
Palestra 11-desenv-farm-metodol-computac-UEZO-2013
50
Maio de 2013 Modelagem Molecular e estudos ADMETox in silico Prof a . Monique A. de Brito Universidade Federal Fluminense Faculdade de Farmácia
Transcript of Palestra 11-desenv-farm-metodol-computac-UEZO-2013

Maio de 2013
Modelagem Molecular e estudos ADMETox
in silico
Profa. Monique A. de Brito
Universidade Federal Fluminense
Faculdade de Farmácia
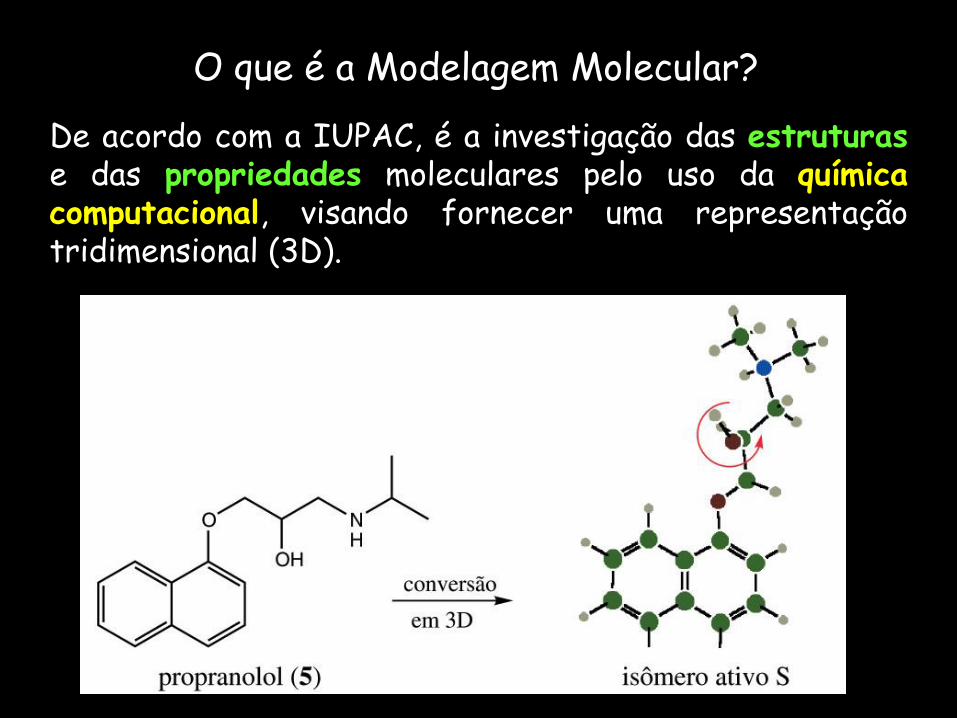
O que é a Modelagem Molecular?
De acordo com a IUPAC, é a investigação das estruturas e das propriedades moleculares pelo uso da química computacional, visando fornecer uma representação tridimensional (3D).
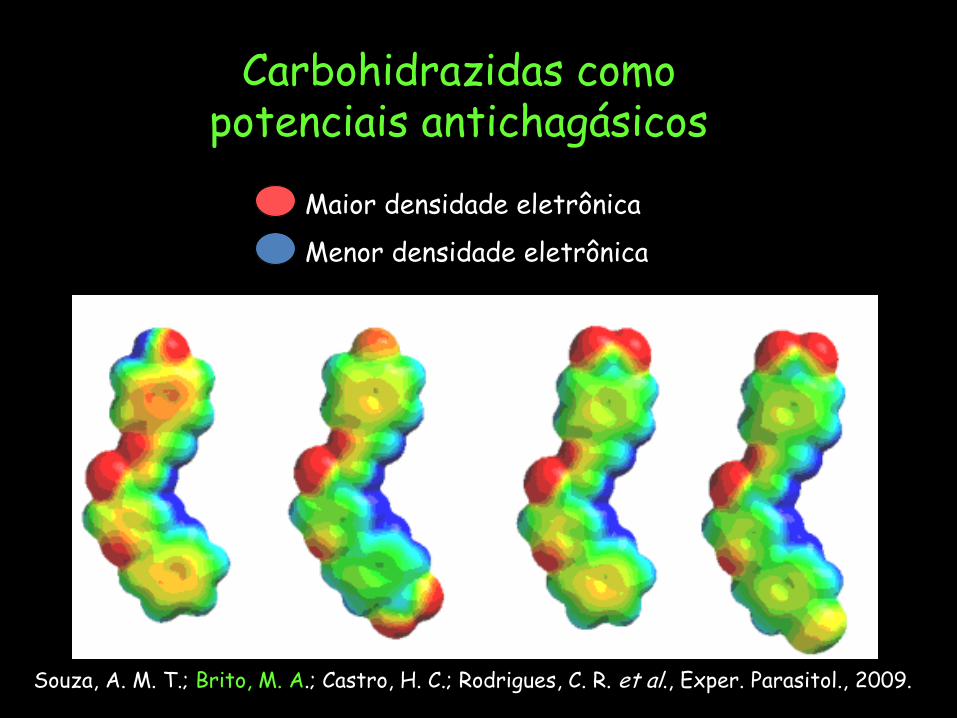
Maior densidade eletrônica
Menor densidade eletrônica
Carbohidrazidas como potenciais antichagásicos
Souza, A. M. T.; Brito, M. A.; Castro, H. C.; Rodrigues, C. R. et al., Exper. Parasitol., 2009.

A importância das cargas atômicas parciais

Mapas de potencial eletrostático de enzimas TR nativa e mutantes do
HIV-1
Ferreira, A. S. R.; Brito, M. A.; Rodrigues, C. R.; Castro, H. C., 2009.
Metodologias computacionais abordadas aqui nesta palestra
***Docking molecular
***Modelagem comparativa
***Relação Estrutura-Atividade (SAR);
***Relação Quantitativa Estrutura-Atividade
(QSAR e QSAR-3D);

Metodologias computacionais abordadas aqui nesta palestra
***Docking molecular
***Modelagem comparativa
***Relação Estrutura-Atividade (SAR);
***Relação Quantitativa Estrutura-Atividade
(QSAR e QSAR-3D);
L+R

