Instituto Superior Técnico - fenix.tecnico.ulisboa.pt · COMPRIMIDO POR ESPECTROFOTOMETRIA DE...
33
1 Instituto Superior Técnico Departamento de Engenharia Química Guia dos Laboratórios III (Bloco de Métodos Ópticos) 2005/2006 M. Amélia Santos
Transcript of Instituto Superior Técnico - fenix.tecnico.ulisboa.pt · COMPRIMIDO POR ESPECTROFOTOMETRIA DE...

1
Instituto Superior Técnico
Departamento de Engenharia Química
Guia dos Laboratórios III
(Bloco de Métodos Ópticos)
2005/2006
M. Amélia Santos

2
Prefácio
Este guia contém as técnicas dos trabalhos práticos do bloco de Métodos Espectroscópicos da disciplina de Laboratórios III (do 3ºano da Licenciatura em Qímica).
Os capítulos introductórios estão reduzidos ao mínimo indispensável. Inclui-se ainda uma cópia da folha de resultados (boletim de análise) a apresentar no final de cada aula, o resumo da constituição de um relatório completo e ainda (em apêndice) notas resumo sobre o tratamento estatístico dos resultados experimentais.

3
Índice
Apresentação dos Métodos 4
Calendário das Aulas 5
Relatório 6
Boletim da Análise 7
I-Técnicas de Métodos Espectroscópicos (UV/Vis) 8
1) Métodos de Absorção Molecular
1-1- Análise de Multicomponentes de um Comprimido 8
1-2- Análise de Ácido Acetylsalicílico ou Paracetamol num Comprimido 12
2) Métodos de Absorção Atómica
2-1- Doseamento do Ferro num Cereal por Espectroscopia de Absorção Atómica
(chama) 16
2-2- Doseamento do Cálcio num Sumo por Espectroscopia de Absorção Atómica
(chama) 19
2-3- Doseamento do Cobre num Cereal por Espectroscopia de Absorção Atómica
(Câmara) 21
3) Métodos de Emissão por Fluorescência Molecular
3-1 - Determinação Fluorimétrica do Quinino numa Água Tónica. 24
3-2 – Determinação da Riboflvina (Vitamina B2)num comprimido 26
II- Anexo: Métodos de Cálculo e Erros em Determinações Analíticas 29
1 Métodos de Cálculo de Concentrações 29
2 Precisão, Exactidão, Sensibilidade e Limite de Detecção 32
3 Análise Estatística dos Erros (Sumário) 33

4
Laboratórios III I- Métodos Espectroscópicos I.1. ESPECTROFOTOMETRIA de ABSORÇÃO MOLECULAR • -Determinação de multicomponentes (ácido acetilsalicílico, paracetamol e
cafeína num comprimido de Anadin-Extra) • - Análise de Ácido Acetilsalicílico ou Paracetamol num Comprimido. • I.2-1. ESPECTROFOTOMETRIA de ABSORÇÃO ATÓMICA (chama) • Determinação do cálcio em sumos • Determinação do ferro num cereal I.2-2. ESPECTROFOTOMETRIA de ABSORÇÃO ATÓMICA
(câmara de grafite) (demonstração) • Determinação do cobre num cereal ou sumo I.3. ESPECTROSCOPIA DE FLUORESCÊNCIA MOLECULAR • -Determinação do quínino numa água tónica • - Doseamento da vitamina B2 num comprimido
5
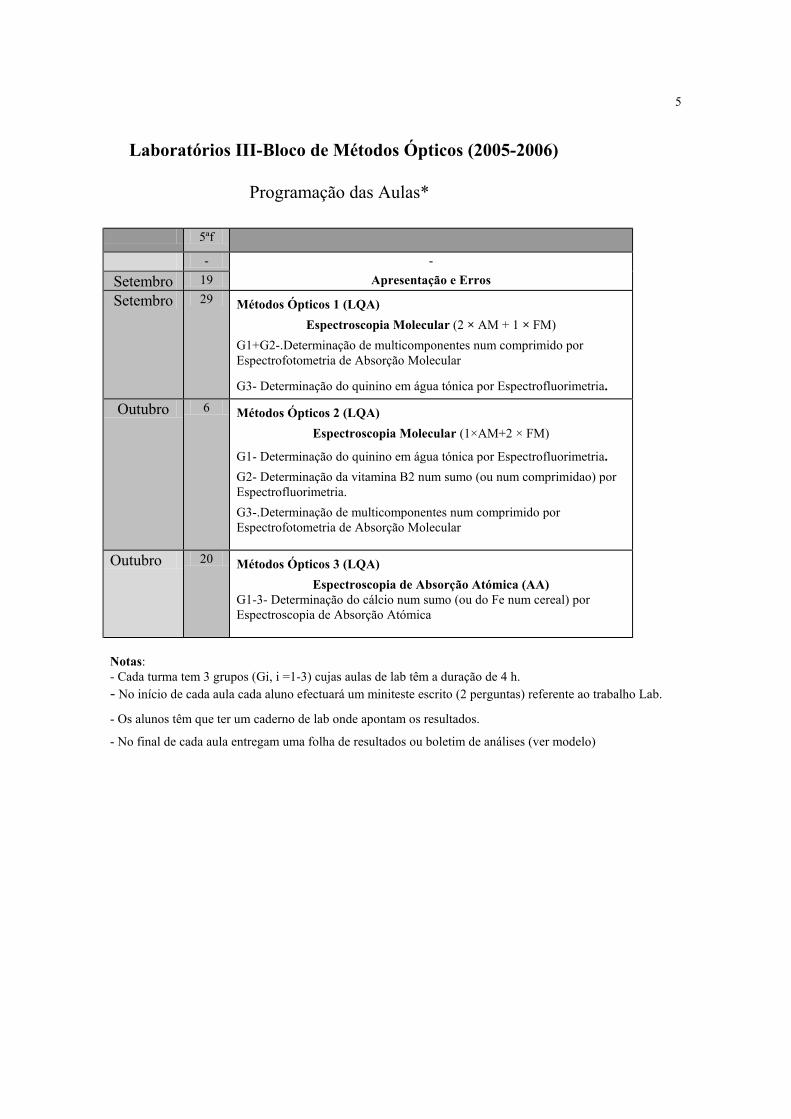
Laboratórios III-Bloco de Métodos Ópticos (2005-2006)
Programação das Aulas*
5ªf
- -
Setembro 19 Apresentação e Erros
Setembro 29 Métodos Ópticos 1 (LQA) Espectroscopia Molecular (2 × AM + 1 × FM)
G1+G2-.Determinação de multicomponentes num comprimido por Espectrofotometria de Absorção Molecular
G3- Determinação do quinino em água tónica por Espectrofluorimetria.
Outubro 6 Métodos Ópticos 2 (LQA) Espectroscopia Molecular (1×AM+2 × FM)
G1- Determinação do quinino em água tónica por Espectrofluorimetria. G2- Determinação da vitamina B2 num sumo (ou num comprimidao) por Espectrofluorimetria. G3-.Determinação de multicomponentes num comprimido por Espectrofotometria de Absorção Molecular
Outubro 20 Métodos Ópticos 3 (LQA)
Espectroscopia de Absorção Atómica (AA) G1-3- Determinação do cálcio num sumo (ou do Fe num cereal) por Espectroscopia de Absorção Atómica
Notas: - Cada turma tem 3 grupos (Gi, i =1-3) cujas aulas de lab têm a duração de 4 h. - No início de cada aula cada aluno efectuará um miniteste escrito (2 perguntas) referente ao trabalho Lab.
- Os alunos têm que ter um caderno de lab onde apontam os resultados.
- No final de cada aula entregam uma folha de resultados ou boletim de análises (ver modelo)

