
Uso de painel de genes para sequenciamento de próxima...
85
Isadora Portelinha Moreira Carneiro Uso de painel de genes para sequenciamento de próxima geração no diagnóstico molecular de osteogênese imperfeita Brasília 2017
Transcript of Uso de painel de genes para sequenciamento de próxima...

Isadora Portelinha Moreira Carneiro
Uso de painel de genes para sequenciamento de próxima
geração no diagnóstico molecular de osteogênese imperfeita
Brasília
2017


Isadora Portelinha Moreira Carneiro
Uso de painel de genes para sequenciamento de próxima
geração no diagnóstico molecular de osteogênese imperfeita
Trabalho de Conclusão de Curso apresentado ao
Departamento de Odontologia da Faculdade de
Ciências da Saúde da Universidade de Brasília,
como requisito parcial para a conclusão do curso
de Graduação em Odontologia.
Orientador: Profa. Dra. Ana Carolina de Acevedo-
Poppe
Co-orientador: Profa. Dra. Pollyanna Almeida
Costa dos Santos
Brasília
2017


Àos meus pais Tânia e Ronaldo e ao Paulo Henrique.


AGRADECIMENTOS
Ao concluir este trabalho um turbilhão de emoções
invadiu o meu sentir, recordei do que me motivou a cursar uma
nova graduação e de todas as pessoas que me ajudaram nessa
decisão. Agradeço especialmente ao Paulo Henrique que foi
fundamental para realização desse sonho. Você soube dizer as
palavras certas quando pensei em desistir de fazer o vestibular.
Impossível não recordar desse momento, sempre confiante em
mim, até mesmo quando me falta essa confiança. A distância
esteve muitas vezes presente, mas ela nunca foi empecilho para
nós. Essa foi apenas uma etapa das muitas que iremos
conquistar juntos. Muito obrigada meu Amor por incentivar os
meus sonhos e por me fazer feliz todos os dias.
Agradeço minha mãe Tânia e meu pai Ronaldo por todo o
incentivo e apoio que me deram durante a realização do curso,
mesmo com toda dificuldade vocês fizeram de tudo para que eu
conseguisse concluir e nos momentos de desespero me
acalmava dizendo que tudo terminaria bem. Vocês não tinham
nenhuma obrigação de me ajudar em uma nova graduação, mas
agarraram a oportunidade como se fossem de vocês e fizeram
desse sonho realidade. Vejo o orgulho que sentem de mim em
seus olhos e poder proporcionar essa felicidade é o meu maior
estímulo para ser cada dia uma pessoa e uma profissional
melhor que faça diferença na vida das pessoas. Obrigada por
serem exemplos de superação, amo vocês infinitamente.
Aos meus irmãos Gustavo e César muito obrigada por
me ajudarem, vocês foram fundamentais para a conclusão desse
sonho. Amo vocês!
Obrigada Cibele e Clarivaldo por me acolher como uma
filha, vocês ocupam um lugar especial na minha vida e no meu
coração, foram afetuosos e sempre presentes com pequenas
atenções, lanchinhos gostosos para estudar, chá para dormir e
até companhia durante a madrugada para fazer os provisórios de

prótese fixa. Agradeço a oportundade de poder fazer parte dessa
família tão especial. Joyce, João Paulo, Ariela e a minha querida
Clarinha muito obrigada pelos momentos felizes compartilhados.
À minha família, tios, tias e primos obrigada por vocês
serem parte da minha história. Obrigada especial ao meu
padrinho Tio Chico que me incentivou e orientou a fazer
odontologia e a Tia Débora de quem sempre irei recordar com
alegria e carinho, você faz muita falta.
Agradeço todos os meus professores pelos
conhecimentos compartilhados. Aos professores Liliana e
Rodrigo, vocês são exemplos para mim, são profissionais
competentes com uma família linda, aprendi com vocês que é
possível ter a vida profissional e a pessoal em harmonia. Ao
professor Lucas Tabata por sempre estar disposto a ajudar no
que precisei durante a graduação, até mesmo para conversar
sobre a profissão. Aos professores Leandro Hilgert e Soraya Leal
por me inspirarem através do exemplo de profissionais
competentes que vocês são. Ao professor Paulo Galvão por todo
encentivo e também por me mostrar que com a odontologia
também podemos ajudar de uma forma diferente os que mais
precisam. Em especial a minha querida orientadora Ana Carolina
que me acolheu com muito carinho em um momento de
desespero no meu primeiro ano de graduação, com quem criei
um vínculo muito especial. Obrigada por sua paciência, por suas
palavras docentes e por acreditar que eu seria capaz de um
desafio tão grande como esse projeto.
Dr. Paulo Yamagutti muito obrigada por compartilhar o
seu conhecimento de uma forma tão especial. Você é exemplo
de dedicação, de competência e de humanidade, um profissional
em quem me espelho. Espero um dia ser um pouquinho do que
você é para esses pacientes.
Obrigada muito especial a minha co-orientadora e amiga
Pollyanna pela ajuda em todos os momentos: tanto nas horas de
realizar os experimentos, analisar os resultados e corrigir o

trabalho, como nas horas de conversas alegres. Que bom que
tive a feliz oportunidade de conhecê-la.
Agradeço aos componentes do laboratório de
Histopatologia Bucal, nele pude aprender muito e fazer muitas
amizades. São pessoas queridas que tive o privilégio de conviver
durante 4 anos. Agradeço especialmente à Lídia e ao Luan pela
participação nas extrações de DNA; a Thaís pelas conversas
divertidas e a querida Ana Luiza pelo carinho, boa vontade e
companhia na Clínica de Anomalias Dentárias. Agradeço aos
pacientes com osteogênese imperfeita que aceitaram participar
dessa pesquisa, sem eles este estudo não seria possível.
A minha querida turma 65 de Odontologia agradeço por
todos os momentos que compartilhamos nesses anos, vocês
estiveram presente em todos os meus dias e contribuíram para
ser uma jornada mais alegre e divertida. Sentirei falta de todos
vocês.
Muito obrigada Tainara, Jéssica, Lucas e Ygor, vocês são
amigos que a odontologia me presenteou para a vida.
A minha querida companheira de atendimentos clínicos
Nathália Maria muito obrigada por ser minha parceira em tudo,
você foi fundamental para que a graduação fosse alegre,
divertida e leve. Sentirei saudade de tê-la todos os dias comigo.
Você é uma pessoa muito especial, com um grande coração.
Sou grata por tudo que compartilhamos e pela amizade que
criamos.
Agradeço aos meus amigos, aos que estão longe e aos
que fiz em Brasília, vocês permitem que a vida não seja feita
apenas de trabalho, mas também de felicidade e bem-estar.
Por fim, agradeço a Deus por me permitir estar em
constante evolução.
Muito Obrigada!


EPÍGRAFE
“Para tudo há soluções, mas é necessário encontrar as mais
felizes. E as mais felizes não podem ser outras do que as que
permitem a mudança em nós mesmos.”
Carlos Gonzáles Pecoche


RESUMO
CARNEIRO, Isadora Portelinha Moreira. Uso de painel de genes
para sequenciamento de próxima geração no diagnóstico
molecular de osteogênese imperfeita. 2017. Trabalho de
Conclusão de Curso (Graduação em Odontologia) –
Departamento de Odontologia da Faculdade de Ciências da
Saúde da Universidade de Brasília.
A osteogênese imperfeita (OI) é um grupo de condições
genéticas heterogêneas, caracterizadas principalmente pela
fragilidade óssea. A OI é considerada um transtorno relacionado
à síntese e estrutura do colágeno e a maioria dos casos (80-
90%) está relacionada a mutações dominantes no COL1A1 e
COL1A2, genes que codificam as cadeias de colágeno tipo 1
(COL1). Na última década, foram relatadas mutações em pelo
menos 16 outros genes associados a casos de OI autossômico
recessivo e um ligado ao cromossomo X. Clinicamente, a OI é
caracterizada por apresentar alterações esqueléticas e extra-
esqueléticas. A dentinogênese imperfeita (DGI), uma alteração
na formação da dentina dentinária, está presente em
aproximadamente 50% dos casos de OI dos tipos I a IV. O
objetivo desse estudo foi identificar, pelo método de
sequenciamento de nova geração (NGS), mutações patológicas
em 33 pacientes diagnosticados com OI e caracterizar as
alterações dentárias clínicas e radiográficas dos pacientes
diagnosticados com DGI, em tratamento com bisfosfonato e em
atendimento na Clínica de Pacientes Portadores de Anomalias
de Desenvolvimento Dentário do Hospital Universitário de
Brasília. Com a finalidade de caracterizar as manifestações
dentárias, os prontuários desses pacientes foram analisados. A
avaliação genética foi realizada por meio do desenvolvimento de
um painel de NGS, composto de 14 genes associados à OI e a
sua análise foi realizada utilizando a plataforma Ion AmpliSeq™.

Por meio do NGS foi possível identificar 22 mutações patológicas
em 33 pacientes diagnosticados com OI, apresentando ou não
DGI. Um total de 18 mutações em heterozigose nos genes
COL1A1 e COL1A2 foram identificadas neste estudo, sendo 9
mutações em COL1A1 previamente relatadas na literatura (7
missense, 1 nonsense e 1 frameshift), e 9 mutações missense
em COL1A2 (6 já descrita anteriormente na literatura e 4 não
relatadas, mas consideradas patogênicas de acordo com as
análises in silico). Em quatro pacientes com história de
consanguinidade foram identificadas mutações missense
homozigóticas nos genes SERPINF1, P3H1 e CRTAP. Não
foram encontradas mutações patogênicas nos genes estudados
em 11 pacientes. Dos pacientes estudados, 21 apresentaram
DGI. Alterações nos genes COL1A1 e COLA1A2 foram as mais
frequentes, concordando com a literatura. Sete mutações não
foram previamente relatas na literatura sugerindo serem
mutações novas. A caracterização clínica revelou que todos os
pacientes com DGI foram do tipo moderada. O estudo permitiu
aos participantes um diagnóstico molecular através da técnica de
NGS, podendo proporcionar para esses pacientes um
aconselhamento genético adequado assim como um
acompanhamento terapêutico mais preciso.

ABSTRACT
CARNEIRO, Isadora Portelinha Moreira. Use of gene panel for next generation sequencing in the molecular diagnosis of imperfect osteogenesis. 2017. Undergraduate Course Final Monograph (Undergraduate Course in Dentistry) – Department of Dentistry, School of Health Sciences, University of Brasília.
Osteogenesis imperfecta (OI) is a group of heterogeneous
genetic conditions characterized mainly by bone fragility. OI is
considered an related disorder and collagen structure and most
cases (80-90%) are related to dominant mutations in COL1A1
and COL1A2, the genes encoding the collagen chains type 1
(COL1). In the last decade, mutations have been reported in at
least 16 other genes associated with cases of autosomal
recessive OI and one associated with chromosome X. Clinically,
OI is characterized by skeletal and extra-skeletal disorder. The
dentinogenesis imperfecta (DGI) is characterize bay dentin
formation anomaly, and it is present in approximately 50% of
cases of OI of types I to IV. The objective of this study was to
identify pathological mutations in 33 patients diagnosed with OI
and to characterize the clinical and radiographic dental alterations
of the patients diagnosed with DGI in treatment with
bisphosphonate and in the Clinical Patients with Dental
Developmental Anomalies of the University Hospital of Brasília. In
order to characterize the dental manifestations, the medical
records of these patients were analyzed. Genetic evaluation was
performed through the development of NGS panel composed of
14 OI associated genes and analyzed using the Ion AmpliSeq ™
platform. Through the NGS it was possible to identify 22
pathological mutations in 33 patients diagnosed with OI,
presenting or not with DGI. A total of 18 heterozygous mutations
in the COL1A1 and COL1A2 genes were identified in this study,
with 9 mutations in COL1A1 previously reported in the literature

(7 missense, 1 nonsense and 1 frameshift), and 9 missense
mutations in COL1A2 (6 previously described in the literature and
4 not reported but considered pathogenic according to in silico
analyzes). In four patients with a history of consanguinity,
homozygous missense mutations were identified in the
SERPINF1, P3H1 and CRTAP genes. No pathogenic mutations
were found in the genes studied in 11 patients. Of the patients
studied, 21 presented with DGI. Changes in the COL1A1 and
COL1A2 genes were the most frequent, in concordance with the
literature. Seven mutations have not previously been reported in
the literature suggesting they are new mutations. Clinical
characterization revealed that all patients with DGI were of the
moderate type. The study allowed the participants a molecular
diagnosis through the NGS technique. Molecular diagnosis will
allow adequate genetic counseling as well as more precise
therapeutic follow-up.

SUMÁRIO
Artigo Científico ........................................................................... 19 Folha de Título ........................................................................ 21 Lista de Abreviaturas .............................................................. 22 Resumo ................................................................................... 24 Abstract ................................................................................... 27 Introdução ............................................................................... 29 Materiais e Métodos ................................................................ 35 Resultados .............................................................................. 38 Discussão ................................................................................ 46 Conclusão................................................................................ 49
Referências ............................................................................. 51
Anexos ......................................................................................... 58
Termo de consentimento livre e esclarecido .......................... 58 Normas da Revista .................................................................. 59


19
ARTIGO CIENTÍFICO
Este trabalho de Conclusão de Curso é baseado no artigo
científico:
CARNEIRO, Isadora Portelinha Moreira; SANTOS, Pollyanna
Almeida Costa; Acevedo-Poppe, Ana Carolina.
Uso de painel de genes para sequenciamento de próxima
geração no diagnóstico molecular de osteogênese imperfeita
Apresentado sob as normas de publicação da revista BONE

20

21
FOLHA DE TÍTULO
Uso de painel de genes para sequenciamento de próxima
geração no diagnóstico molecular de osteogênese imperfeita
Use of gene panel for next generation sequencing in the molecular diagnosis of imperfect osteogenesis
Isadora Portelinha Moreira Carneiro1,4
Pollyanna Almeida Costa dos Santos2
Ana Carolina Acevedo-Poppe3,4
1Aluna de Graduação em Odontologia da Universidade de
Brasília (UnB). 2Universidade Estadual de Ciências da Saúde de Alagoas
(UNCISAL) 3Laboratório de Histopatologia Bucal, Departamento de
Odontologia, Faculdade de Ciências da Saúde, Universidade de
Brasília (UnB). 4Clínica de Atendimento a pacientes portadores de anomalias
dentárias.
Correspondência: Profa. Dra. Ana Carolina Acevedo Poppe
Campus Universitário Darcy Ribeiro - UnB - Faculdade de
Ciências da Saúde - Departamento de Odontologia - CEP:
70910-900 - Asa Norte - Brasília - DF
E-mail: [email protected] / Telefone: (61) 31071977.

