
modulação sinaptica
34
Quais são os fenômenos básicos da “modulação sináptica”? A resposta de um neurônio pós-sináptico depende da que freqüência e da duração com que ele foi estimulado pelo neurônio pré-sináptico. Chamamos de facilitação quando o neurônio pós-sináptico recebe estímulos repetidos, havendo um aumento da resposta pós-sináptica. Na facilitação, cessada a estimulação, o potencial cessa em décimos de segundos. Se a freqüência de estimulação pelo neurônio pré-sináptico aumentar, chamados de aumentação, e neste caso, se houver cessação da estimulação, a geração do potencial poderá durar até 10 segundos. Ainda mais, se aumentarmos mais ainda a freqüência de estimulação, teremos a chamada potenciação pós-tetânica, que cessado a estimulação, poderá gerar potenciais desde décimos de segundo até vários minutos. Uma potenciação pós-tetânica intensa, pode até gerar um potencial que durará horas ou dias após cessada a estimulação, importante na função memória. Se houver estimulação em alta freqüência e persistente, ocorrerá a chamada fadiga sináptica, por redução acentuada (esgotamento), do quanta de neurotransmissor. Agora é hora de entendermos um pouco sobre Neurotransmissores ou simplesmente transmissores químicos! A maioria dos neurotransmissores, podem ser divididos em 3 grupos básicos: aminas, aminoácidos e oligopeptídeos. Particularmente a acetilcolina é utilizada por todos axônios motores que emergem da medula espinhal, nos neurônios pré-ganglionares do sistema nervoso autônomo simpático e parasimpático, nos neurônios pós- ganglionares do sistema nervoso autônomo parasimpático, e finalmente em diversos circuitos centrais, por exemplo participando do sistema de vigília (mesencéfalo). Como representantes das aminas biogênicas temos a norepinefrina, epinefrina e dopamina (compartilham de uma via comum começando com o aminoácido tirosina), serotonina e histamina. A norepinefrina é encontrada nos neurônios pós-ganglionares simpáticos e também em diversos circuitos cerebrais. Já a dopamina é mais encontrada no mesencéfalo, participando da modulação dos movimentos automáticos (substância negra) e auxiliando a acetilcolina na ativação do Sistema Reticular Ativador ascendente da vigília. A deficiência de dopamina na substância negra do mesencéfalo leva a Doença / Síndrome de Parkinson (tremores de repouso, bradicinesia e rigidez). A serotonina é encontrada em muitos núcleos do tronco cerebral (principalmente bulbo), e entre outras funções induz o sono de ondas lentas (fases I a IV). A histamina é encontrada em alguns núcleos hipotalâmicos. Como representantes dos aminoácidos transmissores temos a glicina, glutamato, aspartato e GABA . A glicina é um neurotransmissor inibitório encontrado na medula. O GABA também é um neurotransmissor
-
Upload
jacqueline-quintal -
Category
Documents
-
view
783 -
download
67
Transcript of modulação sinaptica

Quais são os fenômenos básicos da “modulação sináptica”?
A resposta de um neurônio pós-sináptico depende da que freqüência e da duração com que ele foi estimulado pelo neurônio pré-sináptico. Chamamos de facilitação quando o neurônio pós-sináptico recebe estímulos repetidos, havendo um aumento da resposta pós-sináptica. Na facilitação, cessada a estimulação, o potencial cessa em décimos de segundos. Se a freqüência de estimulação pelo neurônio pré-sináptico aumentar, chamados de aumentação, e neste caso, se houver cessação da estimulação, a geração do potencial poderá durar até 10 segundos. Ainda mais, se aumentarmos mais ainda a freqüência de estimulação, teremos a chamada potenciação pós-tetânica, que cessado a estimulação, poderá gerar potenciais desde décimos de segundo até vários minutos. Uma potenciação pós-tetânica intensa, pode até gerar um potencial que durará horas ou dias após cessada a estimulação, importante na função memória.
Se houver estimulação em alta freqüência e persistente, ocorrerá a chamada fadiga sináptica, por redução acentuada (esgotamento), do quanta de neurotransmissor.
Agora é hora de entendermos um pouco sobre Neurotransmissores ou simplesmente transmissores químicos!
A maioria dos neurotransmissores, podem ser divididos em 3 grupos básicos: aminas, aminoácidos e oligopeptídeos.
Particularmente a acetilcolina é utilizada por todos axônios motores que emergem da medula espinhal, nos neurônios pré-ganglionares do sistema nervoso autônomo simpático e parasimpático, nos neurônios pós-ganglionares do sistema nervoso autônomo parasimpático, e finalmente em diversos circuitos centrais, por exemplo participando do sistema de vigília (mesencéfalo).
Como representantes das aminas biogênicas temos a norepinefrina, epinefrina e dopamina (compartilham de uma via comum começando com o aminoácido tirosina), serotonina e histamina. A norepinefrina é encontrada nos neurônios pós-ganglionares simpáticos e também em diversos circuitos cerebrais. Já a dopamina é mais encontrada no mesencéfalo, participando da modulação dos movimentos automáticos (substância negra) e auxiliando a acetilcolina na ativação do Sistema Reticular Ativador ascendente da vigília. A deficiência de dopamina na substância negra do mesencéfalo leva a Doença / Síndrome de Parkinson (tremores de repouso, bradicinesia e rigidez). A serotonina é encontrada em muitos núcleos do tronco cerebral (principalmente bulbo), e entre outras funções induz o sono de ondas lentas (fases I a IV). A histamina é encontrada em alguns núcleos hipotalâmicos.
Como representantes dos aminoácidos transmissores temos a glicina, glutamato, aspartato e GABA . A glicina é um neurotransmissor inibitório encontrado na medula. O GABA também é um neurotransmissor inibitório só que suprasegmentar (desde o tronco cerebral até o córtex cerebral): sabemos que a deficiência de GABA no núcleo caudado leva a chamada Coréia de Huntington (movimentos parasitas). O aspartato e glutamato são neurotransmissores excitatórios encefálicos.
Os neuropeptídeos neuroativos são:
a. Peptídeos intestinais encefálicos
- Polipeptídeo intestinal vasoativo (VIP)

- Colecistocinina octapeptídeo (CCK-8).
- Subst. P.
- Metionina-encefalina
- Leucina-encefalina
- Motilina
- Insulina
- Glucagon
b. Hormônios hipotalâmicos de liberação
- Hormônio liberador de tirotropina (TRH)
- Hormônio de h. luteinizante (LHRH).
- Fator inibidor da liberação de hormônio do crescimento ou SRIF (somatostatina)
c. Peptídeos hiposários
- Corticotropina (ACTH)
- Beta-endorfina
- Hormônio estimulador de melanócitos alfa (alfa-MSH)
d. Outros
- Dinorfina
- Angiotensina II
- Bradicinina
- Vasopressina
- Oxitocina
- Carnosina
- Bombesina
Os peptídeos neuroativos podem atuar como neurotransmissores ou neuromodulares (se deixados próximo à célula alvo) podendo agir na célula pós-sináptica modificando a condutância aos íons (como já exposto), ou na célula pré-sináptica modulando (regulando), o quanto será liberado de neurotransmissor. Sendo assim chegamos a conclusão de que pode haver a coexistência de um neurotransmissor peptídeo com um neurotransmissor não peptídeo na mesma sinapse. Os peptídeos são sintetizados no corpo celular e são conduzidos pelo chamado transporte axônico rápido até o botão sináptico, enquanto os

neurotransmissores não peptídicos são sintetizados no axônio (ex.: Ach). Além do mais, os neurotransmissores peptídicos em geral tem uma ação mais prolongada na emissão do sinal em quantidades bem maiores do que as outras classes de neurotransmissores. Além destas funções, as substâncias peptídicas neuroativas, também podem ser hormônios (são liberadas no sangue para irem agir a distância).
A ação dos fármacos
Os fármacos podem agir, de maneira a alterarem processos metabólicos (sejam eles normais ou já
alterados por qualquer condição ambiental ou patológica), em diversas fases desses processos.
Como é de vital importância que os fármacos se liguem a substratos para terem seu efeito
desencadeado, os possíveis alvos (que servem justamente como ligantes para as moléculas do
fármaco) são, mais comumente: enzimas, moléculas transportadoras, canais iônicos e
receptores de membrana. E o objetivo dessa ligação fármaco-alvo é produzir algum efeito que
pode ser expresso em estimulação ou inibição de processos mediados por esses alvos. Um
exemplo muito comum é a ação de fármacos que mimetizam a ação de transmissores adrenérgicos
em receptores de membrana de endotélio, ligados a proteínas G, que podem resultar em duas
respostas diferentes (dependendo do transmissor, do receptor e da proteína G, como veremos
adiante): vasoconstrição e vasodilatamento vasodilatação. Outros transmissores, como a glicina e o
glutamato, podem se ligar a canais de sódio/cálcio e interferir no transporte transmembrânico
desses íons. Já alguns fármacos utilizados para tratar distúrbios na produção e na liberação de
transmissores simpáticos, agem diretamente nas enzimas sintetizadoras dos neurotransmissores ou
nas moléculas de transporte, sejam dos precursores ou dos neurotransmissores finais (veremos
mais adiante, na parte de transmissão noradrenérgica).
Os fármacos, então, para agir, geralmente mimetizam algum transmissor, íon ou alguma molécula
qualquer que já é natural no organismo. Dessa forma, eles conseguem agir nos receptores destes
transmissores/íons/moléculas, acentuando, reduzindo ou inibindo seus efeitos originais. No
MouseParty, por exemplo, veremos como as moléculas ativas nas drogas agem substituindo certos
neurotransmissores ou inibidores do SNC, resultando em efeitos estimulantes, depressivos ou até
alucinógenos. Sobre essa “substituição” dessas moléculas naturais, nos receptores, pelos fármacos,
conseguimos separar as drogas/fármacos em dois tipos:
Agonistas: aquelas que têm afinidade pelos receptores e substituem a molécula natural,
apresentando eficácia (podendo ser até mais eficazes que a molécula natural, e geralmente
apresentando mais afinidade). O agonismo pode ser total (capaz de produzir uma resposta tecidual
máxima) ou parcial (não é capaz de produzir resposta máxima, só ‘sub-máxima’).
Antagonistas: aquelas que têm afinidade com os receptores mas não apresentam eficácia, logo não
apresentam efeito nenhum sobre o receptor – apenas bloqueia a ação do agonista.
Existem alguns tipos distintos de antagonismos, que não dizem respeito somente à ligação com o
receptor, mas sim à relação entre o antagonista e o agonista ou substância ativa. Os antagonismos
farmacológicos podem ser:
Químico: aquele em que há a incomum interação química entre as substâncias (quelante e metal
pesado, por exemplo), gerando um produto menos ativo ou até inativo em relação à situação inicial
(pelo exemplo, há a redução da toxicidade do metal pesado quando ligado ao quelante).
Farmacocinético: o antagonista acelera os processos farmacocinéticos (que já vimos serem a
absorção, a biotransformação e a eliminação) de uma substância ativa ou de um agonista, causando
uma redução da meia-vida, da concentração circulante e, consequentemente, dos efeitos dessa
substância (ex:drogas que aceleram o metabolismo hepático, aumentando a velocidade de diversas
substâncias).