6
RELATÓRIO
I. RESUMO Determinação de ….. existente em (amostra - referir a origem) pela técnica de ……. . Obteve-se …..± (erro) de produto por …. de amostra. II. INTRODUÇÃO (1pag)
a) Fundamento do método b) Expressão que relaciona a grandeza medida com a
concentração. Definição dos parâmetros envolvidos na equação. c) Limite de detecção da técnica.
III. PARTE EXPERIMENTAL Alterações introduzidas à técnica; referir as massas e aos volumes utilizados ma preparação dos padrões e das amostras; referência a todas as condições experimentais (por exemplo: comprimento de onda da determinação, caudal de aspiração em absorção atómica; tipo de aparelho utilizados e a respectivas marcas. IV. RESULTADOS
a) Tabelas com resultados (por exemplo grandezas medidas versus volume ou concentração).
b) Gráficos (por exemplo rectas de calibração). c) Determinação da quantidade de produto referido à
amostra (em % sempre que possível) d) Cálculo dos erros.
V. DISCUSSÃO E CRÍTICA
a)Dos resultados b) Da técnica
VI. REFERÊNCIAS BIBLIOGRÁFICAS

7
Boletim da Análise Efectuada
Título:
Amostra analisada:
Método/Aparelho:
Resultado Final:
Tabela dos Resultados:
Cp(ppm) L
Amostra
Gráfico A vs S: ver folha em anexo
Cálculos da concentração na amostra inicial (com erros):
a) Equação da recta de calibração:
b) Concentração da amostra tirada da recta
c) Concentração na amostra inicial
Comentários ao trabalho (resultado obtido):
Nomes

8
I-Métodos Espectroscópicos
1-1-ANÁLISE DE MULTICOMPONENTES DE UM
COMPRIMIDO POR ESPECTROFOTOMETRIA DE UV/VIS INTRODUÇÃO
Esta experiência envolve a determinação de quantidade de aspirina, paracetamol e cafeína nos comprimidos de Anadin-extra, um analgésico à venda nas farmácias. A aspirina tem propriedades como analgésico, antipirético e anti-inflamatório. O paracetamol tem maior eficácia do que a aspirina como antipirético. A cafeína é usada devido às suas propriedades diuréticas.1
Existem alguns problemas na análise destas drogas, incluindo a hidrólise da aspirina e a dependência do pH no caso do paracetamol.
O
O
CH3
CO2H
OH
HN
O
CH3
Aspirina Paracetamol (ácido acetilsalicílico) (4-hidroxiacetanilide)
N
CN
C
CC N
N
C
OCH3
H
CH3
O
H3C
Cafeína
Análise Quantitativa de Anadin-extra
Uma vez que a aspirina e o seu produto de hidrólise (ácido salicílico) têm espectros de absorção do ultravioleta diferentes, a solução do comprimido de Anadin-extra deve ser fortemente básica para assegurar que toda a aspirina se hidrolizou e apenas o ácido salicílico está a ser analizado. Do mesmo modo, uma vez que o paracetamol é sensível ao

9
pH e as formas ácida e básica têm diferentes espectros de absorção no UV, o facto da solução estar fortemente básica, assegura que o paracetamol está todo na forma básica.
Portanto, a pH elevado, a solução do comprimido de Anadin-extra contém ácido salicílico (forma básica), a forma básica do paracetamol e a cafeína.
Estes três componentes têm espectros de UV diferentes e por isso eles podem ser analizados simultâneamente, através da medição da absorvância da solução a três comprimentos de onda diferentes e resolvendo o sistema de 3 equações.
Com efeito a absorvância total da solução comprimido a cada comprimento de onda é devida à soma das contribuições dos três componentes presentes. (propriedade da aditividade da Lei de Beer)
A1 = eA1 CA + eP1 CP + eC1 CC
A2 = eA2 CA + eP2 CP + eC2 CC
A3 = eA3 CA + eP3 CP + eC3 CC
em que 1, 2 e 3 são os três comprimentos de onda aos quais se fazem as medições; CA, CP, e Cc são as concentrações de aspirina, paracetamol e cafeína, respectivamente; os e são as nove absortividades das três componentes aos três comprimentos de onda.
O sistema das três equações pode ser resolvido com a calculadora ou usando matemática de matrizes para resolver uma equação matricial:
M = AC ou AC = M
C = A-1 M (A-1 é o inverso de matriz A)
TÉCNICA EXPERIMENTAL
A- Preparação das Soluções
1) Solução 0.1M Na OH (500 mL)
2) Soluções de aspirina
2-1- Solução inicial 1g / L
Pese rigorosamente cerca de 1 g de ácido acetilsalicílico (aspirina) num copo, adicione alguns mL de metanol (cerca de 10mL) para dissolver o sólido, transfira para um balão aferido de 1 L, e dilua com água desionizada atá ao traço e agite bem.
2-2 – Solução de trabalho
Prepare uma solução em que toda a aspirina tenha sido hidrolizada a ácido salicílico pipetando 5 mL da solução de partida para um balão volumétrico de 100 mL, adicionando 40 mL de solução 0.1M NaOH e diluindo até ao traço com água desionizada. Agite bem e deixe em repouso 10 min. para assegurar que toda a aspirina é hidrolizada a ácido salicílico.
3 – Soluções de paracetamol 3-1 – Solução inicial 1g/L

