
Análise de elementos moduladores dos níveis da enzima COX ...‡ÃO... · O conhecimento torna a...
103
UNIVERSIDADE FEDERAL DE OURO PRETO NÚCLEO DE PESQUISAS EM CIÊNCIAS BIOLÓGICAS PROGRAMA DE PÓS-GRADUAÇÃO EM BIOTECNOLOGIA Análise de elementos moduladores dos níveis da enzima COX-2 em células H9c2 infectadas com Trypanosoma cruzi; cepa Berenice-62 Suianne Letícia Antunes Mota Março- 2014
-
Upload
vuongkhanh -
Category
Documents
-
view
213 -
download
0
Transcript of Análise de elementos moduladores dos níveis da enzima COX ...‡ÃO... · O conhecimento torna a...

UNIVERSIDADE FEDERAL DE OURO PRETO
NÚCLEO DE PESQUISAS EM CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO EM BIOTECNOLOGIA
Análise de elementos moduladores dos níveis da enzima COX-2
em células H9c2 infectadas com Trypanosoma cruzi; cepa Berenice-62
Suianne Letícia Antunes Mota
Março- 2014

UNIVERSIDADE FEDERAL DE OURO PRETO
NÚCLEO DE PESQUISAS EM CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO EM BIOTECNOLOGIA
Análise de elementos moduladores dos níveis da enzima COX-2
em células H9c2 infectadas com Trypanosoma cruzi; cepa Berenice-62
Suianne Leticia Antunes Mota
Orientadora
Karen Cristiane Martinez Moraes
Dissertação de Mestrado apresentada ao Programa
de Pós-Graduação em Biotecnologia da
Universidade Federal de Ouro Preto como requisito
parcial para obtenção do Título de Mestre em
Bioquímica e Biologia Molecular.
Ouro Preto- Minas Gerais
Março- 2014



II
Dedico este trabalho a meus pais que mesmo estando longe, me ensinaram a não
desistir e enfrentar as dificuldades de cabeça erguida sem jamais humilhar o outro
para alcançar meus objetivos.

III
O conhecimento torna a alma jovem e diminui a amargura da velhice. Colhe, pois,
a sabedoria. Armazena suavidade para o amanhã.
Leonardo Da Vinci

IV
AGRADECIMENTOS
Primeiramente a Deus, por tua infinita presença e pelas benções derramadas em minha
vida.
Aos meus pais Eurides e Gilberto pelo amor incondicional, conselhos e por me
fortalecerem ainda mais nesta caminhada.
Aos meus irmãos Lívia e Filipe pelo carinho e motivação.
A orientadora Dra Karen Cristiane Martinez de Moraes pela oportunidade e confiança.
Agradecimento especial à FAPEMIG pelos recursos financiados para a execução desse
trabalh
Ao Laboratório de Doença de Chagas, especialmente a professora Dra Terezinha Bahia,
Lívia Diniz e a Ludimila por me receberem de portas abertas e concessão do uso de seus
equipamentos.
Ao professor Luis Crocco pela concessão do uso de seu laboratório e equipamentos.
Aos professores do mestrado que em momento algum omitiram seus conhecimentos
frente as minhas dúvidas.
Aos amigos do LBBM, Walmir, Cintia, Andrea, Brenda, Fabiano, Érica, Victor, Sol e
Esther pelo os bons momentos.
Aos meus colegas de casa, Walmir, Gustavo, Rodrigo, Diogo, Valquíria e Walyson pela
ótima convivência. Sentirei muita falta de vocês.
Aos amigos feitos em Ouro Preto, Patrícia, Rory, Helton, Matheus, Maurício, Hellem,
Bijay, Deena, João, Fabiano e Barbara pelos bons momentos e por tornarem minha
estadia mais divertida.
As amigas de Moc, Pollyana, Sabrina, Amanda, Lorena, Tamyres pelo apoio e torcida.

V
SUMÁRIO
AGRADECIMENTOS ................................................................................................... IV
RESUMO ..................................................................................................................... VIII
ABSTRACT ..................................................................................................................... X
LISTA DE TABELAS ................................................................................................... XII
LISTA DE FIGURAS .................................................................................................. XIII
ABREVIATURAS ....................................................................................................... XIV
1- INTRODUÇÃO ............................................................................................................ 1
1.1 Aspectos gerais da doença de Chagas e do Trypanosoma cruzi ......................... 2
1.2 Ciclo de vida do Trypanosoma cruzi ................................................................... 4
1.3 O processo de adesão e invasão do parasito ........................................................ 6
1.4 Ciclooxigenase-2 (COX– 2) e o processo inflamatório / hipertrófico na doença
de Chagas .................................................................................................................. 8
1.5 Atuação de PTEN e Akt na modulação dos níveis da enzima COX-2 na
hipertrofia cardíaca chagásica ................................................................................ 12
1.6 miRNAs e a regulação da expressão gênica nas hipertrofias cardíacas ............ 15
1.7 A importância do modelo de estudo celular e a busca por marcadores
moleculares na doença de Chagas ........................................................................... 19
1.8 Justificativa ........................................................................................................ 20
2- OBJETIVOS ............................................................................................................... 22
2.1 Objetivo Geral ................................................................................................... 23
2.2 Objetivos específicos ......................................................................................... 23
3- MATERIAIS E MÉTODOS ....................................................................................... 24
3.1 Cultura celular ................................................................................................... 25
3.2 Obtenção de formas tripomastigotas do T. cruzi e infecções celulares ............. 25
3.3 Avaliação da infeção da cepa Be – 62 em células H9c2 por ensaios de ..............
microscopia ............................................................................................................. 26
3.3.1 Microscopia de contraste de fase: coloração com Giemsa ............................. 26
3.3.2 Microscopia de fluorescência: coloração com 4,6 diamino-2-fenilindol ....... 26
3.3.3 Percentual de Infecção .................................................................................... 27
3.4 Analisando a viabilidade celular: Os ensaios de citotoxicidade ........................ 27

VI
3.5 Níveis de Prostaglandina E2 .............................................................................. 27
3.6 Extração do RNA total e transcrição reversa .................................................... 28
3.6.1 Extração do RNA total ................................................................................... 28
3.6.2 Transcrição reversa ......................................................................................... 28
3.7 Análises transcricional de genes em reações de PCR quantitativa em tempo ......
real (qPCR) .............................................................................................................. 29
3.8 Análises da expressão de proteínas por Western blotting ................................. 30
3.8.1 Preparação do extrato proteico ....................................................................... 30
3.8.2 Transferência e revelação das membranas ..................................................... 31
3.9 Análises do padrão de expressão de miRNAs ................................................... 31
3.10 Análises de bioinformática e construções de redes moleculares ..................... 33
3.11 Análises estatísticas ......................................................................................... 33
3.12 Considerações éticas ........................................................................................ 33
4. RESULTADOS .......................................................................................................... 34
4. 1 Análise da infecção da cepa Be – 62 em células H9c2 .................................... 35
4.2 Investigando a sobrevivência celular pós-infecção com a cepa Be-62 .................
do T. cruzi: os ensaios de viabilidade celular .......................................................... 38
4.3 RNAm relacionados com processo hipertrófico ............................................... 38
4.4 A infecção parasitária modula o ambiente pró-inflamatório: a mensuração de
prostaglandina E2 (PGE2) ....................................................................................... 40
4.5 A expressão do RNAm e da proteína COX-2 estão aumentadas durante o..........
processo pró-inflamatório induzido por T. cruzi ..................................................... 40
4.6 A expressão gênica de PTEN encontra-se modulada nos tempos iniciais de ......
infecção por T. cruzi ................................................................................................ 42
4.7 A fosforilação da proteína PTEN é menor no intervalo de 0 horas de infecção ..
com T. cruzi ............................................................................................................. 43
4.8 A expressão do RNAm Akt 1 e Akt 2 está aumentada durante o processo ........
pró-inflamatório induzido por T. cruzi .................................................................... 44
4.9 Análise da expressão de miRNAs ..................................................................... 44
4.9.1 miRNAs com alvos em COX-2 ...................................................................... 45
4.9.2 miRNAs com alvos em PTEN ........................................................................ 46
4.9.3 miRNAs relacionados com o processo hipertrófico cardíaco ........................ 47

VII
4.10 Os miRNAs – 1, -21,-26b podem modular a expressão de genes relacionados
ao processo hipertrófico/ inflamatório .................................................................... 48
5. DISCUSSÃO .............................................................................................................. 50
6. CONCLUSÕES .......................................................................................................... 60
7. REFERÊNCIAS BIBLIOGRÁFICAS ....................................................................... 62

VIII
RESUMO
A doença de Chagas, causada pelo protozoário Trypanosoma cruzi, é responsável por
uma significante mortalidade e morbidade nas Américas Central e do Sul, afetando um
número significativo da população mundial. A doença desenvolveu-se ao longo de
décadas e a presença de mediadores inflamatórios e a hipertrofia cardíaca são
características comuns. Buscando uma compreensão dos mecanismos moleculares que
ocorrem no processo inflamatório e que subsidiem a caracterização de marcadores
moleculares na patologia chagásica, a proteína COX-2 tornou se objeto de estudo. Sabe-
se que a regulação desta proteína pode se dar a nível transcricional e/ou pós traducional,
envolvendo um conjunto de fatores protéicos e incluindo a participação de miRNAs.
Estudos recentes tem mostrado uma possível co-regulação indireta entre os níveis de
PTEN e de COX-2, mas até o momento, os resultados preliminares ainda não
elucidaram esses mecanismos. Particularmente, em relação à Doença de Chagas, poucos
são os estudos que focam na elucidação do processamento do mRNAs na doença e nos
fatores que co-regulam a proteína COX-2. Sendo assim, objetivou se nesse estudo
analisar os moduladores do processo pró-inflamatório/ hipertrófico em células H9c2
infectadas pela cepa Berenice-62 do T.cruzi, buscando a caracterização de marcadores
moleculares do processo inicial da Doença de Chagas. Para isso, os tempos de infecção
de 0, 2, 6, 12, 24 e 48 horas foram estudados, logo após 2 horas de interação com o
parasito. A comprovação da infectividade foi feita por ensaios de microscopia pela
coloração com Giemsa e Dapi. A análise transcricional de genes correlatos ao processo
inflamatório/ hipertrófico e de microRNAs foram realizados por ensaios de qRT-PCRs e
as análises das proteínas COX-2, PTEN, P-PTEN foram investigadas nos ensaios de
Western blots. Outros ensaios bioquímicos, como o de viabilidade celular e o de
mensuração de prostaglandina E2, foram realizados. Com os resultados, conclui-se que
a infecção com a cepa Be-62 diminui a viabilidade celular após 48 horas de infecção e
modula os níveis de expressão dos marcadores de hipertrofia cardíaca Anf, Bnp e β –
myhc, além de modular o ambiente pró-inflamatório nos intervalos iniciais de infecção,
considerando-se o aumento da expressão e da atividade da proteína COX-2 e no
aumento na produção de PGE2. Os resultados também demonstraram uma relação entre
os genes Cox-2, Pten, Akt, sugerindo uma possível correlação cruzada entre COX-2 e

IX
PTEN. Os microRNAs-26b, -463,-1 e -21, respectivamente, indicaram uma possível
regulação pós-traducional das proteínas COX-2 e PTEN, demonstrando um papel
fundamental na instalação do quadro de hipertrofia. Estudos futuros poderão validar o
papel dessas moléculas como marcadores moleculares e espera-se que os resultados do
presente estudo possam direcionar a caracterização de moléculas que auxiliem no
prognóstico e tratamento da doença de maneira direcionada, considerando-se a grande
variabilidade genética das cepas do T. cruzi.

X
ABSTRACT
The Chagas disease, caused by the protozoan Trypanosoma cruzi, is responsible for
significant mortality and morbidity in Central and South America, affecting a
significant number of the world population. The disease develops over decades and the
presence of inflammatory mediators and cardiac hypertrophy are common features.
Searching for an understanding of the molecular mechanisms that occur during the
inflammatory process, and that subsidize the characterization of molecular markers in
Chagas disease, COX - 2 protein became an object of study. It is known that the
regulation of this protein can occur at transcriptional and/ or post- translational level,
involving a number of protein factors and including the involvement of miRNAs.
Recent studies have shown a possible indirect co-regulation in the levels of PTEN and
COX - 2, but so far the preliminary results not yet elucidated these mechanisms.
Particularly in relation to Chagas disease, there are few studies that focus on the
elucidation of the processing of mRNAs in this disease and in the factors that co -
regulate COX - 2 protein. Therefore, this study was aimed to analyze the hypertrophic/
proinflammatory process modulators in H9c2 cells infected with strain Berenice -62 of
the Trypanosoma cruzi, searching for characterize the molecular markers of the initial
process of Chagas disease. For this reason, the time of infection of 0, 2, 6, 12, 24 and 48
hours were studied soon after 2 hours from the interaction with the parasite. The proof
of the infectivity was performed by microscopy assays by the staining with Giemsa and
Dapi. The transcriptional analysis of genes related to the inflammatory / hypertrophic
process and microRNAs were performed by qRT- PCR assays and the analyzes of
COX-2, PTEN, PTEN - P proteins were explored in Western blots assays. Other
biochemical assays such as cell viability and measurement of prostaglandin E2 were
performed. From the results, it is concluded that infection with Be- 62 strain decreases
cell viability after 48 hours of infection and modulates the levels of expression of
markers of cardiac hypertrophy Anf and Bnp β - myhc, in addition to modulate the
environment pro-inflammatory in the initial intervals of infections, considering the
increased expression and COX -2 protein activity and in the increased production of
PGE2. The results also demonstrated a relationship among cox-2, pten, akt genes
suggesting a possible cross- correlation between COX-2 and PTEN. MicroRNAs-26b, -

XI
463, -21 and -1 respectively indicate a possible posttranslational regulation of COX-2
and PTEN protein, demonstrating a crucial role in the installation of hypertrophy.
Future studies could validate the role of these molecules as molecular markers and it is
expected that the results of this study may direct the characterization of molecules that
assist in the prognosis and treatment of disease in targeted way, considering the high
genetic variability of strains of T. cruzi.

XII
LISTA DE TABELAS
Tabela 01: Sequências dos oligonucleotídeos utilizados na análise da expressão dos
gene.
Tabela 02: Sequências dos miRNAs e alvos envolvidos no processo hipertrófico e na
modulação de COX-2 e PTEN.
Tabela-3. miRNAs e genes alvos

XIII
LISTA DE FIGURAS
Figura 01: Morfologia das formas do T. cruzi.
Figura 02: Ciclo evolutivo do Trypanosoma cruzi
Figura 03: Esquema ilustrativo da via do ácido araquidônico
Figura 04: Representação esquemática da via PI3K/ Akt
Figura 05: Relação entre as proteínas PTEN, AKT e COX-2 na modulação do processo
hipertrófico mediado por infamação
Figura 06: Biogênese dos miRNAs
Figura 07: Análise do produto transcricional do gene β-actina.
Figura 08: Análise celular da infecção de células H9c2 pela cepa Berenice-62
Figura 09: Microscopia de fluorescência após 48 h de infecção da célula H9c2 pelo
parasito Be-62.
Figura 10: Índice de Infecção de células cardíacas H9c2 pela cepa Berenice-62
Figura 11: Número de amastigotas por célula infectada.
Figura 12: Viabilidade de células H9c2 infectada com T. cruzi Cepa Be-62.
Figura 13: Níveis de expressão do RNAm de genes relacionados ao processo
hipertrófico.
Figura 14: Níveis de PGE2 produzidos durante a infecção da célula H9c2 pela cepa Be-
62 do T. cruzi
Figura 15: Níveis de expressão da proteína e do RNAm de COX-2
Figura 16. Níveis de expressão da proteína e do RNAm de PTEN
Figura 17. Níveis de fosforilação da enzima- PTEN
Figura 18. Níveis de expressão do RNAm de Akt 1 e Akt 2
Figura 19. Níveis de expressão dos miRNAs com alvo em COX-2.
Figura 20. Níveis de expressão dos miRNAs com alvo em PTEN.
Figura 21. Níveis de expressão dos miRNAs envolvidos no processo hipertrófico.
Figura 22. Rede de interação entre miRNAs e seus alvos relacionados ao processo
hipertrófico mediado por inflamação.

XIV
ABREVIATURAS
AA: Ácido Araquidônico .
AKT: serina-treonina quinase
ANF: Fator natriurético Atrial
ATP: Adenosina Trifosfato
BE-62: Berenice-62
BNP: Peptídeo Natriurético cerebral
Β-MYHC: Cadeia pesada de β-miosina
BSA: Albumina Sérica Bovina
CCC: Cardiomiopatia Chagásica Crônica
CK2: proteína-quinase
COX-2: Ciclooxigenase-2
COX-1: Ciclooxigenase-1
CREB: do Inglês ( cAMP response element binding protein),
cDNA: DNA complementar
DAPI: 4',6-diamidino-2-fenilindol
DMEM : Dulbecco’s Modified Eagle’s Medium
DMSO: Dimetilsulfóxido
DNTP: Desoxirribonucleotídeos Fosfatados
DTT: Ditiotreitol
EDTA: Ácido etilenodiamino tetra-acético
GPI: glicosilfosfatidilinositol
GPS: glicoproteínas
IFN: Interferon
IL: Interleucina
IL-1β: interleucina 1-Beta
IL-8: interleucina 8
MTT: (3-(4,5-Dimetiltiazol-2-yl)-2,5- brometo difeniltetrazolio
miRNA: microRNA
NFAT: Fator Nuclear de Células T Ativadas
NF-IL6: O Fator Nuclear de Interleucina 6

XV
NF-kB: Fator de Transcrição Nuclear κB
PBS: Tampão Fosfato Salino
PCR: Reação em Cadeia da Polimerase
PGIs: prostaciclinas
PGs: prostaglandinas
PGE2: Prostaglandina E2
PIP-3 – Fosfatidil Inositol 3 Fosfato
PIP-2 – Fosfatidil Inositol 2 Fosfato
PI3K – Fosfatidil Inositol 3 Quinase
PDK1 – Quinase 1 dependente de fosfoinositídeos-3
Pri-miRNA – miRNA primário
Pré-miRNA – Precursor de miRNA
PLA2: Fosfolipase A2
PTEN: proteína Fosfatase e Tensina Homóloga
qRT-PCR: do inglês, Quantitative Reverse Transcription PCR
RNA: Ácido Ribonucleico
RNAm: RNA mensageiro
SDS: Sulfato Dodecil de Sódio
SNPs: polimorfismo de nucleotídeo simples
TBS: Tampão de Tris Salina
TNF: Fator de Necrose Tumoral
Tris: Tris(hidroximetil)aminometano
TBXs:tromboxanos

1
1. INTRODUÇÃO

2
1.1 Aspectos gerais da doença de Chagas e do Trypanosoma cruzi
A doença de Chagas é uma enfermidade causada pelo protozoário Trypanosoma
cruzi, o qual pertence à ordem Kinetoplastida e família Trypanosomatidae (CHAGAS;
1909). Essa família é caracterizada pela existência de um único flagelo e de uma
organela denominada cinetoplasto (TELLERIA et al., 2006). Estima-se que dez
milhões de pessoas estejam infectadas em todo mundo e aproximadamente 120.000
encontrem-se em área de risco, sendo que a maioria está distribuída na América Latina
(CORRAL et al; 2013). No Brasil, aproximadamente cinco milhões de pessoas estão
infectadas com o T. cruzi, das quais 60% residem em áreas urbanas (GILBER et al.,
2013).
A principal forma de transmissão da doença se dá por via vetorial; insetos
hematófagos que pertencem à ordem Hemíptera, família Reduviidae, subfamília
Triatominae (GAUNT e MILES, 2000) que depositam as formas infectantes do parasito
juntamente às suas fezes no momento do repasto sanguíneo em um hospedeiro
vertebrado (MAZZA, 1936; BARRETO, 1990; DIAS, 1994). Existem também outros
modos de transmissão, tais como: transfusão de sangue, congênita, via oral, acidentes de
laboratório, manejo de animais infectados, transplante de órgãos e por intermédio do
leite materno (BRASIL, 2008).
A doença de Chagas apresenta duas fases; aguda e crônica. A fase aguda, que
compreende o período de até 3 a 4 meses após a infecção (PRATA et al; 1994), é
caracterizada por intensa proliferação do parasito nos tecidos do hospedeiro com
consequente reação inflamatória e presença de formas tripomastigotas no sangue
periférico. Os pacientes nessa fase são geralmente assintomáticos, entretanto podem
apresentar sinais de porta de entrada do parasito e/ou sintomas como: febre passageira,
linfoadenopatia, esplenomegalia branda e, mais raramente, uma miocardite intensa
(RASSI et al., 2000).
Na fase crônica os pacientes podem permanecer assintomáticos (fase
indeterminada) ou apresentar-se na forma cardíaca, digestiva ou mista. No entanto, a
forma cardíaca é uma das formas mais graves, pois é a principal responsável pela
ocorrência de morte súbita devido à miocardite crônica progressiva e fibrosante e/ou
hipertrofia do coração (BRENER et al., 2000 ).