Docking
R L
L+R

IUPAC Estudos de Ancoramento – São técnicas computacionais para a exploração dos possíveis modos de interação de um substrato a um dado receptor, a uma enzima ou a outro sítio de ligação.
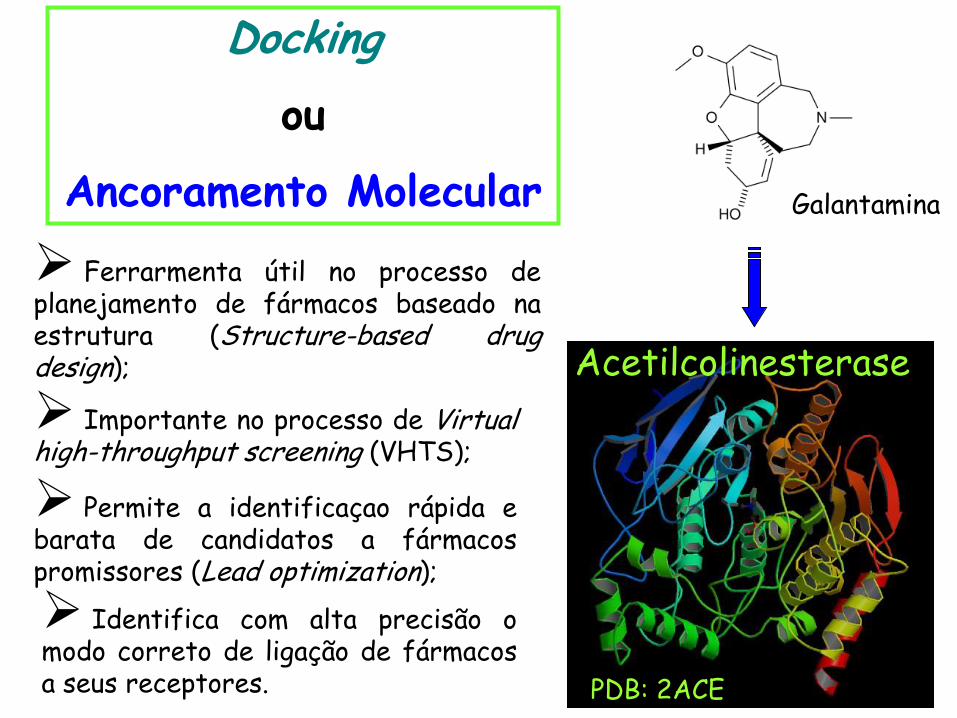
Docking
ou
Ancoramento Molecular
Sant’Anna, C. M. R. Quím. Nova, 25, 505-512, 2002.