10
Pese rigorosamente cerca de 1 g de paracetamol (4-acetamidofenol) para um copo,
adicione alguns mL de metanol (cerca de 10 mL)até dissolver e transfira para o balão de 1L, diluindo com água (agite bem).
3-2 – Solução de trabalho
Prepare uma solução em que todo o paracetamol esteja na forma básica, pipetando 2 ml de solução inicial de paracetamol para um balão volumétrico de 100 ml, adicionando 40 ml de solução 0.1M Na OH, e depois diluindo até ao traço com água.
4 - Soluções de cafeína 4-1 – Solução inicial (Proceda como nas soluções 3) 4-2 – Solução de trabalho (Proceda como nas soluções 3)
5– Soluções de Anadin-Extra 5-1 – Solução inicial (100mg / 100 mL)
Esmague um comprimido num almofariz até obter um pó muito fino. Pese rigorosamente cerca de 100 mg do pó num copo, adicione alguns mL de metanol e agite durante alguns minutos. Transfira então para o balão volumétrico e dilua até ao traço com água.
5-2 - Solução de trabalho Filtre a solução inicial, pipete 2 mL desta soluçaõ para um balão de 100 mL, e
proceda como em 2-2. Esta solução é instável e deve ser usada apenas até 1-2 h depois de ser preparada.
6 - Calcule a média da massa dos comprimidos de Anadin-extra Pese 7 comprimidos e calcule a média.
7 - Solução padrão Prepare em balões volumétricos de 50 mL prepare 4 soluções padrão para a aspirina, paracetamol e cafeína, a partir das correspondentes soluções de trabalho, pipetando 5, 10, 20 e 25 mL para balões volumétricos de 50 mL e diluindo até ao traço com água desionizada. B - Parte Instrumental 1 – Ligue o aparelho e faça acerto do zero, do aparelho com a solução do branco nas 2 células. O aparelho traça uma linha de base recta, (fazer o varrimento de comprimento de onda na gama 230-340 nm e absorvância 0-0.5.
Para um resto das medições deixar a solução de branco na célula de referência (posição posterior). 2 – Para cada um dos componentes, traçar o espectro da solução mais concentrada dos padrões. Determine o comprimento de onda de máxima absorção para cada um dos componentes o qual derverá ser cerca de 296 nm (aspirina, na verdade é ácido salicílico), 257 nm (paracetamol na forma básica) e 273 nm (cafeína). 3.– Para cada componente meça as absorvâncias dos padrões e amostra aos três comprimentos de onda determinados anteriormente. A absorvânciaamostra deverá cair dentro da gama de calibração, diluindo se necessário.
11
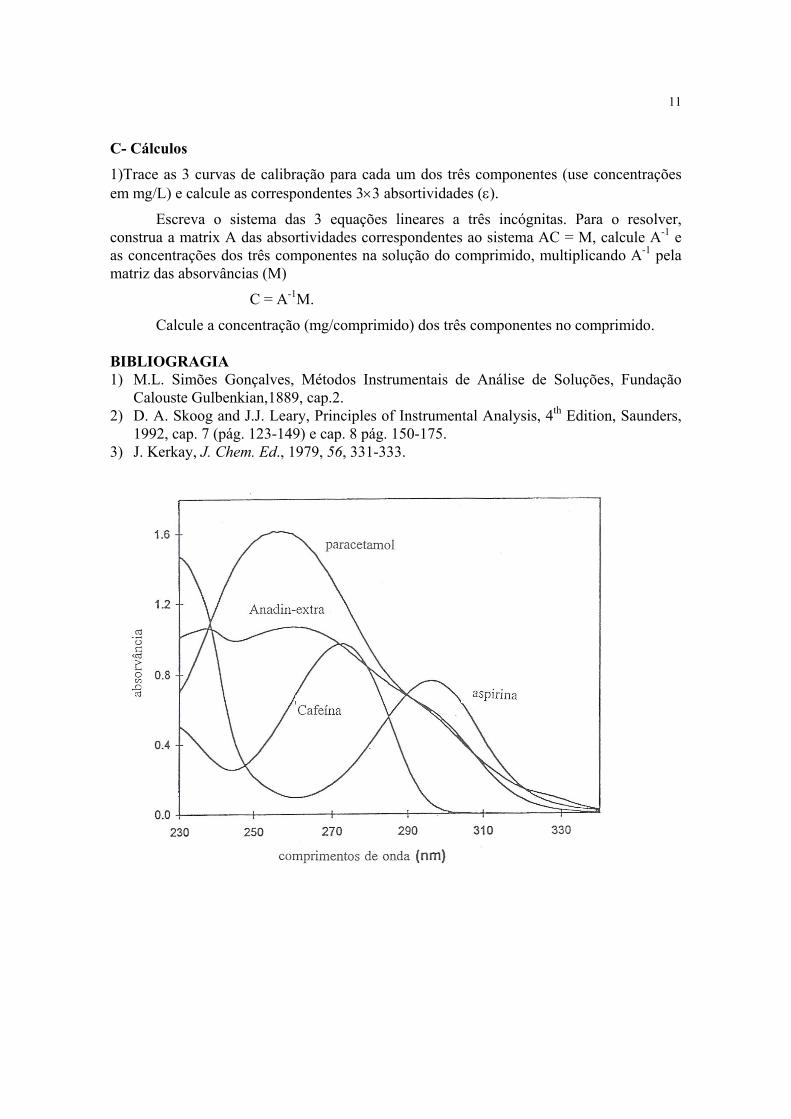
C- Cálculos
1)Trace as 3 curvas de calibração para cada um dos três componentes (use concentrações em mg/L) e calcule as correspondentes 3×3 absortividades (ε).
Escreva o sistema das 3 equações lineares a três incógnitas. Para o resolver, construa a matrix A das absortividades correspondentes ao sistema AC = M, calcule A-1 e as concentrações dos três componentes na solução do comprimido, multiplicando A-1 pela matriz das absorvâncias (M)
C = A-1M.
Calcule a concentração (mg/comprimido) dos três componentes no comprimido. BIBLIOGRAGIA 1) M.L. Simões Gonçalves, Métodos Instrumentais de Análise de Soluções, Fundação
Calouste Gulbenkian,1889, cap.2. 2) D. A. Skoog and J.J. Leary, Principles of Instrumental Analysis, 4th Edition, Saunders,
1992, cap. 7 (pág. 123-149) e cap. 8 pág. 150-175. 3) J. Kerkay, J. Chem. Ed., 1979, 56, 331-333.