3
Aproximadamente 20 a 30% dos doentes desenvolvem alterações no coração e,
apesar dos pacientes na fase indeterminada da doença Chagas terem, geralmente, um
bom prognóstico, essa situação pode mudar para um estágio avançado com
manifestações clínicas graves (ROCHA et al., 2003). A cardiopatia chagásica crônica
(CCC) pode evoluir para uma cardiomiopatia dilatada inflamatória infecciosa
(ANDRADE et al.,1983). No entanto a sua patogenia não está totalmente esclarecida,
sendo propostos mais estudos para explicar a gênese das lesões, mecanismos
inflamatórios ligados ao T. cruzi. (TAFURI, 1985; HIGUSHI, 2001).
A prevalência das diversas formas clínicas da doença tem sido atribuída ao perfil
genético diversificado da população de parasito, uma vez que os mecanismos de invasão
parasitária têm contribuído para a variabilidade genética de cepas do T. cruzi, sendo
uma questão fundamental para a compreensão dessa patologia (PIRES et al; 1996).
Considerando como uma espécie heterogênea, o parasito consiste em várias
subpopulações, que apresentam cepas distribuídas em hospedeiros mamíferos
(ZINGALES et al., 1998). Diversos estudos têm demonstrado que no sangue periférico
de animais infectados com diferentes cepas do T. cruzi são encontradas formas
tripomastigotas delgadas, largas e muito largas (BRENER e CHIARI; 1963).
Em 1909, Carlos Chagas já havia descrito a predominância de formas finas e
largas do parasito circulante (CHAGAS, 1909), mas foi em 1977 que Brener sugeriu a
caracterização do T. cruzi em dois tipos polares baseado na morfologia e no tropismo
tecidual do parasito. Cepas que apresentam a forma fina induzem infecções com altos
níveis de parasitemia e geralmente apresentam suscetibilidade a agentes
quimioterápicos, tendo tropismo maior por macrófagos. Já as formas mais largas
possuem infecções com baixos níveis de parasitemia e normalmente são resistentes a
agentes quimioterápicos, com tropismo maior por células musculares (BRENER et al;
1977).
Paralelamente, Andrade (1985) também sugeriu uma classificação baseada nos
critérios morfológicos e no tropismo tecidual, no qual cepas finas, como a cepa Y,
teriam um tropismo maior por células hematopoiéticas e fibroblastos e, as cepas mais
largas, como as cepas CL, Brasil, Colombiana, teriam um tropismo preferencial por
células cardíacas e esqueléticas. Em estudos feitos por Brener (1965), foi descrito um
tipo de cepa do T. cruzi que até então era desconhecida, a cepa Berenice 62 (Be-62), que

4
havia sido isolada três anos antes, em 1962, através do xenodiagnóstico da paciente
Berenice (primeira paciente infectada observada por Carlos Chagas) em Lassance, MG.
Esta cepa apresenta a forma delgada, induz infecções com altos níveis de parasitemia e
apresenta susceptibilidade a agentes quimioterápicos. No entanto, pouco se conhece
sobre os mecanismos de atuação da mesma envolvida na cardiopatia chagásica, uma vez
que há poucos dados na literatura sobre sua investigação.
Para a confirmação do diagnóstico, é necessário considerar os antecedentes
epidemiológicos do paciente, levando em consideração a possibilidade de contato direto
ou indireto com o parasito e também as evidências clínicas. Porém, mais de 50% dos
pacientes na fase crônica são assintomáticos, o que tem prejudicado muito o
diagnóstico. Para que haja a confirmação, é necessária a detecção do parasito através da
sorologia (L. MURCIA et al.,2013), sendo que a OMS recomenda a utilização de dois
métodos sorológicos diferentes para determinar o diagnóstico de infecção por T. cruzi
(GILBER et al.,2013).
Atualmente, não existem vacinas usadas no controle da doença de Chagas e os
medicamentos utilizados no tratamento de pacientes na fase aguda são o nifurtimox e o
benzonidazol. Contudo, o tratamento com essas drogas é bastante questionado, devido à
ineficiência contra o parasito durante a fase crônica, presença de efeitos colaterais
longos, alto custo do tratamento e surgimento de cepas resistentes às drogas (DIAS et
al., 2006). Sendo assim, estudos bioquímicos e moleculares acerca desse protozoário
são importantes, já que tais estudos proverão informações a respeito de possíveis alvos
para o desenvolvimento de novas drogas ou tratamentos.
1.2 Ciclo de Vida do Trypanosoma cruzi
O T. cruzi apresenta um ciclo de vida heteróxeno que foi descrito há mais de um
século pelo pesquisador Carlos Chagas (Chagas, 1909). Esse ciclo envolve tanto a
participação de hospedeiros invertebrados (triatomínio), que atuam como vetores do
parasito (DIAS, LARANJA, NÓBREGA, 1945), como uma grande variedade de
hospedeiros vertebrados tais como gambás, cachorros, gatos, tatus, cabras, ovelhas,
incluindo o homem, que servem como reservatório da doença (PESSOA, 1958;
COURA, 1966).

5
Conforme mostrado na figura abaixo, o parasito apresenta três formas distintas,
que podem ser encontradas em diferentes hospedeiros. Nos hospedeiros vertebrados são
observadas as formas amastigotas intracelulares e tripomastigotas sanguíneas, enquanto
nos hospedeiros invertebrados ocorrem as formas epimastigotas e tripomastigotas
metacíclicas (TYLER e ENGMAN, 2001).
Figura 01- Morfologia das formas do T. Cruzi. Em (A) Tripomastigota (B)
Epimastigota. (C) Amastigota. Acesso: Imagem: flech.com.ar em 12/11/2013
As formas amastigotas (Figura 1 C) possuem cerca de 5 μm de comprimento,
apresentam flagelo interno, ausência de membrana ondulante e possuem cinetoplasto
visível. As formas epimastigotas (Figura 1 B) apresentam cerca de 20 a 40 μm de
comprimento, membrana ondulante curta, flagelo livre e possuem o cinetoplasto
anterior ao núcleo. Já os tripomastigotas (Figura 1 A) possuem aproximadamente 25 μm
de comprimento, apresentam o cinetoplasto posterior ao núcleo, flagelo livre e presença
da membrana ondulante (BURLEIGH e WOOLSEY, 2002). Estes últimos são
eliminados junto às fezes do inseto vetor, que são liberados sobre a pele lesionada ou
mucosa íntegra do hospedeiro vertebrado durante o repasto sanguíneo, iniciando o ciclo.
Logo após, chegam ao interior das células, onde ocorre a diferenciação em amastigotas,
se multiplicam por divisão binária e se proliferam rapidamente formando ninhos. Após

6
a diferenciação em tripomastigotas, ocorre a ruptura da célula parasitada e a liberação
dos parasitos no meio extracelular (BRENNER e ANDRADE, 1979).
A infecção do hospedeiro invertebrado acontece durante o repasto sanguíneo,
onde em poucas horas após a ingestão, ocorre a diferenciação do tripomastigota em
epimastigota no tubo digestivo do vetor e, em seguida, as epimastigotas se transformam
em tripomastigotas metacíclicos, sendo esta a forma infectante para o hospedeiro
vertebrado (DE SOUZA, 2000; CARLIER e TORRICO, 2003).
Figura 02- Ciclo evolutivo do Trypanosoma cruzi / Acesso:
www.who.int/tdrdiseaseschagas - em 12/ 11/2013 adaptado da Organização Mundial da
Saúde (OMS).
1.3 O processo de adesão e invasão do parasito
A interação do parasito com a célula hospedeira é uma etapa crucial no processo
de invasão e inicia-se com a adesão do parasito à superfície celular. Esse processo
envolve a participação de diversas moléculas no reconhecimento celular, sendo estas
ancoradas em moléculas de glicosilfosfatidilinositol (GPI) (YOSHIDA et al; 2006).
Durante a adesão, ocorre um aumento intracelular de cálcio (Ca2+
) que,
juntamente com as glicoproteínas (Gps) de superfície, atuam favorecendo o processo de
interiorização do parasito (RUIZ et al.; 1998). Estudos recentes sugerem duas vias de

7
invasão diferentes – a dependente de lisossomos e uma alternativa dependente do
citoesqueleto de actina. A dependente de lisossomos compreende a formação de
vacúolos denominados vacúolos parasitóforo, que é formado pela fusão de lisossomos à
membrana celular (DO CAMPO,1996; BURLEIGH e ANDREWS, 1998; YOSHIDA,
1989; 2003). Posteriormente, a membrana sofre ruptura e permite o acesso do parasito
ao citoplasma. O vacúolo no qual o parasito se instala tem pH ácido, condição que pode
ser modificada através de agentes que inibem o processo de acidificação e permitem o
escape do parasito para o citoplasma (KULKARNI et al; 2009).
Já a via dependente do citoesqueleto de actina é estimulada pelo aumento de
cálcio no citoplasma da célula hospedeira que, após a interação com formas
tripomastigotas do T. cruzi, provoca uma reorganização no citoesqueleto de actina,
causando uma despolimerização de actina no local de entrada. As moléculas de F-actina
são acumuladas ao redor da membrana do vacúolo, que se forma no momento da
invasão. Desse modo, forma uma barreira que impede a fusão do vacúolo parasitóforo
com os lisossomos facilitando a invasão do parasito (RAJARAM et al; 2006).
Levando em consideração as particularidades metabólicas de cada cepa e os
mecanismos de invasão celular adotado, as glicoproteínas encontradas em
tripomastigotas metacíclicas vêm sendo amplamente estudadas (YOSHIDA et al., 1990;
RAMIREZ et al., 1993; PREVIATO et al., 1994). Particularmente, a gp82 correlaciona-
se com o aumento da concentração de cálcio intracelular e estão presentes na superfície
das cepas mais infectivas. Já as gp35/50 estão presentes em cepas menos infectivas e
induzem uma cascata de sinalização menos eficiente (DORTA et al., 1995; YOSHIDA
et al., 2000; 2006).
Considerando-se os mecanismos de adesão parasitária e invasão celular, muitos
trabalhos descrevem a relevância de outras proteínas em diversos processos biológicos
do parasito, como a cruzipaína, uma cisteíno-protease que se relaciona na invasão de
células pelo T. cruzi, que é essencial à sobrevivência do parasito. Estudos feitos por
ENGEL et al (1998) com camundongos, demonstraram que a utilização de inibidores da
cruzipaína são capazes de diminuir significativamente a invasão celular, exercendo um
papel importante na imunopatogênese da doença de Chagas experimental.

8
1.4 Ciclooxigenase -2 (COX– 2) e o processo inflamatório / hipertrófico na doença
de Chagas
Nos últimos anos, evidências crescentes demonstram que a inflamação crônica
exerce importante papel nas cardiomiopatias (BARRETO et al., 2009). Baseados nessas
observações, diversos autores já demostraram que, os processos pro-inflamatórios que
conduzem as hipertrofias cardíacas e a outras doenças é marcado principalmente pela
liberação de citocinas que são responsáveis pela modulação de mecanismos celulares
envolvidos na resposta inflamatória (CANDIA, 2007; GARCIA e INCERPI, 2008;
PONTES, 2004).
Entre as citocinas, o fator necrose tumoral (TNFα) é capaz de induzir a
liberação de outras citocinas, tais como a interleucina 1-Beta (IL-1β), a interleucina 8
(IL-8) que, por sua vez, estimula a produção da proteína ciclooxigenase 2 (COX-2),
uma proteína fundamental no controle do processo inflamatório (BASBAUM e
JULIUS, 2006; MOLLACE et al., 2005).
Mediante a presença de um estímulo pró-inflamatório, ocorre a ativação da via
do ácido araquidônico e as proteínas ciclooxigenases (COXs) estão diretamente
envolvidas com essa via metabólica. O ácido araquidônico (AA), presente na membrana
plasmática, é metabolizado pela ação da enzima fosfolipase A2 (PLA2) e, na sequência,
é processado pelas enzimas ciclooxigenases (COX-1 e COX-2), dando origem aos
prostanóides como as prostaglandinas (PGs) e prostaciclinas (PGIs) e aos, tromboxanos
(TBXs), como esquematizado na figura 3 (HINZ e BRUNE, 2002).

9
Figura 03. Esquema ilustrativo da via do ácido araquidônico (adaptado de Mollace et
al., 2005).
As enzimas COXs podem ser encontradas em duas isoformas: a proteína COX-1
é expressa constitutivamente em vários tecidos e está relacionada à produção de
prostanóides relevantes em processos fisiológicos. A proteína COX-2 é induzida em
tecidos inflamados em resposta a agentes pró-inflamatórios e é responsável pela
produção de prostanóides e tromboxanos ligados a eventos patológicos (VANE,1998; O
BANNION, 1999; KUMMER e COELHO, 2002). Em humanos, essas enzimas
compartilham uma homologia de seqüência de aproximadamente 60%, porém os genes
localizam-se em diferentes cromossomos e as enzimas diferem quanto a sua regulação,
expressão e distribuição. COX-1 possui um gene com aproximadamente 22 kb e 11
exons, que transcrevem um RNA mensageiro (RNAm) de 2.8 kb. Já COX-2 possui 8.3
kb, apresenta10 exons e transcrevem um RNAm de 4.6 kb (LIU et al., 2001).
A ativação transcricional do gene cox-2 e sua expressão protéica são reguladas
por uma intrincada via metabólica onde elementos se interconectam. Em resposta a um
estímulo de estresse celular, vários fatores de transcrição parecem se correlacionar com
a ativação do gene cox-2 (MATSUI; 2013), incluindo o Fator Nuclear de Interleucina 6

10
(NF-IL6) , CREB ( cAMP response element binding protein), Fator Nuclear de Células
T Ativadas (NFAT) e Fator Nuclear-kB (NF-kB). Particularmente, NF-kB caracteriza-
se como um regulador de genes que codificam receptores de citocinas e moléculas de
adesão celular que dirigem as respostas imunes e inflamatórias (TANABE e TOHNAI,
2002; GASPARINI et al., 2003; ST-GERMAIN et al., 2004). Além disso, o NF-kB,
também tem sido apontado como um fator responsável pela ativação transcricional de
genes relacionados a processos hipertróficos (CORTEZ et al., 2007).
Estudos feitos por ZHANG et al (2005) demonstraram que os níveis de
expressão do RNAm cox-2 apresentam um perfil diferenciado entre indivíduos em
resposta a agentes ambientais. Esta variabilidade pode ser determinada por fatores
moleculares, como o polimorfismo de nucleotídeo simples (SNPs), nas regiões
funcionais do gene que codifica esta proteína. Essa variação pode alterar a expressão, a
atividade da enzima e/ ou a sua síntese e degradação, influenciando o potencial de
susceptibilidade a doenças de etiologia inflamatória, como as cardiopatias.
Em células normais, cox-2 não é só transcrito em baixos níveis, como o seu
RNAm é degrado rapidamente (SULLY et al., 2004) e a atividade funcional da proteína
pode ainda ser protetora ou deletéria, dependendo da intensidade e do estímulo que
conduz a sua expressão (MUKHERJEE, et al., 2001). MENDEZ e LAPOINTE (2002)
observaram que, durante o processo inflamatório, miócitos ventriculares de ratos
neonatais apresentaram um aumento da proteína COX-2, estimulando assim a síntese de
(PGE2) e, consequentemente, o crescimento celular dos cardiomiócitos. Os mesmos
autores, em 2004, ainda mostraram que a inibição de COX-2 em miocárdios de ratos
neonatos diminuiu a hipertrofia. Já WANG et al, (2009) demonstraram que, em falhas
cardíacas de miocárdio humano, também são identificadas alterações nos níveis da
proteína COX-2. Por outro lado, BATLOUNI (2009) demonstrou que a COX-2 possui
efeitos cardioprotetores pois, ao administrarem aos animais uma droga inibidora
da COX-2, o efeito cardioprotetor foi eliminado e o risco de desenvolverem infarto do
miocárdio foi aumentando.
Interessantemente, evidências crescentes mostram que a infecção pelo
Trypanosoma cruzi produz uma resposta inflamatória intensa em diversos tecidos,
incluindo o coração, e o parasito induz a produção de quantidades acentuadas de
citocinas e quimiocinas em resposta à infecção (MACHADO et al., 2008).

11
Corroborando com esses dados, TAFURI (1973) demostrou que o processo inflamatório
causado pelo o parasito conduz a várias alterações na morfofisiologia do coração,
provocadas por infiltrado de células inflamatórias que, por sua vez, invadem as fibras
miocárdicas, causando lise das células não parasitadas e miocardite, contribuindo assim
para o processo hipertrófico.
Considerando a inflamação por T. cruzi como um precedente de hipertrofia
cardíaca que se instala ao longo da infecção parasitária, observa-se um aumento nos
níveis de PGE2, uma das principais prostaglandinas produzidas durante a resposta
imune pela via das ciclooxigenases, que pode estar correlacionada com a progressão da
doença. Estudos feitos por ABDALLA et al (2008) mostraram que os altos níveis de
PGE2 causam maiores danos teciduais ao coração de camundongos infectados e a
inibição da COX-2 foi capaz de diminuir esses danos. MICHELIN (2005) observou que
as prostaglandinas produzidas principalmente por COX-2 estão envolvidas na
imunossupressão da fase aguda da infecção por T. cruzi, pois, ao administrarem aos
animais o meloxicam (um inibidor da COX-2), a parasitemia reduziu significativamente
e atrasou a mortalidade dos animais infectados.
STERIN e BORDA (1996) também demostraram em camundongos infectados
por T.cruzi um aumento na produção de PGE2 quando comparados com animais não
infectados. BORGES (1998) evidenciou ainda que o aumento dos níveis de PGE2
durante a infecção pode contribuir para a evolução dessa enfermidade.
Baseado nos diversos estudos torna-se evidente que a proteina COX-2 e a PGE2
modulam a resposta do hospedeiro, o que a torna um alvo potencial no tratamento
farmacológico da doença. No entanto, pouco se conhece sobre os mecanismos
moleculares desta proteína nas cardiopatias chagásicas.

12
1.5 Atuação de PTEN e Akt na modulação dos níveis da enzima COX-2 na
hipertrofia cardíaca chagásica
Estudos apontam uma correlação indireta entre o processo inflamatório mediado
pela proteína COX-2 com os mecanismos de ação da proteína Fosfatase e Tensina
Homóloga (PTEN). No entanto, esse mecanismo ainda precisa ser esclarecido (ST-
GERMAIN et al., 2004; LI et al., 2011).
PTEN é uma proteína codificada pelo gene pten, um supressor de tumor que em
humanos está localizado no cromossoma 10q23. Mutações nesse gene estão
relacionadas, na maioria das vezes, ao desenvolvimento de tumores (LI et al., 1997).
PTEN atua na via da proteína Akt/ PI3K, que se caracteriza como uma serina-treonina
quinase. Inicialmente, o fosfatidilinositol (4,5)-bifosfato (PIP2) é convertido pela ação
da enzima fosfatidilinositol 3 quinase (PI3K) em fosfatidilinositol-3,4,5 -trifosfato
(PIP3). A perda da função de PTEN, bem como a ativação PI3K, resulta em acúmulo de
PIP3, que recruta Akt para a membrana, a qual é fosforilada pela proteína PDK1,
ativando-a, como mostra a figura abaixo (CANTLEY, 2002; CARNERO, 2008;
GEORGESCU, 2011).
Figura 04. Representação esquemática da via PI3K/ Akt. A inativação de PTEN resulta
em acúmulo de PIP3 que recruta Akt para a membrana. PDK1 fosforila Akt, que, por
sua vez, interage com várias vias de sinalização celular (adaptado de MOCANU e
YELLON, 2007).

13
Akt, por sua vez, é uma proteína que possui três subtipos: Akt1, Akt2 e Akt3.
Estas são decodificadas por três genes diferentes, que possuem aproximadamente 80%
de homologia e são expressos em níveis variáveis, dependendo do tipo de tecido
(COFFER e WOODGETT, 1991; I. HERS et al., 2011). As isoformas Akt1 e Akt2 são
predominantemente expressas no músculo-esquelético, cérebro, coração e pulmão, e a
isoforma Akt3 é predominante no cérebro e testículo (JONES et al., 1991).
Em condições patofisiológicas, a ativação de PTEN pode ser suficiente para
regular positivamente a via PI3K/ Akt, reduzindo a morte celular associada com a lesão
e impedindo a hipertrofia e a malignidade de tumores (MOCANU e YELLON , 2003;
CHUENKOVA et al, 2011). No tecido cardíaco, PTEN desempenha um papel
importante na regulação do equilíbrio entre a sobrevivência e a morte celular
(SCHWARTZBAUER e ROBBINS, 2001; PROUD, 2004). Em condições normais,
PTEN mantém os níveis de PIP3 baixos; enquanto que, na sua ausência, promove o
aumento da concentração de PIP3 e da sinalização de Akt, que promove a progressão
não coordenada do ciclo celular através de vários estímulos que se intercruzam (DI
CRISTOFANO e PANDOLFI, 2000).
A regulação da atividade de PTEN é complexa. Sabe se que um dos mecanismos
da sua inativação pode ser através da fosforilação (ROSA, 2008; GEORGESCU, 2011).
A principal enzima responsável por este processo é uma proteína-quinase chamada
CK2, expressa ubiquamente que é responsável por regular uma variedade de processos
biológicos, incluindo o crescimento e sobrevivência celular (MEGGIO e PINNA, 2003).
CK2 é super-expressa em células cancerosas e promove o crescimento de tumores
através da regulação da atividade de genes supressores tumorais e a sobrevivência de
células (DRYGIN et al; 2009). Além disso, a super-expressão de CK2 tem sido
associada a vários estados patológicos, inclusive nas patologias cardiovasculares
(GARCIA et al; 2010). De maneira indireta, a proteína COX-2 também tem sido
relacionada à regulação desse processo. Li et al., (2011) demonstram em seus estudos
que a proteína COX-2 favorece a fosforilação de PTEN pela mediação da CK2,
inativando a atividade fosfatásica de PTEN. Por sua vez, PTEN desempenha um papel
importante na regulação do equilíbrio entre a sobrevivência e a morte de muitos tipos de
células, incluindo os cardiomiócitos, como mostrado abaixo na figura 5
(SCHWARTZBAUER e ROBBINS, 2001; PROUD, 2004).

