







Línguas
Páginas
Legal


UNIVERSIDADE PRESBITERIANA MACKENZIE
CENTRO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE
CURSO DE CIÊNCIAS BIOLÓGICAS
FLÁVIA LAGHI CREDIDIO
PADRONIZAÇÃO DA TÉCNICA DE IMUNOPRECIPITAÇÃO DA CROMATINA
(ChIP) PARA ANÁLISE DA ATIVAÇÃO TRANSCRICIONAL DURANTE O
PROCESSO DE DIFERENCIAÇÃO DE iPSC EM CARDIOMIÓCITO
São Paulo
2014

UNIVERSIDADE PRESBITERIANA MACKENZIE
CENTRO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE
CURSO DE CIÊNCIAS BIOLÓGICAS
FLÁVIA LAGHI CREDIDIO
PADRONIZAÇÃO DA TÉCNICA DE IMUNOPRECIPITAÇÃO DA CROMATINA
(ChIP) PARA ANÁLISE DA ATIVAÇÃO TRANSCRICIONAL DURANTE O
PROCESSO DE DIFERENCIAÇÃO DE iPSC EM CARDIOMIÓCITO
Trabalho de Conclusão de Curso
apresentado ao Centro de Ciências
Biológicas e da Saúde da Universidade
Presbiteriana Mackenzie, como requisito
parcial à obtenção do título de Bacharel em
Ciências Biológicas.
Orientador Externo: Dr Alexandre da Costa Pereira
Orientadora Interna: Drª Ana Paula Pimentel Costa
São Paulo
2014

FLÁVIA LAGHI CREDIDIO
PADRONIZAÇÃO DA TÉCNICA DE IMUNOPRECIPITAÇÃO DA CROMATINA
(ChIP) PARA ANÁLISE DA ATIVAÇÃO TRANSCRICIONAL DURANTE O
PROCESSO DE DIFERENCIAÇÃO DE iPSC EM CARDIOMIÓCITO
Trabalho de Conclusão de Curso
apresentado ao Centro de Ciências
Biológicas e da Saúde da Universidade
Presbiteriana Mackenzie, como requisito
parcial à obtenção do título de Bacharel em
Ciências Biológicas.
Aprovada em
BANCA EXAMINADORA
.
Drª Ana Paula Pimentel Costa
.
Dr Alexandre da Costa Pereira
.
Drª Júlia Daher Carneiro Marsiglia

Aos meus pais e irmãos, por acreditarem na
minha capacidade; ao meu noivo, pelo
constante incentivo e apoio.

AGRADECIMENTOS
A Deus, fonte de toda sabedoria, nos guiando para o caminho que nos foi confiado;
À Universidade Presbiteriana Mackenzie e ao Centro de Ciências Biológicas e da
Saúde pela formação a mim oferecida;
Ao Laboratório de Genética e Cardiologia Molecular pela possibilidade de desenvolver
este projeto;
À Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) pela bolsa de
estudos durante este processo;
À minha tia, Drª. Ivani Credidio Trombetta, que acreditou no meu potencial desde o
início e me apresentou ao universo laboratorial;
Ao meu orientador, Dr. Alexandre da Costa Pereira, grande pessoa que me acolheu,
apoiou e incentivou durante todo o meu processo de formação;
Aos meus grandes amigos e colegas do LGCM pelo companheirismo e incentivo
sempre, em especial à microbióloga Graça Rosas, portuguesa que tanto admiro e
onde eu encontrei uma amizade verdadeira;
À Drª Júlia Marsiglia, um agradecimento muito especial, por ter aceitado pegar uma
IC tão nova, ensinando desde o “bê-á-bá” até o que sei hoje, com grande paciência e
maestria;
Ao biólogo Théo Gremen, grande amigo que não hesita em me ajudar e me incentivar
nos momentos mais difíceis, exemplo de amizade e simpatia;
Um agradecimento especial para a enfermeira Carolina Yae, minha grande “japa” que
me faz sorrir até das maiores tragédias. Grande irmã e companheira;
Aos meus grandes amigos do Departamento de Genética Humana da Universidade
de Chicago, Dr Marcelo Nóbrega, Drª Ivy Aneas, Dr Noboru Sakabe, Dr Scott Smemo
e Drª Kathleen Bailey, que me acolheram por maravilhosos 10 meses, me ajudando
nesta nova empreitada. Devo grande parte do meu desenvolvimento profissional e
pessoal a eles;

A todos os meus amigos, grande agradecimento por fazerem dos meus dias algo mais
descontraído;
Ao meu grande pop-pai, por acreditar sempre na minha capacidade. A você, todo o
meu respeito e admiração;
À minha super-mãe, exemplo de incentivo, sempre com um sorriso no rosto. A você
também, todo o meu respeito e admiração;
Aos meus abençoados irmãos, pela paciência, pelas risadas, pelas picuinhas e pelo
amor verdadeiro que temos uns pelos outros;
A toda a minha família, grande agradecimento pela torcida, incentivo e orações;
Ao meu noivo, Felipe Faria, que me apoiou em todos os momentos durante esta
caminhada, acreditando no meu potencial e me fazendo enxergar sempre o lado bom
da vida;
E por fim, um agradecimento mais que especial àqueles que não estão mais presentes
entre nós, mas que permanecem para sempre em meu coração, meu avô Olavo,
grande dentista, mas que sempre teve um “Q” de biólogo, meu tio Júlio, que desde
sempre foi meu ídolo e minha avó Elizabetha, mulher fantástica, exemplo de
perseverança e elegância.

Nossa maior fraqueza está em desistir. O
caminho mais certo de vencer é tentar mais
uma vez (Thomas Edison).

