
UNICAMP ANA ERIKA DIAS FERREIRA EXPRESSÃO … · shed light on the involvement of the FGF pathway...
92
i UNICAMP ANA ERIKA DIAS FERREIRA EXPRESSÃO HIPOCAMPAL DE FATORES DE CRESCIMENTO DE FIBROBLASTOS EM PACIENTES COM EPILEPSIA DO LOBO TEMPORAL HIPPOCAMPAL EXPRESSION OF FIBROBLAST GROWTH FACTORS IN TEMPORAL LOBE EPILEPSY PATIENTS Campinas 2014
Transcript of UNICAMP ANA ERIKA DIAS FERREIRA EXPRESSÃO … · shed light on the involvement of the FGF pathway...

i
UNICAMP
ANA ERIKA DIAS FERREIRA
EXPRESSÃO HIPOCAMPAL DE FATORES DE
CRESCIMENTO DE FIBROBLASTOS EM PACIENTES
COM EPILEPSIA DO LOBO TEMPORAL
HIPPOCAMPAL EXPRESSION OF FIBROBLAST GROWTH
FACTORS IN TEMPORAL LOBE EPILEPSY PATIENTS
Campinas
2014

ii

iii
UNICAMP
UNIVERSIDADE ESTADUAL DE CAMPINAS
Faculdade de Ciências Médicas
ANA ERIKA DIAS FERREIRA
EXPRESSÃO HIPOCAMPAL DE FATORES DE CRESCIMENTO DE
FIBROBLASTOS EM PACIENTES COM EPILEPSIA DO LOBO
TEMPORAL
“HIPPOCAMPAL EXPRESSION OF FIBROBLAST GROWTH FACTORS IN TEMPORAL LOBE EPILEPSY PATIENTS”
Dissertação apresentada à Faculdade de Ciências Médicas da Universidade Estadual de Campinas como parte dos requisitos exigidos para obtenção do título de Mestra em Ciências, na área de concentração Saúde da Criança e do Adolescente.
Masters thesis submitted to the Graduate Program in Child and Adolescent Health of the Faculty of Medical Sciences of the University of Campinas to obtain the title of Master of Science.
Orientador: Lília Freire Rodrigues de Souza Li
Coorientador: Marcelo Ananias Teocchi
Campinas
2014
ESTE EXEMPLAR CORRESPONDE À
VERSÃO FINAL DA DISSERTAÇÃO
DEFENDIDA PELA ALUNA ANA ERIKA DIAS
FERREIRA E ORIENTADA PELA PROF.ª. DR.ª.
LÍLIA FREIRE RODRIGUES DE SOUZA LI.
_______________________________
Assinatura da Orientadora

iv

v

vi

vii
RESUMO
Epilepsia do lobo temporal (ELT) é a forma mais comum de epilepsia em adultos. O
processo de epileptogênese inclui a morte neuronal, brotamento axonal, inflamação,
neurogênese, estresse oxidativo e gliose. No entanto, os mecanismos moleculares
subjacentes não são totalmente compreendidos. Os fatores de crescimento de
fibroblastos (FGFs) são uma família de proteínas com várias funções no organismo,
especialmente no sistema nervoso central. No entanto, o funcionamento dos FGFs no
cérebro humano não é totalmente compreendido. O FGF2 é o membro mais estudado
dessa família e seu papel na fisiopatologia da epilepsia é controversa. Na tentativa de
esclarecer o envolvimento da via de FGF na ELT, nós quantificamos a expressão
hipocampal dos seguintes genes: FGF2, FGF8, FGF22, FGFR1, FGFR2, FGFR3,
ITPR3, PIK3R3 e PIK3R5 em 10 pacientes resistentes a fármacos e quatro controles
post mortem. Além disso, avaliamos a expressão da proteína de FGF2 por
imunofluorescência indireta. Apenas para o FGF2, houve aumento do RNAm no
hipocampo dos pacientes para os dois genes de referência testados, HPRT1 e ENO2
+ TBP em combinação (P = 0,002 e P = 0,036; respectivamente). A porcentagem de
células imunomarcadas para FGF2 no giro dentado foi maior nos pacientes do que
nos controles (P <0,05), mas nenhuma alteração significativa foi encontrada no Corno
de Ammon. O FGF2 pode preservar os neurônios após lesão e atua como um
poderoso fator para a proliferação de células-tronco neurais. Assim, o FGF2 poderia
aliviar os danos induzidos pelas crises, intensificar a reparação e reduzir a
epileptogênese no hipocampo. Por outro lado, evidências têm demonstrado o

viii
envolvimento do FGF2 em mecanismos epileptogênicos, como brotamento de fibras
musgosas e neurogênese. Nossos resultados sugerem a participação do FGF2 na
fisiopatologia da ELT e o indica como um importante alvo para estudos
farmacológicos.

ix
ABSTRACT
Temporal lobe epilepsy (TLE) is the most common form of epilepsy in adults. The
process of epileptogenesis includes neuronal death, axonal sprouting, inflammation,
neurogenesis, oxidative stress and gliosis. However, the molecular mechanisms
behind them are not fully understood. Fibroblast growth factor (FGF) gene family
encodes proteins with several functions in the organism, especially in the central
nervous system. FGF family member functions in the human brain are unclear. To
shed light on the involvement of the FGF pathway in TLE, we quantified the
hippocampal expression of the following genes: FGF2, FGF8, FGF22, FGFR1,
FGFR2, FGFR3, ITPR3, PIK3R3 and PIK3R5 in 10 pharmacoresistant patients and
four post mortem controls. We also assessed the FGF2 protein expression by indirect
immunofluorescence. Only for FGF2, was the mRNA level markedly increased in
patients’ hippocampi for the two reference genes tested, HPRT1 and ENO2+TBP in
combination (P = 0.002 and P = 0.036, respectively). The percentage of FGF2
immunostained cells in the dentate gyrus was higher in patients than in the controls (P
<0.05), but no significant alteration was found in the Ammon’s horn. FGF2 preserves
neurons from ongoing injury and acts as a powerful proliferation factor for neural stem
cells. It could potentially alleviate seizure-induced damage and intensify repair and
reduce epileptogenesis in the hippocampus. On the other hand, evidence has shown
FGF2’s involvement in epileptogenic mechanisms, such as axonal sprouting and

x
neurogenesis. Our results clearly suggest the FGF2 participation in TLE
physiopathology and point it out as an important target for pharmacological studies.

xi
SUMÁRIO
RESUMO ...................................................................................................................................... vii
ABSTRACT .................................................................................................................................. ix
SUMÁRIO ..................................................................................................................................... xi
LISTA DE FIGURAS ................................................................................................................. xix
LISTA DE QUADROS E TABELAS ....................................................................................... xxi
LISTA DE ABREVIATURAS E SIGLAS ............................................................................. xxiii
1. INTRODUÇÃO ...................................................................................................................... 1
1.1 Classificação das Epilepsias .................................................................................................... 2
1.2 Epilepsia do Lobo Temporal .................................................................................................. 4
1.3 Hipocampo ............................................................................................................................... 5
1.4 Reorganização hipocampal na ELT ..................................................................................... 10
1.5 Fatores de Crescimento ......................................................................................................... 13
1.6 Fatores de Crescimento de Fibroblastos ............................................................................. 14
1.7 Fatores de Crescimento de Fibroblastos e Epilepsia .......................................................... 19
2. OBJETIVOS ......................................................................................................................... 25
3. CAPÍTULO 1 ........................................................................................................................ 27
4. CONCLUSÕES .................................................................................................................... 53
5. REFERÊNCIAS ................................................................................................................... 55
ANEXOS ...................................................................................................................................... 63

xii

xiii
A minha mãe Ana e a minha irmã Qelli que me ensinaram o mais
importante: amar.

xiv

xv
AGRADECIMENTOS
A minha orientadora Lília Li pela oportunidade e pelos ensinamentos.
Aos professores membros da banca avaliadora.
A CAPES pela bolsa de estudos e FAPESP pelo apoio financeiro.
Aos pacientes que aceitaram participar desse estudo.
Aos funcionários da secretaria e limpeza do CIPED.
Aos funcionários do LaCTAD pelos serviços prestados.
Aos funcionários e alunos do laboratório de investigação em patologia, laboratório
de imunologia pediátrica e laboratório de biologia molecular por todos os
momentos compartilhados no CIPED e pela ajuda com os experimentos.
Aos estagiários e alunos de IC do laboratório de endocrinologia pediátrica.
A todos meus professores do ensino fundamental, médio, graduação e pós-
graduação que mesmo com todas as dificuldades da profissão compartilharam
comigo um pouco do que sabiam e me ensinaram o gosto pelos estudos.
Aos meus amigos do CIPED: Vinicius Pedroni, Meire Rezende, Paula Hespanholo
e Vanessa Ramalho pelo apoio, pelas conversas, pelos ensinamentos e por toda
ajuda prestada.
A todos os meus amigos, em especial Erica Rodrigues, exemplo de mulher que,
como eu, sabe como é duro para a periferia chegar e se manter até onde estamos.
Meu especial agradecimento ao meu amigo e coorientador Marcelo Teocchi, por
todos os ensinamentos (não foram poucos) técnicos, biológicos, astrológicos e
espirituais; por todas as conversas, por todos os conselhos. Muito obrigada

xvi
mesmo! Sua bondade é uma das coisas que me faz acreditar ainda mais na
humanidade.
Aos meus irmãos: Valdiana, Vânio Flabio, Valdineia, Osvaldo Valerio e Thiago
Fernandes, sei da dedicação e carinho de cada um de vocês. Obrigada pelo
apoio.
Aos meus sobrinhos Jean, Júlio, Bia, Pedro, Anna Julia e Maria Eduarda, por fazer
meus dias mais felizes.
A minha cunhada Ana, pelas correções na dissertação e por todo carinho.
Ao meu companheiro Felipe, pelo amor tranquilo com sabor de fruta mordida. Por
me aconselhar, por me ouvir, pelo carinho, amor e dedicação. Por trazer leveza a
minha vida. Para você todo o meu amor.
Meu agradecimento especial a minha mãe Ana, pelo exemplo de honestidade e
esforço, por ter trabalhado tanto para que os seus filhos estudassem, por ter se
dedicado, por ter amado incondicionalmente. Espero um dia poder retribuir por
tudo isso.
A minha irmã Qelli, por toda amizade, cumplicidade, carinho e amor. Muito
obrigada por ser as flores, o canto dos pássaros, a areia da praia, a brisa do mar e
as estrelas do céu no meu caminho.
A todos aqueles cujo nome não aparece aqui, mas que marcaram e contribuíram
de alguma forma a minha vida.

xvii
“O que vale na vida não é o simples fato de termos vivido. É o que temos
feito de diferença na vida de outras pessoas que irá determinar o
significado da vida que levamos.”
Nelson Mandela.
“Ainda que eu falasse a língua dos homens e falasse a língua dos anjos,
sem amor, eu nada seria.”
Monte Castelo, Legião Urbana.
1 Coríntios 13

xviii

xix
LISTA DE FIGURAS
INTRODUÇÃO
Figura 1. Ilustração do sistema límbico onde está o hipocampo.. .............................. 6
Figura 2. Diagrama de um corte transversal do hipocampo.. ...................................... 7
Figura 3. Regiões do Corno de Ammon.. ....................................................................... 8
Figura 4. Principais circuitos neuronais no hipocampo. . ............................................. 9
Figura 5. Esquema da estrutura do FGFR.. ................................................................. 17
Figura 6. Representação da via de sinalização ativada por FGFR. ......................... 18
Figura 7. Expressão de diferentes isoformas de FGF2 por uso de códon de iniciação
alternativo.. ......................................................................................................................... 21
CAPÍTULO 1
Figura 1. Messenger RNA levels of fibroblast growth factor 2 (FGF2) in
pharmacoresistant temporal lobe epilepsy patients and post mortem
controls……………………………………...……………………………………………50
Figura 2. Immunofluorescence for FGF2 in the hippocampus of TLE patients and post
mortem controls....………………..……………………………………………………..51
Figura S1. Immunofluorescence for FGF2 in the hippocampus of TLE patients and post
mortem controls ………………………….……………………………………….…….52

xx

xxi
LISTA DE QUADROS E TABELAS
INTRODUÇÃO
Quadro 1. Classificação das epilepsias. Extraído de Berg et al., 2010 (7) ............... 3
Quadro 2. FGFs e seus receptores. Adaptado de Zhang et al., 2006 (42). ............ 16
CAPÍTULO 1
Table 1. Clinical data of TLE patients. ........................................................................... 48
Tabela 2. Genes and gene expression assays analyzed in this study. .................... 49

xxii

xxiii
LISTA DE ABREVIATURAS E SIGLAS
AA: Ácido araquidônico
ATP: Adenosine triphosphate
bFGF: basic fibroblast growth factor
CA: Corno de Ammon
CE: Córtex entorrinal
DG: dentate gyrus
DAG: diacilglicerol
DEPC: diethyl pyrocarbonate
EEG: electroencefalograma
ELT: epilepsia do lobo temporal
ENO2: enolase 2 (gamma, neuronal)
EROs: espécies reativas de oxigênio
ERN: espécies reativas de nitrogênio
FGFs: fatores de crescimento de fibroblastos
FGF2: fibroblast growth factor 2
FGF8: fibroblast growth factor 8
FGF22: fibroblast growth factor 22
FGFR1: fibroblast growth factor receptor 1
FGFR2: fibroblast growth factor receptor 2

xxiv
FGFR3: fibroblast growth factor receptor 3
FGFR4: fibroblast growth factor receptor 4
HPRT1: hypoxanthine phosphoribosyltransferase 1
HS: Hippocampal sclerosis
IL-1β: interleukin 1, beta
IL-6: interleukin 6
IL-10: interleukin 10
ILAE: International League Against Epilepsy
IRES – internal ribosome entry site
ITPR1: inositol 1,4,5-trisphosphate receptor, type 1
ITPR2: inositol 1,4,5-trisphosphate receptor, type 2
ITPR3: inositol 1,4,5-trisphosphate receptor, type 3
IP3: inositol-1,4,5-trifosfato
MAPK: mitogen-activated protein kinase
MRI: magnetic resonance imaging
mTOR: mechanistic target of rapamycin
NFκB: nuclear factor kappa B
NMDA: N-Methyl-D-aspartate
PBS: phosphate buffered saline
PCL-ϫ: phospholipase Cγ
PI3K: phosphoinosithide 3-kinase
PIK3C2α: class II PI3K alpha

xxv
PIK3C2β: class II PI3K beta
PIK3C2γ: class II PI3K gama
PIK3R3: phosphoinositide-3-kinase, regulatory subunit 3 (gamma)
PIK3R5: phosphoinositide-3-kinase, regulatory subunit 5
RIN: RNA integrity number
RT-qPCR: reverse transcription-quantitative PCR
STAT: signal transducer and activator of transcription
TBP: TATA box binding protein
TBS: tris-buffered saline
TLE: temporal lobe epilepsy

1
1. INTRODUÇÃO
Segundo a International League Against Epilepsy (ILAE), epilepsia é um distúrbio
cerebral caracterizado por predisposição a crises recorrentes e espontâneas. Crises,
por sua vez, são manifestações clínicas de descargas sincrônicas e excessivas de um
grupo de neurônios no cérebro (1).
Estima-se que 70 milhões de pessoas no mundo tenham epilepsia, o que configura
uma das desordens crônicas mais comuns (2). Oitenta e cinco por cento delas
encontram-se nos países em desenvolvimento, onde doenças parasitárias, como a
neurocisticercose, infecções intracranianas, traumas e tratamento médico ineficiente
são as principais causas da alta taxa de incidência (3, 4). No Brasil, estima-se que três
milhões de pessoas tenham epilepsia, sendo mais frequente em classes
socioeconômicas menos favorecidas e nos idosos (5).
Apesar do grande número de casos, os pacientes com epilepsia são socialmente
estigmatizados. O próprio termo “epilepsia”, de origem grega, remete a condição de ser
possuído ou invadido. Frequentemente, as pessoas com epilepsia experimentam
isolamento social, baixa auto-estima, problemas na família, escola e trabalho. Além
disso, o preconceito, também associado a essa condição, pode levar a pessoa a não
procurar tratamento médico ou não relatar adequadamente seus sintomas o que pode
agravar sua condição (6).

