Caracterização de Misturas de Poliamidas de Elevado ... · Caracterização de Misturas de...
81
Caracterização de Misturas de Poliamidas de Elevado Desempenho para Moldação por Injecção Ana Luísa Varão Dias Moreira Braga Dissertação para obtenção do Grau de Mestre em Engenharia de Materiais Orientador: Professor Doutor António José Boavida Correia Diogo Co-Orientador: Engenheiro Luís Paulo Gonçalves Neves Júri Presidente: Professor Doutor João Carlos Moura Bordado Orientador: Professor Doutor António José Boavida Correia Diogo Vogal: Professora Doutora Maria do Rosário Gomes Ribeiro Novembro 2014 (Imagens/Gráficos)
Transcript of Caracterização de Misturas de Poliamidas de Elevado ... · Caracterização de Misturas de...

Caracterização de Misturas de Poliamidas de Elevado
Desempenho para Moldação por Injecção
Ana Luísa Varão Dias Moreira Braga
Dissertação para obtenção do Grau de Mestre em
Engenharia de Materiais
Orientador: Professor Doutor António José Boavida Correia Diogo
Co-Orientador: Engenheiro Luís Paulo Gonçalves Neves
Júri
Presidente: Professor Doutor João Carlos Moura Bordado
Orientador: Professor Doutor António José Boavida Correia Diogo
Vogal: Professora Doutora Maria do Rosário Gomes Ribeiro
Novembro 2014
(Imagens/Gráficos)

1
“Viver é a coisa mais rara do mundo. A maioria das pessoas apenas existe.”
Oscar Wilde

2
Agradecimentos
Ao Professor A. Correia Diogo e ao Eng.º Luís Neves por terem proporcionado este desafio,
pelo convite e confiança e, acima de tudo, pelos conhecimentos e experiência adquiridos, que valorizo
muito para o meu futuro profissional.
À Sara Lucas e à Ana Elisa pela total disponibilidade em ajudar na parte experimental e em
responder às minhas questões.
Aos meus pais, irmãos, avós e resto da família por todo o apoio durante estes cinco anos, o
que permitiu realizar tudo o que consegui realizar, apesar da distância e das saudades sempre
presentes. Aos meus pais também pela possibilidade de frequentar este curso.
Ao Miguel pela companhia, conselhos e serenidade, que me fizeram crescer como pessoa, ser
mais humilde e lutar para alcançar os meus objectivos.
Aos meus amigos pelo espírito de entreajuda e por todos os momentos memoráveis que
passámos, incluindo as noitadas, fins-de-semana e feriados na casa IST.

3
Resumo
As peças em estudo fazem parte do sistema de refrigeração do motor do automóvel e são
constituídas por uma liga de PA66+PA6 reforçada com fibra de vidro, sendo produzidas por moldação
por injecção. Foram escolhidas e analisadas peças com diferentes características, sendo também
estudado o efeito dos ciclos térmicos a que algumas destas peças foram sujeitas durante o teste do
seu desempenho em serviço e o efeito da alteração do aspecto da superfície das peças. Estas peças
foram caracterizadas por SEM, DMA e DSC, com o objectivo de recolher informações sobre a
morfologia da superfície, o comportamento mecânico em função da temperatura e a composição de
cada peça. Os resultados obtidos por SEM mostram um aumento da rugosidade nas peças com a
alteração do aspecto da superfície e uma evolução na superfície das peças sujeitas aos ciclos térmicos.
A caracterização por DMA evidencia o efeito dos ciclos térmicos na cristalinidade do material das peças.
Os resultados obtidos por DSC mostram também o efeito dos ciclos térmicos na cristalinidade do
material das peças sujeitas a esses ciclos e uma alteração na composição das peças com a alteração
do aspecto da superfície.
Palavras-chave
Poliamidas 66 e 6; Ciclos Térmicos; Aspecto da Superfície; SEM; DMA; DSC.

4
Abstract
The studied components belong to the automobile engine cooling system, they are composed
of a PA66+PA6 blend reinforced with glass fiber and they are produced by injection molding.
Components with different features were selected and analyzed and the effect of the thermal cycles
applied to some of these components (during their usage test) and the effect of the surface aspect
modification were also studied. These components were analyzed by SEM, DMA and DSC with the aim
of getting information about the surface morphology, the mechanical behavior depending on the
temperature and the composition of each component. The results from SEM demonstrate an increase
in the roughness of the components with the surface aspect modification and an evolution of the surface
of the components subject to the thermal cycles. The characterization by DMA shows the effect of the
thermal cycles on the crystallinity of the components’ material. The results from DSC also show the
effect of the thermal cycles on the crystallinity of the material of the components subject to those cycles
and show a change in the composition of the components with the surface aspect modification.
Key words
Polyamides 66 and 6; Thermal Cycles; Surface Aspect; SEM; DMA; DSC.

5
Índice
Agradecimentos ....................................................................................................................................... 2
Resumo ................................................................................................................................................... 3
Palavras-chave ........................................................................................................................................ 3
Abstract.................................................................................................................................................... 4
Key words ................................................................................................................................................ 4
Lista de Figuras ....................................................................................................................................... 7
Lista de Tabelas .................................................................................................................................... 10
Lista de Abreviaturas ............................................................................................................................. 11
Lista de Símbolos .................................................................................................................................. 12
1 Introdução ........................................................................................................................................... 13
1.1 Utilização da Liga PA66+PA6 na Indústria Automóvel ................................................................ 16
1.2 Processo de Moldação por Injecção de Polímeros ..................................................................... 20
1.2.1 Anisotropia Inerente ao Processo de Injecção ..................................................................... 22
2 Técnicas Utilizadas ............................................................................................................................. 25
2.1 Microscopia Electrónica de Varrimento (SEM) ............................................................................ 25
2.1.1 Princípios de Funcionamento ................................................................................................ 25
2.1.2 Procedimento Experimental .................................................................................................. 29
2.1.3 Equipamento ......................................................................................................................... 31
2.2 Análise Dinâmica e Mecânica (DMA) .......................................................................................... 33
2.2.1 Princípios de Funcionamento ................................................................................................ 33
2.2.2 Procedimento Experimental .................................................................................................. 34
2.2.3 Equipamento ......................................................................................................................... 36
2.3 Calorimetria Diferencial por Varrimento (DSC) ........................................................................... 40
2.3.1 Princípios de Funcionamento ................................................................................................ 40
2.3.2 Procedimento Experimental .................................................................................................. 42
2.3.3 Equipamento ......................................................................................................................... 42
3 Apresentação e Discussão dos Resultados ....................................................................................... 45
3.1 SEM ............................................................................................................................................. 45
3.2 DSC.............................................................................................................................................. 48
3.3 DMA ............................................................................................................................................. 55

6
4 Conclusão ........................................................................................................................................... 63
5 Trabalho Futuro .................................................................................................................................. 64
Referências Bibliográficas ..................................................................................................................... 65
Anexos ................................................................................................................................................... 71
Anexo 1 – Ensaio SEM ...................................................................................................................... 71
Anexo 2 – Ensaio DMA ...................................................................................................................... 71
Anexo 3 – Ensaio DSC ...................................................................................................................... 72
Anexo 4 – Análise de Resultados DMA ............................................................................................. 73
Anexo 5 – Análise de Resultados DSC ............................................................................................. 77

7
Lista de Figuras
Figura 1 - Fotografias de dois ângulos de uma peça de PA66+PA6 reforçada com FV, pertencente ao
sistema de arrefecimento do motor de automóveis. ............................................................................. 13
Figura 2 - Fotografias da diferença no aspecto da superfície. (a) Superfície exterior de duas peças, em
que a da direita tem a alteração na superfície. (b) e (c) Superfície interior de duas peças semelhantes
à da figura 1, em que a parte da esquerda apresenta a alteração na superfície. ................................ 14
Figura 3 - Esquema do fluxo de ar num intercooler do tipo ar-ar [1]. ................................................... 15
Figura 4 - Intercooler com o ninho em alumínio e as caixas de entrada e de saída de ar em poliamida
[85]. ........................................................................................................................................................ 15
Figura 5 - A unidade repetitiva de PA6 está representada à esquerda e a de PA66 à direita (-NH- é o
grupo amina, -CO- é o ácido e (-CH2-)n é a cadeia alifática) [89]. ........................................................ 17
Figura 6 - Constituição de uma máquina de injecção de polímeros [86]. ............................................. 20
Figura 7 - Constituição e disposição dos vários componentes de uma unidade de fecho, em que o
enchimento do fundido é feito pelo lado direito quando o bico de injecção contacta a entrada do jito (a
figura da esquerda faz uma descrição desses componentes e a da direita exemplifica a cavidade do
molde preenchida, bem como o seu sistema de alimentação) [87], [88]. ............................................. 21
Figura 8 - Exemplo do desenho de um sistema de alimentação, com o jito, um canal de alimentação e
quatro entradas para quatro peças já processadas [90]. ...................................................................... 21
Figura 9 - Gráfico tensão-extensão para a extensão aplicada na direcção paralela e perpendicular à
direcção das fibras [20]. ........................................................................................................................ 23
Figura 10 - Fonte de fluxo na cavidade de um molde de injecção, em que t4 é o tempo para a frente de
fusão [20]. .............................................................................................................................................. 24
Figura 11 - Fluxo extensional (divergente) após a saída de uma abertura apertada [20]. ................... 24
Figura 12 - Esquema da coluna do SEM e do compartimento da amostra [22]. .................................. 26
Figura 13 - Representação do local de interacção em forma de pêra e do local de cada tipo de emissão
gerada pela interacção do feixe com a amostra [23]. ........................................................................... 27
Figura 14 - Detector de electrões secundários num SEM [23]. ............................................................ 28
Figura 15 - Esquema de um equipamento para revestimento por pulverização normalmente usado [25].
............................................................................................................................................................... 29
Figura 16 – Disco de alumínio onde se fixam os provetes a analisar no SEM [81]. ............................. 30
Figura 17 - (a) Compartimento para colocar o disco de alumínio com os provetes aberto [83]. (b)
Detectores dos vários sinais emitidos pelo provete quando atingido pelo feixe, em que o detector dos
electrões secundários está situado no canto superior esquerdo [79]. .................................................. 30
Figura 18 - SEM S 2400 da Hitachi, no MicroLab do IST [82]. ............................................................. 32
Figura 19 - (a) Representação da diferença de fase entre as curvas sinusoidais da tensão e extensão
para materiais elásticos, viscosos e viscoelásticos. (b) Relação entre os vários parâmetros reológicos,
em que E* está a verde, E’ é o cateto adjacente e E’’ o cateto oposto [36]. ........................................ 34
Figura 20 - (a) Exemplo de um suporte do provete para consola simples e dupla num DMA (para consola
simples só se fixa o provete numa das extremidades e na parte móvel do suporte, que é a do meio)

8
[35]. (b) Fotografia de dois provetes para o ensaio por DMA, ao lado de uma moeda de 5 cêntimos. (c)
Esquema da montagem de um provete em consola simples [80]. ....................................................... 36
Figura 21 - Esquema genérico de um DMA por tensão controlada [34]. .............................................. 36
Figura 22 - Fotografias do DMA Q800 da TA Instruments no laboratório ICTPol do IST, com o forno
fechado (esquerda) e com o forno aberto (direita). ............................................................................... 37
Figura 23 - Disposição dos vários constituintes do DMA Q800 da TA Instruments [35]. ..................... 38
Figura 24 - Fotografias do GCA (esquerda) e da sua ligação ao DMA Q800 da TA Instruments, no
laboratório ICTPol do IST (direita). ........................................................................................................ 39
Figura 25 - Esquema de um DSC de forno simples (fonte de aquecimento para cada cápsula) [43]. . 40
Figura 26 - (a) Variação do calor com a temperatura para Tm. (b) Variação do calor com a temperatura
para Tg [43]. ........................................................................................................................................... 41
Figura 27 - Fotografia de um provete a analisar no DSC ao lado de uma moeda de 5 cêntimos
(esquerda) e exemplo de uma cápsula de alumínio aberta e fechada (direita) [84]. ............................ 42
Figura 28 - Imagem do equipamento DSC 2920 Modulated DSC da TA Instruments. ........................ 43
Figura 29 - Esquema do DSC 2920 Modulated DSC da TA Instruments (esquerda) e da prensa para
vedar as cápsulas de alumínio (direita) [46].......................................................................................... 44
Figura 30 - Imagens obtidas por SEM no modo de electrões secundários para a amostra A. ............ 45
Figura 31 - Imagens obtidas por SEM no modo de electrões secundários para a amostra B. ............ 45
Figura 32 - Imagens obtidas por SEM no modo de electrões secundários para a amostra C. ............ 46
Figura 33 - Imagens obtidas por SEM no modo de electrões secundários para a amostra D. ............ 46
Figura 34 - Curva DSC do 1º ensaio para a amostra A. ....................................................................... 48
Figura 35 - Curva DSC do 1º ensaio para a amostra B. ....................................................................... 48
Figura 36 - Curva DSC do 1º ensaio para a amostra C. ....................................................................... 49
Figura 37 - Curva DSC do 1º ensaio para a amostra D. ....................................................................... 49
Figura 38 - Sobreposição das curvas DSC do 1º ensaio para cada amostra. ...................................... 49
Figura 39 - Curva DSC do 2º ensaio para a amostra A. ....................................................................... 50
Figura 40 - Curva DSC do 2º ensaio para a amostra B. ....................................................................... 50
Figura 41 - Curva DSC do 2º ensaio para a amostra D. ....................................................................... 51
Figura 42 - Curva DSC do 2º ensaio para a amostra C. ....................................................................... 51
Figura 43 - Sobreposição das curvas DSC do 2º ensaio para cada amostra. ...................................... 51
Figura 44 - Curva DSC (aquecimento) típica para os homopolímeros PA66 e PA6 e para uma liga de
PA6+PA66 (60:40 wt%), B (endotérmica para cima) [57]. .................................................................... 53
Figura 45 - Curva DMA para a amostra A, com E' (log), E'' e tan δ em função da temperatura (1Hz). 55
Figura 46 - Curva DMA para a amostra A com a variação de E’ (log) com a temperatura (1Hz). ....... 55
Figura 47 - Curva DMA para a amostra B, com E' (log), E'' e tan δ em função da temperatura (1Hz). 56
Figura 48 - Curva DMA para a amostra B com a variação de E’ (log) com a temperatura (1Hz). ....... 56
Figura 49 - Curva DMA para a amostra C, com E' (log), E'' e tan δ em função da temperatura (1Hz). 57
Figura 50 - Curva DMA para a amostra C com a variação de E’ (log) com a temperatura (1Hz). ....... 57
Figura 51 - Sobreposição das curvas DMA, com E’ (log) em função da temperatura para cada amostra
(1Hz). ..................................................................................................................................................... 58

9
Figura 52 - Curva DMA para a amostra D com a variação de E’ (log) com a temperatura (1Hz). ....... 58
Figura 53 - Curva DMA para a amostra D, com E' (log), E'' e tan δ em função da temperatura (1Hz). 58
Figura 54 - Sobreposição das curvas DMA, com E’’ em função da temperatura para cada amostra (1Hz).
............................................................................................................................................................... 61
Figura 55 - Sobreposição das curvas DMA, com tan δ em função da temperatura para cada amostra
(1Hz). ..................................................................................................................................................... 61
Figura 56 - Fotografia do local das peças de onde foram retirados os provetes para analisar no SEM
(parte mais exterior do lado esquerdo assinalada pelo círculo vermelho). ........................................... 71
Figura 57 - Curva DMA para a amostra A, com E' (log), E'' e tan δ em função da temperatura. ......... 73
Figura 58 - Curva DMA para a amostra B, com E' (log), E'' e tan δ em função da temperatura. ......... 73
Figura 59 - Curva DMA para a amostra D, com E' (log), E'' e tan δ em função da temperatura. ......... 74
Figura 60 - Curva DMA para a amostra C, com E' (log), E'' e tan δ em função da temperatura. ......... 74
Figura 61 - Curva DMA para a amostra B, sem o ajuste vertical para as várias variáveis. .................. 75
Figura 62 - Curva DMA para a amostra B, com o ajuste vertical para as várias variáveis. .................. 75
Figura 63 - Curva DMA para a amostra D, sem o ajuste vertical para as várias variáveis. ................. 76
Figura 64 - Curva DMA para a amostra D, com o ajuste vertical para as várias variáveis. ................. 76
Figura 65 - Curva DSC para a amostra A com os três ciclos do 2º ensaio. ......................................... 77
Figura 66 - Curva DSC para a amostra B com os três ciclos do 2º ensaio. ......................................... 77
Figura 67 - Curva DSC para a amostra D com os três ciclos do 2º ensaio. ......................................... 78
Figura 68 - Curva DSC para a amostra C com os três ciclos do 2º ensaio. ......................................... 78
Figura 69 - Esquema do prolongamento de cada pico de fusão (tracejado a verde) e da área que falta
adicionar ao integral de cada pico (triângulo vermelho). ...................................................................... 79

10
Lista de Tabelas
Tabela 1 - Algumas propriedades mecânicas, térmicas e de processamento de PA6 e PA66 [5], [9]. 17
Tabela 2 - Comparação de algumas propriedades de PA66 com e sem reforço de FV [8]. ................ 19
Tabela 3 - Descrição das amostras de cada peça caracterizadas neste trabalho. .............................. 25
Tabela 4 - Parâmetros de funcionamento durante a obtenção das imagens por SEM. ....................... 30
Tabela 5 - Especificações do SEM S 2400 da Hitachi [26], [27]. .......................................................... 31
Tabela 6 - Características do filamento de tungsténio [28], [22]. .......................................................... 32
Tabela 7 - Parâmetros dos dois ensaios por DMA [38], [37]. ............................................................... 35
Tabela 8 - Especificações do equipamento DMA Q800 da TA Instruments [35], [34]. ......................... 38
Tabela 9 - Parâmetros de funcionamento dos dois ensaios efectuados por DSC [46]. ....................... 42
Tabela 10 - Especificações da célula do forno do DSC 2920 Modulated DSC da TA Instruments, onde
se inserem as cápsulas com o provete e com a referência [46]. .......................................................... 43
Tabela 11 - Valores de ΔHf (o cálculo dos picos separados está explicado no Anexo 5) e de Tm para os
picos da fusão cristalina de cada amostra do 1º ensaio. ...................................................................... 52
Tabela 12 - Valores de %Cx para as amostras do 1º ensaio. .............................................................. 53
Tabela 13 - Valores de ΔHf, Tm e %Cx para as amostras do 2º ensaio. ............................................... 54
Tabela 14 - Valores de E' para cada região da curva de cada amostra. .............................................. 59
Tabela 15 - Tg de PA66 e PA6 (da literatura) e Tg determinada pelos pico de E'' e de tan δ [67]. ....... 60
Tabela 16 - Dimensões dos provetes de cada amostra para os ensaios nas gamas de temperatura de
-60-150ºC e de 35-250ºC. ..................................................................................................................... 71
Tabela 17 - Medidas dos provetes ensaiados por DSC para cada ensaio. .......................................... 72
Tabela 18 - ΔHf para os vários picos e para o triângulo vermelho de cada amostra. .......................... 79
Tabela 19 - Dimensões e áreas do triângulo de cada amostra. ........................................................... 80
Tabela 20 - Dimensões, áreas e ΔHf para os dois picos individuais de cada amostra. ....................... 80
Tabela 21 - ΔHf para cada pico separado no pico de fusão de cada amostra. .................................... 80

11
Lista de Abreviaturas
DMA – Análise dinâmica e mecânica (designa também o equipamento)
DSC – Calorimetria diferencial de varrimento (designa também o equipamento)
FV – Fibra de vidro
GCA – Acessório de arrefecimento por gás
GF – Factor geométrico
LVR – Regime viscoelástico linear
NA – Abertura numérica
PA6 – Poliamida 6
PA66 – Poliamida 66
PA66+PA6 – Liga polimérica da poliamida 66 e da poliamida 6
SEM – Microscopia electrónica de varrimento (designa também o equipamento)
TA Instruments – Thermal Analysis Instruments
N2líq. – Azoto líquido
wt% - Percentagem em peso
%Cx – Percentagem de cristalinidade

12
Lista de Símbolos
E – Módulo elástico (módulo de Young)
E* - Módulo complexo
E’ – Componente elástica
E’’ – Componente dissipativa
Mw – Peso molecular
Wf – Fracção das fibras
�̇� – Taxa de aquecimento
Cp – Calor específico
Tg – Temperatura da transição vítrea
Tm – Temperatura de fusão
ΔHf – Entalpia de fusão
ΔHf100 – Entalpia de fusão do material 100% cristalino
ΔGm – Energia livre de Gibbs de uma mistura
ΔHm – Entalpia de uma mistura
ΔSm – Entropia de uma mistura
δ – Ângulo de fase
Tan δ – Tangente do ângulo de fase
ω – Frequência
t – Tempo
σ0 – Amplitude da tensão máxima
Ɛ0 – Amplitude da extensão máxima
σy – Tensão de cedência
Ɛ – Extensão

13
1 Introdução
O presente trabalho tratou de caracterizar o material de peças pertencentes ao sistema de
arrefecimento do motor de automóveis, em que estas foram disponibilizadas pela empresa JDeus®. As
peças em questão são constituídas por uma liga polimérica de poliamida 66 e poliamida 6 (PA66+PA6),
reforçada com fibra de vidro (FV) curta e são fabricadas pelo processo de moldação por injecção. A
figura seguinte mostra um exemplo dessas peças.
As peças estudadas têm características diferentes, como a percentagem de reforço com FV, o
número de ciclos térmicos a que foram sujeitas e o aspecto da sua superfície. Os ciclos térmicos
consistem num ensaio de pressão pulsada, que tem o objectivo de simular as cargas de pressão ao
longo da vida do automóvel. A pressão é imposta num ciclo sinusoidal, variando entre 10 e 300 kPa, a
uma temperatura de 210ºC durante cerca de 5 a 6 dias, com ciclos de 1 Hz. A alteração no aspecto da
superfície está representada na figura 2. O efeito dos ciclos térmicos relaciona-se com a evolução das
propriedades das peças ao longo do seu uso e a alteração no aspecto da superfície pode ser provocada
para estudar a implicação que uma mudança nas características esperadas pode ter na aplicação das
peças em questão.
Portanto, para além de caracterizar o material destas peças, foi também objectivo deste
trabalho estudar o efeito dos ciclos térmicos e o efeito da alteração no aspecto da superfície nas
propriedades das peças sujeitas a essas variações, uma vez que os componentes com aplicação
automóvel e pertencentes a sistemas complexos, como é o caso do sistema de arrefecimento do motor
de automóveis, têm de responder a requisitos de propriedades térmicas e mecânicas exigentes, bem
como a tolerâncias dimensionais apertadas (entre outros).
Para isso foram escolhidas as técnicas de microscopia electrónica de varrimento (SEM), de
análise dinâmica e mecânica (DMA) e de calorimetria diferencial por varrimento (DSC) para observar a
morfologia da superfície, analisar o comportamento mecânico em função da temperatura e comparar a
composição de cada peça, respectivamente.
Figura 1 - Fotografias de dois ângulos de uma peça de PA66+PA6 reforçada com FV,
pertencente ao sistema de arrefecimento do motor de automóveis.

14
Estas peças em questão fazem parte de um intercooler, que consiste num dispositivo de
entrada e arrefecimento de ar (normalmente usado em motores com turbocompressor). O intercooler
arrefece o ar comprimido pelo turbocompressor, reduzindo a temperatura e aumentando a densidade
do ar fornecido ao motor. Assim que o ar é comprimido pelo turbocompressor fica muito quente, muito
rapidamente e o seu teor (densidade) de oxigénio diminui. Ao arrefecer o ar comprimido, o intercooler
fornece ar mais denso e mais rico em oxigénio ao motor, melhorando a combustão por permitir que
seja queimado mais combustível. Também aumenta a fiabilidade, uma vez que proporciona uma
temperatura mais consistente do ar que entra no motor, o que permite que o rácio entre o ar e o
combustível do motor permaneça num nível seguro. Um intercooler do tipo ar-ar (figura 3) extrai o calor
do ar comprimido ao passá-lo através da sua rede de tubos com alhetas arrefecidas. À medida que o
ar comprimido atravessa o intercooler, o calor é transferido para os tubos e para as alhetas arrefecidas.
O ar que se encontra no exterior do automóvel (mais frio e deslocando-se rapidamente) absorve o calor
das alhetas arrefecidas, reduzindo a temperatura do ar comprimido. O local mais eficiente para se
colocar um intercooler é na dianteira de um automóvel [1].
Figura 2 - Fotografias da diferença no aspecto da superfície. (a) Superfície exterior de duas peças,
em que a da direita tem a alteração na superfície. (b) e (c) Superfície interior de duas peças
semelhantes à da figura 1, em que a parte da esquerda apresenta a alteração na superfície.
(a) (b)
(c)

15
A figura seguinte exemplifica os componentes de um intercooler, em que os componentes em
poliamida são as caixas de entrada e de saída de ar, cravadas ao ninho de alumínio que contém a rede
de tubos com as alhetas arrefecidas.
Figura 3 - Esquema do fluxo de ar num intercooler do tipo ar-ar [1].
Figura 4 - Intercooler com o ninho em alumínio e as caixas de
entrada e de saída de ar em poliamida [85].

