
Roteiros de Aulas de Laboratório de QAnal 2015.1
67
Roteiros de Aulas de Química Analítica 3ª Edição Profº Ricardo França Furtado da Costa CORONEL FABRICIANO / 1° semestre 2015
-
Upload
jamyla-soares -
Category
Documents
-
view
56 -
download
10
description
Práticas de química analítica
Transcript of Roteiros de Aulas de Laboratório de QAnal 2015.1

Roteiros de Aulas de
Química Analítica
3ª Edição Profº Ricardo França Furtado da Costa
CORONEL FABRICIANO / 1° semestre 2015

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
i
SUMÁRIO
Informações Gerais ........................................................................................................... 2
Aula 1 - Preparação e aferição (padronização) de Soluções ............................................... 4
Aula 2 - Introdução à estatística na química analítica e construção de gráficos .................. 8
Aula 3 - Determinação de ácido acético em vinagre. ......................................................... 22
Aula 4 - Determinação da concentração de ácido fosfórico. .............................................. 23
Aula 5 - Determinação de ácido acetilsalicílico em medicamento. .................................... 24
Aula 6 - Determinação do teor de hidróxido de magnésio no leite de magnésia (titulação de retorno). ........................................................................................................................ 25
Aula 7 - Padronização de solução de AgNO3 e determinação de cloreto. (método de Mohr) ................................................................................................................................. 26
Aula 8 - Determinação de cloreto. (método de Volhard) .................................................... 29
Aula 9 - Volumetria de complexação: padronização de solução de ácido etilenodiaminotetraacético (EDTA) e quantificação de metais ........................................... 31
Aula 10 - Volumetria de complexação: Determinação de cálcio na casca do ovo e no leite ........................................................................................................................................... 34
Aula 11 - Volumetria de complexação: Determinação de dureza da água ........................ 35
Anexo 1 - Normas de segurança e boa conduta em laboratórios de química.................... 38
Anexo 2 - Equipamentos Básicos de Laboratório .............................................................. 41

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
2
Informações Gerais
Introdução
As atividades propostas para a disciplina Química Analítica além de proporcionar ao aluno maior autonomia para trabalhar com segurança em um laboratório de química, visam também a verificar diversos métodos analíticos de determinação quantitativa. Dessa forma o aluno estará mais responsável pelo andamento das aulas. Procurar-se-á, para isto, não apenas desenvolver a habilidade no manuseio de reagentes e aparelhagens, mas também criar condições para uma avaliação crítica dos experimentos realizados.
Dinâmica das Aulas Práticas
1. Não desperdice tempo vindo ao laboratório sem estar preparado. Prepare-se para cada experimento estudando-o (tanto a Preparação pré-laboratório, quanto, rapidamente, o Procedimento experimental) antes de chegar ao laboratório.
2. Use sua criatividade e seu bom-senso. Embora as instruções de laboratório sejam bem especificas, elas dão ampla oportunidade para o raciocínio claro, lógico, original e imaginativo. Essa atitude é um pré-requisito para qualquer esforço cientifico.
3. Prepare cuidadosamente seus Relatórios de laboratório para cada experimento. Use um caderno (ou sua apostila) para fazer suas anotações. Todos os dados devem ser registrados diretamente nele, não em folhas soltas ou pedaços de papel. Quando precisar fazer cálculos, faça um cálculo bem organizado para o primeiro conjunto de dados, mas não confunda a parte de cálculos com detalhes aritméticos. Da mesma forma, enquanto você estiver fazendo o experimento, pense e faça perguntas significativas, que lhe deem compreensão dos princípios nos quais o procedimento experimental está fundamentado.
4. Os cientistas aprendem muito discutindo seu trabalho uns com os outros. Você pode, da mesma forma, beneficiar-se de discussões com seus colegas, mas não copie trabalhos. A integridade é a chave do trabalho cientifico. Talvez seja útil também consultar seu próprio texto enquanto estiver trabalhando no laboratório (livros geralmente são fontes de informação ainda mais confiáveis e completas do que seus colegas de classe).
5. Para dados tabelados sobre as propriedades das substâncias, você pode consultar manuais como o Handbook of Chemistry and Physics (CRC Press) ou outros livros na biblioteca da escola.
6. Resumidamente, em nossas aulas haverá uma discussão inicial, com o professor, dos aspectos teóricos e práticos relevantes, seguida da execução pelos alunos dos experimentos utilizando guias práticos e interpretação e discussão dos resultados juntamente com o professor, e, de acordo com os roteiros, apresentação dos resultados em forma de relatório ou semelhante na próxima aula.
Normas de segurança
Familiarize-se com as normas de segurança já vistas nas disciplinas de laboratório de química anteriormente. Para a sua segurança, e a dos outros no laboratório, é essencial que você siga cuidadosamente esses regulamentos, com as modificações ou acréscimos feitos pelo seu instrutor.
Seu instrutor lhe indicará o local dos extintores de incêndio, lava-olhos, chuveiro de emergência e suprimentos de primeiros-socorros. O instrutor também lhe dirá onde você pode obter óculos de segurança.
No Anexo 1 estão listadas algumas normas de segurança que são essenciais para realizar um bom trabalho no laboratório de química.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
3
No Anexo 2 encontram-se diversas ilustrações com os respectivos nomes e funções de diversos equipamentos laboratoriais.
Avaliação
Ao longo do curso, o aluno será avaliado da seguinte forma:
• Participação;
• Questionários;
• Relatórios;
• Trabalhos avaliativos em grupo e/ou individuais;
• Avaliações teóricas individuais;
• Avaliações práticas em grupo.
Procurar-se-á avaliar:
• A correção e clareza na redação de relatórios e questionários;
• A capacidade para trabalhar com independência e eficiência durante as aulas práticas;
• O aproveitamento na associação de conceitos teóricos e práticos através de testes e exercícios escritos;
• Cumprimento dos prazos determinados.
A distribuição detalhada dos pontos consta do plano de aula e cronograma do curso, apresentado ao aluno no início do semestre, que pode ser colado abaixo.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
4
Aula 1 – Preparação e aferição (padronização) de soluções
Objetivos
• Preparar soluções padrão de ácido clorídrico e hidróxido de sódio para posterior aferição ou padronização.
• Calcular massas e/ou volumes necessários para a preparação dessas soluções nas concentrações e volumes indicados.
• Padronizar soluções.
• Calcular o fator de correção (Fc) para as soluções após a padronização
Preparação pré-laboratório
Tanto na área científica como tecnológica (na indústria), frequentemente é necessário determinar a composição e a concentração de um determinado sistema.
Na análise Volumétrica, o reagente de concentração conhecida é o titulante e a substância de concentração desconhecida é o titulado. Assim o processo de se juntar a solução padrão até que a reação esteja completa é a titulação.
O hidróxido de sódio é um padrão secundário por ser uma substância higroscópica, o que pode dificultar a pesagem com exatidão de uma determinada massa dessa substância. Além disso, a substância pode estar contaminada com carbonato de sódio, Na2CO3, pela absorção de dióxido de carbono atmosférico.
O ácido clorídrico concentrado para análise (P.A.) não é substância padrão, pois a sua concentração é função de sua densidade, e varia com o tempo. A solução 0,1 M é preparada pela diluição de certo volume de ácido concentrado. Por ser padrão secundário, a solução precisa ser titulada com uma das substâncias de padrão primário: Bórax (tetraborato de sódio, Na2B4O7. 10 H2O) ou carbonato de sódio anidro (Na2CO3) ou indiretamente com a utilização de uma solução padrão secundário.
O carbonato de sódio anidro é um produto químico adequado para preparar uma solução-padrão (como um padrão primário).
Para conhecermos a concentração real de uma solução de NaOH ou de HCl, devemos titular estas soluções, assim que preparadas, com uma solução de um padrão primário. Para o NaOH utilizamos um ácido, geralmente o biftalato de potássio, sendo que o ácido benzóico, C6H5COOH e o ácido oxálico, HOOCCOOH·2 H2O, também são usados. Para o HCl , podemos utilizar o Na2CO3(s), carbonato de sódio, que em solução libera íons CO3
2–, ou seja, uma base. Podem ser usados também o tetraborato de sódio, Na2B4O7·10 H2O e o oxalato de sódio, Na+ –OOCCOO–Na+ (Na2C2O4).
Os padrões primários devem preencher os seguintes requisitos:
• ser de composição conhecida e de alto teor de pureza, acima de 99,0 %,
• devem ser de baixo custo, fácil obtenção, purificação e secagem,
• não devem ser higroscópicos (não devem absorver água do ambiente),
• devem ter o maior peso molecular possível, para minimizarmos os erros de pesagem,
• deve ser completamente solúvel no meio utilizado para titulação.
Soluções padrão
As soluções padrão são soluções com concentração exatamente conhecida. A exatidão da concentração da solução padrão limita a exatidão do método analítico empregado. Uma solução padrão primário é aquela preparada pela dissolução de uma determinada massa de padrão primário e diluição a volume definido (através de um balão volumétrico), permitindo assim que a concentração seja exatamente calculada.
No entanto, como o número de substâncias padrões primários é limitado, faz-se uso de soluções padrões secundárias. As soluções de HCl e NaOH podem ser padrões secundários, desde que façamos uma padronização (titulação) com uma solução padrão primário. Quando temos as concentrações destas soluções com concentrações conhecidas exatamente, podemos então utilizá-las em outras titulações ácido-base.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
5
Fator de Correção (Fc)
O fator de correção é um valor usado para corrigir a solução. Erros de pesagem e aferição de balão sempre acontecem, então o fator de correção é usado para minimizar os erros durante os cálculos.
Deve-se utilizar a quantidade de matéria, em mols, do padrão primário e pode-se utilizar a fórmula:
FCp x Cp x Vp = nr
onde: nr = quantidade de matéria, em mols, da substância padrão primário
(n = massa x concentração em mol/L) FCp = fator de correção da solução problema (desconhecido) Cp = concentração da solução problema Vp = volume da solução problema utilizado
Se ao invés da solução utilizarmos diretamente a massa do padrão primário, deve-se fazer uma solução com um padrão primário e proceder uma titulação normal, sendo o titulante a solução de concentração conhecida. Anota-se o valor gasto e procede fazendo os cálculos:
FCp x Cp x Vp = FCr x Cr x Vr
onde: FCr = fator de correção da solução padrão primário Cr = concentração da solução padrão primário Vr= volume da solução padrão primário utilizado na titulação
O fator de correção é um número adimensional, ou seja, não tem unidade e quanto mais próximo de 1,00 for o seu valor, mais correta está a concentração da sua solução preparada.
Materiais e Reagentes
• Bureta • balão volumétrico 1000 mL • 3 béquer 100 mL • balão volumétrico 500 mL • bastão de vidro • água destilada
• pera • pipeta graduada de 5 mL e 10 mL • pisseta • bico de Bunsen • 2 vidros de relógio • tripé com tela • espátula • balança analítica • ácido sulfúrico (PA) (H2SO4) • carbonato de sódio anidro (Na2CO3) • hidróxido de sódio PA (NaOH) • fenolftaleína
• biftalato de potássio • alaranjado de metila • vidro para armazenar soluções ácidas 1000 mL • ácido clorídrico PA (HCl)
• recipiente plástico para armazenar soluções de hidróxidos alcalinos 1000 mL
Procedimento
1. Solução de hidróxido de sódio (NaOH) 0,1 M, 1 L
1.1. Pesar no vidro de relógio ou diretamente no béquer (depende do tamanho do último), a quantidade necessária para preparar 1 litro de solução de NaOH na concentração de 0,1 M.
1.2. Dissolver com o auxílio de um bastão de vidro e água fervida ou recentemente destilada. 1.3. Transferir, quantitativamente, para o balão volumétrico de 1 litro e completar até o menisco com
água fervida ou recentemente destilada. 1.4. Após preparar a solução, transferi-la para o recipiente de plástico adequado e rotulado.
Atenção: cuidado com a ordem de colocação do ácido e da água

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
6
2. Solução de ácido clorídrico (HCl) 0,2 M, 500 mL
2.1. Colocar cerca de 100 mL de água destilada no balão volumétrico. 2.2. Transferir para o balão volumétrico o volume adequado do ácido concentrado para preparar 500 mL
de solução 0,2 M. 2.3. Completar o volume com água destilada até o menisco. 2.4. Após o preparar a solução, transferi-la para o recipiente de vidro adequado e rotulado.
3. Solução de ácido sulfúrico (H2SO4) 0,1 M, 250 mL
3.1. Colocar cerca de 100 mL no balão volumétrico. 3.2. Transferir para o balão volumétrico o volume adequado do ácido concentrado para preparar 250 mL
de solução 0,1 M. 3.3. Completar o volume com água destilada até o menisco. 3.4. Transferir a solução para o recipiente de vidro adequado e rotulado.
Procedimentos e cuidados na preparação
Observações:
• Ácidos NÃO devem ser pipetados com a boca, sempre pipetar com auxílio de peras ou pipetadores. Devem ser manipulados na capela em função dos vapores irritantes e corrosivos. Sempre adicionar o ácido concentrado sobre a água. • Rotular os frascos, de preferência, antes de transferir a solução. O rótulo deve conter nome da substância, concentração da solução, identificação do preparador e data do preparo.
4. Padronização de solução de NaOH 0,1 M com padrão primário
4.1. Calcular a massa de biftalato de potássio (C6H4(CO2H)(CO2K) - padrão primário, mm = 204,22 g) necessária para reagir com 25 mL de NaOH 0,1 M.
4.2. Medir a massa calculada de biftalato de potássio, com auxílio de uma espátula e transferir para um erlenmeyer de 250 mL, diluindo a aproximadamente 50 mL com água destilada. A massa medida não precisa ser exata, mas deve-se anotar o valor exato medido, pois as correções serão realizadas nos cálculos
4.3. Adicionar três gotas de fenolftaleína e titular com solução de hidróxido de sódio até o aparecimento da coloração (permanente) levemente rósea. Anotar o volume gasto na titulação. Fazer a análise em triplicata.
5. Padronização de solução de H2SO4 0,1 M com padrão secundário
5.1. Transferir 10,0 mL da solução do ácido preparado, com auxílio de uma pipeta volumétrica, para um erlenmeyer de 250 mL
5.2. Acrescentar cerca de 40,0 mL de água destilada. 5.3. Adicionar três gotas de fenolftaleína. 5.4. Titular com solução padronizada de hidróxido de sódio. 5.5. Anotar o volume gasto na titulação. 5.6. Fazer a análise em duplicata.
6. Padronização de solução de HCl 0,2 M com padrão secundário
6.1. Transferir 10,0 mL da solução do ácido preparado, com auxílio de uma pipeta volumétrica, para um erlenmeyer de 250 mL.
6.2. Acrescentar cerca de 40,0 mL de água destilada. 6.3. Adicionar três gotas de fenolftaleína. 6.4. Titular com solução padronizada de hidróxido de sódio. 6.5. Anotar o volume gasto na titulação. 6.6. Fazer a análise em duplicata.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
7
7. Padronização de solução de HCl 0,2 M com padrão primário
7.1. Pesar duas amostras de aproximadamente 0,265 gramas do padrão primário (Na2CO3) (valor próximo da massa necessária para reagir completamente com um volume correspondente a, aproximadamente, metade da capacidade da bureta, 25 mL). Anotar o valor exato para a realização dos cálculos posteriores.
7.2. Transferir, quantitativamente a massa pesada para um erlenmeyer de 250 mL. Como são duas amostras, significa que serão feitas em duplicata.
7.3. Adicionar cerca de 100 mL de água destilada e agitar até completa dissolução do sal. 7.4. Adicionar 2 gotas de solução de alaranjado de metila e titular a solução com HCl até que a coloração
comece a desviar da que o indicador apresentava inicialmente. 7.5. Aquecer até o início da ebulição e, após resfriar, continuar titulando até que a solução se torne
alaranjada (sem reflexo de amarelo). 7.6. Calcular a concentração de HCl em mol.L-1 pela média dos resultados (desde que os valores estejam
dentro do limite de erro aceito).