RESUMO
Para um bom funcionamento do organismo, é necessária uma regulação gênica
correta, na qual se refere ao controle da quantidade e do momento em que o produto
funcional de um gene é obtido. Para o entendimento dos processos moleculares que
governam os padrões de expressão gênica é importante identificar os elementos
regulatórios, como os enhancers. Os enhancers são elementos cis-regulatórios que
podem ativar a transcrição independentemente da sua localização, distância ou
orientação em relação ao gene que irão ativar. Para que ocorra a transcrição gênica
é necessário que haja a descompactação da cromatina e isso ocorre por meio de
modificações nas histonas (proteínas acopladas ao DNA). Tais modificações ajudam
a identificar as regiões que sofrem ação dos elementos cis- regulatórios. Uma técnica
que vem sendo muito utilizada para mapear a localização genômica de fatores de
transcrição e de modificações de histonas em células vivas é a imunoprecipitação da
cromatina (ChIP –Chromatin Immunoprecipitation). Uma aplicação direta para a
utilização de ChIP é o sequenciamento de nova geração (ChIP-Seq). Seguindo este
contexto, o objetivo do estudo é a padronização da técnica de ChIP para posterior
análise da ativação transcricional durante o processo de diferenciação de iPSCs em
cardiomiócitos.
Palavras-chaves: Regulação Gênica. Enhancers. Modificação de histonas. ChIP.

ABSTRACT
The well function of body is dependent of correct gene regulation which refers
to quantity and the correct time at the final product function is produced. To understand
the molecular process that governs the patterns of gene expression it is important to
identify the genome regulatory elements such as the enhancers. The enhancers are
cys-regulatory elements that can activate the transcription independently of their
location, distance and orientation in relation of the target gene. The gene transcription
occurs only when there are histones modifications and consequently the unpacked of
chromatin (DNA protein coupled). These chromatin modifications can identify specific
regions that suffer cys-regulatory elements actions. Recent technique which has been
widely used to map the genomic location of transcription factors and histones
modifications in vivo is called Chromatin Immunoprecipitation (ChIP). The next-
generation sequencing is a direct application of ChIP (ChIP-Seq). Following this
context, the main objective of this study is optimize ChIP technique for future analysis
of transcriptional activation during differentiation of iPS Cells into cardiomyocytes.
Keywords: Gene Regulation. Enhancers. Histone modifications. ChIP.

LISTA DE ILUSTRAÇÕES
Figura 1. Ilustração esquemática de região regulatória do gene..............................2
Figura 2. Esquema dos elementos regulatórios distais.............................................3
Figura 3. Esquema exemplificando o looping do DNA para ativação da regulação
gênica.........................................................................................................................4
Figura 4. Mecanismo de acetilação das histonas......................................................5
Figura 5. Mecanismo de metilação das histonas.......................................................6
Figura 6. Amostra preparada com apenas uma lavagem de PBS
1X...............................................................................................................................14
Figura 7. Curva de tempo de fragmentação, em minutos, com 3x106
células........................................................................................................................15

LISTA DE GRÁFICOS E TABELAS
Tabela 1. Anticorpos e suas funções..........................................................................10
Tabela 2. Primers para qPCR.....................................................................................12
Gráfico 1. Validação da reação de ChIP por qPCR....................................................16

LISTA DE ABREVIATURAS E SIGLAS
Acetil-CoA- Acetilcoenzima A
ACTN2- alfa actinina 2
BSA- bovine serum albumin ou soro albumina bovina
ChIP- Chromatin Immunoprecipitation ou imunoprecipitação da cromatina
ChIP-Seq- Chromatin Immunoprecipitation Sequencing ou sequenciamento da
imunoprecipitação da cromatina
CpG- citosine-phosphate-guanine ou citosina-fosfato-guanina
Cr- controle
Ct- cycle threshold ou ciclo limiar
dsDNA- Double strand DNA ou DNA dupla fita
DNA- ácido desoxirribonucleico
DPPA4- proteína associada ao desenvolvimento de pluripotência
FT- fator de transcrição
GATA4- fator de transcrição da família GATA
GWAS- genome-wide association ou estudo de associação genômica
h- horas
H3K4me1- monometilação da lisina 4 da histona 3
H3K4me3- trimetilação da lisina 4 da histona 3
H3K27Ac- acetilação da lisina 27 da histona 3
H3K27me3- trimetilação da lisina 27 da histona 3
InCor- Instituto do Coração

IP- material imunoprecipitado
iPSC- induced pluripotent stem cells ou células tronco pluripotentes induzidas
L- ladder ou marcador de bases
M- molar
mL- mililitros
NaCl- cloreto de sódio
Nanog- fator de transcrição nanog
ng- nanogramas
OCT4- proteína POU5F1- POU classe 5 homeobox 1
bp- base pairs ou pares de base
PBS- phosphate buffered saline ou tampão fosfato salino
PMSF- phenylmethylsulfonyl fluoride ou fluoreto de fenilmetilsulfonilo
qPCR- real-time quantitative polymerase chain reaction ou reação quantitativa em
cadeia da polimerase em tempo real
RCLs- Região controladora do locus
RNA- ácido ribonucleico
W- watts
°C- graus Celsius
µg- micrograma
µg/µL- micrograma por microlitro
µL- microlitro

