







Línguas
Páginas
Legal


UNIVERSIDADE DE SÃO PAULO
ESCOLA DE ENGENHARIA DE LORENA
CLÁUDIO AUGUSTO KELLY
Estudo detalhado da sinterização e propriedades mecânicas das cerâmicas
de carbeto de silício
Lorena – SP
2006

CLÁUDIO AUGUSTO KELLY
Estudo detalhado da sinterização e propriedades mecânicas das cerâmicas
de carbeto de silício
Tese apresentada à Escola de Engenharia de
Lorena da Universidade de São Paulo para
obtenção do título de Doutor em Engenharia de
Materiais.
Área de Concentração: Materiais Metálicos,
Cerâmicos e Poliméricos.
Orientador: Dr.Sebastião Ribeiro
Lorena - SP
2006

AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO,
POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E
PESQUISA, DESDE QUE CITADA A FONTE.

FOLHA DE APROVAÇÃO
CLÁUDIO AUGUSTO KELLY
Estudo detalhado da sinterização e propriedades mecânicas das cerâmicas de carbeto de silício
aditivadas com AlN-Y2O3 e/ou AlN-CTR2O3
Tese apresentada à Escola de Engenharia de
Lorena da Universidade de São Paulo para
obtenção do título de Doutor em Engenharia de
Materiais.
Área de Concentração: Materiais Metálicos,
Cerâmicos e Poliméricos.
Aprovado em 07 de Julho de 2006
Banca Examinadora
Prof. Dr. Sebastião Ribeiro – Presidente da Banca - EEL/USP
Prof. Dr. Kurt Strecker - Universidade Federal de São João Del Rey
Prof. Carlos Alberto Alves Cairo – AMR/CTA
Prof. Dr. Olivério Moreira de Macedo e Silva – AMR/CTA
Prof. Dr. Carlos Antonio R. P. Batista – EEL/USP

DEDICATÓRIA
A DEUS, por ser presente em minha vida a todo o momento.
A minha esposa Jaqueline Danielli de Araújo Bento Kelly e a minha filha Tayssa Mychaela
de Araújo Kelly, pessoas muito especiais para mim.
Á minha irmã Cristina Kelly, aos meus pais Francisco Augusto Kelly e Maria Benedita Kelly
por terem me trazido ao mundo e me dado todo amor, carinho e dedicação, pessoas que
sempre estiveram do meu lado, nunca deixando desistir dos meus sonhos.

AGRADECIMENTOS
Ao Prof. Dr. Sebastião Ribeiro, que com dedicação, paciência e incentivo me ajudou a
desenvolver esta tese.
À Escola de Engenharia de Lorena (EEL-USP) por viabilizar o desenvolvimento deste
projeto;
A CAPES pela concessão da bolsa de doutorado e pelo apoio financeiro para realização desta
pesquisa.
Ao Prof. Dr. Cosme R. M. da Silva e Prof. Dr. Carlos A. A. Cairo, do Centro Técnico
Aeroespacial (AMR/CTA), pela doação de carbeto de silício e utilização da prensa isostática;
A Dra Ana Helena Bressiani, do IPEN-USP, pelas análises dilatométricas;
À Hermann C. Starck – Alemanha pela doação de carbeto de silício;
Ao Dr. Gilbert Silva (Laboratório de Difração de Raios X) e ao técnico Francisco Paiva
(Laboratório de Ensaios Mecânicos), respectivamente, pelas ánalises de difração de raios X e
ensaios de flexão;
Aos amigos MSc Marcos Roberto Vargas Moreira, Dr. José Vitor Candido de Souza e Dr.
Claudinei dos Santos que tanto me incentivaram durante o desenvolvimento deste trabalho;
Aos amigos da Escola de Engenharia de Lorena – EEL-USP e do grupo de cerâmica que
contribuíram para realização deste trabalho;

“O Senhor é meu pastor; nada me faltará..
Ele me faz repousar em pastos verdejantes.
Leva-me para junto das águas de descanso; Refrigera-me a alma.
Guia-me pelas veredas da justiça por amor do seu nome.
Ainda que eu ande pelo vale da sombra da morte, não temerei mal nenhum,
porque tu estas comigo; a tua vara e o teu cajado me consolam.
Preparas-me uma mesa na presença dos meus adversários, unges-me
a cabeça com óleo; o meu cálice transborda.
Bondade e misericórdia certamente me seguirão todos os dias da minha vida;
e habitarei na casa do Senhor para todo o sempre”.
Salmo 23
Tu a quem tomei desde os fins da terra, e te chamei
Dentre os seus mais excelentes e te disse: Tu és o meu servo
A ti te escolhi e não te rejeitei.
Não temas, porque eu sou contigo; não te assombres,
Porque eu sou teu Deus; e te esforço e te ajudo, e te sustento
Com a destra da minha justiça.
Eis que envergonhados e confundidos serão todos os que se
Irritaram contra ti; tornar-se-ão nada e os que contenderem
Contigo perecerão.
Buscá-los-ás, mas não os acharás, e os que pelejarem contigo.
Tornar-se-ão nada, e como coisa que não é nada os que
Guerrearem contigo.
Porque eu, o senhor teu Deus, te tomo pela tua mão direita,
E te digo: Não temas que eu te ajudo.
Isaías 41

RESUMO
Kelly, C. A. Estudo detalhado da sinterização e propriedades mecânicas das cerâmicas
de carbeto de silício 2006. 140 f. Tese (Doutorado em Engenharia de Materiais) - Escola de
Engenharia de Lorena, Universidade de São Paulo, São Paulo.
O carbeto de silício, SiC, apresenta excelentes propriedades para aplicações
estruturais. Entretanto é um composto químico de forte caráter covalente, característica que
dificulta a obtenção de cerâmicas densas de SiC pelo processo de sinterização via fase sólida.
O objetivo deste trabalho foi obter cerâmicas densas de SiC por sinterização via fase líquida
com boas propriedades mecânicas. Para tanto, foram utilizadas misturas constituídas de pó α-
SiC e/ou β-SiC mais aditivos. Os aditivos foram AlN/Y2O3 e AlN/CTR2O3 (óxido misto de
ítrio e terras raras). Este trabalho foi dividido em três etapas: na primeira foram realizados
ensaios de dilatometria (IPEN) e aquecimento das amostras em forno com resistência de
grafite (EEL-USP) objetivando adquirir informações de suma importância para as etapas
subseqüentes, ou seja, isoterma (segunda etapa) e re-sinterização das amostras (terceira
etapa). As amostras contendo AlN-CTR2O3 (T10-α e T10-β) apresentaram valores de
densidade e propriedades mecânicas iguais ou superiores às N10-α e N10-β (contendo AlN-
Y2O3) mostrando a viabilidade da substituição do óxido de ítrio pelo óxido misto de ítrio e
terras raras na sinterização via fase líquida do SiC. As amostras contendo α-SiC (N10-α e
T10-α) e β-SiC (N10-β e T10-β) não mostraram mudanças microestruturais significativas,
tanto na etapa de isoterma quanto na re-sinterização.
Palavras chaves: Carbeto de Silício. Sinterização via fase líquida. AlN-Y2O3. AlN-CTR2O3.
Propriedades Mecânicas.

ABSTRACT
Kelly, C. A. Sintering and mechanical properties study detailed of the silicon carbide
ceramics. 2006. 140 f. Thesis (Doctoral in Materials Engineering) – Escola de Engenharia de
Lorena, Universidade de São Paulo, Lorena. 2006
Silicon carcbide, SiC, presents excellents properties for structural applications.
However, it is a strong covalent character chemical compound, characterist that difficult to
obtain SiC ceramic dense by solid phase sintering. The objective of this work was to obtain
dense LPS-SiC ceramics with good mechanical properties. For development of this work were
used mixtures constituted of α-SiC and/or β-SiC powders plus additives. The additives were
AlN-Y2O3 and AlN-CTR2O3 (yttrium and earth rares mixed oxide). This work can be divided
in three stages. In this first stage, dilatometric (IPEN) and heating (EEL-USP) tests have been
realized with objective of to obtain important data for subsequent steps, in others words,
isothermal (second step) and re-sintering (third). The samples content AlN- CTR2O3 (T10-α
and T10-β) showed density and mechanical properties equal or higher to the N10-α and N10-
β (content AlN-Y2O3), presenting the feasibility of substitution of the yttrium oxide for
yttrium and earth rare oxide mixed in the LPS-SiC. The samples content α-SiC (N10-α and
T10-α) and β-SiC (N10-β and T10-β) no showed importants microstructural changes, so as
well as in the isothermal and re-sintering steps.
Keywords: Silicon carbide. Liquid phase sintering. AlN-Y2O3. AlN-CTR2O3. Mechanical
properties.

LISTA DE ILUSTRAÇÕES
Figura 2.1 - Diagrama de fase do sistema silício-carbono a uma pressão total de 1 bar.
25
Figura 2.2 - Estruturas cristalinas do SiC: (a) β-SiC e (b) α-SiC.
26
Figura 2.3 - Estabilidade dos politipos do SiC em relação à temperatura.
28
Figura 2.4 - Produção de carbeto de silício pelo processo “Acheson” modificado: cortes transversais nos sentidos: (a) longitudinal e (b) radial, através do forno.
30
Figura 2.5 - SiC cristalino como recebido a partir do Processo “Acheson”.
31
Figura 2.6 - Fenômeno de molhamento de uma superfície: (a) molhamento desfavorecido (θ >90°); (b) molhamento (θ < 90°) e (c) espalhamento do líquido sobre a superfície sólida (θ =0°) (d) distribuição de energias interfaciais.
34
Figura 2.7 - Ângulo diedral: (a) Contorno de grão em equilíbrio com a fase vapor na superfície; (b) Contorno de grão em equilíbrio com um líquido.
35
Figura 2.8 - Distribuição de fase líquida para diferentes valores de ângulo diedral numa junção tripla.
36
Figura 2.9 - Variação do ângulo diedral em função da relação γSS/γSL.
37
Figura 2.10 - Geometria de uma junção tripla com líquido segregado quando θ < π/6 (2θ = ângulo diedral; r = raio de curvatura e U = raio do canal).
38
Figura 2.11 - Força atrativa entre duas partículas.
38
Figura 2.12 - O efeito de dois valores extremos de ângulo de contato na força capilar entre duas partículas esféricas separadas por um líquido.
39
Figura 2.13 - Influência da solubilidade nos resultados de sinterização.
41
Figura 2.14 - Modelo esquemático da densidade em função do tempo de sinterização representando os estágios da sinterização via fase líquida.
42
Figura 2.15 - Fragmentação das partículas durante a penetração do líquido.
42
Figura 2.16 - Solução-reprecipitação, com crescimento e acomodação de forma dos grãos.
44
Figura 2.17 - Estágios de sinterização via fase líquida usando pó de partida de α-SiC.
45
Figura 2.18 –
Estágios de sinterização via fase líquida usando pó de partida de β-SiC. 45
Figura 2.19 –
Variação de energia livre de Gibbs de formação da reação (2.9) versus a reação (2.10) para vários óxidos metálicos a 2127 ºC.
49
Figura 2.20 –
Definição de espessura, comprimento e razão de aspecto de grãos de SiC. 51

Figura 2.21 –
Curvas de tensão versus deformação para um material frágil e um dúctil
52
Figura 2.22 –
Sistemas de trincas (a) “Palmquist”; (b) “Half-Penny”. 54
Figura 2.23 –
Determinação da resistência mecânica de corpos cerâmicos em forma de barras utilizando as técnicas de resistência a flexão em 3 pontos (a) e 4 pontos (b).
55
Figura 3.1 –
Fluxograma ilustrativo do procedimento experimental e caracterização adotados neste trabalho.
64
Figura 3.2 –
Diagrama de fase do sistema AlN/Y2O3.
65
Figura 4.1 - Curvas de distribuição granulométrica dos pós das misturas.
81
Figura 4.2 - Micrografias realizadas por MEV dos pós das misturas.
82
Figura 4.3 –
Curvas de retração linear e densidade relativa das misturas N10-α, N10-β, T10-α e T10-β.
85
Figura 4.4 –
Curvas de velocidade de retração linear das misturas N10-α, N10-β, T10-α e T10-β.
86
Figura 4.5 -
Densidade relativa das amostras N10-α, N10-β, T10-α e T10-β aquecidas a diferentes temperaturas, sem isoterma.
87
Figura 4.6 -
Perda de massa das amostras N10-α, N10-β, T10-α e T10-β aquecidas a diferentes temperaturas, sem isoterma.
89
Figura 4.7 -
Difratogramas de raios X das misturas N10-α e T10-α aquecidas a diferentes temperaturas, sem isoterma de sinterização.
90
Figura 4.8 -
Difratograma de raios X das misturas N10-β e T10-β aquecidas a diferentes temperaturas, sem isoterma de sinterização.
90
Figura 4.9 -
Micrografias realizadas por MEV da mistura N10-α aquecida em diferentes temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.
93
Figura 4.10 -
Micrografias realizadas por MEV da mistura N10-β aquecida em diferentes temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.
94
Figura 4.11 -
Micrografias realizadas por MEV da mistura T10-α aquecida em diferentes temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.
95
Figura 4.12 -
Micrografias realizadas por MEV da mistura T10-β aquecida em diferentes temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.
96
Figura 4.13 -
Comportamento microestrutural da mistura N10-α após formação da fase líquida.
97

Figura 4.14-
Comportamento microestrutural da mistura N10-β após formação da fase líquida (1900oC).
98
Figura 4.15-
Comportamento microestrutural da mistura N10-β após formação da fase líquida (2000oC).
99
Figura 4.16
Comportamento microestrutural da mistura T10-β após formação da fase líquida.
99
Figura 4.17 -
Resultados de densidade relativa, perda de massa e retração linear das misturas N10-α, N10-β, T10-α e T10-β sinterizadas a 2000oC/1h sob atmosfera de argônio.
100
Figura 4.18 -
Resultados de densidade relativa, perda de massa e retração linear das misturas N10-α, N10-β, T10-α e T10-β sinterizadas a 1800oC/30 minutos e 2000oC/1h sob atmosfera de argônio.
101
Figura 4.19 -
Resultados de densidade relativa, perda de massa e retração linear das misturas N10-α, N10-β, T10-α e T10-β sinterizadas a 1800oC/1h e 2000oC/1h sob atmosfera de argônio.
101
Figura 4.20 -
Difratogramas de raios X das misturas N10-α, N10-β, T10-α e T10-β sinterizadas a diferentes ciclos sob atmosfera de argônio.
103
Figura 4.21 –
Análise quantitativa dos principais politipos de SiC na mistura N10-α sinterizada a diferentes ciclos sob atmosfera de argônio.
105
Figura 4.22 –
Análise quantitativa dos principais politipos de SiC na mistura N10-β sinterizada a diferentes ciclos sob atmosfera de argônio.
106
Figura 4.23 –
Análise quantitativa dos principais politipos de SiC na mistura T10-α sinterizada a diferentes ciclos sob atmosfera de argônio.
107
Figura 4.24 –
Análise quantitativa dos principais politipos de SiC na mistura T10-β sinterizada a diferentes ciclos sob atmosfera de argônio.
107
Figura 4.25 - Micrografias das cerâmicas de SiC sinterizadas e atacadas por NaOH:KOH (1:1) a 500oC por 2 minutos.
108
Figura 4.26 - Caminho percorrido pela trinca na amostra N10-β sinterizada a 2000oC/1h sob atmosfera de argônio
110
Figura 4.27–
Caminho percorrido pela trinca na amostra N10-β sinterizada a 1800oC/1h e 2000oC/1h sob atmosfera de argônio
111
Figura 4.28–
Micrografia da superfície fraturada da mistura N10-β sinterizada a 1800oC/1h e 2000oC/1h sob atmosfera de argônio.
112
Figura 4.29 -
Resultados de densidade relativa e perda de massa da mistura N10-α sinterizada e re-sinterizada.
114
Figura 4.30 -
Resultados de densidade relativa e perda de massa da mistura N10-β sinterizada e re-sinterizada.
115

Figura 4.31 -
Resultados de densidade relativa e perda de massa da mistura T10-α sinterizada e re-sinterizada.
116
Figura 4.32 -
Resultados de densidade relativa e perda de massa ds mistura T10-β sinterizada e re-sinterizada.
116
Figura 4.33 -
Difratogramas de raios X das misturas N10-α, N10-β, T10-α e T10-β sinterizadas e re-sinterizadas.
117
Figura 4.34 -
Análise quantitativa dos principais politipos de SiC na mistura N10-α sinterizada e re-sinterizada.
119
Figura 4.35 -
Análise quantitativa dos principais politipos de SiC na mistura N10-β sinterizada e re-sinterizada.
119
Figura 4.36 -
Análise quantitativa dos principais politipos de SiC na mistura T10-α sinterizada e tratada termicamente pós-sinterização.
120
Figura 4.37 -
Análise quantitativa dos principais politipos de SiC na mistura T10-β sinterizada e re-sinterizada.
120
Figura 4.38 -
Micrografias da mistura N10-β re-sinterizadas e atacadas por NaOH:KOH (1:1) a 500oC por 2 minutos.
121
Figura 4.39 -
Micrografias da mistura T10-β re-sinterizada e atacadas por NaOH:KOH (1:1) a 500oC por 2 minutos.
122
Figura 4.40
Micrografias das superfícies de fratura das misturas N10-β e T10-β sinterizads e re-sinterizadas.
123

LISTA DE TABELAS
Tabela 2.1 – Propriedades cristalográficas dos principais politipos de SiC.
27
Tabela 2.2 - Equações para cálculo da resistência à flexão.
56
Tabela 3.1 - Caracterização física e química do α-SiC.
60
Tabela 3.2 - Caracterização física e química do β-SiC.
61
Tabela 3.3 – Caracterização física e química do AlN.
61
Tabela 3.4 – Caracterização física e química do Y2O3.
62
Tabela 3.5 - Composição Química do CTR2O3, determinada por Espectrometria de Emissão Ótica com Plasma Induzido (ICP-AES), e suas propriedades físicas.
63
Tabela 3.6 - Composição das misturas usadas no trabalho.
66
Tabela 3.7 - Massas específicas utilizadas para os cálculos.
67
Tabela 3.8 - Ciclos de sinterização para isoterma.
73
Tabela 3.9 – Massas específicas teórica, real e densidade relativa a verde das misturas estudadas.
83
Tabela 4.0 – Resultados de densificação, microdureza, tenacidade a fratura e resistência a flexão em 3 pontos das amostras N10-α, N10-β, T10-α e T10-β sinterizadas a diferentes ciclos sob atmosfera de argônio.
113
Tabela 4.1 – Resultados de densificação, microdureza, tenacidade à fratura e resistência a flexão em 3 pontos das amostras N10-α, N10-β, T10-α e T10-β submetidas a tratamento térmico pós-sinterização.
126

LISTA DE SÍMBOLOS
a Parâmetro de rede; metade do comprimento da diagonal da impressão
deixada pelo indentador; tamanho dos defeitos.
ar Razão de aspecto.
b Tamanho da trinca.
c Parâmetro de rede; tamanho do defeito crítico.
D Diâmetro da amostra.
Drel Densidade teórica em percentagem.
dRel Densidade relativa da amostra sinterizada.
dverde Densidade relativa a verde.
E Módulo de elasticidade.
F Força atrativa entre duas partículas; carga aplicada ao corpo de prova.
h Altura da amostra.
HV Dureza Vickers.
KIC Tenacidade à fratura do material.
l Espessura ou largura do grão de SiC
Lo Comprimento inicial
L Comprimento final
mf Massa final da amostra (após tratamento térmico).
mi Massa inicial da amostra (verde).
P Carga aplicada ao corpo de prova.
P.M. Perda de massa.
r Raio de curvatura.
r1, r2 Raios das partículas.

SA Solubilidade do líquido no sólido formado.
SA Solubilidade do líquido no sólido formado.
SB Solubilidade do sólido no líquido.
SR Razão de solubilidade
t Comprimento do grão de SiC.
U Raio do canal.
VT Volume total da mistura.
Y Fator geométrico que caracteriza a forma e a localização dos defeitos.
Z Número de SiC por célula unitária.
WA Massa do α e/ou β-SiC
WB Massa do AlN/CTR2O3 ou AlN/Y2O3.
Wm Massa da amostra.
WT Massa total da mistura.
SLAw
Trabalho de adesão
W1 Massa da amostra seca.
W2 Massa da amostra imersa
∆Go Mudança de energia livre
∆L Variação linear.
∆P Variação da pressão capilar.
φ Ângulo diedral
γSV Energia interfacial sólido–vapor.
γSL Energia interfacial sólido–líquido.
γLV Energia interfacial líquido–vapor.
γi Energia específica de fratura

π Constante Matemática.
θ Ângulo de contato ou de molhamento
ρ Raio da ponta da trinca.
ρA Massa específica do α e/ou β-SiC.
ρB Massa específica do AlN/CTR2O3 ou AlN/Y2O3.
ρgr Massa específica a verde.
ρt Massa específica teórica.
ρm Massa específica da amostra.
ρrel Massa específica relativa.
ρsint Massa específica das amostras sinterizadas.
ρT Densidade total da mistura.
ρH2O Massa específica da água a 20oC.
σa Tensão aplicada.
σf Resistência à fratura.
σ3f Resistência à flexão em três pontos.
σm Tensão máxima na ponta da trinca.
ψ Constante (função da quantidade de líquido, distância entre as partículas e
ângulo de contato).

