
diversidade metagenômica microbiana de de biomas terrestres e ...
269
UNIVERSIDADE FEDERAL DO RIO DE JANEIRO Instituto de Biologia Departamento de Genética DIVERSIDADE METAGENÔMICA MICROBIANA DE DE BIOMAS TERRESTRES E MARINHOS THIAGO BRUCE RODRIGUES Rio de Janeiro, agosto de 2011
Transcript of diversidade metagenômica microbiana de de biomas terrestres e ...

UNIVERSIDADE FEDERAL DO RIO DE JANEIRO
Instituto de Biologia
Departamento de Genética
DIVERSIDADE METAGENÔMICA MICROBIANA DE
DE BIOMAS TERRESTRES E MARINHOS
THIAGO BRUCE RODRIGUES
Rio de Janeiro, agosto de 2011

Universidade Federal do Rio de Janeiro
Instituto de Biologia
Departamento de Genética
Pós-Graduação em Ciências Biológicas (Genética)
DIVERSIDADE METAGENÔMICA MICROBIANA DE
DE BIOMAS TERRESTRES E MARINHOS
Thiago Bruce Rodrigues
Tese submetida ao curso de Pós-Graduação em Ciências Biológicas
(Genética), como parte dos requisitos para a obtenção de grau de
Doutor em Ciências Biológicas (Genética)
Orientador: Dr. Fabiano L. Thompson
Rio de Janeiro, agosto de 2011

Dedico este trabalho à minha Vó, D. Wilma

iv
Para ser grande, sê inteiro: nada
Teu exagera ou exclui.
Sê todo em cada coisa. Põe quanto és
No mínimo que fazes.
Assim em cada lago a lua toda
Brilha, porque alta vive.
-Fernando Pessoa

v
AGRADECIMENTOS
À minha Família, por tudo.
Ao Dr. Fabiano L. Thompson pela oportunidade, parceria e ensinamentos.
Ao Programa de Pós Graduação em Genética.
À Giselle Bussi pelo amor e compreensão.
Aos Colegas do Laboratório de Microbiologia da UFRJ pelos anos de convivência.
Ao Dr. Ricardo Kruger, Alinne Castro e Alessandra Reis pela colaboração na construção das
bibliotecas da Mata Atlântica.
Ao Dr. Nei Pereira e Roberto Maeda pela ajuda com as atividades celulolíticas.
Ao Dr. Robert Edwards e Dra. Elizabeth Dinsdale pela oportunidade e por disponibilizar uma
das plataformas de sequenciamento da San Diego State University.
A Dra. Marina Paez e os alunos Paulo Dick e Felipe Vega pela ajuda nas análises estatísticas.
Á Dra Ana Carolina Vicente, Dr. Allen Hagler e Dra Ana Coelho que compuseram a banca do
meu exame de qualificação.
À Direção do Parque Nacional da Serra dos Órgãos por possibilitar o acesso ao parque.
À Direção do Parque Marinho de Abrolhos por possibilitar o acesso ao parque.
Aos senhores membros da banca.
Ao CNPQ pelo apoio financeiro.
A todos aqueles que contribuíram de forma direta ou indireta durante toda minha formação
acadêmica.

vi
RESUMO
A aplicação de métodos independentes de cultivo para o estudo da diversidade microbiana nos
últimos 20 anos mostrou que parcela significativa da diversidade não é recuperada em meios
de cultura. Cerca de 325 Mb de dados (aproximadamnete 800 mil sequências) foram
analisados neste trabalho. Mais de 700 mil sequências são oriundas de biomas brasileiros.
Este trabalho representa o estudo com maior número de sequências da microbiota Brasileira já
realizado. Quatro estudos sucessivos e complementares foram realizados: (1) Diversidade de
comunidades do bacterioplâncton das águas costeiras da América Latina foi caracterizada
através de pirosequenciamento da região hipervariável V6 do gene RNA 16S a partir de
bibliotecas de amplicons; (2) Diversidade taxonômica e funcional da microbiota planctônica
dos sistemas de recifes de corais do Banco de Abrolhos foi caracterizada através de
pirosequenciamento a partir de bibliotecas de fragmentos aleatórios (método shotgun); (3)
Diversidade de comunidades bacterianas do solo da Mata Atlântica foi caracterizada através
do sequenciamento de fragmentos do gene rRNA 16S e (4) Potencial biotecnológico da
microbiota do solo da Mata Atlântica foi demonstrado através da construção de biblioteca
metagenômica de pequenos insertos (10 Kb) para triagem funcional de clones com atividade
celulolítica. Além disso, foi apresentado um panorama acerca da utilização de métodos
independentes de cultivo para caracterização da diversidade microbiana de ecossistemas
tipicamente brasileiros. Desta forma, foi possível determinar a assinatura microbiana de
diferentes ecossistemas. Os resultados mostram a existência de gradiente latitudinal de
diversidade do bacterioplâncton ao longo da costa da América latina, com predomínio de
organismos cosmopolitas pertencentes a grupos taxonômicos conhecidos (Ex. SAR 11,
Acinetobacter e Flavobacteraceae) e uma pequena fração endêmica, sendo que o maior
endemismo ocorreu em localidades brasileiras. A partir dos resultados obtidos foi possível
concluir que as sequências da região V6 do gene rRNA 16S permitiram a caracterização de

vii
um gradiente latitudinal de diversidade ao longo da costa da América Latina. Já o segundo
estudo, focado na compreensão da diversidade microbiana do Banco de Abrolhos, mostrou
que os recifes protegidos apresentam menor abundância de Arquéias, vírus, patógenos
humanos e patógenos marinhos como os vibrios. As áreas protegidas apresentam maior
contribuição de organismos autotróficos. Foi possível identificar diferentes níveis de
degradação dos recifes do Banco de Abrolhos. Portanto, os metagenomas refletem a tendência
de degradação da região do Banco de Abrolhos, sugerindo que os metagenomas podem ser
usados no monitoramento da saúde recifal. A análise DNA metagenômico da microbiota da
Mata Atlântica permitiu a caracterização de grupos taxonômicos dominantes como
Acidobacteria e Proteobacteria no solo. A estimativa do número de espécies por grama de
solo é de aproximadamente 1500 espécies. Grande número de sequências não classificadas
(Aprox. 15%) reforça a idéia da existência de espécies e filos novos para serem caracterizados
e o potencial da Mata Atlântica para bioprospecção. Estes dados demonstram que existe um
vasto repertório genético que apresenta potencial biotecnológico, mas ainda não foi
completamente explorado. A construção de uma biblioteca metagenômica (aproximadamente
40.000 clones) permitiu a triagem funcional de clones com atividade celulolítica (FPásica e
CMCásica. 20 U/L e 100 U/L, respectivamente), semelhante a observada em isolados
ambientais. Este trabalho de doutoramento. O solo da Mata Atlântica brasileira representa
uma vasta fonte de novas espécies de bactérias com potencial para exploração biotecnológica.
A comparação da diversidade de diferentes ecossistemas evidencia que a microbiota de
biomas terrestres e marinhos é diferente e particular de cada ecossistema.

viii
ABSTRACT
The application of culture independent methods for the study of microbial diversity in the last
20 years has disclosed a unknown majority that not yet was recovered by using of culture
media. About 325 Mb of data (about 800 thousands of sequences) were analyzed in this thesis
(200 Mb correspond to sequences generated in this work). More than 700000 sequences
belonging to Brazilian biomes were analyzed. This work represents the study with the larger
number of sequences of Brazilian microbiota already carried out. Four complementary and
successive studies were performed: (1) Bacterioplankton communities diversity along coastal
waters of Latin America was characterized by means of V6 tag pyrosequencing from
amplicon libraries; (2) Taxonomic and functional diversity of aquatic microbiota of Abrolhos
Bank was characterized through pyrosequencing of shotgun libraries; (3) Microbial
community diversity of Atlantic Forest soils was characterized through rRNA 16S clone
libraries sequencing and (4) Biotechnological potential of microbiota of Atlantic Forest soils
was demonstrated through of a small fragments metagenomic library (10 Kb) for functional
screening of clones with cellulolytic activity. Moreover, an overview on the culture
independent methods for characterization of microbial diversity from typically Brazilian
ecosystems was presented. In such a way, it was possible to determine the microbial signature
of different Brazilian ecosystems. Results showed a latitudinal gradient of diversity for
bacterioplakton communities along Latin America coast with predominance of cosmopolite
organisms belonging to known taxonomic groups (ex. SAR 11, Acinetobacter,
Flavobacteraceae and others) and a small endemic fraction, with higher endemism in
Brazilian samples. The Abrolhos Bank metagenomes showed that protecting reefs present
minor abundance of Archaea, viruses, human patogens and marine patogens as Vibrios and

ix
the protecting areas present greater contribution of autotrophic organisms. It was possible to
identify different degradation levels of Abrolhos reefs. Metagenomes reflected the trend of
degradation of Abrolhos Bank region, suggesting that the metagenomes can be used in the
monitoring of reef health as early warning tool. The strategies based on cloning of the
metagenomic DNA of Atlantic Forest soils allowed the characterization of dominant
taxonomic groups as Acidobacteria and Proteobacteria. The estimate species number per
gram of soil was approximately 1500 species. A great number of unclassified sequences
(approx 15%) reinforce the existence of many species and phyla not yet characterized. These
data demonstrate a vast genetic repertoire that presents biotechnological potential, but not
duly was explored. The construction metagenomic library contend approximately 40000
clones allowed the functional screening of clones with celullolyitic activity (FPásica and
CMCásica; 20 U/L and 100 U/L, respectively), with similar measures observed for
environmental isolates. Brazilian Atlantic Forest soils represent a vast source of new bacteria
with potential for biotechnological exploration. The comparison of microbiotas of different
ecosystems shows that each ecosystem has a typical microbial composition.

x
SUMÁRIO
1. Introdução ...................................................................................................................................................... 1
1.1 Origem da diversidade genética .............................................................................................................. 2
1.1.1 O DNA detem a informação .......................................................................................................... 2
1.1.2 Principais mecanismos de evolução molecular em procariotos ................................................... 4
1.1.2.1 Mutação .................................................................................................................................... 4
1.1.2.2 Duplicação gênica ..................................................................................................................... 5
1.1.2.3 Transferência horizontal de Genes ........................................................................................... 6
1.2 Abordagens de estudo em diversidade microbiana .............................................................................. 10
1.2.1 Métodos dependentes de cultivo ............................................................................................... 10
1.2.2 Métodos independentes de cultivo ............................................................................................ 13
1.2.2.1 Metagenômica ........................................................................................................................ 13
1.2.2.1.1 Análise baseada em função ................................................................................................ 15
1.2.2.1.2 Análise baseada em sequências ......................................................................................... 17
1.2.2.1.3 Desafio computacional ....................................................................................................... 19
1.2.2.1.4 Pirosequenciamento........................................................................................................... 21
1.2.3 Medidas de diversidade .............................................................................................................. 25
1.2.3.1.1 Riqueza de espécies ............................................................................................................ 25
1.2.3.1.2 Índices de diversidade ........................................................................................................ 27
1.2.3.1.3 Estimadores de diversidade ............................................................................................... 27
1.3 Os microrganismos no meio ambiente .................................................................................................. 29
1.3.1 Microbiota aquática e a saúde ambiental ................................................................................... 31
1.3.2 Microbiota terrestre e seu potencial biotecnológico .................................................................. 35
1.4 Biodiversidade ....................................................................................................................................... 40
1.4.1 Biomas aquáticos: zona costeira ................................................................................................. 42
1.4.1.1 Manguezais e lagoas ............................................................................................................... 44
1.4.1.2 Sistemas recifais ...................................................................................................................... 48
1.4.2 Bioma terrestre: o solo da Mata Atlântica .................................................................................. 53
1.4.2.1 Características do solo ............................................................................................................ 54
1.4.2.2 A Mata Atlântica ..................................................................................................................... 57
2. Objetivos ....................................................................................................................................................... 60
2.1 Objetivo geral ........................................................................................................................................ 60

xi
2.2 Objetivos específicos ............................................................................................................................. 60
3. Materiais e métodos ..................................................................................................................................... 61
3.1 Gradiente latitudinal de diversidade do bacterioplâncton da costa da América latina ........................ 61
3.2 Avaliação da saúde do Banco de Abrolhos através da metagenômica ................................................ 63
3.2.1 Área de estudo e coleta .............................................................................................................. 64
3.2.2 Extração do DNA metagenômico da água do mar ...................................................................... 66
3.2.3 Pirosequenciamento dos metagenomas do Banco de Abrolhos ................................................ 66
3.2.3.1 Purificação e quantificação do DNA metagenômico .............................................................. 67
3.2.3.2 Preparação de bibliotecas pelo método shotgun ................................................................... 67
3.2.3.3 Quantificação e checagem da qualidade da fragmentação das bibliotecas ........................... 68
3.2.3.4 Reação em cadeia da polimerase em emulsão (em-PCR) ....................................................... 68
3.2.3.5 Quebra da emulsão (break emulsion)..................................................................................... 68
3.2.3.6 Enriquecimento (Enrichment)................................................................................................. 69
3.2.3.7 Preparação e sequenciamento ............................................................................................... 69
3.2.4 Análises Computacionais dos metagenomas da água dos recifes do Banco de Abrolhos .......... 69
3.2.4.1 Filtragem das sequências de baixa qualidade ......................................................................... 70
3.2.4.2 Anotação dos metagenomas pela tecnologia de subsistemas ............................................... 70
3.2.4.3 Análises comparativas ............................................................................................................ 71
3.3 Diversidade Microbiana do solo da Mata Atlântica............................................................................... 73
3.3.1 Área de estudo e coleta .............................................................................................................. 73
3.3.2 Construção de bibliotecas de rDNA 16S das comunidades microbianas dos solos do PARNASO75
3.3.2.1 Extração do DNA metagenômico ............................................................................................ 75
3.3.2.2 Amplificação do gene rRNA 16S.............................................................................................. 75
3.3.2.3 Clonagem dos genes rRNA 16S ............................................................................................... 76
3.3.2.3.1 Sistema de ligação .............................................................................................................. 77
3.3.2.3.2 Preparação de células eletrocompetentes ......................................................................... 77
3.3.2.3.3 Transformação .................................................................................................................... 78
3.3.2.4 Montagem das bibliotecas e estocagem dos clones .............................................................. 78
3.3.2.4.1 Confirmação dos clones ..................................................................................................... 78
3.3.2.4.2 Extração plasmidial em placa ............................................................................................. 79
3.3.2.5 Sequenciamento dos clones ................................................................................................... 79
3.3.2.6 Análises Computacionais de Sequências do gene rRNA 16S do solo da Mata Atlântica ........ 79
3.3.2.6.1 Filtragem de sequências ..................................................................................................... 80
3.3.2.6.2 Estrutura das comunidades microbianas (classificação taxonômica) ................................ 80
3.3.2.6.3 Estimativa de riqueza e diversidade ................................................................................... 81
3.3.2.6.4 Análises filogenéticas ......................................................................................................... 83

xii
3.3.3 Construção da biblioteca metagenômica de médios insertos .................................................... 84
3.3.3.1 Extração e purificação do DNA metagenômico ...................................................................... 84
3.3.3.2 Preparação do vetor de clonagem pCF430 ............................................................................. 85
3.3.3.3 Preparação dos médios insertos ............................................................................................. 85
3.3.3.4 Análise funcional da Biblioteca Metagenômica do solo da Mata Atlântica ............................ 86
3.3.3.4.1 Triagem de clones com atividade celulolítica ..................................................................... 87
3.3.3.4.2 Determinação da atividade celulolítica .............................................................................. 87
3.3.3.4.2.1 Curva de crescimento................................................................................................... 87
3.3.3.4.2.2 Atividade FPásica (celulose total)................................................................................. 88
3.3.3.4.2.3 Atividade CMCásica (endoglucanásica) ........................................................................ 88
3.3.3.4.2.4 Atividade β-glicosidásica .............................................................................................. 88
3.4 Diversidade microbiana dos biomas brasileiros .................................................................................... 88
4. Resultados e discussão ................................................................................................................................. 90
4.1 Diversidade da comunidade de bacterioplâncton ao longo de uma gradiente latitudinal na costa da
América Latina por meio de pirosequenciamento da região V6....................................................................... 90
4.1.1 Resultados ................................................................................................................................... 90
4.1.2 Discussão ................................................................................................................................... 104
4.2 Avaliação da saúde do Banco de Abrolhos através da metagenômica .............................................. 108
4.2.1 Resultados ................................................................................................................................. 108
4.2.2 Discussão ................................................................................................................................... 124
4.3 Diversidade das comunidades microbianas dos solos da Mata Atlântica e o seu potencial
biotecnológico ................................................................................................................................................. 130
4.3.1 Resultados ................................................................................................................................. 130
4.3.1.1 Diversidade da das comunidades microbianas dos solos da Mata Atlântica........................ 130
4.3.1.2 Potencial biotecnológico do DNA metagenômico do solo da Mata Atlântica ...................... 142
4.3.2 Discussão ................................................................................................................................... 146
4.4 Diversidade microbiana dos biomas brasileiros .................................................................................. 152
4.4.1 Resultados ................................................................................................................................. 152
4.4.2 Discussão ................................................................................................................................... 167
4.4.2.1 Diversidade microbiana nos biomas terrestres .................................................................... 168
4.4.2.2 Diversidade microbiana em ambientes marinhos ................................................................ 172
4.4.2.3 Diversidade microbiana em áreas urbanas costeiras ........................................................... 173
4.4.2.4 Perspectivas .......................................................................................................................... 178
5. Conclusões .................................................................................................................................................. 179
5.1 Conclusões gerais ................................................................................................................................ 179
5.2 Conclusões específicas ........................................................................................................................ 179

xiii
5.2.1 Diversidade da comunidade de bacterioplâncton ao longo de uma gradiente latitudinal na
costa da América Latina por meio de pirosequenciamento da região V6 .................................................. 179
5.2.2 Avaliação da saúde do Banco de Abrolhos através da metageômica ....................................... 180
5.2.3 Diversidade das comunidades microbianas dos solos da Mata Atlântica e o seu potencial
biotecnológico ............................................................................................................................................ 181
5.2.4 Diversidade microbiana dos biomas brasileiros ........................................................................ 182
6. Referências Bibliográficas ........................................................................................................................... 184
7. ANEXOS ....................................................................................................................................................... 214
7.1 Anexo 1. Métodos ............................................................................................................................... 214
7.1.1 Pirosequenciamento dos metagenomas do Banco de Abrolhos .............................................. 214
7.1.1.1 Extração do DNA realizada no interior do Sterivex ............................................................... 214
7.1.1.2 Purificação do DNA metagenômico ...................................................................................... 215
7.1.1.3 Preparação das bibliotecas pelo método shotgun................................................................ 216
7.1.1.3.1 Preparação de esferas magnéticas ................................................................................... 218
7.1.1.3.2 Ligação do DNA as esferas ................................................................................................ 218
7.1.1.3.3 Quantificação e checagem da qualidade da fragmentação das bibliotecas ..................... 219
7.1.1.4 Reação em cadeia da polimerase em emulsão (em-PCR) ..................................................... 219
7.1.1.4.1 Preparação do óleo para emulsões .................................................................................. 220
7.1.1.4.2 Mock Amplification Mix e pré-emulsões .......................................................................... 220
7.1.1.4.3 Preparação do Live Amplification Mix .............................................................................. 220
7.1.1.4.4 Preparação das esferas de captura de DNA (beads) ........................................................ 221
7.1.1.4.5 Captura das bibliotecas .................................................................................................... 222
7.1.1.4.6 Emulsificação .................................................................................................................... 222
7.1.1.5 Quebra da emulsão (break emulsion)................................................................................... 223
7.1.1.5.1 Recuperação das esferas .................................................................................................. 223
7.1.1.5.2 Lavagem das esferas ......................................................................................................... 224
7.1.1.6 Enriquecimento (Enrichment) ............................................................................................... 225
7.1.1.6.1 Enriquecimento indireto .................................................................................................. 225
7.1.1.6.2 Preparação das esferas de enriquecimento (enrichment beads) ..................................... 226
7.1.1.6.3 Enriquecimento das esferas com DNA ............................................................................. 226
7.1.1.6.4 Recuperação das esferas de DNA enriquecidas ............................................................... 227
7.1.1.7 Preparação e sequenciamento ............................................................................................. 228
7.1.1.7.1 Carregamento da Placa PicoTilter Plate (PTP) .................................................................. 229
7.1.1.7.2 Preparação da PTP e DNA beads ...................................................................................... 229
7.1.1.8 Preparação do GS FLX 454 .................................................................................................... 232
7.1.2 Diversidade de comunidades microbianas do solo da Mata Atlântica ..................................... 232
7.1.2.1 Concentração e purificação dos fragmentos do rRNA 16S ................................................... 233

xiv
7.1.2.2 Sistema de ligação ................................................................................................................ 234
7.1.2.3 Preparação de células eletrocompetentes ........................................................................... 235
7.1.2.4 Extração plasmidial em placa ................................................................................................ 236
7.1.3 Análise funcional da Biblioteca Metagenômica ........................................................................ 241
7.1.3.1 Extração e purificação do DNA metagenômico com esferas de vidro .................................. 241
7.1.3.2 Preparação do vetor de clonagem pCF430 ........................................................................... 243
7.1.3.3 Digestão parcial do DNA metagenômico .............................................................................. 244
7.1.3.4 Clonagem dos médios insertos ............................................................................................. 245
7.1.3.5 Reativação e pré - seleção de clones em meio CMC ............................................................ 246
7.1.3.5.1 ............................................................................................................................................... 247
7.1.3.6 Atividade FPásica (celulose total) ......................................................................................... 247
7.1.3.7 Atividade CMCásica (endoglucanásica) ................................................................................ 248
7.1.3.8 Atividade β-glicosidásica ....................................................................................................... 248
7.2 Anexo 2. Lista de publicações .............................................................................................................. 250
7.2.1 Periódicos .................................................................................................................................. 250
7.2.1.1 Artigo 1 ................................................................................................................................. 250
7.2.1.2 Artigo 2 ................................................................................................................................. 250
7.2.1.3 Artigo 3 ................................................................................................................................. 250
7.2.1.4 Artigo 4 ................................................................................................................................. 250
7.2.2 Capítulo de livro ........................................................................................................................ 250
LISTA DE TABELAS
Tabela 1. Principais consórcios que desenvolvem análises a partir de metagenomas baseada em sequenciamento
...................................................................................................................................................................... 21
Tabela 2. Parâmetros biológicos e físico-químicos dos locais de amostragem. .................................................... 91
Tabela 3. Número de sequências, diversidade e cobertura das bibliotecas da região V6 do gene rRNA 16S das
comunidades microbianas ao longo do gradiente latitudinal. ....................................................................... 91
Tabela 4. Distribuição e identificação da fração cosmopolita do gradiente latitudinal. Os números representam a
fração de sequências agrupadas por táxons em relação ao total de seqüências classificadas pelo GAST. A
identificação foi possível em diferentes níveis taxonômicos. ....................................................................... 95
Tabela 5. Distribuição e identificação da fração endêmica ao longo do gradientelatitudinal. *PR – Puerto Rico;
AM –Amazon estuary; GB – Guanabara Bay; LG1 – Laguna do Rocha 1; LG2 –Laguna do Rocha 2; RP –
Rio de la Plata and AS – Argentinian shelf . ................................................................................................ 96
Tabela 6. Características gerais dos locais de coleta e dos metagenomas. Média ± erro padrão. Número de
replicatas entre parênteses. DOC (Carbono orgânico diddolvido); POC (Carbono orgânico particulado). 109

xv
Tabela 7. Diversidade nas areas protegidas e não protegidas. Os valores foram obtidos usando o número total de
sequências classificadas como Bacteria, Virus ou Archeae pelo MG-RAST. Abaixo, o resumo das análises
estatísticas aplicadas aos valores encontrados mostra que houve diferença significativa apenas para os
valores relacionados a riqueza de espécies. NS - diferença não significativa; DS - Diferença significativa.
.................................................................................................................................................................... 118
Tabela 8. Diversidade e abundância dos subsistemas (metabolismo do carboidrato, nitrogênio, virulência,
resposta a estresse e fotossíntese). Os dez primeiros subsistemas (hierarquia 1) de cada subsistemas. ..... 118
Tabela 9. Os grupos mais frequentes encontrados nas bibliotecas e seus vizinhos filogenéticos mais próximos
(cultivados e não cultivados). Os agrupamentos representam pelo menos seis sequências com similaridade
> 97 % entre elas. ....................................................................................................................................... 137
Tabela 10. Distribuição das sequências dos grupos mais frequentes nos diferentes tipos de solo da Mata
Atlântica...................................................................................................................................................... 139
Tabela 11. Ensaios de atividade enzimática. A1, A2 e A3 são clones selecionados da biblioteca de pequenos
insertos. Cel 1A e Cel CAAB são isolados do solo da Mata Atlântica que apresentaram capacidade de
formar halos de degradação no meio de cultura. ........................................................................................ 146
Tabela 12. Lista de estudos de diversidade microbiana nos principais biomas brasileiros por abordagens
independentes de cultivo em ambientes terrestres. ..................................................................................... 156
Tabela 13. Lista de estudos de diversidade microbiana dos principais biomas brasileiros por abordagens
independentes de cultivo em ambientes aquáticos nos últimos anos. ......................................................... 159
Tabela 14. Lista de novos táxons de ambientes terrestres descritos nos últimos 5 anos. .................................... 161
Tabela 15. Lista de novos táxons de ambientes aquáticos descritos nos últimos 5 anos. ................................... 165
LISTA DE FIGURAS
Figura 1. A árvore da vida baseada em filogenia molecular do gene rRNA 16S (adaptado de Pace, 2011). 3
Figura 2. Principais mecanismos de geração de variabilidade genética (adaptado de Alberts, 2008) ..................... 4
Figura 3. Origem de genes parálogos (adaptado de Alberts, 2008). ........................................................................ 6
Figura 4. Processos de intercelulares de troca de material genético . ...................................................................... 7
Figura 5. Regiões de recombinação homóloga das ilhas de patogenicidade, de maneira geral, apresentam maior
conteúdo AT em relação ao restante do genoma. Ilhas genômicas são muitas vezes inseridos em genes
tRNA e genes que codificam fatores envolvidos na mobilidade genética, como integrases, transposases e
sequências de inserção (IS). De acordo com seu conteúdo genético, podem ser descritas como ilhas de
patogenia, simbiose, metabólicas, ilhas de fitness ou de resistência (Juhas, 2009). ....................................... 8
Figura 6. Halo de degradação produzido por bactérias celulolíticas, revelado por vermelho congo. .................... 12
Figura 7. Modelo esquemático de um estudo com abordagem metagenômica.(adaptado de Handelsman, 2004) 15
Figura 8. Representação esqumática do sistema enzimático do pirosequenciamento. O nucleotídeo incorporado
pela polimerase, liberando uma molécula de pirofosfato (PPi). A enzima ATP sulfurilase converte o PPi em
ATP, que atua como substrato para a produção de luz pela luciferase. A luz produzida é detectada,
formando um pirograma que indica a sequência de nucleotídeos incorporados durante o sequenciamento
(retirado de Ahmadian, 2006). ...................................................................................................................... 23

xvi
Figura 9. Preparação da amostra para pirosequenciamento. a) Fragmentos de DNA com adaptadores e acoplados
as esferas submetidas a em-PCR em emulsões de óleo. Após a quebra das emulsões são depositadas em um
slide onde cada esfera contendo os produtos da amplificação são separadas individualmente. b)
Microscopia da emulsão mostrando gotas contendo as esferas. A seta larga representa uma gota de 100 μm
e a fina mostra uma esfera de 28 μm. c) Microscopia eletrônica de um slide (retirada de Margulies, 2005).
...................................................................................................................................................................... 24
Figura 10. Curvas de rarefação mostrando comunidades com diferente diversidade de espécies. É possível
observar que as comunidades presentes em carcaças de baleia são menos diversas que as do solo e por isso
apresentam uma menor inclinação da reta, representando que houve mairo cobertura da diversidade,
indicando um menos número de filotipos em comparação com o solo (Retirado de Tringe, 2005). ............ 26
Figura 11. O modelo DDAM é positivamente retroalimentado quando há aumento de COD, produzido por algas,
para suportar o crescimento de microrganismos associados aos corais. A proliferação de algas ocorre em
função da retirada de peixes, principalmente herbívoros, dos sistemas recifais. A proliferação de
microrganismos promove a morte de corais, aumentando a cobertura de algas. .......................................... 34
Figura 12. Doenças que afetam corais brasileiros. A) Doença da banda-preta em Mussismilia braziliensis, B)
doença da faixa-vermelha em Siderastrea spp, C) doença dark spot like em Siderastrea spp e D) praga
branca em M. braziliensis (retirado de Francini-Filho, 2008) ...................................................................... 35
Figura 13. Estrutura da lignocelulose. O principal componente de lignocelulose é a celulose, ab (1-4) ligada
cadeia de moléculas de glicose. A lignina é sintetizado através da polimerização dos componentes e sua
relação dentro do polímero varia entre diferentes plantas, tecidos e camadas de madeira da parede celular.
Celulose, hemicelulose e lignina formam estruturas chamadas microfibrilas, que são organizados em
macrofibrilas que medeiam a estabilidade estrutural da parede celular vegetal (retirado de Rubin, 2008). . 36
Figura 14. Localização dos hotspots de riqueza de espécies ao redor do mundo considerados áreas prioritárias
para conservação (Meyers, 2000). ................................................................................................................ 41
Figura 15. Áreas no território brasileiro com potencial impacto poluidor, localizadas próximas a corpos d’água.
Os pontos vermelhos indicam as áreas com potencial impacto poluidor. A maior concentração dessas áreas
encontra-se ao longo da costa, onde observamos maior concentração de atividades humanas e industriais.
Fonte: (IBGE, 2000). .................................................................................................................................... 45
Figura 16. Ecossistemas costeiros sob impacto antropogênico. A) Manguezal da Baía de Guanabara (foto: Erick
Aniszewski) e B) Ocupação industrial na Baía de Sepetiba (fonte: http://www.revistaportuaria.com.br). .. 46
Figura 17. A produção primária requerida para sustentar a produção pesqueira mundial de pesca, expresso em
percentagem da produção primária local. Os mapas representam o total de desembarques anuais de 1950
(acima) e 2005 (abaixo) (Swartz, 2010). ...................................................................................................... 50
Figura 18. Mapa com a distribuição dos recifes na costa brasileira. (Fonte: RECOR) ......................................... 52
Figura 19. Os latossolos representam um dos tipos mais comumente encontrados no Mata Atlântica. ................ 56
Figura 20. Cobertura do bioma Mata Atlântica. Aproximadamente 90 % do bioma já foi devastado (Fonte: Atlas
dos Remanescentes Florestais da Mata Atlântica. SOS Mata Atlântica e Instituto Nacional de Pesquisas
Espaciais – INPE. ......................................................................................................................................... 57
Figura 21. Localização geográfica dos pontos de coleta ao longo da costa da America Latina. ........................... 62
Figura 22. Sistema GAST de classificação taxonômica utilizado para implementação de um banco de dados
curados da região V6 do gene rRNA 16S (Retirada de https://vamps.mbl.edu/) .......................................... 63

xvii
Figura 23. Região do Banco de Abrolhos e os pontos de coleta. Em azul, as áreas não protegidas e em vermelho
as áreas protegidas. As linhas vermelhas representam os limites do Parque Marinho de Abrolhos (Foto:
Pedro Meirelles). .......................................................................................................................................... 65
Figura 24. Mergulhador coletando a água dos recifes de Abrolhos a aproximadamente 20 m de profundidade
(Foto: Ronaldo Francini-Filho). .................................................................................................................... 65
Figura 25. Sistem de filtração Niskin. A) Sistema de filtração com os filtros Sterivex já acoplados. B) Filtros
Sterivex durante o processo de filtração (Foto: Rodrigo Moura).................................................................. 66
Figura 26. Mapa do Parque Nacional da Serra dos Órgãos com a localização geográfica dos pontos analisados. 74
Figura 27. Vetor de clonagem pGEMT Easy. ....................................................................................................... 77
Figura 28. Plasmídeo pCF430. .............................................................................................................................. 84
Figura 29. Influência do gradiente latitudinal na equitabilidade e produção primária bacteriana ao longo da costa
da América Latina. A) Os dados indicam uma tendência de diminuição da uniformidade de distribuição das
OTUs no nível de espécie, de acordo com a diminuição da latitude (R2 = 0,9451). B) A produtividade
primária é maior nas regiões tropicais e tende a diminuir em direção as regiões subtropicais (o eixo
Contribuição (%) está em escala logarítimica). ............................................................................................ 92
Figura 30. Estrutura das comunidades bacterianas nos sete locais. Os filos mais comumente encontrados são
indicados. Sequências Unknown representam aquelas as quais não foi possível atribuir uma classificação
taxonômica pelo VAMPS. ............................................................................................................................ 93
Figura 31. Número de sequências identifiicadas na fração conservada do gradiente latitudinal. Nível taxonômico
mais profundo identificados pelo VAMPS. .................................................................................................. 94
Figura 32. Fração de sequências comuns de um local para outro usando 97% de similaridade para definir OTUs.
...................................................................................................................................................................... 94
Figura 33. Análise de agrupamento das sequências. A ) Curvas de rarefação; B) Número de OTUs a diferentes
níveis de similaridade correspondente a diferentes níveis taxonômicos (linhagens, espécies, gêneros e
famílias). ..................................................................................................................................................... 101
Figura 34. Análise baseada na distribuição de filos, demonstrando a existência de táxons dominantes distribuídos
por poucos grupos taxonômicos. Por outro lado, mostra a contribuição de um grande número de táxons em
baixa frequênciaque representa a biosfera rara da microbiota aquática. ..................................................... 102
Figura 35. Análise filogenética demonstrando a resolução taxonômica da região v6 do gene rRNA 16S para os
gênero relacionados Prochlorococcus e Synechococcus. Foi utilizado método neighbor join, corrigido pelo
modelo Juckes-Cantor e bootstrap com 1000 replicatas............................................................................. 104
Figura 36. Comparação dos valores dos parâmetro Carbono Orgânico Dissolvido (COD) (acima) e Clorofila
(abaixo) entre os recifes protegidos e desprotegidos. Foi observada maior concentração de COD nos recifes
protegidos. A) ANOVA e B) Visualização gráfica dos resultados do teste de Tukey. PAB - Parcel dos
Abrolhos; SG - Sebastião Gomes; TIM – Timbebas e CAL – California. . Comparação dos valores do
parâmetro Clrofila entre os recifes mostra maiores concentrações no recife protegido. A) ANOVA e B)
Visualização gráfica dos resultados do teste de Tukey. PAB - Parcel dos Abrolhos; SG - Sebastião Gomes;
TIM – Timbebas ......................................................................................................................................... 110
Figura 37. Estrutura da comunidade microbiana dos recifes de Abrolhos. A). Contribuição dos diferentes
domínios. Enriquecimento de vírus é observada em amostras de recifes desprotegidos. B) Nível trófico dos
microrganismos dos cinco pontos. A classificação foi realizada com base na identificação dos filos.

xviii
Enriquecimento do metabolismo autotrófico é observado nos recifes protegidos. C) Distribuição dos
patógenos comumente encontrados. A classificação foi realizada baseada na identificaçãodas taxonômica
espécies/ linhagens. A contribuição da Vibrios está relacionada a sequências atribuídas como
Gammaproteobacteria. N corresponde ao número total de sequências para cada local ............................. 112
Figura 38. Estrutura das comunidades microbianas nos níveis de A) Filos e B) espécies / linhagens mais
frequentes (contribuição >1% do conjunto de sequências da amostra). A classificação taxonômica foi
realizada usando o servidor Mg-Rast. A identificação taxonômica é realizada baseada no resultado do
BLAST X realizado contra as sequências do banco de dados SEED (banco de genomas completos). A
origem taxonômica do melhor hit para a proteína identificada foi relacionada a abundância do táxon
identificado. N é o número de sequências utilizadas nas análises. ............................................................. 115
Figura 39. Contribuição dos subsistemas (hierarquia 1) nos diferentes recifes. As barras indicam a contribuição
das sequências para cada subsistema nos recifes protegidos e não protegidos. A coluna contribuição é
relativa ao número total de sequências identificadas para cada subsistema. Somente sequências
informativas foram utilizadas para identificação subsistemas. Sequências atribuído como subsistemas
miscelaneus, unknown e clustered based subsystems não foram incluídos nas análises. ............................ 116
Figura 40. Subsistemas do metabolismo de fotossíntese (média dos anos 2009-2010). A) Contribuição dos
subsistemas (hierarquia 3) relativa a todas as sequências anotadas pelo Mg-Rast. Os subsitemas mostraram
diferença (p<0,5; CI 95%) entre os recifes protegidos e não protegidos. B) Contribuição de diferente táxons
para o subsistema das proteorodopsinas. .................................................................................................... 117
Figura 41. Contribuição dos diferentes grupos taxonômicos para seis subsistemas relevantes (carboidrato,
fósforo, nitrogênio, virulência, resposta ao estresse, e fotossíntese), utilizando MG- RAST . As estrelas
coloridas representam maior abundância (p <0,5, IC 95%) de taxa para o respectivo subsistema em áreas
desprotegidas e protegidas. N representa o número de sequências analisadas............................................ 123
Figura 42. Versatilidade metabólica dos Vibrios. O gráfico mostra que os grupos de Vibrios nas áreas não
protegidas apresentam maior comprometimento com metabolismo fototrófico. ........................................ 124
Figura 43. Diversidade de filos encontrados nas bibliotecas do gene rRNA 16S. ................................................ 130
Figura 44. Estrutura das comunidades microbianas do solo da Floresta da Mata Atlântica do PARNASO baseada
no sequenciamento de bibliotecas de clones do gene rRNA 16S de acordo com o RDP (limite de confiança
em 80 %). A) Abundância de filos; B) Contribuição (%) de subgrupos do filo Acidobacteria (Acidobacteria
Gp 1 ao Gp 17) e C) Contribuição (%) das classes de Proteobacteria. N representa o número te sequências
analisadas. ................................................................................................................................................... 133
Figura 45. Análise da cobertura e estimativa da diversidade baseada na análise da bibliotecas de clones do gene
rRNA 16S. Foram utilizados os níveis de corte de 97% (acima), 90% (meio) e 80% (abaixo) de
similaridade entre as sequências.Curvas de rarefação (A, B e C) e curvas coletoras (estimador Chao 1) (D,
E, e F). ........................................................................................................................................................ 136
Figura 46. Análise filogenética baseada nas sequências do gene rRNA 16S dos grupos de clones mais frequentes
nas bibliotecas Os grupos apresentam pelo menos seis sequências com similaridade > 97 % entre elas. Os
círculos pretos representam uma sequência representativa de cada grupo e entre parêntesis o número de
sequências representadas. As linhagens tipo e sequências relacionadas como vizinhos filogenéticos mais
próximos foram determinadas pelos bancos de dados RDP e GenBank. A fonte de identificação das
sequências dos bancos de dados do NCBI estão indicados. ........................................................................ 140
Figura 47. Relação entre diversidade H (índice de Shannon) e parâmetros físico químicos do solo. Unidades:
Matéria Orgânica (%); Alumínio (me/100cm³); Fósforo (mg/L); Potássio (mg/L). ................................... 141

xix
Figura 48. Fração de sequências dos filos Proteobacteria, Firmicutes e Actinobacteria compartilhada pelos
métodos dependentes e independentes de cultivo. ...................................................................................... 142
Figura 49. Extração do DNA metagenômico pelos métodos A) Kit comercial Mo Bio e B) Esferas de vidro. A1)
Mistura de DNA metagenômico dos diferentes tipos de solo da Mata Atlântico do PARNASO; A2)
Resultado da digestão parcial do DNA metagenômico pela enzima PstI, gerando quantidades insuficientes
para a construção da biblioteca metagenômica. B1) Resultado da extração do DNA metagenômico dos
solos ainda com contaminantes em diferentes concentrações (1, 3, 5 e 10 μL); B2)Resultado da purificação
e B3) Resultado da digestão parcial. A linha vermelha mostra a região excisadacontendo os fragmentos
para clonagem no plasmídeo pCF430. ........................................................................................................ 143
Figura 50. Extração plasmidial de colônias aleatórias demonstrando a variedade de insertos com diferentes
tamanhos. 1. pCF430 circular. 2-11. diferentes colônias (5μL). ................................................................. 144
Figura 51. Curva de crescimento dos clones em meio CMC. .............................................................................. 145
Figura 52. Parte superior: análise das componentes principais (PCoA) do pareamenteo unweight através da
matriz de distância do FastUniFrac . Cada ponto corresponde a comparação da presença ou ausência dos
grupos taxonômicos de bactérias em diferentes biomas. Triângulos verdes, círculos roxos e hexágonos
vermelhos representam microrganismos associados a solo, água e organismos marinhos. Comunidades de
(A) Bactérias e (B) Archaea, respectivamente. A porcentagem de variação explicada pelo traçado das
componentes principais está indicada nos eixos. Um total de 6.540 e 1.893 sequências do gene 16S rRNA
foram alinhadas para Bacterias e Archaea, respectivamente. Foram selecionadas sequências referentes as
regiões V1 e V2 do gene rRNA 16S. Sequências curtas menores que 300 pb foram retiradas da análise.
Archeas do solo não foram incluídas. Parte inferior: classifcação taxonômica baseada nas sequências do
gene rRNA 16S através da ferramenta Classifier do RDP. (C) Contribuição dos principais filos bacterianos
e (D) Contribuição de filos do domínio Archeae. ....................................................................................... 155
Figura 53. Análise filogenética de sequências não classificadas, recuperadas da água. As sequências foram
comparadas com sequências tipo a aprtir de comparação com o RDP. Todas as sequências foram alinhadas
usando o programa Muscle. As análises filogenéticas foram realizadas com o programa Mega, usando o
método de neighbor joining. Valores de bootstrap (1000 repetições) > 50 % são mostrados. A linha azul
corresponde a OTUs com alta similaridade (> 97 %) entre os genes rRNA 16S encontrados por diferentes
autores em ambientes aquáticos. É importante salientar os longos ramos, que representam novos grupos
taxonômicos em potencial. ......................................................................................................................... 175
Figura 54. Análise filogenética de sequências não classificadas, recuperadas do solo. As sequências foram
comparadas com sequências tipo a aprtir de comparação com o RDP. Todas as sequências foram alinhadas
usando o software Muscle. As análises filogenéticas foram realizadas com o software Mega, usando o
método de neighbor-joining. Valores de bootstrap (1000 repetições) > 50 % são mostrados. A linha verde
corresponde ao exemplo de sequências singletons, com grande distância evolutiva em comparação com as
sequências do gene 16S rRNA linhagens de tipo, demonstrando a heterogeneidade da diversidade de
Bactérias dos solos. ..................................................................................................................................... 176
Figura 55. Estrutura das comunidades bacterianas associadas ao solo, água e organismos marinhos.A
classificação foi realizada através de comparação com o RDP. A análise foi realizada com 30813
sequências de ecossistemas terrestres e 1864 sequências marinhas. ........................................................... 177

1
1. INTRODUÇÃO
A diversidade microbiana representa o mais vasto repertório genético do planeta. Entretanto,
apenas uma pequena fração dessa diversidade é conhecida. Os microrganismos direcionam os
principais ciclos biogeoquímicos (como o ciclo do carbono) e por isso desempenham funções
ecológicas indispensáveis no ambiente, como o ciclo do carbono. A microbiologia ambiental
foi revolucionada na década de 90 pelo avanço das técnicas moleculares que proporcionaram
a identificação de uma maioria microbiana até então não identificada pelos métodos de cultivo
tradicionais. O sequenciamento massivo tem proporcionado a caracterização de organismos
não cultivados que compõe as comunidades microbianas. Neste trabalho foram utilizadas
técnicas independentes de cultivo para acessar a diversidade microbiana de biomas aquáticos
e terrestres. Os resultados foram sumarizados em 4 capítulos onde apresento os resultados e
discussões.
No primeiro capítulo, a diversidade da comunidade de bacterioplâncton ao longo de uma
gradiente latitudinal na costa da América Latina foi caracterizada por meio de
pirosequenciamento da região V6 através de pirosequenciamento de bibliotecas de amplicons.
O segundo capítulo trata da caracterização do metagenoma da água para avaliação da saúde
do Banco de Abrolhos através de pirosequenciamento de bibliotecas de fragmentos aleatórios
(shotgun).
O terceiro capítulo teve como objetivo avaliar a diversidade das comunidades microbianas
dos solos da Mata Atlântica e o seu potencial biotecnológico através da construção e
sequenciamento de bibliotecas de clones do gene rRNA 16S pelo método de Sanger através
da construção de bibliotecas de pequenos insertos para e avaliação de atividade celulolítica. O
quarto capítulo apresenta um panorama da diversidade microbiana dos biomas brasileiros a

2
demonstração da assinatura microbiana associada aos diferentes ecossistemas terrestres e
aquáticos.
Esta introdução tem como objetivo apresentar os principais aspectos relevantes para o estudo
da diversidade microbiana para compreensão dos resultados que serão apresentados nos
capítulos posteriores. Temas como a origem da diversidade genética e as mudanças na
estruturas das comunidades microbianas em função das alterações das condições ambientais
foram abordados para compreender sua importância no funcionamento dos ecossistemas.
1.1 Origem da diversidade genética
A maioria dos organismos vivos são células simples, outros, como o ser humano, são
formadas por complexos multicelulares que desempenham funções especializadas e
conectadas por sistemas de comunicação. Em ambos os casos, as células, são o veículo para a
informação hereditária. Um dos mistérios em torno da origem da vida é como o material
genético poderia ter sido formado a partir de moléculas mais simples presentes no início
Terra. O DNA surgiu como uma forma permanente de armazenamento, devido à sua maior
estabilidade química. O surgimento da molécula de DNA representa o surgimento de uma
nova era no Planeta.
1.1.1 O DNA detem a informação
O sucesso dos organismos vivos com base em DNA, RNA e proteínas tem sido espetacular. A
capacidade de sobreviver e se perpetuar determinou sua persistência no ambiente (Futuyma,
2002). Células procarióticas vivem em uma ampla variedade de nichos ecológicos, e eles são
incrivelmente versáteis em suas capacidades bioquímicas, sendo capazes de metabolizar uma
ampla variedade de moléculas no ambiente.

3
A elucidação da estrutura da molécula do DNA, a partir de 1953, por Watson e Crick,
forneceu uma melhor compreensão da natureza das mutações e da variação genética. O
conceito de seleção natural foi expandido de modo a incluir não somente a sobrevivência e
reprodução de organismos individuais, mas também aos genes, populações e espécies. Como
a informação genética de cada organismo é escrita na linguagem universal de sequências de
DNA, a sequência de qualquer organismo pode ser obtida através de técnicas moleculares.
Com isso, é possível caracterizar, catalogar e comparar conjunto de organismos vivos a partir
dessas sequências. A partir de tais comparações, podemos estimar o lugar de cada organismo
na árvore genealógica das espécies vivas através de uma ―árvore da vida‖ (Pace, 2009)
(Figura. 1).
As análises do conjunto de genes de uma célula (genoma) é uma poderosa ferramenta para
determinar as relações evolutivas. A sequência completa do DNA de um organismo define a
sua natureza com precisão. Como o DNA está sujeito a mudanças aleatórias que se acumulam
durante longos períodos de tempo, o número de diferenças entre as sequências de DNA de
dois organismos pode proporcionar uma direta, objetiva e quantitativa indicação da distância
evolutiva entre eles.
Figura 1. A árvore da vida baseada em filogenia molecular do gene rRNA 16S (adaptado de Pace, 2011).

4
1.1.2 Principais mecanismos de evolução molecular em procariotos
O DNA é a matéria-prima da evolução. Não existe um mecanismo natural para fazer longos
trechos de sequências aleatórias novos. Sendo assim, nenhum gene é sempre inteiramente
novo. A inovação pode ocorrer através de processos intracelulares como mutação, duplicação
gênica e transferência horizontal de genes (THG) (Figura 2).
1.1.2.1 Mutação
As mutações são a principal origem da variação genética (Nei and Kumar, 2000). Elas geram
variações no conjunto de genes de uma população. Mutações desfavoráveis (ou deletérias)
podem ter sua frequência reduzida ou anulada na população por meio da seleção natural,
Figura 2. Principais mecanismos de geração de variabilidade genética (adaptado de Alberts, 2008)

5
enquanto mutações favoráveis (benéficas ou vantajosas) podem se acumular, resultando em
mudanças evolutivas adaptativas (Futuyma, 2002) (Figura 2).
A maioria das mutações ocorre por mecanismos endógenos, por hidrólise espontânea e erros
durante a replicação. Por exemplo, a DNA polimerase apresenta taxa de erro de 1 para cada
109 nucleotídeos incorporados (Alberts et al., 2008). As substituições podem ocorrer através
das transições, quando a mudança ocorre entre purinas (A ↔ G) ou pirimidinas (T↔C) e
também pode ocorrer através das transversões, quando ocorre a mudança de purina por
pirimidina e vice-versa (A, G ↔ T, C). Substituições também podem ocorrer
espontaneamente através de mudança tautomérica, permitindo o pareamento entre adenina e
citosina ou guanina e timina. Além disso, a duplicação, inserção e deleção de nucleotídeos
podem ocorrer espontaneamente ou através da ação de agentes físico ou químicos e pela
exposição a metabólitos tóxicos como as espécies reativas de oxigênio.
1.1.2.2 Duplicação gênica
Dentro de uma mesma família gênica, ou seja, conjuntos de dois ou mais loci com sequências
similares de DNA, podem surgir genes com novas funções através do processo de duplicação
gênica. Genes não correspondentes, na mesma ou em diferentes espécies, são chamados de
parálogos (Figura 3). Em procariontes, de modo geral, pelo menos 5-10% dos genes são
parálogos. Eventos de duplicação gênica constituem um marco na evolução da complexidade
biológica (Innan and Kondrashov, 2010). A idéia da duplicação gênica como força motriz do
processo evolutivo foi popularizada por Ohno (1970) (Ohno, 1970).
Uma vez duplicado, um gene pode estar sujeito a três destinos: (i) não-funcionalização, em
que uma das duas cópias do gene degenera em um pseudogene que pode ser
subsequentemente perdido do genoma; (ii) sub-funcionalização, que consiste na divisão das
funções originais do gene ancestral entre as duas cópias gênicas duplicadas; (iii) neo-

6
funcionalização, onde a cópia duplicada assume uma nova função (Zhang, 2003). A
duplicação gênica permite que as novas cópias do gene assumam funções mais especializadas
ou mesmo possibilita a origem de novos genes com funções totalmente distintas daquelas dos
genes de origem.
1.1.2.3 Transferência horizontal de Genes
Um pedaço de DNA pode ser transferido do genoma de uma célula para outra. Esse processo
é distinto da transferência vertical da informação genética dos pais para a progênie. Essas
alterações são capazes de deixar um traço característico do DNA exógeno.
Genomas procarióticos são compostos por dois tipos distintos de genes: (i) os
"informacionais", menos transmissíveis e envolvidos no processamento de informação na
célula, tais como a transcrição, tradução e replicação, e (ii) os "operacionais‖, frequentemente
transferíveis e envolvidos no metabolismo, mas com menor restrição funcional (Rivera et al.,
1998). Sendo assim, genomas microbianos podem evoluir através de transferência horizontal
dos genes. Em geral, genomas procariontes têm entre 5-30% de genes transferidos
Figura 3. Origem de genes parálogos (adaptado de Alberts, 2008).

7
lateralmente (Lawrence, 1999). Além dos genes essenciais que codificam as funções
metabólicas, genomas bacterianos também abrigam genes acessórios, adquiridos por
transferência horizontal de genes que podem ser benéficos em determinadas condições
ambientais. No ambiente ocorre transferência horizontal de genes entre vários gêneros e
espécies de bactérias por transformação, conjugação e transdução (Figura 4).
A. Transformação: As células capturam DNA exógeno diretamente do ambiente
ao seu redor.
B. Conjugação: Duas células interagem e uma doadora transfere DNA para a
receptora.
C. Transdução: Fagos transportam DNA de uma célula para outra.
A comparação dos genomas das linhagens de E. coli revelou que o número de genes centrais,
ou que executam funções essenciais naquela espécie, é surpreendentemente baixo (Welch et
al., 2002). Muitos dos genes transferidos recentemente não estão presentes em grupos
filogeneticamente próximos. Os genes ―acessórios‖ contêm um maior percentual de
nucleotídeos A e T (Figura 5), e tem uma utilização de códons que é semelhante à encontrada
Figura 4. Processos de intercelulares de troca de material genético .

8
em fagos e plasmídeos (Gogarten and Townsend, 2005). Muitos dos genes acessórios
adquiridos por transferência horizontal formam blocos chamados de ilhas genômicas. Ilhas
genômicas geralmente são reconhecidas como discretos segmentos de DNA presentes em
estirpes estreitamente relacionadas. Elas promovem a diversificação e adaptação dos
microrganismos, com significativo impacto na plasticidade dos genomas, disseminação de
resistência a antibióticos, genes de virulência e vias catabólicas (Gal-Mor and Finlay, 2006;
Juhas et al., 2009).
Figura 5. Regiões de recombinação homóloga das ilhas de patogenicidade, de maneira geral, apresentam maior
conteúdo AT em relação ao restante do genoma. Ilhas genômicas são muitas vezes inseridos em genes tRNA e
genes que codificam fatores envolvidos na mobilidade genética, como integrases, transposases e sequências de
inserção (IS). De acordo com seu conteúdo genético, podem ser descritas como ilhas de patogenia, simbiose,
metabólicas, ilhas de fitness ou de resistência (Juhas, 2009).

9
Esses processos ocorrem devido à recombinação genética de regiões homólogas do DNA, que
permite o aumento da variabilidade genética. Existem três principais maneiras de ocorrer o
evento de recombinação gênica na natureza. De forma geral, são elas (Ringo, 2004):
Recombinação: Ocorre troca entre moléculas de DNA homólogas, na qual a
localização do sítio de troca não é restrita. Geralmente, porções do DNA dupla-
hélice podem apresentar extensa homologia em regiões que ajudam na ligação de
proteínas (proteínas ligantes do DNA). As duas moléculas trocam segmentos do
DNA em um processo de várias etapas, catalisado por diferentes enzimas.
Recombinação sítio-específica: Ocorre troca entre moléculas de DNA não-
homólogos, ocorrendo apenas em sequências curtas e específicas, restritas a
poucos loci. A integração de plasmídeos ou cromossomos de fagos no DNA
genômico e a inversão de segmentos para expressão de genes alternativos são
exemplos deste tipo de evento.
Transposição: Consiste no movimento de elementos genéticos móveis chamados
transposons. Existem centenas de transposons diferentes. As transposases (enzimas
que promovem a recombinação do transposon) apresentam três diferentes
mecanismos de atuação: não-replicativo (processo de ―cortar-colar‖), replicativo
(processo que integra e resolve) e retrotransposição (via RNA intermediário).
Além disso, diferentes forças evolutivas, como seleção natural, deriva gênica e migração,
também podem interferir no desenvolvimento das comunidades de um ecossistema. Espécies
diferentes, resultantes de processos seletivos em condições ambientais distintas, apresentam
indivíduos com diferenças ainda mais evidentes. Essas diferenças se manifestam não só
através de variáveis estruturais, mas também bioquímicas, fisiológicas e etológicas,
implicando em capacidades de adaptação e habilidades diferentes entre indivíduos. Indivíduos

10
de espécies diferentes podem reunir-se em comunidades que variam conforme as interações
das espécies entre si, fatores históricos e diferentes condições abióticas (clima, solo, relevo).
Assim, a complexidade de uma comunidade está associada a sua diversidade microbiana.
1.2 Abordagens de estudo em diversidade microbiana
As duas abordagens principais no estudo de diversidade microbiana incluem as análises
dependentes e independentes de cultivo. Ambas permitem caracterizar a diversidade, e por
isso ainda são consideradas complementares. Estima-se que apenas 1% dos microrganismos é
detectado em placas com meio de cultura devido às condições seletivas, em função da
composição dos meios de cultura (Torsvik et al., 1990; Torsvik et al., 2002; Whitman et al.,
1998). Por outro lado, a abordagem independente de cultivo indica a predominância de muitas
espécies não cultivadas (Schloss and Handelsman, 2004).
1.2.1 Métodos dependentes de cultivo
As técnicas de cultivo tradicionais desenvolvidas por Pasteur, Koch, Beijerinck e
Winogradsky foram extensivamente utilizadas ao longo do século passado e se mostraram até
certo ponto bem sucedidas diante a falta de outras metodologias para o estudo da diversidade
microbiana. Os métodos tradicionais de análise da diversidade de bactérias envolvem a
caracterização de culturas puras isoladas em laboratório. O isolamento em meio de cultivo
exige que a composição do meio supra as necessidades metabólicas dos microrganismos. Essa
é uma das principais dificuldades para o estudo de diversidade microbiana, pois o repertório
metabólico dos procariontes é vasto. Em solos, por exemplo, já foi estimado que existam pelo
menos cerca de 4000 genomas procariontes por grama de solo (Torsvik et al., 1990). Porém, o
número de espécies que são isoladas de um grama de solo é estimado em 100. Por isso é
necessário adequar as condições de cultivo para conseguir recuperar organismos inicialmente
não cultivados e que são abundantes no ambiente.

11
Durante várias décadas foram concentrados esforços no isolamento de microrganismos
patogênicos, ou relacionados à determinada patogenia, bem como aqueles com capacidade de
sintetizar antimicrobianos, em laboratório. Isso permitiu um enorme avanço para a
microbiologia médica. Por isso, alguns grupos como actinomicetos, enterobactérias e bacilos-
grampositivos apresentam um grande número de espécies identificadas com representantes
cultivados (Schloss and Handelsman, 2004). Por outro lado, a microbiologia ambiental
precisa desenvolver estratégias de aprimoramento dos meios de cultura já estabelecidos, para
uma melhor compreensão da biologia dos microrganismos isolados de amostras ambientais
(Hugenholtz et al., 1998).
O uso de meios com baixa concentração de nutrientes e aumento dos tempos de incubação
permite a obtenção de representantes da cultura pura de várias subdivisões do dos filos
Acidobacteria e Verrucomicrobia (Janssen, 2006; Sait et al., 2002). É importantte ressaltar
que membros destes dois grupos são de difícil obtenção em cultivo mesmo com meios feitos
especificamente para eles. Alteração de condições de cultivo como concentração de oxigênio,
nível de nutrientes, adição de moléculas húmicas e moléculas de sinalização aumentam a
capacidade de cultivo de microrganismos no solo, incluindo esses dois grupos (Stevenson et
al., 2004). A adição de compostos de sinalização também foi relatada para ajudar no cultivo
de organismos aquáticos (Bruns et al., 2002; Bruns et al., 2003).
A utilização de extratos de solo solidificados com ágar é uma alternativa para melhorar a
recuperação de isolados ambientais (Sait et al., 2002; Wirth and Ulrich, 2002). Esta
abordagem tem como objetivo criar condições nutricionais que sejam as mais semelhantes
possíveis àquelas sob as quais os microrganismos se encontram no meio ambiente. O
isolamento e a manutenção em laboratório de microrganismos que ocorrem no meio ambiente,
permite a construção de uma coleção microbiana. É uma metodologia altamente atrativa, pois
permite a determinação de características morfológicas, fisiológicas e bioquímicas da coleção

12
microbiana, além de possibilitar a utilização de microrganismos para fins biotecnológicos.
Estes aspectos permitem a pesquisa e a bioprospecção rápida e pontual de diversos processos
com potencial de aplicação biotecnológica a partir de culturas puras. Para isso os isolados
devem ser crescidos sobre substrato específico em um meio capaz de indicar a ocorrência do
processo metabólico de interesse. Por exemplo, o corante vermelho congo é utilizado para
demonstrar a ocorrência de degradação de celulose através de halos formados pela afinidade
do corante pelas ligações β-1,4 da molécula (Teather and Wood, 1982) (Figura 6). As
propriedades metabólicas de um isolado podem ser utilizadas para inferir o potencial papel
ecológico que esse organismo desempenha em um ecossistema.
Embora os métodos de cultivo representem uma poderosa ferramenta para o estudo da
diversidade (taxonômica e funcional), ele subestima a diversidade taxonômica e metabólica
em função das limitações apresentadas anteriormente. Técnicas moleculares para a análise da
diversidade de comunidades microbianas superam este problema.
Figura 6. Halo de degradação produzido por bactérias celulolíticas, revelado por vermelho congo.

13
1.2.2 Métodos independentes de cultivo
Há pelo menos vinte anos foi verificada a existência de uma vasta diversidade até então não
acessada pelos métodos dependentes de cultivo (Torsvik et al., 1990; Torsvik et al., 2002). A
diversidade microbiana identificada pelos meios de cultivo representa apenas a ponta de um
iceberg de um vasto repertório genético. As análises independentes de cultivo permitem
acesso ao genoma de toda a comunidade microbiana de uma amostra (Handelsman, 2004). As
técnicas como T-RFLP, ARDRA e DGGE diferenciam as comunidades através do padrão de
bandeamento do gene ribossomal sob diferentes tratamentos. Entre as técnicas moleculares, o
sequenciamentos têm se mostrado bastante eficientes na para a identificação e caracterização
de táxons presentes nas comunidades.
Atualmente, as técnicas de sequenciamento são amplamente aplicadas ao estudo da
diversidade microbiana. O desenvolvimento e aprimoramento dos sequenciadores de primeira
geração (baseado no método de Sanger), e mais recentemente, os de segunda
(pirosequenciamento) e terceira (íonsequenciamento) geração permitem o sequenciamento
massivo de amostras ambientais. Os de primeira geração, através do sequenciamento de
bibliotecas de clones. Já os de segunda e terceira geração evitam a distorção inerente à
construção das bibliotecas. Essas metodologias permitem o estudo das populações
microbianas a partir de amostras ambientais.
1.2.2.1 Metagenômica
O termo metagenômica se refere a uma abordagem independente de cultivo baseada na
investigação das moléculas de DNA de uma mistura de populações microbianas, ou seja, é
baseado na análise genômica de DNA microbiano extraído diretamente de amostras
ambientais (Handelsman et al., 1998) (Figura 10). Em grego, a palavra meta significa
"transcendente". Isso significa que essa abordagem vai além das análises genômicas que, de
maneira geral, são aplicadas em microrganismos cultivados. O estudo da diversidade de

14
comunidades microbianas sofreu uma revolução com o estabelecimento da metagenômica.
Com a aplicação dessa abordagem os pesquisadores passaram a ter acesso ao genoma de uma
maior variedade de microrganismos que não haviam sido isolados em meio de cultura em
laboratório. A metagenômica representa um conjunto de técnicas, que inclui muitas
abordagens e métodos relacionados à genômica como o sequenciamento. Sendo assim, a
abordagem metagenômica se caracteriza por contornar a necessidade de cultivo e, em função
da vasta diversidade microbiana, ser conduzida em grande escala (Handelsman, 2005).
Comunidade genômica, genômica ambiental, e genômica populacional são sinônimos para a
mesma abordagem (Handelsman, 2004). As informações obtidas a partir da análise de
metagenomas podem ser usadas para determinar a diversidade de uma comunidade, a
presença de microrganismos específicos ou dominantes, rotas metabólicas, bem como
determinar a simples presença de um gene. A abordagem metagenômica permite a
visualização dos ecossistemas como unidades biológicas com os seus próprios repertórios
genéticos. Desta maneira esta abordagem possibilita a avaliar o que está sendo feito pela
comunidade microbiana. A metagenômica vem sendo aplicada para responder questões
fundamentais da microbiologia ambiental como: quantos são? Quem são? E fazendo o quê?
Por isso, a metagenômica representa uma importante ferramenta para estudo da ecologia
microbiana e prospecção do potencial metabólico através da análise da diversidade
taxonômica e localização de genes e operons responsáveis pela síntese de moléculas com
propriedades de interesse biotecnológico (Handelsman, 2004; Steele and Streit, 2005).
A abordagem metagenômica é conduzida de forma que o DNA metagenômico seja analisado
através da investigação funcional e/ou baseada em sequenciamento (Figura 7). Para isso, o
desenvolvimento da bioinformática tem sido determinante, já que permite o processamento e

15
análise da grande quantidade de dados gerada pelas técnicas de sequenciamento de segunda
geração (pirosequenciamento).
1.2.2.1.1 Análise baseada em função
A metagenômica oferece a possibilidade de localizar e caracterizar uma ampla variedade de
enzimas a partir de amostras ambientais. A detecção dessas moléculas requer uma triagem
funcional que necessita da expressão heteróloga dos genes seguida do sequenciamento dos
fragmentos isolados. Linhagens de E. coli são amplamente utilizadas neste tipo de abordagem.
Figura 7. Modelo esquemático de um estudo com abordagem metagenômica.(adaptado de Handelsman, 2004)

16
Isso ocorre em função do profundo conhecimento já existente sobre a fisiologia e o genoma
desta espécie. A revisão de Daniel (2004) (Daniel, 2004) apresenta um conjunto de trabalhos
que obtiveram sucesso na busca de moléculas bioativas (ex. agarase, lípase, amilase, protease
e outras) através triagem funcional em bibliotecas metagenômicas de amostras de solo.
A frequência de clones que são capazes de expressar uma atividade, de maneira geral é baixa.
Ainda existe dificuldade para melhorar a eficiência na identificação de clones raros em
bibliotecas muito grandes. Em alguns casos foi necessário analisar uma biblioteca de 4 Gb de
DNA ou aproximadamente 1 milhão de genes e assim, sua baixa representatividade torna a
descoberta de novas atividades extremamente laboriosa sem uma triagem prévia. A utilização
de meios indicadores da atividade, onde os clones ativos possam ser facilmente identificados
é uma estratégia bastante utilizada. A descoberta, expressão e caracterização de um novo
antimicrobiano, a partir de DNA metagenômico de solo, representam o primeiro exemplo do
uso bem sucedido da metagenômica para a descoberta de novas moléculas e atividades
(Gillespie et al., 2002). O antibiótico turbomicina foi isolado através da expressão heteróloga
do DNA extraído diretamente do solo por clones de uma biblioteca metagenômica de
microrganismos do solo. Foi necessária a triagem de uma biblioteca metagenômica de 25.000
clones, representando cerca de 1 Gb de DNA, para obter 3clones apresentando níveis
detectáveis da atividade antimicrobiana. Este trabalho representou uma mudança no
paradigma na busca de novas moléculas bioativas.
A utilização de sistemas de clonagem permite a separação e amplificação individual de
fragmentos de DNA clonados, a partir de uma mistura de genes frequentemente
desconhecidos e heterogêneos, oriunda de microrganismos do solo. Existe uma grande
variedade de sistemas de clonagem disponíveis para acomodar diferentes tipos e tamanhos de
fragmentos de DNA em linhagens hospedeiras.

