Regulação das atividades celulares
82
-
Upload
ruan-tcharle -
Category
Documents
-
view
796 -
download
2
Transcript of Regulação das atividades celulares


Mecanismos de ajuste às variações dos meios externo e interno, mantendo
constantes os meios intra e extracelular do organismo, dentro de
limites pouco variáveis e compatíveis com a alta eficiência da
maquinaria celular.

Tendência dos organismos vivos em manter constante o meio interno.
Quando o organismo não consegue manter a homeostase : doença

Ácidos nucléicos e proteínas: moléculas mais importantes nos processos vitais.
Ácidos nucléicos: informação genética
Proteínas: reações enzimáticas

Permitem controlar a atividade das enzimas , adequando-as às necessidades do organismo.
Importante: compreensão das doenças
bases para a terapêutica

Qualitativas e quantitativas nos genes
Nos processos de replicação do DNA
Na transcrição do DNA para RNA
Na tradução do mRNA em proteína
Após a tradução




Constante qualitativa e quantitativamente
Alterações qualitativas: mutações (espontâneas ou provocadas)
Alterações quantitativas: amplificação gênica (ex: tumor; neuroblastoma)

Doenças hereditárias (células germinativas: ovócitos e espermatozóides)
Exemplo: Hemofilia B, distrofia muscular
Transmissão do defeito gênico: direção vertical
Doenças não-hereditárias (células somáticas)
Exemplo: vários tipos de câncer
Transmissão do defeito gênico: direção horizontal

Seqüência de bases: codificação de uma proteína deficiente
Afetar as seqüências de base no DNA que codificam as enzimas responsáveis por modificações pós-traducionais de certas proteínas – numerosas – alta probabilidade – doenças

5% das doenças conhecidas
50% das malformações embrionárias em 3% dos nascimentos

Falhas nos mecanismos de reparação: doenças
Xeroderma pigmentosum
Autossômica e recessiva
Alta sensibilidade aos raios UV
Predisposição ao câncer de pele

Perda dos ribossomos: sinal precoce e inespecífico de degeneração celular
Causa: agentes tóxicos (lesão na tradução)

Doenças podem ser causadas por processos defeituosos de degradação dos componentes celulares
Degradação das macromóleculas: lisosomas
Ausência de enzimas lisosômicas

Degradação incompleta das glicosaminoglicanas e glicoesfingolipídios
A digestão das respectivas moléculas é incompleta
Acúmulo intracelular dos produtos não digeridos completamente
Acúmulo: ≠ células: fígado, músculos, macrófagos etc.

Reações enzimáticas
Informações genéticas


Moléculas protéicas dotadas de atividade da propriedade de acelerar intensamente determinadas reações químicas.
Tanto na síntese quanto na degradação de moléculas.
São as principais responsáveis pela eficiência da maquinaria química intracelular.

São proteínas, produzidas sob o controle do DNA.
São efetores da informação genética contida no DNA
Através dela que o DNA controla todo o metabolismo celular

Substrato: sofre a ação de uma enzima
Centro ativo: parte da molécula da enzima que se combina ao substrato para que seja exercida a ação enzimática
Co-fatores: íons metálicos ou moléculas
Coenzima: quando o co-fator for uma molécula
Holoenzima: complexo enzima + co-fator
Apoenzima: há a remoção da enzima ficando só a enzima

Nome do substrato + sufixo ase
Ex: ácido ribonucléico (substrato) – ribonuclease (enzima)


Catalisadores biológicos
Extremamente eficientes: aceleram 10 9 a 10 12 a velocidade da reação
Transformam de 100 a 1000 moléculas de substrato em produto por minuto de reação
Atuam em concentrações muito baixas e em condições suaves de temperatura e pH

Sensível a diversos agentes
Físicos e químicos
Inibidos
Inibição: competitiva e não-competitiva

Alteram:
Temperatura
Concentração do substrato
Ativadores ou inibidores que alteram a velocidade de atuação das enzimas

Frio: deprime a atividade enzimática retardando
os processos de lise celular e a deterioração
de amostras de tecidos, sangue, urina, etc.