2
1.1 Classificação das Epilepsias
Para fins classificatórios, atualmente, as epilepsias são divididas em mais de 30
tipos (Quadro 1) (7).
Segundo a nova terminologia proposta pela ILAE, quanto à etiologia, as epilepsias
podem ser definidas como: [1] genética (idiopática), quando sua causa é decorrente de
um defeito genético; [2] estrutural-metabólica (sintomática) quando a epilepsia é
causada por lesões no sistema nervoso central tanto genéticas como adquiridas, como,
por exemplo, malformações, acidente vascular cerebral ou traumatismo craniano; e [3]
causa desconhecida (criptogênica) (7).
Este trabalho foca no tipo de epilepsia conhecida como Epilepsia do Lobo
Temporal (ELT) associada à esclerose hipocampal.
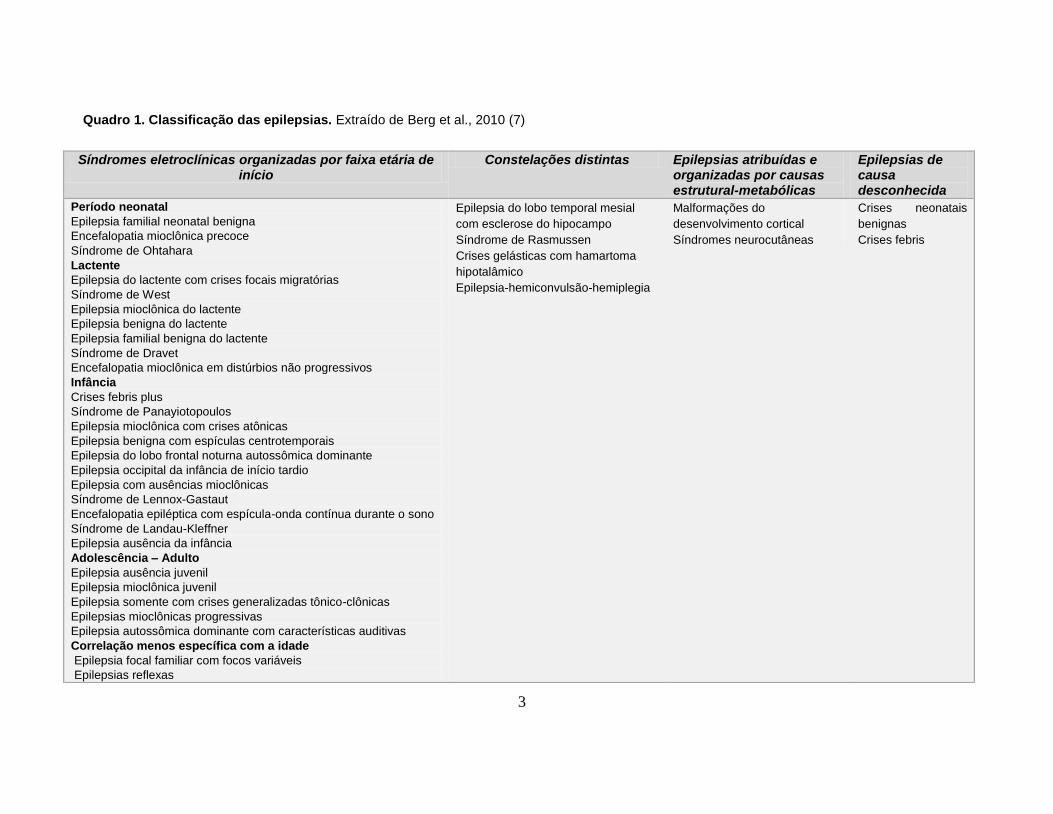
3
Quadro 1. Classificação das epilepsias. Extraído de Berg et al., 2010 (7)
Síndromes eletroclínicas organizadas por faixa etária de início
Constelações distintas Epilepsias atribuídas e organizadas por causas estrutural-metabólicas
Epilepsias de causa desconhecida
Período neonatal
Epilepsia familial neonatal benigna
Encefalopatia mioclônica precoce
Síndrome de Ohtahara
Lactente
Epilepsia do lactente com crises focais migratórias
Síndrome de West
Epilepsia mioclônica do lactente
Epilepsia benigna do lactente
Epilepsia familial benigna do lactente
Síndrome de Dravet
Encefalopatia mioclônica em distúrbios não progressivos
Infância
Crises febris plus
Síndrome de Panayiotopoulos
Epilepsia mioclônica com crises atônicas
Epilepsia benigna com espículas centrotemporais
Epilepsia do lobo frontal noturna autossômica dominante
Epilepsia occipital da infância de início tardio
Epilepsia com ausências mioclônicas
Síndrome de Lennox-Gastaut
Encefalopatia epiléptica com espícula-onda contínua durante o sono
Síndrome de Landau-Kleffner
Epilepsia ausência da infância
Adolescência – Adulto
Epilepsia ausência juvenil
Epilepsia mioclônica juvenil
Epilepsia somente com crises generalizadas tônico-clônicas
Epilepsias mioclônicas progressivas
Epilepsia autossômica dominante com características auditivas
Correlação menos específica com a idade
Epilepsia focal familiar com focos variáveis
Epilepsias reflexas
Epilepsia do lobo temporal mesial
com esclerose do hipocampo
Síndrome de Rasmussen
Crises gelásticas com hamartoma
hipotalâmico
Epilepsia-hemiconvulsão-hemiplegia
Malformações do
desenvolvimento cortical
Síndromes neurocutâneas
Crises neonatais
benignas
Crises febris

4
1.2 Epilepsia do Lobo Temporal
A ELT é a forma de epilepsia mais comum em adultos, com 40% dos casos
(8, 9). É comum o histórico de insultos cerebrais, tais como traumatismos
cranianos, infecções, status epilepticus ou crises febris na infância antecedentes
ao início da epilepsia, com um período ausente de crises de alguns anos após o
insulto e antes do aparecimento de crises recorrentes (8).
Clinicamente, os pacientes com esse tipo de epilepsia podem apresentar
crises parciais simples (envolve somente um hemisfério cerebral sem perda da
consciência) ou complexas (perda total ou parcial da consciência). A ocorrência de
manifestações motoras e sensitivo-sensoriais características, como mal estar
epigástrico, sensação de medo, angústia e déjà-vu são comuns. Após as crises
podem ocorrer um período de amnésia, afasia e confusão mental (10). Há também
perda da autonomia, declínio cognitivo e de memória com a progressão da doença
(11).
O tratamento é realizado com medicação que não previne o desenvolvimento
ou progressão da doença. Além disso, esse é a forma de epilepsia com maior
índice de refratariedade ao tratamento medicamentoso (12). Nestes casos, a
cirurgia é realizada.
A principal estrutura epileptogênica na ELT é o hipocampo (13).

5
1.3 Hipocampo
O hipocampo é uma estrutura cerebral situada na porção medial do lobo
temporal, próximo ao ventrículo lateral e está envolvido, principalmente, nos
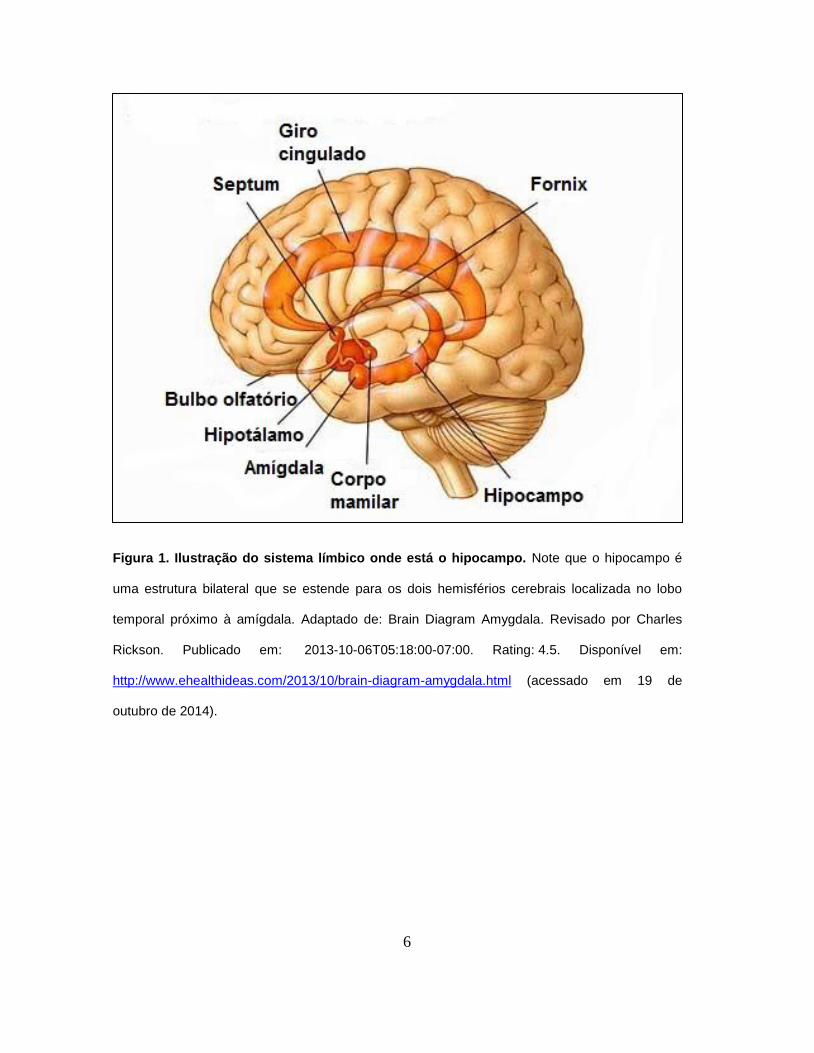
processos de aprendizagem e memória (Figura 1).
Anatomicamente, esse tecido pode ser dividido em duas regiões: o corno de
Ammon (CA) e o giro denteado (14).
O giro denteado pode ser dividido em três camadas: [1] a camada de células
granulares: onde está presente o corpo celular dos principais neurônios dessa
região; [2] camada molecular: contendo os dendritos das células granulares; e [3]
camada polimórfica ou hilo: constituído de axônios das células granulares (fibras
musgosas) e outros neurônios (14).
O corno de Ammon é constituído de seis camadas (Figura 2): [1] alveus:
onde passam as fibras aferentes e eferentes do hipocampo; [2] stratum oriens:
constituído principalmente pelos axônios das células piramidais;[3] stratum
piramidale: onde estão presentes os corpos celulares dos principais neurônios do
corno de Ammon; [4] stratum lucidum: composta pelas fibras musgosas; [5]
stratum radiatum: composta principalmente pelos dendritos das células piramidais;
e [6] stratum lacunosum-moleculare: constituído de fibras aferentes de outras
regiões cerebrais e de interneurônios (14).
Dentro do corno de Ammon se distinguem as regiões CA1, CA2, CA3 e CA4
diferenciadas pela densidade de neurônios e de fibras presentes (Figura 3) (14).
6
Figura 1. Ilustração do sistema límbico onde está o hipocampo. Note que o hipocampo é
uma estrutura bilateral que se estende para os dois hemisférios cerebrais localizada no lobo
temporal próximo à amígdala. Adaptado de: Brain Diagram Amygdala. Revisado por Charles
Rickson. Publicado em: 2013-10-06T05:18:00-07:00. Rating: 4.5. Disponível em:
http://www.ehealthideas.com/2013/10/brain-diagram-amygdala.html (acessado em 19 de
outubro de 2014).

7
(14)
Figura 2. Diagrama de um corte transversal do hipocampo. Na figura estão
representadas as regiões CA1, CA2, CA3 e CA4 e Giro denteado. 1: alveus; 2: stratum
oriens; 3: stratum pyramidale; 3’: stratum lucidum; 4: stratum radiatum; 5: stratum
lacunosum; 6: stratum moleculare; 7: sulco do hipocampo; 7’: cavidade residual; 8:
camada molecular do giro denteado; 9: camada de células granulares; 10: camada
polimórfica ou hilo; 11: fímbria; 12: margo denticulatus; 13: sulco fímbrio-denteado; 14:
sulco hipocampal; 15: subículo. FONTE: adaptado de Duvernoy, 1988.

8
A principal via de comunicação dentro do hipocampo é a via trissináptica.
Nela, o estímulo chega do córtex entorrinal através da via perforante (compostas
de fibras glutamatérgicas) e inervam as células granulares do giro denteado. As
fibras musgosas destas células, por sua vez, fazem conexões com células
piramidais da região CA4 e, principalmente, com as células da região CA3 do
hipocampo. Os axônios de CA3 projetam para as células piramidais de CA1,
constituindo a via colateral de Schaffer. Da região CA1, as fibras saem do
hipocampo em direção ao complexo subicular e, então, para o córtex entorrinal.
Na via temporamônica os neurônios da região CA1 recebem projeções
Figura 3. Regiões do Corno de Ammon. A: CA1; B: CA2; C:
CA3; D: CA4. Adaptado de Durvenoy, 1988 (14).