Docking
Entender e estudar como um inibidor
liga-se a sua enzima-alvo ou como um
antagonista é reconhecido por seu
receptor e consegue ativá-lo é de
grande importância para nós,
farmacêuticos. Sildenafil
Fosfodiesterase 5
Oxazepam
Receptor
GABAA
Etravirina
Transcriptase reversa
Pancurônio
Receptor
colinérgico
nicotínico
Galantamina
Acetilcolinesterase
R L
Docking
ou
Ancoramento Molecular Galantamina
Acetilcolinesterase
PDB: 2ACE
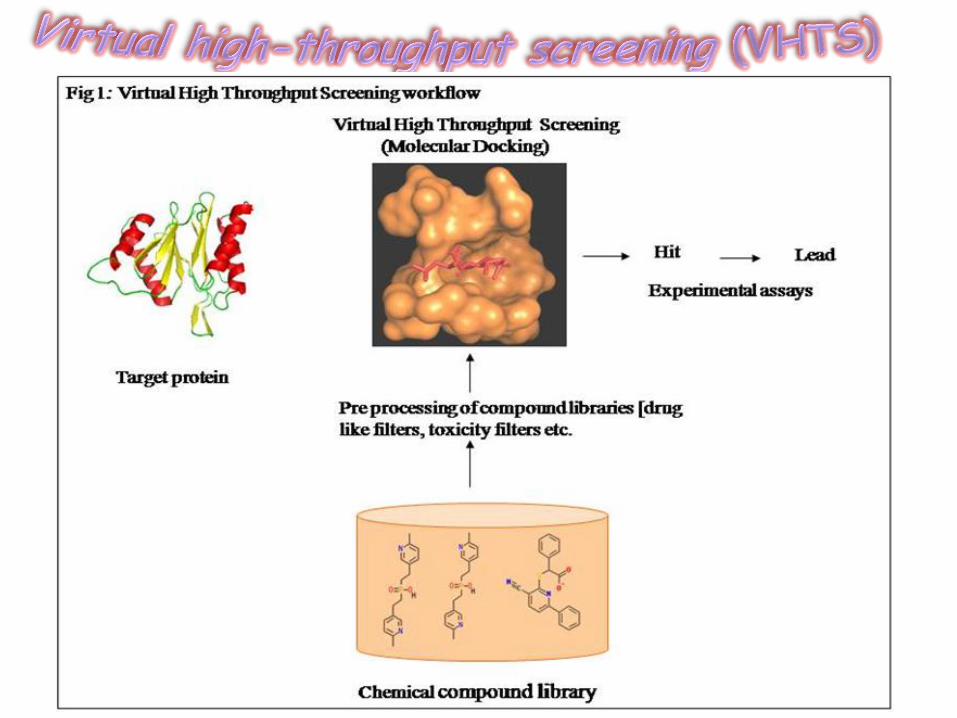
Ferrarmenta útil no processo de planejamento de fármacos baseado na estrutura (Structure-based drug design);
Identifica com alta precisão o modo correto de ligação de fármacos a seus receptores.
Permite a identificaçao rápida e barata de candidatos a fármacos promissores (Lead optimization);
Importante no processo de Virtual high-throughput screening (VHTS);
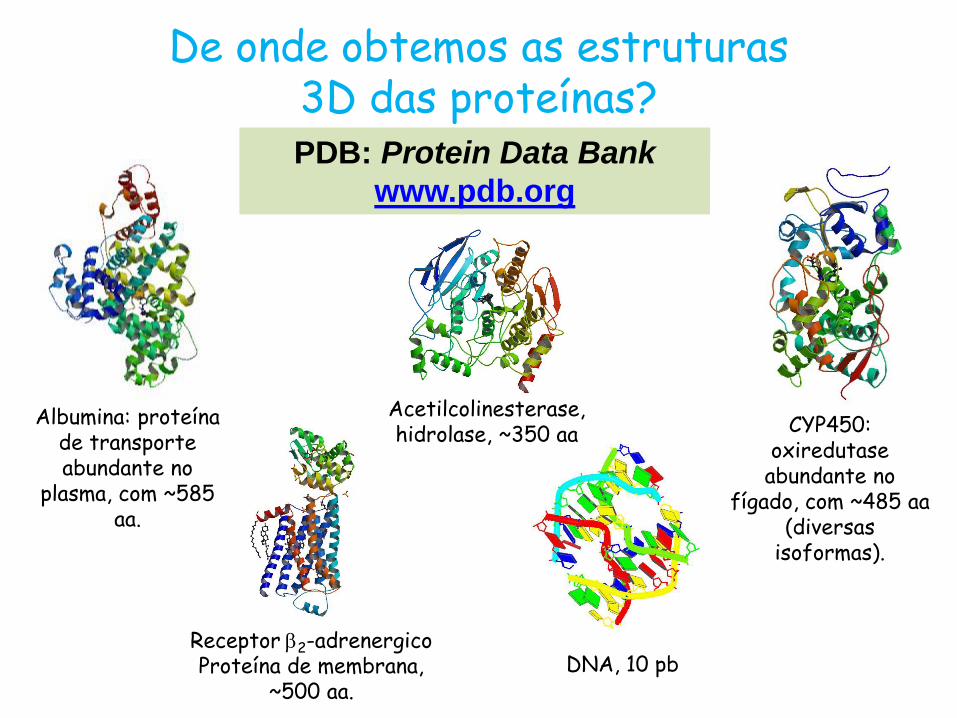
Albumina: proteína de transporte abundante no
plasma, com ~585 aa.
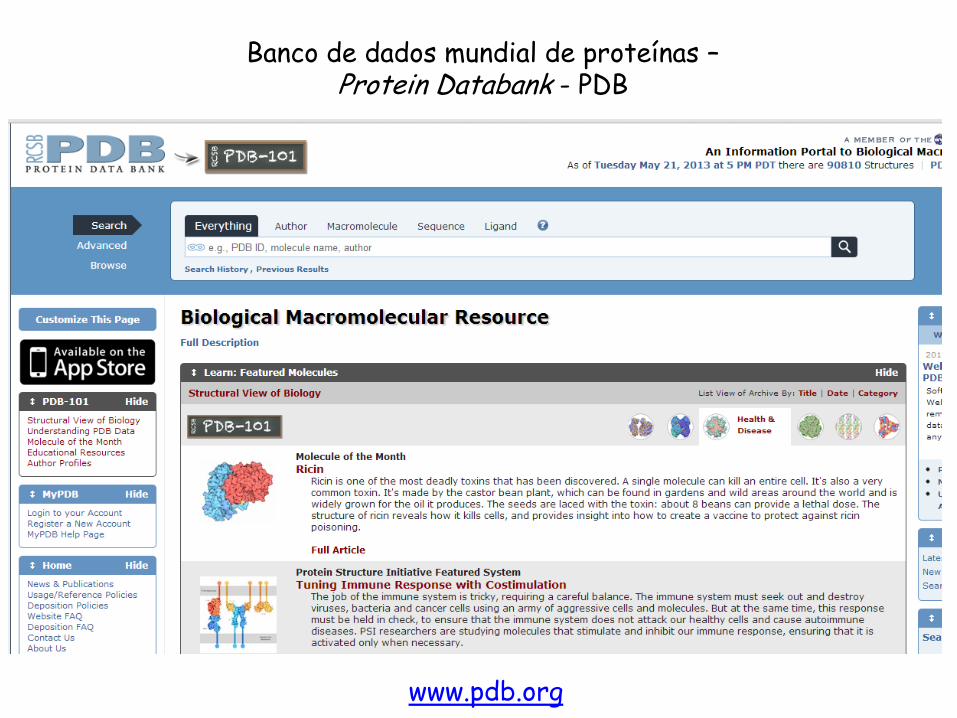
De onde obtemos as estruturas 3D das proteínas? PDB: Protein Data Bank
www.pdb.org
CYP450: oxiredutase
abundante no fígado, com ~485 aa
(diversas isoformas).
Receptor b2-adrenergico Proteína de membrana,
~500 aa.
Acetilcolinesterase, hidrolase, ~350 aa
DNA, 10 pb
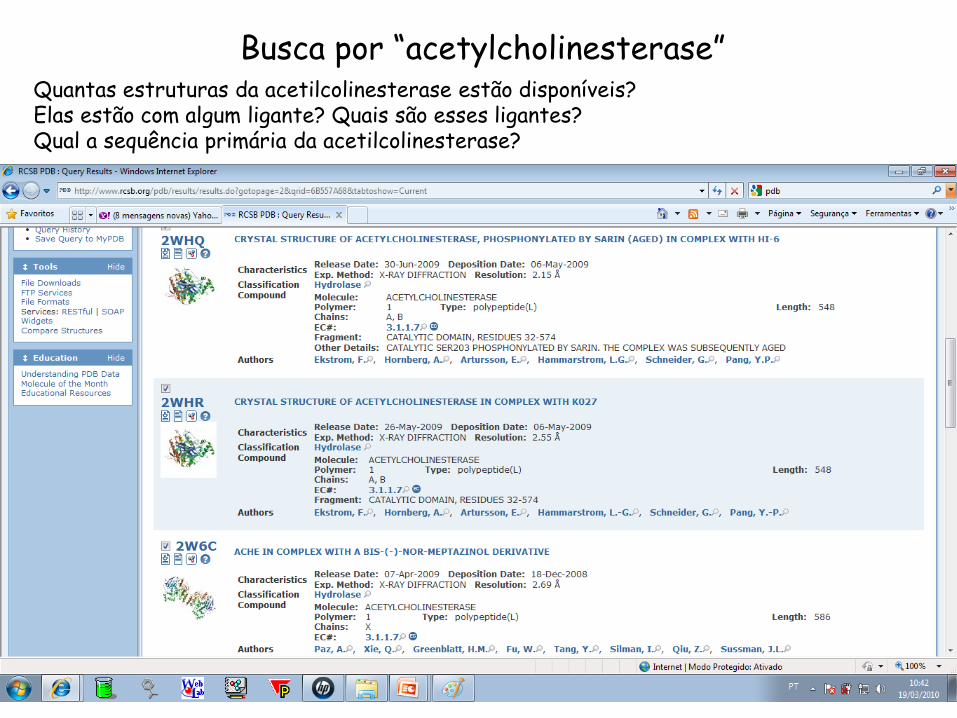
Busca por “acetylcholinesterase” Quantas estruturas da acetilcolinesterase estão disponíveis? Elas estão com algum ligante? Quais são esses ligantes? Qual a sequência primária da acetilcolinesterase?
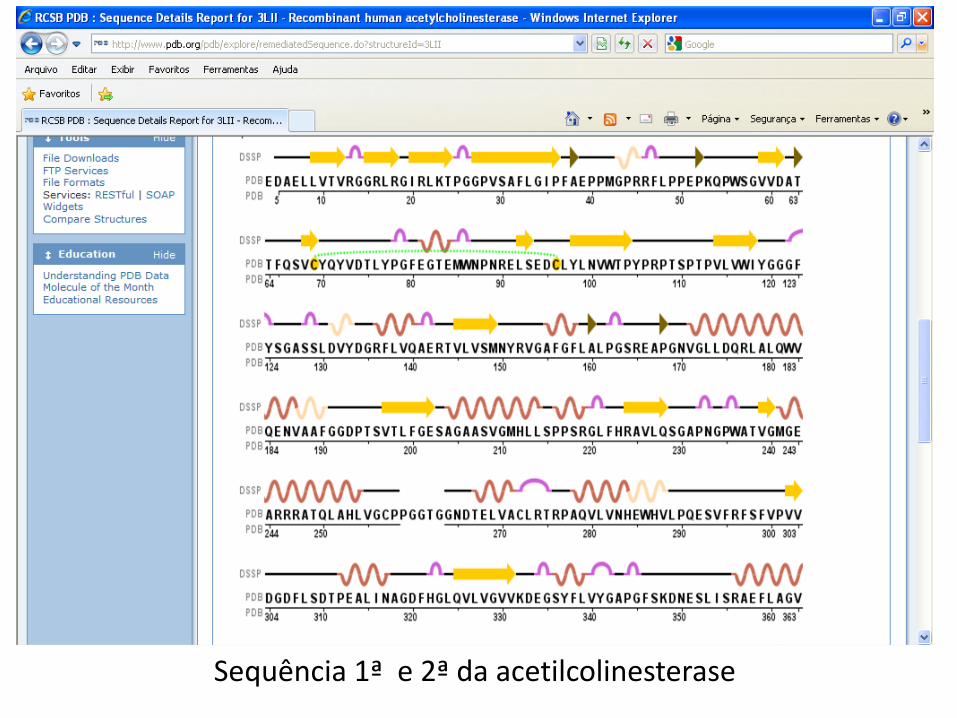
Sequência 1ª e 2ª da acetilcolinesterase
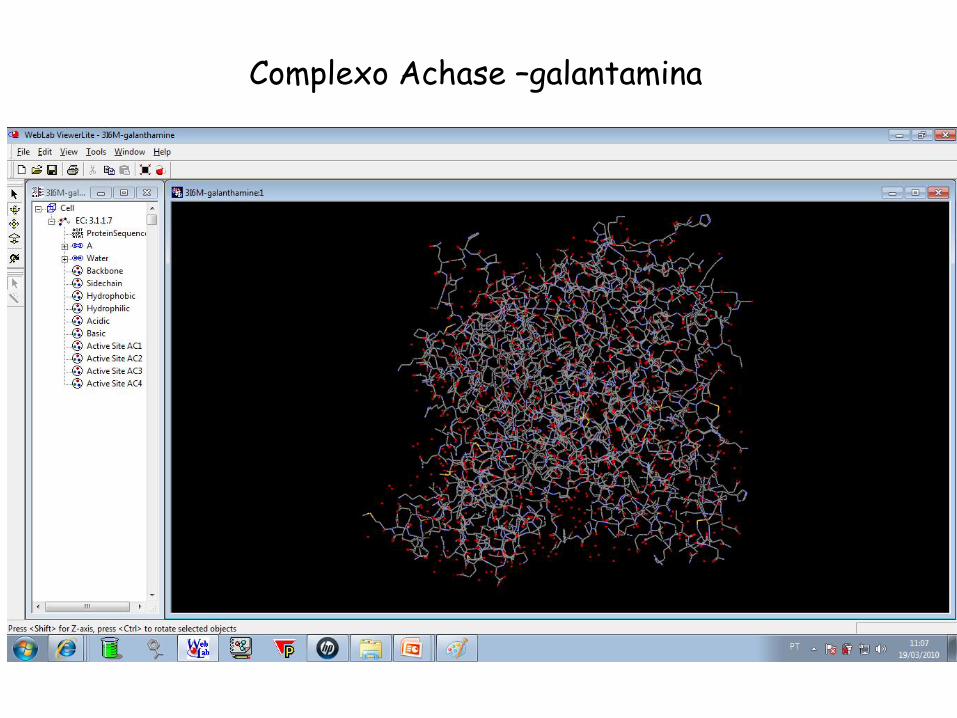
Complexo Achase –galantamina
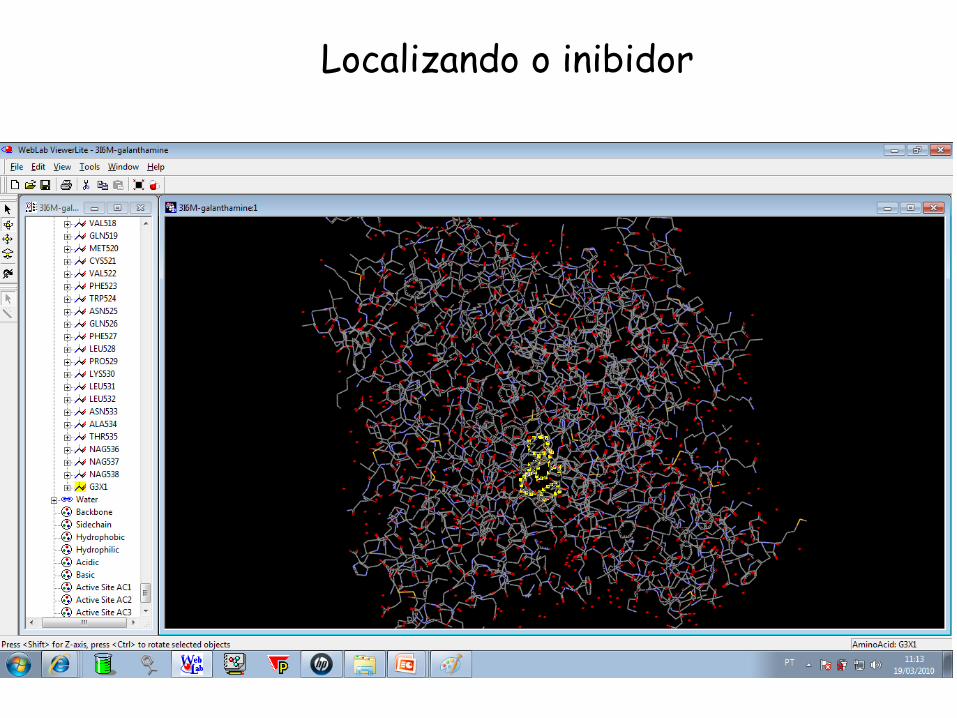
Localizando o inibidor