16
1.1 Utilização da Liga PA66+PA6 na Indústria Automóvel
Com o desenvolvimento de polímeros de elevado desempenho, a utilização de componentes
em plástico na indústria automóvel tem vindo a crescer rapidamente desde as últimas décadas e a
substituir componentes em metal. Uma grande razão deste crescimento é cumprir o objectivo de tornar
os automóveis mais eficientes em termos energéticos pela redução do peso (redução do consumo de
combustível e das emissões gasosas), proporcionando ao mesmo tempo durabilidade, resistência à
corrosão, tenacidade, flexibilidade de desenho dos componentes e possibilidade de integração em
sistemas com outros materiais, resiliência e elevado desempenho a baixo custo. A quantidade de
componentes em plástico presente num automóvel representa cerca de 10 a 15% do seu peso total [2],
[3].
Desde a descoberta das poliamidas (o termo genérico é nylon) em 1939, o seu consumo no
mercado automóvel tem crescido constantemente. As poliamidas normalmente usadas neste sector
são a poliamida 66 (PA66) e a poliamida 6 (PA6), em que a maior aplicação é em componentes que
vão estar debaixo do capô do motor (sendo maioritariamente reforçadas com FV). Estes componentes
podem fazer parte do distribuidor de entrada de ar, do sistema de arrefecimento do motor, do
compartimento do ventilador, do sistema de ar condicionado, do compartimento do corpo do acelerador,
e podem ser também amortecedores de ruído, tampas de válvulas e tubos de combustível, líquido
refrigerante ou ar. Contudo, podem também ser usadas no sistema eléctrico do automóvel, como o
compartimento electrónico, sensores, interruptores e peças periféricas. Nas peças exteriores estão
presentes no compartimento do espelho retrovisor, nos puxadores de porta e nos limpa pára-brisas,
entre outros [4], [5].
Visto a vasta utilização das poliamidas PA66 e PA6 é de interesse prosseguir com a sua
descrição. As poliamidas são termoplásticos, ou seja, uma vez processadas podem ser aquecidas e
reprocessadas novamente. As poliamidas alifáticas (não-aromáticas, em que a cadeia química
constituída por átomos de carbono e hidrogénio é linear) têm um sistema de nomenclatura baseado na
sequência repetitiva e no número de átomos de carbono presentes na cadeia dos monómeros. Um “A”
representa um grupo amina e um “B” representa um grupo ácido. As poliamidas do tipo AB são
designadas por um único número, como PA6 para a poliamida 6 (poli(Ɛ-caprolactama)), visto que tem
seis átomos de carbono entre os átomos de azoto do grupo amina. PA6 é produzida pela polimerização
hidrolítica de Ɛ-caprolactama. As poliamidas formadas por diaminas e diácidos (do tipo AABB) são
designadas por dois números, em que o primeiro representa o número de carbonos na diamina e o
segundo o número de carbonos no diácido, como é o caso de PA66 para a poliamida 66 (polímero feito
pela policondensação de hexametilenodiamina e de ácido adípico), porque tem seis átomos de carbono
entre os átomos de azoto dos dois grupos amina e seis átomos de carbono no diácido (os monómeros
de cada poliamida estão representados na figura seguinte) [6], [5], [7].

17
Estas poliamidas contêm grupos polares –CONH– espaçados em intervalos regulares, que
cristalizam com uma atracção intermolecular elevada (são semicristalinas). Os segmentos alifáticos das
suas cadeias poliméricas dão uma certa flexibilidade às regiões amorfas. Assim, a combinação de uma
elevada atracção intercadeias nas zonas cristalinas e da flexibilidade nas zonas amorfas, leva a que
estes polímeros sejam mecanicamente resistentes acima da sua temperatura de transição vítrea, Tg. A
atracção intermolecular elevada faz com que as poliamidas tenham uma temperatura de fusão (Tm)
elevada. Contudo, acima de Tm a viscosidade do fundido é baixa, devido à flexibilidade do polímero a
temperaturas tão elevadas (que são normalmente 200ºC acima de Tg) e do peso molecular (Mw)
relativamente baixo [8].
A tabela a seguir compara algumas propriedades de PA66 e de PA6.
Tabela 1 - Algumas propriedades mecânicas, térmicas e de processamento de PA6 e PA66 [5], [9].
PA6 PA66
Módulo Elástico, E, antes da
Transição Vítrea (tracção com
0.2% de teor de água)
3 GPa 2 - 3 GPa
Tensão de
Cedência, σy
23ºC 83 MPa 81 MPa
80ºC 50 MPa 52 MPa
Rigidez 1.6 GPa 2 GPa
Dureza (Rockwell) R119 R120
Coeficiente Linear de
Expansão Térmica 8 - 8.3 x 10-5/ºC 8 x 10-5/ºC
Absorção de Água (% de
aumento de peso, saturada) 8.5 - 10% 8.5%
Temperatura de Fusão (Tm) 210 – 220ºC 255 – 265ºC
Temperatura de
Processamento 227 – 288ºC 260 – 327ºC
Pressão de Moldagem 7 – 138 MPa 7 – 173 MPa
Contracção Linear no Molde 0.003 – 0.015 cm/cm 0.007 – 0.018 cm/cm
Pelo que se pode observar na tabela, a maior desvantagem de PA6 em relação a PA66 é que
tem uma tensão de cedência (σy) menor para temperaturas à volta de 80ºC, uma rigidez mais baixa,
Figura 5 - A unidade repetitiva de PA6 está representada à esquerda e a de PA66 à
direita (-NH- é o grupo amina, -CO- é o ácido e (-CH2-)n é a cadeia alifática) [89].

18
uma resistência à abrasão mais baixa (dureza) e pode ter uma maior expansão térmica e maior
absorção de água. No entanto, PA6 tem uma Tm e uma temperatura de processamento mais baixas e
não sofre tanta contracção no molde [5].
Uma liga polimérica é uma combinação de dois ou mais polímeros estruturalmente diferentes
para dar origem a um material com propriedades que não podiam ser obtidas com os componentes
individuais, tanto para melhorar as propriedades técnicas como para ajustar as condições de
processamento e reduzir os custos. As propriedades físicas e as aplicações das ligas poliméricas
dependem em grande escala do grau de miscibilidade dos componentes da liga. As características de
miscibilidade de dois componentes químicos são regidas pela variação da energia livre de Gibbs (ΔGm)
que ocorre na sua combinação, de acordo com a seguinte equação
∆𝐺𝑚 = ∆𝐻𝑚 − 𝑇∆𝑆𝑚 (1)
em que T é a temperatura e ΔHm e ΔSm são a entalpia e a entropia da mistura, respectivamente. Se
ΔGm for negativa, a mistura pode ocorrer espontaneamente e é produzida uma solução como resultado.
No caso de compostos de baixo peso molecular, ΔSm é bastante positiva e, portanto, o termo -TΔSm
vai ser negativo e a mistura particularmente favorecida. ΔHm depende das interacções energéticas entre
as moléculas e é normalmente positiva, não sendo favorável para a mistura. O termo ΔHm positivo é
muitas vezes compensado pelo termo TΔSm negativo e a mistura acaba por acontecer. Quanto mais
semelhante for a natureza química dos componentes, mais baixo é o valor de ΔHm e a mistura torna-
se mais provável. A miscibilidade de polímeros (a determinada temperatura) é termodinamicamente
favorável para polímeros que se atraem um ao outro e mais provável para ligas com polímeros de baixo
peso molecular, apesar de ΔSm ser muito menos positiva devido à diminuição da desordem do sistema,
implicada pela presença de moléculas de grande tamanho [10].
A liga polimérica em questão é a liga PA66+PA6, ou seja, PA66 é o componente presente em
maior quantidade (no entanto, a proporção de cada poliamida não é conhecida). Esta liga também tem
um aditivo para ser termicamente estável. Um rácio de PA66/PA6 igual a 95,0/5,0 wt% até 99,9/0,1
wt% combina durabilidade, resistência ao calor e estabilidade dimensional [11], [12].
Os componentes pertencentes ao intercooler têm de suportar uma variação extensa de
temperaturas, ar quente sob pressão e tensões mecânicas e de vibração. A pressão de serviço
(dinâmica) varia de 0 a 250 kPa, a pressão máxima é de 400 kPa e a temperatura de serviço varia de
-40 a 220ºC [13], [14].
No entanto, estas poliamidas ainda apresentam várias desvantagens em relação aos metais,
tais como, rigidez e resistência à tracção inferiores, instabilidade dimensional devido a um coeficiente
de expansão térmica mais elevado e à maior absorção de água, resistência ao impacto e à fluência
mais baixa, temperatura máxima de serviço mais baixa e dureza inferior. Para minimizar estas
desvantagens é necessário reforçá-las normalmente com FV. Estas fibras são tratadas com uma resina
do tipo poli(vinil acetato) para manter os filamentos da fibra juntos como um fio e um agente de ligação
(silano, por exemplo) para melhorar a ligação entre a matriz e as fibras.
19
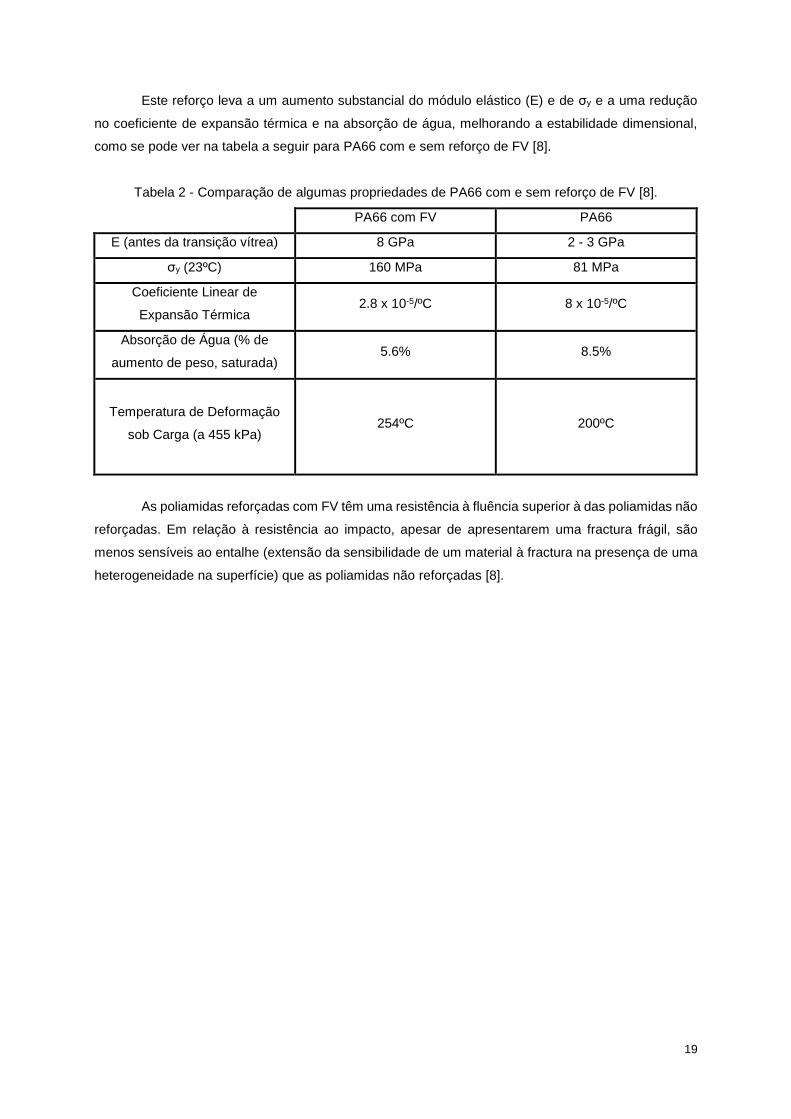
Este reforço leva a um aumento substancial do módulo elástico (E) e de σy e a uma redução
no coeficiente de expansão térmica e na absorção de água, melhorando a estabilidade dimensional,
como se pode ver na tabela a seguir para PA66 com e sem reforço de FV [8].
Tabela 2 - Comparação de algumas propriedades de PA66 com e sem reforço de FV [8].
PA66 com FV PA66
E (antes da transição vítrea) 8 GPa 2 - 3 GPa
σy (23ºC) 160 MPa 81 MPa
Coeficiente Linear de
Expansão Térmica 2.8 x 10-5/ºC 8 x 10-5/ºC
Absorção de Água (% de
aumento de peso, saturada) 5.6% 8.5%
Temperatura de Deformação
sob Carga (a 455 kPa) 254ºC 200ºC
As poliamidas reforçadas com FV têm uma resistência à fluência superior à das poliamidas não
reforçadas. Em relação à resistência ao impacto, apesar de apresentarem uma fractura frágil, são
menos sensíveis ao entalhe (extensão da sensibilidade de um material à fractura na presença de uma
heterogeneidade na superfície) que as poliamidas não reforçadas [8].

20
1.2 Processo de Moldação por Injecção de Polímeros
Como já foi dito, as peças em estudo foram produzidas por moldação por injecção. Uma
máquina de injecção (figura 6) é constituída por dois componentes básicos: a unidade de injecção e a
unidade de fecho (que inclui o molde). A unidade de injecção aquece, funde e mistura (pela rotação de
um parafuso) os grânulos do polímero, injectando uma descarga de volume controlado sob pressão
num molde fechado, com a solidificação do polímero a começar nas paredes da cavidade do molde. O
fundido é injectado por meio do avanço de um parafuso que se encontra dentro do cilindro de injecção,
forçando o polímero fundido a entrar para a cavidade do molde pelo bico de injecção quando este
contacta o jito. A unidade de fecho mantém o polímero injectado sob pressão durante um determinado
período de tempo, para prevenir o retorno do polímero fundido e para compensar o decréscimo de
volume do fundido durante a solidificação, até a peça moldada estar suficientemente rígida para ser
ejectada. Esta unidade recua para se retirar a peça e avança novamente para se iniciar outro ciclo de
injecção [15].
Um molde para injecção é composto por placas metálicas que contêm cavidades e saliências
que definem a forma e a geometria das peças a moldar. Nestas placas estão também incluídos vários
canais que definem o caminho do polímero fundido desde o bico de injecção até à cavidade do molde.
Estes canais são o jito, os canais de alimentação e as entradas, constituindo o sistema de alimentação
do fundido na cavidade do molde. O jito é um canal cónico que liga o bico de injecção aos canais de
alimentação. Os canais de alimentação ligam o jito às entradas e devem ter sempre o menor
comprimento possível e proporcionar um enchimento equilibrado. A entrada é o elemento responsável
pela distribuição do fundido na cavidade do molde. Esta entrada tem a finalidade de sujeitar o fundido
a uma velocidade de corte suficientemente elevada, para que a pequena secção da entrada se
mantenha aberta durante a fase de enchimento e de compactação. Também deve solidificar a tempo
de permitir que o bico de injecção possa recuar sem haver refluxo do material. Para além disso, deve
Figura 6 - Constituição de uma máquina de injecção de polímeros [86].
Molten
Plastic

21
facilitar o controlo do enchimento, principalmente em moldes de injecção de várias peças, e permitir
uma separação fácil da peça do sistema de alimentação. Para expulsar o ar que se encontra dentro da
cavidade do molde à medida que esta é preenchida pelo polímero fundido é necessário um sistema de
escape desse ar (o ar sai por pequenos rasgos maquinados no molde quando é necessária uma
elevada precisão de ajustamento entre as duas ou mais partes da cavidade do molde). Para arrefecer
e solidificar o polímero injectado é necessário um sistema de arrefecimento, sendo um sistema muito
importante já que tem de controlar de forma correcta o arrefecimento do fundido no molde, permitindo
conjugar a rapidez do ciclo de injecção com as especificações pretendidas para a peça ou peças
processadas. O arrefecimento do fundido é feito pela transferência de calor para a superfície do molde,
que se encontra a uma temperatura inferior (são maquinados furos de passagem para um fluido
refrigerante perto da superfície do molde). No final do ciclo de injecção, a peça moldada já atingiu uma
temperatura que garante a sua estabilidade dimensional. O molde abre pelas linhas de partição (através
do movimento dos pinos de ejecção) e o sistema de extracção retira a peça do seu interior [16].
Figura 7 - Constituição e disposição dos vários componentes de uma unidade de fecho, em que o
enchimento do fundido é feito pelo lado direito quando o bico de injecção contacta a entrada do jito (a
figura da esquerda faz uma descrição desses componentes e a da direita exemplifica a cavidade do
molde preenchida, bem como o seu sistema de alimentação) [87], [88].
Figura 8 - Exemplo do desenho de um sistema de alimentação, com o jito, um
canal de alimentação e quatro entradas para quatro peças já processadas [90].

22
Os parâmetros a ter em conta para a projecção de peças a fabricar pelo processo de injecção
e para a concepção do molde são a temperatura de secagem do granulado do material a fundir, a
temperatura do material fundido e a sua viscosidade para essa temperatura, a temperatura do molde,
a pressão de injecção, a pressão de aperto, o binário do parafuso, a velocidade de rotação do parafuso,
o tempo do ciclo de injecção, o sistema de escape de ar necessário, o tipo do bico de injecção e a
contracção do material no molde e depois de extraído [15].
Ao processar poliamidas deve ter-se em consideração a sua tendência para absorver água, o
seu alto ponto de fusão, a baixa viscosidade do fundido e a cristalinidade que o polímero sólido
apresenta, levando a uma contracção extensa durante o arrefecimento. Para evitar estes problemas, o
granulado da poliamida a fundir deve estar seco, o cilindro da máquina de injecção deve ter um
gradiente de temperatura ao longo do seu comprimento e o bico de injecção tem de ter um desenho
especial (afunilamento inverso, por exemplo), já que a viscosidade das poliamidas nas temperaturas
de processamento é baixa e o fundido iria vazar através dos bicos de injecção normais. A contracção
extensa devido à cristalinidade pode ser controlada pela temperatura do fundido, pela temperatura do
molde, pela velocidade de injecção, pela pressão de injecção e pelo tempo que o polímero permanece
no molde [8], [17].
O granulado da liga PA66+PA6 reforçada com FV deve ser seco a uma temperatura de 80ºC,
a temperatura do fundido pode variar de 280 a 305ºC, a temperatura do molde pode variar de 55 a 95ºC
e a pressão de injecção de 70 a 140 MPa. O tempo do ciclo de injecção deve ser de um minuto por 6
mm de espessura da peça a moldar. A percentagem de cristalinidade das poliamidas varia muito com
as condições de processamento, em que para arrefecimentos rápidos e peças de espessura reduzida
pode ser de apenas 10% [18], [17], [8]. É também adicionado um agente desmoldante para facilitar a
remoção da peça da cavidade do molde.
1.2.1 Anisotropia Inerente ao Processo de Injecção
As peças fabricadas por injecção acabam por ter anisotropia nas suas propriedades (variam de
ponto-para-ponto na sua estrutura), que pode ser causada por várias razões, como as diferenças no
fluxo do polímero fundido durante a fase de enchimento da cavidade do molde, a irregularidade da
espessura em certas zonas da peça a moldar e, consequentemente, os diferentes tempos de
arrefecimento nessas zonas (levando a diferentes graus de cristalinidade). Estas causas são
acompanhadas pelo fenómeno de relaxação de tensão e pela orientação das macromoléculas durante
o enchimento, para além das condições de processamento. As propriedades mecânicas são
directamente dependentes da relação entre o eixo da orientação das moléculas do polímero e o eixo
em que é aplicado o esforço sobre essas moléculas, já que têm valores mais elevados na direcção do
alinhamento das moléculas do que na direcção perpendicular (isto acontece porque a força da ligação
entre os átomos da mesma cadeia é mais elevada que as ligações fracas entre as cadeias vizinhas)
[19], [15].
23
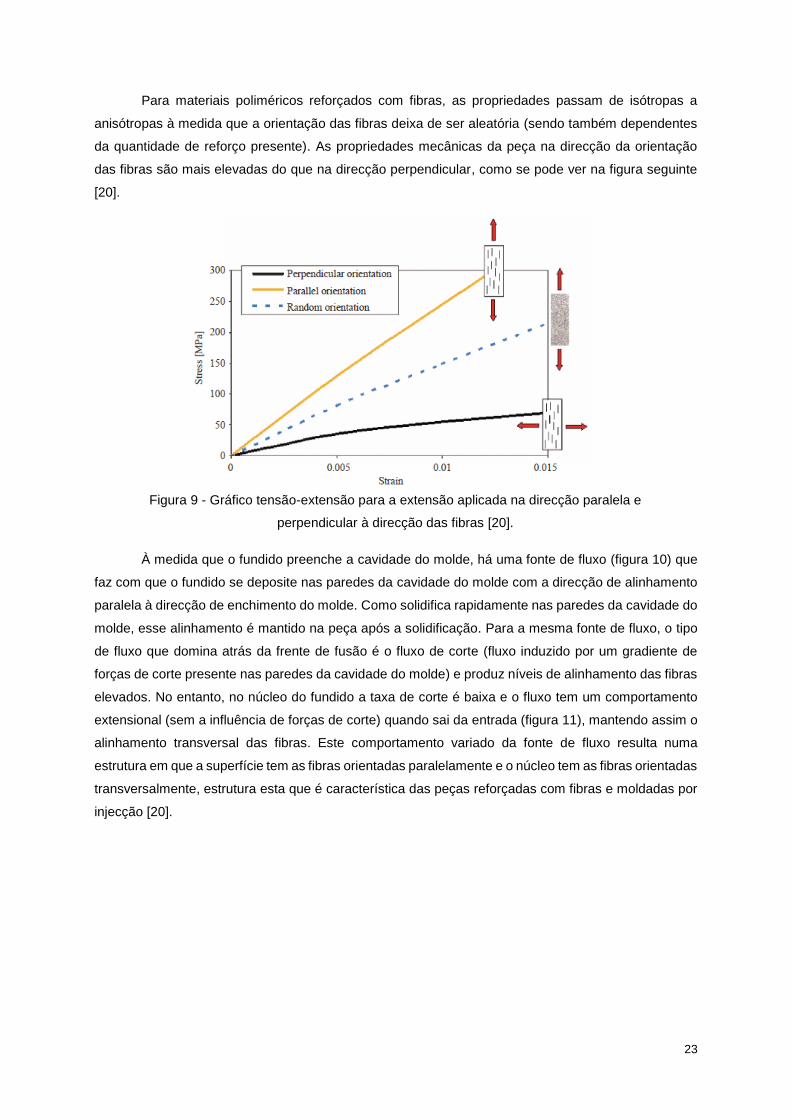
Para materiais poliméricos reforçados com fibras, as propriedades passam de isótropas a
anisótropas à medida que a orientação das fibras deixa de ser aleatória (sendo também dependentes
da quantidade de reforço presente). As propriedades mecânicas da peça na direcção da orientação
das fibras são mais elevadas do que na direcção perpendicular, como se pode ver na figura seguinte
[20].
À medida que o fundido preenche a cavidade do molde, há uma fonte de fluxo (figura 10) que
faz com que o fundido se deposite nas paredes da cavidade do molde com a direcção de alinhamento
paralela à direcção de enchimento do molde. Como solidifica rapidamente nas paredes da cavidade do
molde, esse alinhamento é mantido na peça após a solidificação. Para a mesma fonte de fluxo, o tipo
de fluxo que domina atrás da frente de fusão é o fluxo de corte (fluxo induzido por um gradiente de
forças de corte presente nas paredes da cavidade do molde) e produz níveis de alinhamento das fibras
elevados. No entanto, no núcleo do fundido a taxa de corte é baixa e o fluxo tem um comportamento
extensional (sem a influência de forças de corte) quando sai da entrada (figura 11), mantendo assim o
alinhamento transversal das fibras. Este comportamento variado da fonte de fluxo resulta numa
estrutura em que a superfície tem as fibras orientadas paralelamente e o núcleo tem as fibras orientadas
transversalmente, estrutura esta que é característica das peças reforçadas com fibras e moldadas por
injecção [20].
Figura 9 - Gráfico tensão-extensão para a extensão aplicada na direcção paralela e
perpendicular à direcção das fibras [20].