22
LISTA DE ABREVIATURAS
AD Autossômico dominante
AR Autossômico recessive
BMP1 Proteína morfogenética óssea 1
COL1A1 Colágeno tipo 1 cadeia alfa 1
COL1A2 Colágeno tipo 1 cadeia alfa 1
CREB3L1 Proteína de ligação ao elemento responsivo a cAMP 3 como 1
CRTAP Proteína associada à cartilagem
DGI Dentinogênese imperfeita
FKBP10 Proteína de ligação a FK206
HUB Hospital universitário de Brasília
IFITM5 Proteína transmembranar induzida por interferão 5
LEPRE1 Proteoglicano enriquecido com prolina de leucina 1
MBTPS2 Fator de transcrição ligado à membrana peptidase local 2
NGS Sequenciamento de nova geração
OI Osteogênese imperfeita
P3H1 Prolil 3-hidroxilase 1
P4HB Prolil 4-hidroxilase beta
PLOD2 Procollagen-lisina, 2 oxoglutarato 5-dioxigenase 2
PLS3 Plastin 3

23
PPIB Peptidilprolyl isomerase B
SCN9A Subunidade alfa de canal de tensão de sódio 9
SEC24D SEC24 homólogo D, COPII componente de revestimento complexo
SERPINF1 Serpin família F membro 1
SERPINH1 Serpin família H membro 1
SLC2A2 Família transportadora de soluto 2
SP7 Fator de transcrição 7
SPARC proteína secretada ácida rica em cisteína
TMEM38B Proteína transmembrana 38B
UnB Universidade de Brasília
WNT1 Família de sítios de integração MMTV, membro 1

24
RESUMO
Uso de painel de genes para sequenciamento de próxima
geração no diagnóstico molecular de osteogênese imperfeita
A osteogênese imperfeita (OI) é um grupo de condições
genéticas heterogêneas e caracterizadas principalmente pela
fragilidade óssea. A OI é considerada um transtorno relacionado
à síntese e estrutura do colágeno e a maioria dos casos (80-
90%) está relacionada a mutações dominantes no COL1A1 e
COL1A2, genes que codificam as cadeias de colágeno tipo 1
(COL1). Na última década, foram relatadas mutações em pelo
menos 16 outros genes associados a casos de OI autossômico
recessivo e um ligado ao cromossomo X. Clinicamente, a OI é
caracterizada por apresentar alterações esqueléticas e extra-
esqueléticas. A dentinogênese imperfeita (DGI), uma alteração
na formação da dentina dentinária, está presente em
aproximadamente 50% dos casos de OI dos tipos I a IV. O
objetivo desse estudo foi identificar, pelo método de
sequenciamento de nova geração (NGS), mutações patológicas
em 33 pacientes diagnosticados com OI e caracterizar as
alterações dentárias clínicas e radiográficas dos pacientes
diagnosticados com DGI, em tratamento com bisfosfonato e em
atendimento na Clínica de Pacientes Portadores de Anomalias
de Desenvolvimento Dentário do Hospital Universitário de
Brasília. Com a finalidade de caracterizar as manifestações
dentárias, os prontuários desses pacientes foram analisados. A
avaliação genética foi realizada por meio do desenvolvimento de
um painel de NGS, composto de 14 genes associados à OI e a
sua análise foi realizada utilizando a plataforma Ion AmpliSeq™.
Por meio do NGS foi possível identificar 22 mutações patológicas
em 33 pacientes diagnosticados com OI, apresentando ou não
DGI. Um total de 18 mutações em heterozigose nos genes

25
COL1A1 e COL1A2 foram identificadas neste estudo, sendo 9
mutações em COL1A1 previamente relatadas na literatura (7
missense, 1 nonsense e 1 frameshift), e 9 mutações missense
em COL1A2 (6 já descrita anteriormente na literatura e 4 não
relatadas, mas consideradas patogênicas de acordo com as
análises in silico). Em quatro pacientes com história de
consanguinidade foram identificadas mutações missense
homozigóticas nos genes SERPINF1, P3H1 e CRTAP. Não
foram encontradas mutações patogênicas nos genes estudados
em 11 pacientes. Dos pacientes estudados, 21 apresentaram
DGI. Alterações nos genes COL1A1 e COLA1A2 foram as mais
frequentes, concordando com a literatura. Sete mutações não
foram previamente relatas na literatura sugerindo serem
mutações novas. A caracterização clínica revelou que todos os
pacientes com DGI foram do tipo moderada. O estudo permitiu
aos participantes um diagnóstico molecular através da técnica de
NGS, podendo proporcionar para esses pacientes um
aconselhamento genético adequado assim como um
acompanhamento terapêutico mais preciso.
Palavras-chave
Osteogênese Imperfeita; Dentinogênese Imperfeita; COL1A1;
COL1A2; Sequenciamento de Nova Geração.
Relevância Clínica
Apesar dos avanços nos estudos de biologia molecular, o
diagnóstico de OI ainda é eminentemente clínico. Entretanto, os
dados moleculares são de suma importância para permitir a
classificação do tipo de OI dos pacientes com mais precisão. O
conhecimento do gene envolvido na doença pode auxiliar na
indicação do melhor acompanhamento odontológico. Além disso,
o conhecimento das bases moleculares permite um maior

26
entendimento do padrão de herança da OI na população e
melhor aconselhamento genético à família.

27
ABSTRACT Use of gene panel for next generation sequencing in the molecular diagnosis of imperfect osteogenesis
Osteogenesis imperfecta (OI) is a group of heterogeneous
genetic conditions characterized mainly by bone fragility. OI is
considered an related disorder and collagen structure and most
cases (80-90%) are related to dominant mutations in COL1A1
and COL1A2, the genes encoding the collagen chains type 1
(COL1). In the last decade, mutations have been reported in at
least 16 other genes associated with cases of autosomal
recessive OI and one associated with chromosome X. Clinically,
OI is characterized by skeletal and extra-skeletal disorder. The
dentinogenesis imperfecta (DGI) is characterize bay dentin
formation anomaly, and it is present in approximately 50% of
cases of OI of types I to IV. The objective of this study was to
identify pathological mutations in 33 patients diagnosed with OI
and to characterize the clinical and radiographic dental alterations
of the patients diagnosed with DGI in treatment with
bisphosphonate and in the Clinical Patients with Dental
Developmental Anomalies of the University Hospital of Brasília. In
order to characterize the dental manifestations, the medical
records of these patients were analyzed. Genetic evaluation was
performed through the development of NGS panel composed of
14 OI associated genes and analyzed using the Ion AmpliSeq ™
platform. Through the NGS it was possible to identify 22
pathological mutations in 33 patients diagnosed with OI,
presenting or not with DGI. A total of 18 heterozygous mutations
in the COL1A1 and COL1A2 genes were identified in this study,
with 9 mutations in COL1A1 previously reported in the literature
(7 missense, 1 nonsense and 1 frameshift), and 9 missense
mutations in COL1A2 (6 previously described in the literature and

28
4 not reported but considered pathogenic according to in silico
analyzes). In four patients with a history of consanguinity,
homozygous missense mutations were identified in the
SERPINF1, P3H1 and CRTAP genes. No pathogenic mutations
were found in the genes studied in 11 patients. Of the patients
studied, 21 presented with DGI. Changes in the COL1A1 and
COL1A2 genes were the most frequent, in concordance with the
literature. Seven mutations have not previously been reported in
the literature suggesting they are new mutations. Clinical
characterization revealed that all patients with DGI were of the
moderate type. The study allowed the participants a molecular
diagnosis through the NGS technique. Molecular diagnosis will
allow adequate genetic counseling as well as more precise
therapeutic follow-up.
Keywords
Osteogenesis Imperfecta; Dentinogenesis Imperfecta; COL1A1; COL1A2; New Generation Sequencing.
Clinical Relevance:
Despite advances in molecular biology, the diagnosis of OI and
DGI is still eminently clinical. However, molecular data are of
great importance in order to better classify patients OI type.
Knowledge of the molecular alterations involved in the disease
can assist in the indication of the best dental management. In
addition, knowledge of the molecular basis allows a better
understanding of the inheritance pattern of OI in the population
and better genetic counseling to the family.

29
1.1 INTRODUÇÃO
A Osteogênese Imperfeita (OI) é um grupo de desordens
genéticas raras (1:15.000 a 1:20.000 nascimentos) com grau
variável de alterações congênitas no tecido conjuntivo resultando
em fragilidade e deformidades óssea. As alterações esqueléticas
incluem fragilidade ósseas, deformidades esqueléticas e ossos
wormianos. Além disso, alterações extra-esqueléticas tais como
esclera azulada, frouxidão ligamentar, hipotonia muscular, surdez
precoce e dentinogênese imperfeita (DGI) são observadas em
pacientes com OI. As manifestações clínicas da OI variam de
casos leves com poucas fraturas e estatura normal a casos
graves com letalidade perinatal. Os padrões de herança
autossômico dominante (AD) e recessivo (AR) tem sido
relatados, e mais recentemente herança ligada ao X, sendo o
modo de herança autossômico dominante o mais frequente (90%
AD, 10% AR) [1–3].
Em 1979 Sillence propôs uma classificação baseada nas
características clínicas, radiológicas, hereditárias e na gravidade
da doença em quatro tipos: o tipo I (OMIM # 166200) de OI é a
forma mais leve e a mais comum da doença, geralmente não é
detectada no nascimento, possui deformidades esqueléticas
leves, pode apresentar esclera azulada e perda auditiva. O tipo II
(OMIM # 166210) é letal no período perinatal ou sobrevivem por
alguns meses. O tipo III (OMIM # 259420) é a forma mais grave
dos que sobrevivem o período neonatal, é progressivamente
deformante, possuem múltiplas fraturas, baixa estatura, face
triangular, pode apresentar esclera azulada e DGI. O tipo IV
(OMIM # 16620) apresenta deformidades de moderada a grave,
a DGI pode ou não estar associada, e a esclera azulada podem
estar presentes na infância desaparecendo com o decorrer dos
anos [4].
A classificação de Sillence incluía apenas os casos com
modo de herança autossômico dominante. Dessa forma, Em

30
2004 Rauch e Glorieux diagnosticaram novos tipos de OI e
adicionaram os tipos V (OMIM # 610967), VI (OMIM # 613982) e
VII (OMIM # 610682) à classificação. A partir desses achados,
outros estudos moleculares foram realizados e novos genes com
modo de herança AR foram descobertos e incluídos na
classificação da OI. Esses novos tipos não são causados por
mutações nos genes COL1A1 (OMIM # 120150) e COL1A2
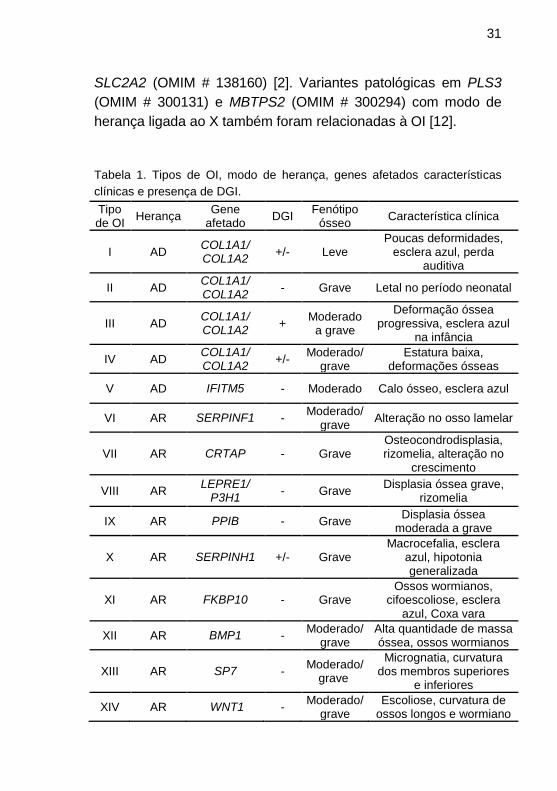
(OMIM # 120160). Na tabela 1 encontram-se todas as formas
descritas, até o momento, de OI, com a forma de herança, genes
envolvidos, fenótipo e características clínicas [5–7].
As evidências dos estudos moleculares tem demonstrado
que os tipos de I a IV estão relacionados a mutações AD em
genes que codificam as cadeias alfa 1 e alfa 2 do colágeno tipo 1
(COL1A1 e COL1A2) [8]. Variantes dominantes em IFITM5
(OMIM # 614757) e P4HB (OMIM # 176790) também foram
descritas em alguns indivíduos afetados com herança
autossômica dominante [1,9,10]. Na última década, foram
relatadas mutações AR nos genes CRTAP (OMIM # 605497),
LEPRE1 (OMIM # 610339), PPIB (OMIM # 123841), FKBP10
(OMIM # 607063), SERPINH1 (OMIM # 600943), PLOD2 (OMIM
# 601865), SEC24D (OMIM # 607186), BMP1 (OMIM # 112264)
e TMEM38B (OMIM # 611236) cujos produtos proteicos
interagem e interferem na síntese do colágeno tipo 1 e no
desenvolvimento ósseo, predispondo a distintos padrões de
fragilidade óssea e evolução clínica [5,11]. Foram descritas
também alterações em outros genes que não estão diretamente
ligados à síntese do colágeno tipo 1, mas atuam na
mineralização ou no desenvolvimento dos osteoblastos, entre
esse grupo de genes estão CREB3L1 (OMIM # 616215),
SERPINF1 (OMIM # 172860), SP7 (OMIM # 606633), SPARC
(OMIM # 182120) e WNT1 (OMIM # 164820). Recentemente, um
estudo identificou mais três mutações relacionadas à OI nos
genes SCN9A (OMIM # 603415), NTRK1 (OMIM # 191315) e
31
SLC2A2 (OMIM # 138160) [2]. Variantes patológicas em PLS3
(OMIM # 300131) e MBTPS2 (OMIM # 300294) com modo de
herança ligada ao X também foram relacionadas à OI [12].
Tabela 1. Tipos de OI, modo de herança, genes afetados características
clínicas e presença de DGI.
Tipo de OI
Herança Gene
afetado DGI
Fenótipo ósseo
Característica clínica
I AD COL1A1/ COL1A2
+/- Leve Poucas deformidades,
esclera azul, perda auditiva
II AD COL1A1/ COL1A2
- Grave Letal no período neonatal
III AD COL1A1/ COL1A2
+ Moderado
a grave
Deformação óssea progressiva, esclera azul
na infância
IV AD COL1A1/ COL1A2
+/- Moderado/
grave Estatura baixa,
deformações ósseas
V AD IFITM5 - Moderado Calo ósseo, esclera azul
VI AR SERPINF1 - Moderado/
grave Alteração no osso lamelar
VII AR CRTAP - Grave Osteocondrodisplasia, rizomelia, alteração no
crescimento
VIII AR LEPRE1/
P3H1 - Grave
Displasia óssea grave, rizomelia
IX AR PPIB - Grave Displasia óssea
moderada a grave
X AR SERPINH1 +/- Grave Macrocefalia, esclera
azul, hipotonia generalizada
XI AR FKBP10 - Grave Ossos wormianos,
cifoescoliose, esclera azul, Coxa vara
XII AR BMP1 - Moderado/
grave Alta quantidade de massa óssea, ossos wormianos
XIII AR SP7 - Moderado/
grave
Micrognatia, curvatura dos membros superiores
e inferiores
XIV AR WNT1 - Moderado/
grave Escoliose, curvatura de
ossos longos e wormiano

