
Universidade de São Paulo Escola Superior de Agricultura ... · Ao meu filho João Victor Viégas...
95
Universidade de São Paulo Escola Superior de Agricultura “Luiz de Queiroz” O genótipo do hospedeiro e as condições ambientais como moduladores da comunidade bacteriana associada Pedro Avelino Maia de Andrade Tese apresentada para obtenção do título de Doutor em Ciências. Área de concentração: Microbiologia Agrícola Piracicaba 2017
Transcript of Universidade de São Paulo Escola Superior de Agricultura ... · Ao meu filho João Victor Viégas...

Universidade de São Paulo Escola Superior de Agricultura “Luiz de Queiroz”
O genótipo do hospedeiro e as condições ambientais como moduladores da comunidade bacteriana associada
Pedro Avelino Maia de Andrade
Tese apresentada para obtenção do título de Doutor em Ciências. Área de concentração: Microbiologia Agrícola
Piracicaba 2017

Pedro Avelino Maia de Andrade Engenheiro Agrônomo
O genótipo do hospedeiro e as condições ambientais como moduladores da comunidade bacteriana associada
versão revisada de acordo com a resolução CoPGr 6018 de 2011
Orientador: Prof. Dr. FERNANDO DINI ANDREOTE
Tese apresentada para obtenção do título de Doutor em Ciências. Área de concentração: Microbiologia Agrícola
Piracicaba 2017

2
Dados Internacionais de Catalogação na Publicação DIVISÃO DE BIBLIOTECA – DIBD/ESALQ/USP
Andrade, Pedro Avelino Maia de
O genótipo do hospedeiro e as condições ambientais como moduladores da comunidade bacteriana associada / Pedro Avelino Maia de Andrade. - - versão revisada de acordo com a resolução CoPGr 6018 de 2011. - - - - Piracicaba, 2017.
94 p.
Tese (Doutorado) - - USP / Escola Superior de Agricultura “Luiz de Queiroz”.
1. Padrões de associação 2. Microrganismo-hospedeiros 3. Anthurium alcatrazense 4. Cianobactérias 5 Comunidade bacteriana I. Título

3
DEDICO
Aos meus familiares,
Hamilton, Mariza (pais), Marina (irmã), João Victor (filho),
Priscila (namorada) e seus microrganismos,
meus holobiontes favoritos.
OFEREÇO
Aos leitores,
que desfrutem dos novos conhecimentos
colocados neste trabalho,
Transforme-os em novas ideias.

4
AGRADECIMENTOS
A Deus, por iluminar meus caminhos e tornar tudo possível.
Aos meus pais, Hamilton Avelino Medeiros de Andrade e Mariza Maia de
Andrade, pelo amor incondicional, confiança e pela compreensão nos meus
momentos de ausência. Acho que sem eles nada seria possível, pois eles são meu
alicerce de vida.
À minha irmã, Dra. Marina Maia de Andrade, pela amizade, confiança e
lealdade, caráter, competência, características que admiro.
Ao meu filho João Victor Viégas de Andrade, pelo amor e compreensão pela
minha ausência. O meu sucesso refletirá diretamente em você e é por isso que luto
por ele.
À minha namorada Priscila Alves Giovani e minha sogra Carmem Ferreira Alves
pelo carinho, amor, apoio e compreensão nos momentos difíceis, e todo apoio na
redação da minha tese e dos artigos.
A todos os meus familiares, por acreditarem em mim e entenderem minha
ausência. A família é o bem mais precioso do mundo.
À Universidade de São Paulo pela oportunidade de estudar em uma das
melhores Universidades do Brasil e à Escola Superior de Agricultura “Luiz de Queiroz”,
pela oportunidade e total disponibilização de recursos e infraestrutura para conclusão
deste trabalho.
Ao Prof. Dr. Fernando Dini Andreote pela confiança e paciência durante todos
esses anos de convivência desde o período de Estágio supervisionado, Mestrado até
Doutorado. Exemplo de profissional, tratando os problemas com garra, sabedoria,
otimismo, caráter, dedicação, perseverança e transparência. Admiro bastante, sua
carreira profissional.
Aos Professores Roberto Gomes de Souza Berlinck e Simone Possedente de
Lira, Marco Antônio de Assis e a Dra. Leticia Poli pelo convite para participar do projeto
Fapesp (processo 2013/50228-8) e também pelo apoio e dedicação para realização
das coletas, toda estruturação do projeto e identificação morfológica das plantas de
Anthurium.

5
À Profa. Dra. Marli de Fátima Fiore, pela, colaboração e disponibilização das
linhagens de cianobactérias e toda a sua estrutura de laboratório. Também à Dra. Ana
Paula Dini Andreote, pela paciência, conversas, colaboração, apoio e dedicação no
desenvolvimento do terceiro capítulo.
À Profa. Dra. Júlia Kuklinsky Sobral, uma pessoa incomparável, merecedora
de todos os elogios, por sua garra, determinação e competência a frente do LGBM-
Garanhuns.
Ao Programa de Pós-graduação em Microbiologia Agrícola em especial a
coordenação do curso por todos os ensinamentos ao longo destes quatro anos e o
apoio pleno à iniciativa de criar o Simpósio de Microbiologia ESALQ-USP, o qual, para
minha felicidade continua sendo organizado com muita dedicação.
Ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)
pelas bolsas de estudo, (142344/2013-3), concedida.
À Marinha do Brasil, à todos os funcionários ICMBio (Estação Ecológica
Tupinambás) pelo apoio logístico nas expedições ao Arquipélago de Alcatrazes.
Também ao mateiro “Beto”, pelo apoio nas coletas. Meu muito obrigado.
Ao Professor Dr. Itamar Soares de Melo por gentilmente ceder espaço para
realização do sequenciamento de DNA, além das boas risadas em nossas conversas.
Ao Dr. Rodrigo Taketani, pelo auxílio na realização do sequenciamento. A todo o
pessoal da Embrapa, Leonardo Silva, Suikinai, Danilo Tosta, Josiane, Camila, Laura,
Martinha, obrigado pelas discussões cientificas, cafezinhos e boas risadas.
Aos meus parceiros de publicação Natália Polesi, Daniela Vega, Daniel Meyer,
Paulo Lopes, Ana Justiniano, Mariana Delgado, Elízio, Jessica Moretto, Franciele,
Gabriela Ferraz, Marcia Leite, obrigado pela confiança, conselhos e risadas.
Aos grandes amigos que fiz na comissão de seminários, Bruno Souza, Felipe
e Gislaine e principalmente Leonardo Silva, o qual compartilhou comigo várias ideias
desde o início da estruturação do Simpósio de Microbiologia Agrícola. Obrigado por
acreditarem que seria possível, realizarmos o simpósio. Parabéns por serem pessoas
tão dedicadas à uma causa.

6
Aos grandes amigos, Diogo Costa, Arthur Prudêncio, Adijailton Souza, Bruno,
Timóteo, obrigado pelo, apoio, risadas e zueiras.
A todos meus amigos do laboratório/salinha, “Polé” (Marcus Venicius),
Alessandra (amiguinho), Daniele, Juliana, Dorotéia, Bruna, Victor Pylro, Kelly, Lucas
Dantas, Michelle, Joelma, Fabio (Dig), Julia Perim, Ademir, Thiago, Cátia, Luana,
Yasmin e German, obrigado pelas risadas e momentos de descontração e os
churrascos. Especialmente à Armando Dias e Simone Cotta (partners), companheiros
super especiais, “pau para toda obra”. Obrigado por tudo.
À três pessoas, as quais, também tenho profunda admiração, Fernando
Baldesin, Denise Mescolotti e Sonia Pires, nunca foram apenas técnicos de
laboratórios, são exemplo de caráter, competência e dedicação. Obrigado por tudo.
E, finalmente, a todos aqueles que, direta ou indiretamente, contribuíram para
a execução deste trabalho, os meus sinceros agradecimentos.
Pedro Andrade

7
“Para se descobrir novas terras, deve-se estar disposto a perder a terra de vista por
um longo tempo. ”
André Gide
“Julgue seu sucesso pelas coisas que você
teve que renunciar para consegui-lo. ”
Dalai Lama

8
SUMÁRIO
RESUMO .................................................................................................................. 10
ABSTRACT ............................................................................................................... 11
1 INTRODUÇÃO ................................................................................................... 13
Referências ............................................................................................................... 18
2 COMPOSIÇÃO DA COMUNIDADE BACTERIANA EM PLANTAS ENDÊMICAS
E NÃO ENDÊMICAS DO GÊNERO ANTHURIUM spp. ........................................... 21
Resumo .................................................................................................................... 21
Abstract ..................................................................................................................... 21
2.1 Introdução ....................................................................................................... 22
2.2 Materiais e Métodos ........................................................................................ 24
2.2.1 Locais de amostragem ................................................................................ 24
2.2.2 Material coletado ......................................................................................... 26
2.2.3 Extração do DNA total ................................................................................. 29
2.2.4 Sequenciamento do gene 16S rRNA total via Ion Torrent. .......................... 29
2.2.5 Análise das sequências ............................................................................... 30
2.2.6 Análises estatísticas estrutura da comunidade bacteriana associada ......... 31
2.3 Resultados ...................................................................................................... 32
2.3.1 Riqueza e diversidade da comunidade bacteriana associada as plantas do
gênero Anthurium ..................................................................................................... 32
2.3.2 Diferenciação da estrutura da comunidade bacteriana nas diferentes
espécies de Anthurium spp. ...................................................................................... 33
2.3.3 Composição taxonômica da comunidade bacteriana associada as plantas de
Anthurium spp. .......................................................................................................... 34
2.3.4 Grupos bacterianos responsáveis pela diferenciação das comunidades
bacterianas associadas à Anthurium spp. ................................................................ 37
2.4 Discussão ....................................................................................................... 38
2.4.1 Relação entre a composição e estrutura da comunidade bacteriana
associada a plantas endêmicas e seus parentes do mesmo gênero. ....................... 41
Referências ............................................................................................................... 47
3 IMPORTÂNCIA DA FILOGENIA E DO AMBIENTE NA COMPOSIÇÃO DA
COMUNIDADE BACTERIANA ASSOCIADA A CIANOBACTÉRIAS ........................ 53
Resumo .................................................................................................................... 53
Abstract ..................................................................................................................... 53
3.1 Introdução ....................................................................................................... 54

9
3.2 Material e Métodos .......................................................................................... 56
3.2.1 Linhagens de cianobactérias utilizadas ........................................................ 56
3.2.2 Extração do DNA e sequenciamento massivo da região V6 do gene 16S
rRNA e análises das sequências ............................................................................... 58
3.2.3 Análise de correlação entre a filogenia das cianobactérias e a estrutura da
comunidade bacteriana associada ............................................................................ 59
3.2.4 Variação das comunidades bacterianas associadas ao longo do cultivo das
cianobactérias ........................................................................................................... 59
3.2.5 Variações nas condições de cultivo das cianobactérias............................... 60
3.3 Resultados....................................................................................................... 61
3.3.1 Alfa e beta-diversidade das comunidades bacterianas associadas aos
gêneros de cianobactérias ........................................................................................ 61
3.3.2 Composição taxonômica da comunidade bacteriana associada as
cianobactérias ........................................................................................................... 62
3.3.3 Relação filogenética entre cianobactérias e a estrutura da comunidade
bacteriana associada ................................................................................................ 66
3.3.3 Alterações na multiplicação celular dos isolados de M aeruginosa e sob
diferentes condições de cultivo ................................................................................. 67
3.3.4 Valores de alfa diversidade das comunidades bacterianas associadas a M.
aeruginosa ao longo de seu desenvolvimento e sob distintas condições de cultivo . 70
3.4 Discussão ........................................................................................................ 80
3.4.1 Relação entre a filogenia das cianobactérias e composição e estrutura das
comunidades bacterianas associadas ....................................................................... 80
3.4.2 Alteração da estrutura e composição da comunidade bacteriana ao longo
das fases de crescimento da cianobactéria .............................................................. 83
3.4.3 Alteração da estrutura e composição da comunidade bacteriana associada a
cianobactérias quando esta é submetida a condições abióticas distintas ................. 85
Referências ............................................................................................................... 88
4. CONSIDERAÇÕES FINAIS .................................................................................. 93

10
RESUMO
O genótipo do hospedeiro e as condições ambientais como moduladores da comunidade bacteriana associada
Sabe-se que humanos, plantas e animais são colonizados por uma elevada diversidade de microrganismos e que esses organismos eucariotos dependem destes microrganismos para manutenção do seu desenvolvimento. Usando dois modelos de associação microrganismo-hospedeiro, foi testado a hipótese de que hospedeiros pertencentes a domínios da vida distintos, apesar de suas particularidades estruturais, genotípicas, filogenéticas e fisiológicas, compartilham similaridades nos modos de associação com a comunidade bacteriana. Sendo assim, o objetivo do presente trabalho foi mapear a comunidade bacteriana associada a plantas do gênero Anthurium endêmicas e/ou não. Paralelamente, mapear a comunidade bacteriana associada a gêneros distintos de cianobactérias, ao longo da curva de crescimento e quando esta é submetida a condições de cultivo distintas. Como resultados, primeiramente, foi observado que plantas Anthurium alcatrazense endêmicas da Ilha apresentam riqueza e diversidade menor que as plantas da espécie Anthurium loefgrenii coletada na ilha de Alcatrazes e também menor que as plantas Anthurium intermedium e Anthurium pentaphyllum coletadas na região de continente. Também foi observado que a estrutura da comunidade bacteriana associada as plantas de A. alcatrazense é distinta quando comparada com as plantas coletadas no continente e também da própria ilha de Alcatrazes. Essa dissimilaridade foi principalmente representada por OTUs afiliadas à Betaproteobacteria e Gammaproteobacteria. Esses resultados sugerem especificidade microrganismo-hospedeiro. Considerando a associação cianobactéria e bactérias heterotróficas, os resultados demonstraram que a comunidade bacteriana associada é especifica de acordo com o gênero de cianobactéria, composta principalmente por classes apresentando abundância relativa de sequencias distintas como, Betaproteobacteria, Gammaproteobacteria, Flavobacteria e Cytophagia. Por outro lado, foi possível observar que ao longo das fases de multiplicação da linhagem Microcystis aeruginosa, ocorre uma sucessão de grupos bacterianos, sendo principalmente representado pela variação da abundância de Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria e Flavobacteria relativo a fase estacionaria de multiplicação. Quando submetida em condições de cultivo distintas, foi possível observar que variações nas taxas de multiplicação da cianobactéria influenciaram uma modulação da estrutura da comunidade bacteriana associada, desta forma sugerindo que rápidas alterações na estrutura da comunidade bacteriana associada a M. aeruginosa, é resultado de processos de auto-regulação entre cianobactéria e bactérias heterotróficas associadas. De forma geral, pode-se sugerir que hospedeiros distintos apresentam padrões de associações com as bactérias similares, podendo estas similaridades sugerir estratégias para um melhor entendimento e manejo dos ecossistemas.
Palavras-chave: Padrões de associação; Microrganismo-hospedeiros; Anthurium alcatrazense; Cianobactérias; Comunidade bacteriana.

11
ABSTRACT
The host genotype and environmental conditions as modulators of the
associated bacterial community
It is known that humans, plants and animals are colonized by a high diversity of microorganisms and that these eukaryotic organisms depend on these microorganisms to maintain their development. Using two microorganism-host association models, we hypothesized that hosts belonging to distinct domains of life, despite their structural, genotypic, phylogenetic and physiological particularities, share similarities in the modes of association with the bacterial community. Thus, the objective of this work was to map the bacterial community associated with plants of the genus Anthurium endemic and / or not. In parallel, map the bacterial community associated with distinct genera of cyanobacteria, along the growth curve of and when it is submitted to different culture conditions. In this context, we observed that Anthurium alcatrazense plants endemic to the Island, present less richness and diversity than the plants of the species Anthurium loefgrenii collected in the island of Alcatrazes and smaller than the plants Anthurium intermedium and Anthurium penthaphyllum collected in the continent. We found that the structure of the bacterial community associated with the plants of A. alcatrazense is distinct when compared to the plants collected in the continent and island of Alcatrazes itself. This dissimilarity was mainly represented by OTUs affiliated with Betaproteobacteria and Gammaproteobacteria. These results suggest microorganism-host specificity. Considering the association cyanobacteria and heterotrophic bacteria, the results demonstrated that the associated bacterial community is specific according to the genus of cyanobacteria, composed mainly by abundance distinct from those of classes, Betaproteobacteria, Gammaproteobacteria, Flavobacteria and Cytophagia. On the other hand, it was possible to observe that during the multiplication stages of the Microcystis aeruginosa strain, a succession of bacterial groups occurs, mainly represented by the variation of the abundance of Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria and Flavobacteria relative to the stationary phase of multiplication. When submitted under different culture conditions, it was possible to observe that variations in cyanobacteria multiplication rates influenced a modulation of the associated bacterial community structure, thus suggesting that rapid changes in the bacterial community structure associated with M. aeruginosa is a result of processes of self-regulation between cyanobacteria and associated heterotrophic bacteria. In general, distinct hosts show patterns of associations with similar bacteria, and these similarities may suggest strategies for a better understanding and management of ecosystems.
Keywords: Association patterns; Microorganism-hosts; A. alcatrazense; Cyanobacteria; Bacterial community

12

13
1 INTRODUÇÃO
Na natureza, a associação entre microrganismos e hospedeiros ocorre de forma
ubíqua, em que todos os organismos envolvidos (Bacteria, Archaea e Eukaria)
(Torsvik et al., 2002; Roesch et al., 2007; Pisa et al., 2011) desenvolveram estratégias
de associação simbiótica para sobreviver as condições bióticas e abióticas que limitam
o desenvolvimento das comunidades e populações (Andrade et al.,1997; Arthusson et
al., 2006; Hess et al., 2011; Mirazhi et al., 2012). O termo simbiose foi incialmente
descrito por Anton deBary, (1879) como uma íntima associação entre organismos
distintos que vivem juntos, normalmente para o benefício de ambos ou no mínimo
benefício de apenas um parceiro. Essas associações simbióticas podem ser
categorizadas como comensalismo, parasitismo e mutualismo e podem ser variáveis
de acordo com processos evolucionários, como mudanças ambientais, mudanças
genéticas e/ou mudanças na saúde da associação microrganismos/hospedeiro
(Zilber-Rosenberg & Rosenberg, 2008). Isso inclui casos de microrganismos
comensais que normalmente, estão presentes em condições naturais no microbioma,
sem apresentar ação patogênica, mas devido a variações nas condições hormonais
e/ou metabólicas que afetam a disponibilidade de nutrientes, estes mudam de hábito
e tornam-se patogênicos (Horn et al., 2004; Serbus et al., 2008). Goodson et al.,
(2009), demonstrou que a presença de uma baixa abundância de patógenos na
cavidade bucal, pode servir como fonte de infecção se o hospedeiro enfrentar
disfunções hormonais ou metabólicas que levem a alteração do equilíbrio microbiano
“disbiose”, aumentando o risco doenças bucais. Por outro ponto de vista, Mendes et
al., (2011) demonstrou que variações na abundância relativa de Proteobacteria e
Actinobacteria estão ligadas a maior ocorrência e severidade de patógenos de plantas
(Mendes et al., 2011).
Em sua definição original de simbiose, deBary também envolveu no conceito do
termo simbionte, microrganismos patógenos/parasitas e mutualistas. Historicamente,
o parasitismo e a patogenicidade microbiana tem sido foco primordial em pesquisas
envolvendo interações microbianas, principalmente devido ao elevado impacto que os
patógenos tem na saúde humana e os efeitos negativos que eles têm na agricultura e
na ciência animal. Todavia os estudos recentes mantêm como foco o emprego do

14
termo simbionte como descrição para uma associação específica, estável e benéfica
a ambos parceiros (Chaparro et a., 2012; Mondo et al., 2012; Hillsland et al., 2014). O
mutualismo como uma relação benéfica para ambas espécies caracterizadas com
relações de sintrofia uma relação onde ocorre uma complementação metabólica entre
ambos organismos, como exemplo, microrganismos degradam materiais mais
complexos, a fim de que outros degradem os mais simples, sinergismo é uma
cooperação entre ambos para a degradação de algum composto e simbiose é uma
relação de dependência entre parceiros onde ocorre a troca mútua de benefícios (van
Elsas et al., 2006).
O mutualismo é uma relação ecológica (+/+), na qual ambos microrganismos e
hospedeiros são beneficiados. Por exemplo, estudos utilizando seres humanos e
animais como modelo de hospedeiros descrevem uma elevada dependência com a
comunidade microbiana, pois estes últimos estão relacionados a funções essenciais
no controle de doenças, nutrição, crescimento e desenvolvimento dos hospedeiros.
Nestes casos, os perfis de grupos microbianos são específicos de acordo com
parâmetros genéticos, metabólicos e características bióticas e abióticas do
ecossistema em que se inserem (Gill et al., 2006). Estudos recentes demonstram que
mamíferos com taxas variáveis de Firmicutes/ Bacteroidetes no intestino possuem
menor capacidade de absorver energia e acumular gordura aumentando sua
predisposição a obesidade (Turnbaugh et al., 2006).
Por outro lado, e semelhantemente, as plantas são colonizadas por
microrganismos epifíticos que se encontram na superfície, como também os
endofíticos que colonizam seus tecidos internos (Chaparro et al., 2014). Esses
microrganismos são descritos por ter um papel crucial no desenvolvimento das
plantas, normalmente desempenhando funções relacionadas a disponibilização de
nutrientes, produzindo hormônios estimuladores do crescimento vegetal como
proteção contra patógenos.
A associação entre plantas e microrganismos é uma das mais estudadas
(Andreote et al., 2014) devido a sua importância tanto na área de conservação de
espécies vegetais, como na agricultura (Andreote & Pereira e Silva, 2017). Teorias
ecológicas recentes descrevem que o conceito de plantas como organismos livre de

15
microrganismos é obsoleto (Vandenkoornhuyse et al., 2015) e sabe-se que as plantas
são colonizadas por uma alta diversidade de microrganismos, principalmente
bactérias (Mendes et al., 2013). As plantas estabelecem estas interações como forma
de se adaptarem as condições adversas e limitantes dos ambientes. Para tanto, a
planta fornece compostos de carbono importantes para multiplicação das bactérias e
estas, desempenham funções essenciais para o crescimento e desenvolvimento da
planta (Azevedo et al., 2000; Lareen et al., 2016), como aquisição de nutrientes,
tolerância à estresses bióticos e abióticos (Hardoim et al., 2008) e proteção contra
patógenos (Berg et al., 2015) e por fim essa composição de comunidade bacteriana
associada é variável de acordo com as espécies das plantas (Turner et al., 2013),
genótipo (Kuklinsky et al., 2004), estágio de desenvolvimento (Chaparro et al., 2014)
e tecidos analisados (Esposito-Polesi et al., 2015).
Salimpour et al., (2010) demonstrou que o uso de inoculante com Thiobacillus sp.
favorece a maior absorção de fósforo pela planta. Neste mesmo caso, Montañez et
al., (2012) demonstrou a inoculação de 10 linhagens de bactérias diazotróficas no
milho promove o crescimento das raízes, e isto possui potencial relação com as
características de fixação de nitrogênio, produção de fitormonios (AIA) e solubilização
de fosfato observadas in vitro. Também outros autores demonstraram que variações
na abundancia relativa de Proteobacteria e Actinobacteria está diretamente
relacionado a proteção das plantas contra infecções de fungos patogênicos (Mendes
et al., 2011). Neste mesmo sentido Costa et al, 2014 demonstrou que a estrutura da
comunidade bacteriana da rizosfera de cana-de-açúcar é diferente de acordo com a
variedade, e esses microrganismos específicos são importantes para funções de
complementação do metabolismo da planta. Neste mesmo contexto, Esposito-Polesi
et al (2015), demonstrou que os tecidos de plantas da mesma espécie hospedam
estruturas de comunidades distintas, e que ao longo do tempo de rejuvenescimento
de plantas micropropagadas, existe uma variação da comunidade bacteriana,
significando maior especificidade na interação quando ela atinge o estágio final de
desenvolvimento para serem levadas a campo.
Avanços recentes no campo do sequenciamento em larga escala,
metagenômica e metabolômica e bioinformática, demonstraram haver muitas
similaridades entre os fatores que determinam a composição da comunidade

16
bacteriana associadas a hospedeiros distintos (Turnbaugh et al., 2006; Gill et al.,
2006, Mendes et al., 2011; Mendes et al., 2013). Alguns autores explicam que plantas,
animais e seres humanos são sistemas abertos, os quais possuem várias áreas e
nichos disponíveis possibilitando a colonização e estabilização de comunidades
microbianas e sendo assim, caracterizados como ecossistemas abundantes. Apesar
das diferenças na composição dos microbiomas oriunda de especificidade genética e
metabólica, estes dois ecossistemas compartilham modos de associação similares
com essas comunidades microbianas. Em que as comunidades microbianas são
determinadas pelas características genotípicas, metabólicas e ambientais dos
hospedeiros. Então, recentemente, estas similaridades entre os padrões de
associação culminaram na teoria do holobiontes de animais e plantas (Zilber-
Rosenberg & Rosenberg, (2008), categorizam plantas e animais e seus microbioma
como uma unidade de seleção natural, apresentando modos de associação
semelhantes. Deste modo, traçando um paralelo entre filogenia e função do
microbioma associado, resultando no melhor entendimento de processos ecológicos
para manutenção da vida dos hospedeiros (Ramírez-Puebla et al., 2013).
Desta forma torna-se importante o uso desses organismos-modelos de
associação como parâmetros para abordagem de outros níveis de classificação dos
organismos. Para alguns autores, explorar os padrões de associação (baseados em
taxonomia ou funcionalidade) entre bactérias e hospedeiros de domínios distintos, nos
levará a uma melhor compreensão dos mecanismos e conceitos envolvidos na
composição das comunidades bacterianas. Assim sendo, esta pode ser uma
estratégia para identificar processos envolvidos na ocorrência destas interações,
auxiliando no entendimento do estabelecimento das interações e no manejo de
ecossistemas (Ramírez-Puebla et al., 2013; Nemergut et al., 2013; Mendes et al.,
2015; Faust et al., 2015).
Sendo assim, comparativamente, mas em outro nível de classificação das
relações ecológicas, um outro modelo de associação recentemente explorado se dá
entre distintos microrganismos, por exemplo, na associação entre cianobactérias e
bactérias heterotróficas (Bagatini et al., 2014). Esta associação é pouco conhecida,
contudo de extrema importância para a biotecnologia, saúde humana e animal. Na
área de biotecnologia, a associação cianobactérias e bactérias heterotróficas é

17
importante porque possui o papel de biorremediação de ambientes contaminados
principalmente por hidrocarbonetos (Abed, 2005; Abed et al., 2009). Na área da
saúde, muitos trabalhos buscam entender essa associação como uma forma de
controlar a multiplicação celular das cianobactérias, e consequentemente a produção
de toxinas. Há também a descrição de que algumas bactérias heterotróficas que
utilizam as moléculas das toxinas para obtenção de energia (Maryuama et al., 2003).
Desta forma, torna-se importante uma completa descrição da associação entre esses
microrganismos.
Similarmente as plantas, estudos recentes descreveram que as cianobactérias
como hospedeiros selecionam microrganismos do meio ambiente (Parveen et al.,
2013; Secker et al., 2016) e necessitam dessa comunidade bacteriana associada
como forma de sobreviver as condições bióticas e abióticas desvantajosas (Dziallas
et al 2011). A composição da comunidade bacteriana associada à cianobactéria é
variável de acordo com sua espécie (Louati et al., 2015), genótipo e condições
ambientais (Eiler et al., 2004).
Sendo assim, nosso trabalho se objetivo em compreender as similaridades nos
modos de associação entre essas modelos plantas– bactérias, e cianobactéria-
bactéria heterotrófica como forma de compreender inter-relações específicas ocorrida
nas associações entre a comunidade bacteriana e hospedeiros de domínios da vida
distintos, Eukaria e Bacteria. Na primeira parte, nós avaliamos a associação entre
plantas endêmicas e bactérias, assumindo que as características de especiação das
plantas e seu genótipo é um fator primordial para a composição da comunidade
bacteriana associada. Na segunda parte, nós buscamos entender como a comunidade
bacteriana está associada a gêneros distintos de cianobactérias e como esta
comunidade se comporta ao longo do crescimento da cianobactéria e ainda, o que
ocorre com a estrutura e composição da comunidade quando o hospedeiro é
submetido a condições de cultivo distintas.
Este trabalho tem como hipótese que hospedeiros distintos, apesar das suas
particularidades estruturais, genotípicas, filogenéticas e fisiológicas, compartilham
similaridades no modo de associação com a comunidade bacteriana. Para isso, nós
mapeamos a comunidade bacteriana de plantas endêmicas (Anthurium alcatrazense)

18
da ilha de Alcatrazes São Paulo - Brasil e plantas do mesmo gênero de ocorrência na
própria ilha de Alcatrazes e no continente, por meio do sequenciamento em larga
escala do gene 16S RNA. Paralelamente, nós mapeamos a comunidade bacteriana
associada a gêneros distintos de cianobactérias, a variação da comunidade ao longo
da multiplicação da cianobactéria e quando esta é submetida a condições de cultivo
distintas.
Referências
Abed R, Köster J. (2005). The direct role of aerobic heterotrophic bacteria associated with cyanobacteria in the degradation of oil compounds. International biodeterioration & biodegradation, 55: 29–37.
Abed, RMM, Dobretsov S, Sudesh K. (2009). Applications of cyanobacteria in biotechnology. Journal of applied microbiology, v. 106, 1:1–12.
Andreote FD et al., (2006); Model plants for studying the interaction of Methylobacterium mesophilicum and Xylela fastidiosa. Canadian Journal of Microbiology. 52: 419–426.
Andreote FD and Silva MCP. (2017) Microbial communities associated with plants: learning from nature to apply it in agriculture. Current Opinion in Microbiology. 37:29-34
Andreote FD, Gumiere T, Durrer A. (2014). Exploring the interactions of plant microbiomes. Scientia Agricola. 71 (6): 528-539.
Arthursson V, Finlay RD, Jansson JK. (2006) Interactions between arbuscular mycorrhizal fungi and bacteria and their potential for stimulating plant growth. Enviromental Microbiology. 8:1-10.
Azevedo JL, Maccheroni MJr, Pereira JO, Araújo WL. (2000) Endophytic microrganisms: a review on insect control and recent advances on tropical plants. Electron J Biotechnol. [WWW document] URL http:// www.ejbiotechnology.info/content/vol3/issue1/full/4/index.html
Bagatini IL, Eiler A, Bertilsson S, Klaveness D, Tessaroli LP, Vieira AAH. (2014). Host-specificity and dynamics in bacterial communities associated with Bloom-forming freshwater phytoplankton. PloS one, 9(1): e85950.
Berg G, Krause R, Mendes R. (2015). Cross-Kingdom similarities in microbiome ecology and biocontrol of pathogens. Frontiers in Microbiology. 6:1311
Bouffaud ML, Poirer MA, Muller D, Moënne-Loccoz Y (2014) Root microbiome relates to plant host evolution. Environmental Microbiology, 16, 2804-2814
Chaparro JM, Badri DV, and Vivanco JM: Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8:790-803.
De Bary. (1879). Die erscheimung der Symbiose. Strassburg Verlag Trubner.