17
Plasmídeos são utilizados para inserção de fragmentos pequenos (< 10kb), assim como os
bacteriófagos (7-10kb), tendo grande aplicação na construção de bibliotecas de fragmentos de
genes ou genes específicos (Henne et al., 1999; Knietsch et al., 2003). Entretanto, estes
vetores não permitem a detecção de clusters genéticos nem operons. Com esse objetivo, a
utilização de cosmídeos (25-35kb) e fosmídeos (até 40 kb) tem sido relatada (Handelsman,
2004). Para bibliotecas de grandes insertos, cromossomas artificiais bacterianos (BAC –
Bacterial Artificial Choromosome) e de leveduras (YAC – Yeast Artificial Chromosome), são
capazes de permitir a inserção de fragmentos de 80-200kb e 200kb-1.5Mb, respectivamente
(Xu, 2006). Esses vetores podem ser utilizados na construção de sistemas de expressão de
enzimas. A utilização de promotores gênicos permite o controle da expressão de um gene em
condições controladas. O desenvolvimento de novas tecnologias que promovam a expressão e
identificação eficiente de clones irá representar um avanço na fronteira da genômica
funcional.
1.2.2.1.2 Análise baseada em sequências
A aplicação de iniciadores universais para amplificação direta de sequências do rRNA 16S,
através de PCR (Polymerase Chain Reaction) do DNA total de uma comunidade microbiana e
combinada com tecnologias de clonagem e sequenciamento têm gerado uma vasta quantidade
de dados sobre a diversidade de procariotos. Woese e Fox (1977) (Woese and Fox, 1977)
foram os primeiros a propor a utilização do gene rRNA 16S para descrever relações
filogenéticas entre bactérias. Já o conceito de clonagem do DNA diretamente de uma amostra
ambiental foi inicialmente sugerido por Pace et al. (1985) (Pace et al., 1985) e primeiramente
implementado por Schmidt et al. (1991) (Schmidt et al., 1991). Ele utilizou fago λ para
construir uma biblioteca de rRNA 16S a partir de amostras de água e comparou a diversidade
picoplanctônica (organismos que medem entre 0.2 e 2 µm) do Oceano Pacífico e com
sequências de organismos cultivados do Oceano Atlântico. Foram identificadas sequências

18
com distante relação filogenética com os táxons de Gammaproteobacteria conhecidos. Essas
sequências foram consideradas representantes de novos organismos profundamente
divergentes filogeneticamente. Isso demonstrou o enorme potencial das abordagens
independentes de cultivo, revolucionando a ecologia microbiana.
O sequenciamento do gene rRNA 16S permite a identificação de células, mesmo em pequenas
quantidades. Com isso, obtemos importantes informações sobre a composição taxonômica da
comunidade microbiana de determinado solo. Desta maneira, a construção de bibliotecas
genômicas de rRNA 16S representa uma poderosa ferramenta para a análise da diversidade
molecular de comunidades de microrganismos representativos do solo. O conjunto de clones
bem como a análise de suas frequências dentro de uma biblioteca pode representar de maneira
geral, uma correlação com as unidades taxonômicas predominantes (Janssen, 2006; Nogales
et al., 2001). Schloss (2004) (Schloss and Handelsman, 2004) avaliou a diversidade
taxonômica de procariotos através de 56.215 sequências curadas do gene rRNA 16S
depositadas no banco de dados de genes ribossômicos RDP (Ribosomal Database Project).
Foram identificados mais de 50 filos, sendo metade deles compostos inteiramente por
membros não cultivados. Levantamentos da diversidade de ecossistemas terrestres e marinhos
revelam sequências dominantes pertencentes a grupos taxonômicos como filo Acidobacteria e
o clado SAR11 (Janssen, 2006; Venter et al., 2004a), que apresentam poucas espécies
cultivadas já isoladas em comparação com o número de espécies identificadas pelos métodos
independentes de cultivo.
Além disso, a abordagem metagenômica tem permitido a identificação de táxons em baixa
frequência, em ambientes terrestres e aquáticos, que não apresentam relação filogenética com
nenhum filo descrito (Elshahed et al., 2008; Schloss and Handelsman, 2006; Sogin et al.,
2006). O sequenciamento parcial do gene marcador filogenético rRNA 16S permite a
identificação de membros de subgrupos do filo Acidobacteria sem representantes cultivados.

19
A abordagem metagenômica permite correlacionar os membros das comunidades microbianas
às sequências de genes funcionais obtidos por diferentes técnicas de sequenciamento. O
sequenciamento do genoma revelou que este grupo apresenta genes envolvidos com o ciclo do
carbono, como por exemplo, celulases (Ward et al., 2009) O padrão de distribuição dos genes
funcionais e marcadores filogenéticos podem ser comparados entre metagenomas de
diferentes ambientes. A metagenômica comparativa possibilita a determinação de habitats
preferidos pelos táxons microbianos de diferentes ecossistemas. A comparação em diferentes
escalas (globais, regionais e locais) ajuda a definir as especificidades dos ecossistemas.
A análise quantitativa do conteúdo gênico revela as assinaturas de habitats que refletem
características específicas de uma amostra ambiental. A composição taxonômica das
comunidades ambientais é um indicador importante da sua ecologia e função. Isto nos permite
comparar a complexidade das comunidades e distingui-las em função do repertório
metabólico representado pelas sequências dos metagenomas (Tringe et al., 2005; von Mering
et al., 2007). Desta forma, podemos correlacionar as características ambientais ao longo de
biomas terrestres e aquáticos com os padrões de distribuição dos táxons microbianos (DeLong
et al., 2006; Dinsdale et al., 2008a; Raes et al., 2011). O Projeto Coleta Global dos Oceanos
(Global Ocean Sampling Project- GOS) coletou um impressionante volume de dados através
do sequenciamento do da comunidade microbiana do Mar Sargasso (Venter et al., 2004a). Foi
obtido um total de 1.045 bilhões de pares de bases não redundantes. Atualmente, os dados da
GOS representam um dos maiores depositórios de dados biológicos, relacionados com
metadados para uma única região (Raes et al., 2011).
1.2.2.1.3 Desafio computacional
Estudos em metagenômica são ricos em dados, tanto na quantidade quanto em complexidade.
Atualmente, após duas décadas de experiência no manuseio e análise de dados da sequências
de DNA baseados no estudo de genomas completos de organismos cultivados, os biólogos

20
estão tendo que lidar com a grande quantidade de dados gerados pela abordagem
metagenômica. Análises computacionais que permitam eficiente processamento e
interpretação desses dados representam uma questão ainda a ser superada pela abordagem
metagenômica. Novas tecnologias de sequenciamento, com o pirosequenciamento, estão
aumentando substancialmente a transferência de dados e representam novos desafios
específicos de plataformas de armazenamento de dados.
Armazenar as sequências que serão geradas pelos projetos que utilizam abordagem
metagenômica será um desafio para os bancos de dados (Ex. GenBank, EMBL-Bank, DDBJ e
outros). Mas além do desafio de arquivamento, fica claro que a comunidade vai exigir novos
bancos de dados secundários para que estes dados possam se utilizados de forma eficaz. A
análise de sequências metagenômicas exige programas computacionais robustos, eficientes e
especializados, como, por exemplo, programas para o agrupamento de sequências ou para
análise de séries temporais. Dados metagenômicos devem ser integrados com dados de
diferentes projetos, tais como dados de satélite sobre as temperaturas oceânicas e dados de
séries temporais sobre as mudanças na salinidade dos oceanos (Sun et al., 2011).
Bancos de dados como o Mg-Rast (Meyer et al., 2008) representam uma forma de armazenar,
disponibilizar e interagir com o grande volume de dados gerados pelos projetos. Os principais
consórcios, envolvidos na caracterização de metagenomas em andamento estão listados na
Tabela 1. O avanço das tecnologias de sequenciamento permite o sequenciamento massivo do
DNA metagenômico, possibilitando a identificação de genes ou famílias de genes
relacionadas a proteínas já conhecidas como as depositadas em bancos de dados. Os
principais projetos de metagenomas têm gerado um significativo aumento no volume de
dados submetidos nos bancos de dados, tornando um desafio para o futuro a capacidade de
armazenamento, processamento e análises dos dados gerados. Esta demanda se trona ainda
mais necessária em função dos avanços das técnicas de sequenciamento, representados pelos

21
sequenciadores de segunda geração. O método de pirosequenciamento tem proporcionado um
aumento significativo na capacidade de geração de sequências nos últimos anos.
Tabela 1. Principais consórcios que desenvolvem análises a partir de metagenomas baseada em sequenciamento
Consórcios Endereço virtual
Genomic Encyclopaedia of Bacteria and Archaea project http://www.jgi.doe.gov/programs/GEBA
Microbial Earth Project http://genome.jgi-psf.org/programs/bacteria-archaea/MEP/index.jsf
Human Microbiome Project http://nihroadmap.nih.gov/hmp
Metagenomics of the Human IntestinalTract consortium http://www.metahit.eu
Terragenome Initiative http://www.terragenome.org
Tara Oceans Expedition http://oceans.taraexpeditions.org
National Ecological Observatory Network -NEON http://www.neoninc.org
International Census of Marine Microbes (ICOMM) http://icomm.mbl.edu
Microbial Inventory Research Across Diverse Aquatic Long-Term
Ecological Research Sites http://amarallab.mbl.edu/mirada/mirada.html
Earth Microbiome Project http://www.earthmicrobiome.org
1.2.2.1.4 Pirosequenciamento
O uso do sequenciamento, através do método de Sanger, e suas variações entre os anos 90 e
2000 representou um avanço significativo no estudo dos sistemas biológicos. A consolidação
do sucesso da aplicação do método veio com o anúncio do sequenciamento do genoma
humano (Lander et al., 2001; Venter et al., 2001). Entretanto, limitações como a clonagem
física de fragmentos e longos tempos de execução incentivaram o desenvolvimento de
técnicas de sequenciamento mais velozes, sensíveis e acuradas. Atualmente, a técnica de
pirosequenciamento vem sendo amplamente aplicada aos estudos da diversidade microbiana
(Dinsdale et al., 2008a; Edwards et al., 2006; Huber et al., 2007; Raes et al., 2011; Rodriguez-
Brito et al., 2010; Sogin et al., 2006; Tringe et al., 2005).
O pirosequenciamento é um método de sequenciamento de DNA, baseado no princípio do
sequenciamento por síntese. A detecção da incorporação das bases difere do sequenciamento

22
de Sanger, pois é baseada na detecção da liberação do pirofosfato, em vez de terminação da
cadeia com dideoxinucleotídeos (Nyrén, 1987). A abordagem desenvolvida para
monitoramento do sequenciamento em tempo real sem a necessidade de eletroforese foi
inicialmente proposto por Ronaghi (1996 e 1998) (Ronaghi et al., 1996; Ronaghi et al., 1998).
O sistema de pirosequenciamento é constituído por um sistema enzimático que consiste de
DNA polimerase, ATP sulfurilase, luciferase e apirase. Estas enzimas, de forma geral,
executam as seguintes funções: a DNA polimerase (EC 2.7.7.7) catalisa a polimerização de
DNA na replicação, reparação e é, portanto, crucial para sobrevivência de todas as células
vivas. A produção de PPi liberado mediante ATP durante a polimerização é catalisada pela
ATP sulfurilase (EC 2.7.7.4). A luciferase (EC 1.13.12.7) catalisa a produção de luz a partir
de ATP que é detectada durante o pirosequenciamento. Já a apirase (EC 3.6.1.5) é incluída no
pirosequenciamento para a degradação dos nucleotídeos não incorporados e ATP em excesso,
entre as adições das bases. O método é baseado na detecção do pirofosfato (PPi) liberado
durante a síntese do DNA. Em uma cascata de reações enzimáticas, a luz visível gerada é
proporcional ao número de nucleotídeos incorporados. O pirofosfato é convertido em ATP. O
ATP fornece a energia necessária para a enzima luficerase catalisar a reação de oxidação da
luciferina, gerando luz. O perfil de emissão da luz resultante da incorporação das bases pode
ser observado através de um pirograma (Figura 8).
O pirosequenciamento massivo em paralelo aliado ao aumento da capacidade computacional e
bioinformática permitiu o desenvolvimento substancial das ciências genômicas. Na metade
dos anos 2000, o custo do sequenciamento foi reduzido por aproximadamente duas ordens de
grandeza e reduziu os esforços dos projetos genomas (Rogers and Venter, 2005).

23
Genomas e metagenomas podem ser sequenciados a partir de bibliotecas geradas pela
fragmentação aleatória do DNA (método de sequenciamento shotgun). A ligação de
adaptadores específicos aos fragmentos aliada a uma quantificação precisa do DNA,
permitem a ligação de peças únicas da molécula a esferas de 28 nm (Figura 9 a). Estas
bibliotecas eliminam a necessidade de sistemas de clonagem utilizados no método de Sanger.
Os fragmentos ligados às esferas são amplificados através de PCR em emulsão (em-PCR),
permitindo a amplificação de centenas de milhares de diferentes fragmentos de DNA em uma
única reação. As esferas são compartimentalizadas em uma emulsão de óleo (Figura 9 b),
criando um ambiente individualizado contendo todos os reagentes necessários para a
amplificação da peça de DNA, gerando milhões de cópias do fragmento ligado a esfera.
Figura 8. Representação esqumática do sistema enzimático do pirosequenciamento. O nucleotídeo
incorporado pela polimerase, liberando uma molécula de pirofosfato (PPi). A enzima ATP sulfurilase
converte o PPi em ATP, que atua como substrato para a produção de luz pela luciferase. A luz produzida
é detectada, formando um pirograma que indica a sequência de nucleotídeos incorporados durante o
sequenciamento (retirado de Ahmadian, 2006).

24
Posteriormente, as emulsões são quebradas e os fragmentos de DNA fita dupla são
desnaturados e as esferas contendo apenas fitas simples são depositados em um slide de fibra
ótica contendo aproximadamente 1,6 milhões de poços, onde ocorrem as reações de
sequenciamento em paralelo (Figura 9 c) (Margulies et al., 2005).
Em janeiro de 2009 o sequenciador 454 Genome sequencer 20 (454 GS20), que apresentava
uma acurácia de 99,5% para sequências não montadas (Huse et al., 2007), foi substituído por
uma nova versão, o equipamento 454 Genome sequencer FLX System (454 GS FLX - Roche,
Life Sciences). O novo sequenciador é capaz de produzir sequências mais longas que o
anterior, alcançando 300 pb, excedendo 1 milhão de pb por dia (> 5GB em 5 dias de trabalho)
Figura 9. Preparação da amostra para pirosequenciamento. a) Fragmentos de DNA com adaptadores e acoplados
as esferas submetidas a em-PCR em emulsões de óleo. Após a quebra das emulsões são depositadas em um slide
onde cada esfera contendo os produtos da amplificação são separadas individualmente. b) Microscopia da
emulsão mostrando gotas contendo as esferas. A seta larga representa uma gota de 100 μm e a fina mostra uma
esfera de 28 μm. c) Microscopia eletrônica de um slide (retirada de Margulies, 2005).

25
(Droege and Hill, 2008). Este melhoramento permitiu maior acurácia da tecnologia 454 de
sequenciamento. Além do desenvolvimento de sequenciadores mais robustos, a tecnologia
454 de sequenciamento também foi otimizada através da alteração da química utilizada nas
reações de sequenciamento. Atualmente, a química 454 GS FLX Titanium fornece cerca de
mais que 1 milhão de sequências em uma única corrida de 10 horas. Estas sequências, com
uma média de leitura de comprimento próxima dos 500 pb para bibliotecas shotgun, geram
muito mais dados do que outras abordagens de sequenciamento disponíveis (Gilles et al.,
2011). Os dados de sequências biológicas são então analisados por meio de fórmulas que
medem a diversidade das comunidades microbianas.
1.2.3 Medidas de diversidade
A diversidade microbiana pode ser avaliada a partir do número de espécies encontradas em
uma amostra. Com isso, podem-se construir curvas de rarefação, que permitem avaliar a o
tamanho da riqueza de espécies amostradas. Amostras de tamanhos diferentes podem ser
comparadas através do uso de índices de diversidade e o tamanho total pode ser estimado a
partir das relações de contribuição das espécies, sem a necessidade de acessar todos os
indivíduos de uma amostra.
1.2.3.1.1 Riqueza de espécies
Obtendo-se várias curvas a partir da adição aleatória das amostras pode-se calcular uma curva
coletora média. As curvas de acumulação de espécies (curvas coletora) permitem avaliar o
quanto um estudo se aproxima de capturar todas as espécies do local. Quando a curva
estabiliza, ou seja, nenhuma espécie nova é adicionada, significa que a riqueza total foi obtida
(Figura 10). A partir disso, novas amostragens não são necessárias.
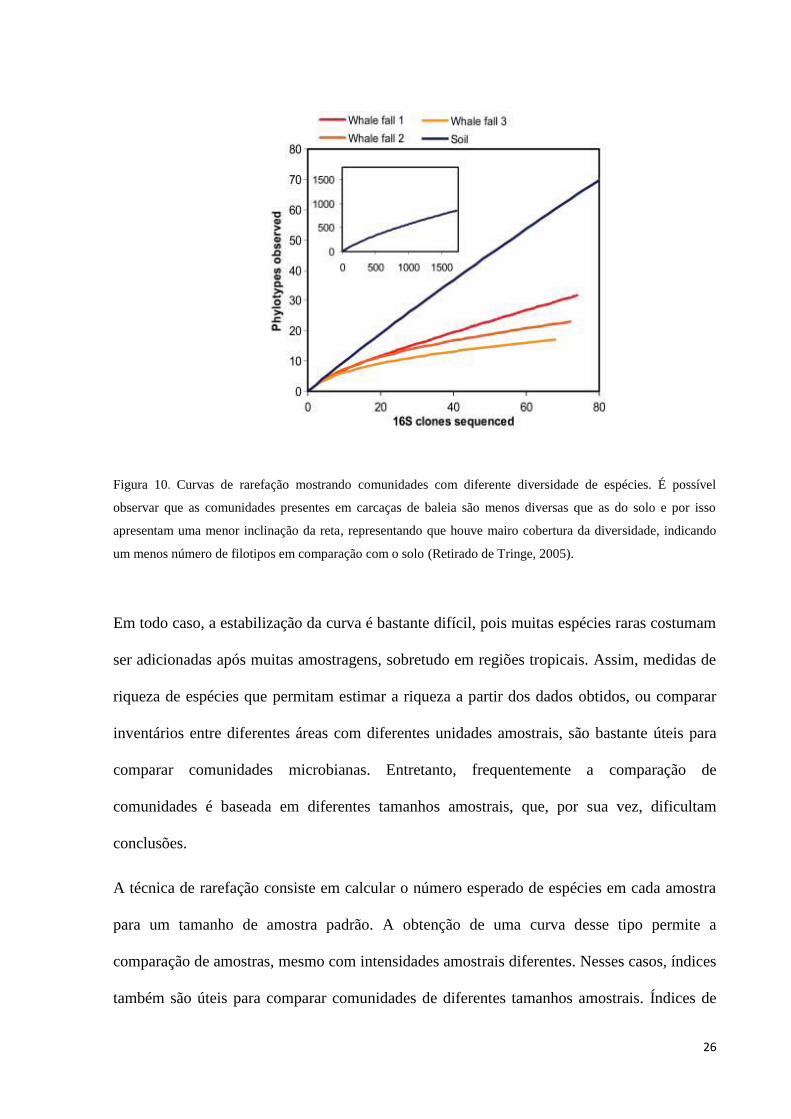
26
Figura 10. Curvas de rarefação mostrando comunidades com diferente diversidade de espécies. É possível
observar que as comunidades presentes em carcaças de baleia são menos diversas que as do solo e por isso
apresentam uma menor inclinação da reta, representando que houve mairo cobertura da diversidade, indicando
um menos número de filotipos em comparação com o solo (Retirado de Tringe, 2005).
Em todo caso, a estabilização da curva é bastante difícil, pois muitas espécies raras costumam
ser adicionadas após muitas amostragens, sobretudo em regiões tropicais. Assim, medidas de
riqueza de espécies que permitam estimar a riqueza a partir dos dados obtidos, ou comparar
inventários entre diferentes áreas com diferentes unidades amostrais, são bastante úteis para
comparar comunidades microbianas. Entretanto, frequentemente a comparação de
comunidades é baseada em diferentes tamanhos amostrais, que, por sua vez, dificultam
conclusões.
A técnica de rarefação consiste em calcular o número esperado de espécies em cada amostra
para um tamanho de amostra padrão. A obtenção de uma curva desse tipo permite a
comparação de amostras, mesmo com intensidades amostrais diferentes. Nesses casos, índices
também são úteis para comparar comunidades de diferentes tamanhos amostrais. Índices de

27
diversidade são expressos por um único número, que pode representar a redução ou a
abundância de um conjunto complexo de táxons. Os índices permitem dimensionar a riqueza,
a igualdade e a diversidade nos diferentes ambientes estudados (Kennedy and Smith, 1995).
1.2.3.1.2 Índices de diversidade
O índice de Shannon-Wiener (H), já tradicionalmente designado como índice de Shannon,
expressa a importância relativa de cada espécie e não apenas a proporção entre espécies e
indivíduos (Shannon, 1948). Este é um índice que atribui um maior peso a espécies raras,
prevalecendo, desta forma, o componente de riqueza de espécies. O índice de Shannon
assume, também, que os indivíduos são amostrados ao acaso de uma população
indefinidamente grande e que todas as espécies estão representadas na amostra coletada,
sendo relativamente independente do tamanho da amostra. Segundo Hutcheson (1970)
(Hutcheson, 1970) os dados obtidos se enquadram em uma distribuição normal, desde que N
seja um número inteiro, de modo que os métodos estatísticos podem ser empregados para
testar a significância da variância.
Já o índice de Simpson (1949) (Simpson, 1949) foi o primeiro a ser usado em estudos
ecológicos e mostra a concentração de dominância, uma vez que, quanto maior o valor, maior
a dominância por uma ou poucas espécies. Este índice exprime, basicamente, a abundância
das espécies mais comuns, sendo, consequentemente, mais sensível a mudanças que ocorrem
nestas espécies (Magurran, 1988). Considerando-se as duas medidas heterogêneas, Shannon e
Simpson, o último é que atribui um peso maior às espécies comuns.
1.2.3.1.3 Estimadores de diversidade
Os estimadores de riqueza fornecem a quantidade de espécies que se pode encontrar em uma
área, sem levar em conta a quantidade de indivíduos por espécie (abundância). Alguns já são
aplicados há três décadas, mas atualmente têm sido mais amplamente aplicados devido ao

28
desenvolvimento de ferramentas computacionais que facilitam seu cálculo. Esses estimadores
são aplicáveis aos dados com diferentes distribuições de abundância, levando-se em conta os
dados relativos às espécies raras (ou aquelas que só aparecem em uma ou em poucas
amostras). O cálculo da riqueza de espécies é mais acurado em comunidades com alta
equitabilidade (as espécies são compostas pelo número semelhante de indivíduos). A
aplicação dos estimadores, em teoria, independe do tamanho da amostra, embora quanto
maior for a quantidade de organismos coletados, maior sua acurácia. O sequenciamento
massivo de DNA tem revelado que a maioria das comunidades microbianas apresenta alto
número de espécies composta por poucos indivíduos, também denominada biosfera rara
(Elshahed et al., 2008; Sogin et al., 2006).
Os estimadores que se baseiam na riqueza das espécies raras compartilhadas entre grupos de
amostras utilizam-se de quatro variáveis: singletons (espécies com somente um indivíduo),
doubletons (espécies com somente dois indivíduos), uniques (espécies que ocorrem em
somente uma amostra) e duplicates (espécies que ocorrem em somente duas amostras). As
estimativas realizadas com espécies representadas por poucos indivíduos são, segundo
(Hellmann and Fowler, 1999), uma função do número de espécies raras encontradas em uma
comunidade.
Os estimadores de riqueza podem calcular: o número de espécies através de curvas coletoras,
de modo que seja possível estimar o esforço empregado para inventariar uma área; número
real de riqueza de espécies baseada em espécies raras compartilhadas entre grupos de
amostras baseadas em incidência e a riqueza de espécies raras compartilhadas entre grupos de
amostras. Amostras amientais costumam apresentar um grande número de espécies. Por
exemplo, as estimativas baseadas em análises não paramétricas apontam um número de
espécie entre 6.400 a 38.000 espécies por grama de solo (Curtis et al., 2002; Schloss and
Handelsman, 2006).

29
1.3 Os microrganismos no meio ambiente
O estudo dos padrões espaciais e processos que estão envolvidos na distribuição da
biodiversidade são conhecidos como a biogeografia (O'Malley, 2007). A maioria dos estudos
sobre padrões de distribuição microbiana tenta avaliar o princípio conhecido como Everything
is everywhere, proposto no fim do século XIX, pelo professor alemão Lourens Gerhard
Marinus Baas Becking (Baas Becking, 1834). Ele se baseou em observações feitas pelo
microbiologista Beijerinck (1813) (Beijerinck, 1913). O princípio diz que ―tudo está em todos
os lugares, mas o ambiente seleciona‖. Vários estudos enfatizam o impacto da composição do
solo e da água na distribuição espacial na diversidade microbiana e na estrutura das
comunidades (Clementino et al., 2008; DeLong et al., 2006; Fuhrman and Azam, 1980;
Galand et al., 2009; Pommier et al., 2007; Schauer et al., 2009). Padrões na distribuição
espacial de organismos fornecem evidências importantes sobre os mecanismos que
influenciam a estrutura das comunidades ecológicas.
Este princípio vem sendo observado e aplicado em alguns trabalhos durante os últimos anos
em comunidades microbianas (Green and Bohannan, 2006; Green et al., 2004; Horner-Devine
et al., 2004; Martiny et al., 2006; Zhou et al., 2008). A relação taxa-área observada em
comunidades microbianas é regida pela Lei de Potência (Power-law). Esta relação é uma das
regras mais difundidas da ecologia: áreas maiores contêm mais espécies do que as pequenas.
A diversidade microbiana pode variar entre locais separados por milímetros ou milhares de
quilômetros, embora o limite de dispersão seja mais aparente em escalas continentais do que
em menores escalas. O pequeno tamanho dos microrganismos pode facilitar sua dispersão.A
capacidade de dispersão por longas distâncias é uma característica fundamental para
distribuição de organismos cosmopolitas. Estudos revelam que a maioria dos táxons
cosmopolitas são também os mais abundantes nas comunidades (Nemergut et al., 2011).

30
Um gradiente latitudinal de diversidade tem sido documentado para organismos (animais e
plantas) marinhos com aumento do número de espécies dos pólos em direção ao Equador. Os
mecanismos que estabelecem esse tipo de padrão estão relacionados com o aumento da
temperatura ao longo do gradiente. Maiores temperaturas e a produtividade primária
influenciam os padrões de distribuição das espécies (Rohde, 1992). Este padrão foi
confirmado recentemente no bacterioplâncton de águas superficiais dos oceanos (Pommier et
al., 2007). A análise dos padrões globais de diversidade de bactérias revela que a salinidade é
um dos principais fatores que influenciam a diversidade global das comunidades microbiana
(Lozupone and Knight, 2007). Fatores climáticos como incidência de raios solares e
temperatura, estão entre os principais fatores que influenciam na diversidade microbiana nos
oceanos (Raes et al., 2011). Entretanto, ainda é pequeno o entendimento sobre a influência de
fatores ambientais na seleção dos grupos funcionais e taxonômicos especialistas que
compõem as comunidades microbianas.
Os métodos independentes de cultivo têm colaborado significativamente para o melhor
entendimento da biogeografia de microrganismos. Sequências de genes podem ser usadas
para atribuir diferenças ou semelhanças entre as comunidades através da similaridade entre
sequências de genes. O gene ribossomal é amplamente utilizado para associar um conjunto de
características que determinam um estilo de vida e estratégias ecológicas gerais com a
identificação de táxons (Green et al., 2008).
Os microrganismos participam de importantes ciclos como do carbono, nitrogênio e enxofre.
Por isso, exercem papéis fundamentais na homeostase de ambientes terrestres e aquáticos,
permitindo o fluxo de energia e a reciclagem de nutrientes (Hackl et al., 2004; Wertz et al.,
2006). O fluxo de matéria e energia paralelo ao fluxo da cadeia trófica clássica é chamado de
alça microbiana. Através dele grande quantidade de matéria orgânica indisponível aos níveis

31
tróficos superiores, pode ser integrada nos ecossistemas aquático e terrestre pela produção
secundária da biomassa microbiana (Fuhrman and Azam, 1980).
A importância da microbiota na manutenção da saúde de ecossistemas aquático e o potencial
biotecnológico da microbiota terrestre são discutidos a seguir.
1.3.1 Microbiota aquática e a saúde ambiental
Microrganismos marinhos estão presentes em milhares de milhões de células por litro de
água do mar. Eles se multiplicam em poucos dias e são consumidos na mesma taxa por
parasitas e predadores protistas e virais. Essas atividades envolvem a captura e processamento
da energia que direcionam os principais ciclos elementares. A diversidade genômica e a
dinâmica evolutiva dos processos ecológicos das populações têm efeitos globais sobre as
taxas e fluxo de energia no mar.
De maneira global, o bacterioplâncton dos oceanos é formado em sua maioria por membros
dos filos Proteobacteria, Cyanobacteria, Bacteroidetes e Actinobacteria (Pommier et al.,
2007). Foram identificados grupos bacterianos como Prochlorococcus; Verrucomicrobiales;
Flexibacteraceae; Gammaproteobacteria (SAR92, OM60, SAR86); Alphaproteobacteria e
Deltaproteobacteria em amostras do Oceano Pacífico. Por outro lado, grupos de bactérias
como Deferribacteres; Planctomycetaceae; Acidobacteriales; Gemmatamonadaceae;
Nitrospina; Alteromonadaeceae SAR202 e SAR11 são encontrados em águas profundas
(DeLong et al., 2006). Entretanto, os fatores que determinam o estabelecimento e manutenção
desses grupos dominantes ainda precisam ser mais bem compreendidos.
A maior parte das Cyanobacteria marinhas está agrupada em dois gêneros estreitamente
relacionados de acordo com suas sequências do gene rRNA 16S: Prochlorococcus e
Synechococcus. Estima-se que essas Cyanobacteria são responsáveis por dois terços da
fotossíntese nos oceanos e elas são amplamente distribuídas nas latitudes equatoriais e

32
temperadas de todos os oceanos (Bouman et al., 2006). Prochlorococcus são os organismos
fotossintéticos mais abundantes nos oceanos do planeta e contribuem significativamente com
a produtividade primária de oceanos tropicais e subtropicais (Partensky et al., 1999). Para
alguns procariotos, as distribuições verticais de profundidade e variação nas propriedades
fisiológicas estão se tornando conhecidas. Diferenças genotípicas e fenotípicas em espécies de
Prochlorococcus foram observadas em populações expostas a diferentes níveis de exposição á
luz (Paul et al., 2010).
A recente descoberta de genes de bacteriorodopsinas em Proteobacteria do clado SAR11, não
cultivadas, revelou a base de sistemas fototróficos em ambientes marinhos (Béjà et al., 2000;
Béja et al., 2001). Bacteriorodopsinas permitem o acoplamento da energia da luz e a ciclagem
de carbono no oceano. As proteorodopsinas são fotoproteínas que são capazes de gerar
energia a partir da luz, realizando a translocação de prótons através da membrana celular.
Esse mecanismo não é baseado no pigmento clorofila como ocorre organismos
fotossintetizantes. A fototrofia baseada em proteorodopsinas é globalmente significante nos
processos microbianos nos oceanos (Béja et al., 2001; Venter et al., 2004b). A presença de
proteorodopsinas em bactérias heterotróficas de crescimento rápido, como as do gênero
Vibrio, está envolvida com o aumento da sobrevida de microrganismos em situação de baixa
disponibilidade de nutrientes (Gómez-Consarnau et al., 2010). Isto significa que as
proteorodopsinas representam uma vantagem ecológica para determinados grupos m
microbianos.
Durante os últimos 30 anos foi demonstrado que as bactérias são dominantes na superfície
terrestre e apresentam alta versatilidade metabólica. Uma grande fração da produção primária
torna-se matéria orgânica dissolvida por vários mecanismos na cadeia alimentar, e esta parte
da produção primária é quase exclusivamente acessível para bactérias heterotróficas e
arqueias (Azam et al., 1983; Ducklow and Carlson, 1992). Como resultado, a absorção de

33
matéria orgânica por bactérias é uma das principais vias de fluxo de carbono e sua
variabilidade pode mudar os padrões globais do fluxo do elemento carbono (Azam, 1998).
A concentração dos gases do efeito estufa tem afetado o equilíbrio em diferentes regiões do
mundo promovendo a acidificação dos oceanos (Caldeira and Wickett, 2003). O equilíbrio
desta molécula determina não só o pH, mas também a disponibilidade de CO2 para a
fotossíntese e carbonato de cálcio usado por organismos marinhos, incluindo corais. O CO2 da
atmosfera é dissolvido em contato com o oceano. O carbono inorgânico dissolvido (CID) é
incorporado à biomassa de algas através de fotossíntese. Na alga ele é convertido em carbono
orgânico particulado (COP) e carbono orgânico dissolvido (COD) (partículas menores que
0,45 µm).
O COD liberado pelas algas promove o crescimento de bactérias que promovem a morte do
coral (Smith et al., 2006). O aumento da concentração de bactérias na superfície do coral
provoca a diminuição da concentração de oxigênio, interferindo no processo de respiração
levando a morte. É observada uma mudança de fase da comunidade bacteriana associada aos
corais, com aumento da concentração de bactérias patogênicas (Mao-Jones et al., 2010). A
correlação entre a retirada de peixes com a morte de corais através do aumento da cobertura
de algas e proliferação de patógenos microbianos é chamada DDAM (do inglês, COD,
Disease, Algae and Microbes) (Rohwer, 2010) (Figura 11).

34
As doenças infecciosas têm sido identificadas como as principais causas da degradação e
extinção de corais em diversas partes do mundo (Bourne et al., 2009). Surtos consecutivos de
doenças no Caribe foram responsáveis por intensa destruição de espécies de corais, levando a
mudança drástica para um ecossistema dominado por algas (Patterson et al., 2002; Weil et al.,
2006). Até este momento, 18 doenças em corais foram descritas, porém somente algumas têm
a doença bem caracterizada (Figura 12). Em uma escala global, o branqueamento tem sido a
doença mais devastadora nas últimas duas décadas (Stone, 2007). Muitas destas doenças em
corais, incluindo o branqueamento, banda preta e banda amarela podem levar a morte.
Figura 11. O modelo DDAM é positivamente retroalimentado quando há aumento de COD, produzido por algas,
para suportar o crescimento de microrganismos associados aos corais. A proliferação de algas ocorre em função
da retirada de peixes, principalmente herbívoros, dos sistemas recifais. A proliferação de microrganismos
promove a morte de corais, aumentando a cobertura de algas.

35
Figura 12. Doenças que afetam corais brasileiros. A) Doença da banda-preta em Mussismilia braziliensis, B)
doença da faixa-vermelha em Siderastrea spp, C) doença dark spot like em Siderastrea spp e D) praga branca
em M. braziliensis (retirado de Francini-Filho, 2008)
1.3.2 Microbiota terrestre e seu potencial biotecnológico
A matéria orgânica no solo é representada pelos restos de animais e vegetais em todos seus
estágios de decomposição. A matéria orgânica decompõe-se até constituir o húmus, que é a
matéria orgânica na forma coloidal, com características benéficas e atribui ao solo uma
coloração escura. Particularmente no solo, os microrganismos desempenham um papel
fundamental na formação do húmus e na degradação da lignocelulose, que representa o
biopolímero mais abundante no nosso planeta. A lignocelulose serve como proteção para as
células de plantas verdes fortalecendo as diferentes estruturas que compõem a planta. A
lignocelulose consiste, de maneira geral, da lignina, uma macromolécula (> 10 KDa)
altamente aromática e hidrofóbica, e da celulose que é um carboidrato polimérico que
compreende cadeias longas de beta-glucose (Figura 13). A hidrólise de polímeros complexos
de celulose permite a disponibilização de glicose para o estabelecimento de outros organismos

36
incapazes de degradá-la. Dessa maneira, microrganismos celulolíticos dirigem o fluxo de
energia no solo.
A celulose é um polissacarídeo formado por unidades residuais de -D-glucose, que
interagem entre si através de ligações -1,4, mantendo uma estrutura linear e plana. A
celulose é muito rígida, podendo ser encontrados em sua estrutura cerca de 4000 a 8000
moléculas de glucose (Aristidou and Penttilä, 2000). A orientação das microfibras de celulose
na parede celular é influenciada por vários fatores que podem influenciar das enzimas
microbianas (celulases) ao substrato (celulose).
Figura 13. Estrutura da lignocelulose. O principal componente de lignocelulose é a celulose, ab (1-4) ligada cadeia de
moléculas de glicose. A lignina é sintetizado através da polimerização dos componentes e sua relação dentro do
polímero varia entre diferentes plantas, tecidos e camadas de madeira da parede celular. Celulose, hemicelulose e
lignina formam estruturas chamadas microfibrilas, que são organizados em macrofibrilas que medeiam a estabilidade
estrutural da parede celular vegetal (retirado de Rubin, 2008).

37
Em um ecossistema tipicamente degradador de celulose como o solo, uma variedade de
microrganismos trabalham em conjunto para converter substratos celulósicos insolúveis em
açúcares solúveis, principalmente celobiose e glicose, que são então assimiladas pelas células.
A decomposição da celulose pode ocorrer tanto em condições aeróbicas quanto anaeróbicas.
Bactérias anaeróbicas desenvolveram um sistema altamente sofisticado e complexo para a
degradação deste polímero chamado de celulossoma (Bayer et al., 2008). Celulossomas são
complexos multienzimáticos produzidos por várias bactérias celulolíticas. O primeiro
exemplo desta estrutura foi descoberto na bactéria anaeróbia termofílica, Clostridium
thermocellum (Bayer et al., 2008; Lamed et al., 1983). A degradação bacteriana aeróbica da
celulose pode ser realizada por bactérias filamentosas (ex: Streptomyces e Micromonospora) e
não filamentosas (ex: Bacillus, Cellulomonas e Ruminococcus). Entretanto, a habilidade de
degradar celulose aerobicamente é muito mais difundida entre fungos. O crescimento em
forma de hifa, apresentado por fungos e actinomicetos, representa uma estratégia que favorece
a degradação da celulose, através da penetração na estrutura cristalina da celulose e secreção
das enzimas necessárias para a degradação do composto. A despolimerização da celulose
representa a etapa que gera o maior rendimento energético para a fermentação. Entretanto,
este polímero encontra-se embebido numa matriz de lignina (10-25%), que apresenta uma
estrutura química bastante complexa, incluindo anéis fenólicos (Lee, 1997).
A exigência de um consórcio de enzimas reflete a dificuldade de acesso aos polissacarídeos
da parede celular das plantas. A degradação completa da celulose requer um complexo
formado principalmente por três classes enzimáticas, que devem atuar de maneira sinérgica.
Este complexo é composto por endoglucanases, que hidrolisam randomicamente a fibra de
celulose nas ligações β- 1,4 internas reduzindo o tamanho do polímero; exoglucanases que
hidrolisam a celulose a partir dos terminais não-redutores, liberando unidades de celobiose; e

38
β glicosidases que clivam os oligossacarídeos ou as celobioses livres, gerando glicose (Han et
al., 1995; Lynd et al., 2002).
A degradação das substâncias orgânicas e das substâncias húmicas é mediada pelos
microrganismos dos grupos Proteobacteria, Firmicutes, Actinobacteria, Verrucomicrobia e
Acidobacteria. Esses grupos taxonômicos são amplamente encontrados em solos e têm papel
importante na ciclagem de biodisponibilização de C e N nestes ambientes. Os filos
Acidobacteria, juntamente com Proteobacteria, são dominantes em solos de florestas (Faoro
et al., 2010a; Janssen, 2006). Os solos ácidos são dominados por poucos táxons,
principalmente Acidobacteria e Alphaproteobacteria.
A distribuição espacial de alguns táxons está relacionada com a heterogeneidade do solo
(Philippot et al., 2009). A diversidade e a riqueza das comunidades bacterianas no solo
mostraram ser influenciadas pelas variáveis edáficas do solo, principalmente o pH, dos
diferentes ecossistemas (Fierer and Jackson, 2006; Griffiths et al., 2011). A diversidade
bacteriana é menor em solos ácidos, como da Amazônia, que foi identificado como o mais
ácido e menos diversificado ao longo das Américas do Norte e do Sul. Os representantes
desses filos apresentam alta versatilidade metabólica e são capazes de degradar fontes
complexas de carbono com a celulose. As Streptomyces e Clostridium, também são
conhecidas produtoras de celulases (Kato et al., 2004; Semedo et al., 2004).
Enzimas produzidas por estes microrganismos são bastante exploradas economicamente em
processos biotecnológicos, em especial pela indústria de energia. Até o momento, vários
laboratórios estão empenhados na construção de microrganismos alterados geneticamente
para a obtenção de enzimas de alto rendimento para a conversão de biomassa em
biocombustível (Galbe and Zacchi, 2002; Lynd et al., 2002; Schubert, 2006). A biomassa
celulósica representa uma matéria-prima alternativa às matrizes energéticas atuais como
combustíveis fósseis, nuclear e hidroelétrica devido à sua disponibilidade em larga, menor

39
custo de produção e produz menor impacto ao meio ambiente (Lynd et al., 1999). Estima-se
que 1011
toneladas de biomassa vegetal, são hidrolisadas anualmente por enzimas microbianas
e a energia liberada equivale a 640 bilhões de barris de petróleo (Ragauskas et al., 2006).
Desta forma, a aplicação a produção de energia com base em biomassa celulósica colabora
com a diminuição do efeito estufa, reduzindo o custo ambiental em função da produção de
energia, pois representa uma fonte mais limpa do que as matrizes energéticas baseadas em
queima de comustíveis fósseis. O principal obstáculo impedindo a produção mais
generalizada de energia a partir de fontes de biomassa é a ausência geral de tecnologias de
baixo custo e de baixo impacto ambiental para superar a recalcitrância desses materiais.
Atualmente tratamentos físico-químicos são bastante utilizados no pré-tratamento da
biomassa, entretanto ainda são métodos caros e que produzem um significativo passivo
ambiental.
Microrganismos capazes de produzir enzimas com aplicação na degradação de resíduos
agrícolas, como a celulose presente no bagaço da cana-de-açúcar, por exemplo, podem reduzir
significativamente os custos associados à produção do combustível e, por isso, apresentam
grande interesse econômico. O emprego de sistemas biológicos, a partir da diversidade
genética das comunidades microbianas do solo da Mata Atlântica, capazes de degradar
compostos celulósicos permite a otimização da produção de energia a partir de biomassa. A
utilização eficiente da celulose permite o aumento da produção de etanol do país, diminuindo
a necessidade de expansão da área de plantio de monoculturas como a cana-de-açúcar. Isso
representa a diminuição da pressão das fronterias agrícolas sobre os biomas brasileiros e seus
recursos naturais.

40
1.4 Biodiversidade
O termo biodiversidade refere-se à variedade de vida no planeta Terra, incluindo a variedade
genética dentro das populações e espécies e suas funções ecológicas desempenhadas pelos
organismos nos ecossistemas (Wilson, 1985; Wilson et al., 2007). O conceito de
biodiversidade envolve dois parâmetros: riqueza e equitabilidade. Riqueza é a quantidade de
espécies e equitabilidade é a quantidade de indivíduos de determinada espécie que ocorre em
um local ou em uma amostra. A maioria das políticas públicas e não-governamentais de
projetos conservacionistas utiliza medidas de diversidade e de riqueza para apoiar os dados
que subsidiarão as medidas de preservação e manejo ambiental. Áreas reconhecidas como
hotspot de diversidade ou ―ecoregiões críticas‖ são usualmente identificadas usando dados
sobre a riqueza de espécies endêmicas (Brooks et al., 2006).
O conceito dos Hotspots, criado em 1999 por Mitermeier e reforçado em 2004 (Mittermeier
and Mittermeier, 2004), estabelece atualmente 25 áreas críticas para conservação em todo o
mundo (Myers et al., 2000) (Figura 14). Essa estratégia foi adotada pela Conservação
Internacional para estabelecer prioridades em seus programas de conservação.
O critério mais importante na determinação dos Hotspots é a existência e o grau de ameaça de
espécies endêmicas, isto é, que são restritas a um ecossistema específico e, portanto, sofrem
maior risco de extinção. Os Hotspots sustentam até metade da diversidade biológica do
planeta em apenas 1% da sua superfície da Terra (Myers et al., 2000). O estabelecimento de
áreas protegidas para conservação da biodiversidade in situ é uma estratégia de manejo,
podendo permitir a preservação de (micro) biota com grande potencial biotecnológico e
ambiental.
A estratégia dos hotspots é justificada pelo fato que os conservacionistas estão longe de
conseguir proteger todas as espécies ameaçadas do mundo. Essa abordagem prioriza as ações
nas áreas mais ricas, como as florestas tropicais da Mata Atlântica brasileira e as savanas do

41
Cerrado brasileiro, os ecossistemas do tipo mediterrâneo e outros, protegendo espécies em
extinção e mantendo o amplo espectro de vida no planeta. Consequentemente, a riqueza
observada nos hotspots pode capturar uma alta diversidade de espécies, mas nem sempre as
espécies raras ocorrem nessas regiões (Prendergast et al., 1993). Muitas áreas mantêm apenas
3 a 8% do que existia inicialmente, como a Mata Atlântica, que hoje guarda entre 7 a 8% de
sua extensão original.
Figura 14. Localização dos hotspots de riqueza de espécies ao redor do mundo considerados áreas prioritárias
para conservação (Meyers, 2000).
Existem diferentes hipóteses que explicam a importância da biodiversidade para a estabilidade
de um ecossistema. Entretanto, todas assumem que a diversidade é fundamental na sua
manutenção. A hipótese da diversidade-estabilidade postula que os ecossistemas são tanto
mais estáveis, quanto maior a diversidade no sistema (Stiling, 1996). A simplificação dos
ecossistemas, em termos de redução de espécies (redução da diversidade), induz uma redução
da estabilidade e aumenta a susceptibilidade a invasões biológicas. Este argumento sustenta
importantes políticas de conservação aplicadas à gestão do meio ambiente. Neste sentido,

42
índices de diversidade poderiam ser ferramentas importantes na delimitação de áreas
estratégicas para a proteção ambiental.
A hipótese da redundância sugere que as espécies se agrupam em unidades funcionalmente
equivalentes (Stiling, 1996). Segundo esta hipótese, o funcionamento dos ecossistemas não é
afetado pela remoção de elementos redundantes, mas sim pelo efeito sobre as espécies com
papel preponderante no funcionamento dos ecossistemas. Por exemplo, com o aumento de
determinados grupos de espécies que tem papel negativo para um determinado bioma, pode
levar a degradação deste bioma.
Nesta seção serão apresentadas as principais características dos biomas aquáticos e terrestres
que foram exploradas neste trabalho. O termo bioma foi utilizado de uma forma ampla para
definir um conjunto de ecossistemas. No caso, abordaremos os ecossistemas de manguezais,
lagoas costeiras, sistemas recifais e solos de floresta.
1.4.1 Biomas aquáticos: zona costeira
Metade do oxigênio existente no ar que respiramos é produzida nos oceanos. Além disso, os
oceanos absorvem grande parte do CO2 lançado na atmosfera. Essas duas funções são
primordiais para a manutenção do equilíbrio ambiental. Por isso, biomas aquáticos como a
Zona Costeira representa ecossistemas prioritários para conservação e preservação dos
recursos naturais.
A Zona Costeira é a região de interface entre o continente e o mar, sendo dominada por
processos oceanográficos e atmosféricos originados nas bacias de drenagem dos rios
afluentes. A elevada concentração de nutrientes e outros fatores ambientais como gradientes
térmicos, salinidade variável e condições para a reprodução e alimentação da maioria das
espécies que habitam os oceanos, fazem com que esse ecossistema desempenhe uma
importante função de ligação e de trocas genéticas entre os ecossistemas terrestres e marinhos.

43
Por ser uma região de transição, a zona costeira registra expressiva sobreposição territorial,
mantendo também interface com os biomas terrestres. Por isso, não se caracteriza como uma
unidade, mas sim como complexos de ecossistemas, onde os fenômenos regionais
determinam as características de cada ecossistema.
Essas regiões englobam menos de 20% da superfície do planeta, mas acomodam mais de 45%
da população humana, hospedando 75% das grandes cidades com mais de 10 milhões de
habitantes e produzindo cerca de 90% da pesca global (Lemay, 1998). Cerca de 60 % dos 475
milhões de habitantes da America Latina vivem em estados costeiros, assim como a maioria
das cidades economicamente mais importantes. A região costeira é uma importante zona de
produção de alimentos e é foco de desenvolvimento industrial. As zonas costeiras são áreas
submetidas à forte pressão ambiental, pois representam áreas intensamente povoadas e com
altos níveis de industrialização. Os efeitos do desenvolvimento desordenado contribuem para
a desestabilização dos ecossistemas tornando a demanda por recursos não sustentável.
A Zona Costeira brasileira ocupa aproximadamente, três milhões de km2, sob jurisdição
brasileira. O país possui uma das maiores faixas costeiras do mundo, com mais de 7.400 km
entre a foz dos rios Oiapoque (04°52’45‖N) e Chuí (33°45’10‖S). Os ecossistemas são
extraordinariamente diversos. Tal fato torna a Zona Costeira brasileira um ambiente complexo
e diversificado, uma vez que a faixa terrestre que compõe a costa do Brasil está inserida em
diferentes biomas (Amazônia, Caatinga, Mata Atlântica, Cerrado e Pampas).
Consequentemente, a diversidade biológica não é igualmente distribuída ao longo dos
diversos ecossistemas costeiros e marinho como as lagoas costeiras, manguezais e recifes de
corais.
Lagoas costeiras e estuários constituem sistemas férteis, servindo de abrigo e região de
criadouro para um grande número de espécies. Os manguezais apresentam elevada
diversidade estrutural e funcional, atuando, juntamente com os estuários, como exportadores

44
de biomassa para os sistemas adjacentes. Finalmente, os recifes de corais comportam uma
variedade de espécies animais próxima àquela observada nas florestas tropicais úmidas e
constituem os ambientes mais diversos do planeta (Reaka-Kudla, 1997). A Zona Costeira
representa um dos principais focos de atenção para a conservação ambiental e manutenção da
biodiversidade, tanto terrestre como aquática. O impacto de rejeitos domésticos e industriais
associados à expansão de atividades predatórias são as maiores ameaças dos ecossistemas
costeiros, principalmente os mangues e recifes de coral que representam os ecossistemas com
maior diversidade.
O crescimento desordenado e o adensamento das populações costeiras no Brasil tiveram sua
origem na colonização do país. O papel de colônia de exploração promoveu a ocupação do
território brasileiro do litoral para o interior, tendo em vista facilitar o escoamento do
comércio e o trânsito de pessoas entre colônia e metrópole. Estas áreas foram alvos de um
processo de ocupação acelerado que teve como vetores básicos a urbanização, o turismo e a
industrialização. Com o surgimento de grandes centros urbanos próximos a faixa litorânea,
têm-se observado mudanças significativas na qualidade das águas costeiras e estuários,
principalmente em função do despejo de esgoto doméstico e efluentes industriais não tratados,
diretamente nos corpos de água em todo o mundo (Bound and Voulvoulis, 2006; Medeiros et
al., 2005; Nakada et al., 2006; Petrovic et al., 2003). Dados do (IBGE, 2000) mostram uma
grande concentração de áreas com potencial poluidor ao longo do litoral brasileiro e próximo
a corpos de água (Figura 15).
1.4.1.1 Manguezais e lagoas
Manguezais e lagoas representam ecossistemas costeiros de transição diretamente
influenciados pelo oceano. Caracterizam-se por serem extremamente dinâmicos em função
das flutuações das características físico-químicas associadas ao encontro da água doce com a

45
água salgada. No planeta existem aproximadamente 172.000 Km² de manguezais, sendo
26.000 Km² distribuídos pela costa brasileira. O manguezal é um ecossistema costeiro,
característico de regiões tropicais e subtropicais, sujeito ao regime das marés, constituído de
organismos adaptados à flutuação de salinidade e capazes de colonizar sedimentos com
baixos teores de oxigênio. Ocorre em regiões abrigadas e apresenta condições propícias para
alimentação, proteção e reprodução de muitas espécies animais.
Figura 15. Áreas no território brasileiro com potencial impacto poluidor, localizadas próximas a corpos d’água.
Os pontos vermelhos indicam as áreas com potencial impacto poluidor. A maior concentração dessas áreas
encontra-se ao longo da costa, onde observamos maior concentração de atividades humanas e industriais. Fonte:
(IBGE, 2000).
O ecossistema manguezal está associado às margens de baías, enseadas, barras,
desembocaduras de rios, lagunas e reentrâncias costeiras, onde haja encontro de águas de rios
com a do mar, ou diretamente expostos à linha da costa.

46
Este é um ecossistema submetido a intenso impacto antropogênico em virtude de sua
proximidade com áreas intensamente urbanizadas e industrializadas. A ocupação urbana,
associada às atividades humanas e industriais, resulta em exploração inadequada dos recursos
disponíveis e a consequente degradação de áreas de mangue (Kennish, 1992; Pelletier et al.,
2006) (Figura 16).
A Baía de Guanabara é localizada no estado do Rio de Janeiro. A baía abrange parte da região
metropolitana do estado do Rio de Janeiro que se caracteriza por ser uma região de intensa
atividade urbana e industrial. Essa região sofreu uma das maiores catástrofes ambientais do
Brasil com o vazamento de cerca de 1,3 milhões de litros de óleo da Refinaria Duque de
Caxias em janeiro de 2000 (Gabardo et al., 2000). A Baía de Sepetiba também está inserida
numa região de intensa atividade industrial, contaminada pela dissolução de metais sulfetados.
Figura 16. Ecossistemas costeiros sob impacto antropogênico. A) Manguezal da Baía de Guanabara (foto: Erick
Aniszewski) e B) Ocupação industrial na Baía de Sepetiba (fonte: http://www.revistaportuaria.com.br).
Já as lagoas costeiras se encontram distribuídas ao redor do mundo e ocupam 13 % das
regiões costeiras no mundo. Uma lagoa costeira é um corpo de água costeira rasa, separada
do oceano por uma barreira e ligada pelo menos de forma intermitente, por restritas conexões
com o oceano (Kjerfve, 1994). Elas apresentam extensões variadas, podendo alcançar 70 km2
como a Lagoa do Rocha no Uruguai até 10000 Km2,
como a Lagoa dos Patos no Rio Grande
do Sul, a maior do Brasil e uma das maiores da América Latina. Lagoas costeiras são

47
utilizadas para recreação e turismo, apresentam valor cultural e o seu valor econômico
(Costanza et al., 1997). Entretanto, assim como os manguezais, lagoas costeiras também se
encontram ameaçadas pelo despejo de efluentes domésticos e industriais.
O intercâmbio entre lagoa e oceano é impulsionado pela ação das marés e das ondas e é
muitas vezes o principal componente do balanço de água da lagoa (Smith, 1994). A
quantidade e qualidade da água de uma lagoa são influenciadas pelas taxas de evaporação,
precipitação, entrada das águas subterrâneas, escoamento superficial e intercâmbio com o
oceano.
A salinidade da água e os nutrientes atuam como os principais reguladores da distribuição de
diversos organismos tais como bactérias, plantas, microcrustáceos e peixes (De Macedo-
Soares et al., 2010; Farjalla et al., 2009; Santangelo et al., 2007; Santos et al., 2006). A
constante variação da salinidade exige que os organismos sejam capazes de se adaptar a esta
condição. Esta é uma das características que influenciam o alto grau de endemismo desses
ecossistemas. A Lagoa de Araruama, maior lagoa hipersalina do mundo, apresenta variação
na salinidade durante o ano entre 45 e 55 % (Kjerfve, 1994). A comunidade microbiana
associada à lagoa apresenta uma diversidade de procariotos adaptados a viver em condições
hipersalinas (Clementino et al., 2008).
Lagoas costeiras também apresentam um alto grau de conectividade aos ecossistemas
terrestres adjacentes, exibindo uma troca significativa de água e material particulados e
dissolvidos. Esse material se acumula nos sedimentos ou são removidos para o oceano.
Lagoas costeiras no norte fluminense (Farjalla, 2002) apresentam altas concentrações de
carbono orgânico dissolvido (COD), comparáveis às concentrações normalmente encontradas
em pântanos e em ecossistemas considerados ricos em matéria orgânica dissolvida (MOD). A
origem desse carbono está relacionada à decomposição parcial de macrófitas e principalmente
pela a decomposição de plantas terrestres nos arredores. Stepenauskas (Stepanauskas, 1999)

48
mostrou que a biodisponibilidade do MOD é reforçada após a mistura do lago e da água do
oceano. No entanto, a MOD do oceano é mais biodisponível que o das lagoas (Søndergaard et
al., 1995). Sendo assim, essas regiões são ecossistemas altamente produtivos que suportam
uma variedade de habitats. As lagoas costeiras representam uma fonte de energia e nutrientes
para as áreas adjacentes. O carbono orgânico dissolvido (COD) derivado de plantas aquáticas
fornece uma importante fonte de energia para bacterioplâncton marinho costeiro (Moran et
al., 1991).
1.4.1.2 Sistemas recifais
Os recifes de corais são estruturas geológicas de origem biogênica, construídas a partir do
crescimento de organismos que secretam esqueletos de carbonato de cálcio. Essas estruturas
representam um habitat que permite a interação de milhares de espécies que dependem da
manutenção das estruturas recifais, permitindo o estabelecimento de relações entre diversos
taxa. Os ecossistemas recifais abrigam quase um terço dos peixes marinhos (McAllister et al.,
1994). Os maiores responsáveis pela edificação dos recifes são os corais escleractíneos e,
secundariamente, os hidróides calcários ou hidrocorais e as algas calcárias (Muscatine, 1990).
Os recifes cobrem, aproximadamente, de 0,1 a 0,5% do assoalho oceânico, com áreas
variando de 255.000 km2 (Spalding and Grenfell, 1997) a 1.500.000 km
2 (Copper, 1994). Os
recifes de corais estão entre os mais diversos, mais complexos e mais produtivos ecossistemas
do planeta (Odum and Odum, 1955).
Os corais são animais pertencentes ao Filo Cnidária, com cerca de 1.300 espécies
pertencentes a 24 famílias (Brusca and Brusca, 2003). A capacidade dos corais de construir
recifes está intimamente relacionada à atividade fotossintética de algas microscópicas
chamadas zooxantelas (gênero Symbiodinium) que vivem em simbiose com o coral. Estas
algas fornecem carbono orgânico para os corais aumentando sua capacidade de secreção de
carbonato de cálcio. Isso permite que estes animais cresçam e forneçam matéria prima para a

49
construção da base da estrutura recifal, em uma velocidade mais rápida do que a taxa de
destruição por erosão.
O fenômeno conhecido como ―branqueamento de coral‖ ocorre quando as algas simbiontes
zooxantelas abandonam o tecido do coral ou quando elas perdem seus pigmentos (Glynn,
1991). O aumento da temperatura da água nos últimos anos, associado ao aumento do efeito
estufa, é apontado como a principal causa do branqueamento (Fitt et al., 2001; Glynn, 1991;
Goreau et al., 2000). O branqueamento de corais representa um impacto significativo nos
recifes, pois os corais, principais construtores, sofrem déficit energético tornando-se
vulneráveis a doenças, predação e morte (Glynn, 1991; Hoegh-Guldberg, 1999). O
branqueamento atinge ainda a saúde dos corais afetando sua reprodução, diminuindo o
crescimento linear e reduzindo a taxa de calcificação do seu exoesqueleto (Nyström et al.,
2000). E como consequência, a manutenção e o desenvolvimento da estrutura recifal pode ser
seriamente comprometida (Goreau et al., 2000). Além das questões globais, os impactos
locais também podem promover prejuízos aos sistemas recifais
As atividades pesqueiras ao redor do mundo mostraram um significativo aumento nos últimos
50 anos (Swartz et al., 2010) (Figura 17). Isso representa uma ameaça aos sistemas recifais,
pois a retirada de organismos que ocupam níveis superiores na cadeia alimentar, provocando
alteração no equilíbrio do ecossistema (Sandin et al., 2008). Mais de 100 países tem recifes de
corais na sua linha de costa, e pelo menos dez milhões de pessoas têm seus recursos ou fonte
protéica oriunda dos recifes (Salvat, 1992). Nestes recifes a base das cadeias alimentares é
seriamente comprometida, afetando toda a estrutura recifal e as comunidades que deles
dependem.

50
A pesca predatória provoca a mudança de uma comunidade recifal dominada por corais para
uma comunidade dominada por algas devido à retirada dos peixes herbívoros que, em
ecossistemas equilibrados, controlam o crescimento das algas (Figura 11). A mudança de fase,
para predomínio de algas ocorre porque a taxa de crescimento das algas e bactérias
heterotróficas é muito superior a do coral. O resultado desta competição é a diminuição da
cobertura de corais e aumento da cobertura de algas. Sendo assim, esses organismos podem
ser utilizados como indicadores de degradação ambiental nos sistemas recifais. A alteração na
relação entre a cobertura de bentos (corais e algas) em sistemas recifais pode ser observada
Figura 17. A produção primária requerida para sustentar a produção pesqueira mundial de pesca, expresso em
percentagem da produção primária local. Os mapas representam o total de desembarques anuais de 1950
(acima) e 2005 (abaixo) (Swartz, 2010).

51
em regiões com maior concentração de atividades humanas (Dinsdale et al., 2008b; Sandin et
al., 2008). Esse fenômeno tem sido observado em vários recifes do mundo, em consequência
não somente da sobre pesca, mas também da poluição marinha e do crescente incidência de
doenças em função do efeito estufa e acidificação dos oceanos (Hughes, 1994; Hughes et al.,
2007).
No Brasil, episódios de doença em corais têm sido relatados nos últimos anos (Leão
(Francini-Filho et al., 2008; Leão et al., 2008). A proximidade dos recifes da costa, onde se
concentra a maior parte da população constitui um fator determinante para o aumento da
incidência de doenças, aliado ao aumento da temperatura global. Estimativas sugerem que a
manutenção do ritmo de degradação dos sistemas recifais pode levar os corais brasileiros à
extinção em torno de 50 anos (Francini-Filho et al., 2008)
Embora raros, os recifes brasileiros ocupam uma área de cerca de 890 km2 sendo que a maior
parte das espécies está localizada na costa do estado da Bahia (Prates, 2003) (Figura 18).
Recifes coralíneos brasileiros são considerados como área prioritária para a conservação da
biodiversidade no Atlântico devido ao tamanho reduzido deles (5% de recifes do Atlântico) e
um alto nível de endemismo (Francini-Filho and de Moura, 2008).