Quando uma substância resistente à ação enzimática , porém de molécula muito parecida com a do substrato da enzima, se fixa nos centros ativos da molécula enzimática
O inibidor compete com o substrato para se localizar no centro ativo
O grau de inibição é influenciado pela concentração do substrato
Quanto maior a concentração do substrato , menor será a probabilidade de o inibidor chocar-se com as moléculas das enzimas e ocupar seus centros ativos

Não é afetada pela concentração do susbtrato
Depende exclusivamente da concentração do inibidor

As cadeias enzimáticas e mais bem organizadas são as que estão ligadas a membranas
Muitas são auto-reguláveis, sobretudo pelo efeito do produto final da cadeia sobre a primeira enzima da seqüência

Retroinibição ou inibição alostérica
Exemplo: L-treonina é transformada em L-isoleucina, através de uma
cadeia de cinco enzimas A primeira enzima desta cadeia é a L-treonina-desaminase – sua
atividade é diminuida ou suprimida pela L-isoleucina A falta de L-isoleucina provoca o funcionamento da cadeia em
toda sua intensidade O excesso de L-isoleucina faz a cadeia diminuir de ritmo ou
mesmo, parar a produção de mais L-isoleucina Assim sendo, a concentração desse aminoácido na célula
permanece dentro dos limites normais
Enzima reguladora: L-treonina-desaminase Substância inibidora (Efetor ou modulador): L-isoleucina

Regulação alostérica
O efetor combina-se com a enzima em um local diferente do centro ativo (centro alostérico)
Conseqüentemente: Ocorre uma modificação no centro ativo da enzima, cuja atividade catalítica é inibida

Izoenzimas: enzimas de uma mesma espécie animal que atacam o mesmo substrato mas que exibem diferenças na atividade, no pH ótimo de ação, na mobilidade eletroforética...
As diferenças de atividades entre as enzimas são conseqüência das diversas proporções dos monômeros em suas moléculas.
A molécula da enzima é constituída por cadeias polipeptídicas (monômeros) diferentes, agrupadas em proporções variáveis.

Izoenzima desidrogenase do ácido láctico
No rato: cada molécula contém quatro cadeias polipeptídicas (monômeros) , de dois tipos:M e H
Conforme a proporção dos monômeros:
1°: 4 cadeias M (M4 H0)
2°: 3 cadeias M + 1 cadeia H (M3 H1)
3°: 2 cadeias M + 2 cadeias H (M2 H2)
4°: 1 cadeia M + 3 cadeias H (M1 H3)
5°: 4 cadeias H (M0 H4)
As cinco desidrogenases forma isoladas puras
Todas atacam o mesmo substrato porém em velocidades diferentes

Biologicamente: a principal distinção entre as izoenzimas é o grau de atividade de cada uma
Um gene: seqüência de aminoácidos de monômeros M
Outro gene: seqüência de aminoácidos de monômeros H
Conforme maior ou menor atividade de cada uma destes genes: maior produção do mRNA para M ou H e os polirribossomos produzirão diferentes quantidades de M e H

Como os monômeros se unem espontaneamente para constituir as enzimas
As atividades de M e H vão depender da atividade daqueles genes
Controle gênico: alterando as proporções dos monômeros produzidos (cadeias polipetídicas), os genes influem na estrutura quaternária das proteínas e podem modular sua atividade enzimática

Células eucariontes: complexo
3 tipos de seqüências nucleotídicas controladoras diferentes: RNA-polimerase
Uma seqüência, ao lado da seqüência codificadora: promotor
Outras duas, distantes: silenciador e reforçador – ativam ou deprimem a transcrição

Transmite para o promotor as informações geradas pelos silenciadores e reforçadores
Diversas proteínas diferentes
Direcionam a RNA-polimerase para os promotores dos genes específicos a serem ativados
Fariam o papel de pontes ligando trechos distantes da cadeia do DNA

A maior quantidade de seqüências reguladoras (silenciadoras e
reguladoras) associadas à grande quantidade de proteínas do
complexo protéico regulador pode explicar a grande diversidade dos
mecanismos reguladores , necessários para orquestrar a
atividade dos 100.000 a 150.000 genes que constituem o genoma
humano.

