
UNIVERSIDADE FEDERAL FLUMINENSE CENTRO DE …§ão de... · 4.3.1 – Calculo do teor de umidade...
79
UNIVERSIDADE FEDERAL FLUMINENSE CENTRO DE ESTUDOS GERAIS INSTITUTO DE QUÍMICA PÓS-GRADUAÇÃO EM GEOCIÊNCIAS – GEOQUÍMICA LÍVIA FERREIRA DE MELO GUEDES SEPARAÇÃO EM FASE SÓLIDA PARA A DETERMINAÇÃO DE ÂNIONS POR CROMATOGRAFIA DE ÍONS EM AMOSTRAS SALINAS, AMBIENTAIS E DA INDÚSTRIA DE PETRÓLEO Niterói 2010
Transcript of UNIVERSIDADE FEDERAL FLUMINENSE CENTRO DE …§ão de... · 4.3.1 – Calculo do teor de umidade...

UNIVERSIDADE FEDERAL FLUMINENSE CENTRO DE ESTUDOS GERAIS INSTITUTO DE QUÍMICA PÓS-GRADUAÇÃO EM GEOCIÊNCIAS – GEOQUÍMICA
LÍVIA FERREIRA DE MELO GUEDES
SEPARAÇÃO EM FASE SÓLIDA PARA A DETERMINAÇÃO DE ÂNIONS POR CROMATOGRAFIA DE ÍONS EM AMOSTRAS SALINAS, AMBIENTAIS E DA
INDÚSTRIA DE PETRÓLEO
Niterói 2010

LÍVIA FERREIRA DE MELO GUEDES
SEPARAÇÃO EM FASE SÓLIDA PARA A DETERMINAÇÃO DE ÂNIONS POR CROMATOGRAFIA DE ÍONS EM AMOSTRAS SALINAS, AMBIENTAIS E DA
INDÚSTRIA DE PETRÓLEO
Dissertação de Mestrado apresentada ao curso de Pós-Graduação em Geociências da Universidade Federal Fluminense, como requisito parcial para obtenção do Grau de Mestre em Geociências, área de concentração: Geoquímica Ambiental.
Niterói 2010

G934 Guedes, Lívia Ferreira de Melo.
Separação em fase sólida para a determinação de ânions por cromatografia de íons em amostras salinas, ambientais e da indústria do petróleo. / Lívia Ferreira de Melo Guedes. – .Niterói: [s.n.], 2010.
76 f. : il. ; 30 cm.
Dissertação (Mestrado em Geociências – Geoquímica Ambiental) - Universidade Federal Fluminense, 2007. Orientador: Prof. Dr.Ricardo Erthal Santelli.
1. Cromatografia. 2. Trocador de íons. 3. Água do mar -
salinidade. 3. Ãnions. 4. Tese. 5. Produção intelectual. I. Título.
CDD 543.0893
CDD 543.0893

LÍVIA FERREIRA DE MELO GUEDES
SEPARAÇÃO EM FASE SÓLIDA PARA A DETERMINAÇÃO DE ÂNIONS POR CROMATOGRAFIA DE ÍONS EM AMOSTRAS SALINAS, AMBIENTAIS E DA
INDÚSTRIA DE PETRÓLEO
Dissertação de Mestrado apresentada ao curso de Pós-Graduação em Geociências da Universidade Federal Fluminense, como requisito parcial para obtenção do Grau de Mestre em Geociências, área de concentração: Geoquímica.
Aprovada em Março de 2010.
BANCA EXAMINADORA
Prof. Dr. Ricardo Erthal Santelli - Orientador UFF
Prof. Dr. John Edmund Lewis Maddock UFF
Prof. Dr. Emmanoel Vieira da Silva Filho UFF
Prof. Dr. Valderi Luiz Dressler UFSM
Dra. Maria de Fátima Batista de Carvalho
Petrobrás
Niterói 2010

AGRADECIMENTOS
Agradeço inicialmente a minha filha que é minha inspiração diária e a Tiago
pelo companheirismo, atenção e, principalmente, paciência. Sem eles nada seria
possível.
Aos meus pais e familiares pela ajuda e empurrão nos momentos difíceis e
de alegria.
À meu orientador que me auxiliou por várias vezes e de diferentes formas.
A Maria de Fátima Batista de Carvalho pela inspiração e auxílio em todo o
trabalho.
A Rede Temática de Geoquímica da Petrobrás.
Ao programa de Pós Graduação em Geoquímica e à Hildete e Nivaldo pelo
socorro em várias horas. Aos professores por todo o ensinamento que levo comigo.
A Aline, pela paciência, amizade e acima de tudo por estar sempre presente.
Ajudou-me muito com as análises e leituras. Valeu amiga!
Aos amigos Ricardo e Aline pelo trabalho escravo voluntário. Vocês são
demais!
A todos os amigos pela ajuda, pelo esforço e os momentos de descontração,
graças a vocês não fiquei louca. Aos companheiros dos laboratórios 403 e 108 pela
ajuda e incentivo.
A todos que de alguma forma contribuíram para este trabalho.
Obrigada.

O simples bater das asas de uma
borboleta pode gerar um furação do
outro lado do planeta.

LISTA DE FIGURAS
Figura 1: Cromatograma de íons obtido para uma amostra com alta salinidade
destacando o sinal elevado devido à presença de alta concentração de cloreto. ..... 16
Figura 2: Estrutura esquemática de uma resina trocadora de íons. Fonte: Helfferich,
1995. ......................................................................................................................... 21
Figura 3: Rota de síntese de resinas trocadoras aniônicas. Fonte: Harland, C.E. .... 23
Figura 4: Reações de síntese de resina trocadora fortemente ácida, na forma
hidrogenada. ............................................................................................................. 24
Figura 5: Reação de síntese da resina trocadora de cátions carboxílica .................. 25
Figura 6: Representação esquemática das estruturas das resinas: (A) Microporosa
(tipo gel); (B) Macroporosa (macroreticular). Fonte: Kressman, T.R.E. .................... 26
Figura 7: Representação esquemática de um sistema de cromatografia de íons com
supressão de condutividade. Fonte: Dionex.............................................................. 30
Figura 8: Representação esquemática do processo de supressão da condutividade
do eluente. Fonte: Dionex ......................................................................................... 31
Figura 9: Representação esquemática da supressão de condutividade em
cromatografia de íons. ............................................................................................... 32
Figura 10: Cromatograma mostrando alguns parâmetros cromatográficos .............. 33
Figura 11: Cromatograma mostrando o modo de cálculo da assimetria do pico. ...... 35
Figura 12: Representação de um plasma indutivamente acoplado, onde H
representa o campo magnético induzido e I, a bobina de indução. Adaptada de
JARVIS et al., 1992. .................................................................................................. 37
Figura 13: Foto de um plasma em operação. (THERMO, 2007) ............................... 38
Figura 14: Esquema mostrando as conficgurações Radia (a) e Axial (b) de
observação do plasma. ............................................................................................. 40

Figura 15: Representação esquemática de um plasma com observação radial e axial
- Dual View (THERMO, 2005) ................................................................................... 40
Figura 16: Fotografia do ICP OES Thermo Scientific Modelo iCAP 6300 ................. 44
Figura 17: Cromatógrafo de Íons utilizado nas análises. ........................................... 45
Figura 18: Fluxograma de etapas para o tratamento das amostras hiper salinas. .... 46
Figura 19: Gráfico mostrando as intensidades da Visão Axial em função da
concentração de Ag+ em mg L-1 ................................................................................ 51
Figura 20: Gráfico mostrando as intensidades da Visão Radial em função da
concentração de Ag+ em mg L-1 ................................................................................ 51
Figura 21: Teste de retenção de cloreto das quatro diferentes resinas ..................... 53
Figura 22: Variação da concentração dos íons cloreto e prata em função do volume
de amostra de matriz salina a ser percolada em mini coluna contendo resina IR 120
.................................................................................................................................. 57
Figura 23: Variação da concentração dos íons cloreto e prata em função do volume
de amostra de matriz salina a ser percolada em mini coluna contendo resina Dowex.
.................................................................................................................................. 58
Figura 24: Variação da retenção de prata referente as quatro resinas em estudo em
função do volume de amostra percolado. ................................................................. 59
Figura 25: Variação da retenção de cloreto pela resina Amberlite IR 120 na forma de
Ag+ e de retenção de prata pela resina Amberlite IR 120 na forma H+ em função do
volume de amostra .................................................................................................... 60
Figura 26: Variação da retenção de cloreto pela resina Amberlite IR 120 na forma
Ag+ e de retenção de prata pela resina Dowex W 50 na forma H+ em função do
volume de amostra. ................................................................................................... 61
Figura 27: Variação da retenção de íons e concentração de prata lançada na
amostra pelos cartuchos de retenção de cloreto comerciais. .................................... 62

Figura 28: Variação da retenção de íons cloreto e prata residual carreada, que é
encontrada após o uso dos cartuchos comerciais. .................................................... 63
Figura 29: Cromatograma do padrão misto de seis ânions utilizado nos testes de
retenção. ................................................................................................................... 64
Figura 30: Variação da retenção dos ânions nitrito, nitrato, brometo e fosfato
referente a primeira fração de 0,5 mL. ...................................................................... 64
Figura 31: Variação da retenção dos ânions fluoreto e sulfato referente à primeira
fração eluída na coluna. ............................................................................................ 65
Figura 32: Variação da retenção dos ânions nitrito, nitrato, brometo e fosfato
referente a segunda fração eluída. ............................................................................ 66
Figura 33: Variação da retenção dos ânions fluoreto e sulfato referente a segunda
fração eluída. ............................................................................................................. 66
Figura 34: Cromatograma de solução com alta concentração de cloreto após o uso
da resina.................................................................................................................... 67
Figura 35: Cromatograma obtido da primeira fração de solução de alta salinidade
contendo os seis ânions de trabalho. ........................................................................ 68

LISTA DE TABELAS
Tabela 1: Comparativo entre resinas do tipo gel e macroporosas. Fonte: Dowex,
2006 .......................................................................................................................... 26
Tabela 2: Parâmetros instrumentais utilizados durante os estudos envolvendo a
determinação de prata nas amostras percoladas nas minicolunas ........................... 44
Tabela 3: Propriedades das resinas trocadoras de cátions ....................................... 48
Tabela 4: Valores de intensidade de emissão obtidos para os diferentes modos de
observação de acordo com a concentração dos padrões. ........................................ 51
Tabela 5: Concentração dos íons contidos no padrão preparado com seis diferentes
ânions. ....................................................................................................................... 53
Tabela 6: Resultados obtidos para a determinação da capacidade de troca catiônica
(CTC) das resinas e seus respectivos percentuais de umidade................................ 55
Tabela 7: Resultados obtidos na determinação das concentrações dos ânions nas
duas frações utilizando cartuchos comerciais. .......................................................... 69

RESUMO
A análise de efluentes hipersalinos ainda é um problema analítico a ser resolvido.
Atualmente, tem ocorrido um grande aumento da demanda para caracterização de
águas de alta salinidade, tendo em vista que esta é um dos maiores descartes da
indústria de petróleo. Devido a isto, técnicas analíticas para sua caracterização têm
sido desenvolvidas amplamente e entre elas temos a cromatografia de íons, onde
diferentes tipos de ânions são quantificados. Contudo, para que seja possível o uso
desta técnica, é imprescindível que haja a retirada do cloreto dessas matrizes
hipersalinas. Essa separação prévia pode ser realizada através do emprego de
cartuchos comercializados, porém de custo elevado. Sendo assim, têm sido
desenvolvidos estudos com trocadores iônicos onde são utilizados no tratamento de
diferentes tipos de matriz de amostra. Para o tratamento de matrizes salinas,
estudos com trocadores catiônicos tratados com prata, como Amberlite IR 120 e
Dowex W50, foram realizados mostrando-se eficientes na remoção do íon cloreto.
Amostras de diferentes salinidades foram eluídas através de mini colunas
preenchidas com estas resinas tratadas com prata. Entretanto, o seu uso leva à
coluna íons de prata que também são retirados de forma eficaz do meio, através de
mini colunas preenchidas com resinas na forma de hidrogênio, de modo que a
coluna analítica não seja afetada. Análises comparativas com cartuchos comerciais
de retenção de cloreto e prata foram realizadas, comprovando a eficiência do
método. Testes para a retenção dos analitos foram realizados e mostraram que a
primeira alíquota de 0,5 mL retém boa parte dos ânions de trabalho tanto nas
resinas de estudo quanto nos cartuchos comerciais.
Palavras - chave: Cromatografia de íons, trocadores catiônicos, águas hipersalinas,
cloreto, ânions.

ABSTRACT
The analysis of hypersaline wastewater is still an analytical problem to be solved.
Currently, there has been an increased demand for characterization of high salinity
water, considering that this is one of the largest discharges of oil industry. Because of
this, analytical methods for their characterization have been developed extensively,
and among them we have the ion chromatography, where different types of anions
are quantified. However, it is possible to use this technique, it is crucial that the
withdrawal of these matrices hypersaline chloride. This separation can be
accomplished in advance through the use of cartridges sold, however costly. Thus,
studies have been developed with ion exchangers which are used to treat different
types of sample matrix. For the treatment of salt matrices studies with cationic
exchangers treated with silver, such as Amberlite IR 120 and Dowex W50 were
performed showing to be efficient in the removal of chloride ion. Samples of different
salinities were eluted through mini columns filled with these resins treated with silver.
However, its use leads to a column of silver ions that are also effectively removed the
medium, using mini columns filled with resins in the form of hydrogen, so that the
analytical column is not affected. Comparisons with commercial cartridges retention
of chloride and silver were performed, proving the efficiency of the method. Tests for
retention of the analytes were performed and showed that the first rate of L retains
much of the work of anions in both resins in the study as 0.5 µL commercial
cartridges.
Keywords: Ion Chromatography, cationic exchangers, high salinity water, chloride,
anions.

SUMÁRIO
Lista de Figuras
Lista de Tabelas
Resumo
Abstract
1 – INTRODUÇÃO .................................................................................................... 13
2 – JUSTIFICATIVA E OBJETIVO ...................... ..................................................... 17
3 – REVISÃO BIBLIOGRÁFICA ......................... ...................................................... 19
3.1 - RESINAS TROCADORAS DE ÍONS................................................................. 19
3.1.1 - Resinas Trocadoras de Ânions ................................................................... 21
3.1.2 – Resinas Trocadoras de Cátions ................................................................. 23
3.1.3 – Resinas Macroporosas e Microporosas .................................................... 25
3.1.4 – Capacidade de Troca ....................... ........................................................... 27
3.2 – HISTÓRICO DA CROMATOGRAFIA ............................................................... 27
3.3 – CROMATOGRAFIA DE ÍONS .......................................................................... 28
3.4 – PARÂMETROS CROMATOGRÁFICOS DE INTERESSE ANALÍTICO ........... 32
3.5 - ESPECTROMETRIA DE EMISSÃO ÓPTICA COM FONTE DE PLASMA
INDUTIVAMENTE ACOPLADO – ICP OES .............................................................. 36
4 - MATERIAIS E MÉTODOS ............................... .................................................... 41
4.1 – SOLUÇÕES E REAGENTES ........................................................................... 41
4.1.1 – Análises Volumétricas ..................... ........................................................... 41
4.1.2 – Determinação dos íons ..................... .......................................................... 42
4.2 – INSTRUMENTAÇÃO ........................................................................................ 43
4.3 – METODOLOGIA UTILIZADA ........................................................................... 45
4.3.1 – Calculo do teor de umidade das resinas util izadas.................................. 47
4.3.2 – Determinação da capacidade de troca das res inas.................................. 47

4.3.3 – Testes de troca iônica das resinas trocador as de cátions ...................... 48
4.3.4 – Aplicação do método em amostras de matriz c om alta salinidade..... Erro!
Indicador não definido.49
4.3.5 – Determinação de prata remanescete na resina por ICP OES .................. 50
4.3.6 -Testes para retenção da prata remanescente a pós passagem pelas
colunas contendo resinas a base de prata........... .................................................51
4.3.7 – Testes de retenção de ânions pelas colunas modificadas com prata .... 52
5 – RESULTADOS E DISCUSSÕES ....................... ................................................. 54
5.1 – TEOR DE UMIDADE E CAPACIDADE DE TROCA DAS RESINAS ................ 55
5.2 – TESTES DE TROCA IÔNICA DAS RESINAS TROCADORAS DE CÁTIONS . 55
5.3 – APLICAÇÃO DO MÉTODO A MATRIZES DE ALTA SALINIDADE E
DETERMINAÇÃO DE PRATA POR ICP OES .......................................................... 56
5.4 – COMPARAÇÃO DA EFICIÊNCIA DAS MINI-COLUNAS COM CARTUCHOS
COMERCIAIS ............................................................................................................ 61
5.5 – POSSÍVEL RETENÇÃO DE ÂNIONS PELAS RESINAS MODIFICADAS ....... 63
6 - CONCLUSÕES .................................................................................................... 70
7 – REFERÊNCIAS BIBLIOGRÁFICAS .................... ............................................... 72


1 INTRODUÇÃO
Atualmente a energia é essencial para a maior parte das atividades humanas.
A produção da energia hoje é feita da exploração de diferentes recursos naturais
que provocam uma série de mudanças ambientais. A necessidade de energia é uma
realidade desde a Revolução Industrial, quando se iniciou o uso intensivo de
combustíveis fósseis, como carvão mineral e o petróleo. Com o auge da revolução, o
uso do petróleo aumentou em larga escala na indústria e o combustível teve o seu
aumento após a Segunda Guerra Mundial, quando o seu uso energético aumentou
ate chegar ao que temos nos dias atuais. Além de grande importância na geração de
energia, o petróleo gera subprodutos que são matéria prima para a manufatura de
inúmeros bens de consumo que têm um papel cada vez mais relevante na vida do
ser humano.
Com base nessa grande necessidade energética, a indústria petrolífera no
Brasil se encontra em franca expansão. Suas atividades, porém, além de grandes
lucros financeiros podem resultar em uma série de custos ambientais. A criação e
exploração de novos campos petrolíferos têm sido cada vez mais intensificadas.
Nas atividades de exploração e produção de óleo e gás é gerada uma grande
quantidade de resíduos e efluentes, dentre os quais podemos destacar a água de
formação e a água produzida. A água de formação (também conhecida como água
conata) esta localizada na região de formação junto com o petróleo. Já a água de
produção, é toda a água produzida (carreada) junto com o óleo ou a água injetada
no reservatório (água de injeção). A água é injetada para aumentar a produção
mantendo as condições de pressão na rocha reservatório (fundamentais para a

migração do petróleo para os poços, sendo efetuada uma operação de injeção de
água nas camadas inferiores da rocha reservatório, e/ou gás nas camadas
superiores).
Plataformas de gás tendem a produzir menor volume de água produzida,
porém com maiores concentrações de contaminantes orgânicos. Plataformas de
óleo, ao contrário, geralmente produzem altos volumes de água produzida. Um
campo novo produz de 5 a 15% de volume de água. À medida que a vida econômica
dos poços se esgota, essa água pode atingir uma faixa de 75 a 90% volume.
Enquanto muitos campos de gás descarregam menos de 10 m3 de água por dia, a
maioria dos campos produtores de óleo descarrega centenas ou até mesmo
milhares de m3 de água por dia (OGP, 2005).
Esta produção excessiva de água se torna um problema sério nos campos de
petróleo maduros, pois dificulta a sua disposição final, tendo em vista que a sua
composição é extremamente complexa, possuindo uma grande diversidade de
contaminantes. Esta composição química varia amplamente e depende
principalmente dos atributos geológicos, do tipo de reservatório e de sua maturidade.
Atualmente, a maior preocupação com relação aos processos de tratamento e
purificação dessas águas diz respeito aos contaminantes que permanecem
dissolvidos, os quais são mais difíceis de serem removidos por técnicas de
tratamentos convencionais.
Deve-se ter em mente que a água produzida é um dos componentes gerados
pela produção de petróleo, e que esta não pode ser usada devido aos fatores
limitados pela qualidade desta água. Um processo óbvio é a reciclagem da água,
com sua reinjeção no reservatório da qual foi produzida. Isto pode ser feito quando
se usam métodos de injeção de água para manutenção de pressão no reservatório,
porém mesmo para este método é requerido um tratamento prévio. Assim sendo, a
água produzida deve ser encarada como resíduo, devendo ser descarregada como
tal, sendo usada toda uma metodologia gerencial, econômica e ambientalmente
aceitável.
Em áreas offshore o descarte é feito em grandes ambientes receptores, como
mar aberto, onde a diluição e a dispersão rápida tomam lugar. Isto poderia ser uma
argumentação para o não tratamento da água produzida. Mas fatores tais como
correntes marítimas, ventos, temperatura da água, mudança de clima, podem

