
Memorando de Entendimento - SNS · PDF fileMemorando de Entendimento ... partilhados pelos...
24
Memorando de Entendimento Entre: Associação Portuguesa da Indústria Farmacêutica (APIFARMA) Associação Portuguesa de Medicamentos Genéricos e Biossimilares (APOGEN) Associação de Grossistas de Produtos Químicos e Farmacêuticos (GROQUIFAR) Associação Nacional de Farmácias (ANF) Associação de Farmácias de Portugal (AFP) Sobre o Modelo de Criação e Gestão de um Sistema de Verificação de Medicamentos da responsabilidade das Partes Interessadas em Portugal Lisboa, 17 de outubro de 2016
Transcript of Memorando de Entendimento - SNS · PDF fileMemorando de Entendimento ... partilhados pelos...

Memorando de Entendimento
Entre:
Associação Portuguesa da Indústria Farmacêutica (APIFARMA)
Associação Portuguesa de Medicamentos Genéricos e Biossimilares (APOGEN)
Associação de Grossistas de Produtos Químicos e Farmacêuticos (GROQUIFAR)
Associação Nacional de Farmácias (ANF)
Associação de Farmácias de Portugal (AFP)
Sobre o Modelo de Criação e Gestão de um Sistema de Verificação de
Medicamentos da responsabilidade das Partes Interessadas em Portugal
Lisboa, 17 de outubro de 2016

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 2 de 24
Definições
Compatível ‐ Capaz de existir ou ocorrer em conjunto sem conflitos.
Constituinte (s) – refere‐se à(s) comunidade(s) das partes interessadas, adiante designadas ‘Partes’,
representando os utilizadores materiais do Sistema, com plenos direitos sobre a sua participação nas
Organizações Nacionais de Verificação de Medicamentos (National Medicines Verification
Organisations – sigla em inglês NMVO), compreendendo (1) as empresas farmacêuticas de
investigação que comercializam medicamentos sujeitos e não sujeitos a receita médica, (2) empresas
farmacêuticas de medicamentos genéricos e biossimilares, (3) distribuidores grossistas de produtos
farmacêuticos (4), farmácias comunitárias (5), farmácias hospitalares e os (6) distribuidores
farmacêuticos paralelos.
Dados Padrão – dados relacionados a um dado artigo/item de venda, sendo igual para todas as
embalagens deste artigo/item (por exemplo, nome, número do artigo, forma de apresentação,
dosagem, tipo de embalagem) que devem ser registados no Sistema.
EMVO (sigla em inglês) ‐ European Medicines Verification Organisation – uma organização sem fins
lucrativos criada pelos parceiros europeus para gerir um repositório eletrónico central (plataforma),
que vai ligar uma série de repositórios de dados nacionais ou supranacionais e que servirá de
plataforma para permitir a verificação da autenticidade dos medicamentos em qualquer ponto da
cadeia de abastecimento no Espaço EEE. De forma integrada, a plataforma central europeia e os
repositórios nacionais podem ser referidos como "Sistema Europeu de Verificação de Medicamentos"
(sigla em inglês, EMVS) ou como "Sistema".
Evento Excepcional – qualquer indicação que dê origem a suspeitas de que um determinado produto
possa ser falsificado ou que o Sistema possa ter sido atacado ou alvo de um outro problema que
impeça o uso normal ou ininterrupto do Sistema. Um evento excepcional pode incluir, a título de
exemplo, uma falha de verificação/autenticação (porque o número de série não está no Sistema ou se
encontra registado como tendo sido dispensado ou desativado devido a uma recolha de lote, por
exemplo), uma tentativa de intrusão por pessoas não autorizadas, ou qualquer outra atividade que
sugira uma eventual intrusão do Sistema. Os Documentos de Fundação irão estabelecer uma escala de
vários níveis para os eventos excepcionais e definir os processos relacionados.
Documentos de Fundação – os documentos técnicos e jurídicos fundamentais, acordados antes do
estabelecimento da NMVO, que serão incorporados no presente Memorando de Entendimento, se e
quando estes forem adotados no âmbito de um acordo entre as partes sobre a arquitetura do sistema,
as regras operacionais, incluindo os princípios acordados sobre o conjunto de casos, a validação de
dados, os tempos de resposta, a gestão de eventos excecionais e os processos relacionados com o
Sistema e com os requisitos operacionais e de segurança.
Interoperabilidade ‐ Diferentes sistemas que podem interagir, mas que têm diferentes modi operandi.
Medicamentos – os produtos que devem ser dotados de dispositivos de segurança em conformidade

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 3 de 24
com a Diretiva sobre Medicamentos Falsificados1 e o respetivo Regulamento Delegado2.
Organização Nacional de Verificação de Medicamentos (sigla em inglês NMVO) – a associação nacional
que será estabelecida pelas Partes para gerir um repositório nacional de dados, que se encontrará
ligado à plataforma central europeia e que servirá como plataforma para permitir a verificação da
autenticidade dos medicamentos em qualquer ponto da cadeia de abastecimento no Espaço EEE. De
forma integrada, a plataforma central europeia e os repositórios nacionais podem ser referidos como
"Sistema Europeu de Verificação de Medicamentos" (sigla em inglês EMVS) ou como "Sistema".
Requisitos da EMVO ‐ documentos essenciais que são expressamente identificados como "EMVO:
requisitos para o sistema EMVS” e que fazem parte dos documentos base da EMVO sobre a repartição
de custos da EMVO e outros princípios e elementos técnicos do Sistema.
TIC ‐ Tecnologia de Informação e Comunicação.
Titular de Autorização de Fabrico – para efeitos do presente documento refere‐se aos fabricantes e
aos distribuidores farmacêuticos paralelos envolvidos em operações de reembalagem, excluindo‐se as
empresas contratadas e subcontratadas que não são responsáveis por colocar o produto farmacêutico
final no mercado. Para evitar dúvidas, um fabricante com contratos desta natureza, em que todas ou
algumas fases do processo de fabrico sejam da responsabilidade de empresas contratadas ou
subcontratadas, deve ser considerado Titular de Autorização de Fabrico.
Titular de Autorização de Introdução no Mercado (AIM): para efeitos do presente documento refere‐
se às entidades detentoras de uma AIM.
Partes – a nível nacional, incluem:
‐ indústria farmacêutica de investigação que comercializa medicamentos sujeitos e não sujeitos
a receita médica;
‐ indústria farmacêutica de medicamentos genéricos e biossimilares;
‐ distribuidores grossistas;
‐ farmácias.
Parceiro ou Parte Interessada – membro individual das Partes acima mencionadas ou qualquer outro
utilizador do sistema, ou órgão interessado incluindo os departamentos e agências governamentais.
Verificação – Ato de confirmação de que um produto farmacêutico existe no repositório nacional com
os mesmos dados que constam da matriz do código 2D da respetiva embalagem.
1 Diretiva 2011/62 de 8 de Junho 2011 que altera a Diretiva 2001/83/CE que estabelece um código comunitário
relativo aos medicamentos para uso humano, no que diz respeito à prevenção da introdução, na cadeia de
abastecimento legal, de medicamentos falsificados (JO 2011 L 174/74, de 1.07.2011).
2 Regulamento Delegado (EU) 2016/161 da Comissão de 2 de Outubro de 2015 (JO 2016 L32/1, de 9.02.2016)