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
8
Aula 2 - Introdução à estatística na química analítica e construção de gráficos
Objetivos
• Realizar um plano de amostragem, empregando confeitos de chocolate.
• Definir as condições experimentais para reduzir a amostra bruta para amostra laboratorial.
• Ilustrar uma distribuição normal através da criação de distribuição de frequências e de histogramas a partir de dados tabelados.
• Realizar cálculos estatísticos para dados experimentais.
• Criar gráficos.
Preparação pré-laboratório
1. Amostragem
“Esta prática é uma adaptação de uma proposta da Profa. Suzane Rath, do Instituto de
Química da UNICAMP (CANAES, L.S. et al. Using Candy Samples to Learn About Sampling Techniques
and Statistical Data Evaluation. Journal of Chemical Education, Washington, DC, v. 8, n. 8, p. 1083-
1088, ago./2008).”
A validade das conclusões derivadas da análise de uma amostra depende, entre outras coisas, dos métodos empregados na obtenção e preservação da amostra.
Sabe-se que a amostragem é a uma das maiores fontes de erro na análise química e, por isso uma amostra deve ter todas as propriedades do material original.
As principais etapas na amostragem compreendem:
• Identificação da população de onde a amostra vai ser retirada. • Seleção e obtenção da “amostra bruta”.
• Redução da “amostra bruta” à amostra laboratorial. • Para efeitos de controle, será utilizada a embalagem azul de 18 g do
confeito de chocolate do tipo “disquete” da marca Dori. Um valor teórico da distribuição média das cores das pastilhas por
pacote é apresentado na Tabela 1 abaixo.
Tabela 1: Valor teórico para a distribuição média das cores de disquete para uma embalagem azul de 18 g.
Cor Vermelho Rosa Amarelo Roxo Azul Verde Laranja
Quant 5 3 3 3 2 2 2 % 25,0 15,0 15,0 15,0 10,0 10,0 10,0
2. Erros em análises químicas
Erros e incertezas: alguns são provenientes a equívocos cometidos pelo analista (padronização). 2.1. Tipos de erros:
a) Erros aleatórios ou indeterminados: afetam a precisão da medida. Não podem ser totalmente eliminados. Não podem ser claramente identificados
b) Erros sistemáticos ou determinados: afetam a exatidão do resultado. Podem ser tanto: constantes (erro absoluto é constante e o erro relativo varia) como proporcionais (erro absoluto varia e o erro relativo é constante)
c) Erros instrumentais: calibrações (equipamentos volumétricos), condições inadequadas. d) Erros de método: proveniente do comportamento químico ou físico não ideal, como
decomposição da amostra ou reagente, tempo de reação (amostras padrões certificadas, preparação de brancos).
Figura 1. Amostra

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
9
e) Erros pessoais: falta de cuidado, atenção ou limitação do analista (amostras padrões certificadas, triplicatas).
3. Precisão e Exatidão
3.1. Precisão: descreve a reprodutibilidade das medidas (concordância), ou seja, proximidade dos resultados obtidos. Pode ser avaliada através do desvio padrão, variância e do coeficiente de variação:
3.2. Exatidão: proximidade da medida com o valor verdadeiro. Pode ser expressa em termos de erro absoluto ou erro relativo.
4. Erro absoluto (E) e erro relativo (Er):
Exemplo: Um analista determinou a concentração de ferro em uma amostra de concentração
conhecida (20,00 mg/L), obtendo-se os seguintes resultados:19,4; 19,5; 19,6; 19,8; 20,1 e 20,3. a) Valor médio: 19,78 mg/L (x = 19,8); b) Mediana: 19,7 (número par de determinações: valor médio entre as análises 3 e 4); c) Erro absoluto: para a amostra 20,1 mg/L, temos: xv = 20,0, seu erro absoluto é +0,1 mg/L de Fe. Para a concentração de 19,5 mg/L, seu erro absoluto é – 0,5 mg/L. d) Erro relativo da leitura 19,8 mg/L é: [(19,8 – 20,0)/20,0]x100 = -1%, ou seja, o valor determina-do apresenta 1% de erro em relação ao valor verdadeiro.
5. Desvio padrão da amostra: empregado como uma medida da precisão.
Outra forma de calcular o desvio padrão em calculadoras que não possuem a função desvio padrão, S.
� = �∑ ����� − �∑ ���� � �� − 1
6. Coeficiente de variância (CV): desvio padrão relativo do porcentual da média fornece uma imagem mais clara da qualidade dos dados que os desvios padrão absolutos.
7. Precisão da medida ou erro padrão da média, de uma série de medidas (N), é inversamente proporcional ao desvio padrão pela raiz quadrada de N:
Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
10
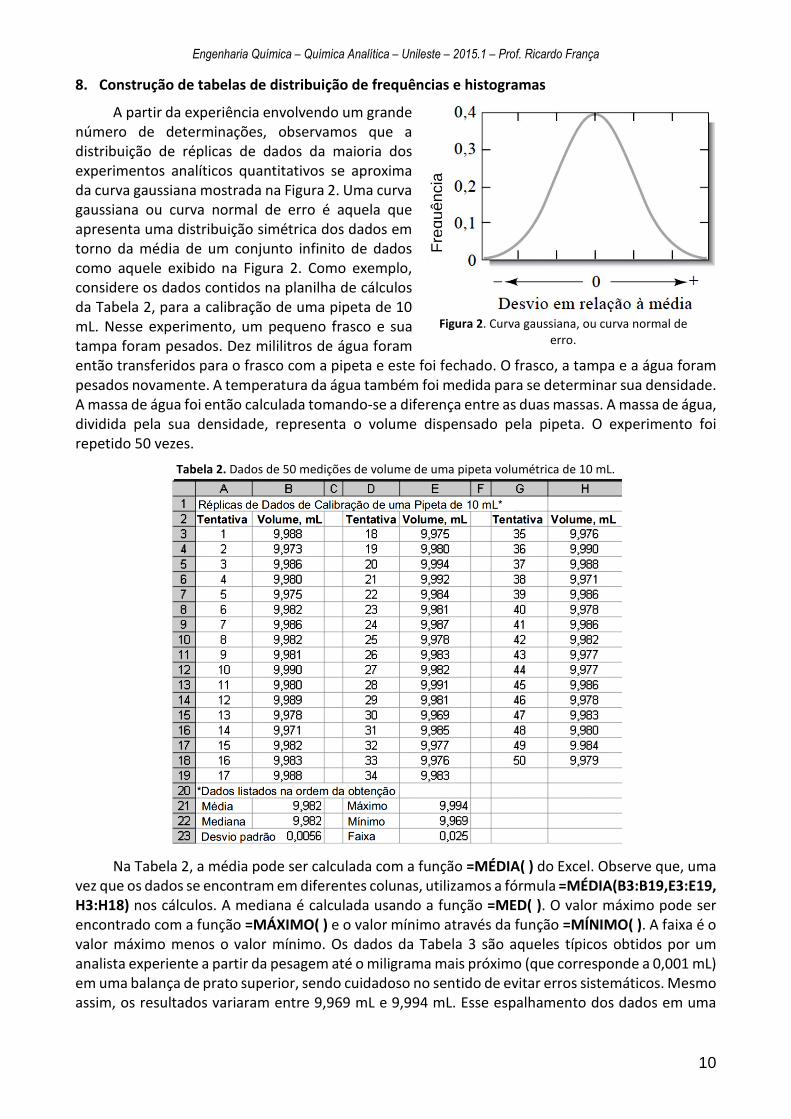
8. Construção de tabelas de distribuição de frequências e histogramas
A partir da experiência envolvendo um grande número de determinações, observamos que a distribuição de réplicas de dados da maioria dos experimentos analíticos quantitativos se aproxima da curva gaussiana mostrada na Figura 2. Uma curva gaussiana ou curva normal de erro é aquela que apresenta uma distribuição simétrica dos dados em torno da média de um conjunto infinito de dados como aquele exibido na Figura 2. Como exemplo, considere os dados contidos na planilha de cálculos da Tabela 2, para a calibração de uma pipeta de 10 mL. Nesse experimento, um pequeno frasco e sua tampa foram pesados. Dez mililitros de água foram então transferidos para o frasco com a pipeta e este foi fechado. O frasco, a tampa e a água foram pesados novamente. A temperatura da água também foi medida para se determinar sua densidade. A massa de água foi então calculada tomando-se a diferença entre as duas massas. A massa de água, dividida pela sua densidade, representa o volume dispensado pela pipeta. O experimento foi repetido 50 vezes.
Tabela 2. Dados de 50 medições de volume de uma pipeta volumétrica de 10 mL.
Na Tabela 2, a média pode ser calculada com a função =MÉDIA( ) do Excel. Observe que, uma vez que os dados se encontram em diferentes colunas, utilizamos a fórmula =MÉDIA(B3:B19,E3:E19, H3:H18) nos cálculos. A mediana é calculada usando a função =MED( ). O valor máximo pode ser encontrado com a função =MÁXIMO( ) e o valor mínimo através da função =MÍNIMO( ). A faixa é o valor máximo menos o valor mínimo. Os dados da Tabela 3 são aqueles típicos obtidos por um analista experiente a partir da pesagem até o miligrama mais próximo (que corresponde a 0,001 mL) em uma balança de prato superior, sendo cuidadoso no sentido de evitar erros sistemáticos. Mesmo assim, os resultados variaram entre 9,969 mL e 9,994 mL. Esse espalhamento dos dados em uma
Figura 2. Curva gaussiana, ou curva normal de erro.
Fre
quên
cia

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
11
faixa de 0,025 mL resulta diretamente do acúmulo de todas as incertezas aleatórias envolvidas no experimento.
A informação contida na Tabela 2 é mais facilmente visualizada se os dados forem rearranjados em grupos de distribuição de frequência, como na Tabela 3. Nesse caso agrupamos o número de dados que se encontram em séries de faixas adjacentes de 0,003 mL e calculamos o percentual de medidas contidas em cada faixa. Observe que 26% dos resultados ocorrem na faixa de volume entre 9,981 e 9,983 mL. Este é o grupo que contém os valores médio e mediano de 9,982 mL. Observe também que mais da metade dos resultados estão na faixa de ±0,004 mL dessa média.
Tabela 3. Distribuição dos dados da Tabela 2.
Os dados da distribuição de frequência da Tabela 3 estão representados como um gráfico de barras, ou histograma (indicado pela letra A na Figura 3). Podemos imaginar, com o aumento do número de medidas, que o histograma aproxima-se do formato de uma curva contínua, apontada como a curva B na Figura 3. Este gráfico mostra uma curva gaussiana, ou curva de erro normal, que se aplica a um conjunto infinitamente grande de dados.
A curva gaussiana tem a mesma média (9,982 mL), a mesma precisão e a mesma área sob a curva que o histograma.
Figura 3. Histograma (A) mostrando a distribuição de 50 resultados contidos na Tabela 4 e uma curva gaussiana (B) para os dados, tendo a mesma média e desvio padrão que os dados do histograma.
As variações em medidas de réplicas, como aquelas indicadas na Tabela 2, resultam de numerosos erros aleatórios pequenos e individualmente indetectáveis que são atribuídos a