9
diretamente do córtex entorrinal (13, 14). Essas vias estão representadas na
Figura 4.
(15)
No hipocampo de pacientes com ELT pode ocorrer um processo de
reorganização caracterizado por alterações estruturais e moleculares, tais como
esclerose hipocampal, brotamento de fibras musgosas, neurogênese, estresse
oxidativo e neuroinflamação que podem contribuir para o desenvolvimento de
crises recorrentes e progressão da doença. Esse processo gradual e dinâmico de
reorganização cerebral (que pode levar ao desenvolvimento de crises recorrentes)
é chamado de epileptogênese (16). As bases moleculares da epileptogênese,
todavia, permanecem desconhecidas.
Figura 4. Principais circuitos neuronais no hipocampo. Via
trissináptica (córtex entorrinal (CE)–giro denteado–CA3–CA1–CE) –
representado pelas setas. Via temporamonica (CE-CA1-CE).
Adaptado de Deng et al 2010 .

10
1.4 Reorganização hipocampal na ELT
Esclerose hipocampal
A esclerose hipocampal é o achado histopatológico mais comum na ELT (8).
Caracteriza-se por uma acentuada perda de neurônios, principalmente nas regiões
CA1, CA3 e hilo do hipocampo, e intensa proliferação de astrócitos reativos
(gliose) (17).
Uma hipótese levantada diz que a perda de células musgosas que excitam
interneurônios inibitórios no hilo na esclerose hipocampal leva a uma desinibição
das células granulares tornando-as hiperexcitáveis. Essa hipótese é chamada de
“hipótese das células em cesto dormentes” (18).
Brotamento de Fibras Musgosas
Na ELT são observados (tanto em modelo animal quanto em humanos)
brotamento e ramificação de fibras musgosas no giro denteado. Estudos mostram
que essa reorganização das fibras pode formar circuitos excitatórios recorrentes
que levam a ativação anormal de circuitos límbicos, o que pode contribuir para o
processo de epileptogênese (19).

11
Neuroinflamação
Diversos fatores associados à inflamação, tais como IL-1β, IL-6, IL-10 e TNF-
α, são hiperexpressos após indução de crises em modelo animal e em tecido
hipocampal de pacientes com ELT (20, 21).
As funções das citocinas inflamatórias no contexto da epilepsia têm sido
intensamente estudadas em roedores. Os resultados mostram que as moléculas
inflamatórias podem levar a um aumento da frequência de crises, redução do
limiar para crises, indução do influxo de Ca2+ mediado por NMDA dentro dos
neurônios, elevação do nível extracelular de glutamato, redução da transmissão
inibitória e alteração da expressão gênica (22).
Dispersão de células granulares
Cerca de 50% dos pacientes com ELT associada à esclerose hipocampal
apresentam dispersão de células granulares (23). A dispersão de células
granulares pode estar associada a mudanças na orientação e distribuição
dendrítica (24) e falha de memória (25). Além disso, as células granulares do giro
denteado podem migrar para o hilo. Nessa região, essas células ectópicas podem
sofrer descargas espontâneas e assim contribuir para a formação de um circuito
excitatório recorrente (26).

12
Estresse Oxidativo
Estresse oxidativo é o desbalanço entre geração e eliminação de espécies
reativas de oxigênio (EROs) e espécies reativas de nitrogênio (ERNs). O acúmulo
de EROS e ERNs pode causar danos a lipídeos, proteínas e ao DNA. Acredita-se
que esses danos possam estar associados à patogênese da epilepsia (27).
As mitocôndrias são as principais organelas envolvidas no processo de
estresse oxidativo das células. Além disso, essas organelas são responsáveis pela
geração de ATP, biossíntese de metabólitos e neurotransmissores, e regulação da
homeostase de cálcio. Todas essas funções podem, direta e indiretamente, alterar
a excitabilidade neuronal e provocar a morte celular (27, 28).
Estudos demonstraram que há alterações nas funções mitocondriais e
estresse oxidativo em tecido humano de pacientes com epilepsia e em diversos
modelos animais (28-30). Por tudo isso, estresse oxidativo e disfunções
mitocôndrias têm sido postulados como possíveis mecanismos epileptogênicos
(29).
Desregulação da expressão gênica
Diversos genes têm a sua expressão alterada após insultos cerebrais.
Essas alterações na expressão gênica podem estar envolvidas nos processos de
reorganização cerebral (31, 32). Deste modo, o conhecimento acerca destes
genes é essencial para a compreensão da epileptogênese e para o

13
desenvolvimento de novas formas de tratamento (11). Consequentemente, há um
considerável interesse na identificação de genes alvos em epilepsia.
1.5 Fatores de Crescimento
Eventos celulares precisam ser extremamente regulados e coordenados para
garantir a completa homeostase do organismo.
Fatores de crescimento são proteínas secretadas pelas células que têm
como função enviar sinais através de receptores (33). Esses fatores podem
controlar uma ampla variedade de funções no organismo como a regulação do
crescimento e diferenciação celular, inflamação, reparo do tecido e angiogênese
(34).
No sistema nervoso central, novas funções para esses fatores têm sido
descobertas, como a regulação de sinapses, plasticidade neuronal e neurogênese
(35). Deste modo, essas moléculas podem ser importantes na fisiopatologia de
doenças cerebrais como doença de Alzheimer, Parkinson e Epilepsia.
Em epilepsia, muitos estudos têm relacionado esses fatores às alterações
encontradas no tecido hipocampal, como neuroinflamação, estresse oxidativo,
brotamento de fibras musgosas, morte neuronal e gliose. Essas características
colocam os fatores de crescimento como potenciais genes alvos (35). Dentre esse
grupo de moléculas, especial atenção tem sido dada aos fatores de crescimento
de fibroblastos (FGFs).

14
1.6 Fatores de Crescimento de Fibroblastos
Os fatores de crescimento de fibroblastos (FGFs) são uma família de
proteínas que compreendem 22 membros em humanos (FGF1-FGF23). FGF15
não é encontrado em humanos (36).
Os membros dessa família são polipeptídeos com cerca de 150 a 300
aminoácidos caracterizados por uma região central conservada com
aproximadamente 120 resíduos de aminoácidos (37).
A maioria dos FGFs possui uma sequência de aminoácidos que sinalizam a
secreção para fora da célula através da via clássica que envolve o retículo
endoplasmático e complexo de Golgi. No entanto, alguns FGFs não possuem esta
sequência sinal e, portanto, residem intracelularmente como no caso dos FGF11-
14, ou são secretados para fora da célula por uma via não convencional, como no
caso do FGF1, FGF2 e FGF3 (38).
Essa família pode ser dividida em três classes: os FGFs intrácrinos,
parácrinos e endócrinos. Enquanto os FGFs intrácrinos (FGF11-14) agem
independentes de receptores, os FGFs parácrinos (FGF1-10, FGF16-18, FGF20 e
FGF22) e endócrinos (FGF19, FGF21 e FGF23) agem fora da célula através da
ligação a receptores de membrana do tipo tirosina quinase, os FGFRs (36). Para a
completa estabilização da ligação de um FGF ao seu receptor é necessária ainda
a ligação de cofatores como α-Klotho, β-Klotho ou glicosaminoglicanos sulfatados,
como heparan sulfato (39).

15
Existem quatro genes para os receptores de FGFs (FGFR1-4) (40). No
entanto, existem sete diferentes isoformas de receptores para essa família
(FGFR1b, 1c, 2b, 2c, 3b, 3c e 4) (40) decorrentes de splicing alternativos no
domínio IgIII da imunoglobulina desses receptores. Essas isoformas conferem
distintas especificidades de ligação aos FGFs (37). A estrutura dos FGFRs está
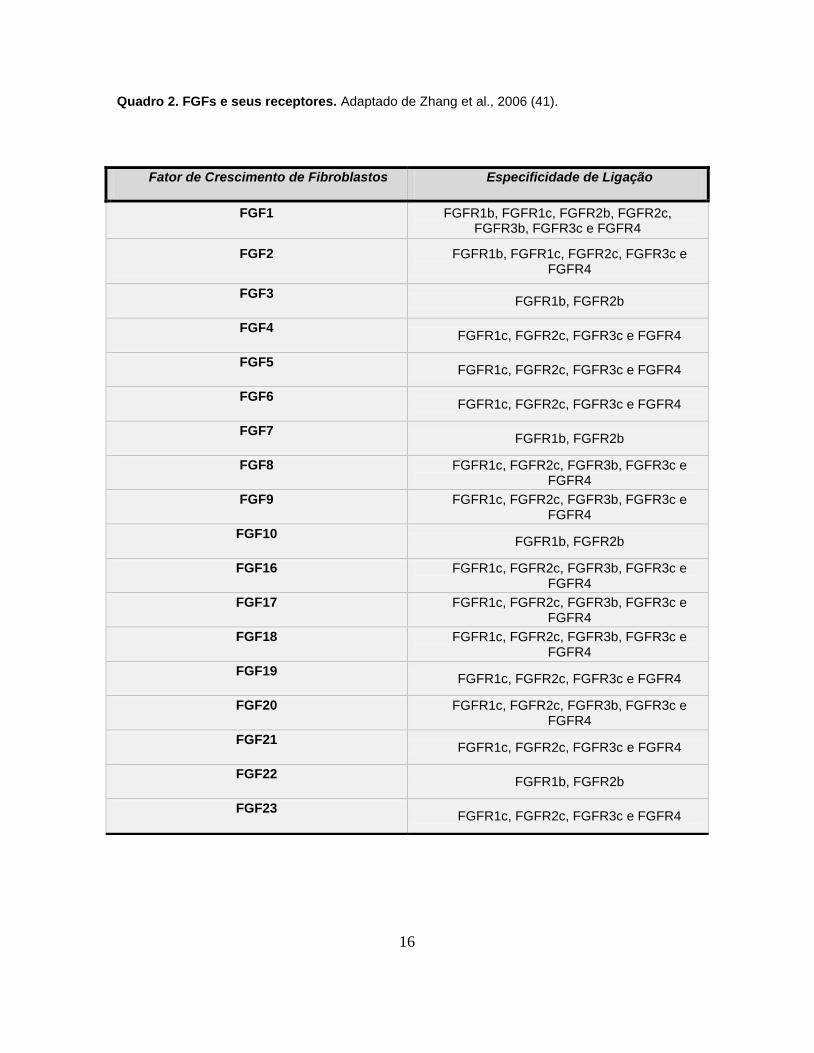
apresentada na Figura 1 e as diferentes especificidades de ligação entre os FGFS
e seus receptores são mostradas no quadro 2.
16
Quadro 2. FGFs e seus receptores. Adaptado de Zhang et al., 2006 (41).
Fator de Crescimento de Fibroblastos Especificidade de Ligação
FGF1 FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF2 FGFR1b, FGFR1c, FGFR2c, FGFR3c e FGFR4
FGF3 FGFR1b, FGFR2b
FGF4 FGFR1c, FGFR2c, FGFR3c e FGFR4
FGF5 FGFR1c, FGFR2c, FGFR3c e FGFR4
FGF6 FGFR1c, FGFR2c, FGFR3c e FGFR4
FGF7 FGFR1b, FGFR2b
FGF8 FGFR1c, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF9 FGFR1c, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF10 FGFR1b, FGFR2b
FGF16 FGFR1c, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF17 FGFR1c, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF18 FGFR1c, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF19 FGFR1c, FGFR2c, FGFR3c e FGFR4
FGF20 FGFR1c, FGFR2c, FGFR3b, FGFR3c e FGFR4
FGF21 FGFR1c, FGFR2c, FGFR3c e FGFR4
FGF22 FGFR1b, FGFR2b
FGF23 FGFR1c, FGFR2c, FGFR3c e FGFR4

17
(40)
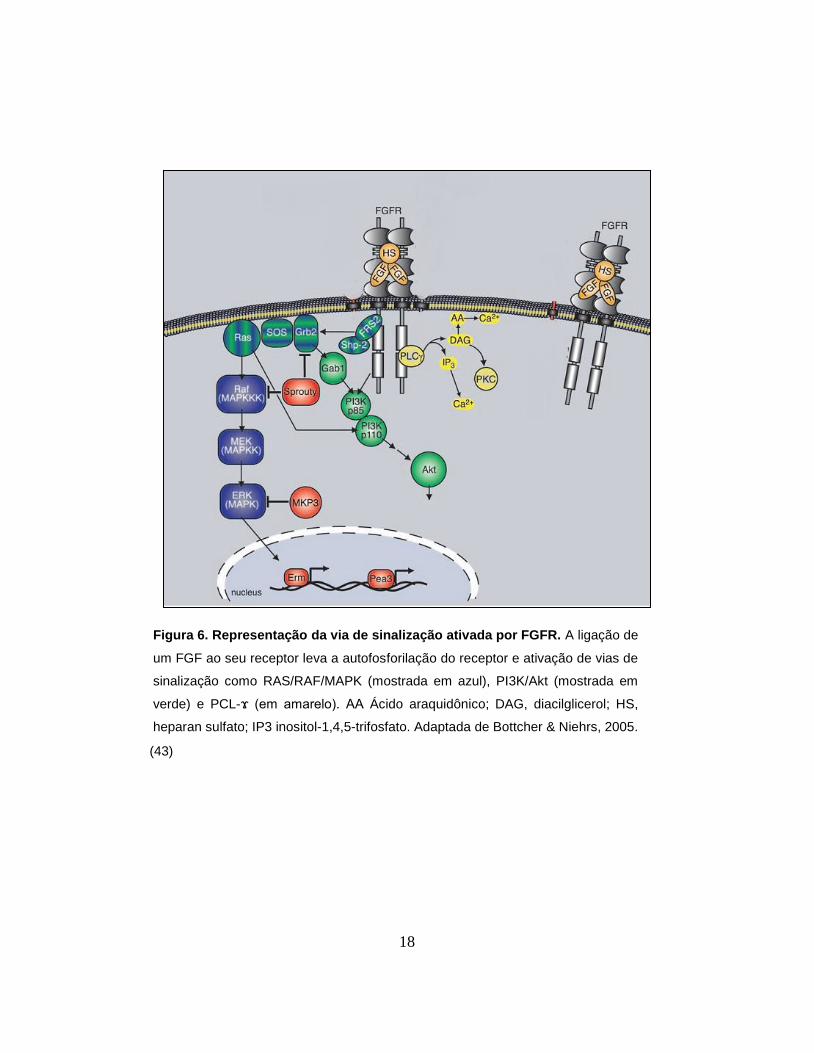
A ligação de um FGF e o seu cofator a um receptor provoca a dimerização e
autofosforilação do receptor, o que leva a ativação de vias de sinalização, como as
vias Ras/Raf/MAPK, PI3K-AKT, signal transducer and activator of transcription
(STAT) e phospholipase Cγ (PCL-ϫ) (42). A ativação dessas vias pode culminar
na ativação de fatores de transcrição que modulam a expressão de diversos
genes levando, assim, a alteração das funções celulares (43). A figura 5 mostra
um exemplo da via de sinalização dos FGFs.
Os FGFs são amplamente expressos no organismo, tanto durante o
desenvolvimento como na vida pós-natal e têm sido intensamente estudados em
diversas doenças como câncer, doenças metabólicas e doenças associadas ao
sistema nervoso central, como a epilepsia (40, 44, 45)
Figura 5. Esquema da estrutura do FGFR. Esquema da estrutura de FGFR. O
receptor consiste de um domínio kinase intracelular na região C-terminal e um
domínio extracelular. O domínio extracelular dos FGFRs consiste de três domínios
IgIII (D1, D2 E D3). Entre D1 e D2 observa-se um segmento característico do
FGFRs composto de ácido glutâmico e aspártico, chamado de caixa ácida.
Adaptada de Beenken & Mohammadi 2009.
18
(43)
Figura 6. Representação da via de sinalização ativada por FGFR. A ligação de
um FGF ao seu receptor leva a autofosforilação do receptor e ativação de vias de
sinalização como RAS/RAF/MAPK (mostrada em azul), PI3K/Akt (mostrada em
verde) e PCL-ϫ (em amarelo). AA Ácido araquidônico; DAG, diacilglicerol; HS,
heparan sulfato; IP3 inositol-1,4,5-trifosfato. Adaptada de Bottcher & Niehrs, 2005.