24
A anisotropia inerente ao processo de injecção pode afectar negativamente a precisão
dimensional exigida pela indústria automóvel e por isso tem de ser controlada. Para controlar o
enchimento do fundido na cavidade do molde projecta-se o desenho do molde e do sistema de
alimentação mais adequados, incluindo as dimensões e inclinação do jito e também as dimensões,
inclinação, forma, localização e número dos canais de alimentação e das entradas [19], [20].
Figura 10 - Fonte de fluxo na cavidade de um molde de injecção, em que
t4 é o tempo para a frente de fusão [20].
Figura 11 - Fluxo extensional (divergente) após a saída
de uma abertura apertada [20].
25
2 Técnicas Utilizadas
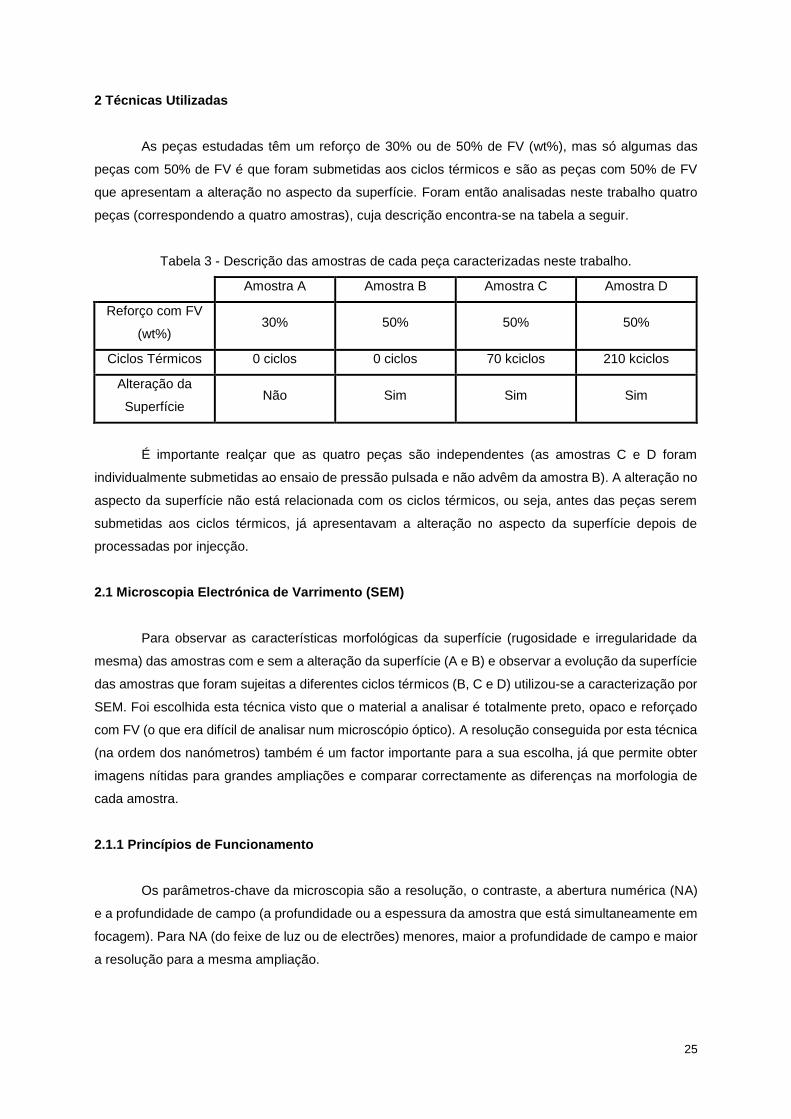
As peças estudadas têm um reforço de 30% ou de 50% de FV (wt%), mas só algumas das
peças com 50% de FV é que foram submetidas aos ciclos térmicos e são as peças com 50% de FV
que apresentam a alteração no aspecto da superfície. Foram então analisadas neste trabalho quatro
peças (correspondendo a quatro amostras), cuja descrição encontra-se na tabela a seguir.
Tabela 3 - Descrição das amostras de cada peça caracterizadas neste trabalho.
Amostra A Amostra B Amostra C Amostra D
Reforço com FV
(wt%) 30% 50% 50% 50%
Ciclos Térmicos 0 ciclos 0 ciclos 70 kciclos 210 kciclos
Alteração da
Superfície Não Sim Sim Sim
É importante realçar que as quatro peças são independentes (as amostras C e D foram
individualmente submetidas ao ensaio de pressão pulsada e não advêm da amostra B). A alteração no
aspecto da superfície não está relacionada com os ciclos térmicos, ou seja, antes das peças serem
submetidas aos ciclos térmicos, já apresentavam a alteração no aspecto da superfície depois de
processadas por injecção.
2.1 Microscopia Electrónica de Varrimento (SEM)
Para observar as características morfológicas da superfície (rugosidade e irregularidade da
mesma) das amostras com e sem a alteração da superfície (A e B) e observar a evolução da superfície
das amostras que foram sujeitas a diferentes ciclos térmicos (B, C e D) utilizou-se a caracterização por
SEM. Foi escolhida esta técnica visto que o material a analisar é totalmente preto, opaco e reforçado
com FV (o que era difícil de analisar num microscópio óptico). A resolução conseguida por esta técnica
(na ordem dos nanómetros) também é um factor importante para a sua escolha, já que permite obter
imagens nítidas para grandes ampliações e comparar correctamente as diferenças na morfologia de
cada amostra.
2.1.1 Princípios de Funcionamento
Os parâmetros-chave da microscopia são a resolução, o contraste, a abertura numérica (NA)
e a profundidade de campo (a profundidade ou a espessura da amostra que está simultaneamente em
focagem). Para NA (do feixe de luz ou de electrões) menores, maior a profundidade de campo e maior
a resolução para a mesma ampliação.

26
O SEM forma uma imagem da amostra a analisar ao percorrer um feixe de electrões focalizado
(que é a sonda de varrimento) através da amostra, em que este feixe interage com a fina camada de
superfície da amostra (no máximo alguns micrómetros) [21], [22].
O esquema da figura 12 mostra os principais componentes de um SEM, que fazem parte de
sete sistemas primários de operação: vácuo, geração do feixe, manipulação do feixe, interacção do
feixe, detecção, processamento do sinal e exibição e registo do sinal e imagem. O sistema de vácuo é
necessário porque os electrões do feixe iriam dispersar rapidamente devido às colisões com outras
moléculas presentes. O sistema de geração do feixe encontra-se no topo da coluna do microscópio e
gera o chamado feixe primário. O sistema de manipulação do feixe é constituído por lentes
electromagnéticas e bobinas localizadas na coluna do microscópio, que controlam o tamanho, forma e
posição do feixe na superfície da amostra. O sistema de interacção entre o feixe e a amostra envolve
o tipo de sinais gerados por esta interacção e que podem ser detectados. O sistema de detecção
consiste em vários detectores, cada um sensível a diferentes emissões de energia que ocorrem na
amostra. O sistema de processamento do sinal é um sistema electrónico que processa o sinal gerado
pelo sistema de detecção e permite a manipulação electrónica adicional da imagem. O sistema de
exibição e registo permite a visualização de um sinal eléctrico usando um tubo de raios catódicos e
permite também o registo dos resultados, usando um meio fotográfico ou magnético [23].
O sistema de geração do feixe é um canhão de electrões e é constituído por três componentes:
um filamento ou cátodo (por exemplo um fio de tungsténio), um cilindro de Whenelt que controla o fluxo
de electrões (polarização) e um ânodo com carga positiva que atrai e acelera os electrões através da
coluna até à amostra. O fio de tungsténio é um filamento de emissão térmico porque usa o aumento de
temperatura para excitar e emitir os electrões.
Figura 12 - Esquema da coluna do SEM e do compartimento da amostra [22].

27
A detecção do sinal começa quando o feixe primário atinge a amostra. O resultado desta
interacção é a formação de um local de interacção com forma de pêra (figura 13). Este local de
interacção é por definição o local na amostra onde se dão todos os efeitos de difracção que os electrões
sofrem quando penetram na superfície da amostra. O volume deste local de interacção depende da
densidade atómica e da topografia da amostra e do potencial de aceleração do feixe primário. A
topografia da amostra vai alterar a quantidade de emissões do local de interacção, em que um aumento
na topografia aumenta a área de superfície do local de interacção, resultando num sinal maior [23],
[22].
Os electrões secundários são talvez o tipo de interacção mais usado. Os electrões secundários
são gerados quando os electrões primários expulsam um electrão da superfície da amostra, até uma
espessura na ordem dos nanómetros (e também podem ser gerados por outros electrões secundários).
Os electrões secundários têm um nível de energia baixo (menor que 50 eV) e por isso só podem ser
detectados quando estão perto da superfície do local de interacção depois de expulsos da amostra.
Por essa razão, os electrões secundários não podem ser expulsos das zonas mais profundas do local
de interacção. Duas razões para operar no modo de imagem por electrões secundários são a obtenção
de informação topográfica e de alta resolução. Outra vantagem deste modo de imagem é que o feixe
primário pode dar origem a vários electrões secundários por múltiplos eventos de difracção,
aumentando potencialmente o sinal. Com um aumento no sinal e uma expulsão de electrões
secundários muito superficial, a resolução espacial pode ser muito maior que outros modos de imagem.
O detector de electrões secundários (figura 14) atrai magneticamente os electrões secundários
emitidos por um potencial de 200 V aplicado a um anel à volta do detector (taça de Faraday). Assim
que os electrões secundários entram no anel, são atraídos e acelerados por um potencial de 10 kV no
cintilador. Os electrões secundários atingem este cintilador causando a emissão de fotões, que vão
viajar pelo tubo do fotomultiplicador até o atingirem. A função do fotomultiplicador é a de amplificar o
sinal [23], [22].
Figura 13 - Representação do local de interacção em forma de pêra e do local de
cada tipo de emissão gerada pela interacção do feixe com a amostra [23].

28
A intensidade do sinal a amplificar depende do número de electrões secundários libertados
pelo local de interacção. Uma vez que a origem destes electrões é limitada a uma fina camada perto
da superfície da amostra porque são electrões de baixa energia, a quantidade dos electrões
secundários gerada é directamente proporcional à área da superfície que os emitiu. A densidade
atómica da superfície emissora vai determinar a intensidade do sinal, em que para materiais mais
densos (como o ouro) vão ser emitidos mais electrões secundários do que para materiais com
densidade mais baixa (como o carbono). As características topológicas (superfícies planas, estruturas
pontiagudas e arestas) vão afectar significativamente a área da superfície emissora. As superfícies
planas vão emitir poucos electrões secundários porque a área de superfície é menor, enquanto
estruturas pontiagudas vão emitir mais electrões secundários porque têm a área de superfície maior.
Quando os electrões secundários são emitidos de depressões, alguns vão ser perdidos devido à
reabsorção pela amostra e o resultado é um efeito de sombra. Contudo, alguns detalhes podem ser
observados porque alguns electrões escapam da área sombreada e são detectados [23], [22].
Na preparação de amostras poliméricas para a análise por microscopia electrónica é
necessário aumentar o contraste e minimizar os danos provocados pela incidência do feixe na amostra.
Os polímeros, que têm densidade e peso atómico mais baixos, têm pouca capacidade de difracção dos
electrões secundários, o que resulta num contraste baixo. Para resolver o problema pode ser aplicado
um revestimento condutor (filme fino), que também protege a amostra dos danos provocados pelo feixe.
Mas o problema maior é que os polímeros são geralmente maus condutores e as amostras a analisar
no SEM têm de ser condutoras eléctricas para produzirem electrões secundários e minimizar a
acumulação de cargas negativas na sua superfície [24].
Figura 14 - Detector de electrões secundários num SEM [23].

29
2.1.2 Procedimento Experimental
Os quatro provetes para analisar no SEM foram cortados da mesma zona de cada peça (que
possibilitasse obter provetes planos), em que a superfície a analisar é perpendicular à zona de corte
(são rectangulares com cerca de 7 mm de comprimento, 5 mm de largura e 3,5 mm de espessura).
Estes provetes não foram polidos nem contrastados, pois não se queriam perder nem alterar as
características morfológicas da superfície dos mesmos. Depois de cortados, os provetes foram
revestidas com um filme fino (cerca de 30 nm de espessura) de uma liga de ouro/paládio condutora.
Como já foi dito, para analisar por SEM materiais electricamente pouco condutores é
necessário revesti-los com um revestimento condutor, caso contrário irá ocorrer acumulação de carga
na superfície do provete durante o bombardeamento com electrões, o que dificulta a análise e obtenção
de uma imagem correcta da superfície do provete em estudo. Esta degradação da imagem acontece
porque os electrões são absorvidos e a rede de carga negativa acumulada repele o feixe seguinte.
Utilizou-se o método de revestimento por pulverização (sputtering) para fazer o filme condutor.
Os revestimentos por pulverização envolvem a erosão dos átomos de um alvo apropriado (neste caso
constituído por uma liga de ouro/paládio) por partículas energéticas (gás ionizado) e a consequente
deposição desses átomos no provete. O tipo de revestimento por pulverização mais básico e que é
normalmente usado em SEM está representado na figura 15 e consiste numa câmara evacuada que
contém um cátodo feito do material alvo, um ânodo e o local para o provete. É drenado um gás inerte
(Ar, N2) para dentro da câmara que vai ser ionizado pela exposição a um campo eléctrico, arrancando
os átomos do alvo quando as partículas de gás ionizado são aceleradas na sua direcção pelo campo
eléctrico. Estes átomos são deflectidos em todas as direcções devido à colisão com as partículas do
gás, ocorrendo a sua deposição nas superfícies frias, incluindo o provete. O desvio geral é em torno do
ânodo, mas o movimento aleatório dos átomos do material a depositar dá origem a uma deposição
multidireccional na superfície do provete e mesmo as superfícies rugosas podem ser revestidas
uniformemente [25], [23].
Figura 15 - Esquema de um equipamento para revestimento por
pulverização normalmente usado [25].

30
Para introduzir os provetes no SEM, estes foram colados num disco de alumínio (figura 16)
com fita-cola de dupla-face de carbono.
As figuras seguintes mostram o local do SEM onde se insere o disco de alumínio e a disposição
dos vários detectores do sinal emitido pelo provete durante a sua análise.
Visto que é necessário abrir o compartimento e expô-lo à atmosfera para colocar o disco de
alumínio com os provetes, o canhão de electrões tem de estar desligado (sem aplicação de tensão)
bem como as válvulas da coluna do SEM, para evitar que esta seja contaminada pela atmosfera.
Quando é fechado o compartimento é ligado o vácuo.
De seguida foram obtidas as imagens da superfície de cada provete, em que os parâmetros de
funcionamento durante a emissão do feixe e da aquisição da imagem estão na tabela seguinte.
Tabela 4 - Parâmetros de funcionamento durante a obtenção das imagens por SEM.
Corrente de Emissão 75 nA
Tensão de Aceleração 20 kV
Ampliação 100x, 500x e 1000x
Figura 16 – Disco de alumínio onde se fixam os provetes a analisar no SEM [81].
Figura 17 - (a) Compartimento para colocar o disco de alumínio com os provetes aberto
[83]. (b) Detectores dos vários sinais emitidos pelo provete quando atingido pelo feixe, em
que o detector dos electrões secundários está situado no canto superior esquerdo [79].
(a) (b)

31
Quanto maior a tensão aplicada, maior a resolução mas também maior é o calor que é gerado
no provete. A corrente do feixe é o fluxo de electrões que atingem o provete, em que aumentá-la faz
com que o número de electrões que atingem a superfície do provete seja maior para uma taxa mais
elevada. Isto resulta numa penetração mais profunda dos electrões e num diâmetro do feixe incidente
maior. Aumentar a corrente do feixe também gera mais calor no provete. Para locais de interacção
pequenos a resolução é maior, enquanto locais de interacção mais largos originam um sinal maior [23].
2.1.3 Equipamento
Utilizou-se um microscópio SEM do modelo S 2400 da Hitachi (figura 18). Este microscópio é
para uso geral, tem um filamento de tungsténio como cátodo para emitir os electrões e o mecanismo
de vácuo usa uma bomba difusora [26]. A aquisição da imagem foi feita através do programa Quantax
Esprit 1.9 da Bruker e o revestimento por pulverização foi realizado pelo modelo E 5100 da South Bay
Technologies (ex-Polaron).
Na tabela seguinte encontram-se as especificações do SEM S 2400 da Hitachi.
Tabela 5 - Especificações do SEM S 2400 da Hitachi [26], [27].
Resolução 10.0 nm
Ampliação 20x – 300 000x
Tensão de Aceleração 0 – 25 kV
Guia da Posição do Disco de Alumínio Manual
Número de Eixos 5
Posição do Disco de
Alumínio
Movimento em X 0 – 80 mm
Movimento em Y 0 – 40 mm
Movimento em Z 5 – 35 mm
Inclinação (não
eucêntrica, isto é, o
ponto focal muda com
os ajustes na posição)
-20 – 90º
Rotação (contínua) 360º
Tamanho Máximo do Disco de Alumínio
(diâmetro) 150 mm
Note-se que a gama tolerável para a movimentação do disco de alumínio depende da distância
deste ao detector e do seu tamanho e ângulo de inclinação [26].

32
São também apresentadas as características do filamento de tungsténio na tabela a seguir:
Tabela 6 - Características do filamento de tungsténio [28], [22].
Brilho 106 A/(cm-2.sr)
Tamanho da Fonte 50 µm
Função de Trabalho 4.5 eV
Vácuo Necessário 10-2 Pa
Vida de Serviço Típica 30 – 100 horas
em que o brilho é definido pela corrente por unidade de área e por unidade de ângulo sólido (em
esferorradiano, sr, isto é, a área da superfície de uma esfera abrangida pelo ângulo a partir do seu
centro) [22].
Figura 18 - SEM S 2400 da Hitachi, no MicroLab do IST [82].

33
2.2 Análise Dinâmica e Mecânica (DMA)
Foi realizada uma análise por DMA para estudar o comportamento mecânico das peças em
função da temperatura, relacionar a microestrutura de cada peça com as suas propriedades
viscoelásticas, determinar as transições térmicas do material das peças (incluindo a transição vítrea) e
observar o efeito dos ciclos térmicos e da alteração do aspeto da superfície no comportamento
mecânico das peças que sofreram essas variações. Os resultados das propriedades viscoelásticas são
tipicamente apresentados com a variação das componentes elástica (E’) e dissipativa (E’’) do módulo
viscoelástico, e de tan δ (coeficiente de amortecimento) em função da temperatura [29].
Os provetes de cada amostra foram ensaiados numa gama de temperaturas compreendida
entre -60ºC e 150ºC (recorrendo ao arrefecimento por azoto líquido, N2líq.) e entre 35ºC e 250ºC, em
que o primeiro ensaio era para observar outras transições que pudessem ocorrer a temperaturas mais
baixas e relacioná-las com as características de cada amostra e o segundo para observar o
comportamento mecânico e viscoelástico deste material para temperaturas mais próximas das
temperaturas de serviço mais elevadas das peças [30].
2.2.1 Princípios de Funcionamento
Na análise por DMA é aplicada à amostra uma tensão (ou extensão) sinusoidal variável,
produzindo uma extensão (ou tensão) sinusoidal que fica atrasada em relação à tensão aplicada por
um ângulo de fase δ. O valor da diferença de fase entre a tensão aplicada e a extensão é dependente
da estrutura do material. No DMA a amostra é encastrada num suporte e a tensão sinusoidal variável
aplicada, de frequência ω, pode ser representada por
𝜎(𝑡) = 𝜎0 sin (𝜔𝑡 + 𝛿) (2)
onde σ0 é a amplitude da tensão máxima e a tensão (em função do tempo t) está adiantada δ em
relação à extensão. A extensão é dada por
휀(𝑡) = 휀0 sin (𝜔𝑡) (3)
onde Ɛ0 é a amplitude da extensão máxima. Estas quantidades estão relacionadas no regime
viscoelástico linear (LVR) por
𝜎(𝑡) = 𝐸∗(𝜔)휀(𝑡) (4)
onde E*(ω) é o módulo dinâmico complexo e
𝐸∗(𝜔) = 𝐸′(𝜔) + 𝑖𝐸′′(𝜔) (5).
E’(ω) e E’’(ω) são a componente elástica dinâmica e a componente dissipativa dinâmica,
respectivamente [31], [32], [33], [34].
Os polímeros comportam-se tanto como um sólido elástico (onde δ é 0º) bem como um líquido
viscoso (onde δ é 90º), sendo por isso viscoelásticos, e por isso vão exibir um δ entre 0º e 90º (figura
19 (a)). A partir de E*(ω) e de δ podem ser calculados E’(ω) e E’’(ω) (figura 19 (b)). Para um polímero
viscoelástico, E’ caracteriza a capacidade do polímero armazenar energia (comportamento elástico),
ou seja, está relacionado com a sua rigidez. Já E’’ revela a tendência do material para dissipar a energia
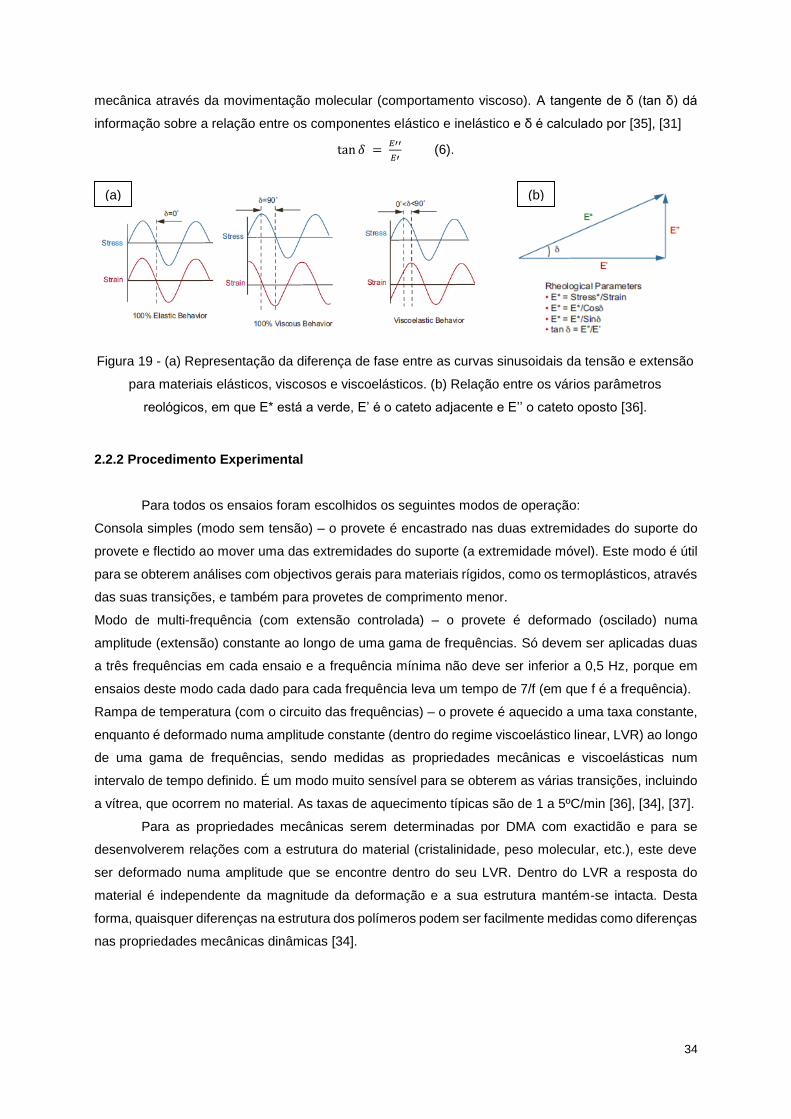
34
mecânica através da movimentação molecular (comportamento viscoso). A tangente de δ (tan δ) dá
informação sobre a relação entre os componentes elástico e inelástico e δ é calculado por [35], [31]
tan 𝛿 = 𝐸′′
𝐸′ (6).
2.2.2 Procedimento Experimental
Para todos os ensaios foram escolhidos os seguintes modos de operação:
Consola simples (modo sem tensão) – o provete é encastrado nas duas extremidades do suporte do
provete e flectido ao mover uma das extremidades do suporte (a extremidade móvel). Este modo é útil
para se obterem análises com objectivos gerais para materiais rígidos, como os termoplásticos, através
das suas transições, e também para provetes de comprimento menor.
Modo de multi-frequência (com extensão controlada) – o provete é deformado (oscilado) numa
amplitude (extensão) constante ao longo de uma gama de frequências. Só devem ser aplicadas duas
a três frequências em cada ensaio e a frequência mínima não deve ser inferior a 0,5 Hz, porque em
ensaios deste modo cada dado para cada frequência leva um tempo de 7/f (em que f é a frequência).
Rampa de temperatura (com o circuito das frequências) – o provete é aquecido a uma taxa constante,
enquanto é deformado numa amplitude constante (dentro do regime viscoelástico linear, LVR) ao longo
de uma gama de frequências, sendo medidas as propriedades mecânicas e viscoelásticas num
intervalo de tempo definido. É um modo muito sensível para se obterem as várias transições, incluindo
a vítrea, que ocorrem no material. As taxas de aquecimento típicas são de 1 a 5ºC/min [36], [34], [37].
Para as propriedades mecânicas serem determinadas por DMA com exactidão e para se
desenvolverem relações com a estrutura do material (cristalinidade, peso molecular, etc.), este deve
ser deformado numa amplitude que se encontre dentro do seu LVR. Dentro do LVR a resposta do
material é independente da magnitude da deformação e a sua estrutura mantém-se intacta. Desta
forma, quaisquer diferenças na estrutura dos polímeros podem ser facilmente medidas como diferenças
nas propriedades mecânicas dinâmicas [34].
Figura 19 - (a) Representação da diferença de fase entre as curvas sinusoidais da tensão e extensão
para materiais elásticos, viscosos e viscoelásticos. (b) Relação entre os vários parâmetros
reológicos, em que E* está a verde, E’ é o cateto adjacente e E’’ o cateto oposto [36].
(a) (b)

35
Durante a preparação dos provetes teve de se ter em consideração alguns factores, como o
local das peças de onde foram retirados os provetes e a redução da espessura dos mesmos. O local
de obtenção dos provetes é importante por causa da anisotropia e, por isso, é para garantir que os
provetes tenham todos a mesma orientação. Estes também tinham de ter uma forma geométrica regular
(neste caso rectangular), tinham de ser planos e não podiam ter zonas de diferente espessura na
superfície, já que esta é considerada constante ao longo do ensaio. Os provetes obtidos directamente
das peças tinham uma espessura de cerca de 5 mm, sendo necessário reduzir esta espessura para
cerca de 2 mm. Esta redução da espessura é necessária, uma vez que os provetes mais espessos vão
retardar mais a taxa de aquecimento do que os provetes mais finos e também porque o material em
estudo tem baixa condutividade térmica, e por essa razão a distribuição da temperatura não é a mesma
no interior e na superfície do provete, o que se agrava para espessuras maiores [34], [38].
Cada provete foi primeiro ensaiado na gama de temperaturas de -60 a 150ºC e depois na gama
de temperaturas de 35 a 250ºC. Seguem-se os parâmetros para os dois ensaios.
Tabela 7 - Parâmetros dos dois ensaios por DMA [38], [37].
Comprimento (modo de consola simples) 10 mm
Binário 6 N.m
Tempo de Estabilização na Temperatura Inicial 5 min
Taxa de Aquecimento 2ºC/min
Frequências 1, 3 e 10 Hz
Amplitude de Deformação 10.0 µm
Uma amplitude de deslocação dinâmica de 10,0 µm vai resultar num pico de extensão de 0,1%
para um provete com 10 mm de comprimento de ensaio, o que mantém o material do provete dentro
do seu LVR. É definida a amplitude de deformação e não a força a aplicar, uma vez que é mais difícil
definir a força exacta a aplicar enquanto a temperatura ainda não estabilizou.
Depois de definir os parâmetros no programa TA AdvantageTM, cada provete foi inserido no
suporte do provete (figura 21) e fixado pela aplicação de um binário nos parafusos de cada extremidade
do suporte, sendo depois fechado o forno e iniciado o ensaio.