32
XV AR TMEM38B - Moderado Costelas finas, ossos
wormianos
XVI AR CREB3L1 - Grave Costelas frisadas,
múltiplas fraturas no período neonatal
Adaptado de Forlino e Marini 2016. AR: Autossômico recessivo; AD: Autossômico dominante; (+): presente; (-): ausente
Manifestações extra-esqueléticas são relatadas nos
estudos da OI, entre essas manifestações, a dentinogênese
imperfeita (DGI) é uma das principais, estando presente em 50%
dos pacientes que possuem OI dos tipos I a IV [13].
A DGI é um grupo de condições hereditárias,
caracterizadas por uma estrutura anormal da dentina devido a
distúrbios na formação, composição ou organização da matriz
dentinária, apresenta modo de herança autossômico dominante e
pode estar ou não associada à OI. A classificação de Shields
(1979) estratificava as condições hereditárias da dentina em DGI
tipo I, II, e III, e displasia dentinária em tipo I e II. Atualmente,
uma classificação mais utilizada propõe que a DGI seja
classificada em forma sindrômica e não-sindrômica [13,14].
A forma sindrômica de DGI está associada à OI, e pode
manifestar-se de forma leve, moderada ou grave. As formas não-
sindrômicas também se manifestam de forma leve a grave e
correspondem, respectivamente, à displasia dentinária tipo II,
DGI tipos II e III de Shields. Apesar dos tipos sindrômicos e não-
sindrômicos de DGI serem geneticamente distintos as
manifestações clínicas observadas na dentina são similares [14–
16].
As dentições decídua e permanente podem ser afetadas,
sendo a dentição decídua mais gravemente atingida. Alguns
estudos mostram que mesmo que a dentição decídua tenha sido
afetada, a permanente pode não apresentar DGI [13,14].

33
Clinicamente a DGI é caracterizada por apresentar
coloração coronária opalescente variando entre cinza, marrom e
amarelo, geralmente com atrições e desprendimento de esmalte.
Radiograficamente, as coroas podem apresentar constrição
cervical, coroas bulbosas, dismorfia da câmara pulpar
(calcificação pulpar, obliteração ou câmara pulpar ampla),
alterações na morfologia radicular (calcificação dos condutos
radiculares, obliteração dos canais ou condutos radiculares
amplos), sendo essas características consideradas
patognomônicas para o diagnóstico de DGI. Essas
características clínicas e radiográficas da DGI podem manifestar-
se de forma leve à grave [13,14].
Na maioria dos casos, a DGI é diagnosticada através de
exames clínicos e radiográficos, entretanto, a ausência de
manifestações clínicas e radiográficas não exclui a sua presença.
Nestes casos, estudos demonstraram que a DGI pode ser
diagnosticada por exames histológicos [17].
Variações no metabolismo do colágeno podem interferir
no desenvolvimento dos ossos faciais alterando o
desenvolvimento craniofacial desses pacientes, podendo
apresentar desde face triangular com testa alargada a
macrocefalia, bem como alterações no tamanho dos maxilares,
resultando em maloclusões como mordida aberta anterior e
posterior, mordida cruzada anterior e posterior, erupção ectópica
e dentes impactados [15,18].
Um estudo realizado por O'Connell e Marini (1999)
relatou que 70% de seus pacientes apresentaram maloclusão de
classe III de Angle, provavelmente uma combinação de
alterações esqueléticas com dentoalveolares. Dentre os
pacientes, 32,5% apresentaram erupção ectópica de molares e
10% agenesia dentária. Recentemente Malmgres e
colaboradores (2017) identificaram em 179 pacientes com OI

34
uma prevalência de 17% de agenesia dentária, sendo os pré-
molares os dentes mais acometidos em 91% dos casos [19].
As abordagens terapêuticas para a OI devem envolver
um tratamento multidisciplinar que possibilite uma melhor
inserção do paciente na sociedade. O ideal é combinar
tratamentos não cirúrgicos para fortalecer a musculatura;
cirúrgicos quando necessários para corrigir posicionamentos
ósseos e prevenir fraturas; e a administração farmacológica de
bisfosfonatos para aumentar a resistência mecânica dos ossos e
diminuir o número de fraturas.
O tratamento farmacológico da OI está relacionado com a
gravidade da doença. Pacientes que apresentam a forma leve,
na maioria dos casos, não necessitam realizar a infusão cíclica
intravenosa de bisfosfonatos. Entretanto, nos casos de moderado
a grave o tratamento padrão é a administração intravenosa de
pamidronato dissódico, uma droga da classe dos bisfosfonatos
introduzido na década de 90 como abordagem terapêutica para a
OI. Este fármaco promove a inibição da reabsorção óssea,
induzindo a apoptose dos osteoclastos. Esta terapia auxilia no
aumento da densidade mineral óssea, reduzindo o número de
fraturas e também melhora a força muscular, além de diminuir as
dores ósseas e aumentar a resistência desses pacientes [3,8].
A portaria 2305/GM de 19 de dezembro de 2001, do
Ministério da Saúde do Brasil (BRASIL, M. D. S. D. Portaria n°
2305/GM), em 19 de dezembro de 2001
(http://dtr2001.saude.gov.br/sas/PORTARIAS/Port2001/GM/GM-
2305.htm) designou à Fundação Universidade de Brasília/
Hospital Universitário de Brasília (FUB/HUB) como um dos
Centros Nacionais de Referência para o tratamento da OI com
administração do pamidronato dissódico. A partir de então, a
equipe médica do Setor de Endocrinologia Pediátrica da Área da
Medicina da Criança e do Adolescente da Faculdade de Medicina
da Universidade de Brasília (FM/UnB/HUB), com o suporte das

35
equipes de genética, otorrinolaringologia e odontologia do HUB e
as equipes de genética e ortopedia da Rede Sarah Kubitschek de
Hospitais, vêm acompanhando crianças e adolescentes
portadores de OI que residem no Distrito Federal e em outros
Estados da Federação referidos para este centro. Desde 2002,
todos os pacientes tratados com infusão cíclica intravenosa de
pamidronato são encaminhados para o Projeto de Extensão de
Ação Continuada "Atendimento a Pacientes Portadores de
Anomalias de Desenvolvimento Dentário", na Divisão de
Odontologia do HUB.
Apesar dos avanços nos estudos moleculares, o
diagnóstico de OI e DGI ainda é eminentemente clínico.
Entretanto, os dados moleculares são de suma importância para
permitir a classificação do tipo de OI dos pacientes com maior
precisão. O conhecimento do gene envolvido na doença pode
influenciar sobremaneira, na indicação do melhor
acompanhamento odontológico, assim como na previsibilidade
de sua evolução. Além disso, o conhecimento das bases
moleculares permite saber o padrão de herança da OI em cada
população e permite também, um melhor aconselhamento
genético à família.
O objetivo do estudo foi identificar, pelo método de
sequenciamento de nova geração, variantes patológicas em
pacientes diagnosticados com OI e caracterizar as alterações
dentárias clínicas e radiográficas dos pacientes diagnosticados
com dentinogênese imperfeita (DGI), em tratamento com
bisfosfonato e em atendimento no Clinica de Anomalias
Dentárias do Hospital Universitário de Brasília.
1.2 MATERIAIS E MÉTODOS
Este estudo foi aprovado pelo comitê de Ética de
Pesquisa em seres humanos (CEP/FS 1.324.282). O Termo de

36
Consentimento Livre e Esclarecido foi apresentado a todos os
sujeitos da pesquisa e assinado por seus responsáveis legais.
Entre os anos de 2002-2016 foram acompanhados 128
pacientes com OI no HUB. Atualmente 63 desses pacientes com
diagnóstico de osteogênese imperfeita, apresentando ou não
dentinogênese imperfeita, em tratamento com pamidronato
dissódico no Serviço de Endocrinologia Pediátrica do HUB são
acompanhados pelo Projeto de Extensão de Ação Continuada
“Atendimento a Pacientes Portadores de Anomalias de
Desenvolvimento Dentário”, na unidade de Saúde Bucal do HUB.
O acompanhamento odontológico é realizado no período
da internação desses pacientes no HUB para o tratamento com
bisfosfonato, que ocorre regularmente a cada 2, 3 ou 4 meses de
acordo com a idade. Informações referentes ao exame físico
(extra e intrabucal), exames complementares (fotografias e
radiografias panorâmicas e periapicais) foram coletadas de seus
prontuários.
Dados referentes à idade do paciente, sexo, tipo de OI, e
manifestações bucais que caracterizam a DGI como: alterações
de cor coronária, atrição, dismorfia da câmara pulpar, alterações
morfológicas radiculares e lesões periapicais foram registradas a
fim de caracterizar o fenótipo dentário daqueles pacientes com
DGI.
Durante a internação para infusão do pamidronato
dissódico amostras de sangue venoso periférico de 63 pacientes
foram coletadas e armazenadas em tubos contendo EDTA e
encaminhadas para o Laboratório de Histopatologia Bucal, onde
foi realizada a extração de DNA genômico pelo método salting
out – Método Puregene, seguida da sua quantificação no
Nanovue Plus (GE®).
Para extração de DNA, as células sanguíneas foram
submetidas à centrifugação e incubadas com dois tampões de

37
lise celular. Após a incubação, as proteínas foram removidas por
precipitação com 7.5M de acetato de amônia. O DNA foi
precipitado com isopropanol, secado ao ar livre e ressuspendido
em tampão de TE (10 mM Tris pH 7.8 e 1 mM EDTA). A
concentração de DNA de cada amostra foi determinada,
invidualmente, por espectrofotômetro (Nanovue Plus, GE
Healthcare Life Sciences, USA).
Um painel de genes por meio de sequenciamento de
nova geração (NGS) foi feito para 14 genes associados à OI até
o momento inicial da pesquisa. (Ion Personal Machine, Life
Technologies) (Tabela 2).
Tabela 2. Genes presentes no NGS, sua localização genômica.
Gene Localização genômica
COL1A1 17q21.33
COL1A2 7q21.3
CRTAP 3p22.3
LEPRE1 1p34.2
PPIB 15q22.31
FKBP10 17q21.2
SERPINF1 17p13.3
SERPINH1 11q13.5
SP7 12q13.13
IFITM5 11p12.5
BMP1 8p21.3
TMEM38B 9q31.2
WNT1 12q13.12
DSPP 4q22.1