transportar ou mesmo concentrar alguns de seus constituintes (fenóis, íon NH4+
etc.).
A água produzida contém quantidades variadas de sais e gases dissolvidos
(CO, CO2, H2S), sólidos em suspensão, altos teores de contaminantes tóxicos como
metais (Cd, Cr, Cu, Pb, Hg, Ag, Ni, Zn), produtos químicos adicionados durante a
injeção (tais como inibidores de corrosão, inibidores de incrustação,
desemulsificantes, metanol, glicol, polieletrólitos), componentes com algum nível de
radiação, uma complexa mistura de compostos orgânicos e inorgânicos (cuja
composição varia com a vida do campo petrolífero) e altas concentrações de
cloretos (GARCIA, 1996). Estes componentes a tornam imprópria para consumo
humano e animal. O impacto ambiental dessas águas é avaliado pela toxicidade dos
constituintes e pela quantidade de compostos presentes. Alguns destes constituintes
permanecerão dissolvidos, enquanto outros são convertidos, seja por decomposição,
evaporação, transformação em outro composto não tóxico, depotando-se no fundo
do mar, etc. Os efeitos mais nocivos ao meio ambiente são aqueles associados aos
compostos que permanecem solúveis após o descarte, por interagirem diretamente
com a vida presente neste meio.
A produção de petróleo offshore origina e faz uso de águas de alta salinidade,
e devido aos grandes volumes gerados e da composição química complexa, faz-se
necessário um estudo detalhado de metodologias analíticas aplicadas a
caracterização destas águas, tendo em vista seus aspectos ecotoxicológicos e de
descarte. Em face à sua composição tão variável quanto a sua toxicidade, torna-se
necessário o estudo sobre a sua disposição, assim como a avaliação dos possíveis
impactos ao meio ambiente. Faz-se necessária uma análise criteriosa para
determinar o melhor tipo de tratamento e definir o método de descarte. A análise
química é um dos melhores meios para se detectar os problemas, portanto deve ser
feita como atividade de rotina em toda água produzida. Para isso, técnicas analíticas
que possam contribuir para a determinação dos constituintes destas águas se
tornam importantes pela necessidade do desenvolvimento de métodos analíticos,
robustos, simples, precisos e acurados, que permitam a determinação das espécies
de interesse nestas águas com a maior confiabilidade possível.
Mediante a importância de se ter o maior conhecimento possível dos
componentes da amostra, a Cromatografia de Íons é um dos métodos
cromatográficos com grande potencial para auxiliar na caracterização química da
água produzida. A Cromatografia de Íons, no modo de troca iônica, consiste na
separação e detecção de íons onde uma coluna de resina trocadora age como uma
fase estacionária, retendo temporariamente os íons que passam juntamente com o
eluente e, de acordo com afinidade diferenciada desses íons pela fase estacionária,
ocorre a separação.
Os íons, depois de separados, chegam até o detector onde os sinais são
registrados e os resultados são obtidos através da avaliação da área ou altura do
sinal onde, em ambos os casos, são proporcionais à concentração do analito.
Quando ocorre de um dos analitos estar em altas concentrações, há uma
sobreposição dos sinais, não ocorrendo a devida separação e, conseqüentemente,
impossibilitando a quantificação.
Este tipo de comportamento pode ser bem observado em águas de alta
salinidade, onde a separação não ocorre de modo eficaz devida à elevada
concentração do ânion cloreto. Neste caso, ocorre a sobreposição dos sinais de
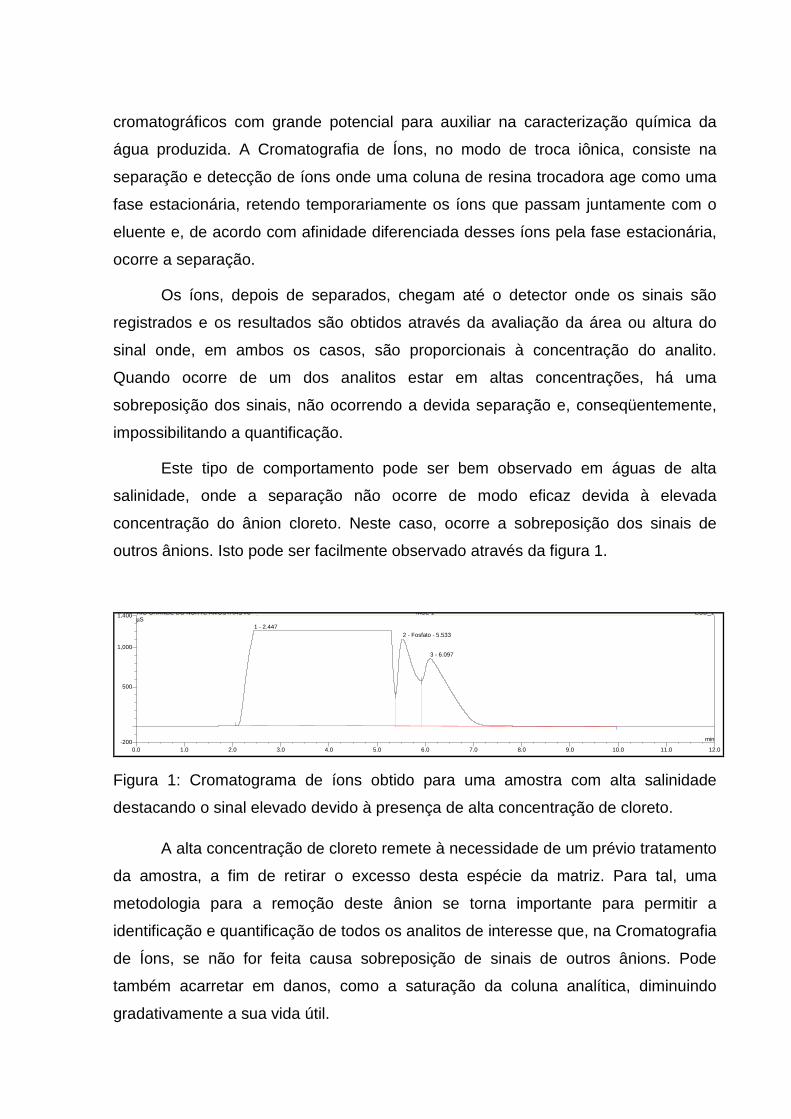
outros ânions. Isto pode ser facilmente observado através da figura 1.
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0-200
500
1,000
1,400 RIO GRANDE DO NORTE AMOSTRAS #8 MCE 1 ECD_1µS
min
1 - 2.4472 - Fosfato - 5.533
3 - 6.097
Figura 1: Cromatograma de íons obtido para uma amostra com alta salinidade
destacando o sinal elevado devido à presença de alta concentração de cloreto.
A alta concentração de cloreto remete à necessidade de um prévio tratamento
da amostra, a fim de retirar o excesso desta espécie da matriz. Para tal, uma
metodologia para a remoção deste ânion se torna importante para permitir a
identificação e quantificação de todos os analitos de interesse que, na Cromatografia
de Íons, se não for feita causa sobreposição de sinais de outros ânions. Pode
também acarretar em danos, como a saturação da coluna analítica, diminuindo
gradativamente a sua vida útil.

2 JUSTIFICATIVA E OBJETIVO
Como visto, a indústria de petróleo, em seu segmento de produção, gera
águas de alta salinidade, cuja caracterização é dificultada pela sua matriz complexa.
Contudo, devido ao grande volume gerado de água produzida durante a extração do
petróleo o estudo de seus constituintes se torna fundamental para que um descarte
adequado seja realizado.
Para isso, o estudo de técnicas analíticas que possam auxiliar na
determinação dos constituintes destas águas se torna importante, visando uma
melhor caracterização das mesmas e auxiliar na melhor forma de descarte ou reuso
destas águas.
O objetivo geral da presente dissertação consiste no desenvolvimento de
metodologias analíticas para a caracterização de águas hipersalinas (de formação e
produzida) da indústria de petróleo em relação a constituintes inorgânicos (ânions).
Como objetivo específico, pretende-se desenvolver métodos analíticos
utilizando colunas de troca iônica para retirada de cloreto e fazer uso da
cromatografia de íons para determinar alguns ânions que estão presentes nestas
águas, após a devida separação do íon cloreto.
Para o tratamento da matriz hipersalina, serão testados diferentes tipos de
materiais (trocadores iônicos) à base de resina catiônica na forma de prata, com a
finalidade de se encontrar uma boa alternativa para a retirada do cloreto existente na
matriz da amostra e viabilizar a posterior caracterização por cromatografia de íons
dos constituintes desta amostra pré tratada. Como trocadores iônicos, serão

testados resinas poliméricas tratadas com íon prata para que haja a retenção de
cloreto e, conseqüentemente, a sua retirada da matriz.

3 REVISÃO BIBLIOGRÁFICA
3.1 RESINAS TROCADORAS DE ÍONS
A troca iônica é um processo de superfície que ocorre entre o contato de um
sólido iônico e uma solução eletrolítica sem mudanças significativas na estrutura do
sólido. Diferentes substâncias naturais (incluindo alguns silicatos, celulose e outros)
possuem propriedades de troca iônica.
As primeiras observações, registradas na literatura, que fazem referência a
troca iônica foram realizadas por Way e Thompson em 1850. Eles descobriram que
o solo, ao ser percolado por soluções de NH4+, tinha a capacidade de remover estes
íons, substituindo-os por quantidades equivalentes de íons Ca2+. Partindo dessas
observações, varias tentativas de produzir trocadores iônicos mais apropriados
foram realizadas e, apesar da dificuldade encontrada, obteve-se êxito na aplicação
da troca iônica para o tratamento de água.
Uma vez que a natureza da troca iônica tinha sido estabelecida por
experiências em trocadores naturais (como aluminossilicatos), explorou-se o
potencial de outros materiais, como certas substâncias orgânicas. Hoje os
trocadores iônicos tornaram-se quase sinônimo de produtos sintéticos de alto poder
de troca iônica: as resinas de troca iônica.
As primeiras resinas sintéticas de troca iônica foram preparadas por Adams e
Holmes em 1935. A base de sua síntese foi a polimerização por condensação do
formaldeído (metanal) com fenol ou compostos de benzeno polissubstituidos,

contudo estas não eram totalmente eficientes. As pesquisas nesta área tiveram
continuidade e, em 1940, foram desenvolvidas resinas trocadoras baseadas na
copolimerização do estireno e divinilbenzeno. Estas resinas eram utilizadas como
um meio de tratamento da água.
Para tornar o polímero ativo, grupos funcionais ácidos e básicos são
quimicamente ligados à estrutura da resina, dando a elas características de
trocadores catiônicos e aniônicos, respectivamente. Os trocadores portadores de
cátions trocáveis são chamados de trocadores catiônicos, já os portadores de ânions
trocáveis são chamados de trocadores aniônicos. Os grupos funcionais mais
utilizados nas estruturas das resinas são ácidos sulfônicos (trocados catiônicos) e
aminas quaternárias (trocadores aniônicos).
Os trocadores iônicos são polímeros portadores de carga elétrica que é
estabilizada pelos contra-íons, como pode ser visto pela figura 2, onde um trocador
catiônico é constituído por um polímero contendo ânions ligados quimicamente
(como os ácidos sulfônicos) e cátions ativos como contra-íons e vice-versa.
Tanto as resinas catiônicas quanto as aniônicas são produzidas a partir dos
mesmos polímeros, diferindo apenas quanto aos grupamentos ionizáveis presos às
cadeias carbônicas. É esse grupo funcional que determina o comportamento químico
da resina.

Figura 2: Estrutura esquemática de uma resina trocadora de íons. Fonte: Helfferich,
1995.
Uma boa resina trocadora de íons deve ter algumas características:
suficientemente hidrofílica; com solubilidade desprezível; fisicamente estável; mais
densa que a água; resistente a ácidos e bases, a oxidação ou redução e a radiação;
constituída por material inerte e conter um número adequado de grupos trocadores
de íons.
A presença de divinilbenzeno (DVB) resulta em ligações cruzadas que
confere estabilidade mecânica à resina. A arrumação das cadeias carbônicas
interconectadas é o que assegura a pouca solubilidade das resinas trocadoras de
íons na maioria dos solventes, o que faz com que elas não sejam destruídas. As
resinas com muitas ligações cruzadas são, em geral, mais duras e mais
impermeáveis do que os materiais com poucas ligações cruzadas.
3.1.1 Resinas trocadoras de ânions
Resinas trocadoras de ânions são usualmente baseadas em grupamentos
amina e amônio com cargas fixas positivas. Sendo trocadores básicos, possuem
grupamentos do tipo amino, amino substituídos e amônio quaternário, onde os dois

primeiros possuem propriedades básicas fracas e a última sendo um trocador de
base forte.
A figura 3 mostra como ocorre a síntese de resinas trocadoras de ânions onde
é possível obter os trocadores aniônicos fracos e fortes.
As resinas de troca aniônica realizam troca entre os ânions da solução com
os ânions livres das resinas – também conhecidos como contra íons. Essas resinas
podem ser fracamente básicas e fortemente básicas. Para resinas fracamente
básicas, o grau de ionização é altamente influenciado pelo pH (ou seja, há poucos
ânions livres para que ocorra a troca aniônica), onde em pH acima de 7,0 a troca
aniônica é mínima. Já para as resinas fortemente básicas, que estão amplamente
ionizadas, a troca aniônica é máxima.
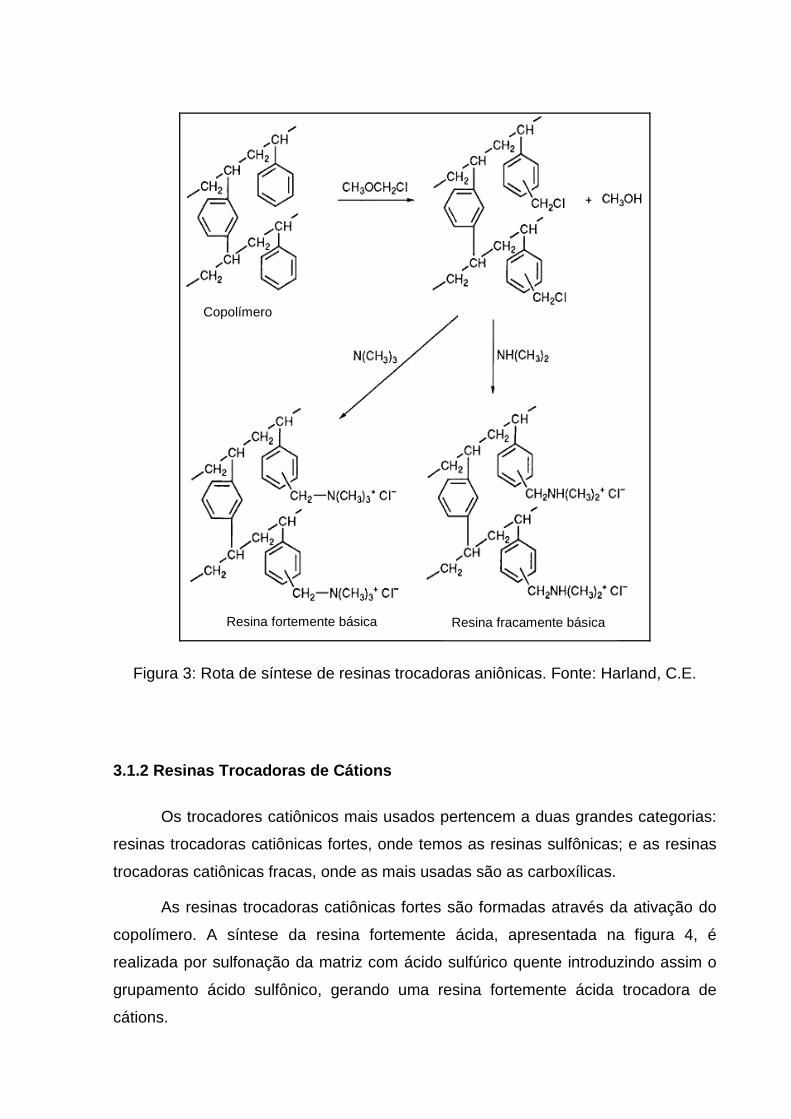
Figura 3: Rota de síntese de resinas trocadoras aniônicas. Fonte: Harland, C.E.
3.1.2 Resinas Trocadoras de Cátions
Os trocadores catiônicos mais usados pertencem a duas grandes categorias:
resinas trocadoras catiônicas fortes, onde temos as resinas sulfônicas; e as resinas
trocadoras catiônicas fracas, onde as mais usadas são as carboxílicas.
As resinas trocadoras catiônicas fortes são formadas através da ativação do
copolímero. A síntese da resina fortemente ácida, apresentada na figura 4, é
realizada por sulfonação da matriz com ácido sulfúrico quente introduzindo assim o
grupamento ácido sulfônico, gerando uma resina fortemente ácida trocadora de
cátions.
Resina fortemente básica Resina fracamente básica
Copolímero
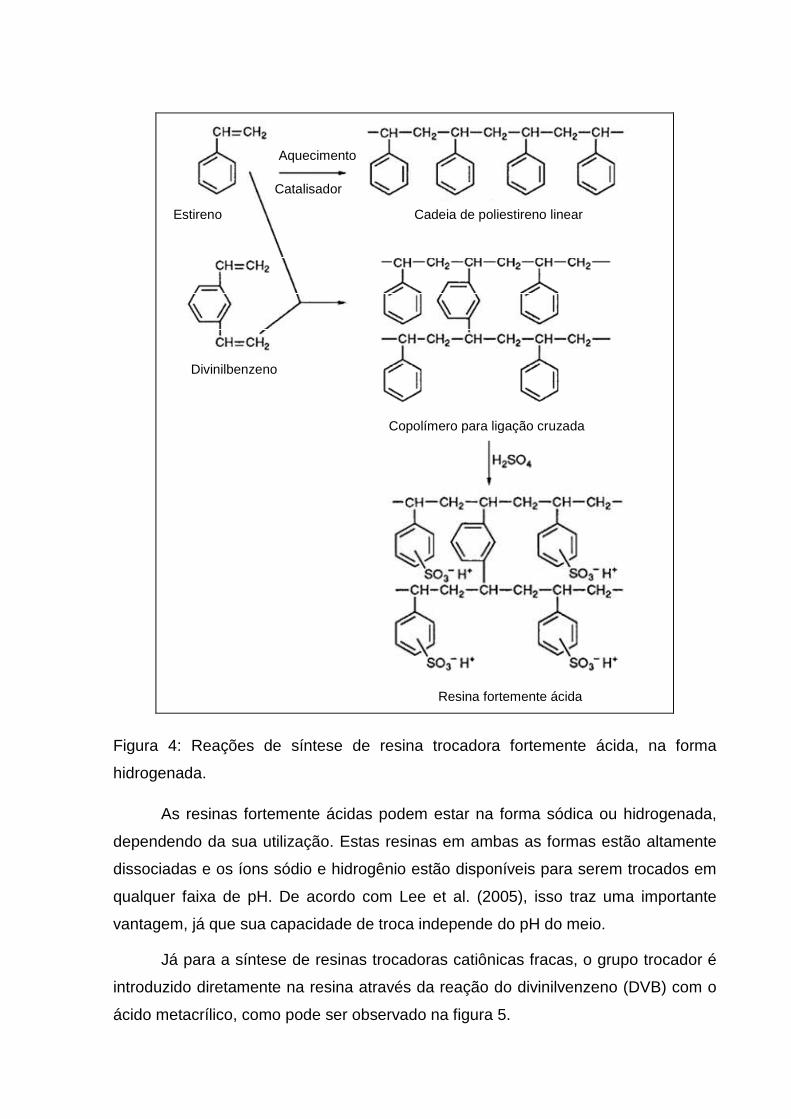
Figura 4: Reações de síntese de resina trocadora fortemente ácida, na forma
hidrogenada.
As resinas fortemente ácidas podem estar na forma sódica ou hidrogenada,
dependendo da sua utilização. Estas resinas em ambas as formas estão altamente
dissociadas e os íons sódio e hidrogênio estão disponíveis para serem trocados em
qualquer faixa de pH. De acordo com Lee et al. (2005), isso traz uma importante
vantagem, já que sua capacidade de troca independe do pH do meio.
Já para a síntese de resinas trocadoras catiônicas fracas, o grupo trocador é
introduzido diretamente na resina através da reação do divinilvenzeno (DVB) com o
ácido metacrílico, como pode ser observado na figura 5.
Resina fortemente ácida
Copolímero para ligação cruzada
Cadeia de poliestireno linear
Divinilbenzeno
Estireno
Catalisador
Aquecimento

