
EM CSL immunews7-09 070709:EM ZLB CSL immunews7-09LR.pdf · Polimiosite e dermatomiosite...
40
A B S T R A C T C O L L E C T I O N immu news 7 2009
Transcript of EM CSL immunews7-09 070709:EM ZLB CSL immunews7-09LR.pdf · Polimiosite e dermatomiosite...

A B S T R A C T C O L L E C T I O N
immunews72009
EM CSL immunews7-09 cop:EM ZLB 3-07-2009 9:47 Pagina I

EM CSL immunews7-09 cop:EM ZLB 3-07-2009 9:47 Pagina II

introduzione 3
immunologia 5Timoma e timectomia
Miastenia gravis e ghiandola timica: revisione storica 7Raica M, Cimpean AM, Ribatti D
Immunoglobuline endovena per una pseudo-ostruzione associata a timoma: 7resoconto di un trattamento con esito positivoGreenburg DL, Mo CC, Hemmer PA
Demenza in un paziente con timoma e ipogammaglobulinemia 8(sindrome di Good)de Jesus NP, Carvalho PM, Dias FM, Gaspar EM, de Moura JJ
Mielodisplasia e sindrome di Good: resoconto di un caso clinico 9Di Renzo M, Pasqui AL, Voltolini L, Gotti G, Pompella G, Auteri A
ematologia 10Porpora trombocitopenica idiopatica
Terapie di prima linea per la porpora trombocitopenica idiopatica: 12 rivalutazione della necessità di trattareRodeghiero F
Meccanismi d’azione delle immunoglobuline endovena e delle 13 immunoglobuline policlonali anti-D nel miglioramento della porpora trombocitopenica idiopatica: che cosa sappiamo veramente?Crow AR, Lazarus AH
Immunoglobuline endovena vs immunoglobuline anti-D endovena 15per il trattamento della porpora trombocitopenica idiopatica acutaShahgholi E, Vosough P, Sotoudeh K, Arjomandi K, Ansari S, Salehi S, Faranoush M, Ehsani MA
La coniugazione del methotrexate con le immunoglobuline induce 16l’apoptosi dei macrofagi attraverso un uptake mediato dal recettore Fc?Wang X, Yao C, Jiang Z
neurologia 17Poliradiculoneuropatia demielinizzante infiammatoria cronicaTrattamenti attuali delle polineuropatie demielinizzanti croniche 19immunomediateBrannagan TH 3rd
Anno III - N.7 - 2009Quadrimestrale di
aggiornamento scientificoReg. Trib. N. 642del 18.10.2007ISSN 1974-4641
Direttore responsabileWubbo Tempel
EditoreElsevier srl
Via Paleocapa, 7 20121 Milano (MI)
Coordinamentoeditoriale
Gloriana Granata
RedazioneIn-folio - Torino
GraficaStudio Sismondo - Roma
StampaGrafiche Ortolan
Opera (MI)
Iniziativa resa possibile grazie ad un contributo
educazionale diCSL Behring
Edizione riservataper i Sigg. Medici
Fuori commercio
© 2009, Elsevier srl - Tutti i diritti riservati.È vietato riprodurre, archiviare in unsistema di riproduzione o trasmettere sottoqualsiasi forma o con qualsiasi mezzo,elettronico, meccanico, per foto copia,registrazione o altro, qualsiasi parte diquesta pubblicazione senza auto ri z zazionescritta dell’Editore. L’Editore non si assumealcuna responsabilità per qual siasi lesionee/o danno a persona o beni in quantoresponsabilità di prodotto, ne gligenza oaltrimenti, oppure a opera zione di qualsiasimetodo, prodotto, istruzione o ideacontenuti nel materiale di cui trattasi. Acausa del rapido progresso nella scienzamedica, l’Editore raccomanda la verificaindipendente delle diagnosi e del dosag -gio dei medicinali.
A B S T R A C T C O L L E C T I O N
72009
EM CSL immunews7-09 130709:EM ZLB 13-07-2009 16:23 Pagina 1

Immunoglobuline endovena per la poliradiculoneuropatia 20demielinizzante infiammatoria cronicaEftimov F, Winer JB, Vermeulen M, de Haan R, van Schaik IN
Risposta motoria acuta dopo un singolo ciclo di trattamento con 21immunoglobuline endovena nella poliradiculoneuropatia demielinizzante infiammatoria cronicaHarbo T, Andersen H, Jakobsen J
Ridotta espressione del recettore inibitorio FcγRIIB sui linfociti B 23nella polineuropatia demielinizzante infiammatoria cronicaTackenberg B, Jelcic I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, Lünemann JD
aggiornamenti 27Polimiosite e dermatomiositeImmunoglobuline endovena in combinazione con mofetil micofenolato 27nel trattamento della miosite severa
Sindrome di Guillain-BarréSindrome di Guillain-Barré: aggiornamento 27
Sindrome di KawasakiValutazione della risposta al trattamento con 1 g/kg di immunoglobuline 27 endovena della sindrome di Kawasaki acuta: metodo efficace e in grado di diminuire i costi
Sindrome da anticorpi antifosfolipidiSindrome da anticorpi antifosfolipidi “catastrofica”: analisi descrittiva 27di una serie di 280 pazienti dal “CAPS Registry”
Miastenia gravisMiastenia gravis giovanile 27
Pemfigo e pemfigoideStudio clinico randomizzato in doppio cieco sull’uso delle 28immunoglobuline endovena nel trattamento del pemfigoStudio clinico crossover controllato con placebo “n-of-1” sulle immunoglobuline endovena in adiuvante nel trattamento del pemfigo volgare refrattario
Encefalomielite acuta disseminataEncefalite postinfettiva negli adulti: diagnosi e trattamento 28
Leucemia linfatica cronicaRiconoscimento e trattamento della porpora trombocitopenica 28immune secondaria associata a malattie linfoproliferative
Neuropatia motoria multifocaleIncremento progressivo della dose di immunoglobuline endovena 29nella neuropatia motoria multifocale: follow-up a sei mesi
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:44 Pagina 2

3
Questo numero di Immunews riprende il discorso su due patologie giàtrattate, la porpora trombocitopenica idiopatica e la poliradiculoneuro-patia demielinizzante infiammatoria cronica, e ne introduce una nuova,il timoma.La porpora trombocitopenica idiopatica è una malattia ben conosciu-ta da lungo tempo, la cui terapia si è classicamente basata sull’uso di far-maci corticosteroidei e sulla splenectomia, trattamenti però entrambigravati da pesanti effetti collaterali, tali da giustificare la ricerca diapprocci terapeutici alternativi. L’articolo qui presentato si concentradunque sulle più recenti acquisizioni sui meccanismi fisiopatologici allabase della malattia e sulle implicazioni terapeutiche di queste scoperte.Vengono in particolare analizzati i meccanismi d’azione delle immuno-globuline endovena (IVIG) e delle immunoglobuline anti-D, apparente-mente simili ma probabilmente significativamente differenti, così comedifferenti possono essere gli effetti terapeutici delle due preparazioni.Il meccanismo d’azione delle IVIG e la recente individuazione di nuovipotenziali target, come il recettore inibitorio FcγRIIB presente sullasuperficie dei linfociti B, vengono trattati anche nella sezione dedicataalla poliradiculoneuropatia demielinizzante infiammatoria cronica,che tuttavia è focalizzata piuttosto sulla gestione clinica di questipazienti. Il caratteristico andamento cronico o recidivante della malattiaimpone infatti un trattamento a lungo termine, basato generalmente sucicli periodici di IVIG. Le modalità più efficaci di monitoraggio e modula-zione della malattia, evitando sia un inutile e costoso sovradosaggio siail peggioramento delle lesioni nervose, fino a renderle irreversibili, costi-tuiscono un problema clinico rilevante e affrontato nelle pubblicazionipiù recenti qui commentate.Infine, il ruolo delle IVIG nel trattamento del timoma, malattia spessoassociata a patologie autoimmuni, fra cui in particolare la miastenia gra-vis, costituisce l’argomento della terza sezione. L’infusione di IVIG svolgeun ruolo terapeutico definito in alcune di queste sindromi paraneopla-stiche ed è particolarmente indicata nei pazienti che vanno incontro arimozione chirurgica della ghiandola (timectomia). Il quadro complessivo che emerge dagli articoli presentati in questauscita di Immunews per quanto riguarda l’utilizzo terapeutico dell’infu-
introduzione
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:45 Pagina 3

sione di IVIG in diverse patologie è ancora una volta quello di uno stru-mento polivalente, la cui stessa indeterminatezza, a tutt’oggi presente,circa i probabilmente multipli meccanismi d’azione ne permette un usoesteso e suscettibile di ulteriori significativi sviluppi.
4
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:46 Pagina 4

5
Timoma e timectomia
Il timoma è un tumore derivato dalle cellule epiteliali del
timo, piuttosto raro, pur costituendo la più frequente neo-
plasia del mediastino anteriore negli adulti. In molti casi la
malattia può essere asintomatica e può essere scoperta
casualmente nel corso di esami radiologici del torace,
oppure può provocare una sintomatologia costituita preva-
lentemente da disfagia, dispnea, tosse, febbre e dolore tora-
cico. La terapia del timoma è essenzialmente chirurgica e
consiste nell’asportazione della ghiandola per via transcer-
vicale o trans-sternale oppure, più recentemente, per via
endoscopica videoguidata. In molti casi la prognosi del
timoma isolato è buona, trattandosi istologicamente di un
tumore non maligno, ma alcune forme possono manifesta-
re un andamento invasivo nei confronti delle strutture cir-
costanti.
Caratteristica del timoma, probabilmente in relazione alla
funzione immunitaria della ghiandola timica, è la frequente
comparsa di una sindrome paraneoplastica, che può com-
prendere patologie come l’artrite reumatoide, l’anemia per-
niciosa, la polimiosite e la tiroidite. Tuttavia, l’associazione
più comune e conosciuta è quella con la miastenia gravis
(MG), presente nel 10% circa dei pazienti con timoma
(mentre quest’ultimo colpisce circa il 20% dei miastenici),
descritta per la prima volta già nel 1901 (Raica). Si ritiene
che il tessuto timico possa partecipare alla genesi della MG,
poiché contiene tutti gli elementi necessari per lo sviluppo
della patologia: cellule mioidi che esprimono il recettore
per l’acetilcolina, target degli autoanticorpi circolanti, cellu-
le presentanti l’antigene professionali, linfociti T e B. I
pazienti con MG, specie se di età inferiore ai 40 anni e con
malattia di nuova insorgenza, vengono spesso sottoposti a
timectomia, indipendentemente dalla presenza di timoma,
ottenendone una remissione della sintomatologia in circa il
25% dei casi e un miglioramento nel 50% dei casi.
L’indicazione all’intervento è ovviamente assoluta nei casi
con timoma, anche se questi ultimi sono quelli che rispon-
dono meno all’asportazione della ghiandola.
L’intervento di timectomia presenta caratteristiche particola-
ri nei pazienti con MG, dovute al rischio di insufficienza
respiratoria e necessità di ventilazione artificiale prolungata
nel periodo postoperatorio. Per questa ragione, viene spes-
so preceduto da un trattamento con plasma exchange, allo
scopo di migliorare le condizioni cliniche generali e ridurre
il tasso di autoanticorpi circolanti. Una possibilità alternati-
va è l’uso delle immunoglobuline endovena (IVIG). Queste
ultime sono largamente utilizzate nel trattamento della MG,
in particolare nelle esacerbazioni acute, dove si sono dimo-
strate in grado di migliorare significativamente la sintoma-
tologia muscolare. L’infusione di IVIG pretimectomia
dovrebbe precedere l’intervento di non più di 10 giorni,
così da sfruttare il massimo beneficio clinico delle immuno-
globuline, che è stato registrato fra 3 e 10 giorni dalla prima
somministrazione. Un recente confronto diretto della tera-
pia preoperatoria con IVIG rispetto al plasma exchange ha
dimostrato un’efficacia clinica comparabile, a fronte di una
maggiore fattibilità della procedura e di minori effetti colla-
terali osservati con le IVIG.
Come già accennato, pur essendo la più frequente la MG
non è l’unica patologia che può presentarsi in associazione
con un timoma. Le malattie descritte sono molteplici e com-
prendono anche patologie come l’aplasia eritroblastica o la
colite ulcerosa. Una forma particolare di sindrome paraneo-
plastica è quella descritta da Greenburg et al. in un paziente
con timoma e una grave forma di pseudo-ostruzione intesti-
nale, con immobilità gastrica e impossibilità all’alimentazione
enterale. Sebbene il paziente, sottoposto a numerosi esami,
non presentasse nessun tipo di autoanticorpi circolanti, in
particolare anticorpi antirecettore dell’acetilcolina, la sinto-
matologia digestiva, resistente all’intervento di timectomia e
alla somministrazione di corticosteroidi ad alte dosi, si è risol-
immunologia
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:47 Pagina 5

ta con l’infusione di IVIG, confermandone l’efficacia nelle
patologie paraneoplastiche associate a timoma.
Una forma particolare di patologia associata a timoma è la sin-
drome di Good, caratterizzata dalla coesistenza di timoma e
immunodeficienza, con ipogammaglobulinemia, riduzione
dei linfociti B e T e inversione del rapporto CD4/CD8. La malat-
tia si manifesta generalmente in soggetti adulti e presenta
una prognosi più sfavorevole rispetto al timoma isolato, poi-
ché spesso l’immunodeficienza non risponde alla timectomia
e predispone a severe infezioni opportunistiche, aggravate
dal deficit dell’immunità cellulare (de Jesus et al.). Come in altre
forme di ipogammaglobulinemia, nella sindrome di Good un
miglioramento delle condizioni cliniche e una riduzione delle
complicanze infettive possono essere ottenuti con l’infusione
regolare di IVIG. Poiché la condizione di immunodeficienza
generalmente non si risolve con l’asportazione della ghiando-
la timica, la terapia con IVIG va attuata sia prima sia dopo l’in-
tervento, come misura terapeutica a lungo termine.
Nel caso descritto da Di Renzo et al., un paziente di 62
anni con sindrome di Good, dopo l’intervento di timecto-
mia, ha ricevuto regolari infusioni mensili di IVIG, otte-
nendo una stabilizzazione delle condizioni cliniche per
più di 7 anni. Complessivamente, il timoma e le patologie
associate costituiscono un gruppo piuttosto eterogeneo
di condizioni, tutte accomunate da disordini del sistema
immunitario, spesso di tipo autoimmune, dovute proba-
bilmente al ruolo cruciale giocato dalla ghiandola nella
regolazione delle risposte immuni. Per molte di queste
condizioni (come la MG e la sindrome di Good) l’infusio-
ne di IVIG rappresenta un’efficace opzione terapeutica,
così come per la preparazione di alcune categorie di
pazienti per l’intervento di timectomia. Sebbene il mec-
canismo d’azione delle IVIG in queste patologie non sia
del tutto conosciuto, è probabile che esso coinvolga non
solo la riduzione dei livelli circolanti di autoanticorpi,
quando presenti, ma anche una regolazione più genera-
le di diversi aspetti della risposta immunitaria, inclusa la
componente cellulare.
6
immunologiaTimoma e timectomia
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:48 Pagina 6

7
immunologiaTimoma e timectomia
Miastenia gravis e ghiandola timica: revisione storica
Raica M, Cimpean AM, Ribatti DClin Exp Med 2008;8(2):61-4
Immunoglobuline endovena per una pseudo-ostruzione associata a timoma: resoconto di un trattamento con esito positivo
Greenburg DL, Mo CC, Hemmer PAEur Neurol 2007;58(2):116-7
Il primo rapporto di un’associazione frala miastenia gravis (MG) e la ghiandolatimica risale al 1901. Nonostante i mec-canismi sottostanti siano ancora poco
conosciuti, le anomalie del timo sonochiaramente associate con la MG. Que -sto articolo di revisione riassume, dalpunto di vista storico, le conoscenze su
questa associazione e il trattamentochirurgico e medico della MG, incluse lagestione dei sintomi, l’immunosop-pressione, le IVIG e il plasma exchange.
• Un timoma è presente nel 10-20% dei pazienti con MG, mentre circa il 70% presentaiperplasia follicolare della ghiandola. L’associazione fra timoma e MG e i possibili tratta-menti medici e chirurgici di queste condizioni costituiscono il fulcro di questa revisione.
• La timectomia viene spesso eseguita nei pazienti con MG, sulla base di una presuntapartecipazione della ghiandola timica alla patogenesi della malattia, ottenendo unabuona percentuale di remissioni.
• Le IVIG rappresentano una delle possibili opzioni terapeutiche nella MG, insieme ai far-maci corticosteroidei e immunosoppressori. Il loro ruolo è particolarmente rilevante neipazienti che vanno incontro a timectomia, i quali richiedono un trattamento preventi-vo con IVIG ad alte dosi.
• L’articolo riporta il caso di un paziente di 37 anni con timoma che presentava sintoma-tologia paraneoplastica associata a una grave forma di pseudo-ostruzione intestinale,con immobilità gastroenterica e impossibilità all’alimentazione enterale.
• I sintomi presentati persistevano anche dopo l’intervento chirurgico di timectomia,nonostante una terapia con piridostigmina e cortisonici ad alte dosi.
• Una risoluzione pressoché completa della sintomatologia e la possibilità di riprendereuna dieta normale hanno invece fatto seguito a un trattamento con IVIG, al dosaggio di2 mg/kg per un periodo di 5 giorni, confermando l’efficacia di questa terapia delle sin-dromi paraneoplastiche associate a timoma e suggerendo la presenza di autoanticorpicircolanti come responsabili della pseudo-ostruzione.
Abstract non disponibile.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:49 Pagina 7

8
immunologiaDemenza in un paziente con timoma e ipogammaglobulinemia (sindrome di Good)
de Jesus NP, Carvalho PM, Dias FM,Gaspar EM, de Moura JJCases J 200813;1(1):90
• L’articolo riporta un caso clinico di timoma associato a immunodeficienza (sindrome diGood), la cui manifestazione paraneoplastica, inusuale, era rappresentata da demenzasecondaria.
• Queste forme di timoma hannouna prognosi favorevole doporimozione chirurgica del tumo-re, tuttavia l’ipogammaglobuli-nemia associata generalmentepersiste dopo la timectomia,come in questo paziente.
• La terapia con IVIG, indicata per iltrattamento dell’ipogammaglo-bulinemia, rappresenta quin diuna misura fondamentale neipazienti con sindrome di Good,sia prima sia dopo l’intervento ditimectomia, per ridurre l’inciden-za di infezioni opportuni stiche.
Timoma e timectomia
La sindrome di Good è estremamenterara e consiste in un’immunodeficien-za acquisita dei linfociti B e T neipazienti con timoma. Gli autori di que-sto articolo presentano il caso clinicodi un paziente di 75 anni, caucasico,
precedentemente sottoposto adaccertamenti per demenza secondariae infezioni ricorrenti, in cui è stataidentificata una sindrome paraneopla-stica derivata da un timoma. Il pazien-te è stato sottoposto a timectomia,
mentre la sindrome da immunodefi-cienza è continuata con frequenti infe-zioni opportunistiche, che hannorichiesto un trattamento costante conIVIG.
1
2
Distanza 1: 6,07 nmDistanza 2: 4,17 nm
Figura. Immagine TAC del timoma nel mediastinoanteriore.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:50 Pagina 8