19
Dziallas C, Grossart H P. (2011). Temperature and biotic factors influence bacterial communities associated with the cyanobacterial Microcystis sp.; Environment Microbiology. 13: 1632-1641.
Eiler A, Bertilsson S. (2004). Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environment Microbiology. 6(12):1228-1243.
Esposito-Polesi NP, Andrade PAM, Almeida CV, Andreote FD, Almeida M. (2015) Endophytic bacterial communities associated with two explant sources of Eucalyptus benthamii Maiden & Cambage. World Journal of Microbiology and Biotechnology. doi 10.1007/s11274-015-1924-0.
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS et al (2006). Metagnomic analysis of the human distal gut microbiome. Science 312: 1355-1359.
Goodson JM, Groopo D, Halem S, Carpino E. (2009). Is Obesity an Oral Bacterial Disease? Journal of Dentristy Research. 88(6): 519-523.
Haichar FZ, Marol C, Berge O, Rangel-Castro JI, Prosser JI, Balesdent J, Heulin T, Achouak W. (2008). Plant host habitat and root exudates shape soil bacterial community structure. The ISME Journal. 2: 1221-1230
Hardoim PR, van Overbeek LS, Elsas, JD, (2008). Properties of bacterial endo- phytes and their proposed role in plant growth. Trends in Microbiology 16: 463 – 471
Hess, M, Sczyrba, A, Egan, R et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 331: 463–467.
Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. (2010). Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent
Kuklinsky-Sobral J, Araújo WL, Mendes R, Geraldi IO, Pizzirani-Kleiner AA, Azevedo JL. (2004). Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environmental microbiology, 6(12): 1244–51
Lareen A, Burton F, Schäfer P. (2016) Plant root-microbe communication in shaping root microbiomes. Plant Molecular Biology. 90: 575-587.
Ley R, Turnbaugh, P, Klein, S, Gordon, J. (2006). Microbial ecology: human gut microbes associated with obesity 444: 1022-1023
Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, et al. (2012) Deep Sequencing of the Oral Microbiome Reveals Signatures of Periodontal Disease. PLoS ONE 7(6): e37919. doi:10.1371/journal.pone.0037919
Louati I, Pascault N, Debroas D, Bernard C, Humbert J F, and Leloup J. (2015). Structural diversity of bacterial communities associated with bloom- forming freshwater cyanobacteria differ according to the cyanobacterial genus. PLoS ONE. 10(11): e0140614. doi:10.1371/journal.pone.0140614
Maruayama T, Kato K, Yokoyama A, Tanaka T, Hiraish. A, Park, HD. (2003); Dynamics of Microcystin- Degrading bacteria in Mucilage of Microrcystis. Microbial Ecology. 46: 279-288.
Mendes R and Raaijmakers JM. (2015). Cross-Kingdom similarities in microbiome functions. The ISME Journal. 1-3

20
Parveen B, Ravet V, Djediat C, Mary I, Quiblier C, Debroas D, and Humbert JF. (2013). Bacterial communities associated with Microcystis colonies differ from free-living communities living in the same ecosystem. Environmental Microbiology. 5: 716–24.
Pisa G, Magnani GS, Weber H, Souza EM, Faoro H, et al. (2011). Diversity of 16S rRNA genes from bacteria of sugarcane rhizosphere soil. Brazilian Journal of Medical and Biological Research. 44(12): 1215–1221.
Ramírez-Puebla ST, Servin-Garcidueñas LE, Jiménez-Marín B, Bolaños LM, Rosenblueth M, Martínez J et al. (2013) Gut and root microbiota commonalities. Applied and Environmental Microbiology. 79: 2-9.
Roesch LFW, Fulthorpe RR, Riva A, Casella G, et al. (2007). Pyrosequence enumerates and contrasts soil microbial diversity. The ISME Journal. 1: 283-290.
Sánchez O, Diestra, E, Esteve I and Mas, J. (2005) Molecular characterization of an oil-degrading cyanobacterial consortium. Microbiological Ecology. 50: 580–588
Secker NH, Chua JPS, McNoe L, Laurie RE, Guy PL, Orlovich A. Summerfiled TC. (2016). Characterization of the cyanobacteria and associated bacterial community from an ephemeral wetland in New Zeland. 52: 761-773.
Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, and Poole PS: Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. The ISME Journal. 2013, 7:2248-2258.
van Elsas JD, Tam L, Finlay RD, Killham K, Trevors JT. (2006); Microbial interactions in soil. In: Van Elsas, J.D.; Jansson JK, Trevors JT. Modern soil microbiology. 2nded.NewYork: CRC press. p 177-204.
Vandenkoornhunyse P, Quaiser A, Duhamed M, Le Van A, Defresne A. (2015). The importance of the microbiome of the plant holobiont. New Phytologist. doi: 10.1111/nph.13312
Zilber-Rosenberg I and Rosenberg E. (2008); Role of microrganisms in the evolution of animals and plants: the hologenomic theory of evolution. FEMS Microbiology reviews. 32: 723-735.

21
2 COMPOSIÇÃO DA COMUNIDADE BACTERIANA EM PLANTAS ENDÊMICAS E NÃO ENDÊMICAS DO GÊNERO ANTHURIUM spp.
Resumo
As plantas hospedam uma elevada diversidade de microrganismos, os quais são selecionados para desenvolverem uma interação mutualística com base nas características genotípicas, por conseguinte funcionais de ambos. Sabe-se que plantas de ocorrência restrita possuem uma composição bacteriana particular, determinada por seu isolamento geográfico e possivelmente associada a diferenciação da comunidade bacteriana ao longo do processo de especiação vegetal. O objetivo deste trabalho foi mapear a comunidade bacteriana associada as folhas de plantas do gênero Anthurium. O estudo foi baseado em 21 plantas distribuídas na ilha de Alcatrazes e no continente (Costão rochoso, Intermédio e Mata). Nesses ambientes, foram coletadas quatro espécies: Anthurium alcatrazense (endêmica da ilha de Alcatrazes), Anthurium loefgrenii (ilha de Alcatrazes), Anthurium penthaphyllum (continente) e Anthurium intermedium (continente). Folhas destas plantas foram utilizadas para a extração do DNA, e posteriormente para o sequenciamento massivo do gene 16S rRNA bacteriano. Os resultados demonstraram que as plantas de A. alcatrazense possuem riqueza e diversidade menor do que as outras espécies do mesmo gênero, estando ou não estas localizadas na ilha de Alcatrazes. A planta de A. alcatrazense mostrou hospedar uma estrutura de comunidade particular, caracterizada por uma maior abundância relativa de Gammaproteobacteria e Betaproteobacteria, enquanto que nas demais espécies foram observadas maiores frequências de sequências afiliadas a classe Alphaproteobacteria. Em conjunto, estes resultados sugerem que plantas endêmicas podem hospedar comunidades microbianas específicas.
Palavras chave: A. alcatrazense; Associação Planta-bactéria; Gene 16S rRNA; Sequenciamento Ion Torrent
Abstract
Plants host a high diversity of microorganisms, which are selected to develop an interaction of mutualism and / or symbiosis based on the functional and genotypic characteristics of both. It is known that plants of restricted occurrence have a particular bacterial composition, determined by their geographic isolation and possibly associated to the differentiation of the bacterial community throughout the process of plant speciation. The objective of this work was to map the bacterial community associated with Anthurium plant leaves. The study was based on 21 plants distributed in the island of Alcatrazes and in the mainland (Rocky Coast, Intermediate and Forest). In these environments, four species were collected: A. alcatrazense (endemic to the island of Alcatrazes), Anthurium loefgrenii (island of Alcatrazes), A. penthaphyllum (continent) and A Intermedium (mainland). Leaves of these plants were used for DNA extraction, and later for the massive sequencing of the bacterial 16S rRNA gene. The

22
results showed that the plants of A. alcatrazense have less richness and diversity than the other species of the same genus, whether or not these are located on the island of Alcatrazes. The plant of A. alcatrazense showed to host a particular community structure, characterized by a greater relative abundance of Gammaproteobacteria and Betaproteobacteria, whereas in the other species frequencies of affiliated sequences the class Alphaproteobacteria. Taken together, these results suggest that endemic plants may also host specific microbial communities.
Keywords: A. alcatrazense; Bacteria profile; Plant-bacteria association; 16S rRNA gene; Ion Torrent sequencing
2.1 Introdução
Os microrganismos podem colonizar diferentes partes das plantas,
estabelecendo interações simbióticas e mutualísticas (Kuklinsky et al 2004).
Comunidades microbianas possuem papel fundamental na associação com plantas,
pois estão envolvidas em funções essenciais, como nutrição, controle de patógenos e
resistência a variações de fatores bióticos ou abióticos (Mendes te al., 2007; Dias et
al., 2009).
O conjunto de microrganismos e seu material genômico, associados à uma
planta recebe o nome de microbioma (Mendes et al., 2013). Este microbioma é
caracterizado por alta diversidade, tanto compondo os grupos de microrganismos
encontrados fora como dentro dos tecidos vegetais (Vandenkoornhuyse et al., 2015).
Esta diversidade está comumente associada a deposição de diversas formas de
carbono que derivam do material assimilado pelas plantas por fotossíntese (Bisseling
et al., 2009). Essa disponibilidade de nutrientes é fortemente relacionada com as
características genotípicas de cada planta, o que resulta numa modulação diferencial
da composição e da estrutura da comunidade bacteriana associada a cada espécie
vegetal (Eilers et al., 2010). Nos últimos anos, a microbiota das plantas tem sido alvo
de pesquisas que atribuem aos microrganismos o papel de reservatório de funções
essenciais para sobrevivência das plantas frente a condições adversas (Zilber-
Rosenberg & Rosenberg, 2008; Kier et al., 2011; Bulgarelli et al., 2013; Werner et al.,
2014; Vandenkoornhuyse et al., 2015). Na agricultura, essas funções têm sido
exploradas biotecnologicamente para aumentar a sustentabilidade de sistemas
agrícolas (Mendes et al., 2007). Em um panorama ambiental, características

23
microbianas importantes têm sido descritas em microrganismos associados as
plantas, sendo estas relacionadas a manutenção de espécies em ambientes restritos
e preservados (Golinska et al., 2015).
Plantas pouco estudadas e de ocorrência restrita, como as endêmicas, são
alvos para a bioprospecção de novos produtos (Debnath et al., 2016). Neste tipo de
exploração, sugere-se que o processo evolutivo ocorrido entre as espécies (plantas e
bactérias) foram primordiais para determinar uma forte e restrita associação entre os
organismos e a sobrevivência de ambos em ambientes com condições bióticas e
abióticas variáveis (Hardoim et al., 2008) e, no entanto, produção de compostos
específicos e pouco explorados. Muitos estudos têm sido realizados no intuito de se
obter um completo entendimento da biologia associativa entre plantas e
microrganismos, incluindo descrições taxonômicas e funcionais (Phillippot et al., 2013;
Shi et al., 2014). Por exemplo, Debnath e colaboradores (2016) descreveram
microrganismos específicos hospedados na planta medicinal Rhododendron
arboreum, endêmica de regiões de elevadas altitudes no sudeste dos alpes Himalaios
(Tawang/China). Esses autores buscaram compreender a associação entre plantas e
a diversidade de microrganismos, e os processos que determinavam a ocorrência de
uma comunidade específica nestas plantas. Um outro estudo mapeou a distribuição e
a diversidade de fungos associados à oito macroalgas endêmicas das regiões geladas
da Antártida, descrevendo nestes fungos um grande potencial biotecnológico
(Godinho et al., 2013).
Desta forma, o presente trabalho se baseia na hipótese de que a planta
Anthurium alcatrazense, endêmica da Ilha de Alcatrazes/ São Paulo, hospeda uma
comunidade bacteriana particular, distinta de outras plantas do gênero Anthurium,
encontradas tanto na própria ilha de Alcatrazes, como também no continente. Levando
em consideração o exposto e face a relevância do tema, este trabalho tem como
objetivo mapear a comunidade bacteriana associada a plantas endêmicas e não
endêmicas do gênero Anthurium, presentes em diferentes ambientes da ilha de
Alcatrazes e plantas do continente, coletadas em três ambientes distintos (Costão
rochoso, intermédio e Mata).

24
2.2 Materiais e Métodos
2.2.1 Locais de amostragem
As amostragens em ambientes de ilha e em regiões de continente foram
realizadas na época de inverno e verão, especificamente nas datas 16/06/2014 e
10/02/2015, respectivamente. Essas amostragens ocorreram na ilha de Alcatrazes -
São Paulo, Brasil e na região de Ubatuba, distantes 97 quilômetros uma da outra. No
continente as coletas foram distribuídas em 3 ambientes distintos, de acordo com a
proximidade do mar. Foram amostradas plantas na região de Costão rochoso, cerca
de 6 metros distante do mar (S 23°27’34,43’’; W 45°01’11,40’’); ambiente de
Intermédio, distante cerca de 600 metros do mar (S 23°27’35,4’’; W 45°02’18,6’’); e
um ambiente de Mata (Mata Atlântica), distante 6 km do mar (S 23°27’34,63’’; W
45°01’11,32’’) (Figura 2.1). Todas as amostragens obtiveram permissão de coleta de
espécimes em ambiente natural sob o número do CISbio (37256-4)

25
Figura 2.1-Visualização e localização das plantas do gênero Anthurium amostradas
para o presente estudo. (A) A. alcatrazense, planta endêmica de ocorrência restrita a
ilha de Alcatrazes. (B) A. loefgrenii, planta não endêmica coletada na ilha de
Alcatrazes. (C) Locais de Amostragem, identificados como, P1- Ilha de Alcatrazes, P2-
Costão rochoso (Continente), P3- Ambiente de intermédio (Continente), P4- Mata. (D)
A. pentaphyllum, espécie coletada no continente, nos ambientes de Costão Rochoso
e Mata. (E) A. intermedium, coletados no Costão rochoso, intermédio e Mata. Em cada
ponto foram amostradas três plantas do gênero Anthurium. Esses pontos estão
localizados na região de São Sebastião/São Paulo (P1) e Ubatuba/São Paulo (P2, P3,
P4). A amostragem no continente foi realizada na região de Ubatuba/São Paulo
(Costão Rochoso - S23° 27'34.43'' /W45°01'11.40'’, Mata- S 23°28'18,5''/
W45°02'18,6'') e a coleta na ilha foi realizada na ilha de Alcatrazes (S24°05’ 55.60’’
/W45°37’ 10.31’’), localizada a 35 km da região de São Sebastião-São Paulo e 97 km
da região dos pontos (P2, P3, P4).

26
2.2.2 Material coletado
No total, foram coletadas vinte e uma amostras de plantas do gênero
Anthurium, pertencentes a 4 espécies distintas e distribuídas dentre os 4 pontos de
amostragem supracitados.
O gênero Anthurium possui características bastante marcantes. São plantas
herbáceas epífitas, semi-epífitas, rupícolas ou terrestre. Possuem lâmina foliar
oblonga, lanceolada, obvoada ou digitada, com ápice agudo, acuminado a
mucronado, base cuneada, obtusa a truncada, margem inteira, venação
peniparalelínea, nervuras medianas achatadas impressas e proeminentes e/ou aguda
à obtusa, dispostas adaxialmente proeminente e arredondadas e carenadas
abaxialmente. Possuem de 10 a 19 pares de nervuras secundárias coletoras saindo
da base da lâmina ou um pouco acima. Inflorescência sempre um por simpódio,
pedúnculo ereto e/ ou pedúnculo cilíndrico (Temponi & Nadruz-Coelho, 2011).
A partir destas características principais do gênero, 4 espécies foram
observadas e amostradas: A. alcatrazense, A. loefgrenii, A penthaphyllum e A.
intermedium. Duas espécies foram coletadas na ilha de Alcatrazes, duas espécies no
Costão rochoso, uma espécie no ambiente de Intermédio, e duas espécies na Mata.
A espécie A. Alcatrazense, descrita por Nadruz-Coelho e Catharino (2008), é
endêmica da ilha de Alcatrazes, possui caule e entrenós curtos, pecíolo esverdeado,
roliço, abaxialmente, 5,1- 27 X 0,2 – 0,5 cm. Genículo curto e intumescido, mais claro
que o pecíolo 3-9 X 4-7 mm Folhas pequenas, lâmina foliar cartácea, lanceolada, com
base aguda. Nervura primária arredondada em ambas as faces à subaguda
adaxialmente; nervuras secundárias numerosas (7-20), pouco visíveis em ambas as
faces. A espécie possui grande plasticidade, expressa por populações tipicamente
heliófilas, a pleno sol, sobre costões rochosos, geralmente em amplas touceiras de
plantas “atarracadas”.
A espécie A. loefgrenii, também amostrada na ilha de Alcatrazes, não possui
característica de endemismo pois já foram observados espécimes habitando regiões
de mata Atlântica, no estado de São Paulo e Paraná (Temponi & Nadruz, 2011). Planta
de hábito terrestre, possui caule e entrenós longos. Folhas simples, grandes; pecíolo

27
3,2 – 37,7 cm comprimento verde e de rara ocorrência verde-avermelhado; cilíndrico.
Lâmina elíptica e lanceolada, 7-42,2 X 2,2-16,5 cm, ereta a patente em relação ao
caule, ápice agudo, obtuso. Nervura secundárias impressas abaxialmente, evidentes
a levemente proeminentes abaxialmente, 7-18 pares, nervuras coletoras inseridas na
base foliar, 0,3 a 2 cm afastada da margem (Rocha et al., 2014).
A espécie A. penthaphyllum, amostradas no ambiente de Costão rochoso e
Mata, é uma planta de hábito hemi-epífita e terrestre; caule e entrenós 0,4- 10,7 cm.
Folhas compostas; pecíolo 21,4 – 65,5 cm, verdes, cilíndricos. Nervura principal reta,
obtusa ou aguda. Nervuras secundárias impressas adaxialmente, proeminentes
abaxialmente, 9-18 pares, nervura coletora na base do folíolo ou um pouco acima
dela, 0,4-2,2 cm afastada da margem. Pedúnculo 1,8- 18,4 cm menos da metade do
comprimento do pecíolo (Rocha et al., 2014).
A espécie A. intermedium, foram amostradas em ambiente de Costão rochoso
e Mata. Planta de hábito terrestre, possui caule e entrenós longos. Folhas simples,
grandes; pecíolo 2,4 – 15,3 cm comprimento verde e de rara ocorrência verde-
avermelhado; cilíndrico. Lâmina elíptica e lanceolada, 3,2-15,3 X 1,1-5,2 cm, ereta em
relação ao caule, ápice agudo, obtuso. Nervura principal aguda ou obtusa. Nervura
secundárias, evidentes a levemente proeminentes abaxialmente, 8-13 pares, nervuras
coletoras inseridas na base foliar, 0,2 a 0,7 cm afastada da margem (Rocha et al.,
2014).
Na ilha de Alcatrazes foram coletadas folhas das espécies A. alcatrazense e
loefgrenii e no continente, inicialmente foram coletadas folhas das espécies A.
intermedium e A. penthaphyllum distribuídas em grandes populações no Costão
rochoso. Posteriormente, foram coletadas folhas da espécie A. intermedium no
ambiente de Intermédio entre o Costão rochoso e Mata. Por fim, foram coletadas
folhas das espécies A. intermedium e A. penthaphyllum no ambiente de Mata. A
amostragem foi realizada em triplicata, e em cada planta foram coletadas três folhas
distintas (Tabela 2.1).
Espécimes de cada espécie foram coletadas e o material botânico foi
herborizado e depositado no herbário HRCB (Herbário Rioclarense) do Instituto de
Biociências (Unesp/ Rio Claro), acrônimo de acordo com o Index Herbarium – (Thiers,
28
2016), sob colaboração do Prof. Dr. Marco Antônio de Assis e Msc. Letícia Peres Poli,
do Instituto de Biociências da Universidade Estadual Paulista “Júlio de Mesquita Filho
“. A identificação taxonômica foi realizada com base em literatura consultada (Coelho,
Waechter e Mayo, 2009; Coelho, 2012; Mamede et al., 2012) (Tabela 2.1).
Tabela 2.1- Descrição das características das amostras e identificação das plantas
pertencentes ao gênero Anthurium.
* Ct. Rochoso – Costão rochoso ** Ambiente intermediário entre Costão rochoso e Mata,
No laboratório, as folhas foram lavadas com água corrente para eliminar a
maior parte das impurezas, e posteriormente submetidas ao protocolo de desinfecção
superficial para eliminação da maior parte dos microrganismos epifíticos. Inicialmente,
foram embebidas em álcool 70% por 1 minuto, posteriormente em hipoclorito de sódio
2% por 2 minutos, álcool 70% por 1 minuto, e depois submetidas à 2 etapas de
lavagem em água destilada previamente esterilizada. Amostras dessas duas etapas
finais foram usadas como controle do processo de desinfecção superficial. Após a
desinfecção superficial, as plantas foram trituradas em nitrogênio líquido, com o auxílio
de cadinhos e pistilos. O material resultante foi então armazenado em “eppendorfs”
de 1,5ml e mantidos em freezer -80°C, até o processamento da extração do DNA total
Descrição Amostragem Coordenadas geográficas
Ident. Gênero Espécies Herbário L. de coleta Latitude Longitude Referência
AA-AA Anthurium A. alcatrazense HRBC 64645
Alcatrazes S 24°05'56.01'' W45°41'32,34'' Nadruz e Catharino, 2008
AI-INT Anthurium A. Intermedium
HRBC 64635 Intermédio*
S 23° 27' 35,4" W 45°02'18,6'' Valadares et al., 2010
AI-CST Anthurium A. Intermedium
HRBC 64637
Costão Rochoso
S 23°27'34.43'' W 45°01'11.40' Valadares et al., 2010
AI-MT Anthurium A. Intermedium
HRBC 64636 Mata
S 23° 28' 18,5'' W 45°10'20,5" Valadares et al., 2010
AL-AL Anthurium A. loefgrenii
HRBC 64647 Alcatrazes
S 24° 05' 55,7'' W 45°41'28,6'' Coelho et al., 2014
AP-CST Anthurium A. penthaphyllum
HRBC 64667
Costão Rochoso
S 23°27'34.63'' W45°01'11.32'' Almeida et al., 2005
AP-MT Anthurium A. penthaphyllum
HRBC 64669 Mata
S 23°27'34.63'' W 45°01'11.32' Almeida et al., 2005

29
2.2.3 Extração do DNA total
Aproximadamente 100 mg do material vegetal foi triturado em nitrogênio líquido,
e cada uma das amostras foi submetida a extração do DNA total utilizando o kit Power
Plant DNA Isolation (MoBio Laboratories, Carlsbab, CA), seguindo as instruções do
fabricante.
A integridade do DNA extraído, assim como sua quantificação, foi determinada
por meio de eletroforese em gel de agarose à 1,0% (m/v), preparado em tampão TAE
(400 mM Tris, 20 mM ácido acético glacial, 1mM EDTA), onde foram aplicados 5µl dos
DNAs extraídos junto a 3µl de um tampão de corrida Loading buffer 6x (Azul de
bromofenol 0,05% (p/v); Sacarose 40% (p/v); EDTA 0,1M (pH 8,0); SDS 0,025% (p/v).
Após a eletroforese, o gel foi corado em solução de brometo de etídeo e visualizado
em luz ultravioleta (DNr Bio-imaging Systems Minibis pro 16mm). As quantidades de
DNA extraídos das amostras variaram entre 5 e 10 ƞg.µl-1 de DNA.
2.2.4 Sequenciamento do gene 16S rRNA total via Ion Torrent.
A geração do fragmento a ser utilizado para o sequenciamento da região
hipervariável V6 do gene 16S rRNA ocorreu em duas etapas. Primeiramente, as
amostras do DNA extraído das folhas foram submetidas à amplificação utilizando os
primers 799F (5’ - AAC MGG ATT AGA TAC CCK G – 3’) e 1492R (5’ – TAC GGY
TAC CTT GTT ACG ACT – 3’) (Chelius & Triplett, 2001), os quais foram utilizados
para evitar a amplificação do DNA Cloroplastidial. As condições de amplificação foram
determinadas para reações de volume final de 50 µl, compostas por 1X Tampão de
PCR, 2,5 mM de MgCl2, 0,2 μM de dNTP, 0,2 μM de cada primer, 0,4 mM BSA, 2 U
de Taq DNA polimerase (Fermentas, São Paulo, Brasil). A ciclagem de amplificação
foi realizada em termociclador Veriti® (Applied Biosystems, Waltham, USA),
programado para fazer o seguinte processo térmico: 95°C por 3 minutos; 35 ciclos de
94°C por 30 segundos, 53°C por 40 segundos e 72°C por 40 segundos; e uma
extensão final a 72°C por 7 minutos. Os produtos da reação foram aplicados em gel
de agarose 1%, e submetidos à eletroforese. Foram geradas duas bandas, uma de

