
Perfil Imunológico de Portadores da síndrome de Down e...
61
1 Perfil Imunológico de Portadores da síndrome de Down e Alopécia Areata. Kalynka Silvia Higino Rio de Janeiro.
Transcript of Perfil Imunológico de Portadores da síndrome de Down e...

1
Perfil Imunológico de Portadores da síndrome de Down e Alopécia Areata.
Kalynka Silvia Higino
Rio de Janeiro.

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

2
PERFIL IMUNOLÓGICO DE PORTADORES DA SÍNDROME DE DOWN E ALOPÉCIA
AREATA.
Kalynka Silvia Higino Dissertação de Mestrado apresentada ao Programa de Pós-graduação em Clínica Médica, Setor da Saúde da Criança e do Adolescente, da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Ciências. Orientadores: Prof. Dr. Antônio José Ledo Alves da Cunha Profa. Dra. Márcia Gonçalves Ribeiro
Rio de Janeiro Dezembro/2006

3
Higino, Kalynka Silvia Perfil Imunológico de Portadores da síndrome de Down e
Alopécia Areata/ Kalynka Silvia Higino.- Rio de Janeiro: UFRJ/IPPMG,2005. VIII, Orientadores: Márcia Gonçalves Ribeiro e Cunha Antônio José Ledo Alves Dissertação de Mestrado – UFRJ/ Programa de Pó-Graduação em Clínica Médica/ Setor de Saúde da Criança e do Adolescente, 2005. Referência Bibliográfica: f. 1. Síndrome de Down 2. Alopécia Areata I. Ribeiro, Márcia Gonçalves e Cunha, Antônio José Ledo Alves. II. Universidade Federal do Rio de Janeiro/ Programa de Pós-Graduação em Clínica Médica/

4
PERFIL IMUNOLÓGICO DE PORTADORES DA SÍNDROME DE DOWN E ALOPÉCIA
AREATA.
Kalynka Silvia Higino Orientadores: Prof. Dr. Antônio José Ledo Alves da Cunha Profa. Dra. Márcia Gonçalves Ribeiro
Dissertação de Mestrado submetida ao Programa de Pós-graduação em Clínica Médica,
Setor da Saúde da Criança e do Adolescente, da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Ciências. Aprovada por: _________________________________________ Presidente, Prof. Dr. Clemax Couto Sant,Anna _____________________________________ Prof. Dr. Gerson Carakushanky _____________________________________ Prof. Dr. Mauro Geller

5
Rio de Janeiro Dezembro/2006
Dedico a meu Deus que me fez parte de suas riquezas, aos meus pais pelo empenho e incentivo; aos meus irmãos; ao meu marido Deneval, parceiro e companheiro; a minha filha Hannah responsável pela experiência maravilhosa de ser mãe. E para todas as crianças portadoras de Síndrome de Down, razão concreta pela qual esta dissertação se tornou realidade.

6
AGRADECIMENTOS Aos meus alunos do PINC José Perrota, Fernanda Nascimento, Carla Salgado Junqueira e
Fabiana Valete Vieira
Ao Prof. Mauro Geller
Ao Prof. Gerson Carakushansky
À Profa Márcia Gonçalves Ribeiro
À Profa. Evelyn kahn
Ao Prof. Isaías Soares de Paiva
Á Dra. Rossana Meneguel
Ao Dr. Marcelo de Paula Coutinho
Ao Dr. Aguinaldo Bonalumi Filho
Ao Prof. Rafael Varella
Ao Prof. Carlos Henrique Nascimento
À Profa. Sandra Alves Pellegrini
Ao Prof. Rodrigo Moura Neto
Ao Laboratório Célula e a todos os seus funcionários(as) que com dedicação e paciência nos
auxiliaram neste trabalho.

7
LISTA DE ILUSTRAÇÕES Figura 1. Criança portadora de Síndrome de Down.............................................................04
Figura 2. Alopécia Areata em couro cabeludo. ...................................................................35
Figura 3. Fotomicrografia mostrando estrutura selecionada de um folículo capilar humano no estágio anágeno. ............................................................................................38
Figura 4. Fatores de regulação do crescimento do cabelo e controle do ciclo do folículo do cabelo. ................................................................................................................39

8
LISTA DE QUADROS QUADRO 1. Tecido Linfóide Organizado. .............................................................................09
QUADRO 2. Classificação do Aglomerado de Diferenciação (CD) das Moléculas Superficiais dos Linfócitos Humanos. ..........................................................................................................11
QUADRO 3. Característica da Ligação dos Antígenos pelas Moléculas de Reconhecimento de Antígenos do Sistema Imunológico. ....................................................................................22
QUADRO 4. Função dos Isótipos de Anticorpos.....................................................................23
QUADRO 5. Variáveis Utilizadas no Estudo..........................................................................63
QUADRO 6. Portadores da SD com Doenças Prévias.............................................................67
QUADRO 7. Alterações Encontradas no Hemograma dos Portadores da SD.........................72
QUADRO 8. Distribuição em Número Absoluto e Percentual de Exames Normais e Anormais da Proteína Total e Frações em Portadores da SD Com e Sem AA..........................................76

9
QUADRO 9. Distribuição do “Cluster of Differentiation” Segundo a Presença e Ausência de AA nos Portadores da SD..........................................................................................................77
QUADRO 10. Distribuição das Imunoglobulinas Segundo a Presença e Ausência de AA nos Portadores da SD. ...............................................................................................................78
QUADRO 11. Distribuição do Complemento Segundo a Presença e Ausência de AA nos Portadores da SD. .....................................................................................................................79
QUADRO 12. Distribuição da Freqüência dos Auto-anticorpos nos Portadores da SD com e sem AA......................................................................................................................................80
QUADRO 13. Distribuição da Média, Mediana e Valores Extremos, das Variáveis Numéricas, nos Portadores da SD. ...........................................................................................84
QUADRO 14. Distribuição da Média, Mediana e Valores Extremos, das Variáveis Numéricas, nos Portadores da SD Com e Sem AA...................................................................85

10
LISTA DE GRÁFICOS
GRÁFICO 1. Distribuição do Gênero Segundo a Presença e Ausência de AA nos Portadores da SD.........................................................................................................................................67
GRÁFICO 2. Distribuição dos Portadores da SD com e sem AA Segundo a Faixa Etária. ...................................................................................................................................................68
GRÁFICO 3. Distribuição do Gênero dos Portadores da SD, Segundo a Faixa Etária...........89
GRÁFICO 4. Distribuição dos Portadores de SD com e sem AA, Segundo as Alterações Cromossômicas. ........................................................................................................................70
GRÁFICO 5. Distribuição do Resultado Geral do Hemograma nos Portadores da SD, Segundo a Presença ou Ausência de AA. .................................................................................71
GRÁFICO 6. Distribuição das Alterações Concomitantes no Hemograma dos Portadores da SD, em números absolutos. ......................................................................................................73

11
GRÁFICO 7. Distribuição dos Portadores da Síndrome de Down, Segundo o Gênero, que Apresentaram Hemograma Completo Normal ou Alterado. ....................................................74
GRÁFICO 8. Distribuição dos Portadores da SD com e sem AA, Segundo o VHS...............75
GRÁFICO 9. Distribuição dos Portadores da Síndrome de Down Com e Sem Alopécia Areata, Segundo a Presença de Auto anticorpos de Acordo com o Gênero..............................80
GRÁFICO 10. Distribuição dos Portadores da Síndrome de Down Com e Sem Alopécia Areata, Segundo a Presença de Auto anticorpos por Faixa Etária, em Anos...........................................................................................................................................81
GRÁFICO 11. Distribuição dos Portadores da Síndrome de Down Com e Sem Alopécia Areata, Segundo a Presença de Auto anticorpos Concomitantes de Acordo com o Gênero.......................................................................................................................................82
GRÁFICO 12. Distribuição dos Portadores da Síndrome de Down Com e Sem Alopécia Areata, Segundo a Presença de Auto anticorpos Concomitantes por Faixa Etária, em Anos...........................................................................................................................................83 LISTA DE ABREVIATURAS AA Alopécia Areata aa Aminoácidos ACA Anticorpo Anticardiolipina AIRE Mutação do Gene Autoimune Regulador ANAs Anticorpo Antinuclear APC Células Apresentadoras de Antígenos APECED Distrofia Autoimune da Poliendocrinopatia-Candidíase-Ectodérmica aPL Anticorpo Antifosfolipídeo APS1 Síndrome Poliglandular Autoimune do Tipo 1 AT Alopécia Total

12
AU Alopécia Universal C 1/2/3/4 Complemento 1/2/3/4 cCD Expressão do CD no Citoplasma CD Cluster Differentiation DMID Diabetes Mellitus Insulino Dependente DNA Ácido Dexoxirribonucleico DSCR Região Crítica da Síndrome de Down EP-GP Glicoproteína Extra Parotídea FI Fator Intrínseco FSH Hormônio Folículo Estimulante HIV Vírus da Imunodeficiência Humana HLA Antígenos de Histocompatibilidade Principal HLA-I Antígenos de Histocompatibilidade Principal de Classe I HLA-II Antígenos de Histocompatibilidade Principal de Classe II ICAM-1 Molécula de Adesão Intracelular 1 IFNγγγγ Fator de Necrose Tumoral γ IFNαααα Fator de Necrose Tumoral α Ig Imunoglobulinas IgA Imunoglobulina A IgD Imunoglobulina D IgE Imunoglobulina E IgG Imunoglobulina G IgM Imunoglobulina M

13
IL-2/3/4/5/6/10/12 Interleucina - 2/3/4/5/6/10/12 LA Lupus Anticoagulant
LES Lúpus Eritematoso Sistêmico LFA-1 Glicoproteina Associada à Função Leucocitária 1 LFA-3 Glicoproteina Associada à Função Leucocitária 3 LGG linfócitos Granulosos Grandes LH Hormônio Luteinizante LLA-T leucemia linfocítica aguda T LLC-T leucemia linfocítica crônica T LT Linfócitos T MHC Complexo de Histocompatibilidade Principal NK Natural Killer RCT Receptor de Célula T RIC Resposta Imune Celular Específica SD Síndrome de Down sIg Imunoglobulinas Superficiais tH Tireoidite de Hashimoto TPO-ab Anticorpo Antitireoideo- peroxidase VHS Velossidade de Hemossedimentação

14
PERFIL IMUNOLÓGICO DE PORTADORES DA SÍNDROME DE DOWN E ALOPÉCIA
AREATA. Kalynka Silvia Higino Orientadores: Prof. Dr. Antônio José Ledo Alves da Cunha Profa. Dra. Márcia Gonçalves Ribeiro
Resumo da dissertação de Mestrado submetida ao Programa de Pós-graduação em Clínica Médica, Setor da Saúde da Criança e do Adolescente, da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Ciências.