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
12
variáveis incontroláveis associadas ao experimento. Esses pequenos erros normalmente tendem a cancelar uns aos outros, tendo assim um efeito mínimo sobre o valor médio. Ocasionalmente, entretanto, ocorrem na mesma direção, para produzir um grande erro líquido positivo ou negativo.
As fontes de incertezas aleatórias na calibração de uma pipeta incluem (1) julgamentos visuais, tais como o nível de água em relação à marca na pipeta e ao nível de mercúrio no termômetro; (2) variações no tempo de escoamento e no ângulo da pipeta, durante seu escoamento; (3) flutuações na temperatura, que afetam o volume da pipeta, a viscosidade do líquido e o desempenho da balança; e (4) vibrações e correntes de ar que causam pequenas variações nas leituras da balança. Indubitavelmente, existem muitas outras fontes de incertezas aleatórias nesse processo de calibração que não listamos aqui. Mesmo o processo simples de calibração de uma pipeta é afetado por muitas variáveis pequenas e incontroláveis. A influência cumulativa dessas variáveis é responsável pela distribuição dos resultados em torno da média.
9. Construindo gráficos
Em muitas circunstâncias a meta de realização das medições é descobrir ou estudar a relação entre duas variáveis. A pressão e o volume de um gás, o volume e a temperatura de uma substância ou a cor de uma solução e a intensidade dessa cor são exemplos de conjuntos de variáveis relacionadas. Quando uma variável muda, a outra também muda.
Muitas vezes empregamos gráficos para visualizar a relação entre duas variáveis. Se existem duas variáveis, o gráfico será um plot bidimensional dos pontos que representam pares de valores dessas duas variáveis. Um exemplo de um gráfico bem desenhado está mostrado na Figura 4. Observe que o gráfico tem vários aspectos que ajudam a esclarecer seu significado.
Figura 4. Este é um exemplo de um gráfico bem feito. Observe a clareza do título, como as legendas dos eixos são adequadas, com os nomes das variáveis e suas unidades, os pontos dos dados visivelmente marcados e a curva suave, mostrando qual a tendência observada e qual a tendência extrapolada.
9.1. Características de um gráfico
1. Título. O título de um gráfico deve ser breve, mas suficiente para fornecer uma descrição clara da relação em estudo. Títulos como ”Laboratório número 1" ou “Volume e temperatura” não são aceitáveis porque o significado fica claro apenas para aqueles que participaram do experimento e será perdido a medida que for esquecido com a passagem do tempo.
2. Eixos rotulados. Cada eixo do gráfico deve ser claramente rotulado para mostrar as quantidades que ele representa e as unidades empregadas para medir as quantidades. Deve-se distinguir entre a quantidade medida (pressão, volume, temperatura, tempo etc.), e as unidades usadas para medir essa quantidade (atmosferas, litros, graus Celsius, segundos etc.).
É conveniente rotular cada eixo com o nome da quantidade medida seguida pela unidade, separando a unidade (geralmente abreviada) da quantidade com uma barra, por exemplo, Volume/L. Portanto, somente números precisam aparecer ao lado de cada eixo, e os eixos não
Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
13
precisam ser poluídos com as unidades em cada marcação. De fato, a barra significa que as variáveis representadas pelos números com unidades foram divididas pelas unidades, deixando números adimensionais marcados ao longo dos eixos.
3. Escalas. A escala de cada eixo deve ser escolhida cuidadosamente, de tal forma que a faixa de variação total dos valores possa ser marcada no gráfico. Por razões práticas 2, 4, 5 ou 10 divisões num gráfico de papel devem representar uma unidade decimal da variável. Essa equivalência facilitará estimar valores que caiam entre as divisões da escala. Para maior exatidão e proporções mais agradáveis, as escalas selecionadas devem ser escolhidas de tal forma que o gráfico praticamente ocupe a página toda. Assegure-se, entretanto, de que nenhum ponto marcado saia das margens do gráfico.
Observe que o canto inferior esquerdo do gráfico não precisa representar zero em nenhum dos eixos. Se a amplitude dos valores marcados se estender para zero, essa pode ser uma boa escolha, mas se não, haverá muito espaço desperdiçado no gráfico.
4. Pontos de dados. É considerado útil marcar a localização de cada dado com um ponto bem pequeno e então fazer um círculo em torno dele para torna-lo mais visível.
5. A curva. Uma curva suave deve ligar os pontos. Ela deve passar o mais perto possível de cada ponto, mas eles não devem ser ligados uns aos outros com curtos segmentos de reta. Se a relação aparentar ser linear, a curva suave deve ser uma linha reta. Se a linha se estender além da amplitude dos valores medidos, essa extensão deve ser indicada por uma linha tracejada e não por uma linha cheia.
9.2. Relações Lineares
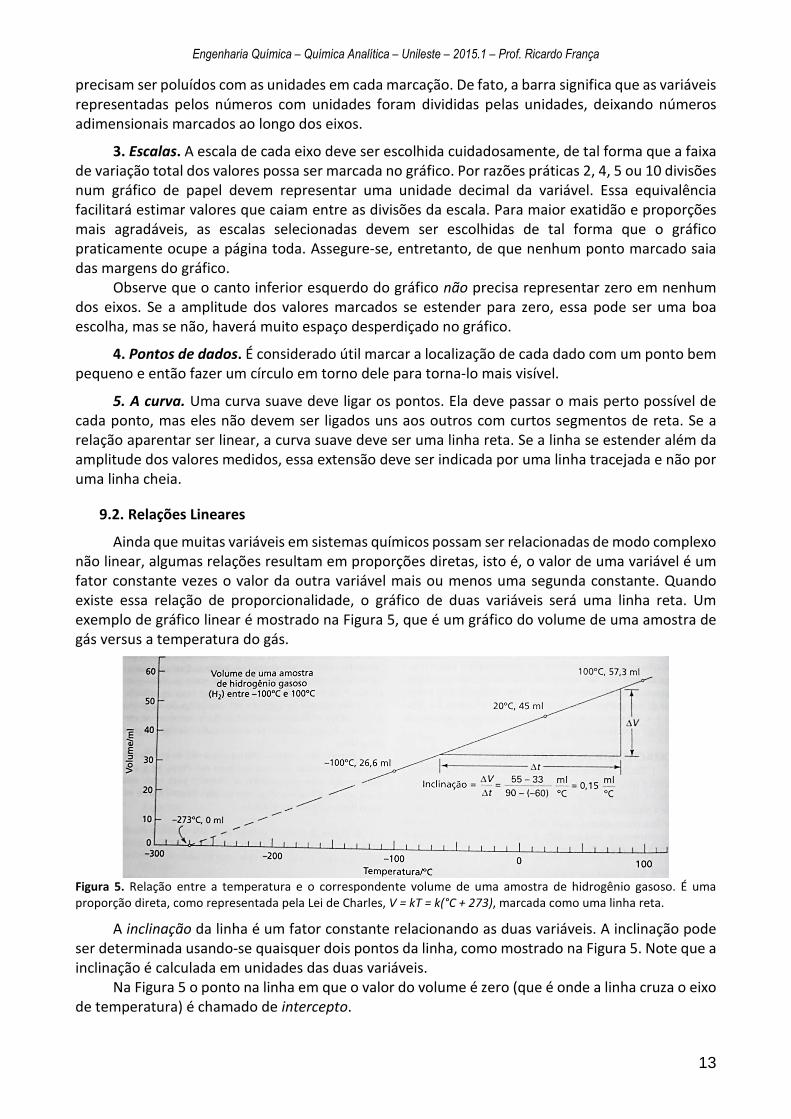
Ainda que muitas variáveis em sistemas químicos possam ser relacionadas de modo complexo não linear, algumas relações resultam em proporções diretas, isto é, o valor de uma variável é um fator constante vezes o valor da outra variável mais ou menos uma segunda constante. Quando existe essa relação de proporcionalidade, o gráfico de duas variáveis será uma linha reta. Um exemplo de gráfico linear é mostrado na Figura 5, que é um gráfico do volume de uma amostra de gás versus a temperatura do gás.
Figura 5. Relação entre a temperatura e o correspondente volume de uma amostra de hidrogênio gasoso. É uma proporção direta, como representada pela Lei de Charles, V = kT = k(°C + 273), marcada como uma linha reta.
A inclinação da linha é um fator constante relacionando as duas variáveis. A inclinação pode ser determinada usando-se quaisquer dois pontos da linha, como mostrado na Figura 5. Note que a inclinação é calculada em unidades das duas variáveis.
Na Figura 5 o ponto na linha em que o valor do volume é zero (que é onde a linha cruza o eixo de temperatura) é chamado de intercepto.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
14
Havendo relação entre as duas variáveis, é possível estimar o valor que uma variável teria para qualquer valor da outra variável usando-se o gráfico. Se o ponto de interesse ficar dentro da amplitude dos valores medidos, essa estimativa chama-se interpolação. Por exemplo, na Figura A-1 podemos determinar por interpolação que a 125°C a pressão de vapor da água é de aproximadamente 1800 torr.
Se a estimativa for feita além da amplitude das medidas, o processo chama-se extrapolação. Na Figura 5 podemos ver que a pressão de vapor da agua a 155°C é de 4000 torr, aproximadamente.
A interpolação e a extrapolação são técnicas úteis, mas ambas são estimativas e assumem que o gráfico seja exato ou que ele se estenda além dos valores medidos. Especialmente na extrapolação essa hipótese pode conduzir a conclusões incorretas
Materiais e Reagentes
• Pacote de 18 g de Disquetes • Moeda
• Calculadora (alunos) • Barbante
• Béqueres • Paquímetro
• Régua • Pipeta volumétrica 10 mL
• Soluções de sacarose a 8% e 16 % • Balança analítica
Procedimento experimental
1. Estatística dos confeitos de chocolate
1.1. Contar o número individual de cada componente do grupo e o total de confeitos no pacote e determinar a quantidade por cor. Anotar os resultados na tabela na parte de “Resultados Experimentais”.
1.2. Transformar todos os resultados do item 1 em porcentagem. Representar o resultado com o número correto de algarismos significativos.
1.3. Verificar se o valor obtido para cada cor difere significativamente do valor teórico do fabricante (erro relativo).
1.4. Calcular o valor percentual de todos os resultados da turma (para cada cor) e a estimativa do desvio padrão.
1.5. Calcular o valor percentual geral, somando os resultados da turma com os resultados de outras turmas (para cada cor) apresentados na Tabela 4 abaixo e a estimativa do desvio padrão.
Tabela 4: Valor percentual total de todas as turmas.
Cor Vermelho Rosa Amarelo Roxo Azul Verde Laranja Total
Quant 500 380 280 380 400 260 200 2400 % 20,8 15,8 11,8 15,7 16,7 10,8 8,4 100%
2. Cara ou coroa
2.1. Jogar uma moeda 10 vezes e anotar os resultados em função do número de caras. 2.2. Repetir o procedimento para cada componente do grupo e anotar na tabela na parte
“Resultados Experimentais”, no item 2.2 2.3. Somar os resultados com os de sua turma e anotar na tabela na parte “Resultados
Experimentais” no item 2.3 2.4. Somar os resultados com os de outras turmas (Tabela 5) anotar na tabela na parte
“Resultados Experimentais” no item 2.4 2.5. Construir uma tabela de distribuição de frequências e um histograma para a média do seu
grupo, para a média de sua turma e para o de sua turma somado ao total de outras turmas (Tabela 5).

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
15
Tabela 5. Resultados experimentais de várias turmas.
Número de caras 0 1 2 3 4 5 6 7 8 9 10 Frequência 1 1 22 42 102 104 92 48 22 7 1
3. Fazendo um gráfico – Circunferência versus. diâmetro de béqueres
3.1. Usando um pedaço de barbante, medir a circunferência de três béqueres diferentes. Puxe o barbante sem apertar em torno do béquer e marque as extremidades do barbante que se superpõem à circunferência com uma caneta.
3.2. Medir a distância entre as marcas com uma régua, para descobrir qual é a circunferência. Fazer em triplicata e registrar os valores.
3.3. Medir o diâmetro de cada béquer três vezes, uma com a régua e três com o paquímetro. Registrar os valores.
3.4. No seu relatório faça um gráfico das circunferências dos béqueres versus seus diâmetros. Desenhar uma linha com os pontos plotados. Pegar dois pontos na linha (localizados perto das extremidades da linha) e determinar sua inclinação:
�������çã� = �����çã������������ê���������çã�����â"�#��
No espaço milimetrado na parte de “Resultados experimentais” ou em outro papel milimetrado, plote a circunferência ao longo do eixo vertical e o diâmetro ao longo do eixo horizontal. Dar um título para o gráfico e rotular as variáveis (com unidades) plotadas em cada eixo.
Calcule a inclinação de seu gráfico, seguindo o exemplo mostrado na Figura 5, e discuta o significado do valor numérico da inclinação. �������çã� = ∆���������ê����∆��â"�#�� =
4. Medição quantitativa da densidade de soluções de açúcar
4.1. Determinação da densidade da solução de sacarose vermelha
4.1.1. Numerar os dois frascos. 4.1.2. Pesar e anotar a massa dos dois frascos limpos e secos. 4.1.3. Colocar, com a pipeta volumétrica, 10 mL de solução de sacarose vermelha em dois
frascos. Lembre-se de retirar qualquer líquido que sobre na ponta da pipeta dentro dos frascos (as letras td ou o anel duplo no alto da pipeta significa que a pipeta está projetada para despejar 10 mL quando todo o líquido tiver sido soprado). Lavar a pipeta com água destilada após
4.1.4. Pesar novamente o frasco para obter a massa da solução de sacarose. 4.1.5. Calcular a densidade da solução de sacarose pela razão massa por volume (g/mL). 4.1.6. Medir e anotar a temperatura da solução. 4.1.7. Repetir todo o procedimento, com uma nova amostra. A média das determinações
duplicadas será mais confiável e possibilitará que você verifique se houve erros grosseiros na medição da massa e na leitura dos volumes.
4.2. Determinação da densidade da solução de sacarose amarela
4.2.1. Repetir completamente o procedimento descrito na parte 3.1 em duplicata empregando a solução de sacarose verde (novamente assegure-se de que se os frascos Erlenmeyer ou béqueres utilizados anteriormente tenham sido lavados, eles devem estar secos antes da colocação da solução de sacarose).
Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
16
4.2.2. Ao terminar de fazer as medições nas duas soluções de sacarose, o(a) professor(a) lhe dirá qual é a solução de sacarose a 8% e a 16% em massa.
4.3. Determinação da densidade da água e calibração da pipeta volumétrica
Como uma verificação final de sua técnica e para lhe dar um meio de determinar o volume de sua pipeta, repita o processo inteiro em duplicata usando água pura. Registre a temperatura da água e compare a densidade que você calculou para a água com o valor (para a mesma temperatura) encontrada na Tabela 7. Se a densidade que você mediu for significativamente diferente (± 0,01 g/mL) do valor da literatura, pode ser uma indicação de que o volume da pipeta era significativamente diferente do seu valor nominal, pois as tolerâncias na fabricação dos volumes das pipetas são de 0,5-1%. Use a massa média de água dividida pelo valor conhecido da densidade da água na mesma temperatura (literatura) para calcular o volume correto da pipeta em todos os cálculos de densidade.
%���"������#���&�&�#��"' = "������á)�����&�������) ���������#�*�������á)���)/"' Tabela 6. Volume que 1 grama de água ocupa a várias a temperaturas.
Temperatura (°C) Volume (mL) Temperatura (°C) Volume (mL)
17.0 1.0022 22.0 1.0033
18.0 1.0024 23.0 1.0035
19.0 1.0026 24.0 1.0037
20.0 1.0028 25.0 1.0040
21.0 1.0030 26.0 1.0043 Obs.: O coeficiente térmico de expansão da água é 0 .00021 por 1° Celsius a 20° Celsius.
Tabela 7. Densidade da água pura (g/cm3) a temperaturas de 15°C até 30,9°C com 0,1°C de variação.
°C 0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9
15 0,999099 0,999084 0,999069 0,999054 0,999038 0,999023 0,999007 0,998991 0,998975 0,998959
16 0,998943 0,998926 0,998910 0,998893 0,998877 0,998860 0,998843 0,998826 0,998809 0,998792
17 0,998774 0,998757 0,998739 0,998722 0,998704 0,998686 0,998668 0,998650 0,998632 0,998613
18 0,998595 0,998576 0,998558 0,998539 0,998520 0,998501 0,998482 0,998463 0,998444 0,998424
19 0,998405 0,998385 0,998365 0,998345 0,998325 0,998305 0,998285 0,998265 0,998244 0,998224
20 0,998203 0,998183 0,998162 0,998141 0,998120 0,998099 0,998078 0,998056 0,998035 0,998013
21 0,997992 0,997970 0,997948 0,997926 0,997904 0,997882 0,997860 0,997837 0,997815 0,997792
22 0,997770 0,997747 0,997724 0,997701 0,997678 0,997655 0,997632 0,997608 0,997585 0,997561
23 0,997538 0,997514 0,997490 0,997466 0,997442 0,997418 0,997394 0,997369 0,997345 0,997320
24 0,997296 0,997271 0,997246 0,997221 0,997196 0,997171 0,997146 0,997120 0,997095 0,997069
25 0,997044 0,997018 0,996992 0,996967 0,996941 0,996914 0,996888 0,996862 0,996836 0,996809
26 0,996783 0,996756 0,996729 0,996703 0,996676 0,996649 0,996621 0,996594 0,996567 0,996540
27 0,996512 0,996485 0,996457 0,996429 0,996401 0,996373 0,996345 0,996317 0,996289 0,996261
28 0,996232 0,996204 0,996175 0,996147 0,996118 0,996089 0,996060 0,996031 0,996002 0,995973
29 0,995944 0,995914 0,995885 0,995855 0,995826 0,995796 0,995766 0,995736 0,995706 0,995676
30 0,995646 0,995616 0,995586 0,995555 0,995525 0,995494 0,995464 0,995433 0,995402 0,995371
Fonte: Adaptado do CRC Handbook of Chemistry and Physics.
4.4. Fazendo um gráfico dos dados
Existe uma relação simples entre a densidade e a massa (%) da sacarose? Para lhe ajudar a visualizar, construa um gráfico de densidade (g/mL) vs massa (%) de sacarose. Inclua um ponto para a água pura (0% de sacarose), de modo que você tenha um total de três pontos. Se você não determinou a densidade da água pura como descrito na parte 6.3, use o valor da literatura para a densidade da água na mesma temperatura de suas soluções de sacarose. Se o seu professor assim

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
17
determinar, plote os valores médios da densidade usando os dados combinados de todos os colegas de sua classe de laboratório (a média de vários valores é estatisticamente uma medida mais confiável do valor verdadeiro de uma quantidade do que qualquer medição isolada dessa quantidade). Trace uma linha reta que pareça se ajustar razoavelmente aos pontos (não interligue os pontos por segmentos de reta). Se você quiser pode utilizar uma planilha eletrônica para traçar o gráfico.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
18
Aula 2 – Introdução à estatística na química analítica e construção de gráficos
Resultados Experimentais
1. Estatística dos confeitos de chocolate
Tabela 1. Resultados do experimento dos confeitos no próprio grupo.
Item Nome Vermelho Rosa Amarelo Roxo Azul Verde Laranja
Estudante 1
Estudante 2
Estudante 3
Estudante 4
Estudante 5
1.1 Número total (grupo)
1.2 Total em % (grupo)
1.3 Erro relativo (%) (grupo)
1.3.1 Desvio padrão (no grupo)
Tabela 2. Resultados do experimento dos confeitos com a turma toda.
Item Nome Vermelho Rosa Amarelo Roxo Azul Verde Laranja
Grupo 1
Grupo 2
Grupo 3
Grupo 4
Grupo 5
1.4.1 Número total (turma)
1.4.2 Total em % (turma)
1.4.2.1 Erro relativo (%) (turma)
1.4.3 Desvio padrão
Tabela 3. Resultados do experimento dos confeitos com a turma toda somados aos dados da Tabela 4 (pag 14).
Item Nome Vermelho Rosa Amarelo Roxo Azul Verde Laranja
Dados Tabela 4
Total em % (Tabela 4)
1.4.1 Número total (turma)
1.4.2 Total em % (turma)
1.5.1 Total geral (turma + Tabela 4)
1.5.1 Tot. geral % (turma + Tabela 4)
1.5.2 Desvio padrão geral