9
Mielodisplasia e sindrome di Good: resoconto di un caso clinico
Di Renzo M, Pasqui AL, Voltolini L, Gotti G, Pompella G, Auteri AClin Exp Med 2008;8(3):171-3
immunologiaTimoma e timectomia
• Il caso clinico riportato in questo articolo riguarda un uomo di 62 anni, sottoposto atimectomia a seguito di diagnosi di sindrome di Good, la cui ipogammaglobulinemia èstata trattata con infusioni mensili di IVIG.
• L’interesse del caso risiede non solo nel notevole miglioramento clinico ottenuto conl’infusione di IVIG, con una notevole riduzione delle infezioni opportunistiche, maanche nel ruolo che le IVIG potrebbero aver giocato nel prosieguo della storia clinicadel paziente. Questi ha infatti sviluppato un’anemia refrattaria, trattata con ciclospori-na fino a risoluzione dei sintomi. Nonostante il trattamento immunosoppressivo, nonsono state osservate infezioni opportunistiche.
• L’immunodeficienza presente nei pazienti con sindrome di Good non è legata solo allariduzione dei livelli di gammaglobuline circolanti, ma anche a un’alterazione dell’immu-nità cellulare, con inversione del rapporto CD4/CD8 e riduzione delle cellule CD4+. Èpossibile che gli effetti benefici della terapia con IVIG in questa patologia siano legatianche ai già descritti effetti di questo trattamento sulle cellule linfocitarie.
La sindrome di Good è una rara immu-nodeficienza a insorgenza nell’adultocaratterizzata da ipogammaglobuline-mia e timoma. Descriviamo qui unpaziente di 72 anni a cui era stata dia-gnosticata la sindrome di Good quan-do ne aveva 62, dopo una storia di 2anni di infezioni respiratorie ricorrenti.
Un esame TAC del torace mostravauna massa mediastinica che è statarimossa chirurgicamente; l’esame isto-logico ha rivelato un timoma. Il pa -ziente era ipogammaglobulinemico ele sue condizioni cliniche sono miglio-rate vistosamente dopo l’inizio di undosaggio appropriato di IVIG. Due anni
fa ha sviluppato un’anemia normocro-mica normocitica che ha richiestodiverse trasfusioni. Una biopsia osseaha rivelato una sindrome mielodispla-stica. Il paziente ha iniziato la ciclospo-rina e l’anemia è gradualmente miglio-rata, fino a raggiungere l’indipendenzadalle trasfusioni.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:51 Pagina 9

10
ematologiaPorpora trombocitopenica idiopatica
La porpora trombocitopenica idiopatica (PTI) rientra nel
novero delle malattie autoimmuni ed è una patologia
caratterizzata dalla produzione di autoanticorpi antipiastri-
ne, prevalentemente diretti verso le glicoproteine IIb/IIIa,
che provocano la distruzione delle piastrine circolanti e
una trombocitopenia pura, non accompagnata da anemia
o leucopenia. Il quadro clinico della malattia è caratterizza-
to da emorragie cutanee e mucose (porpora, gengivorra-
gia, epistassi), mentre le emorragie gastrointestinali e geni-
tourinarie sono poco frequenti e quelle intracraniche sono
rare. Specie negli adulti, la PTI ha decorso cronico, con il
subdolo instaurarsi della trombocitopenia e la tendenza a
frequenti recidive. Classicamente, la gravità del quadro
viene valutata sulla base del numero di piastrine circolanti,
che spesso sono <50.000/mm3; il valore considerato peri-
coloso, in quanto espone il paziente al rischio di sanguina-
menti gravi e potenzialmente letali, è <20.000/mm3.
In realtà, come detto, le emorragie pericolose per la vita, in
particolare quelle intracraniche (prima causa di morte in
questi pazienti), sono piuttosto rare nei soggetti con PTI,
che normalmente vanno incontro a sanguinamenti muco-
cutanei di lieve entità, anche se spesso al prezzo di una
significativa riduzione delle attività possibili cause di trau-
mi ed emorragie. La presenza o meno di una sintomatolo-
gia emorragica e il tipo di quest’ultima dovrebbero quindi
essere il criterio da seguire nella scelta di trattare o no il
paziente con PTI e con quale terapia (Rodeghiero).
Accanto alle classiche opzioni terapeutiche, costituite
essenzialmente da splenectomia e prednisone per via
orale, l’infusione di immunoglobuline endovena (IVIG) e di
immunoglobuline policlonali anti-D si è affermata nelle
ultime decadi come terapia di prima linea in questi pazien-
ti. Sono tutte opzioni spesso efficaci nel breve periodo, ma
che in molti casi, non eliminando la causa della malattia,
non sono curative e pongono il problema del trattamento
a lungo termine. In particolare la terapia corticosteroidea
comporta effetti collaterali, sul lungo periodo, non indiffe-
renti e che rendono necessario in alcuni casi il ricorso alla
splenectomia. La mancanza di cure risolutive della PTI
rende ragione della sperimentazione, attualmente in
corso, di trattamenti alternativi, compresi regimi con desa-
metasone ad alte dosi, l’anticorpo monoclonale anti-CD20
rituximab, le infusioni intermittenti di anti-D e gli analoghi
o gli agonisti della trombopoietina. Nonostante alcuni
risultati promettenti, in particolare per quanto riguarda il
desametasone, questi trattamenti mancano di prove di
efficacia definitive e devono essere considerati ancora in
corso di studio.
L’infusione di IVIG è stata introdotta nel trattamento della
PTI da quasi tre decadi: le IVIG consentono di ottenere un
rapido aumento del numero di piastrine circolanti nella
maggior parte dei pazienti e comportano generalmente
effetti collaterali lievi, consistenti in cefalea, febbre o rash
cutanei, mentre le reazioni avverse gravi sono piuttosto
rare. L’effetto acuto e la rapida normalizzazione della conta
piastrinica le rendono particolarmente indicate per il trat-
tamento di situazioni di emergenza e per la profilassi prima
di manovre o interventi medici.
Nonostante la loro efficacia, il meccanismo d’azione delle
IVIG in questa patologia, come del resto in molte altre, non
è del tutto noto. Nuove conoscenze, derivate anche da
modelli animali, tendono attualmente a mettere in discus-
sione la teoria classica del blocco del recettore Fc sulla
superficie delle cellule fagocitarie mononucleate, respon-
sabili della distruzione delle piastrine, quanto meno come
unico meccanismo d’azione. Evidenze sperimentali in vivo
e in vitro suggeriscono un ruolo anche per gli anticorpi
anti-idiotipo, la modulazione della liberazione di citochine
e dell’attivazione delle cellule del sistema immunitario e
l’interazione con il complemento. Nuovi e più complessi
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:53 Pagina 10

11
ematologiaPorpora trombocitopenica idiopatica
modelli, accanto alla visione forse semplicistica di un bloc-
co competitivo del recettore Fc, coinvolgono vari effettori
della risposta immune e vari tipi di recettori. Uno di questi
(Crow e Lazarus) ipotizza l’intervento di cellule dendritiche
regolatorie che, attivate dalle IVIG e dai complessi immuni
circolanti, agiscono direttamente o indirettamente iniben-
do l’azione fagocitaria dei macrofagi e riducendo così la
distruzione delle piastrine sensibilizzate. Ci si aspetta natu-
ralmente che l’approfondirsi delle conoscenze in questo
campo possa portare al disegno di protocolli di trattamen-
to ancora più efficaci, eventualmente ricorrendo a frazioni
della preparazione di IVIG, individuate come responsabili
dell’azione terapeutica, o a strategie in grado di ridurne la
quantità necessaria e quindi i costi (come la sensibilizzazio-
ne di cellule dendritiche in vitro e l’infusione di queste ulti-
me) (Crow e Lazarus).
È interessante notare come gli stessi studi stiano rivelando
meccanismi d’azione diversi e indipendenti alla base del-
l’efficacia di un altro trattamento utilizzato nella terapia
della PTI, le immunoglobuline policlonali anti-D, una frazio-
ne di immunoglobuline arricchita di specificità diretta
verso l’antigene RhD, efficace nei pazienti con PTI D+ e
non splenectomizzati. Sebbene anche in questo caso sia
stato ipotizzato il blocco competitivo del sistema fagociti-
co mononucleato da parte dei globuli rossi ricoperti di
anticorpi anti-D, sembrano emergere differenze significati-
ve rispetto all’azione delle IVIG. I due tipi di trattamento uti-
lizzerebbero recettori Fc di tipo diverso e non andrebbero
incontro a fenomeni di cross-resistenza, permettendo il
trattamento con IVIG nei pazienti refrattari alle anti-D e
viceversa.
Dal punto di vista clinico, un confronto diretto fra IVIG e
anti-D è stato effettuato in una popolazione di bambini
affetti da PTI da Shahgholi et al. Le forme pediatriche di PTI
hanno spesso un andamento diverso da quelle degli adul-
ti, caratterizzato da esordio acuto, spesso a seguito di un’in-
fezione virale, e andamento benigno. D’altra parte, i farma-
ci corticosteroidei presentano rilevanti effetti collaterali nei
bambini, quindi le infusioni di IVIG o di immunoglobuline
policlonali anti-D costituiscono una valida alternativa tera-
peutica. Un trattamento con 2 g/kg di IVIG suddivisi in due
dosi in giorni successivi ha permesso di ottenere un
aumento della conta piastrinica più rapido e più significa-
tivo rispetto alla terapia con anti-D, ma naturalmente altri
studi saranno necessari per confermare questo dato.
Inoltre, la maggiore facilità di somministrazione delle anti-
D e i costi minori andranno anch’essi presi in considerazio-
ne nella valutazione finale dei due trattamenti.
Le limitazioni ancora esistenti nella terapia della PTI, prece-
dentemente illustrate, stimolano la ricerca di nuove moda-
lità di trattamento. L’efficacia dimostrata dalle IVIG e i loro
scarsi effetti collaterali suggeriscono la possibilità di sfrut-
tarne il meccanismo d’azione potenziandone gli effetti con
modalità e manipolazioni innovative. Una possibilità inte-
ressante, sperimentata in vitro, è quella di coniugare le
immunoglobuline con il methotrexate, un antifolico larga-
mente usato nella terapia dei tumori e delle malattie
autoimmuni, compresa la stessa PTI (Wang et al.). Le mag-
giori limitazioni di questo farmaco risiedono nei rilevanti
effetti collaterali. Il coniugato IVIG-MTX si è dimostrato in
grado di legare specificamente i macrofagi attraverso il
recettore Fc e di provocarne la citotossicità selettiva, fon-
dendo così i vantaggi dei due trattamenti: la potenza di
azione e la specificità d’azione sul tipo cellulare responsa-
bile della distruzione piastrinica, con effetti dannosi poten-
zialmente minori sull’organismo.
Questi dati sperimentali dimostrano come nuovi orizzonti
siano ancora aperti nella terapia della PTI e come una
forma di trattamento quale l’infusione di IVIG, ormai consi-
derata un classico nella terapia di questa malattia, possa
andare incontro a ulteriori evoluzioni e migliore sfrutta-
mento di tutte le sue molteplici potenzialità.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:54 Pagina 11

12
ematologiaPorpora trombocitopenica idiopatica
Terapie di prima linea per la porporatrombocitopenica idiopatica:rivalutazione della necessità di trattare
Rodeghiero FEur J Haematol Suppl 2008;(69):19-26
La PTI può essere una sfida sia nelladiagnosi sia nel trattamento: nono-stante la possibilità di individuare glianticorpi antipiastrine, la diagnosi diPTI rimane una diagnosi di esclusio-ne. Il trattamento della PTI è ugual-mente difficile poiché molte terapiecomportano rischi potenzialmentepeggiori della malattia stessa. È statogeneralmente accettato il principioche la presenza di sanguinamento – enon il numero delle piastrine –dovrebbe costituire il razionale per iltrattamento. Nonostante l’assenza distudi prospettici controllati, vi è con-senso generale sul fatto che i rischi disanguinamento sono significativa-mente maggiori nei pazienti con unaconta piastrinica <20-30 × 109/l e chequindi la terapia sia indicata in questipazienti; nei casi di conta piastrinicamaggiore, ma ancora <50 × 109/l, iltrattamento è anche indicato in pre-senza di sanguinamento significativodelle membrane mucose. La terapia
iniziale standard per la PTI è costituitada corticosteroidi orali per aumentarela conta piastrinica. Anche le IVIG o leimmunoglobuline policlonali anti-Dpossono incrementare la conta pia-strinica e sono particolarmente utiliper indurre un rapido au mento primadi interventi pianificati. La splenecto-mia, che produce una risposta dilunga durata nella maggioranza deipazienti, è ancora comunementeusata per quei casi che non presenta-no una risposta durevole alla terapiasteroidea e dovrebbe rimanere il trat-tamento di riferimento. Tuttavia, lasplenectomia è una procedura invasi-va; inoltre alcuni pazienti recidivanoanche dopo diversi anni. Molto rara-mente, si possono verificare casi diinfezioni pericolose per la vita o letaliin ogni mo mento dopo la splenecto-mia, per cui i medici e i pa zienti sonosempre più riluttanti a consigliare ead accettare questo approccio tera-peutico. Sono stati poi valutati altri
trattamenti per prevenire o ritardarela splenectomia, inclusi il desameta-sone ad alte dosi, le infusioni inter-mittenti di immunoglobuline policlo-nali anti-D e il rituximab, tuttavia ipochi studi randomizzati controllaticon placebo condotti su questiapprocci non consentono attualmen-te di raccomandarli, poiché la loroefficacia e sicurezza rimane non chia-ra. Gli agonisti del recettore dellatrombopoietina sono attualmente insperimentazione clinica per il trat -tamento della PTI e possono rappre-sentare un’opzione terapeutica alter-nativa per il futuro. Ven gono quidiscussi i criteri per trattare la PTI e ibenefici e i rischi delle terapie. Glistudi attualmente in corso e quellifuturi aiuteranno a definire le miglioristrategie per aumentare il numerodelle piastrine e ridurre il rischio disanguinamento nei pazienti con PTI.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:55 Pagina 12

13
ematologiaPorpora trombocitopenica idiopatica
Meccanismi d’azione delle immunoglobulineendovena e delle immunoglobuline policlonali anti-Dnel miglioramento della porpora trombocitopenicaidiopatica: che cosa sappiamo veramente?
Crow AR, Lazarus AHTransfus Med Rev 2008;22(2):103-16
Le IVIG sono state usate per più di 25anni nel trattamento di un numerosempre crescente di malattie autoim-muni, inclusa la PTI. Sebbene l’esattomeccanismo d’azione delle IVIGrimanga indeterminato, sono stateproposte diverse teorie, inclusi il bloc-co/inibizione del sistema fagociticomononucleato, la neutralizzazionedegli anticorpi da parte di anticorpianti-idiotipo, la rimozione degli auto-anticorpi patogeni grazie all’inibizio-ne competitiva del recettore Fc neo-
natale delle immunoglobuline, lamodulazione delle citochine, la neu-tralizzazione del complemento e laformazione di immunocomplessideterminanti l’attivazione delle cellu-le dendritiche. Le immunoglobulinepoliclonali anti-D sono un prodotto diIVIG policlonali arricchite di anticorpidiretti versi l’antigene RhD dei globu-li rossi, che è stato anche usato consuccesso nel trattamento della trom-bocitopenia immune nei pazientiRhD(+). La teoria principale per spie-
gare l’azione delle immunoglobulinepoliclonali anti-D è stata classicamen-te il blocco del sistema fagociticomononucleato, sebbene an che lamodulazione dell’espressione del re -cet tore FcγRIIB e/o l’immunomodula-zione potrebbero avere un ruolo.Que sto articolo descrive il lavorocompiuto su un modello murino diPTI allo scopo di accrescere la com-prensione del meccanismo d’azionedi questi due agenti terapeutici.
• L’articolo di revisione riassume e analizza criticamente le opzioni terapeutiche oggi adisposizione nel trattamento della PTI, sottolineando in particolare come l’obiettivo diaumentare il numero di piastrine circolanti sia ormai da considerare obsoleto e dovreb-be essere sostituito dalla riduzione degli episodi emorragici.
• La presenza o meno di eventi emorragici importanti dovrebbe anche guidare la deci-sione di iniziare o no il trattamento.
• Le terapie di prima linea a disposizione (corticosteroidi, IVIG, anti-D) permettono unabuona risposta immediata nella maggior parte dei casi, ma non intervengono sulla pato-genesi di base della malattia e pongono ancora il problema, a tutt’oggi irrisolto, dellagestione a lungo termine dei pazienti che non vanno incontro a splenectomia (Tabella).
• Fra le terapie alternative attualmente in sperimentazione, risultati incoraggianti sonostati ottenuti col desametasone ad alte dosi, mentre altri approcci (rituximab, anti-Dintermittenti, fattori trombopoietici) richiedono ulteriori conferme di efficacia.
Tabella. Risposte a breve e lungo termine ai trattamenti di prima linea per la PTI
Risposta iniziale* (%) Risposta (%) a lungo Follow-up medianotermine (> 6 mesi) (mesi)
Corticosteroidi 62 13 71IVIG 74 ND NDImmunoglobuline anti-D 71 ND ND
ND: non disponibile. *Pazienti con risposta completa o parziale.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:56 Pagina 13

ematologia
14
Porpora trombocitopenica idiopatica
• Gli autori descrivono dettagliatamente i potenziali meccanismi d’azione delle IVIG edelle immunoglobuline anti-D nella PTI, basandosi sia su modelli animali sia sui datidisponibili in vivo nell’uomo.
• Sebbene le IVIG e le immunoglobuline anti-D agiscano probabilmente attraverso ilblocco del sistema fagocitico mononucleato, responsabile della distruzione delle pia-strine sensibilizzate da autoanticorpi, i relativi meccanismi d’azione appaiono differen-ti e sembrano coinvolgere diversi tipi di recettore Fc delle immunoglobuline (Figura).
Figura. Possibile meccanismo d’azione delle IVIG nella PTI.
• Le IVIG e le immunoglobuline anti-D potrebbero quindi non presentare fenomeni dicross-resistenza, grazie ai diversi meccanismi d’azione coinvolti, e il trattamento conuna di queste due forme di terapia potrebbe essere efficace nei pazienti resistentiall’altra.
2
1
3
4
Activating FcγR FcγR γ-chain IVIG
Soluble immune complex Unknown receptor(s) Antibody-sensitized platelet
Macrophage?DC subser?NK?NKT?NK-DC?Other?
FcγRIIB upregulation
Direct inhibition
Indirect
inhibition
Platelet clearance
MacrophageRegulatory DC
Downregulatedmacrophage
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:57 Pagina 14