Figura 5: Reação de síntese da resina trocadora de cátions carboxílica
As resinas fracamente ácidas possuem comportamento químico semelhante
aos ácidos orgânicos fracos, sendo fracamente ionizáveis. Elas exibem uma
afinidade muito maior por íons hidrogênio do que as resinas fortemente ácidas. O
grau de dissociação dessas resinas é fortemente influenciado pelo pH do meio e, de
acordo com Schuweitzer (1979), elas possuem capacidade de troca muito reduzida
em valores de pH inferiores a 6,0.
3.1.3 Resinas Macroporosas e Microporosas
As resinas trocadoras catiônicas e aniônicas podem ser classificadas como
macroporosas e microporosas (também conhecidas como resinas do tipo gel), onde
a diferença estrutural entre elas pode ser vista na figura 6. A maioria dos trabalhos
desenvolvidos até hoje é realizado com resinas microporosas que contêm, no geral,
cerca de 8% de DVB, o que gera baixo conteúdo de ligações cruzadas, como pode
ser observado na figura 6A.
As resinas macroporosas (também conhecidas como resinas
macroreticulares) possuem uma estrutura rígida, esférica e possuem grande área de
superfície, como pode ser observado na figura 6B. Elas são mais rígidas por
possuírem uma grande quantidade de ligações cruzadas, o que não afeta a sua
capacidade de troca, já que elas possuem poros e canais por onde facilmente
Ácido Metacrílico
Divinilbenzeno Resina acrílica fracamente ácida

penetram os íons. Segundo Godos (2004) as resinas macroreticuladas são
usualmente usadas em processos catalíticos.
Figura 6: Representação esquemática das estruturas das resinas: (A) Microporosa
(tipo gel); (B) Macroporosa (macroreticular). Fonte: Kressman, T.R.E.
A morfologia dos polímeros influencia diretamente no seu desempenho e nas
suas aplicações. Para aplicação como adsorvente, a estrutura química e porosidade
são os parâmetros que afetam a capacidade total e a seletividade da resina.
Atualmente as resinas são comercializadas com teores de DVB que variam de 2 a
20%, sendo este o que atribui à resina o caráter microreticular ou macroreticular. A
tabela 1 mostra um comparativo entre os dois tipos de resinas onde é possivel
observar as suas caracteristicas.
Tabela 1: Comparativo entre resinas do tipo gel e macroporosas. Fonte: Dowex,
2006
Propriedades Gel Macroporosa Comentários
Capacidade
de troca Maior Menor -
Cinética Maior Menor -
Eficiência na
regeneração Maior Menor
O maior grau de ligações cruzadas nas
resinas macroporosas leva a eluições
mais lentas.
Estabilidade à
oxidação Menor Maior
Degradação térmica ou química devido
à quebra das ligações cruzadas e não
devido a sua desfuncionalização.
(A) (B)

Seletividade Menor Maior
Resinas macroporosas são mais
seletivas levando em conta o tamanho
do íon hidratado.
3.1.4 Capacidade de troca
Possivelmente esta é uma das características mais importantes de um
trocador iônico. É definido como o número total de equivalentes de um íon trocável
por quilograma de resina seca, ou seja, o número de miliequivalentes (meq) por
grama de resina seca. Por definição, o equivalente de um íon trocador será a massa
em gramas do íon (ou massa molar) por unidade de carga.
3.2 HISTÓRICO DA CROMATOGRAFIA
O termo “cromatografia” deriva das palavras chrom (cor) e graphe (escrever)
e foi atribuído ao botânico russo Mikhael Semenovich Tswett, que realizou uma
separação de componentes de extratos de folhas em uma fase sólida polar e
interpretou este processo. Apesar do nome, o botânico explicitou que o processo
não dependia da cor, exceto para facilitar o processo de separação.
“Cromatografia” é um termo geral utilizado para uma variedade de técnicas de
separação físico-químicas, das quais todas têm em comum a afinidade entre a fase
móvel e a fase estacionaria.
Em 1938 houve a descoberta da cromatografia em camada fina, que foi
aprimorada e refinada até que, em 1958, se tornou a técnica como é conhecida
atualmente.
Nos anos 1940 Martin e Synge publicaram a descoberta da cromatografia de
partição líquido-líquido. Eles aplicaram a teoria de altura equivalente a um prato e
anteciparam o surgimento de dois métodos cromatográficos: a cromatografia a gás e
a cromatografia a líquido de alta eficiência. A cromatografia a gás foi descrita ainda
nesta década por Hesse e colaboradores que separaram ácidos graxos no vapor a
100ºC, arrastando-os sobre sílica com dióxido de carbono.

Em 1952, Martin e James publicaram o primeiro trabalho com cromatografia a
gás, iniciando um rápido desenvolvimento desta técnica.
Outros desenvolvimentos na década de 1960 aperfeiçoaram os sistemas de
bombeamento e detecção em cromatografia a líquido de alta eficiência,
comprovando que o uso destes equipamentos, operado com fase móvel líquida sob
pressão e com métodos de detecção sensíveis, possibilita análises com rapidez
comparável às da cromatografia a gás, com resultados altamente satisfatórios
(Collins, 1990).
Em 1975 a cromatografia de íons foi introduzida por Small, Stevens e Bauman
como uma nova técnica analítica. Dentro de um curto período de tempo, foi
desenvolvido na cromatografia de íons um novo método de detecção que
selecionava íons inorgânicos, ânions e cátions, se tornando uma técnica analítica
versátil para espécies iônicas em geral. Para melhorar a sensibilidade da detecção
da condutância elétrica uma coluna supressora foi desenvolvida para reduzir o
“background” da condutividade do eluente, aumentando assim a condutividade
mensurável dos analitos iônicos em questão.
Em 1969 Fritz descreveu um método no qual a coluna de separação está
diretamente acoplada à célula de condutividade. Já no fim da década de 1970, o
método da cromatografia de íons foi utilizado para determinar íons orgânicos através
de um processo de exclusão iônica.
3.3 CROMATOGRAFIA DE ÍONS
A técnica denominada cromatografia de íons (IC), proposta em 1975 por
Small, Stevens e Bauman, possibilitou a resolução de muitos problemas
relacionados com a determinação de íons em solução.
Em termos gerais, a cromatografia de íons inclui processos cromatográficos
envolvendo diferentes mecanismos de separação ou combinação entre eles (troca
iônica, exclusão de íons, partição, adsorção, par iônico e fase reversa) com alta
velocidade de separação e alta eficiência com o detector "on line", permitindo a
determinação de espécies iônicas.

Essa técnica pode combinar a capacidade de separação da cromatografia de
troca iônica com a detecção condutimétrica que é ideal para acompanhar as
separações, devido à sua resposta universal aos íons. É um método sensível e
seletivo para a separação, detecção e quantificação de uma ou mais espécies
iônicas em solução.
A moderna cromatografia de íons foi inicialmente aplicada para separação e
determinação de ânions utilizando como eluente (fase móvel) soluções aquosas de
hidróxidos, carbonatos e carbonatos ácidos em um sistema de coluna de troca
aniônica acoplada a uma coluna supressora. Esta permitiu que a medida por
condutividade fosse sensível e seletiva. Trabalhos posteriores aplicaram a
cromatografia de íons para a determinação de cátions dispensando o sistema
supressor.
A coluna supressora tem como função diminuir quimicamente a condutividade
dos íons do eluente (supressão do sinal de fundo, "background") que saem da
coluna separadora convertendo-os em água ou ácidos fracos e, ao mesmo tempo,
converter as espécies de interesse numa forma mais condutiva, como ácidos e
bases fortes, que são então monitorados pela célula condutimétrica. Os resultados
quantitativos são obtidos por cálculo da área ou da altura do pico, que são
proporcionais à concentração da espécie a ser determinada.
Frankenberger Jr. et al. (1990) justificaram o rápido desenvolvimento da
técnica de cromatografia de íons por sua vasta aplicação na área ambiental.
Enfatizaram ainda a especiação de íons e fizeram uma revisão dos trabalhos
publicados aplicando a cromatografia de íons em diferentes tipos de amostras
ambientais (água de chuva, água subterrânea, água de superfície, rejeitos aquosos,
água potável, amostras de gelo, neve, solos, sedimentos, efluentes de esgotos,
plantas, ar, sistemas de exaustão, aerossóis, fumaça de chaminé, fuligem aérea,
óleo combustível e petróleo).
Em muitos casos, o sistema consiste de um eluente líquido, uma bomba de
alta pressão, uma válvula de injeção de amostra, o sistema de separação composto
pelas colunas de guarda e analítica, uma coluna de supressão, uma célula
condutimétrica e um sistema de análise de dados. O esquema deste sistema citado
está mostrado na figura 7.

O líquido que irá carrear a amostra no sistema cromatográfico, ou seja, a fase
móvel, é o eluente. A amostra é introduzida na corrente do eluente, onde seus íons
são separados à medida que se movem pela coluna analítica, que contém a fase
estacionária. Dependendo de sua afinidade com a fase estacionária, o tempo de
saída dos ânions será maior ou menor. Depois de passar pela coluna, os analitos
juntamente com o eluente chegam à coluna supressora, que melhora a detecção
dos íons da amostra enquanto suprime a condutividade do eluente, como pode ser
visto na figura 9. Ao sair do sistema de supressão, os analitos chegam ao detector,
que registra a condutância dos íons transmitindo o sinal para o computador.
Figura 7: Representação esquemática de um sistema de cromatografia de íons com
supressão de condutividade. Fonte: Dionex.
Para o presente trabalho, o tipo de cromatografia de íons utilizado é o de
troca iônica. Este modo de separação é tipicamente utilizado para a separação de
ânions ou cátions. Durante o processo cromatográfico, o contra-íon do eluente é
trocado pelo íon da amostra, e por um curto período este permanece ligado a carga
fixa do material polimérico. Devido a variações de afinidades entre os íons da
amostra e os da fase estacionária, a separação de vários componentes iônicos da
amostra se torna possível.

Para eletrólitos fracos, como ácidos e bases fracas, o principal fator limitante
da linearidade de detecção ena sensibilidade analítica é o grau de dissociação ou
ionização. Eletrólitos fracos não são completamente ionizados em solução. Em
qualquer instante, algumas moléculas estão sob forma não ionizada. As moléculas
não ionizadas não conduzem corrente e, sendo assim, a concentração de íons
detectados é menor que a concentração total de espécies em solução.
Para eletrólitos fortes, como, ácidos e bases fortes e seus sais, que estão, por
definição, completamente dissociados em solução, há condução de corrente elétrica
sendo todos íons detectados.
Com o intuito de reduzir o efeito da condutividade do eluente e melhorar a
sensibilidade, faz-se uso do sistema de supressão. Através da supressão o eluente
se torna um ácido fraco. Através da figura 8 é possível observar um esquema do
processo de supressão da condutividade. Como os ácidos fracos pouco se ionizam,
produzem baixa resposta no detector de condutividade, fazendo com que sua
condutividade seja reduzida e a resposta do analito seja maximizada.
Figura 8: Representação esquemática do processo de supressão da condutividade
do eluente. Fonte: Dionex
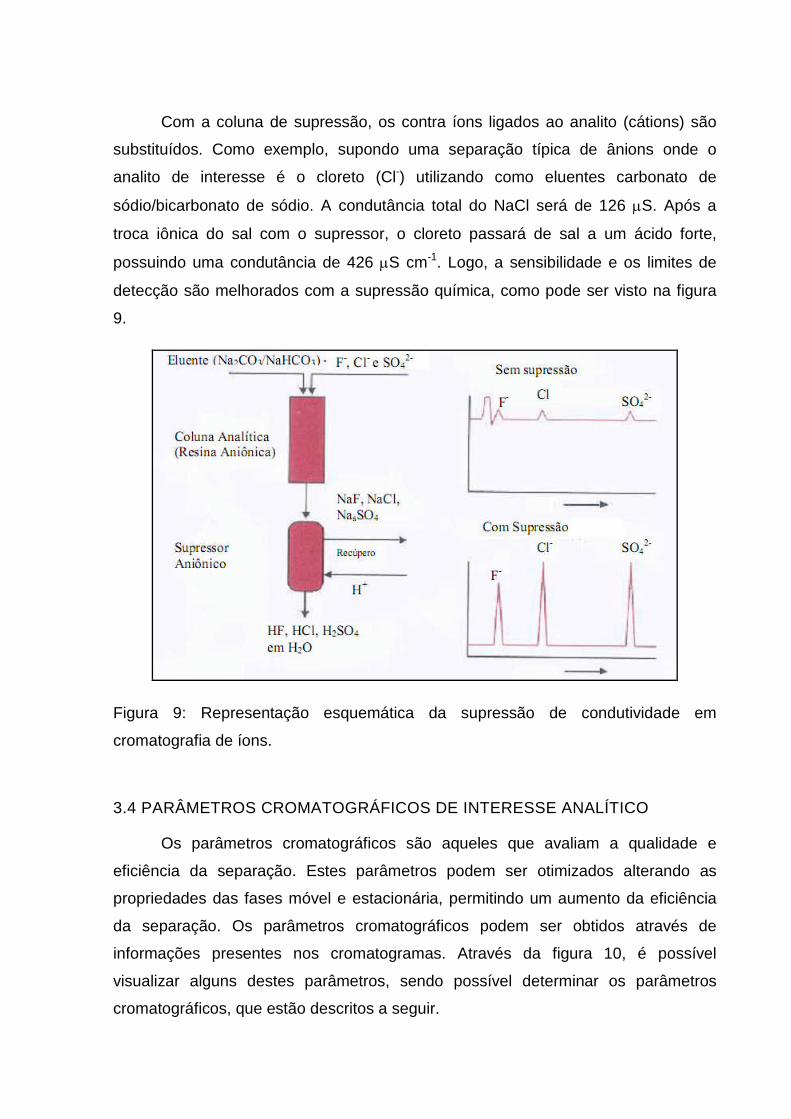
Com a coluna de supressão, os contra íons ligados ao analito (cátions) são
substituídos. Como exemplo, supondo uma separação típica de ânions onde o
analito de interesse é o cloreto (Cl-) utilizando como eluentes carbonato de
sódio/bicarbonato de sódio. A condutância total do NaCl será de 126 µS. Após a
troca iônica do sal com o supressor, o cloreto passará de sal a um ácido forte,
possuindo uma condutância de 426 µS cm-1. Logo, a sensibilidade e os limites de
detecção são melhorados com a supressão química, como pode ser visto na figura
9.
Figura 9: Representação esquemática da supressão de condutividade em
cromatografia de íons.
3.4 PARÂMETROS CROMATOGRÁFICOS DE INTERESSE ANALÍTICO
Os parâmetros cromatográficos são aqueles que avaliam a qualidade e
eficiência da separação. Estes parâmetros podem ser otimizados alterando as
propriedades das fases móvel e estacionária, permitindo um aumento da eficiência
da separação. Os parâmetros cromatográficos podem ser obtidos através de
informações presentes nos cromatogramas. Através da figura 10, é possível
visualizar alguns destes parâmetros, sendo possível determinar os parâmetros
cromatográficos, que estão descritos a seguir.
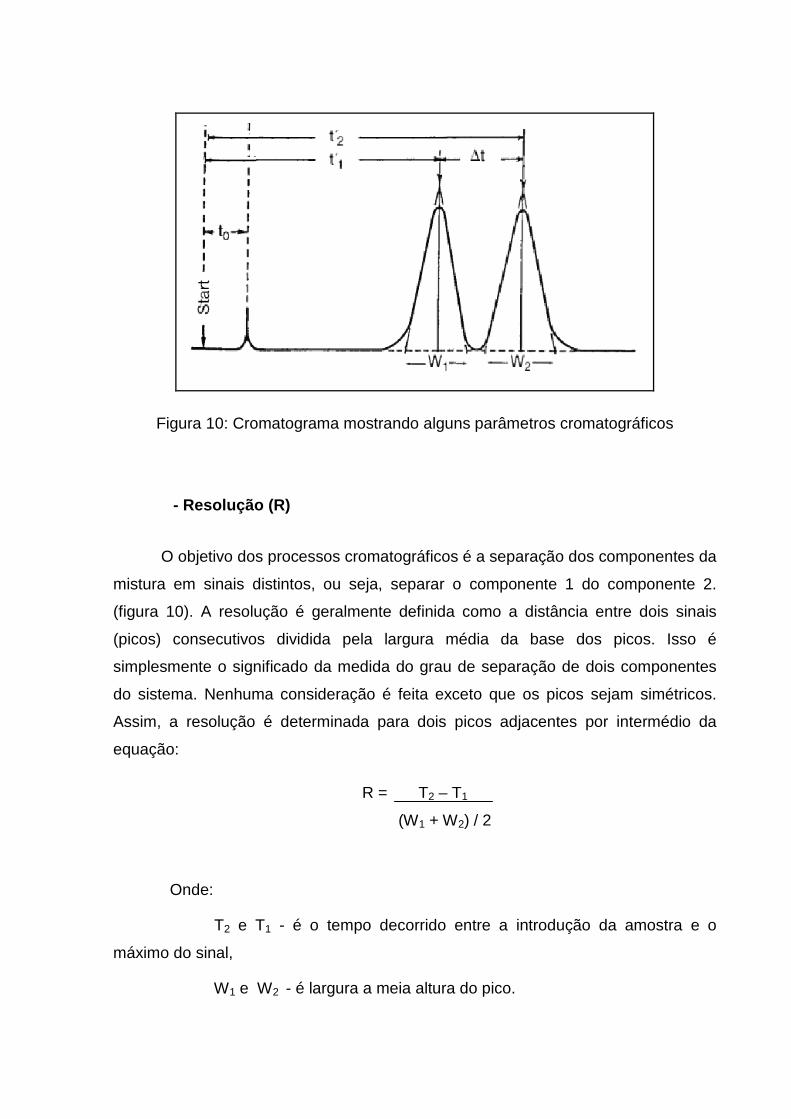
Figura 10: Cromatograma mostrando alguns parâmetros cromatográficos
- Resolução (R)
O objetivo dos processos cromatográficos é a separação dos componentes da
mistura em sinais distintos, ou seja, separar o componente 1 do componente 2.
(figura 10). A resolução é geralmente definida como a distância entre dois sinais
(picos) consecutivos dividida pela largura média da base dos picos. Isso é
simplesmente o significado da medida do grau de separação de dois componentes
do sistema. Nenhuma consideração é feita exceto que os picos sejam simétricos.
Assim, a resolução é determinada para dois picos adjacentes por intermédio da
equação:
R = T2 – T1
(W1 + W2) / 2
Onde:
T2 e T1 - é o tempo decorrido entre a introdução da amostra e o
máximo do sinal,
W1 e W2 - é largura a meia altura do pico.