Sumário 1. INTRODUÇÃO ..................................................................................................... 1
1.1 Elementos Regulatórios do Genoma.............................................................. 1
1.2 Enhancers ...................................................................................................... 3
1.3 Modificação de histonas ................................................................................. 4
1.4 Enhancers e doenças cardíacas .................................................................... 6
1.5 ChIP- Chromatin Immunoprecipitation ........................................................... 7
2. OBJETIVOS ......................................................................................................... 8
2.1 Objetivo Geral ................................................................................................ 8
2.2 Objetivos Específicos ..................................................................................... 8
3. MATERIAL E MÉTODOS ..................................................................................... 9
3.1 Cultura de células .......................................................................................... 9
3.2 Preparação da cromatina para Imunoprecipitação ......................................... 9
3.2.1 Fixação das proteínas no DNA ................................................................ 9
3.2.2 Preparação do núcleo .............................................................................. 9
3.2.3 Fragmentação do DNA ............................................................................ 9
3.2.4 Confirmação da fragmentação no tamanho exato ................................... 9
3.3 Imunoprecipitação da cromatina ...................................................................... 10
3.3.1 Bloqueio dos grânulos (beads) magnéticos ........................................... 10
3.3.2 Incubação dos beads magnéticos com anticorpo .................................. 10
3.3.3 Imunoprecipitação da cromatina ............................................................ 10
3.3.4 Eluição ................................................................................................... 11
3.4 Quantificação e confirmação- Qubit e qPCR ................................................ 11
3.4.1 Desenho dos primers para qPCR .......................................................... 11
4 RESULTADOS E DISCUSSÃO ......................................................................... 13
4.1 Padronização da preparação e fragmentação da cromatina ........................ 13
4.2 Padronização da Imunoprecipitação da Cromatina ...................................... 15

4.3 qPCR ........................................................................................................ 16
5 CONCLUSÃO .................................................................................................... 17
6 PERSPECTIVAS FUTURAS .............................................................................. 18
7 REFERÊNCIAS BIBLIOGRÁFICAS ................................................................... 19
Material suplementar..................................................................................................21

1
1. INTRODUÇÃO
Um organismo atravessa diversos processos biológicos, tais como
desenvolvimento, proliferação, apoptose, envelhecimento, diferenciação, entre tantos
outros. Para um bom funcionamento do organismo é necessária a regulação gênica
correta, pois caso contrário, diversas doenças podem ser desencadeadas. A
regulação gênica é um processo complexo no qual diversos elementos estão
envolvidos desempenhando diferentes funções que na maioria das vezes se
complementam. Sendo assim, a execução dos processos biológicos requer precisão
e cuidado de um conjunto de etapas que dependem da expressão espaço-temporal
adequada dos genes (MATSON et al., 2006).
A regulação da expressão gênica refere-se ao controle da quantidade e do
momento em que o produto funcional de um gene é obtido. O desenvolvimento e
função de um tecido é conduzido por um processo genético complexo que depende
tanto da expressão como da repressão de diversos genes. Essa regulação ocorre pela
ativação de muitos elementos, como potenciadores (enhancers) e fatores de
transcrição (FTs) (SAKABE et al., 2012a; SAKABE et al., 2012b).
As doenças cardíacas congênitas são mais comumente diagnosticadas no
nascimento exibindo prevalência de 4 a 50 por cada 1000 nascidos vivos e são as
principais causas de morbidade e mortalidade, as quais variam de acordo com a
severidade da doença, tanto em crianças como em adultos (BRUNEAU et al., 2008).
As doenças cardíacas congênitas ocorrem por alterações estruturais durante o
desenvolvimento embrionário, originando anormalidades na estrutura e na função do
coração. Estudos de associação genômica (GWAS) indicam que variações em
sequências não codificantes, como enhancers e FTs, influenciam na susceptibilidade
de variados tipos de doença humana (SAKABE et al., 2012b).
1.1 Elementos Regulatórios do Genoma
Para o entendimento dos processos moleculares que governam os padrões de
expressão gênica é importante identificar os elementos regulatórios do genoma.
Nos seres eucariotos, a transcrição de um gene codificante de proteína inicia-se
pela ação de uma enzima chamada RNA-polimerase II. Dentre os genes que essa

2
enzima codifica, há duas famílias de elementos cis-regulatórios do DNA: os
promotores e os elementos regulatórios distais (Figura 1) (MATSON et al., 2006).
Figura 1. Ilustração esquemática de uma região regulatória de gene, composta por elementos regulatórios
proximais e distais (Adaptado de MATSON et al., 2006).
A região promotora contém os elementos regulatórios proximais que são definidos
como regiões exatamente à jusante (upstream) do promotor nuclear dos genes que
irão regular.
Os elementos regulatórios distais são compostos por insuladores, silenciadores,
regiões controladoras do locus (RCLs) e enhancers.
Os insuladores são conhecidos como elementos de contorno e têm a função de
bloquear a transcrição dos genes vizinhos do gene que está sendo regulado. Os
insuladores possuem a capacidade de bloquear a comunicação entre o enhancer e a
região promotora e de evitar a repressão da cromatina (Figura 2a) (MATSON et al.,
2006).
Os silenciadores são elementos de sequência específica que conferem um efeito
negativo à transcrição de um gene alvo, impedindo a sua expressão. Os sítios de
ligação para FTs negativos são conhecidos como repressores (Figura 2b) (MATSON
et al., 2006).

3
As RCLs são regiões que influenciam a regulação da expressão de um locus. A
deleção destas regiões pode interferir na expressão normal de um gene (Figura 2c).
Os enhancers são elementos que atuam na regulação da expressão gênica,
independente de sua localização, distância ou orientação em relação ao gene que irão
regular (Figura 2d).
Figura 2. Esquema dos elementos regulatórios distais. a) Mecanismo de ação dos insuladores; b) Mecanismo de
ação dos silenciadores; c) Mecanismo de ação dos RCLs; d) Mecanismo de ação dos enhancers (Adaptado de
MATSON et al., 2006).
Com isso, podemos observar que os elementos que fazem a regulação da
expressão gênica compreendem disposição espacial linearizada e apresentam
controle rigoroso para que haja uma regulação correta, em que os enhancers, assim
como os outros, são parte essencial do processo.
1.2 Enhancers
Os enhancers são essenciais para a compreensão dos padrões de expressão dos
genes durante o desenvolvimento. Estes são classificados como elementos cis-
regulatórios, ou seja, promovem a expressão de genes localizados na mesma fita de
DNA (SAKABE et al., 2012b). São regiões que podem ativar a transcrição
independentemente da sua localização, distância ou orientação em relação à região