SUMÁRIO
1 INTRODUÇÃO 20
1.1 OBJETIVOS 21
1.2 JUSTIFICATIVAS 22
2 REVISÃO BIBLIOGRÁFICA 24
2.1 - HISTÓRICO DO CARBETO DE SILÍCIO. 24
2.2 - ESTABILIDADE TERMODINÂMICA DO CARBETO DE SILÍCIO. 24
2.3 - ESTRUTURA E POLITIPISMO DO CARBETO DE SILÍCIO. 25
2.4 – PROCESSOS DE OBTENÇÃO DO CARBETO DE SILÍCIO. 28
2.5 - PROCESSAMENTO DE CERÂMICAS DE CARBETO DE SILÍCO. 32
2.6 – ESTABILIDADE QUÍMICA DO CARBETO DE SILÍCIO. 47
2.7 – ASPECTOS MICROESTRUTURAIS E PROPRIEDADES MECÂNICAS
DAS CERÂMICAS DE SIC.
50
3 MATERIAIS E MÉTODOS 60
3.1 –MATERIAIS 60
3.1.1 – Alfa-Carbeto de Silício (α-SiC) 60
3.1.2 – Beta-Carbeto de Silício (β-SiC) 61
3.1.3. - Nitreto de Alumínio (AlN) 61
3.1.4 – Óxido de Ítrio (Y2O3) 62
3.1.5 – Óxido Misto de Ítrio e Terras Raras (CTR2O3) 62
3.1.6 – Álcool isopropílico (C3H8O) 63
3.1.7 – Argônio (Ar) 63
3.2 – MÉTODOS 64
3.2.1 – Preparo dos aditivos 64
3.2.2 – Preparo das misturas 66

3.2.3 – Caracterização das misturas de pós 67
3.2.3.1 - Massa específica teórica 67
3.2.3.2 - Distribuição do tamanho de partículas 68
3.2.3.3 – Microscopia eletrônica de varredura das misturas de pós 68
3.2.3.4 - Análise de fases das misturas de pós 68
3.2.4 – Ensaios de Dilatometria das misturas de pós 69
3.2.5 – Compactação das misturas de pós 70
3.2.6 – Tratamento térmico das amostras 71
3.2.6.1 - Aquecimento das amostras 71
3.2.6.2 - Isoterma de sinterização 72
3.2.6.3 – Re-sinterização 73
3.2.7 – Caracterização dos compactos a verde 73
3.2.7.1 - Massa específica real 73
3.2.7.2 - Densidade relativa a verde 74
3.2.8 – Caracterização das amostras tratadas termicamente 75
3.2.8.1 - Massa específica e densidade relativa 75
3.2.8.2 – Perda de massa 76
3.2.8.3 - Microdureza Vickers (HV) 77
3.2.8.4 - Tenacidade a fratura (KIC) 78
3.2.8.5 - Resistência a flexão (σ3p) 78
3.2.8.6 - Análise de fases 79
3.2.8.7 - Análise da microestrutura 79
4 RESULTADOS E DISCUSSÕES 81
4.1 – CARACTERIZAÇÃO DAS MISTURAS DE PÓS E DOS
COMPACTOS A VERDE
81

4.1.1. – Análise granulométrica 81
4.1.2 – Análise por microscopia eletrônica de varredura 82
4.1.3 – Massas específicas teórica, real e densidade a verde dos compactos. 83
4.2 – ESTUDO DA SINTERABILIDADE DAS MISTURAS DE PÓS 84
4.2.1 – Retração linear e densidade relativa das misturas obtidas via
dilatometria
84
4.3 – CARACTERIZAÇÃO DAS AMOSTRAS TRATADAS
TERMICAMENTE
86
4.3.1. – Aquecimento das amostras 87
4.3.1.1. – Densidade relativa 87
4.3.1.2. – Perda de massa 88
4.3.1.3 – Análise por difratometria de raios X 89
4.3.1.4 – Microscopia eletrônica de Varredura 92
4.3.2. – Isoterma de Sinterização das amostras 100
4.3.2.1 – Densidade relativa, perda de massa e retração volumétrica 100
4.3.2.2 – Análise por difratometria de raios X. 103
4.3.2.3 – Análise quantitativa dos principais politipos de SiC. 105
4.3.2.4 – Análise microestrutural das cerâmicas de SiC 108
4.3.2.5 – Propriedades mecânicas das cerâmicas de SiC. 109
4.3.3. – Re-sinterização das misturas analisadas 114
4.3.3.1 – Densidade relativa, perda de massa e retração volumétrica. 114
4.3.3.2 – Análise por difratometria de raios X das cerâmicas de SiC 117
4.3.3.3 – Análise quantitativa dos principais politipos de SiC. 118
4.3.3.4 - Análise microestrutural das cerâmicas de SiC 121
4.3.3.5 - Propriedades mecânicas das cerâmicas de SiC. 122

5 CONCLUSÕES 127
6 PROPOSTA PARA TRABALHOS FUTUROS 128
REFERÊNCIAS 129

20
1 INTRODUÇÃO
Nos últimos anos, o desenvolvimento cientifico e tecnológico permitiu o surgimento
de novas tecnologias e materiais, objetivando a redução de custos. Dentre esses materiais
estão as cerâmicas de carbeto de silício (SiC) que devido as suas excelentes propriedades
apresentam um amplo campo de aplicações, principalmente estrutural [BLOOR et al, 1994,
DRESSLER, RIEDEL, 1997].
O SiC é um composto químico que apresenta forte caráter covalente e
conseqüentemente baixo coeficiente de autodifusão, características que dificultam a obtenção
de cerâmicas densas de SiC por sinterização via fase sólida [DRESSLER, RIEDEL, 1997,
INOMATA, 1991, JEEPS, PAGE, 1983, FREVEL et al, 1992].
Nos últimos anos, cerâmicas densas de SiC vem sendo obtidas por sinterização via
fase líquida pelo uso de pequenas quantidades de aditivos de sinterização, que facilitam os
fenômenos difusionais, diminuindo a porosidade e aumentando desta maneira a densificação
[IZHEVSKYI, 2000 a, SHAFFER, 1969, KNIPPENBERG, 1963, INOMATA et al, 1968].
No entanto, tal processo apresenta desvantagens, como a instabilidade química do SiC
para com o aditivo, formando compostos voláteis que contribuem para a aumento da perda de
massa e diminuição da densificação [INOMATA et al, 1968]. A perda de massa também é
atribuída a evaporação do SiC e dos aditivos. Uma das técnicas normalmente empregadas
para minimizar a perda de massa, é o uso de leito protetor [CHOI et al, 1995, BELMONTE et
al, 1996, BAKLANOVA, 1997].
Mesmo com os recentes avanços obtidos na densificação do SiC pela utilização da
técnica de sinterização por fase líquida, há necessidade de uma avaliação dos resultados
obtidos até o momento. Segundo Izhevskyi e outros [IZHEVSKYI, 2000 a] existe uma

21
limitada quantidade de dados para a sinterização do SiC usando como aditivos AlN e Y2O3, e
mesmo assim esses dados são contraditórios. Visto que, uma avaliação detalhada do
comportamento da fase líquida para com o SiC é de extrema importância na tentativa de se
obter um cerâmico com alta densidade e boas características microestruturais, proporcionando
deste modo excelentes propriedades mecânicas.
Esse trabalho vem de encontro a essa preocupação, ou seja, analisar o comportamento
de diferentes sistemas de aditivos com o SiC durante processo de sinterização, objetivando
desta maneira obter cerâmicas densas de SiC com boas propriedades mecânicas (tenacidade a
fratura, resistência a flexão e microdureza), contribuindo para o desenvolvimento da ciência e
tecnologia da produção e caracterização de cerâmicas de alto desempenho, viabilizando a
utilização do CTR2O3 como aditivo de sinterização para cerâmicas estruturais.
No entanto, para o desenvolvimento deste trabalho foram preparadas misturas de pós
contendo α e/ou β-SiC mais AlN-CTR2O3 e/ou AlN- Y2O3. As características avaliadas
foram: densidade relativa a verde e final, perda de massa, propriedades mecânicas, fases
presentes, microestruturas e análises de falhas, que foram correlacionadas com os resultados
apresentados na literatura.
1.1 OBJETIVOS
O principal objetivo desse trabalho é analisar o comportamento dos diferentes sistemas
de aditivos (AlN/Y2O3 e/ou AlN/CTR2O3) na sinterização via fase líquida do α-SiC e/ou

22
β-SiC, avaliando de maneira direta a influência dos mesmos nas transformações de fases,
propriedades mecânicas e microestruturais desta cerâmica.
Dentre os vários objetivos secundários que estão inseridos na idéia central desse
trabalho, destaque para o estudo e análise da viabilidade da substituição do Y2O3 pelo
CTR2O3 no sentido de obter um cerâmico de alta densidade e boas propriedades mecânicas,
diminuindo o custo das cerâmicas de SiC.
1.2 JUSTIFICATIVAS
Este trabalho foi proposto com objetivo de obter resultados de densidade relativa,
propriedades mecânicas e microestruturais compatíveis aos encontrados pelo próprio aluno
durante desenvolvimento de seu projeto de mestrado, utilizado como base [KELLY, 2000].
Neste projeto, o aluno trabalhou com amostras em formato de pastilhas e com dois diferentes
sistemas de aditivos Al2O3/Y2O3 e AlN/Y2O3, que foram pré misturados e tratados
termicamente antes de serem adicionados as cerâmicas de SiC. Tal procedimento, melhorou
de maneira significativa os resultados do SiC quando comparados com os trabalhos realizados
anteriores ao seu [STRECKER et al, 1999], trabalhos esses realizados de uma cooperação
internacional (Universidade de Aveiro – Portugal e Faenquil – Brasil) iniciado em 1996, e
tendo o seu término em 1999. Pode-se citar os resultados obtidos pelo próprio aluno como:
uma cerâmica de SiC com alta densidade relativa superior a 98 % D.T., perda de massa
inferior a 5 %, dureza de 16 GPa e tenacidade à fratura superior a 5 MPa.m1/2.

23
Para tanto, as cerâmicas de SiC foram aditivadas com os sistemas AlN/Y2O3 e
AlN/CTR2O3. O uso do primeiro sistema (AlN/Y2O3) foi justificado em função dos resultados
obtidos pelo aluno durante seu projeto de mestrado.
Enquanto que a utilização do sistema AlN/CTR2O3 foi explicado pelo fato do mesmo
ter apresentado bons resultados de densificação e propriedades mecânicas durante sinterização
via fase líquida do nitreto de silício (Si3N4), em substituição ao óxido de ítrio (Y2O3)
[SANTOS, 2004].
A influência da estrutura cristalina do pó de partida de SiC na microestrutura do
mesmo foi avaliada com objetivo de se estudar o comportamento das transformações de fases
ocorridas nas diferentes cerâmicas α e/ou β-SiC durante processo de sinterização via fase
líquida.

24
2 REVISÃO BIBLIOGRÁFICA
2.1 Histórico do Carbeto de Sílicio
O famoso químico francês Henri Moissan foi o primeiro a sintetizar o carbeto de
silício (SiC) no século XIX, no entanto foi o americano A G. Acheson quem produziu o
carbeto de silício em larga escala em 1881. Originalmente, Acheson tinha em mente
diferentes idéias, como a produção de diamantes artificiais por recristalização de grafite em
alumínio silicato fundido. Seus esforços lhe proporcionaram poucos frutos, porém foi
recompensado pela descoberta de mais um material versátil, o SiC. Acreditando que este era
um composto de carbono e “corundum” (corundo), ele denominou a nova substância de
“Carborundo” [DRESSLER, RIEDEL, 1997, BLOOR et al, 1994].
2.2 Estabilidade Termodinâmica do Carbeto de Silício
O carbeto de silício não apresenta um ponto de fusão congruente. Em sistemas
fechados a uma pressão total de 1bar, se decompõe a 2830 ± 40 oC em grafite e um líquido
rico em silício, conforme mostrada Figura 2.1. Enquanto que em sistemas abertos, o carbeto
de silício inicia a sua decomposição a aproximadamente a 2300 oC com a formação de silício
gasoso e um resíduo de grafite [BLOOR et al, 1994].

25
Figura 2.1 - Diagrama de fase do sistema silício-carbono a uma pressão total de 1 bar
[BLOOR et al 1994].
2.3 Estrutura e Politipismo do Carbeto de Silício
O carbeto de silício policristalino apresenta duas estruturas cristalinas: α-SiC (fase
hexagonal e/ou romboédrica) e β-SiC (fase cúbica), conforme apresentado mostrado na
Figura 2.2 [DRESSLER; RIEDEL, 1997].
Atualmente mais de 180 politipos são conhecidos, todos representando a fase α-SiC, o
motivo desse polimorfismo é decorrente das pequenas diferenças de energias livres de
formação existentes entre eles. Em virtude dessa característica, a estabilidade termodinâmica
do SiC pode ser influenciada por vários fatores, tais como: teor de impurezas; temperatura;
atmosfera de sinterização; tempo; pressão; entre outros (BLOOR et al,1994, INOMATA,
1991, JEEPS, PAGE, 1983].

26
(a) (b)
Figura 2.2 - Estruturas cristalinas do SiC: (a) β-SiC e (b) α-SiC [DRESSLER, RIEDEL, 1997].
Os politipos de SiC apresentam um parâmetro de rede a ≅ 3,08 Å, enquanto que o
parâmetro de rede “c” varia com a seqüência de empilhamento das camadas de Si – C em
direções cristalinas preferenciais, [111] ou [0001], conforme equação 2.1 [FREVEL et al,
1992, IZHEVSKYI, et al, 2000, RUSKA, GAUKER, 1979].
Zc .52,2= (2.1)
Em que Z = número de SiC por célula unitária. Dentre essa grande variedade de
politipos, somente 3C (fase beta-estrutura cúbica), 4H, 6H (fase alfa – estrutura hexagonal) e
15R (fase alfa – estrutura romboédrica) são estáveis e fáceis de serem encontrados [JEEPS,
PAGE, 1983]. Algumas propriedades cristalográficas dos principais politipos de SiC são
mostrados na Tabela 2.1 [SHAFFER, 1969].
Toda modificação cristalográfica tem estrutura formada por ligações
predominantemente covalentes e tetraédricas na forma de SiC4 ou CSi4. A estabilidade de
cada politipo está associada com a termodinâmica e a cinética de formação, a qual ainda não
está completamente entendida. A análise das fases do SiC é extremamente complicada devido

27
à superposição de reflexões de difração de raios X causada pelo politipismo do SiC. A fase α-
SiC possui três picos principais (politipo 6H) e a fase β-SiC possui um pico principal (111)3C
que sobrepõe um dos picos da fase α. Através de normalização dos picos é possível
quantificar as reflexões das fases α e β presentes na amostra. Sabe-se que a formação de cada
politipo depende das impurezas, da taxa de aquecimento, da temperatura do processamento
cerâmico, ou seja, o processamento determina o politipo formado, e este determina as
propriedades da cerâmica [IZHEVSKYI, 2000].
Tabela 2.1 – Propriedades cristalográficas dos principais politipos de SiC [SHAFFER, 1969].
Parâmetros de rede Tipo de
SiC
Principais
politipos
Estrutura
Cristalina a (Å) c (Å) α (o)
Grupo
espacial
α-SiC
6H
4H
2H
15R
Hexagonal
Hexagonal
Hexagonal
Romboedrico
3,08
3,08
3,08
12,69
15,08
10,05
5,03
-
13o54-1/2’
-
-
-
C6mc
C6mc
P63mc
R3m
β-SiC 3C Cúbico 4,35 - - F43m
Na Figura 2.3, são apresentados os principais politipos de SiC, bem como suas regiões
de estabilidade em função da temperatura, pesquisados por “Kinipperberg” [KNIPPENBERG,
1963] e “Inomata” e co-autores [INOMATA et al, 1968].
Nesta figura, nota-se que o politipo 6H é o mais estável em temperaturas elevadas,
enquanto que o 3C predomina em temperaturas mais baixas. No entanto, levando em
consideração que esses experimentos foram realizados em sistemas relativamente puros; em
sistemas reais o efeito da pequena quantidade de impureza, não estequiometria entre pressões
de vapores de Si e C, entre outros fatores comentados em parágrafos anteriores, contribuem
para existência mesmo em altas temperaturas, de politipos os quais são estáveis em baixas
temperaturas.
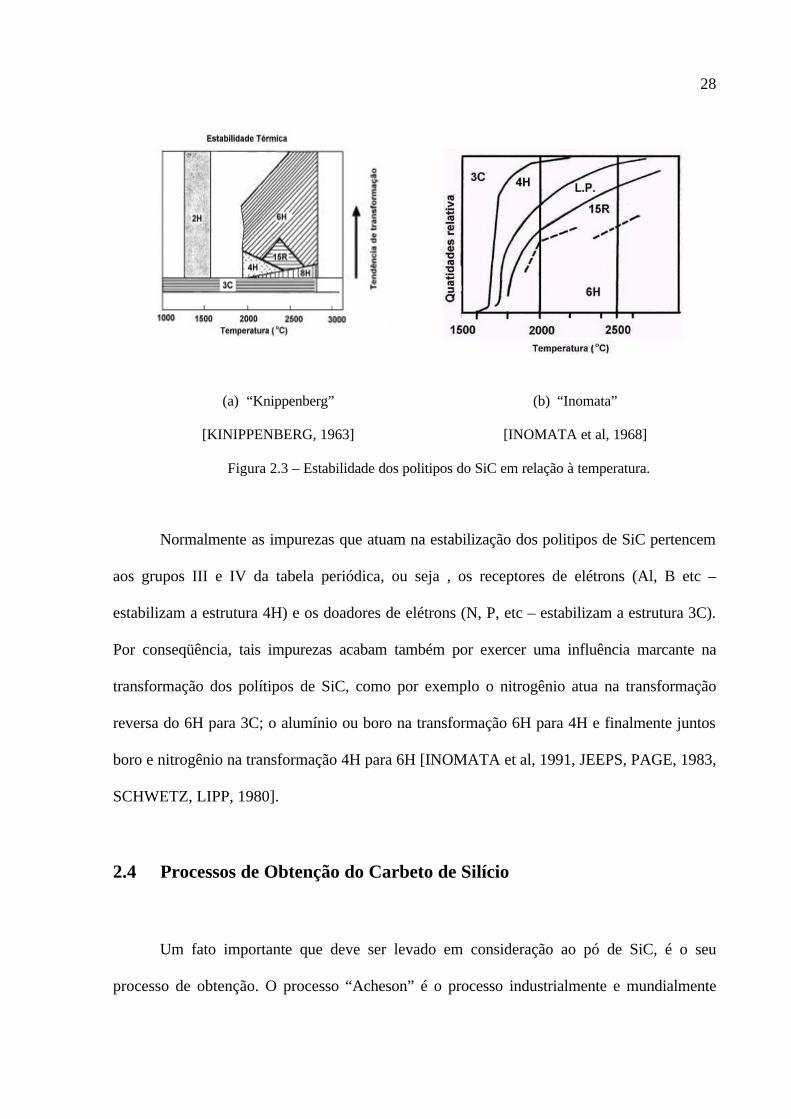
28
(a) “Knippenberg”
[KINIPPENBERG, 1963]
(b) “Inomata”
[INOMATA et al, 1968]
Figura 2.3 – Estabilidade dos politipos do SiC em relação à temperatura.
Normalmente as impurezas que atuam na estabilização dos politipos de SiC pertencem
aos grupos III e IV da tabela periódica, ou seja , os receptores de elétrons (Al, B etc –
estabilizam a estrutura 4H) e os doadores de elétrons (N, P, etc – estabilizam a estrutura 3C).
Por conseqüência, tais impurezas acabam também por exercer uma influência marcante na
transformação dos polítipos de SiC, como por exemplo o nitrogênio atua na transformação
reversa do 6H para 3C; o alumínio ou boro na transformação 6H para 4H e finalmente juntos
boro e nitrogênio na transformação 4H para 6H [INOMATA et al, 1991, JEEPS, PAGE, 1983,
SCHWETZ, LIPP, 1980].
2.4 Processos de Obtenção do Carbeto de Silício
Um fato importante que deve ser levado em consideração ao pó de SiC, é o seu
processo de obtenção. O processo “Acheson” é o processo industrialmente e mundialmente

29
conhecido, utilizado na produção de pós de SiC, sendo que mais de 95% da produção mundial
de SiC é produzido via esse processo.
Nesse processo, o SiC é produzido industrialmente de acordo com a reação 2.1, que
combina quartzo de alta pureza e coque em fornos eletricamente aquecidos [BLOOR et al,
1994, DRESSLER, RIEDEL, 1997]. A mudança de energia livre (∆Go) para a reação 2.1 é
dada pela equação 2.2 [KRSTIC, 1992]:
SiO2 + 3C ⇔ SiC + 2CO (2.1)
∆Go = 609,023 – 0,351T (kJ mol-) (2.2)
A reação é extremamente endotérmica: o calor de formação é 618 kJ.mol-1 SiC,
correspondendo a 4,28 kWh kg-1 SiC. O processo ocorre a temperaturas entre 1600oC a
2500oC e é mais complexa do que a reação 2.1, pois na realidade envolve reações
intermediárias, algumas das quais se dá com a participação de fases sólidas e gasosas,
conforme reações 2.3 – 2.8 [KRISTIC, 1992]:
SiO2 + C → SiO (g) + CO (g) (2.3)
SiO + C → Si (s) + CO (g) (2.4)
Si + C → SiC (s) (2.5)
SiO2 + CO → SiO (g) + CO2 (g) (2.6)
SiO + 2C → SiC (s) + CO (g) (2.7)
2 SiO → Si + SiO2 (2.8)

30
Nos clássicos fornos de “Acheson”, os eletrodos eram posicionados lateralmente
próximos as paredes, e o monóxido de carbono (CO) produzido pelas reações anteriormente
citadas, poluía a atmosfera dentro e fora dos fornos. Na década de 70 tal processo sofreu
algumas alterações recebendo a denominação de “Acheson modificado”, o qual tornou-se
econômico e seguro.
O processo “Acheson modificado” opera com eletrodos fixados ao chão e conectados
diretamente aos condutores de corrente elétrica, conforme mostrada Figura 2.4. Tais eletrodos
entram em contato com a massa a reagir, e o monóxido de carbono produzido é recolhido e
transportado por dutos coletores impermeáveis a gases.
Figura 2.4 - Produção de carbeto de silício pelo processo “Acheson” modificado: cortes transversais
nos sentidos: (a) longitudinal e (b) radial, através do forno [BLOOR et al, 1994].
Os gases recolhidos são convertidos em energia elétrica após queima, essa energia
reciclada representa cerca de 20% da energia necessária para produção de SiC. Os blocos de
SiC localizados dentro dos fornos com resistência em forma linear ou de “U” apresentam
cerca de 50 m de comprimento e pesam aproximadamente 250 toneladas após uma corrida de
10 dias [BLOOR et al, 1994].

31
O SiC resultante do processo “Acheson” (Figura 2.5) apresenta uma granulação
grosseira, e necessita ser moído ao tamanho de partículas desejável e é classificado de acordo
com teor de impurezas e aplicação.
Figura 2.5 - SiC cristalino como recebido a partir do Processo “Acheson”
[DRESSLER, RIEDEL, 1997].
A parte interna do cristal de SiC apresenta uma coloração cinza claro, o qual
representa material mais puro. A quantidade de carbono, alumínio e outras impurezas
aumentam continuamente com relação à distância ao centro do cristal de SiC e é
acompanhada pela mudança da coloração (de cinza claro). Desta maneira para reduzir a
quantidade de resíduos metálicos, o pó de SiC produzido é lavado e lixiviado. Seqüentemente,
o excesso de carbono é oxidado a 400oC e a camada oxida resultante é removida com ácido
fluorídrico [DRESSLER, RIEDEL, 1997].
Nos últimos anos grandes esforços tem sido centrado sobre a síntese de pós-ultrafinos
de SiC (com tamanhos de partículas < 0,1 µm) com a esperança que esse venha aumentar a
cinética de sinterização e conduzir a uma completa densificação sem o uso de aditivos de

32
sinterização [DRESSLER, RIEDEL, 1997, CHOI et al, 1995, BELMONTE et al, 1996,
BAKLANOVA et al, 1997, HASEGAWA et al, 1999].
2.5 Processamento de Cerâmicas de Carbeto de Silíco
A maioria dos materiais cerâmicos clássicos são constituídos principalmente de óxidos,
materiais predominantemente iônicos, em que as ligações iônicas não são direcionais e a
densificação dessas cerâmicas ocorre por difusão volumétrica ou pelo contorno de grão
[GERMAN, 1994, KINGERY et al, 1976]. Ao contrário dessas cerâmicas, o SiC possui maior
quantidade de ligações covalentes, conforme citado anteriormente, fazendo com que esse
composto cerâmico apresente um baixo coeficiente de auto-difusão, dificultando a obtenção
de cerâmicas densas de SiC [GERMAN, 1994, KINGERY et al, 1976, JOHNSON,
PROCHAZKA, 1977].
As cerâmicas de SiC podem ser obtidas utilizando-se diferentes técnicas de
sinterização, tais como: sinterização via fase sólida, prensagem a quente, prensagem
isostática a quente e sinterização via fase líquida. Dentre as técnicas utilizadas, destaque para
sinterização via fase líquida, da qual são obtidas cerâmicas densas de SiC utilizando-se
temperaturas mais baixas de sinterização (1800 – 2000oC), e que também permite um maior
controle da microestrutura e por conseqüência das propriedades mecânicas [NEGITA, 1986,
YE et al, 1999, KIM et al 2002, ZHOU et al, 2003, STRECKER et al, 1999, GERMAN, 1996,
KINGERY, 1957, RAHAMAN, 1995, KELLY, 2000, BISWAS et al 2001].
Na sinterização via fase líquida, a força motriz que comanda todo processo é o excesso
de energia livre superficial, que diminui devido eliminação das interfaces sólido-vapor em

33
detrimento ao surgimento da interface sólido-sólido [GERMAN, 1996, KINGERY, 1957,
RAHAMAN, 1995]. O transporte de massa ocorre por diferentes caminhos:
(a) difusão no estado sólido via rede cristalina, e contorno de grão;
(b) formação de fase líquida, tendo como requisitos básicos, a solubilidade do sólido no
líquido e o molhamento do sólido pelo líquido;
(c) formação de líquido viscoso, mecanismo predominante nos vidros e nas cerâmicas com
alto percentual de fase vítrea.
Os parâmetros relacionados ao comportamento do líquido em relação a matriz
cerâmica são de suma importância durante processo de sinterização via fase líquida. Esses
parâmetros são: molhamento e ângulo diedral, capilaridade e solubilidade.
(a) Molhamento e ângulo diedral
O estudo do balanço de energias interfaciais do sistema sólido-líquido-gasoso
(atmosfera) é responsável pelo fenômeno de molhamento do sólido pelo líquido. A relação
entre superfície e a energia interfacial determina o molhamento de um líquido em uma
superfície sólida Figura 2.6. Se a energia interfacial sólido-líquido (γSL) é alta, o líquido tende
a forma esférica (Figura 2.6-a). Em contraste, se a energia interfacial sólido-vapor (γSV) é alta,
o líquido tende ao espalhamento indefinidamente para eliminar esta interface, como mostra a
Figura 2.6-c. Considerando um desenvolvimento intermediário do estudo das interfaces, tem-
se o molhamento (Figura 2.6-b).

