
Área da Genética Médica em grande ascensão - asic.pt · Cardiogenética 5.º Curso “Genética...
43
+ Cardiogenética 5.º Curso “Genética Médica: Conceitos Elementares e Aplicações Frequentes” Hospital Pediátrico Carmona da Mota 18 a 20 de Novembro de 2013 Maria Margarida Venâncio [email protected] a t t c a g a t g g c t t s e r v i ç o a a t a g g e n é t i c a g g a g m é d i c a t t c c a c a a t c g g t a t g t c h o s p i t a l t c c t p e d i á t r i c o c t c o i m b r a t a c c t t a t g g c a c c t g a t t c a g a t g g c t t s e r v i ç o a a t a g g e n é t i c a g g a g m é d i c a t t c c a c a a t c g g t a t g t c h o s p i t a l t c c t p e d i á t r i c o c t c o i m b r a t a c c t t a t g g c a c c t g
Transcript of Área da Genética Médica em grande ascensão - asic.pt · Cardiogenética 5.º Curso “Genética...

+
Cardiogenética 5.º Curso “Genética Médica:
Conceitos Elementares e Aplicações Frequentes”
Hospital Pediátrico Carmona da Mota 18 a 20 de Novembro de 2013
Maria Margarida Venâncio [email protected]
a t t c a g a t g g c t
t ss ee rr vv ii çç oo a a t a
g gg ee nn éé tt ii cc aa g g a
g mm éé dd ii cc aa t t c c a
c a a t c g g t a t g t
c hh oo ss pp ii tt aa ll t c c
t pp ee dd ii áá tt rr ii cc oo c
t cc oo ii mm bb rr aa t a c c
t t a t g g c a c c t g
a t t c a g a t g g c t
t ss ee rr vv ii çç oo a a t a
g gg ee nn éé tt ii cc aa g g a
g mm éé dd ii cc aa t t c c a
c a a t c g g t a t g t
c hh oo ss pp ii tt aa ll t c c
t pp ee dd ii áá tt rr ii cc oo c
t cc oo ii mm bb rr aa t a c c
t t a t g g c a c c t g

+Cardiogenética
n Área da Genética Médica em grande ascensão
Doenças Cardiovasculares • 1:250 doença monogénica • No Reino Unido
• 100 000 casos/ano MS cardíaca • 10% doença genética
+Cardiogenética
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
CE: Swati; HCO/438; Total nos of Pages: 6;
HCO 438
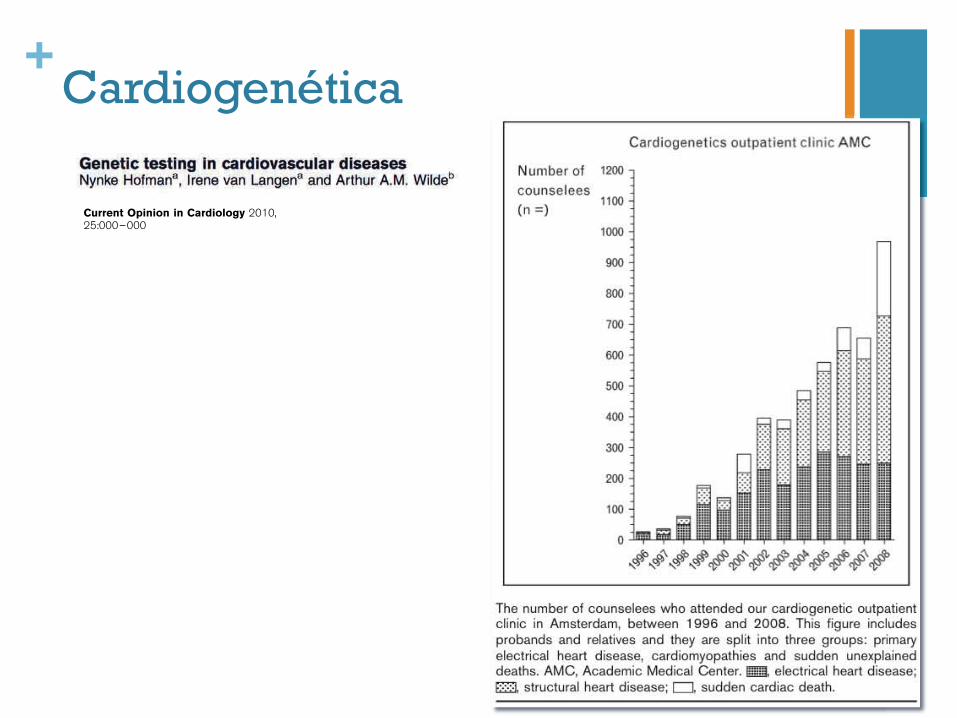
Genetic testing in cardiovascular diseasesNynke Hofmana, Irene van Langena and Arthur A.M. Wildeb
IntroductionIn this article, we will briefly review the (im)possibilities,(dis)advantages and consequences of genetic testing incardiovascular diseases, restricted to inherited arrhyth-mias and cardiomyopathies. We will review and discussrecent developments and new insights, and we will giveour opinion about the dos and don’ts in this era ofmolecular testing, based on literature and our experiencein this field.
General aspects of genetic family testingWith the identification of the molecular basis of animportant part of the inherited cardiovascular diseasesduring the last decades, a lot of pathophysiologicaldetails about the different disease entities have beenunraveled. Increased knowledge of the genetic basis ofthe disease sometimes leads to gene-specific treatmentin known or suspected disease [i.e., the long QT syn-dromes (LQTSs)] or confirms suspected diagnoses,which also can be important when therapeutic optionsare investigated.
Molecular genetic testing usually starts with the proband,the first person in the family usually with symptoms of aninherited disorder. Age, symptoms, triggers of symptoms,results of cardiologic investigation (ECG, echocardiogra-phy, Holter monitoring, exercise testing, provocation
tests or MRI) and medical reports from possibly affectedrelatives can target initial molecular testing. Hence, fromthe start, intensive collaboration between cardiologists,clinical geneticists or genetic counselors and moleculargeneticists is warranted for patient care as well as forresearch purposes. When a pathogenic and thereforecausal mutation is identified in the proband, predictivetesting of family members can be offered. This is pre-ferably performed in a joint cardiogenetics outpatientclinic. Although this seems to be straightforward, thereare a lot of aspects to discuss and explore: the character-istics of the mutation, genotype–phenotype correlations,the indication for further screening when a secondmutation is suspected and, of course, all patient-relatedaspects such as the psychosocial impact of the genetic testresults and insurance-related consequences. After dis-cussing our own experience and results of almost 15 yearsof cardiogenetic counseling in the Academic MedicalCenter (AMC) in Amsterdam, as an example of clinicalpractice based on scientific research in cardiogenetics, wewill briefly discuss the aspects of the inherited cardio-myopathies and the primary inherited arrhythmias,respectively.
Genetic counseling and testing incardiovascular disease in the NetherlandsAll university hospitals in the Netherlands have a clini-cal genetics department, in which a multidisciplinary
aDepartment of Clinical Genetics and bDepartment ofCardiology, Academic Medical Center, Amsterdam, TheNetherlands
Correspondence to Professor Dr Arthur A.M. Wilde,Cardiologist, Academic Medical Center, Meibergdreef9, 1105 AZ Amsterdam, NetherlandsTel: +31 20 5669111; fax: +31 20 6971385;e-mail: [email protected]
Current Opinion in Cardiology 2010,25:000–000
Purpose of reviewTo review the current state and different aspects, including the yield, of geneticcounseling and genetic testing in inherited heart disease.Recent findingsThe number of counselees is growing rapidly all over the world, and the first studiesabout patients’ perspectives and follow-up have been published. Progress has beenmade by gene-specific studies on long QT syndrome to judge the relevance of detectedmutations in the specific domains.SummaryWith the increasing identification of associated genes and available techniques inmolecular testing of the inherited heart diseases, the diagnostic yield of mutationanalysis is growing rapidly. To determine the relevance of all these mutations, ongoingresearch is needed. Furthermore, the process of genetic counseling can be optimizedand extended with cascade screening, which leads to identifying patients at risk andtimely treatment.
Keywordscardiogenetics, genetic counseling, genetic testing
Curr Opin Cardiol 25:000–000! 2010 Wolters Kluwer Health | Lippincott Williams & Wilkins0268-4705
0268-4705 ! 2010 Wolters Kluwer Health | Lippincott Williams & Wilkins DOI:10.1097/HCO.0b013e3283374d69
+
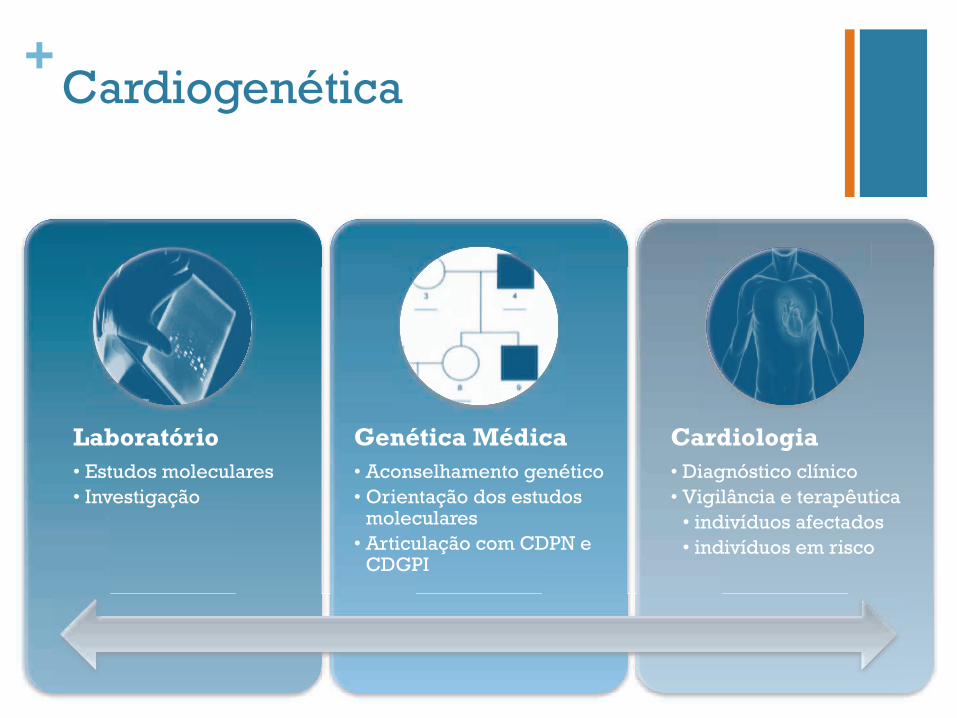
Laboratório • Estudos moleculares • Investigação
Genética Médica • Aconselhamento genético • Orientação dos estudos
moleculares • Articulação com CDPN e
CDGPI
Cardiologia • Diagnóstico clínico • Vigilância e terapêutica
• indivíduos afectados • indivíduos em risco
Cardiogenética