15
ematologiaPorpora trombocitopenica idiopatica
Immunoglobuline endovena vs immunoglobulineanti-D endovena per il trattamento della porporatrombocitopenica idiopatica acuta
Shahgholi E, Vosough P, Sotoudeh K,Arjomandi K, Ansari S, Salehi S,Faranoush M, Ehsani MAIndian J Pediatr 2008. [Epub ahead of print]
• La PTI nei bambini è spesso benigna, ma pone problemi terapeutici specifici, legati aglieffetti collaterali che i corticosteroidi, farmaci di prima linea nel trattamento di questapatologia, hanno in particolare in età pediatrica.
• I due possibili trattamenti alternativi (IVIG e immunoglobuline anti-D) sono stati messia confronto in questo studio: l’efficacia delle IVIG è risultata superiore in termini diaumento del numero delle piastrine circolanti, nonostante l’utilizzo di alte dosi di anti-D (75 μg/kg) (Figura).
• Le risposte al trattamento sono state molto rapide, con la scomparsa dei sintomi emor-ragici circa 16 ore dopo l’infusio-ne di IVIG, confermando chequesta opzione terapeutica è lapiù indicata in caso di necessitàdi recupero rapido della contapiastrinica.
• Pur in presenza di una minoreefficacia terapeutica, l’uso dianti-D presenta, a parere degliautori, i vantaggi di una maggio-re facilità d’uso e di minori costi.
Figura. Conta piastrinica dopo trattamento con IVIG e immunoglobuline anti-D.
������
������
������
������
����
�� � � �
Obiettivi: Lo scopo di questo studio èstato di confrontare l’efficacia e gli ef -fetti collaterali delle IVIG con le immu-noglobuline anti-D endovena per iltrattamento della PTI acuta di nuovadiagnosi nei bambini. Metodi: Pazientipediatrici (da 6 mesi a 14 anni d’età)con PTI acuta di nuova diagnosi e con -ta piastrinica <20.000/mm3 sono statirandomizzati a ricevere una singoladose endovena di anti-D di 75 μg/kg o1 g/kg di IVIG per due giorni consecu-tivi (dose totale: 2 g/kg). La risposta è
stata definita come una conta piastrini-ca >20.000/mm3 72 ore dopo il tratta-mento iniziale. Risultati: Ottantunopazienti (52 maschi e 29 femmine) conun’età mediana di 5 anni e 3 mesi sonostati suddivisi in modo random in ungruppo anti-D (n = 42) e un gruppoIVIG (n = 39). I valori piastrinici medi albasale (prima del trattamento) eranopari a 15.406/mm3 e 15.230/mm3 nelgruppo anti-D e IVIG, rispettivamente.Il tasso di risposta nel gruppo IVIG(98%) è stato significativamente mag-
giore che nel gruppo anti-D (76%) (p =0,017). Dopo 7 giorni la conta piastrini-ca di tutti i pazienti compresi nel grup-po IVIG era >20.000/mm3, mentre nelgruppo anti-D il 12% dei pazienti avevavalori piastrinici <20.000/mm3. Con clu -sioni: Nella PTI acuta dei bambini iltrattamento iniziale con IVIG (2 g/kgdivisi in due dosi) ha aumentato i livellipiastrinici più rapidamente e più signi-ficativamente rispetto alla terapia conanti-D endovena (dose singola di 75μg/kg), entro le prime 72 ore.
Cont
a pi
astr
inic
a (m
m3 )
Giorni dopo il trattamento
IVIG
Basale
Anti-D
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:58 Pagina 15
16
ematologiaLa coniugazione del methotrexate con leimmunoglobuline induce l’apoptosi dei macrofagiattraverso un uptake mediato dal recettore Fc?
Wang X, Yao C, Jiang ZInt J Lab Hematol 2008;30(3):185-90
Lo scopo di questo studio è stato diconiugare il methotrexate (MTX) con leIVIG e di indagare se il coniugato indu-ceva una citotossicità selettiva suimacrofagi, per ottenere una nuova stra-tegia per il trattamento della PTI. Il MTXè stato legato alle IVIG utilizzando albu-
mina sierica umana come intermedia-rio. L’attività di legame del frammentoFc del coniugato è stata testata tramitecitometria di flusso. La citotossicitàselettiva del coniugato è stata determi-nata mediante esclusione con tripanblu. Dopo la coniugazione, l’attività di
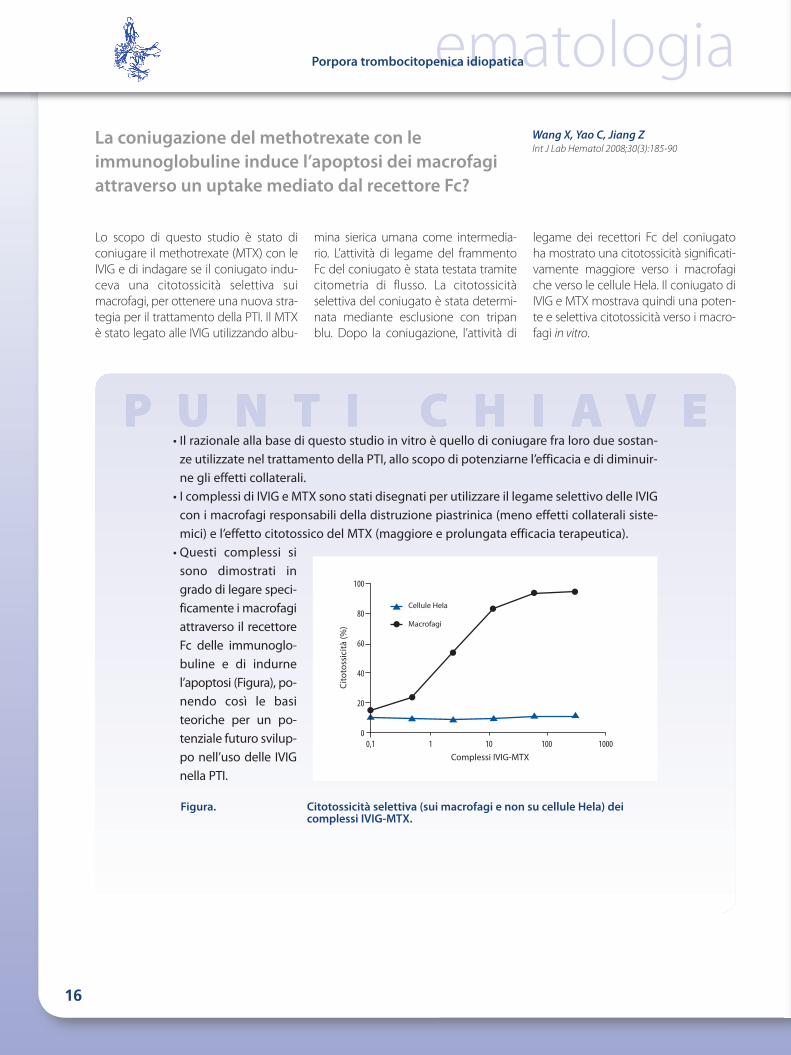
legame dei recettori Fc del coniugatoha mostrato una citotossicità significati-vamente maggiore verso i macrofagiche verso le cellule Hela. Il coniugato diIVIG e MTX mostrava quindi una poten-te e selettiva citotossicità verso i macro-fagi in vitro.
• Il razionale alla base di questo studio in vitro è quello di coniugare fra loro due sostan-ze utilizzate nel trattamento della PTI, allo scopo di potenziarne l’efficacia e di diminuir-ne gli effetti collaterali.
• I complessi di IVIG e MTX sono stati disegnati per utilizzare il legame selettivo delle IVIGcon i macrofagi responsabili della distruzione piastrinica (meno effetti collaterali siste-mici) e l’effetto citotossico del MTX (maggiore e prolungata efficacia terapeutica).
• Questi complessi sisono dimostrati ingrado di legare speci-ficamente i macrofagiattraverso il recettoreFc delle immunoglo-buline e di indurnel’apoptosi (Figura), po -nendo così le basiteoriche per un po -tenziale futuro svilup-po nell’uso delle IVIGnella PTI.
Figura. Citotossicità selettiva (sui macrofagi e non su cellule Hela) deicomplessi IVIG-MTX.
���
�
��
�
��
���� � �� ��� ����
Cito
toss
icità
(%)
Complessi IVIG-MTX
Cellule Hela
Macrofagi
Porpora trombocitopenica idiopatica
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 14:59 Pagina 16

17
Poliradiculoneuropatia demielinizzanteinfiammatoria cronica
neurologia
La poliradiculoneuropatia demielinizzante infiammatoriacronica (CIDP) è una neuropatia a patogenesi autoimmu-ne caratterizzata da comparsa di debolezza muscolaresimmetrica degli arti, coinvolgente sia i muscoli prossima-li sia quelli distali, che progredisce per più di 2 mesi. Sonospesso presenti anche disturbi della sensibilità o alterazio-ne dei riflessi tendinei. La malattia è caratterizzata daisegni elettrofisiologici della demielinizzazione e si ritieneche nella sua patogenesi la risposta umorale, con la pro-duzione di autoanticorpi, giochi un ruolo importante nelmediare il danno nervoso periferico.La CIDP ha un andamento cronico o recidivante; la mag-gior parte dei pazienti rispondenti al trattamento necessi-ta di una terapia di mantenimento a lungo termine perpreservare una buona abilità motoria e impedire l’instau-rarsi di danni neuronali permanenti. Questa caratteristicapone problemi peculiari nelle scelte terapeutiche, poiché,sebbene la malattia sia inclusa fra le neuropatie trattabili,la necessità di un trattamento per lunghi periodi di tempoimpone di prendere in adeguata considerazione costi edeffetti collaterali delle terapie effettuate.Al momento attuale, i trattamenti di prima linea utilizzatinella CIDP comprendono le immunoglobuline endovena(IVIG), i farmaci corticosteroidei e il plasma exchange(Brannagan). Le IVIG vengono spesso considerate la tera-pia di scelta, sulla base della provata efficacia e della scar-sità di effetti collaterali importanti a lungo termine. Lamaggior parte dei pazienti risponde anche al trattamentocon prednisone alla dose di 1-1,5 mg/kg (40-100 mg) al dìper 2-4 settimane. Nei pazienti rispondenti il dosaggioviene poi successivamente ridotto, fino a raggiungere ladose minima efficace. Tuttavia, gli effetti collaterali di unaterapia corticosteroidea per lunghi periodi di tempo nonsono indifferenti e includono fra gli altri iperglicemia, iper-tensione, gastriti, osteoporosi e cataratta. Fino al 70% deipazienti con CIDP recidiva quando la dose di steroidi èridotta e sono quindi stati raccomandati schemi di tratta-mento della durata di almeno 2 anni. Il plasma exchange èun’opzione terapeutica efficace, anche se di gestione piut-tosto complicata e limitata a centri specialistici.
Nei pazienti che non rispondono alla terapia di prima scel-ta vengono impiegati farmaci alternativi, fra cui in partico-lare gli agenti immunosoppressori quali ciclosporina,ciclofosfamide, azatioprina e fludarabina. Più recentemen-te, sono stati sperimentati farmaci immunomodulatorispecifici, come gli anticorpi monoclonali rituximab (anti-CD20) e campath-1H (anti-CD52), per i quali tuttavia man-cano al momento dati definitivi di efficacia, l’anti-TNF eta-nercept, risultato attivo in una piccola coorte di pazienticon CIDP, e l’interferon α e β, con risultati contrastanti(Brannagan). Come già accennato, l’infusione di IVIG èconsiderata la terapia di prima scelta nei pazienti conCIDP. La loro efficacia è stata dimostrata in diversi studirandomizzati, recentemente considerati da una metanali-si Cochrane (Eftimov et al.). Dalla metanalisi di 5 studi ran-domizzati di confronto fra IVIG e placebo nel trattamentodella CIDP, compreso lo studio ICE pubblicato nel 2008, ilpiù vasto finora con 117 pazienti trattati, la capacità dellaterapia con IVIG di migliorare la disabilità dei pazienti conCIDP è emersa in maniera statisticamente significativa.Due piccoli studi hanno anche confrontato le IVIG con icorticosteroidi e il plasma exchange, rispettivamente, otte-nendo dati di efficacia, per quanto parziali per il non otti-male disegno dei trial, sostanzialmente sovrapponibili.Gli autori dell’analisi concludono sulla necessità di valuta-re costi, effetti collaterali, durata del trattamento e facilitàdi somministrazione nella scelta di una terapia di primalinea nei pazienti con CIDP. Sebbene i corticosteroidi pos-sano essere somministrati per via orale e siano economici,in particolare rispetto agli alti costi della terapia con IVIG,viene sottolineato come gli eventi avversi associati con untrattamento corticosteroideo a lungo termine possanorappresentare anch’essi un carico economico non indiffe-rente e andrebbero presi in considerazione. Il plasmaexchange è considerato complicato, costoso e senza realivantaggi rispetto alle IVIG. Uno dei problemi associati al trattamento della CIDP èrappresentato dal monitoraggio periodico della rispostaalla terapia, allo scopo di ridurre il dosaggio, e quindi icosti e gli effetti collaterali, in caso di buona risposta o, al
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:00 Pagina 17

neurologia
18
Poliradiculoneuropatia demielinizzante infiammatoria cronica
contrario, di aumentarlo prima dell’instaurarsi di dannineuronali permanenti, in caso di risposta subottimale. Imetodi proposti sono diversi, compreso quello segnalatoda Harbo et al., basato sulla valutazione della forza musco-lare tramite dinamometria isocinetica. Essi dimostranocome, nei pazienti rispondenti alle IVIG, l’interruzione deltrattamento induca una diminuzione della forza muscola-re di circa il 15%, seguita da un nuovo incremento fino aivalori basali alla ripresa della terapia, e consigliano l’esecu-zione periodica di questo test per valutare la sensibilitàdella malattia all’infusione di IVIG.Indipendentemente dal metodo usato, non vi è dubbioche un monitoraggio periodico dei sintomi e dei segni, ilpiù possibile oggettivo, dei pazienti con CIDP in terapia dimantenimento sia una pratica opportuna e raccomanda-bile, da effettuarsi a intervalli periodici. Occorre infattiricordare che la risposta all’immunoterapia varia conside-revolmente nei diversi casi e nel tempo nel singolopaziente, per cui il trattamento dovrebbe essere aggiusta-to periodicamente e non utilizzato acriticamente sullabase degli schemi riportati nei trial clinici. Tuttavia,un’eventuale interruzione della terapia per valutare l’anda-mento clinico o la potenziale remissione della malattiaandrebbe attuata con criterio, per evitare l’instaurarsi diperdite assonali permanenti, come è stato riportato in let-teratura.Come in altre patologie, il meccanismo d’azione delle IVIGnella CIDP non è completamente noto e include probabil-
mente la riduzione della sintesi di autoanticorpi, la loro ini-bizione o accelerata distruzione e il blocco delle funzionidei fagociti mononucleati tramite il legame ai recettoriper il frammento Fc delle immunoglobuline. Studi clinici ein modelli animali aggiungono tuttavia costantementenuovi elementi alle nostre conoscenze sull’argomento epotrebbero portare in futuro a migliori modalità d’uso diquesta strategia terapeutica. Un’osservazione interessante è quella di una specificariduzione dell’espressione del recettore inibitorio FcγRIIBsui linfociti B dei pazienti con CIDP, espressione che risultainvece aumentata nei pazienti rispondenti all’infusione diIVIG (Tackenberg et al.). L’FCγRIIB è un recettore per il fram-mento Fc delle immunoglobuline a funzione inibitoria, ilcui ruolo potrebbe essere quello di impedire ai linfociti Bcon specificità per il self di maturare e trasformarsi in pla-smacellule. Livelli ridotti o non funzionanti di FcgγRIIBsono risultati associati con patologie autoimmuni, come illupus eritematoso sistemico, e la sua presenza è necessa-ria per l’efficacia clinica delle IVIG.L’osservazione al momento è ovviamente ristretta a unnumero limitato di pazienti. Tuttavia, se confermata, oltrea fornire un nuovo potenziale meccanismo d’azione per leIVIG nella CIDP, e forse in altre patologie autoimmuni,potrebbe aprire la strada a strategie terapeutiche innova-tive per il trattamento di questa malattia, mirate specifica-mente alla modulazione della funzione e dell’espressionedel recettore FcγRIIB.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:01 Pagina 18