4
promotora do gene (BANERJI et al., 1981). Estão geralmente localizadas distantes
dos genes que irão regular e, por isso, se aproximam fisicamente destes através da
formação de um looping no DNA feito entre o promotor e o enhancer. Ao fazer esse
looping, o enhancer estabiliza-se próximo do gene alvo permitindo a interação entre
as proteínas ativadoras, formando um complexo entre elas (Figura 3). Os enhancers
são compostos por um conjunto de sítios de ligação para FTs e são ativados pela
ligação destes, após a modificação da cromatina, pela maquinaria celular.
Figura 3. Esquema exemplificando o looping do DNA para ativação do sistema de regulação gênica (Adaptado de
http://genmol.blogspot.com.br/2011_09_01_archive.html> acessado em 15 de abril, 2014).
Para que ocorra a transcrição gênica é necessário que haja a descompactação
da cromatina, o que ajuda a identificar as regiões que sofrem ação dos elementos cis-
regulatórios, que contribuem para a ligação dos FTs aos enhancers. Essa
descompactação ocorre por meio de modificações em proteínas que estão acopladas
ao DNA, as histonas.
1.3 Modificação de histonas
As modificações das histonas ocorrem após a tradução, no entanto, a questão de
como essas modificações alteram a conformação da cromatina foi algo descoberto
recentemente, com a alta resolução dos raios-X (LUGER et al., 1997).

5
A estrutura da cromatina apresenta muitas histonas básicas com caudas N-
terminais que podem projetar-se de seus próprios nucleossomos e fazer contato com
nucleossomos adjacentes. A modificação dessas caudas afeta as interações
internucleossomais que, consequentemente, afetam a estrutura geral da cromatina
(BANNISTER & KOUZARIDES, 2011). As principais modificações das histonas são
as acetilações e metilações.
As acetilações das lisinas são altamente dinâmicas e controladas pela ação
antagônica de dois tipos de enzimas, as acetiltransferases e deacetilases. As
acetiltransferases utilizam a acetil-CoA como cofator para catalisar a transferência de
um grupo acetil para um grupo amino da cadeia lateral da lisina. Essa transferência
neutraliza a carga positiva da lisina e isso acaba enfraquecendo as interações entre
histonas e DNA, deixando a cromatina mais aberta, sujeita a ações dos FTs e RNA-
polimerase II. Já a deacetilase faz o caminho contrário, compactando a cromatina
(Figura 4). A descompactação e compactação da cromatina ocasionam,
respectivamente, a ativação e o silenciamento gênico.
Figura 4. Mecanismo de acetilação das histonas (Adaptado de http://www.lookfordiagnosis.com> acessado em 26
de setembro, 2014).
As metilações, ao contrário das acetilações, não alteram a carga da histona e
geralmente ocorrem na cadeia lateral das lisinas e argininas. São modificações mais
complexas pois apresentam níveis de metilação, podendo ser classificadas como
mono-, di- ou tri-metiladas. A metilação ocorre nas ilhas CpG por meio da DNA-
metiltransferase, recrutando proteínas específicas que, por sua vez, irão recrutar as
enzimas deacetilases, resultando na perda da acetilação da histona, compactando a
cromatina. Assim, a metilação das histonas ocasiona o silenciamento gênico por meio
da metilação das ilhas CpG e perda da acetilação (Figura 5).

6
Figura 5. Mecanismo de metilação das histonas (Adaptado de http://www.lookfordiagnosis.com> acessado em 26
de setembro, 2014).
As modificações das histonas estão inseridas num contexto complexo de
entendimento da regulação da expressão gênica e consequentemente de eventuais
doenças causadas pelas alterações contidas nos elementos que medeiam tal
regulação (BANNISTER & KOUZARIDES, 2011).
1.4 Enhancers e doenças cardíacas
Um estudo realizado por May e colaboradores, em 2005, identificou 5047
enhancers candidatos no coração fetal e 2233 enhancers candidatos no coração
adulto, sendo que 1082 enhancers candidatos são comuns, ou seja, se mantiveram
após o desenvolvimento. Para analisar a relevância desses enhancers cardíacos, o
estudo se concentrou nos que estão localizados perto de genes relacionados com
diferentes tipos de doenças cardíacas. Através deste estudo, acredita-se que
variações em enhancers que regulam a expressão gênica cardíaca durante o
desenvolvimento embrionário podem conduzir a diversas doenças.
Uma técnica que vem sendo muito utilizada para mapear a localização genômica
de FTs e de modificações de histonas em células vivas para o entendimento das vias
de transcrição é a imunoprecipitação da cromatina (ChIP – Chromatin
Immunoprecipitation).

7
1.5 ChIP- Chromatin Immunoprecipitation
Em ensaios de imunoprecipitação da cromatina (ChIP) fatores de transcrição,
cofatores e outras proteínas de interesse da cromatina são enriquecidas por meio da
imunoprecipitação (LANDTet al., 2012).
A técnica de ChIP consiste na fixação das proteínas, lise dos núcleos e isolamento
da cromatina para a liberação e fragmentação do DNA em segmentos de
aproximadamente 200-500 pares de base (bp). Os fragmentos de DNA são
imunoprecipitados utilizando anticorpos específicos de acordo com o objetivo do
estudo.
Uma aplicação direta para a utilização de ChIP é o sequenciamento de nova
geração (ChIP-Seq) que visa o mapeamento das regiões de interesse que foram
imunoprecipitadas. Seguindo este contexto, o objetivo do estudo é a padronização da
técnica de ChIP em células tronco pluripotentes induzidas (iPSC) para posterior
estudo transcricional através da aplicação de ChIP-Seq.