19
1. 7 Fatores de Crescimento de Fibroblastos e Epilepsia
Os FGFs têm sido associados a uma extensa gama de modificações
encontradas na ELT, como por exemplo, neuroinflamação, neurogênese,
brotamento de fibras musgosas, excitabilidade neuronal, gliose e estresse
oxidativo (46-50). Somado a isso, dados na literatura relatam aumento da
expressão do RNAm e proteína de membros dessa família e seus receptores após
a indução de crises em ratos (51-53). Esses resultados evidenciam que os FGFs
podem estar relacionados às modificações induzidas por crises. No entanto, não
há estudos desses fatores na ELT em humanos.
Os genes que foram estudados nesse trabalho, todos relacionados à família
de FGFs, são descritos a seguir.
Fator de Crescimento de Fibroblasto 2 – FGF2
O FGF2, também conhecido como FGF básico (bFGF), é o membro da
família dos FGFs mais estudado. Essa proteína possui 146 aminoácidos
estruturados em uma única cadeia. Em humanos foram descritas cinco isoformas
da proteína, sendo quatro isoformas de alto peso molecular (22; 22,5, 24 e 34
kDa) e uma isoforma de baixo peso molecular (18 kDa) (54). As isoformas são
geradas por diferentes códons de iniciação. As isoformas de alto peso molecular
(Hmv FGF2) são geradas a partir de códons alternativos de início CUG, enquanto
a isoforma de baixo peso molecular (Lmv FGF2) é gerada pelo códon clássico de
início AUG. O início da tradução por códons alternativos é condicionado por

20
elementos regulatórios presentes no mRNA, como, por exemplo o sítio interno de
entrada do ribossomo (IRES – internal ribosome entry site) (Figura 7) (38).
As isoformas de alto peso molecular são translocadas para o núcleo e podem
regular a expressão gênica. A isoforma de baixo peso molecular é citoplasmática e
excretada para fora da célula, e exerce suas funções através da ligação aos
FGFRs e heparan sulfato (54). O receptor com mais alta afinidade para FGF2 é o
FGFR1 (55).
No sistema nervoso, FGF2 é expresso em neurônios e células gliais (56).
Seu RNAm é amplamente expresso e pode ser encontrado no bulbo olfatório,
córtex, hipocampo, corpo estriado, tálamo, substância negra, ponte, medula
oblonga, e hipófise (55). No hipocampo, FGF2 está relacionado à proliferação
celular, neuroproteção, mecanismos de reparo, excitabilidade neuronal,
neuroinflamação e mecanismos de plasticidade neuronal (55, 57-59).

21
Figura 7. Expressão de diferentes isoformas de FGF2 por uso de códon de iniciação
alternativo. O RNAm de FGF2 contém quatro sítios de iniciação alternativa da tradução através
dos códons CUG e um sítio de iniciação clássica com o códon AUG. As isoformas de alto peso
molecular (34, 24, 22,5 e 22 kD) são geradas pelos códons alternativos, enquanto a isoforma de
baixo peso molecular (18kD) é gerada pelo códon clássico AUG. As isoformas de alto peso
molecular contém uma sequência de localização nuclear (NLS). Todas as isoformas possuem uma
NLS bipartida na região C-terminal. Adaptado de Sorensen, 2006. (38).
Fator de Crescimento de Fibroblastos 8 – FGF8
O fator de crescimento de fibroblasto 8 (FGF8) é um membro da família dos
FGFs com importantes funções no desenvolvimento embrionário. Existem quatro
isoformas de FGF8 em humanos: FGF8a, FGF8b, FGF8e e FGF8f. Essas
proteínas sinalizam através dos receptores FGFR1-4 (60).

22
Poucos estudos analisaram o papel de FGF8 no sistema nervoso. O estudo
de Garcia-Hernandez e colegas mostrou que, em cultura celular de neurônios do
gânglio espiral (coclear), as isoformas FGF8a e FGF8b promovem o crescimento
de neuritos através da ativação de FGFRs e NFκB (60). Já em cultura celular de
neurônios hipocampais de ratos, o FGF8b foi capaz de proteger os neurônios de
insultos oxidativos (47).
Fator de Crescimento de Fibroblastos 22 – FGF22
Ao contrário do FGF2, o FGF22 tem uma expressão mais restrita, sendo
encontrado principalmente na placenta, pele e cérebro (61). Suas ações biológicas
são mediadas principalmente através do FGFR2 (41).
No hipocampo de camundongos, tanto o RNAm quanto a proteína do FGF22
são expressos após o nascimento, principalmente nos neurônios da região CA3,
enquanto seus receptores são amplamente expressos no hipocampo (50).
Ainda em camundongos, o FGF22 tem sido relacionado à organização de
sinapses excitatórias, promovendo a diferenciação de terminais nervosos pré-
sinápticos (62). Sua ausência em camundongos nocautes leva a um aumento da
resistência a indução de crises generalizadas por pentilenetetrazol (50),
diminuição da neurogênese e ausência de células granulares ectópicas (63).

23
Inositol 1,4,5-trisfosfato - ITPR
Receptores de inositol 1,4,5-trisfosfato, os ITPRs, também conhecidos como
IP3Rs, são canais de cálcio intracelulares que estimulam a liberação de cálcio dos
estoques intracelulares do retículo endoplasmático em resposta a estimulação por
fatores de crescimento, hormônios e neurotransmissores (64-66).
No sistema nervoso central a sinalização de cálcio tem um importante papel
nos mecanismos de aprendizado e memória. O cálcio liberado pelos ITPRs pode
ainda estimular a síntese de ATP e iniciar a sinalização de apoptose dentro das
mitocôndrias. Além disso, nos neurônios hipocampais, os ITPRs estão envolvidos
na modulação da transmissão sináptica (67).
Existem três receptores para IP3: ITPR1, ITPR2 e ITPR3, com expressão em
diferentes células no organismo, mas as propriedades funcionais de cada receptor
ainda não estão completamente entendidas. O estudo de Mendes e colaboradores
mostra que o gene ITPR3 tem sido relacionado à indução da morte celular por
apoptose via mitocôndria (68).
Fosfatidilinositol 3’- quinases (PI3Ks)
Fosfatidilinositol 3’-quinases (PI3K) são enzimas que fosforilam o grupamento
3’-hidroxila dos fosfoinositídeos (69). Membros da família PIK3 são agrupados em
três classes. A classe I é dividida em duas subfamílias: classe IA, que são
heterodímeros, compostos por uma subunidade regulatória (p85) e uma
subunidade catalítica (p110), ativados por receptores do tipo tirosina quinase; e

24
classe IB, que são heterodímeros, compostos por uma subunidade regulatória
(p101) e uma subunidade catalítica (p110γ), ativados por receptores acoplados a
proteína-G. Os genes estudados nesse trabalho, PIK3R3 e PIKR5, codificam as
isoformas p55γ da subunidade regulatória da classe IA e a isoforma p101 da
subunidade regulatória da classe IB, respectivamente. A classe II é constituída de
três isoformas: PIK3C2α, PIK3C2β e PIK3C2γ com somente uma subunidade
catalítica. A classe III consiste de somente um membro, Vps34 (69).
A atividade das PI3Ks pode regular a proliferação celular, sobrevivência,
resposta imune e metabolismo (70). No sistema nervoso central, a atividade de
PIK3 está envolvida na sinaptogênese e formação axonal (70, 71). Além dessas
funções, PIK3 pode ainda ativar mTOR que, por sua vez, está envolvido em
mecanismos de morte celular, brotamento axonal e redução da atividade epiléptica
(Berdichevsky, Y et al 2013).

25
2. Objetivos
2.1 Objetivos Gerais
Analisar a expressão gênica e proteica de membros da família FGFs no
hipocampo de pacientes com ELT.
2.2 Objetivos Específicos
Analisar a expressão dos genes FGF2, FGF8 e FGF22 em pacientes com
ELT.
Analisar a expressão gênica dos receptores de FGFR1, FGFR2 e FGFR3
em pacientes com ELT.
Analisar a expressão gênica dos seguintes membros da via de sinalização
de FGFs: ITPR3, PIK3R3, PIK3R5.
Verificar a relação entre a expressão gênica e dados clínicos dos pacientes.
Verificar a expressão e localização celular proteica de FGF2 no hipocampo
de pacientes e controles.

26
Capítulo 1
FGF2 upregulation in
temporal lobe epilepsy

27
3. Capítulo 1
O capítulo 1 corresponde ao artigo intitulado “Fibroblast growth factor 2
upregulation in chronic temporal lobe epilepsy”.
Esse artigo foi submetido para o periódico Epilepsy Research em 12 de
setembro de 2014 e até o momento da conclusão final dessa dissertação,
encontrava-se em revisão.

28
TITLE: HIPPOCAMPAL UPREGULATION OF FIBROBLAST GROWTH FACTOR
2 IN PHARMACORESISTANT TEMPORAL LOBE EPILEPSY PATIENTS
SHORT TITLE: FGF2 UPREGULATION IN TEMPORAL LOBE EPILEPSY
Ana Erika Dias Ferreiraa; Marcelo Ananias Teocchia; Evandro de Oliveirab; Helder
Tedeschib; Lília D’Souza-Lia*
a Laboratory of Pediatric Endocrinology, Center for Investigation in Pediatrics,
Faculty of Medical Sciences, University of Campinas – UNICAMP, Campinas, SP,
Brazil
b Neurology Department - Faculty of Medical Sciences, University of Campinas –
UNICAMP, Campinas, SP, Brazil
*Corresponding Author: Lília D’Souza-Li
E-mail: [email protected]
Address: Center for Investigation in Pediatrics (CIPED) - Faculty of Medical
Science - University of Campinas.
Rua Tessália Vieira de Camargo, 126 - Cidade Universitária “Zeferino Vaz”
Campinas - São Paulo - Brasil - CEP 13083-887
Phone: +55 19 3521 8986 - Fax: +55 19 3521 8964
Grant sponsor: São Paulo Research Foundation (FAPESP); Grant number:
2013/07559-3
Number of text pages: 14 Figures: 2 Tables: 2
Key Words: epileptogenesis; plasticity synaptic; axonal sprouting; neurogenesis,
hippocampal sclerosis.

29
ABSTRACT
Temporal lobe epilepsy (TLE) is the most common form of epilepsy in
adults. The process of epileptogenesis includes neuronal death, axonal sprouting,
inflammation, neurogenesis, oxidative stress and gliosis. However, the molecular
mechanisms behind them are not fully understood. Fibroblast growth factor (FGF)
gene family encodes proteins with several functions in the organism, especially in
the central nervous system. FGF family member functions in the normal and in the
pathological (epileptic) human brain are unclear and even controversial, such as
with FGF2, the most studied of these proteins. To shed light on the involvement of
the FGF pathway in TLE, we quantified the hippocampal expression of the
following genes: FGF2, FGF8, FGF22, FGFR1, FGFR2, FGFR3, ITPR3, PIK3R3
and PIK3R5 in 10 pharmacoresistant patients and four post mortem controls. We
also assessed the FGF2 protein expression by indirect immunofluorescence. Only
for FGF2, was the mRNA level markedly increased in patients’ hippocampi for the
two reference genes tested, HPRT1 and ENO2+TBP in combination (P = 0.002
and P = 0.036, respectively). The percentage of FGF2 immunostained cells in the
dentate gyrus was higher in patients than in the controls (P <0.05), but no
significant alteration was found in the Ammon’s horn. FGF2 preserves neurons
from ongoing injury and acts as a powerful proliferation factor for neural stem cells.
It could potentially alleviate seizure-induced damage and intensify repair and
reduce epileptogenesis in the hippocampus. On the other hand, evidence has
shown FGF2’s involvement in epileptogenic mechanisms, such as axonal sprouting

30
and neurogenesis. Our results clearly suggest the FGF2 participation in TLE
physiopathology and point it out as an important target for pharmacological studies.
INTRODUCTION
All forms of epilepsy are characterized by recurrent unprovoked seizures
resulting in persistent increase of neuronal excitability (McNamara, 1999). In
temporal lobe epilepsy (TLE), which is the most refractory and common form of
epilepsy in adults, the hippocampus is the main epileptogenic region (Engel, 2001).
Hippocampal sclerosis (HS) is the most important pathological finding observed in
resected tissue from refractory TLE patients who underwent an
amygdalohippocampectomy.
Epileptogenesis is a gradual process of molecular and structural changes in
the brain, associated with neurodegeneration, neurogenesis, gliosis, axonal
sprouting, dendritic plasticity, blood–brain barrier (BBB) damage and
neuroinflammation that progressively alters the neuronal excitability leading to
spontaneous seizures (Pitkanen and Lukasiuk, 2009).
Seizure activity is associated with a disturbance in the expression of several
genes in hippocampus (Pitkanen and Lukasiuk, 2009). Recognizing such genes is
crucial for shedding light on the TLE molecular physiopathology and for identifying
new targets for treatment. Growth factors have emerged as therapeutic candidates
since they may be involved in a plethora of effects found in TLE (Jankowsky and

31
Patterson, 2001). Among them, special attention has been given to the fibroblast
growth factor family.
The vast majority of FGF members are widely expressed in the central
nervous system, including the hippocampus, and usually signals through tyrosine
kinase receptors FGFR1-4 (Zechel et al., 2010). The FGF-FGFR complex triggers
several signaling cascades which may activate transcription factors which are
responsible for the expression of numerous genes (Bottcher and Niehrs, 2005).
Despite intensive studies using seizure models, the role of FGFs on human TLE
pathophysiology is not fully understood.
Fibroblast growth factor 2 (FGF2), also termed basic fibroblast growth factor
(bFGF), is one of the most studied FGF family members (Zechel et al., 2010).
FGF2 controls several cellular responses, including proliferation, differentiation and
survival. Studies of the central nervous systems in adult rodents suggest the
involvement of FGF2 in neuroprotection and lesion repair (Zechel et al., 2010).
Conversely, FGF2 could potentially be involved in epileptogenic mechanisms
(Eves et al., 2001; Liu et al., 2014; Palmer et al., 1999; Patel and McNamara,
1995). Nevertheless, the molecular mechanisms that regulate its diverse biological
effects are unknown.
Herein, we present an analysis of the TLE hippocampal mRNA quantification
of three FGF members: FGF2, FGF8 and FGF22; three FGFRs: FGFR1, FGFR2
and FGFR3; and three genes involved in the FGF signaling cascade: ITPR3,
PIK3R3 and PIK3R5. Finally, we studied the hippocampal pattern of FGF2 protein
expression.