38
Um total de 10 ng de DNA por amostra foi usado para
enriquecimento por PCR multiplex. Os amplicons foram ligados
aos adaptadores de sequenciamento com “barcodes” que
permitem misturar as amostras para as etapas subsequentes. As
bibliotecas das amostras com código de barras equalizadas
foram reunidas. O pool de bibliotecas e o sequenciamento
seguiram o protocolo do fabricante. Utilizou-se um Chip Ion
316v2 para sequenciamento das amostras. A realização deste
experimento ocorreu na Universidade Católica de Brasília.
Os dados do sequenciamento do painel foram analisados
usando o programa Torrent Suite (version 5.0.4; Life
Technologies) para as chamadas, alinhamentos e variantes
usando a sequência genômica de referência (hg19) para os
genes alvo. As chamadas de variantes foram anotadas usando
Ion Reporter (version 4.0), que mostrou alterações de acordo
com a localização, tipo de variante, dBSNPs e resultados de
programas “in silico” (Polyphen and SIFT). As variantes foram
filtradas de acordo com os critérios: baixa qualidade da corrida,
regiões homopolímeras de inserções/deleções, e desbalanço
alélico na proporção maior que 80:20. Mutações patogênicas
foram também testadas pelas análises in silico MutationTaster,
Poyphen e SIFT. As regiões sequenciadas tiveram uma
cobertura de 98%.
1.3 RESULTADOS
O sequenciamento de nova geração (NGS) foi realizado
em 33 pacientes, em virtude de ser um experimento com alto
custo e não dispormos de financiamento, no momento, para
realizar em todos os pacientes. A distribuição de acordo com
idade, tipo de OI e presença de DGI dos pacientes que
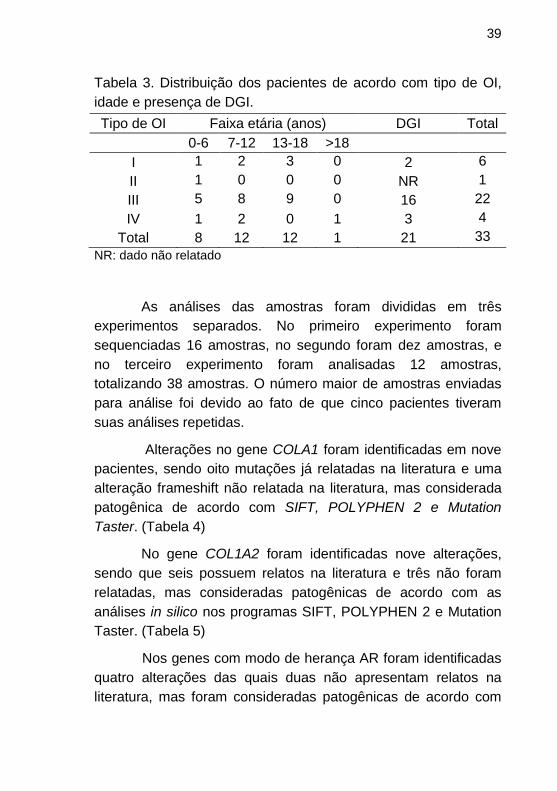
participaram do NGS está resumida na Tabela 3.
39
Tabela 3. Distribuição dos pacientes de acordo com tipo de OI,
idade e presença de DGI.
Tipo de OI Faixa etária (anos) DGI Total
0-6 7-12 13-18 >18
I 1 2 3 0 2 6
II 1 0 0 0 NR 1
III 5 8 9 0 16 22
IV 1 2 0 1 3 4
Total 8 12 12 1 21 33
NR: dado não relatado
As análises das amostras foram divididas em três
experimentos separados. No primeiro experimento foram
sequenciadas 16 amostras, no segundo foram dez amostras, e
no terceiro experimento foram analisadas 12 amostras,
totalizando 38 amostras. O número maior de amostras enviadas
para análise foi devido ao fato de que cinco pacientes tiveram
suas análises repetidas.
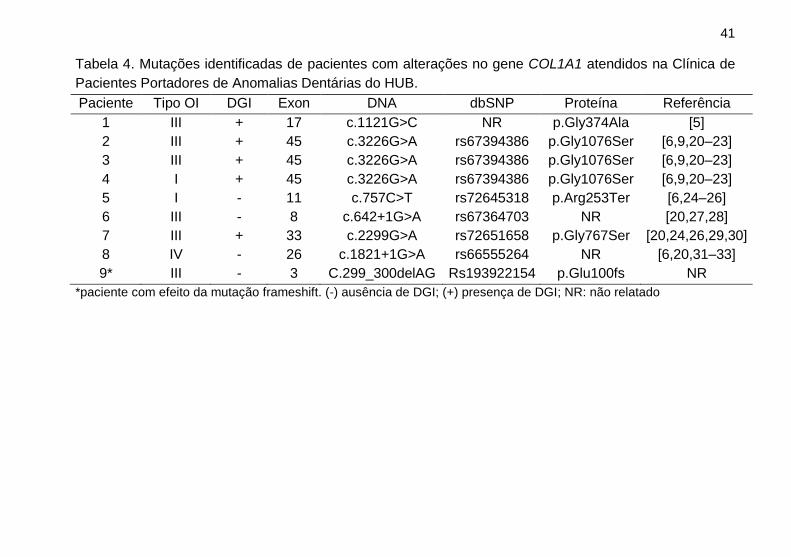
Alterações no gene COLA1 foram identificadas em nove
pacientes, sendo oito mutações já relatadas na literatura e uma
alteração frameshift não relatada na literatura, mas considerada
patogênica de acordo com SIFT, POLYPHEN 2 e Mutation
Taster. (Tabela 4)
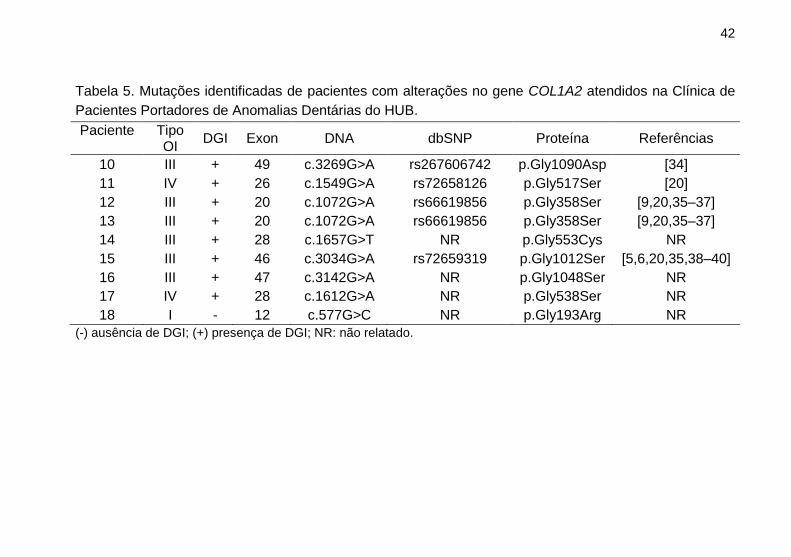
No gene COL1A2 foram identificadas nove alterações,
sendo que seis possuem relatos na literatura e três não foram
relatadas, mas consideradas patogênicas de acordo com as
análises in silico nos programas SIFT, POLYPHEN 2 e Mutation
Taster. (Tabela 5)
Nos genes com modo de herança AR foram identificadas
quatro alterações das quais duas não apresentam relatos na
literatura, mas foram consideradas patogênicas de acordo com

40
as análises in silico nos programas SIFT, POLYPHEN 2 e
Mutation Taster. (Tabela 6)
Em onze pacientes não foram encontradas mutações
patogênicas envolvidas com OI, ou por falha técnica ou por
limitações do painel de genes. Desses, em sete pacientes não foi
possível identificar a mutação causadora da doença por falha da
técnica, uma vez que as mesmas impediram a análise dos dados
por apresentarem baixa cobertura, e em quatro pacientes não
foram encontradas alterações nos genes estudados.
Do total de 33 pacientes testados, 21 apresentaram
características clínicas e radiográficas de DGI, e uma paciente
que foi a óbito aos três meses de idade não foi possível avaliar a
presença de DGI. As alterações dentinárias clínicas e
radiográficas da dentição decídua e permanente dos pacientes
com OI/DGI que participaram do estudo molecular estão
resumida na Tabela 7A e 7B respectivamente.
41
Tabela 4. Mutações identificadas de pacientes com alterações no gene COL1A1 atendidos na Clínica de
Pacientes Portadores de Anomalias Dentárias do HUB.
Paciente Tipo OI DGI Exon DNA dbSNP Proteína Referência
1 III + 17 c.1121G>C NR p.Gly374Ala [5]
2 III + 45 c.3226G>A rs67394386 p.Gly1076Ser [6,9,20–23]
3 III + 45 c.3226G>A rs67394386 p.Gly1076Ser [6,9,20–23]
4 I + 45 c.3226G>A rs67394386 p.Gly1076Ser [6,9,20–23]
5 I - 11 c.757C>T rs72645318 p.Arg253Ter [6,24–26]
6 III - 8 c.642+1G>A rs67364703 NR [20,27,28]
7 III + 33 c.2299G>A rs72651658 p.Gly767Ser [20,24,26,29,30]
8 IV - 26 c.1821+1G>A rs66555264 NR [6,20,31–33]
9* III - 3 C.299_300delAG Rs193922154 p.Glu100fs NR
*paciente com efeito da mutação frameshift. (-) ausência de DGI; (+) presença de DGI; NR: não relatado
42
Tabela 5. Mutações identificadas de pacientes com alterações no gene COL1A2 atendidos na Clínica de
Pacientes Portadores de Anomalias Dentárias do HUB.
Paciente
Tipo OI
DGI Exon DNA dbSNP Proteína Referências
10 III + 49 c.3269G>A rs267606742 p.Gly1090Asp [34]
11 IV + 26 c.1549G>A rs72658126 p.Gly517Ser [20]
12 III + 20 c.1072G>A rs66619856 p.Gly358Ser [9,20,35–37]
13 III + 20 c.1072G>A rs66619856 p.Gly358Ser [9,20,35–37]
14 III + 28 c.1657G>T NR p.Gly553Cys NR
15 III + 46 c.3034G>A rs72659319 p.Gly1012Ser [5,6,20,35,38–40]
16 III + 47 c.3142G>A NR p.Gly1048Ser NR
17 IV + 28 c.1612G>A NR p.Gly538Ser NR
18 I - 12 c.577G>C NR p.Gly193Arg NR
(-) ausência de DGI; (+) presença de DGI; NR: não relatado.
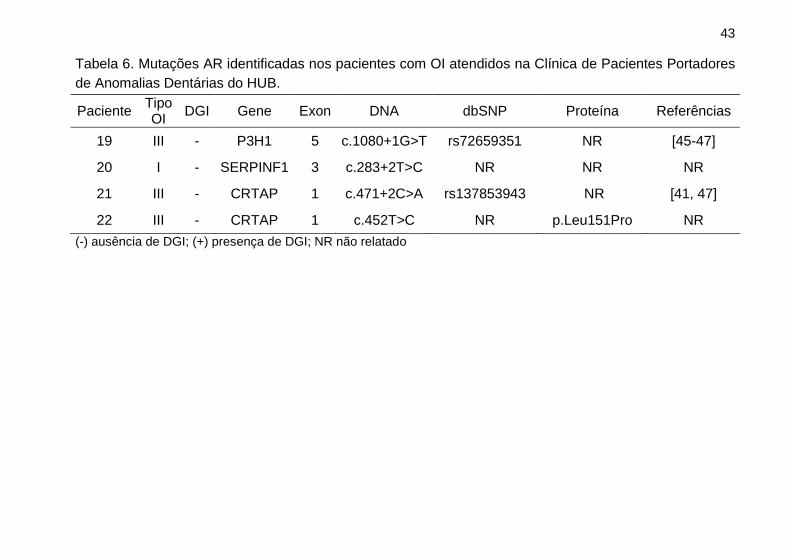
43
Tabela 6. Mutações AR identificadas nos pacientes com OI atendidos na Clínica de Pacientes Portadores
de Anomalias Dentárias do HUB.
Paciente Tipo OI
DGI Gene Exon DNA dbSNP Proteína Referências
19 III - P3H1 5 c.1080+1G>T rs72659351 NR [45-47]
20 I - SERPINF1 3 c.283+2T>C NR NR NR
21 III - CRTAP 1 c.471+2C>A rs137853943 NR [41, 47]
22 III - CRTAP 1 c.452T>C NR p.Leu151Pro NR
(-) ausência de DGI; (+) presença de DGI; NR não relatado
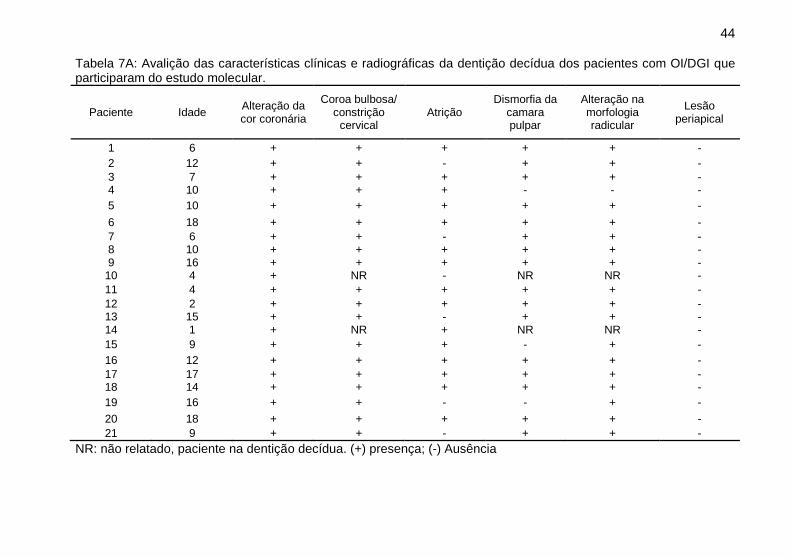
44
Tabela 7A: Avalição das características clínicas e radiográficas da dentição decídua dos pacientes com OI/DGI que participaram do estudo molecular.
Paciente Idade Alteração da cor coronária
Coroa bulbosa/ constrição
cervical Atrição
Dismorfia da camara pulpar
Alteração na morfologia radicular
Lesão periapical
1 6 + + + + + -
2 12 + + - + + -
3 7 + + + + + - 4 10 + + + - - -
5 10 + + + + + -
6 18 + + + + + -
7 6 + + - + + - 8 10 + + + + + - 9 16 + + + + + -
10 4 + NR - NR NR -
11 4 + + + + + -
12 2 + + + + + - 13 15 + + - + + - 14 1 + NR + NR NR -
15 9 + + + - + -
16 12 + + + + + -
17 17 + + + + + - 18 14 + + + + + -
19 16 + + - - + -
20 18 + + + + + -
21 9 + + - + + -
NR: não relatado, paciente na dentição decídua. (+) presença; (-) Ausência
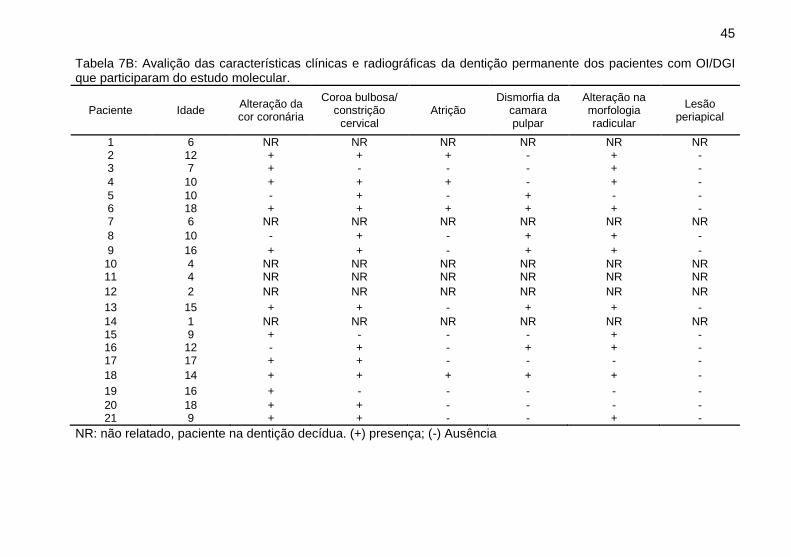
45
Tabela 7B: Avalição das características clínicas e radiográficas da dentição permanente dos pacientes com OI/DGI que participaram do estudo molecular.
Paciente Idade Alteração da cor coronária
Coroa bulbosa/ constrição
cervical Atrição
Dismorfia da camara pulpar
Alteração na morfologia radicular
Lesão periapical
1 6 NR NR NR NR NR NR 2 12 + + + - + - 3 7 + - - - + -
4 10 + + + - + -
5 10 - + - + - - 6 18 + + + + + - 7 6 NR NR NR NR NR NR
8 10 - + - + + -
9 16 + + - + + -
10 4 NR NR NR NR NR NR 11 4 NR NR NR NR NR NR
12 2 NR NR NR NR NR NR
13 15 + + - + + -
14 1 NR NR NR NR NR NR 15 9 + - - - + - 16 12 - + - + + - 17 17 + + - - - -
18 14 + + + + + -
19 16 + - - - - -
20 18 + + - - - - 21 9 + + - - + -
NR: não relatado, paciente na dentição decídua. (+) presença; (-) Ausência

