
UNIVERSIDADE DE SÃO PAULO ESCOLA DE ENGENHARIA DE LORENA WESLEY FELIPE DE...
80
UNIVERSIDADE DE SÃO PAULO ESCOLA DE ENGENHARIA DE LORENA WESLEY FELIPE DE PAULA JOSÉ Validação de Metodologia Analítica para Determinação de Resíduos de Etilenotiouréia (ETU) por Cromatografia Líquida Acoplada a Espectrometria de Massas – CLAE-EM/EM Lorena – SP 2012
Transcript of UNIVERSIDADE DE SÃO PAULO ESCOLA DE ENGENHARIA DE LORENA WESLEY FELIPE DE...

UNIVERSIDADE DE SÃO PAULO
ESCOLA DE ENGENHARIA DE LORENA
WESLEY FELIPE DE PAULA JOSÉ
Validação de Metodologia Analítica para Determinação de Resíduos de
Etilenotiouréia (ETU) por Cromatografia Líquida Acoplada a
Espectrometria de Massas – CLAE-EM/EM
Lorena – SP
2012

Wesley Felipe de Paula José
VALIDAÇÃO DE METODOLOGIA ANALÍTICA PARA DETERMINAÇÃO DE
RESÍDUOS DE ETILENOTIOURÉIA (ETU) POR CROMATOGRAFIA
LÍQUIDA ACOPLADA A ESPECTROMETRIA DE MASSAS – CLAE-EM/EM
Trabalho de Conclusão de Curso II apresentado à Escola de Engenharia de Lorena como requisito para conclusão do curso de graduação em Engenharia Industrial Química, área de
concentração: Química Analítica. Orientador: Prof. Dr. Adilson Roberto Gonçalves
Lorena – SP 2012

DEDICATÓRIA
Dedico o presente trabalho de
conclusão de curso à minha família sempre presente, dedicada e que sempre me deram todo apoio necessário para chegar até aqui.

AGRADECIMENTOS A DEUS, primeiramente.
Ao Prof. Dr. Adilson Roberto Gonçalves, pela valiosa orientação para elaboração
deste trabalho.
À Industria BASF, em especial, à Maria Angélica Duchen, diretora de estudo, Tales
Duque Esteves, estagiário no instrumentação analítica, e José Wellington,
responsável pelo instrumentação analítica, ambos do Laboratório Global de Meio
Ambiente e Segurança Alimentar - GENCS, que estiveram sempre presentes,
prestando uma ajuda essencial para confecção deste trabalho, proporcionando
novos conhecimentos na parte de cromatografia liquida, e sempre dispostos a
sanarem todas as dúvidas de espectrometria de massas
À Simone Freitas Guimarães, que me ajudou dando todas as diretrizes inicias em
relação à escolha do projeto a ser realizado.
À Roberta Leite, Supervisora de Laboratório de resíduos do GENCS, que permitiu a
realização deste trabalho, proporcionando o uso do laboratório para realização dos
experimentos.
A todos os amigos que de alguma maneira contribuíram para realização deste
trabalho.
A todos os professores da Escola de Engenharia de Lorena que contribuíram e
foram responsáveis pela minha formação.

LISTA DE ABREVIATURAS E SIGLAS
ANVISA: Agência Nacional de Vigilância Sanitária
APCI: Ionização Química à Pressão Atmosférica, do inglês atmospheric pressure
chemical ionization
ANOVA: Análise de Variância
B: Branco de Reagentes
BF: Branco Fortificado
CS2: Dissulfeto de carbono
CLAE: Cromatografia Liquida de Alta Eficiência, do inglês High Performance Liquid
Chromatography
CLAE-UV: Cromatografia Liquida de Alta Eficiência com detector UltraVioleta, do
inglês High Performance Liquid Chromatography with Ultra Violet Detector
CG: Cromatografia Gasosa, do inglês Gas Chromatography
CLUE: Cromatografia Liquida de Ultra Eficiência, do inglês Ultra Performance Liquid
Chromatography
CL-EM: Cromatografia Liquida Aclopada à Espectrometria de Massas, do inglês
Liquid Chromatography – Mass Spectrometry
CL: Cromatografia Liquida do inglês Liquid Chromatography
CL-EM/EM: Cromatografia Liquida Aclopada à Espectrometria de Massas em Série,
do inglês Liquid Chromatography with Mass Spectrometry tandem Mass
Spectrometry
CV: Coeficiente de Variação
CID: Dissociação Induzida por Colisão, do inglês Collision Induced Dissociation
DTC: Ditiocarbamatos
DIDT: Etileno Bistioutam Dissulfeto
DPR: Desvio Padrão Relativo
DP: Desvio Padrão
EBDC: Etilenobisditiocarbamatos
ETU: Etilenotiouréia
EU: Etilenouréia
ESI: Ionização por Eletronebulização, do inglês electrospray ionization

FM: Fase Móvel
gl: Grau de Liberdade
IARC: Agência Internacional de Pesquisa com Câncer, do inglês International
Agency for Research on Cancer
ISO: International Standard Organization
LMR: Limite Maximo de Resíduos
LOD: Limite de Detecção
LOQ: Limite de Quantificação
MRM: Monitoramento de Múltiplas Reações, do inglês Multiple-reaction monitoring
MQ: Soma Média Quadrática
m/z: Relação de Massa por Carga
PTU: Propilenotiouréia
PARA: Programa Nacional de Análise de Resíduos de Agrotóxicos em Alimentos
QqQ: Sistemas Quadrupolo
QC: Controle de Qualidade, do inglês Quality Control
RDC: Resolução da Diretoria Colegiada
R: Recuperação
r: Coeficiente de Correlação
SPE: Solução Padrão Estoque
SPC: Solução Padrão de Calibração
STDF: Solução Padrão de Fortificação
SQ: Soma dos Quadrados
SIM: Monitoramento de íons selecionados, do inglês selected íon monitoring
TOF: Analisadores de tempo de vôo, do inglês Time-of-Flight
TIC: Cromatograma de íons totais, do inglês total íon chromatogram
UT: Amostra Testemunha
UV/Vis: Ultravioleta Visível
USP: United States Pharmacopeia
WHO: Organização Mundial de Saúde, do inglês World Health Organization

RESUMO
VALIDAÇÃO DE METODOLOGIA ANALÍTICA PARA DETERMINAÇÃO DE RESÍDUOS DE ETILENOTIOURÉIA (ETU) POR CROMATOGRAFIA LÍQUIDA
ACOPLADA A ESPECTROMETRIA DE MASSAS – CLAE-EM/EM O crescimento contínuo da população mundial tem causado a diminuição das
áreas apropriadas para o cultivo agrícola e, por outro lado, com este crescimento,
observa-se um crescente aumento na demanda por alimentos. Este aumento da
demanda por alimentos tem impulsionado o desenvolvimento de sistemas agrícolas
cada vez mais eficientes e uso cada vez mais constante de defensivos agrícolas,
para se obter uma maior produtividade e manter a qualidade dos produtos, na
agricultura. Porém, exatamente devido a este aumento do uso de defensivos
agrícolas, e ao aumento do risco resultante de uma exposição dos consumidores
aos possíveis resíduos dos agrotóxicos nos alimentos, as agências reguladoras
estabelecem limites máximos de resíduos (LMR) para todos os agrotóxicos. Neste
presente trabalho, efetuou-se a validação de análise de resíduos de etilenotiouréia
(ETU), metabólito da degradação e/ou biotransformação dos fungicidas
etilenobisditiocarbamatos (EBDC), através da técnica cromatográfica de
cromatografia liquida acoplada à espectrometria de massas em série (CL-EM/EM),
em substituição a técnica de cromatografia liquida de alta eficiência com detecção
ultravioleta.
Esta nova metodologia foi validada para este trabalho, utilizando-se a matriz
de pêssego, e os parâmetros utilizados para a validação do método foram:
seletividade, linearidade, limite de detecção, limite de quantificação, exatidão,
precisão e robustez, sendo que todos estes parâmetros foram validados com
obtenção de resultados satisfatórios, dentro das faixas de aceitação recomendada
para cada um destes parâmetros. Os estudos de recuperação foram realizados em
dois níveis de fortificação (1 e 50 LOQ), tendo sido obtidos recuperações médias de
83 e 81% para fortificação de 0,01 mg/kg e 0,50 mg/kg, respectivamente, e
coeficiente de variação de 5 para ambas as fortificações. Além disso, os limites de
quantificação obtidos pela análise em CL-EM/EM mostram que este método pode

ser utilizado para a detecção do ETU em concentrações abaixo dos LMR
estabelecidos pela legislação.
Palavras-chave: Cromatografia liquida acoplada à espectrometria de massas
em série, Etilenotiouréia, Validação.

ABSTRACT
VALIDATION OF ANALYTICAL METHODS FOR DETERMINATION OF ETHYLENETHIOUREA (ETU) RESIDUES BY LIQUID CHROMATOGRAPHY MASS
SPECTROMETRY – LC-MS/MS
The continuing growth of population has caused the decrease of the suitable
areas for agricultural cultivation and on the other hand, with this growth, there is an
increasing demand for food. This increased demand for food has driven the
development of agricultural systems more efficient and increasing use of pesticides
constant, to achieve higher productivity and maintain product quality in agriculture.
But precisely because of this increased use of pesticides, and the increased risk
resulting from a possible consumer exposure to residues of
pesticides in food, regulatory agencies establish maximum residue limits (MRLs) for
all pesticides. In this work, we performed the validation of residue analysis
ethylenethiourea (ETU), a metabolite of the degradation and / or biotransformation of
ethylenebisdithiocarbamates fungicides (EBDC) by chromatographic technique of
liquid chromatography coupled to mass spectrometry in series (LC-MS/MS) to
replace the technique of high performance liquid chromatography with ultraviolet
detection.
This new methodology was validated in this study, using peach, and the
parameters used for the validation of the method were: selectivity, linearity, detection
limit, quantification limit, accuracy, precision and robustness, all of which parameters
were validated with satisfactory results within the recommended ranges of
acceptance for each of these parameters. The recovery studies were performed in
two fortification levels (1 and 50 LOQ) was obtained and an average recovery of 83
to 81% fortification of 0.01 mg / kg and 0.50 mg / kg, respectively, and coefficient of
variation of 5 for both fortifications.
Furthermore, the limit of quantification in LC-MS/MS obtained by the analysis
show that this method can be used for the detection of ETU in concentrations below
the MRL established by law.
Keywords: liquid chromatography coupled to mass spectrometry in series,
ethylenethiourea, Validation.

Sumário
1.Objetivo _______________________________________________________ 15
1.1 Objetivo Geral ________________________________________________ 15
1.2 Objetivos Específicos __________________________________________ 15
2. Justificativa ____________________________________________________ 16
3. Revisão da Literatura ____________________________________________ 17
3.1 Agrotóxicos ________________________________________________ 17
3.2 Definição e Classificação dos Agrotóxicos ________________________ 18
3.3 Ditiocarbamatos _____________________________________________ 18
3.4 Etilenobisditiocarbamatos (EBDC) _______________________________ 20
3.5 Degradação dos EBDC _______________________________________ 20
3.6 Etilenotiouréia (ETU) _________________________________________ 21
3.7 Formação de ETU durante fabricação de EBDC e armazenamento. ____ 22
3.8 Formação de ETU durante preparo dos alimentos __________________ 22
3.9 Cromatografia ______________________________________________ 22
3.10 Cromatografia na análise de resíduos de agrotóxicos ______________ 23
3.11 Cromatografia líquida acoplada à espectrometria de massas ________ 24
3.12 Métodos de análise de resíduos de agrotóxicos em alimentos _______ 29
3.13 Validação ________________________________________________ 29
3.14 Processo de validação ______________________________________ 30
3.15 Parâmetros de Validação ____________________________________ 31
3.16 Validação Curta ___________________________________________ 31
3.17 Precisão _________________________________________________ 31
3.18 Repetitividade _____________________________________________ 31
3.19 Precisão Intermediária ______________________________________ 32
3.20 Seletividade ______________________________________________ 32
3.21 Recuperação _____________________________________________ 32
3.22 Linearidade _______________________________________________ 33
3.23 Limite de Detecção (LOD) ___________________________________ 33

3.24 Limite de Quantificação (LOQ) ________________________________ 34
3.25 Robustez ________________________________________________ 35
3.26 Estabilidade ______________________________________________ 35
4. Validação do Método de Análise ___________________________________ 36
4.1 Números de Amostras para os Ensaios de Validação do Método ________ 36
a) Amostra Testemunha _______________________________________ 36
b) Amostra Fortificada ________________________________________ 36
4.2 Seletividade __________________________________________________ 36
4.3 Linearidade __________________________________________________ 37
4.4 Recuperação _________________________________________________ 37
4.5 Precisão ____________________________________________________ 37
4.6 Robustez ____________________________________________________ 37
5. Materiais e Métodos _____________________________________________ 38
5.1 Materiais de referência _______________________________________ 38
5.2 Análise de Etilenotiouréia (ETU) por CLAE-UV _______________________ 38
5.2.1 Equipamentos, reagentes e solventes, soluções e diversos, materiais e
vidraria. ______________________________________________________ 38
5.2.2 Preparo Típico de Soluções Padrão de Calibração e de Fortificação ___ 40
5.2.3 Preparação da Amostra _____________________________________ 43
5.2.4 Procedimento Analítico ______________________________________ 43
5.2.5 Análise Instrumental ________________________________________ 44
5.2.6 Preparo de Soluções _______________________________________ 44
5.2.7 Análise Quantitativa ________________________________________ 45
5.3 Análise de Etilenotiouréia (ETU) por CLAE-EM/EM ___________________ 45
5.3.1 Equipamentos, reagentes e solventes, soluções e diversos, materiais e
vidraria. ______________________________________________________ 45
5.3.2 Preparo Típico de Soluções Padrão de Calibração e de Fortificação ___ 48
5.3.3 Preparação da Amostra _____________________________________ 50
5.3.4 Procedimento Analítico ______________________________________ 50

5.3.5 Análise Instrumental ________________________________________ 51
5.3.6 Preparo de Soluções _______________________________________ 52
5.3.7 Análise Quantitativa ________________________________________ 54
6. Resultados e Discussões _________________________________________ 55
6.1 Seletividade __________________________________________________ 58
6.2 Linearidade, Faixa Linear de Trabalho, Limite de Detecção e Limite de
Quantificação ___________________________________________________ 61
6.2.1 Resultados fornecidos pelo teste de Linearidade __________________ 62
6.3 Exatidão e Precisão (Intermediária e Repetitividade) __________________ 67
6.4 Robustez ____________________________________________________ 70
6.5 Análise de Impacto da substituição de Metodologia de Análise de CLAE-UV
para CL-EM/EM _________________________________________________ 72
6.5.1 Seletividade ______________________________________________ 72
6.5.2 Tempo de Análise __________________________________________ 73
6.5.3 Valores de Recuperação dos fortificados para CLAE-UV ____________ 73
7. Conclusão _____________________________________________________ 75
8. Referências Bibliografias _________________________________________ 77

Lista de Figuras
Figura 1.Estrutura dos principais ditiocarbamatos de uso autorizado no Brasil ....... 19
Figura 2. Degradação dos EBDC e formação do ETU ............................................. 21
Figura 3. Componentes Básicos de um espectrômetro de massas ......................... 25
Figura 4. Cromatograma para Solução Padrão de Calibração de concentração 25,0
ng/mL injetada no CL-EM/EM ................................................................................... 57
Figura 5. Cromatograma para Solução Padrão de Calibração de concentração 25,0
ng/mL injetada no CLAE-UV ..................................................................................... 57
Figura 6.Cromatograma para Amostra Testemunha (UT) injetada no CL-EM/EM ... 59
Figura 7. Cromatograma para Branco de Reagentes (B) injetada no CL-EM/EM .... 59
Figura 8. Cromatograma para Branco Fortificado (BF) injetada no CL-EM/EM ....... 60
Figura 9. Cromatograma para Quality Control (QC) injetada no CL-EM/EM ............ 60
Figura 10. Curva Sobreposta com Matriz e sem Matriz .......................................... 61
Figura 11. Curva de Calibração ............................................................................... 63
Figura 12. Cromatograma para Solução Padrão de Calibração de concentração 1,25
ng/mL ........................................................................................................................ 64
Figura 13. Cromatograma para Solução Padrão de Calibração de concentração 2,50
ng/mL ........................................................................................................................ 65
Figura 14. Cromatograma para Solução Padrão de Calibração de concentração 6,25
ng/mL ........................................................................................................................ 65
Figura 15. Cromatograma para Solução Padrão de Calibração de concentração 25,0
ng/mL ........................................................................................................................ 66
Figura 16. Cromatograma para Solução Padrão de Calibração de concentração 50,0
ng/mL ........................................................................................................................ 66

