
Apostila_QOII
326
1
-
Upload
tuanny-brito -
Category
Documents
-
view
470 -
download
9
Transcript of Apostila_QOII

1

2
PRESIDENTE DA REPÚBLICA Luiz Inácio Lula da Si lva MINISTRO DA EDUCAÇÃO Fernando Haddad GOVERNADOR DO ESTADO DO PIAUÍ José Well ington Barroso de Araújo Dias REITOR DA UNIVERSIDADE FEDERAL DO PIAUÍ Luiz de Sousa Santos Júnior SECRETÁRIO DE EDUCAÇÃO A DISTÂNCIA DO MEC Carlos Eduardo Bielschowsky COORDENADOR GERAL DA UNIVERSIDADE ABERTA DO BRASIL Celso Costa José da Costa DIRETOR DO CENTRO DE EDUCAÇÃO ABERTA A DISTÂNCIA DA UFPI Gildásio Guedes Fernandes DIRETOR DO CENTRO DE CIÊNCIAS DA NATUREZA Helder Nunes da Cunha COORDENADORA DO CURSO DE QUÍMICA NA MODALIDADE EAD Rosa Lina Gomes do Nascimento Perei ra da Si lva COORDENADORA DE MATERIAL DIDÁTICO DO CEAD/UFPI Cleidinalva Maria Barbosa Olivei ra DIAGRAMAÇÃO Diego Albert COORDENADOR DE REVISÃO DE TEXTO Naziozênio Antonio Larcerda REVISÃO Francisca das Dores Olivei ra Araújo

3
Neste material serão abordados os temas relacionados
ao programa de Educação a Distância da Universidade Aberta
do Piauí (UAPI) vinculada ao consórcio formado pela
Universidade Federal do Piauí (UFPI), Universidade Estadual
do Piauí (UESPI), Centro Federal de Ensino Tecnológico do
Piauí (CEFET-PI), com apoio do Governo do estado do Piauí,
através da Secretaria de Educação.
O texto aborda o conteúdo de química orgânica voltado
para os alunos do curso de Química, está estruturado em 11
unidades e subunidades correspondentes. Todas as unidades
visam contextualizar o aluno com os objetivos apresentados.
Enfatizo que esta não é, de fato, uma obra completa; fazendo-
se necessário buscar informações em outras fontes para um
melhor aprendizado.
Unidade 1 – Serão abordados assuntos que induzem o
aluno a refletir sobre a necessidade desta disciplina para sua
formação profissional plena na área de Química. Conceitos
fundamentais, comumente utilizados em química orgânica,
como estrutura, reatividade e mecanismos: ligações em
compostos de carbono, formação e quebra de ligações, fatores
eletrônicos e efeito estérico, tipos de reagentes.
Unidade 2 – Serão apresentadas a Investigações de
mecanismos: energética e cinética de reação, natureza do
produto, uso de isótopos, estudo de intermediários, critérios de
estereoquímica; capacitando o aluno a compreender melhor os
mecanismos em reações orgânicas.
Unidade 3 – Destacamos a importância das forças de
ácidos e bases orgânicas, origem da acidez em compostos

4
orgânicos, ácidos e bases alifáticas e aromáticas, bases
heterocíclicas e definição de ácido e bases duras e moles.
Estas propriedades das moléculas orgânicas são o foco desta
unidade, e discutiremos os termos fundamentais a ela
relacionados, além de aplicar os conhecimentos dessa área da
Química Orgânica para o melhor entendimento da estrutura
dos compostos orgânicos.
Unidade 4 – Estudaremos a reação de substituição
nucleofílica em átomo de carbono saturado; o mecanismo, a
estereoquímica, e o efeito de grupos que entram e que saem
das moléculas em questão reações com ênfase e derivados
mais importantes, bem como os processos industriais de
obtenção destes serão também abordados.
Unidade 5 – Trataremos da reação de substituição
eletrofílica e nucleofílica em sistemas aromáticos como:
nitração, diazoacoplamento, halogenação, sulfonação,
alquilação, acilação, ataque nucleofílico em espécies
aromáticas, efeito de substituintes, equação de Hammet;
buscando sempre uma interação entre esta importante classe
de reações orgânicas que são fundamentais para os processos
industriais e as principais reações envolvidas.
Unidade 6 – Trata do estudo dos mecanismos de
reações orgânicas pertencentes a Química Orgânica II,
envolvendo íons carbônio e átomos de nitrogênio e oxigênio
deficientes de elétrons. Será dada uma atenção especial para
os métodos de formação, estabilidade, estrutura e reações.
Unidade 7 – Nesta unidade, o aluno terá a oportunidade
de conhecer e aprender algumas reações de adição como:
adição eletrofílica e nucleofílica à ligação dupla carbono-

5
carbono, adição nucleofílica à ligação dupla carbono-oxigênio,
adição a dienos conjugados. Essas reações quando feitas em
laboratório, pela primeira vez, devem ser tomadas algumas
precauções a respeito dos reagentes e produtos envolvidos na
reação.
Unidade 8 – Serão apresentadas as reações de
eliminação e os mecanismo E1, E1cB e E2, eliminação versus
substituição, em compostos orgânicos .
Unidade 9 – Trata do estudo dos mecanismos de
reações orgânicas pertencentes à classe que envolve íons
carbânions; da formação, estabilidade, estereoquímica e das
reações mais importantes desta classe: íons negativos.
Unidade 10 – Será estudada a reação dos compostos
orgânicos com radicais, e ainda, o mecanismo, a
estereoquímica, o processo de formação, detecção e reações
envolvendo radicais.
Unidade 11 – Nesta unidade, trataremos das reações
controladas por simetria de orbitais, das reações eletrocíclicas,
cicloadição e rearranjos sigmatrópicos de carbono e
hidrogênio, buscando de fato entender como as reações
orgânicas ocorrem e quais são permitidas ou proibidas, por
simetria, viabilizando melhor entendimento dos mecanismos e
o porquê de alguns produtos serem formados com maior
prioridade nas reações orgânicas.

6
Unidade I - Estrutura, reatividade e mecanismos ................. 1
Estrutura e reatividade .............................................................. 2
Orbitais atômicos ....................................................................... 2
Hibridização ............................................................................... 4
Ligações em compostos de carbono ......................................... 5
Teoria de Ligação de Valência. ................................................. 5
Teoria do orbital molecular (OM) ............................................... 5
Energias dos orbitais moleculares ........................................... 12
Preenchimento dos orbitais moleculares ................................. 15
Ligações simples carbono-carbono ......................................... 17
Ligações duplas carbono-carbono .......................................... 18
Ligações triplas carbono-carbono ........................................... 20
Ligações carbono-oxigênio e carbono-nitrogênio. ................... 21
Conjugação ............................................................................. 22
Benzeno e aromaticidade ........................................................ 24
Condições necessárias para a deslocalização. ....................... 30
Quebra e formação de ligações .............................................. 31
Fatores que influenciam a disponibilidade eletrônica. ............. 33
Efeitos indutivos ...................................................................... 33
Efeitos mesoméricos ............................................................... 34
Efeitos variáveis com o tempo. ................................................ 36
Hiperconjugação ..................................................................... 37
Efeitos estereoquímicos .......................................................... 39
Tipos de reagentes .................................................................. 40
Tipos de reações ..................................................................... 43
1o roteiro de estudo dirigido ..................................................... 45
Unidade II – Energética, cinética e investigação de
mecanismos ........................................................................... 47
Energética de reações ............................................................ 48

7
Cinética de reações ................................................................ 51
Velocidade de reação e energia livre de ativação ................... 52
Cinética e etapa determinante da velocidade ......................... 54
Controle cinético versus controle termodinâmico .................... 58
Investigação de mecanismos de reações ............................... 59
A natureza dos produtos ......................................................... 59
Dados Cinéticos ...................................................................... 60
Mas o mecanismo real de hidrólise pode muito bem ter mudado
ao se ter alterado o solvente, de tal modo que não ficamos a
saber mais sobre o que de fato se passou na solução aquosa
original. ................................................................................... 61
A utilização de isótopos .......................................................... 62
O estudo de intermediários ..................................................... 64
Critérios estereoquímicos ....................................................... 65
20 Roteiro de estudo dirigido ................................................... 66
Unidade III- Ácidos e bases .................................................. 68
Ácidos ..................................................................................... 69
Força ácida - pKa .................................................................... 69
A origem da acidez em compostos orgânicos ......................... 70
A influência do solvente .......................................................... 72
Ácidos alifáticos simples ......................................................... 73
Ácidos alifáticos substituídos .................................................. 76
Substituintes doadores ou retiradores de elétrons - fenóis ..... 78
Ácidos carboxílicos aromáticos ............................................... 79
Ácidos dicarboxílicos............................................................... 81
Bases ...................................................................................... 82
pKb, pKBH+ e pKa ..................................................................... 82
Bases alifáticas ....................................................................... 83
Bases aromáticas ................................................................... 86
Bases heterocíclicas ............................................................... 90
Conceito de ácidos/ bases duros e moles ............................... 92
Classificação de alguns ácidos e bases de Lewis. ................. 93

8
3º ROTEIRO DE ESTUDO DIRIGIDO..................................... 95
Unidade IV - Substituição nucleófila num átomo de carbono
saturado ................................................................................. 97
Relação entre cinética e mecanismo ....................................... 97
Efeito do solvente .................................................................. 100
Efeito da estrutura ................................................................. 102
Implicações estereoquímicas do mecanismo ........................ 108
Mecanismo SN2, inversão de configuração ........................... 108
Determinação da configuração relativa ................................. 109
Mecanismo SN1: racemização ............................................... 110
Mecanismo SN1: retenção de configuração ........................... 112
Participação de grupos vizinhos: “retenção de configuração” 113
Efeito de nucleófilos e grupos abandonadores...................... 114
Nucleófilo............................................................................... 114
O grupo abandonador ........................................................... 117
4o ROTEIRO DE ESTUDO DIRIGIDO................................... 119
Unidade V - Substituição eletrófilica e nucleófilica em
sistemas aromáticos ........................................................... 123
Ataque eletrofilíco ao benzeno .............................................. 124
Complexos π e σ ................................................................... 124
Nitração ................................................................................. 126
Halogenação ......................................................................... 128
Sulfonação ............................................................................ 130
Reações de friedel-crafts ...................................................... 132
Alquilação de Friedel-Crafts .................................................. 132
Acilação de Friedel-Crafts ..................................................... 135
Acoplamentos diazo .............................................................. 138
Ataque eletrofílico a C6H5Y ................................................... 140
Efeitos eletrônicos do grupo substituinte (y) .......................... 142
Efeitos eletrônicos de grupos substituintes desativantes ...... 142
Efeitos eletrônicos de grupos substituintes ativantes ............ 143
Fatores de velocidade parciais e seletividade ....................... 146

9
Efeitos esteroquímicos e razões orto/para ............................ 148
Controle cinético versus termodinâmico ............................... 150
Substituição eletrofila em outras espécies aromaticas.......... 151
Substituição nucleofílica aromática ....................................... 153
Substituição de hidrogênio .................................................... 153
Substituição de átomos diferentes de hidrogêno .................. 154
Mecanismo unimolecular SN1Ar ............................................ 154
Mecanismo adição-eliminação SN2 ativado .......................... 155
Substituição via intermediários arino ou eliminação-adição .. 157
5o ROTEIRO DE ESTUDO DIRIGIDO .................................. 158
Unidade VI – Rearranjos moleculares envolvendo átomos
de carbono, nitrogênio e oxigênio deficientes de elétrons160
Unidade 6 – Rearranjos moleculares envolvendo átomos de
carbono, nitrogênio e oxigênio deficientes de elétrons ......... 160
Métodos de formação de íons carbônio ................................ 162
Fissão heterolítica de espécies neutras ................................ 162
Adição de cátions a espécies neutras ................................... 163
Obtenção de íons carbônios a partir de outros cátions ......... 164
Estabilidade e estrutura de íons carbônios ........................... 165
Reações de íons carbônio .................................................... 168
Rearranjo de íons carbônios ................................................. 170
Sem alteração da cadeia carbônica ...................................... 170
Rearranjos alílicos ................................................................ 171
Com alteração na cadeia carbônica ...................................... 172
Rearranjos neopentílicos ...................................................... 172
Rearranjo de hidrocarbonetos ............................................... 173
Rearranjos pinacol/pinacolona .............................................. 173
Aspectos estereoquímico ...................................................... 174
Rearranjo de Wolff ................................................................ 177
Cátions diazônios .................................................................. 178
Migração para nitrogênio deficiente de elétron ..................... 180
Reações de Hofmann, Curtius, Lossen e Schmidt ................ 180

10
Rearranjos de Beckman ........................................................ 183
Migração para oxigênio elétron deficiente ............................. 185
Oxidação Baeyer-Villiger ....................................................... 185
Rearranjos de hidroperóxido ................................................. 186
6o ROTEIRO DE ESTUDO DIRIGIDO................................... 188
Unidade VII - Adição eletrófila e nucleófila a C=C e C=O 190
Adição de halogênios ............................................................ 191
Efeito de substituintes na velocidade de adição .................... 194
Orientação da adição ............................................................ 195
Outras reações de adição ..................................................... 197
Outros derivados halogenados .............................................. 197
Hidratação ............................................................................. 198
Íons carbônios ....................................................................... 199
Hidroxilação ........................................................................... 199
Hidrogenação ........................................................................ 201
Adição nucleófila ................................................................... 202
Cianoetilação ......................................................................... 203
Reações de Michael .............................................................. 203
Adição nucleofílica a carbonila (c=o) ..................................... 204
estrutura e reatividade ........................................................... 205
reações de adição simples a aldeídos e cetonas .................. 207
Hidratação ............................................................................. 207
Adição de álcoois – formação de acetal e hemiacetal ........... 209
Adição de tióis ....................................................................... 211
Adição de cianeto de hidrogênio ........................................... 212
Adição de bissulfito ............................................................... 213
Adição de íons hidreto ........................................................... 213
Adição de complexos de íons hidreto com metais ................ 213
Reação de Meerwein-Ponndorf ............................................. 214
Reações de Cannizzaro ........................................................ 215
Adição de elétrons ................................................................. 215
Reações de adição/eliminação .............................................. 216

11
Derivados de NH3 ................................................................. 216
Adição de carbono nucleofílico ............................................. 217
Reagentes de Gringnard ....................................................... 218
Íons acetilênicos ................................................................... 219
Reações aldólicos ................................................................. 219
Reação de Perkin ................................................................. 220
Reações de Knoevenagel e de Stobbe ................................. 221
Condensação de Claisen de ésteres .................................... 222
Reação de Wittig ................................................................... 223
Estereosseletividade em reações de adição a carbonilos..... 224
Reações catalisadas por ácido ............................................. 226
Adição a c≡n ......................................................................... 227
7o ROTEIRO DE ESTUDO DIRIGIDO .................................. 227
Unidade VIII - Reação de eliminação ................................. 230
Reações de eliminação 1,2, eliminação-β............................. 231
Outras eliminações β ............................................................ 231
Mecanismo e1 ....................................................................... 233
Mecanismo e1cb ................................................................... 234
Mecanismo e2 ....................................................................... 235
Estereosseletividade em E2 .................................................. 236
Orientação em E2: Saytzev versus Hofmann ....................... 239
Eliminação versus substituição ............................................. 240
8o ROTEIRO DE ESTUDO DIRIGIDO .................................. 242
Unidade IX - Carbânions ..................................................... 244
Formação de carbânions ...................................................... 245
Estabilidade de carbânions ................................................... 247
Estereoquímica de carbânions .............................................. 250
Reações dos carbânions ....................................................... 252
Reação de adição ................................................................. 252
Reação de adição - carbonação ........................................... 253

12
Reação de eliminação ........................................................... 254
Reação de eliminação - descarboxilação .............................. 254
Reação de substituição ......................................................... 255
Reações de Reimer-Tiemann ................................................ 255
Reações de Kolbe-Schmidt ................................................... 255
Rearranjo............................................................................... 256
Oxidação ............................................................................... 258
Halogenação de cetonas ....................................................... 258
Unidade X - Radicais ........................................................... 260
Formação de radicais ............................................................ 262
Fotólise .................................................................................. 262
Termólise............................................................................... 263
Reações redoxi ..................................................................... 263
Detecção de radicais ............................................................. 264
Forma e estabilização de radicais ......................................... 265
Reações de radicais .............................................................. 267
Adição ................................................................................... 268
Adição de Halogênios ........................................................... 268
Adição de Ácido bromídrico................................................... 270
Outras adições ...................................................................... 273
Polimerização vinílica ............................................................ 273
Substituição ........................................................................... 275
Halogenação ......................................................................... 275
Auto-oxidação ....................................................................... 278
Substituição aromática .......................................................... 280
Rearranjo............................................................................... 282
Bi-radicais.............................................................................. 283
10º ROTEIRO DE ESTUDO DIRIGIDO ................................. 285
Unidade XI - Reações controladas por simetria de orbitais
Introdução ............................................................................. 290

13
Teoria dos orbitais moleculares ............................................ 290
Equação de onda - fase ........................................................ 290
Reações eletrocíclicas .......................................................... 293
Reação térmica ..................................................................... 294
Reação fotoquímica .............................................................. 296
Regras de woodward-hofmann ............................................. 297
Reações de cicloadições ...................................................... 298
Rearranjos sigmatrópicos ..................................................... 302
Migração de hidrogênio......................................................... 303
Migração de carbono ............................................................ 305
Referências ........................................................................... 310
Sites para pesquisa na web .................................................. 311
Sobre os autores: .................................................................. 312


1

2
UNIDADE I - ESTRUTURA, REATIVIDADE E MECANISMOS
ESTRUTURA E REATIVIDADE
Orbitais atômicos
O átomo de carbono tem seis elétrons que
segundo a teoria da estrutura atômica de Bohr, estão dispostos
em órbitas a distância crescente do núcleo. Estas órbitas
correspondem a níveis de energia gradualmente crescentes. A
de energia mais baixa, 1s, acomodando 2 elétrons, a seguinte,
2s, acomodando também dois elétrons e os 2 elétrons
restantes do átomo de carbono indo ocupar o nível 2p, que é
capaz de acomodar um total de 6 elétrons.
O principio da incerteza de Heisenberg e a visão
da mecânica-ondulatória do elétron tornaram necessário
eliminar algo definido com tanta precisão como descritos por
funções de onda ψ, e as órbitas de Bohr, clássicas e exatas,
foram substituídas por orbitais atômicos tridimensionais com
diferentes níveis de energia. O tamanho, forma e orientação
destes orbitais atômicos, regiões em que há máxima
probabilidade de se encontrar um elétron correspondem a um
nível de energia quantificado e são delineados por uma função
de onda, ψA, ψB, ψC, etc. Na verdade, os orbitais são
bastantes semelhantes a mapas de contorno eletrônico
tridimensional, em que ψ2 determina a probabilidade relativa
de encontrar um elétron num dado ponto do orbital.
A teoria dos orbitais moleculares (OM) constitui uma
alternativa para se ter uma visão da ligação. De acordo com
este enfoque, todos os elétrons de valência têm uma influência
na estabilidade da molécula (Elétrons dos níveis inferiores
também podem contribuir para a ligação, mas para muitas
moléculas simples o efeito é demasiado pequeno). A teoria do
OM considera que os orbitais atômicos (OAs) do nível de
valência deixam de existir quando a molécula se forma, sendo

3
substituídos por um novo conjunto de níveis energéticos que
correspondem a novas distribuições da nuvem eletrônica
(densidade de probabilidade). Esses novos níveis energéticos
constituem uma propriedade da molécula como um todo e são
chamados, consequentemente, de orbitais moleculares.
O cálculo das propriedades dos orbitais moleculares é
feito comumente, assumindo que os OAs se combinam para
formar OM. As funções de onda dos OAs são combinados
matematicamente para produzir as funções de onda dos OM
resultantes. O processo é remanescente da mistura de orbitais
atômicos puros para formar orbitais híbridos, exceto que, na
formação de OM, OA de mais de um átomo são combinados
ou misturados. No entanto, como no caso da hibridização, o
número de orbitais novos formados é igual ao número de OA
originários da combinação.
Da mesma maneira que nos orbitais atômicos, estamos
interessados em dois aspectos moleculares:
1) as formas de suas distribuições espaciais da nuvem
eletrônica;
2) suas energias relativas.
O diagrama usual de ψ versus r para o orbital 1s de um
átomo A (Figura 1a) deve, porém, ser modificado para levar em
conta a variação de r entre -∞ e +∞, resultando no diagrama
mostrado na figura 1b.
Figura 1 - Diagramas de ψ versus r para o orbital 1s.
Os seis elétrons do átomo de carbono estão
acomodados em orbitais atômicos de nível de energia
crescente até que todos estejam classificados (principio de Auf

4
bau). Onde dois elétrons, com spins emparelhados, irão para o
orbital 1s, dois para o orbital 2s, no nível 2p os dois elétrons
restantes poderão acomodar-se no mesmo orbital 2px, ou em
orbitais diferentes, 2px e 2py. A regra de Hund determina que
dois elétrons não pode ocupar o mesmo orbital, desde que haja
outro energeticamente equivalente desocupado. Assim, a
configuração eletrônica do átomo de carbono no estado
fundamental com dois elétrons desemparelhado é
1s22s22px12py
1, com o orbital 2pz desocupado. O átomo de
carbono no estado excitado (mais alta energia) possui
configuração eletrônica 1s22s12px12py
12pz1, agora com quatro
elétrons desemparelhados e pode por isso formar quatro
ligações com outros átomos ou grupos de átomos. A grande
quantidade de energia produzida pela formação destas duas
novas ligações contrabalança consideravelmente a energia
necessária para o desacoplamento inicial 2s2 e promoção de 2s
� 2p (≈ 406 kJ)
Hibridização
O átomo de carbono combina-se com outros quatro
átomos, formando entre as ligações ângulos de 109º 28’entre
si, isto pode ser explicado com base na mistura do orbital 2s e
dos três orbitais atômicos 2p de modo a produzir quatro novos
orbitais idênticos capazes de formar quatro ligações mais
fortes. Estes novos OA híbridos sp3 são obtidos pelo processo
chamado de hibridização.
+ +
2s 2px 2py 2pzhibrido sp
3
x
y
z
x
y
z
De modo semelhante, pode-se imaginar que a
hibridização ocorre também quando o átomo de carbono

5
combina-se com três outros átomos, no eteno, três orbitais
atômicos híbridos sp2 dispostos em ângulos de 120º no mesmo
plano (hibridização trigonal plana), e quando o átomo de
carbono combina-se com dois outros átomos de carbono, etino,
são utilizados dois orbitais atômicos híbridos sp1 dispostos a
ângulos de 180º (hibridização diagonal), em cada caso o orbital
s está sempre envolvido, visto que é o de mais baixo nível de
energia.
LIGAÇÕES EM COMPOSTOS DE CARBONO
Teoria de Ligação de Valência.
Uma ligação covalente ocorre quando dois átomos se
aproximam (distância ótima = comprimento da ligação) de
modo que um orbital de um dos átomos, ocupado por um
elétron, se superpõe a um orbital do outro átomo, também com
um elétron.
A força da ligação depende do grau de superposição dos
orbitais.
O grau de superposição entre os OA foi calculado e seu
valor é:
Orbital Grau de superposição s 1,00 p 1,72 sp 1,93 sp2 1,99 sp3 2,00
Teoria do orbital molecular (OM)
A teoria do OM constitui numa alternativa para se ter
uma visão da ligação. De acordo com este enfoque, todos os

6
elétrons de valência têm uma influência na estabilidade da
molécula (elétrons dos níveis inferiores também podem
contribuir para a ligação, mas para muitas moléculas simples o
efeito é pequeno). Além disso, a teoria OM considera que os
OA do nível de valência deixam de existir quando a molécula
se forma, sendo substituídos por um novo conjunto de níveis
energéticos que correspondem a novas distribuições da nuvem
eletrônica. Esses novos níveis energéticos constituem uma
propriedade da molécula como um todo e são chamados,
consequentemente de orbitais moleculares.
Observando os OM que são formados quando dois
átomos idênticos se ligam numa molécula diatômica, usando
um enfoque simples, consideremos que um OA de um átomo
se combina com um OA de um segundo átomo para formar
dois OM. Para que esse processo seja efetivo, duas condições
devem ser favorecidas:
1) os OA devem ter energias comparáveis;
2) eles devem se sobrepor de maneira significativa.
Os cálculos da mecânica quântica para a combinação
dos OA originais consistem em:
1) uma adição das funções de onda do OA;
2) uma subtração das funções de onda do OA.
Quando os dois átomos são diferentes, é incluído um
fator que leva em conta o fato de que os dois OAs não
contribuem igualmente para a formação dos OM. Os
resultados, então, são duas novas funções de onda, uma de
adição e outra de subtração. O quadrado da função de onda
para um elétron nos dá informações acerca da probabilidade
de encontrar este elétron em várias regiões do espaço. Quando
isto é feito para um OM, resultam informações sobre a
densidade da probabilidade para um elétron ocupando aquele
OM e, a partir dessas informações, as superfícies limites
correspondentes (e também os níveis energéticos) podem ser
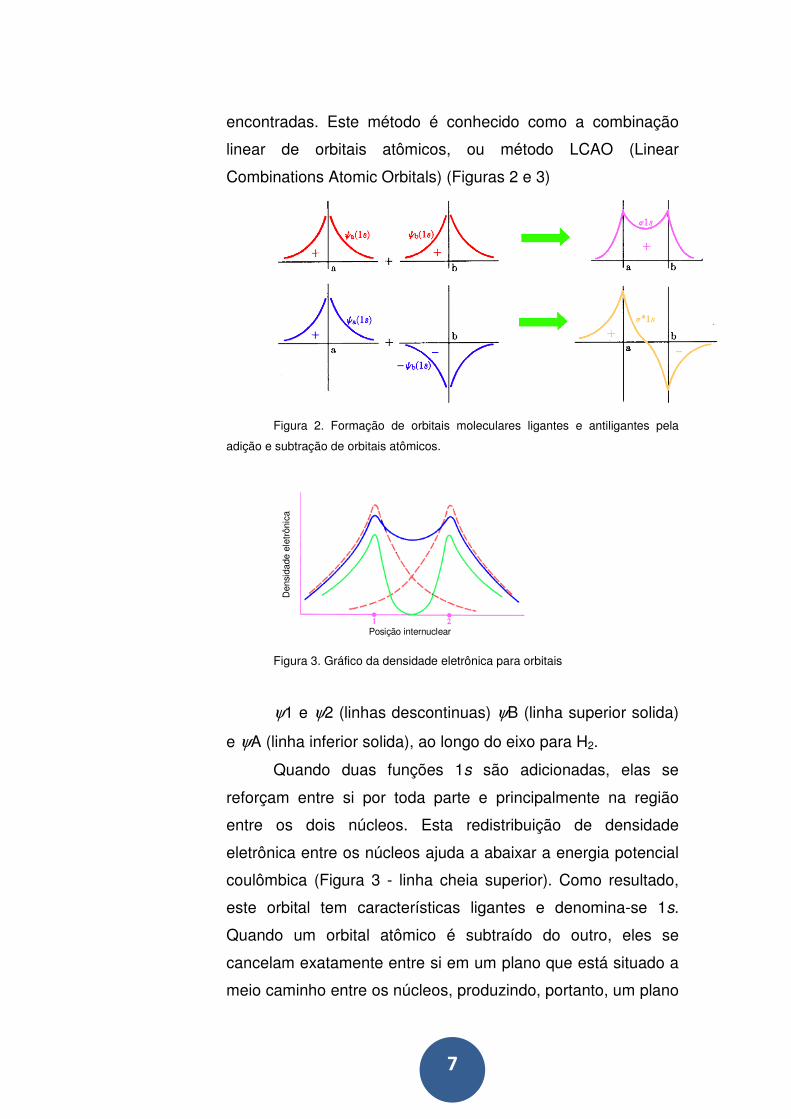
7
encontradas. Este método é conhecido como a combinação
linear de orbitais atômicos, ou método LCAO (Linear
Combinations Atomic Orbitals) (Figuras 2 e 3)
Figura 2. Formação de orbitais moleculares ligantes e antiligantes pela
adição e subtração de orbitais atômicos.
Den
sida
de e
letr
ônic
a
Posição internuclear
Figura 3. Gráfico da densidade eletrônica para orbitais
ψ1 e ψ2 (linhas descontinuas) ψB (linha superior solida)
e ψA (linha inferior solida), ao longo do eixo para H2.
Quando duas funções 1s são adicionadas, elas se
reforçam entre si por toda parte e principalmente na região
entre os dois núcleos. Esta redistribuição de densidade
eletrônica entre os núcleos ajuda a abaixar a energia potencial
coulômbica (Figura 3 - linha cheia superior). Como resultado,
este orbital tem características ligantes e denomina-se 1s.
Quando um orbital atômico é subtraído do outro, eles se
cancelam exatamente entre si em um plano que está situado a
meio caminho entre os núcleos, produzindo, portanto, um plano

8
nodal. A função de onda molecular é de sinal oposto em cada
lado deste plano nodal (Figura 2b). Quando se eleva a função
de onda ao quadrado (Figura 3 - linha cheia inferior), a
densidade de probabilidade resultante é obviamente positiva
em todos os lugares, exceto no plano nodal, onde é zero. Esta
deficiência de densidade eletrônica na região internuclear ajuda
a aumentar a energia potencial coulômbica do sistema, e o nó,
na função de onda, produz um aumento na energia cinética do
elétron. A energia total é, consequentemente, alta, os átomos
não estão ligados, e o orbital é descrito como antiligante.
Deve-se ter em mente que as representações dos OM
são análogas às representações dos OA e podem ser
interpretadas de duas maneiras equivalentes elas mostram:
1) a(s) região(ões) na(s) qual(is) o elétron passa a maior
parte do tempo, isto é, a(s) região(ões) de maior probabilidade
de encontrar-se o elétron ou, alternativamente,
2) a(s) região(ões) na(s) qual(is) a densidade da carga
eletrônica é alta.
Na figura 4 são mostradas as superfícies limites de dois
orbitais moleculares que são formados pela combinação de
dois orbitais atômicos 1s. Vemos à esquerda a sobreposição
dos OAs 1s e, à direita, os OM resultantes. O OM formado pela
subtração de funções de onda OA é representado por σs* (leia:
"sigma asterisco"), enquanto o formado pela adição é
representado por σs. O contraste entre esses dois OM é
grande. Há um aumento da densidade eletrônica de carga
entre os núcleos no orbital σs, mas um decréscimo na mesma
região no orbital σs*. Por essa razão, o orbital σs é chamado
orbital ligante, e o σs*, de orbital antiligante. O primeiro
tende a estabilizar a ligação, enquanto o último tende a
desestabilizá-la. Ambos são chamados orbitais σ porque estão
centrados e são simétricos ao redor do eixo de ligação. Uma

9
secção de cada orbital feita perpendicularmente ao eixo de
ligação apresenta um formato circular.
Figura 4. Combinação de AO 1s para formar OM σ.
Pela combinação linear de um orbital 2s de um átomo A
com um orbital 2s do átomo B, obtemos aproximações dos
orbitais moleculares σ2s, Ligante e antiligante.
O processo é completamente análogo àquele
empregado para σ1s e σ1s*. As quantidades N e N* são fatores
de normalização. O orbital σ2s* possui um plano nodal entre os
dois núcleos, é antiligante e tem energia maior do que o orbital
σ2s que não possui este plano nodal, e é Ligante (Figura 5).
Figura 5. Formação de orbitais σ2s e σ2s
* pela adição e subtração de OA 2s.
Os sinais de mais e menos se referem ao sinal das
funções de onda e não a cargas nucleares ou eletrônicas.
Observa-se que há uma superfície nodal rodeando os
núcleos tanto no orbital σ2s como no orbital σ2s*, o que os
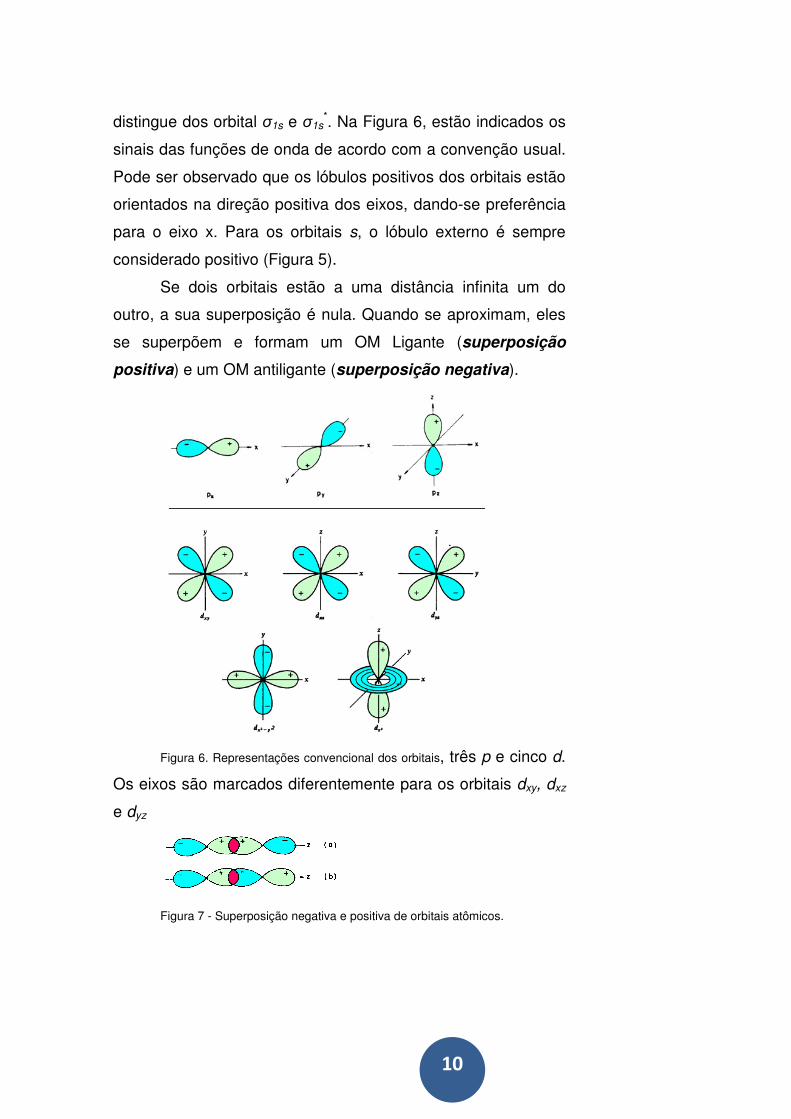
10
distingue dos orbital σ1s e σ1s*. Na Figura 6, estão indicados os
sinais das funções de onda de acordo com a convenção usual.
Pode ser observado que os lóbulos positivos dos orbitais estão
orientados na direção positiva dos eixos, dando-se preferência
para o eixo x. Para os orbitais s, o lóbulo externo é sempre
considerado positivo (Figura 5).
Se dois orbitais estão a uma distância infinita um do
outro, a sua superposição é nula. Quando se aproximam, eles
se superpõem e formam um OM Ligante (superposição
positiva) e um OM antiligante (superposição negativa).
Figura 6. Representações convencional dos orbitais, três p e cinco d.
Os eixos são marcados diferentemente para os orbitais dxy, dxz
e dyz
Figura 7 - Superposição negativa e positiva de orbitais atômicos.

11
A figura 7 mostra que a superposição é positiva se
corresponder à combinação de funções de onda de mesmo
sinal, e será negativa se os sinais forem opostos.
Notamos que, na figura 7a, o orbital 2pz da direita tem
seu lóbulo negativo orientado segundo a direção positiva do
eixo z, o que significa que ele foi multiplicado por menos um.
Isso equivale a dizer que, em vez de soma, foi feita uma
subtração dos OA, enquanto que a figura 7b mostra uma soma
dos OA, pois ambos os OA estão orientados na mesma direção
do eixo z.
A combinação de dois orbitais p pode produzir
resultados diferentes, dependendo de quais orbitais p são
usados. Se o eixo x é o eixo de ligação, então dois orbitais 2px
podem se sobrepor quando eles se aproximarem segundo um
único eixo, como é mostrado na Figura 8. Os OM resultantes
constituem, como antes, um orbital ligante (σx) com carga
eletrônica acumulada entre os núcleos e um OM antiligante
(σx*) com decréscimo de carga entre os núcleos.
Esses orbitais são também classificados como σ, porque
são simétricos ao redor do eixo de ligação e são designados σx
e σx* para indicar que derivaram de OA px.
Figura 8. Combinação de OA 2px para formar OM σ
Quando orbitais 2py e 2pz se sobrepõem para formar
OM, eles o fazem lado a lado, como é apresentado na Figura 9.
Em cada caso, o resultado é um orbital antiligante com quatro

12
lóbulos e um orbital ligante com dois lóbulos. Esses orbitais
não são simétricos em relação ao eixo de ligação, em vez
disso, existem duas regiões, em lados opostos ao eixo da
ligação, nas quais a densidade da nuvem de carga é alta. Isto é
característico de um orbital π.
Observe que, o orbital ligante permite uma alta
concentração da carga eletrônica na região entre os núcleos,
enquanto o orbital antiligante mostra uma diminuição da
densidade de carga nessa região. (Na realidade, cada orbital
antiligante tem um plano nodal entre os dois núcleos).
Figura 9. Combinação de OA 2p na formação de OM π.
Quando OA se combinam, eles passam a compartilhar
uma região do espaço. Se a superposição entre os orbitais é
positiva, os lóbulos envolvidos se fundem e formam um lóbulo
único no orbital molecular resultante. Se a superposição entre
os orbitais é negativa, não ocorre a fusão dos lóbulos,
aparecendo um plano nodal entre eles e a densidade eletrônica
na região internuclear diminui.
Energias dos orbitais moleculares
Um exemplo clássico em estudos é a formação da
molécula H2 a partir dos dois átomos, a uma distância infinita
um do outro. Inicialmente, seus orbitais não se superpõem e a
energia potencial do sistema é considerado igual a zero. À
medida que eles se aproximam, começa a haver interações
entre seus orbitais, formando-se um OM Ligante e um OM
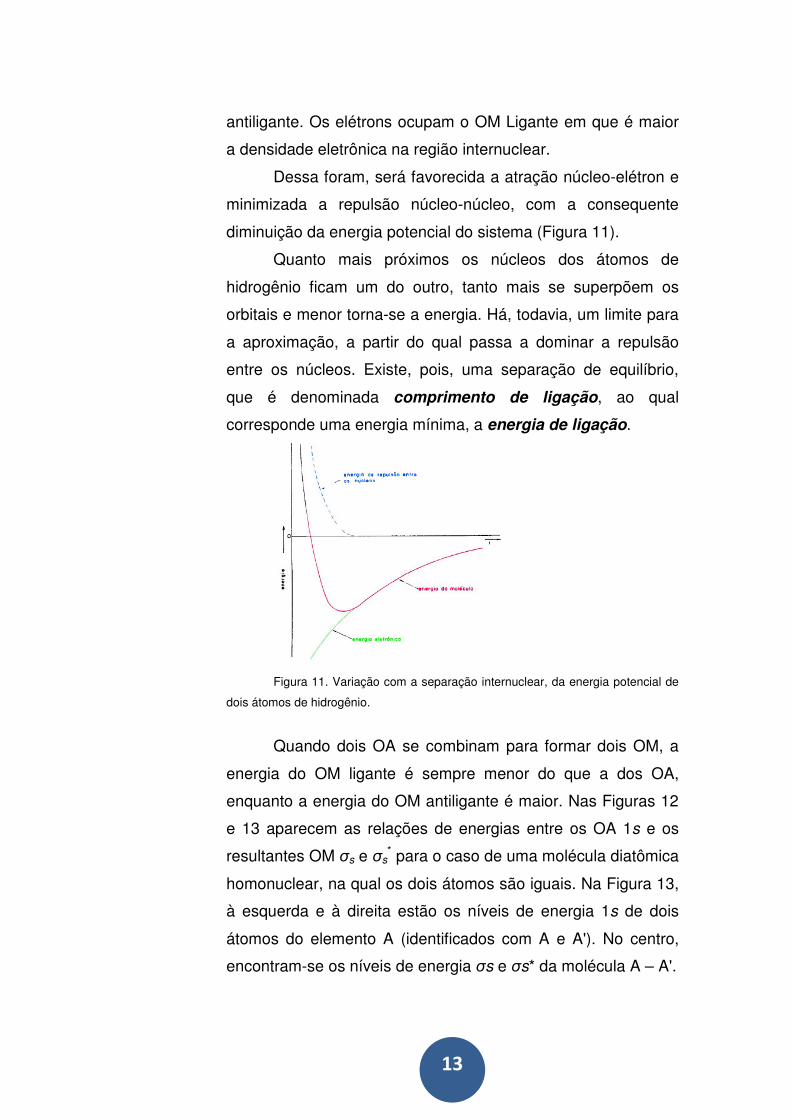
13
antiligante. Os elétrons ocupam o OM Ligante em que é maior
a densidade eletrônica na região internuclear.
Dessa foram, será favorecida a atração núcleo-elétron e
minimizada a repulsão núcleo-núcleo, com a consequente
diminuição da energia potencial do sistema (Figura 11).
Quanto mais próximos os núcleos dos átomos de
hidrogênio ficam um do outro, tanto mais se superpõem os
orbitais e menor torna-se a energia. Há, todavia, um limite para
a aproximação, a partir do qual passa a dominar a repulsão
entre os núcleos. Existe, pois, uma separação de equilíbrio,
que é denominada comprimento de ligação, ao qual
corresponde uma energia mínima, a energia de ligação.
Figura 11. Variação com a separação internuclear, da energia potencial de
dois átomos de hidrogênio.
Quando dois OA se combinam para formar dois OM, a
energia do OM ligante é sempre menor do que a dos OA,
enquanto a energia do OM antiligante é maior. Nas Figuras 12
e 13 aparecem as relações de energias entre os OA 1s e os
resultantes OM σs e σs* para o caso de uma molécula diatômica
homonuclear, na qual os dois átomos são iguais. Na Figura 13,
à esquerda e à direita estão os níveis de energia 1s de dois
átomos do elemento A (identificados com A e A'). No centro,
encontram-se os níveis de energia σs e σs* da molécula A – A'.

14
As linhas tracejadas diagonais mostram que os OM se
formaram dos OA indicados. A Figura 13 poder ser usada para
mostrar a formação dos OM de um par de qualquer orbital s
(2s, 3s, 4s, etc.). Em cada caso, um orbital antiligante e um
orbital ligante são formados.
Figura 12. A idéia do orbital molecular
Figura 13. Energias relativas dos orbitais σs em moléculas diatômicas
homonucleares.
Consideremos a seguir a formação dos OM de um par
de orbitais 2px cujos lóbulos estão dirigidos para o eixo de
ligação, Figura 14, a formação de um par de OM, um ligante
(σx) e um antiligante (σx*).

15
Figura 14. Energias relativas dos orbitais σx em moléculas diatômicas
homonucleares
Observe os OA 2py e 2pz, que se sobrepõem lado a
lado. Os OM formados a partir deles são mostrados na figura
15. A sobreposição py – py é exatamente igual à sobreposição pz
– pz (exceto pela orientação) e assim os OM formam dois
conjuntos de orbitais de mesma energia, os orbitais πy e πz
(ligantes) e os orbitais πy* e πz
* (antiligantes).
Figura 15. Energias relativas dos orbitais πy e πz em moléculas diatômicas
homonucleares
Preenchimento dos orbitais moleculares
O procedimento de Aufbau, pelo qual os elétrons são
adicionados um a um ao diagrama de energia dos OA com o
objetivo de construir a configuração eletrônica dos átomos,
usaremos agora uma técnica semelhante para preencher os
níveis energéticos do diagrama de OM, para construir a
configuração eletrônica de moléculas diatômicas

16
homonucleares no estado fundamental. Os elétrons são
adicionados a partir da base do diagrama para cima, para os
orbitais de maior energia.
A molécula mais simples é a de hidrogênio (H2). A Figura
16 mostra, à esquerda e à direita, elétrons colocados em dois
átomos de H não-ligados e, no meio do diagrama, a molécula
de H2 no estado fundamental. Os dois elétrons 1s vão constituir
um par (spins opostos) no orbital σs (ligante) da molécula. Este
par constitui uma ligação simples. A configuração eletrônica da
molécula de hidrogênio pode ser escrita como: H2 (σs)2
Figura 16. Preenchimento do diagrama OM para H2
Considerando a molécula Hélio e (He2), que poderia ser
formada por dois átomos de hélio em que cada um dos quais é
capaz de fornecer dois elétrons para a molécula. O total é de
quatro elétrons, dois a mais que no H2, de maneira que a
distribuição no OM será a da Figura 17. A configuração
eletrônica no estado fundamental da molécula de He2 deverá
ser:
He2: (σs)2(σs*)2
Figura 17. Preenchimento do diagrama OM para a molécula hipotética He2.

17
Devido ao fato de que o σs* (antiligante) está agora
preenchido e seu efeito desestabilizador cancela o efeito
estabilizador do orbital σs,. O resultado é que não há uma força
de atração entre os átomos de hélio devido ao número igual de
elétrons ligantes e antiligantes e, assim, He2 não existe.
Na teoria dos OM a ordem de ligação é definida como:
Assim, a ordem de ligação na molécula de H2 é:
Enquanto na molécula hipotética de He2 é:
Para a molécula de lítio (Li2) e de berílio (Be2), desde que o
preenchimento de dois OM σ formados de orbitais 1s está
completo, passasse para os dois OM σ formados a partir dos
orbitais 2s.
Ligações simples carbono-carbono
A combinação de dois átomos de carbono no etano
resulta da sobreposição axial de dois orbiatais sp3, um de cada
átomo de carbono, para formar uma ligação sigma forte entre
eles. O comprimento da ligação carbono-carbono é de 0,154
nm. Uma ligação simples semelhante entre dois átomos de
carbonos hibridizados sp2, =CH – CH=, tem em média um
comprimento de ligação de 0,147 nm e entre átomos de
carbono hibridizados sp1, ≡C–C≡, cerca de 0,138 nm. Este fato
ocorre porque um orbital s e qualquer elétron que nele se
encontre são mantidos mais próximos e fortemente pelo núcleo
do que um orbital p e qualquer elétron que nele se encontre. O
mesmo efeito será observado com orbitais híbridos à medida
que aumenta a sua componente s, e para dois átomos de

18
carbonos ligados entre si os núcleos são aproximados à
medida que vai de sp3 – sp3 � sp2 – sp2 � sp1 – sp1.
Entretanto, não é definida uma estrutura única para o
etano, a ligação sigma entre os átomos de carbono é simétrica
em torno de uma linha que une os dois núcleos, teoricamente,
é possível uma variedade infinita de estruturas diferentes
definida pela posição dos hidrogênios num átomo de carbono,
relativamente à posição dos átomos de hidrogênios no outro
átomo de carbono. De todas as estruturas possíveis, os dois
extremos são as formas eclipsadas e em estrela, conhecidas
como projeções em perspectiva de Newman.
HH
H HH
H H
H H
H
H
HHH
H
HH
H
H
H
H
H
H
Eclipisada Em estrela
As formas eclipsadas e em estrela é a infinita variedade
de estruturas possíveis entre esses extremos, são designadas
por conformações da molécula do etano. Conformações são
definidas como diferentes arranjos do mesmo grupo de átomos
que podem ser convertidos uns nos outros sem quebra de
quaisquer ligações.
A conformação em estrela é a mais estável, os átomos
de hidrogênios nos átomos de carbonos adjacentes estão tão
afastados quanto possível uns dos outros (0,310 nm) e
qualquer interação chamada não-ligante entre eles está
portanto num mínimo, enquanto que na conformação eclipsada
estão num máximo de sobreposição (0,230 nm), ligeiramente
menos do que a soma dos seus raios de van der Waals.
Ligações duplas carbono-carbono
No eteno, cada átomo de carbono esta ligado a três
outros átomos, dois hidrogênios e um carbono. Formam-se

19
ligações sigma fortes com estes três átomos pela utilização de
três orbitais derivados por hibridização do orbital 2s e dois
orbitais atômicos 2p do átomo de carbono, um átomo
normalmente só mobiliza tantos orbitais híbridos quantos os
átomos ou grupos com os quais pode formar ligações sigma
fortes. Os orbitais híbridos sp2 resultantes encontram-se todos
no mesmo plano e com ângulos de ligação de 120º, um em
relação aos outros (orbitais trigonais planos). Na formação da
molécula do eteno, dois orbitais sp2 de cada átomo de carbono
são visualizados como sobrepondo-se aos orbitais 1s de dois
átomos de hidrogênios para formar duas ligações C–H sigma
fortes, enquanto que a terceiro orbital sp2 de cada átomo de
carbono se sobrepõe axialmente para formar uma ligação C–C
sigma forte entre eles. Verifica-se experimentalmente que os
ângulos das ligações H–C–H e H–C–C são de fato 116º7’ e
121º6’, respectivamente. O afastamento de 120º não é visto
que estão envolvidos diferentes trios de átomos.
Isto deixa, em cada átomo de carbono, um orbital
atômico não hibridizado 2p com ângulos retos com o plano
contendo os átomos de carbono e de hidrogênio. Estes dois
orbitais 2p paralelos um ao outro, podem sobrepor-se
resultando na formação de um orbital molecular ligante que se
estende sobre ambos os átomos de carbonos, situando-se
acima e abaixo do plano que contêm os dois átomos de
carbonos e os quatro átomos de hidrogênios.
Este novo orbital molecular ligante (também forma um
orbital molecular antiligante) é conhecido como um orbital π, e
os elétrons que ocupam estes são elétrons π. A nova ligação π

20
que se forma tem efeito de fazer aproximar mais os átomos de
carbonos C=C, cuja distância no eteno é de 0,133 nm,
comparada com uma distância C–C de 0,154 nm no etano. A
sobreposição lateral dos orbitais atômicos p axial que ocorre na
formação de uma ligação π, a primeira é mais fraca do que a
última. Isto se reflete no fato de a energia de uma ligação dupla
carbono-carbono, embora superior a de uma ligação simples,
ser inferior ao dobro desta última. Assim a energia da ligação
C–C no etano e de 347 kJ/mol, enquanto que a de C=C no
eteno é de apenas 598 kJ/mol.
A sobreposição lateral dos dois orbitais atômicos 2p, a
força da ligação π, será claramente máxima quando os dois
átomos de carbonos e os quatros átomos de hidrogênios forem
exatamente coplanares, pois só nesta posição é que os orbitais
p são exatamente paralelos entre si, capazes de um máximo
de sobreposição. Qualquer deste estado coplanar, por torção
em torno da ligação σ que une os dois átomos de carbonos,
conduz a redução da sobreposição π, e a um decréscimo na
força da ligação π.
Ligações triplas carbono-carbono
No etino, cada átomo de carbono está ligado a dois
outros átomos, um de hidrogênio e um carbono. Formam
ligações sigma fortes com estes dois átomos pela utilização de
dois orbitais híbridos derivados por hibridização do orbital 2s e
um orbital atômico 2p do átomo de carbono. Os orbitais
híbridos resultantes sp1 diagonais são co-lineares. Assim na
formação da molécula do etino, estes orbitais são utilizados
para formar ligações sigma fortes entre o átomo de carbono e
um átomo de hidrogênio, e entre dois átomos de carbonos
resultando uma molécula linear com dois orbitais atômicos 2p
não híbridos com ângulos retos entre si em cada um dos
átomos de carbonos. Os orbitais atômicos, num átomo de

21
carbono, são paralelos aos do outro, e podem sobrepor-se com
formação de duas ligações π em planos a ângulos retos entre
si.
A molécula de etino está efetivamente envolta num
cilindro de carga negativa. A energia da ligação C≡C e de 812
kJ/mol, de modo que o incremento devido a terceira ligação é
inferior ao que ocorre quando se passa de uma ligação simples
para uma dupla. A distância da ligação C≡C e de 0,120 nm de
modo que os átomos de carbonos se aproximam mais ainda,
aqui novamente o decréscimo ao passar de C=C � C≡C é
inferior ao que ocorre quando se passa de C–C � C=C.
Ligações carbono-oxigênio e carbono-nitrogênio.
Um átomo de oxigênio tem configuração eletrônica
1s22s22px22py12pz1, e também ele, ao combinar-se com outros
átomos, pode ser considerado como utilizando orbitais híbridos
de modo a formar ligações mais fortes. Ao combinar-se com os
átomos de carbono de dois grupos metila, para formar
metoximetano, CH3–O–CH3, o átomo de oxigênio pode utilizar
quatro orbitais híbridos sp3 de cada um dos dois átomos de
carbonos e as outras duas para acomodar os seus dois pares
de elétrons desemparelhados. Verifica-se que o comprimento
da ligação C–O é 0,142 nm, e a energia da ligação de 360
kJ/mol.
O átomo de oxigênio também pode formar uma ligação
dupla com um carbono, assim, na propanona, (CH3)2C=O, o
átomo de oxigênio poderia utilizar três orbitais híbridos sp2, um
para formar uma ligação sigma por sobreposição com o orbital
sp2 do átomo de carbono, e os outros dois para acomodar os

22
dois pares de elétrons desemparelhados. Isto deixa um orbital
não hibridizado p quer no oxigênio quer no carbono, e estes
podem sobrepor-se entre si lateralmente para formar uma
ligação π.
O ângulo da ligação C–C=O é de ≈ 120º, o comprimento
da ligação C=O de 0,122 nm e a energia da ligação 750 kJ/mol.
O fato de esta ser superior ao dobro da energia da ligação C–
O, enquanto que a energia da ligação C=C é inferior ao dobro
da de C–C, em parte, devido ao fato de os pares de elétrons
desemparelhados no oxigênio se encontrarem mais afastados,
e por isso mais estáveis em C=O do que em C–O, não
havendo circunstância equivalente com o carbono. O fato das
ligações carbono-oxigênio, ao contrário das ligações carbono-
carbono, serem polares também desempenha seu papel.
Um átomo de nitrogênio, com a configuração eletrônica
1s2 2s2 2px1 2py1 2pz1, também utiliza orbitais híbridos na
formação de ligações simples, C–N, ligações duplas, C=N, e
nas ligações triplas C≡N. Em cada caso uma desses orbitais é
utilizado para acomodar o par de elétrons desemparelhado do
nitrogênio, na formação de ligações duplas e triplas, são
utilizados orbitais p, não híbridos, do nitrogênio e do carbono.
Os comprimentos e energias médias das ligações são: simples
0,147 nm e 305 kJ/mol, dupla 0,129 nm e 616 kJ/mol e na tripla
0,116 nm e 893 kJ/mol.
Conjugação
Quando se considera moléculas que contêm mais de
uma ligação múltipla, dienos com duas ligações C=C, verifica-
se que compostos em que as ligações são conjugadas
(ligações múltiplas e simples alternadas) são ligeiramente mais
estáveis do que aquelas em que são isoladas.
CH3 CH2 CH2 CH2

23
Esta estabilidade termodinâmica superior de moléculas
conjugadas é comprovada pelo fato de apresentar um calor de
combustão e um calor de hidrogenação inferior ao de duplas
isoladas, e também na observação geral de que ligações
duplas isoladas podem ser induzidas a migrar rapidamente de
modo a tornarem-se conjugadas.
CH3
O
CH3
Base
Catalisadores
CH3
O
CH3
A conjugação não está limitada, é claro, apenas a
ligações múltiplas carbono-carbono.
Quer com duplas conjugadas ou duplas isoladas, a
sobreposição lateral dos OA p em átomos de carbonos
adjacentes pode conduzir a formação de duas ligações π
localizadas, e esperar-se-ia que os compostos se
assemelhassem ao eteno, é de fato o que se verifica com
duplas isoladas, mas com duplas conjugadas apresentam um
comportamento diferente devido sua maior estabilidade,
diferenças de comportamento espectroscópico, e pelo fato de
sofrer reações de adição mais rapidamente do que um dieno
isolado. No entanto, uma observação mais detalhada, mostra
que com duplas conjugadas, mas não com duplas isoladas, a
sobreposição lateral pode ocorrer entre todos os quatro orbitais
atômicos p em átomos de carbonos adjacentes. Tal
sobreposição resultará na formação de quatro orbitais
moleculares, dois ligantes (ψ1 e ψ2) e dois antiligantes (ψ3 e ψ4)
– a sobreposição de n orbitais atômicos dá origem a n orbitais
moleculares.
Observa-se, a partir da figura acima, que acomodando
os quatro elétrons do dieno conjugado em dois orbitais ligantes,
conduzirá a uma energia total para o composto, mais baixa do
que – por analogia com o eteno – acomodando-os em duas
ligações π localizadas. Os elétrons por estarem deslocalizados,

24
estão mantidos pelo conjunto do sistema conjugado em vez de
serem localizados sobre dois átomos de carbonos em ligações
π, como no eteno ou em duplas isoladas. A acomodação dos
quatro elétrons nos orbitais moleculares ψ1 e ψ2 resulta na
distribuição eletrônica numa nuvem de carga numa dada região
da molécula ou nela como um todo.
A deslocalização é muito envolvida na estabilização de
estados excitados de dienos e de polienos, em geral, no
abaixamento do nível de energia dos seus estados excitados.
O seu efeito é reduzir a diferença de energia entre os estados
fundamental e excitado de moléculas conjugadas, por
comparação com aquelas que contêm duplas ligações
isoladas, e esta diferença de energia diminui progressivamente
à medida que aumenta a extensão da conjugação. Os dienos
simples absorvem na região do ultravioleta, mas à medida que
aumenta a extensão da conjugação, a absorção gradualmente
desloca-se para a região do visível e o composto torna-se mais
colorido. Ilustrado pela série de α, ω-difenilpolienos (C6H5–
(CH=CH)n–C6H5).
Série de difenilpolienos (C6H5 – (CH = CH)n – C6H5)
Cor
n = 1 Incolor n = 2-4 Amarelo n = 5 Laranja n = 8 Vermelho
Benzeno e aromaticidade
No início da química orgânica, a palavra aromático foi
utilizada para descrever algumas substâncias que possuíam
fragrâncias, como o Benzaldeído (responsável pelo aroma das
cerejas, pêssegos e amêndoas), o tolueno (bálsamo) e o
benzeno (do carvão destilado). Entretanto, logo se observou
que essas substâncias denominadas aromáticas eram
25
diferentes da maioria dos compostos orgânicos em relação ao
comportamento químico.
C
O
HCH3
Benzeno Benzaldeído Tolueno
Hoje em dia, usamos a palavra aromático para nos
referir ao benzeno e seus derivados estruturais. Assim, os
químicos do século XIX estavam corretos em relação à
diferença entre os compostos aromáticos e os outros, porém a
associação de aromaticidade com fragrância havia se perdido.
Muitos compostos isolados de fontes naturais são, em
parte, aromáticos. Além do benzeno, benzaldeído e tolueno, a
substância hormonal estrona e o bastante conhecido
analgésico morfina têm anéis aromáticos. Muitas drogas
sintéticas também são aromáticas, o tranquilizante diazepam é
um exemplo.
Foi comprovado que a exposição prolongada ao
benzeno causa depressão da medula óssea e
consequentemente leucopenia (diminuição dos glóbulos
brancos). Dessa forma, o benzeno deve ser manuseado
cuidadosamente se utilizado como solvente em laboratório.
Um dos principais problemas da química orgânica
elementar é a estrutura pormenorizada do benzeno. A
conhecida estrutura planar da molécula implica hibridação sp2
OCH3
H
HO
H H
O
N CH3
OH
HO
Estrona Morfina
N
N
OCH3
Cl
Diazepam (valium)

26
com orbitais atômicos p e ângulos retos em relação ao plano
do núcleo, em cada um dos seis átomos de carbono.
A sobreposição poderia ser nas posições 1,2; 3,4; 5,6 ou
1,6; 5,4; 3,2, conduzindo a formulações correspondentes as
estruturas de Kekulé, mas como alternativa todos os orbitais p
adjacentes poderiam sobrepor-se, como com dienos
conjugados, resultando na formação de seis orbitais
moleculares, três ligantes (ψ1 �ψ3) e três antiligantes (ψ4 �
ψ6) com níveis de energia.
O OM ligante de menor energia (ψ1) é cíclico e engloba
todos os seis átomos de carbono deslocalizado. Tem um plano
nodal no plano do anel, de modo que há dois lobos anelares,
um acima e outro abaixo do plano do anel, o de cima está
ilustrado na figura abaixo, onde dois elétrons estão portanto
acomodados. Os dois OM ligantes seguintes (ψ2 e ψ3), de igual
energia, também englobam todos os seis átomos de carbono,
mas cada um tem ainda outro plano nodal, a ângulos retos com
o plano do anel, para além do no plano do anel. Cada OM tem
quatro lobos como ilustrado na figura abaixo, cada um destes
OM acomoda mais dois elétrons, perfazendo um total de seis.
O resultado global é uma nuvem eletrônica anelar, acima
e abaixo do plano do anel.