12
ANÁLISE DE ÁCIDO ACETYLSALICÍLICO OU PARACETAMOL NUM COMPRIMIDO POR ESPECTROFOTOMETRIA DE UV/VIS
INTRODUÇÃO
Esta experiência envolve a determinação da quantidade de aspirina ou paracetamol num comprimido, à venda nas farmácias. A aspirina tem propriedades como analgésico, antipirético e anti-inflamatório. O paracetamol tem maior eficácia do que a aspirina como antipirético. Existem alguns problemas na análise destas drogas, incluindo a hidrólise da aspirina e a dependência do pH no caso do paracetamol.
O
O
CH3
CO2H
OH
HN
O
CH3
Aspirina Paracetamol (ácido acetilsalicílico) (4-hidroxiacetanilide)
Análise quantitativa de um comprimido
Uma vez que a aspirina e o seu produto de hidrólise (ácido salicílico) têm espectros de absorção do ultravioleta diferentes, a solução do comprimido deve ser fortemente básica para assegurar que toda a aspirina se hidrolizou e apenas o ácido salicílico está a ser analizado. Do mesmo modo, uma vez que o paracetamol é sensível ao pH e as formas ácida e básica têm diferentes espectros de absorção no UV, o facto da solução estar fortemente básica, assegura que o paracetamol está todo na forma básica. Portanto, a pH elevado, a solução do comprimido em estudo contém ou o ácido salicílico (forma básica) ou a forma básica do paracetamol.
O método espectrofotométrico que vamos utilizar para a determinação de um destes componentes (acido acetilsalicílico ou paracetamol) baseia-se no facto destes apresentarem absorção de radiação electromagnética UV, devendo a absorvância medida seguir a lei de Lambert-Beer:
A = εbc

13
A - absorvância
ε - absortividade molar
b - espessura da célula
c - concentração molar da espécie
É importante realçar que, neste tipo de determinação, em que apenas se pretende dosear um componente, a amostra a utilizar não poderá conter simultâneamente os dois componentes acima referidos (nem outros constituintes que apresentem absorções de radiação na gama do espectro de absorção do componente em estudo, como ex. a cafeína, ver fig.). Neste caso ter-se-ia que optar por uma determinação simultânea, tendo em conta a aditividade da lei de Beer.
TÉCNICA EXPERIMENTAL
A- Preparação das Soluções
A-1- Soluções para a determinação da aspirina
1) Solução 0.1 M NaOH (500 mL)
2) Soluções de aspirina
2-1- Solução inicial (1g/L)
Pese rigorosamente cerca de 1 g de ácido acetilsalicílico (aspirina) num copo, adicione alguns mL de metanol (cerca de 10 mL) para dissolver o sólido, transfira para um balão aferido de 1 L, e dilua com água desionizada atá ao traço e agite bem.
2-2 – Solução de trabalho
Prepare uma solução em que toda a aspirina tenha sido hidrolizada a ácido salicílico pipetando 5 mL da solução de partida para um balão volumétrico de 100 mL, adicionando 40 mL de solução 0.1 M NaOH e diluindo até ao traço com água desionizada. Agite bem e deixe em repouso 10 min. para assegurar que toda a aspirina é hidrolizada a ácido salicílico.
A-2 - Soluções para a determinação do paracetamol

14
1) Solução 0.1 M NaOH (500 mL)
2) Soluções de paracetamol
2-1 – Solução inicial (1g/L)
Pese rigorosamente cerca de 1 g de paracetamol (4-acetamidofenol) para um copo, adicione alguns mL de metanol (cerca de 10 mL)até dissolver e transfira para o balão de 1L, diluindo com água (agite bem).
2-2 – Solução de trabalho
Prepare uma solução em que todo o paracetamol esteja na forma básica, pipetando 2 mL de solução inicial de paracetamol para um balão volumétrico de 100 mL, adicionando 40 mL de solução 0.1 M NaOH, e depois diluindo até ao traço com água.
A-3– Soluções de Comprimido
1) Solução inicial (100 mg/100 mL)
Esmague um comprimido num almofariz até obter um pó muito fino. Pese rigorosamente cerca de 100 mg do pó num copo, adicione alguns mL de metanol e agite durante alguns minutos. Transfira então para o balão volumétrico e dilua até ao traço com água.
1-2 - Solução de trabalho
Filtre a solução inicial, pipete 2 mL desta soluçaõ para um balão de 100 mL, e proceda como em 2-2. Esta solução é instável e deve ser usada apenas até 1-2 h depois de ser preparada.
A-4– Soluções Padrão
Em balões volumétricos de 50 mL, prepare 5 soluções padrão para a aspirina e paracetamol, a partir das correspondentes soluções de trabalho, pipetando 5, 10, 15, 20 e 25 mL para balões volumétricos de 50 mL e dilua até ao traço com água desionizada.
B - Parte Instrumental

15
1 – Ligue o aparelho e faça o acerto do zero do aparelho com a solução do branco nas duas células. O aparelho traça uma linha de base recta (fazer o varrimento de comprimento de onda na gama 230-340 nm e absorvância 0.0-0.5). Para o resto das medições deixar a solução de branco na célula de referência (posição posterior).
2 – Traçe o espectro da solução mais concentrada dos padrões. Determine o comprimento de onda de absorção máxima do componente em estudo, o qual deverá ser cerca de 296 nm (aspirina, na verdade é ácido salicílico) e 257 nm (paracetamol na forma básica).
3.– Para o componente a dosear, meça as absorvâncias dos padrões e da amostra ao comprimento de onda determinado anteriormente. A absorvância da amostra deverá cair dentro da gama de calibração, diluindo se necessário.
4 – Para a determinação da concentração na amostra, calcule a massa dos comprimidos (pese 7 comprimidos e calcule a média).
BIBLIOGRAFIA
1) M.L. Simões Gonçalves, Métodos Instrumentais de Análise de Soluções, Fundação Calouste Gulbenkian,1989, cap.2.
2) D.A. Skoog and J.J. Leary, Principles of Instrumental Analysis, 4th Edition, Saunders, 1992, cap. 7 (pág. 123-149) e Cap. 8, pág. 150-175.
3) J. Kerkay, J. Chem. Ed., 1979, 56, 331-333.