10 angstrons ao redor do inibidor
Distância muito pequena: 0,0000000001 m

Visualização de possíveis interações ligante-receptor!

Visualização de possíveis interações ligante-receptor!
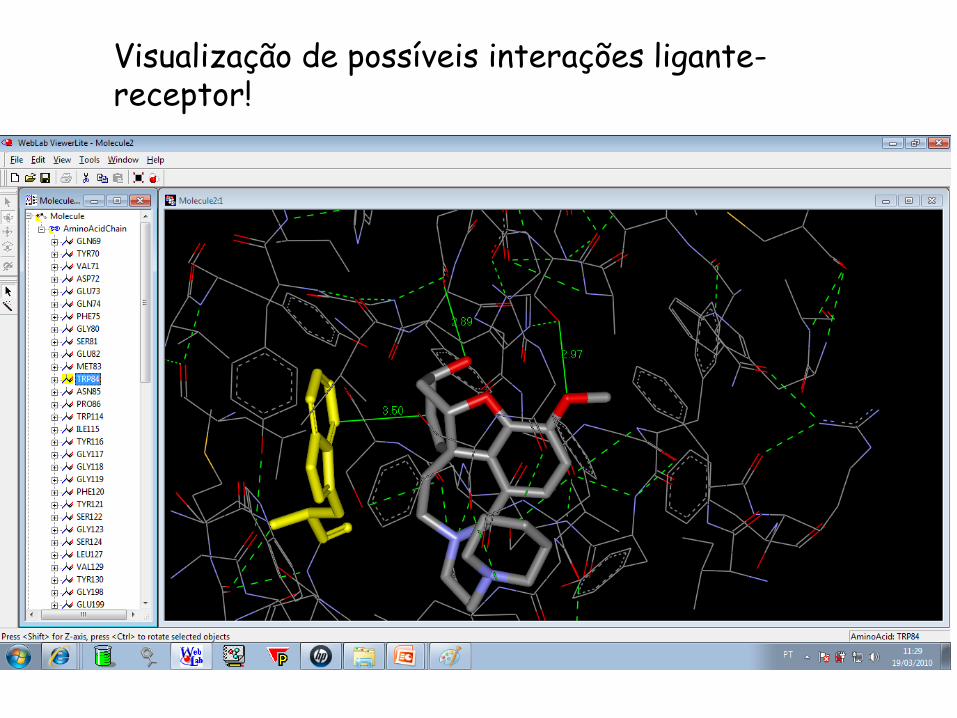
Visualização de possíveis interações ligante-receptor!