34
Figura 2.6. Fenômeno de molhamento de uma superfície: (a) molhamento desfavorecido (θ >90°); (b)
molhamento (θ < 90°) e (c) espalhamento do líquido sobre a superfície sólida (θ =0°) (d) distribuição
de energias interfaciais [KINGERY, 1976].
O balanço das energias interfaciais num sistema sólido-líquido-gasoso em condições
de equilíbrio é dado pelas equações de “Young” (Eq. 2.3) e “Young-Dupré” (Eq. 2.4)
[GERMAN, 1996, RAHAMAN, 1995], em que θ é o ângulo de contato. Observa-se que
quanto menor o ângulo de contato, maior é o molhamento e o trabalho de adesão do líquido.
θγγγ cos.LVSLSV =− (2.3); ( )θγ cos1+= LVSLAw (2.4),
Em que: γsv = energia interfacial sólido-vapor; γsl = energia interfacial sólido-
líquido; γlv = energia da interface líquido-vapor e SLAw é o trabalho de adesão.
Ainda de acordo com a Figura 2.6 observamos as seguintes condições com relação
aos valores atribuídos ao ângulo de contato (θ): para θ > 90o, a energia da interface sólido-
líquido é intensificada, o líquido existente tende a não molhar totalmente a superfície (Figura
2.6-a); para θ < 90o o líquido molha a superfície (Figura 2.6-b); para θ = 0o tem-se o líquido
espalhando completamente sobre a superfície (Figura 2.6–c). Para valor de θ igual a 90o, neste

35
caso tem-se o limite entre os comportamentos de molhamento e não molhamento [KINGERY,
1976].
Outra característica importante do líquido é o ângulo diedral (φ), que mede a
penetrabilidade do mesmo nos contornos de grão, e é expresso através da Equação 2.5
[GERMAN, 1996, KINGERY, 1957, RAHAMAN, 1995].
).2
arccos(sl
ss
γγ
φ = (2.5)
No caso de um sólido policristalino, podemos dizer que quando ele é imerso em uma
fase líquida ou vapor, o ângulo diedral é determinado pela razão da energia de contorno de
grão com a energia de superfície sólido-líquido ou sólido-vapor, como mostrado na Figura 2.7
[GERMAN, 1996].
Figura 2.7. Ângulo diedral: (a) Contorno de grão em equilíbrio com a fase vapor na superfície; (b)
Contorno de grão em equilíbrio com um líquido [GERMAN, 1996].

36
Na sinterização via fase líquida, quando uma certa quantidade de líquido é formada, a
sua distribuição (penetração) entre os grãos da fase sólida depende do ângulo diedral (φ),
como mostrado na Figura 2.8 [RAHAMAN, 1995].
Figura 2.8. Distribuição de fase líquida para diferentes valores de ângulo diedral numa
junção tripla [RAHAMAN, 1995].
Para que o líquido penetre nos contornos, é necessário que o ângulo diedral seja
pequeno. Para que isto ocorra, a energia interfacial γSL deve ser muito menor que γSS, pois se
a relação γSS/γSL é igual ou maior a 2, φ é igual a zero, e no equilíbrio, as faces de todos os
grãos são envolvidas pela fase líquida, ou seja, há penetração total do líquido nos contornos
de grãos. Se a razão entre γSS/γSL varia entre √3 e 2, φ varia entre 0° e 60° e o líquido forma
uma estrutura contínua ao longo das bordas dos grãos. Para γSS/γSL variando entre 1 e √3, φ
situa-se entre 60° e 120°, a fase líquida envolve parcialmente os grãos, porém não forma
canais longos e contínuos.
A Figura 2.9 apresenta um gráfico que relaciona o ângulo diedral com a razão de
energias γSS/γSL [GERMAN, 1996]. Quando a relação entre as energias intefaciais sólido-
sólido e sólido-líquido for igual a 2, o ângulo diedral será zero, com total penetração do
líquido nos contornos de grão.

37
Figura 2.9. Variação do ângulo diedral em função da relação γSS/γSL [GERMAN, 1996].
Estudos termodinâmicos relacionados à estabilidade da fase vítrea formada a partir da
fase líquida mostram que esta fase vítrea aloja-se preferencialmente nos pontos triplos e
contornos de grãos [GERMAN, 1996, KINGERY, 1957, RAHAMAN, 1995]. Reforçando o
estudo feito anteriormente, conclui-se que a estabilidade da fase líquida em um sistema
depende de fatores como: (1) ângulo diedral, que quando inferior a 600, o líquido é estável e
sua cristalização não é possível; (2) o raio de curvatura da interface fase vítrea-cristal; e (3) a
fração volumétrica do vidro. A Figura 2.10 mostra a geometria de uma junção tripla, como
forma de ilustrar uma junção tripla ideal, para preenchimento da fase vítrea.
(b) Capilaridade
Considera-se capilaridade como sendo a relação de forças na interface sólido-líquido.
As forças de adesão sólido-líquido são maiores que as forças de coesão, em meio poroso,
causada pela tensão superficial e dependem das grandezas relativas de coesão e adesão do
líquido na superfície do sólido ou do poro. Assim há molhamento quando a força de adesão
for maior que a força de coesão, caso contrário, o líquido não molhará a fase sólida. Esta

38
diferença de forças está diretamente relacionada com o tamanho dos poros [GERMAN, 1996,
KINGERY, 1957, RAHAMAN, 1995].
Figura 2.10. Geometria de uma junção tripla com líquido segregado quando θ < π/6 (2θ = ângulo
diedral; r = raio de curvatura e U = raio do canal) [GERMAN, 1996, KINGERY, 1957,
RAHAMAN, 1995].
Quando duas partículas são envolvidas por um líquido, pode-se gerar uma força
atrativa entre elas, conforme mostra Figura 2.11, melhorando o rearranjo e aumentando a
densificação final do compacto.
Figura 2.11. Força atrativa entre duas partículas [GERMAN, 1996].
A Força interpartículas é dada pela Equação 2.6 [GERMAN, 1996].

39
ψγππ cos..2.. 2SLxPxF +∆= (2.6)
Em que: ∆P = Variação da pressão capilar; γSL� = [(1/r1) +(1/r2)]; ψ = �� constante [função
da quantidade de líquido, d (distância entre as partículas) e θ (ângulo de contato)]�
A Figura 2.12 apresenta o efeito do ângulo de contato na força gerada entre duas
partículas. Observa-se que no primeiro caso, o bom molhamento facilita a atuação de forças
capilares atrativas, ao passo que no segundo caso, há ação de forças capilares repulsivas,
resultantes de uma maior dificuldade do líquido em molhar as partículas sólidas.
Figura 2.12. O efeito de dois valores extremos de ângulo de contato na força capilar entre duas
partículas esféricas separadas por um líquido [GERMAN, 1996].
(c) Solubilidade
Com o aquecimento do compacto durante a sinterização por fase líquida, há, no
primeiro estágio de sinterização, a formação de um líquido, e os efeitos da solubilidade
começam a ocorrer. Neste processo, há a solubilidade do sólido no líquido (SB) e a

40
solubilidade do líquido formado no sólido (SA). Deve-se esperar que a solubilidade do sólido
no líquido seja maior, para que a razão de solubilidade (SR = SB /SA.) seja superior a 1, porque
uma baixa razão de solubilidade leva a formação de poros no local onde as partículas de
aditivos se encontravam, já que a solubilidade do líquido no sólido e maior que a do sólido no
líquido. A Figura 2.13 mostra como razões de solubilidades diferentes influenciam na
densificação do material durante a sinterização.
Observa-se que há influência direta da solubilidade nos resultados de sinterização.
Assim se a relação entre solubilidade sólido-líquido e solubilidade do líquido–sólido for
maior que um (SB/SA>1), ou seja, uma maior solubilidade do sólido no líquido, ocorre uma
atração entre as partículas, reduzindo a porosidade. Por outro lado, se SA /SB <1, há maior
solubilidade do líquido no sólido gerando expansão e formação de poros entre as partículas
[RAHAMAN, 1995].
Na sinterização via fase líquida as partículas que se encontram somente em contato
físico devido à prensagem, são ativadas termicamente favorecendo seu rearranjo. Ao atingir a
temperatura de fusão do aditivo, há a formação da fase líquida que possibilita o molhamento
na superfície das partículas sólidas. O líquido formado deve solubilizar o sólido de β-SiC e
quando este estiver saturado, ocorre a precipitação de cristais de α-SiC. A partir desta etapa, a
retração não é tão acentuada e os grãos começam a crescer. São definidos três estágios da
sinterização via fase líquida: rearranjo das partículas, solução-precipitação, como também
coalescência dos grãos.

41
Figura 2.13. Influência da solubilidade nos resultados de sinterização [RAHAMAN, 1995].
Estes estágios são mostrados esquematicamente na Figura 2.14. A sinterização via
fase líquida é bastante complexa, pois vários mecanismos ocorrem simultaneamente,
dificultando o estudo destes separadamente. Não há um ponto definido em que termina um
estágio para começar o outro. A maior taxa de densificação ocorre no estágio de rearranjo e
molhamento [GERMAN, 1996].
1º Estágio (Rearranjo e molhamento): O grau de densificação nesse estágio é
significativamente dependente do tamanho e forma do pó de partida, como também da
quantidade e viscosidade da fase líquida que, após solidificação, é chamada fase intergranular.
Portanto, no ínicio da sinterização, o líquido formado se espalha rapidamente entre as
partículas, promovendo o escorregamento das mesmas. Neste caso tem-se o rearranjo e
reempacotamento das partículas, sendo o rearranjo devido à atração capilar entre as partículas.

42
Figura 2.14. Modelo esquemático da densidade em função do tempo de sinterização representando os
estágios da sinterização via fase líquida [GERMAN, 1996].
À medida que o líquido penetra entre as partículas, ele inicia a sua entrada nos
contornos, promovendo deste modo à fragmentação das partículas, conforme mostra a
Figura 2.15 [GERMAN, 1996].
Figura 2.15. Fragmentação das partículas durante a penetração do líquido
[GERMAN, 1996].

43
Deve-se salientar que os fatores mais importantes a serem considerados durante o
rearranjo são o baixo ângulo de contato entre o líquido e o sólido; um pequeno ângulo diedral;
baixo grau de ligação entre as partículas no estado sólido; e individualidade das partículas,
conforme estudado anteriormente.
2º Estágio (solução e reprecipitação): O estágio de solução-reprecipitação pode
ocorrer simultaneamente ao rearranjo, com o decorrer do tempo ele começa a sobrepor ao
processo de rearranjo. A força principal nesse estágio é a alta solubilidade nos pontos de
contato das partículas causadas pela forças capilares, bem como os gradientes de potenciais
químicos entre as pequenas e as grandes partículas, que leva a um aumento da dissolução das
partículas pequenas. A densificação pode ser acelerada por aplicação de pressões externas,
como no caso de prensagem a quente e prensagem isostática a quente. Durante esta ocorrem
os seguintes fenômenos:
- Achatamento do contato interparticulas: Tensões intergranulares devido a forças capilares,
causam a dissolução do sólido no ponto de contato, com reprecipitação nas regiões deslocadas
dos contatos, promovendo aumento da densificação.
- Dissolução dos grãos finos: A fase líquida torna-se um meio de transporte para os átomos da
fase sólida, onde os grãos pequenos dissolvem-se preferencialmente em relação aos grãos
grandes, pois a solubilidade de um grão varia inversamente com o seu tamanho. Os grãos
pequenos dissolvidos no líquido se precipitam sobre a superfície dos grãos maiores, conforme
apresentado na Figura 2.16 [GERMAN, 1996].
- Coalescência: Crescimento do pescoço intergranular através de difusão sólida promovendo
um engrossamento microestrutural.
3º Estágio (coalescência): O estágio final da sinterização não apresenta uma
apreciável contribuição para a densificação dos corpos cerâmicos, porém um aumento na
densificação pode ser atingido a partir da redução da porosidade fechada, sendo altamente

44
dependente das características dos poros e de alguns gases presentes nestes poros. A etapa de
coalescência requer uma permanência considerável na temperatura de sinterização, pois
promove alterações: na distribuição, tamanho e forma dos grãos e dos poros, influenciando
nas propriedades mecânicas do produto sinterizado [GERMAN, 1996, KINGERY, 1957,
RAHAMAN, 1995]. Neste estágio, os grãos coalescem devido à redução da energia
superficial, e a ação dos mecanismos de sinterização por fase sólida predominam [GERMAN,
1996, KINGERY, 1957, RAHAMAN, 1995].
Figura 2.16- Solução-reprecipitação, com crescimento e acomodação
de forma dos grãos [GERMAN, 1996].
No caso específico do SiC, a etapa de solução–difusão-precipitação é de fundamental
importância, pois pode ocasionar uma mudança microestrutural que modificará as
propriedades mecânicas desta cerâmica. O modelo da evolução microestrutural do SiC é
mostrado nas Figuras 2.17 e 2.18 de acordo com os pós de partida� α-SiC e β-SiC,
respectivamente.
Quando o pó de partida é de α-SiC, o processo de solução-difusão-precipitação ocorre
como mostrado na Figura 2.17 onde não irá ocorrer a transformação de fase beta para alfa, e
os grãos grandes crescem em detrimento dos pequenos.

45
Figura 2.17. Estágios de sinterização via fase líquida usando pó de partida
de α-SiC [XU et al, 2001].
Quando o pó de partida é de β-SiC, ocorre a transformação para a fase polimórfica α-
SiC. No estágio inicial ocorre a formação do líquido, e em seguida os grãos menores de β-SiC
dissolvem neste líquido e reprecipitam na forma de grãos de β/α-SiC, crescendo na orientação
do núcleo, como mostrado na Figura 2.18. As próprias partículas de β-SiC que não
solubilizaram no líquido de aditivo são agentes nucleantes de α-SiC. Então, o resultado da
rede é um compósito de grãos de � β/α-SiC na forma de placas, adquirindo maior razão de
aspecto que no caso da Figura 2.17. Aplicando-se tratamento térmico, todo β-SiC pode se
transformar em α-SiC [DESHPANDE et al, 2001].
Figura 2.18. Estágios de sinterização via fase líquida usando pó de partida
de β-SiC [DESHPANDE et al, 2001].

46
Os aditivos mais utilizados na sinterização do SiC via fase líquida são os óxidos de
alumínio, ítrio e terras raras, ou misturas destes [KIM et al, 2002]. Nos últimos anos, os
óxidos de terras raras (Y, Yb, Dy, Ho, e outros) vem sendo utilizado, na forma de óxidos
separados ou mesmo na forma de óxido misto de ítrio e terras raras (solução sólida dos óxidos
de terras raras) em conjunto com o óxido de silício (SiO2), ou mesmo, nitreto de alumínio
(AlN), na sinterização via fase líquida de cerâmicas de carbeto de silício (SiC) e nitreto de
silício (Si3N4), em substituição ao óxido de ítrio (Y2O3). Apresentando um comportamento
igual ou mesmo superior ao óxido de ítrio em relação a densificação e propriedades
mecânicas destas cerâmicas [BALESTRA et al, 2006, KIM et al, 2002, ZHOU et al, 2001,
MENKE et al, 2000, SANTOS, 2004, VERNILLI et al, 2006].
A respeito da sinterização do SiC obtido via fase líquida, foi observado durante a
revisão bibliográfica que o sistema de aditivos, constituído de uma mistura de pós de
Al2O3/Y2O3 é o mais comumente utilizado [SHE, UENO, 1999, BAUD et al, 2003, MULLA,
KRSTIC, 1991, FALK, 1997]. Recentemente, o AlN vem sendo utilizado na sinterização do
SiC, em substituição ao Al2O3, no sistema Al2O3/Y2O3, produzindo também bons resultados
de densidade relativa e propriedades mecânicas [KIM et al, 2002, ZHOU et al, 2003, FALK,
1997, IZHEVSKYI et al, 2000 b]. Essa substituição é decorrente da maior estabilidade
química do AlN para com o SiC, pois o mesmo durante processo de sinterização forma uma
solução sólida com o SiC [MANDAL et al, 2002, PAN et al, 1999, LEE et al, 1996]. Porém, o
AlN apresenta uma desvantagem quando comparado ao Al2O3, no que diz respeito a
transformação de fase do SiC, pois o mesmo tende a estabilizar a fase cúbica do SiC,
resultando numa microestrutura de grãos equiaxiais [IZHEVSKYI et al, 2000 a].
Ao contrário do AlN, a alumina, Al2O3, facilita o processo de transformação de fase do
SiC, resultando numa microestrutura de grãos alongados, com alta razão de aspecto refletindo
diretamente nas propriedades mecânicas do material [IZHEVSKYI et al, 2001, KIM et al,

47
2002]. A formação de uma fase líquida menos viscosa no sistema Al2O3/Y2O3, quando
comparado ao sistema AlN/Y2O3, pode justificar tal comportamento. Pois a presença de uma
fase líquida com baixa viscosidade facilita o transporte de massa através do líquido e
conseqüente crescimento de grãos [GERMAN, 1996, RAHAMAN, 1995]. Biswas e outros
[BISWAS et al, 2001] verificaram o aumento da temperatura de sinterização do SiC com
aumento do teor de AlN no sistema AlN/Y2O3, devido a provável dissolução ou reação do N2
com a fase líquida formada, anterior a decomposição do AlN em N2 (g) e Al (l). Aumentando
a pressão de N2 e conseqüentemente proporcionando o aumento da viscosidade do líquido.
2.6 Estabilidade química do carbeto de silicio
O processo de sinterização via fase líquida apresenta uma série de vantagens, mas
também tem suas desvantagens, umas delas consiste na não estabilidade química do líquido
para com o SiC, pois ambos podem reagir e produzir uma série de compostos voláteis,
contribuindo para a obtenção de cerâmicas com baixa densificação [Hue ET AL, 1997,
SAMANTA et al, 2001, IHLE et al, 2005]. A perda de massa também pode ser atribuída a
evaporação de SiC e dos aditivos [IHLE et al, 2005].
Os compostos voláteis formados durante o processo de sinterização consistem de
subóxidos a base de alumínio (Al2O e AlO), de ítrio (Y2O e YO), além da presença de CO,
CO2 e SiO [HUE et al, 1997, SAMANTA et al, 2001, IHLE et al, 2005]. Uma das técnicas
empregadas para minimizara a perda de massa do SiC durante sinterização por fase líquida, é
o uso de leito de pó [WINN et al, 1999, BAUD et al, 2003 c].

48
Negita analisou a estabilidade do SiC com vários aditivos na forma de metais puros e
compostos óxidos [NEGITA, 1986]. Eles concluíram que se a energia livre de “Gibbs” de
oxidação do SiC for superior às energias de oxidação do metal puro ou carbeto metálico,
menor será a decomposição do SiC pelos aditivos metálicos ou óxidos metálicos. Essas
reações são descritas a seguir:
2 SiC (s) + O2 (g) → 2 Si (s, l) + 2 CO (g) ( oxidação do SiC) (2.8)
b M (s, l) + O2 (g) → a MvOw (s, l) ( oxidação do metal puro) (2.9)
d MxCY (s, l) + O2 (g) → c MvOw (s, l) + f CO (g) (oxidação do carbeto metálico) (2.10)
Na Figura 2.19, as linhas horizontal e vertical do gráfico mostram a variação de
energia livre de Gibbs para a reação de oxidação do SiC. A área hachurada do gráfico é
caracterizada pela variação de energia livre de Gibbs das reações de oxidação do metal puro e
carbeto metálico. Conclui-se que a utilização de óxidos metálicos como aditivos são
preferenciais aos metais, sendo os óxidos mais estáveis em ordem crescente o: Al2O3, BeO,
HfO2, ThO2, Sm2O3, Y2O3, La2O3, Ce2O3, entre vinte e sete pesquisados, por apresentarem as
mais baixas variações de energia livre a 2127 ºC, Figura 2.19.
Ao passo que Hue e co-autores analisaram a perda de massa de compósitos de Al2O3
reforçadas com “whiskers” de SiCw.[HUE et al, 1997]. Em seus estudos descreveram uma
série de reações que ocorrem entre SiCw e Al2O3 a 2000 K, com suas respectivas energia
livres de formação, as quais são descritas pelas seguintes reações:
SiC (s) + Al2O3 (s) → SiO (g) + Al2O (g) + CO (g) ∆G = 286 KJ.mol-1 (2.11)
SiC (s) + 2 Al2O3 (s) → SiO (g) + 2Al2O2 (g) + CO (g) ∆G = 899,7 KJ.mol-1 (2.12)
4 SiC (s) + 2 Al2O3 (s) → Al4SiC4 (s) + 3 SiO2 (s) ∆G = 487,2 KJ.mol-1 (2.13)
SiC (s) + SiO2(s) → 3 SiO (g) + CO (g) ∆G = 94 KJ.mol-1 (2.14)

49
3 Al2O3 (s) + 2 SiO2 (l) → 3 Al2O3.2SiO2 (s) ∆G = 23,97 KJ.mol-1 (2.15)
2 Al2O3 (s) + 9C (s) → Al4C3 (s) + 6 CO (g) ∆G = 283,7 KJ.mol-1 (2.16)
Al2O3 (s) + 3C (s) → 2Al (g) + 3 CO (g) ∆G = 187,3 KJ.mol-1 (2.17)
2 Al2O3 (s) + 2 SiC (s) → 4 Al (g) + 3 CO (g) + 3 SiO (g) ∆G = 607,2 KJ.mol-1 (2.18)
SiO2 (g) + 2C (s) → SiC (s) + CO (g) ∆G = –77,5 KJ.mol-1 (2.19)
Em seus estudos Hue e co-autores concluíram que todas as reações sólido-líquido
(reação: 2.15) podem promover densificação, não conduzindo a perda de massa.
Normalmente as reações que envolvem a formação de espécies gasosas (reações: 2.11, 2.12,
2.14, 2.16 – 2.19) levam degradação das espécimes sólidas (SiC e Al2O3) proporcionando
desta maneira a degradação química ao invés da densificação.
Figura 2.19. Variação de energia livre de Gibbs de formação da reação (2.9) versus a reação (2.10)
para vários óxidos metálicos a 2127 ºC [NEGITA, 1986].