46
1.4 DISCUSSÃO
No presente trabalho foram analisados, por meio do
sequenciamento de nova geração, amostras de 33 pacientes
diagnosticados com OI em acompanhamento no Projeto de
Extensão de Ação Continuada Atendimento a Pacientes
Portadores de Anomalias de Desenvolvimento Dentário, na
divisão de Odontologia do HUB entre os anos de 2002-2016.
No HUB, todos os pacientes com OI em tratamento com
bisfosfonato, são encaminhados do serviço de endocrinologia
pediátrica para acompanhamento na clínica de odontologia. O
acompanhamento odontológico acontece durante o período de
internação para administração do bisfosfonato, que geralmente
ocorre a cada dois, três ou quadro meses de acordo com a idade
dos paciente. Os atendimentos foram realizados em função da
necessidade desses pacientes, os procedimentos mais
realizados foram: profilaxias, orientações sobre a dieta e higiene
oral, adequação do meio bucal e restaurações.
A amostra desse estudo é principalmente de pacientes
com OI do tipo grave ou moderado (tipo III ou IV), isso ocorre por
serem os tipos que mais necessitam de tratamento com o
bisfosfonato.
Nesse estudo, 21 dos pacientes possuem DGI,
entretanto, alguns autores sugerem que a falta de alterações
clínicas e radiográficas da DGI não determina a sua ausência,
pois exames histológicos podem diagnosticar a presença da DGI.
[17]. Para esse estudo, a avaliação foi exclusivamente clínica e
radiográfica e revelou que os pacientes com DGI possuíam
alterações do tipo moderada, com coloração coronária
opalescente, coroas bulbosas e curtas, atrição com

47
desprendimento de esmalte, raízes mais curtas e finas com
obliteração parcial ou total da polpa. Não foi observada presença
de lesões periapicais descritas anteriormente nos casos de DGI
do tipo grave [16]. Os resultados revelaram também que a
dentição decídua dos pacientes foi mais gravemente afetada em
relação à dentição permanente, os dentes decíduos
apresentaram características clínicas e radiográficas com
alterações de moderada a grave, enquanto que os dentes
permanentes apresentaram alterações de leve a grave,
concordando com o descrito na literatura [14–16]. Os casos leves
apresentaram uma discreta alteração na coloração dos dentes,
pouca ou nenhuma atrição e radiograficamente apresentaram
alterações leves ou ausentes.
Numerosos estudos tem demonstrado que diversos
genes estão envolvidos na patogênese da OI. Até o momento,
variantes patogênicas em COL1A1, COL1A2, CRTAP, LEPRE1,
PPIB, FKBP10, SERPINF1, SERPINH1, SP7, IFITM5, BMP1,
TMEM38B e WNT1 tem sido relatados na literatura. Mutações
nos genes COL1A1 e COL1A2 estão associadas a 85-90% dos
casos de OI, sendo estas de herança autossômica dominante
[10]. Variantes dominantes em IFITM5 também têm sido
descritas em alguns indivíduos afetados [1,9,10]. Outro grupo de
genes que não estão implicados diretamente na biossíntese de
colágeno tipo 1, no entanto, são genes importantes nas vias de
mineralização ou desenvolvimento de osteoblastos, como
CREB3L1 e SPARC foram recentemente associados à OI [12,
41]. Mutações em PLS3 e MBTPS2 têm sido associadas a
formas distantes de herança ligada ao X [12,41]. Um estudo
recente ainda identificou mais três mutações relacionadas à OI,
sendo elas nos genes SCN9A, NTRK1 e SLC2A2 [2]. Essas
novas variações foram relatadas na literatura após a elaboração
do painel de genes utilizado nesse estudo.

48
O colágeno tipo 1 consiste em duas cadeias α1 e uma
cadeia α2. Após a tradução, as cadeias pro-α1 e as cadeias pro-
α2 são processadas no retículo endoplasmático rugoso. Estas
cadeias têm de se alinhar para iniciar o processo de dobragem
do pro-colágeno tipo I numa tripla hélice, seguindo do
alinhamento das três cadeias para iniciar a conformação em uma
estrutura helicoidal tripla. Cada cadeia α contém pro-peptídeos
N- (amino) e C- (carboxi) terminais e um domínio central
constituído por 338 repetições. A glicina, como o menor
aminoácido, é o único resíduo que pode ocupar a posição axial
da tripla hélice, de modo que qualquer alteração num resíduo de
glicina resultará na ruptura da estrutura helicoidal do
colágeno. Mutações nos genes COL1A1 e COL1A2 alteram a
estrutura ou a quantidade de colágeno tipo 1, resultando em um
fenótipo esquelético que varia de leve a letal. Quando há uma
substituição da glicina na cadeia α1, o fenótipo dependerá da
posição da substituição: as substituições C-terminais resultam
em fenótipo de doença grave, e as substituições N-terminais
produzem fenótipos mais leves [1,3,42].
As mutações em CO1A1 e COLIA2 encontradas nesse
estudo são quase todas, substituição do aminoácido glicina por
outro, sendo a serina a mais frequente (Tabela 4 e 5). Segundo
Marini (2007) a substituição do aminoácido glicina por outro
aminoácido geralmente causam OI grave ou moderada
resultando em efeito qualitativo. Os resultados desse estudo
estão de acordo com o relatado na literatura, 10 pacientes
apresentaram substituição de glicina por serina, sendo pacientes
portadores dos tipos mais graves de OI (tipo III e IV).
Recentemente, dois estudos foram publicados relatando
os genes SERPINF1, seguido do CRTAP como os genes mais
frequentemente alterados em OI com modo de herança AR.
Nesse estudo foram encontradas mutações AR nos genes
CRTAP (exon1 c.452T>C; exon1 c.471+2C>A) e SERPINF1

49
(exon3 c. 283+2T>C) em três pacientes com histórico de
consanguinidade (Tabela 6), corroborando com os achados da
literatura [2,43]. Inicialmente esses casos foram classificados em
tipo I e III de acordo Sillence. O NGS permitiu redefinir o
diagnóstico para esses pacientes de acordo com Forlino e Marini
(2016) em tipo VII para as alterações em CRTAP e tipo VI para a
variação em SERPINF1.
Devido ao experimento ter um alto custo, não foi possível
analisar os 63 pacientes, os outros 30 pacientes serão
analisados no âmbito do mestrado. Uma vez que a cobertura do
painel de genes para sequenciamento de nova geração possuiu
uma cobertura de 98%, os pacientes que ficaram sem
diagnóstico molecular podem ter alguma alteração nesta região
que não foi contemplada pela técnica utilizada. Uma estratégia
para resolver esta questão seria sequenciar por Sanger apenas
as regiões não cobertas (2%), e verificar a presença ou não de
mutação de ponto. Outra hipótese seria de que esses pacientes
apresentam mutações em outros genes não investigados nesse
estudo e associados recentemente na literatura a mutações
potencialmente causadoras da OI.
1.5 CONCLUSÃO
O presente estudo mostrou que a maioria dos pacientes
atendidos na Clínica de Portadores de Anomalias do
desenvolvimento Dentário entre os anos de 2002-2016 possuíam
OI do tipo III. Por meio do sequenciamento de nova geração foi
possível identificar vinte e duas mutações patológicas em trinta e
três pacientes diagnosticados com OI, sendo que para seis
mutações não foram encontrados relatos na literatura sugerindo
serem mutações novas, entretanto, as outras mutações
encontradas estão de acordo com o relatado na literatura. Foi
possível também caracterizar as alterações clínicas e

50
radiográficas dos pacientes que participaram do NGS mostrando
que 21 pacientes possuíam manifestações clínicas e
radiográficas de DGI e em um paciente não foi possível
identificar clinicamente devido ao seu óbito precoce. A
caracterização revelou também que a amostra estudada é
predominantemente de DGI do tipo moderada, sendo a dentição
decídua mais gravemente afetada em relação a dentição
permanente. A importância de descobrir essas variantes
possibilita tornar o diagnóstico molecular um método mais
sensível e preciso para esses pacientes. Será necessário
investigar as regiões não sequenciadas no painel pelo método
Sanger assim como serão relacionados os sequenciamentos dos
pacientes sem diagnóstico molecular até o presente.

51
REFERÊNCIAS BIBLIOGRÁFICAS
[1] A. Forlino, J.C. Marini, Osteogenesis imperfecta, Lancet. 387 (2016) 1657–1671. doi:10.1016/S0140-6736(15)00728-X.
[2] J.A. Caparros-Martin, M.S. Aglan, S. Temtamy, G.A. Otaify, M. Valencia, J. Nevado, E. Vallespin, A. Del Pozo, C. Prior de Castro, L. Calatrava-Ferreras, P. Gutierrez, A.M. Bueno, B. Sagastizabal, E. Guillen-Navarro, M. Ballesta-Martinez, V. Gonzalez, S.Y. Basaran, R. Buyukoglan, B. Sarikepe, C. Espinoza-Valdez, F. Cammarata-Scalisi, V. Martinez-Glez, K.E. Heath, P. Lapunzina, V.L. Ruiz-Perez, Molecular spectrum and differential diagnosis in patients referred with sporadic or autosomal recessive osteogenesis imperfecta, Mol. Genet. Genomic Med. 5 (2017) 28–39. doi:10.1002/mgg3.257.
[3] J.C. Marini, A. Reich, S.M. Smith, Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation., Curr. Opin. Pediatr. 26 (2014) 500–7. doi:10.1097/MOP.0000000000000117.
[4] D.O. Sillence, A. Senn, D.M. Danks, Genetic heterogeneity in osteogenesis imperfecta., J. Med. Genet. 16 (1979) 101–16. doi:10.1136/jmg.16.2.101.
[5] J. Stephen, K.M. Girisha, A. Dalal, A. Shukla, H. Shah, P. Srivastava, U. Kornak, S.R. Phadke, Mutations in patients with osteogenesis imperfecta from consanguineous Indian families, Eur. J. Med. Genet. 58 (2015) 21–27. doi:10.1016/j.ejmg.2014.10.001.
[6] K. Lindahl, E. Åström, C.-J. Rubin, G. Grigelioniene, B. Malmgren, Ö. Ljunggren, A. Kindmark, Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta., Eur. J. Hum. Genet. 23 (2015) 1042–50. doi:10.1038/ejhg.2015.81.
[7] F. Rauch, F.H. Glorieux, Osteogenesis imperfecta., Lancet (London, England). 363 (2004) 1377–85. doi:10.1016/S0140-6736(04)16051-0.