15
O Portador da síndrome de Down (SD) apresenta alteração do sistema imune: função anormal dos linfócitos, deficiência de Imunoglobulina da classe G (IgG) e diminuição da resposta imune dependente de células T e alterações nas células B. Há relatos de um aumento na freqüência de alopécia areata (AA) na SD que varia de 1 a 8,9% em indivíduos com SD, com predomínio no sexo feminino. A incidência da AA na população é de aproximadamente 0,1%. Os indivíduos com AA apresentam autoanticorpos relacionados à glândula tireóide. Este estudo visa descrever a função imunológica em portadores da síndrome de Down e AA em uma série de casos com grupo de comparação. Em 83 portadores da SD: 21 com AA (14 do sexo masculino e 7 do sexo feminino) e 62 portadores da SD sem AA (pareados por sexo e idade com grupo de portadores da SD com AA), 94,70 % (54/57) apresentou trissomia livre e 5,30% (3/57) mosaicismo, todos eles portadores da SD sem AA. Encontrou-se hipotireoidismo em três portadores da SD com AA. O hemograma nos portadores da SD (com e sem AA) apresentou: policitemia em 21,68% (18/83), leucopenia em 19,27% (16/83) e anemia em 15,66% (13/83). Observou-se aumento de VHS em 86,58% (59/82) dos indivíduos estudados. Nos portadores da SD com e sem AA houve diminuição em 58,9% do CD 19 e 67,46 % (56/83) do C4; e normalidade em 85% das imunoglobulinas e 100% eletroforese de imunoglobulinas. A presença de anticorpos nos portadores da SD com e sem AA foi de 15,66% (13/83) para antitireoglobulinas e de 15,66% para anticardiolipina IgG. A presença do anticorpo antitireoperoxidase foi de 61,90% (13/21) em portadores da SD com AA. Portanto, nos 21 pacientes com SD e AA predominou sexo masculino, os indivíduos mosaicos não apresentaram AA, a doença predominantemente associada a AA foi a alteração tireoideana. O hemograma apresentou alterações concordantes com as da literatura para a SD. O VHS foi aumentado em todos os indivíduos com SD. Quanto à imunidade celular não houve concordância entre os grupos e não se encontrou um padrão específico. Não se observou alteração da imunidade humoral nos dois grupos. O anticorpo antiperoxidase foi positivo percentualmente no grupo com AA.
IMMUNOLOGICAL PROFILE OF CARRIERS OF THE SYNDROME DOWN AND ALOPECIA AREATA.
Kalynka Silvia Higino Orientadores: Prof. Dr. Antônio José Ledo Alves da Cunha Profa. Dra. Márcia Gonçalves Ribeiro

16
Abstract da dissertação de Mestrado submetida ao Programa de Pós-graduação em Clínica Médica, Setor da Saúde da Criança e do Adolescente, da Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Ciências.
ABSTRACT
Down’s syndrome (DS) carriers present immune system alterations: abnormal lymphocyte functions, Immunoglobulin class G (IgG) deficiency and reduction of the immune-dependent T-cell response as well as alterations in B cells. There are reports of an increase in alopecia areata (AA) frequency in DS that vary from 1-8.9% in individuals with SD, predominantly in the female gender. The incidence of AA in the population is approximately 0.1%. Individuals with AA present autoantibodies related to the thyroid gland. This study aims to describe immunologic function in DS patients with AA in series of cases with a comparison group. In 83 DS patients: 21 presented AA (14 male and 7 female patients) and 62 DS patients without AA (pairing for sex and age with the DS and AA patient group), 94.70% (54/57) presented free trisomy and 5.30% presented mosaicism, all of whom presented DS without AA. Hypothyroidism was found in three DS patients with AA. The complete blood count in the DS patients (with and without AA) presented: polycythemia in 21.68% (18/83), leukocytopenia in 19.27% (16/83) and anemia in 15.66% (13/83). Increase in VHS was found in 86.58% (59/82) of the study subjects. In DS patients with and without AA, a 58.9% reduction in CD-19 and 67.46% (56/83) in C4; and normality in 85% of the immunoglobulins and 100% immunoglobulin electrophoresis. In the DS patients with and without AA, thyroglobulin antibodies were detected in 15.66% (13/83) of the patients, while anticardiolipin IgG was detected in 15.66%. The presence of thyroid peroxidase antibodies occurred in 61.90% (13/21) of the DS patients with AA. In summary, male gender was most frequent among the 21 DS patients with AA, mosaic individuals did not present AA, and the predominant previous illness associated with AA was thyroid dysfunction. The complete blood count presented results similar to those in the literature for DS patients. VHS was increased in all DS individuals. Concerning cellular immunity, there was a difference between the groups, and no specific pattern was detected. Neither group presented alteration of humoral immunity. Antiperoxidase antibody was percentually positive in the AA group.
SUMÁRIO
LISTA DE ILUSTRAÇÕES.......................................................................................vii
LISTA DE QUADROS..............................................................................................viii
LISTA DE GRÁFICOS................................................................................................x
LISTA DE ABREVIATURAS....................................................................................xi

17
RESUMO....................................................................................................................xiv
ABSTRACT.................................................................................................................xv
1. INTRODUÇÃO...........................................................................................................01
2. FUNDAMENTOS TEÓRICOS..................................................................................03
2.1 SÍNDROME DE DOWN.........................................................................................03
2.2 SISTEMA IMUNOLÓGICO...................................................................................07
2.2.1 Receptores de Células Te B.........................................................................10
2.2.2 Citocinas......................................................................................................14
2.2.3 Expansão Clonal dos Linfócitos..................................................................15
2.2.4 Papel das Células de Memória.....................................................................16
2.2.5 Células T......................................................................................................16
2.2.6 Células B......................................................................................................19
2.2.7 Interações Celulares na Resposta Imune Normal........................................24
2.2.7.1 Interação Célula T - Célula T e Célula T - Célula B..............................25
2.2.7.2 Interações das CAA - Célula T.............................................................26
2.2.8 O Sistema de Complemento.........................................................................26
2.2.9 Avaliação do Sistema Imunológico.............................................................27
2.2.10 Sistema Imunológico e Síndrome de Down.................................................28
2.2.11 Auto-imunidade e Síndrome de Down........................................................30
2.3 ALOPÉCIA AREATA............................................................................................33
2.3.1 Histórico.......................................................................................................33
2.3.2 Conceito e Epidemiologia............................................................................34
2.3.3 Aspectos Histopatológicos da Alopécia Areata...........................................37
2.3.4 Mecanismo Autoimune para o Início da Alopécia Areata...........................41
2.3.5 Autoanticorpos na Alopécia Areata.............................................................42
2.3.6 Aspectos Genéticos da Alopécia Areata......................................................43
2.3.7 Alopécia Areata na síndrome de Down.......................................................48

18
3. JUSTIFICATIVAS......................................................................................................51
4. OBJETIVOS................................................................................................................52
4.1 OBJETIVO GERAL................................................................................................52
4.2 OBJETIVOS ESPECÍFICOS..................................................................................52
5. METODOLOGIA........................................................................................................53
5.1 TIPO DE ESTUDO.................................................................................................53
5.2 LOCAL DO ESTUDO............................................................................................53
5.3 POPULAÇÃO E AMOSTRA ................................................................................53
5.3.1 Critérios de Inclusão....................................................................................54
5.3.2 Critérios de Exclusão...................................................................................55
5.4 DESCRIÇÃO DAS VARIÁVEIS...........................................................................56
5.4.1 Gênero..........................................................................................................56
5.4.2 Idade.............................................................................................................56
5.4.3 Exame Citogenético.....................................................................................56
5.4.4 Ocorrência de Doença Prévia.......................................................................56
5.4.5 Perfil Imunológico.......................................................................................56
5.4.5.1 Exame inespecíficos...............................................................................57
5.4.5.2 Imunidade Celular..................................................................................59
5.4.5.3 Imunidade Humoral...............................................................................60
5.4.5.4 Auto Anticorpos.....................................................................................61
5.4.5 Frequência das Alterações Laboratoriais.....................................................62
5.5 COLETA DE DADOS...........................................................................................64
5.6 PROCESSAMENTO E ANÁLISE DOS DADOS.................................................64
5.7 QUETÕES ÉTICAS................................................................................................64
6. RESULTADOS............................................................................................................66
6.1 DESCRIÇÃO DA AMOSTRA...............................................................................66

19
6.1.1 Gênero e Idade ............................................................................................67
6.1.2 Citogenética.................................................................................................69
6.2 DESCRIÇÃO DOS EXAMES LABORATORIAIS..............................................70
6.2.1 EXAMES INESPECÍFICOS.......................................................................70
6.2.1.1 Hemograma Completo...........................................................................70
6.2.1.2 VHS........................................................................................................74
6.2.1.3 Proteínas Totais e Frações......................................................................75
6.2.2 IMUNIDADE CELULAR...........................................................................76
6.2.2.1 CD2........................................................................................................76
6.2.2.2 CD3........................................................................................................76
6.2.2.3 CD4........................................................................................................76
6.2.2.4 CD8........................................................................................................76
6.2.2.5 CD19......................................................................................................76
6.2.3 IMUNIDADE HUMORAL.........................................................................77
6.2.3.1 Imunoglobulinas.....................................................................................77
6.2.3.2 Eletroforese de Imunoglobulinas...........................................................78
6.2.3.3 Sistema Complemento...........................................................................78
6.2.4 AUTO ANTICORPOS................................................................................79
6.2.4.1 Antitireoglobulinas................................................................................80
6.2.4.2 Antiperoxidase .....................................................................................80
6.2.4.3 Anticardiolipina IgM.............................................................................80
6.2.4.4 Anticardiolipina IgG..............................................................................80
6.2.4.5 Anticélulas parietais gástricas................................................................80
6.2.4.6 Anticoagulante lúpico............................................................................80
7. DISCUSSÃO................................................................................................................86
8. CONCLUSÕES............................................................................................................93
RECOMENDAÇÕES........................................................................................................94

20
REFERÊNCIAS BIBLIOGRÁFICAS............................................................................95
ANEXOS..........................................................................................................................109
ANEXO A - Exames Laboratoriais ............................................................................109
ANEXO B - Termo de Consentimento Livre e Esclarecido........................................114
APÊNDICES....................................................................................................................116
ANEXO A - Protocolo de Investigação Laboratorial..................................................116
ANEXO B - Banco de Dados Utilizado para Análise dos Resultados........................118
ANEXO C - Artigo Intitulado: ...................................................................................123
Alopécia Areata: Aspectos Clínicos e Imunogenéticos.
ANEXO D - Artigo Intitulado: ...................................................................................136
Perfil Imunológico de Portadores da síndrome de Down e Alopécia Areata.

21
INTRODUÇÃO
A trissomia do cromossomo 21, síndrome de Down (SD) é a cromossomopatia mais
freqüente. A prevalência da SD é de aproximadamente 1:700 nascimentos 1; com uma discreta
preponderância desta síndrome no sexo masculino 5, 6, 7. O fato de estes portadores apresentarem
o cromossomo 21 a mais lhes dá características fenotípicas próprias tais como: base nasal plano
(98%), orelhas pequenas (95%), espaço alargado entre o primeiro e o segundo pododáctilos
(90%), inclinação palpebral superior ou mongólica (90%), diminuição do comprimento da
fissura palpebral, braquicefalia (70%), braquidactilia (70%), clinodactilia do quinto quirodáctilo
(60%), epicanto (50%), prega única palmar (50%), face plana, pescoço largo transversalmente e
curto verticalmente; além de dermatóglifos típicos (99%), pele redundante na nuca (80%),
protrusão da língua (60%) 1, 5. Clinicamente eles apresentam retardo mental (aproximadamente
100%)5, hipotonia muscular (21 a 85%) 9 e estatura abaixo da média 1, 5; além de uma maior
freqüência de infecções da pele e respiratórias, principalmente.
Os portadores da SD apresentam alterações do sistema imune, foram relatadas funções
anormais dos linfócitos, deficiência de Imunoglobulina da classe G (IgG) e diminuição da
resposta imune dependente de células T, bem como alterações nas células B em portadores da
SD 122, 134.
Diversos estudos relatam um aumento da freqüência da alopécia areata (AA) na SD, que
varia de 1 a 8,9% em pacientes com SD, com predominância no sexo feminino. Por outro lado, a
prevalência da AA na população em geral é de aproximadamente 0,1% 75. Já foram descritas
alterações da imunidade e autoimunidade em portadores da SD e AA 16, 36.

