
ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS … · iii V925e Vera-Lozada, María Gabriela. Estudo da...
180
Ministério da Saúde Instituto Nacional de Câncer José Alencar Gomes da Silva Coordenação de Pós-graduação Stricto sensu MARIA GABRIELA VERA LOZADA ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS COMPLEXOS: O LINFOMA DE HODGKIN CLÁSSICO Orientadora: Prof: Dra. Rocio Hassan Co-orientadora: Dra. Paola Cappelletti Rio de Janeiro 2013
Transcript of ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS … · iii V925e Vera-Lozada, María Gabriela. Estudo da...

Ministério da Saúde Instituto Nacional de Câncer José Alencar Gomes da Silva Coordenação de Pós-graduação Stricto sensu
MARIA GABRIELA VERA LOZADA
ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS COMPLEXOS: O LINFOMA DE HODGKIN CLÁSSICO
Orientadora: Prof: Dra. Rocio Hassan Co-orientadora: Dra. Paola Cappelletti
Rio de Janeiro
2013

ii
Ministério da Saúde Instituto Nacional de Câncer José Alencar Gomes da Silva Coordenação de Pós-graduação Stricto sensu
INSTITUTO NACIONAL DE CÂNCER Pós-Graduação em Oncologia
MARIA GABRIELA VERA LOZADA
ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS COMPLEXOS: O LINFOMA DE HODGKIN CLÁSSICO
Dissertação apresentada ao Instituto Nacional de Câncer
como parte dos requisitos para obtenção do título de
Mestre em Oncologia
Orientadora: Prof: Dra. Rocio Hassan Co-orientadora: Dra. Paola Cappelletti
Rio de Janeiro
2013

iii
V925e Vera-Lozada, María Gabriela. Estudo da expressão gênica em modelos complexos: o linfoma Hodgkin clássico. / María Gabriela Vera Lozada. – Rio de Janeiro, 2013. xxvii, 153 f.: il. graf. tab. Dissertação (Mestrado em Oncologia) – Programa de Pós- Graduação em Oncologia - Instituto Nacional de Câncer José Gomes da Silva, 2013. Orientador: Rocio Hassan. 1. Linfoma de Hodgkin . 2. Microambiente tumoral. 3. Vírus Epstein-Barr. 4. Caspase. 5. Sobrevida. I. Hassan, Rocio (Orient.). III. Instituto Nacional de Câncer José Alencar Gomes da Silva. IV. Título.
CDD 616.99446

iv
Ministério da Saúde Instituto Nacional de Câncer José Alencar Gomes da Silva Coordenação de Pós-graduação Stricto sensu
INSTITUTO NACIONAL DE CÂNCER Pós-Graduação em Oncologia
MARIA GABRIELA VERA LOZADA
ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS COMPLEXOS: O LINFOMA DE HODGKIN CLÁSSICO
Orientador: Prof. Dra. Rocio Hassan Co-orientador: Dra. Paola Cappelletti Aprovada em: _ 21 _/_ 03 _/_2013_ EXAMINADORES:
Prof. Dr. Martin Hernan Bonamino Instituto Nacional de Câncer (INCA)
Prof. Dra. Renata Binato Instituto Nacional de Câncer (INCA)
Prof. Dr. Marcelo Pelajo Machado Fundação Oswaldo Cruz (FIOCRUZ)
Prof. Dra. Esmeralda Augusta Jardim Machado Soares – Suplente I Instituto Nacional de Câncer (INCA)
Prof. Dr. Andre Luiz Mencalha – Suplente II Universidade do Estado do Rio de Janeiro (UERJ)
RIO DE JANEIRO
2013

v
À minha Família: Carmen Teresa, Germán e Rafael Daniel.

vi
No importa que tan grande sean tus sueños, son tus sueños, tarde o temprano se dan.
Anônimo

vii
AGRADECIMENTOS
A Rocio, pela oportunidade brindada, pelo apoio, por compartilhar seus
conhecimentos, conselhos, pela amizade e por sempre acreditar em mim. Muito obrigada!
Ao Mário, pela ajuda, pelo apoio nas imunohistoquímicas e por compartilhar seus
conhecimentos para o desenvolvimento deste trabalho.
A Paola pela ajuda, carinho e pelos primeiros ensinamentos recebidos no momento em
que iniciei este caminho.
A Pós-Graduação em Oncologia do Instituto Nacional de Câncer pela oportunidade e
ensinamentos.
A todas as pessoas do Laboratório de Oncovirología do CEMO, Vanessa, Bianca,
Priscilla, Arú, Jorge e Isabella pelo companheirismo e amizade compartilhada.
Ao Dr. Gustavo Stefanoff, pelo apoio, troca de conhecimentos, ajuda concedida desde
o primeiro dia da minha chegada até os experimentos de extração e integridade de RNA
FFPE.
A Dra. Ilana Zalcberg e a todo o pessoal do Laboratório de Biologia Molecular do
CEMO, em especial a Telma, por terem aberto as portas do laboratório, onde foi realizado
grande parte deste trabalho.
A Vanesa Scholl pelo ensino e discussões científicas nos experimentos de PCR em
tempo real e nas análises do ANOVA tipo nested.
A Priscilla pela amizade, solidariedade, ajuda, troca de conhecimentos e revisão deste
trabalho.
A Jackline pelo carinho e a amizade incondicional.
A Dra. Eliana Abdelhay e ao CEMO pela oportunidade de desenvolver este estudo.
A María Belen, parte deste logro foi iniciado por ti.
As equipes de trabalho do Cy0 e Labonnet pela ajuda nas analises de cinética e KOD.
Aos meus pais e irmão, pelo amor, apoio e entendimento da distância e porque mesmo
em todos os momentos me incentivam a superar as dificuldades.

viii
A Jesús, pelo amor, apoio, paciência e por sempre estar presente em todo momento.
A família Flores Silva, pela amizade, conselhos, carinho, apoio cordial e estarem
sempre dispostos a ajudar.
Aos meus amigos, Tállita, Suellen, Raphael Tavares, Carol, Daniel, Edson, Fran pelo
carinho no dia a dia.
A todas as pessoas que, por diferentes motivos, foram fonte de inspiração e coragem
nessa caminhada e que não é possível identificar.
A Coordenação de Ensino e Divulgação Científica (CEDC) – INCA pelo alojamento
concedido, que me permitiu ter concentração nos meus estudos em um ambiente confortável e
acolhedor.
As agências de Fomento, CNPq e FAPERJ, especialmente ao financiamento dado pelo
Instituto Nacional de Ciência e Tecnologia (INCT) para o Controle do Câncer (INCA).

ix
Ministério da Saúde Instituto Nacional de Câncer José Alencar Gomes da Silva Coordenação de Pós-graduação Stricto sensu
ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS COMPLEXOS: O LINFOMA DE HODGKIN CLÁSSICO
RESUMO
DISSERTAÇÃO DE MESTRADO
María Gabriela Vera Lozada
O linfoma de Hodgkin clássico (LHc) caracteriza-se por apresentar 1-3% de células tumorais (Hodgkin-Reed-Sternberg, H-RS), em meio de um infiltrado inflamatório. Alterações em vias celulares, como ciclo celular e apoptose, assim como no microambiente tumoral foram associadas ao desfecho clínico em pacientes adultos. A avaliação quantitativa da expressão gênica é uma ferramenta importante para desenvolvimento de biomarcadores, porém, seu desempenho em sistemas celulares complexos deve ser avaliado para identificar as fontes de variabilidade e lograr maior precisão na quantificação. O objetivo principal deste trabalho foi avaliar perfis de expressão de genes descritos com valor prognóstico em adultos, em um grupo de crianças com LHc, utilizando RNA extraído de material fixado em formalina e impregnado em parafina (FFPE). Para isto, uma metodologia de extração de RNA-FFPE e avaliação por reação de PCR quantitativa em tempo real após retrotranscrição (RT-qPCR) foi aperfeiçoada e avaliada mediante um desenho ANOVA tipo nested que identifica fontes de variabilidade biológica e técnica. Seguidamente, 93 linfonodos de crianças com LHc FFPE (3-18 anos) diagnosticados no Instituto Nacional de Câncer (INCA) foram estudados, realizando extração de RNA, retrotranscrição, pré-amplificação e RT-qPCR em um aparelho ABI7000 utilizando o sistema TaqMan®. Genes do ciclo celular (CENPF, CDK1, CCNA2, CCNE2 e HMMR), do apoptose (BCL2, BCL2L1 e CASP3), monócitos/macrófagos (STAT1 e LYZ) e IRF4 foram analisados; utilizando GUSB e HMBS como genes de referência mediante o método de quantificação 2-∆Cq e analisada em relação ao status do vírus Epstein-Barr (EBV), marcadores das células H-RS, subpopulações celulares do microambiente e sobrevida livre de progressão (SLP) e global (SG). Devido à alta correlação na expressão dos genes, análises de componente principal (PCA) foram realizadas, identificando três PCAs que explicaram 65,7% da variabilidade: PC1, genes do ciclo celular (39%); PC2, microambiente e apoptose (STAT1, LYZ, BCL2 e CASP3) (15,6%); PC3, BCL2L1 (11%). Em relação às subpopulações do microambiente, o número de eosinófilos apresentou correlação inversa com alguns genes do ciclo celular, apoptose e microambiente; células citotóxicas (CD8+, GRZB+, TIA1+) correlacionadas positivamente com CASP3, CDK1 e BCL2 e células TIA1+ ou GRZB+ com STAT1 e/ou LYZ; células CD68+ e CD163+ com os genes STAT1, LYZ e IRF4. Células Ki67+ e CASP3+ associadas a linfócitos CD8+ e CD20+ e à expressão de CENPF, HMMR, CDK1, CASP3 e STAT1. A presença do EBV foi associada com maior expressão de STAT1 (P=0,021) e LYZ (P=0,031). A SLP foi significativamente menor em casos com alta expressão de CASP3 (P=0,04), majoritariamente nos casos EBV- (P=0,012). Nossos resultados indicam que: (1) As fontes de variabilidade biológica e técnica devem ser avaliadas, especialmente em métodos usando amostras FFPE com objetivo de aplicação clínica; (2) Existem padrões de expressão gênica identificáveis e com sentido biológico no LHc pediátrico, associados às células H-RS e ao microambiente. A interação entre estes componentes pode ser melhor identificada por abordagens integradas de expressão gênica e imunohistoquímica; (3) O EBV modula a predição de prognóstico dos genes; (4) Modelos de predição clínica propostos em adultos não são reprodutíveis diretamente, precisando validação antes de serem aplicados no grupo das crianças.

x
Ministério da Saúde Instituto Nacional de Câncer José Alencar Gomes da Silva Coordenação de Pós-graduação Stricto sensu
ESTUDO DA EXPRESSÃO GÊNICA EM MODELOS COMPLEXOS: O LINFOMA DE HODGKIN CLÁSSICO
ABSTRACT
DISSERTAÇÃO DE MESTRADO
María Gabriela Vera Lozada
Classical Hodgkin lymphoma (cHL) is a B-cell neoplasia characterized by 1-3% of tumoral cells (Hodgkin-Reed-Sternberg, H-RS) in the middle of an inflammatory infiltrate. Alterations in cellular pathways, such as cell cycles and apoptosis, as in the tumor microenvironment were associated with clinical outcome in adult patients. The quantitative evaluation of gene expression is an important tool for the development of biomarkers, but their performance in complex cellular systems should be evaluated to identify the sources of variability and achieve greater precision in quantifying. The main objective of this study was to evaluate expression profiles of genes with prognostic value described in adul cHL in a group of children with cHL, using RNA extracted from formalin fixed and paraffin embedded (FFPE) tissues. For this, an optimum method for extraction of RNA-FFPE for quantitative PCR reaction after retrotranscription (RT-qPCR) was chosen and evaluated by a nested-ANOVA design that identifies sources of biological and technical variability. Subsequently, 93 lymph nodes of children with cHL (3-18 years) diagnosed at the Instituto Nacional de Câncer (INCA) were studied by performing RNA extraction, retrotranscription, pre-amplification and RT-qPCR with TaqMan® system in an ABI7000. Cell cycle (CENPF, CDK1, CCNA2, CCNE2 and HMMR), apoptosis (BCL2, BCL2L1 and, CASP3), monocytes/macrophages associated genes (STAT1 and LYZ) and IRF4 were analyzed; using GUSB and HMBS as reference genes using 2-∆Cq as quantification method. Results were analyzed in relation to the status of Epstein-Barr virus (EBV), markers of H-RS cells, lymphocyte subsets of the microenvironment and progression-free survival (PFS) and overall survival (OS). Because of high correlations among the gene expressions, principal component analysis (PCA) were done which identified three PCAs explaining 65.7% of the variability: PC1, cell cycle genes (39%); PC2, microenvironment and apoptosis genes (STAT1, LYZ, BCL2 and CASP3) (15.6%); PC3, BCL2L1 (11%). In relation to cell populations of the microenvironment, the number of eosinophils inversely correlated with several genes of the cell cycle, apoptosis and microenvironment; cytotoxic cells (CD8+, GRZB+, TIA1+) positively correlated with CASP3, CDK1 and BCL2 gene expression; as well with STAT1 and/or LYZ (TIA1+ or GRZB+ alone); macrophages (CD68+ and CD163+) with STAT1, LYZ and IRF4 gene expression. Immunoexpression of the proliferation marker Ki67+ associated with CASP3+, CD8+ and CD20+ cells, and with CENPF, HMMR, CDK1, CASP3 and STAT1 gene expression. EBV+ cases showed increased expression of STAT1 (P=0.021) and LYZ (P=0.031). The SLP was significantly lower in cases with high expression of CASP3 gene (P=0.04), mostly in the EBV- cases (P=0.012). Our results indicate that: (1) Evaluation of the sources of technical and biological variability is of value, especially in methods using FFPE samples with the objective of improving technical reproducibility for clinical application; (2) There are identifiable patterns of gene expression with biological meaning in pediatric cHL, associated with H-RS cells and the microenvironment. The interaction between these components can be better understood by integrated approaches including gene expression and immunohistochemistry; (3) EBV modulates the prognosis prediction of genes; (4) Clinical prediction models proposed in adults are not directly reproducible, requiring validation before be implemented in the group of children.

xi
INDICE
AGRADECIMENTOS VII
RESUMO IX
ABSTRACT X
INDICE XI
LISTA DE TABELAS XVI
LISTA DE FIGURAS XVII
LISTA DE ABREVIATURAS E SIGLAS XIX
LISTA DE GENES E PROTEINAS XXV
1 INTRODUÇÃO 1
1.1 O linfoma de Hodgkin clássico 1
1.2 Classificação histológica do linfoma de Hodgkin clássico 4
1.3 Resposta clínico-terapêutica no linfoma de Hodgkin clássico 5
1.4 O vírus Epstein-Barr no linfoma de Hodgkin clássico 6
1.5 Proliferação e escape da apoptose no linfoma de Hodgkin clássico 8
1.5.1 Prognóstico associado à proliferação e apoptose no linfoma de Hodgkin clássico 11
1.6 O microambiente tumoral no linfoma de Hodgkin clássico 12
1.7 Fatores prognósticos associados ao microambiente tumoral no linfoma de Hodgkin
clássico 16
1.8 Estudos de expressão gênica no linfoma de Hodgkin 17
1.9 Análises da expressão gênica em material fixado e impregnado em parafina 19
1.10 Aplicação de análise da variância tipo nested 19

xii
1.11 PCR quantitativa em tempo real (qPCR) como ferramenta para validação de perfis de
expressão gênica com aplicação clínica 21
1.12 Métodos de determinação do valor de quantificação por PCR em tempo real 22
1.12.1 Método Cy0 25
1.13 Genes de referência e normalização 26
1.14 Estratégia de quantificação utilizada em PCR em tempo real 27
1.15 O linfoma de Hodgkin clássico como doença complexa dos tecidos: abordagem dos
estudos de expressão gênica 28
2 OBJETIVOS 30
2.1 Objetivo geral 30
2.2 Objetivos específicos 30
3 MATERIAIS E MÉTODOS 31
3.1 Amostras 31
3.2 Dados clínicos e epidemiológicos 31
3.3 Método de extração de RNA com o reagente Trizol® 32
3.4 Método de extração de RNA total a partir de amostras fixadas e impregnadas em
parafina 33
3.4.1 Desparafinização 33
3.4.2 Método Master PureTM 33
3.4.3 Método Recover AllTM 34
3.5 Desempenho dos métodos de extração de RNA 35
3.5.1 Eletroforese em gel de agarose 35
3.5.2 Número de integridade do RNA 35

xiii
3.6 Retrotranscrição 35
3.7 Pré-amplificação 36
3.8 Quantificação da expressão gênica 36
3.9 Avaliação da amplificação 38
3.10 Abordagens de quantificação relativa da expressão gênica 39
3.10.1 Método do ciclo threshold 39
3.10.2 Método Cy0 40
3.11 Cálculo dos outliers 40
3.12 Desenho experimental ANOVA tipo nested 41
3.13 Análise do ANOVA tipo nested 41
3.14 Técnica de imunohistoquímica 42
3.15 Análise Microscópica Computacional Assistida 44
3.16 Hibridização in situ para EBER-1 (EBER-ISH) 44
3.17 Definição de respostas e tempo de sobrevida 45
3.18 Análises estatísticas 46
3.19 Critérios Éticos 47
4 RESULTADOS 48
4.1 APERFEIÇOAMENTO DE UMA METODOLOGIA REPRODUTÍVEL PARA
EXTRAÇÃO DE RNA A PARTIR DE MATERIAL EM PARAFINA 48
4.1.1 Desempenho dos métodos de extração de RNA 48
4.1.2 Desenho experimental ANOVA tipo nested 50
4.1.3 Método Cy0 57

xiv
4.1.4 Modificações do método de PCR em tempo real quantitativo 58
4.2 ANÁLISES DE EXPRESSÃO GÊNICA EM UM GRUPO DE PACIENTES COM
LINFOMA DE HODGKIN CLÁSSICO PEDIÁTRICO 60
4.2.1 Características clínicas do grupo de pacientes com linfoma de Hodgkin clássico
pediátrico 60
4.2.2 Análise de expressão gênica em um grupo de pacientes com linfoma de Hodgkin
clássico 63
4.2.2.1 Rendimento da extração de RNA no grupo total de pacientes 63
4.2.2.2 Avaliação da qualidade e determinação dos níveis de expressão gênica 64
4.2.3 Análise não supervisionada de cluster hierárquico 65
4.2.4 Análises de componente principal (PCA) 69
4.2.5 Expressão gênica em relação aos subtipos histológicos do linfoma de Hodgkin clássico
69
4.2.6 Correlação da expressão gênica com marcadores avaliados por imunohistoquímica nas
células de Hodgkin-Reed Sternberg e no microambiente tumoral 71
4.2.6.1 Marcadores das células de Hodgkin-Reed Sternberg e sua associação com a
expressão gênica 71
4.2.6.2 Microambiente tumoral e sua associação com a expressão gênica 72
4.2.6.2.1 Populações linfocitárias 74
4.2.6.2.2 Precursores monocíticos e macrófagos 74
4.2.6.2.3 Eosinófilos 74
4.2.7 Relação entre apoptose e proliferação no microambiente tumoral 78
4.2.8 Associação dos níveis de expressão gênica com a presença do vírus Epstein-Barr 80

xv
4.2.9 Expressão gênica associada ao prognóstico nos pacientes com linfoma de Hodgkin
clássico pediátricos 81
5 DISCUSSÃO 88
5.1 Aperfeiçoamento de uma técnica de extração de RNA a partir de material parafinado
para estudos de expressão gênica por RT-qPCR 88
5.2 Validação de perfis de expressão gênica em um grupo de pacientes com linfoma de
Hodgkin clássico 95
6 CONCLUSÕES 104
7 REFERÊNCIAS BIBLIOGRÁFICAS 106
ANEXO I 127
ANEXO II 128
ANEXO III 133
ANEXO IV 134
ANEXO V 140

xvi
LISTA DE TABELAS
Tabela 3.1
Tabela 4.1
Tabela 4.2
Tabela 4.3
Tabela 4.4
Tabela 4.5
Tabela 4.6
Tabela 4.7
Tabela 4.8
Tabela 4.9
Tabela 4.10
Ensaios utilizados para PCR em tempo real quantitativo.
Quantidade de RNA, pureza e RIN dos métodos comparados em
25 amostras pareadas.
Comparação do desempenho dos métodos Master PureTM e
Recover AllTM em relação à antiguidade da amostra.
Variabilidade estimada entre indivíduos, linfonodos e passos do
processamento das amostras em valores Cq e Cy0.
Variabilidade estimada entre indivíduos, linfonodos e passos do
processamento das amostras em valores ∆Cq e ∆Cy0.
Descrição das características clínicas, histológicas e associação
com o vírus Epstein-Barr no grupo de pacientes com linfoma de
Hodgkin clássico pediátrico.
Correlação entre a expressão gênica e variáveis relacionadas ao
ciclo celular e apoptose nas células de Hodgkin/Reed-Sternberg,
avaliados por marcações de imunohistoquimica (IHQ)
Correlação entre a expressão gênica e o número de células do
microambiente avaliadas por marcações de imunohistoquimica
(IHQ).
Valores de expressão gênica para os grupos de recaída vs. não recaída.
Análise de sobrevida livre de progressão (SLP) e global (SG) em
relação à expressão gênica no grupo de pacientes com linfoma de
Hodgkin clássico pediátricos
Regressão de Cox das variáveis de expressão gênica que
influenciaram a sobrevida livre de progressão em pacientes com
linfoma de Hodgkin clássico pediátricos
37
48
49
54
56
62
76
77
83
84
87

xvii
LISTA DE FIGURAS
Fig. 1.1
Fig. 1.2
Fig. 1.3
Fig. 1.4
Fig. 1.5
Fig. 1.6
Fig. 1.7
Fig. 3.1
Fig. 4.1
Fig. 4.2
Fig. 4.3
Fig. 4.4
Fig. 4.5
Fig. 4.6
Fig. 4.7
Fig. 4.8
Fig. 4.9
Fig. 4.10
Fig. 4.11
Imagem histológica de um linfonodo característico com linfoma
de Hodgkin clássico.
Esquema representando a origem das H-RS a partir de células B
do centro germinativo pré-apoptóticas.
Descrição da via de morte celular (apoptose) dependente de
caspases.
Composição do microambiente tumoral e interações patogênicas
entre células H-RS e células infiltrantes.
Fases de uma curva de reação de PCR quantitativa.
Modelo sigmoide de quatro parâmetros.
Curva de PCR com os cinco parâmetros da função de Richard.
Desenho experimental ANOVA tipo nested para amostras
fixadas e impregnadas em parafina.
Avaliação de qualidade e quantidade de RNA extraído a partir de
amostras FFPE.
Gráfico de amplificação do gene GUSB no Indivíduo 2.
Gráfico de variabilidade interindividual para o gene GUSB.
Variabilidade total em porcentagem observada no desenho
ANOVA tipo nested para valores Cq.
Distribuição da variabilidade em valores desvio padrão
acumulada no ensaio ANOVA tipo nested.
Gráfico de Box plot mostrando os níveis de expressão dos genes
estudados.
Contribuição do Desvio Padrão (DP) acumulado no ensaio
ANOVA tipo nested para cada gene estudado.
Modificações da pré-amplificação no Indivíduo 2.
Modificações da pré-amplificação no Indivíduo 1.
Detecção do vírus Epstein-Barr através da técnica de
hibridização in situ.
Distribuição do rendimento da extração de RNA proveniente de
material FFPE nos casos com linfoma de Hodgkin clássico.
2
3
10
13
22
24
25
41
49
51
51
52
53
57
58
59
60
61
63

xviii
LISTA DE FIGURAS (continuação)
Fig. 4.12
Fig. 4.13
Fig. 4.14
Fig. 4.15
Fig. 4.16
Fig. 4.17
Fig. 4.18
Fig. 4.19
Fig. 4.20
Fig. 4.21
Fig. 4.22
Fig. 4.23
Fig. 4.24
Gráfico de Box plot mostrando a classificação dos níveis de
expressão dos genes estudados.
Gráfico de tipo heat-map da expressão gênica no grupo total de
pacientes.
Gráficos de Box plot das características das populações do
microambiente em pacientes com linfoma de Hodgkin clássico
pediátricos com baixa expressão nos genes do ciclo celular.
Análises de componente principal.
Análise do grau de esclerose nodular do linfoma de Hodgkin
clássico e sua relação com a expressão gênica.
Caracterização imunohistoquímica das células infiltrantes do
microambiente tumoral no linfoma de Hodgkin clássico
pediátrico.
Análise dos eosinófilos no microambiente do linfoma de
Hodgkin clássico pediátrico e suas relações com a expressão
gênica.
Padrão de expressão gênica segundo a expressão de marcadores
de proliferação e apoptose no microambiente celular.
Padrões de associação entre marcadores de proliferação e
apoptose no microambiente tumoral do linfoma de Hodgkin
clássico pediátrico.
Padrão de expressão dos genes LYZ e STAT1 segundo o status do
vírus Epstein-Barr em pacientes com linfoma Hodgkin clássico
pediátrico.
Sobrevida livre de progressão no linfoma de Hodgkin clássico
pediátrico em relação à expressão gênica de CASP3.
Sobrevida livre de progressão (SLP) no grupo de pacientes EBV-
com linfoma de Hodgkin clássico pediátrico.
Sobrevida livre de progressão (SLP) no grupo de alto risco de
linfoma de Hodgkin clássico pediátrico.
65
66
68
69
70
73
75
79
80
81
85
86
87

xix
LISTA DE ABREVIATURAS E SIGLAS
°C Graus centigrados
σ2 Variância
∆(Rn-ROX) Delta do repórter normalizado
∆Cq Delta Cq
∆Cy0 Delta Cy0
λ Comprimento de onda
µg Microgramo
µL microlitro
µm Micrometro
Amp Amplificação
ANOVA Análise da variância (do inglês, analysis of variance)
BCR Receptor de células B (do inglês, B cell receptor)
BSA Albumina de soro bovino
CD Conjunto de diferenciação (do inglês, cluster of differentiation)
cDNA DNA complementar
CEMO Centro de Transplante de Medula Óssea
CG Centro germinativo
CGA Campo de grande aumento
CM Linfoma de Hodgkin clássico subtipo celularidade mista
Cq Ciclo de quantificação

xx
LISTA DE ABREVIATURAS E SIGLAS (continuação)
CTL Linfócitos citotóxicos CD8+
CV Coeficiente de variação
Cy0 Ciclo de quantificação Cy0
DEPC Dietil dicarbonato
dL Decilitro
DL Linfoma de Hodgkin clássico subtipo depleção linfocitária
DNA Ácido desoxirribonucleico
DNTPs Desorribonucleotídeos trifosfatado
DP Desvio padrão
EBER Pequenos RNAs não codificantes do vírus Epstein-Barr
EBNA Antígeno nuclear Epstein-Barr (do inglês, Epstein-Barr nuclear antigen)
EBV Vírus Epstein-Barr
EN Linfoma de Hodgkin clássico subtipo esclerose nodular
Fb Ruído da reação de fluorescência
Fmax Quantidade de fluorescência máxima
FFPE Fixado em formalina e impregnado em parafina (do inglês, formalin-fixed
paraffin-embedded)
Fig. Figura
FITC Fluóroforo Isotiocianato de fluoresceína
GDI Gene de interesse
GDR Gene de referência

xxi
LISTA DE ABREVIATURAS E SIGLAS (continuação)
GRZB Granzima B
H&E Coloração por hematoxilina-eosina
HIV Vírus da imunodeficiência humana
HR Razão de chance (do inglês, Hazard ratio)
H-RS Célula de Hodgkin – Reed Sternberg
IHQ Imunohistoquímica
IFN-γ Interferon gamma (produzido por células Th1)
IL Interleucina
INCA Instituto Nacional de Câncer
IPS Escore Prognóstico Internacional (do inglês, International Prognostic Score)
ISH Hibridização in situ
KMO Do inglês, Kaiser-Meyer-Olkin
KOD Detecção de outliers pela cinética da curva
LHc Linfoma de Hodgkin clássico
LHPLN Linfoma de Hodgkin com predomínio linfocítário nodular
LMP1 Proteína latente de membrana 1 (do inglês, Latent membrane protein 1)
LMP2A Proteína latente de membrana 2A (do inglês, Latent membrane protein 2A)
MAT Microambiente tumoral
mg Miligramo
MHC Complexo principal de histocompatibilidade (MHC; do inglês, major
histocompatibility complex)

xxii
LISTA DE ABREVIATURAS E SIGLAS (continuação)
MIQE Informação mínima para publicação em experimentos de quantificação em
PCR em tempo real
mL Mililitro
mM Milimolar
mm2 Milímetro quadrado
MNI Mononucleose infecciosa
MP Método Master Pure
MSA Medida de amostras adequadas (do inglês, Measures of Sampling Adequacy)
NF-κB Fator nuclear de transcriçao kappa B
NTC Controle sem amostra (do inglês, no template control)
OMS Organização Mundial da Saúde
P Valor de P
PAP Peroxidase-anti-peroxidase
PARP Familia de proteínas polimerase ribosa poli-ADP (do inglês, poly ADP ribose
polymerase)
pb Pares de bases
PBS Tampão fosfato salino
PCA Análises de componente principal (do inglês, principal component analysis)
PCR Reação em cadeia da polimerase
pré-Amp Pré amplificação
qPCR PCR quantitativa

xxiii
LISTA DE ABREVIATURAS E SIGLAS (continuação)
r.p.m. Rotações por minuto
R2 Regressão da curva
RA Método Recover All
RIN Número de integridade do RNA
RL Linfoma de Hodgkin clássico subtipo rico em linfócitos
RNA Ácido ribonucleico
RNAm RNA mensageiro
RT Transcrição reversa ou retrotranscrição
RT-qPCR PCR quantitativa a partir da transcrição reversa
SG Sobrevida global
SLP Sobrevida livre de progressão
SPSS Do inglês, Statistical Package for Social Sciences
TA Temperatura ambiente
TAM Macrófagos associados ao tumor (do inglês, Tumor-associated macrophages)
TBS Tampão tri salino
Th1 Linfócito T auxiliares 1 (do inglês, T helper cell)
Th2 Linfócito T auxiliares 2 (do inglês, T helper cell)
TLDA Do inglês, Taq Man Low Density Array
TMA Microarranjo tecidual (do inglês, tissue microarrays)
TNFR Receptor do fator de necrose tumoral

xxiv
LISTA DE ABREVIATURAS E SIGLAS (continuação)
TUNEL Marcação de Terminações dUTP pela Deoxinucleotidil Transferase Terminal
(do inglês, Terminal deoxynucleotidyl transferase dUTP nick end labeling)
Treg Células T reguladoras
U/µL Unidade por microlitro

xxv
LISTA DE GENES E PROTEINAS
AICDA Proteína “Activation-induced cytidine deaminase”
BAD Proteína “Bcl2 antagonist of cell death”
BAK Proteína “Bcl-2 homologous antagonist/killer”
BAX Proteína “Apoptosis regulator BAX”
BCL2 Gene “B-cell CLL/lymphoma 2”
BCL2 Proteína “Apoptosis regulator Bcl-2”
BCL2L1 Gene “BCL2-like 1”
BCL2L1 Proteína “BCL2-like 1” (antigamente Bcl-XL)
BCL6 Proteína “B-cell lymphoma 6 protein”
BID Proteína “BH3-interacting domain death agonist”
BUB1 Proteína “Mitotic checkpoint serine/threonine-protein kinase BUB1”
CASP3 Gene “caspase 3, apoptosis-related cysteine peptidase”
CASP3 Proteína “Caspase-3”
CCNA2 Gene “cyclin A2”
CCNE2 Gene “cyclin E2”
CDK1 Gene “cyclin-dependent kinase 1”
CDK2 Proteína “Cyclin-dependent kinase 2”
CDK4 Proteína “Cyclin-dependent kinase 4”
CENPF Gene “centromere protein F, 350/400kDa”
CSF1 Proteína “colony stimulating factor 1 (macrophage)”

xxvi
LISTA DE GENES E PROTEINAS (continuação)
CSF1R Proteína “Macrophage colony-stimulating factor 1 receptor”
FAS Proteína “Tumor necrosis factor receptor superfamily member 6”
FASL Proteína “Tumor necrosis factor ligand superfamily member 6”
FOXP3 Proteína “FOXP3” (Marcador de células T reguladoras)
GATA3 Proteína “Trans-acting T-cell-specific transcription factor GATA-3”
(Marcador de células T auxiliares 1)
GRZB Proteína “granzyme B” (Marcador de células citotóxicas)
GUSB Gene “glucuronidase beta”
HDM2 Proteína “E3 ubiquitin-protein ligase Mdm2”
hGAL Proteína “Germinal center-associated signaling and motility protein”
HMBS Gene “hydroxymethylbilane synthase”
HMMR Gene “hyaluronan-mediated motility receptor (RHAMM)”
IL-2 Proteína “Interleukin-2”
IL-4 Proteína “Interleukin-4”
IL-10 Proteína “Interleukin-10”
IL-12 Proteína “Interleukin-12”
IL-21 Proteína “Interleukin-21”
IRF4 Gene “interferon regulatory factor 4”
Ki67 Proteína “Antigen KI-67”
LYZ Gene “lysozyme”
MAF Proteína “Transcription factor Maf”

xxvii
LISTA DE GENES E PROTEINAS (continuação)
MYC Gene “v-myc myelocytomatosis viral oncogene homolog (avian)”
NOTCH1 Gene “notch 1”
p14ARF Proteína “Cyclin-dependent kinase inhibitor 2A, isoform 4”
p16-INK4a Proteína “Cyclin-dependent kinase inhibitor 2A, isoforms 1/2/3”
p21(Cip1/WAF1) Proteína “Cyclin-dependent kinase inhibitor 1”
p27KIP1 Proteína “Cyclin-dependent kinase inhibitor 1B”
p53 Proteína “Cellular tumor antigen p53”
PAX5 Proteína “Paired box protein Pax-5”
PCNA Proteína “Proliferating cell nuclear antigen”
Rb Proteína “Retinoblastoma-associated protein”
STAT1 Gene “signal transducer and activator of transcription 1, 91kDa”
STAT1 Proteína “signal transducer and activator of transcription 1, 91kDa”
TBX21 Proteína “T-box transcription factor TBX21” (antigamente TBET)
TIA1 Proteína “Nucleolysin TIA-1 isoform p40”
TOP2A Proteína “DNA topoisomerase 2-alpha”

1
1 INTRODUÇÃO
1.1 O linfoma de Hodgkin clássico
O linfoma de Hodgkin (LH) representa 1% das neoplasias a nível mundial (PILERI et
al., 2002). Corresponde à terceira neoplasia mais prevalente em idades infantis (entre 10-30%
dos casos) e a aproximadamente 30% dos linfomas em adultos (FARRELL; JARRETT,
2011). No Brasil, os linfomas representam a segunda neoplasia na infância, atrás das
leucemias agudas, sendo que o LH é diagnosticado em aproximadamente 50% do total de
casos com linfoma (INSTITUTO NACIONAL DE CÂNCER JOSÉ ALENCAR GOMES DA
SILVA, 2011; FERREIRA et al., 2012).
A entidade foi descrita pela primeira vez em 1832 por Thomas Hodgkin (HODGKIN,
1832). No entanto, foi só entre 1898 e 1902 que Carl Sternberg e Dorothy Reed descreveram
de maneira independente a célula tumoral como característica patognomônica para o
diagnóstico do LH (STERNBERG, 1898; REED, 1902). Em 1990, a Organização Mundial
da Saúde (OMS) denominou a neoplasia como linfoma.
Segundo a nova classificação das neoplasias linfoides pela OMS (SWERDLOW et al.,
2008), o LH pode ser classificado em duas entidades diferentes: o LH clássico (LHc) e o LH
com predomínio linfocítário nodular (LHPLN), dependendo da célula tumoral identificada.
O LHc é uma doença monoclonal de linfócitos B com fenótipo atípico (KUPPERS et
al., 1994); as células tumorais compõem de 1-3% da massa tumoral e são denominadas de
células de Hodgkin (mononucleadas) e Reed-Sternberg (multinucleadas) (H-RS), sendo que
ambas populações coexistem no mesmo tumor, mas a fração proliferativa está limitada às
células mononucleadas (HODGKIN, 1832; STERNBERG, 1898; REED, 1902). O restante
da massa tumoral está composto por estroma e um grande número e variedade de células
inflamatórias benignas, indispensáveis para a manutenção do estado neoplásico (POPPEMA,
2005) (Fig. 1.1).

2
Fig. 1.1: Imagem histológica de um linfonodo característico com linfoma de Hodgkin clássico. Na imagem pode ser observada a célula de H-RS rodeada de um infiltrado inflamatório. Na imagem ampliada há duas células tumorais, uma uninucleada e outra multinucleada. Coloração realizada com H&E. Crédito: Laboratório de Oncovirología-CEMO (MHM Barros).
A identificação da linhagem de origem da célula H-RS foi particularmente difícil,
devido a dois pontos cruciais: o número baixo destas na massa tumoral e à perda do fenótipo
B (KUPPERS, 2009b). De fato, a célula H-RS perde a expressão dos marcadores da linhagem
B, tais como CD19, CD79 e o receptor de células B (BCR), apresenta baixa expressão do
fator de transcrição PAX5 e, somente entre 20 e 30% dos casos expressam CD20. Em
contraposição, estas células passam a expressar marcadores específicos de outras linhagens,
como CD30, CD15 e o receptor do fator de necrose tumoral (TNFR) (HERTEL et al., 2002;
SCHWERING et al., 2003). Mais especificamente nas crianças com LHc do Instituto
Nacional de Câncer (INCA), um estudo prévio da nossa equipe mostrou que somente 27%
dos casos expressam CD20 (BARROS et al., 2010).
A confirmação de que as células H-RS são oriundas de células B do centro
germinativo (CG) foi possível através de estudos de rearranjos e hiperpermutação somática
dos genes V da imunoglobulina, em células H-RS microdissecadas (KUPPERS et al., 1994;
KANZLER et al., 1996; BRAUNINGER et al., 2003). A perda da diferenciação B foi
atribuída a mutações sem sentido (do inglês, nonsense) em ~25% dos casos, ou a alterações na
transcrição do gene da imunoglobulina nos outros 75% dos casos, as quais deveriam ter

3
levado a célula a apoptose, mas por motivos tais como a presença do vírus Epstein-Barr
(EBV) e/ou alterações genéticas na via NF-κB a célula H-RS conseguiu sobreviver e ser
resgatada da apoptose (MARAFIOTI et al., 2000; KUPPERS, 2009a). Além disso, defeitos
epigenéticos acarretam na desprogramação B das células H-RS (KUPPERS, 2009a) (Fig. 1.2).
Fig. 1.2: Esquema representando a origem das H-RS a partir de células B do centro germinativo pré-apoptóticas. Células B naive ao entrar em contato com o antígeno cognato são ativadas. As células B ativadas migram para dentro do folículo, proliferam e se diferenciam em centroblastos, dando origem ao centro germinativo. As células B do centro germinativo sofrem hipermutação somática, aquelas células portadoras de mutações desfavoráveis são encaminhadas a apoptose via FAS-FASL, enquanto que aquelas células que apresentam o receptor de células B com alta afinidade pelo antígeno sobrevivem, abandonando o centro germinativo para se diferenciar em células B de memória ou células plasmáticas. Células B portadoras de um receptor de células B não funcional são normalmente encaminhadas a apoptose, mas, ao serem resgatadas pela ação de diferentes mecanismos, entre eles pelo vírus Epstein-Barr e/ou alterações genéticas primárias na via de NF-κB, podem dar origem às células H-RS. Adaptado de KAPATAI; MURRAY (2007); SEGGES (2012).

4
Já a entidade conhecida como LHPLN é menos frequente (~5% dos casos), acomete
quase que exclusivamente adultos, a célula tumoral é denominada de célula predominante
linfocítica, expressa marcadores da linhagem B (CD20, CD79), assim como marcadores do
CG (BCL6, hGAL, AICDA) e sua patogênese e relação com as células infiltrantes é diferente
da observada no LHc (SWERDLOW et al., 2008; SCHMITZ et al., 2009; SHANKAR;
DAW, 2012).
1.2 Classificação histológica do linfoma de Hodgkin clássico
Além da sua peculiaridade histogenética, o LHc é uma doença variável do ponto de
vista histopatológico sendo classificado em 4 subtipos: esclerose nodular (EN), celularidade
mista (CM), depleção linfocitária (DL) e rico em linfócitos (RL) (SWERDLOW et al., 2008).
Esta variabilidade pode ser causada por vários motivos, entre elas: a condição imune
específica na transformação maligna, a relação com a idade do paciente no momento de
desenvolvimento da doença e a associação com o EBV (POPPEMA, 2005; BAUMFORTH et
al., 2008; BARROS; HASSAN; NIEDOBITEK, 2011).
A EN representa aproximadamente 70% dos casos de LHc em países desenvolvidos e
em adultos jovens é caracterizada por um padrão de crescimento nodular com bandas de
colágeno circundando, ou tendo a circundar os nódulos (KUPPERS, 2009a). Na EN, o
número de células H-RS é variável, sendo observado muitas vezes, um halo claro ao redor
destas células (células lacunares). As células neoplásicas estão embebidas num infiltrado
inflamatório composto por células inflamatórias (LUKES; BUTLER, 1966; BUTLER;
PUGH, 1993).
A EN pode, por sua vez, ser classificada em dois subgrupos: grau I, caracterizada por
escassas células H-RS junto a um intenso infiltrado inflamatório não neoplásico, e grau II,
caracterizado por apresentar um maior número de células H-RS acompanhado de uma
alteração da morfologia ou redução do número dos linfócitos infiltrantes (MACLENNAN et
al., 1992; SWERDLOW et al., 2008). Originalmente, esta divisão foi correlacionada com a
sobrevida dos pacientes, sendo o grau II associado com um pior prognóstico (BENNETT; TU;
HUDSON, 1981; BENNETT et al., 1983; MACLENNAN et al., 1992), mas na atualidade,
esta graduação perdeu seu impacto prognóstico devido à melhora global da resposta aos
tratamentos poliquimioterápicos (VAN SPRONSEN et al., 1997).