19
neurologiaPoliradiculoneuropatia demielinizzante infiammatoria cronica
• L’articolo rivede in modo approfondito le diverse opzioni di trattamento (IVIG, plasmaexchange, corticosteroidi, chemioterapici e immunosoppressori specifici) per la terapiadelle polineuropatie demielinizzanti croniche, inclusa la CIDP, descrivendo in mododettagliato, per ognuno di questi trattamenti, schemi di somministrazione, risultati edeffetti collaterali.
• Nella CIDP vengono indicate le IVIG come terapia di prima scelta. Possibili alternativesono plasma exchange e corticosteroidi. Nei pazienti che non rispondono a uno di que-sti tre trattamenti possono essere sperimentati immunosoppressori e immunomodula-tori (Tabella).
• Viene in particolare ribadito come la CIDP rappresenti una condizione cronica, che incirca due terzi dei casi richiede una terapia di mantenimento, generalmente istituitavalutando la dose minima di trattamento necessaria per mantenere l’efficacia clinica.Gli effetti collaterali a lungo termine associati ad alcune terapie, come i corticosteroidi,andranno presi in considerazione nella valutazione dei diversi trattamenti.
• La possibilità che si verifichino danni neuronali permanenti sconsiglia di interrompere laterapia di mantenimento per poi riprenderla alla comparsa di un peggioramento clinico.
Tabella. Terapia con IVIG nella CIDP
Trattamento Dose Trial Soggetti (n) Soggetti rispondenti (%)
IVIG 0,4 g/kg per 3 sett., RCT in doppio cieco 10 10 (100%)poi 0,2 g/kg per 3 sett.
IVIG 2 g/kg RCT in doppio cieco 30 19 (63%)controllato con placebo
IVIG 2 g/kg, poi 1 g/kg RCT in doppio cieco 29 22 (76%)controllato con placebo
IVIG 2 g/kg RCT in doppio cieco 16 9 (56%)
IVIG 2 g/kg, 1 g/kg RCT in doppio cieco 59 32 (54%)ogni 3 sett. controllato con placebo
Trattamenti attuali delle polineuropatiedemielinizzanti croniche immunomediate
Brannagan TH 3rd
Muscle Nerve 2009;39(5):563-78
La CIDP, la neuropatia motoria multifo-cale (MMN) e la neuropatia con anti-corpi anti-MAG (Myelin-Associated Gly -co protein) sono neuropatie demieliniz-zanti acquisite con risposte distinteall’immunoterapia. In studi clinici ran-domizzati in doppio cieco controllaticon placebo le IVIG sono risultate effi-caci nella CIDP e nella MMN e il plasma
exchange è risultato efficace nella CIDP.La terapia corticosteroidea ha datorisultati positivi in studi controllati neipazienti con CIDP. Altri agenti, inclusala ciclofosfamide, il rituximab, l’azatio-prina, la ciclosporina, gli interferoni, lafludarabina, il mofetil micofenolato el’etanercept, si sono dimostrati ingrado di produrre benefici nella neu-
ropatia infiammatoria demielinizzantein serie di pazienti e in casi isolati. Larevisione esamina l’uso e la tossicitàassociata con questi trattamenti im -munoterapici nella terapia dei pa zien-ti con polineuropatia demielinizzantecronica immunomediata.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:02 Pagina 19

neurologia
20
Immunoglobuline endovena per lapoliradiculoneuropatia demielinizzanteinfiammatoria cronica
Eftimov F, Winer JB, Vermeulen M, de Haan R, van Schaik INCochrane Database Syst Rev 2009;(1):CD001797
Premesse: La CIDP causa debolezza eintorpidimento progressivi o recidivan-ti degli arti che si sviluppano in un arcodi tempo di almeno 2 mesi. Studi noncontrollati suggeriscono che la sommi-nistrazione di IVIG è efficace. Obiettivi:Rivedere sistematicamente le evidenzeprovenienti da studi randomizzati con-trollati riguardanti l’efficacia e la sicu-rezza delle IVIG nella CIDP. Strategia diricerca: Ricerca in Registro Cochranedei Trial Neuro muscolari, MEDLINE,EMBASE e ISI dal gennaio 1985 al mag-gio 2008. Criteri di selezione: Studirandomizzati controllati di qualunquedosaggio di IVIG vs placebo, plasmaexchange o corticosteroidi nella CIDPsicura o probabile. Raccolta e analisidei dati: Due autori hanno rivisto laricerca della letteratura per identificaregli studi potenzialmente rilevanti, valu-tare la loro qualità ed estrarre i datiindipendentemente. Abbiamo contat-tato gli autori degli studi originali perottenere ulteriori informazioni. Ri sul -tati principali: Sono stati consideratieleggibili 7 studi randomizzati control-
lati, per un totale di 287 partecipanti.Questi studi erano omogenei e la qua-lità complessiva era alta. Cinque studicon 235 partecipanti paragonavano leIVIG con un placebo, uno studio con 20partecipanti paragonava le IVIG con ilplasma exchange e uno le IVIG con ilprednisolone in 32 partecipanti. Unaproporzione significativamente mag-giore di pazienti otteneva un migliora-mento della disabilità entro un mesedalla terapia con IVIG rispetto al place-bo (rischio relativo: 2,40; IC 95% 1,72-3,36). Da questa metanalisi non puòessere dedotto se tutti questi migliora-menti siano ugualmente rilevanti clini-camente, poiché ogni trial ha usato dif-ferenti scale di disabilità e definizionidiverse di miglioramento significativo.In tre studi con 84 partecipanti totali, ladisabilità si è potuta trasformare nelpunteggio modificato di Rankin, sullabase del quale un numero significati-vamente più alto di pazienti è migliora-to di un punto dopo terapia con IVIGrispetto al placebo (rischio relativo:2,40; IC 95 0,98-5,83). Solo uno studio
incluso in questa revisione aveva unfollow-up a lungo termine. I risultati diquesto studio suggeriscono che le IVIGmigliorano la disabilità più del placeboper 24 e 48 settimane. Il punteggiomedio di disabilità non ha rivelato dif-ferenze significative fra IVIG e plasmaexchange a 6 settimane. Non vi eranodifferenze significative in termini diriduzione della disabilità con il predni-solone paragonato alle IVIG dopo 2 o 6settimane. Non vi erano differenze sta-tisticamente significative nella fre-quenza degli effetti collaterali fra questitre tipi di trattamento. Conclusionidegli autori: Le evidenze provenientida studi randomizzati controllatimostrano che l’infusione di IVIG, para-gonata a un placebo, migliora la disabi-lità per almeno 2-6 settimane, connumero di casi da trattare (NTT,Needed-To-Treat) pari a 3,00. Durantequesto periodo, le IVIG presentanoun’efficacia simile al plasma exchange eal prednisolone orale. In un vasto trial, ilbeneficio delle IVIG persisteva per 24 eforse 48 settimane.
• La revisione è l’aggiornamento di una precedente analisi pubblicata nel 2002 e pre-vede un up-date degli studi clinici randomizzati controllati sull’utilizzo della terapiacon IVIG nella CIDP. In particolare, rispetto alla precedente, è stato inserito un largostudio randomizzato di confronto fra IVIG e placebo in 117 pazienti con CIDP (studioICE).
• L’efficacia del trattamento è stata definita come un miglioramento significativo delladisabilità clinica entro 6 settimane dalla terapia (outcome primario).
• L’infusione di IVIG è risultata più efficace rispetto al placebo in 5 studi, giudicati dagliautori omogenei e di buona qualità. Due piccoli studi hanno confrontato le IVIG conil trattamento con prednisolone e con plasma exchange, rispettivamente, a breve ter-mine, senza rilevare differenze significative in termini di efficacia. Tuttavia, i dati ripor-
Poliradiculoneuropatia demielinizzante infiammatoria cronica
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:04 Pagina 20
neurologia
21
È stata studiata nella CIDP la rispostamotoria acuta a seguito della sospen-sione e della ripresa della terapia conIVIG. Undici pazienti con CIDP in tera-pia di mantenimento con IVIG, seguitiprospetticamente, sono stati valutatitramite dinamometria isocinetica,studi di conduzione nervosa e testfunzionali. A breve termine, dopo lasospensione delle IVIG, 8 pazientirispondenti al trattamento hanno
mostrato una perdita pari al 14,2%(8,6-20,0%) della forza isocinetica di 12gruppi muscolari. Tre pazienti sonorimasti stabili senza trattamento esono stati esclusi dalla valutazionesuccessiva. Ai giorni 5 e 10 dopo laripresa della terapia con IVIG la forzamuscolare isocinetica è aumentatadel 5,5% (1,6-9,6%) e dell’11,9% (7,5-16,5%), rispettivamente, ma non sonostati registrati ulteriori incrementi al
giorno 15. I miglioramenti della velo-cità di deambulazione e della funzio-ne della mano coincidevano. La laten-za minima dell’onda F era accorciata,mentre gli altri parametri elettrofisio-logici erano rimasti invariati. In con-clusione, la dinamometria isocineticarappresenta un metodo sensibile e cli-nicamente rilevante per monitorare larisposta acuta al trattamento con IVIGnella CIDP.
Risposta motoria acuta dopo un singolo ciclo di trattamento con immunoglobuline endovenanella poliradiculoneuropatia demielinizzanteinfiammatoria cronica
Harbo T, Andersen H, Jakobsen JMuscle Nerve 2009;39(4):439-470
Poliradiculoneuropatia demielinizzante infiammatoria cronica
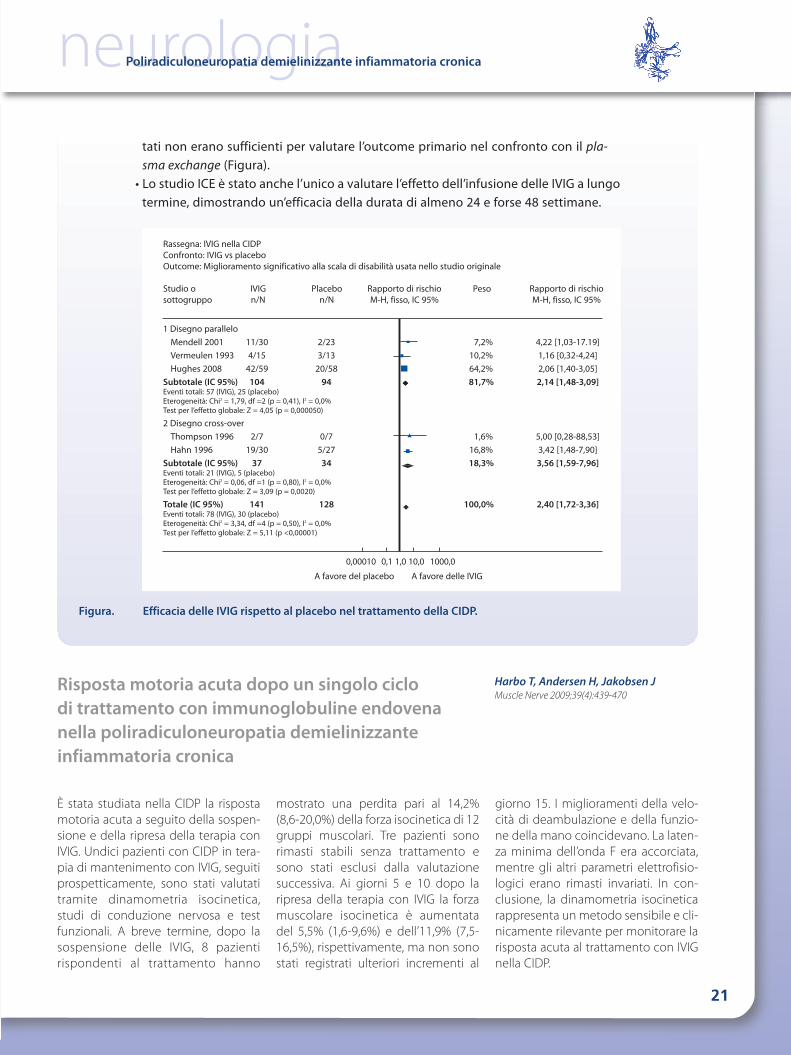
tati non erano sufficienti per valutare l’outcome primario nel confronto con il pla-sma exchange (Figura).
• Lo studio ICE è stato anche l’unico a valutare l’effetto dell’infusione delle IVIG a lungotermine, dimostrando un’efficacia della durata di almeno 24 e forse 48 settimane.
Figura. Efficacia delle IVIG rispetto al placebo nel trattamento della CIDP.
Rassegna: IVIG nella CIDPConfronto: IVIG vs placeboOutcome: Miglioramento significativo alla scala di disabilità usata nello studio originale
Studio o IVIG Placebo Rapporto di rischio Peso Rapporto di rischiosottogruppo n/N n/N M-H, fisso, IC 95% M-H, fisso, IC 95%
1 Disegno paralleloMendell 2001 11/30 2/23 7,2% 4,22 [1,03-17.19]Vermeulen 1993 4/15 3/13 10,2% 1,16 [0,32-4,24]Hughes 2008 42/59 20/58 64,2% 2,06 [1,40-3,05]
Subtotale (IC 95%) 104 94 81,7% 2,14 [1,48-3,09]Eventi totali: 57 (IVIG), 25 (placebo)Eterogeneità: Chi2 = 1,79, df =2 (p = 0,41), I2 = 0,0%Test per l’effetto globale: Z = 4,05 (p = 0,000050)
2 Disegno cross-overThompson 1996 2/7 0/7 1,6% 5,00 [0,28-88,53]Hahn 1996 19/30 5/27 16,8% 3,42 [1,48-7,90]
Subtotale (IC 95%) 37 34 18,3% 3,56 [1,59-7,96]Eventi totali: 21 (IVIG), 5 (placebo)Eterogeneità: Chi2 = 0,06, df =1 (p = 0,80), I2 = 0,0%Test per l’effetto globale: Z = 3,09 (p = 0,0020)
Totale (IC 95%) 141 128 100,0% 2,40 [1,72-3,36]Eventi totali: 78 (IVIG), 30 (placebo)Eterogeneità: Chi2 = 3,34, df =4 (p = 0,50), I2 = 0,0%Test per l’effetto globale: Z = 5,11 (p <0,00001)
0,00010 0,1 1,0 10,0 1000,0
A favore del placebo A favore delle IVIG
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:05 Pagina 21
neurologia
22
Poliradiculoneuropatia demielinizzante infiammatoria cronica
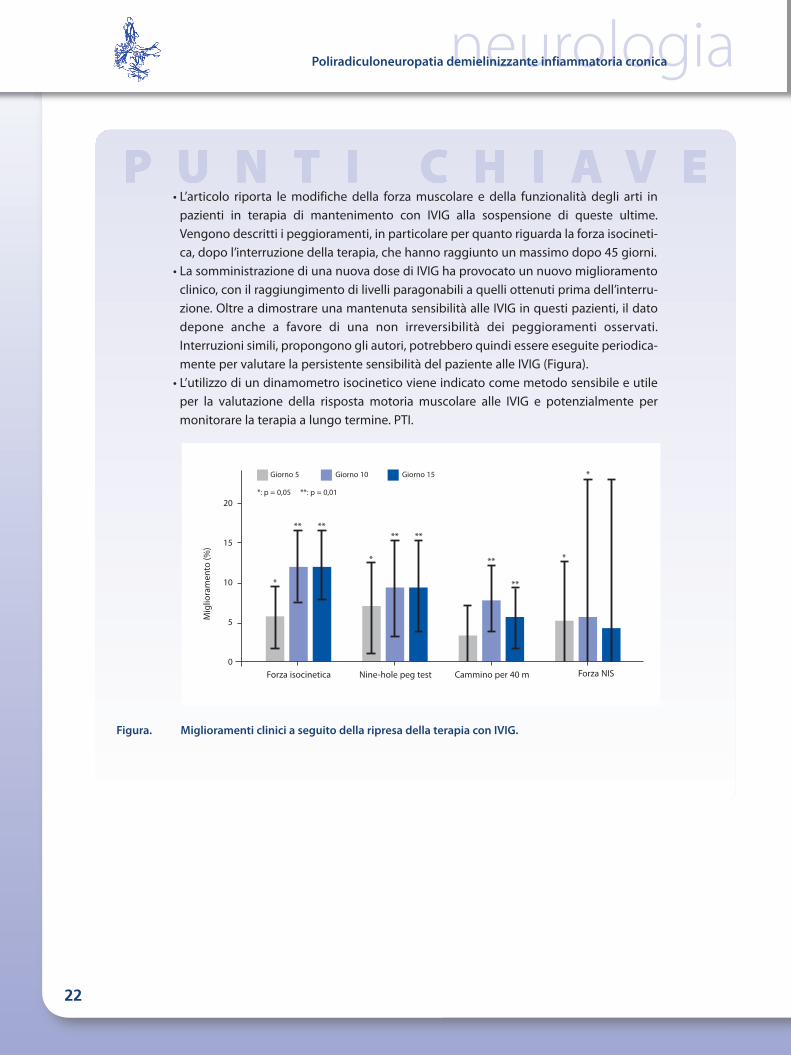
• L’articolo riporta le modifiche della forza muscolare e della funzionalità degli arti inpazienti in terapia di mantenimento con IVIG alla sospensione di queste ultime.Vengono descritti i peggioramenti, in particolare per quanto riguarda la forza isocineti-ca, dopo l’interruzione della terapia, che hanno raggiunto un massimo dopo 45 giorni.
• La somministrazione di una nuova dose di IVIG ha provocato un nuovo miglioramentoclinico, con il raggiungimento di livelli paragonabili a quelli ottenuti prima dell’interru-zione. Oltre a dimostrare una mantenuta sensibilità alle IVIG in questi pazienti, il datodepone anche a favore di una non irreversibilità dei peggioramenti osservati.Interruzioni simili, propongono gli autori, potrebbero quindi essere eseguite periodica-mente per valutare la persistente sensibilità del paziente alle IVIG (Figura).
• L’utilizzo di un dinamometro isocinetico viene indicato come metodo sensibile e utileper la valutazione della risposta motoria muscolare alle IVIG e potenzialmente permonitorare la terapia a lungo termine. PTI.
Figura. Miglioramenti clinici a seguito della ripresa della terapia con IVIG.
��
��
��
�
�
Mig
liora
men
to (%
)
Forza isocinetica Nine-hole peg test Cammino per 40 m Forza NIS
Giorno 10 Giorno 15Giorno 5
*: p = 0,05 **: p = 0,01
*
* *
*
** ****
**
**
**
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:06 Pagina 22

23
• Questo studio identifica una specifica alterazione presente nei pazienti con CIDP, laridotta espressione del recettore inibitorio FcγRIIB sui linfociti B naive e memoria, e nesuggerisce la rilevanza patogenetica, sulla base di modelli sperimentali animali.
• Il recettore FcγRIIB è l’unico a funzione inibitoria fra i recettori per il frammento Fc delleimmunoglobuline e sembra essere implicato nei meccanismi di immunotolleranza, conil ruolo di impedire ai linfociti B con specificità per il self di entrare nei centri germinati-vi e trasformarsi in plasmacellule.
• La ridotta espressione di questo recettore nei pazienti con CIDP e l’aumento dei livellidi proteina a seguito dell’infusione di IVIG suggeriscono una possibile ulteriore moda-lità d’azione delle IVIG inquesta patologia, oltre aquelle già conosciute eipotizzate. È da sottoli-neare come modelli ani-mali abbiano dimostratoche l’efficacia clinica delleIVIG in vari disordini au -toimmuni dipende dallapresenza di un recettoreFcγRIIB integro (Figura).
Figura. Aumento dell’espressione di FcγRIIB nei pazienti con CIDP rispondenti al trattamento con IVIG.
Ridotta espressione del recettore inibitorio FcγRIIBsui linfociti B nella polineuropatia demielinizzanteinfiammatoria cronica
Tackenberg B, Jelcic I, Baerenwaldt A,Oertel WH, Sommer N, Nimmerjahn F,Lünemann JDProc Natl Acad Sci U S A 2009;106(12):4788-92
Il recettore inibitorio FcγRIIB, espressosulle cellule mieloidi e sui linfociti B,gioca un ruolo critico nell’equilibrio fratolleranza e autoimmunità ed è neces-sario per l’attività antinfiammatoriadelle IVIG in diversi modelli murini dipatologie. Tuttavia, la funzione diFcγRIIB e la sua regolazione da partedelle IVIG nelle malattie autoimmuniumane sono meno conosciute. LaCIDP è la più comune fra le polineuro-patie croniche acquisite trattabili e leIVIG sono largamente usate in questapatologia come terapia iniziale e di
mantenimento. Pazienti con CIDP maitrattati, paragonati a controlli sanidemograficamente corrispondenti,mo stravano livelli di espressione diFcγRIIB consistentemente più bassi suilinfociti B naive e non erano in gradodi aumentare l’espressione o mante-nere l’aumento di FcγRIIB quando i lin-fociti B passavano dal compartimentonaive e quello memoria. Con tem -poraneamente, il raro polimorfismo -386C/-120A del promoter di FcγRIIB,risultante in una ridotta attività, prece-dentemente associato con fenotipi
autoimmuni, era maggiormente es pres-so nella CIDP. Inoltre, l’espressionedella proteina FcγRIIB era aumentatasui monociti e sui linfociti B dopo untrattamento clinicamente efficace conIVIG. Quindi, i nostri risultati suggeri-scono che il recettore inibitorio FcγRIIBè alterato in un punto critico nella dif-ferenziazione dei linfociti B nei pazien-ti con CIDP e che la modulazione dellasua espressione potrebbe essere unapproccio promettente per interveni-re efficacemente sull’immunopatolo-gia anticorpo-mediata nella CIDP.
neurologiaPoliradiculoneuropatia demielinizzante infiammatoria cronica
���
��
�
���
���
��
�
���
���
��
�
���
Rego
lazi
one
(%)
CD14+ CD19+ CD27– CD19+ CD27+
Non trattati Post-IVIG Non trattati Post-IVIG Non trattati Post-IVIG
* * *
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:07 Pagina 23

EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:07 Pagina 24

Aggiornamenti
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:07 Pagina 25

EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:08 Pagina 26

27
aggiornamentiPolimiosite e
dermatomiositeImmunoglobuline endovena incombinazione con mofetil micofe-nolato nel trattamento della miosi-te severaDanieli MG, Calcabrini L, CalabreseV, Marchetti A, Logullo F, Gabrielli AAutoimmun Rev 2009 Apr 19 [Epub ahead of print]
L’articolo riporta uno studio eseguitosu 7 pazienti con polimiosite e derma-tomiosite resistenti alla terapia consteroidi e immunosoppressori, trattaticon una combinazione di mofetilmicofenolato (MMF) e IVIG (2 g/kg/mese). Tutti i pazienti hanno mostratomiglioramento clinico, riduzione deglienzimi muscolari sierici e risoluzionedelle alterazioni elettromiografiche. Lerisposte sono state rapide, a causa diun possibile effetto sinergico fra i duetrattamenti, e la somministrazione diIVIG ha ridotto notevolmente i severieffetti collaterali (in particolare ilrischio infettivo) legati all’uso del MMF.La terapia con IVIG in aggiunta al MMFappare dunque un’opzione valida esicura nei pazienti con miosite nonrispondenti ai trattamenti standard.
Sindrome di Guillain-Barré
Sindrome di Guillain-Barré: aggior-namentoVucic S, Kiernan MC, Cornblath DRJ Clin Neurosci 2009;(6):733-741
In questa review vengono riviste le piùrecenti acquisizioni sulla clinica, lapatogenesi e il trattamento dellediverse forme di sindrome di Guillain-Barré. Di particolare interesse, la corre-lazione prospettata dagli autori frapotenziali meccanismi patogenetici erisposta al trattamento, che in questipazienti è basato classicamente su pla-sma exchange e infusione di IVIG. La
rapida (a volte anche ore) risoluzionedei sintomi a seguito di queste terapieimmunomodulanti sembra infatti diffi-cilmente spiegabile con una rimieliniz-zazione assonale, ma depone piutto-sto per la rimozione dal circolo diauto-anticorpi che interferiscono conla funzione dei canali del sodio. Al con-trario, la mancata efficacia dei cortico-steroidi orali potrebbe essere spiegata,almeno in parte, dall’inibizione deimacrofagi responsabili della rimozionedei detriti mielinici.
Sindrome di KawasakiValutazione della risposta al tratta-mento con 1 g/kg di immunoglobu-line endovena della sindrome diKawasaki acuta: metodo efficace ein grado di diminuire i costiIchihashi K, Shiraishi H, Momoi MCardiol Young 2009;19(3):224-227
Lo studio riportato in questo articoloha indagato la possibilità di trattareefficacemente i pazienti con sindromedi Kawasaki con un dosaggio di IVIGinferiore a quello standard (1g/kg inve-ce di 2g/kg). Basandosi su un’indagineretrospettiva delle cartelle cliniche, gliautori hanno individuato alcune carat-teristiche cliniche e di laboratorio ingrado di identificare i pazienti che conmaggiore probabilità possono rispon-dere al dosaggio ridotto. Viene anchefornito un interessante sistema di pun-teggio, la cui sensibilità nell’identificarei pazienti rispondenti risulta del 60% ela cui specificità è del 91%, che puòessere utilmente impiegato in clinicapermettendo, in questi casi, una ridu-zione dei costi terapeutici della metà.
Sindrome da anticorpiantifosfolipidi
Sindrome da anticorpi antifosfolipi-di “catastrofica”: analisi descrittivadi una serie di 280 pazienti dal“CAPS Registry”Cervera R, Bucciarelli S, Plasín MA,Gómez-Puerta JA, Plaza J, Pons-Estel G, Shoenfeld Y, IngelmoM, Espinos G, for the CatastrophicAntiphospholipid Syndrome CapsRegistry Project Group EuropeanForum on AntiphospholipidAntibodiesJ Autoimmun 2009;32(3-4):240-245
Gli autori di questo articolo hannocompiuto un’approfondita analisiretrospettiva delle caratteristiche clini-che e possibilità terapeutiche dellagrave forma, definita “catastrofica”, disindrome da anticorpi antifosfolipidi,la cui percentuale di mortalità, nellaserie di 280 pazienti presa in conside-razione, raggiunge il 44%. Dal puntodi vista terapeutico, le maggiori pro-babilità di risoluzione (con un tasso diguarigione del 69%) risultano dallacombinazione di anticoagulanti esteroidi con trattamenti in gradodi ridurre rapidamente il titoloanticorpale (IVIG in particolare, oplasma exchange).
Miastenia gravisMiastenia gravis giovanileChiang LM, Darras BT, Kang PBMuscle Nerve 2009;39(4):423-431
La review riassume le peculiarità epi-demiologiche, cliniche e terapeutichedelle forme giovanili di miastenia gra-vis. In particolare per quanto riguardail trattamento, l’utilizzo di farmaci cor-ticosteroidei può portare a effetti col-laterali a lungo termine particolar-mente importanti nei bambini e negliadolescenti, mentre gli agenti immu-nosoppressori e i chemioterapici van -no usati con cautela per il rischio di
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:08 Pagina 27

cancerogenicità. Per queste ragioni,l’uso di trattamenti immunomodula-tori, fra cui in particolare le IVIG, asso-ciate a scarsa tossicità, assume unruolo di speciale importanza in questigiovani pazienti rispetto agli adulti.
Pemfigo e pemfigoideStudio clinico randomizzato in dop-pio cieco sull’uso delle immunoglo-buline endovena nel trattamentodel pemfigoAmagai M, Ikeda S, Shimizu H,Iizuka H, Hanada K, Aiba S, Kaneko F,Izaki S, Tamaki K, Ikezawa Z,Takigawa M, Seishima M, Tanaka T,Miyachi Y, Katayama I, Horiguchi Y,Miyagawa S, Furukawa F, Iwatsuki K,Hide M, Tokura Y, Furue M,Hashimoto T, Ihn H, Fujiwara S,Nishikawa T, Ogawa H, Kitajima Y,Hashimoto K, Pemphigus StudyGroupJ Am Acad Dermatol 2009;60(4):595-603
L’efficacia delle IVIG nel trattamentodel pemfigo è stata dimostrata, magli studi randomizzati e controllatisono scarsi. Questo articolo riportauno studio clinico randomizzato indoppio cieco su 61 pazienti conpemfigo resistente ai corticosteroidi,trattati con 400, 200 o 0 mg/kg/die diIVIG per 5 giorni. Nel gruppo trattatocon la dose più alta è stata riscontra-ta un’efficacia terapeutica, con ridu-zione dell’attività della malattia e deilivelli di anticorpi antidesmogleina,significativamente superiore rispettoal placebo, senza incremento deglieffetti collaterali, dimostrando comele IVIG rappresentino una terapia effi-cace e sicura per il trattamento deipazienti con pemfigo resistenti aglisteroidi.
Studio clinico crossover controllatocon placebo “n-of-1” sulle immuno-globuline endovena in adiuvantenel trattamento del pemfigo volga-re refrattarioArnold DF, Burton J, Shine B,Wojnarowska F, Misbah SABr J Dermatol 2009;160(5):1098-1102
Gli autori riportano uno studio cross -over condotto in un unico pazientecon pemfigo volgare grave, trattatocon fasi consecutive di IVIG e di place-bo, allo scopo di valutare la rispostaall’infusione di IVIG dopo una terapia alungo termine (18 mesi con sommini-strazioni cicliche). Sia i livelli sierici diauto-anticorpi sia l’andamento clinicodella malattia hanno mostrato unsignificativo miglioramento a seguitodel trattamento con IVIG rispetto alplacebo. L’interesse di questo articolorisiede nella dimostrazione degli effet-ti benefici dell’infusione di IVIG anchenei pazienti con pemfigo già trattatiper lungo tempo e refrattari alla tera-pia.
Encefalomielite acutadisseminata
Encefalite postinfettiva negli adul-ti: diagnosi e trattamentoSonneville R, Klein I, de Broucker T,Wolff MJ Infect 2009;58(5):321-328
La review riassume tutte le più recen-ti conoscenze sulle forme di encefalo-mielite acuta disseminata che colpi-scono gli adulti. In particolare, vengo-no ricordate le caratteristiche diagno-stiche della malattia, che non possie-de marker biologici specifici, e la cuiidentificazione si basa sull’anamnesi,l’analisi del liquor e l’esame di RMN.Per ottenere la risoluzione dellamalattia, osservabile nella maggiorparte dei casi, viene indicato comeefficace un trattamento basato su
corticosteroidi endovena e IVIG, oplasma exchange, quest’ultimo alloscopo di indurre un rapido migliora-mento clinico.
Leucemia linfaticacronica
Riconoscimento e trattamento dellaporpora trombocitopenica immu-ne secondaria associata a malattielinfoproliferativeLiebman HASemin Hematol 2009;46(1 Suppl 2):S33-36
Le caratteristiche proprie delle formedi porpora trombocitopenica immu-ne (PTI) associate a malattie linfoproli-ferative, in particolare la leucemia lin-fatica cronica (LLC), la più frequente,costituiscono l’argomento di questareview. Sebbene questi casi ricono-scano meccanismi fisiopatologicisimili a quelli delle forme primarie, vipossono essere delle differenze signi-ficative per quanto riguarda il tratta-mento. La terapia standard della PTI(basata su corticosteroidi, splenecto-mia, anti-D e IVIG, quest’ultimoapproccio con il vantaggio di noninterferire con l’immunosoppressionepresente nei pazienti con LLC) puòessere usata con successo in questipazienti. Tuttavia, spesso, una risolu-zione completa della trombocitope-nia si ottiene solo con la remissionedella malattia di base.
aggiornamenti
28
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:08 Pagina 28

aggiornamenti
29
Neuropatia motoriamultifocale
Incremento progressivo della dose diimmunoglobuline endovena nellaneuropatia motoria multifocale:follow-up a sei mesiBaumann A, Hess CW, Sturzenegger MJ Neurol 2009;256(4):608-614
I pazienti con neuropatia motoria mul-tifocale (NMM) vengono trattati con
cicli ripetuti di IVIG, spesso cercando diindividuare la dose minima efficace e ilmaggior intervallo tollerato fra i cicli.Lo studio riportato in questo articoloha voluto valutare se questa strategiapotesse portare a un sottodosaggio alungo termine nei pazienti con NMM,con riduzione dell’efficacia. Nove casidi NMM in terapia con IVIG e sintomistabili e persistenti sono stati dunquesottoposti a uno schema di incremen-to progressivo della dose, fino al mas-simo di 2 g/kg ogni mese. La maggior
parte di essi (8/9) ha mostrato unmiglioramento significativo della fun-zione motoria, con un aumento deglieffetti collaterali transitorio e general-mente modesto. Questi risultati sem-brano indicare che un aumento delladose di IVIG può rappresentare un’op-zione terapeutica in grado di migliora-re la funzione motoria nei pazienti conNMM stabile in trattamento a lungotermine con IVIG, e che andrebbequindi preso in considerazione in que-sti casi.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:09 Pagina 29