22
A prevalência de AA na população de indivíduos portadores da SD no IPPMG entre 2000
a 2003 foi de 4,34%, isto nos motivou a realizar um estudo do perfil imunológico nestes
portadores, uma vez que não há estudos detalhados sobre a imunidade e autoimunidade em
amostras da população brasileira de portadores de SD e AA.

23
1 FUNDAMENTOS TEÓRICOS
2.1 SÍNDROME DE DOWN
A Síndrome de Down (SD) é a cromossomopatia mais freqüente, e causa comum de
retardo mental, representando de 10 a 30% de todos os casos de retardo mental grave 1, 2. Foi
descrita pela primeira vez por John Langdon Haydon Down em 1866 3 e em 1959 a
anormalidade cromossômica responsável por esta condição genética foi identificada 4. A
prevalência da SD é de aproximadamente 1:700 nascimentos 1 e há uma discreta preponderância
desta síndrome no sexo masculino 5, 6, 7. Observa-se que mulheres com idade superior a 35 anos
apresentam um maior risco de ter filhos com SD 1.
Dentre os tipos citogenéticos encontrados na SD, a trissomia livre do cromossoma 21
representa 95% de todos os casos 1. Os 5% restantes são devidos ao mosaicismo de células da
linhagem trissômica e da linhagem normal ou dissômica, à translocação cromossômica e a outras
alterações estruturais cromossômicas menos freqüentes 1.
O quadro clínico da SD é constituído de retardo mental ( aproximadamente 100%)1, 5;
porém Fishler e Koch (1991) encontraram uma diferença significativa entre o nível do Q. I.
(quoeficiente de inteligência) de crianças citogeneticamente trissômicas e portadores de
mosaicismo; as crianças com duas linhagens celulares (mosaicas) mostraram um nível mais
elevado do Q. I., melhores habilidades verbais e mais acurada percepção sensório-motora 8.
Portadores da SD, em geral, apresentam ainda hipotonia muscular (21 a 85%) 9, acompanhado
das seguintes dismorfias: base nasal plano (98%), orelhas pequenas (95%), espaço alargado entre
o primeiro e o segundo pododáctilos (90%), inclinação palpebral superior ou mongólica (90%),

24
diminuição do comprimento da fissura palpebral, braquicefalia (70%), braquidactilia (70%),
clinodactilia do quinto quirodáctilo (60%), epicanto (50%), prega única palmar (50%), face
plana, pescoço largo transversalmente e curto verticalmente; além de dermatóglifos típicos
(99%), pele redundante na nuca (80%), protrusão da língua (60%), estatura abaixo da média,
abdome protruso 1, 5 (Figura 1).
Figura 1. Face de Criança Portadora da SD.
Fonte: adaptado de http://www.google.com/imagens/Down syndrome 10.
A cardiopatia congênita ocorre em 40% dos pacientes e as alterações mais comuns são os
defeitos septais: defeito septal átrio-ventricular e comunicação interventricular. A ocorrência da
cardiopatia congênita na SD é 30 vezes maior que o esperado para a população em geral 3, 11, 12.
As malformações do sistema digestivo ocorrem em 12% das crianças com esta síndrome 12, 13.
Elas são representadas pela atresia duodenal, pâncreas anular, imperfuração anal e fístula
tráqueo-esofágica. A Doença de Hirschuprung, o Divertículo de Meckel e a Doença Celíaca (1 a
4%) são mais freqüentes nos portadores da SD que na população em geral 11, 12, 14. A constipação

25
crônica é um achado constante em 30% desta população 14. Os portadores da SD apresentam
maior freqüência de litíase biliar do que a população geral 15.
Em relação ao sistema ósteo-articular, pode-se encontrar um aumento no risco de
subluxações e deslocamentos articulares pela hipotonia muscular e lassidão ligamentar. Deve-se
ter especial atenção à instabilidade atlanto-axial, que ocorre em 14% dos indivíduos e deste total,
1 a 2 % desenvolvem evidências de compressão medular 11.
No sistema urinário encontra-se um pequeno percentual de alterações anatômicas que
incluem obstrução ao nível da junção urétero-pélvica, com hidronefrose e alterações estruturais
ou na maturação de estruturas parenquimatosas 11, 15.
Em geral, os portadores da SD apresentam pele seca (queratose) e pálida. Uma série de
infecções cutâneas foi descritas como mais freqüentes na SD: queilite angular, foliculite,
furunculose, abscessos, dermatite atópica com ou sem infecção secundária e/ou dermatite
seborréica 16. Existe uma predisposição à escabiose, alta prevalência de tinea pedis (acima de
50%), bem como de onicomicose grave 16. A foliculite por pityrosporun, eritema e erupções
pápulo-foliculares persistentes e crônicas que afetam as regiões pré-esternal e infraescapular,
ocorrem em 50% dos portadores do sexo masculino com SD entre a idade de 20 e 40 anos, sendo
incomum nas portadoras do sexo feminino 16.
Os portadores da SD podem apresentar qualquer tipo de comprometimento dos
componentes hematopoiéticos, no entanto, quatro são as anormalidades mais específicas desta
trissomia: a) Mielo-displasia transitória na infância, b) Macrocitose e eritrocitose, c) Aumento da
susceptibilidade à leucemia e d) Maior susceptibilidade à leucemia megacariocítica aguda. A
policitemia ocorre com relativa freqüência principalmente pela cardiopatia ou por problemas
respiratórios que ao acarretar hipoxemia estimula a eritropoiese. A macrocitose está presente em

26
65% dos portadores da SD e deve ser realizado diagnóstico diferencial com: doença hepática
crônica, hipotireoidismo, deficiência de ácido fólico, deficiência de vitamina B12, anemias
hemolíticas, displasias medulares, toxicidade por anticonvulsivantes e hiperhidratação celular.
As anormalidades dos granulócitos se caracterizam pela evidente susceptibilidade a agentes
infecciosos virais e bacterianos que decrescem com a idade e aparentemente está envolvida com
efeitos de fagocitose e atividade bactericida. Uma das principais enzimas conversoras
intraeritrocitárias é a superóxido desmutase 1 (SOD1), cujo gene que a codifica está localizado
no cromossomo 21, e os portadores da SD por apresentarem uma cópia extra do gene da SOD1,
têm uma atividade intra-eritrocitária aumentada em 50% em diferentes tipos de células:
eritrócitos, leucócitos, plaquetas e fibroblastos. A SOD1 tem um importante papel na fagocitose
bacteriana pelos granulócitos, o excesso da atividade da SOD1 decresce a concentração
intracelular do ânion superóxido e esse desequilíbrio influi nos fatores de defesa intracelular a
determinados patógenos 17.
Podem também ocorrer anormalidade funcional da pálpebra, possível obstrução do ducto
nasolacrimal e blefarite marginal (2 a 70%), além de hipoplasia do estroma da íris, manchas de
Brushfield, catarata (prevalência entre 12 a 46%), ceratocone (10% ou menos), estrabismo (1 a 2
%) e raramente o deslocamento de retina 18. Cabe aqui ressaltar ainda que as causas mais comuns
de perda visual na SD são os erros de refração, principalmente a miopia 18.
A puberdade é freqüentemente tardia e incompleta 11. Na SD há diferenças no
desenvolvimento sexual masculino e feminino. Nos meninos, o início da puberdade ocorre por
volta dos 13 anos e aos 17 anos o desenvolvimento sexual tende a ser completo 19. A
infertilidade masculina é ponto contraditório entre os estudiosos e as causas perpassam por: 1)
oligoespermia ou mesmo ausência de espermatozóides; 2) desenvolvimento do pênis e dos

27
testículos, que segundo alguns autores, é menor nestes pacientes; e 3) dosagens hormonais
elevadas: Hormônio Folículo Estimulante (FSH) e de Hormônio Luteinizante (LH), que
sugeririam hipogonadismo hipergonadotrófico 19. Em relação ao sexo feminino, são raras as
alterações que envolvem a genitália externa. A menarca, em geral se situa entre os 11 e 13 anos
19. Quanto à fertilidade, há vários casos de reprodução em mulheres portadoras da síndrome.
Observou-se que nas portadoras da SD que ficaram grávidas ou tiveram filhos, cerca de 30% das
gestações resultaram em uma criança portadora da SD; cerca de 10% resultaram em
abortamento; e aproximados 60% resultaram em crianças sem a SD, porém cerca de 20% destas
apresentaram comprometimento físico (prematuridade, baixo peso) e/ou mental 19, 20.
Os portadores da SD devem ser acompanhados ambulatorialmente em função da maior
morbidade clínico-cirúrgica 21, 22. As principais causas para o aumento da mortalidade no
primeiro ano de vida são as infecções respiratórias (broncopneumonias) e a presença de
cardiopatia congênita 21.
2.2 SISTEMA IMUNOLÓGICO
A defesa do hospedeiro é composta pelos sistemas imune e não-imune 23. A imunidade se
caracteriza por: a) uma resposta antígeno-específica, contra um antígeno ou patógeno estranho,
que costuma ocorrer em vários dias, e b) memória para o antígeno, que em exposições
subseqüentes produz uma resposta mais rápida e vigorosa 23. O sistema não-imune produz a
inflamação; que é caracterizada por respostas imediatas, não específicas para o antígeno e sem
memória para o estímulo desencadeante 23. Os mediadores do sistema não-imune são os

28
neutrófilos, eosinófilos, basófilos, células destruidoras naturais (Natural Killer), monócitos e
macrófogos 23.
O sistema imunológico é composto pela imunidade humoral e imunidade celular 23. A
imunidade humoral é mediada por anticorpos secretados, e sua função fisiológica é a defesa
contra microorganismos extracelulares e toxinas microbianas, enquanto a imunidade celular é
mediada por linfócitos T e atua para erradicar microorganismos que infectam e vivem dentro das
células do hospedeiro 24. O principal efetor da imunidade celular é o linfócito derivado do timo
(T) e o da imunidade humoral é o linfócito derivado da medula óssea ou equivalente da bursa (B)
23. Os linfócitos T e B derivam de uma célula primordial comum, a célula-tronco hematopoiética
23, 25 (Quadro 1). As outras células efetoras e reguladoras fundamentais do sistema imune são os
linfócitos granulosos grandes (LGG), os monócitos-macrófagos e as células dendríticas/ de
Langherhans 23.