36
2.2.3 Equipamento
Utilizou-se o DMA do modelo Q800 da Thermal Analysis Instruments (TA Instruments) e o
programa que controla os parâmetros do ensaio é o TA AdvantageTM para as séries Q. O DMA Q800 é
um instrumento de tensão controlada (figura 21), mas a amplitude (extensão) é que é o parâmetro
imposto pelo utilizador. Um DMA de tensão controlada é composto por um motor, que aplica a tensão
ao provete, e um sensor de deslocamento que mede a amplitude (extensão). Ao programar o
instrumento para o ensaio, é solicitada uma deformação na forma de amplitude e depois o instrumento
aplica a tensão necessária (o parâmetro que controla) até medir a amplitude que foi solicitada. A
amplitude medida é constante ao longo do teste, enquanto a tensão controlada é alterada à medida
que a rigidez da amostra se altera para manter a amplitude constante [34].
Figura 21 - Esquema genérico de um DMA por tensão controlada [34].
Figura 20 - (a) Exemplo de um suporte do provete para consola simples e dupla num DMA (para
consola simples só se fixa o provete numa das extremidades e na parte móvel do suporte, que é a
do meio) [35]. (b) Fotografia de dois provetes para o ensaio por DMA, ao lado de uma moeda de 5
cêntimos. (c) Esquema da montagem de um provete em consola simples [80].
(a)
(b) (c)

37
De seguida está uma breve descrição da função dos vários constituintes do DMA Q800:
Motor de accionamento – (directo e de não-contacto) fornece a tensão oscilatória necessária.
Rolamentos pneumáticos – recebem a tensão oscilatória do motor de accionamento e estão conectados
ao eixo de transmissão e ao suporte do provete, que vão realizar o movimento vertical durante as
oscilações.
Forno – garante o aquecimento controlado durante o ensaio e tem de ser aberto para se inserir o
provete.
Leitor óptico – mede o deslocamento, baseado em padrões de difracção da luz através de grelhas.
Suportes do provete – são optimizados por uma análise de elementos finitos de modo a fornecer rigidez
elevada (para minimizar a susceptibilidade mecânica deste suporte) com massa reduzida (para garantir
uma calibração da temperatura rápida), estando ligados ao eixo de transmissão.
Compartimento rígido de alumínio – o motor de accionamento, os rolamentos pneumáticos e o leitor
óptico encontram-se dentro deste compartimento, o que minimiza a susceptibilidade mecânica do
sistema e também garante a obtenção de dados precisos porque tem a temperatura controlada [35],
[39].
Seguem-se algumas imagens do DMA Q800, mostrando também um esquema do interior do
equipamento com os vários constituintes.
Figura 22 - Fotografias do DMA Q800 da TA Instruments no laboratório ICTPol do IST, com
o forno fechado (esquerda) e com o forno aberto (direita).

38
Na tabela seguinte estão as especificações do DMA Q800.
Tabela 8 - Especificações do equipamento DMA Q800 da TA Instruments [35], [34].
Força 0.0001 – 18 N
Resolução da Força 0.00001 N
Amplitude (gama de deformação dinâmica da
amostra) +/- 0.5 – 10.0 µm
Resolução da Extensão 1 nm
Módulo 103 – 3x1012 Pa
Precisão do Módulo +/- 1%
Rigidez 100 – 106 N/m
Sensibilidade da Tan δ 0.0001
Resolução da Tan δ 0.00001
Gama de Frequências 0.01 – 200 Hz
Gama de Temperaturas -150 – 600ºC
Taxa de Aquecimento 0.1 – 20ºC/min
Taxa de Arrefecimento 0.1 – 10ºC/min
Estabilidade da Isotérmica +/- 0.1ºC
Figura 23 - Disposição dos vários constituintes do DMA Q800 da TA Instruments [35].

39
Dimensões Máximas do Provete para o Modo
de Deformação em Consola Simples (10 mm de
comprimento)
15 mm de largura e 5 mm de espessura
É o acessório de arrefecimento por gás (GCA) que estende a gama de temperaturas do DMA
Q800 até -150ºC (figura 24). O GCA usa gás de azoto frio gerado pela evaporação controlada de N2líq.,
proporcionando taxas de arrefecimento controladas em toda a gama de temperaturas do DMA Q800
[35].
Figura 24 - Fotografias do GCA (esquerda) e da sua ligação ao DMA Q800 da TA
Instruments, no laboratório ICTPol do IST (direita).

40
2.3 Calorimetria Diferencial por Varrimento (DSC)
Para completar a análise por DMA, as amostras de cada peça foram também caracterizadas
por DSC. Esta técnica permite obter uma análise quantitativa das transições de fase de primeira ordem
(transições que envolvem uma mudança na entalpia e no calor específico, como a cristalização e a
fusão), enquanto a técnica de DMA fornece informação mais detalhada das transições de segunda
ordem, como a transição vítrea (transições que apenas envolvem uma mudança no calor específico).
No entanto, a técnica DSC também permite obter informação sobre a transição vítrea [40].
Foram realizados dois ensaios: o primeiro para obter dados sobre o efeito da história térmica
no comportamento do material de cada amostra (efeitos do processamento e dos ciclos térmicos) e o
segundo para remover a história térmica (dados do segundo aquecimento, depois do primeiro
aquecimento até à fusão), para ser possível comparar directamente a composição do material de cada
amostra.
2.3.1 Princípios de Funcionamento
A técnica de DSC regista a energia necessária (fluxo de calor) para estabelecer a mesma
temperatura entre a amostra e uma referência, em função do tempo ou da temperatura quando
aquecidas a uma taxa controlada. Num DSC com forno simples (figura 25), a cápsula contendo a
amostra e a cápsula de referência, normalmente vazia, são aquecidas no mesmo forno (mas com uma
fonte de aquecimento individual para cada cápsula) [41]. Durante o aquecimento o material da amostra
vai sofrendo transições de fase. As transições endotérmicas (como a fusão) necessitam que a amostra
absorva calor e a cápsula da amostra fica mais fria que a cápsula de referência, resultando numa
temperatura diferencial negativa (pico de depressão na curva obtida por DSC). Já as transições
exotérmicas (como a cristalização) libertam calor e a cápsula da amostra fica mais quente que a cápsula
de referência, resultando numa temperatura diferencial positiva (pico de elevação na curva DSC). Neste
tipo de forno é medida a diferença entre a temperatura da cápsula da amostra e da cápsula de
referência e calculada a maior ou menor quantidade de fluxo de calor necessária a fornecer a uma das
cápsulas para anular essa diferença de temperatura [42], [43], [44].
Figura 25 - Esquema de um DSC de forno simples (fonte de aquecimento para cada cápsula) [43].
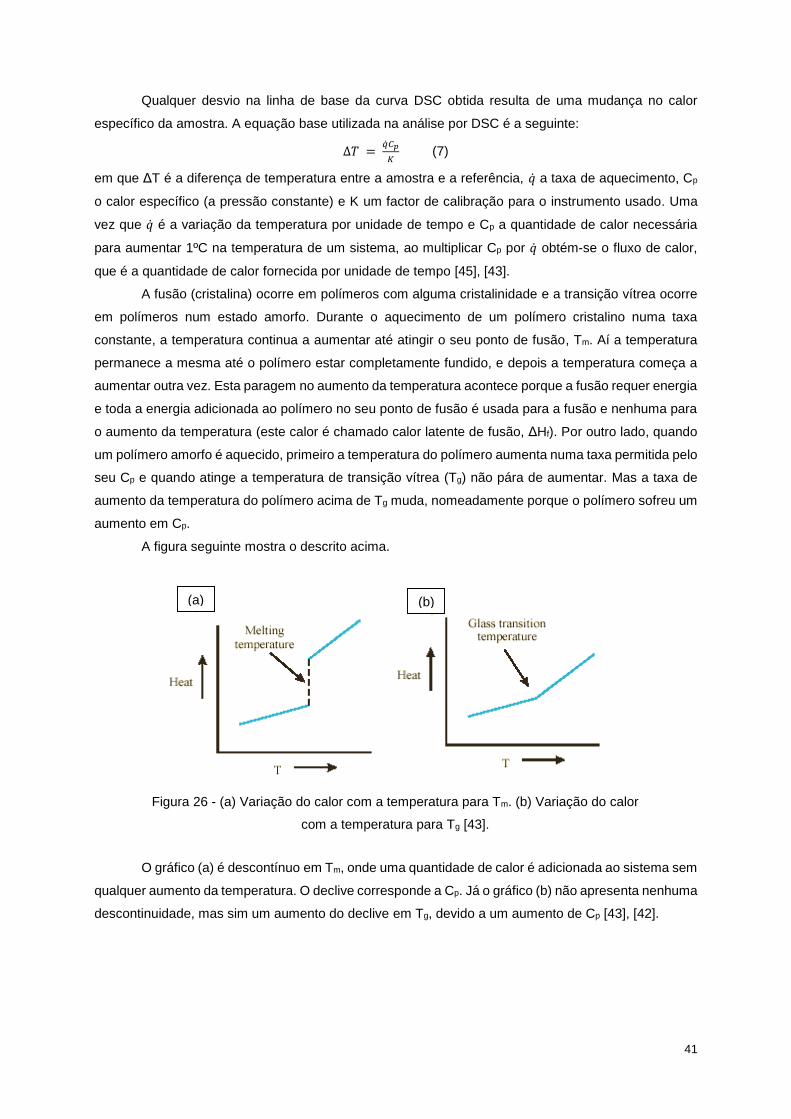
41
Qualquer desvio na linha de base da curva DSC obtida resulta de uma mudança no calor
específico da amostra. A equação base utilizada na análise por DSC é a seguinte:
∆𝑇 = �̇�𝐶𝑝
𝐾 (7)
em que ΔT é a diferença de temperatura entre a amostra e a referência, �̇� a taxa de aquecimento, Cp
o calor específico (a pressão constante) e K um factor de calibração para o instrumento usado. Uma
vez que �̇� é a variação da temperatura por unidade de tempo e Cp a quantidade de calor necessária
para aumentar 1ºC na temperatura de um sistema, ao multiplicar Cp por �̇� obtém-se o fluxo de calor,
que é a quantidade de calor fornecida por unidade de tempo [45], [43].
A fusão (cristalina) ocorre em polímeros com alguma cristalinidade e a transição vítrea ocorre
em polímeros num estado amorfo. Durante o aquecimento de um polímero cristalino numa taxa
constante, a temperatura continua a aumentar até atingir o seu ponto de fusão, Tm. Aí a temperatura
permanece a mesma até o polímero estar completamente fundido, e depois a temperatura começa a
aumentar outra vez. Esta paragem no aumento da temperatura acontece porque a fusão requer energia
e toda a energia adicionada ao polímero no seu ponto de fusão é usada para a fusão e nenhuma para
o aumento da temperatura (este calor é chamado calor latente de fusão, ΔHf). Por outro lado, quando
um polímero amorfo é aquecido, primeiro a temperatura do polímero aumenta numa taxa permitida pelo
seu Cp e quando atinge a temperatura de transição vítrea (Tg) não pára de aumentar. Mas a taxa de
aumento da temperatura do polímero acima de Tg muda, nomeadamente porque o polímero sofreu um
aumento em Cp.
A figura seguinte mostra o descrito acima.
O gráfico (a) é descontínuo em Tm, onde uma quantidade de calor é adicionada ao sistema sem
qualquer aumento da temperatura. O declive corresponde a Cp. Já o gráfico (b) não apresenta nenhuma
descontinuidade, mas sim um aumento do declive em Tg, devido a um aumento de Cp [43], [42].
Figura 26 - (a) Variação do calor com a temperatura para Tm. (b) Variação do calor
com a temperatura para Tg [43].
(a) (b)

42
2.3.2 Procedimento Experimental
Os provetes a analisar por DSC foram retirados do mesmo local de interesse para os provetes
para o DMA, para terem condições de processamento, tempo de arrefecimento e microestruturas
semelhantes. Antes dos provetes serem colocados no forno do DSC, é medida a massa da cápsula de
alumínio vazia (21,2 mg) onde vai ser inserido cada provete. Depois é inserido o provete dentro da
cápsula, esta é vedada (não hermeticamente) e medida novamente. A diferença entre as duas medidas
é a massa do provete. De seguida são colocadas dentro do forno do DSC a cápsula de alumínio com
o provete a analisar e a cápsula de referência vazia. A figura 27 mostra um provete para analisar no
DSC e uma cápsula de alumínio aberta e fechada.
Seguem-se os parâmetros de funcionamento para os dois ensaios (usou-se N2líq. no
arrefecimento de 280ºC para 30ºC no 2º ensaio, a uma taxa de arrefecimento de 10ºC/min). Primeiro é
efectuado um aquecimento até à fusão, seguido de um arrefecimento controlado até à temperatura
inicial e depois a amostra é aquecida novamente até à fusão.
Tabela 9 - Parâmetros de funcionamento dos dois ensaios efectuados por DSC [46].
Temperatura Inicial 30ºC
Temperatura Final 280ºC
Taxa de Aquecimento 10ºC/min
Atmosfera (gás de purga) Hélio
A taxa de aquecimento tem de ser um compromisso entre a sensibilidade das transições obtida
e a resolução conseguida, já que a sensibilidade das transições aumenta com o aumento da taxa de
varrimento mas a resolução diminui com o aumento da mesma [44].
2.3.3 Equipamento
Utilizou-se o equipamento DSC do modelo DSC 2920 Modulated DSC da TA Instruments, a
prensa que vedou as cápsulas de alumínio também é da TA Instruments (figuras 28 e 29) e as medidas
da massa dos provetes e das cápsulas foram efectuadas numa balança da VWR® (mede em gramas,
até quatro casas decimais).
Figura 27 - Fotografia de um provete a analisar no DSC ao lado de uma moeda de 5
cêntimos (esquerda) e exemplo de uma cápsula de alumínio aberta e fechada (direita) [84].

43
O DSC 2920 determina a temperatura e o fluxo de calor associados às transições do material,
em função do tempo e da temperatura. Também fornece dados quantitativos e qualitativos de
processos endotérmicos e exotérmicos que ocorrem nos materiais durante as suas transições físicas.
Este equipamento tem duas partes principais, o instrumento em si que contém o sistema electrónico e
a célula (forno) onde se inserem as cápsulas com os provetes e as cápsulas de referência, e que
contém os termopares para monitorizarem o fluxo de calor e a temperatura. A célula do DSC 2920 é
usada para medir o fluxo de calor diferencial. A cápsula com o provete e a cápsula de referência são
colocadas em plataformas elevadas num disco constante e o calor é transferido através do disco para
o provete e para a referência [46].
A tabela 10 mostra as especificações da célula do DSC 2920 Modulated DSC.
Tabela 10 - Especificações da célula do forno do DSC 2920 Modulated DSC da TA Instruments, onde
se inserem as cápsulas com o provete e com a referência [46].
Massa 1.7 kg
Volume 2 cm3
Gama de Temperaturas 23 – 725ºC (desde -150ºC com AL)
Repetibilidade da Temperatura +/- 0.1ºC
Sensibilidade Calorimétrica 0.2 µW (rms)
Ruído da Linha de Base 0.1 µW (rms)
Massa do Provete 0.5 – 100 mg
Gama de Temperaturas para a Cápsula de
Alumínio -180 – 600ºC
Figura 28 - Imagem do equipamento DSC 2920 Modulated DSC da TA Instruments.

44
Figura 29 - Esquema do DSC 2920 Modulated DSC da TA Instruments (esquerda) e da prensa para
vedar as cápsulas de alumínio (direita) [46].
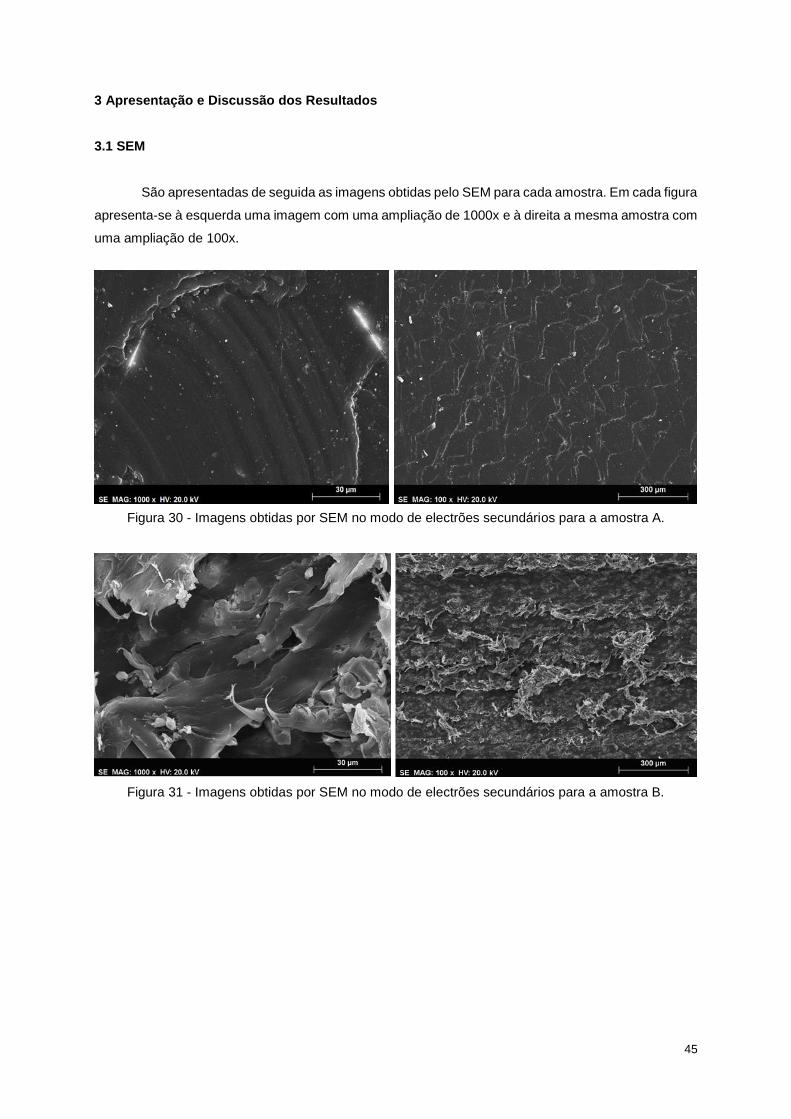
45
3 Apresentação e Discussão dos Resultados
3.1 SEM
São apresentadas de seguida as imagens obtidas pelo SEM para cada amostra. Em cada figura
apresenta-se à esquerda uma imagem com uma ampliação de 1000x e à direita a mesma amostra com
uma ampliação de 100x.
Figura 30 - Imagens obtidas por SEM no modo de electrões secundários para a amostra A.
Figura 31 - Imagens obtidas por SEM no modo de electrões secundários para a amostra B.
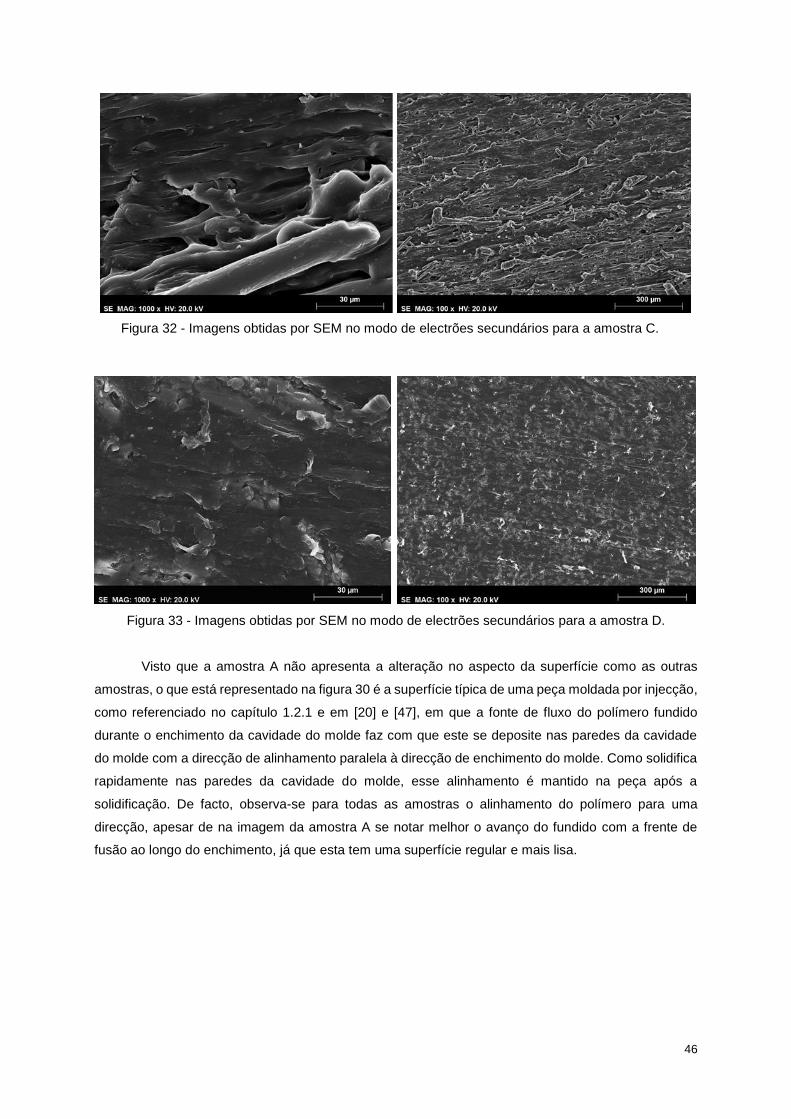
46
Figura 33 - Imagens obtidas por SEM no modo de electrões secundários para a amostra D.
Visto que a amostra A não apresenta a alteração no aspecto da superfície como as outras
amostras, o que está representado na figura 30 é a superfície típica de uma peça moldada por injecção,
como referenciado no capítulo 1.2.1 e em [20] e [47], em que a fonte de fluxo do polímero fundido
durante o enchimento da cavidade do molde faz com que este se deposite nas paredes da cavidade
do molde com a direcção de alinhamento paralela à direcção de enchimento do molde. Como solidifica
rapidamente nas paredes da cavidade do molde, esse alinhamento é mantido na peça após a
solidificação. De facto, observa-se para todas as amostras o alinhamento do polímero para uma
direcção, apesar de na imagem da amostra A se notar melhor o avanço do fundido com a frente de
fusão ao longo do enchimento, já que esta tem uma superfície regular e mais lisa.
Figura 32 - Imagens obtidas por SEM no modo de electrões secundários para a amostra C.