52
O Banco de Abrolhos é um alargamento de 42.000 km2 da plataforma continental dos estados
da Bahia e Espírito Santo, representando o maior e mais rico complexo recifal do Atlântico
Sul (Leão et al., 2003). Os recifes de corais têm sido formados nos últimos cinco mil anos
nesta área e distribuídos sobre a plataforma continental. Mussismilia braziliensis e M. híspida
representam mais de 70% da massa formadora do recife em algumas áreas. Os recifes de
Abrolhos crescem a partir de uma estrutura característica com a forma de cogumelo chamada
de "chapeirão", o qual é construído por uma fauna coralina rica em espécies endêmicas.
Existem chapeirões com diferentes alturas e dimensões laterais (altura < 1 a >25 m, diâmetros
<1 a >50 m) e em diferentes fases de crescimento (Hartt, 1871). Essas estruturas assemelham-
se a cogumelos com o topo mais pronunciado no lado dos ventos dominantes. Com apenas 1
m de altura e 1 a 2 m de diâmetro no topo, podem já apresentar a forma cogumelar desde sua
fase inicial de crescimento, assim como uma simples colônia do coral Mussismilia
Figura 18. Mapa com a distribuição dos recifes na costa brasileira. (Fonte: RECOR)

53
braziliensis com diâmetro de cerca de 20 cm já pode apresentar a forma de um pequeno
cogumelo. Nos chapeirões maduros, os quais podem alcançar alturas de até 25 m com uma
área na superfície de cerca de 50 m, o aumento do diâmetro do topo é acentuado quando estas
estruturas alcançam o nível do mar. Esta forma de crescimento cogumelar dos recifes de
Abrolhos não é uma característica comumente encontrada nos recifes de corais de outros
mares tropicais. Recifes costeiros rasos são sujeitos a uma alta pressão da pesca e acúmulo de
sedimentos, enquanto recifes mais afastados da costa são menos impactados (Francini-Filho
and de Moura, 2008).
O Banco de Abrolhos sustenta uma grande diversidade de vida marinha, potencialmente
desempenhando um papel principal na homeostase de todo o oceano Atlântico Sul como uma
importante área produtora e berçário de espécies. A região hospeda importantes peixes alvos
de pesca como Caranhas (família Lutjanidae), garoupas (família Serranidae) e outros recursos
costeiros valiosos (marisco) (Francini-Filho and de Moura, 2008). De acordo com o Programa
de Avaliação do Potencial Sustentável de Recursos Vivos na Zona Econômica Exclusiva do
Ministério do Meio Ambiente, a produção pesqueira anual no banco de Abrolhos é de aprox.
15 mil toneladas de pescado, gerando divisas na ordem de R$ 150 milhões reais/ano e
envolvendo pelo menos 20 mil famílias de pescadores.
1.4.2 Bioma terrestre: o solo da Mata Atlântica
O solo é a camada mais superficial da crosta terrestre. Ele abriga a maior parte da diversidade
de organismos vivos do planeta. Em consequência, processos ecológicos fundamentais para a
manutenção dos ecossistemas, como os processos biogeoquímicos (Ex. ciclo do carbono e
nitrogênio) ocorrem no solo. O solo desempenha diversas funções primordiais em nossos
ecossistemas, incluindo a capacidade de ciclagem das moléculas orgânicas e inorgânicas. O
solo apresenta grande capacidade para assimilar grandes quantidades de matéria orgânica,
transformando-a em húmus, uma associação de matéria orgânica com minerais em

54
decomposição. Os padrões do processo de decomposição da matéria orgânica se refletem na
forma de húmus. Na Mata Atlântica, são encontradas diferentes formas de húmus (Kindel and
Garay, 2002).
O solo apresenta grande variedade de nichos e habitats. Os poros do solo, aliado aos
agregados particulares oferecem uma ampla variedade de microhabitats, permitindo o
desenvolvimento de organismos como insetos, protozoários, fungos e bactérias. Microzonas
com alguma aeração podem se formar apenas alguns milímetros de distância de regiões
anóxicas. Diferentes áreas podem ser enriquecidas com diferentes compostos orgânicos e
inorgânicos, tornando esses ambientes mais ácidos ou básicos de acordo com a variação de
aporte dessas moléculas no solo.
O solo funciona como um meio de crescimento para as planta superiores, permitindo a fixação
e proteção às raízes, provendo nutrientes. O solo é capaz de reter e armazenar a água da chuva
e disponibiliza os nutrientes inorgânicos em forma de íons dissolvidos, permitindo sua
utilização pelas plantas de forma contínua. O solo funciona como um regulador do suprimento
de água uma vez que, de acordo com suas características, pode reter mais ou menos água. Isto
influencia no controle dos níveis dos corpos d´água como bacias, rios e lagos. Aliado a isto, o
solo também pode influenciar a qualidade da água. Processos de lixiviação e run-off podem
ocasionar o transporte de nutrientes, ou mesmo substâncias tóxicas para os reservatórios de
água. Por outro lado, também podem executar a depuração de águas contaminadas, a partir da
retenção de substâncias tóxicas. Isso irá depender das caractterísticas do solo.
1.4.2.1 Características do solo
Os principais eventos relacionados ao processo de formação do solo são efeitos da atuação do
clima e de organismos vivos sobre um material parental ao longo do tempo, sob a influência
de uma topografia. O solo pode ser considerado uma interface entre quatro componentes

55
fundamentais para a vida. Os quatro principais componentes do solo são matéria orgânica,
matéria mineral, água e ar. A proporção relativa desses componentes pode influenciar nas
características e na produtividade do solo.
A matéria orgânica é representada pelos restos animais e restos vegetais em todos seus
estágios de decomposição, apresentando fundamental importância como fonte de matéria
orgânica para o solo. A matéria orgânica decompõe-se até constituir o húmus, que é a matéria
orgânica na forma coloidal, com características benéficas e atribui ao solo uma coloração
mais escura.
A matéria mineral do solo é representada pelos minerais constituintes da rocha de origem, e
pelos minerais formados como resultado do seu intemperismo. A organização estrutural é
bastante heterogênea, essencialmente formada por agregados resultantes da associação da
matéria orgânica e mineral de diferentes tamanhos e estabilidade (Tisdall and Oades, 1982).
Esses agregados por sua vez, promovem a formação de microhabitats, caracterizados pela
presença de diferentes substratos, concentração de nutrientes e água, bem como variações de
pH (Ladd et al., 1996).
O componente líquido do solo representa uma solução iônica (cátions e ânions). A água fica
retida em microporos e é drenada para as camadas mais profundas do solo, pela ação da
gravidade, através de macroporos, responsáveis pela aeração. Maiores quantidades de matéria
orgânica permitem maior retenção da água no solo. Há uma tendência dos solos mais
argilosos em apresentar maiores quantidades de biomassa microbiana capaz de promover
maior retenção de umidade, formando complexos organo-minerais ou servindo de tampão às
mudanças de pH (Smith and Paul, 1990).
A quantidade de água tem uma relação próxima com o conteúdo de ar no solo, uma vez que
os poros são preenchidos pela água, desalojando o ar do seu interior. A umidade relativa do ar

56
do solo costuma ser próxima de 100 %, a não ser em solos extremamente secos. Além disso, o
conteúdo de CO2 é mais elevado enquanto à concentração de O2 é menor que a observada no
ar atmosférico. Esta tendência é explicada pela atividade metabólica no solo.
Um corte seccional vertical do solo revela a presença de regiões distintas, conhecidas por
horizontes. A camada mais próxima da superfície do solo é conhecida como Horizonte A, que
é rica em matéria orgânica proveniente da decomposição de matéria viva, de plantas e de
raízes. As camadas situadas abaixo do Horizonte A apresentam menores níveis de matéria
orgânica à medida que se distanciam da superfície e se aproximam da rocha de origem. O
material parental influencia em características do solo, como, cor, textura, estrutura,
mineralogia e pH.
Atualmente, os solos brasileiros estão distribuídos em 14 classes de acordo com a Nova
Classificação de Solos (EMBRAPA, 2006). O tipo de solo mais frequente no território
brasileiro é o latossolo (Figura 19). Os latossolos são solos comumente encontrados em áreas
tropicais. São altamente intemperizados e se caracterizam por apresentar um horizonte
mineral composto por uma subsuperfície rica em hidróxidos de ferro e alumínio. Esses tipos
de solo apresentam alto teor de argila e baixa capacidade de troca iônica, além de baixa
concentração de silicato. A maior parte se forma sob a floresta de Mata Atlântica, sendo
submetido às chuvas intensas. Além disso, apresentam baixo teor de nutrientes para plantas,
especialmente após a remoção de parte da cobertura nativa.
Figura 19. Os latossolos representam um dos tipos mais comumente encontrados no Mata Atlântica.

57
1.4.2.2 A Mata Atlântica
A Mata Atlântica é uma formação vegetal brasileira que originalmente acompanhava o litoral
do país do Rio Grande do Sul ao Rio Grande do Norte. Nas regiões Sul e Sudeste, chegava até
Argentina e Paraguai. A Mata Atlântica apresentava cerca de 1.300.000 km², ou cerca de 15%
do território nacional, englobando 17 estados brasileiros (Figura 20). Na porção litorânea, a
maior parte da Mata Atlântica tem áreas de transição com os ecossistemas que compõem a
Zona Costeira. Depois de séculos de exploração madeireira, avanço agrícola e crescimento
urbano, mais de 90% da mata original foi destruída (Figura 20). Atualmente, a Mata Atlântica
encontra-se extremamente reduzida, sendo uma das florestas tropicais mais ameaçadas do
globo.
.
Apesar de reduzida a poucos fragmentos, na sua maioria descontínuos, a biodiversidade de
seu ecossistema é uma dos maiores do planeta e declarada pela UNESCO como Reserva da
Figura 20. Cobertura do bioma Mata Atlântica. Aproximadamente 90 % do bioma já foi devastado (Fonte: Atlas
dos Remanescentes Florestais da Mata Atlântica. SOS Mata Atlântica e Instituto Nacional de Pesquisas Espaciais
– INPE.

58
Biosfera, um Patrimônio da Humanidade. Nas regiões onde ainda existe, a Mata Atlântica
caracteriza-se pela vegetação exuberante, com acentuado higrofitismo. É uma das áreas mais
sujeitas a precipitação no Brasil. As chuvas são orográficas, em função das elevações do
planalto e das serras. Há subdivisões da mata, devidas a variações de latitude e altitude e
possui formações pioneiras. Além disso, vale destacar ainda a existência de sete das nove
maiores bacias hidrográficas brasileiras neste bioma A interface com estas áreas cria
condições particulares de fauna e flora.
Entre as espécies mais comuns encontram-se algumas briófitas, cipós, e orquídeas. Mais de
50% das plantas vasculares conhecidas da Mata Atlântica são endêmicas, ou seja, não
ocorrem em nenhum outro lugar no planeta. Estima-se que há no bioma 1,6 milhão de
espécies de animais, em sua maioria insetos. A fauna endêmica é formada principalmente por
anfíbios, mamíferos e aves das mais diversas espécies. Estima-se que a Mata Atlântica
detenha mais que 30 % das espécies existentes no país.
Apesar desta grande biodiversidade, das 396 espécies de animais consideradas oficialmente
ameaçadas de extinção no Brasil (Instrução Normativa MMA nº 03 de 27 de maio de 2003),
350 são da Mata Atlântica. Do total de 265 espécies de vertebrados ameaçados, 185 ocorrem
na Mata Atlântica (69,8%), sendo 100 (37,7%) deles endêmicos. Das 160 aves da relação, 118
(73,7%) ocorrem nesse bioma, sendo 49 endêmicas. Entre os anfíbios, as 16 espécies
indicadas como ameaçadas são consideradas endêmicas da Mata Atlântica. Das 69 espécies de
mamíferos ameaçados, 38 ocorrem nesse bioma (55%), sendo 25 endêmicas (Ministério do
Meio Ambiente, 2007).
A Mata Atlântica é um dos 5 hotspots de diversidade na lista de áreas prioritárias de
conservação. A maior ameaça ao delicado equilíbrio da biodiversidade é a ação humana
através de sua ocupação e os impactos de suas atividades. Os rios e lagos da Mata Atlântica
abrigam ainda ricos ecossistemas aquáticos, grande parte deles ameaçados pelo desmatamento

59
das matas ciliares e consequente assoreamento dos mananciais, pela poluição da água, e pela
construção de represas sem os devidos cuidados ambientais. Parte considerável da Mata
Atlântica está conservada sob a forma de parques nacionais e estaduais.. O Parque Nacional
da Serra dos Órgãos (PARNASO) é uma região de preservação da Mata Atlântica que foi
criada em 1939 para proteger a biodiversidade de parte do trecho da Serra do Mar na região
serrana do Rio de Janeiro, envolvendo os municípios de Teresópolis, Petrópolis, Magé e
Guapimirim.
O PARNASO apresenta florestas de encosta e campos de altitude que variam de 200 a
2.263m de altitude. A grande e brusca variação de altitude criou ambientes únicos e grande
diversidade biológica (Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais
Renováveis, 2007). A região da Serra dos Órgãos está inserida no domínio morfo-climático
Tropical Atlântico. O clima do Parque é tropical superúmido (com 80 a 90% de umidade
relativa do ar), com média anual variando de 13º a 23º C (atingindo valores de 38ºC a 5ºC
negativos nas partes mais altas). No verão há uma concentração de chuvas, com variação
pluviométrica de 1.700 a 3.600 mm e no inverno, o período de seca é característico (Instituto
Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis, 2007).
A consolidação do ―Encontro de Pesquisadores do PARNASO‖, que ocorre anualmente desde
2007, organizado pela direção do parque, mostra a importância do PARNASO como área de
estudo em diversas áreas da biologia. Entretanto, a literatura sobre a microbiologia da área do
estado fluminense ainda é incipiente e conta com poucos trabalhos abordando a diversidade
de microrganismos do estado fluminense. Alguns dos poucos trabalhos realizados com
amostras coletadas da Mata Atlântica com enfoque microbiológico consideram a diversidade
de bactérias não cultiváveis (Lambais et al., 2006) e o isolamento de grupos bacterianos
específicos (Lima-Bittencourt et al., 2007; Semedo et al., 2004; Souchie et al., 2006)

60
2. OBJETIVOS
2.1 Objetivo geral
Caracterizar a diversidade taxonômica e funcional de comunidades microbianas,
demonstrando o potencial biotecnológico da microbiota de diferentes biomas brasileiros.
2.2 Objetivos específicos
Caracterizar a diversidade do bacterioplâncton de águas costeiras na América Latina (Caribe
ao Atlântico Sul);
Caracterizar a diversidade taxonômica e funcional da microbiota aquática dos sistemas de
recifes de corais do banco de Abrolhos;
Analisar a diversidade de comunidades bacterianas do solo da Mata Atlântica brasileira;
Avaliar o potencial biotecnológico do metagenoma do solo da Mata Atlântica para fins de
produção de celulase;
Estabelecer um panorama sobre o estudo da diversidade microbiana a fim de determinar os
tipos microbianos mais frequentes nos biomas brasileiros.

61
3. MATERIAIS E MÉTODOS
3.1 Gradiente latitudinal de diversidade do bacterioplâncton da costa da América
latina
Foram analisados os dados referentes a Puerto Rico, Brasil, Argentina e Uruguai. As amostras
e suas localizações foram: Bioluminescent Bay (17°58'18 N - 67°00'52 W), Guanabara Bay
(22°55'43 S - 43°08'51 W), Amazon estuary (00 42' 26 S - 50 18' 18 W), Uruguay – Fresh
(34°39'48 N - 54°16'55 W), Uruguay – Brackish (34°39'48 N - 54°16'07 W), Rio de la Plata
(36°06'32 S - 55°14'20 W) e Argentinian Shelf (38°16'59 S - 57°24'36 W) (Figura 21). Os
dados analisados foram depositados no banco do VAMPS (Visualization and Analysis of
Microbial Population Structure) (http://vamps.mbl.edu) e disponibilizados pelo IcoMM
(International Census of Marine Microbes). Todas as amostras foram submetidas à
pirosequenciamento (454) em colaboração com o Laboratório de Biologia Marinha (Woods
Hole, MA, EUA) através do Latin American and Caribbean Cooperative Run.

62
A região V6 do gene rRNA foi amplificada por um conjunto de iniciadores contendo 5
versões do iniciador 967 forward e 4 versões do iniciador 1046 reverse foi utilizado (Huber et
al. 2007). O pirosequenciamento foi realizado na plataforma GS20 de acordo com o protocolo
descrito por Sogin et al. (2006). Sequências de iniciadores e de baixa qualidade foram
removidas, como descrito em Huse et al. (2008).
A metodologia Global Alignment for Sequence Taxonomy (GAST) (Sogin, 2006) foi usada
para atribuir classificação taxonômica das tags de V6 tags (Figura 22). A GAST compreende
um banco de genes rRNA de referência com base no banco de dados SILVA (Al Pruesse al.
2007). Após submeter ao BLAST, os 100 melhores hits de cada sequência gerada foram
utilizados para compor o banco de dados. A classificação taxonômica foi realizada com a
ferramenta Classifier do RDP (Wang, 2007). Sequências com pelo menos 97 % de identidade
foram classificadas como OTUs pertencentes à mesma espécie. Esta classificação foi
realizada para todos os pares alinhados. As OTUs endêmicas foram aquelas que não
apresentaram sequências compartilhadas em outras localidades. Por outro lado, OTUs
cosmopolita são aquelas que têm compartilhado mais de sete sequências, com pelo menos
uma de cada localidade amostrada. As sequências duplicadas, com cópias exatas, foram
removidas das análises por representarem artefatos do sequenciamento
.
Brasil – Baía de Guanabara
Figura 21. Localização geográfica dos pontos de coleta ao longo da costa da America Latina.

63
3.2 Avaliação da saúde do Banco de Abrolhos através da metagenômica
Amostras de água de recifes de corais de áreas não protegidas e protegidas (Parque Marinho
de Abrolhos) foram coletadas entre janeiro e fevereiro de 2009. O DNA foi extraído e o
metagenoma foi pirosequenciado e as sequências foram anotadas através de tecnologia de
subsistemas.
Figura 22. Sistema GAST de classificação taxonômica utilizado para implementação de um banco de dados
curados da região V6 do gene rRNA 16S (Retirada de https://vamps.mbl.edu/)

64
3.2.1 Área de estudo e coleta
Cinco recifes entre 25 e 70 km da costa foram selecionados para este estudo. Os recifes mais
próximos da costa são Sebastião Gomes (17 ° 54'42 .49 "/ 39 ° 7'45 .94‖), Pedra de Leste (17
° 47'01 .3 "/ 39 ° 03'05") e Timbebas (17 ° 28'42 0,3 "/ 39 ° 01'41 .1‖) e os mais distantes da
costa são dos Parcel dos Abrolhos (17 ° 57'32 .7 "/ 38 ° 30'20 .3‖) e California (18 º 06 '7,8
"S/38 º 35' 26,0") foram o foco desta pesquisa (Figura. 23). Os recifes desprotegidos são
locais próximos de comunidades de pescadores e outras comunidades humanas. Por outro
lado, Parcel dos Abrolhos e California estão completamente dentro do Parque Nacional de
Abrolhos, e o acesso é restrito e controlado pelo governo brasileiro. Os três recifes próximos
da costa estão desprotegidos e estão submetidos à pesca intensiva, inclusive o recife de
Timbebas que, teoricamente, está dentro de uma área de proteção marinha do Banco de
Abrolhos. Amostras de água foram coletadas próximas a superfície (<1 m) de corais do recife,
entre 6 e 10 m de profundidade em Sebastião Gomes, e aos 20 m nos recifes de Timbebas
Parcel dos Abrolhos e em California. Cerca de 20 litros de água foram coletados em garrafas
previamente lavadas com água de superfície (Figura. 24). Parte da água coletada foi utilizada
na extração de DNA e outra nas análises físico-químicas, realizadas pelo Laboratório de
Hidrobiologia da UFRJ. Em janeiro de 2010, amostras do recife Pedra de Leste foram
coletadas a 10 m de profundidade em vez do recife California por causa de condições
meteorológicas instáveis.

65
Figura 24. Mergulhador coletando a água dos recifes de Abrolhos a aproximadamente 20 m de profundidade (Foto:
Ronaldo Francini-Filho).
Figura 23. Região do Banco de Abrolhos e os pontos de coleta. Em azul, as áreas não protegidas e em vermelho
as áreas protegidas. As linhas vermelhas representam os limites do Parque Marinho de Abrolhos (Foto: Pedro
Meirelles).

66
3.2.2 Extração do DNA metagenômico da água do mar
Quatro repetições independentes da água do mar foram filtradas através de redes de 100 mM e
20 mM por gravidade. A água pré-filtrada foi filtrada através de Sterivex (0,22 µM)
(Millipore). Entre 2 e 4 litros foram filtradas em cada filtro Sterivex. A filtração foi realizada
por pressão positiva aplicada ao sistema de garrafas Niskin (Figura 25)
O material coletado nos filtros foi preservado com aproximadamente 1,5 mL de tampão SET
(sacarose 20%, 50 mM EDTA e 0,5 mM Tris-HCl, pH 7.6). A extração do DNA foi realizada
no interior do Sterivex .
Os procedimentos de extração do DNA estão no anexo 7.1, item 7.1.1.1
3.2.3 Pirosequenciamento dos metagenomas do Banco de Abrolhos
O sequenciamento dos metagenomas foi realizado utilizando a tecnologia de
pirosequenciamento (Margulies et al., 2005). Bibliotecas foram geradas através de
fragmentação mecânica do DNA em pedaços na faixa de 400 a 2000 pb (shotgun library).
Após a ligação ao adaptador, os fragmentos foram desnaturados e amplificado por PCR em
A B
Figura 25. Sistem de filtração Niskin. A) Sistema de filtração com os filtros Sterivex já acoplados. B) Filtros
Sterivex durante o processo de filtração (Foto: Rodrigo Moura).

67
emulsão (emPCR). Os produtos de amplificação foram sequenciados no sistema 454 GS FLX
(Roche, Life Sciences) através da química de sequenciamento GS FLX Titanium.
As principais etapas de preparação do pirosequenciamento são: purificação e quantificação
do DNA metagenômico, preparação das bibliotecas, emPCR, quebra das emulsões,
enriquecimento e sequenciamento. Todas as reações foram preparadas com kits para de
volume médio (MV). Esta seção apresenta os procedimentos realizados.
3.2.3.1 Purificação e quantificação do DNA metagenômico
A quantificação do DNA metagenômico representa uma etapa crítica para a construção das
bibliotecas. Por isso, a concentração deve ser medida por fluorímetria através do método
PicoGreen (Invitrogen), um corante que apresenta alta sensibilidade e especificidade pelo
DNA de fita dupla (Ahn et al., 1996). Outros métodos (ex. espectrofotometria) podem
superestimar a concentração, levando a uma biblioteca de baixa qualidade. A quantificação
foi realizada utilizando o método PicoGreen.
3.2.3.2 Preparação de bibliotecas pelo método shotgun
O protocolo (Rapid library preparation kit) parte de 500 ng de DNA metagenômico para
produzir fragmentos de fitas através de nebulização com nitrogênio em alta pressão. Esses
fragmentos são reparados por atividade DNA polimerase e adição de fosfato, produzindo
extremidades com cortes retos (blunt end). Dois adaptadores (A e B) são ligados às pontas
reparadas dos fragmentos de DNA. O adaptador A apresenta um iniciador do sequenciamento
e o adaptador B apresenta uma molécula de biotina.
Os procedimentos realizados para a construção das bibliotecas estão no anexo 7.1, item
7.1.1.3.
Após a fragmentação do DNA foi utilizado o Mini eluate PCR purification kit (Qiagen) para
purificar os fragmentos.

68
3.2.3.3 Quantificação e checagem da qualidade da fragmentação das bibliotecas
A quantificação da biblioteca foi feita por fluorometria. É necessário fazer uma curva padrão
utilizando os padrões do kit (rapid library preparation). Com isso, foi determinada a
concentração das bibliotecas e o volume necessário para os ensaios de emPCR.
A checagem da qualidade da fragmentação das bibliotecas foi realizada através de
eletroforese em chip (Bio Analyzer 2100, Agilent Technologies), utilizando DNA LabChip
(Agilent Technologies).
Os procedimentos realizados para a quantificação estão no anexo 7.1, item 7.1.1.3.3.
3.2.3.4 Reação em cadeia da polimerase em emulsão (em-PCR)
O em-PCR ocorre em um ambiente microfluídico onde os reagentes ficam limitados ao
interior de micelas da emulsão. Desta forma, ocorre a reação de amplificação dos fragmentos
de DNA ligados as esferas revestidas por streptavidina pelo adaptador B (biotina). Apenas as
esferas que apresentarem um, e somente um, fragmento de DNA será capaz de gerar
sequências.
Os procedimentos realizados para a montagem das reações para em-PCR de estão no anexo
7.1, item 7.1.1.4.
3.2.3.5 Quebra da emulsão (break emulsion)
Após a amplificação é necessário recuperar as esferas contendo os produtos de amplificação
(haired esferas) para realizar a quebra das emulsões e retirada do óleo emulsificado. As
seções a seguir descrevem como deve ser realizado o processo de quebra das emulsões.
Os procedimentos realizados para a quebra das emulsões estão no anexo 7.1, item 7.1.1.5.

69
3.2.3.6 Enriquecimento (Enrichment)
As esferas recuperadas são contadas (utilizando contador de partículas) no início e após o
procedimento de enriquecimento. Nesta etapa são eliminadas as esferas que falharam na
amplificação e que, portanto não contém DNA amplificado ligado à superfície. A faixa de
enriquecimento utilizada foi de 5 – 20 % das esferas que iniciaram o em-PCR.
Os procedimentos realizados para o enriquecimento estão no anexo 7.1, item 7.1.1.6.
3.2.3.7 Preparação e sequenciamento
A última etapa do sequenciamento pode ser dividida em duas partes principais: a preparação
da placa Pico Tilter Plate (PTP) onde são depositadas as esferas e ocorre o sequenciamento de
fato e a preparação do sequenciador GS FLX 454 e seus reagentes que serão consumidos
durante o procedimento. Foi utilizado ¼ de placa por amostra.
Antes de preparar as DNA e Control beads, foi preciso calcular o volume de esferas (DNA
beads) necessárias para ocupar os 4 quartos da placa, ou seja, aproximadamente 790000
esferas. Foi utilizado o volume de sobrenadante necessário para reduzir o volume de DNA
beads para 20 µL.
Foi utilizado o seguinte programa: 1 ciclo de 4 minutos a 94 ° C; 50 ciclos por 30 segundos a
94 ° C, 4,5 minuto a 58 ° C, 30 segundos a 68 ° C (rampa de aquecimento: 1,5); 10 ° C em
espera
Os procedimentos detalhados para a preparação da corrida estão no anexo 7.1, itens 7.1.1.7 e
7.1.1.8.
3.2.4 Análises Computacionais dos metagenomas da água dos recifes do Banco de
Abrolhos
Os metagenomas foram submetidos no MG-RAST com os seguintes números de acesso: 8181
(Sebastião Gomes 2009); 7673 (Timbebas 2009); 7309 (California); 7376 (Parcel dos

70
Abrolhos 2009); 12979 (Sebastião Gomes 2010); 12911 (Timbebas 2010); 12912 (Pedra de
Leste); 12978 (Parcel do Abrolhos 2010).
3.2.4.1 Filtragem das sequências de baixa qualidade
A checagem da qualidade e filtragem de sequências de baixa qualidade foram realizadas
utilizando programa PRINSEQ (Schmieder and Edwards, 2011), disponível na rede através
do endereço http://prinseq.sourceforge.net/. Arquivos nos formatos FASTA e QUAL são
utilizados como entrada para a análise. O programa oferece um resumo estatístico de
características como tamanho das sequências, conteúdo GC, distribuição dos scores de
qualidade, número de duplicatas, ocorrência de N e cauda poli-A/T.
Os filtros oferecidos pelo programa são capazes de remover sequências curtas, de baixa
qualidade com grande quantidade de N e homopolímeros. As sequências menores que 100 pb
e aquelas que continham N e homopolímeros (repetição de pelo menos 20 nucleotídeos ou
dinucleotídeos) foram retiradas das análises. Além disso, foram retiradas todas as duplicatas,
que são artefatos gerados durante diferentes etapas do processo de sequenciamento. Esses
artefatos podem ocorrer quando uma molécula de DNA amplificado pode se ligar a uma
esfera vazia durante o em-PCR, ou quando o sinal ótico de um poço interfere em um vazio
adjacente (Gomez-Alvarez et al., 2009).
3.2.4.2 Anotação dos metagenomas pela tecnologia de subsistemas
Os metagenomas foram anotados pelo servidor MG-RAST (MetaGenomic Rapid Annotation
by Subsystem Technology) (Meyer et al., 2008). Apesar dos esforços ainda concentrados na
anotação de um genoma único, a análise comparativa das variações de um único componente
da maquinaria celular e seu contexto genômico em uma coleção de genomas completos e
curados permite uma anotação mais rápida e acurada. Um subsistema é um conjunto de papéis
funcionais que, juntos, implementam um determinado processo biológico ou estrutural

71
(Overbeek et al., 2005). Cada subsistema é curado por um grupo de cientistas especializados
em vias metabólicas específicas, estabelecendo quais os principais genes envolvidos e sua
arquitetura genômica.
Dados no formato gerado pelo GS 454 FLX e arquivos FASTA podem ser carregados no
servidor. O fluxo de informações, bem como a interação com os dados e ferramentas de
análise ocorre dentro de um ambiente virtual (SEED). Este ambiente abriga o servidor RAST
que contém um banco de genomas completos anotados, bem como a lista de subsistemas já
caracterizados. As proteínas que compõem os subsistemas formam famílias de homólogos
isofuncionais (FIGfam – Fellowship Interpretation of Genomes family) (Gerlt and Babbitt,
2001). Sendo assim, existem 3 componentes principais que tornam essa estratégia de anotação
rápida e acurada: o servidor RAST, os subsistemas e as FIGfam.
Após carregar os dados, foi gerado um número identificador para cada sequência. Em uma
segunda etapa, as sequências foram submetidas a um BLASTX contra um banco de dados
não-redundante no SEED com e-value 10-5
. A partir desses resultados é possível realizar uma
análise taxonômica e funcional. A análise taxonômica é baseada na correlação entre as
proteínas identificadas e seu organismo de origem nos bancos de dados, permitindo o cálculo
da abundância relativa de cada táxon identificado. A classificação funcional das proteínas de
genes codificantes é calculada pela projeção das FIGfam, baseada na similaridade das buscas.
3.2.4.3 Análises comparativas
As análises comparativas foram realizadas baseadas nos perfis taxonômicos e funcionais
gerados pelo MG-RAST. As tabelas em formato .tsv foram utilizadas como arquivo de
entrada para o programa STAMP (Statistical Analysis of Metagenomic Profiles). O STAMP é
um programa que suporta práticas de análises estatísticas para metagenômica comparativa.

72
Foi realizado teste exato de Fisher (two-side) com valor de p < 0,05 e intervalo de confiança
de 95 %, calculado pelo método Newcombe-Wilson. Neste caso, o teste exato de Fisher visa
verificar se o número de sequências relativas a um subsistema ou táxon é equivalente em dois
metagenomas diferentes. O intervalo de confiança fornece a mesma informação que é
geralmente contido em um teste de significância.
A classificação de patogenicidade foi atribuída com base nas informações disponíveis para
genomas de organismos microbianos no National Center for Biotechnology Information
(NCBI, http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi). A classificação de organismos
autotróficos / heterotróficos não está disponível online em uma única fonte. Portanto, um
sistema de classificação do estado trófico (autotrófica e heterotrófica) foi desenvolvido com
base no filo dos organismos identificados nas amostras. Todos os organismos decorrentes do
mesmo filo foram definidos como tendo o mesmo tipo trófico. Por exemplo, Proteobactérias
podem crescer como heterótrofos ou autótrofos facultativos, dependendo das condições
ambientais, mas eles foram classificados como heterótrofos neste trabalho para permitir uma
classificação única para cada filo.
Os parâmetros físico-químicos foram submetidos a análises estatísticas. A Análise de
Variância (ANOVA de ―ANalysis Of VAriance‖) é utilizada com o propósito de modelar a
média de diferentes grupos. O modelo de Análise de Variância permite testar se as médias
entre os diferentes grupos apresentam diferença significativa. Porém não nos permite indicar
entre quais grupos de fato ocorreu diferença significativa. Desta forma, os testes de
comparação de médias funcionam como um complemento para o estudo da análise de
variância (são chamados de testes post-hoc, ou testes a posteriori, por serem utilizados após a
comparação por ANOVA). Frequentemente é possível determinar quais médias diferem
testando as diferenças entre todos os pares de médias de tratamentos. O teste de Tukey foi o
procedimento utilizado para construir intervalos de confiança de diferenças entre todos os

73
pares de médias. Em alguns casos, os dados coletados não foram suficientes para possibilitar
o uso do método para comparação das localidades, e em alguns outros a variabilidade foi
muito pequena, o que também não possibilita o uso do método estatístico. As análises
estatísticas foram realizadas em colaboração com os alunos Paulo Dick e Felipe Veiga sob
supervisão da Professora Marina Silva Paez do Instituto de Matemática da UFRJ.
3.3 Diversidade Microbiana do solo da Mata Atlântica
O metagenoma dos solos da Floresta da Mata Atlântica foi acessado para avaliação da
diversidade taxonômica e funcional da microbiota. Para isso, foram construídas bibliotecas do
gene rRNA 16S, marcador filogenético, e outra de médios insertos clonados aleatoriamente
para busca de atividade celulolítica. Os clones foram sequenciados automaticamente pelo
método de Sanger através do sistema 3130 (Applied Biosystems). Esta seção apresenta os
experimentos realizados com as amostras coletadas no Parque Nacional da Serra dos Órgãos
(PARNASO) em junho de 2007.
3.3.1 Área de estudo e coleta
O Parque Nacional da Serra dos Órgãos (PARNASO) é uma região de preservação da Mata
Atlântica que fica localizado no estado do Rio de Janeiro, entre 22º 52’ e 22º54’ S e 42º09’ W
(Figura 26). Em julho de 2007, 6 amostras de solo contendo características edáficas distintas
foram coletadas. Foram realizadas coletas em pontos distribuídos ao longo da trilha da
Travessia Petrópolis-Teresópolis localizada no PARNASO (figura 26). Os pontos de coleta
foram definidos baseados na classificação de solos da EMBRAPA. As amostras escolhidas
são representativas dos principais tipos de solo que compõem a Mata Atlântica brasileira
(Martins et al., 2007). Os solos coletados nas regiões 1 e 2 (Bonfim) são os mais prevalentes
no solo do PARNASO. O solo foi obtido de uma profundidade máxima de 11 cm, peneirado
(2 mm de diâmetro) para remoção de partículas grandes e acondicionado em sacos plásticos

74
estéreis, sendo processado menos de 12 h após a coleta. As análises químicas e
microbiológicas foram realizadas a partir das amostras de solo coletadas do campo. As
análises químicas do solo, assim como os mapas georeferenciados, foram realizadas em
colaboração com Professor Eder Martins, na Empresa Brasileira de Pesquisa Agropecuária
(Embrapa), no Centro de Pesquisa Agropecuária dos Cerrados (Cpac), de acordo com padrões
estabelecidos (EMBRAPA, 2006).
Figura 26. Mapa do Parque Nacional da Serra dos Órgãos com a localização geográfica dos pontos analisados.

75
3.3.2 Construção de bibliotecas de rDNA 16S das comunidades microbianas dos solos
do PARNASO
Um dos principais desafios na construção de uma biblioteca de rDNA 16S é a obtenção de
DNA de alta qualidade. Muitos microrganismos do solo estão intimamente associados com a
matriz orgânica das partículas do solo, e produzem substâncias poliméricas extracelulares que
promovem a adesão irreversível das células às partículas do solo (Amorim et al., 2008; Robe
et al., 2003).
3.3.2.1 Extração do DNA metagenômico
A extração do DNA metagenômico dos 6 solos foi realizada utilizando o kit comercial Mo Bio
Power soil. As extrações foram realizadas em triplicata para cada solo, utilizando 0,25g por
amostra e a eluição do material foi realizada em 100µL. A efetividade da extração foi
verificada em gel de agarose 1% em TBE 1X; 90 V / 45 min. A quantificação foi realizada
através de biofotômetro no qual a qualidade do DNA foi verificada através do resultado da
razão dos comprimentos de onda 260/280 nm, que correspondem à contaminação por
proteínas. A concentração média obtida foi de 120 ng/µL de DNA e o grau de pureza variou
entre 1.5 – 2.0.
Os procedimentos detalhados para concentração e purificação do DNA estão no anexo 7.1,
item 7.1.2.1.
3.3.2.2 Amplificação do gene rRNA 16S
A reação de PCR para amplificação do gene rRNA 16S foi realizada utilizando-se iniciadores
desenhados para o anelamento em regiões conservadas dentro de rRNA 16S específicos para
o domínio Eubacteria, gerando fragmentos de aproximadamente 1,5kb. O programa utilizado
foi: 95º C por 3 minutos; seguido de 25 ciclos de 95º C por 1 minuto, 51º C por 1 minuto e 30
segundos, 72º C por 3 minutos. A extensão final ocorre na temperatura de 72º C por 30

76
minutos. Reação: 10, 8 L água milli Q; 2 L Tampão 10X; 2 L dNTP (10mM); 2 L BSA
10X; 1 L PR (5mM); 1 L PF (5mM); 0,2 L Taq (5U/µL); 1 L DNA (50ng) para um
volume total de 20 L. As sequências dos iniciadores são: PF: 27F 5’- AGA GTT TGA TCM
TGG CTC – 3’ e PR: 1492R 5’- GGY TAC CTT GTT ACG ACT – 3’. Para obtenção de
grande quantidade de fragmentos e, assim, maximizar a cobertura da diversidade, 10 reações
para cada solo foram posteriormente concentradas. Foram utilizados 100 ng do DNA
metagenômico por reação.
Após a verificação do material amplificado, uma corrida em gel de agarose 1% TAE 1X foi
realizada para sacar as bandas correspondentes aos produtos de amplificação do gene rRNA
16S. A purificação foi realizada com kit comercial Ultra Clean DNA Purification (Mo Bio).
A efetividade da purificação foi verificada em gel de agarose 1% TBE 1X.
3.3.2.3 Clonagem dos genes rRNA 16S
O vetor de clonagem utilizado foi o pGEM-T easy (Promega) (Figura 27). O vetor comercial
se apresenta linearizado, com uma eficiente região de ligação única 3’- timidina terminal para
produtos de PCR. Este vetor apresenta alto número de cópias e contém os promotores T7 e
SP6 RNA polimerase flanqueando o sítio múltiplo de clonagem, que está localizado em uma
região de codificação de um α-peptídeo da enzima β-galactosidase. Uma vez que as linhagens
de células utilizadas na transformação apresentam deleção da subunidade α da enzima β-
galactosidase, a inativação por inserção do produto de PCR nesta região permite a
identificação de recombinantes. Os recombinantes são selecionados pela resistência a
ampicilina (em meio S-gal + ampicilina (100 µg / µL) A perda de função do gene é indicada
por alterações colorimétricas nas colônias bacterianas, indicando a inserção (colônias brancas)
ou não (colônias pretas) do fragmento de interesse em meio contendo substrato adequado.

77
Figura 27. Vetor de clonagem pGEMT Easy.
3.3.2.3.1 Sistema de ligação
O sistema de ligação consiste nos produto de amplificação do rRNA 16S concentrado e
purificado, ligados ao plasmídeo vetor de clonagem pGEMT easy (Promega). A reação foi
incubada por 16 horas a 16 ° C e depois a enzima foi inativada a 65° C por 20 minutos.
Os procedimentos detalhados para a montagem da reação e precipitação do sistema de ligação
estão no anexo 7.1, item 7.1.2.2.
3.3.2.3.2 Preparação de células eletrocompetentes
Células da linhagem E. coli EPI300 foram utilizadas nos experimentos de transformação e
clonagem. Todo material utilizado durante o procedimento (ponteiras, tubos e soluções) foi
colocado em freezer ou geladeira para garantir a eficiência do procedimento. As células
devem permanecer em gelo enquanto não estiverem sendo manuseadas e todo o procedimento
deve ser realizado em ambiente estéril.
O protocolo detalhado bem como a composição dos meios LB e GYT está no anexo 7.1, item
7.1.2.3.

78
3.3.2.3.3 Transformação
A transformação foi realizada com eletroporação a 1,8kV de corrente, em cubas de 1mm de
distância entre os eletrodos. O tempo de passagem da corrente variou entre 4 e 6
milissegundos. Foram utilizados 5μL do sistema de ligação precipitado (o restante foi
estocado em freezer) em 50 μL de células competentes. Após eletroporação, as células foram
recuperadas em 900 μL meio SOC e incubadas durante 1 h a 37 ° C. Posteriormente, as
culturas foram plaqueadas (100 μL) em meio S-Gal (Sigma – Aldrich) para seleção dos clones
positivos.
3.3.2.4 Montagem das bibliotecas e estocagem dos clones
Após o crescimento dos clones em placa contendo meio S-gal, as colônias brancas foram
coletadas aleatoriamente e inoculadas individualmente em placas de 96 poços utilizando
palitos estéreis. Cada poço continha 130 μL de meio LB com ampicilina numa concentração
final de 300 μg/ml do antibiótico. As placas foram incubadas 37 °C durante a noite. O
crescimento foi observado através da formação de botões no fundo da placa devido à
decantação das células. As células foram ressuspendidas com 50 μL de glicerol 50 %, antes de
estocá-las a -80 °C.
3.3.2.4.1 Confirmação dos clones
Foram escolhidas 10-20 colônias por placa e crescidas em meio LB suplementado com
ampicilina durante 18 horas. A extração plasmidial foi realizada com o kit QIAprep spin
miniprep (Qiagen). A confirmação da presença dos insertos foi realizada pela amplificação de
fragmentos de aproximadamente 1,4 kb utilizando iniciadores que se anelam aos flancos do
plasmídeo vetor.

79
Programa utilizado: 95°C 3 minutos para desnaturação inicial; 95°C 1 minuto; 50°C 1 minuto
e 30 segundos (25 vezes); 72°C 2 minutos 30 segundos; com extensão final a 72°C por 15
minutos.
Reação: 10 μL água milliQ; 2 μL Tampão 10x; 1,6 μL dNTP (10mM); 0,5 μL BSA 10 x; 2 μL
PR (5mM); 2 μL PF (5mM); 0,4 μL Taq (5U/μL); 1 μL DNA (50ng), para um volume total de
20 μL. As sequências dos iniciadores são: PF - SP6 5’- TACGATTAGGGACGTATAG– 3’ e
PR - T7 5’ GTAATAGGACTCACTATAGGG – 3’.
3.3.2.4.2 Extração plasmidial em placa
As extrações plasmidiais para sequenciamento foram realizadas através do método da lise
alcalina em placas de 96 poços.
O procedimento detalhado, bem como a preparação das soluções e meios, está no anexo 7.1,
item 7.1.2.4.
3.3.2.5 Sequenciamento dos clones
A partir dos plasmídeos extraídos por lise alcalina em placa de 96 poços, 1-5 µL da amostra
foram misturados com 1-3 µL de ABI Prism Big Dye Terminator ready reaction mix, versão
3.1 (Applied Biosystems), 3 µL de iniciadores de sequenciamento (4 µM), 1,5 µL de tampão
de diluição (5 X) e 1,5 µL de água milliQ. O programa consiste de 30 ciclos de 15s a 96º C,
1s a 35º C e 4 min. a 60º C. A separação dos fragmentos de DNA foi realizada com ABI
PRISM 3100 genetic analyzer (Applied Biosystems).
3.3.2.6 Análises Computacionais de Sequências do gene rRNA 16S do solo da Mata
Atlântica
As sequências geradas foram depositadas no NCBI com os números de acesso: FJ888655-
FJ889047. As sequências representativas das OTUs mais frequentes forma depositadas com
os números GU170822-GU170837

80
3.3.2.6.1 Filtragem de sequências
Quimeras foram identificadas através do identificador Bellerophon
(http://greengenes.lbl.gov/cgi-bin/nph-bel3_interface.cgi). Quimeras são produtos de PCR
originados por mais de uma fita de DNA molde e se formam durante a amplificação, quando a
síntese das sequências de um modelo é interrompida e continua em outro. Uma janela de 300
pb foi utilizada para detectar as quimeras e o modelo de correção Huber-Hungeholtz foi
aplicado.
Índices de qualidade phred foram calculados para as bases das sequências geradas. Esse
índice determina a probabilidade de uma base ter sido incorporada incorretamente (Ewing and
Green, 1998). As análises foram realizadas com o programa Phred/Prhap disponível em
http://bioinformatica.ucb.br/electro. As sequências com phred menor que 20 (probabilidade de
1 erro a cada 100 bases incorporadas) e com menos que 400 pares de base foram retiradas das
análises.
Além disso, homopolímeros e sequências com mais de 2 % de N (base não identificada)
foram identificados e retiradas manualmente das análises.
3.3.2.6.2 Estrutura das comunidades microbianas (classificação taxonômica)
A classificação taxonômica das sequências do gene rRNA 16S foi realizada através de buscas
no Ribosomal Database Project (RDP) versão 9.21 (Cole et al., 2005). O RDP fornece à
comunidade científica, dados, ferramentas e serviços de análise e inferência filogenética de
sequências de RNA ribossômico através do site http://rdp.cme.msu.edu. O banco de dados é
composto por mais de 100 mil sequências do gene rRNA 16S alinhadas e anotadas.
O RDP oferece a ferramenta Classifier (Wang et al., 2007) , baseado em inferência Bayesiana
simples, que permite a classificação rápida e precisa das sequências de rRNA 16S de acordo
com a taxonomia proposta no Bergey´s Manual. Foi utilizado o limite de 80% de confiança na

81
análise das sequências de rRNA 16S da microbiota dos solos da Mata Atlântica. As
sequências identificadas abaixo do limite de confiança estabelecido foram classificadas como
Unclassified. Esta classificação é qualificada de acordo com o limite do nível taxonômico
identificado (ex. Unclassified Proteobacteria e Unclassified Alphaproteobacteria).
3.3.2.6.3 Estimativa de riqueza e diversidade
A sequências foram alinhadas pelo programa MEGA4 (Tamura et al., 2007) utilizando o
algoritmo clustalW. O alinhamento foi checado e otimizado manualmente e foram retiradas
possíveis sequências de baixa qualidade (contendo N´s, homopolímeros e sequências muitos
gaps). As matrizes de distância dos alinhamentos foram geradas pelo programa BioEdit e
convertidas para o formato .phy, que corresponde ao formato de entrada para o programa
DNADIST do pacote de ferramentas Phylip para análises filogenéticas (Felsenstein, 1989).
As matrizes foram utilizadas como arquivo de entrada para o programa DOTUR (Distance
based OTU Richness) (Schloss and Handelsman, 2005). DOTUR agrupa as sequências em
diferentes níveis de OTUs (Operacional Taxonomic Unity - Unidade taxonômica
Operacional), baseado na homologia entre as sequências do gene rRNA 16S. O agrupamento
foi determinado pelo algoritmo do vizinho mais distante (furthest neighbor clustering). A
frequência das OTUs em diferentes níveis de distância genética (97, 90 e 80% de similaridade
entre as sequências do gene rRNA 16S) foi utilizada para a construção de curvas de rarefação.
A técnica de Rarefação consiste em calcular o número esperado de espécies em cada amostra
para um tamanho de amostra padrão. A obtenção de uma curva desse tipo permite a
comparação de amostras, mesmo que com intensidades amostrais diferentes. O número
esperado de espécies é obtido pela equação:

82
Onde E(S) é o número esperado de espécies em uma amostragem aleatória, S é o número total
de espécies registradas, N é o número total de indivíduos registrados, Ni é o número de
indivíduos da espécie i, e n é o tamanho padronizado da amostra escolhido. As OTUs mais
frequentes foram definidas como grupos de pelo menos 6 sequências com mais que 97 % de
similaridade entre os genes rRNA 16S.
O estimador de riqueza não paramétrico Chao 1 foi calculado nos mesmos níveis de distância
genética das análises de rarefação. É baseado na abundância de OTUs únicas (singletons) e
raras (doubletons) para estimar a diversidade de uma população de tamanho desconhecido. O
cálculo foi realizado de acordo com a equação:
Onde SChao1 é a estimativa de riqueza, Sobs é o número de espécies observadas, n1 é o número
de OTU representada por apenas um indivíduo e n2 é o número de OTU representadas por
apenas dois indivíduos.
O índice de Shannon é utilizado em situações em que uma comunidade inteira não pode ser
inventariada. Seu cálculo leva em consideração que as espécies apresentam abundâncias
diferentes. O índice foi calculado de acordo com a equação:

83
Onde Sobs é o número de espécies observadas, Si é o número de sequências da OTU i e N é o
número de sequências amostradas.
A identidade entre as bibliotecas foi verificada utilizando o programa ∫-LIBSHUFF (Schloss
et al., 2004), que realiza o teste estatístico Cramer-von Mises. A aplicação deste teste em um
par de bibliotecas exige duas comparações para determinar se as bibliotecas são um
subconjunto ou independentes umas das outras. O programa utiliza a matriz de distância
gerada pelo programa DNADIST. As bibliotecas foram comparadas utilizando 10000
sorteios, p < 0.001 e 95 % de intervalo de confiança.
3.3.2.6.4 Análises filogenéticas
Para a reconstrução filogenética foram selecionadas sequências representativa para cada uma
das OTUs mais frequentes definidas pelo DOTUR. Essas sequências foram submetidas à
busca por similaridade no GenBank (http://www.ncbi.nlm.nih.gov) e no RDP para a
determinação dos vizinhos filogenéticos mais próximos. As buscas foram realizadas através
de alinhamento local (BLAST) e através da ferramenta SeqMatch, respectivamente. Foram
identificados tanto os microrganismos mais próximos não cultivados como os cultivados. O
número de acesso das linhagens tipo mais próximas foi obtido com base na lista de nomes
validados publicada no LPSN (List of Prokaryotic names with Standing in Nomenclature)
(Tindall et al., 2006). Isso permitiu o uso de linhagens tipo caracterizadas e validadas
taxonomicamente como âncoras filogenéticas nas árvores.
As sequências das bibliotecas somadas àquelas dos vizinhos filogenéticos mais próximos
foram submetidas a alinhamento no programa MEGA4 através do algoritmo ClustalW
(Thompson et al., 1994). As reconstruções filogenéticas, bem com as matrizes de distância
foram geradas com a opção de deleção completa. O modelo de reconstrução das árvores
filogenéticas utilizado foi o Neighbor Joining (Saitou and Nei, 1987). As árvores foram

84
construídas utilizando a distância correta de Jukes & Cantor, com base em uma matriz de
similaridade calculada através da distância p (análise par a par). A topologia da árvore
filogenética não enraizada resultante foi analisada com um valor de bootstrap baseado em
1000 réplicas.
3.3.3 Construção da biblioteca metagenômica de médios insertos
A biblioteca metagenômica foi construída para a triagem de clones com atividade celulolítica.
Fragmentos na faixa de 4 – 12 Kb foram clonados em plasmídeo pCF430 (Newman and
Fuqua, 1999) que apresenta resistência a tetraciclina e um sítio PstI de clonagem adjacente ao
gene promotor araC (Figura 28). O plasmídeo pCF430 é um vetor de ampla variedade de
hospedeiros de aproximadamente 10 kb (Figura 28) e apresenta uma origem de replicação e
capacidade de conjugação. Ele apresenta um promotor PBAD associado ao regulador araC, que
são comumente utilizados para controlar a expressão de genes em E.coli.
Figura 28. Plasmídeo pCF430.
3.3.3.1 Extração e purificação do DNA metagenômico
O DNA foi extraído de 5 g (aprox. 0,8 g de cada amostra) da mistura dos 6 tipos de solo
coletados através de lise mecânica, utilizando esferas de vidro (150-212 mícrons), aliado ao
tratamento com CTAB (brometo de hexadecilmetilamônio) e SDS (dodecil sulfato de sódio).
A eficiência da extração foi analisada em gel de agarose a 1% em TBE 1X (p/v). Em seguida,
foi construído um gel para a excisão das bandas contendo os fragmentos de DNA com o

85
tamanho esperado. Os fragmentos foram purificados com o kit Ultra Clean DNA purification
(Mo Bio).
O procedimento detalhado de extração e purificação do DNA metagenômico dos solos está no
anexo 7.1, item 7.1.3.1.
3.3.3.2 Preparação do vetor de clonagem pCF430
O plasmídeo pCF430 foi obtido através de Max Prep utilizando Qiagen Plasmid Maxi kit
(Qiagen) a partir de 50ml de cultura de células. A verificação da extração foi realizada através
de gel de agarose 1 % TBE 1X. Após extração o plasmídeo foi linearizado com enzima de
restrição EcoRI, A reação foi incubada a 37 º C durante 4 horas e posteriormente a 65 º C por
20 minutos. A verificação da linearização do plasmídeo foi realizada através de gel de agarose
1 % TBE 1X.
Após linearização, o plasmídeo foi tratado com fosfatase alcalina (SAP – Shrimp alcaline
phosphatase) para retirada dos resíduos de fosfato que permanecem na molécula de DNA após
a ação da endonuclease.
Os, os vetores foram submetidos à eletroforese em gel de agarose Low melting 1% TAE 1X
(sem brometo de etídeo) durante 4 horas e baixa voltagem (aprox. 40 V). A banda referente ao
plasmídeo circularizado é sacada do gel e usando sistema comercial Qiaex II (Qiagen). A
verificação da obtenção do plasmídeo pCF430 tratado e pronto para ser utilizado nos
experimentos de clonagem foi realizada em gel de agarose 1% TBE 1X.
As reações utilizadas para a preparação do vetor estão no anexo 7.1, item 7.1.3.2.
3.3.3.3 Preparação dos médios insertos
O DNA metagenômico foi submetido a ensaios de restrição parcial com a enzima PstI e em
seguida tratado com fosfatase alcalina para a retirada dos resíduos de fosfato, permitindo a
clonagem de fragmentos de aproximadamente 10 kb no plasmídeo vetor pCF430.

86
Após a purificação, o DNA metagenômico foi submetido à digestão parcial pela enzima de
restrição PstI. A região do gel correspondente aos fragmentos de 4 – 10 Kb foi purificada com
Ultra Clean DNA Purification (Mo Bio).
Foi realizada reação de ligação entre os insertos de 4 – 10 Kb purificados com o plasmídeo
pCF430 tratado.
A transformação foi realizada conforme a seção 3.3.2.3.3, e os clones foram plaqueados em
meio LB contendo tetraciclina (40 mg / mL). A qualidade da biblioteca foi avaliada através de
extração plasmidial (QIAprep Spin Miniprep Kit - Qiagen) de colônias escolhidas
aleatoriamente para reações de restrição com EcoRI. A digestão foi verificada em gel de
agarose 1 % TBE 1 X.
Os clones plaqueados em meio LB foram reunidos e fracionadas em alíquotas de 1 mL em
criotubos após adição de 1/3 de glicerol. O banco metagenômico foi estocado a – 80 º C.
As reações de digestão parcial e clonagem dos insertos estão no anexo 7.1, itens 7.1.3.3 e
7.1.3.4.
3.3.3.4 Análise funcional da Biblioteca Metagenômica do solo da Mata Atlântica
Clones de E.coli da linhagem EPI300, transformadas com plasmídeos (pCF430), contendo
insertos variando entre 4 e 10 kb foram selecionados para busca de atividade celulolítica. A
seleção foi realizada em meio mínimo contendo carboximetil celulose (CMC) e a atividade
celulolítica foi avaliada pelo aparecimento de halos de degradação associado ao crescimento
bacteriano. A determinação da atividade enzimática foi realizada para as atividades FPásica
(feita com papel de filtro), β – glicosidásica (feita com celobiose 2%) e CMCásica (ou
endoglucanásica, feita com CMC 2%). As análises foram realizadas a partir dos estoques
preservados a - 80 ° C.

87
3.3.3.4.1 Triagem de clones com atividade celulolítica
Na triagem para verificação da degradação de celulose, o revelador de vermelho congo foi
adicionado a fim de se avaliar a extensão e a intensidade das zonas hidrolíticas formadas
(Teather and Wood, 1982). Foram adicionados aproximadamente 10 mL de solução aquosa de
Vermelho Congo a 0,1%, as quais ficaram em contato com o meio por aproximadamente 5
minutos. Em seguida, o excesso de vermelho congo foi removido, e as placas foram lavadas
por até 3 vezes com 15 mL de tampão fosfato pH 7,0 a 50 mM com 1M de NaCl. As regiões
onde ocorreu a degradação de celulose, foram evidenciadas pelo aparecimento de zonas claras
(halos de degradação) pois o corante vermelho congo deixava de se associar em função de sua
especificidade por ligações do tipo β- 1,4 glucose.
O procedimento de reativação dos clones bem como a composição do meio CMC está no
anexo 7.1, item 7.1.3.5.
3.3.3.4.2 Determinação da atividade celulolítica
As curvas de crescimento foi construída com o objetivo de determinar a fase log de
crescimentos dos clones onde ocorre maior atividade metabólica das células, e com isso maior
produção de celulases.
Os ensaios para determinação das atividades Fpásica, CMcásica e ß-glicosidásica estão nos
anexos 7.1, itens 7.1.3.6, 7.1.3.7 e 7.1.3.8.
3.3.3.4.2.1 Curva de crescimento
A As curvas foram realizadas em triplicata e medições da densidade ótica da cultura e
alíquotas do sobrenadante foram coletados a cada 3 horas em um total de 16 h de crescimento
em paralelo com a contagem das unidades formadoras de colônias (CFU) em meio CMC. A
biomassa foi calculada ao final do crescimento submetendo as culturas (2 mL) à centrifugação
(14000rpm) durante 5 minutos e secagem do material a 55 ° C por aproximadamente 3 dias.

88
3.3.3.4.2.2 Atividade FPásica (celulose total)
Pedaços de folha de papel de filtro Watmann nº 1 (1x6 cm) foram utilizados como substrato.
Os experimentos foram realizados em duplicata e duas reações controle e um branco foram
utilizados nos ensaios.
3.3.3.4.2.3 Atividade CMCásica (endoglucanásica)
O substrato utilizado foi solução CMC 2 %. Os experimentos foram realizados em duplicata e
duas reações controles e um branco foram utilizados nos ensaios e preparados da seguinte
maneira:
3.3.3.4.2.4 Atividade β-glicosidásica
O substrato utilizado foi solução de celobiose 2 %. Os experimentos foram realizados em
duplicata e duas reações controles e um branco foram utilizados nos ensaios e preparados da
seguinte maneira:
3.4 Diversidade microbiana dos biomas brasileiros
As buscas dos trabalhos publicados foram realizadas através do Pub Med
(http://www.ncbi.nlm.nih.gov/pubmed/) e Web of Science
(http://science.thomsonreuters.com) entre os anos 2005 e 2011. As palavras-chave utilizadas
foram: Brazil, microbiology, bacteria, fungi, archea, microbial, community, diversity,
ecology, environment, marine, aquatic, soil, terrestrial, microbial diversity. Diferentes
combinações dos termos foram utilizadas para maior amplitude das buscas
A classificação taxonômica das sequências do gene rRNA 16S foi realizada através de buscas
no Ribosomal Database Project (RDP) versão 9.21 (Cole et al., 2005).

89
As Análises de coordenadas principais (PCoA) foram realizadas usando FastUniFrac matriz
de distância do pareamento não ponderado. Foram utilizadas apenas sequências de trabalhos
que amplificaram a região V1 e V2 maiores que 400 pb.
As árvores filogenéticas de sequências do gene rRNA 16S não classificadas de ecossistemas
terrestres e aquáticos foram baixada e submetidas no banco de dados RDP. Todas as
sequências foram alinhadas utilizando software Muscle. Análises filogenéticas foram
realizadas com o programa Mega4, utilizando o modelo de neighbor joining. Valores de
bootstrap > 50% são mostrados para os ramos que em uma análise de bootstrap de 1000
repetições.

90
4. RESULTADOS E DISCUSSÃO
4.1 Diversidade da comunidade de bacterioplâncton ao longo de uma gradiente
latitudinal na costa da América Latina por meio de pirosequenciamento da
região V6
4.1.1 Resultados
Com exceção do Estuário do Rio Amazonas (salinidade = 0), todos os locais apresentaram
características de águas marinhas (Tabela 2). A Baía de Guanabara mostra maior
concentração de compostos nitrogenados indicando certo nível de eutrofização do sistema. As
altas concentrações estão possivelmente correlacionadas com o impacto antropogênico nesta
região, como por exemplo, por esgoto doméstico. A diversidade de comunidades microbianas
foi avaliada através da tecnologia 454 de pirosequenciamento, aplicada na região V6 do gene
rRNA 16S. Foram observadas temperaturas mais elevadas nos locais de latitudes baixas. A
caracterização da diversidade microbiana confirmou a existência de um gradiente latitudinal
ao longo da costa da América Latina. Foram obtidas 134.197 sequências de alta qualidade,
com tamanho médio de aproximadamente 60 nucleotídeos das 7 localidades (Tabela 3). Foi
utilizada a ferramenta de análise de dados The Visualization and Analysis of Microbial
Population Structures (VAMPS) (http://vamps.mbl.edu/). Foram observadas diferenças
marcantes entre as 7 localidades estudadas.
Foi observada a diminuição da equitabilidade de acordo com o gradiente latitudinal, com
tendência de maior equitabilidade nas regiões tropicais e menor equitabilidade em áreas
subtropicais (Figura 29 A). Além disso, foi possível observar o enriquecimento de organismos
autotróficos no mesmo sentido do gradiente latitudinal de equitabilidade. Esses dados
sugerem que a produção primária microbiana é maior nas regiões tropicais (ex. Puerto Rico),
onde a abundância foi até 100 vezes maior que nas regiões subtropicais (Ex. Argentinian
shelf) (Figura 29 B).