Lista de Tabelas
Tabela 1. Parâmetros que podem causar variações nas respostas do método ....... 35
Tabela 2. Etilenotiouréia (ETU) (utilizada para propósito de calibração e fortificação)
.................................................................................................................................. 38
Tabela 3. Preparo de Soluções Padrão de Fortificação para Análise utilizando-se
CLAE-UV .................................................................................................................. 43
Tabela 4. Preparo de Soluções Padrão de Calibração para Análise utilizando-se
CLAE-UV .................................................................................................................. 44
Tabela 5. Preparo de Soluções Padrão de Fortificação para Análise utilizando-se
CL-EM/EM ................................................................................................................ 51
Tabela 6. Preparo de Soluções Padrão de calibração para Análise utilizando-se CL-
EM/EM ...................................................................................................................... 52
Tabela 7. Parâmetros do CL-EM/EM otimizados após a infusão ............................. 57
Tabela 8. Valores obtidos pela curva de linearidade Erro! Indicador não definido.63
Tabela 9. Valores de Análise de Desvio Padrão e Coeficiente de Variação da Curva
de Calibração ............................................................................................................ 64
Tabela 10. Valores de Análise de Variância Obtidos da Curva de Calibração ..... Erro!
Indicador não definido.65
Tabela 11. Resultados de Recuperação para a metodologia desenvolvida aplicada à
matriz de Pêssego, utilizando-se CL-EM/EM ............................................................ 70
Tabela 12. Valores de recuperação obtidos em 2 dias distintos para avaliação de
exatidão e precisão ...................................................... Erro! Indicador não definido.
Tabela 13. Parâmetros avaliados na robustez ......................................................... 73
Tabela 14. Valores de recuperação no parâmetro de robustez quando não há
variações ao método................................................................................................. 73
Tabela 15. Valores de recuperação no parâmetro de robustez quando há variação
de temperatura do forno em relação método ............................................................ 73
Tabela 16. Valores de recuperação no parâmetro de robustez quando há variação
de proporção da fase móvel em relação método ...................................................... 74
Tabela 17. Valores de Análise de Variância Obtidos da Robustez .......................... 74
Tabela 18. Valores de recuperação para injeção no CLAE-UV ................................ 77

15
1.Objetivo
1.1 Objetivo Geral
Validar Método Analítico por CLAE-EM/EM para determinação de Resíduos
de Etilenotiouréia.
1.2 Objetivos Específicos
Comparar Resultados Obtidos por análise com CLAE-EM/EM com os
resultados pré-determinados por CLAE-UV.
Validar Método em Diferentes Grupos de Matrizes Representativas
Analisar Vantagens da Análise por CLAE-EM/EM

16
2. Justificativa
Com a tendência mundial dos estudos de análise de resíduos em
agroquímicos de se obter uma diminuição dos limites de quantificação para os
métodos analíticos, a fim de se determinar limites máximos de resíduos (LMR) cada
vez menores, técnicas analíticas consideradas mais avançadas em termos de
detecção e separação, como a cromatografia líquida acoplada à espectrometria de
massas em série devem ser empregadas, em função da sua alta seletividade,
sensibilidade e elevado nível de confiabilidade dos resultados (Ion Ratio - razão de
confirmação atráves de detecção do segundo par de massas).

17
3. Revisão da Literatura
3.1 Agrotóxicos
O crescimento contínuo da população mundial tem causado a diminuição das
áreas apropriadas para o cultivo agrícola e, por outro lado, com este crescimento,
observa-se um crescente aumento na demanda por alimentos. Este aumento da
demanda por alimentos tem impulsionado o desenvolvimento de sistemas agrícolas
cada vez mais eficientes. Desta maneira, o uso de agrotóxicos usado para o controle
de pragas durante o cultivo e após a colheita tornou-se um instrumento de essencial
necessidade para garantir o crescimento e a qualidade da produção agrícola.
O Brasil por possuir uma área rural extensa, unido com um clima diversificado
e favorável para o cultivo de diferentes culturas, é um dos maiores produtores e
exportadores de produtos agrícolas de todo o mundo, e por este motivo existe um
controle e cuidado muito grande com o uso de agrotóxicos.
No Brasil, a Agência Nacional de Vigilância Sanitária (ANVISA), do Ministério
da Saúde, é o órgão responsável por estabelecer os limites máximos de resíduos
(LMR) para diversas culturas de alimentos que são cultivados e comercializados no
Brasil.
Também tem como funções verificar se agrotóxicos não registrados estão
sendo utilizados ilegalmente, investigar se os registrados estão sendo utilizados de
forma inadequada e, obter dados mais confiáveis para a estimativa da ingestão
diária de resíduos de agrotóxicos pela população, uma informação fundamental ao
realizar o registro de novos agrotóxicos e/ou renovar o registro dos já existentes
(BASTOS, 2007).

18
3.2 Definição e Classificação dos Agrotóxicos
Segundo o Decreto Federal Brasileiro nº 4.074, de 4 de janeiro de 2002,
entendem-se por agrotóxicos: “produtos e agentes de processos físicos, químicos ou
biológicos, destinados ao uso nos setores de produção, no armazenamento e no
beneficiamento de produtos agrícolas, nas pastagens, na proteção de florestas,
nativas ou plantadas e de outros ecossistemas e de ambientes urbanos, hídricos e
industriais, cuja finalidade seja de alterar a composição da flora ou da fauna, a fim
de preservá-las da ação danosa dos seres vivos considerados nocivos, bem como
substâncias e produtos empregados como desfolhantes,dessecantes, estimuladores
e inibidores do crescimento das plantas” (JARDIM, 2009).
Os agrotóxicos são definidos como substâncias que agem direta ou
indiretamente em um organismo vivo, podendo matá-lo ou controlá-lo de alguma
maneira, por exemplo, interferindo em seu processo reprodutivo. (JARDIM, 2009).
Sendo assim, o termo “agrotóxico” utilizado neste trabalho, refere-se às diferentes
categorias de uso, como inseticidas, pesticidas, fungicidas, herbicidas, dentre as
outras existentes.
Com base na grande quantidade de agrotóxicos utilizados na agricultura e, os
resíduos encontrados nos alimentos, é possível observar uma crescente tendência
na realização de trabalhos científicos envolvendo desenvolvimento, otimização e
validação de métodos analíticos.
3.3 Ditiocarbamatos
Os ditiocarbamatos pertencem a um grupo de agrotóxicos organossulfurados
empregados na agricultura com ação fungicida.
Existem sob a forma de um pó branco ou amarelo-claro, de baixa toxicidade
aguda, baixa volatilidade e solubilidade em solventes orgânicos. O grupo pode ser
dividido em DTC cujos representantes são o ferbam, metam, tiram e ziram, e os
EBDC representados pelo maneb, mancozeb, metiram, propinebe e zineb
(SEIZIOGA, 2003).

19
No Brasil existem registrados seis tipos de substâncias da classe dos
ditiocarbamatos como ingredientes ativos, para quarenta e um diferentes tipos de
cultura (BASTOS, 2007). A Figura 1 apresenta as estruturas químicas dos principais
Ditiocarbamatos de uso autorizado no Brasil.
Os ditiocarbamatos são usados contra vários fitopatologias, com uso
expressivo na agricultura brasileira, nas mais diversas culturas, como hortaliças,
frutas e leguminosas. Estes fungicidas são indicados para o uso contra doenças de
folhagem, no tratamento de sementes e bulbos, contra patógenos de solo e de
madeira, apresentando também uma ação de repelência contra insetos. (SOUSA,
2009).
Fonte – (SOUSA, 2009)
Figura 1. Estrutura dos principais ditiocarbamatos de uso autorizado no Brasil
A relevância toxicológica dessas substâncias deve-se a seus dois maiores
produtos de degradação, etileno tiouréia (ETU) e propilenotiouréia (PTU), suspeitos
de serem bocigênicos, carcinogênicos e mutagênicos em ratos. No Brasil, o uso de
ditiocarbamatos em culturas é intenso, conforme relatado pelo programa gerenciado

20
pela ANVISA, o Programa Nacional de Análise de Resíduos de Agrotóxicos em
Alimentos (PARA) (BASTOS, 2007).
3.4 Etilenobisditiocarbamatos (EBDC)
Os EBDC são sais orgânicos de manganês, zinco ou zinco e sódio, sendo
insolúveis em água e solventes orgânicos.
São produzidos pela reação de dissulfeto de carbono (CS2) com uma amina
em condições alcalinas, podendo ocorrer precipitação com sal de metal pesado de
natureza polimérica e definição incompleta, especialmente na presença de alguns
íons metálicos. São fungicidas usados no mundo todo. Possuem amplo espectro de
ação no controle de moléstias que atacam diversos cultivos, tais como cereais,
frutas e legumes. Na América Latina, mais de 50% de todos os fungicidas utilizados
são do grupo dos EBDC e, no Brasil, esta classe de fungicida supera os 40%,
representando o principal produto contra fungos (LEMES, 2007).
3.5 Degradação dos EBDC
Etilenobisditiocarbamatos são instáveis em meio alcalino ou ácido, na
presença de oxigênio e em sistemas biológicos, e decompõem-se rapidamente em
água. Em meio alcalino ou ácido, decompõem-se em dissulfeto de carbono ou
sulfeto de hidrogênio. A liberação do dissulfeto de carbono depende do meio onde
ocorrerá a hidrólise. Produtos secundários são formados pela degradação oxidativa
dos EBDC (WHO, 1988).
A degradação dos EBDC pode ocorrer durante a manufatura ou o
armazenamento do produto formulado, na cultura após o tratamento e,
principalmente, durante o processamento do alimento (LEMES, 2007)
Microorganismos rapidamente formam ETU a partir do etileno bistioutam
dissulfeto (DIDT), um produto de decomposição espontânea dos EBDC. Essa
conversão ocorre após adição de compostos de redução como cisteína, glutationa

21
ou ácido ascórbico. Consiste na redução da ligação S-S do DIDT, com a
subseqüente ligação com o dissulfeto de carbono para formar ETU (WHO, 1988)
A decomposição metabólica dos EBDC é complexa e ocorre formação de
vários sub-produtos, sendo um deles o ETU, composto de interesse para este
trabalho. A Figura 2 ilustra a decomposição dos EBDC.
Fonte: (WHO, 1988)
Figura 2. Degradação dos EBDC e formação do ETU
3.6 Etilenotiouréia (ETU)
Etilenotiouréia ou 2-imidazoledinona (ETU) é um dos principais produtos da
degradação dos fungicidas ditiocarbamatos (EBDC). Possui fórmula molecular:
C3H6N2S e, massa molecular: 102,16. É solúvel em água, etanol e em metanol.

22
Na água é relativamente estável a hidrólise, mas pode ser facilmente
fotolizado na presença de fotossintetizadores. No solo, o ETU é química e
biologicamente degradado a etilenouréia (EU), com meia-vida de 1-7 dias, sob
condições de campo. Em condições aeróbicas, o ETU e a EU podem ser convertidos
a CO2. O ETU é bastante móvel no solo úmido em geral devido à sua adsorção no
solo fraco e alta solubilidade. O campo de dissipação de meia-vida do ETU é inferior
a uma semana devida à rápida degradação microbiana. Se liberado para a
atmosfera, ETU pode ser facilmente removida pela chuça ou por meio de reações
com radicais hidroxilas. A meia-vida de ETU no ar é de 8-9 dias (Xu, 2000).
3.7 Formação de ETU durante fabricação de EBDC e armazenamento.
ETU pode ser formado durante a elaboração do EBDC, geralmente a ETU
presente nas culturas, logo após a aplicação, são aquelas já presentes durante a
formulação do EBDC, porém pequenas quantidades também podem ser formadas
durante aplicação da calda para aplicação e também devido à degradação do
fungicida nas superfícies da cultura tratada (LEMES,2007).
3.8 Formação de ETU durante preparo dos alimentos
Os resíduos de EBDC nos alimentos podem ser facilmente convertidos em
ETU durante o armazenamento destes alimentos e quando inclui uma etapa de
aquecimento (cozimento ou transformação industrial) (IARC, 2000).
3.9 Cromatografia
A cromatografia é um método físico-químico de separação. Ela está
fundamentada na migração diferencial dos componentes de uma mistura, que ocorre
devido a diferentes interações entre duas fases imiscíveis, a fase móvel e a fase
estacionaria (DEGANI, 1998).

23
A cromatografia liquida de alta eficiência (CLAE) é um importante membro de
toda uma família de técnicas de separação, uma vez que consegue separar misturas
que contêm um grande numero de compostos similares.
A CLAE utiliza instrumentos que podem ser totalmente automatizados. É um
tipo de cromatografia líquida que emprega colunas recheadas com materiais
especialmente preparados e uma fase móvel, eluída sob altas pressões. Ela tem a
capacidade de realizar separações e análises quantitativas de uma grande
variedade de compostos presentes em diversos tipos de amostras, em escala de
tempo de poucos minutos, com alta resolução, eficiência e detectabilidade
(COLLINS, 2006).
Nas ultimas décadas, ocorreu o desenvolvimento de vários detectores
espectrofotômetros que operam em comprimento de onda variável e houve um
aumento na utilização de detectores eletroquímicos, por fluorescência e por
fluorescência induzida por laser, bem como o acoplamento com o espectrômetro de
massas (equipamento utilizado neste trabalho). Com eles, tornou-se possível a
detecção de uma faixa mais ampla de compostos e a análise de compostos em
baixas concentrações presentes em amostras complexas (COLLINS, 2006).
3.10 Cromatografia na análise de resíduos de agrotóxicos
Na determinação de resíduos de agrotóxicos em alimentos, geralmente são
empregados métodos cromatográficos de análise, como a cromatografia gasosa
(CG) e a cromatografia líquida de alta eficiência (CLAE). Também têm sido
desenvolvidas técnicas de análise de agrotóxicos em alimentos cada vez mais
rápidas, seletivas e sensíveis devido à necessidade de se detectar simultaneamente
um grande número de compostos em baixos 2 níveis de concentração (da ordem de
μg L-1 ou μg kg-1) (CHIARADIA, 2009).
Entretanto, nos últimos anos, pode-se observar uma tendência para o uso de
agrotóxicos mais polares, os quais apresentam menor persistência e toxicidade que
os apolares. Os compostos polares iônicos são menos adequados para análise
usando CG e isto implica no uso de técnicas alternativas, (JARDIM, 2009) dentre
elas, pode-se destacar a cromatografia líquida de ultra eficiência (CLUE) e os

24
métodos cromatográficos com detecção por espectrometria de massas (CL-EM)
(DEMOLINER, 2008).
3.11 Cromatografia líquida acoplada à espectrometria de massas
A cromatografia líquida acoplada à espectrometria de massas (CL-EM) é uma
das técnicas mais poderosas para a análise de resíduos de agrotóxicos polares
iônicos, de baixa volatilidade ou instabilidade térmica. A CL é muito efetiva na
separação dos analitos, enquanto a EM permite a sua identificação e/ou confirmação
em nível de traços. Instrumentos modernos de CL-EM empregando ionização à
pressão atmosférica provêm excelentes seletividade e detectabilidade, que habilitam
análises dos compostos-alvo em níveis de traços (JARDIM, 2009).
O acoplamento CL em série com o espectrômetro de massas (CL-EM/EM)
tem se tornando muito importante para análise de resíduos de agrotóxicos nos
alimentos, permitindo a análise de agrotóxicos em níveis de ultra-traços (da ordem
de ng kg-1), mesmo na presença de interferentes. A fragmentação controlada da EM
é uma ferramenta essencial para a identificação confiável do analito de interesse
com maior seletividade; além disso, essa fragmentação gera sinais mais limpos
melhorando a razão sinal/ruído e diminuindo, portanto, os limites de detecção e
quantificação. Desta forma a análises com uso de CL-EM/EM consiste no que há
mais moderno e avançado em análises de resíduos de agrotóxicos.
Quando se utiliza a cromatografia líquida acoplada à espectrometria de
massas (CL-EM), são encontradas incompatibilidades relacionadas à vazão do
eluente do sistema cromatográfico com relação à velocidade de bombeamento do
sistema de vácuo e o projeto da fonte de íons do espectrômetro de massas. As
vazões utilizadas em CLAE são relativamente grandes (da ordem de 1,0 mL min-1),
de maneira que não é possível bombear o eluente de um cromatógrafo a líquido
diretamente para o interior da fonte do espectrômetro, que opera a pressões de
cerca de 1,3 x 10-4 Pa. Assim, uma das mais importantes funções de uma interface
empregada em CL-EM é remover toda ou, pelo menos, uma parte significativa da
fase móvel (FM) (CHIARADIA, 2008).