27
A influência desta nuvem de carga negativa com o
reagente que atacará o benzeno, onde na primeira fase da
reação envolve a interação entre o eletrófilo que se aproxima e
os orbitais π deslocalizados, chama-se complexo π.
O comprimento de todas as ligações carbono-carbono
no benzeno é 0,140 nm, fazendo com que o benzeno seja um
hexágono regular, comprimento intermediário entre os valores
normais para uma ligação simples de 0,154 nm e uma ligação
dupla de 0,133 nm. Esta regularidade pode ser evidenciada se
evitar escrever estruturas de Kekulé para o benzeno, uma vez
que são claramente representações inadequadas, e usando em
vez disso a representação abaixo.
Permanece a questão da estabilidade termodinâmica do
benzeno. Parte desta resulta sem dúvida da disposição das
três ligações σ trigonais planas em torno de cada átomo de
carbono no seu ângulo de 120º, mas grande parte resulta do
uso de OM cíclicas deslocalizados para acomodar os seis
elétrons residuais, este é um arranjo mais estável (energia
mais baixa) do que acomodar os elétrons em três orbitais
moleculares π localizados. A maior estabilização do benzeno,
resulta do fato do benzeno ser um sistema simétrico cíclico.
Uma estimativa aproximada da estabilização do benzeno
(cilcohexa-1,3,5-trieno), comparada com estruturas cíclicas
insaturadas simples, pode ser obtida comparando o seu calor
de hidrogenação com os do ciclohexeno, do ciclohexa-1,3-
dieno.
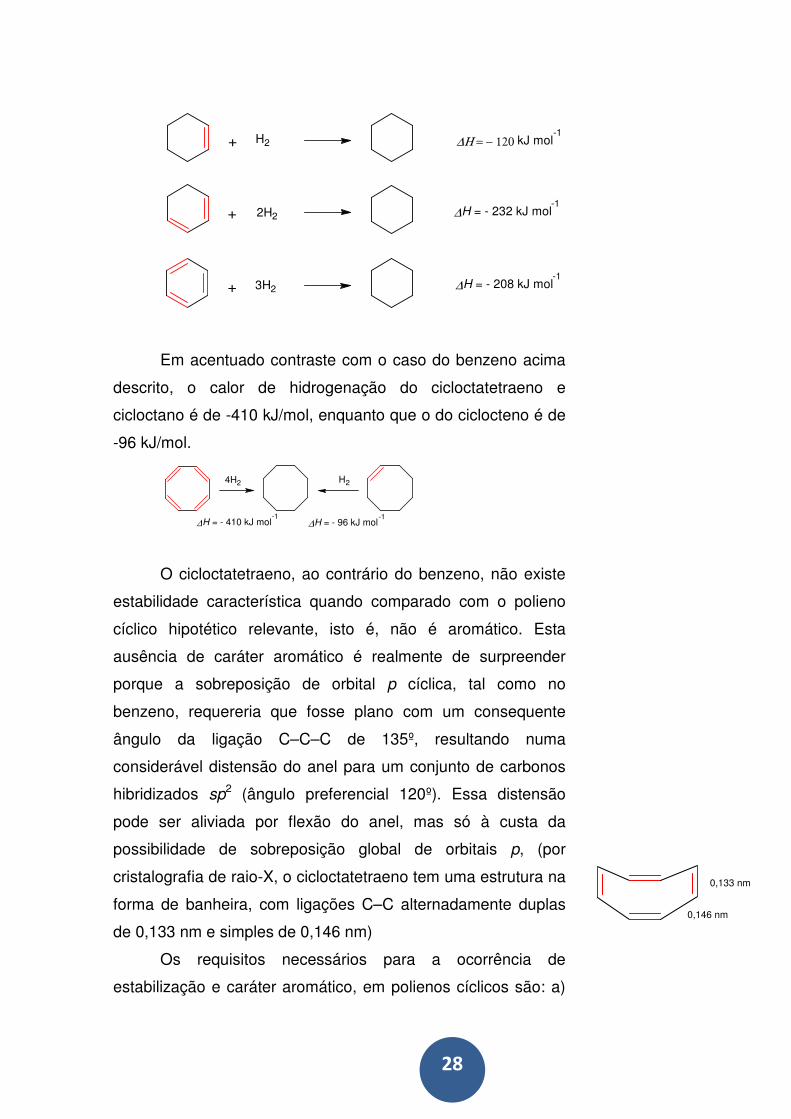
28
+
+
+
H2
2H2
3H2
∆Η = − 120 kJ mol-1
∆H = - 232 kJ mol-1
∆H = - 208 kJ mol-1
Em acentuado contraste com o caso do benzeno acima
descrito, o calor de hidrogenação do cicloctatetraeno e
cicloctano é de -410 kJ/mol, enquanto que o do ciclocteno é de
-96 kJ/mol.
∆H = - 410 kJ mol-1
∆H = - 96 kJ mol-1
4H2 H2
O cicloctatetraeno, ao contrário do benzeno, não existe
estabilidade característica quando comparado com o polieno
cíclico hipotético relevante, isto é, não é aromático. Esta
ausência de caráter aromático é realmente de surpreender
porque a sobreposição de orbital p cíclica, tal como no
benzeno, requereria que fosse plano com um consequente
ângulo da ligação C–C–C de 135º, resultando numa
considerável distensão do anel para um conjunto de carbonos
hibridizados sp2 (ângulo preferencial 120º). Essa distensão
pode ser aliviada por flexão do anel, mas só à custa da
possibilidade de sobreposição global de orbitais p, (por
cristalografia de raio-X, o cicloctatetraeno tem uma estrutura na
forma de banheira, com ligações C–C alternadamente duplas
de 0,133 nm e simples de 0,146 nm)
Os requisitos necessários para a ocorrência de
estabilização e caráter aromático, em polienos cíclicos são: a)
0,133 nm
0,146 nm

29
molécula tem que ser plana; b) todos os orbitais ligantes
completamente preenchidos. Esta condição verifica-se em
sistemas cíclicos com 4n + 2 elétrons π (regra de Hückel), e o
arranjo que ocorre de longe mais vulgarmente em compostos
aromáticos, por exemplo, quando n = 1, isto é, o composto tem
6 eletrons π, 10 eletrons π (n = 2) estão presentes no naftaleno,
e 14 eletrons π (n = 3) no antraceno e fenantraceno.
Naftaleno Antraceno Fenantreno
Embora estas substâncias não sejam monocíclicas
como o benzeno, e a regra de Hückel não deva aplicar-se a
elas, a introdução da ligação transanelar as torna bi e
tricíclicas, parecendo causar uma perturbação relativamente
pequena no que se refere a deslocalizaçao dos elétrons π
sobre o grupo ciclico de dez ou quatorze átomos de carbono.
As estruturas quase-aromáticas são aquelas em
que a espécie cíclica estabilizada é um íon e o cátion
cicloheptatrienilo (tropílio), o ânion ciclopentadienilo, ambos
têm 6 eletrons π (n = 1) e ainda mais surpreendente, o cátion
ciclopropenilo que tem 2 elétrons π (n = 0).
-+ +
Cátion ciclopropeniloÂnion ciclocilcopentadieniloCátion cicloheptatrienilo
Além disso, a estrutura anelar não tem de ser puramente
carbociclica, e a piridina, por exemplo, com um átomo de
nitrogênio no anel e 6 elétrons π (n = 1), encontra-se tão
estabilizada quanto ao benzeno.

30
N
Condições necessárias para a deslocalização.
A dificuldade em encontrar uma representação
satisfatória para a ligação carbono-carbono no benzeno leva-
nos a constatar que a maneira, segundo a qual normalmente
escrevemos as ligações entre átomos com simples, duplas ou
triplas, envolvendo dois, quatro e seis elétrons,
respectivamente, é inadequada. Algumas ligações envolvem
outros números, mesmo fracionários de elétrons. É o que se
pode ver muito claramente no ânion etanoato (acetato), em que
a distância das duas ligações carbono-oxigênio é a mesma,
envolvendo o mesmo número de elétrons.
CH3 C
O
O-
Estas dificuldades conduziram à convenção de
representar moléculas que não podem ser escritas
adequadamente como uma estrutura clássica simples por uma
combinação de duas ou mais estruturas clássicas, as
chamadas estruturas canônicas, ligadas por uma seta com
duas pontas. O modo pelo qual uma destas estruturas pode ser
relacionada com outra é geralmente indicado por meio de setas
encurvadas, indicando a cauda da seta encurvada de onde é
que se move um par de elétrons e a ponta da seta o local para
onde esse par de elétrons se move.
CH3 C
O
O-
CH3 C
O-
O
CH3 C
O
O
-

31
Nunca é de mais acentuar que o ânion etanoato não tem
duas estruturas possíveis e alternativas, que são rapidamente
interconvertidas, mas uma só estrutura real, por vezes
designada como um híbrido, para as estruturas canônicas
menos exatas.
De um modo geral, quanto mais estruturas canônicas
puderem ser escritas para um dado composto, maior será a
deslocalização dos elétrons, e mais estável será o composto.
Estas estruturas não devem diferir muito umas das outras em
conteúdo energético, ou então as de energia mais elevada
contribuirão tão pouco para o híbrido que a sua contribuição se
torna praticamente irrelevante.
As estruturas escritas devem todas elas conter o mesmo
número de elétrons emparelhados, e os átomos constituintes
devem todos eles ocupar essencialmente as mesmas posições
relativamente uns aos outros em cada estrutura canônica. Se a
deslocalização for significativa, todos os átomos ligados a
centros insaturados devem situar-se no mesmo plano, ou
aproximadamente no mesmo plano.
QUEBRA E FORMAÇÃO DE LIGAÇÕES
Uma ligação covalente entre dois átomos pode sofrer
quebra essencialmente dos seguintes modos:
R + X. .R:
-+ X
+
R+
+ X-
R X..
No primeiro caso, cada átomo separa-se com um
elétron, conduzindo a formação de entidades altamente
reativas chamadas de radicais, que devem sua reatividade ao
elétron desemparelhado, e a esta quebra de ligação dá-se o
nome de fissão homolítica da ligação. No segundo caso, um
átomo pode ficar com os dois elétrons, não deixando nenhum

32
para o outro átomo da ligação, resultando, neste caso, um íon
negativo e um íon positivo. Quando R e X não são idênticos, a
fissão pode ocorrer de um ou de outro dos modos, dependendo
se é R ou X que retém o par de elétrons. Ambos estes
processos são designados como fissão heterolítica, sendo o
resultado a formação de um par iônico. A formação de uma
ligação covalente pode ocorrer pelo processo inverso de
qualquer destes processos e, também, pelo ataque dos
radicais ou íons primeiramente formados a outras espécies.
Esses radicais ou pares iônicos formam intermediários
reativos numa grande variedade de reações orgânicas, como
se mostrará mais adiante. As reações envolvendo radicais
tendem a ocorrer na fase gasosa e em solução em solventes
não-polares, catalisadas pela luz ou pela adição de outros
radicais. As reações que envolvem pares iônicos ocorrem mais
rapidamente em solução em solventes, devido a maior
facilidade de separação de carga nessas condições, e
frequentemente devido a estabilização dos pares iônicos
resultantes através da solvatação. Muitos destes intermediários
iônicos podem ser considerados como transportando a sua
carga no átomo de carbono, embora o íon seja muitas vezes
estabilizado por deslocalização da carga, em maior ou menor
extensão, sobre outros átomos e carbono, ou átomos de
elementos diferentes.
Quando o átomo de carbono tem uma carga positiva, é
designado como íon carbônion (carbocátion), e quando é
carregado com uma carga negativa é um carbânion. Embora
tais íons possam formar-se e estar presente apenas em
concentração pequena, são de máxima importância no controle
das reações em que participam.
Estes três tipos, radicais, íons carbônion e
carbânions, de modo algum esgotam as possibilidades de
intermediários transientes em que o carbono é o centro ativo,

33
outros incluem as espécies elétron deficientes carbenos, R2C:,
e benzinos
FATORES QUE INFLUENCIAM A DISPONIBILIDADE
ELETRÔNICA.
Prever quaisquer fatores que influenciem a
disponibilidade relativa de elétrons (densidade eletrônica) em
ligações ou em átomos num composto deve ser considerado a
sua reatividade em relação a um reagente particular, uma
posição de alta densidade eletrônica será atacada com
dificuldade, por exemplo, por OH-, enquanto que uma posição
de baixa densidade eletrônica é provável que seja atacada com
facilidade e vice-versa por um reagente positivamente
carregado.
Efeitos indutivos
Numa ligação covalente simples entre átomos
diferentes, o par eletrônico que forma a ligação sigma nunca é
partilhado de modo absolutamente igual entre os dois átomos,
ele tende a ser atraído para o mais eletronegativo dos dois
átomos. Assim no cloreto de alquila, a densidade eletrônica é
maior, próximo do cloro do que do carbono.
C Cl
H
H
H
δδ+ -C Cl
H
H
H
Se o átomo de carbono ligado ao cloro estiver ele
próprio ligado a outros átomos de carbono, o efeito pode ser
transmitido mais longe.
CH3 CH2 CH2 CH2 Cl
O efeito da apropriação parcial de elétrons pelo átomo
de cloro da ligação carbono-cloro deixa o C1 ligeiramente
elétron-deficiente, ele tenta retificar esta situação, apropriando-

34
se, por sua vez, de um pouco mais do que a sua parte dos
elétrons da ligação sigma que o liga a C2, e assim por diante
ao longo da cadeia. O efeito de C1 em C2 é inferior ao de Cl
sobre C1, no entanto, a transmissão rapidamente se
desvanece numa cadeia saturada, sendo geralmente muito
reduzida para se notar além do C2.
A maior parte dos átomos e grupos ligados ao carbono
exerce estes efeitos indutivos na mesma direção que o cloro,
isto é, são retiradores de elétrons, por serem mais
eletronegativos do que o carbono, sendo a principal exceção os
grupos alquila e elétrons doadores. Embora o efeito seja
quantitativamente pequeno, é responsável pelo aumento de
basicidade que resulta quando um dos átomos de hidrogênio
do amoníaco é substituído por um grupo alquila, pela rápida
substituição do anel aromático no metilbenzeno do que no
benzeno propriamente dito.
Todos os efeitos indutivos são polarizações
permanentes no estado fundamental da molécula, e
manifestam-se nas suas propriedades físicas, por exemplo,
momento de dipolo.
Efeitos mesoméricos
Estes são essencialmente redistribuições de elétrons
que podem ocorrer em sistemas insaturados, especialmente
em sistemas conjugados, via seus orbitais π. Um exemplo é o
grupo carbonilo, cujas propriedades não são explicadas de
modo totalmente satisfatório pela formulação clássica, nem
pelo dipolo extremo obtido pela deslocalizaçao dos elétrons π.
C O
CH3
CH3
C+
O-
CH3
CH3
δδ+ -
C O
CH3
CH3

35
A estrutura real situa-se entre aquelas, um híbrido
resultante das formas canônicas.
Haverá também um efeito indutivo, mas muito menor do
que o efeito mesomérico, visto que os elétrons sigmas são
muito menos rapidamente deslocados do que os elétrons π.
Se o grupo C=O estiver conjugado com C=C, a
polarização acima pode ser transmitida mais além, via elétrons
π.
A deslocalização ocorre de tal modo que resulta num
átomo elétron deficiente em C3, bem como em C1 tal como
num composto carbonílico. A diferença entre esta transmissão
via sistema conjugado, e o efeito indutivo em sistema saturado,
é que aqui o efeito sofre muito menos atenuação após
transmissão, e a polaridade alterna.
A estabilização que pode resultar por deslocalização de
uma carga positiva ou negativa num ânion, via seus orbitais π,
pode ser um fator importante para tornar possível a formação
do íon, em primeiro lugar. A estabilização do ânion fenóxido,
por deslocalização da sua carga via orbitais π deslocalizados
do anel, é responsável pela acidez do fenol.
Uma deslocalização aparentemente semelhante pode
ocorrer no próprio fenol não dissociado, envolvendo um par de
elétrons não compartilhado no átomo de oxigênio, mas isso
envolve separação de cargas e será menos eficaz do que a
CH3 O CH3CH
+O
-CH3
CHO
δ-δ+
OH
+ H2O
O-
O O O
+ H3O+-
-
-

36
estabilização do íon fenóxido, que não envolve separação de
cargas.
OH OH OH OH
-
-
-
:+ + +
Os efeitos mesoméricos, tal como os indutivos, são
polarizações permanentes no estado fundamental de uma
molécula, e manifestam-se nas propriedades físicas dos
compostos em que ocorrem. A diferença essencial entre efeitos
indutivos e mesoméricos é que os primeiros ocorrem
essencialmente em grupos ou compostos saturados, e os
segundos em compostos insaturados e especialmente em
compostos conjugados. Os primeiros envolvem elétrons de
ligações sigma e os segundos os elétrons em ligações π. Os
efeitos indutivos são transmitidos apenas a curtas distâncias
em cadeias saturadas antes de desvanecerem, enquanto que
os efeitos mesoméricos podem ser transmitidos de uma
extremidade a outra da molécula bastante grande desde que
esteja presente conjugação, através da qual podem ser
transmitidos.
Efeitos variáveis com o tempo.
Os fatores variáveis com o tempo, por analogia com os
efeitos com os efeitos permanentes descritos anteriormente,
foram designados por efeitos indutoméricos e efeitos
eletroméricos. Quaisquer destes efeitos podem ser
considerados como polarizabilidades em vez de polaridades,
porque a distribuição dos elétrons reverte a do estado
fundamental da molécula atacada se o reagente for removido
sem que a reação possa ocorrer. Ou se o estado de transição,

37
uma vez atingido, se decompor para dar de novo os materiais
de partida.
Estes efeitos variáveis, com o tempo, sendo apenas
temporários, não se refletirão nas propriedades físicas dos
compostos. Muitas vezes é impossível distinguir
experimentalmente entre efeitos permanentes e efeitos
variáveis com o tempo, mas nunca é de mais acentuar que
apesar das dificuldades em distinguir que proporções de um
dado efeito são devidas a fatores permanentes e a fatores
variáveis com o tempo, de fato a aproximação de um reagente
pode ter um efeito no aumento de reatividade numa molécula
reagente, e assim promover a reação.
Hiperconjugação
A grandeza relativa do efeito indutivo de grupos alquilas
verifica-se seguir normalmente a ordem:
(CH3)3 – C– < (CH3)2 – CH– < CH3 – CH2 – < CH3–
Quando os grupos alquilas estão ligados a um sistema
insaturado, uma ligação dupla ou um grupo benzênico, verifica-
se que aquela ordem é perturbada e, no caso de alguns
sistemas conjugados, é de fato invertida. Parece assim que os
grupos alquilas são capazes, nessas circunstâncias, de dar
origem a um aumento de densidade eletrônica por um
mecanismo diferente do efeito indutivo. Este foi explicado como
ocorrendo através de uma extensão do efeito mesomérico ou
de conjugação, ocorrendo a deslocalização, como segue.
C CH2
H
H
H
C CH2-
H
H
H+
C H
H
H C
H
H C
H
H C
H
HH+
- -
-
H+
H+

38
Este efeito foi designado por hiperconjugação e tem sido
utilizado com sucesso para explicar vários fenômenos de
outros modos não relacionáveis. Deve acentuar-se que não se
está de fato a sugerir que um próton fica livre como mostrado
na figura acima, se deslocasse da sua posição original uma
das condições necessárias para a deslocalização ocorrer, seria
contrariada.
A inversão da ordem esperada (indutiva) de elétron
doação para CH3 > CH3CH2 > (CH3)2CH > (CH3)3C pode ser
explicada com base na hiperconjugação ser dependente da
presença de hidrogênio nos átomos de carbono alfa em relação
ao sistema insaturado. Esta está claramente num máximo com
CH3 e é não existente com (CH3)C, e daí a maior capacidade
elétron doadora dos grupos CH3 nestas condições.
C CH2
H
H
H
C CH2
H
H
CH3
C CH2
CH3
CH3
CH3
C CH2
CH3
CH3
CH3
A hiperconjugação poderia envolver ligações C–C, bem
como ligações C–H, e as diferenças em reatividade relativas
observadas numa série de compostos pode, de fato, resultar da
atuação de efeitos do solvente, bem como de efeitos
hiperconjugativos.
A hiperconjugação também tem sido usada para explicar
a maior estabilidade termodinâmica de alcenos em que a
ligação dupla não é terminal, comparada com compostos
isoméricos em que a dupla é terminal, onde os nove átomos de
hidrogênio alfa hiperconjugáveis, comparados com apenas
cinco átomos no isômero.
CH2CH3
CH3C CH3CH3
CH3

39
Isto resulta na formação preferencial de alcenos não
terminais, em reações que poderiam conduzir a ambos ou aos
seus isômeros terminais por introdução da ligação dupla, e a
isomerização bastante rápida do composto menos estável no
mais estável.
Efeitos estereoquímicos
Até aqui discutimos fatores que podem influenciar a
disponibilidade relativa de elétrons em ligações, ou em átomos
particulares, num composto e assim afetar a reatividade desse
composto. A atuação destes fatores pode, no entanto, ser
modificada ou até mesmo anulada pela influência de fatores
esteroquímicos; assim, a deslocalização efetiva via orbitais π só
pode ter lugar se os orbitais p ou π, nos átomos envolvidos na
deslocalizaçao, puderem tornar-se paralelos ou praticamente
paralelos. Se houver impedimento, não pode ter uma
sobreposição significativa, e a deslocalizaçao pode ser inibida.
Um exemplo é dado pela comparação entre o comportamento
da N,N-dimetilanilina e os seus derivados 2,6-dialquilados. O
grupo N,N-Me2, sendo elétron doador (devido ao par de
elétrons não compartilhado no nitrogênio interagir com os
orbitais π deslocalizados no núcleo), ativa o núcleo em relação
ao ataque pelo cátion diazônio PhN2+, em relação ao
acoplamento azóico, conduzindo a substituição na posição
para.
N
CH3CH3:
PhN2+
N+CH3 CH3
HNNPh
- H+
NCH3 CH3
NNPh
..
O derivado 2,6-dimetílico não sofre acoplamento nessas
condições, apesar do fato de os grupos metila que foram

40
introduzidos estarem afastado um do outro para que o seu
volume interfira diretamente com o ataque a posição para. O
não acoplamento nesta posição é devido aos dois grupos
metilas, nas posições orto a NMe2, interferindo
estereoquimicamente com os dois grupos metilas ligados ao
nitrogênio e impedindo assim que estes se situem no mesmo
plano do anel benzênico. Isto significa que os orbitais p no
nitrogênio e no átomo de carbono do anel ao qual se encontra
ligado são impedidos de se tornarem paralelos entre si, e a
sobreposição é assim inibida. A interação eletrônica com o anel
é impedida, e não ocorre transferência de carga.
..N
CH3 CH3
CH3CH3
O efeito estereoquímico mais comum é a obstrução
estereoquímica clássica, em que o volume dos grupos
influencia a reatividade diretamente num local do composto,
impedindo a aproximação de um reagente do centro reacional,
introduzindo uma obstrução no estado de transição e não
promovendo ou inibindo a disponibilidade eletrônica.
Deve acentuar-se que tal inibição estereoquímica é só
um caso extremo, e quaisquer fatores que perturbem ou inibam
uma orientação particular do reagente relativamente um ao
outro, não permitindo a sua aproximação suficientemente,
podem também afetar profundamente a velocidade de reações,
é o que se verifica frequentemente em reações em sistemas
biológicos.
TIPOS DE REAGENTES
Os grupos elétron doadores e elétron retiradores
possuem efeito de tornar um local de uma molécula rico ou

41
deficiente em elétrons. Isto vai claramente influenciar o tipo de
reagente com o qual o composto reagirá mais facilmente. Uma
espécie rica em elétrons, como o ânion fenóxido,
O-
O
-
tenderá ser mais facilmente atacada por cátions positivamente
carregados como C6H5N2+, um cátion diazônio, ou por outras
espécies que, embora não sendo de fato cátions, possuem um
átomo ou centro que é elétron deficiente, por exemplo, o átomo
de enxofre do trióxido de enxofre na sulfonação.
O S
O
O
δ
δ
δδ
-
-
-
+++
Tais reagentes, porque tendem a atacar o substrato
numa posição de densidade eletrônica elevada, são chamados
reagentes eletrofílicos ou simplesmente eletrófilos.
Inversamente, um centro elétron deficiente, como o
átomo de carbono no clorometano,
CH3 Clδ -δ+
será atacado mais facilmente por ânions (carregados
negativamente) tais como OH-, CN-, etc., ou por outras
espécies que, embora não sendo de fato ânions, possuem um
átomo ou centro que é rico em elétrons; por exemplo, o átomo
de nitrogênio no amoníaco ou em aminas, P3N ou R3N. Estes
reagentes tendem atacar o substrato numa posição de baixa
densidade eletrônica, isto é, onde o núcleo atômico está além
do seu comportamento normal, são chamados reagentes
nucleofílicos ou simplesmente nucleófilos.

42
Esta diferença de eletrófilo/nucleófilo pode ser
considerada como um caso especial da ideia ácido/base. A
definição clássica de ácidos e bases é que os primeiros são
doadores de prótons, e os segundos são aceitadores de
prótons. Esta ideia foi tornada mais geral por Lewis, que definiu
ácidos como compostos preparados para aceitar pares de
eletrons, e bases como substâncias capazes de fornecer esses
pares. Isto incluiria certo número de compostos não
anteriormente considerados como ácidos e bases, exemplo o
trifluoreto de boro, que atua como um ácido aceitando o par de
eletrons do nitrogênio da trimetilamina para formar o complexo
neutro, e é, portanto, chamado de um ácido de Lewis.
F3B + N(CH3)3: F3B N(CH3)3:- +
Os eletrófilos e os nucleófilos em reações orgânicas
podem ser considerados essencialmente como aceitadores e
doadores, respectivamente, de pares eletrônicos, de um para
outro átomo, mais frequentemente carbono. Os eletrófilos e os
nucleófilos podem, é claro, também relacionar-se com agentes
oxidantes e redutores, pois os últimos podem considerar-se
como aceitadores de elétrons e os primeiros como doadores de
elétrons. Alguns eletrófilos e nucleófilos mais comuns
encontram-se listados a seguir.
Exemplos de:
Eletrófilos
H+, H3O
+,
+NO2,
+NO, PhN 2
+, R
3C
+,
+SO3,
+CO2,
+BF3,
+AlCl3,
+ICl, Br 2, O3
Nucleófilos
H-, BH4
-, HSO3
-, HO
-, RS
-, CN
-, RCO2
-, RCC
-, (EtCO2)2CH
-, O:, N:, S:, MgBrR
-, LiR
-
Quando o reagente está marcado com um asterisco,
indica o átomo que aceita elétrons do substrato, ou doa
elétrons ao substrato. Não se pode fazer necessariamente uma
distinção clara entre o que constitui o reagente e qual o

43
substrato, porque embora NO2+, OH-, etc. sejam normalmente
considerados como reagentes, o carbânion ((EtCO2)2CH-) pode
ser o reagente ou substrato quando reage, por exemplo, com
um haleto de alquila. A reação do primeiro com o último é um
ataque nucleofílico, enquanto que a do segundo com o primeiro
é considerado como um ataque eletrofílico, mas qualquer que
seja o ponto de vista, numa reação, é encarado o reagente, a
sua natureza essencial não está por um momento sequer em
dúvida.
Deve-se recordar que as reações que envolvem radicais
como entidades reativas são também conhecidas. Estas são
muito menos susceptíveis a variações em densidade eletrônica
no substrato do que as reações que envolvem intermediários
polares, mas em geral são mais fortemente afetadas pela
adição de pequenas quantidades de substâncias que ou
libertam ou removam radicais. Serão consideradas mais
adiante.
TIPOS DE REAÇÕES
Há quatro tipos de reações que os compostos orgânicos
podem sofrer.
a) Reação de deslocamento (substituição); b) Reação de
adição; c) Reação de eliminação; d) Rearranjo.
Em (a) é a substituição no carbono que normalmente se
refere, mas o átomo substituído pode ser hidrogênio ou outro
grupo.
Na substituição eletrofílica geralmente o hidrogênio é
deslocado, sendo a substituição aromática clássica um bom
exemplo.
H
+-NO2
NO2
+ H+

44
Na substituição nucleofilica é frequentemente, um
átomo, que não é o hidrogênio, é deslocado, porém a
substituição nucleofílica do hidrogênio também pode ocorrer.
NC-
+ R Br CN R + Br-
A substituição induzida por radicais acontece também, e
ocorre, por exemplo, a halogenação de alcanos.
As reações de adição, também, podem ser eletrofílicas,
nucleofílicas ou radicalares, dependendo do tipo de espécie
que inicia o processo. A adição a ligações duplas carbono-
carbono simples é normalmente ou induzida por eletrófilos ou
por radical, exemplo, a adição de HBr, que pode ser iniciada
pelo ataque de ou H+ ou Br- a ligação dupla.
CH3
CH3
CH3
CH3
HBrCH3
CH3
CH3
CH3 H
Br
Pelo contrário, as reações de adição exibidas pela
adição dupla carbono oxigênio, em aldeídos e cetonas simples,
têm geralmente caráter nucleofílico, um exemplo é a formação,
catalisada por base, de cianidrinas em HCN líquido.
As reações de eliminação são essencialmente o inverso
das reações de adição. O tipo mais comum é a perda de
hidrogênio e de outro átomo ou grupo de átomos de carbono
adjacentes para dar alcenos.
Os rearranjos também podem ocorrer via intermediários
que são essencialmente cátions, ânions ou radicais, embora os
que envolvem íons carbônios, ou outras espécies elétron
deficientes, sejam de longe os mais comuns. Podem envolver
CH3
CH3
CH3
CH3 H
Br
- HBrCH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3 H
OH

45
um grande rearranjo do esqueleto carbonado de um composto,
como na conversão de 2,3-dimetilbutan-2,3-diol (pinacol) em
2,2-dimetilbutan-3-ona (pinacolona).
- H2OCH3
CH3
CH3
CH3OH OH
CH3CH3
CH3 O
Nestas transformações a verdadeira etapa do rearranjo
é, muitas vezes, seguida por uma substituição, adição ou
eliminação, antes que se obtenha um produto final estável.
1O ROTEIRO DE ESTUDO DIRIGIDO
1. Porque os DIENOS CONJUGADOS são mais estáveis que os dienos isolados? 2. Sugira uma explicação para as diferenças nos CALORES DE COMBUSTÃO apresentados abaixo:
COMPOSTO CALOR DE COMBUSTÃO (KJ/mol)
Ciclo-hexeno -120 Ciclo-hexadieno -232 Benzeno -208 3. Dê uma explicação para a mudança de coloração observada na série de α,ω-DIFENIL-POLIENOS, abaixo:
C6H5(CH=CH)nC6H5 COR n=1 incolor n=2 amarelo n=3 laranja n=4 vermelho 4. Defina e dê exemplos de: a) efeito indutivo d) efeito eletromérico b) efeito mesomérico e) efeito estérico c) efeito indutomérico f) hiperconjugação 5. Escreva as estruturas de ressonância para: a) fenol b) benzaldeído c) anilina d) nitrobenzeno

46
6. Pesquise sobre a isomerização abaixo:
O
Base
O
7. Justifique porque os ácidos “A” e “B” são difíceis de esterificar e o ”C” não.
Me
CMe
O OH
R3COOH
Me
CH2
Me
CO OH
A B C 8. Embora quase todas as espécies orgânicas estáveis tenham átomos de carbono tetravalente, sabe-se que existem espécies que possuem átomos de carbonos trivalentes. Os carbocátions são uma destas classes de compostos. a) Qual é a relação existente entre um carbocátion e um composto de boro trivalente como o BF3? b) Quantos elétrons de valência possuem o átomo de carbono carregando positivamente? c) Que tipo de hibridação deve possuir esse átomo de carbono? d) Qual é a geometria do carbocátion? 9. Um carbânion é uma espécie que tem um carbono trivalente carregado negativamente. a) Qual é relação existente entre um carbânion e um composto de nitrogênio trivalente como o NH3? b) Quantos elétrons de valência possuem o átomo de carbono carregado negativamente? c) Que tipo de hibridação deve possuir esse átomo de carbono? d) Qual e a geometria do carbânion? 10. Compostos aromáticos sofrem reações de substituição eletrofílica catalisada por ácidos de Lewis anidros. Considerando a piridina e o eletrófilo NO2
+ mostre qual carbono é o mais susceptível de ataque. Idem para o pirrol.

47

48
UNIDADE II – ENERGÉTICA, CINÉTICA E INVESTIGAÇÃO
DE MECANISMOS
ENERGÉTICA DE REAÇÕES
A conversão de reagentes em produtos, que constitui
uma reação orgânica, e “em que extensão ocorrerá à reação
para formar os produtos?” É o queremos saber. Os sistemas
tendem a deslocar-se em direção ao seu estado mais estável,
por isso podemos prever que quanto mais estáveis forem os
produtos, comparados com os reagentes, mais deslocado o
equilíbrio estará do lado dos produtos, quando maior diferença
na estabilidade (∆estabilidade), maior a conversão esperada em
produtos.
A simples variação de energia que ocorre quando se vai
de reagentes para produtos, e que pode ser facilmente medida
como o calor de reação ∆H*, não é uma medida adequada da
diferença de estabilidade entre eles, pois frequentemente se
verifica não existir correlação entre ∆H e a constante de
equilíbrio para a reação, k. Nas reações altamente exotérmicas
com pequenas constantes de equilíbrio (pouca conversão de
reagentes em produtos) e algumas reações com constante de
equilíbrio elevadas, que são endotérmicas (a entalpia dos
produtos é superior a entalpia dos reagentes), é nítido que
algum fator, além da entalpia, deve estar envolvido na
estabilidade relativa de espécies químicas.
Isso é um corolário da Segunda Lei da Termodinâmica
que se relaciona essencialmente com probabilidades e com a

49
tendência dos sistemas ordenados para se tornarem
desordenados, sendo a medida do grau de desordem de um
sistema dada pela sua entropia (S). Ao procurar a sua condição
mais estável, os sistemas tendem para um número de energia
(entalpia, H) e um máximo de entropia (desordem ou acaso),
pelo que uma medida da sua estabilidade relativa deve abarcar
um compromisso entre H e S, e é fornecido pela energia livre
de Gibbs (G) que é definida por:
G = H - TS
em que T é a temperatura absoluta. A variação de
energia livre durante uma reação a uma dada temperatura é
dada por
∆G = ∆H – T∆S,
e verifica-se que a variação de energia livre partindo
dos reagentes para os produtos, ∆G, relaciona-se com a
constante de equilíbrio K, para a reação, pela relação
∆G = 2,303RT logK,
isto é, quanto maior o decréscimo de energia livre,
partindo dos reagentes para produtos, maior será o valor de K,
e mais o equilíbrio estará deslocado a favor dos produtos. A
situação de energia livre mínima corresponde à posição de
equilíbrio para reagentes/ produtos. Para uma reação em que
não há variação de energia livre (∆G = 0), K = 1, corresponde a
50% de reagentes em produtos. Valores positivos crescentes
de ∆G implicam em valores fracionários menores de K,
correspondendo a uma conversão extremamente reduzida em
produtos, enquanto que valores negativos maiores de ∆G
implicam valores maiores de K. Assim um ∆G de -42 kJ
corresponde a uma constante de equilíbrio de 107, e a uma
conversão completa em produtos. O conhecimento das
energias livres de padrão de reagentes e de produtos, foram
determinadas para um grande número de compostos

50
orgânicos, permite-nos prever a extensão de conversão dos
reagentes.
O fator ∆H para a transformação pode ser equacionado
com a diferença de energias entre as ligações nos reagentes e
as ligações nos produtos, e um valor aproximado de ∆H para
uma reação pode, muitas vezes, ser previsto a partir de tabelas
de energias de ligação padrão, visto que foi a partir de valores
de ∆H que as energias médias de ligações foram calculadas.
O fator entropia não pode ser explicado de modo simples, mas efetivamente relaciona-se com o número de modos possíveis em que a energia total, pode ser compartilhada entre um conjunto de moléculas, e também o número de modos em que o quanto de energia de uma molécula pode ser compartilhado para efeitos translacionais, rotacionais e vibracionais, sendo o translacional o de maior magnitude. Assim para uma reação em que há um aumento de espécies moleculares ao passar-se de reagentes para produtos, é provável que haja um acréscimo apreciável de entropia devido ao ganho em liberdade translacional.
A → B + C
O termo -T∆S pode ser maior do que o termo +∆H de
uma reação endotérmica, conduzindo a um valor negativo para
∆G, e a um equilíbrio se encontra a favor dos produtos. De
qualquer modo, se a reação for exotérmica (∆H negativo), ∆G
será ainda mais negativo, e constante de equilíbrio, K, é maior.
Quando o número de espécies decresce ao ir dos reagentes
para os produtos, é provável que haja um decréscimo de
entropia.
∆G- = ∆H - (-)T∆S
Então, a não ser que a reação seja suficientemente
exotérmica (∆H negativo e muito grande) para contrabalancear,
∆G será positivo, e o equilíbrio irá se deslocar muito a favor dos
reagentes.

51
As reações de ciclização podem ter decréscimo de
entropia, embora não seja necessária uma variação de entropia
translacional, é imposta uma restrição à rotação em torno das
ligações simples carbono-carbono, e esta é essencialmente
livre no reagente de cadeia aberta, mas muito restringida no
produto cíclico. Esse termo de entropia rotacional é menor do
que o termo de entropia translacional envolvido em reações em
que o número de espécies participantes decresce ao
formarem-se produtos.
Um fato que se reflete na preferência por ligações
hidrogênio intramoleculares em vez de intermoleculares em
1,2- dióis.
Não se deve minimizar o fato de o termo de entropia
envolver temperatura (T∆S) enquanto que o mesmo não
sucede com o termo de entalpia (∆H), e que as suas
contribuições relativas para a variação de energia livre podem
ser acentuadamente diferentes para a mesma reação realizada
a temperaturas muito diferentes.
CINÉTICA DE REAÇÕES
Embora o valor negativo para ∆G seja uma condição
necessária para que uma reação ocorra para dado um conjunto
de condições, é necessária mais informação, já que o valor de -
∆G nada nos diz quão rápido os reagentes são convertidos em
produtos.
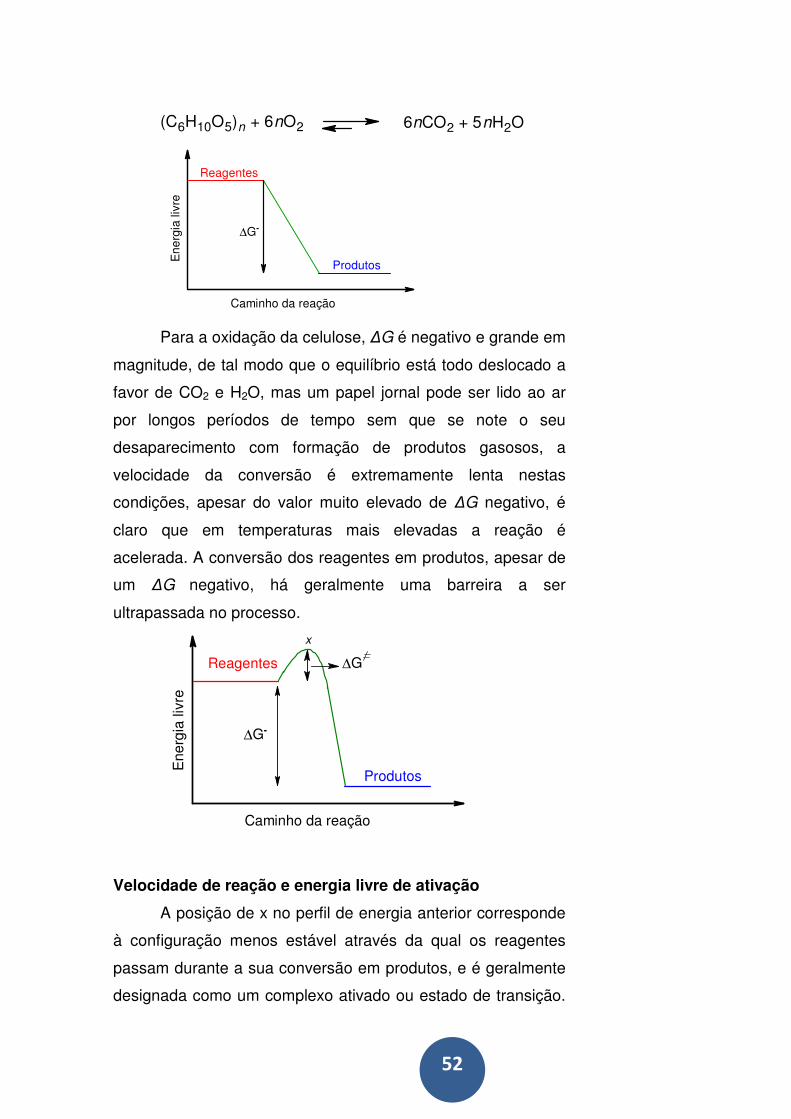
52
(C6H10O5)n + 6nO2 6nCO2 + 5nH2O
Ene
rgia
livr
eReagentes
Produtos
∆G-
Caminho da reação
Para a oxidação da celulose, ∆G é negativo e grande em
magnitude, de tal modo que o equilíbrio está todo deslocado a
favor de CO2 e H2O, mas um papel jornal pode ser lido ao ar
por longos períodos de tempo sem que se note o seu
desaparecimento com formação de produtos gasosos, a
velocidade da conversão é extremamente lenta nestas
condições, apesar do valor muito elevado de ∆G negativo, é
claro que em temperaturas mais elevadas a reação é
acelerada. A conversão dos reagentes em produtos, apesar de
um ∆G negativo, há geralmente uma barreira a ser
ultrapassada no processo.
Ene
rgia
livr
e
Reagentes
Produtos
∆G-
Caminho da reação
∆G
x
Velocidade de reação e energia livre de ativação
A posição de x no perfil de energia anterior corresponde
à configuração menos estável através da qual os reagentes
passam durante a sua conversão em produtos, e é geralmente
designada como um complexo ativado ou estado de transição.

53
Deve acentuar-se que este é apenas um estado altamente
instável, que é atingido durante um processo dinâmico, e não
uma espécie molecular, um intermediário, que pode ser
detectado ou até mesmo isolado. Um exemplo é o estado de
transição na hidrólise alcalina do bromometano, em que a
ligação HO–C forma-se ao mesmo tempo em que a ligação C–
Br está se quebrando, e os três átomos de hidrogênio ligados
ao carbono passam por uma configuração em que todos se
encontram no mesmo plano.
A altura da barreira energética na figura é chamada de
energia livre de ativação para a reação (quanto mais alta, mais
lenta a reação) e pode ser considerada como composta por
termos de entalpia e de entropia.
∆G*= ∆H* - T∆S*
O ∆H≠ corresponde à energia necessária para efetuar
estiramento ou mesmo a quebra de ligações que é um pré-
requisito essencial para que a reação aconteça. Assim, as
moléculas reagentes devem transportar com elas para
qualquer colisão um mínimo de energia para que a reação
aconteça; o aumento da velocidade de uma reação à medida
que a temperatura é aumentada é, de fato, devido à proporção
crescente de moléculas com uma energia acima daquele
mínimo com o aumento de temperatura.
A ordem de grandeza de Eact para uma reação se
calcula a partir de valores de k, constante de velocidade,
determinada experimentalmente a duas temperaturas

54
diferentes T1 e T2, utilizando a expressão de Arrhenius que
relaciona k com T, a temperatura absoluta, (K).
k = Ae-E/RT ou log10k = (Eativação/2,303RT) + log10A
onde R é a constante dos gases, e A é a constante para a
reação, independente da temperatura que está relacionada
com a proporção do número total de colisões entre moléculas
reagentes que resultam numa conversão eficaz em produtos. O
valor de Eativação pode então ser obtido fazendo-se um gráfico
de valores de log10k vs 1/T ou por conversão da equação
anterior em log10 k1/k2 = ((Eativação/2,303 R) x [(1/T1) – (1/T2)]) e
cálculo subsequente.
O termo ∆S#* relaciona-se com uma distribuição, é uma
medida da variação no grau de organização, ou
ordenadamente, de ambas as moléculas reagentes
propriamente ditas e da distribuição de energia dentro delas, ao
ir-se de reagentes para o estado de transição; ∆S≠ está
relacionado com o fator A na equação de Arrhenius. Se a
formação do estado de transição requer a imposição de um alto
grau de organização no modo pelo qual as moléculas
reagentes se devem aproximar uma da outra, e também da
concentração da sua energia em ligações particulares de modo
a permitir finalmente a sua quebra, ao ser atingido o estado de
transição há um decréscimo de entropia, e a possibilidade da
sua formação é menor.
Cinética e etapa determinante da velocidade
Experimentalmente, a medição de velocidades de
reações consiste em investigar a que velocidade os reagentes
desaparecem e/ou os produtos se formam a uma temperatura
particular, e procurar relacionar a velocidade com a
concentração de um ou de todos os reagentes. A reação pode
ser seguida por uma variedade de métodos, diretamente por

55
remoção de alíquotas seguida pela sua determinação
titrimétrica, ou indiretamente por observação de variações
colorimétricas, conductimétricas, espectroscópicas, etc.
Qualquer que seja o método usado, a etapa crucial
normalmente envolve a tentativa de correlacionar os dados
cinéticos não tratados com possíveis funções variáveis de
concentração, quer graficamente, que por cálculo, até que um
resultado que se ajuste razoavelmente seja obtido. Assim, para
a reação,
CH3Br + -OH � CH3OH + Br-
a equação de velocidade é,
Velocidade = k[CH3Br][ -OH]
onde, k é chamado a constante de velocidade para a reação.
A reação de segunda ordem; de primeira ordem em relação ao
CH3Br e ao OH-.
Tal coincidência de estequeometria e lei de velocidade é
bastante rara, a primeira não é geralmente de todo um guia
para a segunda, que só pode ser determinada por
experimentos em laboratórios. A equação da reação de
bromação catalisada por base da propanona é
CH3COCH3 + Br2, (-OH)catalisador � CH3COCH2Br + HBr,
e a equação da velocidade, onde o bromo não aparece,
Velocidade = k[CH3COCH3][ -OH]
É claro que o bromo tem que estar envolvido em alguma
etapa da reação global uma vez que é incorporado no produto
final, mas obviamente não pode estar envolvido em uma etapa
determinante da velocidade de reação. Poucas reações
orgânicas são processos numa só etapa, e isto é evidenciado
na reação de formação de hexamina, em que a probabilidade
de colisão simultânea de seis moléculas de CH2O e de quatro
de NH3 numa colisão de dez corpos é efetivamente nula.
6CH2O + 4NH3 � C6H6N4 + 6H2O
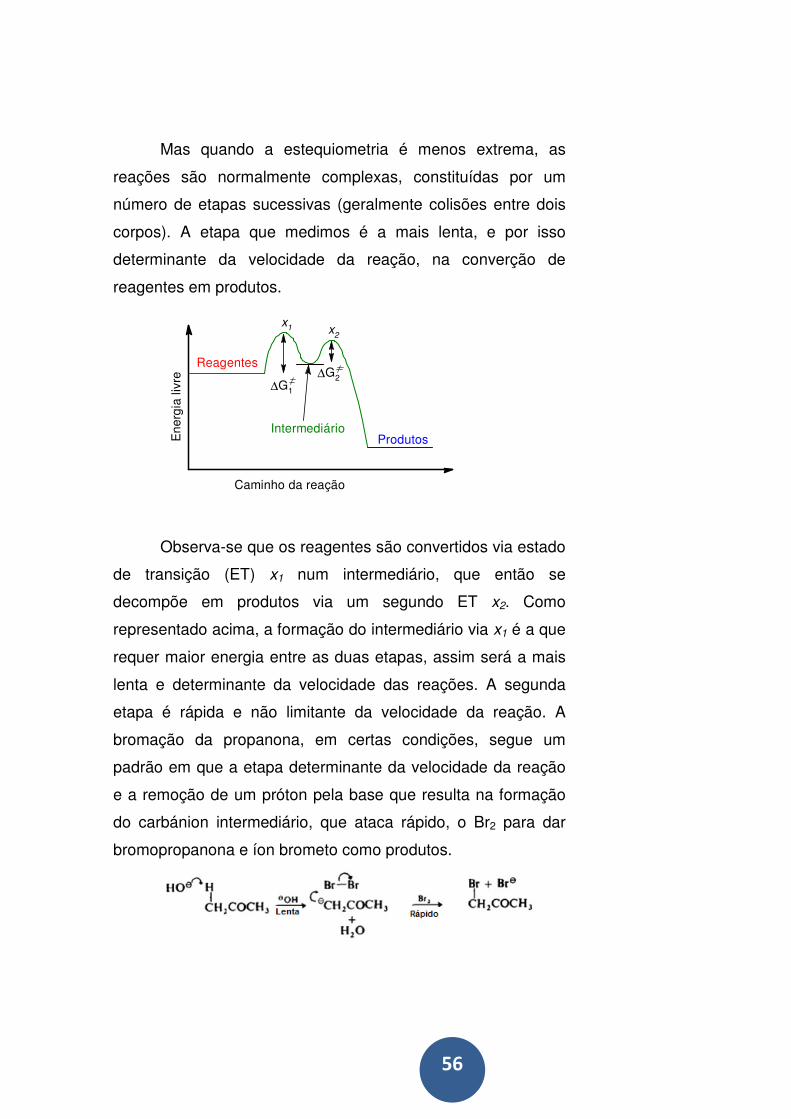
56
Mas quando a estequiometria é menos extrema, as
reações são normalmente complexas, constituídas por um
número de etapas sucessivas (geralmente colisões entre dois
corpos). A etapa que medimos é a mais lenta, e por isso
determinante da velocidade da reação, na converção de
reagentes em produtos.
Ene
rgia
livr
e
Reagentes
Produtos
Caminho da reação
Intermediário
∆G1
∆G2
x1 x
2
Observa-se que os reagentes são convertidos via estado
de transição (ET) x1 num intermediário, que então se
decompõe em produtos via um segundo ET x2. Como
representado acima, a formação do intermediário via x1 é a que
requer maior energia entre as duas etapas, assim será a mais
lenta e determinante da velocidade das reações. A segunda
etapa é rápida e não limitante da velocidade da reação. A
bromação da propanona, em certas condições, segue um
padrão em que a etapa determinante da velocidade da reação
e a remoção de um próton pela base que resulta na formação
do carbánion intermediário, que ataca rápido, o Br2 para dar
bromopropanona e íon brometo como produtos.

57
Deve acentuar-se que embora esta explicação seja uma
dedução razoável da equação de velocidade estabelecida
experimentalmente, esta última não se pode dizer que prova a
primeira. A equação da velocidade determinada
experimentalmente da informação acerca das espécies que
estão envolvidas na etapa determinante da velocidade de uma
reação, e esta sendo incluida, na verdade a equação de
velocidade específica a composição, mas não, a estrutura do
estado de transição para a etapa determinante da velocidade.
Esta etapa não fornece informação direta acerca de
intermediários nem das espécies que estão envolvidas nos
processos rápidos, não determinantes da velocidade.
Ao considerar-se o efeito que uma alteração nas
condições do solvente ou na estrutura do material de partida
pode ter sobre a velocidade de uma reação, precisamos saber
qual o efeito que tais alterações terão sobre a estabilidade do
estado de transição, quaisquer fatores que sirvam para
estabilizar, conduzirão à mais rápida formação, e o inverso
igualmente se aplicará. Raramente é possível obter tal
informação detalhada acerca destes ET de energia elevada,
geralmente podemos tomar os intermediários relevantes como
modelos para eles e ver os efeitos que tais alterações exerçam
sobre estes. Tal modelo é razoável, o intermediário
transientemente formado na da bromação da propanona
assemelha-se muito, em termos de nível de energia livre, ao
ET do que seria o reagente. Em substituições eletrofílicas
aromáticas são utilizados complexos σ como modelos para os
ET que são os seus precursores imediatos.
O efeito de um catalisador é o de aumentar a velocidade
de uma reação, através da diminuição da energia de ativação,
na formação de um novo intermediário mais estável (de menor
energia).

58
Ene
rgia
livr
eReagentes
Produtos
Caminho da reação
E.T. para a reação não catalisada
E.T. para a etapa limitante da velocidade da reação catalisada
Assim a velocidade de hidratação de um alceno,
diretamente com água,
é em geral extremamente lenta, mas pode ser acelerada pela
presença de um catalisador ácido, que efetua a protonação
inicial do alceno para dar um íon (carbônion) positivamente
carregado por uma molécula de água atuando como nucleófilo,
e a liberação de um próton que é capaz de atuar novamente
como um catalisador.
Controle cinético versus controle termodinâmico
Quando um material de partida pode ser convertido em
dois ou mais produtos alternativos, como no ataque eletrofílico
há uma espécie aromática que já possui um substituinte, as
proporções em que os produtos alternativos se formam são
frequentemente determinadas pela sua velocidade de formação
relativa, quanto mais rapidamente se formar um produto, maior
proporção desse produto se encontrará na mistura final, é o
que se chama de controle cinético. No entanto, não é sempre

59
isto o que se observa, porque se uma ou mais das reações
alternativas for reversível, ou se os produtos forem facilmente
interconvertíveis nas condições reacionais, a composição da
mistura final de produtos pode ser ditada não pelas velocidades
de formação relativas dos diferentes produtos, mas sim pelas
suas estabilidades termodinâmicas relativas no sistema
reacional, esta então está em presença de controle
termodinâmico ou de equilíbrio. Na nitração do metilbenzeno, a
reação é cineticamente controlada, enquanto que na alquilação
de Friedel-Crafts da mesma espécie, a reação é controlada em
geral pela termodinamica. A forma de controle que ocorre
também pode ser influenciada pelas condições reacionais,
assim a sulfonação do naftaleno com H2SO4 concentrado a 80°
é cineticamente controlada, enquanto que a 160° é
termodinamicamente controlada.
Investigação de mecanismos de reações
Raramente, é possível fornecer informação completa,
estrutural, energética, regioquímica e estereoquímica, sobre
uma reação química, ou seja, é difícil comprovar que um
mecanismo reacional está completamente correto. No entanto,
e possível reunir um grande número de informações, sobre a
reação e demonstrar teoricamente que, dentre as diversas
alternativas, um determinado mecanismo é viável, e outros
não. Na investigação dos mecanismos de reações, vários
fatores são considerados.
A natureza dos produtos
A informação mais fundamental acerca de uma reação é
fornecida pela estrutura dos produtos que se formam durante o
seu curso, e a relação desta informação com a estrutura do
material de partida. Como é frequentemente o caso com

60
reações orgânicas, se obtém mais do que um produto, então
em geral é necessário conhecer também as proporções
relativas em que os produtos são obtidos com exatidão através
de técnicas de cromatografia e por métodos espectroscópicos,
entre outros, pode ocorrer por controle cinético ou
termodinâmico.
A informação sobre os produtos de uma reação pode ser
informativa quando um deles é totalmente desconhecido.
Assim, a reação de 4-metilclorobenzeno (p-clorotolueno) com
íons amina, -NH2, em amoníaco líquido conduz não só ao
produto previsto, 4-metilfenilamina (p-toluidina), mas também
ao produto bastante inesperado 3-metifenilamina (m-toluidina),
que na realidade é o produto majoritário.
A 3-metifenilamina não pode ser obtida a partir do 4-
metil-clorobenzeno por um simples processo de substituição e
ou se deve formar a partir de 4-metil-clorobenzeno via um
percurso diferente do da 4-metilfenilamina ou, se os dois
produtos se formam através de algum intermidiário comum,
então a 4-metilfenilamina também não pode formar-se por uma
substituição direta.
Dados Cinéticos
A maior quantidade de informação sobre percursos
reacionais provém de estudos cinéticos, mas a interpretação
dos dados cinéticos em termos mecanísticos não é sempre tão
simples como poderia parecer à primeira vista. Assim, as
espécies reagentes efetivas, cuja concentração na verdade
determina a velocidade da reação, podem diferir das espécies
que foram colocadas inicialmente na mistura reacional, e cuja

61
concentração variável estamos de fato a procurar medir. Deste
modo, na nitração aromática a espécie atacante efetiva é
geralmente +NO2, mas é HNO3 que colocamos na mistura
reacional e cuja concentração variável que medimos; a relação
entre as duas pode ser complexa e assim, portanto, também a
relação entre a velocidade da reação e [HNO3]. Apesar de a
reação essencial ser simples, pode não ser fácil deduzir isso a
partir das quantidades que podemos rapidamente medir.
E assim, também, se a hidrólise numa solução aquosa
do haleto de alquilo, RX, se verifica, segui a equação de
velocidade.
Velocidade = k1[RX]
Não é necessariamente seguro concluir que a etapa
determinante da velocidade não envolve a participação de
água, simplesmente com base em que [H2O] não figura na
equação de velocidade; porque se a água está a ser usada
como solvente, estará presente em grande excesso, e a sua
concentração permanecerá virtualmente inalterada quer
participe ou não na etapa determinante da velocidade. O
assunto poderia talvez ser esclarecido se fizesse a hidrólise em
outro solvente, como o HCO2H e usando uma concentração
muito menor de água como um nucleófilo potencial. Pode então
verificar-se que a hidrólise segue a equação de velocidade.
Velocidade = k2[RX][H2O]
Mas o mecanismo real de hidrólise pode muito bem ter
mudado ao se ter alterado o solvente, de tal modo que não
ficamos a saber mais sobre o que de fato se passou na solução
aquosa original.
A grande maioria das reações orgânicas é realizada em
solução, e pequenas alterações no solvente usado podem
produzir as mais profundas alterações nas velocidades e
mecanismos reacionais. Isto sucede particularmente quando

62
intermediários polares, por exemplo, íons carbónio ou
carbânions como constituintes de pares iônicos, estão
envolvidos, pois estas espécies normalmente transportam um
invólucro de moléculas de solvente em torno de si, afetando
enormemente sua estabilidade, e é fortemente influenciado
pela composição e natureza do solvente utilizado,
particularmente a sua polaridade e a capacidade de
solvatação. Pelo contrário, reações que envolvem radicais são
muito menos influenciadas pela natureza do solvente, mas são
grandemente influenciadas pela adição de fontes radicalares
como peróxidos ou absorventes de radicais como quinonas, ou
pela luz que pode iniciar a reação através da produção de
radicais por ativação fotoquímica,
Br2 hv
Br . .Br.
O fato de se verificar que uma reação ocorre
inesperadamente mais rápida ou mais lentamente do que
reações aparentemente semelhantes, em condições
comparáveis, e para compostos de estrutura relacionada,
sugere que está a operar um mecanismo diferente, ou
modificado, do mecanismo geral que de outro modo se
presumiria para essa série de compostos. Assim as
velocidades de hidrólise observadas para a hidrólise dos
clorometanos com bases fortes, em condições comparáveis,
variam como se segue
CH3CL >> CH2Cl2 << CHCl3 >> CCl4
Claramente, sugerindo que o triclorometano sofre hidrólise de
uma maneira diferente dos outros compostos.
A utilização de isótopos
Um assunto que muitas vezes intriga é saber se uma
dada ligação sofreu ou não ruptura, numa etapa ou, incluindo o

63
passo determinante da velocidade de uma reação, os dados
cinéticos simples não nos podem dar estas informações, e por
isso é necessário recorrer técnicas mais abrangentes. Se, por
exemplo, a ligação em causa for C–H, a equação pode ser
resolvida comparando a velocidade de reação, nas mesmas
condições, do composto em que estamos interessados e do
seu análogo em que aquela ligação foi substituída por uma
ligação C–D. As duas ligações terão a mesma natureza
química, uma vez que estão envolvidos isótopos do mesmo
elemento, mas as suas frequências de vibração, e as suas
energias de dissociação, serão ligeiramente diferentes porque
estão envolvidos átomos de massa diferentes, quanto maior for
a massa, mais forte será a ligação. Esta diferença em força de
ligações em condições comparáveis, a ligação mais forte C–D
será quebrada mais lentamente do que a ligação C–H, cálculos
da mecânica quântica sugerem uma diferença de velocidade
máxima, kH/kD, de ≈ 7 a 25°.
Também se podem usar isótopos para resolver
mecanísticos que não são cinéticos. Na hidrólise de ésteres,
para dar um ácido e um álcool, em teoria, ocorre pela quebra
em (a) quebra alquilo/oxigênio, ou em (b) quebra acila/oxigênio:
Se a reação for realizada em água enriquecida no
isótopo mais pesado de oxigênio 18O, conduzirá a um álcool
que é enriquecido em 18O e um ácido que o não é, enquanto
(b) conduzirá a um ácido enriquecido em 18O e a um álcool
normal. Verifica-se que a maior parte dos ésteres simples dá
origem a um ácido enriquecido em 18O, indicando que a
hidrólise, nestas condições, ocorre via (b) ruptura

64
acila/oxigênio. Deve acentuar-se que estes resultados só são
válidos desde que nem o ácido nem o álcool, uma vez
formados, possam eles próprios trocar o seu oxigênio com
água enriquecida e 18O, como de fato se demonstrou ser o
caso.
Uma grande variedade de outros marcadores isotópicos, 3H (ou T),13C, 14C, 15N, 32P, 35S, 37Cl, 131I, etc., têm sido também
utilizados para fornecer informação mecanística importante; os
isótopos do carbono são particularmente úteis uma vez que
este elemento ocorre em todos os compostos orgânicos. As
maiores dificuldades envolvidas nos estudos com marcação
isotópica são: (a) síntese seletiva, de modo que o marcador
isotópico seja introduzido numa posição (ou posições)
conhecida no composto a testar; (b) degradação seletiva do
produto da reação para estabelecer a localização do marcador;
e (c) acompanhamento do conteúdo em marcador isotópico por
medidas de radioatividade (3H, 14C, 32P, 35S, etc.), de
espectroscopia de massa (D, 13C, 15N, 18O, etc.) ou outros
métodos.
O estudo de intermediários
Entre as provas mais concretas que podem ser obtidas
acerca do mecanismo de uma reação, encontra-se a fornecida
pelo isolamento efetivo de um ou mais intermediários da
mistura reacional.
É muito mais comum não isolar quaisquer
intermediários, mas isto não significa que nenhum se forme. A
sua ocorrência pode, muitas vezes, ser inferida de
determinações físicas e espectroscópicas, feitas sobre o
sistema. Assim na formação de oximas a partir de um número
de compostos carbonílicos por reação com hidroxilamina.

65
A banda de absorção no infravermelho característica de
C=O (1780 – 1760 cm-1) no material de partida desaparece
rapidamente e pode ter desaparecido por completo antes que a
banda característica de C=N (1690 – 1635 cm-1) no produto
comece aparecer. Claro que deve formar um intermediário, e
outras provas sugerem que é a carbinolamina que se forma
rapidamente e depois decompõe lentamente para dar os
produtos, a oxima e água.
Quando não temos razão para suspeitar do
envolvimento de uma espécie particular como um intermediário
instável no percurso de uma reação, pode ser possível
confirmar as nossas suspeitas, introduzindo na mistura
reacional uma espécie reativa que se preveja e reaja com
particular rapidez com o intermediário postulado. Pode então
ser possível fazer divergir o intermediário instável do principal
percurso reacional capturando-o e isolar uma espécie estável
no qual ele é incorporado.
O estudo dos intermediários fornece uma ou mais pistas
que auxiliam a definir o caminho de uma reação, e os próprios
intermediários podem fornecer provas acerca dos estados de
transição, os quais são tomados como modelos.
Critérios estereoquímicos
A estereoquímica de uma reação pode fornecer
esclarecimentos sobre o mecanismo sugerido. Assim, a
bromação catalisada por base da PhCOCHMeEt, opticamente
ativa, forma um produto racêmico opticamente inativo, indica
que a reação

66
deve ocorrer através de um intermediário planar, que pode
sofrer ataque de ambos os lados, conduzindo a iguais
quantidades das duas formas do produto que são imagens num
espelho uma da outra.
A adição do ciclopenteno ao bromo em condições
polares da o trans dibrometo, indica que o mecanismo da
reação não pode ser adição numa só etapa da molécula de
bromo à dupla, porque isto conduziria ao dibrometo cis.
A adição deve ser pelo menos um processo em duas
etapas. Reações como estas, que ocorrem para dar
essencialmente um dos estereoisômeros entre as duas
alternativas possíveis, são estereosseletivas.
O grau de certeza com que um mecanismo de uma
reação é sugerido não é determinado apenas por explicar os
fatos conhecidos, e sim pelo êxito com que poderá predizer
uma mudança de velocidade, ou até da natureza de produtos
formados, quando se altera as condições reacionais ou a
estrutura dos reagentes.
20 ROTEIRO DE ESTUDO DIRIGIDO
1. Por que existem reações exotérmicas que apresentam pequenas constantes de equilíbrio e reações endotérmicas que apresentam constantes de equilíbrio elevadas?
2. Qual o valor de K, numa reação onde o ∆G0 é igual a zero, sabendo-se que: ∆G0 = -2,303 RTlogK. O que significa isso, em termos percentuais de transformação de reagentes em produtos?