Riassunto delle caratteristiche del prodotto
1. NOME DELLA SPECIALITÀ MEDICINALE SANDOGLOBULINA 1 g/33 ml Polvere e solvente per soluzione perinfusione SANDOGLOBULINA 3 g/100 ml Polvere e solvente per soluzione perinfusione SANDOGLOBULINA 6 g/200 ml Polvere e solvente per soluzione perinfusione SANDOGLOBULINA 12 g/200 ml Polvere e solvente per soluzione perinfusione
2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA Immunoglobuline umane normali (IgIV). SANDOGLOBULINA 1 g/33 ml Un flacone di polvere contiene: Principio attivo: immunoglobuline umane normali 1,00 g SANDOGLOBULINA 3 g/100 ml Un flacone di polvere contiene: Principio attivo: immunoglobuline umane normali 3,00 g SANDOGLOBULINA 6 g/200 ml Un flacone di polvere contiene: Principio attivo: immunoglobuline umane normali 6,00 g SANDOGLOBULINA 12 g/200 ml Un flacone di polvere contiene: Principio attivo: immunoglobuline umane normali 12,00 g Le IgG costituiscono almeno il 96% delle proteine presenti inSandoglobulina; almeno il 90% delle IgG è presente sottoforma monomerica con piccole quantità di dimeri; sono pre-senti inoltre tracce di IgG polimeriche, IgA ed IgM e frammen-ti di IgG. La distribuzione delle sottoclassi di IgG è così riparti-ta: IgG1 - 57,7%; IgG2 - 35,1%; IgG3 - 3,1%; IgG4 - 4,1%. Per un elenco completo degli eccipienti vedi 6.1
3. FORMA FARMACEUTICA Polvere e solvente per soluzione per infusione
4. INFORMAZIONI CLINICHE 4.1. Indicazioni terapeutiche Terapia sostitutiva in: Sindromi da immunodeficienza primaria quali: - agammaglobulinemia congenita e ipogammaglobulinemia; - immunodeficienza variabile comune; - immunodeficienza combinata grave; - sindrome di Wiskott-Aldrich. Leucemia linfatica cronica. Bambini con AIDS congenito e infezioni ricorrenti. Immunomodulazione - Porpora trombocitopenica idiopatica (PTI), in bambini o adulti ad alto
rischio di emorragia o prima di interventi chirurgici per il ripristinodella conta piastrinica;
- Sindrome di Guillain-Barré. Trapianto allogenico di midollo osseo e altri trapianti. Sindrome diKawasaki
4.2. Posologia e modo di somministrazione Posologia La dose e lo schema terapeutico dipendono dall’indicazione. Nella tera-pia sostitutiva può essere necessario individualizzare il dosaggio perogni paziente in relazione alla risposta farmacocinetica e clinica. Glischemi di trattamento riportati di seguito sono forniti come linee guida. Terapia sostitutiva in sindromi da immunodeficienza primaria Lo schema di trattamento dovrebbe indurre il raggiungimento di unlivello minimo di IgG (misurato prima della successiva infusione) dialmeno 4-6 g/l. Dopo l’inizio della terapia sono necessari da tre a seimesi per il raggiungimento dell’equilibrio. La dose di partenza racco-mandata è 0,4-0,8 g/kg seguita da almeno 0,2 g/kg ogni tre settimane.La dose richiesta per raggiungere un livello di 6 g/l è dell’ordine di 0,2-0,8 g/kg/mese. Una volta raggiunto lo stato stazionario l’intervallo didosaggio varia tra 2 e 4 settimane. Dovrebbero essere misurati i livelliplasmatici in modo da aggiustare la dose e l’intervallo di dosaggio. Terapia sostitutiva in caso di leucemia linfatica cronica con grave ipogam-maglobulinemia secondaria e infezioni ricorrenti: terapia sostitutiva inbambini con AIDS e infezioni ricorrenti. La dose raccomandata è 0,2-0,4 g/kg ogni 3-4 settimane. Porpora trombocitopenica idiopatica Trattamento di un episodio acuto: 0,8-1 g/kg il primo giorno. Il tratta-mento può essere ripetuto per una volta entro tre giorni, oppure pos-sono essere somministrati 0,4 g/kg/die per 2-5 giorni. Il trattamentopuò essere ripetuto in caso di recidiva. Sindrome di Guillain-Barré 0,4 g/kg/die per 3-7 giorni. Nei bambini l’esperienza è limitata. Trapianto allogenico di midollo osseo Il trattamento con immunoglobuline umane normali può essere utiliz-zato come parte della terapia di condizionamento e dopo il trapianto.Per il trattamento delle infezioni e nella profilassi della malattia da tra-
pianto contro ospite, il dosaggio viene adattato individualmente. Ladose iniziale è normalmente 0,5 g/kg/settimana, iniziando sette giorniprima del trapianto e fino a 3 mesi dopo il trapianto. In caso di persi-stente deficit di produzione di anticorpi, è raccomandato il dosaggiodi 0,5 g/kg/mese fino al ritorno alla norma del livello degli anticorpi. Terapia delle infezioni batteriche gravi Almeno 0,2 g/kg di peso corporeo; tale dose può essere ripetuta finoa somministrare, nell’arco di una settimana, una dose totale di 1 g/kgpeso corporeo. Se necessario il trattamento può essere ripetuto. Sindrome di Kawasaki Il dosaggio raccomandato è da 1,6 a 2 g/kg suddiviso in varie dosi in2-5 giorni, oppure 2 g/kg in dose singola. Il paziente deve essere sot-toposto a concomitante terapia con acido acetilsalicilico. I dosaggi raccomandati sono riassunti nella tabella seguente:
Modo di somministrazione Alla prima infusione Sandoglobulina dovrebbe essere infusa per viaendovenosa alla concentrazione del 3% con una velocità di 0,5-1ml/min (corrispondenti a 10-20 gocce/min). Se ben tollerata ed entro15 minuti non si verificano effetti indesiderati, la velocità di sommini-strazione può essere gradualmente aumentata a 1-1,5 ml/min (circa20-30 gocce/min) per altri 15 minuti, e successivamente a 2-2,5ml/min (circa 40-50 gocce/min). Nei pazienti sottoposti a regolareterapia di sostituzione che non hanno presentato effetti indesiderati,l’infusione può essere iniziata a 1-1,5 ml/min (circa 20-30 gocce/min).In pazienti in terapia regolare con Sandoglobulina, che presentanobuona tollerabilità, il farmaco può essere infuso a concentrazioni ele-vate (fino al 12%) ma l’infusione deve sempre essere iniziata a bassavelocità, e il paziente attentamente monitorato quando la velocità diinfusione viene gradualmente incrementata.
4.3. Controindicazioni Ipersensibilità a uno qualsiasi dei componenti. Ipersensibilità alle immunoglobuline omologhe, specialmente in casimolto rari di carenza di IgA quando il paziente ha anticorpi anti-IgA.
4.4. Avvertenze speciali e precauzioni per l’uso Alcune gravi reazioni avverse possono essere correlate alla velocità di infu-sione. La velocità di infusione raccomandata riportata in “4.2 Posologia emodo di somministrazione” deve essere rigorosamente rispettata. Ipazienti devono essere attentamente monitorati e osservati per eviden-ziare la comparsa di qualsiasi sintomo durante il periodo di infusione.Alcune reazioni avverse possono presentarsi più frequentemente: - in caso di alta velocità di infusione; - in pazienti con ipo- o agammaglobulinemia con o senza deficit di IgA; - in pazienti che ricevono immunoglobuline umane normali per la
prima volta o, in rari casi, quando la specialità contenente immuno-globuline umane normali viene sostituita o quando il trattamento èstato sospeso per più di otto settimane. Vere reazioni di ipersensibili-tà sono rare. Queste possono manifestarsi nei rari casi di deficienza diIgA con anticorpi anti-IgA. Raramente, le immunoglobuline umanenormali possono causare una caduta della pressione sanguigna conreazione anafilattica anche in pazienti che precedentemente aveva-no tollerato un trattamento con immunoglobuline umane normali.
Le potenziali complicanze possono essere evitate assicurandosi: - che i pazienti non siano sensibili alle immunoglobuline umane nor-
mali iniettando inizialmente il prodotto lentamente (0,5-1 ml/minpari a 10-20 gocce/min; con una concentrazione di 3%);
- che i pazienti siano attentamente monitorati per evidenziare la com-parsa di eventuali sintomi durante il periodo di infusione. In partico-lare i pazienti che non hanno mai ricevuto in precedenza immuno-
globuline umane normali, i pazienti ai quali una specialità contenen-te immunoglobuline umane normali sia stata sostituita con un’altrao i pazienti in cui sia trascorso un lungo periodo di tempo dall’infu-sione precedente, dovrebbero essere monitorati durante la primainfusione e per la prima ora dopo la prima infusione, per poter evi-denziare eventuali reazioni avverse. Tutti gli altri pazienti dovrebbe-ro essere osservati per almeno 20 minuti dopo la somministrazione.
In pazienti trattati con IVIg sono stati riportati casi di insufficienzarenale acuta. Nella maggior parte dei casi, sono stati individuati fatto-ri di rischio quali preesistente insufficienza renale, diabete mellito, etàsuperiore ai 65 anni, ipovolemia, sovrappeso o assunzione concomi-tante di medicinali nefrotossici. In tutti i pazienti, la somministrazionedi IVIg richiede: - adeguata idratazione prima di iniziare l’infusione di IVIg;
- monitoraggio per la produzione di urina; - monitoraggio dei livelli di creatinina serica; - di evitare l’uso concomitante di diuretici dell’ansa.
In caso di disfunzione renale, dovrebbe essere consideratala sospensione di IVIg. Anche se casi di disfunzione renalee di insufficienza renale acuta sono stati associati all’uso dimolte specialità registrate a base di IVIg, quelle contenentisaccarosio come stabilizzante rappresentano una quotapreponderante dell’intero numero. Nei pazienti a rischio,dovrebbe essere considerato l’uso di IVIg non contenentesaccarosio. In caso di reazioni avverse, è necessario o ridur-re la velocità di infusione o interrompere l’infusione. Il trat-tamento richiesto dipende dalla natura e dalla gravità deglieffetti indesiderati. In caso di shock, il trattamento dovreb-be seguire le linee guida per la terapia dello shock. Quandosi somministrano specialità medicinali ottenute da sangueo plasma umano, non è possibile escludere completamen-te la comparsa di patologie infettive conseguenti alla tra-smissione di agenti infettivi. Ciò risulta applicabile anche apatogeni di natura sconosciuta. Il rischio di trasmissione diagenti infettivi è comunque ridotto da:
- selezione dei donatori mediante visita medica e screeningdelle donazioni per i tre virus maggiormente patogeni, HIV,HCV, HBV;
- verifica dell’eventuale presenza di materiale genomico perHCV nei pool di plasma;
- procedure di rimozione/inattivazione incluse nel processo di produ-zione che siano state validate utilizzando virus modello e siano con-siderate efficaci per HIV, HCV, HAV e HBV.
- il processo produttivo di Sandoglobulina prevede diverse fasi dirimozione ed inattivazione virale che, nel loro complesso, comedocumentato da studi eseguiti su una varietà di modelli sperimen-tali, portano alla rimozione/inattivazione dei virus eventualmentepresenti;
- il procedimento di frazionamento mediante il qualeSandoglobulina viene preparata a partire dal plasma include variefasi, che sono state validate, per l’eliminazione di virus incapsulati enon incapsulati. La sicurezza del prodotto è ulteriormente assicura-ta, durante il procedimento di produzione, da una fase di inattiva-zione virale che prevede il trattamento a pH4 in presenza di pepsi-na. Questo step possiede la proprietà di inattivare i seguenti virus:HIV-1/2 (retrovirus incapsulato), pseudorabies virus (virus a DNAincapsulato), virus della diarrea bovina (virus a RNA incapsulato,modello per HCV) e semiliki forest virus (virus a RNA incapsulato,modello per HCV);
- ad integrazione dei metodi di eliminazione/inattivazione virale giàpresenti nel processo produttivo, è stato introdotto un procedimen-to di nanofiltrazione come ulteriore step di rimozione di virus. Lacapacità di rimozione di virus incapsulati e non incapsulati di taleprocedimento è stata stabilita mediante studi convalidati suiseguenti modelli: HIV-1, virus della diarrea bovina, pseudorabiesvirus, sindbis virus ed entero-virus di origine bovina. Questo ulterio-re step ha la potenzialità di eliminare anche virus di piccole dimen-sioni, come dimostrato per gli entero-virus di origine bovina;
- le procedure di rimozione/inattivazione dei virus potrebbero risulta-re di valore limitato contro virus privi di involucro quali il parvovirusB19.
Nell’interesse dei pazienti, si raccomanda, se possibile, ogni volta cheSandoglobulina viene loro somministrata, di registrare il nome com-merciale del prodotto ed il numero di lotto di produzione.
4.5. Interazioni con altri medicinali e altre forme di interazione Vaccini a base di virus vivi attenuati La somministrazione di immunoglobuline può interferire per unperiodo di 6 settimane e fino ad un massimo di 3 mesi con l’efficaciadi vaccini a base di virus vivi attenuati quali morbillo, rosolia, parotitee varicella. Dopo la somministrazione di questo prodotto, bisogne-rebbe far trascorrere un intervello di 3 mesi prima di procedere a vac-cinazione con vaccini a base di virus vivi attenuati. In caso di morbil-lo, l’interferenza può persistere fino ad un anno. Di conseguenza biso-gnerebbe controllare il titolo anticorpale dei pazienti trattati con ilvaccino per il morbillo.
Indicazione Dose Frequenza di somministrazione Terapia sostitutiva nella dose iniziale: ogni 2-4 settimane per ottenereimmunodeficienza primaria 0,4-0,8 g/kg un livello di IgG di almeno 4-6 g/l
mantenimento: 0,2-0,8 g/kg
Terapia sostitutiva nella 0,2-0,4 g/kg ogni 3-4 settimane per ottenereimmunodeficienza secondaria un livello di IgG di almeno 4-6 g/l Bambini con AIDS 0,2-0,4 g/kg ogni 3-4 settimane Immunomodulazione: Porpora trombocitopenica 0,8-1 g/kg al giorno 1, possibilmente ripetutoidiopatica una sola volta entro 3 giorni per
o 0,4 g/kg/die 2-5 giorni Sindrome di Guillain-Barré 0,4 g/kg/die per 3-7 giorni Trapianto allogenico di midollo osseo: Trattamento delle infezioni 0,5 g/kg ogni settimana dal giorno 7e profilassi della malattia fino a 3 mesi dopo il trapiantoda trapianto contro ospitePersistente deficit 0,5 g/kg ogni mese fino al ritorno alladi produzione di anticorpi norma del livello degli anticorpi Sindrome di Kawasaki 1,6-2,0 g/kg in 2-5 giorni
o 2 g/kg in dose singola
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:10 Pagina 30

Interferenze con analisi sierologiche Dopo l’iniezione di immunoglobuline l’aumento transitorio dei varianticorpi trasferiti passivamente nel sangue dei pazienti può indurrerisultati positivi fuorvianti nelle analisi sierologiche. La trasmissionepassiva di anticorpi contro gli antigeni eritrocitari es.: A,B,D può inter-ferire con alcune analisi sierologiche (conta dei reticolociti, aptoglobi-na, test di Coombs).
4.6. Gravidanza e allattamento La sicurezza di questa specialità medicinale per l’uso durante la gravi-danza non è stata stabilita in studi clinici controllati e, quindi, essadovrebbe essere somministrata con cautela alle donne gravide e allemadri in allattamento. L’esperienza clinica con le immunoglobulinesuggerisce l’assenza di effetti dannosi sul corso della gravidanza o sulfeto e sul neonato. Le immunoglobuline sono escrete nel latte e possono contribuire altrasferimento di anticorpi protettivi al neonato.
4.7. Effetti sulla abilità di guidare e di usare macchine Non sono stati osservati effetti sulla abilità di guidare e di usare mac-chine.
4.8. Effetti indesiderati Occasionalmente possono verificarsi reazioni avverse quali brividi, maldi testa, febbre, vomito, reazioni allergiche, nausea, artralgia, ipotensio-ne e moderato dolore lombare. Raramente le immunoglobulineumane normali possono indurre una riduzione della pressione sangui-gna e, in casi isolati, shock anafilattico, anche in pazienti che nonhanno mostrato ipersensibilità a precedenti somministrazioni. Doposomministrazione di immunoglobuline umane normali sono statiosservati casi di meningite asettica reversibile, isolati casi di anemiaemolitica/emolisi reversibile e rari casi di reazioni cutanee transitorie.Sono stati osservati aumento della creatininemia e/o insufficienzarenale acuta. Eventi trombotici sono stati riportati negli anziani, inpazienti con segni di ischemia cerebrale o cardiaca, e in pazientisovrappeso e marcatamente ipovolemici. Per la sicurezza nei confron-ti di agenti trasmissibili, vedere la sezione 4.4.
4.9. Sovradosaggio Il sovradosaggio può provocare un sovraccarico di fluidi e iperviscosi-tà in particolare in pazienti a rischio, inclusi i pazienti anziani o i pazien-ti con compromissione della funzionalità renale.
5. PROPRIETÀ FARMACOLOGICHE 5.1. Proprietà farmacodinamiche Gruppo farmacoterapeutico: sieri immuni e immunoglobuline: immu-noglobuline umane normali, per somministrazione endovenosa, codi-ce ATC: J06BA02. Le immunoglobuline umane normali contengono principalmenteimmunoglobuline G (IgG), con un ampio spettro di anticorpi controagenti infettivi. Le immunoglobuline umane normali contengono glianticorpi della classe IgG presenti nella popolazione normale. Vengonodi solito preparate da pools di plasma provenienti da non meno di 1000donatori. Posseggono una distribuzione di sottoclassi di immunoglo-buline G strettamente proporzionale a quella del plasma umano nati-vo. Dosi adeguate di questa specialità medicinale possono riportare avalori normali livelli patologicamente ridotti di immunoglobuline G. Ilmeccanismo di azione in indicazioni diverse dalla terapia sostitutivanon è del tutto chiaro, ma include effetti immunomodulatori. Nota: le immunoglobuline umane normali per uso endovenoso (IGIV)possono essere di una certa utilità nella fase acuta nel trattamento dialcune neuropatie periferiche, quali la Neuropatia Motoria Multifocale(NMM), la Poliradiculoneuropatia Infiammatoria Cronica Demielizzante(CIPD), e la Miastenia Gravis (MG). Va tenuto conto, tuttavia, che i risul-tati del trattamento possono essere temporanei e che i dati clinici asostegno dell’impiego delle IGIV in queste indicazioni derivano daesperienze cliniche perlopiù datate e condotte su piccoli numeri dipazienti, mentre non sono disponibili ad oggi studi clinici randomizza-ti controllati condotti in accordo alle norme di buona pratica clinica.
5.2. Proprietà farmacocinetiche Dopo somministrazione, le immunoglobuline umane normali sonoimmediatamente e completamente disponibili nella circolazione delricevente. Esse si distribuiscono in maniera relativamente rapida tra ilplasma e i fluidi extravascolari, l’equilibrio tra compartimenti intra edextravascolari viene raggiunto approssimativamente dopo 3-5 giorni.Le immunoglobuline umane normali hanno una emivita di circa 21giorni. Questa emivita può variare da paziente a paziente, in particola-re nell’immunodeficienza primaria. Le IgG e i complessi IgG vengonodegradati nelle cellule del sistema reticolo-endoteliale.
5.3. Dati preclinici di sicurezza Le immunoglobuline sono costituenti naturali dell’organismo.Nell’animale la prova di tossicità acuta non ha alcuna rilevanza poichédosi più alte provocano un sovraccarico del circolo. Gli studi di tossici-tà ripetuta e quelli di tossicità embrio-fetale non sono fattibili a causadella conseguente produzione ed interferenza di anticorpi contro ideterminanti antigenici umani. Non sono noti gli effetti del farmacosul sistema immunitario del neonato. In base all’esperienza clinica nonsono prevedibili effetti mutageni o oncogenici delle immunoglobuli-
ne: non è stato ritenuto necessario effettuare studi sperimentali, so -prattutto in specie eterologhe.
6. INFORMAZIONI FARMACEUTICHE 6.1. Elenco degli eccipienti Flacone contenente polvere per soluzione per infusione: saccarosioFlacone solvente: acqua per preparazioni iniettabili, cloruro di sodio
6.2. Incompatibilità Sandoglobulina non deve essere miscelata con altri medicinali; som-ministrare sempre Sandoglobulina in una linea di infusione separata.
6.3. Stabilità 3 anni.
6.4. Precauzioni speciali per la conservazione Conservare a temperatura non superiore a 25°C, al riparo dalla luce.Non congelare.
6.5. Natura e contenuto del contenitore Sandoglobulina è disponibile in kits contenenti un flacone di immu-noglobulina umana liofilizzata, un flacone di soluzione fisiologica ste-rile per la ricostituzione e un set per la preparazione e l’infusione dellasoluzione. Entrambi i flaconi sono di vetro tipo II con tappo di gommaclorobutilica privo di lattice. Sono disponibili i seguenti dosaggi: - 1 g/33 ml polvere e solvente per soluzione per infusione; - 3 g/100 ml polvere e solvente per soluzione per infusione; - 6 g/200 ml polvere e solvente per soluzione per infusione; - 12 g/200 ml polvere e solvente per soluzione per infusione.
6.6. Istruzioni per l’uso, la manipolazione e lo smaltimento Seguire attentamente le “Istruzioni per la preparazione della soluzione”qui di seguito riportate: Preparazione di una soluzione al 3%, 6%, 9% o 12% utilizzando il kit: 1) Strappare la capsula protettiva di plastica del flacone del liofilizzatoe di quello contenente il diluente. Disinfettare entrambi i tappi di gomma con alcool.
Rimuovere la guaina protettiva di una delle cannule del dispositivo ditravaso e inserire l’estremità scoperta nel tappo di gomma del flaconecontenente il diluente. 2a) e 2b) Rimuovere la seconda guaina protettiva dell’altra cannula deldispositivo di travaso. Afferrare entrambi i flaconi come illustrato nellafigura 2a, introdurre rapidamente la parte libera del dispositivo di tra-vaso nel tappo del flacone di liofilizzato e contemporaneamente por-tare i flaconi in posizione verticale con l’accortezza di posizionare il fla-cone del diluente nella posizione superiore (figura 2b). In questo modo si otterrà un immediato trasferimento del diluente nelflacone del liofilizzato. 3) Al termine del trasferimento del diluente (figura 3), togliere il flaco-ne superiore dal set di trasferimento. In questo modo si ridurrà la schiuma formatasi col travaso e si facilite-rà la completa soluzione del liofilizzato. Rimuovere completamente il dispositivo di trasferimento dal flaconedi Sandoglobulina. 4) Roteare il flacone vigorosamente senza agitare per evitare il formar-si di schiuma che richiederebbe tempo per essere eliminata. La ricosti-tuzione sarà completa in pochi minuti.
Ricostituzione Sandoglobulina senza impiego del kit o con solventi diversi Per ricostituire Sandoglobulina con solventi diversi, partendo da unflacone da 1 g, 3 g, 6 g o 12 g, prelevare il volume di diluente neces-sario, usando una siringa ipodermica sterile e iniettarlo nel corrispon-dente flacone di Sandoglobulina. A seconda delle necessità possonoessere utilizzati quale solvente, oltre alla soluzione fisiologica conte-nuta nel kit, anche acqua per preparazioni iniettabili o una soluzioneglucosata al 5%, seguendo le indicazioni riportate nella seguentetabella: Di solito la soluzione è trasparente o leggermente opalescente.
Non usare soluzioni torbide o che presentino precipitati. I prodotti disciolti dovrebbero essere controllati visivamente per lapresenza di particelle in sospensione o di colorazione anormale primadella somministrazione. Il prodotto dovrebbe essere portato a tempe-ratura ambiente o temperatura corporea prima dell’uso. Una voltapreparata, utilizzare la soluzione senza ritardi. Il prodotto inutilizzato ei residui dovrebbero essere smaltiti in accordo con le leggi nazionali.
Preparazione per l’infusione - Rimuovere la guaina protettiva dal dispositi-
vo per l’infusione e conficcarla con forza neltappo di gomma del flacone contenente laSandoglobulina (FIG. 5).
- Chiudere bene il tubo flessibile per l’infusio-ne mediante la pinza comandata dalla rotel-la (FIG. 6).
- Esercitare con il pollice e l’indice una legge-ra pressione sulla camera di gocciolamento,in modo che la soluzione penetri in quest’ul-tima (FIG. 7).
- Collegare il dispositivo per l’infusione conl’ago per l’infusione. Aprire la pinza coman-data dalla rotella e riempire di soluzione ilsistema per l’infusione (FIG. 8).
Nei pazienti con cannula a permanenza, latubazione flessibile deve essere disaerataprima di collegarla alla cannula a permanen-za. La somministrazione dell’infusione puòavere inizio. Prodotto e controllato da: CSL Behring AG-Berna Dispositivo di travaso CE 0123 CODAN Il dispositivo medico è conforme alla Direttiva 93/42/CEE
7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
CSL Behring S.p.A. P.le S. Türr, 5 20149 Milano (Mi)
8. NUMERO DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
SANDOGLOBULINA 1 g/33 ml polvere e solvente per soluzione perinfusione 1 flac. polvere da 1 g + 1 flac. solvente da 33 ml + set infusionale A.I.C. n. 025199011 SANDOGLOBULINA 3 g/100 ml polvere e solvente per soluzione perinfusione 1 flac. polvere da 3 g + 1 flac. solvente da 100 ml + set infusionale A.I.C. n. 025199023 SANDOGLOBULINA 6 g/200 ml polvere e solvente per soluzione perinfusione 1 flac. polvere da 6 g + 1 flac. solvente da 200 ml + set infusionale A.I.C. n. 025199035 SANDOGLOBULINA 12 g/200 ml polvere e solvente per soluzione perinfusione 1 flac. polvere da 12 g + 1 flac. solvente da 200 ml + set infusionale A.I.C. n. 025199047
9. DATA DI PRIMA AUTORIZZAZIONE/ RINNOVO DELL’AUTORIZZAZIONE
SANDOGLOBULINA 1 g/33 ml polvere e solvente per soluzione perinfusione SANDOGLOBULINA 3 g/100 ml polvere e solvente per solu-zione per infusione SANDOGLOBULINA 6 g/200 ml polvere e solventeper soluzione per infusione Prima autorizzazione: 17.03.1984 Rinnovodell’autorizzazione: 01.06.2000 Rinnovo dell’autorizzazione: 01.06.2005SANDOGLOBULINA 12 g/200 ml polvere e solvente per soluzione perinfusione Prima autorizzazione: 29.03.1995 Rinnovo dell’autorizzazio-ne: 01.06.2000 Rinnovo dell’autorizzazione: 01.06.2005
10. DATA DI (PARZIALE) REVISIONE DEL TESTO 17 Luglio 2007
VOLUME DI DILUENTE RICHIESTOConcentrazione Flacone 1g Flacone 3g Flacone 6g Flacone 12g
3% 33,0 cc 100 cc 200 cc -6% 16,5 cc 50 cc 100 cc 200 cc 9% 11,0 cc 33 cc 66 cc 133 cc
12% 8,3 cc 25 cc 50 cc 100 cc
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:11 Pagina 31