29
Quadro 1. Tecido Linfóide Organizado.
TECIDO LINFÓIDE ORGANIZADO
Órgão Linfóide
Primário
Timo
T
Medula Óssea
S B
encapsulado Não-encapsulado
Órgão Linfóide
Secundário
linfonodo baço MALT
Resposta Imune
Para antígenos nos tecidos
Para antígenos no sangue
Para antígenos nas superfícies mucosas
Nota: T= timo, S= stem cell (célula tronco), B= baço, MALT= tecido linfóide associado à mucosa. As células-tronco, originadas da medula óssea, diferenciam-se em células B e T imunocompetentes nos órgão linfóides primários (Timo e Medula Óssea). Estas células então colonizam os tecidos linfóides secundários (linfonodo, baço e tecido linfóide associado à mucosa) onde as respostas imunes são organizadas. Fonte: Roitt et al, 2004 25.
Os linfonodos estão envolvidos com respostas a antígenos provenientes dos tecidos,
enquanto o baço está envolvido principalmente com antígenos que a ele chegam via corrente
sanguínea 25. A comunicação entre estes tecidos e o resto do organismo é mantida por um
conjunto de linfócitos recirculantes que passam do sangue para dentro dos linfonodos, baço e
outros tecidos e voltam para o sangue pelos principais canais linfáticos, como o ducto torácico 25.
Esta circulação dos linfócitos entre os tecidos, a corrente sanguínea e os linfonodos capacita as
células sensibilizadas ao antígeno a procurá-lo e a serem recrutadas para locais em que esteja
ocorrendo uma resposta 25. Nos linfonodos, as células T ocupam regiões paracorticais profundas
ao redor dos centros germinativos de células B e, no baço, localizam-se nas áreas periarteriolares
da polpa branca 23.

30
2.2.1 Receptores de Células T e B.
As células T e B podem ser distinguidas por seus marcadores de superfícies 25. À medida
que se diferenciam em populações com funções distintas, as células T e B adquirem moléculas
em sua superfície que refletem essas especializações 25. É possível produzir anticorpos
homogêneos de uma única especificidade - anticorpos monoclonais – capazes de reconhecer
esses marcadores de superfície 25. Em 1982, realizou-se a First International Workshop on
Leukocyte Differentiotion Antigens para definir uma nomeclatura para as moléculas de superfície
dos leucócitos humanos. Surgiu assim, a classificação do aglomerado de diferenciação (CD –
Cluster of Differentiation) 23, (Quadro 2).
Cada linfócito expressa um receptor antigênico de especificidade única em sua
superfície 25. Além dos marcadores de superfície das células T e B (CD), estas células possuem
receptores para identificar antígenos estranhos 25, 26. As células B apresentam receptores da
membrana plasmática, as imunoglobulinas (Ig) de superfície, enquanto o linfócito T apresenta o
receptor de célula T (RCT) 25.
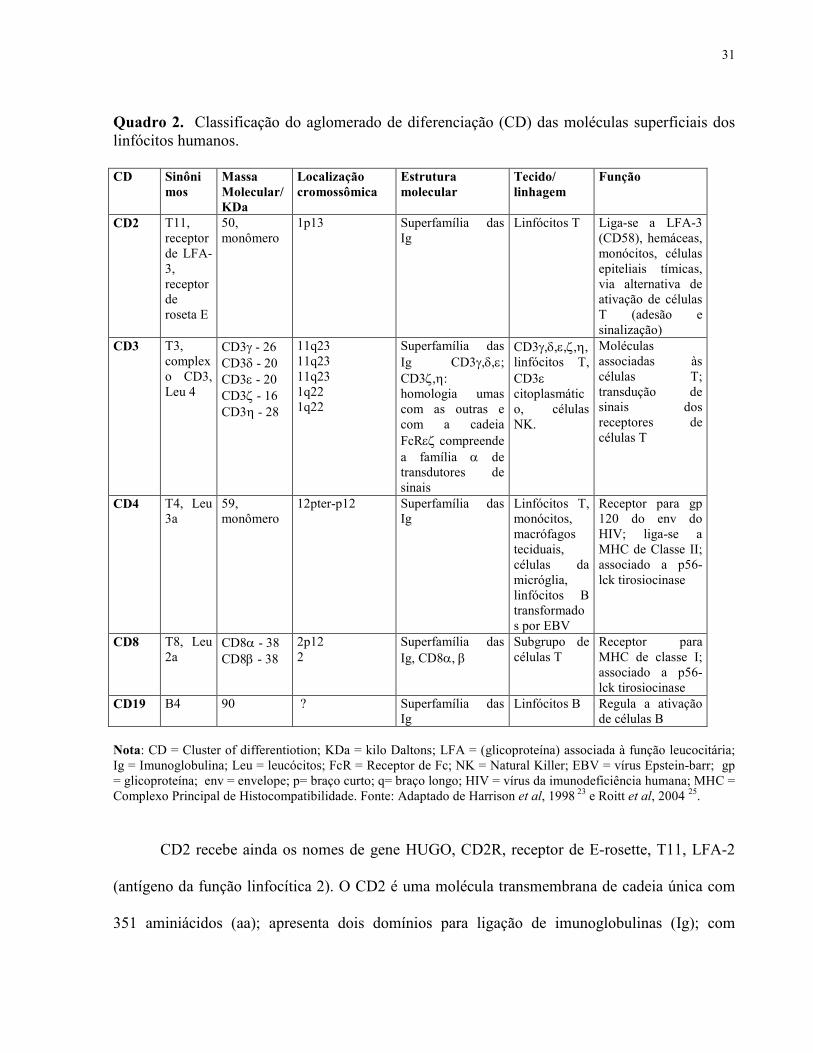
31
Quadro 2. Classificação do aglomerado de diferenciação (CD) das moléculas superficiais dos linfócitos humanos. CD Sinôni
mos Massa Molecular/ KDa
Localização cromossômica
Estrutura molecular
Tecido/ linhagem
Função
CD2 T11, receptor de LFA-3, receptor de roseta E
50, monômero
1p13 Superfamília das Ig
Linfócitos T Liga-se a LFA-3 (CD58), hemáceas, monócitos, células epiteliais tímicas, via alternativa de ativação de células T (adesão e sinalização)
CD3 T3, complexo CD3, Leu 4
CD3γ - 26 CD3δ - 20 CD3ε - 20 CD3ζ - 16 CD3η - 28
11q23 11q23 11q23 1q22 1q22
Superfamília das Ig CD3γ,δ,ε; CD3ζ,η: homologia umas com as outras e com a cadeia FcRεζ compreende a família α de transdutores de sinais
CD3γ,δ,ε,ζ,η, linfócitos T, CD3ε citoplasmático, células NK.
Moléculas associadas às células T; transdução de sinais dos receptores de células T
CD4 T4, Leu 3a
59, monômero
12pter-p12 Superfamília das Ig
Linfócitos T, monócitos, macrófagos teciduais, células da micróglia, linfócitos B transformados por EBV
Receptor para gp 120 do env do HIV; liga-se a MHC de Classe II; associado a p56-lck tirosiocinase
CD8 T8, Leu 2a
CD8α - 38 CD8β - 38
2p12 2
Superfamília das Ig, CD8α, β
Subgrupo de células T
Receptor para MHC de classe I; associado a p56-lck tirosiocinase
CD19 B4 90 ? Superfamília das Ig
Linfócitos B Regula a ativação de células B
Nota: CD = Cluster of differentiotion; KDa = kilo Daltons; LFA = (glicoproteína) associada à função leucocitária; Ig = Imunoglobulina; Leu = leucócitos; FcR = Receptor de Fc; NK = Natural Killer; EBV = vírus Epstein-barr; gp = glicoproteína; env = envelope; p= braço curto; q= braço longo; HIV = vírus da imunodeficiência humana; MHC = Complexo Principal de Histocompatibilidade. Fonte: Adaptado de Harrison et al, 1998 23 e Roitt et al, 2004 25.
CD2 recebe ainda os nomes de gene HUGO, CD2R, receptor de E-rosette, T11, LFA-2
(antígeno da função linfocítica 2). O CD2 é uma molécula transmembrana de cadeia única com
351 aminiácidos (aa); apresenta dois domínios para ligação de imunoglobulinas (Ig); com

32
estrutura similar e uma linhagem genética próxima a do CD58. O CD2 é membro da família de
supergene das imunoglobulinas como o CD48 e o CD58. Bioquimicamente o CD2 se liga
primariamente ao CD 58 das células antigênicas presentes e induz à sinalização das células T;
após estimulação de CD2 há fosforilação de CD3 zeta, CD3 epsilon, p59- fyn, p56-lck, PLC-
gama 1. A função celular do CD2 inclui: ativação de células T alternativas em funções críticas;
molécula regulatória das células T ou citólise mediada por células natural Killer; indução da
apoptose em células T periféricas ativadas; produção de citocinas por células T; regulação da
anergia das células T 27. O CD2 é conhecido, por isso como, pan T e sua utilização a carcterizar a
leucemia linfocítica aguda T (LLA-T), leucemia linfocítica crônica T (LLC-T), síndrome de
Sézary e no estudo de linfomas e imunodeficiências 28.
CD3 Identifica um complexo glicoproteíco que compreende quatro estruturas
polipeptídicas invariáveis designadas: gama (25-28 kDa), delta (20k Da), epsilon (20 kDa), zeta
(21 kDa) e integram a superfície celular e o citoplasma (transmembrânica). É não-covalente, está
associado ao polimórfico RCTα/β e interage com o heterodímero RCTγ/δ. O antígeno CD3
determina a linhagem específica de linfócitos T e sua expressão no citoplasma (cCD3)
caracteriza timócitos/ célula T imatura e, quando na membrana, o linfócito T maduro. Os
anticorpos anti-CD3 são úteis para sondar a região constante dos receptores de células T, os
quais se expressam axclusivamente nos linfócitos T imunocompetentes, no monitoramento de
imunodeficiência, doenças auto-imunes e nas leucemias e linfomas. Imunofenotipagem para
neoplasia hematológica 28.
CD4 ou L3T4 ou W3/25. É uma molécula da família do supergene das imunoglobulinas;
em cuja estrutura molecular tem a região extracelular, ligadas a quatro imunoglobulinas com
domínios de 370 aminoácidos (aa); região transmembrana com 25 aa; região citoplasmática com

33
38 aa. É função celular do CD4: a diferenciação de células do timo, receptor primário do
retrovírus HIV, regulação da adesão dos linfócitos T e B na ausência de antígenos de
reconhecimento. São ligantes extracelulares do CD4: moléculas MHC de classe II, glicoproteína
do envelope do HIV (gp120), interleucina 16, glicoproteína do plasma do semêm humano (gp
17), proteína que induz prolactina, glicoproteína extra-parotídea (EP-GP), ligante intracelular do
CD4: moléculas p56-lcK. A molécula de CD4 está presente principalmente em subtipos de
timócitos; subtipos de linfócitos T, que reconhece antígenos associados com moléculas próprias
do MHC da classe II; monócitos do sangue periférico; macrófagos de tecidos e granulócitos 29. A
determinação de células T CD4+ é utilizada no diagnóstico e prognóstico de imunodeficiências
como: síndrome de DiGeorge (aplasia tímica), agamaglobulinemia e síndrome da
imunodeficiência adquirida (SIDA). Em neoplasias, as células T CD4+ são úteis para
caracterizar: leucemia/ linfoma de células T do adulto, LLA-T, LLC-T, síndrome de Sézary e
micose fungóide 28.
CD8 Identifica uma molécula de membrana com aproximadamente 32-34 kDa. Os
linfócitos T CD8+ funcionam na regulação da resposta imune T e B e também na reação de
células T citotóxicas. Esta molécula interage com o RCT como co-receptor para o MHC classe I
no reconhecimento antigênico. A expressão deste fenótipo é encontrada em subpopulação de
linfócitos T periféricos (citotóxicos/ supressor), timócitos e células natural killer. No estudo de
neoplasias hematológicas contribui para caracterizar leucemia/ linfoma de células T do adulto,
LLA-T e LLC-T. É relatada diminuição dos níveis de linfócitos T CD8+ no sangue periférico de
infecções pelo vírus da hepatite B, Epstein-Barr e citomegalovirus. Imunofenotipagem para
neoplasia hematológica 28.