67
3. Qual a correlação entre energia livre de ativação e velocidade de reação? Escreva a equação de Arrhenius, que correlaciona K (constante de velocidade) com T (temperatura).
4. Faça o gráfico para o perfil energético da halogenação de cetonas, em meio básico.
5. Qual o efeito de um catalisador?
6. O que são produto cinético e produto
termodinâmico de uma reação? 7. Cloreto de t-butila reage com o íon hidróxido de acordo com a seguinte reação: Cloreto de t-butila álcool t-butila Acredita-se que esta reação procede via o seguinte mecanismo: A ordem das constantes de velocidade é: K3>K2>K1>>K4 (a) Construa um diagrama de energia para a reação. (b) A primeira etapa desta reação é endotérmica ou exotérmica? (c) A reação total é endotérmica ou exotérmica? (d) Qual a etapa determinante da reação? 8. Considere a seguinte sequência de reações em que B é um intermediário. Mostre os perfis de energia para cada uma das possíveis relações entre as constantes de velocidade indicada. A parte da reação C é insignificante. (a), K1 e K2 grande; K3 pequeno; (b) K1 grande, K2 e K3 grande, mas K2> K3; (c) K1 e K3 grande; K2 pequeno; (d) pequenos K1, K2 e K3 grande, mas K3> K2; Identificar e determinar a lei de velocidade do estado de transição para cada um dos quatro casos.
(2) (CH3)3OHOH-+(CH3)3+
(1) Cl-+(CH3)3+
OH-+(CH3)3ClK1
K2
K3
K4
AK1
K2
B
B CK3
+ OH-k
(CH3)3OH + Cl-(CH3)3Cl

68

69
UNIDADE III. ÁCIDOS E BASES
Bronsted definiu ácidos como substâncias que perdem
prótons (doadoras de prótons), enquanto que as bases são
aceitadoras de prótons. A primeira ionização do ácido sulfúrico
em solução aquosa, onde a água atua como uma base
aceitando um próton, é convertida no seu ácido conjugado,
H30+, enquanto que o ácido H2SO4, doando um próton, é
convertido na sua base conjugada, HSO4-.
H3O+
Base Ácido conjugado conjugada
Base
+ H2O: HSO4-H2SO4 +
Ácido
Lewis, definiu ácidos como moléculas ou íons capazes
de aceitar pares de elétrons e bases como moléculas ou íons
que doam pares de elétrons disponíveis por compartilhamento.
Exemplo:
(CH3)3N: + BF3 (CH3)3N : BF3-+
ÁCIDOS
Força ácida - pKa
A força de um ácido, HA, em água, e a extensão em que
é dissociado pode ser determinada, considerando-se o
equilíbrio.
H2O + HA H3O+ + A-
A constante de equilíbrio em água é dada por:
[ ][ ][ ]HA
AOHK
a
−+
≈ 3
A concentração da [H2O] está incorporada no Ka ,
porque a sua concentração não varia signficativamente. Deve
acentuar-se que Ka, a constante de acidez do ácido em água,
é apenas aproximada (como acima), se usar concentrações em

70
vez de atividades, o que seria mais correto; é uma premissa
razoável, no entanto, desde que a solução seja bastante
diluída. A constante de acidez é influenciada pelo solvente em
que o ácido é dissolvido e por outros fatores. Para se evitar
escrever potências negativas de 10, o Ka é geralmente
convertido em pKa (pKa = -log10 Ka); assim enquanto que o Ka
para o ácido etanóico em água a 25° é 1,79 x 10-5, o pKa =
4,76. Quanto menor for o valor numérico de pKa, mais forte
será o ácido a que se refere.
Os ácidos muito fracos, aqueles que o pKa é maior que
16, não serão detectáveis como ácidos em água, uma vez que
a [H30+] que produzem nesse meio é inferior à produzida pela
autólise da própria água.
H2O + H2O H3O+
+ OH-
Então o (pKa) não pode ser medido em água. Quando os
ácidos são fortes (pKa baixo), estarão totalmente ionizadas em
água, e assim todos parecem ter a mesma força. Exemplo:
HCl, HNO3, HClO4, etc. Isto é designado como efeito nivelador
da água.
O intervalo de medida de pKa comparativo pode ser
estendido, se no primeiro caso, fornecer uma base mais forte, e
no segundo caso, uma base mais fraca do que a água como
solvente. Efetuando as determinações numa variedade de
solventes de basicidade crescente, é possível fazer as
determinações de forças ácidas até em ácidos tão fracos como
o metano.
A origem da acidez em compostos orgânicos
Entre os fatores que podem influenciar a acidez de um
composto orgânico, HA, encontram-se: (a) A força da ligação
H-A; (b) A eletronegatividade de A; (c) Fatores que estabilizam
A- em relação a HA; (c) A natureza do solvente.

71
Comparando-se esses fatores, evidência –se que (a)
não é normalmente um fator limitante, mas o efeito (b) é
refletido no fato de o pKa do metanol, CH3O-H, ser 16,
enquanto que o do metano, CH3-H é 43, sendo o oxigênio
consideravelmente mais eletronegativo que o carbono. Em
contraste, o pKa do ácido metanóico é 3,77. Isto deve-se em
parte ao grupo carbonilo elétron retirador, que aumenta a
afinidade eletrônica do átomo de oxigênio ao qual está ligado o
hidrogênio, mas muito mais importante é (c), a estabilização do
ânion metanoato resultante, comparada com a molécula de
ácido metanóico não dissociada.
No ânion metanoato a deslocalização é eficaz, e há
estabilização, envolvendo duas estruturas canônicas de
energia idêntica, e embora possa haver deslocalização também
na molécula do ácido metanóico, esta envolve separação de
carga e será menos eficaz como influência estabilizadora. O
efeito dessa estabilização dificulta a recombinação do próton
com o ânion metanoato, o equilíbrio é deslocado para a direita,
e o ácido metanóico é, pelos padrões orgânicos, um ácido
moderadamente forte.
Com alcoóis, não existe um fator deste tipo para
estabilizar o ânion alcóxido (RO-), relativamente os alcoóis são
muito menos ácidos do que os ácidos carboxílicos. Com os
fenóis, há estabilização relativa ao ánion, por deslocalização da
sua carga negativa através de interação com os orbitais π do
anel aromático.

72
O-
O O O
-
-
-
Também ocorre deslocalização na molécula de fenol não
dissociada, mas, envolvendo separação de cargas, o que é
menos eficaz do que no ânion. Verifica- se que os fenóis são
ácidos mais fortes do que os alcoóis (o pKa do fenol é 9,95),
mas consideravelmente mais fracos do que os ácidos
carboxílicos. Isto é devido a deslocalização da carga negativa,
no ânion carboxilato, envolver estruturas de mesmo teor
energético, e dos centros envolvidos, dois são átomos de
oxigênio muito eletronegativos, enquanto que no ânion
fenóxido as estruturas envolvendo carga negativa nos átomos
de carbonos do anel apresentam maior teor energético do que
aquela em que a carga se encontra no oxigênio e dos centros
envolvidos. A estabilização relativa do ânion, relativamente à
molécula não dissociada, é provavelmente menos eficaz com
um fenol do que com um ácido carboxílico, conduzindo a mais
baixa acidez relativa do primeiro.
A influência do solvente
Considerando a água como solvente ionizante, para
compostos orgânicos, tem uma desvantagem, alguns desses
compostos não são solúveis em água na forma não ionizada. A
água é um solvente ionizante eficaz devido (a) à sua elevada
constante dielétrica e (b) à sua capacidade de solvatar íons. A
primeira propriedade justifica que quanto maior a constante
dielétrica de um solvente, menor é a energia eletrostática de
qualquer par de íons nele presente, assim quanto mais
facilmente esses íons se formam, mais estáveis são em
solução, e menos facilmente se recombinam uns com os
outros.

73
Os íons em solução polarizam fortemente as moléculas
de solventes mais próximas, constituindo à sua volta um
envelope de moléculas de solvente, quanto maior a extensão
de formação desse, maior a estabilização do íon, dispersando
ou deslocalizando a sua carga. A eficácia peculiar da água,
como um meio solvatante de íons, resulta do fato de que a
água é polarizada com extrema facilidade e por ser de tamanho
pequeno, pode solvatar, e assim estabilizar, quer cátions ou
ânions. O efeito é acentuado com ânions, pois pode ocorrer
uma solvatação do tipo ligação hidrogênio, e esta não pode em
geral ocorrer com cátions, mas no caso particular de ácidos, o
cátion inicial, H+, também pode solvatar-se através da
formação de ligações hidrogênio com as moléculas de água.
Os alcoóis, desde que não sejam muito volumosos,
como o MeOH, compartilham de algumas das propriedades da
água e, por exemplo, verifica- se que o HCl também é um ácido
forte em metanol. No entanto, não se deve esquecer que o
requisito principal do solvente é que deve ser capaz de atuar
como uma base, quanto mais fraca a base, menor será a
dissociação do ácido.
Ácidos alifáticos simples
Quando ocorre a substituição do átomo de hidrogênio
não hidroxílico do ácido metanóico por um grupo alquila, o
ácido que se forma é mais fraco, devido o efeito indutivo
positivo do grupo alquila reduzir a afinidade eletrônica residual
do átomo de oxigênio pelo hidrogênio ácido, diminuindo a força
do ácido. No ânion com o grupo alquila substituinte, a
disponibilidade eletrônica acrescida no oxigênio serve para
H Y + nH2O O+
H
H
H+Y
H OH
H
O
H
OHHOH
H
-

74
promover a sua recombinação com um próton, por comparação
com o sistema ânion metanoato/ácido metanóico.
CH3 C
O
O
-
H C
O
O
-
Ânion Acetato
Ânion Metanoato
O equilíbrio é deslocado para a esquerda em
comparação com o do ácido metanóico/ânion metanoato, e
verifica- se que o pKa do ácido etanóico é de 4,76 comparado
com o 3,77 para o ácido metanóico. No entanto, a alteração
global na estrutura efetuada numa molécula tão pequena como
o ácido metanóico por substituição de H por CH3 torna
duvidoso que um argumento tão simplista seja realmente
válido, pode muito bem acontecer que as capacidades de
solvatação relativas nos dois casos sejam afetadas pelas
formas diferentes, bem como pela distribuição de carga
relativa, nas duas pequenas moléculas.
O valor da constante de acidez, Ka, de um ácido está
relacionado com a variação de energia livre padrão para a
ionização, ∆G, pela relação,
- ∆G = 2,303RT logKa
e que ∆G- inclui tanto um termo de entalpia como de entropia.
∆G = ∆H -T ∆S
Os valores surpreendentemente baixos de ∆H para os
ácidos etanoico e metanoico quase certamente derivam da
energia requerida para a dissociação da ligação O–H, nos
ácidos carboxílicos não dissociados, ser cancelada pela que é
liberada na solvatação dos íons resultantes.
Os diferentes valores de ∆G, diferentes Ka, para os dois
ácidos resultam dos diferentes valores dos dois termos de
entropia (∆S). Há duas espécies de cada lado do equilíbrio, e
as diferenças em entropia translacional por dissociação serão,
portanto, pequenas. No entanto, as duas espécies são

75
moléculas neutras de um lado do equilíbrio, e íons do outro. O
principal fator que contribui para o ∆S são as camadas de
solvatação de moléculas de água que rodeiam RCO2- e H30
+, e
a restrição, em termos de aumento de ordenação, que é assim
imposta às moléculas de água como solvente, o aumento de
ordenação não é tão grande como seria de esperar já que há
bastante ordenação na própria água líquida. A diferença de
força entre o ácido metanoico e etanoico está assim
relacionada com a solvatação diferencial dos seus ânions.
A substituição de grupos alquila ao ácido etanoico tem
menor efeito do que a primeira substituição, sendo agora
essencialmente um efeito de segunda ordem; a influência na
força do ácido não é sempre tão regular, desempenhando a
sua influência nos fatores estereoquímicos e outros, observam-
se os seguintes valores de pKa.
Ácido orgânico pKa
Me2CHCO2H 4,86 Me3CCO2H 5,05 CH3CO2H 4,76 MeCH2CO2H 4,88 Me (CH2)2CO2H 4,82 Me (CH2)3CO2H 4,86
Se existe um átomo de carbono ligado por uma ligação
dupla adjacente ao grupo carboxila, a força do ácido é
aumentada. Assim, o ácido propenoico CH2CHCO2H tem um
pKa de 4,25 comparado com 4,88 para o análogo saturado,
ácido propanóico. Isto é devido ao átomo de carbono α
insaturado com hibridização sp2, o que significa que os elétrons
se encontram mais próximos do núcleo do carbono do que no
átomo hibridizado sp3, devido a maior contribuição s no híbrido
sp2. O resultado é que os átomos de carbono hibridizados sp2
são menos doadores de elétrons do que os hibridados sp3, e
assim o ácido propenóico, embora ainda mais fraco do que o
ácido metanóico, é mais forte que o propanoico. O efeito é

76
muito mais acentuado com o átomo de carbono hibridizado sp1
de uma ligação tripla, e assim o pKa do ácido propiônico,
HC≡CCO2H, é 1,84. Uma situação análoga ocorre com os
átomos de hidrogênios do eteno e do etino, os do primeiro são
um pouco mais ácidos do que os hidrogênios no etano,
enquanto que os do etino são suficientemente ácidos que
podem ser rapidamente substituídos por um certo número de
metais.
Ácidos alifáticos substituídos
A substituição de hidrogênios por grupos que retiram
elétrons em ácidos alifáticos aumentam a força do ácido.
Exemplos:
O efeito relativo dos diferentes halogênios encontra-se
na ordem prevista, sendo o flúor o mais eletronegativo (elétron
retirador) produzindo um aumento de cem vezes na força do
ácido fluoro etanoico em comparação com o ácido etanoico. O
efeito é muito maior do que o produzido, em sentido oposto,
pela introdução de um grupo alquila, e a introdução de outros
halogênios ainda provoca grandes aumentos na força ácida, o
ácido tricloroetanóico é, por isso, um ácido forte.
O Ka (pKa) está relacionado com ∆G para a ionização, e
∆G inclui o termo ∆S e ∆H. Nesta série de ácidos etanoicos
substituídos com halogênios, verifica-se que ∆H difere pouco
de um composto para outro, sendo a variação de ∆G ao longo
C
O
OH
H
H
H
C
O
OH
H
F
H
C
O
OH
H
Br
H
C
O
OH
H
I
H
C
O
OH
H
Cl
H
C
O
OH
Cl
Cl
H
C
O
OH
Cl
Cl
Cl
pKa = 4,76 pKa = 2,57 pKa = 2,86 pKa = 2,90
pKa = 1,25 pKa = 0,65
pKa = 3,16

77
da série principalmente devida à variação de ∆S. Isto resulta do
fato de o átomo de halogênio substituinte provocar a
deslocalização da carga negativa sobre o conjunto de todo o
ânion, impondo restrição sobre as moléculas de água que o
rodeiam do que o ânion etanoato cuja carga está concentrada,
estando localizada no CO2-.
C C
O
O
F
H
H
-
C C
O
O
H
H
H
-
Há, portanto, um decréscimo menor de entropia na
ionização dos ácidos etanoicos substituídos com halogênio do
que com o ácido etanóico propriamente dito. Isto é acentuado
com CF3CO2H para cuja ionização ∆G =1,3 kJ comparado com
27,2 kJ para CH3CO2H, enquanto que ∆H difere muito pouco
de um ácido para outro.
A introdução de um átomo de halogênio em posição
mais afastada do grupo carboxila do que a posição α tem muito
menos influência, porque o efeito indutivo se desvanece ao
longo de uma cadeia saturada, e como resultado, a carga
negativa se torna menos dispersa, mais concentrada, no ânion
carboxilato. O ácido assemelha-se ao ácido alifático simples,
como mostram os seguintes valores de pKa.
Outros grupos elétrons retiradores, como por exemplo,
NO2, CN, SO2R, CO, CO2R, aumentam a força de ácidos
alifáticos simples, o mesmo sucedendo com os grupos hidroxila
e metoxila. Os elétrons não compartilhados nos átomos destes
CH3 CO
OH
CH3 CC
O
OH
Cl
H
CH3 C CO
OH
Cl
H
C CO
OH
Cl
H
H
pKa = 4,82 pKa = 2,84 pKa = 4,52pKa = 4,06
O2N CO
OH
CO
OHMe3N
pKa = 1,68
pKa = 3,35 pKa = 3,58
pKa = 1,83
EtO2C CO
OH
+
MeCO CO
OH
CO
OHNC
pKa = 3,53
MeO CO
OH
pKa = 3,83
OH CO
OH
pKa = 2,47

78
dois últimos grupos não são capazes de exercer um efeito
mesomérico, no sentido oposto do seu efeito indutivo, devido
aos átomos de carbono saturados. Estes efeitos podem ser
observados nos seguintes valores de pKa.
SUBSTITUINTES DOADORES OU RETIRADORES DE
ELÉTRONS - FENÓIS
Os fenóis substituídos aumentam sua acidez na
presença de grupos elétrons retiradores no anel. No caso de
um substituinte nitro, é de esperar que o efeito indutivo
diminuísse ao ir de o- → m- →p- nitrofenol, mas há também um
efeito elétron retirador mesomérico, quando o grupo nitro está
na posição o- ou p-, mas não na posição m-, isto promoveria
também a ionização por estabilização (através de
deslocalização) do ânion resultante. Portanto o o- e p-
nitrofenóis são mais ácidos que o m-nitrofenol. A introdução de
mais grupos NO2 promove um aumento na acidez, assim o
2,4,6-trinitrofenol (ácido pícrico) é um ácido forte.
Fenol pH
C6H5OH 9,95 o-O2NC6H4OH 7,23 m-O2NC6H4OH 8,35 p-O2NC6H4OH 7,14 2,4-(O2N)2C6H5OH 4,01 2,4,6-(O2N)3C6H2OH 1,02
Como anteriormente, ∆H varia muito pouco entre os o-,
m- e p-nitrofenóis, resultando os diferentes valores para ∆G
para os três compostos de diferenças nos termos T∆S, ou seja,
de variações nos padrões de solvatação dos três ânions,
devido à diferente distribuição de carga negativa nesses
ânions.

79
O efeito da introdução de grupos alquila elétron doador
no anel benzênico é pequeno.
Fenol pKa
C6H5OH 9,95 o-MeC6H4OH 10,28 m-MeC6H4OH 10,08 p-MeC6H4OH 10,19
Os fenóis substituídos resultantes são ácidos
ligeiramente mais fracos, indicando que o efeito desses
substituintes na desestabilização do íon fenóxido, perturbando
a interação da sua carga negativa com os orbitais
deslocalizados no núcleo aromático, é pequeno.
Ácidos carboxílicos aromáticos
O ácido benzóico, com pKa de 4,20, é mais forte do que
o seu análogo saturado, ácido ciclohexano-carboxílico (pKa =
4,87); sugerindo que um grupo fenila, é aqui menos elétron
doador, em comparação com um átomo de carbono saturado,
em relação ao grupo carboxila, devido ao átomo de carbono
hibridado sp2 ao qual o grupo carboxila está ligado.
A introdução de grupos alquilas no anel benzênico tem
pouca influência na força do ácido benzoico, mas grupos
retiradores
Ácido carboxílico pKa
C6H5CO2H 4,20 m-MeC6H4CO2H 4,24 p-MeC6H4CO2H 4,34
de elétrons aumentam a sua força, sendo o efeito mais
pronunciado, quando encontram-se nas posições orto e para.
Ácido carboxílico pKa O O
N+
O O
+
-
- -
C6H5CO2H 4,20 o-O2NC6H4CO2H 2,17 m-O2NC6H4CO2H 3,45 p-O2NC6H4CO2H 3,43 3,5-(O2N)2C6H3CO2H 2,83

80
O efeito particularmente acentuado com o-NO2 pode ser
devido à distância muito curta sobre a qual atua o poderoso
efeito indutivo, mas não se pode excluir mais interação direta
entre os grupos vizinhos.
A presença de grupos como OH, OMe, ou halogênio
possuindo um efeito indutivo elétron retirador, mas um efeito
mesomérico elétron doador quando nas posições o- e p- pode,
no entanto, fazer com que os ácidos p- substituídos sejam mais
fracos do que o m- e, por vezes, ainda mais fracos do que o
próprio ácido não substituído, ex: ácido p- hidroxibenzóico.
pKa de R – C6H5CO2H O O
R+
-
-
O O
R:
H Cl Br OCH3 OH o- 4,20 2,94 2,85 4,09 2,98 m- 4,20 3,83 3,81 4,09 4,08 p- 4,20 3,99 4,00 4,47 4,58
Este efeito compensador torna-se mais pronunciado ao
ir-se de Cl ≈ Br → OH, ou seja, por ordem crescente da
facilidade com que o átomo ligado ao anel se separa com os
seus pares eletrônicos.
O comportamento de ácidos o-substituídos é,
frequentemente, anômalo. Algumas vezes verifica-se que a sua
força é consideravelmente superior ao previsto devido à
interação direta entre os grupos adjacentes. Assim, a ligação
hidrogênio intramolecular estabiliza o ânion em ácido o-
hidroxibenzóico (salicílico) por deslocalização da sua carga,
uma vantagem não partilhada pelos os seus isômeros m- e p-,
nem pelo ácido o- metoxibenzoico.
OH
O
O H
- H+
OH
O
O
-
(3) (4)

81
A ligação de hidrogênio intramolecular pode atuar no
ácido não dissociado tal como no ânion, mas provável que seja
consideravelmente mais eficaz no último do que no primeiro,
porque a carga negativa no oxigênio do ânion conduzirá a uma
ligação hidrogênio mais forte. O efeito é ainda maior quando
pode ocorrer ligação hidrogênio com grupos hidroxilas em
ambas as posições orto, e por isso o ácido 2,6-hidroxibenzóico
tem um pKa = 1,30.
Ácidos dicarboxílicos
O grupo carboxila tem um efeito indutivo elétron
retirador, é de esperar que a presença de um segundo grupo
carboxila num ácido o torne mais forte, diminuindo os valores
de pKa.
Ácido carboxílico pKa Ácido dicarboxílico pKa
HCOOH 3,77 HOOCCOOH 1,23 CH3COOH 2,17 HOOCCH2COOH 2,83 CH3CH2CO2H 4,88 HOOCCH2CH2COOH 4,19 C6H5COOH 4,20 o-HOOCC6H4COOH 2,98 m- HOOCC6H4COOH 3,46 p- HOOCC6H4COOH 3,51
O efeito é muito pronunciado, mas decai à medida que
os grupos carboxilas são separados por mais do que um átomo
de carbono saturado. O ácido cis-butenodioico (maleico), pKa1
= 1,92 é um ácido muito mais forte do que o ácido trans-
butenodioico (fumárico), pKa1 = 3,02, devido à ligação de
hidrogênio intramolecular que pode ocorrer com o primeiro,
mas não com o segundo, conduzindo à estabilização relativa
do mono-ânion cis (maleato).
Já a segunda dissociação do ácido trans-butenodioico
(pKa2 = 4,38) ocorre mais rapidamente do que a do ácido cis
(pKa2 = 6,23), devido a maior dificuldade em remover um próton

82
de um sistema cíclico negativamente carregado no ânion
derivado do último. Os ácidos etanodióicos (oxálico), 1,3-
propanodióico (malônico) e 1,4-butanodióico (succínico) são,
cada um deles, mais fracos nas suas segundas dissociações
do que, respectivamente, os ácidos metanoico, etanoico e
propanoico. Isto se deve ao fato de o segundo próton ter de ser
removido de uma espécie negativamente carregada contendo
um substituinte elétron doador, CO2- , que pode desestabilizar o
ânion relativamente ao ácido não dissociado, quando
comparado com o sistema não substituído.
BASES
pKb, pKBH+ e pKa
A força de uma base (B:) em água, pode ser
determinada considerando-se equilíbrio.
B: + HOH BH2+
+-OH
A constante de equilíbrio, Kb, em água, é então dada
por:
[ ][ ][ ]B
OHBHK
b
−+
=
O termo [H2O] assemelha-se ao caso dos ácidos, é
incorporado em Kb porque a água está presente em tal excesso
que a sua concentração não varia significativamente, também
aqui as concentrações são usadas em vez das atividades mais
corretas, desde que a solução seja razoavelmente diluída.
No entanto, é comum descrever a força de bases
também em termos de Ka e pKa, estabelecendo assim uma
única escala contínua, quer para ácidos ou para bases. Para
tornar isto possível, utiliza-se, como a reação de referência
para bases, no equilíbrio,
BH2+
+ HOH + H3O+
B:
para a qual pode-se escrever,

83
[ ][ ][ ]+
+
=BH
OHBKa
3:
em que Ka (e pKa) é uma medida da força ácida do ácido
conjugado BH+, da base B:. Esta medida da facilidade com que
BH+ perderá um próton é, inversamente, uma medida da
dificuldade com que a base (B:) aceitará um próton: quanto
mais forte for BH+ como um ácido, tanto mais fraca será B:
como base. Quase sempre utiliza pKa para referir a força de
uma base B:, pKBH+ deve ser especificado, ao invés de
escrever simplesmente pKa.
O íon amônio (NH4+) com valor de pKa de 9,25, e
valores de
NH4+
+ H2O NH3 + H3O+
∆G = 52,7 kJ, ∆H = 51,9 kJ, ∆S = -2,9 J e T∆S = - 0,8 kJ
a 25°. Assim, a posição de equilíbrio acima indicada, que é
determinada por ∆H, sendo o efeito de ∆S quase despresível, é
um resultado que contrasta acentuadamente com o
comportamento de muitos ácidos como vimos anteriormente. A
razão de pequeno efeito de ∆S é que aqui há uma espécie
carregada (um íon positivo) de cada lado do equilíbrio, e estes
íons têm efeitos muito comparáveis em restringir as moléculas
de água do solvente que os rodeiam, de modo que as suas
entropias de solvatação tendem a anular-se entre si.
Bases alifáticas
A força crescente das bases nitrogenadas está
relacionada com a facilidade com que estão preparadas para
aceitar prótons e, portanto, com a disponibilidade do par de
elétrons não compartilhado no átomo de nitrogênio, devemos
prever um aumento em força básica na série NH3→ RNH2→
R2NH→ R3N, devido ao crescente efeito indutivo de sucessivos
grupos alquilas que tornam o átomo de nitrogênio mais

84
negativo. Verificou-se que uma série de aminas tem, no
entanto, os valores relativos de pKa que seguem.
Amina NH3 CH3–NH2
(CH3)2–NH
(CH3)3–N
CH3CH2–NH2
(CH3CH2)2–NH
(CH3CH2)3–N
pKa 9,25 10,64 10,77 9,80 10,67 10,93 10,88
A introdução de um grupo alquila no amoníaco aumenta
acentuadamente a força básica como se esperava. A
introdução de um segundo grupo alquila aumenta ainda mais a
força básica, mas o efeito da introdução do segundo grupo
alquila é muito menos acentuado do que o efeito da introdução
do primeiro. No entanto, a introdução de um terceiro grupo
alquila para dar uma amina terciária de fato diminui a força
básica de uma amina, em água, o que justifica a basicidade ser
determinada não só pela disponibilidade eletrônica no átomo
nitrogênio, mas também pela extensão em que o cátion,
formado por aceitação de um próton, pode sofrer solvatação e,
assim, ficar estabilizado. Quanto mais átomos de hidrogênio
estiverem ligados ao nitrogênio no cátion, tanto maiores serão
as possibilidades de solvatação, por ligações de hidrogênio
entre estes e a água.
H2O H
N+
H
HH2O
R OH2 > >
H2O H
N+
R
HH2O
R
H2O H
N+
R
R
R
Decréscimo na estabilidade por solvatação
Ao percorrer a série NH3→ RNH2→ R2NH→ R3N, o
efeito indutivo tenderá a aumentar a basicidade, mas ocorrerá
menor estabilização do cátion por hidratação, que tenderá a
diminuir a basicidade. O efeito final da introdução de
sucessivos grupos alquila diminui a estabilização e ocorre de
fato uma inversão ao passar-se de uma amina secundária para
uma amina terciária. Se esta for a explicação real, essa
inversão não deve ser observada se forem efetuadas

85
determinações de basicidade num solvente em que a formação
de ligações hidrogênio não possa ocorrer. Em clorobenzeno,
verificou-se que a ordem de basicidade das butilaminas é:
BuNH2 > Bu2NH > Bu3N
embora os seus pKa em água sejam 10,61; 11,28; 9,87.
A introdução de grupos retiradores de elétrons, Cl, NO2,
próximo a um centro básico diminui a basicidade, devido ao
efeito indutivo negativo, assim a amina (CF3)3–N é virtualmente
não básica, devido aos três grupos CF3, fortemente elétron
retiradores.
A variação é também pronunciada com C=O, porque não
só o átomo de nitrogênio, com o seu par de elétrons, está
ligado a um grupo elétron retirador através de um átomo de
carbono hibridado sp², mas pode também operar um efeito
mesomérico elétron doador.
R C
NH2
O
R C
NH2+
O-
:
Por isso, as amidas são bases muito fracas em água, e
se estiverem presentes dois grupos C=O, as amidas
resultantes, longe de serem básicas, são suficientemente
ácidas para formar sais de metais alcalinos, 1,2-
benzenodicarboximida.
NH
O
O
OH-
N
O
O
δ−
δ−
δ−
O efeito da deslocalização no aumento da força básica
de uma amina é observado na guanidina, HN=C(NH2)2, que,
com exceção dos hidróxido de tetralquilamônio, se encontra
entre as bases orgânicas nitrogenadas mais fortes que se
NH C
NH2
NH2
NH-
C
NH2
NH2+
NH-
C
NH2+
NH2
NH2+
C
NH2
NH2
NH2 C
NH2
NH2+
NH2 C
NH2+
NH2
Molécula neutra
Cátion
H+

86
conhece, com um pKa de 13,6. Tanto a molécula neutra como o
cátion resultante da sua protonação são estabilizados por
deslocalização.
No cátion, a carga positiva está simetricamente
espalhada por contribuição para o híbrido de três estruturas
exatamente equivalentes, de igual energia. Não ocorre na
molécula neutra um efeito de deslocalização comparável (em
que duas das estruturas contribuintes envolvem separação de
cargas), como resultado de que o cátion é muito mais
estabilizado relativamente à molécula neutra, a guanidina é
uma base extremamente forte.
Uma situação análoga ocorre com as amidinas,
RC(=NH)NH2.
NH C
R
NH2
NH-
C
R
NH2+
NH2+
C
R
NH2
NH2 C
R
NH2+
Molécula neutra
Cátion
H+
Neste caso, a estabilização por deslocalização no cátion
é menos efetiva que com a do cátion da guanidina, verifica-se
que a etanamidina (pKa = 12,4) é uma base mais forte do que a
etilamina (pKa = 10,67).
Bases aromáticas
As aminas aromáticas são bases mais fracas que as
alifáticas em mais fracas que o próprio amoníaco.
Aminas Anilina Metilanilina Amoníaco Ciclohexilamina pKa 4,62 10,67 9,25 10,68
Na anilina, o átomo de nitrogênio está ligado a um átomo
de carbono hibridado sp², mas o que é mais significativo, o par

87
de elétrons não compartilhado no nitrogênio pode interagir com
as orbitais deslocalizadas do anel.
NH2 NH2 NH2 NH2+ + +
--
-
Se a anilina for protonada, essa interação, com
resultante estabilização, no cátion anilínio é proibida, uma vez
que o par de elétrons em N não está disponível.
NH3+
Então, a molécula da anilina é estabilizada em relação
ao cátion anilínio, e é, portanto, energicamente desfavorável
para a anilina aceitar um próton. O efeito de diminuição de
caráter básico é naturalmente mais pronunciado quando são
introduzidos outros grupos fenila no átomo nitrogênio, assim a
difenilamina, Ph2NH, é uma base extremamente fraca (pKa =
0,8), enquanto que a trifenilamina, Ph3N, não é, segundo os
padrões normais, uma base.
Grupos substituintes ligados no átomo de nitrogênio e no
anel Benzênico influenciaram na interação do par não
compartilhado do átomo de nitrogenio com o sistema de
orbitais deslocalizados do anel benzênico. Assim, verifica-se
que as anilinas substituídas tem os seguintes valores de pKa.
Bases Aromáticas pKa
C6H5NH2 4,62 C6H5NHCH3 4,84 C6H5N(CH3)2 5,15 o-CH3C6H4NH2 4,38 m- CH3C6H4NH2 4,67 p- CH3C6H4NH2 5,10

88
O efeito indutivo pequeno, aumentando o caráter básico
que os grupos alquila exercem, não é suficientemente para
influenciar a estabilização da molécula da anilina numa
extensão significativa, e pode mesmo ser modificado por
fatores estereoquímicos e solvatação. Um grupo com efeito
indutivo (elétron retirador) mais poderoso, NO2, tem mais
influência. A retirada de elétrons é intensificada quando um
grupo nitro se encontra na posição o- e p-, porque a interação
do par de elétrons não compartilhado do nitrogênio do grupo
amino com o sistema de orbitais deslocalizados do anel
benzênico é assim acentuada. Deste modo, a molécula neutra
ainda mais estabilizada relativamente ao cátion resulta num
enfraquecimento ainda maior como uma base. Assim, observa-
se que as nitroanilinas têm os seguintes valores de pKa.
Bases Aromáticas pKa NH2
N+
O-
ON
+
O-
O-
NH2
+
C6H5NH2 4,62 o-NO2C6H4NH2 -0,28 m- NO2C6H4NH2 2,45 p- NO2C6H4NH2 0,98
O efeito enfraquecedor de caráter básico adicional
quando o substituinte se encontra na posição o- é em parte
devido à curta distância ao longo do qual atua o efeito indutivo,
e também à interação direta, por estereoquímica e por ligações
de hidrogênio, com o grupo NH2. A o-nitroanilina é uma base
tão fraca que seus sais são quase totalmente hidrolisados em
solução aquosa, enquanto que a 2,4-dinitroanilina é insolúvel
em solução aquosa ácida, e a 2,4,6- trinitroanilina se
assemelha a uma amida, é na verdade conhecida por
picramida e sofre facilmente hidrólise a ácido pícrico (2,4,6-
trinitrofenol).
Substituintes como OH e OMe, que possuem pares de
elétron não compartilhados, podem exercer efeito mesomérico

89
elétron doador, isto é, fortalecedor de caráter básico, das
posições o- e p-, mas não da posição m-, resultando que a
anilina p- substituída é uma base mais forte do que o
correspondente composto m-. O composto m- é uma base mais
fraca do que a própria anilina, devido ao efeito indutivo elétron
retirador exercido pelo átomo de oxigênio, em cada caso.
Como em outros casos, o efeito do substituinte o- permanece
anômalo, devido à interação direta com o grupo NH2 quer por
efeitos estereoquímicos quer por efeitos polares. Verifica-se
que as anilinas substituídas têm os seguintes valores de pKa.
Bases Aromáticas
pKa Bases Aromáticas
pKa
NH2
OCH3 OCH3
NH2
+
-
C6H5NH2 4,62 - -
o-HOC6H4NH2 4,72 o-CH3OC6H4NH2
4,49
m- HOC6H4NH2
4,17 m- CH3OC6H4NH2
4,20
p- HOC6H4NH2
5,30 p- CH3OC6H4NH2
5,29
Um caso interessante é o da 2,4,6-trinitro-N,N-
dimetilanilina e da 2,4,6-trinitroanilina, em que a primeira é
cerca de quarenta mil vezes mais forte como base do que a
segunda. Isso se deve ao fato do grupo NMe2 ser
suficientemente volumoso para interferir estereoquimicamente
com os grupos NO2 muito volumosos em ambas as posições o-
.impedindo a interação da retirada de elétrons, por mesomeria,
pelos grupos nitros, e o esperado enfraquecimento de caráter
básico pelos três grupos nitros, fica restrito aos seus efeitos
indutivos.
N+
O-
O
N+
O-
O
N+
O-
O
NCH3CH3

90
Na 2,4,6-trinitroanilina, o grupo NH2 é suficientemente
pequeno para que seja imposta essa limitação, a formação de
ligações de hidrogênio entre os átomos de oxigênio dos grupos
o-NO2 e os átomos de hidrogênio do grupo NH2 pode de fato
ajudar a manter estes grupos na orientação planar requerida.
As orbitais p podem adquirir uma orientação paralela, e a força
da base é enormemente reduzida pelo efeito mesomérico
elétron retirador pelos três grupos NO2.
N+
O-
O-
N
N+
O
O-
N+
O-
O
HH +
N+
O-
O
N
N+
O
O-
N+
O-
O
HH +
Bases heterocíclicas
A piridina é o protótipo dessa classe. A piridina é um
composto aromático, o átomo de nitrogênio está hibridado sp2 e
contribui com um elétron para o sistema 6eπ (4n = 2, n = 1);
isto deixa um par de elétrons não compartilhado disponível no
nitrogênio (acomodado num orbital híbrido sp²), e verifica-se
que a piridina é básica (pKa = 5,04). No entanto, é uma base
muito mais fraca do que aminas alifáticas terciárias, e nesta
característica de bases, o átomo de nitrogênio está ligado por
uma ligação múltipla. Isto se deve ao fato de à medida que o
átomo de nitrogênio se torna ligado por ligações múltiplas, o
seu par de elétrons não compartilhados é acomodado numa
orbital que tem mais caráter s. O par de elétrons é atraído para
o núcleo do nitrogênio e ligado mais fortemente por ele,
tornando-se assim menos disponível para formar uma ligação
com um próton, com diminuição na basicidade do composto, ao
ir de:

91
NCH3
CH3
CH3
N
CH2
CH3
NCH
em, por exemplo,
NR
R
R
NRCN
os pares não compartilhados estão, respectivamente, em
orbitais sp3, sp2 e sp1, e a basicidade decrescente reflete-se
nos valores de pKa, é o caso da basicidade de cianetos de
alquila ser extremamente baixa.
Na quinuclidina, o par de elétrons não compartilhado
está num orbital sp³ e o seu pKa (10,85) difere muito pouco do
da trietilamina (10,88).
N..
O pirrol exibe algum caráter aromático (embora não tão
pronunciado como o benzeno ou piridina) e não se comporta
como um dieno conjugado.
NH
..NH
..
Pirrol Pirrolidina
Para tal aromaticidade ser conseguida, seis elétrons π
(4n+2, n = 1) dos átomos do anel devem preencher as três
orbitais moleculares ligantes. Isto necessita da contribuição de
dois elétrons por parte do átomo de nitrogenio e, embora a
resultante nuvem eletrônica fique deformada em direção ao
nitrogenio devido à natureza mais eletronegativa desse átomo
em comparação com o dos quatro carbonos, o par de elétrons
do nitrogênio não está disponível para aceitar um próton.
NH
..
H+
NH
+ H
H

92
A protonação se for forçada no pirrol, não tem lugar no
nitrogênio, mas sim no átomo de carbono. Isto acontece porque
a incorporação do par de elétrons não compartilhado do átomo
de nitrogênio no sistema aromático 6eπ deixa o átomo N
positivamente polarizado, os prótons tendem a ser repelidos
por ele, e são assim aceitos pelo átomo de carbono adjacente.
A situação de basicidade assemelha-se bastante à já
encontrada com a anilina pelo fato do cátion estar
desestabilizado relativamente à molécula neutra. O efeito é
muito mais pronunciado com o pirrol, que para funcionar como
uma base tem de perder todo o caráter aromático, e
consequente estabilização, refletindo no seu valor de pKa
comparado com o da anilina. O pirrol é uma base muito fraca e
pode funcionar como um ácido, embora muito fraco, já que o
átomo de H do grupo NH pode ser removido por bases fortes,
como –NH2, sendo o ânion resultante estabilizado por
deslocalização.
NH
..
-NH2
N..
-
Tais considerações não podem, aplicar-se a pirrolidina,
que tem um pKa de 11,27, assemelhando-se muito a
dietilamina.
Conceito de ácidos/ bases duros e moles
Este conceito foi desenvolvido para poder comparar, de
uma maneira qualitativa, a força de ácidos e bases de Lewis.
Especificamente, para poder prever a "força" de um certo ácido
frente a uma certa base.
Para este fim, ácidos e bases de Lewis foram
classificados como duros e moles, dependendo das
características dos seus átomos centrais:
Classificação de Ácidos e Bases Duros e moles:

93
Base mole
EN(↓) P (↑) facilmente oxidável PE não ligado
Base dura
EN (↑)
P (↓) não oxidável PE muito ligado
Ácido mole
EN (↓)
P (↑) densidade de carga + (↓)
PE orbitais p ou d (átomo central grande)
Ácido duro
EN (↑)
P (↓) densidade de carga + (↑)
não tem PE (átomo central pequeno)
EN: Eletronegatividade; P: Polarizabilidade; (↓): baixo;
(↑): alto; PE: Par de Elétron
A utilidade desta classificação é baseada no
princípio de que um ácido duro interage melhor com uma base
dura, e um ácido mole interage melhor com uma base mole, ou
seja, as interações entre um ácido de Lewis / base de Lewis
duro/duro e mole/mole possuem constantes de equilíbrio altas:
ALmole + BLmole ALmole BLmole K
ALduro + BLduro ALduro BLduro K
ALmole + BLduro ALmole BLduroK
KALduro + BLmole ALduro BLmole
Este princípio é chamado de "Hard Soft Acid Base
Principle - HSAB", ou seja, o "Princípio de Ácidos e Bases
Duras e Moles - ABDM".
Classificação de alguns ácidos e bases de Lewis.
Tabela 2. Ácidos e Bases Duros e Moles:
Bases Duras Bases Moles Bases Intermediárias
H2O OH- F
-
AcO- SO42
- Cl
-
CO32- NO3
- ROH
RO- R2O NH3
RNH2
R2S RSH RS-
I- R3P (RO)3P
CN- RCN CO
C2H4 C6H6
H- R
-
ArNH2 C5H5N
N3- Br
-
NO2-

94
Ácidos Duros Ácidos Moles Ácidos Intermediários
H+ Li
+ Na
+
K+ Mg2+ Ca2+
Al3+ Cr3+ Fe3+
BF3 B(OR)3 AlMe3
AlCl3 AlH3 SO3
RCO+ CO2
HX (moléculas com ponte de hidrogênio)
Cu+ Ag
+ Pd 2+
Pt2+ Hg2+ BH3
GaCl3 I2 Br2
CH2 (carbenos)
Fe3+ Co2+ Cu2+
Zn2+ Sn2+ Sb3+
BMe3 SO2 R3C+
NO+ C6H5+
Com respeito à tabela anterior, chama-se a atenção para
alguns pontos:
� A "moleza" das bases diminui na seguinte ordem:
I- > Br- > Cl- > F- e CH3- > NH2- > OH- > F-
De acordo com o aumento de eletronegatividade e
diminuição da polarizabilidade.
� Alcenos e anéis aromáticos são bases moles
(formam complexos com Ag+, Pt2+ e Hg2+, mas não com Na+,
Mg2+ e Al3+).
� Carbânions e o hidreto são bases moles.
� Bases com oxigênio e nitrogênio como átomo
central são duros; com fósforo, enxofre e carbono como átomo
central são moles.
� Anilinas e piridinas são bases intermediárias.
� Cátions pequenos são ácidos duros; cátions
grandes são ácidos moles.
� Chama-se atenção ao fato de que o próton é um
ácido de Lewis duro.
� Carbenos são ácidos moles, e carbocâtions são
ácidos intermediários.

95
3º ROTEIRO DE ESTUDO DIRIGIDO
1. Que fatores contribuem para as diferenças nos pKa a seguir: etano (pKa=45); etanol (pKa=18) e ácido acético (pKa=4,76) 2. A acidez do 2,2,2- trifluoro-etanol (pKa=12,4) é cerca de 1000 vezes maior que a do etanol (pKa=18). Explique esta diferença. 3. Os fenóis abaixo relacionados têm os valores de pKa iguais a: 1, 4, 7, 9 e 10. Determine o valor de pKa correspondente a cada fenol e explique. a) 2,4-dinitrofenol, b) 4-nitrofenol, c) 2,4,6-trinitrofenol, d) fenol e e) 3-clorofenol 4. Em cada um dos itens abaixo, indique (e justifique) qual a substância mais ácida: a) fenol ou ciclo-hexano? b) fenol ou ácido benzóico? c) Ácido propanóico, ácido propenóico (acrílico) ou ácido propinóico? d) Ácido o-hidroxibenzóico (salicílico), seu isômero meta ou seu isômero para? e) Ácido maleico ou ácido fumárico? f) Meta ou para-cianofenol? g) Ciclopentadieno ou penta-1,4-dieno? 5. A quinuclidina é uma base mais forte que a trietilamina, frente ao trimetil-boro; mas, a basicidade destas duas aminas frente a prótons é praticamente a mesma. Explique. 6. Explique a basicidade das aminas primárias, secundárias e terciárias: a) em água e b) em clorobenzeno
7. Escreva as estruturas de ressonância da guanidina e de seu cátion. Com base nestas estruturas, responda: a guanidina é uma base forte ou fraca? 8. Compare as basicidades da piridina, do pirrol, da quinuclidina e da pirrolidina.
HC CH2 C CHa) , b) CH2 CON(Et)2 , [(CH3)2CH]2N-
c) d) p-nitrofenolato e m-nitrofenolatoCH3O- , (CH3)3CO-
96
9. Para cada um dos seguintes pares indique a base mais forte. 10. Para cada um dos seguintes compostos indique qual o próton é mais facilmente removido: 11.A quinina é um fármaco de origem natural, utilizado no tratamento da malária. Quantos equivalentes de HCl este alcaloide pode reagir?Desenhe a estrutura do sal formado.
N
MeON
HOH
H
12.Mostre, através de exemplo de reações, por que o álcool benzílico (PhCH2OH) pode ser considerado um ácido ou base de Bronsted-Lowry. 13.Utilizando a teoria ácido-base mole–duro de Pearson, explique a razão da quimios- seletividade das reações a seguir:
O
O
EtONa EtOH
Na OOEt
O
O
O
NaCNEtOH
NCO
O
Na
O
O
EtSNaEtOH
EtSO
O
Na
a) b)CH3COCH2COCH3 H2NCH2CH2OH
c)
O
d) HN NNH
NH2

97
14. Justifique a diferença de acidez dos seguintes isômeros:
UNIDADE IV - SUBSTITUIÇÃO NUCLEÓFILA NUM ÁTOMO DE CARBONO SATURADO
Uma reação clássica é a conversão de um haleto de alquila num álcool em soluão aquosa básica. Esta reação foi muito estudada por Ingold e sua escola.
R XOH-
+ R OH + X-
Determinações cinéticas em reações em que haletos de
alquila são atacados por uma grande variedade de diferentes
nucleófilos, Nu:, revelaram dois tipos essencialmente extremos:
um em que a velocidade é dependente de [Nu:],
Velocidade = k2[R – X] [HO-]
e outro em que, a velocidade é independente de [Nu:],
Velocidade = k1[R – X].
Em alguns casos verifica-se que as equações de
velocidade são “mistas” ou então complexas, e outros que
seguem exatamente as relações simples de lei de velocidade.
Relação entre cinética e mecanismo

98
Demonstrou-se que a hidrólise do haleto primário
clorometano (cloret de metila) em base aquosa se dá de
acordo com a equação (Velocidade = k2[R–X][HO-]), e esta foi
interpretada como envolvendo a participação de ambos o
haleto de alquila e o íon hidroxila na etapa determinante da
velocidade (mais lenta) da reação. Ingold tinha sugerido um
estado de transição em que o íon hidroxila ataca ficando
parcialmente ligado ao átomo de carbono reagente antes que o
íon cloreto tenha dele desligado. A energia necessária para
efetuar a quebra da ligação C–Cl é fornecida pela energia que
é produzida na formação da ligação HO–C . Cálculos de
mecânica quântica mostram que a aproximação pelo íon
hidroxila ao longo da linha dos centros dos átomos de carbono
e de cloro é a que apresenta requisitos energéticos mais
baixos. Isto pode ser representado como se indica a seguir.
A carga positiva é dispersa no estado de transição no
decurso da sua transferência do hidroxila para o cloro, e os
átomos de hidrogênio ligados a átomo de carbono passam por
uma posição em que todos eles se encontram num plano (a
ângulos retos com a folha de papel). Este tipo de mecanismo
foi designado por Ingold como SN2, significando Substituição
Nucleófila bimolecular.
Em contraste, a hidrólise do haleto terciário 2-cloro-2-
metilpropano (cloreto de t-butila) em base cineticamente segue
a equação (Velocidade = k1[R–X]). Como a velocidade é
independente de [-OH], este não pode desempenhar qualquer
papel na etapa determinante da velocidade, indicando que o
haleto sofre ionização lenta (nesta molécula existe completa
polarização R→Cl) como a etapa determinante da velocidade
OH-
+ C Cl
H
HH
C ClHO
HH
Hδ
-δ
-
CHO
H
HH
+ Cl-δ
+δ
-

99
para dar o par iônico R+Cl-, seguida pelo ataque rápido, não
determinante da velocidade, por –OH ou pelo solvente.
Cl
CH3
CH3
CH3
H2O
Lento
CH3
C+
CH3 CH3
+ Cl-
Este tipo de mecanismo foi designado por SN1,
significando Substituição Nucleófila Unimolecular. A energia
necessária para efetuar a ionização inicial é recuperada pela
energia de solvatação dos íons formados. O cátion, no par
iônico, em que o átomo de carbono possui uma carga positiva
é um íon carbônio, e durante a sua formação o átomo de
carbono inicialmente tetraédrico encontra-se num estado planar
mais estável em que os três grupos metila se encontram tão
afastados quanto possível, o ataque por –OH ou solvente (H2O)
pode então ocorrer pelo outro lado. Se o estado planar for
inibido por fatores estereoquímicos ou outros, o íon carbônio
será formado com dificuldade, e a reação pelo mecanismo SN1,
pode então não ocorrer.
No entanto, determinações cinéticas simples podem ser
um guia inadequado para qual dos dois mecanismos
anteriores, SN1 ou SN2, ocorra, por exemplo, na hidrólise de um
haleto. Assim, onde o solvente pode atuar como um nucleófilo
(solvólise), no caso da H2O, é de esperar uma reação do tipo
SN2,
Velocidade = k2 [RX] [H2O]
CH3
C+
CH3 CH3
rápido
rápido
H2O
-OH
OH
CH3
CH3
CH3
OH2
CH3
CH3
CH3
+OH
CH3
CH3
CH3
- H+
Cl-

100
mas como [H2O] permanece efetivamente constante, a
equação de velocidade de fato observada será
Velocidade = kobs [RX],
e determinações cinéticas simples em solução aquosa
sugerirão erradamente que a reação é do tipo SN1.
Efeito do solvente
Mudandça no solvente em uma reação exerce um efeito
profundo na sua velocidade e pode, de fato, resultar mesmo
numa mudança do seu percurso mecanístico. Assim, para um
haleto que sofre hidrólise pelo mecanismo SN1, um aumento na
polaridade do solvente (aumento de ε, constante dielétrica)
e/ou sua capacidade de solvatar íons resulta num aumento
acentuado da velocidade de reação. Por isso, a velocidade de
solvólise do haleto terciário Me3CBr é 3 x 104 vezes mais
rápida em etanol aquoso 50% do que só em etanol. Isto sucede
porque, no mecanismo SN1, a carga é desenvolvida e
concentrada no estado de transição em comparação com o
material de partida.
R XR X
X-
R+δ
+ δ-
A energia requerida para efetuar o processo decresce à
medida que aumenta ε; o processo também é facilitado pela
solvatação crescente, e consequente estabilização, do par
iônico em desenvolvimento em comparação com o material de
partida. O efeito de solvatação é evidenciado pelo fato de que
as reações do tipo SN1 são extremamente raras em fase
gasosa.
No mecanismo SN2, verifica-se que aumentando a
polaridade do solvente, o efeito é menos acentuado,
resultando, algumas vezes num leve decréscimo na velocidade
da reação. Isto ocorre porque neste exemplo particular não se
desenvolve nova carga, e a carga existente é dispersa no
Nu-
+ R X R XNuδ
-δ
-
RNu + X-

101
estado de transição, em comparação com os materiais de
partida.
A solvatação do estado de transição é provavelmente
algo menos eficaz do que a do nucleófilo inicial, daí o leve
decréscimo. Este diferente comportamento dos mecanismos
SN1 e SN2 às variações de solvente pode até certo ponto ser
usado como diagnóstico o tipo mecanismo da reação.
Contudo, consegue-se um efeito maior sobre a
velocidade de uma reação SN2 quando se passa de solventes
polares próticos para solventes polares não-próticos. Assim a
velocidade de reação do haleto primário, Iodeto de Metila,
(MeI), com N3- a 0° C aumentou 4,5 x 104 vezes ao ser
transferida de metanol (ε = 33) para N,N-dimetilmetanamida
(dimetil formamida, DMF), HCONMe2, que tem praticamente a
mesma polaridade (ε = 37). Esta grande diferença de
velocidade provém do N3-, ser altamente solvatado através de
ligações de hidrogênio no MeOH, enquanto que é menos
solvatado, e não por ligações de hidrogênio, em HCONMe2. O
ânion N3- largamente não solvatado em (HCONMe2) é um
nucleófilo muito mais forte do que quando rodeado (MeOH)
efeito solvatante tornando o muito menos nucleófilo, e daí o
aumento da velocidade de reação. Aumento de velocidade da
ordem de 109 tem sido observado ao transferir reações de SN2,
em MeOH, para outro solvente polar não-prótico,
dimetilsulfóxido (DMSO), Me2SO (ε = 46).
Quando há alterações mecanísticas com a
mudança de solvente, um aumento na polaridade do solvente e
capacidade de solvatar íons pode (mas não necessariamente)
alterar o mecanismo SN2 → SN1. A transferência de solventes
hidroxílicos para solventes polares não-próticos (DMSO) pode,
e muitas vezes o faz, mudar o mecanismo da reação de SN1 →
SN2 por aumentar a eficácia do nucleófilo no sistema.
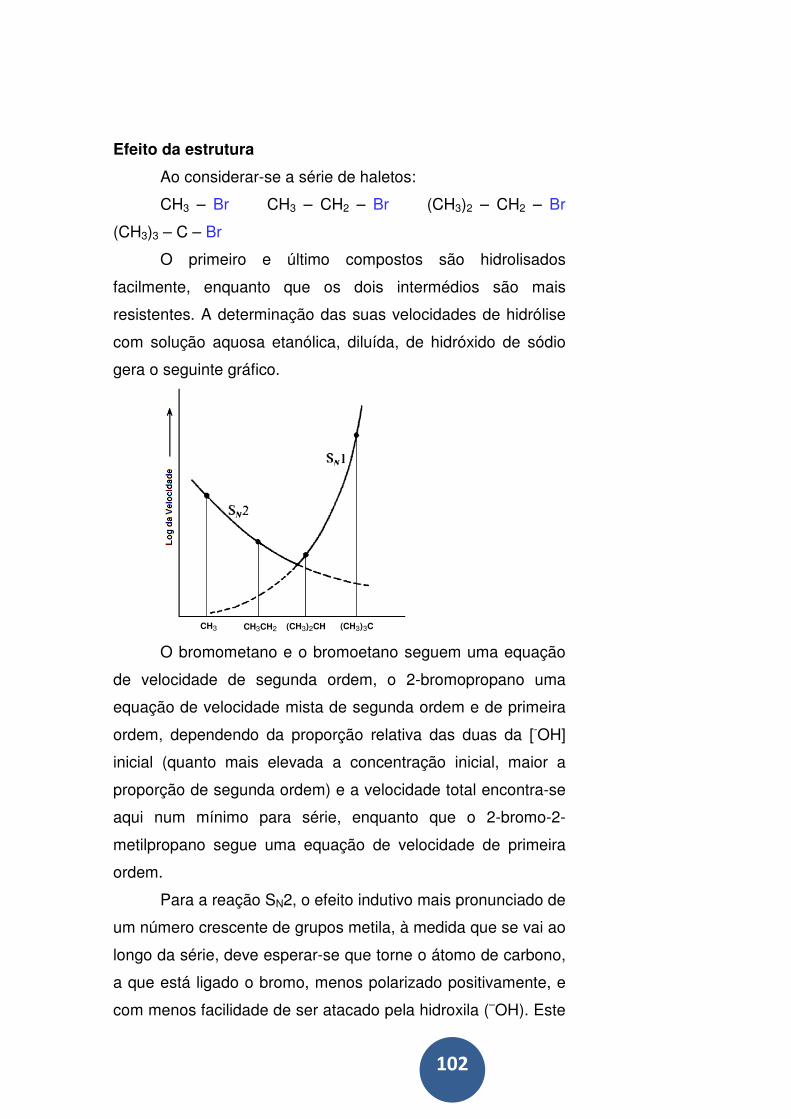
102
Efeito da estrutura
Ao considerar-se a série de haletos:
CH3 – Br CH3 – CH2 – Br (CH3)2 – CH2 – Br
(CH3)3 – C – Br
O primeiro e último compostos são hidrolisados
facilmente, enquanto que os dois intermédios são mais
resistentes. A determinação das suas velocidades de hidrólise
com solução aquosa etanólica, diluída, de hidróxido de sódio
gera o seguinte gráfico.
CH3 CH3CH2 (CH3)2CH (CH3)3C
O bromometano e o bromoetano seguem uma equação
de velocidade de segunda ordem, o 2-bromopropano uma
equação de velocidade mista de segunda ordem e de primeira
ordem, dependendo da proporção relativa das duas da [-OH]
inicial (quanto mais elevada a concentração inicial, maior a
proporção de segunda ordem) e a velocidade total encontra-se
aqui num mínimo para série, enquanto que o 2-bromo-2-
metilpropano segue uma equação de velocidade de primeira
ordem.
Para a reação SN2, o efeito indutivo mais pronunciado de
um número crescente de grupos metila, à medida que se vai ao
longo da série, deve esperar-se que torne o átomo de carbono,
a que está ligado o bromo, menos polarizado positivamente, e
com menos facilidade de ser atacado pela hidroxila (–OH). Este

103
efeito provavelmente é pequeno, e os fatores estéreoquímicos
são mais importantes, A hidroxila (–OH) terá mais dificuldade
em atacar o átomo de carbono que está ligado o bromo à
medida que esse átomo se encontra mais substituído. Se o
estado de transição da reação de SN2 resultante tiver cinco
grupos em torno desse átomo de carbono (comparado com
apenas quatro no haleto inicial), haverá um aumento da
obstrução espacial ai ir-se do haleto inicial para o estado de
transição, e esta obstrução relativa aumentará à medida que
aumenta o tamanho dos substituintes originais (H→Me).
Quanto maior a obstrução no estado de transição relativo aos
materiais de partida, maior será a sua energia, e portanto
formar-se-á mais lentamente. A velocidade da reação SN2
diminuirá à medida que se prossegue ao longo da série.
Relação CH3I CH3CH2Cl (CH3)2CHCl (CH3)3CCl Velocidade ( SN2) 1 2,7 x 10-2 4,9 x 10-4 2,2 x 10-5
Para o ataque SN1, a separação de carga no estado de
transição, e o par iônico intermediário a que dá origem, são
muitas vezes tomados como modelo para o estado de
transição. À medida que se prossegue ao longo série de
haletos, há uma crescente estabilização do íon carbônio do par
iônico, aumentando a velocidade de formação do estado de
transição. Esta estabilização crescente resulta da atuação
simultânea de um efeito indutivo,
e de hiperconjugação,
CH2
CH3CH3
H
+
CH2
CH3CH3
+H
H
HH
CH3
HH
CH3
CH3H
CH3
CH3CH3+ + + +< < <

104
dos átomos de hidrogênio ligados aos carbonos α, tendo a
referida série de íons carbônio respectivamente, 3, 6 e 9
desses átomos de hidrogênio.
A interação das ligações H–C com o átomo de carbono
transportando a carga positiva é obtido ao se substituir H por D
no haleto original, verificando-se então que a velocidade de
formação do par iônico diminui em ≈ 10% por átomo de
deutério incorporado, um resultado só é compatível se as
ligações H–C estiverem envolvidas na ionização. Isto é
conhecido por efeito cinético isotópico secundário,
secundário porque é uma ligação que não aquela que
transporta o marcador isotópico que sofre ruptura. Em
termos estereoquímicos, há um alívio da obstrução ao passar-
se do haleto inicial para uma conformação tetraédrica dos
quatro substituintes em torno do átomo de carbono hibridizado
sp3, para o íon carbônio, com uma conformação planar de três
substituintes (cinco para o E.T. SN2) em torno do átomo de
carbono agora com hibridização sp2. Os três substituintes estão
afastados uns dos outros no íon carbônio planar, e o efeito
relativo diminui o impedimento e aumentará à medida que os
substituintes aumentam de tamanho (H→Me). A velocidade de
reação SN1 aumentará à medida que se percorre a série.
Uma vez que a velocidade SN2 diminui e a velocidade
SN1 aumenta ao longo da série de haletos, tornam-se evidentes
as razões da alteração de mecanismo.
Uma alteração mecanística semelhante é observada,
embora mais rápido, ao percorrer-se a série.
CH3 – Cl, C6H5 – CH2 – Cl, (C6H5)2 – CH – Cl, (C6H5)3 –
C – Cl
Assim, para a hidrólise em acetona em água a 50%,
observa-se para o fenilclorometano (cloreto de benzilo) uma
equação de velocidade mista de segunda e primeira ordem,
passando quase completamente para o mecanismo SN1

105
quando apenas em água. A principal razão para a maior
promoção de ionização, com consequente passagem para o
mecanismo SN1 nesta série, é a considerável estabilização do
íon carbônio, por deslocalização da sua carga positiva, que é
agora possível.
CH2 CH2 CH2 CH2+
++
+
Semelhante estabilização de íon carbônio pode também
ocorrer com haletos de alila, 3-cloropropeno.
O ataque SN1 é assim promovido e os haletos de alila,
tal como os de benzila, são em geral extremamente reativos,
quando comparados com, CH3CH2CH2Cl e C6H5CH2CH2CH2Cl,
onde estabilização de íon carbônio não ocorre. O ataque SN2 é
acelerado pela interação, no ET, da orbital p não hibridizado no
átomo de carbono sp2 atacado com a orbital π da ligação dupla.
Verifica-se que as proporções da reação total que ocorrem por
cada um dos mecanismos dependem das condições,
promovendo os nucleófilos mais poderosos o mecanismo SN2.
Ao contrário, haletos de vinila como o cloroeteno,
CH2=CHCl, e halobenzenos são muito pouco reativos em
relação aos nucleófilos. Isto resulta do fato de o átomo de
halogênio estar agora ligado a um carbono hibridado sp2, com
o resultado que o par de elétrons da ligação C–Cl são
deslocalizado para mais perto do carbono do que na ligação a
um carbono sp3. A ligação C–Cl é mais forte e, por isso, é mais
difícil de sofrer ruptura do que por exemplo CH3CH2Cl com
momento de dipolo C–Cl menor, com menos tendência a
ionização (SN1) e um carbono menos positivo para o ataque
por –OH (SN2). Os elétrons da ligação dupla também inibem a
aproximação de um nucleófilo atacante. A ligação dupla não
CH2 ClCH2
CH2 H2C CH2
+ +Cl
-

106
ajudaria a estabilizar quer o estado de transição SN2 quer o íon
carbônio envolvido no mecanismo SN1. Muitas das mesmas
considerações aplicam-se aos halobenzenos, como carbonos
hibridizado sp2 e o sistema orbital π do anel benzênico.
A influencia de fatores estereoquímicos no mecanismo
da reação é particularmente observado quando ocorre
substituição na posição β.
Relação CH3 – CH2–Br
CH3CH2 – CH2–Br
(CH3)2CH – CH2–Br
(CH3)3C – CH2–Br
Velocidade ( SN2) 1 2,8 x 10-1 3,0 x 10-2 4,2 x 10-6
Assim, para a série acima, os valores indicados são
velocidades de reação relativas para SN2, com etóxido em
etanol a 55 °C. Quaisquer diferenças no efeito eletrônico dos
grupos metila através de dois átomos de carbono saturados
seriam muito pequenas, e a razão para as diferenças de
velocidade é estereoquímica crescente, dificultando
aproximação de etóxido “por trás” do átomo de carbono ao qual
o Br está ligado, e crescente impedimento estereoquímico no
ET resultante. A razão para a diminuição da velocidade entre 1-
bromo-2-metilpropano e 1-bromo-2,2-dimetilpropano (brometo
de neopentilo) é que o ET do primeiro, mais obstruído, pode,
por rotação em torno da ligação Cα–Cβ, adaptar uma
conformação em que o etóxido sofre interferência pelo H,
enquanto que esta desobstrução não é possível no ET do
segundo.
C
CHH
CH3
CH3
H
BrH3CH2CO
C
C
HH
CH3
CH3
CH3
BrH3CH2CO

107
O E.T. do 1-bromo-2,2-dimetilpropano estará num nível
de energia mais elevado, ∆G será maior e a velocidade de
reação correspondente mais baixa.
O efeito da estrutura na reatividade relativa pode ser
observado com particular clareza quando um átomo de
halogênio está localizado na cabeça de ponte de um sistema
bicíclico. Assim, observaram-se as seguintes velocidades para
a solvólise em etanol aquoso 80% a 25°C:
Relação
Br
CH3
CH
Br Br
Velocidade
( SN2) 1
≈
10-6
≈
10-14
Todos são haletos terciários; não é de esperar que
ocorra reação pelo mecanismo SN2 em bicilco[2,2,2]1-
bromoctano ou no biciclo [2,2,1] 1-bromoheptano, mais ocorre
em 1-bromo-2,2-dimetiletano. O ataque SN2 “por trás” no átomo
de carbono a que está ligado Br é, em qualquer dos casos,
impedido em bicilco[2,2,2]1-bromoctano ou no biciclo[2,2,1]1-
bromoheptano, quer pela estereoquímica de sua estrutura em
gaiola, ou pela sua estrutura rígida no ET, sem conformação
planar de ligações de átomo de carbono da ponte. A solvólise
via SN1, a etapa determinante da velocidade, é a formação do
par iônico, como acontece com 1-bromo-2,2-dimetiletano, está
também inibida, porque o íon carbônio resultante de
bicilco[2,2,2]1-bromooctano ou no biciclo[2,2,1]1-bromoheptano
(estrutura rígida), estabilizão um estado planar estável. Os
intermediários íon cabônio tem um nível de energia mais alto
do que o normal, e só se formam lentamente e com relutância.
A velocidade de solvólise, enormemente reduzida de
biciclo[2,2,1]1-bromoheptano em comparação com a de
bicilco[2,2,2]1-bromoctano, reflete a maior rigidez em torno do

108
átomo de carbono em cabeça de ponte (íon carbônio para uma
ponte com um carbono biciclo[2,2,1]1-bromoheptano do que
para uma com dois carbonos bicilco[2,2,2]1-bromooctano).
Esta rigidez no 1-bromotripticeno faz com que o átomo
de bromo seja inerte a nucleófilos.
Relação
Br
Br
Velocidade ( SN2) 1 ≈ 10-23
Apesar da semelhança formal da vizinhança do átomo
de bromo em 1-bromotripticeno com o trifenilbromometano,
verifica-se que as suas velocidades de reação, em condições
comparáveis, diferem por um fator de 10-23:1. Isto resulta da
estabilização do íon carbônio no trifenilbromometano ocorrer
por deslocalização da sua carga, através dos sistemas orbitais
π dos três anéis de benzeno, enquanto que no 1-
bromotripticeno a coplanaridade necessária no íon carbônio
não pode ser atingida como resultado da sua estrutura
extremamente rígida.
IMPLICAÇÕES ESTEREOQUÍMICAS DO MECANISMO
A hidrólise de uma forma opticamente ativa de um haleto
quiral apresenta algumas características estereoquímicas
interessantes. Consideremos então cada um dos mecanismos.
Mecanismo SN2, inversão de configuração
O átomo de carbono sofre inversão da sua configuração,
quando o arranjo espacial dos três grupos ligados ao átomo de
carbono após a reação for invertido. O que se pode observar
no mecanismo de reação abaixo.
OH-
+ C Br
H
HH
C BrHO
HH
Hδ
-δ
-
CHO
H
HH
+ Br-δ
+δ
-

109
Verificar-se que o arranjo espacial dos três grupos
residuais ligados ao átomo de carbono atacado foi invertido. Na
verdade, se o produto pudesse ser o brometo, em vez de o
álcool, o plano de polarização giraria, e a luz polarizada iria na
direção oposta, (-), à do material de partida, (+), seria a sua
imagem num espelho. No entanto, o produto que se obtém é o
álcool e infelizmente não somos capazes, apenas por
observação da sua rotação óptica, de dizer se tem ou não a
mesma configuração do brometo. Assim, para confirmar na
prática que a reação SN2 acima indicada é acompanhada por
uma inversão de configuração, como requer a teoria, é
necessário dispor de um método independente para relacionar
a configuração do reagente e do produto, o brometo e o álcool
correspondente indicados.
Determinação da configuração relativa
Esta se baseia no fato de que se um composto quiral
sofre uma reação que quebra uma ligação entre um dos grupos
e o centro quiral, então o centro pode sofrer inversão de
configuração. Por outro lado, se o composto reagir sem que
haja quebra da ligação, então a configuração do centro quiral
se conserva.
Assim, na série de reações no álcool opticamente ativo
(+),
OH
R
R'R" (+)
OSO2Ar
CH3
CH3CH3
Br-ArSO2Cl
Br
R"
R
R'
CH3CO2-
CH3CO O
R"
R
R'CH3
CH3OH-
OH
R"
R
R'(-)
R = PhCH2
R' = CH3
R" = HAr = p-CH3C6H4

110
sabe-se que a formação de um éster com cloreto de 4-
metilbenzenosulfonila (tosila) não quebra a ligação C–O do
álcool, pelo que o tosilato deve ter a mesma configuração que
o álcool original. Sabe-se que a reação deste éster com
CH3CO2- é uma substituição em que ArSO3
-(Ar = p-MeC6H4) sai
e é introduzido MeCO2-, donde a ligação C–O quebra nesta
reação, e, assim, pode ocorrer inversão de configuração na
formação de acetato. Na hidrólise alcalina do acetato, não
envolve quebra da ligação alquil-oxigênio C–O, porque o álcool
tem a mesma configuração que o acetato. Como se verifica
que álcool opticamente inativo (-) é a imagem num espelho do
material de partida, o álcool opticamente ativo (+), direção
oposta de rotação óptica, durante a série de reações, tem de
ter ocorrido uma inversão de configuração, e só pode ter
ocorrido durante a reação de MeCO2- com tosilato. A reação
deste tosilato com um número de outros íons mostrou que
ocorre inversão de configuração na reação com Br- , para dar o
brometo, e este como o acetato tem configuração oposta à do
álcool de partida.
Mecanismo SN1: racemização
Um íon carbônio formado na etapa lenta, determinante
da velocidade da reação, é planar, e o ataque subsequente por
um nucleófilo como –OH, ou o solvente (H2O), ocorre com igual
facilidade por ambos os lados do íon carbônio planar,
produzindo uma mistura 50:50 de espécies com a mesma
configuração e com configuração oposta à do material de
partida, a racemização daria origem a um produto (±)
opticamente inativo.

