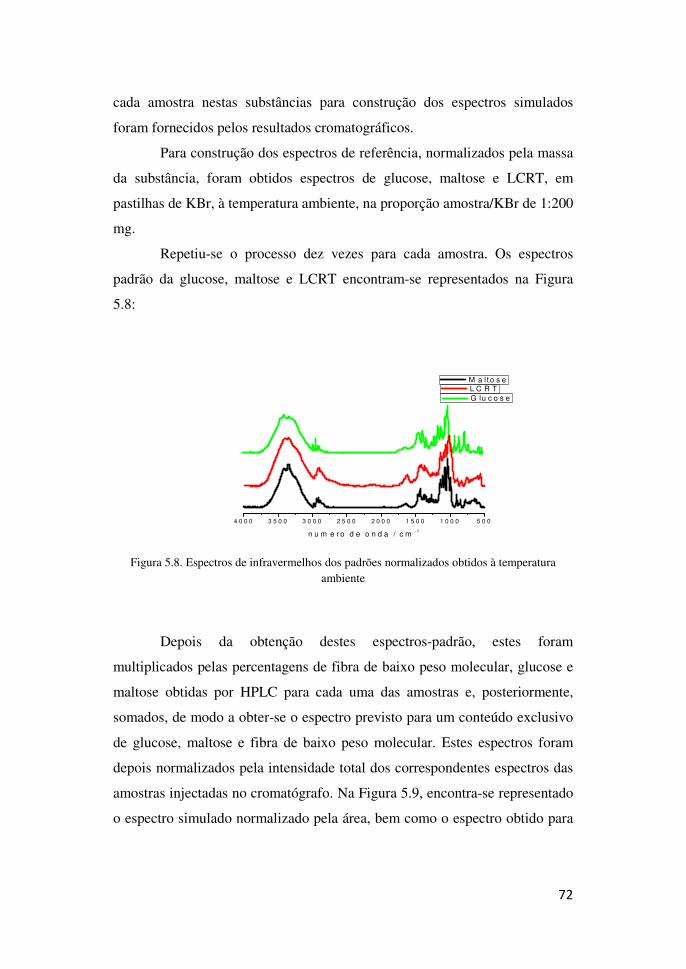
72
cada amostra nestas substâncias para construção dos espectros simulados
foram fornecidos pelos resultados cromatográficos.
Para construção dos espectros de referência, normalizados pela massa
da substância, foram obtidos espectros de glucose, maltose e LCRT, em
pastilhas de KBr, à temperatura ambiente, na proporção amostra/KBr de 1:200
mg.
Repetiu-se o processo dez vezes para cada amostra. Os espectros
padrão da glucose, maltose e LCRT encontram-se representados na Figura
5.8:
Figura 5.8. Espectros de infravermelhos dos padrões normalizados obtidos à temperatura ambiente
Depois da obtenção destes espectros-padrão, estes foram
multiplicados pelas percentagens de fibra de baixo peso molecular, glucose e
maltose obtidas por HPLC para cada uma das amostras e, posteriormente,
somados, de modo a obter-se o espectro previsto para um conteúdo exclusivo
de glucose, maltose e fibra de baixo peso molecular. Estes espectros foram
depois normalizados pela intensidade total dos correspondentes espectros das
amostras injectadas no cromatógrafo. Na Figura 5.9, encontra-se representado
o espectro simulado normalizado pela área, bem como o espectro obtido para
4 0 0 0 3 5 0 0 3 0 0 0 2 5 0 0 2 0 0 0 1 5 0 0 1 0 0 0 5 0 0
G lu c o s e
L C R T
n u m e r o d e o n d a / c m-1
M a l t o s e
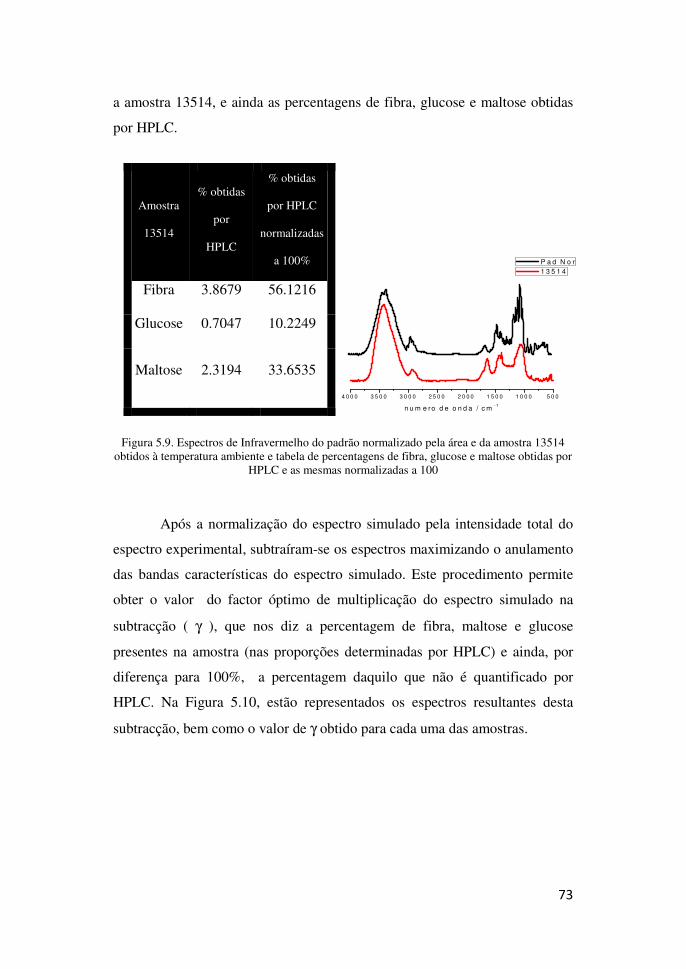
73
a amostra 13514, e ainda as percentagens de fibra, glucose e maltose obtidas
por HPLC.
Figura 5.9. Espectros de Infravermelho do padrão normalizado pela área e da amostra 13514 obtidos à temperatura ambiente e tabela de percentagens de fibra, glucose e maltose obtidas por
HPLC e as mesmas normalizadas a 100
Após a normalização do espectro simulado pela intensidade total do
espectro experimental, subtraíram-se os espectros maximizando o anulamento
das bandas características do espectro simulado. Este procedimento permite
obter o valor do factor óptimo de multiplicação do espectro simulado na
subtracção ( γ ), que nos diz a percentagem de fibra, maltose e glucose
presentes na amostra (nas proporções determinadas por HPLC) e ainda, por
diferença para 100%, a percentagem daquilo que não é quantificado por
HPLC. Na Figura 5.10, estão representados os espectros resultantes desta
subtracção, bem como o valor de γ obtido para cada uma das amostras.
Amostra
13514
% obtidas
por
HPLC
% obtidas
por HPLC
normalizadas
a 100%
Fibra 3.8679 56.1216
Glucose 0.7047 10.2249
Maltose 2.3194 33.6535
4 0 0 0 3 5 0 0 3 0 0 0 2 5 0 0 2 0 0 0 1 5 0 0 1 0 0 0 5 0 0
P a d N o r
n u m e ro d e o n d a / c m-1
1 3 5 1 4