Três mecanismos:
i. Modulação por fatores protéicos que participam da síntese de proteínas, principalmente os fatores de iniciação
ii. Modulação pela variação e estabilidade do mRNA
iii. Modulação através da inativação do mRNA no citoplasma

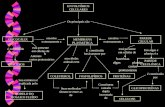
Doenças devidas a mutações em diferentes classes de proteínas
Alterações das proteínas nas doenças genéticas

Mutação↓
Proteína alterada↓
Função anormal↓
Doença

Defeitos Enzimáticos:AminoacidopatiasDefeitos no metabolismo das purinasDoenças do armazenamento lisossômicoDefeitos de proteínas receptorasDefeitos do transporte de membranaDistúrbios que afetam proteínas estruturaisMutações heterogênicas nos genes do colágeno

Anormalidades que afetam as taxas de síntese de mRNA e proteínas
Anormalidades que afetam a estrutura da proteína
Anormalidades que afetam a localização subcelular ou montagem de múltiplas proteínas
Mutações que alteram a associação de proteínas a outras proteínas
Mutações que comprometem a ligação, disponibilidade ou remoção de cofatores ou grupos prostéticos
Mutações que afetam principalmente a função da proteína

Defeitos Enzimáticos

Hiperfenilalaninemias:Hiperfenilalaninemias:
Aumento de fenilalanina no sangue: Fenilcetonúria (PKU)
Fenilcetonúria: protótipo dos erros inatos do metabolismo

Autossômico recessivo do catabolismo da fenilalanina
É resultado: mutação da fenilalanina-hidroxilase
Fenilalanina-hidroxilase: enzima que converte a fenilalanina em tirosina

Fenilananina: Não é degradada por pacientes com PKU
Acumula-se nos líquidos corporais, lesando o SNC em desenvolvimento no início da infância, interferindo na função do cérebro maduro → retardamento mental
Pequena fração: metabolizada por vias alternativas
Produz quantidades aumentadas de ácido fenilpirúvico e outros metabólitos: excretados na urina

Triagem neonatal: Teste do Pezinho
TTO: se precoce, eficaz sem ele, retardamento
intenso inevitável

Síndrome de Lesch-NyhanSíndrome de Lesch-Nyhan
Características: Coreoatetose (distúrbios do movimento) Espasticidade Retardamento mental variável Superprodução de ác. úrico: gota Automutilação
Anormalidade neurológicas: ↑ purinas
Deficiência da enzima ligada ao X hipoxantina-guanina-fosforribosiltransferase (HPRT) – ausência de atividade

Lisossomos: organelas ligadas à membrana
Contêm uma série de enzimas hidrolíticas envolvidas na degradação de várias macromoléculas biológicas
Defeitos genéticos dessas hidrolases: acúmulo dos seus substratos dentro do lisossomo → disfunção → morte celular

Única característica clínica: progressão inexorável (aumento da massa dos tecidos e órgãos afetados)
Cérebro: neurodegeneração
Heterogeneidade clínica e genética Gene: diferentes defeitos em um gene Fenótipos intensos: lactância e distúrbios com início na
idade adulta Lócus: defeitos em enzimas codificadas separadamente
que atuam em etapas diferentes de uma via catabólica Clínica: pequenas variações – atividade enzimática residual

Gangliosidoses GM2 : grupo de doenças do armazenamento lisossômico heterogêneas
Incapacidade de degradar um esfingolipídio: gangliosídio GM2
Lesão bioquímica: deficiência de hexosamina A (hex A)
Impacto clínico: cérebro (local de predominância da síntese de gangliosídio GM2)
Curso clínico: trágico

Lactentes: parecem normais até 3 a 6 meses
Deterioração neurológica progressiva até a morte aos 2 a 4 anos
Efeito da morte celular neuronal: manchas vermelho-cereja na retina
Adulto: disfunção do neurônio motor inferior e ataxia → devido à degeneração espinocerebelar (com visão e inteligências normais)

Mucopolissacarídios (glicosaminoglicanas - GAGs): cadeias de polissacarídios sintetizadas por células do tecido conjuntivo
Mucopolissacaridoses: mucopolissacarídios acumulam-se nos lisossomos como resultado de deficiência de uma das enzimas essenciais à sua degradação
Uma ou mais GAGs acumulam-se se a enzima deficiente for necessária ao seu catabolismo

GAGs não degradadas: urina – detectáveis pelos testes de triagem
“Gargoilismo” – feições grosseiras
Crianças: retardamento mental, anormalidades esqueléticas, baixa estatura
São: Síndrome de Hunter – recessiva ligada ao X Síndrome de Hurler – autossômica recessiva (deficiência de α-L-iduronidase)

Hipercolesterolemia familial:Hipercolesterolemia familial:
Caráter autossômico dominante
Elevação do colesterol plasmático transportado pela LDL
LDL: principal proteína transportadora de colesterol no plasma
Mutações no gene estrutural que codificada o receptor da LDL
Receptor de LDL: proteína de superfície celular responsável pela ligação da LDL e transferência desta para o interior da célula