+Cardiogenética
n Etiologia genética
n Tipo de hereditariedade (avaliação do risco de recorrência)
n Clínica e prognóstico da doença
n Possibilidades e limitações do estudo molecular
n Opções reprodutivas (DPN ou DGPI)
n Indicação para estudo dos familiares
n Orientação para associações de doentes
n Fontes de informação médica sobre a doença

+Estudos moleculares em Cardiogenética
+Por quem começar o estudo molecular?

+Resultados possíveis
Identificação de mutação
Ausência de identificação de mutação
Identificação de variação sequência de significado desconhecido

+Quando se identifica uma mutação - Confirmação molecular do diagnóstico
Estudos familiares
Resultado “positivo” • Confirmação molecular • Risco de recorrência • Estudos familiares • Protocolo de vigilância
Resultado “negativo” • Sem risco de doença • Sem risco de recorrência • Sem necessidade de
vigilância específica

+Na ausência de confirmação molecular do diagnóstico
n Não exclui a doença n Heterogeneidade genética
n Limitações da técnica utilizada n Novas metodologias de estudo
n Impossibilita a realização de estudos familiares n No entanto, aos familiares em 1.º grau deve ser
oferecida vigilância cardíaca

+Quando se identifica variação sequência de significado desconhecido
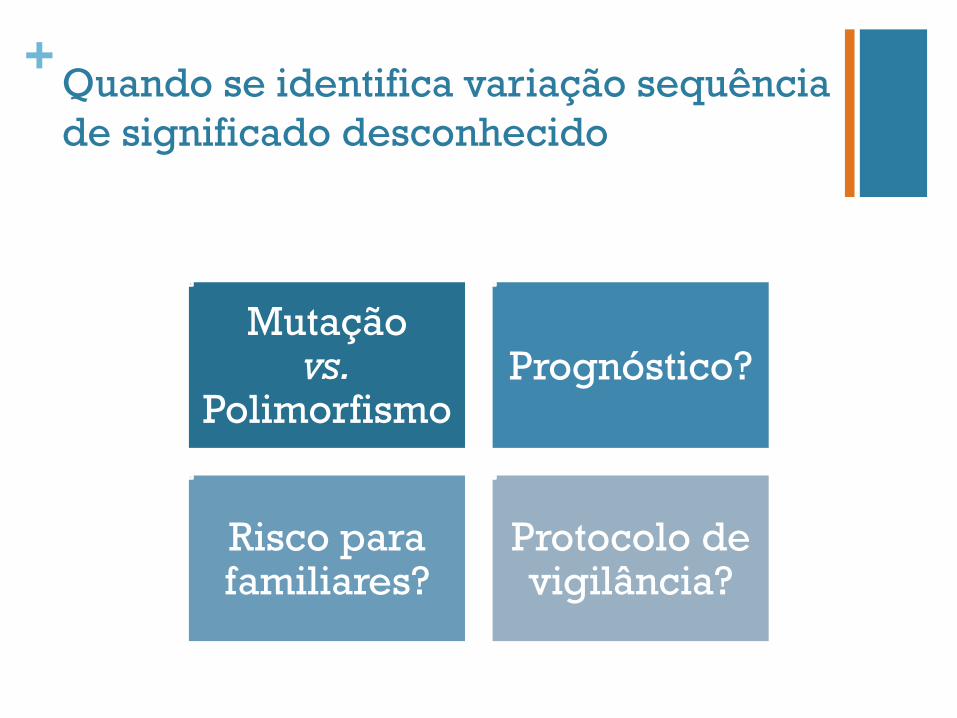
+Quando se identifica variação sequência de significado desconhecido
Mutação vs.
Polimorfismo Prognóstico?
Risco para familiares?
Protocolo de vigilância?
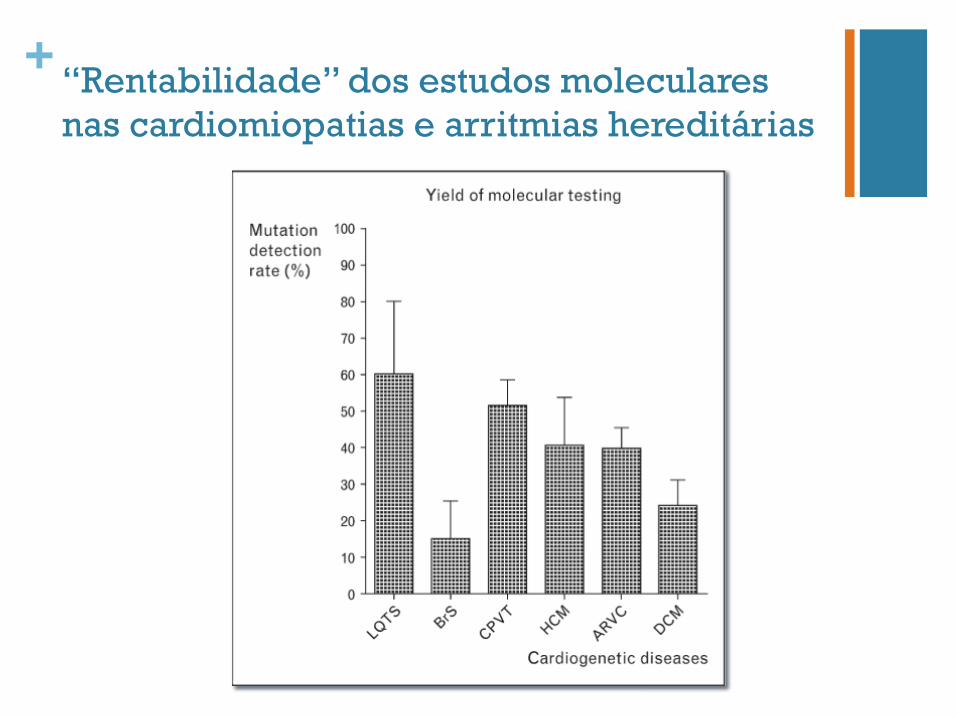
+ “Rentabilidade” dos estudos moleculares nas cardiomiopatias e arritmias hereditárias