52
[8] F.S. Van Dijk, J.M. Cobben, A. Kariminejad, A. Maugeri, P.G.J. Nikkels, R.R. Van Rijn, G. Pals, Osteogenesis imperfecta: A review with clinical examples, Mol. Syndromol. 2 (2011) 1–20. doi:10.1159/000332228.
[9] F. Rauch, L. Lalic, F.H. Glorieux, P. Moffatt, P. Roughley, Targeted sequencing of a pediatric metabolic bone gene panel using a desktop semiconductor next-generation sequencer., Calcif. Tissue Int. 95 (2014) 323–31. doi:10.1007/s00223-014-9897-9.
[10] H. Kang, S. Aryal A.C., J.C. Marini, Osteogenesis imperfecta: new genes reveal novel mechanisms in bone dysplasia, Transl. Res. (2016). doi:10.1016/j.trsl.2016.11.005.
[11] J.C. Marini, A.R. Blissett, New Genes in Bone Development: What’s New in Osteogenesis Imperfecta, J. Clin. Endocrinol. Metab. 98 (2013) 3095–3103. doi:10.1210/jc.2013-1505.
[12] U. Lindert, W.A. Cabral, S. Ausavarat, S. Tongkobpetch, K. Ludin, A.M. Barnes, P. Yeetong, M. Weis, B. Krabichler, C. Srichomthong, E.N. Makareeva, A.R. Janecke, S. Leikin, B. Röthlisberger, M. Rohrbach, I. Kennerknecht, D.R. Eyre, K. Suphapeetiporn, C. Giunta, J.C. Marini, V. Shotelersuk, MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta., Nat. Commun. 7 (2016) 11920. doi:10.1038/ncomms11920.
[13] M. Chetty, T. Roberts, L.X.G. Stephen, P. Beighton, Hereditary dentine dysplasias: terminology in the context of osteogenesis imperfecta, Bdj. 221 (2016) 727–730. doi:10.1038/sj.bdj.2016.915.
[14] M. de La Dure-Molla, B. Philippe Fournier, A. Berdal, Isolated dentinogenesis imperfecta and dentin dysplasia: revision of the classification., Eur. J. Hum. Genet. 23 (2014) 15–21. doi:10.1038/ejhg.2014.159.
[15] a C. O’Connell, J.C. Marini, A.C.O. Connell, B. Sc, J.C. Marini, Evaluation of oral problems in an osteogenesis imperfecta population., Oral Surg. Oral Med. Oral Pathol. Oral

53
Radiol. Endod. 87 (1999) 189–196. doi:10.1016/S1079-2104(99)70272-6.
[16] E.D. Shields, D. Bixler, A.M. El-Kafrawy, A proposed classification for heritable human dentine defects with a description of a new entity, Arch. Oral Biol. 18 (1973) 543–553. doi:10.1016/0003-9969(73)90075-7.
[17] J. Waltimo, A. Ojanotko-Harri, P.L. Lukinmaa, Mild forms of dentinogenesis imperfecta in association with osteogenesis imperfecta as characterized by light and transmission electron microscopy, J. Oral Pathol. Med. 25 (1996) 256–264. http://dx.doi.org/10.1111/j.1600-0714.1996.tb01381.x.
[18] J. Rizkallah, S. Schwartz, F. Rauch, F. Glorieux, D.D. Vu, K. Muller, J.M. Retrouvey, Evaluation of the severity of malocclusions in children affected by osteogenesis imperfecta with the peer assessment rating and discrepancy indexes, Am. J. Orthod. Dentofac. Orthop. 143 (2013) 336–341. doi:10.1016/j.ajodo.2012.10.016.
[19] B. Malmgren, K. Andersson, K. Lindahl, A. Kindmark, G. Grigelioniene, V. Zachariadis, G. Dahllöf, E. Åström, Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes, Oral Dis. 23 (2017) 42–49. doi:10.1111/odi.12568.
[20] J.C. Marini, W.A. Cabral, A.M. Barnes, W. Chang, Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development, Cell Cycle. 6 (2007) 1675–1681. doi:10.4161/cc.6.14.4474.
[21] Z.L. Zhang, H. Zhang, Y.H. Ke, H. Yue, W.J. Xiao, J.B. Yu, J.M. Gu, W.W. Hu, C. Wang, J.W. He, W.Z. Fu, The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta, J. Bone Miner. Metab. 30 (2012) 69–77. doi:10.1007/s00774-011-0284-6.
[22] E. Barkova, U. Mohan, D. Chitayat, S. Keating, A. Toi, J. Frank, R. Frank, G. Tomlinson, P. Glanc, Fetal skeletal

54
dysplasias in a tertiary care center: Radiology, pathology, and molecular analysis of 112 cases, Clin. Genet. 87 (2015) 330–337. doi:10.1111/cge.12434.
[23] a M. Lund, a C. Nicholls, M. Schwartz, F. Skovby, Parental mosaicism and autosomal dominant mutations causing structural abnormalities of collagen I are frequent in families with osteogenesis imperfecta type III/IV., Acta Paediatr. 86 (1997) 711–8. doi:10.1111/j.1651-2227.1997.tb08573.x.
[24] L. Ries-Levavi, T. Ish-Shalom, M. Frydman, D. Lev, S. Cohen, G. Barkai, B. Goldman, P. Byers, E. Friedman, Genetic and biochemical analyses of Israeli osteogenesis imperfecta patients., Hum. Mutat. 23 (2004) 399–400. doi:10.1002/humu.9230.
[25] G. Venturi, E. Tedeschi, M. Mottes, M. Valli, M. Camilot, S. Viglio, F. Antoniazzi, L. Tatò, Osteogenesis imperfecta: clinical, biochemical and molecular findings., Clin. Genet. 70 (2006) 131–139. doi:10.1111/j.1399-0004.2006.00646.x.
[26] L. Mauri, S. Uebe, H. Sticht, U. Vossmerbaeumer, N. Weisschuh, E. Manfredini, E. Maselli, M. Patrosso, R.N. Weinreb, S. Penco, A. Reis, F. Pasutto, Expanding the clinical spectrum of COL1A1 mutations in different forms of glaucoma, Orphanet J. Rare Dis. 11 (2016) 108. doi:10.1186/s13023-016-0495-y.
[27] U. Schwarze, B.J. Starman, P.H. Byers, Redefinition of exon 7 in the COL1A1 gene of type I collagen by an intron 8 splice-donor-site mutation in a form of osteogenesis imperfecta: influence of intron splice order on outcome of splice-site mutation., Am. J. Hum. Genet. 65 (1999) 336–344. doi:10.1086/302512.
[28] H.-Y. Lin, C.-K. Chuang, Y.-N. Su, M.-R. Chen, H.-C. Chiu, D.-M. Niu, S.-P. Lin, Genotype and phenotype analysis of Taiwanese patients with osteogenesis imperfecta., Orphanet J. Rare Dis. 10 (2015) 152. doi:10.1186/s13023-015-0370-2.
[29] A. Forlino, F. Zolezzi, M. Valli, P.F. Pignatti, G. Cetta, P.C.

55
Brunelli, M. Mottes, Severe (type III) osteogenesis imperfecta due to glycine substitutions in the central domain of the collagen triple helix, Hum. Mol. Genet. 3 (1994) 2201–2206. doi:10.1093/hmg/3.12.2201.
[30] H. Zhang, R. Yang, Y. Wang, J. Ye, L. Han, W. Qiu, X. Gu, A pilot study of gene testing of genetic bone dysplasia using targeted next-generation sequencing., J. Hum. Genet. 60 (2015) 769–76. doi:10.1038/jhg.2015.112.
[31] M.L. Stover, D. Primorac, S.C. Liu, M.B. McKinstry, D.W. Rowe, Defective splicing of mRNA from one COL1A1 allele of type I collagen in nondeforming (type I) osteogenesis imperfecta, J. Clin. Invest. 92 (1993) 1994–2002. doi:10.1172/JCI116794.
[32] J. Körkkö, H. Kuivaniemi, P. Paassilta, J. Zhuang, G. Tromp, A. DePaepe, D.J. Prockop, L. Ala-Kokko, Two new recurrent nucleotide mutations in the COL1A1 gene in four patients with osteogenesis imperfecta: About one-fifth are recurrent, Hum. Mutat. 9 (1997) 148–156. doi:10.1002/(SICI)1098-1004(1997)9:2<148::AID-HUMU7>3.0.CO;2-5.
[33] C. Johnson, D. Primorac, M. McKinstry, J. McNeil, D. Rowe, J.B. Lawrence, Tracking Col1a1 RNA in Osteogenesis Imperfecta, J. Cell Biol. 150 (2000) 417–431.
[34] E. Faqeih, P. Roughley, F.H. Glorieux, F. Rauch, Osteogenesis Imperfecta Type III With Intracranial Hemorrhage and Brachydactyly Associated With Mutations in Exon 49 of COL1A2, (2009) 461–465. doi:10.1002/ajmg.a.32653.
[35] K.-S. Lee, H.-R. Song, T.-J. Cho, H.J. Kim, T.-M. Lee, H.-S. Jin, H.-Y. Park, S. Kang, S.-C. Jung, S.K. Koo, Mutational spectrum of type I collagen genes in Korean patients with osteogenesis imperfecta, Hum. Mutat. 27 (2006) 599–599. doi:10.1002/humu.9423.
[36] W. Kishta, F.H. Abduljabbar, M. Gdalevitch, F. Rauch, R. Hamdy, F. Fassier, Hip Dysplasia in Children With

56
Osteogenesis Imperfecta: Association With Collagen Type I C-Propeptide Mutations, J. Pediatr. Orthop. 0 (2015) 1–5. doi:10.1097/BPO.0000000000000644.
[37] A. Taillandier, C. Domingues, C. De Cazanove, V. Porquet-Bordes, S. Monnot, T. Kiffer-Moreira, A. Rothenbuhler, P. Guggenbuhl, C. Cormier, G. Baujat, F. Debiais, Y. Capri, M. Cohen-Solal, P. Parent, J. Chiesa, A. Dieux, F. Petit, J. Roume, M. Isnard, V. Cormier-Daire, A. Linglart, J.L. Mill??n, J.P. Salles, C. Muti, B. Simon-Bouy, E. Mornet, Molecular diagnosis of hypophosphatasia and differential diagnosis by targeted Next Generation Sequencing, Mol. Genet. Metab. 116 (2015) 215–220. doi:10.1016/j.ymgme.2015.09.010.
[38] R. Sztrolovics, F.H. Glorieux, M. Van Der Rest, P.J. Roughley, Identification of type I collagen gene (COL1A2) mutations in nonlethal osteogenesis imperfecta, Hum. Mol. Genet. 2 (1993) 1319–1321. doi:10.1093/hmg/2.8.1319.
[39] a Forlino, E. D’amato, M. Valli, G. Camera, E. Hopkins, J.C. Marini, G. Cetta, D. a Coviello, Phenotypic comparison of an osteogenesis imperfecta type IV proband with a de novo alpha2(I) Gly922 --> Ser substitution in type I collagen and an unrelated patient with an identical mutation., Biochem. Mol. Med. 62 (1997) 26–35. doi:10.1006/bmme.1997.2620.
[40] H. Hartikka, K. Kuurila, J. Körkkö, I. Kaitila, R. Grénman, S. Pynnönen, J.C. Hyland, L. Ala-Kokko, Lack of correlation between the type of COL1A1 or COL1A2 mutation and hearing loss in osteogenesis imperfecta patients, Hum. Mutat. 24 (2004) 147–154. doi:10.1002/humu.20071.
[41] F.S. van Dijk, P.H. Byers, R. Dalgleish, F. Malfait, A. Maugeri, M. Rohrbach, S. Symoens, E.A. Sistermans, G. Pals, EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta., Eur. J. Hum. Genet. 20 (2012) 11–9. doi:10.1038/ejhg.2011.141.
[42] E.R. Valadares, T.B. Carneiro, P.M. Santos, A.C. Oliveira, B. Zabel, What is new in genetics and osteogenesis imperfecta classification?, J. Pediatr. (Rio. J). 90 (2014) 536–541.

57
doi:10.1016/j.jped.2014.05.003.
[43] G. Bardai, P. Moffatt, F.H. Glorieux, F. Rauch, DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum, Osteoporos. Int. 27 (2016) 3607–3613. doi:10.1007/s00198-016-3709-1.
58
ANEXOS
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO ANEXOS

59
NORMAS DA REVISTA
BONE - GUIDE FOR AUTHORS
Your Paper Your Way We now differentiate between the
requirements for new and revised submissions. You may choose
to submit your manuscript as a single Word or PDF file to be used
in the refereeing process. Only when your paper is at the revision
stage, will you be requested to put your paper in to a 'correct
format' for acceptance and provide the items required for the
publication of your article. To find out more, please visit the
Preparation section below. Types of article Types of articles Bone
accepts include:
1) Memoriam
2) Editorial or Commentary
3) Review
4) Original Articles
5) Rapid Communication
6) Case Report
7) Technical Note
8) Letters and Response to Letter to the Editor
9) Erratum/Corrigendum
10) Announcements

60
There are no length or format requirements other than
those already shown on the GFA under Article Structure.
Submission checklist
You can use this list to carry out a final check of your
submission before you send it to the journal for review. Please
check the relevant section in this Guide for Authors for more
details.
Ensure that the following items are present: One author has been
designated as the corresponding author with contact details:
• E-mail address
• Full postal address
All necessary files have been uploaded: Manuscript:
• Include keywords
• All figures (include relevant captions)
• All tables (including titles, description, footnotes)
• Ensure all figure and table citations in the text match the files
provided
• Indicate clearly if color should be used for any figures in print
Graphical Abstracts / Highlights files (where applicable)
Supplemental files (where applicable) Further considerations
• Manuscript has been 'spell checked' and 'grammar checked'

61
• All references mentioned in the Reference List are cited in the
text, and vice versa
• Permission has been obtained for use of copyrighted material
from other sources (including the Internet)
• Relevant declarations of interest have been made
• Journal policies detailed in this guide have been reviewed
• Referee suggestions and contact details provided, based on
journal requirements
For further information, visit our Support Center.
BEFORE YOU BEGIN
Ethics in publishing
Please see our information pages on Ethics in publishing
and Ethical guidelines for journal publication.
Human and animal rights
If the work involves the use of human subjects, the author
should ensure that the work described has been carried out in
accordance with The Code of Ethics of the World Medical
Association (Declaration of Helsinki) for experiments involving
humans; Uniform Requirements for manuscripts submitted to
Biomedical journals. Authors should include a statement in the
manuscript that informed consent was obtained for
experimentation with human subjects. The privacy rights of
human subjects must always be observed.