91
Tabela 2. Parâmetros biológicos e físico-químicos dos locais de amostragem.
# dados não disponíveis; * Dados de Artigas, 2008
Tabela 3. Número de sequências, diversidade e cobertura das bibliotecas da região V6 do gene rRNA 16S das
comunidades microbianas ao longo do gradiente latitudinal.
Puerto
Rico
Guanabara
Bay
Amazonas
Estuary
Uruguay-
Fresh
Uruguay-
Brackish
Rio de
la Plata
Argentinian
shelf
Número de sequências 8384 17631 7220 28666 21468 33145 17817
Estimador Chao (0.03) 2322 2795 1649 4850 3931 2660 1323
Cobertura de Good (%) 92 95 93 95 94 97 97
As sequências foram distribuídas em 36 filos. Os filos mais comuns nos sete locais foram
Proteobacteria, Bacteroidetes, Cyanobacteria, e Actinobacteria (Figura 30), correspondendo
a aproximadamente 80% do número total de sequências. Cyanobacteria e Bacteroidetes
Puerto Rico
Amazon
estuary Guanabara Bay
Laguna de
Rocha (1 e 2)
Rio de la
Plata
Argentinia
n Shelf
Data 22/09/2008 26/11/2007 22/09/2008 25/04/2008 29/06/2006 01/09/2006
Latitude 17°97’N 0°70’S 22°56’S 34°39’S 36°17’S 38°69’S
Longitude 67°01’W 50°29’W 43°09’W 54°16’W 55°26’W 57°68’W
Profundidade (m) 0.1 0.1 0.5 0.5-0.6 2 5
Temperatura (°C) 26.0 27.5 23.0 19.0-20.0 12.9 10.6
Salinidade 35.5 0.0 34.0 14.7-16.0 32.0 34.0
NH4 (µM) 3.1* 3.89 23.4 0.0-1.7 3.13** < 2.0
NO2+NO3 (µM) 0.25* 0.648 5.1 1.61-0.245 < 1.0** 2.13
Nitrogênio total (µM) # # 66.3 7.13-23.63 # #
PO4 (µM) 0.05* 3.79 2.2 1.097-0.54 0.6* 0.77
Fósforo total (µM) # # 2.9 5.16-2.69 # #
Silicato (µM) 2.3* # 19.8 65-30 3.0* 4.02
Chlorofila a (µg/L) 2.00 1.27 4.0 7.7-13.4 0.5* #
Contagem de bactérias (x106
células.mL-1
) 1.50 0.67 (1.34) 3.5 2.4-1.8 1.00 0.75
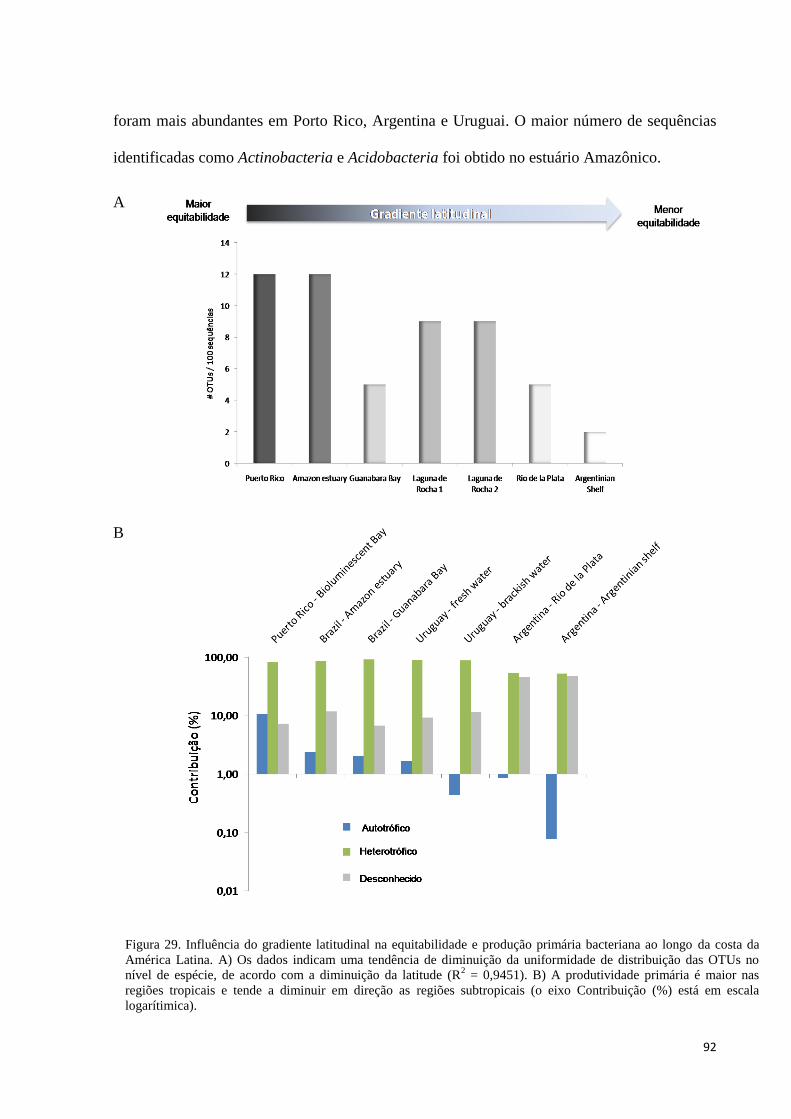
92
foram mais abundantes em Porto Rico, Argentina e Uruguai. O maior número de sequências
identificadas como Actinobacteria e Acidobacteria foi obtido no estuário Amazônico.
A
B
Figura 29. Influência do gradiente latitudinal na equitabilidade e produção primária bacteriana ao longo da costa da
América Latina. A) Os dados indicam uma tendência de diminuição da uniformidade de distribuição das OTUs no
nível de espécie, de acordo com a diminuição da latitude (R2 = 0,9451). B) A produtividade primária é maior nas
regiões tropicais e tende a diminuir em direção as regiões subtropicais (o eixo Contribuição (%) está em escala
logarítimica).

93
A fração de sequências que se conserva ao longo do gradiente latitudinal corresponde a
aproximadamente 58% de todas as sequências identificadas. Vinte e um grupos foram
identificados em todas as amostras (Figura 31; Tabela 4). Alguns destes táxons foram
tentativamente classificados em grupos taxonômicos conhecidos como Acinetobacter,
Flavobacteriaceae, SAR11 e Enterobacteriaceae, de acordo com a classificação taxonômica
do banco de dados VAMPS. A classificação definiu grupos mais profundos mas nenhum
desses táxons foi identificado comoespécies.
A fração de OTUs (OTU é definida com um grupo de sequências da região V6 do rRNA 16S
com > 97 % de similaridade) comuns entre os sete locais é de pelo menos 0,6 %, chegando até
93 % (entre as duas amostras da Argentina) da sua microbiota, sugerindo ampla dispersão
microbiana no meio marinho (Figura 32). A fração endêmica das sequências, ou seja, OTUs
Figura 30. Estrutura das comunidades bacterianas nos sete locais. Os filos mais comumente encontrados são
indicados. Sequências Unknown representam aquelas as quais não foi possível atribuir uma classificação
taxonômica pelo VAMPS.

94
com ocorrência limitada a apenas uma localidade, correspondeu a apenas uma pequena
parcela (2%) de todas as sequências. Os táxons endêmicos foram distribuídos em um total de
245 grupos. Os primeiros 40 táxons endêmicos mais abundantes em todo o conjunto de dados
são listados (Tabela 5).
Figura 31. Número de sequências identifiicadas na fração conservada do gradiente latitudinal. Nível
taxonômico mais profundo identificados pelo VAMPS.
Figura 32. Fração de sequências comuns de um local para outro usando 97% de similaridade para definir OTUs.

95
Tabela 4. Distribuição e identificação da fração cosmopolita do gradiente latitudinal. Os números representam a
fração de sequências agrupadas por táxons em relação ao total de seqüências classificadas pelo GAST. A
identificação foi possível em diferentes níveis taxonômicos.
Identificação
Taxonômica
Puerto
Rico
(%)
Brazil
Amazon
estuary
(%)
Brazil
Guanabara
Bay
(%)
Urugauy
Laguna do
Rocha 1
(%)
Uruguay
Laguna do
Rocha 2
(%)
Argentina
Rio de la
Plata
(%)
Argentina
Argentinian
shelf
(%)
Unknown 0,211 0,171 0,268 4,429 4,604 1,132 1,068
Gammaproteobacteria
(Classe) 1,810 0,008 3,146 2,161 1,403 1,627 1,644
Flavobacteriaceae
(Família) 0,354 0,001 0,427 0,853 2,425 3,125 2,061
Alphaproteobacteria
(Classe) 0,519 0,885 1,000 0,816 0,286 3,305 2,148
Actinobacteria
(Filo) 0,220 0,589 0,416 0,167 0,006 0,987 0,508
Flavobacteriales
(Ordem) 0,148 0,013 0,208 0,136 0,386 0,429 0,660
SAR11
(Ordem) 0,104 0,183 0,179 0,618 0,399 0,154 0,057
Bacteroidetes
(Filo) 0,126 0,099 0,155 0,232 0,009 0,369 0,334
Methylophilus
(Gênero) 0,082 0,001 0,172 0,107 0,006 0,538 0,389
Proteobacteria
(Filo) 0,035 0,118 0,144 0,157 0,004 0,338 0,287
Rhodospirillales
(Ordem) 0,068 0,013 0,327 0,151 0,052 0,227 0,165

96
Tabela 5. Distribuição e identificação da fração endêmica ao longo do gradientelatitudinal. *PR – Puerto Rico;
AM –Amazon estuary; GB – Guanabara Bay; LG1 – Laguna do Rocha 1; LG2 –Laguna do Rocha 2; RP – Rio
de la Plata and AS – Argentinian shelf .
Identificação taxonômica
Número de
sequências
Número de
unique Localização*
Fração do conjunto de
dados (%)
Neisseria
(Gênero) 492 50 GB 0,3916
Enterobacteriaceae
(Família) 0,002 0,004 0,410 0,381 0,034 0,064 0,041
Cyanobacteria
(Filo) 0,390 0,043 0,132 0,087 0,005 0,219 0,033
Bacteria
(Domínio) 0,053 0,116 0,214 0,357 0,032 0,075 0,048
Burkholderiales
(Ordem) 0,007 0,197 0,060 0,001 0,003 0,428 0,175
Acinetobacter
(Gênero) 0,001 0,019 0,045 0,227 0,264 0,007 0,012
Fluviicola
(Família) 0,020 0,010 0,004 0,012 0,055 0,250 0,039
Sphingobacteriales
(Ordem) 0,008 0,074 0,032 0,081 0,117 0,046 0,008
Opitutus
(Gênero) 0,021 0,042 0,051 0,011 0,004 0,018 0,004
Ralstonia
(Gênero) 0,004 0,015 0,077 0,001 0,001 0,003 0,003
Prochlorales
(Ordem) 0,047 0,015 0,017 0,001 0,002 0,003 0,009
Planctomycetaceae
(Família) 0,036 0,001 0,014 0,003 0,003 0,016 0,012

97
Gp8
(Gênero) 204 31 AM 0,1623
Curvibacter
(Gênero) 87 10 AM 0,0692
Rubrobacter
(Gênero) 60 8 GB 0,0477
Cryobacterium
(Gênero) 53 6 LR1 0,0421
Solibacteraceae
(Família) 39 5 LR1 0,0310
Neisseria gonorrhoeae
(Espécie) 35 1 GB 0,0278
Terrimonas
(Gênero) 30 3 AM 0,0238
Herbaspirillum
(Gênero) 25 4 AM 0,0199
Owenweeksia hongkongensis
(Espécie) 24 3 GB 0,0191
Tenacibaculum
(Família) 20 7 LR2 0,0159
Roseateles
(Gênero) 13 3 AM 0,0149
Pseudoalteromonas elyakovii
(Espécie) 12 3 LR1 0,0095
Chitinophaga sancti
(Espécie) 10 4 AM 0,0079
Dorea
(Gênero) 9 4 GB 0,0071
Thalassobacter
(Gênero) 9 4 LR2 0,0071
Chloroflexi 8 1 PR 0,0063
98
(Filo)
Kordiimonas
(Gênero) 8 1 RP 0,0063
Dietzia
(Gênero) 7 3 GB 0,0055
Fusibacter
(Gênero) 7 2 AS 0,0055
Turneriella
(Gênero) 6 4 LR1 0,0047
Aquimarina brevivitae
(Espécie) 5 3 RP 0,0039
Faecalibacterium
(Gênero) 5 2 GB 0,0039
Cystobacteraceae
(Família) 5 4 AM 0,0039
Klebsiella
(Gênero) 5 3 GB 0,0039
Gemmata
(Ordem) 4 1 AM 0,0031
Beijerinckiaceae
(Família) 4 3 AM 0,0031
Hydrogenophilus
(Gênero) 4 3 AM 0,0031
Desulfosarcina
(Gênero) 4 1 PR 0,0031
Methylococcaceae
(Família) 4 2 AS 0,0031
Pseudomonas fluorescens
(Espécie) 4 5 LR1 0,0031
Kocuria
(Gênero) 3 3 GB 0,0023

99
Parabacteroides
(Gênero) 3 3 GB 0,0023
Myroides
(Gênero) 3 2 AS 0,0023
Wautersiella
(Gênero) 3 1 GB 0,0023
Atopococcus tabaci
(Espécie) 3 4 AM 0,0023
Lactobacillus luminis
(Espécie) 3 1 GB 0,0023
A identificação taxonômica fornecida pelo VAMPS indicou que as OTUs identificadas como
Acidobacteriaceae GP8, Methylophilaceae, Curvibacter, Terrimonas, Aquitalea e
Hongkongensis Fangia só aparecem no estuário do Rio Amazonas. Bactérias potencialmente
patogênicas, tais como Neisseria foram observadas na Baía de Guanabara. A maioria dos
endêmicos foi tentativamente identificada no nível de gênero, e somente oito táxons foram
identificados em espécies conhecidas. A grande maioria dos endêmicos foi obtida no Brasil
(Tabela 5). O estuário Amazônico é muito diferente dos outros locais, compartilhando a
menor fração de suas sequências com outras localidades.
Foram construídas curvas de rarefação para verificação da cobertura da diversidade amostrada
para o nível taxonômico de espécie (Figura 33A). Além disso, os estimadores não
paramétricos de diversidade Good e Chao também foram utilizados para verificar a cobertura
da diversidade estimada pela amostragem. A estimativa da cobertura variou entre 92 e 97%
com o número de espécies estimado entre 1323 (Argentinian Shelf) e 4850 (Uruguay –
Laguna do Rocha 1) (Tabela 3). O número de OTUs no nível de espécie (similaridade > 97
%) encontradas variou entre 1000 e 2500, enquanto o número de famílias oscilou entre 287 e
966 (Figura 33B). Os locais de água doce do estuário do Uruguai e do Rio de la Plata

100
apresentaram maior número de sequências únicas. Esses locais também tiveram o maior
número de sequências analisadas. Em todos os sete locais, observou-se cauda nas curvas de
abundância de classificação, indicando a presença de um grande número de organismos raros
em todos os locais, corroborando que as comunidades do bacterioplâncton apresentam alta
equitabilidade, de uma forma geral (Figura 34).

101
Figura 33. Análise de agrupamento das sequências. A ) Curvas de rarefação; B) Número de OTUs a diferentes
níveis de similaridade correspondente a diferentes níveis taxonômicos (linhagens, espécies, gêneros e famílias).

102
A capacidade de resolução taxonômica da região V6 foi avaliada através da análise
filogenética comparativa de dois gêneros de Cyanobacteria filogeneticamente relacionados,
Procholrococcus e Synechococcus (Figura 35). Foram comparadas as sequências das
diferentes cópias do gene rRNA 16S de ambos os gêneros, observando-se baixa resolução
taxonômica. Sequências de referência de Synechococcus agruparam com sequências de
Prochlorococcus. Além disto, as sequências obtidas neste estudo e identificadas pelo VAMPS
como Prochlorococcus também agruparam de maneira difusa em ambos os gêneros. Apesar
da resolução taxonômica da região V6 ser limitada para alguns grupos taxonômicos, vários
estudos vem demonstrando a utilidade desta estratégia para estudos abrangentes de
diversidade.
Figura 34. Análise baseada na distribuição de filos, demonstrando a existência de táxons dominantes
distribuídos por poucos grupos taxonômicos. Por outro lado, mostra a contribuição de um grande número de
táxons em baixa frequênciaque representa a biosfera rara da microbiota aquática.

103
Pela primeira vez na America Latina, foi determinada a diversidade das comunidades
microbianas em um gradiante latitudinal empregando pirosequenciamento da região V6. Foi
observado que a microbiota da água tem alta dispersão, com grande parte da microbiota tendo
distribuição cosmopolita na água. A maior proporção de endêmicos na região Amazônica se
deve possivelmente a contribuição de microrganismos de origem terrestre.
P ro c h l o ro c o c c u s T a g 1 4
P ro c h lo r o c o c c u s T a g 1 5
P ro c h l o ro c o c c u s T a g 5
P ro c h l o ro c o c c u s T a g 1 1
P ro c h l o ro c o c c u s T a g 6
P r o c h lo ro c o c c u s T a g 7
P r o c h lo ro c o c c u s T a g 8
P ro c h l o ro c o c c u s m a ri n u s s t r. M IT 9 3 0 1
P r o c h lo r o c o c c u s m a ri n u s s t r . A S 9 6 0 1
P ro c h l o ro c o c c u s T a g 2
P ro c h l o ro c o c c u s T a g 4
P ro c h l o ro c o c c u s T a g 3
P r o c h lo r o c o c c u s T a g 1 2
P r o c h lo r o c o c c u s T a g 1 3
P ro c h l o ro c o c c u s m a ri n u s s t r. M IT 9 2 1 1
P ro c h l o ro c o c c u s m a ri n u s s t r. N A T L 1 A
C y a n o b a c te ri a T a g 7
P ro c h lo r o c o c c u s T a g 1
P ro c h lo r o c o c c u s T a g 1 0
S y n e c h o c o c c u s s p . R C C 3 0 7
S y n e c h o c o c c u s s p . W H 7 8 0 3
S y n e c h o c o c c u s s p . W H 7 8 0 3 (2 )
C y a n o b a c te r ia T a g 1 4
P r o c h l o ro c o c c u s m a ri n u s s t r. M IT 9 3 0 3
C y a n o b a c te ri a T a g 9
C y a n o b a c te ra T a g 1 5
C y a n o b a c te r i a T a g 6
S y n e c h o c o c c u s s p . C C 9 3 1 1
S y n e c h o c o c c u s s p . C C 9 3 1 1 (2 )
S y n e c h o c o c c u s s p . C C 9 6 0 5 (2 )
S y n e c h o c o c c u s s p . C C 9 6 0 5
C y a n o b a c te ri a T a g 3
C y a n o b a c te ri a T a g 8
C y a n o b a c te r ia T a g 4
C y a n o b a c te r ia T a g 4 (2 )
P ro c h lo r o c o c c u s T a g 9
S y n e c h o c o c c u s e l o n g a tu s P C C 7 9 4 2
S y n e c h o c o c c u s e l o n g a tu s P C C 7 9 4 2 (2 )
C y a n o b a c te r i a T a g 1
C y a n o b a c te ri a T a g 2
S y n e c h o c o c c u s s p . P C C 7 0 0 2
S y n e c h o c o c c u s s p . P C C 7 0 0 2 ( 2 )
C y a n o b a c te ri a T a g 1 1
C y a n o b a c te r ia T a g 1 0
C y a n o b a c te r ia T a g 1 2
C y a n o b a c te r ia T a g 1 3
A n a b a e n a va ri a b i l is A T C C 2 9 4 1 3
A n a b a e n a va ri a b i l is A T C C 2 9 4 1 3 (3 )
A n a b a e n a va ri a b i l is A T C C 2 9 4 1 3 (2 )
A n a b a e n a va ri a b i l is A T C C 2 9 4 1 3 (4 )
A c i d o b a c t e ri u m c a p s u la t u m A T C C 5 1 1 9 6
B o rr e li a d u t to n i i L y9 9
9 9
1 0 0
8 8
9 6
9 3
5 4
7 6
7 8
6 0
5 7
7 2
6 0
7 1
7 6
5 65 9
8 4
8 9
6 2
0 . 1

104
Figura 35. Análise filogenética demonstrando a resolução taxonômica da região v6 do gene rRNA 16S para os
gênero relacionados Prochlorococcus e Synechococcus. Foi utilizado método neighbor join, corrigido pelo
modelo Juckes-Cantor e bootstrap com 1000 replicatas.
4.1.2 Discussão
Este estudo traz novas informações sobre a diversidade de bacterioplâncton de águas
costeiras da América Latina. Nós descrevemos a estrutura do bacterioplâncton costeiro
ao longo de um gradiente latitudinal, abrangendo sete locais distintos, principalmente no
meio marinho. O pirosequenciamento da região hipervariável V6 correspondentes às
posições 984-1047 (sequência rRNA 16S de Escherichia coli) resultou em um número
ineditamente elevado de sequências que foram utilizadas para identificar a diversidade de
bactérias planctônicas da América Latina.
Padrões globais de distribuição de microrganismos ainda são pouco conhecidos e os fatores
envolvidos ainda precisam ser mais profundamente investigados. Sabe-se que os aspectos
ecológicos e evolutivos são os principais fatores que influenciam nos padrões de distribuição
de microrganismos. A existência de um gradiente latitudinal foi identificada recentemente
para o bacteriplâncton de águas superficiais dos oceanos (Fuhrman et al., 2008; Pommier et
al., 2007). Os principais mecanismos envolvidos para o estabelecimento de um gradiente
latitudinal são a temperatura e a produtividade primária. Esses mecanismos não são
excludentes. Os dados deste trabalho mostram que ambos estão atuando sobre as
comunidades de bacterioplâncton ao longo da costa da América Latina. Por um lado, as
maiores temperaturas nas regiões tropicais influenciam na cinética dos processos
biológicos, agindo no estabelecimento de grupos microbianos. Por outro, a maior
abundância de microrganismos autotróficos nos trópicos sugere uma maior
produtividade primária microbiana. Entretanto, medidas de produtividade (como taxa de
respiração e fotossintética) precisam ser realizadas para confirmação desta hipótese.

105
Ecossistemas mais produtivos são capazes de suportar um maior número e tipos de
organismos adaptados as condições locais. A energia é um bom preditor de riqueza de
espécies de plantas e animais (Rohde, 1992). Os resultados mostram que a predominância
de organismos autotróficos no bacterioplâncton varia de acordo com o gradiente
latitudinal de diversidade. Isso sugere que a produção primária pode ser utilizada como
um preditor de diversidade ao longo da costa da America Latina.
Em geral, a mais profunda resolução taxonômica obtida foi no nível de gênero. Estudos
anteriores também sugeriram que a resolução taxonômica das sequências V6 pode permitir a
discriminação de gêneros afins, mas não de espécies relacionadas (Huber et al., 2007; Huse et
al., 2007). O número de OTUs, correspondente a espécies, em sete locais variou entre 937
e 1946. Estas estimativas podem ser consideradas conservativas, pois espécies diferentes
podem ter sequências idênticas nas regiões V6. Apenas um número reduzido de sequências
foi atribuído a espécies formalmente conhecidas. Aproximadamente, 11% do número total de
sequências conservadas foram classificadas como Gammaproteobacteria sem espécies
conhecidas ou identidade de gênero. À primeira vista, os resultados sugerem que existe
uma grande diversidade de táxons novos que circulam nas áreas de estudo à espera de
ser formalmente descrita, mas claramente, mais sequências rRNA 16S mais longas são
necessárias para estabelecer a posição exata dos novos grupos taxonômicos, evitando os
inconvenientes da atual metodologia quanto à sua relativamente baixa resolução taxonômica.
As sequências de região V6 identificadas como Prochlorococcus foram indistinguíveis
daquelas sequências tentativamente identificadas como Synechococcus e Cyanobacteria,
sugerindo que uma fração das sequências de V6 de Cyanobacteria pode ser realmente
relacionada com Prochlorococcus. Não é inesperado que esses grupos não possam ser
diferenciados porque o gênero Synechococcus é um dos vizinhos mais próximos de
Prochlorococcus são diferenciados através da aplicação da metodologia ARISA (Automated

106
rRNA Intergenic Spacer Analysis) (Rocap et al., 2002). O fato de a região V6 não permitir
a diferenciação de gêneros pode ilustrar uma resolução taxonômica limitada. Por outro
lado, os filos proximamente relacionados (Acidobacteria, Proteobacteria) foram
claramente diferenciados dos grupos Prochlorococcus / Cyanobacteria a partir das
sequências da região V6.
Uma parcela significativa das OTUs parece ser conservada ao longo do gradiente
latitudinal, abrangendo entre Puerto Rico e Argentina, de estuários de águas salobras a
marinhas. Todos os sete locais analisados neste estudo podem ser considerados ricos em
nutrientes. Entretanto, o aporte de nitrogênio na Baía de Guanabara é aparentemente
enriquecido pelo despejo de efluentes domésticos e industriais.
A temperatura parece ser o parâmetro mais variável entre os pontos, variando entre 10,6 e
27,5°C. Alguns táxons conservados foram classificados em grupos taxonômicos
conhecidos, ou seja, SAR11. Este grupo é dominante nos ambientes marinhos (Brown et al.,
2009; Morris et al., 2002). Acinetobacter também foi observada entre os grupos conservados
que apresentam versatilidade metabólica e foi recuperado em diferentes biomas, incluindo do
Mar Antártica, Região sub-saariana, e Amazônia (Demba Diallo et al., 2004; O’Neill et al.,
2009; Van Trappen et al., 2002). Acinetobacter também tem sido implicado com infecções
nosocomiais em diferentes países (Towner, 2009).
Um estudo recente sobre a diversidade de Bactérias e Arquéias do Oceano Ártico mostrou que
as espécies raras (ou então chamadas filotipos) não têm uma distribuição conservada (Galand
et al., 2009). No presente estudo, utilizando critérios mais rigorosos para a definição do
táxon (97% de identidade em vez de 94% de identidade do estudo no Ártico),
mostramos que várias OTUs consideradas conservadas (tentativamente identificado
como Fuviicola, Sphingobacteriales, Cyanobacteria, e Prochlorales) aparecem em baixa
frequência (<1%). A baixa freqüência pode ser explicada, em parte, por causa da amplitude

107
latitudinal de nosso conjunto de dados, a partir de 10 N a 38 S, e assim com uma grande
mudança nos regimes de temperatura e ambiente. A ampla distribuição destas bactérias pode
ser explicada pela sua plasticidade genômica, permitindo sua sobrevivência em condições
ambientais variáveis.
OTUs endêmicas representaram apenas 1,96% de todas as sequências. A maioria destas
sequências foi tentativamente identificada apenas no nível de gênero. Apesar da baixa
abundância, OTUs endêmicas podem desempenhar um papel fundamental no meio
ambiente. Mudanças nos regimes biogeoquímicos podem determinar variações na
comunidade microbiana, e os grupos dominantes podem surgir a partir da biosfera rara.
O maior endemismo foi observado em locais brasileiros. Várias OTUs endêmicas
(tentativamente identificada como Acidobacteriaceae GP8 e Terrimonas), que apareceram
apenas nas regiões estuarinas amazônicas, são microrganismos típicos do solo, possivelmente
levados pelo rio Amazonas em direção ao Oceano Atlântico. Outros parâmetros físicos
(temperatura, por exemplo) também podem influenciar a diversidade da comunidade
bacteriana. Uma OTU tentativamente designada como pertencente ao gênero psicrófilo
Cryobacterium foi a mais abundante nos locais com as temperaturas mais baixas ao longo do
gradiente latitudinal.
Algumas OTUs endêmicas encontradas na Baía de Guanabara tentativamente
identificadas como Neisseria refletem um ambiente impactado por ação antropogênica.
A Baía de Guanabara é uma conhecida área impactada e intensamente urbanizada, onde
vivem aproximadamente 8 milhões de pessoas em seu entorno. Este local teve a maior
quantidade de nutrientes no presente estudo. Descargas de nutrientes orgânicos e inorgânicos
podem promover o crescimento de bactérias de crescimento rápido. Ambientes ricos em
nutrientes tendem a favorecer o crescimento de bactérias patogênicas oportunistas e resultam
na perda de homeostase (Dinsdale et al., 2008b).

108
Além de parâmetros químicos, o estado autotrófico dos diferentes locais pode influenciar a
riqueza de espécies (Fuhrman et al., 2008). Aumento da produtividade primária resulta na
entrada de matéria e energia no sistema, permitindo o crescimento microbiano e
diversificação. Houve um aumento acentuado nos autótrofos (Cyanobacteria) em Porto
Rico, sugerindo uma maior atividade fotossintética.
Apesar das controvérsias em relação ao pirosequenciamento da região V6 para avaliar a
diversidade bacteriana e identificação das espécies (Claesson et al., 2009; Quince et al.,
2009), o presente estudo é uma primeira tentativa de levantamento da diversidade de
bactérias em áreas costeiras do Caribe e no Atlântico Sul. A divulgação de conservação de
grupos de bactérias é um achado importante que sugere novos estudos. Outro aspecto
interessante deste trabalho é o possível aparecimento de organismos indicadores dentro da
fração endêmica, sugerindo que áreas impactadas, como a Baía de Guanabara, podem
influenciar a composição da comunidade bacteriana.
Trabalhos futuros serão necessários para determinar as relações entre os parâmetros
ambientais (por exemplo, nutrientes) e mudanças temporais da comunidade microbiana de
composição no Atlântico Sul Ocidental.
4.2 Avaliação da saúde do Banco de Abrolhos através da metagenômica
4.2.1 Resultados
Neste segundo capítulo foi realizado o estudo da diversidade microbiana em uma área
geográfica mais restrita do que a abordada no capítulo anterior: o Banco dos Abrolhos. Foi
determinada a diversidade microbiana em um dos principais hotspots de diversidade do
Atlântico Sul, bem como o efeito de áreas marinhas protegidas na abundância e diversidade
microbiana.

109
A abundância de bactérias totais variou entre 4,88 x 105 e 6,62 x 10
5 células / mL (Tabela 6).
A menor contagem de células ocorreu nas áreas protegidas (California e Parcel dos Abrolhos).
As contagens de Vibrios foram maiores nos recifes desprotegidos, variando entre 102 e 10
4
UFC / mL. A contagem de Vibrios variou entre 0 e 102 UFC / mL nos recifes protegidos. Os
recifes protegidos (Parcel dos Abrolhos e California) apresentam maiores concentrações de
COD que as áreas desprotegidas (p < 0,001) (Figura 36).
Tabela 6. Características gerais dos locais de coleta e dos metagenomas. Média ± erro padrão. Número de replicatas entre
parênteses. DOC (Carbono orgânico diddolvido); POC (Carbono orgânico particulado).

110
A B
C D
Figura 36. Comparação dos valores dos parâmetro Carbono Orgânico Dissolvido (COD) (acima) e Clorofila
(abaixo) entre os recifes protegidos e desprotegidos. Foi observada maior concentração de COD nos recifes
protegidos. A) ANOVA e B) Visualização gráfica dos resultados do teste de Tukey. PAB - Parcel dos Abrolhos;
SG - Sebastião Gomes; TIM – Timbebas e CAL – California. . Comparação dos valores do parâmetro Clrofila
entre os recifes mostra maiores concentrações no recife protegido. A) ANOVA e B) Visualização gráfica dos
resultados do teste de Tukey. PAB - Parcel dos Abrolhos; SG - Sebastião Gomes; TIM – Timbebas

111
O nível de clorofila é um indicativo do nível de autotróficos na área. O recife Parcel dos
Abrolhos apresentou maiores concentrações que os recifes desprotegidos, principalmente
entre Parcel dos Abrolhos e Sebastião Gomes (p < 0,001) (Figura 36 C). Através do teste de
Tukey, observa-se que os três recifes são diferente entre eles (Figura 36 D).
O número total de sequências geradas variou entre 10.906 (Sebastião Gomes em 2009) e
167.513 (California, em 2009) (Tabela 6). O número total de sequências identificadas variou
entre 4.224 e 67.018, através de Blastx com um valor de corte (E-value) de 10-5
, contra o
banco de dados MG-RAST. Foi observada maior abundância relativa de sequências
identificadas como Arquéia (3-7% de contribuição) e vírus (3-7% de participação) nos recifes
desprotegidos, em comparação com os recifes protegidos (1% de contribuição) (Figura 37 A).
A Arquéia Maripaludis Methanococcus foi identificada como a espécie mais frequente em
Sebastião Gomes (N = 75; 42% das Arquéias). Methanococcoides burtonii foi abundante em
Timbebas (N = 148; 5%), enquanto Archaeoglobus fulgidus foi abundante na California (N =
47; 5%). A fração de vírus identificada nos metagenomas consistiu principalmente de
Cyanophages (N = 95, 84% em Sebastião Gomes, N = 404, 61% em Pedra de Leste; N = 693,
38% em Timbebas; N = 731, 68% no Parcel dos Abrolhos, e N = 610, 70% na California). Os
principais tipos de fagos foram fagos de Prochlorococcus. É possível observar maior
contribuição de sequências associadas a vírus nos recifes não protegidos (Figura 37 A).

112
A
B
C
Figura 37. Estrutura da comunidade microbiana dos recifes de Abrolhos. A). Contribuição dos diferentes
domínios. Enriquecimento de vírus é observada em amostras de recifes desprotegidos. B) Nível trófico dos
microrganismos dos cinco pontos. A classificação foi realizada com base na identificação dos filos.
Enriquecimento do metabolismo autotrófico é observado nos recifes protegidos. C) Distribuição dos patógenos
comumente encontrados. A classificação foi realizada baseada na identificaçãodas taxonômica espécies/
linhagens. A contribuição da Vibrios está relacionada a sequências atribuídas como Gammaproteobacteria. N
corresponde ao número total de sequências para cada local

113
O número de sequências identificadas como bactérias variou entre 92% (N = 5.297) em
Timbebas e 97% (N = 87.683), em California. Proteobacteria foi o filo mais abundante em
cada local: Sebastião Gomes (63%, N = 1.212), Pedra de Leste (76%, N = 15.160), Timbebas
(76%, N = 79.740), Parcel dos Abrolhos (57%, N = 49.756) e California (68%, N = 58.812)
(Figura 38 A). Gammaproteobacteria correspondeu a 21-45% das sequências em todos os
recifes. Deltaproteobacteria foram mais abundantes (8%) no recife de Sebastião Gomes
desprotegidos do que em proteger recife California (1%). A Proteobacteria Pelagibacter
ubique foi a mais abundante nos recifes desprotegidos (11 a 16%, N = 1311-12758), mas
contribuiu com apenas 2-8% em locais protegidos (Figura 38 B). Pelagibacter ubique,
Alteromonas macleodii, Synechococcus sp. CC9605 e Gammaproteobacteria KT71 estavam
presentes nos cinco recifes. Alteromonas macleodii foi a espécie mais abundante nos recifes
protegidos (10 a 24% no Parcel dos Abrolhos e da California, N = 8.729 e 20.757,
respectivamente). Por outro lado, se encontra em baixa frequência em Sebastião Gomes
(0,2%, N = 238). Não foram observadas diferenças significativas entre os índices de
diversidade entre a composição das comunidades de Arquéia, vírus e Bactéria dos recifes
protegidos e não protegidos. Porém, houve tendência de menor riqueza de espécies em
Sebastião Gomes (área não protegida), sugerindo que a diversidade microbiana tem papel
importante nas áreas marinhas protegidas, promovendo a saúde ambiental.
Sequências relacionadas aos sistemas autotróficos foram mais abundantes nos recifes
protegidos (9 a 21%, no Parcel dos Abrolhos e da California, N = 7.856 a 18.162) do que nos
recifes desprotegidos (5-7% em Pedra de Leste e Timbebas, N = 1101 a 5581) (Figura 37 B).
Em comparação, a contribuição de sequências relacionadas ao metabolismo heterotrófico foi
maior nos recifes desprotegidos, alcançando 93% em Pedra de Leste. Sequências classificadas
como Bacteroidetes foram encontradas em todos os cinco recifes, mas a contribuição mais
baixa foi no recife California (3%, N = 2594). A sua contribuição chegou a 24% (N = 20.950)

114
em Parcel dos Abrolhos. Actinobacteria correspondeu a cerca de 3% nos recifes
desprotegidos e 2% em recifes protegidos (Figura 38 A). A maior abundância de sequências
relacionadas a patógenos humanos (7-11%), patógenos animais (6-8%), e Vibrios (5-7%)
foram observadas nos metagenomas dos recifes desprotegidos (Fig. 37 C). Entre 5 e 8% de
todas as sequências de patógenos humanos foram identificados como Pseudomonas
mendocina em todos os locais.
Os metagenomas foram classificados em 24 subsistemas envolvidos com ampla diversidade
de funções metabólicas (Figura 39). Os cinco processos mais abundantes (carboidratos,
aminoácidos e derivados, proteínas, co-fatores e virulência) contribuíram com mais de 50%
de todas as sequências do DNA metagenômico anotado. O número total de sequências
identificadas variou entre 4.224 a 67.018 em Sebastião Gomes e California (Tabela 3).
Diferença entre os recifes desprotegidos e protegidos ocorreu apenas para a síntese de
macromoléculas e a fotossíntese.
Os recifes protegidos mostraram maior abundância de subsistemas envolvidos na fotossíntese
(Figura 40 A). A contribuição total de subsistemas identificados envolvidos com a
fotossíntese variou de 0,3 a 0,97% no conjunto de dados (Tabela 8). Fotossistemas I e II
foram mais abundantes nos recifes protegidos (0,17-0,18 e 0,28 e 0,4%, respectivamente) do
que nos recifes desprotegidos (0,02 a 0,07% e 0,17 a 0,14, respectivamente). Sequências do
subsistema ficobilisoma foram mais abundantes nos recifes protegidos (0,07 a 0,3%) do que
nos recifes desprotegidas (entre 0,005 e 0,03%). Por outro lado, a contribuição de sequências
do subsistema proteorodopsina foi semelhante em todos os locais (0,04 a 0,1%). A maioria
das sequências de proteorodopsinas foram relacionadas com Pelagilbacter ubique (SAR11),
Vibrionaceae e Flavobacteriales (Figura 40 B).

115
A
B
Figura 38. Estrutura das comunidades microbianas nos níveis de A) Filos e B) espécies / linhagens mais
frequentes (contribuição >1% do conjunto de sequências da amostra). A classificação taxonômica foi
realizada usando o servidor Mg-Rast. A identificação taxonômica é realizada baseada no resultado do
BLAST X realizado contra as sequências do banco de dados SEED (banco de genomas completos). A
origem taxonômica do melhor hit para a proteína identificada foi relacionada a abundância do táxon
identificado. N é o número de sequências utilizadas nas análises.

116
A fim de determinar a contribuição dos diferentes microrganismos para os diferentes tipos de
subsistemas, seis subsistemas (carboidrato, fósforo, nitrogênio, virulência, resposta ao
estresse, e fotossíntese), foram submetidos à identificação taxonômica utilizando MG RAST
(Figura 41). Esses subsistemas foram selecionados porque eles são relevantes para o
funcionamento do recife de coral. Alguns subsistemas (como carboidratos e resposta ao
estresse) foram mais generalistas, em diferentes grupos taxonômicos, enquanto outros
subsistemas (por exemplo, de fotossíntese) ficaram restritos a poucos grupos taxonômicos. As
principais vias de cada um dos seis subsistemas pertenciam a diferentes processos celulares
(Tabela 8).
Figura 39. Contribuição dos subsistemas (hierarquia 1) nos diferentes recifes. As barras indicam a contribuição das
sequências para cada subsistema nos recifes protegidos e não protegidos. A coluna contribuição é relativa ao
número total de sequências identificadas para cada subsistema. Somente sequências informativas foram utilizadas
para identificação subsistemas. Sequências atribuído como subsistemas miscelaneus, unknown e clustered based
subsystems não foram incluídos nas análises.

117
Figura 40. Subsistemas do metabolismo de fotossíntese (média dos anos 2009-2010). A) Contribuição dos
subsistemas (hierarquia 3) relativa a todas as sequências anotadas pelo Mg-Rast. Os subsitemas mostraram
diferença (p<0,5; CI 95%) entre os recifes protegidos e não protegidos. B) Contribuição de diferente táxons para
o subsistema das proteorodopsinas.
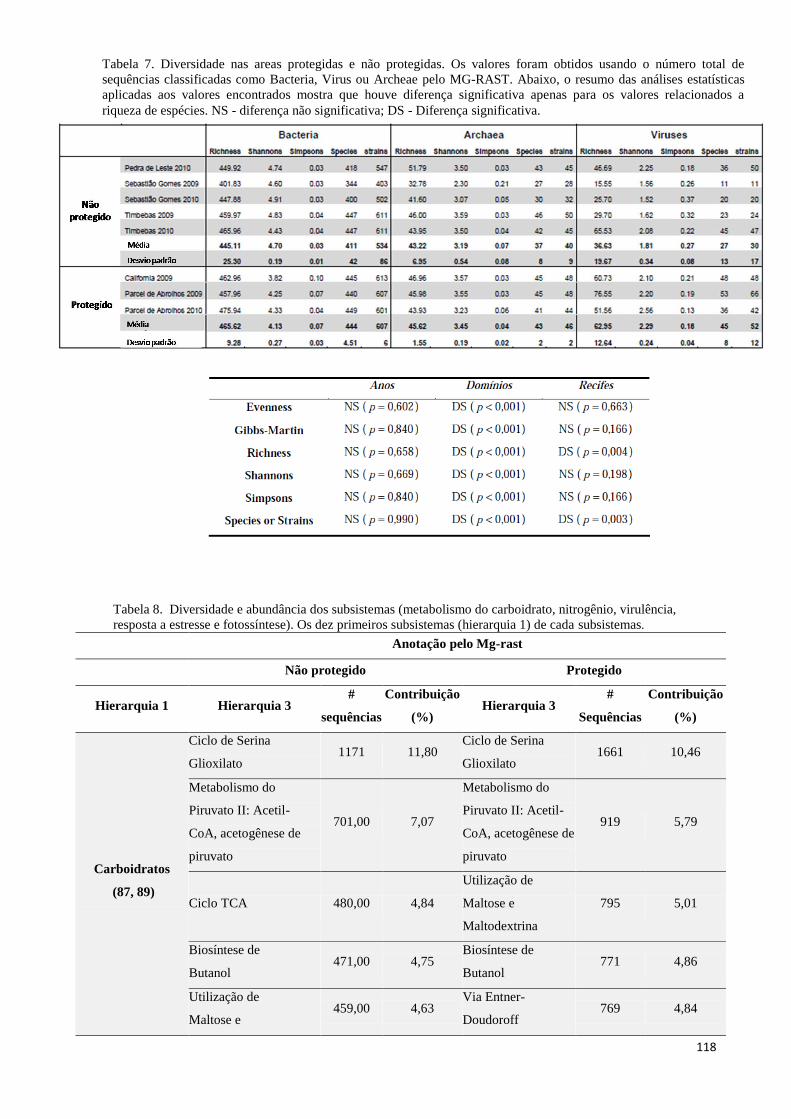
118
Tabela 8. Diversidade e abundância dos subsistemas (metabolismo do carboidrato, nitrogênio, virulência,
resposta a estresse e fotossíntese). Os dez primeiros subsistemas (hierarquia 1) de cada subsistemas.
Anotação pelo Mg-rast
Não protegido Protegido
Hierarquia 1 Hierarquia 3 #
sequências
Contribuição
(%) Hierarquia 3
#
Sequências
Contribuição
(%)
Carboidratos
(87, 89)
Ciclo de Serina
Glioxilato 1171 11,80
Ciclo de Serina
Glioxilato 1661 10,46
Metabolismo do
Piruvato II: Acetil-
CoA, acetogênese de
piruvato
701,00 7,07
Metabolismo do
Piruvato II: Acetil-
CoA, acetogênese de
piruvato
919 5,79
Ciclo TCA 480,00 4,84
Utilização de
Maltose e
Maltodextrina
795 5,01
Biosíntese de
Butanol 471,00 4,75
Biosíntese de
Butanol 771 4,86
Utilização de
Maltose e 459,00 4,63
Via Entner-
Doudoroff 769 4,84
Tabela 7. Diversidade nas areas protegidas e não protegidas. Os valores foram obtidos usando o número total de
sequências classificadas como Bacteria, Virus ou Archeae pelo MG-RAST. Abaixo, o resumo das análises estatísticas
aplicadas aos valores encontrados mostra que houve diferença significativa apenas para os valores relacionados a
riqueza de espécies. NS - diferença não significativa; DS - Diferença significativa.

119
Maltodextrina
Via Entner-
Doudoroff 435 4,39 Ciclo TCA 613 3,86
Complexos da
desidrogenase 403 4,06
Complexos da
desidrogenase 581 3,66
Metabolismo do
Piruvato I: reações
anapleróticas, PEP
375 3,78 Metabolismo da
sacarose 542 3,41
Fermentação de
Acetil-CoA a
Butirato
346 3,49
Metabolismo do
Piruvato I: reações
anapleróticas, PEP
492 3,10
Metabolismo da
sacarose 327 3,30
Metabolismo de D-
gliconato e ceto-
gliconato
456 2,87
Nitrogênio
(9, 8)
Assimilação de
Amônia 215 36,50
Assimilação de
Amônia 327 34,49
Degradação de
Alantoína 188 31,92
Amonificação de
Nitrito e de Nitrato 249 26,27
Amonificação de
Nitrito e de Nitrato 103 17,49
Degradação de
Alantoína 231 24,37
Óxido nítrico
sintetase 58 9,85
Óxido nítrico
sintetase 77 8,12
Desnitrificação 11 1,87 Desnitrificação 28 2,95
Hidrólise de Cianato 6 1,02 Hidrólise de Cianato 27 2,85
Estresse Nitrosativo 5 0,85 Fixação de
Nitrogênio 7 0,74
Fixação de
Nitrogênio 2 0,34 Estresse Nitrosativo 2 0,21
Síntese de novo da
Pirimidina 1 0,17 - - -
Fósforo
(6, 8)
Metabolismo do
Fosfato 1200 91,39
Metabolismo do
Fosfato 1879 92,61
Transportadores de
Fosfato de Alta
Afinidade e controle
de PHO Regulon
40 3,05 Utilização de
Alquilfosfonato 46 2,27
Utilização de
Alquilfosfonato 33 2,51
Transportadores de
Fosfato de Alta
Afinidade e controle de
33 1,63

120
PHO
Regulon
Captação de Fósforo
(Cyanobacteria) 14 1,07
Captação de Fósforo
(Cyanobacteria) 32 1,58
Fosfoenolpiruvato
Fosfomutase 13 0,99
Metabolismo do
Fosfonato 24 1,18
Metabolismo do
Fosfonato 13
Fosfoenolpiruvato
Fosfomutase 13 0,64
- - - Modificação de
Lipídio A 1 0,05
- - - Biosíntese Global de
Cofator NAD e NADP 1 0,05
Virulência
(73, 70)
Sistemas de
Transporte Tol e Ton 1241 0,99
Sistemas de
Transporte Tol e
Ton
2791 32,31
Grupo Resistente à
Acriflavina 400 30,03
Resistência Cobalto-
Zinco-Cádmio 924 10,70
Resistência Cobalto-
Zinco-Cádmio 379 9,68
Grupo Resistente à
Acriflavina 762 8,82
Resistência às
Fluoroquinolonas 343 9,17
Bombas de Efluxo
para Multidroga
Resistência
722 8,36
Bombas de Efluxo
para Multidroga
Resistência
290 8,30 Resistência às
Fluoroquinolonas 477 5,52
Transporte de Ferro 205 7,02 Transporte de Ferro 380 4,40
Beta-lactamase 188 4,96 Beta-lactamase 346 4,01
Pilus Tipo IV 161 4,55 Pilus Tipo IV 320 3,70
Ilhas de
Patogenicidade de
Estafilococos (SaPI)
97 3,90
Ilhas de
Patogenicidade de
Estafilococos (SaPI)
173 2,00
Nova Biosíntese de
Pioverdine 87 2,35
Pilus Hemaglutinina
Tipo 4 Manose-
sensíveis
146 1,69
Resposta a
estresse
(29, 30)
Estresse Oxidativo 317 2,11 Homeostase de
Cobre 596 18,70
Homeostase de
Cobre 274 19,18 Estresse Oxidativo 590 18,51
Metabolismo 182 16,58 Metabolismo Redox 418 13,12

121
Redox da
Glutationa
da Glutationa
Gene do choque
térmico DNA K 158 11,01
Gene do choque
térmico dnaK 292 9,16
Operon HFL 127 9,56 Resposta ao Estresse
Periplasmático 202 6,34
Resposta ao Estresse
Periplasmático 118 7,68
Mecanismos de
Resistência de Acido 180 5,65
Flavo Hemoglobina 76 7,14 Flavo Hemoglobina 137 4,30
Mecanismos de
Resistência de Acido 64 4,60 HFL Operon 137 4,30
Regulação da
Resposta ao estresse
Sigma B
44 3,87
Regulação da
Resposta ao estresse
Sigma B
104 3,26
Metabolismo da
Dimetilarginina 37 2,66 Estarvação 97 3,04
Fotossíntese
(6, 6)
Fotosistema II 117 40,77 Fotosistema II 480 42,40
Proteorodopsina 72 25,09 Ficobilissoma 296 26,15
Fotosistema I 56 19,51 Fotosistema I 259 22,88
Ficobilissoma 29 10,10 Proteorodopsina 78 6,89
Fotosistema tipo II-
Centro de Reação
Fotossintética
11 3,83
Fotosistema tipo II-
Centro de Reação
Fotossintética
18 1,59
Proteínas
Bacterianas de
captação de Luz
2 0,70
Proteínas
Bacterianas de
captação de Luz
1 0,09
Um total de 14 grupos taxonômicos teve uma maior contribuição em pelo menos um dos
subsistemas nos recifes desprotegidos em relação aos recifes protegidos. Alphaproteobacteria
e Gammaproteobacteria tiveram maior contribuição nos subsistemas envolvidos em
processos fototróficos nos recifes desprotegidos do que nos recifes protegidos (Figura 41). O
metabolismo fotototrófico ocorre nesses organismos em situação de limitação de recursos,
reforçando o valor ecológico de genes como as proteorodopsinas. Alphaproteobacteria
também apresentaram maior contribuição para a virulência e Bacteroidetes tiveram uma

122
maior contribuição à virulência, carboidratos e resposta ao estresse. Entre os
Gammaproteobacteria, Vibrios contribuíram com apenas 1-2% das sequências de subsistemas
de metabolismo de nitrogênio e de resposta ao estresse, mas até 6% dos subsistemas de
fotossíntese nos recifes desprotegidos.
Seis grupos taxonômicos tiveram maior contribuição nos recifes protegidos, em pelo menos
um dos subsistemas, do que nos recifes não protegidos (Figura 41). Entre os mais abundantes,
Gammaproteobacteria apresentou uma maior contribuição para a composição dos
subsistemas de virulência, metabolismo nitrogênio, fósforo e de resposta ao estresse.
Cyanobacteria contribuíram para os seis subsistemas, enquanto Actinobacteria mostrou maior
contribuição para o subsistema de virulência nos recifes protegidos. Viridiplantae contribuiu
para as diferenças nos subsistemas de carboidratos, fotossíntese e subsistemas de resposta ao
estresse. Stramenopiles apresentou maior contribuição para os subsistemas de carboidratos e
fostossíntese. Apesar dobaixo número de sequências, foi identificada maior contribuição de
seqüências de Korarcheota para o subsistema de carboidratos nos recifes protegidos.
Os dados sugerem que diferentes populações de Vibrios estão associadas aos recifes
protegidos e desprotegidos. Foi possível observar um comprometimento metabólico
diferencial entre os metagenomas para os subsistemas de nitrogênio, fósforo, resposta a
estresse e fotossíntese entre áreas protegidas e não protegidas. Os Vibrios das áreas não
protegidas apresentaram maior abundância de genes dos subsistemas de nitrogênio, resposta a
estresse e fotossíntese enquanto os de áreas protegidas apresentam maior abundância de genes
envolvidos nos subsistema de fósforo (Figura.42) A contribuição de Vibrios para estes
subsistemas foi indetectável nos recifes protegidos, mas eles contribuíram com 1,5% para o
subsistema de fósforo nesses locais.
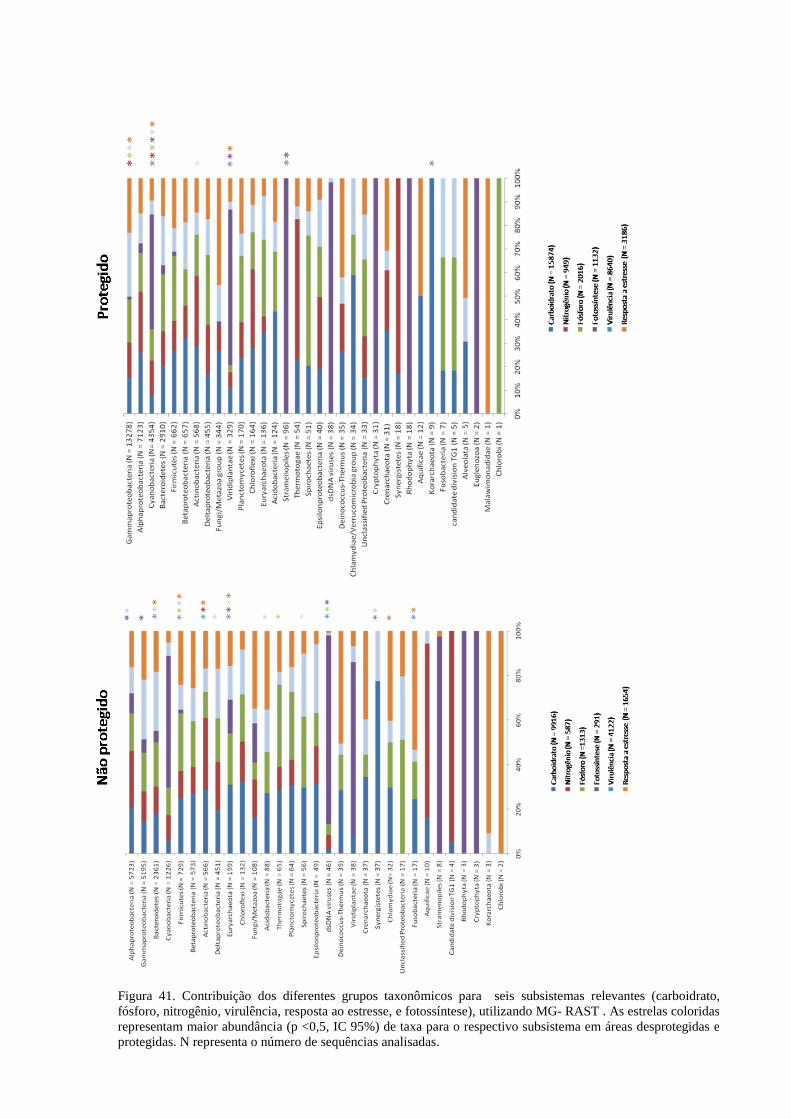
123
Figura 41. Contribuição dos diferentes grupos taxonômicos para seis subsistemas relevantes (carboidrato,
fósforo, nitrogênio, virulência, resposta ao estresse, e fotossíntese), utilizando MG- RAST . As estrelas coloridas
representam maior abundância (p <0,5, IC 95%) de taxa para o respectivo subsistema em áreas desprotegidas e
protegidas. N representa o número de sequências analisadas.

124
Em suma, neste capítulo está demonstrado, pela primeira vez, a diversidade metagenômica no
Banco de Abrolhos. Este estudo levou a uma maior compreensão do impacto de áreas
marinhas protegidas na abundância e diversidade microbiana. A maior abundância de
sequências metagenômicas oriundas de patógenos potenciais e de bactérias de crescimento
rápido em áreas não protegidas reflete a tendência de degradação destas áreas. O recife de
Timbebas aparece em posição intermediária em termos de uso e degradação, não sendo
totalmente protegido ou desprotegido. Deste forma, podemos concluir que a diversidade
microbiana identificada nos metagenomas pode ser ferramenta importante no manejo de áreas
marinhas protegidas, apresentando potencial comoestratégia de biomonitoramento.
4.2.2 Discussão
Nossos resultados vão ao encontro dos achados que sugerem que recifes de corais
saudáveis como California, por exemplo, apresentam maior cobertura de corais que
macroalgas. Coral e cobertura de macroalgas em recifes desprotegidos em todo o mundo
variam de forma significativa. A proliferação de macroalgas pode resultar no aumento de
bactérias com potencial patogênico através da produção de COD (carbono orgânico
Figura 42. Versatilidade metabólica dos Vibrios. O gráfico mostra que os grupos de Vibrios nas áreas não
protegidas apresentam maior comprometimento com metabolismo fototrófico.

125
dissolvido). O contato físico entre algas carnudas e coral pode causar descoloração ou necrose
próximo à interface devido a toxinas (metabólitos lipossolúveis), especialmente na ausência
de peixes herbívoros (Barott et al., 2009; Rasher and Hay, 2010). A proliferação de
macroalgas no recife desprotegido dentro do Banco Abrolhos pode ser uma consequência da
diminuição de peixes herbívoros nestes locais. Compostos solúveis (carbono orgânico
dissolvido), liberados pelas algas podem promover a mortalidade de coral através da atividade
microbiana (Sekar et al., 2006a).
A menor concentração de COD observada nos recifes desprotegidos neste estudo sugere
que este conjunto de nutrientes é convertido rapidamente em biomassa bacteriana pelo
rápido crescimento das bactérias heterotróficas. Assim, as maiores contagens Vibrios
foram observadas nos recifes desprotegidos. Padrões similares de baixa concentração de COD
e alta contagem de UFC de heterotróficos (Vibrios) em recifes degradados foram observadas
em recifes no Oceano Pacífico (Dinsdale et al., 2008b). O COD biodisponível pode ser
necessário para a degradação do COD semi-lábil. Estes compostos são resistentes ao rápido
ataque microbiano (Cherrier et al., 1996; Sondergaard et al., 2000). Um aumento de nutrientes
inorgânicos por si só não é suficiente para permitir que as comunidades bacterianas utilizem o
COD refratário (Carlson et al., 2002). Assim, a baixa concentração de COD em recifes
desprotegidos do Banco dos Abrolhos pode ser um efeito combinado do aumento da
produtividade de algas fotossintéticas e maior consumo de COD lábil pelos
heterotróficos. Além disso, a maior número relativo de heterótrofos nos recifes desprotegidos
pode contribuir para a maior concentração de CO2 nestes sítios. Ambientes ricos em CO2
parecem reforçar a capacidade competitiva das macroalgas sobre os corais (Diaz-Pulido et al.,
2011).
Os elevados níveis de nutrientes dissolvidos detectados no sistema de recifes de
Abrolhos, em especial NID (11,3 mM) e FID (0,6 mM), sugerem um processo de

126
eutrofização nesta área. A biomassa do fitoplâncton deduzida a partir da concentração de
clorofila (0,23-0,34 mg / L) também foi alta em todos os cinco recifes e se aproximam do
limiar que define a eutrofização de sistemas recifais (Bell et al., 2007; Costa et al., 2008). Os
níveis desses nutrientes foram até dez vezes maiores do que a limiar de concentração sugerida
para NOD (1,0 mM) e FOD (0,2 m) na Grande Barreira de Corais (Bell, 1992). Estes
limiares determinam o processo de eutrofização não só neste recife, mas também em
outros recifes (por exemplo, Barbados e Florida Keys). As concentrações de nutrientes
observadas neste estudo são comparáveis com os obtidos em estudos anteriores
realizados em uma área próxima, a norte do Banco de Abrolhos, em Porto Seguro (Costa
et al., 2006a). Eles encontraram até 8,19 mM e 1,42 mM NIP e FID e concluíram que pode
haver uma fonte permanente de fósforo nessa área, enquanto o crescimento de
microrganismos pode ser limitado por nitrogênio (Costa et al., 2006a). Nosso estudo também
sugere que os recifes do Banco de Abrolhos (Timbebas, Sebastião Gomes, Parcel dos
Abrolhos e California) estão sob impacto de fontes de fósforo e nitrogênio, indicada pelos
altos teores destes nutrientes. As verdadeiras fontes de nutrientes são desconhecidas, mas
podem ser de escoamento da costa (ou seja, efluentes domésticos e agrícolas), o acoplamento
bento-pelágico, ou da descarga de águas subterrâneas. A descarga de águas subterrâneas
parece ser uma fonte importante no norte do Banco de Abrolhos (Costa et al., 2006a). A alta
concentração de nitrogênio na Sebastião Gomes e de silicatos nos recifes desprotegidos pode
ser devido à proximidade do rio Caravelas e da cidade de Caravelas. Na Ilha Kiritimati, uma
das ilhas que compõem um atol no Oceano Pacífico, os mais altos níveis de nitrogênio foram
encontrados em áreas com maior densidade humana (Dinsdale et al., 2008b). Altas cargas de
nutrientes podem promover o crescimento massivo de macroalgas. Mudanças na fase de
recifes de coral para macroalgas têm sido observadas em diferentes locais, incluindo Kaneohe
Bay (Smith 1981), Brasil (Costa et al., 2000), Bahamas (Lapointe et al., 2004), e a Grande

127
Barreira de Corais (Bell et al., 2007). Proliferação de algas pode por sua vez, promover o
crescimento microbiano, devido à produção de matéria orgânica dissolvida altamente
lábil (Barott et al., 2009; Rasher and Hay, 2010; Smith et al., 2006).
A contagem total de bactérias (4,88 e 5,63 x 105 x 105 células / mL) ficou dentro da faixa
observada para outros sistemas recifais (Dinsdale et al., 2008b). A menor contagem foi
observada no recife California e as mais altas no recife de Sebastião Gomes. Observamos
maior contagem de Vibrios (tanto de UFC como nas sequências metagenômicas) nos recifes
desprotegidos (Sebastião Gomes, Timbebas e Pedra de Leste), possivelmente em resposta a
pulsos de nutrientes. Microrganismos com taxas de crescimento rápido, como Vibrios
podem ser bom indicador da saúde do recife, pois podem responder rapidamente a
eutrofização. Como alguns Vibrios são capazes de fixar N2 é esperado que eles não sejam
limitados por nitrogênio. Além disso, Vibrios são capazes de gerar a energia a partir da luz
usando proteorodopsinas (Gómez-Consarnau et al., 2010). Genes de proteorodopsinas
encontrados nos recifes de Abrolhos pareciam pertencer a Vibrios, indicando a
plasticidade genômica dessas bactérias.
As bactérias patogênicas são abundantes em áreas de recifes desprotegidos. O
predomínio de diferentes tipos de microrganismos pode resultar em diferentes níveis tróficos
nos sistemas recifais do Banco dos Abrolhos. As bactérias heterotróficas dominam as
comunidades microbianas dos recifes desprotegidos: a análise metagenômica revelou uma
maior abundância de bactérias patogênicas humanas e animais (Bacteroidetes,
Pseudoalteromonas e Alteromonas macleodii), e menos genes fotossintéticos. Em
contrapartida, uma maior abundância de Picocyanobacteria fotossintética foi observada
nos recifes protegidos. Picocyanobacteria marinha são os organismos fotossintéticos mais
abundantes na Terra, com apenas dois gêneros, Prochlorococcus e Synechococcus,
numericamente dominantes na maioria das águas oceânicas (Johnson et al., 2006; Partensky et

128
al., 1999). Os recifes protegidos tiveram um número maior de sequências associadas com
microrganismos autotróficos.
Vários estudos têm detectado um enriquecimento de Bacteroidetes em diferentes espécies de
corais doentes e nos sistemas de recifais (de Castro et al., 2010a; Jones et al., 2004; Pantos et
al., 2003; Sekar et al., 2006a; Thurber et al., 2009). Estes organismos foram mais
representados em dois dos metagenomas desprotegidos, e em um dos metagenomas dos
recifes protegidos, de modo que nenhum padrão foi identificado no nível de classe para estes
recifes. As áreas protegidas apresentaram maior número de bactérias, Arquéia, e táxons virais.
Sugerimos que os recifes de coral protegidos têm uma maior riqueza microbiana e homeostase
mais elevada (Tabela 7), enquanto que os recifes desprotegidos criam condições favoráveis
que promovam o crescimento de patógenos de coral e patógenos oportunistas. O recife de
Sebastião Gomes, o mais impactado entre os analisados neste trabalho, apresentou menor
riqueza de espécies do que os outros recifes.
Foi estabelecido um panorama sobre a metagenômica e funcionamento do Banco de
Abrolhos. O recife de Abrolhos está em um acelerado processo de deterioração, com recifes
desprotegidos apresentando baixa biomassa de peixes e alta contagem de microrganismos
patogênicos de coral. Em todos os pontos amostrados houve uma alta carga de NID e FID.
Esses parâmetros, isoladamente, não explicam as grandes mudanças de fase observadas entre
recifes desprotegidos (Sebastião Gomes, Pedra de Leste e Timbebas) e protegidos (Parcel dos
Abrolhos e California). A ocorrência de cobertura mais elevada das algas e menor cobertura
de peixes com maior contagem de Vibrios sugerem que a remoção dos peixes pode promover
a proliferação de algas e crescimento de bactérias. O maior número de microrganismos
heterotróficos nos recifes desprotegidos, particularmente, as de rápido crescimento (por
exemplo, Vibrios e outras bactérias patogênicas para humanos), sugere uma rápida
conversão de energia dentro dos recifes desprotegidos. A maior abundância de vírus

129
identificada no metagenoma dos recifes desprotegidos reforça a sugestão de uma rápida
mudança da comunidade microbiana e uso de energia nestes recifes. Os recifes desprotegidos
têm sofrido com a pesca predatória na última década. O recife de Timbebas, que oficialmente
pertence à área do Parque Nacional de Abrolhos protegida pela marinha, é na verdade uma
área sujeita à pesca, um exemplo de má gestão que leva à mudança das linhas de base do
sistema recifal. Por outro lado, a pesca nos recifes protegidos é rara devido à aplicação dos
regulamentos por parte da guarda do parque.
Nossos dados indicam que as áreas protegidas também estão em diferentes estágios do
processo de degradação. Observa-se que a microbiota do recife de Timbebas apresenta
características de recifes impactados em áreas não protegidas (Sebastião Gomes) como o
enriquecimento de vírus, archaea e os organismos heterotróficos. No entanto, Timbebas ainda
preserva biomassa de peixes e cobertura bentônica de áreas protegidas menos degradadas
(California), em comparação com Sebastião Gomes, que representa uma área desprotegida
com o maior grau de impacto.
Embora em níveis mais baixos, também foi possível detectar no recife de Parcel dos
Abrolhos apresenta uma fase inicial de degradação. Assim como Timbebas, Parcel dos
Abrolhos também apresentou biomassa de peixes e cobertura bentônica compatíveis com
áreas menos degradadas como a California. No entanto, a microbiota revelou uma alta
abundância de Bacteroidetes. O enriquecimento deste filo bacteriano tem sido observado nos
corais doentes (de Castro et al., 2010a). Além disso, o de dados metagenômicos apontam para
uma redução significativa de genes envolvidos na produtividade primária (fotossíntese).
Genes de proteorodopsinas foram amplamente distribuídos pelas bactérias
heterotróficas relacionados à família Vibrionaceae.
Nossa análise sugere que os dados moleculares são mais precisos na detecção de
alterações em curto prazo da qualidade da saúde dos recifes do Banco dos Abrolhos. A

130
análise integrada em diferentes níveis sistêmicos atua como um eficiente marcador e preditor
de processos ecológicos (Raes et al., 2011). A criação e execução de áreas marinhas
protegidas parecem ser ferramentas valiosas para manter a homeostase do sistema de
recife dos Abrolhos. Como foi visto nas Northern Line Islands (Dinsdale et al., 2008b), as
modificações na cadeia alimentar do Banco dos Abrolhos, com a redução de peixes
herbívoros e aumento de macroalgas, parece afetar a diversidade microbiana e a abundância
desse bioma. As comunidades microbianas, por sua vez influenciam a saúde de todo o
bioma através de seu metabolismo, o que ilustra a importância das AMPs como
instrumentos para manter a homeostase dos recifes de corais em Abrolhos.
4.3 Diversidade das comunidades microbianas dos solos da Mata Atlântica e o seu
potencial biotecnológico
4.3.1 Resultados
Neste capitulo descrevo os resultados da análise da diversidade microbiana de um bioma
típico brasileiro, a Mata Atlântica. Este estudo permite estabelecer diferenças entre o grau de
endemismo da microbiota da Mata Atlântica e o meio marinho, onde foi observada aparente
fração microbiana ubíqua como foi observado nos capítulos anteriores. Além disso, foi
possível demonstrar o potencial biotecnológico da microbiota do solo da Mata Atlântica
através da triagem funcional para a busca de genes envolvidos na degradação de celulose.
4.3.1.1 Diversidade da das comunidades microbianas dos solos da Mata Atlântica
A extração do DNA metagenômico dos diferentes tipos de solo do PARNASO permitiu o
estabelecimento de bibliotecas de clones do gene rRNA 16S. Um total de 13 filos foram
delineados com base em 894 sequências parciais do gene rRNA 16S das seis localidades
através de comparação com o banco de dados RDP (Figura 42).