5
A CM é o subtipo mais comum nos países em desenvolvimento, nas crianças e nos
adultos mais velhos e nos casos EBV+ (CARBONE et al., 2011). A CM é caracterizada pela
ausência de bandas colágenas e nódulos, assim como do espessamento capsular, embora possa
haver a presença de fibrose. Assim como na EN, o número de células H-RS é pequeno e estas
células estão embebidas num infiltrado inflamatório composto principalmente por linfócitos,
plasmócitos, eosinófilos, neutrófilos e macrófagos (SWERDLOW et al., 2008).
Os subtipos RL e DL são os menos frequentes dentre os subtipos histológicos do LHc.
O RL representa 5% dos casos e exibe escassas células H-RS, fundo nodular (mais comum)
ou difuso de pequenos linfócitos, com pouca quantidade de neutrófilos e eosinófilos. Uma
proporção de células H-RS pode se assemelhar às células predominantes linfocitárias ou às
células lacunares mononucleares (SWERDLOW et al., 2008). Já o subtipo DL é uma forma
difusa do LHc e representa menos que 1% dos casos. Caracteriza-se por ser rico em células H-
RS e uma por apresentar uma diminuição do número de linfócitos não neoplásicos. Assim
como na CM, apresenta-se com maior frequência nos países em desenvolvimento e nos
indivíduos HIV positivos (SWERDLOW et al., 2008).
Em relação às diferenças histopatogênicas entre os dois subtipos principais do LHc,
EN e CM, BIRGERSDOTTER et al. (2009), através de análises de expressão gênica
comparativas, sugeriu que os dois subtipos têm características que recapitulam as diferentes
fases de um processo de cicatrização normal; enquanto que na EN é observado um perfil de
remodelamento da matriz extracelular, na CM observa-se a presença de uma resposta
citotóxica semelhante a outros linfomas.
1.3 Resposta clínico-terapêutica no linfoma de Hodgkin clássico
O LHc é caracterizado por ser uma doença heterogênea tanto do ponto de vista
histopatológico quanto clínico. O estadiamento Ann Arbor (CARBONE, et al., 1971) para o
LH, gradua esta doença em 4 níveis de acordo com número e a localização de cadeias
linfonodais envolvidas, sendo caracterizados os estádios I e II como doença localizada, e os
estádios III e IV, como doença avançada.
Na tentativa de se estabelecer uma terapia risco-específica, o LH é dividido em grupos
de risco, combinando-se o estadiamento clínico com a presença dos sintomas “B” (febre
maior que 38ºC por pelo menos 3 dias consecutivos, sudorese noturna e a perda de peso
inexplicada maior que 10% do peso corpóreo nos últimos 6 meses). Dois grandes grupos são

6
reconhecidos pela maioria dos protocolos de tratamento: desfavorável (estadiamentos IIB,
IIIB e IV) e favorável (demais estadiamentos) (VASSILAKOPOULOS et al., 2005).
O tratamento é baseado em quimioterapia, podendo ou não ser utilizada a radioterapia
na doença localizada (SPECHT et al., 1992). Os regimes quimioterápicos atuais são baseados
na utilização de antraciclinas e agentes alquilantes em baixas doses. A utilização destes
protocolos tem garantido uma sobrevida livre de eventos (SLE) em 5 anos que variam entre
75 a 97% (VASSILAKOPOULOS et al., 2005). Entretanto, cerca de 20 a 30% dos pacientes
podem apresentar uma recaída ou progressão da doença, sendo que para estes casos não
existem opções terapêuticas definidas e eficazes (DIEHL; THOMAS; RE, 2004).
Um sistema de pontuação clínica (escore), denominado escore de prognóstico
internacional (IPS: do inglês, International Prognostic Score) foi desenvolvido para predizer
o prognóstico de pacientes com estádios avançados, utilizando dados clínicos em conjunto
com dados laboratoriais e demográficos, como a idade do paciente, sexo, estádio da doença,
níveis séricos de albumina, hemoglobina, contagem de leucócitos e linfócitos
(HASENCLEVER; DIEHL, 1998). Este sistema é utilizado em adultos, entretanto, sua
aplicação no grupo de crianças não foi validado até o momento.
Diferentes estudos têm sido conduzidos visando desenvolver marcadores biológicos
capazes de identificar o grupo de pacientes com alto risco de recaída ou de progressão da
doença, permitindo uma intensificação da terapia adaptada a risco. Da mesma maneira, é
extremamente necessário beneficiar o grupo de menor risco com terapias menos agressivas,
diminuindo assim o risco de toxicidade e efeitos colaterais, característicos do esquema de
tratamento quimioterápico do LHc. Dentre os marcadores biológicos, há diversos tipos que
podem ser classificados em marcadores séricos, fatores genéticos ou marcadores tumorais
focados tanto na célula H-RS quanto nas células do infiltrado tumoral (HSI, 2008).
1.4 O vírus Epstein-Barr no linfoma de Hodgkin clássico
O EBV é um γ-herpesvírus que infecta assintomaticamente mais de 90% da população
humana (CRAWFORD, 2001). Na infecção primária, seguida à sua transmissão via a saliva, o
EBV ingressa no organismo pelo epitélio da orofaringe infectando linfócitos B circulantes e
estabelecendo latência nas células B de memória. Esta primo-infecção ocorre em idades
variáveis, dependendo das condições sócio-econômicas das populações, e embora a infecção
seja quase sempre controlada ao nível subclínico, quando ocorre durante a adolescência e

7
juventude pode ter como consequência uma doença conhecida como mononucleose infecciosa
(MNI) (FIELDS et al., 2001). No entanto, o EBV pode também estar associado causalmente
a processos neoplásicos como o linfoma de Burkitt endêmico, carcinoma de nasofaringe,
doença linfoproliferativa pós-transplante e uma fração dos casos com LHc. Por isto, a OMS
considera este vírus como carcinógeno humano do grupo I (IARC WORKING GROUP ON
THE EVALUATION OF CARCINOGENIC RISKS TO HUMANS, 1997; GANDHI;
TELLAM; KHANNA, 2004).
A hipótese da associação causal entre o EBV e o LHc foi baseada nas evidências
epidemiológicas de que uma história anterior de MNI aumentava em 5 vezes o risco de
desenvolvimento de LHc (HJALGRIM et al., 2000; MASSINI; SIEMER; HOHAUS, 2009),
e na detecção da natureza clonal da infecção das células H-RS pelo EBV em uma fração dos
casos (CRAWFORD, 2001). Em 2005, o papel causal do EBV na transformação das células
de H-RS foi demonstrado em modelos experimentais (BECHTEL et al., 2005; MANCAO et
al., 2005) passando a ser reconhecido como agente etiológico da doença.
O modelo mais aceito de manutenção do estado latente do EBV propõe que este vírus
explora a biologia da célula B para chegar ao comportamento de memória onde estabelece
latência, atravessando o CG (THORLEY-LAWSON, 2001). No CG, para que haja uma
reação folicular de sucesso, a célula B precisa dos estímulos provenientes da célula T auxiliar
(Th: do inglês T helper) através do CD40L, além de sinais tônicos fornecidos pelo BCR
(imunoglobulina de superfície) (VIKSTROM; TARLINTON, 2011). Quando um destes sinais
falha, a célula B é enviada para apoptose (KUPPERS, 2009a; NUTT; TARLINTON, 2011).
O EBV é capaz, por si só, de fornecer estes sinais, através de suas duas proteínas de
membrana, LMP1 e LMP2A (KAPATAI; MURRAY, 2007; KUPPERS, 2009b). No caso de
uma célula B destinada a apoptose, o vírus pode “enganar” o sensor de apoptose levando à
sobrevida destas células, com alto potencial de transformação. Este é o mecanismo proposto
para a origem do LHc EBV+.
O padrão de latência no CG é conhecido como latência tipo II, que inclui a expressão
das proteínas EBNA1, LMP1 e LMP2A, além dos RNAs não traduzidos EBER1 e 2. As
proteínas virais de membrana LMP1 e LMP2A possuem a capacidade de mimetizar a proteína
receptora de membrana CD40 (TNFR) e o BCR, respectivamente (THORLEY-LAWSON,
2001). Sendo assim, tanto LMP1 quanto CD40, através de seu ligante a nível extracelular
(CD40L), são capazes de induzir a ativação da via de fatores de transcrição nuclear κB (via

8
NF-κB), o que torna esta via permanentemente ativa nas células H-RS, seja de uma maneira
T-dependente no caso de CD40 ou T-independente (constitutiva) na LMP1 (POPPEMA,
2005)
A via NF-κB regula a expressão de diversos genes envolvidos na proliferação celular e
apoptose na resposta inflamatória, resistência a drogas, citocinas, entre outras (HINZ et al.,
2002) e é observada constitutivamente ativada tanto em pacientes EBV positivos como
negativos, indicando sua importância para o desenvolvimento da doença (YOUNG;
RICKINSON, 2004).
Ainda com relação à latência II do EBV, recentemente foi mostrado que a manutenção
deste padrão de latência no LHc é regulada pela interação da célula tumoral com estímulos
das células do microambiente, especificamente células T CD4+, mediado por IL-21 (KIS et
al., 2010).
Em relação aos subtipos de LHc, o vírus EBV tem sido associado majoritariamente
com o subtipo de CM (CARBONE et al., 2009) e a idades inferiores a 10 anos
(ARMSTRONG et al., 1993; CHABAY et al., 2008).
No que se refere à resposta terapêutica, aparentemente o EBV tem efeitos diferentes
no LHc de acordo com a idade do paciente: nas crianças apresenta um papel favorável
enquanto que uma pior resposta é observada no grupo de adultos (CLAVIEZ et al., 2005;
JARRETT et al., 2005; KEEGAN et al., 2005).
1.5 Proliferação e escape da apoptose no linfoma de Hodgkin clássico
Os estudos no LHc trazem junto de si um grande desafio metodológico, visto a
dificuldade de abordar de maneira teórica e funcional uma doença em que a população
tumoral não ultrapassa 2% da massa tumoral, e além disso, está composta por duas
populações neoplásicas, uma constituída por células que mantém sua capacidade proliferativa,
e uma população que perdeu a capacidade de proliferar, como as RS. De fato, as células H-RS
expressam marcadores de proliferação celular (Ki67, PCNA, TOP2A, etc.) em diversas
proporções indicando a entrada no ciclo celular; porém, não há correlação entre o nível de
expressão destes marcadores e o número de células tumorais (BAI et al., 2006; BARROS et
al., 2010). Isto sugere que a indução da replicação do DNA está desacoplada da divisão
celular. De fato, sabe-se que a multinucleação e poliploidia das células H-RS é devida à
parada mitótica seguida por defeitos na citocinese e/ou endoreduplicação do material

9
genético, assim como estádios mitóticos abortivos e defeitos na passagem de metáfase para
anáfase (KUPPERS, 2001; PARK et al., 2008; BARROS; ZALCBERG; HASSAN, 2009).
As células H-RS exibem alterações nas principais vias do ciclo celular. Os principais
mecanismos descritos até o momento são a desregulação de genes relacionados aos
checkpoints G1/S (por exemplo, ciclinas D1, D2 e D3, ciclina E, CDK2) e G2/M (por
exemplo, ciclinas A, B1, C, CDK4, BUB1) (BAI et al., 2005), assim como a inativação da
supressão tumoral através das vias p14ARF/HDM2/p53/p21(Cip1/WAF1), p16-INK4a/Rb e
p27KIP1 (TZANKOV et al., 2005; SANCHEZ-AGUILERA; MONTALBAN; et al., 2006).
Mesmo com grandes alterações na maquinaria do ciclo celular, mutações em p53 são
significativamente menores no LHc do que em outras neoplasias (ELENITOBA-JOHNSON
et al., 1996; MONTESINOS-RONGEN et al., 1999), e o modelo que vem sendo proposto
para a inativação da via de p53 nas células H-RS seria a perturbação estrutural ou topológica
de proteínas nucleares que regulam a estabilidade de p53 (GARCIA et al., 2002), tais como
HDM2 e p14ARF.
Em relação à apoptose, é sabido que no LHc há duas vias principais ativas,
tipicamente envolvidas no controle da apoptose nos linfócitos, como a via FAS-FASL e a via
endógena da família de proteínas BCL2 (BAI et al., 2005). As proteínas da família BCL2
podem ser indutoras e repressoras de morte celular programada e participam ativamente da
sua regulação. Proteínas como BCL2 e BCL2L1 (Bcl-XL) inibem a apoptose pois previnem a
liberação de citocromo c e são consideradas reguladores anti-apoptóticos. Por outro lado,
BAX, BID e BAK são proteínas pró-apoptóticas. Dada a sua importância no controle da
apoptose nos linfócitos, BCL2 e BCL2L1 estão constantemente alteradas em diversos tipos de
neoplasias hematológicas. No LH, um aumento nos níveis de BCL2, BCL2L1, BAX, BAD e
uma diminuição de BAK e BID tem sido descritos nas células de H-RS (GARCIA et al.,
2003; SANCHEZ-AGUILERA; MONTALBAN; et al., 2006; BAI et al., 2007). Apesar do
foco dos estudos sobre apoptose nas neoplasias hematológicas ser em BCL2, a alta expressão
de BCL2L1, BAX e BAD nas células H-RS indicam que estas proteínas podem ter papeis
predominantes na regulação da apoptose no LHc (BAI et al., 2007).
A execução do apoptose depende do funcionamento adequado de seus efetores,
especialmente das caspases. As caspases (cysteine-aspartic-acid-proteases) são um grupo de
proteínas que pertencem à família das cisteínas proteases (caracterizadas pela presença de
uma cisteína no sítio ativo) capazes de reconhecer e clivar substratos que possuem um resíduo
10
de ácido aspártico (D'AMELIO; CAVALLUCCI; CECCONI, 2010). São descritas 14
caspases humanas, entretanto, apenas seis (caspases -3, -6, -7, -8, -9 e -10) participam do
processo de apoptose. As outras caspases tem suas funções descritas na maturação de
citoquinas e seu papel na apoptose ainda não esta esclarecido (DENAULT; SALVESEN,
2002; BOATRIGHT; SALVESEN, 2003). A Caspase-3 (CASP3) é a principal efetora da
apoptose, clivando (ou modificando a função) de substratos celulares e sendo um importante
ponto de convergência das vias de apoptose extrínseca (iniciada por receptores na superfície
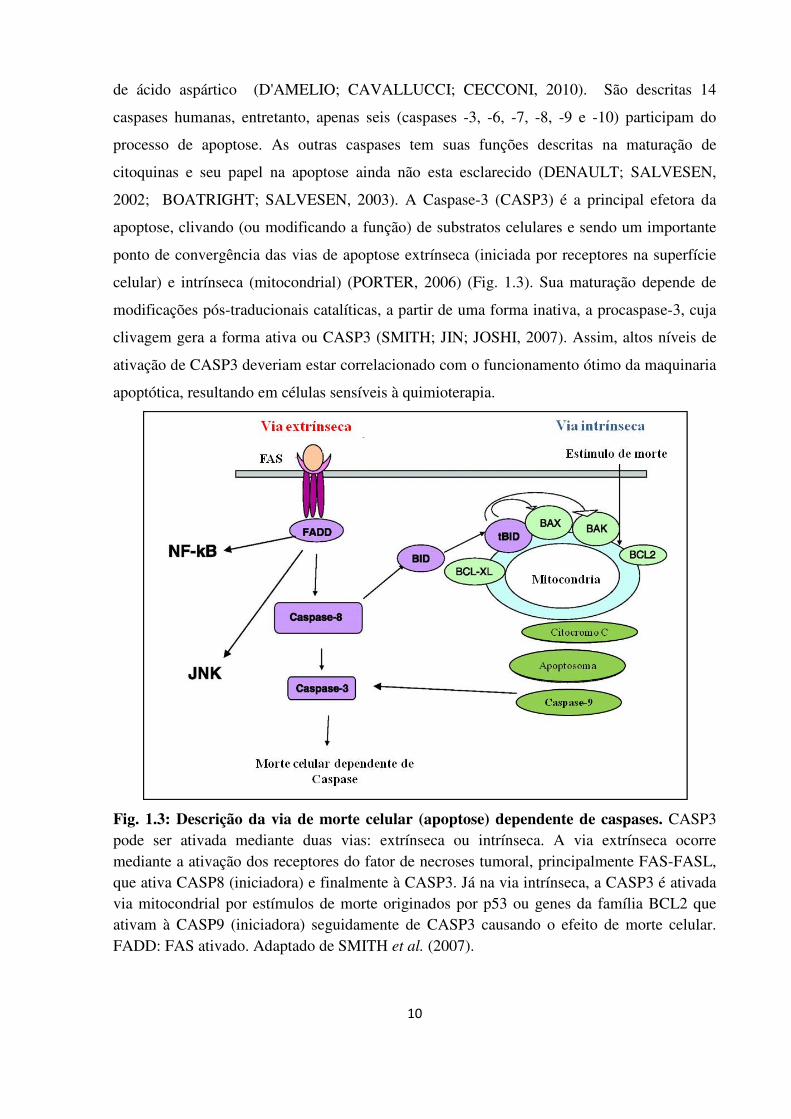
celular) e intrínseca (mitocondrial) (PORTER, 2006) (Fig. 1.3). Sua maturação depende de
modificações pós-traducionais catalíticas, a partir de uma forma inativa, a procaspase-3, cuja
clivagem gera a forma ativa ou CASP3 (SMITH; JIN; JOSHI, 2007). Assim, altos níveis de
ativação de CASP3 deveriam estar correlacionado com o funcionamento ótimo da maquinaria
apoptótica, resultando em células sensíveis à quimioterapia.
Fig. 1.3: Descrição da via de morte celular (apoptose) dependente de caspases. CASP3 pode ser ativada mediante duas vias: extrínseca ou intrínseca. A via extrínseca ocorre mediante a ativação dos receptores do fator de necroses tumoral, principalmente FAS-FASL, que ativa CASP8 (iniciadora) e finalmente à CASP3. Já na via intrínseca, a CASP3 é ativada via mitocondrial por estímulos de morte originados por p53 ou genes da família BCL2 que ativam à CASP9 (iniciadora) seguidamente de CASP3 causando o efeito de morte celular. FADD: FAS ativado. Adaptado de SMITH et al. (2007).

11
O papel das caspases não tem sido relacionado unicamente à morte celular, tendo sido
envolvidas em processos não apoptóticos como proliferação ou diferenciação nas células
hematopoiéticas, musculares e do sistema nervoso central (SCHWERK; SCHULZE-
OSTHOFF, 2003; D'AMELIO et al., 2010).
Em relação à expressão de CASP3 pelas células tumorais do LHc, diferentes resultados
tem sido relatados, desde uma alta expressão na maioria das células (67% de células
expressando CASP3) (BAI et al., 2007) a um menor número de células expressando-a (0-
13%) (DUKERS et al., 2002). Sua expressão foi correlacionada com outros marcadores da
apoptose, como TUNEL (do inglês, Terminal deoxynucleotidyl transferase dUTP nick end
labeling), clivagem de PARP (família de proteínas poly ADP ribose polymerase), entre outros
(DUKERS et al., 2002; BAI et al., 2005; BAI et al., 2007). No entanto, até o momento não
existe uma caracterização da expressão de CASP3 e sua relação com a apoptose nas
subpopulações celulares do microambiente tumoral do LHc.
1.5.1 Prognóstico associado à proliferação e apoptose no linfoma de Hodgkin
clássico
Numa visão clássica do câncer, considera-se que as alterações da proliferação e
apoptose das células tumorais são determinantes na resposta destas células aos tratamentos.
No LHc, a busca por marcadores terapêuticos associados às células H-RS tem sido intensa,
para uso na estratificação de pacientes ao diagnóstico e adequação da intensidade das terapias
administradas, visto a baixa idade da maioria dos pacientes e a toxicidade das drogas básicas
utilizadas no tratamento (SCHWARTZ, 2003).
A desregulação de genes relacionados aos checkpoints G1/S e G2/M foi associado ao
prognóstico, como será descrito com mais detalhe na secção 1.8. Um conceito importante, e
ainda não provado, na biologia do LHc é que os defeitos do ciclo celular estariam associados
de forma intrínseca à aquisição do fenótipo de resistência a drogas (SANCHEZ-AGUILERA
et al., 2006).
Por outro lado, a positividade na imunoexpressão de p53 no LHc pode variar de 30% a
93% (MORENTE et al., 1997; GARCIA et al., 2003; MONTALBAN et al., 2004) e o
prognóstico desta expressão é controverso, principalmente devido à ausência de um ponto de
corte padronizado para a distinção entre casos positivos e negativos (MONTALBAN et al.,
2004; SPECTOR et al., 2005).

12
BCL2 tem sido alvo de vários estudos a fim de elucidar se a presença desta proteína
anti-apoptótica poderia influenciar a sobrevida no LHc com resultados contraditórios. Em
alguns estudos, a imunoexpressão de BCL2 esteve associada a uma pior sobrevida (BRINK et
al., 1998; SMOLEWSKI et al., 2000; SUP et al., 2005), enquanto em outros, não houve
diferenças na resposta ao tratamento (SMOLEWSKI et al., 1998; SPECTOR et al., 2005).
Em relação a CASP3, sua expressão nas células H-RS determinada por
imunohistoquímica (IHQ) foi correlacionada com o prognóstico; altos números de células
tumorais expressando CASP3 (≥5%) está correlacionada com uma boa resposta suportando a
noção que uma ativação da cascata da apoptose é essencial para o efeito da quimioterapia na
morte celular (DUKERS et al., 2002). Já BAI et al. (2007) não encontraram associação dos
níveis de expressão de CASP3 com o prognóstico da doença.
É possível que a combinação de marcadores relacionados ao funcionamento do ciclo
celular e a capacidade de morte celular possam ser mais hábeis no delineamento prognóstico
dos pacientes com LHc. Neste sentido, MONTALBAN et al. (2004) mostraram que a
expressão pelas células H-RS de p53, BCL2 e apoptose (TUNEL) poderia ser agrupada num
escore prognóstico capaz de definir grupos de risco biológicos com diferenças na sobrevida. É
importante ressaltar que, apesar de todos os esforços de pesquisa, nenhum destes marcadores
encontra-se validado para sua aplicação clínica nos dias de hoje.
1.6 O microambiente tumoral no linfoma de Hodgkin clássico
O recrutamento de células do sistema imune observado em muitos tumores,
especialmente nas neoplasias linfóides e, dentro delas, de maneira excepcional no LHc, indica
um papel primário dos processos inflamatórios e de vigilância tumoral na constituição do
microambiente, onde é possível enxergar pelo menos dois processos em conflito. Por um lado,
a célula H-RS produz diferentes tipos de citocinas e quimiocinas cuja função pode ser
autócrina (para seu próprio sustento) ou pode servir para recrutar células não malignas do
sistema imune, causando um processo inflamatório onde estas células atraídas contribuem à
manutenção e proliferação das células H-RS (Fig. 1.4) (ALDINUCCI et al., 2010). Por outro
lado, antígenos tumorais, ou, no caso do LHc EBV+, virais, podem estar deflagrando uma
resposta imune citotóxica tendente a combater as células tumorais. Mecanismos de contra-
insurgência destinados a suprimir, anergizar ou tolerizar esta resposta podem ser montados
pela célula tumoral. O resultante destes processos é o microambiente tumoral (MAT), que
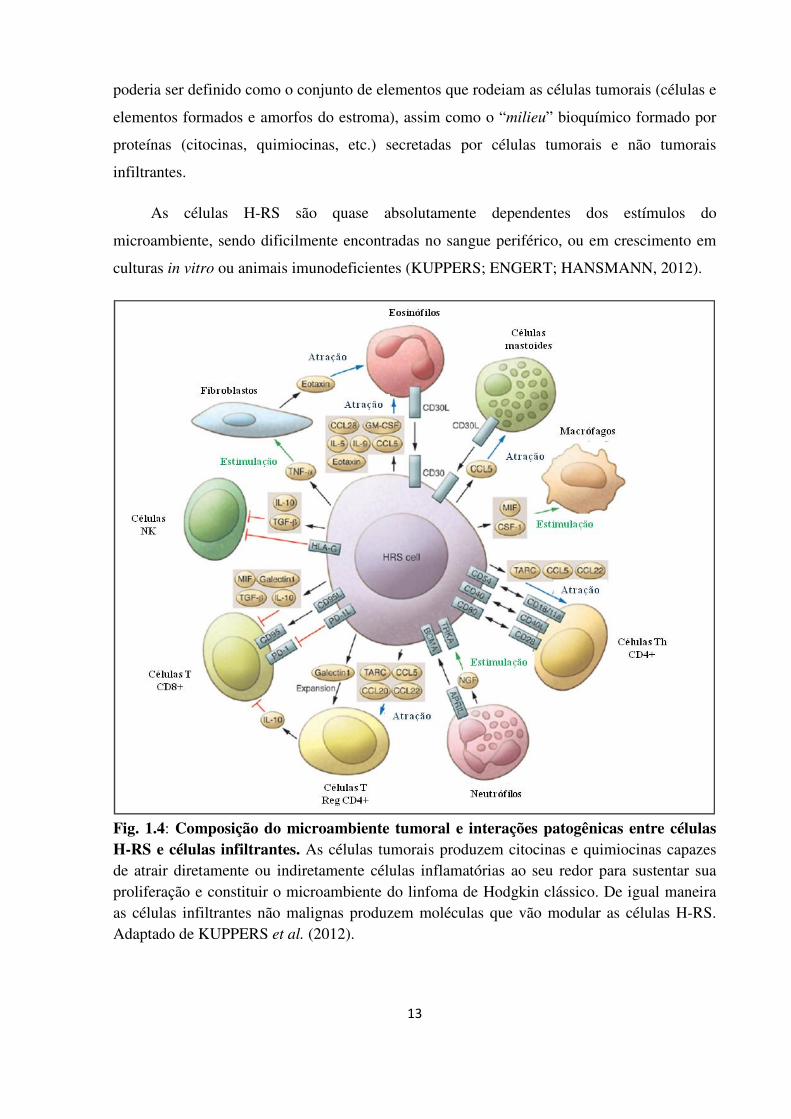
13
poderia ser definido como o conjunto de elementos que rodeiam as células tumorais (células e
elementos formados e amorfos do estroma), assim como o “milieu” bioquímico formado por
proteínas (citocinas, quimiocinas, etc.) secretadas por células tumorais e não tumorais
infiltrantes.
As células H-RS são quase absolutamente dependentes dos estímulos do
microambiente, sendo dificilmente encontradas no sangue periférico, ou em crescimento em
culturas in vitro ou animais imunodeficientes (KUPPERS; ENGERT; HANSMANN, 2012).
Fig. 1.4: Composição do microambiente tumoral e interações patogênicas entre células H-RS e células infiltrantes. As células tumorais produzem citocinas e quimiocinas capazes de atrair diretamente ou indiretamente células inflamatórias ao seu redor para sustentar sua proliferação e constituir o microambiente do linfoma de Hodgkin clássico. De igual maneira as células infiltrantes não malignas produzem moléculas que vão modular as células H-RS. Adaptado de KUPPERS et al. (2012).

14
O MAT é formado por uma grande diversidade de células inflamatórias infiltrantes
como células T (reguladora, citotóxicas, auxiliares), células B, plasmócitos, monócitos,
macrófagos, eosinófilos, neutrófilos, células mastocitas, entre outras (ALDINUCCI et al.,
2010), todas em quantidades variáveis. Foi mostrado que a composição do MAT depende do
subtipo histológico, da idade do paciente e da presença do EBV (MAGGIO et al., 2002;
ALDINUCCI et al., 2010; STEIDL; CONNORS; GASCOYNE, 2011)
Os linfócitos T são uma parte importante do infiltrado inflamatório do LHc. Estas
células podem ser classificadas em diferentes subtipos dependendo da sua função: células T
citotóxicas, células Th tipo 1, 2, 17 (existem outros tipos que escapam ao objetivo desta
revisão), células T reguladoras e células T de memória.
Os linfócitos citotóxicos CD8+ (CTLs) tem um papel na resposta imune antiviral,
assim como também são responsáveis pela vigilância e imunidade antitumoral (SUN;
LANIER, 2011). Sua ação é exercida de maneira direta através da citotoxicidade mediada por
sustâncias citolíticas contra as células alvo, como perforina, TIA1 e granzima B (GRZB)
(ANIKEEVA; SYKULEV, 2011; SANT; MCMICHAEL, 2012).
As células Th, CD4+ tem a função geral de ativar as células apresentadoras de
antígenos (células dendríticas, macrófagos ou células B) após reconhecimento do antígeno
restrito pelo complexo principal de histocompatibilidade (MHC; do inglês, major
histocompatibility complex) de classe II, que levam em primeiro lugar ao aumento de
expressão de co-receptores para ativação das células T efetoras, e em segundo lugar, à
polarização da resposta imune para pro-inflamatória ou supressora, dependendo das citocinas
secretadas. As células Th1 são produtoras de interleucina (IL) 2, interferon gamma (IFN-γ),
TNF e IL-12, e podem ser identificadas pela expressão nuclear do fator de transcrição (FT)
TBX21 (TBET). O papel funcional destas células é principalmente polarizar a resposta pró-
inflamatória acompanhada de uma ativação citotóxica e de macrófagos de tipo M1
(DENARDO; ANDREU; COUSSENS, 2010; VESELY et al., 2011). Já as Th2 são
produtoras principalmente de IL-4 e IL-10 sendo reconhecida principalmente pelo fator de
transcrição GATA3. O FT MAF é responsável pela regulação da expressão de IL10 e pode ser
considerado marcador de célula T CD4+ Th2/reguladora (ZHU; PAUL, 2008).
As células T reguladoras (Treg) caracterizam-se pelo imunofenótipo CD4+ CD25+
FOXP3+ e por apresentar uma função de imunoregulação, via principalmente a expressão de

15
IL10, suprimindo a proliferação de outros tipos celulares como CTLs. O FT FOXP3 é capaz
de inativar a via NF-κB assim como atuar na repressão de IL-2, IL-4 ou IFN-γ evitando a
polarização de outras células T (OBERG et al., 2011).
Um linfonodo normal está composto por um grande número de linfócitos B. No LHc,
estas células fazem parte do MAT, mas ainda seu papel e envolvimento no tumor é
desconhecido. Os linfócitos B podem atuar como células efetoras da imunidade humoral,
expressar citocinas e quimiocinas importantes nos processos de diferenciação, proliferação e
resposta inflamatória da célula B, ou ser apresentadoras de antígeno nas reações do centro
germinativo (STEIDL; CONNORS; et al., 2011). O desenvolvimento de melhores
marcadores celulares de diferenciação permitirá compreender melhor o envolvimento desta
linhagem nos processos tumorais.
Os monócitos/macrófagos são células caracterizadas por atuar na primeira linha de
defesa do organismo frente a patógenos e apresentam um papel importante tanto nos processo
de inflamação como na resposta imune do hospedeiro. Seu papel na resposta imune inata é
caracterizado por sua capacidade de reconhecimento de antígenos, enquanto que atuam como
células apresentadoras de antígenos para as células T na resposta imune adaptativa (BISWAS;
MANTOVANI, 2010; MANTOVANI et al., 2013).
No contexto tumoral, os monócitos são recrutados e transformados em macrófagos
para atuar como células efetoras em diferentes situações como na inflamação, na reparação
tecidual, e para produzir uma resposta ou erradicar uma infecção (MANTOVANI et al.,
2002). Por outro lado, as células macrofágicas se caracterizam por apresentar uma alta
plasticidade (MANTOVANI; LOCATI, 2009), podendo ser polarizadas dependendo do
estímulo encontrado: patógenos, tecido danificado, sub-populações de células T, mudando sua
função imunoreguladora para pró-inflamatória ou vice-versa (GUIDUCCI et al., 2005).
Os macrófagos podem reprogramar sua função e ser polarizados para diferentes
fenótipos:
1 Função pró-inflamatória causada por uma resposta Th1 a IFN-γ com a fosforilação de
STAT1 (MANTOVANI et al., 2002; SAHA et al., 2010).
2 Função imunoreguladora de remodelação tecidual, angiogênese e progressão tumoral
modulada por uma resposta Th2 (CAO et al., 2005; BISWAS; MANTOVANI, 2010).

16
No modelo do LHc, os macrófagos associados ao tumor (TAM) são considerados uma
população especial que expressam a proteína de membrana CD163, e desenvolvem funções
na resposta inflamatória e imune adaptativa, na remodelamentoo tecidual e no reparo
(MANTOVANI et al., 2002; ALLAVENA et al., 2008; BISWAS; MANTOVANI, 2010). O
estado de polarização intratumoral dos macrófagos no LHc não foi descrita até o momento.
Os eosinófilos são leucócitos com características multifuncionais que participam da
resposta imune inata, mas podem engajar-se em um “cross-talk” com a resposta adaptativa,
participando da resposta inflamatória, da regulação da resposta imune e da homeostase
tecidual. No caso do LHc, eles podem ser encontrados em grandes quantidades ao redor da
célula tumoral, sendo atraídos principalmente pela eotaxina produzida pelos fibroblastos e
interagem com a célula H-RS através do ligante da proteína de membrana CD30 (CD30L),
que reconhece o receptor CD30 na célula tumoral (ALDINUCCI et al., 2010; GATAULT et
al., 2012).
1.7 Fatores prognósticos associados ao microambiente tumoral no linfoma de
Hodgkin clássico
A composição do MAT no LHc tem sido relacionada à resposta terapêutica. Em
termos gerais, um maior número de CTLs (CD8+, TIA1+, GRZB+) e um menor número de
células Treg (CD4+ CD25+ FOXP3+) estão associadas a um pior prognóstico (ALVARO-
NARANJO et al., 2005; ALVARO et al., 2005; KELLEY et al., 2007; TZANKOV et al.,
2008). Isto é o oposto ao observado na maioria dos tumores sólidos, onde um pior prognóstico
é descrito associado a um maior número de células Treg (DELEEUW et al., 2012), em
concordância com um modelo de resposta imune citotóxica suprimida pelas Treg. Nas
neoplasias hematológicas, o observado aponta para a necessidade de refinar os conceitos da
resposta imune antitumoral baseado na imunovigilância.
O papel dos TAMs ainda é controverso, visto que a maioria dos trabalhos observou
uma associação desfavorável entre macrófagos CD68+ ou CD163+ e a sobrevida dos
pacientes adultos (SANCHEZ-AGUILERA; MONTALBAN; et al., 2006; STEIDL et al.,
2010; TZANKOV; MATTER; DIRNHOFER, 2010; KAMPER et al., 2011; TAN et al.,
2012; DEAU et al., 2013), porém, vários outros estudos não identificaram uma associação
com o prognóstico (AZAMBUJA et al., 2012; HARRIS et al., 2012). Nas crianças, um pior

17
prognóstico foi associado à presença de um número maior de macrófagos CD163+
(BARROS; HASSAN; NIEDOBITEK, 2012).
Quanto ao papel dos eosinófilos infiltrantes, um número maior foi associado a um pior
prognóstico no subtipo EN, não havendo impacto aparente na sobrevida nos outros subtipos
histológicos (GATAULT et al., 2012). Em contraste, nos tumores sólidos um número maior
destas células está associado a um melhor prognóstico (MURDOCH et al., 2008; GATAULT
et al., 2012).
1.8 Estudos de expressão gênica no linfoma de Hodgkin
Nos últimos anos, muitos estudos de expressão gênica global foram conduzidos no
câncer com o intuito de identificar marcadores moleculares que possam diferenciar perfis de
expressão e, consequentemente, auxiliar no diagnóstico, subclassificação, e/ou escolha
terapêutica. Todavia, são poucos os estudos que conseguiram ser validados para aplicação na
rotina clínica (VAN 'T VEER et al., 2002; VAN DE VIJVER et al., 2002; MALUMBRES et
al., 2008; XIE et al., 2011). Metodologias como a PCR (reação em cadeira da polimerase)
quantitativa a partir de RNA (RT-qPCR, reação de transcrição reversa seguida por PCR)
permitem quantificar com maior precisão a expressão gênica e por isto são candidatas a serem
validadas na rotina clínica (TANG et al., 2012).
No LHc, visto a complexidade do tumor, os estudos de expressão gênica em grande
escala tem sido realizados por diferentes autores, tendo como foco tanto a célula H-RS como
o MAT. DEVILARD et al. (2002), realizaram o primeiro estudo de identificação de perfis de
expressão gênica, utilizando RNA obtido da massa tumoral total e “chips” caseiros em
suporte de nylon para aproximadamente 1000 genes, visando diferenciar níveis de expressão
gênica com papel na predição da evolução clínica de 21 pacientes. Os autores encontraram as
vias da ativação dos fibroblastos e angiogênese associadas a um desfecho clínico
desfavorável.
Outro estudo em pacientes com LHc avançado foi realizado para identificar perfis
diferenciais, utilizando um chip de microarray com 9348 genes. Neste estudo, as vias
relacionadas à resposta imune do hospedeiro, ao microambiente (macrófagos, células T e
dendríticas), ao ciclo celular e à apoptose estiveram associadas ao grupo com desfecho
desfavorável, enquanto que genes relacionados às células B, células apresentadoras de

18
antígenos, remodelamento tecidual e fibroblastos foram expressos pelo grupo com desfecho
favorável (SANCHEZ-AGUILERA et al., 2006).
Ainda através da metodologia de microarray (Affymetrix), outro estudo realizado com
63 pacientes adultos observou que genes relacionados às células B e apoptose estavam
associados a um melhor prognóstico, enquanto que os genes da matriz extracelular foram
relacionados a uma pior sobrevida como genes relacionados ao colágeno, entre outros
(CHETAILLE et al., 2009).
Em relação à identificação de perfis de expressão específicos da célula H-RS, dois
estudos de microarray foram realizados mediante o uso de microdissecção da célula tumoral.
O primeiro estudo visou identificar padrões de expressão das células H-RS, sem qualquer
preocupação com o prognóstico clínico, e identificou um grupo de casos cuja células H-RS
expressavam genes relacionados à diferenciação citotóxica, receptores de sinalização de
superfície e apoptose na célula tumoral, concomitante a baixa expressão de genes de
diferenciação B. Genes relacionados à função dos macrófagos, a angiogênese e da via de
sinalização NF-κB estiveram associadas à falha do tratamento primário. A superexpressão de
CSF1R (receptor para CSF1) foi observada nas células H-RS. Em diferenças de perfis de
expressão foram atribuídos a presença do EBV, embora não tenha sido identificado uma via
específica. (STEIDL et al., 2012).
O segundo estudo, apesar de não observar uma assinatura diferenciada para os casos
EBV+ e EBV-, identificou 14 genes diferencialmente expressos apenas nos casos EBV
positivos. Por outro lado, detectou dois grupos de pacientes diferenciados pela assinatura de
genes relacionados a MYC, IRF4 e NOTCH1, assim como diferenças entre células H-RS
provenientes do subtipo EN em relação às de CM (TIACCI et al., 2012).
Recentemente, um estudo utilizando a metodologia de RT-qPCR em array (TaqMan
Low Density Array, TLDA) a partir de RNA da massa tumoral total identificou um desfecho
desfavorável associado a super-expressão de genes das vias do ciclo celular, apoptose
mitocondrial e checkpoint mitótico (células H-RS) e de genes relacionados às células T,
monócitos e macrófagos (MAT) (SANCHEZ-ESPIRIDION et al., 2009). A partir deste
estudo, foi proposto um escore clinico para ser aplicado em pacientes adultos com LHc
avançado, cujo valor prognóstico foi baseado na alteração combinada da expressão gênica de
quatro vias funcionais (ciclo celular, apoptose, ativação de monócitos/macrófagos e o fator

19
regulador de interferon 4), onde uma superexpressão dos genes relacionados ao ciclo celular
e apoptose foram considerados como um fator negativo, enquanto que os relacionados ao
monócitos/macrófagos como um fator de proteção (SANCHEZ-ESPIRIDION et al., 2010).
1.9 Análises da expressão gênica em material fixado e impregnado em parafina
Tecidos fixados em formalina e impregnados em parafina (FFPE) são uma fonte
inestimável de material genético para estudos moleculares e uma maneira fácil e econômica
de guardar as amostras para que possam ser usadas em pesquisa ou refinamento diagnóstico.
O principal problema com estes materiais é que os ácidos nucleicos sofrem degradação, e a
etapa da fixação com o formaldeído produz ligações entre DNA, RNA e proteínas e adiciona
grupamentos monometil que comprometem a amplificação do RNA isolado (MASUDA et al.,
1999).
Em amostras provenientes de linfonodos, a extração de RNA de boa qualidade
apresenta maior dificuldade porque estes tecidos são muito frágeis e a morte celular começa
logo após a separação do tecido, e antes mesmo da ação do fixador ter início (ANTICA et al.,
2010). Como regra geral, nos RNAs provenientes de amostras FFPE, a amplificação das
reações de PCR ocorre com valores altos nos ciclos de quantificação (Cq), quando
comparados aos RNAs de amostras provenientes de tecidos frescos. Esta situação é causada
principalmente pela presença de inibidores que são coextraídos com o RNA (GODFREY et
al., 2000; KOCH et al., 2006; BAR; KUBISTA; TICHOPAD, 2012), afetando as análises de
qPCR no grupo de genes com baixo nível de expressão gênica.
Por tanto, vários estudos tem sido realizados visando a otimização tanto da extração
como da amplificação de RNA de material FFPE provenientes de amostras de linfonodos
(CHEN; BYRNE; LOSSOS, 2007; BOHMANN et al., 2009; SANCHEZ-ESPIRIDION et
al., 2010; ELLSWORTH et al., 2011; LENEHAN et al., 2012). Em termos gerais, estes
estudos são focados na avaliação de qualidade e amplificação do RNA.
1.10 Aplicação de análise da variância tipo nested
Uma abordagem muito interessante para avaliar a variabilidade introduzida em cada
etapa experimental de uma metodologia utilizada em biologia molecular foi proposta por
TICHOPAD et al. (2009) e KITCHEN; KUBISTA; TICHOPAD (2010) através de um
desenho ANOVA tipo nested que permite identificar a variação introduzida em cada etapa da

20
metodologia, fornecendo bases experimentais para realizar ajustes no desenho do estudo e nos
procedimentos técnicos.
ANOVA (do inglês, analysis of variance) é um método paramétrico usado para
comparar as médias de um grupo de amostras. A ideia subjacente é calcular as medias das
observações dentro de cada grupo e então comparar as variâncias entre essas médias à
variância media dentro de cada grupo. Sob a hipótese nula de que as observações nos
diferentes grupos tem a mesma média, a variância entre grupos não será diferente que as
observadas dentro de cada grupo. O estatístico calculado Fs é a razão da variância entre as
médias dos grupos dividida pela variância dentro dos grupos. No ANOVA de um fator há
uma variável contínua e uma variável nominal (fator). Múltiplas observações da variável
contínua devem existir para cada valor tomado pela variável nominal.
ANOVA tipo nested é uma extensão do ANOVA de um fator, onde cada grupo é
divido em subgrupos. No momento das análises os subgrupos são escolhidos de maneira
aleatória a partir de um conjunto maior de subgrupos possíveis (MCDONALD, 2009). O
primeiro desenho experimental foi desenvolvido por Ronald A. Fisher (FISHER, 1926), em
um método estruturado e organizado. Este desenho hierárquico permite conhecer os erros
introduzidos em cada nível onde é possível determinar as relações entre diferentes fatores e o
efeito dos processos experimentais em cada passo.
O desenho ANOVA tipo nested quando é aplicado ao desenho experimental em qPCR
permite quantificar os ruídos no nível biológicos e técnicos incorporados no processamento de
amostras e assim determinar a precisão dos ensaios da expressão gênica. Uma diferença real
nos níveis de expressão causados pela condição clínica ou por manipulações experimentais
somente poderia ser percebida se conseguem superar as variabilidades técnicas e biológicas
(TICHOPAD et al., 2009).
Não existem dados na literatura avaliando os processamentos utilizados para extração
de RNA a partir de material FFPE, assim como a variabilidade biológica subjacente a
amostras complexas como é o caso do LHc.