62
All animal experiments should comply with the ARRIVE
guidelines and should be carried out in accordance with the U.K.
Animals (Scientific Procedures) Act, 1986 and associated
guidelines, EU Directive 2010/63/EU for animal experiments, or
the National Institutes of Health guide for the care and use of
Laboratory animals (NIH Publications No. 8023, revised 1978)
and the authors should clearly indicate in the manuscript that
such guidelines have been followed.
Declaration of interest
All authors must disclose any financial and personal
relationships with other people or organizations that could
inappropriately influence (bias) their work. Examples of potential
conflicts of interest include employment, consultancies, stock
ownership, honoraria, paid expert testimony, patent applications/
registrations, and grants or other funding. If there are no conflicts
of interest then please state this: 'Conflicts of interest: none'.
More information.
Submission declaration and verification
Submission of an article implies that the work described
has not been published previously (except in the form of an
abstract or as part of a published lecture or academic thesis or as
an electronic preprint, see 'Multiple, redundant or concurrent
publication' section of our ethics policy for more information), that
it is not under consideration for publication elsewhere, that its
publication is approved by all authors and tacitly or explicitly by
the responsible authorities where the work was carried out, and
that, if accepted, it will not be published elsewhere in the same
form, in English or in any other language, including electronically
without the written consent of the copyright-holder. To verify

63
originality, your article may be checked by the originality detection
service CrossCheck.
Authorship
All authors should have made substantial contributions to
all of the following: (1) the conception and design of the study, or
acquisition of data, or analysis and interpretation of data, (2)
drafting the article or revising it critically for important intellectual
content, (3) final approval of the version to be submitted.
Changes to authorship
Authors are expected to consider carefully the list and
order of authors before submitting their manuscript and provide
the definitive list of authors at the time of the original submission.
Any addition, deletion or rearrangement of author names in the
authorship list should be made only before the manuscript has
been accepted and only if approved by the journal Editor. To
request such a change, the Editor must receive the following from
the corresponding author: (a) the reason for the change in author
list and (b) written confirmation (e-mail, letter) from all authors
that they agree with the addition, removal or rearrangement. In
the case of addition or removal of authors, this includes
confirmation from the author being added or removed. Only in
exceptional circumstances will the Editor consider the addition,
deletion or rearrangement of authors after the manuscript has
been accepted. While the Editor considers the request,
publication of the manuscript will be suspended. If the manuscript
has already been published in an online issue, any requests
approved by the Editor will result in a corrigendum.
Article transfer service

64
This journal is part of our Article Transfer Service. This
means that if the Editor feels your article is more suitable in one
of our other participating journals, then you may be asked to
consider transferring the article to one of those. If you agree, your
article will be transferred automatically on your behalf with no
need to reformat. Please note that your article will be reviewed
again by the new journal. More information.
Copyright
Upon acceptance of an article, authors will be asked to
complete a 'Journal Publishing Agreement' (see more information
on this). An e-mail will be sent to the corresponding author
confirming receipt of the manuscript together with a 'Journal
Publishing Agreement' form or a link to the online version of this
agreement. Subscribers may reproduce tables of contents or
prepare lists of articles including abstracts for internal circulation
within their institutions. Permission of the Publisher is required for
resale or distribution outside the institution and for all other
derivative works, including compilations and translations. If
excerpts from other copyrighted works are included, the author(s)
must obtain written permission from the copyright owners and
credit the source(s) in the article. Elsevier has preprinted forms
for use by authors in these cases.
For open access articles: Upon acceptance of an article, authors
will be asked to complete an 'Exclusive License Agreement'
(more information). Permitted third party reuse of open access
articles is determined by the author's choice of user license.
Author rights
As an author you (or your employer or institution) have
certain rights to reuse your work. More information. Elsevier

65
supports responsible sharing Find out how you can share your
research published in Elsevier journals.
Role of the funding source
You are requested to identify who provided financial
support for the conduct of the research and/or preparation of the
article and to briefly describe the role of the sponsor(s), if any, in
study design; in the collection, analysis and interpretation of data;
in the writing of the report; and in the decision to submit the article
for publication. If the funding source(s) had no such involvement
then this should be stated.
Funding body agreements and policies
Elsevier has established a number of agreements with
funding bodies which allow authors to comply with their funder's
open access policies. Some funding bodies will reimburse the
author for the
Open Access
Publication Fee. Details of existing agreements are
available online.
Open access
This journal offers authors a choice in publishing their
research:
Open access
• Articles are freely available to both subscribers and the wider
public with permitted reuse.

66
• An open access publication fee is payable by authors or on their
behalf, e.g. by their research funder or institution.
Subscription
• Articles are made available to subscribers as well as developing
countries and patient groups through our universal access
programs.
• No open access publication fee payable by authors.
Regardless of how you choose to publish your article, the
journal will apply the same peer review criteria and acceptance
standards.
For open access articles, permitted third party (re)use is
defined by the following Creative Commons user licenses:
Creative Commons Attribution (CC BY) Lets others
distribute and copy the article, create extracts, abstracts, and
other revised versions, adaptations or derivative works of or from
an article (such as a translation), include in a collective work
(such as an anthology), text or data mine the article, even for
commercial purposes, as long as they credit the author(s), do not
represent the author as endorsing their adaptation of the article,
and do not modify the article in such a way as to damage the
author's honor or reputation.
Creative Commons Attribution-NonCommercial-NoDerivs
(CC BY-NC-ND): For non-commercial purposes, lets others
distribute and copy the article, and to include in a collective work
(such as an anthology), as long as they credit the author(s) and
provided they do not alter or modify the article. The open access
publication fee for this journal is USD 2150, excluding taxes.

67
Learn more about Elsevier's pricing policy:
https://www.elsevier.com/openaccesspricing.
Green open access: Authors can share their research in a
variety of different ways and Elsevier has a number of green open
access options available. We recommend authors see our green
open access page for further information. Authors can also self-
archive their manuscripts immediately and enable public access
from their institution's repository after an embargo period. This is
the version that has been accepted for publication and which
typically includes author-incorporated changes suggested during
submission, peer review and in editor-author communications.
Embargo period: For subscription articles, an appropriate amount
of time is needed for journals to deliver value to subscribing
customers before an article becomes freely available to the
public. This is the embargo period and it begins from the date the
article is formally published online in its final and fully citable form.
Find out more. This journal has an embargo period of 12 months.
Elsevier Publishing Campus: The Elsevier Publishing Campus
(www.publishingcampus.com) is an online platform offering free
lectures, interactive training and professional advice to support
you in publishing your research. The College of Skills training
offers modules on how to prepare, write and structure your article
and explains how editors will look at your paper when it is
submitted for publication. Use these resources, and more, to
ensure that your submission will be the best that you can make it.
Language (usage and editing services): Please write your
text in good English (American or British usage is accepted, but
not a mixture of these). Authors who feel their English language
manuscript may require editing to eliminate possible grammatical
or spelling errors and to conform to correct scientific English may

68
wish to use the English Language Editing service available from
Elsevier's WebShop.
Submission
Our online submission system guides you stepwise
through the process of entering your article details and uploading
your files. The system converts your article files to a single PDF
file used in the peer-review process. Editable files (e.g., Word,
LaTeX) are required to typeset your article for final publication. All
correspondence, including notification of the Editor's decision and
requests for revision, is sent by e-mail. Referees Please submit
the names and institutional e-mail addresses of several potential
referees. For more details, visit our Support site. Note that the
editor retains the sole right to decide whether or not the
suggested reviewers are used.
PREPARATION
NEW SUBMISSIONS
Submission to this journal proceeds totally online and you
will be guided stepwise through the creation and uploading of
your files. The system automatically converts your files to a single
PDF file, which is used in the peer-review process. As part of the
Your Paper Your Way service, you may choose to submit your
manuscript as a single file to be used in the refereeing process.
This can be a PDF file or a Word document, in any format or
layout that can be used by referees to evaluate your manuscript.
It should contain high enough quality figures for refereeing. If you
prefer to do so, you may still provide all or some of the source
files at the initial submission. Please note that individual figure
files larger than 10 MB must be uploaded separately.

69
References
There are no strict requirements on reference formatting
at submission. References can be in any style or format as long
as the style is consistent. Where applicable, author(s) name(s),
journal title/book title, chapter title/article title, year of publication,
volume number/book chapter and the pagination must be
present. Use of DOI is highly encouraged. The reference style
used by the journal will be applied to the accepted article by
Elsevier at the proof stage. Note that missing data will be
highlighted at proof stage for the author to correct.
Formatting requirements
There are no strict formatting requirements but all
manuscripts must contain the essential elements needed to
convey your manuscript, for example Abstract, Keywords,
Introduction, Materials and Methods, Results, Conclusions,
Artwork and Tables with Captions. If your article includes any
Videos and/or other Supplementary material, this should be
included in your initial submission for peer review purposes.
Divide the article into clearly defined sections.
Figures and tables embedded in text Please ensure the
figures and the tables included in the single file are placed next to
the relevant text in the manuscript, rather than at the bottom or
the top of the file. The corresponding caption should be placed
directly below the figure or table.
REVISED SUBMISSIONS
Use of word processing software Regardless of the file
format of the original submission, at revision you must provide us
with an editable file of the entire article. Keep the layout of the

70
text as simple as possible. Most formatting codes will be removed
and replaced on processing the article. The electronic text should
be prepared in a way very similar to that of conventional
manuscripts (see also the Guide to Publishing with Elsevier). See
also the section on Electronic artwork. To avoid unnecessary
errors you are strongly advised to use the 'spell-check' and
'grammar-check' functions of your word processor.
Article structure
Subdivision - numbered sections
your article into clearly defined and numbered sections.
Subsections should be numbered 1.1 (then 1.1.1, 1.1.2, ...), 1.2,
etc. (the abstract is not included in section numbering). Use this
numbering also for internal cross-referencing: do not just refer to
'the text'. Any subsection may be given a brief heading. Each
heading should appear on its own separate line.
Introduction
State the objectives of the work and provide an adequate
background, avoiding a detailed literature survey or a summary of
the results.
Material and Methods
Provide sufficient detail to allow the work to be
reproduced. Methods already published should be indicated by a
reference: only relevant modifications should be described. Since
there are significant differences in skeletal structure and
remodeling between the sexes and strains, it is essential that all
animal studies report the age, sex, and strain (e.g., C57BL/6) of
animals used.

71
Theory/calculation
A Theory section should extend, not repeat, the
background to the article already dealt with in the Introduction
and lay the foundation for further work. In contrast, a Calculation
section represents a practical development from a theoretical
basis.
Results
Results should be clear and concise.
Discussion
This should explore the significance of the results of the
work, not repeat them. A combined Results and Discussion
section is often appropriate. Avoid extensive citations and
discussion of published literature.
Conclusions
The main conclusions of the study may be presented in a
short Conclusions section, which may stand alone or form a
subsection of a Discussion or Results and Discussion section.
Essential title page information
• Title. Concise and informative. Titles are often used in
information-retrieval systems. Avoid abbreviations and formulae
where possible.
• Author names and affiliations. Please clearly indicate the
given name(s) and family name(s) of each author and check that
all names are accurately spelled. Present the authors' affiliation

72
addresses (where the actual work was done) below the names.
Indicate all affiliations with a lowercase superscript letter
immediately after the author's name and in front of the
appropriate address. Provide the full postal address of each
affiliation, including the country name and, if available, the e-mail
address of each author.
• Corresponding author. Clearly indicate who will handle
correspondence at all stages of refereeing and publication, also
post-publication. Ensure that the e-mail address is given and that
contact details are kept up to date by the corresponding author.
• Present/permanent address. If an author has moved since the
work described in the article was done, or was visiting at the time,
a 'Present address' (or 'Permanent address') may be indicated as
a footnote to that author's name. The address at which the author
actually did the work must be retained as the main, affiliation
address. Superscript Arabic numerals are used for such
footnotes.
Abstract
A concise and factual abstract is required. The abstract
should state briefly the purpose of the research, the principal
results and major conclusions. An abstract is often presented
separately from the article, so it must be able to stand alone. For
this reason, References should be avoided, but if essential, then
cite the author(s) and year(s). Also, non-standard or uncommon
abbreviations should be avoided, but if essential they must be
defined at their first mention in the abstract itself.
Graphical abstract

73
Although a graphical abstract is optional, its use is
encouraged as it draws more attention to the online article. The
graphical abstract should summarize the contents of the article in
a concise, pictorial form designed to capture the attention of a
wide readership. Graphical abstracts should be submitted as a
separate file in the online submission system. Image size: Please
provide an image with a minimum of 531 × 1328 pixels (h × w) or
proportionally more. The image should be readable at a size of 5
× 13 cm using a regular screen resolution of 96 dpi. Preferred file
types: TIFF, EPS, PDF or MS Office files. You can view Example
Graphical Abstracts on our information site. Authors can make
use of Elsevier's Illustration and Enhancement service to ensure
the best presentation of their images and in accordance with all
technical requirements: Illustration Service.
Highlights
Highlights are mandatory for this journal. They consist of
a short collection of bullet points that convey the core findings of
the article and should be submitted in a separate file in the online
submission system. Please use Highlights in the file name and
include 3 to 5 bullet points (maximum 20 words per bullet point).
See http://www.elsevier.com/highlights for examples.
Keywords
Immediately after the abstract, provide a maximum of 6
keywords, using American spelling and avoiding general and
plural terms and multiple concepts (avoid, for example, 'and', 'of').
Be sparing with abbreviations: only abbreviations firmly
established in the field may be eligible. These keywords will be
used for indexing purposes.