+Cardiomiopatias hereditárias - Cardiomiopatia hipertrófica - Cardiomiopatia dilatada - Cardiomiopatia arritmogénica do ventrículo direito
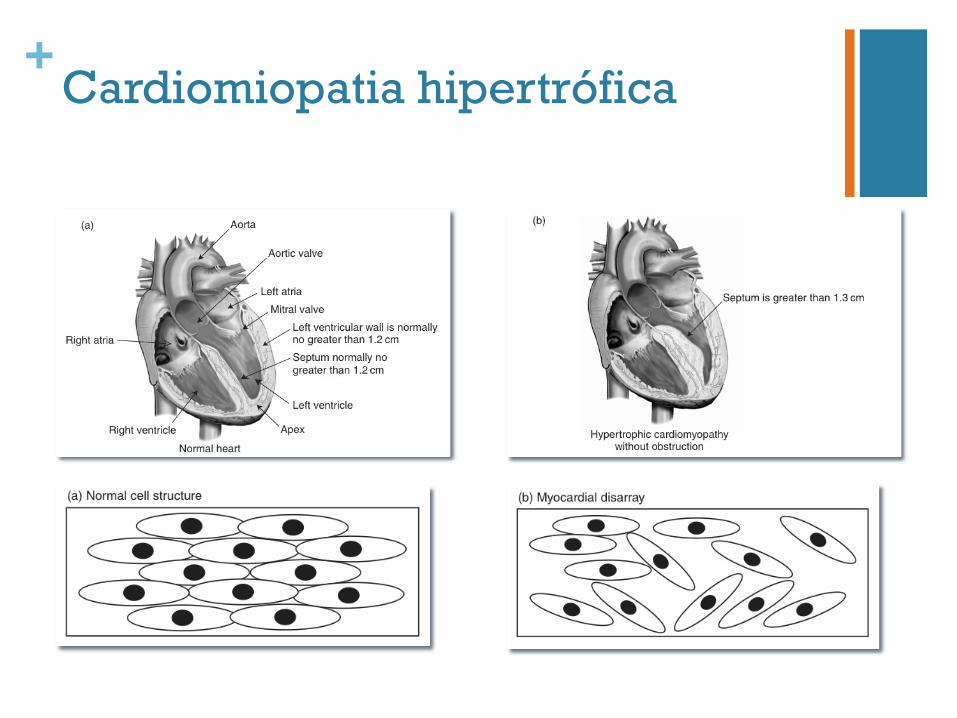
+Cardiomiopatia hipertrófica