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 4 de 24
1. Introdução
A Directiva sobre Medicamentos Falsificados introduz dispositivos de segurança harmonizados e
obrigatórios a nível europeu sob a forma de dispositivos de prevenção de adulteração e de um
identificador único como parte da embalagem exterior dos medicamentos sujeitos a receita médica,
embora se apliquem derrogações baseadas numa avaliação de risco. Esta medida tem como objetivo
impedir a introdução de medicamentos falsificados na cadeia de abastecimento legal e, em última
análise, que estes cheguem aos doentes. O Regulamento Delegado, que será aplicável a partir de
9.02.2019, define as características e as especificações técnicas do identificador único, permitindo
identificar cada embalagem e garantir a acessibilidade às bases de dados nacionais ou sistema de
repositórios que permitem verificar a autenticidade de cada embalagem dispensada.
As Partes apoiam inteiramente os princípios da Directiva e do Regulamento Delegado e gostariam de
trabalhar com a Comissão Europeia e as autoridades competentes nacionais no estabelecimento de
um sistema eficaz, no interesse da segurança dos doentes. Neste contexto, as partes têm vindo a
colaborar na elaboração do presente Memorando de Entendimento com vista a promover,
conjuntamente, o desenvolvimento de um sistema de verificação e autenticação de produtos custo
efetivo e escalonável, a ser criado e gerido pelas partes interessadas de acordo com o estabelecido no
art.º 31.º do Regulamento Delegado.
Este Memorando de Entendimento entre as partes nacionais tem como objectivo:
• descrever as condições para a execução conjunta de um sistema nacional tendo em conta os
requisitos da Directiva e do Regulamento Delegado,
• apoiar os processos internos e gerir o desenvolvimento e/ou as adaptações que se identifiquem
como necessários,
• constituir a base para uma tomada de decisão,
• apoiar e facilitar a comunicação sobre estas questões.
Este Memorando de Entendimento não se destina a ser juridicamente vinculativo para as Partes,
sendo sua única intenção a de apoiar o desenvolvimento atempado de uma organização nacional
(NMVO), a qual possa implementar um sistema de verificação e autenticação de produtos custo‐
efetivo para as necessidades nacionais.
Os custos do sistema de verificação e autenticação de produtos serão suportados pelos Titulares de
Autorização de Fabrico de medicamentos dotados de dispositivos de segurança mencionados no
artigo 54.º‐A, n.º 2, alínea e) da Directiva Europeia e no n.º 5 do art.º 31.º do Regulamento Delegado e
partilhados pelos mesmos tendo por base os princípios acordados no modelo de alocação de custos
definidos pela EMVO em relação ao modelo Blueprint. O sistema não poderá, em caso algum, ser
gerido para fins comerciais.
As Partes Participantes acordam sobre a necessidade de criar um quadro global para a implementação
da Directiva e do Regulamento Delegado, que irá determinar, entre outras questões, o acesso aos
dados, os respectivos controlos e as regras de funcionamento de uma base de dados nacional
(Enquadramento). No entanto, este Memorando de Entendimento não é, em si, a base de tal
enquadramento, devendo este ser acordado separadamente entre as Partes.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 5 de 24
A base de dados nacional vai ser ligada ao sistema Europeu, o qual é composto por uma plataforma
central europeia de dados e informações, que por sua vez está ligada a um conjunto de outras bases
de dados nacionais. Estas bases de dados são a plataforma para a verificação da autenticidade de cada
embalagem pelas partes interessadas. O sistema europeu será interoperável entre os diferentes
estados membros e irá funcionar para todos os medicamentos sujeitos a receita médica e não sujeitos
a receita médica dotados de dispositivos de segurança, nos termos do anexo II do Regulamento
Delegado.
O sistema deverá ser capaz de responder às diferentes necessidades dos países, com base na
plataforma comum que estabelece um sistema de codificação harmonizado a nível europeu.
Assente no sistema de repositórios, a verificação da autenticidade do medicamento desde o
fabricante até ao ponto de dispensa, i.e, a verificação e a autenticação de extremo a extremo pode ser
realizada por:
1. fabricantes que inserem os dados padrão na plataforma central europeia,
2. intervenientes de toda a cadeia de abastecimento (distribuidores grossistas, farmácias,
distribuidores farmacêuticos paralelos, etc.) que utilizem equipamentos de leitura ótica para
verificar a autenticidade das embalagens,
3. distribuidores farmacêuticos paralelos responsáveis pela informação sobre os identificadores
únicos carregada ou desativada no sistema de repositórios,
4. intervenientes na cadeia de abastecimento, responsáveis por alterarem a informação quanto
ao estado da embalagem na base de dados numa situação de dispensa ao doente.
Outros aspetos a acordar entre as Partes e que podem promover potenciais ganhos de eficiência
incluem (funcionalidades adicionais e não propriamente requisitos do Regulamento Delegado):
• permitir a verificação automática dos prazos de validade,
• melhorar as atividades de farmacovigilância,
• reduzir o risco de fraude,
• maior efetividade nos procedimento de recolha dos produtos do mercado, prevenindo que
estes entrem novamente na cadeia de abastecimento e sejam dispensados ao doente,
• tratamento mais eficiente dos procedimentos de devolução dos produtos.
As Partes aceitam inteiramente a necessidade de um enquadramento global para a implementação da
Directiva e do Regulamento Delegado, que irá determinar, entre outras questões, o acesso aos dados,
os respectivos controlos e as regras de funcionamento de uma base de dados nacional. A utilização
dos acessos terá de ser acordada por todas as Partes, tendo em conta a situação jurídica existente em
matéria de acesso e propriedade de dados.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 6 de 24
2. Um Modelo Europeu de Verificação de Medicamentos ‐ 10 Princípios
Fundamentais
As Partes concordam que o quadro para a implementação da Directiva e do Regulamento Delegado
deve reflectir os seguintes princípios fundamentais:
1. Garantir a continuidade da protecção ao longo de toda a cadeia de abastecimento:
• Os distribuidores farmacêuticos paralelos devem, aquando da substituição dos dispositivos de
segurança, verificar a autenticidade do identificador único que contém o código do produto e
o número de série, desativá‐lo ao nível da plataforma central europeia, para posteriormente
ser gerado um novo número de série. Na base de dados, este novo número deve ficar ligado
ao número original do produto para o respetivo lote, de forma a permitir a sua rastreabilidade
em situações de recolha do mercado ou outras situações relacionadas com a segurança.
2. Garantir um sistema único de identificação e de codificação em cada embalagem na UE:
• Considerando a circulação de medicamentos ao nível das fronteiras, qualquer sistema de
codificação e de identificação eficiente deve ser capaz de permitir as trocas de informação
entre os Estados‐Membros. Deve, portanto, ser um sistema de codificação harmonizado em
toda a UE, mas que permita a inclusão dos códigos nacionais do produto relevantes.
• As Partes propõem a adopção do código bidimensional3, contendo um número de série único
para codificar todos os produtos que devem ser dotados de dispositivos de segurança. Este
código pode ser verificado na respectiva base de dados. Tal significa que o farmacêutico pode
rapidamente verificar qual o estado de cada embalagem antes de a dispensar ao doente. Além
do número de série, o código pode incluir o prazo de validade, juntamente com a identificação
do produto (incluindo o código nacional), o número de lote e eventuais sufixos que sejam
exigidos pelos distribuidores farmacêuticos paralelos, proporcionando uma melhoria acrescida
na protecção da segurança do doente.
3. Garantir a articulação dos sistemas de base de dados de verificação dos produtos num regime de
interoperabilidade na UE:
• Além de utilizar um padrão comum para a identificação das embalagens na Europa, todos os
sistemas de bases de dados nacionais devem também ser capazes de operar em conjunto e
trocar informações, permitindo desta forma que qualquer farmacêutico e distribuidor
grossista possa, quando considerado necessário, verificar em qualquer Estado Membro se a
embalagem foi anteriormente dispensada, independentemente do seu país de origem.
• Deverá haver flexibilidade suficiente para implementar as soluções nacionais no Sistema.
• Os sistemas de bases de dados nacionais devem respeitar requisitos de garantia da qualidade
equivalentes.
• Sem essa interoperabilidade, é introduzida a possibilidade de os falsificadores explorarem as
lacunas entre os sistemas nacionais e, desta forma, introduzirem os medicamentos falsificados
na cadeia legal de abastecimento.
3 Data matrix ECC 200