14
Figura 05: Relação entre as proteínas PTEN, AKT e COX-2 na modulação do processo
hipertrófico mediado por inflamação (MOTA, 2014).
A perda de PTEN desempenha um papel chave na supressão do
desenvolvimento patológico da hipertrofia, considerando-se que PTEN está associado a
uma grande variedade de respostas adaptativas hipertróficas em cardiomiócitos
(CRACKOWER et al; 2002). Estudos feitos por OUDIT e PENNINGER (2008), com
camundongos nocaute, mostraram que, no coração, a perda de PTEN leva a um
aumento da concentração de Akt1/ PI3K, resultando em hipertrofia e resistência a
apoptose. Em estudos subsequentes realizados pelos mesmos autores (2009), com ratos
neonatos, demonstrou-se que a atividade de PI3K é essencial para o crescimento das
células, tanto em respostas adaptativas (fisiológicas), quanto patológicas, como na
hipertrofia. Os inibidores de PI3K são capazes de reduzir o tamanho dos cardiomiócitos,
enquanto que a super-expressão de PI3K aumenta o volume dessas células.
Paralelamente, SCHWARTZBAUER e ROBBINS (2001) demonstraram que
PTEN mutante conduz a hipertrofia dos cardiomiócitos, devido ao significativo
aumento na síntese de proteínas e, consequentemente, no volume das células. De modo

15
semelhante, MATSUI et al (2002) demonstraram que a super-expressão de Akt1 em
camundongos resulta em um aumento tecidual e consequentemente hipertrofia do
músculo esquelético.
Por sua vez, CHUENKOVA (2001) relacionou a via Akt/ PI3K com
sobrevivência das células, na infecção por T. cruzi, sugerindo um papel importante na
patogênese da doença de Chagas. Corroborando com esses dados PETERSEN et al
(2006) demostraram que, o início do processo infeccioso, desencadeado pela infecção,
ativa várias vias de sinalização celular, incluindo a via Akt/ PI3K e NF-kB, que
contribui para estimulação de hipertrofia de cardiomiócitos e o próprio processo pró-
inflamatório. Além disso, a ativação de via bioquímica controlada por Akt, quando da
invasão pelo T. cruzi, modula positivamente o processo de invasão celular (BURLEIGH
e WOOLSEY , 2002).
No entanto, os mecanismos correlacionados com inativação de PTEN e com a
ativação da proteina COX-2, sobretudo na hipertrofia cardiaca chagásica, são pouco
esclarecidos, sendo necessários mais estudos para uma melhor compreensão de tais
mecanismos. PTEN aparece como um co-regulador da atividade de COX-2, cabendo
uma maior atenção a esse estudo e, futuramente, poderá subsidiar o desenvovimento de
drogas pela indústria farmacêutica.
1.6 miRNAs e a regulação da expressão gênica nas hipertrofias cardíacas
O controle da expressão gênica é vital para permitir que uma célula mantenha
sua homeostase. Considerando os elementos que regulam e/ou modulam a expressão
gênica, os microRNAs (miRNAs) tem ganhado posição de destaque na comunidade
cientifica. São pequenas moléculas de RNAs não-codificantes, de 18-24 nucleotídeos,
que modulam a expressão de genes (XIN DUAN e XIAOHUA WANG, 2011).
Os miRNAs foram descobertos quando LEE e colaboradores (1993)
identificaram o miRNA lin-4 em Caenorhabidits elegans, seguido pelo miRNA (let-7),
também em nematóide. E hoje sabe se que os miRNAs são reguladores fundamentais da
expressão gênica de plantas e animais. Essas moléculas foram caracterizadas em
diferentes espécies, apresentando-se relativamente bem conservados e já foram
relacionadas com diversos processos biológicos, tais como: o desenvolvimento,

16
proliferação, diferenciação do crescimento celular e apoptose (LEWIS, 2003; HE e
HANNON, 2004; KIMURA, 2006; ZHANG, 2008; ALMEIDA, 2011 ).
Realizam a repressão da síntese proteica através do emparelhamento da região 3‘
não traduzida (3’ UTR) do RNA mensageiro da molécula alvo. Este mecanismo permite
a redução dos níveis protéicos de seus genes-alvo, seja pela degradação do RNA
mensageiro ou pela inibição da tradução (JI et al, 2009). Até o momento, identificaram-
se mais de 700 miRNAs sequenciado em seres humanos e cerca de 150 a 200 expressos
no coração e, análises bioinformáticas, indicam que o número de genes seja mais de
1000 (KUKREJA, 2011; SILVA, 2012).
Os miRNAs são inicialmente transcritos (figura 6) em miRNAs primário (pri-
miRNAs), que são processados no núcleo pela enzima Drosha (LEE et al., 2004). Estes
precursores originam os (pré-miRNAs), que são similares à estrutura de um grampo, e
que são exportados para o citoplasma pela enzima exportina 5 (HAVENS et al., 2012).
No citoplasma, os pré-miRNAs são processadas pela enzima Dicer, formando um
duplex de RNA com 22 pares de nucleotídeos (VARGAS, 2013; SCHOFIELD, 2011).
O complexo proteico denominado RISC separa o duplex, sendo que uma das fitas é
degradada enquanto a outra é direcionada para regiões alvo nos RNA, desencadeando o
silenciamento do RNA (BATISTA e VERBISCK, 2009).
Figura 06: Biogênese dos miRNAs. Adaptado por BATISTA e VERBISCK (2009).

17
Estudos apontam o envolvimento dessas pequenas moléculas nas patologias
cardiovasculares (BARRY, 2008; ZHANG, 2008). Alguns miRNAs estão presentes
especificamente em determinados tipos de tecidos ou células, incluindo o tecido
cardíaco. Estudos sugerem que os miRNAs desempenham um papel crucial na
patogênese da insuficiência cardíaca, pois alguns miRNAs estão altamente expressos
no coração como o miR-1, miR-21, miR-133 e miR-208 e alguns deles estão associados
ao desenvolvimento da hipertrofia cardíaca ( ZHANG, 2008; CARVALHO, 2012).
MATKOVICH e colaboradores (2009) avaliaram o perfil de expressão de
miRNAs em pacientes com insuficiência cardíaca, antes e depois do tratamento com
dispositivos de assistência ventricular esquerda e concluíram que, 71,4% dos miRNAs
diferencialmente regulados na insuficiência cardíaca foram normalizados após o
tratamento. Esses resultados sugerem que os miRNAs podem servir como marcadores
de recuperação do miocárdio em pacientes com insuficiência cardíaca avançada. Por
outro lado, THUM e colaboradores (2008) induziram ratos à hipertrofia cardíaca por
meio de sobrecarga de pressão e, após três semanas, administraram uma droga chamada
antagomir, que foi desenvolvida para inibir funcionalmente o miR-21. Como resultado,
eles observaram que os ratos apresentaram uma regressão significativa da hipertrofia
cardíaca e fibrose, além da atenuação do comprometimento da função cardíaca.
Considerando a relevância de miRNAs no equilíbrio celular, a análise de
expressão dessas pequenas moléculas poderá ser de grande utilidade como ferramenta
diagnóstica e prognóstica em processos patológicos, pois existem evidências de que elas
possam estar regulando COX-2 e que também funcionem como um marcador molecular
em diversas patologias. Em estudos feitos, JI et al. (2009) demostraram que células de
carcinoma nasofaríngeo tratados com desferrioxamine (DFON), os miR- 26a e miR-
26b regulam a expressão de COX-2. Paralelamente, STRILLACCI et al., (2009)
demostraram que o miR-101 também podem modular os níveis de COX-2
Levando em consideração a regulação cruzada entre COX-2 e PTEN, análises de
bioinformática demonstram a presença de pelo o menos 20 sítios na porção 3’-UTR
desses genes, que são possíveis de ligação de miRNAs comuns (LI et al; 2011).
Reforçando essas observações, HUSE et al. (2013) demonstraram que o miR- 26a
regulam a expressão de PTEN.

18
A regulação pós-transcricional de PTEN por miRNAs também tem sido
relacionada com desenvolvimento de patologias, inclusive na hipertrofia. MENG (2008)
demonstrou que, em células de carcinoma hepatocelular, a inibição do miR-21 promove
o aumento da expressão da proteína PTEN, sendo um alvo direto do miR-21. Por fim,
WEIYUN WU (2013) também mostrou um local de ligação do miR-32, na região 3`-
UTR de PTEN. Já em cardiomiócitos, PTEN foi identificado como sendo um alvo do
miR-22, sugerindo que o aumento da regulação do miR-22 pode ser responsável pela
diminuição da regulação de PTEN durante hipertrofia. (X U et al; 2011)
Embora a maioria das pesquisas evidencie o envolvimento dos miRNAs em
células cancerígenas, trabalhos recentes têm demostrado a importância dos miRNAs
expressos nas patologias cardiovasculares e crescentes são as evidências que sugerem
que os miRNAs representam um papel fundamental na regulação da diferenciação
celular cardiovascular (KUKREJA, 2001; VAN, 2011; CARVALHO, 2012). Caso
ocorra alguma desregulação da função do miRNA, doenças cardiovasculares podem se
manifestar, tais como: a hipertrofia cardíaca, a formação da lesão vascular e arritmias
cardíacas ( ZHANG, 2008; YANG, 2011).
Assim como o aumento da expressão de alguns miRNAs pode estar relacionado
ao desencadeamento de processos patogênicos, a diminuição da expressão dessas
moléculas também pode levar a um estado patológico. Sabe-se que miRNAs podem
permanecer por longos períodos estáveis no plasma e a abundância dos miRNAs
circulantes pode variar de acordo com o tipo da doença, possibilitando assim o uso de
miRNAs extracelulares como marcadores de diversas doenças humanas, inclusive no
diagnóstico e prognóstico de doenças cardíacas (MITCHELL, 2008; SMALL,2010;
LATERZA,2009; TURCHINOVICH, 2011; VARGAS, 2013).
Está cada vez mais evidente o potencial dos miRNAs como nova ferramenta
biotecnológica. Entretanto, muitos estudos são ainda necessários para compreender o
papel do miRNAs na hipertrofia cardíaca e, em particular, na hipertrofia mediada pela
inflamação decorrente da infecção por T. cruzi, que também pode ser regulada por vias
bioquímicas moduladas por COX-2 e PTEN.

19
1.7 A importância do modelo de estudo celular e a busca por marcadores
moleculares na doença de Chagas
Com os recentes avanços na tecnologia, a utilização de técnicas que permitem
uma compreensão mais detalhada dos mecanismos moleculares da célula auxilia no
desenvolvimento de novas drogas. Particularmente, o que assegura o desenvolvimento
da doença de Chagas são as etapas iniciais da invasão e infecção celular. Sendo assim,
um modelo de cultura de células que simule um comportamento in vivo se faz
necessário. A cultura celular permite a manutenção de células vivas independente do
organismo que a originou, além de garantir uma reprodutibilidade dos experimentos e
redução do uso de animais (MIN-HSIEN, SONG-BIN, GWO-BIN, 2010)
O modelo celular utilizado no nosso estudo foram células H9c2 (tecido cardíaco
de embrião de rato recém-nascidos), utilizadas como modelo experimental ex vivo,
também considerada por alguns autores como in vitro. Estudos já demonstram a
importância da utilização dessas células em diferentes patologias cardíacas, sendo,
portanto, consideradas bons modelos para potenciais estudos moleculares em patologias
do coração (BESHAY, 2007; VILMA, 2008; SARAH, 2011).
Nos últimos anos, o desenvolvimento de modelos experimentais no estudo
da doença de Chagas proporcionou um avanço importante na medicina. No entanto,
a maioria dos estudos da infecção por Trypanosoma cruzi faz o uso de modelos animais,
sendo a cultura de células um modelo alternativo para se estudar essa patologia (JORGE
e CASTRO; 2000). A busca por marcadores moleculares correlatos à doença é
constante. Entretanto, para tal, a caracterização molecular das vias de sinalização dessa
doença se faz necessária.
Sabe-se que a agressão ao músculo cardíaco é mediada principalmente pela
resposta imune do hospedeiro (CUNHA-NETO, 2005; MACHADO, 2008). Uma
deficiência na regulação da resposta imune pode levar a um intenso processo
inflamatório e, consequentemente, a um dano tecidual. Com o objetivo de evitar o dano,
várias vias regulatórias atuam no sentido de controlar a inflamação. As células
regulatórias e as citocinas tem papel importante nesta modulação e, entre estas, as
interleucinas, que tem efeito modulador da resposta imune e, por sua vez, promovem a
ativação de diversas proteínas, entre as quais a proteína COX-2 (BASBAUM e JULIUS,
2006).

20
Interessantemente, a regulação e expressão da proteína COX-2 podem ocorrer
em nível transcricional e envolver a participação de fatores de transcrição, ou pós-
transcricional, tais como o envolvimento de miRNAs e através de fosforilação de
proteínas como a supressora tumoral PTEN, sugerindo assim uma possível modulação
dos níveis de expressão da mesma. Entretanto, um dos maiores desafios na doença de
Chagas tem sido a busca por marcadores moleculares, pois com o advento da tecnologia
genética e da biologia molecular, surgiram diversas técnicas que utilizam marcadores
moleculares. Porém, a maioria dos estudos está voltado para o diagnóstico de doenças
auto-imunes, doenças inflamatórias crônicas e neoplasias. Estudos mais recentes
poderão ser úteis no diagnóstico precoce de doenças infecciosas e parasitárias, bem
como na doença de Chagas (DALE e CHAPARRO, 1997; SCHRIEFER,2008). A
caracterização de marcadores moleculares na doença de Chagas poderá direcionar um
tratamento mais direcionado ao paciente.
A descoberta de mecanismos moleculares envolvidos no desenvolvimento da
Doença Chagas é essencial para que marcadores moleculares possam ser identificados.
No entanto, a diversidade genética de populações de T.cruzi tem sido caracterizada
como um desafio à compreensão de alterações que ocorrem durante o processo
infeccioso e, sobretudo, na determinação das diferentes formas clínicas. Até hoje, pouco
se conhece sobre os mecanismos de atuação da cepa Berenice 62 e a sua relação com a
proteína COX-2 e como seu RNAm é modulado na hipertrofia chagásica, sendo
necessários estudos que busquem potenciais marcadores moleculares que poderão, no
futuro, permitir uma melhor compressão dessa patologia (BORGES et al; 1998;
MACHADO et al., 2008). Espera-se, portanto, que os resultados do presente estudo
possam direcionar a caracterização de novos marcadores que auxiliem no prognóstico e
tratamento direcionado da doença de Chagas, bem como no desenvolvimento de novas
drogas.
1.8- JUSTIFICATIVA
A doença de Chagas constitui um grave problema de saúde pública,
especialmente na América Latina, onde é considerada como uma das principais causas
de problemas cardíacos. Embora essa enfermidade tenha sido amplamente estudada,
muitos aspectos ainda carecem de investigação. Considerando os mecanismos pró-

21
inflamatórios desencadeados pela proteína COX-2 como um precedente das
hipertrofias, a atenção especial tem sido dada a essa proteína. Sabe-se que a atividade
pró-inflamatória da COX-2 é apontada como importante elemento na regulação da
hipertrofia cardíaca mediado pela inflamação. No entanto, os mecanismos de ação da
proteína COX-2 desencadeados pela infecção causada por T. cruzi e particularidades
relacionadas ao processamento do seu RNAm da doença precisam ser esclarecidos.
Sendo assim, o objetivo desse trabalho foi analisar moduladores do processo pró-
inflamatório/ hipertrófico em células H9c2 infectadas pela cepa Berenice-62 do T.cruzi,
buscando a caracterização de marcadores moleculares do processo inicial da patologia.
Além disso, buscar maiores detalhes sobre a infecção da cepa em tecido cardíaco poderá
colaborar para um maior conhecimento dos mecanismos moleculares que asseguram o
desenvolvimento da doença, tornando-se uma ferramenta importante na busca de
marcadores moleculares e na determinação de potenciais alvos terapêuticos.

22
2. OBJETIVOS

23
2.1 Objetivo Geral
Analisar moduladores do processo pró-inflamatório/ hipertrófico em células H9c2
infectadas pela cepa Berenice-62 do T.cruzi, buscando a caracterização de marcadores
moleculares do processo inicial da doença de Chagas experimental.
2.2 Objetivos específicos
- Avaliar a infectividade da cepa Berenice-62 do T. cruzi em células H9c2 nos tempos
de infecção de 0, 2, 6, 12, 24 e 48 horas, logos após 2 horas de interação com o parasito;
- Analisar a viabilidade celular após a infecção de H9c2 pela cepa Berenice-62 nos
diferentes tempos de infecção;
-Análisar o nível transcricional dos genes Bnp,Anf e B-myhc correlacionados com o
processo pró-inflamatório/ hipertórfico;
- Acompanhar os níveis de PGE2 produzidos durante o processo inflamatório causado
por T. cruzi ;
- Investigar a associação entre as proteínas COX-2 e PTEN;
-Investigar níveis transcricionais de microRNAs caracterizados como possíveis
moduladores da expressão de COX-2 e PTEN.

24
3. MATERIAIS E MÉTODOS

25
3.1 Cultura celular
Para a execução deste estudo, dois tipos de células foram utilizadas: a linhagem
celular H9c2 (ATCC CRL-1446) oriundas de tecido cardíaco de embrião de rato (Rattus
norvergicus) e células Vero (ATCC: CCL81™) oriundas de rim de macaco verde
(Cercopithecus aethiops). Essas linhagens foram cultivadas em garrafas apropriadas de
175 cm2 em meio Dulbecco’s Modified Eagle’s Medium (DMEM - Life Technologies)
suplementado com 10% de soro fetal bovino. As culturas foram incubadas em estufa,
contendo 5% de CO2 e mantidas à temperatura de 37°C.
3.2 Obtenção de formas tripomastigotas do T. cruzi e infecções celulares
Para que ocorresse a infecção, camundongos do tipo Swiss foram infectados
com a cepa Berenice-62 do T. cruzi (Protocolo nº 2011/76) e, posteriormente, foi feito a
coleta de sangue dos animais no dia do pico de parasitemia (8º dia de infecção). Logo
após, os parasitos (107
parasitos) foram utilizados na infecção de células VERO e
incubados em estufa a 37ºC. Após 48 horas, as células VERO foram lavadas
extensivamente com solução salina de fosfato (PBS – 1X = 2,7 mM de KCl, 1,5 mM de
KH2PO4, 137 mM NaCl, 8 mM de Na2HPO4, pH 7,4) e, em seguida, foi adicionado
novo meio de cultura. As culturas foram então mantidas a 33 ºC para obtenção das
formas tripomastigotas. Após a liberação das mesmas para o meio extracelular, todo o
meio de cultura foi aspirado e centrifugado a 600 rpm, durante 3 minutos, a 26 ºC, para
abaixar as células VERO. O sobrenadante foi coletado e centrifugado novamente, a
1300 rpm, durante 10 minutos, a 4 ºC, para precipitação das formas tripomastigotas.
Estas foram contadas e, aproximadamente, 1 x 107 parasitos foram incubados com 1 x
106 células H9c2 durante 2 horas em estufa a 37°C e 5% CO2 para permitir a interação
da célula com o parasito . Em seguida, as tripomastigotas que não entraram nas células
foram removidas com PBS e novo meio de cultura foi adicionado à garrafa infectada, a
qual foi incubada novamente a 37°C e 5% CO2. As garrafas de cultura,
independentemente infectadas, foram analisadas em diferentes intervalos: 0 hora, 2, 6,
12, 24 e 48 horas após interação de 2 horas.