Por bloqueio de receptores: esse, sim, se encaixa na definição inicial de antagonista – se liga ao
receptor impedindo a ligação do agonista ou da substância ativa. Também chamado de
antagonismo competivivo, e pode ser reversível ou irreversível (se a ligação entre ele e o receptor
for covalente, por exemplo).
Não-competitivo: o antagonista não compete com o agonista no sítio de ligação do receptor, mas
bloqueia algum passo posterior (na transdução de sinais, por exemplo), reduzindo ou até
extinguindo os efeitos celulares do agonista.
Fisiológico: o antagonista tem ações contrárias ao do agonista (o que muitas vezes ocorre entre os
sistemas Simpático e Parassimpático, por exemplo). Um exemplo seria o antagonismo entre
histamina, que estimula a secreção ácida gástrica, e o omeprazol, que inibe a bomba de prótons.
OBS importante! Os quadros resumos (azuis: pontos-chave ou amarelos: usos clínicos) do Rang e
Dale são muito bons para a memorização de certos aspectos da matéria, inclusive esses diferentes
tipos de antagonismo, nas páginas 17 e 19 (da 6ª edição).
Os fármacos, em geral, sejam eles ligados a receptores, a transportadores, a canais iônicos ou a
enzimas, sofrem aqueles processos famacocinéticos que já comentamos (absorção, distribuição,
eliminação e/ou biotransformação). Como vimos, alguns antagonistas podem acelerar esses
processos, em especial os de eliminação e/ou biotransformação. Mas não só antagonistas fazem
isso. Um processo natural que ocorre frequentemente quando há administração contínua ou
repetida e que é responsável por acelerar a metabolização ou eliminação das moléculas dos
fármacos e consequente redução de seu efeito ou ao menos da duração desse efeito é o fenômeno
chamado de dessensibilização ou taquifilaxia (não confundir com a tolerância, que é um processo
mais demorado e necessita de maior continuidade na administração do fármaco). A ocorrência da
dessensibilização se dá por diversos mecanismos, entre eles:
Alteração ou perda de receptores: com a contínua exposição ao fármaco, podem ocorrer reações de
mudança de conformação dos receptores – que leva ao término da ligação com o fármaco ou da
ativação do sistema receptor-efetor (como é o caso de muitos receptores ligados a proteínas G) -,
ou até mesmo pode haver redução do número de receptores, muitas vezes por um processo
chamado down-regulation (como vimos em Endócrino), que é a endocitose, mediada pela própria
ligação fármaco-receptor, dos receptores ligados ao fármaco.
Depleção de mediadores: certos mediadores, moléculas intermediárias, são necessárias para a
ação de determinados fármacos. Como não há produção na mesma taxa de utilização, os estoques
desses mediadores são rapidamente esvaziados e o fármaco perde rapidamente o efeito.
Aumento da degradação metabólica e adaptação fisiológica: ocorre aumento da biotransformação
do fármaco ou o organismo consegue se adaptar às concentrações de fármaco presente com
respostas intrínsecas (homeostáticas) – como as taquicardias reflexas quando da administração de
fármacos bradicárdicos.
Extrusão (expulsão) do fármaco das células: processo relevante no tratamento de câncer com
quimioterapia.
Agora que já vimos como ocorre a ligação (e em alguns casos a dessensibilização) dos fármacos
com seus sítios de ação, vejamos alguns complexos e sistemas que levam a ativação de sistemas
efetores, segundos mensageiros, fosforilações e, geralmente, a efeitos celulares e biológicos. Esses
complexos são os que vimos em Endócrino, os complexos de receptores ligados a proteínas G e
toda a sua maquinaria celular. Vejamos rapidamente como funcionam…
Os receptores acoplados à proteína G funcionam, como vimos em Endócrino, a partir da ligação
receptor-ligante (fármaco ou substância ativa, no caso), que gera a mudança conformacional da

proteína, com ativação da atividade GTPásica da subunidade alfa, que se desprende do complexo e
ativa um sistema efetor. O sistema efetor, geralmente uma enzima, por sua vez, gera a produção de
segundos mensageiros, que levarão, por meio de vias celulares, a efeitos biológicos (seja de
transcrição, de inibição, de abertura/fechamento de canais, etc).
Dois tipos principais de proteína G devem ser abordados aqui: a proteína Gs e a proteína Gi,
estimuladora e inibitória, respectivamente. A proteína Gs é responsável pela ativação dos sistemas
adenilato ciclase/AMPc e fosfolipase C/fosfato de inositol, enquanto a Gi inibe esses sistemas,
bloqueando seus efeitos. Um exmplo prático e bem comum na nossa matéria é a diferença entre
alguns receptores adrenérgicos: enquanto os receptores α1 endoteliais se ligam a proteínas Gs que
ativam o sistema de fosfolipase C e produzem, a partir do IP3, vasoconstrição, os
receptores α2inibem a adenilato ciclase, diminuindo a concentração de AMPc e inibindo a própria
liberação nas terminações nervosas noradrenérgicas, como veremos na parte de modulação
sináptica.
Agora que vimos a teoria geral de fármacos, farmacocinética, antagonismos farmacológicos,
receptores, sistemas efetores e segundos mensageiros, veremos como isso se dá no organismo,
estudando inicialmente o Sistema Nervoso Autônomo (que já estamos cansados de revisar desde o
Ensino Médio, cada vez com um enfoque diferente).
Sistema Nervoso Autônomo
O Sistema Nervoso Autônomo (daqui pra frente = SNA), com todas as suas características
funcionais de regulação e balanceamento de funções homeostáticas (manutenção da pressão
arterial média, do ciclo respiratório, de resposta do organismo em situações de stress, entre outras),
pode ser dividido em três partes funcionalmente e anatomicamente distintas que trabalham
conjuntamente. São elas:
Sistema Nervoso Simpático: responsável pelas respostas extremas de “fuga e luta”;
Sistema Nervoso Parassimpático: responsável pelos extremos do respouso e da digestão;
Sistema Nervoso Entérico: constituído de fibras de ambos os anteriores e plexos entéricos (no TGI)
que funciona coordenadamente com os outros dois, mas pode funcionar independente de controle
central, diferentemente do Simpático e do Parassimpático.
Nos ateremos aqui (e nesse início da Farmacologia) nos dois primeiros, alvos de ação de uma
extensa gama de drogas/fármacos. Suas diferenças são tanto anatômicas quanto funcionais.
Vejamos:
Anatomicamente:
O Sistema Nervoso Simpático tem origem no corno lateral da medula espinhal, que é encontrado
nos seguimentos tóraco-lombares da medula. Dá origem a pares de gânglios
paravertebrais (ligados à coluna vertebral em toda a sua extensão e não apenas na região tóraco-
lombar) egânglios (sem pares) pré-vertebrais, ligados a ramos da artéria aorta abdominal.
Suas fibras são caracterizadas por serem, antes dos gânglios,curtas (pré-ganglionares) e, após
os gânglios, (pós-ganglionares) longas, alcançando os órgãos alvo – a exceção importante é a
inervação simpática da adrenal, que tem fibras pré-ganglionares extensas até o órgão, considerado
ele mesmo o gânglio.
O Sistema Nervoso Parassimpático tem origem nos pólos cervical (além de nervos cranianos, como
o vago) e sacral. Seus gânglios encontram-se em íntimo contato com os órgãos-alvo, sendo então
as fibras pré-ganglionares longas e as pós-ganglionares curtíssimas.
Neurotransmissores:

Tanto o SNSimpático quanto o SNParassimpático utilizam, comoneurotransmissor principal pré-
ganglionar, a Acetilcolina (através de receptores nicotínicos, como veremos posteriormente),
inclusive na exceção da adrenal (inervação simpática pré-ganglionar longa). As diferenças são
encontradas nas terminações pós-ganglionares: enquanto oSimpático utiliza, principalmente
(com exceção das gls. sudoríparas), o neurotransmissor Noradrenalina, enquanto
oParassimpático utiliza, exclusivamente, a Acetilcolina.
Existem também diferentes receptores para os diferentes neurotransmissores, além de existirem
subtipos diferentes para o mesmo transmissor. Mas isso veremos detalhadamente em cada tipo de
transmissão (noradrenérgica ou colinérgica).
Funcionalmente:
“As ações simpáticas e parassimpáticas, em geral, se opõem umas às outras.” Mas, nem sempre
isso ocorre. No homem, por exemplo, na genitália, os dois sistemas se complementam: o
parassimpático promove a ereção e o simpático, ao invés de produzir o “brochamento”, leva à
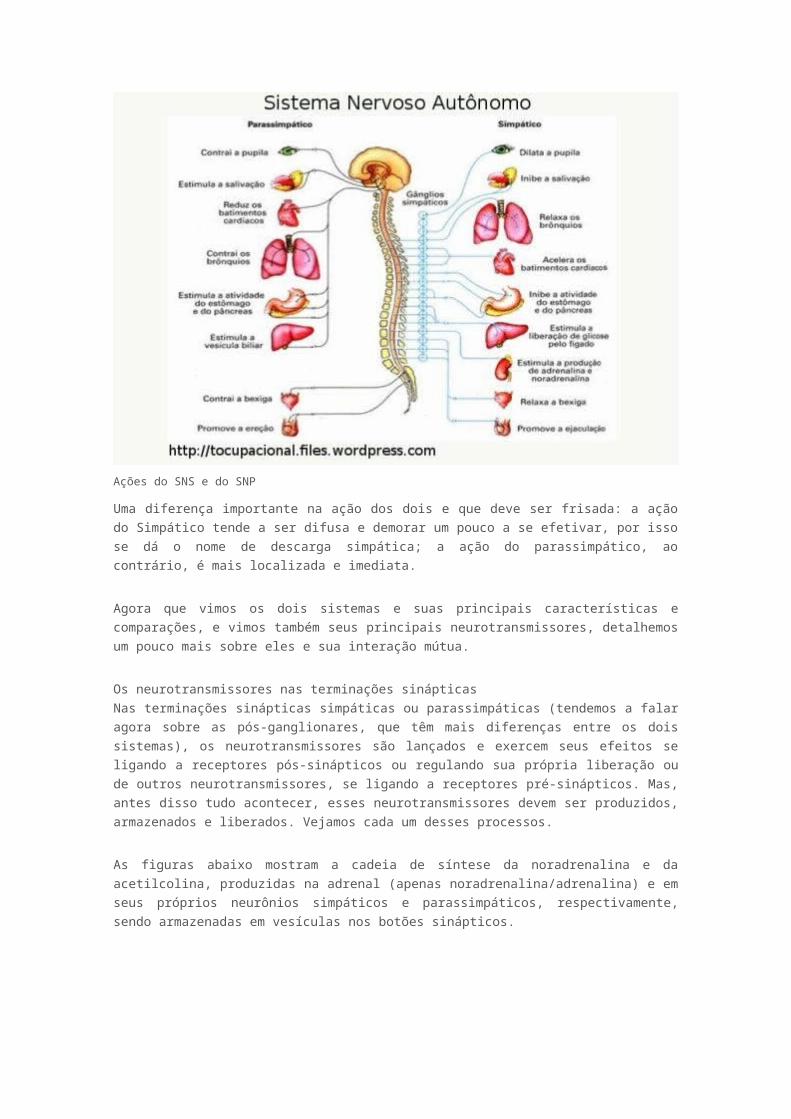
ejaculação. Algumas das principais ações dos dois sistemas são mostrados na imagem abaixo:
Ações do SNS e do SNP
Uma diferença importante na ação dos dois e que deve ser frisada: a ação do Simpático tende a ser
difusa e demorar um pouco a se efetivar, por isso se dá o nome de descarga simpática; a ação do
parassimpático, ao contrário, é mais localizada e imediata.
Agora que vimos os dois sistemas e suas principais características e comparações, e vimos também
seus principais neurotransmissores, detalhemos um pouco mais sobre eles e sua interação mútua.
Os neurotransmissores nas terminações sinápticas

Nas terminações sinápticas simpáticas ou parassimpáticas (tendemos a falar agora sobre as pós-
ganglionares, que têm mais diferenças entre os dois sistemas), os neurotransmissores são lançados
e exercem seus efeitos se ligando a receptores pós-sinápticos ou regulando sua própria liberação ou
de outros neurotransmissores, se ligando a receptores pré-sinápticos. Mas, antes disso tudo
acontecer, esses neurotransmissores devem ser produzidos, armazenados e liberados. Vejamos
cada um desses processos.
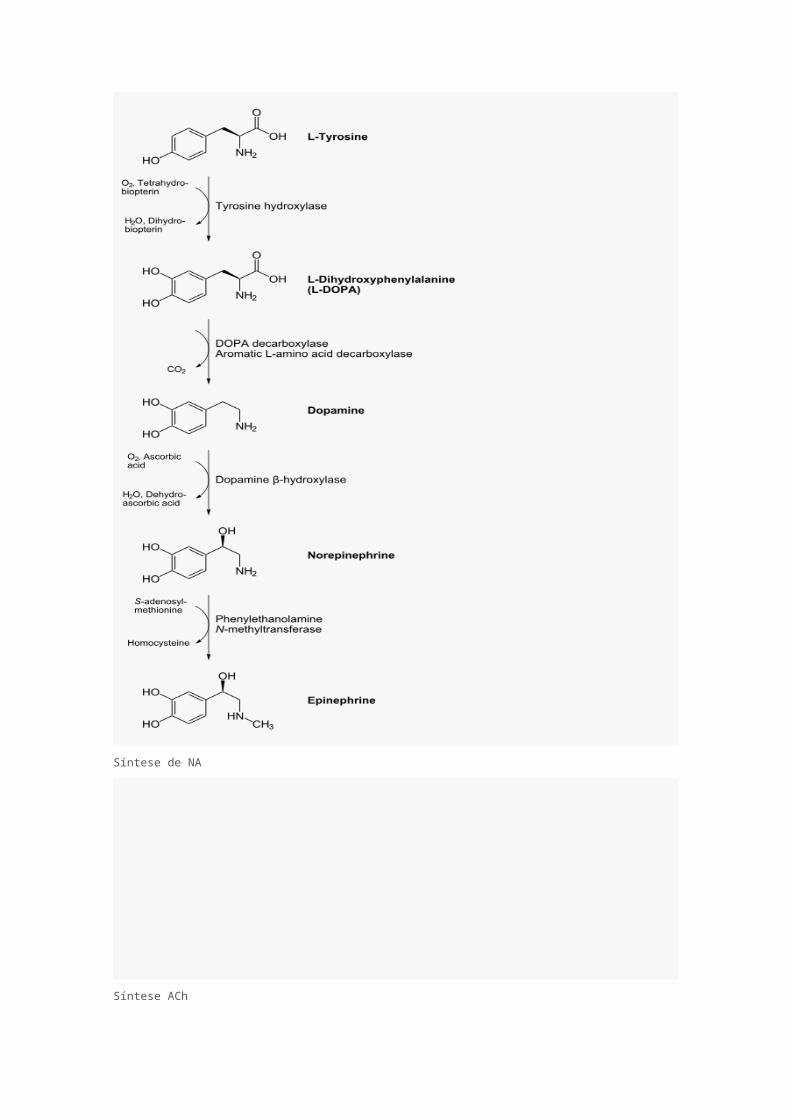
As figuras abaixo mostram a cadeia de síntese da noradrenalina e da acetilcolina, produzidas na
adrenal (apenas noradrenalina/adrenalina) e em seus próprios neurônios simpáticos e
parassimpáticos, respectivamente, sendo armazenadas em vesículas nos botões sinápticos.
Síntese de NA
Síntese ACh
A síntese da adrenalina (esq.) é limitada pela primeira reação, pela enzima Tirosina Hidroxilase, que
é controlada pela quantidade de Noradrenalina na fenda sináptica (uma espécie de feedback
negativo muito bem controlado). Já a síntese da acetilcolina (abaixo) é limitada pelo transporte da
colina do meio extracelular para o botão sináptico.
A produção da adrenalina é contínua mas é especialmente estimulada por stress (responsável pela
“descarga simpática”).
Depois da produção, as moléculas de NA e ACh são armazenadas em altas concentrações em
vesículas sinápticas. Sua liberação depende de potenciais pré-sinápticos que abrem canais de
cálcio mediados por voltagem. O aumento da concentração intracelular de Ca++ leva à exocitose
das vesículas e liberação dos neurotransmissores na fenda sináptica. No caso da NA, além dela
própria, suas vesículas armazenam co-transmissores, como o ATP e a β – dopamina.
A liberação, que ocorre normalmente por esse influxo de Cálcio, pode ocorrer por algumas
substâncias que mimetizam os neurotransmissores e penetram nas vesículas em seus lugares,
expulsando-os para o meio intracelular (e daí logo para a fenda sináptica). Esse mecanismo ocorre
com muitas das drogas do Mouse Party, como veremos depois.
Após sua liberação, os neurotransmissores atingem a fenda sináptica e ali podem ligar-se a
receptores pós-sinápticos e levarem aos efeitos celulares esperados, podem se ligar a receptores
pré-sinápticos da própria terminação nervosa ou de outro neurônio, modulando a produção/liberação
de neurotransmissores (deles próprios ou de outros tipos) e podem (e são!) ser degradados para
que sua ação tenha um fim. Falemos sobre esses dois últimos processos: a modulação pré-
sináptica e a degradação de neurotransmissores.
Modulação Pré-sináptica:
Existem receptores nas terminações nervosas pré-sinápticas que reconhecem certas substâncias,
entre elas os próprios neurotransmissores liberados por aquelas terminações. Esses receptores,
quando estão ligados a esses neurotransmissores, são capazes de gerar alterações intracelulares
que regulam a síntese e/ou a liberação de neurotransmissores por aquela terminação – seja por
alterações enzimáticas, por alterar concentrações iônicas e levar à endocitose ou à exocitose de
vesículas com neurotransmissores. É assim que a liberação de NA nos músculos lisos é capaz de
inibir maior liberação da própria NA, possibilitando um efeito mais curto e que pode sofrer repetições
mais frequentes. Da mesma maneira, a ação da ACh nas glândulas exócrinas reduz a liberação de
ACh pro modulação pré-sináptica.
Essa modulação que acabei de exemplificar é chamada interação homotrópica, onde o próprio
neurotransmissor se liga à sua terminação pré-sináptica e interfere na sua liberação. Mas pode
acontecer o que se denomina interação heterotrópica, que acontece quando um transmissor afeta a
liberação de outro. Usando os mesmos exemplos: uma terminação parassimpática nos músculos
lisos libera ACh que inibe a liberação de NA (juntamente com a própria NA); assim como uma
terminação simpática em uma glândula exócrina libera NA que inibe a liberação de ACh (juntamente
com a própria ACh).
Esses receptores seguem a mesma lógica daqueles pós-sinápticos: se forem ligados a proteínas G
inibidoras ou estimuladoras, podem gerar modulações inibitórias ou estimuladoras. A
terminação simpática liberadora de NA citada acima, por exemplo, tem receptores α2 adrenérgicos
que se acoplam a proteínas Gi.
Degradação de neurotransmissores:
A degradação dos neurotransmissores é processo similar à biotransformação dos fármacos e tem a
mesma função: liberar os receptores e encerrar a ação das moléculas ativas. No caso da NA (ou de
outras catecolaminas, como a adrenalina, a dopamina ou a isoprenalina), as enzimas MAO
(monoaminoxidase) e COMT (catecol-o-metil transferase) são as responsáveis. No caso da ACh, a
enzima acetilcolinesterase (de ação extremamente rápida) é a responsável. Essas enzimas
degradadoras são alvos de fármacos que têm o objetivo de prolongar a ação dos
neurotransmissores endógenos.
Já falamos então de toda a parte teórica dos transmissores adrenérgicos e colinérgicos: como são
produzidos, como são armazenados e liberados, como podem se ligar após a liberação e como são
degradados e recaptados para o reinício do ciclo. Vejamos então detalhadamente como funcionam
as etapas específicas de cada tipo de transmissor e como os fármacos existentes podem afetá-las.
Transmissão Noradrenérgica
A Noradrenalina (NA) é uma das catecolaminas endógenas, produzida na terminações nervosas
pós-ganglionares do Sistema Nervoso Simpático.[ A adrenalina, com estrutura muitíssimo similar e
ações também similares no sistema simpático, é produzida na medula da adrenal.] Sua produção,
armazenamento e liberação já foram vistos anteriormente, então vamos nos focar nas suas ações
após sua liberação. Para tanto, precisamos dar um enfoque e entender claramente os diferentes
tipos de receptores adrenérgicos e suas diferentes ações.
Foram identificados receptores adrenérgicos do tipo alfa e beta, que diferem de acordo com a sua
potência (relativa à potência da ligação dos diferentes agonistas – ver pag. 168). O tipo alfa é
subdividido em alfa-1 e alfa-2 e o tipo beta nos subtipos beta-1, beta-2 e beta-3. Todos eles são
típicos receptores acoplados à proteína-G (alfa-1 estimulador da fosfolipase C, alfa-2 inibidor da
adenilato ciclase e todos os beta estimuladores da adenilato ciclase).
Os efeitos mediados por cada um desses subtipos são:
receptores α1-adrenérgicos: vasoconstrição, relaxamento do m. liso gastrintestinal, aumento da
secreção salivar e da glicogenólise hepática.