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 7 de 24
4. Verificação sistemática de cada embalagem serializada ao nível da farmácia:
• É da responsabilidade de todos os intervenientes na cadeia de abastecimento verificar a
autenticidade e a integridade dos dispositivos de segurança dos medicamentos.
• A verificação ao nível da farmácia, no momento da dispensa, é uma forma robusta e custo‐
efectiva de melhorar a protecção dos doentes.
• Contudo, a menos que cada embalagem serializada seja verificada de forma individual no
momento da dispensa, os doentes não beneficiarão inteiramente da protecção conferida
pelos dispositivos de segurança. O número de série único apenas poderá garantir essa
protecção se for rotineiramente verificado ao nível de uma base de dados central e o estado
da embalagem for alterado para "dispensado" no momento da sua entrega ao doente.
• Os sistemas devem ser configurados de forma a possibilitar que os farmacêuticos realizem as
verificações necessárias no momento da entrada dos medicamentos no stock da farmácia,
bem como no momento da sua dispensa. Uma vez que podem existir desafios técnicos quanto
à verificação das embalagens dos medicamentos no ponto de dispensa, os farmacêuticos
podem, inicialmente, adoptar um sistema de verificação apenas à entrada na farmácia, até
que todas as questões técnicas em relação à verificação no ponto de dispensa sejam
resolvidas.
• O processo de verificação na farmácia deve ser virtualmente instantâneo, assegurando um
eficiente fluxo de trabalho e evitando atrasos. Para garantir que os produtos são verificados
numa única acção de leitura pelos equipamentos de leitura óptica, o respetivo software de
verificação deve estar integrado com os atuais softwares existentes na farmácia.
• O processo de verificação ao nível do distribuidor grossista é feito nos procedimentos de
devolução de medicamentos previstos no art.º 20.º do Regulamento Delegado, com as
derrogações estabelecidas do seu art.º 21.º, sem que o estado do medicamento seja alterado
ao nível da base de dados.
• O processo de verificação pelos distribuidores farmacêuticos paralelos deve, igualmente,
permitir a verificação dos produtos à sua entrada sem alterar o estado do medicamento na
base de dados.
• Os parceiros devem trabalhar em conjunto para definir os procedimentos normalizados para o
tratamento de eventos excepcionais, tais como os relacionados com falhas de verificação,
falhas do sistema, etc.
5. Maximizar todos os benefícios potenciais de uma serialização em massa:
• A serialização em massa proporciona outros benefícios para além das melhorias ao nível da
prevenção contra os medicamentos falsificados. A sua maximização poderá incentivar à
utilização generalizada destes sistemas de identificação e apoiar todos os parceiros.
• O sistema de codificação permite que o número do lote, número de série e prazo de validade
sejam legíveis por meios eletrónicos, aumentando significativamente a segurança do doente e
melhorando os procedimentos de recolha de produtos.
6. Garantir a segurança do doente e proteger a sua privacidade:
• Os sistemas de verificação servem para prevenir falsificações, e não para aceder aos dados
individuais dos parceiros.
• Os fabricantes não pretendem nem irão ter acesso a informações sobre o perfil individual do
doente/prescrição.
• Os dados transaccionais pertencem ao farmacêutico ou, em relação à verificação no

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 8 de 24
distribuidor grossista, ao próprio distribuidor grossista ou, em relação aos distribuidores
farmacêuticos paralelos, ao titular da autorização de fabrico que realiza esta actividade.
Contudo, as partes interessadas podem necessitar de ter acesso a certos dados para as ajudar
em investigações quando existem falhas no sistema de verificação, numa situação de recolha
de produto ou quando possa existir um nível de actividade pouco comum relacionado com um
dado número de série, de acordo com as circunstâncias nacionais.
• Qualquer utilização adicional de dados transacionais terá de ser acordada entre as partes
interessadas, de acordo com as circunstâncias nacionais.
7. Combinar embalagens invioláveis com um número de série único:
• As Partes apoiam a exigência de que os dispositivos de segurança devem conter um número
de série único e um dispositivo de prevenção de adulterações na embalagem dos
medicamentos abrangidos pela Directiva e pelo Regulamento Delegado.
• A verificação de um número de série único, randomizado, colocado em cada embalagem, a
partir de uma base de dados central, no ponto de dispensa, é actualmente uma das formas
mais seguras de verificar a autenticidade de um produto. Contudo, um sistema de verificação
de produto só pode garantir total proteção em relação ao conteúdo da respetiva embalagem,
se esta se mantiver selada em todos os momentos /pontos da cadeia de abastecimento. A
utilização de dispositivos de prevenção de adulterações é, portanto, um complemento
essencial ao sistema de verificação de produto.
• O âmbito e a aplicação dos dispositivos de segurança serão determinados de acordo com as
disposições da Directiva e respectivo Regulamento Delegado.
8. Utilizar dispositivos de segurança que sejam simples, robustos e custo‐efectivos:
• A solução proposta para a verificação de medicamentos deve ser prática, acessível e
disponível. Entende‐se que soluções desnecessariamente complexas e dispendiosas devem ser
evitadas.
• A Directiva e o Regulamento Delegado definem que os custos do sistema de repositórios
deverão ser suportados pelo Titulares de Autorização de Fabrico. Os custos para o
desenvolvimento completo do Sistema devem ser estabelecidos com base num modelo de
taxa fixa a ser acordada por unanimidade pelos seus Constituintes. Para evitar dúvidas, no
caso em que o Titular da Autorização de Fabrico e o Titular de Autorização de Introdução no
Mercado não sejam a mesma entidade jurídica, os custos deverão ser assumidos pela
entidade responsável pelo carregamento dos dados no Sistema.
9. Trabalhar em conjunto na defesa da segurança dos Doentes:
• Como principais parceiros envolvidos no processo de verificação de medicamentos, as Partes
estão empenhadas em trabalhar conjuntamente no estabelecimento de um sistema eficiente,
viável e efectivo para proteger os doentes contra a ameaça que representam os
medicamentos falsificados.
• A criação e a gestão de sistemas de verificação de produto devem respeitar o estabelecido no
art.º 31.º do Regulamento Delegado, tendo por base o actual enquadramento em matéria de
codificação nos diversos países, os quais devem responder às necessidades dos doentes e de
todos os intervenientes na cadeia de abastecimento.
• Cada Constituinte será individualmente responsável pelo Sistema.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 9 de 24
10. Envolver outros parceiros:
• As Partes irão trabalhar com as autoridades competentes e apoiam a participação de outras
organizações parceiras relevantes que desempenhem um papel activo na cadeia de
abastecimento do medicamento, em relação à sua intervenção no sistema de verificação do
produto. De forma conjunta será, assim, possível garantir um sistema robusto e abrangente
para fazer face aos falsificadores.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 10 de 24
3. Arquitectura e Propriedades do Sistema
3.1. Introdução
As Partes apoiam a criação de um sistema europeu, conforme ilustrado na figura abaixo.
A fim de permitir a interoperabilidade e a implementação de um sistema de verificação nacional
custo‐efectivo, o repositório nacional deve ser implementado em conformidade com os requisitos da
organização EMVO, considerando um modelo Blueprint.
Este sistema compreende uma plataforma única central europeia ligada a uma série de repositórios de
informação nacionais que funcionam como uma plataforma de verificação que as farmácias ou outras
entidades autorizadas podem utilizar para atestar a autenticidade de um produto. Os dados ao nível
da farmácia ou do distribuidor grossista não ficarão disponíveis na plataforma central europeia. Os
repositórios nacionais são criados e utilizados sob a gestão das respectivas organizações das partes
interessadas nacionais (as Partes). O sistema "Blueprint" refere‐se, por sua vez, aos repositórios
nacionais, operacionalizados pela EMVO em nome dos respetivos parceiros nacionais, adiante
designado por "sistemas nacionais operacionalizados centralmente". As farmácias comunicam com o
repositório nacional através do “portal de farmácias”, que tem como única função anonimizar a
farmácia, assegurando que apenas os dados necessários à validação e autenticação dos medicamentos
são transmitidos ao repositório, garantindo o cumprimento da Directiva e Regulamento Delegado.
Todas as referências futuras a "sistemas nacionais" incluirão tais sistemas. Ao longo do tempo, os
sistemas podem migrar entre os modelos nacional, regional ou operacionalizados centralmente, de
acordo com as pretensões das partes interessadas e as regras das suas respetivas organizações.
3.2. Responsabilidades da EMVO
Portal de
Farmácias
Portal de
Farmácias