Se os picos se exibem na forma de uma gaussiana, a resolução de R=2,0 é
suficiente para análise. Contudo, altos valores de R, indicam um tempo prolongado
de análise. Já valores muito baixos, como R = 0,5, indicam que alguns sinais de
componentes da amostra podem não ser reconhecidos como picos realmente
separados.
- Eficiência
A eficiência é a capacidade de uma coluna em reter um componente sem
difundi-lo. É a medida da dispersão de um sinal quando um analito se move pela
coluna. A eficiência é uma medida relacionada a coluna de separação e é
encontrada calculando-se o número de pratos teóricos (N), através da equação que
segue:
N = 5,54 ( T )2
W1/2
Onde:
T - é o tempo decorrido entre a injeção e o máximo do sinal,
W1/2 - é a largura da meia altura do pico.
N é proporcional ao comprimento da coluna logo, quanto maior a coluna,
maior o número de pratos teóricos.
A eficiência de uma coluna pode ser alterada pela vazão dos eluentes.
Quanto menor a vazão, maior a eficiência, ao passo que existe um tempo maior para
o analito na fase móvel interagir com a fase estacionária. Contudo, isto pode levar
também a um alargamento dos picos.
- Assimetria
Esta é uma importante propriedade pois trata-se das distorções frontais e
posteriores (caudas) dos picos que podem induzir a uma sobreposição. O fator
assimetria é calculado a 10 % da altura dos picos em relação à linha de base, sendo
uma medida da dispersão dos componentes da amostra no sistema cromatográfico.
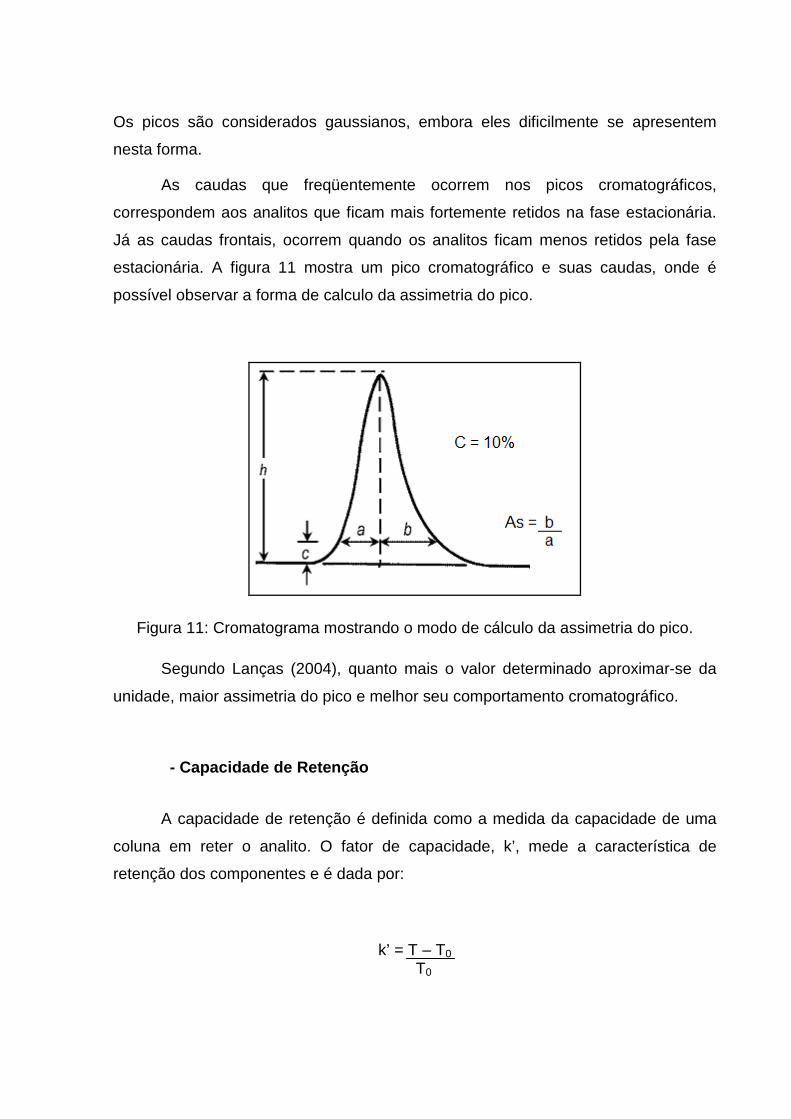
Os picos são considerados gaussianos, embora eles dificilmente se apresentem
nesta forma.
As caudas que freqüentemente ocorrem nos picos cromatográficos,
correspondem aos analitos que ficam mais fortemente retidos na fase estacionária.
Já as caudas frontais, ocorrem quando os analitos ficam menos retidos pela fase
estacionária. A figura 11 mostra um pico cromatográfico e suas caudas, onde é
possível observar a forma de calculo da assimetria do pico.
Figura 11: Cromatograma mostrando o modo de cálculo da assimetria do pico.
Segundo Lanças (2004), quanto mais o valor determinado aproximar-se da
unidade, maior assimetria do pico e melhor seu comportamento cromatográfico.
- Capacidade de Retenção
A capacidade de retenção é definida como a medida da capacidade de uma
coluna em reter o analito. O fator de capacidade, k’, mede a característica de
retenção dos componentes e é dada por:
k’ = T – T0 T0

Onde:
- T é o tempo entre o pico do analito e o ponto de injeção;
- T0 é o tempo do Volume Morto.
Em outras palavras, o fator capacidade de retenção nos dá uma idéia de
quanto tempo o analito permaneceu na fase estacionária e na fase móvel. Ele é
utilizado para determinar a vida útil de uma coluna, e, portanto o momento de trocá-
la ou limpá-la. Quando uma coluna está deteriorada, existem poucos sítios
disponíveis na resina, devido ao leito da resina ter sido degradado ou saturado.
3.5 ESPECTROMETRIA DE EMISSÃO ÓPTICA COM FONTE DE PLASMA
INDUTIVAMENTE ACOPLADO – ICP OES
Os métodos espectrométricos abrangem um grupo de procedimentos
baseados na espectroscopia atômica e molecular. Uma parte da espectrometria se
refere às medidas de intensidade da radiação usando transdutores fotoelétricos ou
outros tipos de dispositivos eletrônicos. Os métodos espectrométricos mais
amplamente utilizados fazem uso da radiação eletromagnética (SKOOG et al.,
2006).
Para detecção, os constituintes de uma amostra devem ser convertidos em
átomos gasosos ou átomos ionizados, que podem ser determinados por medidas de
espectros de emissão, de absorção, de fluorescência ou de massa.
O plasma indutivamente acoplado é uma fonte efetiva para emissão de
radiação luminosa (emissão atômica) que pode, em princípio, ser usada para a
determinação de todos os elementos com energia de excitação menor que o gás
que suporta o plasma, o argônio (Thompson; Walsh, 1983). O plasma é um gás
parcialmente ionizado, produzido a partir de uma descarga em uma corrente de gás
inerte (argônio), mediante aquecimento por indução em uma tocha de quartzo
localizada dentro de uma bobina de indução ligada a um gerador de radiofreqüência,
operando com freqüência e potência apropriadas. O ICP é atualmente o mais usado
para fins analíticos devido a sua boa sensibilidade analítica (Hill, 1999).
O sistema usado para gerar o plasma é formado por três componentes
principais: uma tocha, uma bobina de indução magnética e uma fonte de

radiofreqüência. Na Figura 12 é mostrado um esquema de um plasma utilizado em
ICP.
Figura 12: Representação de um plasma indutivamente acoplado, onde H
representa o campo magnético induzido e I, a bobina de indução. Adaptada de
JARVIS et al., 1992.
A tocha consiste de três tubos concêntricos de quartzo. Na parte externa da
tocha, argônio é introduzido tangencialmente com vazões que variam de 8 a 20 L
min-1, em torno das paredes do tubo, provocando o isolamento térmico do cilindro
mais externo, que resfria as paredes internas do tubo central e centraliza
radialmente o plasma, sendo esta vazão de gás responsável pela sustentação do
mesmo (GINÉ, 1998). Na seção intermediária é introduzido o gás auxiliar, com
vazões que variam de 0,1 a 1,5 L min-1 o qual tem a função de dar um formato
adequado ao plasma, mantendo-o na ponta da tocha e evitando assim que a mesma
sofra processo de fusão. O tubo central é o que conduz a amostra em forma de
aerossol para o plasma (0,7 - 1,5 L min-1) que é chamado de gás de arraste.
Em torno da tocha encontra-se a bobina de indução (Figura 12) a qual é
refrigerada internamente ou o próprio argônio por água. A bobina de indução é
responsável por manter o plasma devido ao acoplamento da radiofreqüência (RF),
cuja freqüência usada em plasma de argônio é 27 ou 40 MHz, enquanto que a
potência pode variar de 800 a 1500 W. A fonte de radiofreqüência fornece corrente

elétrica que circula pelas espirais da bobina, induzindo um campo magnético
oscilante com linhas de força orientadas axialmente dentro do tubo, formando
elipses fechadas conforme indica a Figura 12. O campo magnético induzido acelera
os elétrons, os quais fluem em trajetórias anulares dentro da tocha produzindo
ionização por colisão, iniciando reação em cadeia com transferência energética que
produz aquecimento. O aquecimento gerado nessas colisões pode elevar a
temperatura do plasma a 10.000 K, o que garante a completa atomização e/ou
ionização da maioria dos elementos presentes (SKOOG, 2006). A Figura 13 mostra
um plasma em operação.
Figura 13: Foto de um plasma em operação. (THERMO, 2007)
O sistema de introdução de amostra é composto, em geral, por um
nebulizador e uma câmara de nebulização. O nebulizador produz um aerossol da
amostra que é conduzido ao plasma pela câmara, a qual favorece a introdução
apenas das gotículas menores, sendo as gotículas com tamanho médio maior do
que 10 a 20 µm descartadas. Geralmente somente uma fração (da ordem de 2 a
5%) da amostra atinge o plasma. Nele ocorrem os processos de dessolvatação
(evaporação do solvente), volatilização e atomização/ionização. Durante o processo
de transporte da amostra até o plasma, podem ocorrer interferências não espectrais
relacionadas às propriedades físicas como viscosidade da solução e/ou à presença
de constituintes combustíveis que podem alterar a temperatura do plasma (SKOOG,
2006).
A Espectrometria de Emissão Óptica com Plasma Indutivamente Acoplado
(ICP OES) se destaca por ser uma técnica de grande precisão e exatidão, com
capacidade de analisar simultaneamente e/ou sequecialmente elementos em faixas
Tocha
Bobina de RF

de concentrações muito diferentes. A Emissão Óptica com Plasma (ICP OES) é uma
técnica analítica moderna e poderosa para determinação de metais e outros
elementos nas mais diversas amostras ambientais devido à alta sensibilidade (ppm a
ppb) e seletividade alcançada.
A capacidade multielementar da técnica de ICP OES é uma das mais
importantes características, juntamente com a espectrometria de massa com fonte
de plasma indutivamente acoplado (ICP-MS), podendo ser utilizada para a
determinação de, aproximadamente, 70 elementos em uma ampla variedade de
amostras (NÖLTE, 2003).
A técnica de ICP OES é baseada na medida da intensidade da radiação
emitida, quando um átomo ou íon excitado pelo plasma volta a seu estado
fundamental. Cada elemento emite radiação em comprimentos de onda
característicos e estas linhas de emissão podem ser usadas para análise
quantitativa depois de uma calibração (SKOOG et al., 2006).
Essa técnica é utilizada no Brasil desde a década de 70, quando foram
instalados os primeiros equipamentos de ICP OES. Desde então, seus componentes
ópticos e sistemas de detecção vem sendo aprimorados, a fim de se obter
resultados mais exatos e precisos. Um exemplo é o emprego da visão axial do
plasma, que proporciona melhores LDs em relação à visão radial, em cerca de uma
ordem de grandeza, embora os efeitos de matriz sejam mais acentuados (SILVA et
al., 2002).
A diferença principal entre as duas configurações (axial e radial) consiste
basicamente na composição da faixa espectral observada. Instrumentos com visão
axial têm a tocha posicionada horizontalmente em relação ao sistema óptico,
enquanto que em instrumentos com visão radial a tocha tem posicionamento vertical
(Figura 14). Os aparelhos com configuração da visão axial apresentam limites de
detecção (LD) 2 a 20 vezes melhores que os de visão radial, apesar de sofrerem
mais com as interferências de matriz e recombinação na cauda do plasma (região
mais fria) (TREVISAN, 2007).

Figura 14: Esquema mostrando as conficgurações Radia (a) e Axial (b) de
observação do plasma.
Existem também alguns instrumentos que combinam os dois modos de
observação (dual view - DV) em uma única unidade onde a radiação é coletada por
um foco ótico, com lentes convexas ou côncavas. A escolha do modo de observação
mais adequado é controlada pelo operador (Figura 15).
Figura 15: Representação esquemática de um plasma com observação radial e axial
- Dual View (THERMO, 2005)

4 MATERIAIS E MÉTODOS
4.1 SOLUÇÕES E REAGENTES
4.1.1 Análises volumétricas
Para os testes de capacidade de troca das resinas, utilizou-se soluções
aquosas de cloreto de sódio (NaCl, Merck, Darmstadt, Alemanha) 1 mol L-1
preparadas adequadamente em balões volumétricos devidamente aferidos e
armazenadas em frascos de polietileno. O cloreto de sódio foi previamente seco em
estufa por 2 horas a 120°C, utilizando água ultra p ura para o preparo da solução.
Os ácidos utilizados nas análises volumétricas foram padronizados com
solução de hidróxido de sódio (NaOH, Merck, Darmstadt, Alemanha) previamente
titulado com padrão primário de hidrogenoftalato de potássio (C8H5KO4, Vetec
Química Fina, Rio de Janeiro, Brasil) dessecado por 1 hora em estufa à 110°C.
Para a determinação da concentração de cloreto pelo método de Mohr, o
titulante utilizado foi uma solução aquosa de nitrato de prata (AgNO3, Vetec Química
Fina, Rio de Janeiro, Brasil), preparado com reagente PA e água ultra pura. O
reagente sólido foi devidamente seco em estufa a 150°C por duas horas e, após
preparo, padronizado com solução de cloreto de sódio (NaCl). O reagente foi
estocado em frasco de vidro âmbar. Para a titulação, utilizou-se como indicador do
ponto de equivalência, cromato de potássio 5% (Riedel-de Haën, Seelze,
Alemanha), preparado dissolvendo cromato de potássio em água ultra pura.

4.1.2 Determinação dos íons
Para a determinação de prata por ICP OES utilizou-se soluções aquosas de
Ag+ que foram preparadas em balões volumétricos, a partir de diluição de solução-
padrão estoque de concentração 1.000 mg L-1 (CertiPrep SPEX, Metuchen, USA),
até as concentrações necessárias para a obtenção de curva analítica (preparada
sempre no dia da determinação). Para preservação dos íons de prata, foi utilizado
ácido nítrico (Merck, Darmstadt, Germany).
Tubos graduados de polietileno foram utilizados em todas as etapas. Os
mesmos foram mantidos por 24 horas em solução de ácido nítrico 10% v/v na etapa
de determinação de prata por ICP OES e mantidos em solução de ácido clorídrico
1% v/v (HCl, Merck, Darmstadt, Alemanha) na etapa de determinação de ânions por
cromatografia de íons. Antes do uso, os frascos foram rinsados com água ultra.
Para a quantificação dos ânions por cromatografia de íons, fez-se uso de um
eluente composto por uma solução mista de 4,5 mmol L-1 de carbonato de sódio
(Na2CO3) e 0,8 mmol L-1 de bicarbonato de sódio (NaHCO3), sendo ambos de grau
analítico (Acros Organics, Geel, Bélgica). A calibração foi feita através do método de
curva analítica, onde fez-se uso do padrão estoque de sete ânions da empresa
DIONEX (Dionex Corporation, Sunnyvale, CA, EUA), que foi diluído com água ultra
pura, até as concentrações desejadas para a obtenção da curva analítica, sendo
todos preparados em balões volumétricos devidamente aferidos.
Para teste de retenção de ânions nas resinas, fez-se uso de soluções dos
ânions fluoreto (fluoreto de sódio, NaF, Vetec Química Fina, Rio de Janeiro, Brasil),
nitrito (nitrito de sódio, NaNO2, Merck, Darmstadt, Alemanha), brometo (brometo de
potássio, KBr, Merck, Darmstadt, Alemanha), nitrato (nitrato de sódio, NaNO3,
Merck, Darmstadt, Alemanha), fosfato (hidrogenofosfato (ou bifosfato) de sódio,
NaHPO3, Merck, Darmstadt, Alemanha) e sulfato (sulfato de magnésio, MgSO4,
Reagen, Rio de Janeiro, Brasil) em diferentes concentrações. As soluções-padrão
estoque foram preparadas a partir dos reagentes sólidos de grau analítico, em
balões volumétricos aferidos e com água ultra pura, sendo estocados em frascos de
polietileno.

4.2 INSTRUMENTAÇÃO
Para a determinação de prata, fez-se uso de um espectrômetro de emissão
ótica com plasma indutivamente acoplado (ICP OES - Thermo Fisher Scientific,
Bremen, Alemanha) modelo iCAP 6300. Os parâmetros instrumentais para a
determinação de prata podem ser encontrados na tabela 2.
Tabela 2: Parâmetros instrumentais utilizados durante os estudos envolvendo a
determinação de prata nas amostras percoladas nas minicolunas
Parâmetro Valor
Potência do gerador de Radiofreqüência (W) 1350
Vazão do gás auxiliar (L min-1) 1,5
Tipo de nebulizador Mira Mist
Pressão do nebulizador (bar) 0,2
Tempo de integração (s) 1
Tempo de estabilização (s) 5
Gás de purga Ar
Comprimento de onda (nm) para Ag 328,068
Número de replicatas 2
A observação do plasma foi feita em duas configurações: na vista axial e
radial. Através de ambos os resultados, avaliou-se qual modo de observação
forneceu a melhor resposta. Como método de quantificação, utilizou-se curva de
calibração. As amostras foram introduzidas sem previa filtração, via nebulizador Mira
Mist (Mira Mist CE, Burgener Research Inc., Ontario, Canadá) com câmara ciclônica.
A figura 16 mostra uma fotografia do equipamento utilizado nas análises.

Figura 16: Fotografia do ICP OES Thermo Scientific Modelo iCAP 6300
As análises cromatográficas foram efetuadas com sistema cromatográfico de
íons DIONEX (Dionex Corporation, Sunnyvale, CA, EUA), modelo ICS 2500 com
detecção condutimétrica equipado com uma bomba gradiente GP 50, uma célula de
condutividade com estabilizador de detecção DS3 controlada por um módulo ED 50,
uma célula de supressão ASRS ultra II 4 mm, uma coluna analítica AS 23 (4 x 250
mm) e uma coluna de guarda AG 23 (4 x 50mm). O controle da instrumentação,
assim como o controle dos dados analíticos, foi efetuado com o auxilio do software
Chromeleon 6.5. A figura 17 mostra uma fotografia do equipamento utilizado.

Figura 17: Cromatógrafo de Íons utilizado nas análises.
Todos os reagentes e soluções foram preparados com água ultra pura obtida
de um sistema Milli-Q (Elix e Synergy, Millipore, Bedford, MA, USA). Um banho
ultrassom Branson 1510 foi utilizado para a retirada de gases solúveis dos eluentes
preparados. Filtros 0,22 µm Millex da Millipore foram utilizados para filtrar as
amostras que passaram pelo cromatógrafo de íons, assim como seringas
hipodérmicas descartáveis de 1 mL utilizadas para introdução das amostras.
Cartuchos comerciais Onguard II – Ag (2,5 cc) e OnGuard II – H (1 cc) (DIONEX)
foram utilizados para a retirada de cloreto da amostra e da prata proveniente do
cartucho Onguard II – Ag, respectivamente, das matrizes de amostras.
4.3 METODOLOGIA UTILIZADA
A metodologia utilizada pode ser representada através da figura 18, onde
tem-se as etapas realizadas.
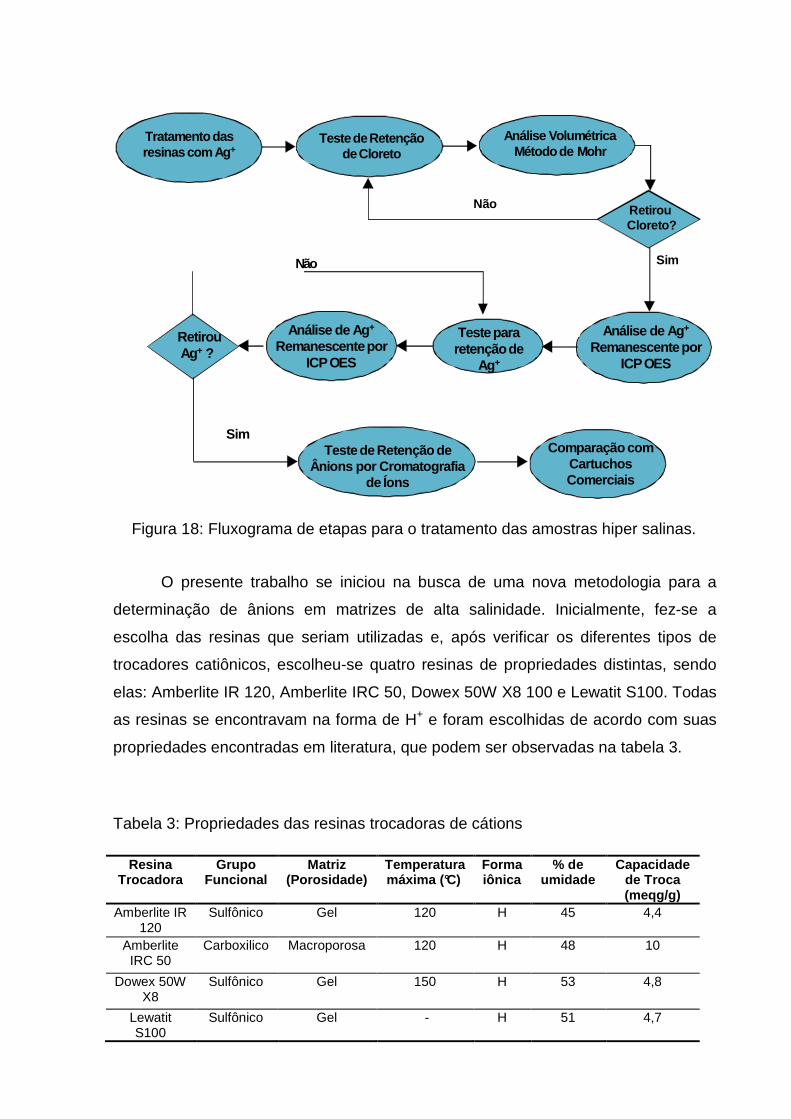
O presente trabalho se iniciou na busca de uma nova metodologia para a
determinação de ânions em matrizes de alta salinidade. Inicialmente, fez-se a
escolha das resinas que seriam utilizadas e, após verificar os diferentes tipos de
trocadores catiônicos, escolheu-se quatro resinas de propriedades distintas, sendo
elas: Amberlite IR 120, Amberlite IRC 50, Dowex 50W X8 100 e Lewatit S100. Todas
as resinas se encontravam na forma de H+ e foram escolhidas de acordo com suas
propriedades encontradas em literatura, que podem ser observadas na tabela 3.
Tabela 3: Propriedades das resinas trocadoras de cátions
Resina Trocadora
Grupo Funcional
Matriz (Porosidade)
Temperatura máxima (°C)
Forma iônica
% de umidade
Capacidade de Troca (meqg/g)
Amberlite IR 120
Sulfônico Gel 120 H 45 4,4
Amberlite IRC 50
Carboxilico Macroporosa 120 H 48 10
Dowex 50W X8
Sulfônico Gel 150 H 53 4,8
Lewatit S100
Sulfônico Gel - H 51 4,7
RetirouCloreto?
Sim
Não
Análise Volumétrica Método de Mohr
Teste de Retenção de Cloreto
Tratamento das resinas com Ag +
RetirouAg+ ?
Teste para retenção de
Ag+
Análise de Ag +
Remanescente por ICP OES
Análise de Ag +
Remanescente por ICP OES
Não
Teste de Retenção de Ânions por Cromatografia
de Íons
SimComparação com
Cartuchos Comerciais
Figura 18: Fluxograma de etapas para o tratamento das amostras hiper salinas.