26
3.3 Avaliação da infeção pela cepa Be – 62 em células H9c2 por ensaios de
microscopia
3.3.1 Microscopia de contraste de fase: coloração por Giemsa
Para confirmar a infecção das células H9c2 pela cepa Be-62 de T. cruzi e avaliar
a sua eficiência, 3,0 x 104 células H9c2 foram cultivadas sobre lamínulas de vidro
circulares em placas contendo 24 poços. Após atingirem 80% de confluência, as células
foram infectadas com as formas tripomastigota do T. cruzi, conforme descrito acima, na
proporção de 10:1 (parasitos: célula). Durante 2 horas, as células foram incubadas com
o parasito a 37°C e 5% CO2. Após esse intervalo, as células foram lavadas com PBS
para remoção das formas tripomastigotas não internalizadas. Em seguida, meio de
cultura fresco foi adicionado às células, que novamente foram incubadas nas condições
previamente descritas. A infecção celular foi avaliada em diferentes intervalos de
infecção (0, 2, 6, 12, 24 e 48 horas). Após os tempos de incubação, as células foram
lavadas com PBS 1 X e fixadas em metanol por 7 minutos. Em seguida, foram
mergulhadas em solução de Giemsa (Merck) diluída na proporção 1:10 (1mL de
Giemsa para 9 mL de agua) durante 10 minutos e à temperatura ambiente. As lamínulas
foram lavadas com água e, após a coloração, foram montadas sobre as lâminas com
Entellan (Merck). Posteriormente, foram feitas fotografias do material em microscópio
óptico modelo Nikon, ECLIPSE E200, utilizando-se câmera OLYMPUS LENS.
3.3.2 Microscopia de fluorescência: coloração com 4,6 diamino-2-fenilindol
A infecção também foi analisada por microscopia de fluorescência após
coloração do material nuclear com 4,6 diamino-2-fenilindol (DAPI, Sigma-Aldirch). Os
mesmos procedimentos para a coloração com Giemsa foram adotados, com exceção de
que, após os respectivos intervalos de infecção (0, 2, 6, 12, 24 e 48 horas), as células
foram lavadas com PBS 0,5 X e fixadas com solução de 3.8 % de paraformaldeído,
contendo PBS 0,5 X, 0,2% de Triton X-100 (Sigma-Aldrich), durante 7 minutos, a 37ºC
e, em seguida, foram lavadas novamente com PBS 0,5 X. Posteriormente, as lamínulas
foram incubadas com solução de albumina bovina a 1%, durante 2 h, para impedir
marcações inespecíficas. Em seguida, foram processadas as marcações do núcleo celular
com a concentração final de 3,33 mg/mL diluído em PBS por um intervalo de 3
minutos. Após sucessivas lavagens com PBS 0,5 X, as lâminas foram montadas em

27
meio contendo 200 mM de n-propil-galato (Sigma-Aldrich) e glicerol a 90%.
Fotografias do material foram feitas utilizando microscópio modelo Leica DMLB.
3.3.3 Percentual de Infecção
O percentual de células infectadas e o número de amastigotas por célula foram
avaliados pela contagem do número de células e do número de amastigotas, após
tempos variados de infecção. Para isso, as imagens foram capturadas no aumento de 100
X e contadas em microscópio óptico modelo Nikon, ECLIPSE E200, logo após a
coloração com Giemsa e DAPI. Esses valores foram obtidos a partir da contagem de
100 células por lamínula, incluindo infectadas e não infectadas.
3.4 Análise da viabilidade celular: ensaios de citotoxicidade
Para analisar a viabilidade das células infectadas pelo T. cruzi, a atividade
metabólica das células foi investigada pelo ensaio de MTT (Mosman, 1983). Esse
clássico ensaio avalia a redução do MTT (Brometo de 3-[4,5-dimetil-tiazol-2-]-2,5-
difeniltetrazólio - Sigma - Aldrich) pela atividade de desidrogenases mitocondriais,
resultando na formação de cristais de formazan no interior das células. Para esses
ensaios,
1,5 x 104 células foram cultivadas em placas de 96 poços a 37°C e 5% CO2.
Quando as culturas atingiram aproximadamente 80% de confluência, elas foram
infectadas com as formas tripomastigota do T. cruzi, na relação 10:1 (parasito / célula) e
as células foram novamente incubadas por diferentes intervalos. Logo após, foi
adicionado 0.5 mg/mL do reagente MTT e posteriormente, a placa foi incubada por 2
horas a 37°C e 5% CO2, seguindo os tempos de infecção 0 hora, 2, 6, 12, 24 e 48 horas.
Após esse período, o MTT foi removido e foi adicionado a placa 100 um do reagente
DMSO e após 30 minutos, foi feita a leitura em comprimento de onda 550 nm no leitor
de ELISA / Molecular Devices.
3.5 Níveis de Prostaglandina E2
Para a mensuração dos níveis de Prostaglandina E2 (PGE2), foi utilizado o
sobrenadante das células H9c2 infectadas com a cepa Be-62 do T. cruzi em diferentes
tempos de infecção (0 hora, 2, 6, 12, 24 e 48 horas). Posteriormente, foram coletados

28
para análise, utilizando-se o produto comercial Prostaglandin E2 EIA- Monoclonal®
(Cayman Chemical Company). O método é baseado na competição entre PGE2 da
amostra e PGE2 conjudada à acetilcolinesterase, por uma quantidade limitada de
anticorpo de PGE2 monoclonal presente no kit. A amostra foi pipetada em placa de
Elisa com 96 poços contendo o anticorpo policlonal anti IgG de rato, no qual se liga o
anticorpo monoclonal de PGE2. Logo após, a placa foi lavada para remover os reagentes
não ligados, e então incubada com 200 µL do reagente Ellman´s. A densidade ótica das
soluções foi avaliada por espectrofotometria e a análise da absorbância foi feita em
leitor de ELISA / Molecular Devices em um comprimento de onda de 412 nm.
3.6 Extração do RNA total e Transcrição reversa
3.6.1 Extração do RNA total
Células H9c2 foram infectadas com a cepa Be-62 e, posteriormente, coletadas
por tripsinização. Após lavagens apropriadas com PBS 1 x, as amostras foram
ressuspensas em 1ml de Trizol®
(Life Technologies) e centrifugadas a 12.000 g por 10
minutos a 4ºC. O sobrenadante foi coletado e transferido para um novo tubo.
Posteriormente, foi mantida a temperatura ambiente durante 5 minutos. Logo após,
adicionou-se 200 ul de clorofórmio (Sigma-Aldrich). A mistura foi homogeneizada
vigorosamente por 15 segundos e, depois, foi mantida à temperatura ambiente por 3
minutos. Em seguida, realizou-se nova centrifugação de 12.000 g por 10 minutos a 4ºC.
A fase aquosa foi transferida para um novo tubo, cuidadosamente, para evitar
contaminação da fase fenólica. Foi adicionado 500 ul de álcool isopropílico seguido de
nova centrifugação ( 10 minutos, a 12.000g a 4ºC). O precipitado foi lavado com álcool
70% e, posteriormente, foi rapidamente seco à temperatura ambiente. As amostras
foram quantificadas com o comprimento de onda de 260 nm, utilizando
espectrofotômetro, e a pureza das amostras foi medida pela razão entre as absorbâncias
de 260, 280 e 230 nm.
3.6.2 Transcrição reversa
Após a extração do RNA total, foi realizada a transcrição reversa para a
obtenção do cDNA, utilizando o kit High Capacity cDNA Reverse Transcription
(Applied Biossystems - Life Technologies), Foi pipetado um volume final de 10µL,

29
sendo utilizados 1µg de RNA total nas reações, 10 mM de oligonucleotídeos (10x RT
Random primer), 10 mM de dNTPs(25x dNTP), 1,25 U da enzima Muiti ScriptTM
Reverse Transcriptase, e água livre de RNAse . A mistura foi incubada a 25ºC por 10
minutos, posteriormente 120 minutos a 37ºC e 85º por 5 minutos, em um termociclador
para inativação da atividade da trasncriptase. Para avaliar a eficiência da RNA total, foi
feita a reação de PCR utilizando β-actina (NM_031144.3) como normalizador. A
Análise do produto transcricional é observada na (Figura 07).
Figura 7: Análise do produto transcricional do gene β-actina, obtido por extração do
RNA total em gel de agarose 1 %.
3.7 Análises transcricional de genes em reações de PCR quantitativa em tempo real
(qPCR)
A expressão dos transcritos cox-2, pten e akt e de genes relacionados com o
processo hipertrófico; Fator natriurético atrial (Anf) peptídeo natriurético cerebral (Bnp)
e miosina de cadeia pesada do tipo beta (β-Myhc) foram analisados em ensaios de PCR
em tempo real (qPCR). As reações, de 10 µL foram montadas em placas de 96 poços
0,75 µM de conjunto de oligonucleotídeos específicos para cada gene (sense e reverso)
(Tabela 1), 2 µL de cDNA diluído 10 vezes e 5µL do produto SYBR® Green PCR
Master Mix (Applied Biosystes - Life Technologies). As análises foram realizadas em
triplicata e a expressão transcricional do gene da β-actina foi utilizada como
normalizador. As reações foram processadas no aparelho ABI 7500 (Applied Biosystes
- Life Technologies). A análise da quantificação relativa da expressão gênica foi feita
pelo (ΔCT) e os valores foram ajustados da baseline em 3 -15 ciclos e do threshold em
0,2 para todas as amostras. Os oligonucleotídeos utilizados foram desenhados
utilizando-se o programa Gene Runner (Version 3.05), e foram utilizadas sequências
dos RNAm de Rattus norvegicus disponíveis em bancos de dados do NCBI.
30
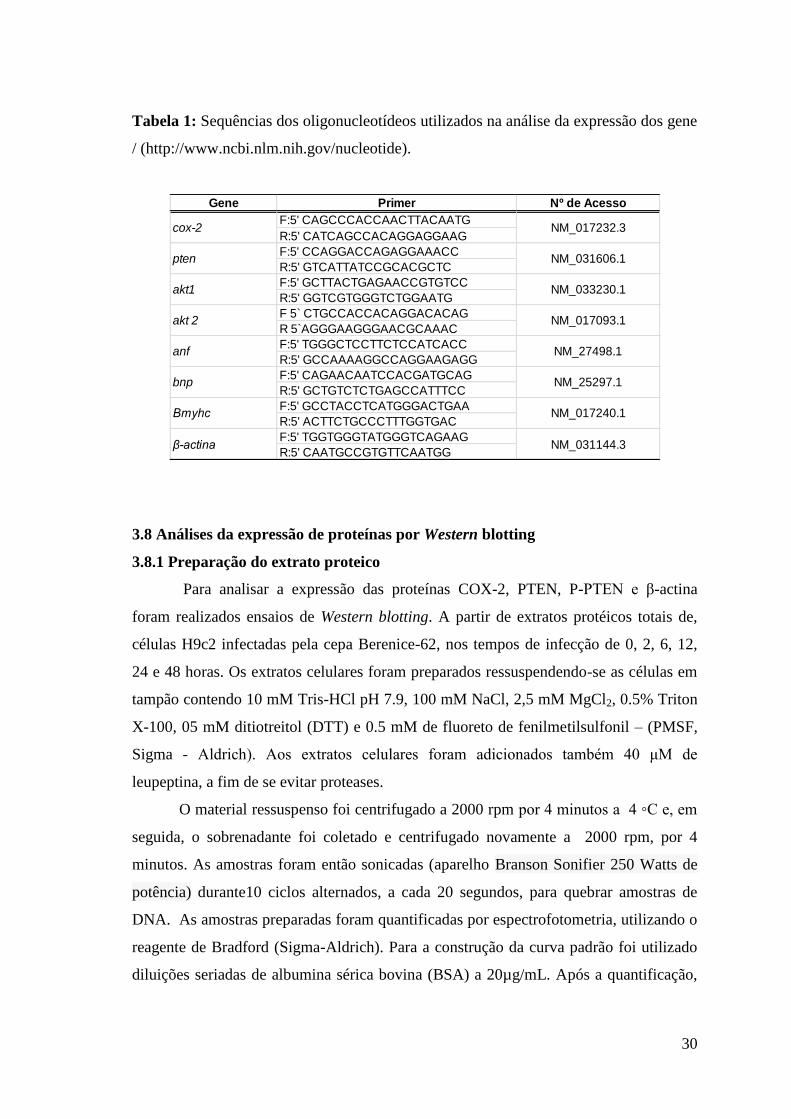
Tabela 1: Sequências dos oligonucleotídeos utilizados na análise da expressão dos gene
/ (http://www.ncbi.nlm.nih.gov/nucleotide).
Gene Primer Nº de Acesso
F:5' CAGCCCACCAACTTACAATG
R:5' CATCAGCCACAGGAGGAAG
F:5' CCAGGACCAGAGGAAACC
R:5' GTCATTATCCGCACGCTC
F:5' GCTTACTGAGAACCGTGTCC
R:5' GGTCGTGGGTCTGGAATG
F 5` CTGCCACCACAGGACACAG
R 5`AGGGAAGGGAACGCAAAC
F:5' TGGGCTCCTTCTCCATCACC
R:5' GCCAAAAGGCCAGGAAGAGG
F:5' CAGAACAATCCACGATGCAG
R:5' GCTGTCTCTGAGCCATTTCC
F:5' GCCTACCTCATGGGACTGAA
R:5' ACTTCTGCCCTTTGGTGAC
F:5' TGGTGGGTATGGGTCAGAAG
R:5' CAATGCCGTGTTCAATGG
NM_27498.1
cox-2
pten
akt1
akt 2
anf
NM_017232.3
NM_031606.1
NM_033230.1
NM_017093.1
NM_25297.1
NM_031144.3
bnp
β-actina
Βmyhc NM_017240.1
3.8 Análises da expressão de proteínas por Western blotting
3.8.1 Preparação do extrato proteico
Para analisar a expressão das proteínas COX-2, PTEN, P-PTEN e β-actina
foram realizados ensaios de Western blotting. A partir de extratos protéicos totais de,
células H9c2 infectadas pela cepa Berenice-62, nos tempos de infecção de 0, 2, 6, 12,
24 e 48 horas. Os extratos celulares foram preparados ressuspendendo-se as células em
tampão contendo 10 mM Tris-HCl pH 7.9, 100 mM NaCl, 2,5 mM MgCl2, 0.5% Triton
X-100, 05 mM ditiotreitol (DTT) e 0.5 mM de fluoreto de fenilmetilsulfonil – (PMSF,
Sigma - Aldrich). Aos extratos celulares foram adicionados também 40 μM de
leupeptina, a fim de se evitar proteases.
O material ressuspenso foi centrifugado a 2000 rpm por 4 minutos a 4 ◦C e, em
seguida, o sobrenadante foi coletado e centrifugado novamente a 2000 rpm, por 4
minutos. As amostras foram então sonicadas (aparelho Branson Sonifier 250 Watts de
potência) durante10 ciclos alternados, a cada 20 segundos, para quebrar amostras de
DNA. As amostras preparadas foram quantificadas por espectrofotometria, utilizando o
reagente de Bradford (Sigma-Aldrich). Para a construção da curva padrão foi utilizado
diluições seriadas de albumina sérica bovina (BSA) a 20µg/mL. Após a quantificação,

31
foi adicionado 60 μg de proteína ao tampão de corrida concentrado 6X SDS (100 mM
Tris-HCl, pH 6.8, 20% (w/v), glicerol, 4% (w/v) SDS, 0.001% (w/v) azul de
bromofenol), juntamente com 1% de betamercaptoetanol (Sigma-Aldrich). As amostras
foram posteriormente desnaturadas a 95ºC, em banho-maria, durante 5 minutos e,
posteriormente, submetidas à eletroforese em gel desnaturante de poliacrilamida a 100
V e 25 mA, por aproximadamente 1 h e 30 minutos.
3.8.2 Transferência e revelação das membranas
As proteínas presentes no gel foram transferidas para as membranas de
nitrocelulose (Santa Cruz Biotechnology) em sistema de transferência elétrica semi -
úmida, utilizando solução de transferência (25mM Tris-HCl, 0,192 M de glicina, 10%
metanol). A transferência foi realizada durante 1 hora e 20 minutos, sob corrente
elétrica de 100 V e 300mA . Logo após, as membranas foram bloqueadas, utilizando-se
solução de Blotto (leite em pó desnatado 5%) diluídos em TBS (150mM NaCl, 50mM
Tris-HCl pH 7.6 ), por 12 horas. Em seguida, as membranas foram lavadas 3 x com
TBS, durante 7 minutos, para retirar o excesso de leite. Após o bloqueio, as membranas
foram incubadas separadamente com os anticorpos primários policlonais de coelho de
COX-2 (Cayman), PTEN e P-PTEN (Cell Signalling) e b-actina (Cell Siganlling),
diluídos em TBS, na concentração de 1:250, durante 24 horas, a 4◦C . As membranas
foram lavadas 3 x com TBS, durante 7 minutos. Posteriormente, a membrana foi
incubada com o anticorpo secundário policlonal anti IgG de Coelho (Caymam), diluído
em TBS na concentração de 1:6.000, durante 4 horas, sob temperatura ambiente. As
membranas foram lavadas novamente e, em seguida, foram reveladas utilizando-se o
produto ECL Plus Western Blot detection Reagents (GE Healthcare), na concentração
1:1 e visualizadas pela sensibilização de filme de raios-X expostos sob a membrana por
intervalos variados. Em seguida, foi revelado. Após a revelação, foi feito a
quantificação das bandas através da densidade ótica, utilizando-se o programa Quantity
One® (Biorad).
3.9 Análises do padrão de expressão de miRNAs
A análise de expressão dos miRNAs foi analisada. Para isso, 1 x 106
células
H9c2 foram infectadas pela a cepa Be- 62 do T. cruzi nos tempos de infecção de 0, 2, 6,

32
12, 24 e 48 horas. Posteriormente, foram utilizadas na preparação de extratos de RNAm
total, enriquecidos com miRNAs, com o reagente miRNAse mini kit (Qiagen®). Os
miRNAs foram isolados com o auxílio do produto comercial miRNAse mini kit
(Qiagen®). Depois de devida quantificação em espectrofotômetro, os miRNAs isolados
foram utilizados em reações de transcrição reversa com os reagentes Miscript II RT Kit
(Qiagen®
), seguindo as recomendações do fornecedor.
A expressão dos miRNAs foi obtida através da quantificação por PCR em tempo
real, utilizando-se o MiScript SYBR® Green PCR Kit (Qiagen®) com os
oligonucleotídeos descritos abaixo. Os oligonucleotídeos utilizados foram desenhados
utilizando o banco de dados Blast, disponíveis em: (www.ncbi.lm.nih.gov/Blast), mirDB
(http://mirdb.org/miRDB/), MiRBase (http://www.mirbase.org/), TargetScan
(http://www.targetscan.org/). Os resultados foram analisados através do método do ΔCt
para cálculo da expressão gênica relativa dos miRNAs, seguindo as recomendações do
fornecedor do aparelho de qPCR 7500 (Applied Biossystems - Life Technologies). O
gene U6 foi utilizado como normalizador.
Tabela 02. Sequências dos miRNAs e alvos envolvidos no processo hipertrófico e na
modulação de COX-2 e PTEN
miRNA Sequencia Alvo
COX-2
rno-miR-26A
rno-miR-26B
rno-miR-16-5P
rno-miR-7F-2-3P
COX-2
COX-2
COX-2
5'-TTCAAGTAATCCAGGATAGGCT
5'- TTCAAGTAATTCAGGATAGGT
5´:TAGCAGCACGTAAATATTGGCG
rno-miR-21
5´:CTATACAGTCTACTGTCTTTC
Normalizadorrno-miR-U6
rno-miR-873 PTEN5´:GCAGGAACTTGTGAGTCTCCT
5´:TGGCCCCTGCGCAAGGATG
rno-miR-463* 5´: TACCTAATTTGTTGTCCATCA PTEN
rno-miR-1 5' TGGAATGTAAAGAAGTGTGTAT Hipertrofia
5' TAGCTTATCAGACTGATGTTGA Hipertrofia

33
3.10 – Análises de bioinformática e construções de redes moleculares
Foram utilizadas técnicas de bioinformáticas disponíveis no programa
Cytoscape®
Versão 2.8.1. Foi construído um diagrama mostrando redes moleculares de
interação entre os miRNAs escolhidos com os alvos relacionados a COX-2, PTEN, Akt
e ao processo hipertrófico/ inflamatórios.
3.11 Análises estatísticas
As análises estatísticas foram realizadas utilizando-se o software GraphPad
Prism 5.0. Os resultados foram relatados pela média ± desvio padrão e avaliado por
análise de variância One-Way ANOVA, seguido por teste de Dunnett, sendo
considerado significante p<0,05.
3.11 Considerações éticas
Para a realização deste experimento, foram utilizados camundongos mantidos no
biotério da Universidade Federal de Ouro Preto, sob - responsabilidade do laboratório
de Doença de Chagas da mesma Universidade. A utilização desses animais foi aprovada
pelo Comitê de Ética em Uso de animal da Universidade Federal de Ouro Preto, sob o
Protocolo nº 2011/76 e “Manutenção de cepas do T. Cruzi in vivo” válido até 10/2015.

34
4. RESULTADOS

35
4. 1 Análise da infecção pela cepa Be – 62 em células H9c2
Para avaliar o padrão da infecção celular pela cepa Be-62 do T. cruzi, foram
investigados o perfil celular e bioquímico das células H9c2 imediatamente após 2 horas
de interação com o parasito. Os tempos de infecção de 0, 2, 6, 12, 24 e 48 horas foram
investigados. As células foram coradas pelo Giemsa e analisadas em microscópio óptico
modelo Leica DMLB. Em cada análise 100 células foram contadas e a infecção foi
verificada pela presença das formas amastigotas do parasito no interior do citoplasma. A
Figura 7 ilustra a infecção das células H9c2 pela cepa Be-62 nas células H9c2, e as
formas amastigotas são apontadas no final das setas.
Figura 8. Análise celular da infecção de células H9c2 pela cepa Berenice-62: a
Figura representa a evolução temporal da infecção. A) células não infectadas, B), C) ,
D, E) e F) representam os intervalos de infecção celular de 0, 2, 6,12, 24 e 48 horas.
Considerando-se o maior número de amastigotas no intervalo de 48 h de
infecção, devido a multiplicação do parasito no interior da célula, análises de
microscopia de fluorescência utilizando o corante 4',6-Diamidino-2-phenylindole
(DAPI – Sigma-Aldrich) foram realizadas. Para isso, 100 células foram novamente

36
analisadas e contadas e o número de amastigotas foi verificado em microscópio modelo
Leica DMLB.
Figura 9. Microscopia de fluorescência após 48 h de infecção da célula H9c2 pela cepa
Be-62 do T. cruzi.
Baseado nas análises de microscopia, o percentual de infeção foi estimado
através da contagem do número de células infectadas, o que permitiu a determinação da
taxa de infecção celular. Uma taxa de infecção de 9% foi verificada em todos os tempos
estudados. As análises foram realizadas em triplicata.

37
T e m p o d e in fe c ç ã o
Pe
rc
etu
al
de
in
feç
ão
0 H
2 H
6 H
12 H
24 H
48 H
0
5
1 0
1 5
Figura 10. Índice de Infecção de células cardíacas H9c2 pela cepa Berenice-62. O
teste estatístico One-Way ANOVA foi realizado.
Paralelamente, o número de amastigotas intracelulares, foi determinado em
diferentes tempos por meio da contagem das amastigotas do parasito dentro das células
infectadas. Observamos que esses valores aumentaram no tempo de 48 horas em
decorrência da proliferação do parasito.
T e m p o d e in fe c ç ã o
Nú
me
ro
de
am
as
tig
ota
s/c
elú
la
0 H
2 H
6 H
12 H
24 H
48 H
0
1 0
2 0
3 0
4 0***
Figura 11. Número de amastigotas por célula infectada. Foi aplicado teste estatístico
One-Way ANOVA.