30
maior tamanho aproximadamente 1090pb, correspondente ao gene 16S rRNA
mitocondrial das plantas; e uma de menor tamanho aproximadamente 735pb
correspondente ao gene 16S rRNA bacteriano desejado.
A banda menor foi excisada do gel e purificada utilizando o Kit Wizard®
Genomic DNA purification (Promega, USA). Após a purificação, estas amostras foram
utilizadas como DNA molde em uma nova reação utilizando o conjunto primers 967F
(CAA CGC GAA GAA CCT TAC C) (Sogin et al., 2006) e 1193R (CGT CRT CCC CRC
CTT CC) (Wang; Qian, 2009), os quais, geram fragmentos de aproximadamente
230pb. O primer forward 967F foi adicionado de sequências “barcodes” contendo um
conjunto de 5 nucleotídeos, os quais foram sintetizados separadamente e utilizados
como marcadores para cada uma das amostras (http://vamps.mbl.edu/).
As condições de amplificação foram determinadas para reações de volume final
de 50 µl, compostas por 1X Tampão de PCR, 3 mM de MgCl2, 200 μM de dNTP, 0,2
μM de cada primer, 0,02 U/μL de Taq DNA polimerase (Fermentas, São Paulo, Brasil)
de acordo com (Kavamura et al, 2013). Após amplificação, todos fragmentos contendo
os “barcodes” foram misturados em concentrações equimolares e purificados com o
kit Charge Switch PCR Clean-UP (Invitrogen, Brasil). Posteriormente, foi realizada
uma PCR de emulsão usando o Ion OneTouch 2™ com o Ion Template PGM™ OT2
400 Kit (Life Technologies) de acordo com instruções do fabricante. As bibliotecas de
fragmentos do gene 16S rRNA foram sequenciadas utilizando um Ion Chip 316™ Kit
v2, usando o sistema Ion Torrent (Personal Genome Machine™).
Todos os procedimentos de preparação das bibliotecas, desde a PCR de
emulsão até a obtenção do arquivo de sequências (. fastq), foram realizadas pela
equipe do Doutor Itamar Soares de Melo, no Laboratório de Microbiologia Ambiental
da Embrapa Meio Ambiente (Jaguariúna, SP).
2.2.5 Análise das sequências
As análises do arquivo (. fastq) contendo as sequências brutas foram realizadas
utilizando o software Quantitative Insights Into Microbial Ecology (QIIME) versão 1.9
(Caporaso et al., 2010a). Inicialmente, as sequências foram separadas por amostra
de acordo com “barcode” inserido na PCR. Posteriormente, as sequências foram

31
separadas dos primers e foram filtradas por qualidade (Qual.score = 25, tamanho dos
barcodes = 5, número máximo de primer mismatch = 2 e uma janela de qualidade 50
e número máximo de homopolímeros = 6). Adicionalmente, sequências menores que
180pb foram descartadas.
As sequências válidas foram agrupadas em OTUs (Operational Taxonomic
Units) à 97% de similaridade usando o método Uclust (Edgar, 2010), e alinhadas pelo
método PyNAST (Caporaso et al., 2010). A afiliação taxonômica de cada sequência
representativa das OTUs foi realizada por comparação com as sequências disponíveis
no banco de dados Greengenes (DeSantis et al., 2006). Posteriormente, sequências
quimeras foram removidas usando o método UCHIME (Edgar et al., 2011). OTUs de
baixa abundância (ex., singletons e doubletons) foram removidas do conjunto de
dados. No entanto, mesmo após todos esses procedimentos de curadoria das
sequências, ainda restaram sequências da classe “Chloroplast”. O conjunto de dados
foi então filtrado de todas as sequências referentes à classe “Chloroplast”, as quais
não foram objeto de estudo. Após esses passos, foi realizada a normalização das
amostras, utilizando o comando do Qiime “single_rarefaction”, o qual realiza uma re-
amostragem dos dados, gerando um novo arquivo normalizado, onde todas as
amostras são analisadas com o mesmo número de sequências. No final da análise foi
gerada uma tabela otu_table, descrito por OTUs X amostras X taxonomia.
2.2.6 Análises estatísticas estrutura da comunidade bacteriana associada
A compreensão das mudanças na composição da comunidade bacteriana
associadas as plantas coletadas foi obtida por meio do cálculo de alfa-diversidade e
beta-diversidade das amostras. A alfa-diversidade compreende as características
ecológicas de cada uma das amostras, enquanto a beta-diversidade faz uma análise
comparativa da diversidade entre as amostras (Lozupone et al., 2007).
O cálculo da alfa-diversidade foi realizado utilizando o software Qiime, onde
foram obtidos os índices de Riqueza de grupos (Chao1 - S’), Diversidade (Shannon –
H’) e índices de cobertura do sequenciamento Good’s Coverage. Para entender a
variação entre os valores obtidos, foi realizado um teste de análise de variâncias

32
(ANOVA) (p < 0,05), seguido do teste de comparação de médias de Tukey,
considerando o valor de significância à 5%.
Para o cálculo da beta-diversidade foram geradas matrizes de distâncias
baseadas no algoritmo de Bray-Curtis (Bray & Curtis, 1957), as quais sustentaram a
análise de coordenadas principais (PCoA). Esta então, permitiu a observação da
distribuição dos agrupamentos entre as amostras numa escala bidimensional. Ainda
assim, foram empregadas análises de PERMANOVA e ANOSIM (p < 0,05), baseadas
em 9.999 permutações randômicas da tabela de OTUs, para testar a significância dos
agrupamentos encontrados na PCoA. Por fim, foi realizado um teste de dissimilaridade
de agrupamentos (SIMPER), baseado no resultado da (PCoA), o qual teve como
objetivo destacar as OTUs mais responsivas para a dissimilaridade entre os
agrupamentos encontrados. Este teste foi desenvolvido utilizando o software PAST
versão 2.17 (Hammer et al., 2001).
2.3 Resultados
2.3.1 Riqueza e diversidade da comunidade bacteriana associada as plantas
do gênero Anthurium
A partir do sequenciamento massivo do gene 16S rRNA foi obtido um total de
572.330 sequências válidas após processo de filtragem das mesmas por qualidade
de reads. Depois da exclusão das sequências oriundas da classe “Chloroplast”, a
otu_table foi normalizada em 1.085 sequências por amostra, as quais apresentaram
índices de cobertura de sequenciamento Good’s Coverage com valor médio de 0,83
± 0,15.
Na análise de alfa-diversidade, apenas as plantas endêmicas A. alcatrazense
(AA-AL) apresentaram índices estimativos de riqueza e diversidade de bactérias
associadas, significativamente menores do que as demais plantas do gênero
Anthurium (S’ = 160,22; H’ = 3,83) (p < 0,05). Adicionalmente, foi observado que as
plantas coletadas no ambiente de intermédio e mata apresentaram valores maiores
de riqueza e diversidade do que as demais plantas coletadas nos ambientes próximos
ao mar (Figura 2.2A, B).
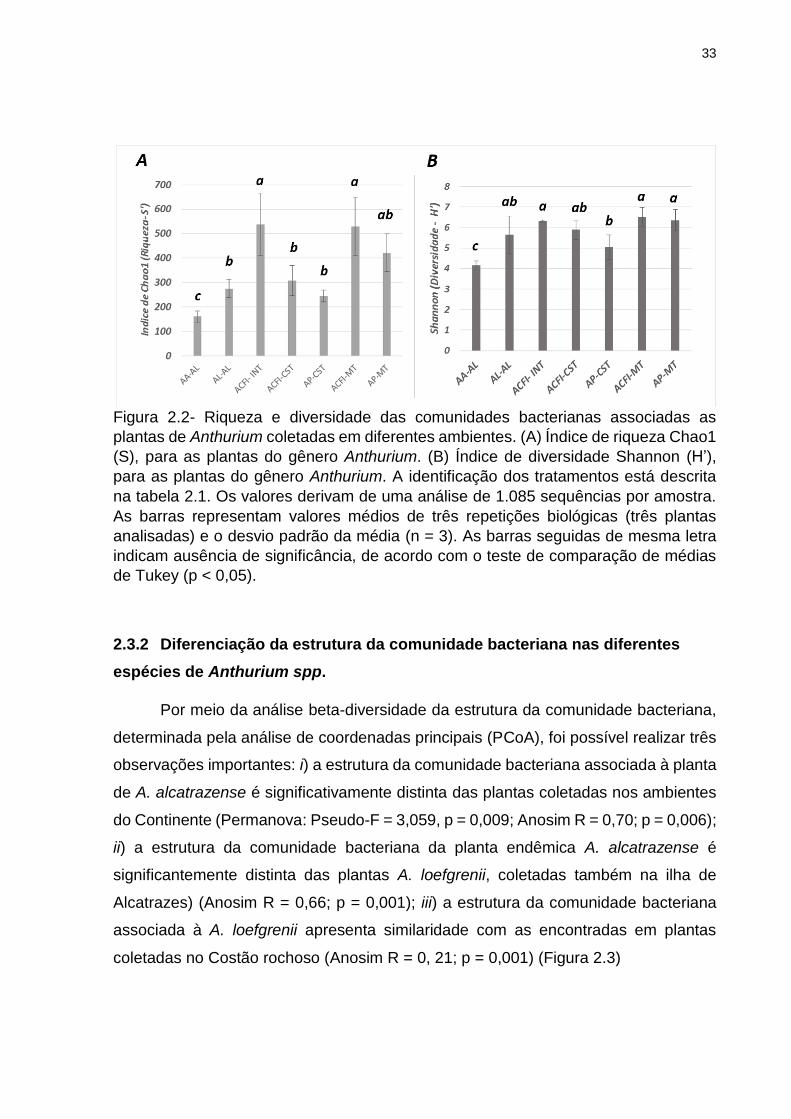
33
Figura 2.2- Riqueza e diversidade das comunidades bacterianas associadas as
plantas de Anthurium coletadas em diferentes ambientes. (A) Índice de riqueza Chao1
(S), para as plantas do gênero Anthurium. (B) Índice de diversidade Shannon (H’),
para as plantas do gênero Anthurium. A identificação dos tratamentos está descrita
na tabela 2.1. Os valores derivam de uma análise de 1.085 sequências por amostra.
As barras representam valores médios de três repetições biológicas (três plantas
analisadas) e o desvio padrão da média (n = 3). As barras seguidas de mesma letra
indicam ausência de significância, de acordo com o teste de comparação de médias
de Tukey (p < 0,05).
2.3.2 Diferenciação da estrutura da comunidade bacteriana nas diferentes
espécies de Anthurium spp.
Por meio da análise beta-diversidade da estrutura da comunidade bacteriana,
determinada pela análise de coordenadas principais (PCoA), foi possível realizar três
observações importantes: i) a estrutura da comunidade bacteriana associada à planta
de A. alcatrazense é significativamente distinta das plantas coletadas nos ambientes
do Continente (Permanova: Pseudo-F = 3,059, p = 0,009; Anosim R = 0,70; p = 0,006);
ii) a estrutura da comunidade bacteriana da planta endêmica A. alcatrazense é
significantemente distinta das plantas A. loefgrenii, coletadas também na ilha de
Alcatrazes) (Anosim R = 0,66; p = 0,001); iii) a estrutura da comunidade bacteriana
associada à A. loefgrenii apresenta similaridade com as encontradas em plantas
coletadas no Costão rochoso (Anosim R = 0, 21; p = 0,001) (Figura 2.3)
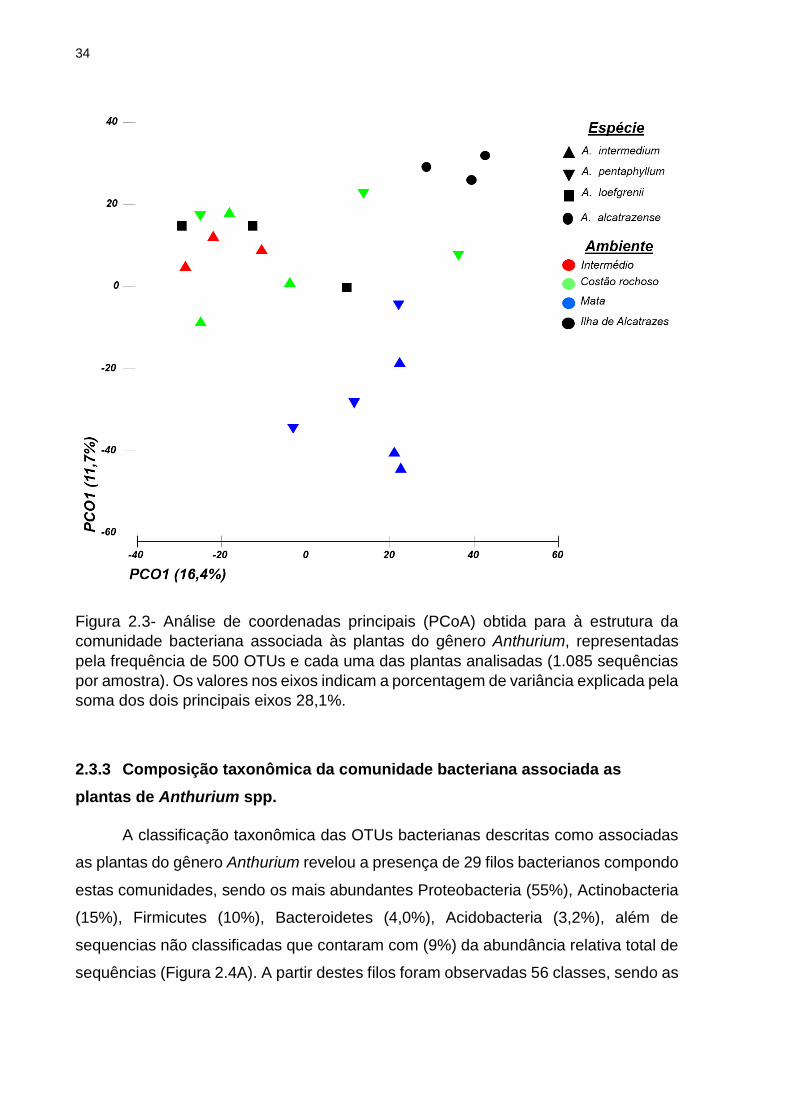
34
Figura 2.3- Análise de coordenadas principais (PCoA) obtida para à estrutura da
comunidade bacteriana associada às plantas do gênero Anthurium, representadas
pela frequência de 500 OTUs e cada uma das plantas analisadas (1.085 sequências
por amostra). Os valores nos eixos indicam a porcentagem de variância explicada pela
soma dos dois principais eixos 28,1%.
2.3.3 Composição taxonômica da comunidade bacteriana associada as
plantas de Anthurium spp.
A classificação taxonômica das OTUs bacterianas descritas como associadas
as plantas do gênero Anthurium revelou a presença de 29 filos bacterianos compondo
estas comunidades, sendo os mais abundantes Proteobacteria (55%), Actinobacteria
(15%), Firmicutes (10%), Bacteroidetes (4,0%), Acidobacteria (3,2%), além de
sequencias não classificadas que contaram com (9%) da abundância relativa total de
sequências (Figura 2.4A). A partir destes filos foram observadas 56 classes, sendo as

35
classes mais abundantes: Gammaproteobacteria (25%); Actinobacteria (18%);
Alphaproteobacteria (13%); Betaproteobacteria (12%); Bacilli (6%); Clostridia (3%) e
Deltaproteobacteria (2%); Bacteroidia (2%) e Acidobacteria (2%). As outras classes
taxonômicas representaram por uma abundância geral menor que 2% (Figura 2.4B).
Destacadamente, as plantas de A. alcatrazense (AA-AL) possuíram uma maior
abundância de Proteobacteria (85%) e menor abundância de Actinobacteria (5%) e
Firmicutes (2%), estes grupos apresentaram ocorrência estatisticamente distinta,
comparado as plantas de A. loefgrenii e as plantas coletadas no continente.
Especificamente, estas últimas não apresentaram diferença estatística sob a
ocorrência dos filos. Considerando a classificação ao nível de classe, foi possível
observar que as frequências relativas de Gammaproteobacteria e Betaproteobacteria
foram de (44%) e (23%) e foram estatisticamente maiores nas plantas de A.
alcatrazense (p < 0,05). As outras classes não apresentaram diferença estatística
quando comparadas entre as plantas de A. alcatrazense e as outras plantas
coletadas.
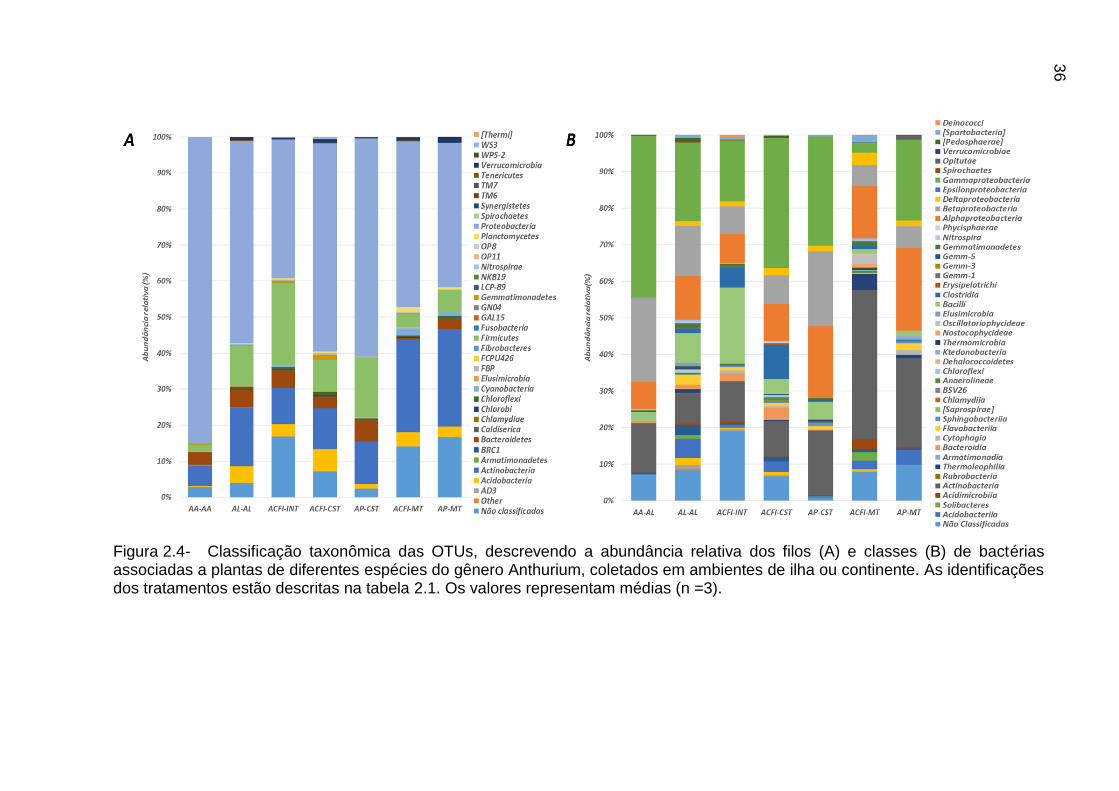
Figura 2.4- Classificação taxonômica das OTUs, descrevendo a abundância relativa dos filos (A) e classes (B) de bactérias associadas a plantas de diferentes espécies do gênero Anthurium, coletados em ambientes de ilha ou continente. As identificações dos tratamentos estão descritas na tabela 2.1. Os valores representam médias (n =3).
36

37
2.3.4 Grupos bacterianos responsáveis pela diferenciação das comunidades
bacterianas associadas à Anthurium spp.
Por meio do teste de Simper, baseado nas inferências da PCoA, foi possível
descrever onze táxons bacterianos principais, o quais, contribuíram com 45% da
dissimilaridade geral (em média) observada entre as plantas de A. alcatrazense das
demais plantas do gênero Anthurium, ou comparando a comunidade de A.
alcatrazense e A. loefgrenii (“t” Student; p< 0,05).
A partir disso, foi possível fazer duas inferências importantes: i) A afiliação
taxonômica destas OTUs revelou a presença de cinco OTUs contribuindo com mais
de 2%, (abundância relativa) para a dissimilaridade entre as plantas de A.
alcatrazense e as plantas coletadas no Continente: denovo 13665
(Betaproteobacteria) (18,2% de contribuição a dissimilaridade), denovo 5450
(Gammaproteobacteria) (10,2%), denovo 8378 (Gammaproteobacteria) (6,2%),
denovo 6870 (Alphaproteobacteria) (4,8%) e denovo 10827 (Gammaproteobacteria)
(2,5%). Por meio do teste de comparação de médias, foi possível observar que apenas
duas dessas OTUs, ocorreram diferencialmente (p < 0,05) nas plantas de A.
alcatrazense e nas plantas coletadas no Continente, denovo 13665
(Betaproteobacteria) e denovo 5450 (Gammaproteobacteria) (Figura 2.5A) ii) A
afiliação taxonômica das OTUs revelou a presença cinco OTUs contribuindo com mais
de 2% para a dissimilaridade entre plantas de A. alcatrazense e A. loefgrenii: denovo
13665 (Betaproteobacteria) (19,2% de contribuição a dissimilaridade), denovo 6870
(Alphaproteobacteria) (6,1%), denovo 5450 (Gammaproteobacteria) (3,0%), denovo
8378 (Gammaproteobacteria) (2,2%), denovo 4168 (Gammaproteobacteria) (2,0%).
Por meio do teste de comparação de médias, foi possível observar que quatro dessas
OTUs, ocorreram diferencialmente (p < 0,05) nas plantas (Figura 2.5B).
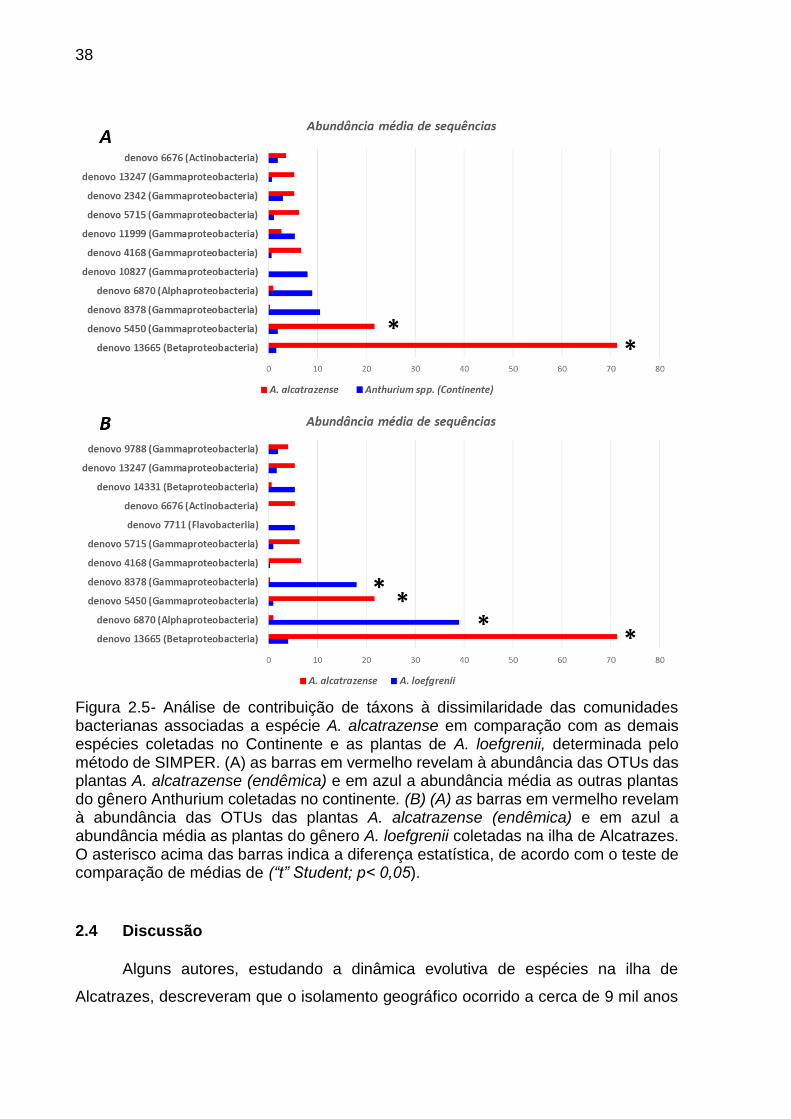
38
Figura 2.5- Análise de contribuição de táxons à dissimilaridade das comunidades bacterianas associadas a espécie A. alcatrazense em comparação com as demais espécies coletadas no Continente e as plantas de A. loefgrenii, determinada pelo método de SIMPER. (A) as barras em vermelho revelam à abundância das OTUs das plantas A. alcatrazense (endêmica) e em azul a abundância média as outras plantas do gênero Anthurium coletadas no continente. (B) (A) as barras em vermelho revelam à abundância das OTUs das plantas A. alcatrazense (endêmica) e em azul a abundância média as plantas do gênero A. loefgrenii coletadas na ilha de Alcatrazes. O asterisco acima das barras indica a diferença estatística, de acordo com o teste de comparação de médias de (“t” Student; p< 0,05).
2.4 Discussão
Alguns autores, estudando a dinâmica evolutiva de espécies na ilha de
Alcatrazes, descreveram que o isolamento geográfico ocorrido a cerca de 9 mil anos

39
(era do Pleistoceno) teve papel importante na especiação dos organismos que
permaneceram na ilha, levando a alterações genotípicas e fenotípicas (Furtado et al.,
1992; Marques et al., 2002b). Dentro desse panorama, Nadruz-Coelho & Catharino
(2008) descreveram as plantas da espécie A. alcatrazense como exemplos deste
processo. Estas plantas possuem características morfológicas peculiares, as quais
diferenciam-nas de outras plantas de mesmo gênero, como tamanho da planta, hábito
heliófilo e epífita. As espécies mais próximas a esta, encontrados em ambientes do
continente, são plantas maiores, habitando ambientes sombreados e sobrevivendo
sobre a serapilheira, características também observadas na própria ilha de Alcatrazes
para a planta A. loefgrenii (Coelho et al., 2004; Almeida et al., 2005; Valadares et al.,
2010; Rocha et al., 2014). Esse padrão de especiação tem sido observado também
para outras espécies de plantas em Arquipélagos (Comes, Tribsch e Bittkau, 2008;
Chase e Myers, 2011). Ainda assim, Marques et al. (2002b) descreveram um processo
de especiação semelhante para outros organismos endêmicos da ilha de Alcatrazes,
correlacionando o tamanho dos organismos com o tipo de nutrição na ilha
Neste mesmo contexto, Nadruz-Coelho & Catharino (2008) sugerem que a
evolução geológica da ilha e as flutuações no nível do mar, gerou um cenário ideal
para estudar os efeitos do isolamento geográfico na especiação alopátrica dirigida por
deriva genética. Por exemplo, no caso de A. alcatrazense, pode-se sugerir que a
limitação nutricional levou a geração de plantas menores, e a busca de uma maior
obtenção de energia via processo de fotossíntese, encontrada pela colonização dos
costões rochosos.
Estudos teóricos e empíricos abordando sistemas de dispersão de
macrorganismos, sugerem que os mesmo processos e parâmetros de distribuição
destes podem ser atribuídos à ocorrência de microrganismos associados a seus
hospedeiros (Martiny et al., 2006; Lankau et al., 2012; Peralta et al., 2014). Este
sistema assume que organismos com distribuição restrita, podem hospedar uma
estrutura de comunidade bacteriana particular. Essas sugestões servem de base para
dar suporte a nossa hipótese de que plantas endêmicas, restritas à determinados
ambientes, hospedam uma estrutura e composição de comunidade bacteriana
particular.