Molegro Virtual Docker (MVD) (http://www.molegro.com/)
eHiTS (http://www.simbiosys.ca/ehits/)
Autodock The Scripps Research Institute http://autodock.scripps.edu/
GOLD (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/)
Programas Computacionais de Docking

Programas Computacionais para Visualização de Estruturas 3D
Rasmol - Molecular Visualization Freeware http://www.umass.edu/microbio/rasmol/ Pymol - DeLano Scientific LLC http://www.pymol.org/ VMD - Theoretical and Computational Biophysics Group (TCBG), an NIH Resource for Macromolecular Modeling and Bioinformatics http://www.ks.uiuc.edu/Research/vmd/ Marvin beans – Chemaxon, Inc. http://www.chemaxon.com/ Molegro molecular viewer – Bioinformatics software solutions http://www.molegro.com/mmv-product.php

Metodologias computacionais abordadas aqui nesta palestra
***Docking molecular
***Modelagem comparativa
***Relação Estrutura-Atividade (SAR);
***Relação Quantitativa Estrutura-Atividade
(QSAR e QSAR-3D);

Modelagem Comparativa Proteína
conhecida Proteína que se quer
modelar
Alinhamento de sequência
Modelo construido
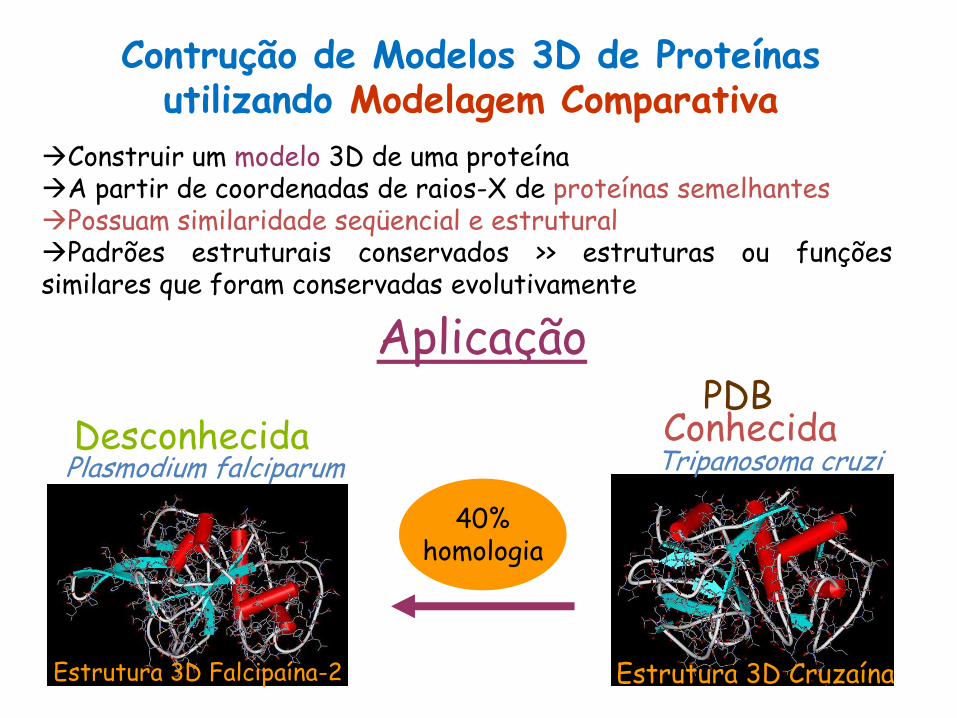
Contrução de Modelos 3D de Proteínas utilizando Modelagem Comparativa
Construir um modelo 3D de uma proteína A partir de coordenadas de raios-X de proteínas semelhantes Possuam similaridade seqüencial e estrutural Padrões estruturais conservados >> estruturas ou funções similares que foram conservadas evolutivamente
Aplicação
Estrutura 3D Falcipaína-2 Estrutura 3D Cruzaína
40% homologia
Conhecida Desconhecida Plasmodium falciparum Tripanosoma cruzi
PDB

E T A P A S
do P R O C E S S O
M O D E L A G E M C O M P A R A T I V A

Metodologias computacionais abordadas aqui nesta palestra
***Docking molecular
***Modelagem comparativa
***Relação Estrutura-Atividade (SAR);
***Relação Quantitativa Estrutura-Atividade
(QSAR e QSAR-3D);

Os estudos de SAR são importantes
para se determinar o grupo farmacofórico
ou farmacóforo de uma classe de
fármacos.
Estudos de SAR (Structure-Activity Relationship)
Relação Estrutura Química-Atividade
Nesses estudos são geradas
informações que auxiliam o planejamento
de fármacos.
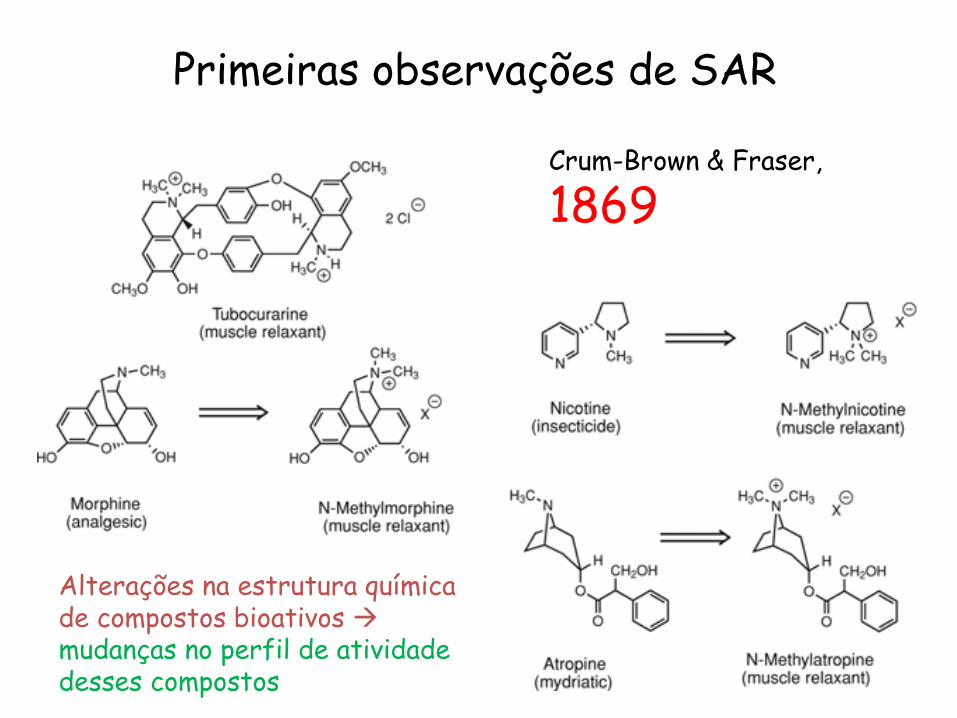
Crum-Brown & Fraser,
1869
Primeiras observações de SAR
Alterações na estrutura química de compostos bioativos mudanças no perfil de atividade desses compostos

Os dez fármacos mais vendidos no mundo em 2008.