Hipercolesterolemia familial:Hipercolesterolemia familial:
Hetero e homozigotos: cardiopatia prematura em decorrência de ateromas, xantomas e arcos das córneas.
Ateromas: depósitos de colesterol da LDL nas artérias coronárias
Xantomas: depósitos de colesterol na pele e tendões
Arcos das córneas: depósitos de colesterol ao redor da periferia da
córnea

Fibrose Cística
Distúrbio genético autossômico recessivo fatal
+ comum em crianças (brancas)
Incidência: 1 em 2000 RNs
Freqüência de portadores: 1 em 22

Defeito fisiológico básico: não foi inteiramente determinado
Pulmões e pâncreas
Doença pulmonar obstrutiva crônica (secreções espessas)
Infecções recorrentes
Deficiência de enzimas pancreáticas (lipase, tripsina, quimiotripsina) – impedem a digestão normal
Tto intensivo da doença pulmonar: prolongamento da vida

Digestão e nutrição: restabelecidas por suplementos de enzimas pancreáticas
Morte: insuficiência pulmonar e infecção
Hoje: apenas metade dos pacientes sobrevive até 26 anos de idade
Curso clínico variável

Proteína RTFC: reguladora da condutância transmembrana da FC
Polipetídio codificado pelo gene FC
Defeito bioquímico: desconhecido
Embora: mutações da RTFC devam ser diretamente responsáveis pela FC
Ainda não está claro como são produzidos os defeitos na permeabilidade ao cloro
A função da RTFC não é ainda inteiramente compreendida

Distrofias musculares Duchenne e Becker:
defeitos da distrofina

Severa, intratável e relativamente comum
Associada a uma deterioração clínica inexorável
Gene ligado ao X
Proteína: distrofina (mutação)

Meninos: Normais até 1 ou 2 anos de vida Fraqueza muscular – 3 e 5 anos Dificuldade para subir escadas e levantar-se da posição
sentada Cadeira de rodas: 12 anos Improvável que sobreviva aos 20 anos de idade
Morte: insuficiência respiratória e cardíaca (miocárdio)
CK: elevada – 50 a 100x o limite superior do normal
Cérebro: redução do QI cerca de 20 pontos

Tb: mutações no gene da distrofina
Fenótipo + leve
Pacientes deambulando após 16 anos de idade: DMB
Significativa variação na progressão da doença.

Osteogênese Imperfeita
Grupo de distúrbios hereditários do Colágeno Tipo I
Predispõem o paciente a fraturas ósseas (mesmo traumatismos leves) e deformidade esquelética
Heterogeneidade clínica: devido a qual cadeia do precolágeno Tipo I é afetada e o tipo e a localização da mutação no lócus

Colágeno Tipo I: principal proteína estrutural do osso e outros tecidos fibrosos
+ de 50 mutações
Molécula de procolágeno Tipo I pro α 1 (no cromossomo 17) 2 cadeias pro α 2 ( no cromossomo 7)
Colágeno: helicoidal tríplice

Glicina: único resíduo compacto o bastante para ocupar a posição axial da hélice
Em conseqüência: as mutações que acarretam substituições por outros resíduos são altamente desorganizadoras da estrutura helicóidal

Portanto:
1. Quando a montagem da hélice é diminuída por uma mutação, as seções não montadas das cadeias aminoterminais ao defeito são altamente modificadoras, reduzindo sua secreção para o espaço extracelular e contribuindo para sua instabilidade.
2. A modificação excessiva também pode interferir na formação das fibras de colágeno.

Portanto:
3. Não apenas o n° de fibrilas é reduzido como muitas das fibrilas secretadas são defeituosas.
4. Ossos: cadeias anormais + n° reduzido = mineralização
deficiente

Alterações das proteínas Alterações das proteínas nas doenças genéticasnas doenças genéticas

Anormalidades que afetam as taxas de síntese de mRNA e proteínas
Anormalidades que afetam a estrutura da proteína
Anormalidades que afetam a localização subcelular ou montagem de múltiplas proteínas
Mutações que alteram a associação de proteínas a outras proteínas
Mutações que comprometem a ligação, disponibilidade ou remoção de cofatores ou grupos prostéticos
Mutações que afetam principalmente a função da proteína

Anormalidades primárias da síntese de mRNA ou proteína
Anormalidades secundárias da síntese de mRNA ou proteína