40
Estudos recentes descrevem que a comunidade bacteriana associada a plantas
do gênero Anthurium apresentam uma redução na riqueza e diversidade, quando
comparada com a rizosfera e o solo adjacente, sendo, portanto, ativo o processo
exercido pela planta para selecionar microrganismos do ambiente adjacente para
compor sua comunidade bacteriana, o que deve resultar em benefícios da saúde da
planta (Sarria-Guzmán et al., 2016). Poucos e raros trabalhos foram publicados,
descrevendo a comunidade microbiana associada a plantas do gênero Anthurium.
Sendo assim, este último trabalho corrobora e serve de base para o nosso trabalho,
sugerindo, portanto, que as plantas do gênero Anthurium selecionam microrganismos
parceiros do ambiente, e existe uma especificidade e uma redução de grupos
bacterianos quando avaliado a comunidade bacteriana de acordo com os tecidos das
plantas desde a superfície das raízes até os tecidos mais internos da planta, e até as
folhas. No presente trabalho foi observado que a estimativa da riqueza e da
diversidade bacteriana associada às folhas das plantas endêmicas da espécie A.
alcatrazense, foi significativamente menor que os valores encontrados para a folhas
das plantas encontradas no Continente (Costão rochoso, Intermédio e Mata). Este fato
também ocorreu ao compararmos os valores para a planta endêmica e a outra espécie
do mesmo gênero que ocorre na ilha (A loefgrenii).
Ao desenvolver o nosso trabalho e durante todas as buscas na literatura, não
foram encontrados relatos sobre a distribuição da comunidade bacteriana associada
à A. alcatrazense e A. loefgrenii. Essa falta de estudos comparativos, reforça o
pioneirismo do presente trabalho.
Alguns autores nomeiam o processo de redução da diversidade bacteriana
relacionado a processos evolutivo do hospedeiro endêmico de ilhas localizadas
distantes do continente, como processo de deriva ecológica (Lankau et al., 2012).
Neste processo grupos bacterianos de baixa abundância e não essenciais para
manutenção das condições adaptativas do hospedeiro são extintos, levando a
permanência de um menor número de simbiontes ao longo do tempo (Ezenwa et al.,
2012; Wong et al 2015; Adair et al., 2017). De fato, isso nos leva a acreditar que essa
menor diversidade associada as plantas de A. alcatrazense seja resultado de uma
íntima relação bactéria-planta ao longo do processo de especiação da planta. Esse
processo de associação entre organismos endêmicos e comunidades microbianas, já

41
foi descrito para outras espécies de plantas (Prakamhang et al., 2009; Debnath et al.,
2016).
É conhecido que a interação entre bactérias e plantas é baseada na troca de
benefícios entre os organismos, onde a planta oferece compostos de carbono à
bactéria e as bactérias auxiliam as plantas a sobreviverem, suprindo estas com
nutrientes e proteção. Ainda assim, levando em consideração que ilhas restritas do
continente são considerados ambientes preservados, com baixa taxa de impacto
ambiental (Zilber-Rosenberg & Rosenberg, 2008; Rosindell et al., 2012),
principalmente antropológico, podemos reforçar a ideia de que essa redução de
diversidade bacteriana associada à A. alcatrazense esteja relacionado à um poder
tampão do ambiente, explicando assim que a planta apenas se associa com
microrganismos necessários a sua sobrevivência, sem necessidade de aquisição de
novos microrganismos para auxiliar a tolerância as condições do ambiente (Dinsdale
et al., 2008; Kier et al., 2009; Konopka et al., 2015).
Essas sugestões estão de acordo com os resultados observados no presente
trabalho, no qual plantas coletadas em ambientes mais preservados (ilha de
Alcatrazes e ambiente de Costão rochoso), apresentam relação diretamente
proporcional a menor riqueza e diversidade bacteriana, comparativamente as plantas
coletadas na Mata, pois estas habitam ambientes de alto impacto ambiental,
normalmente com ocorrência de civilização humana (como o ambiente observado na
Mata em Ubatuba São Paulo – Brasil), hospedando assim uma maior riqueza e
diversidade de bactérias.
2.4.1 Relação entre a composição e estrutura da comunidade bacteriana
associada a plantas endêmicas e seus parentes do mesmo gênero.
Neste trabalho nós observamos que a espécie endêmica A. alcatrazense
(Nadruz-Coelho e Catharino, 2008) é colonizada por uma comunidade bacteriana
particular mesmo quando comparada com a espécie A. loefgrenii coletada na mesma
ilha. Estes resultados podem sugerir que a especiação tem levado à um parâmetro de
seleção de microrganismos distintos. Também o mesmo padrão de isolamento

42
geográfico observado para os macrorganismos pode ocorrer para a estrutura da
comunidade bacteriana habitando as folhas dessas plantas, mesmo necessitando de
mais trabalhos baseados na composição gênica desses seres vivos para confirmar a
hipótese de endemismo da comunidade bacteriana associada (Martiny et al., 2006).
Em um estudo recente, Debnath et al., (2016), observaram haver uma comunidade
bacteriana particular associada à Rhododendron arboreum, uma planta medicinal
endêmica do sudeste himalaio. Neste trabalho foi assumido que a distribuição das
plantas endêmicas tem alta correlação com os parâmetros do solo (pH, nitrogênio total
e matéria orgânica), nicho de ocupação e assim concomitantemente ocorre seleção
diferencial da composição da comunidade associada as plantas endêmicas. Esses
padrões determinísticos de distribuição da comunidade bacteriana associada aos
seus hospedeiros, tem sido descrito por muitos autores (Green et al., 2004; Martiny et
al., 2006). Todavia, no nosso modelo de estudo foi observado que plantas não
endêmicas como A. loefgrenii, também coletadas na ilha de Alcatrazes, apresentam
uma comunidade bacteriana similar as plantas coletadas na região do Continente,
mesmo estando distantes uma das outras. Esses resultados rejeitam a hipótese de
distribuição espacial e reforçam a sugestão de que as características genotípicas e
fenotípicas das plantas determinam o perfil da comunidade bacteriana associada.
Também pode-se sugerir que a particularidade da comunidade bacteriana seja um
efeito conjunto do isolamento geográfico e todo o período de especiação e ajuste da
comunidade bacteriana e o desenvolvimento da planta. Essas sugestões corroboram
as pressuposições dos padrões de associação da teoria do hologenoma onde
variações genéticas no holobionte, podem advir de variações genéticas no hospedeiro
e na comunidade bacteriana associada, sendo esta originária de seleção de novas
linhagens de bactérias e/ou transferência horizontal de genes (Dinsdale et al 2008;
Zilber-Rosenberg & Rosenberg, 2013; Moran et al., 2015).
A maior parte dos estudos relacionados a análises da microbiota associada a
plantas têm utilizado apenas um marcador molecular (genes ou regiões intergênicas)
como base para inferir sobre a associação envolvendo a dinâmica de plantas e
microrganismos. Os padrões observados pela identificação e ocorrência dos seres
vivos, têm sido caracterizados como um dos mecanismos para entender processos
naturais evolutivos, relacionados a dinâmica de comunidades e/ou população em um

43
ecossistema (evolução, especiação, imigração, extinção, declínio de populações)
(Lankau et al., 2012; Nemergut et al., 2013 Debnath et al., 2016; Souza et al., 2016).
A maior parte dos estudos envolvendo à associação entre bactérias e plantas
tem utilizado o gene 16S rRNA como marcador molecular padrão para identificação
dos microrganismos, sugerindo que conhecer quem são os microrganismos
envolvidos nesse processo de associação com a planta é o primeiro passo no
entendimento da evolução do estabelecimento das associações (Pommier et al., 2007;
Li et al., 2011; Godinho et al., 2013).
As plantas não podem mais ser consideradas como entidades simples, pois
elas possuem uma grande diversidade de microrganismos associados tanto dentro
como fora de seus tecidos (Vandenkoornhusen et al., 2015). Em uma busca extensiva
pelo entendimento da diversidade bacteriana associada a plantas, foi observado que
96% de um total (n = 7,348 sequências de microrganismos associados a plantas),
estão distribuídos entre os quatro filos mais abundantes (54% Proteobacteria, 20%
Actinobacteria, 16% Firmicutes e 6% Bacteroidetes) (Hardoim et al., 2015; Vorholt et
al., 2012; Bulgarelli et al., 2013). Estudos recentes demonstram que planta da espécie
A. andraeanum são colonizadas por Proteobacteria (39,8% de
Gammaproteobacteria), Firmicutes (26,9%) e Actinobacteria (2,9%) (Sarria-Guzmán
et al., 2016). Nossos resultados estão de acordo com estes resultados citados ao
ponto que, de uma forma geral, foi observado que Proteobacteria responde por 54%
da abundância de bactérias associadas, seguido de Actinobacteria (16%), Firmicutes
(11%), Bacteriodetes (4%), além de terem sido obtidas muitas sequências não
identificadas. De acordo com os procedimentos de qualificação/cura do nosso banco
de dados (Caporaso et al., 2010b), essas sequências não classificadas podem ser
consideradas como sequências ainda não conhecidas, ao invés de sequências de
baixa qualidade. Um alto número de sequências não classificadas também foi
observado por Charnock et al., (2016), estudando ambientes extremos, citando que
essas podem ser oriundas de microrganismos não conhecidos. Ainda assim, essa
diversidade de microrganismos não conhecidos podem se tornar foco de novas
pesquisas (Langarica-Fuentes et al., 2015).

44
Examinando em um nível taxonômico mais profundo, Gammaproteobacteria,
Actinobacteria, Alphaproteobacteria, Betaproteobacteria e Bacilli foram os membros
mais abundantes associados às plantas. Membros dessas classes, supracitadas,
estão associados à uma diversidade de organismos que desempenham relações
ecológicas que variam de mutualísticas à patogênicas, mesmo que em sua maioria
são benéficos aos seus hospedeiros (Pini et al., 2011; Mendes et al., 2011; Hardoim
et al., 2013; Golinska et al., 2015; Klann et al., 2016; Köberl et al., 2017).
Um resultado interessante observado no presente trabalho foi que plantas do
gênero A. alcatrazense possuíram elevada abundância de Proteobacteria,
representado por Gammaproteobacteria e Betaproteobacteria (44% e 23% da
microbiota associada, respectivamente), os quais ocorreram em diferentes
frequências nas demais plantas analisadas. Adicionalmente, foi possível observar que
apenas duas OTUs afiliadas à Betaproteobacteria e Gammaproteobacteria ocorreram
diferencialmente nas plantas de A. alcatrazense comparativamente as plantas
coletadas no continente e a espécie A. loefgrenii, sugerindo que essas sequências
são oriundas de bactérias que estão intimamente associadas as plantas endêmica
desenvolvendo alguma função específica na sobrevivência e adaptação da planta.
Betaproteobacteria e Gammaproteobacteria têm sido identificados como parte
importante do microbioma das plantas (Köberl et al., 2017), normalmente descritos
como antagonistas no biocontrole de patógenos bacterianos (Fürnkranz et al., 2012).
Considerando que as plantas do gênero A. alcatrazense só ocorre na ilha em um local
específico (Nadruz-Coelho e Catharino et al., 2008), sugere-se que essa
biodiversidade, possua um papel crucial na interação e adaptação da planta as
condições do ambiente (Berendsen et al., 2012). Por exemplo, em ambiente agrícolas,
muito organismo pertencente a essas classes tem sido descrito como bactérias
promotoras de crescimento vegetal, atuando como fixadores de nitrogênio,
solublizadores de fosfato, produtores de fito- hormônios e no biocontrole de patógenos
(Kuklinsky et al., 2004) características importantes para a manutenção da
sobrevivência e elevada produção. Por outro lado, outros trabalhos têm descrito que
organismos associados a essas classes, participam do microbioma core de alguns
hospedeiros, sendo assim, participando das funções basais para garantir o
desenvolvimento desses organismos (Horn et al., 2002; Neave et al., 2017).

45
Estudando microbiomas, foi observado que bactérias afiliadas à
Gammaproteobacteria, estão intimamente associados a insetos e plantas
desenvolvendo a processos basais como funções biogênese de células e resposta às
condições de estresse (Hardoim et al., 2011; Bennett et al., 2014). Portanto, esses
trabalhos corroboram as sugestões do nosso resultado ao ponto que, esses
microrganismos são específicos do microbioma dessas plantas endêmicas
desempenhando funções cruciais para a sobrevivência e adaptação na ilha de
Alcatrazes.
Por outro lado, é interessante notar que as plantas coletadas no ambiente de
mata apresentaram uma alta abundância de membros relacionados à classe
Actinobacteria (32,34% em média), em comparação com as plantas coletadas em
outros ambientes. Actinobacteria também é considerada parte importante do
microbioma de plantas (Bulgarelli et al., 2013), contudo diferentemente de
Gammaproteobacteria, são caracterizadas como bactérias Gram-positivas, capazes
de produzir estruturas de resistência e tem sido observado em relativa elevada
abundância em ambientes de Mata Atlântica, essas bactérias desempenham papel
importante na decomposição de materiais complexos e reciclagem de nutrientes
resultantes da formação de húmus. São microrganismos que produzem uma vasta
gama de compostos antimicrobianos (Castillo et al., 2002; Ding et al., 2011) e quando
em associação com a planta são descritos por controlar fungos patogênicos (Kloepper
& Ryu, 2006). Apesar de não ser o foco do presente estudo, a composição das
comunidades de Actinobacteria, descritas neste trabalho podem servir como fonte
para pesquisas futuras de bioprospecção.
Muito trabalhos descrevem que a associação entre plantas e bactérias tem sido
baseada nas características genotípicas do hospedeiro, podendo variar de simbiótica
à mutualística e de comensal à patogênica (Kuklinsky et al., 2004; Mendes et al 2007;
Hardoim et al., 2008; Andreote et al., 2010; Hardoim et al., 2015). A planta, tende a
modular a seleção dos microrganismos para colonizar sua superfície ou seus tecidos
internos de acordo com a sua composição de exsudatos (Vandenkoornhusen et al.,
2015). A composição da exsudação é diferente entre as espécies de plantas, e
variável quando estas são submetidas a condições ambientais distintas (Bulgarelli et
al., 2013). Mendes e colaboradores (2011), trabalhando com solos supressivos e

46
condutivos, demonstraram que em condições de estresses a planta seleciona uma
comunidade específica na rizosfera, para favorecer o controle de patógenos afetando
a planta.
Alguns autores sugerem que as bactérias e plantas desempenham uma relação
simbiótica muito forte e de alta dependência entre ambos (Moran & Sloan. 2015), no
qual, variações nas abundâncias relativas dos grupos microbianos são mecanismos
de adaptação e evolução do holobionte as condições do ambiente (Zilber-Rosenberg
& Rosenberg, 2008; Vandenkoornhusen et al., 2015; Köberl et al., 2017). Neste
mesmo contexto, nossos resultados demonstraram que a maior parte dos grupos de
bactérias (OTUs), estão presentes em todas as plantas analisadas. Contudo, estes
ocorrem em abundâncias distintas revelando a dissimilaridade entre as comunidades,
principalmente relacionadas às OTUs afiliadas as classes Gammaproteobacteria,
Alphaproteobacteria e Betaproteobacteria. Esses resultados que indicam que plantas
submetidas a processos evolutivos distintos caracterizado por ambientes
geograficamente isolados um do outro, tendem a selecionar microrganismos distintos
como forma de melhor se adaptar as condições limitantes.
Conclui-se, portanto que plantas que habitam regiões restritas, particularmente
plantas do gênero A. alcatrazense, hospedam uma estrutura e composição da
comunidade bacteriana associada específica. Esta característica de particularidade
pode ser oriunda de processos bióticos e abióticos naturais de especiação e
adaptação do genótipo dos microrganismos- genótipo do hospedeiro ao ambiente,
podendo sugerir que podem uma melhor seleção de microrganismos que auxiliam a
sobrevivência e adaptação hospedeiro as condições vigentes podem dirigir a
associação simbiótica. Também pode-se concluir que Betaproteobacteria e
Gammaproteobacteria podem ser grupos, bacterianos associados, (97%
similaridade), de relativa importância para os processos metabólicos das plantas de
A. alcatrazense, pois estes apresentaram abundância maior que as plantas de A.
loefgrenii coletadas na mesma ilha.
Assim, por fim pode-se sugerir que uma abordagem exploratória de
comunidades bacterianas associadas a planta endêmicas, pode desvendar padrões
de associação bactérias-planta importantes e não conhecidos. Ainda assim, podem

47
ser utilizados como embasamento teórico para trabalhos cujo o objetivo é estudar
processos genéticos dessa associação, assim como para bioprospecção de
metabólitos inéditos, importantes para as áreas de biotecnologia.
Referências
Adair KL, Douglas AE. (2017). Making a microbiome: the many determinants of host-associated microbial community composition. Current Opinion in Microbiology. 35: 23-29.
Andreote FD, da Rocha UN, Araujo WL, Azevedo JL, van Overbeek LS. (2010). Effect of bacterial inoculation, plant genotype and developmental stage on root-associated and endophytic bacterial communities in potato (Solanum tuberosum). Antonie van Leeuwenhoek. 97: 389-399.
Bennett GM, McCutcheon JP, MacDonald BR, Romanovicz D, Moran NA. (2014). Differential genome evolution between companion symbionts in an insect-bacterial symbiosis. Molecular Biology. Bio 5(5):e01697-14.
Berendsen RL, Pieterse CMJ, Bakker P. (2012). The rhizosphere microbiome and plant health. Trends Plant Science 17:478–486
Bisseling T, Dangl J L, and Schulze-Lefert P. (2009). Next-generation communication. Science 324:691.
Bulgarelli D, Rott M, Schlaeppi K, van Themaat EVL, Ahmadinejad N, et al. (2012). Revealing structure and assembly cues for Arabidopsis root- inhabiting bacterial microbiota. Nature. 488: 91-95.
Caporaso JG, Bittinger, K, Bushman FD, DeSantis TZ, Andersen GL, et al. (2010a) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. Canada 26: 266–267.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello. E, K et al. (2010b) QIIME allows analysis of high-throughput community sequencing data. Nature Methodologies. 7: 335-336
Castillo UF, Strobel GA, Ford EJ, Hess WM, Porter H, Jensen JB, Albert H, Robison R, Condron MAM, Teplow DB, Stevens D, Yaver D. (2002). Munumbicins, wide-spectrum antibiotics produced by Streptomyces NRRL 30562, endophytic on Kennedia nigriscans. Microbiology. 148:2675–2685.
Charnock C, Nordille AL. (2016). Proteobacteria, extremophiles and unassigned species dominate in tape-like showrhead biofilm. Brazilian Journal of Microbiology. 47: 345-351.
Debnath R, Yadav A, Gupta VK, Singh BP, Handique PJ, Saikia R. (2016). Rhizospheric bacterial community of Endemic Rhododendron arboreum Sm. Ssp. Delavayi along Eastern Himalayan slope in Tawang. Frontiers in plant science 7: 1345.

48
DeSantis TZ, Hugenholtz P, Larsen, N, Rojas M, Brodie, EL, et al. (2006). Greengenes, a chimera-checked 16 S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology 72: 5069–5072
Dias ACF, Costa FEC, Andreote FD, Lacava PT, Teixeira M A, Assumpção LC, Araújo WL, Azevedo JL, Melo I S. (2009) Isolation of micropropagated strawberry endophytic bacteria and assessment of their potential for plant growth promotion. World Journal of Microbiology and Biotechnology, 189-195.
Ding L, Maier A, Fiebig H-H, Lin W-H, Hertweck C. 2011. A family of multicyclic indolosesquiterpenes from a bacterial endophyte. Organic Biomolecular Chemistry 9:4029–4031.
Dinsdale EA, Edwards RA, Hall D et al. (2008) Functional metagenomics profiling of nine biomes. Nature 452: 629–632.
Easson CG. Thacker RW (2014). Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Frontiers in Microbiology. 5(532)
Edgar RC, Haas BJ, Clemente J.C, Quince C, Knight R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16): 2194-2200,
Edgar RC. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19). 2460-2461.
Ezenwa VO, Gerardo NM, Inouye DW, Medina M, Xavier JB: Animal behavior and the microbiome. Science 2012, 338:198-199.
Godinho VM, Furbino LE, Santiago IF, Pelizzare FM, et al. (2013). Diversity and bioprospecting of fungal communities associated with endemic and cold-adapted macroalgae in Antarctica. The ISME Journal. 7: 1434-1451.
Golinska P, Wypij M, Agarkar G, Rathod D, Dahm H, Rai M. (2015). Endophytic actinobacteria of medicinal plants: diversity and bioactivity. Antonie van Leeuwenhoek. 108: 267-289.
Green J, Bohannan BJM. (2006). Spatial scaling of microbial biodiversity. Trends in Ecology and Evolution. 21(9): 501-507.
Hammer O, Harper DAT, Ryan PD. (2001). PAST: paleontological statistics software package for education and data analysis. Palaeontologica Electronica 4:1–9
Hardoim PR, van Overbeek LS, Berg G, Pirttillä AM, et al. (2015). The hidden world within plants: Ecological and Evolutionary considerations for defining Functioning of |Microbial Endophytes. Microbiology and Molecular Reviews. 79(3): 293-320.
Hardoim PR, van Overbeek LS, van Elsas JD. (2008). Properties of bacterial endophytes and their proposed role in plant growth. Cell press. 16(10): 463-471
Kavamura VN. Taketani RG. Lançoni MD, Andreote FD, Mendes, R, Melo IS. (2013)
Water regime influences bulk soil and Rhizosphere of Cereus jamacaru bacterial
communities in the Brazilian Caatinga biome. PLoS One, 8(9) e73606.
doi:10.1371/journal.pone.0073606

49
Kier G, Kreft H, Lee TM, Jetz W, Ibisch PL, Nowichi C, Mutke J, Bethlott W. (2009). Global diversity assessment of endemism and species richness across island and mainland regions. Proceedings of National Academy of Science. 106(23): 9322-9327.
Klann J, Mchenry A, Mentelongo C, Goffredi SK. (2016). Decomposition of plant-sourced carbon compounds by heterotrophic betaproteobacteria isolated from tropical Costa Rica bromeliad. Microbiology Open. 5(3): 479–489.
Kloepper JW, Ryu CM. 2006. Bacterial endophytes as elicitors of induced systemic resistance, p 33–52. In Schulz BJE, Boyle CJC, Sieber TN (Ed), Microbial root endophytes. Springer-Verlag, Berlin, Germany.
Köberl M, Dita M, Martinuz A, Staver C, Berg G. (2017). Members of Gammaproteobacteria as indicator species of healthy banana plants on Fusarium wilt-infested fields in Central America. Scientifc Reports. 7:45318. doi: 10.1038/srep45318
Konopka A, Lindemann S, Fredrickson J. (2015). Dynamics in microbial communities: unraveling mechanisms to identify principles. The ISME Journal. 9: 1488-1495.
Kuklinsky-Sobral J, Araújo WL, Mendes R, Geraldi IO, Pizzirani-Kleiner AA, Azevedo JL. (2004). Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environmental microbiology, 6(12): 1244–51
Lankau EW, Hong PY, Mackie RI. (2012). Ecological drift and local exposures drive enteric bacterial community differences within species of Galápagos iguanas. Molecular Ecology. 21: 1779-1788.
Liu G, Chater KF, Chandra G, Niu G, Tan H. 2013. Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiology Molecular Biology Reviews 77:112–143.
Mamede MCH, Silva SJG, Jacques EL, Arenque BC, (2012) In Flora Fanerogâmica do Estado de São Paulo. São Paulo: Instituto de Botânica, 132p
Marques OAV, Martins M, Sazima I. (2002b). A new insular species of pitviper from Brazil, with comments on evolutionary Biology and conservation of Bothrops jararaca group (Serpentes, VIperidae), Herpetologica. 58(3): 303-312.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Martiny, J. B. H. et al. Microbial biogeography: putting microorganisms on the map. Nature reviews. Microbiology, v. 4, n. 2, p. 102–12, fev. 2006.
Mendes R, Kruijt, M, De Bruijn, I et al (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 332:1097–1100.
Mendes, R, Pizzirani-Kleiner AA, Araujo WL, Raaijmakers JM. (2007). Diversity of cultivated endophytic bacteria from sugarcane: genetic and biochemical characterization of Burkholderia cepacia complex isolates. Applied and environmental microbiology, 73(22): 7259–67.
Mendes R, Garbeva P, Raaijmakers JM. 2013. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiology Reviews 37: 634-663.

50
Moran NA, Sloan DB (2015). The Hologenome Concept: Helpful or Hollow? PLoS Biology 13(12): e1002311.
Nadruz-Coelho MA, Catharino ELM. (2008). Duas espécies de Anthurium (Araceae) endêmicas do Litoral de São Paulo, Brasil. Rodriguésia. 59(4): 829-833.
Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinsky TM, et al. (2013). Patterns and processes of microbial community assembly. Microbiology and Molecular Reviews 77(3): 342-356.
Pedrós-Alió C. (2006). Marine microbial diversity: can it be determined? Trends in Microbiology. 14(6): 257-263.
Peralta AL, Mathews JW, Kent AD. (2014). Habitat specialization along a wetland moisture gradient differs between Ammonia-oxidizing and denitrifying microorganisms. Microbial Ecology. doi 10.1007/s00248-014-0407-4.
Phillippot L, Raaijmakers JM, Lemanceau P, van der Putten WH. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nature Reviews in Microbiology. 11: 780-799.
Pini F, Galardini M, Bazzicalupo M, Mengoni A. (2011). Plant-Bacteria Association and symbiosis: Are there common genomic traits in Alphaproteobacteria. Genes. 2: 1017-1032.
Pommier T, Canbäck B, Riemann L, Boström KH, et al. (2007). Global patterns of diversity and community stricture in marine bacterioplankton. 16: 867-880.
Prakamhang J, Minamisawa K, TeamTaisong K, Bookerd N, Teaumroong N. (2009). The communities of endophytic diazotrophic bacteria in cultivated rice (Oryza sativa L.). Applied Soil Ecology. 42: 141-149.
Rocha LCF, Smidt EC, Nadruz-Coelho MA, Temponi LG. (2014). The genus Anthurium (Araceae) in Parana State, Brazil
Rosindell J, Hubbell SP, He F, Harmon LJ, Etienne RS. (2012). The case for ecological neutral theory. Cell Press. 27(2): 203-208.
Sarria-Guzmán Y, Chávez-Romero Y, Gómez-Acata S, Montes-Molina A, et al (2016). Bacterial communities associated with different Anthurium andraeanum L.
Shi YW, Yang H, Zhang T, Sun J, Lou K. (2014). Illumina-based analysis of endophytic bacterial diversity and space-time dynamics in sugar beet on the north slope of Tianshan Mountain. Applied Microbiology and Biotechnology. DOI: 10.1007/s00253-014-5720-9.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM.; Herndl
GJ. (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”.
Proceedings of the National Academy of Sciences of the USA. 103(32): 12115-12120.
Souza DT, Genuário DB, Silva FSP, Pansa CC, et al. (2017). Analysis of the bacterial composition in marine sponges reveals the influence of host phylogeny and environment. FEMS Microbiology Ecology. 93(1): doi: 10.1093/femsec/fiw204.

51
Temponi LG, Nadruz-Coelho MA. (2011). Two new species of Anthurium sect.. Urospadix (Araceae) for Brazil. Rodriguésia. 62(2): 315-320.
Thiers B, Index herbarium: A global directory of public herbaria and associated staff. New York: Botanical Garden’s Virtual Herbarium. Available in: http://sweetgum.nybg.org/ih/. Access in: April 15th 2017
Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, Poole PS (2013). Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. The ISME Journal. 7:2248-2258.
Van Elsas JD, Tam, L.; Finlay RD, Killham K, Trevors JT. (2006); Microbial interactions in soil. In: Van Elsas, J.D.; Jansson JK, Trevors JT. Plant-Bacteria associated lifestyle and Molecular interactions. 2nded.NewYork: CRC press, p 177-204.
Vandenkoornhunyse P, Quaiser, A, Duhamed, M, Le Van, A, Defresne A. (2015). The importance of the microbiome of the plant holobiont. New Phytologist. doi: 10.1111/nph.13312
Vorholt JA. (2012). Microbial life in the phyllosphere. Nature Reviews in Microbiology 10:828–840. http://dx.doi.org/10.1038/nrmicro2910.
Wang Y, Qian P-Y. (2009) Conservative fragments in bacterial 16S rRNA genes and
primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One,
4(10). e7401, 2009.
Zilber-Rosenberg I and Rosenberg. E. (2008); Role of microrganisms in the evolution of animals and plants: the hologenomic theory of evolution. FEMS Microbiology reviews. 32: 723-735.