131
Acidobacteria, Proteobacteria e Verrucomicrobia foram os filos mais abundantes,
correspondendo a 70% de todas as sequências analisadas (Figura 44 A). A contribuição de
Acidobacteria, variou entre 29% em Campo Úmido e 54% no site Cavalinho. As 379
sequências identificadas como Acidobacteria foram distribuídas em 154 grupos (definida
como "grupos" com pelo menos três sequências, com similaridade de pelo menos 97% entre
elas), 81 singletons e 29 doubletons. Nove subgrupos foram identificados em Acidobacteria.
Os subgrupos Gp1 s e GP2 foram os mais frequente, representando mais de 70% das
sequências identificadas como Acidobacteria (Figura 44 B). Sequências de Acidobacteria do
grupo GP3 estavam presentes em seis localidades, variando entre 2%e 16%.

132
Figura 43. Diversidade de filos encontrados nas bibliotecas do gene rRNA 16S. O tamanho dos triângulos
corresponde ao número de sequências identificadas. Unclassifieddesigna as sequências para as quais não foi
possível obter uma classificação taxonômica através do RDP (Ribosomal Database Project).

133
A
B
C
Figura 44. Estrutura das comunidades microbianas do solo da Floresta da Mata Atlântica do PARNASO
baseada no sequenciamento de bibliotecas de clones do gene rRNA 16S de acordo com o RDP (limite de
confiança em 80 %). A) Abundância de filos; B) Contribuição (%) de subgrupos do filo Acidobacteria
(Acidobacteria Gp 1 ao Gp 17) e C) Contribuição (%) das classes de Proteobacteria. N representa o número
te sequências analisadas.

134
O segundo grupo mais frequentemente recuperado em todos os locais foi Proteobacteria
(Figura 44 C). A classe Alphaproteobacteria correspondeu a 28% das sequências de Bonfim
Vermelho e 74% das sequências do site do Ajax. Vários outros filos, por exemplo,
Actinobacteria, Bacteroidetes, Planctomycetes, Gemmatimonadetes e Verrucomicrobia,
ocorreram em uma frequência relativamente baixa, geralmente abaixo de 10%. Cerca de 15%
de todas as sequências do gene rRNA 16S (N = 134) foram atribuído às bactérias não
classificada pela RDP (Unclassified). Estas sequências foram distribuídas em 105 e 41 OTUs
com 3% e 20% de distância genética como níveis de corte, respectivamente, e representando
potenciais espécies e filos novos.
As curvas de rarefação mostraram que a taxa de cobertura atingiu uma assíntota nos níveis de
filo e de classe em todos os locais, mas não atingiu a saturação ao nível de espécie (Figura
45). O número de filos variou entre 54 e 98, utilizando o limite de 80 % de similaridade
(Figura 45 C). As curvas de rarefação referente às espécies mostraram números entre 93 (no
Ajax solo) e 123 (no solo Vermelho Bonfim) (Figura 45 A). O estimador de riqueza não
paramétricos Chao1 indicou a existência de 263 a 446 espécies (Figura 45 D) e 41-152 filos
(Figura 45 F).
O número de sequências foi suficiente para fornecer uma estimativa aproximada do número
de filos, mas não foi o suficiente para ter uma estimativa robusta do número de espécies no
solo da Mata Atlântica. O baixo número de grupos delineados (N = 155) frente ao estimados
pode muito bem ilustrar esse fato. Dezoito grupos mais frequentes, correspondente às
espécies, foram delineados usando todas as 894 sequências de gene rRNA 16S (Figura 46;
Tabela 9 e 10). Os grupos mais frequentes foram definidos como grupos contendo pelo menos
seis sequências com mais de 97% similaridade entre elas. A comparação com sequências
disponíveis no GenBank mostrou que esses grupos estão relacionados com clones de
diferentes eocssitemas. Esses grupos representaram apenas 17% de todas as sequências do

135
rRNA 16S. Onze grupos (1, 3, 4, 5, 6, 7, 8, 9, 12, 16 e 17) foram divididos em Acidobacteria.
Quatro grupos (2, 10, 11 e 13) foram divididos em Proteobacteria e apenas um grupo (14) foi
identificado como Verrucomicrobia. Dois grupos (15 e 18) não apresentaram similaridade
significativa de sequência de rRNA 16S com qualquer outro bacteriano já formalmente
descrito. O baixo nível de semelhança entre as sequências do gene rRNA 16S dos grupos
delineados e as sequências de banco de dados impede a sua identificação em espécies
conhecidas. Seis grupos (1, 2, 3, 8, 12 e 18) apresentaram mais de 97% similaridade entre as
sequência do rRNA 16S com sequências de bactérias não cultivadas e não classificadas
taxonomicamente. A maioria dos grupos apareceu em quatro locais diferentes (Tabela 10).
Em contrapartida, os grupos 7 e 18 apareceram em apenas uma localidade. Uma baixa co–
relação entre as características físico químicas, por exemplo o pH do solo, e o índice de
diversidade Shannon (H) foi verificada. A maior correlação correu com o aumento da
concentração de potássio (R2= 0,395 (Figura 47).
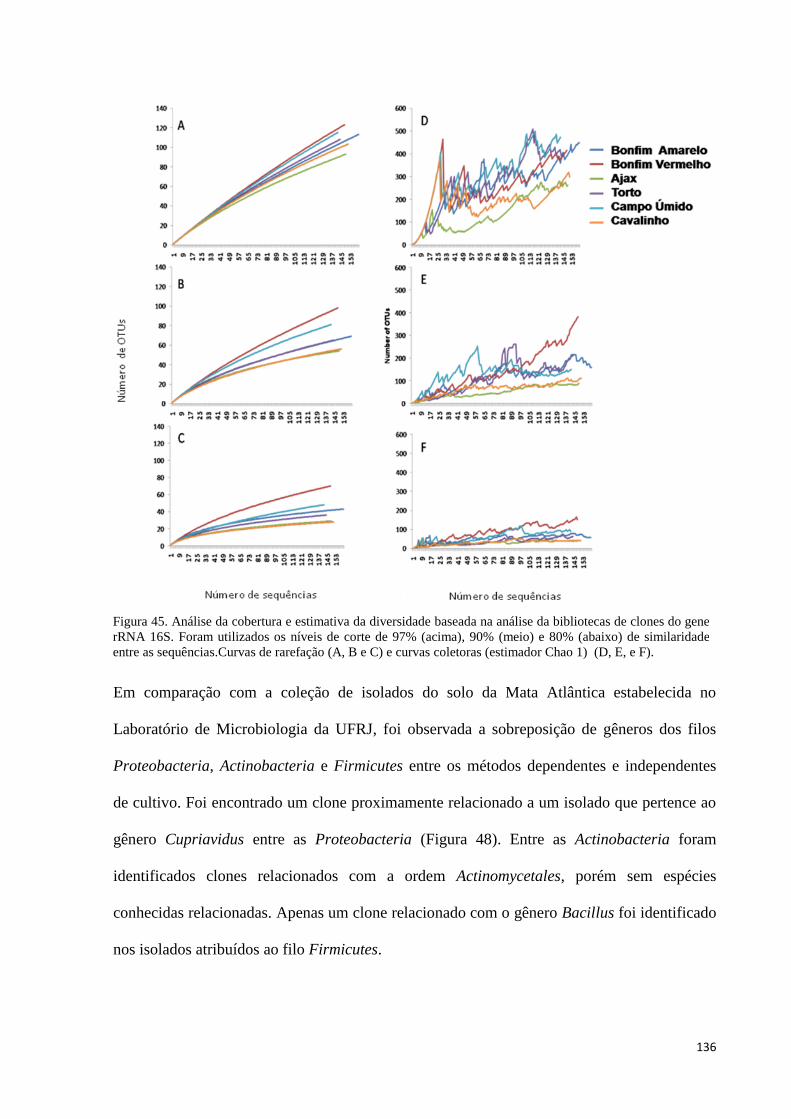
136
Em comparação com a coleção de isolados do solo da Mata Atlântica estabelecida no
Laboratório de Microbiologia da UFRJ, foi observada a sobreposição de gêneros dos filos
Proteobacteria, Actinobacteria e Firmicutes entre os métodos dependentes e independentes
de cultivo. Foi encontrado um clone proximamente relacionado a um isolado que pertence ao
gênero Cupriavidus entre as Proteobacteria (Figura 48). Entre as Actinobacteria foram
identificados clones relacionados com a ordem Actinomycetales, porém sem espécies
conhecidas relacionadas. Apenas um clone relacionado com o gênero Bacillus foi identificado
nos isolados atribuídos ao filo Firmicutes.
Figura 45. Análise da cobertura e estimativa da diversidade baseada na análise da bibliotecas de clones do gene
rRNA 16S. Foram utilizados os níveis de corte de 97% (acima), 90% (meio) e 80% (abaixo) de similaridade
entre as sequências.Curvas de rarefação (A, B e C) e curvas coletoras (estimador Chao 1) (D, E, e F).

137
Tabela 9. Os grupos mais frequentes encontrados nas bibliotecas e seus vizinhos filogenéticos mais próximos
(cultivados e não cultivados). Os agrupamentos representam pelo menos seis sequências com similaridade > 97
% entre elas.
Grupo
Classificação pelo
RDP
Vizinho filogenético mais próximo
Linhagem tipo
(% de similaridade)
Número de
acesso
Resultado BLAST
(% de similaridade)
Número de
acesso
1 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (95) D26171
Uncultured soil bacterium
clone 1209-2 (97) AY326560
2 Alphaproteobacteria
Bradyrhizobium
japonicum
ATCC 10324
(94) U69638
Bradyrhizobium
sp. KO14 (97) AB367695
3 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (92) D26171
Uncultured Acidobacteria
bacterium clone FAC9 (97) DQ451448
4 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (85) D26171
Uncultured bacterium clone
AS66 (95) AY963428
5 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (84) D26171
Uncultured bacterium clone
PW290 (95) GQ402730
6 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (93) D26171
Uncultured bacterium clone
JH-WH215 (97) EF492903
7 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (94) D26171
Uncultured bacterium clone
S7-82 (95) EU680420
8 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (93) D26171
Uncultured bacterium clone
FCPT602 (96) EF515877
9 Acidobacteria Acidobacterium
Capsulatum ATCC D26171
Uncultured bacterium clone
Amb_16S_775 (94) EF018517

138
51196 (85)
10 Alpharoteobacteria
Rhodoplanes elegans
ATCC 51906 (90) D25311
Uncultured bacterium clone
KGB200711-106 (97) EU881284
11 Deltaproteobacteria
Stigmatella erecta
DSM 16858T (86) AJ970180
Uncultured delta
proteobacterium clone 437T3
(91) DQ110129
12 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (95) D26171
Uncultured Acidobacteriaceae
bacterium clone
Elev_16S_571 (96) EF019354
13 Gammaproteobacteria
Methylosarcina fibrata
ATCC 700909 (82) AF177296
Uncultured bacterium clone
AS44 (96) AY963408
14 Verrucomicrobia
Candidatus
Xiphinematobacter
rivesi (95) AF217461
Uncultured bacterium clone
FCPS466 (95) EF516262
15 Unclassified bacteria N/A N/A
Uncultured soil bacterium
clone FACE.R4.AC.D01 (95) FJ621116
16 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (93) D26171
Uncultured bacterium clone
GASP-38KB-91-C04 (98) EU044112
17 Acidobacteria
Acidobacterium
Capsulatum ATCC
51196 (92) D26171
Uncultured
bacterium clone
AS63 (96) AY963426
18
Unclassified
bacteria N/A N/A
Uncultured
bacterium clone
BacC-u_068 (93) EU335405
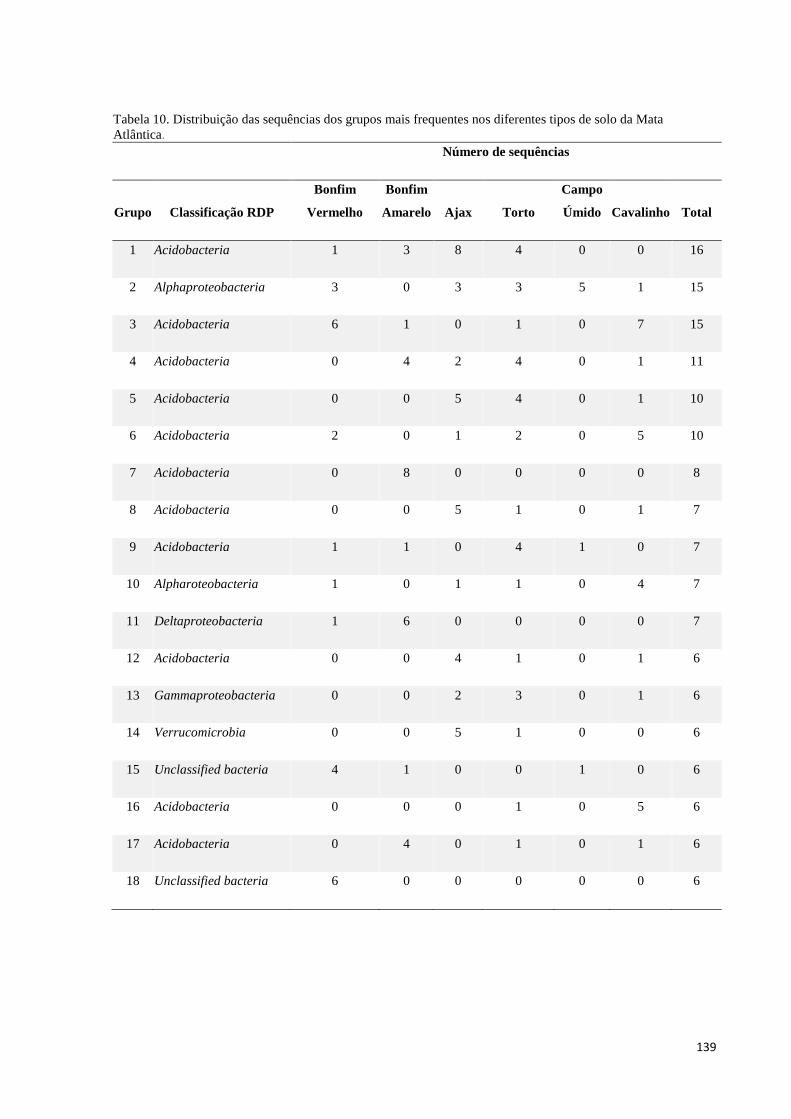
139
Tabela 10. Distribuição das sequências dos grupos mais frequentes nos diferentes tipos de solo da Mata
Atlântica.
Número de sequências
Grupo Classificação RDP
Bonfim
Vermelho
Bonfim
Amarelo Ajax Torto
Campo
Úmido Cavalinho Total
1 Acidobacteria 1 3 8 4 0 0 16
2 Alphaproteobacteria 3 0 3 3 5 1 15
3 Acidobacteria 6 1 0 1 0 7 15
4 Acidobacteria 0 4 2 4 0 1 11
5 Acidobacteria 0 0 5 4 0 1 10
6 Acidobacteria 2 0 1 2 0 5 10
7 Acidobacteria 0 8 0 0 0 0 8
8 Acidobacteria 0 0 5 1 0 1 7
9 Acidobacteria 1 1 0 4 1 0 7
10 Alpharoteobacteria 1 0 1 1 0 4 7
11 Deltaproteobacteria 1 6 0 0 0 0 7
12 Acidobacteria 0 0 4 1 0 1 6
13 Gammaproteobacteria 0 0 2 3 0 1 6
14 Verrucomicrobia 0 0 5 1 0 0 6
15 Unclassified bacteria 4 1 0 0 1 0 6
16 Acidobacteria 0 0 0 1 0 5 6
17 Acidobacteria 0 4 0 1 0 1 6
18 Unclassified bacteria 6 0 0 0 0 0 6

140
Figura 46. Análise filogenética baseada nas sequências do gene rRNA 16S dos grupos de clones mais frequentes nas bibliotecas Os
grupos apresentam pelo menos seis sequências com similaridade > 97 % entre elas. Os círculos pretos representam uma sequência
representativa de cada grupo e entre parêntesis o número de sequências representadas. As linhagens tipo e sequências relacionadas
como vizinhos filogenéticos mais próximos foram determinadas pelos bancos de dados RDP e GenBank. A fonte de identificação
das sequências dos bancos de dados do NCBI estão indicados.

141
Figura 47. Relação entre diversidade H (índice de Shannon) e parâmetros físico químicos do solo.
Unidades: Matéria Orgânica (%); Alumínio (me/100cm³); Fósforo (mg/L); Potássio (mg/L).

142
Proteobacteria Firmicutes Actinobacteria
Portanto, foi demonstrado que Acidobacteria e Proteobacteria são abundantes no solo da
Mata Atlântica. A existência de novos filos e espécies potenciais reflete a tremenda
diversidade microbiana presente no solo da Mata Atlântica, representando um vasto repertório
genético para descobertas biotecnológicas.
4.3.1.2 Potencial biotecnológico do DNA metagenômico do solo da Mata Atlântica
O potencial biotecnólogico do DNA metagenômico da Mata Atlântica foi demonstrado por
meio de procedimentos de extração, clonagem e expressão heteróloga, visando descobrir
sequências de DNA metagenômico com atividade enzimática. Foram realizados dois métodos
Figura 48. Fração de sequências dos filos Proteobacteria, Firmicutes e Actinobacteria compartilhada pelos
métodos dependentes e independentes de cultivo.

143
de preparação do DNA metagenômico dos solos que compõem o PARNASO para o
estabelecimento de uma biblioteca metagenômica visando a prospecção de moléculas
bioativas. O método de extração através de esferas de vidro mostrou-se mais eficaz que o kit
comercial Power Soil da Mo Bio (Figura 49) .
A
B
Entretanto, foi necessário realizar uma etapa adicional de purificação do DNA metagenômico
para os ensaios de digestão parcial. Os volumes de inserto e vetor foram determinados após a
Figura 49. Extração do DNA metagenômico pelos métodos A) Kit comercial Mo Bio e B) Esferas de vidro.
A1) Mistura de DNA metagenômico dos diferentes tipos de solo da Mata Atlântico do PARNASO; A2)
Resultado da digestão parcial do DNA metagenômico pela enzima PstI, gerando quantidades insuficientes para
a construção da biblioteca metagenômica. B1) Resultado da extração do DNA metagenômico dos solos ainda
com contaminantes em diferentes concentrações (1, 3, 5 e 10 μL); B2)Resultado da purificação e B3)
Resultado da digestão parcial. A linha vermelha mostra a região excisadacontendo os fragmentos para
clonagem no plasmídeo pCF430.

144
purificação de ambos. A eficiência da clonagem foi alta conforme o número de clones
estocados (estimativa de 40.000 clones) e a diversidade de fragmentos clonados (Figura 50).
Apesar de parecer trivial, a extração de DNA metagenômico de alta qualidade é um grande
desafio pois este DNA esta associados com material húmico e metais do solo que resultam na
sua rápida degradação.
Figura 50. Extração plasmidial de colônias aleatórias demonstrando a variedade de insertos com diferentes
tamanhos. 1. pCF430 circular. 2-11. diferentes colônias (5μL).
A partir da biblioteca metagenômica foram triados clones com atividade celulolítica em meio
mínimo de sais suplementado com carboximetil celulose (CMC), um polímero solúvel
derivado da celulose, para a triagem da atividade celulolítica. A atividade foi avaliada através
da ocorrência de halos de degradação revelados pelo reagente vermelho congo, capaz de se
associar as ligações tipo β- 1,4 glucose (Teather and Wood, 1982). A formação de discretos
halos foi observada em torno da maioria dos clones selecionados, após três etapas de repique.
Aproximadamente mil clones foram triados e três clones foram escolhidos aleatoriamente
para avaliação da atividade enzimática. Estes clones apresentaram crescimento em meio
mínimo suplementado com CMC (carboxi metil celulose), como única fonte de carbono.
Após três passagens em meio mínimo, ficou evidente que estes clones possuíam DNA
metagenômico com sequências gênicas codantes de genes envolvidos na degradação de
celulose. Curvas de crescimento foram avaliadas para a determinação e padronização da fase
1 2 3 4 5 6 7 8 9 10 11

145
ótima de produção de celulases da cultura bacteriana ao longo de 16 horas. A fase estacionária
foi alcançada aproximadamente após 4 horas (Figura 51).
Em aproximadamente 2 horas foi observada maior atividade enzimática para as atividades
Cmcásica (ligação ß, 1-4). A atividade máxima alcançada corresponde com o início da fase
log de crescimento dos clones em cultura.
Além de 3 clones, 3 isolados ambientais da Floresta da Mata Atlântica foram selecionados em
meio mínimo de sais suplementado com CMC para determinação da atividade celulolítica. Os
resultados da atividade enzimática dos clones e isolados foram comparados com fungo
filamentoso Penicillium funiculosum, reconhecido produtor de celulases com alta atividade
enzimática. As atividades apresentadas na Tabela 11 mostram que foi detectada atividade
celulolítica nos clones e isolados analisados. Um importante aspecto do clone Cel 1A foi a
presença de atividade -glucosidásica, em proporção equilibrada em relação às atividades
FPásica e CMCásica. Além disso é importante destacar que as atividades FPásica e CMCásica
dos clones alcançaram níveis bem próximos das observadas nos isolados.Desta forma,
demonstramos que existem clones com sequências gênicas codantes de enzimas relacionadas
com a degradação da celulose.
Figura 51. Curva de crescimento dos clones em meio CMC.

146
Tabela 11. Ensaios de atividade enzimática. A1, A2 e A3 são clones selecionados da biblioteca de pequenos
insertos. Cel 1A e Cel CAAB são isolados do solo da Mata Atlântica que apresentaram capacidade de formar
halos de degradação no meio de cultura.
Clones
Atividades (U/L)
FPásica CMCásica -glucosidásica
A1 21,66 ± 2,56 92,19 ± 1,94 0
A2 19,84 ± 0,40 111,44 ± 4,54 0
A4 20,12 ± 0,40 98,15 ± 5,18 0
Cel 1A 24,04 ± 1,98 148,09 ± 7,13 80,79 ± 5,21
Cel CAAB 20,40 ± 1,59 134,35 ± 1,94 0
Penicillium
funiculosum 500 6900 1850
4.3.2 Discussão
A utilização de uma abordagem independente de cultivo não mostrou uma sobreposição
significativa com o método dependente de cultivo, demonstrando que esses métodos são
capazes de recuperar diferentes frações da diversidade de bactérias do solo da Mata Atlântica..
Os grupos bacterianos identificados neste estudo são habitantes típicos do solo (Janssen,
2006). A fisiologia e as características de microrganismos pouco estudados do solo
apresentam grande relevância para a microbiologia ambiental, seja para estudos de
diversidade filogenética ou funcional (Gray and Head, 2001; Hugenholtz, 2002; Palleroni,
1997; Rappé and Giovannoni, 2003; Zinder and Salyers, 2005).
A abordagem metagenômica permite relacionar a identificação dos principais grupos
microbianos e seu papel ecológico no solo da Mata Atlântica. Por isso, não é possível
desconsiderar a possibilidade de que a fração até então não bem caracterizada pode apresentar
papel relevante nos processos ecológicos que ocorrem no solo. Embora a atribuição de

147
funções e de genes associados a marcadores filogenéticos, seja possível na ausência de
culturas (Gray and Head, 2001; Liesack and Dunfield, 2003), ela pode ser mais bem avaliada
em bactérias cultivadas
Acidobacteria são genética e metabolicamente versáteis, apresentam capacidade de degradar a
celulose e outros compostos normalmente encontrados em solos da floresta (Davis et al.,
2005). A abundância relativa de grupos Acidobacteria GP1 e GP2 sugere que estas
bactérias de crescimento lento podem desempenhar um papel fundamental nos
processos microbianos no solo. Acidobacteria também são capazes de crescer em baixo pH e
ambientes com baixa tensão de oxigênio como solos florestais, o que talvez explique a
predominância destas bactérias nas bibliotecas de clones de rRNA 16S. O pH parece ser um
dos principais fatores que afetam a diversidade de bactérias do solo (Fierer and Jackson,
2006). Entretanto, a estreita faixa de pH observada no presente estudo pode ter
obscurecido uma clara correlação entre distâncias genéticas e geográficas entre as seis
localidades.
Acidobacteria e Proteobacteria têm distribuição ubíqua no solo. O solo do tipo Terra Preta
da Amazônia indicou uma predominância de Proteobacteria e Acidobacteria em bibliotecas
de clones de rRNA 16S, o que compreende aproximadamente 30% das sequências do último
(Kim et al., 2007). Em outro estudo sobre a diversidade de bactérias do solo do Cerrado
brasileiro, a contribuição de Acidobacteria e Proteobacteria foi de 22% e 30%,
respectivamente (Quirino et al., 2009). No entanto, a contribuição da Acidobacteria foi baixa
nos solos de canaviais do sul do Brasil (Roesch et al., 2007). Um número significativo de
rRNA 16S (82,6%) foram agrupados como singletons ou doubletons (similaridade > 97 %).
Eles pertencem a Acidobacteria e Proteobacteria, bem como a grupos com baixa frequência
de ocorrência (Gemmatimonadetes, Planctomycetes, Cyanobacteria, Chloroflexi, DO1 e

148
OP10). Estas sequências podem ser consideradas membros da biosfera rara da microbiota do
solo da Mata Atlântica.
A análise de rarefação indica que apenas uma fração de toda a diversidade de espécies
foi recuperada (N = 93-123). Os números encontrados são extremamente conservadores
e parecem mostrar uma subestimativa da diversidade microbiana no solo da Mata
Atlântica. A complexidade genética da comunidade microbiana de solo, estimada por
experimentos de reassociação de DNA, mostrou que o tamanho da comunidade de uma
amostra de solo é equivalente a cerca de 10.000 genomas de E., coli (Torsvik et al., 1990;
Torsvik et al., 2002). Outro estudo baseado em análises moleculares mostrou que 30 g do solo
podem conter mais de 500.000 espécies (Doolittle, 1999). As estimativas baseadas em
análises não paramétricas apontam um número de espécie entre 6.400 e 38.000 espécies por
grama de solo (Curtis et al., 2002; Schloss and Handelsman, 2006). Estes resultados mostram
que a totalidade dos genomas microbianos encontrados em amostras de solo, pode ser
extremamente variado e na maioria das vezes a comunidade é altamente diversa. Estudos
utilizando sequenciamento de massivo serão necessários para descobrir a diversidade da
biosfera rara e o número exato de espécies por grama de solo da Mata Atlântica.
É importante perceber que as bibliotecas de rRNA 16S podem não representar uma
imagem completa ou exata da comunidade bacteriana. Entretanto, são capazes de
estabelecer um sólido panorama sobre os grupos dominantes. A diversidade de espécies é
tão grande que, geralmente, as bibliotecas de com cerca de 1.000 sequências clonadas são
capazes de representar apenas uma amostragem incompleta. Mesmo as análise do banco de
dados RDP (cerca de 70.000 sequências) não foi capaz de constituir um censo completo de
todos os genes rRNA 16S no planeta (Schloss and Handelsman, 2004). O número
significativo de sequências atribuída às bactérias não classificadas, de acordo com RDP

149
e GenBank no presente estudo, reforça a visão de que o solo da Mata Atlântica brasileira
é um grande reservatório de novos grupos de bactérias.
Dois grupos potencialmente novos foram identificados entre as OTUs mais frequentes. A
sequência completa do rRNA 16S do grupo 15 mostrou 98% de similaridade com sequências
do solo de relva na California e em agregados de solos agrícolas (Cruz-Martinez et al., 2009;
Hansel et al., 2008). Por outro lado,o grupo 18 mostrou apenas 95% de similaridade para as
sequências obtidas em solos do tipo Tundra (Costello and Schmidt, 2006; Nemergut et al.,
2008). Esses resultados sugerem ampla distribuição desses grupos no ambiente.
O bioma Mata Atlântica pode ser uma fonte potencial de novas biomoléculas, especialmente
aqueles com atividade celulolítica. O presente estudo é o primeiro levantamento global
sobre a diversidade da comunidade bacteriana em solos de Mata Atlântica. Este estudo
mostra claramente que o bioma Mata Atlântica representa um grande reservatório para
a descoberta da diversidade bacteriana. Isso representa um forte indício de que pode
existir alta riqueza de espécies que desempenham papel no ciclo do carbono através da
degradação de celulose da matéria orgânica depositada nos solos de Mata Atlântica.
No entanto, o solo também é rico em ácidos húmicos, que desnaturam o DNA e se ligam a
grupos fenólicos. Esta interações prejudicam a qualidade do DNA extraído por métodos
enzimáticos (Bertrand et al., 2005; Liles et al., 2008; Lombard et al., 2011; Robe et al., 2003;
Rondon et al., 2000). O DNA de alto peso molecular e livre dessas substâncias produz bons
resultados para reações enzimáticas como, amplificação, digestão e ligação. Esses processos
são cruciais na construção de uma biblioteca metagenômica para busca de moléculas bioativas
(Bertrand et al., 2005). Os resultados apresentados mostram que os melhores resultados
foram alcançados através da aplicação de um método de extração direta do DNA,
utilizando esferas de vidro, para obtenção de DNA de qualidade em concentração
necessária para a construção da biblioteca metagenômica do solo. Métodos de extração

150
direta de DNA são mais rápidos que indiretos e alcançam rendimentos até 100 vezes maiores
DNA (Gabor et al., 2003).
Embora grande parte das buscas por celulases através de abordagem metagenômica se
concentre em ambientes extremos (por exemplo, rúmen bovino), a busca em ambientes não
extremos, como o solo da Mata Atlântica, oferecem maior diversidade genética de celulases
altamente estáveis e adequadas para aplicações industriais (Voget et al., 2006). Considerando
a estimativa de 104 espécies de bactérias presentes no solo (Torsvik et al., 2002), uma
biblioteca metagenômica de milhões de clones (com tamanho médio de inserção de 30-40 kb)
é necessária para obtenção de pelo menos um representante de cada espécie bacteriana
(utilizando o tamanho do genoma de E.coli como referência). Neste sentido, a biblioteca
construída nesse trabalho precisa ser expandida para obter uma maior cobertura da
diversidade, uma vez que foi construída com um número estimado de 40.000 clones. As
técnicas de extração de DNA atualmente em uso dão acesso, principalmente, às populações
dominantes no solo. Foi possível recuperar cerca de milhares de clones com potencial
atividade celulolítica através do enriquecimento da biblioteca em meio mínimo contendo
CMC.
Estratégias de triagem funcional têm como objetivo detectar a reação biocatalítica
primeiro e, em seguida, caracterizar o gene responsável. A expressão heteróloga em
linhagens de E. coli pode limitar a atividade enzimática em caso de possíveis diferenças de
repertório metabólico e mecanismos de regulação moleculares em relação ao microrganismo
de origem. Uma pesquisa com 32 genomas procarióticos constatou que apenas 40% dos
genes podem ser expressas utilizando E. coli (Gabor et al., 2004). Além disso, mesmo quando
um gene é expresso, o nível de expressão pode ser tão baixo que não pode ser detectada
através de triagem funcional. Alguns trabalhos mostram um enorme esforço de triagem para a
detecção de poucos clones positivos (Daniel, 2004). As atividades avaliadas mostram que

151
foi detectada atividade celulolítica nos clones e isolados analisados. Um importante
aspecto do clone Cel 1A foi a presença de atividade -glucosidásica, em proporção
equilibrada em relação às atividades FPásica e CMCásica. Alguns trabalhos mostraram
sucesso no isolamento de genes envolvidos na degradação da celulose a partir de amostras de
solo (Kim et al., 2008; Voget et al., 2006; Wang et al., 2009).
A utilização de promoters de indução pode aumentar a probabilidade de encontrar enzimas
através da seleção funcional. Entretanto, não são capazes de influenciar na triagem caso a
atividade celulolítica de um gene dependa da atividade de outras enzimas para
disponibilização de substrato específico, quando em seu ambiente natural. Neste trabalho foi
utilizado um vetor com promotor induzido por arabinose, entretanto não realizamos sua
indução, testando o potencial de degradação do gene apenas em celulose como fonte de
carbono. Testes de indução devem ser realizados com os clones selecionados para otimização
da capacidade de degradação em experimentos posteriores.
Embora as abordagens independentes de cultivo permitam o isolamento de genes de celulases
a partir de amostras ambientais, incluindo metagenomas do solo, a maioria dos estudos relata
o isolamento de endocelulases (Feng et al., 2007; Healy et al., 1995; Kim et al., 2008; Voget
et al., 2006; Wang et al., 2009). Até o momento, o isolamento e caracterização de genes de
exocelulase da microbiota ruminal têm sido realizado com sucesso (Ferrer et al., 2005). No
entanto, como o taxa de descoberta de genes de celulase nesses estudos alcançou apenas de
0,001% a 0,5%. O isolamento de exocelulases a partir de metagenomas do solo parece uma
promissora fonte de novos genes celulolíticos (Fujii et al., 2011).
A detecção de atividade celulolítica em uma amostragem relativamente pequena de
clones demonstra o enorme potencial do metagenoma da microbiota do solo na busca de
moléculas bioativas. A possibilidade de manipulação da expressão de genes triados na
Floresta de Mata Atlântica brasileira representa um enorme potencial biotecnológico no que

152
diz respeito à utilização de enzimas celulolíticas na produção de biocombustíveis. Portanto,
demonstrado que o DNA metagenômico do solo da Mata Atlântica é uma fonte rica para
a descoberta e desenvolvimento biotecnológico.
O número limitado de linhagens que geralmente podem ser simultaneamente testados na
triagem com base em métodos de cultivo em grande volume inviabiliza uma triagem mais
ampla, eficaz e rápida de isolados ambientais e clones de bibliotecas metagenômicas. O
desenvolvimento de estratégias destinadas a permitir a miniaturização e avaliação paralela de
um grande número de linhagens representa uma importante ferramenta na busca de novos
genes codantes de enzimas microbianas com atividade celulolítica (Cianchetta et al., 2010).
4.4 Diversidade microbiana dos biomas brasileiros
4.4.1 Resultados
Neste capitulo realizei uma análise exploratória da diversidade microbiana de ecossistemas
tipicamente brasileiros abordada por métodos independente de cultivo. Foi realizada uma
busca bibliográfica baseada nos principais bancos de dados de publicações científicas (Pub
Med e Web os Science) para levantamento das publicações e sequências disponíveis no NCBI
(National Center for Biotechnology Information). Por meio da análise de sequencias de
rRNA 16S oriundas de diferentes estudos foi possivel estabelecer um panorama geral sobre os
principais grupos microbianos associados aos biomas brasileiros. Foi demonstrado que a
microbiota associada é característica e adaptada as condições ambientais. Foram analisados
mais de 70 trabalhos sobre o estudo da diversidade microbiana utilizando métodos
independentes de cultivo em todos os sete principais biomas brasileiros (Amazônia, Mata
Atlântica, Cerrado, Pantanal, Caatinga e Zona Costeira) nos últimos 5 anos (Tabelas 12 e 13).
Além disso, foi listada a produção sobre descrição de espécies microbianas através de

153
métodos moleculares (aproximadamente 30 artigos). A compilação desses dados fornece um
panorama geral sobre a diversidade microbiana identificada no Brasil nos últimos anos.
Existe diferenciação clara entre a microbiota (Bactéria e Arquéia) de biomas terrestres e
aquáticos de acordo com analises de sequências do gene ribossomal rRNA 16S (Figuras 52 A
e B). As análises de componentes principais mostraram que as sequências agruparam de
acordo com o ecossitema associado (Figura 52 C e D). Foram distinguidos três grupos. O
primeiro grupo consistiu de três bibliotecas de amostras de solo (Araujo and Kruger; Bruce et
al., 2010; Faoro et al., 2010b). O segundo grupo, constituído de quatro bibliotecas da água do
mar (Clementino et al., 2008; Cury et al., 2011; de Castro et al., 2010b; Reis et al., 2009) e o
terceiro grupo foi composto por quatro bibliotecas de microorganismos associados com
animais marinhos (de Castro et al., 2010b; Hardoim et al., 2009; Lins-de-Barros et al., 2010b;
Reis et al., 2009). As diferenças mais evidentes entre os ambientes terrestres e de água do mar
estão relacionadas ao fato de que Acidobacteria, Actinobacteria e Firmicutes foram mais
comuns em solos, enquanto que Cyanobacteria e Bacteroidetes foram mais abundantes em
água (Figura 52 C e D). Grande número de rRNA 16S (cerca de 10% no solo e 20% em água)
não foram classificadas e portanto representam diversidade desconhecida, possivelmente
novas espécies ou até mesmo famílias ou filos. Entre as Arquéias, Euryarchaeota
predominam em ecossitemas terrestres, enquanto Crenarchaeota são dominantes nos
ecossitemas marinhos e uma grande fração (> 40 %) é desconhecida.
Um total de 18 espécies bacterianas, 32 de fungos 12 protozoários foram descritas no Brasil
nos últimos 5 anos (Tabela 14 e 15). A descrição taxonômica de espécies de protozoários é
escassa e é realizada principalmente baseada em características morfológicas (Dias et al.,
2010b; Siqueira-Castro et al., 2009). Enquanto a descrição de novas espécies de bactérias e
fungos é submetida à taxonomia polifásica. Os novos táxons foram isolados de diferentes
fontes e localidades de biomas brasileiros terrestres e aquáticos.

154
Atualmente, existem mais de 1.532 genomas completos de procarioto sequenciados e cerca
4.726 projetos genoma em andamento de acordo com o NCBI
(http://www.ncbi.nlm.nih.gov/sites/genome). Cerca de 35 genomas estão depositados no
NCBI por projetos brasileiros. Mais de 20 genomas estão publicados. Há 52 genomas de
Leptospira oriundos da FIOCRUZ que foram sequenciadas pelo J. Craig Venter Institute.

155
Figura 52. Parte superior: análise das componentes principais (PCoA) do pareamenteo unweight através da matriz de distância
do FastUniFrac . Cada ponto corresponde a comparação da presença ou ausência dos grupos taxonômicos de bactérias em
diferentes biomas. Triângulos verdes, círculos roxos e hexágonos vermelhos representam microrganismos associados a solo,
água e organismos marinhos. Comunidades de (A) Bactérias e (B) Archaea, respectivamente. A porcentagem de variação
explicada pelo traçado das componentes principais está indicada nos eixos. Um total de 6.540 e 1.893 sequências do gene 16S
rRNA foram alinhadas para Bacterias e Archaea, respectivamente. Foram selecionadas sequências referentes as regiões V1 e V2
do gene rRNA 16S. Sequências curtas menores que 300 pb foram retiradas da análise. Archeas do solo não foram incluídas.
Parte inferior: classifcação taxonômica baseada nas sequências do gene rRNA 16S através da ferramenta Classifier do RDP. (C)
Contribuição dos principais filos bacterianos e (D) Contribuição de filos do domínio Archeae.

156
Tabela 12. Lista de estudos de diversidade microbiana nos principais biomas brasileiros por abordagens
independentes de cultivo em ambientes terrestres.
Bioma Local Abordagem Molecular Amostras Referências
Solo Agrícola Paraná PCR e DGGE do gene rRNA 16S Solo (Nakatani et
al., 2011)
Mangue Bahia PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S
Rizosfera e
Sedimentosos
(Peixoto et
al., 2011)
Cerrado Mato Grosso do Sul ITS, PCR e sequenciamento do gene
rRNA 16S Solo
(Alves-Prado
et al., 2010)
Mata Atlântica Rio de Janeiro Sequenciamento de biblioteca de clones
do gene rRNA 16S Solo
(Bruce et al.,
2010)
Mangue São Paulo PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Sedimentos
(Dias et al.,
2010a)
Mata Atlântica Paraná Sequenciamento de biblioteca de clones
do gene rRNA 16S Solo
(Faoro et al.,
2010b)
Mangue Rio de Janeiro Pirosequenciamento do gene rRNA 16S Rizosfera (Gomes et
al., 2010a)
Mangue Rio de Janeiro
PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S;
PCR, DGGE e sequenciamento de
biblioteca de clones do gene ndo
Rizosfera (Gomes et
al., 2010b)
Amazônia Amazonas
PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S
e análise por T-RFLP
Solo (Grossman et
al., 2010)
Campo de Cana
de Açúcar São Paulo
Sequenciamento dos genes rRNA 16S e
gyrB
Rizosfera e
Raiz
(Luvizotto et
al., 2010)
Mangue São Paulo
PCR, DGGE e sequenciamento de
biblioteca de clones dos genes dsrB e
mcrA
Sedimentos (Taketani et
al., 2010b)
Mangue Bahia PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Sedimentos
(Taketani et
al., 2010a)
Amazônia Amazonas T-RFLP e sequenciamento de biblioteca
de clones do gene rRNA 16S; Real-
Solo (Taketani and

157
Time PCR e sequenciamento do clone
do gene amoA
Tsai, 2010)
Cultivo de
Eucalipto
Transgênico
São Paulo
Real-Time PCR quantitativo, PCR,
DGGE e sequenciamento de biblioteca
de clones do gene rRNA 16S
Rizosfera (Andreote et
al., 2009)
Amazônia Rondônia PCR e DGGE do gene rRNA 16S Solo (Cenciani et
al., 2009)
Amazônia Amazonas T-RFLP e sequenciamento de biblioteca
de clones do gene rRNA 16S Solo
(Jesus et al.,
2009)
Amazônia Amazonas Sequenciamento de clones do gene
rRNA 16S
Nódulos de
raízes
(Lima et al.,
2009)
Mata Atlântica Bahia PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Solo
(Maciel et al.,
2009)
Caatinga Sergipe PCR, ARDRA, DGGE e
sequenciamento do gene rRNA 16S Rizosfera
(Monteiro et
al., 2009)
Mata Atlântica Rio de Janeiro
PCR quantitativo, PCR e
sequenciamento de biblioteca de clones
dos genes rpoB e gyrB
Solo
Sedimentos
(Rodrigues et
al., 2009)
Mata Atlântica Rio de Janeiro PCR e DGGE do gene rpoB Solo (Aboim et al.,
2008)
Transgenic
Tobacco
cultivar
São Paulo PCR, ARDRA, DGGE e
sequenciamento do gene rRNA 16S Rizosfera
(Andreote et
al., 2008)
Campo de
Trigo Rio Grande do Sul
PCR, RFLP do gene nifH e
sequenciamento do gene rRNA 16S
Rizosfera e
massa de
solos
(Beneduzi et
al., 2008a)
Campo de
Arroz Rio Grande do Sul
PCR, RFLP do gene nifH e
sequenciamento do gene rRNA 16S
Rizosfera e
massa de
solos
(Beneduzi et
al., 2008b)
Mangue Rio de Janeiro PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Sedimentos
(Gomes et
al., 2008)
Campo de Rio Grande do Sul
Sequenciamento de biblioteca de clones Rizosfera, (Roesch et

158
Milho do gene nifH Raízes e
Caule
al., 2008)
Cerrado Minas Gerais PCR, DGGE e sequenciamento do gene
rpoB Rizosfera
(Coelho et
al., 2007)
Amazônia Amazonas Sequenciamento de biblioteca de clones
do gene rRNA 16S Solo
(Kim et al.,
2007)
Campo de Cana
de Açúcar São Paulo
BOX PCR do gene rRNA 16S e
sequenciamento do gene RecA
Rizosfera e
Raiz
(Mendes et
al., 2007)
Plantio de Soja Paraná PCR, DGGE ; RFLP e BOX - PCR do
gene rRNA 16S Solo e nódulo
(Pereira et
al., 2007)
Campo de Cana
de Açúcar Rio Grande do Sul Pirosequenciamento do gene rRNA 16S Solo
(Roesch et
al., 2007)
Mata Atlântica Paraná e São Paulo BOX-PCR do gene nifH e
sequenciamento do gene rRNA 16S Raízes
(Albino et al.,
2006)
Mangue Rio de Janeiro T-RFLP e sequenciamento do gene
rRNA 16S Sedimentos
(Brito et al.,
2006)
Campo de
Milho Rio de Janeiro PCR e DGGE do gene rRNA 16S Rizosfera
(Costa et al.,
2006b)
Cerrado São Paulo ITS, PCR, DGGE e sequenciamento do
clone do gene rRNA 16S Solo
(de Oliveira
et al., 2006)
Solo Agrícola PR, RS, DF, AM, MG e
GO
ERIC-REP PCR, RAPD PCR, RFLP e
sequenciamento de biblioteca de clones
do gene rRNA 16S
Solo (Hungria et
al., 2006)
Mata Atlântica São Paulo Sequenciamento de biblioteca de clones
do gene rRNA 16S Filosfera
(Lambais et
al., 2006)
Mangue Rio de Janeiro Sequenciamento do gene rRNA 16S Rizosfera
(Maciel-
Souza et al.,
2006)
Mata Atlântica Rio de Janeiro Sequenciamento do gene rRNA 16S Solo (Souchie et
al., 2006)
Pantanal Mato Grosso do Sul PCR e ARDRA do gene rRNA 16S Raízes (Brasil et al.,
159
2005)
Cultivo de
Milho Minas Gerais
PCR, DGGE e sequenciamento de
biblioteca de clones do gene rpoB Solo
(da Mota et
al., 2005)
Amazônia Amazonas Sequenciamento do gene rRNA 16S Sedimentos (Fiore et al.,
2005)
Tabela 13. Lista de estudos de diversidade microbiana dos principais biomas brasileiros por abordagens
independentes de cultivo em ambientes aquáticos nos últimos anos.
Bioma Local Abordagem Molecular Amostras Referências
Zona de ressurgências
de Cabo Frio Rio de Janeiro
PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Água
(Cury et al.,
2011)
Baía de Guanabara Rio de Janeiro Pirosequenciamento do gene rRNA 16S Água (Thompson
et al., 2011)
Lagoa da Conceição Santa Catarina PCR e DGGE do gene rRNA 16S Água (Fontes et
al., 2011)
Banco de Abrolhos Bahia Sequenciamento do gene pyrH Muco de Coral (Alves et
al., 2010)
Banco de Abrolhos Bahia PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Coral
(de Castro
et al.,
2010b)
Bacia de Campos Rio de Janeiro PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Água
(Korenblum
et al., 2010)
Praia da Tartaruga Rio de Janeiro PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Coral
(Lins-de-
Barros et
al., 2010b)
Praia de Guaecá, Ilhota
da Prainha e Ilha de
Toque-Toque
São Paulo ITS, PCR, ARDRA e sequenciamento do
gene rRNA 16S
Esponjas,
ascídias e algas
(Menezes et
al., 2010)
Arquipélago das
Cagarras Praia
Vermelha
Rio de Janeiro PCR e sequenciamento do gene rRNA
16S Esponjas
(Fontana et
al., 2010)
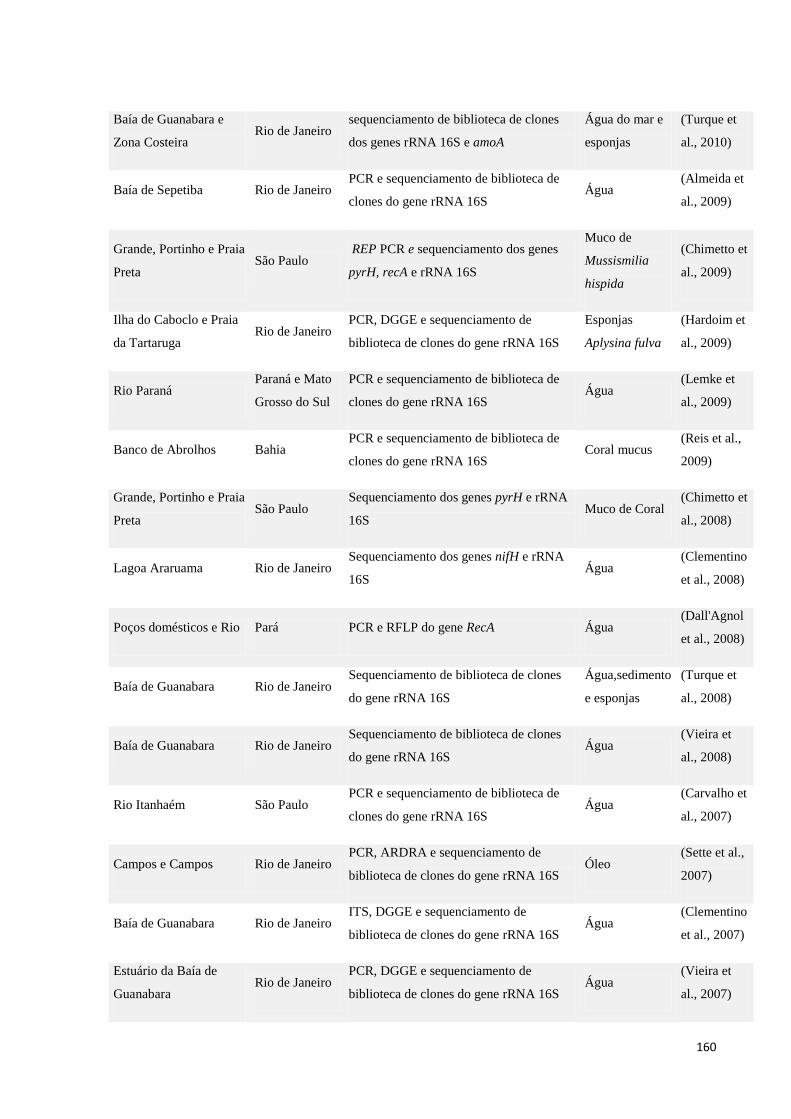
160
Baía de Guanabara e
Zona Costeira Rio de Janeiro
sequenciamento de biblioteca de clones
dos genes rRNA 16S e amoA
Água do mar e
esponjas
(Turque et
al., 2010)
Baía de Sepetiba Rio de Janeiro PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Água
(Almeida et
al., 2009)
Grande, Portinho e Praia
Preta São Paulo
REP PCR e sequenciamento dos genes
pyrH, recA e rRNA 16S
Muco de
Mussismilia
hispida
(Chimetto et
al., 2009)
Ilha do Caboclo e Praia
da Tartaruga Rio de Janeiro
PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S
Esponjas
Aplysina fulva
(Hardoim et
al., 2009)
Rio Paraná Paraná e Mato
Grosso do Sul
PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Água
(Lemke et
al., 2009)
Banco de Abrolhos Bahia PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Coral mucus
(Reis et al.,
2009)
Grande, Portinho e Praia
Preta São Paulo
Sequenciamento dos genes pyrH e rRNA
16S Muco de Coral
(Chimetto et
al., 2008)
Lagoa Araruama Rio de Janeiro Sequenciamento dos genes nifH e rRNA
16S Água
(Clementino
et al., 2008)
Poços domésticos e Rio Pará PCR e RFLP do gene RecA Água (Dall'Agnol
et al., 2008)
Baía de Guanabara Rio de Janeiro Sequenciamento de biblioteca de clones
do gene rRNA 16S
Água,sedimento
e esponjas
(Turque et
al., 2008)
Baía de Guanabara Rio de Janeiro Sequenciamento de biblioteca de clones
do gene rRNA 16S Água
(Vieira et
al., 2008)
Rio Itanhaém São Paulo PCR e sequenciamento de biblioteca de
clones do gene rRNA 16S Água
(Carvalho et
al., 2007)
Campos e Campos Rio de Janeiro PCR, ARDRA e sequenciamento de
biblioteca de clones do gene rRNA 16S Óleo
(Sette et al.,
2007)
Baía de Guanabara Rio de Janeiro ITS, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Água
(Clementino
et al., 2007)
Estuário da Baía de
Guanabara Rio de Janeiro
PCR, DGGE e sequenciamento de
biblioteca de clones do gene rRNA 16S Água
(Vieira et
al., 2007)

161
Mangue Ceará PCR e DGGE do gene rRNA 16S Água (Sousa et
al., 2006)
Rio Negro Manaus REP PCR e sequenciamento dos genes
16S e 23S rRNA Água
(Hungria et
al., 2005)
Tabela 14. Lista de novos táxons de ambientes terrestres descritos nos últimos 5 anos.
Espécies Bioma Local Abordagem Amostra Referências
Bactéria
Burkholderia
mimosarum Mata Atlântica
Sudeste
brasileiro Polifásica
Nódulos de raízes
(Mimosa pigra e M.
scabrella)
(Chen et al.,
2006)
Paenibacillus
rioGrandensis Rizosfera
Rio Grande do
Sul Polifásica
Rizosfera de trigo
(Triticum
aestivum)
(Beneduzi
et al., 2010)
Stenotrophomonas
pavanii
Cana de
Açúcar Paraná Polifásica
Caule de Cana de
Açúcar (Saccharum
officinarum)
(Ramos et
al., 2010)
Streptomyces
lunalinharesii Cerrado Brasil central Polifásica Acid orthic ferralsol
(Souza et
al., 2008)
Burkholderia sabiae Cultivo
Agrícola Brasil Polifásica
Árvore Leguminosa
(Mimosa
caesalpiniifolia)
(Chen et al.,
2008)
Metschnikowia
cerradonensis Cerrado Tocantins Polifásica
Flores (Ipomoea
carnea) e besouro
(Conotelus beetle)
(Rosa et al.,
2007a)
Burkholderia nodosa Mata Atlântica Sudeste Brasil Polifásica
Nódulos de raízes
(Mimosa
bimucronata e
Mimosa scabrella)
(Chen et al.,
2007)
Ceidatus
Magnetoglobus
multicellularis
Lagoa de
Araruama Rio de Janeiro
Morfologia e
gene 16S
rDNA
Lagoa costeira
hipersalina
(Abreu et
al., 2007)

162
Azorhizobium
doebereinerae Raízes
Minas Gerais
e Rio de
Janeiro
Polifásica
Nódulos de raízes de
espécies lenhosas
(Sesbania virgata)
(Maria de
Souza
Moreira et
al., 2006)
Burkholderia ferrariae Minério de
Ferro Minas Gerais Polifásica
Suspensão de
minério de Ferro rica
em Fósforo
(Valverde et
al., 2006)
Burkholderia
silvatlantica
Cana de
Açúcar e
Cultivo de
Milho
Rio de Janeiro Polifásica Rizosfera (Perin et al.,
2006)
Fungi
Ceida queiroziae Mata Atlântica Minas Gerais
Análise da
região D1/D2
da subunidade
maior do gene
rRNA do gene
rDNA e
Morfologia
Madeira podre e
insetos de madeira
(Santos et
al., 2011)
Lachancea mirantina Processo de
Fermentação Pernambuco
Análise da
região D1/D2
da subunidade
maior dos
genes rRNA e
rDNA, região
ITS e gene
EFA1
Processo de
fermentação na
produção de cachaça
(Pereira et
al., 2011)
Wickerhamiella
pagnoccae e Ceida
jalapaonensis
Cerrado Tocantins
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Néctar de flores de
plantas brácteas
(Heliconia
psittacorum)
(Barbosa et
al., 2011)
Ophiocordyceps
rufipedis, O. balzani,
O. melanotici e
O.novogranadensis
Mata Atlântica Minas Gerais Morfologia Formigas
carpinteiras
(Evans et
al., 2011)

163
Hypochnella
verrucospora Mata Atlântica
Rio Grande do
Sul Morfologia
Madeira de árvore
angiosperma
apodrecida
(Piptadenia
gonoacanthae)
(Coelho et
al., 2010)
Trichosporon chiarellii Formigueiro São Paulo
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e ITS e
Morfologia
Formiga Saúva (Atta
capiguara)
(Pagnocca
et al., 2010)
Ceida golubevii Pantanal Mato Grosso
do Sul
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Flor de Ipomoea sp. (Rosa et al.,
2009b)
Ceida aechmeae e
Ceida vrieseae Parque Itapuã
Rio Grande do
Sul
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Folhas de bromélias
(Aechmea recurvata
e Billbergia nutans)
e água de tanque de
bromélia Vriesea
gigantea
(Landell et
al., 2010)
Spathaspora
arborariae
Mata Atlântica
e cerrado Minas Gerais
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Madeira Podre (Cadete et
al., 2009)
Wickerhamomyces
queroliae e Ceida
jalapaonensis
Cerrado Tocantins
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Larva de Anastrepha
mucronata de frutos
maduros de
Peritassa campestris
e flores de
Centropogon
cornutus
(Rosa et al.,
2009c)
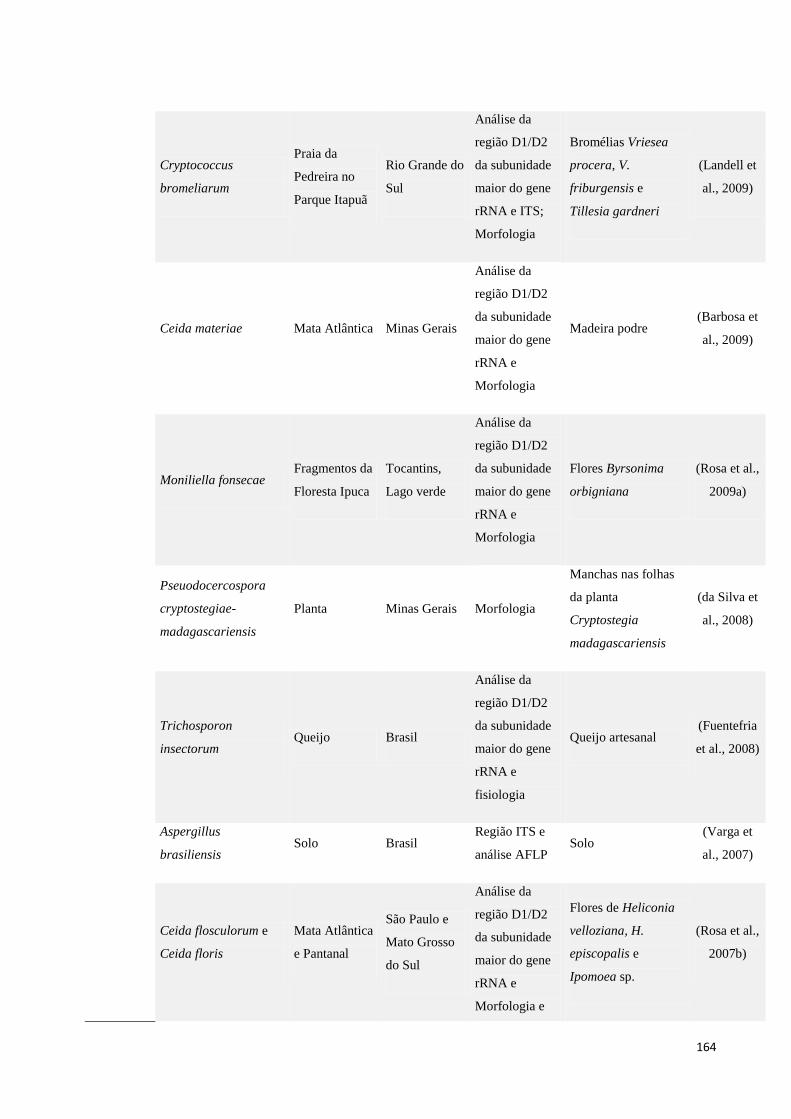
164
Cryptococcus
bromeliarum
Praia da
Pedreira no
Parque Itapuã
Rio Grande do
Sul
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e ITS;
Morfologia
Bromélias Vriesea
procera, V.
friburgensis e
Tillesia gardneri
(Landell et
al., 2009)
Ceida materiae Mata Atlântica Minas Gerais
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Madeira podre (Barbosa et
al., 2009)
Moniliella fonsecae Fragmentos da
Floresta Ipuca
Tocantins,
Lago verde
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia
Flores Byrsonima
orbigniana
(Rosa et al.,
2009a)
Pseuodocercospora
cryptostegiae-
madagascariensis
Planta Minas Gerais Morfologia
Manchas nas folhas
da planta
Cryptostegia
madagascariensis
(da Silva et
al., 2008)
Trichosporon
insectorum Queijo Brasil
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
fisiologia
Queijo artesanal (Fuentefria
et al., 2008)
Aspergillus
brasiliensis Solo Brasil
Região ITS e
análise AFLP Solo
(Varga et
al., 2007)
Ceida flosculorum e
Ceida floris
Mata Atlântica
e Pantanal
São Paulo e
Mato Grosso
do Sul
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia e
Flores de Heliconia
velloziana, H.
episcopalis e
Ipomoea sp.
(Rosa et al.,
2007b)
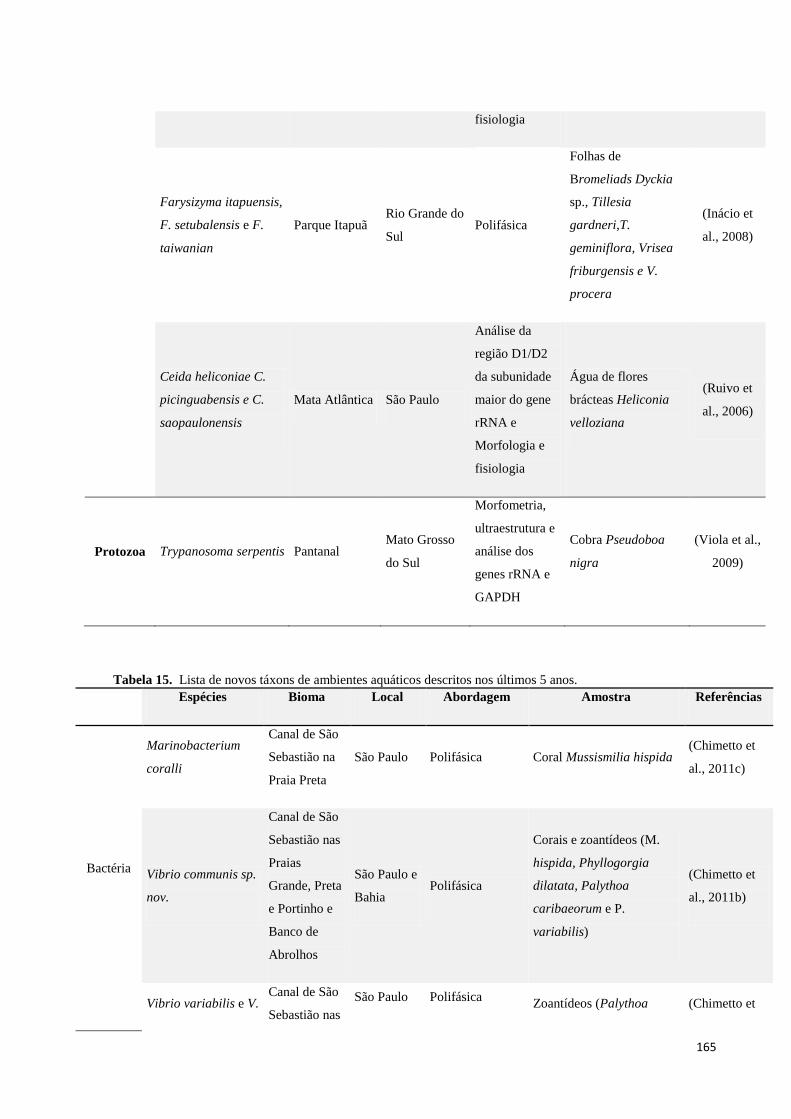
165
fisiologia
Farysizyma itapuensis,
F. setubalensis e F.
taiwanian
Parque Itapuã Rio Grande do
Sul Polifásica
Folhas de
Bromeliads Dyckia
sp., Tillesia
gardneri,T.
geminiflora, Vrisea
friburgensis e V.
procera
(Inácio et
al., 2008)
Ceida heliconiae C.
picinguabensis e C.
saopaulonensis
Mata Atlântica São Paulo
Análise da
região D1/D2
da subunidade
maior do gene
rRNA e
Morfologia e
fisiologia
Água de flores
brácteas Heliconia
velloziana
(Ruivo et
al., 2006)
Protozoa Trypanosoma serpentis Pantanal Mato Grosso
do Sul
Morfometria,
ultraestrutura e
análise dos
genes rRNA e
GAPDH
Cobra Pseudoboa
nigra
(Viola et al.,
2009)
Tabela 15. Lista de novos táxons de ambientes aquáticos descritos nos últimos 5 anos.
Espécies Bioma Local Abordagem Amostra Referências
Bactéria
Marinobacterium
coralli
Canal de São
Sebastião na
Praia Preta
São Paulo Polifásica Coral Mussismilia hispida (Chimetto et
al., 2011c)
Vibrio communis sp.
nov.
Canal de São
Sebastião nas
Praias
Grande, Preta
e Portinho e
Banco de
Abrolhos
São Paulo e
Bahia Polifásica
Corais e zoantídeos (M.
hispida, Phyllogorgia
dilatata, Palythoa
caribaeorum e P.
variabilis)
(Chimetto et
al., 2011b)
Vibrio variabilis e V. Canal de São
Sebastião nas
São Paulo Polifásica Zoantídeos (Palythoa (Chimetto et