47
As imagens da superfície das amostras B, C e D evidenciam uma alteração da rugosidade e
um aumento da irregularidade da superfície das peças com 50% de FV, não sendo possível observar
uma superfície típica de uma peça moldada por injecção.
Com estas imagens obtidas por SEM não é possível fazer uma análise quantitativa da
rugosidade, no entanto, a imagem da superfície da amostra B mostra que esta é muito irregular numa
escala de 30 µm. Com o aumento dos ciclos térmicos nota-se uma evolução da superfície das peças
sujeitas a esses ciclos, que é confirmada pelas imagens das amostras C e D. À medida que estas peças
vão estando em serviço vão sofrendo desgaste provocado pelo atrito, o que vai diminuir a rugosidade
da sua superfície, tornando-a mais plana, para além das peças estarem sujeitas a temperaturas
elevadas [48], [49]. Esta alteração no aspecto da superfície pode levar a problemas relacionados com
as tolerâncias dimensionais nas zonas de ligação das peças com outros componentes do sistema de
refrigeração do motor de automóveis.
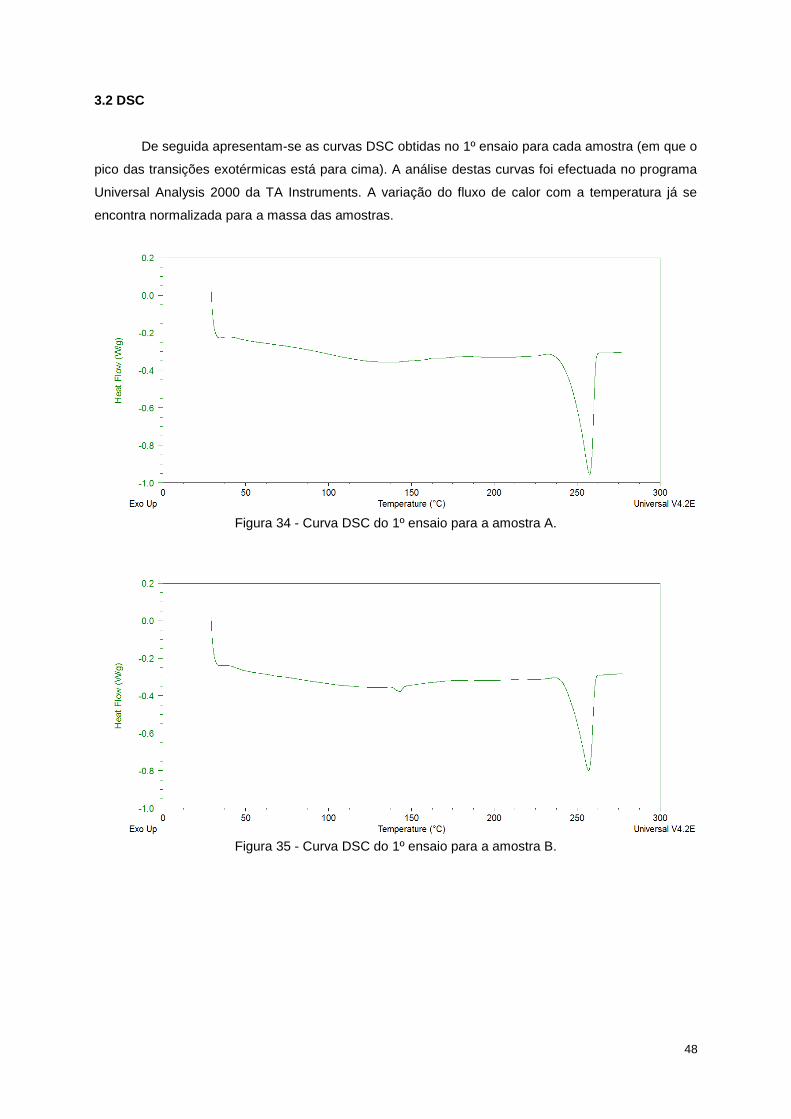
48
3.2 DSC
De seguida apresentam-se as curvas DSC obtidas no 1º ensaio para cada amostra (em que o
pico das transições exotérmicas está para cima). A análise destas curvas foi efectuada no programa
Universal Analysis 2000 da TA Instruments. A variação do fluxo de calor com a temperatura já se
encontra normalizada para a massa das amostras.
Figura 35 - Curva DSC do 1º ensaio para a amostra B.
Figura 34 - Curva DSC do 1º ensaio para a amostra A.
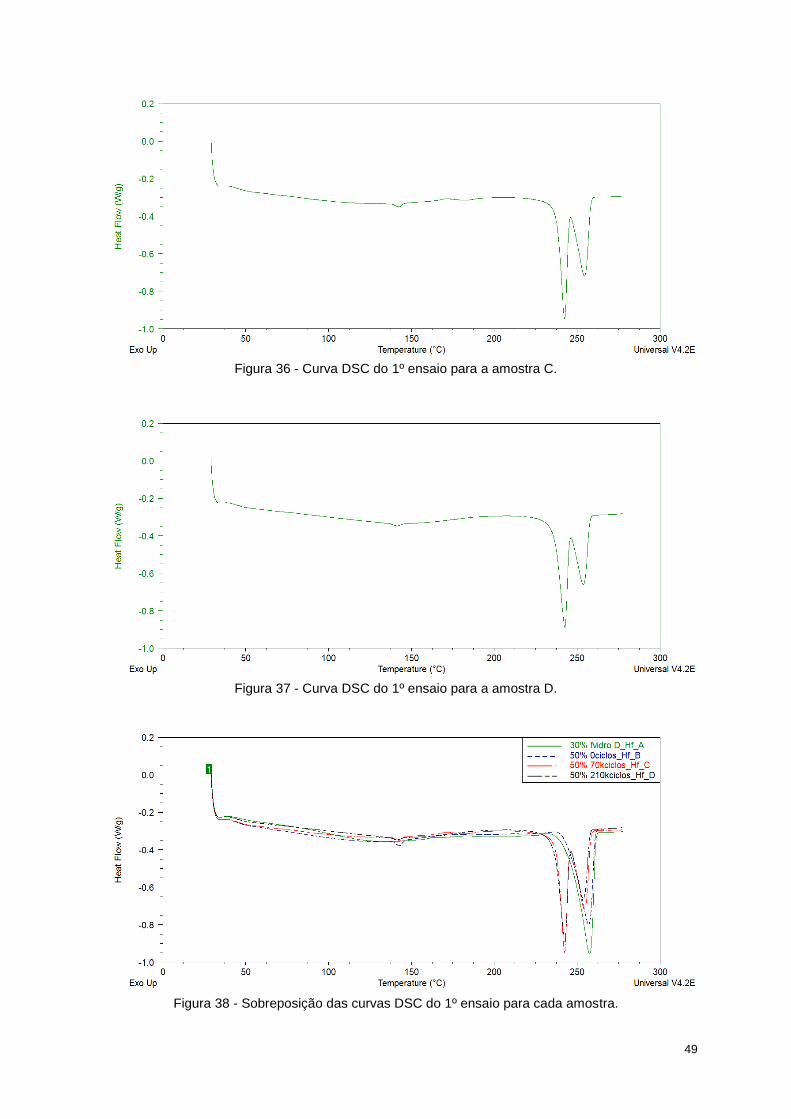
49
Figura 37 - Curva DSC do 1º ensaio para a amostra D.
Figura 38 - Sobreposição das curvas DSC do 1º ensaio para cada amostra.
Figura 36 - Curva DSC do 1º ensaio para a amostra C.

50
De seguida apresentam-se as curvas DSC obtidas para o 2º ensaio de cada amostra,
mostrando apenas o resultado do segundo aquecimento.
Figura 39 - Curva DSC do 2º ensaio para a amostra A.
Figura 40 - Curva DSC do 2º ensaio para a amostra B.
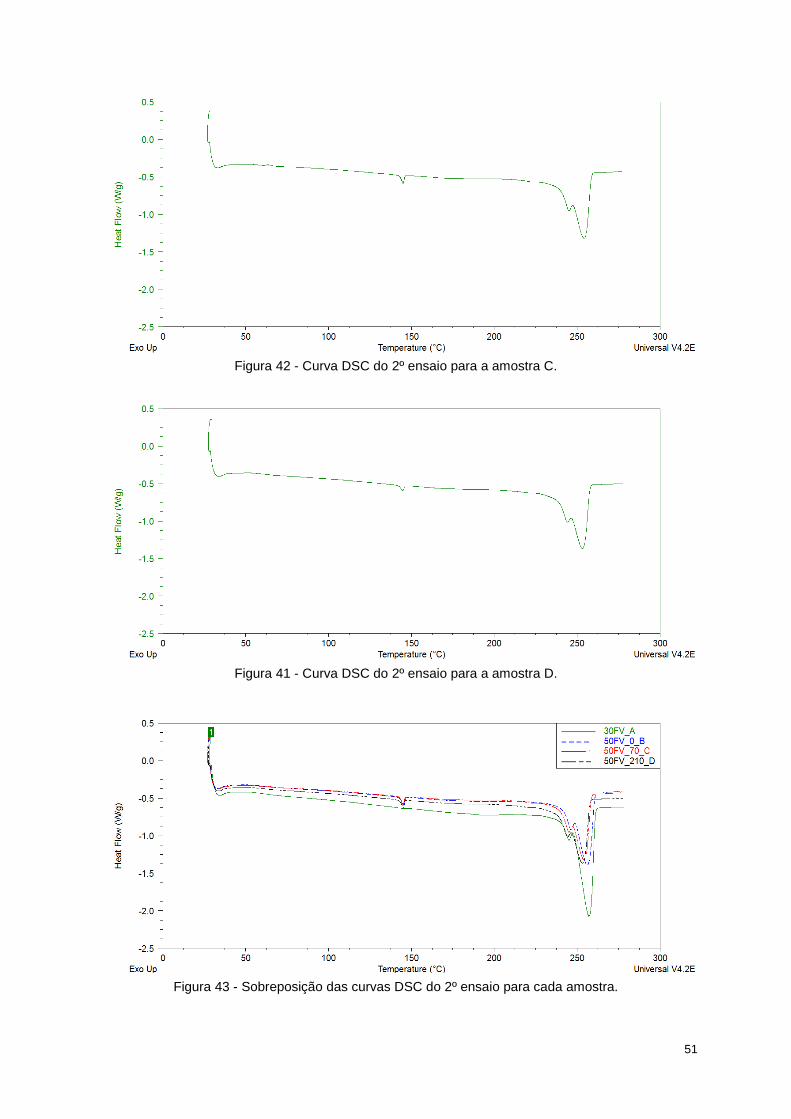
51
Figura 42 - Curva DSC do 2º ensaio para a amostra C.
Figura 43 - Sobreposição das curvas DSC do 2º ensaio para cada amostra.
Figura 41 - Curva DSC do 2º ensaio para a amostra D.
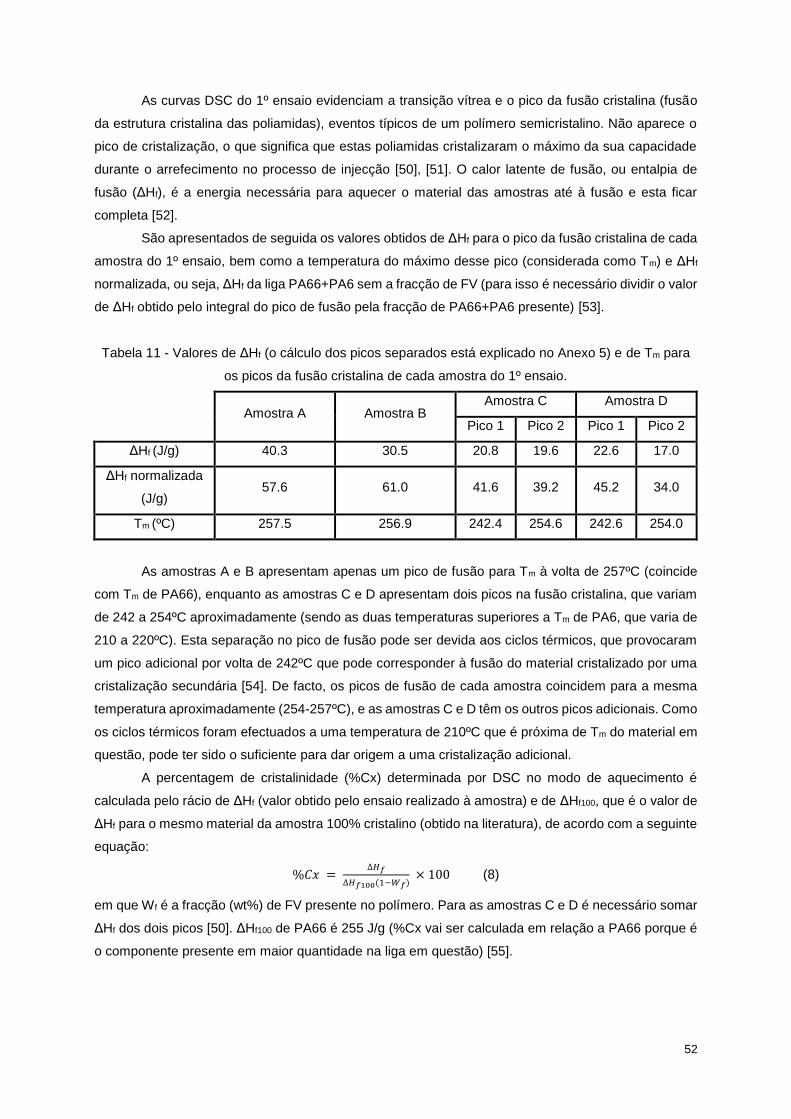
52
As curvas DSC do 1º ensaio evidenciam a transição vítrea e o pico da fusão cristalina (fusão
da estrutura cristalina das poliamidas), eventos típicos de um polímero semicristalino. Não aparece o
pico de cristalização, o que significa que estas poliamidas cristalizaram o máximo da sua capacidade
durante o arrefecimento no processo de injecção [50], [51]. O calor latente de fusão, ou entalpia de
fusão (ΔHf), é a energia necessária para aquecer o material das amostras até à fusão e esta ficar
completa [52].
São apresentados de seguida os valores obtidos de ΔHf para o pico da fusão cristalina de cada
amostra do 1º ensaio, bem como a temperatura do máximo desse pico (considerada como Tm) e ΔHf
normalizada, ou seja, ΔHf da liga PA66+PA6 sem a fracção de FV (para isso é necessário dividir o valor
de ΔHf obtido pelo integral do pico de fusão pela fracção de PA66+PA6 presente) [53].
Tabela 11 - Valores de ΔHf (o cálculo dos picos separados está explicado no Anexo 5) e de Tm para
os picos da fusão cristalina de cada amostra do 1º ensaio.
Amostra A Amostra B Amostra C Amostra D
Pico 1 Pico 2 Pico 1 Pico 2
ΔHf (J/g) 40.3 30.5 20.8 19.6 22.6 17.0
ΔHf normalizada
(J/g) 57.6 61.0 41.6 39.2 45.2 34.0
Tm (ºC) 257.5 256.9 242.4 254.6 242.6 254.0
As amostras A e B apresentam apenas um pico de fusão para Tm à volta de 257ºC (coincide
com Tm de PA66), enquanto as amostras C e D apresentam dois picos na fusão cristalina, que variam
de 242 a 254ºC aproximadamente (sendo as duas temperaturas superiores a Tm de PA6, que varia de
210 a 220ºC). Esta separação no pico de fusão pode ser devida aos ciclos térmicos, que provocaram
um pico adicional por volta de 242ºC que pode corresponder à fusão do material cristalizado por uma
cristalização secundária [54]. De facto, os picos de fusão de cada amostra coincidem para a mesma
temperatura aproximadamente (254-257ºC), e as amostras C e D têm os outros picos adicionais. Como
os ciclos térmicos foram efectuados a uma temperatura de 210ºC que é próxima de Tm do material em
questão, pode ter sido o suficiente para dar origem a uma cristalização adicional.
A percentagem de cristalinidade (%Cx) determinada por DSC no modo de aquecimento é
calculada pelo rácio de ΔHf (valor obtido pelo ensaio realizado à amostra) e de ΔHf100, que é o valor de
ΔHf para o mesmo material da amostra 100% cristalino (obtido na literatura), de acordo com a seguinte
equação:
%𝐶𝑥 = ∆𝐻𝑓
∆𝐻𝑓100(1−𝑊𝑓) × 100 (8)
em que Wf é a fracção (wt%) de FV presente no polímero. Para as amostras C e D é necessário somar
ΔHf dos dois picos [50]. ΔHf100 de PA66 é 255 J/g (%Cx vai ser calculada em relação a PA66 porque é
o componente presente em maior quantidade na liga em questão) [55].
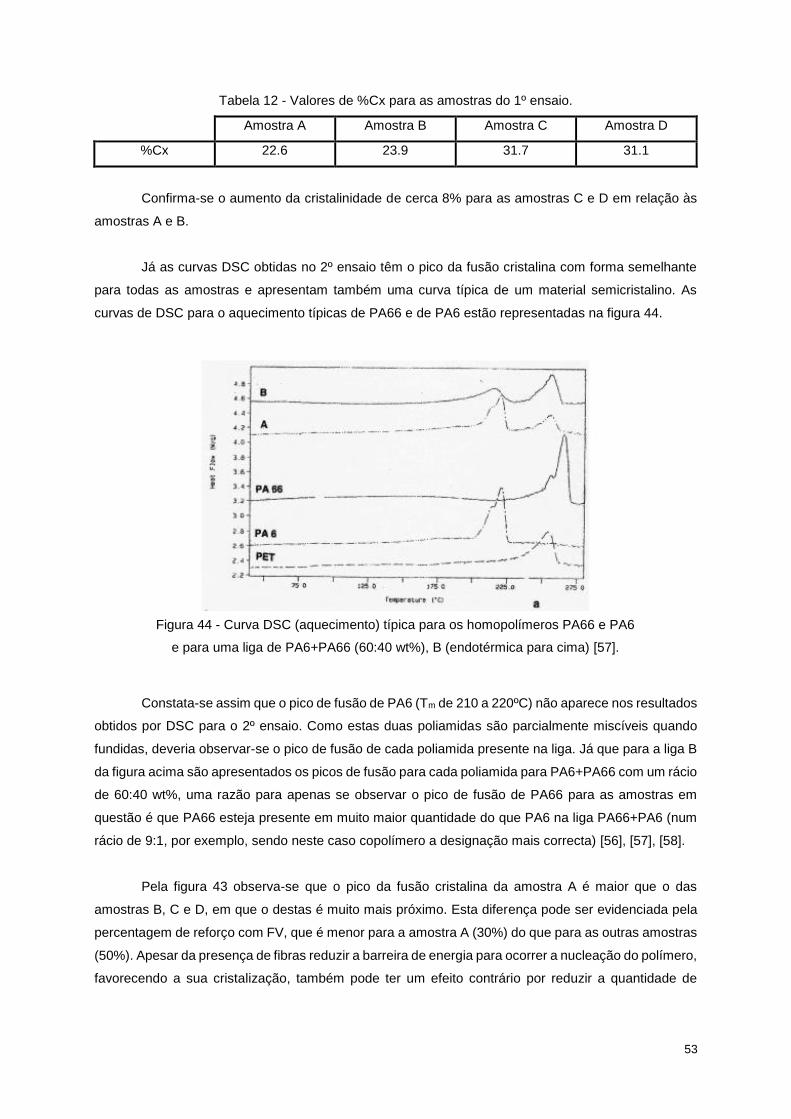
53
Tabela 12 - Valores de %Cx para as amostras do 1º ensaio.
Amostra A Amostra B Amostra C Amostra D
%Cx 22.6 23.9 31.7 31.1
Confirma-se o aumento da cristalinidade de cerca 8% para as amostras C e D em relação às
amostras A e B.
Já as curvas DSC obtidas no 2º ensaio têm o pico da fusão cristalina com forma semelhante
para todas as amostras e apresentam também uma curva típica de um material semicristalino. As
curvas de DSC para o aquecimento típicas de PA66 e de PA6 estão representadas na figura 44.
Constata-se assim que o pico de fusão de PA6 (Tm de 210 a 220ºC) não aparece nos resultados
obtidos por DSC para o 2º ensaio. Como estas duas poliamidas são parcialmente miscíveis quando
fundidas, deveria observar-se o pico de fusão de cada poliamida presente na liga. Já que para a liga B
da figura acima são apresentados os picos de fusão para cada poliamida para PA6+PA66 com um rácio
de 60:40 wt%, uma razão para apenas se observar o pico de fusão de PA66 para as amostras em
questão é que PA66 esteja presente em muito maior quantidade do que PA6 na liga PA66+PA6 (num
rácio de 9:1, por exemplo, sendo neste caso copolímero a designação mais correcta) [56], [57], [58].
Pela figura 43 observa-se que o pico da fusão cristalina da amostra A é maior que o das
amostras B, C e D, em que o destas é muito mais próximo. Esta diferença pode ser evidenciada pela
percentagem de reforço com FV, que é menor para a amostra A (30%) do que para as outras amostras
(50%). Apesar da presença de fibras reduzir a barreira de energia para ocorrer a nucleação do polímero,
favorecendo a sua cristalização, também pode ter um efeito contrário por reduzir a quantidade de
Figura 44 - Curva DSC (aquecimento) típica para os homopolímeros PA66 e PA6
e para uma liga de PA6+PA66 (60:40 wt%), B (endotérmica para cima) [57].
54
polímero presente que pode cristalizar [59], [60], [61]. No entanto, ao retirar W f, confirma-se que %Cx
é superior para as amostras com 50% de FV.
De seguida são apresentados os valores de ΔHf, ΔHf normalizada, Tm e %Cx para as amostras
do 2º ensaio.
Tabela 13 - Valores de ΔHf, Tm e %Cx para as amostras do 2º ensaio.
Amostra A Amostra B Amostra C Amostra D
ΔHf (J/g) 101.4 80.6 77.3 77.4
ΔHf normalizada
(J/g) 144.9 161.2 154.6 154.8
Tm (ºC) 257.2 255.9 254.2 253.1
%Cx 56.8 63.2 60.6 60.7
Pelos valores da tabela acima verifica-se que %Cx obtida no 2º ensaio tem um valor adicional
de cerca de 30% em relação ao valor de %Cx do 1º ensaio, que pode ser explicado pelo facto da taxa
de arrefecimento do processo de injecção ser superior à taxa de arrefecimento do ensaio DSC
(10ºC/min, desde os 280ºC até aos 30ºC).
As amostras do 1º ensaio com 50% de FV apresentam um pequeno pico endotérmico por volta
dos 150ºC que a amostra com 30% de FV não evidencia, o que é confirmado pelos resultados do 2º
ensaio, onde este pico é mais nítido. Este pico pode representar a fusão do ácido adípico (Tm de 152ºC).
Este ácido pode ser o ácido adípico de PA66 ou então o ácido presente em lubrificantes usados no
processamento de componentes em poliamida (neste caso tinha entrado na formulação da composição
da liga PA66+6 das amostras com 50% de FV, já que o pico mantém-se constante para todas as
amostras com 50% de FV nos dois ensaios). Este pico adicional sugere uma composição da liga
PA66+PA6 da amostra A diferente da composição da liga das amostras B, C e D [62], [55], [63].
Os resultados obtidos pela técnica DSC mostram que os ciclos térmicos têm efeito no aumento
da cristalinidade das amostras sujeitas a esses ciclos e mostra também que as amostras das peças
sujeitas à alteração no aspecto da superfície apresentam um pico endotérmico adicional por volta dos
150ºC, evidenciando uma composição diferente.
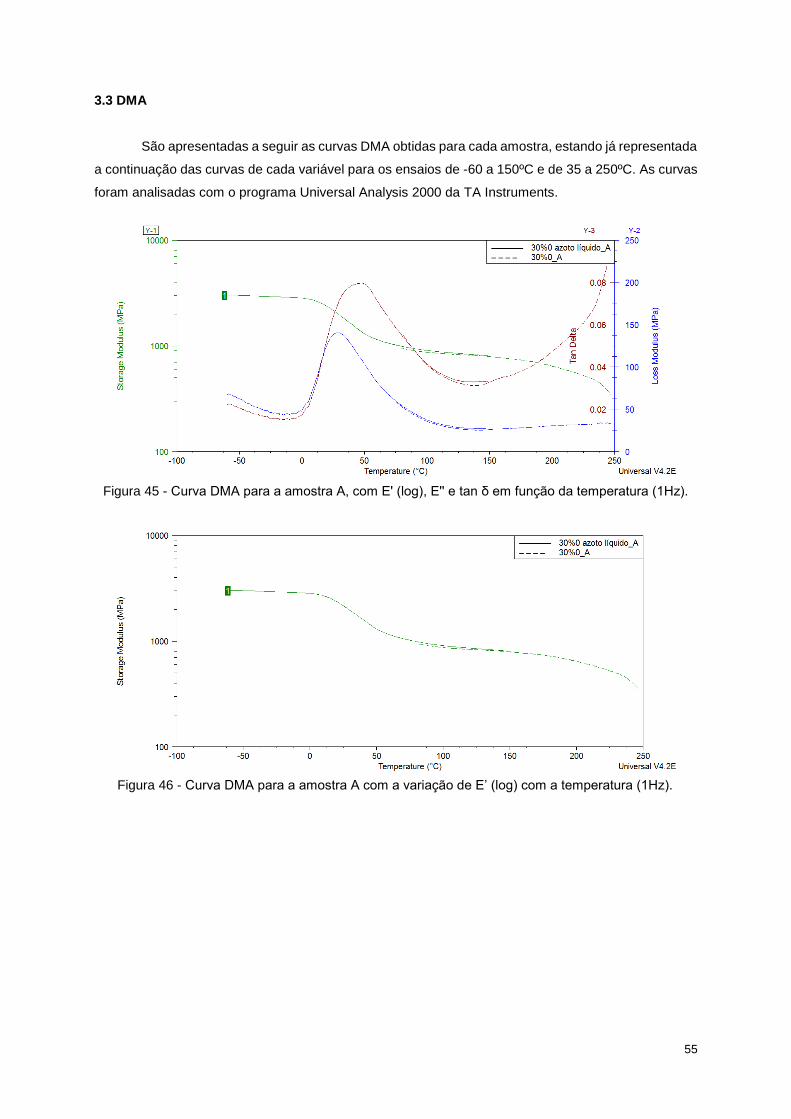
55
3.3 DMA
São apresentadas a seguir as curvas DMA obtidas para cada amostra, estando já representada
a continuação das curvas de cada variável para os ensaios de -60 a 150ºC e de 35 a 250ºC. As curvas
foram analisadas com o programa Universal Analysis 2000 da TA Instruments.
Figura 45 - Curva DMA para a amostra A, com E' (log), E'' e tan δ em função da temperatura (1Hz).
Figura 46 - Curva DMA para a amostra A com a variação de E’ (log) com a temperatura (1Hz).
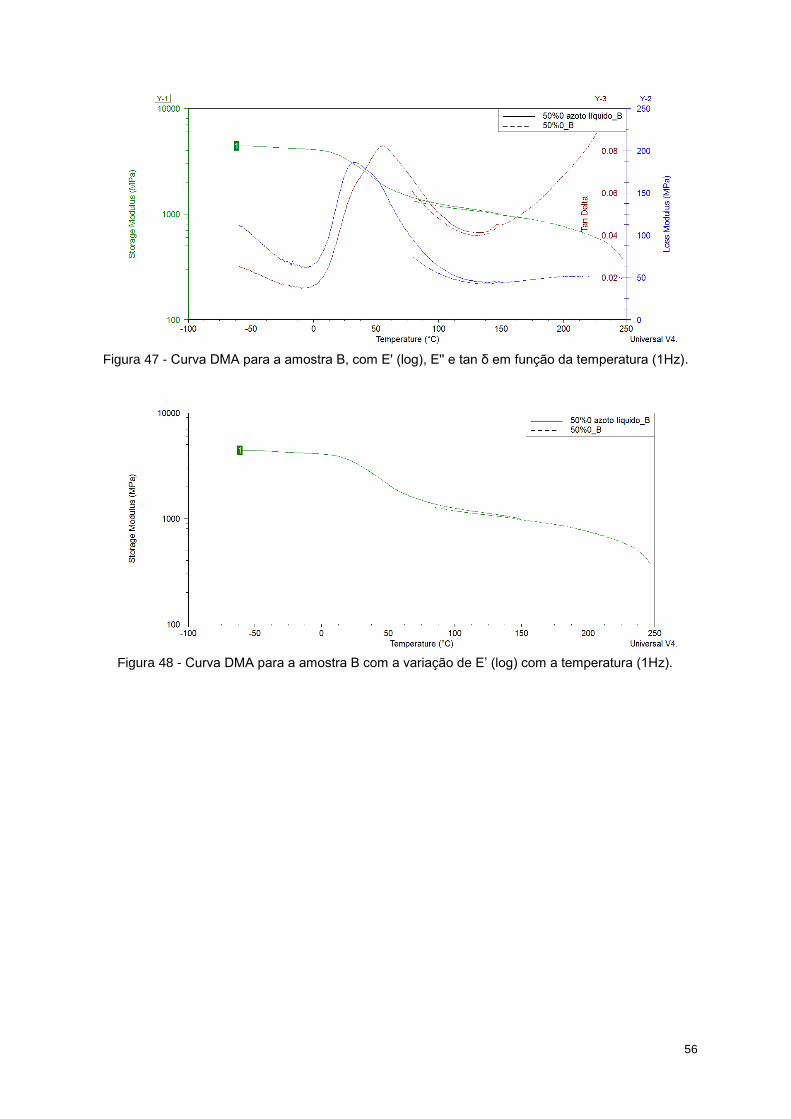
56
Figura 47 - Curva DMA para a amostra B, com E' (log), E'' e tan δ em função da temperatura (1Hz).
Figura 48 - Curva DMA para a amostra B com a variação de E’ (log) com a temperatura (1Hz).
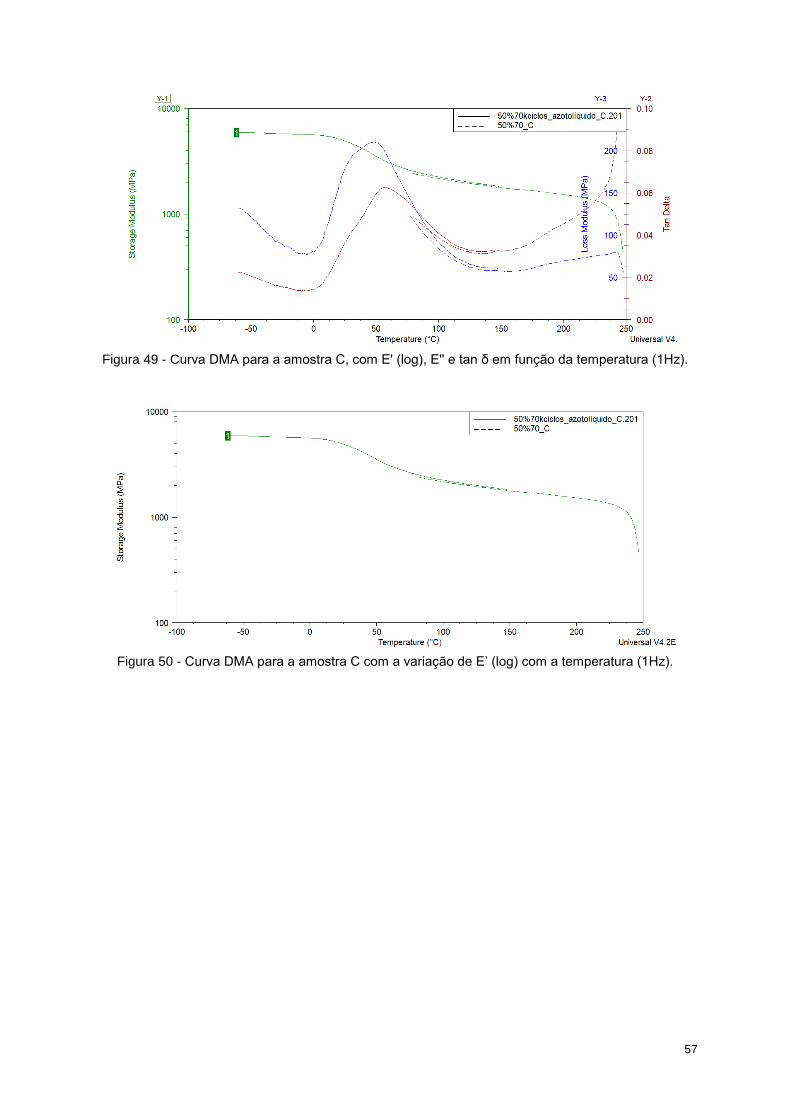
57
Figura 49 - Curva DMA para a amostra C, com E' (log), E'' e tan δ em função da temperatura (1Hz).
Figura 50 - Curva DMA para a amostra C com a variação de E’ (log) com a temperatura (1Hz).
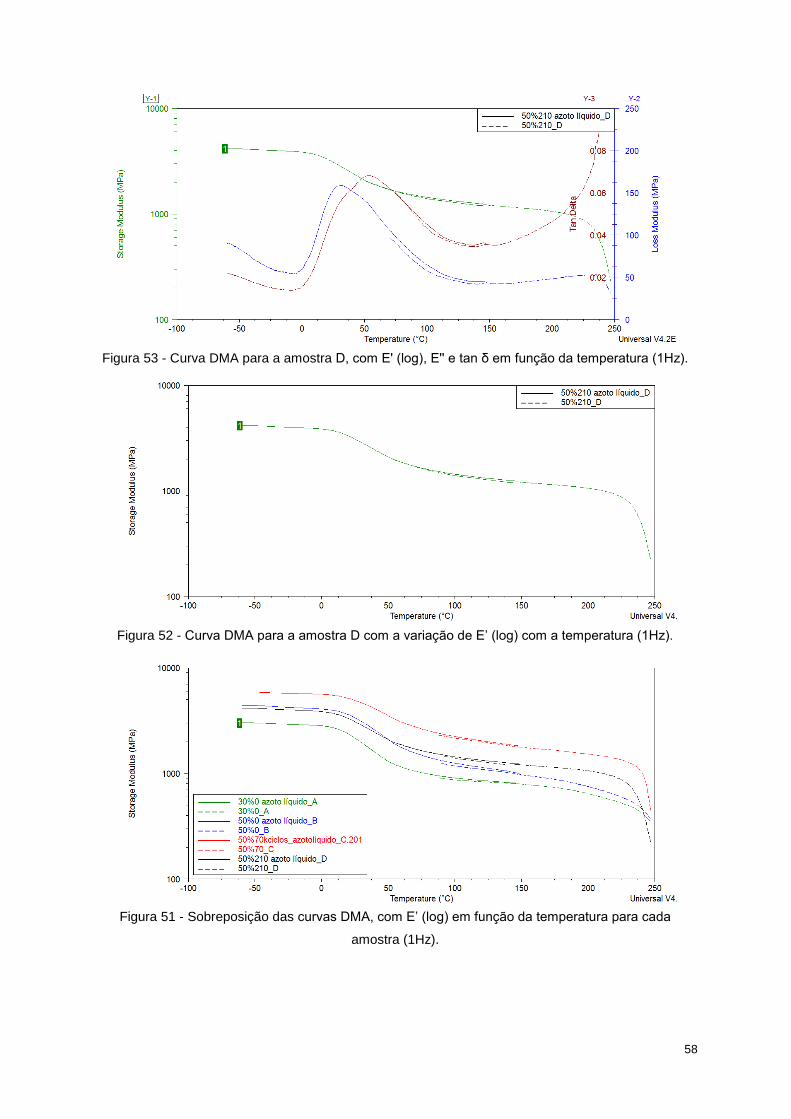
58
Figura 53 - Curva DMA para a amostra D, com E' (log), E'' e tan δ em função da temperatura (1Hz).
Figura 52 - Curva DMA para a amostra D com a variação de E’ (log) com a temperatura (1Hz).
Figura 51 - Sobreposição das curvas DMA, com E’ (log) em função da temperatura para cada
amostra (1Hz).
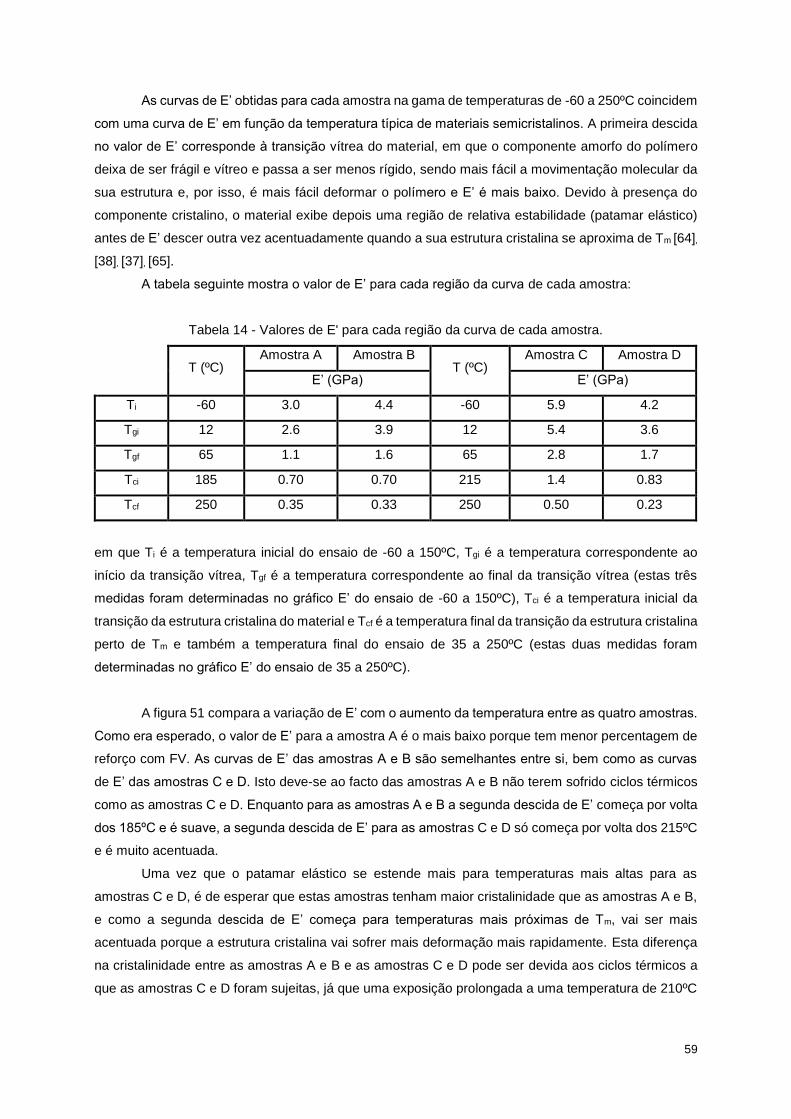
59
As curvas de E’ obtidas para cada amostra na gama de temperaturas de -60 a 250ºC coincidem
com uma curva de E’ em função da temperatura típica de materiais semicristalinos. A primeira descida
no valor de E’ corresponde à transição vítrea do material, em que o componente amorfo do polímero
deixa de ser frágil e vítreo e passa a ser menos rígido, sendo mais fácil a movimentação molecular da
sua estrutura e, por isso, é mais fácil deformar o polímero e E’ é mais baixo. Devido à presença do
componente cristalino, o material exibe depois uma região de relativa estabilidade (patamar elástico)
antes de E’ descer outra vez acentuadamente quando a sua estrutura cristalina se aproxima de Tm [64],
[38], [37], [65].
A tabela seguinte mostra o valor de E’ para cada região da curva de cada amostra:
Tabela 14 - Valores de E' para cada região da curva de cada amostra.
T (ºC) Amostra A Amostra B
T (ºC) Amostra C Amostra D
E’ (GPa) E’ (GPa)
Ti -60 3.0 4.4 -60 5.9 4.2
Tgi 12 2.6 3.9 12 5.4 3.6
Tgf 65 1.1 1.6 65 2.8 1.7
Tci 185 0.70 0.70 215 1.4 0.83
Tcf 250 0.35 0.33 250 0.50 0.23
em que Ti é a temperatura inicial do ensaio de -60 a 150ºC, Tgi é a temperatura correspondente ao
início da transição vítrea, Tgf é a temperatura correspondente ao final da transição vítrea (estas três
medidas foram determinadas no gráfico E’ do ensaio de -60 a 150ºC), Tci é a temperatura inicial da
transição da estrutura cristalina do material e Tcf é a temperatura final da transição da estrutura cristalina
perto de Tm e também a temperatura final do ensaio de 35 a 250ºC (estas duas medidas foram
determinadas no gráfico E’ do ensaio de 35 a 250ºC).
A figura 51 compara a variação de E’ com o aumento da temperatura entre as quatro amostras.
Como era esperado, o valor de E’ para a amostra A é o mais baixo porque tem menor percentagem de
reforço com FV. As curvas de E’ das amostras A e B são semelhantes entre si, bem como as curvas
de E’ das amostras C e D. Isto deve-se ao facto das amostras A e B não terem sofrido ciclos térmicos
como as amostras C e D. Enquanto para as amostras A e B a segunda descida de E’ começa por volta
dos 185ºC e é suave, a segunda descida de E’ para as amostras C e D só começa por volta dos 215ºC
e é muito acentuada.
Uma vez que o patamar elástico se estende mais para temperaturas mais altas para as
amostras C e D, é de esperar que estas amostras tenham maior cristalinidade que as amostras A e B,
e como a segunda descida de E’ começa para temperaturas mais próximas de Tm, vai ser mais
acentuada porque a estrutura cristalina vai sofrer mais deformação mais rapidamente. Esta diferença
na cristalinidade entre as amostras A e B e as amostras C e D pode ser devida aos ciclos térmicos a
que as amostras C e D foram sujeitas, já que uma exposição prolongada a uma temperatura de 210ºC