74
Abbreviations: Define abbreviations that are not standard in this
field in a footnote to be placed on the first page of the article.
Such abbreviations that are unavoidable in the abstract must be
defined at their first mention there, as well as in the footnote.
Ensure consistency of abbreviations throughout the article.
Acknowledgements: Collate acknowledgements in a separate
section at the end of the article before the references and do not,
therefore, include them on the title page, as a footnote to the title
or otherwise. List here those individuals who provided help during
the research (e.g., providing language help, writing assistance or
proof reading the article, etc.).
Formatting of funding sources: List funding sources in this
standard way to facilitate compliance to funder's requirements:
Funding: This work was supported by the National Institutes of
Health [grant numbers xxxx, yyyy]; the Bill & Melinda Gates
Foundation, Seattle, WA [grant number zzzz]; and the United
States Institutes of Peace [grant number aaaa].
It is not necessary to include detailed descriptions on the
program or type of grants and awards. When funding is from a
block grant or other resources available to a university, college, or
other research institution, submit the name of the institute or
organization that provided the funding.
If no funding has been provided for the research, please
include the following sentence: This research did not receive any
specific grant from funding agencies in the public, commercial, or
not-for-profit sectors.
Genbank

75
Footnotes: should be used sparingly. Number them consecutively
throughout the article. Many word processors build footnotes into
the text, and this feature may be used. Should this not be the
case, indicate the position of footnotes in the text and present the
footnotes themselves separately at the end of the article.
Electronic artwork General points:
• Make sure you use uniform lettering and sizing of your original
artwork. • Preferred fonts: Arial (or Helvetica), Times New Roman
(or Times), Symbol, Courier.
• Number the illustrations according to their sequence in the text.
• Use a logical naming convention for your artwork files.
• Indicate per figure if it is a single, 1.5 or 2-column fitting image.
• For Word submissions only, you may still provide figures and
their captions, and tables within a single file at the revision stage.
• Please note that individual figure files larger than 10 MB must
be provided in separate source files. A detailed guide on
electronic artwork is available. You are urged to visit this site;
some excerpts from the detailed information are given here.
Formats
Regardless of the application used, when your electronic
artwork is finalized, please 'save as' or convert the images to one
of the following formats (note the resolution requirements for line
drawings, halftones, and line/halftone combinations given below):
EPS (or PDF): Vector drawings. Embed the font or save the text
as 'graphics'. TIFF (or JPG): Color or grayscale photographs

76
(halftones): always use a minimum of 300 dpi. TIFF (or JPG):
Bitmapped line drawings: use a minimum of 1000 dpi. TIFF (or
JPG): Combinations bitmapped line/half-tone (color or grayscale):
a minimum of 500 dpi is required. Please do not:
• Supply files that are optimized for screen use (e.g., GIF, BMP,
PICT, WPG); the resolution is too low.
• Supply files that are too low in resolution.
• Submit graphics that are disproportionately large for the content.
Color artwork
Please make sure that artwork files are in an acceptable
format (TIFF (or JPEG), EPS (or PDF), or MS Office files) and
with the correct resolution. If, together with your accepted article,
you submit usable color figures then Elsevier will ensure, at no
additional charge, that these figures will appear in color online
(e.g., ScienceDirect and other sites) regardless of whether or not
these illustrations are reproduced in color in the printed version.
For color reproduction in print, you will receive information
regarding the costs from Elsevier after receipt of your accepted
article. Please indicate your preference for color: in print or online
only. Further information on the preparation of electronic artwork.
Illustration services
Elsevier's WebShop offers Illustration Services to authors
preparing to submit a manuscript but concerned about the quality
of the images accompanying their article. Elsevier's expert
illustrators can produce scientific, technical and medical-style
images, as well as a full range of charts, tables and graphs.
Image 'polishing' is also available, where our illustrators take your

77
image(s) and improve them to a professional standard. Please
visit the website to find out more.
Figure captions
Ensure that each illustration has a caption. A caption
should comprise a brief title (not on the figure itself) and a
description of the illustration. Keep text in the illustrations
themselves to a minimum but explain all symbols and
abbreviations used.
Tables
Please submit tables as editable text and not as images.
Tables can be placed either next to the relevant text in the article,
or on separate page(s) at the end. Number tables consecutively
in accordance with their appearance in the text and place any
table notes below the table body. Be sparing in the use of tables
and ensure that the data presented in them do not duplicate
results described elsewhere in the article. Please avoid using
vertical rules and shading in table cells.
References
Citation in text
Please ensure that every reference cited in the text is also
present in the reference list (and vice versa). Any references cited
in the abstract must be given in full. Unpublished results and
personal communications are not recommended in the reference
list, but may be mentioned in the text. If these references are
included in the reference list they should follow the standard
reference style of the journal and should include a substitution of
the publication date with either 'Unpublished results' or 'Personal

78
communication'. Citation of a reference as 'in press' implies that
the item has been accepted for publication.
Reference links
Increased discoverability of research and high quality
peer review are ensured by online links to the sources cited. In
order to allow us to create links to abstracting and indexing
services, such as Scopus, CrossRef and PubMed, please ensure
that data provided in the references are correct. Please note that
incorrect surnames, journal/book titles, publication year and
pagination may prevent link creation. When copying references,
please be careful as they may already contain errors. Use of the
DOI is encouraged.
A DOI can be used to cite and link to electronic articles
where an article is in-press and full citation details are not yet
known, but the article is available online. A DOI is guaranteed
never to change, so you can use it as a permanent link to any
electronic article. An example of a citation using DOI for an article
not yet in an issue is: VanDecar J.C., Russo R.M., James D.E.,
Ambeh W.B., Franke M. (2003). Aseismic continuation of the
Lesser Antilles slab beneath northeastern Venezuela. Journal of
Geophysical Research, https://doi.org/10.1029/2001JB000884.
Please note the format of such citations should be in the same
style as all other references in the paper.
Web references
As a minimum, the full URL should be given and the date
when the reference was last accessed. Any further information, if
known (DOI, author names, dates, reference to a source
publication, etc.), should also be given. Web references can be

79
listed separately (e.g., after the reference list) under a different
heading if desired, or can be included in the reference list.
Data references
This journal encourages you to cite underlying or relevant
datasets in your manuscript by citing them in your text and
including a data reference in your Reference List. Data
references should include the following elements: author
name(s), dataset title, data repository, version (where available),
year, and global persistent identifier. Add [dataset] immediately
before the reference so we can properly identify it as a data
reference. The [dataset] identifier will not appear in your
published article.
References in a special issue
Please ensure that the words 'this issue' are added to any
references in the list (and any citations in the text) to other articles
in the same Special Issue.
Reference management software
Most Elsevier journals have their reference template
available in many of the most popular reference management
software products. These include all products that support
Citation Style Language styles, such as Mendeley and Zotero, as
well as EndNote. Using the word processor plug-ins from these
products, authors only need to select the appropriate journal
template when preparing their article, after which citations and
bibliographies will be automatically formatted in the journal's style.
If no template is yet available for this journal, please follow the
format of the sample references and citations as shown in this
Guide. Users of Mendeley Desktop can easily install the

80
reference style for this journal by clicking the following link:
http://open.mendeley.com/use-citation-style/bone When
preparing your manuscript, you will then be able to select this
style using the Mendeley plugins for Microsoft Word or
LibreOffice.
Reference formatting
There are no strict requirements on reference formatting
at submission. References can be in any style or format as long
as the style is consistent. Where applicable, author(s) name(s),
journal title/book title, chapter title/article title, year of publication,
volume number/book chapter and the pagination must be
present. Use of DOI is highly encouraged. The reference style
used by the journal will be applied to the accepted article by
Elsevier at the proof stage. Note that missing data will be
highlighted at proof stage for the author to correct. If you do wish
to format the references yourself they should be arranged
according to the following examples:
Reference style Text: Indicate references by number(s) in
square brackets in line with the text. The actual authors can be
referred to, but the reference number(s) must always be given.
Example: '..... as demonstrated [3,6]. Barnaby and Jones [8]
obtained a different result ....' List: Number the references
(numbers in square brackets) in the list in the order in which they
appear in the text. Examples: Reference to a journal publication:
[1] J. van der Geer, J.A.J. Hanraads, R.A. Lupton, The art of
writing a scientific article, J. Sci. Commun. 163 (2010) 51–59.
Reference to a book: [2] W. Strunk Jr., E.B. White, The Elements
of Style, fourth ed., Longman, New York, 2000. Reference to a
chapter in an edited book: [3] G.R. Mettam, L.B. Adams, How to
prepare an electronic version of your article, in: B.S. Jones, R.Z.
Smith (Eds.), Introduction to the Electronic Age, E-Publishing Inc.,

81
New York, 2009, pp. 281–304. Reference to a website: [4]
Cancer Research UK, Cancer statistics reports for the UK.
http://www.cancerresearchuk.org/
aboutcancer/statistics/cancerstatsreport/, 2003 (accessed
13.03.03). Reference to a dataset: [dataset] [5] M. Oguro, S.
Imahiro, S. Saito, T. Nakashizuka, Mortality data for Japanese
oak wilt disease and surrounding forest compositions, Mendeley
Data, v1, 2015. https://doi.org/10.17632/ xwj98nb39r.1. Journal
abbreviations source Journal names should be abbreviated
according to the List of Title Word Abbreviations.
Video
Elsevier accepts video material and animation sequences
to support and enhance your scientific research. Authors who
have video or animation files that they wish to submit with their
article are strongly encouraged to include links to these within the
body of the article. This can be done in the same way as a figure
or table by referring to the video or animation content and noting
in the body text where it should be placed. All submitted files
should be properly labeled so that they directly relate to the video
file's content. In order to ensure that your video or animation
material is directly usable, please provide the files in one of our
recommended file formats with a preferred maximum size of 150
MB. Video and animation files supplied will be published online in
the electronic version of your article in Elsevier Web products,
including ScienceDirect. Please supply 'stills' with your files: you
can choose any frame from the video or animation or make a
separate image. These will be used instead of standard icons and
will personalize the link to your video data. For more detailed
instructions please visit our video instruction pages. Note: since
video and animation cannot be embedded in the print version of
the journal, please provide text for both the electronic and the

82
print version for the portions of the article that refer to this
content.
Supplementary material
Supplementary material such as applications, images
and sound clips, can be published with your article to enhance it.
Submitted supplementary items are published exactly as they are
received (Excel or PowerPoint files will appear as such online).
Please submit your material together with the article and supply a
concise, descriptive caption for each supplementary file. If you
wish to make changes to supplementary material during any
stage of the process, please make sure to provide an updated
file. Do not annotate any corrections on a previous version.
Please switch off the 'Track Changes' option in Microsoft Office
files as these will appear in the published version.
Data linking
If you have made your research data available in a data
repository, you can link your article directly to the dataset.
Elsevier collaborates with a number of repositories to link articles
on ScienceDirect with relevant repositories, giving readers access
to underlying data that give them a better understanding of the
research described. There are different ways to link your datasets
to your article. When available, you can directly link your dataset
to your article by providing the relevant information in the
submission system. For more information, visit the database
linking page. For supported data repositories a repository banner
will automatically appear next to your published article on
ScienceDirect. In addition, you can link to relevant data or entities
through identifiers within the text of your manuscript, using the
following format: Database: xxxx (e.g., TAIR: AT1G01020;
CCDC: 734053; PDB: 1XFN).

83
ARTICLE ENRICHMENTS
AudioSlides
The journal encourages authors to create an AudioSlides
presentation with their published article. AudioSlides are brief,
webinar-style presentations that are shown next to the online
article on ScienceDirect. This gives authors the opportunity to
summarize their research in their own words and to help readers
understand what the paper is about. More information and
examples are available. Authors of this journal will automatically
receive an invitation e-mail to create an AudioSlides presentation
after acceptance of their paper.
3D radiological data
You can enrich your online article by providing 3D
radiological data in DICOM format. Radiological data will be
visualized for readers using the interactive viewer embedded
within your article, and will enable them to: browse through
available radiological datasets; explore radiological data as 2D
series, 2D orthogonal MPR, 3D volume rendering and 3D MIP;
zoom, rotate and pan 3D reconstructions; cut through the volume;
change opacity and threshold level; and download the data.
Multiple datasets can be submitted. Each dataset will have to be
zipped and uploaded to the online submission system via the '3D
radiological data' submission category. The recommended size of
a single uncompressed dataset is 200 MB or less. Please provide
a short informative description for each dataset by filling in the
'Description' field when uploading each ZIP file. Note: all datasets
will be available for download from the online article on
ScienceDirect. So please ensure that all DICOM files are
anonymized prior to submission.

84
Interactive plots
This journal enables you to show an Interactive Plot with
your article by simply submitting a data file. Full instructions.
AFTER ACCEPTANCE
Online proof correction
Corresponding authors will receive an e-mail with a link
to our online proofing system, allowing annotation and correction
of proofs online. The environment is similar to MS Word: in
addition to editing text, you can also comment on figures/tables
and answer questions from the Copy Editor. Web-based proofing
provides a faster and less error-prone process by allowing you to
directly type your corrections, eliminating the potential
introduction of errors. If preferred, you can still choose to
annotate and upload your edits on the PDF version. All
instructions for proofing will be given in the e-mail we send to
authors, including alternative methods to the online version and
PDF. We will do everything possible to get your article published
quickly and accurately. Please use this proof only for checking
the typesetting, editing, completeness and correctness of the text,
tables and figures. Significant changes to the article as accepted
for publication will only be considered at this stage with
permission from the Editor. It is important to ensure that all
corrections are sent back to us in one communication. Please
check carefully before replying, as inclusion of any subsequent
corrections cannot be guaranteed. Proofreading is solely your
responsibility.
Offprints

85
The corresponding author will, at no cost, receive a
customized Share Link providing 50 days free access to the final
published version of the article on ScienceDirect. The Share Link
can be used for sharing the article via any communication
channel, including email and social media. For an extra charge,
paper offprints can be ordered via the offprint order form which is
sent once the article is accepted for publication. Both
corresponding and co-authors may order offprints at any time via
Elsevier's Webshop. Corresponding authors who have published
their article open access do not receive a Share Link as their final
published version of the article is available open access on
ScienceDirect and can be shared through the article DOI link.
AUTHOR INQUIRIES
Visit the Elsevier Support Center to find the answers you
need. Here you will find everything from Frequently Asked
Questions to ways to get in touch. You can also check the status
of your submitted article or find out when your accepted article
will be published.


