+Cardiomiopatia hipertrófica
n Frequência 1:500-1 000
n Diagnóstico clínico, electro e ecocardiográfico n Morte súbita – 1º “sinal”
n Hereditariedade autossómica dominante
n Heterogeneidade genética
n Penetrância
n Dependente da idade
n Expressividade variável
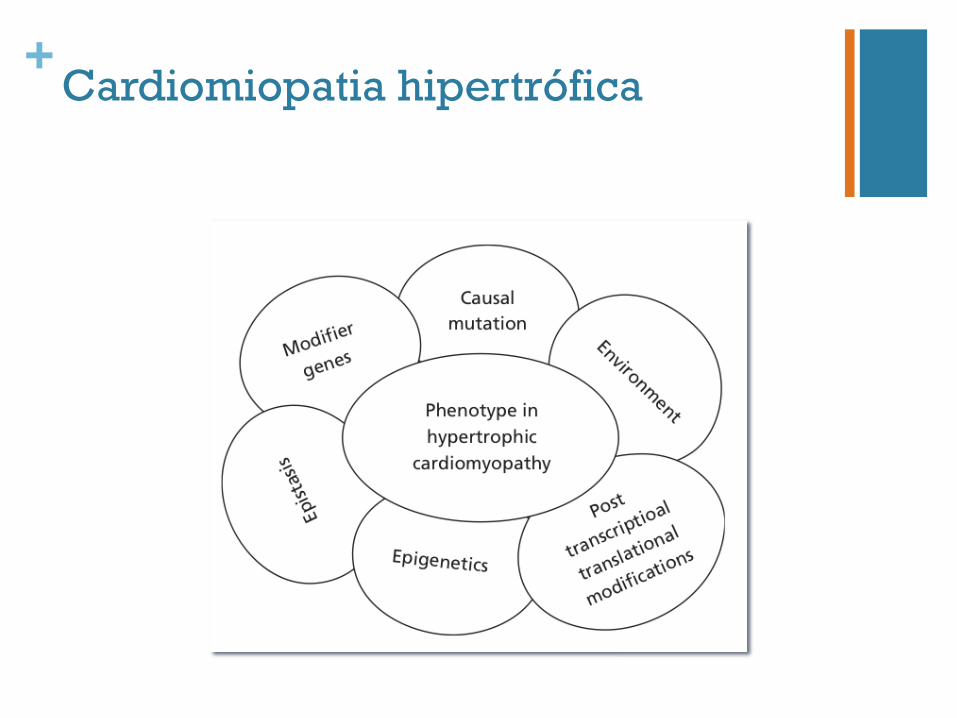
+Cardiomiopatia hipertrófica
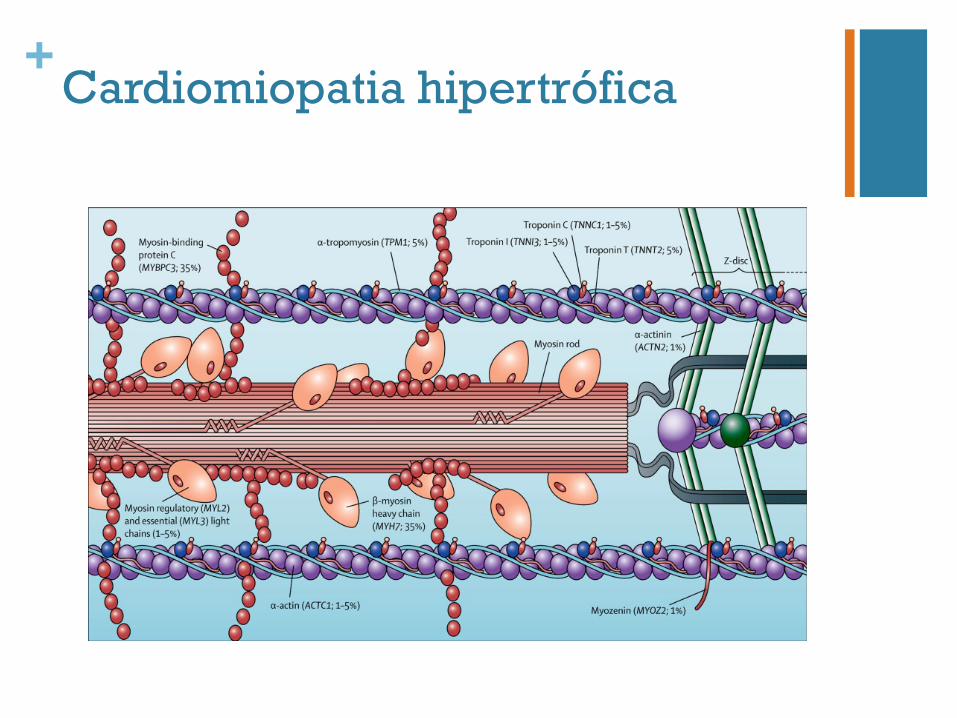
+Cardiomiopatia hipertrófica
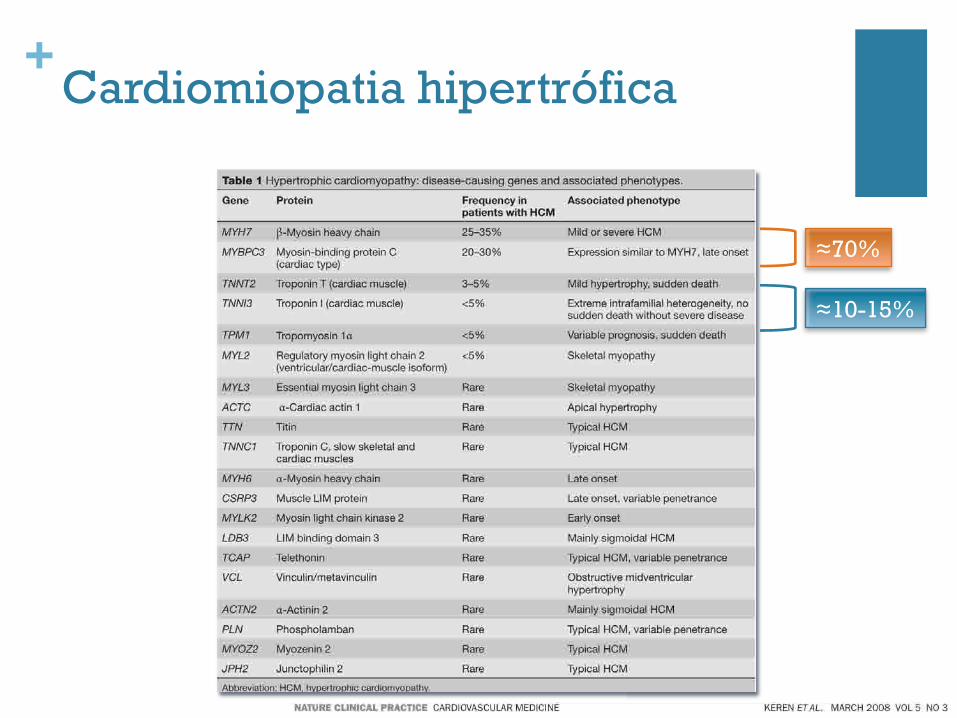
+Cardiomiopatia hipertrófica
≈70%
≈10-15%
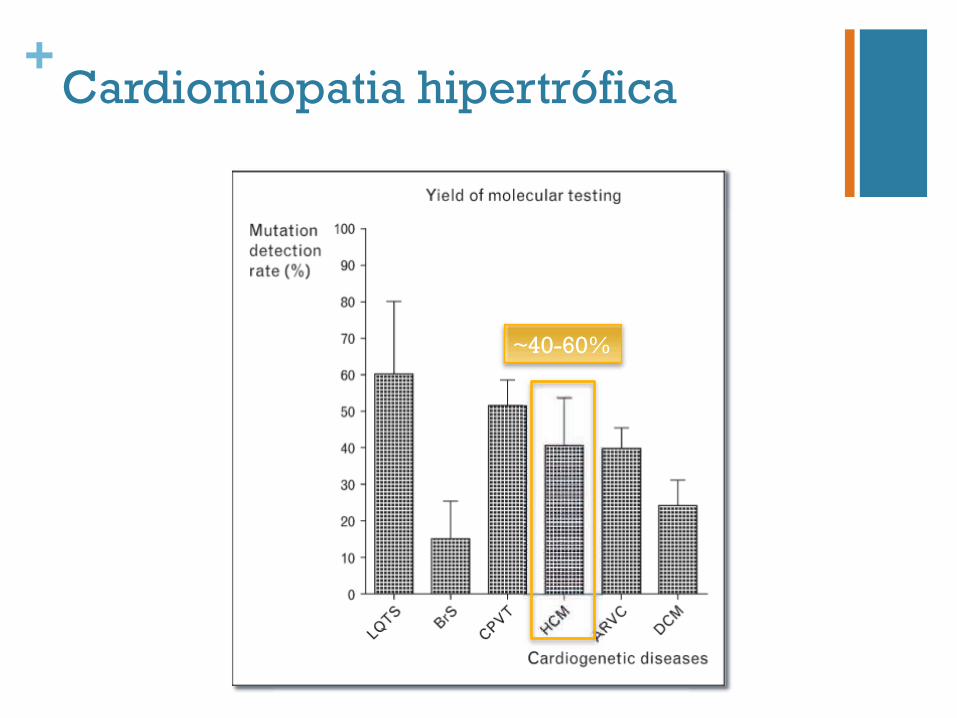
+Cardiomiopatia hipertrófica
~40-60%

+Cardiomiopatia hipertrófica
n Correlação genótipo-fenótipo n Papel limitado na avaliação do prognóstico e no acompanhamento
n 5% (4 - 15%) dos doentes n Duplos Heterozigotos
n Heterozigotos Compostos
n Homozigotos
Mutação A Mutação A
Mutação A Mutação B
Mutação
Mutação

+Cardiomiopatia hipertrófica
• Algumas mutações (R430Q, R453C, R719W) estão associadas a um fenótipo mais grave
• Outras mutações estão associadas a uma esperança média de vida praticamente normal
MYH7
• Início mais tardio, menor hipertrofia, menor penetrância e melhor prognóstico
MYBPC3
• Prognóstico mais benigno e menor taxa de mortalidade MYL3
• Hipertrofia ligeira mas maior proporção de arritmias e de MS TNNT2
• Fenótipo heterogéneo de gravidade intermédia TPM1
• Início precoce dos sintomas, hipertrofia marcada e risco elevado de arritmias
MYOZ2
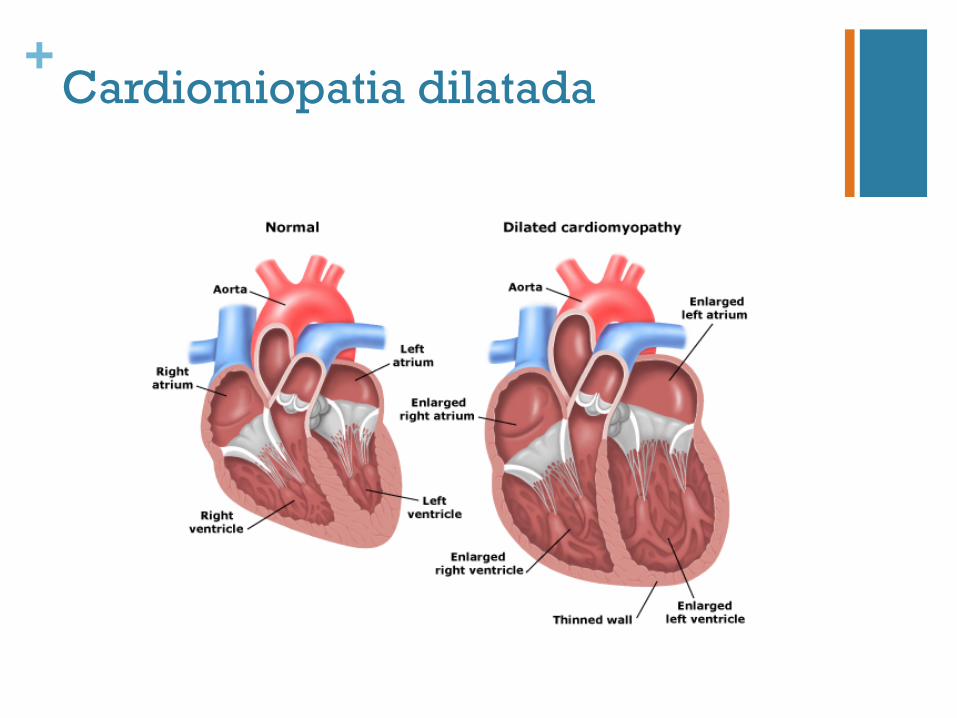
+Cardiomiopatia dilatada