52

53
3 IMPORTÂNCIA DA FILOGENIA E DO AMBIENTE NA COMPOSIÇÃO DA COMUNIDADE BACTERIANA ASSOCIADA A CIANOBACTÉRIAS
Resumo
O sucesso evolutivo das cianobactérias sempre foi atribuído a suas características genômicas. Contudo, estudos recentes demonstraram que as cianobactérias sempre estiveram associadas a outras bactérias, as quais participavam da ecologia destes organismos, podendo ser cruciais na ocupação de diferentes habitats. No entanto, pouco se sabe sobre a associação entre cianobactérias e outras bactérias, principalmente considerando cianobactérias oriundas de ambientes tropicais. Para isso, foram estudadas, por meio do sequenciamento massivo do gene 16S rRNA, as comunidades de bactérias associadas a diferentes linhagens dos gêneros Brasilonema, Cylindrospermopsis, Leptolyngbya, Limnothrix, Microcystis e Nostoc. Primeiramente, foi observada uma correlação maior entre a estruturação da comunidade microbiana e a filogenia dos isolados estudados do que entre o local de isolamento e o meio de cultivo, onde estas foram mantidas em cultivo. Posteriormente, estudando apenas os isolados afiliados ao gênero Microcystis, foi demonstrada a variação na comunidade ao longo das fases de cultivo dos isolados, caracterizada por maior dominância de grupos bacterianos na fase estacionária do cultivo. Adicionalmente, foram encontradas distinções na composição da comunidade bacteriana quando as cianobactérias foram submetidas a condições de cultivo limitantes na concentração de nitrogênio, alteração da luminosidade e alteração na temperatura. Foi possível observar que os tratamentos com alteração de luminosidade obtiveram maiores valores de densidade ótica para o cultivo, resultando em uma estruturação de comunidade bacteriana distinta. Em conjunto, estas observações indicam uma íntima associação entre as cianobactérias e a comunidade bacteriana associada a estas.
Palavras-chave: Associação microrganismo-microrganismos; Cianobactérias-bactérias; Sequenciamento Ion Torrent; Gene 16S rRNA
Abstract
The evolutionary success of cyanobacteria has always been attributed to their genomic characteristics. However, recent studies have shown that cyanobacteria have always been associated with other bacteria, which participate in the ecology of these organisms, and may be crucial to occupy different niches. However, little is known about the association between cyanobacteria and other bacteria, especially considering cyanobacteria from tropical environments. For this, we studied the bacterial communities associated with different strains of the genera Brasilonema, Cylindrospermopsis, Leptolyngbya, Limnothrix, Microcystis and Nostoc, through the massive sequencing of the 16S rRNA gene. Firstly, there was a greater correlation between the structure of the microbial community and the phylogeny of the isolates studied than between the isolation site and the culture medium in which they were kept in culture. Subsequently, only the isolates belonging to the genus Microcystis were used to study, the variation in the community during the cultivation phases of the

54
isolates was characterized by a greater dominance of bacterial groups in the stationary phase of culture. In addition, distinctions were found in the composition of the bacterial community when cyanobacteria were submitted to limiting culture conditions in nitrogen concentration, alteration of luminosity and temperature change. It was possible to observe that the treatments with alteration of luminosity obtained higher values of optical density for the culture, resulting in a structuring of different bacterial communities. Taken together, these observations indicate an intimate association between cyanobacteria and the bacterial community associated with them.
Keywords: Association microorganisms-microorganisms; Cyanobacteria-bacteria; Ion Torrent Sequencing; 16S rRNA gene
3.1 Introdução
As cianobactérias são organismos procarióticos, uni e multicelulares
pertencentes ao filo Cyanobacteria, considerado como um grupo de organismos, que
possuem uma longa história evolutiva (Whitton and Potts, 2000). A origem desses
microrganismos data de cerca de 3,5 bilhões de anos (Schopf and Walter, 1982),
período no qual a atmosfera terrestre era livre de oxigênio. Evolutivamente, sugere-se
que essas bactérias tiveram papel fundamental na mudança da composição
atmosférica terrestre, por possuir a capacidade de realizar a fotossíntese oxigênica
(Whitton and Potts, 2000; Blankenship, 2002; Bekker et al 2004). Além disso,
espécimes desse grupo também são capazes de realizar à fotossíntese não oxigênica
(Shilo, 1980), resistir à presença de metais, baixa disponibilidade de oxigênio
(Robinson et al., 2000) e altas concentrações de sulfureto (Padan & Cohen, 1982;
Cohen et al., 1986); além de alguns grupos poderem usar H2S como doador de
elétrons (Cohen et al., 1975) e serem fixadores de nitrogênio (Farnelid et al., 2010).
Essas estratégias de crescimento, e a versatilidade metabólica, foram algumas das
particularidades que garantiram o sucesso evolutivo das cianobactérias (Garcia-Pichel
& Pringault, 2001, Schirrmeister et al., 2011; Schirrmeister et al., 2013).
Esses organismos podem ser encontrados habitando quase todos os
ambientes terrestre, desde ambientes comuns, tais como água doce, solo e ambientes
epifíticos, até ambientes extremos, como águas termais, solos congelados, solos com
alta temperatura, solos com faixa limites de pH, entre outros (Whitton & Potts, 2000;
Komárek et al., 2012; Rigonato et al., 2013; Hoff-Risseti et al., 2013; Andreote et al.,
2014; Genuário et al., 2015).

55
Por muitos anos, alguns autores sugeriram que o sucesso evolutivo das
cianobactérias em habitar ambientes distintos estaria apenas relacionado às
características fisiológicas e metabólicas e estruturais da própria cianobactéria (Paerl
et al., 1996; Sanchez-Baracaldo et al., 2005; Genuário et al., 2013). Contudo, alguns
trabalhos posteriores descreveram a comunidade bacteriana associada a estas
cianobactérias, a qual anteriormente era considerada como contaminante dos cultivos
destes organismos (Abed et al., 2005; Sánchez et al., 2005). A ocorrência destes
organismos associados de forma ubíqua sugere sua participação no metabolismo das
cianobactérias, e indica a possível ocorrência de importantes processos de
complementação entre a célula das cianobactérias e as demais bactérias a essas
associadas (Farnelid et al., 2010). Esse fato trouxe luz à importância da compreensão
da interação mutualística entre cianobactérias e bactérias associadas (Al-Hasan et al.,
2002; Brauer et al 2015). Atualmente, sabe-se que essas bactérias possuem papel
fundamental como facilitadoras do desenvolvimento das cianobactérias frente a
condições estressantes abióticas e bióticas, promovendo o ajustes metabólico do
hospedeiro; enquanto as cianobactérias, por sua vez, fornecem abrigo em suas
estruturas celulares e nutrição aos simbiontes, por meio de compostos orgânicos
simples derivados da fotossíntese e nitrogênio oriundo da fixação biológica (Sanchez
et al., 2005; Leloup et al., 2009; Parveen et al., 2013).
Estudos recentes trouxeram grandes avanços na compreensão dos fatores
ecológicos que modulam a interação entre cianobactérias e bactérias heterotróficas.
Trabalhos distintos mostraram que: (i) as cianobactérias são “hot-spots” para o
desenvolvimento e atividade de comunidades bacterianas associadas (Worm and
Sondergaard, 1998); (ii) a diversidade associada à cianobactérias em geral é
composta por membros dos grupos Alphaproteobacteria, Betaproteobacteria,
Bacteroidetes, Actinobacteria, CFB (Cytophaga-Flavobacterium-Bacteroides) e
Verrucomicrobia (Berg et al., 2009); (iii) o perfil bacteriano pode ser modulado pelo
tipo de toxina produzida pela cianobactéria (Kolmonen, Sivonen, Rapala, & Haukka,
2004); (iv) a estrutura, composição e diversidade da comunidade bacteriana
associada são distintas da comunidade bacteriana presente no ambiente adjacente à
cianobactéria (Parveen et al., 2013); (v) a estrutura da comunidade associada também
é variável de acordo com o hospedeiro e as condições ambientais (Dziallas &

56
Grossart, 2011; Bagatini et al., 2014; Louati et al., 2015; Zhu et al., 2016). Apesar
desses estudos recentes, informações sobre bactérias heterotróficas associadas a
cianobactérias ainda são escassas, principalmente quando consideradas
cianobactérias de ambientes tropicais e linhagens cultivadas. Portanto, as hipóteses
deste estudo são: (a) a estrutura e composição das comunidades de bactérias
associadas às cianobactérias cultivadas são determinadas pela filogenia do
hospedeiro; (b) essa comunidade se altera em relação as fases de desenvolvimento
do cultivo das cianobactérias; e (c) alterações nas condições de cultivo das
cianobactérias promovem alterações no seu crescimento e consequentemente na
estrutura da comunidade bacteriana associada. Para tanto, três objetivos foram
delineados: (1°) correlacionar a composição das comunidades de bactérias
associadas às cianobactérias aos grupos taxonômicos/distâncias filogenéticas
analisadas e aos locais de origem dos isolados estudados; (2°) determinar a
diferenciação da comunidade bacteriana nas fases de desenvolvimento de uma
cultura de cianobactéria; (3°) verificar alterações na composição das comunidades
bacterianas devido a alterações nas condições de cultivo das cianobactérias
3.2 Material e Métodos
3.2.1 Linhagens de cianobactérias utilizadas
Foi realizada uma seleção de 18 linhagens de cianobactérias, compondo 6
gêneros, dos quais foram escolhidas 3 linhagens representantes para cada gênero,
oriundos de diferentes ambientes (Tabela 3.1). Todas estas linhagens foram
previamente identificadas filogeneticamente com base no sequenciamento do gene
16S rRNA. Estas sequências foram utilizadas no presente estudo para calcular as
distâncias filogenéticas entre as linhagens, determinada pelo número de substituição
de bases, derivado da aplicação do modelo de máximo verossimilhança sobre o
alinhamento das sequências realizado no software MEGA 7 (Kumar et al., 2016).
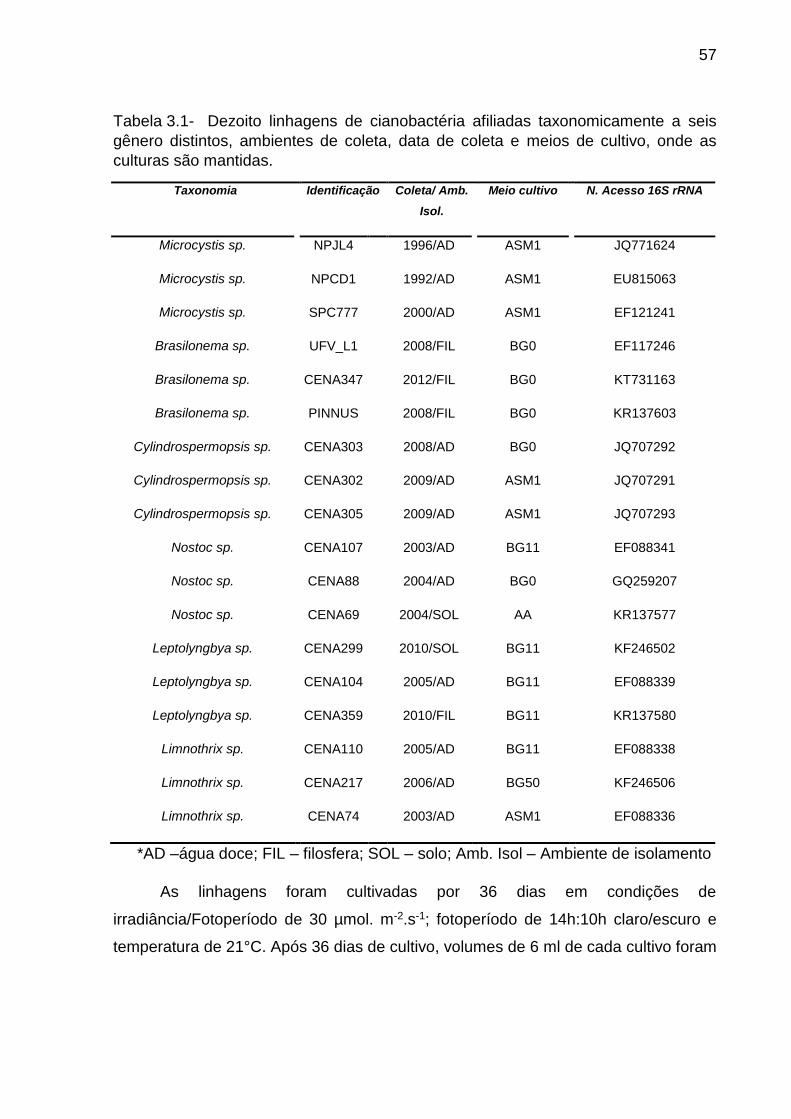
57
Tabela 3.1- Dezoito linhagens de cianobactéria afiliadas taxonomicamente a seis
gênero distintos, ambientes de coleta, data de coleta e meios de cultivo, onde as
culturas são mantidas.
Taxonomia Identificação Coleta/ Amb.
Isol.
Meio cultivo N. Acesso 16S rRNA
Microcystis sp. NPJL4 1996/AD ASM1 JQ771624
Microcystis sp. NPCD1 1992/AD ASM1 EU815063
Microcystis sp. SPC777 2000/AD ASM1 EF121241
Brasilonema sp. UFV_L1 2008/FIL BG0 EF117246
Brasilonema sp. CENA347 2012/FIL BG0 KT731163
Brasilonema sp. PINNUS 2008/FIL BG0 KR137603
Cylindrospermopsis sp. CENA303 2008/AD BG0 JQ707292
Cylindrospermopsis sp. CENA302 2009/AD ASM1 JQ707291
Cylindrospermopsis sp. CENA305 2009/AD ASM1 JQ707293
Nostoc sp. CENA107 2003/AD BG11 EF088341
Nostoc sp. CENA88 2004/AD BG0 GQ259207
Nostoc sp. CENA69 2004/SOL AA KR137577
Leptolyngbya sp. CENA299 2010/SOL BG11 KF246502
Leptolyngbya sp. CENA104 2005/AD BG11 EF088339
Leptolyngbya sp. CENA359 2010/FIL BG11 KR137580
Limnothrix sp. CENA110 2005/AD BG11 EF088338
Limnothrix sp. CENA217 2006/AD BG50 KF246506
Limnothrix sp. CENA74 2003/AD ASM1 EF088336
*AD –água doce; FIL – filosfera; SOL – solo; Amb. Isol – Ambiente de isolamento
As linhagens foram cultivadas por 36 dias em condições de
irradiância/Fotoperíodo de 30 µmol. m-2.s-1; fotoperíodo de 14h:10h claro/escuro e
temperatura de 21°C. Após 36 dias de cultivo, volumes de 6 ml de cada cultivo foram

58
concentrados por centrifugação à 20.000 x g por 10 min. O material precipitado foi
armazenado à – 20 ºC para obtenção do DNA.
3.2.2 Extração do DNA e sequenciamento massivo da região V6 do gene 16S
rRNA e análises das sequências
Para obtenção do DNA genômico (cianobactéria e bactérias heterotróficas),
1,5mL de solução tampão (Tris-HCl 10mM + EDTA 1mM, pH 8,0) foi adicionado aos
precipitados, os quais foram então submetidos à um choque térmico, utilizando
nitrogênio líquido e banho aquecido (70° C), repetido por 3 vezes. A partir da
suspensão formada procedeu-se a extração do DNA genômico, utilizando o kit Power
Soil DNA Extraction (MoBio Laboratories, Carslab, CA), seguindo as instruções do
fabricante.
A integridade do DNA extraído, assim como sua quantificação foi avaliada por
meio de eletroforese em gel de agarose a 1,0% (m/v) em tampão TAE (400 mM Tris,
20 mM ácido acético glacial, 1mM EDTA), onde foram aplicados 5µl dos DNAs
extraídos junto a 3µl de um tampão de corrida Loading buffer 6x (Azul de bromofenol
0,05% (p/v); Sacarose 40% (p/v); EDTA 0,1M (pH 8,0); SDS 0,025% (p/v). Após a
eletroforese, o gel foi corado em solução de brometo de etídeo e visualizado em luz
ultravioleta (DNr Bio-imaging Systems Minibis pro 16mm).
Os procedimentos de sequenciamento massivo da região V6 do gene 16S
rRNA, análise das sequências obtidas, análises alfa e beta-diversidade e taxonomia
da estrutura e composição de grupos bacterianos associados à cianobactéria foram
realizadas como descrito no item 2.2.4, 2.2.5 e 2.2.6 presentes no capítulo 2. As
principais diferenças entre os estudos estão no fato de que não houve uma
amplificação (reação de PCR inicial), para eliminação do 16S rRNA Cloroplastidial e
os bancos de sequências obtidos no presente trabalho, foram filtrados em relação ao
filo “Cyanobacteria”. Estas sequências foram removidas e apenas as afiliadas a outros
filos bacterianos foram utilizadas nas análises posteriores.

59
3.2.3 Análise de correlação entre a filogenia das cianobactérias e a estrutura da
comunidade bacteriana associada
Para validar a conexão entre dos grupos bacterianos em associação com os
determinados gêneros de cianobactéria, foi determinada a correlação entre as
matrizes de distâncias filogenéticas entre os isolados de cianobactérias e a matriz de
dissimilaridade das comunidades bacterianas associadas (Kembel et al., 2010;
Easson and Tracker, 2014). O teste de correlograma de Mantel foi realizado utilizando
a matriz de distância de Bray-Curtis da comunidade bacteriana e a matriz de distância
filogenética entre as linhagens de cianobactéria, utilizadas no presente estudo. Os
dados foram previamente transformados em Log (X+1) para se adequarem à
normalidade.
3.2.4 Variação das comunidades bacterianas associadas ao longo do cultivo
das cianobactérias
Para esta análise foram utilizadas quatro linhagens de Microcystis aeruginosa
(NPCD-1; NPJL-4; SPC 777; SPC 759). Inicialmente, foi definida uma curva padrão
de multiplicação celular para estas linhagens, inoculando (2 mL; 2,86 mL; 1,66 mL;
3,46 mL) do cultivo das cianobactérias, respectivamente, com a finalidade de ajustar
os volumes de inóculo de acordo com a densidade óptica (680nm) de cada isolado.
Posteriormente, as linhagens foram mantidas em câmara de cultivo sob
irradiância/fotoperíodo de 30 µmol. m-2.s-1; 14:10h claro/escuro e temperatura de
21°C. Estas linhagens foram cultivadas por um período de 43 dias. A densidade ótica
(OD) foi determinada em espectrofotômetro à 680nm em intervalos de 3 em 3 dias
para determinação da curva padrão de multiplicação de cada isolado.
Com base nestas curvas, foram definidos ao longo do cultivo das cianobactérias,
três pontos de amostragem: fase Lag (10 dias), fase Log (36 dias) e fase estacionária
(43 dias). Em cada uma destas fases, cada uma das quatro culturas, realizadas em 3
repetições, foram amostradas, onde 6 ml foram coletados e submetidos aos
procedimento de extração do DNA, como descrito acima nos itens (3.2.2),
posteriormente, os procedimentos de sequenciamento massivo da região V6 do gene

60
16S rRNA, análise das sequências obtidas, e análises alfa e beta-diversidade e
taxonomia da estrutura e composição de grupos bacterianos associados à
cianobactéria foram realizadas como descrito no item 2.2.4, 2.2.5 e 2.2.6 presente no
capítulo 2, e descrita anteriormente no item 3.2.2.
3.2.5 Variações nas condições de cultivo das cianobactérias
No intuito de verificar as variações nas comunidades bacterianas associadas
quando as cianobactérias experimentam diferentes condições ambientais, foram
alteradas algumas condições de cultivo dos isolados alvos de estudo e as
comunidades bacterianas presentes em cada um dos tratamentos foram então
estudadas. As condições de cultivo foram alteradas em quantidade de nitrogênio,
irradiância/luminosidade e temperatura (Tabela 3.2).
Tabela 3.2- Cinco condições distintas de cultivos para as quatro linhagens de M.
aeruginosa. A variação das condições de cultivo, foram baseadas nas condições
controle de cultivo, descrita no tratamento C1 (Control).
Tratamento Meio de Cultura Irradiância
(µmol.fóton.m-2.s-1) Temperatura
(°C)
(Control) - Controle BG11 30 21
(10% N) BG11 10% Nitrogênio 30 21
(50% N) BG11 50% Nitrogênio 30 21
(60 light) BG11 60 21
(25°C ) BG11 30 25
Previamente ao início do experimento, as quatro linhagens de M. aeruginosa
foram aclimatadas nas condições de cada tratamento por um período de 36 dias, até
atingirem o estágio de maior desenvolvimento celular, compreendendo o final da fase
Log. Amostras da fase de aclimatação foram coletadas para servir de inóculo para o
desenvolvimento do experimento. Além da amostragem que deu origem ao inóculo,
aos 36 dias de cultivo foram realizadas amostragens em todas as condições de cultivo
distintas, gerando um total de 84 amostras.
Da mesma maneira, seis mililitros de cada cultivo foram coletados e
processados conforme descrito nos itens (3.2.2; 3.2.4).

61
3.3 Resultados
3.3.1 Alfa e beta-diversidade das comunidades bacterianas associadas aos
gêneros de cianobactérias
Os valores de estimativos dos índices de diversidade (Riqueza, Diversidade e
Dominância) demonstraram não haver diferenças significativas, considerando a
médias dos tratamentos (n = 3), entre as comunidades associadas aos gêneros de
cianobactérias (Tabela 3.3).
Tabela 3.3- Descrição da estimativa de alfa-diversidade, valore dos índices de Chao1, Shannon e Simpson, para os gêneros de cianobactérias analisados. Os valores indicam as médias (n = 3) seguidas dos valores de desvio padrão. A letras indicam diferenças estatística (p< 0,05) entre os gêneros.
Gênero Chao1 (S’) Shannon (H’) Simpson (1-D’)
Brasilonema spp. 293,62 (20,36) a 3,89 (0,37) a 0,84 (0,04) a
Cylindrospermopsis spp. 376,63 (129,29) a 3,77 (0,98) a 0,80 (0,11) a
Leptolyngbya spp. 372,40 (74,31) a 3,86 (0,14) a 0,84 (0,02) a
Limnothrix spp. 334,03 (122,82) a 3,51 (1,36) a 0,81 (0,15) a
Microcystis spp. 354,97 (223,00) a 2,94 (1,49) a 0,67 (0,23) a
Nostoc spp. 199,89 (121,81) a 2,04 (1,05) a 0,46 (0,32) a
Na análise de beta-diversidade, representada pela PCoA, foi possível observar
que a estrutura da comunidade bacteriana é distinta entre os gêneros de
cianobactérias analisados (Permanova = 1,55; p= 0,001: Anosim; R = 0,70; p=
0,0001). Contudo, alguma sobreposição ocorre principalmente entre os gêneros
Brasilonema, Nostoc, Leptolyngbya, e Limnothrix, ao passo que os gêneros
Microcystis e Cylindrospermopsis apresentaram a maior dissimilaridade, dentre os
analisados
Não foi observado separação significativa quando considerado o meio de
cultivo original no qual a linhagem é mantida sendo cultivada desde o período de sua
coleta no ambiente (Permanova; Pseudo-F = 1,06, p= 0,226; Anosim; R = 0,10; p=
0,222), e também não houve uma separação das comunidades com validade
estatística para os ambientes de origem das linhagens (Permanova; Pseudo-F = 0,66,
p= 0,126; Anosim; R = 0,04; p= 0,32) (Figura 3.1).
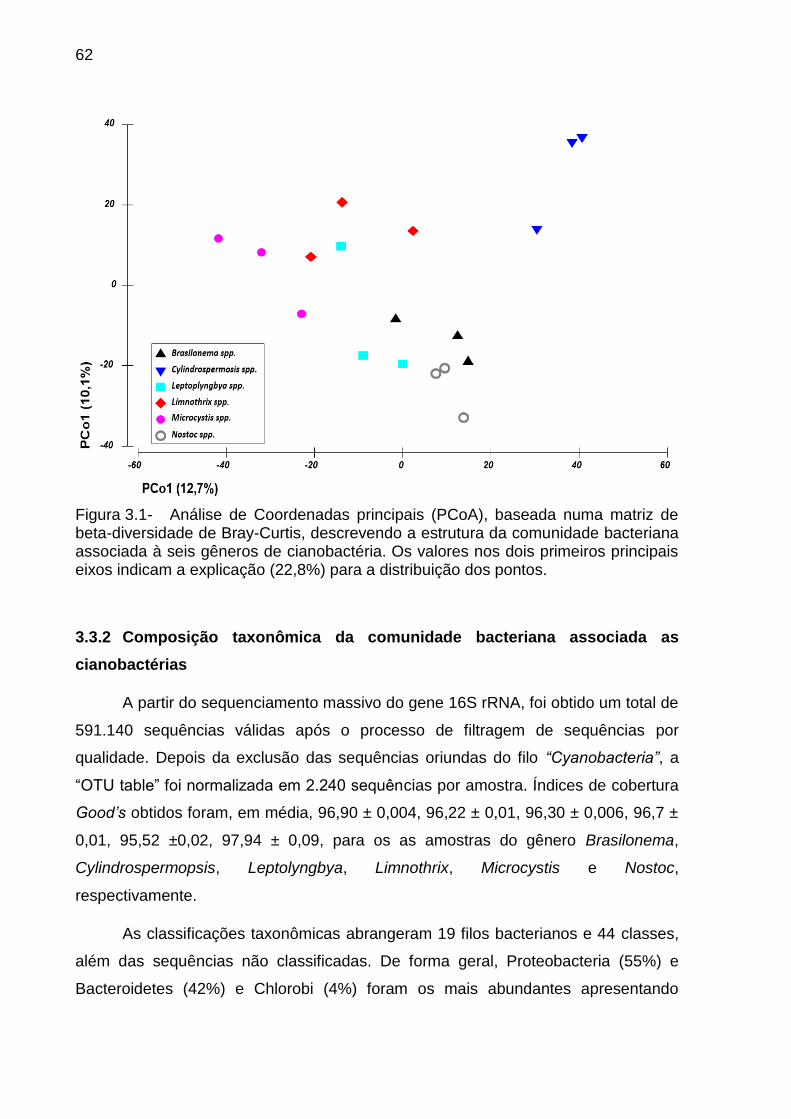
62
Figura 3.1- Análise de Coordenadas principais (PCoA), baseada numa matriz de beta-diversidade de Bray-Curtis, descrevendo a estrutura da comunidade bacteriana associada à seis gêneros de cianobactéria. Os valores nos dois primeiros principais eixos indicam a explicação (22,8%) para a distribuição dos pontos.
3.3.2 Composição taxonômica da comunidade bacteriana associada as
cianobactérias
A partir do sequenciamento massivo do gene 16S rRNA, foi obtido um total de
591.140 sequências válidas após o processo de filtragem de sequências por
qualidade. Depois da exclusão das sequências oriundas do filo “Cyanobacteria”, a
“OTU table” foi normalizada em 2.240 sequências por amostra. Índices de cobertura
Good’s obtidos foram, em média, 96,90 ± 0,004, 96,22 ± 0,01, 96,30 ± 0,006, 96,7 ±
0,01, 95,52 ±0,02, 97,94 ± 0,09, para os as amostras do gênero Brasilonema,
Cylindrospermopsis, Leptolyngbya, Limnothrix, Microcystis e Nostoc,
respectivamente.
As classificações taxonômicas abrangeram 19 filos bacterianos e 44 classes,
além das sequências não classificadas. De forma geral, Proteobacteria (55%) e
Bacteroidetes (42%) e Chlorobi (4%) foram os mais abundantes apresentando