74
Figura 5.10. Espectros de infravermelho obtidos à temperatura ambiente dos resíduos das amostras e tabela com as percentagens de fibra, glucose e maltose obtida por HPLC e o valor
de γ obtido da subtracção dos espectros de referência normalizado à área com a amostra
Desta análise, concluiu-se que 50% dos espectros correspondem à
fibra, maltose e glucose presentes nas amostras e quantificadas por HPLC, e
os restantes 50% a outras substâncias não precipitáveis pela solução etanólica
e que estão, por isso, na amostra a injectar no cromatógrafo. Por simples
análise dos espectros-resíduo, estas substâncias são de difícil caracterização,
exigindo um estudo detalhado, a realizar no futuro. De acordo com a
bibliografia consultada [29], um tipo de substância com espectros compatíveis
com algumas das bandas mais intensas observadas nos espectros-resíduo são
ácidos carboxilicos, uma vez que aos picos mais intensos se podem fazer
corresponder elongações C-H e OH (entre 3000 e 2920 cm-1) e C=O (1724
cm-1).
Amostras
% Gama
(γ )
11594
Fibra 2.55 0.51
Glucose 0.28
Maltose 0.79
11595
Fibra 3.74 0.48
Glucose 1.24
Maltose 0.00
15188
Fibra 5.51 0.50
Glucose 0.00
Maltose 1.12
13514
Fibra 3.87 0.42
Glucose 0.70
Maltose 2.32
4000 3500 3000 2500 2000 1500 1000 500
11595
15188 13514
numero de onda / cm-1
11594