+Cardiomiopatia dilatada
n Doença clínica e geneticamente heterogénea
n Frequência desconhecida n Japão - 14,0/100.000
n EUA – 36,4/100.000
n Factores genéticos - 30-50%
n Heterogeneidade genética
n Penetrância incompleta
n Expressividade variável
AD ≥60%
AR ≥15%
XL ≥10%
Mito.
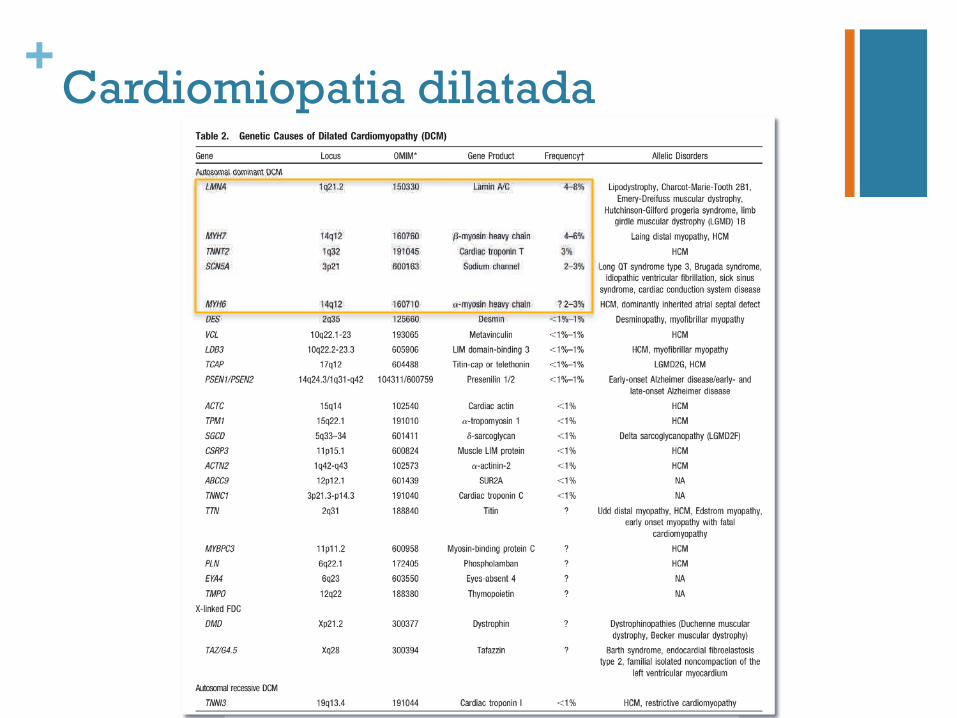
+Cardiomiopatia dilatada
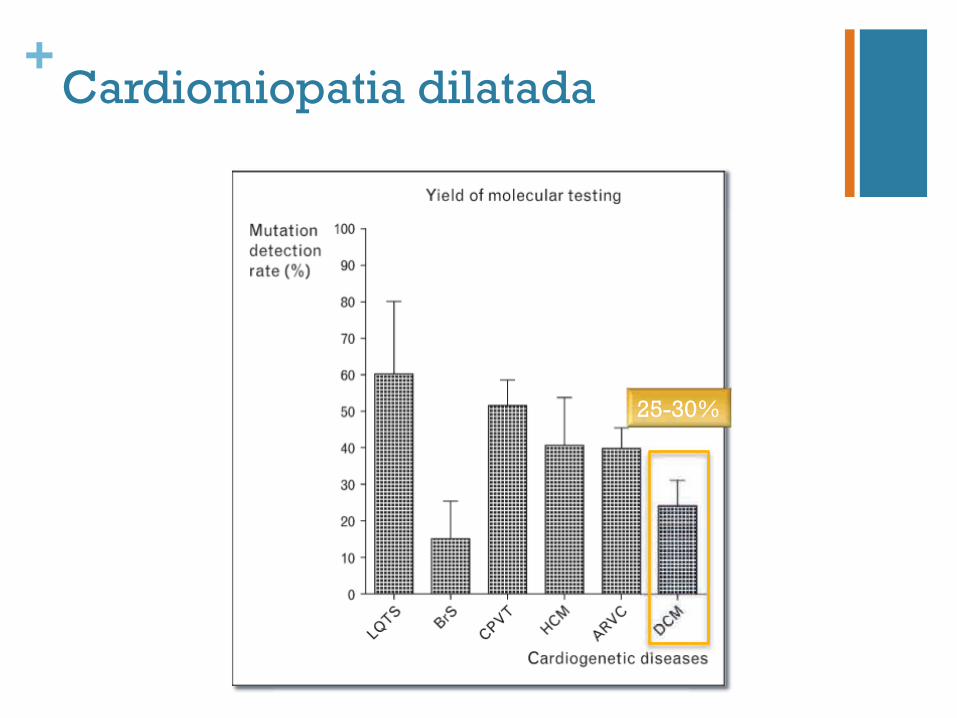
+Cardiomiopatia dilatada
25-30%
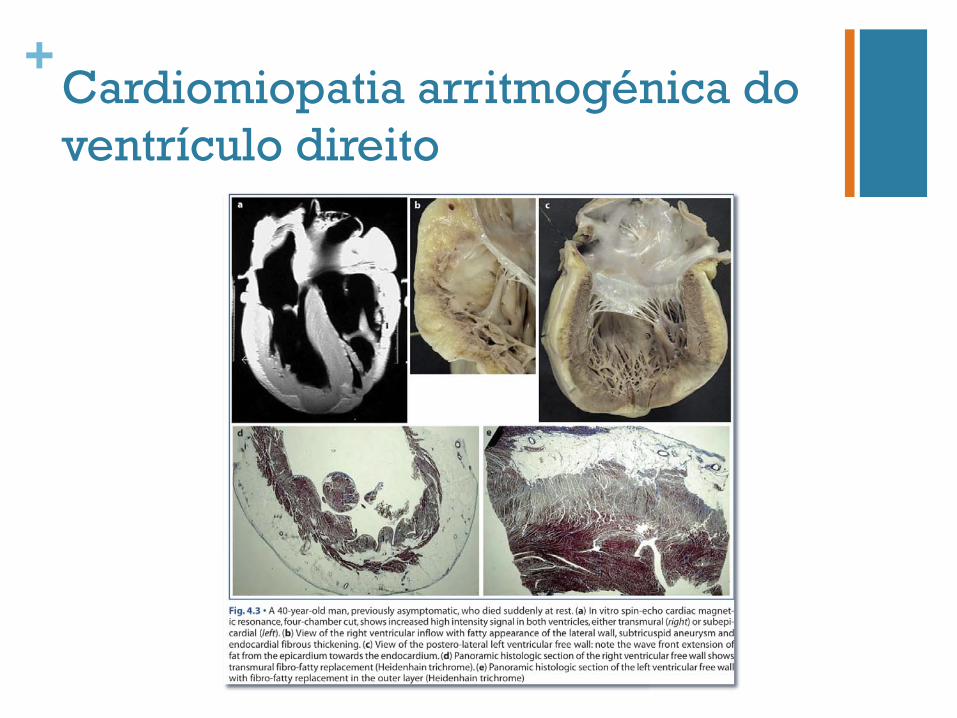
+Cardiomiopatia arritmogénica do ventrículo direito