63
abundância acima de 2%. Considerando ao nível taxonômico de classe observou-se
que os principais grupos associados aos gêneros de cianobactérias analisados foram
Alphaproteobacteria (35%), Betaproteobacteria (17%), Gammaproteobacteria (11%),
Flavobacteria (10%), Cytophaga (7%), Sphingobacteria (5%), Saprospirae (4%) e
OPB56 (4%). As demais classes apresentaram abundância relativa menor do que 2%
(Figura 3.2A).
Em relação à distribuição dos grupos mais abundantes, ao nível de classe, foi
possível observar que não houve diferença significativa para Alphaproteobacteria.
Contudo, Betaproteobacteria foi mais abundante nos isolados pertencentes aos
gêneros Cylindrospermopsis, Leptolyngbya e Microcystis (27%; 28%; 34%) (p< 0,05)
comparativamente à abundância em Brasilonema, Limnothrix, Nostoc (5,0%; 4,4%;
1,2%). De forma análoga, sequências afiliadas a classe Gammaproteobacteria foram
mais abundantes em Brasilonema, Limnothrix, Nostoc (16%; 11%; 30%),
Flavobacteria foi mais abundante em Brasilonema, Cylindrospermopsis e Nostoc
(11%; 22%; 28%). A classe Cytophagia foi mais abundante em Brasilonema,
Leptolyngbya e Nostoc (11%;11%;13%). Sphingobacteria foi mais abundante em
Limnothrix e Leptolyngbya (11%; 17%), a classe OPB56 foi mais abundante em
Microcystis (18%) (Figura 3.2B). Não houveram diferenças significativas para a classe
Saprospirae.
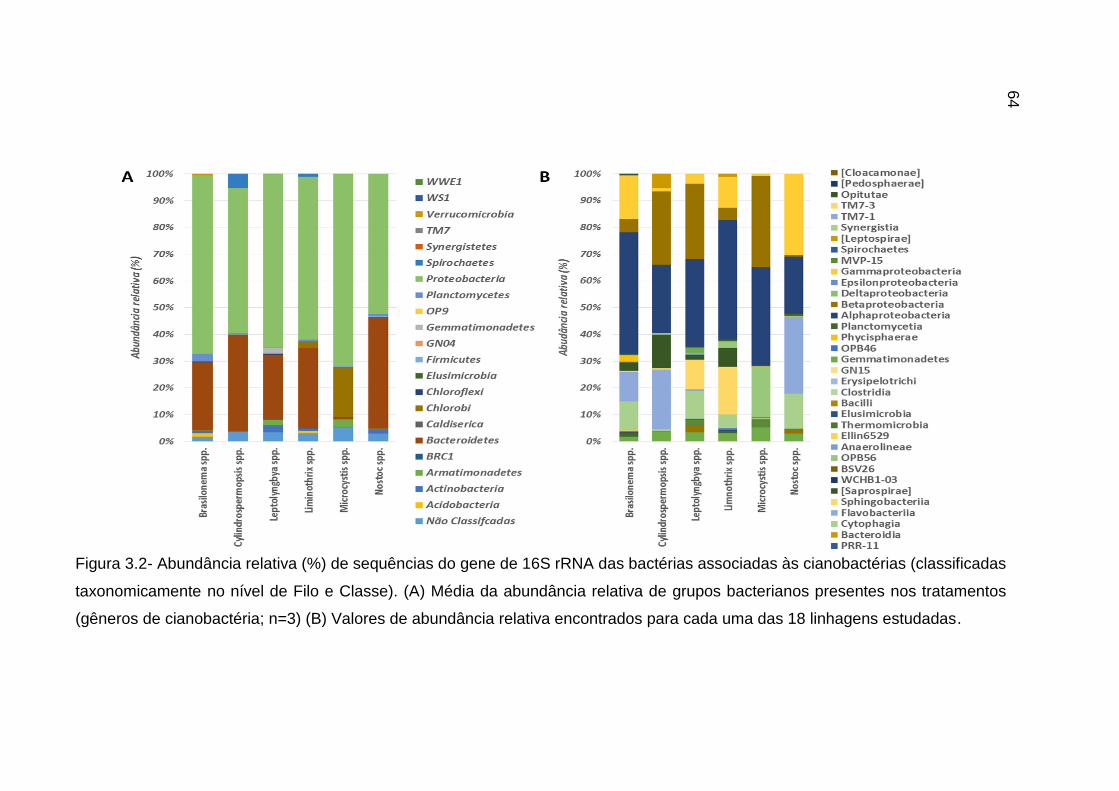
64
Figura 3.2- Abundância relativa (%) de sequências do gene de 16S rRNA das bactérias associadas às cianobactérias (classificadas
taxonomicamente no nível de Filo e Classe). (A) Média da abundância relativa de grupos bacterianos presentes nos tratamentos
(gêneros de cianobactéria; n=3) (B) Valores de abundância relativa encontrados para cada uma das 18 linhagens estudadas.

65
Ao examinarmos a composição da comunidade bacteriana associada em nível
taxonômico mais profundo, como por exemplo ao nível de OTUs (geradas a 97% de
similaridade), identificamos a ocorrência de alguns grupos específicos em cada
gênero de cianobactéria (Figura 4). Por exemplo, as OTUs denovo3883
(Betaproteobacteria), denovo 6182 (OPB56), são mais abundantemente
representadas por sequências oriundas de Microcystis em relação aos demais
gêneros avaliados; denovo4083 (Flavobacteria) e denovo2073
(Gammaproteobacteria) foram mais representativas no gênero Nostoc e denovo5473
(Betaproetobacteria) foi mais representativa no gênero Cylindrospermopsis (Figura
3.3). De maneira geral, cada gênero de cianobactéria apresentou OTUs que foram
específicas e em elevada abundância.
Figura 3.3- A abundância de sequências afiliadas as principais OTUs influenciando a
dissimilaridade entre as comunidades bacterianas associadas aos diferentes gêneros
de cianobactéria, foi descrita com base no teste de Simper. A soma da contribuição
dessas OTUs correspondeu à 90% da dissimilaridade total. Os valores indicam a
abundância de sequências, e a significância dos testes estatísticos (p < 0,05). As
cores indicam uma escala de abundância de sequências, rosa escuro alta abundância
variando até azul escuro baixa abundância.

66
3.3.3 Relação filogenética entre cianobactérias e a estrutura da comunidade
bacteriana associada
Por meio de um teste de Mantel as distâncias filogenéticas entre as linhagens
de cianobactérias foram correlacionadas com a dissimilaridade das estruturas das
comunidades bacterianas associadas. Foi encontrada uma correlação positiva entre
a filogenia dos hospedeiros e a estrutura da comunidade bacteriana associada (p =
0,002). Usando o teste de correlograma de Mantel, observou-se que a correlação foi
confirmada para curtas distâncias filogenéticas entre os hospedeiros (R² = 0,669; p =
0,002), ou seja, quanto menor as distâncias filogenéticas entre as linhagens, maior a
explicação da estruturação da comunidade bacteriana associadas as cianobactérias
(Figura 3.4). Estes resultados indicam que existe dissimilaridade significativa de
acordo com o gênero da cianobactéria e possivelmente com a espécie de
cianobactéria.
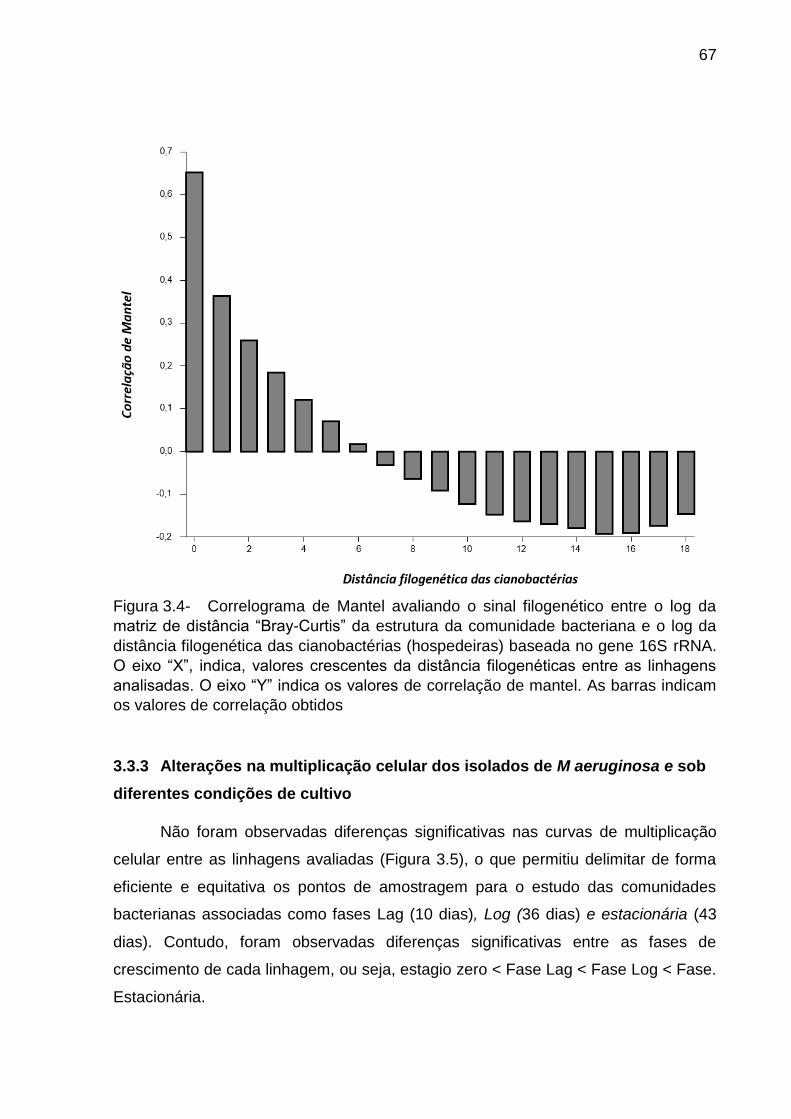
67
Figura 3.4- Correlograma de Mantel avaliando o sinal filogenético entre o log da
matriz de distância “Bray-Curtis” da estrutura da comunidade bacteriana e o log da
distância filogenética das cianobactérias (hospedeiras) baseada no gene 16S rRNA.
O eixo “X”, indica, valores crescentes da distância filogenéticas entre as linhagens
analisadas. O eixo “Y” indica os valores de correlação de mantel. As barras indicam
os valores de correlação obtidos
3.3.3 Alterações na multiplicação celular dos isolados de M aeruginosa e sob
diferentes condições de cultivo
Não foram observadas diferenças significativas nas curvas de multiplicação
celular entre as linhagens avaliadas (Figura 3.5), o que permitiu delimitar de forma
eficiente e equitativa os pontos de amostragem para o estudo das comunidades
bacterianas associadas como fases Lag (10 dias), Log (36 dias) e estacionária (43
dias). Contudo, foram observadas diferenças significativas entre as fases de
crescimento de cada linhagem, ou seja, estagio zero < Fase Lag < Fase Log < Fase.
Estacionária.
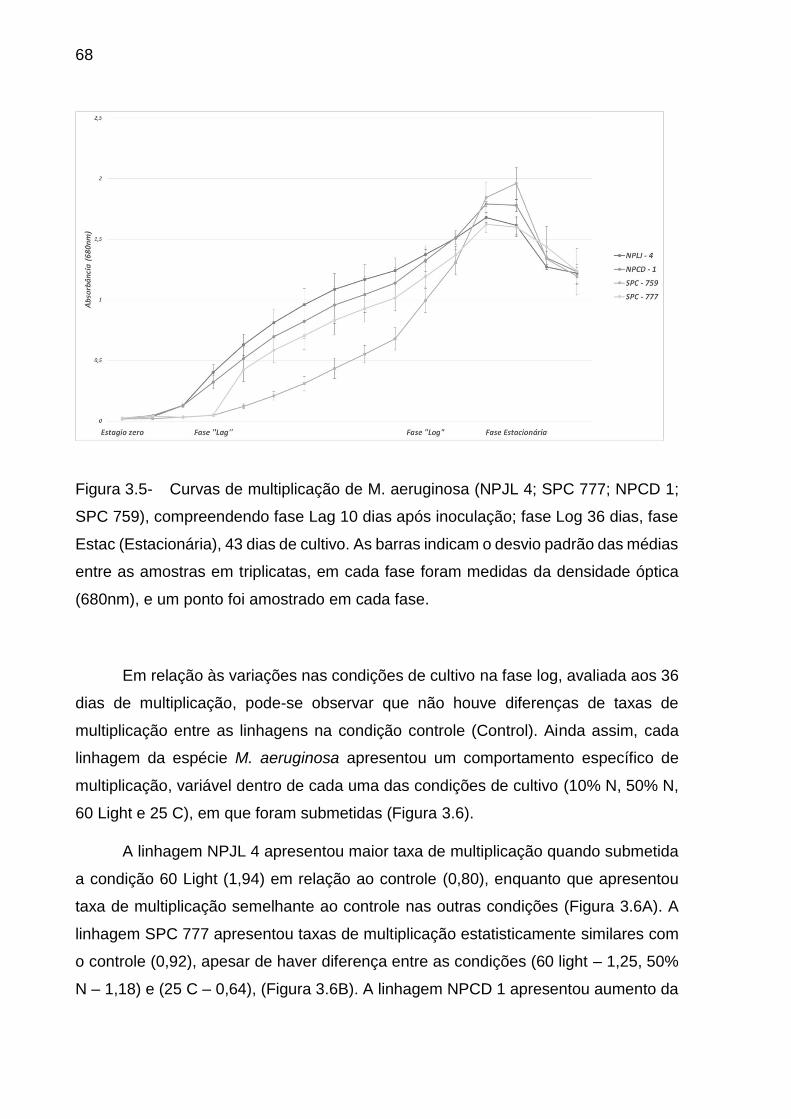
68
Figura 3.5- Curvas de multiplicação de M. aeruginosa (NPJL 4; SPC 777; NPCD 1;
SPC 759), compreendendo fase Lag 10 dias após inoculação; fase Log 36 dias, fase
Estac (Estacionária), 43 dias de cultivo. As barras indicam o desvio padrão das médias
entre as amostras em triplicatas, em cada fase foram medidas da densidade óptica
(680nm), e um ponto foi amostrado em cada fase.
Em relação às variações nas condições de cultivo na fase log, avaliada aos 36
dias de multiplicação, pode-se observar que não houve diferenças de taxas de
multiplicação entre as linhagens na condição controle (Control). Ainda assim, cada
linhagem da espécie M. aeruginosa apresentou um comportamento específico de
multiplicação, variável dentro de cada uma das condições de cultivo (10% N, 50% N,
60 Light e 25 C), em que foram submetidas (Figura 3.6).
A linhagem NPJL 4 apresentou maior taxa de multiplicação quando submetida
a condição 60 Light (1,94) em relação ao controle (0,80), enquanto que apresentou
taxa de multiplicação semelhante ao controle nas outras condições (Figura 3.6A). A
linhagem SPC 777 apresentou taxas de multiplicação estatisticamente similares com
o controle (0,92), apesar de haver diferença entre as condições (60 light – 1,25, 50%
N – 1,18) e (25 C – 0,64), (Figura 3.6B). A linhagem NPCD 1 apresentou aumento da
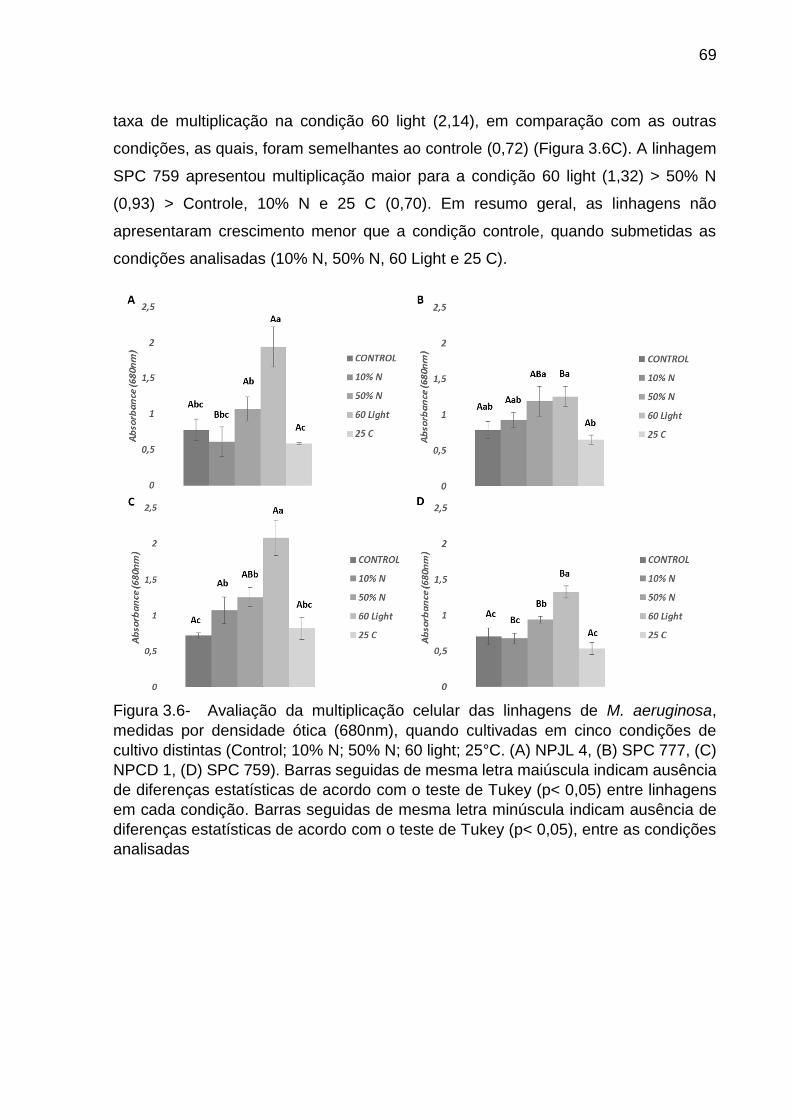
69
taxa de multiplicação na condição 60 light (2,14), em comparação com as outras
condições, as quais, foram semelhantes ao controle (0,72) (Figura 3.6C). A linhagem
SPC 759 apresentou multiplicação maior para a condição 60 light (1,32) > 50% N
(0,93) > Controle, 10% N e 25 C (0,70). Em resumo geral, as linhagens não
apresentaram crescimento menor que a condição controle, quando submetidas as
condições analisadas (10% N, 50% N, 60 Light e 25 C).
Figura 3.6- Avaliação da multiplicação celular das linhagens de M. aeruginosa,
medidas por densidade ótica (680nm), quando cultivadas em cinco condições de
cultivo distintas (Control; 10% N; 50% N; 60 light; 25°C. (A) NPJL 4, (B) SPC 777, (C)
NPCD 1, (D) SPC 759). Barras seguidas de mesma letra maiúscula indicam ausência
de diferenças estatísticas de acordo com o teste de Tukey (p< 0,05) entre linhagens
em cada condição. Barras seguidas de mesma letra minúscula indicam ausência de
diferenças estatísticas de acordo com o teste de Tukey (p< 0,05), entre as condições
analisadas

70
3.3.4 Valores de alfa diversidade das comunidades bacterianas associadas a
M. aeruginosa ao longo de seu desenvolvimento e sob distintas condições de
cultivo
O sequenciamento massivo do 16S rRNA utilizado nesta etapa do estudo gerou
um total de 1.072.848 sequências válidas do gene 16S rRNA após trimagem por
qualidade. Depois da exclusão das sequências oriundas do filo “Cyanobacteria”, a
OTU table foi normalizada em 4.240 sequências por amostra. Os índices de cobertura
de Good’s foram: NPJL 4 - 96,0 ± 0,04, SPC 777 - 96,56 ± 0,01, NPCD 1 - 97,30 ±
0,006, SPC 759 - 95,52 ±0,02.
Considerando as fases de desenvolvimento das cianobactérias, pode-se
observar que a dinâmica diversidade bacteriana foi diferente para cada linhagem de
cianobactéria, refletido pelos índices de Chao1, Shannon e Simpson. A linhagem
NPJL 4 apresentou menor de riqueza de grupos bacterianos no início do
desenvolvimento fase lag, seguido do aumento dos valores de Chao1 para a fase log
e estacionária, esse aumento foi também observado para os índices de Shannon e
Simpson. A linhagem SPC 777, também apresentou menores valores dos índices de
Chao1 e Shannon, Simpson nas primeiras fases de multiplicação, comparado com os
valores maiores na fase estacionária. A linhagem NPCD 1, apresentou valores de
Chao1, Shannon e Simpson mais similares entre as fases de multiplicação (p< 0,05).
A linhagem SPC 759, apresentou valores de Chao1, Shannon e Simpson maiores na
primeira fase em comparação com a fase estacionária (p< 0,05) (Tabela 3.4). De
forma geral pode-se observar uma relação positiva dos valores do índice de Shannon
(dominância de grupos bacterianos) com o desenvolvimento das culturas das
cianobactérias (R = 0,66; p< 0,003).
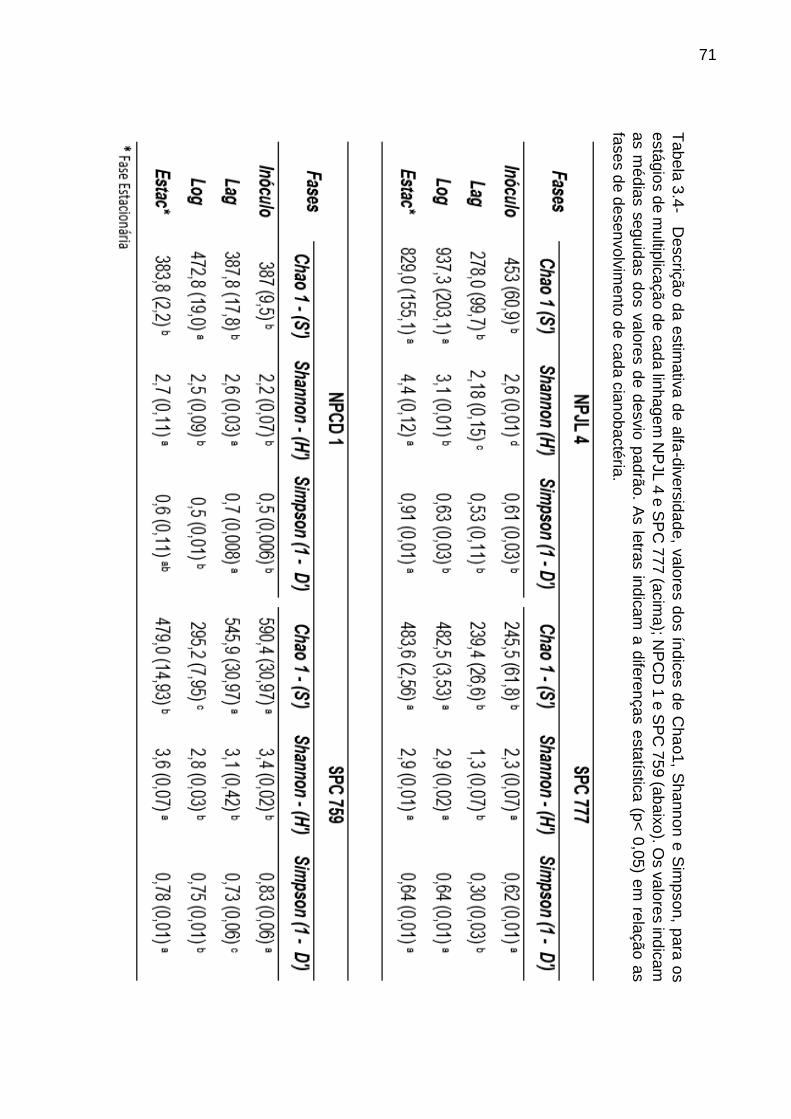
71
Tab
ela
3.4
- D
escriç
ão
da e
stim
ativ
a d
e a
lfa-d
ive
rsid
ade
, va
lore
s d
os ín
dic
es d
e C
ha
o1
, Sh
ann
on
e S
imp
son
, pa
ra o
s
está
gio
s d
e m
ultip
licaçã
o d
e c
ada
linha
ge
m N
PJL
4 e
SP
C 7
77
(acim
a); N
PC
D 1
e S
PC
75
9 (a
ba
ixo
). Os v
alo
res in
dic
am
as m
éd
ias s
egu
ida
s d
os v
alo
res d
e d
esvio
pa
drã
o. A
s le
tras in
dic
am
a d
ifere
nça
s e
sta
tístic
a (p
< 0
,05
) em
rela
çã
o a
s
fase
s d
e d
esen
vo
lvim
en
to d
e c
ad
a c
ian
ob
acté
ria.

72
Considerando as condições de cultivos as quais as cianobactérias foram
submetidas, foi possível observar diferenças significativas entre os valores dos índices
de riqueza (Chao1 – S’), diversidade (Shannon - H’) e dominância (Simpson – 1-D’)
da comunidade bacteriana associada, comparativamente com a comunidade
bacteriana associada na condição controle. Não houveram diferenças estatísticas
para os índices de diversidade analisados para a linhagem NPJL 4, em relação ao
controle. Contudo, para a linhagem SPC 777, foi observado uma diversidade e
dominância estatisticamente (p < 0,05) maior ao controle quando submetida à
condição 25° C (Tabela 3.5).
Para as linhagens NPCD 1 e SPC 759 apenas foram observados valores de
Shannon e Simpson maiores ou iguais nas condições 10% N; 50% N; 60 light; 25°C
em relação ao controle (Tabela 3.5).
Estes resultados demonstram que cada linhagem de cianobactéria apresenta
padrão distinto de relação ecológica com a diversidade bacteriana associada. Ainda
assim, analisando comparativamente os índices de diversidade e as taxas de
multiplicação celular destas linhagens (Figura 3.6), os resultados indicam que, em
situações adversas ao controle, como as quais os hospedeiros foram submetidos, a
cianobactéria promove uma modulação da diversidade bacteriana favorecendo o
desenvolvimento de mais grupos de bactérias a se tornarem dominantes.

73
C
on
diç
õe
s
NP
JL
4
S
PC
777
C
ha
o 1
- (S')
Sh
an
no
n -
(H')
Sim
ps
on
(1 -
D')
C
ha
o 1
- (S')
Sh
an
no
n -
(H')
Sim
ps
on
(1 -
D')
C
on
trol
80
4,6
2 (2
55
,3) a
b 2
,83 (0
,6) a
b 0
,75 (0
,03
) a
6
37
,65
(91
,9) a
1,9
4 (0
,28
) b
0,4
3 (0
,05
) b
c
1
0%
N
88
5,4
6 (4
66
,2) a
b 3
,46 (0
,94
) a
b 0
,78 (0
,04
) a
4
24
,79
(97
,3) a
b 1
,36 (0
,14
) c 0
,33 (0
,03
) c
5
0%
N
90
4,6
4 (5
98
,3) a
2,5
4 (0
,52
) b
0,5
7 (0
,09
) b
3
69
,36
(13
0) a
b 1
,99 (0
,66
) b
c 0
,48 (0
,18
) bc
6
0 lig
ht
28
0,5
(108
) b 3
,13 (0
,53
) a
b 0
,78 (0
,01
) a
2
88
,05
(12
0,4
) b
2,0
6 (0
,36
) b
0
,58 (0
,14
) b
2
5 ° C
5
93
,61
(23
8,8
) a
b 3
,61 (0
,31
) a
0,7
5 (0
,17
) a
6
45
,81
(19
8) a
3,1
3 (0
,26
) a
0,7
7 (0
,00
8) a
C
on
diç
õe
s
NP
CD
1
S
PC
759
C
ha
o 1
- (S')
Sh
an
no
n -
(H')
Sim
ps
on
(1 -
D')
C
ha
o 1
- (S')
Sh
an
no
n -
(H')
Sim
ps
on
(1 -
D')
C
on
trol
53
4,6
1 (3
20
) a
2,4
4 (0
,12
) b 0
,59 (0
,02
) c
3
63
,51
(11
8) a
b 2
,84 (0
,01
3) d
0,7
5 (0
,01
) b
1
0%
N
33
6,6
1 (2
1,8
) a
3,2
7 (0
,32
) a
0,7
8 (0
,04
) a
b
47
6,5
0 (7
0) a
3,2
0 (0
,02
) c 0
,74 (0
,02
) b
5
0%
N
45
7,5
1 (1
08
) a
3,1
4 (0
,3) a
0,7
7 (0
,01
) b
2
46
,61
(27
) b
2,5
5 (0
,01
6) e
0,7
1 (0
,01
) b
6
0 lig
ht
63
7,1
6 (1
58
) a
3,4
7 (0
,28
) a
0,8
0 (0
,01
) a
2
65
,17
(75
,5) b
3
,69 (0
,09
) a
0,7
6(0
,06
) a
b
2
5 ° C
8
97
,19
(69
7,9
) a
3,4
76
(0,5
) a
0,7
7 (0
,17
) a
bc
3
45
,70
(73
) ab
3,2
7 (0
,01
) b 0
,81 (0
,01
) a
Tab
ela
3.5
- D
escriç
ão
da e
stim
ativ
a d
e a
lfa-d
ive
rsid
ad
e, v
alo
res d
os ín
dic
es d
e C
ha
o1
, Sh
an
non
e S
imp
son
, pa
ra a
co
mu
nid
ade
ba
cte
rian
a a
sso
cia
da
à N
PJL 4
e S
PC
77
7 (a
cim
a); N
PC
D 1
e S
PC
75
9 (a
ba
ixo
), su
bm
etid
as a
s c
inco c
on
diç
õe
s d
e c
ultiv
o d
istin
tas
(Co
ntro
l; 10
% N
; 50
% N
; 60
ligh
t; 25
°C). O
s v
alo
res in
dic
am
as m
éd
ias s
egu
ida
s d
os v
alo
res d
e d
esvio
pa
drã
o. A
s le
tras in
dic
am
a
dife
ren
ça
s e
sta
tístic
a (p
< 0
,05
) em
rela
ção
as fa
se
s d
e d
ese
nvo
lvim
ento
de
ca
da c
iano
ba
cté
ria.