38
4.2 Investigando a sobrevivência celular pós-infecção pela cepa Be-62 do T. cruzi:
os ensaios de viabilidade celular
Ensaios de viabilidade celular foram realizados para analisar o efeito do parasito
na sobrevivência celular. Os resultados demonstraram que nos intervalos de 0, 2 e 6
horas não houve alteração significativa na viabilidade celular, sendo observada uma
pequena diminuição nos tempos de 12 e 24 horas. No entanto, no intervalo de 48 horas
de infecção parasitária, houve uma redução de 50 % na viabilidade celular quando
comparado com o controle.
T e m p o d e in fe c ç ã o
Via
bil
ida
de
Ce
lula
r
C o n tr o le0 H 2 H 6 H 1 2 H 2 4 H 4 8 H
0
5 0
1 0 0
1 5 0
***
Figura 12: Viabilidade de células H9c2 infectada com T. cruzi Cepa Be-62. Os
tempos de infecção de 0, 2, 6, 12,24 e 48 horas foram analisados. Foi feito o teste
estatístico One-Way ANOVA com pós-teste de Dunnett. Os experimentos foram
realizados em triplicata biológica e técnica, onde *p<0,05; **p<0,01 e ***p<0,001.
4. 3 RNAm relacionados com processo hipertrófico
A expressão do RNAm de genes relacionados ao processo hipertrófico foi
realizadas pelo método de qPCR após a infecção de células H9c2 por T. cruzi. Os
resultados demostraram uma diminuição nos níveis de expressão do RNAm de Anf em
todos os tempos de infecção sendo que nos intervalos de 0, 2 e 6 horas esses valores
caíram cerca de 65% quando comparado com o controle. Os níveis de expressão do
RNAm de Myhc também mantiveram diminuídos nos intervalos entre 0 a 24 horas,
tendo um pequeno aumento no tempo de 48 horas de aproximadamente 20% em relação
ao controle. Por outro lado, a análise do RNAm de Bnp (Figura 17C) revelou uma

39
oscilação nos seus níveis entre os intervalos 0 a 24 horas, sendo que em 6 horas esses
valores tiveram um aumento de aproximadamente sete vezes em relação ao controle,
seguido por uma diminuição significativa em 48 horas quando comparado com o grupo
controle, sugerindo um controle do mecanismo de estresse celular.
B N P
T e m p o d e in fe c ç ã o
Ex
pre
sã
o G
ên
ica
Re
lati
va
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
2
4
6
8
1 0
***
A N F
T e m p o d e in fe c ç ã o
Ex
pre
sã
o G
ên
ica
Re
lati
va
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0 .0
0 .5
1 .0
1 .5
*** ******
Figura 13. Níveis de expressão do RNAm de genes relacionados ao processo
hipertrófico. Expressão do RNAm obtido pelo método de qPCR . O método teve a β-
actina como gene normalizador. Foi aplicado teste estatístico One-Way ANOVA com
pós-teste de Dunnett. Os experimentos foram realizados em triplicata biológica e
técnica, onde *p<0,05; **p<0,01 e ***p<0,001.
A
)
B
)
C
)

40
4.4 A infecção parasitária modula o ambiente pró-inflamatório: a mensuração de
prostaglandina E2 (PGE2)
Os níveis de PGE2 produzidos durante a infecção de células H9c2 pela cepa Be-
62 do T. cruzi foram mensurados e foi observado, que nas condições estudadas, a cepa
Be-62 induz aumento da síntese de PGE2 significativamente em todos os tempos de
infecção estudados. A análise dos resultados demonstrou que no intervalo de 0 hora de
infecção ocorreu um aumento de aproximadamente 80 % nos níveis de PGE2 quando
comparado com o controle. Interessantemente, a presença do parasito manteve os níveis
de PGE2 mais elevados que a condição controle em todos os tempos de infecção
analisados.
Co
ntr
ole 0
H2H
6H
12H
24H
48H
0
5 0
1 0 0
1 5 0
2 0 0 ***
PG
E2
(p
g/
ml)
T e m p o d e in fe c ç ã o
Figura 14. Níveis de PGE2 produzidos durante a infecção da célula H9c2 pela cepa
Be-62 doT. cruzi. Foram estudados os tempos de infecção de 0, 2, 6,12, 24 e 48 horas.
Foi aplicado teste estatístico One-Way ANOVA com pós-teste de Dunnett. Os
experimentos foram realizados em triplicata biológica e técnica, onde *p<0,05;
**p<0,01 e ***p<0,001.
4.5 A expressão do RNAm e da proteína COX-2 estão aumentadas durante o
processo pró-inflamatório induzido por T. cruzi
A Figura 13 A e 13 B, representam os gráficos da expressão do RNAm de Cox-
2e da sua proteína durante o processo pró-inflamatório celular modulado pelo T. cruzi.
As análises realizadas por qPCR (Figura 13 A) demonstram um aumento significativo

41
dos níveis transcricionais do RNAm de Cox-2 no intervalo de 0 horas, e esse aumento
foi 16 vezes maior quando comparado ao controle, decaindo na sequência dos intervalos
analisados. Sendo que nos intervalos de 24 e 48h, os níveis de Cox-2 mantiveram
abaixo do controle. Nas análises feitas pelo o método de Western blotting, os valores
também permaneceram aumentados. A Figura 13 B, mostra um aumento significativo
da expressão nos tempos iniciais de infecção de 0, 2 e 6 horas (140 %, 90 % e 76%
respectivamente), em comparação ao o controle. Nos intervalos seguintes esses valores
foram reduzidos.
R N A m d e c o x -2
T e m p o d e in fe c ç ã o
Ex
pre
sã
o G
ên
ica
Re
lati
va
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
5
1 0
1 5
2 0
***
*
***
Figura 15. Níveis de expressão da proteína e do RNAm de COX-2. A) Expressão do
RNAm obtido pelo método de qPCR . B) Expressão da proteína obtida pelo método de
Western blotting. Ambos os métodos tiveram a β-actina como gene normalizador. Foi
aplicado teste estatístico One-Way ANOVA com pós-teste de Dunnett. Os experimentos
foram realizados em triplicata biológica e técnica, onde *p<0,05; **p<0,01 e
***p<0,001.
P r o te ín a C O X -2
Co
ntr
ole 0
H2H
6H
12H
24H
48H
0
1
2
3
***
*****
T e m p o d e in fe c ç ã o
Ex
pre
ss
ão
Pro
teic
a
A
)
B
)

42
4.6 A expressão gênica de PTEN encontra-se modulada nos tempos iniciais de
infecção por T. cruzi
Considerando os objetivos centrais desse projeto, análises da expressão do
RNAm e da proteína PTEN também foram realizadas. As análises feitas por qPCR
(Figura 14 A) demonstraram uma aumento significativo no nível transcricional de Pten
em todos os tempos de infecção. Um aumento mais expressivo foi observado após 6
horas de infecção aproximadamente três vezes mais que o controle. A Figura 14 B
apresenta resultados obtidos pelo método de Western blotting, uma diminuição da
expressão proteica de PTEN nos tempos iniciais de 0, 2 e 6 horas foram observadas,
sendo que esses valores foram de aproximadamente a metade do valor encontrado na
condição controle. No entanto, houve um aumento significativo da expressão de PTEN
nos intervalos de 12 e 24 horas de infecção (126 e 152% respectivamente em relação ao
controle). A análise ainda demonstrou que no intervalo de 48 horas a expressão proteica
de PTEN diminui quando da comparação com o intervalo de 24 h. Entretanto, foi
observado um aumento global de 23% nos níveis da proteína, em relação ao controle.
R N A m d e p te n
T e m p o d e in fe c ç ã o
Ex
pre
sã
o G
ên
ica
Re
lati
va
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
1
2
3
4***
Figura 16. Níveis de expressão gênica de pten. A) Expressão do RNAm obtido pelo
método de qPCR . B) Expressão da proteína obtida pelo método de Western blotting.
Ambos os métodos tiveram a β-actina como gene normalizador. Foi aplicado teste
P ro te ín a P T E N
Co
ntr
ole 0
H2H
6H
12H
24H
48H
0
1
2
3
******
***
*
T e m p o d e in fe c ç ã o
Ex
pre
ss
ão
Pro
teic
a
A
)B
)

43
estatístico One-Way ANOVA com pós-teste de Dunnett. Os experimentos foram
realizados em triplicata biológica e técnica, onde *p<0,05; **p<0,01 e ***p<0,001.
4.7 A fosforilação da proteína PTEN é menor no intervalo de 0 horas de infecção
com T. cruzi
A expressão da proteína P-PTEN também foi analisada por ensaios de Western
blotting para saber o quanto dessa proteína esta sendo fosforilada. Observou- se que o
nível de fosforilação da proteína diminui em 67% do valor no intervalo de 0 hora,
quando comparado ao controle. Nos intervalos seguintes (2, 6 e 12 h) um aumento de
35% nos níveis de fosforilação da proteína foi detectado quando comparado com o
controle. No intervalo de 24 horas ocorreu uma queda nos níveis de fosforilação da
proteína para novamente ter esses níveis aumentados em 48h (33% maior em relação ao
controle).
P -P T E N
Co
ntr
ole 0
H2H
6H
12H
24H
48H
0 .0
0 .5
1 .0
1 .5 ***
Ex
pre
ss
ão
Pro
teic
a
Figura 17. Níveis de fosforilação da enzima- PTEN. Obtido pelo método de Western
Blot. A β-actina foi escolhida como gene normalizador. Foi aplicado teste estatístico
One-Way ANOVA com pós-teste de Dunnett. Os experimentos foram realizados em
triplicata biológica e técnica, onde *p<0,05; **p<0,01 e ***p<0,001.

44
4.8 A expressão do RNAm Akt 1 e Akt 2 está aumentada durante o processo pró-
inflamatório induzido por T. cruzi
O gráfico seguinte mostra a expressão gênica de Akt 1 e Akt 2 pelo o método de
qPCR. A Figura 16 A mostra a expressão gênica de Akt 1; observou-se um aumento na
expressão do RNAm de Akt 1 em todos os tempos de infecção, sendo mais expressivo
no intervalo de 0 h (176%) quando comparado com o controle. No entanto, no tempo de
48 horas de infecção a expressão não apresentou diferença significativa em relação ao
controle, porem apresentou expressão significante menor em relação aos outros tempos.
A figura 16 B mostra a expressão gênica de Akt 2, onde se observa um aumento
significativo da expressão (maior que 400%) em todos os tempos e analisados quando
comparado ao controle.
T e m p o d e in fe c ç ã o
Ex
pre
sã
o G
ên
ica
Re
lati
va
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
1
2
3
R N A m d e A k t 1
_ _ _ _ _ _ _ _ * * * _ _ _ _ _ _ _ _
T e m p o d e in fe c ç ã o
Ex
pre
sã
o G
ên
ica
Re
lati
va
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
2
4
6
8
R N A m d e A k t 2
_ _ _ _ _ _ _ _ _ * * * _ _ _ _ _ _ _ _ _
Figura 18. Níveis de expressão do RNAm de Akt 1 e Akt 2 . Expressão do RNAm
obtido pelo método de qPCR . O método teve a β-actina como gene normalizador. Foi
aplicado teste estatístico One-Way ANOVA com pós-teste de Dunnett. Os experimentos
foram realizados em triplicata biológica e técnica, onde *p<0,05; **p<0,01 e
***p<0,001.
4.9 Análise da expressão de miRNAs
miRNAs candidatos a modulação dos níveis transcricionais e proteicos de
COX-2 e PTEN e de genes corrrelacionados ao processo hipertrófico foram analisados
em ensaio de qPCR. Buscas em banco de dados (mirDB http://mirdb.org/miRDB,
A
)
B
)

45
MiRBase http://www.mirbase.org) nortearam nossa investigação e considerando-se os
objetivos específicos os miRNA -1, -21, -26a, -26b, -463, -873, -16-5p e - let 7f-23p de
Rattus norvergicus foram analisados. A análise foi realizada buscando-se identificar
possíveis marcadores moleculares moduladores do processo inflamatório/ hipertrófico
mediado pela infecção parasitária.
4.9.1 miRNAs com alvos em COX-2
A análise dos resultados demonstram que os miRNAs 16-5p e let 7f-2-3p
tiveram seus níveis de expressão alterados em quase todos os tempos de infecção
célula/parasito quando comparados com o controle. Para o miRNA 16-5p (Figura 19 A),
os intervalos de 2 e 24 h foram os mais expressivos (140 e 500 % respectivamente em
relação ao controle) e para o miRNA - let 7f-23p (Figura 19 B) os aumentos foram
significativos, em todos os tempos analisados com exceção do intervalo de 12 h (que
teve redução de 23 % em relação ao controle. A Figura 19 C) demostraram que o
miRNA 26a teve seus níveis reduzidos 68% ao longo das 48 horas de infecção. Por
outro lado o miRNA -26b (Figura 19 D) mostrou um interessante padrão de oscilação
que se correlaciona ao RNA cox-2, com nível significativamente reduzido em 0 h (89%
menor que o nível de expressão transcricional quando comparado ao controle)

46
m iR -1 6 -5 p
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
2
4
6
8
***
***
Ex
pre
sã
o R
ela
tiv
a
m iR -le t-7 f-2 -3 p
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0 .0
0 .5
1 .0
1 .5
**
*
***
Ex
pre
sã
o R
ela
tiv
a
m iR 2 6 a
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0 .0
0 .5
1 .0
1 .5
Ex
pre
sã
o R
ela
tiv
a
***
m iR -2 6 b
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0 .0
0 .5
1 .0
1 .5
Ex
pre
sã
o R
ela
tiv
a
****
Figura 19. Níveis de expressão dos miRNAs. Os miRNAs-16-5p (A) – let 7f-2-3p (B)
- 26a (C), -26b (D) foram obtidos pelo ensaio de qPCR . O método teve a β-actina
como gene normalizador. Foi aplicado teste estatístico One-Way ANOVA com pós-teste
de Dunnett. Os experimentos foram realizados em triplicata biológica e técnica, onde
*p<0,05; **p<0,01 e ***p<0,001.
4.9.2 miRNAs com alvos em PTEN
Os resultados obtidos da análise do perfil de expressão gênica mostraram que o
miR-873 (Figura 20A) teve um aumento da expressão apenas no intervalos de 0, 12 e 24
horas de infecção, sendo mais expressivo no intervalo de 12 horas que apresentou um
valor oito vezes maior que a condição controle. A análise também revelou uma aumento
na expressão do miR-463* (Figura 20B) porém esse aumento foi mais significativo nos
intervalos de 0, 2, 6 e 24 horas de infecção, sendo que em 2 horas esse aumento foi dez
maior que o controle.
A
)
B
)
CD
)

47
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
2
4
6
8
1 0
***
***
**Ex
pre
sã
o R
ela
tiv
a m iR 8 7 3
m iR - 4 6 3 *
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0 .0 0
0 .0 5
0 .1 0
0 .1 5
***
*
15
10
5
0
Ex
pre
sã
o R
ela
tiv
a
Figura 20. Níveis de expressão dos miRNAs. Os miRNAs-873 (A) – 463 (B) foram
obtidos pelo ensaio de qPCR . O método teve a β-actina como gene normalizador. Foi
aplicado teste estatístico One-Way ANOVA com pós-teste de Dunnett. **p<0.01 e
***p<0.001.
4.9.3 miRNAs relacionados com o processo hipertrófico cardíaco
Os níveis de expressão relativa do miR-21 (Figura 21A), tiveram uma queda na
expressão no intervalo de 0, 2 e 6 de aproximadamente 10% quando comparado ao
controle e logo em seguida, no intervalo de 12, 24 e 48 horas de infecção houve um
aumento significativo da expressão cerca de 70 % quando comparado ao controle . Já o
miR-1 (Figura 21B) mostraram uma expressão diminuída em todos os intervalos de
infecção analisados .
A B

48
Co
ntr
ole
0 H
2 H
6 H
12 H
24 H
48 H
0
5
1 0
1 5
2 0
2 5 ***
m iR -2 1
Ex
pre
sã
o R
ela
tiv
a
C
on
tro
le0 H
2 H
6 H
12 H
24 H
48 H
0 .0
0 .5
1 .0
1 .5 m iR -1
***
Ex
pre
sã
o R
ela
tiv
a
Figura 21. Níveis de expressão dos miRNAs. Os miRNAs-21 (A) –1 (B) foram
obtidos pelo ensaio de qPCR . O método teve a β-actina como gene normalizador. Foi
aplicado teste estatístico One-Way ANOVA com pós-teste de Dunnett. Os experimentos
foram realizados em triplicata biológica e técnica, onde *p<0,05; **p<0,01 e
***p<0,001.
4.10 Os miRNAs – 1, -21, -26b podem modular a expressão de genes relacionados
ao processo hipertrófico/ inflamatório
Foram utilizadas ferramentas de bioinformáticas disponíveis na internet tais
como Blast (www.ncbi.lm.nih.gov/Blast), mirDB (http://mirdb.org/miRDB/), MiRBase
(http://www.mirbase.org/), TargetScan (http://www.targetscan.org/) para busca de
potenciais alvos dos miRNAs -1, -21, -26b. Os alvos selecionados foram àqueles
relacionados à via do processo hipertrófico mediado por inflamação. Estes alvos podem
ser visualizados na Tabela 3 e são representados na Figura 22.
Tabela-3. miRNAs e genes alvos
miRNA Score Alvo
rno-miR-26b 86 Pten
rno-miR-1 66 Pla2
rno-miR-1 70 Akt
Akt
54 Ptgs 2
rno-miR-21 50 P13k
rno-miR-21 62 Pla2
rno-miR-26b
rno-miR-26b
56
A B

49
Figura 22. Rede de interação entre miRNAs e seus alvos relacionados ao processo
hipertrófico mediado por inflamação.

50
5. DISCUSSÃO

51
A doença de Chagas, causada pelo agente etiológico Trypanosoma cruzi,
constitui um grave problema de saúde pública, que se distribui predominantemente no
continente Americano (RASSI et al; 2010). A cardiopatia é uma das formas mais graves
da doença de Chagas, sendo a hipertrofia cardíaca uma característica comum no estágio
crônico da doença (TANOWITZ, 2009; MARTINS-MELO, 2012).
Muitos estudos apontam a presença da proteína COX-2 no processo hipertrófico
(MENDEZ e LAPOINTE; 2002). Buscando uma maior compressão, o presente estudo
investigou mecanismos moleculares inerentes a COX-2 no processo hipertrófico
induzido pela inflamação causada por T. cruzi, visando à caracterização de marcadores
moleculares da patogenia. Sabe-se que a regulação da expressão gênica se dá em nível
transcricional e, até mesmo, pós-traducional, com o envolvimento de diferentes
elementos. Particularmente, a regulação da expressão da COX-2 ainda é obscura, mas
estudos apontam uma regulação indireta entre COX-2 e a proteína supressora tumoral
PTEN (LI et al; 2011) e o envolvimento de alguns miRNAs (JI et al, 2009;
STRILLACCI et al, 2009). Considerando-se o desafio de entender as particularidades
mecanísticas da regulação fina de COX-2 e a presença do estímulo pró-inflamatório na
doença de Chagas, objetivamos nesse estudo investigar a regulação da expressão de
COX-2, visando à identificação de alguns de seus possíveis moduladores. Para isso,
células H9c2 (células cardíacas de ratos neonatos) foram infectadas com o T. cruzi
(Figura 8 e 9) e os tempos iniciais de infecção foram investigados, focando-se na análise
da fase aguda da doença.
A cepa escolhida para execução dos experimentos foi a cepa Berenice-62. Esta é
uma cepa pouco estudada. É caracterizada por ser delgada e é semelhante à cepa Y, em
relação à morfologia e sensibilidade a agentes quimioterápicos, motivo pelo o qual nos
levou a estudá-la. Para analisarmos o comportamento dessa cepa em o nosso modelo de
estudo, as células H9c2 infectadas foram analisadas, através de microscopia óptica, para
avaliar a porcentagem de células infectadas e o número de amastigotas por célula nos
intervalos de infecção de 0, 2, 6, 12, 24 e 48 horas. Nossos resultados demonstraram
uma baixa infectividade em todos os intervalos analisados, verificando-se uma taxa de
infecção celular de aproximadamente 9% (Figura 10). Interessantemente, a análise
microscópica demonstrou que o número de amastigotas intracelulares teve um aumento
significativo após 48 horas de infecção, provavelmente devido o processo de

52
proliferação do parasito (Figura 11). O aumento ou a diminuição da carga parasitária
tem sido relacionado com a variabilidade genética das cepas do T. cruzi, que parece
exercer um papel fundamental no processo, podendo apresentar uma maior ou menor
capacidade multiplicativa (BRENER; 1978). Corroborando com esses dados,
SCHARFSTEIN (1999) demostrou em seus estudos que camundongos infectados com
diferentes cepas do T. cruzi apresentaram diferenças significativas na infectividade,
dependendo da virulência, patogenicidade e tropismo de cada uma. Além disso, o
sistema imune também tem sido apontado como fundamental na progressão da doença,
pois pode estar envolvido tanto na redução da taxa parasitária, bem como no
aparecimento de lesões na fase crônica da doença (CUNHA NETO; 2008).
O efeito modulador do parasito na sobrevivência celular também foi investigado.
Para isso, ensaios de citotoxidade foram realizados utilizando o teste MTT, que avalia a
atividade das desidrogenases mitocondriais à partir da redução do MTT, produzindo um
sal de formazan insolúvel e que, posteriormente, é solubilizado para quantificação em
espectrofotômetro (MOSMANN; 1983). Nossos resultados indicaram uma diminuição
da viabilidade celular em, aproximadamente, 50% no período de 48 horas de infecção,
quando comparado com o controle (Figura 12), período este que coincide com o
aumento do número de amastigotas intracelulares devido a multiplicação do parasito por
divisão binária (Figura 11). Interessantemente, estudos demonstram que a resposta
imune causada pela presença do parasita ou antígenos do próprio T.cruzi induz à
apoptose das células cardíacas (ANDRADE, 1985; BRENER, 1997; MACHADO,
2000; HENRIQUES-PONS, 2002; SOUZA, 2003; KIERSZENBAUM, 2005; CUNHA-
NETO, 2008; MELO, 2008). Por outro lado, a presença do parasito no interior da célula
tem sido apontada como crucial na inibição da apoptose. Para sobreviver o tempo
suficiente para reproduzir- se, o parasito tem a capacidade de modular vias apoptóticas
e, dessa maneira, garantir a sua sobrevivência ou disseminação no hospedeiro (BROW,
1996; GREN, 2004). Corroborando com esses achados, estudos in vitro feitos por
PETERSEN, et al,(2006) demostraram que a modulação das vias de apoptose está
correlacionada com a presença do parasito intracelular, que parece ser uma estratégia do
T. cruzi para promover a sua própria sobrevivência e crescimento.
De maneira semelhante, estudos vêm demonstrando que, durante a infecção,
tanto o parasito como proteínas produzidas por ele, como a cruzipaína, são capazes de