34
CD19 ou B4: é uma molécula da família do supergene das imunoglobulinas de cerca de
280 aa no domínio extracelular e um domínio citoplasmático de 240 aa. O CD19 é uma molécula
crítica de transdução de sinal que regula o desenvolvimento, ativação e diferenciação dos
linfócitos B 30. É o antígeno mais precocemente expresso na diferenciação da linhagem B (
estágio pré-pré-B), porém ausente em plasmócitos. É o principal pan B e está presente em todos
os linfócitos B periféricos e em aproximadamente 5% de todas as células da medula óssea. O
anticorpo anti-CD19 é restrito à linhagem B. É utilizado na avaliação de LMC em crise blástica
linfóide, linfomas não-hodgkin, leucemia linfoblástica aguda, leucemia linfocítica crônica, mas
ausente em mieloma múltiplo. Normalmente não se expressa em neoplasias T precursoras ou
pós-tímicas e assim sua importância é na diferenciação entre neoplasias B e T 28.
2.2.2 Citocinas
As citocinas são proteínas solúveis produzidas por uma ampla variedade de tipos
celulares hematopoiéticos e não-hematopoiéticos 23. Estão envolvidas na regulação do
crescimento, do desenvolvimento e da ativação das células do sistema imune e na mediação da
resposta inflamatória 23. Em geral as citocinas são redundantes: citocinas diferentes têm as
mesmas funções; e pleotópicas: são capazes de atuar em muitos tipos celulares diferentes 23. A
ação das citocinas pode ser: 1- autócrina, quando a célula alvo é a mesma que secreta a citocina;
2- parácrina, quando a célula alvo está próxima; 3- endócrina, quando a citocina é secretada na
circulação e atua longe da origem 23.
Uma classificação empírica divide as citocinas em três grupos: 1- citocinas
imunorreguladoras envolvidas na ativação, no crescimento e na diferenciação dos linfócitos e

35
monócitos; 2- citocinas pró-inflamatórias, que são produzidas predominantemente por fagócitos
mononucleares em resposta a agentes infecciosos; e 3- citocinas que regulam o crescimento e a
diferenciação de leucócitos imaturos 23.
As citocinas exercem seus efeitos influenciando a ativação de genes, que resulta em
ativação, crescimento, diferenciação, expressão de moléculas funcionais superficiais e função
efetora das células 23.
2.2.3 Expansão Clonal dos Linfócitos
Quando um microorganismo entra no hospedeiro (organismo humano), seus antígenos
constituintes se combinam apenas com os linfócitos B cujos receptores de superfície sejam
complementares ao formato destes antígenos. As células B que se ligam ao antígeno se tornam
ativadas e proliferam clonalmente, sob a influência das citocinas e formam uma grande
população de células derivadas das originais 25.
No caso das células B, uma grande parte dos linfócitos expandidos clonalmente se
transformam em plasmócitos dedicados à síntese e secreção de anticorpos. Esses plasmócitos são
derivados de uma célula parenteral comprometida com a produção de apenas um anticorpo
específico. Dessa forma, o produto final é idêntico à molécula que estava expressa na superfície
da célula original que reconheceu o antígeno 25.
Um processo semelhante de seleção e expansão clonal ocorre com as células T,
produzindo um grande número de células T efetoras com a mesma especificidade da célula-mãe
original. De significado crucial é o fato de que no caso de ambas as células B e T, uma fração da
população expandida clonalmente se diferencia em células de memória em repouso. Assim, estas

36
células de memória são capazes de reconhecer o antígeno microbiano em qualquer infecção
subseqüente 25.
2.2.4 O Papel das Células de Memória
As células de memória são mais prontamente estimuladas por uma dada dose de
antígeno que as células virgens 25. Nas células B, esse maior poder de combinação ocorre através
de mutação e seleção durante a resposta primária. Enquanto nas células T há mutação de
afinidade que ocorre através da expressão aumentada de moléculas de adesão acessórias: CD2,
LFA-1, LFA-3 e da molécula de adesão intercelular-1 (ICAM-1), que permitem que o linfócito
se ligue mais firmemente às células especializadas que apresentam o antígeno. Estes fatores
combinados com o número de linfócitos específicos para um dado antígeno, produzido pela
resposta primária, resultam em uma resposta muito mais intensa do anticorpo ou da célula T no
segundo contato com o antígeno. A memória geralmente tem vida mais longa e se estende por
muitos anos 25.
2.2.5 Células T
Os linfócitos T (LT) maduros constituem 70 a 80% dos linfócitos sanguíneos periféricos
normais (somente 2% dos linfócitos corporais totais estão contidos no sangue periférico), 90%
dos linfócitos do ducto torácico, 30 a 40% das células nos linfonodos e 20 a 30% das células
linfóides esplênicas 23. Diversas citocinas regulam o processo de regulação e diferenciação das
células T 23. As primeiras células da linhagem T (células pró-T CD34+) são encontradas no

37
fígado fetal, saco vitelino e na medula óssea após o nascimento 23. No timo, os precursores
CD71+, CD34+ das células T iniciam a síntese citoplasmática de componentes do complexo
CD3 das moléculas associadas aos receptores de células T. Nos precursores de CD7+, CD2+ e
cCD3+, o rearanjo do gene do receptor da célula T (RCT) por influência da IL-7, origina duas
linhagens de linfócitos T que expressam cadeias RCTαβ ou RCTγδ. As cadeias que expressam
RCTαβ compreendem a maioria dos linfócitos T periféricos no sangue, nos linfonodos e no baço
e sofrem diferenciação terminal CD4+ (Linfócitos T auxiliares) ou CD8+ (Linfócito T
citotóxico). Os linfócitos que expressam as cadeias RCTγδ circulam como uma pequena
população no sangue; embora suas funções não sejam totalmente conhecidas 23. Os timócitos
corticais ao atingirem a maturidade deixam de expressar CD1 e passa a ocorrer a expressão
recíproca de CD4 e CD8 23. Os linfócitos CD4+ RCTαβ+ maduros induzem a diferenciação das
células B e a proliferação de linfócitos T CD8+ citotóxicos, produzem diversas citocinas e
regulam determinados estágios da eritropoiese 23. Um subgrupo de linfócitos CD4+ também
funciona como linfócitos efetores citotóxicos, reconhecendo fragmentos peptídicos antigênicos
estranhos fisicamente associados a moléculas MHC de classe II em células apresentadoras de
antígenos (APC) 23. Os linfócitos CD8+ RCTαβ+ atuam como células T efetoras citotóxicas ou
como células imunorreguladoras que modulam a função das células B e T; os linfócitos CD8+
reconhecem fragmentos de peptídeos antigênicos estranhos associados à molécula MHC de
classe I 23.
Funcionalmente os linfócitos são divididos em LT auxiliares (LT helper), LT citotóxico,
LT supressor. Cada um deles possui receptores característicos, além do RCT padrão para as
células T, identificados por técnicas imunológicas e com funções específicas. Entretanto, todas

38
as células T possuem os receptores RCT e o CD3 31. O receptor CD3 dos linfócitos participa do
mecanismo de ativação intrínseca do linfócito 31.
O LT auxiliar (CD3/ RCTαβ, CD4+) tem a função de reconhecer o macrófago ativado e
é o principal alvo do vírus HIV. Esta célula é o mensageiro mais importante do sistema imune.
Ela envia mensagens de ataque para os diversos leucócitos para realizar a “guerra imunológica”
contra o agente agressor. O LT auxiliar interage com os macrófagos, reconhecendo o epítopo
que lhe é apresentado. A interleucina-1 (IL-1) estimula a expansão clonal de LT auxiliares
monoclonais a secretar diversas interleucinas, sendo, portanto, dividido em LT auxiliar 1 (TH1)
e LT auxiliar 2 (TH2). Esses subtipos de LT auxiliares secretam interleucinas distintas, cada uma
com uma função específica: as interleucinas produzidas pelo LT TH1 (IL-2 e interferon gama)
estão relacionadas com a resposta imune celular e as produzidas pelo LT TH2 (IL-4, IL-5, IL-6 e
IL-10) estão relacionadas à resposta imune humoral 23, 25.
A função do LT auxiliar é reguladora e atua na: a) estimulação do crescimento e
proliferação de LT citotóxicos e supressores contra o antígeno; b) estimulação do crescimento e
diferenciação dos Linfócitos B em plasmócitos para produzir anticorpos contra o antígeno; c)
ativação dos macrófagos e d) autoestimulação (um LT auxiliar pode estimular o crescimento da
população de LT auxiliares) 31.
O Linfócito T citotóxico (CD3/ RTCαβ, CD8+) tem a função de reconhecer o complexo
de histocompatibilidade principal (MHC - “Major Histocompatibility Complex”) de classe I
expresso por células rejeitadas. Todas as células do organismo possuem genes próprios para o
MHC denominados de HLA (antígeno de histocompatibilidade principal). Quando uma célula
estranha esta presente no organismo, como nos transplantes e enxertos, a mesma expressa o HLA
de classe I (HLA-I) em sua superfície, cuja expressão é ampliada por estímulos como o

39
interferon gama. O HLA de classe II (HLA-II) é produzido por macrófagos e linfócitos B, cuja
função é ligá-los aos linfócitos T auxiliares para lhes apresentar o antígeno, através da interação
CD4-HLA-II e RCT- epítopo 31.
O linfócito T citotóxico (LTc) é a principal defesa do sistema imune, pois lisa
diretamente as células estranhas que expressam o HLA- I. A resposta imune celular específica
(RIC) se baseia na ativação e agressão das células CD8 31. Esta célula também participa de
reações de hipersensibilidade tardia (tipo IV), como as reações que caracterizam os testes
intradérmico tipo PPD na pele. O seu principal estimulador é a interleucina 2 (produzida pelo LT
TH1), que causa a expansão clonal de linfócitos T citotóxicos monoclonais na RIC 31.
Os linfócitos T supressores são linfócitos que têm a função de modular a resposta imune
através da inibição da mesma. Pouco se conhece a respeito desta célula, mas sabemos que ela
age através da inativação dos linfócitos T citotóxicos e auxiliares, limitando a ação destes no
organismo numa reação imune. Sabemos que o LT auxiliar ativa o LT supressor que controla a
atividade dos LT auxiliares, impedindo que eles exerçam suas atividades excessivamente. Os LT
supressores também participam da chamada “tolerância imunológica”, que é o mecanismo pelo
qual o sistema imune usa para impedir que os leucócitos ataquem as próprias células do
organismo. Portanto, se houver deficiência na produção ou ativação dos linfócitos T supressores,
poderá haver uma agressão autoimune ao organismo 31.
2.2.6 Células B
As células B maduras compreendem 10 a 15% dos linfócitos do sangue periférico
humano, 50% dos linfócitos esplênicos e aproximadamente 10% dos linfócitos da medula óssea.