8
2. OBJETIVOS
2.1 Objetivo Geral
O objetivo deste trabalho é a padronização da técnica de imunoprecipitação da
cromatina (ChIP) para analisar a ativação transcricional durante a diferenciação de
iPSC em cardiomiócito.
2.2 Objetivos Específicos
- Estabelecer os buffers apropriados para cada etapa da técnica;
- Estabelecer a quantidade de material necessária para a reação de
imunoprecipitação;
- Determinar o tempo ideal de fragmentação do material;
- Identificar as regiões e desenhar primers específicos para cada uma delas para
validação da imunoprecipitação.

9
3. MATERIAL E MÉTODOS
3.1 Cultura de células
As células foram cultivadas e fornecidas pelo grupo de iPSC do Laboratório de
Genética e Cardiologia e Molecular do Instituto do Coração (InCor/HC-FMUSP). 3x106
células foram ressuspendidas em 1 mL de PBS 1x gelado + 1 M de PMSF (inibidor de
protease).
3.2 Preparação da cromatina para Imunoprecipitação
3.2.1 Fixação das proteínas no DNA
Para a reação de fixação, o pellet foi ressuspendido em solução de fixação (Fixing
Buffer) com 1 % de formaldeído a cada 3x106 células. O tubo foi mantido em um
oscilador (Rocker), à temperatura ambiente, por 8-10 minutos. Para interromper a
reação de fixação foram adicionados 0,125 M de glicina e as amostras foram
colocadas no Rocker por 5 minutos à temperatura ambiente.
3.2.2 Preparação do núcleo
O pellet de células já fixado foi ressuspendido em solução de lise (Lysis Buffer) e
o tubo foi colocado no Rocker por 10 minutos, à 4°C. Em seguida, o pellet foi lavado
com PBS 1x gelado.
Após preparação dos núcleos estes foram ressuspendidos em 160 µL de solução
específica para o processo de fragmentação (Shearing Buffer).
3.2.3 Fragmentação do DNA
A fragmentação do DNA foi realizada no equipamento Bioruptor® (Life
Technologies, USA) em aproximadamente 200-500 bp. Os parâmetros foram
ajustados de acordo com as células utilizadas.
3.2.4 Confirmação da fragmentação no tamanho exato
Para confirmar se o material foi fragmentado corretamente foram coletados 10 µL
de cada amostra e submetidos à reversão da fixação (uncrosslink) (adição de 85 µL

10
de água e 4 µL de NaCl 5 M, por 16h à 65 °C). Após o tratamento com 2 µL de RNase
A (10 µg/µL) e 4 µL de proteinase K (20 µg/µL), as amostras foram purificadas com o
kit de purificação de PCR (Qiagen, Germany) e quantificadas no equipamento
NanoDrop 1000 (Thermo Scientific, USA). Cerca de 200 ng de cada amostra foram
submetidas a uma eletroforese em gel de agarose 1,5 %, por 45 minutos, à 90 W, e
através do fotodocumentador QUANTUM ST5 (Fisher Biotec, Australia), o padrão de
bandas pode ser observado, confirmando a fragmentação (Figura 6).
3.3 Imunoprecipitação da cromatina
3.3.1 Bloqueio dos grânulos (beads) magnéticos
Para realizarmos a imunoprecipitação da cromatina foram utilizados beads
magnéticos (Dynabeads® Protein G, Life Technologies, USA) previamente
bloqueados com BSA 0,5 %, por três incubações de 5 minutos no misturador (shaker).
3.3.2 Incubação dos beads magnéticos com anticorpo
Nesta etapa, 8 µL de anticorpo específico para a região de interesse foi adicionado
aos beads magnéticos, em shaker, à 4 ºC por 16h, para a formação de um complexo
bead + anticorpo, para posterior reação com DNA. Foram utilizados os anticorpos
H3K4me1, H3K27Ac, H3K27me3e H3K4me3 (Tabela 1) que têm como objetivo a
identificação da ativação e repressão de diversos elementos no momento em que se
encontra a célula.
Tabela 1. Anticorpos e suas funções.
Anticorpo Função
H3K4me1 Identificação da presença de enhancer ativo ou inativo
H3K4me3 Identificação de regiões promotoras de genes ativos
H2K27me3 Identificação de enhancers inativos
H3K27Ac Identificação de enhancers ativos
3.3.3 Imunoprecipitação da cromatina
A cromatina fragmentada foi diluída 10x com solução de diluição (ChIP dilution
buffer). Foram coletados 100 µL da cromatina diluída para controle do experimento
(armazenada em freezer -20 ºC até o momento do uso).