53
manter a viabilidade celular, por meio da ativação de via PI3K/Akt, garantindo a sua
permanência no interior da célula e impedindo temporariamente a morte celular
(BURLEIGH e WOOLSEY, 2002; AOKI, 2004). Estudos feitos por WILKOWSKY et
al. (2001) demonstraram que a via PI3K/Akt é um importante regulador do processo de
invasão por T. cruzi. Além disso, também estimula mecanismos que favorecem a sua
própria sobrevivência nas células parasitadas (PROCÓPIO, 1999; CHUENKOVA,
2001). A indução ou a inibição da apoptose pode estar relacionada com o controle da
carga parasitária ou mecanismo de escape do parasito da resposta imunológica
produzida durante a infecção. Embora ROSA DA SILVA (2009) sugira que alterações
nas mensurações de absorbâncias devam-se provavelmente à presença de proteínas do
próprio parasito que interfiram nas análises do MTT e, não especificamente,
correlacionando com a diminuição da viabilidade celular. No entanto, é importante
salientarmos que não foram feitos estudos que avaliem o mecanismo de morte. Mais
estudos precisam ser feitos a fim de esclarecermos tais mecanismos.
Considerando que durante o processo pró-inflamatório ocorre o aumento da
expressão de marcadores fenotípicos da hipertrofia (HUNTER e CHIEN; 1996), o nível
transcricional dos genes Anf, Bnpe β-Myhc foram investigados após a infecção de
células H9c2 com o T. cruzi. Particularmente, os peptídeos natriuréticos são
considerados marcadores de hipertrofia e insuficiência cardíaca e podem ser utilizados
tanto no diagnóstico como no prognóstico das cardiopatias (RICHARDS, 2007). De
modo relevante, nossos resultados demonstraram uma diminuição da expressão de Anf
(Figura 13 A) em todos os intervalos de infecção analisados. Um padrão semelhante de
expressão foi verificado nas análises de β-Myhc (Figura 13 B), com exceção do
intervalo de 48 horas, que teve um pequeno aumento em relação ao controle. Estes
resultados corroboram com os estudos de FRANCHINI (2001), que demonstraram que
em corações de ratos submetidos a uma sobrecarga de pressão, ocorre o aumento dos
níveis do RNAm de β – Myhc apenas entre o segundo e o terceiro dias e que as
alterações no RNAm desse gene depende da intensidade da hipertrofia cardíaca e da
espécie estudada. Paralelamente, GOODMAN (1993) demonstrou em seus estudos que
no coração chagásico a expressão de Anf depende da intensidade da patologia, pois pode
se manifestar de forma intensa em alguns indivíduos e de baixa intensidade em outros.

54
Por outro lado, os níveis de expressão de Bnp (Figura 13C) revelaram uma
oscilação nos seus níveis entre os intervalos 0 a 24 horas, sendo que em 48 horas
ocorreu diminuição significativa dos seus níveis, quando comparado com o controle. O
processo de remodelação cardíaca é influenciado por diversos estímulos e, entre eles, o
aumento da concentração de oxido nítrico, cálcio e estresse oxidativo (OGAWA; 2002).
Esse processo é marcado principalmente pela modificação do padrão de expressão de
varias proteínas, com a re-expressão de genes do período fetal, tais como o Bnp
(KOGLIN; 2001). Provavelmente, o processo de multiplicação do T. cruzi no interior da
célula pode ter gerado espécies reativas do oxigênio que podem ter contribuído para
oscilações nos níveis do RNAm desse gene, sugerindo um controle do mecanismo de
estresse celular desencadeado pela infecção. A fase aguda da doença é bastante
complexa e nela está envolvida a presença do parasito, a sua multiplicação, morte
celular e a resposta imunológica que levam à evolução da enfermidade (MELO; 2005).
As alterações dos níveis de Bnp podem estar envolvidas com as formas clínicas da
doença e diversos estudos já demonstraram que, na circulação de pacientes chagásicos
com cardiomiopatia, ocorre um aumento de Bnp, contudo, esse aumento tem sido
relacionado com a evolução da doença (PIAZZA,1994; DAVIDSON;1996;
RIBEIRO,2002 ; BENVENUTI, 2003).
Analisamos também os níveis de PGE2 produzidos durante a infecção de células
H9c2 pela cepa Be-62 do T. cruzi, considerando-se que a prostaglandina tem um efeito
imunomodulador na resposta imune na infecção por Trypanosoma cruzi (ABDALLA et
al., 2008). Observamos o aumento dos níveis da PGE2 durante todos os intervalos
estudados (Figura 14), sendo mais expressivo no intervalo de 0 hora, período este que
coincide com o aumento da expressão de COX-2 (Figura 15 A), sugerindo que a
infecção parasitária modula o ambiente pró-inflamatório. Na literatura, vários trabalhos
descrevem o aumento da produção de PGE2 durante a infecção com o T. cruzi e os
resultados apresentam uma correlação com a evolução da enfermidade (STERIN e
BORDA 1996; BORGES 1998; MICHELIN 2005; ABDALLA, 2008). Isso pode ser
explicado pelo fato de que processo inflamatório produzido em resposta ao hospedeiro
pode ter desencadeado uma reação exacerbada em resposta ao estímulo desencadeador,
conduzindo, assim, para uma expressão aumentada dos níveis de PGE2 e,
consequentemente, da expressão de COX-2.

55
A COX-2 é uma enzima chave na síntese de prostaglandinas e, em condições
normais, é expressa em pequenas quantidades (SULLY et al., 2004). Porém, durante o
processo inflamatório, ocorre aumento dos níveis transcricionais e também aumento da
estabilidade do RNAm (CHAOUKIet al., 2009). Estes resultados sugerem que a
inibição da COX-2, bem como da síntese da prostaglandina, tem um papel importante
na reversão da imunossupressão dessa patologia. Estudos feitos por CORRAL (2013)
demonstraram que a proteina COX-2 é expressa em corações e em cardiomiócitos
infectados pelo T. Cruzi, sugerindo um papel importante na patogênese da
cardiomiopatia chagásica crônica. ABDALLA (2008) demonstrou que o tratamento
com drogas inibidoras de COX-2 é capaz de diminuir danos no tecido cardíaco de
animais infectados por T. cruzi. Além disso, MUKHERJEE (2011) evidenciou que a
inibição da síntese de PGE2 reduz os infiltrados inflamatórios e a fibrose cardíaca em
corações de camundongos infectados por T. cruzi. Acreditamos que a compreensão da
mecanística da regulação da enzima COX-2 possa ser utilizada como alvo terapêutico
capaz de modular os danos cardíacos causados pela infecção do T. Cruzi no processo
inicial da Doença de Chagas.
Investigar a regulação cruzada de COX-2 e PTEN, na busca das particularidades
mecanísticas que apontem a presença de marcadores moleculares nessa via bioquímica,
torna-se relevante. Sendo assim, o nível transcricional dos genes Cox-2, Pten e Akt
foram analisados através de ensaios de PCR em tempo real. A análise da expressão
proteica de COX-2, PTEN e P-PTEN também foi investigada pelo o método Western
blotting. Nossos resultados demonstraram que a invasão parasitária induziu uma
modulação na expressão do RNAm e da expressão proteica de COX-2, nos intervalos
iniciais de infecção, uma vez que observamos um aumento do RNAm deste gene nos
intervalos de 0, 2 e 6 horas de interação célula/parasito (Figura 15 A). A expressão da
proteína COX-2 também aumentou nos intervalos iniciais de infecção, de 0, 2 e 6 horas
(Figura 15 B), retornando aos níveis basais nos demais intervalos. No entanto esse
aumento foi mais significativo no tempo de 0 hora. Vários fatores podem promover o
aumento da expressão da Cox-2, entre eles, a liberação de citocinas, fatores de
crescimento, hormônios e oncogenes (BURLEIGH; 2002). No entanto, esta indução é
quase sempre de curta duração e pode regressar aos valores basais num intervalo de 24 a
48 horas, caso o estímulo inicial seja retirado (STACK e DUBOIS, 2001).

56
Ao correlacionarmos a expressão proteica de COX-2 com os níveis de expressão
de PTEN, observamos um aumento do RNAm de PTEN em todos os intervalos de
infecção (Figura16 A) e uma diminuição da proteína nos intervalos iniciais de 0, 2 e 6
horas (Figura 16 B). De uma maneira interessante, notamos que, onde os níveis da
proteína COX-2 encontram-se elevados (Figura15 B), os níveis da proteína PTEN
encontram-se diminuídos (Figura 16 B). Concordando com esses dados notamos ainda
que, em relação ao controle, a fosforilação de PTEN foi menor no intervalo de 0 hora
(Figura 17), sugerindo uma correlação de expressão entre PTEN e COX-2. Nossa
hipótese é que a expressão de COX-2 pode estar envolvida na fosforilação de PTEN
através da ativação da Caseína quinase-2 (CK-2) que, por sua vez, suprime atividade de
PTEN, desempenhando um papel significativo no desenvolvimento de hipertrofia
através da ativação da via PI3K/AKT.
Na literatura, encontram-se descritos alguns estudos que demonstraram uma
relação entre o aumento dos níveis de COX-2 e a diminuição de PTEN. LI et al. (2011)
sugeriram em seus estudos que a expressão de COX-2 pode regular a expressão de
PTEN. COVEY et al.(2007) demonstraram ainda que PTEN correlaciona-se de maneira
indireta com a expressão de COX-2, modulando a atividade de Akt. Estudos feitos por
ST- GERMAIN et al.(2004) também demonstraram que a perda de PTEN, bem como
ativação de Akt, exerce uma função regulatória sobre a expressão de NF-KB (Fator de
transcrição) aumentando, assim, os níveis de COX- 2. De maneira semelhante, KUNDU
(2006) sugeriu que a expressão alterada dos níveis da COX-2 pode estar relacionada
com a fosforilação do IkB que, por sua vez, transloca NF-KB para o núcleo,
contribuindo para a ativação de genes como o da COX-2.
Paralelamente, nossos resultados demostraram um aumento da expressão gênica
de Akt1 e Akt2 (Figura 18 A e 18 B) em todos os intervalos de infecção quando
comparados com a condição controle, sugerindo uma co-relação direta com COX-2.
Tais achados encontram concordância na literatura, uma vez que diversos trabalhos vêm
demostrando que a via de sinalização de Akt está relacionada com a ação de COX-2
(CHUENKOVA, 2001; BURLEIGH e WOOLSEY , 2002; O`CONNOR, 2004). Além
disso, estudos feitos por HIRAOKA (2001) demonstraram que cardiomiócitos de ratos
neonatos infectados com o T. cruzi, na presença da via de sinalização PI3K/Akt, pode
ativar o NF-KB, culminando, assim, para o desenvolvimento de hipertrofia cardíaca.

57
Contudo, para elucidarmos tais mecanismos, futuros estudos deverão ser realizados a
fim de esclarecermos como COX-2 poderia interferir na fosforilação PTEN. Na
literatura, são poucos os estudos que investigam essa regulação. Estudos
complementares poderão contribuir na compreensão da doença de Chagas a partir do
conhecimento da expressão e regulação da COX-2 na fase inicial da infecção por T.
cruzi, pois o nosso trabalho demonstrou uma relação entre os genes Cox-2, PTEN e Akt,
sugerindo uma possível participação desses elementos na instalação do processo
hipertrófico.
Considerando-se os objetivos centrais do presente estudo, buscamos investigar a
participação de moléculas efetoras na regulação da expressão gênica e, entre esses
elementos, procuramos investigar o perfil de expressão de microRNAs, uma vez que
vários estudos apontam o papel dessas moléculas no desenvolvimento de patologias
cardiovasculares (ZHANG, 2008; WANG, 2010; LI, 2011; CARVALHO, 2012). Sendo
assim, para investigarmos o possível envolvimento de miRNAs na regulação pós-
transcricional de COX-2, PTEN e dos pró-inflamatórios/hipertróficos, ensaios de qPCR
foram realizados. Cabe a observação de que a escolha dos miRNAs investigados
respalda-se numa busca em banco de dados.
Com relação aos miRNAs, com alvo em COX-2, nossos resultados
demonstraram que a infecção das células com a cepa Be-62 induziu um aumento dos
miR-16-5p e -let 7f-2-3p (Figura 19 A e 19 B) e uma diminuição do miR-26a (Figura 19
C). A análise do conjunto desses resultados infere que esses miRNAs parecem não estar
envolvidos na regulação de COX-2 no períodos de infecção analisados, pois os níveis
de expressão dos mesmos não estão correlacionados com alterações nos níveis da
proteína COX-2. No entanto, o miR - 26b parece estar envolvido na regulação de COX-
2, pois, os níveis destes miRNAs encontram-se baixos onde os níveis de expressão de
COX-2 apresentam-se aumentados, sugerindo uma possível regulação dos níveis da
proteína COX-2 por miRNA. Concordando parcialmente com esses dados, JI et al.
(2009) demonstraram que o miR- 26b regula a expressão de COX-2 em células de
carcinoma nasofaríngeo. No entanto, o papel desses reguladores na doença de Chagas
ainda é pouco explorado, considerando-se que no plasma sanguíneo essas moléculas são
estáveis (VARGAS, 2013). Um estudo mais detalhado poderá levar à caracterização de

58
miRNAs como marcadores moleculares da Doença de Chagas de maneira precisa,
direcionando assim para o tratamento.
Ao analisarmos os miRNAs, com alvo em PTEN, observamos que o miR-873
(Figura 20 A) teve um aumento da expressão nos tempos de 0, 2 e 24 horas. Já o miR-
463 (Figura 20 B) teve um aumento da expressão nos tempos de 0, 2, 6 e 24 horas.
Tomando em conjunto com os níveis da proteína PTEN, podemos verificar que parece
haver regulação da expressão de PTEN pelo miR-463 nos tempos iniciais de 0, 2 e 6
horas, pois os níveis destes miRNAs apresentam-se altos onde os níveis da proteína
COX-2 foram diminuídos. Entretanto, o papel deste miRNA na regulação de PTEN não
se encontra descrito na literatura e, os resultados apresentados, portanto, são pioneiros e
muito promissores. Para um maior esclarecimento, serão necessários mais estudos que
investiguem essa regulação.
De maneira interessante, o miR-1 (Figura 21 B) teve uma diminuição da
expressão em todos os tempos estudados. Já o miR-21(Figura 21 A) teve um aumento
da expressão nos tempos finais de 12, 24 e 48 horas, sugerindo um possível
envolvimento desses miRNAS na instalação do processo pró-inflamatório/ hipertrófico.
Esta situação encontra se em concordância com a literatura, uma vez que o aumento da
expressão dos miRNAs podem estar relacionados ao desencadeamento de processos
patogênicos e, a diminuição da expressão dessas moléculas, também pode levar a um
estado patológico. Estudos demonstram que a sobre-expressão do miR-1 atenua a
hipertrofia dos cardiomiócitos. Em contrapartida, a sua sub-expressão causa hipertrofia
cardíaca (SAYED et al., 2007; IKEDA et al., 2009). A diminuição dos níveis de
expressão destes miRNAs nas patologias cardiovasculares já foi referida por outros
autores, o que sugere que os miRNAs desempenham um importante papel nas doenças
cardíacas, uma vez que a desregulação pode conduzir à hipertrofia cardíaca (CARÈ et
al., 2007; SAYED et al., 2007). Por outo lado, o miR-21 teve um aumento da expressão
nos tempos finais de infecção por T. cruzi. Diversos autores já demonstraram que a
sobre-expressão do miR-21 leva à hipertrofia dos cardiomiócitos (VAN ROOIJ et al.,
2008; THUM, 2008; SMALL e OLSON, 2011), reforçando a veracidade dos resultados
obtidos com o nosso modelo de investigação celular da Doença de Chagas.
Considerando-se os resultados obtidos na investigação da expressão de miRNAs
em ensaios de qPCR, onde focamos em Cox-2 e Pten, ferramentas básicas de

59
bioinformática disponíveis em banco de dados da internet foram utilizadas para
identificar outros possíveis alvos dos miRNAs de interesse. Para essas análises
selecionamos os miRNAs -1, -21, -26b (Figura 22) e construímos uma pequena e
hipotética rede de interação entre os miRNAs escolhidos. Seus alvos foram desenhados
utilizando o programa TargetScan (Tabela 3). As análises demonstraram que o miR-1 e
o miR-21 tem como alvo a via Akt/P13k e a Fosfolipase A2 (Pla2). Já miR-26b foi
identificado como alvo envolvido indiretamente na expressão de COX-2, PTEN e Akt,
demonstrando um papel fundamental na instalação do quadro de hipertrofia.
O estudo desses miRNAs é de grande importância devido ao envolvimento
dessas pequenas moléculas na modulação da regulação da expressão gênica que
subsidia o processo inflamatório/ hipertrófico. Contudo, a participação das mesmas na
doença de Chagas ainda não está completamente esclarecida. Os resultados
apresentados nesse trabalho podem ser caracterizados como um primeiro passo no
esclarecimento da regulação fina da expressão gênica. Desta forma, estudos que
permitam um maior detalhamento sobre as vias de sinalização envolvidas na infecção
parasitária, bem como a identificação dos mecanismos pelos quais a expressão das
proteínas correlacionadas é modulada na doença Chagas, colaborarão com a
caracterização de marcadores moleculares da patologia, auxiliarão em métodos
diagnósticos mais precisos e no tratamento pertinente aos seus pacientes.

60
6. CONCLUSÃO

61
- A infecção de células H9c2 pela cepa Berenice-62 diminui as taxas de viabilidade
celular após 48 horas.
- A infecção pela cepa Berenice-62 modula os níveis de expressão dos marcadores de
hipertrofia cardíaca Anf, Bnp e β -myhc, bem como o ambiente pró-inflamatório nos
tempos iniciais de infecção considerando-se o aumento da expressão e da atividade da
proteína COX-2 e o aumento na produção de PGE2.
- Nossos resultados também demostraram uma relação entre os genes Cox-2, Pten, Akt
sugerindo uma possível correlação cruzada entre COX-2 e PTEN.
- O miRNA -26b parece está envolvido na regulação pós-traducional da proteína COX-
2. Já os microRNAs -16-5p, -let 7f-2-3p,-26a parecem não estar envolvidos nessa
regulação.
- O miRNA -463 parece está envolvido na regulação pós-traducional da proteína PTEN.
Já O miRNA -873 parece não estar envolvidos nessa regulação.
- A expressão alterada dos microRNAs, -1 e -21 sugere um possível envolvimento
desses miRNAS na instalação do processo pró-inflamatório/ hipertrófico ;
- A caracterização de marcadores moleculares na doença de Chagas é relevante, espera-
se que os resultados do presente estudo possam direcionar a caracterização de novos
marcadores que auxiliem no prognóstico e tratamento direcionado da doença, bem
como no desenvolvimento de novas drogas.

62
7. REFERÊNCIAS BIBLIOGRÁFICAS

63
ABDALLA, G.; K., FARIA, G. E.; SILVA, K. T.; CASTRO, E. C.; REÍS, M. A.;
MICHELIN, M. A. (2008). Trypanosoma cruzi: the role of PGE2 in immune response
during the acute phase of experimental infection. Exp Parasitol 118, 514-21.
ALMEIDA, M. I.; REIS, R. M.; CALIN, G. A. (2011) MicroRNA history: discovery,
recent applications, and next frontiers. Mutation Research/Fundamental and Molecular
Mechanisms of Mutagenesis.
ANDRADE, S.G. et al. (1985). Immunological response of Swiss mice to infection with
three different strains of Trypanosoma cruzi. Ann Med Trop Parasitol. v. 79, n. 4, p.
397-407.
ANDRADE, Z.A. (1983). Mechanisms of myocardial damage in T. cruzi infection. In
Cytophatology of Parasitic Diseases, Ciba Foundation Symposium n.99 London.
AOKI, M. P.; GUIÑAZÚ, N.L.; PELLEGRINI, A.V.; GOTOH, D. T.; MASIH, E. S.;
GEA, D.E. (2004). Cruzipaína, um importante antígeno de Trypanosoma cruzi,
promove arginase-2 expressão e sobrevivência dos cardiomiócitos neonatais do
mouse. Am. J. Physiol. 286. c206-c212.
BARRETO, A.C.P et al. (1990). Forma Indeterminada da doença de Chagas: uma
doença polimorfica. Arqu Bras Cardiol, v55, p.347-353.
BARRETTO, A.C.P.; DEL CARLO, C.H.; CARDOSO, J.N.; OCHIAL, M.E.;
OLIVEIRA-JR.M.T.; SCIPIONI, A.R. et al. (2009). Mortality in heart failure is going
down even in patients with inotropics (abstract). Eur J Heart Fail., v. 7, p. 66-66.
BARRY, S.; DAVIDSON, S. M.; TOWNSEND, P. A. (2008). Molecular regulation of
cardiac hypertrophy. The International Journal of Biochemistry & Cell Biology.
40:2023–2039.