166
marinus praias
Grande, Preta
e Portinho
caribaeorum) al., 2011a)
Marinomonas
brasilensis
Canal de São
Sebastião na
Praia Grande
São Paulo Polifásica Coral (M. hispida) (Chimetto et
al., 2010a)
Photobacterium
jeanii
Banco de
Abrolhos e
Canal de São
Sebastião nas
Praias
Portinho e
Preta
Bahia e São
Paulo Polifásica
Corais e zoantídeos
(Phyllogorgia dilatata e
Palythoa caribaeorum)
(Chimetto et
al., 2010b)
Limnohabitans
australis Lagoa São Paulo Polifásica Lagoa de água doce
(Hahn et al.,
2010)
Fungi
Kabatana rondoni Peixe da
Amazônia Amazônia
Ultra-estrutural e
molecular
Peixe
Gymnorhamphichthys
rondoni
(Casal et al.,
2010)
Potaspora
morhaphis
Rio
Amazonas Pará
Microscopia
ótica, ultra
estrutura e região
ITS e18S rDNA
Peixe Potamorhaphis
guianensis
(Casal et al.,
2008)
Protozoa
Myxobolus brycon Pantanal
Mato
Grosso do
Sul
Morfologia e
ultra-estrutural Peixe Brycon hilarii
(Azevedo et
al., 2011)
Myxobolus oliveirai Pantanal
Mato
Grosso do
Sul
Morfologia,
ultra-estruturais,e
gene 18S rDNA
Peixe Brycon hilarii (Milanin et al.,
2010)
Myxobolus sciades Rio Poti Piauí Microscopia
ótica e eletrônica Peixe Sciades herzbergii
(Azevedo et
al., 2010)
Henneguya
hemiodopsis Rio Poti Piauí Ultra-estrutural
Peixe Hemiodopsis
microlepes
(Azevedo et
al., 2009c)
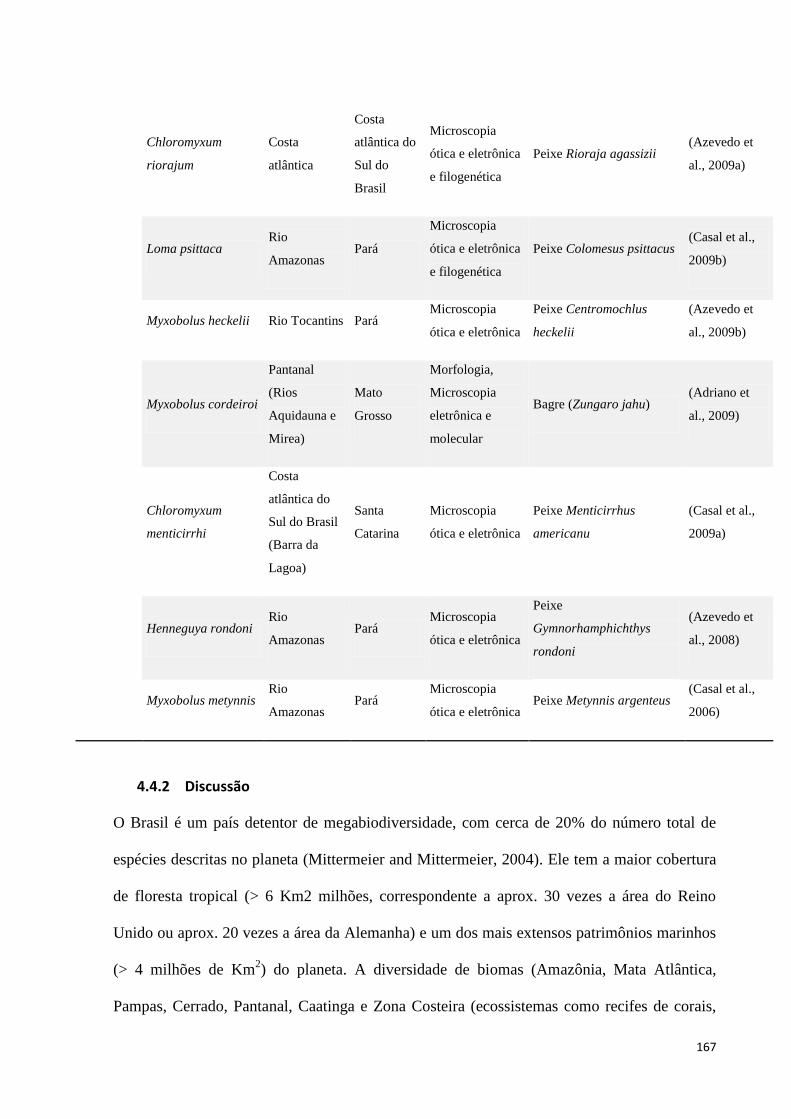
167
Chloromyxum
riorajum
Costa
atlântica
Costa
atlântica do
Sul do
Brasil
Microscopia
ótica e eletrônica
e filogenética
Peixe Rioraja agassizii (Azevedo et
al., 2009a)
Loma psittaca Rio
Amazonas Pará
Microscopia
ótica e eletrônica
e filogenética
Peixe Colomesus psittacus (Casal et al.,
2009b)
Myxobolus heckelii Rio Tocantins Pará Microscopia
ótica e eletrônica
Peixe Centromochlus
heckelii
(Azevedo et
al., 2009b)
Myxobolus cordeiroi
Pantanal
(Rios
Aquidauna e
Mirea)
Mato
Grosso
Morfologia,
Microscopia
eletrônica e
molecular
Bagre (Zungaro jahu) (Adriano et
al., 2009)
Chloromyxum
menticirrhi
Costa
atlântica do
Sul do Brasil
(Barra da
Lagoa)
Santa
Catarina
Microscopia
ótica e eletrônica
Peixe Menticirrhus
americanu
(Casal et al.,
2009a)
Henneguya rondoni Rio
Amazonas Pará
Microscopia
ótica e eletrônica
Peixe
Gymnorhamphichthys
rondoni
(Azevedo et
al., 2008)
Myxobolus metynnis Rio
Amazonas Pará
Microscopia
ótica e eletrônica Peixe Metynnis argenteus
(Casal et al.,
2006)
4.4.2 Discussão
O Brasil é um país detentor de megabiodiversidade, com cerca de 20% do número total de
espécies descritas no planeta (Mittermeier and Mittermeier, 2004). Ele tem a maior cobertura
de floresta tropical (> 6 Km2 milhões, correspondente a aprox. 30 vezes a área do Reino
Unido ou aprox. 20 vezes a área da Alemanha) e um dos mais extensos patrimônios marinhos
(> 4 milhões de Km2) do planeta. A diversidade de biomas (Amazônia, Mata Atlântica,
Pampas, Cerrado, Pantanal, Caatinga e Zona Costeira (ecossistemas como recifes de corais,

168
ilhas oceânicas, manguezais, marismas e de mar profundo)) (IBGE, 2004), permite a
diversificação de uma variedade de formas de vida. Há de fato uma quantidade considerável
de literatura sobre a biodiversidade de plantas e animais no Brasil. Sua diversidade é usada
como um parâmetro fundamental na implementação das ações de gestão em áreas prioritárias
para a conservação (Myers et al., 2000). Estudos sobre a diversidade microbiana brasileira
são relativamente escassos. Por exemplo, só recentemente a diversidade microbiana dos
solos da Amazônia e do Cerrado e do meio marinho tem sido estudada de maneira
sistemática. O objetivo deste capítulo foi estabelecer uma visão geral sobre os estudos de
diversidade microbiana (cerca de 100 trabalhos publicados) realizados no Brasil,
principalmente nos últimos cinco anos, incluindo estudos taxonômicos baseados em
microrganismos cultivados e não cultivados (baseada em perfis moleculares e rRNA 16S
bibliotecas de clones). O capítulo se concentrou principalmente em procariontes ambientais
(meio aquático e associados a organismos marinhos), aplicados à agricultura (culturas de
promotores de crescimento de plantas) e de importância biotecnológica (biorremediação).
Entre os mais de 70 estudos sobre comunidades microbianas levantados, destacamos os
ecossistemas mais amostrados nos últimos anos. Este levantamento permitiu a obtenção de
um panorama da pesquisa nacional sobre a diversidade microbiana dos principais
biomas brasileiros.
4.4.2.1 Diversidade microbiana nos biomas terrestres
Pelo menos um estudo foi realizado em cada grande brasileiro Bioma brasileiro (Tabela 12).
A Amazônia é um dos principais hostspots da biodiversidade global. No entanto, a
atenção para a biodiversidade microbiana foi rara nas últimas décadas (Borneman and
Triplett, 1997). Na região da Amazônia Ocidental um tipo de solo, conhecido como "Terra
Preta" é capaz de acumular matéria orgânica estável (Lima et al., 2002). Este tipo de solo tem
sido manejado com sucesso por comunidades indígenas pré-colombianas (cerca de 800 anos

169
atrás) (Mann, 2002). Este solo caracteriza-se pela alta fertilidade e armazenamento de
carbono. Estas características fazem desta região interessante para a investigação de
microrganismos promotores de crescimento de plantas. A indução do crescimento ocorre
pela síntese de compostos microbianos (por exemplo, vitaminas, antibióticos) que facilita a
absorção de nutrientes. Um total de 14 filos foram identificados na Terra Preta e apenas 9
filos foram identificadas em solos adjacentes inalterados, com uma prevalência de
Acidobacteria (Kim et al., 2007). O tipo de solo Terra Preta apresenta riqueza de espécies
25% maior que no solo da floresta. Crenarchaeota apresentou predominância no solo
adjacente inalterado (Cenciani et al., 2009; Grossman et al., 2010; Jesus et al., 2009). Este
grupo de Arquéia tem uma contribuição significativa no ciclo de nitrogênio do solo por meio
de oxidação da amônia (Taketani and Tsai, 2010). Até agora, o isolamento e caracterização
taxonômica microbiana não foram realizados neste solo, a fim de encontrar possíveis novos
microrganismos que possam ser utilizados na fertilização de solos, como já foi visto para
outros biomas (Adesemoye et al., 2009; Beneduzi et al., 2008a; Miransari, 2011).
Claramente, a pesquisa visando à biodiversidade microbiana na Floresta Amazônica,
tem apenas começado a divulgar o enorme potencial do reservatório inexplorado da
diversidade microbiana.
A Mata Atlântica é também um hotspot de diversidade, e apenas 10% de sua cobertura
original ainda está preservada (Myers et al., 2000). A diversidade de comunidades
microbianas da Mata Atlântica nos estados do Rio de Janeiro, Paraná e São Paulo
foram analisadas. Há um claro domínio do filo Acidobacteria (Bruce et al., 2010; Faoro et
al., 2010b). Bactérias como Enterobacteriaceae e Bacillus sp. e fungos micorrízicos como
Aspergillus sp., Glomus macrocarpum e Glomus etunicatum foram identificados e parecem
executar funções essenciais para a saúde do solo da Mata Atlântica (Souchie et al., 2006). A
microbiota do solo da Mata Atlântica pode reduzir sulfatos e promover a reabilitação de

170
ambientes contaminados por metais pesados (Muyzer and Stams, 2008). Sulfatos de metais
(por exemplo, cádmio, cobalto, cobre, ferro, níquel e zinco) são altamente solúveis, enquanto
que os sulfetos de metais correspondentes de baixa solubilidade permitem a precipitação e
remoção de contaminantes por microrganismos do solo. Ecossistemas de mangue estão
incluídos no bioma Mata Atlântica, e são encontrados em zonas de transição entre os
ecossistemas terrestres, marinhos e de água doce por mais de 6 mil quilômetros de costa. Os
manguezais são considerados berçários de diversidade, embora estes ecossistemas estejam
sujeitos à intensa interferência antrópica modificação no ecossistema através das atividades
urbanas e industriais (Amado-Filho et al., 2008).
Em janeiro de 2000, as áreas de mangue da Baía de Guanabara sofreram uma das maiores
catástrofes ambientais da região devido ao vazamento de cerca de 1,3 milhões de litros de
petróleo (Gabardo et al., 2000). A caracterização da diversidade bacteriana e biorremediação
pelos microorganismos que habitam o manguezal representam importantes abordagens que
podem melhorar a gestão desses ambientes. As comunidades microbianas do manguezal
refletem a variação espacial da composição dos sedimentos (Peixoto et al., 2011; Taketani
et al., 2010a). A maior abundância de plasmídeos (por exemplo, RIPC-1a, 1b-RIPC, RIPC
RIPC-7 e-9) e genes funcionais (naftaleno e intradiol dioxigenases extradiol) envolvidos na
degradação de hidrocarbonetos foram identificados nas comunidades microbianas após a
exposição ao petróleo (Gomes et al., 2010b). Aparentemente, os níveis elevados de petróleo
estão significativamente associados com Betaproteobacteria, enquanto os níveis de
hidrocarbonetos aromáticos policíclicos estão associados com Actinobacteria. Gêneros
aeróbios envolvidos na degradação do óleo e anaeróbias, incluindo Alteromonadales,
Burkholderiales, Pseudomonadales, Rhodobacterales, Rhodocyclales, Marinobacter,
Alcanivorax, Microbulbifer, Sphingomonas, Micrococcus, Cellulomonas, Dietzia e Gordonia
foram detectados em áreas impactadas (Brito et al., 2006; Gomes et al., 2008). Áreas de

171
mangue não impactado apresentam comunidade dominada por Alphaproteobacteria,
Gammaproteobacteria e Acidobacteria, enquanto os componentes secundários das
comunidades foram identificados como Betaproteobacteria e Actinobacteria (Dias et al.,
2010a). Além disso, as baixas concentrações de oxigênio e alta concentração de matéria
orgânica criam condições propícias para o estabelecimento de organismos anaeróbicos.
Bactérias redutoras de enxofre e Arquéia metanogênicas foram identificadas em
diferentes profundidades de sedimentos. Os padrões de distribuição de genes
relacionados a esses processos metabólicos (redutase sulfato - dsrB e metil-coenzima A
redutase M-mcrA) mostraram uma maior diversidade em sedimentos de profundidades
superiores (Taketani et al., 2010b).
Estudos sobre os campos de Cerrado incidiram sobre os grupos de microrganismos que
desempenham um papel na ciclagem de nutrientes em sistemas agrícolas. O Cerrado é um
campo de tipo savana, que está sendo convertida em culturas como soja, milho e café (Marris,
2005) A adubação química muitas vezes resulta em efeitos ambientais inesperados e
prejudiciais, incluindo a lixiviação de nitrato para águas subterrâneas, o escoamento
superficial de fósforo e nitrogênio e eutrofização dos ecossistemas aquáticos. A fixação
biológica do nitrogênio mediada por microrganismos permite uma redução significativa
da entrada de fertilizantes nitrogenados. A diversidade de Paenibacillus é mais
influenciada pelo tipo de plantio que pela adubação nitrogenada (Coelho et al., 2007). As
subpopulações foram identificadas por métodos moleculares, o que representa uma ferramenta
útil para monitorar o efeito das práticas agrícolas (de Oliveira et al., 2006). Comunidades
fúngicas apresentam 50% de perda da diversidade em solos convertidos em plantações
de soja (de Castro et al., 2008). Esse achado reforça a necessidade de preservação do
Cerrado, a fim de evitar a perda da diversidade microbiana pela transformação dos

172
solos originais em de pastagem. Atualmente, o bioma Cerrado tem apenas cerca de 20% de
sua cobertura original e, portanto, está entre os hotspots de conservação (Myers et al., 2000).
4.4.2.2 Diversidade microbiana em ambientes marinhos
Ambientes marinhos como recifes de coral, águas costeiras, zonas de ressurgência lagoa
hipersalina foram analisados nos últimos anos (Tabela 13).
Os primeiros estudos sobre a microbiota do ambiente marinho, centrado na análise do
coral holobionte Mussismilia. O gênero Mussismilia compreende 3 espécies (M. hispida, M.
braziliensis and M. hartii). Um dos primeiros estudos que investigaram a diversidade
microbiana deste coral endêmico do litoral brasileiro, utilizando o gene rRNA 16S, foi
realizado por Reis et al. (2009) (Reis et al., 2009). As análises revelaram a dominância dos
membros do Alphaproteobacteria em M. hispida saudável. A prevalência desse grupo de
bactérias do gênero Favia Siderastrea e recifes no Caribe e no Mar Vermelho estão
relacionados à doença da banda preta (Barneah et al., 2007; Sekar et al., 2006b). Os
gêneros mais comumente encontrados na M. hispida foram Azospirillum, Fabibacter,
Blastochloris, Stella, Vibrio, Flavobacterium, Ochrobactrum, Terasakiella, Alkalibacter,
Azospirillum, Propionibacterium, Arcobacter e Paenibacillus (de Castro et al., 2010b).
Observou-se um enriquecimento de Bacteroidetes em M. braziliensis doentes em
comparação com os corais saudáveis e água (de Castro et al., 2010b). A microbiota de
corais saudáveis e água do mar circundante apresentam composições diferentes. Além
disso, um estudo abrangente sobre a diversidade microbiana das três espécies de Mussismilia
por meio de pirosequenciamento da região intergênica dos genes ribossomais revelou uma
microbiota residente associada para cada espécie de Mussismilia (em fase de submissão). Esta
microbiota residente pode representar endossimbiontes da Mussismilia que co-evoluíram em
estreita relação com este holobionte. Esses dados reforçam o modelo de mudança de fase da

173
comunidade microbiana em indivíduos saudáveis e doentes (Mao-Jones et al., 2010). Estudos
recentes mostraram também que a maioria das comunidades de Arquéia associada à M.
hispida parecem incluir novas espécies do filo Crenarchaeota, indicando a necessidade de
isolamento microbiano e mais estudos taxonômicos (Lins-de-Barros et al., 2010a; Wegley et
al., 2007).
4.4.2.3 Diversidade microbiana em áreas urbanas costeiras
A Baía de Guanabara abrange parte da região metropolitana do Rio de Janeiro, que é
caracterizada por uma zona de atividade urbana e industrial intensa, com impactos na
microbiota associada (Clementino et al., 2007). Os principais parâmetros bióticos
envolvidos na formação dessas comunidades microbianas não são completamente
compreendidos. Aparentemente, o vazamento de óleo e de esgoto influencia a estrutura da
comunidade de bactérias e arquéias, com a dominância de microrganismos indicadores em
potencial (Vieira et al., 2007; Vieira et al., 2008).
Na Baía de Sepetiba (localizado nas proximidades da cidade do Rio de Janeiro), houve uma
redução da diversidade microbiana, possivelmente devido ao despejo de efluentes industriais
contaminados por metais (Ex. zinco). Os principais grupos são formados por organismos
acidófilos como Acidocella, Acidosphaera e Acidiphilium. A maioria das Arquéias
pertence ao filo Crenarchaeota, relacionados aos gêneros Sulfolobus e Metallosphaera
(Almeida et al., 2009). Esta lagoa fica nas proximidades da Baía de Guanabara.
O primeiro estudo sobre a diversidade de comunidades microbianas da Lagoa de
Araruama mostrou os principais grupos taxonômicos e possíveis padrões espaciais
relacionadas com a concentração de sal. Os gêneros Methanomicrobia e
Methanothermococcus (Arquéia Euryarcheota) foram encontrados em áreas de menor
concentração de sal (Clementino et al., 2008). Por outro lado, os gêneros Haloarcula

174
(Arquéia) e Salinibacter (bactérias) foram dominantes nas zonas hipersalinas. A comunidade
bacteriana da água da lagoa mostrou predomínio de sequências relacionadas com
Gammaproteobacteria, Actinobacteria e Synechococcus (Clementino et al., 2008). Estes
microrganismos podem estar envolvidos na fixação de nitrogênio e carbono nesta lagoa. O
ambiente hipersalino da Lagoa de Araruama mostrou influência sobre o procarionte
magnetotático multicelular Candidatus Magnetoglobus multicellularis (Martins et al.,
2009). É uma bactéria Gram-negativa, filogeneticamente relacionada às Deltaproteobacteria
redutoras de sulfato que foi descrita pela primeira vez na Lagoa de Araruama (Abreu et al.,
2007). Essa bactéria se caracteriza por ser capaz de formar um agregado de células
bacterianas flageladas, organizado em forma de esfera, que nadam em trajetórias helicoidais
ou retas. A composição e abundância dos magnetossomos e o fato de que os magnetossomos
podem ser digeridas por seus predadores ciliados sugerem um papel para o Ca. M.
multicellularis nos ciclos biogeoquímicos de ferro e enxofre.
Sequências de rRNA 16S de sequências não classificadas (pelo RDP) foram utilizadas na
reconstrução filogenética, a fim de revelar os vizinhos filogenéticos mais próximos.
Amostras de solo foram mais heterogêneas do que as sequências da água (Figura 53 e 54).
Foram observadas sequências altamente relacionadas filogeneticamente, porém
identificadas em diferentes trabalhos. Esse achado reforça a idéia de que existem grupos
microbianos desconhecidos que são frequentes e, possivelmente, estão envolvidos com
funções ecológicas primordiais nesses ambientes. Porém, esses grupos ainda não foram
caracterizados taxonomicamente e merecem novos estudos, pois representam novos filos
ou classes em potencial.
A abundância relativa de filos dominantes como as Cyanobacteria da microbiota
aquática e Bacteroidetes associados a organismos aquáticos é heterogênea entre os
ecossitemas analisados (Figura 55). Por outro lado, filos dominantes em biomas terrestres

175
como Acidobacteria e Actinobacteria são distribuídos de maneira relativamente uniforme
entre os solos da Mata Atlântica e Cerrado.
Figura 53. Análise filogenética de sequências não classificadas, recuperadas da água. As sequências foram comparadas com
sequências tipo a aprtir de comparação com o RDP. Todas as sequências foram alinhadas usando o programa Muscle. As
análises filogenéticas foram realizadas com o programa Mega, usando o método de neighbor joining. Valores de bootstrap
(1000 repetições) > 50 % são mostrados. A linha azul corresponde a OTUs com alta similaridade (> 97 %) entre os genes
rRNA 16S encontrados por diferentes autores em ambientes aquáticos. É importante salientar os longos ramos, que
representam novos grupos taxonômicos em potencial.

176
Figura 54. Análise filogenética de sequências não classificadas, recuperadas do solo. As sequências foram comparadas com
sequências tipo a aprtir de comparação com o RDP. Todas as sequências foram alinhadas usando o software Muscle. As
análises filogenéticas foram realizadas com o software Mega, usando o método de neighbor-joining. Valores de bootstrap
(1000 repetições) > 50 % são mostrados. A linha verde corresponde ao exemplo de sequências singletons, com grande
distância evolutiva em comparação com as sequências do gene 16S rRNA linhagens de tipo, demonstrando a
heterogeneidade da diversidade de Bactérias dos solos.
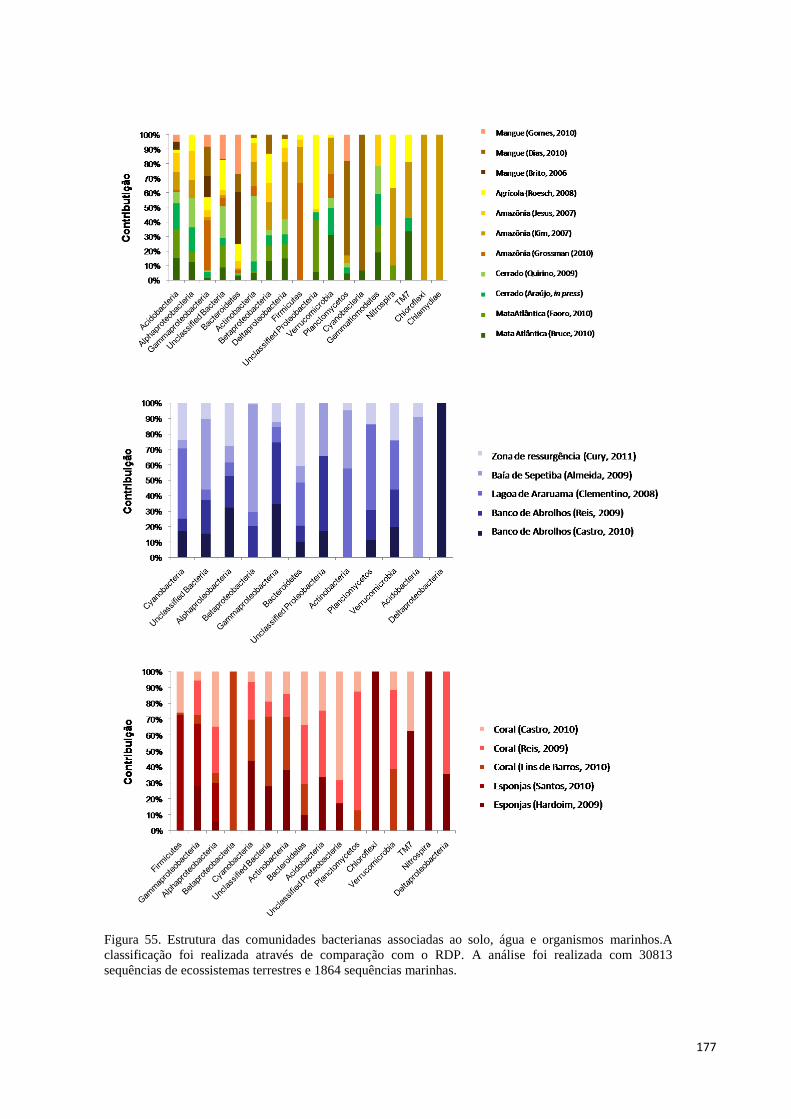
177
Figura 55. Estrutura das comunidades bacterianas associadas ao solo, água e organismos marinhos.A
classificação foi realizada através de comparação com o RDP. A análise foi realizada com 30813
sequências de ecossistemas terrestres e 1864 sequências marinhas.

178
4.4.2.4 Perspectivas
A enorme diversidade microbiana de biomas brasileiros representa um reservatório
inexplorado de novos genes e metabolismos que desempenham um papel fundamental na
saúde ambiental. Além disso, a diversidade microbiana representa um grande reservatório
para a descoberta e aplicações biotecnológicas. Descobertas baseadas em abordagem
metagenômica, através de sequenciamento massivo e isolamento de microrganismos-alvo
podem representar abordagens adequadas para usar esta vasta biodiversidade. Identificar a
biodiversidade cultivável pela taxonomia genômica é agora também um esforço viável. Além
disso, estudos de diversidade microbiana estão implementando tecnologias de
sequenciamento de segunda (454 - Roche) e terceira (Ion Torrent – Life) geração em estudo
de diversidade microbiana de ambientes terrestres e marinhos.
Atualmente, o grupo do Laboratório de Microbiologia está realizando o mapeamento da
diversidade metagenômica das ilhas oceânicas brasileiras (São Pedro e São Paulo, Trindade e
Martin Vaz) e recifes de profundidade no talude do Banco de Abrolhos. Os estudos são
integrados a fim de estabelecer um panorama da diversidade metagenômica em todo o sul do
Oceano Atlântico. Além disso, a biblioteca metagenômica, bem como a coleção de isolados
do solo da Mata Atlântica, representa um enorme repertório genético que permitirá a
construção de vetores de expressão de celulases de alto rendimento. Esses estudos permitirão
a exploração do potencial biotecnológico (Ex. novas celulases) da diversidade microbiana.
Claramente, o tempo é emocionante e desafiador para novas descobertas da biodiversidade no
Brasil.

179
5. CONCLUSÕES
5.1 Conclusões gerais
A abordagem metagenômica determinou a assinatura microbiana de diferentes ecossistemas e
permitiu a exploração da diversidade funcional a partir de amostras ambientais.
5.2 Conclusões específicas
Foram listadas as conclusões de cada um dos capítulos apresentados na seção Resultados e
discussão
5.2.1 Diversidade da comunidade de bacterioplâncton ao longo de uma gradiente
latitudinal na costa da América Latina por meio de pirosequenciamento da
região V6
Foi identificado um gradiente latitudinal de diversidade microbiana ao longo da costa
da América Latina. A diversidade microbiana varia conforme a latitude, com
tendência de maior equitabilidade nas regiões tropicais e menor equitabilidade em
áreas sub-tropicais. O número de espécies (grupo com > 97 % de similaridade na V6
rRNA 16S) variou entre 12 e 2 (para cada 100 sequências da V6 rRNA 16S), com
maiores valores nas regiões tropicais. Parâmetros ambientais tais como intensidade
luminosa, temperatura, e nutrientes, podem exercer papel fundamental no
estabelecimento destes padrões latitudinais.
Os cosmopolitas correspondem a grupos taxonômicos conhecidos, tais como,
Acinetobacter, Flavobacteriaceae, SAR11 e Enterobacteriaceae, representando
aproximadamente 58% de todas as sequências identificadas. A fração cosmopolita ao
longo da costa da America Latina sugere ampla dispersão microbiana no meio
marinho;

180
A fração endêmica das sequências correspondeu a apenas 2% de todas as sequências
identificadas, indicando baixo grau de endemismo;
A região V6 apresenta resolução taxonômica satisfatória para níveis de gênero e
acima, demonstrando a utilidade desta estratégia para estudos abrangentes de
diversidade microbiana. Entretanto, a resolução taxonômica da região V6 é limitada
para a diferenciação de espécies de grupos filogeneticamente próximos, tais como
Proclorococcus e Synechococcus.
5.2.2 Avaliação da saúde do Banco de Abrolhos através da metageômica
Arquéias e vírus foram mais abundantes nos recifes desprotegidos, em comparação
com os recifes protegidos, com dominância de fagos de Procholrococcus.
Proteobacteria e Cyanobacteria foram os filos mais abundantes na água dos recifes do
Banco de Abrolhos;
Foi observada tendência de maior diversidade e abundância microbiana dentro das
áreas protegidas do que fora das áreas protegidas, sugerindo que áreas marinhas
protegidas têm papel importante na abundância e diversidade microbiana, promovendo
a saúde ambiental. As áreas protegidas estão em diferentes estágios de degradação,
porém com aparente menor nível de impacto, quando comparadas com as áreas não
protegidas. Parcel dos Abrolhos apresenta estágio inicial de degradação, enquanto que
California representa o recife menos impactado.
Sequências relacionadas aos sistemas autotróficos foram mais abundantes nos recifes
protegidos, enquanto que sequências relacionadas ao metabolismo heterotrófico foram
mais abundantes nos recifes desprotegidos. Os cinco processos metabólicos mais
abundantes foram os de carboidratos, aminoácidos e derivados, proteínas, co-fatores e
virulência. Houve diferença entre os recifes desprotegidos e protegidos apenas para a

181
síntese de macromoléculas e a fotossíntese. Alguns subsistemas, tais como,
carboidratos e resposta ao estresse tiveram a contribuição de vários tipos microbianos,
enquanto que os subsistemas de fotossíntese tiveram contribuições predominantes de
poucos grupos taxonômicos.
A maioria das sequências de proteorodopsinas foi relacionada com as SAR11 e
Vibrionaceae, indicando a contribuição destes heterótrofos na produção primária no
ambiental recifal.
As sequências dos metagenomas refletem a tendência de degradação da região do
Banco de Abrolhos. Sebastião Gomes representa uma área desprotegida em estágio de
degradação acelerada, enquanto Timbebas é uma área protegida em estágio de
degradação intermediário. Portanto, a abordem metagenômica apresenta potencial de
aplicação como ferramenta de biomonitoramentoda saúde recifal.
5.2.3 Diversidade das comunidades microbianas dos solos da Mata Atlântica e o seu
potencial biotecnológico
A extração do DNA metagenômico com esferas de vidro seguida de purificação se
mostrou mais eficiente do que a extração por kit comercial para a construção de
bibliotecas metagenômicas. Foi estabelecidauma bibliotecade pequenos insertos,
contendo um número estimado de 40.000 clones, contendo fragmentos de
aproximadamente 10 kb.
Aproximadamente 1.000 clones foram triados (após enriquecimento) e alguns deles
tiveram sua atividade celulolítica avaliada. Os níveis de atividade FPásica e
CMCásicados clones foram próximos aos observados nos isolados ambientais.

182
Foram recuperados 13 filos. Os filos Acidobacteria, Proteobacteria e
Verrucomicrobia foram os mais abundantes no solo da Mata Atlântica. Acidobacteria
e Verrucomicrobia não são facilmente recuperados através de isolamento clássico.
As curvas de rarefação mostraram que a taxa de cobertura das bibliotecas foi
satisfatória para filos e classes em todos os locais, mas não atingiu a saturação ao nível
de espécie, demonstrando a alta diversidade de espécies nos solos da Floresta da Mata
Atlântica. A estimativa do número de filos presentes no solo da Mata Atlântica variou
entre 41-152 e de espécies entre 876-1486 por grama de solo.
Os grupos 15 e 18 não apresentaram similaridade significativa de sequência de rRNA
16S com qualquer outro grupo bacteriano e, portanto possivelmente representam dois
filos novos.
5.2.4 Diversidade microbiana dos biomas brasileiros
Os mais de 70 trabalhos de grupos brasileiros, publicados sobre o tema da diversidade
microbiana e utilizando métodos independentes de cultivo, englobaram todos os sete
principais biomas brasileiros nos últimos cinco anos. Os biomas mais intensamente
estudos são Mata Atlântica, Cerrado, Amazônia. Já para a Caatinga e o Pantanal existe
apenas um trabalho.
Cada bioma apresenta microbiota específica. Existe diferenciação clara entre a
microbiota (Bactéria e Arquéia) de biomas terrestres e aquáticos de acordo com
análises de sequências do gene ribossomal rRNA 16S. Euryarchaeota é mais
abundante em ambientes aquáticos, enquanto que Crenarchaeota é mais abundante no
solo. Acidobacteria, Actinobacteria e Firmicutes são predominantes no solo, enquanto
que Cyanobacteria e Bacteroidetes são mais abundantes em água.

183
Os biomas brasileiros são fonte rica para descoberta de novos grupos taxonômicos
(espécies, famílias e até mesmo filos). A partir do isolamento microbiano e taxonomia
polifásica será possível descrever estes novos taxa. Sequências não classificadas a
partir de amostras de solo foram mais heterogêneas do que as sequências não
classificadas da água, indicando a tremenda diversidade em solos.
Os biomas brasileiros apresentam enorme potencial para exploração biotecnológica da
diversidade microbiana como na produção de biocombustíveis, biomonitoramento,
biorremediação e aplicação de microrganismos promotores de crescimento de plantas

184
6. REFERÊNCIAS BIBLIOGRÁFICAS
Aboim, M.C.R., Coutinho, H.L.C., Peixotoc, R.S., Barbosa, J.C., and Rosado, A.S. (2008).
Soil bacterial community structure and soil quality in a slash-and-burn cultivation system in
Southeastern Brazil. Applied Soil Ecology 38, 100-108.
Abreu, F., Martins, J.L., Silveira, T.S., Keim, C.N., de Barros, H.G.P.L., Filho, F.J.G., and
Lins, U. (2007). 'Candidatus Magnetoglobus multicellularis', a multicellular, magnetotactic
prokaryote from a hypersaline environment. Int J Syst Evol Microbiol 57, 1318-1322.
Adesemoye, A., Torbert, H., and Kloepper, J. (2009). Plant Growth-Promoting Rhizobacteria
Allow Reduced Application Rates of Chemical Fertilizers. Microbial Ecology 58, 921-929.
Adriano, E.A., Arana, S., Alves, A.L., Silva, M.R.M., Ceccarelli, P.S., Henrique-Silva, F.,
and Maia, A.A.M. (2009). Myxobolus cordeiroi n. sp., a parasite of Zungaro jahu
(Siluriformes: Pimelodiade) from Brazilian Pantanal: Morphology, phylogeny and
histopathology. Veterinary Parasitology 162, 221-229.
Ahn, S.J., Costa, J., and Rettig Emanuel, J. (1996). PicoGreen Quantitation of DNA: Effective
Evaluation of Samples Pre-or Psost-PCR. Nucleic Acids Research 24, 2623-2625.
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2008). Molecular
Biology of the Cell. Garland science, Taylor & Francis Group 5ª Edição.
Albino, U., Saridakis, D.P., Ferreira, M.C., Hungria, M., Vinuesa, P., and Andrade, G. (2006).
High diversity of diazotrophic bacteria associated with the carnivorous plant Drosera villosa
var. villosa growing in oligotrophic habitats in Brazil. Plant and Soil 287, 199-207.
Almeida, W.I., Vieira, R.P., Cardoso, A.M., Silveira, C.B., Costa, R.G., Gonzalez, A.M.,
Paranhos, R., Medeiros, J.A., Freitas, F.A., Albano, R.M., et al. (2009). Archaeal and
bacterial communities of heavy metal contaminated acidic waters from zinc mine residues in
Sepetiba Bay. Extremophiles 13, 263-271.
Alves-Prado, H.F., Pavezzi, F.C., Leite, R.S.R., de Oliveira, V.M., Sette, L.D., and DaSilva,
R. (2010). Screening and Production Study of Microbial Xylanase Producers from Brazilian
Cerrado. Applied Biochemistry and Biotechnology 161, 333-346.
Alves, N., Neto, O.S.M., Silva, B.S.O., de Moura, R.L., Francini, R.B., Castro, C.B.E.,
Paranhos, R., Bitner-Mathe, B.C., Kruger, R.H., Vicente, A.C.P., et al. (2010). Diversity and
pathogenic potential of vibrios isolated from Abrolhos Bank corals. Environmental
Microbiology Reports 2, 90-95.
Amado-Filho, G., Salgado, L., Rebelo, M., Rezende, C., Karez, C., and Pfeiffer, W. (2008).
Heavy metals in benthic organisms from Todos os Santos Bay, Brazil. Brazilian Journal of
Biology 68, 95-100.
Amorim, J.H., Macena, T.N.S., Lacerda, G.V., Rezende, R.P., Dias, J.C.T., Brendel, M., and
Cascardo, J.C.M. (2008). An improved extraction protocol for metagenomic DNA from a soil
of the Brazilian Atlantic Rainforest. Genetics and Molecular Research 7, 1226-1232.

185
Andreote, F., Carneiro, R., Salles, J., Marcon, J., Labate, C., Azevedo, J., and Araújo, W.
(2009). Culture-Independent Assessment of Rhizobiales-Related Alphaproteobacteria and the
Diversity of Methylobacterium in the Rhizosphere and Rhizoplane of Transgenic Eucalyptus.
Microbial Ecology 57, 82-93.
Andreote, F., Mendes, R., Dini-Andreote, F., Rossetto, P., Labate, C., Pizzirani-Kleiner, A.,
van Elsas, J., Azevedo, J., and Araújo, W. (2008). Transgenic tobacco revealing altered
bacterial diversity in the rhizosphere during early plant development. Antonie van
Leeuwenhoek 93, 415-424.
Araujo, J., and Kruger, R. Characterization of cerrado soils' bacterial diversity. unplubished.
Aristidou, A., and Penttilä, M. (2000). Metabolic engineering applications to renewable
resource utilization. Current Opinion in Biotechnology 11, 187-198.
Azam, F. (1998). Microbial Control of Oceanic Carbon Flux: The Plot Thickens. Science 280,
694-696.
Azam, F., Fenchel, T., Field, J.G., Gray, J.S., Meyerreil, L.A., and Thingstad, F. (1983). The
ecological role of water column microbes in the sea. Marine Ecology-Progress Series 10, 257-
263.
Azevedo, C., Casal, G., Garcia, P., Matos, P., Teles-Grilo, L., and Matos, E. (2009a).
Ultrastructural and phylogenetic data of Chloromyxum riorajum sp nov (Myxozoa), a parasite
of the stingray Rioraja agassizii in Southern Brazil. Diseases of Aquatic Organisms 85, 41-51.
Azevedo, C., Casal, G., Marques, D., Silva, E., and Matos, E. (2011). Ultrastructure of
Myxobolus brycon n. sp. (Phylum Myxozoa), Parasite of the Piraputanga Fish Brycon hilarii
(Teleostei) from Pantanal (Brazil). Journal of Eukaryotic Microbiology 58, 88-93.
Azevedo, C., Casal, G., Matos, P., Ferreira, I., and Matos, E. (2009b). Light and Electron
Microscopy of the Spore of Myxobolus heckelii n. sp. (Myxozoa), Parasite from the Brazilian
Fish Centromochlus heckelii (Teleostei, Auchenipteridae). Journal of Eukaryotic
Microbiology 56, 589-593.
Azevedo, C., Casal, G., Matos, P., and Matos, E. (2008). A New Species of Myxozoa,
Henneguya rondoni n. sp. (Myxozoa), from the Peripheral Nervous System of the Amazonian
Fish, Gymnorhamphichthys rondoni (Teleostei). Journal of Eukaryotic Microbiology 55, 229-
234.
Azevedo, C., Casal, G., Mendonça, I., Carvalho, E., Matos, P., and Matos, E. (2010). Light
and electron microscopy of Myxobolus sciades n. sp. (Myxozoa), a parasite of the gills of the
Brazilian fish Sciades herzbergii (Block, 1794) (Teleostei: Ariidae). Memórias do Instituto
Oswaldo Cruz 105, 203-207.
Azevedo, C., Casal, G., Mendonça, I., and Matos, E. (2009c). Fine structure of Henneguya
hemiodopsis sp. n. (Myxozoa), a parasite of the gills of the Brazilian teleostean fish
Hemiodopsis microlepes (Hemiodontidae). Memórias do Instituto Oswaldo Cruz 104, 975-
979.
Baas Becking, L.G.M. (1834). Geobiologie of inleiding tot de milieukunde. In, V. Stockum,
ed. (Hague).

186
Barbosa, A., Morais, C., Morais, P., Rosa, L., Pimenta, R., Lachance, M.-A., and Rosa, C.
(2011). Wickerhamiella pagnoccae sp. nov. and Candida tocantinsensis sp nov., two
ascomycetous yeasts from flower bracts of Heliconia psittacorum (Heliconiaceae). Int J Syst
Evol Microbiol, ijs.0.032466-032460.
Barbosa, A.C., Cadete, R.M., Gomes, F.C.O., Lachance, M.-A., and Rosa, C.A. (2009).
Candida materiae sp. nov., a yeast species isolated from rotting wood in the Atlantic Rain
Forest. Int J Syst Evol Microbiol 59, 2104-2106.
Barneah, O., Ben-Dov, E., Kramarsky-Winter, E., and Kushmaro, A. (2007). Characterization
of black band disease in Red Sea stony corals. Environmental Microbiology 9, 1995-2006.
Barott, K., Smith, J., Dinsdale, E., Hatay, M., Sandin, S., and Rohwer, F. (2009).
Hyperspectral and Physiological Analyses of Coral-Algal Interactions. PLoS One 4, e8043.
Bayer, E.A., Lamed, R., White, B.A., and Flint, H.J. (2008). From cellulosomes to
cellulosomics. The Chemical Record 8, 364-377.
Beijerinck, M.W. (1913). De infusies en de ontdekking der backteriën. In, W. Müller, ed.
(Amsterdam, Jaarboek van de Koninklijke Akademie).
Béjà, O., Aravind, L., Koonin, E.V., Suzuki, M.T., Hadd, A., Nguyen, L.P., Jovanovich, S.B.,
Gates, C.M., Feldman, R.A., Spudich, J.L., et al. (2000). Bacterial Rhodopsin: Evidence for a
New Type of Phototrophy in the Sea. Science 289, 1902-1906.
Béja, O., Spudich, E.N., Spudich, J.L., Leclerc, M., and DeLong, E.F. (2001).
Proteorhodopsin phototrophy in the ocean. Nature 411, 786-789.
Bell, P.R.F. (1992). Eutrophication and coral reefs--some examples in the Great Barrier Reef
lagoon. Water Research 26, 553-568.
Bell, P.R.F., Lapointe, B.E., and Elmetri, I. (2007). Reevaluation of ENCORE: Support for
the eutrophication threshold model for coral reefs. Ambio 36, 416-424.
Beneduzi, A., Costa, P.B., Parma, M., Melo, I.S., Bodanese-Zanettini, M.H., and Passaglia,
L.M.P. (2010). Paenibacillus riograndensis sp. nov., a nitrogen-fixing species isolated from
the rhizosphere of Triticum aestivum. Int J Syst Evol Microbiol 60, 128-133.
Beneduzi, A., Peres, D., da Costa, P.B., Bodanese Zanettini, M.H., and Passaglia, L.M.P.
(2008a). Genetic and phenotypic diversity of plant-growth-promoting bacilli isolated from
wheat fields in southern Brazil. Research in Microbiology 159, 244-250.
Beneduzi, A., Peres, D., Vargas, L.K., Bodanese-Zanettini, M.H., and Passaglia, L.M.P.
(2008b). Evaluation of genetic diversity and plant growth promoting activities of nitrogen-
fixing bacilli isolated from rice fields in South Brazil. Applied Soil Ecology 39, 311-320.
Bertrand, H., Poly, F., Van, V.T., Lombard, N., Nalin, R., Vogel, T.M., and Simonet, P.
(2005). High molecular weight DNA recovery from soils prerequisite for biotechnological
metagenomic library construction. Journal of Microbiological Methods 62, 1-11.

187
Borneman, J., and Triplett, E. (1997). Molecular microbial diversity in soils from eastern
Amazonia: evidence for unusual microorganisms and microbial population shifts associated
with deforestation. Appl Environ Microbiol 63, 2647-2653.
Bouman, H.A., Ulloa, O., Scanlan, D.J., Zwirglmaier, K., Li, W.K.W., Platt, T., Stuart, V.,
Barlow, R., Leth, O., Clementson, L., et al. (2006). Oceanographic Basis of the Global
Surface Distribution of Prochlorococcus Ecotypes. Science 312, 918-921.
Bound, J.P., and Voulvoulis, N. (2006). Predicted and measured concentrations for selected
pharmaceuticals in UK rivers: Implications for risk assessment. Water Research 40, 2885-
2892.
Bourne, D.G., Garren, M., Work, T.M., Rosenberg, E., Smith, G.W., and Harvell, C.D.
(2009). Microbial disease and the coral holobiont. Trends in Microbiology 17, 554-562.
Brasil, M.D., Baldani, J.L., and Baldani, V.L.D. (2005). Occurrence and diversity
diazotrophic bacteria associated to forage grasses of the Pantanal in the state of Mato Grosso
do Sul. Revista Brasileira De Ciencia Do Solo 29, 179-190.
Brito, E.M.S., Guyoneaud, R., Goñi-Urriza, M., Ranchou-Peyruse, A., Verbaere, A., Crapez,
M.A.C., Wasserman, J.C.A., and Duran, R. (2006). Characterization of hydrocarbonoclastic
bacterial communities from mangrove sediments in Guanabara Bay, Brazil. Research in
Microbiology 157, 752-762.
Brooks, T.M., Mittermeier, R.A., da Fonseca, G.A.B., Gerlach, J., Hoffmann, M., Lamoreux,
J.F., Mittermeier, C.G., Pilgrim, J.D., and Rodrigues, A.S.L. (2006). Global Biodiversity
Conservation Priorities. Science 313, 58-61.
Brown, M.V., Philip, G.K., Bunge, J.A., Smith, M.C., Bissett, A., Lauro, F.M., Fuhrman,
J.A., and Donachie, S.P. (2009). Microbial community structure in the North Pacific ocean.
ISME J 3, 1374-1386.
Bruce, T., Martinez, I., Maia Neto, O., Vicente, A., Kruger, R., and Thompson, F. (2010).
Bacterial Community Diversity in the Brazilian Atlantic Forest Soils. Microbial Ecology 60,
840-849.
Bruns, A., Cypionka, H., and Overmann, J. (2002). Cyclic AMP and Acyl Homoserine
Lactones Increase the Cultivation Efficiency of Heterotrophic Bacteria from the Central Baltic
Sea. Appl Environ Microbiol 68, 3978-3987.
Bruns, A., Nubel, U., Cypionka, H., and Overmann, J. (2003). Effect of Signal Compounds
and Incubation Conditions on the Culturability of Freshwater Bacterioplankton. Appl Environ
Microbiol 69, 1980-1989.
Brusca, R.C., and Brusca, G.J. (2003). Invetebrates, 2 edn (Sunderland, Sinauer Associates
Publishers).
Cadete, R.M., Santos, R.O., Melo, M.A., Mouro, A., Gonçalves, D.L., Stambuk, B.U.,
Gomes, F.C.O., Lachance, M.-A., and Rosa, C.A. (2009). Spathaspora arborariae sp. nov., a
d-xylose-fermenting yeast species isolated from rotting wood in Brazil. FEMS Yeast
Research 9, 1338-1342.

188
Caldeira, K., and Wickett, M.E. (2003). Oceanography: Anthropogenic carbon and ocean pH.
Nature 425, 365-365.
Carlson, C.A., Giovannoni, S.J., Hansell, D.A., Goldberg, S.J., Parsons, R., Otero, M.P.,
Vergin, K., and Wheeler, B.R. (2002). Effect of nutrient amendments on bacterioplankton
production, community structure, and DOC utilization in the northwestern Sargasso Sea.
Aquatic Microbial Ecology 30, 19-36.
Carvalho, F., Vazoller, R., Foronda, A., and Pellizari, V. (2007). Phylogenetic Study of
Species in Pristine and Polluted Aquatic Samples from a Tropical Atlantic Forest Ecosystem.
Current Microbiology 55, 288-293.
Casal, G., Garcia, P., Matos, P., Monteiro, E., Matos, E., and Azevedo, C. (2009a). Fine
structure of Chloromyxum menticirrhi n. sp. (Myxozoa) infecting the urinary bladder of the
marine teleost Menticirrhus americanus (Sciaenidae) in Southern Brazil. European Journal of
Protistology 45, 139-146.
Casal, G., Matos, E., and Azevedo, C. (2006). A new mixozoan parasite from the amazonian
fish Metynnis argenteus (Teleostei, characidae): light eletronic microscope observations.
Journal of Parasitology 92, 817-821.
Casal, G., Matos, E., Teles-Grilo, L., and Azevedo, C. (2010). Ultrastructural and Molecular
Characterization of a New Microsporidium Parasite from the Amazonian Fish,
Gymnorhamphichthys rondoni (Rhamphichthyidae). Journal of Parasitology 96, 1155-1163.
Casal, G., Matos, E., Teles-Grilo, M.L., and Azevedo, C. (2008). A new microsporidian
parasite, Potaspora morhaphis n. gen., n. sp. (Microsporidia) infecting the Teleostean fish,
Potamorhaphis guianensis from the River Amazon. Morphological, ultrastructural and
molecular characterization. Parasitology 135, 1053-1064.
Casal, G., Matos, E., Teles-Grilo, M.L., and Azevedo, C. (2009b). Morphological and
genetical description of Loma psittaca sp n. isolated from the Amazonian fish species
Colomesus psittacus. Parasitology Research 105, 1261-1271.
Cenciani, K., Lambais, M.R., Cerri, C.C., de Azevedo, L.C.B., and Feigl, B.J. (2009).
Bacteria diversity and microbial biomass in forest, pasture and fallow soils in the
southwestern Amazon basin. Revista Brasileira De Ciencia Do Solo 33, 907-916.
Chen, W.-M., de Faria, S.M., Chou, J.-H., James, E.K., Elliott, G.N., Sprent, J.I., Bontemps,
C., Young, J.P.W., and Vandamme, P. (2008). Burkholderia sabiae sp. nov., isolated from
root nodules of Mimosa caesalpiniifolia. Int J Syst Evol Microbiol 58, 2174-2179.
Chen, W.-M., de Faria, S.M., James, E.K., Elliott, G.N., Lin, K.-Y., Chou, J.-H., Sheu, S.-Y.,
Cnockaert, M., Sprent, J.I., and Vandamme, P. (2007). Burkholderia nodosa sp. nov., isolated
from root nodules of the woody Brazilian legumes Mimosa bimucronata and Mimosa
scabrella. Int J Syst Evol Microbiol 57, 1055-1059.
Chen, W.-M., James, E.K., Coenye, T., Chou, J.-H., Barrios, E., de Faria, S.M., Elliott, G.N.,
Sheu, S.-Y., Sprent, J.I., and Vandamme, P. (2006). Burkholderia mimosarum sp. nov.,
isolated from root nodules of Mimosa spp. from Taiwan and South America. Int J Syst Evol
Microbiol 56, 1847-1851.

189
Cherrier, J., Bauer, J.E., and Druffel, E.R.M. (1996). Utilization and turnover of labile
dissolved organic matter by bacterial heterotrophs in eastern north Pacific surface waters.
Marine Ecology-Progress Series 139, 267-279.
Chimetto, L., Cleenwerck, I., Moreira, A.P.B., Brocchi, M., Willems, A., De Vos, P., and
Thompson, F. (2011a). Vibrio variabilis sp. nov. and Vibrio marinus sp. nov., isolated from
Palythoa caribaeorum. Int J Syst Evol Microbiol, ijs.0.026997-026990.
Chimetto, L.A., Brocchi, M., Gondo, M., Thompson, C.C., Gomez-Gil, B., and Thompson,
F.L. (2009). Genomic diversity of vibrios associated with the Brazilian coral Mussismilia
hispida and its sympatric zoanthids (Palythoa caribaeorum, Palythoa variabilis and Zoanthus
solanderi). Journal of Applied Microbiology 106, 1818-1826.
Chimetto, L.A., Brocchi, M., Thompson, C.C., Martins, R.C.R., Ramos, H.R., and Thompson,
F.L. (2008). Vibrios dominate as culturable nitrogen-fixing bacteria of the Brazilian coral
Mussismilia hispida. Systematic and Applied Microbiology 31, 312-319.
Chimetto, L.A., Cleenwerck, I., Alves, N., Jr, Silva, B.S., Brocchi, M., Willems, A., De Vos,
P., and Thompson, F.L. (2011b). Vibrio communis sp. nov., isolated from the marine animals
Mussismilia hispida, Phyllogorgia dilatata, Palythoa caribaeorum, Palythoa variabilis and
Litopenaeus vannamei. Int J Syst Evol Microbiol 61, 362-368.
Chimetto, L.A., Cleenwerck, I., Brocchi, M., Willems, A., De Vos, P., and Thompson, F.L.
(2010a). Marinomonas brasiliensis sp. nov. isolated from the coral Mussismilia hispida and
reclassification of Marinomonas basaltis as a later synonym of Marinomonas communis. Int J
Syst Evol Microbiol, ijs.0.024661-024660.
Chimetto, L.A., Cleenwerck, I., Brocchi, M., Willems, A., De Vos, P., and Thompson, F.L.
(2011c). Marinobacterium coralli sp. nov., isolated from mucus of coral (Mussismilia
hispida). Int J Syst Evol Microbiol 61, 60-64.
Chimetto, L.A., Cleenwerck, I., Thompson, C.C., Brocchi, M., Willems, A., De Vos, P., and
Thompson, F.L. (2010b). Photobacterium jeanii sp. nov., isolated from corals and zoanthids.
Int J Syst Evol Microbiol 60, 2843-2848.
Cianchetta, S., Galletti, S., Burzi, P.L., and Cerato, C. (2010). A novel microplate-based
screening strategy to assess the cellulolytic potential of Trichoderma strains. Biotechnology
and Bioengineering 107, 461-468.
Claesson, M.J., O'Sullivan, O., Wang, Q., Nikkilä, J., Marchesi, J.R., Smidt, H., de Vos,
W.M., Ross, R.P., and O'Toole, P.W. (2009). Comparative Analysis of Pyrosequencing and a
Phylogenetic Microarray for Exploring Microbial Community Structures in the Human Distal
Intestine. PLoS ONE 4, e6669.
Clementino, M.M., Fernandes, C.C., Vieira, R.P., Cardoso, A.M., Polycarpo, C.R., and
Martins, O.B. (2007). Archaeal diversity in naturally occurring and impacted environments
from a tropical region. Journal of Applied Microbiology 103, 141-151.
Clementino, M.M., Vieira, R.P., Cardoso, A.M., Nascimento, A.P.A., Silveira, C.B., Riva,
T.C., Gonzalez, A.S.M., Paranhos, R., Albano, R.M., Ventosa, A., et al. (2008). Prokaryotic

190
diversity in one of the largest hypersaline coastal lagoons in the world. Extremophiles 12,
595-604.
Coelho, G., Douanla-Meli, C., Langer, E., and Langer, G. (2010). Hypochnella verrucospora
(Basidiomycota, Atheliales), a neotropical new species with ornamented basidiospores.
Mycologia 102, 1158-1162.
Coelho, M.R.R., Da Mota, F.F., Carneiro, N.P., Marriel, I.E., Paiva, E., Rosado, A.S., and
Seldin, L. (2007). Diversity of Paenibacillus spp. in the rhizosphere of four sorghum
(Sorghum bicolor) cultivars sown with two contrasting levels of nitrogen fertilizer assessed by
rpoB-based PCR-DGGE and sequencing analysis. Journal of Microbiology and
Biotechnology 17, 753-760.
Cole, J., Chai, B., Farris, R., Wang, Q., Kulam, S., McGarrell, D., Garrity, G., and Tiedje, J.
(2005). The ribosomal database project (RDP): sequences and tools for high-throughput
rRNA analysis. Nucleic Acids Res 33, D294 - 296.
Copper, P. (1994). Ancient Reef Ecosystem Expansion and Collapse. Coral Reefs 13, 3-11.
Costa, O.S., Attrill, M.J., and Nimmo, M. (2006a). Seasonal and spatial controls on the
delivery of excess nutrients to nearshore and offshore coral reefs of Brazil. Journal of Marine
Systems 60, 63-74.
Costa, O.S., Leao, Z., Nimmo, M., and Attrill, M.J. (2000). Nutrification impacts on coral
reefs from northern Bahia, Brazil. Hydrobiologia 440, 307-315.
Costa, O.S., Nimmo, M., and Attrill, M.J. (2008). Coastal nutritication in Brazil: A review of
the role of nutrient excess on coral reef demise. Journal of South American Earth Sciences 25,
257-270.
Costa, R., Gomes, N.C.M., Peixoto, R.S., Rumjanek, N., Berg, G., Mendonca-Hagler, L.C.S.,
and Smalla, K. (2006b). Diversity and antagonistic potential of Pseudomonas spp. associated
to the rhizosphere of maize grown in a subtropical organic farm. Soil Biology & Biochemistry
38, 2434-2447.
Costanza, R., d'Arge, R., de Groot, R., Farber, S., Grasso, M., Hannon, B., Limburg, K.,
Naeem, S., O'Neill, R.V., Paruelo, J., et al. (1997). The value of the world's ecosystem
services and natural capital. Nature 387, 253-260.
Costello, E.K., and Schmidt, S.K. (2006). Microbial diversity in alpine tundra wet meadow
soil: novel Chloroflexi from a cold, water-saturated environment. Environmental
Microbiology 8, 1471-1486.
Cruz-Martinez, K., Suttle, K.B., Brodie, E.L., Power, M.E., Andersen, G.L., and Banfield,
J.F. (2009). Despite strong seasonal responses, soil microbial consortia are more resilient to
long-term changes in rainfall than overlying grassland. ISME J 3, 738-744.
Curtis, T.P., Sloan, W.T., and Scannell, J.W. (2002). Estimating prokaryotic diversity and its
limits. Proceedings of the National Academy of Sciences 99, 10494-10499.

191
Cury, J.C., Araujo, F.V., Coelho-Souza, S.A., Peixoto, R.S., Oliveira, J.A.L., Santos, H.F.,
Dávila, A.M.R., and Rosado, A.S. (2011). Microbial Diversity of a Brazilian Coastal Region
Influenced by an Upwelling System and Anthropogenic Activity. PLoS ONE 6, e16553.
da Mota, F.F., Gomes, E.A., Paiva, E., and Seldin, L. (2005). Assessment of the diversity of
Paenibacillus species in environmental samples by a novel rpoB-based PCR-DGGE method.
FEMS Microbiology Ecology 53, 317-328.
da Silva, J., Barreto, R., and Pereira, O. (2008). Pseudocercospora cryptostegiae-
madagascariensis sp. nov. on Cryptostegia madagascariensis, an Exotic Vine Involved in
Major Biological Invasions in Northeast Brazil. Mycopathologia 166, 87-91.
Dall'Agnol, L., Martins, R., Vallinoto, A., and Ribeiro, K. (2008). Diversity of
Chromobacterium violaceum isolates from aquatic environments of state of Pará, Brazilian
Amazon. Memorias Do Instituto Oswaldo Cruz 103, 678-682.
Daniel, R. (2004). The soil metagenome - a rich resource for the discovery of novel natural
products. Current Opinion in Biotechnology 15, 199-204.
Davis, K.E.R., Joseph, S.J., and Janssen, P.H. (2005). Effects of Growth Medium, Inoculum
Size, and Incubation Time on Culturability and Isolation of Soil Bacteria. Appl Environ
Microbiol 71, 826-834.
de Castro, A., Araújo, S., Reis, A., Moura, R., Francini-Filho, R., Pappas, G., Rodrigues, T.,
Thompson, F., and Krüger, R. (2010a). Bacterial Community Associated with Healthy and
Diseased Reef Coral Mussismilia hispida from Eastern Brazil. Microbial Ecology 59, 658-
667.
de Castro, A.P., Araujo, S.D., Reis, A.M.M., Moura, R.L., Francini, R.B., Pappas, G.,
Rodrigues, T.B., Thompson, F.L., and Kruger, R.H. (2010b). Bacterial Community
Associated with Healthy and Diseased Reef Coral Mussismilia hispida from Eastern Brazil.
Microbial Ecology 59, 658-667.
de Castro, A.P., Quirino, B.F., Pappas, G., Kurokawa, A.S., Neto, E.L., and Kruger, R.H.
(2008). Diversity of soil fungal communities of Cerrado and its closely surrounding
agriculture fields. Archives of Microbiology 190, 129-139.
De Macedo-Soares, P.H.M., Petry, A.C., Farjalla, V.F., and Caramaschi, E.P. (2010).
Hydrological connectivity in coastal inland systems: lessons from a Neotropical fish
metacommunity. Ecology of Freshwater Fish 19, 7-18.
de Oliveira, V.M., Manfio, G.P., da Costa Coutinho, H.L., Keijzer-Wolters, A.C., and van
Elsas, J.D. (2006). A ribosomal RNA gene intergenic spacer based PCR and DGGE
fingerprinting method for the analysis of specific rhizobial communities in soil. Journal of
Microbiological Methods 64, 366-379.
DeLong, E.F., Preston, C.M., Mincer, T., Rich, V., Hallam, S.J., Frigaard, N.-U., Martinez,
A., Sullivan, M.B., Edwards, R., Brito, B.R., et al. (2006). Community Genomics Among
Stratified Microbial Assemblages in the Ocean's Interior. Science 311, 496-503.
Demba Diallo, M., Willems, A., Vloemans, N., Cousin, S., Vandekerckhove, T.T., De
Lajudie, P., Neyra, M., Vyverman, W., Gillis, M., and Van der Gucht, K. (2004). Polymerase

192
chain reaction denaturing gradient gel electrophoresis analysis of the N2-fixing bacterial
diversity in soil under Acacia tortilis ssp. raddiana and Balanites aegyptiaca in the dryland
part of Senegal. Environmental Microbiology 6, 400-415.
Dias, A.C.F., Andreote, F.D., Rigonato, J., Fiore, M.F., Melo, I.S., and Araujo, W.L. (2010a).
The bacterial diversity in a Brazilian non-disturbed mangrove sediment. Antonie Van
Leeuwenhoek International Journal of General and Molecular Microbiology 98, 541-551.
Dias, R.J.P., Cabral, A.F., Siqueira-Castro, I.C.V., da Silva-Neto, I.D., and D'Agosto, M.
(2010b). Morphometric study of a Brazilian strain of Carchesium polypinum (Ciliophora:
Peritrichia) attached to Pomacea figulina (Mollusca: Gastropoda), with notes on a high
infestation. Zoologia 27, 483-488.
Diaz-Pulido, G., Gouezo, M., Tilbrook, B., Dove, S., and Anthony, K.R.N. (2011). High CO2
enhances the competitive strength of seaweeds over corals. Ecology Letters 14, 156-162.
Dinsdale, E.A., Edwards, R.A., Hall, D., Angly, F., Breitbart, M., Brulc, J.M., Furlan, M.,
Desnues, C., Haynes, M., Li, L., et al. (2008a). Functional metagenomic profiling of nine
biomes. Nature 452, 629-632.
Dinsdale, E.A., Pantos, O., Smriga, S., Edwards, R.A., Angly, F., Wegley, L., Hatay, M.,
Hall, D., Brown, E., Haynes, M., et al. (2008b). Microbial Ecology of Four Coral Atolls in the
Northern Line Islands. PLoS One 3, e1584.
Doolittle, W.F. (1999). Phylogenetic Classification and the Universal Tree. Science 284,
2124-2128.
Droege, M., and Hill, B. (2008). The Genome Sequencer FLX(TM) System--Longer reads,
more applications, straight forward bioinformatics and more complete data sets. Journal of
Biotechnology 136, 3-10.
Ducklow, H.W., and Carlson, C.A. (1992). Oceanic bacterial production. Advances in
Microbial Ecology 12, 113-181.
Edwards, R., Rodriguez-Brito, B., Wegley, L., Haynes, M., Breitbart, M., Peterson, D., Saar,
M., Alexander, S., Alexander, E.C., and Rohwer, F. (2006). Using pyrosequencing to shed
light on deep mine microbial ecology. BMC Genomics 7, 57.
Elshahed, M.S., Youssef, N.H., Spain, A.M., Sheik, C., Najar, F.Z., Sukharnikov, L.O., Roe,
B.A., Davis, J.P., Schloss, P.D., Bailey, V.L., et al. (2008). Novelty and Uniqueness Patterns
of Rare Members of the Soil Biosphere. Appl Environ Microbiol 74, 5422-5428.
EMBRAPA (2006). Sistema Brasileiro de Classificação de Solos, Empresa Brasileira de
Pesquisa Agropecuária edn (Rio de Janeiro, EMBRAPA-SPI).
Evans, H.C., Elliot, S.L., and Hughes, D.P. (2011). Hidden Diversity Behind the Zombie-Ant
Fungus Ophiocordyceps unilateralis: Four New Species Described from Carpenter Ants in
Minas Gerais, Brazil. PLoS ONE 6, e17024.
Ewing, B., and Green, P. (1998). Base-Calling of Automated Sequencer Traces UsingPhred.
II. Error Probabilities. Genome Research 8, 186-194.