11
O restante da cromatina diluída foi adicionada ao complexo beads + anticorpo
para que ocorresse a imunoprecipitação das regiões específicas do DNA. Estes foram
mantidos por 16h no shaker, à 4 ºC.
Os beads foram precipitados utilizando uma estante magnética, o sobrenadante
foi descartado e os mesmos foram submetidos a uma série de lavagens para eliminar
as regiões que não foram imunoprecipitadas.
3.3.4 Eluição
As amostras imunoprecipitadas passaram por um processo de eluição com um
tampão específico (ChIP elution buffer). O processo consiste em ressuspender os
beads em 100 µL de tampão e incubar 3 vezes, por 5 minutos cada, à 65 ºC,
ressuspendendo os beads em cada intervalo. Após a incubação, salvar o
sobrenadante, pois nele estão contidas as regiões de interesse para o estudo. Esse
passo de eluição com ChIP elution buffer foi realizado duas vezes tendo como volume
final da amostra, 200 µL.
As amostras eluídas, juntamente com a amostra controle, foram submetidas ao
processo de uncrosslink e, ao final da purificação, foram eluídas em 20 µL de água
livre de DNase.
3.4 Quantificação e confirmação- Qubit e qPCR
3.4.1 Desenho dos primers para qPCR
Nesta etapa, as amostras foram quantificadas através do aparelho Qubit com o kit
Qubit® dsDNA BR Assay (Life Technologies, USA). Em seguida, foram congeladas à
-20 ºC para futura reação de qPCR como validação do experimento de
imunoprecipitação. O desenho dos primers foi realizado pelo software Primer 3
(http://bioinfo.ut.ee/primer3-0.4.0/> acessado em 10 de julho, 2014). Para a
localização das regiões promotoras em células iPS utilizamos o banco de dados
UCSC Genome Browser (https://genome.ucsc.edu/> acessado em 10 de julho, 2014)
(NANOG e DPPA4). Para a localização das regiões de enhancer inativo em iPSC,
utilizamos o banco de dados VISTA Enhancer Browser (http://enhancer.lbl.gov/>
acessado em 10 de julho, 2014) (ACTN2 e GATA4, genes ativos em cardiomiócito), e
para os enhancers ativos em iPSC e regiões intergênicas (Inter 1 e 2) utilizamos as

12
regiões citadas em um recente trabalho de Iglesias e colaboradores, 2011 (NAG e
OCT).
Tabela 2: Primers para qPCR
Forward Reverse
NANOG 5´-GTCCAGCCTGTTCCAAAAA-3´ 5´-TCCCTGTCCCATTGTGTCT-3´
DPPA4 5´-CCCTTCACACATCCTGCTC-3´ 5´-ATTCCTGGCCCTTTTCTGT-3´
ACTN2 5´-AAAATCCGTCCCAGACCTC-3´ 5´-TTCCAACCTCAACAAAACCA-3´
GATA4 5´-CCCCACCATCCTCTTCTCT-3´ 5´-AGAGGCAAGGGCAGATTTT-3´
NAG 5´-CCCAGGTTCAAGGGATTCT-3´ 5´-GCGAAACCCCCTCTCTACT-3´
OCT 5´-CCTCGGAAGTACCCAGTGA-3´ 5´-CCTTGGCAGTGGACCTTAG-3´
Inter 1 5'-CTCCCAAATTGCTGGGATTA-3' 5'-ATTCCAGGCACCACAAAAAG-3'
Inter 2 5'-AAAGCTGGACTGGTGAATGC-3' 5'-TCAAAGGCTCATCTTTGCAG-3'

13
4 RESULTADOS E DISCUSSÃO
4.1 Padronização da preparação e fragmentação da cromatina
Para a padronização da fase inicial da técnica de ChIP, nós avaliamos a
quantidade necessária de células para a preparação e fragmentação da cromatina.
Iniciamos o processo com cerca de 1x106 células para realizarmos a preparação
e fragmentação da cromatina e concluímos, pela quantificação após o uncrosslink,
que havia perda total de material. Desta forma, optamos por alterar o protocolo padrão
na fase anterior à fragmentação, substituindo as duas lavagens com solução de
lavagem (Wash Buffer) por apenas uma lavagem com PBS 1x gelado, sem
homogeneizar. Esta substituição permitiu a diminuição da degradação e consequente
perda do material durante a preparação do mesmo para fragmentação. Isso nos fez
concluir que o reagente responsável por tal acontecimento é o Tris-HCl por apresentar
capacidade de permeabilizar a membrana celular, servindo como um emulsificante já
que nossa membrana é composta por uma bicamada lipídica. O problema que nós
encontramos não foi somente atribuído à degradação que este buffer gera na
membrana celular, mas também por sua manipulação durante o processo, pela
formação excessiva de bolhas. Com a substituição, a quantidade de material obtido
aumentou cerca de 10x (~45 ng/µL).
Para determinarmos o melhor tempo de fragmentação do material, nós fizemos
uma curva para obtenção de fragmentos de 200-500 bp. No entanto, a partir da
observação da eletroforese em gel de agarose 1,5%, observamos que a amostra
coletada no tempo 0’ (sem passar pelo processo de fragmentação) estava degradada
(Figura 6a). Com isso, refizemos as soluções do processo de preparação da cromatina
com maior precisão na concentração, utilizamos um kit novo de purificação das
amostras e concluímos que o DNA estava totalmente íntegro (Figura 6b). Sem
degradação do material, prosseguimos com a curva de fragmentação, no qual o
melhor tempo observado para fragmentar o DNA de 1x106 células no tamanho
pretendido foi de 12’ (Figura 6c).

14
Figura 6. Amostra preparada com apenas uma lavagem de PBS 1x; L (ladder- 100 bp). a) Amostra 0´
degradada. b) Amostra 0´ sem degradação. c) Curva tempo de fragmentação.
No entanto, após a quantificação, nós obtivemos uma média de 45 ng/uL para
1x106 células, o que corresponde a, aproximadamente, 15 µg de material total
(Material Suplementar). Assumindo que começaríamos a padronização de reação de
imunoprecipitação com 10 µg de material para cada anticorpo, concluímos que 1x106
células não eram suficientes para a reação com os 4 anticorpos.
Sendo assim, utilizamos 3x106 células para realizarmos uma nova curva de tempo
de fragmentação, pois a quantidade de células é diretamente proporcional à
quantidade de ondas sonoras necessárias para a fragmentação do DNA, ou seja,
quanto maior o número de células, maior o tempo de fragmentação (Figura 7).
Observamos, através de eletroforese em gel de agarose 1,5 %, que o melhor tempo
obtido foi de 18 minutos uma vez que a maioria do material estava entre 200-500 bp.
Esse tempo foi escolhido, pois as amostras com o tempo de 20 minutos parecem estar
hiperfragmentadas, ou seja, por um excesso de exposição das amostras às ondas
sonoras houve a perda do epítopo da proteína o que prejudica a reação de
imunoprecipitação, já que uma vez perdido o epítopo, o anticorpo não tem local para
se ligar. A quantidade de material final aumentou para aproximadamente 100 µg, o
que nos possibilitou utilizar 20 µg de material para cada anticorpo na reação de
imunoprecipitação, nos deixando ainda uma sobra de material caso fosse necessário
repetir alguma reação.