21
1.11 PCR quantitativa em tempo real (qPCR) como ferramenta para validação de
perfis de expressão gênica com aplicação clínica
A invenção da metodologia de PCR em tempo real por HIGUCHI et al. (1992) teve
um papel muito importante na pesquisa biológica e na biologia molecular. Esta ferramenta
tem uma grande aplicação na medicina, e em particular na oncologia, para pesquisa, busca e
desenvolvimento de marcadores biológicos para o diagnóstico, seguimento e predição de
doenças, como para patógenos associados.
A PCR quantitativa é conhecida pelo termo de qPCR, ou RT-qPCR quando é referida
à amplificação a partir de RNA retrotranscrito. Esta metodologia permite a monitoração da
reação de PCR enquanto esta ocorre, medindo a cinética da reação desde as fases iniciais,
ciclo após ciclo, o que possibilita a quantificação mediante o uso de corantes fluorescentes.
Existem duas tecnologias principalmente utilizadas nas reações de qPCR: 1)
TaqMan®, baseada no fenômeno do FRET mediante a utilização de uma sonda e; 2) corantes
intercalantes onde a emissão de fluorescência é evidenciada pela ligação a duplas fitas
(exemplo: Syber Green) (KUBISTA et al., 2006).
As reações de RT-qPCR para análises de expressão gênica podem ser afetadas pela
pureza e integridade do RNA inicial, sendo a qualidade do mesmo uma condição importante
para o desenvolvimento de uma metodologia ótima (FLEIGE; PFAFFL, 2006). A presença de
contaminantes como DNA, proteínas ou inibidores (fenol, sais, etanol) nas amostras altera o
rendimento tanto das reações de retrotranscrição (RT) como da PCR (STAHLBERG et al.,
2004; TICHOPAD; DIDIER; PFAFFL, 2004; SUSLOV; STEINDLER, 2005)
A cinética de uma reação de qPCR está composta por diferentes fases de acordo à
disponibilidade dos reagentes na reação, considerando a eficiência de duplicação de produtos
em cada ciclo: línea de base (do inglês, baseline), exponencial, linear e platô (do francês,
plateau) (KUBISTA et al., 2006) (Fig. 1.5).
• Fase de línea de base: se localiza nos primeiros ciclos da reação e a fluorescência
produzida não é capaz de se diferenciar do ruído da reação.
• Fase exponencial: durante esta fase da reação o produto acumulado é exatamente o
dobro do produto amplificado em cada ciclo, podendo ser considerada que a reação se
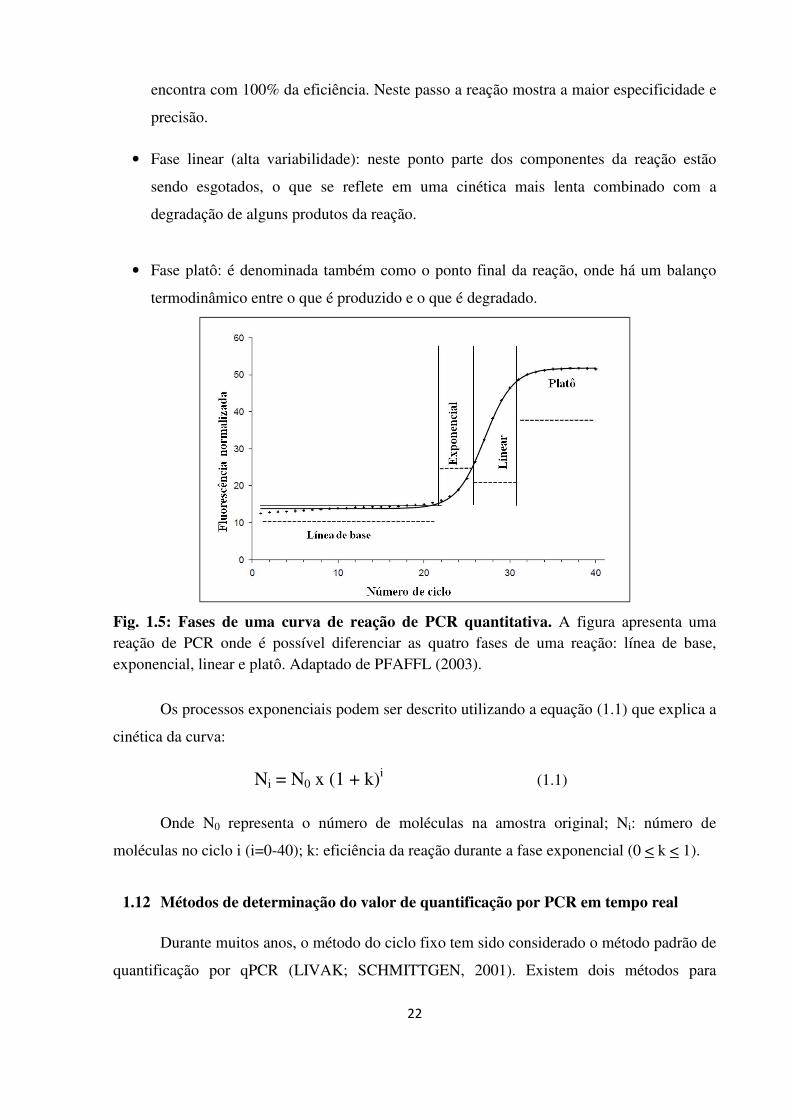
22
encontra com 100% da eficiência. Neste passo a reação mostra a maior especificidade e
precisão.
• Fase linear (alta variabilidade): neste ponto parte dos componentes da reação estão
sendo esgotados, o que se reflete em uma cinética mais lenta combinado com a
degradação de alguns produtos da reação.
• Fase platô: é denominada também como o ponto final da reação, onde há um balanço
termodinâmico entre o que é produzido e o que é degradado.
Fig. 1.5: Fases de uma curva de reação de PCR quantitativa. A figura apresenta uma reação de PCR onde é possível diferenciar as quatro fases de uma reação: línea de base, exponencial, linear e platô. Adaptado de PFAFFL (2003).
Os processos exponenciais podem ser descrito utilizando a equação (1.1) que explica a
cinética da curva:
Ni = N0 x (1 + k)i (1.1)
Onde N0 representa o número de moléculas na amostra original; Ni: número de
moléculas no ciclo i (i=0-40); k: eficiência da reação durante a fase exponencial (0 < k < 1).
1.12 Métodos de determinação do valor de quantificação por PCR em tempo real
Durante muitos anos, o método do ciclo fixo tem sido considerado o método padrão de
quantificação por qPCR (LIVAK; SCHMITTGEN, 2001). Existem dois métodos para

23
determinar este valor: 1) Método fit point, onde o sinal de fluorescência é capaz de atravessar
uma línea arbitrária denominada threshold que se encontra situada na região logarítmica (fase
exponencial da reação) (PFAFFL, 2004). O ciclo threshold (Ct) é definido como o número de
ciclo fracionar na região logarítmica-linear da reação, onde esta atinge um ponto pré-
estabelecido; 2) Método da segunda derivativa máxima, em que o ponto de quantificação é
situado onde a segunda derivativa da curva de intensidade de fluorescência atinge um valor
máximo calculado, conhecido na literatura como Cp (LUU-THE et al., 2005). Há na
atualidade uma tendência a denominar qualquer um dos mencionados como ciclo de
quantificação Cq (BUSTIN et al., 2009). Uma condição essencial para a adoção deste método
é que a eficiência das reações deve ser similar entre amostras padrões vs. desconhecidas, ou
gene de interesse vs. gene de referência.
Novas estratégias de determinação do ponto de quantificação baseados na cinética da
curva vem surgindo nos últimos anos, fundamentadas na observação da falta de similaridade
das cinéticas dos padrões em relação às amostras, principalmente quando estas apresentam
matrizes diferentes das dos controles (por exemplo, controles plasmidias e amostras de RNA
de parafina) que levam a diferenças no desempenho da reação de PCR, as quais não são
consideradas pelos métodos Cq. Os métodos baseados na cinética da curva de PCR buscam
superar a quantificação obtida a partir de um ponto único, visando capturar características da
cinética em todas as suas fases. A principal limitação é que a cinética da PCR não é
totalmente conhecida e não há modelo matemático satisfatório que possa descrever a reação
desde o início até a fase platô (SCHNELL; MENDOZA, 1997; KAINZ, 2000).
Por isto, o modelo clássico exponencial pode ser aplicado somente à parte inicial da
trajetória cinética da PCR. Para remediar isto, vários modelos empíricos não lineares foram
propostos, a maioria deles baseada numa eficiência constante na fase exponencial da reação,
mas com diferentes maneiras de realizar os cálculos da linha de base da fluorescência, a fase
exponencial, a eficiência, o limite de quantificação e o Cq. Alguns dos algoritmos propostos
são MAK2, FPK-PCR, LinRegPCR, Cy0 e Real time PCR-Miner (RUIJTER et al., 2013).
Neste trabalho, escolhemos comparar o método clássico de Cq (LIVAK; SCHMITTGEN,
2001) com o modelo denominado Cy0 (GUESCINI et al., 2008). Para entender este último,
apresentaremos inicialmente o modelo sigmoide de quatro parâmetros, estabelecido por
TICHOPAD et al. (2003).
Neste modelo, definido pela equação (1.2):
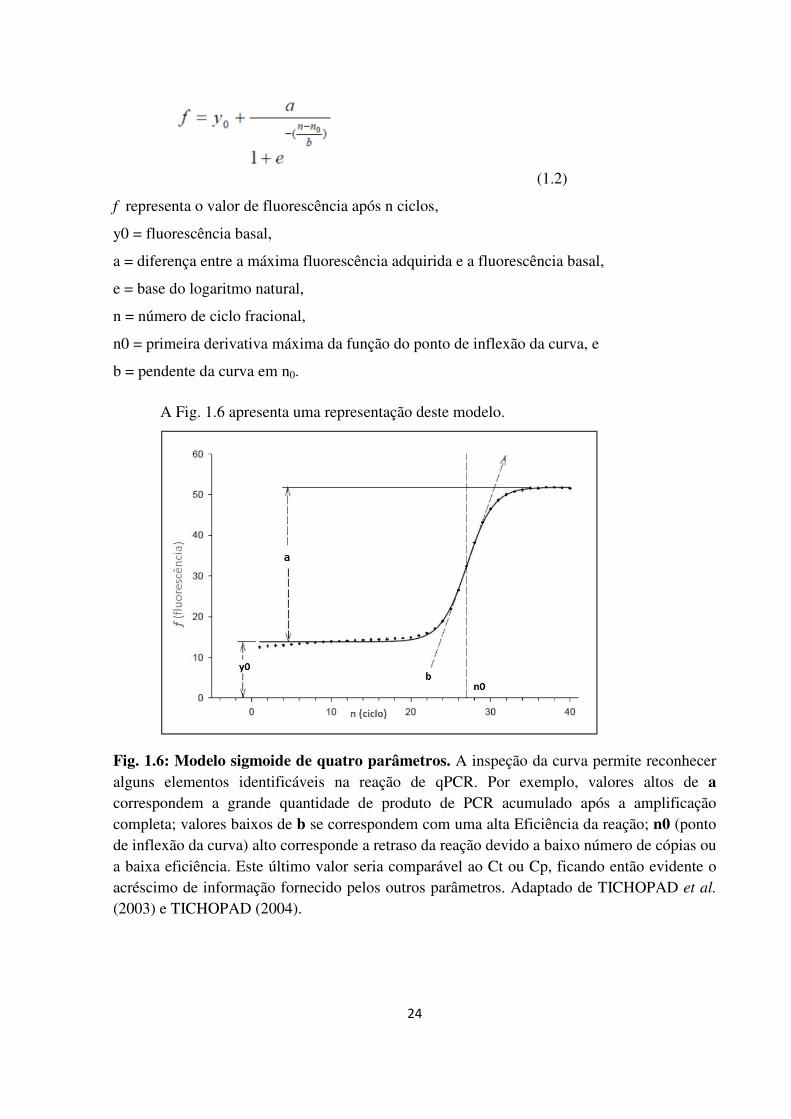
24
(1.2)
f representa o valor de fluorescência após n ciclos,
y0 = fluorescência basal,
a = diferença entre a máxima fluorescência adquirida e a fluorescência basal,
e = base do logaritmo natural,
n = número de ciclo fracional,
n0 = primeira derivativa máxima da função do ponto de inflexão da curva, e
b = pendente da curva em n0.
A Fig. 1.6 apresenta uma representação deste modelo.
Fig. 1.6: Modelo sigmoide de quatro parâmetros. A inspeção da curva permite reconhecer alguns elementos identificáveis na reação de qPCR. Por exemplo, valores altos de a correspondem a grande quantidade de produto de PCR acumulado após a amplificação completa; valores baixos de b se correspondem com uma alta Eficiência da reação; n0 (ponto de inflexão da curva) alto corresponde a retraso da reação devido a baixo número de cópias ou a baixa eficiência. Este último valor seria comparável ao Ct ou Cp, ficando então evidente o acréscimo de informação fornecido pelos outros parâmetros. Adaptado de TICHOPAD et al. (2003) e TICHOPAD (2004).
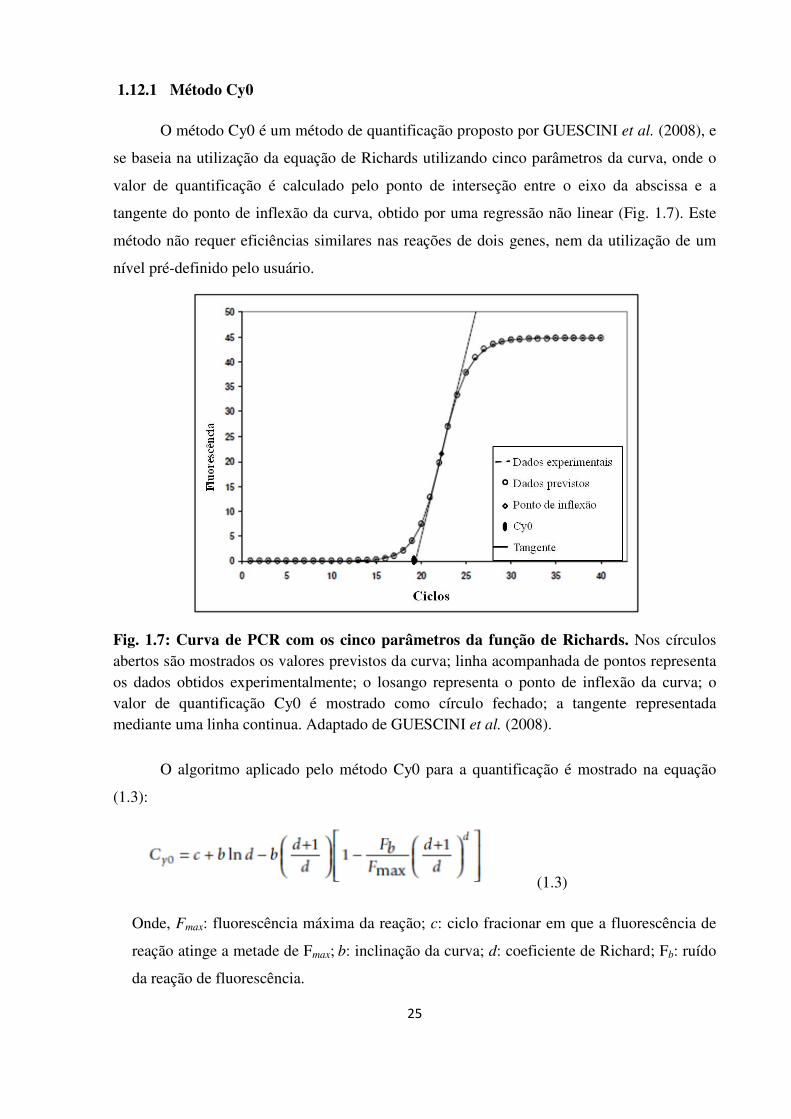
25
1.12.1 Método Cy0
O método Cy0 é um método de quantificação proposto por GUESCINI et al. (2008), e
se baseia na utilização da equação de Richards utilizando cinco parâmetros da curva, onde o
valor de quantificação é calculado pelo ponto de interseção entre o eixo da abscissa e a
tangente do ponto de inflexão da curva, obtido por uma regressão não linear (Fig. 1.7). Este
método não requer eficiências similares nas reações de dois genes, nem da utilização de um
nível pré-definido pelo usuário.
Fig. 1.7: Curva de PCR com os cinco parâmetros da função de Richards. Nos círculos abertos são mostrados os valores previstos da curva; linha acompanhada de pontos representa os dados obtidos experimentalmente; o losango representa o ponto de inflexão da curva; o valor de quantificação Cy0 é mostrado como círculo fechado; a tangente representada mediante uma linha continua. Adaptado de GUESCINI et al. (2008).
O algoritmo aplicado pelo método Cy0 para a quantificação é mostrado na equação
(1.3):
(1.3)
Onde, Fmax: fluorescência máxima da reação; c: ciclo fracionar em que a fluorescência de
reação atinge a metade de Fmax; b: inclinação da curva; d: coeficiente de Richard; Fb: ruído
da reação de fluorescência.

26
Como pode ser visto, as maiores diferenças com o método sigmoide é a utilização de
um quinto parâmetro e a necessidade de que a reação tenha atingido um platô. Os autores
propõem que este método é útil para detectar a presença de inibidores e, devido a recolher um
maior número de informação da curva, fornecer uma quantificação mais precisa nestes casos,
em que claramente as amostras não têm a mesma eficiência que os controles usados para
calcular esta eficiência.
1.13 Genes de referência e normalização
Erros na quantificação por qPCR vêm da acumulação de variações no material inicial
desde o processamento da amostras até a realização da reação de qPCR (diferenças de
amostragem, integridade de RNA, eficiências de RT, quantidade de cDNA colocada na
reação, entre outros). Uma ferramenta importante para diminuir esta variabilidade é o uso da
quantificação de um gene de referência (GDR), definidos como aqueles cuja variação entre
diferentes tecidos é mínima e que não se vejam afetados pelos tratamentos utilizados
experimentalmente. A normalização não é mais que a relativização da quantificação do gene
de interesse (GDI) frente ao GDR, calculado da seguinte maneira (equação 1.4):
Normalização = (Cq GDI – Cq GDR) (1.4)
Este passo apresenta grande importância quando as amostras são conservadas por
tempo variável, tomadas em diferentes tempos e indivíduos, diferentes tecidos e diferentes
situações experimentais, assim, é fundamental para compensar variações intra e inter qPCR
(amostras e corridas).
Diferentes modelos matemáticos têm sido propostos para escolher os melhores GDR
como (VANDESOMPELE; KUBISTA; PFAFFL, 2009):
• geNorm: se fundamenta no algoritmo matemático que determina um par GDR mais
estável de um grupo proposto, através do M-value de todos os genes candidatos,
eliminando o gene com maior M-value. Para poder ser aplicado este algoritmo é
necessário a comparação do mínimo três genes (VANDESOMPELE et al., 2002).
• Bestkeeper: se baseia no melhor par de GDR selecionados por analise de correlação e
regressão dos genes de a pares (PFAFFL et al., 2004).

27
• NormFinder: este algoritmo encontra o melhor GDR baseado em um teste de
ANOVA, calculando a variação intragrupo e intergrupo (ANDERSEN; JENSEN;
ORNTOFT, 2004).
1.14 Estratégia de quantificação utilizada em PCR em tempo real
As análises de expressão gênica podem ser realizadas mediante o uso de duas
estratégias: a quantificação absoluta ou a quantificação relativa. A quantificação absoluta é
realizada por calibração reversa em relação a uma curva padrão construída com diluições
seriadas com concentrações conhecidas do mesmo alvo a ser estudado, utilizando geralmente
um plasmídeo (PFAFFL; HAGELEIT, 2001). O valor de quantificação é expresso em número
de copias do transcrito (BUSTIN, 2000). A sua utilização principal é no contexto clínico, em
que a expressão de um alvo molecular pode ser assimilada ao valor da carga tumoral
(RENAULT et al., 2011). Para a análise de expressão gênica, prefere-se usar a estratégia de
quantificação relativa. Esta é utilizada para determinar as mudanças da expressão do RNAm
de um GDI em relação aos níveis de RNAm de um controle interno (GDR). Esta estratégia é
muito utilizada na procura de alterações fisiológicas no nível de expressão gênica (PFAFFL,
2004). Os valores de quantificação são expressos em fold change. Duas maneiras têm sido
propostas para calcular o valor da expressão relativa:
1 Sem correção da eficiência: denominado “método do Cq comparativo”, este método
não utiliza a correção da eficiência da reação, pois se baseia no requisito de que tanto
GDI como GDR apresentam uma eficiência ~100% e similares entre elas, com limites
de tolerância de 5-10% (LIVAK; SCHMITTGEN, 2001). Para este cálculo,
estabelece-se uma amostra, linhagem celular, equivalente tecidual normal ou valor
grupal (por exemplo, média do grupo controle) como calibrador (valor 1). O cálculo é
realizado mediante a equação (1.5):
Fold change = 2-∆∆Cq (1.5)
Onde ∆∆Cq = [(Cq GDI amostra – Cq GDR amostra) - (Cq GDI calibrador – Cq GDR calibrador)]
e o número 2 representa uma eficiência de 100%.
Esta forma é utilizada quando o interesse é comparar dois grupos de amostras
pudendo ser um grupo de pacientes tratados vs. não tratados ou doentes vs. sadios.

28
SCHMITTGEN; LIVAK (2008) propõem que os dados de RT-qPCR também
podem ser apresentados como dado único, dispensando a calibração, onde o fold
change é calculado como 2-∆Cq.
2 Com correção de eficiência: este método é utilizado quando as eficiências entre o
GDI e GDR são diferentes e é baseado na seguinte equação (1.6) (PFAFFL, 2004):
Quantificação relativa = (Eficiência GDI) ∆Cq GDI (calibrador - amostra) (1.6)
(Eficiência GDR) ∆Cq GDR (calibrador - amostra)
Recentemente foi publicada uma guia sobre a mínima informação necessária para
publicação de experimentos usando RT-qPCR denominada MIQE (do inglês, Minimum
Information for Publication of Quantitative Real-Time PCR Experiments), com a finalidade
de unificar os conceitos metodológicos, assim como a consistência e transparência dos
experimentos realizados (BUSTIN et al., 2009; BUSTIN et al., 2010).
1.15 O linfoma de Hodgkin clássico como doença complexa dos tecidos: abordagem
dos estudos de expressão gênica
A passagem para a multicelularidade a partir do estado unicelular da vida foi possível
a partir da aquisição evolutiva de mecanismos que articulam o controle molecular da divisão
celular e a decisão intrínseca da entrada em apoptose (e as outras formas de morte celular
programada) com estímulos vindos do meio extracelular, incluindo os que garantem a
homeostase tecidual no tempo (desenvolvimento) e no espaço (manutenção do estado
diferenciado). No nível organísmico, o desenvolvimento de sistemas de integração (nervoso,
imune, endócrino) permitiu a expansão da vida tal como hoje a conhecemos. A subversão
destes mecanismos requintados de controle trouxe como risco o câncer, que deste ponto de
vista deve ser considerado uma doença dos tecidos e não somente um processo de
transformação celular. Desta maneira, não é surpreendente encontrar que, entre os pilares
fundamentais que definem o estado neoplásico em geral, encontram-se as alterações no ciclo
celular e na resposta aos estímulos indutores de apoptose (HANAHAN; WEINBERG,
2000;2011), e mais recentemente, a inclusão do conceito de que não existe tumor que surja
alheio às condições de sustentação fornecidas pelo seu microambiente particular
(HANAHAN; WEINBERG, 2011). O LHc, com seu delicado balanço entre célula tumoral e

29
ambiente, representa um dos expoentes mais expressivos do conceito do câncer como doença
complexa dos tecidos.
De fato, o estudo destes fenômenos no LHc é extremamente difícil pelo baixo número
de células H-RS no tumor e pela dependência estrita da célula tumoral primária aos estímulos
bioquímicos e físicos do microambiente. Uma limitação importante para entender os defeitos
que levam ao fenótipo maligno das células H-RS é a inadequação dos modelos in vitro da
doença. Existem pouco mais de 10 linhagens celulares estabelecidas, todas elas EBV-, e
estabelecidas a partir de células circulantes (sangue periférico ou efusões de cavidades) de
pacientes com estádios avançados da doença, resistente a quimioterapia, onde as células cuja
ancoragem com o microambiente foi perdida. Quanto aos modelos animais, apesar do esforço
da comunidade científica, ainda não se chegou a um modelo tumoral que recrute um
microambiente similar ao do LHc.
Assim, a aplicação de modelos de pesquisa baseados na experimentação in vitro ou in
vivo para gerar hipóteses demostráveis em tumor primário de pacientes não é uma opção
válida hoje nesta doença. Os modelos aplicados, em geral, baseiam-se em inferir relações
patogênicas a partir do material primário, para depois confirmar mecanisticamente nos
modelos in vitro. Neste cenário, a transferência para a clínica se torna difícil; por exemplo,
nos últimos 30 anos, somente uma droga nova foi aprovada para tratamento da recaída da
doença (anticorpo monoclonal anti-CD30, brentuximabe vedotina), refletindo em parte a falta
de conhecimento dos mecanismos histopatogênicos básicos. As abordagens por IHQ tem sido
as de maior importância para caracterizar estes processos, apesar das limitações destes
métodos. Os estudos de expressão gênica, por sua vez, adolescem de falta de
reprodutibilidade, que pode ser devido à grande heterogeneidade biológica, evidente para
quem inspeciona os tumores no microscópio, mesmo sem ser patologista.
Esta dissertação apresenta uma trajetória de pesquisa para desenvolver métodos
reprodutíveis de análise da expressão gênica a partir de amostras sub-ótimas (RNA de
parafina), que começa com a extração da amostra e termina com a avaliação de métodos
alternativos de quantificação adequados ao material analisado. Apresentamos também a
aplicação dos métodos de expressão gênica padronizados em um grupo de pacientes com LHc
infantil visando identificar padrões de expressão com significado biológico, assim como
marcadores de prognóstico da doença.

30
2 OBJETIVOS
2.1 Objetivo geral
Avaliar perfis de expressão gênica relacionados à célula tumoral e ao microambiente
em um grupo de crianças diagnosticadas com linfoma de Hodgkin clássico, após o
desenvolvimento de uma metodologia para a extração de RNA a partir material fixado em
formalina e impregnado em parafina (FFPE) e PCR quantitativa em tempo real, visando
identificar padrões de importância biológica e clínica.
2.2 Objetivos específicos
• Desenvolver e aperfeiçoar metodologias para extração de RNA, retrotranscrição e
quantificação da expressão gênica a partir de linfonodos FFPE, no linfoma de
Hodgkin clássico.
• Avaliar as fontes de variabilidade (biológicas e técnicas) introduzidas na metodologia
desenvolvida para extração de RNA de parafina utilizando um desenho ANOVA tipo
nested.
• Comparar diferentes métodos para a quantificação de reações de PCR em tempo real
(ciclo pontual Cq vs. cinética da curva).
• Analisar os níveis de expressão de genes relacionados às vias do ciclo celular,
apoptose, ativação de monócitos/macrófagos e fator regulador de interferon 4 em um
grupo de pacientes com linfoma de Hodgkin clássico pediátrico.
• Correlacionar os níveis de expressão gênica com as características das células
tumorais, os subtipos das células do microambiente tumoral e a resposta terapêutica
dos pacientes.

31
3 MATERIAIS E MÉTODOS
3.1 Amostras
Foram incluídos pacientes com LHc com idade até 18 anos e cujo diagnóstico e
tratamento tenham sido feitos entre 1999 e 2006, no INCA (Rio de Janeiro). Só foram
incluídos os casos que dispunham do bloco de parafina contendo o material tumoral do
diagnóstico; negativos para o vírus da imunodeficiência humana (HIV) e que tivessem
material genético amplificável para realização dos estudos moleculares. Ao todo, 93 casos
preencheram os critérios de inclusão.
Este grupo de pacientes pertence a um grupo total de 100 pacientes com LHc bem
caracterizados tanto do ponto de vista histológico, quanto imunológico. Todos os casos foram
revisados pelos patologistas M.H.M. Barros e G. Niedobitek (BARROS et al., 2010;
BARROS, 2011). A subclassificação histológica foi realizada segundo o estabelecido pela
OMS em EN, CM, DL e RL (SWERDLOW et al., 2008). Para a realização do estudo piloto
utilizando o desenho ANOVA tipo nested, três indivíduos com LHc do subtipo histológico
EN que dispunham de mais de um bloco do tumor com similar proporção tumor-estroma
foram avaliados e selecionados pelo patologista M.H.M. Barros.
O grupo controle não tumoral foi constituído por 6 linfonodos diagnosticados com
hiperplasia folicular reativa. Como controles de qualidade das reações de qPCR, um pool de
doadores sadios formado por 10 indivíduos e linhagens celulares de LHc (KMH2, L1236 e
L428), leucemia mielomonocítica (U937) e mieloma múltiplo (U266) foram incluídas.
3.2 Dados clínicos e epidemiológicos
Para cada paciente, os dados de procedência, história clínica, e exames clínico-
radiológico-laboratorial ao diagnóstico, durante o tratamento, ao final do tratamento e durante
o acompanhamento pós-tratamento, incluindo as informações a respeito da resposta
terapêutica foram obtidos a partir dos prontuários médicos.
Os pacientes foram estadiados, seguindo os critérios Ann Arbor (CARBONE et al.
1971). Pacientes com estádios IIB, IIIB e IV foram estratificados como doença desfavorável,
e pacientes com estadiamento I e IIA foram considerados como doença favorável
(VASSILAKOPOULOS et al., 2005).

32
O número de células tumorais, de eosinófilos e de células tumorais em mitose foi
determinado usando um microscópio Nikon modelo Eclipse-E2000 com um aumento total de
10x das lentes oculares e 40x da lente objetiva como previamente mostrado (BARROS,
2011).
Pacientes com o subtipo EN foram classificados segundo os critérios do Grupo
Nacional Britânico de Investigação de Linfoma (BENNETT et al., 1981) em EN grau I ou II,
de acordo ao número de células H-RS relacionadas à presença de quantidades de linfócitos,
inflamatório misto ou fibrohistiocítico (SWERDLOW et al., 2008).
3.3 Método de extração de RNA com o reagente Trizol®
O local de trabalho e instrumentos a utilizar foram mantidos em condições livres de
RNAses mediante limpeza escrupulosa com isopropanol 70% e uma solução comercial
(RNAse away). Foram utilizados micropipetas e material plástico especialmente dedicados
para a extração de RNA, assim como soluções e reagentes especialmente preparados e
armazenados.
A extração de RNA foi realizada a partir de 1 mL de Trizol® (Invitrogen, Life
Technologies, Carlsbad, CA) que continha um lisado celular de 5-8 x 106 células, foram
adicionados 200 µL de clorofórmio, agitados por inversão por 15 segundos e incubados por 3
minutos a temperatura ambiente (TA). Seguidamente, os tubos foram centrifugados durante
15 minutos a 12.000 rotações por minuto (r.p.m.) em uma centrifuga refrigerada a 4°C. A fase
aquosa que continha o RNA foi transferida para um tubo novo de microcentrifuga e
precipitado com 500 µL de isopropanol (Merck). Os tubos foram agitados de 30 a 40 vezes
por inversão e incubados por 1 hora a -20°C. Sucessivamente, as amostras foram
centrifugadas a 4°C por 10 minutos a 12.000 r.p.m. e o sobrenadante retirado e descartado
cuidadosamente ficando o pellet de RNA. Uma lavagem ao pellet de RNA foi realizada com
900 µL de etanol (Merck) 75% diluído em água tratada com dietil dicarbonato (DEPC)
seguida de uma centrifugação de 10.000 r.p.m. por 5 minutos a 4°C. O sobrenadante e restos
de etanol foram retirados cuidadosamente e o pellet de RNA ressuspendido em 12 µL de
água-DEPC, solubilizado no termobloco a 65°C durante 30 segundos, seguido de 15 minutos
no gelo. O armazenamento do RNA foi realizado a -70°C.