50
2.7 Aspectos Microestruturais e Propriedades Mecânicas das Cerâmicas
de SiC
Sabemos, que as propriedades dos diferentes materiais estão diretamente relacionados
com suas respectivas microestruturas. Assim como os demais materiais de engenharia, as
cerâmicas em geral, dentre elas as cerâmicas de carbeto de silíco, também não fogem a essa
realidade.
Microestruturas, tipo duplex (compósito de grãos), constituídos de grãos alongados e
grãos equiaxiais, podem ser obtidos mediante um controle microestrutural baseado na
transformação de fase β → α-SiC durante a sinterização via fase líquida. Esse controle
microestrutural pode ocorrer durante a sinterização, ou mesmo, mediante um tratamento
térmico pós-sinterização, ministrando-se vários parâmetros, como: composição dos pós de
partida (α-SiC, β-SiC, ou misturas destes), tipo e composição do sistema de aditivo a ser
utilizado, temperatura, atmosfera, taxa de aquecimento e resfriamento entre outros [NADER
et al, 1999, KIM et al, 1998, KIM et al, 1997, LEE et al, 1996, RIXECKER et al, 2001,
STRECKER et al, 2004]. A finalidade de obter esse tipo de microestrutura é controlar tanto a
tenacidade a fratura quanto a dureza, isto é, obter um material cerâmico com uma boa
tenacidade e um valor de dureza compatível, destinado a uma determinada aplicação
específica.
Conforme comentado em parágrafos anteriores, para se obter uma cerâmica de SiC
com boas propriedades mecânicas, há necessidade de se controlar a microestrutura do mesmo,
a qual está diretamente relacionada a razão de aspecto e que pode ser medida conforme o
desenho esquemático na Figura 2.20 [LEE et al, 2001].

51
Figura 2.20. Definição de espessura, comprimento e razão de aspecto
de grãos de SiC [LEE et al, 2001].
As propriedades mecânicas mais comuns avaliadas num material cerâmico são: dureza
(HV), tenacidade a fratura (KIC), módulo de elasticidade (E) e resistência a flexão (σf -3
pontos ou 4 pontos) [RICHERSON, 1992]. Para um curto prazo de carregamento, a grande
maioria das cerâmicas comporta-se elasticamente, apresentando fratura frágil com pouca ou
nenhuma deformação plástica, conforme mostrado na Figura 2.2.1. Tal comportamento das
cerâmicas pode ser atribuído as ligações covalentes e iônicas que resultam num impedimento
dos movimentos de discordâncias, vistos com bastante clareza nas ligas metálicas
[RICHERSON, 1992].
Quando um material cerâmico é tensionado mecanicamente abaixo da temperatura de
amolecimento da fase intergranular, alguns mecanismos de falhas podem vir a ocorrer, como:
(1) fratura rápida, decorrente da baixa deformação; (2) propagação de trincas sub críticas (tipo
de quebra por corrosão sob tensão do contorno de grão) e (3) fadiga mecânica [PETZOW et
al, 1991]. Em virtude destas características, a tensão de fratura pode ser descrita conforme
Equação 2.7 [ZANOTO, 1991, SANTOS, 2001].
2
1
..21
=
c
E
Yi
f
γσ (2.7)

52
Onde: σf = resistência à fratura, γi = energia específica de fratura, E = módulo de
elasticidade, c = tamanho do defeito crítico, Y = fator dependente da geometria da amostra e
da trinca.
Figura 2.21. Curvas de tensão versus deformação para um material frágil
e um dúctil [RICHERSON, 1992].
A energia específica de fratura (γi) é relacionada com a tenacidade à fratura, pela
equação de Irwin ( EK iIC ..22 γ= ), que substituindo na Equação 2.7 obtém-se a relação da
tenacidade à fratura com a resistência à fratura dada pela Equação 2.8 [ZANOTO, 1991,
SANTOS, 2001]:
2
1
.. cYK fIC σ= (2.8)

53
Observando a Equação 2.8, verifica-se que a resistência à fratura ( fσ ) é controlada
pela energia de fratura (γi), a qual representa a resistência do material contra a propagação de
trincas, e é uma propriedade intrínseca do material sem defeitos (hipotético). Essa resistência
à fratura depende de parâmetros microestruturais, tais como morfologia e tamanho de grãos.
Porém, como indica a quantidade de energia consumida pelo processo de fratura, pode ser
aumentada através de outras fases que dificultam a propagação de trincas.
A dureza é definida como a reação do material ao tipo de tensão imposta e medida por
meio do tamanho da impressão plástica residual deixada na superfície do material. Essa
impressão plástica residual pode apresentar várias geometrias, dependendo dos testes de
dureza empregado, Vickers (piramidal), Knoop (piramidal) e Rockwell (esférico). Cargas até
1 kgf são utilizadas para medidas de microdureza, já para cargas entre 1-5 kgf tem-se a baixa
dureza e para dureza padrão ou real, cargas acima de 5 kgf [MCCOLM, 1990]. Com
indentador piramidal, somente no descarregamento são geradas as tensões, evitando um largo
anel de trincas em volta da superfície [MARCHI, 1999, PONTON, RAWLINGS, 1989,
COOK, 1990, DUSZA, 1992]. É por essa razão que a dureza Vickers é baseada no
comprimento das duas diagonais formadas pelo indentador, e é dada pela equação (2.10)
[MARCHI, 1999]. Neste caso tem-se indentador piramidal com ângulos de 136o, em que 2a é
o comprimento da diagonal da impressão deixada pelo indentador (µm); e P é a carga dada
em (N).
22
464.0
136
322.0
a
P
sena
PH v ==
o (2.10)
Quando as amostras são indentadas, microtrincas são desenvolvidas abaixo do
indentador, chamadas de “trincas Half-Penny”, e na superfície chamadas de “trincas

54
Palmqvist”, conforme ilustrada Figura 2.22. As trincas geradas pelo indentador Vickers são
geralmente mais superficiais, e formadas durante o descarregamento, no contorno entre a zona
elástica (material deformado não permanente), e a plástica (material deformado plasticamente
próximo ao indentador). A interface entre estas duas regiões é a fonte do campo de tensão. No
descarregamento, a tensão é maior que no carregamento, podendo propagar trincas ao longo
da direção do indentador ou paralelo ao plano da superfície. Após a nucleação da trinca ocorre
sua propagação instável, que termina na zona plástica. O comprimento da trinca final é
geralmente maior que a diagonal da indentação [MARCHI, 1999].
Figura 2.22 – Sistemas de trincas (a) “Palmquist”; (b) “Half-Penny” [MARCHI, 1999].
Os cálculos de tenacidade à fratura, baseados nos ensaios de dureza Vickers são
realizados utilizando-se a equação 2.11. O uso desta é justificado, pois as trincas se formam
como resultado de indentação Vickers e se desenvolvem em sistemas do tipo “Half-Penny”,
em que a relação c/a ≥ ou ≈ 2 [PONTON, RAWLINGS, 1989].

55
2/32/1 ..).(016,0 −= bFH
EK IC (2.11)
Em que: KIC = tenacidade à fratura; E = módulo de elasticidade do material; HV =
dureza Vickers; b = tamanho da trinca (metade do comprimento total da trinca, ou seja,
metade de 2c); F = carga aplicada ao corpo de prova.
A resistência mecânica de materiais cerâmicos geralmente é determinada por meio de
ensaios de resistência a flexão, em 3 ou 4 pontos, conforme mostrada Figura 2.23. Os corpos
de prova utilizados para a realização destes ensaios podem exibir seção transversal com
formato circular, quadrado ou retangular, apresentando-se de modo uniforme por todo
comprimento do corpo cerâmico. Os custos para obter esses corpos de prova são menores se
comparados com os utilizados em testes de tração [KELLY, 2000].
(a)
(b)
Figura 2.23 - Determinação da resistência mecânica de corpos cerâmicos em forma de barras
utilizando as técnicas de resistência a flexão em 3 pontos (a) e 4 pontos (b) [KELLY, 2000].
A Tabela 2.2 mostra as Equações 2.12 – 2.15 para cálculo da resistência à flexão (F),
com carregamentos em 3 e 4 pontos e seção de amostra retangular e circular [TAGUCHI,

56
2005]. Fa é a força aplicada para fraturar a amostra, L1 é a distância entre os suportes, L2 é a
distância entre o rolete superior e o inferior no teste de flexão de 3 pontos e a distância dos
dois roletes superiores da aplicação da carga em 4 pontos, b é a largura da amostra retangular,
h é a altura da amostra retangular, e D é o diâmetro da amostra circular. As unidades usuais
são psi (lb/in2), ksi (1000 lb/in2), ou MPa.
Tabela 2.2 - Equações para cálculo da resistência à flexão [TAGUCHI, 2005].
Seção retangular Seção circular
Carregamento em 3
pontos
21
2
3
bh
FaLF =
(2.12)
318
D
FaLF
π=
(2.13)
Carregamento em 4
pontos
( )2
21
2
3
bh
LLFaF
−=
(2.14)
( )3
228
D
LLFaF
π−
=
(2.15)
Os parâmetros mais importantes no controle da determinação da resistência flexão são
o alinhamento da amostra, a taxa de carregamento, e a razão L1/h, que deve ser pelo menos
10:1 [TAGUCHI, 2005]. Método padrão deve ser utilizado para realizar o teste de resistência
a flexão, ou seja, à posição dos roletes e o tamanho da amostra devem estar de acordo com as
normas: JIS 1601 (Japão), MIL STD 1942 (MR) – ASTM C 1161 (Estados Unidos), DIN 51-
110-1 (Alemanha) e B41-104 (França).
Os cálculos de tenacidade a fratura nos ensaios de flexão é determinado utilizando-se
a equação 2.16 [RIBEIRO, 1997].
4/33/18/1 )..().(59,0 pHV
EK IC σ= (2.16)

57
Em que: KIC = tenacidade a fratura; E = módulo de elasticidade do material; HV =
dureza Vickers; σ = tensão de ruptura; p = carga de ruptura.
A tenacidade à fratura de materiais frágeis pode ser determinada com a utilização de
ensaios de indentações Vickers (indentatação e medida de trincas), método ICL (Indentation
Crack Length). Outro método que apresenta maior confiabilidade em relação ao ICL é o IBS
(Indentation Strength Bending), que consiste na indução de trinca e posterior quebra das
amostras. Em ambos os casos são aplicados cargas que geram trincas nas extremidades da
impressão [DUSZA, STEEN, 1999].
Um balanço geral de energia para o processo de impressão Vickers pode ser
formulado onde o trabalho externo aplicado pelo indentador é consumido na deformação e no
processo de fratura do material. O trabalho é convertido em uma componente de energia
superficial, proporcional a área de contato da pirâmide [MARCHI, 1999]. Vários parâmetros
influenciam um ensaio de dureza, como: tamanho e morfologia de grãos; carga aplicada;
superfície e porosidade da amostra [PETZOW et al, 1991, MCCOLM, 1990]. A presença de
falhas tais como: trinca; poros ou inclusões num material cerâmico resultam em
concentradores de tensão, diminuindo a resistência mecânica do material, podendo resultar em
falhas catastróficas [PETZOW et al, 1991].
A equação (2.17) exibe a relação entre os concentradores de tensão na ponta de uma
trinca elíptica e a resistência mecânica num material não dúctil [PETZOW, 1991].
σm/σa = 2 (c/ρ)1/2 (2.17)
σm = tensão máxima na ponta da trinca; σa = tensão aplicada; c = comprimento do maior
eixo da trinca; ρ = raio da ponta da trinca.

58
De maneira geral, a redução da resistência de um material cerâmico é afetada por uma
combinação de fatores, como: formato do poro; a presença de trincas ou contornos de grãos
adjacentes aos poros; distância entre poros ou entre um poro e a superfície; tamanho e formato
de uma inclusão e diferenças entre os módulos de elasticidade e coeficientes de expansão
térmica das inclusões e matriz [ZANOTO, 1991].
No intuito de melhorar a performance das cerâmicas, principalmente a altas
temperaturas, vários mecanismos são utilizados, como: tratamento térmico pós-sinterização
para cristalização (devitrificação) da fase intergranular e melhora da microestrutura; adição de
“whiskers”, os quais atuam como verdadeiros escudos das pontas das trincas, impedindo de
uma certa forma a propagação das mesmas, por meio da absorção de energia da ponta da
trinca, dificultando sua continuidade [PETZOW et al, 1991, DUSZA, STEEN, 1999].
Geralmente os mecanismos mais comuns de absorção de energia e conseqüentemente,
paralisação da propagação da trinca presenciados nas cerâmicas de Si3N4 e SiC são: “pull
out”, onde os grãos de alta razão de aspecto são puxados da matriz provocando uma
transformação de energia da ponta da trinca em energia de superfície, “crack bridging”, onde
a ponta da trinca desvia por baixo ou por cima dos grãos; e “crack deflection”, onde as trincas
desviam pelos contornos de grãos [PETZOW et al, 1991, ZANOTO, 1991, DUSZA, STEEN,
1999]. Para que isso seja realmente efetivo as propriedades das fases das cerâmicas
(composição, quantidade, estrutura cristalina da fase secundária, aliada à morfologia, a razão
de aspecto, ao tamanho e distribuição dos grãos) são fundamentais [CHARLIE, McGILL,
1994].
Uma ferramenta de extrema importância utilizada na engenharia, em especial nos
materiais cerâmicos, com objetivo de determinar se a falha ou dano ocorreu devido a uma
deficiência na forma ou no material é a análise de falha ou fractografia. Tal técnica consiste
em analisar o material fraturado, em especial a região fraturada, num esforço voltado para

59
determinar a causa da fratura. Aspectos presentes na superfície fraturada permitem determinar
a posição em que a fratura iniciou (origem da fratura), a causa do inicio da fratura (impacto,
tensão sobrecarregada, choque térmico, etc) e tensão local aproximada que causou a fratura
[RICHERSON, 1992, ZANOTO, 1991].

60
3 MATERIAIS E MÉTODOS
3.1 MATERIAIS
As caracterizações e procedências dos materiais utilizados para o desenvolvimento
deste trabalho são descritos neste item.
3.1.1 – Alfa-Carbeto de Silício (αα-SiC)
Foi utilizado alfa-Carbeto de Silício (α-SiC), tipo UF-15 (Lonza AG) para execução
deste trabalho, cujas as características químicas e físicas fornecidas pelo fabricante são
apresentadas na Tabela 3.1.
Tabela 3.1 – Caracterização física e química do α-SiC.
Composição Química
(% em massa) Características Físicas
C 39 Ti 0,1 Superfície específica (m2/g) 9 - 11
O 0,81 Na 0,7 Tamanho médio de partículas (µm) 0,7
Si livre 0,12 Mg 0,6 Politipo predominante 6H
Fe 0,04 Ca 0,6
Al 0,05 N 0,9

61
3.1.2 – Beta-Carbeto de Silício (ββ-SiC)
O beta-Carbeto de Silício (β-SiC), tipo GRADE BF-12, utilizado para execução deste
trabalho foi fabricado pela HCST (Alemanha), e as características químicas e físicas
fornecidas pelo fabricante são apresentadas na Tabela 3.2.
Tabela 3.2 - Caracterização física e química do β-SiC.
Composição Química Características Físicas
C 30,33 (% em massa) Superfície específica (m2/g) 11,52
O 1,20 (% em massa) Tamanho médio de partículas (µm) 0,73
Si-livre < 0,10 (% em massa) α-SiC (%) 4,4
Fe 450 (ppm) β-SiC (%) 95,6
Al 290 (ppm)
Ca 20 (ppm)
3.1.3. - Nitreto de Alumínio (AlN)
O nitreto de alumínio (AlN), tipo GRADE FINE, utilizado para execução deste
trabalho foi fabricado pela HCST (Alemanha), e as características químicas e físicas
fornecidas pelo fabricante são apresentadas na Tabela 3.3.
Tabela 3.3 – Caracterização física e química do AlN.
Composição Química
(% em massa) Características Físicas
C 0,10 máx Superfície específica (m2/g) 4 - 8
O 2,50 máx Tamanho médio de partículas 98 % < 8 µm
N 29,50 máx
Fe 0,005 máx
Outras imprurezas
metálicas
0,01 máx

62
3.1.4 – Óxido de Ítrio (Y2O3)
O óxido de ítrio (Y2O3), tipo FINE, utilizado para execução deste trabalho foi
fabricado pela HCST (Alemanha), e as características químicas e físicas fornecidas pelo
fabricante são apresentadas na Tabela 3.4.
Tabela 3.4 – Caracterização física e química do Y2O3.
Composição Química
(ppm) Características Físicas
Al < 2 Superfície específica (m2/g) 12,76
C < 0,5 Tamanho médio de partículas (µm) 0,8
Fe < 15
Y2O3 (%) 99,98
3.1.5 – Óxido Misto de Ítrio e Terras Raras (CTR2O3)
O óxido misto de ítrio e terras raras, utilizado neste trabalho, foi produzido na
EEL/USP, a partir do minério “Xenotima”.
Algumas propriedades físicas, a análise química quantitativa e composição global do
CTR2O3 são mostrados na Tabela 3.5 [SANTOS, 2004, VERNILLI et al, 2006], observando
como principais óxidos: Y2O3, Yb2O3, Er2O3 e Dy2O3, porém com predominância do Y2O3.

63
Tabela 3.5 - Composição Química do CTR2O3, determinada por Espectrometria de Emissão Ótica com
Plasma Induzido (ICP-AES), e suas propriedades físicas [SANTOS, 2004, VERNILLI et al, 2006].
Composição Química (% massa)
Y2O3 43,90 Pr2O3 0,27
Yb2O3 17,00 CeO2 0,18
Er2O3 13,60 Nd2O3 0,18
Dy2O3 11,00 Eu2O3 0,06
Ho2O3 3,10 La2O3 0,04
Tm2O3 2,50 ZrO2 1,06
Tb2O3 2,41 SiO2 0,09
Lu2O3 2,00 Fe2O3 <lim.detecção
Gd2O3 1,73 Insolúveis 0,56
Sm2O3 0,41
Características Físicas
Superfície específica (m2/g) 8,0
Tamanho médio de partículas (µm) 1,89
Massa específica (g/cm3) 6,51
3.1.6 – Álcool isopropílico (C3H8O)
Foi utilizado álcool isopropílico tipo p.a. fornecido pela ICIBRA.
3.1.7 – Argônio (Ar)
Foi utilizado argônio gasoso tipo B50, fornecido pela White Martins, com pureza
mínima de 99,999 %.

64
3.2 – MÉTODOS
As etapas realizadas durante desenvolvimento deste trabalho, são descritas nesse item,
e resumidas esquematicamente na Figura 3.1.
3.2.1 – Preparo dos aditivos
Normalmente os aditivos são adicionados ao SiC de forma tradicional, ou seja, mistura
direta dos componentes. Nesse trabalho foi realizada uma pré mistura dos sistemas de aditivos
AlN/Y2O3 e AlN/CTR2O3, com a finalidade de melhorar a homogeneidade da mistura final.
Figura 3.1 - Fluxograma ilustrativo do procedimento experimental e caracterização adotados neste
trabalho.
CTR2O3
AlN
Y2O3
AlN
Mistura eMoagem
Mistura eMoagem
Moinho de atriçãoÁlcool isopropílico
1000 rpm/1h
SecagemAlN/CTR2O3
AlN/Y2O3
Pesagem
αα-SiC e/ou ββ-SiC
Moinho de atriçãoÁlcool isopropílico
1000 rpm/10h
Mistura emoagemSecagem
Tamanho e distribuiçãode partículasMEVDifração de raios X
Prensagem
Dilatometria
Aquecimento
Pastilhas cilíndricasTaxa de aquecimento: 20oC/minIsoterma: 0hRetração e velocidade de retraçãoversus temperatura
Pastilhas cilíndricasTaxa de aquecimento: 20oC/minTemperatura máxima: 1600, 1800,1900 e 2000oC.Isoterma: 0h.
CaracterizaçãoPerda de massaDensidadeMEVDifração de raios X.
Isoterma desinterização
Barras (120x25x12,5 mm)Taxa de aquecimento: 20oC/minTemperatura intermediária: 1800oCIsoterma: 0,5 e 1h.Temperatura máxima: 2000oCIsoterma: 1h
Caracterização
Perda de massaDensidadeMEVDifração de raios XPropriedades Mecânicas
Re-sinterização
Barras (120x25x12,5 mm)Taxa de aquecimento: 20oC/minTemperatura intermediária: 1800oCIsoterma: 1h.Temperatura máxima: 2000oCIsoterma: 3h e 6h
Caracterização
Perda de massaDensidadeMEVDifração de raios XPropriedades Mecânicas
CTR2O3
AlN
Y2O3
AlN
Mistura eMoagem
Mistura eMoagem
CTR2O3
AlN
Y2O3
AlN
Mistura eMoagem
Mistura eMoagem
Moinho de atriçãoÁlcool isopropílico
1000 rpm/1h
SecagemAlN/CTR2O3
AlN/Y2O3
Pesagem
αα-SiC e/ou ββ-SiC
Moinho de atriçãoÁlcool isopropílico
1000 rpm/10h
Mistura emoagemSecagem
Tamanho e distribuiçãode partículasMEVDifração de raios X
Prensagem
Dilatometria
Aquecimento
Pastilhas cilíndricasTaxa de aquecimento: 20oC/minIsoterma: 0hRetração e velocidade de retraçãoversus temperatura
Pastilhas cilíndricasTaxa de aquecimento: 20oC/minTemperatura máxima: 1600, 1800,1900 e 2000oC.Isoterma: 0h.
CaracterizaçãoPerda de massaDensidadeMEVDifração de raios X.
Isoterma desinterização
Barras (120x25x12,5 mm)Taxa de aquecimento: 20oC/minTemperatura intermediária: 1800oCIsoterma: 0,5 e 1h.Temperatura máxima: 2000oCIsoterma: 1h
Caracterização
Perda de massaDensidadeMEVDifração de raios XPropriedades Mecânicas
Caracterização
Perda de massaDensidadeMEVDifração de raios XPropriedades Mecânicas
Re-sinterização
Barras (120x25x12,5 mm)Taxa de aquecimento: 20oC/minTemperatura intermediária: 1800oCIsoterma: 1h.Temperatura máxima: 2000oCIsoterma: 3h e 6h
Caracterização
Perda de massaDensidadeMEVDifração de raios XPropriedades Mecânicas
Caracterização
Perda de massaDensidadeMEVDifração de raios XPropriedades Mecânicas

65
Os sistemas de aditivos foram moídos e homogeneizados em moinho de atrito em
meio álcool isopropílico a 1000 rpm durante 1 h, secos em evaporador rotativo a 80oC e
passados em peneira ABNT 40. Foi utilizada uma proporção molar entre AlN e Y2O3 igual a
3:2, correspondente a composição do menor eutético, conforme mostrado Figura 3.2
[KELLY, 2000]. A critério de comparação e também devido a ausência de dados sobre as
relações de fases entre AlN e CTR2O3, a mesma proporção molar foi escolhida para
AlN/CTR2O3 (3:2).
O óxido misto de ítrio e terras raras foi utilizado pela primeira vez num trabalho de
tese de doutorado, no DEMAR/EEL - USP, como aditivo de sinterização do SiC. Sendo que o
mesmo já havia sido utilizado com sucesso como aditivo de sinterização do nitreto de silício,
Si3N4, em trabalhos de dissertação de mestrado e tese de doutorado, no mesmo departamento
[KELLY, 2000].
Nesse trabalho, foram analisadas e comparadas as influências dos aditivos CTR2O3 e
Y2O3 quanto às propriedades físicas, microestruturais e mecânicas dos produtos finais
(cerâmicas de α-SiC e β-SiC).
Figura 3.2 - Diagrama de fase do sistema AlN/Y2O3 [KELLY, 2000].