16
2-1- DOSEAMENTO DO FERRO NUM CEREAL POR
ESPECTROSCOPIA DE ABSORÇÃO ATÓMICA 1. INTRODUÇÃO A absorção atómica é um método de chama em que esta é usada para atomizar uma solução, mas não para excitar os átomos a um estado de energia mais elevado, ao invés do que acontece na fotometria de chama. O facto de a chama ser apenas utilizada para atomizar a solução elimina grande parte das interferências presentes nesta última técnica. A excitação é, no caso da absorção atómica, produzida por uma lâmpada cujo cátodo emissor é oco e feito do elemento a determinar, emitindo radiações com as frequências características desse elemento. Os átomos neutros, livres, no estado de mais baixa energia, existentes na chama podem absorvê-las, incluindo a frequência de ressonância correspondente à transição do estado fundamental para o nível excitado de mais baixa energia. Estes átomos excitados voltam depois ao estado fundamental, podendo emitir energia radiante com a frequência de ressonância, mas esta é transmitida em todas as direcções e a fracção detectada pelo espectrofotómetro não produz perturbação notável na medida da radiação não absorvida. A radiação que passou através da chama, com o elemento a determinar e com o solvente, vai em seguida a um sistema de selecção de comprimentos de onda e, depois, a sua intensidade é medida num detector, determinando-se qual a percentagem que foi absorvida. Num espectrofotómetro de absorção atómica, munido dum registador, mede-se a absorção em relação à linha de base traçada com o solvente. A absorvância e a concentração estão relacionadas pela lei de Lambert-Beer
- log T = - log I / Io = A = a b c
em que I - intensidade de radiação transmitida Io - intensidade de radiação incidente T - transmitância A - absorvância

17
a - absortividade (constante para cada espécie de solvente) b - espessura do meio absorvente (neste caso a chama) c - concentração 2. PARTE EXPERIMENTAL 2.1 Material e aparelhos - Material corrente de laboratório - Espectrofotómetro de absorção atómica - Lâmpada de cátodo oco de ferro 2.2 Soluções a preparar - 1 l de HCl (1 M) - 250 ml de uma solução 100 ppm em ferro a partir de FeCl3 usando como solvente a solução de HCl (1 M); por diluição desta solução preparar 100 ml de uma solução 10 ppm em ferro usando novamente o HCl (1M) como solvente, por diluição desta prepare 50 ml de uma solução 1 ppm em ferro novamente em HCl (1M). 2.3 Técnica Preparação dos padrões Para balões de 25 ml pipete 10, 20, da solução 1 ppm em ferro e 5, 10, 15 ml da solução 10 ppm em ferro; perfaça ao traço com HCl (1M). Prepare também um branco. Preparação da amostra Pese rigorosamente cerca de 2 gramas do produto a analisar num Erlenmeyer de 250 ml e adicione-lhe 50 ml da solução de HCl (1M). Tape o copo com um vidro de relógio e leve a ferver durante 30 minutos. Agite, de vez em quando, com uma vareta, não deixando que haja projecções da amostra para fora do copo. Se a evaporação for muito intensa, reduza o aquecimento e, se necessário, adicione mais alguns mililitros de HCl (1M).

18
Deixe arrefecer e filtre então a solução para um balão de 100 ml. Perfaça o volume com a solução de HCl (1M). Se tiver indicação do fabricante sobre o teor em ferro no produto a analisar, faça uma ou duas diluições de modo a ter uma concentração em ferro na solução resultante compreendida entre as concentrações dos padrões. Caso não tenha aquela indicação proceda por tentativas até obter esta situação. Use sempre HCl (1M) como solvente. Leve as soluções padrão e amostra ao aparelho de absorção atómica, depois de escolhidas as condições experimentais para a sua utilização, meça as absorvâncias das várias soluções. Use o método da recta de calibração para determinar a concentração da amostra na solução levada ao aparelho e a partir daí calcule o erro estatístico associado a esta determinação. 3. BIBLIOGRAFIA LASWICK, P. H. , J. Chem. Ed., 50 (1973), 132.

19
2-2- DETERMINAÇÃO DO CÁLCIO EM SUMOS POR
ESPECTROSCOPIA DE ABSORÇÃO ATÓMICA
1. INTRODUÇÃO A absorção atómica é um método de chama em que esta é usada para atomizar uma solução, mas não para excitar os átomos a um estado de energia mais elevado, ao invés do que sucede na fotometria de chama. O facto de a chama ser apenas utilizada para atomizar a solução elimina parte das interferências presentes nesta última técnica. A excitação é, no caso da absorção atómica, produzida por uma lâmpada cujo cátodo emissor é feito do elemento a determinar e emite radiações com as frequências caracteristicas desse elemento. Os átomos neutros, livres, no estado de mais baixa energia existentes na chama podem absorvê-las, incluindo a frquência de ressonância correspondente à transição do estado fundamental para o nível excitado de mais baixa energia. Estes átomos excitados voltam depois ao estado fundamental, podendo emitir energia radiante com a frequência de ressonância, mas esta é transmitida em todas as direcções e a fracção detectada pelo espectrofotómetro não produz perturbação notável na medida da radiação não absorvida. A radiação que passou através da chama, com o elemento a determinar e com o solvente, vai em seguida a um sistema de selecção de comprimentos de onda e depois a sua intensidade é medida num detector, determinando-se qual a percentagem que foi absorvida. Num espectrofotómetro de absorção atómica, munido dum registador, mede-se a absorção em relação à linha de base traçada com o solvente. A absorvância e a concentração estão relacionadas pela lei de Lambert-Beer.
− = − = =log logT II
A a bc0
em que I - intensidade de radiação transmitida I - intensidade de radiação incidente T - transmitância A - absorvância a - absortividade (constante para cada espécie absorvente) b - espessura do meio absorvente (neste caso a chama) c - concentração

20
2. PARTE EXPERIMENTAL 2.1. Material e aparelhagem - material corrente de laboratório - espectrofotómetro de absorção atómica - lâmpada de cátodo oco de cálcio - centrífuga 2.2. Soluções a preparar - 1 l solução 500 µg/ml em Ca, a partir de carbonato de cálcio (adicione HCl gota a gota até dissolver). - 250 ml solução 100 µg/ml em Ca, a partir da solução anterior. - 100 ml solução 5 % em La, a partir de óxido de lantâneo (La2O3 , adicione gota a gota até se dissolver). - HCl (6 M). 2.3. Técnica Preparação da amostra Filtre cerca de 50 ml de sumo por um filtro de poros apertados. Se necessário centrifuge previamente a amostra de modo a obter um líquido límpido. Determinação do cálcio Em balões de 100 ml prepare uma gama de 5 soluções-padrão de concentração compreendida entre 0 e 7 ppm em Ca a partir da solução 100µg/ /ml. Adicione a todas elas 10 ml da solução de La, 20 ml da solução de HCl e 5 ml de sumo. Prepare ainda um branco para calibrar o aparelho. Determine o teor em Ca no sumo pelo método de adição de padrão. 3. BIBLIOGRAFIA . A. N. Strohl, J. Chem. Educ., vol. 62, nº 4, pág. 343 (1985). . M. Lurdes S. S. Gonçalves, Métodos Instrumentais para Análise de Soluções, Ed. Fundação Calouste Gulbenkian, Lisboa (1983).