32
METHODS
Tissue collection and all procedures were carried out with the approval of the
local research ethics committee (CEP #470/2003).
Patients and Controls
Autopsy control tissues were kindly provided by the “Instituto Médico Legal of
Campinas” from four individuals with no clinical evidence of dementia or history of
brain disease. Collections were performed in March 2009 and May 2010 and the
post mortem delay averaged 7.8 h (range: 6.0 – 9.0 h). The control group
consisted of one female and three males with the age mean of 22.75 ± 5.56 years
ranging from 19 to 31.
The patient group included hippocampal specimens from 10 TLE patients with
hippocampal sclerosis (HS) who underwent a therapeutical surgery due to the
epilepsy pharmacoresistance. HS was confirmed in all patients by
electroencephalogram (EEG) video monitoring/telemetry and magnetic resonance
imaging (MRI). Surgeries occurred in the “Hospital de Clínicas of the University of
Campinas” in December 2005 (TLE16), May 2007 (TLE15) and between March
2009 and September 2010 (for the other patients). Each patient signed an
informed consent agreement to allow scientific use of the tissue.

33
Patient’s clinical data included in this study are described in Table 1. Part of
patients was reported in a recent study (Teocchi et al., 2013).
Tissue preparation
All hippocampal tissue samples were immediately collected and divided into
two parts. One part was frozen in liquid nitrogen and kept at - 80° C until RNA
extraction. The other part was fixed in paraformaldehyde 4% solution diluted in
PBS (pH 7.4) for 24 hours for histopathological analysis. After fixation, the tissue
was immersed in paraffin. Paraffin-embedded hippocampi were cut in successive 5
μm cross sections.
Gene Expression
RNA Extraction and cDNA Synthesis
Total RNA was harvested from tissues using 1 mL of TRIzol® reagent (Life
Technologies, Foster City, USA) per 75 mg of frozen tissue sample and processed
according to the manufacturer’s instructions. Sterilized and filtered DEPC treated
water was used in all RNA procedures.
The RNA integrity was checked by Agilent 2100 Bioanalyzer instrument. RNA
integrity number (RIN) means in the control and patient groups were 7.67 ± 1.02
(n=4) and 5.72 ± 1.42 (n= 10), respectively.

34
Subsequently, 1μg of total RNA of each sample was reverse transcribed into
cDNA using the commercial High Capacity cDNA Reverse Transcription Kit (Life
Technologies, USA) following the manufacturer’s instructions.
Reverse transcription-quantitative PCR (RT-qPCR)
Messenger RNA was quantified using an ABI PRISM 7500 Sequence
Detection System (Life Technologies, USA) and TaqMan Gene Expression Assays
with FAM dye-labeled hydrolysis probes (Life Technologies, USA) using the
method of ∆∆Ct. All genes in this study had their amplification efficiencies near to
100%. Information about the assays used in this study is shown on Table 2. We
used as reference genes: HPRT1, and ENO2 and TBP in combination as
suggested by Wierschke et al. (Wierschke et al., 2010).
The real-time reaction mixture was prepared in a total volume of 12.5 μl with
10 ng of cDNA sample diluted in 5μl of purified H2O, 6.25 μl TaqMan Gene
Expression Master Mix (Life Technologies, USA), 0.625 μl of the respective
probe/primer mix (Life Technologies, USA), and 0.625 μl purified H2O. All samples
were run as triplicates.

35
Protein Pattern
Immunofluorescence
Immunofluorescence was performed on hippocampal paraffin-embedded
sections from patients and autopsy controls. Antigen retrieval was achieved by the
citrate buffer method, in which slides were placed in boiling citrate buffer solution
1X (Diagnostic Biosystem, USA) for 25 min followed by 30 min incubation at room
temperature. Tissue sections were blocked followed by overnight incubation at 4ºC
with goat polyclonal anti-FGF2 (1:40; RD System, USA) diluted in Tris-buffered
saline (TBS), pH 7.4 containing 0.2% Triton X-100 (TBST) Sections were washed 3
times with TBST and then incubated with anti-goat secondary antibody conjugated
Alexa Fluor 488 (1:2000, Life Technologies, USA). TO-PRO3 was used for nuclei
staining (1:400; Life Technologies, USA). Sections were washed, mounted and
cover slipped on glass slides. Incubation with the primary antibody was omitted in
negative controls. The slides were analyzed on Leica TCS SP5 II confocal
microscope.
Data analysis
Statistical differences for the gene expression data were verified by the Mann-
Whitney U test using the GraphPad Prism software (San Diego, USA). The
Spearman test was used for correlation analyses.

36
Eight-bit images of the Ammon's horn and the dentate gyrus from controls
and TLE patients were analyzed by the ImageJ software (NIH, Bethesda, MD,
USA). Percentages of cells were stipulated by automated counting of total amount
of cell nuclei and stained cells. Again, statistical analyses were performed by the
GraphPad Prism software (San Diego, USA) with an unpaired t test (one-tail).
Differences of P <0.05 were considered significant for all analyses.
RESULTS AND DISCUSSION
The first aim of this study was to evaluate and compare the hippocampal
gene expression of FGF-related targets in pharmacoresistant TLE patients and
post mortem controls. The nine target genes were divided into the following
categories: (1) FGF family members: FGF2, FGF8 and FGF22; (2) the three major
FGFRs: FGFR1, FGFR2 and FGFR3; and three proteins involved in the FGF
signaling pathway: ITPR3, PIK3R3 and PIK3R5. FGF family members are key
players in a number of biological processes, particularly in the central nervous
system.
We found that the relative expression level of the FGF2 mRNA was higher in
the TLE group when compared with the control group for the two reference genes
analyzed, HPRT1 and ENO2+TBP (P = 0.002 and P = 0.036, respectively) (Figure
1). There was no correlation between the FGF2 expression and age, age at
epilepsy onset or disease duration. In addition, the expression of FGF2 did not
differ in respect to the gender, status epilepticus occurrence and the side of HS.

37
Our second aim was to explore the FGF2 protein expression in our patient
and control hippocampal tissue samples and, as expected, the FGF2 mRNA
hyperexpression was also accompanied by an increase in the FGF2 protein
expression. The role of FGF2 in epileptogenesis is controversial and this is the first
study conducted in human TLE hippocampal samples.
FGF2 increases in response to seizure in several acute phase rodent models
(Bugra et al., 1994; Gall et al., 1994; Gomez-Pinilla et al., 1995a; Humpel et al.,
1993; Riva et al., 1994). Although FGF2 mRNA expression appears within 3 to 24
h after seizure induction (Bugra et al., 1994; Gall et al., 1994; Gomez-Pinilla et al.,
1995a; Humpel et al., 1993; Riva et al., 1994), protein levels may remain elevated
for 30 days, showing that even rapid changes in gene expression can lead to
prolonged effects (Ballabriga et al., 1997).
Only one study, in a rat epilepsy model, evaluated the FGF2 expression in the
chronic phase, and contrary to our finding, there was a decrease in the FGF2
protein expression (Hattiangady et al., 2004). These contrary findings are hard to
reconcile and differences in methods should be carefully considered.
Indeed, the significance of FGF2 overexpression in epilepsy is far from being
elucidated. On the one hand, FGF2 may function as a neuroprotective response to
seizure-induced brain insult. This hypothesis is supported by results of in vivo and
in vitro experiments. In vivo experiments showed that FGF2 infusion protected
hippocampal neurons from cell death induced by seizures (Liu et al., 1993),
reduced the number of spontaneous seizures and increased the seizure threshold
(Liu and Holmes, 1997b). In addition, for both lesioned middle-aged and aged

38
hippocampus from a kainate-induced TLE model, the pre-treatment and
transplantation of CA3 cell grafts with FGF2 resulted in an augmentation of
surviving cells from grafts (Zaman and Shetty, 2003). In vitro, FGF2 promoted
survival in hippocampal neurons cultured under serum-free and low-insulin
conditions (Johnson-Farley et al., 2007).
On the other hand, the harmful effects of FGF2 have been reported, inciting
several features associated with epileptogenesis. FGF2 supplementation in
neuronal hippocampal cell culture caused the increase of excitatory synapses (Li et
al., 2002). FGF2 overexpression in transgenic mice intensified excitability and
increased the number of glutamatergic synaptic vesicles in the hippocampus,
although cell loss was been decreased (Zucchini et al., 2008). Hippocampal FGF2
injection in mice produced seizures in seven out of nine mice tested (Liu and
Holmes, 1997a).
It is well documented that the hippocampus of TLE patients has an important
inflammatory component with clinical significance, which is believed to be involved
in the disease progression (Yang et al., 2010). Using rat hippocampal cells, Eves
and colleagues found an association between FGF2 and an increase in the
neuronal TNF level, implicated in the induction of apoptosis through death TNF
receptor (Eves et al., 2001). This finding is reinforced by the fact that FGF2 triggers
the transcription factor ELK1 (Chung et al., 1998), which can activate the TNF
promoter (Tsai et al., 2000). TNF level augmentation has already been reported in
seizure models and in hippocampal tissues from TLE patients (Balosso et al.,

39
2013; Teocchi et al., 2013), highlighting this cytokine as a major inflammatory
mediator in TLE.
In our study, hippocampal tissues from controls and patients showed a FGF2
cytoplasmatic staining in morphologically similar neuronal and glial cells (Figure 2).
The percentage of FGF2 staining cells was higher in the patient group when
compared to controls in the dentate gyrus (DG) (P = 0.0458), but not in the
Ammon’s horn (P = 0.1182) (Figure 2). No immunereactivity was seen in negative
controls when the primary antibody was omitted (data not shown).
Our result on FGF2 protein overexpression is supported by other studies in
rodents and humans (Eckenstein et al., 1991; Weickert et al., 2005). FGF2 may
have distinct roles in different cells. In neuronal cells, FGF2 may be involved in
synaptic plasticity and neuroprotection; while in glial cells FGF2 may participate in
proliferation and contribute to astrocyte reactivity (Eclancher et al., 1990; Gomez-
Pinilla et al., 1995b). Reactive glial cells are extremely important in
epileptogenesis, since they regulate ions, neurotransmitters and act on
inflammatory mechanisms (Jabs et al., 2008). The dual role of FGF2 in neuronal
ad glial cells may be in part explained by differences in receptor expression.
FGFR1 is expressed mainly in neuronal cells; FGFR2, in glial cells (Asai et al.,
1993).
Animal studies have shown the well-defined role of FGF2 in both neuronal
reorganization (Palmer et al., 1999; Patel and McNamara, 1995; Rai et al., 2007;
Tao et al., 1997) and neuroprotection (Johnson-Farley et al., 2007; Liu et al.,
1993). Our result on FGF2 upregulation in the DG of hippocampus from TLE

40
patients highlights these two well-defined roles of FGF2 and suggests its
participation in TLE physiopathology. Axonal sprouting and neurogenesis, two
important epileptogenic features, contributed to the neuronal reorganization
mechanism in the DG, identifying this hippocampal region as an important site
involved in TLE physiopathology. Moreover, this region shows a considerable
preservation of neurons. It is plausible that FGF2 may play reorganization and
neuroprotection roles in the DG of TLE patients, and functional studies are required
to prove this hypothesis.
Notwithstanding, FGF2 role in neuroprotection and neurogegeneration in
epilepsy is far from being demystified. Its effects may depend of its concentration,
isoform, cellular localization, activated receptors, and the triggering event. Also, the
combination with other neurotrophic factors may be important for its role. The use
of a vector to supplement FGF2 and BDNF in the rat hippocampus after
pilocarpine-induced status epilepticus attenuated neuroinflammation, increased
neurogenesis, reduced neuronal loss and the occurrence of spontaneous seizures
(Bovolenta et al., 2010; Paradiso et al., 2009). Nevertheless, the progressive
condition of TLE associated with an isolated FGF2 upregulation, especially in the
chronic phase, would suggest a damaging scenario, with its harmful outcomes
greater than any possible neuroprotection.
A better comprehension of the FGF signaling pathways in TLE patients would
provide more clues regarding the controversy of FGF effects in the brain. To shed
light on this issue, we evaluated the mRNA expression of genes encoding the three
major FGF receptors: FGFR1, FGFR2 and FGFR3; three proteins related with the

41
FGF signaling pathway: ITPR3, PIK3R3 and PIK3R5; and two other members of
the FGF family: FGF8 and FGF22. Despite several studies showing the role of
these FGF related proteins in a variety of hippocampal process such as
neurogenesis and synaptic plasticity (Kelly et al., 2005; Lee and Umemori, 2013;
Zhao et al., 2007), synaptogenesis (Cuesto et al., 2011; Tohda et al., 2006), and
oxidative stress (Mark et al., 1999), no difference in the expression of these genes
was found when we compared controls and patients (Table 2). For statistical
significance, we considered a P value lower than 0.05 for the gene expression
relative to the both reference genes. For PIK3R5 we found a significant
upregulation (P = 0.0240), but only when ENO2/TBP was the reference gene. Also,
we did not look at the phosphorylated status of these tyrosine cascade-signaling
proteins to completely rule out their relevance in TLE.
In conclusion, this is the first report on the hippocampal FGF2 upregulation in
TLE patients. We showed that the FGF2 protein was overexpressed in the DG,
speculating on its involvement in axonal sprounting and neurogenesis, two
important epileptic features of TLE. Since the DG neurons are preserved in
hippocampal sclerosis, it is plausible to infer the neuroprotective effect of FGF2 in
this hippocampal region. Our work is in agreement with seizure models and our
results emphasize the preponderant role of this multifunctional growth factor in
TLE. FGF2 is a potential therapeutic target; however, as a pleiotropic growth
factor, further investigation is required to delineate its beneficial and harmful
outcomes.