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 11 de 24
A Organização Europeia de Verificação de Medicamentos (sigla em inglês EMVO), uma organização
internacional sem fins lucrativos gerida pelos parceiros, irá desenvolver uma plataforma central
europeia, estabelecer normas em relação aos requisitos da EMVO e garantir a qualidade e a
disponibilidade do sistema.
Para obter informações específicas sobre a EMVO, que irá liderar e dirigir a referida plataforma central
europeia, p.f consultar o Memorando de Entendimento referente às partes interessadas europeias
(ver Anexo).
3.3. Principais tarefas da NMVO
A NMVO irá liderar e gerir o sistema nacional que estará ligado à plataforma central europeia e
incluirá os seguintes elementos /atribuições:
a verificação e a autenticação das embalagens nacionais de medicamentos,
uma localização central para armazenamento de dados nacionais de produtos,
um ponto nacional a partir do qual são gerados os alertas , caso o sistema detecte um evento
excepcional.
3.4. Sistema Nacional
Para que o sistema funcione de forma custo‐efectiva deverá cumprir com os requisitos do utilizador da
EMVO e com os princípios de um acordo legalmente vinculativo entre as Partes nacionais (NMVO).
O principal objectivo do Sistema Nacional é servir como plataforma de verificação que farmácias ou
outras entidades registadas, como os distribuidores grossistas ou as farmácias hospitalares, podem
usar para verificar a "autenticidade" de um produto, desactivar os identificadores únicos ou reverter o
estado de um identificador único desactivado. Todos os dados necessários para realizar esta e outras
transacções relevantes encontram‐se armazenados nos respectivos Sistemas Nacionais e devem
cumprir as disposições legais relativas à protecção de dados pessoais e à propriedade dos dados do
Sistema.
O Sistema Nacional “Blueprint” serve como uma plataforma custo‐efectiva de verificação “off‐the‐
shelf” (imediatamente disponível), que se encontra ao dispor das organizações nacionais interessadas.
O Sistema Nacional utiliza a interface com a plataforma central europeia e cujas funcionalidades se
encontram descritas nos requisitos dos utilizadores da EMVO.
As funções principais do Sistema Nacional são:
ser o repositório dos dados relevantes de serialização de produtos,
receber novos dados ou dados revistos de serialização de produtos a partir da plataforma
central europeia,
servir de plataforma de verificação para farmácias (através de portal de farmácias), ou outros
entidades registadas, tais como os distribuidores grossistas, para atestar a autenticidade dos
produtos,
servir de plataforma em que as farmácias (através de portal de farmácias), assinalam a
embalagem de um medicamento como dispensada antes da sua entrega ao doente,
servir como plataforma em que as entidades registadas, como as farmácias (através de portal
de farmácias), e os distribuidores grossistas, desactivam uma dada embalagem ou a assinalam
como tendo sido alvo de exportação para fora da UE.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 12 de 24
4. Verificação do produto no Sistema Nacional
4.1. Dispositivo de Segurança ‐ Identificador Único
O sistema de identificação de cada embalagem individual deve incluir, pelo menos:
1. Código do produto,
2. Número do lote e, se necessário, um sufixo / código interno para os produtos de distribuição
paralela, para que tenham ligação ao lote original do produto,
3. Prazo de validade,
4. Número de série,
5. Número nacional para efeitos de comparticipação, nos casos em que este é utilizado,
6. O estado /situação actual da embalagem original na base de dados,
7. Data e hora da alteração da situação /estado na base de dados.
Estes dados são divididos em dois tipos:
• Estáticos: por exemplo, o código do produto, o prazo de validade (ou seja, os pontos
1‐4 acima descritos) que não se alteram ao longo do tempo.
• Dinâmicos: por exemplo, os elementos que reflectem a alteração de estado de um
dado número de série (ou seja, os pontos 5‐7 acima descritos).
4.2. Propriedade e acesso aos dados
A segurança da cadeia de abastecimento não poderá ser gerida de forma eficaz se não for permitido o
acesso aos dados em determinadas circunstâncias. A fim de maximizar os benefícios de segurança dos
doentes, tendo em conta o artigo 54.ºA da Directiva, é importante garantir que a efectividade do
sistema não venha a ser comprometida por restrições excessivas de acesso aos dados.
Para tal, deverá ser feita uma distinção entre o que é a geração de dados, a propriedade e a licença de
utilização e as respectivas atribuições de direitos de acesso. É necessário que seja estabelecido um
conjunto de regras que rejam estas questões, no sentido de garantir os adequados direitos de acesso
dos utilizadores e que sejam implementadas outras disposições técnicas e organizacionais.
O sistema não irá registar ou gerar quaisquer dados pessoais.
4.2.1 Geração e propriedade de dados
Todas as partes interessadas com acesso ao sistema serão proprietários dos seus próprios dados, i.e
dos dados de verificação gerados por si na interação com o sistema.
As partes reconhecem a natureza sensível deste tipo de informação e propõem um Sistema que seja
altamente seguro e que permita o acesso aos dados, em condições estritas e bem definidas.
4.2.2 Cenários e condições de acesso das partes interessadas
As Partes identificaram os seguintes cenários, em particular as situações em que os parceiros
relevantes poderão procurar ter acesso a determinado tipo de informação relacionada com os dados
de serialização e de verificação do produto por motivos de segurança do doente.
4.2.2.1 Verificação negativa, dispensa e actividades invulgares
O acesso à informação no sistema sobre os números de série suspeitos permitiria uma investigação
mais rápida sobre a ocorrência de Eventos Excepcionais relacionados com o risco de falsificação. Os