25
Atualmente, a abordagem mais popular usa uma técnica de ionização à
pressão atmosférica de baixa vazão (HOLLER, 2009). A Figura 3 mostra um
diagrama de blocos de um sistema típico de cromatografia líquida acoplado com
espectrometria de massas, sendo constituído por um sistema de injeção da amostra,
fonte de ionização, analisador/separador de massas, detector e sistema de
aquisição de amostra.
Fonte: (DEMOLINER, 2008)
Figura 3. Componentes Básicos de um espectrômetro de massas
Em CL-EM, a amostra é inicialmente separada e depois detectada, o
espectrômetro de massas faz isso produzindo partículas carregadas (íons) a partir
dos analitos da amostra, e usando campos elétricos e/ou magnéticos para separar
partículas carregadas de acordo com sua relação massa/carga (m/z) (HARRIS,
2001).
Na tentativa de minimizar os problemas encontrados no interfaceamento do
sistema CL com EM foram desenvolvidas varias interfaces, nas quais, muitas vezes,
também é realizada a ionização do analito por métodos que permitem a obtenção de
íons a partir de moléculas sensíveis à temperatura e/ou pouco voláteis. Por este
motivo muitos autores referem-se a algumas dessas interfaces simplesmente como
fonte de ionização (DEMOLINER, 2008).

26
As fontes de ionização mais comuns são a ionização por eletronebulização
(“electrospray ionization”) – ESI e a ionização química à pressão atmosférica
(“atmospheric pressure chemical ionization”) - APCI. A combinação da CLAE com a
espectrometria de massas proporciona alta seletividade, uma vez que picos não
resolvidos podem ser isolados monitorando-se somente um valor de massa
selecionado (HOLLER, 2009) além de permitir análises de substâncias não voláteis,
o que não é possível na cromatografia gasosa sem a etapa de derivatização
(COLLINS, 2006).
Na ionização por eletronebulização, o liquido no qual o analito de interesse se
encontra dissolvido (na FM, no caso do eluente da CLAE) passa através de um
capilar, a pressão atmosférica, mantido sob alta voltagem. Na saída do capilar são
formadas pequenas gotas altamente carregadas (“spray”) que são dessolvatadas ao
se deslocarem em sentido contrário ao posicionamento de um eletrodo em uma
região de pressão atmosférica. A dessolvatação é assistida por um fluxo continuo de
gás seco (geralmente N2) na região do “spray”. À medida que ocorre a
dessolvatação, o tamanho das gotas é reduzido até o ponto em que a força de
repulsão entre as cargas similares fica maior que as forças de coesão da fase
líquida (tensão superficial). Neste momento, ocorre a chamada “explosão
coulômbica” que gera gotas com tamanhos equivalentes a 10% do tamanho das
gotas a partir das quais se originaram. Uma série de explosões passa então a
ocorrer até que são produzidos os íons do analito a partir destas gotas, os quais são
transferidos para o interior do espectrômetro de massas por uma série de
dispositivos de focalização (CHIARADIA, 2009).
Graças ao modo de obtenção dos íons nesta fonte de ionização, ela pode ser
utilizada na análise de compostos sensíveis a temperatura e com elevadas massas
molares. Em ESI, compostos sensíveis à temperatura podem ser ionizados sem
sofrer degradação, já que a ionização ocorre diretamente na solução. Já os
compostos com massas molares relativamente grandes podem ser analisados
utilizando-se ESI, porque esta fonte de ionização é capaz de gerar íons com
múltiplas cargas. Assim, como o espectrômetro de massas mede a razão
massa/carga (m/z) dos íons, o intervalo de “massa” de aplicabilidade do instrumento
pode ser expandido por um fator equivalente ao numero de cargas do íon, isto é, um

27
íon com m/z 1000 e com 20 cargas representa um composto com uma massa molar
de 20000 Da (CHIARADIA, 2009).
Como a ESI é dependente da concentração do analito no eluente da coluna
cromatográfica, o uso de divisores de fluxo, para diminuir a vazão do eluente para o
interior da interface, não afeta, de forma notável, sua detectabilidade. Há a
necessidade de uso de divisores de fluxo somente quando a vazão utilizada no
cromatógrafo ultrapassar 1 mL min-1 ou 0,5 mL min-1 no caso da FM com elevada
porcentagem de água em sua composição (CHIARADIA, 2009).
O modo de ionização que ocorre na ESI é denominado ionização suave,
porque a energia empregada nesta fonte de ionização não é suficiente para gerar
uma fragmentação significativa das moléculas do analito, de maneira que são
formados íons pseudo-moleculares intactos, do tipo [M+H]+ no modo positivo ou [M-
H]- no modo negativo. O modo de operação positivo ou negativo é estabelecido, na
ESI, pelos modificadores adicionados à FM. No modo de ionização positivo é
adicionado à FM um ácido orgânico, geralmente ácido fórmico. Já no modo negativo,
adiciona-se à FM uma base orgânica, geralmente trietilamina (CHIARADIA, 2009).
Na EM/EM, ao invés de utilizar apenas um analisador de massas para
separar os íons de mesma razão m/z gerados na fonte de ionização, como na EM,
utiliza dois estágios de espectrometria de massas (EM1 e EM2), em que um deles é
usado para isolar o íon de interesse e o outro é usado para estabelecer uma relação
entre íon de interesse isolado e o outro é usado para estabelecer uma relação entre
este íon de interesse isolado e outros íons que foram gerados a partir da
decomposição induzida (CHIARADIA, 2009). O analisador de massas opera sob
vácuo, para assegurar que os íons se desloquem com eficiência máxima. Existem
vários tipos de analisadores de massas, tais como:
Sistemas Quadrupolo (QqQ): Os analisadores de massa quadrupolo usam
quatro eletrodos em forma de bastão paralelos organizados em um quadrado para
gerar campos elétricos que filtram os íons com base em sua relação m/s enquanto
se deslocam pelos eletrodos. Em determinadas magnitudes e freqüências, apenas
íons com a massa selecionada atingem o detector. Alterando os campos elétricos,

28
as massas de todos os íons podem ser varridas seqüencialmente, de baixa para alta
ou vice-versa, gerando um espectro de massas (SKOOG & LEARY, 1992).
Analisadores de tempo de vôo (Time-of-Flight – TOF): Os analisadores
TOF baseiam-se no principio de que, como os íons são gerados na mesma fonte de
ionização do espectrômetro de massas, elas possuem a mesma energia cinética, de
maneira que as sua velocidades serão apenas diferenciadas pelas suas massas
(velocidade é inversamente proporcional à raiz quadrada da massa do íon). Por isso,
neste analisador de massas, os íons produzidos na fonte de ionização do
espectrômetro são acelerados através de um tubo de vôo para serem identificados,
uma vez que o tempo que levam para atravessá-lo esta relacionado com a razão
m/z de cada íon (DEMOLINER, 2008).
Sistemas Ion-trap: O íon-trap é um quadrupolo tridimensional que “captura”
todos os íons que são introduzidos em seu interior e os mantêm “aprisionados” até
que uma determinada radiofreqüência seja aplicada e torna os íons de certa razão
m/z instáveis, de forma que são liberados do trap (ARDREY, 2003).
Triplo Quadrupolo: Em um espectrômetro de massas do tipo triplo
quadrupolo é formado pela junção de três quadrupolos em seqüência. No primeiro
um íon selecionado é separado da corrente de íons vinda da fonte de íons. No
segundo quadrupolo este íon sofre nova fragmentação por colisão com íons de N2
ou Ar. O terceiro quadrupolo seleciona então um dos fragmentos iônicos formados
para enviar ao detector (HARRIS, 2001).
Sistemas de EM em tandem (EM-EM): A espectrometria de massas
seqüencial (Tandem, EM/EM, é a técnica espectrométrica que, ao invés de utilizar
apenas um analisador de massas para separar os íons de mesma razão m/z
gerados na fonte de ionização, utiliza dois ou mais estágios de analise de massa
(Q1 e Q2). O compartimento Q1 funciona como um filtro de massas usado para
estabelecer uma relação entre o íon precursor isolado e outros íons que foram
gerados a partir da sua decomposição induzida, que é o processo de fragmentação
ocorrido na célula de colisão, com auxilio de um gás inerte, argônio ou hélio
(ARDREY, 2003).

29
Na EM/EM podem ser utilizadas varias técnicas de varredura para realizar a
análise, tais como a varredura dos íons produto (“product-ion scan”), varredura dos
íons precursores (“precursor-ion scan”), varredura da constante perda de íons
neutros (“constant-neutral-loss scan”) e o monitoramento seletivo de reação
(selected-reaction monitoring”). Quando se faz o acoplamento da cromatografia com
a EM obtém-se o chamado cromatograma de massas que é assim denominado por
se tratar de um cromatograma constituído de todos os íons produzidos pelo
espectrômetro de massas ou apenas pelos íons de interesse produzidos por este. O
cromatograma contendo todos os íons produzidos pelo espectrômetro de massas é
denominado cromatograma de íons totais (“total íon chromatogram”) - TIC. Já o
cromatograma constituído apenas pelos íons de interesse pode ser obtido pelo
monitoramento de íons selecionados (“selected íon monitoring”) – SIM, ajustando-se
o detector de massas para que sejam observados os íons de razão m/z de interesse
ou selecionando-os a partir de um banco de dados que contenha os espectros de
massas completos (CHIARADIA, 2009).
3.12 Métodos de análise de resíduos de agrotóxicos em alimentos
Os métodos de análise de resíduos de agrotóxicos em alimentos
compreendem, basicamente, três etapas: A primeira etapa consiste na amostragem,
que deve ser representativa de um todo. A segunda é o preparo da amostra, isto é, a
extração do analito da matriz, remoção de interferentes (“clean-up”) e concentração;
a terceira etapa trata-se da separação e determinação dos analitos (CHIARADIA,
2009).
3.13 Validação
O desenvolvimento de um método analítico, a adaptação ou implementação
de método conhecido, envolve processo de avaliação que estime sua eficiência na
rotina do laboratório. Esse processo costuma ser denominado de validação (BRITO,
2003).

30
O guia para validação de métodos analíticos e bioanalíticos, proposto pela
resolução nº 899/2003 da ANVISA, determina que a validação deve garantir, por
meio de estudos experimentais, que o método atenda às exigências das aplicações
analíticas, assegurando a confiabilidade dos resultados (ANVISA,2003).
Vários autores definem validação de métodos e pode-se dizer que os
conceitos continuam evoluindo e estão constantemente sob consideração pelas
agências reguladoras. É possível encontrar várias outras definições para validação
descritas na literatura:
A validação é a “confirmação por testes e apresentação de evidências
objetivas de que determinados requisitos são preenchidos para um dado uso
internacional” (ISO/IEC 17025, 1999).
O objetivo da validação consiste em demonstrar que o método analítico é
adequado para o seu propósito (WALSH, 1999).
A validação de métodos assegura a credibilidade destes durante o uso
rotineiro, sendo algumas vezes mencionado como o processo que fornece uma
evidência documentada de que o método realiza aquilo para o qual é indicado para
fazer. (USP U.S. PHARMACOPEIA).
Avaliação sistemática de um procedimento analítico para demonstrar que está
sob as condições nas quais deve ser aplicado (WHO, 1988).
3.14 Processo de validação
É essencial que os estudos de validação sejam representativos e conduzidos
de modo que a variação da faixa de concentração e os tipos de amostras sejam
adequados. Um método para um composto majoritário requer um critério de
aceitação e uma abordagem diferente de um método desenvolvido para análise de
traços. A freqüência que o método será utilizado (muitas vezes em um dia, uma vez
em um dia para um estudo rápido, uma vez em um mês, etc.) também influencia o

31
tipo de estudo de validação que é necessário. Os parâmetros analíticos devem ser
baseados na intenção do uso do método (RIBANI, 2004).
3.15 Parâmetros de Validação
Os parâmetros de validação que serão utilizados neste método são referentes
ao requisitado pela ANVISA, através da RDC nº 216, sendo analisados os
parâmetros de: seletividade, linearidade, faixa linear, precisão, exatidão, limite de
detecção, limite de quantificação e robustez.
3.16 Validação Curta
Em casos onde um método analítico validado por outro laboratório for ser
utilizado, a metodologia será considerada apropriada para uso, desde que seja
realizada uma validação curta. Na validação curta devem ser avaliados os
parâmetros mínimos de linearidade, precisão e recuperação.
3.17 Precisão
Precisão é um termo geral para avaliar a dispersão de resultados entre
ensaios independentes, repetidos de uma mesma amostra, amostras semelhantes
ou padrões em condições definidas. É normalmente determinada para
circunstâncias especificas de medição e as duas formas mais comuns de expressá-
la são por meio da repetitividade e da precisão intermediária. O desvio padrão
relativo (DPR), também conhecido como coeficiente de variação (CV), será o calculo
estatístico para expressar os resultados.
3.18 Repetitividade
A repetitividade representa a concordância entre os resultados de medições
sucessivas de um mesmo método, efetuadas sob as mesmas condições de
medição, chamadas condições de repetitividade: mesmo procedimento; mesmo
analista; mesmo instrumento usado sob as mesmas condições; mesmo local;
repetições em um curto intervalo de tempo.

32
3.19 Precisão Intermediária
Indica o efeito das variações dentro do laboratório devido a eventos como
diferentes dias ou diferentes analistas ou diferentes equipamentos ou uma
combinação destes fatores. A precisão intermediária é reconhecida como a mais
representativa da variabilidade dos resultados em um único laboratório e, como tal,
mais aconselhável de ser adotada.
O objetivo da validação da precisão intermediária é verificar que no mesmo
laboratório o método fornecerá os mesmos resultados.
3.20 Seletividade
A seletividade define a capacidade do método em detectar o analito de
interesse na presença de outros componentes da matriz.
Um método seletivo é um método capaz de responder a vários analitos, mas
que pode distinguir a resposta de um analito das respostas dos outros analitos.
A seletividade de um método analítico pode ser avaliada de duas formas:
(a) Comparando-se a matriz isenta dos agrotóxicos com a matriz adicionada
com os padrões dos agrotóxicos para verificar a existência de coeluição
destes compostos junto a interferentes da matriz;
(b) Utilizando-se detectores modernos (arranjos de diodos e espectrômetros
de massas) que permitem comparar o espectro do pico obtido na amostra
com o espectro do padrão puro e, no caso da espectrometria de massas
em série, é possível selecionar as transições MRM entre os íons dos
analitos e seus íons fragmentos específicos (RIBANI, 2004).
3.21 Recuperação
A recuperação (R) é definida como razão entre os valores de concentração
obtidos e os teóricos, e geralmente é expressa em porcentagem.
O intervalo aceitável de recuperação para a análise de resíduos de
agrotóxicos é de 70 a 120%, com precisão de 20%.

33
A recuperação pode ser calculada utilizando-se a equação abaixo.
R (%) = (C1 / C2) x 100
Onde,
C1 é a concentração determinada na amostra fortificada
C2 é a concentração adicionada à amostra
3.22 Linearidade
A linearidade corresponde à capacidade do método em fornecer resultados
diretamente proporcionais à concentração da substância em exame, dentro de uma
determinada faixa de aplicação.
A faixa linear de um método é definida como o intervalo entre os níveis inferior
e superior de concentração do analito no qual foi demonstrado ser possível a
determinação com precisão, exatidão e linearidade exigidos sob as condições
especificas para o ensaio.
Recomenda-se que para a validação a linearidade seja determinada com a
utilização de, no mínimo, cinco concentrações diferentes. Estas concentrações
devem possuir recuperação entre 70%-130%, segundo a RDC nº216.
Havendo relação linear, determina-se o coeficiente de correlação (r), sendo
que um valor maior ou igual a 0,99 é requerido.
3.23 Limite de Detecção (LOD)
O limite de detecção (LOD) representa a menor concentração da substância
em exame que pode ser detectada, mas não necessariamente quantificada,
utilizando um determinado procedimento experimental.
O LOD pode ser calculado de três maneiras diferentes: método visual, método
relação sinal-ruído, método baseado em parâmetros da curva analítica.