21
2-3- DOSEAMENTO DO COBRE NUM CEREAL POR ESPECTROSCOPIA DE ABSORÇÃO ATÓMICA (CÂMARA DE GRAFITE)
1. INTRODUÇÃO
A absorção atómica com câmara de grafite é um método de absorção em que a câmara (tubo de grafite) aquecida electricamente) é usada para atomizar uma solução, mas não para excitar os átomos a um estado de energia mais elevado. O facto de o tubo de grafite (atomizador) ter reduzidas dimensões (l = 3 cm), de a amostra não ser aspirada e nublizada e atomizada em meio comburente, como no método de chama, elimina grande parte das interferências presentes nesta última técnica e tenha várias vantagens. Entre estas destacam-se o menor volume de amostra, o mais baixo limite de detecção e menores interferências.
A excitação é produzida por uma lâmpada cujo cátodo emissor é oco e feito do elemento a determinar, emitindo radiações com as frequências características desse elemento. Os átomos neutros, livres, no estado de mais baixa energia, existentes na câmara podem absorvê-las, incluindo a frequência de ressonância correspondente à transição do estado fundamental para o nível excitado de mais baixa energia.
A radiação que passou através do tubo de grafite, com o elemento a determinar, vai em seguida a um sistema de selecção de comprimentos de onda e, depois, a sua intensidade é medida num detector, determinando-se qual a percentagem que foi absorvida.
A amostra que é pipetada para a câmara é sujeita a vários passos de variação de temperatura com o tempo cuja função é evaporação ao solvente, redução a cinzas e atomização.
A absorvância e a concentração na solução estão relacionadas pela lei de Lambert-Beer (válida no percurso óptico).
-log T = -log I/Io = A = A = a b c
em que I - intensidade de radiação transmitida Io - intensidade de radiação incidente T - transmitância

22
A - absorvância a - absortividade (constante para cada espécie de solvente) b - espessura do meio absorvente (neste caso a câmara) c - concentração 2. PARTE EXPERIMENTAL
2.1 Material e aparelhos
- Material corrente de laboratório - Espectrofotómetro de absorção atómica com câmara de grafite (ver Fig.1) - Lâmpada de cátodo oco de cobre
2.2 Soluções a preparar
- 11 de HCl (1 M) - A partir de uma solução stock 1000 ppm em cobre (Cu(NO)2) prepare uma
solução 1000 ppb em cobre em solução de HCl (1 M); preparar 100 ml de uma solução 50 ppb em cobre usando novamente a solução de HCl (1M) como solvente.
2.3 Técnica
• Preparação de padrões (0, 5, 10, 15, 20, 25 ppb) Esta preparação é feita automaticamente pelo aparelho, usando o branco (HCl,
1M) e a solução mais concentrada (50 ppm), após prévia programação de concentração dos padrões no aparelho.
• Preparação da amostra
Pese rigorosamente cerca de 1,5 gramas do cereal a analisar num Erlenmyer de 250 ml e adicione-lhe 50 ml da solução de HCl (1M). Tape o copo com um vidro de relógio e leve a ferver durante 30 minutos. Agite, de vez em quando, com uma vareta, não deixando que haja projecções da amostra para fora do copo. Se a evaporação for muito intensa, reduza o aquecimento e, se necessário, adicione mais alguns mililitros de HCl (1M).
Deixe arrefecer e filtre então a solução para um balão de 100 ml. Perfaça o volume com a solução de HCl (1M). Se tiver indicação do fabricante sobre o teor em

23
cobre no produto a analisar, faça uma ou duas diluições de modo a ter uma concentração em cobre na solução resultante compreendida entre as concentrações dos padrões.
Faça o programa das concentrações dos padrões, conforme indicação no aparelho.
Faça em seguida o programa no que concerne às temperaturas dos vários passos (evaporação, redução a cinzas e atomização e limpeza) velocidade e tempo de aquecimento e fluxo de azoto de um modo semelhante ao representado na Tabela I.
Introduza o branco e a solução mais concentrada e a amostra em cuvetes do tambor de doseamento automático ("autosampler").
Recolha os dados das absorvâncias referentes aos padrões e amostras e use o método da recta de calibração para determinar a concentração da amostra na solução levada ao aparelho e a partir daí calcule o erro estatístico associado a esta determinação.
3. BIBLIOGRAFIA
LASWICK, P.H., J. Chem. Ed. 50 (1973), 132.
B. WELZ, Atomic Absorption Spectroscopy, VCH, Germany, 1985.
Tabela I. Programação da câmara de grafite.
Temperatura (oC) Tempo (s) Fluxo de N2 Leitura (ON/OFF) 85 5 3.0 OFF 95 40 3.0 OFF 120 10 3.0 OFF 1000 5 3.0 OFF 1000 1 3.0 OFF 1000 2 0 OFF 2300 1.1 0 ON 2300 2 0 ON 2300 2 3.0 OFF

24
3.1-DETERMINAÇÃO FLUORIMÉTRICA DO QUININO EM ÁGUA TÓNICA
1 - Introdução O presente trabalho ilustra as potencialidades da fluorescência molecular como método analítico. O quinino numa solução de H2SO4 (0,05 M) apresenta duas bandas de absorção centradas em 250 e 350 nm. Qualquer que seja o comprimento da onda de excitação o máximo da emissão situa-se sempre a 450 nm.
N
H3CO
N
CH2
HO
Quinino Espectro de excitação ( ) e emissão ( ) de fluorescência do sulfato de quinino em H2SO4 (0,05M) O quinino é uma substância muito utilizada como padrão e para muitos fluorímetros comerciais a sua sensibilidade é definida em termos de fluorescência de quinino.
Interferências - O quinino pode existir na água tónica sob a forma de hidrocloreto ou sulfato de quinino até um conteúdo máximo de 65,4 ppm de quinino.
A única interferência que pode aparecer na dosagem do quinino na água tónica é o ião cloreto que actua como agente de extinção da fluorescência do quinino. No entanto esse efeito pode ser desprezado se a concentração em ião cloreto for inferior a 0,4.10-3 M como acontece vulgarmente. Para a concentração de ião cloreto indicada o erro cometido é inferior a 0,4%. 2 - Parte Experimental 2.1 - Reagentes . sulfato de quinino dihidratado: (C20H24N2O2)2.H2SO4.2H2O . ácido sulfúrico concentrado, pró-análise. 2.2 - Material e aparelhagem