receptores α2-adrenérgicos: modulação sináptica inibitória* da liberação de neurotransmissores
(interações homo e heterotrópicas), agregação plaquetária, contração do m. liso vascular, inibição
da liberação de insulina pancreática.
receptores β1-adrenérgicos: aumento da frequência e força de contração cardíacas (aumento da
cronotropia e da inotropia, respectivamente – sendo frequentes causadores de fibrilações
ventriculares)
receptores β2-adrenérgicos: broncodilatação, vasodilatação, relaxamento da musculatura lisa
visceral, glicogenólise hepática e tremor muscular.
receptores β3-adrenérgicos: lipólise.
Podemos ver que essas ações (com exceção de algumas mediadas pelosreceptores β2 **) são
coerentes com as funções simpáticas de liberação de energia rápida para as situações de “fuga ou
luta”, além de desviar a energia usada na digestão para os músculos estriados.
*= A modulação sináptica inibitória que a NA faz sobre ela mesma é chamada também
de retroalimentação auto-inibitória.
Outro aspecto que temos que analisar acerca da transmissão adrenérgica é a captura ou captação
dos neurotransmissores de volta à terminação nervosa pré-sináptica, que atua juntamente com a
degradação para encerrar a ação da molécula ativa e também manter um estoque do
neurotransmissor. A captura ou captação pode ocorrer por dois mecanismos: o neuronal (captura 1)
e o extraneuronal (captura 2). [As diferentes formas de captura (ou captação) estão resumidas nos
quadros do Rang e Dale (pág.173) e nos slides da aula 4 da Vânia.]
Captura 1: responsável pela captura de cerca de 75% da noradrenalina liberada pelos neurônios
simpáticos, a captura 1 consiste em um transporte ativo responsável por encurtar a ação do
neurotransmissor e reciclá-lo para dentro das vesículas sinápticas, mantendo um estoque razoável.
Captura 2: responsável pelo restante da captura da noradrenalina, também por transporte ativo,
evitando/limitando sua disseminação tecidual fora dos neurônios.
Ambas as capturas utilizam moléculas transportadoras. O NET (norepinephrine transporter) é
responsável pela captura 1 enquanto o VMAT (vesicular monoamine transporter) é responsável pela
2. Suas diferenças estruturais são expressas em seus diferentes substratos, resumidos nas tabelas.
A captura 1, por ser a principal forma de encurtamento da ação noradrenérgica, é alvo principal de
drogas como a cocaína, anfetaminas e antidepressivos tricíclicos (inibem a ação da NET ou
substituem a NA na NET, levando à liberação da NA em maior quantidade).
Agora, tendo visto toda a teoria específica sobre a transmissão noradrenérgica (ou simplesmente
adrenérgica), vejamos as etapas em que agem as drogas (e quais são essas drogas e suas ações).
Síntese de NA
Drogas podem afetar a síntese de NA especialmente agindo na primeira reação da síntese, mediada
pela enzima tirosina hidroxilase, ou secundariamente em qualquer uma das outras reações,
substituindo seus substratos por moléculas inativas ou defeituosas. Essas drogas podem ser a α-
metiltirosina, a α-metildopa, entre outros (além dos que falaremos no ítem 3 logo abaixo, que são
agonistas α2).
2. Liberação de NA
Pode ser afetada por drogas que impeçam a exocitose das vesículas sinápticas, que induzam a
liberação da NA sem necessidade do potencial despolarizante (pré-sináptico), que interajam com
receptores pré-sinápticos (modulação pré-sináptica) potencializando ou reduzindo a liberação de NA
(dopamina e agonistas de receptores α2, por exemplo) ou ainda que aumentem/diminuam as