Após identificar os trocadores, iniciou-se uma pesquisa para a determinação
de suas propriedades, em especial a capacidade de troca catiônica.
4.3.1 Cálculo do teor de umidade das resinas utiliz adas
Pesou-se em balança analítica uma massa aproximada de 0,200g de resina
trocadora catiônica. Esta foi levada a estufa a uma temperatura de 105 °C até se
obter peso constante. O calculo do teor de umidade das resinas foi realizado para
poder dar prosseguimento na determinação da capacidade de troca.
4.3.2 Determinação da capacidade de troca das resin as
Em um bécher, pesou-se em balança analítica 0,200 g de resina trocadora de
cátions. Adicionou-se 25 mL de NaOH 0,1 mol L-1 deixando sob agitação constante
por 24 horas. Depois de decorrido o tempo determinado, retirou-se duas alíquotas
de 10 mL deste sobrenadante e adicionou-se 1 gota do indicador fenolftaleína. Em
seguida essas alíquotas foram tituladas com uma solução de HCl 0,1 mol L-1.
O cálculo da capacidade de troca catiônica (CTC) das resinas foi determinado
através da equação abaixo:
CTC = VNaOH * CNaOH – VHCl*CHCl
Massa de resina seca (g)
Onde: VNaOH = Volume de sobrenadante utilizado na titulação
CNaOH = Concentração da solução de NaOH utilizado no teste
VHCl = Volume de HCl gasto na titulação
CHCl = Concentração da solução de HCl utilizado na titulação
Realizou-se esse mesmo procedimento para as quatro diferentes resinas,
determinando a capacidade de troca de cada uma delas.

4.3.3 Testes de troca iônica das resinas trocadoras de cátions
O íon cloreto precipita com o íon de prata sob a forma de AgCl. Sendo assim,
decidiu-se tratar as resinas com íons de prata, de modo que o hidrogênio desses
trocadores catiônicos fossem substituídos por prata. Através da capacidade de troca
obtida para cada resina, calculou-se a quantidade de prata necessária para que
todos os hidrogênios ligados à resina fossem substituídos por prata. Esta resina
tratada foi então chamada de resina de prata.
Uma massa de 2,000 g de cada tipo de resina seca foi previamente pesada
em balança analítica e, para cada tipo de resina, uma massa de nitrato de prata foi
adicionado. Colocou-se então 10 mL de água para a dissolução do nitrato de prata,
agitando para uma perfeita homogeneização. Deixou-se a resina em solução de
nitrato de prata por 24h.
Após tratamento, lavou-se intensamente as resinas com água ultra-pura para
certificar-se que toda a prata que não foi trocada não ficasse adsorvida na resina.
Reservou-se um volume de 5,0 mL dessas águas de lavagem para verificar a
presença ou não de prata por ICP OES. Depois da lavagem final, as resinas eram
levadas à estufa para secagem a uma temperatura de 70 °C até peso constante.
Com as resinas devidamente secas, as mesmas foram estocadas em frascos
recobertos com papel alumínio para não permitir o contato com a luz do ambiente,
evitando assim que a resina tratada com prata fosse degradada.
Com esta resina, montou-se mini colunas com tubos do tipo Tygon, que foram
pesadas em balança analítica e, em seguida, preenchidas com resina tratada até
que atingisse uma massa de resina de cerca de 0,200 g. Com a mini coluna pronta
para uso, iniciou-se os testes para verificar se havia a retenção de cloreto pelas
colunas ou não.
Para isso, preparou-se uma solução de cloreto de sódio 5,8 g L-1 e, com uma
bomba peristáltica, passou-se a solução do sal pela coluna a uma vazão de 0,5 mL
min-1. A solução salina que eluiu pela coluna foi coletada a cada fração de 0,5 mL e
levada a um volume final de 5,0 mL onde foi realizada a determinação de cloreto
pelo método de Mohr.

O método de Mohr é uma análise volumétrica que emprega a titulação de
cloreto utilizando como titulante uma solução de nitrato de prata 0,01 mol L-1 como
reagente. A solução neutra é titulada com AgNO3, em presença de K2CrO4, que atua
como indicador. O cloreto precipita na forma de AgCl e, na presença do primeiro
excesso de prata no meio, o indicador reage formando um precipitado vermelho –
alaranjado de AgCrO4 mostrando o fim da titulação. Através do volume de titulante e
de sua concentração, pode-se quantificar o cloreto presente na amostra.
4.3.4 Aplicação do método em amostras de matriz com alta salinidade
Comprovada a retenção de cloreto pelas mini colunas de resina tratada com
prata, preparou-se novas resinas para testes com amostras de alta salinidade. As
amostras utilizadas eram de água do mar, onde a concentração média de cloreto
esta em torno de 30 g L-1.
Seguindo o mesmo procedimento dos testes de troca iônica, montou-se mini
colunas com massa de aproximadamente 0,200 g de resina trocadora catiônica.
Percolou-se as amostras de água do mar pelas colunas com a bomba peristáltica a
uma vazão de 0,5 mL min-1. Assim como nos testes as amostras foram coletadas em
frações de 0,5 mL e diluídas a um volume final de 5,0 mL. Estas soluções coletadas
foram então separadas em duas frações onde 3,0 mL foi reservado para a
determinação de prata sendo acidificada com acido nítrico para a sua conservação
até a análise por ICP OES. Aos outros 2,0 mL, adicionou-se 1 gota do indicador
cromato de potássio 5% sendo esta amostra titulada contra uma solução de nitrato
de prata 0,01 mol L-1, como descrito pelo método de Mohr.
4.3.5 Determinação de prata remanescente na resina por ICP OES
Para a determinação da prata remanescente na resina que chega aos eluídos
das amostras, utilizou-se um espectrômetro de emissão óptica com plasma
indutivamente acoplado (ICP OES). A observação do plasma foi realizada tanto no
modo axial quanto na radial, podendo assim verificar qual das duas visões
apresentava melhor sensibilidade.
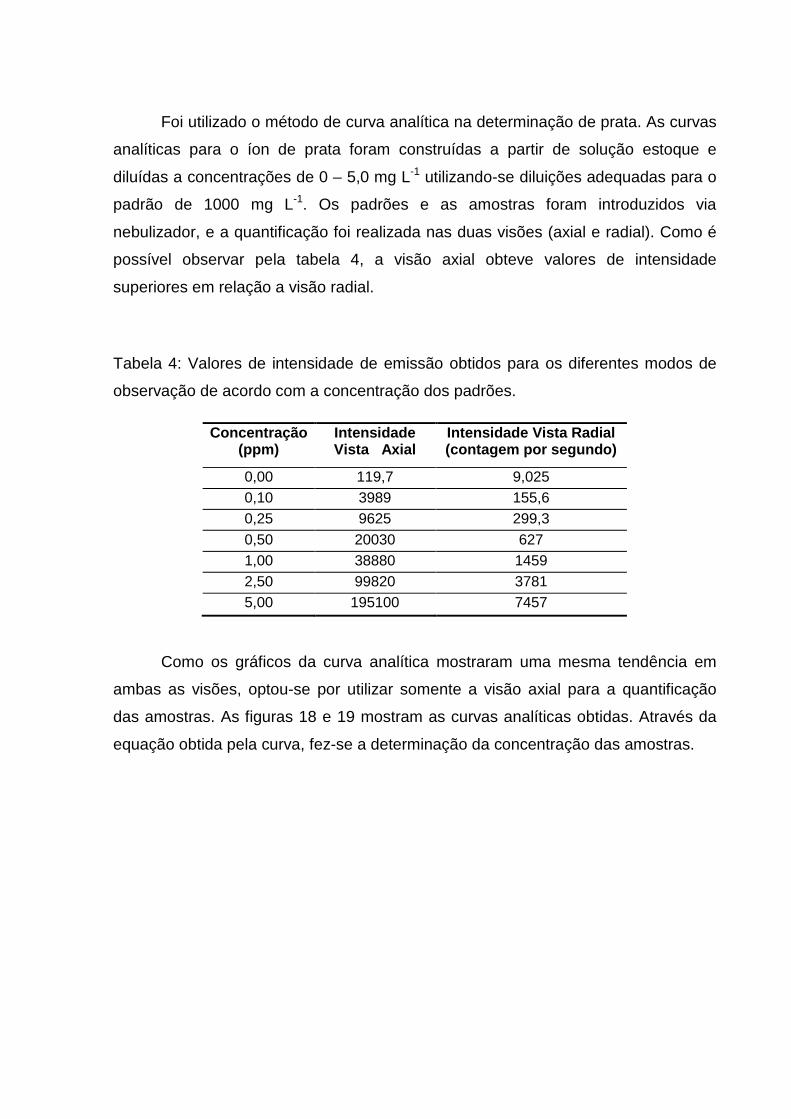
Foi utilizado o método de curva analítica na determinação de prata. As curvas
analíticas para o íon de prata foram construídas a partir de solução estoque e
diluídas a concentrações de 0 – 5,0 mg L-1 utilizando-se diluições adequadas para o
padrão de 1000 mg L-1. Os padrões e as amostras foram introduzidos via
nebulizador, e a quantificação foi realizada nas duas visões (axial e radial). Como é
possível observar pela tabela 4, a visão axial obteve valores de intensidade
superiores em relação a visão radial.
Tabela 4: Valores de intensidade de emissão obtidos para os diferentes modos de
observação de acordo com a concentração dos padrões.
Concentração (ppm)
Intensidade Vista Axial
Intensidade Vista Radial (contagem por segundo)
0,00 119,7 9,025 0,10 3989 155,6 0,25 9625 299,3 0,50 20030 627 1,00 38880 1459 2,50 99820 3781 5,00 195100 7457
Como os gráficos da curva analítica mostraram uma mesma tendência em
ambas as visões, optou-se por utilizar somente a visão axial para a quantificação
das amostras. As figuras 18 e 19 mostram as curvas analíticas obtidas. Através da
equação obtida pela curva, fez-se a determinação da concentração das amostras.
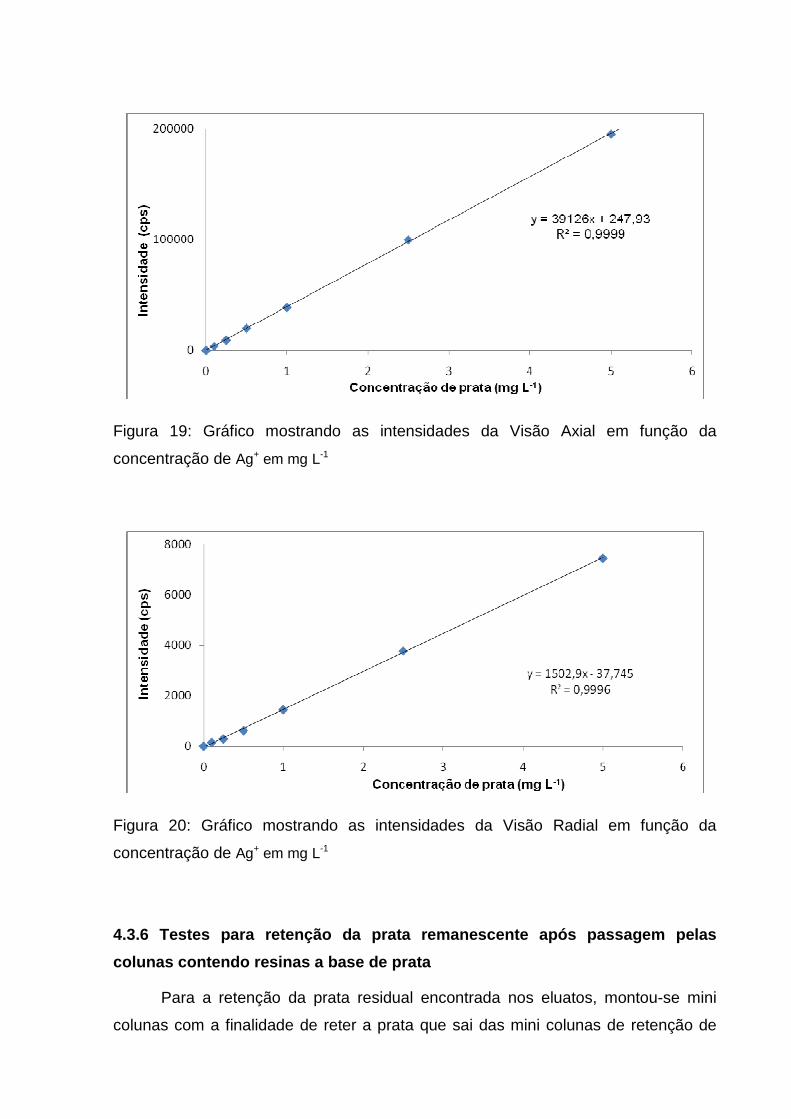
Figura 19: Gráfico mostrando as intensidades da Visão Axial em função da
concentração de Ag+ em mg L-1
Figura 20: Gráfico mostrando as intensidades da Visão Radial em função da
concentração de Ag+ em mg L-1
4.3.6 Testes para retenção da prata remanescente ap ós passagem pelas
colunas contendo resinas a base de prata
Para a retenção da prata residual encontrada nos eluatos, montou-se mini
colunas com a finalidade de reter a prata que sai das mini colunas de retenção de

cloreto. As mini colunas de retenção de prata foram preparadas com tubos do tipo
Tygon e preenchidas com aproximadamente 0,100 g de resina catiônica na forma de
hidrogênio seca. As resinas utilizadas para retenção de prata são as mesmas
utilizadas nas mini colunas de retenção de cloreto. Contudo estas resinas não foram
pré-tratadas com prata, elas foram apenas lavadas e secas em estufa a 100 °C até
peso constante. Por não terem sido previamente tratadas, estas se encontram na
forma de hidrogênio, podendo dessa forma reter a prata remanescente na resina de
prata.
Sendo assim, montou-se em sequência a mini coluna de resina na forma de
prata seguida da mini coluna de resina na forma de hidrogênio. Montado este
esquema, fez-se a percolação da amostra de matriz salina de forma que essa
amostra passasse pelas duas resinas. Coletou-se frações a cada 0,5 mL de amostra
eluida, levando-se a um volume final de 5 mL. Essas amostras foram então
novamente separadas em duas alíquotas, onde uma alíquota de 3,0 mL foi
reservada e acidificada para posterior quantificação de prata por ICP OES. A
segunda alíquota contendo 2,0 mL, foi titulada pelo método de Mohr com solução de
nitrato de prata 0,01 mol L-1 utilizando 1 gota de solução de dicromato de potássio
5% como indicador.
4.3.7 Testes de retenção de ânions pelas colunas mo dificadas com prata
Para verificar a eficiência de trabalho da resina proposta, realizou-se testes
para verificar a possível retenção dos ânions de trabalho pelas resinas modificadas
com prata utilizando a Cromatografia de Íons. Para tal, preparou-se um padrão
contendo seis ânions, onde suas respectivas concentrações podem ser observadas
pela tabela 5.
Tabela 5: Concentração dos íons contidos no padrão preparado com seis diferentes
ânions.
Ânions Concentração (mg L -1)
Fluoreto 1,0
Cloreto 5,0