74
A composição das comunidades bacterianas associadas aos isolados de M.
aeruginosa é variável de acordo com a fase de crescimento destes organismos. As
classificações taxonômicas das sequências geradas encontraram 19 filos e 44
classes, além das sequências não classificadas. Dentro desse panorama, a
composição das comunidades bacterianas foi descrita com base nas classes que
apresentaram abundância relativa geral ≥ 2%. Para a linhagem NPJL 4
(Betaproteobacteria 55%; Alphaproteobacteria 33%; OPB56 6% e Fimbriimonada 3%)
(Figura 3.7A) foram as mais abundantes. Para a linhagem SPC 777
(Betaproteobacteria 58%; Alphaproteobacteria 24% e Flavobacteria 15% e
Actinobacteria 2%) (Figura 3.7B); para a linhagem NPCD 1 (Betaproteobacteria 45%;
Alphaproteobacteria 25%; Flavobacteria 21%, Sphingobacteria 4%,
Gammaproteobacteria 2% e Fimbriimonada 2%) (Figura 3.7C); e para a linhagem SPC
759 (Betaproteobacteria 33%; Flavobacteria 26%; Alphaproteobacteria 19%;
Gammaproteobacteria 12%; Opitutae 5% e Sphingobacteria 2%) (Figura 3.7D).
Em resumo, foi possível observar uma dinâmica de grupos ao longo das fases
de multiplicação das cianobactérias. Por exemplo, o grupo Betaproteobacteria
apresenta uma alta abundância nas fases iniciais de cultivo das cianobactérias, com
subsequente redução na fase estacionária. A redução de Betaproteobacteria sempre
esteve acompanhada de um aumento significativo de, especificamente
Alphaproteobacteria para linhagem NPJL 4, Flavobacteria para a linhagem SPC 777,
NPCD 1 e SPC 759. Por fim, para a linhagem SPC 759, foi observado o aumento da
abundância de Gammaproteobacteria (Figura 3.7D).
Foi interessante notar que a classe Flavobacteria apresentou baixa abundância
relativa na fase lag para as linhagens SPC 777 e NPCD 1 e SPC 759 e tornou-se
dominante na fase estacionária. Pode-se também inferir que as linhagens SPC 777 e
NPCD 1 e SPC 759 apresentam similaridades em relação a dinâmica de grupos
relacionado a suas fases de desenvolvimento.
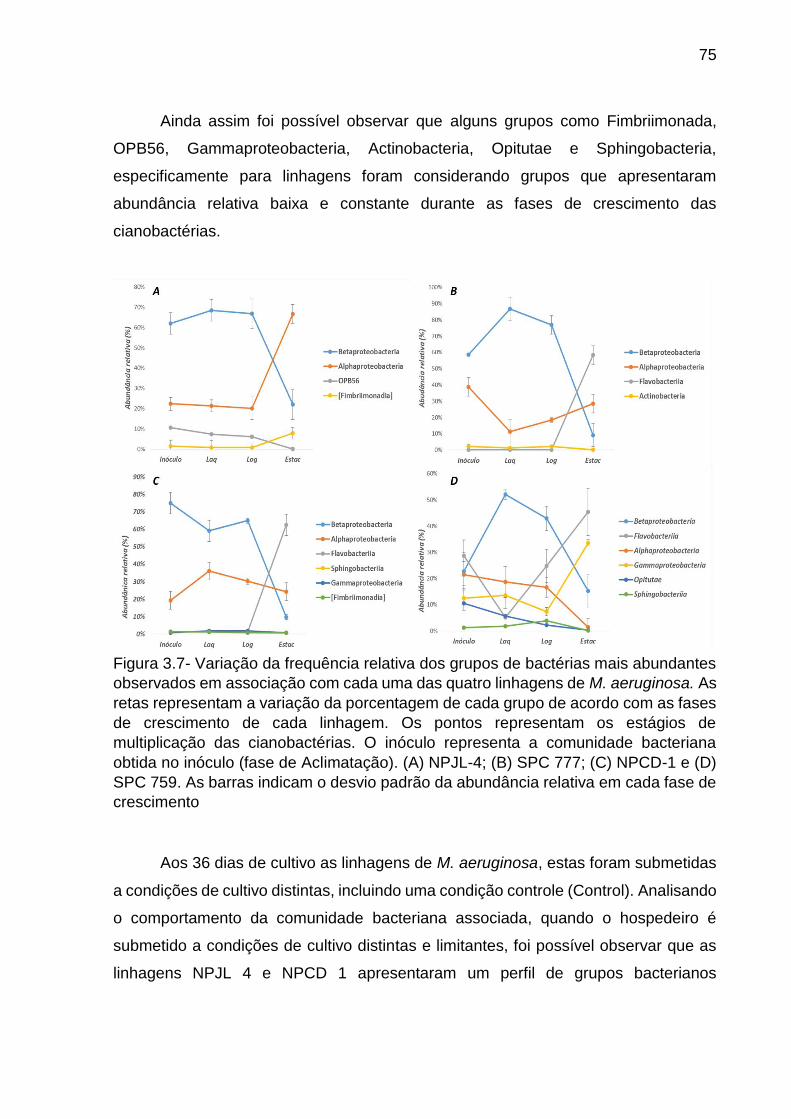
75
Ainda assim foi possível observar que alguns grupos como Fimbriimonada,
OPB56, Gammaproteobacteria, Actinobacteria, Opitutae e Sphingobacteria,
especificamente para linhagens foram considerando grupos que apresentaram
abundância relativa baixa e constante durante as fases de crescimento das
cianobactérias.
Figura 3.7- Variação da frequência relativa dos grupos de bactérias mais abundantes
observados em associação com cada uma das quatro linhagens de M. aeruginosa. As
retas representam a variação da porcentagem de cada grupo de acordo com as fases
de crescimento de cada linhagem. Os pontos representam os estágios de
multiplicação das cianobactérias. O inóculo representa a comunidade bacteriana
obtida no inóculo (fase de Aclimatação). (A) NPJL-4; (B) SPC 777; (C) NPCD-1 e (D)
SPC 759. As barras indicam o desvio padrão da abundância relativa em cada fase de
crescimento
Aos 36 dias de cultivo as linhagens de M. aeruginosa, estas foram submetidas
a condições de cultivo distintas, incluindo uma condição controle (Control). Analisando
o comportamento da comunidade bacteriana associada, quando o hospedeiro é
submetido a condições de cultivo distintas e limitantes, foi possível observar que as
linhagens NPJL 4 e NPCD 1 apresentaram um perfil de grupos bacterianos

76
semelhante ao controle, quando submetidas as condições 25°C e 50% N (Anosim, r
= 0,25, p = 0,001). Ao passo, que as linhagens apresentaram o mesmo padrão de
estruturação da comunidade bacteriana quando as cianobactérias foram submetidas
as condições 10% N (Anosim, r = 0,78, p= 0,0001) e 60 light (Anosim, r = 0,86, p=
0,0001) (Figura 3.8A e C).
Estes padrões de estruturação da comunidade bacteriana associada não foram
observados para as outras linhagens de cianobactérias (SPC 777 e SPC 759). Estas
últimas apresentaram uma estruturação de comunidade particular e especifica de
acordo com as condições de cultivo (50% N e 60 light). Especificamente para a
linhagem SPC 777, a estrutura da comunidade bacteriana associada foi descrita por
uma maior similaridade quando o hospedeiro foi submetido as condições Control,
25°C e 10% N e 25°C (r = 0,35, p = 0,001) e comparativamente com o controle, maior
especificidade quando submetida na condição 50% N (r = 0,72, p= 0,0001) e 60 light
(r = 0,70, p= 0,0001) (Figura 3.8B).
Para a linhagem SPC 759, foi possível observar uma maior dissimilaridade
entre a comunidade bacteriana associada as amostras Control, 10% N, 25°C e 50%
N (r = 0,32, p = 0,001), em relação a amostras do tratamento 60 light (Figura 3.8D).
Ainda assim relacionando os dados apresentados na figura 3.6 com a estrutura
da comunidade bacteriana associada, pode-se observar que o tratamento 60 light
influenciou o crescimento da cianobactéria e também favoreceu uma maior
dissimilaridade destas amostras com todas as outras. Por outro lado, a condição 25°C,
foi limitante para o crescimento das cianobactérias, pois todas a as linhagens
apresentaram crescimento semelhante ao controle, e correlatamente, a estrutura da
comunidade bacteriana foi semelhante a comunidade bacteriana das amostras
controle.
Esses dados foram validados por um teste de Mantel (R = 0,70; p < 0,002).
Ainda assim a condição com alteração da porcentagem de nutrientes no meio tem
uma influência variável, pois a dissimilaridade da comunidade não foi semelhante para
todas as linhagens.
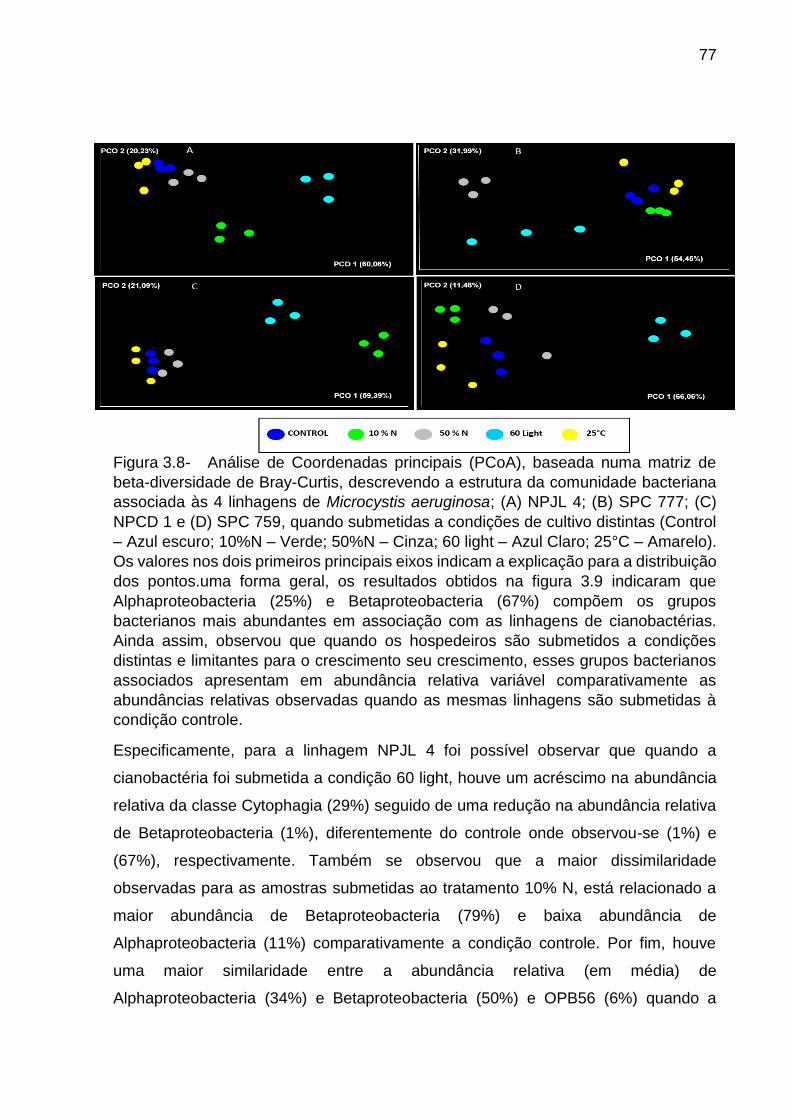
77
Figura 3.8- Análise de Coordenadas principais (PCoA), baseada numa matriz de
beta-diversidade de Bray-Curtis, descrevendo a estrutura da comunidade bacteriana
associada às 4 linhagens de Microcystis aeruginosa; (A) NPJL 4; (B) SPC 777; (C)
NPCD 1 e (D) SPC 759, quando submetidas a condições de cultivo distintas (Control
– Azul escuro; 10%N – Verde; 50%N – Cinza; 60 light – Azul Claro; 25°C – Amarelo).
Os valores nos dois primeiros principais eixos indicam a explicação para a distribuição
dos pontos.uma forma geral, os resultados obtidos na figura 3.9 indicaram que
Alphaproteobacteria (25%) e Betaproteobacteria (67%) compõem os grupos
bacterianos mais abundantes em associação com as linhagens de cianobactérias.
Ainda assim, observou que quando os hospedeiros são submetidos a condições
distintas e limitantes para o crescimento seu crescimento, esses grupos bacterianos
associados apresentam em abundância relativa variável comparativamente as
abundâncias relativas observadas quando as mesmas linhagens são submetidas à
condição controle.
Especificamente, para a linhagem NPJL 4 foi possível observar que quando a
cianobactéria foi submetida a condição 60 light, houve um acréscimo na abundância
relativa da classe Cytophagia (29%) seguido de uma redução na abundância relativa
de Betaproteobacteria (1%), diferentemente do controle onde observou-se (1%) e
(67%), respectivamente. Também se observou que a maior dissimilaridade
observadas para as amostras submetidas ao tratamento 10% N, está relacionado a
maior abundância de Betaproteobacteria (79%) e baixa abundância de
Alphaproteobacteria (11%) comparativamente a condição controle. Por fim, houve
uma maior similaridade entre a abundância relativa (em média) de
Alphaproteobacteria (34%) e Betaproteobacteria (50%) e OPB56 (6%) quando a

78
cianobactéria foi submetida a condição 10% N e 25° C e estas foram similares a
condição controle (Figura 3.9A).
Para a linhagem SPC 777, foi possível observar que quando a cianobactéria foi
submetida as condições 50% N e 60 light, apenas foi observado a presença de
Alphaproteobacteria e Betaproteobacteria, apesar de esses grupos apresentarem
abundância relativas distintas em cada condição de cultivo. Ainda assim, ocorreu uma
maior similaridade entre a composição bacteriana associada a cianobactérias,
considerando as condições Control, 10% N e 25° C (Figura 3.9B). Isto foi representado
por abundancia similares das classes Alphaproteobacteria (~25%),
Betaproteobacteria (~67%) e Actinobacteria (~8%)
Analisando a figura 3.8A e 3.8C, pode demonstrar que ambas as cianobactérias
apresentam um padrão de estruturação da comunidade bacteriana semelhante.
Analogamente, analisando a figura 3.9A e 3.9C, mostra-se que apesar de
apresentarem padrões de estruturação de comunidade semelhantes, estas
cianobactérias apresentam composições de comunidades distintas. Neste caso, não
houveram diferenças significativas entre as abundâncias relativas de
Alphaproteobacteria e Betaproteobacteria nas condições Control, 50% N e 25° C. Por
outro lado as maiores diferenças foram obtidas para as condições 10% N,
representada Flavobacteria (23%) e Sphingobacteria (19%), valores estatisticamente
distintos dos valores obtidos para a condição controle. Também para a condição 60
light foi observado uma alta porcentagem de Betaproteobacteria (76%) e
Fimbriimonada (5%) distintos do controle (p< 0,05). Estes resultados podem sugerir
uma especificidade metabólica entre os grupos bacterianos e as linhagens de
cianobactéria.
Para a linhagem SPC 759, foi observado que houve uma elevada abundância
do grupo Flavobacteria em todas as condições de cultivo. Este grupo apresentou
maior abundância apenas em associação com esta linhagem de cianobactéria
diferentemente (p< 0,05) das outras linhagens. Também foi observado que as
condições Control, 10% N, 50% N e 25° C apresentaram uma maior similaridade entre
elas relacionado aos grupos Alphaproteobacteria, Betaproteobacteria,
Gammaproteobacteria, Opitutae. Por fim, houve uma maior diferença quando a
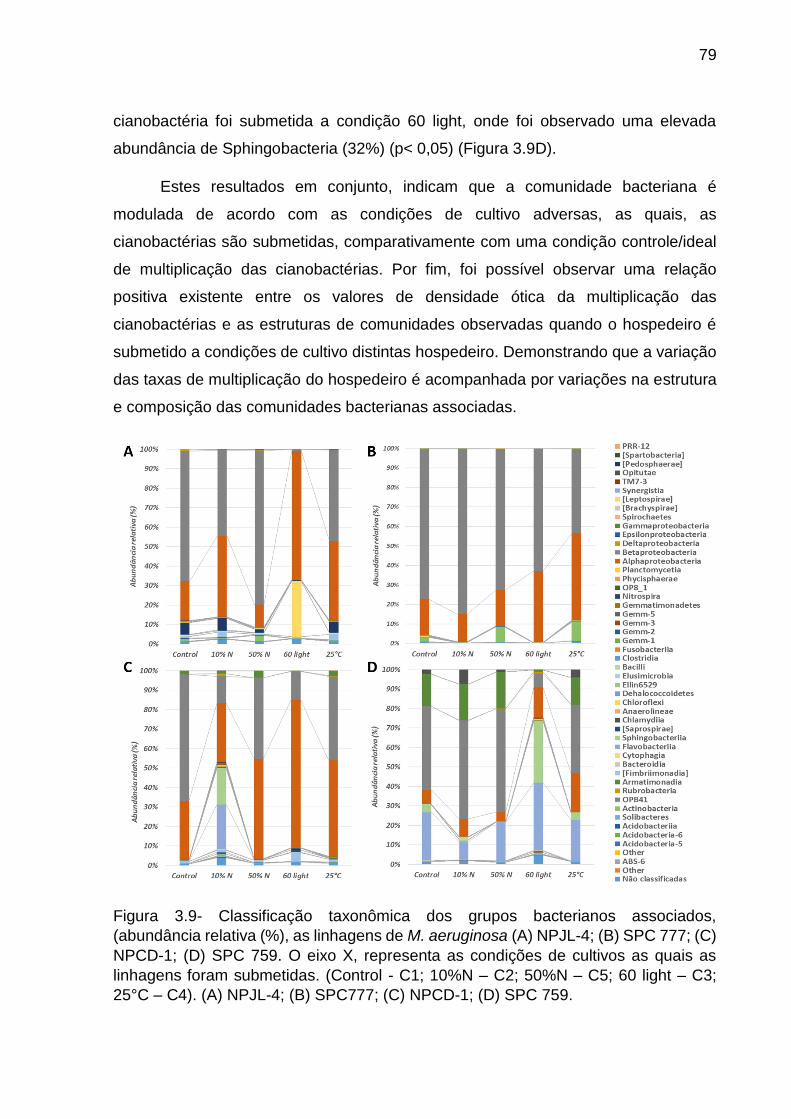
79
cianobactéria foi submetida a condição 60 light, onde foi observado uma elevada
abundância de Sphingobacteria (32%) (p< 0,05) (Figura 3.9D).
Estes resultados em conjunto, indicam que a comunidade bacteriana é
modulada de acordo com as condições de cultivo adversas, as quais, as
cianobactérias são submetidas, comparativamente com uma condição controle/ideal
de multiplicação das cianobactérias. Por fim, foi possível observar uma relação
positiva existente entre os valores de densidade ótica da multiplicação das
cianobactérias e as estruturas de comunidades observadas quando o hospedeiro é
submetido a condições de cultivo distintas hospedeiro. Demonstrando que a variação
das taxas de multiplicação do hospedeiro é acompanhada por variações na estrutura
e composição das comunidades bacterianas associadas.
Figura 3.9- Classificação taxonômica dos grupos bacterianos associados,
(abundância relativa (%), as linhagens de M. aeruginosa (A) NPJL-4; (B) SPC 777; (C)
NPCD-1; (D) SPC 759. O eixo X, representa as condições de cultivos as quais as
linhagens foram submetidas. (Control - C1; 10%N – C2; 50%N – C5; 60 light – C3;
25°C – C4). (A) NPJL-4; (B) SPC777; (C) NPCD-1; (D) SPC 759.

80
3.4 Discussão
3.4.1 Relação entre a filogenia das cianobactérias e composição e estrutura
das comunidades bacterianas associadas
No presente trabalho foi observada uma correlação positiva entre a estrutura
da comunidade bacteriana e as caraterísticas filogenéticas dos hospedeiros, ambas
baseadas no gene 16S rRNA. Esses resultados demonstram pode haver um sinal
filogenético, ou uma relação do gênero do hospedeiro com sua comunidade
associada, sugerindo que todas as características fenotípicas e genotípicas e
fisiológicas obtidas ao longo da história evolutiva das cianobactérias possuíram papel
chave para a modulação da estrutura da comunidade bacteriana associada.
Adicionalmente, também foi mostrado que quanto menor a distância filogenética entre
as linhagens, aqui utilizadas, maior é esse sinal filogenético, sugerindo que
possivelmente a espécie da cianobactéria também seja um fator determinante para a
estrutura da comunidade bacteriana associada. O gene 16S rRNA ou ITS são
considerados genes conservados entre o grupo de microrganismos e têm sido
normalmente utilizados como meio para descrever a filogenia entre grupos distintos
(XX). Neste caso, recentemente com os avanços dos estudos de interação entre
microrganismos e hospedeiros aliado aos avanços nas tecnologias de
sequenciamento, pesquisadores tem sugerido que correlações positivas entre a
comunidade microbiana e as distâncias filogenéticas dos hospedeiros podem indicar
um panorama de co-evolução entre ambos microrganismos e hospedeiros, podendo
apenas sugerir estatiscamente sobre a biologia da relação ecológica. Neste contexto,
Easson e Thacker (2014) observaram um forte sinal filogenético entre a comunidade
bacteriana associada a esponjas com a filogenia e identidade dos hospedeiros. Ainda
assim, Bouffaud et al 2014, mostrou uma relação positiva entre as distâncias
filogenéticas de plantas da família Poaceae com a estrutura da comunidade
bacteriana da rizosfera. Portanto, esses dados corroboram nossa sugestão de que
cianobactérias sempre apresentaram uma comunidade bacteriana especifica e está
despenhando algum papel importante de complementação de seu metabolismo.

81
Neste estudo foi observado que a comunidade bacteriana associada às
cianobactérias, de uma forma geral, é composta por alta abundância dos grupos
taxonômicos Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria,
Flavobacteria, Sphingobacteria, Saprospirae, Cytophagia e OPB56. A maioria das
classes identificadas neste estudo foram semelhantes àquelas observadas em
trabalhos anteriores de comunidades bacterianas associadas com cianobactérias
(Maruyama et al., 2006; Lemes et al., 2008; Manage et al., 2009b; Manage et al.,
2010). No entanto, foram observadas algumas diferenças na composição da
comunidade comparativamente a esses trabalhos citados, o que por sua vez, podem
estar relacionadas com as estratégias de sequenciamento, ou por estarem sendo
analisadas cianobactérias oriundas de ambientes tropicais.
Muitos trabalhos sugerem que existe uma íntima associação simbiótica entre
cianobactérias e bactérias heterotróficas (Abed et al., 2005; Fitzsimons et al., 2007),
sugerindo certa especificidade entre cianobactéria-comunidade bacteriana associada
(Grossart et al., 2005; Bagatini et al., 2014), e que essa especificidade é dependente
da identidade do hospedeiro. Esses trabalhos corroboram os nossos resultados, que
demonstram que a identidade da cianobactéria é um fator determinante na
composição da comunidade de bactérias heterotróficas. Neste contexto Louati et al.
(2015) mostrou que cianobactérias afiliadas a Microcystis spp. e Anabaena spp.
apresentaram perfis distintos das comunidades bacterianas associadas.
Neste mesmo contexto, vale ressaltar no presente trabalho o enriquecimento
da classe OPB56, representado pela OTU denovo6182, observado especificamente
nas linhagens de Microcystis spp., o que não foi relatado em nenhum dos trabalhos
prévios relacionados a este tema. OPB56 é uma classe pertencente ao filo Chlorobi,
a qual não possui membros cultivados, o que dificulta inferências sobre sua
funcionalidade nos ambientes em que se encontram (Hiras et al., 2015). Esses
autores, baseado numa análise filogenética e funcional, demonstraram que a classe
OPB56 forma uma clado monofilético com as Chlorobea e Iganvibacteria e as
bactérias verde-sulfurosas, apresentando uma relação evolutiva e filogenética com o
filo Bacteroidetes. É descrito que estas bactérias possuem clusters gênicos
relacionados à ciclagem de C e N, podendo atuar na complementação de processos

82
metabólicos relacionados as transformações de C e N (Zhu et al., 2016), auxiliando o
desenvolvimento da cianobactéria.
Validando as sugestões acima citadas e corroborando as sugestões, alguns
outros autores relatam que as bactérias heterotróficas do complexo bacteriano
Cytophaga/Flavobacterium/Bacteroides tem papel fundamental em consumir parte da
microcistina produzida por algumas linhagens de cianobactérias tóxicas, como forma
de aliviar as condições dentro da célula evitando a sua morte (Brunberg, 1999;
Maryuama et al., 2003). Louati et al. (2015) sugere que a composição comunidade
bacteriana é dependente do gênero da cianobactéria, potencialmente da espécie das
cianobactérias e suas capacidades metabólicas.
Liu et al. (2016), estudando a relação entre filogenia de árvores subtropicais e de sua
comunidade de fungos patógenos associada, explicitam que essa associação
patogênica pode ser oriunda tanto de uma co-evolução entre a ocorrência da
associação entre plantas e fungos, como espécies de fungos se a associaram a
hospedeiros espacialmente distribuídos, mas possuindo parâmetros filogenéticos
semelhantes e preferências por habitat. Por outro lado, Sison-Magnus et al., 2014
analisando à associação entre diatomáceas marinhas e sua comunidade microbiana,
sugeriu um processo de especificidade de hospedeiros e co-adaptação,
demonstrando que diferentes genótipos de Pseudo-nitzschia hospedam estrutura e
composição de comunidades microbianas distintas. Ainda assim, Bouffaud et al.
(2014) observaram uma correlação positiva entre a distância filogenética e a estrutura
da comunidade bacteriana da rizosfera de plantas da família Poaceae. Essa relação
positiva pode ser o elo explicativo da co-evolução entre filogenia e função, na
interação entre hospedeiros e microrganismos (Provorov & Vorobyov, 2009; Kinkel et
al., 2011; Gilbert & Webb, 2007). Apesar de serem avaliados em organismos distintos,
estes últimos trabalhos suportam as sugestões levantadas no presente estudo, de que
a história evolutiva das cianobactérias pode ter dirigido a associação com a
comunidade microbiana.