34
(a) Visual: O LOD é determinado por meio da adição de concentrações
conhecidas do analito à matriz, de tal modo que se possa determinar o
menor nível em que o analito realmente pode ser detectado
(b) Relação Sinal/Ruído: O LOD é determinado através da comparação entre
a medição dos sinais de amostras com baixas concentrações conhecidas
do analito na matriz e um branco dessas amostras. Assim é estabelecida
uma concentração mínima na qual a substancia pode ser facilmente
detectada.
(c) Baseado em Parâmetros da Curva Analítica: O LOD é determinado
através de parâmetros da curva analítica de acordo com a equação a
seguir:
LOD = 3,3 x (s / a)
Onde,
s é a estimativa do desvio padrão da resposta do coeficiente linear da
equação da reta da curva de calibração
a é a inclinação ou coeficiente angular da equação da reta da curva analítica
Neste trabalho, utilizou-se como LOD o valor fixo de 20% do LOQ, visto que
tanto para a amostra testemunha quanto para o branco de reagentes pode-se ter um
valor máximo de 20% do LOQ do método de resíduos utilizado, este valor foi usado
como base para todas as analises.
3.24 Limite de Quantificação (LOQ)
O limite de quantificação (LOQ) representa a menor concentração da
substância em exame que pode ser medida, utilizando um determinado
procedimento experimental.
Como o LOD, o LOQ é expresso como uma concentração, sendo que a
precisão e exatidão das determinações também devem ser registradas. Esse critério
é uma boa regra a ser seguida, porém não se deve esquecer que a determinação do
LOQ representa um compromisso entre a concentração, a precisão e a exatidão

35
exigidas. Isto significa que, quando decresce o nível de concentração do LOQ, a
medição torna-se menos precisa. Se houver necessidade de maior precisão, uma
concentração maior deve ser registrada para o LOQ. O método analítico e seu
respectivo uso ditam esse compromisso (RIBANI, 2004).
3.25 Robustez
Robustez de um método analítico é a medida de sua capacidade em resistir a
pequenas variações. Diz-se que um método é robusto quando ele não é afetado por
uma modificação pequena e deliberada em seus parâmetros. Indica sua confiança
durante o uso normal. A Tabela 1 relaciona alguns dos principais parâmetros que
podem resultar em variações na resposta do método.
Tabela 1. Parâmetros que podem causar variações nas respostas do método
Preparo das Amostras Estabilidade das soluções analíticas
Tempo de retenção
Espectrometria
Variação do pH da solução
Temperatura
Diferentes fabricantes de solventes
Cromatografia Liquida
Variação do pH da fase móvel
Variação da composição da fase móvel
Diferentes lotes ou fabricantes de colunas
Temperatura
Fluxo da fase móvel
Fonte: (MORAES, 2010)
3.26 Estabilidade
Para gerar resultados confiáveis e reprodutíveis, as amostras, os padrões e
reagentes usados devem ser estáveis por um período razoável (por ex. um dia, uma
semana, um mês, dependendo da necessidade).

36
4. Validação do Método de Análise
4.1 Números de Amostras para os Ensaios de Validação do Método
Serão utilizados amostras de matriz representativa, analisadas com 5
repetições e em 2 níveis de fortificação, além de que também será injetada uma
amostra testemunha.
a) Amostra Testemunha
A amostra testemunha, utilizada na validação do método analítico,
corresponde à matriz de mesma espécie, a qual se sabe que não possui o
agrotóxico a qual será analisado.
b) Amostra Fortificada
A amostra fortificada, utilizada na validação do método analítico, corresponde
a amostra testemunha, na qual foi adicionada determinada concentração da solução
de fortificação.
4.2 Seletividade
Para a avaliação deste parâmetro será preparado 4 amostras:
Branco: No branco adicionam-se todos os reagentes e soluções
utilizados na rota analítica sem a presença da amostra e/ou analito de
interesse, para verificar possíveis interferências do método.
Branco Fortificado: No branco fortificado adicionam-se todos os
reagentes e soluções utilizados na rota analítica e uma concentração
conhecida do analito.
Amostra Testemunha: Amostra controle da matriz, a qual serve para
comprovar a existência ou não de algum interferente na matriz em análise.

37
Quality Control (QC): É uma amostra testemunha, na qual no final da
rota adiciona-se uma concentração conhecida do analito.
4.3 Linearidade
A determinação da linearidade é efetuada por meio da análise do analito de
interesse em diferentes concentrações. É feita uma curva de calibração construída
com no mínimo 3 repetições e, em 5 concentrações diferentes.
4.4 Recuperação
A RDC 216 (ANVISA 2006) considera valores de variabilidade para
determinação da aceitabilidade de métodos, recuperações entre 70% e 120%.
4.5 Precisão
A precisão é determinada por meio do coeficiente de variação percentual em
relação à média, sendo este valor de 20% conforme estabelecido pela ANVISA
(2006).
Para a analise da precisão intermediaria e repetitividade realiza-se no mínimo
5 repetições do ensaio.
4.6 Robustez
A robustez é determinada através de algumas pequenas modificações, como
mudança de coluna, equipamento para quantificação, composição da fase móvel.
Para isto, identificar-se-á uma ou mais variáveis no procedimento analítico
que possam ter efeito significativo no desempenho do método. Executar-se-á
analises na amostra para monitorar o efeito da mudança. Realizar o ensaio com no
mínimo 5 repetições para cada variável estabelecida.

38
5. Materiais e Métodos
Nos próximos itens, estão descritos todos os materiais que serão utilizados
para a realização deste trabalho, tais como equipamentos, solventes, soluções,
padrões vidrarias e softwares, os quais serão imprescindíveis para a realização do
mesmo.
5.1 Materiais de referência
Tabela 2. Etilenotiouréia (ETU) (utilizada para propósito de calibração e fortificação)
Código BASF Reg. No. Fórmula empírica
Pureza (certificado)
BF 222-ETU 146099 C3H6N2S 99,9%
Nome químico (IUPAC)
imidazolidine-2-thione
Fórmula estrutural
Homogeneidade Peso molecular Pressão de vapor
Obtida 102,2 g/mol Não disponível
Validade Origem Lote Certificado de análise
01/07/2012 BASF SE 01815-177 ASAP11_002
Estabilidade Não disponível.
Solubilidade Não disponível.
5.2 Análise de Etilenotiouréia (ETU) por CLAE-UV
5.2.1 Equipamentos, reagentes e solventes, soluções e diversos, materiais e vidraria.
Equipamentos
Cromatógrafo Líquido Agilent Technologies, 1100 Series
Detector de Ultra Violeta (UV/Vis) Agilent Technologies, Modelo G1314A

39
Coluna para cromatografia líquida Lichrosorb RP-18, (250 x 4)mm, 10m
Balança Semi-Analítica Mettler Toledo, PG 5002-S; Sartorius,
LE6202S
Balança Analítica Mettler Toledo, XS205DU
Sistema de Purificação de Água Millipore, Milli-Q Gradient
Homogeneizador -
Banho de Ultra-som -
Centrifuga -
Dispensete Brand, Dispensette III
Pipetador Automático Gilson, M1000, Eppendorf, Research
variável (1-10 mL)
Mesa agitadora -
Conjunto Rotavapor-Banho -
Reagentes e Solventes
Fabricante Referência Lote
Metanol J T. Baker 9093 K06C15
Água Milli-Q Millipore QTUM000EX F1DA42701
Acetonitrila J T. Baker 9012-03 K08C75
Diclorometano Mallinckrodt H485-10 J32J00
Hidróxido de Sódio
(NaOH)
QHEMIS QHH023 Q0036
Cloreto de Sódio (NaCl) J T. Baker 3624-19 E08C55
Óxido de alumínio Merck 815020 664512510
Soluções e diversos
NaOH 1 mol/L (Solução 1) 8g de NaOH para 200 mL com Água
Fase Móvel 0,2 % de Acetonitrila em Água
Óxido de Alumino atividade Água/Alumina 15% p/p: Ativar o óxido de alumínio por

40
5 (Alumina Ativada) 12 horas a cerca de 140 °C em erlenmeyer com
tampa e resfriar no dessecador. Adicionar 15% de
água e agitar em mesa agitadora por no mínimo 1
hora.
Materiais
Fabricante Referência Lote
Filtro Sartorius 17821 1106002VS
Extrelut NT3 Merck 1150950001 HC111124
Outros Materiais
Seringa Plástica 3 mL
Espátula -
Vidraria
Balão Volumétrico 5 mL
Balão Volumétrico Calibrado 25, 50, 100 mL
Balão de fundo chato 125 mL
Béquer 10, 50, 100, 250 mL
Erlenmeyer 250 mL
Proveta 50, 100, 250, 1000 mL
Recipiente para Extração 250 mL
Vias cromatográficas 3 mL
Vidro âmbar Diversos volumes.
5.2.2 Preparo Típico de Soluções Padrão de Calibração e de Fortificação
Solução Padrão Estoque (SPE)
Número do Lote: 01815-177
Pureza: 99,9 %
Peso Líquido: 0,01000 g

41
Volume Final: 10 mL
Concentração: 1,00 x 106 ng/mL
Solvente: Metanol
Código: SPE-1
Soluções Padrão de Fortificação (STDF)
Pipetou-se 2,5 mL da “Solução Padrão Estoque”, com concentração igual a
1,00 x 106 ng/mL, para um balão volumétrico de 100 mL, completando-se o volume
do balão com metanol, obtendo-se desta forma a Solução Padrão de Fortificação
STDF “F”, com concentração de 2,50 x 104 ng/mL. Com esta solução, preparou-se a
Solução Padrão de Fortificação STDF “A”, conforme descrito na tabela 3.
Tabela 3. Preparo de Soluções Padrão de Fortificação para Análise utilizando-se
CLAE-UV
Nome da
Solução
Concentração
de ETU na
Solução
(ng/mL)
Alíquota
Tomada
(mL)
Volume
Final
(mL)
Solvente
de
Diluição
Solução
Padrão de
Fortificação
Concentraçã
o de ETU na
Solução
(ng/mL)
Peso da
Amostra
(g)
Fortificaçã
o
(mg/kg)
SPE
ETU 1,00 x 106 2,5 100
Metano
l
STDF “F” 2,50 x 104 5,0 5,0
STDF
“F” 2,50 x 104 1,0 100 STDF “A” 250 5,0 0,050
SPE
ETU 1,00 x 106 0,250 100 STDF “G” 2500 5,0 0,50
A alíquota tomada da STDF para a fortificação de amostras testemunhas é de
1 mL.
O cálculo do nível de fortificação (mg/kg) é dado pela razão entre a
concentração da respectiva STDF e o peso da amostra, multiplicada pela alíquota
tomada da respectiva STDF para fortificação de amostras testemunhas, dividida por
1000.
Os níveis de fortificação de 0,050 mg/kg, 0,50 mg/kg e 5,0 mg/kg foram
42
usados para determinar as recuperações das amostras testemunhas fortificadas
com ETU nos grupos de análise.
Soluções Padrão de Calibração (SPC)
Pipetou-se 1 mL da Solução Padrão de Fortificação STDF “F”, com
concentração igual a 2,50 x 104 ng/mL, para um balão volumétrico de 100 mL,
completando-se o volume do balão com Fase Móvel, obtendo-se desta forma a
“Solução Padrão de Calibração E” (SPC E) com concentração de 250 ng/mL.
As Soluções Padrão de Calibração (SPC) foram preparadas com a solução
STDF “F” e STDF “G”, avolumadas com Fase Móvel, conforme descrito na tabela 4.
Tabela 4. Preparo de Soluções Padrão de Calibração para Análise utilizando-se
CLAE-UV
Nome da
Solução
Concentração
de ETU na
Solução
(ng/mL)
Alíquota
Tomada
(mL)
Volume
Final
(mL)
Concentração de ETU
na solução (ng/mL)
Solução de
Calibração
STDF “F” 2,50 x 104 1,00 100 250 SPC “E”
STDF “F” 2,50 x 104 0,25 50 125 SPC “D”
STDF “G” 2500 0,5 25 50 SPC “C”
STDF “G” 2500 0,5 50 25 SPC “B”
STDF “G” 2500 0,25 50 12,5 SPC “A”
SPC “E” 250 0,5 25 5,0 SPC “F”
As Soluções Padrão de Calibração foram usadas para checar a linearidade da
resposta do detector do sistema usado, na faixa de 5,0 ng/mL a 250 ng/mL para
ETU, durante a injeção dos grupos de análise.

43
5.2.3 Preparação da Amostra
A amostra congelada foi homogeneizada, misturando-se bem todas as partes
representativas. Triturou-se a amostra com gelo seco, retirou-se sub-amostras, sem
utilização de luvas de borracha, rotulou-se e estocou-se na Câmara Fria
(Temperatura operacional de -20°C ou inferior).
5.2.4 Procedimento Analítico
A amostra analisada foi retirada do local de estoque apenas no início da
marcha analítica e retornada ao local originário após a etapa de pesagem.
Fortificação (quando aplicável)
Pesou-se (5 0,1) g da amostra testemunha, a ser fortificada, em um
recipiente de extração. Adicionou-se, com pipeta volumétrica, a solução padrão de
fortificação apropriada para o nível de fortificação desejado.
Método de Análise (todas as amostras)
Adicionou-se 10 mL de água à massa de (5 ± 0,1) g de amostra e acertou-se
o pH utilizando-se papel indicador universal, entre 8 a 10 com NaOH 1 mol/L. As
amostras permaneceram em repouso por 5 minutos e, após isso, adicionou-se 0,5g
de cloreto de sódio seguido de 90 mL de metanol. Após agitação no
homogeneizador por 5 minutos sob gelo, centrifugou-se o extrato por 10 minutos a
4500 rpm. Retirou-se uma alíquota de 20 mL em uma vidraria para concentração de
amostras previamente pesada e evaporou-se o resíduo aquoso em turbo-vap até
restar cerca de 2 mL. Acertou-se o peso para 2 g (quando necessário) e ajustou-se o
pH para 8-9. Preparou-se uma coluna cromatográfica de vidro contendo 4 g de
alumina ativada e 3 g de Extrelut. Transferiu-se o extrato para o topo da coluna
cromatográfica e deixou-se a amostra percolar por cerca de 10 minutos.
Eluiu-se o resíduo da coluna com 50 mL de diclorometano a uma vazão de 1-
2 gotas/segundo em vidraria para concentração de amostras previamente pesada.
Adicionou 2 mL de fase móvel e evaporou-se no conjunto rotavapor-banho até

44
restar somente o resíduo aquoso. Ajustou-se o peso para 2g com Fase Móvel
(quando necessário). Filtrou-se as amostras em unidade filtrante para injeção
Cromatográfica (Detector UV/VIS).
Observação: As amostras foram mantidas em banho de gelo até o término da
filtração.
5.2.5 Análise Instrumental
Condições Cromatográficas Cromatografia Líquida
Detector Ultra Violeta (UV/VIS)
Comprimento de Onda 240 nm
Coluna Lichosorb RP-18;(250x4)mm, 10m
Volume de Injeção 100 L
Fase Móvel 0,2% de Acetonitrila em Água
Gradiente / Fluxo
Tempo
(min)
Fluxo
(L/min)
Fase Móvel
(%)
0,00 1000 100
12,00 1000 100
Tempo de Retenção aproximado: 4 minutos.
Tempo de Corrida: 12 minutos.
5.2.6 Preparo de Soluções
- Preparo da Solução de NaOH 1 mol/L
Pesou-se 8 gramas de Hidróxido de Sódio dissolveu-o e avolumou-o em 200
mL de água.