25
. espectrofluorímetro*/ fluorímetro . material corrente de laboratório 2.3 - Preparação de soluções 100 ml - solução de ácido sulfúrico 1 M. Fazer uma toma de 14 ml de H2SO4
concentrado (36 N) para um balão de 50 ml contendo cerca de 25 ml de água destilada e depois perfazer ao traço (a concentração desta solução é 5M). Diluir a solução de 1/5.
500 ml - solução de ácido sulfúrico 0,05 M por diluição da solução anterior.
1 l - solução padrão de sulfato de quinino, 100 ppm em quinino. Pesar 120,7 mg de sulfato de quinino dihidratado e transferir para um balão de 1000 ml. Juntar 50 ml de H2SO4 1 M e perfazer ao traço com água destilada.
50 ml - solução intermédia 10 ppm quinino. Diluir de 1/10 a solução anterior, perfazendo o volume com H2SO4 0,05 M.
Solução amostra (água tónica) - Fazer uma toma de 5 ml de água tónica para um balão de 250 ml e perfazer ao traço com H2SO4 0,05 M. 2.4 Técnica Preparar padrões de concentração 0,2 ; 0,5 ; 0,8 ; 1,2 ; 1,6 e 2,0 ppm de quinino em balões de 25 ml, perfazendo sempre o volume com H2SO4 0,05 M. Prepare ainda um branco. Após a calibração do aparelho, ler as intensidades de emissão para cada uma das soluções. Tendo em conta a diluição da amostra determinar o teor de quinino na água tónica analisada e comparar o resultado obtido com as indicações do rótulo. Nota: 1)-O hidrocloreto de quinino comercial contém duas moléculas de água: C20H24N2O2.HCl.2H2O. 2)- Caso use um espectrofluorímetro use para os padrões concentrações mais diluidas (10 vezes ) do que as indicadas na técnica. 3 - Bibliografia
O'Reilly, J. E., J. Chem. Educ. 52 (9) , 610 (1975)

26
3.2-DETERMINAÇÃO FLUORIMÉTRICA DA RIBOFLAVINA (VITAMINA B2) NUM COMPRIMIDO
1 – Introdução A riboflavina (vitamina B2) é uma vitamina essencial ao metabolismo dos animais,
(catalização de reacções químicas) e, embora não são sintetizada pelos mesmos, é
abundantemente distribuída em géneros alimentícios animais e vegetais, tais como fígado,
leite, rim, carne, ovos, ostras, nabos, beterraba e farelo de arroz. A ausência dessa vitamina
no organismo pode causar queda de cabelo, lesões na pele, olhos, lábios, boca e orgãos
genitais entre outras.
A riboflavina (7,8-dimetil-10-(ribitil)isoaloxazina) é uma molécula aromática cuja sua estrutura molecular está abaixo indicada.
N
N
N H
N
H O
H OO H
O H
O
O
Na sua forma cristalina é um pó amarelo-alaranjado (p.f. = 285ºC), levemente solúvel em água (≈ 0,1 g/l) e em álcool, e insolúvel em éter etílico e clorofórmio. Em solução aquosa, a riboflavina exibe uma fluorescência na região do visível, que ao olho humano se apresenta como verde-amarelada (λ = 530-545 nm), pelo que a Farmacopeia Americana (USP-23) e a A.O.A.C. recomendam que o seu doseamento seja efectuado pelo método fluorimétrico, com excitação ao comprimento de onda de 444 nm e medida da intensidade de fluorescência a 530 nm.
No estado anidro, a riboflavina é estável à luz, porém, em soluções alcalinas decompõe-se rapidamente. Na presença de oxigénio, a riboflavina é transformada irreversivelmente pela luz em luminoflavina, lumicromo e compostos de menor importância. 2 –Parte experimental 2.1 – Reagentes

27
Riboflavina ácido acético concentrado, proanálise.
2.2 – Material/Aparelhagem
Espectrofluorímetro/Fluorímetro Material corrente de laboratório Folha de alumínio
2.3 – Preparação de Soluções 1 L - Solução de ácido acético 5% (v/v) 500 mL - Solução mãe em riboflavina (20 ppm)
Dissolve-se 10,0 mg de riboflavina num balão volumétrico de 500 mL usando a solução de ácido acético a 5% (V/V).
50 mL - Solução intermédia de riboflavina (2,0 ppm) por diluição da solução anterior em
ácido acético 5% (V/V).
Solução amostra - Esmague um comprimido contendo vitamina B2 num almofariz até obter um pó fino. Pese rigorosamente cerca de 50 mg do pó num copo e adicione solução de ácido acético 5% (V/V) até se dissolver (caso exista precipitado insolúvel proceda a uma filtração). Transfira, então, para um balão volumétrico de 500 mL e dilua até ao traço com a solução de ácido.
2.4 – Técnica
1. Em balões de 25 mL, prepare 5 padrões de concentração entre 0,02 e 0,2 ppm de riboflavina em ácido acético 5% (V/V). Prepare ainda um branco.
2. Após a calibração do aparelho, selecione os comprimentos de onda de excitação e de emissão experimental e leia as intensidades de emissão para cada uma das soluções padrão e amostra. (Se a absorvância da amostra sair do intervalo dado pelos padrões proceda a uma nova diluição desta).
3. Tendo em conta a diluição da amostra, determine o teor de riboflavina no comprimido e compare o resultado obtido com as indicações do rótulo.
ATENÇÃO: Uma vez que a riboflavina é sensível à luz, os balões volumétricos que contêm soluções dessa vitamina devem ser envolvidos com folha de alumínio. 3 – Bibliografia 1) http://www.chembio.niu.edu/eletrochem/lab3.htm

28
2) C.H. Whitnah, B.L. Bernice, M.M. Kramer, J. Am. Soc. 1937, 59, 1153. 3) M.L. Gonçalves, “Métodos Instrumentais para Análise de Soluções”, 4ª edição,
Fundação Calouste Gulbenkian, 2001, Lisboa.