reservas disponíveis de NA (ex: inibidores da degradação pela MAO; reserpina – que age no
armazenamento; guanetidina, que é bloqueadora de neurônios noradrenérgicos).
3. Ligação com os Receptores Adrenérgicos
A maior parte das drogas adrenérgicas e anti-adrenérgicas se concentra nessa parte, podendo ser
agonistas(adrenérgicas ou simpatomiméticas) ou antagonistas adrenérgicos (antiadrenérgicas ou
simpatolíticas).
E dentro dessas categorias ainda temos os antagonistas e agonistas seletivos para determinado
subtipo de receptor e não-seletivos. Tabelas de usos clínicos muito úteis estão nas páginas 179 e
182 do Rang e Dale.
Agonistas Adrenérgicos: (quadro da pág. 177)
- Noradrenalina e adrenalina: não seletivos.
- Felilefrina e oximetazolina são agonistas α1-seletivos, mimetizam a ação dos receptores α1
– contração da musculatura lisa, contração da musculatura radial da íris e estimulação do SNC.
- Clonidina e α-metilnoradrenalina são agonistas α2-seletivos, mimetizando a ação dos
receptores α2 – diminuição da síntese e da liberação de neurotransmissores adrenérgicos.
- Dobutamina é um dos agonistas β1-seletivos, mimetizando a ação dos receptores β1 – aumento
da frequência e da força de contração cardíacas. Obs: todas as drogas dessa classe são
arritmogênicas.
- Salbutamol, terbutalina e salmeterol são agonistas β2 -seletivos, mimetizando a ação dos
receptores β2 – vasodilatação e forte broncodilatação. São, pelo último motivo, utilizados
principalmente na asma.
- Não se conhecem agonistas β3-seletivos, mas seu uso poderia incluir o controle da obesidade, já
que os receptores β3 estão envolvidos na ativação da lipólise.
Antagonistas Adrenérgicos não-seletivos:
Labetalol e carvedilol são exemplos de fármacos antagonistas tanto de receptores α quanto de
receptores β-adrenérgicos. São utilizados para o tratamento de hipertensão durante a gravidez e
hipertensão/insuficiência cardíaca, respectivamente.
Antagonistas (ou bloqueadores) α- Adrenérgicos: (quadro pág. 179)
- Fenoxibenzamina e fentolamina são fármacos antagonistas α-adrenérgicos não-seletivos e já
foram usados para produzir vasodilatação no tratamento de doenças vasculares periféricas, mas
esse uso está obsoleto (a tendência é o uso de drogas seletivas para evitar efeitos colaterais
indesejados).
- Prazosina, doxazosina e terazosina são antagonistas α1-seletivos, usados no tratamento da
hipertensão por antagonizar os efeitos vasoconstritores dos receptores α1.
- A iombina é um antagonista α2-seletivo não utilizado clinicamente. Teoricamente, causa aumento
da liberação de NA por inibir a modulação pré-sináptica (pode-se pensar que o motivo de não ser
utilizado é que essa modulação pré-sináptica não é específica para a NA e pode influenciar na
liberação de diversos neurotransmissores, causando algum desbalanço nervoso).
Antagonistas (ou bloqueadores) β- Adrenérgicos: (quadro pág. 182)
- Propanolol, alprenolol, oxprenolol e sotalol são fármacos antagonistas β-adrenérgicos não-
seletivos e antagonizam o efeito dos receptores β, especialmente no aparelho cardiovascular
(gerando redução da força e da frequência cardíacas, além de leve vasoconstrição) e na árvore
brônquica (causando constrição). Alprenolol e oxprenolol podem ter atividade agonista parcial.

- Atenolol, practolol e nebivolol são antagonistas β1-seletivos, com ação mais específica sobre o
coração.
- Butoxamina é um antagonista β2-seletivo, com ação mais específica sobre a árvore brônquica e
pouco visível vasoconstrição.
Todos os antagonistas β-adrenérgicos têm riscos importantes como broncoconstrição e bradicardia
com insuficiência cardíaca, mas todos têm muitos usos clínicos, como mostrado no quadro da
página 182. Alguns deles são: na angina de peito, no infarto do miocárdio, nas arritmias/insuficiência
cardíaca, no glaucoma e no tremor familiar benigno – entre outros. Um efeito colateral comum no
uso de beta-bloqueadores é o cansaço (eu mesmo já tive que tomar o bendito propanolol por um
tempo, e o cansaço era uma constante no tratamento).
Transmissão NA e ação de drogas
Falemos agora, terminando com a parte noradrenérgica/simpática, sobre o caso clínico apresentado
pela professora e que diz respeito ao item 12 do questionário direcionador: “Odontologia: uso de
propanolol e atenolol com/sem vasoconstritores”. Cito agora o caso clínico:
“A.C.R., de 60 anos, necessitou de exodontia de dois restos radiculares, com vistas à instalação de
prótese total. Na anamnese, o paciente referiu ser hipertenso leve e ter eventuais distúrbios do ritmo
cardíaco, condições tratadas com beta-bloqueador (atenolol, 50 mg/dia). O dentista resolveu, então,
anestesiá-lo com lidocaína a 2% sem vasoconstritor. Necessitou-se de suplementação anestésica e

houve maior sangramento local, dificultando o procedimento. Assim, para a segunda extração, o
dentista solicitou ao médico do paciente um laudo de liberação para usar anestésico com
vasoconstritor.”
Resumindo: o paciente é hipertenso leve e toma atenolol diariamente. O dentista pediu um laudo de
liberação para o uso do vasoconstritor com medo de que a vasoconstrição pudesse desencadear
uma hipertensão grave.
Lembremos que o atenolol é um beta-bloqueador seletivo de receptoresβ1; ou seja, seu remédio
tem efeito sobre os receptores cardíacos e não leva a vasoconstrição. Sendo assim, o receio do
dentista era infundado, pois os pacientes hipertensos leves tomando beta-bloqueadores só podem
ser acometidos por hipertensão grave caso estejam tomando beta-bloqueadores não-seletivos, que
já causam certa vasoconstrição.
(Conclusão: o dentista fez certo e passou o problema pra quem realmente poderia resolver algo
assim: um médico! Ok, ok, sem preconceitos inter-profissionais.)
Agora que analisamos a transmissão adrenérgica, passemos para a transmissão colinérgica…
Transmissão Colinérgica
Descoberta por seu efeito redutor da pressão quando retiraram a adrenalina de um extrato de
adrenal, a acetilcolina (ACh) foi dividida, por Dale, em dois tipos de ação: as ações que são
reproduzidas pela muscarina e as ações que são semelhantes às produzidas pela nicotina. Sendo
assim, Dale postulou que haveriam dois tipos de receptores diferentes para a Ach, os receptores
muscarínicos e os nicotínicos, o que foi provado ser verdade com o avanço das técnicas de estudo
de receptores. O experimento de Dale pode ser visto no gráfico da pág. 145 do livro ou nos slides da
profª Vânia, que é representado abaixo:

Experimento de Dale
Dale sabia que a atropina bloqueava as ações muscarínicas da ACh (A e B: queda brusca da
pressão por vasodilatação e bradicardia por diminuição do cronotropismo), mesmo com dose mais
alta (C). Mas uma dose muito alta de ACh após a administração de atropina levava a dois picos de
pressão, que resultam dos efeitos nicotínicos da ACh, ou seja, nos gânglios autônomos simpáticos
(os parassimpáticos também são estimulados, mas seu efeito final não é expresso pois a atropina
bloqueia os seus receptores muscarínicos pós-ganglionares). O primeiro pico é de ação direta do
sistema simpático, ou seja, a vasoconstrição e a taquicardia; o segundo é decorrente da liberação
reflexa de adrenalina pela adrenal, em resposta à alta dose de ACh nos receptores nicotínicos.
A ACh, então, é o neurotransmissor geral dos gânglios autônomos e nos músculos esqueléticos,
agindo em receptores nicotínicos. E é também o neurotransmissor do sistema nervoso
parassimpático, agindo em receptores muscarínicos. As exceções são:
Os efeitos de vasodilatação generalizada causados por prevalência parassimpática não são
mediados diretamente pela ACh, mas sim por uma via indireta: a ação da ACh no endotélio leva à
liberação de NO (óxido nítrico), que é quem causa os efeitos vasodilatadores.
Por mais que a ACh seja responsável pela secreção das glândulas sudoríparas, a inervação destas
é simpática.
Vejamos então onde se encontram os receptores muscarínicos, quais seus tipos e quais seus
efeitos, já que os nicotínicos nós já sabemos que se encontram nos gânglios autônomos e nas
junções neuromusculares e são responsáveis pela transmissão sináptica excitatória rápida nesses
locais.
Receptores Colinérgicos Muscarínicos:
Podem ser dos tipos M1, M2, M3, M4 e M5, sendo que os três primeiros já foram descritos e já se
sabe a sua importância
- M1 (neurais): responsáveis pelo relaxamento do músculo liso gastrintestinal (se contrapondo aos
alfa-1 adrenérgicos), aumento da secreção ácida gástrica, entre outros.
- M2 (cardíacos): presentes no coração (duur!) e nas terminações pré-sinápticas dos neurônios
autônomos (como os alfa-2 adrenérgicos). São responsáveis pela inibição colinérgica do coração,
assim como pela modulação pré-sináptica inibitória (homo e heterotrópica) no SNC e no SNA.
- M3 (glandulares/do músculo liso): aumentam a lipólise (por meio da excitação glandular) e
medeiam a vasodilatação, por estarem presentes no endotélio e comandarem a produção de óxido
nítrico.
Assim como fizemos na transmissão adrenérgica, outro aspecto que temos que analisar acerca da
transmissão colinérgica é como ocorre a desativação da ligação ACh-receptor. E isso, como já
comentamos, é feito por colinesterases (especialmente a AcetilColinesterase – AChE), que age de
maneira super rápida e quebra a ACh em colina e acetato em questão de milisegundos. Por isso, a
etapa de quebra da ACh pela AChE é um importante sítio de ação de fármacos, chamados
anticolinesterásicos, que veremos em breve.