75
5.3- Resultados obtidos por Espectroscopia de Raman
Foram realizados ensaios preliminares de caracterização do padrão
LCRT e ainda da amostra 13514. Na Figura 5.11, encontram-se representados
os espectros obtidos para o padrão referido e respectiva amostra.
Figura 5.11. Espectros de Raman obtidos para um tempo de exposição de 100 s e 50 espectros
De acordo com a bibliografia consultada, a região entre 3000 e 3100
cm-1 corresponde às vibrações dos grupos C-H presentes tanto nas moléculas
de glucose, como as de maltose e de fibra de baixo peso molecular. Entre
1400 e 1470 cm-1, surgem as bandas devidas às as elongações dos grupos
CH3 e CH2 também estes presentes nas moléculas de glucose, maltose e fibra.
As restantes zonas espectrais são de difícil caracterização, contudo
contribuições das vibrações C-O-C dos anéis de glucose, maltose e fibra, entre
outras.
Tal como os espectros de infravermelho discutidos na secção anterior,
os espectros de Raman da amostra e do LCRT (bem como os de glucose e
maltose constante de literatura [30] são muito semelhantes entre si. Sugere-se
então aqui para a análise destes resultados a aplicação de uma metodologia
3000 2500 2000 1500 1000 500 0
LCRT
13514
cm-1

76
semelhante à descrita atrás para o tratamento dos resultados obtidos por
espectroscopia de infravermelho. Da conjugação dos resultados obtidos pelas
duas técnicas espectroscópicas pode esperar-se uma maior certeza nos
resultados obtidos e, eventualmente, a determinação de elementos que
permitam a identificação de pelo menos algumas das substâncias-resíduo
presentes nas amostras e não identificadas cromatográficamente. Este é um
trabalho que fica em aberto para exploração futura.

77
VI- Conclusão
Esta dissertação reflecte um trabalho de investigação sobre fibra
alimentar (alto e baixo peso molecular), por recurso a três técnicas, sendo que
a mais utilizada, durante o desenvolvimento deste trabalho foi a cromatografia
líquida de alta eficiência.
Com esta técnica e seguindo os procedimentos do método AOAC
2009.01 foi possível quantificar a percentagem de fibra alimentar de baixo
peso molecular (fibra sintética), presente em oito amostras de gelados, tendo-
se obtido valores de acordo com os valores rotulados, um requisito muito
exigente em segurança alimentar. Este método foi validado tendo-se obtido
como limite de quantificação valores de 0.2 g/mL.
O método analítico passou assim a fazer parte dos recursos do
laboratório de química da empresa ControlVet. Tratou-se da primeira
implementação deste método à escala industrial em Portugal.
As amostras foram ainda submetidas à espectroscopia de
infravermelho, da qual se pode concluir que 50% do espectro obtido pertence
à percentagem de fibra de baixo peso molecular, maltose e glucose
quantificadas por HPLC e os outros 50% a outro tipo de substâncias não
precipitáveis pela solução etanólica e que estão, por isso, na amostra a injectar
no cromatógrafo. A determinação da natureza precisa, destas substâncias foi
deixada em aberto para futura investigação.
A utilização da espectroscopia de Raman conjuntamente com a
espectroscopia de infravermelho é aqui proposta para garantir uma maior
fidelidade das determinações analíticas semi-quantitativas por espectroscopia.