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 13 de 24
parâmetros para definir uma actividade invulgar deverão ser definidos no âmbito do próprio sistema.
Existe um conjunto de informação que pode fundamentar essas suspeitas, aqui referidas a título de
exemplo:
• O número de série não está no sistema de verificação.
• Os elementos /dados recolhidos por leitura óptica não correspondem às informações que
constam da base de dados.
• O número de série encontra‐se já registado na base de dados como dispensado.
• O número de série já foi desactivado (por exemplo, quando o produto é reembalado e lhe é
atribuído um novo número de série).
• O número de série já foi desactivado devido a uma recolha de lote.
• Qualquer actividade relacionada com um número de série ou um conjunto de números de
série que reflicta uma impossibilidade prática no tempo, em termos geográficos ou
relacionada com a capacidade do fabricante ou, ainda, que sugira um ataque ao sistema.
Embora se reconheça que as autoridades nacionais competentes e outras autoridades reguladoras
locais irão, naturalmente, ser envolvidas no caso de ocorrência de um Evento Excepcional, cada
fabricante afectado poderá solicitar ter acesso a alguns dados para os ajudar a rastrear a origem ou o
ponto de inserção do produto ilícito. O desenho do sistema irá garantir que, apenas os dados
acordados e considerados relevantes serão disponibilizados sob estas condições, a partir da
plataforma europeia, que será responsável por solicitar e receber esses dados provenientes do(s)
sistema(s) nacional(ais).Desta forma garante‐se um adequado nível de abstracção dos dados e que o
Titular da Autorização de Fabrico possa cumprir com as suas obrigações em relação aos reguladores
nacionais.
4.2.2.2 Recolha de produto
Usando as informações relativas a cada uma das embalagens, o sistema permitiria a identificação
quase em "tempo real" dos lotes afectados e uma gestão mais eficiente das recolhas.
Num cenário de recolha de produto do mercado, as partes interessadas necessitariam de ter acesso à
informação sobre todos os números de série envolvidos, incluindo os detalhes sobre aqueles que
foram dispensados ou reembalados em conformidade com os procedimentos acordados para a gestão
de Eventos Excepcionais e com a legislação relevante em sede de protecção de dados.
Para os produtos de distribuição paralela reembalados, a informação relativa ao número original do
fabricante e aos novos números de lote a serem aplicados pelos distribuidores farmacêuticos paralelos
deve estar ligada à plataforma central europeia para permitir uma rápida e eficiente recolha de
mercado, quando necessário. Mais uma vez, a plataforma central europeia irá permitir um adequado
nível de abstracção dos dados, tendo por base os dados que podem ser obtidos a partir dos
repositórios nacionais e disponibilizados a cada um dos fabricantes.
4.2.2.3 Manutenção do sistema
Ocasionalmente, poderá ser necessário verificar se uma transação(ões) ocorreu ou foi concluída com
êxito ou ser necessário alterar dados aquando da ocorrência de erros. Nestas circunstâncias deverá
existir a capacidade para executar um relatório ou, pelo menos, para aceder aos dados. O acesso aos
dados ficaria então limitado a empresas de TIC autorizadas, sujeito às devidas salvaguardas.
Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 14 de 24
5. Disponibilidade e utilização do Sistema Nacional
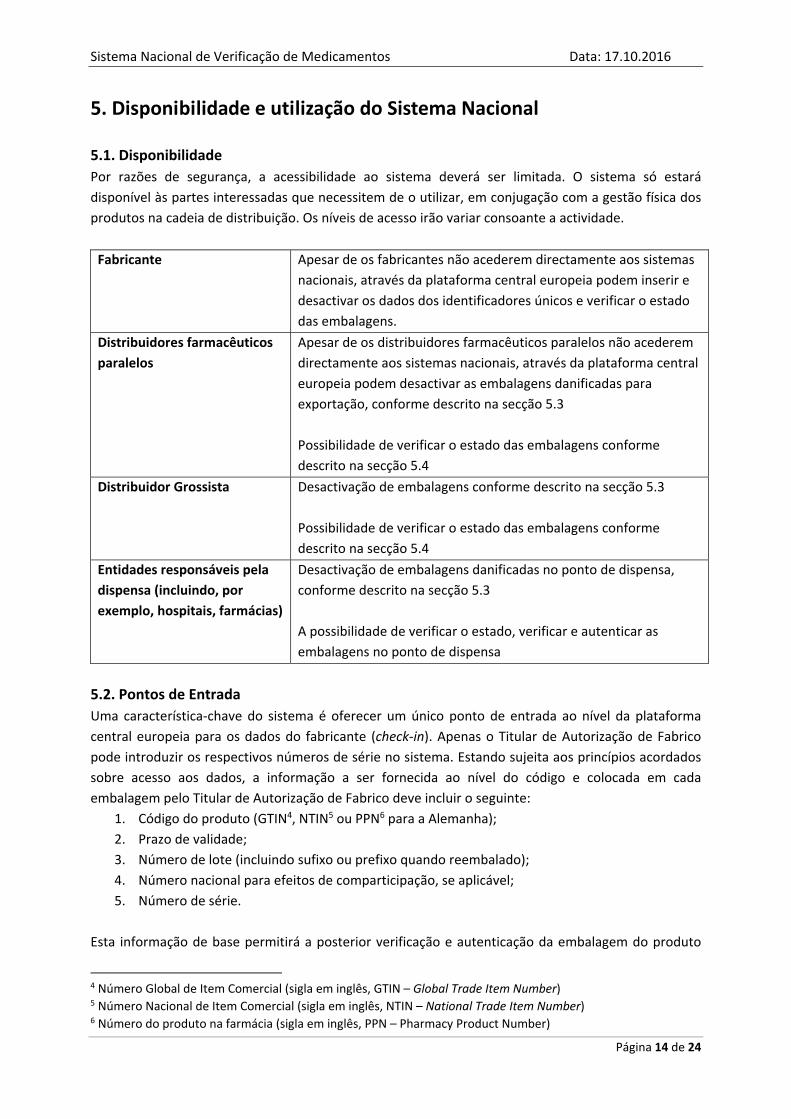
5.1. Disponibilidade
Por razões de segurança, a acessibilidade ao sistema deverá ser limitada. O sistema só estará
disponível às partes interessadas que necessitem de o utilizar, em conjugação com a gestão física dos
produtos na cadeia de distribuição. Os níveis de acesso irão variar consoante a actividade.
Fabricante Apesar de os fabricantes não acederem directamente aos sistemas
nacionais, através da plataforma central europeia podem inserir e
desactivar os dados dos identificadores únicos e verificar o estado
das embalagens.
Distribuidores farmacêuticos
paralelos
Apesar de os distribuidores farmacêuticos paralelos não acederem
directamente aos sistemas nacionais, através da plataforma central
europeia podem desactivar as embalagens danificadas para
exportação, conforme descrito na secção 5.3
Possibilidade de verificar o estado das embalagens conforme
descrito na secção 5.4
Distribuidor Grossista Desactivação de embalagens conforme descrito na secção 5.3
Possibilidade de verificar o estado das embalagens conforme
descrito na secção 5.4
Entidades responsáveis pela
dispensa (incluindo, por
exemplo, hospitais, farmácias)
Desactivação de embalagens danificadas no ponto de dispensa,
conforme descrito na secção 5.3
A possibilidade de verificar o estado, verificar e autenticar as
embalagens no ponto de dispensa
5.2. Pontos de Entrada
Uma característica‐chave do sistema é oferecer um único ponto de entrada ao nível da plataforma
central europeia para os dados do fabricante (check‐in). Apenas o Titular de Autorização de Fabrico
pode introduzir os respectivos números de série no sistema. Estando sujeita aos princípios acordados
sobre acesso aos dados, a informação a ser fornecida ao nível do código e colocada em cada
embalagem pelo Titular de Autorização de Fabrico deve incluir o seguinte:
1. Código do produto (GTIN4, NTIN5 ou PPN6 para a Alemanha);
2. Prazo de validade;
3. Número de lote (incluindo sufixo ou prefixo quando reembalado);
4. Número nacional para efeitos de comparticipação, se aplicável;
5. Número de série.
Esta informação de base permitirá a posterior verificação e autenticação da embalagem do produto
4 Número Global de Item Comercial (sigla em inglês, GTIN – Global Trade Item Number) 5 Número Nacional de Item Comercial (sigla em inglês, NTIN – National Trade Item Number) 6 Número do produto na farmácia (sigla em inglês, PPN – Pharmacy Product Number)