+Cardiomiopatia arritmogénica do ventrículo direito
n Frequência: 1: 2.500-5.000 n >50% dos casos são familiares
n Tipo de hereditariedade mais frequente: AD
n Penetrância incompleta
n Expressividade variável
n Percentagem significativa de duplos heterozigotos
n Taxa de mortalidade: 2-4%/ano

+Cardiomiopatia arritmogénica do ventrículo direito
n Heterogeneidade genética
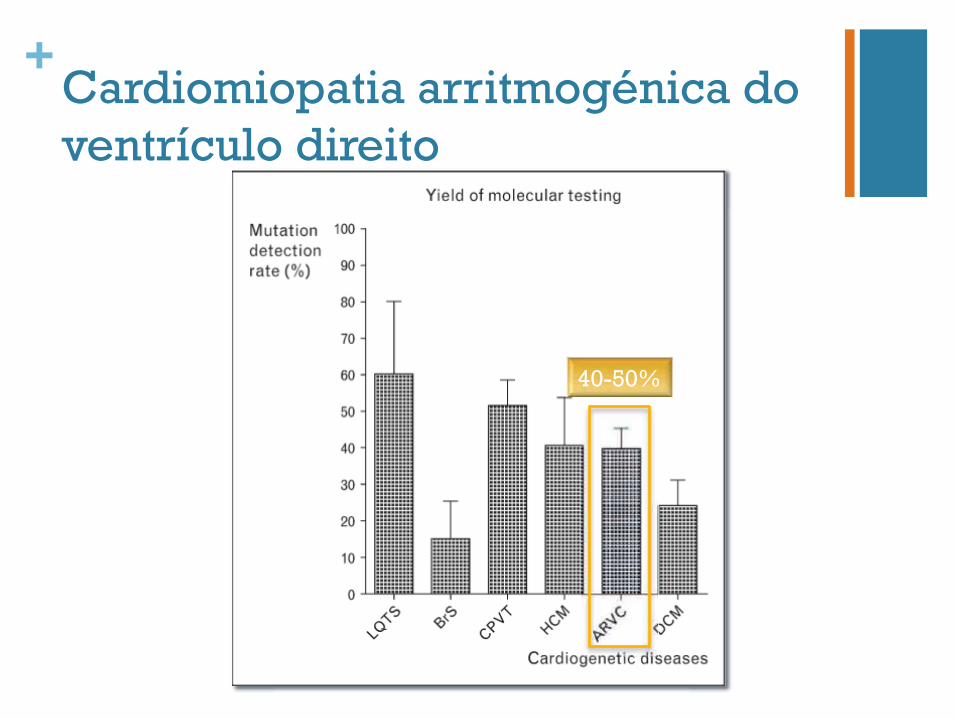
+Cardiomiopatia arritmogénica do ventrículo direito
40-50%

+Arritmias hereditárias Síndrome do QT longo Síndrome de Brugada Taquicardia Ventricular Polimórfica Catecolaminérgica

+Síndrome do QT longo
n Frequência: 1:5.000-6.000
n Autossómica dominante – Síndrome Romano-Ward n AR – Síndrome de Jervell e Lange-Nielsen
n doença cardíaca grave + surdez neuro-sensorial
n Risco de morte súbita: 4%
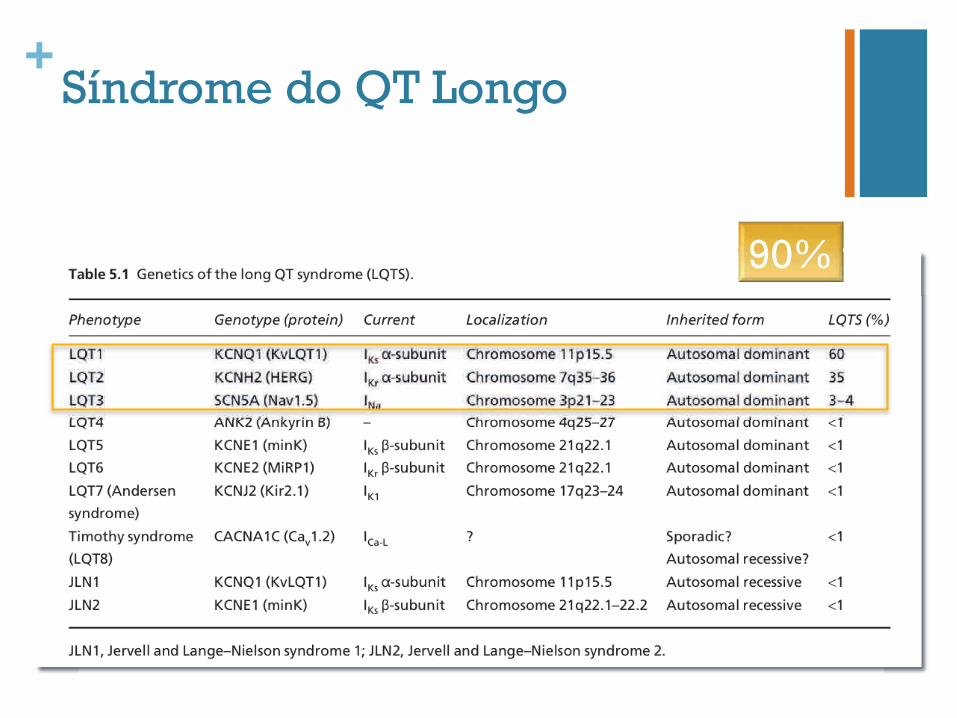
+Síndrome do QT Longo
90%
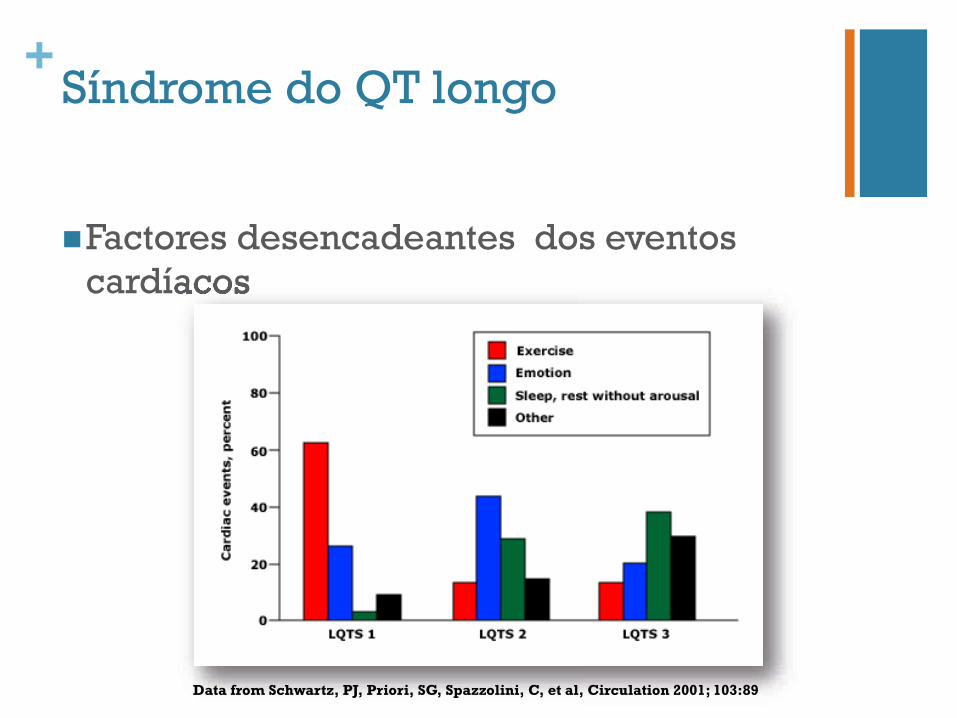
+Síndrome do QT longo
n Factores desencadeantes dos eventos cardíacos cardíacos
Data from Schwartz, PJ, Priori, SG, Spazzolini, C, et al, Circulation 2001; 103:89
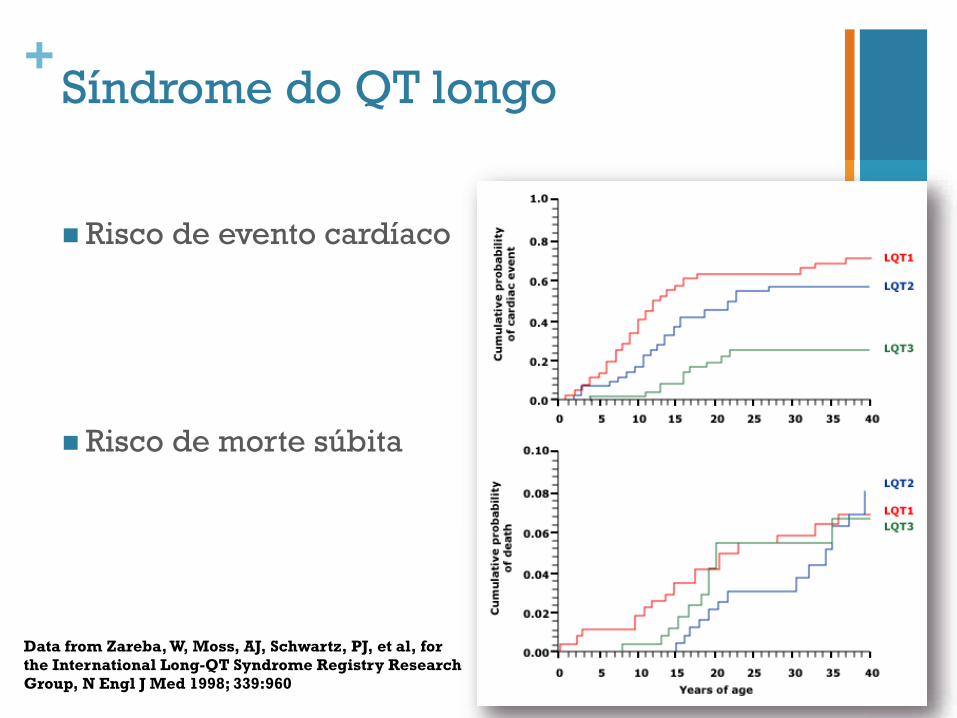
+Síndrome do QT longo
n Risco de evento cardíaco
n Risco de morte súbita
Data from Zareba, W, Moss, AJ, Schwartz, PJ, et al, for the International Long-QT Syndrome Registry Research Group, N Engl J Med 1998; 339:960