60
pode ser suficiente para provocar uma cristalização adicional (como se constatou nos resultados de
%Cx para o pico da fusão cristalina da curva DSC do 1º aquecimento).
Ao passar a transição vítrea, o valor de E’ da amostra D é ligeiramente superior ao da amostra
B devido ao aumento na cristalinidade, que torna a estrutura mais resistente à deformação. A amostra
C é a que apresenta um valor de E’ mais elevado, mesmo em relação às amostras B e D (cerca de 2
GPa superior) que têm a mesma percentagem de reforço de FV (e a amostra D também sofreu ciclos
térmicos).
Tg pode ser determinada pelo pico de E’’ ou pelo pico de tan δ. O pico de E’’ ocorre para a
temperatura a meio da transição vítrea e está mais relacionado com as mudanças nas propriedades
físicas do polímero devido à transição vítrea (reflecte o processo molecular e admite Tg como a
temperatura inicial da movimentação molecular da estrutura amorfa). O pico de tan δ ocorre para a
temperatura mais próxima da temperatura final da transição vítrea e é o valor normalmente usado na
literatura (é uma boa medida para saber o ponto de transição entre o estado vítreo e elástico de um
polímero), em que a sua altura e forma mudam com o conteúdo amorfo [66].
A tabela 15 mostra os valores da literatura de Tg para PA66 e PA6 e os valores de Tg para cada
amostra, calculados pelos picos de E’’ e de tan δ.
Tabela 15 - Tg de PA66 e PA6 (da literatura) e Tg determinada pelos pico de E'' e de tan δ [67].
Tg
(literatura) Tg Amostra A Amostra B Amostra C Amostra D
PA66 60 – 70ºC Pico E’’ 40.20ºC 48.06ºC 55.40ºC 48.66ºC
PA6 50 – 60ºC Pico tan δ 55.02ºC 63.84ºC 68.15ºC 69.15ºC
Dá para verificar que os valores de Tg obtidos pelo pico de tan δ são mais próximos dos valores
da literatura destas poliamidas. No entanto, os valores de Tg para o pico de E’’ são muito semelhantes
para as amostras B e D e praticamente 8ºC acima do valor de Tg da amostra A, o que pode estar
relacionado com as restrições à movimentação molecular impostas pela maior quantidade de FV [68].
A amostra C é a que apresenta um valor de Tg maior. Tg também é dependente do peso molecular (Mw)
para polímeros lineares, em que aumentar o peso molecular diminui o número de terminais das cadeias
(ou seja, as cadeias moleculares do polímero são maiores), sendo necessária uma temperatura maior
para se iniciar a movimentação molecular. Este aumento da temperatura necessária à movimentação
molecular está também relacionado com a energia de activação requerida para ocorrer essa transição
(quanto maior a energia requerida para ocorrer uma transição, maior a temperatura necessária para se
atingir e ultrapassar essa energia) [38], [69]. Visto que a amostra C tem a mesma quantidade de reforço
com FV que as amostras B e D, pode ter Mw superior às outras amostras (o que também pode explicar
o facto de ter o maior valor de E’, apesar de não se poder descartar a influência de uma possível
variação nas condições de processamento, nomeadamente temperatura, durante a injecção de cada
peça e também a influência da preparação dos provetes, como ligeiras diferenças no local da sua
obtenção).
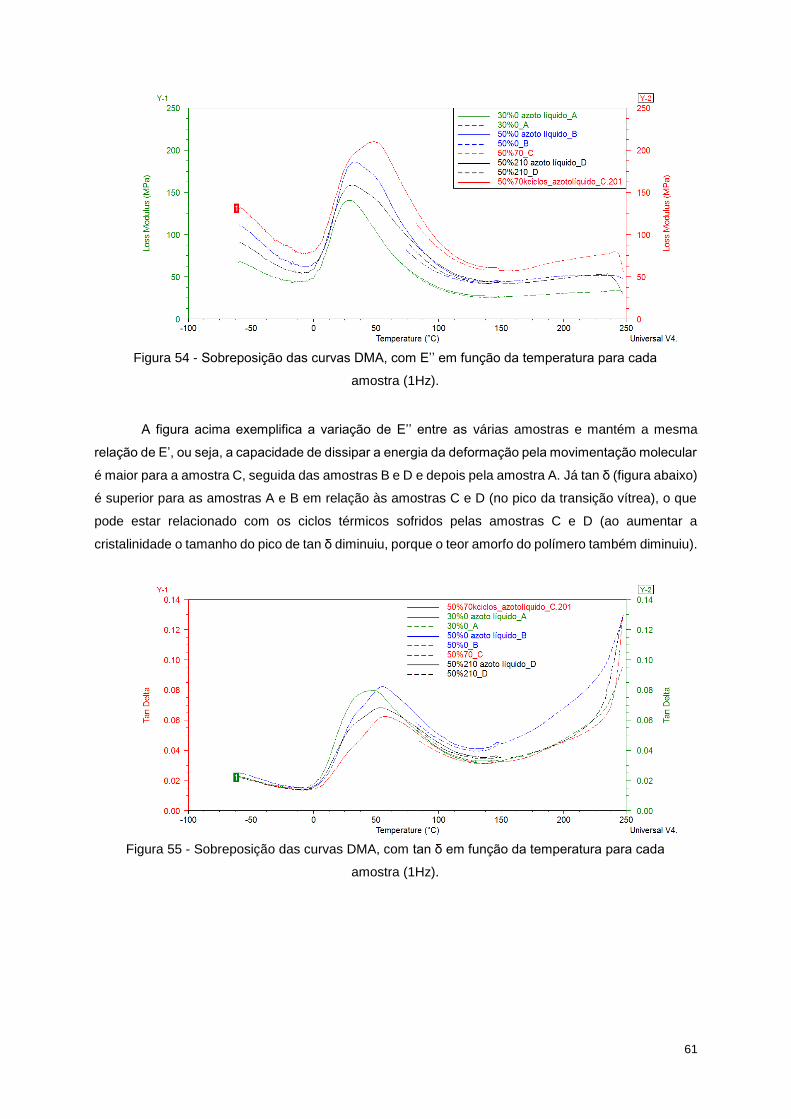
61
A figura acima exemplifica a variação de E’’ entre as várias amostras e mantém a mesma
relação de E’, ou seja, a capacidade de dissipar a energia da deformação pela movimentação molecular
é maior para a amostra C, seguida das amostras B e D e depois pela amostra A. Já tan δ (figura abaixo)
é superior para as amostras A e B em relação às amostras C e D (no pico da transição vítrea), o que
pode estar relacionado com os ciclos térmicos sofridos pelas amostras C e D (ao aumentar a
cristalinidade o tamanho do pico de tan δ diminuiu, porque o teor amorfo do polímero também diminuiu).
Figura 54 - Sobreposição das curvas DMA, com E’’ em função da temperatura para cada
amostra (1Hz).
Figura 55 - Sobreposição das curvas DMA, com tan δ em função da temperatura para cada
amostra (1Hz).

62
A técnica DMA mostra que a alteração no aspecto da superfície não afecta o comportamento
mecânico das peças sujeitas a essa alteração. Já os ciclos térmicos aumentam a cristalinidade do
material das peças que sofreram esses ciclos e, consequentemente, aumentam a estabilidade da
estrutura do material dessas peças para temperaturas mais elevadas (o valor de E’ da amostra D é
cerca de 200 MPa superior ao valor de E’ da amostra B, por volta dos 215ºC).
Era esperado que a curva de E’ da amostra C estivesse muito próxima da curva de E’ da
amostra D, já que tem a mesma percentagem de reforço com FV e também sofreu ciclos térmicos. No
entanto, esta técnica mostra que o material da amostra C deve ter características diferentes do material
das outras amostras, visto que o seu valor de E’ é cerca de 2 GPa superior (constante em toda a gama
de temperaturas) ao valor de E’ das amostras B e D que têm a mesma percentagem de reforço com
FV (50%). Esta diferença pode ser justificada por uma alteração em Mw. A resistência de um polímero
à deformação aumenta com o aumento de Mw (cadeias moleculares maiores) porque as forças de
ligação entre os átomos de uma cadeia molecular são mais elevadas do que as forças de ligação entre
as várias cadeias constituintes do polímero. Assim, um polímero com Mw maior apresenta cadeias
moleculares com uma força de ligação mais elevada, levando a uma estrutura com propriedades
mecânicas superiores, tais como a tenacidade e a resistência mecânica a altas temperaturas [70]. O
facto de Tg da amostra C também apresentar um valor superior ao das amostras B e D favorece a ideia
do material da amostra C ter Mw maior que o das outras amostras (como já foi dito acima).

63
4 Conclusão
Pode concluir-se neste trabalho que a rugosidade e irregularidade da superfície da amostra B
não permite visualizar uma superfície típica de uma peça moldada por injecção, como é possível
observar na imagem da superfície da amostra A, e que há uma evolução da superfície das amostras
B, C e D com o aumento dos ciclos térmicos, ficando esta mais lisa.
Os ciclos térmicos provocaram uma cristalização secundária e, consequentemente, maior
cristalinidade (cerca de 8%) nas amostras C e D, que é visível no patamar elástico de E’ (que se mantém
estável até cerca de 215ºC) e pelos dois picos separados no pico da fusão cristalina obtido no 1º ensaio
de DSC.
Depois de um aquecimento até à fusão, seguido de um arrefecimento controlado, as curvas
DSC do 2º ensaio mostram uma transição vítrea e um pico de fusão de típico de PA66. As amostras
com 50% de FV (amostras B, C e D, que têm a alteração no aspecto da superfície) apresentam também
um pico endotérmico por volta dos 150ºC, que pode estar relacionado com a fusão do ácido adípico
presente em PA66 ou num lubrificante próprio para o processamento de poliamidas, resultando numa
composição diferente destas amostras em relação à amostra A.
A técnica DMA evidencia uma diferença entre a amostra C e as outras amostras, já que a
amostra C apresenta um valor de E,’ ao longo de todo o ensaio, cerca de 2 GPa superior às outras
amostras com 50% de FV (amostras B e D, em que esta também sofreu ciclos térmicos). Esta diferença
pode ser explicada por um valor de Mw mais elevado para a amostra C, uma vez que esta amostra
apresenta também um valor de Tg mais alto do que o das outras amostras (no entanto não se pode
descartar a influência de variações nas condições do processo de injecção das peças e na preparação
dos provetes, porque os resultados da técnica DSC não evidenciam alteração do material da amostra
C em relação ao das amostras B e D).

64
5 Trabalho Futuro
Como trabalho futuro seria interessante determinar a variação da rugosidade ao longo das
peças (tanto no interior como no exterior), conhecer a composição da liga PA66+PA6 (nomeadamente
o rácio de PA66 e de PA6), contrastar amostras de cada peça para analisar no SEM e observar a
microestrutura da liga PA66+PA6 e a disposição das fibras, e realizar ensaios de DMA a temperaturas
inferiores a -60ºC para detectar outras transições que não foram detectadas no ensaio realizado com
N2líq. para o arrefecimento até -60ºC.
A medição da rugosidade pode ser feita através de um perfilómetro mecânico (perfilómetro com
estilete) que contacta e percorre a superfície da amostra, medindo as variações máximas e mínimas
da rugosidade dessa superfície. É uma técnica simples, consegue analisar praticamente todas as
superfícies e abrange uma variação da rugosidade dos nanómetros aos milímetros [71]. Para
determinar a composição da liga PA66+PA6 pode ser utilizada uma técnica de espectroscopia de
massa, que identifica a quantidade e o tipo de componentes presentes numa amostra ao medir o rácio
entre a massa e a carga de iões e ao medir também a abundância dos mesmos iões (estes iões são
gerados pela ionização das moléculas do material das amostras, que se separam em fragmentos
carregados). Estes iões são separados de acordo com o seu rácio massa/carga ao serem sujeitos a
um campo eléctrico ou magnético, mesmo que os componentes da liga tenham composição semelhante
[72].
Uma vez que estas peças foram produzidas pelo processo de moldação por injecção e sendo
este um processo complexo, também não se podia deixar de referir que era importante estudar a
viscosidade destas poliamidas e a sua variação com a temperatura, cuja alteração poderia levar a
complicações no enchimento do fundido.
A viscosidade de PA6 é menor que a de PA66 e a viscosidade destas poliamidas varia muito
com a temperatura. Uma maneira de estudar a viscosidade desta liga é com a extrusão do seu
granulado, controlando a temperatura e a velocidade de corte [73], [74]. Um parâmetro importante para
a morfologia de uma liga polimérica é o rácio entre as viscosidades dos constituintes dessa liga, já que
para o caso em que o componente minoritário tem uma viscosidade menor que o componente
maioritário, esse vai ficar disperso finamente e de forma homogénea no componente maioritário. A
compatibilidade de uma liga polimérica também é dependente da temperatura [75], [76]. Ao determinar
a viscosidade desta liga pela extrusão do seu granulado, consegue estimar-se também a distribuição
de Mw pela relação de proporcionalidade da viscosidade com Mw [77].