193
Faoro, H., Alves, A.C., Souza, E.M., Rigo, L.U., Cruz, L.M., Al-Janabi, S.M., Monteiro,
R.A., Baura, V.A., and Pedrosa, F.O. (2010a). Influence of Soil Characteristics on the
Diversity of Bacteria in the Southern Brazilian Atlantic Forest. Appl Environ Microbiol 76,
4744-4749.
Faoro, H., Alves, A.C., Souza, E.M., Rigo, L.U., Cruz, L.M., Al-Janabi, S.M., Monteiro,
R.A., Baura, V.A., and Pedrosa, F.O. (2010b). Influence of Soil Characteristics on the
Diversity of Bacteria in the Southern Brazilian Atlantic Forest. Applied and Environmental
Microbiology 76, 4744-4749.
Farjalla, V., Amado, A., Suhett, A., and Meirelles-Pereira, F. (2009). DOC removal
paradigms in highly humic aquatic ecosystems. Environmental Science and Pollution
Research 16, 531-538.
Farjalla, V.F.F., Bias M.; Esteves, Francisco A. (2002). The relationship between DOC and
planktonic bacteria in tropical coastal lagoons. Archiv für Hydrobiologie 156, 97-119.
Felsenstein, J. (1989). PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics 5,
164-166.
Feng, Y., Duan, C.-J., Pang, H., Mo, X.-C., Wu, C.-F., Yu, Y., Hu, Y.-L., Wei, J., Tang, J.-L.,
and Feng, J.-X. (2007). Cloning and identification of novel cellulase genes from uncultured
microorganisms in rabbit cecum and characterization of the expressed cellulases. Applied
Microbiology and Biotechnology 75, 319-328.
Ferrer, M., Golyshina, O.V., Chernikova, T.N., Khachane, A.N., Reyes-Duarte, D., Santos,
V.A.P.M.D., Strompl, C., Elborough, K., Jarvis, G., Neef, A., et al. (2005). Novel hydrolase
diversity retrieved from a metagenome library of bovine rumen microflora. Environmental
Microbiology 7, 1996-2010.
Fierer, N., and Jackson, R.B. (2006). The diversity and biogeography of soil bacterial
communities. Proceedings of the National Academy of Sciences of the United States of
America 103, 626-631.
Fiore, M.d.F., Neilan, B.A., Copp, J.N., Rodrigues, J.L.M., Tsai, S.M., Lee, H., and Trevors,
J.T. (2005). Characterization of nitrogen-fixing cyanobacteria in the Brazilian Amazon
floodplain. Water Research 39, 5017-5026.
Fitt, W.K., Brown, B.E., Warner, M.E., and Dunne, R.P. (2001). Coral bleaching:
interpretation of thermal tolerance limits and thermal thresholds in tropical corals. Coral Reefs
20, 51-65.
Fontana, L.F., Mendonça Filho, J.G., Pereira Netto, A.D., Sabadini-Santos, E., de Figueiredo
Jr, A.G., and Crapez, M.A.C. (2010). Geomicrobiology of cores from Suruí Mangrove -
Guanabara Bay - Brazil. Marine Pollution Bulletin 60, 1674-1681.
Fontes, M., Suzuki, M., Cottrell, M., and Abreu, P. (2011). Primary Production in a
Subtropical Stratified Coastal Lagoon—Contribution of Anoxygenic Phototrophic Bacteria.
Microbial Ecology 61, 223-237.

194
Francini-Filho, R.B., and de Moura, R.L. (2008). Dynamics of fish assemblages on coral reefs
subjected to different management regimes in the Abrolhos Bank, eastern Brazil. Aquatic
Conservation: Marine and Freshwater Ecosystems 18, 1166-1179.
Francini-Filho, R.B., Moura, R.L., Thompson, F.L., Reis, R.M., Kaufman, L., Kikuchi,
R.K.P., and Leão, Z.M.A.N. (2008). Diseases leading to accelerated decline of reef corals in
the largest South Atlantic reef complex (Abrolhos Bank, eastern Brazil). Marine Pollution
Bulletin 56, 1008-1014.
Fuentefria, A.M., Suh, S.-O., Landell, M.F., Faganello, J., Schrank, A., Vainstein, M.H.,
Blackwell, M., and Valente, P. (2008). Trichosporon insectorum sp. nov., a new anamorphic
basidiomycetous killer yeast. Mycological Research 112, 93-99.
Fuhrman, J.A., and Azam, F. (1980). Bacterioplankton secondary production estimates for
coastal waters of Britsh-Columbia, Antarctica and California. Applied and Environmental
Microbiology 39, 1085-1095.
Fuhrman, J.A., Steele, J.A., Hewson, I., Schwalbach, M.S., Brown, M.V., Green, J.L., and
Brown, J.H. (2008). A latitudinal diversity gradient in planktonic marine bacteria.
Proceedings of the National Academy of Sciences 105, 7774-7778.
Fujii, K., Kuwahara, A., Nakamura, K., and Yamashita, Y. (2011). Development of a simple
cultivation method for isolating hitherto-uncultured cellulase-producing microbes. Applied
Microbiology and Biotechnology, 1-10.
Futuyma, D.J. (2002). Biologia Evolutiva. FUNPEC Editora 2ª Edição.
Gabardo, I., Meniconi, M., Falcão, L., Vital, N., Pereira, R., and Carreira, R. (2000).
Hydrocarbon and ecotoxicity in seawater and sediment samples of Guanabara Bay after the
oil spill in january 2000, , American Petroleum Institute Publ. Proceedings 2001 International
Oil Spill Conference, 941-950.
Gabor, E.M., Alkema, W.B.L., and Janssen, D.B. (2004). Quantifying the accessibility of the
metagenome by random expression cloning techniques. Environmental Microbiology 6, 879-
886.
Gabor, E.M., de Vries, E.J., and Janssen, D.B. (2003). Efficient recovery of environmental
DNA for expression cloning by indirect extraction methods. FEMS Microbiology Ecology 44,
153-163.
Gal-Mor, O., and Finlay, B.B. (2006). Pathogenicity islands: a molecular toolbox for bacterial
virulence. Cellular Microbiology 8, 1707-1719.
Galand, P.E., Casamayor, E.O., Kirchman, D.L., and Lovejoy, C. (2009). Ecology of the rare
microbial biosphere of the Arctic Ocean. Proceedings of the National Academy of Sciences
106, 22427-22432.
Galbe, M., and Zacchi, G. (2002). A review of the production of ethanol from softwood.
Applied Microbiology and Biotechnology 59, 618-628.

195
Gerlt, J.A., and Babbitt, P.C. (2001). Divergent evolution of enzymatic function:
Mechanistically Diverse Superfamilies and Functionally Distinct Suprafamilies. Annual
Review of Biochemistry 70, 209-246.
Gilles, A., Meglecz, E., Pech, N., Ferreira, S., Malausa, T., and Martin, J.-F. (2011). Accuracy
and quality assessment of 454 GS-FLX Titanium pyrosequencing. BMC Genomics 12, 245.
Gillespie, D.E., Brady, S.F., Bettermann, A.D., Cianciotto, N.P., Liles, M.R., Rondon, M.R.,
Clardy, J., Goodman, R.M., and Handelsman, J. (2002). Isolation of Antibiotics Turbomycin
A and B from a Metagenomic Library of Soil Microbial DNA. Appl Environ Microbiol 68,
4301-4306.
Glynn, P.W. (1991). Coral reef bleaching in the 1980s and possible connections with global
warming. Trends in Ecology & Evolution 6, 175-179.
Gogarten, J.P., and Townsend, J.P. (2005). Horizontal gene transfer, genome innovation and
evolution. Nat Rev Micro 3, 679-687.
Gomes, N.C.M., Borges, L.R., Paranhos, R., Pinto, F.N., Mendonca-Hagler, L.C.S., and
Smalla, K. (2008). Exploring the diversity of bacterial communities in sediments of urban
mangrove forests. FEMS Microbiology Ecology 66, 96-109.
Gomes, N.C.M., Cleary, D.F.R., Pinto, F.N., Egas, C., Almeida, A., Cunha, A., Mendonça-
Hagler, L.C.S., and Smalla, K. (2010a). Taking Root: Enduring Effect of Rhizosphere
Bacterial Colonization in Mangroves. PLoS ONE 5, e14065.
Gomes, N.C.M., Flocco, C.G., Costa, R., Junca, H., Vilchez, R., Pieper, D.H.,
Krogerrecklenfort, E., Paranhos, R., Mendonca-Hagler, L.C.S., and Smalla, K. (2010b).
Mangrove microniches determine the structural and functional diversity of enriched
petroleum hydrocarbon-degrading consortia. Fems Microbiology Ecology 74, 276-290.
Gomez-Alvarez, V., Teal, T.K., and Schmidt, T.M. (2009). Systematic artifacts in
metagenomes from complex microbial communities. ISME J 3, 1314-1317.
Gómez-Consarnau, L., Akram, N., Lindell, K., Pedersen, A., Neutze, R., Milton, D.L.,
González, J.M., and Pinhassi, J. (2010). Proteorhodopsin Phototrophy Promotes Survival of
Marine Bacteria during Starvation. PLoS Biol 8, e1000358.
Goreau, T., McClanahan, T., Hayes, R., and Strong, A. (2000). Conservation of coral reefs
after the 1998 global bleaching event. Conservation Biology 14, 5-15.
Gray, N.D., and Head, I.M. (2001). Linking genetic identity and function in communities of
uncultured bacteria. Environmental Microbiology 3, 481-492.
Green, J., and Bohannan, B.J.M. (2006). Spatial scaling of microbial biodiversity. Trends in
Ecology & Evolution 21, 501-507.
Green, J.L., Bohannan, B.J.M., and Whitaker, R.J. (2008). Microbial Biogeography: From
Taxonomy to Traits. Science 320, 1039-1043.

196
Green, J.L., Holmes, A.J., Westoby, M., Oliver, I., Briscoe, D., Dangerfield, M., Gillings, M.,
and Beattie, A.J. (2004). Spatial scaling of microbial eukaryote diversity. Nature 432, 747-
750.
Griffiths, R.I., Thomson, B.C., James, P., Bell, T., Bailey, M., and Whiteley, A.S. (2011). The
bacterial biogeography of British soils. Environmental Microbiology 13, 1642-1654.
Grossman, J.M., O'Neill, B.E., Tsai, S.M., Liang, B.Q., Neves, E., Lehmann, J., and Thies,
J.E. (2010). Amazonian Anthrosols Support Similar Microbial Communities that Differ
Distinctly from Those Extant in Adjacent, Unmodified Soils of the Same Mineralogy.
Microbial Ecology 60, 192-205.
Hackl, E., Zechmeister-Boltenstern, S., Bodrossy, L., and Sessitsch, A. (2004). Comparison
of Diversities and Compositions of Bacterial Populations Inhabiting Natural Forest Soils.
Appl Environ Microbiol 70, 5057-5065.
Hahn, M.W., Kasalicky, V., Jezbera, J., Brandt, U., and Simek, K. (2010). Limnohabitans
australis sp. nov., isolated from a freshwater pond, and emended description of the genus
Limnohabitans. Int J Syst Evol Microbiol 60, 2946-2950.
Han, S.J., Yoo, Y.J., and Kang, H.S. (1995). Characterization of a Bifunctional Cellulase and
Its Structural Gene. Journal of Biological Chemistry 270, 26012-26019.
Handelsman, J. (2004). Metagenomics: Application of Genomics to Uncultured
Microorganisms. Microbiol Mol Biol Rev 68, 669-685.
Handelsman, J. (2005). Metagenomics or Megagenomics? Nat Rev Micro 3, 457-458.
Handelsman, J., Rondon, M.R., Brady, S.F., Clardy, J., and Goodman, R.M. (1998).
Molecular biological access to the chemistry of unknown soil microbes: a new frontier for
natural products. Chemistry & Biology 5, R245-R249.
Hansel, C.M., Fendorf, S., Jardine, P.M., and Francis, C.A. (2008). Changes in Bacterial and
Archaeal Community Structure and Functional Diversity along a Geochemically Variable Soil
Profile. Appl Environ Microbiol 74, 1620-1633.
Hardoim, C.C.P., Costa, R., Araujo, F.V., Hajdu, E., Peixoto, R., Lins, U., Rosado, A.S., and
van Elsas, J.D. (2009). Diversity of Bacteria in the Marine Sponge Aplysina fulva in Brazilian
Coastal Waters. Applied and Environmental Microbiology 75, 3331-3343.
Hartt, C.F. (1871). The Geology and Physical Geography of Brazil. The American Naturalist
5, 33-36.
Healy, F.G., Ray, R.M., Aldrich, H.C., Wilkie, A.C., Ingram, L.O., and Shanmugam, K.T.
(1995). Direct isolation of functional genes encoding cellulases from the microbial consortia
in a thermophilic, anaerobic digester maintained on lignocellulose. Applied Microbiology and
Biotechnology 43, 667-674.
Hellmann, J.J., and Fowler, G.W. (1999). Bias, precision, and accuracy of four measures of
species richness. Ecological Applications 9, 824-834.

197
Henne, A., Daniel, R., Schmitz, R.A., and Gottschalk, G. (1999). Construction of
Environmental DNA Libraries in Escherichia coli and Screening for the Presence of Genes
Conferring Utilization of 4-Hydroxybutyrate. Appl Environ Microbiol 65, 3901-3907.
Hoegh-Guldberg, O. (1999). Climate change, coral bleaching and the future of the world's
coral reefs. Marine and Freshwater Research 50, 839-866.
Horner-Devine, M.C., Lage, M., Hughes, J.B., and Bohannan, B.J.M. (2004). A taxa-area
relationship for bacteria. Nature 432, 750-753.
Huber, J.A., Mark Welch, D.B., Morrison, H.G., Huse, S.M., Neal, P.R., Butterfield, D.A.,
and Sogin, M.L. (2007). Microbial Population Structures in the Deep Marine Biosphere.
Science 318, 97-100.
Hugenholtz, P. (2002). Exploring prokaryotic diversity in the genomic era. Genome Biology
3, reviews0003.0001 - reviews0003.0008.
Hugenholtz, P., Goebel, B.M., and Pace, N.R. (1998). Impact of Culture-Independent Studies
on the Emerging Phylogenetic View of Bacterial Diversity. J Bacteriol 180, 4765-4774.
Hughes, T.P. (1994). Catastrophes, phase-shifts and large-scale degradation of Caribean coral
reef. Science 265, 1547-1551.
Hughes, T.P., Rodrigues, M.J., Bellwood, D.R., Ceccarelli, D., Hoegh-Guldberg, O.,
McCook, L., Moltschaniwskyj, N., Pratchett, M.S., Steneck, R.S., and Willis, B. (2007).
Phase Shifts, Herbivory, and the Resilience of Coral Reefs to Climate Change. Current
Biology 17, 360-365.
Hungria, M., Astolfi-Filho, S., Chueire, L.M.O., Nicolás, M.F., Santos, E.B.P., Bulbol, M.R.,
Souza-Filho, A., Nogueira Assunção, E., Germano, M.G., and Vasconcelos, A.T.R. (2005).
Genetic characterization of Chromobacterium isolates from black water environments in the
Brazilian Amazon. Letters in Applied Microbiology 41, 17-23.
Hungria, M., Chueire, L.M.O., Megias, M., Lamrabet, Y., Probanza, A., Guttierrez-Manero,
F.J., and Campo, R.J. (2006). Genetic diversity of indigenous tropical fast-growing rhizobia
isolated from soybean nodules. Plant and Soil 288, 343-356.
Huse, S., Huber, J., Morrison, H., Sogin, M., and Mark Welch, D. (2007). Accuracy and
quality of massively parallel DNA pyrosequencing. Genome Biology 8.
Hutcheson, K. (1970). A test for comparing diversities based on the shannon formula. Journal
of Theoretical Biology 29, 151-154.
IBGE (2000). Pesquisa Nacional de Saneamento Básico 1989/2000., D.d.P.e.I.S. Diretório de
Pesquisas, ed. (Rio de Janeiro, Atlas nacional do Brasil).
IBGE (2004). Mapas de biomas do Brasil. Instituto Brasileiro de Geografia e Estatística
Inácio, J., Landell, M.F., Valente, P., Wang, P.-H., Wang, Y.-T., Yang, S.-H., Manson, J.S.,
Lachance, M.-A., Rosa, C.A., and Fonseca, Á. (2008). Farysizyma gen. nov., an anamorphic
genus in the Ustilaginales to accommodate three novel epiphytic basidiomycetous yeast
species from America, Europe and Asia. FEMS Yeast Research 8, 499-508.

198
Innan, H., and Kondrashov, F. (2010). The evolution of gene duplications: classifying and
distinguishing between models. Nat Rev Genet 11, 97-108.
Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis (2007).
http://www.ibama.gov.br/parnaso/.
Janssen, P.H. (2006). Identifying the Dominant Soil Bacterial Taxa in Libraries of 16S rRNA
and 16S rRNA Genes. Appl Environ Microbiol 72, 1719-1728.
Jesus, E.D., Marsh, T.L., Tiedje, J.M., and Moreira, F.M.D. (2009). Changes in land use alter
the structure of bacterial communities in Western Amazon soils. Isme Journal 3, 1004-1011.
Johnson, Z.I., Zinser, E.R., Coe, A., McNulty, N.P., Woodward, E.M.S., and Chisholm, S.W.
(2006). Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental
gradients. Science 311, 1737-1740.
Jones, R.J., Bowyer, J., Hoegh-Guldberg, O., and Blackall, L.L. (2004). Dynamics of a
temperature-related coral disease outbreak. Marine Ecology-Progress Series 281, 63-77.
Juhas, M., Van Der Meer, J.R., Gaillard, M., Harding, R.M., Hood, D.W., and Crook, D.W.
(2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS
Microbiology Reviews 33, 376-393.
Kato, S., Haruta, S., Cui, Z.J., Ishii, M., Yokota, A., and Igarashi, Y. (2004). Clostridium
straminisolvens sp. nov., a moderately thermophilic, aerotolerant and cellulolytic bacterium
isolated from a cellulose-degrading bacterial community. International Journal of Systematic
and Evolutionary Microbiology 54, 2043-2047.
Kennedy, A., and Smith, K. (1995). Soil microbial diversity and the sustainability of
agricultural soils. Plant and Soil 170, 75-86.
Kennish, M. (1992). Ecology of Estuaries: Anthropogenic Effects (Boca Raton, FL).
Kim, J.S., Sparovek, G., Longo, R.M., De Melo, W.J., and Crowley, D. (2007). Bacterial
diversity of terra preta and pristine forest soil from the Western Amazon. Soil Biology &
Biochemistry 39, 684-690.
Kim, S.-J., Lee, C.-M., Han, B.-R., Kim, M.-Y., Yeo, Y.-S., Yoon, S.-H., Koo, B.-S., and Jun,
H.-K. (2008). Characterization of a gene encoding cellulase from uncultured soil bacteria.
FEMS Microbiology Letters 282, 44-51.
Kindel, A., and Garay, I. (2002). Humus form in ecosystems of the Atlantic Forest, Brazil.
Geoderma 108, 101-118.
Kjerfve, B. (1994). Coastal Lagoon Processes. In Elsevier Oceanography Series, B. Kjerfve,
ed. (Amsterdam, Elsevier).
Knietsch, A., Waschkowitz, T., Bowien, S., Henne, A., and Daniel, R. (2003). Construction
and Screening of Metagenomic Libraries Derived from Enrichment Cultures: Generation of a
Gene Bank for Genes Conferring Alcohol Oxidoreductase Activity on Escherichia coli. Appl
Environ Microbiol 69, 1408-1416.

199
Korenblum, E., Valoni, E., Penna, M., and Seldin, L. (2010). Bacterial diversity in water
injection systems of Brazilian offshore oil platforms. Applied Microbiology and
Biotechnology 85, 791-800.
Ladd, J., Foster, R., Nannipieri P., and JM., O. (1996). Soil structure and biological activity.
Soil Biochem 9, 23–78.
Lambais, M.R., Crowley, D.E., Cury, J.C., Büll, R.C., and Rodrigues, R.R. (2006). Bacterial
Diversity in Tree Canopies of the Atlantic Forest. Science 312, 1917.
Lamed, R., Setter, E., Kenig, R., and Bayer, E.A. (1983). Cellulosome: a discrete cell surface
organelle of Clostridium thermocellum which exhibits separate antigenic, cellulose-binding
and various cellulolytic activities.
Landell, M.F., Billodre, R., Ramos, J.P., Leoncini, O., Vainstein, M.H., and Valente, P.
(2010). Candida aechmeae sp. nov. and Candida vrieseae sp. nov., novel yeast species isolated
from the phylloplane of bromeliads in Southern Brazil. Int J Syst Evol Microbiol 60, 244-248.
Landell, M.F., Inacio, J., Fonseca, A., Vainstein, M.H., and Valente, P. (2009). Cryptococcus
bromeliarum sp. nov., an orange-coloured basidiomycetous yeast isolated from bromeliads in
Brazil. Int J Syst Evol Microbiol 59, 910-913.
Lander, E.S., Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar , K,
D.M., FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, , Lehoczky
J, L.R., McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C,, Morris W, N.J.,
Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C,, Stange-Thomann N, S.N.,
Subramanian A, Wyman D, Rogers J, Sulston J,, Ainscough R, B.S., Bentley D, Burton J,
Clee C, Carter N, Coulson A, Deadman R,, Deloukas P, D.A., Dunham I, Durbin R, French L,
Grafham D, Gregory S, Hubbard, T, H.S., Hunt A, Jones M, Lloyd C, McMurray A,
Matthews L, Mercer S, Milne , S, M.J., Mungall A, Plumb R, Ross M, Shownkeen R, Sims S,
Waterston RH,, Wilson RK, H.L., McPherson JD, Marra MA, Mardis ER, Fulton LA,
Chinwalla, et al. (2001). Initial sequencing and analysis of the human genome. Nature 409,
860-921.
Lapointe, B.E., Barile, P.J., Yentsch, C.S., Littler, M.M., Littler, D.S., and Kakuk, B. (2004).
The relative importance of nutrient enrichment and herbivory on macroalgal communities
near Norman's Pond Cay, Exumas Cays, Bahamas: a "natural" enrichment experiment.
Journal of Experimental Marine Biology and Ecology 298, 275-301.
Lawrence, J.G. (1999). Gene transfer, speciation, and the evolution of bacterial genomes.
Current Opinion in Microbiology 2, 519-523.
Leão, Z., Kikuchi, R., and Oliveira, M. (2008). Branqueamento de corais nos recifes da Bahia
e sua relação com eventos de anomalias térmicas nas águas superficiais do oceano. Biota
Neotropica 8, 0-0.
Leão, Z.M.A.N., Kikuchi, R.K.P., and Testa, V. (2003). Corals and coral reefs of Brazil. In
Latin America Coral Reefs, J. Cortês, ed. (Elsevier).
Lemay, M. (1998). Banco Inter-Americano (BID) (Washington, DC, No. Env.).

200
Lemke, M., Lienau, E., Rothe, J., Pagioro, T., Rosenfeld, J., and DeSalle, R. (2009).
Description of Freshwater Bacterial Assemblages from the Upper Paraná River Floodpulse
System, Brazil. Microbial Ecology 57, 94-103.
Liesack, W., and Dunfield, P.F. (2003). Biodiversity in Soils: Use of Molecular Methods for
its Characterization. In Encyclopedia of Environmental Microbiology (John Wiley & Sons,
Inc.).
Liles, M.R., Williamson, L.L., Rodbumrer, J., Torsvik, V., Goodman, R.M., and Handelsman,
J. (2008). Recovery, Purification, and Cloning of High-Molecular-Weight DNA from Soil
Microorganisms. Appl Environ Microbiol 74, 3302-3305.
Lima-Bittencourt, C.I., Astolfi-Filho, S., Chartone-Souza, E., Santos, F.R., and Nascimento,
A.M.A. (2007). Analysis of Chromobacterium sp natural isolates from different Brazilian
ecosystems. Bmc Microbiology 7.
Lima, A.S., Nobrega, R.S.A., Barberi, A., da Silva, K., Ferreira, D.F., and Moreira, F.M.D.
(2009). Nitrogen-fixing bacteria communities occurring in soils under different uses in the
Western Amazon Region as indicated by nodulation of siratro (Macroptilium atropurpureum).
Plant and Soil 319, 127-145.
Lima, H.N., Schaefer, C.E.R., Mello, J.W.V., Gilkes, R.J., and Ker, J.C. (2002). Pedogenesis
and pre-Colombian land use of "Terra Preta Anthrosols" ("Indian black earth") of Western
Amazonia. Geoderma 110, 1-17.
Lins-de-Barros, M., Vieira, R., Cardoso, A., Monteiro, V., Turque, A., Silveira, C., Albano,
R., Clementino, M., and Martins, O. (2010a). Archaea, Bacteria, and Algal Plastids
Associated with the Reef-Building Corals Siderastrea stellata and Mussismilia hispida from
Búzios, South Atlantic Ocean, Brazil. Microbial Ecology 59, 523-532.
Lins-de-Barros, M., Vieira, R., Cardoso, A., Monteiro, V., Turque, A., Silveira, C., Albano,
R., Clementino, M., and Martins, O. (2010b). Archaea, Bacteria, and Algal Plastids
Associated with the Reef-Building Corals Siderastrea stellata and Mussismilia hispida from
Búzios, South Atlantic Ocean, Brazil. Microbial Ecology 59, 523-532.
Lombard, N., Prestat, E., van Elsas, J.D., and Simonet, P. (2011). Soil-specific limitations for
access and analysis of soil microbial communities by metagenomics. FEMS Microbiology
Ecology, no-no.
Lozupone, C.A., and Knight, R. (2007). Global patterns in bacterial diversity. Proceedings of
the National Academy of Sciences 104, 11436-11440.
Luvizotto, D., Marcon, J., Andreote, F., Dini-Andreote, F., Neves, A., Araújo, W., and
Pizzirani-Kleiner, A. (2010). Genetic diversity and plant-growth related features of
Burkholderia spp. from sugarcane roots. World Journal of Microbiology and Biotechnology
26, 1829-1836.
Lynd, L.R., Weimer, P.J., van Zyl, W.H., and Pretorius, I.S. (2002). Microbial Cellulose
Utilization: Fundamentals and Biotechnology. Microbiol Mol Biol Rev 66, 506-577.
Lynd, L.R., Wyman, C.E., and Gerngross, T.U. (1999). Biocommodity Engineering.
Biotechnology Progress 15, 777-793.

201
Maciel-Souza, M.D., Macrae, A., Volpon, A.G.T., Ferreira, P.S., and Mendonca-Hagler, L.C.
(2006). Chemical and microbiological characterization of mangrove sediments after a large
oil-spill in Guanabara Bay, RJ, Brazil. Brazilian Journal of Microbiology 37, 262-266.
Maciel, B.M., Santos, A.C.F., Dias, J.C.T., Vidal, R.O., Dias, R.J.C., Gross, E., Cascardo,
J.C.M., and Rezende, R.P. (2009). Simple DNA extraction protocol for a 16S rDNA study of
bacterial diversity in tropical landfarm soil used for bioremediation of oil waste. Genetics and
Molecular Research 8, 375-388.
Magurran, A.E. (1988). Ecological diversity and its measurements. Princeton: Princeton
University press.
Mann, C.C. (2002). The Real Dirt on Rainforest Fertility. Science 297, 920-923.
Mao-Jones, J., Ritchie, K.B., Jones, L.E., and Ellner, S.P. (2010). How Microbial Community
Composition Regulates Coral Disease Development. PLoS Biol 8, e1000345.
Margulies, M., Egholm, M., Altman, W., Attiya, S., Bader, J., Bemben, L., Berka, J.,
Braverman, M., Chen, Y., and Chen, Z. (2005). Genome sequencing in microfabricated high-
density picolitre reactors. Nature 437, 376 - 380.
Maria de Souza Moreira, F., Cruz, L., Miana de Faria, S., Marsh, T., Martínez-Romero, E., de
Oliveira Pedrosa, F., Maria Pitard, R., and Peter W. Young, J. (2006). Azorhizobium
doebereinerae sp. Nov. Microsymbiont of Sesbania virgata (Caz.) Pers. Systematic and
Applied Microbiology 29, 197-206.
Marris, E. (2005). Conservation in Brazil: The forgotten ecosystem. Nature 437, 944-945.
Martins, E., de Carvalho-Júnior, O., de Souza, V., Couto-Júnior, A., de Oliveira, S., Gomes,
R., and Reatto, A. (2007). Relação solo-relevo em vertentes assimétricas no Parque Nacional
da Serra dos Órgãos, RJ. Revista Brasileira de Geomorfologia 8, 45-62.
Martins, J.L., Silveira, T.S., Silva, K.T., and Lins, U. (2009). Salinity dependence of the
distribution of multicellular magnetotactic prokaryotes in a hypersaline lagoon. International
Microbiology 12, 193-201.
Martiny, J.B.H., Bohannan, B.J.M., Brown, J.H., Colwell, R.K., Fuhrman, J.A., Green, J.L.,
Horner-Devine, M.C., Kane, M., Krumins, J.A., Kuske, C.R., et al. (2006). Microbial
biogeography: putting microorganisms on the map. Nat Rev Micro 4, 102-112.
McAllister, D., Schueler, F., Roberts, C., and Hawkins, J. (1994). Mapping and GIS analysis
of the global distribution of coral reef fishes on an equal-area grid. In Mapping the diversity
of nature, R.I. Miller, ed. (London, Chapman & Hall), pp. 155-175.
Medeiros, P.M., Bicego, M.C., Castelao, R.M., Del Rosso, C., Fillmann, G., and Zamboni,
A.J. (2005). Natural and anthropogenic hydrocarbon inputs to sediments of Patos Lagoon
Estuary, Brazil. Environment International 31, 77-87.
Mendes, R., Pizzirani-Kleiner, A.A., Araujo, W.L., and Raaijmakers, J.M. (2007). Diversity
of Cultivated Endophytic Bacteria from Sugarcane: Genetic and Biochemical Characterization
of Burkholderia cepacia Complex Isolates. Appl Environ Microbiol 73, 7259-7267.

202
Menezes, C.B.A., Bonugli-Santos, R.C., Miqueletto, P.B., Passarini, M.R.Z., Silva, C.H.D.,
Justo, M.R., Leal, R.R., Fantinatti-Garboggini, F., Oliveira, V.M., Berlinck, R.G.S., et al.
(2010). Microbial diversity associated with algae, ascidians and sponges from the north coast
of São Paulo state, Brazil. Microbiological Research 165, 466-482.
Meyer, F., Paarmann, D., D'Souza, M., Olson, R., Glass, E., Kubal, M., Paczian, T.,
Rodriguez, A., Stevens, R., Wilke, A., et al. (2008). The metagenomics RAST server - a
public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC
Bioinformatics 9, 386.
Milanin, T., Eiras, J.C., Arana, S., Maia, A.A., Alves, A.L., Silva, M.R., Carriero, M.M.,
Ceccarelli, P.S., and Adriano, E.A. (2010). Phylogeny, ultrastructure, histopathology and
prevalence of Myxobolus oliveirai sp. nov., a parasite of Brycon hilarii (Characidae) in the
Pantanal wetland, Brazil. Memórias do Instituto Oswaldo Cruz 105, 762-769.
Ministério do Meio Ambiente (2007). Áreas Prioritárias para Conservação, Uso Sustentável e
Repartição de Benefícios da
Biodiversidade Brasileira: Atualização - Portaria MMA n°9, de 23 de janeiro de 2007,
S.d.B.e. Florestas, ed. (Brasília).
Miransari, M. (2011). Arbuscular mycorrhizal fungi and nitrogen uptake. Archives of
Microbiology 193, 77-81.
Mittermeier, R.A., Robles-Gil, P., Hoffmann, M., Pilgrim, J. D., Brooks, T. M.,, and
Mittermeier, C.G., Lamoreux, J. L. & Fonseca, G. (2004). Hotspots Revisited: Earth’s
Biologically Richest and Most Endangered Terrestrial Ecoregions. CEMEX, Mexico City.
Monteiro, J., Vollú, R., Coelho, M., Alviano, C., Blank, A., and Seldin, L. (2009).
Comparison of the bacterial community and characterization of plant growth-promoting
rhizobacteria from different genotypes of <i>Chrysopogon zizanioides</i> (L.)
Roberty (Vetiver) rhizospheres. The Journal of Microbiology 47, 363-370.
Moran, M.A., Pomeroy, L.R., Sheppard, E.S., Atkinson, L.P., and Hodson, R.E. (1991).
Distribution of Terrestrially Derived Dissolved Organic Matter on the Southeastern U.S.
Continental Shelf. Limnology and Oceanography 36, 1134-1149.
Morris, R.M., Rappe, M.S., Connon, S.A., Vergin, K.L., Siebold, W.A., Carlson, C.A., and
Giovannoni, S.J. (2002). SAR11 clade dominates ocean surface bacterioplankton
communities. Nature 420, 806-810.
Muscatine, L. (1990). The role of symbiotic algae in carbon and energy flux in reef corals. In
Ecosystems of the world: Coral Reefs, Z. Dubinsky, ed. (New York, Elsevier).
Muyzer, G., and Stams, A.J.M. (2008). The ecology and biotechnology of sulphate-reducing
bacteria. Nat Rev Micro 6, 441-454.
Myers, N., Mittermeier, R.A., Mittermeier, C.G., da Fonseca, G.A.B., and Kent, J. (2000).
Biodiversity hotspots for conservation priorities. Nature 403, 853-858.

203
Nakada, N., Tanishima, T., Shinohara, H., Kiri, K., and Takada, H. (2006). Pharmaceutical
chemicals and endocrine disrupters in municipal wastewater in Tokyo and their removal
during activated sludge treatment. Water Research 40, 3297-3303.
Nakatani, A.S., Martines, A.M., Nogueira, M.A., Fagotti, D.S.L., Oliveira, A.G., Bini, D.,
Sousa, J.P., and Cardoso, E.J.B.N. (2011). Changes in the genetic structure of Bacteria and
microbial activity in an agricultural soil amended with tannery sludge. Soil Biology and
Biochemistry 43, 106-114.
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. Oxford University
Press, New York.
Nemergut, D.R., Costello, E.K., Hamady, M., Lozupone, C., Jiang, L., Schmidt, S.K., Fierer,
N., Townsend, A.R., Cleveland, C.C., Stanish, L., et al. (2011). Global patterns in the
biogeography of bacterial taxa. Environmental Microbiology 13, 135-144.
Nemergut, D.R., Townsend, A.R., Sattin, S.R., Freeman, K.R., Fierer, N., Neff, J.C.,
Bowman, W.D., Schadt, C.W., Weintraub, M.N., and Schmidt, S.K. (2008). The effects of
chronic nitrogen fertilization on alpine tundra soil microbial communities: implications for
carbon and nitrogen cycling. Environmental Microbiology 10, 3093-3105.
Newman, J.R., and Fuqua, C. (1999). Broad-host-range expression vectors that carry the -
arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227,
197-203.
Nogales, B., Moore, E.R.B., Llobet-Brossa, E., Rossello-Mora, R., Amann, R., and Timmis,
K.N. (2001). Combined Use of 16S Ribosomal DNA and 16S rRNA To Study the Bacterial
Community of Polychlorinated Biphenyl-Polluted Soil. Appl Environ Microbiol 67, 1874-
1884.
Nyrén, P. (1987). Enzymatic method for continuous monitoring of DNA polymerase activity.
Analytical Biochemistry 167, 235-238.
Nyström, M., Folke, C., and Moberg, F. (2000). Coral reef disturbance and resilience in a
human-dominated environment. Trends in Ecology & Evolution 15, 413-417.
O'Malley, M.A. (2007). The nineteenth century roots of 'everything is everywhere'. Nat Rev
Micro 5, 647-651.
O’Neill, B., Grossman, J., Tsai, M., Gomes, J., Lehmann, J., Peterson, J., Neves, E., and
Thies, J. (2009). Bacterial Community Composition in Brazilian Anthrosols and Adjacent
Soils Characterized Using Culturing and Molecular Identification. Microbial Ecology 58, 23-
35.
Odum, H.T., and Odum, E.P. (1955). Trophic structure and productivity of a windward coral
reef community on Eniwetok atoll. Ecological Monographs 25, 291-320.
Ohno, S. (1970). Evolution by duplication. Berlin: Springer, 160 p.
Overbeek, R., Begley, T., Butler, R., Choudhuri, J., Chuang, H., Cohoon, M., de Crecy-
Lagard, V., Diaz, N., Disz, T., Edwards, R., et al. (2005). The subsystems approach to
genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res.

204
Pace, N.R. (2009). Mapping the Tree of Life: Progress and Prospects. Microbiol Mol Biol
Rev 73, 565-576.
Pace, N.R., Stahl, D.A., and Olsen, G.J. (1985). Analyzing natural microbial populations by
rRNA sequences.
Pagnocca, F.C., Legaspe, M.F.C., Rodrigues, A., Ruivo, C.C.C., Nagamoto, N.S., Bacci, M.,
Jr, and Forti, L.C. (2010). Yeasts isolated from a fungus-growing ant nest, including the
description of Trichosporon chiarellii sp. nov., an anamorphic basidiomycetous yeast. Int J
Syst Evol Microbiol 60, 1454-1459.
Palleroni, N.J. (1997). Prokaryotic diversity and the importance of culturing. Antonie van
Leeuwenhoek 72, 3-19.
Pantos, O., Cooney, R.P., Le Tissier, M.D.A., Barer, M.R., O'Donnell, A.G., and Bythell, J.C.
(2003). The bacterial ecology of a plague-like disease affecting the Caribbean coral
Montastrea annularis. Environmental Microbiology 5, 370-382.
Partensky, F., Hess, W.R., and Vaulot, D. (1999). Prochlorococcus, a Marine Photosynthetic
Prokaryote of Global Significance. Microbiol Mol Biol Rev 63, 106-127.
Patterson, K.L., Porter, J.W., Ritchie, K.B., Polson, S.W., Mueller, E., Peters, E.C., Santavy,
D.L., and Smith, G.W. (2002). The etiology of white pox, a lethal disease of the Caribbean
elkhorn coral, Acropora palmata. Proceedings of the National Academy of Sciences 99, 8725-
8730.
Paul, S., Dutta, A., Bag, S., Das, S., and Dutta, C. (2010). Distinct, ecotype-specific genome
and proteome signatures in the marine cyanobacteria Prochlorococcus. BMC Genomics 11,
103.
Peixoto, R., Chaer, G., Carmo, F., Araújo, F., Paes, J., Volpon, A., Santiago, G., and Rosado,
A. (2011). Bacterial communities reflect the spatial variation in pollutant levels in Brazilian
mangrove sediment. Antonie van Leeuwenhoek 99, 341-354.
Pelletier, E., Sargian, P., Payet, J., and Demers, S. (2006). Ecotoxicological effects of
combined UVB and organic contaminants in coastal waters: A review. Photochemistry and
Photobiology 82, 981-993.
Pereira, A.A., Hungria, M., Franchini, J.C., Kaschuk, G., de Oliveira, L.M., Campo, R.J., and
Torres, E. (2007). Qualitative and quantitative changes in soil microbiota and biological
nitrogen fixation under different soybean managements. Revista Brasileira De Ciencia Do
Solo 31, 1397-1412.
Pereira, L.F., Costa, C.R.L., Jr, Brasileiro, B.T.R.V., and de Morais, M.A., Jr (2011).
Lachancea mirantina sp. nov., an ascomycetous yeast isolated from the cachaca fermentation
process. Int J Syst Evol Microbiol 61, 989-992.
Perin, L., Martinez-Aguilar, L., Paredes-Valdez, G., Baldani, J.I., Estrada-de los Santos, P.,
Reis, V.M., and Caballero-Mellado, J. (2006). Burkholderia silvatlantica sp. nov., a
diazotrophic bacterium associated with sugar cane and maize. Int J Syst Evol Microbiol 56,
1931-1937.

205
Petrovic, M., Gonzalez, S., and Barceló, D. (2003). Analysis and removal of emerging
contaminants in wastewater and drinking water. TrAC Trends in Analytical Chemistry 22,
685-696.
Philippot, L., Bru, D., Saby, N.P.A., Čuhel, J., Arrouays, D., Šimek, M., and Hallin, S.
(2009). Spatial patterns of bacterial taxa in nature reflect ecological traits of deep branches of
the 16S rRNA bacterial tree. Environmental Microbiology 11, 3096-3104.
Pommier, T., CanbÄCk, B., Riemann, L., BostrÖM, K.H., Simu, K., Lundberg, P., Tunlid,
A., and HagstrÖM, Å. (2007). Global patterns of diversity and community structure in marine
bacterioplankton. Molecular Ecology 16, 867-880.
Prates, A.P.L. (2003). Atlas dos recifes de coral nas unidades de conservação
brasileirasBrasília, M.d.M. Ambiente, ed. (Brasília).
Prendergast, J.R., Quinn, R.M., Lawton, J.H., Eversham, B.C., and Gibbons, D.W. (1993).
Rare species, the coincidence of diversity hotspots and conservation strategies. Nature 365,
335-337.
Quince, C., Lanzen, A., Curtis, T.P., Davenport, R.J., Hall, N., Head, I.M., Read, L.F., and
Sloan, W.T. (2009). Accurate determination of microbial diversity from 454 pyrosequencing
data. Nat Meth 6, 639-641.
Quirino, B.F., Pappas, G.J., Tagliaferro, A.C., Collevatti, R.G., Neto, E.L., da Silva,
M.R.S.S., Bustamante, M.M.C., and Krüger, R.H. (2009). Molecular phylogenetic diversity of
bacteria associated with soil of the savanna-like Cerrado vegetation. Microbiological
Research 164, 59-70.
Raes, J., Letunic, I., Yamada, T., Jensen, L.J., and Bork, P. (2011). Toward molecular trait-
based ecology through integration of biogeochemical, geographical and metagenomic data.
Mol Syst Biol 7.
Ragauskas, A.J., Williams, C.K., Davison, B.H., Britovsek, G., Cairney, J., Eckert, C.A.,
Frederick, W.J., Hallett, J.P., Leak, D.J., Liotta, C.L., et al. (2006). The Path Forward for
Biofuels and Biomaterials. Science 311, 484-489.
Ramos, P.L., Van Trappen, S., Thompson, F.L., Rocha, R.C.S., Barbosa, H.R., De Vos, P.,
and Moreira-Filho, C.A. (2010). Screening for endophytic nitrogen-fixing bacteria in
Brazilian sugarcane varieties used in organic farming and description of Stenotrophomonas
pavanii sp. nov. Int J Syst Evol Microbiol, ijs.0.019372-019370.
Rappé, M.S., and Giovannoni, S.J. (2003). The uncultured microbial mojority. Annual
Review of Microbiology 57, 369-394.
Rasher, D.B., and Hay, M.E. (2010). Chemically rich seaweeds poison corals when not
controlled by herbivores. Proceedings of the National Academy of Sciences of the United
States of America 107, 9683-9688.
Reaka-Kudla, M. (1997). The Global Biodiversity of Coral Reefs: A Comparison with Rain
Forests (Washington DC, Joseph Henry Press).

206
Reis, A.M.M., Araújo Jr, S.D., Moura, R.L., Francini-Filho, R.B., Pappas Jr, G., Coelho,
A.M.A., Krüger, R.H., and Thompson, F.L. (2009). Bacterial diversity associated with the
Brazilian endemic reef coral Mussismilia braziliensis. Journal of Applied Microbiology 106,
1378-1387.
Ringo, J. (2004). Fundamental genetics. Cambridge University press 1 ª Edição.
Rivera, M.C., Jain, R., Moore, J.E., and Lake, J.A. (1998). Genomic evidence for two
functionally distinct gene classes. Proceedings of the National Academy of Sciences 95,
6239-6244.
Robe, P., Nalin, R., Capellano, C., Vogel, T.M., and Simonet, P. (2003). Extraction of DNA
from soil. European Journal of Soil Biology 39, 183-190.
Rocap, G., Distel, D.L., Waterbury, J.B., and Chisholm, S.W. (2002). Resolution of
Prochlorococcus and Synechococcus Ecotypes by Using 16S-23S Ribosomal DNA Internal
Transcribed Spacer Sequences. Appl Environ Microbiol 68, 1180-1191.
Rodrigues, D.F., da C Jesus, E., Ayala-del-Rio, H.L., Pellizari, V.H., Gilichinsky, D.,
Sepulveda-Torres, L., and Tiedje, J.M. (2009). Biogeography of two cold-adapted genera:
Psychrobacter and Exiguobacterium. ISME J 3, 658-665.
Rodriguez-Brito, B., Li, L., Wegley, L., Furlan, M., Angly, F., Breitbart, M., Buchanan, J.,
Desnues, C., Dinsdale, E., Edwards, R., et al. (2010). Viral and microbial community
dynamics in four aquatic environments. ISME J 4, 739-751.
Roesch, L.F., Fulthorpe, R.R., Riva, A., Casella, G., Hadwin, A.K.M., Kent, A.D., Daroub,
S.H., Camargo, F.A.O., Farmerie, W.G., and Triplett, E.W. (2007). Pyrosequencing
enumerates and contrasts soil microbial diversity. Isme Journal 1, 283-290.
Roesch, L.F.W., Camargo, F.A.O., Bento, F.M., and Triplett, E.W. (2008). Biodiversity of
diazotrophic bacteria within the soil, root and stem of field-grown maize. Plant and Soil 302,
91-104.
Rogers, Y.-H., and Venter, J.C. (2005). Genomics: Massively parallel sequencing. Nature
437, 326-327.
Rohde, K. (1992). Latitudinal Gradients in Species Diversity: The Search for the Primary
Cause. Oikos 65, 514-527.
Rohwer, F. (2010). Coral reef in the microbial seas: the influence of fishing, nutrients,
bacteria, viruses and climate change on nature's most wondrous constructs. In (Basalt,
Colorado, Plaid Press).
Ronaghi, M., Karamohamed, S., Pettersson, B., Uhlen, M., and Nyren, P. (1996). Real-time
DNA sequencing using detection of pyrophosphate release. Anal Biochem 242, 84 - 89.
Ronaghi, M., Uhlen, M., and Nyren, P. (1998). A sequencing method based on real-time
pyrophosphate. Science 281, 363, 365.
Rondon, M.R., August, P.R., Bettermann, A.D., Brady, S.F., Grossman, T.H., Liles, M.R.,
Loiacono, K.A., Lynch, B.A., MacNeil, I.A., Minor, C., et al. (2000). Cloning the Soil

207
Metagenome: a Strategy for Accessing the Genetic and Functional Diversity of Uncultured
Microorganisms. Appl Environ Microbiol 66, 2541-2547.
Rosa, C.A., Jindamorakot, S., Limtong, S., Nakase, T., Lachance, M.-A., Fidalgo-Jimenez,
A., Daniel, H.-M., Pagnocca, F.C., Inacio, J., and Morais, P.B. (2009a). Synonymy of the
yeast genera Moniliella and Trichosporonoides and proposal of Moniliella fonsecae sp. nov.
and five new species combinations. Int J Syst Evol Microbiol 59, 425-429.
Rosa, C.A., Jindamorakot, S., Limtong, S., Nakase, T., Pagnocca, F.C., and Lachance, M.-A.
(2009b). Candida golubevii sp. nov, an asexual yeast related to Metschnikowia lunata. Int J
Syst Evol Microbiol, ijs.0.014050-014050.
Rosa, C.A., Lachance, M.-A., Teixeira, L.C.R.S., Pimenta, R.S., and Morais, P.B. (2007a).
Metschnikowia cerradonensis sp. nov., a yeast species isolated from ephemeral flowers and
their nitidulid beetles in Brazil. Int J Syst Evol Microbiol 57, 161-165.
Rosa, C.A., Morais, P.B., Lachance, M.-A., Santos, R.O., Melo, W.G.P., Viana, R.H.O.,
Braganca, M.A.L., and Pimenta, R.S. (2009c). Wickerhamomyces queroliae sp. nov. and
Candida jalapaonensis sp. nov., two yeast species isolated from Cerrado ecosystem in North
Brazil. Int J Syst Evol Microbiol 59, 1232-1236.
Rosa, C.A., Pagnocca, F.C., Lachance, M.-A., Ruivo, C.C.C., Medeiros, A.O., Pimentel,
M.R.C., Fontenelle, J.C.R., and Martins, R.P. (2007b). Candida flosculorum sp. nov. and
Candida floris sp. nov., two yeast species associated with tropical flowers. Int J Syst Evol
Microbiol 57, 2970-2974.
Ruivo, C.C.C., Lachance, M.-A., Rosa, C.A., Bacci, M., Jr, and Pagnocca, F.C. (2006).
Candida heliconiae sp. nov., Candida picinguabensis sp. nov. and Candida saopaulonensis sp.
nov., three ascomycetous yeasts from Heliconia velloziana (Heliconiaceae). Int J Syst Evol
Microbiol 56, 1147-1151.
Sait, M., Hugenholtz, P., and Janssen, P.H. (2002). Cultivation of globally distributed soil
bacteria from phylogenetic lineages previously only detected in cultivation-independent
surveys. Environmental Microbiology 4, 654-666.
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for
reconstructing phylogenetic trees. Molecular Biology and Evolution 4, 406-425.
Salvat, B. (1992). Coral reefs - A challenging ecosystem for human societies. Global
Environmental Change-Human and Policy Dimensions 2, 12-18.
Sandin, S.A., Smith, J.E., DeMartini, E.E., Dinsdale, E.A., Donner, S.D., Friedlander, A.M.,
Konotchick, T., Malay, M., Maragos, J.E., Obura, D., et al. (2008). Baselines and
Degradation of Coral Reefs in the Northern Line Islands. PLoS ONE 3, e1548.
Santangelo, J.M., de M. Rocha, A., Bozelli, R.L., Carneiro, L.S., and de A. Esteves, F.
(2007). Zooplankton responses to sandbar opening in a tropical eutrophic coastal lagoon.
Estuarine, Coastal and Shelf Science 71, 657-668.
Santos, A., Amado, A., Minello, M., Farjalla, V., and Esteves, F. (2006). Effects of the Sand
Bar Breaching on Typha domingensis (PERS.) in a Tropical Coastal Lagoon. Hydrobiologia
556, 61-68.

208
Santos, R., Cadete, R., Badotti, F., Mouro, A., Wallheim, D., Gomes, F., Stambuk, B.,
Lachance, M.-A., and Rosa, C. (2011). Candida queiroziae sp. nov., a cellobiose-fermenting
yeast species isolated from rotting wood in Atlantic Rain Forest. Antonie van Leeuwenhoek
99, 635-642.
Schauer, R., Bienhold, C., Ramette, A., and Harder, J. (2009). Bacterial diversity and
biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J 4, 159-170.
Schloss, P.D., and Handelsman, J. (2004). Status of the microbial census. Microbiology and
Molecular Biology Reviews 68, 686-+.
Schloss, P.D., and Handelsman, J. (2005). Introducing DOTUR, a Computer Program for
Defining Operational Taxonomic Units and Estimating Species Richness. Appl Environ
Microbiol 71, 1501-1506.
Schloss, P.D., and Handelsman, J. (2006). Toward a Census of Bacteria in Soil. PLoS Comput
Biol 2, e92.
Schloss, P.D., Larget, B.R., and Handelsman, J. (2004). Integration of Microbial Ecology and
Statistics: a Test To Compare Gene Libraries. Appl Environ Microbiol 70, 5485-5492.
Schmidt, T.M., DeLong, E.F., and Pace, N.R. (1991). Analysis of a marine picoplankton
community by 16S rRNA gene cloning and sequencing. J Bacteriol 173, 4371-4378.
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic
datasets. Bioinformatics.
Schubert, C. (2006). Can biofuels finally take center stage? Nat Biotech 24, 777-784.
Sekar, R., Mills, D.K., Remily, E.R., Voss, J.D., and Richardson, L.L. (2006a). Microbial
communities in the surface mucopolysaccharide layer and the black band microbial mat of
black band-diseased Siderastrea siderea. Applied and Environmental Microbiology 72, 5963-
5973.
Sekar, R., Mills, D.K., Remily, E.R., Voss, J.D., and Richardson, L.L. (2006b). Microbial
Communities in the Surface Mucopolysaccharide Layer and the Black Band Microbial Mat of
Black Band-Diseased Siderastrea siderea. Appl Environ Microbiol 72, 5963-5973.
Semedo, L., Gomes, R.C., Linhares, A.A., Duarte, G.F., Nascimento, R.P., Rosado, A.S.,
Margis-Pinheiro, M., Margis, R., Silva, K.R.A., Alviano, C.S., et al. (2004). Streptomyces
drozdowiczii sp nov., a novel cellulolytic streptomycete from soil in Brazil. International
Journal of Systematic and Evolutionary Microbiology 54, 1323-1328.
Sette, L.D., Simioni, K.C.M., Vasconcellos, S.P., Dussan, L.J., Neto, E.V.S., and Oliveira,
V.M. (2007). Analysis of the composition of bacterial communities in oil reservoirs from a
southern offshore Brazilian basin. Antonie Van Leeuwenhoek International Journal of
General and Molecular Microbiology 91, 253-266.
Shannon, C.E. (1948). A mathematical theory of communication. Bell System Technical
Journal 27, 379-423.
Simpson, E.H. (1949). Measurement of diversity. Nature 163, 688-688.

209
Siqueira-Castro, I.C.V., Paiva, T.d.S., and Silva-Neto, I.D.d. (2009). Morphology of
Parastrongylidium estevesi comb. nov. and Deviata brasiliensis sp. nov. (Ciliophora:
Stichotrichia) from a sewage treatment plant in Rio de Janeiro, Brazil. Zoologia (Curitiba,
Impresso) 26, 774-786.
Smith, J., and Paul, E. (1990). The Significance of soil biomass estimates ( New York, Marcel
Decker).
Smith, J.E., Shaw, M., Edwards, R.A., Obura, D., Pantos, O., Sala, E., Sandin, S.A., Smriga,
S., Hatay, M., and Rohwer, F.L. (2006). Indirect effects of algae on coral: algae-mediated,
microbe-induced coral mortality. Ecology Letters 9, 835-845.
Smith , K.W., Laws EA, Brock RE, Walsh TW (1981). Kaneohe Bay Sewage Diversion
Experiment: Perspectives on Ecosystem Responses to Nutritional Perturbation. Pac Sci 35,
279-395.
Smith, N.P. (1994). Water, Salt and Heat Balance of Coastal Lagoons. In Elsevier
Oceanography Series, K. Björn, ed. (Elsevier), pp. 69-101.
Sogin, M.L., Morrison, H.G., Huber, J.A., Welch, D.M., Huse, S.M., Neal, P.R., Arrieta,
J.M., and Herndl, G.J. (2006). Microbial diversity in the deep sea and the underexplored ―rare
biosphere‖. Proceedings of the National Academy of Sciences 103, 12115-12120.
Søndergaard, M., and Middelboe (1995). A cross-system analysis of labile dissolved organic
carbon. Marine Ecology Progress Series 118, 283-294.
Sondergaard, M., Williams, P.J.L., Cauwet, G., Riemann, B., Robinson, C., Terzic, S.,
Woodward, E.M.S., and Worm, J. (2000). Net accumulation and flux of dissolved organic
carbon and dissolved organic nitrogen in marine plankton communities. Limnology and
Oceanography 45, 1097-1111.
Souchie, E.L., Saggin-Junior, O.J., Silva, E.M.R., Campello, E.F.C., Azcon, R., and Barea,
J.M. (2006). Communities of P-solubilizing bacteria, fungi and arbuscular mycorrhizal fungi
in grass pasture and secondary forest of Paraty, RJ Brazil. Anais Da Academia Brasileira De
Ciencias 78, 183-193.
Sousa, O.V., Macrae, A., Menezes, F.G.R., Gomes, N.C.M., Vieira, R.H.S.F., and Mendonça-
Hagler, L.C.S. (2006). The impact of shrimp farming effluent on bacterial communities in
mangrove waters, Ceará, Brazil. Marine Pollution Bulletin 52, 1725-1734.
Souza, R.F.d., Coelho, R.R.R., Macrae, A., Soares, R.M.A., Nery, D.d.C.M., Semedo,
L.T.d.A.S., Alviano, C.S., and Gomes, R.C. (2008). Streptomyces lunalinharesii sp. nov., a
chitinolytic streptomycete isolated from cerrado soil in Brazil. Int J Syst Evol Microbiol 58,
2774-2778.
Spalding, M.D., and Grenfell, A.M. (1997). New estimates of global and regional coral reef
areas. Coral Reefs 16, 225-230.
Steele, H.L., and Streit, W.R. (2005). Metagenomics: Advances in ecology and
biotechnology. FEMS Microbiology Letters 247, 105-111.

210
Stepanauskas, R. (1999). Differential Dissolved Organic Nitrogen Availability and Bacterial
Aminopeptidase Activity in Limnic and Marine Waters. Microbial Ecology 38, 264-272.
Stevenson, B.S., Eichorst, S.A., Wertz, J.T., Schmidt, T.M., and Breznak, J.A. (2004). New
Strategies for Cultivation and Detection of Previously Uncultured Microbes. Appl Environ
Microbiol 70, 4748-4755.
Stiling, P.D. (1996). Ecology. Theories and applications. 2 ª Edição. New York Jersey
Prentice-Hall.
Stone, R. (2007). A world without corals? Science 316, 678-681.
Sun, S., Chen, J., Li, W., Altintas, I., Lin, A., Peltier, S., Stocks, K., Allen, E.E., Ellisman, M.,
Grethe, J., et al. (2011). Community cyberinfrastructure for Advanced Microbial Ecology
Research and Analysis: the CAMERA resource. Nucleic Acids Research 39, D546-D551.
Swartz, W., Sala, E., Tracey, S., Watson, R., and Pauly, D. (2010). The Spatial Expansion and
Ecological Footprint of Fisheries (1950 to Present). PLoS ONE 5.
Taketani, R., Franco, N., Rosado, A., and van Elsas, J. (2010a). Microbial community
response to a simulated hydrocarbon spill in mangrove sediments. The Journal of
Microbiology 48, 7-15.
Taketani, R., and Tsai, S. (2010). The Influence of Different Land Uses on the Structure of
Archaeal Communities in Amazonian Anthrosols Based on 16S rRNA and
<i>amo</i>A Genes. Microbial Ecology 59, 734-743.
Taketani, R., Yoshiura, C., Dias, A., Andreote, F., and Tsai, S. (2010b). Diversity and
identification of methanogenic archaea and sulphate-reducing bacteria in sediments from a
pristine tropical mangrove. Antonie van Leeuwenhoek 97, 401-411.
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: Molecular Evolutionary
Genetics Analysis (MEGA) Software Version 4.0. Molecular Biology and Evolution 24,
1596-1599.
Teather, R.M., and Wood, P.J. (1982). Use of Congo red-polysaccharide interactions in
enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl
Environ Microbiol 43, 777-780.
Thompson, F., Bruce, T., Gonzalez, A., Cardoso, A., Clementino, M., Costagliola, M.,
Hozbor, C., Otero, E., Piccini, C., Peressutti, S., et al. (2011). Coastal bacterioplankton
community diversity along a latitudinal gradient in Latin America by means of V6 tag
pyrosequencing. Archives of Microbiology 193, 105-114.
Thompson, J.D., Higgins, D.G., and Gibson, T.J. (1994). CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting, position-
specific gap penalties and weight matrix choice. Nucleic Acids Research 22, 4673-4680.
Thurber, R.V., Willner-Hall, D., Rodriguez-Mueller, B., Desnues, C., Edwards, R.A., Angly,
F., Dinsdale, E., Kelly, L., and Rohwer, F. (2009). Metagenomic analysis of stressed coral
holobionts. Environmental Microbiology 11, 2148-2163.

211
Tindall, B.J., Kämpfer, P., Euzéby, J.P., and Oren, A. (2006). Valid publication of names of
prokaryotes according to the rules of nomenclature: past history and current practice.
International Journal of Systematic and Evolutionary Microbiology 56, 2715-2720.
Tisdall, J.M., and Oades, J.M. (1982). Organic matter an dwater stable aggregates in soils.
Journal of Soil Science 33, 141-163.
Torsvik, V., Goksoyr, J., and Daae, F.L. (1990). High diversity in DNA of soil bacteria. Appl
Environ Microbiol 56, 782-787.
Torsvik, V., Øvreås, L., and Thingstad, T.F. (2002). Prokaryotic Diversity--Magnitude,
Dynamics, and Controlling Factors. Science 296, 1064-1066.
Towner, K.J. (2009). Acinetobacter: an old friend, but a new enemy. Journal of Hospital
Infection 73, 355-363.
Tringe, S., von Mering, C., Kobayashi, A., Salamov, A., Chen, K., Chang, H., Podar, M.,
Short, J., Mathur, E., Detter, J., et al. (2005). Comparative metagenomics of microbial
communities. Science 308, 554 - 557.
Turque, A.S., Batista, D., Silveira, C.B., Cardoso, A.M., Vieira, R.P., Moraes, F.C.,
Clementino, M.M., Albano, R.M., Paranhos, R., Martins, O.B., et al. (2010). Environmental
Shaping of Sponge Associated Archaeal Communities. PLoS ONE 5, e15774.
Turque, A.S., Cardoso, A.M., Silveira, C.B., Vieira, R.P., Freitas, F.A.D., Albano, R.M.,
Gonzalez, A.M., Paranhos, R., Muricy, G., and Martins, O.B. (2008). Bacterial communities
of the marine sponges Hymeniacidon heliophila and Polymastia janeirensis and their
environment in Rio de Janeiro, Brazil. Marine Biology 155, 135-146.
Valverde, A., Delvasto, P., Peix, A., Velazquez, E., Santa-Regina, I., Ballester, A.,
Rodriguez-Barrueco, C., Garcia-Balboa, C., and Igual, J.M. (2006). Burkholderia ferrariae sp.
nov., isolated from an iron ore in Brazil. Int J Syst Evol Microbiol 56, 2421-2425.
Van Trappen, S., Mergaert, J., Van Eygen, S., Dawyndt, P., Cnockaert, M.C., and Swings, J.
(2002). Diversity of 746 Heterotrophic Bacteria Isolated from Microbial Mats from Ten
Antarctic Lakes. Systematic and Applied Microbiology 25, 603-610.
Varga, J., Kocsube, S., Toth, B., Frisvad, J.C., Perrone, G., Susca, A., Meijer, M., and
Samson, R.A. (2007). Aspergillus brasiliensis sp. nov., a biseriate black Aspergillus species
with world-wide distribution. Int J Syst Evol Microbiol 57, 1925-1932.
Venter, J., Remington, K., Heidelberg, J., Halpern, A., Rusch, D., Eisen, J., Wu, D., Paulsen,
I., Nelson, K., Nelson, W., et al. (2004a). Environmental genome shotgun sequencing of the
Sargasso Sea. Science 304, 66 - 74.
Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O.,
Yandell, M., Evans, C.A., Holt, R.A., et al. (2001). The Sequence of the Human Genome.
Science 291, 1304-1351.
Venter, J.C., Remington, K., Heidelberg, J.F., Halpern, A.L., Rusch, D., Eisen, J.A., Wu, D.,
Paulsen, I., Nelson, K.E., Nelson, W., et al. (2004b). Environmental Genome Shotgun
Sequencing of the Sargasso Sea. Science 304, 66-74.