15
Figura 7. Curva de tempo de fragmentação, em minutos, com 3x106 células. L (ladder- 100 bp).
4.2 Padronização da Imunoprecipitação da Cromatina
Para a padronização da imunoprecipitação da cromatina utilizamos cerca de 20
µg de material fragmentado para cada anticorpo.
Utilizamos os quatro anticorpos previstos para imunoprecipitação, sendo que, tal
como a literatura sugere, o anticorpo H3K4me3 serviu como controle (SCHMIDT et
al., 2009). A dosagem destas amostras foi feita por fluorescência captando apenas
sinal de dsDNA.
H3K4me3 IP- 1,23 ng/µL x 20 µL finais de amostra= 24,6 ng totais
H3K27me3 IP- 1,12 ng/µL x 2 0µL finais de amostra= 22,4 ng totais
H3K4me1 IP- 0,74 ng/µL x 20 µL finais de amostra= 14,8 ng totais
H3K27Ac IP- 1,12 ng/µL x 20 µL finais de amostra= 22,4 ng totais
Com este resultado, levando em consideração a comparação do valor obtido com
o anticorpo considerado controle positivo da reação (H3K4me3), podemos concluir
que o material foi imunoprecipitado, embora não possamos excluir a hipótese de
termos imunoprecipitado regiões do DNA que não são do nosso interesse, por meio
de ligações inespecíficas do anticorpo. Sendo assim, prosseguiremos com o teste de
qPCR para validação da reação.
16
4.3 qPCR
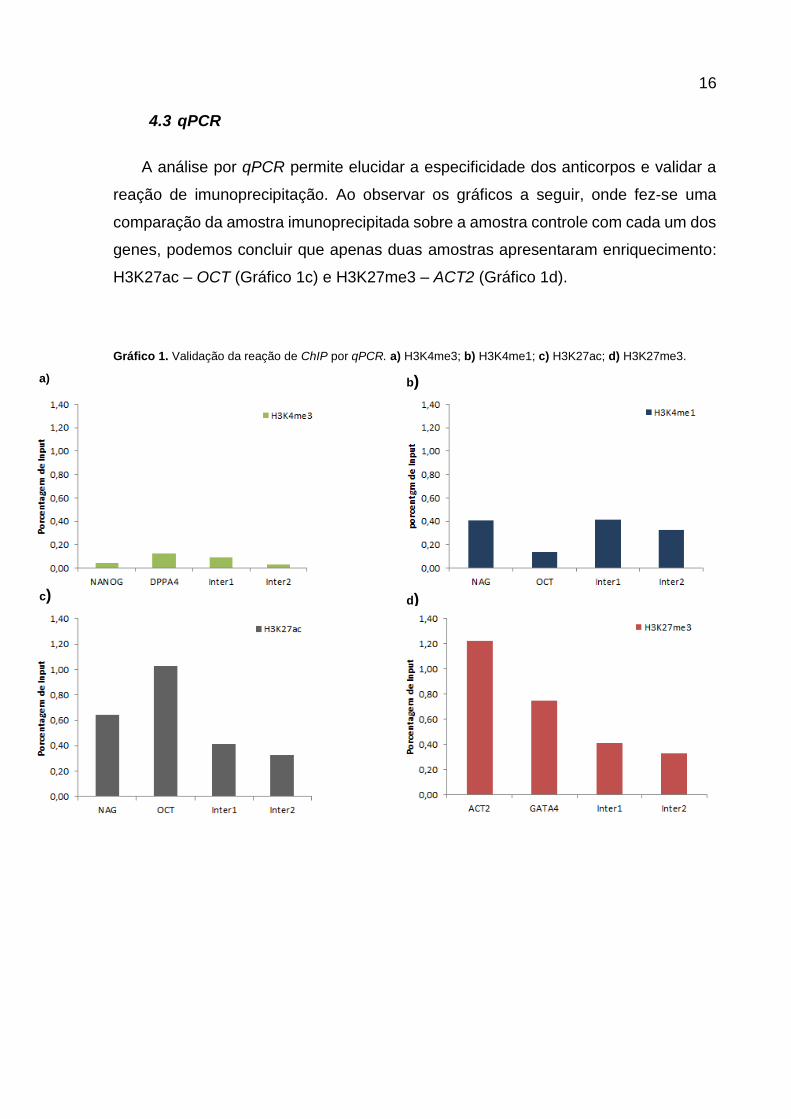
A análise por qPCR permite elucidar a especificidade dos anticorpos e validar a
reação de imunoprecipitação. Ao observar os gráficos a seguir, onde fez-se uma
comparação da amostra imunoprecipitada sobre a amostra controle com cada um dos
genes, podemos concluir que apenas duas amostras apresentaram enriquecimento:
H3K27ac – OCT (Gráfico 1c) e H3K27me3 – ACT2 (Gráfico 1d).
Gráfico 1. Validação da reação de ChIP por qPCR. a) H3K4me3; b) H3K4me1; c) H3K27ac; d) H3K27me3.
a) b)
c) d)