+Síndrome do QT longo
n Terapêutica farmacológica
LQT1
Beta-bloqueantes
(81%)
LQT2
Beta-bloqueantes
(59%)
LQT3
Beta-bloqueantes
Pouco eficazes
LQT4-8
Beta-bloqueantes
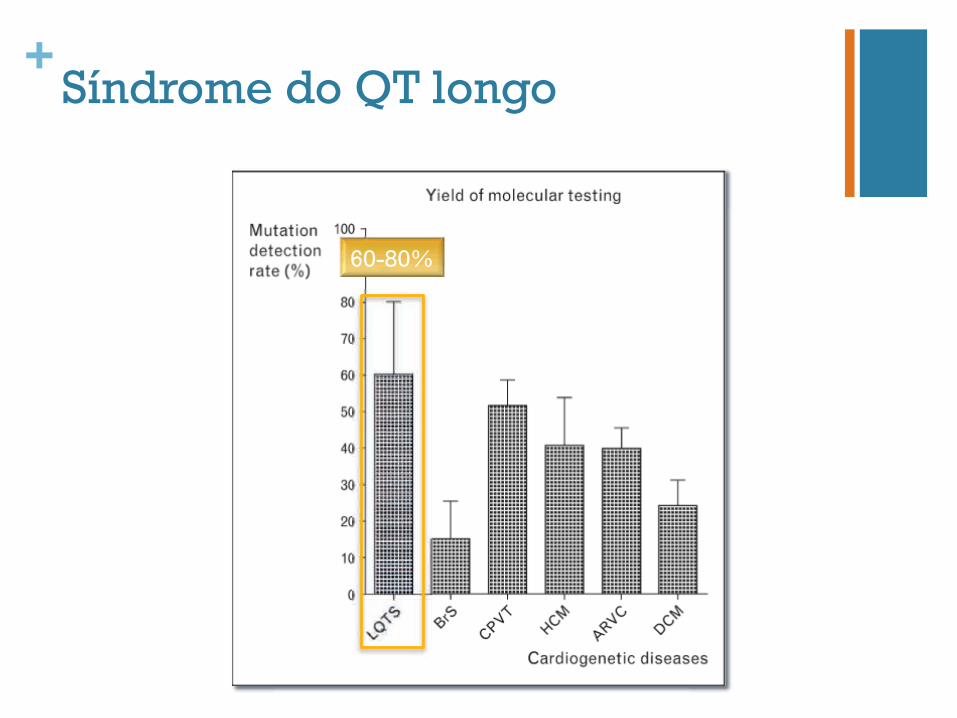
+Síndrome do QT longo
60-80%