Nitrito 5,0
Brometo 5,0
Nitrato 5,0
Fosfato 5,0
Sulfato 4,0
Inicialmente, percolou-se, com o auxílio de uma bomba peristáltica a uma
vazão de 0,5 mL min-1, a solução com os padrões de ânions pela coluna de prata e
pela coluna de hidrogênio, com o objetivo de observar se a resina estava retendo ou
não os ânions de interesse.
Após esta avaliação, realizou-se um estudo de como esses ânions eram
retidos com a variação da salinidade do meio. Sendo assim, preparou-se diferentes
soluções-padrão de ânions, mantendo-se a concentração dos seis ânions de
trabalho (tabela 5), variando-se apenas a concentração do íon cloreto no meio. Fez-
se um estudo em duas partes, onde a primeira visava avaliar a retenção desses seis
ânions em concentrações muito baixas de cloreto e a segunda visava avaliar
retenção desses ânions em concentrações mais altas. As concentrações de cloreto
estudadas variavam de 1 a 40.000 mg L-1.
As soluções foram injetadas no cromatógrafo de três diferentes formas: a
primeira, sendo a solução de padrões com a sua respectiva concentração de cloreto;
a segunda referente à primeira fração coletada após a percolação pelas duas
colunas, ou seja, os primeiros 0,5 mL eluídos; e a terceira referente à segunda
fração percolada pelas duas colunas ( 0,5 mL).
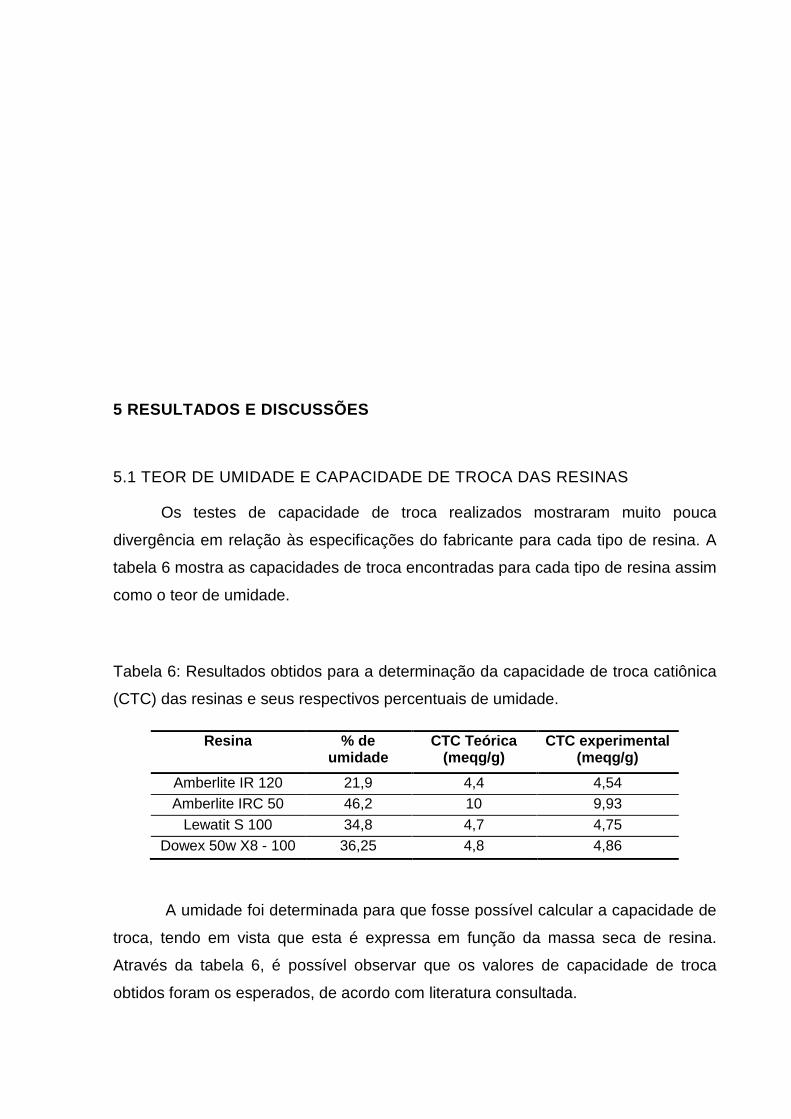
5 RESULTADOS E DISCUSSÕES
5.1 TEOR DE UMIDADE E CAPACIDADE DE TROCA DAS RESINAS
Os testes de capacidade de troca realizados mostraram muito pouca
divergência em relação às especificações do fabricante para cada tipo de resina. A
tabela 6 mostra as capacidades de troca encontradas para cada tipo de resina assim
como o teor de umidade.
Tabela 6: Resultados obtidos para a determinação da capacidade de troca catiônica
(CTC) das resinas e seus respectivos percentuais de umidade.
Resina % de umidade
CTC Teórica (meqg/g)
CTC experimental (meqg/g)
Amberlite IR 120 21,9 4,4 4,54 Amberlite IRC 50 46,2 10 9,93
Lewatit S 100 34,8 4,7 4,75 Dowex 50w X8 - 100 36,25 4,8 4,86
A umidade foi determinada para que fosse possível calcular a capacidade de
troca, tendo em vista que esta é expressa em função da massa seca de resina.
Através da tabela 6, é possível observar que os valores de capacidade de troca
obtidos foram os esperados, de acordo com literatura consultada.
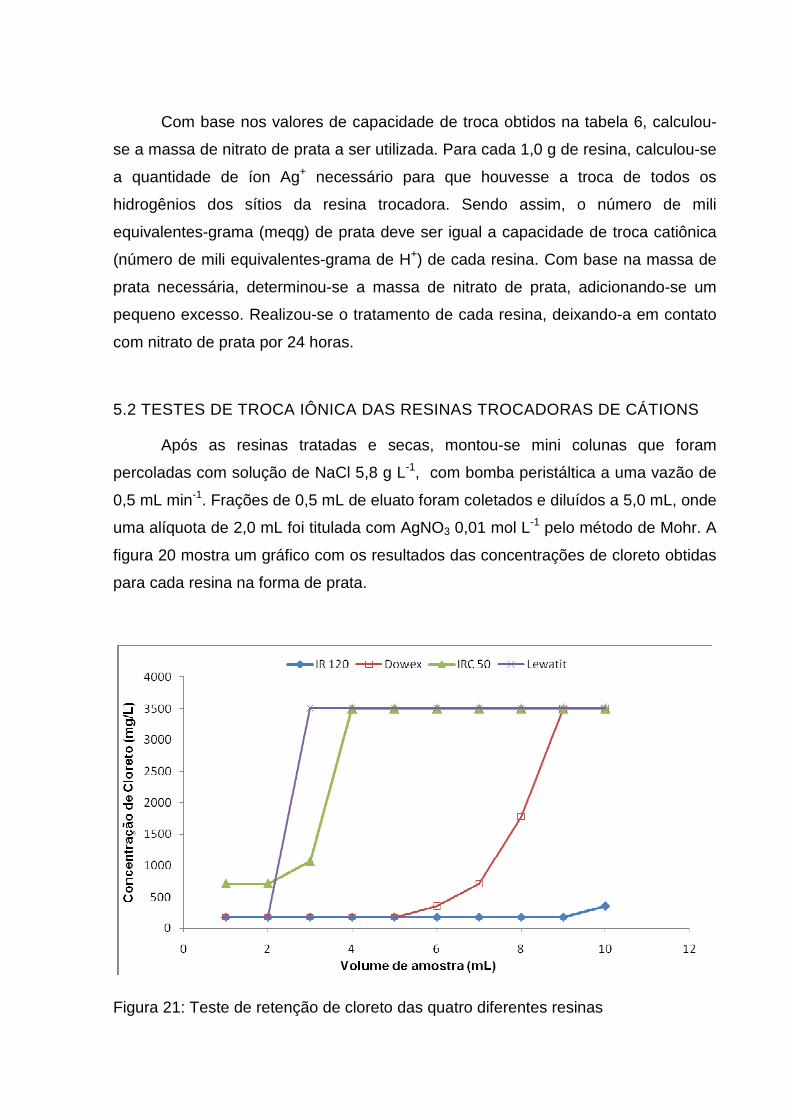
Com base nos valores de capacidade de troca obtidos na tabela 6, calculou-
se a massa de nitrato de prata a ser utilizada. Para cada 1,0 g de resina, calculou-se
a quantidade de íon Ag+ necessário para que houvesse a troca de todos os
hidrogênios dos sítios da resina trocadora. Sendo assim, o número de mili
equivalentes-grama (meqg) de prata deve ser igual a capacidade de troca catiônica
(número de mili equivalentes-grama de H+) de cada resina. Com base na massa de
prata necessária, determinou-se a massa de nitrato de prata, adicionando-se um
pequeno excesso. Realizou-se o tratamento de cada resina, deixando-a em contato
com nitrato de prata por 24 horas.
5.2 TESTES DE TROCA IÔNICA DAS RESINAS TROCADORAS DE CÁTIONS
Após as resinas tratadas e secas, montou-se mini colunas que foram
percoladas com solução de NaCl 5,8 g L-1, com bomba peristáltica a uma vazão de
0,5 mL min-1. Frações de 0,5 mL de eluato foram coletados e diluídos a 5,0 mL, onde
uma alíquota de 2,0 mL foi titulada com AgNO3 0,01 mol L-1 pelo método de Mohr. A
figura 20 mostra um gráfico com os resultados das concentrações de cloreto obtidas
para cada resina na forma de prata.
Figura 21: Teste de retenção de cloreto das quatro diferentes resinas