66
3.2.2 – Preparo das misturas
Dentre os mais variados sistemas de aditivos utilizados na sinterização do SiC, o
sistema Al2O3/Y2O3 é o mais comumente utilizado, portanto, recentemente o AlN vem sendo
utilizado na sinterização do SiC em substituição ao Al2O3, no sistema Al2O3/Y2O3,
produzindo também bons resultados de densidade relativa e propriedades mecânicas. Essa
substituição é decorrente da maior estabilidade química do AlN para com o SiC, pois durante
processo de sinterização forma uma solução sólida com o SiC.
Após moagem e homogeneização, os sistemas de aditivos, AlN/Y2O3 e AlN/CTR2O3,
foram adicionados às cerâmicas de α e/ou β-SiC, na quantidade de 10 % em volume,
utilizando o mesmo procedimento de preparação da mistura de aditivo, com tempo de
moagem igual a 10h. Essa quantidade de aditivo utilizada, em relação ao sistema AlN/Y2O3 é
justificado pelos bons resultados obtidos pelo próprio aluno em sua dissertação de mestrado.
Enquanto que, para o sistema AlN/CTR2O3 essa quantidade se justifica como critério de
comparação aos resultados encontrados no sistema AlN/Y2O3.
As quantidades volumétricas da mistura, bem como as suas designações e composições
mássicas estão representadas na Tabela 3.6.
Tabela 3.6 - Composição das misturas usadas no trabalho.
Composição (% em massa) Designação das
misturas αα-SiC ββ-SiC AlN Y2O3 CTR2O3
N10-α 85,88 - 1,52 12,60 -
T10-α 83,05 - 1,83 - 15,12
N10-β - 85,88 1,52 12,6 -
T10-β - 83,05 1,83 - 15,12

67
3.2.3 – Caracterização das misturas de pós
3.2.3.1 - Massa específica teórica
A massa específica teórica das misturas de pós foi determinada utilizando-se as
informações contidas na Tabela 3.7 [KELLY, 2000, SANTOS, 2004] as quais foram
aplicadas a equação 3.1 [SANTOS, 2004].
Tabela 3.7 - Massas específicas utilizadas para os cálculos [SANTOS, 2004].
Material αα- � SiC ββ-� SiC AlN Y2O3 CTR2O3
Massa específica (g/cm 3) 3,21 3,20 3,26 5,01 6,49
)/()/(
)(
BBAA
BA
T
Tt WW
WW
V
W
ρρρ
++
== (3.1)
Onde:
WT = massa teórica da mistura [ g ]
VT = volume teórico da mistura [ cm3 ]
WA = massa do α e/ou β-SiC [ g ]
WB = massa do aditivo [ g ]
ρA = massa específica do α e/ou β-SiC [ g/cm3 ]
ρB = massa específica do aditivo [ g/cm3 ]

68
3.2.3.2 - Distribuição do tamanho de partículas
Para verificar o tipo de distribuição, tamanho médio e variação do tamanho de
partículas foram realizadas as análises de distribuição granulométricas. Após preparação das
misturas em moinho de atrito e secagem em evaporador rotativo e estufa, as análises de
distribuição do tamanho de partículas foram realizadas num equipamento da marca
“Sympatec Helos” (Universidade de Karlsruhe – Alemanha), utilizando-se o método de
espalhamento de luz laser.
Para a realização desta análise, as diferentes misturas de pós foram desaglomeradas
em ultra-som durante 3 minutos, utilizando como dispersante metafosfato de sódio (3 gotas
em cada teste).
3.2.3.3 – Microscopia eletrônica de varredura das misturas de pós
A homogeneidade das misturas de pós, ou seja, a distribuição das partículas de
aditivos em relação às de SiC e também o formato destas partículas foram analisados
mediante microscopia eletrônica de varredura, utilizando-se de elétrons retroespalhados para
obtenção das imagens. Portanto foram preparadas suspensões de pó, com diluição de 0,05g do
pó por 10ml de acetona, sem adição de agente dispersante. Cada suspensão foi deixada por 25
minutos em ultra-som, em seguida depositada em porta amostra para evaporação do solvente.
3.2.6.4 - Análise de fases
A realização das análises de difração de raios X nas misturas de pós como preparadas
teve por objetivos, verificar as fases inicialmente presentes nas diferentes misturas e compará-

69
las às alterações ocorridas com as mesmas durante as etapas de tratamento térmico realizadas
num forno de grafite.
Por intermédio de um difratômetro, empregando-se uma radiação Cu-Kα com tubo
emissor de filamento de cobre (λ=1,54439 Å), num intervalo de 10 a 80o, com tempo de
interação igual a 1 segundo e passo de 0,05o, em conjunto com fichas de identificação
compiladas pela JCPDS - International Centre for Diffraction Data, foi possível detectar e
analisar as fases presentes nas diferentes misturas de pós. As condições de execução das
análises foram similares para todas composições, utilizando-se um difratômetro de raios X da
Rich Seiferst & Co, mod. Iso-DEBYEFLEX 1001 Co (EEL - USP).
3.2.4 – Ensaios de Dilatometria das misturas de pós
Para a realização desta etapa, as amostras N10-α, N10-β, T10-α e T10-β em forma de
pastilhas foram aquecidas a uma taxa de 20 oC/min até temperatura máxima de 2000 oC, sem
isoterma de sinterização. Os ensaios foram realizados sob fluxo de argônio em dilatômetro
tipo DIL 402 E/7 - Netzsch (IPEN-USP). Com posse dos resultados de dilatometria, as curvas
de retração linear e velocidade de retração foram plotadas em função da temperatura, cujas
informações foram analisadas e subseqüentemente aplicadas nas etapas realizadas no forno
com resistência de grafite.
As curvas de retração linear foram plotadas com os dados fornecidos pelo próprio
equipamento, o qual baseia-se na equação (3.1) [RIBEIRO, 1997]
x100LÄL
x100L
LLdl
oo
o=
−= (3.1)

70
Em que: L = comprimento final [mm] e Lo = comprimento inicial [mm]. As curvas de
velocidade de retração foram plotadas utilizando os dados de retração linear e um programa
computacional, o qual permitiu realizar a derivada desses dados. Enquanto que as curvas de
densidade relativa foram obtidas utilizando os dados de retração linear fornecidos pelo
equipamento e as equações 3.2 e 3.3 [RIBEIRO, 1997], respectivamente:
3]LoÄL
[1
grñ
relñ
−= (3.2)
Em que: ρrel = massa específica relativa [g/cm3]; ρgr = massa específica a verde
[g/cm3]; ∆L/Lo = retração linear.
Drel = (ρrel/ρteórico) x 100 (3.3)
Em que: Drel = % D.T.; ρrel = massa específica relativa [g/cm3]; ρteórico = massa
específica teórica [g/cm3].
3.2.5 – Compactação das misturas de pós
A compactação das misturas de pós para sinterização no forno com elemento resistivo
de grafite foi realizada em dois estágios. No 1o estágio, as amostras foram prensadas
uniaxialmente a 30 MPa, em prensa da SCHUMS, tipo Mex PH515t (DEMAR-FAENQUIL).
A prensagem uniaxial foi realizada em matrizes com diferentes dimensões, ou seja, para
realização das etapas de aquecimento, isoterma de sinterização e tratamento térmico pós-
sinterização. As matrizes foram confeccionadas de aço VC 130 temperadas e retificadas. As

71
amostras em forma cilíndricas prensadas em matriz com dimensões de 10 mm de diâmetro e 2
mm de altura foram utilizadas na etapa de aquecimento. Enquanto que, as amostras em forma
de barras foram prensadas em matriz com dimensões de 120 mm de comprimento e 25 mm de
largura e utilizadas na etapa de isoterma de sinterização e tratamento térmico pós-sinterização.
Anterior a compactação, as paredes das matrizes foram lubrificadas com estearina
dissolvida em querosene, com objetivo de impedir ou diminuir o atrito das mesmas com o pó
compactado. No segundo estágio de compactação, as amostras prensadas uniaxialmente foram
acondicionadas num invólucro de borracha flexível, submetido a vácuo primário e prensadas
isostaticamente a frio (AMR-CTA), utilizando pressão de 300 MPa. Durante prensagem
isostática, as amostras ficaram imersas em banho de óleo, permitindo que a pressão fosse
distribuída igualmente na amostra.
3.2.6 – Tratamento térmico das amostras
Essa etapa teve como objetivo analisar e avaliar um conjunto de informações oriundas
das diferentes caracterizações, visando obter uma cerâmicas de SiC com boas características
microestruturais e mecânicas. O tratamento térmico englobou as etapas de aquecimento,
isoterma e re-sinterização, todas realizadas num forno de resistência de grafite, tipo 1000-
4560-FP20 da Thermal Tecnology (EEL - USP).
3.2.6.1 - Aquecimento das amostras
O objetivo desta etapa foi estudar de modo detalhado, as regiões próximas das
inflexões ou alterações no comportamento das curvas de dilatometria. Portanto, com base nos

72
resultados das análises de dilatometria, as amostras foram sinterizadas nas temperaturas de
1600 oC, 1800 oC, 1900 oC e 2000 oC, sem isoterma.
Anterior as sinterizações, as amostras em forma de pastilhas foram acondicionadas em
cadinho de grafite e envolvidas em leito de pó constituído de 70 % em massa de SiC + 30 %
em massa de mistura de aditivos (AlN/Y2O3 e/ou AlN/CTR2O3). Essa técnica denominada de
“powder bed” tem como objetivo diminuir a perda de massa das amostras, que ocorre devido
a formação de compostos voláteis oriundos da reação do SiC com aditivos. A perda de massa
também pode ser atribuída a vaporização do SiC mais aditivos.
Por meio das modificações físicas e microestruturais ocorridas durante o
desenvolvimento desta etapa, um conjunto de dados foram extraídos e explorados nas etapas
subseqüentes.
3.2.6.2 – Isoterma de sinterização
Nessa etapa as informações extraídas durante processo de aquecimento das amostras
foram analisadas no sentido de levantar ciclos de sinterização para obter uma cerâmica de SiC
com alta densidade, baixa perda de massa e conseqüentemente boas propriedades mecânicas.
Na Tabela 3.8 são mostrados de modo esquemático os ciclos de sinterizações
utilizados durante desenvolvimento desta etapa. Anterior as sinterizações, as amostras em
forma de barras foram acondicionadas em cadinho de grafite e envolvidas em leito de pó
constituído de 70 % em peso de SiC + 30 % em peso de mistura de aditivos (AlN/Y2O3 e/ou
AlN/CTR2O3). Dentre os vários ciclos de sinterização analisados, o que permitiu obter uma
cerâmica de SiC com características desejáveis, foi o ciclo 3.

73
Tabela 3.8 - Ciclos de sinterização para isoterma.
Ciclo 1 Ciclo 2 Ciclo 3
T. amb – 1800 oC (20 oC/min)
1800 – 2000 oC (10 oC/min)
2000 oC/1h
2000 oC – T. amb (20 oC/ min)
T. amb – 1800 oC (20oC/min)
1800 oC/0,5h
1800 - 2000oC (10 oC/min)
2000 oC/1h
2000 oC – T .amb. (20oC/min)
T. amb – 1800 oC (20oC/min)
1800 oC/1h
1800 - 2000oC (10 oC/min)
2000 oC/1h
2000 oC – T .amb. (20oC/min)
3.2.6.3 – Re-sinterização
A re-sinterização foi realizada utilizando-se as amostras obtidas no ciclo 3 da etapa de
isoterma, variando-se o tempo de sinterização (3h e 6h). O objetivo desta etapa é analisar a
influência do tempo de isoterma na evolução microestrutural e propriedades mecânicas do
SiC.
Anterior às sinterizações, as amostras em forma de barras foram acondicionadas em
cadinho de grafite e envolvidas em leito de pó constituído de 70 % em peso de SiC + 30 % em
peso de mistura de aditivos (AlN/Y2O3 e/ou AlN/CTR2O3). Os ciclos de sinterização
utilizados durante essa etapa podem ser visualizados na Tabela 3.9.
3.2.7 – Caracterização dos compactos a verde
3.2.7.1 - Massa específica real
A massa específica real foi determinada pelo método geométrico, a partir da pesagem
das amostras em formato de pastilhas cilíndricas. As amostras foram medidas com

74
micrômetro de precisão de 0,001 mm, e posteriormente pesadas em balança analítica de
precisão (10–6 g), Sartorius Werke GMBH (Demar-Faenquil). Para obtenção de uma maior
precisão, foram realizadas 15 medições de cada amostra para se obter valores médios. A partir
destes dados utilizou-se a equação 3.2 [KELLY, 2000, SANTOS, 2004] para obter o valor da
massa específica real das amostras.
volume
massaamostrav =ρ [g/cm3] (3.2)
3.2.7.2 - Densidade relativa a verde
A partir da equação 3.3 [KELLY, 2000], obteve-se a massa específica teórica,
utilizada posteriormente para cálculo da densidade relativa a verde, que por sua vez, é obtida a
partir da relação entre as massas específicas a verde (real) das amostras e as massas
específicas teóricas das misturas.
100xdt
rverde ρ
ρ= (3.3)
Onde:
ρr = massa específica real [g/cm3]
ρt = massa específica teórica [g/cm3]

75
3.2.8 – Caracterização das amostras tratadas termicamente
As amostras provenientes das etapas realizadas no forno de grafite, ou seja,
aquecimento, isoterma e re-sinterização, foram caracterizadas, conforme propriedades
descritas a seguir:
3.2.8.1 - Massa específica e densidade relativa
A massa específica aparente das amostras em formato de pastilhas cilíndricas (Etapa
de aquecimento) e barras (Etapas de isoterma e tratamento pós-sinterização), foram
determinadas utilizando-se o método geométrico (amostras porosas, equação 3.2) [KELLY,
2000] e princípio de Arquimedes (amostras densas, equação 3.3) [SANTOS, 2004].
A determinação destas propriedades nas amostras porosas foi realizada de acordo com
procedimento descrito no item 3.2.7. Enquanto que, nas amostras densas tais propriedades
foram determinadas seguindo-se a norma ASTM C20-87 [SANTOS, 2004], e um número
mínimo de 10 medições em balança de precisão (10 –5 g).
)( 21
1 2
f
OHSINT WWW
W
−−
×=
ρρ (3.3)
Onde:
ρSint = Massa específica das amostras sinterizadas [g/cm3]
W1 = massa da amostra seca [g]
ρH2O = massa específica da água a 20 0C [g/cm3]
W2 = massa da amostra imersa [g]
WF = massa do fio imerso [g]

76
O valor da massa específica da água ( ρH2O ) foi obtido utilizando a equação 3.4
[KELLY, 2000, SANTOS, 2004].
TOH .0002315,00017,12
−=ρ (3.4)
A densidade relativa foi calculada pela relação entre a massa específica da cerâmica
sinterizada (ρSint) e a massa específica teórica de cada composição estudada (ρT), como
mostrado na equação 3.5 [SANTOS, 2004].
100)( intRe ×=
T
sld
ρρ
[%] (3.5)
ρsint = massa específica do material sinterizado [g/cm3]
ρT = massa específica teórica [g/cm3]
3.2.8.2 – Perda de massa
A perda de massa das amostras sinterizadas foi determinada pela diferença de massa
entre a inicial (verde) e final (após tratamento térmico), conforme mostrado equação 3.6
[KELLY, 2000].
P.M. = [(mi – mf)/mi]x100 [%] (3.6)
onde:
P.M. = perda de massa [%]

77
mi = massa inicial da amostra (verde) [g ]
mf = massa final da amostra (após tratamento térmico) [g]
3.2.8.3 - Microdureza Vickers (HV)
A técnica de impressão Vickers é normalmente empregada para a determinação da
dureza superficial em materiais cerâmicos, e baseia-se no tamanho da deformação permanente
ou elástica provocada no material pelo penetrador piramidal de diamante, com seção
quadrada. A microdureza Vickers foi determinada utilizando-se a equação 2.10 (revisão
bibliográfica).
Os valores obtidos de microdureza Vickers em kgf/mm2 foram convertidos para GPa,
utilizando a equação 3.7 [SANTOS, 2004].
1 GPa = 1,02 x 10-2 kgf/mm2 (3.7)
Antes da realização dos ensaios de microdureza Vickers, uma das superfícies planas
das amostras em formato de barras (Etapas: isoterma e pós-sinterização) foi preparada
utilizando-se a seguinte seqüência de lixas: 74, 40 e 20 µm; e panos de polimento: 15, 9, 6, 3
e 1 µm. A preparação ceramografica das amostras foi realizada numa politriz automática,
marca IGAN WURTZ, mod. Phoenix 4000 (Demar-Faenquil).
Os ensaios de microdureza Vickers foram realizados num microdurômetro tipo
Micromet 2004, da BUEHLER (EEL - USP). As amostras foram submetidas à diferentes
cargas e tempos de indentação, e em cada condição foram realizadas um número mínimo de
20 medições, objetivando obter um valor médio com pequeno desvio padrão. A variação das

78
condições dos ensaios de microdureza teve como finalidade obter uma carga e tempo ideal de
indentação.
3.2.8.4 - Tenacidade à fratura (KIC)
A tenacidade a fratura das amostras sinterizadas foram determinadas por diferentes
métodos, ou seja, da impressão Vickers ou “ICL” (Indentation Crack Length), e flexão ou
“ISB” (Indentation Strength Bending), com objetivo de comparar e avaliar os resultados e a
confiabilidade dos mesmos.
Para determinar os valores de tenacidade a fratura por meio do método de impressão
Vickers ou “ICL” foi utilizada a equação 2.11 (revisão bibliográfica).
Enquanto que, a determinação da mesma propriedade mecânica pelo método da flexão
ou “ISB” foi realizada utilizando-se da equação 2.16 (revisão bibliográfica). Para realização
deste método foram utilizadas amostras em formato de barras. Tais amostras foram retificadas
por rebolos diamantados, utilizando uma retificadora com regulagem de avanço micrométrico,
marca Yadoya, mod. RMG-320 (EEL – USP).
3.2.8.5 - Resistência a flexão (σσ3p)
Os testes de flexão foram realizados em máquina universal (EEL – USP), com
dispositivo de espaçamentos de 20/10, velocidade de carregamento igual a 0,5 mm/min, à
temperatura ambiente. Anterior aos ensaios de flexão as amostras com dimensões de 3x4x40
mm3 tiveram umas de suas faces planas lixadas e polidas, utilizando-se a seguinte seqüência
de lixas: 74, 40 e 20 µm e panos de polimentos: 15, 9, 6, 3 e 1 µm. Em cada condição de
sinterização, foram utilizadas no mínimo um total de 10 amostras.

79
A resistência à flexão das amostras foi determinada utilizando-se da equação 2.12
(revisão bibliográfica).
3.2.8.6 - Análise de fases
As análises de fases das amostras tratadas termicamente (amostras densas e porosas)
foram realizadas por difração de raios X, empregando radiação Cu-Kα, no intervalo de 10 -
80o, com tempo de interação igual a 1 segundo e passo de 0,05o, num difratômetro da Rich
Seiferst & Co, mod. Iso-DEBYEFLEX 1001 Co (EEL - USP). Os resultados desta análise
permitiram avaliar as fases secundárias e cristalinas do SiC, dando ênfase aos principais
politipos de SiC (3C, 4H, 6H e 15R). Tais politipos foram determinados de maneira
quantitativa, utilizando um programa computacional, que tem como base o método de
Rietveld, utilizado para refinar parâmetros de rede de diferentes estruturas cristalinas. Este
programa foi desenvolvido por Ruska et al (1979). Neste caso, as intensidades dos picos dos
principais politipos de SiC nas posições 33.7, 34.2, 34.9, 35.7, 38.2 e 41.5o são determinadas
e computadas no programa computacional.
3.2.8.7 - Análise da microestrutura
As análises das microestruturas das amostras tratadas termicamente foram realizadas
com objetivo de avaliar e comparar o comportamento das diferentes misturas de pós durante
aquecimento, e as características microestruturais das mesmas durante etapas de isoterma de
sinterização e tratamento térmico pós-sinterização, ou seja, morfologia e distribuição de fases
presentes (SiC e secundárias).

80
Nas amostras porosas (etapa de aquecimento) foram analisadas as superfícies
fraturadas, enquanto que as amostras densas (etapas de isoterma e tratamento pós-
sinterização) obtidas dos ensaios de flexão foram lixadas e polidas com pasta de diamante de
até 1 µm. Para revelar a microestrutura, as amostras foram atacadas por uma mistura de
NaOH:KOH proporção 1:1 a 500oC durante 2 minutos.
Assim como as superfícies lixadas e polidas, as superfícies fraturadas resultantes da
etapa de aquecimento e ensaios de flexão foram avaliadas por microscopia eletrônica de
varredura, utilizando uma fina camada de ouro (6 nm) para tornar o material condutor. As
imagens destas superfícies foram obtidas utilizando elétrons retroespalhados. A finalidade
dessa análise foi auxiliar na avaliação do defeito causador da fratura, modelo de fratura
presente, ou seja, intergranular e/ou transgranular.

81
4 - RESULTADOS E DISCUSSÕES
4.1 – Caracterização das misturas de pós e dos compactos a verde
Esta etapa tem como objetivo levantar informações importantes relacionadas às
características das diferentes misturas de pós, como distribuição do tamanho de partículas,
tamanho médio de partículas, morfologia das partículas, presença e distribuição dos
aglomerados (o modo como estes aglomerados estão distribuídos dentro das misturas de pós)
e distribuição das partículas de aditivos com relação às de SiC.
4.1.1. – Análise granulométrica
Os resultados de distribuição granulométrica das misturas de pós são apresentados na
Figura 4.1.
0,1 1 10
0
20
40
60
80
100
N10-α N10-β T10-α T10-β
Por
cent
agem
acu
mul
ada
Diâmetro esférico equivalente [µm]
Figura 4.1 – Curvas de distribuição granulométrica dos pós das misturas.

82
Nesta figura nota-se para todas as misturas de pós: distribuições do tipo monomodal,
com tamanho de partículas variando de 0,18 a 2,1 µm e tamanho médio de partículas de
0,57 µm.
4.1.2. – Análise por microscopia eletrônica de varredura
As fotomicrografias das misturas de pós N10-α, N10-β, T10-α e T10-β obtidas por
microscopia eletrônica de varredura são apresentadas na Figura 4.2. Para obtenção das
imagens foram utilizados elétrons retroespalhados, com objetivo de avaliar a distribuição dos
aditivos e a morfologia das partículas.
Figura 4.2 – Fotomicrografias realizadas por MEV dos pós das misturas.