Anormalidades primárias que alteram a conformação da proteína
Anormalidades secundárias que afetam a estrutura da proteína: defeitos nas modificações pós-tradução

Defeitos primários que comprometem o trânsito
Defeitos secundários que comprometem o trânsito

Mutações primárias que comprometem a montagem ou interação das subunidades em proteínas multiméricas
Deficiências funcionais secundárias devidas à ausência de formação de complexos de múltiplas proteínas

Mutações primárias que comprometem a ligação do cofator à proteína
Anormalidades secundárias da função da proteína devidas a inadequação da síntese, transporte, fixação ou remoção de moléculas associadas
Defeitos na síntese ou transporte de moléculas pequenas associadas
Defeitos na fixação ou remoção de ligantes

Anormalidades primáriasnas etapas diretamente dependentes da seqüência de DNA do gene estrutural
Anormalidades secundáriasnos eventos modificadores, ou na síntese de proteínas associadas essenciais à
função
Exemplo da doença Etapa afetada Evento modificador Exemplo da doença
SEQUÊNCIA DE NUCLEOTÍDIOS
Talassemias, nas quais a redução do mRNA resulta de deleções, ou defeitos nos sítios reguladores ou da emendaPHHF: transcrição aumentada
Transcrição, emenda do RNA
↓Regulação da transcrição Porfiria intermitente aguda: drogas que
induzem o citocromo P450 reduzem o heme livre – indução da ALA-sintetase-
sintomas
Talassemias devidas a mRNA não funcionantes com mutações sem sentido ou por mudança da matriz de leitura
Tradução
↓Regulação da síntese da
proteínaPorfiria intermitente aguda: o heme afeta a transcrição e tradução da ALA-
sintetase
Hb Hammersmith: o bolso do heme é deformado → instabilidade da configuraçãoMutantes do receptor da LDL da classe 2°: dobramento anormal
Dobramento do polipeptídio (estrutura secundária e terciária) ↓
Modificações pós-tradução (por ex., glicosilação,
hidroxilação)
Síndrome de Ehlers-Danlos tipo VI: deficiência de lisil-hidroxilase→ colágeno
entrelaçado de maneira insuficiente
Acidúria metilmalônica: defeito da seqüência condutora em um alelo da metilmalonil-CoA-mutase
Localização subcelular devida a informações na seqüência de
aminoácidos ↓Localização subcelular
devida a modificaçõe pós-traducionais do polipeptídio
Doença da Célula I: ausência de acréscimo de um marcador de reconhecimento para enzimas
lisossômicas
Muitos defeitos do colágeno → OI por comprometimento da montagem da hélice de colágenoHb Kansas, Hb Kempsey: interação das subunidades comprometida
Para proteínas multiméricas:- associação de subunidades- interação das subunidades ↓ Formação de complexos
de múltiplas proteínas e organelas
Síndrome de Zellweger, um defeito da biogênese dos peroxissomos
Deficiência de cistationa-sintase: ligação fraca ao piridoxal-fosfato→ homocistinúria em 50% dos pacientes
Ligação (não covalente ou covalente) a cofator ou grupo prostético ↓
Síntese ou transporte do cofator ou grupo prostético
Ligação ou remoção (covalente) do cofator
Mutações no metabolismo da vitamina B12 → acidúria metilmalônica, homocistinúria, ou ambasDeficiência de holocarboxilase-sintase; deficiência de biotinidase
Alelo B1 da doença de Tay-Sachs: as mutações podem afetar apenas a função da proteína, como mutações no sítio ativo de enzimasDeficiência de G6PD, variante A: muitas mutações alteram a conformação →instabilidade→degradação→material de reação cruzada reduzido
Degradação proteolítica↓ Regulação da degradação
da proteínaNenhum exemplo conhecido até o
presente
RNA MENSAGEIRO
POLIPEPTÍDIO NÃO DOBRADO
CONFORMAÇÃO TRIDIMENSIONAL
LOCALIZAÇÃO E MONTAGEM
FUNÇÃO BIOLÓGICA
PROTEÍNA DEGRADADA
*Adaptado de Thompson & Thompson, 1993
Quadro 12.9 Como as proteínas são alteradas nas doenças genéticas *

1. Mutações com perda da função
2. Mutações com ganho da função
3. Mutações com propriedades novas
4. Mutações nas proteínas específicas de tecidos
5. Mutações nas proteínas de manutenção

Fim !