42
ACKNOWLEDGEMENTS
The São Paulo Research Foundation (FAPESP process 05/565778-4 and
2013/07559-3) and the Coordenação de Aperfeiçoamento de Pessoal de Nível
Superior (CAPES) financially supported this study. Quantitative PCRs were carried
out in the Multiuser Laboratory (www.laboratoriomultiusuario.com.br) and RIN
detection and confocal images were performed in the Central Laboratory of High
Performance Technologies (LaCTAD; www.lactad.unicamp.br)
REFERENCES
Asai T, Wanaka A, Kato H, Masana Y, Seo M, Tohyama M. 1993. Differential
expression of two members of FGF receptor gene family, FGFR-1 and FGFR-
2 mRNA, in the adult rat central nervous system. Brain Res Mol Brain Res
17(1-2):174-8.
Ballabriga J, Pozas E, Planas AM, Ferrer I. 1997. bFGF and FGFR-3
immunoreactivity in the rat brain following systemic kainic acid administration
at convulsant doses: localization of bFGF and FGFR-3 in reactive astrocytes,
and FGFR-3 in reactive microglia. Brain research 752(1-2):315-8.
Balosso S, Ravizza T, Aronica E, Vezzani A. 2013. The dual role of TNF-alpha and
its receptors in seizures. Experimental neurology 247:267-71.
Bottcher RT, Niehrs C. 2005. Fibroblast growth factor signaling during early
vertebrate development. Endocrine reviews 26(1):63-77.
Bovolenta R, Zucchini S, Paradiso B, Rodi D, Merigo F, Navarro Mora G, Osculati
F, Berto E, Marconi P, Marzola A and others. 2010. Hippocampal FGF-2 and
BDNF overexpression attenuates epileptogenesis-associated

43
neuroinflammation and reduces spontaneous recurrent seizures. J
Neuroinflammation 7:81.
Bugra K, Pollard H, Charton G, Moreau J, Ben-Ari Y, Khrestchatisky M. 1994.
aFGF, bFGF and flg mRNAs show distinct patterns of induction in the
hippocampus following kainate-induced seizures. The European journal of
neuroscience 6(1):58-66.
Chung KC, Gomes I, Wang D, Lau LF, Rosner MR. 1998. Raf and fibroblast
growth factor phosphorylate Elk1 and activate the serum response element of
the immediate early gene pip92 by mitogen-activated protein kinase-
independent as well as -dependent signaling pathways. Molecular and
cellular biology 18(4):2272-81.
Cuesto G, Enriquez-Barreto L, Carames C, Cantarero M, Gasull X, Sandi C, Ferrus
A, Acebes A, Morales M. 2011. Phosphoinositide-3-kinase activation controls
synaptogenesis and spinogenesis in hippocampal neurons. The Journal of
neuroscience : the official journal of the Society for Neuroscience 31(8):2721-
33.
Eckenstein F, Woodward WR, Nishi R. 1991. Differential localization and possible
functions of aFGF and bFGF in the central and peripheral nervous systems.
Ann N Y Acad Sci 638:348-60.
Eclancher F, Perraud F, Faltin J, Labourdette G, Sensenbrenner M. 1990. Reactive
astrogliosis after basic fibroblast growth factor (bFGF) injection in injured
neonatal rat brain. Glia 3(6):502-9.
Engel J, Jr. 2001. Mesial temporal lobe epilepsy: what have we learned?
Neuroscientist 7(4):340-52.
Eves EM, Skoczylas C, Yoshida K, Alnemri ES, Rosner MR. 2001. FGF induces a
switch in death receptor pathways in neuronal cells. The Journal of
neuroscience: the official journal of the Society for Neuroscience 21(14):4996-
5006.

44
Gall CM, Berschauer R, Isackson PJ. 1994. Seizures increase basic fibroblast
growth factor mRNA in adult rat forebrain neurons and glia. Brain research.
Molecular brain research 21(3-4):190-205.
Gomez-Pinilla F, van der Wal EA, Cotman CW. 1995a. Possible coordinated gene
expressions for FGF receptor, FGF-5, and FGF-2 following seizures.
Experimental neurology 133(2):164-74.
Gomez-Pinilla F, Vu L, Cotman CW. 1995b. Regulation of astrocyte proliferation by
FGF-2 and heparan sulfate in vivo. The Journal of neuroscience : the official
journal of the Society for Neuroscience 15(3 Pt 1):2021-9.
Hattiangady B, Rao MS, Shetty AK. 2004. Chronic temporal lobe epilepsy is
associated with severely declined dentate neurogenesis in the adult
hippocampus. Neurobiology of disease 17(3):473-90.
Humpel C, Lippoldt A, Chadi G, Ganten D, Olson L, Fuxe K. 1993. Fast and
widespread increase of basic fibroblast growth factor messenger RNA and
protein in the forebrain after kainate-induced seizures. Neuroscience
57(4):913-22.
Jabs R, Seifert G, Steinhauser C. 2008. Astrocytic function and its alteration in the
epileptic brain. Epilepsia 49 Suppl 2:3-12.
Jankowsky JL, Patterson PH. 2001. The role of cytokines and growth factors in
seizures and their sequelae. Progress in neurobiology 63(2):125-49.
Johnson-Farley NN, Patel K, Kim D, Cowen DS. 2007. Interaction of FGF-2 with
IGF-1 and BDNF in stimulating Akt, ERK, and neuronal survival in
hippocampal cultures. Brain Res 1154:40-9.
Kelly PT, Mackinnon RL, 2nd, Dietz RV, Maher BJ, Wang J. 2005. Postsynaptic
IP3 receptor-mediated Ca2+ release modulates synaptic transmission in
hippocampal neurons. Brain research. Molecular brain research 135(1-
2):232-48.

45
Lee CH, Umemori H. 2013. Suppression of epileptogenesis-associated changes in
response to seizures in FGF22-deficient mice. Front Cell Neurosci 7:43.
Li AJ, Suzuki S, Suzuki M, Mizukoshi E, Imamura T. 2002. Fibroblast growth
factor-2 increases functional excitatory synapses on hippocampal neurons.
The European journal of neuroscience 16(7):1313-24.
Liu X, Albano R, Lobner D. 2014. FGF-2 induces neuronal death through
upregulation of system xc. Brain research 1547:25-33.
Liu Z, D'Amore PA, Mikati M, Gatt A, Holmes GL. 1993. Neuroprotective effect of
chronic infusion of basic fibroblast growth factor on seizure-associated
hippocampal damage. Brain research 626(1-2):335-8.
Liu Z, Holmes GL. 1997a. Basic fibroblast growth factor-induced seizures in rats.
Neuroscience letters 233(2-3):85-8.
Liu Z, Holmes GL. 1997b. Basic fibroblast growth factor is highly neuroprotective
against seizure-induced long-term behavioural deficits. Neuroscience
76(4):1129-38.
Mark RJ, Fuson KS, Keane-Lazar K, May PC. 1999. Fibroblast growth factor-8
protects cultured rat hippocampal neurons from oxidative insult. Brain Res
830(1):88-93.
McNamara JO. 1999. Emerging insights into the genesis of epilepsy. Nature
399(6738 Suppl):A15-22.
Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. 1999. Fibroblast growth
factor-2 activates a latent neurogenic program in neural stem cells from
diverse regions of the adult CNS. The Journal of neuroscience : the official
journal of the Society for Neuroscience 19(19):8487-97.
Paradiso B, Marconi P, Zucchini S, Berto E, Binaschi A, Bozac A, Buzzi A,
Mazzuferi M, Magri E, Navarro Mora G and others. 2009. Localized delivery
of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces

46
spontaneous seizures in an epilepsy model. Proc Natl Acad Sci U S A
106(17):7191-6.
Patel MN, McNamara JO. 1995. Selective enhancement of axonal branching of
cultured dentate gyrus neurons by neurotrophic factors. Neuroscience
69(3):763-70.
Pitkanen A, Lukasiuk K. 2009. Molecular and cellular basis of epileptogenesis in
symptomatic epilepsy. Epilepsy & behavior : E&B 14 Suppl 1:16-25.
Rai KS, Hattiangady B, Shetty AK. 2007. Enhanced production and dendritic
growth of new dentate granule cells in the middle-aged hippocampus
following intracerebroventricular FGF-2 infusions. The European journal of
neuroscience 26(7):1765-79.
Riva MA, Donati E, Tascedda F, Zolli M, Racagni G. 1994. Short- and long-term
induction of basic fibroblast growth factor gene expression in rat central
nervous system following kainate injection. Neuroscience 59(1):55-65.
Tao Y, Black IB, DiCicco-Bloom E. 1997. In vivo neurogenesis is inhibited by
neutralizing antibodies to basic fibroblast growth factor. Journal of
neurobiology 33(3):289-96.
Teocchi MA, Ferreira AE, da Luz de Oliveira EP, Tedeschi H, D'Souza-Li L. 2013.
Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B
and tumor necrosis factor in temporal lobe epilepsy patients. Journal of
neuroinflammation 10:53.
Tohda C, Nakanishi R, Kadowaki M. 2006. Learning deficits and agenesis of
synapses and myelinated axons in phosphoinositide-3 kinase-deficient mice.
Neuro-Signals 15(6):293-306.
Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton
MJ, Gordon DC, Dunn IF, Goldfeld AE. 2000. A lipopolysaccharide-specific
enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and

47
p300 is recruited to the tumor necrosis factor alpha promoter in vivo.
Molecular and cellular biology 20(16):6084-94.
Weickert CS, Kittell DA, Saunders RC, Herman MM, Horlick RA, Kleinman JE,
Hyde TM. 2005. Basic fibroblast growth factor and fibroblast growth factor
receptor-1 in the human hippocampal formation. Neuroscience 131(1):219-33
Wierschke S, Gigout S, Horn P, Lehmann TN, Dehnicke C, Brauer AU, Deisz RA.
2010. Evaluating reference genes to normalize gene expression in human
epileptogenic brain tissues. Biochem Biophys Res Commun 403(3-4):385-90.
Yang T, Zhou D, Stefan H. 2010. Why mesial temporal lobe epilepsy with
hippocampal sclerosis is progressive: uncontrolled inflammation drives
disease progression? Journal of the neurological sciences 296(1-2):1-6.
Zaman V, Shetty AK. 2003. Fetal hippocampal CA3 cell grafts enriched with
fibroblast growth factor-2 exhibit enhanced neuronal integration into the
lesioned aging rat hippocampus in a kainate model of temporal lobe epilepsy.
Hippocampus 13(5):618-32.
Zechel S, Werner S, Unsicker K, von Bohlen und Halbach O. 2010. Expression
and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation.
The Neuroscientist : a review journal bringing neurobiology, neurology and
psychiatry 16(4):357-73.
Zhao M, Li D, Shimazu K, Zhou YX, Lu B, Deng CX. 2007. Fibroblast growth factor
receptor-1 is required for long-term potentiation, memory consolidation, and
neurogenesis. Biological psychiatry 62(5):381-90.
Zucchini S, Buzzi A, Barbieri M, Rodi D, Paradiso B, Binaschi A, Coffin JD,
Marzola A, Cifelli P, Belluzzi O and others. 2008. Fgf-2 overexpression
increases excitability and seizure susceptibility but decreases seizure-induced
cell loss. The Journal of neuroscience : the official journal of the Society for
Neuroscience 28(49):13112-24.