29
II-Anexo: Métodos de Cálculo e Erros em Química Analítica 1. Métodos de Cálculo de Concentrações
(Calibração com Padrões)
a) Método da recta de calibração Lpadrão = K × Cpadrão K (L = valor de Leitura)
Lamostra = K × Camostra Cam.= Lamostra/K
b) Método da adição de padrão
. . . +CP1 CP2 CPi
amos-tra
Lam
CamCp
- Determinação da concentração da amostra pelo método da recta de calibração

30
Usa-se quando MATRIZ AMOSTRA ≠ MATRIZ PADRÕES, afectando o
valor de K que relaciona o valor de Leitura com a amostra e os padrões.
Lmedido = K' × (Cpadrão + Camostra) (Camostra = const)
L = 0 Cam. = - Cpad.
c) Método do padrão interno Lpadrão = K × Cpadrão = K' × Vinjectado × Cpadrão
CPiVam Vam
CP2 . . . CPiVam
Cp
- Determinação da concentração da amostra pelo método da adição de padrão
Cam = -(CP)L=O
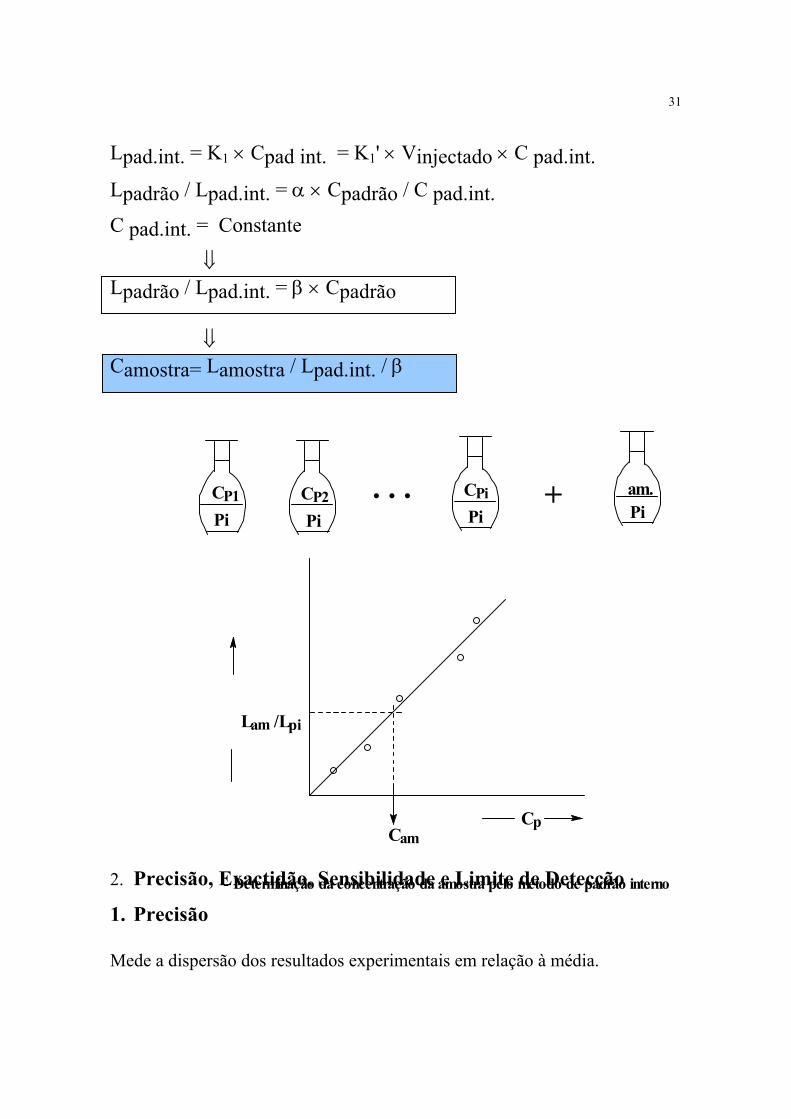
31
Lpad.int. = K1 × Cpad int. = K1' × Vinjectado × C pad.int.
Lpadrão / Lpad.int. = α × Cpadrão / C pad.int.
C pad.int. = Constante ⇓
Lpadrão / Lpad.int. = β × Cpadrão
⇓ Camostra= Lamostra / Lpad.int. / β
2. Precisão, Exactidão, Sensibilidade e Limite de Detecção
1. Precisão Mede a dispersão dos resultados experimentais em relação à média.
- Determinação da concentração da amostra pelo método de padrão interno
CpCam
Lam /Lpi
CPi. . .CP2PiCP1
Pi Pi+
Piam.

32
(⇔ ∃ Erros aleatórios)
Avaliação- Métodos Estatísticos x
xxi )( −
a) Exactidão Mede o desvio em relação ao valor real. (⇔ ∃ Erros sistemáticos)
Avaliação- (?) r
rcx
xx )( −
b) Sensibilidade Razão entre o acréscimo do valor lido, ∆L, e a variação da concentração AC, correspondente aquele acréscimo (∆L/∆C).
Se L = F(c) ⇔ relação linear ⇒ (∆L/∆C = declive da recta de calibração) c) Limite de Detecção Menor valor que se pode distinguir do zero
⇓ Menor valor de concentração que um dado método consegue medir. Avaliação: limite de detecção operacional (L.D.)
L.D. = 3 σy/x/a
=σ=
estimativadapadrãodesviorectadaangularecoeficienta
xy /
3- Análise estatística dos Erros (Sumário)
a) Recta dos mínimos quadrados e erros associados (programa Excell)
y = (a ± σa) × (b ± σa)

33
a: declive; σa erro associado.
b: ordenada na origem; σb erro associado.
Qualquer destes parâmetros é calculado pelo Excell
b) Cálculo do erro na concentração da amostra σx0, a partir da recta dos
mínimos quadrados:
→Método da recta de calibração
σx02 = (σx/y2 /a2) {(1/n) + (1/L) + (yo-y)2/[a2Σ(xi-x)2]}
σy/x2 : desvio padrão da estimativa (standard error
dado na folha de cálculo do Excell),
n = número de pontos experimentais
L = número de leituras para cada padrão ou amostra;
x = média dos valores de xi; y = média dos valores
de yi
xi = cada valor de x
→Método de adição de padrão
σx2 = (σx/y2 /a2) {(1/n) + y2/[a2Σ(xi-x)2]}




![Apostila de Espectrofotometria Uv-Vis[1]](https://static.fdocumentos.com/doc/165x107/557201ab4979599169a212be/apostila-de-espectrofotometria-uv-vis1.jpg)