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
19
2. Cara ou Coroa
Tabela 4. Resultados do experimento de ”cara ou coroa” do grupo.
Item Número de caras 0 1 2 3 4 5 6 7 8 9 10
2.2 Estudante 1 2.2 Estudante 2 2.2 Estudante 3 2.2 Estudante 4 2.2 Estudante 5 2.2 Total
Tabela 5. Resultados do experimento de ”cara ou coroa” da turma.
Item Número de caras 0 1 2 3 4 5 6 7 8 9 10
2.3 Grupo 1 2.3 Grupo 2 2.3 Grupo 3 2.3 Grupo 4 2.3 Grupo 5 2.3 Total
Tabela 6. Resultados do experimento de ”cara ou coroa” da turma somados ao da Tabela 5 (pag 15).
Item Número de caras 0 1 2 3 4 5 6 7 8 9 10
2.4 Grupo 1 2.4 Grupo 2 2.4 Grupo 3 2.4 Grupo 4 2.4 Grupo 5 2.4 Total
3. Circunferência vs diâmetro
Tabela 7. Resultados experimentais das circunferências dos béqueres.
Volume do béquer (mL)
Circunferência (cm) Diâmetro (cm)
Régua Paquímetro
�������çã� = ∆���������ê����∆��â"�#�� =

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
20
Figura 1. Gráfico da variação do volume vs variação do diâmetro.
4. Densidade de soluções de açúcar
Calculando a densidade, use o volume nominal (10 mL) ou preferivelmente o volume corrigido, determinado a partir da calibração da pipeta na parte 4.3.
%���"������#���&�&�#��"' = "������á)�����&�������) �����������á)���)/"'
Volume calculado a partir dos dados da parte 4.3 = _____________ mL
Tabela 8. Resultados experimentais da medida da densidade de soluções de açúcar.
(4.1) solução vermelha
de sacarose (4.2) solução amarela de
sacarose (4.2) água
Temperatura °C
Experimento 1 Experimento 2 Experimento 1 Experimento 2 Experimento 1 Experimento
2 Massa do frasco
vazio (g)
Massa do frasco + solução (g)
Massa líquida da solução (g)
Massa média da solução (g)
Volume da Solução (mL)
Densidade calculada (g/mL)

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
21
4.1. Fazendo um gráfico de densidade vs. massa (%) de sacarose
Fazer um gráfico da densidade (g/mL) vs. massa (%) de sacarose. Considere adequado que a escala de densidade varie de 0,99 a 1,08 g/mL e a de massa (%) de sacarose de 0 a 20%.
Deverão existir três pontos, um para água pura (massa (%) de sacarose = 0) e os pontos para as soluções vermelha e amarela de sacarose. Se possível, faça seu gráfico usando a média dos dados de toda a sua turma de laboratório. Trace a reta de melhor ajuste para os pontos.
O uso de um programa de planilha eletrônica (como Microsoft Excel) torna o desenho dos gráficos e a adição da linha de melhor ajuste (chamada “tendência” no Excel) mais fáceis. Entretanto, existe um espaço milimetrado para a plotagem dos valores e realização do gráfico manualmente.
Figura 2. Gráfico da densidade vs porcentagem em massa de sacarose.
Questionário sobre a verificação experimental
1. A análise gravimétrica do ferro apresentou os seguintes valores: 298 mg; 302 mg; 286 mg e 297 mg. Determine: a) a média b) o desvio padrão c) CV d) precisão da medida
2. Os seguintes resultados foram obtidos para réplicas da determinação de chumbo em uma amostra de sangue: 0,752; 0,756; 0,752 e 0,760 ppm de Pb. Calcule a média e o desvio padrão.
3. No valor numérico da inclinação (item 3), qual é a razão da circunferência para o diâmetro do círculo? Qual deveria ser o valor encontrado e porquê?

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
22
Aula 3 - Determinação de ácido acético em vinagre.
Objetivos
• Determinar a quantidade de ácido acético presente em uma amostra de vinagre comercial.
Introdução
Ácido acético ou ácido etanóico tem fórmula molecular CH3COOH. Em uma solução aquosa atua como ácido fraco tendo um Ka de 1,8 x 10-5. O ácido acético puro recebe o nome de ácido acético glacial (densidade de 1,053 g/cm3 e 99,8 % m/V). devido ao fato de congelar-se a temperaturas ligeiramente mais baixas que a temperatura ambiente (16-17 °C). Quando misturado com água solidifica a temperaturas muito mais baixas. O ácido acético pode ser misturado com água e com numerosos dissolventes orgânicos.
Pode ser obtido pela ação do ar sobre soluções de álcool, em presença de certa classe de bactérias, como a Bacterium aceti. As soluções diluídas (de 3,5 a 8%) preparadas deste modo a partir do vinho, sidra ou malta constituem o que conhecemos como vinagre. O ácido acético concentrado se prepara industrialmente mediante distintos processos, como a reação de metanol (álcool metílico) e de monóxido de carbono (CO) em presença de um catalisador, ou pela oxidação do etanal (acetaldeído). Tem um ponto de ebulição de 118 °C e um ponto de fusão de 17 °C.
Por sua ação desincrustante, o ácido acético é utilizado na lavagem química de equipamentos de diálise (em diluições que vão de 2,5% a 5% dependendo da recomendação do fabricante do equipamento). Como ação complementar, o ácido acético funciona como bactericida.
Um dos controles de qualidade deste tipo de alimento é feito através de uma titulação ácido-base, para verificar a quantidade de ácido acético presente. A reação é:
CH3COOH + NaOH CH3COONa + H2O
A quantidade de ácido acético (HAc) presente no vinagre deve atender às exigências do Ministério da Agricultura
Materiais e Reagentes
• 2 Erlenmeyer 250 mL • água destilada • solução de NaOH 0,1 M padronizada • pipeta volumétrica de 10 mL • fenolftaleína • bureta • suporte universal • garra dupla para buretas
• 3 béqueres • amostra comercial de vinagre • balão volumétrico 100 mL
1. Procedimento
1. Pipetar 10 mL de vinagre, com o auxílio de uma pipeta volumétrica, para um balão volumétrico de 100 mL e completar o volume até a marca com água destilada.
2. Transferir uma alíquota de 10 mL do balão, com o auxílio de uma pipeta volumétrica, para um erlenmeyer.
3. Adicionar aproximadamente 30 mL de água destilada e de 3 a 5 gotas de indicador fenolftaleína. 4. Preparar a bureta (fazer ambiente e zerar) com a solução de NaOH 0,1 M previamente padronizada para
titular a solução de ácido acético. 5. Titular cuidadosamente a mistura com solução padrão de NaOH 0,1 mol/L até o aparecimento de uma
leve coloração rósea que persista por mais de 30 segundos. 6. Anotar o volume gasto. 7. Fazer a determinação em duplicata. 8. Calcular em mol/L, g/L e em % m/v a concentração de ácido acético na solução ORIGINAL (vinagre
comercial).

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
23
Aula 4 - Determinação da concentração de ácido fosfórico.
Objetivos
• Determinar a concentração real de ácido fosfórico em amostra comercial.
• Fazer a titulação de um ácido poliprótico.
Introdução
O ácido fosfórico é um ácido triprótico, que pode ser titulado diretamente apenas como mono e diprótico:
H3PO4 + NaOH � Na+ + H2PO4ᅳ + H2O Ka1 = 7,5 x 10ᅳ3 pKa1 = 2,12 H2PO4ᅳ + NaOH � Na+ + HPO42ᅳ + H2O Ka2 = 6,2 x 10ᅳ8 pKa2 = 7,21 HPO42ᅳ + NaOH � Na+ + PO43ᅳ + H2O Ka3 = 4,8 x 10ᅳ13 pKa2 = 12,30
Materiais e Reagentes
• 2 Erlenmeyer 125 mL • água destilada • solução de NaOH 0,1 M padronizada • pipeta volumétrica de 10 mL
• timolftaleína • bureta
• alaranjado de metila • garra para buretas
• 3 béqueres • solução de H3PO4 0,05 M
1. Procedimento
1. Prepare 250 mL de uma solução de ácido fosfórico 0,05 M sem fazer ajuste nenhum com relação à pureza do reagente, isto é, considerando-o 100% puro.
2. Transferir 20,0 mL de uma solução de ácido fosfórico 0,05 M para um erlenmeyer de 125 mL, com o auxílio da pipeta volumétrica.
3. Adicionar cerca de 50 mL de água destilada. 4. Titular contra uma solução de hidróxido de sódio 0,1 M usando alaranjado de metila como indicador
para o primeiro ponto de equivalência, até a mudança de coloração vermelha para amarelo claro. 5. Anotar o volume gasto. 6. Adicionar ao erlenmeyer 3 gotas de timolftaleína para titulação do segundo ponto de equivalência. 7. No ponto final observa-se uma mudança de coloração de amarelo para verde. 8. Anotar o volume gasto. 9. Repetir o procedimento titulando-se o ácido como se fosse monoprótico, utilizando-se a timolftaleína
como indicador. 10. Anotar o volume gasto. 11. Fazer todas as análises em triplicata. 12. Comparar os resultados obtidos pelos diferentes métodos com o resultado teórico.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
24
Aula 5 - Determinação de ácido acetilsalicílico em medicamento.
Objetivos
• Determinar o teor de ácido acetilsalicílico em amostras disponíveis comercialmente
Preparação pré-laboratório
O ácido acetilsalicílico é um dos medicamentos mais antigos em uso em todo o mundo. É feito a partir da reação entre o ácido salicílico e o anidrido acético. É uma molécula pouco solúvel em água, o que torna difícil a sua titulação direta com o NaOH em meio aquoso. No entanto, pode-se quantificá-lo através de uma titulação de retorno, ou titulação pelo excesso. Nesta titulação trata-se a amostra contendo o ácido acetilsalicílico com um excesso conhecido de NaOH. A seguinte reação se processa:
C6H4(COCH3)COOH(s) + 2 NaOH(aq) � C6H4(OH)(COONa)(aq) + CH3COONa(aq) ácido acetilsalicílico salicilato de sódio acetato de sódio
Esta reação consome todo o ácido acetilsalicílico da amostra, que não consome todo o NaOH colocado no meio de reação, pois este está em excesso. O passo seguinte é titular, com uma solução padrão de HCl, o NaOH que sobrou. Como conhecemos a quantidade de NaOH que foi adicionada e quanto sobrou ao término da reação, podemos calcular quantos mols de NaOH reagiram com o ácido acetilsalicílico da amostra e obter portanto a quantidade do ácido presente na amostra.
A pequena solubilidade em água do ácido acetilsalicílico é contornada pelo fato de que durante a ocorrência da reação são formados produtos solúveis. Verifique que os sais formados tem carga, o que facilita a solubilidade em água.
Materiais e Reagentes
• 2 Erlenmeyer 250 mL • água destilada • Proveta 100 mL • solução de NaOH 0,1 M padronizada
• fenolftaleína • bureta
• suporte universal • garra dupla para buretas
• 3 béqueres • bico de Bunsen
• solução de HCl 0,2 M padronizada • amostra comercial de AAS
1. Procedimento Experimental
1.1. Triturar 01 comprimido de ácido acetilsalicílico em um béquer e junte exatamente 100 mL de solução NaOH 0,1 mol/L padronizado. Fazer o procedimento em duplicata.
1.2. Transferir quantitativamente a mistura para um erlenmeyer. 1.3. Ferver a mistura brandamente por 10 minutos. 1.4. Colocar no erlenmeyer o indicador fenolftaleína (3 gotas). 1.5. Preparar a bureta (fazer ambiente e zerar) com a solução de HCl 0,2 M previamente padronizada.
1.6. Titular o excesso de NaOH (NaOH que não reagiu) com a solução padrão de HCl 0,2 M. 1.7. Anote os volumes gastos na titulação e calcule a quantidade de ácido acetilsalicílico em cada
comprimido. Verifique se há concordância com a quantidade rotulada no medicamento.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
25
Aula 6 - Determinação do teor de hidróxido de magnésio no leite de magnésia (titulação de retorno).
Objetivos
• Determinar a concentração de hidróxido de magnésio no leite de magnésia.
• Fazer a titulação de uma suspensão básica.
Preparação pré-laboratório
O leite de magnésia é constituído de uma suspensão de hidróxido de magnésio, com uma especificação média estabelecida de 7% em peso. A titulação direta de uma alíquota da amostra de leite de magnésia é difícil de ser realizada, pois é uma suspensão branca e opaca. Além disso, as partículas de hidróxido de magnésio em suspensão podem causar erros ao aderirem às paredes do frasco erlenmeyer, ficando fora de contato com o ácido titulante. Outro problema que pode surgir em consequência de a amostra ser opaca é a difícil percepção de uma mudança precisa da cor do indicador no ponto final da titulação. Para contornar tais problemas, adiciona-se um volume definido e que proporcione concentração em excesso de uma solução-padrão de ácido clorídrico para dissolver e neutralizar todas as partículas suspensas de hidróxido de magnésio, resultando em uma solução transparente. Em seguida, o ácido clorídrico em excesso é titulado com uma solução padrão de hidróxido de sódio.
Mg(OH)2(S) + 2 H+(excesso) � 2 H2O + Mg2+
H+(que não reagiu) + OH−
(titulante) � H2O
Como a amostra já se encontra como uma suspensão, não será necessária a etapa de abertura da mesma. O produto da reação é uma solução neutra, portanto, o pH do ponto final da reação é igual a 7. Assim, a fenolftaleína, com zona de viragem entre 8,0 e 10,0, passando de incolor para rosa, será utilizada como indicador.
Materiais e Reagentes
• 3 Erlenmeyer 125 mL • solução de NaOH 0,1 M padronizada
• solução de HCl 0,2 M padronizada • pipeta volumétrica de 10 mL
• fenolftaleína • bureta
• Leite de magnésia comercial • garra para buretas
• 3 béqueres • Pipeta de Pasteur
1. Procedimento
1.1. Agitar vigorosamente o frasco que contém o leite de magnésia para homogeneizar a suspensão. 1.2. Em um vidro de relógio ou béquer, medir, com o auxílio de um conta gotas, não mais que 0,150 g
da amostra previamente homogeneizada. Transferir quantitativamente para um erlenmeyer de 125 mL.
1.3. Adicionar aproximadamente 25mL de água destilada ao erlenmeyer contendo a amostra. 1.4. Pipetar 5,0 mL de solução de HCl aproximadamente 0,2 mol/L padronizada, acrescentar ao
erlenmeyer e homogeneizar a solução resultante. 1.5. Adicionar ao erlenmeyer duas (2) gotas de fenolftaleína como indicador. 1.6. Titular com solução padronizada de NaOH aproximadamente 0,1 mol/L até o aparecimento de
uma coloração rósea. 1.7. Anotar o volume de NaOH consumido. 1.8. Repetir a titulação para mais duas amostras.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
26
Aula 7 - Padronização de solução de AgNO3 e determinação de cloreto (método de Mohr).
Objetivos
• Realizar a padronização de uma solução de AgNO3.
• Determinar a quantidade de cloreto em uma amostra pelo método da precipitação com prata
Preparação pré-laboratório
A argentimetria envolve o uso de soluções padrões de nitrato de prata e tem como principal campo de aplicação a determinação de haletos e outros ânions que formam sais de prata pouco solúveis. A argentimetria compreende diferentes métodos, que podem ser classificados conforme a titulação seja direta ou indireta. Nos métodos diretos, a solução que contém a substância a determinar é titulada com solução padrão de nitrato de prata ao ponto de equivalência. O ponto final pode ser identificado de várias maneiras: adição de nitrato de prata até que não mais se observe a formação de precipitado ou mudança de coloração de um indicador. Os métodos usuais da argentimetria com titulação direta fazem uso de indicadores para localizar o ponto final. O método de Mohr é um método argentimétrico direto, que usa cromato de potássio como indicador. Também são usados indicadores de adsorção em titulações argentimétricas diretas. Já o método de Volhard é um método argentimétrico indireto, que consiste em precipitar o haleto com um excesso de nitrato de prata e, então, titular a prata residual em meio ácido com uma solução padrão auxiliar de tiocianato usando íon Fe3+ como indicador. O método de Mohr é aplicável à determinação de cloreto ou brometo. A solução neutra do haleto é titulada com nitrato de prata em presença de cromato de potássio como indicador. Os haletos são precipitados como sais de prata: o cloreto de prata é branco e o brometo de prata branco-amarelado. O ponto final é assinalado pela formação de cromato de prata, vermelho. O método baseia-se, pois, na precipitação fracionada: precipitam, primeiro, o haleto de prata e, depois, o cromato de prata. Considere o caso da titulação de cloreto de sódio com nitrato de prata em presença de cromato. Obviamente, as condições da titulação devem ser tais que o cloreto seja quantitativamente precipitado como cloreto de prata branco antes que a precipitação de cromato de prata vermelho se torne perceptível: por outro lado, é preciso que o indicador acuse a mudança de coloração com apenas um leve excesso de prata. Estando as duas fases sólidas, cloreto de prata e cromato de prata, em equilíbrio com a solução, têm-se:
[Ag+] [Cl-] = Kps AgCl = 1,8 x 10-10 [Ag+]2 [CrO4
2-] = Kps Ag2CrO4 = 1,1 x 10-12 no ponto de equivalência,
[Ag+] = [Cl-] = AgClK = 1,35x10-5
Para o cromato de prata poder precipitar exatamente neste ponto, a concentração de íon cromato teria de ser a seguinte:
325
12
224 100,6
)1035,1(
101,1
][][ 42 −
−
−
=− === x
x
x
Ag
KCrO
CrOAg
Portanto, teoricamente, a concentração de cromato de potássio na solução deveria ser igual a 0,006 mol L-1. Entretanto, na prática, faz-se uso de cromato em concentração algo mais baixa, aproximadamente 0,002 mol L-1 (no ponto final), pois a coloração amarela das soluções mais concentradas dificultaria a observação do ponto final. Então, o cromato de prata começará a precipitar quando
512
24
104,23100,2
101,1
][][ 42 −
−
−+ =
−== x
x
x
CrO
KAg
CrOAg
Esta concentração de íon prata é atingida além do ponto de equivalência. Quando a concentração de Ag+ = 2,4 x 10-5 a concentração de Cl- = 8 x 10-6. Portanto, haverá precipitação de uma quantidade adicional de cloreto de prata além do ponto de equivalência, que corresponde a um consumo de íon prata igual a (1,35x10-5) - (8x10-6) = 5,5x10-6 mol L-1. De fato, o ponto de equivalência deve ser sobrepassado ainda mais,