111
C Br
R
R'R" (+)
C
R
R' R"Br-
+OH
-
OH-
OH
R"
R
R'
OH
R
R'R"
50%
50%
CH3 CH3+
Na prática, a racemização prevista raramente é
observada, sendo acompanhada por certo grau de inversão.
Verifica-se que as proporções relativas das duas dependem:
(a) da estrutura do haleto, estabilidade reativa do íon carbônio,
(b) do solvente, sua capacidade como nucleófilo.
Quanto mais estável for o íon carbônio, maior é a
proporção de racemização, quanto mais nucleófilo for o
solvente, maior é a proporção de inversão. Estas observações
tornam-se compreensíveis se a ionização SN1 é determinante
da velocidade.
Onde (1) é um par iônico íntimo em que os contra-íons
solvatados estão fortemente associados sem que entre eles
haja moléculas do solvente; (2) é um par iônico separado pelo
solvente; e (3) representa o par de íons dissociados e
solvatados separadamente.
Numa reação de solvólise, é provável que o ataque em
R+ por uma molécula de solvente, H2O, em (1) conduza a
inversão, dado que o ataque pode ocorrer “por trás” de R+, mas
não pela “frente” onde não há moléculas de solvente, e que
está impedido pelo contra-íon Br-. Em (2) é mais provável que o
ataque ocorra por ambos os lados, conduzindo a racemização,
enquanto que ataque em (3) pode ocorrer sem dúvida com
igual facilidade por ambos os lados. Assim, quanto mais longa
R Br R+ Br-
R+ Br-
Br-
R+ +δ
+δ
-
(1) (2) (3)

112
for vida de R+, mais difícil é o ataque pelo nucleófilo, e maior a
proporção de racemização
Assim, a solvólise de (+)C6H5CHMeCl, que pode formar
um íon carbônio estabilizado de tipo benzílico, conduz a 98%
de racemização, enquanto que a de (+)C6H13CHMeCl, em que
não há estabilização comparável, conduz apenas a 34% de
racemização. A solvólise de (+)C6H5CHMeCl em 80% de
acetona e 20% de água conduz a 98% de racemização, mas
em água apenas, observa-se somente 80% de racemização.
As mesmas considerações gerais aplicam-se a reações de
substituição nucleofílicas por Nu: tal como à solvólise, exceto
quando R+ tiver tempo de vida maior, pelo menos em parte, o
solvente que tem de ser removido antes que o Nu: possa
atacar R+. É importante notar que a racemização é nitidamente
um requisito estereoquímico menos importante para reações
SN1 do que a inversão é para as reações SN2.
Mecanismo SN1: retenção de configuração
As reações de substituição que conduzem a inversão de
configuração, a racemização, ou a uma mistura de ambas, são
comuns, mas conhecem-se diversos casos de reações que
ocorrem com retenção de configuração. Uma reação para a
qual se demonstrou ocorrer essa retenção de configuração é a
substituição de OH por Cl, através do uso de cloreto de tionilo,
SOCl2, na presença de solvente nucleofílico.
C OH
Me
PhH
SOCl2C Cl
Me
PhH
+ SO2 + HCl
Dois métodos, com base nos reagentes inorgânicos e
cloreto de tionilo, tribrometo fósforo, ostenta uma menção
especial. O cloreto de tionilo reage com álcoois para formar

113
cloretos de alquila. Os subprodutos da reação inorgânicas,
dióxido de enxofre e ácido clorídrico, ambos são gases à
temperatura ambiente e são facilmente removidos, tornando-se
fácil isolar o cloreto de alquila.
Os álcoois terciários são facilmente convertidos em
cloretos de alquila primário e secundário na presença de ácido
clorídrico e cloreto de tionilo. As reações com cloreto de tionila
normalmente são realizadas na presença de carbonato de
potássio ou piridina.
Participação de grupos vizinhos: “retenção de
configuração”
A retenção de configuração em reações de substituição
nucleofílica em que a característica comum é um átomo ou
grupo, próximo do carbono que sofre o ataque, tem um par
eletrônico disponível. Este grupo vizinho participa utilizando o
seu par eletrônico para impedir a “parte de trás” do átomo de
carbono, que sofre substituição, do ataque pelo reagente
nucleófilo, assim o ataque só pode ocorrer “pela frente”,
conduzindo a retenção de configuração. Verifica-se que a
hidrólise básica da 1,2-cloridina (10) dá o 1,2-diol (14) com a
ClCH3
H
OH
CH2
CH3
CH2
CH3
OH-
* ClCH3
H
O
CH2
CH3
CH2
CH3
*-
C
CH3H
O
CH2
CH3
CH2
CH3
*
-
OH-
CCH3
H
O
CH2
CH3
CH2
CH3
OH*H2O
OHCH3
H
OH
CH2
CH3
CH2
CH3
*
Inversão (1)
Inversão (2)
OH-
(10) (11)
(12)
(13)(14)

114
mesma configuração.
O ataque inicial pela base em (10) dá o ânion alcóxido
(11), o ataque interno por este RO- dá então o epóxido (12)
com inversão de configuração em C* (estes intermediários
cíclicos podem ser isolados em muitos casos), este átomo de
carbono, sofre ataque normal SN2 por –OH, com uma segunda
inversão de configuração em C*. Finalmente, este segundo
ânion alcóxido (13) abstrai um próton do solvente para dar o
produto 1,2-diol (14) com a mesma configuração que o material
de partida (10). Esta retenção aparente de configuração foi
resultante de duas inversões sucessivas.
O oxigênio, enxofre e nitrogênio, também foram estudos
como grupos vizinhos em reaçoes e em situações em que,
embora não esteja em causa qualquer aspecto estereoquímico,
velocidades inesperadamente elevadas sugerem uma
alteração mecanística.
EFEITO DE NUCLEÓFILOS E GRUPOS ABANDONADORES
Nucleófilo
A alteração do reagente utilizado, o grupo de entrada
(nucleófilo), não alterará diretamente a velocidade de uma
reação de substituição SN1, porque este reagente não toma
parte na etapa determinante da velocidade da reação. No
entanto, numa substituição SN2, quanto mais forte for o
reagente nucleofílico, mais rápido a reação ocorre. Pode talvez
esperar-se que um reagente nucleofílico se correlacione com a
sua basicidade, ambas envolvem a disponibilidade de pares
eletrônicos e as facilidade com que são doados. O paralelo não
é de modo algum exato, a basicidade envolve a doação de
pares eletrônicos ao hidrogênio, enquanto que a nucleofilia
envolve a doação de pares eletrônicos para outro átomo,

115
frequentemente o carbono. A basicidade envolve uma situação
de equilíbrio (termodinâmico), ∆G, enquanto que nucleofilia
envolve normalmente um equilíbrio cinético, ∆G≠≠≠≠, a basicidade
é provável que seja pouco afetada por influências
esteroquímicas, enquanto que a nucleofilia pode ser muito
afetada.
Esta distinção resulta em certa medida que foi
introduzida entre bases moles e bases duras.
Base dura é aquela em que o átomo doador é muito
eletronegativo, de baixa polarizabilidade e difícil de oxidar, –OH, –OR, R3N:.
Base macia é aquela em que o átomo doador tem baixa
eletronegatividade, polarizabilidade elevada e é fácil oxidar,
RS-, I-, SCN-. Para um dado grau de basicidade, a moleza
promove a nucleofilia. Os dados relativos a basicidade são em
geral os mais acessíveis e podem ser utilizados como guias
para a nucleofilia, desde que comparados com análogos.
Quando o átomo atacante for o mesmo, os dois são
razoavelmente paralelos, e quanto mais forte é a base, mais
poderoso é o nucleófilo.
EtO- > PhO- > MeCO2- > NO3
-
Também pode ocorrer variação no tipo mecanístico com
a mudança de nucleófilo, assim uma substituição que é SN1,
em H2O, HCO3-, MeCO2
-, etc., pode tornar-se SN2 com –OH ou
EtO-.
Verifica-se que a nucleofilia é muito afetada pelo
tamanho do átomo atacante no nucleófilo, pelo menos em
comparação dentro do mesmo grupo ou subgrupo da tabela
periódica, observa-se:
I- > Br- > Cl- RS- > RO-
Tanto o tamanho como a eletronegatividade governam a
polarizabilidade, à medida que o átomo aumenta de tamanho,

116
menor a influência exercida pelo núcleo sobre os elétrons
periféricos, que assim se tornam mais facilmente polarizáveis,
conduzindo à iniciação de ligação a separações nucleares
crescentes. Também quanto maior o íon ou grupo nucleófilo,
menor a sua energia de solvatação, mais rapidamente é
convertido no nucleófilo eficaz largamente não solvatado. É a
combinação destes fatores que torna o íon iodeto, I-, grande,
altamente polarizável e pouco solvatado, num nucleófilo muito
melhor do que o íon fluoreto, F-, pequeno, dificilmente
polarizável e muito solvatado (ligações de hidrogênio com um
solvente hidroxilado), apesar de este ser a base mais forte.
Neste caso, devemos esperar que a velocidade da
reação, ao passar de um solvente hidroxilado para um solvente
não-prótico polar, aumente muito menos com I- do que com Br-
ou Cl-.
Outro aspecto também interessante surge com
nucleófilos que tem mais do que um, geralmente dois átomos
adequados dos quais podem atacar o substrato, nucleófilos
ambidentados. -X Y X Y
-
Para as reações SN1 (altamente polares) o ataque
ocorre no íon carbônio, R+, através do átomo do nucleófilo que
tem a densidade eletrônica mais elevada. Por exemplo, com
haletos que não reagem facilmente por SN1, este pode ser
induzido pelo uso do sal de prata do ânion, AgCN, dado que
Ag+ promove a formação de R+ através da precipitação de AgX.
R Br + Ag+CN
- Lento Br + R+ + CN- Rápido R N C
+ -Ag
C N- C N
-.. ..
..
..
..

117
Na ausência daquela indução por Ag+, com Na+[CN]-,
verifica-se que a reação SN2 resultante ocorre com ataque
preferencial no átomo mais polarizável do nucleófilo.
Isto é compreensível se, ao contrário de SN1, a formação
da ligação ocorrer agora no ET para a etapa determinante da
velocidade, a fácil polarizabilidade do átomo ligante no
nucleófilo, o inicio da ligação e uma separação internuclear tão
grande quanto possível. Analogamente, verifica-se que o íon
nitrito [NO2]- resulta na formação de nitritos de alquila, R-O-
N=O, em condições SN1 (oxigênio é o átomo de densidade
eletrônica maior) e nitroalcanos, R-NO2, em condições SN2 (N é
o átomo mais facilmente polarizável).
O grupo abandonador
A alteração do grupo abandonador afetará a velocidade
das reações SN1, e SN2, desde que em ambas, a quebra da
ligação ao grupo abandonador está envolvida na etapa
determinante da velocidade. Em ambos os casos, a força da
base é sem dúvida um aspecto com algum significado, mas
nas reações SN2 a redistribuição de cargas negativas no ET e,
tal como com o grupo de entrada, a polarizabilidade no átomo
ligante do grupo de saída estará em cheque na formação do
ET. Esta é a principal razão para a sequência de reatividade
SN2 de haletos.
R – I > R – Br > R – Cl > R – F
A polarizabilidade elevada torna I- tanto um bom grupo
de entrada (nucleófilo) como um bom grupo de saída
NC-
+ R Br ................NCδ
-δ
-
BrR N C R + Br-

118
(abandonador), e por isso é frequentemente utilizado para
induzir (substituir) numa reação de substituição lenta.
OH2 R Cl+ +
+
+
LentoOH R H
+Cl
-:
I-
R Cl+ Rápido
Rápido
I R Cl-
R OHH+I-
Este efeito é designado catálise nucleofílicla. Quanto
mais forte e dura, como base, como grupo de saída, menos
facilmente pode ser substituído, por isso, grupos como: –OH, –
OR, NH2 ligados a carbono por átomos pequenos, muito
eletronegativos de baixa polarizabilidade, não podem
normalmente ser diretamente substituídos por outros
nucleófilos. Verifica-se que os melhores grupos abandonadores
são os ânions de ácidos fortes, os ânions haleto indicados
acima e espécies como p-MeC6H4SO3- (ânion tosilato), p-
BrC6H4SO3- (ânion brosilato), etc. Os requisitos SN1 são muito
semelhantes, observa-se a mesma frequência de reatividade
para R-X, e os principais requisitos para o grupo de saída são a
sua fraqueza como uma base, ânion de ácido forte e de fácil
solvatação.
Reações de substituição difíceis, ou mesmo impossíveis
de ocorrer diretamente podem ser efetuadas por modificação
do grupo de saída, por protonação, de modo a torná-lo numa
base mais fraca e/ou mole. O grupo -OH não pode ser
diretamente substituído por Br-, mas é facilmente substituído se
for primeiro protonado.

119
+Br-
+
+R OH Br R OH-
X
H+
+Br-
R O
H
H +Br R OH2
Existem duas razões principais para que isto aconteça:
(a) Br-, agora ataca uma espécie positivamente carregada, e
não neutra, e (b) a H2O é uma base mais fraca, é um grupo de
saída muito melhor do que o –OH fortemente básico. A bem
conhecida utilização de HI para a clivagem de éteres resulta do
fato de I- ser das espécies mais nucleófilas que podem ser
geradas na solução fortemente ácida requerida para a
protonação inicial.
++
R OPhH
+
R OPh
H
I-
R I PhOH
A ordem de reatividade relativa dos grupos
abandonadores segue:
OH -
→ NH2
-
→ OR -
→ F- → Cl- → Br -
→ I- → TosO
- 1 1 1 1 200 10000 30000 60000
4o ROTEIRO DE ESTUDO DIRIGIDO
1. Escreva a equação da lei da velocidade para a reação de hidrólise de haletos primários e secundários. A lei de velocidade é a mesma para os haletos terciários? 2. De onde provém a energia necessária para a heterólise dos haletos terciários?

120
3. Qual a correlação entre a polaridade do solvente e a velocidade de uma reação que se processa via mecanismos SN1 e SN2? 4. Dada a reação SN2: H3C – CH2I + N3
- � H3C –CH2N3 + I- Explique em que solventes ela ocorre mais rapidamente: metanol ou DMSO? 5. Explique o efeito da estrutura dos haletos (primários, secundários e terciários) na reação de hidrólise. 6. Dê exemplos de haletos que são difíceis de hidrolisar pelos mecanismos SN1 e SN2.
7. Explique a estereoquímica dos produtos formados via mecanismos SN1 e SN2. 8. Como se faz determinação da configuração relativa?
9. Em que mecanismo de substituição nucleofílica observa-se a retenção de configuração? Explique. 10. Quando se deixa 2-iodo-octano opticamente ativo, em solução de acetona com NaI, observa-se que o haleto apresenta perda de atividade ótica. Mostre o que acontece.
11. A reação, CH3CH2CH2Br + NaCN � CH3CH2CH2CN + NaBr deve ocorrer mais rapidamente em DMF ou em metanol? 12. Explique as afirmações abaixo: a) Quando brometo de t-butila sofre solvólise, em uma mistura de metanol e água, a velocidade de solvólise aumenta, quando o percentual de água aumenta. b) Quando cloreto de etila reage com iodeto de potássio, em metanol e água, a velocidade da reação diminui, quando o percentual de água aumenta. 13. Partindo-se de (S)-2-bromo-butano mostre como preparar: a)(R)-CH3CH(OCOCH3)CH2CH3, b) (R)-CH3CH(SCH3)CH2CH3 14. Em cada par indique que haleto de alquila reage mais rapidamente pelo mecanismo SN2: a) Brometo de iso-butila ou brometo de n-propila? b) Cloreto de n-propila ou brometo de n-propila?

121
c) Cloreto de etila ou cloreto de vinila? 15. O cloreto de (R)-α-fenil-etila (C6H5CHClCH2CH3), opticamente puro, apresenta (α)= -109; o álcool (R)-α-fenil-etila (C6H5CHOHCH2CH3), opticamente puro, apresenta (α)= -42,3. Por tratamento do cloreto (α)= -34, com solução diluída de NaOH, obtém-se o álcool de (α)= +1,7. Calcule: a) a pureza ótica do reagente e do produto da reação; b) a porcentagem de retenção e de inversão; c) a porcentagem de racemização. E de retenção ou de inversão. 16. Quando p-(2-bromoetil)-fenol é cuidadosamente tratado com base, um composto C8H8O é isolado. Este composto não mostra bandas de absorção no IV acima de 3200 cm-1, mas apresenta uma banda forte em 1670 cm-1 e UV máximo em cerca de 230 nm. Em tratamento com HBr o composto reverte facilmente ao fenol original. Considere estas observações e explique o que realmente acontece. 17. Em cada caso, indique o mecanismo mais provável da reação de SN1 ou SN2. Escreva o mecanismo que leva ao produto principal (incluindo a estereoquímica onde for apropriado) e indique também produtos secundários (sub-produtos) possíveis de serem formados: a) 3-fenil-1-bromo-propano + NaCN em dimetilformamida. b) (R)-3-fenil-1-bromo-butano + NaCN em dimetilformamida. c) (S)-1-fenil-1-bromo-butano + NaCN em dimetilformamida. d) (S)-1-fenil-1-bromo-butano + AgOAc em etanol. e) γ,γ-dimetil-alil-tosilato + (C2H5)3N em metanol. f) Ácido-(R)-4-cloropentanóico + NaNO2 em água. g) Ácido-(R)-4-cloropentanóico + em ebulição com água. h) Cloreto de benzila + Na2CS3 em acetona. i) (S)(-)1,3-dibromo-1-fenil-propano em solução aquosa de ácido acético a quente. j) (S)(-)1,3-dibromo-1-fenil-propano + KOAc em dimetilsulfóxido. 18. Considere a transformação representada pelo esquema abaixo:
O tipo de reação e a expressão da lei de velocidade são:

122
Tipo de reação Expressão da lei de velocidade
a) Substituição nucleofilica de primeira ordem
v = k[CH3Cl]
b) Substituição nucleofilica de sugunda ordem
v = k[CH3Cl][OH-]
c) Adição eletrofilica de primeira ordem v = k[OH-] d) Adição eletrofilica de segunda ordem
v = k[CH3Cl]2
e) Adição eletrofilica seguida de eliminação
v = k[CH3Cl][OH-]2
19. Na reação do but-1-eno com HBr, o intermediário e o tipo de mecannismo operante, respectivamente, são:
Intermediário Tipo de mecanismo a) Carbânion Substituição nucleofílica b) Carbânion Adição nucleofílica c) Carbocátion Adição eletrofílica d) Carbocátion Substituição eletrofílica e) Radical livre Eliminação
20. Derivados halogenados são bons substratos para a preparação de éteres, em laboratório, através de reação com reagentes nucleofílicos fortes como o metóxido de sódio. A velocidade dessas reações pode ser drasticamente aumentada pela escolha apropiada do solvente. Considerando os substratos 3-bromo-3-metilhexano e brometo de metila, qual o tipo de mecanismo e o solvente adequado para a reação desses substratos halogenados com o metóxido de sódio? Dado: DMSO corresponde a (CH3)2S = O
3-bromo-3-metilhexano Brometo de metila Mecanismo Solvente Mecanismo Solvente a) SN1 DMSO SN2 DMSO b) SN1 Hexano SN2 Hexano c) SN1 Hexano SN1 Metanol d) SN2 DMSO SN1 Metanol e) SN2 Metanol SN2 Hexano

123

124
UNIDADE V - SUBSTITUIÇÃO ELETRÓFILICA E NUCLEÓFILICA EM SISTEMAS AROMÁTICOS
ATAQUE ELETROFILÍCO AO BENZENO
Complexos ππππ e σ
A primeira etapa da reação envolve a interação entre o
eletrófilo que se aproxima e as orbitais π deslocalizadas,
chamados complexos π.
Por exemplo, o metilbenzeno (tolueno) forma um
complexo 1:1 com o ácido clorídrico (HCl) a -78 °C, sendo a
reação facilmente reversível, não se forma uma ligação entre
um átomo de carbono do anel e o próton de HCl que é
confirmado quando a reação é feita em DCl. Este também dá
origem a um complexo π, mas a sua formação e decomposição
não faz a troca de deutério com qualquer dos átomos de
hidrogênio do anel, mostrando que no complexo não se formou
nenhuma ligação C–D. Os hidrocarbonetos aromáticos formam
complexos π com espécies como os halogênios, Ag+, o ácido
pícrico, 2,4,6-(O2N)3C6H2OH, formando-se adutos estáveis,
corados e cristalinos, cujos pontos de fusão podem ser usados
na caracterização desses hidrocarbonetos. Esses adutos são
também conhecidos como complexos de transferência de
carga.
Na presença de um composto que tenha um
orbital vazio, um ácido de Lewis como AlCl3, forma-se um
complexo diferente. A troca de HCl, por DCl, origina uma troca
rápida de deutério com os átomos de hidrogênio do anel, o que
indica a formação de um complexo σ, também chamado
intermediário de Wheland, no qual H+ ou D+, está ligado por
covalência a um átomo do anel.

125
+ DCl
AlCl3
H D
+AlCl4
-
(-H+)
D
Os complexos π e σ com, metilbenzeno e HCl, saõ
diferentes uns dos outros. Assim, a formação do complexos π
conduz a solução não condutora de eletricidade, sem alteração
de cor e a uma pequena modificação no espectro ultravioleta,
indicando pouca alteração na distribuição eletrônica do
metilbenzeno inicial. Na presença de AlCl3, a solução torna-se
verde, condutora de eletricidade e o espectro ultravioleta é
diferente do metilbenzeno inicial, indicando a formação de um
complexo σ, não há provas de que o cloreto de alumínio forme
complexos do tipo H+AlCl-4.
A reação pode ser completada pela remoção de um
próton do complexo σ pela espécie AlCl-4 [(2) → (4)]. Assim,
ocorre a troca de átomos de hidrogênio na presença de HCl,
mas há substituição de hidrogênio por deutério quando se
utiliza DCl, a reação é uma substituição eletrofíliica.
Expulsando H+, por substituição em vez de adição, são retidas
no produto (4) as orbitais π deslocalizados e preenchidos, e a
aromaticidade do composto é recuperada.
O aumento de estabilização que se obtém quando se
passa (2) → (4) fornece a energia requerida para quebrar a
ligação C–H, necessária para a expulsão de H+, como na

126
reação de HCl com alcenos, não existe este fator para
favorecer a substituição, e a reação é de adição.
Nitração
Esta é uma reação de substituição aromática, muito
estudada, e, em consequência, é a reação que provavelmente
fornece um quadro mecanístico mais detalhado. A reação de
nitração é feita com uma mistura de ácidos nítrico e sulfúrico
concentrados, chamada mistura nitrante.
O ácido sulfúrico reage com o ácido nítrico e não sobre o
benzeno, na mistura reacional. Isto é evidenciado, porque as
soluções de ácido nítrico em ácido sulfúrico puro apresentam
uma depressão quádrupla do ponto de congelação molecular, o
que foi interpretado como sendo devido à formação dos quatro
íons.
OH NO2
H2SO4
O+
NO2
H
H
H2SO4H3O
+ + HSO4- + +NO2
HNO3 + 2 H2SO4+NO2 + H3O
+ + HSO4-
ou
A presença de +NO2, o íon nitrônio, foi confirmada pelo
espectro de Raman em 1400 cm-1, como uma espécie linear e
triatômica. O próprio ácido nítrico, em ácido sulfúrico
concentrado, é quase totalmente convertido em +NO2, e não há
dúvidas de que é este o eletrófilo na nitração. A pouca eficácia
do próprio ácido nítrico na nitração do benzeno é explicada por
conter pouco +NO2. A pequena quantidade que se encontra
presente é obtida pelo processo em duas etapas em que o
ácido nítrico é primeiro rapidamente convertido no seu ácido
conjugado e este, mais lentamente, no íon nitrônio.

127
Muitas nitrações estão de acordo com uma lei de
velocidade, Velocidade = k[Ar–H][+NO2], mas na prática, por
diversas razões, a cinética da reação nem sempre é fácil de
acompanhar ou de interpretar.
A lei de velocidade para a reação de nitração do
benzeno é compatível com o seguinte mecanismo postulado. A
nitração ocorre pelo mecanismo em etapas, envolvendo um
estado de transição simples, onde a formação da ligação C–
NO2 é lenta e determinante da velocidade, passando por um
intermediário, e após ocorre a quebra rápida da ligação C–H.
+ +NO2
H NO2 NO2
+ + H+Lenta Rápida
A quebra da ligação C–H, perda de H+, do intermediário
restabelece o caráter aromática no nitrobenzeno formado. O
próton, H+, aqui é removido do intermediário pelo ataque pela
base, HSO-4, da mistura nitrante.
No que se refere às velocidades das reações de
substituição aromática, é a formação do estado de transição
(ET1) que precede o intermediário, determina a velocidade da
reação, conforme a figura abaixo:
ET1ET2
Caminho da reação
Ene
rgia
livr
e
+ +NO2
H NO2
+NO2
+ H+
Muitas vezes é difícil obter informação detalhada acerca
das espécies envolvidas nos estados de transição e dos
intermediários de reação. Por isso, tomado como seus
modelos, pode justificar-se com base no princípio de

128
Hammond, segundo o qual, numa dada sequência, as espécies
que se sucedem imediatamente se assemelham em termos
energéticos também se assemelham quanto à estrutura. Na
figura acima é provável que o intermediário seja um modelo
melhor para o ET do que o material de partida.
Compostos aromáticos extremamente reativos, como o
fenol, sofrem nitração rápida mesmo em ácido nítrico diluído, e
a uma velocidade muito maior do que pode ser explicado com
base na concentração de +NO2 presente na mistura.
Demonstrou-se ser isto devido à presença de ácido nitroso no
sistema, o qual efetua a nitrosação do núcleo reativo através
do íon nitrosônio, +NO (ou outra espécie capaz de realizar a
nitrosação).
HNO3 + 2 HNO3H3O
+ + 2NO3- + +NO
OH
+NO
OH
NOH
+
OH
NO
NO3- HNO3
OH
NO2
+ HNO2
O nitrosofenol obtido é oxidado muito rápido pelo
ácido nítrico, dando nitrofenol e ácido nitroso; é produzido mais
ácido nitroso, e o processo reacional é acelerado. Não é
necessária a presença inicial de ácido nitroso no ácido nítrico,
dado que uma pequena quantidade deste último oxida o fenol
com formação de HNO2. A etapa determinante da velocidade é
novamente a formação do intermediário.
Halogenação
Ao contrário da nitração, a halogenação pode envolver
vários eletrófilos diferentes no ataque ao sistema aromático. Os
halogênios livres, Cl2 e Br2, atacam rapidamente um núcleo
ativado como o fenol, mas são incapazes de substituir o próprio
benzeno (contudo, a ativação fotoquímica pode conduzir a
adição por ação dos átomos livres de halogênio), sendo, para

129
tal, necessário um ácido de Lewis catalisador, como AlCl3, que
polariza a molécula de halogênio, tornando-a com uma
extremidade eletrofílica.
A lei de velocidade para a halogenação é geralmente da
forma:
Velocidade = k[Ar–H][X2][ácido de Lewis]
É provável que o benzeno forme um complexo π com,
por exemplo, Br2, e que o ácido de Lewis interague então com
esse complexo. O catalisador provavelmente polariza a ligação
Br–Br, assiste na formação de uma ligação σ entre a molécula
de bromo, agora com uma extremidade eletrofílica, e um átomo
de carbono do anel remove o íon brometo na formação do
complexo σ.
BrBrHBr Br
Br2
FeBr3
- FeBr4-
+ HBr + FeBr 3+
O ânion FeBr-4 remove o próton do complexo σ (positivo.
Nas reações de halogenação, não foram observados
efeitos isotópicos cinéticos, de hidrogênio, para a cloração, e
raramente para a bromação, porque estas reações de cloração
e bromação seguem normalmente o mecanismo proposto para
a nitração, em que a etapa determinante da velocidade da
reação é formação do complexo σ.
Na reação de iodação, que ocorre com o próprio iodo em
espécies ativadas, é observado o efeito isotópico cinético, de
hidrogênio, porque a reação é facilmente reversível (contrário
às outras halogenações) com perda de I pelo complexo σ, em
vez de perda de H, onde k-1 > k2.

130
Na reação de iodação do fenol e de 2,4,6-trideuteriofenol
kH/kD ≈ 4, sugerindo um mecanismo onde há um equilíbrio
entre as espécie dos reagentes e do estado de transição. A
iodação é geralmente assistida pela presença de bases ou de
agentes oxidantes, que removem HI e assim deslocam o
equilíbrio para a direita. Os agentes oxidantes também tendem
a produzir, a partir de I2, I+ ou um complexo contendo iodo
positivamente polarizado e proporcionando um eletrófilo mais
eficaz. A halogenação também pode ocorrer utilizando-se
compostos inter-halogenados, Brδ+–Clδ-, Iδ+–Clδ-, etc.,
ocorrendo o ataque através do halogênio menos eletronegativo
sendo esta a extremidade eletrófila da molécula. Verifica-se
que as duas espécies referidas efetuam, respectivamente, a
bromação e a iodação.
Sulfonação
A reação de sulfonação do benzeno é uma reação na
qual um átomo de hidrogênio em um anel aromático é
substituído por um grupo funcional ácido sulfônico.
O benzeno é sulfonado de modo relativamente lento por
ácido sulfúrico concentrado, a quente, mas rapidamente por
ácido sulfúrico fumegante (estando a velocidade da reação
relacionada com o concentração de SO3 no ácido) ou por SO3
em solventes inertes. O eletrófilo atacante pode ser o SO3 ou o
seu ácido conjugado, +SO3H, o qual pode variar com as
condições reacionais, mas em H2SO4 em geral deve ser SO3,
que existe em pequenas concentrações no equilíbrio.
O ataque ocorre através de enxofre (S), que encontra-se
polarizado positivamente, muito deficiente de elétrons.

131
A reação de benzeno com ácido sulfúrico para produzir
ácido benzenossulfónico, é reversível, mas pode ocorrer por
várias técnicas. Retirar a água formada na reação, por
exemplo, permite benzenossulfónico ser obtido em maior
quantidade. Quando uma solução de trióxido de enxofre em
ácido sulfúrico é usada como agente de sulfonação, a
velocidade de sulfonação é muito mais rápida, e o equilíbrio é
deslocado totalmente para o lado dos produtos, de acordo com
a equação.
Entre uma variedade de espécies eletrofílicas presentes
em ácido sulfúrico concentrado, o trióxido de enxofre é
provavelmente o eletrófilo real em sulfonação aromática. O
mecanismo proposto da sulfonação do benzeno por trióxido de
enxofre ocorre em etapas:
Etapa 1. O benzeno ataca o trióxido de enxofre, e esta é
a etapa determinante da velocidade da reação.
Etapa 2. O carbono hibridizado, sp3, do intermediário,
perde um próton e a aromaticidade do anel é restaurada. A
espécie que abstrai o próton é o íon hidrogênio sulfato formado
pela ionização do ácido sulfúrico.

132
Etapa 3. Uma transferência rápida de próton do oxigênio
do ácido sulfúrico ao oxigênio do benzenossulfonato conclui o
processo.
REAÇÕES DE FRIEDEL-CRAFTS
Estas reações podem ser subdividas em reações de
alquilação e acilação.
Alquilação de Friedel-Crafts
O átomo de carbono dos haletos de alquila, Rδ+–Xδ-, é
eletrofílico, mas os haletos de alquila reagem com benzeno na
presença de cloreto de alumínio, AlCl3, (um ácido de Lewis)
para produzir alquilbenzenos.
O fato dos haletos de alquila reagirem com ácidos de
Lewis foi demonstrado pela troca de bromo radioativo em EtBr,
quando misturado com AlBr3 e re-isolado, e pelo isolamento de
complexos 1:1, sólidos, do CH3Br·AlBr3 a baixas temperaturas -
78 °C. Estes complexos, embora polares, são condutores
fracos. Quando R é capaz de formar um íon carbônio
particularmente estável, Me3C–Br, é provável que na alquilação
seja o próprio íon carbônio, Me3C+.

133
Haletos de alquila por si só não são suficientemente
eletrofílicos para reagir com o benzeno. Cloreto de alumínio,
ácido de Lewis, serve como um catalisador para melhorar a
eletrofilicidade do agente alquilante. Com haletos de alquila
secundário e terciário, a adição de cloreto de alumínio leva à
formação de carbocátions, que atacam em seguida o anel
aromático.
Os haletos de alquila secundário reagem por um
mecanismo semelhante envolvendo ataque do benzeno a um
carbocátion secundário. Haletos de etila e metila não formam
carbocátions quando tratados com cloreto de alumínio, mas
reagem frente ao benzeno sob condições da alquilação de
Friedel-Crafts.
O mecanismo da reação do ataque do anel benzênico
ao cátion terc-butílico ocorre em duas:
Etapa 1. A reação do cloreto de terc-butilo com o cloreto
de alumínio gera o cátion terc-butílico, que é atacado pelos
elétrons do benzeno, e uma ligação de carbono-carbono é
formada
Etapa 2. A perda de um próton do intermediário o cátion
ciclohexadienil, produz o terc-butilbenzeno.

134
O mecanismo da reação é compatível com a lei de
velocidade:
Velocidade = k[Ar – H][RX][MX3]
A ordem de eficácia dos ácidos de Lewis como
catalisadores na alquilação de Friedel-Crafts é:
AlCl3 > FeCl3 > BF3 > TiCl3 > ZnCl2 > SnCl4
Em certo número de casos de alquilação de Friedel-
Crafts, verifica-se que o produto final contém um grupo alquilo
rearranjado. Assim, a reação de (CH3)3CCH2Cl/AlCl3 com
benzeno dá quase exclusivamente o produto rearranjado,
PhCMe2CH2Me, o que pode ser explicado com base no fato de
o complexo eletrófilo inicial ser polarizado para permitir o
rearranjo de [Me3CCH2]δ+...Cl...AlCl3
δ- a
[Me3CCH2Me]δ+...Cl...AlCl3δ- mais estável (estabilidade relativa
do íons carbônio). Pelo contrário, verifica-se que a reação de
Me3CCH2Cl/FeCl3 com benzeno dá quase exclusivamente o
produto não-rearranjado, Me3CCH2Ph, pressupondo-se que
isso se deve ao fato de o complexo com o ácido de Lewis não
se encontrar polarizado para permitir que ocorra isomerização.
Verifica-se que a temperatura também influi, para um dado
haleto/ácido de Lewis.
Os ácidos de Lewis catalisadores podem efetuar a
desalquilação, ou seja, a reação é reversível. Assim, verifica-se
que o etilbenzeno, com BF3 e HF, o produto da reação é
desproporcional. ET
HF
BF3
+
ET
+
ET
ET
45 % 10 % 45 %
Esta reação deve ser intermolecular, mas também há
rearranjos que envolvem alteração das posições relativas dos
substituintes no anel benzênico e que se verificou serem
intramoleculares. Assim, aquecendo p-dimetilbenzeno (p-
xileno) com AlCl3 e HCl, dá-se a sua conversão maioritária em

135
m-dimetilbenzeno (m-xileno), mais estável. A presença de HCl
é essencial, e a alteração envolve a migração de um grupo Me
na espécie incialmente protonada. CH3
CH3
H+
CH3
H CH3
CH3
CH3
H
CH3
CH3
++
p-Xileno m-Xileno
Além da possibilidade de rearranjar, a principal
desvantagem da utilização, para fins preparativos, desta
reação de Friedel-Crafts é a polialquilação. A presença de um
substituinte elétron retirador é geralmente suficiente para inibir
a alquilação de Friedel-Crafts e, por exemplo, o nitrobenzeno é
usado frequentemente como solvente que dissolve o AlCl3
muito rápido, evitando desse modo uma reação heterogênea.
Acilação de Friedel-Crafts
Uma reação de Friedel-Crafts que usa haletos de acila,
em vez de haletos de alquila, produz acilbenzenos conhecida
com reação de acilação de Friedel-Crafts. A acilação de
Friedel-Crafts, segue a mesma lei de velocidade geral da
alquilação.
Velocidade = k[Ar–H][RCOCl][AlCl3]
Também aqui se põe a questão de o eletrófilo efetivo ser
o íon acílio, como constituinte de um par iônico, ou um
complexo polarizado.
Muitos íons acílios foram detectados como complexos
sólidos e líquidos, MeCOCl e AlCl3, em solução nos solventes
polares (por espectrocopia no IV), e nos casos em que R é
muito volumoso.
O eletrófilo em uma reação de acilação de Friedel-Crafts
é um cátion acila (íon acílio), que é estabilizado por

136
ressonância. O cátion acila derivado de cloreto propanoíla é
representado por duas formas de ressonância.
Contudo, em solventes menos polares e em outras
condições, os íons acílios não são detectados e deverão ser o
complexo polarizado que atua como eletrófilo.
As reações químicas diretas indicam que íon acílio ou o
complexo polarizado podem estar envolvidos, dependendo das
condições da reação. Assim, na benzoilação do tolueno,
obtém-se a mesma mistura de produtos (1% m-, 9% o- e 90%
p-) com qualquer catalisador, ácido de Lewis, usando o cloreto
de benzoíla ou o brometo de benzoíla; isto sugere uma espécie
reagente comum para todos os casos, é o cátion Ph–C+=O. Por
outro lado, em muitos casos a proporção do produto orto é
muito reduzida, quando comparada com outras reações de
substituição eletrofílica, por exemplo na nitração, sugerindo o
envolvimento de um eletrófilo muito volumoso, obtendo
rendimento pelo complexo polarido do que pelo íon acila R–
C+=O linear. A natureza do eletrófilo, em cada caso, depende
muito das condições.
A reação pode então ser representada do modo
seguinte.
Uma diferença significativa entre a acilação e a
alquilação é que na acilação mais de 1 mol de ácido de Lewis é

137
usado. Isto porque, na acilação, ocorre a complexação do
ácido de Lewis com a cetona à medida que esta é formada,
impedindo a participação subsequente do ácido de Lewis na
reação.
Na reação de acilação, não ocorre poliacilação; a cetona
é muito menos reativa do que o benzeno. Não se observa o
rearranjo de R, ao contrário do que se verifica na alquilação,
mas pode ocorrer descarbonilação, e em especial quando R
pode formar um íon carbônio estável, de tal modo que o
resultado final é a alquilação em vez da acilação esperada.
(CH3)3C–C+=O � CO + (CH3)3C+ + C6H6 � C6H5–
C(CH3)3
Pode fazer a formilação utilizando CO, HCl e AlCl3
(reação de Gatterman-Koch), o eletrófilo mais provável é o íon
acilíon, HC+=O.
Na reação, o equilíbrio é desfavorável à formação do
produto, mas é deslocado para a direita pela complexação de
aldeído com o catalisador, o ácido de Lewis.
A acilação também pode ser efetuada com anidridos de
ácidos, (RCO)2O, em presença de ácidos de Lewis
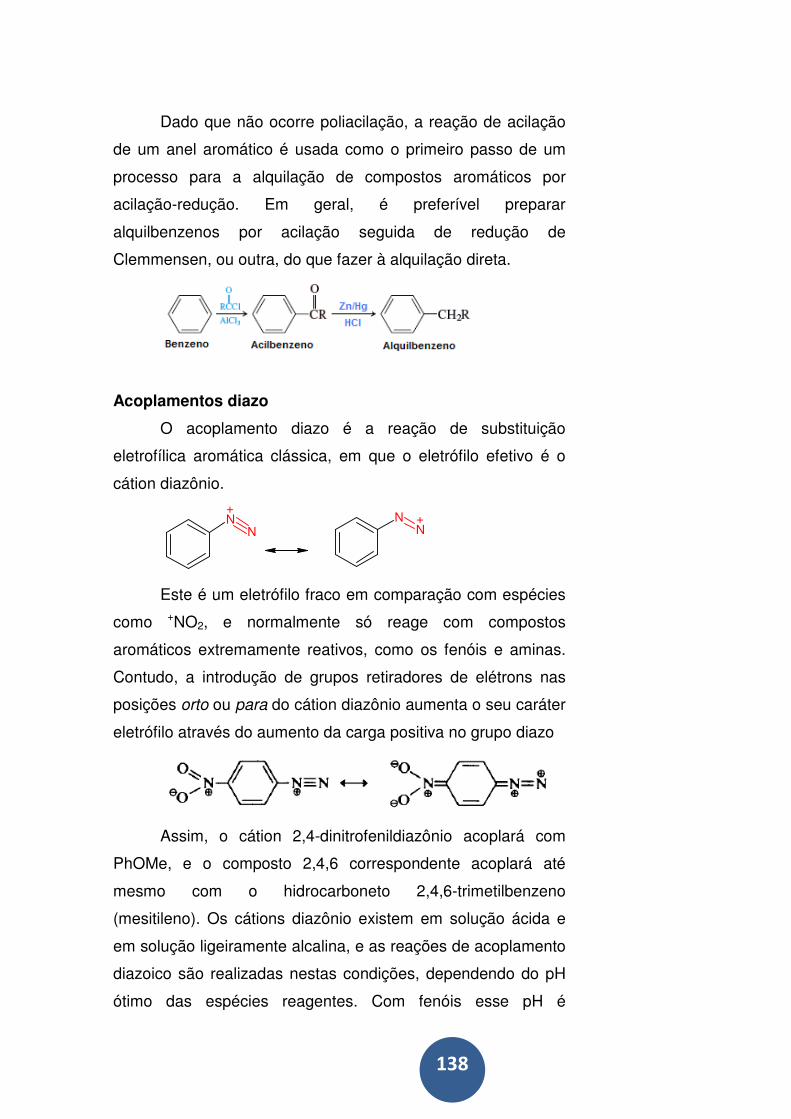
138
Dado que não ocorre poliacilação, a reação de acilação
de um anel aromático é usada como o primeiro passo de um
processo para a alquilação de compostos aromáticos por
acilação-redução. Em geral, é preferível preparar
alquilbenzenos por acilação seguida de redução de
Clemmensen, ou outra, do que fazer à alquilação direta.
Acoplamentos diazo
O acoplamento diazo é a reação de substituição
eletrofílica aromática clássica, em que o eletrófilo efetivo é o
cátion diazônio.
NN
NN+
+
Este é um eletrófilo fraco em comparação com espécies
como +NO2, e normalmente só reage com compostos
aromáticos extremamente reativos, como os fenóis e aminas.
Contudo, a introdução de grupos retiradores de elétrons nas
posições orto ou para do cátion diazônio aumenta o seu caráter
eletrófilo através do aumento da carga positiva no grupo diazo
Assim, o cátion 2,4-dinitrofenildiazônio acoplará com
PhOMe, e o composto 2,4,6 correspondente acoplará até
mesmo com o hidrocarboneto 2,4,6-trimetilbenzeno
(mesitileno). Os cátions diazônio existem em solução ácida e
em solução ligeiramente alcalina, e as reações de acoplamento
diazoico são realizadas nestas condições, dependendo do pH
ótimo das espécies reagentes. Com fenóis esse pH é

139
ligeiramente alcalino, dado ser PhO-, e não PhOH, que reage
com ArN+2, seguindo a lei de velocidade da reação.
Velocidade = [ArN2+][PhO-]
O acoplamento diazócio com o íon fenóxido forma o
produto substituído, após a abstração do próton H+.
A remoção do próton (normalmente não determinante da
velocidade) é assistida por uma das espécies básicas
presentes na solução. O acoplamento normalmente ocorre na
posição para, em vez de na posição orto, devido ao volume
considerável do eletrófilo reagente, ArN+2.
As aminas aromáticas são, em geral, menos facilmente
atacadas do que os fenóis, e o acoplamento diazoico
normalmente ocorre em soluções levemente ácidas,
assegurando assim uma protonada [PhN+2], sem conversão na
amina, ArNH2, o cátion protonado inerte, ArN+H3, são bases
muito fracas. A diazotação inicial de aminas primárias
aromáticas é efetuada em meio fortemente ácido para garantir
que a amina que ainda não reagiu seja convertida no cátion,
evitando deste modo o acoplamento com o sal de diazônio à
medida que se forma.
Com aminas aromáticas, a reação ocorre principalmente
no nitrogênio, com aminas primárias e secundárias (N-
alquilanilinas), para dar compostos diazoamino, após a
abstração do próton.

140
Com a maioria das aminas primárias, este é o produto
da reação, mas com as aminas secundárias (N-alquilanilinas)
também pode ocorrer acoplamento no átomo de carbono do
anel, enquanto que com aminas terciárias (N,N-dialquilanilinas)
só se obtém o produto acoplado ao carbono.
Verifica-se que a reação segue normalmente a lei geral
de velocidade.
Velocidade = k[ArN2+][PhNR2]
Em alguns casos, a reação de acoplamento é catalisada
por bases, e verifica-se que é acompanhada por um efeito
isotópico cinético, k-1 > k2, e a quebra da ligação, C–H participa
da etapa determinante da velocidade da reação.
Ataque eletrofílico a C6H5Y
Quando um derivado monossubstituído do benzeno,
C6H5Y, sofre substituição eletrofílica, nitração, o novo
substituinte pode ser incorporado na posição orto, meta ou
para, e a velocidade realtiva da reação de substituição pode
ser maior ou menor do que a velocidade da correspondente
reação de substituição no benzeno.
A reação de substituição ocorre de modo a formar, o
isômero meta ou uma mistura dos isômeros orto e para. Nos
compostos formados na posição meta do anel, em relação ao

141
substituinte, a velocidade relativa da reação é sempre menor
do que a da correspondente reação no benzeno.
Y
Y
NO2
Y
NO2
Y
NO2
NO2
+NO2
+NO2+
No caso, onde os produtos são formados nas posições
orto e para, a velocidade relativa da reação é normalmente
maior do que a velocidade da reação correspondente com o
benzeno. O grupo substituinte Y é quem controla a posição em
que vai ocorrer a reação com o segundo grupo substituinte, o
que pode ser explicado com base nos efeitos eletrônicos que Y
pode exercer. O grupo substituinte Y pode também exercer um
efeito estereoquímico, mas a influência deste fator está limitada
ao ataque na posição orto.
Os substituintes Y são classificados como diretores meta
ou orto/para. Se a reação é mais rápida do que o do benzeno,
os grupos substituintes são ativadores; se o ataque for mais
lento, são desativadores. Deve salientar que estes efeitos
diretores são mais relativos do que absolutos, numa reação de
substituição praticamente formam-se sempre os três isômeros,
embora a proporção de produto meta com um substituinte Y
diretor orto e para, ou de produtos orto/para com um
substituinte Y diretor meta possam ser muito pequenas. Assim,
verifica-se que a nitração do nitrobenzeno (Y = NO2) resulta
numa mistura de 93% de isômero meta, 6% de isômero orto e
1% de isômero para, assim, o grupo NO2 é classificado como
um substituinte desativador e diretor meta. Pelo contrário, a
nitração do metoxibenzeno (Y = MeO-) origina 56% do isômero

142
para, 43% do isômero orto e 1% do isômero meta, então o
grupo MeO- é um substituinte ativador e diretor orto e para.
EFEITOS ELETRÔNICOS DO GRUPO SUBSTITUINTE (Y)
O efeito que um dado substituinte Y no anel aromático
exerce sobre a velocidade da reação nas posições orto/para e
meta, respectivamente, será discutidos a seguir.
Efeitos eletrônicos de grupos substituintes desativantes
Os grupos substituintes desativantes, Y = +NR3, CCl3,
NO2, CHO, CO2H, SO3H, CN, etc., têm em comum um átomo
carregado positivamente, ou polarizado positivamente,
adjacente a um átomo de carbono do anel benzênico.
Por isso, são retiradores de elétrons (efeito indutivo
negativo) do anel benzênico.
Quando o grupo substituinte é o NO2, um grupo
desativante e meta-dirigente apresenta as seguintes formas de
ressonância, como exemplo de complexos σ correspondentes
para reagir com um segundo eletrófilo, E+, na posição, meta
em relação ao substituinte inicial NO2.
O efeito desativante de Y sobre o anel aromático
aumenta a velocidade relativa da reação de substituição, na
seguinte ordem aproximada, +NR3 < NO2 < CN < SO3H < C=O
< CO2H.
Os substituintes como +NR3 serão particularmente
desativantes em reações de substituição em que o próprio

143
eletrófilo reage estando este carregado positivamente, +NO2,
(kC6H5Y/kC6H6 = 1,5 x 10-8 para a nitração quando R=CH3).
Efeitos eletrônicos de grupos substituintes ativantes
Os grupos substituintes ativantes, como grupo alquila e
fenila, em relação ao hidrogênio, são doadores de elétrons
(efeito indutivo positivo). As estruturas canônicas
correspondentes ao ataque orto e para, nos quais uma carga
positiva está localizada no átomo de carbono adjacente, são
estabilizadas.
Esses fatores não atuam no complexo σ correspondente
ao ataque, e assim a substituição orto e para é favorecida,
devido ao efeito doador de elétron, o ataque em qualquer
posição será mais rápido do que no benzeno (kC6H5Y/kC6H6 = 3,4
x 102, para a cloração).
O grupo CMe3 exerce um efeito indutivo doador de
elétron maior do que CH3, verifica-se que o tolueno sofre um
ataque orto e para mais rápido do que o 2,2-dimetilbenzeno.
Isto é devido ao fato de a doação de elétrons de CH3 poder
ocorrer por hiperconjugação, assim como através de um efeito
indutivo, enquanto que com CMe3 só o efeito indutivo pode
atuar.

144
A estabilização específica das formas canônicas dos
complexos σ correspondentes ao ataque orto e para também
pode ser efetuada por um grupo fenila, e a velocidade relativa
da reação com o bifenil é maior do que no caso do benzeno
(kC6H5Y/k C6H6 = 4,2 x 102 para a cloração).
Os casos em que grupos substituintes (Y) têm em
comum um átomo adjacente ao núcleo que pode exercer efeito
indutivo doador de elétron, e um par eletrons (Y = OCOR,
NHCOR, OR, OH, NH2, NR2) que pode estabilizar
especificamente os complexos σ correspondentes ao ataque
orto e para.
A estabilização é particularmente acentuada não só por
estar envolvido mais um estado canônico na estabilização dos
complexos σ orto e para, mas porque as formas, em que a
carga positiva está localizada no oxigênio, são mais estáveis
do que as outras, em que a carga positiva está localizada no
carbono. Este efeito é suficientemente pronunciado para se
sobrepor muito ao feito indutivo retirador de elétron, que
também atua nestes dois complexos σ, e assim a substituição
é quase complemente orto e para (obtêm-se << 1% do
isômero meta na nitração de PhOMe) e muito mais rápido do
que com o benzeno (kC2H5OMe /kC6H6 = 0,7 X 106 para a
cloração).
A ação do efeito indutivo retirador de elétron pode,
contudo, ser observada na pequena quantidade de ataque em
meta (para o qual não há estabilização específica do complexo

145
σ por deslocalização dos elétrons), que ocorre mais lentamente
que no caso do benzeno.
O efeito ativador de grupo substituinte (Y) no anel
aromático aumenta a velocidade relativa de substituição, na
seguinte ordem aproximada.
OCOR < NHCOR < OR < OH < NH2 < NR2
O grupo NR2 é um ativante mais forte do que NH2,
devido ao efeito doador de elétron dos grupos R. Contudo, não
se deve esquecer que em solução ácida, a reação de nitração,
estes dois grupos serão convertidos em +NHR2 e +NH3, o
núcleo estará assim desativado, e a substituição será
predominantemente meta. Os grupos OCOR e NHCOR são
ativantes mais fracos do que OH e NH2, devido à redução de
disponibilidade eletrônica no O e N, por deslocalização sobre o
grupo carbonila elétron atraente adjacente.
Os halobenzenos que tem o grupo substiuinte (F,Cl, Br,
e I) têm um átomo adjacente ao núcleo que transporta um par
eletrônico, assim, pode ocorrer a estabilização específica dos
complexos σ correspondentes ao ataque orto e para.
Os halogênios são desativantes, e orto e para diretores.
O efeito indutivo (negativo) retirador de elétron dos halogênios
é tal que o ataque é mais lento do que no caso do benzeno.
São substituintes desativantes (kC6H5Cl/kC6H6 = 3 x 10-2 para a
nitração). Esta atração de elétrons por halogênios reflete-se no
estado fundamental por um dipolo no clorobenzeno, com a
extremidade positiva no núcleo, em comparação com o
metoxibenzeno, no qual o dipolo tem orientação oposta.

146
O efeito nos complexos σ carregados positivamente não
é assim tão simples, apresentando a seguinte ordem de
velocidade relativa para o ataque eletrofílico nos halobenzenos.
C6H5F > C6H5Cl ≈ C6H5Br > C6H5I
O fluorbenzeno reage com uma velocidade muita
próxima a do benzeno.
Fatores de velocidade parciais e seletividade
Métodos cinéticos refinados e a capacidade de
determinar com exatidão as proporções relativas dos isômeros
orto, meta e para formados, por métodos espectroscópicos,
permitem uma abordagem quantitativa da substituição
eletrofílica aromática. Um conceito útil é o de fatores de
velocidade parciais, a velocidade em que uma posição, em
C6H5Y é
atacada,
compara
da com
a
velocida
de de
ataque de uma posição do benzeno, e que se representa por
fposição.
Os fatores de velocidade parciais podem ser obtidos por
determinações cinéticas independentes das constantes de
velocidade realtivas kC6H5Y e kC6H6. Em condições análogas (ou
por uma experiência competitiva, em que quantidades
equimolares de C6H5Y e de C6H6 competem por uma
quantidade insuficiente de um eletrófilo, dando assim a razão

147
kC6H5Y/kC6H6) e análises das quantidades relativas de produtos
orto, meta e para obtidos a partir de C6H5Y – a distribuição
isomérica (geralmente referida como a porcentagem do produto
de substituição total obtido). Assim, recordando que existem 6
posições disponíveis para ataque em C6H6, comparadas com
as posições 2orto-,2meta- e 1para- em C6H5Y, tem-se:
para a nitração do tolueno pelo ácido nítrico em anidrido
acético a 0 °C, kC6H5Me/kC6H6 é 27, e a porcentagem da
distribuição isomérica é: 6,5 de orto; 1,5 de meta e 37,0 de
para; e os fatores de velocidade parciais para a nitração,
nestas condições são:
A comparação destes fatores de velocidade parciais
para a nitração do tolueno com os da cloração e da bromação
mostra que diferem, em termos absolutos ou em termos
relativos, com o eletrófilo reagente, assim como de Y. Os
valores absolutos dos fatores de velocidade parciais, kY/kH,
aumentam na ordem (Nitração < Cloração < Bromação), à
medida que diminui a reatividade do eletrófilo atacante.
O uso dos fatores de velocidades parciais permite uma
investigação mais exata dos efeitos diretores do que era
possível até agora. Assim, todos os fatores de velocidade
parciais para o tolueno são >1, indicando que o grupo CH3
ativa todas as posições do núcleo em relação ao benzeno. O
mesmo se verifica para Y = CMe3, mas neste caso fm- para a
nitração é 3,0, em comparação com 1,3 para o tolueno,
indicando que CMe3 exerce um efeito indutivo (polar) elétron
doador maior do que o grupo CH3. Ao contrário, quando Y =

148
C6H5 no bifenila, fm- para a cloração é 0,7, a reação nesta
posição é mais lenta do que no benzeno (embora kC6H5Y/kC6H6 =
4,2 x 102), dado que o átomo de carbono sp2 pelo qual o
substituinte C6H5 está ligado ao anel benzênico exerce um
efeito indutivo (polar) reterador de elétron.
Observa-se um efeito semelhante com substituintes
ativantes e diretores orto e para, quando é possível investigar
uma reação que produz o produto meta em quantidade
suficiente para ser medida, a troca de deutério como ácido forte
CF3CO2D em C6H5OPh.
Os valores muito elevados de fo- e fp- refletem a
capacidade do par eletrônico em O de estabilizar seletivamente
os estados de transição para o ataque orto e para, enquanto
que o valor de fm- < 1 reflete a desestabilização do estado de
transição para o ataque meta pelo indutivo (polar) retirador de
elétron do átomo de oxigênio.
Os fatores de velocidade parciais e a distribuição
isomérica, numa reação de substituição, também são afetados
pela temperatura.
Efeitos esteroquímicos e razões orto/para
A razão dos produtos orto/para em uma reação
raramente é estatística de 2:1. Para qualquer diferença nos
efeitos eletrônicos que possam atuar nos estados de transição
(ou nos seus modelos, os intermediários de Wheland) para os

149
ataques orto e para, o estado de transição corresponde ao
ataque orto é ainda suscetível a efeitos estereoquímicos, que
podem ser pequenos ou insignificantes no estado de transição
correspondente ao ataque para. Verifica-se em geral que a
proporção de isômero orto diminui à medida que aumenta o
volume de Y ou do volume do eletrófilo atacante.
Assim, verifica-se que, para a nitração de alquil
benzenos (Y = CH3 → CMe3) em condições análogas,
Y % o- % p- fo- /fp-
CH3 58 37 1,57
CH2Me 45 49 0,92
CHMe2 30 62 0,48
CMe3 16 73 0,22
Analogamente, para vários ataques eletrofílicos
diferentes no clorobenzeno observam-se os seguintes valores:
Reação % o- % p- fo- /fp-
Cloração 39 55 0,71
Nitração 30 70 0,43
Bromação 11 87 0,14
Sulfonação 1 99 0,01
A cloração versus sulfonação corresponde ao aumento
crescente do volume da espécie eletrofílica reagente. Estes
resultados podem ser explicados com base no aumento
crescente do impedimento no estado de transição para o
ataque orto, o que representa o nível de energia mais elevado
(maior ∆G) e resulta numa velocidade corresponde mais lenta
de formação do isômero orto.
O fator estereoquímico pode não ser o único a atuar,
observa-se na nitração dos halobenzenos.