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 15 de 24
por outras partes implicadas em processos de manipulação física ao longo da cadeia de
abastecimento.
O titular da Autorização de Fabrico deve especificar o(s) país(es) onde a embalagem será vendida pelo
distribuidor grossista /fabricante, com o número de série para esse país. Desta forma, os números de
série ficam ligados à base de dados nacional.
Os novos números de série serão listados em processos de reembalagem e ligados ao(s) país(es) onde
a embalagem será vendida. É necessário que haja uma relação clara entre o identificador único (I.U.)
na embalagem original e o I.U. que ligue os antigos e os novos números de lote na plataforma central
europeia. Este processo deve ser automático e simples.
5.3. Pontos de Saída
O sistema só funcionará se os pontos de saída estiverem identificados e respeitarem os princípios do
Sistema em conformidade com os procedimentos acordados.
O esquema actual prevê que a embalagem possa existir em diferentes estados, como se apresenta:
• Disponível,
• Dispensada, incluindo embalagens divididas nos países onde tal é aplicável,
• Dispensada noutro mercado,
• Desactivada,
• Desactivada noutro sistema,
• Comércio fora do espaço EEE,
• Sujeito a recolha,
• Reembalada.
Neste documento, o termo "desactivação" é um termo colectivo para descrever o estado de uma
embalagem que se encontre em qualquer um dos estados acima referidos, excepto numa situação de
"disponível".
Parte Interessada /Parceiro Desactivação de embalagens da responsabilidade dos parceiros,
processo de check‐out.
Fabricante / Distribuidores
farmacêuticos paralelos
Expedição/devolução do produto, cancelamento, acidente,
embalagem danificada, correcção de erros no registo inicial,
ajustamentos logísticos não previstos, roubo de números de série
ou de embalagens.
Distribuidores farmacêuticos
paralelos
Desactivação de embalagens anterior ao processo de
reembalagem do produto, com o subsequente registo de um novo
número de série.
Entidades responsáveis pela
dispensa (incluindo, por
exemplo, hospitais, farmácias)
Autenticação e verificação.
Desactivação de embalagens danificadas no ponto de dispensa,
conforme descrito na secção 5.3.1.
Distribuidor Grossista Desactivação de embalagens nas situações previstas no art.º 22.º
e 23.º do Regulamento Delegado
Em algumas circunstâncias, pode ser necessário reverter um registo de autenticação. Por exemplo, em
caso de um produto ser autenticado incorrectamente, ou se o doente não levantar os medicamentos
previamente encomendados. Nestas situações deve ser possível inverter a autenticação em qualquer

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 16 de 24
ponto, dentro do prazo de validade do medicamento.
5.3.1 Desactivação
A desactivação deve ser obrigatória para que a integridade do sistema seja mantida e que tal aumente
a segurança do doente, reduzindo o risco dos medicamentos contrafeitos entrarem na cadeia legal de
abastecimento. Sem um mecanismo que garanta que cada embalagem individual, com um número de
série único, seja desactivada de forma adequada a partir do sistema, não será possível garantir os
benefícios de segurança deste Sistema para os doentes. O número de série só poderá servir como
protecção segura contra os medicamentos contrafeitos, se os produtos forem desactivados de forma
sistemática. O “estado” do número de série tem de ser alterado na base de dados, quando um
produto é dispensado ao doente ou quanto este é reembalado.
5.4. Disponibilidade em relação aos Distribuidores Grossistas
Os distribuidores grossistas devem verificar os dispositivos de segurança dos medicamentos que
estejam na sua posse física por (i) devolução por pessoas autorizadas ou habilitadas a fornecer
medicamentos ao público ou por outro grossista; (ii) medicamentos que recebe de um grossista que
não é nem o fabricante nem o grossista titular da autorização de introdução no mercado, nem um
grossista designado pelo titular da autorização de introdução no mercado, através de um contrato
escrito, para armazenar e distribuir em seu nome os medicamentos abrangidos pela sua autorização
de introdução no mercado.
A verificação da autenticidade do dispositivo de segurança nos termos da alínea (ii) não é exigível nas
seguintes situações: (a) quando o medicamento em causa muda de proprietário, mas permanece na
posse física do mesmo grossista; (b) quando o medicamento em causa é distribuído no território
de um Estado‐Membro entre dois entrepostos pertencentes ao mesmo grossista ou à mesma
entidade legal e não se verifica qualquer venda.
As Partes acordam em avançar com o diálogo sobre o desenvolvimento de práticas normalizadas para
as situações que exijam a retirada dos produtos da cadeia de abastecimento (tais como bens
danificados) e para os quais os números de série devem ser desactivados ao nível do Sistema.
5.5. Entidades responsáveis pela dispensa (Farmácias, Hospitais)
As entidades responsáveis pela dispensa (farmácias, hospitais) poderão verificar os dados no sistema.
5.6. Outras situações referentes à utilizações dos dados
A implementação do Sistema proposto, além de fornecer informações detalhadas aos parceiros sobre
os produtos falsificados encontrados no mercado, tem o potencial de gerar outros benefícios que
incluem (sujeito a acordo entre as partes interessadas a nível nacional): uma redução do número de
pedidos de comparticipação fraudulentos; maior eficácia na prevenção de dispensa ao doente de
produtos retirados do mercado; maior eficiência nos procedimentos de devolução de produtos e uma
maior facilidade nos processos de gestão de stocks nas farmácias.
Qualquer outra utilização dos dados transaccionais está sujeita a negociação e a acordo entre as
partes interessadas, caso a caso, em função das circunstâncias nacionais e em conformidade com a
legislação pertinente.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 17 de 24
****
Os vários requisitos acima descritos são necessários para evitar o risco de operadores sem escrúpulos
terem acesso ao sistema a nível regional e, dessa forma, possam distorcer os dados para facilitar a
entrada de produtos falsificados na cadeia de abastecimento legal. Estes requisitos constituem
garantias sistémicas mais fiáveis do que as auditorias nacionais realizadas ad hoc. Além disso, estes
fornecem um enquadramento para um Sistema dito bloqueado, que facilite a rápida identificação de
qualquer tentativa de distorção do mesmo, salvaguardando os interesses de segurança dos doentes e
que, no futuro, possa evoluir para atender as necessidades e outros desafios.

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 18 de 24
6. Estrutura Organizacional e de Governação: Parâmetros‐chave
Prevê‐se que a NMVO será estabelecida como uma organização sem fins lucrativos, de duração
ilimitada. Os seus estatutos irão formalizar o seu estatuto legal, a forma de financiamento, a estrutura
organizacional e os processos de tomada de decisão. Sem prejuízo de uma decisão final sobre a
estrutura jurídica que seja mais custo‐efectiva e quaisquer outros requisitos legais obrigatórios, as
Partes prevêem que a NMVO será gerida pelos princípios enunciados no presente capítulo.
A NMVO irá deter activos mínimos e a gestão tecnológica do projecto será subcontratada a um ou
mais fornecedores de TIC, tendo por base um projecto financiado por programa tipo, regido por um
acordo de nível de serviço.
6.1. Mandato
A NMVO irá estabelecer e gerir o Sistema Nacional de Repositórios que será interoperável com a
plataforma central europeia e, como tal, com outros repositórios nacionais que servem como
plataformas de verificação e onde estão armazenados os dados necessários para permitir às
farmácias, ou a outras entidades autorizadas, a verificação da autenticidade de um produto. A NMVO
irá cooperar com as partes interessadas na implementação da Directiva e do Regulamento Delegado
sobre medicamentos falsificados.
A NMVO irá facilitar as negociações entre as partes interessadas com vista à celebração de acordos
normalizados que regem a sua relação com a EMVO. Estes acordos devem assegurar que os princípios
de boa governação são aplicados através de um Sistema que é totalmente interoperável, permitindo
aos participantes, de forma mais eficaz, identificar, monitorizar e, sempre que possível, reduzir os
riscos específicos e comuns à segurança dos doentes decorrentes dos produtos contrafeitos.
A NMVO será responsável por:
a. Aplicar os requisitos da EMVO e assegurar a qualidade geral (em questões como a limpeza de
dados, a disponibilidade e a capacidade de resposta do Sistema, o nível apropriado de
segurança, etc.),
b. Definir os termos e as condições que regem o acesso ao Sistema, os quais devem ser
objectivos, transparentes e abertos a qualquer parte devidamente autorizada a operar na
cadeia de abastecimento legal, em qualquer parte do Espaço EEE,
c. Gerir as TI, das interfaces contratuais e humanas entre a NMVO e a EMVO,
d. Fornecer relatórios de actividade regular sobre as várias questões relacionadas com o
funcionamento e o desempenho do Sistema e gerar relatórios estatísticos com o objectivo de
auxiliar as comunicações sobre o funcionamento do Sistema,
e. Conduzir revisões estratégicas periódicas para garantir que o Sistema evolui ao longo do
tempo, no interesse da segurança dos doentes e em linha com a evolução da infra‐estrutura
dos cuidados de saúde na Europa,
f. Facturar e cobrar as taxas nos termos da Directiva e do Regulamento Delegado e quaisquer
outras quantias devidas pelos membros,
g. Concluir e gerir os acordos de utilização e de disposições relativa a remunerações e
modalidades de pagamento, tendo em conta o estabelecido no art.º 31.º do Regulamento
Delegado e o acordado entre os Constituintes,