17
5 CONCLUSÃO
A padronização de ChIP em células tronco pluripotentes induzidas (iPSC)
apresentou diversas limitações, pela inexistência de estudos que relatou a aplicação
desta técnica neste tipo celular. Desta forma, o processo foi repetido diversas vezes,
variando a quantidade de material e os buffers a serem utilizados em cada etapa.
Concluímos que a substituição da lavagem com Wash Buffer por PBS 1x gelado
foi essencial para evitar perdas ou degradação do material e que a utilização de 3x106
células fornece a quantidade ideal para obter 20 µg de material fragmentado para
cada anticorpo, o que permite realizar a reação de imunoprecipitação e montagem da
biblioteca para sequenciamento de nova geração (ChIP-Seq).
Os resultados obtidos por qPCR podem ser explicados por uma possível
hiperfragmentação do material no qual provocou a perda do epítopo e consequente
falha na reação de imunoprecipitação, pela inespecificidade dos primers desenhados
para a validação da mesma ou por uma variação nos lotes dos anticorpos utilizados.
A padronização desta técnica é fundamental para a futura investigação da
atividade transcricional da iPSC e da sua diferenciação, proporcionando o
entendimento de mecanismos naturais de doenças congênitas.

18
6 PERSPECTIVAS FUTURAS
Realizar uma nova curva de tempo de fragmentação e seguir com um Westernblot
para ver se epítopo se mantém na amostra;
Pegar novas referências de lotes dos anticorpos para a reação de
imunoprecipitação;
Verificar a especificidade dos primers utilizados para a validação e redesenhar
aqueles utilizados para controle da reação de qPCR (regiões intergênicas);
Terminar a padronização da técnica.

19
7 REFERÊNCIAS BIBLIOGRÁFICAS
BANERJI, J., RUSCONI, S. & SCHAFFNER, W. Expression of a beta-globin gene is
enhanced by remote SV40 DNA sequences. Cell, v.27, p. 299–308,1981.
BANNISTER, A.J.; KOUZARIDES, T. Regulation of chromatin by histone
modifications.CellResearche, v. 21, p. 381-395, 2011.
BRUNEAU, B. G. The development genetics of congenital heart disease.Nature, v.
451, p. 943-948, 2008
IGLESIAS, A. R.; BAJPAI, R.; SWIGUT, T.; et al. A unique chromatin signature
uncovers early developmental enhancers in humans. Nature, v. 470, p. 279-283, 2011
LANDT, S. G.; MARINOV, G. K.; KUNDAJE, A.; et al. ChIP-seq guidelines and
practices of the ENCODE and modENCODE consortia. Genome Res., v. 22, p. 1813-
1831, 2012
LUGER K, MADER AW, RICHMOND RK, SARGENT DF, RICHMOND TJ. Crystal
structure of the nucleosome core particle at 2.8 A resolution. Nature, v. 389, p. 251-
260, 1997.
MATSON, G. A.; EVANS, S. K.; GREEN, M. R. Transcriptional Regulatory Elements
in the Human Genome.Annu Rev Genomics Hum Genet., v. 7, p. 29-59, 2006
MAY, D.; BLOW, M. J.; KAPLAN, T. et al. Large-scale discovery of enhancers from
human heart tissue.Nat.Genet., v. 44, p. 89-93, 2012
SAKABE, N. J.; ANEAS, I; SHEN, T. et al. Dual transcriptional activator and repressor
roles of TBX20 regulate adult structure and fuction. Hum MolGenet., v. 21, p. 22194-
2204, 2012
SAKABE, N. J.; SAVIC, D.; NOBREGA, M. A. Transcriptional enhancers in
development and disease. Genome Biol., v. 13, p. 238, 2012
SCHMIDT, D.; WILSON, M. D.; SPYROU, C.; et al. ChIP-Seq: Using high-throughput
sequencing to discover protein-DNA interactions. Elsevier, v. 48, p. 240-248, 2009

20
http://genmol.blogspot.com.br/2011_09_01_archive.html> Acessado em 15 de abril,
2014
http://www.lookfordiagnosis.com> Acessado em 26 de setembro, 2014
http://www.lookfordiagnosis.com> Acessado em 26 de setembro, 2014
http://bioinfo.ut.ee/primer3-0.4.0/> Acessado em 10 de julho, 2014
https://genome.ucsc.edu/> Acessado em 10 de julho, 2014
http://enhancer.lbl.gov/> Acessado em 10 de julho, 2014

21
Material Suplementar
Fixing Buffer
50 mM HEPES, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% formaldeído.
Lysis Buffer
50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 10 % glicerol, 0.5 % NP-40, 0.25 %
TRITON X-100, inibidor de proteína.
PBS 1x
8 g NaCl, 0,2 g KCl, 1,44 g Na2HPO4, 0,24 g KH2PO4.
Wash Buffer
10 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA, 0,5 mM EGTA, inibidor de proteína.
Shearing Buffer
1 mM EDTA, 10 mM Tris-HCl, 0.1 % SDS, inibidor de proteína.
ChIP dilution Buffer
Basic Lysis Buffer, TRITON X-100, dTT
Basic Lysis Buffer
140 mM NaCl, 15 mM HEPES, 1 mM EDTA, 0,5 mM EGTA, 0,1 % deoxicolato de
sódio
ChIP elution Buffer
1 % SDS, 0,1 M NaHCO3, água livre de DNase e RNase.

22
Cálculo quantidade final de material após quantificação no NanoDrop
45 ng---------------- 1 µL
X ---------------- 20 µL (eluição pós-purificação)= 10 µL de amostra pós-fragmentação
X= 0,9 µg de material em 10 µL de amostra
0,9 µg ---------------- 10 µL
X’ ---------------- 160 µL
X’= 14,4 µg de material total (~15 µg)
Top Related