65
Referências Bibliográficas
[1] “How an Intercooler Works,” TurboSmart, [Online]. Available:
http://www.turbosmart.com.au/technical-articles/how-an-intercooler-works/. [Acedido em 29
Setembro 2014].
[2] K. Szeteiová, “Automotive Materials, Plastics in Automotive Markets Today,” Bratislava, 2010.
[3] E. Ghassemieh, “Materials in Automotive Application, State of the Art and Prospects,” em New
Trends and Developments in Automotive Industry, Sheffield, InTech, 2011, pp. 365-390.
[4] N. Strumberger, A. Gospocic e C. Bartulic, “Polymeric Materials in Automobiles,” Traffic
Engineering, vol. 17, nº 3, pp. 149-160, 2005.
[5] D. K. Platt, “Polyamide (PA),” em Engineering and High Performance Plastics, Shawbury, Rapra
Technology Limited, 2003, pp. 11-14.
[6] F. W. B. Jr., “Heterochain Thermoplastics,” em Textbook of Polymer Science, Nova Iorque, John
Wiley & Sons, Inc., 1984, pp. 407-411.
[7] I. B. Page, “Polyamides as Engineering Thermoplastic Materials,” Rapra Review Reports, vol. 11,
nº 1, pp. 3-10, 2000.
[8] J. A. Brydson, “Polyamides and Polymides,” em Plastics Materials, Londres, Whitefriars Press
Ltd., 1975, pp. 392-414.
[9] D. E. Floyd, “General Properties of the Polyamide Resins,” em Polyamide Resins 2nd Edition,
Nova Iorque, Chapman & Hall, Ltd., 1966, pp. 12-21.
[10] B. H. Stuart, “Blends,” em Polymer Analysis, Sidney, John Wiley & Sons, Inc., 2002, pp. 134-135.
[11] ISO 1874-1: 2010 - Polyamides Designation System and Basis for Specification.
[12] T. Kurokawa, “Gear from a PA66/PA6 Polymer Blend”. Europa Patente EP2476933 A1, 18 Julho
2012.
[13] “Materials that Withstand Today's Hotter Running Engine Systems,” DuPont, 2014. [Online].
Available: http://www.dupont.com/industries/automotive/powertrain-engine-
system/articles/performance-polymers-solutions.html. [Acedido em 3 Outubro 2014].
[14] “ProLine IC,” Setrab, [Online]. Available: http://www.setrab.com/products/proline/technical-
specifications/ic/. [Acedido em 3 Outubro 2014].
[15] D. V. Rosato, D. V. Rosato e M. G. Rosato, “The Complete Injection Molding Process,” em Injection
Molding Handbook 3rd Edition, Massachusetts, Kluwer Academic Publishers , 2000, pp. 1-28.
[16] T. A. Oswald, L. S. Turng e P. J. Gramann, Injection Molding Handbook, Munique: Hanser Gardner
Publications, 2008.
[17] D. E. Floyd, “Molding, Casting and Extrusion,” em Polyamide Resins 2nd Edition, Nova Iorque,
Reinhold Publishing Corporation, 1966, pp. 138-142.
[18] Injection Moulding of Polyamides, DSM Engineering Plastics.

66
[19] P. Postawa e A. Gnatowski, “Anisotropy of Physical Properties Injection Moulded Parts and Its
Analysis,” Journal of Achievements on Materials and Manufacturing Engineering, vol. 23, nº 2, pp.
35-38, 2007.
[20] S. Shaharuddin, M. Salit e E. Zainudin, “A Review of the Effect of Moulding Parameters on the
Performance of Polymeric Composite Injection Moulding,” Tübitak, Selangor, 2006.
[21] S. L. Hsu, “Microscopy,” em Characterization and Analysis of Polymers, Nova Jérsia, John Wiley
& Sons, Inc., 2008, pp. 600-649.
[22] L. C. Sawyer, D. T. Grubb e G. F. Meyers, “Scanning Electron Microscopy,” em Polymer
Microscopy 3rd Edition, Nova Iorque, Springer, 2008, pp. 35-41.
[23] M. Dunlap e D. J. E. Adaskaveg, Introduction to the Scanning Electron Microscope, Theory,
Practice & Procedures, 1997.
[24] L. C. Sawyer, D. T. Grubb e G. F. Meyers, “Polymer Characterization,” em Polymer Microscopy
3rd Edition, Nova Iorque, Springer, 2008, pp. 17-21.
[25] S. M. Mukhopadhyay, “Sample Preparation for Microscopic and Spectroscopic Characterization of
Solid Surfaces and Films,” em Sample Preparation Techniques in Analytical Chemistry, Dayton,
John Wiley & Sons, Inc., 2003, pp. 377-389.
[26] “Hitachi Scanning Electron Microscope/S 2400,” Nanotech, 2009. [Online]. Available:
http://www.nanotech.ucsb.edu/index.php/component/content/article/79-uncategorised/107-
hitachi-scanning-electron-microscope-s-2400. [Acedido em 11 Setembro 2014].
[27] “Hitachi SEM S-2400,” Azom.com, [Online]. Available:
http://www.azom.com/Myclassified/classifieddetails.aspx?jobID=437. [Acedido em 11 Setembro
2014].
[28] Cathodes for Electron Microscopes: CeBix and LaB6 Filaments, Standard Tungsten Loop
Filaments, Hatfield: Electron Microscopy Sciences.
[29] “Thermal Characterization of Polymers: Thermoplastics, Thermoplastics Elastomers, Elastomers
and Thermosets,” Netzsch, Selb.
[30] D. C. Prevorsek, R. H. Butler e H. K. Reimschuessel, “Mechanical Relaxations in Polyamides,”
Journal of Polymer Science, vol. 9, nº Part A-2, pp. 867-886, 1971.
[31] T. Hatakeyama e F. X. Quinn, “Mechanical Analysis,” em Thermal Analysis, Fundamentals and
Applications to Polymer Sciences, Second Edition, John Wiley & Sons, Inc., 1999, pp. 125-133.
[32] J. D. Menczel e R. B. Prime, “Dynamic Mechanical Analysis (DMA),” em Thermal Analysis of
Polymers, Fundamentals and Applications, Nova Jérsia, John Wiley & Sons, Inc., 2009, pp. 387-
497.
[33] B. H. Stuart, “Dynamic Mechanical Analysis,” em Polymer Analysis, Sydney, John Wiley & Sons,
Inc., 2002, pp. 157-161.
[34] Dynamic Mechanical Analyzer DMA 2980 Getting Started Guide, Nova Iorque: TA Instruments,
1997.

67
[35] Thermal Analysis, TA Instruments, 2010.
[36] RSA-G2 Solids Analyzer, TA Instruments, 2011.
[37] Dynamic Mechanical Analysis (DMA), a Beginner's Guide, Waltham: Perkin Elmer, Inc., 2008.
[38] J. Ducan, “Principles and Applications of Mechanical Thermal Analysis,” em Principles and
Applications of Thermal Analysis, Blackwell Publishing Ltd., 2008, pp. 119-163.
[39] J. Foreman e K. Reed, “Dynamic Mechanical Analyzers: How do They Work,” TA Instruments.
[40] Differential Scanning Calorimetry: First and Second Order Transitions in PETE.
[41] R. L. Danley e P. A. Caulfield, “DSC Baseline Improvements Obtained by a New Heat Flow
Measurement Technique,” TA Instruments, New Castle, 2001.
[42] J. D. Menczel, L. Judovits, R. B. Prime, H. E. Bair, M. Reading e S. Swier, “Differential Scanning
Calorimetry (DSC),” em Thermal Analysis of Polymers: Fundamentals and Applications, New
Jersey, John Wiley & Sons, Inc., 2009, pp. 7-225.
[43] Differential Scanning Calorimetry Investigation of Polymers, Berlim: Humboldt-Universität.
[44] Differential Scanning Calorimetry (DSC) a Beginner's Guide, Waltham: PerkinElmer, Inc., 2010.
[45] B. H. Stuart, “Differential Scanning Calorimetry,” em Polymer Analysis, Sidney, John Wiley & Sons,
Inc., 2002, pp. 55-58.
[46] “DSC 2920 CE, Differential Scanning Calorimeter Operator's Manual,” TA Instruments, New
Castle, 1998.
[47] L. C. Sawyer, D. T. Grubb e G. F. Meyers, “Polymer Morphology,” em Polymer Microscopy 3rd
Edition, Nova Iorque, Springer, 2008, pp. 3-12.
[48] E. Charrault, C. Gauthier, P. Marie e R. Schirrer, “Structural Recovery (Physical Ageing) of the
Friction Coefficient of Polymers,” Wiley InterScience, Estrasburgo, 2008.
[49] L. W. McKeen, “Introduction to the Effect of Heat Aging on Plastics,” em The Effect of Long Term
Thermal Exposure on Plastics and Elastomers, Waltham, Elsevier Inc., 2014, pp. 17-42.
[50] S. Mutlur, Thermal Analysis of Composites Using DSC, 2004.
[51] J. A. Brydson, “Melt Processing of Thermoplastics,” em Plastics Materials 7th Edition, Butterworth-
Heinemann, 1999, pp. 159-176.
[52] P. Gabbott, “Principles of DSC and Types of Measurements Made,” em Principles and Applications
of Thermal Analysis, Blackwell Publishing Ltd., 2008, pp. 2-5.
[53] E. Espigulé, X. Puigvert, F. Vilaseca, J. A. Mendez, P. Mutjé e J. Girones, “Thermoplastic Starch-
Based Composites Reinforced with Rape Fibers: Water Uptake and Thermomechanical
Properties,” Bioresources.com, vol. 8, nº 2, pp. 2620-2630, 2013.
[54] J. C. Salamone, Polymeric Materials Encyclopedia, Florida: CRC Press, Inc., 1996.
[55] Polymer DSC, Mettler Toledo, Inc..

68
[56] F. Rybnikar e P. H. Geil, Melting and Recrystallization of PA-6/PA-66 Blends, Illinois: Air
Conditioning & Refrigeration Center, Mechanical & Industrial Engineering Dept., University of
Illinois, 1992.
[57] M. Evstatiev, S. Petrovich e B. Krasteva, “Calorimetric Studies on PET/PA6 and PA66/PA6 Drawn
Blends with Various Thermal Histories,” Bulg. J. Phys., vol. 30, pp. 70-79, 2003.
[58] Y. Li e G. Yang, “Studies on Molecular Composites of Polyamide 6/Polyamide 66,” Macromolecular
Rapid Communications Journal, vol. 25, pp. 1714-1718, 2004.
[59] E. Plorkowska, “Fiber-Reinforced Composites,” em Handbook of Polymer Crystallization, Nova
Jérsia, John Wiley & Sons, Inc., 2013, pp. 382-385.
[60] W. Feng, A. Ait-Kadi, J. Brisson e B. Riedl, “Polymerization Compounding Composites of Nylon-
6,6/Short Glass Fiber,” Polymer Composites, vol. 24, nº 4, pp. 512-524, 2003.
[61] G. Bogoeva-Gaceva e A. Grozdanov, “Crystallization of Isotactic Polypropylene: The Effect of
Fiber Surface,” J. Serb. Chem. Soc., vol. 71, nº 5, pp. 483-499, 2006.
[62] Adipic Acid CAS Nº:124-04-9, Paris: UNEP Publications, 2004.
[63] G. Wypych, “Polyamide,” em Handbook of Plasticizers, Toronto, Elsevier, Inc., 2012, pp. 298-299.
[64] J. Foreman, “Dynamic Mechanical Analysis of Polymers,” TA Instruments, 1997.
[65] B. H. Stuart, “Thermal Behaviour,” em Polymer Analysis, Sidney, John Wiley & Sons, Inc., 2002,
pp. 135-139.
[66] Thermal Solutions, Measurement of the Glass Transition Temperature Using Dynamic Mechanical
Analysis, TA Instruments.
[67] R. J. Mehta, “Physical Constants of Various Polyamides,” em Polymer Handbook 4th Edition, J.
Brandrup, E. H. Immergut, E. A. Grulke, 1998, pp. V/121-V/135.
[68] D. Romanzini, A. Lavoratti, H. L. O. Jr., S. C. Amico e A. J. Zattera, “Influence of Fiber Content on
the Mechanical and Dynamic Properties of Glass/Ramie Polymer Composites,” Materials and
Design, pp. 9-15, 22 Dezembro 2012.
[69] J. Charles, G. R. Ramkumaar, S. Azhagiri e S. Gunasekaran, “FTIR and Thermal Studies on
Nylon-66 and 30% Glass Fiber Reinforced Nylon-66,” E-Journal of Chemistry, pp. 23-33, 20 Julho
2009.
[70] B. Sivasankar, “Structure and Properties of Polymers, Size of the Polymer Molecule Chain,” em
Engineering Chemistry, Nova Deli, Tata McGraw-Hill Publishing Company Ltd., 2008, p. 229.
[71] T. Chi, T. Ballinger, R. Olds e M. Zecchino, “Surface Texture Analysis Using Dektak Stylus
Profilers,” Bruker, 2010.
[72] B. W. Schueler, “Time-of-Flight Mass Analysers,” em ToF-SIMS : Materials Analysis by Mass
Spectrometry 2nd Edition, UK, IM Publications LLP and SurfaceSpectra Ltd., 2013, pp. 247-271.
[73] Materials and Databases, Autodesk Moldflow Communicator, 2012.

69
[74] B. K. Nguyen, G. McNally e A. Clarke, “Real Time Measurement and Control of Viscosity for
Extrusion Processes Using Recycled Materials,” Polymer Degradation and Stability, vol. 102, pp.
212-221, 2014.
[75] S. Sánchez-Valdes, L. F. R.-d. Valle e O. Manero, “Polymer Blends,” em Handbook of Polymer
Synthesis, Characterization and Processing, John Wiley & Sons, Inc., 2013, pp. 505-519.
[76] A. Franck, “Understanding Rheology of Thermoplastic Polymers,” TA Instruments.
[77] Polymer Properties, Viscosity, Aldrich, 1990.
[78] Universal Analysis 2000, New Castle: TA Instruments, 2003.
[79] K. Hollocher, “Backscattered Electrons (BSE) Imaging,” SEM web Page, [Online]. Available:
http://minerva.union.edu/hollochk/sem/bse.htm. [Acedido em 14 Setembro 2014].
[80] J. Gotro, “Thermoset Characterization Part 15: Experimental Aspects of Dynamic Mechanical
Analysis (DMA),” Polymer Innovation, [Online]. Available:
http://polymerinnovationblog.com/thermoset-characterization-part-15-experimental-aspects-
dynamic-mechanical-analysis-dma/. [Acedido em 19 Setembro 2014].
[81] “Stubs and Mounts for SEM Specimens (Scanning Electron Microscopy) - Agar Scientific,”
Azom.com, 2000. [Online]. Available: http://www.azom.com/equipment-
details.aspx?EquipID=2673. [Acedido em 14 Setembro 2014].
[82] “MicroLab Electron Microscopy Laboratory,” Instituto Superior Técnico, [Online]. Available:
http://groups.ist.utl.pt/microlab/index.html. [Acedido em 11 Setembro 2014].
[83] [Online]. Available: http://www.sft66.com/fungi/html/sem.html. [Acedido em 14 Setembro 2014].
[84] “PerkinElmer(R) DSC Aluminum Sample Pans and Covers,” Spectrum Chemical MFG Corp, 2014.
[Online]. Available: https://www.spectrumchemical.com/OA_HTML/lab-supplies-
products_Aluminum-Sample-Pans-and-Covers-
Pkg400_312230.jsp?minisite=10020&respid=22372. [Acedido em 27 Setembro 2014].
[85] “GT2 RS Turbo Intercooler Set,” SunCoast Parts.com, 2014. [Online]. Available:
http://www.suncoastparts.com/product/SKU997TTRSC.html. [Acedido em 29 Setembro 2014].
[86] “Plastic Injection Moulding Machine,” Rutland Plastics Limited, 2013. [Online]. Available:
http://www.rutlandplastics.co.uk/advice/moulding_machine.html. [Acedido em 29 Setembro 2014].
[87] “The Injection Mold,” Injection Molding Expertise.com, [Online]. Available:
http://www.injectionmoldingexpertise.com/injectionmold.html. [Acedido em 30 Setembro 2014].
[88] “7 Tips for Choosing Great Gate Locations,” QuickParts Custom Tips for Injection Molding , 2014.
[Online]. Available: http://injectionmolding.blog.quickparts.com/2011/03/07/7-tips-for-choosing-
great-gate-locations/. [Acedido em 30 Setembro 2014].
[89] E. S. Clark, “Unit Cell Information on Some Important Polymers,” em Physical Properties of
Polymers Handbook 2nd Edition, Nova Iorque, Springer, 2007, pp. 619-625.

70
[90] “Gates & Runners - Plastic Injection Molding,” Xcentric Mold & Engineering, Inc., [Online].
Available: http://www.xcentricmold.com/resource/injection-mold-gates.php. [Acedido em 4
Outubro 2014].
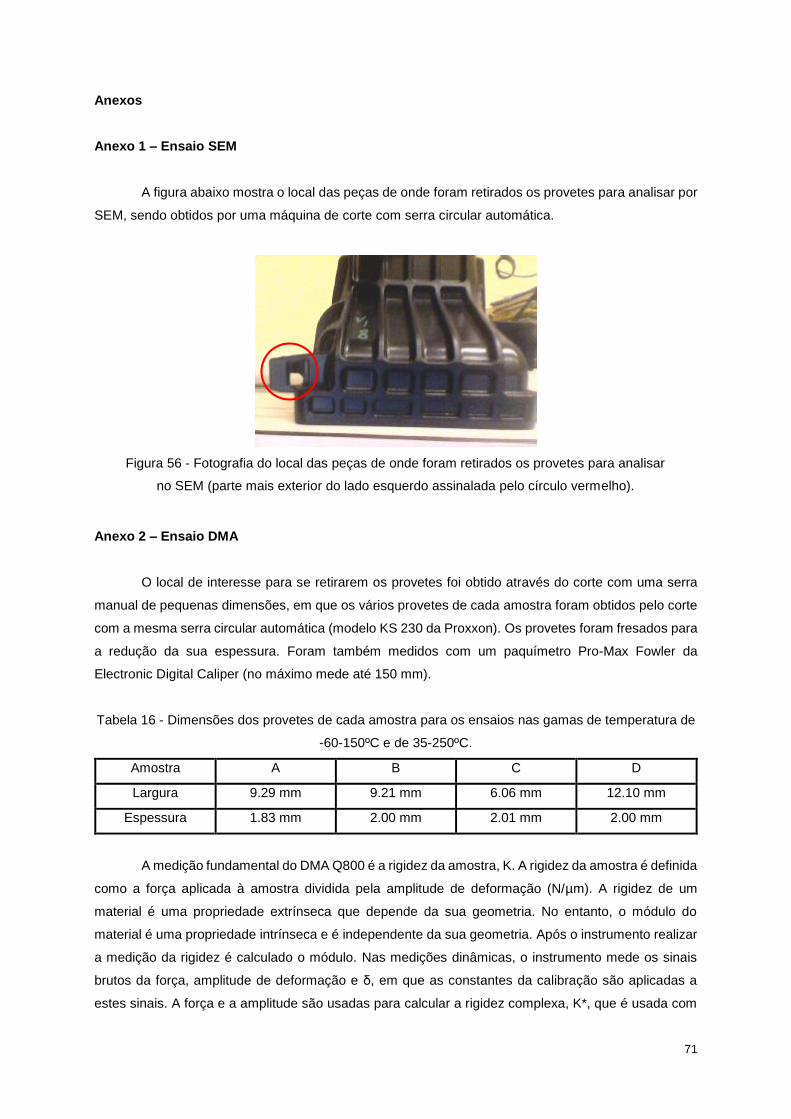
71
Anexos
Anexo 1 – Ensaio SEM
A figura abaixo mostra o local das peças de onde foram retirados os provetes para analisar por
SEM, sendo obtidos por uma máquina de corte com serra circular automática.
Anexo 2 – Ensaio DMA
O local de interesse para se retirarem os provetes foi obtido através do corte com uma serra
manual de pequenas dimensões, em que os vários provetes de cada amostra foram obtidos pelo corte
com a mesma serra circular automática (modelo KS 230 da Proxxon). Os provetes foram fresados para
a redução da sua espessura. Foram também medidos com um paquímetro Pro-Max Fowler da
Electronic Digital Caliper (no máximo mede até 150 mm).
Tabela 16 - Dimensões dos provetes de cada amostra para os ensaios nas gamas de temperatura de
-60-150ºC e de 35-250ºC.
Amostra A B C D
Largura 9.29 mm 9.21 mm 6.06 mm 12.10 mm
Espessura 1.83 mm 2.00 mm 2.01 mm 2.00 mm
A medição fundamental do DMA Q800 é a rigidez da amostra, K. A rigidez da amostra é definida
como a força aplicada à amostra dividida pela amplitude de deformação (N/µm). A rigidez de um
material é uma propriedade extrínseca que depende da sua geometria. No entanto, o módulo do
material é uma propriedade intrínseca e é independente da sua geometria. Após o instrumento realizar
a medição da rigidez é calculado o módulo. Nas medições dinâmicas, o instrumento mede os sinais
brutos da força, amplitude de deformação e δ, em que as constantes da calibração são aplicadas a
estes sinais. A força e a amplitude são usadas para calcular a rigidez complexa, K*, que é usada com
Figura 56 - Fotografia do local das peças de onde foram retirados os provetes para analisar
no SEM (parte mais exterior do lado esquerdo assinalada pelo círculo vermelho).