40
As células B expressam moléculas de imunoglobulinas superficiais (sIg) intramembrana que
atuam como receptores antigênicos das células B em um complexo de moléculas α e β de
sinalização associadas à Imunoglobulinas (Ig) com ocorrência de sinalização intracelular
semelhantes àquelas observadas nas células T 23.
O desenvolvimento dos linfócitos B pode ser dependente ou independente de antígenos.
O desenvolvimento das células B independente de antígenos ocorre nos órgãos linfóides
primários, e inclui todos os estágios da maturação da célula B até a célula madura sIg+ 23; esta
molécula age como receptores de antígenos para as células B, assim como, as moléculas do
MHC e os receptores de antígenos de células T26, (Quadro 3). A maturação dependente de
antígenos, que ocorre nos órgãos linfóides secundários, é impulsionada pela interação do
antígeno com a sIg da célula B, o que gera indução das células B de memória, modificação da
classe de Ig e formação de plasmócitos, que produzem imunoglobulinas (anticorpos) que são
secretados e atuam na eliminação dos antígenos 26. A eliminação dos antígenos geralmente
requer a interação do anticorpo com componentes do sistema imunológico natural, proteínas do
complemento, células fagocitárias e eosinófilos. As funções efetoras mediadas pelos anticorpos
incluem a neutralização de microorganismos ou de toxinas microbianas, ativação do sistema de
complemento, opsonização de antígenos para aumentar a fagocitose, citotoxicidade celular
dependente de anticorpo e hipersensibilidade imediata 26.
As glicoproteínas do plasma ou soro são separadas pela sua solubilidade em albuminas
e globulinas. A maioria dos anticorpos é encontrada no terceiro grupo de globulinas de migração
mais rápida, as gamaglobulinas. Todas as moléculas de anticorpos possuem as mesmas
características estruturais básicas, mas apresentam uma grande variedade nas regiões que ligam
os antígenos. As funções efetoras e as propriedades físico-químicas comuns dos anticorpos estão

41
associadas às regiões que não se ligam a antígenos e que demonstram muito pouca variação entre
os diferentes anticorpos. Uma molécula de anticorpo possui uma estrutura básica simétrica
composta de duas cadeias leves idênticas e duas cadeias pesadas idênticas 26. Tanto as cadeias
pesadas quanto as leves possuem uma região aminoterminal variável (V) que participa no
reconhecimento dos antígenos, e de regiões constantes (C) carboxiterminais; as regiões C das
cadeias pesadas possuem as funções efetoras 26; e diferentes isótipos de cadeia pesada de Ig que
desempenham funções efetoras distintas 24 (Quadro 4).
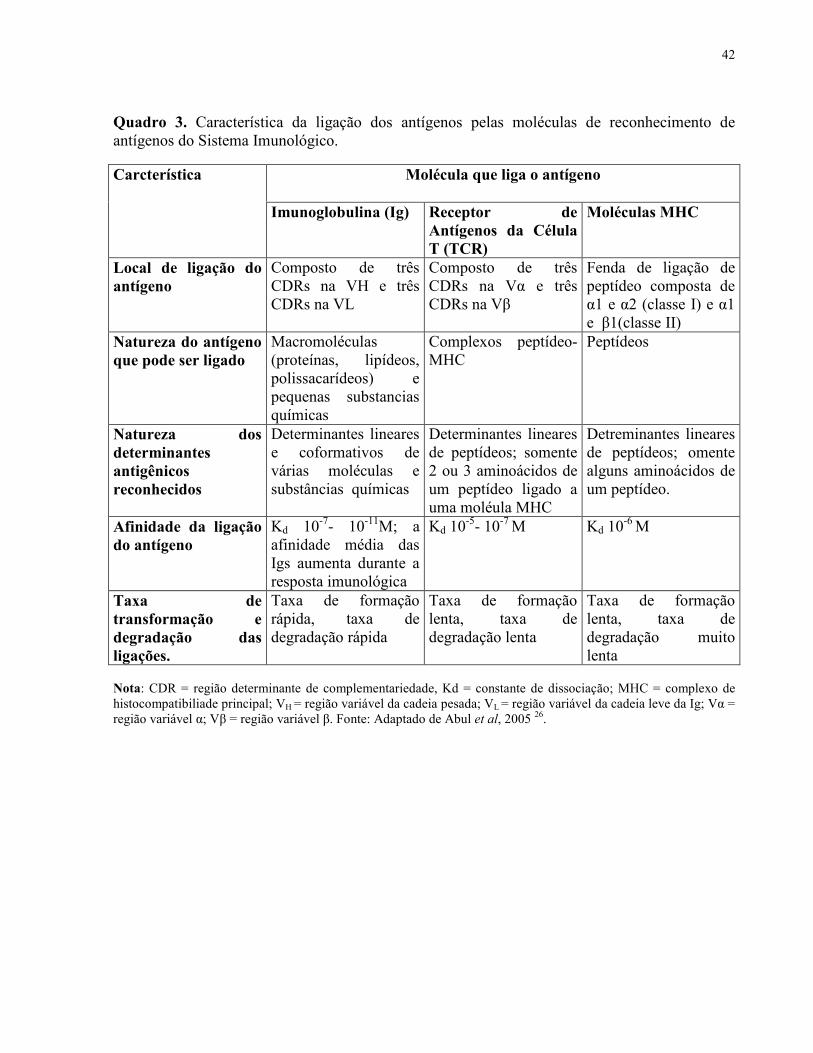
42
Quadro 3. Característica da ligação dos antígenos pelas moléculas de reconhecimento de antígenos do Sistema Imunológico.
Nota: CDR = região determinante de complementariedade, Kd = constante de dissociação; MHC = complexo de histocompatibiliade principal; VH = região variável da cadeia pesada; VL = região variável da cadeia leve da Ig; Vα = região variável α; Vβ = região variável β. Fonte: Adaptado de Abul et al, 2005 26.
Molécula que liga o antígeno Carcterística
Imunoglobulina (Ig) Receptor de Antígenos da Célula T (TCR)
Moléculas MHC
Local de ligação do antígeno
Composto de três CDRs na VH e três CDRs na VL
Composto de três CDRs na Vα e três CDRs na Vβ
Fenda de ligação de peptídeo composta de α1 e α2 (classe I) e α1 e β1(classe II)
Natureza do antígeno que pode ser ligado
Macromoléculas (proteínas, lipídeos, polissacarídeos) e pequenas substancias químicas
Complexos peptídeo-MHC
Peptídeos
Natureza dos determinantes antigênicos reconhecidos
Determinantes lineares e coformativos de várias moléculas e substâncias químicas
Determinantes lineares de peptídeos; somente 2 ou 3 aminoácidos de um peptídeo ligado a uma moléula MHC
Detreminantes lineares de peptídeos; omente alguns aminoácidos de um peptídeo.
Afinidade da ligação do antígeno
Kd 10-7- 10-11M; a afinidade média das Igs aumenta durante a resposta imunológica
Kd 10-5- 10-7 M Kd 10
-6 M
Taxa de transformação e degradação das ligações.
Taxa de formação rápida, taxa de degradação rápida
Taxa de formação lenta, taxa de degradação lenta
Taxa de formação lenta, taxa de degradação muito lenta

43
Quadro 4. Funções dos Isótipos de Anticorpos. Isótipo do Anticorpo
Funções Efetoras Isótipo Específicas
IgG Opsonização de antígenos para fagocitose por macrófagos e neutófilos
Ativação da via clássica do complemento
Citotoxicidade mediada por células e dependente de anticorpos, mediada por células natural Killer e macrófagos Imunidade neonatal; transferência de anticorpos maternos através da placenta e do intestino Inibição da ativação da célula B por feedback
IgM Ativação da via clássica do complemento
Receptor antigênico de linfócitos B naive*
IgA Imunidade das mucosas: secreção de IgA nos lumens dos tratos gastrointestinal e respiratório.
IgE Citotoxicidade mediada por células e dependente de anticorpo envolvendo eosinófilo Desgranulação de mastócito (reações de hiperensibilidade imediata)
IgD Receptor antigênico de linfócitos B naive*
Nota: IgG = Imunoglobulina G; IgA = Imunoglobulina A; IgM = Imunoglobulina M; IgE = Imunoglobulina E; IgD = Imunoglobulina D; *naive estas funções são mediadas por anticorpos ligados à membrana e não secretados. Fonte: Adaptado de Abul et al., 2005 26.
IgA (Imunoglobulina A): Representa 15-20% das imunoglobulinas do soro humano. No
homem, mais de 80% da IgA ocorre sob a forma monomérica e está presente no sangue desta
forma. O principal papel da IgA é proteger o organismo de invasão viral ou bacteriana através
das mucosa 32.
IgG (Imunoglobulina G): É uma imunoglobulina monomérica simples de 150.000
daltons, cadeias pesadas tipo gama, que perfaz 80% das imunoglobulinas do organismo. É o

44
anticorpo principal nas respostas imunes secundárias e a única classe antitoxinas. A resposta de
anticorpos secundária é mais rápida e mais abundante que a resposta primária. A região FC
realiza ativação de complemento (quando unida ao antígeno) e auxilia a fagocitose por se ligar a
macrófagos. Com a ativação do complemento, há geração de quimiotaxia de neutrófilos,
aumento da permeabilidade vascular e amplificação da resposta inflamatória 32.
IgM (Imunoglobulina M): Perfaz aproximandamente 10% do conjunto de
imunoglobulinas. A IgM é encontrada principalmente no intravascular, sendo uma classe de
anticorpos "precoces" (são produzidas nas fases agudas iniciais das doenças que desencadeiam
resposta humoral). É encontrada também na superfície dos linfócitos B de forma monomérica,
realizando a função de receptor de antígenos. É o anticorpo produzido após a exposição inicial a
um antígeno 32.
2.2.7 Interações Celulares na Resposta Imune Normal
O resultado final da ativação do sistema imune humoral e celular por um antígeno
estranho é a eliminação do antígeno por células efetoras, ou em combinação com anticorpos
específicos. Além disso, é ativada uma série de células reguladoras, que modulam a ativação das
células T efetoras e a produção de anticorpos pelas células B 23.