45
- Preparo da Fase Móvel
Mediu-se 2 mL de Acetonitrila HPLC e avoluma-se com água até 1000 mL.
- Preparo de Alumina Atividade 5
Ativou-se o Óxido de Aluminio por 12 horas a uma temperatura de
aproximadamente 140 ºC. Resfriar no dessecador até atingir a temperatura
ambiente. Pesa a alumina em um erlenmeyer com tampa. Adicionou 15% de água e
agitar vigorosamente na mesa agitadora, no mínimo 1 hora.
- Preparo da Coluna de Extretut NT3
Removeu o filtro superior e o recheio da coluna Extrelut NT3 (3g). Adicionou 4
gramas de alumina atividade 5. Adicionar o recheio previamente removido e o filtro
superior.
5.2.7 Análise Quantitativa
A determinação foi feita e quantificada pela comparação com a curva analítica
obtida pelos padrões externos (substância de referência).
5.3 Análise de Etilenotiouréia (ETU) por CLAE-EM/EM
5.3.1 Equipamentos, reagentes e solventes, soluções e diversos, materiais e vidraria.
Equipamentos
Cromatógrafo Líquido Agilent Technologies (HP), 1100 Series
Detector de massas/massas (EM/EM) Applied Biosystems, API 4000
Coluna para cromatografia líquida Eclipse XDB C8, (150 x 4,6)mm, 5m
Balança Semi-Analítica Mettler Toledo, PG 5002-S; Sartorius,
LE6202S

46
Balança Analítica Mettler Toledo, XS205DU
Sistema de Purificação de Água Millipore, Milli-Q Gradient
Homogeneizador -
Banho de Ultra-som -
Centrifuga -
Dispensete Brand, Dispensette III
Pipetador Automático Gilson, M1000, Eppendorf, Research
variável (1-10 mL)
Mesa agitadora -
Conjunto Rotavapor-Banho -
Reagentes e Solventes
Fabricante Referência Lote
Metanol J T. Baker 9093 K06C15
Água Milli-Q Millipore QTUM000EX F1DA42701
Formiato de Amônio J T. Baker - -
Diclorometano Mallinckrodt H485-10 J32J00
Hidróxido de Sódio
(NaOH)
QHEMIS QHH023 Q0036
Cloreto de Amônio J T. Baker 0660-01 J49C22
Etilenouréia
(2-imidazolidona)
ALDRICH I601 STBB6541V
Ascorbato de Sódio ALDRICH A7631 079K0355
Ácido Ascórbico Mallinckrodt M4407-02 G02H00
Soluções e diversos
Óxido de Alumino atividade 5
(Alumina Ativada)
Água/Alumina 15% p/p: Ativar o óxido de alumínio por
12 horas a cerca de 140 °C em erlenmeyer com
tampa e resfriar no dessecador. Adicionar 15% de

47
água e agitar em mesa agitadora por no mínimo 1
hora.
15 g/L etilenouréia em metanol
1,5 g de etilenouréia em 100 mL de metanol
1.0% cloreto de amônio 1 g de cloreto de amônio em 100 mL de água
Solução de Ascorbato de sódio
99,2 g de ascorbato de sódio e 2 g de ácido
ascorbico, dissolvidos em 1 L de água
Metanol/ Água, (50/ 50, v/v) Metanol/ Água, (10/ 90, v/v)
50 mL de metanol e 50 mL de água
10 mL de metanol e 90 mL de água
Solução de extração 100 mL de solução 1.0% cloreto de amônio + 100 mL
de 15g/L de etilenouréia em metanol + 250 mL de
solução ascorbato de sódio + 550 mL de
metanol/Água , (50/50, v/v)
Fase Móvel A
0,02 mol/L de formiato de amônio em água
Fase Móvel B NaOH 1 mol/L
Metanol
8 gramas de NaOH em 200 mL de água
Materiais
Fabricante Referência Lote
Filtro Sartorius 17821 1106002VS
Extrelut NT3
Merck 1150950001 HC111124
Outros Materiais
Seringa Plástica 3 mL
Espátula
-
Vidraria

48
Balão Volumétrico 5 mL
Balão Volumétrico Calibrado 25, 50, 100 mL
Balão de fundo chato 125 mL
Béquer 10, 50, 100, 250 mL
Erlenmeyer 250 mL
Proveta 50, 100, 250, 1000 mL
Recipiente para Extração 250 mL
Vias cromatográficas 3 mL
Vidro âmbar Diversos volumes.
5.3.2 Preparo Típico de Soluções Padrão de Calibração e de Fortificação
Solução Padrão Estoque (SPE)
Número do Lote: 01815-177
Pureza: 99,9 %
Peso Líquido: 0,01000 g
Volume Final: 10 mL
Concentração: 1,00 x 106 ng/mL
Solvente: Metanol
Código: SPE-1
Soluções Padrão de Fortificação (STDF)
Pipetou-se 1,25 mL da “Solução Padrão Estoque”, com concentração igual a
1,00 x 106 ng/mL, para um balão volumétrico de 100 mL, completando-se o volume
do balão com metanol, obtendo-se desta forma a Solução Padrão de Fortificação
STDF “F”, com concentração de 1,25 x 104 ng/mL. Com esta solução, preparou-se a
Solução Padrão de Fortificação STDF “A”, conforme descrito na tabela 5.
Tabela 5. Preparo de Soluções Padrão de Fortificação para Análise utilizando-se

49
CL-EM/EM
Nome da
Solução
Concentração
de ETU na
Solução
(ng/mL)
Alíquota
Tomada
(mL)
Volume
Final
(mL)
Solvente
de
Diluição
Solução
Padrão de
Fortificação
Concentração
de ETU na
Solução
(ng/mL)
Peso
da
Amost
ra
(g)
Fortificação
(mg/kg)
SPE
ETU 1,00 x 106 1,25 100
Metano
l
STDF “F” 1,25 x 104 12,5 1,0
STDF
“F” 1,25 x 104 1,0 100 STDF “A” 125 12,5 0,01
SPE
ETU 1,00 x 106 1,25 200 STDF “G” 6250 12,5 0,50
A alíquota tomada da STDF para a fortificação de amostras testemunhas é de
1 mL.
O cálculo do nível de fortificação (mg/kg) é dado pela razão entre a
concentração da respectiva STDF e o peso da amostra, multiplicada pela alíquota
tomada da respectiva STDF para fortificação de amostras testemunhas, dividida por
1000.
Os níveis de fortificação de 0,01 mg/kg, 0,50 mg/kg e 1,0 mg/kg foram usados
para determinar as recuperações das amostras testemunhas fortificadas com ETU
nos grupos de análise.
Soluções Padrão de Calibração (SPC)
Pipetou-se 0,40 mL da Solução Padrão de Fortificação STDF “F”, com
concentração igual a 1,25 x 104 ng/mL, para um balão volumétrico de 100 mL,
completando-se o volume do balão com Fase Móvel, obtendo-se desta forma a
“Solução Padrão de Calibração E” (SPC E) com concentração de 50 ng/mL.
As Soluções Padrão de Calibração (SPC) foram preparadas com a solução
STDF “F” e STDF “A”, avolumadas com Fase Móvel, conforme descrito na tabela 6.

50
Tabela 6. Preparo de Soluções Padrão de calibração para Análise utilizando-se CL-
EM/EM
Nome da
Solução
Concentração
de ETU na
Solução
(ng/mL)
Alíquota
Tomada
(mL)
Volume
Final
(mL)
Concentração de
ETU na solução
(ng/mL)
Solução de
Calibração
STDF “F” 1,25 x 104 0,40 100 50 SPC “E”
STDF “F” 1,25 x 104 0,20 100 25 SPC “D”
STDF “F” 1,25 x 104 0,10 100 12,5 SPC “C”
STDF “A” 125 1,0 20 6,25 SPC “B”
STDF “A” 125 0,5 25 2,5 SPC “A”
SPC “E” 50 1,25 50 1,25 SPC “F”
As Soluções Padrão de Calibração foram usadas para checar a linearidade da
resposta do detector do sistema usado, na faixa de 1,25 ng/mL a 50 ng/mL para
ETU, durante a injeção dos grupos de análise.
5.3.3 Preparação da Amostra
A amostra congelada foi homogeneizada, misturando-se bem todas as partes
representativas. Triturou-se a amostra com gelo seco, retirou-se sub-amostras, sem
utilização de luvas de borracha, rotulou-se e estocou-se na Câmara Fria
(Temperatura operacional de -20°C ou inferior).
5.3.4 Procedimento Analítico
A amostra analisada foi retirada do local de estoque apenas no início da
marcha analítica e retornada ao local originário após a etapa de pesagem.
Fortificação (quando aplicável)

51
Pesou-se (12,5 0,1) g da amostra testemunha, a ser fortificada, em um
recipiente de extração. Adicionou-se, com pipeta volumétrica, a solução padrão de
fortificação apropriada para o nível de fortificação desejado.
Método de Análise (todas as amostras)
Adicionou-se 50 mL de solução de extração à massa de (12,5 ± 0,1) g de
amostra e acertou-se o pH utilizando-se papel indicador universal, entre 8 a 10 com
NaOH 1 mol/L. Após agitação no homogeneizador por 2 minutos a 10000 rpm,
centrifugou-se o extrato por 10 minutos a 4500 rpm. Retirou-se uma alíquota de 20
mL em uma vidraria para concentração de amostras previamente pesada e
evaporou-se o resíduo aquoso em conjunto rotavapor até restar cerca de 2 mL.
Acertou-se o peso para 2 g (quando necessário) e ajustou-se o pH para 8-9.
Preparou-se uma coluna cromatográfica de vidro contendo 2 g de alumina ativada e
3 g de Extrelut. Transferiu-se o extrato para o topo da coluna cromatográfica e
deixou-se a amostra percolar por cerca de 20 minutos.
Eluiu-se o resíduo da coluna com 70 mL de diclorometano a uma vazão de 1-
2 gotas/segundo em vidraria para concentração de amostras previamente pesada.
Adicionou 2 mL de solução metanol/água (10/90, v/v) e evaporou-se no
conjunto rotavapor-banho até restar somente o resíduo aquoso. Ajustou-se o peso
para 2g com solução metanol/água (10/90, v/v) (quando necessário). Filtrou-se as
amostras em unidade filtrante para injeção Cromatográfica (CLAE-EM/EM).
5.3.5 Análise Instrumental
Condições Cromatográficas Cromatografia Líquida
Detector CLAE-EM/EM
Temperatura de Forno 20.0 ºC
Coluna Eclipse XDB C8, (150 x 4,6)mm, 5m
Volume de Injeção 50 L
Tipo de Scan MRM (Multiple Reaction Monitoring)
Tipo de Ionização Positiva

52
Fase Móvel A 0,02 mol/L de formiato de amônio em
Água
Fase Móvel B Metanol
Gradiente / Fluxo
Tempo
(min)
Fluxo
(L/min)
Fase Móvel A
(%)
Fase Móvel B
(%)
0,00 1000 85 15
5,00 1000 85 15
Tempo de Retenção aproximado: 3,7 minutos.
Tempo de Corrida: 5 minutos.
5.3.6 Preparo de Soluções
- Preparo da Solução de NaOH 1 mol/L
Pesou-se 8 gramas de Hidróxido de Sódio e dissolveu-o e avolumou-o em
200 mL de água.
- Preparo da Solução de 15 g/L de etilenouréia em metanol
Pesou-se 15 gramas de etilenouréia e dissolveu-o e avolumou-o em 1000 mL
de metanol.
- Preparo da Solução de 1,0 % de cloreto de Amônio
Pesou-se 1 grama de cloreto de amônio e dissolveu-o e avolumou-o em 100
mL de água.
- Preparo da Solução de Ascorbato de Sódio
Pesou-se 99,2 gramas de ascorbato de sódio e dissolveu-o em um pouco de
água, então se adicionou a ele 2 g de ácido ascórbico e avolumou-o em 1 L de água.

53
- Preparo da Solução Metanol/Água, (50/50, v/v)
Misturou-se 50 mL de metanol juntamente com 50 mL de água.
- Preparo da Solução Metanol/Água, (10/90, v/v)
Misturou-se 10 mL de metanol juntamente com 90 mL de água.
- Preparo da Solução de Extração
Misturou-se 100 mL da solução de 1,0% de cloreto de amônio juntamente
com 100 mL solução 15 g/L de etilenouréia em metanol, 250 mL de solução de
ascorbato de sódio e 550 mL de solução metanol/Água (50/50, v/v), levando a
mistura ao banho de ultra-som para total homogeneização.
- Preparo da Fase Móvel A
Pesou-se 1,26 gramas de formiato de amônio e dissolve-o em 1 L de água.
- Preparo de Alumina Atividade 5
Ativou-se o Óxido de Aluminio por 12 horas a uma temperatura de
aproximadamente 140 ºC. Resfriar no dessecador até atingir a temperatura
ambiente. Pesou a alumina em um erlenmeyer com tampa. Adicionar 15% de água e
agitar vigorosamente na mesa agitadora, no mínimo 1 hora.
- Preparo da Coluna de Extretut NT3
Removeu o filtro superior e o recheio da coluna Extrelut NT3 (3g). Adicionou 4
gramas de alumina atividade 5. Adicionou o recheio previamente removido e o filtro
superior.

54
5.3.7 Análise Quantitativa
A determinação foi feita e quantificada pela comparação com a curva analítica
obtida pelos padrões externos (substância de referência).

55
6. Resultados e Discussões
Na CLAE-EM/EM, devido à seletividade do sistema de detecção é possível
determinar a concentração do etilenotiouréia sem grandes interferências de matriz.
O modo de varredura utilizado para este trabalho foi o MRM, sendo inicialmente
otimizados os parâmetros da fonte de ionização para que se pudesse obter o íon
molecular e também do íon produto mais abundante através da infusão via seringa,
diretamente no espectrômetro de massas, de solução 50 ng/mL de padrão
etilenotiouréia dissolvidos em uma solução metanol/0,02 mol/L de formiato de
amônio em água (50/50, v/v). A vazão de bombeamento desta solução através da
seringa para o interior do espectrômetro foi feito com auxilio de uma bomba injetora
com uma vazão constante de 10 L min-1.
A adição de formiato de amônio à solução utilizada para a infusão, tal como a
utilizada na fase móvel, faz com que a ionização deste padrão de etilenotiouréia se
desse no modo positivo, isto quer dizer, sendo formados íons [M + H]+.
As transições MRM selecionadas para o monitoramento do etilenotiouréia
foram aquelas entre o íon precursor [M + H]+ e o seu respectivo íon produto de
maiores intensidades, as quais são apresentadas na tabela 7 juntamente com os
parâmetros de ionização da fonte otimizados por meio da infusão, como a voltagem
de cone e a energia de colisão, sendo que o íon produto de massa 44,2 apresentou
maior intensidade e por isso será utilizado como transição de quantificação do
método e o íon produto de massa 60,1 apresentou segunda maior intensidade e por
este motivo será utilizada como transição de confirmação para o método de
validação.
Após haver sido otimizado os parâmetros da fonte de ionização, realizou-se o
acoplamento do sistema cromatográfico ao espectrômetro de massas para
otimização de alguns parâmetros de fonte para melhor intensidade na quantificação
da quebra dos dois pares de massas selecionados pela infusão, sendo realizada
esta otimização da fonte e também da melhor escolha do gradiente através do FIA,
garantindo-se desta maneira uma maior detectabilidade do analito analisado e
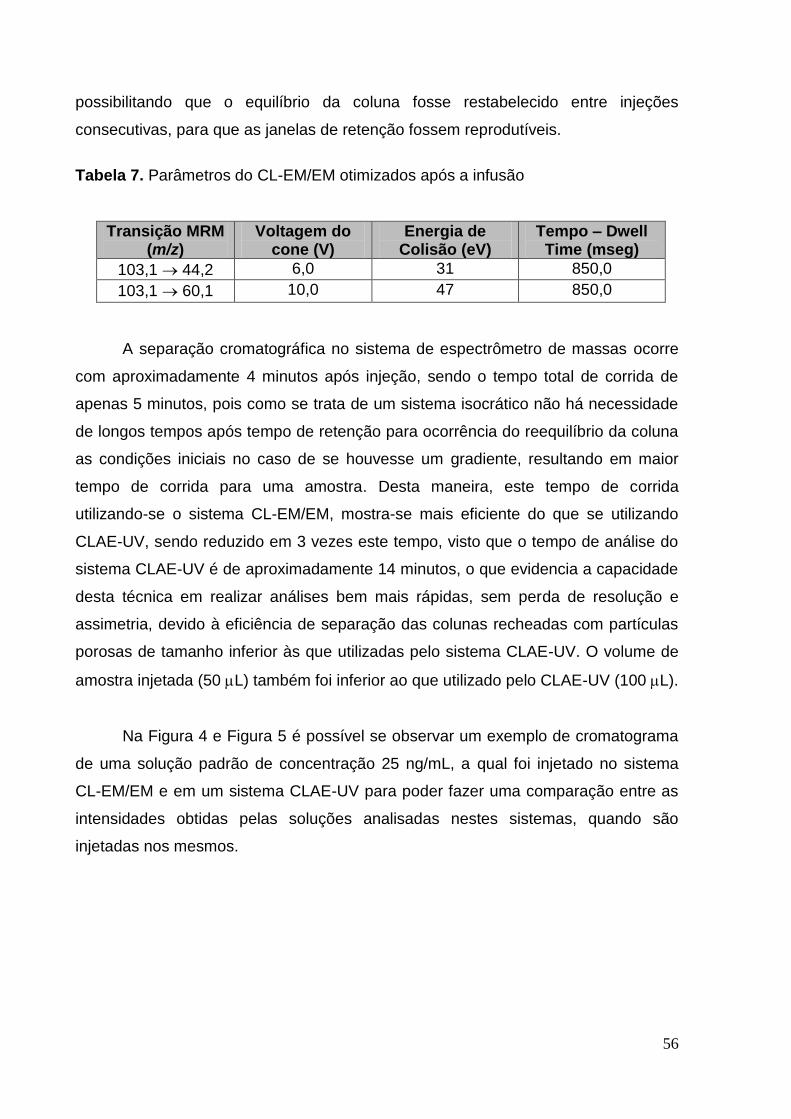
56
possibilitando que o equilíbrio da coluna fosse restabelecido entre injeções
consecutivas, para que as janelas de retenção fossem reprodutíveis.
Tabela 7. Parâmetros do CL-EM/EM otimizados após a infusão
Transição MRM (m/z)
Voltagem do cone (V)
Energia de Colisão (eV)
Tempo – Dwell Time (mseg)
103,1 44,2 6,0 31 850,0
103,1 60,1 10,0 47 850,0
A separação cromatográfica no sistema de espectrômetro de massas ocorre
com aproximadamente 4 minutos após injeção, sendo o tempo total de corrida de
apenas 5 minutos, pois como se trata de um sistema isocrático não há necessidade
de longos tempos após tempo de retenção para ocorrência do reequilíbrio da coluna
as condições iniciais no caso de se houvesse um gradiente, resultando em maior
tempo de corrida para uma amostra. Desta maneira, este tempo de corrida
utilizando-se o sistema CL-EM/EM, mostra-se mais eficiente do que se utilizando
CLAE-UV, sendo reduzido em 3 vezes este tempo, visto que o tempo de análise do
sistema CLAE-UV é de aproximadamente 14 minutos, o que evidencia a capacidade
desta técnica em realizar análises bem mais rápidas, sem perda de resolução e
assimetria, devido à eficiência de separação das colunas recheadas com partículas
porosas de tamanho inferior às que utilizadas pelo sistema CLAE-UV. O volume de
amostra injetada (50 L) também foi inferior ao que utilizado pelo CLAE-UV (100 L).
Na Figura 4 e Figura 5 é possível se observar um exemplo de cromatograma
de uma solução padrão de concentração 25 ng/mL, a qual foi injetado no sistema
CL-EM/EM e em um sistema CLAE-UV para poder fazer uma comparação entre as
intensidades obtidas pelas soluções analisadas nestes sistemas, quando são
injetadas nos mesmos.