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
27
para que se forme cromato de prata em quantidade suficiente para tornar a mudança de coloração perceptível. Experimentalmente, verificou-se que a quantidade mínima de cromato de prata, necessária para uma mudança de coloração bem definida corresponde ao consumo de aproximadamente, 2 x 10-5 mol L-1 de íon prata. O erro da titulação será maior com o aumento da diluição da solução e será bem apreciável (cerca de 0,4%) em soluções diluídas, quando a concentração de cromato é da ordem de 0,003 a 0,005 mol L-1. Elimina-se este erro mediante um ensaio em branco com o indicador; neste ensaio se mede o volume da solução padrão de nitrato de prata que é necessário para atribuir uma cor perceptível à água destilada com a mesma quantidade de indicador que a usada na titulação. Este volume é subtraído do volume consumido da solução padrão. Deve-se observar que a titulação deve ser feita em solução neutra, ou em solução levemente alcalina, isto é, no intervalo de pH 6,5 a 9. Em solução ácida, ocorre a seguinte reação:
O HCrO4− é um ácido fraco e por isso a concentração do íon cromato se reduz e é possível que o
produto de solubilidade do cromato de prata não seja excedido. Em soluções muito alcalinas, é possível a precipitação do hidróxido de prata (Ksol. = 2,3x10-8). Um procedimento simples de tornar neutra uma solução ácida é o de adicionar um excesso de carbonato de cálcio ou de hidrogenocarbonato de sódio(bicarbonato de sódio) puros. Uma solução alcalina pode ser acidificada com ácido acético e então se acrescenta um pequeno excesso de carbonato de cálcio. O produto de solubilidade do cromato de prata cresce com a elevação de temperatura; por isso a titulação deve ser feita na temperatura ambiente. Por outro lado, em
pH muito alto a presença da alta concentração de íons OH- ocasiona a formação do hidróxido de prata. Como consequência, o método de Mohr é um bom processo para se determinar cloretos em soluções neutras ou não tamponadas, tal como em água potável. Para ser usado como padrão primário, o cloreto de sódio PA deve ser previamente aquecido a 120-130 °C por uma hora, deixar esfriar em dessecador. Pesar, exatamente, 0,5844 g de cloreto de sódio necessário para preparar 100,0 mL de solução 0,1 M. Caso a massa de cloreto de sódio não seja exata, fazer a correção posteriormente. Transferir quantitativamente para um balão volumétrico de 100,0 mL, completar o volume e homogeneizar. Em função da massa pesada, corrigir a molaridade e rotular.
Materiais e Reagentes
• 2 Erlenmeyer 125 mL • água destilada • NaCl seco • pipeta graduada de 1 mL
• cromato de potássio • bureta
• sol K2CrO4 5% • garra para buretas
• 3 béqueres • proveta 100 mL
• solução fisiológica (soro) • pipeta volumétrica de 10 mL
• KCl • AgNO3
1. Procedimento para padronização da solução de AgNO3 0,1 M (Mohr)
1.1. Pesar a massa de AgNO3 (previamente seca em estufa a 150 °C por 2h) necessária para preparar 250 mL de solução 0,1 mol/L.
1.2. Para a padronização utilizar aproximadamente 0,17 g de NaCl em um erlenmeyer, adicionando 80 mL de água e 1 mL de cromato de potássio 5% (indicador).
1.3. Titular com a solução de nitrato de prata, lentamente, agitando o frasco até que a cor avermelhada formada pela adição de cada gota desapareça cada vez mais lentamente (isto é indicação de que a maior parte do cloreto está precipitada).
1.4. Continuar a adição gota a gota até que ocorra uma mudança de cor fraca mas distinta, que deve persistir após agitação forte.
2 CrO42- + 2H+ 2HCrO4
- Cr2O72-+H2O
2 Ag+ + 2OH- 2AgOH 2Ag2O + H2O

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
28
1.5. Fazer uma correção de branco do indicador, adicionando 0,5 g de carbonato de cálcio e 1 mL do indicador a um volume de água igual ao volume final da titulação.
1.6. Titular com a solução de nitrato de prata até que a cor do branco fique igual a da solução titulada. A correção do branco não deve ser muito maior que 0,1 mL e esse volume deve ser deduzido do volume gasto na titulação.
1.7. Fazer no mínimo em triplicata.
2. Determinação de cloreto em amostra de KCl de concentração conhecida
2.1. Pipetar 10 mL da solução de KCl 0,1M. 2.2. Adicionar 1 mL de cromato de potássio a 5% p/v e homogeneizar. 2.3. Titular lentamente com o nitrato de prata segundo o procedimento anterior. Fazer no mínimo em
duplicata. 2.4. Calcular a concentração da amostra em mols por litro e em gramas por litro.
3. Determinação da porcentagem de NaCl em uma solução injetável (Mohr)
3.1. Pipetar 10 mL da solução injetável de NaCl (soro fisiológico) para um erlenmeyer de 125 mL.
3.2. Adicionar 20 mL de água destilada, 2 g de bicarbonato de sódio e 1 mL de solução do indicador de cromato de potássio
3.3. Titular com a solução de AgNO3 até obter uma coloração vermelha persistente.
3.4. Repetir o procedimento até obter dois resultados concordantes.
3.5. Calcular a concentração do NaCl em % m/v e em mol/L

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
29
Aula 8 - Determinação de cloreto (método de Volhard).
Objetivos
• Determinar a porcentagem de cloreto em uma determinada amostra, pelo método de Volhard.
Preparação pré-laboratório
No método de Volhard, com formação de um complexo solúvel, a solução nítrica contendo o íon prata é titulada com tiocianato de potássio, em presença de íon ferro (III), que é adicionado em forma de solução saturada de sulfato de amônio e ferro (III) em ácido nítrico. Consideremos a forma mais simples de aplicação do método de Volhard, que é a determinação de prata por meio de titulação da solução nítrica com tiocianato. A prata precipita com o tiocianato de prata muito pouco solúvel:
Ag+ + SCN- � AgSCN Um leve excesso de tiocianato é identificado pela formação de um complexo solúvel de ferro, [FeSCN]2+ , intensamente corado de vermelho. Trata-se de uma reação extremamente sensível. Assim, uma ou duas gotas da solução saturada de sulfato de amônio e ferro (III), adicionadas a ácido nítrico dão uma coloração alaranjada fraca com tiocianato; uma quantidade um pouco maior da solução de tiocianato muda a coloração para vermelho - alaranjado. O erro do indicador é praticamente nulo. Deve-se, entretanto, observar que a mudança de coloração do indicador aparece cerca de 1% antes do ponto estequiométrico. É que o tiocianato de prata absorve íons prata, que são removidos da solução. Por isso, depois de observada a primeira mudança de coloração, a titulação deve prosseguir sob vigorosa agitação até que a coloração alaranjada não mais desapareça. Desta maneira, toda a prata é precipitada e os resultados apresentam elevada exatidão. As mais importantes aplicações do método de Volhard são as que se relacionam com a determinação de cloretos, brometos e iodetos em meio ácido. A solução nítrica contendo o halogeneto é tratada com nitrato de prata em excesso e esse excesso titulado com solução de tiocianato. O cloreto começa a precipitar com excesso de prata:
Ag+ + Cl- � AgCl O excesso de prata é, então, titulado com a solução de tiocianato:
Ag+ + SCN- � AgSCN Entretanto, o tiocianato de prata é menos solúvel do que o cloreto de prata, assim, é preciso evitar que, uma vez completada a titulação do excesso de prata, um excesso de tiocianato reaja com o cloreto de prata precipitado.
AgCl + SCN- � Cl- + AgSCN Se o excesso de prata fosse titulado simplesmente em presença do precipitado de cloreto de prata, a titulação estaria sujeita a um erro considerável, pois, após a precipitação de toda a prata, o excesso de tiocianato reagiria com o cloreto de prata. Por essa razão utilizam-se alguns artifícios para contornar a dificuldade apontada. Um dos artifícios consiste em efetuar a filtração do cloreto de prata precipitado antes de realizar a titulação com a solução de tiocianato. Outro artifício é o de cobrir a suspensão aquosa com uma camada de éter, tolueno, ligroína ou outro solvente orgânico não miscível em água e que não apresente toxicidade. O cloreto de prata aglomera-se na superfície de separação das duas fases, livrando-se da ação dissolvente do tiocianato. Finalmente, a outra alternativa é a adição de pequena quantidade de nitrobenzeno na suspensão de AgCl. O nitrobenzeno é insolúvel em água e forma uma película sobre as partículas, impedindo-as de reagirem com o SCN-; este procedimento assegura um ponto final nítido e permanente.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
30
Materiais e Reagentes
• 3 Erlenmeyer 125 mL • água destilada
• solução fisiológica (soro) • pipeta graduada de 5 mL
• pipeta volumétrica 25 mL • bureta
• balão volumétrico 250 mL • garra para buretas
• 3 béqueres • pera
• papel de filtro • HNO3 6 mol/L
• NH4Fe(SO4)2·12 H2O – 40% • AgNO3 0,1 mol/L
1. Determinação da porcentagem de NaCl em uma solução injetável (Volhard)
1.1. Pesar cerca de 2,43 g de KSCN seco por 1 – 2h 150ºC, em estufa resfriando-se e mantendo-se em descarregador.
1.2. Dissolver em água e completar o volume em balão volumétrico de 250 mL com água destilada. 1.3. Medir, em pipeta, 15 mL de uma solução de soro fisiológico (0,9%) e transferir-se para um
erlenmeyer de 125 mL. 1.4. Adicionar 25 mL de nitrato de prata 0,1 mol/L. 1.5. Filtrar o precipitado formado e recolher o filtrado.
1.6. Adicionar 1,0 mL do indicador (solução saturada de sulfato de ferro amonical 40%) acidificando-se o meio com 5,0 mL de HNO3 6,0 mol/L.
1.7. Titular com solução de tiocianato de potássio 0,1 mol/L padrão até aparecer uma coloração marrom- avermelhada.
Obs. A primeira mudança perceptível de cor para o avermelhado ocorre cerca de 1% antes do ponto de equivalência, porque os íons de prata ainda estão presentes na superfície do precipitada, por adsorção. Após o aparecimento da primeira mudança de cor, continua-se a titulação sob forte agitação, até o aparecimento de uma coloração marrom- avermelhado, que persiste mesmo sob forte agitação. 1.8. Anotar o volume gasto do titulante e repetir o procedimento mais duas vezes. 1.9. Calcular a porcentagem de NaCl e Cl- no soro fisiológico

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
31
Aula 9 - Volumetria de complexação: padronização de solução Na2EDTA e quantificação de metais
Objetivos
• Realizar a padronização de uma solução de EDTA assim como utilizá-la para realizar titulações volumétricas na determinação do teor de metais em soluções.
Preparação pré-laboratório
Titulação de Complexação Muitos íons metálicos formam complexos suficientemente estáveis. Este fato serve de base para um método barato, e de comprovada eficácia na determinação de íons metálicos e de seus complexantes.
Por bastante tempo a complexometria foi limitada pela baixa estabilidade dos complexos conhecidos e pelo fato da formação de vários complexos ocorrer em estágios gerando complexos do tipo MLn , onde M = metal, L = ligante, n = assume vários valores, dependendo do íon e dos reagentes em uso. Isto gerava muitas discussões pois, com tantos produtos formados, as teorias criadas acabavam perdendo credibilidade devido aos resultados diversos.
Tais limitações somente foram superadas em 1945, quando foi introduzido o ácido etilenodiaminotetracético (EDTA), um poderoso reagente, que complexa com vários íons, incluindo metais pesados e alcalino terrosos, formando estruturas estáveis do tipo 1:1.
Figura 1. Fórmula estrutural do EDTA livre e na sua forma complexada.
Titulações complexométricas são extremamente úteis para a determinação de grande números de metais. Esta técnica tem alcance de milimol (10-3 mol) e pelo uso de agentes auxiliares e controle do pH, a seletividade necessária pode ser alcançada.
A complexação é uma atração eletrostática entre um íon e um agente quelante de modo que não há transferência de elétrons entre estes. Quanto as cargas, a estrutura final terá como carga a somatória das cargas individuais de cada participante do complexo.
Agente quelante: Qualquer estrutura, da qual façam parte dois ou mais átomos possuidores de pares de elétrons não utilizados em ligações químicas primárias, mas sim, usados como "imãs" eletrostáticos para se prenderem a íons metálicos.
Dentre os complexantes mais comuns podemos citar a água, responsável (ligada ao íons cobre) pela cor azul das soluções de sais de cobre, a amônia (quando substitui a água ao redor do cobre, produz cor azul mais intensa) e o EDTA (ácido etilenodiaminotetracético (que com o cobre, rivaliza a amônia).