83
Uma distribuição uniforme das partículas de aditivos (REGIÕES CLARAS - AlN/Y2O3
e/ou AlN/CTR2O3) em relação às de SiC (REGIÕES ESCURAS) tem-sido observado.
Definem-se as regiões claras como sendo partículas de aditivos, por apresentarem maior peso
molecular quanto comparado ao SiC, contribuindo para um maior espalhamento dos elétrons
[CHARLIE; McGILL, 1994]. As partículas de SiC possuem um formato mais arredondado,
enquanto que as de aditivos são em sua grande parte irregulares e angulares, apresentando
desta forma maior pontos de contato que contribuem para o aumento do atrito entre as
partículas de aditivos e SiC, ou seja, maior amarramento entre elas [KINGERY, 1976].
4.1.3. – Massas específicas teórica, real e densidade a verde dos compactos.
Os resultados das massas específicas teórica, real e densidade relativa a verde das
misturas N10-α, T10-α, N10-β e T10-β são apresentados na Tabela 3.9.
Tabela 3.9 – Massas específicas teórica, real e densidade relativa a verde das misturas estudadas.
Massa específica (g/cm3)
Mistura Teórica Real
Densidade relativa
(% D.T.)
N10-α 3,36 1,95 ± 0,01 58,04 ± 0,78
N10-β 3,35 1,86 ± 0,03 55,52 ± 0,63
T10-α 3,48 2,04 ± 0,02 61,15 ± 0,63
T10-β 3,47 1,95 ± 0,01 56,44 ± 0,51
Os valores de densidade relativa a verde estão compatíveis com a literatura [KELLY,
2000]. As misturas de pós contendo α-SiC (N10-α e T10-α) apresentaram valores de

84
densidade relativa superiores aos das misturas N10-β e T10-β (contendo β). Tal condição
pode ser justificada devido as características físicas dos pós de partida, α-SiC e β-SiC, em que
o pó de α-SiC apresenta uma superfície específica maior que o pó de β-SiC, contribuindo
desta forma para um maior contato entre as partículas (α-SiC e aditivos) e diminuição da
porosidade.
4.2. Estudo da sinterabilidade das misturas de pós
Nesta etapa foi observado o comportamento das diferentes misturas de pós durante
processo de aquecimento sem isoterma de sinterização, sob atmosfera de argônio. Para tanto,
foram levantadas curvas de retração linear, densidade relativa e velocidade de retração em
função da temperatura. As informações extraídas desta etapa foram de suma importância para
o desenvolvimento do trabalho, no intuito de desenvolver ciclos de sinterização para obter
cerâmicas densas de SiC com boas propriedades mecânicas. Evitando consumo exagerado de
matérias-primas e tempo.
4.2.1 - Retração linear e densidade relativa das misturas obtidas via
dilatometria
Os resultados de retração linear e densidade relativa das amostras submetidas às
análises por dilatometria são mostrados na Figura 4.3.

85
600 800 1000 1200 1400 1600 1800 2000-0,20
-0,18
-0,16
-0,14
-0,12
-0,10
-0,08
-0,06
-0,04
-0,02
0,00
0,02
reg
ião
III
região IIregião I
(a)
dL/L
o
Tempeartura (oC)
N10-α N10-β T10-α T10-β
400 600 800 1000 1200 1400 1600 1800 2000
45
50
55
60
65
70
75
80
85
90
(b)
N10-α N10-β T10-α T10-β
Den
sida
de r
elat
iva
(% D
.T.)
Temperatura (oC)
Figura 4.3 – Curvas de retração linear (a) e densidade relativa (b) das misturas
N10-α, N10-β, T10-α e T10-β.
Analisando as curvas de retração linear para as diferentes misturas de pós (Figura 4.3 -
a) nota-se para as misturas aditivadas com o sistema AlN-CTR2O3 (T10-α e T10-β) retrações
lineares um pouco superior as encontradas nas misturas NT10-α e N10-β (contendo AlN-
Y2O3). Com base neste resultado pode-se dizer que o líquido formado no sistema AlN-
CTR2O3 apresentou uma melhor eficiência quanto aos estágios de sinterização via fase líquida
do SiC [BALESTRA et al, 2006].
Em relação ao comportamento das curvas de retração linear, nota-se que praticamente
todas as misturas apresentam o mesmo comportamento ou tendência, exceto a mistura N10-β.
Pois tal mistura foi a que apresentou o menor valor de densidade relativa a verde.
Comparando as diferentes curvas de retração das misturas N10-α e T10-α com N10-β
e T10-β, nota-se uma retração linear um pouco superior, devido ao fato de tais amostras
apresentam maiores valores de densidade relativa a verde.
Na Figura 4.4 são mostrados os resultados de velocidade de retração linear das
diferentes misturas de pós em função da temperatura. Analisando os resultados contidos nesta

86
figura nota-se que as máximas velocidades de retração para todas as misturas ocorreram
praticamente na mesma temperatura, ou seja, próximo de 1800oC, indicando o ínicio de
formação da fase líquida para todas as misturas analisadas, conforme Figura 4.3 - b.
600 800 1000 1200 1400 1600 1800 2000
2
0
-8
-6
-2
-4
-14
-16
-10
-18
-12d(dL
/Lo)
/dT
x 1
0-4
Temperatura (oC)
N10-α N10-β T10-α T10-β
Figura 4.4 - Curvas de velocidade de retração linear das misturas N10-α, N10-β, T10-α e T10-β.
4.3 – Caracterização das amostras tratadas termicamente
Assim como a etapa anterior (análise dilatométrica das misturas de pós), esta etapa
teve como objetivo verificar o comportamento das diferentes misturas de pós durante
processo de aquecimento sem isoterma de sinterização num forno com resistência de grafite,
sob atmosfera de argônio. Nesta etapa é mostrado de modo detalhado por meio de
micrografias, o comportamento microestrutural das diferentes misturas de pós, reforçando
ainda mais as informações obtidas destas misturas de pós durante ensaios de dilatometria. As
informações obtidas das análises de dilatometria e desta etapa foram utilizadas para

87
desenvolver ciclos de sinterização, que foram de grande importância nas etapas subseqüentes
do trabalho, ou seja, isoterma e re-sinterização.
4.3.1. – Aquecimento das amostras
4.3.1.1. – Densidade relativa
Os resultados de densidade relativa das misturas N10-α, N10-β, T10-α e T10-β
aquecidas a diferentes temperaturas, sem isoterma, são mostrados na Figura 4.5.
1100 1200 1300 1400 1500 1600 1700 1800 1900 2000 210050
55
60
65
70
75
80
85
90
95
100
N10-α
N10-β
T10-α
T10-β
Den
sida
de r
elat
iva
(% D
.T.)
Temperatura (oC)
Figura 4.5 – Densidade relativa das amostras N10-α, N10-β, T10-α e T10-β aquecidas a diferentes
temperaturas, sem isoterma.
Analisando os resultados contidos nesta figura, nota-se uma pequena mudança nas
curvas de densidade relativa das diferentes misturas para faixa de temperatura compreendendo
1600 – 1800oC. Provavelmente, essa região do gráfico corresponda ao primeiro estágio da

88
sinterização via fase líquida, em que a retração não é tão significativa quando comparado com
o segundo estágio. Nesta etapa tem-se a mudança no formato dos poros devido ao
espalhamento do líquido, que molha e envolve as partículas de SiC, promovendo a
aproximação das partículas. O grau de densificação deste estágio depende das características
dos pós de partida (tamanho e forma), tão bem como da quantidade, viscosidade e
molhabilidade da fase líquida em relação às partículas de SiC.
Acima de 1800oC nota-se uma mudança significativa nas curvas de densidade relativa
das diferentes misturas. Essa região do gráfico corresponde ao segundo estágio de
sinterização, responsável por grande parte da retração das misturas. Neste estágio tem-se a
solubilização das partículas pequenas e reprecipatação das mesmas sobre as partículas
grandes. Portanto, pode-se dizer que essa etapa depende da solubilidade das partículas de SiC
na fase líquida formada.
As misturas T10-α e T10-β apresentaram comportamento de densidade relativa um
pouco melhor quando comparado as N10-α e N10-β, conforme observado e comentado
anteriormente. Resumindo, pode-se concluir que os resultados mostrados na Figura 4.4
refletem basicamente os fenômenos ocorridos nos ensaios de dilatometria, mostrando a
grande importância desta análise no estudo da sinterabilidade de um cerâmico em relação a
um determinado sistema de aditivo, antes de submete-lo a um ciclo de sinterização.
4.3.1.2. – Perda de massa
Na Figura 4.6 são mostrados os resultados de perda de massa das amostras N10-α,
N10-β, T10-α e T10-β aquecidas a diferentes temperaturas, sem isoterma.

89
1550 1600 1650 1700 1750 1800 1850 1900 1950 2000 2050
2
4
6
8
10
12
14
16
18
N10-α N10-β T10-α T10-β
Perd
a de
mas
sa (
%)
Temperatura (oC)
Figura 4.6 – Perda de massa das amostras N10-α, N10-β, T10-α e T10-β aquecidas
a diferentes temperaturas, sem isoterma.
Nesta figura nota-se que as misturas N10-α e T10-α apresentaram maiores perda de
massa quando comparadas às N10-β e T10-β, devido provavelmente a uma maior quantidade
de impurezas presentes no pó de partida α-SiC, que podem auxiliar nas reações entre os
aditivos e o SiC, resultando na formação de compostos voláteis [HUE, 1997].
4.3.1.3 – Análise por difratometria de raios X
Os resultados das análises por difratometria de raios X das amostras N10-α, N10-β,
T10-α e T10-β aquecidas a diferentes temperaturas, sem isoterma, são mostrados nas Figuras
4.7 – 4.8, respectivamente.
Nas misturas contendo AlN-Y2O3 (N10-α e N10-β) são observados como fases
principais α-SiC (4H e 6H) e β-SiC (3C), e AlN, Y2O e Al2Y4O9. Nas misturas contendo
AlN-CTR2O3 (T10-α e T10-β), as fases α-SiC (4H e 6H) e β-SiC (3C) também são

90
observadas como principais, enquanto que as fases secundárias são constituídas por AlN,
CTR2O e Al2CTR4O9.
upl
vv
upn
mm
upn
lmmll
up
up
l
upn
u
mv
ml
N10-αα/1600oC-0h
up
lll
N10-αα/1800oC-0h
p
p
lup
uuppm
m
lll lll
llllll
l l
l
ll
l
l
l
N10-αα/1900oC-0h
10 20 30 40 50 60 70 80
ul
pl
n -3C m -Y2O
3
p -4H v -AlN u -6H lAl
2Y
4O
9
ul
upll l
ml
2θ (graus)
N10-αα/2000oC-0h
u
u
u
vmp
l l
vvlll
mlll
up
up
upn
mvm
upn
mlvmmm
lu
up
l
upn
p
vm
ml
l l
T10-αα/1600oC-0h
ulll l
mm
mmmm
2θ (graus)
T10-αα/1800oC-0h
l
lllvm
upl
lm
lll uupp
mv
p
p
T10-αα/1900oC-0h
10 20 30 40 50 60 70 80
n -3C m -CTR2O
3
p -4H v -AlN u -6H l-Al
2CTR
4O
9
Obs: CTR = Y, Yb, Er, Dy e/ou Ho
upl
l
l
ml
mv
m
l ll
ll lm
T10-αα/2000oC-0h
Figura 4.7 – Difratogramas de raios X das misturas N10-α e T10-α aquecidas a diferentes
temperaturas, sem isoterma de sinterização.
p
u upv
np
ll
pu
upn
upn
mvl
pn
n
up
lml
n -3C m -Y2O
3
p -4H v -AlN u -6H l-Al
2Y
4O
9
N10-ββ/1800oC-0h
N10-ββ/1600oC-0h
m llmlp
m
p
ul
ull l
l
mvl
pl
lll
m
2θ (graus)
N10-ββ/2000oC-0h
N10-ββ/1900oC-0h
10 20 30 40 50 60 70 80
pull
u
up
p
up
p
l
m
m
l
n
n
nvv
T10-ββ/1600oC-0h
muplmllll
lv
mlll l
up
pu
up
lmllll
pupl
upn
upll
m
lll
2θ (graus)
T10-ββ/1800oC-0h
p
upv
m
l
pu
l lmll
vl
pll
l l l
T10-ββ/1900oC-0h
10 20 30 40 50 60 70 80
n -3C m -CTR2O
3
p -4H v -AlN u -6H l - Al
2CTR
4O
9
Obs: CTR = Y, Yb, Er, Dy e/ou Ho
v
u
l l l
mll pu
lv ll m l
up
v
T10-ββ/2000oC-0h
Figura 4.8 - Difratograma de raios X das misturas N10-β e T10-β sinterizadas a diferentes
temperaturas, sem isoterma de sinterização.

91
Em relação às fases CTR2O3 e Al2CTR4O9, nota-se que ambas ocupam praticamente as
mesmas posições cristalográficas (2θ) quando comparadas às fases Y2O3 e Al2Y4O9,
respectivamente. Mostrando um comportamento semelhante do óxido misto de ítrio e terras
raras (CTR2O3) para com o Y2O3, levando a crer que este óxido se trata de uma solução sólida
de óxidos de terras raras em matriz de óxido de ítrio. Tal veracidade foi analisada e
comprovada por Vernilli e colaboradores [VERNILLI et al, 2006].
Em relação às fases principais (α-SiC e β-SiC) não é observado uma significativa
transformação de β-SiC para α-SiC, principalmente nas misturas N10-β e T10-β, em que as
intensidades dos picos relacionados aos polítipos 6H (33,25o e 34,75o) e 4H (34,10o e 38,10o)
mantiveram constante.
Em todas as misturas, com aumento da temperatura tem-se a diminuição nas
quantidades das fases AlN, Y2O3 e CTR2O3. Tal comportamento pode ser explicado devido ao
aparecimento de uma maior quantidade das fases Y2Al4O9 e CTR2Al4O9. No entanto, a
presença destes compostos (Al2Y4O9 – N10-α e Al2CTR4O9 – T10-α) pode ser explicada pela
reação do Al(l) com Y2O3 ou CTR2O3, que se encontra em excesso em ambas as misturas
analisadas. A presença de Al(l) é devido a decomposição do AlN, que resulta em Al(l) e N2
(g) [BISWAS, et al, 2001]. O Al (l) pode também vir a reagir com os sub-óxidos de ítrio (YO
e Y2O) e produzir tais aluminatos (Al2Y4O9 e Al2CTR4O9). Sendo que esses sub-óxidos são
produtos das reações do SiC com Y2O3 [HUE et al, 1997].
Nas misturas N10-α e T10-α nota-se a presença de um pico intenso do politipo 6H (2θ
= 33,25), pois no pó de partida α-SiC, tem-se uma quantidade predominante de 6H.

92
4.3.1.4 – Microscopia eletrônica de Varredura
O comportamento microestrutural das misturas N10-α, N10-β, T10-α e T10-β
sinterizadas a diferentes temperaturas, sem isoterma, são apresentados nas Figuras 4.9 – 4.12,
respectivamente.
Em algumas fotomicrografias nota-se a presença de uma grande quantidade de regiões
claras, no entanto, nem todas essas regiões são constituídas de aditivos, pois como a superfície
é irregular, tem-se a influência do efeito topográfico sobre as imagens obtidas por elétrons
retroespalhados.
Analisando as fotomicrografias das diferentes misturas de pós, tem-se para as
temperaturas de 1600oC e 1800oC uma mudança microestrutural não muito significativa, que
ocorre principalmente na quantidade e formato dos poros, devido ao espalhamento do líquido
formado que envolve as partículas de SiC, promovendo a aproximação das mesmas, estando
tais resultados de acordo com os observados nos ensaios de dilatometria.
Acima de 1800oC nota-se uma significante mudança microestrutural, devido a uma
maior taxa de espalhamento do líquido e aproximação das partículas, proporcionando uma
diminuição na quantidade dos poros e aumento da retração do material. Nesta etapa tem-se
praticamente a presença do estágio de solução-reprecipitação, grande responsável pela
retração no material.
Comparando o comportamento microestrutural das diferentes misturas durante
aquecimento, nota-se que nas misturas aditivadas com AlN-CTR2O3 (T10-α e T10-β)
apresentou um melhor espalhamento do líquido, contribuindo para maior retração das
amostras, condição também observada e discutida nos ensaios de dilatometria.

93
Figura 4.9 – Micrografias realizadas por MEV da mistura N10-α aquecida em diferentes temperaturas,
sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.

94
Figura 4.10 – Micrografias realizadas por MEV da mistura N10-β aquecida em diferentes
temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.

95
Figura 4.11 – Micrografias realizadas por MEV da mistura T10-α aquecida em diferentes
temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.

96
Figura 4.12 – Micrografias realizadas por MEV da mistura T10-β aquecida em diferentes
temperaturas, sem isoterma: (a) verde, (b) 1600oC, (c) 1800oC, (d) 1900oC e (e) 2000oC.

97
Enquanto que nas Figuras 4.13 – 4.16 é mostrado o comportamento microestrutural
das diferentes misturas após formação da fase líquida. Em todas misturas nota-se a presença
de ilhas de líquidos localizados em vários pontos da amostra, criando regiões com gradientes
de densidade. A não homogeneidade entre os aglomerados grandes e pequenos, comentado
anteriormente, permitiu o aparecimento destes defeitos, que são prejudiciais no objetivo de se
obter cerâmicas de SiC densas, com microestrutura homogênea e boas propriedades
mecânicas.
Figura 4.13 – Comportamento microestrutural da mistura N10-α após formação da fase líquida.
Nas Figuras 4.13 (1900oC) e 4.14 (2000o) relacionadas à mistura N10-β é mostrado
duas regiões bem distintas, uma localizada próxima à borda da amostra e outra na região
central da amostra. Na temperatura de 1900oC (Figura 4.13), região da interface é mostrado
na borda da amostra uma maior quantidade de áreas cinzas (SiC) se comparado a áreas claras

98
(fase líquida ou aditivos), enquanto que na região central a quantidade de áreas claras é maior.
Ainda na borda da amostra tem-se a presença de ilhas de líquidos (contendo partículas de SiC
mais aditivos) com alta densificação quando comparado às outras regiões. O aumento da
temperatura (Figura 4.14) diminui essa diferença de densificação existente entre as regiões da
amostra, devido ao decréscimo do gradiente de fase líquida existente entre as mesmas.
Figura 4.14 – Comportamento microestrutural da mistura N10-β após Formação da fase líquida
(1900oC).
Mesmo assim, podemos observar que a borda da amostra é mais densa que a região
central, devido a diferença de temperatura existente entre as mesmas, que permitiu um melhor
espalhamento do líquido na borda da amostra. Tais resultados microestruturais apresentados
nas figuras comentadas anteriormente podem ser devido à difusão de material do leito de pó
para amostra, pois o mesmo apresenta uma quantidade muito maior de aditivo quando
comparado à amostra (Materiais e Métodos).

99
Figura 4.15 – Comportamento microestrutural da mistura N10-β após Formação da fase líquida
(2000oC).
Figura 4.16 – Comportamento microestrutural da mistura T10-β após formação da fase líquida.

100
4.3.2. – Isoterma de sinterização das amostras
Nesta etapa foram analisados diferentes programas (ciclos) de sinterização, com
objetivo de obter cerâmicas de SiC densas com boas propriedades mecânicas. Esses ciclos de
sinterização foram levantados baseados nas informações extraídas das etapas anteriores, como
dilatometria e aquecimento sem isoterma.
4.3.2.1 – Densidade relativa, perda de massa e retração volumétrica.
Os resultados de densidade relativa, retração volumétrica e perda de massa das
misturas N10-α, N10-β, T10-α e T10-β sinterizadas em diferentes programas de sinterização
sob atmosfera de argônio são mostrados nas Figuras 4.17 – 4.19. Durante a sinterização das
misturas de pós a 2000oC por 1h (Figura 4.16) não foram obtidas amostras com alta densidade
(≥ 98% D.T.).
Figura 4.17 – Resultados de densidade relativa, perda de massa e retração linear das misturas N10-α,
N10-β, T10-α e T10-β sinterizadas a 2000oC/1h sob atmosfera de argônio.

101
Como os resultados obtidos no ciclo 1 de sinterização (2000oC/1h) não foram
satisfatórios; com base nas informações obtidas nas etapas de dilatometria e aquecimento,
foram propostos ciclos de sinterização com isoterma intermediária de 1800oC, variando os
tempos em 0,5 h e 1h.
Figura 4.18 – Resultados de densidade relativa, perda de massa e retração linear das misturas N10-α,
N10-β, T10-α e T10-β sinterizadas a 1800oC/30 minutos e 2000oC/1h sob atmosfera de argônio.
Figura 4.19 – Resultados de densidade relativa, perda de massa e retração linear das misturas N10-α,
N10-β, T10-α e T10-β sinterizadas a 1800oC/1h e 2000oC/1h sob atmosfera de argônio.

102
Os ciclos de sinterizações com temperatura intermediária contribuíram para uma
melhora na densidade, retração linear e perda de massa das diferentes misturas,
principalmente com tempo maior de isoterma intermediária, ou seja, 1800 oC por 1h.
Essa melhora significativa nos resultados de densidade, retração linear e perda de
massa das amostras quando submetidas a uma isoterma intermediária de 1800 oC é
argumentada devido a formação da fase líquida, que se espalhou por entre as partículas de SiC
promovendo uma maior aproximação das mesmas, conforme comentado nos ensaios de
dilatometria e aquecimento das amostras.
As amostras contendo AlN-CTR2O3 (T10-α e T10-β) apresentaram valores de
densidade, retração linear e perda de massa maiores quando comparadas às N10-α e N10-β
(contendo AlN-Y2O3). Tais resultados podem ser justificados pelo fato do líquido formado no
sistema AlN-CTR2O3 apresentar uma boa molhabilidade sobre o SiC [109].
Ainda analisando os resultados contidos nas Figuras 4.17 – 4.18, observa-se uma
diminuição significativa nos valores de perda de massa das amostras. Provavelmente, o
acréscimo da isoterma intermediária possibilitou que os sistemas de aditivos (AlN-Y2O3 e/ou
AlN-CTR2O3) fossem respectivamente transformados nas fases Al2Y4O9 e Al2CTR4O9, as
quais possivelmente apresentam uma maior estabilidade química para o SiC.
De acordo com resultados apresentados nas Figuras 4.17 – 4.18 pode-se avaliar a
importância dos ensaios de dilatometria e aquecimento das amostras no sentido de permitir
alteração nos programas de sinterização visando uma melhora das propriedades do material
cerâmico em questão.

103
4.3.2.2 – Análise por difratometria de raios X.
Os resultados das análises por difratometria de raios X das amostras N10-α, N10-β,
T10-α e T10-β sinterizadas em diferentes programas de sinterização são mostrados na Figura
4.19, respectivamente.
Nas misturas contendo AlN-Y2O3 (N10-α e N10-β) são observados como fases
principais α-SiC (4H e 6H) e β-SiC (3C), e Al2Y4O9 como fase secundária. Nas misturas
contendo AlN-CTR2O3 (T10-α e T10-β), as fases α-SiC (4H e 6H) e β-SiC (3C) também são
observadas como principais, enquanto que a fase secundária é constituída somente por
CTR4O9.
ll
u
oo
u
o
u u
l
l
u
on
N10-β
2000oC-1h
N10-α
1800oC-1h/2000
oC-1h
N10-α
1800oC-30 min/2000
oC-1h
N10-α
2000oC-1h
ll
ll
u
u
o
u
o
oo
o
ou
ul
l
l
o
u
u
u
u
ul
ll
l
u
u
u
o
o
o
lu
N10-β
1800oC-30 min/2000
oC-1h
30 35 40 45 50
n -3C l -Al2Y
4O
9
p -4H u -6H
lo
o
uu
2θθ (graus)
N10-β
1800oC-1h/2000oC-1h
uo
o
ll ll
o
oo
uuu
l
u
o
u
u
o
n
lll
o
l l
llluouu
l
2θθ (graus)
T10-β
2000oC1h
T10-α
1800oC-1h/2000
oC-1h
T10-α
1800oC-30 min/2000
oC-1h
T10-α
2000oC-1h
o
u
o
ul
uuu
ul o
T10-β
1800oC-30 min/2000
oC-1h
30 35 40 45 50
n -3C l - Al2CTR
4O
9
p -4H u -6H Obs: CTR = Y, Yb, Er, Dy e/ou Ho
o
uu
ouu ll
ll
l
T10-β
1800oC-1h/2000
oC-1h
Figura 4.20 – Difratogramas de raios X das misturas N10-α, N10-β, T10-α e T10-β sinterizadas a
diferentes ciclos sob atmosfera de argônio.