57
Sample Name: "SPC D" Sample ID: "Conc.: 25.0 ng/mL" File: "Teste de Linearidade e Repetitibilidade.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "CCL:111 Eclipse XDB-C8" Annotation: ""Sample Index: 54
Sample Type: Standard
Concentration: 25.0 ng/mL
Calculated Conc: 25.9 ng/mL
Acq. Date: 4/1/2012
Acq. Time: 5:44:20 AM
Modified: Yes
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 0.72 cps
Area Threshold: 3.62 cps
Num. Smooths: 3
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.73 min
Use Relative RT: No
Int. Type: Manual
Retention Time: 3.74 min
Area: 102000. counts
Height: 10400. cps
Start Time: 3.45 min
End Time: 4.36 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0.00
500.00
1000.00
1500.00
2000.00
2500.00
3000.00
3500.00
4000.00
4500.00
5000.00
5500.00
6000.00
6500.00
7000.00
7500.00
8000.00
8500.00
9000.00
9500.00
1.00e4
Inte
ns
ity
, c
ps
3.74
Figura 4. Cromatograma para Solução Padrão de Calibração de concentração 25,0 ng/mL injetada no CL-EM/EM
Figura 5. Cromatograma para Solução Padrão de Calibração de concentração 25,0 ng/mL injetada no CLAE-UV Pode-se notar visivelmente que a intensidade obtida pelo CL-EM/EM é
superior em relação ao CLAE-UV, sendo desta forma possível reduzir o LOQ do
método visto que é possível injetar no sistema CL-EM/EM soluções com

58
concentrações de até 1,25 ng/mL a qual não seria possível se quantificar quando
injetado em um sistema CLAE-UV.
6.1 Seletividade
Pela infusão, conforme apresentado nos resultados e discussões, pode-se
selecionar as transições entre o íon precursor e seu respectivo íon produto com
maior intensidade, a fim de poder se realizar a identificação e quantificação do
etilenotiouréia, com base nesta transição. Isso ocorre, pois o sinal gerado pelo
detector massa/massa, ao empregar-se uma varredura MRM, corresponde às
transições selecionadas, que devem ser especificas deste composto. Além disso,
pode-se determinar duas transições de íon produto, sendo o de maior intensidade a
de transição de quantificação e a de segunda maior intensidade a de transição de
confirmação, fazendo desta forma que este método seja altamente seletivo.
Para confirmar a especificidade do método, injetaram-se amostras de branco,
branco fortificado, amostra testemunha e quality control, sendo que para critérios de
validação deste parâmetro não pode haver interferência dos componentes da matriz
e não pode ser detectados picos interferentes acima do limite de detecção com o
mesmo tempo de retenção do ativo de etilenotiouréia nos cromatogramas da
amostra testemunha e do branco.

59
Sample Name: "UT" Sample ID: "Amostra Testemunha" File: "Teste ETU_11abr12.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "" Annotation: ""Sample Index: 4
Sample Type: Blank
Concentration: 0.00 ng/mL
Calculated Conc: N/A
Acq. Date: 4/11/2012
Acq. Time: 10:21:56 AM
Modified: No
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 1.20 cps
Area Threshold: 6.01 cps
Num. Smooths: 2
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.82 min
Use Relative RT: No
Int. Type: Valley
Retention Time: 3.81 min
Area: 48.3 counts
Height: 5.30 cps
Start Time: 3.71 min
End Time: 4.05 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
Inte
ns
ity
, c
ps
0.35 1.16 4.322.56 4.570.57 4.151.84 3.482.25 3.09 4.831.65
Figura 6. Cromatograma para Amostra Testemunha (UT) injetada no CL-EM/EM
Sample Name: "B" Sample ID: "Branco de Reagentes" File: "Teste ETU_11abr12.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "" Annotation: ""Sample Index: 23
Sample Type: Double Blank
Concentration: 0.00 ng/mL
Calculated Conc: N/A
Acq. Date: 4/11/2012
Acq. Time: 1:35:49 PM
Modified: No
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 1.20 cps
Area Threshold: 6.01 cps
Num. Smooths: 2
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.82 min
Use Relative RT: No
Int. Type: Valley
Retention Time: 3.84 min
Area: 44.0 counts
Height: 4.55 cps
Start Time: 3.76 min
End Time: 4.02 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
Inte
ns
ity
, c
ps
1.030.89 4.141.17 1.920.48 3.21 4.342.71 4.642.161.51 3.51 3.840.05
Figura 7. Cromatograma para Branco de Reagentes (B) injetada no CL-EM/EM

60
Sample Name: "BF" Sample ID: "Branco Fortificado" File: "Teste ETU_11abr12.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "" Annotation: ""Sample Index: 17
Sample Type: QC
Concentration: 25.0 ng/mL
Calculated Conc: 30.9 ng/mL
Acq. Date: 4/11/2012
Acq. Time: 11:56:53 AM
Modified: No
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 1.20 cps
Area Threshold: 6.01 cps
Num. Smooths: 2
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.82 min
Use Relative RT: No
Int. Type: Valley
Retention Time: 3.73 min
Area: 72700. counts
Height: 4350. cps
Start Time: 3.28 min
End Time: 4.50 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
3600
3800
4000
4200
Inte
ns
ity
, c
ps
3.73
Figura 8. Cromatograma para Branco Fortificado (BF) injetada no CL-EM/EM
Sample Name: "QC" Sample ID: "Quality Control" File: "Teste ETU_11abr12.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "" Annotation: ""Sample Index: 11
Sample Type: QC
Concentration: 25.0 ng/mL
Calculated Conc: 30.0 ng/mL
Acq. Date: 4/11/2012
Acq. Time: 11:13:02 AM
Modified: No
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 1.20 cps
Area Threshold: 6.01 cps
Num. Smooths: 2
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.82 min
Use Relative RT: No
Int. Type: Valley
Retention Time: 3.72 min
Area: 70700. counts
Height: 4320. cps
Start Time: 3.31 min
End Time: 4.39 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
3600
3800
4000
4200
Inte
ns
ity
, c
ps
3.72
Figura 9. Cromatograma para Quality Control (QC) injetada no CL-EM/EM

61
Não se detectou nenhum pico interferente acima do limite de detecção no
mesmo tempo de retenção do ativo de etilenotiouréia para as soluções de branco e
amostra testemunha, e fazendo-se uma comparação entre os picos do branco
fortificado e do quality control, pode-se observar que não houve interferência dos
componentes da matriz do quality control no ativo, sendo que houve uma
intensidade muito próxima entre eles.
Desta forma, o critério de aceitação para este parâmetro foi considerado
satisfatório e este parâmetro será dado como validado.
6.2 Linearidade, Faixa Linear de Trabalho, Limite de Detecção e Limite de Quantificação
Foi preparado, curva de calibração em 5 níveis de concentração e em pelo
menos 3 replicatas independentes para cada nível, nas concentrações de 1,25
ng/mL, 2,50 ng/mL, 6,25 ng/mL, 25,0 ng/mL e 50,0 ng/mL. Foi-se preparada também
uma curva de mesmas concentrações a partir dos padrões no extrato da matriz, ou
seja, padronização externa com superposição da matriz, que se pode observar na
figura 10. Com base nestas curvas, com e sem matriz, pode-se observar que a
matriz não causa grande influência para uma variação da curva de calibração, e por
este motivo será utilizado para os cálculos de validação de linearidade apenas a
curva sem matriz.
0
50000
100000
150000
200000
250000
0 20 40 60
Área
Concentração
Curva sem Matriz
Curva com Matriz
Figura 10. Curva Sobreposta com Matriz e sem Matriz
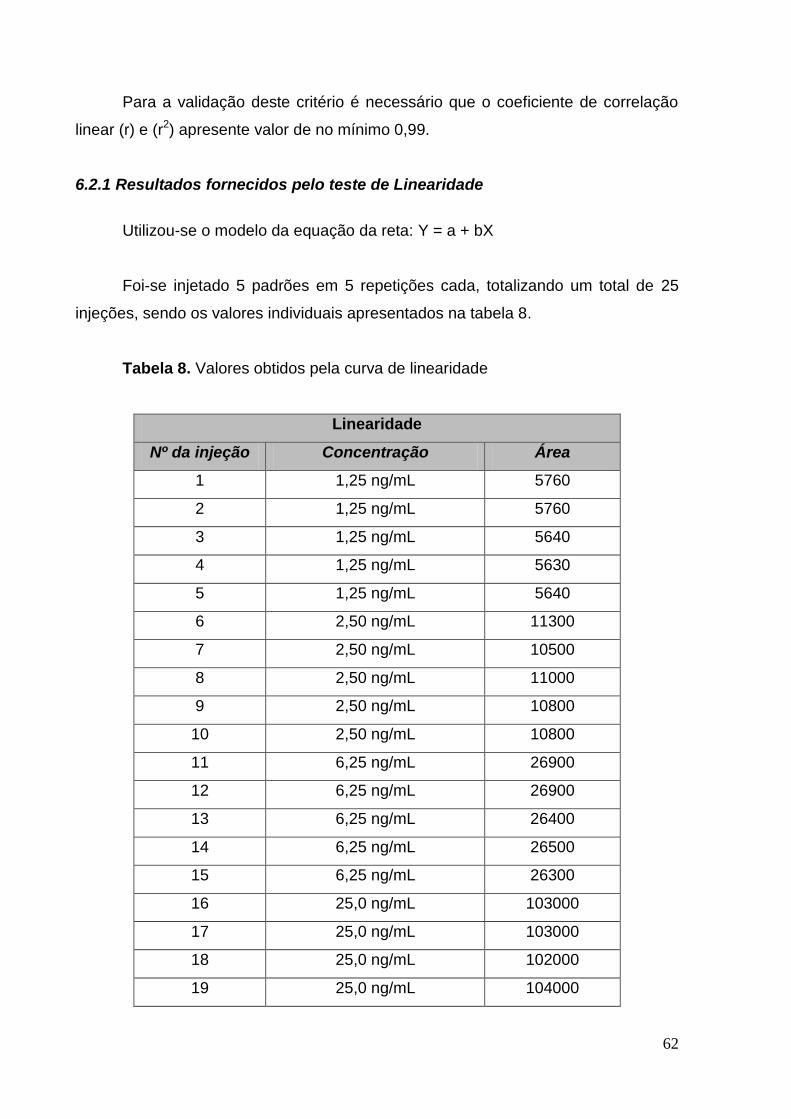
62
Para a validação deste critério é necessário que o coeficiente de correlação
linear (r) e (r2) apresente valor de no mínimo 0,99.
6.2.1 Resultados fornecidos pelo teste de Linearidade
Utilizou-se o modelo da equação da reta: Y = a + bX
Foi-se injetado 5 padrões em 5 repetições cada, totalizando um total de 25
injeções, sendo os valores individuais apresentados na tabela 8.
Tabela 8. Valores obtidos pela curva de linearidade
Linearidade
Nº da injeção Concentração Área
1 1,25 ng/mL 5760
2 1,25 ng/mL 5760
3 1,25 ng/mL 5640
4 1,25 ng/mL 5630
5 1,25 ng/mL 5640
6 2,50 ng/mL 11300
7 2,50 ng/mL 10500
8 2,50 ng/mL 11000
9 2,50 ng/mL 10800
10 2,50 ng/mL 10800
11 6,25 ng/mL 26900
12 6,25 ng/mL 26900
13 6,25 ng/mL 26400
14 6,25 ng/mL 26500
15 6,25 ng/mL 26300
16 25,0 ng/mL 103000
17 25,0 ng/mL 103000
18 25,0 ng/mL 102000
19 25,0 ng/mL 104000

63
20 25,0 ng/mL 102000
21 50,0 ng/mL 196000
22 50,0 ng/mL 189000
23 50,0 ng/mL 191000
24 50,0 ng/mL 192000
25 50,0 ng/mL 192000
Figura 11. Curva de Calibração
Tabela 9. Valores de Análise de Desvio Padrão e Coeficiente de Variação da Curva de Calibração
Concentração Numero de
Repetições
Média Desvio
Padrão
% CV
1,25 ng/mL 5 5686 67,6756 1,19
2,50 ng/mL 5 10880 294,9576 2,71
6,25 ng/mL 5 26600 282,8427 1,06
25,0 ng/mL 5 102800 836,6600 0,81
50,0 ng/mL 5 192000 2549,5098 1,33

64
Tabela 10. Valores de Análise de Variância Obtidos da Curva de Calibração RESUMO DOS RESULTADOS
Estatística de regressão
R múltiplo 0.99932114
R-Quadrado 0.99864273 R-quadrado ajustado 0.99858372
Erro padrão 0.7138126
Observações 25
ANOVA
gl SQ MQ F F de
significação
Regressão 1 8622.655846 8622.655846 16922.81605 1.74695E-34
Resíduo 23 11.71915383 0.509528427
Total 24 8634.375
Coeficientes Erro padrão Stat t valor-P 95%
inferiores 95%
superiores
Interseção -
0.59459565 0.196657519 -
3.023508331 0.006046829 -
1.001412718 -0.187778576
Variável X 1 0.0002603 2.00097E-06 130.0877244 1.74695E-34 0.000256162 0.000264441
SQ: Soma dos Quadrados; gl: Grau de Liberdade; MQ: Soma Média Quadrática;
Com base nos resultados obtidos pode-se observar que o método apresenta
linearidade, e observa-se também um elevado valor de F, que significa que há uma
pequena probabilidade de ocorrer de forma aleatória, e assim pode-se concluir que
está é uma regressão significativa, sendo que desta maneira este parâmetro esta
validado.
Nas figuras 12 até 16 são dados exemplos de cromatogramas de cada uma
das concentrações utilizadas para a curva de calibração.