33
3.4 Método de extração de RNA total a partir de amostras fixadas e impregnadas em
parafina
Seguindo todos os cuidados para trabalho em ambiente livre de RNAses, cinco seções
com espessura de 3µm foram obtidas a partir de cada bloco de parafina, usando um
micrótomo Leica Biosystems, escrupulosamente limpo com soluções livres de RNases antes e
depois do trabalho. Os cortes foram colocados em um tubo de microcentrifuga previamente
rotulado e submetidos ao processo de desparafinização, seguido pelos métodos específicos
para extração de RNA.
3.4.1 Desparafinização
Ao tecido previamente cortado, foi adicionado 1 mL de xilol 100% e colocado no
agitador à 37°C durante 20 minutos, seguido de centrifugação por 5 minutos à 13.000 r.p.m.
O sobrenadante foi descartado. Este passo foi realizado 3 vezes. Após o tratamento com xilol,
foram realizadas três incubações repetidas com 1,2 mL de etanol 100% no agitador à 37°C
durante 30 minutos, sempre descartando o sobrenadante. Ao final, foram eliminados os restos
de etanol e o pellet foi deixado a temperatura ambiente por 5 minutos, permitindo a
evaporação da maior quantidade de etanol.
Para a extração de RNA a partir de material FFPE, dois métodos comerciais foram
utilizados: Master PureTM (Epicentre, Madison, WI) (MP) e Recover AllTM Total Nucleic
Acid Isolation Kit (Ambion, Life Technologies) (RA).
3.4.2 Método Master PureTM
A extração de RNA foi realizada pelo método de purificação de RNA Master PureTM
seguindo a instruções do fabricante com modificações na etapa de digestão, como sugerido
por CHEN et al. (2007). A etapa de digestão com proteases foi realizada a partir do pellet do
tecido, adicionando 480 µL de solução de lise celular e de tecido (do inglês, Tissue and Cell
Lysis Solution) como recomendado pelo fabricante e 60 µL de proteinase K (Invitrogen) a 60
mg/mL, colocando o tubo no banho-maria a 65°C durante 20 horas ou até que o tecido
estivesse completamente disperso. Em seguida, a amostra foi colocada no gelo por 5 minutos.
Para a separação dos ácidos nucleicos das proteínas, 315 µL do reagente de precipitação de
proteínas MPC (do inglês, MPC Protein Precipitation Reagent) foram adicionados à amostra,
lisada e vortexada vigorosamente durante 10 segundos, continuamente centrifugadas por 10

34
minutos a 13.000 r.p.m. O sobrenadante (RNA) foi transferido para um tubo de
microcentrifuga novo e colocado em gelo por 5 minutos. Para a precipitação do RNA, 500 µL
de isopropanol foram adicionados ao sobrenadante e os tubos invertidos durante 30-40 vezes,
centrifugando a 4°C durante 10 minutos a 13.000 r.p.m. Em seguida, retirou-se o
sobrenadante com cuidado e o tratamento com DNAse I (200 µL do tampão e 5 µL da enzima
fornecida pelo fabricante) foi aplicado ao resíduo (RNA) e incubado durante 30 minutos à
37°C. Posteriormente, 200 µL da solução de lise C e T 2X (do inglês, 2X T and C Lysis
solution) foram adicionadas à amostra, vortexando por 5 segundos, acrescentando 200 µL do
reagente de precipitação de proteínas MPC e misturando por 10 segundos com o vortex. As
amostras foram colocadas no gelo por 5 minutos e centrifugadas a 4°C por 10 minutos a
13.000 r.p.m.. A fase aquosa foi transferida para um tubo novo de microcentrifuga,
adicionando 500 µL de isopropanol, invertendo os tubos por 30-40 vezes e centrifugando-os
novamente a 4°C por 10 minutos a 13.000 r.p.m.. Duas lavagens foram realizadas com etanol
(Merck) 75% diluído em água-DEPC, seguidas de centrifugações a 4°C, 10.000 r.p.m. por 5
minutos. O tubo foi deixado aberto a TA por 5 minutos para permitir a evaporação dos restos
de etanol. Finalmente o pellet foi ressuspendido em 12 µL de água-DEPC e colocado 30
segundos no termobloco a 65°C para sua solubilização, seguido de 15 minutos no gelo. O
armazenamento do RNA foi realizado a -70°C.
3.4.3 Método Recover AllTM
A extração de RNA a partir de amostras FFPE com o método Recover AllTM foi
realizada seguindo as indicações do fabricante com modificações na etapa de digestão. Após a
desparafinização, o pellet que contém o tecido foi dissolvido em 200 µL de tampão de
digestão com 60 µL de proteinase K (Invitrogen) a uma concentração de 60 mg/dL, incubado
durante 15 minutos a 50°C, seguido de 20 horas a 65°C no banho-maria. Em seguida, foi
adicionado 240 µL do aditivo de isolamento (do inglês, Isolation Additive) fornecido pelo
fabricante com 550 µL de etanol 100%. Após a homogeneização, 700 µL da mistura foram
transferidos para um filtro de captura e centrifugado por 30 segundos a 10.000 r.p.m. As
etapas de lavagens foram realizadas adicionando 700 µL da solução de lavagem 1 e
centrifugando a 10.000 r.p.m. por 30 segundos, descartando o sobrenadante. O tratamento
com DNaseI foi aplicado nesta etapa, pela adição de 60 µL da solução de tratamento
composta por 6 µL do tampão de DNase, 4 µL de DNaseI e 50 µL de água livre de nucleases;
o tempo de incubação foi de 30 minutos a temperatura ambiente. Após o tratamento com

35
DNase I uma etapa de lavagem foi realizada com 700 µL da solução de lavagem 1 e duas com
500 µL da solução de lavagem 2/3, em cada etapa centrifugações a TA foram realizadas por
30 segundos a 10.000 r.p.m. Finalmente, a solubilização do RNA foi feita com 30 µL da
solução de eluição, centrifugando a 13.000 r.p.m. durante 1 minuto a TA. O RNA foi
armazenado a -70°C.
3.5 Desempenho dos métodos de extração de RNA
A quantificação e o grau de pureza do RNA foram avaliados por espectrofotometria
(Nanodrop, Wilmington, Delaware USA) a partir de uma alíquota de 1 µL da amostra, no
comprimento de onda (λ) 260, 280 e 230, que quantificam ácidos nucleicos, proteínas e
contaminação com álcool, fenol e EDTA, respectivamente. Uma amostra foi considerada de
qualidade ótima na presença de razões λ260/280 entre 1,8 - 2,0; e razões λ260/230 entre 2,0 -
2,2.
A integridade do RNA foi avaliada pela conservação relativa das frações do RNA
ribossômico (28S, 18S e 5S) através de duas metodologias:
3.5.1 Eletroforese em gel de agarose
O RNA (1 µL) junto com tampão de corrida para RNA foi aplicado em um gel de
agarose 1,2% preparado com tampão fosfato (Na2HPO4 0,01M), com coloração de brometo de
etídio (0,5 µg/mL) (Sigma), em condições livres de RNases. A eletroforese foi realizada por
30 minutos com voltagem constante de 80 volts. A avaliação das amostras foi realizada sob a
luz ultravioleta.
3.5.2 Número de integridade do RNA
A avaliação da integridade do RNA foi realizada com o kit RNA 6000 Nano na
plataforma Agilent (2100 Bioanalyzer, Agilent Technologies), seguindo as indicações do
fabricante. O número da integridade do RNA (RIN) apresenta uma faixa entre 1-10, em que 1
corresponde a amostras altamente degradas e 10, a RNAs de alta integridade.
3.6 Retrotranscrição
A transcrição reversa (RT) foi realizada com o conjunto de reagentes High-Capacity
cDNA Archive kit (Applied Biosystems) a partir de 1 µg de RNA total para o ensaio inicial
ANOVA tipo nested. Para o restante dos ensaios, foi utilizado 0,5 µg de RNA, devido às

36
melhoras observadas e a diminuição de erro introduzido durante o processamento técnico
(descrito na seção 4.1.4). O volume final da reação foi de 20 µL (10 µL de RNA diluído e 10
µL de mistura RT). A mistura RT foi preparada com 2 µL de tampão RT 10X, 0,8 µL de
dNTPs 25X, 2 µL de iniciadores randômicos (do inglês, Random primers) 10X, 1 µL da
enzima MultiScribe RT 50U/ul e 4,2 µL de água livre de nucleases. Esta reação foi incubada a
25°C por 10 minutos e a 37°C por 120 minutos e armazenada a -70°C.
3.7 Pré-amplificação
A pré-amplificação (pré-Amp) foi realizada com o conjunto de reagentes TaqMan
PreAmp Master Mix (Applied Biosystems) em reações de 10 µL cujo volume final era
composto por: 5 µL de Master Mix 2X; 2,5 µL do conjunto iniciadores/sonda 20X diluídos
em tampão TE (tris-EDTA) e 2,5 µL de DNA complementar (cDNA). O passo inicial foi de
95°C por 10 minutos, seguido de 14 ciclos a 95°C por 15 segundos e 60°C por 4 minutos em
um termociclador Veriti. O produto foi diluído na proporção de 1:20 em água livre de
nucleases e usado para o análise na qPCR. Tanto os produtos como as diluições foram
armazenados a -70°C.
3.8 Quantificação da expressão gênica
As reações de RT-qPCR foram realizadas na plataforma ABI 7000 (Applied) usando
sondas de hidrólise (TaqMan) marcadas 5' 6-FAM-3'MGB e dois iniciadores específicos
para cada alvo. As reações de qPCR continham 1X TaqMan PCR Master Mix Universal
(Applied Biosystems), 1X do ensaio TaqMan de expressão gênica específico (Applied
Biosystems) e 4 µL de pré-Amp cDNA diluído na proporção 1:20 em 15 µL de volume final,
incubadas a 50°C por 2 minutos e 95°C por 10 minutos, seguido de 40 ciclos por 15 segundos
à 95°C e 60°C por 1 minuto.
Todas as corridas foram realizadas com os controles de qualidade recomendados para
qPCR. As reações de qPCR foram preparadas em 3 ambientes separados (um para extração e
manipulação de ácidos nucleicos, um para realização das reações e um terceiro para
incorporação dos ácidos nucleicos à reação de qPCR). Controles da reação com ausência de
cDNA chamados de NTC (do inglês, no template control) foram colocados em cada placa (2
NTC por cada gene avaliado), o qual não deveria amplificar até o Cq <40 ciclos. Todas as
reações de PCR foram realizadas em duplicata. No desenho das placas, os genes de interesse
(GDI) e genes de referencia (GDR) foram colocados na mesma corrida, para evitar o uso de
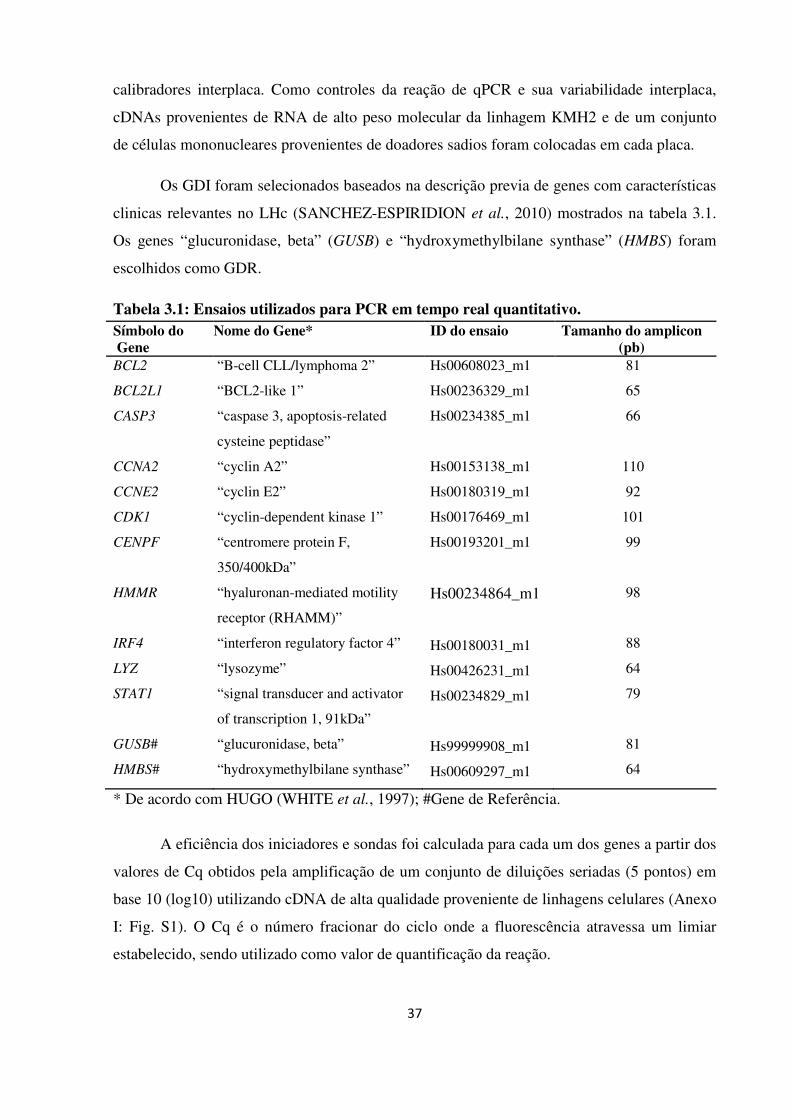
37
calibradores interplaca. Como controles da reação de qPCR e sua variabilidade interplaca,
cDNAs provenientes de RNA de alto peso molecular da linhagem KMH2 e de um conjunto
de células mononucleares provenientes de doadores sadios foram colocadas em cada placa.
Os GDI foram selecionados baseados na descrição previa de genes com características
clinicas relevantes no LHc (SANCHEZ-ESPIRIDION et al., 2010) mostrados na tabela 3.1.
Os genes “glucuronidase, beta” (GUSB) e “hydroxymethylbilane synthase” (HMBS) foram
escolhidos como GDR.
Tabela 3.1: Ensaios utilizados para PCR em tempo real quantitativo. Símbolo do Gene
Nome do Gene* ID do ensaio Tamanho do amplicon (pb)
BCL2
BCL2L1
CASP3
CCNA2
CCNE2
CDK1
CENPF
HMMR
IRF4
LYZ
STAT1
GUSB#
HMBS#
“B-cell CLL/lymphoma 2”
“BCL2-like 1”
“caspase 3, apoptosis-related
cysteine peptidase”
“cyclin A2”
“cyclin E2”
“cyclin-dependent kinase 1”
“centromere protein F,
350/400kDa”
“hyaluronan-mediated motility
receptor (RHAMM)”
“interferon regulatory factor 4”
“lysozyme”
“signal transducer and activator
of transcription 1, 91kDa”
“glucuronidase, beta”
“hydroxymethylbilane synthase”
Hs00608023_m1
Hs00236329_m1
Hs00234385_m1
Hs00153138_m1
Hs00180319_m1
Hs00176469_m1
Hs00193201_m1
Hs00234864_m1
Hs00180031_m1
Hs00426231_m1
Hs00234829_m1
Hs99999908_m1
Hs00609297_m1
81
65
66
110
92
101
99
98
88
64
79
81
64
* De acordo com HUGO (WHITE et al., 1997); #Gene de Referência.
A eficiência dos iniciadores e sondas foi calculada para cada um dos genes a partir dos
valores de Cq obtidos pela amplificação de um conjunto de diluições seriadas (5 pontos) em
base 10 (log10) utilizando cDNA de alta qualidade proveniente de linhagens celulares (Anexo
I: Fig. S1). O Cq é o número fracionar do ciclo onde a fluorescência atravessa um limiar
estabelecido, sendo utilizado como valor de quantificação da reação.

38
O valor da eficiência foi calculado utilizando o software Genex enterprise (MultiD)
através da seguinte fórmula matemática (3.1):
Eficiência = [10 (-1/pendente)] – 1 * 100 (3.1)
Onde a pendente foi calculada utilizando a equação da reta
y = ax + b
Em que a = pendente; b = ponto na qual a reta cruza o eixo y= 0
O valor ideal aceito para a pendente foi entre – (3,3 – 3,4) e do coeficiente de
determinação de regressão da curva (R2) 0,997 < R2 < 1, o que explica que toda variação
apresentada por y é explicada pela variação de x.
Após observações realizadas nas reações, identificando o momento de ocorrência das
mínimas mudanças do sinal de fluorescência, valores fixos para a linha de base para
quantificação foram estabelecidos no instrumento, antes do qual os produtos produzidos
foram considerados ruído. A línea de base para reações particulares foi estabelecida entre os
ciclos da reação 3-15.
A lista das informações mínimas para publicação de experimentos em qPCR (MIQE),
de acordo com o recomendado por BUSTIN et al. (2009), encontra-se no Anexo II.
3.9 Avaliação da amplificação
A amplificação das amostras foi avaliada pela amplificação dos genes de referência
em Cq ˂35, assim como pela visualização da cinética da curva em escala exponencial.
Amostras com amplificação em Cq> ciclo 35 e uma cinética da curva diferente da curva
sigmoide característica de uma reação de qPCR (fases exponencial, linear e platô) foram
consideradas como não amplificáveis. As amostras com inclinação à direita na curva de
reação (alteração na fase exponencial) foram suspeitas de presença de inibidores que
poderiam estar interferindo na reação de RT-qPCR, consequentemente, novas extrações de
RNA, sínteses de cDNA e novos produtos com suas diluições de pré-Amp foram obtidos e as
reações repetidas.

39
3.10 Abordagens de quantificação relativa da expressão gênica
A estratégia escolhida foi a da quantificação relativa que determina as mudanças de
expressão de um GDI em relação à expressão de um GDR. Dois métodos foram utilizados
para calcular o valor de quantificação das reações de RT-qPCR: o método do ciclo threshold e
o método do Cy0.
3.10.1 Método do ciclo threshold
A quantificação foi subministrada pelo instrumento ABI7000 em um arquivo
compatível com Microsoft Excel. Este método exige a determinação de um limiar ou
threshold (línea paralela ao eixo X, estabelecido na fase exponencial da reação), onde o valor
de quantificação é determinado no número de ciclo ao momento que a quantidade de
fluorescência da reação alcance sobrepassar o limiar estabelecido. O valor do threshold foi
estabelecido em 0,376 e mantido em todas as corridas realizadas. As reações foram realizadas
em duplicatas, aceitando como valor máximo diferencial de desvio padrão (DP) < +0,15
ciclos.
A quantificação foi realizada utilizando o modelo matemático proposto por LIVAK;
SCHMITTGEN (2001), sem a utilização de um calibrador (equação 3.2):
Quantificação relativa = 2-∆Cq (3.2)
Onde ∆Cq= Cq GDI – Cq GDR
A unidade de ∆Cq é a normalização da expressão de um GDI com a expressão de
GDR (controle interno), no qual a expressão não varia entre os tecidos, células ou na resposta
aos tratamentos experimentais. No caso da avaliação da expressão gênica no escore clínico, o
valor médio da quantificação dos genes GUSB e HMBS foi utilizado como Cq GDR, como
recomendado (VANDESOMPELE et al., 2002).
Para ser aplicado o algoritmo de quantificação relativa aos valores de quantificação
obtidos por este método é pré-requisito que as eficiências de reação dos GDI e GDR devem
ser similares. Experimentos de diluição para avaliação da eficiência foram realizados, e estas
não variaram em mais que +10% (Anexo I: Fig. S1 e Anexo II). O uso da terminologia Cq ao
longo do trabalho será referente às quantificações realizadas pelo método do ciclo threshold.

40
3.10.2 Método Cy0
Valores de quantificação das reações de RT-qPCR foram obtidos usando o método do
Cy0, em que o ponto de quantificação é denominado Cy0 (GUESCINI et al., 2008), baseado
na análise dos parâmetros da cinética da curva de qPCR.
Os dados iniciais para esta análise foram os valores do repórter normalizado [∆(Rn-
ROX)] para cada ciclo de cada amplificação de GDIs e GDRs para cada amostra. Estes
valores foram armazenados em planilhas Excel. A análise foi realizada com os resultados
obtidos no desenho de ANOVA tipo nested, para comparar o desempenho dos métodos Cq e
Cy0 e sua capacidade de identificar a possível presença de inibidores como causa da
variabilidade obtida nos níveis biológicos.
O uso da terminologia Cy0 ao longo do trabalho será referente às quantificações
realizadas pelo método da equação de Richards. Resultados do ∆Cy0 refere-se à normalização
realizada com resultados Cy0s (∆Cy0 = Cy0 GDI – Cy0 GDR).
3.11 Cálculo dos outliers
Um outlier é um valor dos resultados que se encontra afastado da distribuição do
conjunto total. Neste trabalho, a definição de outlier foi aplicada em relação aos parâmetros
técnicos, como elemento para avaliar a qualidade dos dados primários.
A determinação de outliers nos ensaios de expressão gênica foi realizada de duas
maneiras:
1 Software Genex enterprise (MultiD), mediante o teste de Grubbs que considera
valores outliers aqueles que se encontram dentro da área de 1% da distribuição
normal (média + 3 DP).
2 Software Kineret, baseado no método de estatística KOD (detecção de outliers pela
cinética da curva) (BAR et al., 2003), que realiza uma comparação da eficiência de
uma amostra desconhecida com a média das eficiências das curvas de qPCR de um
grupo de amostras de referência predeterminadas como de boa qualidade. Arquivos
com os dados do ∆(Rn-ROX) para cada ciclo de cada reação das amostras foram
enviados para a equipe de trabalho do Labonnet (http://labonnet.gene-
quantification.info/).

41
3.12 Desenho experimental ANOVA tipo nested
Um desenho experimental de tipo nested de seis níveis (3x2x2x2x2x2) foi usado para
investigar as fontes de variabilidade (biológicas e técnicas) em ensaios de expressão gênica.
Para tanto, foram utilizados 3 casos com LHc com 2 linfonodos, que foram analisados com
duplicatas de extração de RNA, seguidas de duplicatas de RT, pré-Amp e qPCR (Fig. 3.1).
Este desenho experimental produziu ao todo 96 valores de Cq para cada um dos 7 genes
avaliados (GDI: BCL2, CASP3, IRF4, LYZ e STAT1; GDR: GUSB e HMBS). As placas foram
desenhadas para incluir um GDI e o gene de referência para cada indivíduo na mesma placa.
O RNA foi extraído com o kit MP, selecionado por apresentar o melhor rendimento.
Quantificações de ∆Cq (Cq GDI – Cq GDR) utilizando os valores de quantificação do GUSB.
Níveis do desenho tipo nested Reações
Fig. 3.1. Desenho experimental ANOVA tipo nested para amostras fixadas e impregnadas em parafina. σ2: variância; RT: retrotranscrição; pré-Amp: pré-amplificação; qPCR: PCR quantitativa em tempo real.
3.13 Análise do ANOVA tipo nested
O modelo linear dos efeitos de processamento biológicos e técnicos foi calculado
como descrito por TICHOPAD et al. (2009) (equação 3.3):
Cqijklmn = µ + ai + bj(i) + ck(ji) + dl(kji) + em(lkji) + fn(mlkji) (3.3)
σσσσ
2+2σσσσ
2E(D)+4σσσσ
2D(C)+8σσσσ
2C(B)+16σσσσ
2B(A)
σσσσ
2+2σσσσ
2E(D)+4σσσσ
2D(C)+8σσσσ
2C(B)
Indivíduo 1 Indivíduo 2 Individuo 3
Indivíduos (n = 3)
Linfonodos (n = 3x2 = 6)
Extração RNA n (n = 3x2x2 = 12)
RTs (n= 3x2x2x2
= 24)
pré-Amps (n= 3x2x2x2x2
= 48)
qPCRs (n= 3x2x2x2x2 x2
= 96 )
σσσσ
2+2σσσσ
2E(D)+4σσσσ
2D(C)+8σσσσ
2C(B)+16σσσσ
2B(A)+32σσσσ
2A
B)
σσσσ2+2σσσσ2
E(D)+4σσσσ2
D(C)
σσσσ
2+2 σσσσ 2E(D)
σσσσ2
3333
6666
12121212
24242424
48484848
1152115211521152

42
Onde Cqijklmn é a quantificação individual de qPCR expressado em número de ciclos de
amplificação, incorporando o efeito randômico do sujeito (i), linfonodo (j), RNA (k) RT
(l) pré-Amp (m) e qPCR (n). µ é a média total do grupo; ai: efeito randômico do indivíduo
i; bj(i) efeito randômico do linfonodo j dentro de indivíduo i; ck(ij): efeito randômico dos
RNAs k do linfonodo j dentro do indivíduo i; dl(ijk) efeito randômico das RT l dentro do
RNA k, das amostras j dentro do sujeito i, e assim por diante.
As análises foram realizadas por ANOVA tipo nested de 6 fatores com efeito
randômico.
A partição da variância foi calculada através da seguinte fórmula (3.4) (KITCHEN et
al., 2010):
Variância = 100 x σ2x/σ
2Cq´ (3.4)
onde x= i, j, k, l, m, n;
A variabilidade biológica total foi calculada de maneira independente empregando a
somatória dos DP dos níveis interindividuais e linfonodos. De igual maneira, a variabilidade
técnica total foi calculada somando as variabilidades dos níveis RNA, RT, pré-Amp e qPCR.
O cálculo da variabilidade total do método foi realizado através da somatória da variabilidade
biológica total e a variabilidade técnica total, expressa em valores de DP.
O ruído total foi calculado como a variação acumulada em valores de DP de todas as
medidas de Cq, ∆Cq, Cy0 ou ∆Cy0 dos três indivíduos como se houvessem sido obtidas de
maneira independente. Já o ruído individual foi calculado pela somatória dos DP dos valores
Cq, ∆Cq, Cy0 ou ∆Cy0 por indivíduo (dividido por 3, número de indivíduos totais).
3.14 Técnica de imunohistoquímica
A partir de lâminas de microarranjo tecidual de 1 mm2 (TMA: do inglês, Tissue micro
array) foram realizadas imunomarcações para caracterizar as células H-RS e células
infiltrantes do MAT, com posterior análise microscópica computacional assistida. A análise
foi realizada pelo patologista M.H.M. Barros. No Anexo IV: Tabela S1 estão descritos os
anticorpos utilizados, clones, diluições e marcações esperadas.
Os ensaios de IHQ foram realizados com a técnica do complexo peroxidase-anti-
peroxidase (PAP), resumida nos seguintes passos:

43
1. As lâminas contendo as seções de material FFPE foram incubadas em estufa a 60°C
por 30 minutos, seguido pela desparafinização realizada por incubação em xilol por 10
minutos por duas vezes;
2. A hidratação foi realizada com concentrações de etanol descendentes (100% e 70%)
em banhos contínuos de 5 minutos. A seguir, os cortes foram lavados em água
corrente por 5 minutos.
3. A recuperação antigênica foi realizada em panela de pressão ou em um forno de
microondas na potência máxima, usando os tampões descritos no Anexo IV: Tabela
S1.
4. O bloqueio da peroxidase endógena foi realizado com peróxido de hidrogênio (10%
v/v) por 5 minutos, seguido de três lavagens de 5 minutos em água corrente.
5. As proteínas inespecíficas foram bloqueadas por incubação em uma solução de leite
em pó desnatado 3% por 20 minutos.
6. A incubação com 300 µL do anticorpo primário diluído em solução de albumina de
soro bovino (BSA) 3% foi realizada em câmara úmida por 1 hora a TA. Ao término, as
lâminas foram lavadas por 5 minutos em tampão fosfato salino (PBS).
7. O anticorpo secundário foi colocado e incubado por 30 minutos sobre cada corte.
Posteriormente, o excesso foi removido por três lavagens de 5 minutos em PBS,
seguido por incubação com streptavidina peroxidase por 20 minutos. Após este tempo,
as lâminas foram lavadas com três banhos de 5 minutos em PBS.
8. Como cromógeno, foi utilizada solução de DAB (DakoCytomation) por 5 minutos.
Após o tempo de incubação as lâminas foram lavadas em água corrente por 5 minutos
seguida da contra-coloração com hematoxilina de Harris por 30 segundos e lavagem
em água corrente por 5 minutos.
9. Por fim, as lâminas foram desidratadas em banhos contínuos de 5 minutos no etanol a
concentrações crescentes (70%, 100%, 100%), seguido de um banho final de xilol por
5 minutos.
10. As lâminas foram montadas com meio não aquoso e cobertas com lamínula.

44
3.15 Análise Microscópica Computacional Assistida
Cada caso foi fotografado em um aumento de 200x com a câmera AxioCam MRc
(Zeiss, Alemanha), as fotos foram transferidas para um computador e analisadas com o
programa HISTO (Biomas, Elangen, Alemanha), a fim de se determinar o número de células
imunomarcadas por mm2. Para esta análise, foi escolhido o core com o maior número de
células H-RS. Casos com negatividade dos marcadores em ambos os cores foram
desconsiderados.
3.16 Hibridização in situ para EBER-1 (EBER-ISH)
A técnica de hibridização in situ (ISH) foi utilizada para investigar a presença do EBV
nas amostras de LHc. Foram usadas sondas biotiniladas para os RNAs virais não codificantes
EBERs, e condições estritas para evitar a presença de RNAses, como descrito anteriormente
(HASSAN et al., 2006). Em resumo:
1. As lâminas foram colocados na estufa à 60°C por 30 minutos, seguidas de
desparafinização em xilol por 10 minutos, por 2 vezes.
2. Posteriormente, os cortes foram hidratados em banhos sucessivos de etanol com
concentrações decrescente (etanol à 100%, etanol à 95% e etanol à 70%) e água
destilada por 5 minutos cada um.
3. O processo de digestão do tecido foi realizado com proteinase K (20µg/mL) em
câmara úmida à 65º C por 30 minutos, seguido de duas lavagens de 3 minutos em água
destilada.
4. A seguir, os cortes foram desidratados através de banhos em etanol com concentrações
crescentes (etanol à 70%, etanol à 95%, etanol à 100%), por um período de 5 minutos
cada um. As lâminas foram deixadas a TA para a secagem dos cortes.
5. 10 µL da ribosonda biotinilada contra EBER-1 (Novocastra) foram utilizadas em cada
corte. As lâminas foram acondicionadas em câmara úmida e incubadas a 65°C por 15
minutos, seguidas de incubação de à 37°C por 2 horas.
6. A seguir, foram realizados três banhos com tampão tri salino (TBS) 0,1% Triton X-
100, 3 minutos cada. O bloqueio inespecífico foi feito com 100 µL de solução
bloqueante (TBS 0,1%; Triton X-100; BSA 3%) durante 10 minutos à TA.

45
7. Posteriormente, 100 µL da solução de anticorpo secundário (anti-FITC conjugado com
fosfatase alcalina, Novocastra) diluído em solução bloqueante (1:200) foi encubado
por 20 minutos. Ao final, duas lavagens de 3 minutos com TBS foram realizadas,
seguida de incubação com solução de fosfatase alcalina (pH 9,0) por 5 minutos.
8. Como solução cromógena, foi utilizada 100 µL de uma solução composta de 1 mL de
solução de fosfatase alcalina, 8 µL do BCIP/NBT e 1 µL de Levamisole (Novocastra).
As lâminas com a solução cromogênica foram acondicionadas em câmara úmida
escura e encubadas durante 16 horas à TA.
9. A seguir, os cortes foram lavados em água corrente durante 5 minutos, seguidos de
contra-coloração com hematoxilina de Harris por 30 segundos e lavagem em água
corrente por 5 minutos.
10. As lâminas foram montadas em meio aquoso e armazenadas a 4ºC.
Como controle positivo, foi utilizado um caso com linfoma de Burkitt EBV+. A
presença de marcações enegrecidas no núcleo das células H-RS significou positividade da
reação. Os casos de LHc foram considerados associados ao EBV quando qualquer célula H-
RS foi EBER-ISH positiva.
3.17 Definição de respostas e tempo de sobrevida
Para avaliação do tipo de resposta e determinação do tempo de sobrevida, foram
utilizadas as informações clínico-radiológico-laboratoriais existentes nos prontuários médicos
de cada paciente incluído no estudo.
Resposta completa foi definida como ausência de tumor ao final do tratamento,
mantida por pelo menos 4 semanas. A resposta parcial foi considerada quando houve redução
de 50 a 99% de todas as lesões tumorais ao final do tratamento. A resposta menor foi
considerada como redução de 25 a 49% de todas as lesões tumorais ao final do tratamento. A
doença estável foi definida como indução de menos de 25% das lesões tumorais, sem o
surgimento de novas lesões ao final do tratamento. Já a progressão de doença foi definida
como o surgimento de novas lesões ou aumento em 25% de alguma das lesões iniciais ao final
do tratamento.
A sobrevida livre de progressão (SLP) foi considerada como o período, em meses,
compreendido entre o diagnóstico e progressão, ou recaída, ou início um tratamento não

46
planejado, ou último seguimento em casos de resposta completa. Já a sobrevida global (SG)
foi definida como o período compreendido, em meses, entre o diagnóstico e o óbito ou último
seguimento (do estudo).
Todos os pacientes foram tratados com protocolos baseados na administração de
antracíclicos e foram avaliados como um único grupo terapêutico, nos estudos de sobrevida,
conforme feito na literatura (EICH et al., 2010; BAUER et al., 2011; MEYER et al., 2012).
3.18 Análises estatísticas
Os testes não paramétricos de Mann-Whitney e Kruskal-Wallis foram usados para
análises de associação entre variáveis categóricas e continuas, Wilcoxon para análises de
relação entre duas amostras relacionadas e o teste de Spearman para correlações de variáveis
continuas.
Análise de componente principal (PCA) foi utilizada para detectar as relações entre as
variáveis de expressão gênica. A PCA é um método multivariado que visa a redução de dados,
onde transforma um grupo de variáveis correlacionadas a outro não correlacionadas
representadas em valores de covariância. A PCA foi realizada nos valores de expressão de 10
genes (exceto para IRF4) obtidos de 84 pacientes usando o programa SPSS. A relação entre
casos e número de genes avaliados (variáveis) foi de 8,4 para 1, superando os requerimentos
estabelecidos para o PCA que é de 5 para 1. Correlações substanciais na matriz de correlações
entre as variáveis foram identificadas. Para identificar se a amostra era adequada para PCR a
medida de amostra adequada (MAS: do inglês, Measures of Sampling Adequacy), foi avaliada
através do teste Kaiser-Meyer-Olkin (KMO) na matriz anti-imagem para cada variável
individual, assim como para o set de variáveis, obtendo-se valores maiores a 0,5. A
identificação dos componentes principais foi realizada tendo em conta valores eigen maiores a
1, identificando posteriormente as variáveis que se carreavam com valores superiores a 0,5 em
cada componente. A validação dos resultados se realizou através das análises de
comunalidades (do inglês, communalities) com resultados maiores a 0,5.
A distribuição das sobrevidas foi estimada mediante o método Kaplan-Meier e as
diferenças foram comparadas usando o teste log-rank. O método de regressão de Cox para
análises multivariadas foi utilizado para determinar o valor prognóstico independente das
variáveis com valores P < 0,2 nas análises univariadas

47
Valores de P < 0,05 e P < 0,01 foram considerados significativos no testes
bicaudados. As análises estatísticas foram realizadas com os softwares GENEX enterprise
(MultiD), SPC (BPI Consulting, LLC) e SPSS 20.0.
3.19 Critérios Éticos
Este estudo obedece às normas regulamentadoras de pesquisas envolvendo seres
humanos do Conselho Nacional de Saúde, em sua resolução nº 196 de 10 de outubro de 1996,
visando assegurar os direitos e deveres que dizem respeito à comunidade científica, aos
sujeitos da pesquisa e ao Estado. Medidas que proporcionam confidencialidade e privacidade
dos pacientes, foram adotadas, assim como sigilo e a segurança dos dados obtidos. Este
projeto foi aprovado pelo Comitê de Ética em Pesquisa do INCA (Protocolo 37/05) (Anexo
III).

48
4 RESULTADOS
4.1 APERFEIÇOAMENTO DE UMA METODOLOGIA REPRODUTÍVEL PARA
EXTRAÇÃO DE RNA A PARTIR DE MATERIAL EM PARAFINA
4.1.1 Desempenho dos métodos de extração de RNA
Os métodos de extração de RNA: MP (baseado em precipitação) e RA (baseado em
colunas) foram comparados para selecionar o melhor método. Diferentes concentrações e
tempos de digestão foram testados obtendo-se melhores resultados com 60 µL de proteinase
K (Invitrogen) concentrada a 60 mg/dL por 20 horas a 65 °C comparadas com os protocolos
propostos pelos kits.
Para comparar os resultados dos métodos de extração de RNA entre os dois kits
utilizados, foram analisadas 25 amostras de maneira pareada, observando-se uma melhor
quantidade de RNA, assim como maior pureza no método MP que no método RA. Diferenças
nos valores médios do RIN não foram observadas nas 10 amostras pareadas avaliadas,
indicando que o RNA proveniente de material FFPE é altamente degradado
independentemente da metodologia utilizada para a extração (Tabela 4.1 e Fig. 4.1).
Tabela 4.1: Quantidade de RNA, pureza e RIN dos métodos comparados em 25 amostras pareadas.
Desempenho Master PureTM
Média + DP Recover AllTM
Média + DP P-valor
Quantificação absoluta (µg) 40,06 + 16,87 14,31 + 4,96 0,016
Pureza OD 260/280 1,84 + 0,15 1,77 + 0,20 0,094
Pureza OD 260/230 1,83 + 0,33 1,39 + 0,57 0,000
RIN 2,46 + 0,10 2,30 + 0,10 0,293
O valor P foi obtido através do teste de Wilcoxon
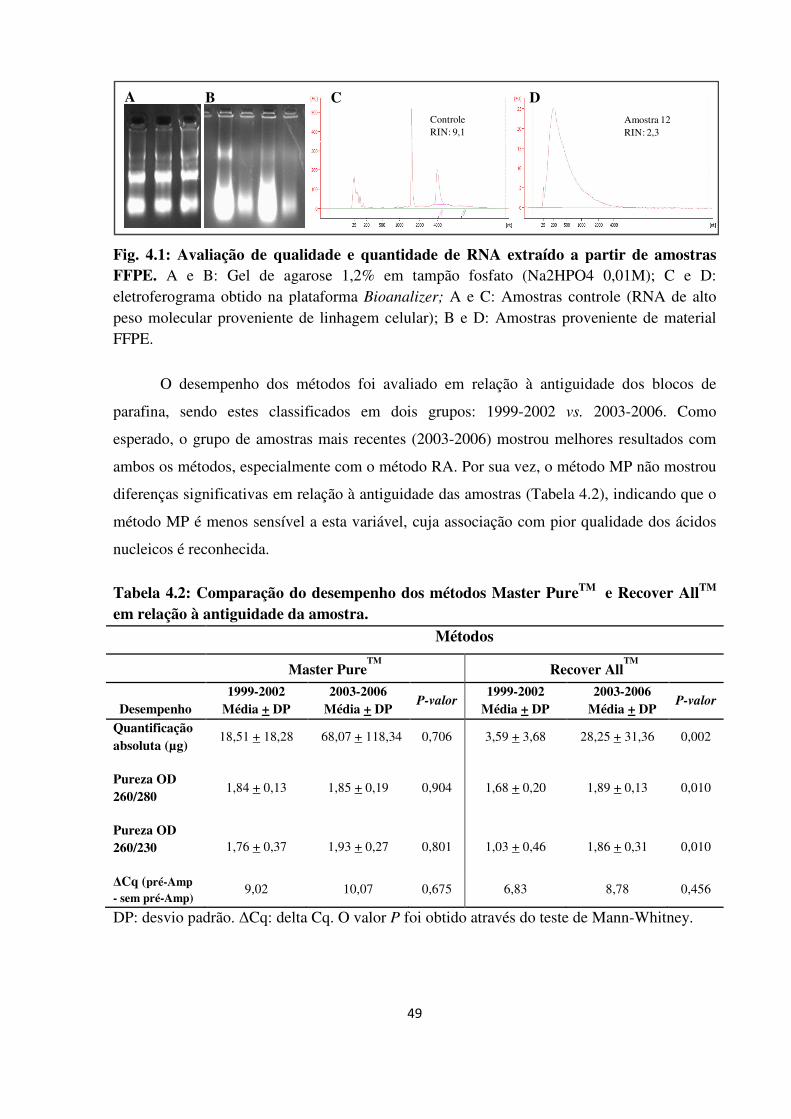
49
Fig. 4.1: Avaliação de qualidade e quantidade de RNA extraído a partir de amostras FFPE. A e B: Gel de agarose 1,2% em tampão fosfato (Na2HPO4 0,01M); C e D: eletroferograma obtido na plataforma Bioanalizer; A e C: Amostras controle (RNA de alto peso molecular proveniente de linhagem celular); B e D: Amostras proveniente de material FFPE.
O desempenho dos métodos foi avaliado em relação à antiguidade dos blocos de
parafina, sendo estes classificados em dois grupos: 1999-2002 vs. 2003-2006. Como
esperado, o grupo de amostras mais recentes (2003-2006) mostrou melhores resultados com
ambos os métodos, especialmente com o método RA. Por sua vez, o método MP não mostrou
diferenças significativas em relação à antiguidade das amostras (Tabela 4.2), indicando que o
método MP é menos sensível a esta variável, cuja associação com pior qualidade dos ácidos
nucleicos é reconhecida.
Tabela 4.2: Comparação do desempenho dos métodos Master PureTM e Recover AllTM em relação à antiguidade da amostra. Métodos
Master PureTM
Recover AllTM
Desempenho 1999-2002
Média + DP 2003-2006
Média + DP P-valor
1999-2002 Média + DP
2003-2006 Média + DP
P-valor
Quantificação absoluta (µg)
18,51 + 18,28 68,07 + 118,34 0,706 3,59 + 3,68 28,25 + 31,36 0,002
Pureza OD 260/280
1,84 + 0,13 1,85 + 0,19 0,904 1,68 + 0,20 1,89 + 0,13 0,010
Pureza OD 260/230 1,76 + 0,37 1,93 + 0,27 0,801 1,03 + 0,46 1,86 + 0,31 0,010
∆Cq (pré-Amp - sem pré-Amp)
9,02 10,07 0,675 6,83 8,78 0,456
DP: desvio padrão. ∆Cq: delta Cq. O valor P foi obtido através do teste de Mann-Whitney.
ControleRIN: 9,1
Amostra 12RIN: 2,3
AC DA B