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
32
O EDTA pode ser considerado o reagente complexométrico (agente quelante) padrão, daí a necessidade do conhecimento de sua estrutura e suas titulações devem ser realizadas sob pH controlado, optando pelo menor valor possível segundo cada complexo desejado, de modo a impedir a ionização da molécula de EDTA e até a competição pelos cátion metálicos devido aos íons OH- (que existem em maior ou desprezível quantidade conforme pH>7 ou pH<7).
Na complexação de íons metálicos com EDTA, a sua espécie ativa é o íon y4-,cuja concentração depende do pH. Pois, somente em solução nitidamente alcalina o EDTA se encontra presente predominantemente na forma de íon y4-, com o aumento da concentração de íon de hidrogênio, cai a concentração da espécie y4- ou seja, a concentração do íon y4- diminui com o decréscimo do pH. (OHLWELER, O. A.;1981)
Além do titulante (EDTA), certas substâncias presentes em solução podem formar complexos com íons metálicos e, como consequência, competir com a reação básica da titulação. Na titulação pelo EDTA usa-se um indicador sensível ao íon metálico para detectar as modificações do pM.
O cálcio está presente na casca de ovo na forma de carbonato de cálcio e na proporção de cerca de 40% do produto em pó. Além do aspecto econômico, o cálcio da casca de ovo apresenta vantagens nutricionais, pois não está associado a elevadas quantidades de proteína e sódio (como acontece, por exemplo, nos queijos), que podem induzir a um aumento da excreção renal de cálcio (Food, 1997; Weinsier & Krumdieck, 2000).
Um novo estudo demonstrou que as propriedades físico-químicas do carbonato de cálcio extraído da casca do ovo apresentam maior estabilidade e resistência térmica quando comparado ao carbonato de cálcio produzido industrialmente - amplamente utilizado como diluente sólido em produtos farmacêuticos, odontológicos, cosméticos e em suplementos alimentares. (Revista - Ciência e Tecnologia de Alimentos 2007)
O composto obtido da farinha de ossos não tem a mesma disponibilidade do cálcio extraído de fontes sintéticas. Nas conchas de ostras, o carbonato tem vestígios de chumbo, entre outros elementos potencialmente tóxicos como alumínio, cádmio e mercúrio. Nesse cenário, a casca de ovo tem a vantagem de não conter elementos tóxicos. Há grande interesse no mercado em encontrar novas fontes puras de carbonato de cálcio. (Revista - Ciência e Tecnologia de Alimentos 2007)
Materiais e Reagentes
• CaCO3 • balão volumétrico 100 mL • Tampão pH 10 • 2 pipeta volumétrica 5 mL
• negro de eriocromo T • proveta 100 mL
• Casca ovo calcinada • garra para buretas
• leite de magnésia • bureta
• 3 erlenmeyer 250 mL • 3 béquer
• HCl • espátula PREPARAÇÃO DE SOLUÇÕES a) Solução padrão 0,01 M de EDTANa2. (1 L). Guarde a solução em frasco de polietileno.
b) Solução de NaOH a 4 % . (100 mL).
c) Solução da amostra de Zinco: Pesar 4,3986 g de ZnSO4⋅7 H2O e diluir com água destilada a 1 L.
d) Solução tampão pH 10: (NH4Cl/NH4OH). Dissolver 6,4 g de NH4Cl em 58 mL de NH4OH e completar com água até 100 mL. CUIDADO, VAPORES TÓXICOS � CAPELA. Guardar a solução em frasco de polietileno para evitar a passagem de íons metálicos do vidro à solução-tampão e o frasco deve permanecer bem fechado para impedir a perda de NH3 e a entrada de CO2.
e) SOLUÇÃO DE CÁLCIO PADRÃO Pesar exatamente 0,5000 g de CaCO3 anidro e P.A. e transferir para um Erlenmeyer de 250 mL de capacidade. Adicionar aos poucos, solução de HCl (1:1) até dissolver todo o carbonato (adicionar o mínimo de solução
ácido possível). Em seguida juntar 100 mL de água e levar à ebulição por alguns minutos para eliminar todo o CO2. Esfriar. Adicionar 2 a 3 gotas de vermelho de metila e ajustar a cor laranja intermediária pela adição de solução NH4OH ou HCl (1:1). Transferir toda a mistura para um balão volumétrico de 500 mL e completar
com água destilada. Agitar para homogeneizar. Cada mL dessa solução equivale a 1,00 mg de CaCO3.
f) Preparação de Solução de EDTA 0,01 mol/L

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
33
O sal dissódico (Na2H2C10H12O8N2 .2H2O) dihidrogenoetilenodiamino tetraacetato de sódio contém 0,3% de umidade absorvida e sua massa molar é 372,3 g. Pode-se dessecá-lo mediante aquecimento a 80°C durante 2 ou 3 horas em uma estufa a 110°C e as duas moléculas de água de hidratação permanecem intactas nessas condições. Essa solução deve ser conservada em frasco de polietileno.
Pesar exatamente 1,8615 g do sal dissódico e transferir para um béquer de 500 mL de capacidade. Juntar 250 mL de água redestilada e dissolver por completo o reagente.
Transferir essa solução para um balão volumétrico de 500 mL de capacidade e completar seu volume com água redestilada. Agitar para homogeneizar. Transferir imediatamente a solução preparada para um frasco de poletileno e, nestas condições, pode ser considerada como solução padrão. Entretanto, periodicamente as soluções de EDTA devem ser repadronizadas, seguindo métodos definidos em literatura.
1. Procedimento para padronização da solução de EDTA 0,01 mol/L (padrão primário)
1. Transferir 20,0 mL de uma solução de CaCO3 0,01 mol/L para um erlenmeyer de 250 mL com o auxílio de uma pipeta volumétrica.
2. Adicionar cerca de 50 mL de água destilada.
3. Ajustar o pH para 10 adicionando cerca de 10 mL de solução tampão de hidróxido de amônio/cloreto de amônio e acrescentar uma pitada do indicador de negro de eriocromo T (mistura de 0,1 g de Negro de Eriocromo T com 10 g de NaCl guardada em frasco bem fechado).
4. Titular com uma solução de EDTA 0,01 mol/L até o aparecimento da coloração azul. 5. Anotar o volume. Fazer a análise em triplicata.
2. Determinação complexométrica de zinco
1- Transferir para um erlenmeyer exatamente 10 mL da amostra de Zinco preparada no item “c” e 20 mL de água destilada.
2- Adicionar 2 gotas de NaOH a 4 %. 3- Acrescentar aproximadamente 100 mg do indicador Eriocromo T. 4- Adicionar 5 mL de tampão pH10. 5- Carregar a bureta com EDTA.Na2 preparada no item “a”. 6- Proceder a titulação até mudança de coloração do indicador (agite a solução titulada constantemente). 7- Fazer o procedimento em triplicata
1 mL de EDTA 0,01 M � 0,0006537 g de Zn

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
34
Aula 10 - Volumetria de complexação: Determinação de cálcio na casca do ovo e no leite
Objetivos
Determinar o teor de cálcio na casca do ovo de galinha e no leite em pó.
Preparação pré-laboratório
Ovos de galinha contém de 25 a 30 mg de cálcio, 90% do qual encontra-se na gema. Entretanto, a casca contém muito mais cálcio e pode ser usada como uma amostra adequada para a análise de cálcio por titulação com EDTA. Nos últimos anos, o teor de cálcio nos ovos de pássaros tem se tornado objeto de interesse ambiental, pois pesticidas organoclorados, como DDT e dieldrina, entraram na cadeia alimentar de aves de rapina. Como estes compostos têm efeitos adversos sobre o metabolismo de cálcio nas aves, as cascas de seus ovos se tornam muito finas e, portanto, mais frágeis. O resultado é que poucos ovos atingem a maturidade. Materiais e Reagentes
• 2 erlenmeyer de 250 mL
• Béquer
• Proveta
• Bureta
• Pipeta volumétrica
• Pera
• Solução de EDTA-Na (sal dissódico de EDTA) 0,01 mol/L
• Solução tampão NH4OH/NH4Cl
• Papel indicador universal
• Solução de NaOH (hidróxido de sódio) 1 mol/L
• Cascas de ovos de galinha.
• Indicador Negro de eriocromo -T
Procedimento Experimental 1. Determinação do teor de cálcio na casca do ovo.
1.1. Pesar cerca de 0,5 g de casca de ovo seca, transfira-a para um béquer de 250 mL e dissolva-a em 25 mL de HCl 6 M. Se necessário, remova qualquer material orgânico restante através de filtração.
1.2. Transferir quantitativamente para um balão volumétrico de 250 mL. 1.3. Transferir uma alíquota de 10 mL da amostra para um erlenmeyer de 125 mL com uma pipeta
volumétrica. 1.4. Adicionar 10 mL de solução tampão de pH 10 e 2 ou 3 pontas de espátula do indicador negro de
eriocromo T junto com NaCl. 1.5. Titular com solução 0,01 mol.L-1 de EDTA até que um azul puro seja alcançado. 1.6. Certificar-se de titular através do azul escuro (arroxeado) até um azul médio. 1.7. Realizar o procedimento em triplicata. 1.8. Reportar a massa da casca de ovo analisada e os teores de Ca e CaCO3 encontrados.
2. Determinação do cálcio no leite em pó 1.1. Pesar 0,500 g da amostra de leite em pó e transferir quantitativamente para um erlenmeyer. 1.2. Dissolver a amostra com aproximadamente 30 mL de água destilada (deve-se evitar deixar qualquer
quantidade, por menor que seja, do leite em pó aderido nas paredes do frasco, sem dissolver). Se necessário pode-se aquecer levemente e resfriar antes de prosseguir a análise.
1.3. Adicionar 8 mL da solução tampão pH 10 e o indicador de negro de eriocromo T. 1.4. Titular com EDTA 0,01 M, até o aparecimento da coloração azul do indicador livre. 1.5. Realizar o procedimento em triplicata. 1.6. Calcular o teor de cálcio na amostra de leite, expressando o resultado em ppm de Ca. 1.7. Compare com o valor teórico.
1 ppm = 1 mg/L

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
35
Aula 11 - Volumetria de complexação: Determinação de dureza da água
Objetivos
Determinar a dureza total, o cálcio e o magnésio em amostras de água para abastecimento público e águas naturais em geral, através do método titulométrico do EDTA-Na.
Preparação pré-laboratório
DEFINIÇÃO: Característica conferida à água, pela presença de sais alcalino-terrosos (cálcio, magnésio, e outros) e de alguns metais, em menor intensidade. Quando a dureza é devida aos sais bicarbonatos e carbonatos (de cálcio, magnésio, e outros), denomina-se temporária, pois pode ser eliminada quase totalmente pela fervura; quando é devida a outros sais, denomina-se permanente. As águas duras, em função de condições desfavoráveis de equilíbrio químico, podem incrustar nas tubulações e dificultar a formação de espumas com o sabão. (NBR 9896/1993). A dureza total de uma amostra de água é a concentração total de cátions bivalentes, principalmente de cálcio e magnésio, expressa em termos de CaCO3. Exprime a dureza da água obtida pela soma das durezas de carbonatos (dureza temporária) e de não-carbonatos (dureza permanente). MÉTODOS:
1) Método titulométrico do EDTA-Na (NBR 12621/Set 1992). 2) Método complexométrico (NBR 5761/Dez 1984).
Princípio do Método titulométrico do EDTA-Na para determinação da Dureza Total: Os íons Ca2+ e Mg2+ de umas solução formam um complexo vermelho vinho com o corante negro de eriocromo-T, em pH= 10,0. Pela adição de EDTA-Na à solução colorida, ocorre a formação de um complexo estável e não dissociado do EDTA-Na com íons Ca++ e Mg++, separando-se o corante. Quando a quantidade de EDTA-Na for suficiente para complexar todo o cálcio e magnésio, a solução toma a cor azul original do corante, o que indica o fim da titulação.
Princípio do Método titulométrico do EDTA-Na para determinação do Cálcio: O EDTA quando adicionado a uma amostra de água contendo Ca2+ e Mg2+ combina principalmente com o cálcio que pode ser determinado diretamente, com o EDTA. Para tanto, deve-se elevar o pH, a fim de precipitar o magnésio como hidróxido, e usar um indicador que se combine somente com o cálcio. Alguns indicadores provocam uma visível troca de cor quando todo o cálcio é complexado pelo EDTA, em pH na faixa de 12-13.
Materiais e Reagentes
• 2 erlenmeyer de 250 mL
• Béquer
• Proveta
• Bureta
• Pipeta volumétrica
• Pera
• Solução de EDTA-Na (sal dissódico de EDTA) 0,01 mol/L
• Solução tampão NH4OH/NH4Cl
• Papel indicador universal
• Solução de NaOH (hidróxido de sódio) 1 mol/L
• Indicador murexida.
• Indicador Negro de eriocromo -T
Procedimento Experimental 1. Determinação da dureza total da amostra
1.1. Transferir 50 mL da amostra para um erlenmeyer, utilizando pipeta volumétrica. 1.2. Adicionar 10 mL da solução de tampão pH 10 e indicador de negro de eriocromo-T. 1.3. Titular com EDTA-Na 0,01mo/L, lentamente e com agitação constante até mudança da coloração de
vermelho vinho para azul, anotar o volume gasto. 1.4. Realizar o procedimento em triplicata. 1.5. Efetuar uma prova em branco com igual volume de água destilada para facilitar a observação da
viragem e corrigir possível contaminação da água destilada com Ca2+ e Mg2+. Anotar o volume gasto. 2. Determinação do Cálcio
2.1. Pipetar 50 mL da água a analisar num erlenmeyer de 250 mL e adicionar 2 mL de NaOH 1 mol/L, para elevar o pH entre 12 e 13 testando com o papel indicador universal.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
36
2.2. Juntar aproximadamente 0,2 mg de murexida à amostra e titular lentamente com EDTA 0,01 mol/L até mudança na coloração de rósea para púrpura.
2.3. Anotar o volume gasto na titulação 2.4. Efetuar uma prova em branco com igual volume de água destilada para facilitar a observação da
viragem e corrigir possível contaminação da água destilada com Ca2+. Anotar o volume gasto. 3. EXPRESSÃO DOS RESULTADOS A dureza total é expressa por: mg./.01/2 = �% − %3 × �� × 0,01 × 100 × 1000%7
onde, V1= Volume (mL) de solução de EDTA-Na gasto na titulação da amostra. Vb= Volume (mL) de solução de EDTA-Na gasto na titulação do branco. fc= fator de correção volumétrica da solução de EDTA-Na. Va= Volume (mL) da amostra.
O teor de cálcio é determinado por: mg.//2 = �%� − %3 × �� × 0,01 × 40,08 × 1000%7
onde, V2= Volume (mL) de solução de EDTA-Na gasto na titulação da amostra. Vb= Volume (mL) de solução de EDTA-Na gasto na titulação do branco. fc= fator de correção volumétrica da solução de EDTA-Na. Va= Volume (mL) da amostra.
Pela diferença de volume gasto para titular a dureza total e o cálcio obtém-se o teor de Mg2+: mg:;/2 = �% − %� × �� × 0,01 × 24,31 × 1000%7
onde, V1= Volume (mL) de solução de EDTA-Na gasto na determinação da dureza total. V2= Volume (mL) de solução de EDTA-Na gasto na determinação do cálcio. fc= fator de correção volumétrica da solução de EDTA-Na. Va= Volume (mL) da amostra.
Referências Bibliográficas 1. Associação Brasileira de Normas Técnicas- ABNT/ NBR 12621/Set 1992, NBR 13799/ Abr 1997 e NBR 9896/1993.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
37
Aula 12 - Volumetria de Oxirredução: Permanganometria
Objetivos
Padronização de uma solução de permanganato de potássio com solução de oxalato de sódio e determinação da concentração de água oxigenada em % m/v e em volumes de oxigênio.
Preparação pré-laboratório
A permanganometria é um tipo de volumetria de oxidação-redução que utiliza permanganato de potássio como titulante, o qual apresenta alto poder de oxidação. As reações de oxirredução ocorrem em meio ácido, no qual MnO4
- é reduzido a Mn2+. As soluções aquosas de KMnO4 não são completamente estáveis, porque o íon MnO4
- tende a oxidar a água e se decompõe com exposição à luz, portanto, devem ser estocadas em fracos escuros e padronizadas periodicamente.
Comercialmente, a concentração de água oxigenada é referida a volume de oxigênio, ou seja, o volume de oxigênio gerado por uma determinada concentração de água oxigenada.
Assim, 1,00 mL H2O2 a 100 volumes liberará 100,00 mL de O2 nas CNPT.
2 H2O2(aq) � O2(g) + 2 H2O(l) A água oxigenada apesar de ser um agente oxidante pode ser oxidada pelo permanganato em meio ácido.
2 KMnO4(aq) + 3 H2SO4(aq) + 5 H2O2(aq) � K2SO4(aq) + 2 MnSO4(aq) + 5 H2O(l)
Procedimento Experimental 1. Padronização de uma solução de permanganato de potássio 0,02 mol/L com solução de oxalato de
sódio 0,05 mol/L (padrão primário) 1.1. Transferir uma alíquota de 5,00 mL, de uma solução padrão de oxalato de sódio 0,05 mol/L, com
auxílio de uma pipeta volumétrica, para um erlenmeyer de 125 mL. 1.2. -Acrescentar 50,00 mL de água destilada e 2,00 mL de ácido sulfúrico 20% (v/v) ao erlenmeyer
contendo a solução de oxalato de sódio 1.3. -Aquecer a solução de oxalato de sódio à no máximo 80ºC. 1.4. -Titular com uma solução de permanganato de potássio (KMnO4) até o aparecimento de uma
coloração violeta clara, tendendo a rósea, permanente por 30 segundos. No final da titulação, a temperatura da solução titulada deverá ser, no mínimo, de 60ºC. Anote o volume gasto.
1.5. O íon permanganato reage com o íon oxalato numa proporção de 2:5, nessa ordem, conforme reação abaixo:
2 MnO4−
(aq) + 5 H2C2O4(aq) + 6 H+ �2 Mn+(aq) + 10 CO2(g) +8 H2O(l)
O aquecimento da solução acelera a reação, porém não deve atingir 100ºC, temperatura na qual o oxalato é decomposto. O controle da temperatura deve ser feito com termômetro.
O H2SO4 é o reagente apropriado para acidificar a solução porque o íon sulfato não sofre a ação do permanganato. Também pode ser utilizado o ácido perclórico. O ácido clorídrico pode sofrer oxidação do íon cloreto.
O ponto final do permanganato não é permanente porque o excesso de íons permanganato reage lentamente com os íons manganês(II) presentes em concentração relativamente elevada no ponto final, de acordo com a reação:
2 MnO4−
(aq) + 3 Mn2+(aq) + 2 H2O(l) � 5 MnO2(s) + 4 H+
(aq) 2. Determinação da concentração de água oxigenada em % m/v e em volumes de oxigênio.
2.1. Transferir 1,00 mL de uma amostra de água oxigenada, com auxílio de uma pipeta volumétrica, para um erlenmeyer de 125 mL.
2.2. Acrescentar 5,0 mL de ácido sulfúrico 20% (v/v) e cerca de 50,00 mL de água destilada. 2.3. Titular com uma solução de permanganato de potássio ~0,02mol/L até o aparecimento de uma
coloração rósea persistente por 30 segundos. Anote o volume gasto durante o procedimento de titulação.
2.4. -Repetir esse procedimento mais duas vezes
5 H2O2(l) + 2 MnO4−
(aq) + 6 H+ � 5 O2(g) + 2 Mn2+(aq) + 8 H2O(l)