64
BASBAUM, A. I.; JULIUS, D. (2006). Novos alvos contra a dor. Scientific American
Brasil, v.50, p.76-83.
BATISTA, M.S.; VERBISK, N.V. (2009). Estudo molecular sobre a aplicabilidade
terapeutica dos miRNAs regulatorios na expressão genicas em processos patologicos.
BATLOUNI, M .(2009). Nonsteroidal Anti-Inflammatory Drugs: Cardiovascular,
Cerebrovascular and Renal Effects. Arq Bras Cardiol 2010;94(4): 556-563.
BENVENUTI, L.A.; AIELLO, V.D.; PALOMINO, S.A.; HIGUCHI, M.L. Ventricular
expression of atrial natriuretic peptideo in chonic chagasic cardiomyopathy is not
induced by myocarditis. Int J Cardiol. 2003; 88(1):57-61
BESHAY, N.M.; ZORDOKY.; AYMAN, O.S.; EL-KADI. (2007). H9c2 cell line is a
valuable in vitro model to study the drug metabolizing enzymes in the heart, Journal of
Pharmacological and Toxicological Methods, Volume 56, Issue 3, November–
December ,Pages 317–322.
BORGES, M.M.; KLOETZEL, J.K.; ANDRADE, H.F.; TADAKORO,C.E.; PINGE-
FILHO, P.; ABRAHAMSOHN, I. (1998). Proteglandin and nitric oxide regulate TNF-
alpha production during trypanosoma cruzi. Immunology Letters; Netherlands, v. 63,p,
1-8.
BRADFORD, M.M. (1976). A rapid and sensitive method for the quantitation o
microgram quantities of proteins utilizing the principle of protein-dye binding.
Analytical Biochemistry, v.72, p.248-254.
BRASIL. Ministério da Saúde, Fundação Osvaldo Cruz. Brasília. (2008).
Disp.nívelem<www.fiocruz.br/chagas/cgi/cgilua.exe/sys/start.htm?sid=130

65
BRENER, Z.; CHIARI, E. (1963). Variações morfológicas observadas em diferentes
amostras de Trypanosoma cruzi. Rev. Inst. Med. Trop. São Paulo, v. 5, p. 220-224.
BRENER, Z. (1965). Comparative studies of different strains of Trypanosoma cruzi.
Ann. Trop. Parasitol., n. 59, p. 19-26.
BRENER, Z. (1973). Biology of Trypanosoma cruzi. Annual Review of Microbiology,
v. 27, p.347-382.
BRENER, Z.(1977). Intraspecific variation of tripasoma cruzi, two types of pasasite
populations of distinct characteristics; PAHO; Sci publ – 347; 11, 21.
BRENER, Z.; ANDRADE, Z. (1979). (Org.) - Trypanosoma cruzi e Doença de Chagas.
Rio de Janeiro: Guanabara Koogan.
BRENER, Z.; GAZZINELLI, R. T. (1997). Immunological control of Trypanosoma
cruzi infection and pathogenesis of Chagas' disease. International Archives Allergy
Immunology. v. 114, n. 2, p. 103-110.
BRENER, Z.; ANDRADE, Z.; BARRAL-NETTO, M. (2000). Trypanosoma cruzi e
doença de Chagas, 2ª edição. Rio de janeiro: Guanabara Koogan.
BROWN, L.; JACKSON, D.; RIFFKI, M.; SEOW, H.; WOOD, P. (1996). Defense
against the immune barrage: Helminth survival strategies, Immunology and Biology,
74, pp. 564-574.
BURLEIGH, B.; ANDREWS, N.W.; (1998). Signaling and host cell invasion by
Trypanosoma cruzi. Current Opinion in Microbiology, v.1, p.461–465.
BURLEIGH, B.A.; WOOLSEY, A.M. (2002). Cell signalling and Trypanosoma cruzi
invasion. Cellular Microbiology, v.4, n.11, p.701-711.

66
BURLEIGH, M.E. (2002). Cyclooxigenase-2 promotes early atherosclerotic lesion
formation in LDL receptor-deficient mice. Circulation, v.105, n.53-60, p. 1816- 1823.
CANDIA, A. M; VILLACORTA, H. J; MESQUITA, E. T. (2007). Ativação imune-
inflamatória na insuficiência cardíaca. Arq. Bras. Cardiol. vol.89 no.3 São Paulo Sept.
CANTLEY, L.C. (2002). The phosphoinositide 3-kinase pathway. Science, v. 31;296, n.
5573, p. 1655-1667.
CARÈ, A.; CATALUCCIC, D.; FELICETTI, F.; BONCI, D.; ADDARIO, A.;
GALLO, P.; BANG, M.L.; SEGNALINI, P.; GU, Y.; DALTON, N.D.; ELIA, L.;
LATRONICO, M.V.; HØYDAL, M.; AUTORE, C.; RUSSO, M.A.; DORN, G.W. 2ND
;
ELLINGSEN, O.; RUIZ-LOZANO, P.; PETERSON, K.L.; CROCE, C.M.; PESCHLE,
C.; CONDORELLI, G. (2007). MicroRNA-133 controls cardiac hypertrophy. Nature
Medicine 13(5): 613-618.
CARLIER, Y.; TORRICO, F. (2003). Congenital infection with T. Cruzi: from
mechanisms of transmission to strategies for diagnosis and control. Rev. Soc. Bras.
Med. Trop, v. 36, n.6, p.767-771.
CARNERO, A. (2008). The PTEN/PI3K/AKT Signalling Pathway in Cancer,
Therapeutic Implications; Current Cancer Drug Targets, 8, 187-198.
CARVALHO, V. (2012). MicroRNAs: Um Novo Paradigma no Tratamento e
Diagnóstico da Insuficiência Cardíaca; Arq Bras Cardiol ;98(4):362-370.
CHAGAS, C. (1909). Nova tripanozomase humana. Estudos sobre a morfologia e o
ciclo evolutivo do Schizotrypanum cruzi n. gen. n. sp, agente etiológico de nova
entidade mórbida do homem. Memórias do Instituto Oswaldo Cruz, v.1, p.159-218.
CHAOUKI, W.; LEGER, D. Y.; LIAGRE, B.; BENEYTOUT, J. L.; HMAMOUCHI,
M. (2009). Citral inhibits cell proliferation and induces apoptosis and cell cycle arrest
in MCF-7 cells. Fundam. Clin. Pharmacol., v. 23, p.549-556.

67
CHUENKOVA, M. V. Trypanosoma cruzi trans sialidase: A potent and specific
survival factor for human Schwann cells by means of phosphatidylinositol 3-kinaseyAkt
signaling; PNAS, v 161298398. 2001
COFFER, P.J.; WOODGETT, J.R. (1991). Molecular cloning and characterization of a
novel putative protein-serine kinase related to the cAMP-dependent and protein kinase
C families. Eur. J. Biochem., v. 201, 475-481.
CORRAL, R.S.; GUERRERO, N.A.; CUERVO, H.; GIRONÈS, N.; FRESNO, M.
(2013) Trypanosoma cruzi Infection and Endothelin-1 Cooperatively Activate
Pathogenic Inflammatory Pathways in Cardiomyocytes. PLoS Negl Trop Dis 7(2): e 2
034.
CORTEZ, D.M.; FELDMAN, M.D.; MUMMIDI, S.; VALENTE, A.J.; STEFFENSEN,
B.; VINCENTI, M.; BARNES, J.L.; CHANDRASEKAR, B. (2007). IL-17 stimulates
MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK-and ERK1/2-
dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ
Physiol. 293(6):H3356-65.
COURA, J. R.; ABREU, L.; PEREIRA, J. B.; WILCOX, H. P. (1966). Morbidade da
doença de Chagas. IV Estudo longitudinal de 10 anos em Pains e Iguatama, Minas
Gerais. Mem. Inst. Oswaldo Cruz, v. 18, n.2, p. 09-98.
COVEY, T.M.; EDES, K.; FITZPATRICK, F.A. (2007). Akt activation by arachidonic
acid metabolism occurs via oxidation and inactivation of PTEN tumor suppressor.
26(39): 5784-92.
CRACKOWER M.A.; OUDIT G.Y.; KOZIERADZKI I.; SARAO R.; SUNH, SASAKI
T, HIRSCH, E.; SUZUKI, A,; SHIOI, T.; IRIE-SASAKI, J.; SAH, R.; CHENG, H.Y.;
RYBIN, V.O.; LEMBO, G.; FRATTA, L.; OLIVEIRA-DOS-SANTOS, .A.J;
BENOVIC, J.L.; KAHN, C.R.; IZUMO, S.; STEINBERG, S.F.; WYMANN, M.P.;
BACKX, P.H.; PENNINGER, J.M. (2002). Regulation of myocardial contractility and
cell size by distinct PI3K-PTEN signaling pathways. Cell 110:737–749.

68
CUNHA-NETO, E.; DZAU, V.J.; ALLEN, P.D., STAMATIOU, D., BENVENUTTI,
L., HIGUCHI, M.L., KOYAMA, N.S., SILVA, J.S., KALIL, J., LIEW, C-C. (2005).
Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular
mechanism in Chagas’ disease cardiomyopathy. Am. J.Pathol. 167(2): 305-313.
CUNHA-NETO E.; NOGUEIRA, L.G.; TEIXEIRA, P.C.; RAMASAWMY, R.;
DRIGO, S.A.; GOLDBERG, A.C.; et al. (2009). Immunological and non-
immunological effects of cytokines and chemokines in the pathogenesis of chronic
Chagas disease cardiomyopathy. Mem Inst Oswaldo Cruz.104(Suppl 1):252-8.
DAHIA, P.L; PTEN is inversely correlated with the cellsurvival factor Akt, Pkb and is
inactivated via multiple mechanismsin haematological malignancies. Hum Molec
Genet. 8;185 – 93. 1999
DALE, G.; CHAPARRO, J. Integration of molecular markers into tree breeding and
improvent programs.Proceedings Colombo: Embrapa/ CNPJ,1997. V2, p80.
DAVIDSON, N; NAAB, A; HANSON, J; KENNEDY,N; CONTIE,W. (1966).
Comparison of atrial natriuretic peptide as indicators of left ventricular systolic
dysfunction. Am J Cardiol.77:828-831
DE SOUZA, W. O; BRENNER, Z.; ANDRADE, Z.A.; BARRAL-NETO, M.
Trypanossoma cruzi e doença de Chagas. 2.ed. Rio de Janeiro: Guanabara Koogan,
2000. p. 88-126
DIAS, E.; LARANJA, F, S; NOBREGA, G. Doença de Chagas. Mem. Inst. Oswaldo
Cruz, v.43, n.3, p.495-581, dez. 1945.
DIAS, J.C.P. Doença de Chagas: sucessos e desafios. Cadernos de Saúde Pública, Rio
de Janeiro, v.22(10), p.2020-2021, 2006.

69
DI CRISTOFANO A;. PANDOLFI, P.P. (2000). The multiple roles of PTEN in tumor
supression. Cell; 100:387- 90.
DO CAMPO R.; SCOTT, D.A.; VERCESI A.E.; MORENO S.N.J. (1996). The role of
Ca2+ in the process of cell invasion by intracellular parasites. Parasitology Today, v.12,
p.61–65.
DORTA, M. L.; FERREIRA, A. T.; OSHIRO, M. E. M.; YOSHIDA, N. (1995). Ca +2
signal induced by Trypanosoma cruzi metacyclic trypomastigote surface molecules
implicated in mammalian cell invasion. Mol. Biochem. Parasitol., v.73, p.285-289.
DRYGIN, D. J. B; CAROLINE, H.O.; ADAM S.-J; SEAN, O.B; MAYUKO, O.N.H;
CHRIS PROFFITT; NICOLE, S.W. G. R.; KENNA, A. (2009). Abstract C198: CX-
4945, a novel small molecule inhibitor of CK2 protein kinase, reduces hyperactivated
Akt signaling and synergizes with Akt inhibitors in breast cancer cells. Molecular
Cancer Therapeutics: December; Volume 8, Issue 12, Supplement 1 doi: 10.1158/1535-
7163.TARG-09-C198
ENGEL, J. C.; DOYLE, P. S.; HSIEH, I.; MCKERROW, J. H. (1998). Cysteine
protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med., v.
188, p. 725-734.
FRANCHINI, K.G. (2001). Hipertrofia cardíaca: mecanismos moleculares; Rev Bras
Hipertens 8: 125-42.
GARCIA, J.A.D; INCERPI.E.K. (2008). Factors and mechanisms involved in left
ventricular hypertrophy and the anti-hypertrophic role of nitric oxide. Arq. Bras.
Cardiol. vol.90 no.6 São Paulo.
GARCIA.; VICTOR, M.; BOLANOS. (2010). 'Caseine kinase' CK2. SciTopics.
Retrieved March 16, 2012, from http://www.scitopics.com/Caseine_kinase_CK2.html

70
GASPARINI, G; LONGO, R; MORABITO. (2003) A.Inhitors of cyclo-oxigenase2: A
new classo of anticancer agents. The lancet Oncology, v 4,p605-615.
GAUNT, M.W.; YEO, M.; FRAME, I.A.; STOTHARD, J.R.; CARRASCO, H.J.;
TAYLOR, M.C.; MENA, S.S.; VEAZEY, P.; MILES, G.A.; ACOSTA, N.; ARIAS,
A.R.; MILES, M.A. (2003). Mechanism of genetic exchange in American
trypanosomes. Nature, v.421, p.936-939.
GEORGESCU, M.M. (2011). PTEN Tumor Suppressor Network in PI3K-Akt Pathway
Control. Genes & Cancer 1(12) 1170–1177.
GILBER, S.R.L. (2013). Comparison of conventional serology and PCR methods for
the routine diagnosis of infectionTrypanosoma cruzi. Revista da Sociedade Brasileira de
Medicina Tropical 46(3):310-315.
GOODMAN, J.M.; LOGAN, A.G.; MCLAUGHLIN, P.R.; LAPRADE, A.; LIU,
P.P.(1993). Atrial Natriuretic Peptide during Acute and Prolonged Exercise in Well-
Trained Men. Int J Sports Med, v. 14, p. 185-190.
GREEN, D. R; JAMES, E. R. (2004). Manipulation of apoptosis in the host-parasite
interaction, trends in parasitology, 20, pp. 280-287.
HAVENS, M. A.; REICH, A. A.; DUELLI, D. M.; HASTINGS, M. L. (2012).
Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic
acids research, Jan 23 2012.
HE, L.; HANNON, G.J. (2004). microRNA: small with a big role in gene
regulation.Nat Rev Genet, v 5, n 7, p 522-31.
HENRIQUES-PONS, A.; OLIVEIRA, G.M; PAIVA, M.M; CORREA, A.F.;
BATISTA, M.M; BISAGGIO, R.C; LIU, C.C; COTTA-DE-ALMEIDA, V;

71
COUTINHO, C.M; PERSECHINI, P.M; ARAÚJO-JORGE, T.C. (2002). Evidence for
a perforin-mediated mechanism controlling cardiac inflammation in Trypanosoma
cruzi infection. Int J Exp Pathol 83: 67-79.
HIGUSHI, M.L. (2001).A patogenia da ICC na forma crônica da DC. Jornal da IC, v2,
p.3-5.
HINZ, B. BRUNE, K. (2002). Cyclooxygenase-2–10 years later. J. Pharmacol. Exp.
Ther., 300, 367–375 Review
HIRAOKA, E.; KAWASHIMA, S.; TAKAHASHI, T.; RIKITAKE, Y.; KITAMURA,
T.; OGAWA, W.; YOKOYAMA, M. (2001). TNF-alpha induces protein synthesis
trough PI3-kinase-akt/PKB pathway in cardiacmyocytes. American Journal of
Physiology: Heart and Circulatory Physiology, 280, H1861-H1868.
HUNTER, C.A; SLIFER, T; ARAUJO, F. (1996). Interleukin-12 mediated resistance to
Trypanosoma cruzi is dependent on tumor necrosis factor alpha and gamma interferon.
Infection and Immunity.;64 (7):2381-6
HUSE, J.T. (2009). The PTEN-regulating microRNA miR-26a is amplified in high-
grade glioma and facilitates gliomagenesis in vivo. GENES & DEVELOPMENT
23:1327–1337 by Cold Spring Harbor Laboratory Press ISSN 0890-9369/09;
www.genesdev.org
I, HERS. (2011). Akt signaling in health and disease.Cellular signaling v,23, 1515-
1527.
IKEDA, S.; KONG, S.W.; LU, J.; BISPING, E.; ZHANG, H.; ALLEN, P.D.; GOLUB,
T.R.; PIESKE, B. E; PU, W.T. (2007). Altered microRNAexpression in human heart
disease. Physiol Genomics 31(3): 367-373.

72
JI, Y.; HE, Y.; LIU, L.; ZHONG, X. (2010). MiRNA-26b regulates the expression of
cyclooxygenase-2 indesferrioxamine-treated CNE cells. FEBS Letters. 584(5): 961-967.
JONES, P.F.; JAKUBOWICZ, T.; PITOSSI, F.J.; MAURER, F., HEMMINGS, B.A.
(1991). Molecular cloning and identication of a serine/threonine protein kinase of the
second-messenger subfamily. Proc. Natl. Acad. Sci. U.S.A., v. 88, 4171-4175.
JORGE, T.C.A.; CASTRO, S.L., orgs. (2000). Doença de chagas: manual para
experimentação animal [online]. Rio de Janeiro: Editora FIOCRUZ. 368 p.
Antropologia e Saúde collection. ISBN 85-85676-75-2.
KIERSZENBAUM, F. (2005). Where do we stand on the autoimmunity hypothesis of
Chagas disease? Trends Parasitology. v. 21, n. 11, p. 513-516.
KIMURA, E.T. (2006). MicroRNAs: novel class of gene regulators involved in
endocrine function and cancer. Arq Bras Endocrinol Metab vol.50 no.6 São Paulo Dec.
KOGLIN, J; PEHLIVANLI, S; SCHWAIBLMAIR, M, et al. (2001) Role of brain
natriuretic peptide in risk stratification of patients with congestive heart failure. J Am
Coll Cardiol., 38:1934-41
KROEMER,G; GALLUZZI, L; BRENNER, C. (2007). Mitochondrial membrane
permeabilization in cell death. Physiological Reviews, Washington, v 87, n 1,p. 99-163.
KUKREJA, R.C; CHANG, YIN; FADI, N.; SALLOUM. (2011). MicroRNAs: New
Players in Cardiac Injury and Protection.Molecular pharmacology. Received May
10,accepted July 7.
KULKARNI, M. M.; OLSON, C. L.; ENGMAN, D. M.; BRADFORD, S.; MCGWIRE,
B. S. (2009). Trypanosoma cruzi GP63 Proteins Undergo Stage-Specific Differential
Posttranslational Modification and Are Important for Host Cell Infection. Infection and
Immunity, v. 77, p. 2193-2200.

73
KUNDU, J.K; CHOI,K.Y; SURH, Y.J. (2006). Beta- catenin-mediated signaling: a
novel molecular target for chemoprevention with anti-inflammatory substances.
Biochim Biophys;1765:14-24.
KUMMER, C.L.; COELHO, T.C.R.B. (2002). Antiinflamatórios não esteróides
inibidores da ciclooxigenase II (COX II): aspectos atuais. Rev Bras Anestesiol, v. 5, n.
4.
L. MURCIA et al. (2013). Diagnóstico y tratamiento de la enfermedad de Chagas.
Enferm Infecc Microbiol Clin. ;31(Supl 1):26-34.
LATERZA, O.F.; LIM, L.; GARRETT-ENGELE, P.W.; VLASAKOVA, K.;
MUNIAPPA, N.; TANAKA, W.K.; JOHNSON, J.M.; SINA, J.F.; FARE, T.L.;
SISTARE, F.D.; GLAAB, W.E. (2009). Plasma MicroRNAs as Sensitive and Specific
Biomarkers of TissueInjury. ClinicalChemistry 55 (11):1977-1989.
LEE, Y.; KIM, M.; HAN, J. et al. (2004). MicroRNA genes are transcribed by RNA
polymerase II. EMBO J. 23(20): 4051-4060.
LEWIS, B.P; SHIH, I.H. (2003). Prediction of mammalian microRNA targets. Cell, v
115, n 7, p 787-98.
LI, J.; YEN, C.; LIAW, K. et al. (1997). PTEN, a putative protein tyrosine phosphatase
gene mutated in human brain, breast, and prostate cancer. Science. 275:1943-1947.
LI, C.J.; CHANG, J.K.; WNG, G.J.; HO, M.L. (2011). Constitutively expressed COX-2
in osteoblasts positively regulates Akt signal transduction via suppression of PTEN
activity. Bone. 48: 286-297.
LIU, CH; CHANG, S-H; NARKO, K; TRIFAN, O.C; WU, M-T; SMITH, E;
HAUDENSCHILD, C; LANE, TF; HLA, T. (2001). J. Biol. Chem. 276, 18563-18569.