Riassunto delle caratteristiche del prodotto
1. DENOMINAZIONE DEL MEDICINALE Vivaglobin, soluzione di 160 mg/mL per iniezione (uso sottocutaneo).
2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA 1 mL contiene: immunoglobulina umana normale (sottocutanea) 160 mg* *Corrispondenti al contenuto di proteine totali di cui almeno il 95% IgG. Distribuzione delle sottoclassi di IgG: IgG1 ca. 61% IgG2 ca. 28% IgG3 ca. 5% IgG4 ca. 6% IgA max. 1,7 mg/mL Per gli eccipienti, vedere paragrafo 6.1.
3. FORMA FARMACEUTICA Soluzione per iniezione (uso sottocutaneo).
4. INFORMAZIONI CLINICHE 4.1 Indicazioni terapeutiche Terapia sostitutiva negli adulti e nei bambini affetti da sindromi diimmunodeficienza primitiva (PID) quali: • agammaglobulinemia e ipogammaglobulinemia congenite, • immunodeficienza comune variabile, • immunodeficienza combinata grave, • carenza di sottoclassi IgG con infezioni ricorrenti.
Terapia di sostituzione nel mieloma o nella leucemia linfaticacronica, con grave ipogammaglobulinemia secondaria e infezio-ni ricorrenti.
4.2 Posologia e modo di somministrazione Posologia Il dosaggio va determinato singolarmente per ciascun paziente,tenendo conto dei parametri farmacocinetici e della risposta clini-ca. I dosaggi qui di seguito riportati sono da ritenere come indicati-vi. Con somministrazione per via sottocutanea, il dosaggio deveessere scelto in modo tale da conseguire un livello sostenuto di IgGnel plasma. Può essere necessaria una dose di carico di almeno 0,2-0,5 g/kg (1,3-3,1 mL/kg) di peso corporeo, ripartita in più giorni, conuna dose massima giornaliera di 0,1 fino a 0,15 g/kg di peso corpo-reo, e secondo quanto indicato dal medico curante. Dopo che ilivelli di IgG abbiano raggiunto lo stato stazionario, le dosi di man-tenimento si somministreranno a intervalli successivi, preferibil-mente con cadenza settimanale tali da raggiungere una dose men-sile complessiva compresa fra circa 0,4 e 0,8 g/kg (2,5-5 mL/kg) dipeso corporeo. Per la regolazione della dose e degli intervalli didosaggio di Vivaglobin vanno misurati i livelli minimi di IgG. Modo di somministrazione Vivaglobin deve essere somministrato per via sottocutanea.L’infusione sottocutanea nel trattamento domiciliare deve essereavviata da un medico esperto nel trattamento dell’immunodefi-cienza e nell’orientamento dei pazienti in tema di terapia domici-liare. I pazienti saranno istruiti sull’impiego della pompa a siringa,sulle tecniche di infusione, sulla compilazione di un diario di trat-tamento e sui provvedimenti da adottare in caso di gravi reazioniavverse. La velocità di infusione raccomandata è pari a 22 mL/h. Inuna sperimentazione clinica, nel corso della quale sono stati valu-tati 53 pazienti, la velocità di infusione di Vivaglobin è stata porta-ta - nella fase di addestramento sotto la supervisione di un medi-co - dagli iniziali 10 mL/h a 22 mL/h. Vivaglobin deve essere prefe-ribilmente iniettato nella parete addominale, nella coscia e/o nelgluteo. In ogni singolo sito di iniezione non devono essere inietta-ti più di 15 mL. Dosi di quantità superiore a 15 mL devono essereiniettate ripartendole in più punti.
4.3 Controindicazioni Ipersensibilità accertata nei confronti di qualsiasi componente delprodotto. Vivaglobin non deve essere iniettato per via intravasco-lare. Non deve essere somministrato per via intramuscolare incaso di trombocitopenia di grado severo e in altri disturbi dellacoagulazione.
4.4 Avvertenze speciali e opportune precauzioni d’impiego Non iniettare per via endovascolare! In caso di iniezione acciden-tale di Vivaglobin in un vaso sanguigno, è possibile che il pazien-te sviluppi uno shock anafilattico. La velocità di infusione racco-mandata per Vivaglobin è indicata al paragrafo “4.2 Posologia emodo di somministrazione” e deve essere rispettata. I pazientidevono essere tenuti sotto stretto monitoraggio ed attentamentecontrollati durante l’infusione per accertare tempestivamente
l’eventuale insorgenza di qualsiasi effetto avverso. Alcune reazioniavverse possono presentarsi con maggiore frequenza nei pazien-ti ai quali l’immunoglobulina umana normale è somministrata perla prima volta, oppure, ma raramente, quando si cambia prodottoo se il trattamento è stato interrotto per più di 8 settimane. Vere reazioni di ipersensibilità sono rare. Possono manifestarsi inrarissimi casi di carenza di IgA con anticorpi anti-IgA: questipazienti devono essere trattati con cautela. Raramente, Vivaglobin può causare caduta pressoria accompa-gnata da reazione anafilattica anche in pazienti che hanno bentollerato un precedente trattamento con immunoglobulinaumana normale. Le potenziali complicanze possono essere sovente evitate, accer-tandosi: • che i pazienti non siano sensibili alle immunoglobuline umane
normali, infondendo loro, la prima volta, il prodotto lentamente(vedere paragrafo ”4.2 Posologia e modo di somministrazione”);
• che i pazienti siano attentamente monitorati per accertare contempestività l’insorgenza di qualsiasi sintomo nel corso dell’infu-sione. In particolare, si raccomanda di monitorare i pazienti nelcorso della prima infusione e per la prima ora successiva, al finedi potere subito individuare potenziali reazioni avverse cheinsorgano nelle seguenti situazioni:- pazienti non precedentemente trattati con immunoglobulina
umana normale,- pazienti in precedenza trattati con un altro prodotto, oppure- quando è intercorso molto tempo dalla precedente infusione.
Tutti gli altri pazienti devono essere comunque tenuti sotto osser-vazione per almeno 20 minuti dopo la somministrazione. In casodi sospetta reazione allergica o anafilattica si dovrà sospendereimmediatamente la somministrazione del prodotto. In caso dishock devono essere adottate le procedure correnti standard peril trattamento dello shock. Le procedure standard per prevenireinfezioni che risultino dall’uso di prodotti derivati da sangue o pla-sma umano comprendono la selezione dei donatori, il controllodelle singole donazioni e dei pool di plasma per la presenza dispecifici marcatori di infezione e l’adozione di fasi di produzioneefficaci per l’inattivazione/la rimozione dei virus. Ciò nonostante,quando vengono somministrati prodotti derivati da sangue o pla-sma umano, non può essere totalmente esclusa la possibilità ditrasmissione di agenti infettivi. Ciò vale anche per virus sconosciu-ti o emergenti e per altri patogeni. I provvedimenti adottati sonoconsiderati efficaci nei confronti di virus capsulati come HIV, HBVe HCV, e nei confronti dei virus non capsulati HAV e parvovirusB19. Esiste una rassicurante esperienza clinica in merito alla nontrasmissione dell’epatite A o del parvovirus B19 con la sommini-strazione di immunoglobuline e si ritiene anche che il contenutoanticorpale rappresenti un importante contributo alla sicurezzacontro i virus. Si raccomanda in modo particolare che, ogni qualvolta si somministri Vivaglobin, si registrino sia il nome del pazien-te che il numero di lotto del prodotto stesso, in modo da stabilireun collegamento fra il nome del paziente e il numero del lotto.
4.5 Interazioni con altri medicinali ed altre forme d’interazioneVaccini con virus vivi attenuati La somministrazione di immunoglobulina può compromettere, inun periodo compreso fra 6 settimane e 3 mesi dalla vaccinazione,l’efficacia di vaccini vivi attenuati, come i vaccini contro il morbil-lo, la rosolia, la parotite e la varicella. Dopo la somministrazione diVivaglobin deve intercorrere un intervallo di almeno 3 mesi primadi procedere alla vaccinazione con vaccini contenenti virus viviattenuati. Nel caso del morbillo, questo effetto di indebolimentodella vaccinazione può durare fino a 1 anno. Pertanto, nei pazien-ti vaccinati contro il morbillo si deve controllare la specifica situa-zione anticorpale. Interazioni con analisi sierologiche È opportuno tenere presente all’atto dell’interpretazione dei risul-tati di test sierologici che il transitorio aumento degli anticorpi tra-sportati passivamente in seguito ad iniezioni di immunoglobulinepuò rendere positivi i risultati dei test. La trasmissione passiva dianticorpi per gli antigeni eritrocitari, ad es. A, B e D, può interferirecon alcuni test sierologici per la ricerca di allo-anticorpi eritrocita-ri (ad es. test di Coombs), con la conta dei reticolociti e con l’apto-globina.
4.6 Gravidanza ed allattamento La sicurezza di questo medicinale in donne gravide non è statastabilita in sperimentazioni cliniche controllate, pertanto, occorreporre particolare attenzione nel decidere se somministrare questaspecialità medicinale durante la gravidanza o nella fase di allatta-mento al seno. L’esperienza clinica acquisita nell’impiego dellegammaglobuline non porta a ritenere la comparsa di effetti peri-colosi in caso di somministrazione delle stesse durante la gravi-danza né per la madre, né per il feto o per il neonato.
4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Non vi sono indicazioni che Vivaglobin possa compromettere lacapacità di guidare veicoli o di usare macchinari.
4.8 Effetti indesiderati In uno studio clinico eseguito con somministrazione sottocutaneain 60 soggetti, sono stati riportati i seguenti effetti indesiderati: rea-zioni al sito di infusione molto comuni e in gran parte di intensitàlieve (gonfiore, irritazione, arrossamento, indurimento, sensazionelocalizzata di calore, prurito, ecchimosi) all’inizio del trattamentosottocutaneo e con riduzione molto rapida entro le prime dieciinfusioni, quando i soggetti si abituano a questo tipo di trattamen-to. (Le reazioni al sito di iniezione non sono state segnalate in unostudio in cui i pazienti erano stati trattati con immunoglobulinasottocutanea per anni prima della sperimentazione). In singoli casi:• reazioni allergiche comprendenti caduta della pressione,• reazioni generalizzate come brividi, febbre, cefalea, malessere,
moderata lombalgia, sincope, capogiri, disturbi cutanei, bronco-spasmo.
Durante la sorveglianza post-marketing di prodotti somministratiper via intramuscolare o sottocutanea, sono stati segnalati rara-mente i seguenti effetti indesiderati: • reazioni allergiche comprendenti caduta della pressione,
dispnea, reazioni cutanee che, in casi isolati, sono progreditefino allo shock anafilattico, anche quando il paziente non avevapresentato reazioni di ipersensibilità in occasione di sommini-strazioni precedenti,
• reazioni generalizzate come brividi, febbre, cefalea, malessere,nausea, vomito, artralgia e moderata lombalgia,
• reazioni cardiovascolari, in particolare nei casi di accidentalesomministrazione del prodotto per via endovascolare,
• reazioni locali nel sito di infusione/iniezione: gonfiore, irritazione,arrossamento, indurimento, sensazione localizzata di calore, pru-rito, ecchimosi o rash.
Per informazioni in merito al rischio di malattie infettive, vedereparagrafo 4.4 “Avvertenze speciali e opportune precauzionid’impiego”.
4.9 Sovradosaggio Non sono note conseguenze da sovradosaggio.
5. PROPRIETÀ FARMACOLOGICHE 5.1 Proprietà farmacodinamiche Gruppo farmacoterapeutico: sieri immuni ed immunoglobuline;immunoglobuline umane normali, per somministrazione extrava-scolare. Codice ATC: J06B A01 L’immunoglobulina umana normale contiene principalmenteimmunoglobulina G (IgG), caratterizzata da un ampio spettro anti-corpale verso vari agenti infettivi. Vivaglobin contiene gli anticor-pi dell’immunoglobulina G che sono presenti nella popolazionenormale. Per la sua preparazione si impiegano pool di plasmaottenuti da almeno 1.000 donatori. Vivaglobin presenta una distri-buzione di sottoclassi di immunoglobulina G strettamente pro-porzionale a quella del plasma umano nativo. La somministrazio-ne di dosi adeguate di questa specialità medicinale consente diriportare alla norma bassi valori di immunoglobulina G.
5.2 Proprietà farmacocinetiche Mediante somministrazione sottocutanea dell’immunoglobulinaumana normale sono stati raggiunti nel circolo del ricevente valo-ri di picco con un ritardo di circa 2 giorni. I dati ottenuti da unasperimentazione clinica (n = 60) hanno evidenziato che, nel pla-sma, possono essere mantenuti livelli di 8-9 g/L (n = 53), sommi-nistrando ogni settimana dosi di Vivaglobin comprese fra 0,05 e0,15 g per kg di peso corporeo. Ciò è paragonato a un dosaggiocumulativo mensile di 0,2-0,6 g per kg di peso corporeo. La IgG ei complessi di IgG vengono catabolizzati nelle cellule del sistemareticolo-endoteliale
5.3 Dati preclinici di sicurezza Non esistono dati considerati rilevanti per la sicurezza clinica oltreai dati inclusi in altre sezioni del Riassunto delle caratteristiche delprodotto.
6. INFORMAZIONI FARMACEUTICHE 6.1 Elenco degli eccipienti Glicina, sodio cloruro, acido idrocloridrico o idrossido di sodio (inpiccole quantità, per la regolazione del pH), acqua per preparazio-ni iniettabili.
6.2 Incompatibilità In assenza di studi di compatibilità questo prodotto medicinalenon deve essere miscelato ad altri prodotti medicinali.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:12 Pagina 32

6.3 Periodo di validità Il periodo di validità è di 3 anni. Il prodotto deve essere utilizzatoimmediatamente dopo l’apertura della fiala o del flacone.
6.4 Speciali precauzioni per la conservazione Vivaglobin va conservato in frigorifero (+2° C e +8° C) nella confe-zione. Non congelare!
6.5 Natura e contenuto del contenitore Flaconcino (vetro Tipo I) da 3 mL di soluzione con tappo (clorobutile) - confezione da 1 o 10 flaconcini; Fiala (vetro Tipo I) da 5 mL di soluzione - confezione da 1 o 10 fiale; Flaconcino (vetro Tipo I) da 10 mL di soluzione con tappo (clorobutile) - confezione da 1, 2, 10 o 20 flaconcini; Flaconcino (vetro Tipo I) da 20 mL di soluzione con tappo (clorobutile) - confezione da 1 flaconcino. Solo la confezione da 2 flaconcini × 10 mL contiene i seguenti dispositivi: 1 siringa da 20 mL, 1 tubo-perfusore con ago, 2 aghi ipodermici, 2 aghi areatori, 3 tamponi con alcool. È possibile che non tutte le confezioni siano commercializzate.
6.6 Speciali precauzioni per lo smaltimentoVivaglobin è una soluzione pronta per l’uso e deve essere sommi-nistrata a temperatura corporea. Vivaglobin è una soluzione limpi-da. Il colore può variare da trasparente a giallo pallido fino a mar-rone chiaro entro il periodo di validità. Non usare soluzioni chesono torbide o che presentano depositi. Il prodotto non utilizzatoed i materiali di scarto devono essere smaltiti in conformità airequisiti di legge locali.
7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
CSLBehring GmbH - Emil-von-Behring-Str. 76 D-35041 Marburg - Germania
8. NUMERO(I) DELL’AUTORIZZAZIONE (DELLE AUTORIZZAZIONI) ALL’IMMISSIONE IN COMMERCIO
037882014/M - 160 mg/mL soluzione per infusione sottocutanea 1 fiala da 5 mL037882026/M - 160 mg/mL soluzione per infusione sottocutanea 10 fiale da 5 mL037882038/M - 160 mg/mL
soluzione per infusione sottocutanea 1 flaconcino 10 mL037882040/M - 160 mg/mL soluzione per infusione sottocutanea 10 flaconcini 10 mL037882053/M - 160 mg/mL soluzione per infusione sottocutanea 20 flaconcini 10 mL037882065/M - 160 mg/mL soluzione per infusione sottocutanea 1 flaconcino 3 mL037882077/M - 160 mg/mL soluzione per infusione sottocutanea 10 flaconcini 3 mL037882089/M - 160 mg/mL soluzione per infusione sottocutanea 1 flaconcino 20 ml
9. DATA DELLA PRIMA AUTORIZZAZIONE/RINNOVO DELL’AUTORIZZAZIONE
28 settembre 2007
10. DATA DI REVISIONE DEL TESTO Aprile 2008
CSL Behring - P.zza S. Tuerr, 5 - 20149 Milano - Tel. 02 349641
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:13 Pagina 33