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 19 de 24
h. Garantir a comunicação com as autoridades reguladoras nacionais sobre a utilização do
Sistema para facilitar os procedimentos de recolha de produtos e gerir outras questões
relacionadas com a segurança dos doentes,
i. Prestar serviços aos parceiros no cumprimento dos princípios mutuamente acordados sobre o
acesso aos dados, bilateral ou multi‐partido, conforme acordado caso‐a‐caso.
A NMVO deverá ser autorizada a realizar todas as actividades que directa ou indirectamente se
relacionem com a condução do seu mandato. Para este efeito, será autorizada a contratar pessoal
relevante e fornecedores, e a exercer outros direitos nos termos dos respetivos e Estatutos.
A NMVO pode optar por alocar as responsabilidades a terceiros (pontos c. a i.), neste caso à EMVO,
sob um acordo de nível de serviço.
6.2. Afiliação
A NMVO será composta por membros efectivos e membros associados sem direito a voto.
Admissão. Os candidatos a membros podem ser admitidos pela Assembleia Geral, sob recomendação
da Direcção após comunicação à Direcção, por escrito, quanto à sua elegibilidade e aceitação e adesão
aos estatutos da NMVO.
Membros Efectivos. Podem ser Membros Efectivos, directamente ou através das respectivas
organizações associativas, os fabricantes e titulares de autorizações de introdução no mercado de
medicamentos dotados de dispositivos de segurança e, caso decidam participar, os grossistas e
pessoas autorizadas ou habilitadas a fornecer medicamentos ao público.
O procedimento para candidatura ou anulação da adesão será regulamentado nos Estatutos da
NMVO. Quaisquer disputas quanto ao preenchimento dos critérios de adesão serão pronunciadas por
um auditor terceiro independente a ser designado pela Direcção.
Os membros efectivos têm os seguintes direitos e obrigações:
• O direito de participar e votar nas Assembleias Gerais,
• O direito de participar e votar nos grupos de trabalho/task‐forces que possam vir a ser
estabelecidos,
• O direito de solicitar uma auditoria independente à segurança e desempenho do Sistema,
desde que essas auditorias sejam apenas realizadas com uma periodicidade razoável e
suportadas financeiramente pelo membro requerente,
• A obrigação de pagar uma taxa anual, nos termos da Directiva e do Regulamento Delegado, ou
outras que venham ser acordadas entre os Constituintes,
• A obrigação de agir em conformidade com os Estatutos,
• Qualquer outro direito ou obrigação que possa vir a ser decidido(a) pela Assembleia Geral ou
Direcção.
Membros associados. As seguintes entidades são elegíveis para membros associados da NMVO:
1. Outras associações de partes interessadas que representem os utilizadores ou os potenciais
utilizadores do sistema para fins de autenticação.
Os membros associados têm os seguintes direitos e obrigações:

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 20 de 24
• O direito de serem notificados de todas as Assembleias Gerais e o direito a assistir a tais
reuniões na qualidade de observador,
• O direito de serem consultados sobre as actividades da NMVO, conforme for decidido,
periodicamente pela Assembleia Geral,
• A obrigação de pagar uma taxa anual de associado, tal como poderá vir a ser previsto, nos
termos da secção 6.4 abaixo,
• A obrigação de agir em conformidade com os Estatutos,
• Qualquer outro direito ou obrigação que possa a vir ser decidido(a) pela Assembleia Geral ou
Direcção.
6.3. Governação da NMVO
O trabalho da NMVO será desenvolvido por:
6.3.1 A Assembleia Geral
Reuniões dos Membros. A Assembleia Geral reúne‐se em sessão ordinária uma vez por ano. Podem
ser convocadas Assembleias Gerais Extraordinárias a pedido de pelo menos dois terços dos votos dos
membros Constituintes.
Quórum. Deverá haver um quórum para a realização de qualquer Assembleia Geral, quando uma
maioria de dois terços dos membros Constituintes estiverem representados pessoalmente ou por
procuração escrita à data da reunião. Cada membro Constituinte deve dar a conhecer a identidade do
seu representante com poderes para votar na Assembleia Geral, pelo menos cinco dias antes de cada
Assembleia Geral.
Gestão pelos Membros. A Assembleia Geral terá plenos poderes para determinar as políticas globais,
objectivos, procedimentos, métodos e definição de acções necessários para concretizar o mandato da
NMVO. A Assembleia Geral determinará quais as decisões que poderão ser delegadas à Direcção. A
mesma deverá rever anualmente, como ponto de agenda e com base num relatório da Direcção, a
adequação da estrutura da NMVO e os recursos disponíveis, à luz dos seus objectivos.
Decisões. As decisões da Assembleia Geral devem incluir:
1. Aprovação do orçamento anual e das contas anuais,
2. Alteração dos estatutos,
3. Nomeação e demissão de um Presidente, um Vice‐Presidente e um Tesoureiro sob proposta
da Direcção,
4. A admissão de novos membros e a revogação dos direitos de membro em conformidade com
o procedimento previsto no secção 6.5 abaixo, em caso de (i) liquidação ou falência, ou (ii) não
pagamento das taxas devidas, ou (iii) ausência de um membro suficientemente
representativo, ou (iv) outras condutas não compatíveis com os objectivos da NMVO,
5. O calendário e a forma de efectuar a dissolução e liquidação da NMVO.
Votação. Cada membro Constituinte terá direito a um voto em qualquer Assembleia Geral, mas é
permitida a presença de tantos representantes quantos os considerados necessários para a
Assembleia Geral. No caso em que qualquer um dos membros Constituintes esteja representado por

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 21 de 24
mais de um representante de pleno direito, o membro Constituinte deverá chegar a uma solução justa
e razoável quanto à forma do exercício da votação e fornecer ao Presidente da Direcção da NMVO
uma descrição completa da solução acordada.
As decisões em Assembleia Geral devem ser aprovadas por, pelo menos, dois terços dos votos
expressos pelos membros presentes ou validamente representados, a menos que qualquer membro
Constituinte exerça legitimamente o direito de veto em qualquer das circunstâncias referidas abaixo.
A falta de votação será considerada uma abstenção.
Direitos de veto. Cada membro terá os seguintes direitos de veto em relação ao poder de decisão da
Assembleia Geral, após devida consulta e análise e, desde que o exercício de tais direitos resulte numa
situação que esteja em conformidade com as leis aplicáveis:
Deflexão/Desvio aos requisitos da EMVO (sem prejuízo das exigências da Directiva ou
Regulamento Delegado), incluindo alterações aos princípios acordados em matéria de acesso
e gestão de dados, desde que estas últimas alterações digam respeito aos próprios dados do
respectivo Membro;
Aumento do valor das taxas pagas à NMVO acima de 15% numa base anual;
Cada membro Constituinte que represente os Titulares de Autorização de Fabrico, estes sendo
(a) as empresas farmacêuticas de investigação que comercializam medicamentos sujeitos e
não sujeitos a receita médica, (b) as empresas farmacêuticas de medicamentos genéricos e
biossimilares, e (c) os distribuidores farmacêuticos paralelos, devem ter direito de veto em
relação a aumentos no orçamento anual da NMVO acima de 20% numa base anual, salvo se
esse aumento for necessário para cumprir com os requisitos da EMVO.
Direitos de presença. Os Membros associados serão admitidos à Assembleia Geral, na qualidade de
observadores, bem como os representantes da Comissão Europeia e as autoridades nacionais
competentes. A Direcção poderá decidir admitir outros observadores externos.
6.3.2 A Direcção
Reuniões. A NMVO será gerida por uma Direcção que reunirá pelo menos três vezes por ano. Uma
sessão extraordinária da Direcção será convocada, se pelo menos metade dos directores solicitar tal
reunião.
Composição. A Direcção é composta por um delegado de cada membro de pleno direito ou qualquer
outro número que possa ser fixado pela Assembleia Geral sob proposta da Direcção. Nenhum membro
poderá ser representado por mais de um delegado.
Quórum. A Direcção irá validamente reunir e deliberar quando pelo menos a maioria dos membros
Constituintes estiverem representados pessoalmente ou por procuração escrita.
Gestão pela Direcção. A Direcção deve assegurar que a NMVO opera em conformidade com todas as
leis aplicáveis e com os seus Estatutos. A Direcção terá todos os poderes, excepto aqueles reservados
à Assembleia Geral, para implementar políticas globais, objectivos, procedimentos, métodos e acções
da NMVO que devem incluir o seguinte:

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 22 de 24
1. Propor à Assembleia Geral um Presidente, um Vice‐Presidente e um Tesoureiro de entre os
seus membros de pleno direito,
2. Assegurar que as actas de todas as reuniões da Direcção e comunicar as suas decisões a todos
os membros,
3. Preparar o orçamento e as contas anuais,
4. Fazer recomendações à Assembleia Geral, como sobre o pagamento das quotizações pelos
membros de pleno direito e membros associados,
5. Delegar a gestão diária ou parte de seus poderes a um ou mais directores, incluindo a
capacidade de nomear um Director‐Geral ou Gerente da NMVO, ou delegar a terceiros
projectos claramente definidos, tais como à EMVO,
6. Publicar, na medida do necessário, regras e procedimentos internos compatíveis com os
Estatutos, a fim de garantir o adequado funcionamento da NMVO,
7. Supervisionar a implementação, e monitorizar numa base contínua, os problemas de
desempenho do Sistema, gestão de incidentes, alterações operacionais, gestão da
configuração, segurança de acesso a dados e reportar sobre tais questões à Assembleia Geral,
8. Propor políticas, à Assembleia Geral, em relação à implementação e desenvolvimento do
Sistema,
9. Fazer recomendações por escrito e devidamente fundamentadas, à Assembleia Geral, sobre a
admissão de novos membros e a cessação da qualidade de membro que considere como
adequado.
O mandato dos membros da Direcção será de dois anos. O mandato é renovável duas vezes e não é
remunerado. Os directores têm o dever de administração para com os interesses comuns dos
membros.
Decisões. Cada membro Constituinte terá direito a um voto em qualquer reunião da Direcção e deve
dar a conhecer a identidade do seu representante com poderes para votar na Direcção.
Votação. Os seguintes actos estão sujeitos a aprovação por maioria de dois terços dos membros
Constituintes:
1. Aprovar as despesas de capital acima de € 100.000 (excluindo quaisquer pagamentos devidos
ao prestador do(s) sistema(s) de TIC que é regido por contrato separado),
2. Alterar os termos dos acordos de serviços com a plataforma central europeia,
3. Nomeação e destituição do Presidente e do Vice‐Presidente / Director‐Geral / Tesoureiro
Para todas as outras decisões, é necessária uma maioria simples dos votos dos membros presentes ou
validamente representados numa reunião da Direcção.
A falta de votação será considerada uma abstenção.
Executivos.
A NMVO terá como executivos um Presidente, Vice‐Presidente e Tesoureiro.
O Presidente e o Vice‐Presidente devem assegurar que a Direcção é efectiva quanto às suas
responsabilidades de definição e implementação da estratégia. Estes irão presidir as reuniões de
Direcção e da Assembleia Geral, e executar as políticas e as instruções de ambos. As suas principais
responsabilidades incluirão:
1. Convocar todas as reuniões da Assembleia Geral e da Direcção,

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 23 de 24
2. Realizar as políticas decididas pela Direcção e propor à Direcção planos adequados e gerir a
sua execução, uma vez aprovados,
3. Estabelecer e manter uma comunicação adequada com todos os membros,
4. Representar a NMVO e os seus membros a terceiros, nomeadamente organismos
governamentais e regulamentares.
A Direcção poderá nomear um Director‐Geral, o qual será responsável por executar as actividades de
gestão delegadas pela Direcção.
6.4. Receita/Assinaturas
Cada membro Constituinte, de acordo com os Estatutos do NMVO pagará, durante o período de
implementação do sistema, uma taxa anual como determinado pela Assembleia Geral, sob
recomendação da Direcção, para cobrir as despesas de organização e de funcionamento da NMVO. No
caso e que qualquer um dos membros Constituintes seja representado por mais de um representante
de pleno direito, o Constituinte deverá chegar a uma solução justa e razoável sobre como será
atribuída a sua taxa anual entre os seus membros.
Os membros associados poderão ser convidados a pagar uma taxa anual para cobrir os custos da sua
participação na Assembleia Geral.
O pagamento deverá ser efectuado com um prazo não superior a 60 dias desde a data em que é
emitido o pedido de pagamento.
A Direcção poderá aprovar novas ou adicionais subscrições ou contribuições, aos membros de pleno
direito obrigados a suportar os custos do sistema de repositório para financiar projectos especiais,
para cobrir a presença dos membros na Assembleia Geral, ou outros valores ad hoc para aspectos não
cobertos pelo orçamento anual, bem como aumentos no valor da taxa anual de afliação e de
utilização, desde que estas sejam proporcionais e em consonância com os objectivos da NMVO.
6.5. Resolução de Disputas, Lei Aplicável e Jurisdição
A Direcção poderá propor, embora não seja obrigada, uma forma de resolução amigável de qualquer
controvérsia ou disputa relativa à validade, interpretação, execução, desempenho ou rescisão dos
Estatutos, afiliação, e os direitos de utilização, através da mediação ou por qualquer outra forma
alternativa de resolução de litígios.
Todas as questões, dúvidas e litígios sobre a validade, interpretação, execução, desempenho ou
rescisão dos Estatutos da NMVO que não possam ser resolvidos pela Direcção devem ser geridos e
interpretados de acordo com a legislação nacional. Nenhum efeito deve ser dado a qualquer outra
escolha da lei aplicável ou de quaisquer outras normas de conflitos de leis ou disposições que
resultariam na aplicação de leis de qualquer outro país.
A NMVO pode tomar toda e qualquer acção perante qualquer tribunal competente para recuperar as
verbas devidas por utilizadores ou membros. Caso contrário, qualquer diferendo que não possa ser
resolvido pela Direcção, sobre a validade, interpretação, execução, desempenho ou rescisão dos
Estatutos, de associação, ou dos direitos de utilização, será exclusiva e definitivamente resolvido por
arbitragem de acordo com as Regras de Arbitragem vinculativa, por três árbitros nomeados de acordo
com essas regras. O idioma da arbitragem será Português. O local da arbitragem será Portugal. Nada
contido nesta cláusula deverá limitar o direito de qualquer membro ou utilizador em procurar junto de

Sistema Nacional de Verificação de Medicamentos Data: 17.10.2016
Página 24 de 24
qualquer tribunal de jurisdição competente, na pendência da nomeação de um tribunal arbitral,
medidas provisórias de apoio à arbitragem ou para proteger ou fazer valer os seus direitos.
*****
Assinado em Lisboa, em 17 de Outubro de 2016, ficando cada uma das entidades signatárias com um
exemplar,
O Presidente da Direcção da APIFARMA
_______________________________
Dr. João Pedro Almeida Lopes
O Presidente da Direcção da APOGEN
________________________________
Dr. Paulo Clímaco Lilaia
O Presidente da Divisão Farmacêutica da GROQUIFAR
________________________________
Dr. Diogo Miguel Parreira de Gouveia
O Presidente da Direcção da ANF
________________________________
Dr. Paulo Jorge Cleto Duarte
A Presidente da Direcção da AFP
________________________________
Dra. Maria Manuela Hortas Silva Pacheco


