50
Referente à amplificação dos genes de referência (GDR), os valores de Cqs foram
menores naquelas amostras que apresentaram uma relação ótima RNA/proteínas (λ260/280
entre 1,80-2,00), independentemente do método de extração utilizado e do GDR testado
(GUSB-MP, GUSB-RA, HMBS-RA, P<0,05; HMBS-MP P<0,005; teste de Mann-Whitney).
A análise comparativa dos métodos permitiu identificar o método MP como o de
melhor desempenho, devido à quantidade, qualidade, reprodutibilidade e relação custo-
benefício (Tabela 4.2).
Em um grupo de 10 amostras cujo RNA não foi amplificável com nenhuma das duas
metodologias após duas extrações e, havendo disponibilidade de um segundo bloco, a
amplificação do RNA foi realizada nesta segunda amostra. Este resultado indicou que a falta
inicial de amplificação é principalmente devida a problemas intrínsecos da amostra (por
exemplo, fixação) e levou a utilizar um novo bloco de parafina sempre que disponível,
quando ocorresse a falta de amplificação na primeira tentativa.
O procedimento de pré-Amp resultou em um aumento significativo de sensibilidade
comparada com as amostras sem pré-amplificação. O incremento médio em GUSB foi de 8,7
ciclos de valores Cq (10,1+1,5 ciclos para MP vs. 7,3+3,8 ciclos para RA).
4.1.2 Desenho experimental ANOVA tipo nested
O método MP foi selecionado para dar continuidade aos experimentos. Um desenho
experimental ANOVA tipo nested (hierárquico) foi realizado para avaliar as fontes de
variabilidade introduzidas em cada etapa da metodologia aplicada, para a análise de expressão
gênica a partir de material FFPE.
O primeiro experimento consistiu em avaliar os GDR e mostrou que GUSB
apresentava um melhor desempenho comparado com HMBS [Cq médio 23,085 vs. 24,017;
DP +0,120 vs. +0,139 e coeficiente de variação (CV), 0,520 vs. 0,581 respectivamente] (Fig.
4.2). Por isto¸ GUSB foi selecionado para normalizar os seguintes experimentos do desenho
ANOVA tipo nested. Foi observado que os Indivíduos 1 e 2 mostraram valores Cq similares
(faixa 21-23 ciclos) enquanto que o Individuo 3 mostrou valores Cqs maiores (24-26 ciclos)
(Fig. 4.3). Esta variabilidade poderia ter sido causada por diferentes quantidades de RNA
incluídas no ensaio de RT, apesar de terem sido usadas quantidades iniciais de RNA total de 1
µg para os 3 Indivíduos que tinham sido submetidas a digestão com DNAseI. Outra possível
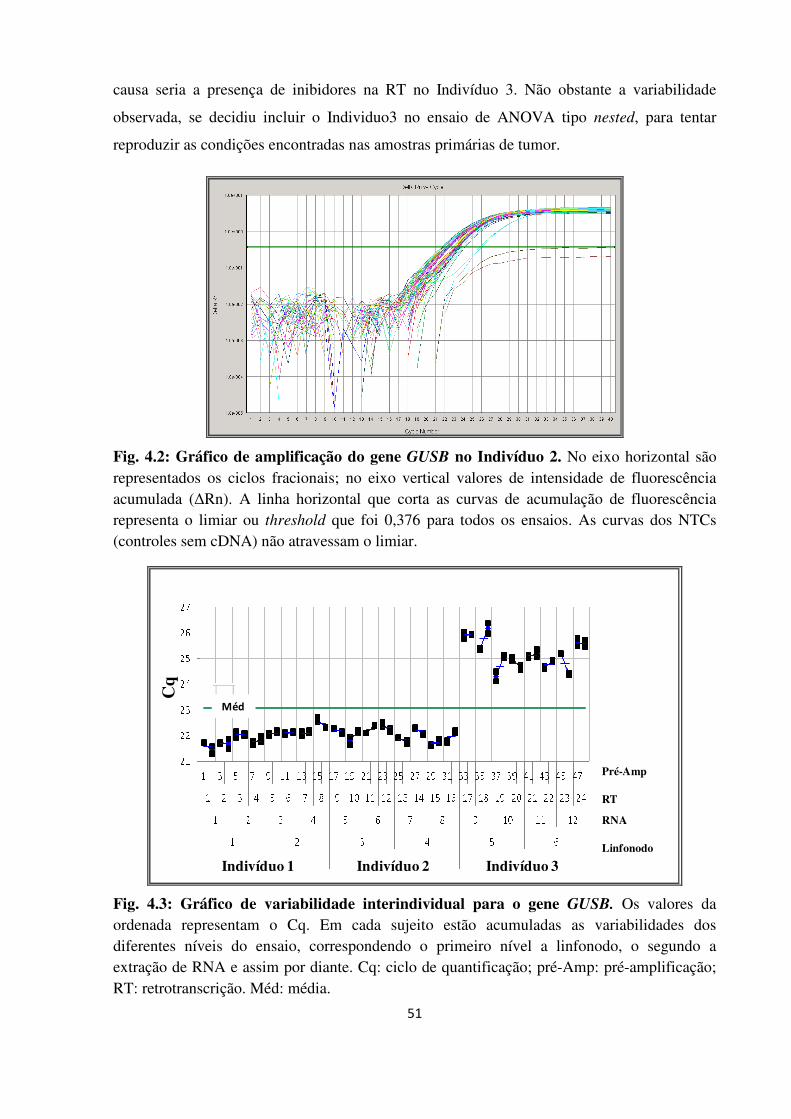
51
causa seria a presença de inibidores na RT no Indivíduo 3. Não obstante a variabilidade
observada, se decidiu incluir o Individuo3 no ensaio de ANOVA tipo nested, para tentar
reproduzir as condições encontradas nas amostras primárias de tumor.
Fig. 4.2: Gráfico de amplificação do gene GUSB no Indivíduo 2. No eixo horizontal são representados os ciclos fracionais; no eixo vertical valores de intensidade de fluorescência acumulada (∆Rn). A linha horizontal que corta as curvas de acumulação de fluorescência representa o limiar ou threshold que foi 0,376 para todos os ensaios. As curvas dos NTCs (controles sem cDNA) não atravessam o limiar.
Fig. 4.3: Gráfico de variabilidade interindividual para o gene GUSB. Os valores da ordenada representam o Cq. Em cada sujeito estão acumuladas as variabilidades dos diferentes níveis do ensaio, correspondendo o primeiro nível a linfonodo, o segundo a extração de RNA e assim por diante. Cq: ciclo de quantificação; pré-Amp: pré-amplificação; RT: retrotranscrição. Méd: média.
Cq
Subject 1 Subject 2 Subject 3
Aver
Pre-Amp
RT
RNA
Lymph node
Sujeito 1 Sujeito 2 Sujeito 3
Pré-Amp
RT
RNA
Linfonodo
Gráfico de Variabilidade
Méd
Indivíduo 1 Indivíduo 2 Indivíduo 3
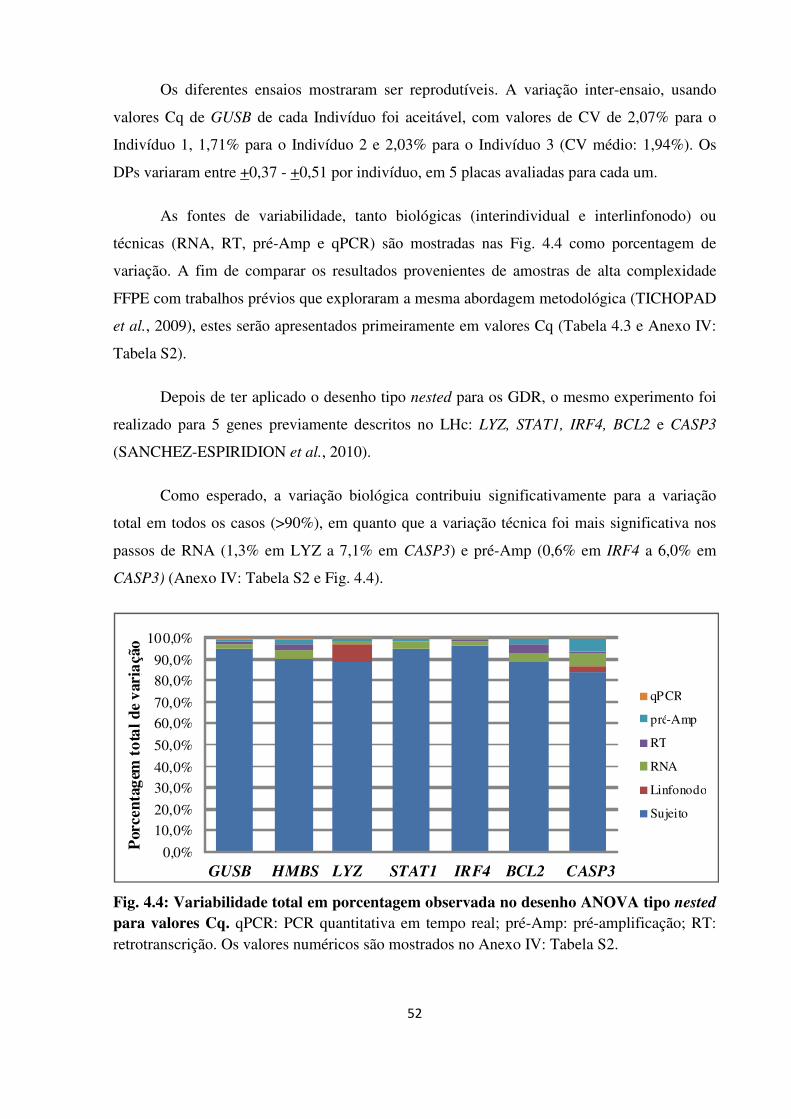
52
Os diferentes ensaios mostraram ser reprodutíveis. A variação inter-ensaio, usando
valores Cq de GUSB de cada Indivíduo foi aceitável, com valores de CV de 2,07% para o
Indivíduo 1, 1,71% para o Indivíduo 2 e 2,03% para o Indivíduo 3 (CV médio: 1,94%). Os
DPs variaram entre +0,37 - +0,51 por indivíduo, em 5 placas avaliadas para cada um.
As fontes de variabilidade, tanto biológicas (interindividual e interlinfonodo) ou
técnicas (RNA, RT, pré-Amp e qPCR) são mostradas nas Fig. 4.4 como porcentagem de
variação. A fim de comparar os resultados provenientes de amostras de alta complexidade
FFPE com trabalhos prévios que exploraram a mesma abordagem metodológica (TICHOPAD
et al., 2009), estes serão apresentados primeiramente em valores Cq (Tabela 4.3 e Anexo IV:
Tabela S2).
Depois de ter aplicado o desenho tipo nested para os GDR, o mesmo experimento foi
realizado para 5 genes previamente descritos no LHc: LYZ, STAT1, IRF4, BCL2 e CASP3
(SANCHEZ-ESPIRIDION et al., 2010).
Como esperado, a variação biológica contribuiu significativamente para a variação
total em todos os casos (>90%), em quanto que a variação técnica foi mais significativa nos
passos de RNA (1,3% em LYZ a 7,1% em CASP3) e pré-Amp (0,6% em IRF4 a 6,0% em
CASP3) (Anexo IV: Tabela S2 e Fig. 4.4).
Fig. 4.4: Variabilidade total em porcentagem observada no desenho ANOVA tipo nested para valores Cq. qPCR: PCR quantitativa em tempo real; pré-Amp: pré-amplificação; RT: retrotranscrição. Os valores numéricos são mostrados no Anexo IV: Tabela S2.
0,0%
10,0%
20,0%
30,0%
40,0%
50,0%
60,0%
70,0%
80,0%
90,0%
100,0%
GUSB HMBS LYZ STAT1 IRF4 BCL2 CASP3
( %)
qPCR
pré-Amp
RT
RNA
Linfonodo
Sujeito
Por
cent
agem
tota
l de
vari
ação
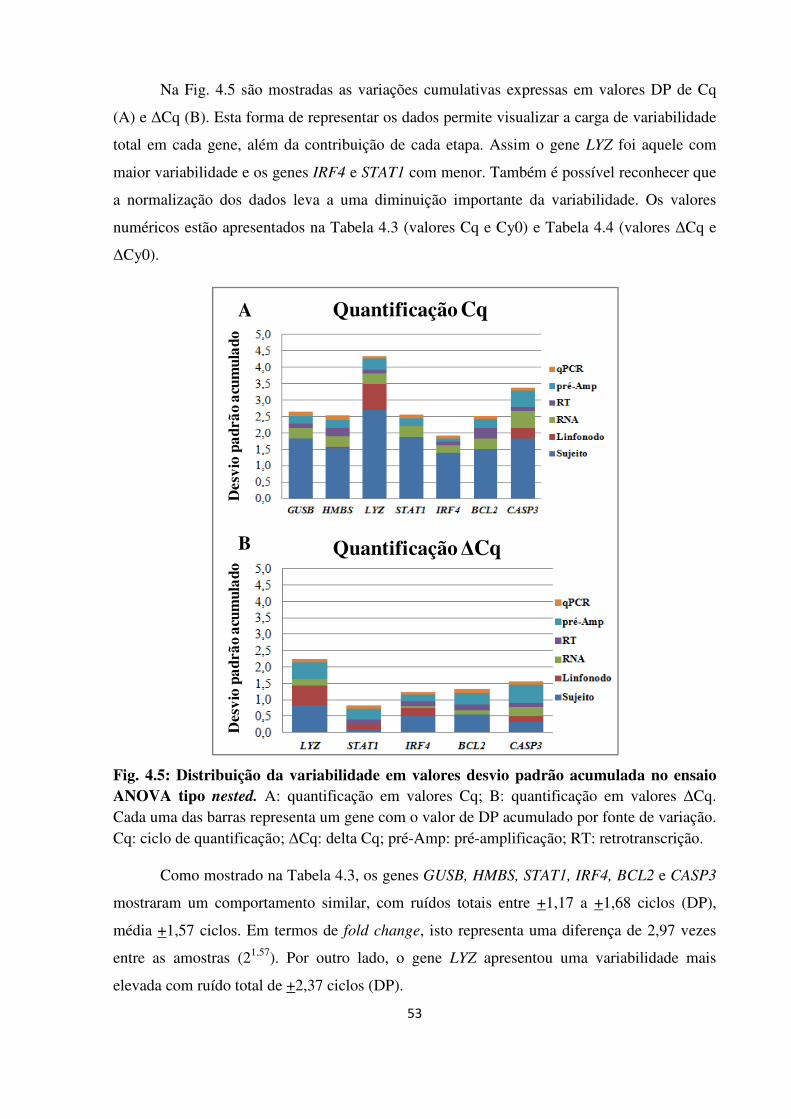
53
Na Fig. 4.5 são mostradas as variações cumulativas expressas em valores DP de Cq
(A) e ∆Cq (B). Esta forma de representar os dados permite visualizar a carga de variabilidade
total em cada gene, além da contribuição de cada etapa. Assim o gene LYZ foi aquele com
maior variabilidade e os genes IRF4 e STAT1 com menor. Também é possível reconhecer que
a normalização dos dados leva a uma diminuição importante da variabilidade. Os valores
numéricos estão apresentados na Tabela 4.3 (valores Cq e Cy0) e Tabela 4.4 (valores ∆Cq e
∆Cy0).
Fig. 4.5: Distribuição da variabilidade em valores desvio padrão acumulada no ensaio ANOVA tipo nested. A: quantificação em valores Cq; B: quantificação em valores ∆Cq. Cada uma das barras representa um gene com o valor de DP acumulado por fonte de variação. Cq: ciclo de quantificação; ∆Cq: delta Cq; pré-Amp: pré-amplificação; RT: retrotranscrição.
Como mostrado na Tabela 4.3, os genes GUSB, HMBS, STAT1, IRF4, BCL2 e CASP3
mostraram um comportamento similar, com ruídos totais entre +1,17 a +1,68 ciclos (DP),
média +1,57 ciclos. Em termos de fold change, isto representa uma diferença de 2,97 vezes
entre as amostras (21,57). Por outro lado, o gene LYZ apresentou uma variabilidade mais
elevada com ruído total de +2,37 ciclos (DP).
Quantificação∆Cq
Des
vio
padr
ão a
cum
ulad
o
Quantificação Cq
Des
vio
padr
ão a
cum
ulad
o
A
B
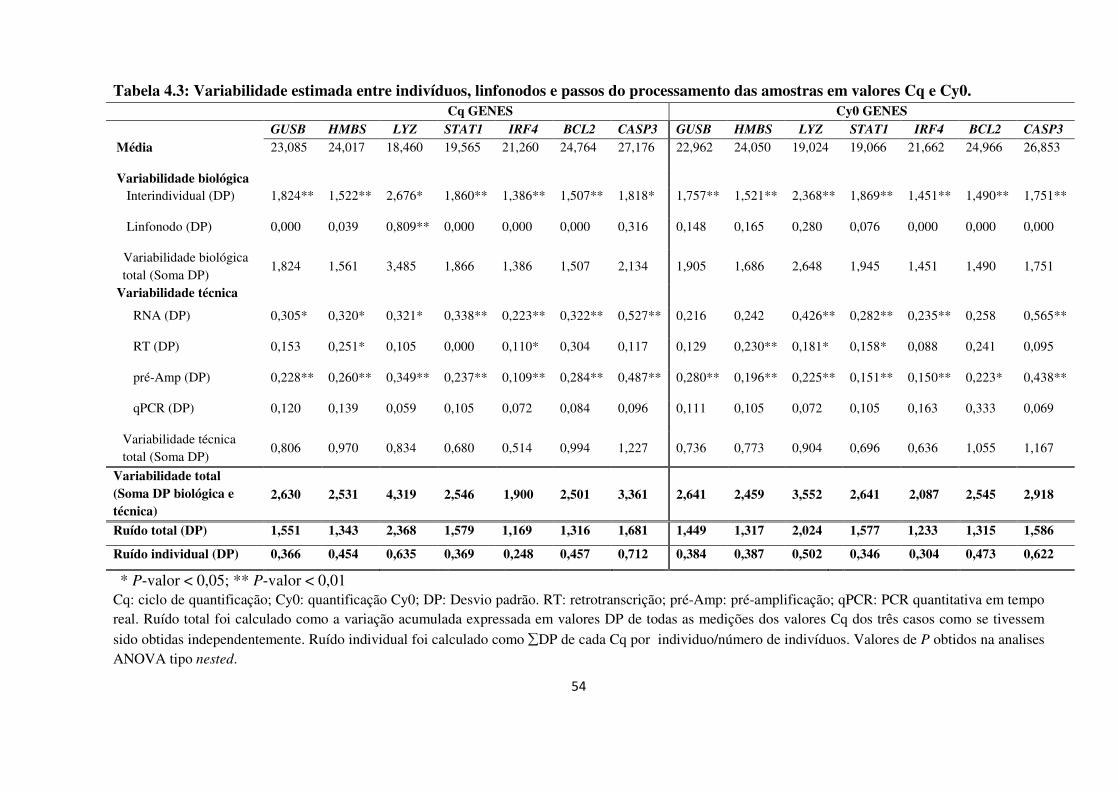
54
Tabela 4.3: Variabilidade estimada entre indivíduos, linfonodos e passos do processamento das amostras em valores Cq e Cy0. Cq GENES Cy0 GENES
GUSB HMBS LYZ STAT1 IRF4 BCL2 CASP3 GUSB HMBS LYZ STAT1 IRF4 BCL2 CASP3
Média 23,085 24,017 18,460 19,565 21,260 24,764 27,176 22,962 24,050 19,024 19,066 21,662 24,966 26,853
Variabilidade biológica Interindividual (DP) 1,824** 1,522** 2,676* 1,860** 1,386** 1,507** 1,818* 1,757** 1,521** 2,368** 1,869** 1,451** 1,490** 1,751**
Linfonodo (DP) 0,000 0,039 0,809** 0,000 0,000 0,000 0,316 0,148 0,165 0,280 0,076 0,000 0,000 0,000
Variabilidade biológica total (Soma DP)
1,824 1,561 3,485 1,866 1,386 1,507 2,134 1,905 1,686 2,648 1,945 1,451 1,490 1,751
Variabilidade técnica
RNA (DP) 0,305* 0,320* 0,321* 0,338** 0,223** 0,322** 0,527** 0,216 0,242 0,426** 0,282** 0,235** 0,258 0,565**
RT (DP) 0,153 0,251* 0,105 0,000 0,110* 0,304 0,117 0,129 0,230** 0,181* 0,158* 0,088 0,241 0,095
pré-Amp (DP) 0,228** 0,260** 0,349** 0,237** 0,109** 0,284** 0,487** 0,280** 0,196** 0,225** 0,151** 0,150** 0,223* 0,438**
qPCR (DP) 0,120 0,139 0,059 0,105 0,072 0,084 0,096 0,111 0,105 0,072 0,105 0,163 0,333 0,069
Variabilidade técnica total (Soma DP)
0,806 0,970 0,834 0,680 0,514 0,994 1,227 0,736 0,773 0,904 0,696 0,636 1,055 1,167
Variabilidade total (Soma DP biológica e técnica)
2,630 2,531 4,319 2,546 1,900
2,501 3,361 2,641 2,459 3,552 2,641 2,087
2,545 2,918
Ruído total (DP) 1,551 1,343 2,368 1,579 1,169 1,316 1,681 1,449 1,317 2,024 1,577 1,233 1,315 1,586
Ruído individual (DP) 0,366 0,454 0,635 0,369 0,248
0,457 0,712 0,384 0,387 0,502 0,346 0,304
0,473 0,622
* P-valor < 0,05; ** P-valor < 0,01
Cq: ciclo de quantificação; Cy0: quantificação Cy0; DP: Desvio padrão. RT: retrotranscrição; pré-Amp: pré-amplificação; qPCR: PCR quantitativa em tempo real. Ruído total foi calculado como a variação acumulada expressada em valores DP de todas as medições dos valores Cq dos três casos como se tivessem
sido obtidas independentemente. Ruído individual foi calculado como ∑DP de cada Cq por individuo/número de indivíduos. Valores de P obtidos na analises ANOVA tipo nested.

55
A interpretação dos valores ∆Cq foi considerada mais informativa. Primeiro, nas
condições metodológicas deste estudo, a variabilidade diminuiu sistematicamente depois da
normalização com o GDR. Isto pode ser observado na comparação dos ruídos totais dos
valores Cqs vs. ∆Cqs (Tabelas 4.3 e 4.4), e foi confirmado com o cálculo do DP usando a
covariância. Na análise ∆Cq, os ruídos totais variaram entre +0,39 e +0,70 (DP) para STAT1,
IRF4, BCL2 e CASP3, média do ruído total +0,65 (DP) que representa 1,57 fold change entre
as amostras. Entretanto LYZ manteve uma alta variabilidade, com +1,02 DP.
Em segundo lugar, considerando a variabilidade introduzida durante os passos
técnicos em valores Cq, uma contribuição significante na etapa de RNA foi registrada para
todos os genes, que desapareceu após a normalização. Por outro lado, a etapa de pré-Amp foi
uma fonte significante de variabilidade introduzida para todos os genes antes e depois da
normalização.
Com respeito à variabilidade biológica total em valores ∆Cq, LYZ mostrou o maior
valor (DP +1,43 ciclos); enquanto que STAT1 apresentou a menor variação biológica (DP
+0,24 ciclos). BCL2 mostrou uma variabilidade interindividual significante, por outro lado
IRF4 e LYZ manifestaram variabilidade interlinfonodo, isto poderia ser causado por um
número de células diferentes representadas nas amostras que expressam estes genes.
Uma vez que trabalhos prévios mostraram um efeito do nível de expressão gênica na
variabilidade (TICHOPAD et al., 2009), os genes de interesse (GDI) foram caracterizados de
acordo com os níveis de expressão: LYZ, STAT1 e IRF4 foram considerados genes de alta
expressão (∆Cq médios de -4,218, -3,029 e -1,422, respectivamente), BCL2 como
intermediário (∆Cq médio de 1,679) e CASP3 como gene de baixa expressão (∆Cq médio de
4,340) (Fig. 4.6). Genes com expressão intermediaria e baixa sofreram mais os efeitos da
manipulação técnica que os genes de alta expressão (valor médio do DP 0,930 vs. 0,541,
respectivamente, excluindo os valores de LYZ). STAT1 e IRF4 foram os menos afetados pela
variabilidade técnica, enquanto que CASP3 exibiu um profundo efeito técnico (Tabela 4.4).
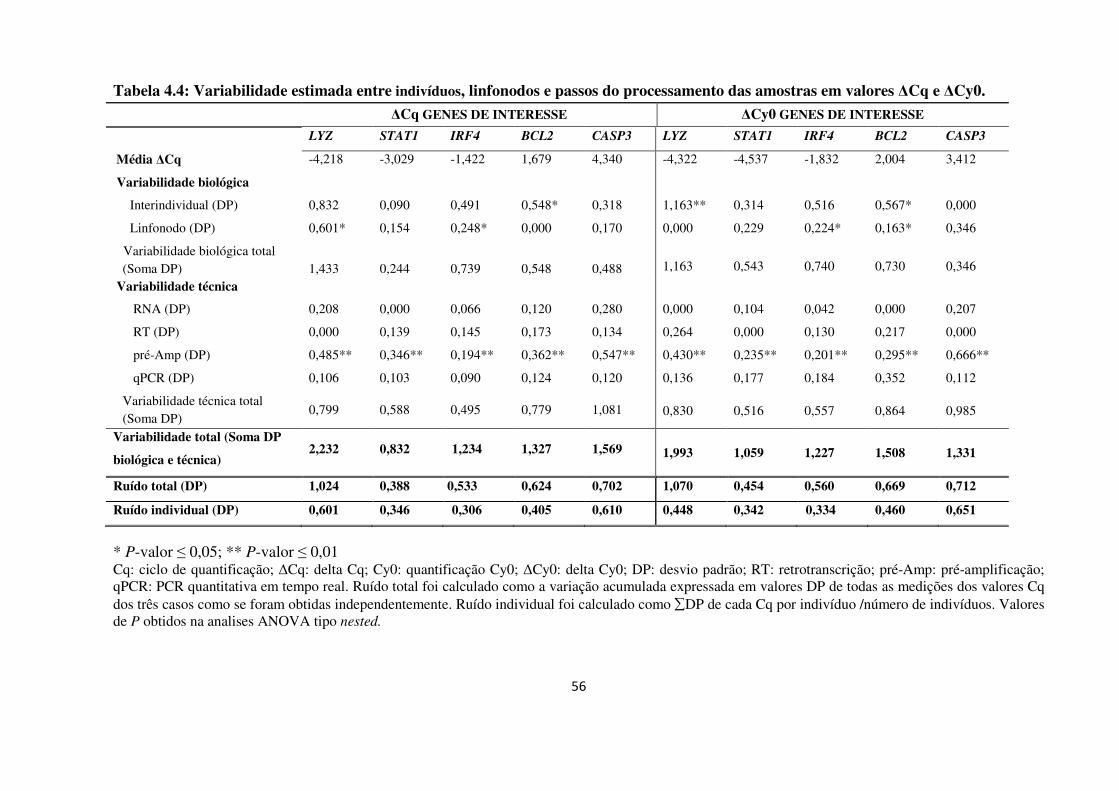
56
Tabela 4.4: Variabilidade estimada entre indivíduos, linfonodos e passos do processamento das amostras em valores ∆Cq e ∆Cy0.
* P-valor ≤ 0,05; ** P-valor ≤ 0,01 Cq: ciclo de quantificação; ∆Cq: delta Cq; Cy0: quantificação Cy0; ∆Cy0: delta Cy0; DP: desvio padrão; RT: retrotranscrição; pré-Amp: pré-amplificação; qPCR: PCR quantitativa em tempo real. Ruído total foi calculado como a variação acumulada expressada em valores DP de todas as medições dos valores Cq dos três casos como se foram obtidas independentemente. Ruído individual foi calculado como ∑DP de cada Cq por indivíduo /número de indivíduos. Valores de P obtidos na analises ANOVA tipo nested.
∆Cq GENES DE INTERESSE ∆Cy0 GENES DE INTERESSE
LYZ STAT1 IRF4 BCL2 CASP3 LYZ STAT1 IRF4 BCL2 CASP3
Média ∆Cq -4,218 -3,029 -1,422 1,679 4,340 -4,322 -4,537 -1,832 2,004 3,412
Variabilidade biológica
Interindividual (DP) 0,832 0,090 0,491 0,548* 0,318 1,163** 0,314 0,516 0,567* 0,000
Linfonodo (DP) 0,601* 0,154 0,248* 0,000 0,170 0,000 0,229 0,224* 0,163* 0,346
Variabilidade biológica total (Soma DP)
1,433
0,244
0,739
0,548
0,488
1,163
0,543
0,740
0,730
0,346
Variabilidade técnica
RNA (DP) 0,208 0,000 0,066 0,120 0,280 0,000 0,104 0,042 0,000 0,207
RT (DP) 0,000 0,139 0,145 0,173 0,134 0,264 0,000 0,130 0,217 0,000
pré-Amp (DP) 0,485** 0,346** 0,194** 0,362** 0,547** 0,430** 0,235** 0,201** 0,295** 0,666**
qPCR (DP) 0,106 0,103 0,090 0,124 0,120 0,136 0,177 0,184 0,352 0,112
Variabilidade técnica total (Soma DP)
0,799 0,588 0,495 0,779 1,081 0,830 0,516 0,557 0,864 0,985
Variabilidade total (Soma DP
biológica e técnica) 2,232 0,832 1,234
1,327 1,569 1,993 1,059 1,227 1,508 1,331
Ruído total (DP) 1,024 0,388 0,533 0,624 0,702 1,070 0,454 0,560 0,669 0,712
Ruído individual (DP) 0,601 0,346 0,306
0,405 0,610 0,448 0,342 0,334
0,460 0,651
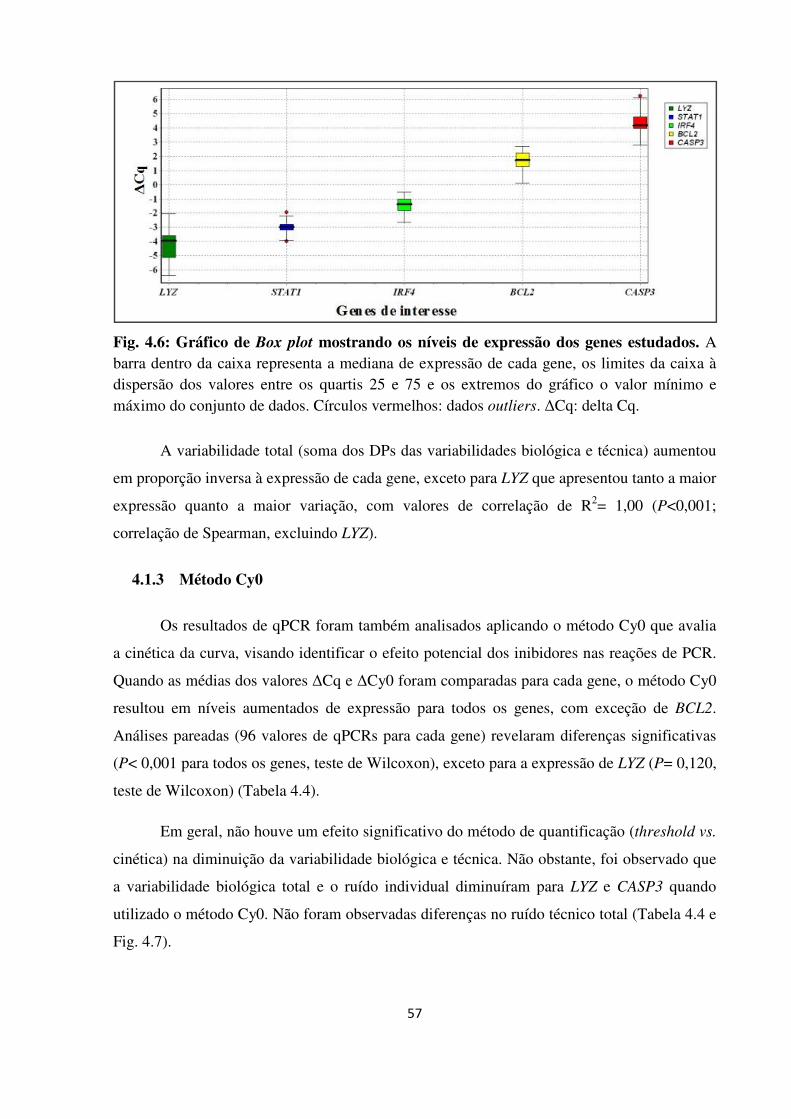
57
Fig. 4.6: Gráfico de Box plot mostrando os níveis de expressão dos genes estudados. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico o valor mínimo e máximo do conjunto de dados. Círculos vermelhos: dados outliers. ∆Cq: delta Cq.
A variabilidade total (soma dos DPs das variabilidades biológica e técnica) aumentou
em proporção inversa à expressão de cada gene, exceto para LYZ que apresentou tanto a maior
expressão quanto a maior variação, com valores de correlação de R2= 1,00 (P<0,001;
correlação de Spearman, excluindo LYZ).
4.1.3 Método Cy0
Os resultados de qPCR foram também analisados aplicando o método Cy0 que avalia
a cinética da curva, visando identificar o efeito potencial dos inibidores nas reações de PCR.
Quando as médias dos valores ∆Cq e ∆Cy0 foram comparadas para cada gene, o método Cy0
resultou em níveis aumentados de expressão para todos os genes, com exceção de BCL2.
Análises pareadas (96 valores de qPCRs para cada gene) revelaram diferenças significativas
(P< 0,001 para todos os genes, teste de Wilcoxon), exceto para a expressão de LYZ (P= 0,120,
teste de Wilcoxon) (Tabela 4.4).
Em geral, não houve um efeito significativo do método de quantificação (threshold vs.
cinética) na diminuição da variabilidade biológica e técnica. Não obstante, foi observado que
a variabilidade biológica total e o ruído individual diminuíram para LYZ e CASP3 quando
utilizado o método Cy0. Não foram observadas diferenças no ruído técnico total (Tabela 4.4 e
Fig. 4.7).
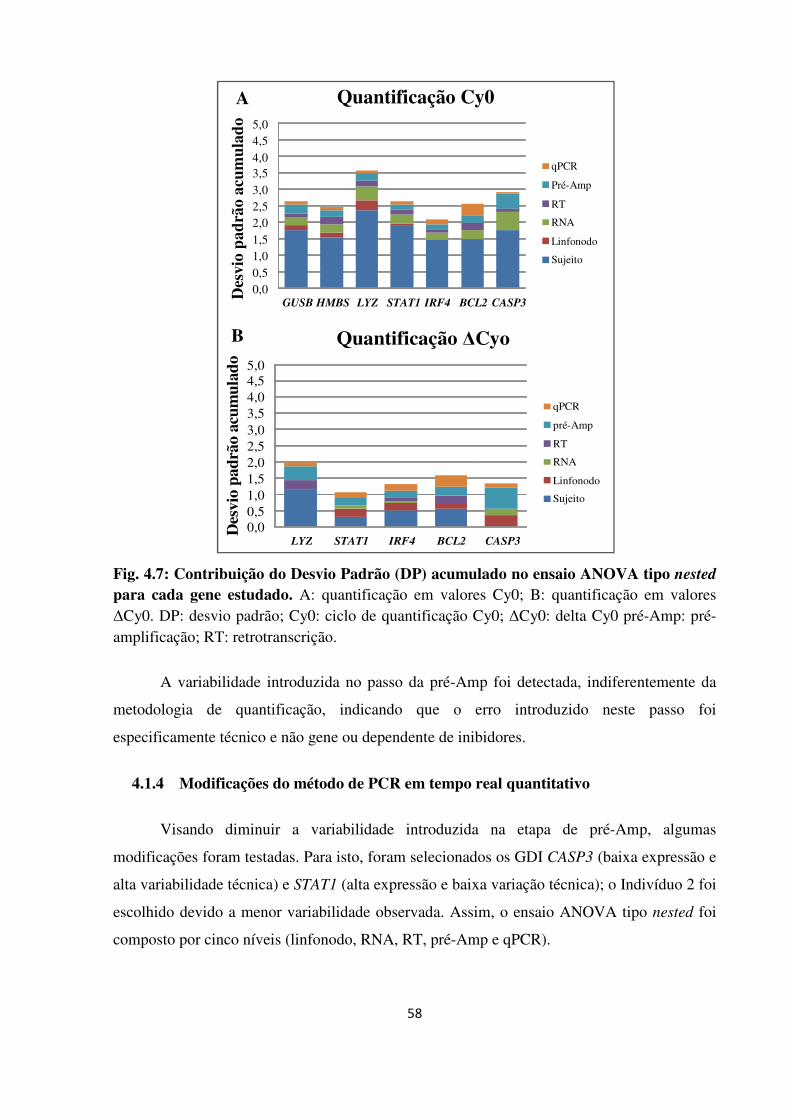
58
Fig. 4.7: Contribuição do Desvio Padrão (DP) acumulado no ensaio ANOVA tipo nested para cada gene estudado. A: quantificação em valores Cy0; B: quantificação em valores ∆Cy0. DP: desvio padrão; Cy0: ciclo de quantificação Cy0; ∆Cy0: delta Cy0 pré-Amp: pré-amplificação; RT: retrotranscrição.
A variabilidade introduzida no passo da pré-Amp foi detectada, indiferentemente da
metodologia de quantificação, indicando que o erro introduzido neste passo foi
especificamente técnico e não gene ou dependente de inibidores.
4.1.4 Modificações do método de PCR em tempo real quantitativo
Visando diminuir a variabilidade introduzida na etapa de pré-Amp, algumas
modificações foram testadas. Para isto, foram selecionados os GDI CASP3 (baixa expressão e
alta variabilidade técnica) e STAT1 (alta expressão e baixa variação técnica); o Indivíduo 2 foi
escolhido devido a menor variabilidade observada. Assim, o ensaio ANOVA tipo nested foi
composto por cinco níveis (linfonodo, RNA, RT, pré-Amp e qPCR).
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0
GUSB HMBS LYZ STAT1 IRF4 BCL2 CASP3
Quantificação Cy0
qPCR
Pré-Amp
RT RNA
Linfonodo Sujeito
A
Des
vio
padr
ão a
cum
ulad
o
0,00,51,01,52,02,53,03,54,04,55,0
LYZ STAT1 IRF4 BCL2 CASP3
Quantificação ∆Cyo
qPCR
pré-Amp
RT
RNA
Linfonodo Sujeito
B
Des
vio
padr
ão a
cum
ulad
o
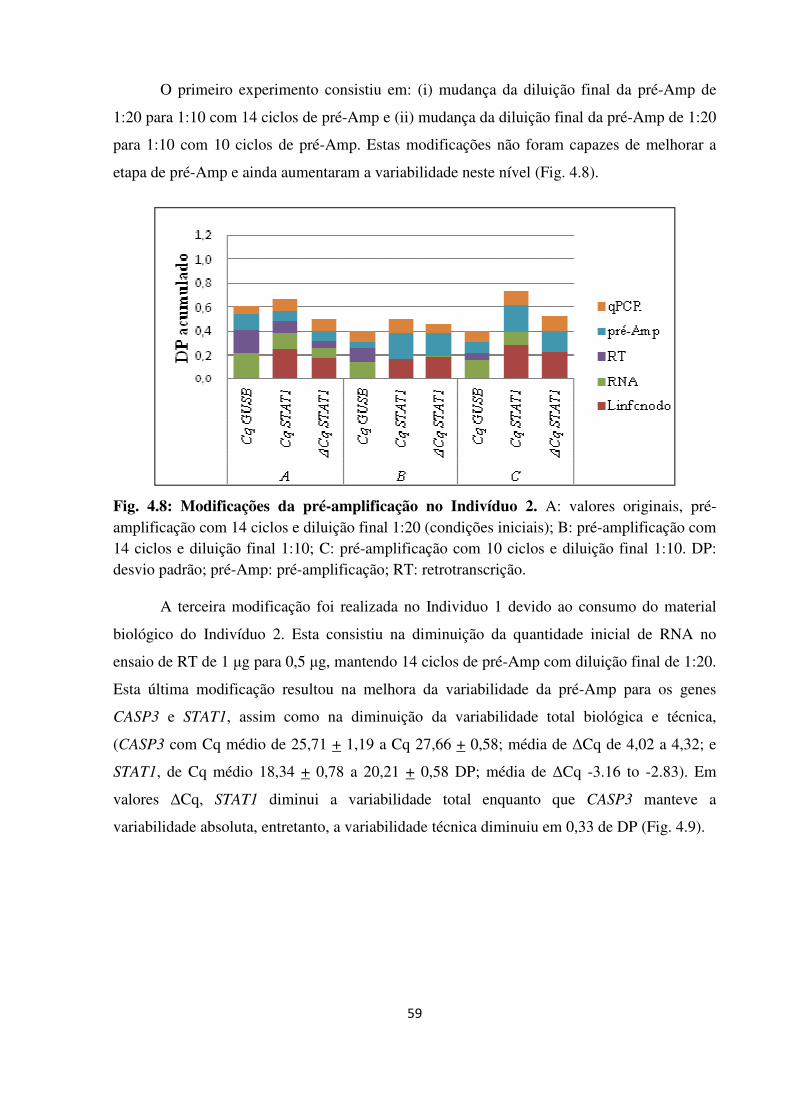
59
O primeiro experimento consistiu em: (i) mudança da diluição final da pré-Amp de
1:20 para 1:10 com 14 ciclos de pré-Amp e (ii) mudança da diluição final da pré-Amp de 1:20
para 1:10 com 10 ciclos de pré-Amp. Estas modificações não foram capazes de melhorar a
etapa de pré-Amp e ainda aumentaram a variabilidade neste nível (Fig. 4.8).
Fig. 4.8: Modificações da pré-amplificação no Indivíduo 2. A: valores originais, pré-amplificação com 14 ciclos e diluição final 1:20 (condições iniciais); B: pré-amplificação com 14 ciclos e diluição final 1:10; C: pré-amplificação com 10 ciclos e diluição final 1:10. DP: desvio padrão; pré-Amp: pré-amplificação; RT: retrotranscrição.
A terceira modificação foi realizada no Individuo 1 devido ao consumo do material
biológico do Indivíduo 2. Esta consistiu na diminuição da quantidade inicial de RNA no
ensaio de RT de 1 µg para 0,5 µg, mantendo 14 ciclos de pré-Amp com diluição final de 1:20.
Esta última modificação resultou na melhora da variabilidade da pré-Amp para os genes
CASP3 e STAT1, assim como na diminuição da variabilidade total biológica e técnica,
(CASP3 com Cq médio de 25,71 + 1,19 a Cq 27,66 + 0,58; média de ∆Cq de 4,02 a 4,32; e
STAT1, de Cq médio 18,34 + 0,78 a 20,21 + 0,58 DP; média de ∆Cq -3.16 to -2.83). Em
valores ∆Cq, STAT1 diminui a variabilidade total enquanto que CASP3 manteve a
variabilidade absoluta, entretanto, a variabilidade técnica diminuiu em 0,33 de DP (Fig. 4.9).
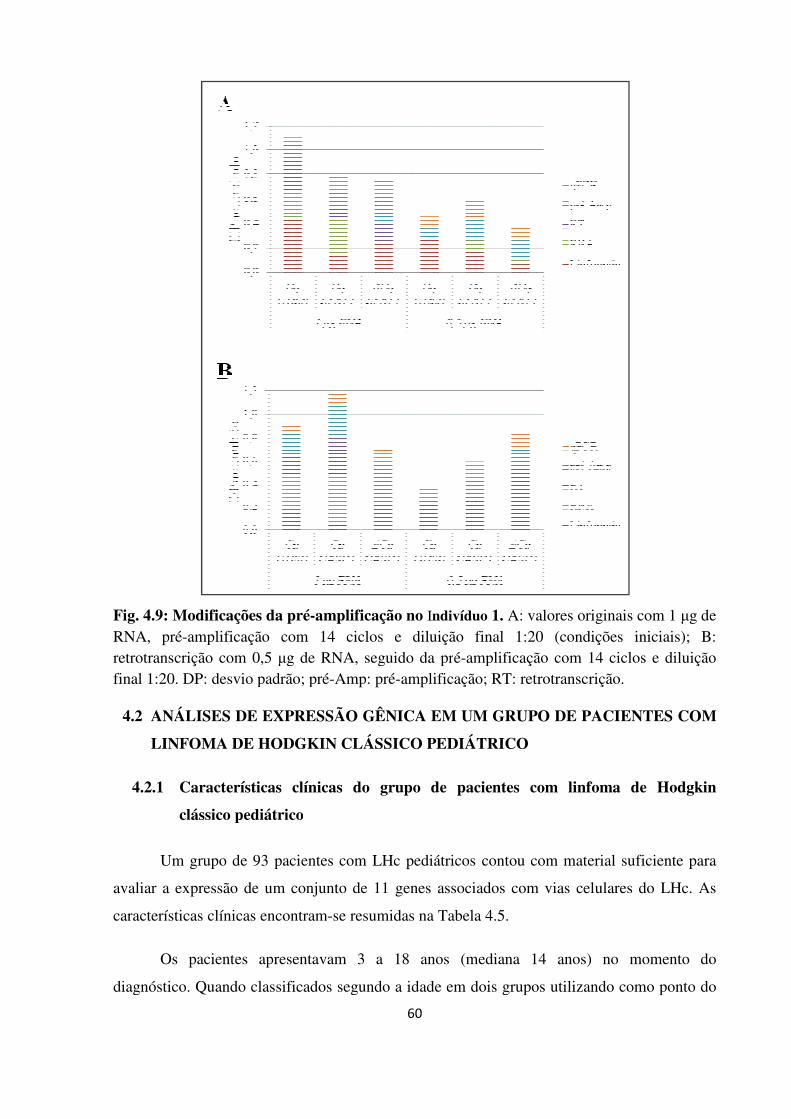
Fig. 4.9: Modificações da préRNA, pré-amplificação com 14 ciclos e diluição final 1:20 (condições iniciais); B: retrotranscrição com 0,5 µg de RNA, seguido da prfinal 1:20. DP: desvio padrão; pré
4.2 ANÁLISES DE EXPRESSÃO GÊNICA EM UM GRUPO DE PACIENTES COM
LINFOMA DE HODGKIN CLÁSSICO PEDIÁTRICO
4.2.1 Características clínicas do grupo de pacientes com linfoma de Hodgkin
clássico pediátrico
Um grupo de 93 pacientes
avaliar a expressão de um conjunto de 11 genes associados com vias celulares do LHc. As
características clínicas encontram
Os pacientes apresentavam 3 a 18 anos (mediana 14 anos) no mome
diagnóstico. Quando classificados segundo a idade em dois grupos utilizando como ponto do
60
4.9: Modificações da pré-amplificação no Indivíduo 1. A: valores originais com 1 amplificação com 14 ciclos e diluição final 1:20 (condições iniciais); B:
com 0,5 µg de RNA, seguido da pré-amplificação com 14 ciclos e diluição final 1:20. DP: desvio padrão; pré-Amp: pré-amplificação; RT: retrotranscrição.
ANÁLISES DE EXPRESSÃO GÊNICA EM UM GRUPO DE PACIENTES COM
LINFOMA DE HODGKIN CLÁSSICO PEDIÁTRICO
Características clínicas do grupo de pacientes com linfoma de Hodgkin
clássico pediátrico
Um grupo de 93 pacientes com LHc pediátricos contou com material suficiente para
avaliar a expressão de um conjunto de 11 genes associados com vias celulares do LHc. As
características clínicas encontram-se resumidas na Tabela 4.5.
apresentavam 3 a 18 anos (mediana 14 anos) no mome
diagnóstico. Quando classificados segundo a idade em dois grupos utilizando como ponto do
A: valores originais com 1 µg de amplificação com 14 ciclos e diluição final 1:20 (condições iniciais); B:
amplificação com 14 ciclos e diluição icação; RT: retrotranscrição.
ANÁLISES DE EXPRESSÃO GÊNICA EM UM GRUPO DE PACIENTES COM
Características clínicas do grupo de pacientes com linfoma de Hodgkin
ricos contou com material suficiente para
avaliar a expressão de um conjunto de 11 genes associados com vias celulares do LHc. As
apresentavam 3 a 18 anos (mediana 14 anos) no momento do
diagnóstico. Quando classificados segundo a idade em dois grupos utilizando como ponto do
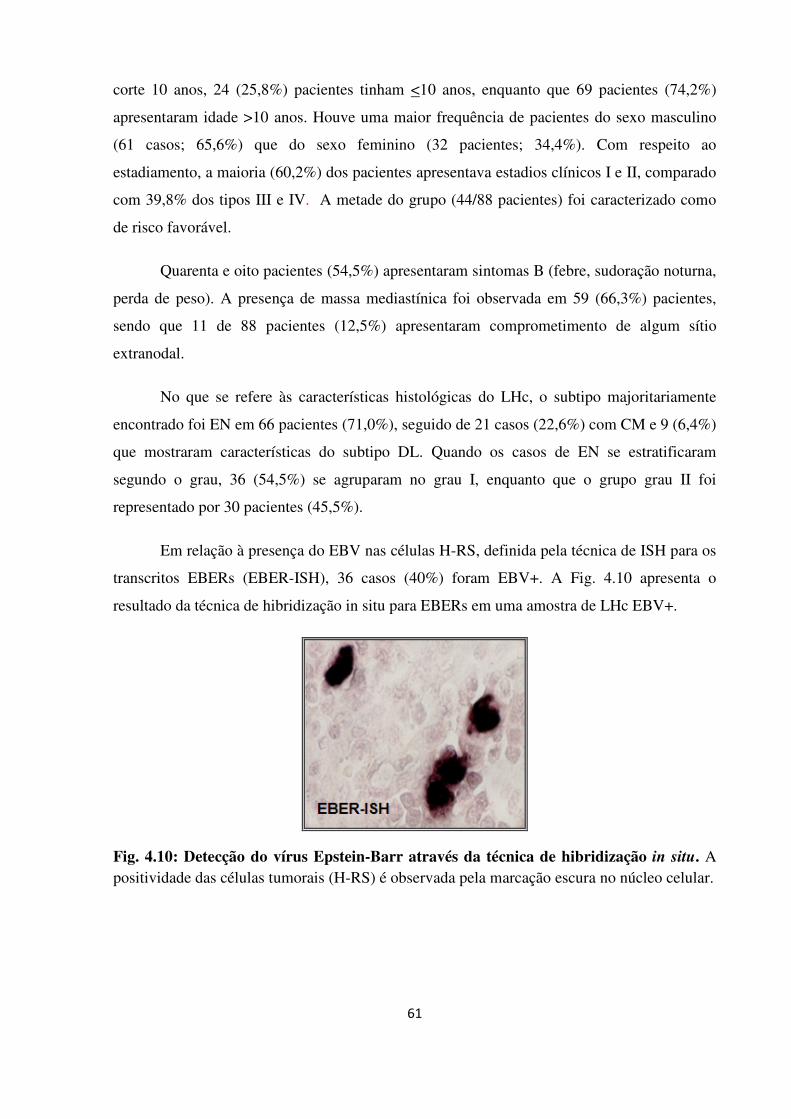
61
corte 10 anos, 24 (25,8%) pacientes tinham <10 anos, enquanto que 69 pacientes (74,2%)
apresentaram idade >10 anos. Houve uma maior frequência de pacientes do sexo masculino
(61 casos; 65,6%) que do sexo feminino (32 pacientes; 34,4%). Com respeito ao
estadiamento, a maioria (60,2%) dos pacientes apresentava estadios clínicos I e II, comparado
com 39,8% dos tipos III e IV. A metade do grupo (44/88 pacientes) foi caracterizado como
de risco favorável.
Quarenta e oito pacientes (54,5%) apresentaram sintomas B (febre, sudoração noturna,
perda de peso). A presença de massa mediastínica foi observada em 59 (66,3%) pacientes,
sendo que 11 de 88 pacientes (12,5%) apresentaram comprometimento de algum sítio
extranodal.
No que se refere às características histológicas do LHc, o subtipo majoritariamente
encontrado foi EN em 66 pacientes (71,0%), seguido de 21 casos (22,6%) com CM e 9 (6,4%)
que mostraram características do subtipo DL. Quando os casos de EN se estratificaram
segundo o grau, 36 (54,5%) se agruparam no grau I, enquanto que o grupo grau II foi
representado por 30 pacientes (45,5%).
Em relação à presença do EBV nas células H-RS, definida pela técnica de ISH para os
transcritos EBERs (EBER-ISH), 36 casos (40%) foram EBV+. A Fig. 4.10 apresenta o
resultado da técnica de hibridização in situ para EBERs em uma amostra de LHc EBV+.
Fig. 4.10: Detecção do vírus Epstein-Barr através da técnica de hibridização in situ. A positividade das células tumorais (H-RS) é observada pela marcação escura no núcleo celular.
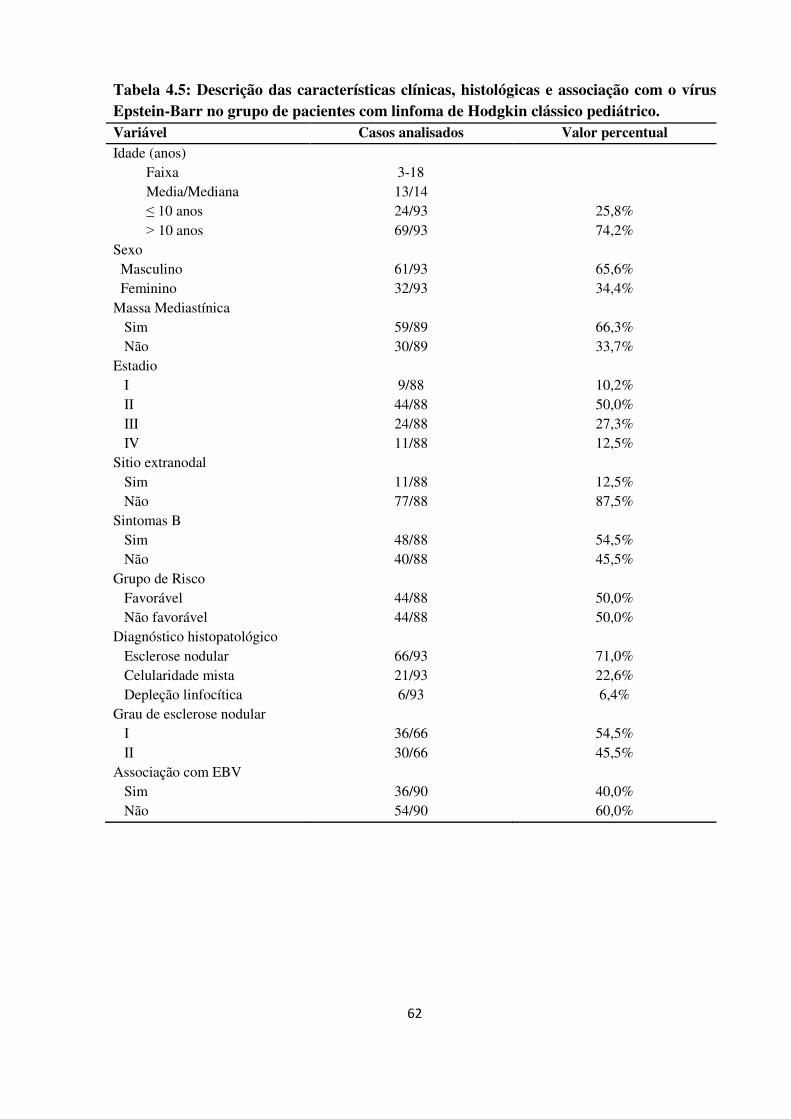
62
Tabela 4.5: Descrição das características clínicas, histológicas e associação com o vírus Epstein-Barr no grupo de pacientes com linfoma de Hodgkin clássico pediátrico. Variável Casos analisados Valor percentual Idade (anos) Faixa 3-18 Media/Mediana 13/14 ≤ 10 anos 24/93 25,8% ˃ 10 anos 69/93 74,2% Sexo Masculino 61/93 65,6% Feminino 32/93 34,4% Massa Mediastínica Sim 59/89 66,3% Não 30/89 33,7% Estadio I 9/88 10,2% II 44/88 50,0% III 24/88 27,3% IV 11/88 12,5% Sitio extranodal Sim 11/88 12,5% Não 77/88 87,5% Sintomas B Sim 48/88 54,5% Não 40/88 45,5% Grupo de Risco Favorável 44/88 50,0% Não favorável 44/88 50,0% Diagnóstico histopatológico Esclerose nodular 66/93 71,0% Celularidade mista 21/93 22,6% Depleção linfocítica 6/93 6,4% Grau de esclerose nodular I 36/66 54,5% II 30/66 45,5% Associação com EBV Sim 36/90 40,0% Não 54/90 60,0%
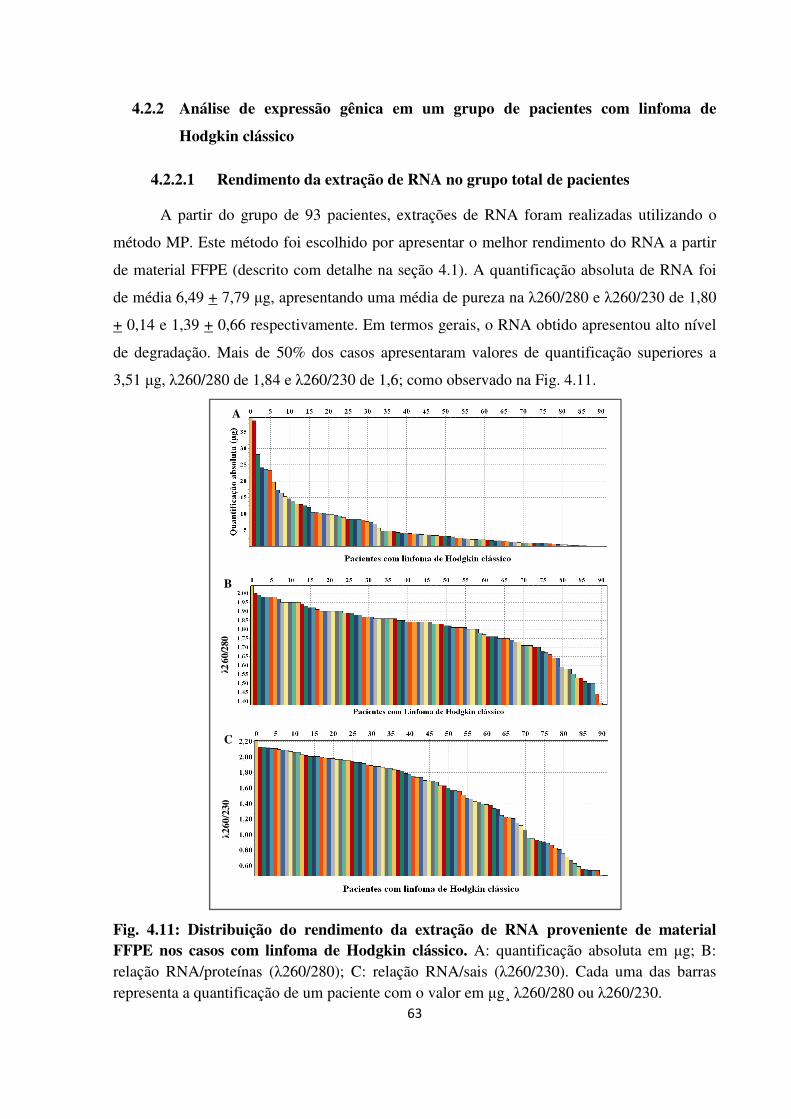
63
4.2.2 Análise de expressão gênica em um grupo de pacientes com linfoma de
Hodgkin clássico
4.2.2.1 Rendimento da extração de RNA no grupo total de pacientes
A partir do grupo de 93 pacientes, extrações de RNA foram realizadas utilizando o
método MP. Este método foi escolhido por apresentar o melhor rendimento do RNA a partir
de material FFPE (descrito com detalhe na seção 4.1). A quantificação absoluta de RNA foi
de média 6,49 + 7,79 µg, apresentando uma média de pureza na λ260/280 e λ260/230 de 1,80
+ 0,14 e 1,39 + 0,66 respectivamente. Em termos gerais, o RNA obtido apresentou alto nível
de degradação. Mais de 50% dos casos apresentaram valores de quantificação superiores a
3,51 µg, λ260/280 de 1,84 e λ260/230 de 1,6; como observado na Fig. 4.11.
Fig. 4.11: Distribuição do rendimento da extração de RNA proveniente de material FFPE nos casos com linfoma de Hodgkin clássico. A: quantificação absoluta em µg; B: relação RNA/proteínas (λ260/280); C: relação RNA/sais (λ260/230). Cada uma das barras representa a quantificação de um paciente com o valor em µg¸ λ260/280 ou λ260/230.
λ260
/280
λ
260/
230
A
B
C