48
Table 1. Clinical data of TLE patients.
The TLE patient group consisted of five males and five females. The age mean for the patient group was 43.13 ± 12.16 years and
the age mean for their epilepsy onset was 3.20 ± 2.71 years. The duration of disease was 39.93 ± 14.03 years. Childhood febrile seizures
were present in only one case (TLE 13). HS, hippocampal sclerosis; M, male; F, female; B, bilateral; R, right; L, left; SE, status epilepticus;
FS, febrile seizure; ND, not determined; AED,antiepileptic drug; CBZ, carbamazepine; CLB, clobazam; PHT, phenytoin; OXC,
oxcarbazepine; LTG, lamotrigine; VPA, valproate; nd, not determined.
Patients Age
(Years)
Gender
Age at
onset of
epilepsy
(years)
Duration
of Illness
(years)
SE Side of
HS
FS in
childhood
AEDs before
surgery
Last
Seizure(days
before surgery)
TLE1 34.6 F 6.0 28.6 No L No PHT, OXC, CBZ nd
TLE5 41.2 M 7.0 34.2 No L No CBZ 5
TLE6 50.8 M 2.0 48.8 No R No CBZ 7
TLE8 43.8 F 2.0 41.8 Yes B No CBZ, LTG, CLB 3
TLE9 58.2 F 0.5 57.7 No R No VPA, OXC nd
TLE10 54.9 F 4.0 50.9 Yes L No CBZ, CLB nd
TLE13 54.1 F 0.75 53.3 Yes L Yes OXC, CLB nd
TLE14 34.4 M 0.25 34.2 No R No OXC nd
TLE15 44.1 M 1.5 42.6 No R No PHT, CLB, OXC nd
TLE16
15.2 M 8.0 7.2 No L No LMG, CLB nd
49
.
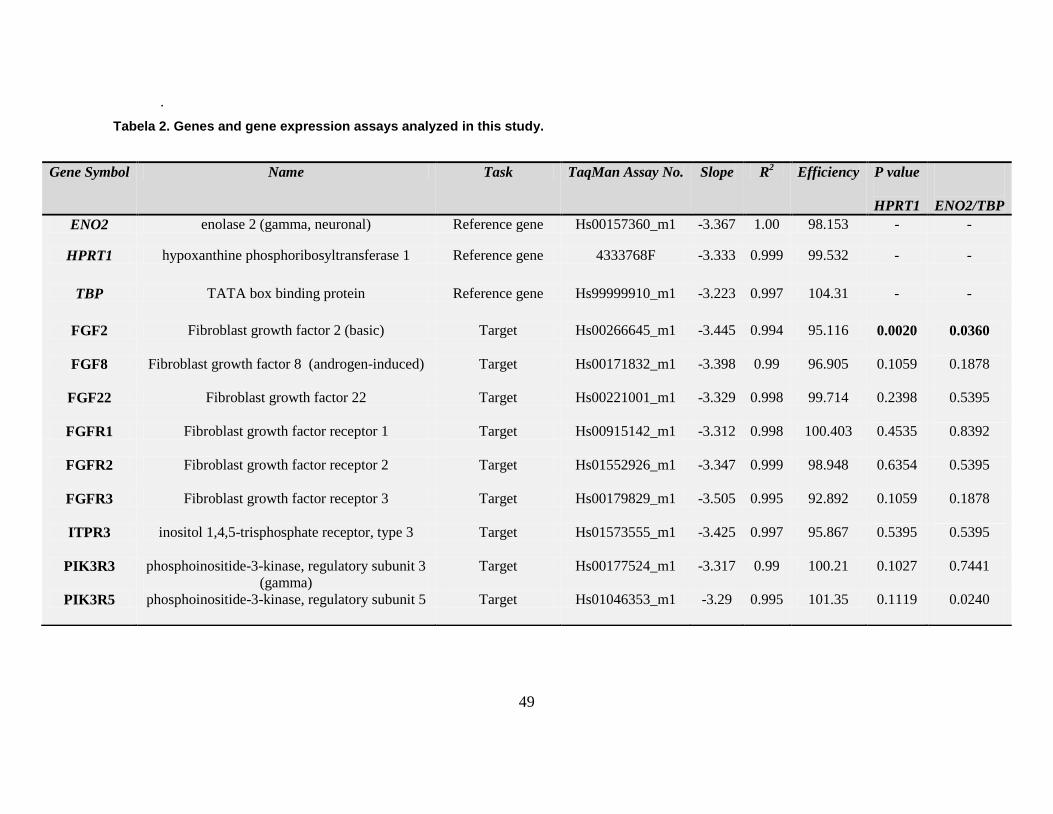
Tabela 2. Genes and gene expression assays analyzed in this study.
Gene Symbol Name Task TaqMan Assay No. Slope R2 Efficiency P value
HPRT1
ENO2/TBP
ENO2 enolase 2 (gamma, neuronal) Reference gene Hs00157360_m1 -3.367 1.00 98.153 - -
HPRT1 hypoxanthine phosphoribosyltransferase 1 Reference gene 4333768F -3.333 0.999 99.532 - -
TBP TATA box binding protein Reference gene Hs99999910_m1 -3.223 0.997 104.31 - -
FGF2 Fibroblast growth factor 2 (basic) Target Hs00266645_m1 -3.445 0.994 95.116 0.0020 0.0360
FGF8 Fibroblast growth factor 8 (androgen-induced) Target Hs00171832_m1 -3.398 0.99 96.905 0.1059 0.1878
FGF22 Fibroblast growth factor 22 Target Hs00221001_m1 -3.329 0.998 99.714 0.2398 0.5395
FGFR1 Fibroblast growth factor receptor 1 Target Hs00915142_m1 -3.312 0.998 100.403 0.4535 0.8392
FGFR2 Fibroblast growth factor receptor 2 Target Hs01552926_m1 -3.347 0.999 98.948 0.6354 0.5395
FGFR3 Fibroblast growth factor receptor 3 Target Hs00179829_m1 -3.505 0.995 92.892 0.1059 0.1878
ITPR3 inositol 1,4,5-trisphosphate receptor, type 3 Target Hs01573555_m1 -3.425 0.997 95.867 0.5395 0.5395
PIK3R3 phosphoinositide-3-kinase, regulatory subunit 3
(gamma)
Target Hs00177524_m1 -3.317 0.99 100.21 0.1027 0.7441
PIK3R5 phosphoinositide-3-kinase, regulatory subunit 5 Target Hs01046353_m1 -3.29 0.995 101.35 0.1119 0.0240
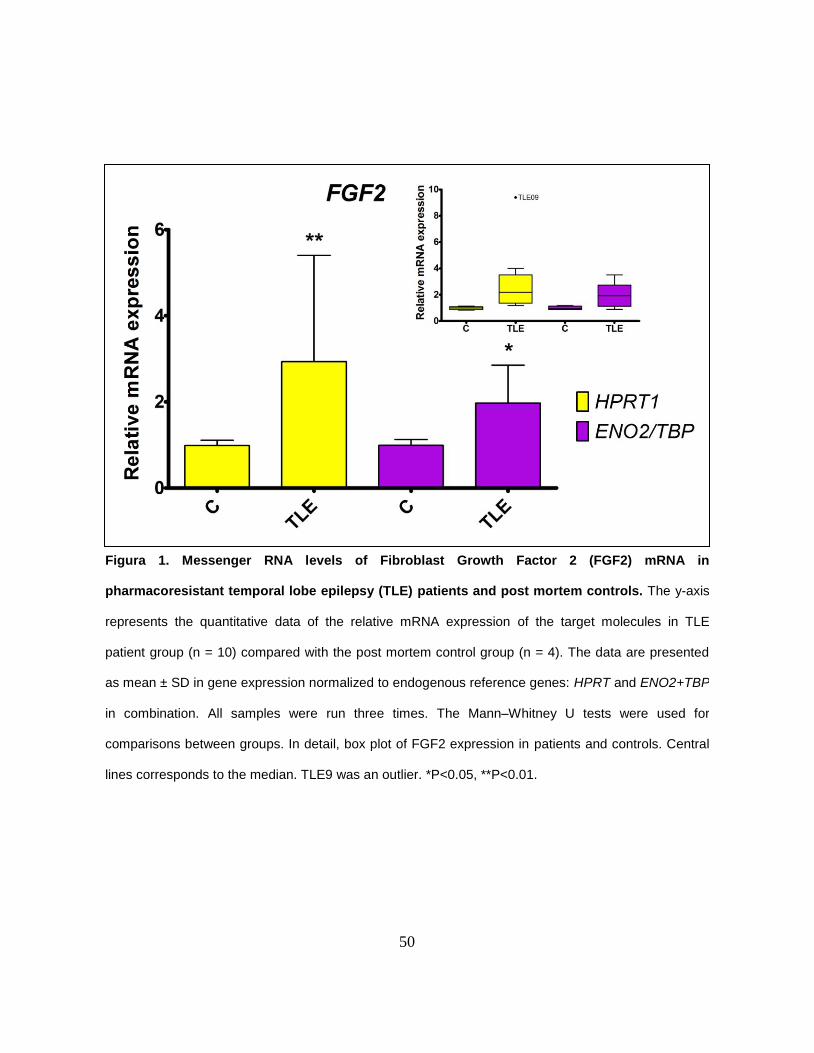
50
Figura 1. Messenger RNA levels of Fibroblast Growth Factor 2 (FGF2) mRNA in
pharmacoresistant temporal lobe epilepsy (TLE) patients and post mortem controls. The y-axis
represents the quantitative data of the relative mRNA expression of the target molecules in TLE
patient group (n = 10) compared with the post mortem control group (n = 4). The data are presented
as mean ± SD in gene expression normalized to endogenous reference genes: HPRT and ENO2+TBP
in combination. All samples were run three times. The Mann–Whitney U tests were used for
comparisons between groups. In detail, box plot of FGF2 expression in patients and controls. Central
lines corresponds to the median. TLE9 was an outlier. *P<0.05, **P<0.01.

51
Figura 2. Immunofluorescence for FGF2 (green) in the hippocampus of
TLE patients (C and D) and post-mortem controls (A and B). TO-PRO3 was
used for nuclei staining (blue). A and C shows the FGF2 expression in dentate
gyrus. B and D describes the FGF2 expression in Ammon’s horn. FGF2 is
expressed in morphologically similar neuronal and glial cells. The percentage of
immunostaining cells in dentate gyrus was higher in TLE patients than controls
(P = 0.0458) and no significance was found in Ammon’s horn (P = 0.1182).
Differences between groups were analyzed by the Test t.

52
Supplementary Material
Figura S1. Immunofluorescence for FGF2 (green) in the hippocampus of TLE patients
(C,D,G) and post-mortem controls (A,B,E,F). TO-PRO3 was used for nuclei staining (blue).
(A-D) shows the FGF2 expression in dentate gyrus. E-G describes the FGF2 expression in
Ammon’s horn. FGF2 is expressed in morphologically similar neuronal and glial cells.

53
4. Conclusões
A principal descoberta deste estudo consiste na hiperexpressão gênica e
proteica do FGF2 no hipocampo de pacientes com ELT. Esta conclusão é
baseada nos seguintes fatos:
A quantificação relativa do RNAm do FGF2 no grupo de pacientes com
ELT foi maior em relação ao grupo de controles post mortem (P = 0,002
para o gene de referência HPRT1; e P = 0,036 para o gene de referência
ENO2+TBP).
A porcentagem de células imunomarcadas para o FGF2 no giro dentado foi
maior nos pacientes do que nos controles (P <0,05), e nenhuma alteração
significativa foi encontrada no Corno de Ammon.
A hiperexpressão gênica e proteica do FGF2 parece não estar associada a
alterações na expressão de outros genes da família do FGF (FGF8 e
FGF22) e fatores envolvidos na via de sinalização (ITPR3, PIK3R3 e
PIK3R5).
Este é o primeiro estudo a avaliar a expressão do FGF2 no hipocampo de
pacientes com ELT. Nosso achado sobre a hiperexpressão desse fator de
crescimento reforça o envolvimento deste na fisiopatologia da ELT, especialmente
em pacientes crônicos e refratários aos medicamentos.

54
O gene do FGF2 codifica uma proteína com múltiplas funções no organismo,
especialmente no hipocampo. Evidências experimentais demonstraram a
participação do FGF2 não só em mecanismos anticonvulsivantes, mas também
em proconvulsivantes, o que denota contrariedades sobre seus efeitos nos
pacientes com ELT. A indução de crises em modelos murinos associou o FGF2 a
neuroproteção, excitabilidade neuronal, reorganização sináptica, neuroinflamação
e neurodegeneração.
É sabido que o giro denteado de pacientes com ELT apresenta intensa
reorganização sináptica e preservação neuronal, ao contrário de áreas como o
CA1 e CA3, onde ocorre grande perda neuronal acompanhada de gliose. A
hiperexpressão do FGF2 no giro denteado dos pacientes sugere seu envolvimento
nessas duas importantes características – reorganização sináptica e
neuropreservação.
Dessa forma, nossos achados tornam ainda mais relevante a atuação do FGF2
como alvo crítico relacionado à epileptogênese, elegendo-o como uma importante
linha para estudos funcionais e farmacológicos na ELT.

55
5. REFERÊNCIAS
1. Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, et al.
Epileptic seizures and epilepsy: definitions proposed by the International League
Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia.
2005;46(4):470-2.
2. Ngugi AK, Kariuki SM, Bottomley C, Kleinschmidt I, Sander JW, Newton CR.
Incidence of epilepsy: a systematic review and meta-analysis. Neurology.
2011;77(10):1005-12.
3. Newton CR, Garcia HH. Epilepsy in poor regions of the world. Lancet.
2012;380(9848):1193-201.
4. De Boer HM. "Out of the shadows": a global campaign against epilepsy.
Epilepsia. 2002;43 Suppl 6:7-8.
5. Noronha AL, Borges MA, Marques LH, Zanetta DM, Fernandes PT, de Boer
H, et al. Prevalence and pattern of epilepsy treatment in different socioeconomic
classes in Brazil. Epilepsia. 2007;48(5):880-5.
6. Fernandes PT, Li LM. Percepção de estigma na epilepsia. Journal of
Epilepsy and Clinical Neurophysiology. 2006;12:207-18.
7. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas
W, et al. Revised terminology and concepts for organization of seizures and
epilepsies: report of the ILAE Commission on Classification and Terminology,
2005-2009. Epilepsia. 2010;51(4):676-85.
8. Engel J, Jr. Mesial temporal lobe epilepsy: what have we learned? The
Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry.
2001;7(4):340-52.

56
9. McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms
underlying epileptogenesis. Science's STKE : signal transduction knowledge
environment. 2006;2006(356):re12.
10. Engel J, Jr. Introduction to temporal lobe epilepsy. Epilepsy Res.
1996;26(1):141-50.
11. Pitkanen A, Sutula TP. Is epilepsy a progressive disorder? Prospects for
new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol.
2002;1(3):173-81.
12. Engel J, Jr. Etiology as a risk factor for medically refractory epilepsy: a case
for early surgical intervention. Neurology. 1998;51(5):1243-4.
13. Lothman EW, Bertram EH, 3rd, Stringer JL. Functional anatomy of
hippocampal seizures. Prog Neurobiol. 1991;37(1):1-82.
14. Duvernoy HM. The human hippocampus : an atlas of applied anatomy.
M nchen: J.F. Bergmann; 1988. 166 p. p.
15. Deng W, Aimone JB, Gage FH. New neurons and new memories: how does
adult hippocampal neurogenesis affect learning and memory? Nature reviews
Neuroscience. 2010;11(5):339-50.
16. Pitkanen A, Lukasiuk K. Molecular and cellular basis of epileptogenesis in
symptomatic epilepsy. Epilepsy Behav. 2009;14 Suppl 1:16-25.
17. Fisher PD, Sperber EF, Moshe SL. Hippocampal sclerosis revisited. Brain &
development. 1998;20(8):563-73.
18. Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA,
Frotscher M. "Dormant basket cell" hypothesis revisited: relative vulnerabilities of
dentate gyrus mossy cells and inhibitory interneurons after hippocampal status
epilepticus in the rat. The Journal of comparative neurology. 2003;459(1):44-76.

57
19. Dudek FE, Sutula TP. Epileptogenesis in the dentate gyrus: a critical
perspective. Progress in brain research. 2007;163:755-73.
20. Silveira G, de Oliveira AC, Teixeira AL. Insights into inflammation and
epilepsy from the basic and clinical sciences. Journal of clinical neuroscience :
official journal of the Neurosurgical Society of Australasia. 2012;19(8):1071-5.
21. Teocchi MA, Ferreira AE, da Luz de Oliveira EP, Tedeschi H, D'Souza-Li L.
Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B and
tumor necrosis factor in temporal lobe epilepsy patients. J Neuroinflammation.
2013;10:53.
22. Vezzani A. Epilepsy and inflammation in the brain: overview and
pathophysiology. Epilepsy currents / American Epilepsy Society. 2014;14(1
Suppl):3-7.
23. Wieser HG, Epilepsy ICoNo. ILAE Commission Report. Mesial temporal
lobe epilepsy with hippocampal sclerosis. Epilepsia. 2004;45(6):695-714.
24. Freiman TM, Eismann-Schweimler J, Frotscher M. Granule cell dispersion in
temporal lobe epilepsy is associated with changes in dendritic orientation and
spine distribution. Experimental neurology. 2011;229(2):332-8.
25. Neves RS, de Souza Silva Tudesco I, Jardim AP, Caboclo LO, Lancellotti C,
Ferrari-Marinho T, et al. Granule cell dispersion is associated with memory
impairment in right mesial temporal lobe epilepsy. Seizure : the journal of the
British Epilepsy Association. 2012;21(9):685-90.
26. Scharfman HE, McCloskey DP. Postnatal neurogenesis as a therapeutic
target in temporal lobe epilepsy. Epilepsy Res. 2009;85(2-3):150-61.
27. Aguiar CC, Almeida AB, Araujo PV, de Abreu RN, Chaves EM, do Vale OC,
et al. Oxidative stress and epilepsy: literature review. Oxidative medicine and
cellular longevity. 2012;2012:795259.