150
Y % o- % p- fo- /fp-
F 12 88 0,14
Cl 30 69 0,44
Br 37 62 0,60
I 38 60 0,63
Apesar do aumento do tamanho do substituinte Y
quando se passa de F → I, verifica-se que aumenta a
proporção de isômero orto e da razão orto/para. Não há
motivos para supor que um aumento de efeitos estereoquímico,
como o observado nos alquilbenzenos, não atue, e o seu efeito
deve, neste caso, ser menor do que o efeito indutivo (polar)
elétron retirador de Y. Este diminuirá com a distância, sendo
menos forte na posição para do que na posição orto; este
efeito será forte na posição orto relativo ao Flúor, muito
eletronegativo, assim o ataque em orto é pequeno apesar do
tamanho reduzido do substituinte. O efeito indutivo retirador de
elétron diminui consideravelmente do Fluor → Iodo (alteração
maior quando passa do F → Cl), resultando no aumento do
ataque à posição orto apesar do aumento do volume de Y.
Controle cinético versus termodinâmico
Nas reações anteriores, considerou-se que as
proporções de produtos formados numa reação são os
isômeros orto, meta e para, determinados por suas
velocidades de formação relativas, isto é, controle é cinético.
Contudo, nem sempre é o que se observa na prática. Na
alquilação de Friedel-Crafts do metilbenzeno com brometo de
benzila e GaBr3 (ácido de Lewis) a 25 ºC verifica-se que a
distribuição isomérica é:
Tempo (s) % o- % m- % p-
0,01 40 21 39

151
10 23 46 31
Mesmo após um tempo de reação muito curto (0,01
segundos) é duvidoso se a distribuição isomérica (na pequena
quantidade de produto reacional formado até então) é
puramente controlada pela cinética, a proporção de isômero
meta já é bastante elevada, e a após 10 segundos, já a reação
não é cineticamente controlada.
É importante salientar que as proporções relativas dos
produtos alternativos formados serão definidas pelas suas
estabilidades termodinâmicas relativas nas condições de
reação, que podem diferir das estabilidades termodinâmicas
das moléculas isoladas.
Substituição eletrofila em outras espécies aromáticas
A piridina, assim como o benzeno, tem 6 elétrons π
(sendo um deles fornecido pelo nitrogênio) nos orbitais π
deslocalizadas, mas, ao contrário do benzeno, os orbitais são
deformados pela atração em direção ao átomo de nitrogênio
por este ser mais eletronegativo do que o carbono. Isto reflete
no dipolo da piridina, que tem a extremidade negativa no N e a
extremidade positiva no anel.
A piridina é, por isso, descrita como um heterocíclico
deficiente em elétrons π, e, por analogia com o anel benzênico
que contenha um substituinte elétron retirador, NO2, pode
prever-se que esteja desativada frente ao reagente eletrofílico.
A substituição ocorre, com dificuldade, na posição meta por ser
a que conduz ao intermediário Wheland mais estável, ambos

152
os intermediários resultantes dos ataques nas posições orto e
para, a forma canônica em que a carga positiva está localizada
no nitrogênio, altamente instável e com energia elevada.
Esta dificuldade de ataque é devida, em parte, ao fato de
a piridina ter um par de elétrons livres no átomo de nitrogênio,
e pode, por isso, protonar ou interatuar com um eletrófilo.
A carga positiva irá desestabilizar ainda mais qualquer
dos complexos σ para a substituição eletrofílica, como
acontece o substituinte +NR3, a carga positiva está agora num
átomo do próprio anel e não simplesmente num substituinte.
O pirrol também tem 6 elétrons π em orbitais π
deslocalizadas, mas, neste caso, o átomo de nitrogênio tem de
contribuir com dois elétrons para completar o sexteto (por isso
não é básico durante o processo) e verifica-se que o dipolo do
pirrol se encontra em direção oposta ao da piridina, com a
extremidade positiva no nitrogênio e a extremidade negativa no
anel.
O pirrol é um heterocíclico excedente em elétrons π e
comporta-se de modo semelhante a um derivado do benzeno
ativado (anilina), reagindo com um eletrófilo com muita
facilidade. Isto pode não ocorrer em solução fortemente ácida

153
porque a protonação é forçada mesmo no pirrol fracamente
básico, a protonação ocorre no átomo de carbono 2 em vez do
nitrogênio.
Perde-se, assim, o caráter aromático, e o cátion
comporta-se como um dieno conjugado, sofrendo
polimerização com grande facilidade.
SUBSTITUIÇÃO NUCLEOFÍLICA AROMÁTICA
Substituição de hidrogênio
O ataque por nucleófilos no anel benzênico sem
substituintes é muito mais difícil de ocorrer do que o ataque por
eletrófilos, visto que:
(a) a nuvem de elétrons π do anel está apta a repelir um
nucleófilo que se aproxime e;
(b) o seu sistema orbital π é menos capaz de
deslocalizar os dois elétrons extras, complexo e carregado
negativamente, do que no intermediário de Wheland, carregado
positivamente.
As dificuldades, dos ítens (a) ou (b) podem ser vencidas
se estiver presente um substituinte retirador de elétron para
facilitar o ataque pelo nucleófilo. O nitrobenzeno quando
aquecido com KOH, na presença de ar, produz o o-nitrofenol e
uma pequena quantidade de p-nitrofenol.

154
O substituinte NO2 retirador de elétron, orienta
substituição nucleofílica nas posições orto e para.
A piridina não requer mais do que a sua própria
capacidade elétron retiradora, e é atacada por nucleófilos
fortes, –NH2, em amoníaco conhecida como reação de
Tschitschibabin.
O grupo H- é removido formando o produto, 2-
aminopiridina, que foi durante muito tempo usado como
material de partida na síntese da sulfapiridina, um
medicamento do grupo das sulfonamidas.
SUBSTITUIÇÃO DE ÁTOMOS DIFERENTES DE
HIDROGÊNO
Mecanismo unimolecular SN1Ar
As reações de substituição nucleófila aromática são
mais comuns e úteis, quando um átomo de hidrogênio ou grupo
é substituído por outro grupo de saída melhor do que H+, como:
Cl-, Br-, N2, SO32-, –NR2, etc..
Um exemplo é a substituição de N2 nas reações dos sais
de diazônio, ArN2+:
ArN2+ + Y- � ArY + N2
Seguindo a lei de velocidade, Velocidade = k[ArN2+], em
que a velocidade é independente de [Y-]. A lei de velocidade
observada foi interpretada em termos da etapa lenta, e
determinante da velocidade, de um cátion arilo, seguida pela
etapa rápida com o nucleófilo.

155
A semelhança com a reação SN1 em carbono saturado,
é reforçada ao verificar que os nucleófilos adicionados Cl-,
MeOH, etc., afetam a posição do produto, mais não a
velocidade da reação.
A formação do cátion fenila é extremamente instável (a
carga positiva não pode ser deslocalizada pelo sistema orbital
π), e a força diretora é fornecida pela eficácia de N2 como
grupo de saída. O mais importante desta reação é poder gerar
o cátion arila simples em solução. Os cátions arilas são
extremamente reativos, e, por isso, não-seletivos frente a
nucleófilos, a seletividade entre Cl- e H2O (kCl-/kH2O) é de
apenas 3 para C6H5+ em comparação com 180 para Me3C
+. A
reatividade muito elevada de C6H5+ reflete-se na sua
capacidade de se combinar com N2, a decomposição do cátion
diazônio é reversível, o que foi demonstrado pela observação
da dispersão parcial com nitrogênio marcado, 15N.
As reações de sais de diazônio, em solventes menos
polares, podem ocorrer através da formação inicial de um
radical arila.
Mecanismo adição-eliminação SN2 ativado
As reações de substituição aromática nucleófila mais
comuns envolvem substituição de X- de um haleto ativado por
grupo retirador de elétron, esses mecanismos podem ser
denominados: adição-eliminação ou SN2 ativado.

156
Essas reações seguem, geralmente, a seguinte lei de
velocidade, velocidade = k[ArX][Y-], com semelhança a reação
de SN2 no carbono saturado. O mecanismo difere do
mecanisno SN2, porque o ataque por Y- não ocorre por trás no
átomo de carbono que transporta o grupo de saída, ocorrendo
pelo mesmo lado. E por isso, a reação de substituição
bimolecular, SN2 aromática, não é sincronizada, ocorre por um
mecanismo em duas etapas. Via complexo σ ou intermediário
de Meisenheimer.
A reação ocorre através de dois estados de transição,
ET1 e ET2 entre os quais não há o complexo intermediário (CI),
o esquema proposto para indicar como a reação ocorre é:
A
X
B
Nu-
A
X
B-
Nu
A
X
B-
Nu
A
X
B-
Nu
A
Nu
B
+ X-
ET1 ET2CI
O mecanismo é facilitado por:
1 - A existência de um bom grupo abandonador,
facilmente deslocável com seu par de elétron e;
2 – A presença de substituintes ou heteroátomos
retiradores de elétron no anel aromático.
Nestas reações a etapa lenta pode ser a 1ª ou a 2ª como
ilustrados pelos gráficos dos perfis energéticos:

157
ET1
ET2
Coordenada da reaçãoE
nerg
ia li
vre
EI
EF
ET1
ET2
EI
EF
CI CI
Substituição via intermediários arino ou eliminação-adição
Sugerindo que o ataque inicial do –NH2 deve ser como
base em vez de ser como nucleófilo, retirando o hidrogênio orto
ao grupo abandonador; já que este hidrogênio possui acidez
suficiente para levar a uma eliminação-β, na primeira etapa:
A perda do próton do p-clorometilbenzeno pode ser
síncrona com, ou seguida por, perda de Cl- para dar o
intermediário arino. Este tem duas posições alternativas que
podem sofrer ataque por –NH2; a adição de NH3 de dois modos
alternativos. Não se deve esperar que as proporções dos dois
produtos alternativos sejam as mesmas, porque o arino não é
um intermediário simétrico, as duas posições de ataque por –
NH2 são diferentes.
Este é um mecanismo de eliminação/adição (em
contraste com a adição/eliminação de SN2 aromática).
Os arinos apresentam características estruturais
interessante. Não podem ser acetilênicos, dado que isso
requeria uma deformação enorme do anel benzênico para que
acomodasse o ângulo de 180° exigido pelos carbonos
Cl
CH3 H
+-NH2
CH3 NH2
H
CH3
NH2
H
Cl
CH3 CH3-
-NH2 /NH3
-NH2/NH3
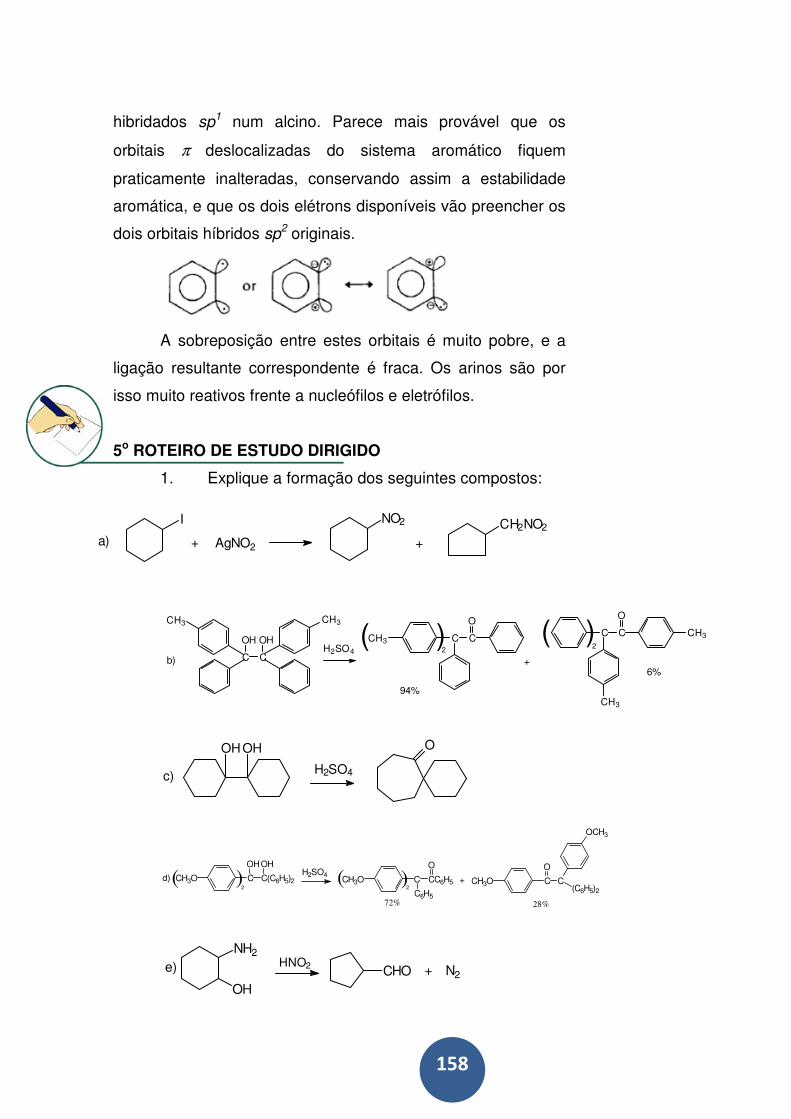
158
hibridados sp1 num alcino. Parece mais provável que os
orbitais π deslocalizadas do sistema aromático fiquem
praticamente inalteradas, conservando assim a estabilidade
aromática, e que os dois elétrons disponíveis vão preencher os
dois orbitais híbridos sp2 originais.
A sobreposição entre estes orbitais é muito pobre, e a
ligação resultante correspondente é fraca. Os arinos são por
isso muito reativos frente a nucleófilos e eletrófilos.
5o ROTEIRO DE ESTUDO DIRIGIDO
1. Explique a formação dos seguintes compostos:
a)
I
+ AgNO2
NO2 CH2NO2
+
b)
CH3CH3
C C
OH OHH2SO4
C C
O
CH3( )2
C C
O
CH3
CH3
)(2
+
94%
6%
OH OH
H2SO4
O
c)
d) CH3O CC
O
(C6H5)2
OCH3
+H2SO4
72% 28%
C C(C6H5)2
OHOH
CH3O( )2
C CC6H5CH3O
C6H5
O
)(2
NH2
OHCHO + N2
e) HNO2
159
CH2NH2HNO2 CH2OH
OH
H2C CHCH2CH2OHf) + +
NH3 NH
O
O
NaOClO
O
O
CO2H
NH2
g)
CH3CH2CONH2NaOBr
Br2 + NaOHCH3CH2NH2 + CO2h)
O
RCO3H
H+
OOi)
O
HN3
H2SO4
NO
H
j)
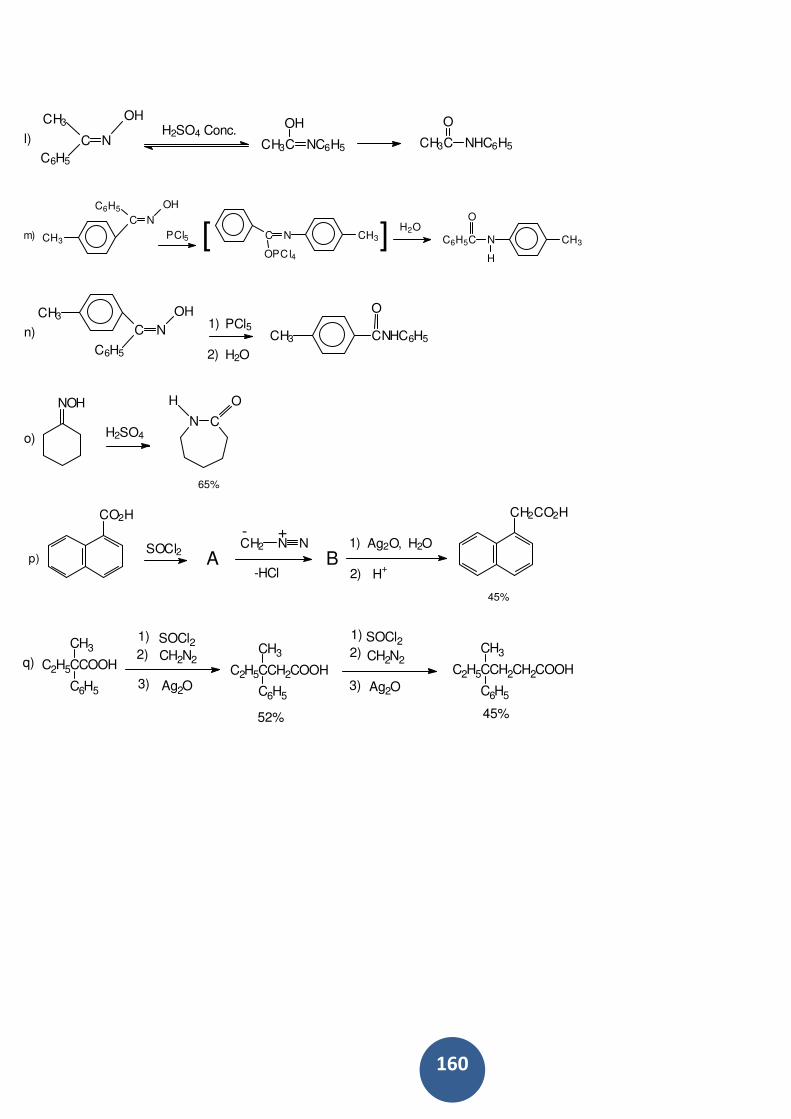
160
C N
OH
C6H5
CH3H2SO4 Conc.
CH3C NHC6H5
O
CH3C NC6H5
OHl)
C N
OHC6H5
CH3PCl5 C N
OPCl4
CH3 C6H5C N
O
H
CH3
H2Om) [ ]
C NOH
C6H5
CH3PCl51)
2) H2O
CH3 CNHC6H5
O
n)
NOH
H2SO4CN
OH
o)
65%
SOCl2 ACH2 N N
-HClB
1) Ag2O, H2O
2) H+
CO2H CH2CO2H
45%
p)
+-
C2H5CCOOH
CH3
C6H5
1)2)
3)
SOCl2CH2N2
Ag2OC2H5CCH2COOH
CH3
C6H5
C2H5CCH2CH2COOH
CH3
C6H5
q)
Ag2O
CH2N2
SOCl2
3)
2)1)
52% 45%

161

162
UNIDADE VI – REARRANJOS MOLECULARES
ENVOLVENDO ÁTOMOS DE CARBONO, NITROGÊNIO E
OXIGÊNIO DEFICIENTES DE ELÉTRONS
MÉTODOS DE FORMAÇÃO DE ÍONS CARBÔNIO
Fissão heterolítica de espécies neutras
Em cada caso é geralmente necessário um meio
altamente polar (ε elevado), com elevada capacidade
solvatante. Num contexto semelhante, foi já mencionado o
efeito de Ag+ na catálise de rações, geralmente por uma
mudança de mecanismo SN2 → SN1.
Ag+
+ R Br Ag + R+
Br
O efeito catalítico de Ag+ pode, contudo, ser complicado
pelo fato de o haleto de prata precipitado poder atuar como um
catalisador heterogênio.
A ionização também pode ser reduzida por ácido de
Lewis, por exemplo, o BF3,
+CCH3 OF BF3 CH3 C O+
BF4-
para dar um cátion acila, e sendo o equilíbrio
consideravelmente influenciado pela enorme estabilidade do
ânion, BF4-. Também com AlCl3,
++
+AlCl3 AlCl4-
CH3
C+
CH3CH3
AlCl4-
CO
CH3
CCH3
CH3
C
O
Cl
CH3
CCH3
CH3
C
O
onde o cátion acilo relativamente instável se decompõe,
originando Me3C+ muito estável, e sendo o equilíbrio deslocado
para a direita pela liberação de gás monóxido de carbono (CO).
Exemplos particularmente ilustrativos são fornecidos
pelos trabalhos de Olah com SbF5 como ácido de Lewis, em
SO2 líquido ou com excesso de SbF5 como solventes.
+R F SbF3 R+SbF6
-

163
A utilização dos “super ácidos”, daquele mesmo
investigador __ como SbF5/FSO3H, por exemplo __ permite a
formação de cátions alquilo, mesmo a partir de alcanos.
A estabilidade relativa de Me3C+ é evidenciada pelo fato
de se ter verificado que o íon carbônio isométrico,
MeC+HCH2Me, obtido de MeCH2CH2Me, rearranja de modo
instantâneo a Me3C+. A relação entre a estabilidade relativa de
íons carbônio e a sua estrutura é discutida mais adiante.
Adição de cátions a espécies neutras
O cátion mais comum é H+, que se adiciona a ligações
insaturadas, i. e. protonação, por exemplo na hidratação
catalisada por ácidos e alcenos.
A reação é reversível, e a inversa é a desidratação de
álcoois, catalisada por ácido. A protonação também pode
ocorrer no oxigênio de uma ligação dupla carbono-oxigênio,
conduzindo, assim, a um átomo de carbono mais positivo para
ataque subsequente por um nucleófilo, neste caso H2O, na
hidratação catalisada por derivados de ácidos carbonílicos.
A protanação ocorre na ausência de água, pela dupla
depressão do ponto de congelação observado com cetonas em
ácido sulfúrico concentrado.
+CH CHCH3 CH3 CH CHCH3 CH3
H
CH CHCH3 CH3
H
+OH2
CH CHCH3 CH3
H
OH
H+
OH2 - H+
+ +
CH3
CCH3
CH3
H SbF5/FSO3H H2
CH3
C+
CH3CH3
SbF5FSO3-
++ +C O
CH3
CH3H
+
C OH
CH3
CH3
C+
OH
CH3
CH3 OH2C
OHCH3
CH3 OH
H+δ
+δ
-
+C O
CH3
CH3 δ+
δ-
H2SO4 +C+
OH
CH3
CH3
HSO4-

164
Também podem ser gerados íons carbônios por
protonação de pares eletrônicos livres, neste caso, o átomo
protonado é convertido num bom grupo de saída e a ionização
é favorecida.
++ ++
Ph3C OHH2SO4
Ph3C OH
H
HSO4- H2SO4 Ph3C
+H3O
+ 2HSO4-
As ácidos de Lewis, também podem ser usados,
C O
CH3
CH3 δ+
δ-
+ +AlCl3 C O
CH3
CH3
AlCl3-
e outros cátions, por exemplo, o +NO2 na nitração do benzeno,
++
+
H
NO2
CH
NO2
em que o intermediário carregado positivamente é um íon
carbônio deslocalizado.
Obtenção de íons carbônios a partir de outros cátions
Os íons carbônios podem ser obtidos a partir da
decomposição de outros cátions, por exemplo, o cátion
diazônio proveniente da reação de NaNO2/HCl com RNH2,
R N N+
++
R N N
e também pela utilização de um íon carbônio facilmente
acessível para gerar outro de mais difícil obtenção.
+ +Ph3C+
HH
Ph3C H +

165
Estabilidade e estrutura de íons carbônios
O íon carbônio de grupo alquila simples apresentam a
seguinte sequência de estabilidades:
Me3C+ > Me2CH+ > MeCH2
+ > CH3+
A crescente substituição no átomo de carbono do íon
carbônio aumenta a deslocalização da carga positiva, por ação
de efeitos indutivos e de hiperconjugação. A estabilidade
particular de Me3C+ provém do fato de poder muitas vezes ser
formado, em condições vigorosas, por isomerização de outros
íons carbônio primeiro formados, e por que se mantém
inalterado após aquecimento a 170 °C em SbF5/FSO3H durante
4 semanas.
A estabilidade do íon carbônio acontece porque este é
planar, e nesta configuração pode ocorrer deslocalização
eficaz. Cálculos de mecânica quântica para cátions alquilo
simples sugerem, de fato, que a configuração planar (sp2) é
mais estável do que a piramidal (sp3) em cerca de 84 kJ (20
kcal)mol-1. À medida que este se afasta da planaridade,
aumenta a instabilidade do cátion e a consequente dificuldade
da sua formação. Isto foi observado para areação do 1-
bromotripticeno ao ataque SN1, devido à incapacidade de fomar
uma configuração planar, o que impede a formação do íon
carbônio. A estrutura planar observada por RMN e IV de
espécies como MeC+SbF5- é semelhante aos trialquilboranos,
R3B, com os quais são isoeletrônicas.
Um dos principais fatores que influencia a estabilidade
de cátions menos simples é a possibilidade de deslocalização
da carga, quando esta pode envolver orbitais π.
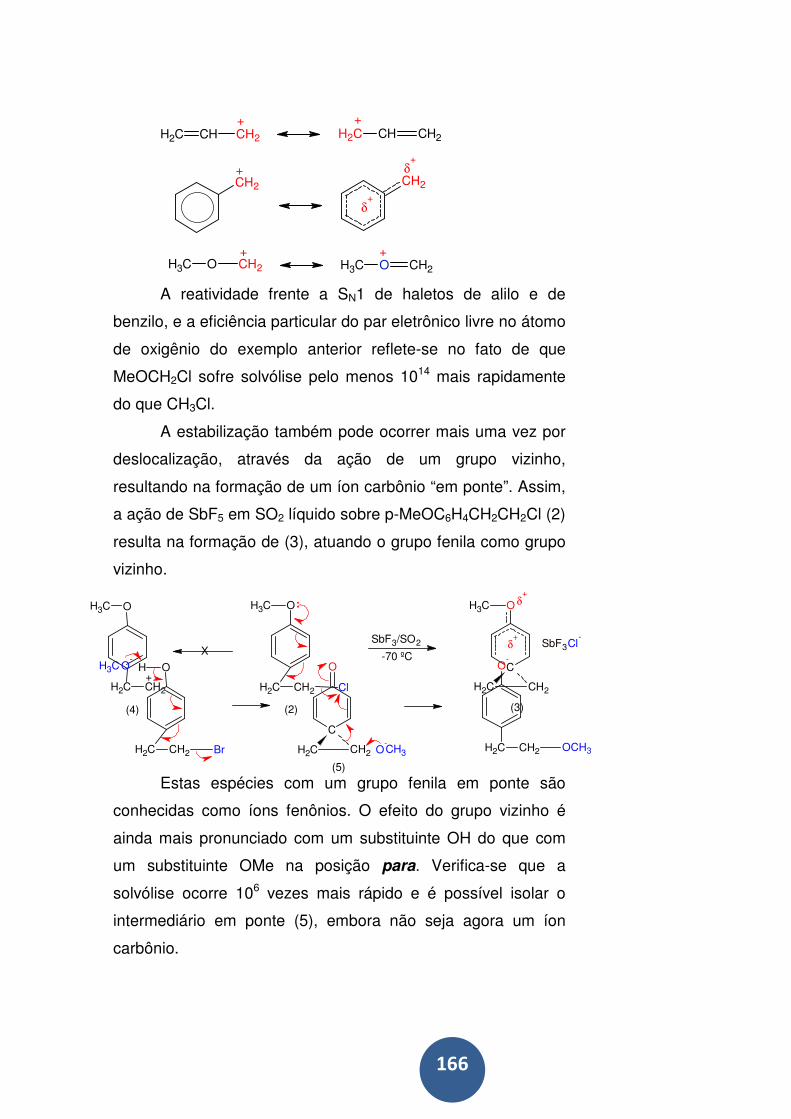
166
CH2 CH CH2 H2C CH CH2
+
++
+
CH2 CH2
δ+
δ+
CH3 O CH2
+CH3 O CH2
A reatividade frente a SN1 de haletos de alilo e de
benzilo, e a eficiência particular do par eletrônico livre no átomo
de oxigênio do exemplo anterior reflete-se no fato de que
MeOCH2Cl sofre solvólise pelo menos 1014 mais rapidamente
do que CH3Cl.
A estabilização também pode ocorrer mais uma vez por
deslocalização, através da ação de um grupo vizinho,
resultando na formação de um íon carbônio “em ponte”. Assim,
a ação de SbF5 em SO2 líquido sobre p-MeOC6H4CH2CH2Cl (2)
resulta na formação de (3), atuando o grupo fenila como grupo
vizinho.
Estas espécies com um grupo fenila em ponte são
conhecidas como íons fenônios. O efeito do grupo vizinho é
ainda mais pronunciado com um substituinte OH do que com
um substituinte OMe na posição para. Verifica-se que a
solvólise ocorre 106 vezes mais rápido e é possível isolar o
intermediário em ponte (5), embora não seja agora um íon
carbônio.
OCH3
CH2 CH2
+
OCH3
CH2 CH2 Cl
XSbF3/SO2
-70 ºC
:
C
OCH3
CH2 CH2
δ+
δ+
SbF3Cl-
(4) (2) (3)
OH
CH2 CH2 Br
C
O
CH2 CH2
CH3 O-
CH3O-
O-
CH2 CH2 OCH3
(5)

167
A estabilização, por meio de deslocalização,
ocorre em sistemas aromáticos. O 1-bromociclohepta-2,4,6-
trieno (brometo de tropílio, 6), que é isométrico com CH5CH2Br,
é um sólido cristalino (p.f 208 °C), extremamente solúvel em
água dando íons brometo em solução, um par iônico. A razão
deste comportamento reside no fato de o cátion cíclico (7)
apresentar 6 elétrons π, que podem ser acomodados em três
orbitais moleculares deslocalizados sobre os sete átomos.
Trata-se de um sistema que obedece a regra de Huckel 4n+2
(n=1) como o benzeno e exibe estabilidade quase aromática,
sendo o íon carbônio planar estabilizado, por aromatização.
BrH
(6)
H
H
HH
H
H
H
+ Br-
(7)
A aromatização deslocalizada indicada é confirmada
pelo fato de seu espectro de RMN apresentar um único sinal de
próton dos sete átomos de hidrogênio, reflete no fato de (7) ser
cerca de 1011 vezes mais estável do que o Ph3C+ altamente
deslocalizado.
Um caso particularmente interessante de estabilização
de um íon carbônio pode ser observado nos sistemas de
Huckel (4n + 2 em que n = 0), em sistemas cíclicos com 2
elétrons π. Verifica-se assim que os derivados do 1,2,3-
tripropilciclopropeno (8) dissocia ao correspondente cátion
ciclopropenilo (9) com extrema facilidade.
++
Pr Pr
YPr
Pr Pr
Pr
Y- SbCl6
-
(8) (9) (10)
Observa-se que (10) é mais estável (≈ 103 vezes) do que
(7) e encontra-se presente como íon carbônio numa extensão
de cerca de 50% em água a pH 7. Mais recentemente também

168
se mostrou ser possível isolar um par iônico o próprio cátion
ciclopropenilo (10) como um sólido branco cristalino.
Reações de íons carbônio
Os íons carbônio podem reagir por quatro tipos básicos
de reações. (a) Combinação com um nucleófilo, (b) Eliminação
de um próton, (c) Adição a uma ligação insaturada e (d)
Rearranjo da sua estrutura e seguindo (a) ou (b).
Os dois primeiros tipos de reação (a) e (b) conduzem
frequentemente à formação de produtos finais estáveis, mas (c)
e (d) dão origem à formação de novos íons carbônio, para os
quais são possíveis seguir (a) ou (b). A maior parte destas
possibilidades está bem ilustrada na reação de 1-
aminopropano (11) com nitrito de sódio e ácido clorídrico
diluído, formando cátion diazônio.
++
+
+CH3 CH2 CH2 N N NaNO2
HClCH3 CH2 CH2 NH2
N2 CH3 CH2 CH2
CH3 CH2 CH2 OH
CH3 CH CH2
CH3 CH CH3
CH3 CH CH3
OH
(11)(12)
(13)
(14)
(15)
(16)
(17)
H2O
H2O
- H+
A reação do cátion 1-propilo (13) com água (reação tipo
(a), combinação com um nucleófilo) forma o propan-1-ol (14), a
eliminação de um próton de (13) forma o propeno (15, reação
do tipo (b) Eliminação de um próton), enquanto que o rearranjo
(13, reação do tipo (d) Rearranjo da sua estrutura), neste caso
migração de H+ forma o cátion 2-propilo (16). A reação do tipo
(b) neste cátion rearranjado (16) forma o propeno (15),
enquanto que uma reação do tipo (a) com água dará propan-2-
ol (17). A mistura de produtos obtida foi de 7% de propan-1-ol,

169
28% de propeno e 32% propan-2-ol, refletindo as proporções
relativas de propan-1-ol e de propan-2-ol a estabilidade relativa
dos dois cátions, (13) e (16).
A soma dos produtos referidos representa apenas 67%
de conversão do 1-aminopropino inicial. Há outros nucleófilos
presentes no sistema, como Cl- e NO2-, capazes de reagir com
qualquer dos cátions, (13) ou (16), com à possível formação
tanto de RNO2 como de RONO (podem também formar-se
nitritos por esterificação direta do ROH primeiramente
formado). Os cátions (13) e (16) podem também reagir com o
ROH inicialmente formado para dar éteres, ROR, ou com RNH2
para dar RNHR (que, pode sofrer alquilação ou nitosação).
Finalmente, qualquer dos cátions pode adicionar à ligação
dupla do propeno formado inicialmente, MeCH=CH2 (reação do
tipo (c)), para dar outros cátions , MeC+H__CH2R, que podem
reagir. A mistura de produtos que se obtêm é influenciada
pelas condições reacionais, mas esta reação não é um método
de reação satisfatório para a conversão RNH2 → ROH.
As reações do tipo (d) Rearranjo da sua estrutura,
dificulta a reação de alquilação de Friedel-Crafts do benzeno
(uma reação do tipo (c)/(b)) por 1-bromopropano,
MeCH2CH2Br, na presença de brometo de gálio, GaBr3, ácido
de Lewis. Aqui o eletrófilo atacante é um complexo altamente
polarizado, Rδ++GaBr4δ-- e a maior estabilidade do complexo em
que Rδ++ transporta a sua carga positiva principalmente no
átomo de carbono secundário, em vez de num primário,
Me2Cδ++HGaBr4
-- em vez de MeCH2Cδ++HGaBr4
--, resulta de
novo numa transferência de hidreto, de tal modo que o principal
produto da reação é Me2CHC6H5.
Os rearranjos não são simples como parecem ser uma
simples migração de H+, isto é ilustrado pelo comportamento
de 13CH3CH2CH3 com AlBr3, em que o carbono marcado fica
disperso. O produto consiste de 2 partes de 13CH3CH2CH3 e 1
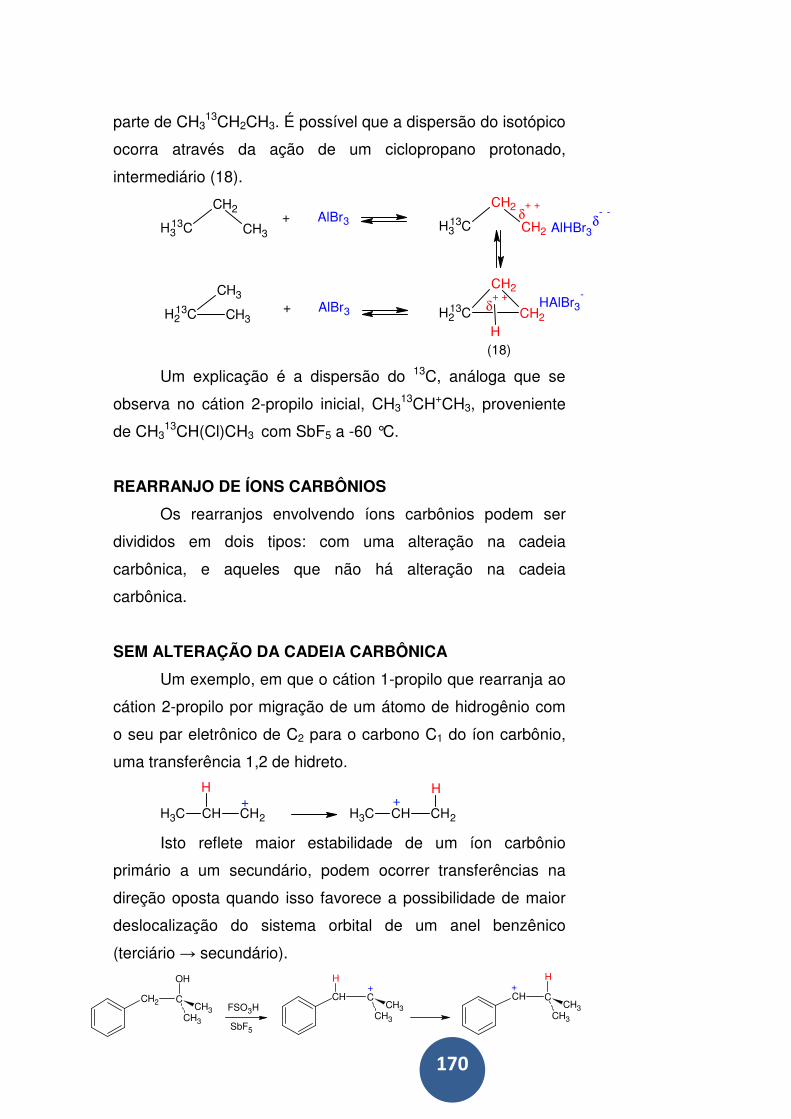
170
parte de CH313CH2CH3. É possível que a dispersão do isotópico
ocorra através da ação de um ciclopropano protonado,
intermediário (18).
+
C
CH2
CH3H313
C
CH3
CH3H213
C
CH2
CH2H313
C
CH2
CH2H213
+ AlBr3
AlBr3HAlBr3
-
AlHBr3 δ
+ +
δ+ +
H
δ- -
(18)
Um explicação é a dispersão do 13C, análoga que se
observa no cátion 2-propilo inicial, CH313CH+CH3, proveniente
de CH313CH(Cl)CH3 com SbF5 a -60 °C.
REARRANJO DE ÍONS CARBÔNIOS
Os rearranjos envolvendo íons carbônios podem ser
divididos em dois tipos: com uma alteração na cadeia
carbônica, e aqueles que não há alteração na cadeia
carbônica.
SEM ALTERAÇÃO DA CADEIA CARBÔNICA
Um exemplo, em que o cátion 1-propilo que rearranja ao
cátion 2-propilo por migração de um átomo de hidrogênio com
o seu par eletrônico de C2 para o carbono C1 do íon carbônio,
uma transferência 1,2 de hidreto.
CH3 CH CH2
H+
CH3 CH CH2
H+
Isto reflete maior estabilidade de um íon carbônio
primário a um secundário, podem ocorrer transferências na
direção oposta quando isso favorece a possibilidade de maior
deslocalização do sistema orbital de um anel benzênico
(terciário → secundário).
+ +CH2 C
CH3
OH
CH3
CH C
CH3
CH3
H
CH C
CH3
CH3
H
FSO3H
SbF5

171
Há possibilidades de diferentes rearranjos, os mais
interessantes são aqueles em que os catiões estão
deslocalizados, por exemplo em rearranjos alílicos.
Rearranjos alílicos
Na solvólise via SN1 de 3-clorobut-1-eno (19) em EtOH
obtém-se não um, mas uma mistura de dois éteres isoméricos;
obtém-se a mesma mistura (os éteres, aproximadamente nas
mesmas proporções) a partir da solvólise de 1-clorobut-2-eno
(20), em condições semelhantes.
Isto reflete a formação do mesmo cátion alílico
deslocalizado (23) como um par iônico intermediário a partir de
cada haleto, capaz de sofrer subsequentemente um ataque
rápido por um nucleófilo, EtOH no C1 ou no C3.
Quando o EtO-, em concentração relativamente elevada,
é utilizado como o nucleófilo em vez de EtOH, a reação de (19)
é do tipo Sn2 e dá apenas um éter (21). Contudo, foram
observados rearranjos alílicos no decurso de reações de
substituição que ocorrem por um processo bimolecular. Essas
reações são designadas por SN2 e ocorre segundo o
mecanismo.
+CH3 CH
Cl
CH CH2
CH3 CH CH CH2
OEt
CH3 CH
OEt
CH CH2
CH3 CH CH CH2
ClEtOH EtOH
(19) (20)
(21)
(22)
+CH3 CH CH CH2
(23)
+CH3 CH CH CH2 Cl
-
+ +CH2 CH CH
R
ClNu-: CH2 CH CH
R
Nu Cl-

172
Este processo tende a ocorrer quando substituintes (R)
no átomo de carbono α são suficientemente volumosos para
reduzir de modo acentuado a velocidade de substituição SN2
direta em Cα. Os rearranjos alílicos são bastante comuns, mas
o mecanismo detalhado pelo qual ocorrem é assunto bastante
complexo.
COM ALTERAÇÃO NA CADEIA CARBÔNICA
Rearranjos neopentílicos
A hidrólise via SN2 de 1-bromo-2,2-dimetilpropano
(brometo de neopentilo, 24) é lenta devido ao impedimento
estereoquímico. Efetuando a reação em condições que
favoreçam o mecanismo SN1 pode resultar numa velocidade de
reação maior, mas verifica-se que o álcool produzido é o 2-
metilbutan-2-ol (26), e não o produto previsto, 2,2-
dimetilpropanol (álcool neopentílico, 25), por ocorrer o rearranjo
neopentílico.
A maior estabilidade do íon carbônio terciário (28),
comparada com a do primário inicialmente formado (27),
fovorece a quebra da ligação C–C, envolvida na migração do
grupo Me. Estas alterações na cadeia carbônica, envolvendo o
íon carbônio, são conhecidas por rearranjo de Wagner-
Meerwein. Confirmação subsequente do envolvimento de (28)
é a formação simultânea do alceno 2-metilbut-2-eno (29) por
+
+
CH3 C
CH3
CH3
CH2 Br CH3 C
CH3
CH3
CH2
CH3C
CH3
C
CH3
H
CH3 C
CH3
CH2
CH3
XH2O
CH3 C
CH3
CH3
CH2 OH
- H+
H2O CH3 C
CH3
CH2 CH3
OH
(24) (25)(27)
(26)(28)(29)

173
perda de próton, um produto que não poderia ser obtido a partir
de (27).
É interessante observar que , enquanto que o brometo
de tipo neopentilo (30) sofre rearranjo durante a hidrólise SN1,
tal rearranjo não ocorre com o seu análogo fenólico (31).
Isso reflete na maior estabilidade do cátion benzílico
(32), embora seja apenas secundário, ao cátion terciário (33)
que não é formado pelo seu rearranjo.
Rearranjo de hidrocarbonetos
O rearranjo de alcenos ocorre facilmente na presença de
ácido.
Este arranjo relativamente fácil pode ser um
inconveniente na adição preparativa de ácidos a alcenos, por
exemplo, ácidos hologenídricos a alcenos ou na hidratação
catalisada por ácido podem formar-se misturas de produtos
difíceis de separar ou, em casos menos favoráveis, pode não
se obter o produto desejado. Pode também ocorrer a adição de
íons carbônios aos alcenos iniciais ou a alcenos formados.
Rearranjos pinacol/pinacolona
+CH3 C
CH3
CH CH3
CH3
Br
SN1CH3 C
CH3
CH CH3
CH3+
CH3 C
CH3
CH CH3
CH3
Produtos
(30)
CH3 C
CH3
CH Ph
CH3
Br
+SN1CH3 C
CH3
CH Ph
CH3+
CH3 C
CH3
CH Ph
CH3
X
(31) (32) (33)
Produtos
CH3 C CH CH2
CH3
CH3
CH3 C CH CH3
CH3
CH3
CH3 C CH CH3
CH3
CH3 CH3
C C
CH3CH3
CH3H+
H+-+ +

174
Outro exemplo de migração de um grupo, metila, para
um átomo de carbono de um íon carbônio é observado no
rearranjo catalisado por ácido, de 1,2-dióis, por exemplo, o
pinacol Me2C(OH)Me2 (34) e cetonas, pinacolona, MeCOCMe3
(35).
O fato de em (36), que já é um íon carbônio terciário,
ocorrer uma transferência-1,2 de metila, resulta da
estabilização adicional conferida ao íon carbônio rearranjado
(37) por deslocalização de carga através de um par eletrônico
no átomo de oxigênio (37) que pode também perder facilmente
um próton para dar um produto final estável (35). Também
ocorre analogia com outros compostos capazes de formar o íon
carbônio como (36).
Aspectos estereoquímico
Há três aspectos de interesse estereoquímico nos
rearranjos de íons carbônio, a configuração no átomo de
carbono a partir do qual ocorre a migração (a origem da
migração), a configuração do átomo de carbono para o qual
ocorre a migração (o carbono do íon carbônio, o fim da
migração), e a configuração do grupo migrante. O que é
interessante é que para estas três questões nunca foi
encontrada resposta para um mesmo composto, apesar do
+
+
+
+
CH3 C C CH3
CH3
OH OH
CH3 H+
CH3 C C CH3
CH3
OH OH2
CH3- H2O
CH3 C CCH3
CH3
OH
CH3
CH3 C C CH3
O CH3
CH3 H+
CH3 C C CH3
O CH3
CH3
H
CH3 C C CH3
OH CH3
CH3
34
3537
36

175
enorme volume de trabalho que tem sido feito sobre íon
carbônio.
O último aspecto nunca foi diretamente estabelecido
experimentalmente para um rearranjo exclusivamente de íon
carbônio. Em consequência, estabeleceu-se que o grupo
migrante nunca fica livre durante o rearranjo. Isto é
demonstrado se tomar, por exemplo, dois pinacóis (48 e 49),
de estrutura muito semelhante (e que rearrangem a
velocidades muito semelhantes), mas tendo diferentes grupos
migrantes, e se forem rearranjos simultaneamente na mesma
solução (uma experiência cruzada), nunca se observa
migração cruzada.
Analogamente, se rearranjos em que há uma
transferência de hidreto forem realizados num solvente
deuterado (D2O, MeOD, Etc.), não há incorporação de deutério
na nova ligação C–H(D) no produto final rearranjado. Portanto,
em ambos os casos o rearranjo é estritamente intramolecular,
isto é, o grupo migrante nunca fica desligado do resto da
molécula, ao contrário dos rearranjos intermoleculares, em que
isso acontece.
Isto sugere uma associação muito próxima do grupo
migrante, R, com o término da migração antes que tenha
quebrado completamente a sua ligação com o átomo de origem
da migração, com retenção de configuração em R. Este fato foi
experimentalmente estabelecido numa reação envolvendo a
migração de um carbono elétron deficiente, no rearranjo de
+
Ph C C CH3
Ph
OH OH
CH3
Ph C C C2H5
Ph
OH OH
C2H5
+
Ph C C CH3
Ph O
CH3
Ph C C C2H5
Ph O
C2H5
H+
Ph C C CH3
Ph O
CH3
Ph C C C2H5
Ph O
C2H5
ouNão
48
49
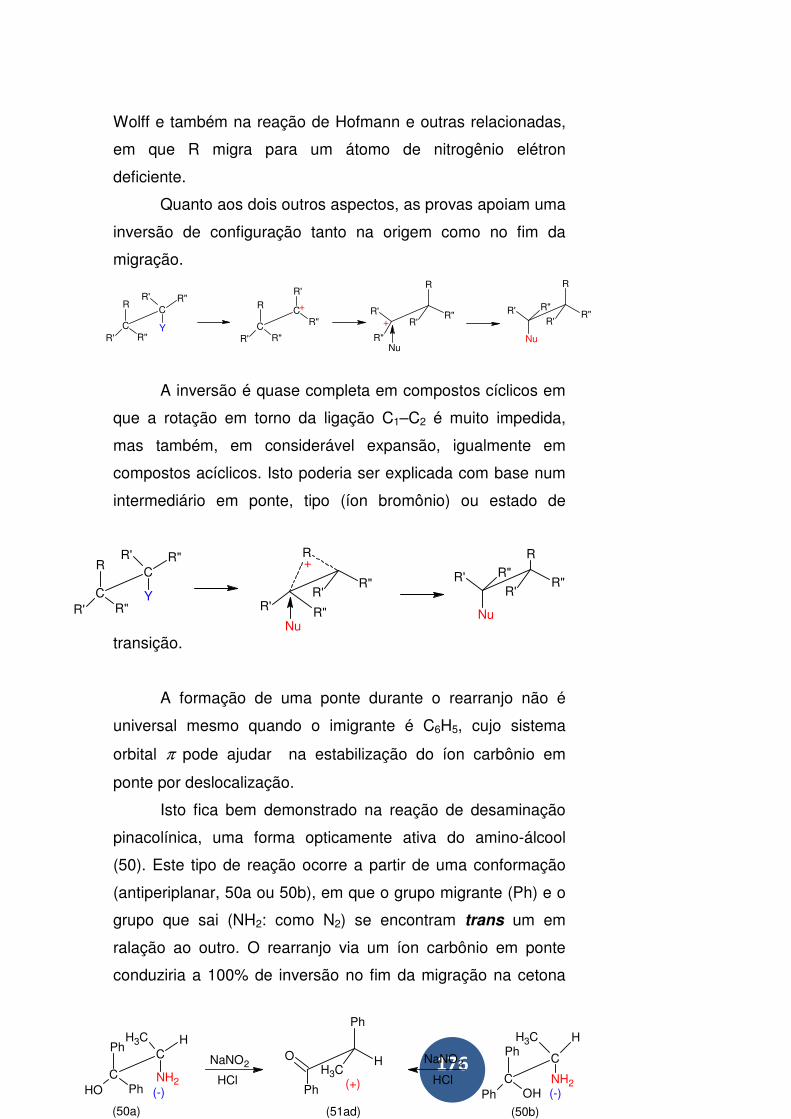
176
Wolff e também na reação de Hofmann e outras relacionadas,
em que R migra para um átomo de nitrogênio elétron
deficiente.
Quanto aos dois outros aspectos, as provas apoiam uma
inversão de configuração tanto na origem como no fim da
migração.
A inversão é quase completa em compostos cíclicos em
que a rotação em torno da ligação C1–C2 é muito impedida,
mas também, em considerável expansão, igualmente em
compostos acíclicos. Isto poderia ser explicada com base num
intermediário em ponte, tipo (íon bromônio) ou estado de
transição.
A formação de uma ponte durante o rearranjo não é
universal mesmo quando o imigrante é C6H5, cujo sistema
orbital π pode ajudar na estabilização do íon carbônio em
ponte por deslocalização.
Isto fica bem demonstrado na reação de desaminação
pinacolínica, uma forma opticamente ativa do amino-álcool
(50). Este tipo de reação ocorre a partir de uma conformação
(antiperiplanar, 50a ou 50b), em que o grupo migrante (Ph) e o
grupo que sai (NH2: como N2) se encontram trans um em
ralação ao outro. O rearranjo via um íon carbônio em ponte
conduziria a 100% de inversão no fim da migração na cetona
+
+C
CR
R' R"
R' R"
Y C
CR
R' R"
R'
R"R'
R"
R
R"R'
Nu
R' R"
R
R"R'
Nu
C
CR
R' R"
R' R"
Y
+
R' R"
R
R"R'
Nu
R' R"
R
R"R'
Nu
C
CPh
OH Ph
CH3 H
NH2 C
CPh
Ph OH
CH3
NH2
H
O
Ph
Ph
HCH3
NaNO2
HCl
NaNO2
HCl(-)
(+)(-)
(50a) (50b)(51ad)

177
formada (51ab), qualquer que fosse a conformação inicial
envolvida (50a ou 50b).
No entanto, verificou-se que, embora a inversão
predominasse (51ab, 88%), a cetona formada teria uma
quantidade da imagem num espelho do (51d, 12%), porque
12% da reação não ocorreu via íon carbônio em ponte. A
explicação mais simples é que pelo menos parte do rearranjo
total ocorre via íon carbônio, não em ponte (52c), em que pode
ocorrer rotação em torno da ligação C1–C2 (52c → 52d),
formando a cetona (51d) em que houve retenção da
configuração.
+
C
CPh
OH Ph
CH3
HO
Ph
Ph
H CH3
(-)
(52c) (51d)
C
CPh
OH PhCH3
H
(-)
(52d)
+
A razão entre a inversão (51ab) e a retenção (51d) na
cetona formada é determinada pela velocidade relativa de
rotação em torno de C1–C2 (52c), comparada com a velocidade
de migração do Ph.
Rearranjo de Wolff
Este rearranjo foi separado dos rearranjos de íon
carbônio porque envolve migração para um carbono
semelhante a um carbeno, não carregado, apesar de elétron
deficiente em vez de migrar para um átomo de carbono
carregado positivamente. A reação envolve a perda de
nitrogênio a partir da α-diazocetona (53) e rearranjo a cetenos
muito reativo (54).
C CH
R
O
N N+- - N2
C CH
R
O
O C C
R
H..
(53) (55) (54)

178
Os cetenos reagem facilmente com quaisquer
nucleófilos presentes no sistema, por exemplo, H2O. A reação
pode ser por fotólise, termólise ou por tratamento com óxido de
prata. Nos casos de fotólise e termólise, forma-se um carbeno
intermediário (55). Na reação catalisa pela prata, a perda de
nitrogênio e a migração de R podem ser simultâneas. No caso
em que R é C4H9C(Me)Ph, demonstrou-se que migra com
retenção de configuração.
A obtenção de diazocetonas (53) ocorre por reação de
diazometano, CH2N2, com cloretos de acila, seguindo-se com,
um rearranjo de Wolff na presença de água, constitui parte do
processo Arndt-Eistert, por meio do qual um ácido pode ser
convertido no seu homólogo.
Além da água, outros nucleófilos como amoníaco ou
álcool podem ser adicionados à ligação C=C do ceteno para
formar amida ou éster.
O rearranjo de Wolff é muito semelhante com a reação
de Hofmann, em que a migração ocorre para um átomo de
nitrogênio deficiente de elétron para formar o intermediário,
isocianato RN=C=O.
CÁTIONS DIAZÔNIOS
A nitrosação de aminas primárias, RNH2 com nitrito de
sódio e ácido diluído conduz à formação de cátions diazônios
(56).
RC
O
OH SOCl2 R C
O
ClCH2N2 R C
O
CHN2
- N2
Ag2ORCH C O
OH2R CH
H
C O
OH(53) (54)
+
+
+ ++
+
-
N
H
R
H
N O
X
: N
H
R
H
N O N
H
R N O
NR N OH
NR N
NR N
R N N(1) + H
(2) - H2O
- H+
(56)
X
+

179
O verdadeiro agente nitrosante não é HNO2, e sim o
anidrido N2O3 (X=ONO) obtido por 2 mols de HNO2.
+2HNO2 ONO NO H2O
Com aminas alifáticas simples, o cátion diazônio inicial
(56) decompõe-se de modo extremamente rápido para dar íons
carbônio que são mais reativos do que os obtidos por outros
processos de fissão, por exemplo, RBr→R+Br-.
A instabilidade de cátions diazônio alifáticos, na
ausência de uma estrutura estabilizada, é devida em grande
parte à eficácia de N2 como grupo de saída, já em cátions
diazônios aromáticos, a estabilidade é fornecida pelo sistema
de orbitais π do anel aromático.
-++N N
+N N
As aminas primárias aromáticas são bases/nucleófilos
mais fracos do que as alifáticas (devido à interação do par
eletrônico do N com o sistema de orbitais π do anel aromático),
portanto é necessário um agente nitrosante muito poderoso,
sendo a reação feita em meio com acidez elevada. Permanece
uma concentração no equilíbrio suficiente de ArNH2 não
protonada, mas a concentração é suficientemente baixa para
impedir que a amina ainda não diazotada sofra uma reação de
acoplamento com ArN2+ formado. Os cloretos, sulfatos, nitratos,
etc., de diazônio aromáticos são relativamente estáveis em
solução aquosa à temperatura ambiente ou mais baixa, mas
não são facilmente isoladas sem decomposição. Os
fluoroboratos, ArN2+BF4
-, são mais estáveis e podem ser
isolados no estado sólido. A termólise do sólido seco é um
método preparativo de fluorarenos.
-++ArN2 BF4 Ar F N2 BF3∆
+

180
Os substituintes no núcleo aromático têm um efeito
acentuado na estabilidade de ArN2+, e os que são doadores de
elétron têm efeito estabilizador.
-+N NMe2N
+N NMe2N
.. +
A nitrosação também ocorre em aminas secundárias,
mas para na etapa N-nitroso, R2N–N=O. As aminas alifáticas
terciárias são inicialmente convertidas no cátion
nitrosotrialquilamônio, R3N+–N=O, mas este sofre facilmente
ruptura da ligação C–N para dar produtos bastante complexos.
Com aminas terciárias aromáticas, ArNR2, a nitrosação pode
ocorrer, não no N, mas na posição para ativada do anel para
dar um composto C nitroso.
R2N N O
MIGRAÇÃO PARA NITROGÊNIO DEFICIENTE DE ELÉTRON
Os rearranjos, até aqui considerados, todos têm uma
característica comum, a migração de um grupo alquila ou arila,
com seu par eletrônico para um átomo de carbono, íon
carbônio ou não, deficiente de elétron. O átomo de nitrogênio
deficiente de elétron, exemplo, R2N+ (íon nitrônio) ou RN
(nitreno), e a migração de grupo alquila ou arila para esses
centros também acontece como com o carbono em:
+R3C R2 C
..
..e
Reações de Hofmann, Curtius, Lossen e Schmidt
A característica comum destas reações é a presença no
composto, de um bom grupo de partida que facilita o rearranjo.
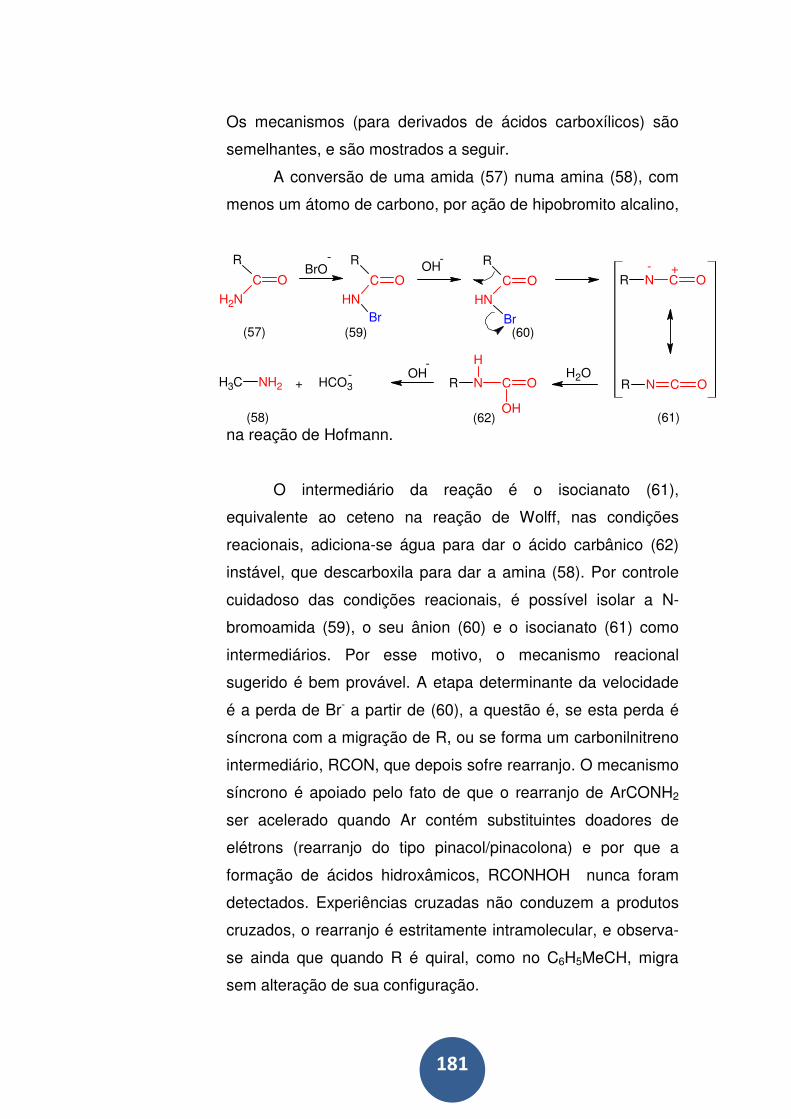
181
Os mecanismos (para derivados de ácidos carboxílicos) são
semelhantes, e são mostrados a seguir.
A conversão de uma amida (57) numa amina (58), com
menos um átomo de carbono, por ação de hipobromito alcalino,
na reação de Hofmann.
O intermediário da reação é o isocianato (61),
equivalente ao ceteno na reação de Wolff, nas condições
reacionais, adiciona-se água para dar o ácido carbânico (62)
instável, que descarboxila para dar a amina (58). Por controle
cuidadoso das condições reacionais, é possível isolar a N-
bromoamida (59), o seu ânion (60) e o isocianato (61) como
intermediários. Por esse motivo, o mecanismo reacional
sugerido é bem provável. A etapa determinante da velocidade
é a perda de Br- a partir de (60), a questão é, se esta perda é
síncrona com a migração de R, ou se forma um carbonilnitreno
intermediário, RCON, que depois sofre rearranjo. O mecanismo
síncrono é apoiado pelo fato de que o rearranjo de ArCONH2
ser acelerado quando Ar contém substituintes doadores de
elétrons (rearranjo do tipo pinacol/pinacolona) e por que a
formação de ácidos hidroxâmicos, RCONHOH nunca foram
detectados. Experiências cruzadas não conduzem a produtos
cruzados, o rearranjo é estritamente intramolecular, e observa-
se ainda que quando R é quiral, como no C6H5MeCH, migra
sem alteração de sua configuração.
C O
R
NH2
C O
R
NHBr
C O
R
NH
Br
R N C O
R N C O
+-
C O
OH
N
H
RHCO3CH3 NH2
(58)
(59) (60)
(61)(62)
(57)
BrO OH
OH H2O
- -
-
+-

182
Há um grupo de reações relacionadas com a reação de
Hofmann, e em que todas elas envolvem a formação de um
isocianato (61) por rearranjo de um intermediário análogo a
(60).
A reação de Lossen envolve a ação de uma base sobre
derivados O-acilados (63) de ácidos hidroxâmicos, RCONHOH,
e R’CO2- como o grupo de saída do intermediário (64), em
comparação com Br- de (60).
C O
NH
R
OCOR'
C O
NH
R
OCOR'
CH3 N C O
C O
NH
R
NH2
C O
OH
R
HN3
NaNO2
HCl
C O
N
R
N N
OH-
-
- +
(63) (64)
(61)
(65)
(66)
(67)
A reação também ocorre com os ácidos hidroxâmicos,
mas não com a mesma facilidade com que ocorre para os seus
derivados O-acilados que R’CO2- é um melhor grupo de saída
do que o OH. A natureza síncrona do rearranjo é comprovada
pelo fato de que não só a reação é facilitada por substituintes
doadores de elétrons em R (Hofmann), mas também por
substituintes retiradores de elétrons em R’, isto é, ambos estão
envolvidos na etapa determinante da velocidade da reação.
As reações de Curtius e de Schmidt envolvem ambas N2
como grupo de saída da azida intermediaria (67), e aqui
novamente a migração de R ocorre num processo síncrono. A
azida pode ser obtida quer por nitrosação de uma hidrazina

183
ácida (65), reação de Curtius, quer pela reação de ácido
hidrazóico, HN3, com um ácido carboxílico (66), reação de
Schmidt.
Rearranjos de Beckman
O rearranjo de Beckman é uma transformação
catalisada por ácido para a conversão de cetoximas em amidas
N substituídas.
RR’C = NOH � R’CONHR ou RCONHR’
A reação é catalisada por uma grande variedade de
reagentes ácidos como: H2SO4, SO3, SOCl2, P2O5, BF3, etc., e
ocorre não só com cetoximas, mas também com os seus O-
ésteres. O uso do ácido polifosfórico como catalisador faz com
que ocorra rearranjo em muitas aldóximas. A característica
deste rearranjo, quanto ao grupo migrante é o arranjo
estereoquímico dos grupos R e R’ que determina qual deles é
que migra. Verifica-se, quase sem exceção, que o grupo que
migra C�N é o grupo R anti em relação ao grupo OH. A
reação é altemente estereoespecífica.
C
N
R'R
OH
C
N
R'OH
R
C
N
R'O
R H
(i.e. somente R'CONHR)ácido
Como, claramente demonstrado que é o grupo R anti
que migra nos rearranjos de Beckmann, a estrutura da amida
formada é usada para estabelecer a configuração da oxima
inicial. Assim, verificou-se que (68) só dá a benzamida N-metil
substituída (71), enquanto que (69) dá unicamente a
acetanilida aril substituída (72).
C
N
MeAr
OH
Ar C
O
NHMeC
N
MeAr
OH
Me C
O
NHAr
(68) (69)(71) (72)

184
O rearranjo se processa segundo o mecanismo:
Protonando o grupo –HO formando (73a), seguida
por perda de água para dar (74), enquanto que com cloretos de
ácidos, PCl5, etc., se forma o éster intermediário (73b); que
elimina o ânion –OCl, um bom grupo de saída, formando (74).
Na série em que XO– é CH3CO2- < ClCH2CO2
- < PhSO3-, a
ionização é a etapa determinante da velocidade no rearranjo,
também é sugerido pela observação de que a velocidade de
reação aumenta à medida que aumenta a polaridade do
solvente.
A perda do grupo de saída e a migração de R’ são
síncronizadas na conversão de (73) em (74), mostrando que o
rearranjo é intramalecular de elevada estereosseletividade.
Quando R’ é quiral, PhCHMe retém a sua configuração durante
a migração. Isto também reflete em maior estabilidade do íon
carbônio intermediário, RC+=NR’ (74) do que a do ânion
nitrônio, RR’C=N+, que seria obtido se a perda do grupo de
saída precedesse a migração de R’.
O rearranjo de Beckmann tem aplicação industrial na
síntese do polímero têxtil Nylon-6 a partir da oxima da
ciclohexanona via a amida cíclica (caprolactama).
N
R
R'
OH H+
PCl5 ou SOCl2
N
R
R'
+OH2
N
R
R'
OCl
R C N R'+
R C N R'+
N
R
O+
R'
H
H
N
R
O R'H
NH
R
O R'
- H+
(75) (76) (77)
(73a)
(73b)
(74)
-H2O
-OCl
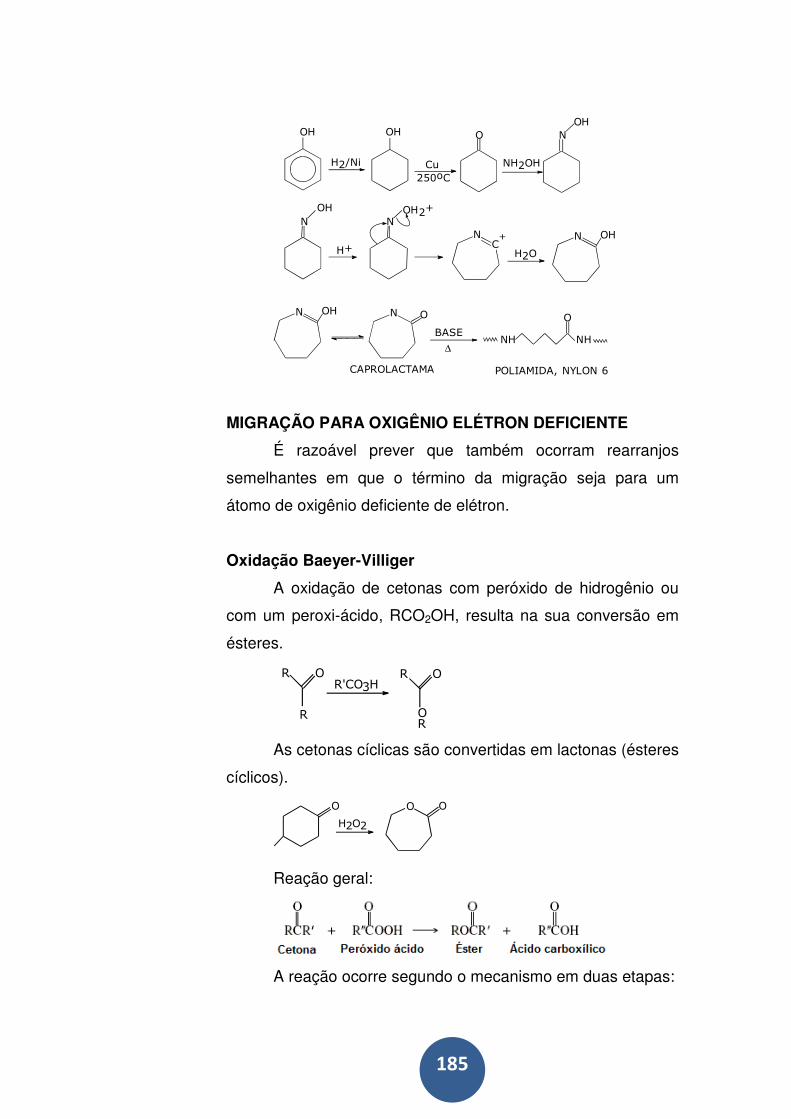
185
OH OH O N
OH
H2/Ni Cu
250oC
NH2OH
N
OH
H+
OH2+
N
H2O
OH
BASE
∆NH NH
O
CAPROLACTAMA POLIAMIDA, NYLON 6
C+N
O
N
N OH N
MIGRAÇÃO PARA OXIGÊNIO ELÉTRON DEFICIENTE
É razoável prever que também ocorram rearranjos
semelhantes em que o término da migração seja para um
átomo de oxigênio deficiente de elétron.
Oxidação Baeyer-Villiger
A oxidação de cetonas com peróxido de hidrogênio ou
com um peroxi-ácido, RCO2OH, resulta na sua conversão em
ésteres.
R O
R
R'CO3H
OR
OR
As cetonas cíclicas são convertidas em lactonas (ésteres
cíclicos).
O
H2O2
O O
Reação geral:
A reação ocorre segundo o mecanismo em duas etapas:
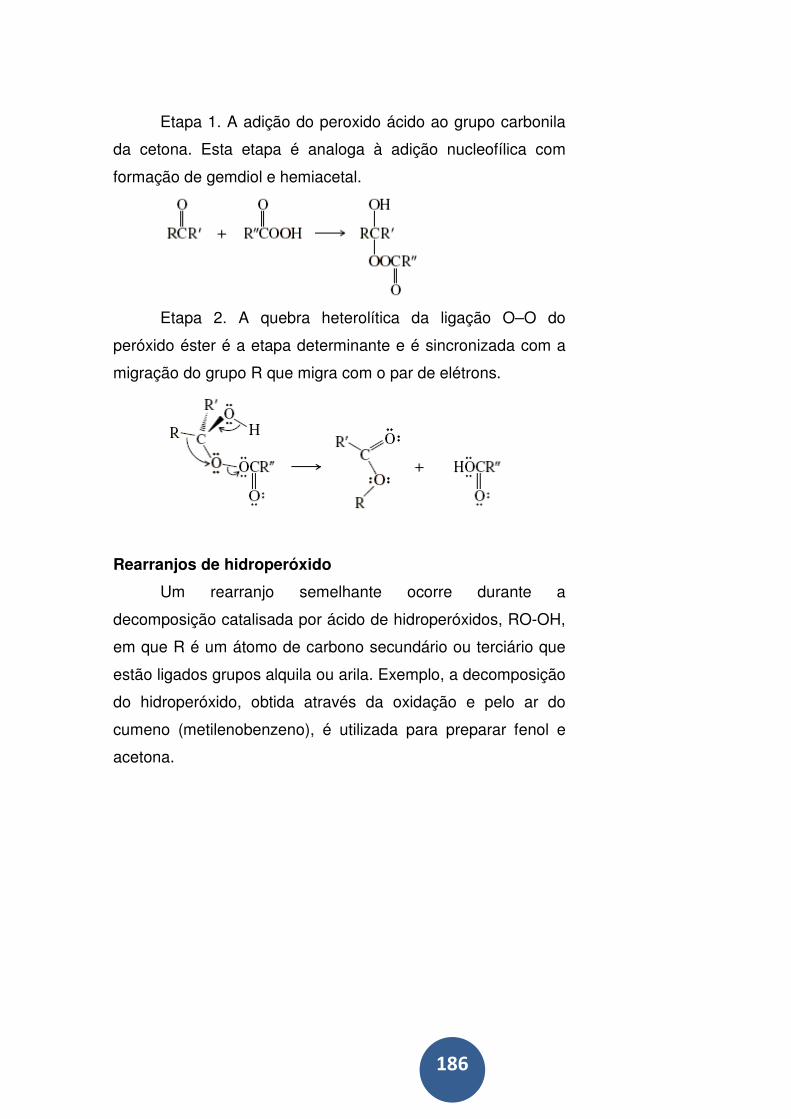
186
Etapa 1. A adição do peroxido ácido ao grupo carbonila
da cetona. Esta etapa é analoga à adição nucleofílica com
formação de gemdiol e hemiacetal.
Etapa 2. A quebra heterolítica da ligação O–O do
peróxido éster é a etapa determinante e é sincronizada com a
migração do grupo R que migra com o par de elétrons.
Rearranjos de hidroperóxido
Um rearranjo semelhante ocorre durante a
decomposição catalisada por ácido de hidroperóxidos, RO-OH,
em que R é um átomo de carbono secundário ou terciário que
estão ligados grupos alquila ou arila. Exemplo, a decomposição
do hidroperóxido, obtida através da oxidação e pelo ar do
cumeno (metilenobenzeno), é utilizada para preparar fenol e
acetona.