64
4.2.2.2 Avaliação da qualidade e determinação dos níveis de expressão gênica
Um grupo de 11 genes pertences às vias do ciclo celular (CENPF, CDK1, CCNA2,
CCNE2 e HMMR), apoptose (BCL2, BCL2L1 e CASP3), genes do microambiente das células
relacionados à ativação de macrófagos (LYZ e STAT1) e IRF4 (células H-RS e
microambiente) foram caracterizados por ser altamente expressos em um grupo de pacientes
adultos com LHc avançado (SANCHEZ-ESPIRIDION et al., 2010). A avaliação por RT-
qPCR foi realizada seguindo os métodos com melhor desempenho previamente definidos (ver
secção 4.1.4: modificações do método de RT-qPCR).
Dois critérios de qualidade foram utilizados para a exclusão dos casos das análises
estatísticas: 1) falta de amplificação dos GDR (Cq>35 ciclos), o que levou à exclusão de 6
casos e; 2) quando 5 ou mas GDI amplificaram com um Cq>35 ciclos, nesta situação 3 casos
adicionais foram excluídos. Assim, 84 pacientes tiveram critérios de qualidade para ser
incluídos no estudo de expressão gênica.
Em relação aos níveis de expressão gênica no grupo avaliado, foram identificados dois
grupos, utilizando como valor de quantificação a mediana de fold change de cada gene: o
grupo de genes de alta expressão composto por IRF4 (0,54); BCL2L1 (1,11); STAT1 (2,51) e
LYZ (3,17) e um grupo de genes de baixa expressão formados por CCNA2 (-5,40); HMMR (-
4,83); CDK1 (-4,75); CASP3 (-4,47); CCNE2 (-3,97); CENPF (-2,78) e BCL2 (-2,46) (Fig.
4.12).
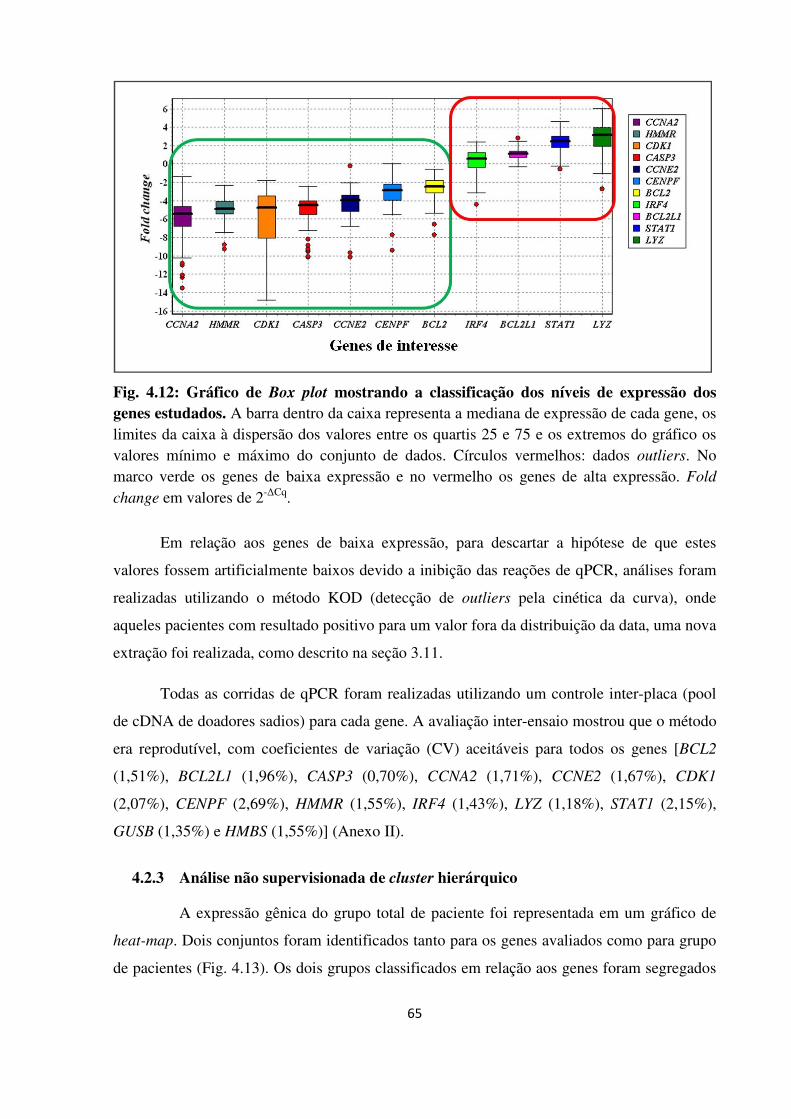
65
Fig. 4.12: Gráfico de Box plot mostrando a classificação dos níveis de expressão dos genes estudados. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. Círculos vermelhos: dados outliers. No marco verde os genes de baixa expressão e no vermelho os genes de alta expressão. Fold change em valores de 2-∆Cq.
Em relação aos genes de baixa expressão, para descartar a hipótese de que estes
valores fossem artificialmente baixos devido a inibição das reações de qPCR, análises foram
realizadas utilizando o método KOD (detecção de outliers pela cinética da curva), onde
aqueles pacientes com resultado positivo para um valor fora da distribuição da data, uma nova
extração foi realizada, como descrito na seção 3.11.
Todas as corridas de qPCR foram realizadas utilizando um controle inter-placa (pool
de cDNA de doadores sadios) para cada gene. A avaliação inter-ensaio mostrou que o método
era reprodutível, com coeficientes de variação (CV) aceitáveis para todos os genes [BCL2
(1,51%), BCL2L1 (1,96%), CASP3 (0,70%), CCNA2 (1,71%), CCNE2 (1,67%), CDK1
(2,07%), CENPF (2,69%), HMMR (1,55%), IRF4 (1,43%), LYZ (1,18%), STAT1 (2,15%),
GUSB (1,35%) e HMBS (1,55%)] (Anexo II).
4.2.3 Análise não supervisionada de cluster hierárquico
A expressão gênica do grupo total de paciente foi representada em um gráfico de
heat-map. Dois conjuntos foram identificados tanto para os genes avaliados como para grupo
de pacientes (Fig. 4.13). Os dois grupos classificados em relação aos genes foram segregados
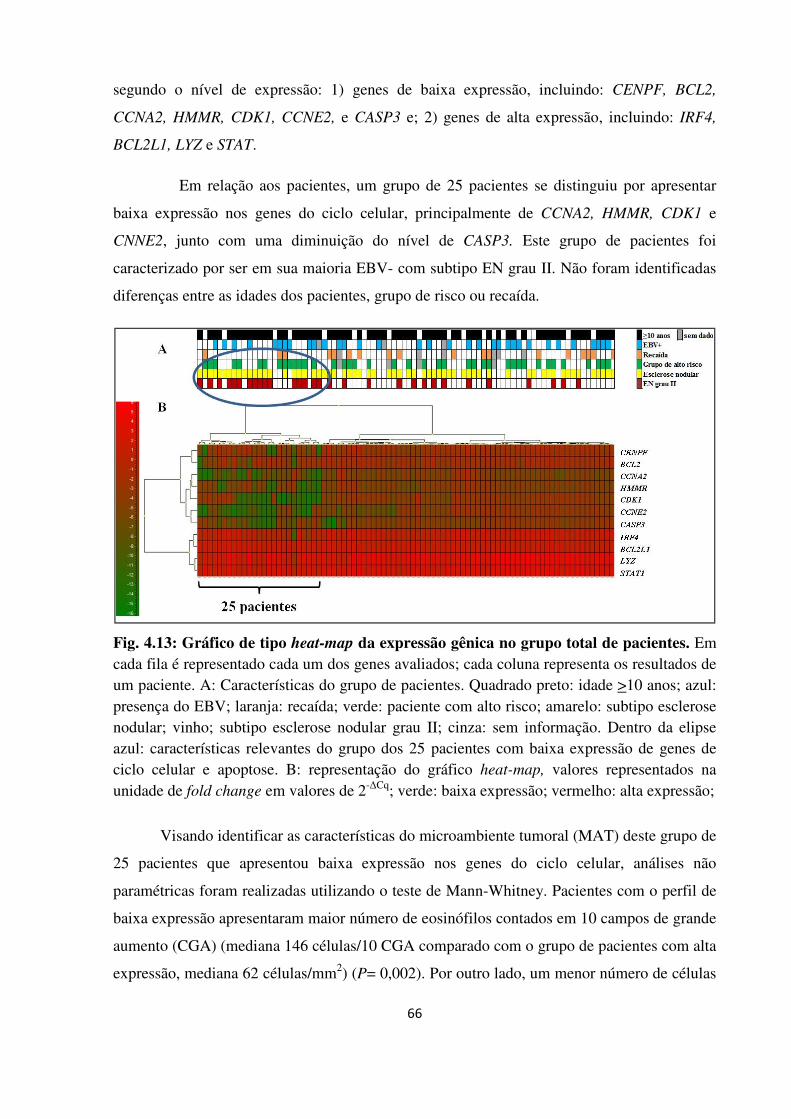
66
segundo o nível de expressão: 1) genes de baixa expressão, incluindo: CENPF, BCL2,
CCNA2, HMMR, CDK1, CCNE2, e CASP3 e; 2) genes de alta expressão, incluindo: IRF4,
BCL2L1, LYZ e STAT.
Em relação aos pacientes, um grupo de 25 pacientes se distinguiu por apresentar
baixa expressão nos genes do ciclo celular, principalmente de CCNA2, HMMR, CDK1 e
CNNE2, junto com uma diminuição do nível de CASP3. Este grupo de pacientes foi
caracterizado por ser em sua maioria EBV- com subtipo EN grau II. Não foram identificadas
diferenças entre as idades dos pacientes, grupo de risco ou recaída.
Fig. 4.13: Gráfico de tipo heat-map da expressão gênica no grupo total de pacientes. Em cada fila é representado cada um dos genes avaliados; cada coluna representa os resultados de um paciente. A: Características do grupo de pacientes. Quadrado preto: idade >10 anos; azul: presença do EBV; laranja: recaída; verde: paciente com alto risco; amarelo: subtipo esclerose nodular; vinho; subtipo esclerose nodular grau II; cinza: sem informação. Dentro da elipse azul: características relevantes do grupo dos 25 pacientes com baixa expressão de genes de ciclo celular e apoptose. B: representação do gráfico heat-map, valores representados na unidade de fold change em valores de 2-∆Cq; verde: baixa expressão; vermelho: alta expressão;
Visando identificar as características do microambiente tumoral (MAT) deste grupo de
25 pacientes que apresentou baixa expressão nos genes do ciclo celular, análises não
paramétricas foram realizadas utilizando o teste de Mann-Whitney. Pacientes com o perfil de
baixa expressão apresentaram maior número de eosinófilos contados em 10 campos de grande
aumento (CGA) (mediana 146 células/10 CGA comparado com o grupo de pacientes com alta
expressão, mediana 62 células/mm2) (P= 0,002). Por outro lado, um menor número de células

67
citotóxicas GRZB+ (mediana 5 vs. 17 células/ mm2) (P= 0,035), linfócitos Ki67+ (mediana 61
vs. 105 células mm2) (P= 0,008), MAF+ (mediana 26 vs. 82 células mm2) (P= 0,008) e
macrófagos CD163+ (mediana 76 vs. 202 células mm2) (P= 0,005) foram observadas no
grupo de alta expressão vs. o grupo de baixa expressão, respectivamente (Fig. 4.14).

Fig. 4.14: Gráficos de Box
pacientes com linfoma de Hodgkin clássico ciclo celular. A: em relação à mediana do número de eosinófilos quantificados em 10 campos de grande aumento (CGA); B: em relação à mediana do número de células GRZB+/mmem relação à mediana do número de linfócitos Ki67+/mmnúmero de células MAF+/mmA barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo máximo do conjunto de dados. * e **: diferença estatisticamente significativa aos níveis P<0,05 e P<0,01; teste Mann
68
Box plot das características das populações do microambiente em com linfoma de Hodgkin clássico pediátricos com baixa expressão nos genes do
em relação à mediana do número de eosinófilos quantificados em 10 campos de grande aumento (CGA); B: em relação à mediana do número de células GRZB+/mmem relação à mediana do número de linfócitos Ki67+/mm2; D: em relação à mediana do
+/mm2; F: em relação à mediana do número de células CD163+/mmA barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo máximo do conjunto de dados. * e **: diferença estatisticamente significativa aos níveis
0,01; teste Mann-Whitney.
das características das populações do microambiente em pediátricos com baixa expressão nos genes do
em relação à mediana do número de eosinófilos quantificados em 10 campos de grande aumento (CGA); B: em relação à mediana do número de células GRZB+/mm2; C:
; D: em relação à mediana do ; F: em relação à mediana do número de células CD163+/mm2.
A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. * e **: diferença estatisticamente significativa aos níveis
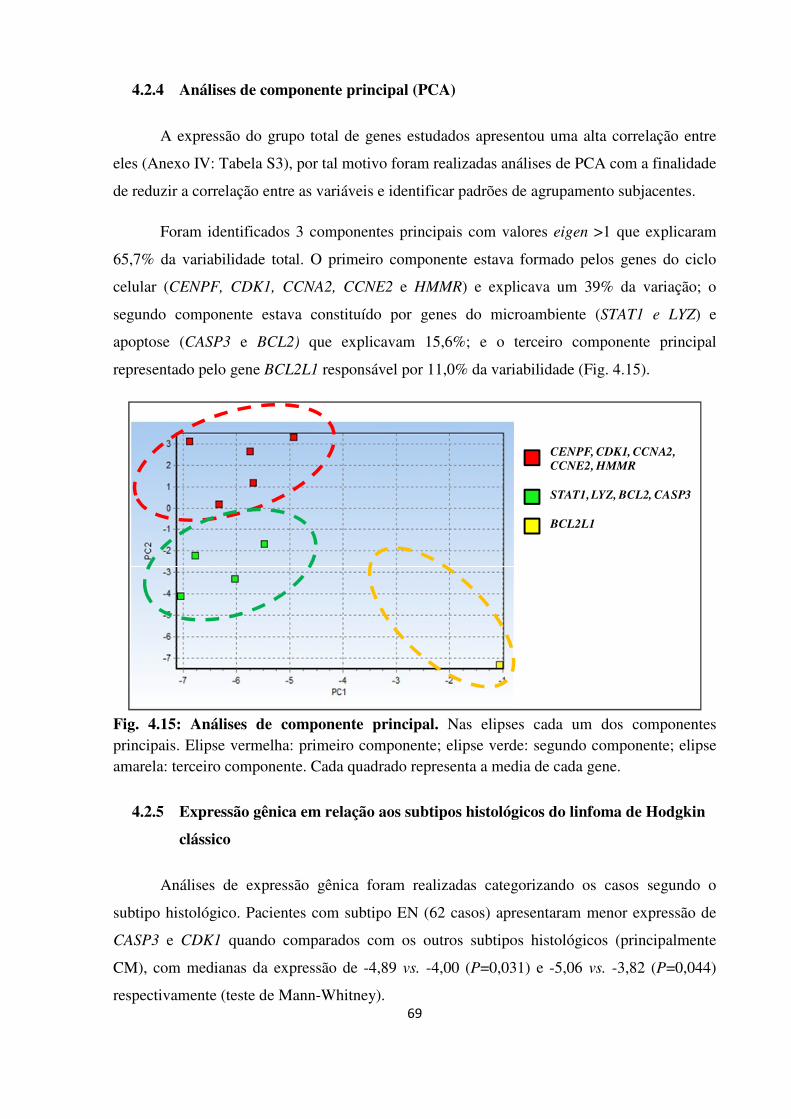
69
4.2.4 Análises de componente principal (PCA)
A expressão do grupo total de genes estudados apresentou uma alta correlação entre
eles (Anexo IV: Tabela S3), por tal motivo foram realizadas análises de PCA com a finalidade
de reduzir a correlação entre as variáveis e identificar padrões de agrupamento subjacentes.
Foram identificados 3 componentes principais com valores eigen >1 que explicaram
65,7% da variabilidade total. O primeiro componente estava formado pelos genes do ciclo
celular (CENPF, CDK1, CCNA2, CCNE2 e HMMR) e explicava um 39% da variação; o
segundo componente estava constituído por genes do microambiente (STAT1 e LYZ) e
apoptose (CASP3 e BCL2) que explicavam 15,6%; e o terceiro componente principal
representado pelo gene BCL2L1 responsável por 11,0% da variabilidade (Fig. 4.15).
Fig. 4.15: Análises de componente principal. Nas elipses cada um dos componentes principais. Elipse vermelha: primeiro componente; elipse verde: segundo componente; elipse amarela: terceiro componente. Cada quadrado representa a media de cada gene.
4.2.5 Expressão gênica em relação aos subtipos histológicos do linfoma de Hodgkin
clássico
Análises de expressão gênica foram realizadas categorizando os casos segundo o
subtipo histológico. Pacientes com subtipo EN (62 casos) apresentaram menor expressão de
CASP3 e CDK1 quando comparados com os outros subtipos histológicos (principalmente
CM), com medianas da expressão de -4,89 vs. -4,00 (P=0,031) e -5,06 vs. -3,82 (P=0,044)
respectivamente (teste de Mann-Whitney).
CENPF, CDK1, CCNA2,
CCNE2, HMMR
STAT1, LYZ, BCL2, CASP3
BCL2L1
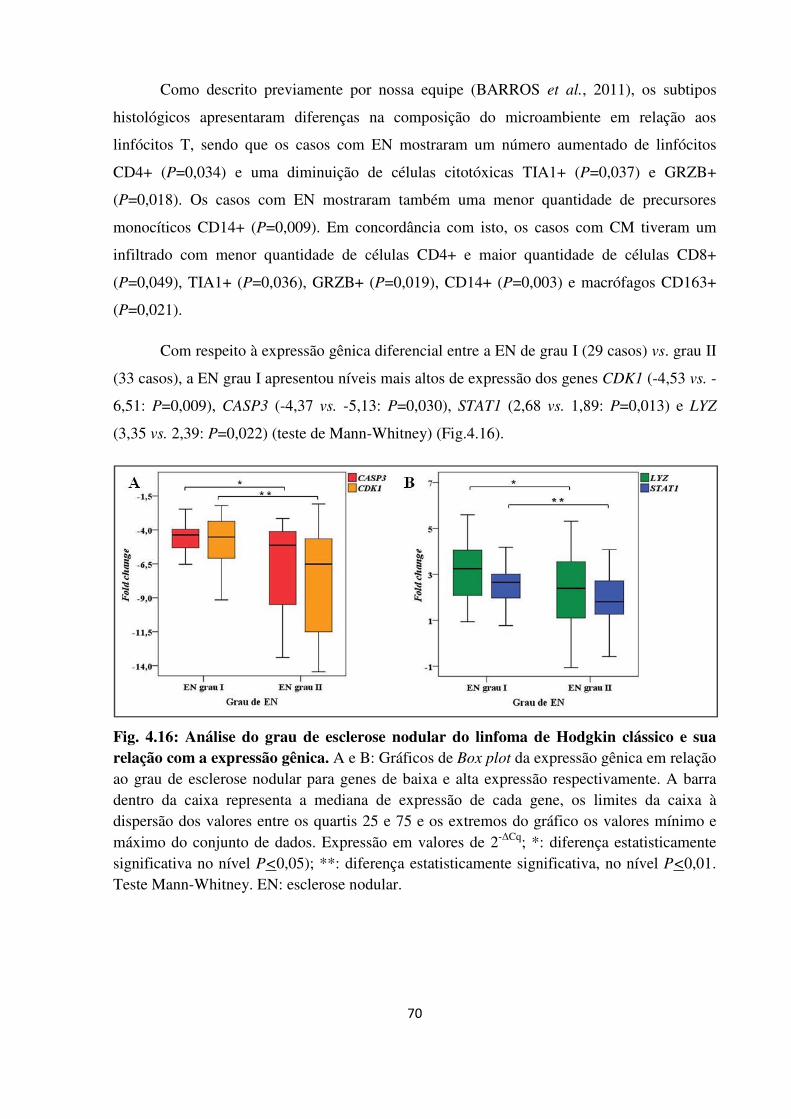
70
Como descrito previamente por nossa equipe (BARROS et al., 2011), os subtipos
histológicos apresentaram diferenças na composição do microambiente em relação aos
linfócitos T, sendo que os casos com EN mostraram um número aumentado de linfócitos
CD4+ (P=0,034) e uma diminuição de células citotóxicas TIA1+ (P=0,037) e GRZB+
(P=0,018). Os casos com EN mostraram também uma menor quantidade de precursores
monocíticos CD14+ (P=0,009). Em concordância com isto, os casos com CM tiveram um
infiltrado com menor quantidade de células CD4+ e maior quantidade de células CD8+
(P=0,049), TIA1+ (P=0,036), GRZB+ (P=0,019), CD14+ (P=0,003) e macrófagos CD163+
(P=0,021).
Com respeito à expressão gênica diferencial entre a EN de grau I (29 casos) vs. grau II
(33 casos), a EN grau I apresentou níveis mais altos de expressão dos genes CDK1 (-4,53 vs. -
6,51: P=0,009), CASP3 (-4,37 vs. -5,13: P=0,030), STAT1 (2,68 vs. 1,89: P=0,013) e LYZ
(3,35 vs. 2,39: P=0,022) (teste de Mann-Whitney) (Fig.4.16).
Fig. 4.16: Análise do grau de esclerose nodular do linfoma de Hodgkin clássico e sua relação com a expressão gênica. A e B: Gráficos de Box plot da expressão gênica em relação ao grau de esclerose nodular para genes de baixa e alta expressão respectivamente. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. Expressão em valores de 2-∆Cq; *: diferença estatisticamente significativa no nível P<0,05); **: diferença estatisticamente significativa, no nível P<0,01. Teste Mann-Whitney. EN: esclerose nodular.

71
4.2.6 Correlação da expressão gênica com marcadores avaliados por
imunohistoquímica nas células de Hodgkin-Reed Sternberg e no
microambiente tumoral
Uma vez que o estudo da expressão gênica não é capaz de identificar as células
expressando os RNAm específicos, nós visamos identificar associação entre os níveis de
expressão gênica e a expressão de algumas proteínas envolvidas em vias celulares específicas
ou com marcadores de sub-populações celulares infiltrantes no MAT.
4.2.6.1 Marcadores das células de Hodgkin-Reed Sternberg e sua associação com
a expressão gênica
As células H-RS tinham sido caracterizadas em relação ao número, presença de
divisão mitótica, expressão de Ki67 (marcador clássico de proliferação), assim como à
expressão de p21, p53, e BCL2 em um trabalho da equipe previamente publicado (BARROS
et al., 2010). As correlações entre as expressões proteicas e gênicas são mostradas na Tabela
4.6.
O número de células de H-RS e o número de células em mitoses não tiveram
correlação com os níveis de expressão dos genes analisados. Considerando P<0,01 como
valor significativo para as análises de correlação para mitigar os efeitos das comparações
múltiplas.
Quando categorizados os casos com ponto de corte em 50% de células H-RS
expressando as proteínas, foi observado que 35/77 (45,5%) dos casos apresentaram
positividade para p53 e 39/74 (52,7%) expressavam p21. Pacientes com baixos níveis de p53
(<50%) apresentaram um aumento da expressão de RNAm de CASP3 (P=0,04; teste de
Mann-Whitney). Não houve correlação entre a expressão de p21 e qualquer um dos genes
investigados.
Devido ao fato de a imunomarcação de BCL2 ter sido registrada através da
porcentagem de células H-RS, esta foi categorizada em positiva: BCL2 > 10% das células H-
RS e negativa: <10% das células H-RS. Não houve diferença na expressão de RNAm de
BCL2 para estas duas categorias (P=0,63; teste de Mann-Whitney).