104
Em relação às fases principais (α-SiC e β-SiC) é observado uma significativa
transformação de β-SiC para α-SiC, principalmente nas misturas N10-β e T10-β, em que as
intensidades dos picos relacionados aos polítipos 6H (33,25o e 34,75o) e 4H (34,10o e 38,10o)
apresentaram um aumento considerável. Essa transformação é observada somente nos ciclos
de sinterização com isoterma intermediária de 1800oC (0,5h e 1h). De acordo com as etapas
de dilatometria e aquecimento foi observado nesta temperatura o início da formação de
líquido. Portanto, a permanência das amostras na isoterma de 1800oC por 0,5 e 1h
provavelmente permitiu um maior espalhamento do líquido e envolvimento das partículas de
SiC, contribuindo de forma significativa para os estágios de rearranjo e principalmente
solução-reprecipitação (crescimento de grãos).
Enquanto que nas misturas N10-α e T10-α as mudanças em relação aos picos das
fases principais não tem sido observado em nenhuma das condições analisadas. A presença do
polítipo 4H nestas misturas pode ser explicada devido à transformação de 6H para 4H.
Segundo Schwetz e Lipp [SCHWETZ; LIPP, 1980], que observaram essa transformação num
SiC prensado a quente, e aditivado com B e Al, tal condição pode ser estendida ao processo
de sinterização via fase líquida do SiC contendo elementos do grupo III da tabela periódica,
que tendem a estabilizar a estrutura 4H. A presença do polítipo 6H é devido a grande
quantidade deste no pó de partida de α-SiC. A transformação de β-SiC para α-SiC permite a
presença de grãos na forma de placas, que possuem a característica de melhorar as
propriedades mecânicas do material [STRECKER, 2004].
Analisando a influência dos diferentes sistemas de aditivos (AlN-Y2O3 e/ou
AlN/CTR2O3) nas cerâmicas de α-SiC e β-SiC, nota-se praticamente o mesmo
comportamento, com relação a intensidade dos principais picos de SiC (6H: 33,25o e 34,75o;
4H: 6H: 34,10o e 38,10o). Tal característica do sistema AlN-CTR2O3 pode ser atribuída a sua
boa molhabilidade sobre o SiC, quando comparado ao AlN-Y2O3.

105
4.3.2.3 – Análise quantitativa dos principais politipos de SiC.
Os resultados das análises quantitativas dos principais politipos de SiC realizados nas
misturas N10-α, N10-β, T10-α e T10-β sinterizadas em diferentes programas de sinterização
são mostrados nas Figuras 4.21 – 4.24.
Figura 4.21 – Análise quantitativa dos principais politipos de SiC na mistura N10-α sinterizada a
diferentes ciclos sob atmosfera de argônio.
Analisando os resultados presentes nas Figuras 4.21 e 4.23 nota-se nas misturas N10-α
e T10-α a predominância do polítipo 6H, devido a sua maior presença nos pós de α-SiC e sua
pequena taxa de transformação a 4H.
Nas Figuras 4.22 e 4.24 referentes às misturas N10-β e T10-β também é observado
uma predominância do polítipo 6H, devido à transformação do 3C para 6H, visto que o 3C é
estável até 1600oC. Segundo Jeeps e Page [4] os elementos do grupo III da tabela periódica,
ou seja, os aceptores de elétrons (Al, B, etc) tendem a estabilizar camadas em ambientes

106
hexagonais. Além disso, a própria característica do líquido, como boa molhabilidade e
viscosidade, também podem ter influenciado nestes resultados.
Figura 4.22 – Análise quantitativa dos principais politipos de SiC na mistura N10-β sinterizada a
diferentes ciclos sob atmosfera de argônio.
A transformação do politipo 3C para 6H, permite a presença de grãos em forma de
placas, melhorando as propriedades mecânicas das cerâmicas de SiC, como tenacidade a
fratura e resistência a flexão (3 ou 4 pontos). No entanto, a quantidade e a razão de aspecto
(relação L/D = comprimento/diâmetro) desta forma microestrutural de grãos depende de uma
série de fatores, como: temperatura e tempo de sinterização; tipo e quantidade de aditivo;
processo de sinterização (sinterização a baixa pressão, prensagem a quente ou isostática a
quente) entre outros.

107
Figura 4.23 – Análise quantitativa dos principais politipos de SiC na mistura T10-α sinterizada a
diferentes ciclos sob atmosfera de argônio.
Figura 4.24 – Análise quantitativa dos principais politipos de SiC na mistura T10-β sinterizada a
diferentes ciclos sob atmosfera de argônio.

108
4.3.2.4 – Análise microestrutural das cerâmicas de SiC
As análises microestruturais das cerâmicas de SiC foram realizadas somente nas
misturas N10-β e T10-β sinterizadas a 1800oC/1h e 2000oC/1h sob atmosfera de argônio,
devido aos resultados anteriores, como difração de raios X e análise de politipos, que
revelaram transformação de β-SiC (3C) para α-SiC (4H e 6H). Enquanto que nas outras
misturas, N10-α e T10-α, partiu-se de um pó que contém uma grande quantidade do polítipo
6H, o qual praticamente se manteve estável nos diferentes programas de sinterização. Neste
caso provavelmente a miscroestrutura da cerâmica de SiC possa se apresentar na forma de
grãos placas.
Analisando as micrografias presentes na Figura 4.25, nota-se para mistura N10-β uma
morfologia constituída em grande parte de grãos aciculares ou arredondados, com baixa razão
de aspecto (relação L/D = comprimento/diâmetro). E de uma pequena quantidade de grãos na
forma de placas.
Figura 4.25 – Micrografias das cerâmicas de SiC sinterizadas a 1800oC/1h e 2000oC/1h sob atmosfera
de argônio e atacadas por NaOH:KOH (1:1) a 500oC por 2 minutos.

109
Enquanto que para mistura T10-β grãos alongados uma maior quantidade de grãos na
forma de placas. Essas diferenças microestruturais estão diretamente relacionadas com a
temperatura de sinterização, e também com as características e comportamento do líquido
formado nos diferentes sistemas de aditivos para com as partículas de SiC durante processo de
sinterização [TAGUCHI, 2005]. Características como boa molhabilidade e capilaridade do
líquido, baixa viscosidade as quais influenciam diretamente no processo de solução-
reprecipitação, etapa responsável pela grande parte da densificação do material cerâmico e
transformação de fase do material [KINGERY, 1997].
4.3.2.5 – Propriedades mecânicas das cerâmicas de SiC.
Os resultados de propriedades mecânicas, como microdureza, tenacidade a fratura
(métodos ICL e ISB) e resistência a flexão (3 pontos) das misturas N10-α, N10-β, T10-α e
T10-β submetidas a tratamentos térmicos pós-sinterização são mostrados na Tabela 4.0.
Em todas as amostras estudadas nota-se um aumento da dureza e resistência a flexão
em 3 pontos com aumento da densidade, diminuindo a quantidade de poros nas amostras e
conseqüentemente presença de defeitos, pois se sabe que os mesmos podem atuar como
concentradores de tensões.
Nas amostras sinterizadas a 2000oC por 1h, os altos valores de tenacidade à fratura
medida pelo método ICL (indentação seguida da medida do comprimento da trinca) pode ser
justificado pela baixa densidade das amostras, pois os poros presentes podem atuar como
absorvedores de energia contidos nas pontas das trincas, impedindo desta maneira a
propagação da trinca, conforme mostrada Figura 4.26.

110
Figura 4.26 – Caminho percorrido pela trinca na amostra N10-β sinterizada
a 2000oC por 1h sob atmosfera de argônio.
As amostras submetidas às sinterizações com isoterma intermediária (1800oC por 0,5 e
1h) e 2000oC por 1h apresentaram um aumento nos valores de microdureza, resistência à
flexão e tenacidade a fratura pelo método ICL. O aumento da microdureza pode ser devido às
altas densidades das diferentes misturas de pós. Enquanto que, o aumento da resistência
flexão e tenacidade à fratura estão relacionados às características microestruturais do material,
permitindo o aparecimento de mecanismos de tenacificação (como “crack deflection” –
deflexão da trinca ou desvio da trinca devido a presença de um obstáculo), conforme
mostrado Figura 4.27.

111
Figura 4.27 – Caminho percorrido pela trinca na amostra N10-β sinterizada
a 1800o/1h e 2000oC/1h sob atmosfera de argônio.
Nas amostras sinterizadas com isoterma intermediária, o aumento da mesma não
proporcionou um aumento significativo em relação às propriedades mecânicas analisadas,
provavelmente o tempo de isoterma não tenha sido suficiente para promover grandes
mudanças microestruturais das amostras.
Analisando a influência do pó de partida α-SiC e/ou β-SiC nas propriedades
mecânicas analisadas pode-se verificar uma pequena variação em relação as mesmas, isso
ocorre provavelmente devido ao fato das microestruturas das amostras contendo α-SiC e/ou
β-SiC apresentarem características similares.
As amostras contendo AlN-CTR2O3 (T10-α e T10-β) apresentaram valores de
propriedades mecânicas iguais ou superiores às N10-α e N10-β (contendo AlN-Y2O),
mostrando a eficácia do líquido formado neste sistema (AlN-CTR2O3) com relação as
mudanças microestruturais de α-SiC e β-SiC.
Para as diferentes amostras, em todas as condições analisadas nota-se baixos valores
de resistência à flexão. Tal comportamento pode ser explicado devido a presença de defeitos

112
no material, como poros (residuais ou decorrentes da saída de material da amostra),
aglomerados de vidros (provavelmente decorrentes da presença de gradientes de fase líquida
no material), conforme mostrado Figura 4.28.
Figura 4.28 – Micrografia da superfície fraturada da mistura N10-β sinterizada a 1800oC/1h e
2000o/1h sob atmosfera de argônio.

113
Tabela 4.0 – Resultados de densidade, microdureza, tenacidade à fratura e resistência a flexão em 3 pontos das amostras
N10-α, N10-β, T10-α e T10-β sinterizadas a diferentes ciclos sob atmosfera de argônio.
Condições de sinterização Propriedades Mecânicas
KIC (MPa.m1/2) Amostras Leito de pó
Temperatura/tempo
(oC/h)
Drelativa
(%D.T.)
HV
(GPa) ICL ISB
σσ3P
(MPa)
2000/1 95 ± 0,4 17 ± 0,4 5,2 ± 0,6 4,4 ± 0,1 194 ± 1,8
1800/0,5 e 2000/1 98 ± 0,4 18 ± 0,4 5,3 ± 0,3 5,0 ± 0,2 203 ± 4,3 N10-α
1800/1 e 2000/1 99 ± 0,5 18,7 ± 0,9 5,0 ± 0,1 4,8 ± 0,5 204 ± 8,2
2000/1 93 ± 0,5 16,6 ± 0,8 5,7 ± 0,2 4,0 ± 0,1 183 ± 4,4
1800/0,5 e 2000/1 95 ± 0,3 17,3 ± 0,9 5,5 ± 0,6 4,3 ± 0,1 200 ± 3,9 N10-β
SiC + AlN-Y2O3
70:30
1800/1 e 2000/1 97 ± 0,2 17,7 ± 0,5 5,4 ± 0,9 4,5 ± 0,2 204 ± 2,7
2000/1 97 ± 0,4 17,3 ± 0,1 5,7 ± 0,6 4,5 ± 0,3 203 ± 5,2
1800/0,5 e 2000/1 99 ± 0,1 17,8 ± 0,5 5,3 ± 0,8 4,6 ± 0,1 210 ± 4,3 T10-α
1800/1 e 2000/1 99 ± 0,6 18,8 ± 0,4 5,4 ± 0,5 4,7 ± 0,1 224 ± 1,8
2000/1 97 ± 0,3 16,4 ± 0,2 5,8 ± 0,6 4,5 ± 0,4 225 ± 8,9
1800/0,5 e 2000/1 97 ± 0,2 18,3 ± 0,4 5,4 ± 0,3 4,6 ± 0,2 247 ± 2,2 T10-β
SiC + AlN-CTR2O3
70:30
1800/1 e 2000/1 98 ± 0,7 18,7 ± 0,7 5,1 ± 0,6 5,2 ± 0,3 254 ± 7,0

114
4.3.3. – Re-sinterização das misturas analisadas
Nesta etapa foi utilizado o ciclo 1 de sinterização, ou seja, 1800oC/1h e 2000oC/1h. Tal
ciclo ou programa de sinterização permitiu obter cerâmicas densas de SiC com melhora nas
propriedades mecânicas. As amostras foram re-sinterizadas variando os tempo em 3h e 6h à
2000oC, objetivando analisar a influência do tempo de sinterização na densidade, perda de
massa, propriedades mecânicas e microestrurturas das cerâmicas de SiC.
4.3.3.1 – Densidade relativa e perda de massa.
Os resultados de densidade relativa, retração volumétrica e perda de massa das
misturas N10-α, N10-β, T10-α e T10-β submetidas a tratamento térmico pós-sinterização são
mostrados nas Figuras 4.29 – 4.32.
Figura 4.29 – Resultados de densidade relativa e perda de massa da mistura N10-α
sinterizada e re-sinterizada.

115
Em todas as misturas são observadas as diminuições das densidades relativas e
retrações linear em função do aumento da temperatura e perda de massa. Mesmo com
aumento do tempo de sinterização, a densidade relativa das amostras teve um pequena
variação (cerca de 2 a 3% em relação ao valor original). A perda de massa das amostras
também apresentou comportamento similar. Tais resultados apresentados podem ser
explicados provavelmente devido a possibilidade de reação da fase intergranular com o SiC,
gerando compostos voláteis [HUE et al, 1997, TAGUCHI, 2005].
Figura 4.30 – Resultados de densidade relativa e perda de massa da mistura N10-β
sinterizada e re-sinterizada.

116
Figura 4.31 – Resultados de densidade relativa e perda de massa da mistura T10-α
sinterizada e re-sinterizada.
Figura 4.32 – Resultados de densidade relativa e perda de massa da mistura T10-β
sinterizada e re-sinterizada.

117
4.3.3.2 – Análise por difratometria de raios X
Os resultados das análises por difratometria de raios X das amostras N10-α, N10-β,
T10-α e T10-β submetidas a re-sinterização são mostrados na Figura 4.33.
o
o
l
o
l
u o
u
o
u
u
l
l
l
l
l
l
l
l
u
o
N10-ββ
1800oC-1h/2000oC-3h
N10-ββ
1800oC-1h/2000oC-1h
N10-αα
1800oC-1h/2000oC-6h
N10-αα
1800oC-1h/2000oC-3h
N10-αα
1800oC-1h/2000oC-1h
l
u
u
o
o
u
u
u
l
o
o
o
u
u
u
o
o
u
o
ol
o
oo
l
u
u
u
u
uu
uu
30 35 40 45 50
o
l
l
u
nn -3C l -Al2Y
4O
9
o -4H u -6H
2θθ (graus)
N10-ββ
1800oC-1h/2000oC-6h
uo
u
u
u
u
l
lu
on
uo
u
u
T10-ββ
1800oC-1h/2000oC-1h
T10-αα
1800oC-1h/2000oC-6h
T10-αα
1800oC-1h/2000oC-3h
T10-αα
1800oC-1h/2000oC-1h
uo
u
uu
l
l
l
l
l
ll
l
uo
u
uo
u
u
u
u
2θθ (graus)
T10-ββ
1800oC-1h/2000oC-6h
T10-ββ
1800oC-1h/2000oC-3h
30 35 40 45 50
uou
uu
l
n -3C l -Al2CTR
4O
9
o -4H u -6H
Figura 4.33 – Difratogramas de raios X das misturas N10-α, N10-β, T10-α e T10-β sinterizadas e
re-sinterizadas sob atmosfera de argônio.
Nas misturas contendo AlN-Y2O3 (N10-α e N10-β) são observados como fases
principais α-SiC (4H e 6H) e β-SiC (3C), e Al2Y4O9 como fase secundária. Nas misturas
contendo AlN-CTR2O3 (T10-α e T10-β), as fases α-SiC (4H e 6H) e β-SiC (3C) também são
observadas como principais, enquanto que a fase secundária é constituída somente por
CTR4O9.

118
Comparando as intensidades dos picos relacionados aos polítipos 6H (33,25o e 34,75o)
e 4H (34,10o e 38,10o), entre as amostras N10-β e T10-β, nota-se uma maior intensidade dos
mesmos na amostra T10-β, quando comparado aos das amostras N10-β. Provavelmente tal
comportamento esteja diretamente relacionada com a fase líquida formada no sistema AlN-
CTR2O3, que deve possuir boa molhabilidade e viscosidade, características primordiais no
estágio de solução-reprecipitação, responsável pela maior taxa de densificação e
transformação de fase do material.
Enquanto que nas misturas N10-α e T10-α as mudanças em relação aos picos das
fases principais não tem sido observado em nenhuma das condições analisadas. A presença do
polítipo 4H nestas misturas pode ser explicada devido à transformação de 6H para 4H.
Segundo Schwetz e Lipp [SCHWETZ, LIPP, 1980], que observaram essa transformação num
SiC prensado a quente, e aditivado com B e Al, tal condição pode ser estendida ao processo
de sinterização via fase líquida do SiC contendo elementos do grupo III da tabela periódica,
que tendem a estabilizar a estrutura 4H. A presença do polítipo 6H é devido a grande
quantidade deste no pó de partida de α-SiC.
Ainda analisando a Figura 4.33 nota-se que aumento do tempo de sinterização não
permitiu uma transformação de β-SiC para α-SiC de modo significativo, pois os picos
correspondentes aos politpos 6H e 4H permaneceram constantes. Talvez tal comportamento
possa ser devido à temperatura de sinterização ou processo de sinterização empregado.
4.3.3.3 – Análise quantitativa dos principais politipos de SiC.
Os resultados das análises quantitativas dos principais politipos de SiC realizados nas
misturas N10-α, N10-β, T10-α e T10-β submetidas a tratamentos térmicos pós-sinterização
são mostrados nas Figuras 4.34 – 4.37.

119
Figura 4.34 – Análise quantitativa dos principais politipos de SiC na mistura N10-α
sinterizada e re-sinterizada.
Figura 4.35 – Análise quantitativa dos principais politipos de SiC na mistura N10-β
sinterizada e re-sinterizada.
Analisando os resultados presentes nas Figuras 4.34 e 4.36 nota-se nas misturas N10-α
e T10-α a predominância do polítipo 6H, comentado anteriormente. Nas Figuras 4.35 e 4.37

120
referentes às misturas N10-β e T10-β também é observado uma predominância do polítipo
6H, comentado anteriormente.
Em todas as misturas é observado um pequeno acréscimo na quantidade dos politipos
6H, mostrando a pequena influência do tempo de re-sinterização e da fase líquida nas
transformações dos politipos 4H → 6H e 3C → 6H.
Figura 4.36 – Análise quantitativa dos principais politipos de SiC na mistura T10-α
sinterizada e re-sinterizada.
Figura 4.37 – Análise quantitativa dos principais politipos de SiC na mistura T10-β
sinterizada e re-sinterizada.

121
4.3.2.4 – Análise microestrutural das cerâmicas de SiC
As micrografias das misturas N10-β e T10-β re-sinterizadas em diferentes tempos são
mostrados nas Figuras 4.38 e 4.39. Em ambas as amostras, o aumento do tempo de re-
sinterização permitiu um acréscimo na quantidade de grãos em forma de placas,
provavelmente devido a uma pequena variação na quantidade do politipo 6H. Porém, na
mistura T10-β (Figura 4.31) uma maior quantidade de grãos em forma de placas tem sido
observado, possivelmente devido a eficiência do líquido quanto ao processo de sinterização
do SiC.
Figura 4.38 – Micrografias da mistura N10-β re-sinterizada e atacadas por NaOH:KOH (1:1)
a 500oC por 2 minutos.

122
Figura 4.39 – Micrografias da mistura T10-β re-sinterizada e atacadas por NaOH:KOH (1:1)
a 500oC durante 2 minutos.
Na Figura 4.40 são mostradas as superfícies fraturadas das misturas N10-β e T10-β
submetidas à re-sinterização. Analisando as micrografias das diferentes misturas observa-se a
presença de defeitos (poros) (Figura 4.34 a). Praticamente em todas as amostras nota-se
presença de fratura transgranular, ou seja, com rompimento da fase matricial (SiC). Tal
comportamento pode ser explicado devido a presença de poros, que atuam como
concentradores de tensão acelerando o processo de crescimento da trinca e ruptura brusca do
corpo cerâmico [ZANOTO, 1991].
4.3.2.5 – Propriedades mecânicas das cerâmicas de SiC.
Os resultados de propriedades mecânicas, como microdureza, tenacidade a fratura
(métodos ICL e ISB) e resistência a flexão (3 pontos) das misturas N10-α, N10-β, T10-α e
T10-β submetidas a re-sinterização são mostrados na Tabela 4.1.

123
Figura 4.40 – Micrografias das superfícies de fratura das misturas N10-β e T10-β
sinterizadas e re-sinterizadas.
Em todas as amostras estudadas nota-se um aumento da dureza e resistência a flexão
em 3 pontos com aumento da densidade, diminuindo a quantidade de poros nas amostras e
conseqüentemente presença de defeitos, pois se sabe que os mesmos podem atuar como
concentradores de tensões.

124
Nas amostras sinterizadas a 2000oC por 1h, os altos valores de tenacidade à fratura
medida pelo método ICL (indentação seguida da medida do comprimento da trinca) pode ser
justificado pela baixa densidade dos materiais, pois os poros presentes podem atuar como
absorvedores de energia contidos nas pontas das trincas, impedindo desta maneira a
propagação da trinca.
As amostras submetidas às sinterizações com isoterma intermediária (1800oC por 0,5 e
1h) e 2000oC por 1h apresentaram um aumento nos valores de microdureza, resistência à
flexão e tenacidade a fratura pelo método ISB (indentação seguida de flexão das amostras em
3 pontos), devido provavelmente ao aumento da densidade e mudanças microestruturais
ocorridas.
Nas amostras sinterizadas com isoterma intermediária, o aumento da mesma não
proporcionou um aumento significativo em relação às propriedades mecânicas analisadas,
provavelmente o tempo de isoterma não tenha sido suficiente para promover grandes
mudanças microestruturais das amostras.
Analisando a influência do pó de partida α-SiC e/ou β-SiC nas propriedades
mecânicas analisadas pode-se verificar uma pequena variação em relação as mesmas, isso
ocorre provavelmente devido ao fato das microestruturas das amostras contendo
α-SiC e/ou β-SiC apresentarem características similares.
As amostras contendo AlN-CTR2O3 (T10-α e T10-β) apresentaram valores de
propriedades mecânicas igual ou superiores às N10-α e N10-β (contendo AlN-Y2O),
mostrando a eficácia do líquido formado neste sistema (AlN-CTR2O3) com relação as
mudanças microestruturais de α-SiC e β-SiC.
Para todas as misturas analisadas, o aumento do tempo de sinterização proporcionou
uma diminuição na microdureza do material, devido ao decréscimo da densidade do mesmo.
Enquanto que, a tenacidade à fratura (ICL e ISB) e resistência a flexão em 3 pontos

125
apresentaram um pequeno aumento com acréscimo do tempo de sinterização, devido
provavelmente ao maior engrossamento microestrutural das amostras, conforme comentado
item 4.3.2.4 (análise microestrutural das cerâmicas de SiC).
De modo geral pode-se concluir que a re-sinterização das diferentes misturas
analisadas neste trabalho não proporcionou uma mudança microestrutural significante, devido
provavelmente ao tempo, a temperatura ou mesmo processo de sinterização adotado. Em
trabalhos realizados por Strecker e colaboradores [STRECKER et al, 2004] notou-se uma
grande mudança microestrutural quando da sinterização de cerâmicas de SiC aditivadas com
AlN-Y2O3 por prensagem a quente variando-se os tempos de sinterização.