Tendo visto a teoria específica sobre a transmissão colinérgica, vejamos as etapas em que agem as
drogas (e quais são essas drogas e suas ações).
Ação do Botox (Toxina Botulínica)
Liberação da ACh
As drogas que atuam nessa fase podem potencializar a sua liberação (como a 4-aminopiridina),
podem impedir a captação do precursor colina (como o hemicolínio) ou podem inibir o mecanismo
de liberação (exocitose) de ACh, como é mostrado na figura a seguir.
2. Destruição da ACh
Nessa fase entram os chamados anticolinesterásicos, que inibem as colinesterases. Entre eles,
temos a fisostigmina, a neostigmina (ambas de ação média), o edrofônio (de ação curta) e o
ecotiopato (de ação longa, usado em colírios no tratamento do glaucoma). Depois de falarmos de
bloqueio neuromuscular, falamos de novo nos anticolinesterásicos.
3. Ligação com os receptores Colinérgicos
Grande parte dos fármacos entra nessa fase, da mesma maneira como foi na transmissão
adrenérgica. Aqui se encaixam os agonistas e antagonistas colinérgicos,principalmente
muscarínicos, e os estimuladores e bloqueadores ganglionares, principalmente nicotínicos; além dos
bloqueadores neuromusculares, que veremos à parte.
Agonistas Muscarínicos (drogas colinérgicas ou parassimpatomiméticas):
Podem ser vistos efeitos cardiovasculares (como a diminuição da frequência e do débito cardíacos,
por redução da força de contração atrial*, além da vasodilatação generalizada mediada por NO),
efeitos sobre a musculatura lisa (contração**, exceto no músculo vascular), efeitos glandulares
(aumento do volume de secreção) e efeitos oculares (acomodação visual, regulação da pressão
intra-ocular***).
- Acetilcolina, carbacol, metacolina, betanecol, a própria muscarina, pilocarpina e oxotremorina são
agonistas muscarínicos.
*=Os efeitos cardíacos são devidos à invervação parassimpática vagal, principalmente nos nodos
SA e AV, reduzindo a frequência de disparo; além disso, a inervação atrial leva à redução da força

de contração, que reduz o débito cardíaco. Quase não há alteração direta na força de contração
ventricular.
**= Drogas como o betanecol são utilizadas para auxiliar no esvaziamento da bexiga e na
motricidade gastrintestinal, por atuação nos receptores colinérgicos M1.
*** = Ações importantes dos receptores muscarínicos no olho; motivos pelos quais são usados
agonistas muscarínicos e fármacos anticolinesterásicos no tratamento do glaucoma. A pilocarpina,
por exemplo, ultrapassa a membrana conjuntival e é muito utillizada por ser um composto estável
(dura cerca de um dia).
Antagonistas Muscarínicos (drogas anticolinérgicas ou parassimpatolíticas):
No geral, os antagonistas produzem efeitos glandulares (inibição das secreções), efeitos cardíacos
(taquicardia moderada sem vasoconstrição), efeitos oculares (perda do efeito de acomodação
visual, midríase, irresponsividade a luz), efeitos gastrintestinais (inibição da motilidade do TGI, não
muito grave), efeitos sobre a musculatura lisa (relaxamento) e efeitos sobre o SNC (excitação, no
caso da atropina: inquietação, agitação, desorientação – esses efeitos sobre o SNC são devido ao
bloqueio dos receptores muscarínicos, o que pode ser revertido pela fisostigmina; sedação, no caso
da escopolamina, o que pode ser útil na redução dos movimentos involuntários e na rigidez
provocada pela doença de Parkinson).
- Atropina e escopolamina (também chamada de hioscina) são alcalóides de ocorrência natural
(vegetal) que são utilizados como adjuvantes de anestesias, por reduzirem as secreções e
causarem broncodilatação. Também são utilizados para tratamento na bradicardia e da
hipermotilidade gastrintestinal (são antiespasmódicos). A diferença entre os dois é que a atropina é
estimulante do SNC enquanto a escopolamina é depressor. Além disso, a atropina é melhor
absorvida por via oral, enquanto a escopolamina é mais usada transcutaneamente, e geralmente
como remédio para enjôo de movimento (cinetose).
- Pirenzepina e Darifenacina são fármacos seletivos para receptores M1 e M3, respectivamente.
Suas ações respectivas são a redução da secreção gástrica nas células glandulares (no tratamento
de úlcera péptica) e a redução da contração da musculatura lisa da bexiga (no tratamento de
incontinência urinária).
Estimulantes Ganglionares (ou agonistas nicotínicos):
Geralmente, os estimulantes ganglionares também são estimulantes da placa muscular. Seus
efeitos são complexos, pois a estimulação é tanto simpática quanto parassimpática. Geralmente o
quadro é de taquicardia e hipertensão, efeitos variados sobre a motilidade gastrintestinal e
geralmente aumento da secreção brônquica, salivar e sudorípara. A estimulação dos gânglios pode
ser seguida de bloqueio por despolarização. Com exceção da nicotina, que é utilizada em terapias
para o abandono do tabagismo, não apresentam uso terapêutico.
- Nicotina, lobelina, DMPP (dimetilfenilpiperazínio – ô trem difícil!) são estimulantes ganglionares que
não afetam a placa muscular. A nicotina gera estimulação dos gânglios, seguida de bloqueio por
despolarização. (Isso excluindo o efeito estimulador sobre o SNC.)

- Suxametônio e decametônio são estimulantes ganglionares que atuam justamente na placa
muscular e o primeiro é utilizado clinicamente como relaxante muscular.
Bloqueadores Ganglionares (ou antagonistas nicotínicos):
São usados com frequência em estudos experimentais sobre o SNA, mas com pouca utilidade
clínica. São exemplos de bloqueadores ganglionares o hexametônio e a trimetafano (que pode
chegar a ser usado em raros casos de hipotensão induzida controlada durante anestesia).
Bloqueadores Neuromusculares (agonistas ou antagonistas nicotínicos):
Agem bloqueando os receptores nicotínicos pós-sinápticos da ACh (sendo agonistas ou
antagonistas). Do ponto de vista clínico, o bloqueio neuromuscular só é utilizado como adjuvante da
anestesia – sendo necessária ventilação artificial do paciente.
A descoberta do mecanismo dos bloqueadores veio com o estudo de venenos utilizados pelos
índios para a paralisação e captura de animais. O “veneno” mais comum era o curare, depois
descoberto como sendo o fármaco tubocurarina, que veio a ser substituído pelos fármacos sintéticos
vecurônio, atracúrio e pancurônio (só diferem na duração do efeito). Por não atravessarem a
placenta, chegaram a ser utilizados como anestésicos na obstetrícia. Em geral, os bloqueadores
neuromusculares podem ser divididos em não-despolarizantes (adespolarizantes) e despolarizantes.
- Os não-despolarizantes atuam como antagonistas competitivos da ACh nos receptores da placa
muscular, impedindo a contração mediada por ACh (dependendo da quantidade de bloqueador,
visto que a quantidade de ACh nas junções neuromusculares é sempre grande). Além da ação de
antagonista, alguns estudos mostram que alguns bloqueadores não-despolarizantes também se
ligam em auto-receptores colinérgicos pré-sinápticos (modulação pré-sináptica), inibindo a liberação
de ACh na placa muscular durante a estimulação muscular repetitiva. Os efeitos são em série:
primeiramente, os músculos faciais (oculares extrínsecos, pequenos músculos faciais) são afetados;
depois, as extremidades e a faringe (dificuldade de deglutição); por último, os músculos respiratórios
– que são também os primeiros a serem recuperados.
Anticolinesterásicos aumentam a força muscular e ajudam na reversão do quadro de paralisia, por
aumentarem a disponibilidade de ACh na placa muscular, especialmente com uma maior frequência
de disparo na junção neuromuscular. Daí a resposta do “como a neostigmina e a fisostigmina agem
no organismo para reverter os efeitos dos bloqueadores adespolarizantes?” (questão 18 do
questionário).
- Os despolarizantes são agonistas da ACh, como o suxametônio. A despolarização ocorre com a
permanência da ACh ou de um agonista como o suxametônio nos receptores da placa muscular (é
como se o músculo não conseguisse relaxar após ter se contraído; na não-despolarização, ele nem
chega a contrair). Em situações fisiológicas isso não ocorre, haja vista que a colinesterase age
rapidamente e impede a contínua despolarização.Mas, em certas situações, podem advir efeitos
indesejados, citados abaixo (respostas da questão 19)
Fatores como o uso de drogas anticolinesterásicas, certas variantes genéticas que alteram a
funcionalidade da colinesterasa e baixa atividade da colinesterase plasmática (em recém-nascidos e
hepatopatas, por exemplo), favorecem o prolongamento da despolarização – podendo causar
apnéia prolongada.