45
2.2.7.1 Interação Célula T – Célula T e Célula T – Célula B
A expressão da função das células imunes é o resultado de uma complexa série de
eventos imunorreguladores que ocorrem em fases. Os linfócitos T e B medeiam funções imunes
e cada um desses tipos celulares, quando recebem sinais apropriados, passam por estágios, da
ativação e indução à proliferação, diferenciação e finalmente, execução de funções efetoras. A
função efetora expressa pode ser o ponto final de uma resposta (a secreção de anticorpos por um
plasmócito diferenciado), ou exercer uma função reguladora que modula outras funções
(linfócitos T auxiliares CD4+ ou citotóxicas CD8+) que modulam a diferenciação das células B e
a ativação de linfócitos T citotóxicos CD8+ ou CD4+ 23.
Os linfócitos T auxiliares CD4 são subdivididos com base nas citocinas produzidas. Os
linfócitos TH1 CD4+ fornecem células T para gerar linfócitos T citotóxicos e, em geral,
respondem a antígenos que originam tipos de respostas imunes de hipersensibilidade tardia. Ao
contrário, os linfócitos TH2 regulam a intensidade das respostas imunes mediante a secreção de
uma citocina, IL-10, que inibe a produção de outras citocinas. Além disso, os linfócitos T CD4+
TH2 ajudam as células B a produzirem Ig específica e a responderem a antígenos que necessitem
de níveis elevados de anticorpos para a eliminação do antígeno estranho (nas parasitoses, por
exemplo). Citocinas diferentes são capazes de impelir a resposta imune preferencialmente na
direção de uma resposta TH1 (IL-12) ou de uma resposta TH2 (IL-4) 23.
Os linfócitos T ao serem ativados pelas células apresentadoras de antígenos, geram
subgrupos reguladores de linfócitos T que produzem IL-2, IL-3, IFNγ e/ou IL-4, IL-5, IL-6 e IL-
10 que exercem influências positivas e negativas sobre os linfócitos T e B efetores. No caso das
células B, os efeitos tróficos são mediados por citocinas (IL-3, IL-4, e IL-5) derivados de
linfócitos T, que atuam em estágios seqüenciais da maturação das células B e resultam em

46
proliferação, diferenciação e, por fim, secreção de anticorpos. Para os linfócitos T citotóxicos, os
fatores tróficos incluem a secreção de linfócitos T indutores de IL-2, IFNγ, IL-12 e IL-15. Além
disso, as células B são capazes de atuar como células apresentadoras de antígenos, processando e
apresentando antígenos para os linfócitos T e secretando FNTα e IL-6 23.
Embora as células B reconheçam antígenos nativos através receptores de sIg, as células
B necessitam de ajuda dos linfócitos T para produzir anticorpos de alta afinidade de múltiplos
isótipos, para uma resposta imune mais eficaz contra antígenos estranhos. Essa dependência dos
linfócitos T provavelmente atua para regular as respostas das células B e proteger contra a
produção excessiva de auto-anticorpos 23.
2.2.7.2 Interações das Células Apresentadoras de Antígenos – Linfócitos/Células T
Os três tipos celulares básicos de fragmentos peptídicos antigênicos das células T são as
células dendríticas/ de Langerhans, os monócitos-macrófagos e os linfócitos B. Muitos dos
efeitos de ativação e da regulação desses tipos celulares ocorrem por meio de citocinas que
regulam a maturação de células T e B, após contato direto entre as células apresentadoras de
antígenos e a célula T 23.
2.2.8 O Sistema de Complemento
O sistema de complemento corresponde a uma série em cascata de enzimas plasmáticas,
proteínas reguladoras e proteínas capazes de lise celular, cujo principal local de síntese é o
fígado. Existem duas vias de ativação para o sistema de complemento: a ativação da via clássica

47
por meio de C1, C4 e C2 e a ativação da via alternativa por meio do fator D, C3 e do fator B que
conduz à clivagem e ativação de C3. C3 é uma proteína, cujos fragmentos de ativação, quando
ligados à superfície-alvo de bactérias ou outros antígenos estranhos, são crucias para a
opsonização na preparação da fagocitose 23. O aumento de C3 é observado em câncer e colite
ulcerativa. A diminuição da atividade de C3 é observada em: angioedema hereditário, infecções
bacterianas (principamente por neisseria), cirrose, glomerulonefrite, hepatite, nefrite por lúpus,
desnutrição, rejeição a transplante renal e lúpus eritematoso sistêmico 32.
2.2.9 Avaliação do Sistema Imunológico
A função imunológica pode ser avaliada através de anamnese e exame físico detalhados,
associados a exames latoratoriais das condições de defesa do hospedeiro, como hemograma
completo com contagem diferencial e níveis de imunoglobulinas séricas IgM, IgG, IgA, IgE,
para triagem inicial 33. Outros exames complementares incluem 34:
a) Quantificação das populações de células mononucleares sanguíneas por imunofluorescência
empregando anticorpos marcadores monoclonais para:
Células T (CD3, CD4, CD8, RCTαβ, RCTγδ);
Células B (CD 19, CD 20, CD 21, Ig [µ, δ, γ, α, κ, λ] e moléculas associadas a Ig [αβ]);
Células NK (CD16);
Monócitos (CD15);
b) Marcadores de ativação (HLA-DR, CD25, CD80);
c) Avaliação funcional das células T:
1.Provas de hipersensibilidade cutânea tardia

48
2. Resposta proliferativa a mitógenos
3. Produção de citocinas
d) Avaliação funcional de células B:
1. Anticorpos naturais ou comumente adquiridos
2. Resposta à imunização com antígenos protéicos e carboidratos
3. Determinação quantitativa das subclasses de IgG
e) Complemento
1. Ensaios de CH50
2. C3, C4 e outros componentes
f) Função fagocitária
1. Redução do tetrazólio nitroazul
2. Ensaios de quimiotaxia
3. Atividade bactericida
2.2.10 Sistema Imunológico e Síndrome de Down
Portadores da síndrome de Down apresentam alteração do sistema imune 35. Foram
relatadas funções anormais dos linfócitos, deficiência de Imunoglobulina da classe G (IgG) e
diminuição da resposta imune dependente de células T 16, 36, bem como alterações nas células B
35. Um estudo de caso-controle, realizado na Itália, com 20 crianças portadoras de SD, não
institucionalizadas, e 20 controles adultos não portadores da SD, revelou uma porcentagem de
células T CD3+ drasticamente aumentada nos indivíduos com SD; nestas crianças, houve
correlação positiva entre a idade e o aumento da porcentagem de células CD3+ circulantes 35. A

49
expressão dos antígenos CD3 é considerada uma ativação do marcador de células T-helper-2
(TH2); as células TH2+ estão associadas a certas patologias, o que explica em parte a
susceptibilidade aumentada de portadores da SD a infecções 16, 35, doenças malignas e auto-
imunidade 35. Dados de experimentos evidenciaram que CD3 é também co-expressado por
clones de células T citotóxicas CD8+, as quais exibem uma secreção de citocinas semelhante à
TH2, bem como por células T ou clones causados por antígenos, os quais induzem a resposta
imune mediada por TH1 35.
Há evidências sugerindo que o defeito imune é principalmente devido ao desarranjo no
timo do portador da SD, isto é, por depleção de linfócitos, diminuição do córtex, perda da
demarcação corticomedular e aumento dos corpúsculos císticos de Hassal dentro da medula
tímica. Como conseqüência, há uma maturação anormal no meio tímico, resultando em
anormalidades funcionais e fenotípicas nos linfócitos T sanguíneos 35. Estas alterações do
sistema imune podem estar implicadas no desenvolvimento da alopécia areata (AA) 35, 37.
A deficiência de zinco também pode explicar uma parte da susceptibilidade aumentada
para infecções e AA existentes na SD 38. O zinco está envolvido em vários processos celulares
tais como fenômenos de membrana, metabolismo do RNA mensageiro e reações de várias
enzimas (transferases, hidrolases, liases, isomerases, oxirredutases) e fatores de transcrição 38, 39.
A carência deste oligoelemento está associada, ainda, a deficiências imunológicas caracterizadas
por diminuição da quimiotaxia dos neutrófilos, hipersensibilidade da pele ao dinitroclorobenzeno
16, 38, tricofitina e/ou candidina 16. As células com deficiência de zinco não se dividem nem se
diferenciam; assim, tecidos com altas taxas de renovação celular incluindo pele, mucosa
gastrointestinal, condrócitos, espermatogônia e timócitos são comprometidos. As manifestações

50
dermatológicas da deficiência do zinco incluem hiperceratose, paraceratose, acrodermatite e
alopécia 39, 40.
2.2.11 Autoimunidade e Síndrome de Down
Algumas vezes, o sistema imune identifica erroneamente os tecidos do organismo
humano como estranhos (“not self”) e os “destrói”, resultando em uma reação autoimune 31.
Os portadores da SD apresentam uma prevalência aumentada de desordens autoimunes
afetando órgãos endócrinos (Tireoidite de Graves 41, Diabetes Mellitus tipo I 43) e não endócrinos
(vitiligo, anemia perniciosa, Lupús Eritematoso Sistêmico, Doença de Moyamoya) 36, 40, 42, 44.
A disfunção da tireóide em pacientes com SD é comum na infância, e a doença
autoimune da tireóide ocorre mais freqüentemente após os oito anos de idade 43. Observa-se uma
prevalência elevada dos níveis de auto-anticorpos anti-tiroideanos, descrita em 13 a 34% dos
portadores da SD 42, 43. Alguns estudos sugerem herança de traço autossômico dominante para a
propensão de desenvolver auto-anticorpos 45, 46. Foi observado em estudo do tipo caso-controle
de famílias de portadores da SD, um aumento na freqüência de anticorpos anti-tireoideanos e
anormalidades da tireóide nas mães das crianças portadoras da SD 45. Ainda em relação ao
acometimento da glândula tireóide temos as seguintes desordens auto-imunes relacionados à
presença de seus respectivos auto anticorpos:
a) tireoidite de Hashimoto (tH) 41, 45, 47, 48: anticorpo antiperoxidase (TPO-ab) 49 e HLA de
classe II DR4 41. Para a tH do tipo atrófica temos HLA da classe II DR3 47 e para a do tipo bócio
temos o HLA DR5 41, 47. A tH coexiste com certa freqüência com outras doenças autoimunes

51
como anemia perniciosa, LES, Doença de Graves, diabetes mellitus, insuficiência de supra-renal,
atrite reumatóide, síndrome de Sjogren e hepatite crônica ativa autoimune 33, 48.
b) tireoidite Autoimune Hipotireóide: é a forte associação com o alelo HLA DQA 0301
da classe II na SD 50.
c) tireoidite de Graves 41: a presença de anticorpos dirigidos contra antígenos da tireóide e
expressão do HLA DR3 da classe II na superfície das células tireóideas.
Vitiligo: A prevalência na população geral é de 1% 51, porém Carter e Jegasothy (1976) 36
encontraram uma prevalência de 1,9% no estudo com indivíduos portadores de SD. Os pacientes
com vitiligo apresentam maior incidência de distúrbios autoimunes incluindo hipotireoidismo,
Doença de Graves, anemia perniciosa, alopécia areata, doença de Addison, uveíte, candidíase
mucocutânea crônica e as síndromes autoimunes poliglandulares (tipos I e II) 40. As doenças da
glândula tireóide ocorrem em até 30% dos pacientes com vitiligo 40.
Lúpus Eritematoso Sistêmico (LES) é uma doença inflamatória sistêmica crônica, que
segue uma evolução de exacerbações e remissões alternadas. Ocorre comprometimento de
múltiplos sistemas orgânicos durante os períodos de atividade da doença. O LES apresenta as
seguintes características imunológicas: presença de anticorpos antinucleares, de anticorpos anti-
DNA de duplo filamento e anticorpos anti-Sm (antígeno Smith) e de numerosos outros
anticorpos, redução dos níveis séricos do complemento, deposição de imunoglobulinas e
complemento ao longo da membrana basal glomerular e na junção dermo-epidérmica 44.
Pacientes com epítopos HLA-DR2 tendem a produzir anticorpos anti-DNA de duplo filamento;
aqueles com HLA-DR3 produzem anticorpos anti-SS-A e anti-SS-B, e aqueles com HLA-DR4 e
HLA-DR5 produzem anticorpos anti-Sm e anti-RNP (ribonucleoproteína) 44.