65
Sample Name: "SPC F" Sample ID: "Conc.: 1.25 ng/mL" File: "Teste de Linearidade e Repetitibilidade.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "CCL:111 Eclipse XDB-C8" Annotation: ""Sample Index: 51
Sample Type: Standard
Concentration: 1.25 ng/mL
Calculated Conc: 0.860 ng/mL
Acq. Date: 4/1/2012
Acq. Time: 5:22:08 AM
Modified: Yes
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 0.72 cps
Area Threshold: 3.62 cps
Num. Smooths: 3
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.73 min
Use Relative RT: No
Int. Type: Manual
Retention Time: 3.74 min
Area: 5640. counts
Height: 557. cps
Start Time: 3.25 min
End Time: 4.50 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
Inte
ns
ity
, c
ps
3.74
Figura 12. Cromatograma para Solução Padrão de Calibração de concentração 1,25 ng/mL
Sample Name: "SPC A" Sample ID: "Conc.: 2.50 ng/mL" File: "Teste de Linearidade e Repetitibilidade.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "CCL:111 Eclipse XDB-C8" Annotation: ""Sample Index: 52
Sample Type: Standard
Concentration: 2.50 ng/mL
Calculated Conc: 2.24 ng/mL
Acq. Date: 4/1/2012
Acq. Time: 5:29:31 AM
Modified: No
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 0.72 cps
Area Threshold: 3.62 cps
Num. Smooths: 3
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.73 min
Use Relative RT: No
Int. Type: Base To Base
Retention Time: 3.74 min
Area: 11000. counts
Height: 1170. cps
Start Time: 3.48 min
End Time: 4.16 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
Inte
ns
ity
, c
ps
3.74
Figura 13. Cromatograma para Solução Padrão de Calibração de concentração 2,50 ng/mL
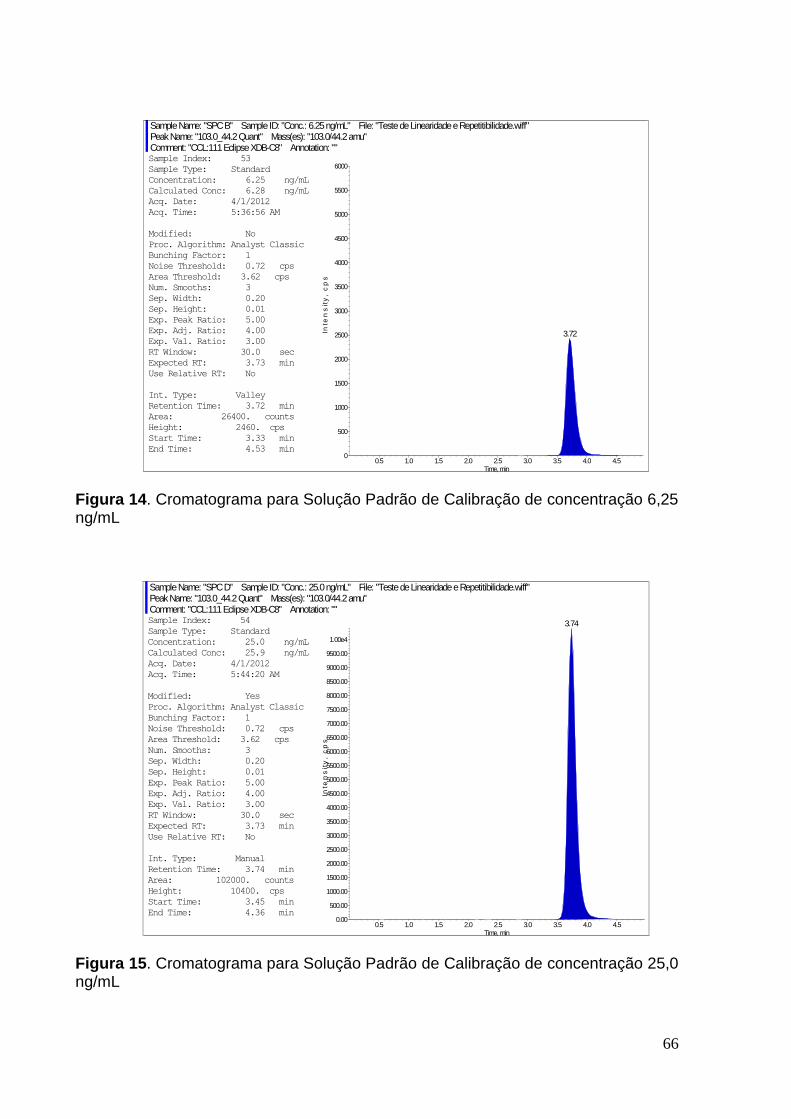
66
Sample Name: "SPC B" Sample ID: "Conc.: 6.25 ng/mL" File: "Teste de Linearidade e Repetitibilidade.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "CCL:111 Eclipse XDB-C8" Annotation: ""Sample Index: 53
Sample Type: Standard
Concentration: 6.25 ng/mL
Calculated Conc: 6.28 ng/mL
Acq. Date: 4/1/2012
Acq. Time: 5:36:56 AM
Modified: No
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 0.72 cps
Area Threshold: 3.62 cps
Num. Smooths: 3
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.73 min
Use Relative RT: No
Int. Type: Valley
Retention Time: 3.72 min
Area: 26400. counts
Height: 2460. cps
Start Time: 3.33 min
End Time: 4.53 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
Inte
ns
ity
, c
ps
3.72
Figura 14. Cromatograma para Solução Padrão de Calibração de concentração 6,25 ng/mL
Sample Name: "SPC D" Sample ID: "Conc.: 25.0 ng/mL" File: "Teste de Linearidade e Repetitibilidade.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "CCL:111 Eclipse XDB-C8" Annotation: ""Sample Index: 54
Sample Type: Standard
Concentration: 25.0 ng/mL
Calculated Conc: 25.9 ng/mL
Acq. Date: 4/1/2012
Acq. Time: 5:44:20 AM
Modified: Yes
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 0.72 cps
Area Threshold: 3.62 cps
Num. Smooths: 3
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.73 min
Use Relative RT: No
Int. Type: Manual
Retention Time: 3.74 min
Area: 102000. counts
Height: 10400. cps
Start Time: 3.45 min
End Time: 4.36 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0.00
500.00
1000.00
1500.00
2000.00
2500.00
3000.00
3500.00
4000.00
4500.00
5000.00
5500.00
6000.00
6500.00
7000.00
7500.00
8000.00
8500.00
9000.00
9500.00
1.00e4
Inte
ns
ity
, c
ps
3.74
Figura 15. Cromatograma para Solução Padrão de Calibração de concentração 25,0 ng/mL

67
Sample Name: "SPC E" Sample ID: "Conc.: 50.0 ng/mL" File: "Teste de Linearidade e Repetitibilidade.wiff"Peak Name: "103.0_44.2 Quant" Mass(es): "103.0/44.2 amu"Comment: "CCL:111 Eclipse XDB-C8" Annotation: ""Sample Index: 55
Sample Type: Standard
Concentration: 50.0 ng/mL
Calculated Conc: 49.1 ng/mL
Acq. Date: 4/1/2012
Acq. Time: 5:51:47 AM
Modified: Yes
Proc. Algorithm: Analyst Classic
Bunching Factor: 1
Noise Threshold: 0.72 cps
Area Threshold: 3.62 cps
Num. Smooths: 3
Sep. Width: 0.20
Sep. Height: 0.01
Exp. Peak Ratio: 5.00
Exp. Adj. Ratio: 4.00
Exp. Val. Ratio: 3.00
RT Window: 30.0 sec
Expected RT: 3.73 min
Use Relative RT: No
Int. Type: Manual
Retention Time: 3.74 min
Area: 191000. counts
Height: 18400. cps
Start Time: 3.42 min
End Time: 4.73 min
0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5Time, min
0.0
1000.0
2000.0
3000.0
4000.0
5000.0
6000.0
7000.0
8000.0
9000.0
1.0e4
1.1e4
1.2e4
1.3e4
1.4e4
1.5e4
1.6e4
1.7e4
1.8e4
Inte
ns
ity
, c
ps
3.74
Figura 16. Cromatograma para Solução Padrão de Calibração de concentração 50,0 ng/mL 6.3 Exatidão e Precisão (Intermediária e Repetitividade)
A exatidão do método desenvolvido foi determinada por meio de ensaios de
recuperação, conforme sugerido pela ANVISA. Os resultados dos ensaios de
recuperações apresentados na tabela 11 encontram-se entre 75 e 89 para a matriz
de Pêssego, sendo que estes valores de recuperação encontram-se dentro do
intervalo sugerido para as amostras complexas. A precisão foi avaliada em termos
de repetitividade e precisão intermediaria, em 2 níveis de fortificação (0,01 mg/kg e
0,50 mg/kg) e em 5 preparos distintos para cada nível de fortificação, sendo que a
precisão intermediaria foi avaliada realizando-se em 2 dias distintos.
Foi-se obtido coeficientes de variação menores do que 7,0 e 5,0 para a
repetitividade e precisão intermediária, respectivamente, na matriz de pêssego,
conforme observado na tabela 12.

68
Tabela 11. Resultados de Recuperação para a metodologia desenvolvida aplicada à
matriz de Pêssego, utilizando-se CL-EM/EM
Dia Nº Concentração (ng/mL)
Concentração Calculada (ng/mL)
Recuperação (%)
1
1 6,25 5,30 85
2 6,25 5,14 82
3 6,25 4,88 78
4 6,25 4,95 79
5 6,25 5,19 83
1 25,0 21,8 87
2 25,0 21,2 85
3 25,0 21,1 84
4 25,0 19,9 80
5 25,0 19,5 78
2
1 6,25 5,41 87
2 6,25 4,87 78
3 6,25 5,53 89
4 6,25 5,08 81
5 6,25 5,52 88
1 25,0 19,4 78
2 25,0 19,8 79
3 25,0 18,7 75
4 25,0 21,5 86
5 25,0 18,8 75
A partir dos valores obtidos de recuperação para a matriz de pêssego obtidos
na tabela 11, pode-se obter os valores de coeficiente de variação, desvio padrão,
incerteza de medição e cálculos de análise de variância disponíveis na tabela 12.
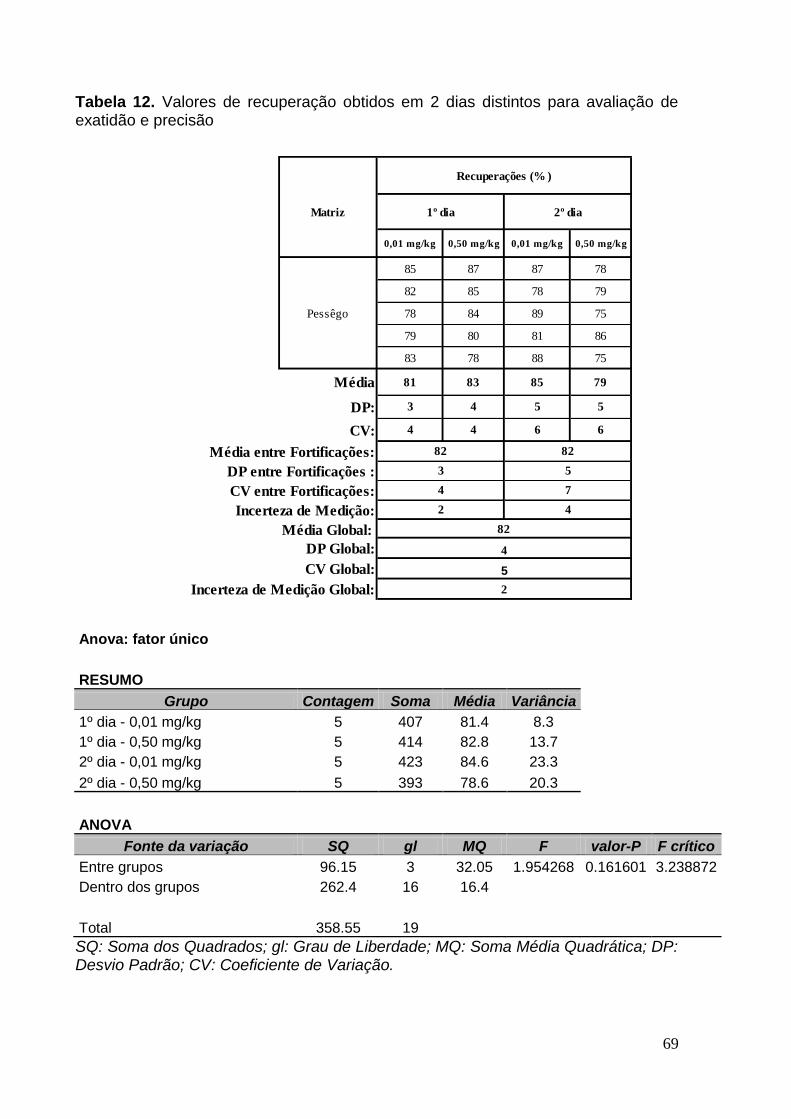
69
Tabela 12. Valores de recuperação obtidos em 2 dias distintos para avaliação de exatidão e precisão
Matriz
Média
DP:
CV:
DP entre Fortificações :
CV entre Fortificações:
Incerteza de Medição:
Média Global:
DP Global:
CV Global:
Incerteza de Medição Global:
Média entre Fortificações:
5
82
4
2
4
82
81 83
80
2
3 4
4
5
82
6
7
3
4
2º dia
0,01 mg/kg
Recuperações (% )
1º dia
0,01 mg/kg 0,50 mg/kg 0,50 mg/kg
78 79
85 87 87 78
81 86
82 85
6
5
7985
5
75
79
Pessêgo
8878
4
78 84 89 75
83
Anova: fator único
RESUMO
Grupo Contagem Soma Média Variância
1º dia - 0,01 mg/kg 5 407 81.4 8.3
1º dia - 0,50 mg/kg 5 414 82.8 13.7
2º dia - 0,01 mg/kg 5 423 84.6 23.3
2º dia - 0,50 mg/kg 5 393 78.6 20.3
ANOVA
Fonte da variação SQ gl MQ F valor-P F crítico
Entre grupos 96.15 3 32.05 1.954268 0.161601 3.238872
Dentro dos grupos 262.4 16 16.4
Total 358.55 19
SQ: Soma dos Quadrados; gl: Grau de Liberdade; MQ: Soma Média Quadrática; DP: Desvio Padrão; CV: Coeficiente de Variação.
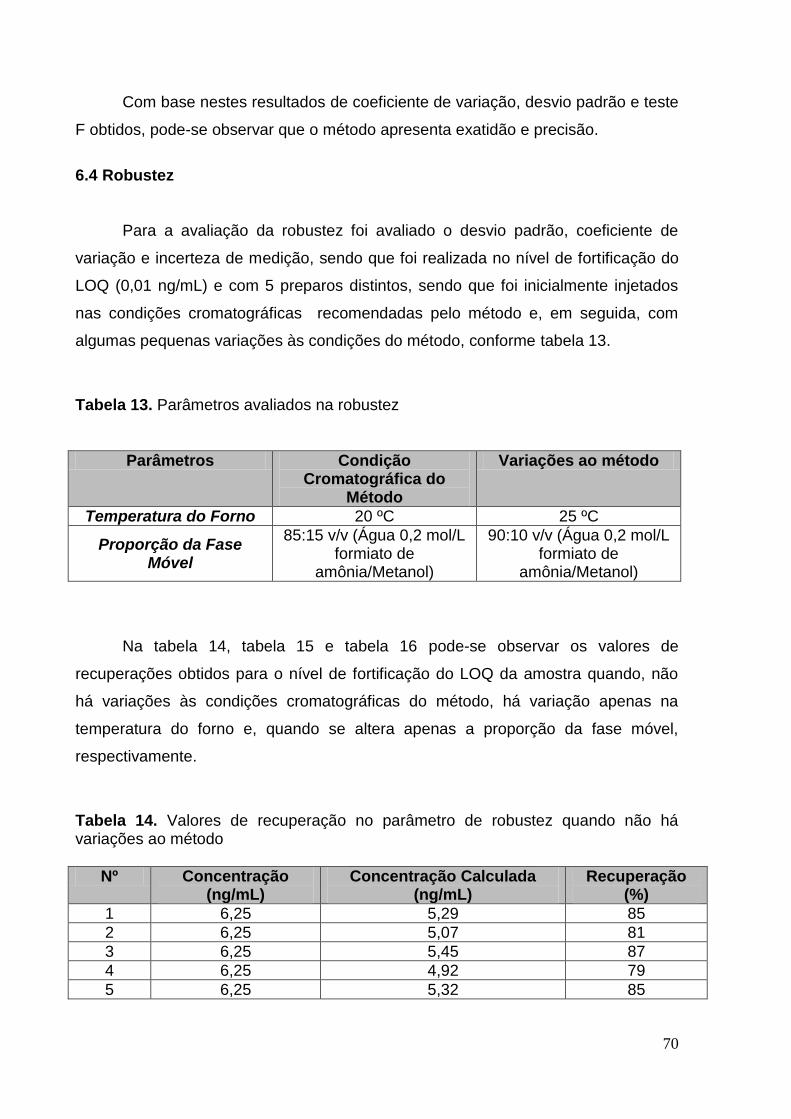
70
Com base nestes resultados de coeficiente de variação, desvio padrão e teste
F obtidos, pode-se observar que o método apresenta exatidão e precisão.
6.4 Robustez Para a avaliação da robustez foi avaliado o desvio padrão, coeficiente de
variação e incerteza de medição, sendo que foi realizada no nível de fortificação do
LOQ (0,01 ng/mL) e com 5 preparos distintos, sendo que foi inicialmente injetados
nas condições cromatográficas recomendadas pelo método e, em seguida, com
algumas pequenas variações às condições do método, conforme tabela 13.
Tabela 13. Parâmetros avaliados na robustez
Parâmetros Condição Cromatográfica do
Método
Variações ao método
Temperatura do Forno 20 ºC 25 ºC
Proporção da Fase Móvel
85:15 v/v (Água 0,2 mol/L formiato de
amônia/Metanol)
90:10 v/v (Água 0,2 mol/L formiato de
amônia/Metanol)
Na tabela 14, tabela 15 e tabela 16 pode-se observar os valores de
recuperações obtidos para o nível de fortificação do LOQ da amostra quando, não
há variações às condições cromatográficas do método, há variação apenas na
temperatura do forno e, quando se altera apenas a proporção da fase móvel,
respectivamente.
Tabela 14. Valores de recuperação no parâmetro de robustez quando não há variações ao método
Nº Concentração (ng/mL)
Concentração Calculada (ng/mL)
Recuperação (%)
1 6,25 5,29 85
2 6,25 5,07 81
3 6,25 5,45 87
4 6,25 4,92 79
5 6,25 5,32 85