Miastenia gravis, depois da apnéia prolongada, é outro efeito indesejável dos bloqueadores
despolarizantes. A eficácia dos anticolinesterásicos na melhora da força muscular em indivíduos
com miastenia gravis é um fato, descoberto antes mesmo de se desvendar a causa da doença (o
que é parte da resposta da questão 20). De certa forma é um certo paradoxo tratar a miastenia com
anticolinesterásicos, sendo que acabamos de ver que os anticolinesterásicos juntamente com
bloqueadores despolarizantes podem levar à apnéia prolongada – acho que é só uma questão de
dosagem para evitar os malefícios sem abrir mão dos benefícios.
Depois de miastenia gravis e apnéia prolongada, outro efeito adverso é a hipertermia maligna. É o
resultado de uma condição genética rara herdada, que causa fortes espamos musculares e súbita
hipertermia quando são administradas drogas como o suxametônio (bloqueador despolarizante) e
halotano, além de várias outras drogas. O tratamento consiste na administração do inibidor da
liberação de cálcio dos retículos sarcoplasmáticos, o dantroleno (outra parte da resposta da questão
20).
A colinesterase e sua relação com os anticolinesterásicos, como já citado nas devidas seções,
inclusive no tratamento de certos efeitos adversos do uso de alguns bloqueadores neuromusculares,
é importantíssima para a regulação dos níveis de ACh. Prova disso é que, quando por
envenenamento por anticolinesterásicos (inseticidas ou gases que atacam os nervos), os efeitos
autônomos do excesso de ACh nas fendas sinápticas envolvem bradicardia, hipotensão,
broncoconstrição, hipermotilidade gastrintestinal e redução da pressão intraocular. Nos músculos,
ocorre fasciculação e maior força nos espasmos (por causa da falta de controle da contração). Um
importante fármaco quando há esse tipo de envenenamento (ou inclusive no caso da apnéia
prolongada) é a pralidoxima, que reativa a colinesterase plasmática no indivíduo envenenado (parte
que faltava da resposta da questão 20).
Pronto, de matéria é isso. Agora um adendo sobre o programa MouseParty (questão 7) e sobre o
filme, que fala de todos as substâncias citadas na questão 15 – a parte do House é mais em relação
à drogas anticolinesterásicas e a outras drogas já citadas no resumo.
Mouse Party: falarei dos ratinhos brevemente de acordo com a ação das drogas que cada um
consumiu…
Cocaína: age bloqueando os receptores pré-sinápticos de dopamina, em especial na área de
controle dos movimentos voluntários (iniciativa) e na área septal (recompensa, vício).
LSD: age como agonista serotoninérgico, em especial no locus cerúleos, que faz sinapses com
regiões diversas do córtex, criando sensações variadas e alucinógenas.
Álcool: auxilia a inibição GABAérgica e é antagonista glutamatérgico (impede a excitação pelo
glutamato), age especialmente na área pré-frontal do córtex, impedindo a formação de memórias e
inibindo o o comportamento sociável (o bêbado perde a noção do que deve ou não deve fazer, mas
vocês já devem saber bem disso).
Maconha: mimetiza a ação e é mais durável que o canabinóide natural do SNC ( a anandamida,
liberada como inibidora dos inibidores normais da dopamina. Causa relaxamento, lentidão e
remoção de memória recente.
Heroína: ação mimética do opióide natural do SNC (liberado como inibidor dos inibidores normais da
dopamina), age nas áreas responsáveis pelo processamento da dor, pela resposta ao stress e pelas
respostas com caráter emocional. É da mesma família da morfina.
Metanfetamina: mímico da dopamina, expulsa a dopamina das vesículas (por captura 1). Age na
área pré-septal, como estimulante.

Ecstasy: o ratinho mais comédia do programa; mímico pré-sináptico da serotonina, é capturado
pelos transportadores de serotonina e leva à extrusão da serotonina das vesículas (similar à
metanfetamina). Age na área pré-septal e em diversas áreas responsáveis pelo humor, pelo sono,
pela percepção e pelo apetite (o cara fica doidão, aew!).
Sobre as substâncias no filme Linha Mortal (mucho doido e mucho sem noção):
Adrenalina: já vimos as respostas adrenérgicas no resumo, mas recapitulando –
hormônio simpaticomimético de ação sobre os receptores alfa-adrenérgicos (gerando
vasoconstrição) e beta-adrenérgicos (gerando aumento da força e da frequência cardíacas. É
arritmogênico.
Bretílio: agente anti-arrítmico, é anti-adrenérgico, além de bloquear canais de potássio. Pode
gerar hipertensão seguida de hipotensão e extrassístoles. Usado em taquicardia e fibrilação
ventricular, mas recomendado apenas em UTI’s, pelo grande potencial hipotensivo.
Atropina: como vimos, é antagonista colinérgico muscarínico. Usado como hipertensivo, em
casos de bradicardia. Pode gerar aumento da temperatura corporal (inibindo a sudorese). Pode
gerar alucinações e delírio por ação no SNC, se administrado em altas doses. A estimulação
cardíaca pode ser seguida por depressão reflexa: colapso circulatório e insuficiência
respiratória.
Lidocaína: antiarrítmico e anestésico por ser bloqueador dos canais rápidos de sódio de
miócitos e nervos periféricos.
Potássio: indução de hipercalemia, que leva a acentuada despolarização – pode gerar parada
cardíaca, fraqueza muscular, etc.
Cálcio: indução de hipernatremia, que leva a acentuada hiperpolarização – pode gerar parada
cardíaca em sístole, tetania muscular, etc.
O potássio pode ser utilizado no tratamento da hipernatremia e o cálcio pode ser usado no
tratamento da hipercalemia, o que foi feito no filme – por possuírem características contrárias.
Abraços e boa prova. Desculpem não ter feito antes, os próximos eu acelero.
Félix
1) ORGANIZAÇÃO GERAL DO SISTEMA NERVOSO AUTÔNOMO
- Ativado por centros localizados na medula espinhal, no tronco cerebral e no hipotálamo;- Opera também por meio de reflexos viscerais;
A) SUBDIVISÕES:
SISTEMA NERVOSO SIMPÁTICO

SISTEMA NERVOSO PARASIMPÁTICO
2) ANATOMIA E FISIOLOGIA:
Antigamente acreditava-se que era um sistema isolado do sistema nervoso central, hoje sabemos que o SN vegetativo está intimamente ligado ao SNC.
2.1) Revisão do Sistema Nervoso Neurovegetativo:
2.1.1) Sistema Nervoso Simpático: grego (sympatheia – harmonia)
- Inerva todas as vísceras e a maioria dos vasos de todo o organismo;
- Os neurônios deste sistema se dividem em duas partes, a primeira, fibra pré-ganglionar, que parte da medula e vai em direção a uma cadeia de gânglios e a Segunda fibra é chamada pós-ganglionar, que sai do gânglio e vai em direção ao tecido. No final da pré-ganglionar (Curta) ocorre a liberação de Ach e no final da pós-ganglionar (longa) ocorre a liberação de Noradrenalina. Todas as fibras simpáticas passam por cadeias de gânglios: cadeia paravertebral e cadeia pré-vertebral (tronco simpático).
- Anatomicamente as raízes simpáticas são chamadas Tóraco – Lombares.
2.1.2) Sistema Nervoso Parassimpático:
- Cerca de 75% de todas as fibras parassimpáticas estão no nervo vago
- Se distribue em locais menos amplos do que o simpático e exerce funções bem específicas, freqüentemente (mas nem sempre) opostas ao simpático.
- Pré-ganglionar (longa) – libera Ach
- Pós- ganglionar (curta) – libera Ach
- As fibras do parassimpático não passam por cadeias de gânglios e a sinapse das duas fibras irá ocorrer no próprio tecido ou muito próximo dele.
- Anatomicamente as raízes parassimpáticas são chamadas de Crânio – Sacrais.
3) CARACTERÍSTICAS BÁSICAS DA FUNÇÃO SIMPÁTICA E PARASSIMPÁTICA:

Acetilcolina (Ach) – Colinérgicas;
Noradrenalina (Nor)/Norepinefrina – Adrenérgicas;
Todos os neurônios pré-ganglionários são colinérgicos;
Todos ou quase todos os neurônios pós-ganglionários do sistema parassimpático são também colinérgicos, por outro lado a maioria das fibras pós-ganglionárias do simpático são adrenérgicas, exceto para as glândulas sudoríparas, músculos piloeretores e alguns vasos sangüíneos são colinérgicos;
4) MECANISMOS DE SECREÇÃO E REMOÇÃO DO NEUROTRANSMISSOR
Inicia-se um potencial de ação pelas fibras terminais, aumenta a permeabilidade da membrana aos íons cálcio, permitindo a entrada desses para o interior das terminações nervosas, aí os íons de cálcio interagem com as vesículas secretoras, fazendo com que essas difundam seu material para o exterior.
4.1) Síntese de Acetilcolina
Acetil- CoA + Colina (ação da colina acetil transferase) -> Acetilcolina
Dura segundos na fenda sináptica e o excesso é degradada pela acetilcolinesterase (AchE).
Transformada em Acetato + colina, geralmente a colina retorna para o terminal sináptico e serve de substrato para nova Ach.
4.2) Síntese de Noradrenalina
Tirosina -> dopa -> Dopamina -> transportada para dentro das vesículas
Dopamina -> Noradrenalina
4.3) Enzima que degrada a Nor – MAO
Na medula da adrenal essa reação prossegue, onde cerca de 80% da Nor é transformada em Adrenalina.

4.4) Receptores Colinérgicos
Muscarínicos – encontrados nas células estimuladas pelos pós ganglionares do parassimpático;
Nicotínicos – encontrados nas sinapses do pré com o pós-ganglionar;
4.5) Receptores Adrenérgicos
Alpha –
Beta – 1 e 2 -
O Sistema Nervoso Autônomo (SNA) faz parte do Sistema Nervoso Periférico (SNC), sendo ele constituído por vias eferentes (efetora, motora).
O Sistema Nervoso Autônomo (SNA) têm suas fibras nervosas dividida em dois grupos, Sistema Nervoso Autônomo Simpático e Parassimpático.
O SNA é responsável pelo controle visceral (controle involuntário). Função do músculo liso, cardíaco e controle visceral. O SNA é responsável pelo homeostase corporal.
Morfologicamente:
Fibras Simpáticas = Região Torácica e Lombar.
Fibras Parassimpáticas = Região do Tronco encefálico e Sacral.
As fibras possuem padrão bineural ( 2 neurônios) Pré e pós-ganglionar.
Simpático = Adrenalina/Noradrenalina (liberada após ativação da supra-renal). Noradrenalina/Norepinefrina (Principal transmissor – 80%). Dopamina (SNC – Encéfalo)
Receptores simpáticos = Alfa (Alfa-1, Alfa-2) e Beta ( Beta-1, Beta-2, Beta-3)
Alfa-1 = Localizado nos órgãos efetuadores (membrana pós-sináptica). Localizado principalmente nos músculos lisos das arteríolas (vasoconstricção).
Alfa-2 = Está localizado na membrana pré-sináptica. Inibe a liberação de mais noradrenalina. Localizado no pâncreas (inibe secreção de insulina)
Beta-1 = Coração (Aumenta todas as propriedades cardíadas), Fígado (Gliconeogênese).
Beta-2 = Pulmão (Aumento da frequência respiratória) > Mais oxigênio no sangue. Vasodilatação dos vasos dós músculos esqueléticos ativados durante atividade física. Contração (tremor fino) dos músculos.