Através dos testes de retenção de cloreto, verificou-se que dentre as quatro
resinas utilizadas, as resinas Lewatit e Amberlit IRC 50 não apresentavam uma
retenção do cloreto satisfatória. A quantidade de cloreto retida em ambas resinas foi
muito pequena ao se avaliar o volume de solução percolada. Nas duas resinas, o
volume máximo de solução que obteve retenção de cloreto foi de 3,0 mL. Este
volume foi considerado insatisfatório tendo em vista que as amostras de alta
salinidade possuem uma concentração de cloreto muito superior a solução utilizada
no teste (NaCl 0,1 mol L-1).
Sabe-se que resinas trocadoras catiônicas fracas não possuem uma ampla
troca pois, por possuírem grupamentos de ácido fraco os hidrogênios não estão
todos disponíveis para troca. Ou seja, os hidrogênios da IRC 50 não estão
totalmente dissociados para que possam ser trocados pela prata. Isso também
restringe o pH de trabalho, o que faz com que não atuem em toda a faixa de pH.
Logo, deu-se continuidade ao trabalho com apenas as outras duas resinas: Dowex e
Amberlite IR 120, que foram as que apresentavam retenções de cloreto significativas
para o presente estudo.
5.3 APLICAÇÃO DO MÉTODO A MATRIZES DE ALTA SALINIDADE E
DETERMINAÇÃO DE PRATA POR ICP OES
Após comprovada a retenção de cloreto pelas mini colunas de prata, aplicou-
se esta metodologia a matrizes com alta salinidade. Para tal, utilizou-se amostras de
água do mar, com salinidade em torno de 35 g L-1. Percolou-se as amostras de água
do mar com uma bomba peristáltica, coletando-se frações a cada 0,5 mL e diluindo a
um volume final de 5,0 mL.
Titulou-se o eluato proveniente da mini coluna de resina de prata, onde foi
possível observar uma redução considerável da concentração de cloreto da amostra,
como pode ser verificado pelas figuras 21 e 22. Para a resina Amberlite IR 120, foi
possível observar uma maior retenção de cloreto ao ser comparada com a Dowex
50W, onde esta maior retenção na primeira resina pode ser atribuída ao fato de
possuir granulometria mais fina do que a segunda, tendo uma maior área superficial
e, conseqüentemente, sítios ativos mais disponíveis na IR 120 do que na Dowex.
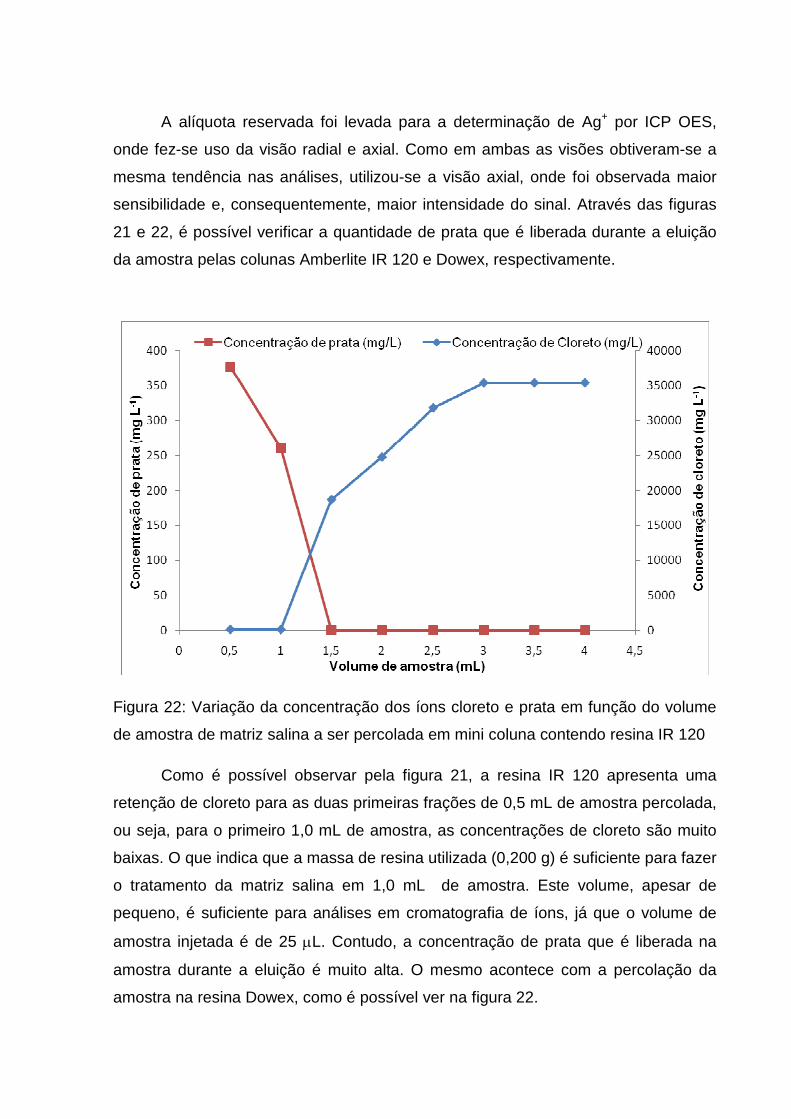
A alíquota reservada foi levada para a determinação de Ag+ por ICP OES,
onde fez-se uso da visão radial e axial. Como em ambas as visões obtiveram-se a
mesma tendência nas análises, utilizou-se a visão axial, onde foi observada maior
sensibilidade e, consequentemente, maior intensidade do sinal. Através das figuras
21 e 22, é possível verificar a quantidade de prata que é liberada durante a eluição
da amostra pelas colunas Amberlite IR 120 e Dowex, respectivamente.
Figura 22: Variação da concentração dos íons cloreto e prata em função do volume
de amostra de matriz salina a ser percolada em mini coluna contendo resina IR 120
Como é possível observar pela figura 21, a resina IR 120 apresenta uma
retenção de cloreto para as duas primeiras frações de 0,5 mL de amostra percolada,
ou seja, para o primeiro 1,0 mL de amostra, as concentrações de cloreto são muito
baixas. O que indica que a massa de resina utilizada (0,200 g) é suficiente para fazer
o tratamento da matriz salina em 1,0 mL de amostra. Este volume, apesar de
pequeno, é suficiente para análises em cromatografia de íons, já que o volume de
amostra injetada é de 25 µL. Contudo, a concentração de prata que é liberada na
amostra durante a eluição é muito alta. O mesmo acontece com a percolação da
amostra na resina Dowex, como é possível ver na figura 22.
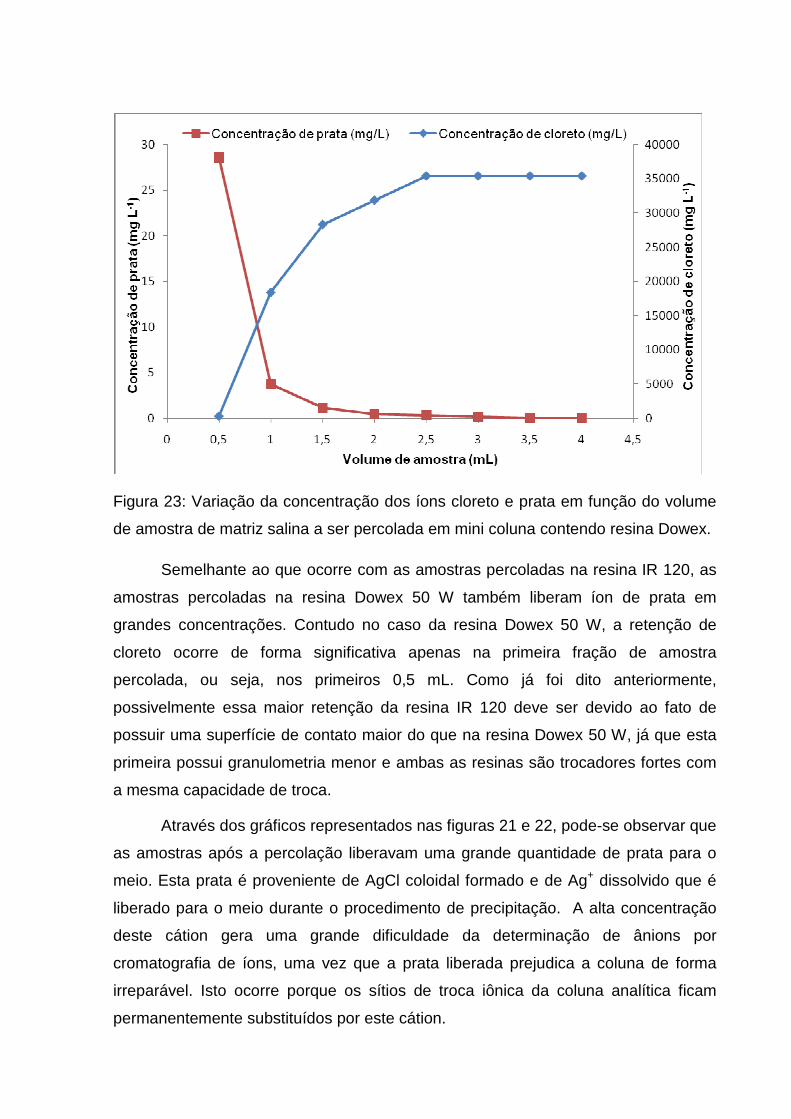
Figura 23: Variação da concentração dos íons cloreto e prata em função do volume
de amostra de matriz salina a ser percolada em mini coluna contendo resina Dowex.
Semelhante ao que ocorre com as amostras percoladas na resina IR 120, as
amostras percoladas na resina Dowex 50 W também liberam íon de prata em
grandes concentrações. Contudo no caso da resina Dowex 50 W, a retenção de
cloreto ocorre de forma significativa apenas na primeira fração de amostra
percolada, ou seja, nos primeiros 0,5 mL. Como já foi dito anteriormente,
possivelmente essa maior retenção da resina IR 120 deve ser devido ao fato de
possuir uma superfície de contato maior do que na resina Dowex 50 W, já que esta
primeira possui granulometria menor e ambas as resinas são trocadores fortes com
a mesma capacidade de troca.
Através dos gráficos representados nas figuras 21 e 22, pode-se observar que
as amostras após a percolação liberavam uma grande quantidade de prata para o
meio. Esta prata é proveniente de AgCl coloidal formado e de Ag+ dissolvido que é
liberado para o meio durante o procedimento de precipitação. A alta concentração
deste cátion gera uma grande dificuldade da determinação de ânions por
cromatografia de íons, uma vez que a prata liberada prejudica a coluna de forma
irreparável. Isto ocorre porque os sítios de troca iônica da coluna analítica ficam
permanentemente substituídos por este cátion.
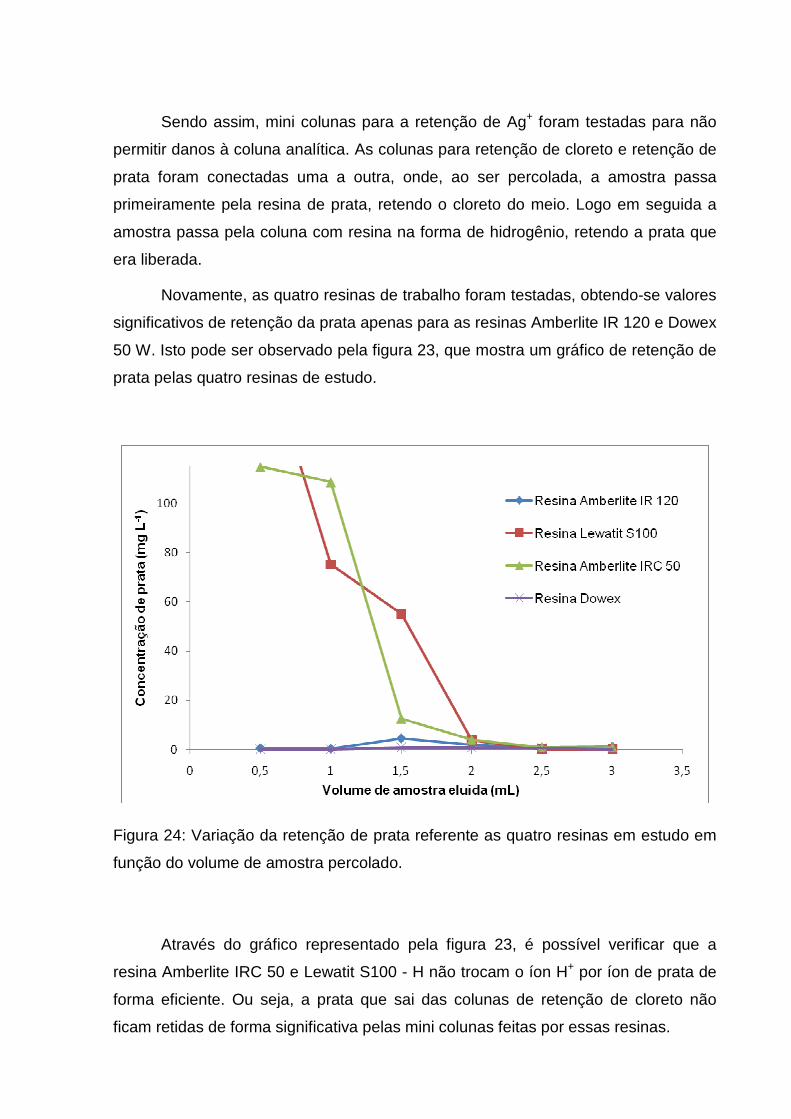
Sendo assim, mini colunas para a retenção de Ag+ foram testadas para não
permitir danos à coluna analítica. As colunas para retenção de cloreto e retenção de
prata foram conectadas uma a outra, onde, ao ser percolada, a amostra passa
primeiramente pela resina de prata, retendo o cloreto do meio. Logo em seguida a
amostra passa pela coluna com resina na forma de hidrogênio, retendo a prata que
era liberada.
Novamente, as quatro resinas de trabalho foram testadas, obtendo-se valores
significativos de retenção da prata apenas para as resinas Amberlite IR 120 e Dowex
50 W. Isto pode ser observado pela figura 23, que mostra um gráfico de retenção de
prata pelas quatro resinas de estudo.
Figura 24: Variação da retenção de prata referente as quatro resinas em estudo em
função do volume de amostra percolado.
Através do gráfico representado pela figura 23, é possível verificar que a
resina Amberlite IRC 50 e Lewatit S100 - H não trocam o íon H+ por íon de prata de
forma eficiente. Ou seja, a prata que sai das colunas de retenção de cloreto não
ficam retidas de forma significativa pelas mini colunas feitas por essas resinas.
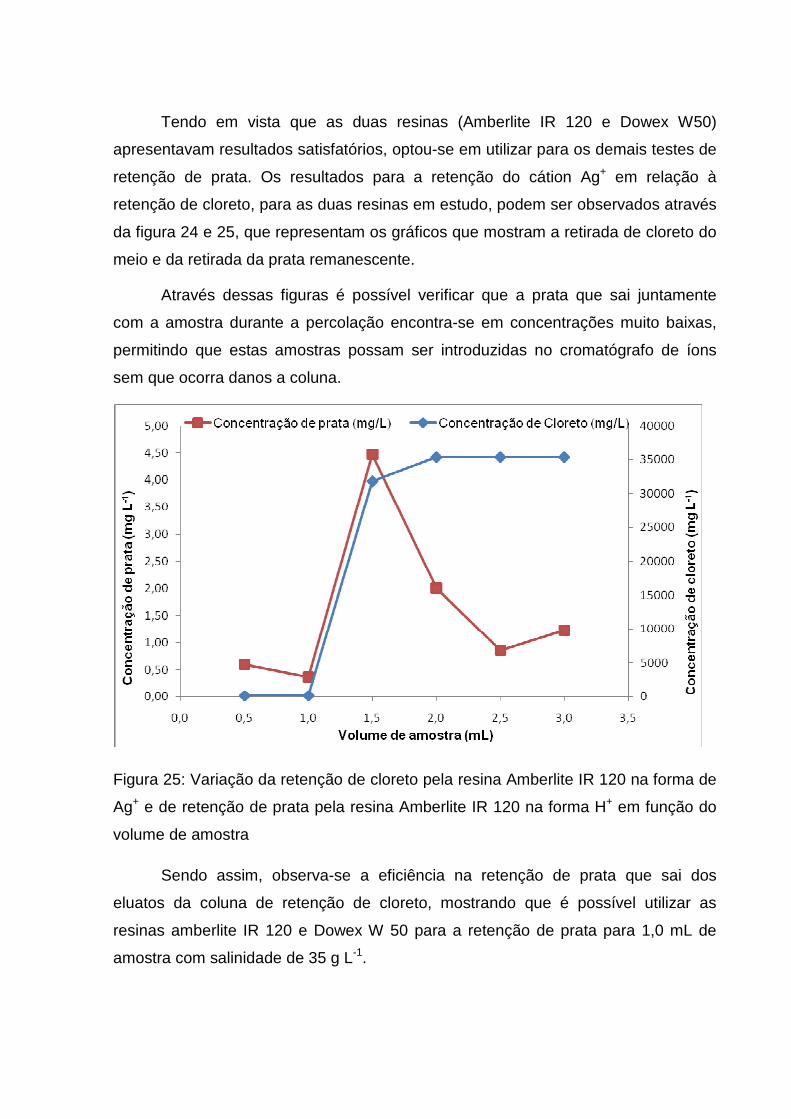
Tendo em vista que as duas resinas (Amberlite IR 120 e Dowex W50)
apresentavam resultados satisfatórios, optou-se em utilizar para os demais testes de
retenção de prata. Os resultados para a retenção do cátion Ag+ em relação à
retenção de cloreto, para as duas resinas em estudo, podem ser observados através
da figura 24 e 25, que representam os gráficos que mostram a retirada de cloreto do
meio e da retirada da prata remanescente.
Através dessas figuras é possível verificar que a prata que sai juntamente
com a amostra durante a percolação encontra-se em concentrações muito baixas,
permitindo que estas amostras possam ser introduzidas no cromatógrafo de íons
sem que ocorra danos a coluna.
Figura 25: Variação da retenção de cloreto pela resina Amberlite IR 120 na forma de
Ag+ e de retenção de prata pela resina Amberlite IR 120 na forma H+ em função do
volume de amostra
Sendo assim, observa-se a eficiência na retenção de prata que sai dos
eluatos da coluna de retenção de cloreto, mostrando que é possível utilizar as
resinas amberlite IR 120 e Dowex W 50 para a retenção de prata para 1,0 mL de
amostra com salinidade de 35 g L-1.
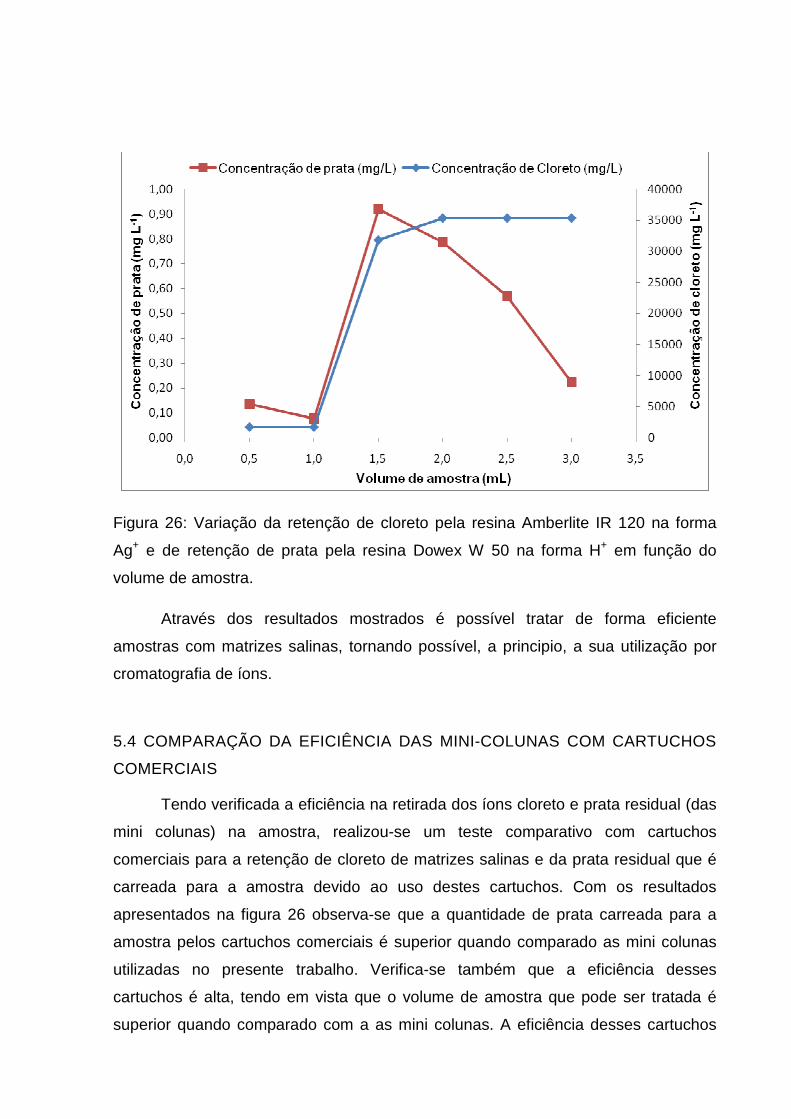
Figura 26: Variação da retenção de cloreto pela resina Amberlite IR 120 na forma
Ag+ e de retenção de prata pela resina Dowex W 50 na forma H+ em função do
volume de amostra.
Através dos resultados mostrados é possível tratar de forma eficiente
amostras com matrizes salinas, tornando possível, a principio, a sua utilização por
cromatografia de íons.
5.4 COMPARAÇÃO DA EFICIÊNCIA DAS MINI-COLUNAS COM CARTUCHOS
COMERCIAIS
Tendo verificada a eficiência na retirada dos íons cloreto e prata residual (das
mini colunas) na amostra, realizou-se um teste comparativo com cartuchos
comerciais para a retenção de cloreto de matrizes salinas e da prata residual que é
carreada para a amostra devido ao uso destes cartuchos. Com os resultados
apresentados na figura 26 observa-se que a quantidade de prata carreada para a
amostra pelos cartuchos comerciais é superior quando comparado as mini colunas
utilizadas no presente trabalho. Verifica-se também que a eficiência desses
cartuchos é alta, tendo em vista que o volume de amostra que pode ser tratada é
superior quando comparado com a as mini colunas. A eficiência desses cartuchos
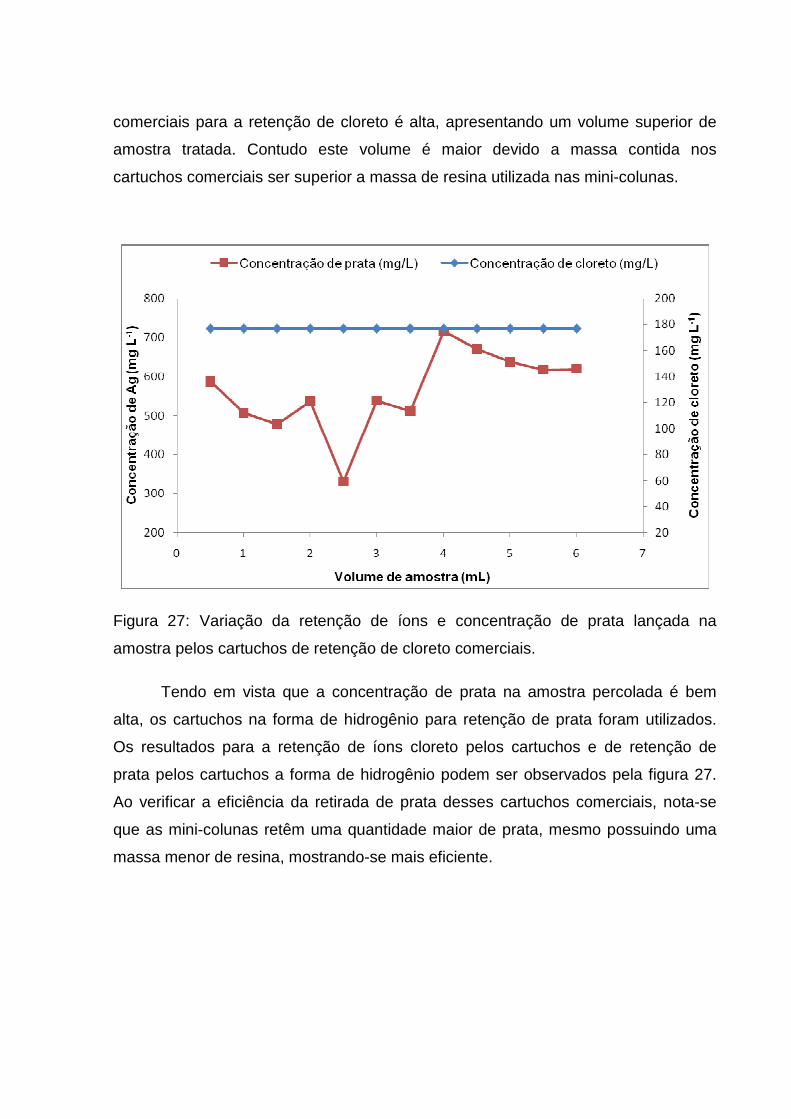
comerciais para a retenção de cloreto é alta, apresentando um volume superior de
amostra tratada. Contudo este volume é maior devido a massa contida nos
cartuchos comerciais ser superior a massa de resina utilizada nas mini-colunas.
Figura 27: Variação da retenção de íons e concentração de prata lançada na
amostra pelos cartuchos de retenção de cloreto comerciais.
Tendo em vista que a concentração de prata na amostra percolada é bem
alta, os cartuchos na forma de hidrogênio para retenção de prata foram utilizados.
Os resultados para a retenção de íons cloreto pelos cartuchos e de retenção de
prata pelos cartuchos a forma de hidrogênio podem ser observados pela figura 27.
Ao verificar a eficiência da retirada de prata desses cartuchos comerciais, nota-se
que as mini-colunas retêm uma quantidade maior de prata, mesmo possuindo uma
massa menor de resina, mostrando-se mais eficiente.
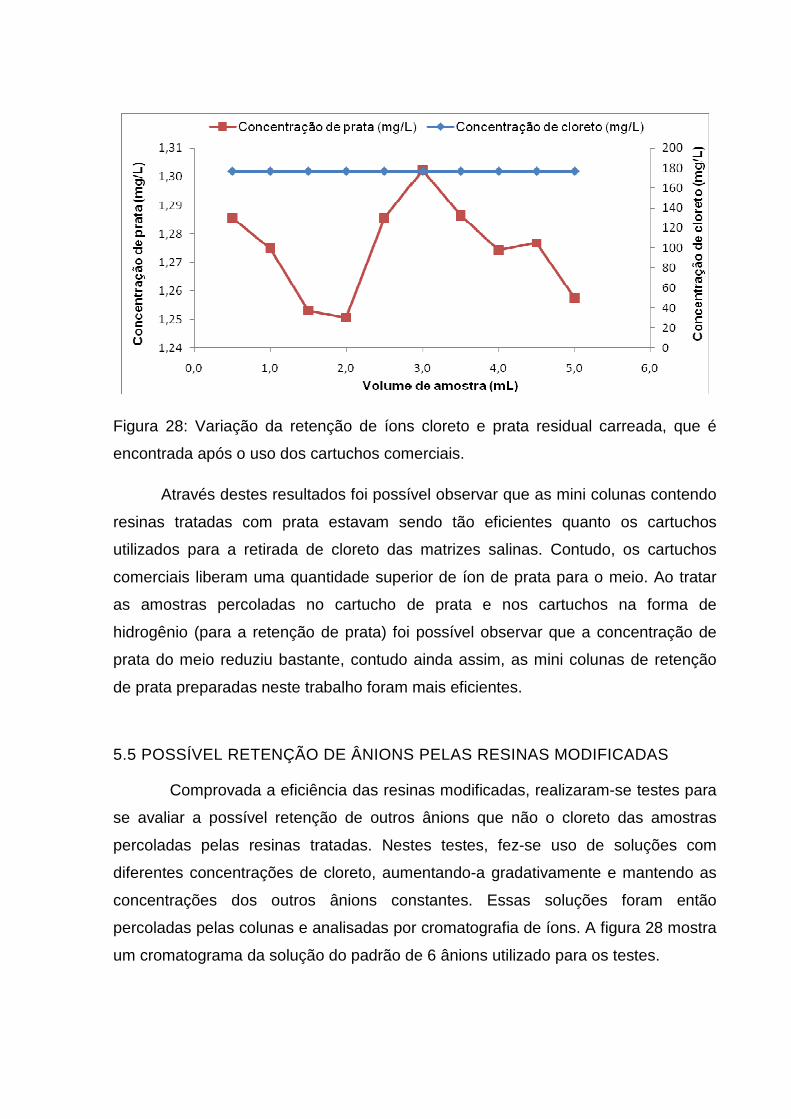
Figura 28: Variação da retenção de íons cloreto e prata residual carreada, que é
encontrada após o uso dos cartuchos comerciais.
Através destes resultados foi possível observar que as mini colunas contendo
resinas tratadas com prata estavam sendo tão eficientes quanto os cartuchos
utilizados para a retirada de cloreto das matrizes salinas. Contudo, os cartuchos
comerciais liberam uma quantidade superior de íon de prata para o meio. Ao tratar
as amostras percoladas no cartucho de prata e nos cartuchos na forma de
hidrogênio (para a retenção de prata) foi possível observar que a concentração de
prata do meio reduziu bastante, contudo ainda assim, as mini colunas de retenção
de prata preparadas neste trabalho foram mais eficientes.
5.5 POSSÍVEL RETENÇÃO DE ÂNIONS PELAS RESINAS MODIFICADAS
Comprovada a eficiência das resinas modificadas, realizaram-se testes para
se avaliar a possível retenção de outros ânions que não o cloreto das amostras
percoladas pelas resinas tratadas. Nestes testes, fez-se uso de soluções com
diferentes concentrações de cloreto, aumentando-a gradativamente e mantendo as
concentrações dos outros ânions constantes. Essas soluções foram então
percoladas pelas colunas e analisadas por cromatografia de íons. A figura 28 mostra
um cromatograma da solução do padrão de 6 ânions utilizado para os testes.
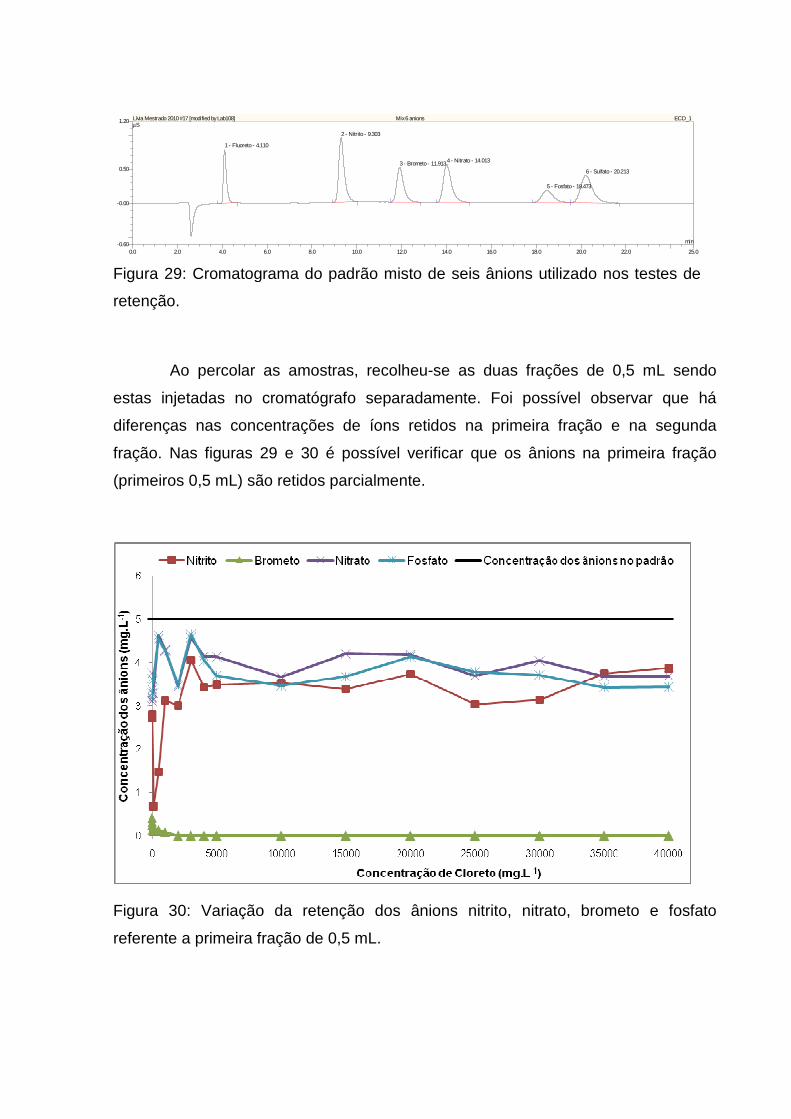
Ao percolar as amostras, recolheu-se as duas frações de 0,5 mL sendo
estas injetadas no cromatógrafo separadamente. Foi possível observar que há
diferenças nas concentrações de íons retidos na primeira fração e na segunda
fração. Nas figuras 29 e 30 é possível verificar que os ânions na primeira fração
(primeiros 0,5 mL) são retidos parcialmente.
Figura 30: Variação da retenção dos ânions nitrito, nitrato, brometo e fosfato
referente a primeira fração de 0,5 mL.
0.0 2.0 4.0 6.0 8.0 10.0 12.0 14.0 16.0 18.0 20.0 22.0 25.0-0.60
-0.00
0.50
1.20 Lívia Mestrado 2010 #17 [modified by Lab108] Mix 6 anions ECD_1µS
min
1 - Fluoreto - 4.110
2 - Nitrito - 9.303
3 - Brometo - 11.9134 - Nitrato - 14.013
5 - Fosfato - 18.473
6 - Sulfato - 20.213
Figura 29: Cromatograma do padrão misto de seis ânions utilizado nos testes de
retenção.
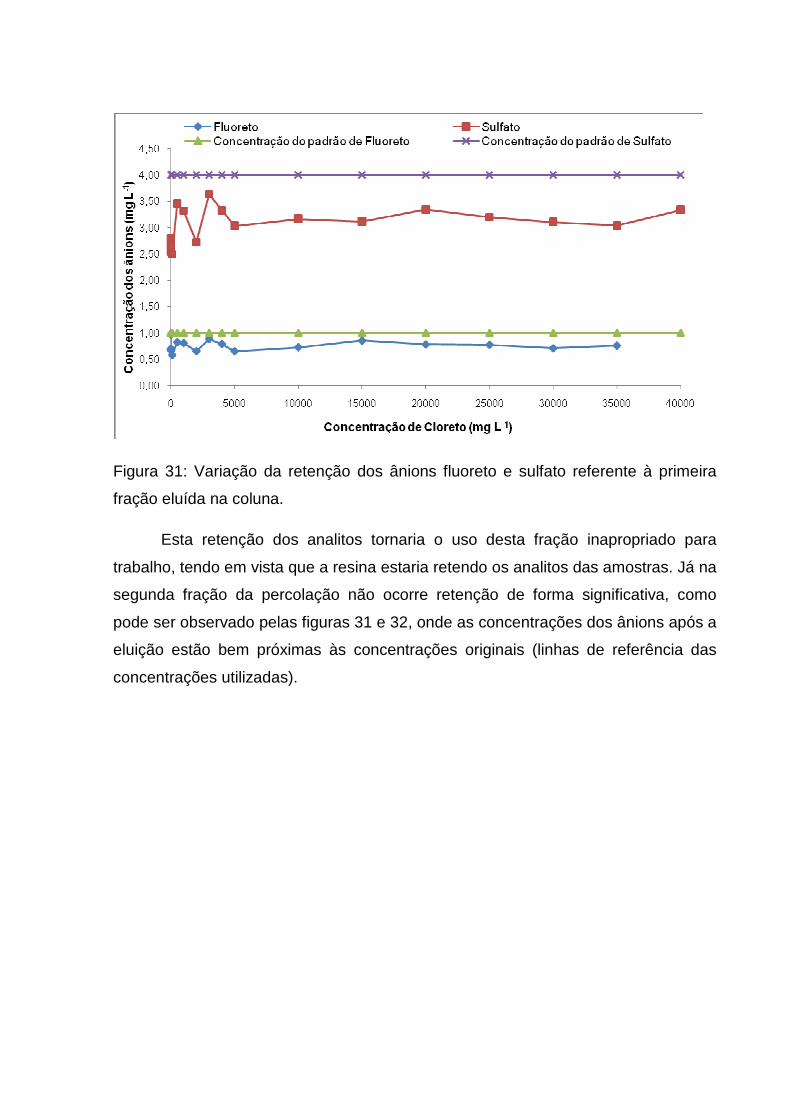
Figura 31: Variação da retenção dos ânions fluoreto e sulfato referente à primeira
fração eluída na coluna.
Esta retenção dos analitos tornaria o uso desta fração inapropriado para
trabalho, tendo em vista que a resina estaria retendo os analitos das amostras. Já na
segunda fração da percolação não ocorre retenção de forma significativa, como
pode ser observado pelas figuras 31 e 32, onde as concentrações dos ânions após a
eluição estão bem próximas às concentrações originais (linhas de referência das
concentrações utilizadas).
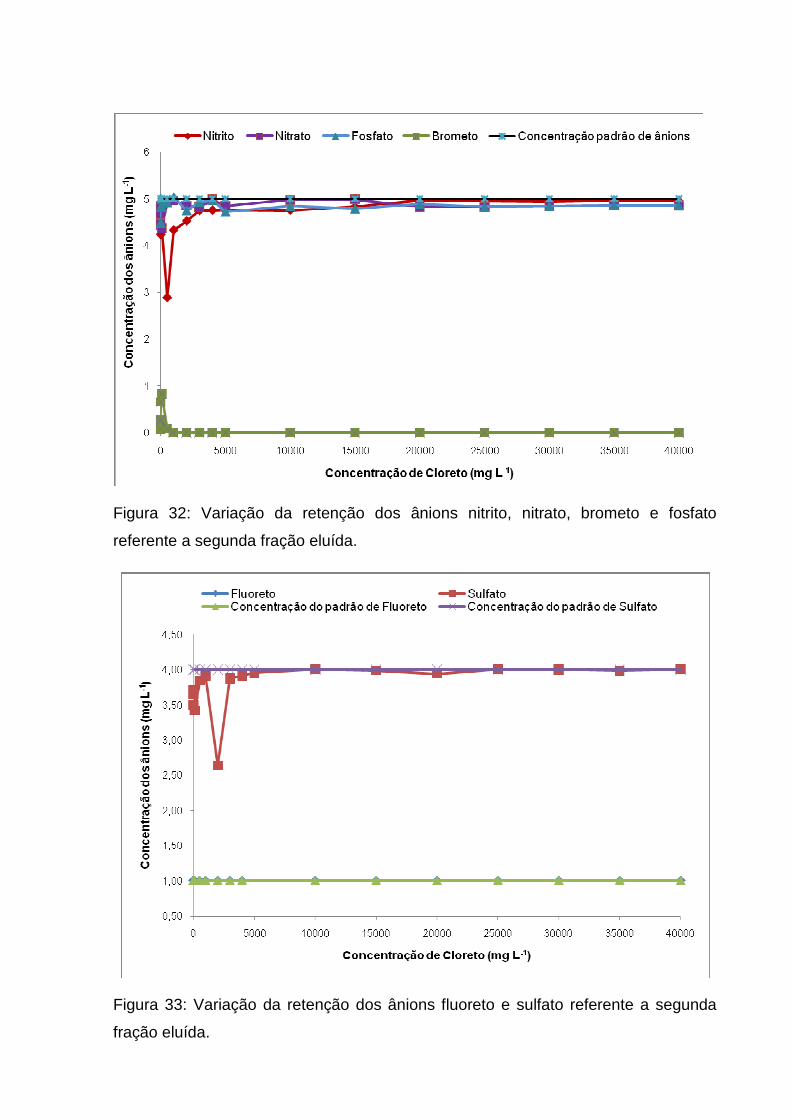
Figura 32: Variação da retenção dos ânions nitrito, nitrato, brometo e fosfato
referente a segunda fração eluída.
Figura 33: Variação da retenção dos ânions fluoreto e sulfato referente a segunda
fração eluída.