52
O Diabetes Mellitus tipo I ou diabetes melitus insulino dependente (DMID) tem uma
prevalência aumentada em pacientes com SD 52. Em um estudo do tipo caso-controle realizado
com 18 pacientes portadores de SD e DMID (casos) e um grupo controle de 99 familiares de
portadores SD mostraram que a média de idade de aparecimento do DMID é de
aproximadamente nove anos de idade 52. O diagnóstico de DMID em portadores de SD é
realizado mais precocemente que na população em geral (média de idade de 6,7 versus 8,0 anos,
respectivamente). É significativamente alta a proporção de crianças que com SD que
desenvolvem DMID antes do segundo ano de vida (22% das crianças com SD x 7% das crianças
em geral) 52.
Não está claro se a Doença de Moyamoya 42 representa uma displasia arterial congênita
ou é uma síndrome causada por uma reação vascular não específica 42, que acomete grandes
artérias intracranianas 53. Recentemente, tem-se postulado que uma proteína encontrada no
cromossomo 21 pode estar relacionada à patogênese da Doença de Moyamoya. Por outro lado, a
presença de processos autoimunes e de auto-anticorpo na trissomia do cromossomo 21 nos faz
suspeitar de uma freqüência maior da Doença de Moyamoya na SD pode ser resultado de um
distúrbio imune. O diagnóstico laboratorial da doença de Moyamoya associada à SD é realizado
através da dosagem de aPL (anticorpos antifosfolipídeos) no plasma. Em um relato de caso de
SD e Doença de Moyamoya verificaram-se dosagens de tiroxina e T3 total aumentadas 42.
Quanto à anemia perniciosa a causa mais comum é a deficiência de cobolamina 54. A
ausência ou escassez de fator intrínseco (FI) devido à atrofia da mucosa gástrica dificulta a
assimilação da cobolamina, resultando em anemia 54. Observa-se que 60% dos pacientes com
anemia perniciosa apresentam anticorpo anti-FI e 90% apresentam anticorpo anticélula parietal
(este anticorpo também está presente em 50% dos pacientes com atrofia gástrica, mas sem

53
anemia perniciosa). Outro fator frequentemente associado à da anemia perniciosa é a presença do
antígeno DR5 do HLA 54. Clinicamente os sinais e sintomas incluem anemia (fraqueza,
vertigem, tonteira, zumbido, palpitações, angina, sintomas de insuficiência cardíaca congestiva),
torpor, parestesia de extremidades, irritabilidade branda e esquecimento, podendo vir a
manifestar demência grave ou psicose franca. O quadro neurológico, quando estabelecido, é
irreversível 54.
2.3 ALOPÉCIA AREATA
2.3.1 Histórico
Cornelius de Celsus em 30 A. D. foi o primeiro a descrever a queda de cabelo, enquanto
doença. Ele descreveu duas formas de queda de cabelo: a primeira foi relatada como calvície
completa, que ocorria em pessoas de todas as idades e a segunda foi chamada de “ophiasis”,
devido a uma ampla área calva distribuída através do couro cabeludo 55. Porém, foi Hipócrates o
primeiro a utilizar o termo alopécia aplicado como característica da doença, queda de cabelo, tal
como nós a conhecemos hoje: alopécia areata 55. Alopécia significa "queda de cabelo" e Areata,
do grego, significa "temporária" 56. Entretanto, o termo atual alopécia areata (AA) foi
primeiramente utilizado por Sauvages em "Nosologica Medica", publicado em 1760 em Lyons,
França 57. Apartir do século dezoito houve vários debates sobre as causas da alopécia areata.
Duas principais hipóteses se destacaram: uma baseada em infecção parasitaria 58, 59 e a outra
baseada em uma alteração nervosa 60. A hipótese de infecção parasitária foi desconsiderada, uma
vez que várias tentativas de isolar o organismo infectante e transferir AA por inoculação

54
falharam 61-63. Enquanto isso, o desenvolvimento da AA como desordem neurológica,
amplamente conhecida como trofoneurótica, neurotrófica, ou hipótese neuropática, foi
sustentada com base aparentemente na freqüência das observações clínicas de traumas e estresse
físico e emocional, frequentemente relatados em jornais médicos da época 64, 65. Esta idéia
circulou entre dermatologistas por muitos anos, pois foi, e ainda é muito difícil provar ou negar
que a AA seja uma desordem neurológica 64, 65. Esta hipótese ainda é sustentada por alguns
dermatologistas atualmente 64, 65. No início do século XX, a AA ficou conhecida por estar
associada a desordens das glândulas endócrinas, principalmente a tireóide 66. Por volta da década
de 1920, muitos dermatologistas acreditavam nas teorias trofoneuróticas e endócrina, e
frequentemente na combinação de ambas 66. Ainda no século XX surgiu a hipótese de que a AA
seria induzida por agentes tóxicos, porém esta hipótese não teve aceitação no meio médico 67, 68,
69. Estudos realizados há mais de 100 anos mostraram que folículos capilares afetados pela AA
estavam envolvidos por células inflamatórias 69. No entanto, está hipótese só foi aceita depois de
1960 e atualmente acreditamos que a AA seja uma lesão que esteja relacionada com a
autoimunidade 70, 71.
2.3.2 Conceito e Epidemiologia
A AA é um tipo de queda de cabelos súbita, temporária, por vezes recorrente, não-
cicatricial 72, de áreas circulares bem circunscrita, de perda de cabelos, de dois a cinco
centímetros de diâmetro 73, que pode afetar qualquer área portadora de pêlos (do couro cabeludo
ou outra parte do corpo) 72. O tamanho e o número das placas, o lapso de tempo entre perda do

55
cabelo e crescimento normal, bem como a duração dos episódios pode ocorrer clinicamente de
várias formas 72 (Figura. 2).
Figura 2. Alopécia Areata em couro cabeludo.
Fonte: Adaptado de htpp://www.keratin.com/Alopecia areata 74.
A incidência da AA na população em geral é de aproximadamente 0,1% 75. A história
familiar positiva está presente em 25% dos casos 73. Encontrou-se uma prevalência de 1,5% de
AA em pacientes dermatológicos não hospitalizados e em 0,1% na população de indivíduos com
retardo mental (RM) sem SD 36, 37. A AA pode ocorrer em homens e mulheres de qualquer idade,
sendo que o surgimento é mais freqüente na infância. O prognóstico da AA é menos favorável
quando o início ocorre na infância, e na AA do tipo ophiasis 76-79 e em alguns relatos quando
associada à doença atópica 79. Estima-se que 1% da população já teve episódios de AA 80. Cerca
de 34 a 50% dos portadores de AA apresentam remissão do quadro em um ano, embora quase
todos experimentem mais de um episódio de AA; e 14 a 25% progridem para perda total de
cabelos no couro cabeludo (Alopecia Total, AT) ou perda de cabelos do corpo inteiro (Alopécia
Universal, AU), nesta a recuperação total é incomum 76, 81. Anderson (1950) relata que 35% dos

56
homens desenvolvem a primeira placa de AA na área occipital, visto que somente 15% das
mulheres apresentam a primeira placa de AA neste local. A primeira placa de AA nas mulheres
ocorre na região fronto-parietal. A localização da primeira placa de perda de cabelo (AA) não é
significativa para predizer como percorrerá o desenvolvimento das futuras placas de AA 77. Em
geral portadores de AA apresentam concomitantemente acometimento da unha de 25%, a parada
de crescimento ou distrofia ungueal pode acometer todas as unhas ou somente uma delas. A
distrofia ungueal varia de pontos difusos a graves alterações em poucos casos 82.
O fato de que os pacientes com AA ocasionalmente se queixem de prurido ou dor nas
áreas afetadas levanta a possibilidade de alterações no sistema nervoso periférico. Os níveis
circulantes do peptídeo relacionado ao gene da calcitonina (CGRP), um neuropeptídeo,
diminuíram em 3 pacientes com AA, comparados aos controles. O CGRP tem múltiplos efeitos
sobre o sistema imune, incluindo a quimiotaxia e a inibição da apresentação de antígenos às
células de Langerhans e da proliferação de linfócitos T estimulada por mitógenos.
O CGRP também aumenta a vasodilatação e a proliferação endotelial. Foram relatados achados
semelhantes em outro estudo, no qual foi verificada em biópsias do couro cabeludo a diminuição
dos níveis cutâneos de substância P e de CGRP, mas não do polipeptídeo intestinal vasoativo. O
estudo também observou um fluxo sangüíneo basal inferior e maior vasodilatação após injeção
intradérmica de CGRP em pacientes com AA, comparados aos controles. São necessários mais
estudos para lançar luz sobre a significância desses achados 83.
Como recursos terapêuticos têm se os esteróides 36, 37, tiroxina 84, reposição de sulfato de
zinco 38, antralina tópica e sensibilizante tópico de contato 73.

57
2.3.3 Aspectos Histopatológicos da Alopécia Areata
Sob circunstâncias normais, o crescimento de cada folículo capilar ocorre em ciclos. Há
três fases principais do ciclo do crescimento do cabelo: anágena (crescimento), catágena
(repouso) e telógena (queda) 85. A fase anágena é a mais longa, encontram-se neste estágio até
90% dos folículos do couro cabeludo humano normal. Na fase telógena, 9% dos folículos. A taxa
média do crescimento da fibra do cabelo é próxima de 0,35mm por dia, mas há variações nesta
taxa de crescimento dependendo do local do folículo capilar e da idade do indivíduo 86. A
duração da fase anágena no couro cabeludo é entre 4 e 10 anos; a catágena de duas semanas e a
fase telógena de 30 a 90 dias 87. O número total de folículos capilares no ser humano adulto é
estimado em 5 milhões, com 1 milhão no couro cabeludo 88 (Figuras 3 e 4).
58
Figura 3. Fotomicrografia mostrando estrutura selecionada de um folículo capilar humano no estágio anágeno, corado pela hematoxilina – eosina.
Nota: (a) secção longitudinal do comprimento total do folículo fotografado pouco ampliado. (b) Fotografia da região do bulbo capilar altamente ampliado. Abreviações: APM= músculo pelo-eretor; B=Bulbo; CTS= tecido conjuntivo frouxo; CTX= córtex do eixo do pelo; CU= cutícula do eixo do pelo; DP= papila dérmica; E= epiderme; HS= eixo do pelo; IRS=bainha interna da raiz; M= matriz; ORS= bainha externa da raiz; S= glândula sebácea. Modificado de Cotsarelis e Millar, 2001 89.
Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas
Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo


