Estudos de SAR (Structure-Activity Relationship)
Alterações na estrutura química de compostos bioativos mudanças no perfil de atividade desses compostos
Corwin Hansch e Toshio Fujita, década
de 1960
Relação Estrutura Química-Atividade
Requisitos para um estudo de SAR O maior número de compostos estruturalmente semelhantes que atuem no mesmo alvo Suas estruturas químicas Suas atividades biológicas
Hansch, C.; Fujita, T. J. Am. Chem. Soc., 86, 1616-1626, 1964. Hansch, C. A. Acc. Chem. Res., 2, 232-239, 1969.
Opcional
Descritores
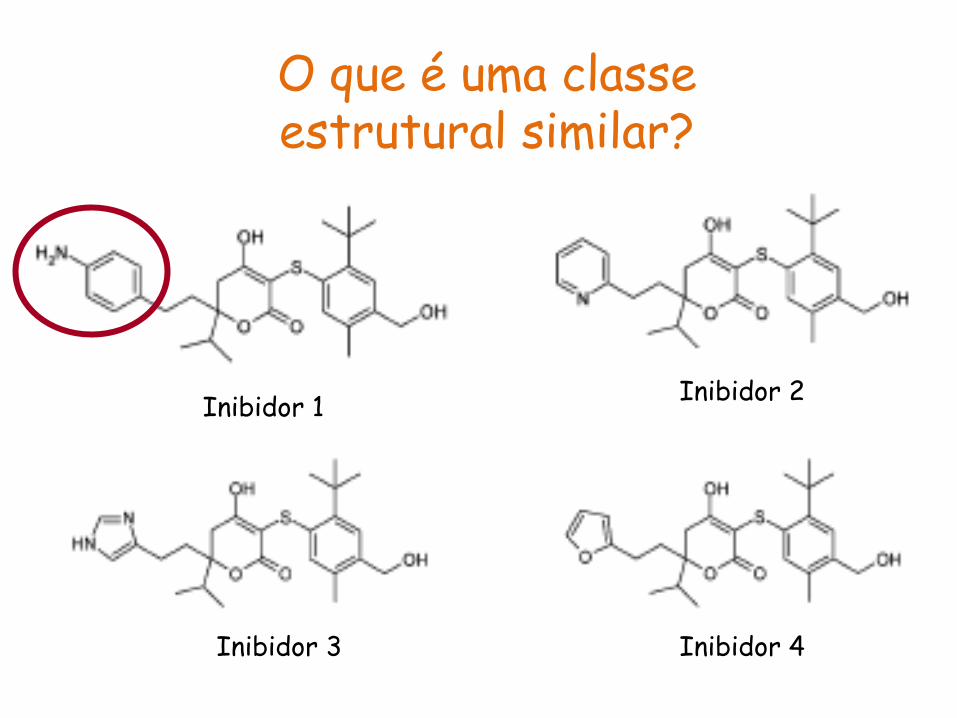
Inibidor 1 Inibidor 2
Inibidor 3 Inibidor 4
O que é uma classe estrutural similar?
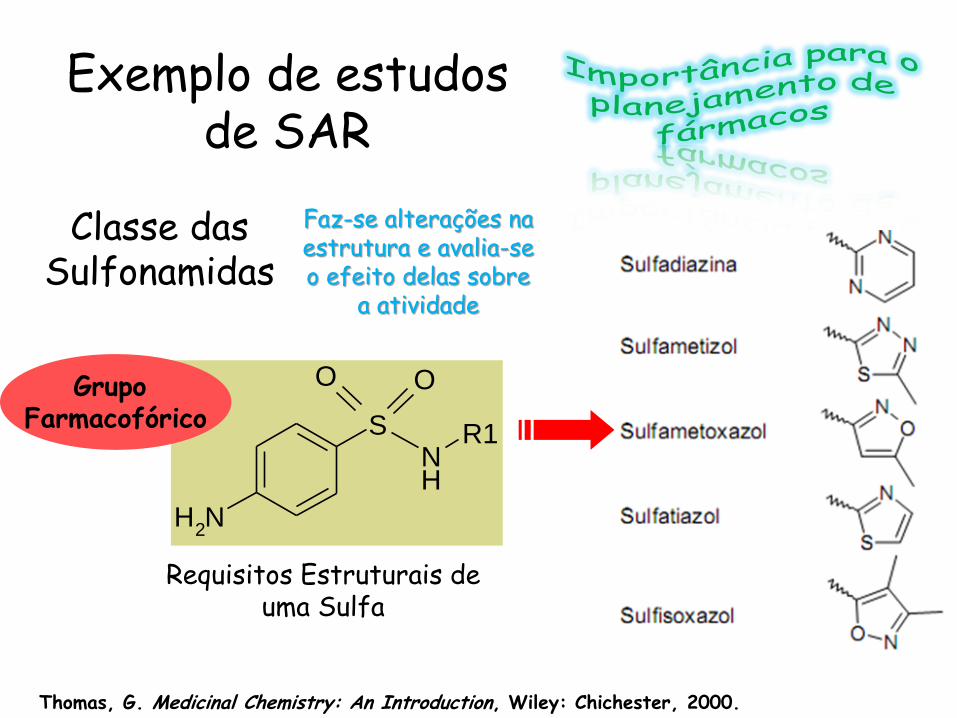
NH2
SNH
R1
OO
Requisitos Estruturais de uma Sulfa
Exemplo de estudos de SAR
Grupo Farmacofórico
Thomas, G. Medicinal Chemistry: An Introduction, Wiley: Chichester, 2000.
Classe das Sulfonamidas
Faz-se alterações na estrutura e avalia-se o efeito delas sobre
a atividade

Exemplo de estudos de SAR

Metodologias computacionais abordadas aqui nesta palestra
***Docking molecular
***Modelagem comparativa
***Relação Estrutura-Atividade (SAR);
***Relação Quantitativa Estrutura-
Atividade (QSAR e QSAR-3D);