72
4.2.6.2 Microambiente tumoral e sua associação com a expressão gênica
Em relação ao MAT, a caracterização das sub-populações celulares T infiltrantes se
encontra publicada em BARROS et al. (2012) (Anexo V). Adicionalmente, foram
caracterizadas as populações de macrófagos (CD68+ e CD163+), monócitos (CD14+), assim
como o estado de proliferação das células infiltrantes (Ki67+) e a imunoexpressão de
CASP3+ ativa por estas células (Fig. 4.17; Anexos IV: Tabelas S4 e S5).
Os resultados de RT-qPCR para cada um dos genes avaliados foram correlacionados
com o número de células do microambiente identificadas por IHQ por mm2. Na Tabela 4.7
são mostrados os valores de correlação (R2) entre o número de células e a expressão gênica,
aceitando P< 0,01 como valor significativo da correlação (teste de Spearman).
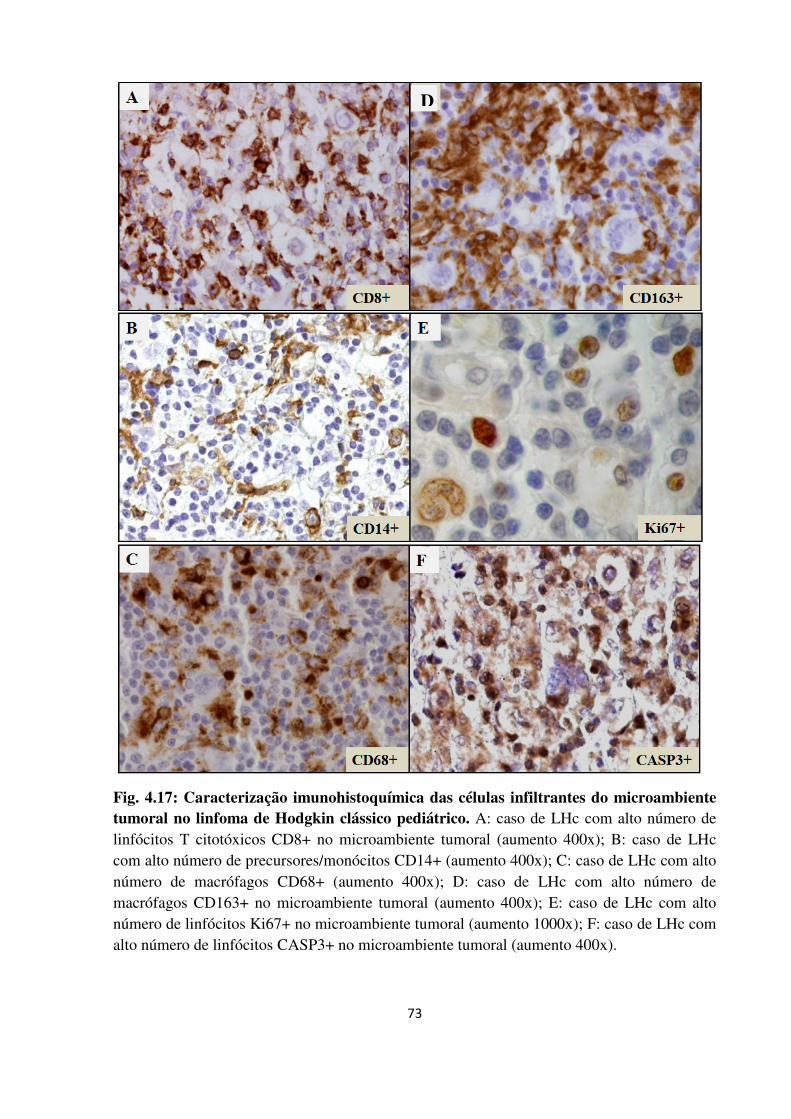
Fig. 4.17: Caracterização imunohistoquímica das células infiltrantes do microambiente tumoral no linfoma de Hodgkin clássico pediátrico.linfócitos T citotóxicos CD8+ no microambiente tumoral (aumento 40com alto número de precursores/número de macrófagos CD68+ (aumento 4macrófagos CD163+ no microambiente tumoral (aumento 40número de linfócitos Ki67+ no microambiente tumoral (aumento alto número de linfócitos CASP3+ no microambiente tumoral (aumento 40
73
4.17: Caracterização imunohistoquímica das células infiltrantes do microambiente tumoral no linfoma de Hodgkin clássico pediátrico. A: caso de LHc com alto número de linfócitos T citotóxicos CD8+ no microambiente tumoral (aumento 40
alto número de precursores/monócitos CD14+ (aumento 400x); C: caso de LHc com alto número de macrófagos CD68+ (aumento 400x); D: caso de LHc com alto número de macrófagos CD163+ no microambiente tumoral (aumento 400x); E: caso de LHc com alto
nfócitos Ki67+ no microambiente tumoral (aumento 1000x); F: caso de LHc com alto número de linfócitos CASP3+ no microambiente tumoral (aumento 40
D
4.17: Caracterização imunohistoquímica das células infiltrantes do microambiente A: caso de LHc com alto número de
linfócitos T citotóxicos CD8+ no microambiente tumoral (aumento 400x); B: caso de LHc x); C: caso de LHc com alto
0x); D: caso de LHc com alto número de x); E: caso de LHc com alto
00x); F: caso de LHc com alto número de linfócitos CASP3+ no microambiente tumoral (aumento 400x).

74
4.2.6.2.1 Populações linfocitárias
O número de células citotóxicas (CD8+, GRZB+, TIA1+), mostrou uma correlação
positiva com a expressão de CASP3, CDK1 e BCL2. O número de células expressando
proteínas citotóxicas foi também associado com os níveis de expressão de genes relacionados
à ativação de macrófagos (STAT1 e/ou LYZ).
Um alto número de células expressando o fator de transcrição MAF+ (linfócitos Th2)
teve correlação com alta expressão de RNAm de CASP3, sendo que um menor número de
linfócitos Th2 foi correlacionado com maior expressão de BCL2L1.
4.2.6.2.2 Precursores monocíticos e macrófagos
No que se refere aos monócitos e macrófagos intratumorais, nos 84 casos avaliados
houve um número maior de macrófagos CD68+ (mediana: 284 células/mm2; faixa: 44-601
células/mm2) e CD163+ (mediana: 137 células/mm2; faixa 0-413 células/mm2) que de
monócitos/precursores CD14+ (mediana: 49 células/mm2; faixa 0-275 células/mm2).
O número de monócitos (células CD14+) mostrou correlação positiva com a expressão
de STAT1, e de maneira importante a expressão de STAT1, LYZ e IRF4 foi associada a um
aumento no número de macrófagos CD68+ e CD163+ (Tabela 4.7).
4.2.6.2.3 Eosinófilos
O número de eosinófilos no MAT foi variável nos 84 casos analisados, com infiltração
de entre 0 e 3500 células em 10 CGA (mediana de 27 células/10 CGA) (Fig. 4.18). Houve
uma correlação inversa com a expressão dos genes das vias do ciclo celular (CENPF e
CDK1), apoptose (CASP3 e BCL2), genes do microambiente relacionados a macrófagos
(STAT1 e LYZ) e o gene IRF4 (Tabela 4.7). Adicionalmente, testes de Mann-Whitney
mostraram diferenças significativas entre maior número de eosinófilos (> 27 eosinófilos/10
CGA) e menor expressão de CENPF (P <0,001), CDK1 (P=0,001), CASP3 (P=0,007), LYZ
(P=0,007), STAT1 (P=0,001) e IRF4 (P<0,001) (Fig. 4.19).
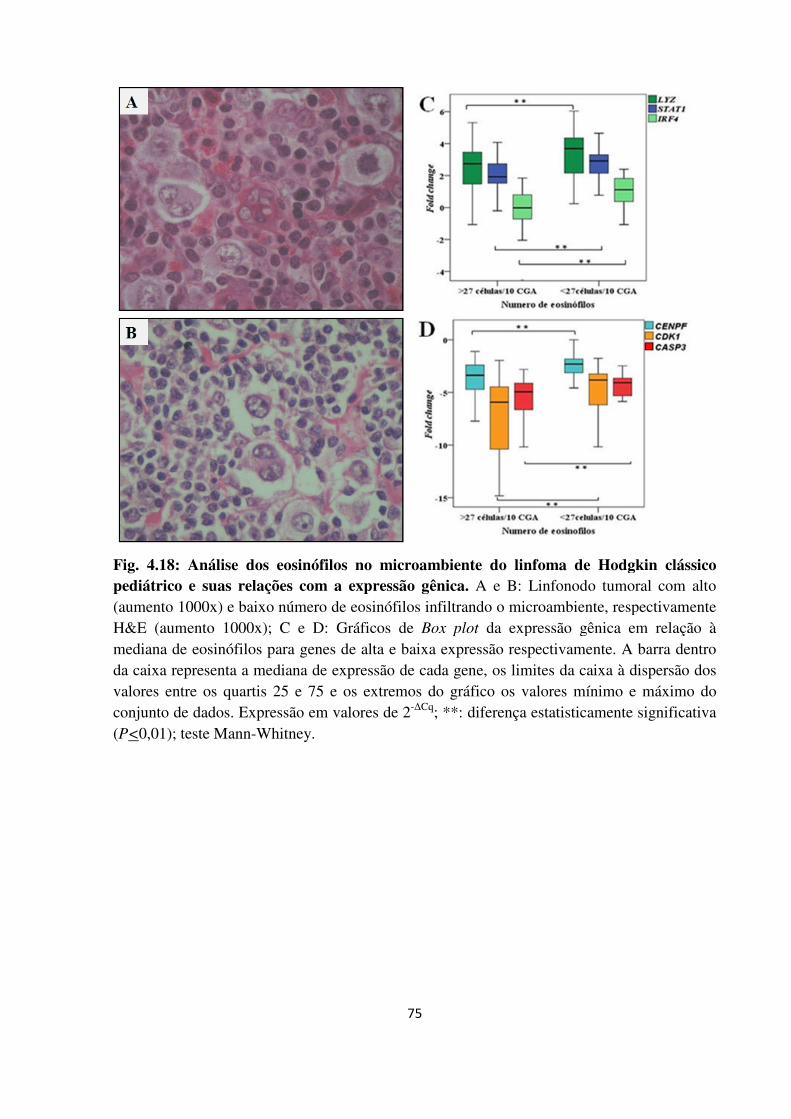
Fig. 4.18: Análise dos eosinófilos no microambiente do linfoma de Hodgkin clássico pediátrico e suas relações com a expressão gênica. (aumento 1000x) e baixo núH&E (aumento 1000x); C e D: mediana de eosinófilos para genes de alta e baixa expressão respectivamente. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispvalores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. Expressão(P<0,01); teste Mann-Whitney.
75
4.18: Análise dos eosinófilos no microambiente do linfoma de Hodgkin clássico pediátrico e suas relações com a expressão gênica. A e B: Linfonodo tumoral com alto
e baixo número de eosinófilos infiltrando o microambiente, respectivamente 0x); C e D: Gráficos de Box plot da expressão gênica em relação à
mediana de eosinófilos para genes de alta e baixa expressão respectivamente. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispvalores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. Expressão em valores de 2-∆Cq; **: diferença estatisticamente significativa
Whitney.
4.18: Análise dos eosinófilos no microambiente do linfoma de Hodgkin clássico nodo tumoral com alto
mero de eosinófilos infiltrando o microambiente, respectivamente da expressão gênica em relação à
mediana de eosinófilos para genes de alta e baixa expressão respectivamente. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do
; **: diferença estatisticamente significativa
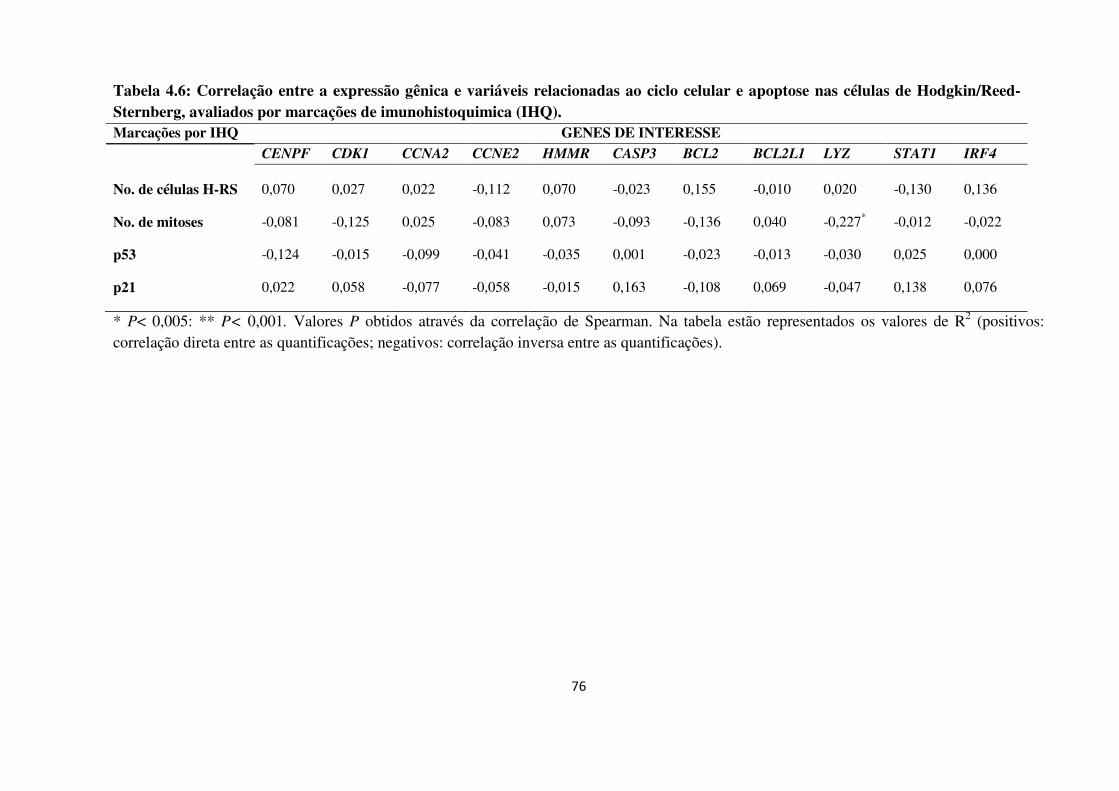
76
Tabela 4.6: Correlação entre a expressão gênica e variáveis relacionadas ao ciclo celular e apoptose nas células de Hodgkin/Reed-Sternberg, avaliados por marcações de imunohistoquimica (IHQ). Marcações por IHQ GENES DE INTERESSE CENPF CDK1 CCNA2 CCNE2 HMMR CASP3 BCL2 BCL2L1 LYZ STAT1 IRF4
No. de células H-RS 0,070 0,027 0,022 -0,112 0,070 -0,023 0,155 -0,010 0,020 -0,130 0,136
No. de mitoses -0,081 -0,125 0,025 -0,083 0,073 -0,093 -0,136 0,040 -0,227* -0,012 -0,022
p53 -0,124 -0,015 -0,099 -0,041 -0,035 0,001 -0,023 -0,013 -0,030 0,025 0,000
p21 0,022 0,058 -0,077 -0,058 -0,015 0,163 -0,108 0,069 -0,047 0,138 0,076
* P< 0,005: ** P< 0,001. Valores P obtidos através da correlação de Spearman. Na tabela estão representados os valores de R2 (positivos: correlação direta entre as quantificações; negativos: correlação inversa entre as quantificações).
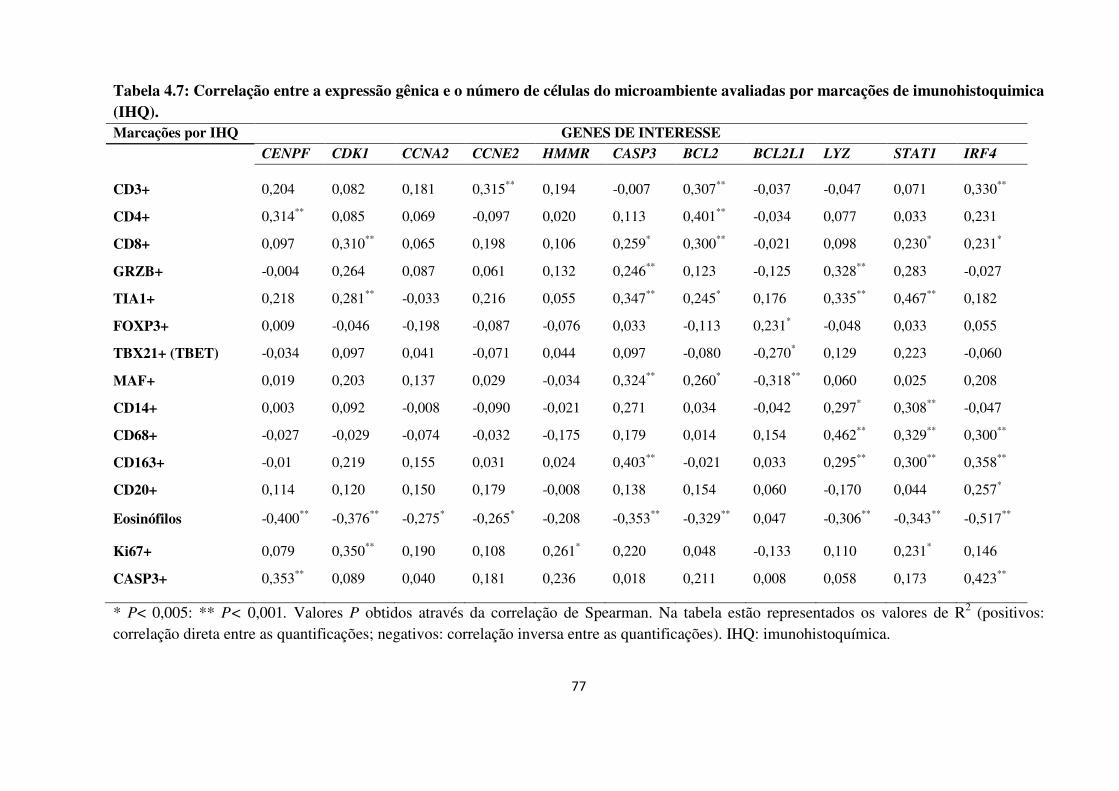
77
Tabela 4.7: Correlação entre a expressão gênica e o número de células do microambiente avaliadas por marcações de imunohistoquimica (IHQ). Marcações por IHQ GENES DE INTERESSE CENPF CDK1 CCNA2 CCNE2 HMMR CASP3 BCL2 BCL2L1 LYZ STAT1 IRF4
CD3+ 0,204 0,082 0,181 0,315** 0,194 -0,007 0,307** -0,037 -0,047 0,071 0,330**
CD4+ 0,314** 0,085 0,069 -0,097 0,020 0,113 0,401** -0,034 0,077 0,033 0,231
CD8+ 0,097 0,310** 0,065 0,198 0,106 0,259* 0,300** -0,021 0,098 0,230* 0,231*
GRZB+ -0,004 0,264 0,087 0,061 0,132 0,246** 0,123 -0,125 0,328** 0,283 -0,027
TIA1+ 0,218 0,281** -0,033 0,216 0,055 0,347** 0,245* 0,176 0,335** 0,467** 0,182
FOXP3+ 0,009 -0,046 -0,198 -0,087 -0,076 0,033 -0,113 0,231* -0,048 0,033 0,055
TBX21+ (TBET) -0,034 0,097 0,041 -0,071 0,044 0,097 -0,080 -0,270* 0,129 0,223 -0,060
MAF+ 0,019 0,203 0,137 0,029 -0,034 0,324** 0,260* -0,318** 0,060 0,025 0,208
CD14+ 0,003 0,092 -0,008 -0,090 -0,021 0,271 0,034 -0,042 0,297* 0,308** -0,047
CD68+ -0,027 -0,029 -0,074 -0,032 -0,175 0,179 0,014 0,154 0,462** 0,329** 0,300**
CD163+ -0,01 0,219 0,155 0,031 0,024 0,403** -0,021 0,033 0,295** 0,300** 0,358**
CD20+ 0,114 0,120 0,150 0,179 -0,008 0,138 0,154 0,060 -0,170 0,044 0,257*
Eosinófilos -0,400** -0,376** -0,275* -0,265* -0,208 -0,353** -0,329** 0,047 -0,306** -0,343** -0,517**
Ki67+ 0,079 0,350** 0,190 0,108 0,261* 0,220 0,048 -0,133 0,110 0,231* 0,146
CASP3+ 0,353** 0,089 0,040 0,181 0,236 0,018 0,211 0,008 0,058 0,173 0,423**
* P< 0,005: ** P< 0,001. Valores P obtidos através da correlação de Spearman. Na tabela estão representados os valores de R2 (positivos: correlação direta entre as quantificações; negativos: correlação inversa entre as quantificações). IHQ: imunohistoquímica.

78
4.2.7 Relação entre apoptose e proliferação no microambiente tumoral
Visando obter informações sobre a proliferação e morte celular das células do MAT,
marcações por IHQ para Ki67 e CASP3 respectivamente, foram realizadas e as células no
MAT expressando estes marcadores foram contadas.
O número de linfócitos intratumorais expressando o marcador de proliferação Ki67+
variou entre 3 e 352 (mediana 91 células Ki67+/mm2), enquanto que o número de linfócitos
com compromisso CASP3+ variou entre 41 e 864 (mediana 397 células CASP3+/mm2). Em
termos gerais, não houve correlação entre os números de linfócitos em proliferação e
apoptose.
Uma vez que duplas marcações não foram ainda realizadas para identificar as
populações responsáveis por estes números, foram realizados testes de correlação, os quais
mostraram que a quantidade de linfócitos CASP3+ teve uma forte correlação positiva com o
número de linfócitos T (CD3+) especialmente T CD8+, assim como com linfócitos B
(CD20+). Monócitos (CD14+), linfócitos T citotóxicos (CD8+ e GRZB+), Th1 (TBX21+)
mostraram correlação positiva com o marcador de proliferação. Todas estas correlações
tiveram um valor de significação P< 0,01 (Anexo IV: Tabela S5)
Referente à expressão gênica, foi encontrada uma correlação entre alto número de
linfócitos CASP3+ no microambiente com altos níveis de expressão de RNAm de CENPF e
IRF4; no mesmo sentido, houve correlação do número de linfócitos Ki67+ com a expressão
de CDK1 (Tabela 4.7).
Seguidamente, os níveis de expressão gênica foram comparados, utilizando como
valor de corte a mediana de células expressando Ki67+ no microambiente (91 células/mm2).
Um alto índice proliferativo no microambiente foi associado com uma maior expressão de
STAT1 (P=0,007), CDK1 (P<0,001), CASP3 (P=0,039) e HMMR (P=0,010) (teste de Mann-
Whitney). Por outro lado, um maior número de linfócitos CASP3+ (valor de corte, mediana
397 células/mm2) foi associado a um aumento de RNAm de CENPF (P=0,042), HMMR
(P=0,047) e IRF4 (P=0,009) (teste de Mann-Whitney) (Fig. 4.19).
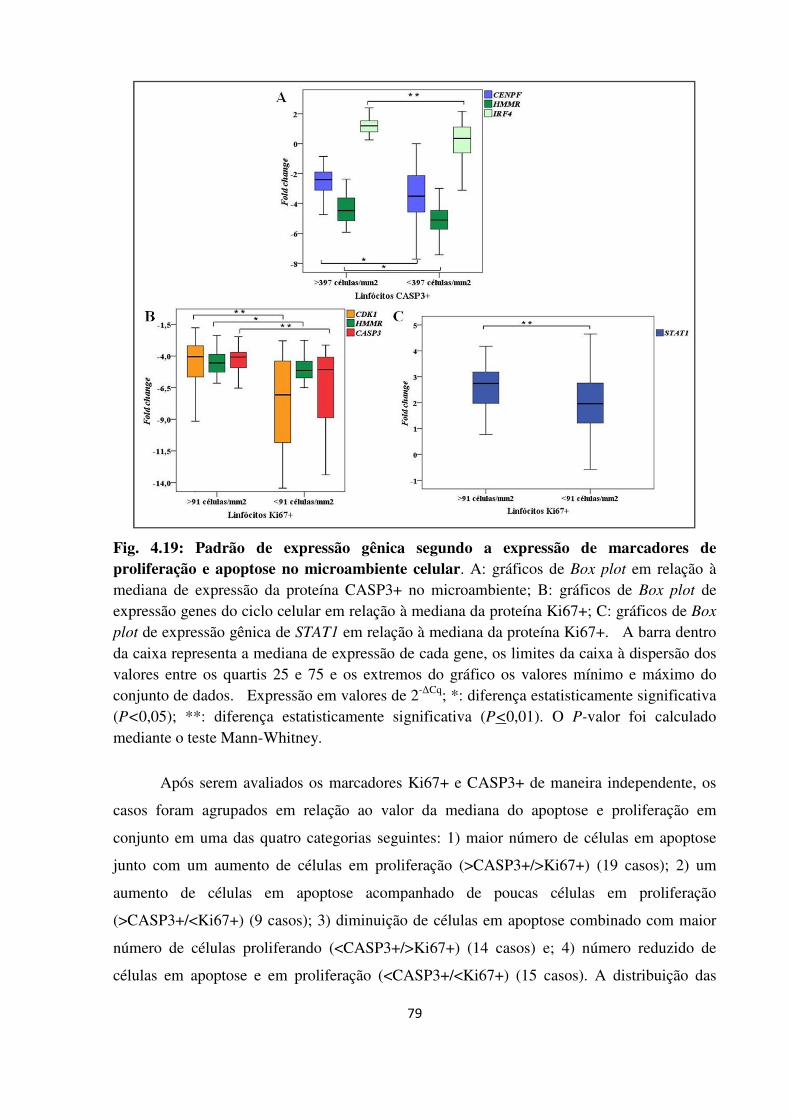
79
Fig. 4.19: Padrão de expressão gênica segundo a expressão de marcadores de proliferação e apoptose no microambiente celular. A: gráficos de Box plot em relação à mediana de expressão da proteína CASP3+ no microambiente; B: gráficos de Box plot de expressão genes do ciclo celular em relação à mediana da proteína Ki67+; C: gráficos de Box plot de expressão gênica de STAT1 em relação à mediana da proteína Ki67+. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. Expressão em valores de 2-∆Cq; *: diferença estatisticamente significativa (P<0,05); **: diferença estatisticamente significativa (P<0,01). O P-valor foi calculado mediante o teste Mann-Whitney.
Após serem avaliados os marcadores Ki67+ e CASP3+ de maneira independente, os
casos foram agrupados em relação ao valor da mediana do apoptose e proliferação em
conjunto em uma das quatro categorias seguintes: 1) maior número de células em apoptose
junto com um aumento de células em proliferação (>CASP3+/>Ki67+) (19 casos); 2) um
aumento de células em apoptose acompanhado de poucas células em proliferação
(>CASP3+/<Ki67+) (9 casos); 3) diminuição de células em apoptose combinado com maior
número de células proliferando (<CASP3+/>Ki67+) (14 casos) e; 4) número reduzido de
células em apoptose e em proliferação (<CASP3+/<Ki67+) (15 casos). A distribuição das
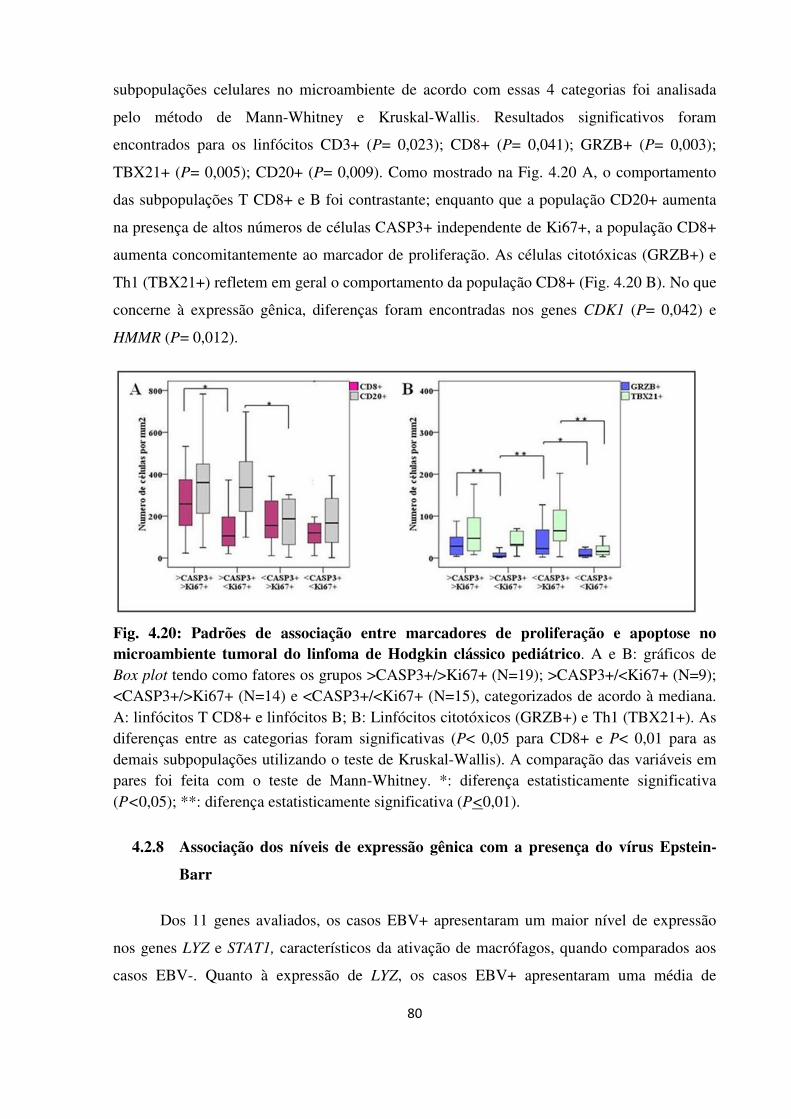
80
subpopulações celulares no microambiente de acordo com essas 4 categorias foi analisada
pelo método de Mann-Whitney e Kruskal-Wallis. Resultados significativos foram
encontrados para os linfócitos CD3+ (P= 0,023); CD8+ (P= 0,041); GRZB+ (P= 0,003);
TBX21+ (P= 0,005); CD20+ (P= 0,009). Como mostrado na Fig. 4.20 A, o comportamento
das subpopulações T CD8+ e B foi contrastante; enquanto que a população CD20+ aumenta
na presença de altos números de células CASP3+ independente de Ki67+, a população CD8+
aumenta concomitantemente ao marcador de proliferação. As células citotóxicas (GRZB+) e
Th1 (TBX21+) refletem em geral o comportamento da população CD8+ (Fig. 4.20 B). No que
concerne à expressão gênica, diferenças foram encontradas nos genes CDK1 (P= 0,042) e
HMMR (P= 0,012).
Fig. 4.20: Padrões de associação entre marcadores de proliferação e apoptose no microambiente tumoral do linfoma de Hodgkin clássico pediátrico. A e B: gráficos de Box plot tendo como fatores os grupos >CASP3+/>Ki67+ (N=19); >CASP3+/<Ki67+ (N=9); <CASP3+/>Ki67+ (N=14) e <CASP3+/<Ki67+ (N=15), categorizados de acordo à mediana. A: linfócitos T CD8+ e linfócitos B; B: Linfócitos citotóxicos (GRZB+) e Th1 (TBX21+). As diferenças entre as categorias foram significativas (P< 0,05 para CD8+ e P< 0,01 para as demais subpopulações utilizando o teste de Kruskal-Wallis). A comparação das variáveis em pares foi feita com o teste de Mann-Whitney. *: diferença estatisticamente significativa (P<0,05); **: diferença estatisticamente significativa (P<0,01).
4.2.8 Associação dos níveis de expressão gênica com a presença do vírus Epstein-
Barr
Dos 11 genes avaliados, os casos EBV+ apresentaram um maior nível de expressão
nos genes LYZ e STAT1, característicos da ativação de macrófagos, quando comparados aos
casos EBV-. Quanto à expressão de LYZ, os casos EBV+ apresentaram uma média de
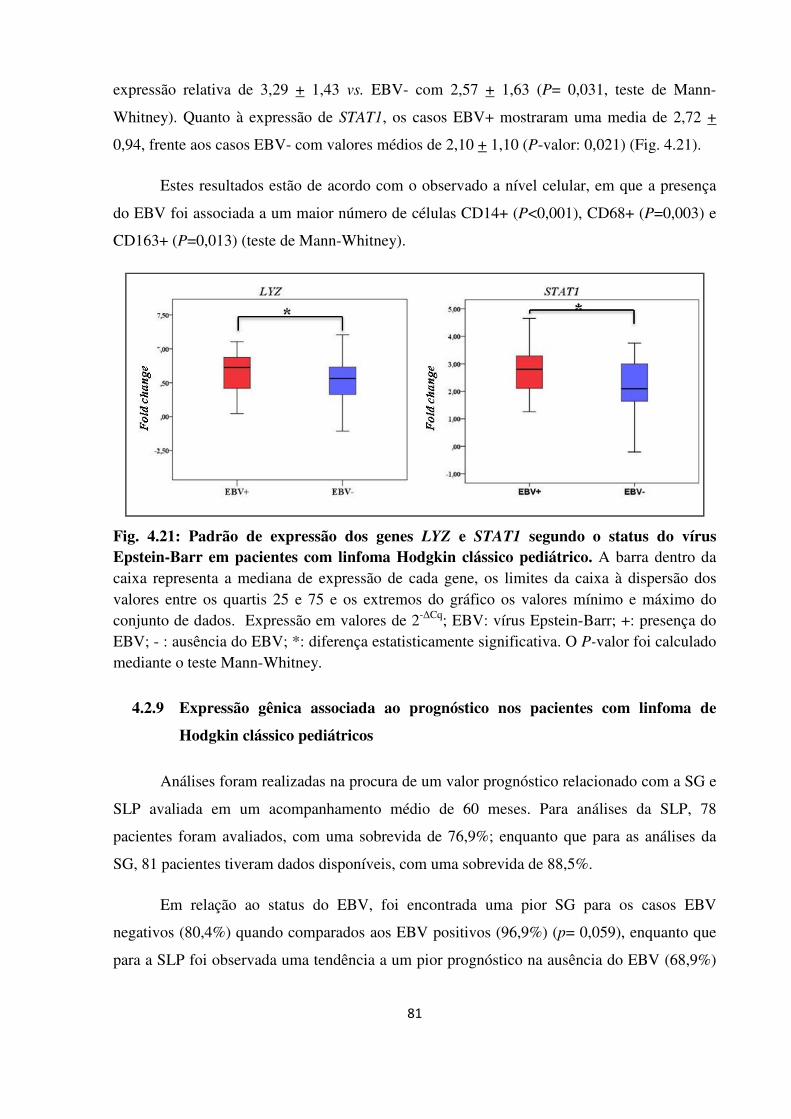
81
expressão relativa de 3,29 + 1,43 vs. EBV- com 2,57 + 1,63 (P= 0,031, teste de Mann-
Whitney). Quanto à expressão de STAT1, os casos EBV+ mostraram uma media de 2,72 +
0,94, frente aos casos EBV- com valores médios de 2,10 + 1,10 (P-valor: 0,021) (Fig. 4.21).
Estes resultados estão de acordo com o observado a nível celular, em que a presença
do EBV foi associada a um maior número de células CD14+ (P<0,001), CD68+ (P=0,003) e
CD163+ (P=0,013) (teste de Mann-Whitney).
Fig. 4.21: Padrão de expressão dos genes LYZ e STAT1 segundo o status do vírus Epstein-Barr em pacientes com linfoma Hodgkin clássico pediátrico. A barra dentro da caixa representa a mediana de expressão de cada gene, os limites da caixa à dispersão dos valores entre os quartis 25 e 75 e os extremos do gráfico os valores mínimo e máximo do conjunto de dados. Expressão em valores de 2-∆Cq; EBV: vírus Epstein-Barr; +: presença do EBV; - : ausência do EBV; *: diferença estatisticamente significativa. O P-valor foi calculado mediante o teste Mann-Whitney.
4.2.9 Expressão gênica associada ao prognóstico nos pacientes com linfoma de
Hodgkin clássico pediátricos
Análises foram realizadas na procura de um valor prognóstico relacionado com a SG e
SLP avaliada em um acompanhamento médio de 60 meses. Para análises da SLP, 78
pacientes foram avaliados, com uma sobrevida de 76,9%; enquanto que para as análises da
SG, 81 pacientes tiveram dados disponíveis, com uma sobrevida de 88,5%.
Em relação ao status do EBV, foi encontrada uma pior SG para os casos EBV
negativos (80,4%) quando comparados aos EBV positivos (96,9%) (p= 0,059), enquanto que
para a SLP foi observada uma tendência a um pior prognóstico na ausência do EBV (68,9%)