212
Vieira, R., Clementino, M., Cardoso, A., Oliveira, D., Albano, R., Gonzalez, A., Paranhos, R.,
and Martins, O. (2007). Archaeal Communities in a Tropical Estuarine Ecosystem: Guanabara
Bay, Brazil. Microbial Ecology 54, 460-468.
Vieira, R.P., Gonzalez, A.M., Cardoso, A.M., Oliveira, D.N., Albano, R.M., Clementino,
M.M., Martins, O.B., and Paranhos, R. (2008). Relationships between bacterial diversity and
environmental variables in a tropical marine environment, Rio de Janeiro. Environmental
Microbiology 10, 189-199.
Viola, L.B., Attias, M., Takata, C.S.A., Campaner, M., De Souza, W., Camargo, E.P., and
Teixeira, M.M.G. (2009). Phylogenetic Analyses Based on Small Subunit rRNA and
Glycosomal Glyceraldehyde-3-Phosphate Dehydrogenase Genes and Ultrastructural
Characterization of Two Snake Trypanosomes: Trypanosoma serpentis n. sp. from Pseudoboa
nigra and Trypanosoma cascavelli from Crotalus durissus terrificus. Journal of Eukaryotic
Microbiology 56, 594-602.
Voget, S., Steele, H.L., and Streit, W.R. (2006). Characterization of a metagenome-derived
halotolerant cellulase. Journal of Biotechnology 126, 26-36.
von Mering, C., Hugenholtz, P., Raes, J., Tringe, S.G., Doerks, T., Jensen, L.J., Ward, N., and
Bork, P. (2007). Quantitative Phylogenetic Assessment of Microbial Communities in Diverse
Environments. Science 315, 1126-1130.
Wang, F., Li, F., Chen, G., and Liu, W. (2009). Isolation and characterization of novel
cellulase genes from uncultured microorganisms in different environmental niches.
Microbiological Research 164, 650-657.
Wang, Q., Garrity, G.M., Tiedje, J.M., and Cole, J.R. (2007). Naive Bayesian Classifier for
Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ
Microbiol 73, 5261-5267.
Ward, N.L., Challacombe, J.F., Janssen, P.H., Henrissat, B., Coutinho, P.M., Wu, M., Xie, G.,
Haft, D.H., Sait, M., Badger, J., et al. (2009). Three Genomes from the Phylum Acidobacteria
Provide Insight into the Lifestyles of These Microorganisms in Soils. Appl Environ Microbiol
75, 2046-2056.
Wegley, L., Edwards, R., Rodriguez-Brito, B., Liu, H., and Rohwer, F. (2007). Metagenomic
analysis of the microbial community associated with the coral Porites astreoides.
Environmental Microbiology 9, 2707-2719.
Weil, E., Smith, G., and Gil-Agudelo, D.L. (2006). Status and progress in coral reef disease
research. Diseases of Aquatic Organisms 69, 1-7.
Welch, R.A., Burland, V., Plunkett, G., Redford, P., Roesch, P., Rasko, D., Buckles, E.L.,
Liou, S.-R., Boutin, A., Hackett, J., et al. (2002). Extensive mosaic structure revealed by the
complete genome sequence of uropathogenic Escherichia coli. Proceedings of the National
Academy of Sciences 99, 17020-17024.
Wertz, S., Degrange, V., Prosser, J.I., Poly, F., Commeaux, C., Freitag, T., Guillaumaud, N.,
and Roux, X.L. (2006). Maintenance of soil functioning following erosion of microbial
diversity. Environmental Microbiology 8, 2162-2169.

213
Whitman, W.B., Coleman, D.C., and Wiebe, W.J. (1998). Prokaryotes: The unseen majority.
Proceedings of the National Academy of Sciences of the United States of America 95, 6578-
6583.
Wilson, E.O. (1985). Time to Revive Systematics. Science 230, 1227.
Wilson, K.A., Underwood, E.C., Morrison, S.A., Klausmeyer, K.R., Murdoch, W.W., Reyers,
B., Wardell-Johnson, G., Marquet, P.A., Rundel, P.W., McBride, M.F., et al. (2007).
Conserving Biodiversity Efficiently: What to Do, Where, and When. PLoS Biol 5, e223.
Wirth, S., and Ulrich, A. (2002). Cellulose-Degrading Potentials and Phylogenetic
Classification of Carboxymethyl-cellulose Decomposing Bacteria Isolated from Soil.
Systematic and Applied Microbiology 25, 584-591.
Woese, C.R., and Fox, G.E. (1977). Phylogenetic structure of the prokaryotic domain: the
primary kingdoms. Proceedings of the National Academy of Sciences 74, 5088-5090.
Xu, J. (2006). Microbial ecology in the age of genomics and metagenomics: concepts, tools,
and recent advances. Molecular Ecology 15, 1713-1731.
Zhang, J. (2003). Evolution by gene duplication: an update. Trends in Ecology & Evolution
18, 292-298.
Zhou, J., Kang, S., Schadt, C.W., and Garten, C.T. (2008). Spatial scaling of functional gene
diversity across various microbial taxa. Proceedings of the National Academy of Sciences
105, 7768-7773.
Zinder, S.H., and Salyers, A.A. (2005). Microbial Ecology—New Directions, New
Importance. In Bergey’s Manual® of Systematic Bacteriology, D.J. Brenner, N.R. Krieg, J.T.
Staley, and G.M. Garrity, eds. (Springer US), pp. 101-110.

214
7. ANEXOS
7.1 Anexo 1. Métodos
Este anexo contém procedimentos detalhados citados na seção materiais e métodos.
7.1.1 Pirosequenciamento dos metagenomas do Banco de Abrolhos
Esta seção apresenta os principais protocolos utilizados para realização do
pirosequenciamento das amostras do Banco de Abrolhos
7.1.1.1 Extração do DNA realizada no interior do Sterivex
-Adicionar 100 µL de lisozima (concentração 1mg/ml final) e incubar a 37 ° C por
45 min.,
- Adicionar 20 µL de proteinase K (concentração final de 0,2 mg / ml) e 100 µL de
dodecil sulfato de sódio (SDS) (concentração final 1%) e incubar a 55 ° C, com
agitação suave durante 60 minutos.
O lisado foi transferido e recuperado para um novo tubo com a adição de 1 mL de tampão
SET para extração orgânica com um volume de fenol: clorofórmio: álcool isoamílico
(25:24:1):
-Adicionar um volume de fenol: clorofórmio: álcool isoamílico,
-Vortexar e centrifugar 14000 rpm por 5 minutos,
-Coletar a fase superior aquosa e transferir para um tubo novo,
-Adicionar um volume de clorofórmio: álcool isoamílico (24:1),
-Adicionar 500 µL de fenol: clorofórmio: álcool isoamílico, vortexar, e centrifugar
por 5 minutos,
-Coletar a fase superior aquosa e transferir para um tubo novo,
–Precipitar com etanol absoluto e 3 M acetato de sódio (0,3 M final) a - 20 ° C
durante a noite.
-Centrifugar 14000 rpm por 30 minutos
-Adicionar 1 mL de etanol 70 % e centrifugar por 5 minutos e descartar o
sobrenadante (duas vezes)

215
-Secar o precipitado em temperatura ambiente e ressuspender em 30 µL de água
milliQ.
O resultado das extrações de DNA foi verificado em gel de agarose 1 % TBE 1 X.
7.1.1.2 Purificação do DNA metagenômico
Foi utilizado o High pure PCR template preparation kit (Roche) para preparação das
bibliotecas de acordo com o protocolo a seguir:
- Pré aquecer uma alíquota de elution buffer a 70°
- Adicionar elution buffer (sem ser o pré-aquecido) até a amostra atingir 200µL
- Adicionar 200µL de binding buffer. Misturar bem
- Adicionar 100 µL de isopropanol. Misturar bem
- Transferir a amostra para a coluna em um tubo coletor
- Centrifugar 1min / 8000g
- Remover o filtro. Descartar o filtrado e colocá-lo em um tubo novo
-Adicionar 500 µL de inhibitor removal buffer no filtro
- Centrifugar 1min / 8000g
-Transferir coluna para tubo novo
-Adicionar 500 µL de wash buffer
- Centrifugar 1min / 8000g
- Descartar o filtrado e adicionar novamente 500 µL de wash buffer
- Centrifugar 1min / 8000g
- Descartar o filtrado e agora centrifugar apenas 10s
- Transferir a coluna para tubos 1.5 mL
-Adicionar 150µL de elution buffer pré-aquecido no filtro e incubar a temperatura
ambiente por 15 minutos
-Centrifugar 1min / 8.000g
-Recuperar o que foi filtrado e aplicar novamente no filtro
- Incubar 15minutos

216
- Centrifugar 1min / 8000g para eluição final do DNA
É preciso preparar uma curva padrão de concentração do DNA padrão de acordo com a tabela
a seguir após diluir o TE 20X (tampão Tris-EDTA) para 1X em um total de 20 mL, o padrão
(100 ng/µL) para 2 ng / µL (1:50) em um total de 1 mL e o corante Picogreen (1:200) em um
total de 10 mL. As diluições foram preparadas de acordo com os valores a seguir:
Tabela 1. Diluição dos reagentes para a curva de concentração do padrão do reagente PicoGreen.
Em seguida, foram preparadas as amostras:
- Adicionar 346,5µL TE 1X + 3,5µL de amostra + 350µL de PicoGreen
- Vortexar e dar um giro (minicentrífuga) em todos os tubos
- Utilizar 200µL das amostras para as leituras. Os comprimentos de onda utilizados no
ensaio foram 480 nm de excitação e 520 nm de emissão.
- Plotar a curva padrão em tabela Excel e determinar as concentrações de cada amostra
utilizando a curva da equação.
7.1.1.3 Preparação das bibliotecas pelo método shotgun
As amostras devem estar diluídas em TE e apresentar um volume máximo de 100 µL. A
fragmentação do DNA e purificação dos fragmentos foi realizada de acordo com o protocolo:
- Aplicar os 100µL no fundo do tubo de nebulização
- Adicionar 500µL da solução nebulizer buffer e misturar por pipetagem
-Aplicar 30 psi (2,1 bar) de pressão por 1 minuto
Diluições
(ng/µL)
PicoGreen
(µL)
TE 1X
(µL)
padrão
(µL)
1000 500 0 500
100 500 450 50
10 500 495 5
1 500 499 0,5
Branco 500 500 0

217
Após a fragmentação do DNA foi utilizado o Mini eluate PCR purification kit (Qiagen) para
purificar os fragmentos.
- Aplicar 2,5 mL PBI buffer que contendo indicador de pH. A suspensão de DNA
deve permanecer na cor amarelo claro.
- Aplicar nas colunas, centrifugar 13000 rpm por 25 segundos e descartar o
sobrenadante.
-Repetir até filtrar todo o volume de dentro do tubo. A última centrifugação deve
levar 1 minuto para secar o filtro.
- Adicionar 750 µL de Wash buffer (PE) e centrifugar a 13000 rpm por 1 minuto.
Rodar os tubos 180° e centrifugar novamente.
- Adicionar 16 µL de TE no centro do filtro e esperar 2 minutos para então
centrifugar a 13000 rpm por 2 minutos.
O DNA está pronto para ser reparado. Segue a reação de reparação e o programa utilizado:
Tabela 2 Reação de reparo do DNA
Programa:
25°C 20min
72°C 20 min.
Manter a 4 °C
Reagentes
Volume
(µL)
Tampão 10X 2,5
ATP 2,5
T4 polimerase 1
PNK (polinucleotídeo
kinase) 1
Taq polimerase 1
Total 8

218
7.1.1.3.1 Preparação de esferas magnéticas
Os fragmentos de DNA são ligados a esferas magnéticas. As esferas são preparadas de acordo
com o protocolo:
- Vortexar as esferas para ressuspendê-las completamente
- Adicionar 125 µL da suspensão de esferas e concentrador de partículas
magnéticas
- Remover o sobrenadante
- Adicionar 73 µL de TE e vortexar por 5 segundos
- Adicionar 500 µL de sizing solution e manter no gelo até a sua utilização
7.1.1.3.2 Ligação do DNA as esferas
As esferas magnéticas são utilizadas para selecionar os fragmentos de DNA com tamanho
entre 400 e 2000 pb. Foram seguidas as seguintes e etapas:
- Adicionar 1 µL de cada adaptador (A e B) + 1 µL de ligase
- Incubar 25 ° C por 10 minutos
- Adicionar a reação às esferas preparadas e vortexar 5 segundos, baixar as esferas
em minicentrífuga girar o tubo e incubar 5 minutos
- Colocar no concentrador de partículas magnéticas e remover o sobrenadante
- Adicionar 100 µL de TE vortexar 5 segundos
- Adicionar 500 µL de Sizing solution
- Realizar os dois passos anteriores mais uma vez, ainda no concentrador de
partículas magnéticas
- Adicionar 1 mL de etanol 70 % e depois remover o etanol. Fazer duas vezes
- Manter os tubos no concentrador de partículas magnéticas e deixar secando com a
tampa aberta por aproximadamente 5minutos
- Remover do concentrador de partículas magnéticas e adicionar 53 µL de TE.
Vortexar e baixar as esferas
- Colocar no concentrador de partículas magnéticas e transferir 50 µL do
sobrenadante contendo a biblioteca para um tubo novo.

219
7.1.1.3.3 Quantificação e checagem da qualidade da fragmentação das bibliotecas
A quantificação da biblioteca foi feita por fluorometria. É necessário fazer uma curva padrão
utilizando os padrões do kit (rapid library preparation). Com isso, foi determinada a
concentração das bibliotecas e o volume necessário nos ensaios de emPCR. A quantificação
foi realizada de acordo com o protocolo:
- Adicionar 90 µL do padrão em 90µL TE
- Preparar sete tubos com 60µL de TE para fazer diluição seriada do padrão
- Transferir 120 µL do padrão (após vortexar para homogeneizar a suspensão) para
o próximo tubo, num total de sete diluições
- Adicionar 50 µL de cada diluição (em duplicata) em placa preta de 96 poços para
fluorometria
- Em seguida, aplicar 50 µL da biblioteca na placa e realizar a leitura
- Os comprimentos de onda utilizados para emissão e excitação foram 465 e 515
nm, respectivamente
-Após a leitura recuperar as bibliotecas e estocá-las a - 20 ° C
- Exportar o arquivo para Excel e montar a curva padrão para determinação da
concentração. Também existe um programa disponível no sítio da Roche
(http://454.com/)
-As bibliotecas devem ser diluídas para obter a concentração em 107
moléculas /
µL.
A checagem da qualidade da fragmentação das bibliotecas foi realizada através de
eletroforese em chip (Bio Analyzer 2100, Agilent Technologies), utilizando DNA LabChip
(Agilent Technologies).
7.1.1.4 Reação em cadeia da polimerase em emulsão (em-PCR)
O em-PCR ocorre em um ambiente microfluídico onde os reagentes ficam limitados ao
interior de micelas da emulsão. Desta forma, ocorre a reação de amplificação dos fragmentos
de DNA ligados as esferas revestidas por streptavidina pelo adaptador B (biotina). Apenas as
esferas que apresentarem um, e somente um, fragmento de DNA será capaz de gerar

220
sequências. Por isso, o volume (µL) da biblioteca que foi usada como modelo na reação foi
calculado de acordo com a equação:
2 (Número de moléculas por esfera) X 6,8 X106 (número de esferas para em-PCR
107
(número de moléculas) / μL ([ ] da biblioteca)
As etapas de preparo da reação de em-PCR (GS FLX Titanium emPCR Breaking Kits MV)
estão descritas nas seções a seguir.
7.1.1.4.1 Preparação do óleo para emulsões
O óleo precisa ser agitado em alta velocidade para a produção das emulsões:
- Instale os tubos de emulsão de óleo de forma segura no misturador Tissue Lyser II
(Qiagen). As tampas devem ficar voltadas para fora
- Agitar a emulsão de óleo 28 Hz por 2 minutos
- O óleo emulsificado será utilizado na próxima etapa
7.1.1.4.2 Mock Amplification Mix e pré-emulsões
A mistura que cria o ambiente microfluídico para os reagentes da reação de amplificação deve
ser preparada da seguinte forma:
- Diluir 2 mL de Mock Amplification Mix 5 × (tubos laranja), com 8 ml de água
para fazer uma solução de trabalho (adicionar água primeiro) e vortexar.
- Adicionar 1 mL de solução Mock Amplification Mix diluída a cada tubo de óleo
emulsificado
- Inverter (não agite) os tubos 2 - 3 vezes para misturar
- Agitar no Tissue Lyser II a 28 Hz por 5 minutos
- Retire as emulsões da Tissue Lyser II e reservá-las (não agitar)
7.1.1.4.3 Preparação do Live Amplification Mix
Reagentes da reação de amplificação. Adicionar os reagentes em tubos cônicos na ordem em
que estão listadas abaixo:

221
Tabela 3 Reagentes para a preparação do Live Amplification mix
Vortexar o Live Amplification Mix por 5 segundos, e armazenar no gelo.
7.1.1.4.4 Preparação das esferas de captura de DNA (beads)
As esferas de captura são revestidas por estreptavidina onde os fragmentos se ligam através
do adaptador B, que contém biotina. Os fragmentos ligados as esferas são utilizados como
molde na reação de amplificação. A preparação seguiu as seguintes etapas:
- Preparar Capture Bead Wash Buffer TW 1 X misturando 1 mL de Capture Bead
Wash buffer TW 10 X em de água
- Ressuspender as os tubos de DNA Esferas de captura
- Transferir 920μL de DNA Esferas de captura para 2 tubos de 1,5 mL
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos
- Cuidadosamente remover e eliminar o sobrenadante sem perturbar as esferas.
- Lavar as esferas duas vezes com 1 ml de Capture Bead Wash Buffer TW 1x.
Vortexar para ressuspender as esferas. Baixar as esferas em mini-centrífuga por 10
Reagentes (cor)
Volume
(µL)
Água 2400
emPCR Aditive (Preto - 2 tubos) 3000
5x mix amplification (verde claro - 2
tubos) 1560
Primer amplification (amarelo) 460
emPCR Enzyme Mix (verde escuro - 2
tubos) 400
PPiase (rosa) 10
Total 7830

222
segundos, girar o tubo 180 ° e centrifugar novamente por 10 segundos e desprezar o
sobrenadante após cada lavagem.
- Ressuspender cada pellet de DNA Capture Esferas em 320 μL de Capture Bead
Wash Buffer TW 1x.
- Medir o volume total de beads (aproximadamente 600 μL) e distribuí-las
uniformemente entre 4 tubos de tubos de 1,5 mL
- Manter as esferas preparadas no gelo.
7.1.1.4.5 Captura das bibliotecas
Nesta etapa os fragmentos de DNA metagenômico se ligam às esferas. Para isso, foram
seguidas as seguintes etapas:
- Descongelar uma alíquota da biblioteca (concentração de 107 moléculas /µL) a ser
amplificada.
- Colocar 10 μL da biblioteca em tubo de 0.2 µL
- Desnaturar o DNA em termociclador a 95 ° C por 2 minutos
- Baixar as esferas em mini-centrífuga por 10 segundos, gire o tubo 180 ° e
centrifugue novamente por 10 segundos. Remover e descartar o sobrenadante.
-Para cada tubo de contendo as esferas lavadas, adicionar 1.36 μL da biblioteca e
vortexar 5 segundos
- Adicionar 875 μL do Live Amplification Mix em cada tubo contendo a biblioteca
capturada e vortexar.
7.1.1.4.6 Emulsificação
Nesta etapa ocorre a interiorização das esferas para o ambiente limitado contendo os reagentes
para a amplificação.
- Pipetar o conteúdo de cada tubo de biblioteca capturada pelas esferas em um tubo
de emulsão preparada
- Inverter os tubos de 3 vezes para misturar (não agitar)
- Agitar no Tissue Lyser II em 12 Hz por 5 minutos

223
- Distribuir a emulsão em placa de 96 poços (dois tubos de emulsão por placa).
Dispensar 100 μL por poço
7.1.1.5 Quebra da emulsão (break emulsion)
Após a amplificação é necessário recuperara as esferas contendo os produtos de amplificação
(haired esferas) para realizar a quebra das emulsões e retirada do óleo emulsificado. As
seções a seguir descrevem como deve ser realizado o processo de quebra das emulsões.
7.1.1.5.1 Recuperação das esferas
É utilizado o GS FLX Titanium emPCR Breaking Kit
-Retirar da geladeira o Enhancing Fluid XT e Annealing Buffer XT, e manter em
temperatura ambiente.
- Inserir o conector azul na abertura superior da estrutura de 8 pinos (Figura
estrutura.). Conecte a outra extremidade do tubo a uma fonte de vácuo de acordo
com a figura.
Figura 1. Estrutura de oito pinos utilizada na recuperação das esferas utilizadas no em-PCR.
- Aspirar as emulsões de cada amostra utilizando o vácuo. Após aspirar todas as
emulsões, virar a estrutura de cabeça para baixo para ajudar a drenar o máximo de
material. Aspirar um pouco de isopropanol ajuda a drenar as esferas do interior da
estrutura.
- Lavar os poços duas vezes com 100 μL de isopropanol por poço e aspirar o
restante do material.
- Aspirar um adicional (aproximadamente 5 mL) de isopropanol a recolher todas as
esferas que podem ter permanecido na tubulação.
- Troque a posição dos tubos de 50 mL, de modo que o recipiente cheio seja
conectado ao vácuo e o vazio a estrutura de aspiração e repita o procedimento para
a placa seguinte

224
7.1.1.5.2 Lavagem das esferas
Nesta etapa é realizada a retirada do restante do óleo da reação. A lavagem foi realizada
acordo com as seguintes etapas:
- Vortexar o tubo de coleta de 50 mL
- Adicionar isopropanol até o volume final de 40 mL em cada tubo de coleta e
vortexar
- Centrifugar a 930 × g por 5 minutos e remover o sobrenadante cuidadosamente
- Adicionar 20 mL de Enhancing Fluid XT e vortexar. Caso a amostra empedrar,
usar um bastão de vidro para desmanchá-la
- Centrifugar e desprezar o sobrenadante como anteriormente
- Adicionar 35 mL de isopropanol e vortexar
- Centrifugar e desprezar o sobrenadante como anteriormente
- Adicionar 35 mL de isopropanol e vortexar
- Centrifugar e desprezar o sobrenadante como anteriormente
- Adicionar 35 mL de etanol e vortexar
- Centrifugar e desprezar o sobrenadante como anteriormente.
- Adicionar 20 mL de Enhancing Fluid XT e vortexar
- Centrifugar e desprezar o sobrenadante como anteriormente, deixando cerca de 2
mL de Enhancing Fluid XT.
- Transferir a suspensão para tubos de 1,5 mL. Um tubo novo para cada tubo de
coleta de 50 mL
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante
- Repetir as duas últimas etapas até a toda a suspensão de todo ser transferida
- Lavar cada tubo de coleta de 50 mL com 600 μL de Enhancing Fluid XT, e
transferir para o tubo de 1,5 mL para recuperar o máximo de amostra possível
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante

225
- Lavar as esferas duas vezes com 500 μL de Enhacinhg Fluid XT. Baixar as
esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e centrifugar
novamente por 10 segundos e desprezar o sobrenadante
- Adicionar 500 μL de Enhancing Fluid XT em cada tubo vortexar
7.1.1.6 Enriquecimento (Enrichment)
As esferas recuperadas são contadas (utilizando contador de partículas, ou pesando o material)
no início e após o procedimento de enriquecimento. Nesta etapa são eliminadas as esferas que
falharam na amplificação e que, portanto não contém DNA amplificado ligado à superfície. A
faixa de enriquecimento utilizada foi de 5 – 20 % das esferas que iniciaram o em-PCR. O
enriquecimento é realizado de acordo com as seções a seguir detalham o procedimento.
7.1.1.6.1 Enriquecimento indireto
Nesta primeira etapa ocorre a preparação das haired esferas para o enriquecimento.
-Preparar Melt Solution misturando 125 μL de NaOH (10 N) em 9,875 ml de água
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante
- Adicione 1 mL de Melt Solution as esferas e vortexar. Incubar por 2 minutos em
temperatura ambiente
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante
- Repetir os dois últimos passos.
- Adicionar 1 mL de Annealing buffer XT e vortexar. Baixar as esferas em mini-
centrífuga por 10 segundos, girar o tubo 180 ° e centrifugar novamente por 10
segundos e desprezar o sobrenadante
- Repita o passo anterior
- Para determinar o número de esferas recuperadas na etapa anterior (quebra das
emulsões) ao enriquecimento:
a. Adicionar 500 μL de Annealing buffer XT e vortexar.
b. Transferir uma alíquota de 3 μL da suspensão de esferas para a cubeta de
contagem contendo 10ml de óleo para contagem. A recuperação foi calculada

226
de acordo com a equação: Número de esferas recuperadas / 13,6 X 106) × 100,
(sendo 13,6 X 106, número de esferas para volume médio)
c. Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante.
- Adicionar 30 μL de Annealing buffer XT e 24 μL de Enrichment Primer e vortexar
- Incubar a 65 ° C por 5 minutos e resfriar imediatamente em gelo por 2 minutos.
- Adicionar 500 μL de Enhancing Fluid XT e vortexar. Baixe as esferas em mini-
centrífuga por 10 segundos, gire o tubo 180 ° e centrifugue novamente por 10
segundos e desprezar o sobrenadante
- Repita o último passo mais duas vezes
- Adicionar 800 μL de Enhancing Fluid XT por tubo e vortexar.
7.1.1.6.2 Preparação das esferas de enriquecimento (enrichment beads)
As esferas de enriquecimento são capazes de reter as esferas que obtiveram sucesso na
amplificação (haired esferas) e foram submetidas ao sequenciamento. Elas foram preparadas
da seguinte forma:
- Ressuspender completamente as Enrichment Beads (esfera marrom) por 1 minuto
-Concentrar 320 μL de Enrichment Beads utilizando o Concentrador de partículas
magnéticas (MPC)
- Desprezar o sobrenadante, tomando cuidado para não perturbar o sedimento
- Adicionar 1 mL de Enhancing Fluid XT e vortexar
- Concentrar as esferas no MPC
- Desprezar o sobrenadante, tomando cuidado para não perturbar o sedimento.
- Repetir os três últimos passos
- Após descartar o sobrenadante, remova o tubo do MPC
- Adicionar 320ul μL (40 μL por tubo de emulsão) de Enhancing Fluid XT
7.1.1.6.3 Enriquecimento das esferas com DNA
Nesta etapa as dot beads são eliminadas após o procedimento:

227
- Adicionar 80 μL de Enrichment Beads lavadas a cada tubo de esferas com DNA
amplificado e vortexar
- Misturar as beads de enriquecimento e beads com DNA usando Labquake, em
temperatura ambiente por 5 minutos
- Colocar os tubos no MPC e esperar 3 - 5 minutos para concentrar as esferas.
- Inverter o MPC 5 vezes.
- Descartar cuidadosamente o sobrenadante de cada tubo.
- Lavar as esferas com o Enhancing Fluid XT até que não haja beads visíveis
restantes no sobrenadante, da seguinte maneira:
a. Adicionar 1 mL de Enhancing Fluid XT por tubo
b. Remover os tubos do MPC e vortexar
c. Colocar os tubos no MPC. Inverter o MPC 5 vezes
d. Descartar cuidadosamente para não perturbar as esferas
e. Repetir 6 a 10 vezes até retirar todas as esferas brancas (dot beads)
7.1.1.6.4 Recuperação das esferas de DNA enriquecidas
A Melt Solution promove o desligamento entre as haired esferas e as esferas de
enriquecimento. As etapas deste procedimento foram:
- Retirar os tubos do MPC e ressuspender em 700 µL de Melt solution.
- Vortexar por 5 segundos e colocar os tubos no MPC. Nesta etapa é importante não
demorar muito para recuperar as esferas pois a Melt Solution interfere no
magnetismo das esferas de enriquecimento e pode haver mistura com as esferas
contendo o DNA.
- Transferir o sobrenadante contendo as esferas de DNA enriquecidas para um tubo
novo
- Baixar as esferas em mini-centrífuga por 10 segundos, gire o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante
- Adicionar 700 µL de Melt Solution.

228
- Vortexar por 5 segundos e colocar no MPC para eliminar as possíveis esferas
magnéticas transferidas junto com as esferas de DNA.
- Transferir o sobrenadante contendo esferas de enriquecimento com DNA para um
novo tubo
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante
- Adicionar 500 µL de Annealing buffer XT e vortexar por 5 segundos.
- Baixar as esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e
centrifugar novamente por 10 segundos e desprezar o sobrenadante
- Repetir os dois últimos passos mais duas vezes.
- Ressuspender as esferas em 120 μL de Annealing Buffer XT e vortexar.
- Adicionar 24 μL de Sequencing Primer e vortexar.
- Incubar a 65 ° C por 5 minutos e transferir imediatamente para o gelo e manter
por 2 minutos
- Adicionar 1 mL de Annealing Buffer XT e vortexar por 5 segundos. Baixar as
esferas em mini-centrífuga por 10 segundos, girar o tubo 180 ° e centrifugar
novamente por 10 segundos e desprezar o sobrenadante
- Repita o último passo duas vezes
- Adicione 1 mL de Annealing buffer XT e vortexar
-Transferir uma alíquota de 3 μL das esferas para a cubeta de contagem
-Calcular a porcentagem de enriquecimento utilizando a seguinte equação:
(Número de beads enriquecidas / 13,6 X 106) × 100
-Conservar as esferas, entre 2 ° C e 8 ° C. Elas podem ser mantidas por
aproximadamente de duas semanas
7.1.1.7 Preparação e sequenciamento
A última etapa do sequenciamento pode ser dividida em duas partes principais: a preparação
da placa Pico Tilter Plate (PTP) onde é depositada as esferas e ocorre o sequenciamento de
fato e a preparação do sequenciador GS FLX 454 e seus reagentes que serão consumidos
durante o procedimento. Foi utilizado ¼ de placa por amostra.

229
7.1.1.7.1 Carregamento da Placa PicoTilter Plate (PTP)
Os principais componentes depositados na placa e sua função são descritos a seguir:
-DNA esferas (DNA beads): carregam o DNA da biblioteca para ser sequenciado
(amostra)
-Esferas controle (Control beads): carregam sequências conhecidas. Podem ser
utilizadas para corrida teste em separado
- Esferas de enzima (Enzime beads): carregam os componentes imobilizados de
enzimas do sistema de quimiluminescência (sulfurilase e luciferase)
-Esferas PPiase (Ppiase beads): retira o PPi inorgânico para reduzir as
interferências cruzadas entre poços, durante cada fluxo de nucleotídeo, assim como
ruído residual entre os fluxos
-Esferas de empacotamento (Packing beads): estabiliza e mantém todos os
componentes do sistema imobilizados dentro dos poços do dispositivo PTP, durante
a execução do sequenciamento.
Esses componentes foram preparados de acordo com as seções a seguir.
7.1.1.7.2 Preparação da PTP e DNA beads
A preparação do tampão Bead Buffer 2 (BB2) foi realizada da seguinte forma:
-Adicionar 1,2 mL de Titanium Supplement CB para a garrafa de 200 mL de
Titanium Bead Buffer de pré-refrigerados. Re-tampão e misture suavemente por
inversão
-Remova o tubo de Apyrase da tira de reagentes e descongelar completamente.
Deixar os outros componentes em gelo até serem utilizados
-Adicione 34 ml de solução Apyrase na garrafa de Titanium Bead Buffer acrescido
de Titanium Supplement CB.
-Feche a garrafa e o rotule a nova reação como Bead Buffer 2 (BB2). Misturar
suavemente por inversão, e retornar a garrafa para o gelo. Também retornam o
restante do de Titanium Supplement CB e da apirase
-Aliquotar 15 ml de BB2 em novo tubo.

230
A seguir, retirar a PTP do envelope e anotar o código identificador de 6 dígitos na placa e
retornar a placa para o envelope e submergi-la em BB2 e deixá-la em repouso por, pelo
menos, 10 minutos. Manter o restante da solução no gelo.
Antes de prepara as DNA e as Control beads, foi preciso calcular o volume de esferas (DNA
beads) necessárias para ocupar os 4 quartos da placa, ou seja, aproximadamente 790000
esferas. O volume deve ser calculado de acordo com a equação:
A partir deste cálculo, foi retirado o volume de sobrenadante necessário para reduzir o volume
de DNA beads para 20 µL. Após a medição, foram baixadas as esferas, giradas 180 ° e
centrifugadas novamente e foi adicionado 10 µL de Control beads para cada tubo, de modo
que o volume final foi de 30 µL.
Separadamente, preparar o DNA Bead Incubation Mix (DBIM) em um tubo novo de 15mL de
acordo com a tabela abaixo. Identificar o tubo e vórtex com cuidado para assegurar que o
Cofator e a DNA polimerase sejam bem misturados.
Tabela 4. Preparação do DNA Incubation Mix (DBIM).
Adicionar 320 µL de DBIM em cada tubo de DNA beads e misturar por rotação em
temperatura ambiente por 15 minutos. Durante este tempo, é realizada a lavagem das Packing
beads. As esferas foram lavadas 3 vezes com 1 mL de BB2 por centrifugação a 10000 rpm
por 5 minutos, vortexando após a adição do tampão. Após a terceira lavagem adicionar 550
µL de BB2 por tubo, vortexar e manter em gelo
As Enzime beads foram preparadas conforme as etapas a seguir:
Volume de DNA beads (µL) =
790000
x 100 Número de esferas enriquecidas
Reagente Volume (μL)
BB2 1570
Polimerase Cofator 150
DNA polimerase 300
Total 2020

231
-Vortexar e baixar as Enzime e PPiase Beads usando um concentrador de partículas
magnéticas (MPC) por 30 segundos. Inverter os MPC várias vezes e aguarde 30
segundos de novo. Retire cuidadosamente o sobrenadante e remover os tubos da
MPC
- Lave as PPiase Beads três vezes com 1 ml de BB2 as Enzime beads com 500 μL
de BB2s. Vortexar e lavar as esferas usando o MPC
-Após a terceira lavagem, adicionar 1 ml de BB2 a cada tubo de Enzime beads e
500 μL do BB2 ao tubo de Ppiase beads. Vortexar as esferas e manter em gelo
-Preparar as esferas para as camadas 1, 3 e 4 em três tubos de 15 mL separados e
devidamente rotulados conforme a tabela abaixo. Isto é feito através da diluição da
Enzime beads (camada 1: Enzime beads da camada pré-e camada 3: Enzime beads
pós camada) e as Enzime PPiase (camada 4) no BB2, conforme listado na Tabela.
Vortexar as enzime e Ppiase beads lavadas para obter uma suspensão uniforme
antes da transferência.
Tabela 5. Preparação das camadas 1, 3 e 4.
Camada Kit
BB2
(μL)
Enzyme Beads
(μL)
PPiase Beads
(μL)
Total Volume
(μL)
1
XLR70
3250 550 --- 3800
3 2500 1300 --- 3800
4 3340 --- 460 3800
A camada 2 é composta pelas DNA beads. Ao fim da incubação foi adicionado 100 μL de
Packing beads em cada tubo com DNA e em seguida adicionar 210 μL de DBIM. O volume
final de 660 μL foi vortexado e misturado durante 5 minutos em temperatura ambiente.
O dispositivo de carregamento e os divisores de borracha foram previamente lavados com
solução detergente alcalina (Sparkleen). A aplicação das camadas ocorreu na ordem 1, 2, 3 e
4, sendo necessária centrifugação de 5 minutos a 4000 rpm e descarte do sobrenadante antes
da aplicação da nova camada. Após a aplicação das 4 camadas, a placa foi colocada no
sequenciador GS FLX 454.

232
7.1.1.8 Preparação do GS FLX 454
Realizar uma pré-lavagem, descartando os reagentes utilizados e limpeza do cassete de
reagentes. Os reagentes da bandeja de sequenciamento devem ser descongelados, mas
mantidos em geladeira.
- Adicionar 6,6 mL de Titanium Supplement CB e 1mL de DTT em cada garrafa de
Titanium Buffer CB e misturar
- Diluir 5 µL de PPiase (tubo rosa) em 45 µL de Inhibitor TW em um tubo de 1.5
mL e vortexar.
- Suplementar os 4 reagentes da bandeja do sequenciamento com seus aditivos
adequados, conforme listado abaixo. Adicione os suplementos na ordem
estabelecida, seguindo as instruções:
a. Adicionar uma pastilha de cloreto de sódio no tubo Post Run Wash (posição
1, tampa branca) e deixar efervescer por 2 minutos. Mudar as luvas
b. Adicionar 13.2 μL a diluição de PPiase ao Inhibitor TW (posição 9, tampa
rosa). Mudar as luvas
c. Adicionar 260 μL de Apyrase ao Apyrase buffer (posição 11, tampa
amarela). Mudar as luvas
d. Adicionar 3000 μL de dATP ao Buffer for ATP (A) (posição 10, tampa cor
lavanda). Mudar as luvas
-Inverter a bandeja de 20 vezes para misturar uniformemente o conteúdo de todos
os tubos.
- Remover o cassete de pré-lavagem cassete e limpe a área
- Remover os tubos de pré-lavagem e descartá-los. Os pré-tubos de lavagem devem
ser descartados após cada uso. Nunca reutilizar os tubos de pré-lavagem
- Limpar a superfície exterior do cassete de reagentes com uma toalha de papel
- Limpe a superfície da plataforma com etanol 50%, sem tocar nos Tubes Sipper
- Posicione o cassete de reagentes e dar início ao sequenciamento
7.1.2 Diversidade de comunidades microbianas do solo da Mata Atlântica

233
Esta seção apresenta os principais protocolos e composição dos meios e soluções utilizados
para a construção das bibliotecas do gene rRNA 16S e de pequenos insertos.
7.1.2.1 Concentração e purificação dos fragmentos do rRNA 16S
Para obtenção de grande quantidade de fragmentos e, assim, maximizar a cobertura da
diversidade, 10 reações para cada solo foram posteriormente concentradas. Foram utilizados
100 ng do DNA metagenômico por reação. O material foi precipitado com acetato de sódio e
etanol seguindo as seguintes etapas:
- Adição de 10% do volume total de acetato de sódio 3M
- Adição de 3X o volume de etanol 100%
- Centrifugação a 14000 rpm por 30 minutos a 4°C
- Descarte do sobrenadante;
- Adição de 1mL etanol 70%;
- Centrifugação a 14000 rpm 20 min.
- Descarte do sobrenadante;
- Esperar secar
- Ressuspender o material (20μL).
Após a verificação do material amplificado, uma corrida em gel de agarose 1% TAE 1X foi
realizada para sacar as bandas correspondentes aos produtos de amplificação do gene rRNA
16S. A purificação foi realizada com kit comercial Ultra Clean DNA Purification (Mo Bio). O
protocolo apresenta as seguintes etapas:
- Pesagem das bandas para determinar o volume de solução Ultra Salt (NaI 6M). Adicionar
3X volume (µL) correspondente ao peso das bandas (g) (ex: 3 µL: 1 g)
- Mistura e incubação das bandas a 55°C até derreter o gel (5-10 minutos)
- Adição de 10 µL da resina
- Incubação em temperatura ambiente por 5 minutos. Agitar suavemente por inversão
continuamente ou a cada minuto
- Centrifugação por 5 segundos a 14000 rpm (todas as centrifugações deste protocolo)

234
- Transferência do sobrenadante para outro tubo e guardá-lo em geladeira caso não
haja uma boa recuperação no final. Descartar após confirmação da eficiência do procedimento
- Ressuspender com 1mL da solução Ultra Wash (etanol 90%). Descarta o
sobrenadante. Repetir a lavagem.
- Ressuspender em 20 µL de água milliQ.
- Incubação por 5 minutos a temperatura ambiente.
- Centrifugação por 1 minuto.
- Transferência do sobrenadante para novo tubo e repetir a centrifugação.
- Recuperação do sobrenadante em tubo novo do DNA purificado.
A efetividade da purificação foi verificada em gel de agarose 1% TBE 1X.
7.1.2.2 Sistema de ligação
O sistema de ligação consiste nos produto de amplificação do rRNA 16S concentrado e
purificado, ligados ao plasmídeo vetor de clonagem pGEMT easy (Promega). A relação
Produto: Vetor utilizado nas reações foi no mínimo de 4:1. As reações de ligação foram
montadas da seguinte maneira:
Tabela 6. Reação de preparação do sistema de ligação.
Reagente Volume (µL)
DNA 3
Tampão (5x) 5
T4 DNA ligase 1
Água 1
Total 10
A reação foi incubada por 16 horas a 16 ° C e depois a enzima foi inativada a 65° C por 20
minutos. O material foi precipitado de acordo com o protocolo:
- 5 μL do sistema de ligação (guardar o restante no freezer)
- Adicionar 1 μL de tRNA (1 μg / μL).

235
- 14 μL de água milliQ (misturar)
- 50 μL de etanol 100% gelado
- Centrifugar 10 minutos a1400 rpm
- Adicionar etanol 70% (400μL). Centrifugar 5 minutos a 14000 rpm
- Esperar secar e ressuspender em 10μL.
7.1.2.3 Preparação de células eletrocompetentes
Células da linhagem E. coli EPI300 foram utilizadas nos experimentos de transformação e
clonagem. Todo material utilizado durante o procedimento (ponteiras, tubos e soluções) foi
colocado em freezer ou geladeira para garantir a eficiência do procedimento. As células
devem permanecer em gelo enquanto não estiverem sendo manuseadas e todo o procedimento
deve ser realizado em ambiente estéril. Foi utilizado o seguinte protocolo de preparação de
células competentes para eletroporação:
- Preparar um pré inóculo de 5 mL em meio LB da célula e incubar a 37°C durante a noite;
- Passar o pré inóculo (5 mL) para 500mL de meio LB novo;
- Incubar a 37 °C até alcançar densidade ótica na faixa 0.5 - 0.6 (cerca de três horas);
- Incubar em gelo por 30 minutos;
- Transferir para tubos novos;
- Centrifugar 20 minutos/ 4000 rpm/ 4 °C;
- Descartar sobrenadante e ressuspender em 5mL de água milliQ e depois completar
para 50 mL;
- Centrifugar 20 minutos/ 4000 rpm/ 4°C. Descartar o sobrenadante;
- Ressuspender em 5mL de glicerol 10 % e completar para 25 mL;
- Juntar de dois em dois tubos;
- Centrifugar 15 minutos/ 4000 rpm/ 4°C. Descartar o sobrenadante;
- Ressuspender o precipitado em 15 ml de glicerol 10%;
- Juntar dois tubos, cuidadosamente, sem perturbar o precipitado;

236
- Centrifugar 15 minutos / 4000 rpm/ 4°C. Descartar sobrenadante e aspirar o restante
com pipeta;
- Ressuspender o precipitado em 500μL de meio GYT;
- Juntar os dois tubos e distribuir 50μL por tubo;
- Armazenar -80°C
Meio LB (Luria-Bertani) caldo: 1 g de triptona; 0,5 g de extrato de levedura; 1 g de NaCl;
volume final de 100 mL; pH 7.0.
Meio GYT (Glicerol, yeast extract and triptone): 10% (v/v) glicerol; 0.125% (p/v) extrato de
levedura; 0.25% (p/v) triptona.
7.1.2.4 Extração plasmidial em placa
As extrações plasmidiais foram realizadas através do método da lise alcalina em placas de 96
poços de acordo com o protocolo a seguir:
-Retirar a placa contendo os clones a serem preparados para extração de DNA para
sequenciamento do freezer -80° e deixar descongelar no gelo por alguns minutos
-Adicionar, com o auxílio da pipeta de repetição (PR), 1 mL do meio de cultura LB
com ampicilina em cada poço da microplaca de cultura (deep wells)
-Utilizando o replicador (carimbo), através de técnicas assépticas, inocular os poços da
placa de crescimento com a cultura bacteriana certificando-se que todos os 96 poços foram
inoculados
-Selar a placa com um adesivo e furar o adesivo que cobre cada poço da placa com
uma agulha esterilizada para permitir a aeração e crescimento bacteriano
-Colocar a placa em um agitador horizontal ajustado a 240 rpm, 37 ° C. Deixe crescer
por 22 horas.
-Remover a placa do agitador. Conferir a uniformidade do crescimento e anotar
qualquer anormalidade após checar cada poço
-Centrifugar a 4000 rpm durante 15 minutos (23ºC) a microplaca de cultura para obter
um precipitado das células
-Remover o adesivo que recobre a placa de crescimento e descartar o sobrenadante em
um recipiente com lisorfórmio. Deixar escorrer o excesso de meio invertendo a placa sobre

237
um papel toalha por alguns minutos. Certifique-se que todo o meio de cultura foi removido
passando a placa com cuidado para outro papel toalha seco
-Adicionar ao sedimento bacteriano 240 µL da Solução I (GET = Tris/EDTA/Glicose).
Selar a placa com o mesmo adesivo e vórtex até ressuspender as células: 8 ponteiras amarelas
+ micropipeta multicanal + recipiente + GET + vórtex
-Centrifugar 6 minutos a 4000 rpm para obter um precipitado das células
-Remover a placa da centrífuga, descartar sobrenadante e secar a placa sobre papel
absorvente
-Ressuspender o sedimento bacteriano em 70 μL de GET com RNAse adicionada na
hora do uso (ver quantidade abaixo) com vórtex:
Para 4 placas: 28 ml de GET + 300 μL de RNAse (10 mg/ml)
Para 2 placas: 14 ml de GET + 150 μL de RNAse (10 mg/ml)
-Transferir 70 μL das suspensões de células para microplaca de fundo U, devidamente
identificada: caixa c/ 96 ponteiras amarelas + micropipeta multicanal + microplaca de fundo
U
-Adicionar 70 μL da Solução 2 (Solução de Lise = NaOH 0,2 M / SDS 1%, ver
quantidade abaixo, preparar na hora do uso), selar com adesivo novo e homogeneizar por
inversão 40 vezes:
Para 2 placas: NaOH 4M ................... 750 μL
H2O ............................ 10 ml
SDS 10% .................... 1,5 ml
H2O ............................ q.s.p 15 ml
-Incubar no gelo por 10 minutos
-Centrifugar 1 minuto a 4000 rpm para a solução soltar do adesivo antes de removê-lo
da placa
-Adicionar 70 μL da solução 3 gelada (Solução de Neutralização = KOAc 3 M), selar
com adesivo e homogeneizar por inversão 40 vezes

238
-Incubar no gelo por 10 minutos
-Centrifugar 20 minutos a 4000 rpm, 20 ° C. Enquanto isso, fixar com fita adesiva a
microplaca de filtro (Millipore) sobre a microplaca de fundo V já identificada, certificando-se
que os poços estão alinhados
-Transferir 120 μL do sobrenadante para microplaca de filtro (cuidado para não
transferir o precipitado)
-Centrifugar/Filtrar 6 min. a 4000 rpm
-Remover a microplaca-filtro. Precipitar os plasmídeos com a adição de 100 μL de
isopropanol (temperatura ambiente) a microplaca-fundo V, selar com adesivo novo e
homogeneizar por inversão 20 vezes
-Centrifugar 45 minutos a 4000 rpm, 20 ° C
-Descartar o sobrenadante e manter a placa invertida em papel filtro por 5 minutos.
-Lavar com adição de 200 µL de etanol 70% (temperatura ambiente). Selar com o
mesmo adesivo
-Centrifugar 15 minutos a 4000 rpm e descartar o sobrenadante
-Centrifugar 30 segundos a 600 rpm a microplaca invertida sobre papel toalha para a
remoção de traços de etanol
-Deixar a placa secar por 1 hora em temperatura ambiente (placa virada para cima,
tampada com papel toalha)
-Ressuspender o precipitado de plasmídeos em 30 μL de água Milli-Q e solubilizar
durante a noite
-O DNA deve ser estocado em ultrafreezer ou freezer convencional.
As amostras foram corridas em gel de agarose 1 % TBE 1 X para verificar a efetividade do
procedimento. A seguir, os meios e soluções utilizados:
Meio LB: NaCl ...................................................... 10 g
Bacto peptone (tryptone) ...................... 10 g
Bacto yeast extract ................................ 5 g
H2O ....................................................... 1000 ml

239
Autoclavar.
Ampicilina 100 mg/ml (estoque):
Ampicilina ............................................. 1 g
H2O Milli-Q autoclavada ...................... 1000 ml
Esterilizar por filtração (0,2 µm) e estocar a -20ºC – Não autoclavar.
Glicose 20%:
Glicose .................................................. 20 g
H2O Milli-Q autoclavada ...................... 1000 ml
Esterilizar por filtração (0,2 µm) e estocar a temperatura ambiente – Não autoclavar.
Solução EDTA – 0,5 M – pH 8,0:
EDTA 2 H2O ......................................... 186,1 g
H2O Milli-Q autoclavada ....................... 700 ml
Ajustar pH 8,0 com 10 M de NaOH
H2O Milli-Q autoclavada ....................... q.s.p 1000 ml
Estocar a 4ºC - Não autoclavar.
TRIS-HCl 1M pH 7,4:
TRIS base ................................................ 121 g
H2O Milli-Q autoclavada ........................ 800 ml
Ajustar pH 7,4 com HCl concentrado
H2O Milli-Q autoclavada ......................... q.s.p 1000 ml
Estocar a 4ºC – Não autoclavar.
Solução 1 (GET = Glicose/EDTA/TRIS):

240
Glicose 20% (filtrada) ................................. 46 ml
EDTA 0,5 M pH 8,0 ................................... 20 ml
TRIS-HCl 1M pH 7,4 ................................ 26 ml
H2O Milli-Q autoclavada ............................ q.s.p 1.000 ml
Estocar a 4ºC – Não autoclavar.
NaOH 4 M:
NaOH ......................................................... 16 g
H2O Milli-Q autoclavada ........................... 100 ml
Estocar a 4ºC – Não autoclavar.
SDS 10%:
SDS ............................................................. 10 g
H2O Milli-Q autoclavada ............................ 100 ml
Estocar a temperatura ambiente.
Solução 2 (NaOH 0,2 M / SDS 1%):
NaOH 4 M ................................................... 750 μL
H2O .............................................................. 10 ml
SDS 10% ...................................................... 1,5 ml
H2O .............................................................. q.s.p 15 ml
Preparar na hora do uso.
KOAc 5 M (estoque):
KOAc ............................................................ 46,87 g
H2O Milli-Q autoclavada .............................. 500 ml

241
Esterilizar por filtração (0,2 µm) e estocar a 4ºC.
Solução 3 (KOAc 3 M):
KOAc 5 M ..................................................... 60 ml
Ácido Acético Glacial ................................... 11,5 ml
H2O Milli-Q autoclavada ............................... 28,5 ml
Estocar a 4ºC
RNAse 10 mg/ml (estoque):
RNAse A ....................................................... 100 mg
H2O Milli-Q autoclavada ............................... 10 ml
Ressuspender em um tubo falcon de 50 ml, ferver por 15 minutos (a partir da fervura) em
banho-maria, esfriar a temperatura ambiente, aliquotar e estocar a -20 º C.
Etanol 70 %:
Etanol absoluto ............................................... 350 ml
H2O Milli-Q autoclavada ................................ 150 ml
Estocar a 4ºC.
7.1.3 Análise funcional da Biblioteca Metagenômica
7.1.3.1 Extração e purificação do DNA metagenômico com esferas de vidro
O DNA foi extraído de 5 g (aprox. 0,8 g de cada amostra) da mistura dos 6 tipos de solo
coletados através de lise mecânica, utilizando esferas de vidro (150-212 mícrons), aliado ao
tratamento com CTAB (brometo de hexadecilmetilamônio) e SDS (dodecil sulfato de sódio).
A extração de DNA do solo foi realizada conforme o seguinte protocolo:
-Adicionar 5 g de solo + 14 mL de Tampão A + 2 g de esferas de vidro

242
-Vortexar por 3 vezes por 90 s em intervalos de 10 minutos
-Colocar 1,5 mL de SDS 20 % + 100 μL de proteinase K
-Incubar 1 hora a 65 º C leve agitação a cada 15 minutos
-Centrifugar 15 minutos a 5000 rpm
-Remover o sobrenadante
-Precipitar com 1 volume de isopropanol e deixar 15 minutos em temperatura
ambiente
-Centrifugar 15 minutos a 12000 rpm e descartar o sobrenadante
-Ressuspender em 800 µL de TE (200 µL cada tubo, lavando o precipitado e passando
para outro tubo)
-Adicionar 1 mL de PEG 8000 13 % Na Cl 1.6 M
-Incubar 1 hora a temperatura ambiente
-Centrifugar 20 minutos a 12000 rpm e remover o sobrenadante
-Ressuspender em 400 µL de TE (200 µL cada tubo, lavando o precipitado e passando
para outro tubo)
-Adicionar acetato de potássio (KAc) na concentração final de 0,5 M (0,98 g em 10
mL de água). Incubar em gelo por 5 minutos
-Centrifugar 20 minutos a 12000 rpm
-Transferir o sobrenadante e extrair 4 vezes com fenol, com centrifugações de 2
minutos a 12000 rpm e transferir a fase superior para novo tubo
-Lavar 2 vezes com 1 volume de clorofórmio: álcool isoamílico e centrifugar 2
minutos
-Transferir a fase aquosa par tubo novo e precipitar com 1 volume de isopropanol
-Incubar 1 hora a temperatura ambiente
-Centrifugar 20 minutos a 12000 rpm
-Adicionar 1 volume de etanol 70 % e centrifugar 5 minutos a 12000 rpm
-Secar e ressuspender em 200 µL de água

243
A eficiência da extração foi analisada em gel de agarose a 1% em TBE 1X (p/v). Em seguida,
foi construído um gel para a excisão das bandas contendo os fragmentos de DNA com o
tamanho esperado. Os fragmentos foram purificados com o kit Ultra Clean DNA purification
(Mo Bio).
Tampão A: 100 mM Tris-HCl pH 8.0; 100 mM de fosfato de sódio pH 8.0; 100 mM sódio
EDTA; 1.5 M HCl; 1 % CTAB.
7.1.3.2 Preparação do vetor de clonagem pCF430
Após extração o plasmídeo foi linearizado com enzima de restrição EcoRI, seguindo a
seguinte reação:
Tabela 7: Reação de digestão do vetor pCF430.
A reação foi incubada a 37 º C durante 4 horas e posteriormente a 65 º C por 20 minutos.
A verificação da linearização do plasmídeo foi realizada através de gel de agarose 1 % TBE
1X. Após linearização, o plasmídeo foi tratado com fosfatase alcalina (SAP – Shrimp alcaline
phosphatase) para retirada dos resíduos de fosfato que permanecem na molécula de DNA
após a ação da endonuclease. A seguinte reação foi incubada a 37 ° C durante a noite.
Tabela 8: Tratamento do vetor pCF430com SAP.
Reagentes Volume (µL)
pCF430 70
Tampão10 x 10
BSA 10x 10
PstI 10
Total 100
Reagentes
Volume
(µL)
SAP (Shrimp Alkaline 5

244
A reação foi precipitada com acetato
de sódio e álcool e ressuspendido em 16 μL,
que foram utilizados para
montagem da reação de religação do plasmídeo. A reação foi incubada a 16 ° C durante a
noite.
Tabela 9 Reação de ligação do vetor pCF430.
Após os tratamentos, os vetores foram submetidos à eletroforese em gel de agarose Low
melting 1% TAE 1X (sem brometo de etídeo) durante 4 horas e baixa voltagem (aprox. 40 V).
A banda referente ao plasmídeo circularizado é sacada do gel e usando sistema comercial
Qiaex II (Qiagen). A verificação da obtenção do plasmídeo pCF430 tratado e pronto para ser
utilizado nos experimentos de clonagem foi realizada em gel de agarose 1% TBE 1X.
7.1.3.3 Digestão parcial do DNA metagenômico
A reação de digestão parcial do DNA metagenômico otimizada foi:
phosphatase)
Tampão 5x 5
Água 45
pCF430 95
Total 150
Reagentes
Volume
(µL)
pCF430 tratado 16
T4 DNA ligase (Invitrogen) 2
Tampão 10 x 2
Total 20

245
Tabela 10 Reação de digestão parcial do DNA metagenômico
pela enzima PstI.
A reação foi incubada a 37 ° C por 2 horas e posteriormente a 65 ° C por 20 minutos.
O resultado da digestão foi verificado em gel de agarose low melting 1,5% TAE 1X (sem
brometo de etídeo). A região do gel correspondente aos fragmentos de 4 – 10 Kb foi
purificada com Ultra Clean DNA Purification (Mo Bio).
7.1.3.4 Clonagem dos médios insertos
A reação de ligação entre os insertos de 4 – 10 Kb purificados com o plasmídeo pCF430
tratado foi feita da seguinte maneira:
Tabela 11 Reação de ligação entre o vetor pCF430 e os fragmentos de DNA metagenômico
Reagentes Volume (µL)
DNA (0,5 µg / µL) 4
BSA 10 x 1
Tampão H 1
PstI 1
Água 3
Total 10
Reagentes
Volume
(µL)
DNA (30 ng / µL) 21
pCF430 (100 ng /µL) 4
Tampão 10x 3

246
A reação foi incubada a 16 ° C durante a noite e posteriormente a 65 ° C por 20 minutos.
Após a inativação o sistema de ligação foi diluído 2 vezes e dialisado em membrana Type-VS
Millipore (0.025 µm) durante meia hora.
7.1.3.5 Reativação e pré - seleção de clones em meio CMC
Foram seguidas as seguintes etapas a partir da biblioteca metagenômica estocada a – 80 º C:
- Uma alíquota de 10 µL da cultura foi inoculada em 50 mL de LB +Tc (40 mg / mL) e
incubada a 37 º C durante 16 horas;
- Após crescimento, duas passagens foram realizadas em 50 ml de meio CMC + Tc (40
mg / mL)
- 100 µL da diluição 10 -2
foram plaqueados em placa de petri contendo meio CMC + Tc
(40 mg / mL).
- As colônias foram separadas e isoladas em placas de 96 poços de CMC+Tc (40 mg /
mL). e estocadas a – 80 º C após adição de 50 µL de glicerol 50 %.
Tabela 12 Composição do meio mínimo de sais com carboxi metil celulose (CMC).
Reagentes Peso (g)
Nitrato de sódio (NaNO3) 1,2
Fosfato monobásico de potássio (KH2PO4) 3
Fosfato dibásico de fosfato (K2HPO4) 6
Ligase 2
Total 30

247
Sulfato de magnésio (MgSO4.7H2O) 0,2
Cloreto de cálcio (CaCl2.2H2O) 0,05
Sulfato de zinco (ZnSO4.7H2O 0,001
Sulfato de manganês (MnSO4,7H2O) 0,01
Extrato de levedura 1
Carboxi metil celulose (CMC) 10
Ágar 10
7.1.3.5.1
7.1.3.6 Atividade FPásica (celulose total)
Pedaços de folha de papel de filtro Watmann nº 1 (1x6 cm) foram utilizados como substrato.
Os experimentos foram realizados em duplicata e duas reações controle e um branco foram
utilizados nos ensaios, preparados da seguinte maneira:
Amostra: Papel + 1 mL de tampão + 500 µL de amostra;
Controle 1 (enzima): 500 µL de amostra (enzima) + 1 mL de tampão;
Controle 2 (substrato): Papel + 1,5 mL de tampão;
Branco: 1,5 mL de tampão.
- Incubar a 50 º C por 1hora
- Transferir imediatamente para banho fervente (100 º C) e incubar por 10 min.
- Adicionar 3 mL de reagente DNS (ácido dinitrosalicílico) e incubarem banho
fervente por 5 minutos.
- Colocar 20 mL de água destilada em todos os tubos.
- A leitura das amostras deve ser feita no comprimento de onda 540nm.

248
7.1.3.7 Atividade CMCásica (endoglucanásica)
O substrato utilizado foi solução CMC 2 %. Os experimentos foram realizados em duplicata e
duas reações controles e um branco foram utilizados nos ensaios e preparados da seguinte
maneira:
Amostra: 50µL de CMC2% e 50µL de amostra
Controle 1 (enzima): 50 µL de amostra + 50 µL de tampão
Controle 2 (substrato): 50µL de CMC 2 % e 50 µL de tampão
Branco – 100µL de tampão
- Incubar a 50ºC por 15min.
- Transferir imediatamente para banho fervente (100ºC) e incubar por 10min.
- Adicionar 300 µL de reagente DNS Voltar para o banho fervente por 5min.
- Adicionar 1,5 ml de água destilada
- A leitura das amostras deve ser feita no comprimento de onda 540nm.
7.1.3.8 Atividade β-glicosidásica
O substrato utilizado foi solução de celobiose 2 %. Os experimentos foram realizados em
duplicata e duas reações controles e um branco foram utilizados nos ensaios e preparados da
seguinte maneira:
Amostra: 50µL de celobiose 2 % + 50 µL de amostra.
Controle 1 (enzima): 50 µL de amostra + 50 µL de tampão
Controle 2 (substrato): 50 µL de celobiose 2 % e 50 µL de tampão.
Padrão: Solução padrão de glicose
- Incubar a 50ºC por 15min
- Transferir imediatamente para o banho fervente (100ºC) e encubar por 10min.
- Adicionar 1mL de solução GOD (glicose oxidase)
- Levar tudo para o banho a 37ºC, onde fica por 15min.

249

250
7.2 Anexo 2. Lista de publicações
Trabalhos produzidos durante o doutorado.
7.2.1 Periódicos
7.2.1.1 Artigo 1
de Castro AP, Araújo SD Jr, Reis AM, Moura RL, Francini-Filho RB, Pappas G Jr,
Rodrigues TB, Thompson FL, Krüger RH. Bacterial community associated with healthy
and diseased reef coral Mussismilia hispida from eastern Brazil. Microb Ecol. 2010
May;59(4):658-67.
7.2.1.2 Artigo 2
Bruce T, Martinez IB, Maia Neto O, Vicente AC, Kruger RH, Thompson FL. Bacterial
community diversity in the Brazilian Atlantic forest soils. Microb Ecol. 2010
Nov;60(4):840-9.
7.2.1.3 Artigo 3
Thompson FL, Bruce T, Gonzalez A, Cardoso A, Clementino M, Costagliola M, Hozbor
C, Otero E, Piccini C, Peressutti S, Schmieder R, Edwards R, Smith M, Takiyama LR,
Vieira R, Paranhos R, Artigas LF. Coastal bacterioplankton community diversity along a
latitudinal gradient in Latin America by means of V6 tag pyrosequencing. Arch
Microbiol. 2011 Feb;193(2):105-14.
7.2.1.4 Artigo 4
Bruce T, Meirelles PM, Garcia G, Paranhos R, Rezende CE, de Moura RL, Francini-Filho
R, Vasconcelos AT, Amado-Filho G, Hatay M, Edwards R, Schmieder R, Dinsdale L and
Thompson FL. Abrolhos reef Bank health evaluated by means of water quality, microbial
diversity, benthic cover, and fish biomass data. Plos One. Aceito para publicação.
7.2.2 Capítulo de livro
Bruce T, Castro A, Thompson CC Kruger RH, Thompson FL. Microbial diversity of
Brazilian biomes. Genomics Applications and the Developing World. Springer. J. Craig
Venter Institute. Em fase de edição.


