187
O2 O-OHH+
O-OH 2+
O
+
+
OH2O
-H+
O OH
OHOH3O
+OH
+
O
A perda do grupo de saída (H2O) e a migração de Ph
para o átomo de oxigênio deficiente de elétron resultante são
processos síncronizados. A adição de água ao íon carbônico
forma o hemicetal, que é facilmente hidrolisado nas condições
reacionais para gerar fenol e acetona. É possível que a
capacidade migratória superior do Ph seja resultante da
migração via um estado de transição em ponte.
Ph
O
Me
Me
OH 2+
δ+
δ+δ+
δ+
Neste exemplo, considerou-se fissão heterolítica de
ligação peróxido, −O:O− → −O Oө−, em solventes polares; em
condições apropriadas, também pode ocorrer fissão homolítica
para gerar radicais,−O:O− → −O· ·O−.
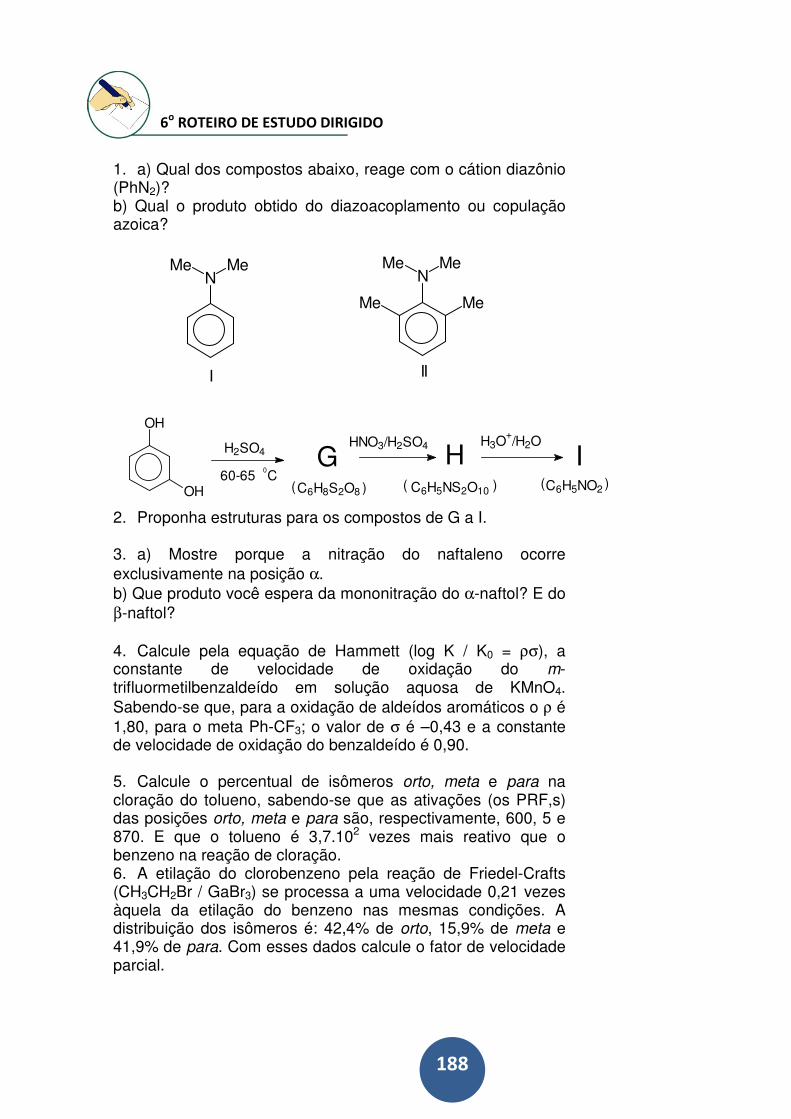
188
6o ROTEIRO DE ESTUDO DIRIGIDO
1. a) Qual dos compostos abaixo, reage com o cátion diazônio (PhN2)? b) Qual o produto obtido do diazoacoplamento ou copulação azoica?
2. Proponha estruturas para os compostos de G a I. 3. a) Mostre porque a nitração do naftaleno ocorre exclusivamente na posição α. b) Que produto você espera da mononitração do α-naftol? E do β-naftol? 4. Calcule pela equação de Hammett (log K / K0 = ρσ), a constante de velocidade de oxidação do m-trifluormetilbenzaldeído em solução aquosa de KMnO4. Sabendo-se que, para a oxidação de aldeídos aromáticos o ρ é 1,80, para o meta Ph-CF3; o valor de σ é –0,43 e a constante de velocidade de oxidação do benzaldeído é 0,90. 5. Calcule o percentual de isômeros orto, meta e para na cloração do tolueno, sabendo-se que as ativações (os PRF,s) das posições orto, meta e para são, respectivamente, 600, 5 e 870. E que o tolueno é 3,7.102 vezes mais reativo que o benzeno na reação de cloração. 6. A etilação do clorobenzeno pela reação de Friedel-Crafts (CH3CH2Br / GaBr3) se processa a uma velocidade 0,21 vezes àquela da etilação do benzeno nas mesmas condições. A distribuição dos isômeros é: 42,4% de orto, 15,9% de meta e 41,9% de para. Com esses dados calcule o fator de velocidade parcial.
NMe Me
Me Me
NMeMe
I II
OH
OH
H2SO4
60-65 C0G H I
HNO3/H2SO4 H3O+/H2O
(C6H5NO2 )( C6H5NS2O10 )(C6H8S2O8 )
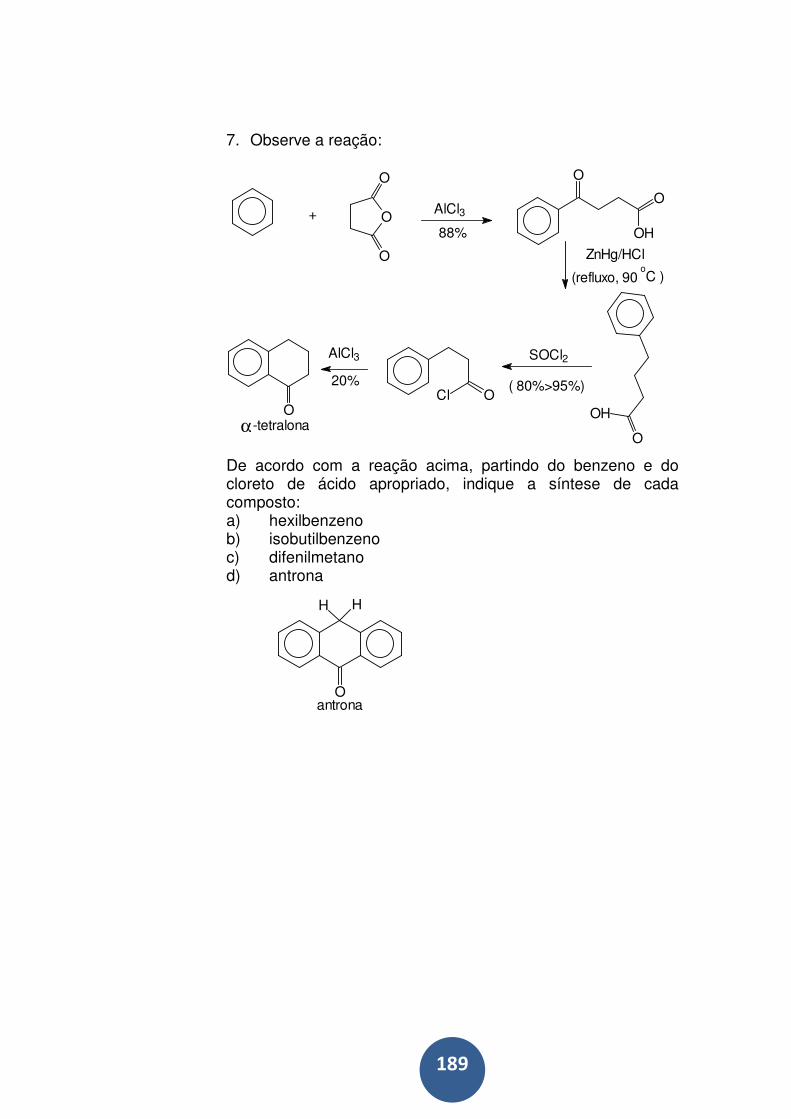
189
7. Observe a reação:
De acordo com a reação acima, partindo do benzeno e do cloreto de ácido apropriado, indique a síntese de cada composto: a) hexilbenzeno b) isobutilbenzeno c) difenilmetano d) antrona
+ O
O
O
O
OH
O
O
OH
SOCl2
( 80%>95%)Cl O
AlCl3
88%
AlCl3
20%
O
ZnHg/HCl
(refluxo, 90oC )
α-tetralona
H H
Oantrona

190

191
UNIDADE VII - ADIÇÃO ELETRÓFILA E NUCLEÓFILA A
C=C E C=O
O par de elétrons na orbital π está mais difuso e menos
ligado aos núcleos de carbono; é mais facilmente polarizável
do que o par da ligação σ, conduzindo à reatividade
característica destes compostos insaturados. Os elétrons π são
características às ligações duplas carbono-carbono, e
protegem a molécula do ataque por reagentes nucleófilos. As
reações mais características do sistema são iniciadas por
espécies deficientes de elétrons, tais como X+ e X. (os radicais
podem ser considerados espécies deficientes de elétron dado
que necessitam de um elétron adicional para formar uma
ligação), cátions induzindo fissão heterolítica, e radicais fissão
homolítica da ligação π. Verifica-se que a primeira predomina
normalmente em solventes polares, e a última em solventes
não polares, especialmente na presença de luz.
ADIÇÃO DE HALOGÊNIOS
A descoloração de bromo, em solução de CCl4, é um
dos testes de insaturação entre as reações de adição de
alcenos. Normalmente ocorre rapidamente na ausência de
catalisadores externos.
Se a adição de bromo for realizada na presença de
nucleófilos externos Y: ou Y- (Cl-, NO3-, H2O), então o 1,2-
dibromo (3) é formado, e os produtos nos quais um átomo de
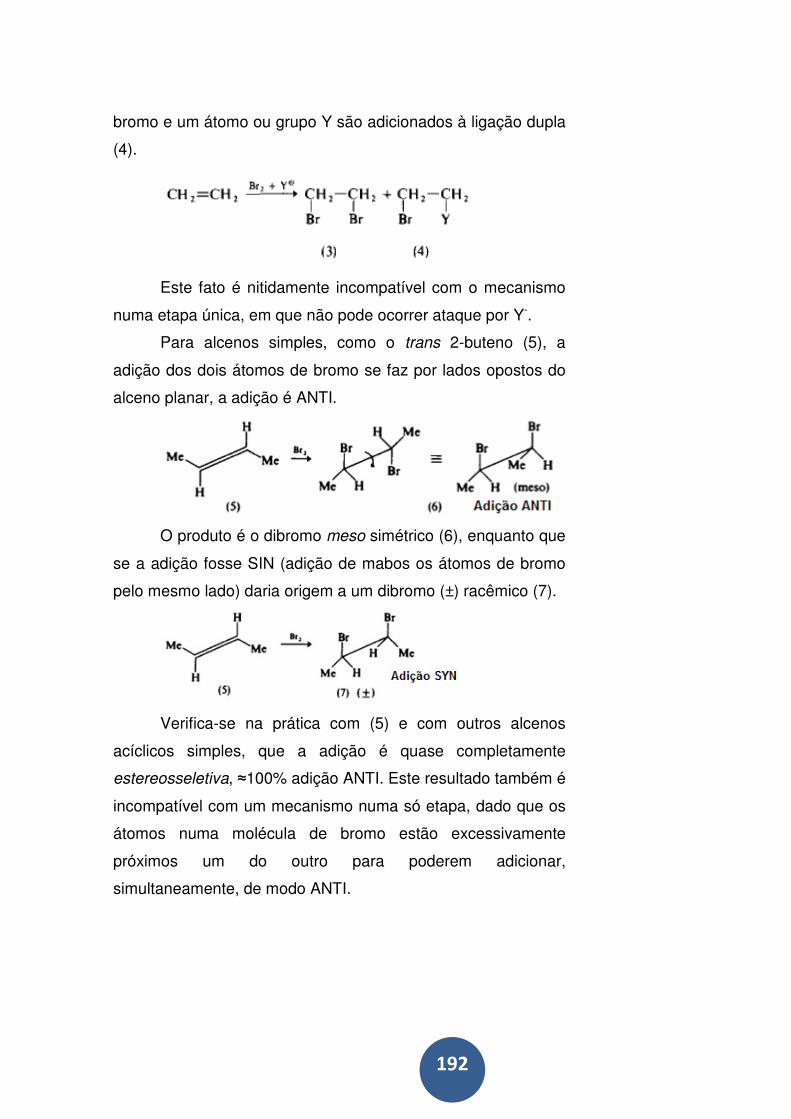
192
bromo e um átomo ou grupo Y são adicionados à ligação dupla
(4).
Este fato é nitidamente incompatível com o mecanismo
numa etapa única, em que não pode ocorrer ataque por Y-.
Para alcenos simples, como o trans 2-buteno (5), a
adição dos dois átomos de bromo se faz por lados opostos do
alceno planar, a adição é ANTI.
O produto é o dibromo meso simétrico (6), enquanto que
se a adição fosse SIN (adição de mabos os átomos de bromo
pelo mesmo lado) daria origem a um dibromo (±) racêmico (7).
Verifica-se na prática com (5) e com outros alcenos
acíclicos simples, que a adição é quase completamente
estereosseletiva, ≈100% adição ANTI. Este resultado também é
incompatível com um mecanismo numa só etapa, dado que os
átomos numa molécula de bromo estão excessivamente
próximos um do outro para poderem adicionar,
simultaneamente, de modo ANTI.

193
Estas observações são explicáveis por um mecanismo
em que um dos extremos de uma molécula de bromo fica
polarizado positivamente através da repulsão eletrônica pelos
elétrons π do alceno, formando, assim, um complexo π (8). Esta
quebra então para formar um íon bromônio cíclico (9), uma
forma canônica alternativa do íon carbônio (10). A adição
ocorre pelo ataque nucleofílico do Br- (ou Y-) a um dos átomos
de carbono que originou a ligação dupla pelo lado oposto do
íon bromônio Br+ volumoso, para dar o dibromo meso (6).
Íons bromônios intermediários tais como (9), explicam a
elevada estereosseletividade da adição (ANTI) frequentemente
observada com alcenos acíclicos simples.
A cloraçao não é tão estéreosseletiva quanto a
bromação, mas tende a seguir o mesmo padrão. A formação
do íon halônio é mais dificultada, devido a menor
polarizabilidade do cloro. Também se pode prever que as
características estruturais que conduzem a estabilização
específica de íons carbônio, devem igualmente conduzir a uma
menor estereosseletividade ANTI, isto observa-se com, por
exemplo, trans 1-fenilpropeno (11).

194
A possível formação de um íon carbônio (12), resulta
numa estereoespecificidade ANTI muito menor (70%) do que
com trans but- 2-eno (5, ≈100% ANTI), onde não é possível a
deslocalização. Verifica-se também que o aumento de
polaridade do solvente e da sua capacidade de solvatação de
íons estabiliza o íon carbônio intermediário, relativamente ao
íon bromônio, com consequente decréscimo de
estereosseletividade ANTI. Assim, verifica-se que a adição de
bromo a 1,2-difeniletileno (estilbeno) é 90-100% ANTI em
solventes de constante dielétrica baixa, mas só ≈50% ANTI
num solvente com ε = 35.
EFEITO DE SUBSTITUINTES NA VELOCIDADE DE ADIÇÃO
O intermediário na reação de bromação, se é um íon
bromônio ou um íon carbônio, é certamente carregado
positivamente. Sua formação, o intermediário, é determinante
da velocidade, e o estado de transição que o procede, é
estabilizado por substituintes doadores de elétrons (13) que
acelerem a velocidade da adição eletrofílica.
O aumento de velocidade observada provém da doação
de elétrons por introdução dos grupos alquila e é menor do que
se esperava, devido ao aumento do impedimento estéreo no

195
estado de transição. Um grupo fenilo também aumenta
consideravelmente a velocidade da adição eletrofílica (4 x 103),
devido à estabilização que pode ocorrer no intermediário (15),
e no estado de transição.
Orientação da adição
Quando um eletrófilo é adicionado a um alceno não-
simétrico, por exemplo, propeno, qual é orientação da adição?
Uma reação é escrita como regiosseletiva se na adição a um
alceno não simétrico houver a predominância de um dos
possíveis produtos de adição, e é regioespecífica se for
formado exclusivamente um produto. Estas reações são
regioespecíficas. Seguem a regra de Markownikov.
Na adição de ácidos hologenídricos, verifica-se que a
ordem de velocidades é: HF < HCl < HBr < HI, que é a ordem
da força ácida. A adição do próton ao alceno é o passo
determinante da velocidade, seguindo-se o ataque pelo
nucleófilo rápido por X-. Em solventes não-polares o próton é,
sem dúvida, fornecido por HX, mas em solventes polares,
especialmente nos hidroxílicos, é fornecido pelo seu ácido
conjugado H3O+ em H2O.
Para a reação do propeno com HX, forma um
intermediário como um íon carbônio e é a estabilidade relativa
de possíveis íons carbônio (17 que 18) determina a orientação
da adição, na reação de adição de HBr ao propeno (16) em
condições polares.

196
Os íons carbônio secundários são mais estáveis do que
os primários, assim como os estados de transição que os
precedem, (18) será formado de preferência a (17), e o único
produto de adição obtido é o 2-bromopropano (19). A adição,
em que o halogênio (ou o agrupamento mais negativo de
qualquer outro aduto assimétrico) fica ligado ao mais
substituído dos dois átomos de carbono do alceno, é conhecida
como adição de Markownikov.
Essa regiosseltividade ocorre, porque a formação de um
carbocátion intermediário apresenta a seguinte ordem de
estabilidade: +CH2CH2R < CH3
+CHR < CH3+CHAr < CH3
+CR2 <
CH3+CAr2
A orientação da adição eletrofílica a 1-haloalceno (20) é
controlada pelos pares eletrônicos no Bromo.
O íon corbônio (22) é estabilizado em relação a (21), e
por isso é formado preferencialmente o 1,1-dibromoetano (23)
como único produto. A velocidade da adição é controlada pelo
efeito indutivo negativo do átomo de halogênio. A reação de

197
adição de HBr ao bromoeteno (20) é cerca de 30 vezes mais
lenta do que o eteno, e (22) é menos estável, e sua formação é
mais lenta do que (24).
OUTRAS REAÇÕES DE ADIÇÃO
Outros derivados halogenados
Os compostos inter-halogenados adicionam a alcenos
como os halogênios, tendo sido observada a seguinte ordem
de reatividade, BrCl > Br2 > ICl > IBr > I2.
A adição é iniciada pela extremidade polarizada
positivamente da molécula assimétrica, e daí resulta
provavelmente um íon halônio intermediário, cíclico. Com um
alceno assimétrico, o 2-metilpropeno (25), a distribuição de
carga no intermediário será assimétrica, o carbono mais
alquilado será mais positivo (26) e será atacado pelo haleto
nucleofílico residual. Deste modo, determinará a orientação
(Markownikov) da adição (27).
Os ácidos hipohalogenoso, HOδ-–Brδ+ (água de bromo),
adicionam-se de modo semelhante, sendo o átomo de bromo o
eletrófilo, formando a 1,2-bromo-hidrina (29) que é obtida por
competição de Br- e H2O pelo íon bromônio intermediário inicial
(28).

198
Hidratação
A hidratação de um alceno, catalisada por ácido, é o
inverso da desidratação de álcois a alcenos, catalisada por
ácido (mecanismo de reação E1).
A formação do íon carbônio intermediário (30), via
complexo π inicial, é determinante da velocidade, e a adição é
como Markownikov e a estereosseletividade é ANTI, mas
depende do alceno e das condições reacioanais.
A hidratação anti-Markownikov de alcenos pode
ocorrer por adição de B2H6 (hidroboração), seguida de
oxidação do trialquilborano resultante (31) com H2O2 alcalino.
O diborano é gerado (in situ, ou separadamente, a partir
de NaBH4 e Et2O-, BF3
-), tal como o BH3, com solvente etéreo
usado na reação. BH3 é um ácido de Lewis e adiciona-se ao
átomo de carbono menos substituído do alceno (adição anti-
Markownikov), sendo a adição global completada por
transferência de hidreto para o átomo de carbono adjacente,
polarizado positivamente.
É provável que (33) tenha algum caráter cíclico na a
adição de BH3, e a estereosseletividade é SIN. O RBH2
formado reage depois com o alceno para dar o trialquilborano,
R3B (31). A oxidação com H2O2 resulta na ruptura da ligação
C__B para dar o álcool (32). A hidratação é anti-Markownikov,
com estereosseletividade SIN.

199
Íons carbônios
A protonação de alcenos origina íons carbônios, na
ausência de outros nucleófilos eficazes (H2O), estes íons
reagem como eletrófilos frente a alcenos não protonados como
o 2-metilpropeno (34).
O cátion (35) reage com uma segunda molécula de 2-
metilpropeno (34) para dar o novo cátion (36), este, pode
perder um próton para dar o alceno (37), ou, adicionar uma
terceira molécula de alceno para dar o cátion (38), e assim
sucessivamente.
O 2-metilpropeno pode ser tratado de modo a continuar
o processo para dar altos polímeros, polimerização catiônica,
mas a maior parte dos alcenos simples não vai além de
estruturas di- ou tri-monoméricas. Os principais alcenos
monoméricos usados em larga escala são o 2-metilpropeno
(borracha butílica) e éteres vinílicos, ROCH=CH2 (adesivos). A
polimerização catiônica é frequentemente iniciada por ácidos
de Lewis, como o BF3, mais uma fonte de prótons iniciail, como
co-catalisador, que pode ser traços de H2O, nestas condições a
polarização ocorre facilmente a temperaturas baixas e é, em
geral, muito rápida. Muitos outros alcenos são polimerizados
por um mecanismo induzido por radicais.
Hidroxilação
Os reagentes que adicionam dois grupos OH aos
alcenos, como o tetróxido de ósmio, OsO4, adiciona de modo a
formar ésteres ósmicos cíclicos (40), que podem sofrer rápida
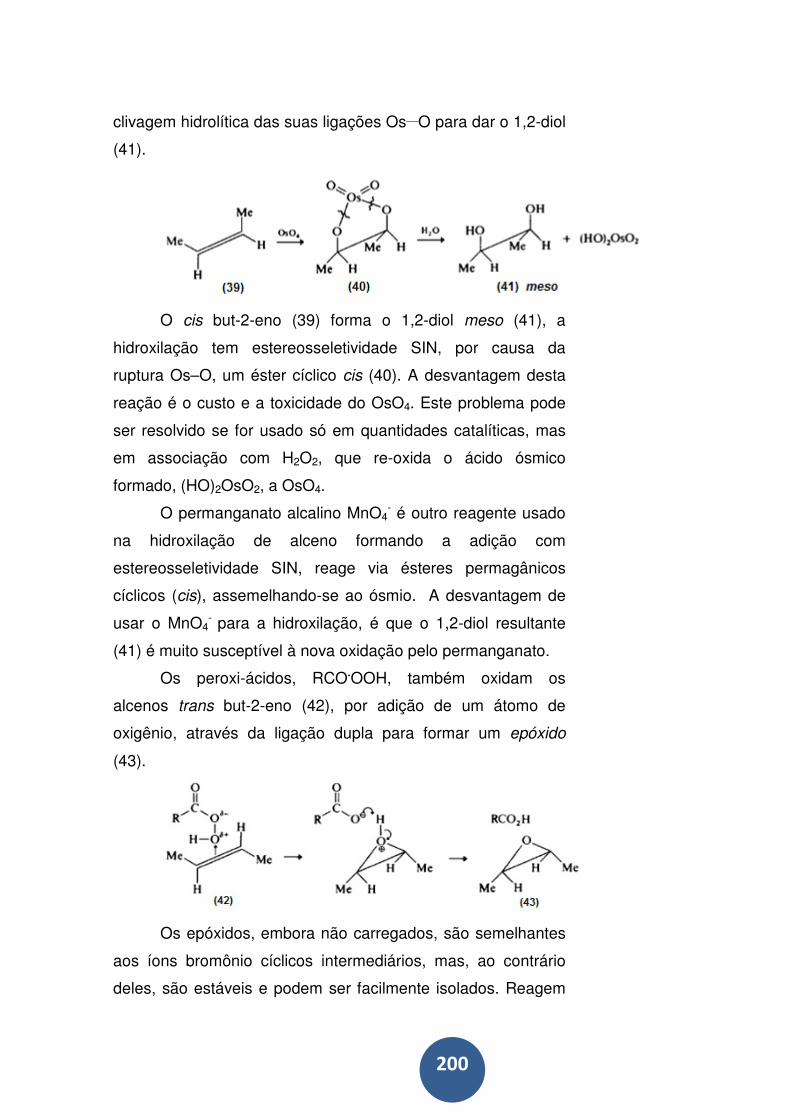
200
clivagem hidrolítica das suas ligações Os__O para dar o 1,2-diol
(41).
O cis but-2-eno (39) forma o 1,2-diol meso (41), a
hidroxilação tem estereosseletividade SIN, por causa da
ruptura Os–O, um éster cíclico cis (40). A desvantagem desta
reação é o custo e a toxicidade do OsO4. Este problema pode
ser resolvido se for usado só em quantidades catalíticas, mas
em associação com H2O2, que re-oxida o ácido ósmico
formado, (HO)2OsO2, a OsO4.
O permanganato alcalino MnO4- é outro reagente usado
na hidroxilação de alceno formando a adição com
estereosseletividade SIN, reage via ésteres permagânicos
cíclicos (cis), assemelhando-se ao ósmio. A desvantagem de
usar o MnO4- para a hidroxilação, é que o 1,2-diol resultante
(41) é muito susceptível à nova oxidação pelo permanganato.
Os peroxi-ácidos, RCO.OOH, também oxidam os
alcenos trans but-2-eno (42), por adição de um átomo de
oxigênio, através da ligação dupla para formar um epóxido
(43).
Os epóxidos, embora não carregados, são semelhantes
aos íons bromônio cíclicos intermediários, mas, ao contrário
deles, são estáveis e podem ser facilmente isolados. Reagem

201
frente a um nucleófilo, em condições de catálise ácida ou
básica, formando o 1,2-diol. Em qualquer caso, o ataque pelo
nucleófilo num átomo de carbono será pelo lado oposto à ponte
de oxigênio em (43), resultando em inversão de configuração.
O ataque ocorre em um dos dois átomos de carbono
possíveis de (43) e (44) e, em cada caso, conduzirá ao mesmo
produto o meso 1,2-diol (45). Comparando a configuração de
(45) com a do alceno original (43) o e ocorrendo a hidroxilação,
esta aconteceu com estereosseletividade ANTI. Dependendo
da escolha do reagente, a hidroxilação de alcenos pode formar
produtos SIN ou ANTI.
Hidrogenação
A adição de hidrogênio a compostos insaturados pode
ocorrer pela adição direta de hidrogênio catálisada por metais,
como Ni, Pt, Pd, Ru, Rh, catálise heterogênia.
O catalisador é usado em pequena quantidade em
relação aos outros reagentes. O catalisador adsorve fortemente
o alceno, e dessorvem imediatamente o alcano resultante,
ficando livres para nova adsorção de alceno.
A hidrogenação ocorre com estereoespecificidade SIN,
predominante.
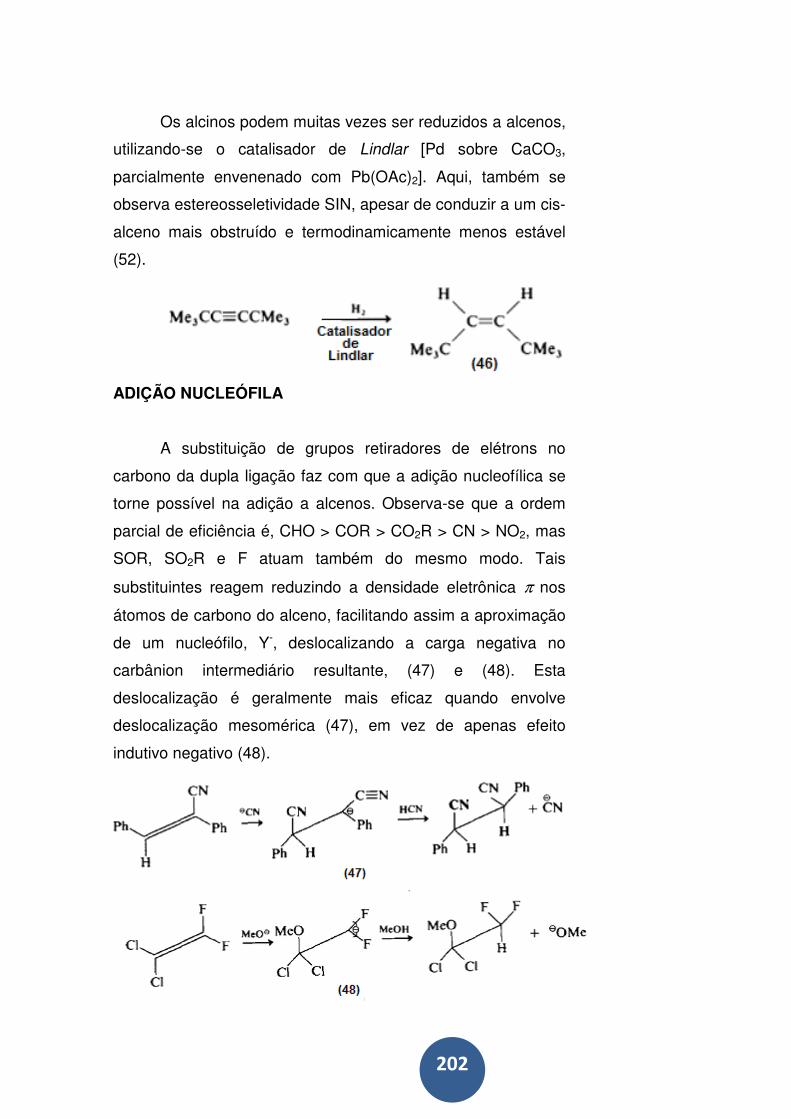
202
Os alcinos podem muitas vezes ser reduzidos a alcenos,
utilizando-se o catalisador de Lindlar [Pd sobre CaCO3,
parcialmente envenenado com Pb(OAc)2]. Aqui, também se
observa estereosseletividade SIN, apesar de conduzir a um cis-
alceno mais obstruído e termodinamicamente menos estável
(52).
ADIÇÃO NUCLEÓFILA
A substituição de grupos retiradores de elétrons no
carbono da dupla ligação faz com que a adição nucleofílica se
torne possível na adição a alcenos. Observa-se que a ordem
parcial de eficiência é, CHO > COR > CO2R > CN > NO2, mas
SOR, SO2R e F atuam também do mesmo modo. Tais
substituintes reagem reduzindo a densidade eletrônica π nos
átomos de carbono do alceno, facilitando assim a aproximação
de um nucleófilo, Y-, deslocalizando a carga negativa no
carbânion intermediário resultante, (47) e (48). Esta
deslocalização é geralmente mais eficaz quando envolve
deslocalização mesomérica (47), em vez de apenas efeito
indutivo negativo (48).

203
A orientação da adição de um aduto assimétrico, HY ou
XY, a um alceno substituído assimétrico será definida pela
formação preferencial do carbânion mais estabilizado. A adição
nucleofílica também ocorre com alcinos convenientes e
geralmente mais facilmente do que com os alcenos
correspondentes.
Cianoetilação
Dentre as reações importantes, em síntese, ocorrem
com o eteno com um substituinte ciano (acrilonitrila, 49). O
ataque de Y- ou Y: ao átomo de carbono não substituído,
seguida por abstração de um próton do solvente, conduz à
ligação de um grupo 2-cianoetila ao nucleófilo inicial.
O processo é então referido como cianoetilação. A
reação é frequentemente realizada na presença de base, de
modo a converter HY no nucleófilo poderoso Y-. A utilidade
sintética da cianoetilação reside na incorporação de uma
unidade de três carbonos, em que o grupo ciano terminal pode
ser modificado por redução, hidrólise, etc., podendo ser
utilizado em operações de síntese subsequentes.
Reações de Michael
Quando o nucleófilo que ataca o alceno substituído é um
carbânion, o processo é conhecido como reação de Michael, e
a sua utilidade em síntese reside no fato de ser um método
geral para a formação de ligações carbono-carbono (50).

204
A reação é favorecida por uma variedade de bases, em
quantidades catalíticas, que geram uma concentração no
equilíbrio do carbânion (51) que é reversível, sendo a etapa
determinante da velocidade a formação da ligação carbono-
carbono, a reação do carbânion (51) com o alceno (50). A sua
utilidade geral em síntese provém da grande variedade de
alcenos substituídos e de carbânions que podem ser
empregados, principalmente provenientes de CH2(CO2Et)2,
MeCOCH2CO2Et, NCCH2CO2Et, RCH2NO2, etc.
ADIÇÃO NUCLEOFÍLICA A CARBONILA (C=O)
Os compostos carbonílicos têm momentos dipolares (µ)
porque o átomo de oxigênio da carbonila (C=O) é mais
eletronegativo do que o átomo de carbono.
O efeito indutivo C→O que atua na ligação σ, que une
os dois átomos, afeta também os elétrons π mais facilmente
polarizáveis, de tal modo que o grupo carbonila é melhor
representado pela estrutura híbrida.
A ligação C=O, devido à sua natureza bipolar, pode ser
atacado inicialmente pelo eletrófilo X+ ou X (ácido ou ácido de
Lewis) no oxigênio ou pelo ataque nucleofílico de Y- ou Y no
carbono.

205
A protonação aumentará nitidamente o caráter positivo
do átomo de carbono carbonílico,
C O
R
R'
C OH
R
R'
C OH
R
R'
H+
+ +
e facilitará assim o ataque pelo nucleófilo no átomo de carbono.
Uma ativação semelhante, embora menos pronunciada, ocorre
por ligações de hidrogênio entre o átomo de oxigênio da
carbonila e um ácido, ou até mesmo um solvente hidroxílico.
Na ausência deste tipo de ativação, os nucleófilos
fracos, H2O, podem reagir apenas muito lentamente, mas os
fortes, –CN, não necessitam daquela ativação para que a
reação ocorra. As adições também podem ser catalisadas por
base, a qual converte o nucleófilo fraco HY no Y- mais forte,
HCN + base → –CN. Enquanto que os ácidos podem ativar o
átomo de carbono carbonílico para o ataque nucleofílico e
simultaneamente podem reduzir a concentração do nucleófilo, –
CN + HA → HCN + A+, RNH2 + HA → RNH3+ + A-. Assim, as
reações de adição nucleofílica a compostos carbonílicos
ocorrem em um pH ótimo.
ESTRUTURA E REATIVIDADE
Nas adições nucleofílicas simples, em que a etapa
determinante da velocidade é o ataque por Y-, o caráter
positivo do átomo de carbono carbonílico é reduzido ao passar-
se do material de partida para o estado de transição.
R2C O R2C O
Y
R2C O
Y
R2C OH
Yδ
δ δ δ δ-+ -
-
+Y+
HY --Y -

206
Os grupos substituintes (R) doadores elétrons diminuem
a velocidade de adição enquanto que os grupos substituintes
(R) retiradores de elétrons aumentam, o que é evidenciado
pela seguinte sequência:
Os grupos R em que o grupo C=O está conjugado com
C=C ou com um anel benzênico, também apresentam reações
de adição mais lentas do que os seus análogos saturados, por
causa da estabilização, através de deslocalização:
Nos exemplos referidos, as velocidades de reação
relativas podem ser influenciadas por efeitos estereoquímicos e
eletrônicos.
Em geral, esses fatores influenciam na velocidade da
reação (k), na posição do equilíbrio (K), isto por que o estado
de transição para reações de adição simples se assemelha
muito mais ao aduto do que ao próprio composto carbonílico
original. Assim, verifica-se que os valores de K para a formação
de cianidrina refletem a influência tanto de fatores
estereoquímicos como eletrônicos.
Reagentes Constante de equilíbrio (K) CH3CHO Muito grande p-NO2C6H4CHO 1420 C6H5CHO 210 p-CH3OC6H4CHO 32 CH3COCH2CH3 38

207
C6H5COCH3 0,8 C6H5COC6H5 Muito pequeno
Cetonas extremamente impedidas, como Me3CCOCMe3,
não reagem, exceto, com nucleófilos muito pequenos e
altamente reativos.
REAÇÕES DE ADIÇÃO SIMPLES A ALDEÍDOS E CETONAS
Hidratação
Muitos compostos carbonílicos sofrem hidratação
reversível em solução aquosa,
sendo os valores de K a 20° C para H2C=O, MeHC=O e
Me2C=O de 2, 1,4 e 2 x 10-3, respectivamente, esta sequência
reflete a ação progressiva do efeito doador de elétron
crescente. A reversibilidade da hidratação reflete-se no fato de
H2C=O podem ser destilado a partir de uma solução aquosa. A
reação em presença de H218O demonstrou que com Me2C=O
se estabelece um equilíbrio dinâmico, embora a concentração
do hidrato no meio seja muito reduzida.
Em pH 7, praticamente não há incorporação de 18O na
cetona, mas na presença de traços de ácido ou de base, a
incorporação é extremamente rápida. O fato de um composto
carbonílico se encontrar hidratado não influenciará as adições
nucleófilas que são irreversíveis, mas pode influenciar a
posição de equilíbrio em reações de adição reversíveis, e

208
também a velocidade da reação, já que a concentração real de
composto carbonílico livre [R2C=O] é naturalmente reduzida.
Verifica-se que a hidratação é suscetível quer de catálise
ácida geral, quer de catálise básica geral. A etapa determinante
da velocidade da reação envolve ou a protonação do composto
carbonílico, ou conversão de H2O no –OH mais nucleófilo .
O H2CO hidrata facilmente a pH 7, o átomo de carbono
carbonílico é positivo para sofrer ataque por H2O, mas este
composto hidrata muito mais rapidamente a pH 4 ou 11.
Os substituintes doadores de elétrons inibem a formação
de hidratos, mas os substituintes retiradores de elétrons
favorecem a hidratação. Assim, para a hidratação de Cl3CCHO
com K igual a 3,7 x 104, e este aldeído (tricloroetanal, cloral)
forma de fato um hidrato cristalino que pode ser isolado. Os
átomos de cloro fortemente elétron retiradores desestabilizam o
composto carbonílico inicial, mas não o hidrato formado.

209
Para que o hidrato reverta ao composto carbonílico
inicial, tem que perder –OH ou H2O, o que é dificultado pela
presença dos grupos elétron retiradores. Os grupos carbonila
são eficientes na estabilização de hidratos, por ligações de
hidrogênio, e por ação de efeitos elétron retiradores, como a
difenilpropanotriona que cristaliza em água na forma de hidrato.
Outro exemplo de um hidrato fácil de isolar é o hidrato
obtido a partir de ciclopropanona, devido a diminuição da
tensão angular das ligações ao passar-se (C–C–C de 60°,
comparado com o valor normal de 120°) para o hidrato (C–C–C
de 60°, comparado com o valor normal de 109°).
Adição de álcoois – formação de acetal e hemiacetal
Em síntese orgânica, por vezes acontece que um dos
reagentes contém grupos funcionais incompatíveis com as
condições de reação. Assim, faz-se necessário introduzir
grupos protetores, fáceis de introduzir e que mantêm-se
inalterados quando necessário e fáceis de remover. Estes
grupos protetores são conhecidos como acetais ou hemiacetal.
Muitas das reações de adição nucleofílica a aldeídos e
cetonas com alcoóis, em condições de catálise ácida, implicam
na transformação do produto inicial em um hemiacetal. O
produto da reação, no entanto, quando a reação é feita com um
mol de aldeído e dois mols de álcool, forma diéteres geminal
conhecido como acetais.

210
As reações globais ocorrem em duas etapas. O
hemiacetal é formado na primeira etapa por adição nucleofílica
do álcool ao grupo carbonila. O mecanismo de formação
hemiacetal é exatamente análogo ao de hidratação catalisada
por ácido, de aldeídos e cetonas.
Sob as condições ácidas a formação do hemiacetal é
convertida para um acetal por meio de um intermediário,
carbocátion.
O carbocátion é estabilizado pela deslocalização de
elétrons do oxigênio, uma forma de ressonância
particularmente estável, ambos carbono e oxigênio têm octetos
de elétrons.
A adição nucleofílica no carbocátion intermediário por
uma molécula de álcool leva a um acetal.

211
A formação acetal é reversível em ácido. Um equilíbrio é
estabelecido entre os reagentes, ou seja, o composto
carbonílico e do álcool, e o produto acetal. A posição de
equilíbrio é favorável para a formação de acetal da maioria dos
aldeídos, especialmente quando o álcool em excesso está
presente na reação como solvente. Para a maioria das reações
com cetonas, a posição de equilíbrio é desfavorável, e os
acetais são formados mais facilmente em presença de dióis,
por exemplo, a reação de uma cetona com o 1,2-etanodiol para
formar acetais cíclicos.
O fato de a reação se dar com o diol, mas não com
álcool simples, é devido o valor de ∆S para diol ser mais
favorável do que o álcool mais simples, que envolve um
decréscimo no número de moléculas ao passar de reagente
para produto.
Adição de tióis
Os compostos carbonílicos reagem com tióis, RSH, para
formar hemitioacetais e tioacetais, mais fácil do que reagem
com álcool, isto reflete a maior nucleofilicidade do enxofre em
comparação com oxigênio. Como os acetais, os tioacetais
oferecem proteção para o grupo C=O, já que são relativamente
estáveis a ácidos diluídos, e decompoem-se rápido por ação de
HgCl2/CdCO3. Sofrem também dessulfurização com: hidrogênio

212
e níquel como catalisador, úteis no processo de síntese para a
conversão indireta de C=O→CH2.
R2C OR'SH
R2C(SR')2
H2/NiR2CH2
Esta é uma conversão geralmente difícil de
efetuar de modo direto.
Adição de cianeto de hidrogênio
A adição de HCN deve envolver um carbânion. Trata-se
de uma reação de interesse em síntese orgânica e seu
mecanismo estabelecido (Lapworth, 1903). O HCN não é um
nucleófilo poderoso para atacar C=O, e a reação deve ser feita
em catálise básica de modo que HCN seja convertido em –CN
mais nucleofílico, com a seguinte lei de velocidade.
Velocidade = k[R2C=O][-CN]
A adição de –CN é reversível, e o equilíbrio tende a
deslocar-se no sentido dos reagentes, se estiver presente um
doador de prótons; este desloca o equilíbrio para a direita, já
que o equilíbrio que envolve a cianidrina é geralmente mais
favorável do que o que envolve o ânion.
O ataque por –CN é lento (determinante da velocidade),
ao passo que a transferência de próton de HCN ou de um
solvente prótico, H2O, é rápida.
A reação é reversível, o problema é a toxicidade do HCN
que tem afinidade pelo Fe(III) inibindo a cadeia de transporte
da mitocôndria, na etapa mediada pela enzima citocromo
oxidase. Dessa forma, cessam a respiração mitocondrial e a
produção de energia pelas células, levando a morte. Daí há

213
outras alternativas para produzir cianidrinas, via bissulfito
(somente para aldeídos) e via cianeto de trimetilsilila.
Adição de bissulfito
A adição do íon bissulfito ocorre com aldeídos,
metilcetonas e cetonas cíclicas, formando adutos cristalinos.
Os sais de ácidos sulfônicos formados refletem numa maior
nucleofilicidade do enxofre em relação ao oxigênio. O nucleófilo
que reage é o SO32-, e não HSO3
-, (HO- + HSO3- H2 + SO3
2-).
O ânion atacante está presente na solução, o SO32-, é
um nucleófilo poderoso e não requer ativação (por protonação)
do grupo carbonilo, não sendo necessária catálise ácida ou
básica.
Adição de íons hidreto
Os grupos carbonilos podem ser reduzidos
cataliticamente tal como o são as ligações carbono-carbono
insaturadas. No entanto, normalmente é mais difícil efetuar a
redução catalítica de grupo C=O do que os grupos, C=C, C≡C,
C=N ou C≡N. A maioria dos compostos carbonílicos são
reduzidos por reagentes de boro ou alumínio por transferência
de hidretos. Essas reduções seletivas podem, no entanto, ser
efetuadas com diversos hidretos metálicos, geralmente
complexos.
Adição de complexos de íons hidreto com metais
O hidreto de lítio e alumínio, Li+AlH4-, é entre os
complexos um dos mais potentes, que reduz o grupo C=O em
aldeídos, cetonas, ácidos, ésteres e amidas a CH2OH nos

214
primeiros, e CH2 nas amidas, deixando intacta qualquer ligação
C=C ou C≡C que também esteja presente no composto (as
ligações C=C conjugadas com C=O são, por vezes, afetadas).
O verdadeiro agente redutor atua como um poderoso doador
de íon hidreto, H-, a redução não pode ser efetuada em
solventes próticos, H2O, ROH, dado que então haveria uma
abstração de próton preferencial. Os éteres, em grande número
dos quais Li+AlH4- é solúvel, são por isso vulgarmente utilizados
como solventes.
O AlH4- nucleófilo doa H-, de modo irreversível, ao átomo
de carbono carbonílico, e o AlH3 residual complexa então com
o seu átomo de oxigênio para formar o ânion, que forma o
complexo
e AlH4-, única espécie de fato envolvida na doação de
hidreto. O complexo é convertido no produto, o álcool, por
tratamento com um solvente prótico.
Reação de Meerwein-Ponndorf
Esta reação é um exemplo clássico de redução de
cetonas, com Al(OCHMe2)3 em propan-2-ol, pela transferência
de hidreto do carbono para um átomo de carbono carbonílico,
de modo reversível, estabelecendo-se um equilíbrio.
A propanona é o constituinte do sistema com o
ponto de ebulição mais baixo, o equilíbrio é deslocado, para a
direita por destilação contínua da propanona, removendo-a do
sistema. É utilizado um excesso de propano-2-ol, que troca

215
com o alcóxido misto de alumínio produzido, libertando o
produto de redução desejado R2CHOH, onde o átomo de
hidrogênio é obtido por transferência de hidreto do carbono no
estado de transição.
Reações de Cannizzaro
Esta reação envolve a transferência de hidreto de uma
molécula de aldeído sem átomos de hidrogênio em posição α,
como HCHO, R3CCHO, ArCHO, para uma segunda molécula
quer do mesmo aldeído, quer para uma molécula de um
aldeído diferente (reação de Cannizzaro cruzada). A reação
requer a presença de bases fortes e com, por exemplo,
PhCHO, segue a lei de velocidade, Velocidade = k[PhCHO]2[-
OH].
A reação apresenta o seguinte mecanismo:
A adição rápida e reversível de –OH a PhCHO dá
origem ao grupo doador do hidreto, pela transferência, lenta e
determinante da velocidade, de hidreto para o átomo de
carbono carbonílico de uma segunda molécula de PhCHO. A
reação é completada por uma troca rápida de próton para dar o
par estável, com o equilíbrio deslocado para a direita. Ocorre
uma oxidação/redução mútua de duas moléculas do aldeído
para dar uma molécula do ânion carboxilato e do álcool
primário.
Adição de elétrons
Os átomos dos metais mais eletropositivos, Na, K, Li,
etc., podem em determinadas condições, produzir elétrons
solvatados.

216
Estes elétrons podem atuar como nucleófilos e
adicionar-se a átomos de carbono carbonílicos C=O, para dar
um ânion radicalar, como um par iônico com o cátion metálico,
M+.
A propanona é facilmente convertida por magnésio em
2,3-dimetilbutan-1,2-diol, chamado pinacol:
Reações de adição/eliminação
As reações de adições nucleofílicas a C=O, em que o
nucleófilo adicionado carrega um próton ácido, tornam possível
uma eliminação subsequente, o resultado é uma substituição.
Os exemplos mais comuns observam-se com os
derivados de NH3, como HONH2, NH2COCNHNH2, PhNHNH2,
etc., usados para converter compostos carbonílicos líquidos em
derivados sólidos.
Derivados de NH3
A reação a pH 7
entre o ânion piruvato e
hidroxilamina, NH2OH,
para formar a oxima,
ocorre via formação de um intermediário:

217
Aumentando a acidez da mistura reacional diminui a
velocidade adição (desaparecimento da absorção de CO)
porque a hidroxilamina NH2OH é convertida em HN+H2OH, que
não é nucleofílico, mas aumenta a velocidade à qual aparece a
absorção de C=N, devido a catálise ácida da desidratação.
Estas observações são compatíveis com um mecanismo
reacional da seguinte forma.
Nucleófilos fortes tais como NH2OH (Y=OH) não
requerem catálise para a adição inicial a C=O, mas outros mais
fracos, como PhNHNH2 (Y=PhNH) e NH2CONHNH2
(Y=NHCONH2), frequentemente requerem catálise ácida para
ativar o grupo C=O. Dependendo do pH da reação, a etapa de
adição inicial ou a etapa de desidratação é determinante da
velocidade da reação. A pH neutro ou alcalino geralmente é a
desidratação, que é etapa lenta, determinante da velocidade,
enquanto que a pH mais ácidos, a etapa lenta e determinante
da velocidade, é a adição inicial do nucleófilo. Assim, para a
formação de uma oxima a partir de propanona, Me2CO, pH
ótimo é 4,5.
Adição de carbono nucleofílico
As reações adição de carbono nucleofílico serão
consideradas um grupo que resultam na formação de ligações
carbono-carbono, que têm utilidade e importância nas reações
em síntese orgânica.

218
Reagentes de Gringnard
A estrutura do reagentes de Grignard, RMgX, depende
da natureza de R e também do solvente em que o reagente é
dissolvido. O reagente de Grignard reage como fonte de
carbono polarizado negativamente, como δ-RMgXδ+. A
complexação do átomo de Mg do reagente de Grignard com o
átomo de oxigênio carbonílico, em alguns casos, estão
envolvidas duas moléculas de RMgX na reação de adição, via
um estado de transição cíclico como:
A segunda molécula de RMgX, como um catalisador
ácido de Lewis, aumenta a polarização positiva do átomo de
carbono carbonílico através da complexação com oxigênio.
Os reagentes de Grignard que apresentam átomos de H
no carbono β (RCH2CH2MgX) tendem a reduzir C=O → CHOH,
formando alceno (durante o processo é uma transferência de
H, em vez de transferência de RCH2CH2).
As cetonas estereoquimicamente impedidas, contendo
átomos de H nos seus carbonos α, tendem a ser convertidas
nos seus enóis, sendo o reagente de Grignard RMgX perdido
no processo sob a forma de RH.

219
Íons acetilênicos
Os acetilenos, RC≡CH e HC≡CH, são ácidos e podem
ser convertidos, por bases fortes, como -NH2, em amoníaco,
em ânions correspondentes, que são mais nucleofílicos do que –CN. Assim podem adicionar-se a C=O, e a reação tem
utilidade em síntese pelo fato de a ligação C≡≡≡≡C presente poder
sofrer redução ao alceno por H2 com o catalisador de Lindlar.
Reações aldólicos
Nestas reações, o carbânion, obtido pela base (-OH) por
retirar um hidrogênio α de uma molécula de um composto
carbonílico, adiciona-se ao átomo de carbono carbonílico de
outra molécula para dar um composto β-hidroxicarbonílico.
Com etanal, CH3CHO, o produto é o 3-hidroxibutanal, um aldol:
No caso de CH3CHO, verifica-se que o equilíbrio se
encontra deslocado para a direita, a favor do aldol. A reação
direta da etapa (2) é muito mais rápida do que a reação inversa
da etapa (1), ambas competindo entre si pelo carbânion.
Em condições ácidas ocorre desidratação, e a reação é
denominada condensação aldólica.
As reações aldólicas cruzadas, em que ambos os
aldeídos têm átomos de H em posição α, podem resultar numa

220
mistura de quatro produtos diferentes. N as reações aldólicas
cruzadas de um aldeído que não tem α-H, só pode reagir
aceitando carbânion, como a condensação de Claisen-Schmidt
de aldeídos aromáticos com aldeídos alifáticos simples ou
cetonas (geralmente metil cetonas), em presença de 10% KOH
aquoso (a desidratação ocorrre, nestas condições, depois da
adição de carbânion).
Os substituintes doadores de elétrons em aromáticos
tornam a reação mais lenta, o p-MeOC6H4CHO reage com uma
velocidade que é cerca de 1/7 da velocidade de reação de
C6H5CHO. É claro que a autocondensação do aldeído alifático
é uma reação competitiva nestas condições, mas a reação de
Cannizzaro de ArCHO é uma reação tão lenta que não chega a
poder competir significativamente.
As reações aldólicas ocorrem com compostos
dicarbonílicos apropriados, intramoleculares e com ciclizações.
Reação de Perkin
Nesta reação, o carbânion é obtido por remoção de um
próton de átomo α-H de uma molécula de um andrido de ácido
e reage com uma carbonila de um aldeído aromático. Os
produtos obtidos são ácidos α,β-insaturados, como na síntese
de ácido 3-fenilpropenóico (ácido cinâmico)

221
Nas condições reacionais (excesso de anidrido como
solvente a ~140 °C), ocorre a desidratação do aduto inicial,
com formação do anidrido misto. Colacando a mistura reacional
em água, ocorre a hidrólise do anidrido misto aos ácidos
correspondentes, PhCH=CHCO2H + MeCO2H.
Reações de Knoevenagel e de Stobbe
Este tipo de adições envolve carbânions provenientes de
uma variedade de compostos do tipo CH2XY, mas
particularmente aqueles em que X e/ou Y são grupos CO2R,
CH2(CO2Et)2, na presença de bases orgânicas como
catalisadores. Na maioria das reações, o aldol intermediário é
desidratado ao produto α,β-insaturado, o éster. Os carbânions,
derivados de ésteres do ácido 1,4-butanodióico (ácido
succínico, CH2CO2Et)2), e aldeídos ou cetonas, com íons
alcóxido como bases catalisadoras é conhecida como
condensação de Stobbe. Um mecanismo reacional sugerido,
envolve um intermediário cíclico, uma lactona, que pode ser
isolada.

222
Condensação de Claisen de ésteres
Esta reação envolve carbânions derivados de ésteres,
que são adicionados a átomos de carbono carbonílico de outra
molécula de éster. O início, da reação é semelhante, à
condensação aldólica.
No entanto, uma diferença significativa em relação à
reação aldólica simples é que o aduto inicial, que possui agora
um bom grupo de saída (OEt), perde –OEt para dar um β-ceto-
éster, 3-cetobutanona de etila (acetoacetato de etila). Este é
finalmente convertido por ação de uma base (-OEt) no seu
carbânion estabilazado (deslocalização).
As reações de Claisen cruzadas com dois ésteres
diferentes, cada um deles com átomos de H em posição α,

223
formam quatro produtos possíveis. Mas as reações de Claisen
cruzadas quando um dos dois ésteres não tem átomos α-H,
HCO2Et, ArCO2Et, (CO2Et)2, etc., reagem como aceitador de
carbânions. Estas espécies são, boas aceitadoras de
carbânions e não ocorre a reação de autocondensação. O
OC2H5
benzoato deetila (aceptor)
H3C C
O
OC2H5
acetato de etila(doador)
NaF/THF
H3O+
O
OC2H5
O
β-ceto-éster
As reações de Claisen intramoleculares, em que ambos
os grupos CO2Et são parte da mesma molécula, são chamadas
ciclizações de Dieckmann. Ocorre em condições simples, com
formação de ânions de β-ceto-ésteres cíclicos com 5, 6 ou 7
átomos com EtO2C(CH2)nCO2Et em que n = 4 – 6.
Reação de Wittig
Esta é uma reação útil para transformar C = O em C = C.
Esta reação envolve a adição de um ilídio de fosfônio,
(Ph3P=CRR’), que no seu estado fundamental, tem cargas de
sinais opostos em átomos adjacentes, a um aldeído ou a uma
cetona. o ilídio é um carbânion envolvendo também um
heteroátomo. Estas espécies são formadas pela reação de um
haleto de alquila, RR′CHX, com trifenil-fosfina Ph3P, para
formar um sal de fosfônio, que perde um próton ao reagir com
uma base muito forte, PhLi.

224
A adição do reagente de Wittig a C=O, segue o
mecanismo seguinte:
A etapa (1) pode, ou não, ser um equilíbrio, a etapa (1)
ou as etapas (2) e (3) podem ser determinantes da velocidade,
e a etapa (2) pode de fato ser concertada, em alguns casos
isolados, foi possível isolar o zwitterion intermediário.
Dependendo das condições reacionais, pode gerar olefinas cis
ou trans.
Estereosseletividade em reações de adição a carbonilos
As reações de adição nucleofílica de HY a C=O, ocorre
com formação de produtos idênticos, devido à possibilidade de
rotação livre em torno das ligações C─O.
A adição de HY a RR′C=O introduz um centro quiral no
aduto, mas o produto será sempre a forma (±), o racemato,
porque o ataque nucleofílico incial por cima do plano de (a) ou
por baixo (b), do composto carbonílico planar é igual.