126
Tabela 4.1 – Resultados de densificação, microdureza, tenacidade à fratura e resistência a flexão em 3 pontos das amostras N10-α,
N10-β, T10-α e T10-β submetidas à re-sinterização.
Condições de sinterização Propriedades Mecânicas
KIC (MPa.m1/2) Amostra Leito de pó
Temperatura/tempo
(oC/h)
Drelativa
(%D.T.)
HV
(GPa) ICL ISB
σσ3P
(MPa)
1800/1 e 2000/1 99 ± 0,5 18 ± 0,4 5,0 ± 0,1 4,9 ± 0,5 203 ± 8,2
1800/1 e 2000/3 97 ± 0,3 17,7 ± 0,5 5,5 ± 0,9 5,0 ± 0,2 226 ± 2,6 N10-α
1800/1 e 2000/6 95 ± 0,4 15,7 ± 1,3 7,5 ± 0,8 5,1 ± 0,2 229 ± 2,1
1800/1 e 2000/1 97 ± 0,3 17 ± 0,5 5,4 ± 0,9 4,5 ± 0,1 204 ± 2,7
1800/1 e 2000/3 95 ± 0,2 16,1 ± 0,7 6,3 ± 0,5 4,5 ± 0,1 204 ± 3,9 N10-β
SiC + AlN-Y2O3
70:30
1800/1 e 2000/6 95 ± 0,5 15,2 ± 0,2 6,6 ± 0,4 5,4 ± 0,6 269 ± 8,9
1800/1 e 2000/1 99 ± 0,6 18,8 ± 0,4 5,4 ± 0,5 4,7 ± 0,1 224 ± 1,8
1800/1 e 2000/3 97 ± 0,6 17,4 ± 0,5 5,7 ± 0,4 4,9 ± 0,1 234 ± 2,3 T10-α
1800/1 e 2000/6 95 ± 0,5 15,7 ± 0,4 6,4 ± 0,4 5,1 ± 0,2 234 ± 3,5
1800/1 e 2000/1 98 ± 0,8 18,7 ± 0,7 6,1 ± 0,6 5,0 ± 0,4 247 ± 12,2
1800/1 e 2000/3 96 ± 0,4 16,7 ± 0,7 5,8 ± 0,5 5,4 ± 0,3 253 ± 5,5 T10-β
SiC + AlN-CTR2O3
70:30
1800/1 e 2000/6 95 ± 0,5 15,7 ± 0,4 6,8 ± 0,5 5,9 ± 0,2 304 ± 8,1

127
5 - CONCLUSÕES
As análises de dilatometria foram de suma importância para o desenvolvimento deste
trabalho, pois permitiu analisar de modo detalhado o comportamento do material durante
aquecimento e propor um ciclo de sinterização que permitisse a obtenção de cerâmicas densas
de carbeto de silício;
A utilização da temperatura intermediária (1800oC/0,5h e 1h) no processo de
sinterização via fase líquida do carbeto de silício permitiu uma melhora considerável na
densidade e propriedades mecânicas do mesmo;
De um modo geral, as amostras contendo AlN-Y2O3 (N10-α e N10-β) e/ou AlN-
CTR2O3 (T10-α e T10-β) sinterizadas a 1800oC/1h e 2000oC/1h exibiram os melhores
resultados de densidade e propriedades mecânicas;
As amostras aditivadas com AlN-CTR2O3 (T10-α e T10-β) apresentaram resultados de
densidade e propriedades mecânicas praticamente similar às contendo AlN-Y2O3 (N10-α e
N10-β), mostrando a eficácia do sistema AlN-CTR2O3 quanto à sinterização via fase líquida
do SiC; e também a viabilidade da substituição do óxido de ítrio (Y2O3) pelo óxido misto de
ítrio e terras raras (CTR2O3) produzido na Escola de Engenharia de Lorena (EEL-USP) à
partir do mineral Xenotima.
Nas amostras submetidas às etapas de isoterma e re-sinterização não foram observadas
mudanças microestruturais significantes quanto à transformação de fase do carbeto de silício,
β-SiC (3C) para α-SiC (6H, 4H); havendo portanto a necessidade de aumentar a temperatura
ou tempo de sinterização, ou mesmo utilizar outro processo de sinterização, como prensagem
a quente ou isostática a quente;
De um modo geral, pode-se concluir que esse trabalho vem a contribuir de maneira
relevante com o desenvolvimento cientifico e tecnológico de cerâmicas de carbeto de silício

128
sinterizado via fase líquida, pela gama de informações contidas nas diferentes etapas do
mesmo.
6 - PROPOSTAS PARA TRABALHOS FUTUROS
Nos últimos anos, com o desenvolvimento cientifico e tecnológico, novos materiais
foram criados, principalmente ligas metálicas utilizadas nas indústrias automobilísticas,
aeroespaciais e outras, havendo a necessidade de se desenvolver novas ferramentas de corte
ou mesmo aperfeiçoar as utilizadas comercialmente para usinagem destes materiais. Com
base nestas informações são propostos os seguintes trabalhos:
F Sinterização e caracterização de cerâmicas a base de carbeto de silício obtidas via fase
líquida visando produção de ferramentas de corte para usinagem de ligas a base de Fe-C
(Ferro fundido cinzento, vermicular, nodular); a base de níquel; base de cromo e a base de
titânio (utilizadas em implantes dentários ou mesmo na indústria automobilística).

129
REFERÊNCIAS
BALESTRA, R. M., RIBEIRO, S., TAGUCHI, S. P., MOTTA, F. V., BORMIO-NUNES, C.
Wetting behaviour of Y2O3/AlN additive on SiC ceramics. Journal of the European
Ceramic Society, v. 26, n 16, p. 3881 - 3886, 2006.
BAKLANOVA, N. I., KYLYUKIN, N., LYAZHOV, N. Z., TURIKINA, G. YU, YAROSH,
O. G., VORONKOV, M. G. Polysilethenesilethyne – A Promising Precursor to Silicon
Carbide. Journal of Materials Synthesis and Processing, v. 5, n. 6, p. 443 – 448, 1997.
BAUD, S, THÉVENOT, F., CHATILLON, C. High temperature sintering of SiC with oxide
additives: I. Analysis in the SiC-Al2O3 and SiC-Al2O3-Y2O3 systems. Journal of the
European Ceramic Society, v. 23, p. 1 - 8, 2003 a.
BAUD, S., THÉVENOT, F., CHATILLON, C. High temperature sintering of SiC with oxide
additives: II. Vaporization process in powder beds and gas-phase analysis by mass
spectrometry. Journal of the European Ceramic Society, v. 23, p. 9 – 18, 2003 b.
BAUD, S., THÉVENOT, F., CHATILLON, C. High temperature sintering of SiC with oxide
additives: III. Quantitative vaporization of SiC-Al2O3 powder beds as revealed by mass
spectrometry. Journal of the European Ceramic Society, v. 23, p. 19 – 27, 2003 c.
BAUD, S., THÉVENOT, F., CHATILLON, C. High temperature sintering of SiC with oxide
additives: IV. Powder beds and the influence of vaporization on the behavior of SiC
compacts. Journal of the European Ceramic Society, v. 23, p. 29 – 36, 2003 d.

130
BELMONTE, T., BONNETAIN, L., GINOUX, J. L. Synthesis of silicon carbide whiskers
using the vapour-liquid-solid mechanism in a silicon-rich droplet. Journal of Materials
Science, v. 31, p. 2367 – 2371, 1996.
BISWAS, K.; RICECKER, G.; WEDMANN, I.; SCHIWEIZER, M.; UPADHYAYA, G. S.;
ALDINGER, F. Liquid phase sintering and microstructure-property relationships of silicon
carbide ceramics with oxynitrides additives. Materials Chemistry and Physics, v. 67, p. 180
– 191, 2001.
BLOOR, D. The Encyclopedia of Advanced Materials, v.4. 1994, 2455 p.
CHARLIE, R. B., McGILL, B. L., The application of Scanning Electron Microscopy to
Fractograph. Materials Characterization, v. 33, p. 195 - 243, 1994.
CHOI, HEON-J.and LEE, J. G., Continuous synthesis of silicon carbide whiskers. Journal of
Materials Science, v. 30, p. 1982 – 1986, 1995.
COOK, R F, Direct observation and analysis of indentation cracking in glasses and ceramics.
Journal of the American Ceramic Society, v. 73, p. 783 – 817, 1990.
DRESSLER, W., RIEDEL, R. Progress in Silicon-Based Non-Oxide Structural Ceramics.
International Journal of Refractary Metals && Hard Materials, v. 15, p. 13-47, n. 1-3,
1997.

131
DAI, J, LI, J., CHEN, Y. The phase transformation behavior of Si3N4 with single Re2O3
(Re=Ce, Nd, Sm, Eu, Gd, Dy, Er, Yb) additive. Materials Chemistry and Physics, v. 80, n.
1, p. 356 - 359, 2003.
DESHPANDE, S. A., BHATIA, T., XU, H., N. P. PADTURE, ORTIZ, A. L., CUMBRERA,
F. L. Microstructural evolution in liquid-phase-sintered SiC: Part II, Effects of planar defects
and seeds in the starting powder, Journal of the American Ceramic Society, v. 84, n. 7, p.
1585 – 1590, 2001.
DUSZA, J, Comparison of fracture toughness testing methods applied to Si3N4 + Si3N4 –
whiskers system. Scripta Metalurgicall and Materials, v. 26, p. 337 – 342, 1992.
DUSZA, J, STEEN, M. Fractography and fracture mechanics property assessment of
advanced structural ceramics, International Materials Reviews, v. 44, n. 5, p. 165 – 216,
1999.
FALK, L. K. L. Microstrutural Development during Liquid Phase Sintering of Silicon
Carbide Ceramics. Journal of the European Ceramic Society, v.17, p. 983-994, 1997.
FREVEL, L. K., PETERSEN, D. R., SAHA, C. K. Polytype Distribution in Silicon Carbide.
Journal of Materials Science, v.27, p. 1913 – 25, 1992.
GERMAN, M.R. Powder Metallurgy Science. 2nd ed. Princeton, New Jersey - USA: Ed.
Metal Powder Industries Federation, 1994. 472 p.

132
GERMAN, M. R. Sintering theory and practice, John Wiley & Sons, Inc., N. York, 550p,
1996.
HASEGAWA, I., FUJII, Y., YAMADA, K., KARIYA, C., TAKAYAMA, T. Lignin-Silica
Hybrids as Precursors for Silicon Carbide. Journal of Applied Polymer Science, v. 73, n. 7,
p. 1321 – 1328, 1999.
HUE, F., JORAND, Y., DUBOIS, J., FANTOZZI, G. Analysis of the weight loss during
sintering of silicon carbide whisker-reinforced alumina composites. Journal of the European
Ceramic Society., v. 17, n. 4, p. 557-563, 1997.
IHLE, J., HERRMANN, M., ADLER, J. Phase formation in porous liquid phase sintered
silicon carbide: Part I: Interaction between Al2O3 and SiC. Journal of the European
Ceramic Society, v. 25, n. 7, p. 987 – 995, 2005 a.
IHLE, J., HERRMANN, M., ADLER, J. Phase formation in porous liquid phase sintered
silicon carbide: Part II: Interaction between Y2O3 and SiC. Journal of the European
Ceramic Society, v. 25, n. 7, p. 997 – 1003, 2005 b.
IHLE, J., HERRMANN, M., ADLER, J. Phase formation in porous liquid phase sintered
silicon carbide: Part III: Interaction between Al2O3-Y2O3-SiC. Journal of the European
Ceramic Society, v. 25, n. 7, p. 1005 – 1013, 2005 c.

133
INOMATA, Y., Crystal chemistry of Silicon Carbide. p. 1 – 11 In: Silicon Carbide Ceramics
– 1 Fundamental and Solid Reaction., edited by S. SOMIYA, Y. INOMATA. Elsevier Apllied
Science – London and New York, 1991.
INOMATA, Y., INOUE, A., MITOMO, M. and SUZUKI, H. Relation between growth
temperature and the structure of silicon carbide crystals grown by the sublimation method.
Yogyo-Kyokai-Shi, v. 76, p. 313 – 319, 1968.
IZHEVSKYI, V.A., GENOVA, L. A., BRESSIANI, J. C., BRESSIANI, A. H. A. Review
article: Silicon Carbide. Structure, Properties and Processing. Cerâmica, v.46, n. 297, p. 4 –
14, 2000 a.
IZHEVSKYI, V. A., GENOVA, L. A., BRESSIANI, A. H. A., BRESSIANI, J. C. Liquid
phase sintered SiC. Processing and transformation controlled microstructure tailoring.
Materials Research, v.3, n.4, p.131-138, 2000 b.
IZHEVSKYI, V A, GENOVA, V. A., BRESSIANI, A.H.A., BRESSIANI, J. C.
Microstructure and properties tailoring of liquid-phase sintered SiC. International Journal of
Refractory Metals and Hard Materials, v. 19, n. 4 – 6, p. 409-417, 2001.
JEEPS, N. W., PAGE, T. F., “Polytypic transformation in silicon carbide”. pp. 259 – 307 In:
Crystal growth and characterization of polytype structures, vol. 7. Edited by P. Krishma.
Pergamon Press, Oxford, U. K., 1983.

134
JOHNSON, C A, PROCHAZKA, S. Ceramic Microstructures ’76 edited by Fulrath and
Park, p. 366-378, 1977.
KELLY, C. A., Sinterização via fase líquida e propriedades mecânicas do carbeto de
silício. 2000. 111 f. Dissertação (Mestrado em Engenharia de Materiais) –
DEMAR/FAENQUIL, Lorena - SP, 2000.
KINGERY, W. D., BOWEN, H. K., UHLMANN, D. R. Introduction to Ceramics. 2nd ed.
New York: John Wiley e Sons 1976. 1032 p.
KINGERY, W. D. Sintering in the presence of a liquid phase. W. D. Kingery, Editor MIT
Press, 235p, 1957.
KIM, Y-W, MITOMO, M., NISHIMURA, T. High-temperature strength of liquid–phase-
sintered SiC with AlN and Re2O3 (RE=Y, Yb). Journal of the American Ceramic Society,
v. 85, n. 4, p. 1007 – 1009, 2002.
KIM, Y. W., MITOMO, M., HIROTSURU, H. Microstructual development of silicon carbide
containing large seed grains. Journal of the American Ceramic Society, v. 80, n. 1, p. 99 –
105, 1997.
KIM, Y. W., MITOMO, M., EMOTO, H., LEE, J. G. Effect of initial α-phase content on
microstructure and mechanical properties of sintered silicon carbide. Journal of the
American Ceramic Society, v. 81, p. 3136 – 3140, 1998.

135
KNIPPENBERG, W. F. Growth phenomena in silicon carbide. Philips Res. Rep. 18, 161 –
274, 1963.
LEE, J. K., TANAKA, H., KIM, H. Formation of solid solutions between SiC and AlN during
liquid-phase sintering. Materials Letters, v.29, p.1 - 6, 1996.
LEE, J. K., TANAKA, H., KIM, H., KIM, D. J. Microstructural Changes in Liquid-Phase-
Sintered α-silicon carbide. Materials Letters, v. 29, n. 1196, p.135 - 142, 1996.
LEE, S-G, KIM, Y-W, MITOMO, M. Relationship between Microstructure and Fracture
Toughness of Toughened Silicon Carbide Ceramics. Journal of the American Ceramic
Society, v. 84, n. 6, p. 1347 – 1353, 2001.
MANDAL, S., DHARGUPTA, K. K., GHATAK, S. Gas pressure sintering of SiC-AlN
composites in nitrogen atmosphere. Ceramics International, v. 28, p.145 - 151, 2002.
MARCHI, J. Estudo de sinterização de cerâmicas a base de nitreto de silício utilizando-se
como aditivos óxido de cério e alumínio. São Paulo: IPEN, 1999. 139f. Dissertação
(Mestrado em Engenharia de Materiais).
MCCOLM, J. Ceramic Hardness. New York: Plenum Press, 1990. p. 324.
MENKE, Y. Effect of rare-earth on properties of sialon glasses. Journal of Non-Crystalline,
v. 276, p. 145 - 150, 2000.

136
MULLA, M. A., KRSTIC, D. V. Low temperature pressureless sintering of β-silicon carbide
with aluminum oxide and yttrium oxide additions. Ceramic Bulletin, v.70, n.3, p. 439 - 443,
1991.
NADER, M., ALDINGER, F., HOFFMAN, M. J. Influence of the α/β-SiC phase
transformation on microstructure development and mechanical properties of liquid phase
sintered carbide, Journal of Materials Science, v. 34, n. 6, p. 1197-1204, 1999.
NEGITA, K. Effective Sintering Aids for Silicon Carbide Ceramics: Reactivities of Silicon
Carbide with Various Additives. Journal of the American Ceramic Society, v. 69, n. 12. p.
308 – 310, 1986.
PAN, B. Y., QIU, J. K., HAWAGOE, M., MORITA, M., TAN, S. H., JIANG, D. SiC-AlN
Particulate Composite. Journal of the European Ceramic Society, v. 19, p. 1789-1793,
1999.
PETZOW, G.; TELLE, R.; DANZER, R. Microstructural Defect and Mechanical Properties
of High-Performance Ceramics. Material Characterization, v. 26, p. 289 - 302, 1991.
PONTON, C B, RAWLINGS, R. D. Vickers indentation fracture toughness test part I:
Review of literature and formulation of standardized indentation toughness equations.
Materials Science. and Technology, v.5, p. 865 - 871, 1989.
RAHAMAN, M. N. Ceramic processing and sintering. N.Y.: Marcel Dekker, 1995. p. 770.

137
RIBEIRO, S. Estudo comparativo das propriedades do nitreto de silício sinterizado com
óxidos de ítrio/óxido de silício concentrado de óxido de ítrio e terras raras/óxido de
silício. 1997. 148 f. Tese (Doutorado em Engenharia de Materiais) – DEMAR/FAENQUIL,
Lorena - SP, 1997.
RICHERSON, D. W. Modern Ceramic Engineering: Properties, processing and use in
design. New York: Marcel Dekker, 1992. p. 162 - 203, p. 680 - 728.
RIXECKER, G., WIEDMANN, I., ROSINUS, A., ALDNGER, F. High-temperature effects
in the fracture mechanical behaviour of silicon carbide liquid-phase sintered with AlN-Y2O3
additives. Journal of the European Ceramic Society, v. 21, p. 1013 – 1019, 2001.
RUSKA, J., GAUCKLER, L. J., LORENZ, J., REXER, H. U. The quantitative calculation of
SiC polytypes from measurements of X-ray diffraction peak intensities, Journal of the
Materials Science, v. 14, n. 8, p. 2013 – 2017, 1979.
SHAFFER, P. T. B. A Review of the Structure of Silicon Carbide. Acta Crystallographica,
v. B25, p. 477 – 488, 1969.
SHE, J. H., UENO, K. Effect of additive content on liquid-phase on silicon carbide ceramics.
Materials Research Bulletin, v.34, n. 10 - 11, p.1629 - 1636, 1999.
STRECKER, K., RIBEIRO, S., CAMARGO, D., SILVA, R., VIEIRA, J., OLIVEIRA, F.
Liquid phase sintering of silicon carbide with AlN/Y2O3, Al2O3/Y2O3, and SiO2/Y2O3
additions. Materials research, v. 2, n. 4, p. 249-254, 1999.

138
STRECKER, K., RIBEIRO,S., OBERACKER, R., HOFMANN, M.-J. Influence of
microstructural variation on fracture toughness of LPS-SiC ceramics. International Journal
of Refractory Metals & Hard Materials, v. 22, p. 169-175, 2004.
SAMANTA, A. K., DHARGUPTA, K. K., GHATAK, S., Decomposition reactions in the
SiC-Al-Y-O system during gas pressure sintering. Ceramics International, v. 27, p.123-133,
2001.
SANTOS, C, Prensagem uniaxial à quente de cerâmicas de Si3N4, com misturas de
CTR2O3/AlN e CTR2O3/Al2O3 como aditivos: caracterização microestrutural,
comportamento mecânico e resistência à fluência. 2004. 140 f. Tese (Doutorado em
Engenharia de Materiais) – DEMAR/FAENQUIL, Lorena, 2004.
SANTOS, C. Estudo da sinterização, propriedades mecânicas e resistência à corrosão
por alumínio líquido, de cerâmicas de Si3N4 com misturas de aditivos de Al2O3/Y2O3 e
Al2O3/CTR2O3 2001. 99 f. Dissertação (Mestrado em Engenharia de Materiais) - DEMAR-
FAENQUIL, Lorena -SP, 2001.
SCHWETZ, K. A., LIPP, A. In Proceedings of the Tenth Symposium on the Science of
Ceramics. Ed. H. Hausner. Deutsche Keram. Ges., 1980. p. 149–158.
TAGUCHI, S., Estudo das interações dos sistemas SiC + Al2O3 e SiC + AlN/Y2O3 em
temperaturas elevadas visando melhor entendimento de cerâmicas de SiC. 2005. 170 f.
Tese (Doutorado em Engenharia de Materiais) DEMAR - FAENQUIL, Lorena - SP, 2005.

139
VERNILLI, F, VERNILLI, D. C., FERREIRA, B., SILVA, G. Characterization of a rare earth
oxide obtained from Xenotime mineral. Materials Characterization, v. 58, n. 1, p. 1–7,
2007.
XU, H., BHATIA, T., DESHPANDE, S. A., PACTURE, N. P., ORTIZ, A. L. Microstructural
evolution in liquid-phase-sintered SiC: Part I, Effect of starting powder, Journal of the
American Ceramic Society, v. 84, n. 7, p. 1578-84, 2001.
YE, H., PUJAR, V. V., PADTURE , N. P. Coarsening in liquid-phase-sintered α-SiC. Acta
Materialia, v. 47, n 2, p. 481-487, 1999.
ZANOTO, E. D., MIGLIORE JR., A. R. Propriedades mecânicas de materiais cerâmicos:
uma introdução. Cerâmica, v. 37, n. 247, p. 7-16, 1991.
ZHOU, Y, HIRAO, K., YAMAUCHI, Y., KANZAKI, S. Tailoring the mechanical properties
of silicon carbide ceramics by modification of the intergranular phase chemistry and
microstructure, Journal of the European Ceramic Society, v. 22, n. 14-15, p. 2689-2696,
2002.
ZHOU, Y, HIRAO, K., TORIYAMA, M., YAMAUCHI, Y., KANZAKI, S. Effects of
intergranular phase chemistry on the microstructure and mechanical properties of silicon
carbide ceramics densified with rare-earth oxide and alumina additions. Journal of the
American Ceramic Society, v. 84, n 7, p. 1642-44, 2001.

140
WINN, E. J., CLEGG, W. J. Role of the powder bed in the densification on silicon carbide
sintered with yttria and alumina additives. Journal of the American Ceramic Society, v. 82,
n. 12, p.3466-3470, 1999.
Top Related