74
MACHADO, F.S.; MARTINS, G.A.; ALIBERTI, J.C.S.; MESTRINER, F.L.A.C.;
CUNHA, F.Q. (2000). Trypanosoma cruzi infected cardiomyocytes produce
chemokines and cytokines that trigger potent NO-dependent trypanocidal activity.
Circulation, v. 102, p.3003-3008.
MACHADO, F.S.; SOUTO, J.T.; ROSSI, M.A.; ESPER, L.; TANOWITZ, H.B.;
ALIBERTI, J.; SILVA, J.S. (2008). Nitric oxide synthase-2 modulates chemokine
production by Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 10,
1558–156.
MARTINS-MELO, F. R.; et al. (2012). Epidemiology of Mortality Related to Chagas’
Disease in Brazil, 1999–2007. PLoS Neglected Tropical Diseases, v. 6, n. 2, e1508, p.
1-8.
MATKOVICH, S.J; VAN BOOVEN, D.J; YOUKER, K.A; TORRE-AMIONE, G;
DIWAN, A; ESCHENBACHER, W.H; et al. (2009). Reciprocal regulation of
myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of
the microRNA signature by biomechanical support. Circulation. 119(9):1263-71.
MATSUI, T.; LI, L.; WU, J. C.; COOK, S.A.; NAGOSHI, T.; PICARD, M. H.; LIAO,
R.; ROSENZWEING. (2002). A. Phenotypic spectrum caused by transgenic
overexpression of activated Akt in the heart. J. Biol. Chem., v. 277, 22896-22901.
MATSUI, M.Y; CHU, ZHANG H, GAGNON KT, SHAIKH S, KUCHIMANCHI S, M
MANOHARAN; COREY, D.R; JANOWSKI, B.A. (2013). Promoter RNA links
transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res . 41
(22):10086-109. doi: 10.1093/nar/gkt777.
MAZZA, S. (1949). La enfermidad de Chagas em La Republica Argentina. Mem. Inst.
Osvaldo Cruz,v 47, p.273-278.

75
MEGGIO, F; PINNA, L.A. (2009). One-thousand-and-one substrates of protein kinase
CK2. FASEB J 17, 349–368.
MELO; BEZERRA, R; PARENTE, G.B.O; VICTOR,E.G. (2005). Determinação do
peptídeo natriurético cerebral humano em portadores da doença de Chagas; Arq. Bras.
Cardiol. vol.84 no.2 São Paulo.
MELO, R. C. N. (2008). Acute Heart Inflammation: ultrastructural and functional
aspects of macrophages elicited by Trypanosoma cruzi infection. Journal of Cellular
and Molecular Medicine. ìPostprintî;10.1111/j.1582-4934.2008.00388.x,
MENDEZ, M. ; LAPOINTE, M.C. (2002). Trophic effects of the cyclooxygenase-2
product prostaglandin E(2) in cardiac myocytes. Hypertension. 39 (2 Pt2): 382-388.
MENG, F; HENSON, R; WEHBE-JANEK, H; GHOSHAL, K; JACOB, S.T; PATEL
T. (2007). MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in
human hepatocellular cancer. Gastroenterology, volume 133,issue 2,pages 647-658.
MICHELIN MA, SILVA JS, CUNHA FQ. (2005). Inducible cyclooxygenase released
prostaglandin mediates immunosuppression in acute phase of experimental
Trypanosoma cruzi infection. Exp Parasitol.;111(2):71-9.
MIN-HSIEN, W; SONG-BIN, H. E; GWO-BIN, L. (2010). Microfluidic cell culture
systems for drug research Lab Chip,10, 939-956.
miRanda (http://www.microRNA.org)
miRBase – The microRNA database (http://www.mirbase.org/)
miRTarBase (mirtarbase.mbc.nctu.edu.tw)

76
MITCHELL, P. S.; et al. (2008) Circulating microRNAs as stable blood-based markers
for cancer detection. Proc Natl Acad Sci U S A, v. 105, n. 30, p. 10513-8.
MOCANU, M.M.; YELLON, D.M. (2007). PTEN, the Achilles’ heel of myocardial
ischaemia/reperfusion injury? Br. J.Pharmacol.. 150: 833–838.
MOLLACE, V.; MUSCOLI, C.; MASINI, E.; CUZZOCREA, S.; SALVEMINI, D.
(2005). Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide
donors. Pharmacol Rev 57, 217-252.
MOSMANN, T. (1983). Rapid colorimetric assay for cellular growth and survival:
application to proliferation and cytotoxicity assays. Journal of Immunological Methods
65 (1–2): 55–63.
MUKHERJEE, D.; NILSEN, S.E.; TOPOL, E.J. (2001). Cox-2 inhibitors and
cardiovascular risk: we defend our data and suggest caution. Cleve. Clin. J. Med.
68(11): 963-4.
O´BANNION, M. K. (1999). Cyclooxygenase-2: molecular biology, pharmacology, and
neurobiology. Crit. Rev. Neurobiol., v.13, p.45-82.
O` CONNOR, J.K; AVENT, J.; LEE, R.J; FISCHBACH, J; GAFFNEY, D.K. (2004).
Cyclooxygenase-2 correlates with dimished survivel in invasive breast treated with
mastectomy and radiotherapy. Int J Radiation Oncol Biol Phya.58(4):1034-40.
OGAWA, K; OIDA, A; SUGIMARA, H; et al. (2002). Clinical significance of blood
brain natriuretic peptide level measurement in the detection of heart disease in untreated
outpatients: comparison of electrocardiography, chest radiography and
echocardiography. Circ Res, 66:122-6

77
OUDIT, G. Y; PENNINGER, J. F.M. (2009). Cardiac regulation by phosphoinositide
3-kinases and PTEN; Cardiovascular Research; v 82, 250–260.
PESSOA, S.B. (1958) Hospedeiros vertebrados (não humanos) do Trypanossoma cruzi .
Ver. Goiânia. Méd., v.4, n. 2, p. 83-101.
PETERSEN, C.A.; KRUMHOLZ, K.A.; CARMEN J, SINAI AP, BURLEIGH BA
(2006). Trypanosoma cruzi infection and nuclear factor kappa B activation prevent
apoptosis in cardiac cells. Infect Immun 74:1580-1587.
PIAZZA, L.A, de BOLD, A.J; SANTAMARINA, N; HILBA, E; RUBIOLO,
E.R.(1994). Atrial natriuretic factor in experimental acute Chagas disease. Parasitol
Research. 80(1):78-80.
PIRES, L.; BOGLIOLO, A. R.; TEIXEIRA, A. R. L. (1996). Diversity of Trypanosoma
cruzi stocks and clones derived from Chagas’ disease patients. II. Isoenzyme and RFLP
characterizations. Exp. Parasitol., v. 82, p. 182-190.
PONTES, M.R.N; LEAES,P.E. (2004). Remodelamento Ventricular: dos Mecanismos
Moleculares e Celulares ao Tratamento. Revista da Sociedade de Cardiologia do Rio
Grande do Sul -Ano XIII nº 03.
PRATA, A. (2001). Clinical and epidemiological aspects of Chagas disease . The
Lancet Infectious Diseases , Volume 1 , Issue 2 , Pages 92 - 100 A.
PREVIATO, J.O.; JONES, C.; GONÇALVES, L.P.; WAIT, R.; TRAVASSOS, L.R.;
MENDONÇA-PREVIATO, L. (1994).O-glycosidically linke Nacetylglucosaminebound
oligosaccharides from glycoproteins of Trypanosoma cruzi. The Biochemical Journal,
v.301, p.151–159.

78
PROCOPIO D.O., BARROS H.C., MORTARA R.A. (1999). Actin-rich structures
formed during the invasion of cultured cells by infective forms of Trypanosoma cruzi.
Eur J Cell Biol; 78: 911-924.
PROUND, C.G. (2004). Ras, P13- Kinase mTOR signaling in cardiac hypertrophy.
Cardiovasc. 15; 63(3):403-13.
RAJARAM, M. V. S; GANESIA, L. P; PARSA, K. V,L; BUTCHAR, J. P; GUNN,
J.S; TRIDANDAPANI, S. (2006). Akt/ protein kinase B modulates macrophage
inflammatory response to francisella infection and cofers a survival advantage in mice.
The journal of immunology. V. 177, n, 9, p. 6317-6324.
RAMIREZ, M.I., RUIZ, R.C., ARAYA, J.E., FRANCO DA SILVEIRA,
J.E.,YOSHIDA, N. (1993). Involvement of the stage-specific 82-kilodalton adhesion
molecule of Trypanosoma cruzi metacyclic trypomastigotes in host cell invasion.
Infection and immunity, v.61, p.3636-3641.
RASSI, A., RASSI, A. J., LITTLE, W.C (2000). Chagas’ heart disease. Clinical
Cardiology, v.23, p.883–889.
RASSI JUNIOR, A., RASSI, A., et al. (2010). Chagas disease. Lancet 375(1388): 402.
RIBEIRO, A.L.P; REIS,A.M; BARROS, M. V. L et al. (2002). Brain natriuretic peptide
and left ventricular dysfunction in Chagas disease. Lancet. 360:461-462.
RICHARDS, A.M. (2007). Natriuretic peptides. Update on peptide release, bioactivity,
and clinical use. Hypertension, v.50, n.1,p.25-30, Disponível em:
<http://hyper.ahajournals.org/cgi/reprint/50/1/25>. Acesso em: 24 set. 2007. doi:
10.1161/HYPERTENSIONAHA.106.069153
ROCHA, M.O.C; RIBEIRO, A.L.;TEIXEIRA, M.M. Clinical managment of chronic
Chagas cardiomyopathy.Front Biosci. 8 e 44 e 54.

79
ROMAN-CAMPOS D, DUARTE HL, SALES PA JR, NATALI AJ, ROPERT C,
GAZZINELLI RT, CRUZ JS. (2009). Changes in cellular contractility and cytokines
profile during Trypanosoma cruzi infection in mice. Basic Research in Cardiology, 104,
238-246.
ROSA MP et al. (2003). Contribution to prevention of cerebral ischemia by cilostazol, a
phosphodiesterase III inhibitor: a review. Jornal Vascular Brasileiro 2008, Vol. 7, Nº 1
ROSA DA SILVA, M.(2009). Padronização de método colorimétrico para avaliação de
atividade biológica de substâncias sob formas traquizoítas do Toxoplasma gondi, com a
avaliação de triterpenos ácidos sobre o parasito. 46f. Dissertação (Mestrado em
Biociências Biológicas) – Faculdade de Ciências Farmacêuticas, USP, Ribeirão Preto.
RUIZ, R. C.; FAVORETO Jr., S.; DORTA, M. L.; OSHIRO, M. E. M.;FERREIRA, A.
T.;MANQUE , P. M.; YOSHIDA, N. Infectivity of Trypanosoma cruzi isolates is
associated with differential expression of surface glycoproteins with differential Ca2+
signaling activity. Biochem. J., v. 330, p.505–511.
SARAH J,W ; GILLIAN M. BORTHWICK; HELEN M. ARTHUR. (2011). The
H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic
responses in vitro. In Vitro Cellular & Developmental Biology - Animal
February, Volume 47, Issue 2, pp 125-131.
SAYED, D., HONG, C., CHEN, I. -Y., LYPOWY, J. E ABDELLATIF, M. (2007).
MicroRNAs Play an Essential Role in Development of Cardiac Hypertrophy.
Circulation Research 100: 416-424.
SCHARFSTEIN, A.C., J. (1992). Inhibitors of the major cysteinyl proteinase (GP57/51)
impair host cell invasion and arrest the intracellular development of Trypanosoma cruzi
in vitro. Mol Biochem Parasitol. v.2, n. 52:175-184.

80
SCHARFSTEIN, J; MORROT, A. (1999). A role for extracellular amastigotes in the
immunopathology of Chagas disease.Mem Inst Oswaldo Cruz. V. 94, p.51-63.
SCHARFSTEIN, J.; SCHMITZ, V.; MORANDI, V.; CAPELLA, M. M. A.; LIMA, A.
P. C. A.; MORROT, A.; JULIANO, L.; MULLER-ESTER, W. (2000). Host cell
invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B2 receptors.
J. Exp. Med., v.192. p.1289–1299.
SCHOFIELD, C. M.; HSU, R.; BARKER, A. J.; GERTZ, C. C.; BLELLOCH, R.;
ULLIAN, E. M. (2011). Monoallelic deletion of the microRNA biogenesis gene Dgcr8
produces deficits in the development of excitatory synaptic transmission in the
prefrontal cortex. Neural development, v. 6, p. 11.
SCHRIEFER, A e EDGAR M. CARVALHO.(2008). Biomarkers in Medicine; Gaz.
méd. Bahia;78 (Suplemento 1):47-51.
SCHWARTZBAUER, G., ROBBINS, J . (2001). The tumor suppressor gene PTEN can
regulate cardiac hypertrophy and survival. J. Biol. Chem. 276(38) 35786-35793.
SILVA, M. M,F. (2012). Exercício físico e microRNAs: novas fronteiras na
insuficiência cardíaca; Arq. Bras. Cardiol. vol.98 no.5 São Paulo
SOUZA E.M., ARAÚJO-JORGE T.C., BAILLY C., LANSIAUX A., BATISTA M.M.,
OLIVEIRA G.M., SOEIRO M.N.C. (2003). Host and parasite apoptosis following
Trypanosoma cruzi infection in in vitro and in vivo models. Cell Tissue Res.314: 223-
235.
SMALL, E.M. e OLSON, E.N. (2011). Pervasive roles of microRNAs in cardiovascular
biology. Nature 469(7330): 336-342.

81
SMITH, W. L.; GARAVITO, R. M.; DEWITT, D. L. (1996). Prostaglandin
endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem., 1996; v. 271, p.
33157–33160,
STACK, E. E DUBOIS, R.N. (2001). Regulation of cyclo-oxygenase-2. Best practice &
research - Clinical gastroenterology, 15(5), 787-800
STERIN-BORDA L., CREMASCHI G., GENARO A., VILA ECHAGUE A., GOIN
J.C., BORDA E. (1996). Involvement of NO and PKC activation on chagasic antibody
activity upon myocardial contractility. Mol. Cell Biochem. 160/161:75–82.
ST-GERMAIN, M.E., GAGNON, V., MATHIEU, I., PARENT, S., ASSELIN, E.
(2004). Akt regulates COX-2 mRNA and protein expression in mutated-PTEN human
endometrial cancer cells. Intern. Journ. Oncology. 24 (5): 1311-1324.
STRILLACCI, A., GRIFFONI, C., SANSONE, P., PATERINI, P., PIAZZI, G.,
SPISNI, E., PANTALEO, M.A., BIASCO, G., TOMASI, V. (2009). MiR-101
downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer
cells. Exp. Cell Res. 315(8): 1439-1447.
SULLY, G.; DEAN, J.L.; WAIT, R.; RAWLINSON, L.; SANTALUCIA, T.;
SAKLATVALA, J.; CLARK, A.R. (2004). Structural and functional dissection of a
conserved destabilizing element of cyclo-oxygenase-2 mRNA: evidence against the
involvement of AUF-1 [AU-rich element/poly(U)-binding/degradation factor-1], AUF-
2, tristetraprolin, HuR (Hu antigen R) or FBP1 (far-upstream-sequence-elementbinding
protein 1). Biochem. J. 377: 629-639.
TAFURI, W.L.(1985). Patogênese. In: Cançado JR, Chuster M. (ed.) Cardiopatia
Chagásica. Belo Horizonte: Fundação Carlos Chagas.p. 1-9.
TANABE, T. e TOHNAI, N. (2002). Cyclooxygenase isozymes and their gene
structures and expression. Prostaglandins Other Lipid Mediat, 68-69, 95-114.

82
TANOWITZ, H. B.; et al. (2009). Perspectives on Trypanosoma cruzi-induced heart
disease (Chagas Disease). Progress in Cardiovascular Disease, v. 51, n. 6, p. 524-539.
TELLERIA, J.; LAFAY, B.; VIRREIRA, M.; BARNABÉ, C.; TIBAYREBC, M.;
SVOBODA, M. (2006). Trypanosoma cruzi: Sequence analysis of the variable region of
kinetoplast minicircles. Experimental parasitology, v.114, p.279- 288.
THUM T, GROSS C, FIEDLER J, FISCHER T, KISSLER S, BUSSEN M, et al.
(2008); MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase
signalling in fibroblasts. Nature.456(7224):980-4.
TURCHINOVICH, A.; WEIZ, L.; LANGHEINZ, A.; BURWINKEL, B. (2011)
Characterization of extracellular circulating microRNA. Nucleic acids research, v. 39, n.
16, p. 7223-7233.
TYLER, K.M.; ENGMAN, D.M. (2001). The life cycle of Trypanosoma cruzi revisited.
International Journal for Parasitology, v.1, p.472-81.
VAN DE VRIE. (2011).MicroRNA Involvement in Immune Activation During Heart
Failure ; Cardiovasc Drugs Ther; 25:161–170
VANE, J.R.; BAKHLE, Y.S.; BOTTING, R.M. (1998). Cyclooxygenases 1 and 2.
Annu Rev Pharmacol Toxicol, 38:97-120. ISSN 0362-1642
VAN ROOIJ, E. E OLSON, E.N. (2008). MicroRNAs: powerful new regulators of heart
disease and provocative therapeutic targets. J. Clin. Invest. 117(9): 2369-76.
VARGAS,M V. (2013). Application of microRNAs in the clinical practice .Rev Bras
Clin Med. São Paulo, jan-mar;11(1):62-6

83
VILMA A. SARDÃO e PAULO J. OLIVEIRA e JON HOLY e CATARINA R.
OLIVEIRA & KENDALL B. WALLACE. (2008). Morphological alterations induced
by doxorubicin on H9c2 myoblasts: nuclear, mitochondrial, and cytoskeletal targets,
Cell Biol Toxicol. DOI 10.1007/s10565-008-9070-1.
XIAN.DUAN; B.J. and X.W. (2012). Expression of MicroRNA-1 and MicroRNA-21
in Different Protocols of Ischemic Conditioning in an Isolated Rat Heart Model;
Cardiology; v 122:36–43,2012;122:36–43 DOI: 10.1159/00033
XU-DONG e t a l. (2011). Attenuation of MicroRNA-22 Derepressed PTEN to
Effectively Protect Rat Cardiomyocytes From Hypertrophy. JOURNAL OF
CELLULAR PHYSIOLOGY.
Y. Ji. (2010). MiRNA-26b regulates the expression of cyclooxygenase-2 in
desferrioxamine-treated CNE cells. FEBS Letters, V. 584 961–967.
YANG, B. LIN, H. XIAO, J. (2011). The muscle-specific microRNA miR-1 regulates
cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nature Medicine.
13(4): 486-91.
YANG, T. T.; WENG, S. F.; ZHENG, N.; PAN, Q. H.; CAO, H. L.; LIU, L.; ZHANG,
H. D.; MU DA, W. (2011). Histopathology mapping of biochemical changes in
myocardial infarction by Fourier transform infrared spectral imaging. Forensic science
international, v. 207, n. 1-3, p. e34-9, Apr 15.
YOSHIDA, N.; ARAYA, J.E.; FRANCO DA SILVEIRA, J.; GIORGIO, S. (1989).
Antibody production and T cell response induced by stage-specific surface
glycoproteins purified from metacyclic trypomastigotes. Experimental Parasitology,
v.77, p.405-413.

84
YOSHIDA, N., BLANCO, S.A., ARAGUTH, M.F., RUSSO, M., GONZALEZ, J.
(1990).The stage-specific 90-kilodalton surface antigen of metacyclic trypomastigotes
of Trypanosoma cruzi. Molecular and Biochemical Parasitology, v.39, p.39–46.
YOSHIDA, N. (2003). Trypanosoma cruzi cell invasion mechanisms. In: World Class
Parasites: American Trypanosomiasis. TYLER, K.M., MILES, M.A. (Eds), Kluwer
Academic Publishers, v.7, p.69–79.
YOSHIDA, N. (2006). Molecular basis of mammalian cell invasion by Trypanosoma
cruzi. Anais da Academia Brasileira de Ciências, v.78, n.1, p.87-111.
WANG, H.J.; RUAN, H.J.; HE, X.J.; MA, Y.Y.; JIANG, X.T.; XIA, Y.J.; et al. (2010).
MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and
invasion. European Journal of Cancer. 46(12):2295–2303.
WEIYUN WU, et al.( 2013). MicroRNA-32 (miR-32) regulates phosphatase and tensin
homologue (PTEN) expression and promotes growth, migration, and invasion in
colorectal carcinoma cells. Molecular Cancer, 12:30 http://www.molecular-
cancer.com/content/12/1/30
WILKOWSKY S.E., BARBIERI M.A., STAHL P., ISOLA E.L.D.( 2001).
Trypanosoma cruzi phosphatidylinositol 3-kinase and protein kinase B activation is
associated with parasite invasion. Exp Cell Res; 264: 211-218.
ZHANG, C. (2009). MicroRNAs: role in cardiovascular biology and disese. Clinical
Science. 114: 699-706.
ZHANG, X., MIAO, X., TAN, M., et al. (2005). Identification of Functional Genetic
Variants in Cyclooxygenase-2 and Their Association With Risk of Esophageal Cancer
Gastroenterology,129:565–576.

85
ZINGALES, B., SOUTO, R.P. MANGIA, R.H., LISBOA C.V., CAMPBELL,D.A.,
COURA, J.R., JANSEN, A., FERNANDES, O. (1998). Molecular epidemiology of
American trypanosomiasis in Brazil based dimorphisms of rRNA and miniexon gene
sequences. International Journal for Parasitology, v.28, n.1, p.105-112.















![MARIANA MORATO MARQUES LEUCOTRIENOS COMO …€¦ · RESUMO Morato-Marques M. Leucotrienos como moduladores da imunidade inata a fungos. [tese (Doutorado em Imunologia)]. São Paulo:](https://static.fdocumentos.com/doc/165x107/607d5b01cdd286033623efde/mariana-morato-marques-leucotrienos-como-resumo-morato-marques-m-leucotrienos-como.jpg)