58
28. Rowley S, Patel M. Mitochondrial involvement and oxidative stress in
temporal lobe epilepsy. Free radical biology & medicine. 2013;62:121-31.
29. Chang SJ, Yu BC. Mitochondrial matters of the brain: mitochondrial
dysfunction and oxidative status in epilepsy. Journal of bioenergetics and
biomembranes. 2010;42(6):457-9.
30. Frantseva MV, Perez Velazquez JL, Tsoraklidis G, Mendonca AJ, Adamchik
Y, Mills LR, et al. Oxidative stress is involved in seizure-induced
neurodegeneration in the kindling model of epilepsy. Neuroscience.
2000;97(3):431-5.
31. Scharfman HE. The neurobiology of epilepsy. Curr Neurol Neurosci Rep.
2007;7(4):348-54.
32. Lowenstein DH. Recent advances related to basic mechanisms of
epileptogenesis. Epilepsy research Supplement. 1996;11:45-60.
33. Kopec AM, Carew TJ. Growth factor signaling and memory formation:
temporal and spatial integration of a molecular network. Learning & memory.
2013;20(10):531-9.
34. Pimentel E. Handbook of growth factors. Boca Raton: CRC Press; 1994.
35. Jankowsky JL, Patterson PH. The role of cytokines and growth factors in
seizures and their sequelae. Prog Neurobiol. 2001;63(2):125-49.
36. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to
roles in development, metabolism and disease. J Biochem. 2011;149(2):121-30.
37. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends in
genetics : TIG. 2004;20(11):563-9.

59
38. Sorensen V, Nilsen T, Wiedlocha A. Functional diversity of FGF-2 isoforms
by intracellular sorting. BioEssays : news and reviews in molecular, cellular and
developmental biology. 2006;28(5):504-14.
39. Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor
signaling in physiology and pathology. Cold Spring Harbor perspectives in biology.
2013;5(6).
40. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and
therapy. Nature reviews Drug discovery. 2009;8(3):235-53.
41. Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM.
Receptor specificity of the fibroblast growth factor family. The complete mammalian
FGF family. The Journal of biological chemistry. 2006;281(23):15694-700.
42. Turner N, Grose R. Fibroblast growth factor signalling: from development to
cancer. Nature reviews Cancer. 2010;10(2):116-29.
43. Bottcher RT, Niehrs C. Fibroblast growth factor signaling during early
vertebrate development. Endocr Rev. 2005;26(1):63-77.
44. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their
receptors in cancer. The Biochemical journal. 2011;437(2):199-213.
45. Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their
receptors in the central nervous system. Cell and tissue research.
2003;313(2):139-57.
46. Eclancher F, Perraud F, Faltin J, Labourdette G, Sensenbrenner M.
Reactive astrogliosis after basic fibroblast growth factor (bFGF) injection in injured
neonatal rat brain. Glia. 1990;3(6):502-9.

60
47. Mark RJ, Fuson KS, Keane-Lazar K, May PC. Fibroblast growth factor-8
protects cultured rat hippocampal neurons from oxidative insult. Brain Res.
1999;830(1):88-93.
48. Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth
factor-2 activates a latent neurogenic program in neural stem cells from diverse
regions of the adult CNS. J Neurosci. 1999;19(19):8487-97.
49. Patel MN, McNamara JO. Selective enhancement of axonal branching of
cultured dentate gyrus neurons by neurotrophic factors. Neuroscience.
1995;69(3):763-70.
50. Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA,
Umemori H. Distinct FGFs promote differentiation of excitatory and inhibitory
synapses. Nature. 2010;465(7299):783-7.
51. Bugra K, Pollard H, Charton G, Moreau J, Ben-Ari Y, Khrestchatisky M.
aFGF, bFGF and flg mRNAs show distinct patterns of induction in the
hippocampus following kainate-induced seizures. Eur J Neurosci. 1994;6(1):58-66.
52. Gomez-Pinilla F, van der Wal EA, Cotman CW. Possible coordinated gene
expressions for FGF receptor, FGF-5, and FGF-2 following seizures. Experimental
neurology. 1995;133(2):164-74.
53. Riva MA, Donati E, Tascedda F, Zolli M, Racagni G. Short- and long-term
induction of basic fibroblast growth factor gene expression in rat central nervous
system following kainate injection. Neuroscience. 1994;59(1):55-65.
54. Delrieu I. The high molecular weight isoforms of basic fibroblast growth
factor (FGF-2): an insight into an intracrine mechanism. FEBS letters.
2000;468(1):6-10.
55. Zechel S, Werner S, Unsicker K, von Bohlen und Halbach O. Expression
and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation. The

61
Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry.
2010;16(4):357-73.
56. Eckenstein F, Woodward WR, Nishi R. Differential localization and possible
functions of aFGF and bFGF in the central and peripheral nervous systems. Annals
of the New York Academy of Sciences. 1991;638:348-60.
57. Zucchini S, Buzzi A, Barbieri M, Rodi D, Paradiso B, Binaschi A, et al. Fgf-2
overexpression increases excitability and seizure susceptibility but decreases
seizure-induced cell loss. J Neurosci. 2008;28(49):13112-24.
58. Eves EM, Skoczylas C, Yoshida K, Alnemri ES, Rosner MR. FGF induces a
switch in death receptor pathways in neuronal cells. J Neurosci. 2001;21(14):4996-
5006.
59. Gomez-Pinilla F, Vu L, Cotman CW. Regulation of astrocyte proliferation by
FGF-2 and heparan sulfate in vivo. J Neurosci. 1995;15(3 Pt 1):2021-9.
60. Garcia-Hernandez S, Potashner SJ, Morest DK. Role of fibroblast growth
factor 8 in neurite outgrowth from spiral ganglion neurons in vitro. Brain Res.
2013;1529:39-45.
61. Nakatake Y, Hoshikawa M, Asaki T, Kassai Y, Itoh N. Identification of a
novel fibroblast growth factor, FGF-22, preferentially expressed in the inner root
sheath of the hair follicle. Biochimica et biophysica acta. 2001;1517(3):460-3.
62. Umemori H, Linhoff MW, Ornitz DM, Sanes JR. FGF22 and its close
relatives are presynaptic organizing molecules in the mammalian brain. Cell.
2004;118(2):257-70.
63. Lee CH, Umemori H. Suppression of epileptogenesis-associated changes in
response to seizures in FGF22-deficient mice. Frontiers in cellular neuroscience.
2013;7:43.

62
64. Mikoshiba K. The IP3 receptor/Ca2+ channel and its cellular function.
Biochemical Society symposium. 2007(74):9-22.
65. Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms.
Biochimica et biophysica acta. 2009;1793(6):933-40.
66. Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked
mitochondrial calcium signals. The EMBO journal. 1999;18(22):6349-61.
67. Kelly PT, Mackinnon RL, 2nd, Dietz RV, Maher BJ, Wang J. Postsynaptic
IP3 receptor-mediated Ca2+ release modulates synaptic transmission in
hippocampal neurons. Brain Res Mol Brain Res. 2005;135(1-2):232-48.
68. Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, et
al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits
apoptotic Ca2+ signals into mitochondria. The Journal of biological chemistry.
2005;280(49):40892-900.
69. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-
kinases as regulators of growth and metabolism. Nature reviews Genetics.
2006;7(8):606-19.
70. Tohda C, Nakanishi R, Kadowaki M. Learning deficits and agenesis of
synapses and myelinated axons in phosphoinositide-3 kinase-deficient mice.
Neurosignals. 2006;15(6):293-306.
71. Cuesto G, Enriquez-Barreto L, Carames C, Cantarero M, Gasull X, Sandi C,
et al. Phosphoinositide-3-kinase activation controls synaptogenesis and
spinogenesis in hippocampal neurons. J Neurosci. 2011;31(8):2721-33.

63
ANEXOS
Anexo 1: Termo de Consentimento Livre e Esclarecido
FORMULÁRIO DE CONSENTIMENTO PARA PESQUISA MÉDICA, Página 1 de 3
Título do projeto: Caracterização neuropatológica da epilepsia de lobo temporal mesial
familiar
Investigadores principais:
Profa. Cláudia V. Maurer Morelli, Prof. Dr. Luciano de Souza Queiroz, Prof. Dr. Fernando
Cendes e Profa. Dra. Iscia Lopes Cendes
OBJETIVO DA PESQUISA:
Eu entendo que fui convidado (a) a participar em um projeto de pesquisa envolvendo
pacientes e famílias de indivíduos com epilepsia. O objetivo geral do estudo é comparar se
a lesão do tecido nervoso cerebral encontrada em pacientes com epilepsia familiar são
iguais àquelas encontradas em pacientes com epilepsia na forma esporádica (sem
antecedentes familiares). Esse estudo pode eventualmente melhorar o diagnóstico e levar a
um melhor tratamento dessa doença. Tanto as amostras de tecido nervoso como a
informação médica a meu respeito, bem como a respeito de minha família que forem
obtidas para esse estudo, poderão ser compartilhadas com outros pesquisadores que
trabalham com epilepsia. O sigilo será mantido em todos os estudos colaborativos através
da utilização de um número de código para a identificação dos indivíduos participantes.
PROCEDIMENTO:
Eu entendo que se concordar em participar desse estudo, os pesquisadores participantes
farão perguntas a respeito dos meus antecedentes médicos e familiais. O meu
consentimento se refere ao uso do material que será retirado durante a cirurgia para a
realização de estudos científicos. É importante lembrar que a cirurgia mencionada acima,
faz parte do tratamento médico. A pesquisa laboratorial utilizando as amostras de tecido
poderá ser feita durante um período máximo de 30 anos após a coleta. Após o término desta
pesquisa com prazo de duração de cerca de 4 anos, pretende-se armazenar este material
durante no máximo 30 anos caso haja a necessidade de se dar continuidade a pesquisas
complementares. Neste caso será feito novo protocolo para submissão à aprovação do
Comitê de Ética em Pesquisa local e se necessário ao Conselho Nacional de Ética em Saúde
(CONEP).

64
RISCO E DESCONFORTO:
Não há quaisquer riscos ou prejuízos adicionais para os sujeitos da pesquisa, já que o
material utilizado para a pesquisa (pequena amostra de tecido nervoso) será obtido de
pacientes submetidos à retirada cirúrgica do tecido lesado, como forma de tratamento.

65
FORMULÁRIO DE CONSENTIMENTO PARA PESQUISA MÉDICA, Página 2 de 3
Título do projeto: Caracterização neuropatológica da epilepsia de lobo temporal mesial
familiar
Investigadores principais:
Profa. Cláudia V. Maurer Morelli, Prof. Dr. Luciano de Souza Queiroz, Prof. Dr. Fernando
Cendes e Profa. Dra. Iscia Lopes Cendes
VANTAGENS:
Eu entendo que não terei nenhuma vantagem pessoal com a participação neste estudo e que
o diagnóstico e o tratamento de possíveis familiares com a epilepsia familiar ou esporádica
não serão modificados. Contudo, os resultados desse estudo podem, em longo prazo,
oferecer vantagens para os indivíduos com epilepsia e suas famílias, possibilitando um
melhor diagnóstico e um tratamento mais adequado. Caso, no futuro, pesquisas com o
material venham a trazer resultados com algum tipo de benefício aos familiares envolvidos,
estes serão prontamente informados.
SIGILO:
Eu entendo que toda informação médica, assim como os resultados dos estudos decorrentes
desse projeto de pesquisa, serão submetidos aos regulamentos do HC-UNICAMP referentes
ao sigilo da informação médica, o que garante que os dados pessoais dos participantes não
serão de livre acesso. Se os resultados ou informações fornecidas forem utilizados para fins
de publicação científica, nenhum nome será revelado.
FORNECIMENTO DE INFORMAÇÃO ADICIONAL:
Eu entendo que posso requisitar informações adicionais relativas ao estudo a qualquer
momento. A Dra. Iscia Lopes Cendes, tel (019) 3788-8907 estará disponível para responder
minhas questões e preocupações. Em caso de recurso, dúvidas ou reclamações contactar a
secretaria da comissão de ética da Faculdade de Ciências Médicas-UNICAMP, tel. (019)
3788-7232.
RECUSA OU DESCONTINUAÇÃO DA PARTICIPAÇÃO:
Eu entendo que a minha participação é voluntária e que eu posso me recusar a participar ou
retirar meu consentimento e interromper a minha participação no estudo a qualquer
momento sem comprometer os cuidados médicos que recebo atualmente ou receberei no
futuro no HC-UNICAMP. Eu reconheço também que a Dra. Iscia Lopes Cendes pode
interromper a minha participação nesse estudo a qualquer momento que julgarem
apropriado.

66
FORMULÁRIO DE CONSENTIMENTO PARA PESQUISA MÉDICA, Página 3 de 3
Título do projeto: Caracterização neuropatológica da epilepsia de lobo temporal mesial
familiar
Investigadores principais:
Profa. Cláudia V. Maurer Morelli, Prof. Dr. Luciano de Souza Queiroz, Prof. Dr. Fernando
Cendes e Profa. Dra. Iscia Lopes Cendes
Eu confirmo que o(a) Dr(a)._______________________________________________ me
explicou o objetivo do estudo, os procedimentos aos quais serei submetido e os riscos,
desconforto e possíveis vantagens advindas desse projeto de pesquisa. Eu li e/ou me foi
explicado, assim como compreendi esse formulário de consentimento e estou de pleno
acordo em participar desse estudo.
_______________________________________________________________________
Nome do participante ou responsável
_________________________________________________ __________________
Assinatura do participante ou responsável data
________________________________________________________________________
Nome da testemunha
__________________________________________________ ___________________
Assinatura da testemunha data
RESPONSABILIDADE DO PESQUISADOR:
Eu expliquei a _____________________________________________________o objetivo
do estudo, os procedimentos requeridos e os possíveis riscos e vantagens que poderão advir
do estudo, usando o melhor do meu conhecimento. Eu me comprometo a fornecer uma
cópia desse formulário de consentimento ao participante ou responsável.
________________________________________________________________________
Nome do pesquisador ou associado
___________________________________________________ _______________
Assinatura do pesquisador ou associado data

67
Anexo 2: Parecer do Comitê de Ética


