+Síndrome de Brugada
n Frequência – 5:10 000
n M:F – 8:1
n Hereditariedade autossómica dominante
n Penetrância de ~30%
n Expressividade variável
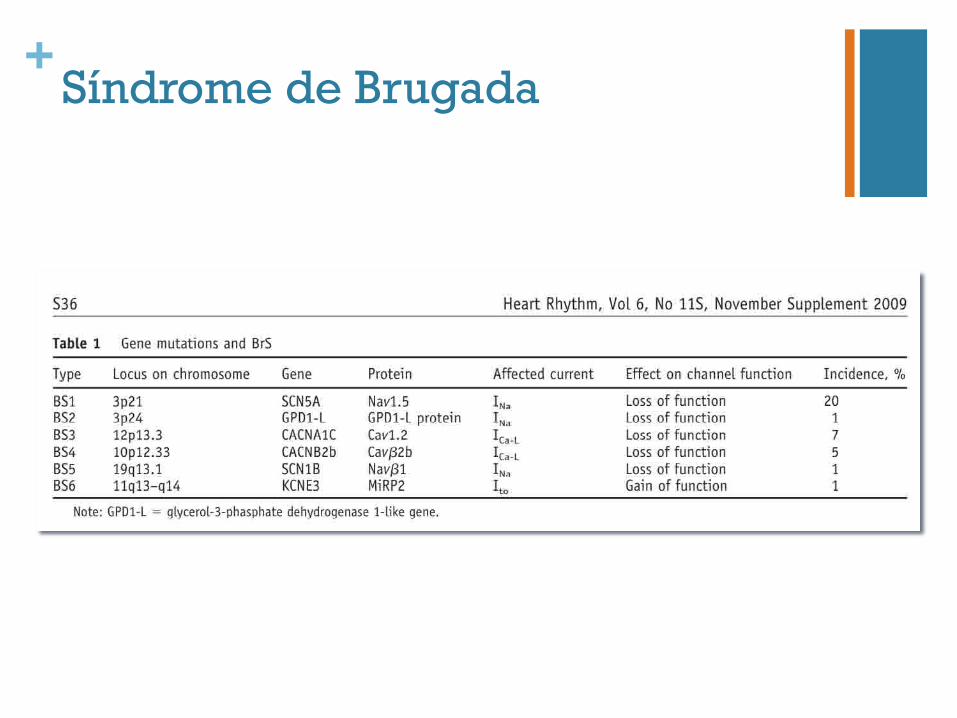
+Síndrome de Brugada
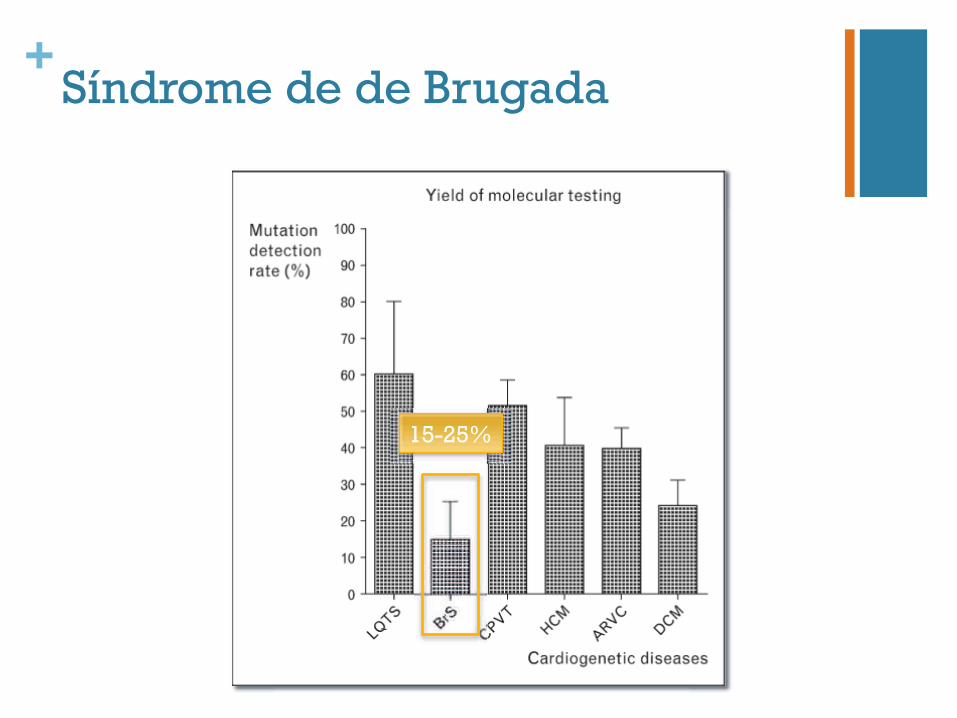
+Síndrome de de Brugada
15-25%

+Taquicardia Ventricular Polimórfica Catecolaminérgica
n Episódios de síncope no contexto de exercício físico ou emoção aguda
n Prevalência: 1:7.000-10.000
n Idade média de diagnóstico: 7-14 anos
n Taxa de mortalidade aos 40 anos: 30-50%
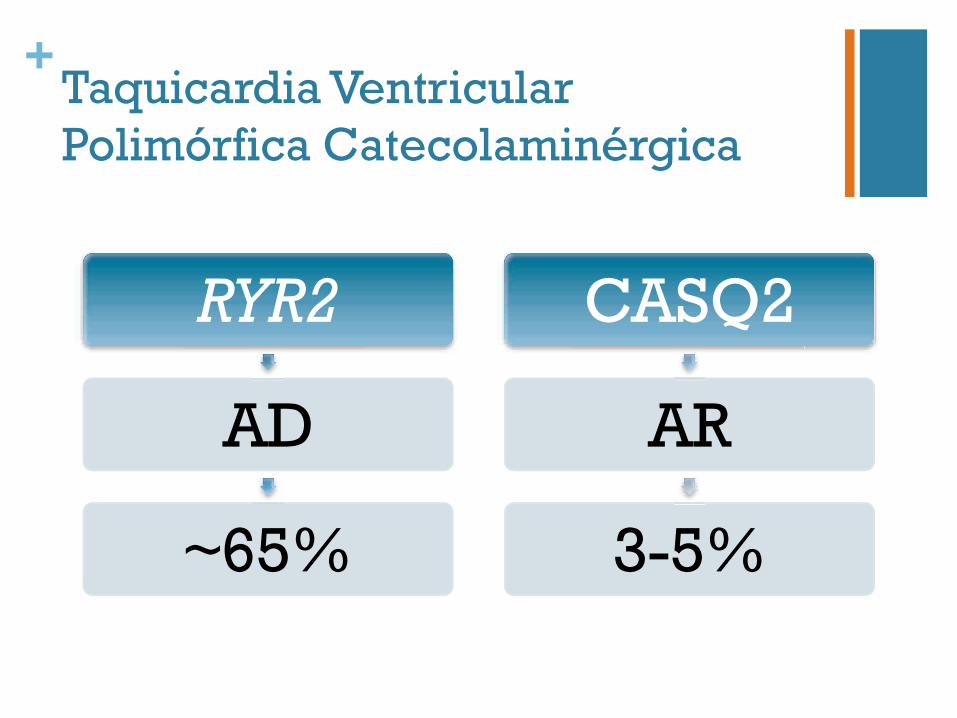
+Taquicardia Ventricular Polimórfica Catecolaminérgica
RYR2
AD
~65%
CASQ2
AR
3-5%
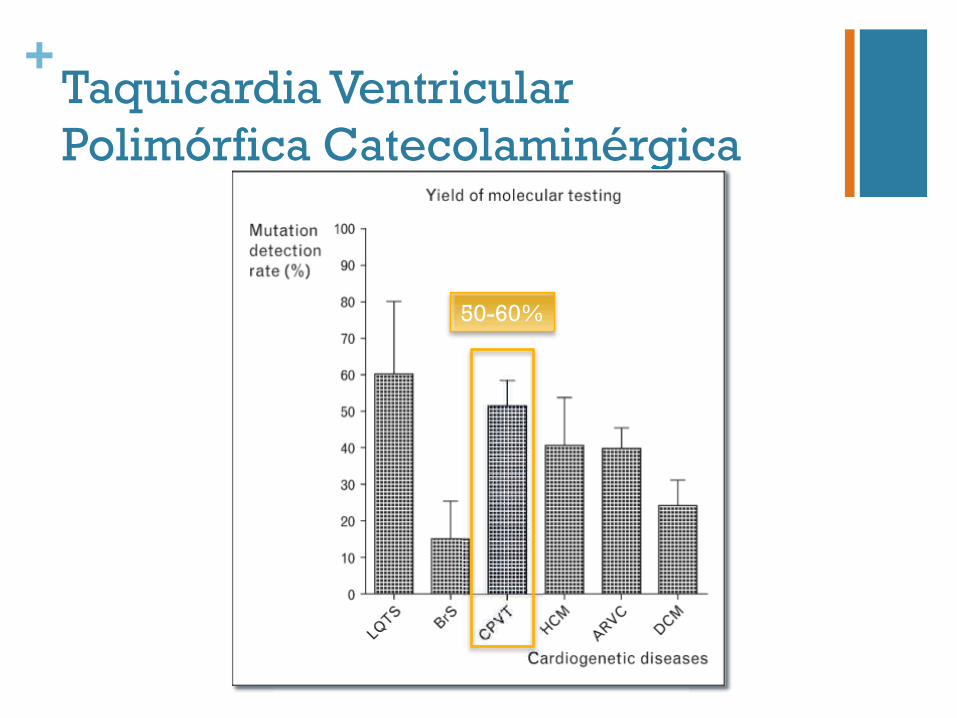
+Taquicardia Ventricular Polimórfica Catecolaminérgica Catecolaminérgica
50-60%


