Descritores
Relação Quantitativa Estrutura Química-Atividade
Parâmetros Estéricos
Substituintes Hidrofóbicos
Parâmetros Eletrônicos
Log P
Propriedades Físico-Químicas
Métodos Estatísticos (r2,SD)
IUPAC: São modelos matemáticos que relacionam a estrutura química e a atividade farmacológica de um modo quantitativo para uma série de compostos.
Parâmetros Energéticos
Parâmetros Toxicológicos
Atividade = f {parâmetros}
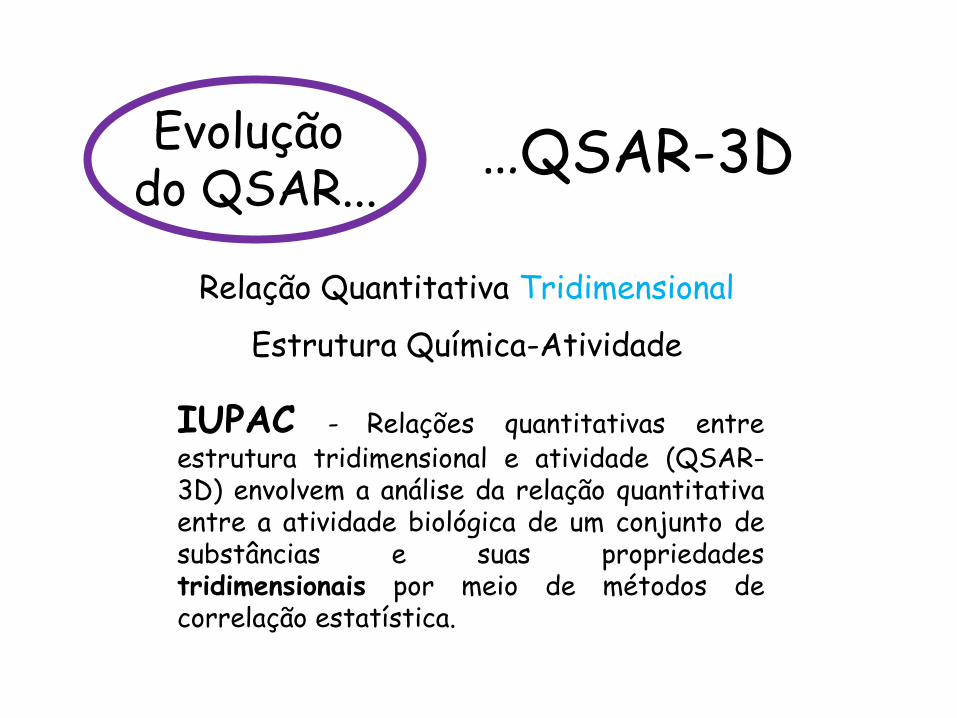
IUPAC - Relações quantitativas entre estrutura tridimensional e atividade (QSAR-3D) envolvem a análise da relação quantitativa entre a atividade biológica de um conjunto de substâncias e suas propriedades tridimensionais por meio de métodos de correlação estatística.
…QSAR-3D
Relação Quantitativa Tridimensional
Estrutura Química-Atividade
Evolução do QSAR...

Estudos de QSAR são realizados por diversos grupos de pesquisa e pela indústria farmacêutica para a otimização de fármacos;
A química computacional dispõe de diversos softwares de QSAR, como o CoMFA, CoMSIA e o Catalyst;
Os softwares acima geram mapas coloridos ao redor das moléculas, indicando se há influência de campos estéricos, eletrostáticos, hidrofóbicos, e com capacidade de fazer ligação hidrogênio em cada região dos compostos.
Essas informações permitem ao químico medicinal que otimize aquela classe de compostos.
Estudos de QSAR-3D

CoMFA
Campo Estérico Campo Eletrostático
Estudos de QSAR-3D CoMFA
Região Favorável - Grupos Volumosos
Região Desfavorável - Grupos Volumosos
Região Favorável - Grupos PE Negativos
Região Desavorável - Grupos PE Negativos

Campo Estérico
Estudos de QSAR-3D CoMFA
Região Favorável - Grupos Volumosos
Região Desfavorável - Grupos Volumosos

Estudos de QSAR-3D CoMFA
Campo Eletrostático Região Favorável - Grupos PE Negativos
Região Desavorável - Grupos PE Negativos