Beta-3 = Lipólise (queima de gordura)
Parassimpático = Dimunui dronotropismo, cronotropismo e ionotropismo.
Receptores parassimpáticos = M1, M2 e M3:
M1: SNC (memória/excitação)
M2: Coração (diminui as propriedades do músculo cardíaco)
M3: Trato Genito Urinário (TGU)/ Trato Gastro Intestinal (TGI): Aumento do peristaltismo e segmentar, aumenta a secreção de glândulas (pâncreas – suco pancreático, estômago – HCl). Aumento da contração do músculo detrusor e relaxamento dos esfíncteres (micção).
O corpo em homeostase se encontra com o SNA simpático e parassimpático em equilíbrio.
Postado por DOCTOR FISIO às 13:37
SISTEMA NERVOSO AUTÔNOMO
- Sistema responsável pelo controle das funções viscerais como pressão arterial, motilidade do trato gastrointestinal, vesical e sudorese.
- Organização do sistema nervoso autônomo: este sistema é regulado por centros medulares, pelos núcleos do tronco encefálico como também pelo hipotálamo.
- Anatomicamente fazemos a distinção de três sistemas que compõe o sistema nervoso autônomo: Simpático, Parassimpático e Entérico.
Sistema Nervoso Autônomo Simpático
- Conta com uma cadeia de gânglios simpáticos paravertebrais situadas bilateralmente ao lado da coluna vertebral torácica e lombar.
- Os neurônos pré-ganglionares, isto é, aqueles que interligam a medula espinhal com o gânglio nervoso, é curto, eferindo do corno lateral do H medular. As fibras pós-ganglionares, aquelas que partem dos gânglios, são longas atingindo os órgãos alvo do sistema nervoso autônomo simpático.
- As fibras pré-ganglionares simpáticas fazem sinapses num gânglio nervoso liberando acetilcolina (ACh), atuando sobre receptores nicotínicos. Já as fibras pós-ganglionares simpáticas fazem sinapses com os órgãos alvo com liberação de noradrenalina e ou adrenalina, atuando sobre receptores adrenérgicos alpha e ou beta, dependendo do tecido envolvido.
- Apesar dessas considerações, há exceções: As fibras que inervam as glândulas sudoríparas fazem sinapses ganglionares e terminais (nos órgãos alvo) mediadas por ACh. Outra exceção as considerações anteriores é a inervação da glândula supra-renal: há fibras pré-ganglionares curtas liberando ACh porém não há sinapse ganglionar nervosa típica, a própria glândula supra-renal atua como gânglio nervoso com produção e liberação de catecolaminas.
Sistema Nervoso Autônomo Parassimpático
- Anatomicamente o sistema nervoso autônomo parassimpático situa-se na porção cranial e caudal da coluna vertebral.
- Geralmente as fibras pré-ganglionares são longas (contrário ao SNA Simpático) e as fibras pós-ganglionares são curtas já que os gânglios nervosos, neste sistema, situam-se próximos ao tecido alvo.

- A maior parte das fibras (75%) do sistema nervoso autônomo parassimpático são provenientes do nervo vago.
- Outros pares de nervos cranianos deixam o sistema nervoso compondo o sistema parassimpático: III (nervo óculomotor), VII (nervo facial) e IX (nervo glossofaríngeo).
- Os receptores ganglionares, a exemplo do sistema nervoso autônomo simpático, são colinérgicos nicotínicos enquanto que nas terminações (órgãos alvo) os receptores são muscarínicos (subdivididos ainda em diversos tipos, dependendo do órgão envolvido).

0,
Secreção e Remoção dos Neurotransmissores
- As fibras simpáticas e parassimpáticas apenas tocas as células efetoras, apresentam uma dilatação bulbosa denominada varicosidades contendo vesículas de noradrenalina e acetilcolina.
- O aumento da permeabilidade aos íons cálcio permite difusão do neurotransmissor para o interior do neurônio.
- Síntese de ACh: maior parte da síntese ocorre no axoplasma. A acetil-CoA une-se à colina na presença da enzima colina acetiltransferase.
- Na fenda sináptica ocorrerá remoção do neurotransmissor por difusão, por recaptação pelas vesículas ou pela degradação enzimática (acetilcolinesterase – AChE).

- Síntese de Noradrenalina: inicia-se no axoplasma sendo completada nas vesículas presentes nas terminações sinápticas. Diversas enzimas participam desta síntese: tirosina hidroxilase (converte a tirosina em DOPA), DOPA descarboxilase (converte DOPA em Dopamina), dopamina beta-hidroxilase (converte dopamina em noradrenalina) e, finalmente, a feniletanolamina n-metil transferase (converte a noradrenalina em adrenalina – sendo esta conversão exclusiva da medula da glândula supra-renal).
- A remoção desse neurotransmissor da fenda também ocorre por difusão, recaptação pela vesículas ou ainda pelas enzimas (MAO e COMT).
Atividade Fisiológica dos Receptores Autonômicos
- Alpha 1: vasoconstrição, midríase, glicogenólise hepática, relaxamento da musculatura lisa do trato gastrointestinal, secreção salivar espessa, secreção de suor nas extremidades (suor frio).
- Alpha 2: inibem a liberação do neurotransmissor, atuando como um mecanismo de feedback negativo. Controlam a liberação de insulina pelo pâncreas endócrino.
- Beta 1: taquicardia, lipólise e relaxamento da musculatura lisa do trato gastrointestinal.
- Beta 2: vasodilatação, broncodilatação, relaxamento da musculatura lisa do trato gastrointestinal e glicogenólise hepática.
- Beta 3: lipólise.
FARMACOLOGIA: O SISTEMA ADENILATO CICLASE E AMPCpor: Colunista Portal - Educação
O viagra pode aumentar as concentrações de AMPc nas células
A descoberta do papel do AMPc (3´5´-adenosina-monofosfato-cíclico) como mediador intracelular introduziu o conceito de segundos mensageiros na transdução de sinais. O AMPc é um nucleotídeo sintetizado no interior das células a partir do ATP, sob ação de uma enzima ligada à membrana, a adenilato ciclase. O AMPc é continuamente produzido e inativado por hidrólise da 5´-AMP, por meio de uma família de enzimas conhecidas como fosfodiesterases.
O AMPc regula muitos aspectos da função celular, incluindo enzimas envolvidas no metabolismo energético, divisão e diferenciação celulares, transporte de íons, canais iônicos e proteínas contráteis. Entretanto, estes efeitos são produzidos por um mecanismo comum, isto é, a ativação de proteínas-quinases pelo AMPc.

As proteínas-quinases regulam a função de muitas proteínas celulares diferentes ao catalisarem a fosforilação de resíduos de serina e treonina, utilizando o ATP como fonte de fosfato. A fosforilação pode ativar ou inibir enzimas-alvo ou canais iônicos.
A produção aumentada de AMPc, em resposta à ativação dos receptores β-adrenérgicos, afeta várias enzimas envolvidas no metabolismo do glicogênio e da gordura no fígado, adipócitos e células musculares. O resultado consiste em uma resposta coordenada, em que a energia armazenada na forma de glicogênio e gordura torna-se disponível como glicose para suprir a contração muscular.
O AMPc é hidrolisado no interior das células por fosfodiesterases, uma família de enzimas inibidas por drogas como a teofilina, cafeína e o sildenafil (Viagra®), aumentam as concentrações de AMPc nas células.
Monofosfato cíclico de guanosina (GMPc) é um nucleotídeo cíclico derivado da guanosina trifosfato (GTP). A GMPc atua como umsegundo mensageiro assim como o AMP cíclico, mais notavelmente por ativar as proteínas cinases intracelulares em resposta à ligação dehormônios peptídeos à membrana celular.
Na farmacologia, agonista refere-se às ações ou estímulos provocados por uma resposta, referente ao aumento (ativação) ou diminuição (inibição) da atividade celular. Sendo uma droga receptiva.

Agonista inverso refere-se à interação deste agonista com outro agonista para a produção de diferentes efeitos.
Os denominados antagonistas tem a função contrária dos agonistas. Sendo estes bloqueadores dos receptores, ou seja, diminuem as respostas dos neurotransmissores, presentes no organismo. O antagonismo pode diminuir ou anular o efeito do agonista.
Os medicamentos sem prescrição médica, se misturados podem ser perigosos, podendo os mesmos interagir entre si.
Os antagonistas são classificados em:
parcial/total
reversível/irreversível
competitivo/alostérico
O antagonista parcial não anula totalmente o efeito de um agonista, sendo este mais utilizado, já o total atua somente no problema, não interferindo nas partes que

estão funcionando. Em caso de intoxicação é aconselhável o antagonista total, pois protege melhor o organismo.
No antagonista reversível/irreversível, o agonista tem ao poder de reverter e o outro de inibir os efeitos do antagonista. Já o antagonista competitivo impede o agonista de se encaixar, competindo com o mesmo e o alostérico atua em receptores que tem o efeito diminuidor liberado pelo agonista.
O efeito de um antagonista sobre o agonista se torna inferior devido as constantes estimulações.
Fontes:http://pt.wikipedia.org/wiki/Agonistahttp://www.manualmerck.net/?id=33&cn=562


