71
Tabela 15. Valores de recuperação no parâmetro de robustez quando há variação de temperatura do forno em relação método
Nº Concentração (ng/mL)
Concentração Calculada (ng/mL)
Recuperação (%)
1 6,25 5,27 84
2 6,25 4,98 80
3 6,25 5,32 85
4 6,25 5,02 80
5 6,25 5,32 85
Tabela 16. Valores de recuperação no parâmetro de robustez quando há variação de proporção da fase móvel em relação método
Nº Concentração (ng/mL)
Concentração Calculada (ng/mL)
Recuperação (%)
1 6,25 5,01 80
2 6,25 4,76 76
3 6,25 5,19 83
4 6,25 4,96 79
5 6,25 4,94 79
Tabela 17. Valores de Análise de Variância Obtidos da Robustez Anova: fator único
RESUMO
Grupo Contagem Soma Média Variância DP %CV
Sem Variações ao Método 5 417 83.4 10.8 3.29 3.94 Variação da Temperatura de Forno 5 414 82.8 6.7 2.59 3.13
Variação da proporção da FM 5 397 79.4 6.3 2.51 3.16
ANOVA
Fonte da variação SQ gl MQ F valor-P F crítico
Entre grupos 46.53333 2 23.26667 2.932773 0.091831 3.885294
Dentro dos grupos 95.2 12 7.933333
Total 141.7333 14
SQ: Soma dos Quadrados; gl: Grau de Liberdade; MQ: Soma Média Quadrática; DP: Desvio Padrão; CV: Coeficiente de Variação.

72
É possível observar com base nos resultados obtidos, que o coeficiente de
variação entre os métodos para análise de robustez variou cerca de 3 a 4% sendo
considerado um valor baixo, em comparação com o valor de 20% estipulado como
valor máximo para CV pela ANVISA para matrizes complexas e também pose-se
observar que todas as recuperações estão dentro da faixa de aceitação de
recuperação de 70-120% segundo a RDC nº 216. Além disso, observa-se que o
valor F calculado foi menor que o valor F critica, comprovando que não há diferença
significativa entre os resultados, sendo este método considerado robusto.
6.5 Análise de Impacto da substituição de Metodologia de Análise de CLAE-UV para CL-EM/EM 6.5.1 Seletividade
Com relação à seletividade, pode-se considerar que a cromatografia liquida
com espectrômetro de massas e muito mais vantajosa que quando utilizada CLAE-
UV. A espectrometria de massas em série, no qual se utilizou um detector do tipo
triplo quadrupolo, permite que apenas os íons dos analitos formados na fonte de
ionização sejam transmitidos pelo primeiro quadrupolo à cela de colisão, onde estes
íons são fragmentados por CID e, posteriormente detectados no terceiro quadrupolo.
Desta forma, além de se detectar o íon do analito, é também possível detectar os
íons dos fragmentos gerados a partir deles. A informação estrutural fornecida pelos
fragmentos gerados permite que, mesmo quando analisados agrotóxicos de mesma
massa molar, eles possam ser diferenciados entre si. Esta propriedade torna-se
muito importante no caso da análise de matrizes complexas, como é o caso de
análise de agrotóxicos nos alimentos, como neste caso, no qual se se utilizou da
matriz de pêssego, pois faz com que a obtenção de falsos positivos, gerados pela
matriz que tenham a mesma massa molar do agrotóxico seja praticamente anulada,
tais como falsos positivos gerados pela matriz que possua comprimento de onda
parecido ou idêntico ao absorvido pelo agrotóxico. Por este motivo, algumas
agências reguladoras estabelecem que a espectrometria de massas em série é a
técnica de detecção mais adequada para ser empregada em métodos oficiais de
determinação de resíduos de agrotóxicos em alimentos, devido a sua seletividade
(CHIARADIA, 2009).

73
Devido à comprovada seletividade do CL-EM/EM, os limites de quantificação
e de detecção utilizada para esta técnica foram muito inferiores do que quando
utilizado CLAE-UV, sendo que desta maneira, caso não apresente níveis inferiores
de resíduos de agrotóxicos é possível até mesmo diminuir a LMR determinados
pelas agências regulamentadoras.
6.5.2 Tempo de Análise
O parâmetro tempo é de grande importância nas análises, pois quanto mais
rápida for à análise deste produto no equipamento, é possível colocar várias outras
seqüências para injeção e obtenção de resultados em um menor tempo, agilizando o
tempo de finalização dos estudos.
Ambos os métodos de análise apresentados (CLAE-UV e CL-EM/EM) gastam
o mesmo tempo em relação ao preparo das amostras para injeção, e um tempo
muito próximo em relação à estabilização do equipamento, porém o tempo de
análise no CL-EM/EM é de apenas 5 minutos de corrida para cada injeção, enquanto
o tempo de análise no CLAE-UV chega a 15 minutos de corrida para cada injeção.
6.5.3 Valores de Recuperação dos fortificados para CLAE-UV
Na tabela 18 pode-se observar os resultados obtidos para a recuperação em
dois níveis de fortificação para a amostra de pêssego quando injetados no CLAE-
UV, podendo-se dessa maneira fazer uma comparação entre estes resultados e os
obtidos quando injetados no CL-EM/EM.
Para o caso da injeção no CLAE-UV foi-se preparado amostras fortificados no
nível de 0,05 mg/kg e 0,50 mg/kg, enquanto no sistema CL-EM/EM as amostras
fortificadas foram dos níveis de 0,01 mg/kg e 0,50 mg/kg, sendo que o nível de maior
fortificação foi o mesmo em ambos os casos, porém como o CL-EM/EM é mais
seletivo que o CLAE-UV, foi possível preparar amostras fortificadas no nível de 0,01
mg/kg como nível do LOQ, 5 vezes menor do que o LOQ empregado para o detector
de ultravioleta.
Porem vendo-se a tabela 18 é possível fazer uma comparação destas
injeções com a do CL-EM/EM e observar que as recuperações de ambos foram bem
parecidas, sendo elas em torno de 74-86 % para o caso do CLAE-UV, e de 75-89%

74
para as injeções no espectrômetro de massas, mostrando desta forma que existe a
possibilidade de substituição do sistema CLAE-UV pelo CL-EM/EM.
Tabela 18. Valores de recuperação para injeção no CLAE-UV

75
7. Conclusão
Ao efetuar-se a comparação entre o método utilizando-se CLAE-UV e o
método utilizando-se CL-EM/EM, pode-se concluir que ambos os métodos são
robustos, apresentando precisão e exatidão, dentro dos previstos pela legislação.
Além disso, ambos apresentam faixas de recuperação bem semelhantes na análise
da matriz de pêssego, sendo que se obteve recuperações variando entre 75-89%
para fortificações de 0,01 mg/kg no CL-EM/EM e 74-82% para fortificações de 0,05
mg/kg no CLAE-UV e, entre 75-87% e 72-86% para fortificações de 0,50 mg/kg para
CLAE-UV e CL-EM/EM, respectivamente.
Esta substituição para a nova metodologia de análise tem como um impacto
positivo o tempo de análise das amostras, pois o tempo de análise utilizando-se o
CL-EM/EM é de apenas 5 minutos de corrida para cada injeção, enquanto o tempo
de análise no CLAE-UV chega a 15 minutos de corrida para cada injeção.
Pode-se observar também como vantajosa, devido à comprovada seletividade
do CL-EM/EM, os limites de quantificação e de detecção utilizada para esta técnica
foram muito inferiores do que quando utilizado CLAE-UV, sendo que não apresentou
interferência para a matriz de pêssego analisada neste estudo, sendo inclusive
utilizado apenas o íon fragmento de maior intensidade, não havendo necessidade de
se utilizar a transição de confirmação do mesmo, diferentemente no caso de se
utilizar o CLAE-UV, que permite apenas identificar se o sinal é puro, através dos
espectros de absorção no UV, porém não permite separar o sinal do analito do sinal
gerado por uma possível interferência gerada pela matriz quando esta ocorre no
mesmo tempo de absorção.
Para o sistema CL-EM/EM, as condições cromatográficas de separação e
detecção otimizadas foram: fase móvel composta por uma solução aquosa de 0,20
mols L-1 de formiato de amônio (Fase Móvel A) e metanol (Fase Móvel B), com
vazão de 1 mL min-1, eluição isocrático com composição 85:15 v/v (Fase Móvel
A/Fase Móvel B), volume de injeção de 50 L, coluna cromatografica analitica
Eclipse XDB C8, (150 x 4,6)mm, 5m de diâmetro de particula e detecção por

76
espectrometria de massas em série, com ionização por eletronebulização no modo
positivo e varredura por monitoramento de múltiplas reações com transição de
massa 103,1 44,2 como transição de quantificação e 103,1 60,1 como
transição de confirmação, porém que não foi utilizada visto que na transição de
quantificação obteve-se uma boa seletividade.
Ambas as transições foram monitoradas sob valores ajustados de voltagem
de cone e energia de colisão otimizada, através de infusão direta do ativo no
espectrômetro de massas.

77
8. Referências Bibliografias
[ANVISA] – Agencia Nacional de Vigilância Sanitária. Guia para Validação de
Métodos Analíticos e Bioanalíticos, RE nº 899 de 29/05/2003.
[ANVISA] – Agencia Nacional de Vigilância Sanitária. Resolução da Diretoria
Colegiada RDC nº 216 de 15 de dezembro de 2006.
ARDREY, R. E. Liquid Chromatography-Mass Spectrometry: An Introduction, Wiley:
Huddersfield, 2003
BASTOS, L. H. P.; GÓES, H. C. A.; CARDOSO, M. H. W. M.; GOUVÊA, A. V.; DIAS,
D. P.; ALMEIDA, R. R. R.; NÓBREGA, A.; ABRANTES, S. Ensaio de Proficiência
para Análise de Ditiocarbamatos em Polpa de Banana. Química Nova vol. 30, nº 1,
São Paulo, 2007.
BARROS, Cleide Bassini. Validação de Métodos Analíticos. Biológico, vol. 64, nº 2, p
175-177, São Paulo, 2002.
BRITO, N. M.; AMARANTE JUNIOR, O. P. de; POLESE, L.; RIBEIRO, M. L.
Validação de Métodos Analíticos: Estratégia e Discussão. Pesticidas: Ecotoxicologia
e Meio Ambiente. R., vol. 13, p 130-146, Curitiba, 2003.
CHIARADIA, Mariza Campagnolli. Desenvolvimento, Validação e Aplicação de
Métodos para Análise Multirresidual de Agrotóxicos em Suco de Laranja e Tangerina
Utilizando CLAE-DAD, CL-EM-EM e CLUE-DAD. Campinas, SP, 2009
COLLINS, C. H.; GUIMARÃES, L. F. L.; Cromatografia Líquida de Alta Eficiência. In:
Collins, C. H.; BRAGA, G. L. Introdução a Métodos Cromatográficos, ed. 3, Editora
UNICAMP,São Paulo, 2006
COSTA NETO, Pedro Luís de Oliveira. Estatística. Edgard Bluncher, São Paulo,
1977

78
De AQUINO NETO, F. R.; NUNES, D. S. S. Cromatografia. Princípios Básicos e
Técnicas Afins. Rio de Janeiro: Interciência, 2003
DEMOLINER, Adriana. Otimização e Validação de Metodologia Analítica
Empregando SPE e LC-ESI-MS/MS para determinação de Multiclasses de
Agrotóxicos e Metabólitos em Água de Superfície e de Abastecimento Publico, Rio
Grande, RS, 2008.
[IARC] International Agency for Research on Cancer, Genetic and Related Effects.
Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man, vol 79,
2000.
HARRIS, Daniel, C. Análise Química Quantitativa. 5. Ed. Rio de Janeiro: LTC
Editora, 2001
HOLLER, F. J.; SKOOG, D. A.; CROUCH, S. R. Princípios de Análise Instrumental.
Tradução: Celio Pasquini, ET al., ed 6, Porto Alegre: Bookman, 2009.
International Standard Organization ; General Requirements for the Competence of
Testing and Calibration Laboratories, ISO/IEC 17025, 1999.
JARDIM, I. C. S. F.; ANDRADE, J. A.; QUEIROZ, S. C. N. Resíduos de Agrotóxicos
em Alimentos: Uma Preocupação Ambiental Global – Um Enfoque as Maças.
Química Nova, vol. 32, nº 4, São Paulo, 2009.
MORAES, A. M. E.; PEREIRA, L, M.; TODELO, T. M. R.; SILVA, V. A. C.
Substituição da Análise de Teor de Amistraz no Produto X Intervet Schering-plough
Realizada por CLAE Fase Normal por CLAE Fase Reversa, 2010.
LEMES, Vera Regina Rossi. Avaliação de Resíduos de Etilenotiouréia (ETU) em
Frutos Comercializados na Cidade de São Paulo, 2007.

79
KAZOS, ELIAS A., CONSTANTINE D. Determination of Dithiocarbamate fungicide
propineb and its main metabolite propylenethiourea in airborne samples.
Chemosphere. V. 68, p. 2104-2110. 2007
KOLANKAYA, D.; Ogus, A.; Ayas, Z.; Akay, M. T. Manganese
Ethylenedithiocarbamate (Maneb) and Ethylenethiourea (ETU) Residues in Different
Parts of Tomato Plant and Soil. FoodChemistry 34, p. 181-186, 1989
RIBANI, M.; BOTTOLI, C. B. G.; COLLINS, C.H.; JARDIM, I. C. S. F.; MELO, L. F. C.
Validação em Métodos Cromatográficos e Eletroforéticos. Química Nova, vol. 27, nº
5, p. 771-780, 2004.
SEIZIOGA. Fundamentos de Toxicologia. 2.ed. São Paulo: Atheneu. Editora, 2003
SKOOG, Douglas A.; WEST, Donald M,; HOLLER, F. James; CROUCH, Stanley R.
Fundamentos da Química Analítica. Cengage Learning, Tradução da 8ª ed. Norte-
Americana, 2006
SOUSA, Leandro Augusto Ferreira de. Validação de Metodologia Analítica para
Determinação de Resíduos de Fungicidas Ditiocarbamatos em Tomate por
Espectrofotometria, 2009.
SPIEGEL, MURRAY RALPH. Probabilidade e Estatística. McGraw-Hill do Brasil, São
Paulo, 1978
SUE XU, Environmental Fate of Ethylenethiourea.
http://www.cdpr.ca.gov/docs/emon/pubs/fatememo/etu.pdf. Acessado em:
21/05/2011
[USP] United States Pharmacopeia Convention; US Pharmacopeia 24, Validation of
Compendial Methods, Rockville, 1999.

80
[WHO] World Health Organization. Environmental Health Criteria 78 –
Dithiocarbamate Pesticides, Ethylenethiourea, and Propylenethiourea: A General
Introductio. Geneva, 1988.
WALSH, M. C. Moving from Official to Traceable Methods: Trends in Analytical
Chemistry, vol. 18, p. 616-623, 1999.


