225
Quando R ou R′ é quiral, as duas faces do composto
carbonílico não são equivalentes, e a adição por cima e por
baixo não é igual. Quando a reação é reversível, haverá maior
proporção do produto termodinamicamente mais. Para reações
essencialmente irreversíveis, com RMgX, LiAlH4, etc., o
produto que se forma mais rapidamente é provavelmente
aquele que predomina (controle cinético), das duas
possibilidades é possível prever, a partir de regra de Cram, que
uma cetona reagirá na conformação em que o O do grupo C=O
é anti em relação ao mais volumoso dos três substituintes do
átomo de carbono α. O ataque nucleofílico preferencial de
R′MgBr será pelo lado menos impedido do átomo de carbono
carbonílico em (a). Isto pode observar-se com mais facilidade,
recorrendo a fórmulas de projeção de Newman.
A reação ocorre preferencialmente via o estado de
transição menos impedido (de menor energia), e a razão x/y
aumenta à medida que aumenta a diferença de tamanho entre
M e S e à medida que aumenta o tamanho de R′ em R′MgBr.
As letras S, M e L são as iniciais das palavras inglesas: short,

226
medium e large, indicando os grupos: pequeno, médio e
grande.
Reações catalisadas por ácido
É difícil efetuar o ataque ao carbono carbonílico de
RCO2H, com nucleófilos do tipo geral Y-, porque estes
nucleófilos abstraem um próton, e o RCO2 resultante não é
suscetível ao ataque nucleófilo. Os nucleófilos mais fracos da
forma YH e ROH não apresentam esta limitação, mas as suas
reações com o átomo de carbono carbonílico, RCO2H, são
lentas. O caráter eltrofílico da carbonila pode ser aumentado
por protonação, via catálise ácida, por exemplo, na reação de
esterificação.
A protonação do átomo de oxigênio do ácido na reação
direta (esterificação), e do éster na reação inversa (hidrólise). A
catálise ácida também tem efeito sobre a perda do grupo de
saída, mais fácil perder H2O ou EtOH do que perder –OH ou –
OEt na reação de esterificação. O equilíbrio pode normalmente
ser deslocado no sentido pretendido, utilizando-se para tal um
excesso de ROH (ou de H2O para a hidrólise). Este mecanismo
é conhecido como AAC2 (catalise ácida, com quebra acil-
oxigênio, bimolecular). Nestas condições, a reação de R′OH
com RCO2R′′ resulta em transesterificação, sendo a posição do
equilíbrio determinada pelas proporções relativas de R′OH e

227
R′′OH. Os anidridos de ácidos e as amidas sofrem hidrólise
catalisada por ácido de modo muito semelhante aos ésteres.
ADIÇÃO A C≡N
A ligação C≡N apresenta uma semelhança com C=O e
pode reagir de modo semelhante a um certo número de
reações de adição nucleofílica análogas.
Os reagentes de Grignard regem com as nitrilas
formando sais de cetiminas que podem ser hidrolisadas em
meio ácido para formar cetona.
A adição de H2O (hidrólise) pode ser catalisada por
ácidos e por bases.
O produto inicial é uma amida, mas esta sofre hidrólise
fácil catalisada por ácidos ou por bases, e o produto da reação
é, muitas vezes, o ácido carboxílico, RCO2H, ou o seu ânion.
7o ROTEIRO DE ESTUDO DIRIGIDO
1. Compare o momento de dipolo do formaldeído e da acetona. 2. Justifique os valores de k (constante de velocidade) na formação de cianidrinas. Escreva as reações químicas:

228
Composto K (cte de velocidade) CH3CHO Muito grande p-NO2C6H4CHO 1420 C6H5CHO 210 p-MeOC6H4CHO 32 CH3COCH2CH3 38 C6H5COCH3 08 C6H5COC6H5 Muito pequeno
3. Qual a influência do pH na hidratação da acetona? Escreva as reações químicas em pH 4,7 e 11. 4. Escreva as interações intramoleculares que estabilizam o cloral hidratado e a ninidrina (revelador específico de aminoácidos).
5. Mostre a reação de formação de: a) acetal b) hemi-acetal c) tio acetal d) hemi-tio acetais 6. Justifique a resistência de hidrólise dos acetais em meio alcalino. Como os tioacetais são decompostos? 7. Fazer a hidrólise de um éster: a) Catalisada por um ácido. b) Catalisada por um base. c) Qual das duas reações é irreversível? d) Fazer a hidrólise ácida de um éster de um álcool terciário. e) Fazer a hidrólise da etanonitrila até ácido carboxílico. 8. O ácido mandélico {C6H5CH(OH)CO2H} pode ser isolado do óleo de amêndoas amargas, e é usado na medicina no tratamento de infeções urinárias. Sugira uma síntese em duas etapas, a partir do benzaldeído. 9. Fazer a adição do reagente de Grignard, seguindo o modelo de cram: a) C6H5(Me)CHCHO + MeMgI � X + Y X/Y = 2:1 b) C6H5(Et)CHCHO + MeMgI � X + Y X/Y = 2,5:1 c) C6H5(Me)CHCHO + C6H5MgI � X + Y X/Y > 4:1 10. Um composto A reage com brometo de metilmagnésio para fornecer, após hidrólise, o composto B. A oxidação de B com ácido crômico fornece C (C5H10O), o qual forma um produto cristalino pela reação com 2,4-dinitrofenilidrazina, e dá teste de iodofórmito positivo. Dê as estruturas possíveis de A até C, e as equações das reações mecionadas.
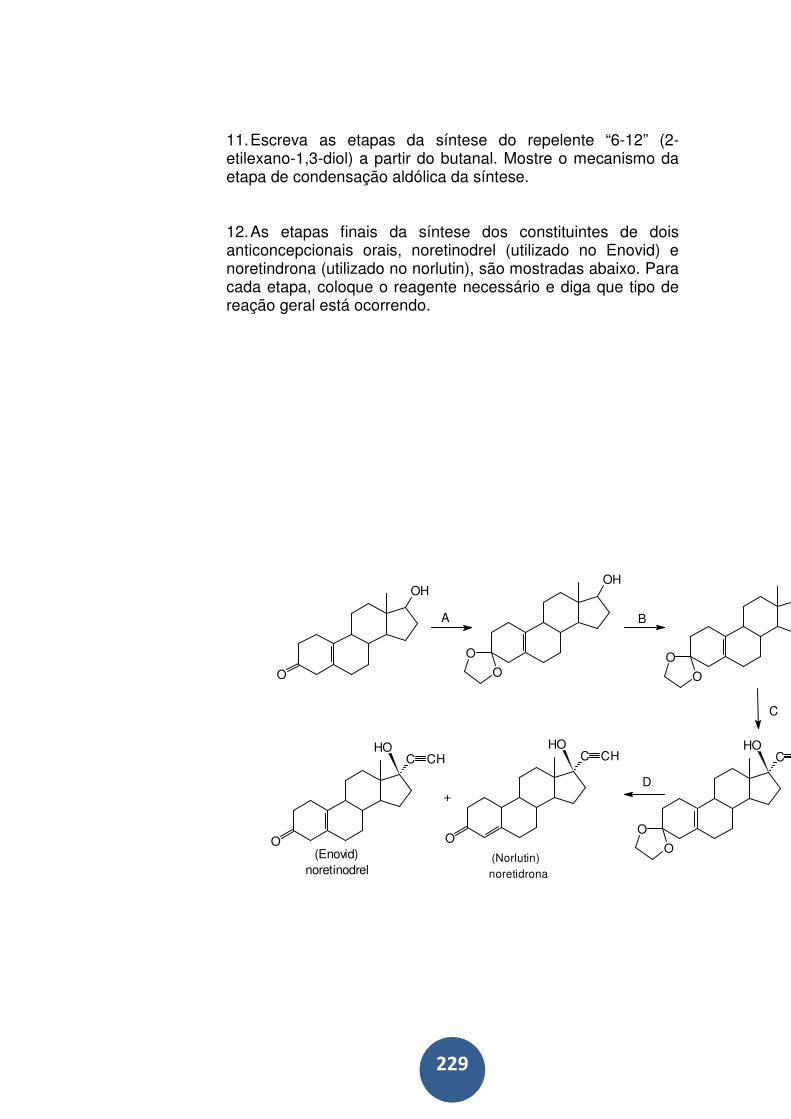
229
11. Escreva as etapas da síntese do repelente “6-12” (2-etilexano-1,3-diol) a partir do butanal. Mostre o mecanismo da etapa de condensação aldólica da síntese.
12. As etapas finais da síntese dos constituintes de dois anticoncepcionais orais, noretinodrel (utilizado no Enovid) e noretindrona (utilizado no norlutin), são mostradas abaixo. Para cada etapa, coloque o reagente necessário e diga que tipo de reação geral está ocorrendo.
noretidrona(Norlutin)
noretinodrel(Enovid)
+
HOC CH
O
HOC CH
O
D
O
O
HOC
C
O
O
BA
O
O
OHOH
O

230

231
UNIDADE VIII - REAÇÃO DE ELIMINAÇÃO
Ocoore também eliminações de átomos ou grupos de
átomos ligados a átomos do carbono diferentes, e as
eliminações no mesmo átomo (eliminações 1,1 ou eliminações-
α), eliminações em átomos mais afastados do que 1,2, são as
eliminações 1,5 e 1,6 conduzindo a ciclizações.
REAÇÕES DE ELIMINAÇÃO 1,2, ELIMINAÇÃO-β.
Nas reações de eliminação 1,2 envolvendo átomos de
carbono, o átomo que perde Y é o carbono-1 (α), e o que perde
H é o carbono-2 (β).
A reação de eliminação, induzida por base, de ácido
hologenídrico de haletos de alquila, para a obtenção de
alcenos, a mais comum é a reação de eliminação de brometos,
Outras eliminações β
Desidratação de alcoóis catalisada por ácido,
Degradação de Hofmann de hidróxidos de alquilamônio
quartenários.

232
Contudo, conhecem-se muitas outras espécies Y: ou Y-
como grupos de saída, como: SR2, SO2R, OSO2Ar, etc.
Há três mecanismos diferentes para as eliminações-1,2,
diferindo entre si pelo instante em que ocorre a quebra das
ligações H–C e C–Y. Esta quebra pode ser (a) simultânea, um
mecanismo numa só etapa, através de um único ET, e que é
designado mecanismo E2 (Eliminação bimolecular), semelhante
ao mecanismo da SN2.
As ligações H–C e C–Y podem sofrer quebra
separadamente num processo, em duas etapas. Se a ligação
C–Y for a primeira a romper-se, forma um íon carbônio como
intermediário,
Este mecanismo é E1 (Eliminação unimolecular),
semelhante ao mecanismo SN1 e os íons carbônio
intermediários para SN1 e E1 são idênticos.
Quando a ligação H–C quebra primeiro, há um processo
em duas etapas envolvendo um carbânion intermediário.
Este mecanismo é E1cB (eliminação da base
conjugada). Dentre os três tipos de mecanismos descritos,
E1cB é o mais raro, e E2 o mais comum. E os três mecanismos
são discutidos separadamente a seguir.
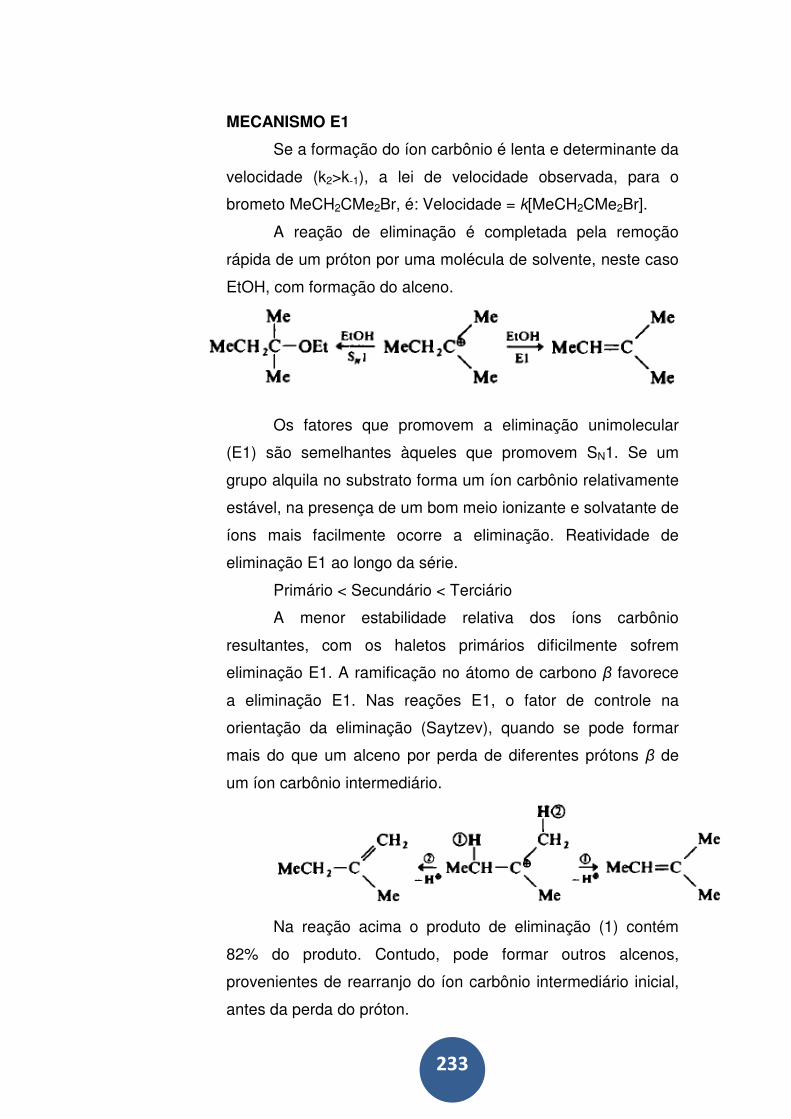
233
MECANISMO E1
Se a formação do íon carbônio é lenta e determinante da
velocidade (k2>k-1), a lei de velocidade observada, para o
brometo MeCH2CMe2Br, é: Velocidade = k[MeCH2CMe2Br].
A reação de eliminação é completada pela remoção
rápida de um próton por uma molécula de solvente, neste caso
EtOH, com formação do alceno.
Os fatores que promovem a eliminação unimolecular
(E1) são semelhantes àqueles que promovem SN1. Se um
grupo alquila no substrato forma um íon carbônio relativamente
estável, na presença de um bom meio ionizante e solvatante de
íons mais facilmente ocorre a eliminação. Reatividade de
eliminação E1 ao longo da série.
Primário < Secundário < Terciário
A menor estabilidade relativa dos íons carbônio
resultantes, com os haletos primários dificilmente sofrem
eliminação E1. A ramificação no átomo de carbono β favorece
a eliminação E1. Nas reações E1, o fator de controle na
orientação da eliminação (Saytzev), quando se pode formar
mais do que um alceno por perda de diferentes prótons β de
um íon carbônio intermediário.
Na reação acima o produto de eliminação (1) contém
82% do produto. Contudo, pode formar outros alcenos,
provenientes de rearranjo do íon carbônio intermediário inicial,
antes da perda do próton.

234
Mecanismo E1cB
No mecanismo E1cB, ocorre a formação do íon
carbânion intermediário, rápida e reversível, e se a perda
subsequente do grupo de saída Y- for lenta e determinante da
velocidade, k-1 > k2, esta reação obedecerá então à lei de
velocidade, e fica cineticamente indistinguível do mecanismo
concertado, E2, com lei de velocidade igual a velocidade =
k[RY][B]. Nesse caso, ocorre troca isotópica entre o substrato
inicial e o solvente, e isso acontece durante a formação rápida
e reversível do carbânion sendo possível distingui-los, fato que
não pode ocorrer no mecanismo E2, concertado, de etapa
única.
No PhCH2CH2Br, o grupo Ph no átomo de carbono β
promove a acidez nos átomos β-H, porque estabiliza o
carbânion por deslocalização.
Quando a reação é feita com –OEt em EtOD, o produto
da reação, o alceno, não contém deutério, se o produto fosse
formado por eliminação a partir do reagente inicial deuterado.
Este caso potencialmente favorável não ocorre por um
mecanismo E1cB da forma descrita anteriormente. Aqui a
velocidade de k2>>k-1, a formação de carbânion irreversível,
onde a velocidade da reação depende da concentração do íon
carbânion formado.

235
As reações que ocorrem por este mecanismo
carbâniônico são raras e são favorecidas quando no reagente
tem grupos substituintes reatiradores de elétrons como na
reação do X2CHCF3 em meio básico.
O composto, X2CHCF3, com: (a) átomos de halogênio
eletronegativos no átomo de carbono β para tornar o β-H mais
ácido, (b) estabilização do carbânion através da atração de
elétrons pelos átomos de halogênio no átomo de carbono do
carbânion, e (c) um grupo de saída ruim, F, favorece o
mecanismo E1cB. Outra característica estrutural que favorece
E1cB é (d) um substituinte positivamente carregado no átomo
de carbono α, contribuindo também para a acidez dos átomos
β-H
Mecanismo E2
O mecanismo de eliminação E2 é um mecanismo
concertado, numa só etapa então não sofre rearranjo.
A reação de eliminação de HX, induzida por base, do
haleto RCH2CH2X, tem a lei de velocidade igual a Velocidade =
k[RCH2CH2X][B].
Como B é muitas vezes tanto um nucleófilo como uma
base, a eliminação é acompanhada frequentemente pela
substituição nucleófila concertada numa só etapa (SN2).
A ruptura da ligação C–H na etapa determinante da
velocidade, como requer um mecanismo concertado, é dada
pela observação de um efeito isotópico cinético primário
quando H é substituído por D no carbono β.

236
Um dos fatores que afeta a velocidade das reações E2 é
a força da base usada. Assim, verifica-se que: -NH2 > -OR > -
OH.
O solvente exerce influência sobre o grupo saída Y: ou
Y-, via ligação de hidrogênio ou de solvatação no ET. Para um
substrato com uma dada estrutura, variando Y, quanto melhor
for o grupo de saída, mais rápida é a reação. Os melhores
grupos de saída são os ânions (bases conjugadas) dos ácidos
fortes HY, e a sua capacidade como grupos de saída pode ser
relacionada em parte com a força relativa de HY, por exemplo,
o p-MeC6H4SO3- (tosilato) é um grupo de saída muito melhor
do que –OH.
As características estruturais do substrato que
favorecem a eliminação E2 são aquelas que servem para
estabilizar o alceno formado (R2C=CR2 > R2C=CHR >
R2C=CH2 > RCH=CHR > RCH=CH2 > H2C=CH2) ou o ET que
o precede. Tais características incluem aumento da
substituição com grupos alquilo em ambos os átomos de
carbono α e β (formando alcenos de estabilidade
termodinâmica crescente) ou a introdução de um grupo fenilo
que possa entrar em conjugação com a ligação dupla em
desenvolvimento.
Estereosseletividade em E2
A eliminação em moléculas acíclicas pode ocorrer numa
das duas conformações limite, a anti ou a syn.
Há uma vantagem na eliminação com conformação em
que o H, Cβ, Cα e Y estão no mesmo plano das orbitais p em Cβ
e Cα, quando H+ e Y- saem, se tornam paralelas entre si, com
uma sobreposição máxima na formação da ligação π. Será

237
energeticamente favorável para uma base B que ataca nesse
plano comum. Estabelecendo a preferência da eliminação a
partir de uma conformação planar, põe-se a questão de qual
delas, syn ou anti, é mais fovorável em relação à outra.
Deste modo, dois fatores justificam a preferência pela
eliminação antiperiplanar:
(a) o par eletrônico formado em Cβ por perda de H+
próximo do Cα pelo lado oposto do grupo de saída do par
eletrônico com Y-;
(b) a eliminação ocorre na conformação em estrela de
menor energia (anti), em lugar de ocorrer na conformação
eclipsada de maior energia (syn).
Assim a eliminação E2, tem uma conformação
preferencial de eliminação Anti (lado oposto).
Quando os Cβ e Cα são quirais, a eliminação a partir das
suas conformações forma produtos diferentes, o alceno cis ou
trans. Assim, conhecendo a configuração do diastereômero
original, e estabelecendo a configuração do(s) isômero(s)
geométrico(s) que se forma(m), podemos estabelecer o grau
de estereosseletividade do processo de eliminação.
Nos casos acíclicos mais simples, verifica-se que a
eliminação Anti é preferencial.
Em compostos cíclicos a conformação da eliminação
pode ser determinada pela rigidez relativa da estrutura cíclica.
Um experimento compara as velocidades da eliminação
dos isómeros cis e trans do brometo de 4-terc-butilciclohexil.

238
Embora o produto da reação seja o alceno
estereoisômero 4-terc-butyiciclohexeno, as velocidades de
reação são muito diferentes. O isômero cis reage mais de 500
vezes mais rápido do que o trans. A diferença nas velocidades
de reação é devido ao grau de desenvolvimento da ligação π
no estado de transição E2. A sobreposição de orbitais π exige
que sejam paralelos, a formação da ligação pi é formada mais
facilmente quando os quatro átomos H–C–C–X estão no
mesmo plano, no estado de transição. As duas conformações
que permitam esta relação são denominadas syn periplanar e
anti periplanar.
Assim, para uma série de eliminações em anéis de
diferentes dimensões, foram observados os seguintes graus de
estereosseletividade.
Tamanho
do ciclo
% da
eliminação syn

239
Cilcobutil 90
Ciclopentil 46
Ciclohexil 3
Cilcoheptil 37
Orientação em E2: Saytzev versus Hofmann
Em substratos que possuem átomos de hidrogênio β
alternativos disponíveis, é possível obter mais do que um
alceno por eliminação, onde existem duas possibilidades.
CH3CH2CHYCH3 + KOH � CH3CH=CHCH3 +
CH3CH2=CH2
Saytzev Y = Br 81% 19%
Hofmann Y = +SMe2 26% 74%
Hofmann Y = +NMe3 5% 95%
Para ajudar a prever qual o alceno mais provável,
existem duas regras empíricas que podem ser resumidas do
modo seguinte: (a) Hofmann (Y=+NMe3) afirmou que o alceno
predominante será o que tem o menor número de substituintes
alquil nos carbonos da ligação dupla; (b) Satzev (Y=Br) afirmou
que o alceno que predomina é aquele com maior número de
substituintes alquil nos carbonos da ligação dupla. Ambas a
generalizações são válidas, ficando claro que a composição da
mistura de alcenos obtida por eliminação é influenciada por Y.
Se a ligação C–Y sofrer ruptura com relativa facilidade,
então, a medida que a base atacante começar a quebrar a
ligação H–Cβ, a ligação C–Y também começará a romper. A
ligação dupla começa, assim, a formar-se cedo no processo
global, com o ET, com acentuado caráter de ligação dupla e
será, pois, estabilizado por quaisquer características que
estabilizem o alceno resultante. Uma destas características é a
substituição por grupos alquil nos carbonos da ligação dupla:

240
quanto mais substituído for um alceno, normalmente mais
estável ele é. O que isto significa é que a base atacante
tenderá a remover preferencialmente o β-H que conduzirá ao
ET mais estável (semelhante a um alceno) e este forma o
alceno resultante mais estável, trata-se de uma eliminação de
Saytzev. É isto que sucede em quando Y = Br. A preferência é
pela eliminação do tipo Saytzev no mecanismo E1.
Se a ligação C–Y for mais difícil de quebrar, então a
remoção de um β-H pela base atacante ocorrerá muito antes
da ligação C–Y começar a sofrer ruptura, tendência esta que
pode ser reforçada pela presença de uma carga positiva em Y.
O ET terá agora um caráter menos semelhante ao alceno, e
mais semelhante a carbânion; será por isso estabilizado por
quaisquer fatores que estabilizem carbânions, com estabilidade
relativa de carbânions na sequência seguinte: primário >
secundário > terciário. A base atacante tenderá a remover
preferencialmente o β-H que conduzir ao ET mais estável
semelhante a carbânion, que, por sua vez, resultará na
formação do alceno menos substituído. Trata-se de uma
eliminação de Hofmann.
Eliminação versus substituição
As reações de eliminação E1 são normalmente
acompanhadas por reações de substituição SN1, ambas têm
um intermediário comum, um íon carbônio, ainda que este seja
convertido em produtos de eliminação ou de substituição via
ET diferentes, numa etapa rápida, não determinante da
velocidade.
A eliminação E2 é acompanhada muitas vezes por
substituição SN2, embora, neste caso, os processos
concertados paralelos envolvam mecanismos inteiramente
independentes. Assim, considerando a eliminação via
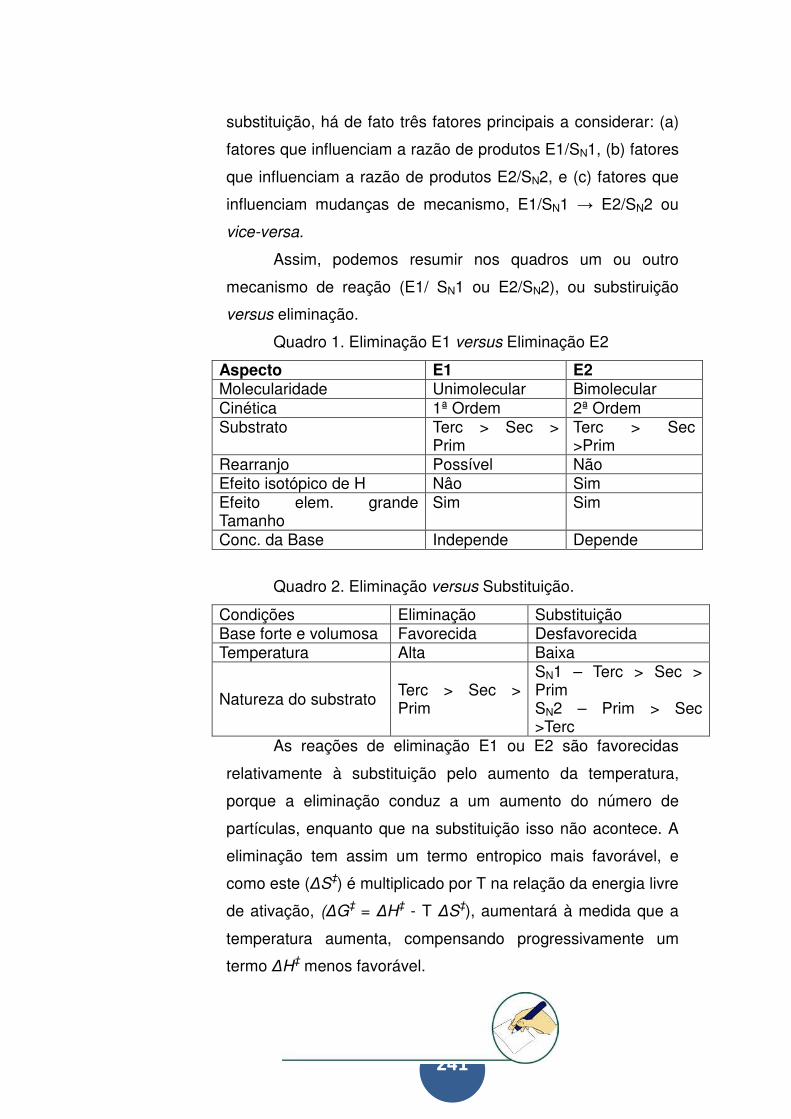
241
substituição, há de fato três fatores principais a considerar: (a)
fatores que influenciam a razão de produtos E1/SN1, (b) fatores
que influenciam a razão de produtos E2/SN2, e (c) fatores que
influenciam mudanças de mecanismo, E1/SN1 → E2/SN2 ou
vice-versa.
Assim, podemos resumir nos quadros um ou outro
mecanismo de reação (E1/ SN1 ou E2/SN2), ou substiruição
versus eliminação.
Quadro 1. Eliminação E1 versus Eliminação E2
Aspecto E1 E2 Molecularidade Unimolecular Bimolecular Cinética 1ª Ordem 2ª Ordem Substrato Terc > Sec >
Prim Terc > Sec >Prim
Rearranjo Possível Não Efeito isotópico de H Nâo Sim Efeito elem. grande Tamanho
Sim Sim
Conc. da Base Independe Depende
Quadro 2. Eliminação versus Substituição.
Condições Eliminação Substituição Base forte e volumosa Favorecida Desfavorecida Temperatura Alta Baixa
Natureza do substrato Terc > Sec > Prim
SN1 – Terc > Sec > Prim SN2 – Prim > Sec >Terc
As reações de eliminação E1 ou E2 são favorecidas
relativamente à substituição pelo aumento da temperatura,
porque a eliminação conduz a um aumento do número de
partículas, enquanto que na substituição isso não acontece. A
eliminação tem assim um termo entropico mais favorável, e
como este (∆S‡) é multiplicado por T na relação da energia livre
de ativação, (∆G‡ = ∆H‡ - T ∆S‡), aumentará à medida que a
temperatura aumenta, compensando progressivamente um
termo ∆H‡ menos favorável.
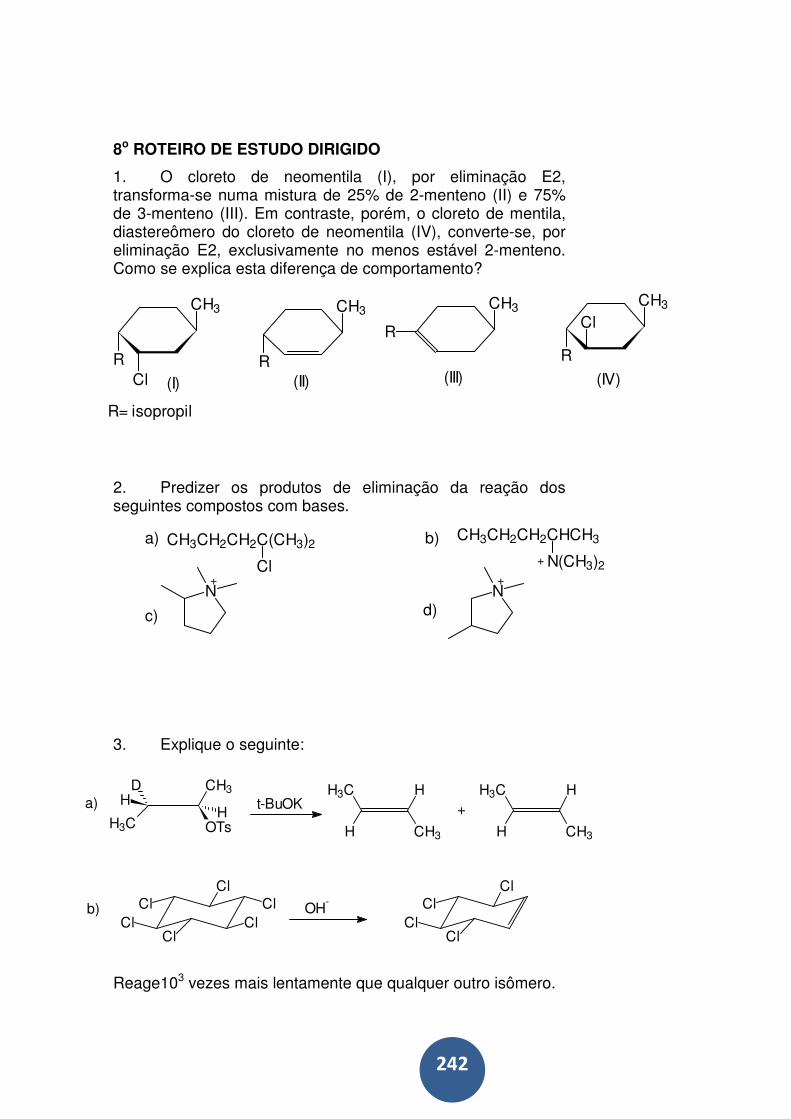
242
8o ROTEIRO DE ESTUDO DIRIGIDO
1. O cloreto de neomentila (I), por eliminação E2, transforma-se numa mistura de 25% de 2-menteno (II) e 75% de 3-menteno (III). Em contraste, porém, o cloreto de mentila, diastereômero do cloreto de neomentila (IV), converte-se, por eliminação E2, exclusivamente no menos estável 2-menteno. Como se explica esta diferença de comportamento?
2. Predizer os produtos de eliminação da reação dos seguintes compostos com bases. 3. Explique o seguinte: Reage103 vezes mais lentamente que qualquer outro isômero.
(I)
R= isopropil
CH3
RCl
CH3
R
CH3
R
CH3
R
Cl
(IV)(III)(II)
CH3CH2CH2C(CH3)2
Cl
N+
N+
a) b)
c) d)
CH3CH2CH2CHCH3
N(CH3)2+
CH3
H3CH
DH
OTs
t-BuOK +a)
b)Cl
ClCl
Cl
ClCl
OH-Cl
Cl
ClCl
H
H3C
CH3
H
H
H3C
CH3
H

243
4. Explique os resultados obtidos nas duas reações de eliminação mostradas abaixo. Baseie sua resposta em seus conhecimentos de análise conformacional relativos ao ciclo-hexano. 5. Nas reações de eliminação, mostre, apresentando exemplos, quando há predomínio da olefina de Hoffmanm e quando há predomínio da olefina de Saytzev. 6. As reações de eliminação 1,1 (alfa) são raras. Mostre este tipo de eliminação na preparação do ciclopropano, halogenado.
7. Descreva as reações de Cope e de Chugaev e dê exemplos. Qual a vantagem destas reações pirolíticas? 8. Se as reações de eliminação e substituição são competitivas, indique as condições que favorecem cada uma destas reações.
9. Explique a regra de Bredt.
Br Br
t-BuOK t-BuOK
fácil difícil

244

245
UNIDADE IX - CARBÂNIONS
A acidez relativa de carbonos ácidos tais como em R3C
– H, a velocidade à qual o próton é transferido para a base
pode ser lenta de modo a constituir um fator limitante, e a
acidez de R3C – H é assim controlada cinteticamente e não
termodinamicamente.
Há outros métodos de gerar carbânions, além da
remoção do próton. A formação do carbânion é importante,
dada a sua participação numa grande variedade de reações
em síntese, muita das quais de grande interesse por resultarem
na formação de ligações carbono-carbono.
Formação de carbânions
O método mais comum para a formação de carbânions
é por remoção de um átomo ou grupo X de um carbono,
deixando X o seu par de eletrôns de ligação.
O grupo de saída mais comum é X = H, onde um próton
é removido, R3C – X � R3C–, embora se conheçam também
outros grupos de saída, como o Cl no cloreto de trifenilmetilo.
A tendência de alcanos perderem um próton e formar
carbânions, como o CH4 foi calculado um valor de pKa de ≅ 43,
em comparação com 4,76 para MeCO2H. Os métodos normais
para determinação do pKa não funcionam, numa escala de
acidez tão baixa, e estas determinações foram feitas a partir de
medidas no equilíbrio.

246
Parte-se do princípio de que quando mais forte for um
ácido, RH, maior será a sua proporção na forma RM
relativamente a RI. A determinação da constante de equilíbrio
K permite uma determinação da acidez relativa de RH e R’H e,
por escolha conveniente de pares, pode subir-se na escala de
pKa até ser possível uma comparação direta com um composto
RH cujo pKa tenha sido determinado por outros meios.
Assim , encontra-se para Ph3C-H um valor de pKa de um
ácido muito mais forte do que CH4, e o carbanion Ph3C-Na+
pode ser obtido por ação do amideto de sódio, -NH2Na+, em
amoníaco.
O carbânion, Ph3C-, pode ser obtido por ação de sódio
metálico sobre Ph3C-H em solvente inerte, e a solução de
trifenilmetilo de sódio resultante é usada como uma base
orgânica muito forte com capacidade de capturar próton do
carbanion. Os alcenos são ácidos ligeiramente mais fortes do
que os alcanos –CH2=CH2 tem um valor de pka 37, mas os
alcinos são mais ácidos, tendo HC≡CH um valor de pKa de 25.
O carbânion HC≡C- (ou RC≡C-) pode ser gerado a partir do
hidrocarboneto, por ação de –NH2 em amoníaco; estes anions
acetilênicos são de importância sintética.
A introdução de grupos substituintes retiradores de
elétrons aumenta a acidez dos átomos de hidrogênio no
carbono. Assim, a formação de um carbânion instável, -CCl3,
por ação de bases fortes em clorofórmio, encontrando-se para
HCF3 e HC(CF3)3 os valores de pKa de ≅ 28 e 11,
respectivamente. O efeito com substituintes que podem
deslocalizar uma carga negativa e exercem também um efeito
indutivo negativo é ainda mais acentuado, e os valores de pKa
de CH3CN, CH3COCH3 e CH3NO2 são, respectivamente, 25, 20

247
e 10,2. Com CH3NO2, o carbânion correspondente –CH2NO2
pode ser obtido por ação de –OEt em EtOH, ou mesmo de –OH
em H2O, em pequenas concentrações do carbânion em
solução aquosa, nos compostos carbonílicos menos ácidos,
permitindo que ocorra a reação aldólica.
Estabilidade de carbânions
As características estruturais em R–H que favorecem a
remoção de H por bases, e fatores que servem para estabilizar
o carbânion resultante, R-, ambos os efeitos resultantes de um
mesmo fator. Os fatores principais que servem para estabilizar
carbânions são: (a) aumento do caráter s no carbono do
carbânion, (b) efeitos indutivos negativos, (c) conjugação do
par de eletrôns isolado do carbanion com uma ligação múltipla,
e (d) através de aromização.
O efeito de (a) observa-se no aumento da acidez dos
átomos de hidrogênio na sequência: CH3-CH3 < CH2=CH2 <
HC≡CH, refletindo no aumento de caráter s do orbital híbrido
envolvido na ligação σ do H, sp3 < sp2 < sp1. Os orbitais s estão
mais próximos do núcleo, isto facilita a saída do átomo H sem o
seu par de elétrons, tornando–o ácido, estabilizando o
carbânion formado.
O efeito de (b) é observado em HCF3 (pKa = 28) e
HC(CF3)3 (pKa = 11), onde a alteração em relação a CH4 (pKa ≅
43) resulta do efeito indutivo negativo do átomo de flúor, que
torna o átomo H mais ácido, e estabiliza os carbanios
formados, -CF3 e –C(CF3)3, por atração de elétrons. O efeito é
mais acentuado em HC(CF3)3, onde estão envolvidos nove
átomos de flúor, em comparação com apenas três em HCF3,
apesar de não estarem, neste caso, atuando diretamente no
átomo de carbono do carbânion. O mesmo efeito ocorre na
formação de –CCl3 a partir de HCCl3, onde ocorre efeito
indutivo negativo semelhante. Este efeito é provavelmente

248
menos eficaz com Cl do que com o F mais eletronegativo, mas
a deficiência pode ser superada em parte pela deslocalização
do par eletrônico do carbânion para as orbitais d livres do cloro,
elemento do segundo período, o que evidentemente não é
possível para o flúor, elemento do primeiro período.
A influência desestabilizadora do efeito indutivo negativo
de grupos alquila, observa-se na seguinte sequência de
estabilidade de carbânions:
CH3- > RCH2
- > R2CH- > R3C-
Esta sequência é exatamente inversa à da estabilidades
observada para os íons carbônions.
O efeito de (c) é uma característica estabilizadora mais
comum, com CH2–CN, (CH3)2–C=O, CH2–NO2, CH3–CO2Et,
etc..
O efeito indutivo negativo, em cada caso, aumenta a
acidez dos átomos de H no átomo de carbono incipiente, mas a
estabilização do carbânion formado por deslocalização tem
maior significado. Em geral, o NO2 é o mais forte. O efeito
acentuado da introdução de mais um desses grupos no átomo
de carbono observa-se pelos valores de pKa; assim, CH(CN)3 e
CH(NO2)3 são ácidos tão fortes em água como HCl, HNO3, etc.
O grupo carboxilato no CH3–CO2Et é menos eficaz na
estabilização do carbânion do que o grupo C=O em aldeídos e
cetonas simples, como se pode observar pela sequência de

249
valores de pKa: CH2(CO2Et)2 = 13,3; MeCOCH2CO2Et = 10,7 e
CH2(COMe)2 = 8,8, resultante da capacidade de conjugação
doadora de elétron dos elétrons do par isolado no átomo de
oxigênio do grupo OEt.
Com elementos do segundo período, qualquer efeito
indutivo pode ser complementada pela deslocalização, através
do uso das orbitais d livres para acomodar o par elétron do
átomo de carbono do carbânion, isto acontece com o enxofre
num substituinte ArSO2 e também com o fósforo em, R3P+.
O efeito de (d) é observado no ciclopentadieno, que
apresenta um valor de pKa de 16, enquanto que para o alceno
simples esse valor é de pKa ≅ 37. Isto deve-se ao fato de o
carbânion formado, o ânion ciclopentadienilo, ser um sistema
eletrônico 6p deslocalizado, um sistema de Huckel 4n + 2 onde
n = 1. Os 6 elétrons preenchem três orbitais moleculares π
estabilizados, como o benzeno, e o ânion apresenta assim
estabilização quase-aromática; e estabilizado por
aromatização.
A sua aromaticidade não pode ser testada por
substituição eletrofílica, porque o ataque por X+ conduziria
simplesmente à combinação direta com o ânion. Porém, o
caráter verdadeiro (uma reação de Friedel Grafts) demonstra-
se na notável série de compostos neutros extremamente
estáveis obtidos a partir de ciclopentadienilo, chamados
metalocenos, ferroceno, no qual o metal está ligado por
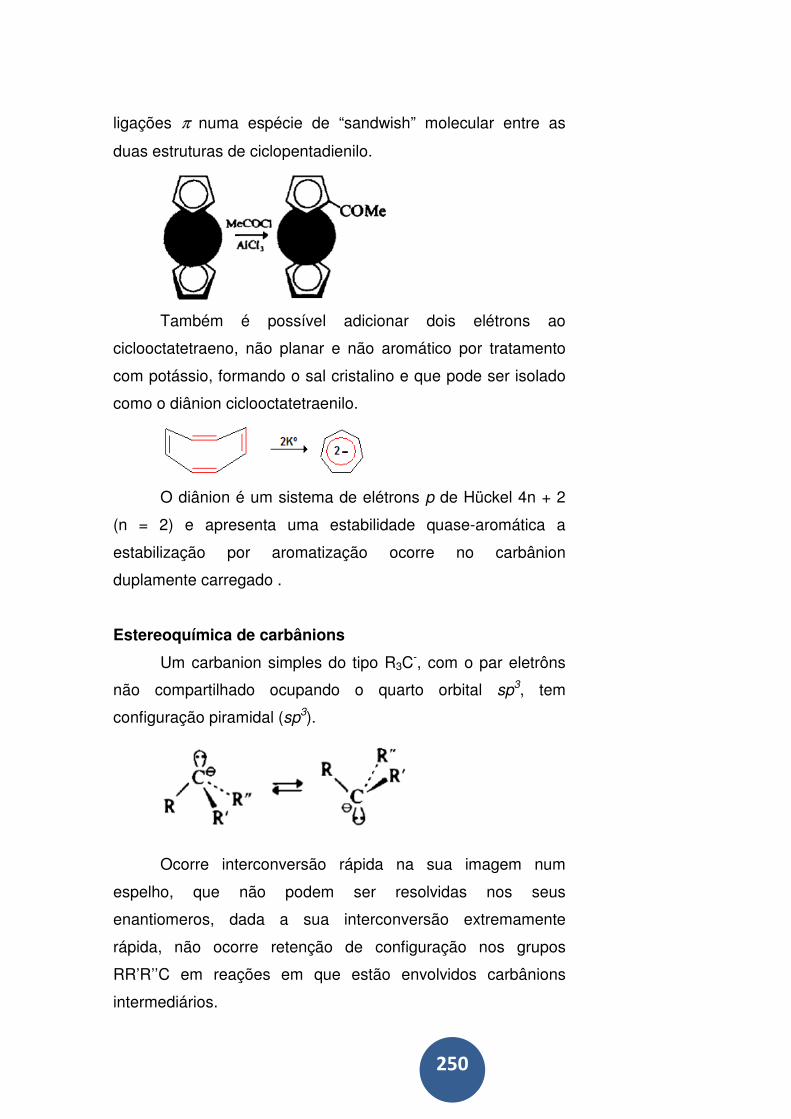
250
ligações π numa espécie de “sandwish” molecular entre as
duas estruturas de ciclopentadienilo.
Também é possível adicionar dois elétrons ao
ciclooctatetraeno, não planar e não aromático por tratamento
com potássio, formando o sal cristalino e que pode ser isolado
como o diânion ciclooctatetraenilo.
O diânion é um sistema de elétrons p de Hückel 4n + 2
(n = 2) e apresenta uma estabilidade quase-aromática a
estabilização por aromatização ocorre no carbânion
duplamente carregado .
Estereoquímica de carbânions
Um carbanion simples do tipo R3C-, com o par eletrôns
não compartilhado ocupando o quarto orbital sp3, tem
configuração piramidal (sp3).
Ocorre interconversão rápida na sua imagem num
espelho, que não podem ser resolvidas nos seus
enantiomeros, dada a sua interconversão extremamente
rápida, não ocorre retenção de configuração nos grupos
RR’R’’C em reações em que estão envolvidos carbânions
intermediários.

251
Em compostos organometálicos da forma RR’R’’C-M, as
ligações de todas as espécies, desde a ligação essencialmente
covalente até à iônica, RR’R’’C-M+, e pela ligação covalente-
polar em RR’R’’Cδ-Mδ+. Nas reações destes compostos
observa-se retenção, racemização e inversão de configuração.
O resultado para cada caso particular depende não só do
substituinte alquila, mas também do metal e particularmente do
solvente. Mesmo com os exemplos mais iônicos, parece pouco
provável que se trate de um carbânion simples, na reação do
Etil com [PhCOCHMe]-M+, as velocidades relativas, em
condições análogas, diferem na ordem de ≅ 104 para M = Li,
Na e K.
Carbânions com substituintes suscetíveis de
deslocalização conjugada do par de eletrôns são planares
(sp2), de modo a permitir a sobreposição orbital máxima da
orbital p com as do substituinte.
Quando características estruturais ou estereoquímicas
impedem este alinhamento, pode não ocorrer a estabilização.
Assim, enquanto a pentan-2,4-diona, com um valor de pKa de
8,8 e a ciclohexan-1,3-diona são ambas muito solúveis em
NaOH aquoso (embora não sejam em água) e dão cor
vermelha com solução de FeCl3, o biciclo[2,2,2]octan-2,6-diona
comporta-se de modo totalmente oposto.

252
O átomo de H rodeado pelos dois grupos C=O no
biciclo[2,2,2]octan-2,6-diona dificilmente exibe caráter mais
ácido do que o correspondente H no hidrocarboneto análogo. O
comportamento diferente provém do fato do orbital que contém
o par eletrônico no carbânion formado estar num plano
virtualmente perpendicular ao que contém os orbitais p dos
átomos de carbono carbonílicos, não ocorrendo estabilização
no carbânion, que por isso não se forma.
REAÇÕES DOS CARBÂNIONS
Os carbânions podem ocorrer na maior parte das
reações de adição, eliminação, substituição, rearranjo, dentre
outras. Muitas reações já foram abordadas, como reação de
Dieckmann, reação aldólica, eliminação E1cB, rearranjo do
ácido benzílico, reação de Michael, etc.. Será agora
considerada uma seleção de reações em que participam
carbanions; muitas delas têm utilidade sintética especial, visto
resultarem na formação de ligações carbono-carbono.
Reação de adição
Um grande grupo de reações em que os carbânions
adicionam ao grupo C=O, incluindo exemplos de adição
intramolecular de carbânion como a reação aldólica, a reação
de Dieckmann, o rearranjo do ácido benzílico, e reações de
adições ao sistema C=C–C=O, a reação de Michael.

253
Reação de adição - carbonação
Uma reação de carbânions interessante e de utilidade
sintética, envolve compostos organometálicos atuando como
fontes de carbono negativo, é a adição ao eletrófilo muito fraco
CO2, para formar o correspondente ânion carboxilato é a
carbonação.
Esta reação ocorre com ãnions alquilas, arilas ou
acetiletos de metais, incluindo os reagentes de Grignard. É
frequentemente realizada por adição de uma solução do
composto organometálico num solvente inerte em um grande
excesso de CO2, é um método útil para a preparação de ácidos
acetilênicos. A reação de Kolbe-Schmidt é outro exemplo de
carbonação do carbânion.
A retenção de configuração num carbânion no carbono
da dupla de um alceno, foi demonstrada na reação do trans-2-
bromo but-2-eno com lítio, com formação do produto, o trans-2-
acetato de lítio but-2-eno.
O rendimento do produto é cerca de 75%, enquanto que
o do seu isômero geométrico é menor que 5%.

254
Reação de eliminação
As reações de eliminação, que ocorrem pelo mecanismo
E1cB, os intermediários envolvidos, são exemplos de
carbânions, por exemplo.
Reação de eliminação - descarboxilação
A perda de CO2 de ânions carboxilatos envolve como
intermediário um carbânion que adquire um próton do solvente
ou de outra fonte.
A perda de CO2 é normalmente limitante da velocidade,
Velocidade = K[RCO2-]
e a captura do próton é rápida. A descarboxilação é favorecida
por substituintes retiradores de elétrons em R que possam
estabilizar o carbânion intermediário por deslocalização da sua
carga negativa, o que é evidenciado pela descarboxilação
muito mais fácil do ânion carboxilato nitrossubstituído do que a
do próprio Me2CHCO2-.

255
REAÇÃO DE SUBSTITUIÇÃO
Os carbãnions, ou espécies semelhantes, estão
envolvidos nas diversas reações de substituição, como
intermediários, ou como nucleófilos reagentes. As reações de
susbistiuição mais importantes são:
Reações de Reimer-Tiemann
Esta reação envolve um carbanion arílico com
deslocalização (íon fenóxido) e com –CCl3, proveniente da
reação de bases fortes sobre HCCl3, embora o –CCl3 seja uma
espécie transitória, decompondo-se em CCl2, um eletrófilo
altamente deficiente em elétrons que reage com o anel
aromático.
O produto formado é o o-hidroxi benzalldeído
(saliçaldeído), mais uma pequena quantidade do isômero p
(para).
Reações de Kolbe-Schmidt
A reação de Kolbe-Schmidt, é análoga a anterior,
envolve CO2 como eletrófilo no ataque sobre íon fenóxido de
sódio.
O produto é o-hidroxibenzoato de sódio (salicilato),
obtendo-se pequenos vestígios do isômero para, mas se a
reação for feita com fenóxido de potássio, o sal do ácido para
passa a ser o produto majoritário.

256
O ataque orto proferencial com fenóxido de sódio
poderia resultar da estabilização do ET, através da quelação
pelo Na+ no par iônico,
O cátion K+ é maior e por isso o ataque na posição para
se torna mais favorável.
Rearranjo
Os rearranjos envolvendo carbânions, nos quais o grupo
migrante se desloca para o átomo de carbono do carbânion
sem o par de elétrons, são menos comuns do que os que
envolvem íons carbônio (carbocátion), em que o grupo
migrante transporta consigo um par de elétrons. Contudo,
conhecem-se deslocamentos-1,2 de grupos arila ligados a
átomos de carbono, por ação de sódio metálico sobre o cloreto
do Ph3C–CH2Cl.
O produto é um carbânion de sódio, mas a protonação e
carbonação originam os produtos rearranjados,
respectivamente.
Contudo, com Li em lugar de Na, é possível formar o
carbânion de lítio sem rearranjo, correspondente, observado

257
pelos produtos da sua protonação e carbonação. A tendência
para o rearranjo em reações de Ph3C–CH2Cl com metais, ou
derivados metálicos, diminui na ordem seguinte, decrescente
do caráter iônico da ligação carbono-metal.
K ≈ Na > Li > Mg
Os deslocamentos-1,2 simples de carbono aquil para o
carbono com caráter carbaniônico. Os grupos podem migrar de
um átomo diferente do carbono como, N e S, para um átomo
carbaniônico, trata-se do rearranjo de Stevens.
São necessárias bases muito fortes, PhLi, para remover
um próton da espécie positivamente carregada como o
(Me)2N+–CH2Ph, a menos que esteja presente um substituinte
retirador de elétrons, como C=O, em Me(PhCH2)S+–CH2COPh.
Verifica-se que PhCH2 migra preferencialmente em relação a
Me.
Éteres Benzílicos e alílicos, sofrem o rearranjo de Wittig.
Os rearranjos de carbânions induzidos por base que
envolvem uma eliminação-1,3 para formar uma ciclopropanona
intermediária, trata-se do rearranjo de Favorskii de 1-(α)-
halocetonas (PhHCCOCH2Cl).

258
A ciclopropanona intermediária sofre adição
subsequente de –OH, seguida de abertura do anel para dar o
mais estável dos dois carbânions.
Oxidação
Os carbânions podem, em determinado condições, ser
oxidados, por exemplo, o ânion trifenilmetila é lentamente
oxidado pelo ar.
O radical resultante (Ph3C.) pode ser reduzido,
retornando a carbânion, por agitação com amálgama de sódio.
Halogenação de cetonas
A reação ocorre via carbânion como intermediários e
ocorre com os hidrogênios mais ácidos.
A reação de bromação da acetona na presença de base
aquosa obedece à lei de velocidade, que é independente da
concentração de bromo [Br2].
Velocidade = k[MeCOMe][-OH]
A terceira halogenação é mais fácil, e o produto normal
de halogenação catalisada por base com o RCH2COCX3. Em
CX3 temos um bom grupo de saída, e a adição de base ao
grupo C=O da cetona resulta na quebra da ligação C–C.

259
O CX3 é um bom grupo de saída e devido ao efeito
indutivo negativo dos três átomos de halogênio, o que ativa o
átomo de carbono carbonílico em RCH2COCX3 para o ataque
nucleofílico, também estabiliza o carbânion que sai –CX3. O
produto final é anion carboxilato e o halofórmio, e o processo
global, RCH2COCH3 � RCH2CO2- + HCX3, é conhecido como
reação do halofórmio. Tem sido utilizada como teste
diagnóstico para metilcetonas, usando I2 e base aquosa, dado
que o CHI3 resultante (‘iodofórmio’) é amarelo, de cheiro
característico e insolúvel no meio reacional.

260

261
UNIDADE X- RADICAIS
Mas, também pode ocorrer fissão homolítica, gerando
espécies que possuem um elétron não emparelhado,
chamados de radicais.
As reações que envolvem radicais ocorrem
frequentemente em fase gasosa. A combustão de qualquer
composto orgânico é quase sempre uma reação por radicais, e
a quebra oxidativa de alcanos em máquinas de combustão
interna é a reação química que se dá em maior escala.
As reações radicalares podem ocorrer em solução, em
solventes não polares ou catalisadas pela luz ou na
decomposição simultânea de substâncias conhecidas
produzindo radicais, por exemplo, os peróxidos orgânicos. Os
radicais, formados em solução, são menos seletivos ao ataque
a outras espécies, ou em posições alternativas na mesma
espécie do que os íons carbônio e carbânions.
As reações com radicais, uma vez iniciadas, procedem
com grande rapidez, devido as reações serem em cadeia e
com pouca exigência energética, como na halogenação de
alcanos
Neste caso, o radical obtido, Br., gera outro, R., na
reação com o substrato neutro, R–H. Este radical reage por
sua vez com uma molécula neutra Br2, gerando Br. mais uma
vez. O ciclo continua sem a necessidade de geração posterior
de Br. fotoquimicamente.

262
FORMAÇÃO DE RADICAIS
Há varias maneiras de gerar radicais a partir de
moléculas neutras; as mais importantes são (a) fotólise, (b)
termólise, e (c) reação redox por íons inorgânicos, metais ou
eletrólitos, que envolvem a transferência de um elétron.
Fotólise
A capacidade de uma molécula absorver radiação na
região do ultravioleta ou visível, como a acetona, na fase de
vapor, é decomposta pela luz com comprimento de onda de
320 nm. Isto acontece porque os compostos carbonilicos têm
uma banda de absorção nesta região.
A decomposição fotoquímica origina o par de radicais
inicial, decompondo-se para formar outro radical metilo e a
espécie CO estável. Outras espécies que sofrem fotólise rápida
são os hipocloritos de alquila RO–Cl e nitrilos RO–NO,
podendo ambos ser usados para obtenção de radicais alcóxi
A quebra por homólise fotolítica é muito útil em
moléculas de halogênios para formar átomos radicalares, que
podem iniciar a halogenação de alcanos ou adição de alcenos.
As duas vantagens de fotólise sobre a termólise
(pirólise) para a geração de radicais são: (a) a possibilidade de
quebrar ligações fortes que não quebram facilmente, ou nem
quebram as temperaturas razoáveis, como nos azoalcanos; e
(b) a transferência para a molécula de uma dada quantidade de
energia, de modo que este é um método específico de efetuar

263
a homólise, forma reações mais limpas que a pirólise
(termólise), que forma reações laterais.
Termólise
Baseia-se na fragilidade, facilidade de fissão térmica, da
ligação carbono-metal, por exemplo, Pb-R. Os radicais são
gerados em solução em solventes inertes, ou através da
termólise de ligações fracas, com energia de dissociação < 165
kJ e ocorre na fase de vapor.
Tais ligações envolvem frequentemente átomos
diferentes do carbono, e a maior fonte de radicais em solução é
a termólise de peróxidos (O2) adequados e compostos azo
(N2).
A formação de radicais através da fissão da ligação
carbono-carbono é observada na indução do radical a 600° de
alcanos de cadeias longas. Os radicais introduzidos
inicialmente no sistema atuam capturando um átomo de
hidrogênio do grupo CH2 da cadeia, o radical não-terminal de
cadeia mais longa sofre fissão β em relação ao átomo de
carbono radicalar para originar um alceno de peso molecular
inferior mais um radical, que mantém a reação em cadeia.
O termino da reação por interação radical/radical não
ocorre em extensão significativa, até a concentração do alcano
de cadeia longa cair num nível mais baixo.
Reações redoxi
As reações redoxi envolvem transferência de um elétron
na geração do radical e envolve íons metálicos do tipo Fe2+

264
/Fe3+ e Cu+/Cu2+. Os íons Cu+ aceleram a decomposição de
peróxidos de acilo, como o (ArCO2)2.
Este é um método útil para gerar o radical ArCO2.,
porque na termólise de (ArCO2)2 a decomposição pode formar
os radicais Ar. + CO2.. O Cu+ reage com conversão de sais de
diazônio, ArN2+Cl-, a ArCl + N2 (reação de Sandmeyer, onde
Ar. se forma, como intermediário).
O Fe2+ é utilizado para catalisar reações de oxidação de
solução aquosa de peróxido de hidrogênio, para formar o
radical HO..
A mistura é conhecida como reagente de Fenton e o
agente oxidante efetivo no sistema é o radical hidroxilo (.OH).
Este é um bom captador de H, e também pode ser usado para
gerar um radical do tipo .CH2CMe2OH para preparar compostos
por dimerização.
A formação de um radical por um processo oxidativo
ocorre provavelmente na iniciação da auto-oxidação, que é
catalisada por um número de íons metálicos pesados
susceptíveis de transferência de um elétron.
Detecção de radicais
A alta reatividade química de radicais de vida curta é
aproveitada para ajudar na sua detecção através da
capacidade de originar espelhos metálicos. O fato da transição

265
de um elétron não emparelhado entre os níveis de energia de
um radical envolver menos energia que a transição de um par
de elétrons na molécula precursora, estável, significa que o
radical tende a absorver radiação de comprimentos de onda
superiores. Um número de radicais são corados e podem ser
detectados pela absorção de radiação em certos comprimentos
de onda. Esses radicais podem ser detectados pelo
desaparecimento rápido da cor em soluções contendo espécies
como o 1,1-N,N-difenil-2-N-picril-hidrazilo.
Forma e estabilização de radicais
Os radicais simples do tipo R3C., contêm um elétron
desemparelhado numa orbital p ou num orbital híbrido sp3 tem
forma piramidal.
A evidência física direta para CH3. provém do espectro
de ressonância de spin de elétron (e.s.r.) de 13CH3., A análise
das linhas, resultantes da interação entre o elétron
desemparelhado e o núcleo de 13C paramagnético, fornece
informação acerca do grau de caráter s do orbital que contém o
elétron desemparelhado.
Em 13CH3. parece ter um pouco ou nenhum, e o radical é
essencialmente planar; uma conclusão que é suportada pela
evidência encontrada nos espectros de UV e IV. O caráter s do
orbital semipreenchido aumenta ao longo da série.
CH3. < CH2F
. < CHF2. < CF3
.

266
No radical .CF3 essencialmente sp3, este radical é
piramidal. Os radicais .CH2OH e .CMe2OH não são planares.
Comparando a facilidade de formação e a reatividade de
radicais em ponte, como no bicilco[2,2,2]octil e biciclo
[2,2,1]hept-2-enil, com os seus equivalentes acíclicos, porque
os radicais alquilas exibem preferência pelo estado planar.
E contrariamente aos íons carbônios, no
bicilco[2,2,2]octil e biciclo [2,2,1]hept-2-enil, é fácil formar
radicais em posições em ponte.
Observa-se que a estabilidade relativa de radicais
alquilas simples segue a sequência seguinte.
R3C. > R2CH.> RCH2
. > CH3.
Esta reflete na facilidade relativa com a qual a ligação C-
H no precursor alcano sofre fissão homolítica, diminuição de
estabilização, por hiperconjugação ou outros fatores à medida
que se percorre a série. Há também uma diminuição da tensão
(quando R é grande) quando se passa do precursor de
hibridação sp3 para o radical hibridizado sp2, à medida que a
série é percorrida. Contudo, a diferença relativa na estabilidade
é muito menor do que com os íons carbônios correspondentes.
Os radicais do tipo alílico, RCH=CHCH2. e benzílico,
PhCHR, são mais estáveis e menos reativos que radicais
alquilo simples dada a deslocalização dos elétrons
desemparelhado sobre o sistema orbital π.
Ambos são essencialmente planares, hibridização sp2 no
átomo de carbono radicalar, e só nesta configuração a

267
sobreposição orbital p/π máxima é possível, com a
estabilização consequente.
A estabilidade de um radical aumenta à medida que
aumenta a extensão da deslocalização; o radical Ph2CH. é
mais estável que PhCH2. , e Ph3C
. é um radical bastante
estável.
A geometria de Ph3C. tem uma extensão de
deslocalização do elétron desemparelhado e com a
estabilização consequente. O átomo do carbono radical
encontra-se hibridizado em sp2 em Ph3C., em que as ligações
que o ligam aos três anéis benzênicos estão todas no mesmo
plano, mas a estabilização máxima só ocorre se os três
núcleos benzênicos estiverem coplares.
Nesta conformação o orbital p do átomo de carbono
central pode interagir ao máximo com o sistema de orbitais π
dos três anéis benzênicos.
Os radicais triarilmetil apresentam, por determinações
espectroscópicas e cristalográficas de raio X, uma forma em
hélice do Ph3C., onde os anéis benzênicos fazem um ângulo de
cerca de 30° com o plano médio comum. Embora ocorra
deslocalização em Ph3C. como o indicado pelo espectro de
E.S.R., ela não é máxima, e a sua extensão não é muito maior
em Ph3C. do que em Ph2CH. ou mesmo PhCH2
..
REAÇÕES DE RADICAIS
As reações de radicais podem ser classificadas de
acordo com o radical: (a) reações unimoleculares, exemplo,
fragmentação, rearranjo; (b) reações bimoleculares entre

268
radicais, exemplo, dimerização, disproporção; e (c) reações
bimoleculares entre radicais e moléculas, exemplo, adição,
substituição, abstração de um átomo.
ADIÇÃO
As adições a ligações dupla cabono-carbono, C=C, são
as reações mais importantes envolvendo radicais, entre elas, a
polimerização de adição, a adição de halogênios e de
hidrácidos.
Adição de Halogênios
A adição de halogênios a alcenos pode ocorrer via
radicais intermediários por solventes não polares (fase
gasosa); é favorecida pela luz solar ou radiação UV e pela
adição de precursores radicalares (iniciadores) como
catalisadores.
Adição de cloro ao tetracloroetano por catálise
fotoquímica envolve uma reação em cadeia.
Cada molécula de cloro, por fissão fotoquímica, originará
dois radicais de átomos de cloro, cada um dos quais é capaz
de iniciar uma reação em cadeia contínua. A cada quantum de
energia absorvida conduz, na verdade, à iniciação de duas
reações em cadeia. E isso é confirmado pela relação seguinte:
Os átomos de cloro são eletrófilos (eletronegativo, e o Cl.
atrairá rapidamente um elétron para completar o octeto) e
assim adiciona-se rapidamente à dupla ligação do tetracloro
etano para dar o radical pentacloroetano. Este, por sua vez,
pode retirar um átomo de cloro de uma segunda molécula para
formar o produto final de adição; o hexacloroetano, mais um
radical de cloro para continuar a reação em cadeia. Uma

269
reação em cadeia contínua muito rápida é iniciada por cada
átomo de cloro iniciador, gerado fotoquimicamente.
A cada quantum de energia absorvida leva à conversão
de várias centenas de moléculas de tetracloroetano em
hexacloroetano. Até ao final da reação, quando quase todo o
tetracloroetano e Cl2 foram consumidos, as concentrações do
radical pentacloroetano e de Cl. serão muito pequenas em
relação às dos materiais de partida; a colisão de um radical
com outro radical é mais frequente. A terminação da cadeia
ocorrerá principalmente através da colisão radical/radical que,
geralmente, envolve dois radicais de pentacloroetano formando
o decaclorobutano
A reação de adição radicalar é inibida pela presença do
oxigênio, porque a molécula de oxigênio tem dois elétrons
desemparelhados e comporta-se como um dirracal, ·O–O·.
Pode, contudo, combinar-se com radicais intermediários
altamente reativos na adição anterior, convertendo-os nos
radicais peróxido, RaO-O. muito menos reativos, incapazes de
continuar a cadeia é assim um inibidor eficiente. Que o
oxigênio reage largamente com radicais pentacloroetilo é
mostrado pela formação do tricloro de cloreto ácido, quando a
adição normal é inibida por oxigênio.
A sequência de reatividade para a adição homolítica dos
diferentes halogênios a alcenos é a mesma da adição eletrófila,
F2 > Cl2 > Br2 > I2.