ITC 30
8037
1 - D
epos
itato
AIFA
il 09
/10/
2007
Riassunto delle caratteristiche del prodotto
1. DENOMINAZIONE DEL MEDICINALE Rhophylac 300 microgrammi/2 ml, soluzione iniettabile in siringapre-riempita.
2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA Immunoglobulina umana anti-D. 2 ml di soluzione iniettabile in siringa pre-riempita contengono1500 UI (300 microgrammi) di immunoglobulina umana anti-Dcorrispondenti ad una concentrazione di 750 UI (150 microgram-mi)/ml. Il prodotto contiene un massimo di 30 mg/ml di proteine plasma-tiche umane, di cui 10 mg/ml sono costituiti da albumina umanacome stabilizzante. Almeno il 95% delle altre proteine plasmaticheè costituito da IgG. Rhophylac non contiene più di 5 microgram-mi/ml di IgA. Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
3. FORMA FARMACEUTICA Soluzione iniettabile in siringa pre-riempita.
4. INFORMAZIONI CLINICHE 4.1 Indicazioni terapeutiche Prevenzione della immunizzazione Rh(D) in donne Rh(D)-negative - Gravidanza e parto di un feto/neonato Rh(D)-positivo- Aborto/minaccia di aborto, gravidanza ectopica o mola idatiforme - Emorragia transplacentare conseguente ad una emorragia ante-
parto, ad una amniocentesi, ad una biopsia dei villi coriali o aprocedure ostetriche di manipolazione, come ad esempio laversione cefalica esterna, o ad un trauma addominale
Trattamento di soggetti Rh(D)-negativi dopo trasfusioni incompatibilidi sangue Rh(D)-positivi od altri prodotti contenenti eritrociti.
4.2 Posologia e modo di somministrazione PosologiaLo schema posologico seguente viene raccomandato sulla basedegli studi clinici condotti con Rhophylac; tuttavia, occorre tenerein considerazione le linee guida professionali per l’impiego delleIgG anti-D nei vari Stati Membri dell’UE.
Prevenzione della immunizzazione Rh(D) in donne Rh(D)-negative:• Profilassi ante-parto: la dose raccomandata è una dose singola
pari a 300 microgrammi (1500 UI) somministrata per via endove-nosa od intramuscolare alla 28° – 30° settimana di gravidanza.
• Profilassi post-parto: per somministrazione endovenosa, si ritie-ne che 200 microgrammi (1000 UI) siano una dose sufficiente,mentre vengono raccomandati da 200 (1000 UI) a 300 micro-grammi (1500 UI) per somministrazione intramuscolare.Rhophylac deve essere somministrato prima possibile entro 72ore dal parto. La dose post-parto deve essere somministrataanche quando sia stata effettuata una profilassi ante-parto. Se sisospetta una emorragia materno-fetale massiva [maggiore di 4ml (0,7-0,8% delle donne )], ad esempio in caso di anemia fetaleo di morte fetale intrauterina, deve essere determinata la suaentità con metodi appropriati, ad esempio il test di Kleihauer-Betke, e devono essere somministrate ulteriori dosi di anti-Dcome indicato (20 microgrammi/100 UI per ciascun ml di ema-zie fetali).
• Profilassi delle complicazioni della gravidanza: - Interventi ed incidenti che avvengono fino alla 12° settimana di
gravidanza: devono essere somministrati 200 microgrammi(1000 UI) per via endovenosa od intramuscolare non appenapossibile e comunque non oltre le 72 ore dall’evento che rap-presenta rischio emorragico;
- Interventi ed incidenti che avvengono dopo la 12° settimana digravidanza: devono essere somministrati non meno di 200microgrammi (1000 UI) per via endovenosa od intramuscolarenon appena possibile e comunque non oltre le 72 ore dal-l’evento che rappresenta rischio emorragico;
- Biopsia dei villi coriali: devono essere somministrati 200 micro-grammi (1000 UI) per via endovenosa od intramuscolare nonappena possibile e comunque non oltre le 72 ore dall’eventoche rappresenta rischio emorragico.
Trasfusioni incompatibili La dose raccomandata è di 20 microgrammi (100 UI) di immuno-globulina anti-D ogni 2 ml di sangue Rh(D)-positivo trasfuso odogni 1 ml di concentrato eritrocitario. Si raccomanda la sommini-strazione per via endovenosa. Se viene impiegata la via intramu-scolare, occorre somministrare dosi elevate per un periodo didiversi giorni. In caso di trasfusioni incompatibili più ampie, è suf-ficiente una dose massima di 3000 microgrammi, indipendente-mente dal fatto che il volume di trasfusione sia maggiore di 300ml di sangue Rh(D)-positivo.
Modo di somministrazione Rhophylac può essere somministrato sia per iniezione endoveno-sa che per iniezione intramuscolare. In caso di malattie emorragi-che ove sia controindicata la iniezione intramuscolare, Rhophylacdeve essere somministrato per via endovenosa. Se sono richiestedosi totali elevate (>5 ml) da somministrarsi per via intramuscola-re, è consigliabile la somministrazione di dosi divise in differentisiti di iniezione.
4.3 Controindicazioni Ipersensibilità ad uno qualsiasi dei componenti. La via intramuscolare è controindicata in soggetti con tromboci-topenia grave o altri disordini dell’emostasi.
4.4 Avvertenze speciali e precauzioni d’impiego Nel caso di uso post-partum, l’immunoglobulina anti-D è riserva-ta alla somministrazione materna. Essa non deve essere iniettata alneonato. Il prodotto non deve essere usato in soggetti Rh(D) positivi. Le pazienti devono essere attentamente osservate per almeno 20minuti dalla somministrazione. Se insorgono sintomi di reazioni allergiche o di tipo anafilattico, lasomministrazione deve essere immediatamente interrotta. Possono determinarsi risposte allergiche alla immunoglobulinaanti-D. I pazienti devono essere informati circa i sintomi precoci ditali reazioni di ipersensibilità, che comprendono orticaria, orticariageneralizzata, senso di oppressione al torace, difficoltà respirato-rie, ipotensione ed anafilassi. Il trattamento richiesto dipende dallanatura e dalla severità dell’evento avverso. In caso di shock, devo-no essere osservati gli standard medici per il trattamento delloshock. Rhophylac contiene una concentrazione di IgA al di sotto del limi-te analitico di 5 microgrammi/ml. Il prodotto, tuttavia, può conte-nere tracce di IgA. Sebbene l’immunoglobulina anti-D sia stataimpiegata con successo per il trattamento di pazienti selezionaticarenti di IgA, i soggetti con deficit di IgA sono a rischio per svilup-pare anticorpi IgA e possono andare incontro a reazioni anafilatti-che dopo somministrazione di componenti del sangue contenen-ti IgA. Pertanto, il medico deve attentamente valutare il beneficiodel trattamento con Rhophylac verso i rischi potenziali di reazionidi ipersensibilità.
Informazioni sulla sicurezza nei confronti di agenti trasmissibiliProvvedimenti standard per prevenire infezioni che risultino dal-l’uso di medicinali derivati da sangue o plasma umano compren-dono la selezione dei donatori, il controllo delle singole donazio-ni e dei pool di plasma per la presenza di specifici marcatori diinfezione e l’adozione di fasi di produzione efficaci per l’inattiva-zione/la rimozione dei virus. Ciò nonostante, quando vengonosomministrati medicinali derivati da sangue o plasma umano, nonpuò essere totalmente esclusa la possibilità di trasmissione diagenti infettivi. Ciò vale anche per virus sconosciuti o emergenti eper altri patogeni. I provvedimenti adottati sono considerati efficaci nei confronti divirus capsulati come HIV, HBV e HCV. Tali provvedimenti possonoessere di valore limitato nei confronti di virus non capsulati comeHAV o parvovirus B19. Esiste una rassicurante esperienza clinica in merito alla non tra-smissione dell’epatite A o del parvovirus B19 con la somministra-zione di immunoglobuline e si ritiene anche che il contenuto anti-corpale rappresenti un importante contributo alla sicurezza con-tro i virus. Si raccomanda in modo particolare che, ogni qual volta si sommi-nistra Rhophylac, si registrino sia il nome del paziente stesso che ilnumero di lotto del prodotto, in modo da stabilire un collegamen-to fra il nome del paziente e il numero del lotto.
4.5 Interazioni con altri medicinali ed altre forme d’interazioneLe interazioni di Rhophylac con altri medicinali non sono state stu-diate. Pertanto, le informazioni contenute in questo paragrafoderivano dalla letteratura scientifica e dalle linee guida attuali. L’immunizzazione attiva con vaccini contenenti virus vivi (adesempio, morbillo, parotite, rosolia o varicella) deve essere differi-ta di almeno 3 mesi dall’ultima somministrazione di immunoglo-bulina anti-D, in quanto può essere compromessa l’efficacia delvaccino con virus vivo. Se vi è necessità di somministrare l’immu-noglobulina anti-D entro 2-4 settimane da una vaccinazione convirus vivo, l’efficacia di tale vaccinazione potrebbe essere compro-messa. Dopo l’iniezione di immunoglobulina, il transitorio aumento divari anticorpi trasferiti passivamente nel sangue delle pazienti puòcausare un risultato falso positivo nei test sierologici per gli anti-corpi anti-emazie, ad esempio il test di Coomb nel neonato. Rhophylac può anche contenere anticorpi ad altri antigeni Rh, adesempio anticorpi anti-Rh(C), che possono essere rilevati conmetodi sierologici sensibili dopo la somministrazione del prodotto.
4.6 Gravidanza ed allattamento Questo medicinale viene usato in gravidanza. Non sono stati segnalati eventi avversi correlabili con il farmaco inneonati di 432 pazienti che hanno ricevuto una somministrazionedi Rhophylac prima del parto.
4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari
Non sono stati osservati effetti sulla capacità di guidare veicoli esull’uso di macchinari.
4.8 Effetti indesiderati Quando le immunoglobuline anti-D vengono somministrate pervia intramuscolare, possono essere osservati dolore locale ed ipe-restesia al sito di iniezione. Occasionalmente, possono insorgerefebbre, malessere, cefalea, reazioni cutanee e brividi. In rari casisono stati segnalati nausea, vomito, ipotensione, tachicardia, ereazioni allergiche o di tipo anafilattico, inclusi dispnea e shock,anche in pazienti che non avevano mostrato alcun segno di iper-sensibilità ad una precedente somministrazione. Vedere il paragrafo 4.4 per quanto riguarda la sicurezza nei con-fronti di agenti trasmissibili.
4.9 Sovradosaggio Non sono disponibili dati riguardo il sovradosaggio. I pazienti chehanno ricevuto una trasfusione di sangue incompatibile ed a cuisono state somministrate dosi molto elevate di immunoglobulinaanti-D devono essere attentamente monitorati sia dal punto divista clinico che da quello dei parametri biologici per il rischio direazioni emolitiche. In altri individui Rh(D)-negativi, un sovrado-saggio non dovrebbe causare effetti indesiderati più frequenti opiù gravi rispetto a quelli osservabili dopo una dose normale.
5. PROPRIETÀ FARMACOLOGICHE 5.1 Proprietà farmacodinamiche Categoria farmacoterapeutica: Immunoglobuline e sieri immuni:immunoglobulina anti-D. Codice ATC: J06BB01 Rhophylac contiene anticorpi IgG specifici contro l’antigene Rh(D)degli eritrociti umani. Durante la gravidanza, ed in particolare al momento del parto, leemazie fetali possono penetrare nella circolazione materna.Quando la madre è Rh(D)-negativa ed il feto Rh(D)-positivo, lamadre può venire immunizzata all’antigene Rh(D) e può quindiprodurre anticorpi anti-Rh(D) che attraversano la placenta e cau-sano una malattia emolitica neonatale. L’immunizzazione passivacon gammaglobuline anti-D, se somministrate in quantità appro-priate e ad un momento sufficientemente precoce dopo l’esposi-zione alle emazie fetali Rh(D)-positive, previene, nel 99% dei casi,l’immunizzazione Rh(D). Il meccanismo d’azione attraverso il quale l’immunoglobulinaanti-D sopprime l’immunizzazione alle emazie Rh(D)-positivenon è noto. Tale soppressione può essere correlata alla clearanceeritrocitaria dalla circolazione sistemica prima che esse raggiun-gano siti immunocompetenti, o potrebbe essere dovuta a mec-canismi più complessi che coinvolgono il riconoscimento dell’an-tigene estraneo e la presentazione dell’antigene da parte dellecellule appropriate ai siti appropriati, in presenza o in assenza dianticorpo. In volontari sani di sesso maschile Rh(D)-negativi, la somministra-zione di 200 microgrammi (1000 UI) di Rhophylac sia per viaendovenosa che intramuscolare, dopo 48 ore dalla iniezione di 5ml di emazie Rh(D)-positive, ha determinato entro 24 ore unaclearance delle emazie Rh(D)-positive quasi completa. Mentre lasomministrazione endovenosa di Rhophylac ha determinato unascomparsa istantanea delle emazie Rh(D)-positive, la loro elimi-nazione dopo somministrazione del prodotto per via intramu-scolare è stata ritardata, in quanto le IgG anti-D devono primaessere assorbite dal sito di iniezione. In media, il 70% delle ema-zie iniettate era stato eliminato dopo 2 ore dalla somministrazio-ne endovenosa di Rhophylac. Dopo somministrazione intramu-scolare, un simile grado di clearance delle emazie veniva misura-to dopo 12 ore.Inoltre, l’efficacia, la sicurezza ed il profilo farmacocinetico diRhophylac sono supportati dai risultati di tre studi clinici condottiin pazienti. Rhophylac 200 microgrammi (1000 UI) è stato sommi-nistrato post-partum in 139 pazienti. Rhophylac 300 microgrammi(1500 UI) è stato somministrato sia prima che dopo il parto in 446ed in 256 pazienti, rispettivamente. Nessuno dei soggetti arruola-ti in questi studi ha sviluppato anticorpi contro l’antigene Rh(D). Non sono stati eseguiti studi clinici con Rhophylac a dosi inferiori a200 microgrammi (1000 UI).
5.2 Proprietà farmacocinetiche Concentrazioni anticorpali misurabili vengono rilevate dopo circa4 ore dalla somministrazione intramuscolare. I livelli sierici di piccosi osservano normalmente dopo 5 giorni dalla somministrazione.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:14 Pagina 34

Dopo somministrazione endovenosa, vengono immediatamenteottenute concentrazioni misurabili di anticorpi. L’emivita medianella circolazione di donne in gravidanza con normali livelli di IgGè stata di 17 giorni. Le IgG ed i complessi IgG vengono degradatidalle cellule del sistema reticoloendoteliale.
5.3 Dati preclinici di sicurezza Non vi sono dati preclinici rilevanti per l’immunoglobulina anti-D.Le prove di tossicità per dose ripetuta e di tossicità embrio-fetalenon sono state condotte né sono praticabili, date l’induzione di el’interferenza con anticorpi. Non è stato studiato il potenzialemutageno delle immunoglobuline.
6. INFORMAZIONI FARMACEUTICHE 6.1 Elenco degli eccipienti Albumina umana Glicina Sodio cloruro
6.2 Incompatibilità In assenza di studi di compatibilità questo medicinale non deveessere miscelato con altri medicinali.
6.3 Periodo di validità 3 anni.
6.4 Precauzioni particolari per la conservazione Conservare in frigorifero (2°- 8°C). Non congelare. Tenere la siringa(blister originale) nell’imballaggio esterno per tenerla al riparodalla luce. Conservare fuori dalla portata e dalla vista dei bambini.
6.5 Natura e contenuto della confezione Siringa di vetro (Tipo I) pre-riempita con 2 ml di soluzione inietta-bile (1500 UI anti-D-IgG). Confezione: 1 blister contenente 1 sirin-ga pre-riempita e 1 ago per iniezione.
6.6 Precauzioni particolari per lo smaltimento e la manipola-zione
Rhophylac deve essere portato a temperatura ambiente o a tem-peratura corporea prima dell’uso. La soluzione deve presentarsilimpida o solo lievemente opalescente. Non usare soluzioni torbi-de o che mostrano depositi. Rhophylac è monouso (una siringa -un paziente). Il prodotto non utilizzato ed i rifiuti derivati da talemedicinale devono essere smaltiti in conformità ai requisiti dilegge locali.
7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
CSL Behring GmbH - Emil-von-Behring-Str. 76 D-35041 Marburg - Germania Distribuito da: CSL Behring S.p.A. P.le S. Türr, 5 -20149 Milano
8. DATA DELLA PRIMA AUTORIZZAZIONE/ RINNOVO DELL’AUTORIZZAZIONE
5 novembre 2004.
9. NUMERO DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
036161026/M 1 siringa pre-riempita da 300 mcg/2 ml
10. DATA DI REVISIONE DEL TESTO Settembre 2007.
EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:15 Pagina 35

EM CSL immunews7-09 070709:EM ZLB 7-07-2009 15:15 Pagina 36

Doppia opzione terapeuticanel trattamentocon immunoglobuline
Immunoglobulina umana normale(infusione endovenosa)
Immunoglobulina umana normale(uso sottocutaneo)
A17
89. D
epo
sito
AIF
A d
el 0
9/09
/200
8
EM CSL immunews7-09 cop:EM ZLB 3-07-2009 9:47 Pagina III

Immunoglobulina umana normaleper somministrazione sottocutanea
Dep
osi
tato
AIF
A il
13/
11/2
008
Nuova modalità di conservazione: conservare tra 2° C - 8° C;si può anche conservare fino a 3 mesi
a temperatura ambiente (non superiore a 25° C)
Novità: flacone da 3 mLPer i pazienti più piccoli e per una maggiore precisione posologica
AXXXX
EM CSL immunews7-09 cop:EM ZLB 3-07-2009 9:47 Pagina IV


