78
VII- Bibliografia
[1] Champ M., Langkilde A.M., Brouns F., Kettlitz B. e Collet Y.L.B.,
Nutrition Research Reviews,16, 2003,71-82.
[2] McCleary, B. V., Cereal Foods World, 55, 2010,24-28.
[3] Report of the Dietary Fiber Definition Committee to the Board of
Directors of the American Association Of Cereal Chemists, Cereal Foods
World,46, 2001,112-126.
[4] McCleary, B.V., Proceedings of the Nutrition Society, 62, 2003, 3–9.
[5] Lunn J. e Buttriss J.L., British Nutrition Foundation Nutrition
Bulletin, 32, 2007, 21-64.
[6] DeVries J.W., Prosky L., Li B., e Cho S., Cereal Foods
World,44,1999, 367-369.
[7] http://www.fao.org/ (Acedido em: 05-01-2011)
[8] http://www.who.int/en/ (Acedido em: 19-12-2010)
[9] Johnson, I.T., e Southgate, D.A.T., Dietary fibre and related
substances. 1.ª.ed. Chapman & Hall. London, 1994
[10] Wakbayashi, K., Sakurai, N., e Kuraishi, S., Plant Cell Physiol, 30,
1989, 99-105.
[11] Knudsen, K.E, Animal Feed Sci Technol, 90, 2001, 3-20.

79
[12] Castilho, A.C. et al. A Importância das Fibras Alimentares para o
Paciente Diabético. Support, 2005.
[13] Coppini, L. Z., Waitzberg, D. L., Campos, F.G., Harb.Gama, As
Fibras Alimentares e Ácidos Gordos de Cadeia Curta, Nutrição Oral, Enteral
e Parenteral na Prática Clinica, 3ª ed. Atheneu, São Paulo, 2004, 79 – 94.
[14] Mahan, L.K., Arlin, M.T. Krause: Alimentos Nutrição e
Dietoterapia. 8ª ed, Roca, São Paulo, 1995, 38 – 40.
[15] Coppini, L.Z. Introdução à fibra terapêutica: características e
funções. Byk Química.
[16] Fibras em Nutrição Enteral. Tópicos em Nutrição Clinica. Nestlé
Nutrition Services, 2000.
[17] Lineback, D.R., The chemistry of complex carbohydrates. Complex
Carbohydrates in Food . Marcel Dekker, Inc New York,1999.
[18] Gray J.,Dietary fibre. Definition, Analysis, Phisiology and Health.
ILSI Europe Concise Monography Series. Brussels: ILSI
[19] Mataix J., e Gassull, M.A., Nutrición y alimentación humana.
1.ºed.,. Ergon, Madrid, 2002 119-135.
[20] Simpson CF: Practical High Perfomance Liquid Chromatography.
Edited by Simpson CF. London: Heyden & Son; 1978
[21] Schwedt G: The Essential Guide to Analytical Chemistry edn 2ª.
Chichester, England: John Wiley & Sons Ltd.; 1997

80
[22] Prichard E, MacKay GM, Points J: Trace analysis: a structured
approach to obtaining reliable results. Cambridge: The Royal Society of
Chemistry; 1996
[23] Sentell KB, Dorsey JG: Retention mechanism in Reverse-Phase
Liquid Cromatography. Stationary-Phase bonding density and partitioning.
Anal. Chem. 1989, 61:930-934.
[24] Neue UD: HPLC Trubleshooting, Waters Corporation; 1999
[25]http://www.chromatography-online.org/HPLC/Refractive-
Index/rs33.html (Acedido em: 15-03-11)
[26]- Padilha, D.F.N. – Estudos sobre betaxolol. Polimorfismo e
partição entre octanol e água. Coimbra: Universidade de Coimbra, Faculdade
de Ciências e Tecnologias, 2006-2007. Relatório de estágio
[27] Willard H.; L. Merritt, Jr; Dean J.: Análise Instrumental; 2ºedição,
Fundação Calouste Gulbenkian
[28] Fausto, R., Burrows, H. D.; Pereira, M.: Química Síntese e
Estrutura, Escolar Editora
[29] Fausto, R., Pineiro, Marta, Kus, Nihal, Espectroscopia Vibracional
de proteínas, lipidos e hidratos de carbono:Tópicos Seleccionados,em
Aspectos Probióticos Y Tecnologicos de Las Bactérias Lácticas Novel Probio,
2010.
[30]- Mathlouthi M., Carbohydrate Research, 284, 1996, 145-147.


