270
A adição de cloro ou bromo ao benzeno, uma das
poucas reações de simples adição ao anel benzênico, ocorre
por mecanismo radicalar. A reação é catalisada pela luz e pela
adição de peróxidos e é retardada ou evitada pelos inibidores.
A adição de cloro procede da seguinte froma.
O produto é uma mistura de alguns dos oito isômeros
geométricos possíveis do hexaclorociclohexano. A reação não
ocorre na ausência de luz ou peróxidos, enquanto que na
presença de ácidos de Lewis a substituição eletrofílica ocorre
via adição/eliminação com radicais diferentes do Cl..
O ataque do radical de cloro ao metilbenzeno (tolueno) é
de preferência pela abstração de um hidrogênio pelo radical
(Cl.) conduzindo a substituição no grupo CH3, e à adição de
preferência ao anel. Devido a maior estabilidade do radical
benzil inicialmente formado (deslocalizado), PhCH2., em
relação ao radical hexadienil, no qual perde a estabilização
aromática do material de partida.
Adição de Ácido bromídrico
A adição de HBr a propeno, MeCH=CH2, em solventes
polares, forma o 2-bromopropano. Em presença de peróxido

271
(ou em outras condições que promovam a formação de
radicais), a adição ocorre via uma reação em cadeia rápida e
forma o 1-bromopropano, este é geralmente referido como o
efeito peróxido conduzindo a uma adição anti- Markownikov.
Esta orientação diferente da adição de HBr é devida ao fato de
no primeiro caso (polar) ser iniciada por H+ e ocorrer via o íon
carbônion (secundário) mais estável, enquanto no segundo
caso é iniciada pelo radical Br. e procede via o radical
(secundário) mais estável.
A iniciação é feita por Br. dado a abstração de hidrogênio
por RO. de HBr ser energeticamente mais favorável que a
abstração de bromo, para formar ROBr + H.. A adição de Br. ao
propeno (MeCH=CH2) forma o MeCH(Br)CH2. e não ocorre
com radicais secundários Me.CHCH2Br, por serem mais
estáveis que os primários.
O HBr é o único dos quatro haletos de hidrogênio que se
adiciona facilmente a alcenos pelo mecanismo radicalar. A
razão é dada pelos valores de ∆H em kJ mol -1, para as duas
etapas da reação em cadeia para a adição de HX a CH2=CH2,
por exemplo.
Hidrácidos Etapa 1, ∆H (kJ mol -1) Etapa 2, ∆H (kJ mol -1) 2X. + CH2 = CH2 XCH2CH2
. + HX H – F - 188 +155 H – CL -109 +21 H – Br -21 -46 H – I +29 -113
Para o HBr, as duas etapas da reação em cadeia são
exotérmicas; para HF, a segunda etapa é altamente
endotérmico, refletindo a força de ligação H–F e a dificuldade
da sua quebra; para HCl o segundo passo é endotérmico;
enquanto para HI é o primeiro passo que é endotérmico,
refletindo na energia liberada na formação da ligação C–I fraca
por quebra da ligação dupla C=C.

272
As reações adição de HBr em cadeia tendem a ser muito
rápidas. O processo de auto-oxidação do alceno pelo ar, gera
peróxido suficiente no alceno para iniciar a reação de adição
radicalar de HBr no alceno, e predominará o produto anti-
Markownikov. Se pretender o produto Markownikov, por
exemplo, MeCH(Br)CH3 do propeno, é necessário purificar
rigorosamente o alceno ou adicionar inibidores (fenóis,
quinonas, etc.) para remover quaisquer radicais, ou radicais
potenciais presentes. O controle da orientação da adição de
HBr, pode ser pela introdução de peróxidos (iniciadores de
radicais) ou inibidores de radicais na mistura reacional. Assim,
o alceno CH2=CHCH2Br, por exemplo, pode ser convertido em
1,2- ou 1,3-dibromopropano.
A baixas temperaturas ou pelo uso de concentração
elevada de HBr. A adição de HBr líquido a 80 °C ao cis-2-
bromobut-2-eno forma com elevada estereoespecificidade o
isômero TRANS.
Nestas mesmas condições a adicção radicalar de HBr
no trans-2-bromobut-2-eno, também ocorre adição ANTI quase
exclusiva de HBr com formação do trans-2,3-dibromobutano.
Em alcenos cíclicos, caso do ciclo-hexeno, a adição
radicalar ocorre com formação do isômero TRANS, em outros
alcenos a adição ocorre com preferência, mas não há
exclusividade do isômero TRANS.

273
OUTRAS ADIÇÕES
Radicais tiil, RS., podem ser obtidos pela abstração de
hidrogênio de RSH, e adiconar-se-ão rapidamente a alcenos
por uma reação em cadeia análoga à de HBr. Esta reação de
adição é usada para sintetizar sulfitos dialquilo, sendo que esta
reação é reversível.
Os cloretos de sulfenila, por exemplo, Cl3CSCl, também
podem ser usados como fontes de radicais tiil, mas aqui a
adição é iniciada por Cl. e o R’S. ligar-se-á ao outro átomo de
carbono da ligação dupla.
As ligações carbono-carbono podem ser formadas pela
adição, entre outras, de radicais halometil a alcenos. O .CX3
(X=Br, Cl) pode ser gerado pela ação de peróxidos em CX4 ou
por fotólise.
O CCl4 relativamente inerte se adiciona, porque os
valores de ∆H para ambos os passos da cadeia reacional são
exotérmicos: -75 e -17 kJ mol-1. O primeiro radical formado
pode, contudo, competir com .CCl3 na adição a RCH=CH2 de
modo a formarem-se polímeros de baixo peso molecular, sob
condições determinadas além do produto normal de adição.
Polimerização vinílica
A reação de polimerização vinílica é de grande
importância para preparar polímeros com inúmeras aplicações
industriais e comerciais. O mecanismo reacional é semelhante

274
as outras reações envolvendo radicais nas quais envolve três
etapas, (a) iniciação (b) propagação e (c) terminação.
a) Iniciação
Formação do iniciador a partir de peróxidos ou
compostos azoicos
b) Propagação
Normalmente muito rápida.
c) Terminação – reação entre os radicais
Os monômeros podem adsorver oxigênio do ar,
formando peróxido, cuja decomposição rápida pode efetuar a
auto-inicação de polimerização, é comum adicionar-se uma
pequena quantidade de inibidor, (quinona), para estabilizar o
monômero durante o armazenamento. Quando se pretende
efetuar a polimerização, deve-se adicionar radical iniciador
suficiente para saturar o inibidor antes de iniciar a
polimerização. A eficiência de alguns iniciadores pode ser
tão elevada que, após qualquer período de indução, todos os
radicais gerados conduzem a uma cadeia polimérica.
O crescimento de uma cadeia pode ser
interrompido pela colisão com o radical iniciador ou com outra
cadeia, ocorrendo dimerização. A captura de H pode ocorrer
pelo ataque de uma cadeia em crescimento num polímero
“morto” (que cessou de crescer), conduzindo a um ponto de
crescimento e por consequência, à ramificação.

275
A extensão com que ocorre a ramificação pode ter um
efeito nas propriedades físicas e mecânicas do polímero
resultante.
SUBSTITUIÇÃO
As reações e alguns casos obtêm-se um radical a partir
do substrato por perda, e este radical efetuará substituiição ou
adição em diversas espécies. Em alguns casos, contudo,
consegue-se a substituição direta por meio de
adição/substituição.
Halogenação
A substituição direta que ocorre no carbono e na
cloração, por exemplo, de alcanos consiste na abstração de H
de RH por Cl., seguido de abstração de Cl de Cl–Cl por R., os
dois passos alternando-se numa reação em cadeia muito
rápida.
O comprimento da cadeia, RH→RCl por Cl. produzido
por fotólise, é 106 para CH4, e a reação pode ser explosiva
quando exposta á luz solar. A cloração também pode ser
iniciada termoliticamente, mas não são necessárias
temperaturas elevadas para converter Cl2 → 2Cl., e a
velocidade de cloração de C2H6, no escuro a 120° não é
praticamente detectável. Torna-se extremamente rápida
quando adicionam-se resíduos de PbEt4, porque decompõem-
se em radicais etilo, atuando como iniciadores. A cloração de
alcanos simples, por este método é bastante útil para a

276
preparação de derivados monoclorados, porque o reagente
pode sofrer ataque facilmente pelo cloro altamente reativo,
obtendo-se misturas complexas.
A facilidade de ataque nos diferentes átomos de
hidrogênio num alcano aumenta na ordem do enfraquecimento
da ligação C–H , e no aumento de estabilidade do produto
radicalar.
H3C – H < H3C – R < H2C – R2 < HC – R3 Metano Primário
1 Secundário 4,4
Terciário 6,7
Esta diferença pode-se opor frequentemente ao número
relativo dos diferentes tipos de hidrogênio presentes; assim em
(CH3)3CH existem 9 átomos de hidrogênio primários acessíveis
para um átomo de hidrogênio terciário. Na cloração de
(CH3)3CH verifica-se que se formam produtos monoclorados na
razão de 65% de (CH3)2CHCH2Cl para 35% de (CH3)3CCl-. Se
a cloração ocorrer em solução, verifica-se que a distribuição do
produto depende da natureza do solvente e, particularmente,
da sua capacidade para complexar com Cl., estabiizando-o e
aumentando a sua seletividade.
A cloração, contrario à maior parte das reações de
halogenação radicalares, é influenciada pela presença no
substrato de substituinte polares; devido ao fato do radical Cl.,
cloro muito eletronegativo, atacar onde a densidade eletrônica
é elevada. A cloração pode ser inibida pela presença de grupos
receptores de elétrons, como se observa nas quantidades
relativas de substituição nos quatro átomos de carbono
diferentes do 1-clorobutano na cloração de iniciação
fotoquímica a 35°.
CH3 – CH2 – CH2 – CH2 – Cl
25% 50%
17%
3%

277
A variação dos três grupos CH2 mostra a diminuição com
a distância do efeito indutivo negativo do Cl.. O grupo γ-(3-)CH2
comporta-se de modo análogo ao de CH3CH2CH3, enquanto
que o menor número encontrado para o grupo CH3 reflete a
maior dificuldade de quebra da ligação C–H em CH3 do que em
CH2.
Com o propeno, CH3CH=CH2, há a possibilidade da
adição do cloro à ligação dupla ou no grupo CH3. Em
temperaturas elevadas, 450°, a substiuição ocorre sem
qualquer adição. Isto resulta porque o radical alila é obtido por
subtração de H estabilizado por deslocalização, enquanto o
readical 1-cloropropano obtido na adição de Cl. não é
hibridizado, e a sua formação é reversível a altas temperaturas,
estando o equilíbrio deslocado para a esquerda.
No ciclohexeno ocorre cloração alílica análoga ao
propeno pelas mesmas razões.
Os valores de ∆H em kJ mol-1 para a substituição
radicalar para os hologênios para as duas etapas da reação em
cadeia de hologenação no CH4 são os seguintes:
Halogênios Etapa 1 (X. + H – CH3) ∆H (kJ mol-1)
Etapa 2 (CH3. + X2)
∆H (kJ mol-1)
F2 -134 -292 Cl2 -4 -96 Br2 +63 -88 I2 +138 -75

278
Os valores para a fluoração refletem na fraca ligação F–
F, e a força da ligação H–F. A fluoração ocorre via mecanismo
radicalar e não requer iniciação específica; é explosiva a não
ser em solução muito diluída. A bromação é mais lenta que a
cloração em condições comparáveis, porque a etapa 1,
abstração de H por Br., é endotérmica. Para a reação de
iodação de alcanos, a etapa 1 tem um valor de ∆H (kJ mol-1)
grande, reação endotérmica, de modo que não ocorre reação
com o radical I..
A reatividade mais baixa do Br. em relação ao Cl., quanto
à abstração de H significa que a bromação é muito mais
seletiva que a cloração. Visto que na reação de bromação do
(CH3)3CH, forma só o (CH3)3CBr.
H3C – R < H2C – R2 < HC – R3
Primário
1
Secundário
80
Terciário
1600
O efeito é mais pronunciado quando estão presentes
substituintes que podem estabilizar o radical inicial. Na série,
CH4, PhCH3, Ph2CH2 e Ph3CH, as velocidades relativas de
bromação diferem na ordem de 109, mas para a cloração, na
ordem de 103 , porém, a seletividade diminui com o aumento de
temperatura.
A halogenação de um alcano quiral RR’R”CH forma
normalmente um haleto racêmico, um resultado que não diz
nada a cerca da conformação preferencial do radical
intermediário RR’R”C., porque a racemização pode ocorrer com
uma estrutura planar ou uma estrutura piramidal de
interconversão rápida
Auto-oxidação
A auto-oxidação é a oxidação à baixa temperatura de
compostos orgânicos pelo O2, envolvendo uma reação

279
radicalar em cadeia. O passo inicial é normalmente a formação
de hidroperóxidos, RH→ ROOH, de forma que é na sua
globalidade uma substituição, embora a verdadeira via envolva
a abstração de H e adição de O2. Os hidroperóxidos formados
inicialmente sofrem frequentemente outras reções. A auto-
oxidação tem importância no endurecimento das tintas, onde
os ésteres insaturados dos óleos formam normalmente
hidroperóxidos, cuja decomposição é a RO. Inicia a
polimerização das moléculas insaturadas para formar um filme
superficial polimérico e protetor. Mas a auto-oxidação é
também responsável pelas alterações prejuciais,
particularmente em materiais com ligações insaturadas, por
exemplo, ranso nas gorduras e deteriorização ds borrachas. Na
realidade, a decomposição gradual de muitos compostos
orgânicos expostos ao ar e à luz é devido à auto-oxidação
fotosensível. A auto-oxidação pode ser iniciada por resíduos de
íons metálicos, assim como pela luz ou por iniciadores de
radicais comuns.
O mecanismo reacional mais importante é a cadeia com
um dos passos envolvendo a abstração de H.
Em certas condições o próprio hidroperóxido sofre
ruptura para formar radicais RO. + .OH que podem atuar como
iniciadores, e a auto-oxidação torna-se a autocatalítica. Os
radicais peróxi são normalmente de reatividade relativamente
baixa e são assim muito específicos relativamente às posições
de onde capturam os H. Assim, o C–H alílico e benzílico são
relativamente fáceis de ser atacados, dado as ligações C–H
serem um pouco mais fracas e os radicais resultantes ficarem
estabilizados por deslocalização, enquanto em alcanos simples

280
são geralmente unicamente os C–H terciários, por exemplo, a
posição alílica do hidroperóxido de cilco pent-2-eno e a posição
terciária do hidroperóxido do biciclo[4,4,0]-1-hidroperóxido.
A reatividade relativa, da captura de H por RO2. a 30 °C,
segue a seguinte ordem: PhCH3, Ph2CH2, e PhCH2CH=CH2.
Com alcenos, mais do que com alcanos, a auto-
oxidação pode envolver adição de RO. à dupla ligação ou a
subtração de H ligações C–H alílicas, benzílicas ou terciárias.
O efeito da presença de tais peróxidos nos alcenos na
orientação da adição HBr a estes já foi referida anteriormente.
Os éteres são menos propensos a auto-oxidação, ocorrendo o
ataque inicial na ligação α-, C–H ao átomo de hidrogênio para
dar um radical estabilizado; o hidroperóxido formado
inicialmente reage para dar peróxidos de dialquilo que são
altamente explosivos quando aquecidos. Os peróxidos
acumulados, no éter armazenado, devem ser decompostos
antes do seu uso, por lavagem com uma solução de um agente
redutor, por exemplo, FeSO4.
Substituição aromática
As reações podem ocorrer com espécies aromáticas
com radicais, assim como com os eletrófilos e nucleófilos, do
memso modo como acontece com necleofilos, a substituição
aromática homolítica ocorre por via mecanismo de
adição/eliminação.

281
A perda de um átomo de hidrogênio do radical
intermediário deslocalizado ciclohexadienilo forma o produto
substituído, não ocorre espontaneamente, mas requer a
intervenção de outro radical. A reação entre dois radicais, o
ciclohexadienilo e o H subtraído, é provavelmente rápida, e não
determina a velocidade e não se observa efeito isotópico
significativo, sendo o radical Ra. ao substrato aromático
determinante da velocidade da reação.
Quanto à reação de substituição, notam-se diferenças
marcantes entre o ataque eletrofílico e nucleofílico, quando se
toma em consideração o comportamento de derivados
benzênicos substituídos (C6H5Y). Assim o ataque homolítico a
C6H5Y é mais rápido do que C6H6, não interessa se Y é elétron
retirador ou elétron doador. O ataque preferencial ocorre
orto/para a Y. O elétron cedido pelo radical de ataque, do
intermediário, ciclohexadienilo, pode ser deslocalizado daí
também o intermediário formado por substituintes doadores ou
receptores elétrons.
Os valores de velocidade parcial abaixo em que: (a) o
ataque na posição meta a Y é mais rápido que o ataque ao
benzeno, e (b) o ataque é mais rápido na posição orto em
relação Y, a menos que Y seja tão volumoso que impeça a
aproximação do reagente, por exemplo, CMe3.
PhY orto meta para
PhOMe 5,60 1,23 2,31 PhNO2 5,50 0,86 4,90 PhMe 4,70 1,24 3,55 PhCl 3,90 1,65 2,12 PhBr 3,05 1,70 1,92

282
PhCMe3 0,70 1,64 1,81
Os dados anteriores referem-se à fenilação por Ph.
gerado a partir de (PhCO2)2.
Rearranjo
No que diz respeito a rearranjos, os radicais
assemelham-se muito mais que os carbânions do que os íons
carbônios. Assim, como acontece com carbânions,
desconhecem-se migrações 1,2 de alquilos de carbono para
carbono, assim como migrações 1,2 de hidrogênio, mas
conhecem-se algumas migrações 1,2 de arilos que envolvem
estado de transição em ponte estabilizado. Por exemplo, o
aldeído (RCHO) abaixo, sofre abstração de H do grupo CHO
pelo Me3CO. formando o radical acil, que perde rapidamente
CO para formar um novo radical. Esta pode, por sua vez, retirar
H de RCHO formando o hidrocarboneto, proveniente do radical
rearranjado.
O radical rearranjado é mais estável que o original, não
só por o primeiro ser terciário e o segundo primário, mas
também devido se encontrar estabilizado por deslocalização do
elétron não emparelhado no sistema orbital π do anel
benzênico e a migração do Me conduzir a um radical ainda
mais estabilizado, Ph2.CH2Me, refletindo numa vantagem
energética da migração via um E.T. deslocalizado. Quando não

283
existe grupo Ph, como em EtMe2CCH2. de EtMe2CCH2CHO,
não ocorre qualquer migração, e o produto final EtMe2CCH3.
As migrações dos radical aril não estão limitadas a
rearranjos carbono/ carbono como o observado pelo
comportamento de (Ph3CO)2 por aquecimento.
Esta reação também ocorre via um ET em ponte, devido
ao rearranjo gerar uma maior estabilidade do radical. Assim,
como as migrações 1,2 do radical aril, também ocorre
migrações semelhantes de grupos vinil, acil e acilóxi, ocorrendo
via estados de transição ou intermediários em ponte e também
migrações 1,2 de cloro, na qual, uma orbital d vazia do
halogênio é usada para acomodar o elétron desemparelhado
do intermediário em ponte.
Bi-radicais
A molécula de oxigênio, uma espécie paramagnética
com um elétron desemparelhado em cada átomo é um
birradical, pouco ativo. A excitação fotoquímica de um
antraceno a um birradical ou qualquer espécie afim pode
ocorrer na ausência de ar ou oxigênio; em lugar de um
peróxido trans-anelar, forma-se um fotodímero.
Os birradicais ocorrem como intermediários nas
reduções com Mg de cetonas a pinacóis e, como ânions

284
radicais, na condensação de aciloína dos ésteres. A termólise
de ciclopropano a propeno à 500 °C também envolve
intermediários birradicalares.
Para formar o birradical, a molécula de ciclopropano
torna-se vibracionalmente excitada por colisão com outra
molécula; a ligação C–C pode ser quebrada desde que a
energia extra não se perca rapidamente, por posterior colisão.
Existe uma força condutora aqui para migração 1,2 de
hidrogênio contrária ao que acontece em monorradicais, dada
a oportunidade do par de eletrônico formar ligação π (com
liberação de energia).
Os birradicais, com exceção da molécula de
oxigênio, são todos altamente instáveis, porém, existem
algumas espécies mais estáveis que mostram caráter
birradical. O hidrocarboneto abaixo existe, em solução como
um birradical.
Este comporta-se como Ph3C. existindo fora da solução
como um sólido incolor, provavelmente um polímero e não um
dímero como com Ph3C.. Os elétrons desemparelhados na
forma birradical não podem reagir uns com os outros para
formar uma espécie emparelhada, diamagnética, dado que tal
interação através do núcleo central benzênico requereria
formas m-quinoides que não podem existir, pois os elétrons
estão “isolados internamente”. Este isolamento interno nos

285
birradicais pode também provir de efeitos estereoquímicos e
não eletrônicos. Por exemplo, a espécie abaixo existe em
solução como birradical na extensão de 17%, estando em
equilíbrio com o polímero.
Aqui não existe qualquer impedimento eletrônico
formal à interação entre os elétrons, mas na realidade não
acontece, dado os átomos de cloro volumosos em posição orto
não permitirem aos anéis benzênicos adquirir uma
conformação coplanar, única posição onde ocorre
sobreposição significativa das orbitais p, requisito necessário
para o emparelhamento eletrônico.
10º ROTEIRO DE ESTUDO DIRIGIDO
1. Os processos reacionais que envolvem a quebra e a formação de uma ligação simétrica são chamados reações radicalares. Um radical é uma espécie química que contém um número ímpar de elétrons e assim tem um único elétron desemparelhado em um de seus orbitais. a) Uma característica de muitas reações de radicais é que, uma vez iniciadas, procedem com grande rapidez. Explique por que isso acontece baseando-se no mecanismo de halogenação radilar de alcanos. b) Existem radicais que são menos reativos, ou seja, apresentam uma maior estabilidade e, portanto, maior existência em condições normais. Cite exemplos de alguns desses compostos e explique a razão da sua estabilidade.
Formação de Radicais 2. As maneiras mais importantes de se gerar radicais a partir de moléculas neutras são através de: (a) fotólise; (b) termólise; e (c) reação redox. 3. Como ocorre a fotólise? Cite exemplos de compostos que podem sofrer decomposição fotolítica. Apresente o mecanismo. 4. Em que condições ocorre termólise? Mostre reações. 5. Quais as vantagens da fotólise sobre a termólise?

286
6. Mostre como acontece a reação de Sandmeyer, a formação do reagente de Fenton, e como se processa a síntese eletrolítica de Kolbe de hidrocarbonetos. Detecção de radicais Alguns radicais apresentam uma alta reatividade química, tendo por isso uma existência extremamente curta. Entretanto, existem testes químicos e análises instrumentais que tornam possível a sua detecção. 7. Como é feita essa detecção de radicais? Formação e estabilização de radicais
A estabilização relativa de radicais alquilo simples segue a seguinte seqüência:
Os radicais alílico e benzílico são mais estáveis e, portanto, menos reativos que esses radicais alquilo simples. 8. Justifique a ordem de estabilidade mostrada acima, e diga
por que os radicais alílico e benzílico são menos reativos.
9. O que são heterorradicais? Quais fatores influenciam na sua estabilidade relativa?
Reações de radicais As reações radicalares são classificadas em: (a) Reações unimoleculares (fragmentação, rearranjo) (b) Reações bimoleculares entre radicais (dimerização, disproporção) (c) Reações bimoleculares entre radicais e moléculas (adição, substituição, abstração de um átomo)
Adição: Halogêneos A adição de halogêneos a alcenos pode ocorrer via radicais intermediários. 10. Mostre o mecanismo de adição de cloro a tetracloroetano por catálise fotoquímica. De que forma ocorre a terminação desta reação em cadeia? Ácido bromídrico
Em presença de peróxidos, a adição de HBr a alcenos ocorre via uma reação em cadeia rápida, conhecida como efeito peróxido, conduzindo a uma adição anti-Markownikov.

287
11. A adição de HBr líquido a -80 °C ao cis-2-bromobut-2-eno procede com elevada estereoespecificidade trans, explique através de mecanismos por que isso acontece.
Polimerização vinílica 12. Discuta as etapas das reações envolvendo radicais baseado na reação de polimerização vinílica. Substituição: Halogenação A facilidade de ataque nos diferentes átomos de H num alcano aumenta na sequência:
13. Justifique essa ordem. Explique ainda por que a bromação radicalar é mais seletiva que a cloração. A halogenação radicalar por meio de outros reagentes é frequentemente de maior utilidade sintética do que a efetuada pelos próprios halogêneos. 14. Mostre o mecanismo de uma halogenação utilização N-bromo-succinimida. Auto-oxidação A auto-oxidação é a oxidação a baixas temperaturas de compostos orgânicos pelo O2, envolvendo uma reação radicalar em cadeia. O mecanismo reacional mais importante é a cadeia com dois passos envolvendo a abstração de H:
15. Os aldeídos são muito susceptíveis a auto-oxidação, sendo convertidos em ácido benzóico. Faça o mecanismo desta oxidação utilizando íons metálicos. Como a auto-oxidação de aldeídos e de outros compostos orgânicos pode ser inibida?
Substituição aromática O ataque a espécies aromáticas por radicais ocorre via adição/eliminação.

288
16. Mostre o mecanismo do ataque de .
Ph em espécies aromáticas. 17. A reação de Pschorr constitui uma arilação radicalar intramolecular. Como ela ocorre? Rearranjo 18. A adição fotocatalisda de HBr a CCl3CH = CH2 não produz o composto esperado CCl3CH2CH2Br, mas 100 % de CHCl2CHCl CH2Br. Justifique essa afirmação.

289
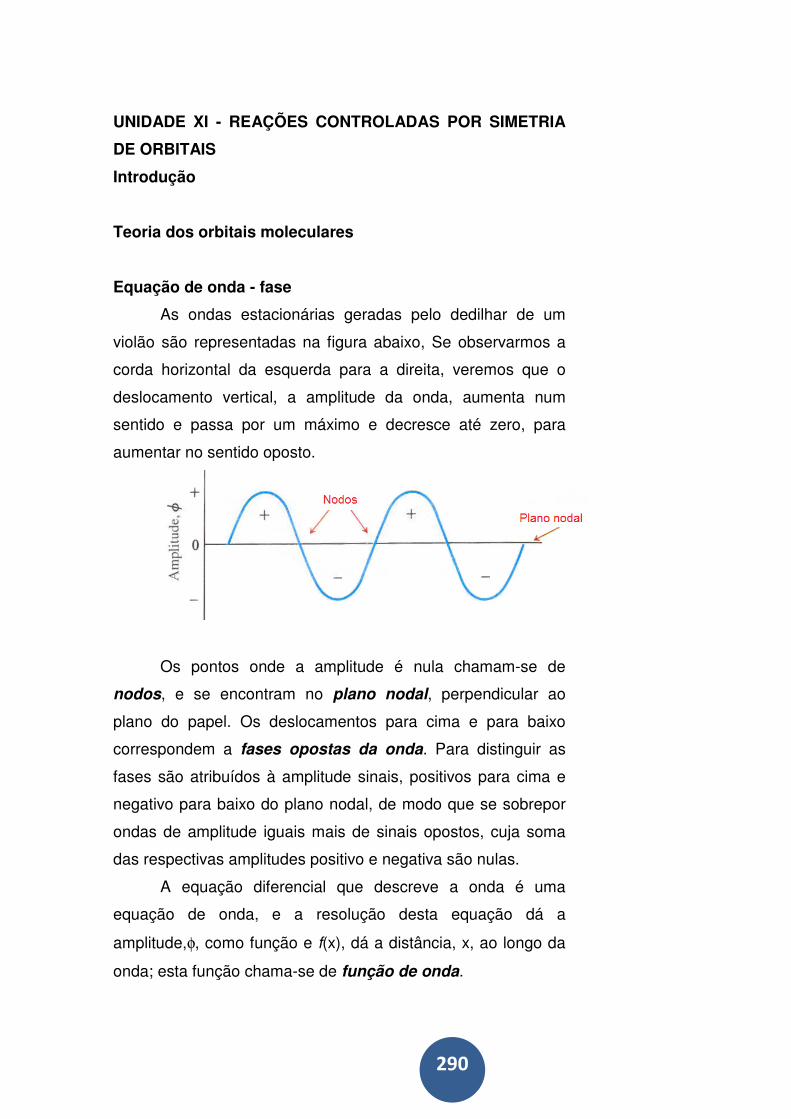
290
UNIDADE XI - REAÇÕES CONTROLADAS POR SIMETRIA
DE ORBITAIS
Introdução
Teoria dos orbitais moleculares
Equação de onda - fase
As ondas estacionárias geradas pelo dedilhar de um
violão são representadas na figura abaixo, Se observarmos a
corda horizontal da esquerda para a direita, veremos que o
deslocamento vertical, a amplitude da onda, aumenta num
sentido e passa por um máximo e decresce até zero, para
aumentar no sentido oposto.
Os pontos onde a amplitude é nula chamam-se de
nodos, e se encontram no plano nodal, perpendicular ao
plano do papel. Os deslocamentos para cima e para baixo
correspondem a fases opostas da onda. Para distinguir as
fases são atribuídos à amplitude sinais, positivos para cima e
negativo para baixo do plano nodal, de modo que se sobrepor
ondas de amplitude iguais mais de sinais opostos, cuja soma
das respectivas amplitudes positivo e negativa são nulas.
A equação diferencial que descreve a onda é uma
equação de onda, e a resolução desta equação dá a
amplitude,φ, como função e f(x), dá a distância, x, ao longo da
onda; esta função chama-se de função de onda.

291
As ondas correspondentes aos elétrons são descritas
por equações de mesma forma geral que as de ondas geradas
por uma corda vibrando, mas as funções de onda que são
soluções possíveis desta equação dão a amplitude,φ, que não
são representadas apenas em uma coordenada, mas em três
coordenadas necessárias para descrever o movimento em três
dimensões. Esta função de onda dos elétrons chamamos de
orbitais.
A onda do elétron pode ter nós, onde a amplitude é
zero. Em lado oposto de um nó, a amplitude tem sinais opostos
e encontra-se em fases opostas, e entre dois lobos de um
orbital p se encontra um plano nodal, perpendicular ao eixo do
orbital. Os dois lobos estão em fases opostas, indicados pelo
sinal positido e negativo, ou lobo achuriado ou não achuriado.
Estes sinais utilizados não têm nada a ver com carga
elétrica, o sinal matemático positivo ou negativo, já que não há
probilidade negativa de encontrar o elétron no orbital, indicam
apenas que os dois lobos apresentam fases opostas.
A combinação de orbitais atômicos para formar OM, só é
efetiva se:
a) Sobreporem mutuamente numa considerável extensão;
b) Forem de energia comparável;
c) Tiverem a mesma simetria relativamente ao eixo da ligação.

292
Com relação a configuração eletrônica das moléculas,
vamos exemplificar usando a molécula do etileno, onde no
estado fundamental só pode haver dois elétrons de spins
opostos em cada orbital, sendo os orbitais de mais baixa
energia preenchidos primeiro. Se houver vários orbitais de
mesma energia, cada um recebe um elétron antes de qualquer
um deles receber um par de elétrons.
Utilizaremos neste estudo apenas os elétrons de
ligações π que encontram-se em plano perpendicular aos
elétrons σ, por isso funcionam com um sistema independente.
Para os elétrons π do etileno, há dois orbitais
moleculares, visto existir duas combinações lineares de dois
orbitais p componentes. Onde tem o orbital ligante π e o orbital
não-ligante π*, conforme figura abaixo.
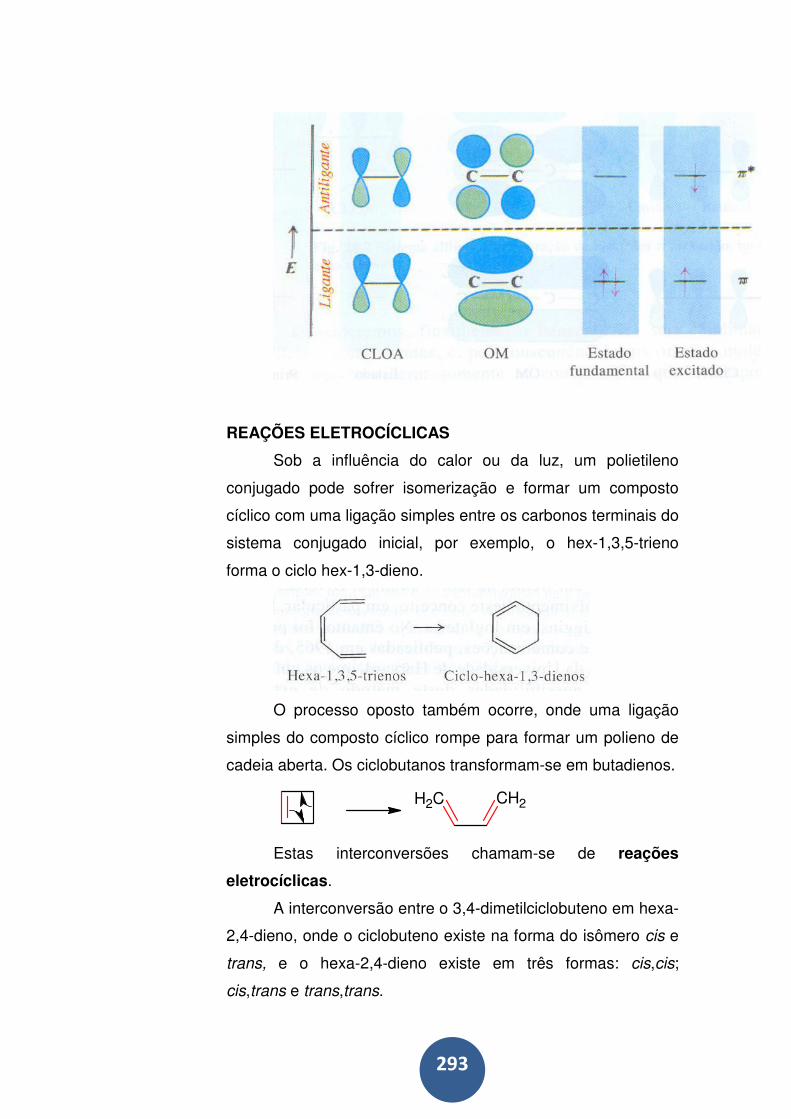
293
REAÇÕES ELETROCÍCLICAS
Sob a influência do calor ou da luz, um polietileno
conjugado pode sofrer isomerização e formar um composto
cíclico com uma ligação simples entre os carbonos terminais do
sistema conjugado inicial, por exemplo, o hex-1,3,5-trieno
forma o ciclo hex-1,3-dieno.
O processo oposto também ocorre, onde uma ligação
simples do composto cíclico rompe para formar um polieno de
cadeia aberta. Os ciclobutanos transformam-se em butadienos.
CH2 CH2
Estas interconversões chamam-se de reações
eletrocíclicas.
A interconversão entre o 3,4-dimetilciclobuteno em hexa-
2,4-dieno, onde o ciclobuteno existe na forma do isômero cis e
trans, e o hexa-2,4-dieno existe em três formas: cis,cis;
cis,trans e trans,trans.

294
O cis-ciclobuteno produz só um dos três isômeros e o
isômero trans forma um isômero diferente.
A reação de interconversão do ciclobuteno é, portanto,
completamente estéreosseletiva e completamente
estereoespecífica. A ciclização fotoquímica do isômero trans,
trans-dieno forma o ciclobuteno diferente daquele a partir do
qual ele se formou por abertura térmica do ciclo.
As reações eletrocíclicas, por consequência, são
completamente estereoespecíficas. A estereoquímica exata
dependede de:
a) O número de ligações duplas no polieno;
b) Ser a reação térmica ou fotoquímica.
A simetria de orbital justifica todos os fatos das reações
formarem um ou outro isômero, e a maioria dos exemplos de
reações conhecidas hoje em dia foram previstos por Woodward
e Hoffmann.
Reação térmica
A ciclização de trans,cis,trans-octa-2,4,6-trieno ocorre
termicamente formando apenas o cis-5,6-dimetilciclohexa-1,3-
dieno e fotoquimicamente forma só o isômero trans-5,6-

295
dimetilciclohexa-1,3-dieno; em ambos os casos, o equilíbrio
encontra-se quase totalmente deslocado no sentido da forma
cíclica. De fato, o grau de estereosseletividade é tão grande
que a ciclização térmica forma menos de 0,1% do isômero
trans, apesar de este ser termodinamicamente mais estável do
que cis.
Nas reações de ciclizações, dois elétrons π formam a
nova ligação σ do cicloalcano. Esses dois elétrons π
encontram-se no orbital molecular ocupado de mais alta
energia (HOMO), são os elétrons de valência da molécula, são
menos firmes em posição e os mais facilmente deslocáveis de
um lado para outro durante a reação.
Na reação de ciclização térmica de um butadieno
dissubstituido RCH=CH–CH=CHR, o orbital molecular ocupado
de mais alta energia de um dieno conjugado é ψ2. São os
elétrons neste orbital que vão formar a ligação que fecha o
ciclo. A formação de ligações requer sobreposição, neste caso,
sobreposição de lobos no C1 e C4 do dieno. E para ocorrer
sobreposição mútua dos lobos, tem que haver rotação em
torno de duas ligações: C1–C2 e C3–C4. Este movimento pode
ser co-rotatório ou disrotatório.
O movimento co-rotatório (giro no mesmo sentido)
resulta em sobreposição de orbitais com a mesma fase, uma
interação ligante formando a ligação de ciclização do
ciclobutadieno em que os dois grupos R ficarão cis, enquanto

296
que o movimento disrotatório (giro no sentido oposto) resulta
em orbitais de fases opostas, uma interação antiligante e
repulsiva.
O movimento co-rotatório forma a estereoquímica que
na realidade se observa para o butadieno dissubstituido abaixo.
Reação fotoquímica
Na reação de ciclização fotoquímica, a irradiação resulta
na promoção de um elétron à orbital do nível de energia mais
baixa, onde ψ2 hv
ψ3 e o LUMO (orbital molecular ocupado
de mais baixa energia) do estado fundamental (ψ2) torna-se
agora o HOMO (orbital molecular ocupado de mais alta
energia) do estado excitado ψ3.
Agora, em ψ3 a simetria relativa dos carbonos terminais
é oposta à que se verifica em ψ2 e o movimento disrotatório
que aproxima lobos da mesma fase, situação ligante, com
formação do isômero trans e com estereoquímica invertida.

297
Para a reação fotoquímica é interessante fazer o
contraponto entre este caso e o do 4-cis, 2-trans hexa-2,4-
dieno e o 4-trans, 2-trans hexa-2,4-dieno
Aqui observa-se exatamente as inter-relações
estereoquímicas opostas. O trans,trans-hexa-2,4-dieno está
termicamente associado ao trans-3,4-dimetilciclobuteno e
fotoquimicamente ao isômero cis-3,4-dimetilciclobuteno.
Regras de woodward-hofmann
Alternativamente, as condições reacionais
térmica/fotoquímica encontram-se sumarizadas nas
generalizações das Regras de Woodward-Hofmann.

298
Número de eletrone ππππ Reação Movimento 4n Térmica Co-rotatório 4n Fotoquímica Disrotatório 4n + 2 Térmica Disrotatório 4n + 2 Fotoquímica Co-rotatório
As reações eletrocíclicas têm importância considerável
em síntese, na formação de ligações carbono-carbono devido a
sua estereoespecificidade rígida, que é muito maior do que a
observada na grande maioria de outras reações não-
síncronizadas.
REAÇÕES DE CICLOADIÇÕES
Nas reações de cicloadições, estão geralmente
envolvidas dois componentes, e a possibilidade de ser posto
em prática um processo particular será determinada pelo fato
de poder haver sobreposição entre o HOMO de uma
componente e a LUMO do outro.
A reação de Diels-Alder, em que um dieno conjugado e
um alceno substituído, dienófilo, reagem para formar um
ciclohexeno, um exemplo de reação de cicloadição.
Esta reação envolvendo duas moléculas insaturadas se
combinam para formar um composto cíclico, criando novas
ligações σ a partir de elétrons π dos reagentes.
A reação de Diels-Alder é uma cicloadição [4 + 2],
implica em um sistema de 4 eletrons π e um sistema de dois
elétrons π, reagindo rigorosamente com estereoespecificidade
SIN, tanto em relação ao dieno como em relação ao dienófilo,
por que há duas combinações possíveis: sobreposição do
HOMO do butadieno (ψ2) com o LUMO do etileno (π*) e a
sobreposição do HOMO do etileno (π) com o LUMO do
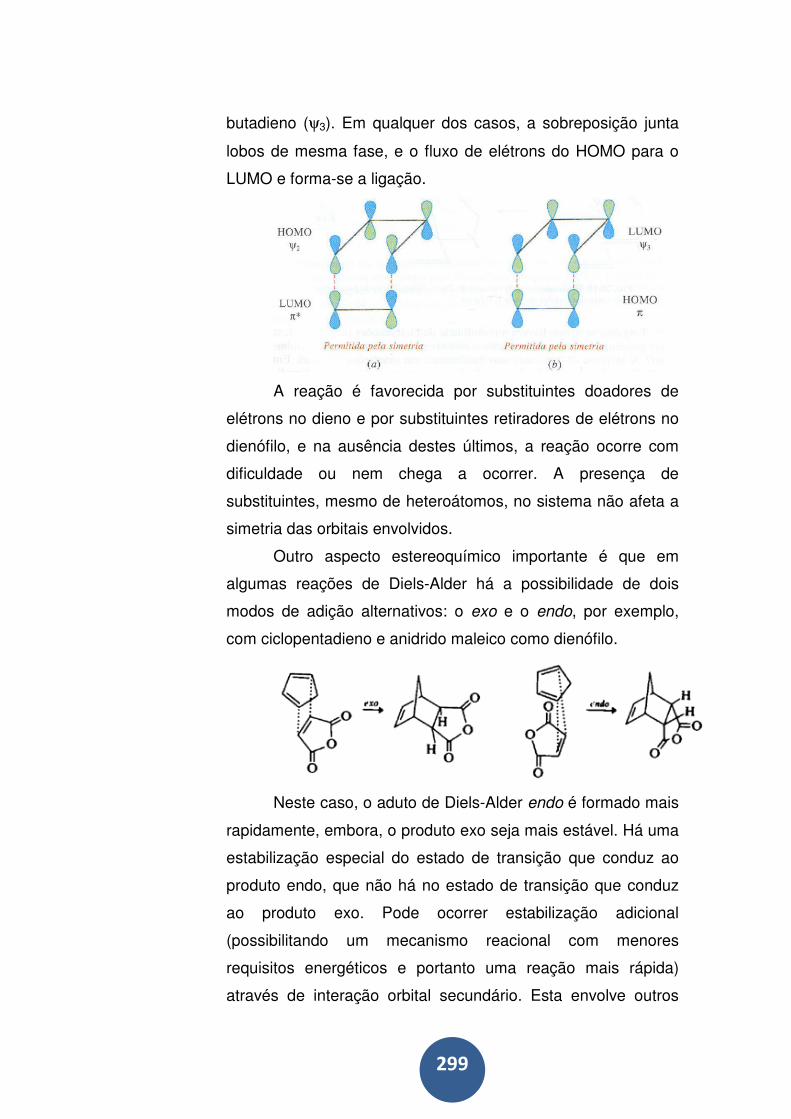
299
butadieno (ψ3). Em qualquer dos casos, a sobreposição junta
lobos de mesma fase, e o fluxo de elétrons do HOMO para o
LUMO e forma-se a ligação.
A reação é favorecida por substituintes doadores de
elétrons no dieno e por substituintes retiradores de elétrons no
dienófilo, e na ausência destes últimos, a reação ocorre com
dificuldade ou nem chega a ocorrer. A presença de
substituintes, mesmo de heteroátomos, no sistema não afeta a
simetria das orbitais envolvidos.
Outro aspecto estereoquímico importante é que em
algumas reações de Diels-Alder há a possibilidade de dois
modos de adição alternativos: o exo e o endo, por exemplo,
com ciclopentadieno e anidrido maleico como dienófilo.
Neste caso, o aduto de Diels-Alder endo é formado mais
rapidamente, embora, o produto exo seja mais estável. Há uma
estabilização especial do estado de transição que conduz ao
produto endo, que não há no estado de transição que conduz
ao produto exo. Pode ocorrer estabilização adicional
(possibilitando um mecanismo reacional com menores
requisitos energéticos e portanto uma reação mais rápida)
através de interação orbital secundário. Esta envolve outros

300
orbitais apropriados no ciclopetadieno e os orbitais disponíveis
(além dos da ligação dupla carbono-carbono) no anidrido
maleico. Esta sobreposição secundária não pode ocorrer no
estado de transição para a adição exo pelo fato de os orbitais
relevantes se encontrarem afastados.
Uma ciclização do tipo [2 + 2] pode ocorrer via adição de
calor, mas não é uma reação concertada ou via fotoquímica,
reação concertada.
Na reação de dimerização térmica do etileno, uma
reação de ciclização térmica [2 + 2] implica na sobreposição do
HOMO, π, de uma molécula com o LUMO, π*, da outra. Mas o
π e π* são de simetria oposta e, portanto, lobos de fase oposta
que se aproximam um do outro. A interação é antiligante e
repulsiva, e a reação que ocorre não é concertada. A reação
não é permitida por simetria.
As ciclizações fotoquímicas [2 + 2] são permitida por
simetria. A sobreposição do HOMO, π*, de uma molécula
excitada com o LUMO, π*, da molécula no estado fundamental,
por exemplo, duas molécuas de etileno, uma no estado
fundamental e outra no estado excitado, a interação é ligante.

301
Em uma cicloadição, o sistema pi pode ser atacado de
duas maneiras distintas. Se o sistema pi é atacado na mesma
face, a reação é dita ser suprafacial. Se o sistema pi é
atacado em faces opostas, a reação é dita ser antarafacial
neste componente.
Nas reações de cicloadição [4 + 2], a estereoquímica
mostra que a reação é suprafacial. Na cicloadição térmica [2 +
2], pela simetria dos orbitais, pode ocorrer ataque suprafacial
relativamente a um componente e antarafacial ao outro. Neste
caso, o processo supra, antara é impossível por razões
geométricas.
Quando o anel ou o ciclo que vai ser formado for grande,
ambos os processos supra, supra e supra, antara são
geometricamente possíveis, a simetria de orbitais determina
não se a cicloadição ocorre, mas sim como ela ocorre, e são
determinadas pelas regras de Woodward-Hofmann para
cicloadição.

302
i + j Térmica Fotoquímica
4n Supra-antara Supra-supra Antara-supra Antara-antara
4n + 2 Supra-supra Supra-antara Antara-antara Antara-supra
As cicloadições são reversíveis. Estas ciclorreversões
(retroreação de Diels-Alder) seguem as mesmas regras de
simetria que as cicloadições, visto ocorrerem através do
mesmo estado de transição.
Rearranjos sigmatrópicos
São reações em que um substituinte com ligação σ
migra através de um sistema de elétrons π, pode ocorrer em
um alceno simples ou em um polieno. Os exemplos mais
simples envolvem a migração de uma ligação σ transportando
um átomo de hidrogênio.
Nas designações [1,3] e [1,5], o “3 e o 5” referem-se ao
número de ordem do átomo de carbono (terminal da migração)
para onde migra o grupo G. O número “1” não se refere so ao
ponto de partida da migração, apenas indica que tanto no
reagente como no produto da reação a ligação se efetua no
mesmo átomo do grupo migrante.
O rearranjo de Cope dos hexa-1,5-dienos, por exemplo,
é uma reação sigmatrópica [3,3], na qual ocorre uma mudança
na posição da ligação de G, onde aqui o próprio G é o
esqueleto π.

303
C
C
C
C
C
C
C
C
C
C
C
C
C
C
C
C
C
C
Hexa-1,5-dieno
Reaçãosigmatrópica [3,3]
1
2
3
1
2
3
No estado de transição de uma reação sigmatrópica, o
grupo migrante está ligado tanto ao ponto de partida da
migração como ao ponto de chegada, é a natureza deste
estado de transição das reações sigamtrópicas resultam da
sobreposição entre um orbital de um átomo ou radical (G) e um
orbital de um radical alílico (o esqueleto π).
No estado de transição, existe sobreposição entre o
HOMO de um componente o HOMO do outro, cada um deles
está ocupado por um só elétron, em conjunto eles fornecem um
par de elétrons.
O HOMO de um radical alílico depende do
número de carbonos no esqueleto π.
O grupo migrante passa de uma extremidade do radical alílico
para a outra e são os carbonos terminais que estão em causa,
e a simetria nestes carbonos terminais alterna quando se
passa de C3 para C5, C7, etc.. O HOMO do grupo migrante
depende da natureza do grupo.
Migração de hidrogênio
A migração de hidrogênio é o caso mais simples de
rearranjo sigmatrópico, e a estereoquímica desta transposição
pode ser suprafacial ou antarafacial.

304
No estado de transição, é necessária uma ligação de
três centros, o que implica na sobreposição entre um orbital s
do hidrogênio e lobos dos orbitais p dos dois carbonos
terminais. A natureza da transposição permitida, suprafacial ou
antarafacial, depende da simetria destes orbitais terminais, mas
a viabilidade de um rearranjo sigmatrópico não depende só da
simetria, mas também da geometria do sistema.
As transposições antara [1,3] e [1,5] devem ser
extremamente difícieis porque exigem uma forte torsão do
esqueleto π comprometendo a planaridade necessária à
deslocalização de elétrons.
As reações sigmatrópicas [1,3] e [1,5] são limitadas a
transposição supra. Uma transposição supra [1,3] de
hidrogênio é proibida por simetria, como o orbital s do
hidrogênio tem que sebrepor a lobos p de fase oposta, o
hidrogênio não pode ligar-se simultaneamente a ambos os
carbonos. A transposição supra [1,5] de hidrogênio é permitida
por simetria.

305
Em esqueletos π maiores, as transposições supra e
antara são possíveis do ponto de vista da geometria, e que a
estereoquimica depende simplesmente da simetria dos orbitais.
Uma transposição [1,7] de hidrogênio deve ser antara, e uma
transposição [1,9] de hidrogênio é supra. Para uma reação
fotoquímica as previsões são opostas as anteriormente
escritas.
Migração de carbono
A migração de carbono envolve duas espécies possíveis
de ligações ao grupo migrante. Uma delas é semelhante à que
acabamos de descrever para a migração do hidrogênio, ligação
de ambas as extremidades do esqueleto π ao mesmo lobo do
carbono, consoante com a simetria do esqueleto π, assim a
migração permitida por simetria pode ser suprafacial ou
antarafacial.
No caso do carbono, a estereoquimica no grupo
migrante ao formar a ligação através do mesmo lobo no
carbono significa fixação à mesma face do átomo, envolvendo
retenção de configuração no grupo migrante.
A outra espécie é a de se formar ligações com as duas
extremidades do esqueleto π através de diferentes lobos de um
orbital p. Estes lobos estão em faces opostas do carbono,
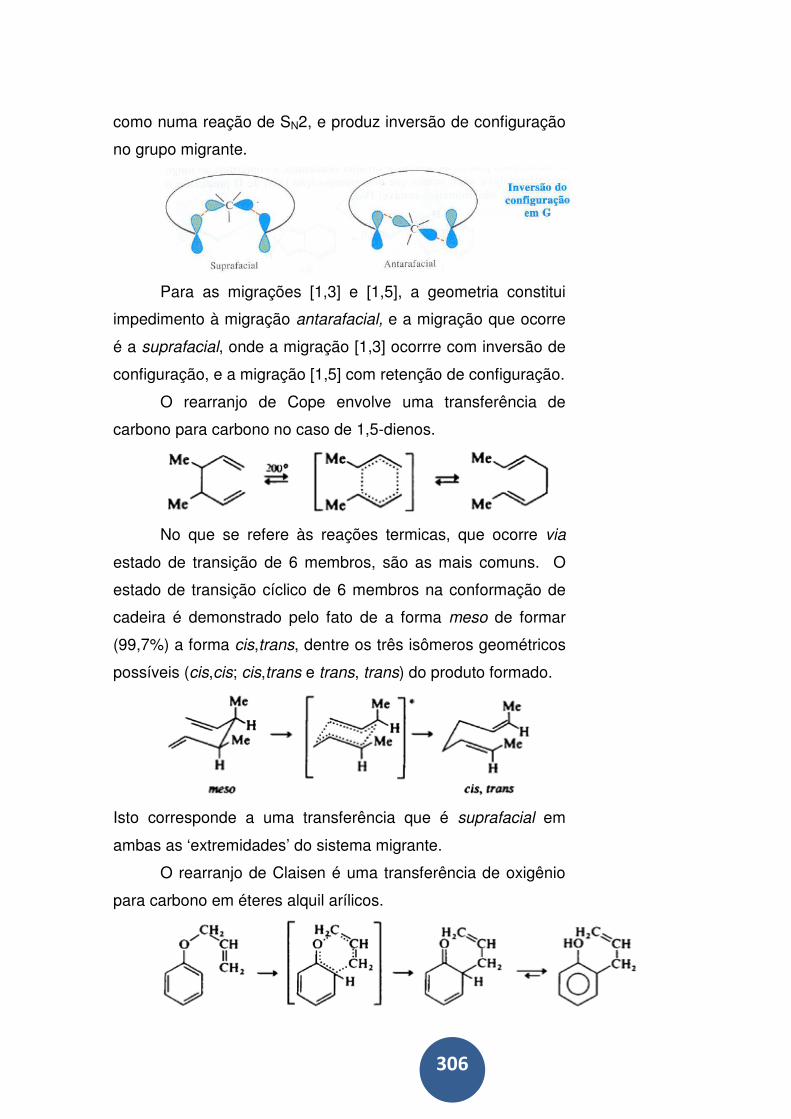
306
como numa reação de SN2, e produz inversão de configuração
no grupo migrante.
Para as migrações [1,3] e [1,5], a geometria constitui
impedimento à migração antarafacial, e a migração que ocorre
é a suprafacial, onde a migração [1,3] ocorrre com inversão de
configuração, e a migração [1,5] com retenção de configuração.
O rearranjo de Cope envolve uma transferência de
carbono para carbono no caso de 1,5-dienos.
No que se refere às reações termicas, que ocorre via
estado de transição de 6 membros, são as mais comuns. O
estado de transição cíclico de 6 membros na conformação de
cadeira é demonstrado pelo fato de a forma meso de formar
(99,7%) a forma cis,trans, dentre os três isômeros geométricos
possíveis (cis,cis; cis,trans e trans, trans) do produto formado.
Isto corresponde a uma transferência que é suprafacial em
ambas as ‘extremidades’ do sistema migrante.
O rearranjo de Claisen é uma transferência de oxigênio
para carbono em éteres alquil arílicos.

307
O rearranjo de Claisen é estritamente intramolecular e
apresenta um elevado valor negativo para ∆S, característico do
grau de ordenação requerido por um estado de transição
cíclico. Este último requisito é também evidenciado por
marcação com 14C, que indica que a posição do átomo 14C no
grupo alilo é ‘invertida’ durante a migração do reagente e o
produto.
A dienona intermediária, além de enolizar ao fenol, é
capaz de sofrer, ela própria, um rearranjo de Cope para dar
uma segunda dienona, cujo enol é o fenol p-substituído. A
enolização normalmente predomina, mas quando o éter alquil
arílicos tem substituintes em posição orto, a ‘o-enolização’ não
pode ocorrer e então só é obtido o p-fenol. Este produto não é
formado por migração direta do grupo alilo, mas sim por duas
transferências sucessivas, é sugerido pela “dupla inversão” da
posição do 14C que se observa no grupo alilo.
Finalmente, deve acentuar-se que quando, em qualquer
das reações eletrocílcicas, cicloadições ou rearranjos
sigmatrópicos, uma reação é como simetricamente proibida, se
aplica apenas no mecanismo concertado, pode acontecer

308
ainda que esteja disponível em um mecanismo não concertado,
energeticamente possível, envolvendo intermediários
zwiteriônicos ou birradicalares. Igualmente, a afirmação de que
uma reação é simetricamente permitida não garante
necessariamente que na prática ocorra facilmente; a geometria
requerida no estado de transição pode bem ser inibida pela
dimensão do anel, pela presença de determinados
substituintes, ou por outros motivos.
Nas reações eletrocíclicas, um anel é sempre quebrado
ou formado. Anéis podem, no curso da reação, ser formados
por cicloadição, mas a diferença com as reações eletrocíclicas
é que apenas uma ligação sigma nova é formada (ou
quebrada) entre as extremidades de um sistema único π
conjugado. Em uma cicloadição, duas ligações σ novas são
formadas (ou quebradas), em um rearranjo sigmatrópico uma
forma σ, enquanto a outra é quebrada.
O tipo de reaçôes periciclicas é caracterizada pelo
número de ligações σ formada ou quebrada.
Cicloadições Rearranjos sigmatrópico
Reações eletrocíclicas
CH2
CH2
CH2
CH2
CH2
CH2
CH2
CH2
CH2
CH2
Duas novas ligações σ são formadas
Uma nova ligação σ é formada como a outra quebrada
Uma nova ligação σ é formada
CH2
CH2
CH2
CH2
CH
CH
Duas ligações σ são quebradas
Uma ligação σ é quebrada
∆σ = ±2 ∆σ = 0 ∆σ = ±1
Uma das reações elecrocíclicas mais simples ocorre
quando hexatrieno é aquecido a 500 ºC.

309
Numa reação pericíclica os elétrons giram em um anel
(você pode também girar as setas indo para o outro lado),
porque na reação eletrocíclica uma nova ligação σ é formada
finalizando um sistema π. A reação ocorre porque a ligação
sigma que é formada é mais forte que a ligação π que é
quebrada. O oposto é verdadeiro para a reação eletrocíclica
mostrada, limitando-se na tensão do anel de quatro membros,
significando que o inverso da reação, abertura do anel, é a
preferida.

310
Referências Bibliográficas
1. CLAYDEN, J.; GREEVES, N.; WARREN, S.;
WOTHERS, P. Organic Chemistry. New York: Oxford
University Press. 2001.
2. McMURRAY, J. Organic Chemistry. 6. ed. New York:
Brooks/Cole, 2005.
3. MORRISON, R. T.; BOYD, R. N. Química Orgânica. 14.
ed. Lisboa: Fundação Calouste Goulbenkian, 2005.
4. SYKES, P. A Guidebook to Mechanism in Organic
Chemistry, 6th. ed., Longman Scientific & Technical, Londres,
1991.
5. SOLOMONS, T. W. G. Organic Chemistry. 5. ed. New
York: John Wiley & Sons , 1992.
6. VOLLHARDT, K. P. C.; SCHORE N. E. Química
Orgânica: estrutura e função. 4 ed. Porto Alegre: Bookman,
2004
Revistas e periódicos
7. Boletim da Sociedade Brasileira de Química, SBQ – Instituto
de Química da USP. São Paulo.
8. Revista de Aditivos & Ingredientes – Insumos para Alimentos
e Bebidas São Paulo, Editora Insumo Ltda.
9. Ciência Hoje – Revista de Divulgação Científica da
Sociedade Brasileira para o Progresso da Ciência (SBPC). Rio
de Janeiro.
10. Química Nova na Escola - Revista da Sociedade Brasileira
de Química, SBQ, com uma periodicidade trimestral, propõe-se
a subsidiar o trabalho, a formação e a atualização da
comunidade do Ensino de Química brasileiro – Instituto de
Química da USP. São Paulo.

311
SITES PARA PESQUISA NA WEB
http://www.chemfinder.com/>
http://www.hazard.com/msds/>
http://webbook.nist.gov/>
http://www.epa.gov/>
http://www.capes.gov.br/portal de periodicos

312
Sobre os autores:
A Professora doutora Antônia Maria das Graças Lopes
Citó conclui o doutorado em Química pela Universidade
Estadual de Campinas em 1994. Atualmente é professora
associado II da Universidade Federal do Piauí. Publicou 17
artigos em periódicos especializados e 160 trabalhos em anais
de eventos científicos. Orientou 4 dissertações de mestrado,
além de ter orientado 29 projetos de iniciação científica na área
de Química/Farmácia. Recebeu 7 prêmios e/ou homenagens.
Entre 1997 e 2008 participou de 7 projetos de pesquisa.
Atualmente coordenada 1 projeto de pesquisa. Atua na área de
Química, com ênfase em Química de Produtos Naturais. Em
suas atividades profissionais interagiu com 110 colaboradores
em co-autorias de trabahlos científicos. Trabalha com ênfase
em: Óleos Essenciais, Própolis, Mel, Pólen, Atividade
Microbiana, Triterpenóides, Anarcadium Occidentale e LCC
(Líquido da Castanha de Caju).
O professor doutor Davi da Silva conclui a graduação
em Bacharel e Licenciatura em Química pela Fundação
Universidade Regional de Blumenau (FURB, 1999 e 2000),
mestrado em Química pela Universidade Federal de Santa
Catarina (UFSC, 2002) e doutorado em Química pela
Universidade Federal de Santa Catarina (UFSC, 2006). Desde
fevereiro de 2008, está desenvolvendo projeto na área de
energia alternativa (biodiesel) na UFPI, com bolsa DCR -
CNPq/FAPEPI/UFPI. Atua nas áreas de Química dos Produtos
Naturais, Polímero e Energia Renovável. Orienta e/ou Co-
orienta alunos Iniciação Científica (UFPI) e Co-orienta dois
alunos do Programa de Doutorado da Rede Nordeste de
Biotecnologia (RENORBIO).


