83
3.4.2 Alteração da estrutura e composição da comunidade bacteriana ao
longo das fases de crescimento da cianobactéria
A segunda hipótese deste trabalho é que a comunidade bacteriana associada
é alterada ao longo das fases de crescimento do hospedeiro quando este é submetido
à diferentes condições abióticas. Para tal, seguindo os resultados observados sobre
a comunidade associada a gêneros de cianobactérias já apresentados, nós
selecionamos apenas um gênero para responder as hipóteses seguintes. Neste caso
foi selecionado o gênero Microcystis spp., pois compreendem cianobactérias capazes
de formar florações em água paradas de reservatórios e também são capazes de
produzir diferentes metabólitos secundários, tais como a toxina microcistina e outros
(Nicolaus et al., 1999; Kirkwood et al., 2006). Além disso, tem sido o foco de diversos
estudos ecológicos (Humbert et al., 2009; Kaplan et al., 2015) devido a sua alta
prevalência em muitos ecossistemas ao redor do mundo (Paerl, 1996; Eiler et al.,
2004). O gênero Microcystis apresenta uma organização colonial e uma matriz
mucilaginosa ao redor da sua célula, composta de uma vasta gama de compostos
carbonáceos e polissacarídeos como: glicose, manose, fucose, xilose, galactose e
ramnose (Anemiya et al., 1990; Worm and Sondergaard, 1998;), os quais oferecem
um nicho em potencial para comunidades de bactérias heterotróficas e um suporte
físico para o estudo de interação microrganismo- hospedeiro (Zhu et al., 2016).
Mesmo assim, informações sobre a dinâmica de grupos bacterianas associados a
cianobactérias mantidas em coleções de culturas, são escassas e podem apresentar
um panorama de processos de dependência microrganismos-hospedeiros.
Os resultados obtidos, no presente trabalho demonstraram que a diversidade
bacteriana é variável de acordo com as fases de desenvolvimento da cianobactéria,
desde a fase de adaptação (fase lag) até a fase estacionária. Também foi observado
uma maior dominância de grupos bacterianos na fase estacionária. Estes resultados
podem sugerir que o comportamento da comunidade bacteriana está relacionado ao
processo de estabilização das principais mudanças físicas e metabólicas ocorrida nas
primeiras fases de crescimento da cianobactéria, relativas a multiplicação celular e a
taxa de produção de metabólitos secundários, inferências citadas por Shi et al., 2012;
Sheng et al., 2013; Leloup et al., 2013; Brauer et al., 2015, Bagatini et al., 2014; Louati.

84
et al 2015; Zhu et al., 2016 e nós usamos com embasamento teórico para nossas
sugestões.
Também pode-se observar, que os grupos de bactérias flutuaram entre alta
abundância para baixa abundância e vice-versa. Estudos prévios corroboram os
resultados encontrados no presente trabalho. Por exemplo Bagatini et al., (2014)
mostrou que Betaproteobacteria foi um dos grupos mais abundantes, nas fases iniciais
do multiplicação de linhagens de M. aeruginosa, mas sua contribuição relativa
decresceu até a última fase de desenvolvimento da cianobactéria, em contraste a
classe Alphaproteobacteria que foi o segundo grupo mais abundante, e aumentou em
abundância, desde as fases inicias ao longo da curva de crescimento da
cianobactéria, além de outros grupos bacterianos que apresentavam abundancia <
1% em média, e passaram à apresentar elevada abundância nas fases finais e vice e
versa.
Pedros-Aliós, 2006 descreve que a biodiversidade total de um ecossistema é
formada por táxons abundantes e raros e que o balanço do ecossistema é
determinado pela flutuação desses dois grupos de acordo com as condições abióticas
vigentes. Esse contexto permeia a associação entre cianobactérias e bactérias
heterotróficas apresentada neste trabalho, ao ponto que a presença transitória de
alguns grupos bacterianos de baixa abundância durante o desenvolvimento das
cianobactérias, como por exemplo Flavobacteria (SPC 777; NPCD-1;SPC 759),
OPB56 (NPJL 4), Sphingobacteria (NPCD 1; SPC 759), Opitutae (SPC 759) e
Fimbriimonada (NPJL 4). Alguns autores sugerem que essa dinâmica de flutuação
entre grupos transitórios e abundantes está relacionada a microrganismos
especialistas que mudam de acordo com os estágios de desenvolvimento do
hospedeiro e fatores bióticos, tais como composição de exsudatos, em resposta às
características genotípicas específicas dos hospedeiros em cada etapa de seu
desenvolvimento (Haukka et al 2006; Becker et al., 2014; McFrederick et al., 2014).
Estes resultados em conjunto podem sugerir que ocorreu um processo de
sucessão ecológica ao longo do desenvolvimento do hospedeiro, consequentemente
levando a uma estabilização da estrutura da comunidade, correlacionado com o
período de maior crescimento da cianobactéria, sendo este o início da fase

85
estacionária. Esses processos de sucessão ecológica e estabilização de comunidades
microbianas e seleção de grupos de acordo com a fisiologia do hospedeiro são bem
fundamentados em ecologia de comunidades microbianas em ambientes em transição
(Dini-Andreote et al., 2016), na associação de patógeno-hospedeiro (Thrall et al.,
2003) e na manutenção de patógenos no solo em relação a dinâmica da diversidade
bacteriana (van Elsas et al., 2012). Não foram observados trabalhos que
descrevessem o papel da sucessão ecológicas de grupos bacterianos associados ao
desenvolvimento de cianobactérias cultivadas oriundas de coleção de culturas. Assim
demonstrando o pioneirismo do trabalho.
3.4.3 Alteração da estrutura e composição da comunidade bacteriana
associada a cianobactérias quando esta é submetida a condições abióticas
distintas
Os resultados observados neste trabalho corroboram a hipótese de que
alterações nas condições de cultivo de M. aeruginosa, promovem alterações no seu
crescimento e consequentemente promovem alterações na sua estrutura da
comunidade bacteriana associada.
Paerl, (1996) comparou florações de cianobactérias em três ambientes
aquáticos distintos, e elucidou que alguns fatores ambientais como principalmente,
temperatura (>20°C e < 25°C), quantidade excessiva ou limitantes de (N) e Irradiância,
são fatores cruciais que influenciam diferentemente, limitando ou favorecendo o
crescimento e desenvolvimento das cianobactérias e produção de toxinas, referente
à sua capacidade metabólica e fisiológica (Reynolds et al., 1987; Bentley &
Meganathan, 1991; Wasby 1992; Paerl & MiIllie, 1996).
Segundo a World Healthy Organization (1998), a formação de florações de
Microcystis aeruginosa são influenciadas por fatores físicos, químicos e biológicos e
que esses fatores determinam os níveis de cianobactéria e de suas toxinas no
ambiente. Por exemplo cita-se que a temperatura ótima para o desenvolvimento de
Microcystis aeruginosa inclui uma faixa entre 20 e 25°C, sendo essa faixa ideal para
a produção de toxinas e que essas temperaturas citadas são limítrofes para o

86
crescimento das cianobactérias (Chorus & Bartram, 1999), justificando a escolha de
uma temperatura ideal para avaliação das variações do crescimento da cianobactéria.
O aumento da intensidade luminosa acima de 40 microeisnteins/ m². s¹ está
diretamente relacionado ao aumento das taxas de multiplicação de M. aeruginosa,
também relacionado ao aumento da biomassa e produção de toxinas no ambiente
(Mitrovic et al., 2001; Brookes et al., 2003). Muitas cianobactérias estão associadas
com quantidades elevadas de nutrientes, e isso leva a uma redução nas proporções
das florações (Chorus & Bartram, 1999). Como já descrito na literatura e corroborado
nos nossos dados, as cianobactérias respondem as variações ambientais as quais
são submetidas e essas respostas são acompanhadas por variações em seu
microbioma associado.
Trabalhos prévios corroboram os resultados observados neste presente
trabalho, afirmando que a comunidade bacteriana associada à Microcystis spp. são
significantemente diferente quando a cianobactéria é submetida a temperaturas
distintas (Dziallas & Grossart, 2011). Neste mesmo contexto, Eiler et al., (2004), já
tinha demonstrado que florações de cianobactérias em ambientes que possuem
condições abióticas distintas, apresentam comunidade bacterianas distintas. Parveen
et al., (2013), demonstrou que as cianobactérias selecionam microrganismos
específicos do ambiente adjacente com o intuito de promover o seu desenvolvimento
e a sua tolerância as condições limitantes do meio.
Neste mesmo contexto, Louati et al., (2015), sugeriu que cianobactérias
fixadoras e não fixadoras de nitrogênio possuem comunidades bacterianas distintas,
e que a seleção bacteriana é direcionada para auxiliar a cianobactérias em seus
processos metabólicos. Em trabalho, subsequente, Zhu et al., (2016), por meio de
análise metagenômica, confirmou a presença de genes relacionados à dinâmica de
carbono e nitrogênio, e que esses genes estavam associados à comunidade
bacteriana. Esses trabalhos exemplificam a íntima relação ecológica que ocorre entre
bactérias heterotróficas e cianobactérias em ambientes naturais e como a comunidade
se comporta em relação a sobrevivência do hospedeiro (Abed et al., 2005; 2009).
Sugerindo que cianobactérias de ambientes naturais quando em situações
estressantes selecionam microrganismos específicos na sua ficosfera, termo análogo

87
a rizosfera ou micosfera, para auxiliar em seu desenvolvimento e garantir a
sobrevivência do hospedeiro (Bagatini et al., 2014).
Entretanto, no presente trabalho foram utilizadas linhagens da espécie M.
aeruginosa oriundas de uma coleção de cultivo, subentendendo que não há fonte de
seleção de novos microrganismos do ambiente, e os microrganismos em associação
são aqueles que foram adquiridos verticalmente por meio dos sub-cultivos. Sendo
assim, sugere-se que as rápidas alterações na estrutura da comunidade bacteriana
associada a cianobactérias cultivadas em condições distintas, é resultado de
processos de auto regulação entre comunidade microbiana e hospedeiros envolvidos
na interação simbiótica onde processos de auto regulação permeiem a associação
entre linhagens de M. aeruginosa e bactérias heterotróficas. (van Elsas, Jansson &
Trevors, 2006). Desta forma, observa-se uma modulação em termos de abundância
dos microrganismos associados como forma de garantir a sobrevivência da
associação ou aliviar as condições abióticas limitantes do ambiente, mantendo o
balanço do ecossistema formado pela interação entre cianobactérias e bactérias.
Estas sugestões se baseiam no fato de que as cianobactérias não apresentaram
redução no processo de multiplicação em relação a condição controle (ideal), ao ponto
de indicar que a cianobactéria estaria morrendo. Neste contexto, nosso trabalho está
de acordo também com as observações feitas para corais e esponjas marinhas,
descrevendo que variações na estrutura e composição de comunidade bacterianas
como mecanismo para garantir a resiliência de espécies de corais (Webster et al.,
2001; Buddemeier et al., 2004; Rosenberg et al., 2007).
Este processo de interação mutualística microrganismo-hospedeiros tem sido
majoritariamente descrito para a associação entre procariotos e eucariotos (Animais
e Plantas). Essas unidades de interação são definidas pelo termo Holobionte (Zilber-
Rosenberg & Rosenberg, 2008; Moran & Sloan, 2015; Vandenkoornhusen et al.,
2015). A teoria do hologenoma de onde deriva o termo holobionte ou
“superorganismos”, sugere que quando ocorrem variações na fisiologia do hospedeiro
em resposta as variações nas condições bióticas ou abióticas, as comunidades
microbianas associadas irão acompanhar essas alterações, ao ponto de favorecer
adaptação e sobrevivência do holobionte como um todo, garantindo um balanço no
ecossistema (Youle et al., 2013; Mendes et al., 2013).

88
A luz do nosso conhecimento, estes conceitos têm apenas sido aplicados para
a associação entre eucariotos e procariotos (Singh et al.,2013; Zilber-Rosenberg &
Rosenberg, 2013; Krediet, Ritchie & Paul, 2013; Berg, Krause & Mendes, 2015; Sweet
& Bulling, 2017). Sendo assim, de acordo com os resultados observados e toda a
literatura considerada, nós sugerimos que esta teoria citada também possa abordar
as interações entre microrganismos-microrganismos, procariotos-procariotos,
especificamente cianobactérias e bactérias.
Referências
Abed RMM, Köster J. (2005). The direct role of aerobic heterotrophic bacteria associated with cyanobacteria in the degradation of oil compounds. International Biodeterioration & Biodegradation. 55: 29-37.
Abed RMN, Bobrestov S, Sudesh K. (2009); Applications of cyanobacteria in Biotechnology. Journal of Applied Microbiology. 106: 1-12.
Adam, B., Klawonn, I., Svedén, J. B., Bergkvist, J., Nahar, N., Walve, J., et al. (2016). N2-fixation, ammonium release and N-transfer to the microbial and classical food web within a plankton community. The ISME Journal. 10: 450–459.
Al-Hasan, RH, Sorkhoh, NA, Al-Bader, DA, Radwan, SS (1994) Utilization of hydrocarbon by cyanobacteria from microbial mats on oily coasts of the Gulf. Applied Microbiology and Biotechnology 41: 615–619
Bagatini, I. L., Eiler, A., Bertilsson, S., Klaveness, D., Tessarolli, L. P., and Vieira, A. A. H. (2014). Host-specificity and dynamics in bacterial communities associated with bloom-forming freshwater phytoplankton. PLoS ONE 9:e85950.
Bekker A, et al. (2004) Dating the rise of atmospheric oxygen. Nature 427(6970):117–120.
Berg KA, Lyra C, Sivonen K, Paulin L, Suomalainen S, Tuomi P, Rappala J. (2009). High diversity of cultivable heterotrophic bacteria in association with cyanobacterial water blooms. The ISME Journal. 3: 314-325.
Blankenship RE (2002) Molecular Mechanisms of Photosynthesis (Blackwell Science, Oxford).
Bouffaud, ML, Poirer MA, Muller D, Moënne-Loccoz Y (2014) Root microbiome relates to plant host evolution. Environmental Microbiology, 16, 2804-2814
Brauer, V. S., Stompe, M., Bouvier, T. Fouilland E. Leboulanger C. Confurius-Guns, V. et al. (2015). Competition and facilitation between the marine nitrogen-fixing cyanobacterium Cyanothece and its associated bacterial community. Frontiers in Microbiology 5:795.
Brookes, J.D., Regel, R.H. and Ganf, G.G. (2003) ‘Changes in the photochemistry of

89
Buddemeier RW, Baker AC, Fautin DG & Jacobs JR (2004). The adaptive hypothesis of bleaching. Coral Health and Disease (Rosenberg E & Loya Y, Eds), pp. 427–444. Springer-Verlag, Berlin.
Chorus I, Bartram J (Eds) (1999). Toxic cyanobacteria in water. A guide to their public health consequences, monitoring and management. E & FN Spon, London, on behalf of the World Health Organisation
Dini-Andreote F, Pylro VS, Baldrian P, van Elsas JD, Salles JF. (2016). Ecological succession reveals potential signatures of marine-terrestrial transition in salt marsh fungal communities. The ISME Journal.
Dziallas, C., and Grossart, H. P. (2011). Temperature and biotic factors influence bacterial communities associated with the cyanobacterium Microcystis sp. Environmental Microbiology. 13: 1632–1641.
Easson CG. Thacker RW (2014). Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Frontiers in Microbiology. 5(532)
Eiler, A., and Bertilsson, S. (2004) Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ Microbiol 6: 1228– 1243
Farnelid, H, Tarangkoon W, Hansen G, Hansen PJ, Riemann L. (2010). Putative N2-fixing heterotrophic bacteria associated with dinoflagellate-Cyanobacteria consortia in the low-nitrogen Indian Ocean. Aquatic Microbial Ecology. 61: 105-117.
Fitzsimons AG, Smith RV. (2007). The isolation and growth of axenic cultures of planktonic blue-green algae. British Phycological Journal. 19(2): 157-162.
Garcia-Pichel, F, Pringault O. (2001) Cyanobacteria track water in desert soils. Nature 413: 380–381.
Genuário DB, Corrêa DM, Komarek J, Fiore MF. (2013). Characterization of freshwater benthic biofilm-forming Hydrocoryne (Cyanobacteria) isolates form Antarctica. Journal of Phycology. (49): 1142-1153.
Gerphagnon M, Macarthur DJ, Latour D, Gachon CMM, Ogtrop FV, Gleason FH, Sime-Ngando T. (2015). Microbial players involved in the decline of filamentous and colonial cyanobacterial blooms with a focus on fungal parasitism. Environmental Microbiology. doi: 10.1111/1462-2920.12860
Gilbert GS, Webb CO. (2007). Phylogenetic signal in plant pathogen-host range. Proceedings of National Academy of Sciences. 104(12): 4979-4983.
Grossart HP, Levold F, Allgaier M, Simon M, Brinkhoff T (2005) Marine diatom species harbor distinct bacterial communities. Environmental Microbiology 7: 860–873
Hoff-Risseti C, Dörr, FA, Schaker PDC, Pinto E, Werner VR, Fiore MF. (2013). Cylindrospermopsin and saxitoxin synthetase genes in Cylindrospermopsis raciborskii strains from Brazilian freshwater. PLoS ONE 8:e74238.
Humbert J F, Dorigo U, Cecchi P, Le Berre B, Debroas D, and Bouvy M. (2009). Comparison of the structure and composition of bacterial communities from temperate and tropical freshwater ecosystems. Environmental Microbiology. 11: 2339–2350.

90
Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP and Webb C O. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26:1463–1464.
Kimura M. (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16:111-120.
Kinkel LL, Bakker MG, and Schlatter DC. (2011) A coevolutionary framework for managing disease- suppressive soils. Annuals Reviews in Phytopathology 49: 47–67.
Kirkwood AE, Nalewajko C, and Fulthorpe RR. (2006). The effects of cyanobacterial exudates on bacterial growth and biodegradation of organic contaminants. Microbial Ecololy 51: 4–12.
Kolmonen E; Sivonen K J, Rapala J, Hauka K. (2004). Diversity of cyanobacteria and heterotrophic bacterial in cyanobacterial blooms in lake Joutikas Finland. Aquatic Microbial Ecology. 36: 201-211.
Komárek J, Nedbalová L, Hauer T. (2012). Phylogenetic position and taxonomy of three heterocystous cyanobacteria dominating the litoral of deglaciated lakes, James Ross Island, Antarctica. Polar Biology. 35: 759-774.
Krediet CJ, Ritchie KB, Paul VJ, and Teplitski M. (2013). Coral-associated micro-organisms and their roles in promoting coral health and thwarting diseases. Proceedings in Biological Science. 280, 20122328–20122328.
Kumar S., Stecher G., and Tamura K. (2015). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution (submitted). Genuário DB, Corrêa DM, Komárek J, Fiore MF. (2013), Characterization of freshwater benthic biofilm-Forming Hydrocoryne (Cyanobacteria) isolates from Antarctica. Journal of Phycology. 49: 1142-1153.
Leloup, M., Nicolau, R., Pallier, V., Yéprémian, C., and Feuillade-Cathalifaud, G. (2013). Organic matter produced by algae and cyanobacteria: quantitative and qualitative characterization. Journal of Environmental Science. 25: 1089–1097.
Louati, I., Pascault, N., Debroas, D., Bernard, C., Humbert, J. F., and Leloup, J. (2015). Structural diversity of bacterial communities associated with bloom- forming freshwater cyanobacteria differ according to the cyanobacterial genus. PLoS ONE. 10(11): e0140614.
Maruayama T, Kato K, Yokoyama A, Tanaka T, Hiraish. A, Park, HD. (2003); Dynamics of Microcystin- Degrading bacteria in Mucilage of Microrcystis. Microbial Ecology. 46: 279-288.
Maruyama THD, Park K, Ozawa Y, Tanaka T, Sumino K, Hamana A, Hirashi and K. Kato (2006): Sphingosinicella microcystinivorans gen. nov., sp. nov., a microcystin degrading bacterium. Int. J. Systematic and Evolutive Microbiology, 56, 85–89.
Maruyama, T., Kato, K., and Park, H. D. (2004). Population dynamics of free- living bacteria related to the microcystin-degrading strain Y2 in Lake Suwa and in microcystin amended enrichments. Microbes and Environment. 19: 137–146.

91
Mitrovic, S.M., Howden, C.G., Bowling, L.C. and Buckney, R.T. (2003) “Unusual allometry between in situ growth of freshwater phytoplankton under static and fluctuating light environments: possible implications for dominance’. Journal of Plankton Research. 25(5): 517-526.
Nicolaus, B., Panico, A., Lama, L., Romano, I., Manca, M.C., De Guilio, A., and Gambacorta, A. (1999) Chemical composition and production of exopolysaccharides from representative members of heterocystous and non- heterocystous cyanobacteria. Phytochemistry 52: 639– 647.
Paerl HW. (1996), A comparison of cyanobacterial bloom dynamics in freshwater, estuarine and marine environments. Phycologia. 35(6): 25-35.
Parveen B, Ravet V, Djediat C, Mary I, Quiblier C, Debroas D, Humbert JF. (2013). Bacterial communities associated with Microcystis colonies differ from free-living communities living in the same ecosystem. Environmental Microbiology. 5: 716–24.
Parveen B, Ravet V, Djediat C, Mary I, Quiblier C, Debroas D, et al. (2013). Bacterial communities associated with Microcystis colonies differ from free– living communities living in the same ecosystem. Environmental Microbiology. Reports. 5: 716–724.
Pedrós-Alió, C. (2006). Marine microbial diversity: can it be determined? Trends in Microbiology. 14: 257–263.
Ploug, H., Adam, B., Musat, N., Kalvelage, T., Lavik, G., Wolf-Gladrow, D., et al. (2011). Carbon, nitrogen and O2 fluxes associated with the cyanobacterium Nodularia spumigena in the Baltic Sea. The ISME Journal. 5: 1549–1558.
Provorov, NA., and Vorobyov, NI. (2009) Host plant as an organizer of microbial evolution in the beneficial symbioses. Phytochemistry Reviews 8: 519–534.
Radwan, SS, Al-Hasan, RH, Salamah, S, Al-Dabbous, S. (2002) Bioremediation of oily sea water by bacteria immobilized in biofilms coating macroalgae. International Biodeterioration and Biodegradation 50: 55–59.
Rigonato J, Alvarenga DO, Andreote FD, Dias AFC, et al., (2012). Cyanobacterial diversity in the phyllosphere of a mangrove forest. FEMS Microbial Ecology. 80: 312-322.
Sánchez O, Diestra E, Esteve I, Mas J. (2005). Molecular characterization of an Oil-Degrading Cyanobacterial consortium. Microbial ecology.
Sanchez-Baracaldo P, Hayes, PK., Blank, CE. (2005). Morphological and habitat evolution in the Cyanobacteria using a compartmentalization approach. Geobiology, 3(145) - 165. 2005. 50: 580-588.
Schirrmeister BE, De Vos JM; Antonelli A; Bagheri H. (2013). Evolution of multicellularity coincided with the increased diversification of cyanobacteria and the Great Oxidation Event. PNAS. 110(5): 1791-1796.
Schirrmeister BE; Antonelli A; Bagheri H. (2011). The origin of multicellularity in cyanobacteria. BMC Evolutionary Biology. 11(45).

92
Schopf JM, Walter MR. (1982) Origin and early evolution of cyanobacteria: The geological evidence. In: Carr NG, Whitton BA (Eds). The Biology of Cyanobacteria, 543-564, Blackwell, Oxford, University of California Press, Berkeley
Shen, H., Niu, Y., Xie, P., Tao, M., and Yang, X. (2011). Morphological and physiological changes in Microcystis spp. as a result of interactions with heterotrophic bacteria. Freshwater Biology. 56: 1065–1080.
Shi, L., Cai, Y., Kong, F., and Yu, Y. (2012). Specific association between bacteria and buoyant Microcystis colonies compared with other bulk bacterial communities in the eutrophic Lake Taihu, China. Environment. Microbiology. Reports. 4: 669–678.
Shilo, M. (1980) Strategies of Adaptation to Extreme Conditions in Aquatic Microorganisms. Naturwissenshafen. 67: 384-389.
Sison-Magnus. MP; Jiang S; Tran KN; Kudela RM. (2014). Host-specific adaptation governs the interaction of the marine diatom, Pseudo-nitzschia and their microbiota. The ISME journal. (8). 63-76.
Sweet MJ, Bulling MT. (2017). On the importance of the Microbiome and Path biome in coral health and Disease. Frontiers in Marine Science. 4(9): 1-11.
Thrall PH, Burdon JJ. (2003). Evolution of virulence in plant host-pathogen metapopulation. Science. 299:1735-1737.
Van Elsas JD, Chiurazzi M, Mallon CA, Elhottova D, Kristufek V, Salles JF. (2012). Microbial diversity determines the invasion of soil by a bacterial pathogen. Proceedings in National Academy of Science. 109: 1159–1164.
Whitton, B A, Potts M. Introduction to cyanobacteria. In Whitton, B A, Potts M. (2000) (Ed.). The ecology of cyanobacteria. Dordrecht; Kluwer Academic.
Worm J, Sondergaard M. (1998). Dynamics of heterotrophic bacteria attached to Microcystis spp. (Cyanobacteria). Aquatic Microbial Ecology. 14: 19-28.
Worm J, Sondergaard M. (1998). Dynamics of heterotrophic bacteria attached to Microcystis sp. (Cyanobacteria). Aquatic Microbial Ecology. (14): 19-28.
Zhu L, Zancarini A, Louati I, De Cesare S, Duval C, Tambosco K, Bernard C, Debroas D, Song L, Leloup J, Humbert J –F. (2016). Bacterial Communities Associated with Four Cyanobacterial Genera Display Structural and Functional Differences: Evidence from an Experimental Approach. Frontiers of Microbiology. 7:1662.

93
4. CONSIDERAÇÕES FINAIS
O presente estudo elucidou, dois panoramas distintos de associação
microrganismos – hospedeiros, com o intuito de elucidar que apesar das
dissimilaridades entre os hospedeiros e serem organismos alocados em Domínios da
vida distintos, existem similaridades entre os padrões de associação ou fatores que
determinam a associação desses seres com a comunidade bacteriana.
Ao avaliar o efeito do endemismo das plantas em sua comunidade bacteriana
associada podemos observar que as características fenotípicas dessas plantas
desempenham papel crucial na seleção de microrganismos para compor sua
microbiota. Pode-se também sugerir que o isolamento geográfico e as condições de
restrição das ilhas que dirigiram a especiação das plantas podem ter influenciado a
estrutura e composição de sua comunidade bacteriana. Além disso, o principal grupo
associado à essas plantas endêmicas foi Proteobacteria, mais especificamente
Gammaproteobacteria e Betaproteobacteria.
De maneira similar ao observado nas plantas endêmicas, as comunidades
bacterianas hospedadas pelas cianobactérias são influenciadas diretamente por suas
características filogenéticas e fenotípicas. Assim como em relações estabelecidas
entre microrganismos e organismos superiores, foi possível observar que ao
ocorrerem modificações na fisiologia do hospedeiro em decorrência de modificações
no ambiente, o microbioma associado responde a essa variação e que essa resposta
é essencial para o desenvolvimento e a adaptação das cianobactérias a essa nova
condição. Os principais grupos associados são Betaproteobacteria, Flavobacteria,
Alphaproteobacteria, Gammaproteobacteria, OPB56 e Opitutae.
De forma geral, pode-se considerar que tanto as plantas quanto as
cianobactérias seguem padrões semelhantes no estabelecimento de suas
associações com a comunidade bacteriana. Ambos os hospedeiros são capazes de
interagir com uma vasta gama de bactérias, visto os elevados índices de diversidade
apresentados, e que dentro dessa diversidade, alguns grupos são preferencialmente
encontrados (Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria,
Actinobacteria e Flavobacteria. Além disso, observou-se que o microbioma associado

94
a esses hospedeiros é espécie/gênero específico e que essa interação é vital para a
sobrevivência de ambos frente as variações ambientais.


