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
38
Anexo 1 - Normas de segurança e boa conduta em laboratórios de química
É muito importante que todas as pessoas que lidam num laboratório tenham uma noção bastante clara dos riscos existentes e de como diminuí-los. Nunca é demais repetir que o melhor combate aos acidentes é a sua prevenção. O descuido de uma única pessoa pode por em risco todos os demais no laboratório. Por esta razão, as normas de segurança descritas a seguir terão seu cumprimento exigido. Acima disto, porém, espera-se que todos tomem consciência da importância de se trabalhar em segurança, do que só resultarão benefícios para todos.
1. É altamente recomendado o uso de jaleco ou guarda-pó no laboratório por estudantes e
professores. A não observância desta norma gera roupas furadas por agentes corrosivos, queimaduras, etc.
2. Nunca despejar água num ácido, mas sim o ácido sobre a água. Além disso, o ácido deve ser adicionado lentamente, com agitação constante.
3. Os alunos não devem tentar nenhuma reação não especificada pelo professor. Reações desconhecidas podem causar resultados desagradáveis.
4. É terminantemente proibido fumar em qualquer laboratório.
5. É proibido trazer comida ou bebida para o laboratório, por razões óbvias. Da mesma forma, não se deve provar qualquer substância do laboratório, mesmo que inofensiva.
6. Cuidado com lentes de contato quando estiver trabalhando em laboratórios, devido ao perigo de, num acidente, ocorrer a retenção de líquido corrosivo entre a lente e a córnea.
7. Não cheirar um reagente diretamente. Os vapores devem ser abanados em direção ao nariz, enquanto se segura o frasco com a outra mão.
8. Não usar sandálias no laboratório. Usar sempre algum tipo de calçado que cubra todo o pé.
9. Não usar bermudas ou saias no laboratório, utilizar preferencialmente uma vestimenta tipo calça comprida (jeans ou outro tecido)
10. Nunca acender um bico de gás quando alguém no laboratório estiver usando algum solvente orgânico. Os vapores de solventes voláteis, como éter etílico, podem se deslocar através de longas distâncias e se inflamar facilmente.
11. Não deixar livros, blusas, etc., jogadas nas bancadas. Ao contrário, colocá-los longe de onde se executam as operações.
12. Não pipetar nenhum tipo de produto com a boca (exceto se orientado pelo professor).
13. Não levar as mãos à boca ou aos olhos quando estiver trabalhando com produtos químicos.
14. Fechar cuidadosamente as torneiras dos bicos de gás depois de seu uso.
15. Não aquecer tubos de ensaio com a boca virada para o seu lado, nem para o lado de outra pessoa.
16. Usar equipamentos apropriados nas operações que apresentarem riscos potenciais.
17. Não aquecer reagentes em sistemas fechados.
18. Fechar todas as gavetas e portas que abrir.
19. Planejar o trabalho a ser realizado.
20. Verificar as condições da aparelhagem. Não trabalhar com material imperfeito ou defeituoso, principalmente com vidro que tenha trincas, pontas ou arestas cortantes.
21. Conhecer a periculosidade dos produtos químicos.
22. Aprender a localização e a utilização do extintor de incêndio existente.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
39
23. Manter as bancadas sempre limpas e livres de materiais estranhos ao trabalho.
24. Jogar papéis usados e materiais inservíveis no lixo somente quando não apresentar riscos.
25. Utilizar a capela ao trabalhar com reações que liberem fumos venenosos ou irritantes.
26. Em caso de derramamento de produtos tóxicos, inflamáveis ou corrosivos, tomar as seguintes precauções:
• Parar o trabalho, isolando na medida do possível a área;
• Advertir pessoas próximas sobre o ocorrido;
• Só efetuar a limpeza após consultar a ficha de emergência do produto;
• Alertar o professor;
• Verificar e corrigir a causa do problema;
• No caso de envolvimento de pessoas, lavar o local atingido com água corrente e procurar o serviço médico.
27. Saber tomar certas iniciativas em caso de pequenos acidentes. Exemplos:
• Todas as vezes que ocorrer um acidente com algum aparelho elétrico (centrífuga, por exemplo), retirar imediatamente o pino da tomada;
• Cuidado com mercúrio entornado (de termômetros quebrados, por exemplo). O mercúrio, além de corrosivo, é muito tóxico. Deve-se coletá-lo ou cobri-lo com enxofre ou zinco em pó;
• Procurar conhecer a toxidez dos vários reagentes usados e tratá-los com a devida seriedade;
• Lembrar que em caso de incêndio, na ausência de um extintor, um guarda-pó pode servir como um cobertor para abafar as chamas.
28. Comunicar imediatamente ao professor qualquer acidente ocorrido.
29. Finalmente, lembrar que a atenção adequada ao trabalho evita a grande maioria dos acidentes. É muito importante ter a certeza de que se sabe perfeitamente bem o que se está fazendo.
Materiais de vidro e conexões
1. Ao usar material de vidro, verifique sua condição. Lembre-se que o vidro quente tem a mesma aparência que a do vidro frio. Qualquer material de vidro trincado deve ser rejeitado e comunicado ao professor ou monitor.
2. Vidros quebrados devem ser descartados em local apropriado, nunca no lixo comum.
3. Usar sempre um pedaço de pano protegendo a mão quando estiver cortando vidro ou introduzindo-o em orifícios. Antes de inserir tubos de vidro em tubos de borracha ou rolhas lubrifique-os.
4. Ter cuidado especial ao trabalhar com sistemas sob vácuo ou pressão. Dessecadores sob vácuo devem ser protegidos com fita adesiva e colocados em grades de proteção próprias.
Os Resíduos
Os resíduos aquosos ácidos ou básicos comuns devem ser neutralizados antes e só então descartados na pia. Para ácidos, bases ou sais que são prejudiciais ao meio ambiente e seres vivos (metais tóxicos, por exemplo), o descarte deve ser realizado nos recipientes adequados.
Limpeza
É importante que o usuário do laboratório habitue-se a limpar o material, principalmente vidro e cerâmica, logo após o término do experimento, enquanto a natureza do resíduo é conhecida. O material de vidro após o uso deve ser lavado com água e detergente com o auxílio de uma escova. Depois de bem enxaguado com água da torneira, enxaguar duas vezes com água destilada. Depois