72
δ para calcular a rigidez de conservação e de perda (K’ e K’’, respectivamente). Tan δ é calculada pelo
rácio entre K’’ e K’. E’ e E’’ são depois calculados ao multiplicar a rigidez medida pelo factor geométrico
apropriado, GF.
Para o modo de operação de consola simples, o módulo pode ser determinado por
𝑀ó𝑑𝑢𝑙𝑜 = 𝐾 × 𝑤 (𝑡
𝑙)
3
(A1)
em que w é a largura da amostra, t a espessura da amostra e l o comprimento da consola simples (10
mm). Esta relação permite saber as dimensões mais adequadas da amostra, de acordo com o seu
módulo, para se enquadrar na gama de rigidez permitida pelo equipamento [34], [38].
Anexo 3 – Ensaio DSC
Os provetes a analisar por DSC foram obtidos pelo corte com a mesma serra manual de
pequenas dimensões. Estes devem ser planos para garantir um bom contacto com a superfície da base
da cápsula de alumínio onde estão inseridos, e assim um aquecimento uniforme do provete. A tabela
seguinte especifica as medidas dos provetes de cada amostra para os dois ensaios.
Tabela 17 - Medidas dos provetes ensaiados por DSC para cada ensaio.
1º Ensaio 2º Ensaio
Amostra A B C D A B C D
Massa (mg) 5.70 4.90 6.40 5.60 6.10 5.20 5.50 5.70
Comprimento (mm) 4.54 3.85 3.87 3.97 3.63 4.59 3.85 3.32
Largura (mm) 3.22 3.30 3.44 2.62 2.93 4.23 3.13 2.79
Espessura (mm) 0.51 0.34 0.26 0.48 0.63 0.46 0.47 0.68
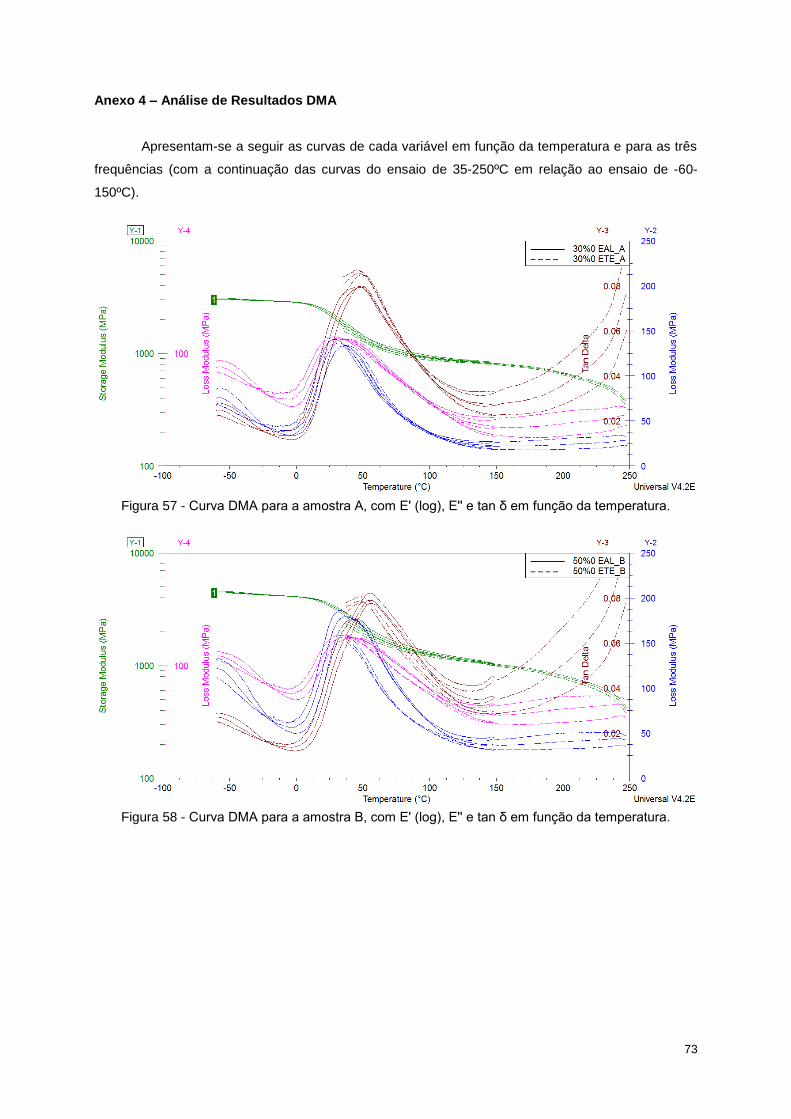
73
Anexo 4 – Análise de Resultados DMA
Apresentam-se a seguir as curvas de cada variável em função da temperatura e para as três
frequências (com a continuação das curvas do ensaio de 35-250ºC em relação ao ensaio de -60-
150ºC).
Figura 57 - Curva DMA para a amostra A, com E' (log), E'' e tan δ em função da temperatura.
Figura 58 - Curva DMA para a amostra B, com E' (log), E'' e tan δ em função da temperatura.
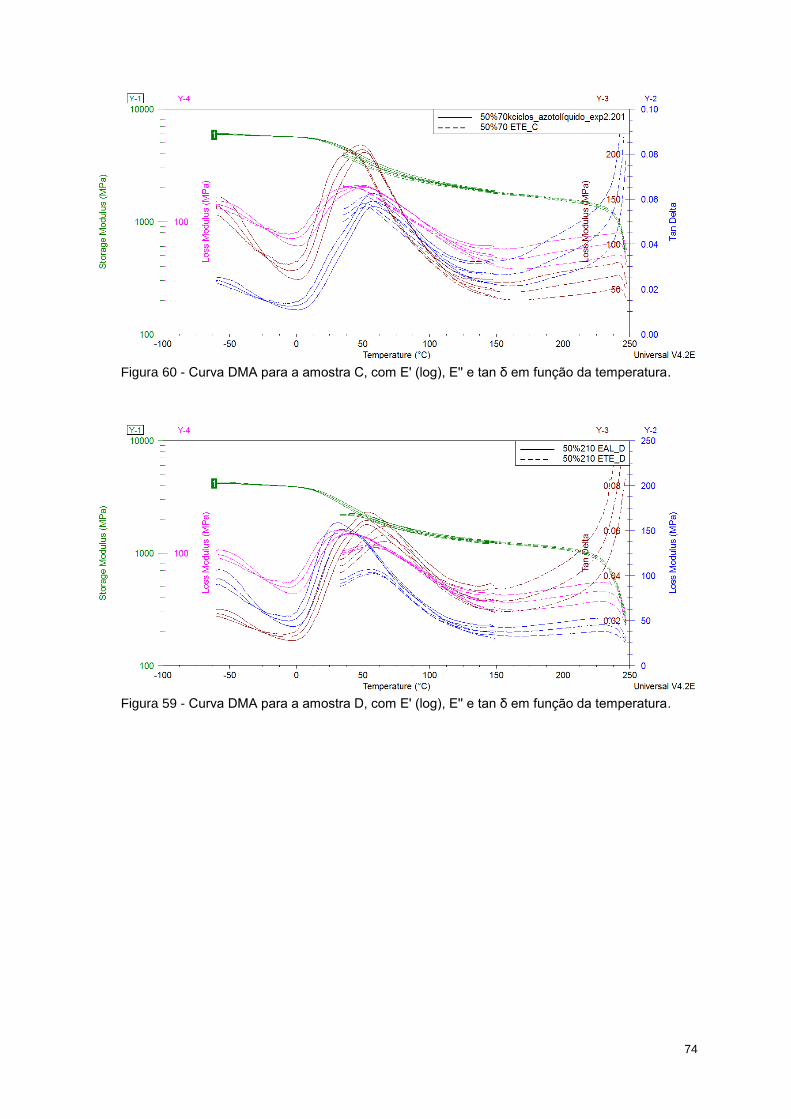
74
Figura 60 - Curva DMA para a amostra C, com E' (log), E'' e tan δ em função da temperatura.
Figura 59 - Curva DMA para a amostra D, com E' (log), E'' e tan δ em função da temperatura.
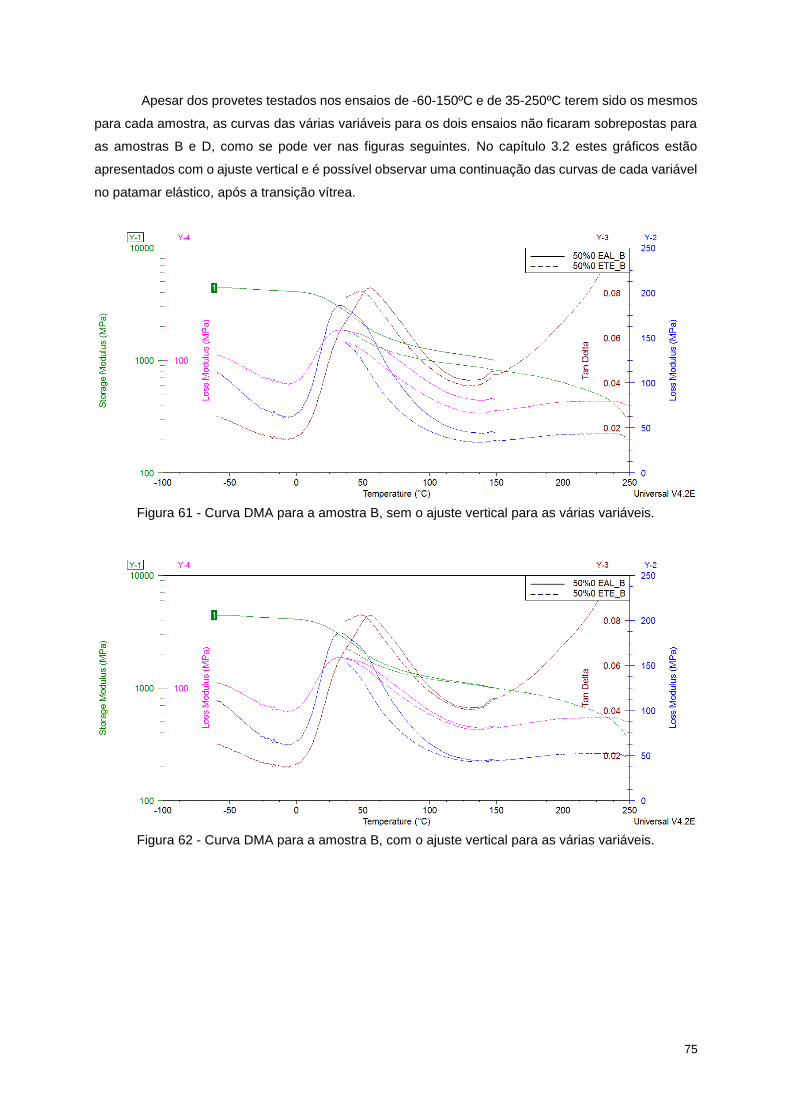
75
Apesar dos provetes testados nos ensaios de -60-150ºC e de 35-250ºC terem sido os mesmos
para cada amostra, as curvas das várias variáveis para os dois ensaios não ficaram sobrepostas para
as amostras B e D, como se pode ver nas figuras seguintes. No capítulo 3.2 estes gráficos estão
apresentados com o ajuste vertical e é possível observar uma continuação das curvas de cada variável
no patamar elástico, após a transição vítrea.
Figura 61 - Curva DMA para a amostra B, sem o ajuste vertical para as várias variáveis.
Figura 62 - Curva DMA para a amostra B, com o ajuste vertical para as várias variáveis.
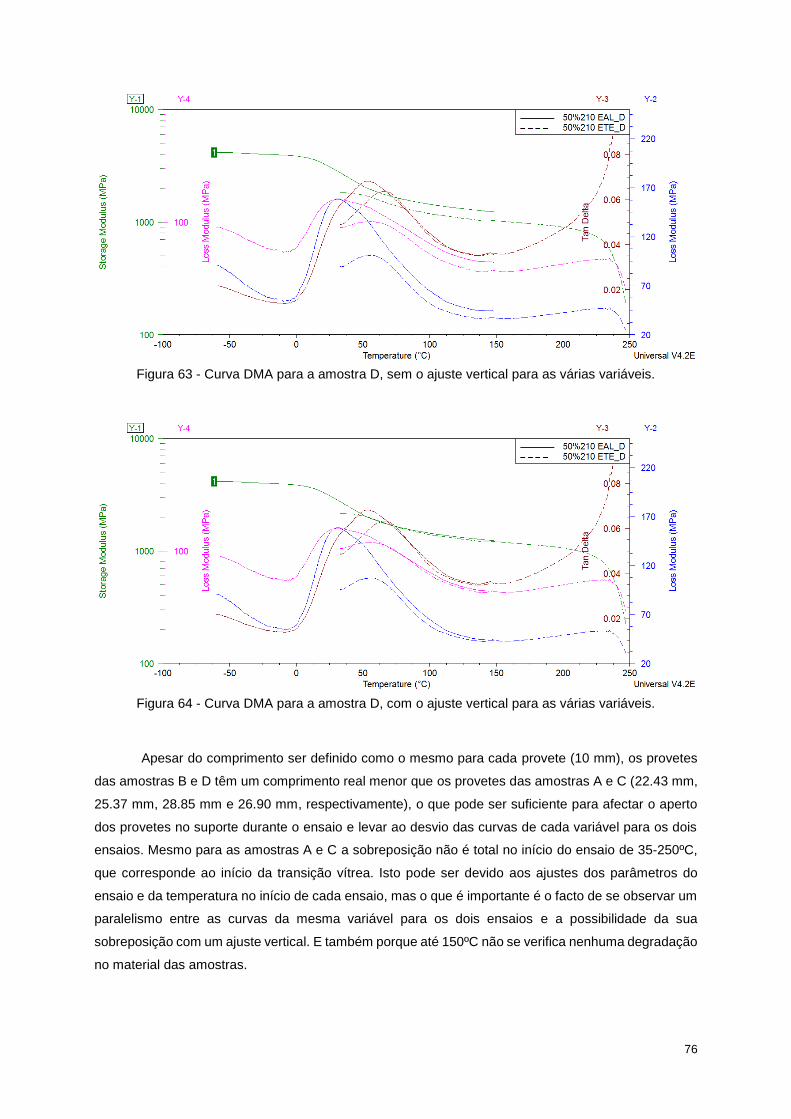
76
Apesar do comprimento ser definido como o mesmo para cada provete (10 mm), os provetes
das amostras B e D têm um comprimento real menor que os provetes das amostras A e C (22.43 mm,
25.37 mm, 28.85 mm e 26.90 mm, respectivamente), o que pode ser suficiente para afectar o aperto
dos provetes no suporte durante o ensaio e levar ao desvio das curvas de cada variável para os dois
ensaios. Mesmo para as amostras A e C a sobreposição não é total no início do ensaio de 35-250ºC,
que corresponde ao início da transição vítrea. Isto pode ser devido aos ajustes dos parâmetros do
ensaio e da temperatura no início de cada ensaio, mas o que é importante é o facto de se observar um
paralelismo entre as curvas da mesma variável para os dois ensaios e a possibilidade da sua
sobreposição com um ajuste vertical. E também porque até 150ºC não se verifica nenhuma degradação
no material das amostras.
Figura 63 - Curva DMA para a amostra D, sem o ajuste vertical para as várias variáveis.
Figura 64 - Curva DMA para a amostra D, com o ajuste vertical para as várias variáveis.
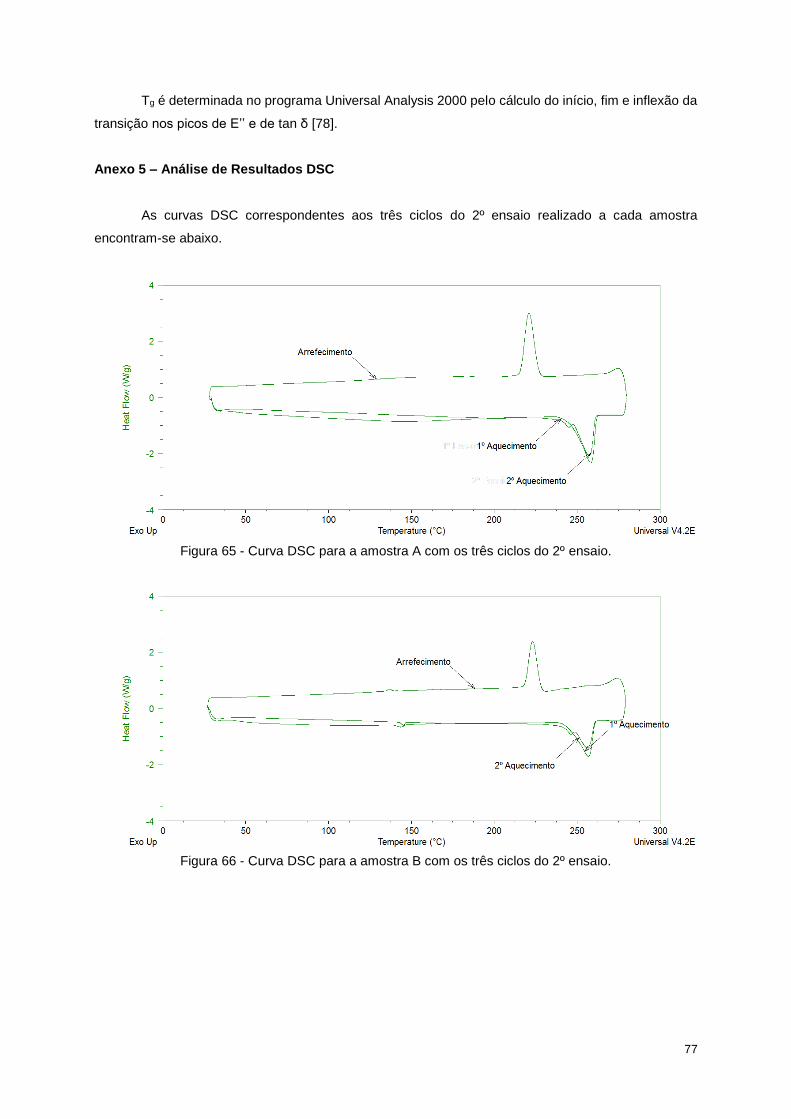
77
Tg é determinada no programa Universal Analysis 2000 pelo cálculo do início, fim e inflexão da
transição nos picos de E’’ e de tan δ [78].
Anexo 5 – Análise de Resultados DSC
As curvas DSC correspondentes aos três ciclos do 2º ensaio realizado a cada amostra
encontram-se abaixo.
Figura 65 - Curva DSC para a amostra A com os três ciclos do 2º ensaio.
Figura 66 - Curva DSC para a amostra B com os três ciclos do 2º ensaio.
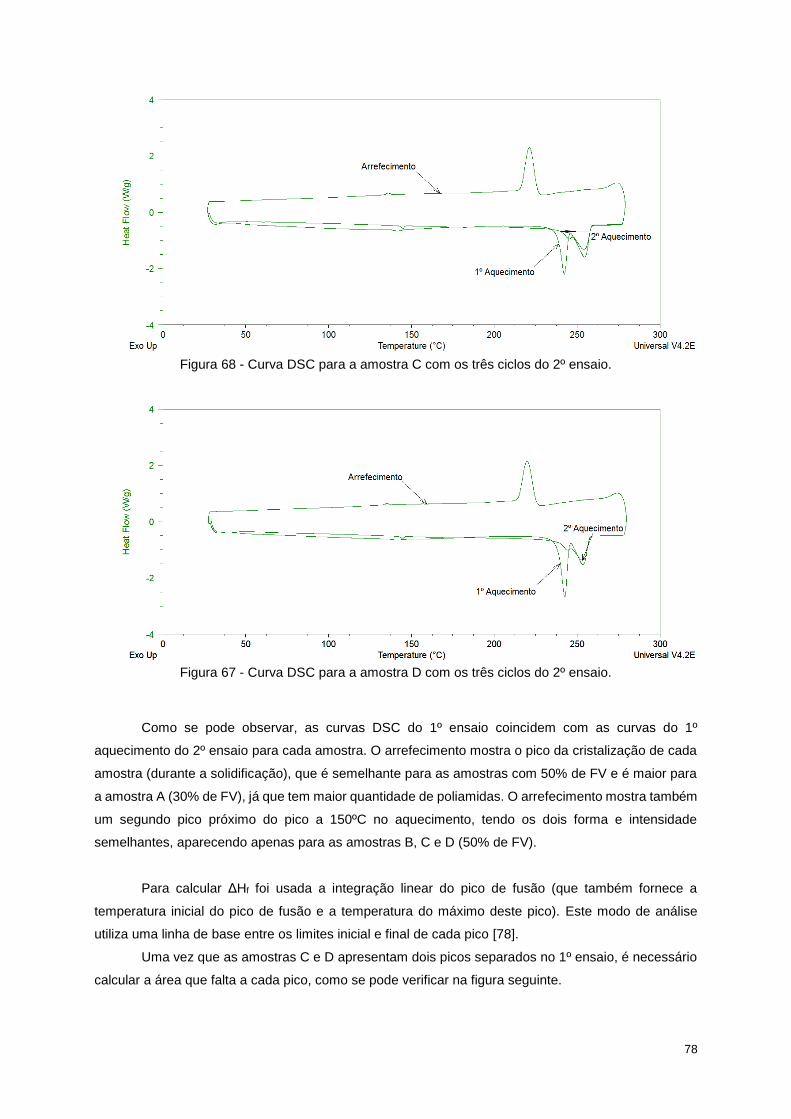
78
Como se pode observar, as curvas DSC do 1º ensaio coincidem com as curvas do 1º
aquecimento do 2º ensaio para cada amostra. O arrefecimento mostra o pico da cristalização de cada
amostra (durante a solidificação), que é semelhante para as amostras com 50% de FV e é maior para
a amostra A (30% de FV), já que tem maior quantidade de poliamidas. O arrefecimento mostra também
um segundo pico próximo do pico a 150ºC no aquecimento, tendo os dois forma e intensidade
semelhantes, aparecendo apenas para as amostras B, C e D (50% de FV).
Para calcular ΔHf foi usada a integração linear do pico de fusão (que também fornece a
temperatura inicial do pico de fusão e a temperatura do máximo deste pico). Este modo de análise
utiliza uma linha de base entre os limites inicial e final de cada pico [78].
Uma vez que as amostras C e D apresentam dois picos separados no 1º ensaio, é necessário
calcular a área que falta a cada pico, como se pode verificar na figura seguinte.
Figura 67 - Curva DSC para a amostra D com os três ciclos do 2º ensaio.
Figura 68 - Curva DSC para a amostra C com os três ciclos do 2º ensaio.

79
O tracejado a verde representa a continuação (pela tangente) de cada pico de fusão à linha de
base. O modo de integração dos picos segue a linha (cheia) a vermelho para definir os limites inicial e
final de cada pico, o que não engloba a área limitada pela linha de base (a preto), pela linha cheia a
vermelho e pela linha tracejada vermelha correspondente a cada pico (note-se que o pico 1 entra no
domínio do pico 2 e vice-versa).
Calcula-se então o integral (ΔHf) total dos dois picos e subtrai-se o integral individual de cada
pico determinado pelo modo de integração escolhido, obtendo-se o integral do triângulo limitado pela
linha de base e pelas duas linhas cheias a vermelho. Sabendo as dimensões deste triângulo consegue
associar-se a sua área ao integral obtido pelo modo de integração. Desta forma, conhecendo as
dimensões de cada triângulo correspondente a cada pico, determina-se a parte de ΔHf que falta ao
integral de cada pico. Uma vez que a altura é a mesma para cada triângulo (da curva de junção dos
dois picos até à linha de base), o que varia é o comprimento da linha de base abrangido. As tabelas a
seguir exemplificam as dimensões e as áreas de cada triângulo e os integrais da curva DSC
correspondentes.
Tabela 18 - ΔHf para os vários picos e para o triângulo vermelho de cada amostra.
ΔHf Total ΔHf Pico 1 ΔHf Pico 2 ΔHf Triângulo
Amostra C – 1º
Ensaio 40.35 J/g 14.73 J/g 14.72 J/g 10.90 J/g
Amostra D – 1º
Ensaio 39.04 J/g 15.33 J/g 11.88 J/g 11.83 J/g
Figura 69 - Esquema do prolongamento de cada pico de fusão (tracejado a verde)
e da área que falta adicionar ao integral de cada pico (triângulo vermelho).
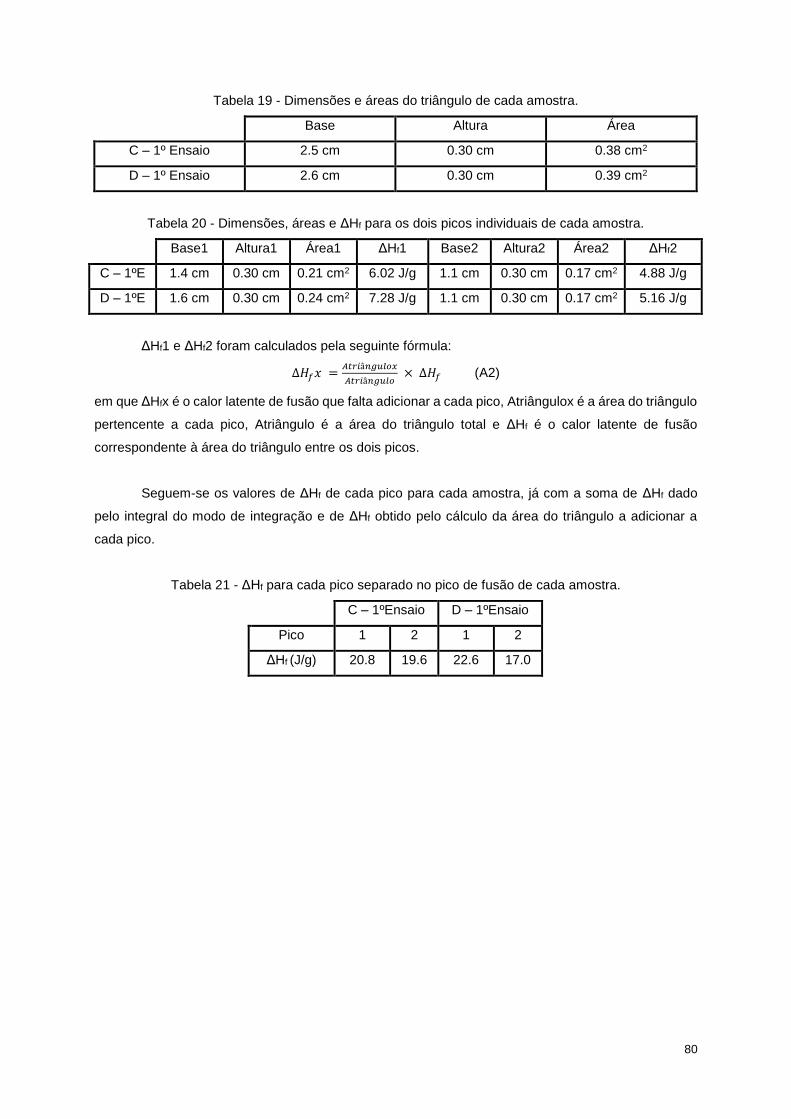
80
Tabela 19 - Dimensões e áreas do triângulo de cada amostra.
Base Altura Área
C – 1º Ensaio 2.5 cm 0.30 cm 0.38 cm2
D – 1º Ensaio 2.6 cm 0.30 cm 0.39 cm2
Tabela 20 - Dimensões, áreas e ΔHf para os dois picos individuais de cada amostra.
Base1 Altura1 Área1 ΔHf1 Base2 Altura2 Área2 ΔHf2
C – 1ºE 1.4 cm 0.30 cm 0.21 cm2 6.02 J/g 1.1 cm 0.30 cm 0.17 cm2 4.88 J/g
D – 1ºE 1.6 cm 0.30 cm 0.24 cm2 7.28 J/g 1.1 cm 0.30 cm 0.17 cm2 5.16 J/g
ΔHf1 e ΔHf2 foram calculados pela seguinte fórmula:
∆𝐻𝑓𝑥 =𝐴𝑡𝑟𝑖â𝑛𝑔𝑢𝑙𝑜𝑥
𝐴𝑡𝑟𝑖â𝑛𝑔𝑢𝑙𝑜 × ∆𝐻𝑓 (A2)
em que ΔHfx é o calor latente de fusão que falta adicionar a cada pico, Atriângulox é a área do triângulo
pertencente a cada pico, Atriângulo é a área do triângulo total e ΔHf é o calor latente de fusão
correspondente à área do triângulo entre os dois picos.
Seguem-se os valores de ΔHf de cada pico para cada amostra, já com a soma de ΔHf dado
pelo integral do modo de integração e de ΔHf obtido pelo cálculo da área do triângulo a adicionar a
cada pico.
Tabela 21 - ΔHf para cada pico separado no pico de fusão de cada amostra.
C – 1ºEnsaio D – 1ºEnsaio
Pico 1 2 1 2
ΔHf (J/g) 20.8 19.6 22.6 17.0