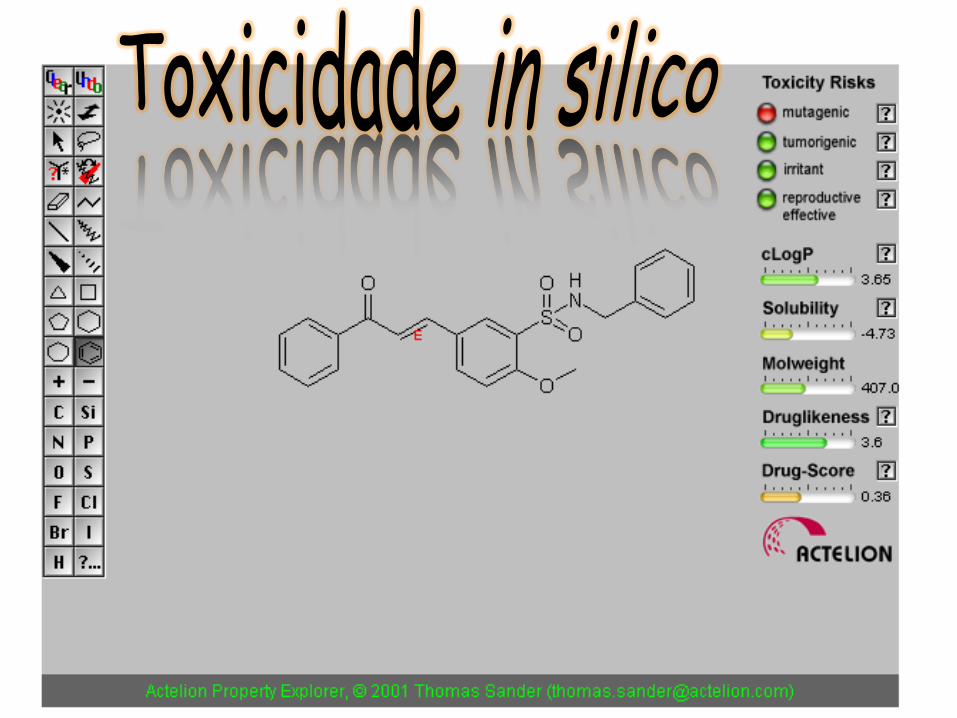
Farmacocinética = principal razão para o insucesso de fármacos
DAVIS, A. M. & RILEY, R. J. Predictive ADMET studies, the challenges and the opportunities. Curr. Op. Chem. Biol., 8, 378–386, 2004.
Durante o processo de P&D de um fármaco, um estudo de ADME eficiente é essencial para que se tenham compostos promissores, com maiores probabilidades de não serem descartados na fase clínica.
Estudos farmacocinéticos
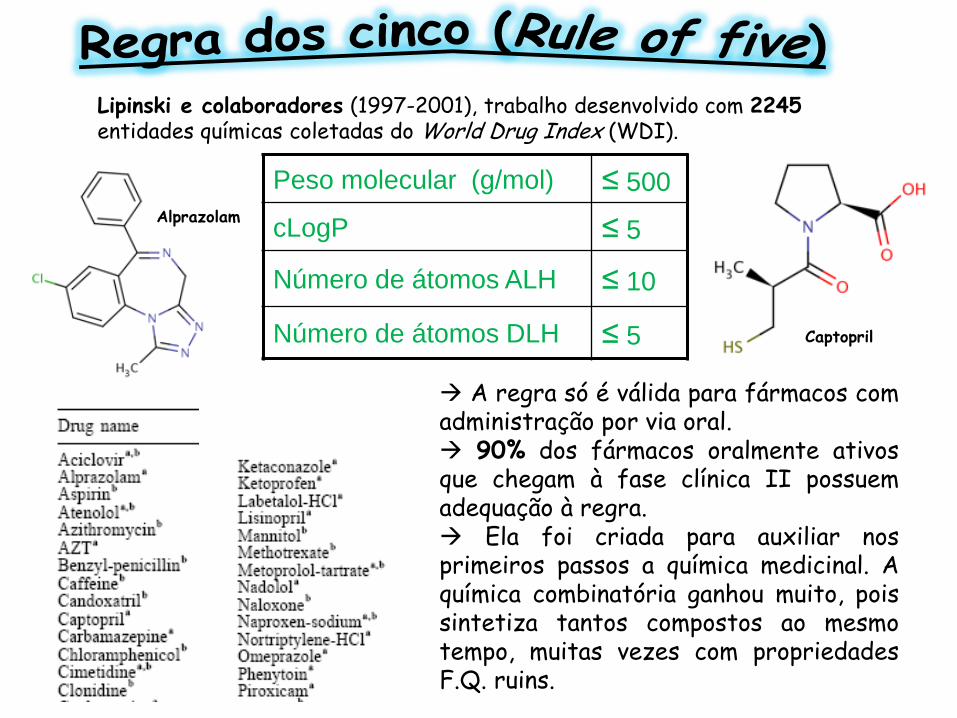
A regra só é válida para fármacos com administração por via oral. 90% dos fármacos oralmente ativos que chegam à fase clínica II possuem adequação à regra. Ela foi criada para auxiliar nos primeiros passos a química medicinal. A química combinatória ganhou muito, pois sintetiza tantos compostos ao mesmo tempo, muitas vezes com propriedades F.Q. ruins.
Peso molecular (g/mol) ≤ 500
cLogP ≤ 5
Número de átomos ALH ≤ 10
Número de átomos DLH ≤ 5
Alprazolam
Captopril
Lipinski e colaboradores (1997-2001), trabalho desenvolvido com 2245 entidades químicas coletadas do World Drug Index (WDI).
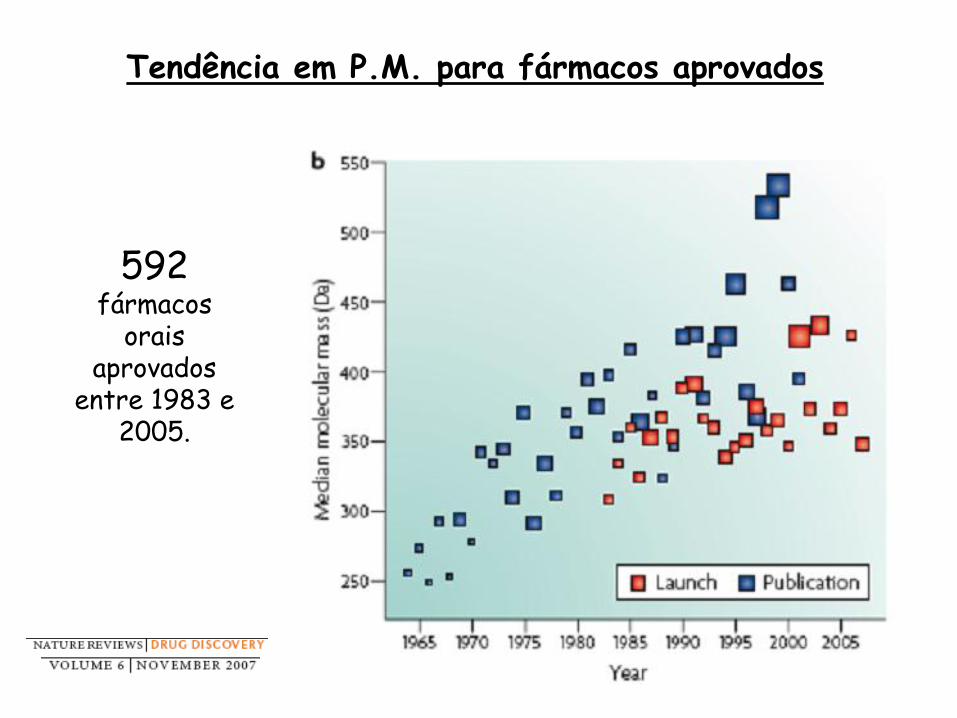
592 fármacos
orais aprovados
entre 1983 e 2005.
Tendência em P.M. para fármacos aprovados
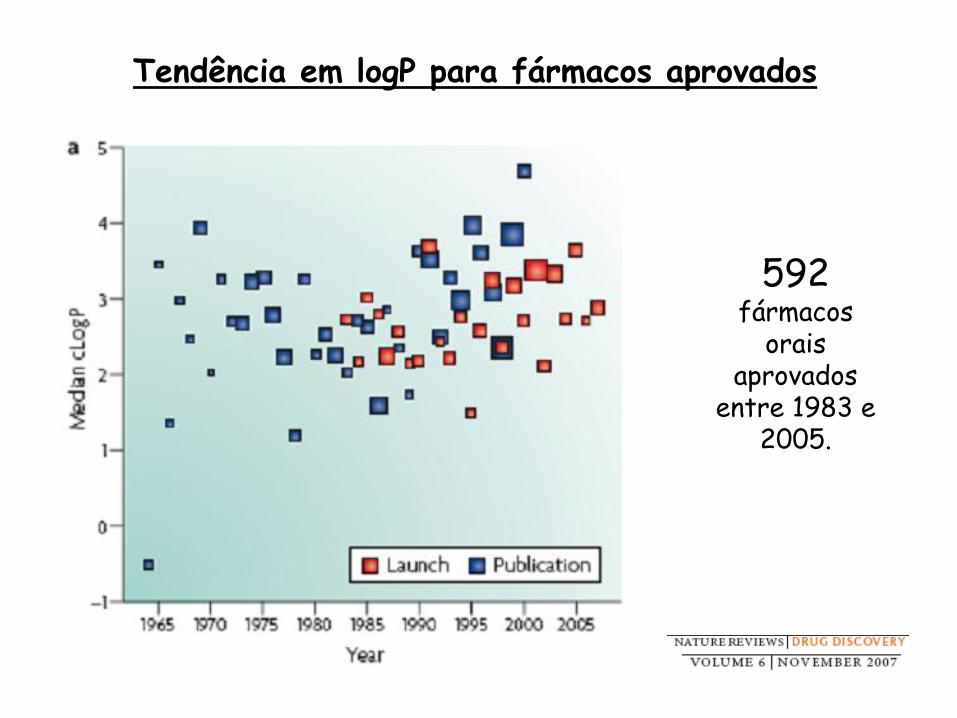
Tendência em logP para fármacos aprovados
592 fármacos
orais aprovados
entre 1983 e 2005.

"Um dia é preciso parar de sonhar e, de algum modo, partir“.
Amyr Klink


