Como é possível observar, a primeira fração de amostra que percola pela
coluna retém boa parte dos ânions de trabalho, contudo já na segunda fração, a
retenção é bem menor. A figura 33 mostra um cromatograma obtido através da
percolação de uma matriz de alta salinidade na mini coluna empacotada com a
resina IR 120 na forma de prata e na mini coluna na forma de hidrogênio. Nesta
figura é possível verificar a boa separação entre os ânions de trabalho.
Através do teste de retenção, pode-se constatar que a variação da
concentração do cloreto influi diretamente na retenção dos ânions, tendo em vista
que, em concentrações menores de cloreto, os ânions são mais retidos pela coluna
do que quando a concentração de cloreto é alta. Ou seja, a medida que a
concentração de aumenta, menos analitos são retidos pela resina da mini coluna.
Assim sendo, pode-se concluir que a resina tem maior afinidade pelo cloreto, mas
que, em sua ausência, os outros ânions são retidos, provavelmente na forma de sais
de prata.
Com o objetivo de comparar a eficiência das resinas modificadas com os
cartuchos comerciais, utilizaram-se cartuchos comerciais de retenção de cloreto e de
retenção de prata comercializados pela DIONEX. Os cartuchos foram conectados
em série e então uma solução de alta salinidade contendo os seis ânions foi
percolada. A figura 34 mostra o cromatograma obtido utilizando os cartuchos
comerciais.
0.3 2.5 3.8 5.0 6.3 7.5 8.8 10.0 11.3 12.5 13.8 15.0 16.3 17.5 18.8 20.0 21.3 22.5 24.7-0.21
0.50
1.00
1.55 Lívia Mestrado 2010 #75 [modified by Lab108, 1 peak manually assigned] Mix 6 anions + Cl- 35g/L res 2 ECD_1µS
min
1 - Fluoreto - 4.103
2 - Cloreto - 7.850
3 - Nitrito - 9.3534 - Nitrato - 13.983
5 - Fosfato - 18.533
6 - Sulfato - 20.427
Figura 34: Cromatograma de solução com alta concentração de cloreto após o uso
da resina.
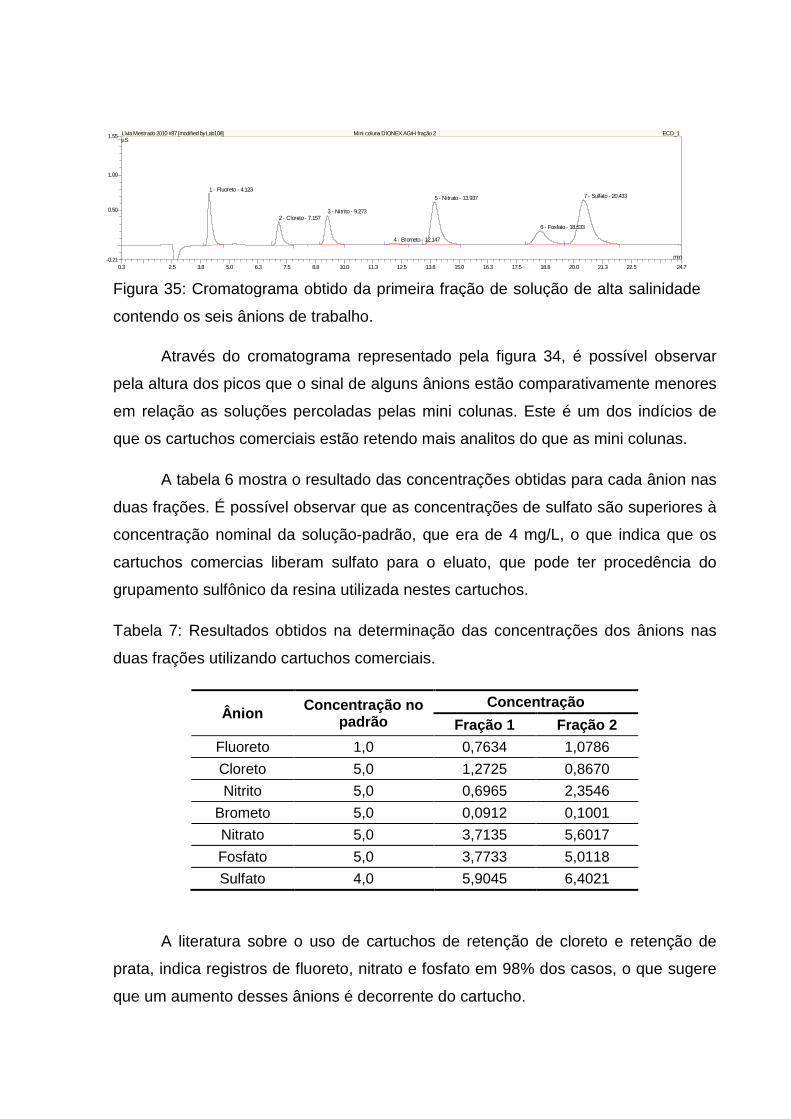
Através do cromatograma representado pela figura 34, é possível observar
pela altura dos picos que o sinal de alguns ânions estão comparativamente menores
em relação as soluções percoladas pelas mini colunas. Este é um dos indícios de
que os cartuchos comerciais estão retendo mais analitos do que as mini colunas.
A tabela 6 mostra o resultado das concentrações obtidas para cada ânion nas
duas frações. É possível observar que as concentrações de sulfato são superiores à
concentração nominal da solução-padrão, que era de 4 mg/L, o que indica que os
cartuchos comercias liberam sulfato para o eluato, que pode ter procedência do
grupamento sulfônico da resina utilizada nestes cartuchos.
Tabela 7: Resultados obtidos na determinação das concentrações dos ânions nas
duas frações utilizando cartuchos comerciais.
Ânion Concentração no padrão
Concentração Fração 1 Fração 2
Fluoreto 1,0 0,7634 1,0786 Cloreto 5,0 1,2725 0,8670 Nitrito 5,0 0,6965 2,3546
Brometo 5,0 0,0912 0,1001 Nitrato 5,0 3,7135 5,6017 Fosfato 5,0 3,7733 5,0118 Sulfato 4,0 5,9045 6,4021
A literatura sobre o uso de cartuchos de retenção de cloreto e retenção de
prata, indica registros de fluoreto, nitrato e fosfato em 98% dos casos, o que sugere
que um aumento desses ânions é decorrente do cartucho.
Figura 35: Cromatograma obtido da primeira fração de solução de alta salinidade
contendo os seis ânions de trabalho.
0.3 2.5 3.8 5.0 6.3 7.5 8.8 10.0 11.3 12.5 13.8 15.0 16.3 17.5 18.8 20.0 21.3 22.5 24.7-0.21
0.50
1.00
1.55 Lívia Mestrado 2010 #87 [modified by Lab108] Mini coluna DIONEX AG/H fração 2 ECD_1µS
min
1 - Fluoreto - 4.123
2 - Cloreto - 7.1573 - Nitrito - 9.273
4 - Brometo - 12.147
5 - Nitrato - 13.937
6 - Fosfato - 18.533
7 - Sulfato - 20.433

Um outro agravante observado durante os testes com estes cartuchos é que
um pico no tempo retenção em torno de 5 minutos aparece. Esta impureza é
problemática durante as analises, tendo em vista que ela demora para sair da
coluna, o que gera um “efeito de memória” nas amostras seguintes.

6 CONCLUSÕES
Dada a importância da identificação dos componentes das águas de alta
salinidade, é possível observar que o desenvolvimento de métodos analíticos que
podem auxiliar na caracterização dessas águas. Através do presente trabalho foi
possível desenvolver uma nova metodologia para o pré tratamento e posterior
análise de águas hipersalinas, para a determinação de ânions baseado no uso da
Cromatografia de Íons.
Um pré-tratamento da amostra é necessário, tendo em vista que a alta
concentração de cloreto presente na matriz afeta a identificação e quantificação dos
outros ânions. Para tal, empregou-se resinas tratadas com prata, que se mostraram
eficientes nessa função.
Contudo, a retenção de cloreto gera um grande inconveniente, que é a
liberação de prata das mini colunas para o eluato das mesmas que, se atingir a
coluna analítica cromatográfica, permanece retida nos sítios ativos, inutilizando-os e
impedindo que ocorra a separação. Para solucionar este problema, uma mini coluna
para reter a prata é colocada logo após a mini coluna de retenção de cloreto. Logo,
ao percolar a amostra por ambas as mini colunas, ocorre seqüencialmente a
retenção do cloreto e da prata no mesmo processo.
Testes realizados com as resinas modificada com prata mostraram que outros
ânions ficam parcialmente retidos apenas na primeira fração, enquanto que as
seguintes não apresentaram este comportamento. A eficácia do método foi
comprovada através do estudo comparativo com cartuchos comerciais de prata e

hidrogênio, mostrando resultados mais satisfatórios quando se utiliza as resinas
modificadas com prata.
Deste modo, os resultados obtidos indicam que as resinas estudadas podem
ser utilizadas em análises de águas com matriz salina, permitindo assim, a
identificação e quantificação de ânions por Cromatografia de Íons.

7 REFERÊNCIAS BIBLIOGRÁFICAS
ALEXANDRATOS, S. D. Ion – Exchange Resins: A Retrospective from Industrial and Engineering Chemistry Research. Industrial & Engineering Chemistry Research , v.48, p.388-398, 2009.
ALI, S. A.; DARLINGTON, L. W.; OCCAPINTI, J. New filtration process cuts contaminants from offshore produced water. Oil & Gas Journal. Novembro 1998, p. 73.
BACCAN, N. et al. Química Analítica Quantitativa Elementar. 3.ed. Campinas: Universidade Federal de Campinas. 2001, 308p.
BLIESNER, D.M. Validating Chromatographic Methods – A Paratical Guide. New Jersey: John Wiley & Sons. 2006, 297p.
BRAITHWATIH, A.; SMITH, S.J. Chromatographic Methods . 5. ed. Kluwer Academic Publishers.1999, 571p.
BRUNO, P. et al. Determination of nutrients in the presence of high chloride concentrations by column-switching ion chromatography. Journal of Chromatography A , v.1003, p.133-141, 2003.
COLLINS, C. H.; BRAGA, G. L.; BONATO, P. S. Fundamentos de Cromatografia. Campinas, SP. Editora Unicamp. 2006. 453p.

COSTA, E. C. T. A.; FROTA, T. M. P. Use of ion chromatography to determine simultaneously high chloride and low íons concentrations in produced water. Brazilian Journal of Petroleum and Gas. v.3, n 1, p 29-34, 2009.
CROMPTON, T.R. Cromatography of Natural, Treated and Wate Waters . NY. Spon Press. 2003. 460p.
DEAN, J. R. Methods for Environmental Trace Analysis. England: John Wiley & Sons Ltd. 2003. 265p.
Dowex Company: Gel x macroporous resins. Uma breve relação das principais características das resinas. Disponível em: <http://www.dow.com/liquidseps/prod/mac_gel.htm> Acesso em: 20/02/2010
DYSON, N. Chromatographic Integration Methods . 2.ed. Cornwall: The Royal Society of Chemistry. 1998.
FARIAS, R. F. Introdução à Química do Petróleo. Rio de Janeiro: Editora Ciência Moderna, 2008. 106p.
FRITZ, J. S. Alook at contemporary ion chromatography. Journal Chromatography, v.439, p 3-11, 1988.
FRITZ, J.S.; GJERDE, D.T. Ion Chromatography . 4.ed. Weinheim: Wiley – VCH. 2009, 385p
GINÉ, M.F. Espectrometria de Emissão Atômica com Plasma Acopla do Indutivamente ICP-AES. Piracicaba: CENA, 1998, 143 p.
GODOS, J.M. Estudio de procesos de adsrción/desorción de iones em resinas encapsuladas: aplicaciones a la remineralización de tejidos dentales. 2004. (Tese de Doutorado). Universitat Autonoma de Barcelona, Barcelona, 2004.

GROS, N. et al. Ionic Composition of seawaters and derived saline solutions determined by ion chromatography and its relation to other water quality parameters. Journal of Chromatography A , v.1210, p.92-28, 2008.
HANSEN, B. R., Davies, S. R. H. Trans. Inst. Chem. Eng. 1994, 72, 176.
HARLAND, C.E. Ion Exchange: Theory and Pratice. 2. ed. Cambridge, UK: Royal Society of chemistry. 1994. 306p.
HELFFERICH, F. Ion Exchange. Nova York. Macgray- Hill, 1995. 624p.
HILL, S.J. Inductively coupled plasma spectrometry and its app lications. England: Sheffield Academic Press, 1999, 370 p.
HUANG, J. S.; Varadaraj, R. J. Colloid Interface Sci. 1996, 1, 535.
INTERNATIONAL ASSOCIATIONS OF OIL AND GAS PRODUCERS (OGP). Fate and effects of naturally occurring substances in pr oduced water on the marine environment. Report n.364, February 2005, 42p.
LANÇAS, F. M. Validação de métodos cromatográficos de análise. São Carlos: Rima, 2004, p.47
LEE, C.H. et al. Development of íon Exchange system for plating wastewater treatment. Desalination. n.180, p 163-172, 2005.
LUCAS, E. F.; Barbosa, C. C. R.; Carvalho, M. S. Estudo do Tratamento de Água Produzida com Membranas Poliméricas. (Relatório final do projeto CT-PETRO-1301), 2001.
OLIVEIRA, A. J. B.; AGUIAR, A. P.; AGUIAR, M.R.M.P. How to mantain the morphology of styrene-divinylbenzene copolymer beads during the sulfonation reaction. Materials letters, 2004.
NETO, F. R. A.; NUNES, D. S. S. Cromatografia: Princípios Básicos e Técnicas Afins. Rio de Janeiro, Interciência, 2003. 187p.

NÖLTE, J.; ICP Emission Spectrometry: a Pratical Guide. Weinheim: Willey-VCH, 2003, 267 p.
PATERSON, R. An Introduction to Ion Exchange. London, Heyden/Sadtler. 1970. 109p
RABELO, D. Formação da estrutura porosa em copolímeros à bas e de estireno e divinilbenzeno. (Dissertação de Mestrado). Universidade Federal do Rio de Janeiro. 1993.
RAZPOTNIK, P. et al. Efficiency and characteristic of solid-phase (ion-exchange) extraction for removal of Cl- matrix. Journal of Chromatography A , v.991, p.23-29, 2003.
SAMUELSON, O. Ion Exchange Separations in Analytical Chemistry . New York. John Wiley & Sons, 1963, 473p.
SCHUWEITZER, P. A. Handbook of separation Techniques for Chemical Engineers . Nova York: McGrawHill, 1979
SILVA, F.V.; TREVIZAN, L.C.; SILVA, C.S.; NOGUEIRA, A.R.A; NÓBREGA, J.A.; Evaluation of inductively coupled plasma optical emission spectrometers with axially and radially viewed configurations. Spectrochimica Acta Part B , v. 57, p.1905-1913, 2002.
SKOOG, D. A. Princípios de Análise Instrumental. 8.ed. Porto Alegre: Bookmam, 2006.
SLINGSBY, R.; KISER, R. Sample Treatment Techniques and methodologies for ion chromatography. Trac trends analytical chemistry , v. 20, p.288-295. 2001
STEPHENSON, M. T. J. Petr. Tech. 1992, 44, 548.
THERMO FISHER SCIENTIFIC INC. iCAP 6000 Series ICP-OES Spectrometer – Operator Manuals, 2005.

THERMO FISHER SCIENTIFIC INC. X SERIES 2 ICP-MS Getting Started Guide – Revision B., 2007.
THOMAS, J. E.; Fundamentos de Engenharia de Petróleo . Rio de Janeiro: Interciência, 2001, v.9, p. 255-267.
THOMPSON, M.; WALSH, J.N. A Handbook of Inductively Coupled Plasma Spectrometr y. New York: Blackie, 1983, 149p. TREVIZAN, L. C.; Nóbrega, J. A. Inductively Coupled Plasma Optical Emission Spectrometry with Axially Viewed Configuration: an Overview of Applications. Journal of Brazilian Chemical Society , v. 18, n. 4, p. 678-690, 2007.
VALCÁRCEL, M.; CASTRO, M. D. Non-Chromatographic continuous separation techniques. Cambridge: The Royal Society of Chemistry, 1991. 290p.
WEISS, J. Handobook of Ion Chromatpgraphy . 3.ed. Weinheim: Wiley-VCH. 2004. 902p




![4.3.2. A Primeira República [11º ano]³dulo 7c.pdf · História A | 12º ano | 2015/2016 1 4.3.2. A Primeira República [11º ano] - Tomada do poder pelo Partido Republicano, de](https://static.fdocumentos.com/doc/165x107/5c002e6a09d3f2ad078bfe65/432-a-primeira-republica-11o-ano-dulo-7cpdf-historia-a-12o-ano.jpg)