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
40
de lavado, o vidro deve permitir o escoamento de água sobre sua superfície, sem formar gotas, que indicam a presença de matéria gordurosa. Deixar secar sobre a bancada em papel toalha ou na estufa, lembrando-se de avisar o técnico responsável sobre o material dentro da estufa.
Equipamentos básicos de laboratório
A execução de qualquer experimento na Química envolve geralmente a utilização de uma variedade de equipamentos de laboratório, a maioria muito simples, porém com finalidades específicas. O emprego de um dado equipamento ou material depende dos objetivos e das condições em que a experiência será executada. No Anexo 1 encontram-se diversas ilustrações com os respectivos nomes e funções de diversos equipamentos laboratoriais.
A T E N Ç Ã O – I M P O R T A N T E Em caso de acidente de qualquer natureza, é indispensável manter a calma e agir com rapidez e precisão. É preferível evitar que os acidentes aconteçam, observando sempre as medidas de segurança. Lembre-se de avisar o professor ou técnico o mais rápido possível.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
41
Anexo 2 - Equipamentos Básicos de Laboratório
Materiais de vidro
1. Tubo de ensaio: utilizado principalmente para efetuar reações químicas em pequena escala. 2. Béquer: recipiente com ou sem graduação utilizado para o preparo de soluções, aquecimento de líquidos, recristalizações.
3. Erlenmeyer: frasco utilizado para aquecer líquidos ou para efetuar titulações.
4. Kitassato: frasco de paredes espessas, munido de saída lateral e usado em filtrações sob vácuo.
5. Balão volumétrico: recipiente calibrado, de precisão, destinado a conter um determinado volume de líquido, a uma dada temperatura; utilizado no preparo de soluções de concentrações definidas.
6. Proveta: frasco cm graduações, destinado a medidas aproximadas de volume de líquidos.
7. Bureta: equipamento calibrado para medida precisa de volume de líquidos. Permite o escoamento do líquido e é muito utilizada em titulações.
Pipeta: equipamento calibrado para medida precisa de volume de líquidos. Existem dois tipos de pipetas: pipeta graduada (8) e pipeta volumétrica (9). A primeira é utilizada para escoar volumes variáveis e a segunda para escoar volumes fixos de líquidos.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
42
10. Funil: utilizado na transferência de líquidos de um frasco para outro ou para efetuar filtrações simples.
11. Vidro de relógio: usado geralmente para cobrir béqueres contendo soluções e finalidades diversas.
12. Dessecador: utilizado no armazenamento de substâncias quando se necessita de uma atmosfera com baixo teor de umidade. Também pode ser utilizado para manter as substâncias sob pressão reduzida. Existem vários tipos.
13. Pesa-filtro: recipiente destinado à pesagem de sólidos.
14. Bastão de vidro: usado na agitação e transferência de líquidos.
15. Funil de separação: equipamento para separar líquidos não miscíveis. Existem vários modelos.
Condensador: Equipamento destinado à condensação de vapores, em destilações ou aquecimento sob refluxo. Existem três tipos básicos, condensador reto ou liso (16), de bola (17) e espiral (18). O primeiro mais utilizado em destilações e os últimos para refluxo.
19. Termômetro: Usado para medidas de temperatura.
Materiais de porcelana
20. Funil de Büchner: utilizado em filtrações por sucção, devendo ser acoplado a um kitassato. 21. Cápsula de porcelana: usada para efetuar evaporação de líquidos.
22. Cadinho: usado para calcinação de substâncias.
23. Almofariz (gral) e pistilo: destinados à pulverização de sólidos. Além de porcelana, podem ser feitos de ágata, vidro ou metal.
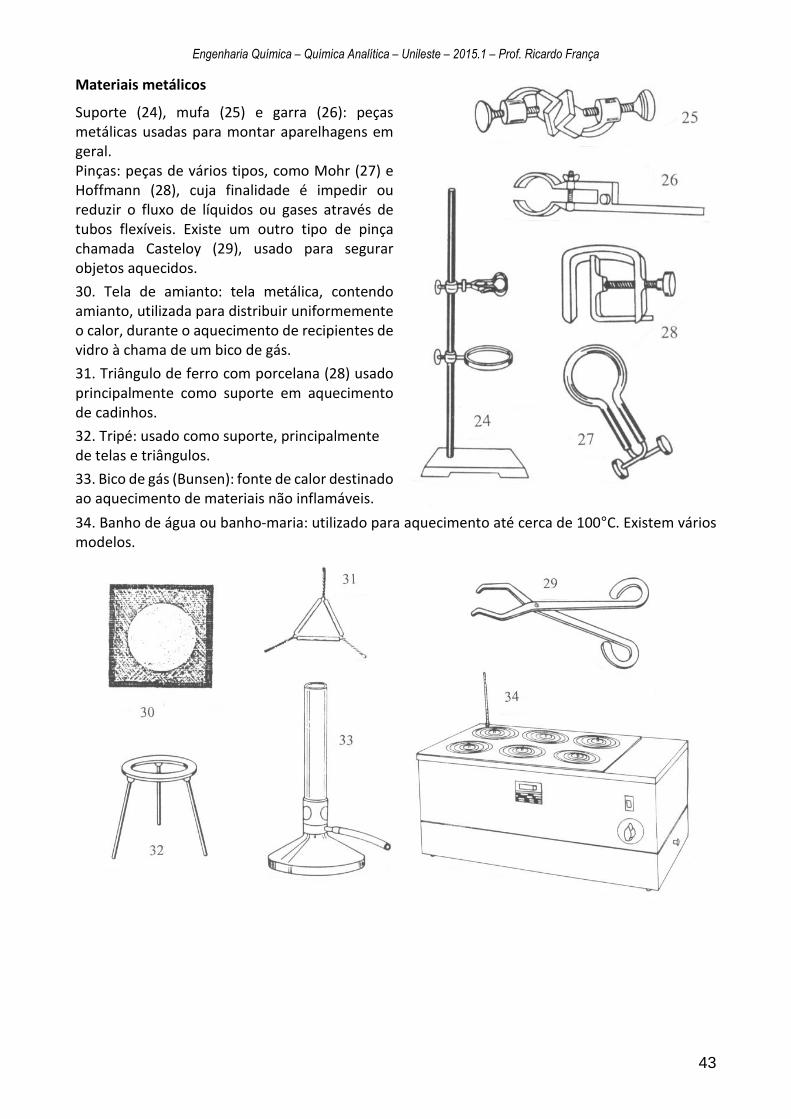
Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
43
Materiais metálicos
Suporte (24), mufa (25) e garra (26): peças metálicas usadas para montar aparelhagens em geral. Pinças: peças de vários tipos, como Mohr (27) e Hoffmann (28), cuja finalidade é impedir ou reduzir o fluxo de líquidos ou gases através de tubos flexíveis. Existe um outro tipo de pinça chamada Casteloy (29), usado para segurar objetos aquecidos.
30. Tela de amianto: tela metálica, contendo amianto, utilizada para distribuir uniformemente o calor, durante o aquecimento de recipientes de vidro à chama de um bico de gás.
31. Triângulo de ferro com porcelana (28) usado principalmente como suporte em aquecimento de cadinhos.
32. Tripé: usado como suporte, principalmente de telas e triângulos.
33. Bico de gás (Bunsen): fonte de calor destinado ao aquecimento de materiais não inflamáveis.
34. Banho de água ou banho-maria: utilizado para aquecimento até cerca de 100°C. Existem vários modelos.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
44
35. Argola: usada como suporte para funil de vidro ou tela metálica. 36. Espátula: usada para transferir substâncias sólidas. Existem vários modelos. 37. Furador de rolhas: utilizado na perfuração de rolhas de cortiça ou borracha.
Materiais diversos
38. suporte para tubos de ensaio. Existem vários modelos de diversos tamanhos. 39. Pinça de madeira: utilizada para segurar tubos de ensaio. 40. Pisseta: frasco geralmente contendo água destilada, álcool ou outros solventes, usado para efetuar a lavagem de recipientes ou materiais com jatos do líquido nele contido. 41. Trompa de água: dispositivo para aspirar o ar e reduzir a pressão no interior de um frasco; muito utilizado em filtrações por sucção. 42. Estufa: equipamento empregado na secagem de materiais, por aquecimento, em geral até
200°C.
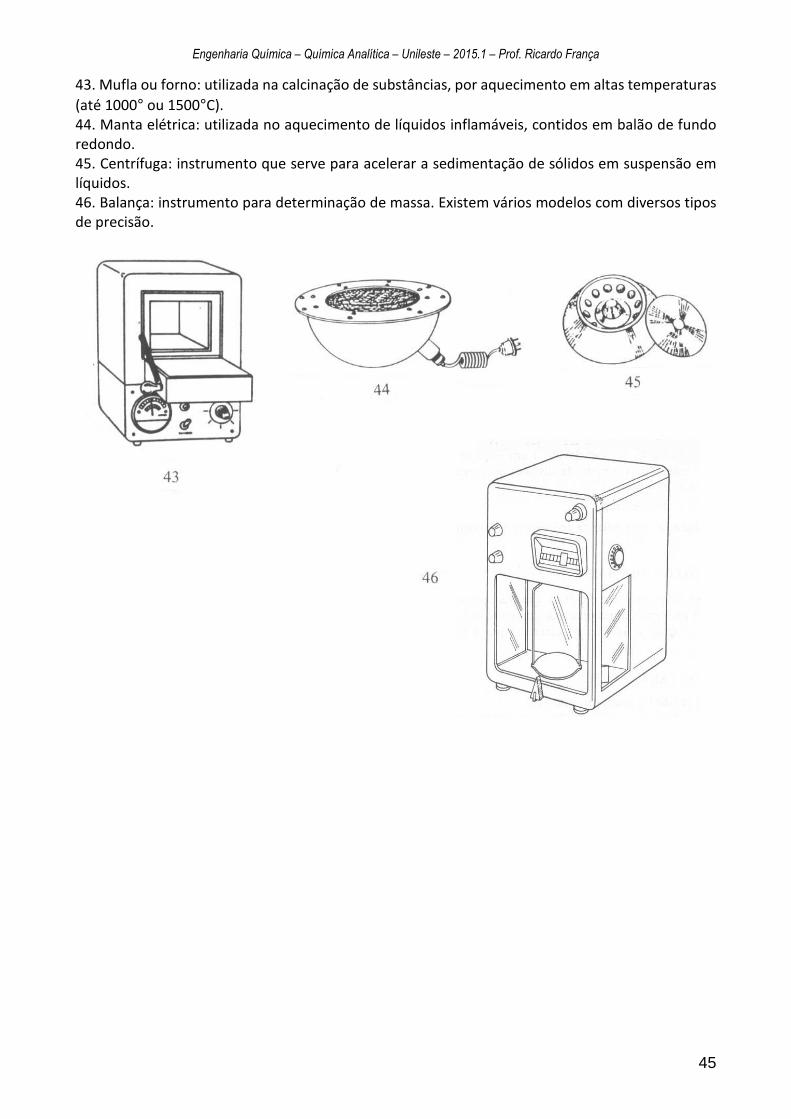
Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
45
43. Mufla ou forno: utilizada na calcinação de substâncias, por aquecimento em altas temperaturas
(até 1000° ou 1500°C). 44. Manta elétrica: utilizada no aquecimento de líquidos inflamáveis, contidos em balão de fundo redondo. 45. Centrífuga: instrumento que serve para acelerar a sedimentação de sólidos em suspensão em líquidos. 46. Balança: instrumento para determinação de massa. Existem vários modelos com diversos tipos de precisão.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
46
Anexo 3 - Folhas de respostas
Aula 1 – Preparação e aferição de soluções
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Massas necessárias para o preparo das soluções.
Nº Solução Teor (pureza) em % Massa Volume
1 NaOH 0,1 M
---
2 HCl 0,2 M
3 H2SO4 0,1 M
Tabela 2. Padronização das soluções.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) Fator de
correção (Fc)
1 NaOH 0,1 M
2 H2SO4 0,1 M
3 HCl 0,2 M
(Padrão 2º)
4 HCl 0,2 M
(Padrão 1º)
Questionário sobre a verificação experimental 1. Qual é o motivo para as soluções dos hidróxidos de metais alcalinos serem armazenadas em frascos de
plástico? 2. Qual é o motivo para o ácido clorídrico PA ter uma pureza, ou teor, tão baixo (36,5 - 38 %) comparado
com os outros ácidos? 3. Qual é o motivo para a utilização de água fervida ou recentemente destilada para a preparação de
soluções de hidróxidos de metais alcalinos? 4. Faça um desenho, aproximado, da fórmula estrutural das substâncias padrão primário abaixo:
a) biftalato de potássio; b) carbonato de sódio; c) ácido oxálico; d) ácido benzoico; e) oxalato de sódio.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
47

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
48
Aula 3 – Determinação de ácido acético em vinagre
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela1. Determinação de ácido acético em vinagre. Nº Solução 1ª réplica (mL) 2ª réplica (mL) Média (mL)
1 Amostra de vinagre
Cálculos
1. Quantidade de ácido acético presente na amostra de vinagre: Devido à estequiometria da reação temos:
n moL de HAc ––––––––– n de NaOH onde n= número de mols
Como n = (Fc x concentração) x volume temos que a quantidade de matéria de NaOH é:
n = Y = (Fc x 0,1) x volume gasto na titulação (em mL) x 0,001 = Y mol de NaOH = Y mol de HAc
Assim temos 1,0 moL de HAc ––––––––– ____ g de HAc Y moL de HAc ––––––––––– X g de HAc
2. Cálculo das concentrações: 2.1 em mol/L Y mol de HAc ––––––––––– 1 mL Z mol de HAc ––––––––––– 1000 mL � Z = mol/L de HAc 2.2 em g/L X g de HAc –––––––––––– 1 mL W g de HAc ––––––––––– 1000 mL � W = g/L de HAc 2.3 em % m/v X g de HAc –––––––––––– 1 mL Q g de HAc –––––––––––– 100 mL � Q = % m/V de HAc
Tabela2. Concentração de ácido acético no vinagre. Nº Solução mol/L g/L % m/v
1 Ácido acético em
vinagre
Questionário sobre a verificação experimental 1. Porque o ácido acético PA também é chamado de ácido acético glacial? 2. Industrialmente, o ácido acético pode ser obtido pela carbonilação do metanol. Porque,
mundialmente, o ácido acético utilizado na produção de vinagre (para consumo alimentício) é obtido através do método de fermentação acética (com o microorganismo Mycoderma aceti)?

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
49

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
50
Aula 4 – Determinação da concentração de ácido fosfórico
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Determinação de ácido fosfórico.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) Teor (%)
1 H3PO4 0,05 M
(dois indicadores)
2 H3PO4 0,05 M (um indicador)
Questionário sobre a verificação experimental
1. Explique qual dos dois métodos, de acordo com o seu trabalho experimental, pode ser considerado o mais correto, o que trata o ácido como diprótico (dois hidrogênios – dois pontos de viragem – dois indicadores) ou o que o trata como monoprótico (um hidrogênio – um ponto de viragem – um indicador) explicando seus motivos.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
51

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
52
Aula 5 – Determinação de ácido acetilsalicílico em medicamento
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Determinação de AAS.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) Média (mL)
1 AAS
Cálculos
NaOH adicionado (mol) - NaOH que não reagiu (mol) = NaOH que reagiu (mol)
O ácido acetilsalicílico presente na amostra, em número de moles, corresponde exatamente à quantidade do NaOH que reagiu, de acordo com a proporção estequiométrica da reação ácido base.
Quantidade de AAS em cada comprimido (500 mg) = _______________________ .
Erro % = ________________ .
Questionário sobre a verificação experimental 1 - Qual é o nome do procedimento utilizado nesta titulação? 3 - Qual é o motivo da necessidade do aquecimento da solução contendo o AAS?

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
53

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
54
Aula 6 – Determinação do teor de hidróxido de magnésio no leite de magnésia
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Determinação de Mg(OH)2 em leite de magnésia.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL)
1 Mg(OH)2
Concentração de Mg(OH)2 em % m/m = ________________ .
Questões a serem respondidas durante a elaboração do relatório 1. Escreva as equações químicas envolvidas na titulação.
2. Calcular a concentração em gL−1 de hidróxido de magnésio em leite de magnésia

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
55

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
56
Aula 7 – Padronização de solução de AgNO3 e determinação de cloreto (método de Mohr)
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Padronização de solução de AgNO3.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) Fc
1 AgNO3
Tabela 2. Concentração de cloreto em amostra de KCl 0,1M.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) Média (mL) C (mol/L) C (g/L)
1 KCl 0,1M
Tabela 3. Concentração de cloreto em amostra de NaCl.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) Média (mL) C (mol/L) C (% m/v)
1 NaCl
QUESTÕES PÓS-LABORATÓRIO 1. Quais os fatores a serem observados na escolha de uma titulação por precipitação? 2. Por que a titulação por precipitação de Mohr não pode ser realizada em meio ácido ou meio muito
alcalino? 3. Escreva todas as reações da análise. 4. A padronização da solução de AgNO3 pode ser efetuada em meio fortemente ácido ou fortemente
básico? Justifique. 5. Se o KPS do Ag2CrO4 é menor que o KPS do AgCl, por que o AgCl precipita primeiro?

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
57

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
58
Aula 8 – Padronização de solução de AgNO3 e determinação de cloreto (método de Volhard)
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Concentração de cloreto em amostra de NaCl.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) C NaCl
(% m/v) C Cl−
(% m/v)
1 NaCl
QUESTÕES PÓS-LABORATÓRIO 1. Explique resumidamente o método de Volhard. 2. Qual a função de adicionarmos HNO3 à solução inicial? 3. Qual a razão de colocarmos AgNO3 em excesso? 4. Em quantidades maiores, usamos o nitrobenzeno como película protetora do NaCl, justifique esse
procedimento.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
59

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
60
Aula 9 – Volumetria de complexação: padronização de solução de Na2EDTA e quantificação de metais
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Padronização de solução de Na2EDTA 0,01 M.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) Fc
1 Na2EDTA 0,01 M
Tabela 2. Concentração de cloreto em amostra de NaCl.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) C Zn2+
(% m/v) C Zn2+
(mol/L)
1 ZnSO4
QUESTÕES PÓS-LABORATÓRIO 1. Na determinação de zinco por complexometria, utilizou-se EDTA 0,01 M, na titulação foram
consumidos 20 mL para 10 mL da amostra. Determine: a) Quantos g de zinco contém a amostra b) Quantos mols de zinco corresponde a massa encontrada em a.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
61

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
62
Aula 10 – Volumetria de complexação: Determinação de cálcio na casca do ovo e no leite
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Concentração de cálcio em amostra de casca de ovo.
Nº Solução 1ª réplica
(mL) 2ª réplica
(mL) 3ª réplica
(mL) Média (mL)
Massa de casca de ovo (g)
C Ca2+ (% m/m)
C CaCO3 (% m/m)
1 casca de
ovo
Tabela 2. Concentração de cálcio em amostra de leite em pó.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) C Ca2+ (ppm)
1 leite em
pó

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
63

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
64
Aula 11 – Volumetria de complexação: Determinação de dureza da água
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Determinação da dureza total em amostra de água.
Nº Solução 1ª réplica
(mL) 2ª réplica
(mL) 3ª réplica
(mL) Média (mL)
C CaCO3 mg/L
1 Água (sais de Ca2+ e Mg2+)
Tabela 2. Concentração de cálcio em amostra de água.
Nº Solução 1ª réplica
(mL) 2ª réplica
(mL) 3ª réplica
(mL) Média (mL)
C Ca2+ mg/L
1 Água (sais de Ca2+ e Mg2+)
Tabela 3. Concentração de magnésio em amostra de água.
Nº Solução Vol. médio CaCO3 (mL)
Vol. médio Ca2+ (mL)
C Mg2+ mg/L
1 Água (sais de Ca2+ e Mg2+)
QUESTÕES PÓS-LABORATÓRIO
Questionário 1. Uma água é considerada dura quanto possui acima de quantos ppm de CaCO3? 2. Expresse a dureza da água do item 4 em ppm de CaCO3. 3. A água que você analisou é dura? 4. Uma amostra de água utilizada em caldeiras foi enviada ao seu laboratório para análise da sua dureza
em termos de ppm de cálcio. Você partiu de 15 mL da amostra, 50mg de Eriocromo T, tamponou a pH 10 e gastou 5,9 mL de solução padrão de EDTA 0,02 M. Você recomendaria a utilização desta água em caldeiras? Explique sua resposta.
5. Na determinação da dureza da água, gastou-se 13,3 mL de solução 0,02 M de EDTA para titular 100 mL da amostra, em presença de ERIO T e pH 10. Calcular a dureza em mg/L na forma de CaCO3.

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
65

Engenharia Química – Química Analítica – Unileste – 2015.1 – Prof. Ricardo França
66
Aula 12 – Volumetria de oxirredução: Permanganometria
Unileste Química Analítica Professor:
Data:
Grupo:
Tabela 1. Padronização de solução de KMnO4 0,02 M.
Nº Solução 1ª réplica (mL) 2ª réplica (mL) 3ª réplica (mL) Média (mL) Fc
1 KMnO4 0,02 M
Tabela 2. Concentração de H2O2 em amostra de água oxigenada.
Nº Solução 1ª réplica
(mL) 2ª réplica
(mL) 3ª réplica
(mL) Média (mL)
C H2O2 (mol/L)
C H2O2 (% m/v)
C H2O2 (volumes)
1 H2O2
QUESTÕES PÓS-LABORATÓRIO 1. Quais são os agentes oxidante e redutor na reação?


