82
comparada com a presença do vírus (87,1%) (P=0,083). Não houve sobrevida
significativamente diferente por grupo de risco da doença.
Para categorizar o grupo de pacientes segundo o nível da expressão gênica em alta e
baixa expressão, o valor da mediana do grupo foi estabelecido como ponto de corte. Na
Tabela 4.8 são mostrados os valores em média + DP e mediana da expressão gênica para os
grupos que mostraram recaída vs. não recaída. As análises de sobrevida para cada um dos
genes avaliados são apresentadas na Tabela 4.9.
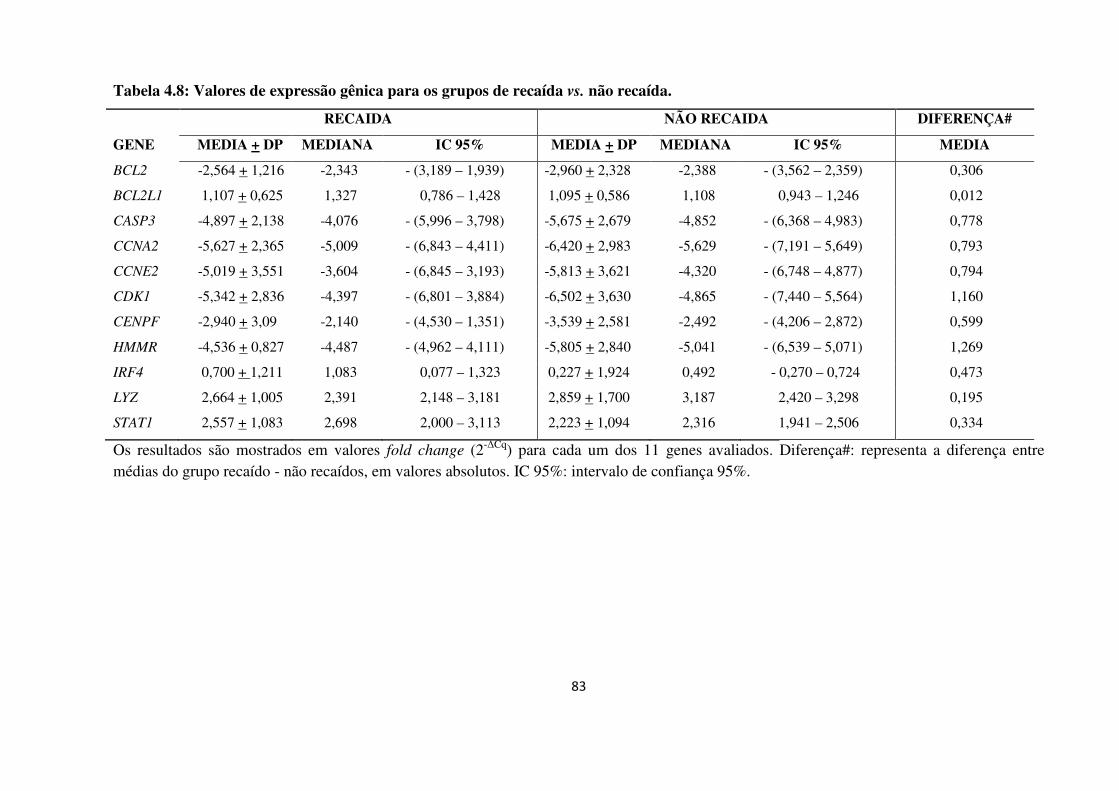
83
Tabela 4.8: Valores de expressão gênica para os grupos de recaída vs. não recaída.
RECAIDA NÃO RECAIDA DIFERENÇA#
GENE MEDIA + DP MEDIANA IC 95% MEDIA + DP MEDIANA IC 95% MEDIA
BCL2 -2,564 + 1,216 -2,343 - (3,189 – 1,939) -2,960 + 2,328 -2,388 - (3,562 – 2,359) 0,306
BCL2L1 1,107 + 0,625 1,327 0,786 – 1,428 1,095 + 0,586 1,108 0,943 – 1,246 0,012
CASP3 -4,897 + 2,138 -4,076 - (5,996 – 3,798) -5,675 + 2,679 -4,852 - (6,368 – 4,983) 0,778
CCNA2 -5,627 + 2,365 -5,009 - (6,843 – 4,411) -6,420 + 2,983 -5,629 - (7,191 – 5,649) 0,793
CCNE2 -5,019 + 3,551 -3,604 - (6,845 – 3,193) -5,813 + 3,621 -4,320 - (6,748 – 4,877) 0,794
CDK1 -5,342 + 2,836 -4,397 - (6,801 – 3,884) -6,502 + 3,630 -4,865 - (7,440 – 5,564) 1,160
CENPF -2,940 + 3,09 -2,140 - (4,530 – 1,351) -3,539 + 2,581 -2,492 - (4,206 – 2,872) 0,599
HMMR -4,536 + 0,827 -4,487 - (4,962 – 4,111) -5,805 + 2,840 -5,041 - (6,539 – 5,071) 1,269
IRF4 0,700 + 1,211 1,083 0,077 – 1,323 0,227 + 1,924 0,492 - 0,270 – 0,724 0,473
LYZ 2,664 + 1,005 2,391 2,148 – 3,181 2,859 + 1,700 3,187 2,420 – 3,298 0,195
STAT1 2,557 + 1,083 2,698 2,000 – 3,113 2,223 + 1,094 2,316 1,941 – 2,506 0,334
Os resultados são mostrados em valores fold change (2-∆Cq) para cada um dos 11 genes avaliados. Diferença#: representa a diferença entre médias do grupo recaído - não recaídos, em valores absolutos. IC 95%: intervalo de confiança 95%.
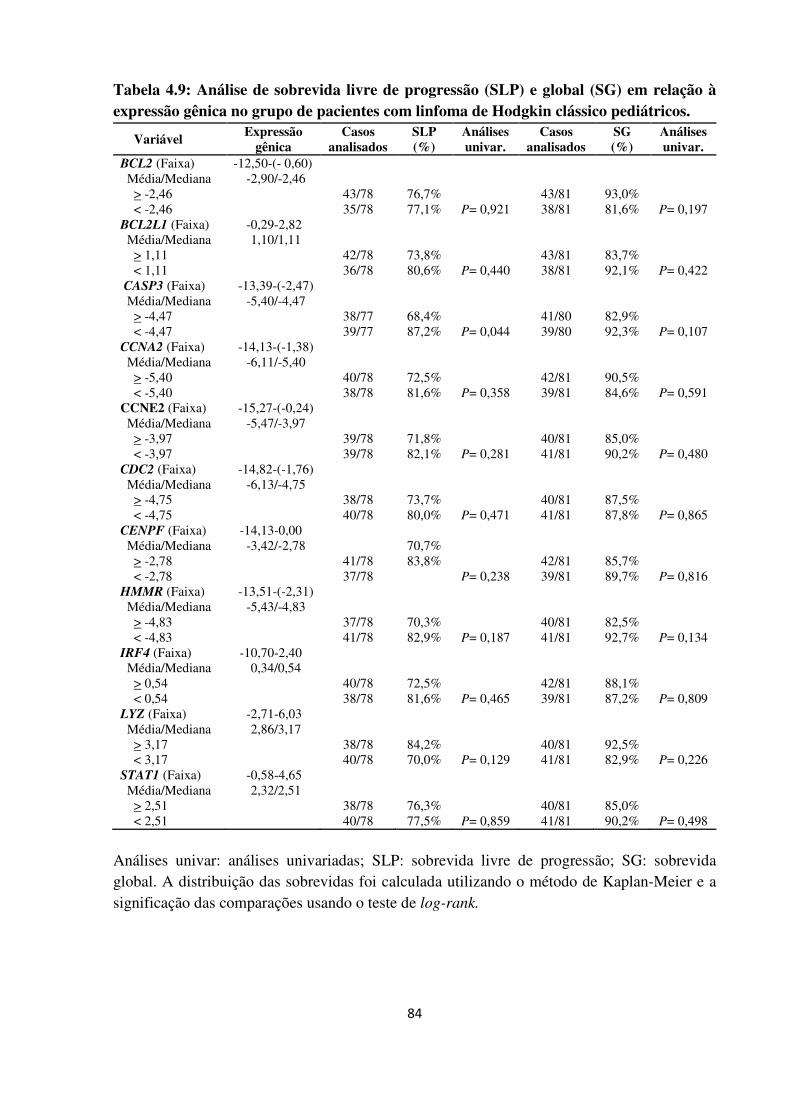
84
Tabela 4.9: Análise de sobrevida livre de progressão (SLP) e global (SG) em relação à expressão gênica no grupo de pacientes com linfoma de Hodgkin clássico pediátricos.
Variável Expressão
gênica Casos
analisados SLP (%)
Análises univar.
Casos analisados
SG (%)
Análises univar.
BCL2 (Faixa) Média/Mediana > -2,46 < -2,46 BCL2L1 (Faixa) Média/Mediana > 1,11 < 1,11 CASP3 (Faixa) Média/Mediana > -4,47 < -4,47 CCNA2 (Faixa) Média/Mediana > -5,40 < -5,40 CCNE2 (Faixa) Média/Mediana > -3,97 < -3,97 CDC2 (Faixa) Média/Mediana > -4,75 < -4,75 CENPF (Faixa) Média/Mediana > -2,78 < -2,78 HMMR (Faixa) Média/Mediana > -4,83 < -4,83 IRF4 (Faixa) Média/Mediana > 0,54 < 0,54 LYZ (Faixa) Média/Mediana > 3,17 < 3,17 STAT1 (Faixa) Média/Mediana > 2,51 < 2,51
-12,50-(- 0,60) -2,90/-2,46
-0,29-2,82 1,10/1,11
-13,39-(-2,47) -5,40/-4,47
-14,13-(-1,38) -6,11/-5,40
-15,27-(-0,24) -5,47/-3,97
-14,82-(-1,76) -6,13/-4,75
-14,13-0,00 -3,42/-2,78
-13,51-(-2,31) -5,43/-4,83
-10,70-2,40 0,34/0,54
-2,71-6,03 2,86/3,17
-0,58-4,65 2,32/2,51
43/78 35/78
42/78 36/78
38/77 39/77
40/78 38/78
39/78 39/78
38/78 40/78
41/78 37/78
37/78 41/78
40/78 38/78
38/78 40/78
38/78 40/78
76,7% 77,1%
73,8% 80,6%
68,4% 87,2%
72,5% 81,6%
71,8% 82,1%
73,7% 80,0%
70,7% 83,8%
70,3% 82,9%
72,5% 81,6%
84,2% 70,0%
76,3% 77,5%
P= 0,921
P= 0,440
P= 0,044
P= 0,358
P= 0,281
P= 0,471
P= 0,238
P= 0,187
P= 0,465
P= 0,129
P= 0,859
43/81 38/81
43/81 38/81
41/80 39/80
42/81 39/81
40/81 41/81
40/81 41/81
42/81 39/81
40/81 41/81
42/81 39/81
40/81 41/81
40/81 41/81
93,0% 81,6%
83,7% 92,1%
82,9% 92,3%
90,5% 84,6%
85,0% 90,2%
87,5% 87,8%
85,7% 89,7%
82,5% 92,7%
88,1% 87,2%
92,5% 82,9%
85,0% 90,2%
P= 0,197
P= 0,422
P= 0,107
P= 0,591
P= 0,480
P= 0,865
P= 0,816
P= 0,134
P= 0,809
P= 0,226
P= 0,498
Análises univar: análises univariadas; SLP: sobrevida livre de progressão; SG: sobrevida global. A distribuição das sobrevidas foi calculada utilizando o método de Kaplan-Meier e a significação das comparações usando o teste de log-rank.
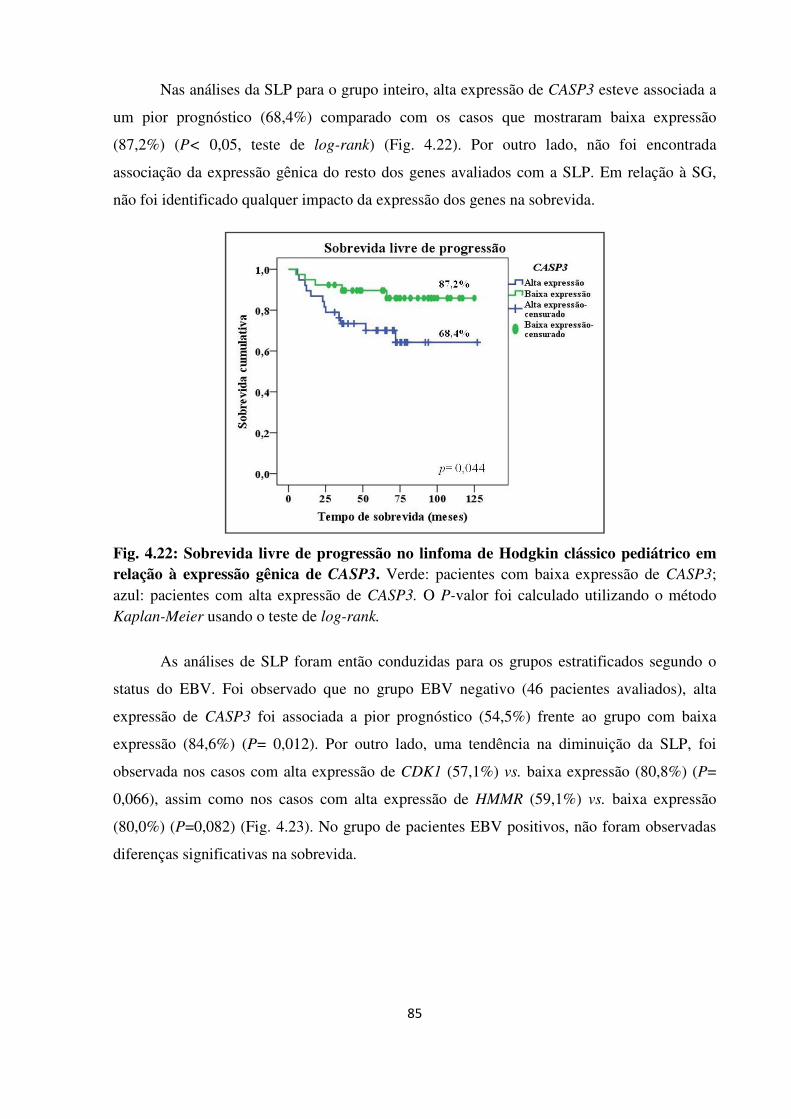
85
Nas análises da SLP para o grupo inteiro, alta expressão de CASP3 esteve associada a
um pior prognóstico (68,4%) comparado com os casos que mostraram baixa expressão
(87,2%) (P< 0,05, teste de log-rank) (Fig. 4.22). Por outro lado, não foi encontrada
associação da expressão gênica do resto dos genes avaliados com a SLP. Em relação à SG,
não foi identificado qualquer impacto da expressão dos genes na sobrevida.
Fig. 4.22: Sobrevida livre de progressão no linfoma de Hodgkin clássico pediátrico em relação à expressão gênica de CASP3. Verde: pacientes com baixa expressão de CASP3; azul: pacientes com alta expressão de CASP3. O P-valor foi calculado utilizando o método Kaplan-Meier usando o teste de log-rank.
As análises de SLP foram então conduzidas para os grupos estratificados segundo o
status do EBV. Foi observado que no grupo EBV negativo (46 pacientes avaliados), alta
expressão de CASP3 foi associada a pior prognóstico (54,5%) frente ao grupo com baixa
expressão (84,6%) (P= 0,012). Por outro lado, uma tendência na diminuição da SLP, foi
observada nos casos com alta expressão de CDK1 (57,1%) vs. baixa expressão (80,8%) (P=
0,066), assim como nos casos com alta expressão de HMMR (59,1%) vs. baixa expressão
(80,0%) (P=0,082) (Fig. 4.23). No grupo de pacientes EBV positivos, não foram observadas
diferenças significativas na sobrevida.
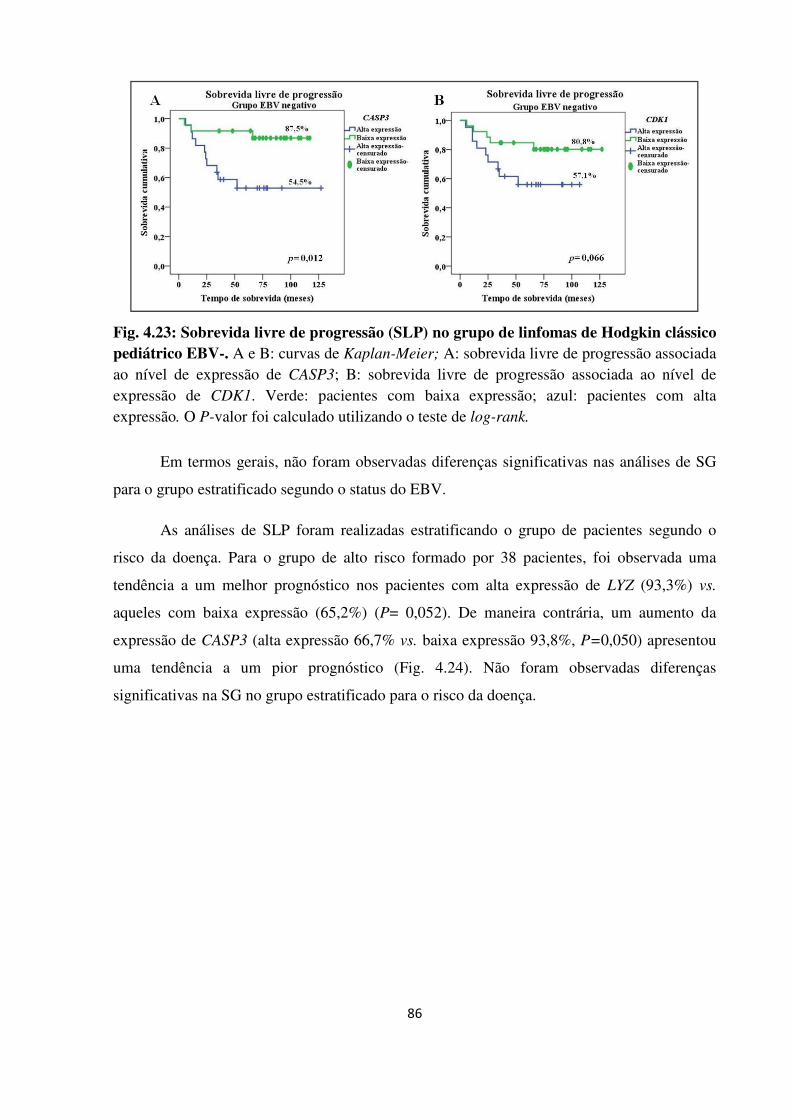
86
Fig. 4.23: Sobrevida livre de progressão (SLP) no grupo de linfomas de Hodgkin clássico pediátrico EBV-. A e B: curvas de Kaplan-Meier; A: sobrevida livre de progressão associada ao nível de expressão de CASP3; B: sobrevida livre de progressão associada ao nível de expressão de CDK1. Verde: pacientes com baixa expressão; azul: pacientes com alta expressão. O P-valor foi calculado utilizando o teste de log-rank.
Em termos gerais, não foram observadas diferenças significativas nas análises de SG
para o grupo estratificado segundo o status do EBV.
As análises de SLP foram realizadas estratificando o grupo de pacientes segundo o
risco da doença. Para o grupo de alto risco formado por 38 pacientes, foi observada uma
tendência a um melhor prognóstico nos pacientes com alta expressão de LYZ (93,3%) vs.
aqueles com baixa expressão (65,2%) (P= 0,052). De maneira contrária, um aumento da
expressão de CASP3 (alta expressão 66,7% vs. baixa expressão 93,8%, P=0,050) apresentou
uma tendência a um pior prognóstico (Fig. 4.24). Não foram observadas diferenças
significativas na SG no grupo estratificado para o risco da doença.
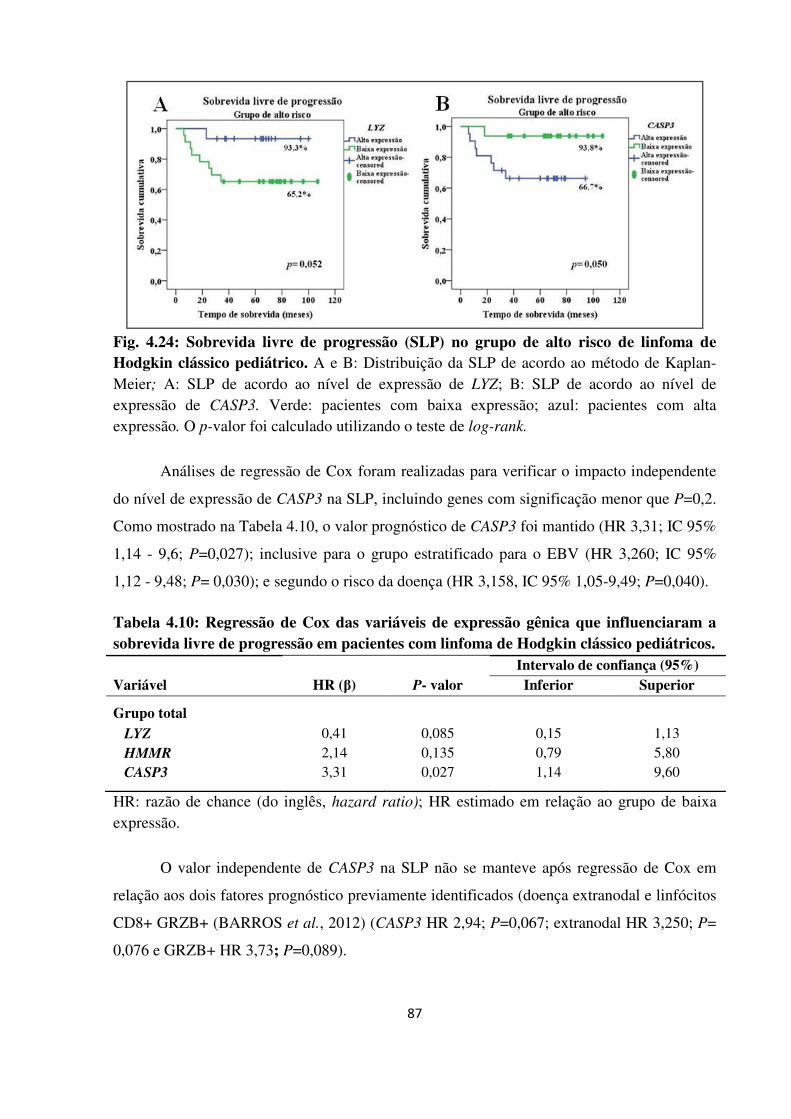
87
Fig. 4.24: Sobrevida livre de progressão (SLP) no grupo de alto risco de linfoma de Hodgkin clássico pediátrico. A e B: Distribuição da SLP de acordo ao método de Kaplan-Meier; A: SLP de acordo ao nível de expressão de LYZ; B: SLP de acordo ao nível de expressão de CASP3. Verde: pacientes com baixa expressão; azul: pacientes com alta expressão. O p-valor foi calculado utilizando o teste de log-rank.
Análises de regressão de Cox foram realizadas para verificar o impacto independente
do nível de expressão de CASP3 na SLP, incluindo genes com significação menor que P=0,2.
Como mostrado na Tabela 4.10, o valor prognóstico de CASP3 foi mantido (HR 3,31; IC 95%
1,14 - 9,6; P=0,027); inclusive para o grupo estratificado para o EBV (HR 3,260; IC 95%
1,12 - 9,48; P= 0,030); e segundo o risco da doença (HR 3,158, IC 95% 1,05-9,49; P=0,040).
Tabela 4.10: Regressão de Cox das variáveis de expressão gênica que influenciaram a sobrevida livre de progressão em pacientes com linfoma de Hodgkin clássico pediátricos. Intervalo de confiança (95%) Variável HR (β) P- valor Inferior Superior
Grupo total
LYZ 0,41 0,085 0,15 1,13 HMMR 2,14 0,135 0,79 5,80 CASP3 3,31 0,027 1,14 9,60
HR: razão de chance (do inglês, hazard ratio); HR estimado em relação ao grupo de baixa expressão.
O valor independente de CASP3 na SLP não se manteve após regressão de Cox em
relação aos dois fatores prognóstico previamente identificados (doença extranodal e linfócitos
CD8+ GRZB+ (BARROS et al., 2012) (CASP3 HR 2,94; P=0,067; extranodal HR 3,250; P=
0,076 e GRZB+ HR 3,73; P=0,089).

88
5 DISCUSSÃO
Durante os últimos anos, os estudos de expressão gênica tomaram relevância no
diagnóstico e na pesquisa biomédica, e uma ênfase cada vez maior vem sendo colocada na
necessidade de controle das etapas pré-analiticas e analíticas, desde a obtenção de material
genético de qualidade, até o controle de qualidade dos resultados, visando o desenvolvimento
de metodologias que possam capturar com precisão a complexidade dos sistemas biológicos e
que possam ser aplicadas com resultados reprodutíveis. Este trabalho foi desenvolvido em
duas partes: 1) aperfeiçoamento de uma metodologia eficiente para extração de RNA a partir
de material FFPE, e aplicação de um desenho experimental para conhecer as variabilidades
introduzidas, tanto biológicas (inerentes à diversidade de indivíduos e doença), como a
técnica para seu uso nas análises de expressão gênica e; 2) a aplicação da metodologia
anteriormente desenvolvida em um grupo de pacientes, visando validar um conjunto de genes
associados a um pior prognóstico descrito em pacientes adultos com LHc.
5.1 Aperfeiçoamento de uma técnica de extração de RNA a partir de material
parafinado para estudos de expressão gênica por RT-qPCR
Neste trabalho é mostrada a importância de selecionar cuidadosamente o método de
extração de RNA quando se trabalha com ácidos nucleicos de baixa qualidade. Diferentes
fatores podem afetar a qualidade e quantidade de RNA extraído, como por exemplo,
qualidade e tempo dos processos de fixação e antiguidade do bloco de parafina (MASUDA et
al., 1999; SRINIVASAN; SEDMAK; JEWELL, 2002; CRONIN et al., 2004). Dos dois
métodos comerciais comparados neste estudo, o método baseado na precipitação (MP)
superou em rendimento ao método baseado em purificação através de colunas (RA),
basicamente pelo rendimento estável do primeiro, independente da antiguidade dos blocos de
parafina. Diferentes trabalhos vêm mostrando um alto rendimento com o uso do método RA
aplicado ao material FFPE, que é de fato, o mais amplamente usado na literatura
(DOLESHAL et al., 2008; LI et al., 2008; ROBERTS et al., 2009; OKELLO et al., 2010).
ARZT et al. (2011) reportam alta qualidade na relação dos ácidos nucleicos/proteínas
(λ260/280), similares aos resultados obtidos neste trabalho. Entretanto, nenhum dos trabalhos
prévios compara a utilização dos métodos e seu desempenho em relação à qualidade e
antiguidade das amostras. Desta maneira, nós contribuímos com a observação de que o
método MP tem um melhor desempenho e é mais estável quando se pretende estudar uma

89
série de amostras obtidas ao longo do tempo, em que os métodos de fixação e o uso de
formalina tamponada podem ter variado.
Um segundo motivo que estimulou este trabalho é a característica da extrema
complexidade do LHc, em que se observam não somente diferentes subtipos histológicos,
com suas particulares relações entre quantidade de fibrose e células H-RS (SWERDLOW et
al., 2008), mas também a grande variabilidade interindividual na composição da massa
tumoral em relação às células inflamatórias (linfócitos T, linfócitos B, macrófagos, entre
outras). Esta grande variabilidade biológica é a causa da falta de reprodutibilidade de padrões
na expressão gênica global entre diferentes estudos (SANCHEZ-ESPIRIDION et al., 2009;
SANCHEZ-ESPIRIDION et al., 2010; STEIDL et al., 2012), e afeta diretamente o poder
estatístico para detectar diferenças no prognóstico clínico. Assim, uma diferença real nos
níveis de expressão gênica associados à doença somente poderia ser percebida se estes níveis
forem superiores à variabilidade introduzida durante o processamento técnico e à
heterogeneidade biológica de base da doença.
TICHOPAD et al. (2009) abordaram o problema da variabilidade nos estudos de
expressão gênica e conseguiram mostrar experimentalmente que a variabilidade biológica é
maior nos tecidos sólidos (fígado bovino, com ruído total: + 1,4 desvio padrão médio, DP),
que no sangue periférico (+ 0,6 DP médio), que nas linhagens celulares (+ 0,44 DP médio).
Do ponto de vista da variabilidade técnica nos tecidos sólidos, esta é incluída
predominantemente nas etapas de RT, sendo que também há um componente gene-específico.
O estudo da expressão de células simples foi o que mostrou maior variabilidade (+ 2 DP
médio), em acordo com um modelo proposto de distribuição de Poisson, em comparação com
uma distribuição normal da variação na expressão gênica nos outros tecidos (AUDIC;
CLAVERIE, 1997).
Sendo assim, esta abordagem seria adequada para responder as nossas perguntas sobre
o impacto da variabilidade no LHc e não tinha, até o momento, sido aplicada na avaliação de
extrações de RNA de parafina. Neste estudo, o desenho ANOVA tipo nested foi expandido e
adaptado para sua aplicação em amostras de RNA FFPE em LHc usando seis níveis, pela
incorporação de mais um nível biológico (diferenças entre linfonodos de um mesmo paciente)
e mais um nível técnico (pré-Amp).

90
A primeira análise foi realizada considerando os valores Cq e sua variabilidade
expressa em DP, a efeitos de comparação com os resultados previamente descritos
(TICHOPAD et al., 2009). O valor do ruído total capturado pelos genes de referência (GDR)
usados neste trabalho (GUSB e HMBS) foi similar e menor ao descrito pelos autores para
ACTB em tecidos sólidos frescos (fígado) (média dos DP, 23,55 Cq + 1,45 ciclos neste
trabalho vs. 20,41 +1,63 para ACTB em TICHOPAD et al. (2009). A escolha dos GDR para
este trabalho foi guiada por um trabalho prévio que mostrou uma maior estabilidade destes no
LHc (SANCHEZ-ESPIRIDION et al., 2009), e os nossos resultados com uma abordagem
diferente confirmam a sua adequação como GDR nesta doença.
Em relação aos genes de interesse (GDI) avaliados, STAT1, IRF4, BCL2 e CASP3
mostraram ruídos totais entre + (1,17 - 1,68) ciclos (DP), média + 1,57 ciclos, o que foi
similar ao publicado previamente para tecidos a fresco (1,0 a 1,7 DP, média + 1,3), indicando
que a utilização de RNA de parafina não acarreta em uma variabilidade adicional intolerável
para estudos com sentido biológico. Uma comparação entre RNAs extraídos de linfonodos
FFPE e “frescos”, que não foi feita neste trabalho, permitiria confirmar esta afirmação.
Entretanto, visto a complexidade do LHc, parece improvável que a variabilidade dos
linfonodos a fresco seja significativamente menor que a relatada para o fígado no trabalho
tomado como referência.
Em relação à distribuição da variabilidade biológica, houve diferenças entre os dois
estudos, sendo que a variabilidade biológica em nosso estudo, integrada pelas diferenças entre
casos (N=3) e linfonodos dentro de casos (N=3x2) deu conta de entre 83,8% e 96,0% da
variabilidade observada, com contribuição mínima do linfonodo. No estudo de TICHOPAD et
al. (2009), a variabilidade entre indivíduos foi desprezível, enquanto que a etapa que eles
chamaram de “amostragem” (3 biopsias por cada fígado), teve variabilidade entre 35,0% e
94,0%. Isto faz sentido, na medida em que os indivíduos do estudo relatado eram mais
homogêneos (gado) em comparação aos humanos estudados no presente trabalho. Mais ainda,
aponta para a manutenção das características tumorais nos diferentes linfonodos acometidos
pela doença, minimizando então o efeito de “amostragem” na seleção entre linfonodos para
estudo.
Quanto à variabilidade técnica, os nossos valores foram superiores aos relatados no
estudo de referência (média + 0,86 vs. + 0,67 DP), o que é justificável se se tem em conta que
o desenho para parafina incluiu 4 níveis de processamento técnico vs. somente 2 (RT e qPCR)

91
no trabalho de referência, porém o nosso gene de maior variabilidade técnica teve níveis
comparáveis com os do gene com maior variabilidade do referido estudo (CASP3 vs. FGF7, +
1,23 e + 1,29 DP, respectivamente).
Diferentes estudos têm mostrado a importância do uso de valores normalizados em
estudos de expressão gênica por qPCR (VANDESOMPELE et al., 2002; BUSTIN et al.,
2009), o que corresponde no nosso trabalho ao cálculo de ∆Cq. Entretanto, não é claro qual é
o melhor valor para descrever a distribuição de variabilidade num ensaio ANOVA tipo
nested. É intuitivo pensar que a utilização de ∆Cq, no qual a expressão do GDI referencia-se
ao nível de RNAm incluído na reação, levaria a uma redução da variância. No entanto, uma
inspeção das ferramentas estatísticas usadas no cálculo permite ver que este não é sempre o
caso. O DP do ∆Cq é calculado como a razão entre: (raiz quadrada) da somatória das
variâncias de alvo (GDI) e endógeno (GDR) e a covariância dos Cqs do GDI e GDR. A
covariância é a medida de como as variações em cada nível afetam o GDI e GDR em
conjunto. Se a variação sempre afeta ambos os genes no mesmo sentido, então a covariância é
alta e o DP do ∆Cq será menor. Se a covariância é menor que a soma das variações de GOI e
GDI, então a normalização vai realmente aumentar o ruído. Se o processamento afeta a ambos
os genes de maneira diferente ou independente, a normalização não tem sentido e pode até
afetar a qualidade dos dados (TICHOPAD et al., 2009).
Em nosso estudo foi observado que, após normalização, a variabilidade total diminui
com valores em todos os genes avaliados no desenho ANOVA tipo nested. Isto está em
contraste com o mostrado em outro estudo, no qual os genes de baixa expressão não se
beneficiaram da normalização (KITCHEN et al., 2010), sugerindo que a fonte do material
genético (parafina vs. fresco) seja mais importante que o nível de expressão gênica na
normalização.
De maneira importante, a variação na etapa de extração de RNA foi reduzida após a
normalização, indicando que as diferenças significativas encontradas na análise do Cq
obedeceram a diferenças na quantidade de RNA inicial. Tendo em conta que a mesma
quantidade de RNA foi incluída em todas as reações, é possível que as diferenças sejam
devidas à presença de DNA que aumentou artificialmente a densidade óptica a 260nm, e que
nas amostras de RNA provenientes de parafina, a contaminação com DNA de baixo peso
molecular seja mais difícil de eliminar com tratamentos de DNase I. Em contraste, a
variabilidade da pré-Amp não diminuiu significativamente com a normalização, indicando

92
que, para diminuir a variabilidade nesta etapa, seriam necessárias modificações nas condições
técnicas de processamento. Em conclusão, os resultados das comparações feitas neste trabalho
indicam o uso do ∆Cq para avaliar os dados de um ensaio ANOVA tipo nested em RNA de
parafina.
Daqui em diante, serão discutidas algumas características observadas no ensaio
ANOVA tipo nested utilizando os valores de ∆Cq. TICHOPAD et al. (2009) descreve que a
variabilidade introduzida numa metodologia pode variar de acordo ao nível de expressão
gênica, sendo esta variação dependente de cada gene em particular. Neste trabalho, nós
estendemos esta descrição a um maior número de genes. Quando classificados segundo o
nível de expressão gênica, foi observado que genes de alta expressão apresentaram menores
valores de ruído total (exceto LYZ), comparado com os genes de intermediaria e baixa
expressão. O gene CASP3 foi o que apresentou a maior variabilidade técnica total dentre os
genes estudados. Vários motivos podem ser apontados, em primeiro lugar, as conhecidas
limitações do método de quantificação para baixos níveis, decorrentes da perda de linearidade
(PFAFFL, M. W.; HAGELEIT, 2001). Além disso, esta situação faz com que o viés
introduzido pela presença de inibidores seja amplificado. Os níveis técnicos superiores (RNA
e RT) são muito sensíveis à inibição, seja pela própria enzima transcriptase reversa
(SUSLOV; STEINDLER, 2005), como por inibidores co-extraídos com o RNA (BAR et al.,
2003). Este efeito não é bem capturado pela análise de ANOVA, uma vez que o erro
estatístico não se propaga de maneira linear para os níveis inferiores do modelo hierárquico.
Poderia ser especulado que CASP3, como gene de baixa expressão, sofra diferencialmente o
efeito técnico nestes níveis.
No extremo oposto tanto no nível de expressão como na contribuição da variabilidade,
encontra-se o gene LYZ. Este gene, o de maior expressão, apresentou uma variabilidade muito
elevada, com DP de + 2,37 ciclos (Cq), compatível com o relatado para single cells. Esta
variabilidade persistiu mesmo após normalização, com ruído total de + 1,02 DP, que
representa uma variabilidade de ~2 vezes (fold change) de caso a caso. Como a variabilidade
é majoritariamente biológica, ou seja, subjacente ao processamento técnico, o emprego deste
conjunto iniciador/sonda para quantificar o gene LYZ com propósitos diagnósticos ou de
estratificação de risco não seria recomendável. O comportamento particular do gene LYZ
neste estudo mostra características próprias que não são explicáveis pelas condições do
ensaio.

93
A pré-Amp é uma estratégia utilizada para obter uma melhor amplificação de RNAm
a partir de tecidos FFPE, incrementando a sensibilidade na quantificação de genes de baixa
expressão (DENNING et al., 2007; LI et al., 2008; NOUTSIAS et al., 2008). Os trabalhos
que utilizam esta metodologia não avaliam a variabilidade introduzida nesta etapa, ou o fazem
com ensaios de baixo poder estatístico (SANCHEZ-ESPIRIDION et al., 2010). No mesmo
sentido, as descrições do fabricante são assertivas sobre a reprodutibilidade desta metodologia
(APPLIED, 2007). A variabilidade introduzida no nível de pré-Amp foi significativa para os
sete genes avaliados durante o ensaio ANOVA tipo nested, e persistiu após normalização.
Esta etapa foi eficiente para melhorar os valores de Cq, e, em alguns casos, amostras que
poderiam ser consideradas não amplificáveis (Cq > 35), foram recuperas pela aplicação desta
técnica. Isto foi logrado às custas de introduzir maior quantidade de variabilidade técnica.
Esta poderia ser causada pela presença de inibidores que diminuem a eficiência a
retrotranscrição refletindo na variabilidade.
Referente às duplicatas de qPCR, foi observado que em todos os genes avaliados
apresentaram uma variação menor de DP< + 0,15 ciclos (incluindo o gene de baixa
expressão), similar ao relatado na literatura e recomendado pelas abordagens atuais de
quantificação (STAHLBERG et al., 2004; TICHOPAD et al., 2009). De fato, para os ensaios
posteriores foi estabelecido um valor de DP <+ 0,15 ciclos como limite aceitável das
duplicatas das reações de qPCR.
Um conjunto de abordagens novas tem sido descritas, que aplicam algoritmos
matemáticos para estudar as cinéticas das curvas de qPCR, evitando assim o estabelecimento
de um limiar e o uso da curva de eficiência (ZHAO; FERNALD, 2005; GUESCINI et al.,
2008; RUIJTER et al., 2009; BOGGY; WOOLF, 2010; BAR et al., 2012; RUIJTER et al.,
2013). Durante o desenho ANOVA tipo nested foi proposto aplicar o método Cy0, com o
intuito de comparar ambas as abordagens. Em primeiro lugar, as analises comparativas entre
Cq e Cy0 confirmaram que o primeiro método subestima a expressão gênica (PFAFFL, M.
W., 2004; BAR et al., 2012; RUIJTER et al., 2013), sendo a expressão quantificada pelo
método Cy0 significativamente de maior magnitude. Além disso, esta abordagem permitiu
diminuir, mesmo que levemente, a variabilidade de LYZ e CASP3 (GUESCINI et al., 2008),
nos níveis biológicos e técnicos. É possível que a alta variabilidade nestes genes tenha sido
devida a um efeito diferencial de inibidores, que sabidamente apresentam um maior efeito nos
níveis superiores comparados com os níveis inferiores da metodologia, onde o efeito de

94
diluição reduz a quantidade de inibição em cada etapa pelos processos de diluição (BAR et
al., 2012). Comparações mais extensas do desempenho de ambos os métodos e aplicação para
um grupo maior de amostras encontram-se em curso.
Visto que as análises de expressão gênica a partir de material FFPE são cada vez mais
frequentes, e que são sistematicamente afetadas pela presença de inibidores, os novos
métodos de quantificação mediante a cinética da curva podem ser uma alternativa mais
eficiente, entretanto, comparações que mostrem superioridade significativa são necessárias,
dada a relativa dificuldade do cálculo, que requer a manipulação de valores crus de
fluorescência e aplicação do algoritmo específico, antes do pré-processamento rotineiro dos
valores de quantificação. Até o momento, não existem estudos na literatura utilizando a
metodologia do Cy0 em amostras FFPE, e sua avaliação mediante o desenho ANOVA tipo
nested poderá contribuir com informação relevante.
Que modificações podem ser propostas a partir da experiência realizada com o LHc, e
que possam ser aplicadas a qualquer processo envolvendo extração de RNA e quantificação
da expressão gênica a partir de linfonodos FFPE?
Em primeiro lugar, o incremento na concentração e tempo de digestão da Proteinase K
foi considerado a modificação mais importante para melhorar a quantidade de RNA para
ambos os métodos. Isto foi previamente relatado (CHEN et al., 2007), sugerindo que os
protocolos de extração de RNA a partir de amostras de parafina podem ser beneficiados com
esta mudança.
Em relação aos fatores que afetam a amplificação do RNA, foi observado que há um
efeito associado à peça patológica (bloco de parafina) e não ao tumor em si, indicando que
defeitos na fixação ou processamento do bloco são irreversíveis, requerendo a utilização de
novo material. Isto foi anteriormente relatado para DNA obtido de material FFPE
(STEFANOFF et al., 2003), e é um fenômeno bem conhecido na prática do diagnóstico
patológico: quando uma amostra apresenta defeitos de fixação, o diagnóstico se vê
prejudicado, desde a observação microscópica até a biologia molecular.
Visto que a etapa de pré-Amp foi a que sistematicamente introduziu variabilidade
técnica, foram realizadas modificações na metodologia. As alterações que visaram concentrar
o produto para diminuir o efeito randômico em genes de baixa expressão (diminuição da

95
diluição final e aumento do número de ciclos usados na pré-Amp), resultaram em um
incremento ou manutenção da variabilidade na etapa de pré-Amp.
Com a ideia de que a variabilidade poderia ser causada nas etapas anteriores, a
quantidade de RNA colocada na etapa de RT foi diminuída para desta maneira atenuar o
efeito de possíveis inibidores. Observamos então que a variabilidade introduzida na pré-Amp
diminuiu (porém manteve-se significativa) e que esta poderia ser causada pela propagação de
variabilidade das etapas superiores. O resultado dos experimentos indicou a vantagem de se
utilizar menor quantidade de RNA para as próximas etapas. Outros experimentos estão em
curso para melhorar o desempenho desta importante etapa do processamento de amostras
FFPE.
Em suma, diferentes fontes de variabilidade foram identificadas neste trabalho nas
amostras trabalhadas: (i) a presença de uma marcada heterogeneidade biológica causada por
cada condição individual (paciente); (ii) a variabilidade técnica inerente a cada etapa, com
ênfase na etapa de RT e pré-Amp; (iii) a presença de sustâncias inibidoras que interferem de
maneira não linear e; (iv) variações inerentes às reações de PCR que são especificas de cada
gene e que ainda são desconhecidas.
A conclusão geral desta parte do trabalho é que o conhecimento das variabilidades
biológicas e técnicas que resultam da complexidade dos sistemas biológicos ou que são
introduzidas durante as etapas do processamento técnico é essencial para um desenho
experimental apropriado toda vez que a detecção de uma diferença de expressão gênica é o
objetivo principal do estudo.
5.2 Validação de perfis de expressão gênica em um grupo de pacientes com linfoma
de Hodgkin clássico
Neste trabalho, foi avaliada a expressão de 11 genes previamente identificados como
fator prognóstico no LHc avançado de adultos (SANCHEZ-ESPIRIDION et al., 2010). O
conjunto de genes foi avaliado em um grupo de 84 crianças com LHc, que assim serviria
como coorte de validação independente, visando determinar a aplicabilidade desse escore
prognóstico nas crianças com esta doença. No LHc, nesse grupo etário há uma grande
necessidade de biomarcadores para estratificação das terapias, visto que mesmo o escore
clínico internacional (IPS) amplamente usado em adultos não e aplicável nas crianças. O

96
tratamento do LHc é considerada um balanço de riscos entre a recaída, se o tratamento não for
o suficientemente intenso, e a ocorrência de efeitos tardios devido a um excesso de
quimioterápicos (KLUMB et al., 2007; MORAIS et al., 2009).
Para validar o escore baseado na expressão de 11 genes, os mesmos conjuntos de
iniciadores e sondas, assim como as mesmas abordagens metodológicas propostos no estudo
original foram aplicados (SANCHEZ-ESPIRIDION et al., 2010). Apesar da unificação
metodológica, neste estudo identificamos apenas a expressão gênica de CASP3 associada ao
prognóstico no grupo pediátrico; com associações similares as descritas para os adultos.
Dentre as possíveis razões para a falta de reprodutibilidade dos resultados podemos ressaltar
(i) a complexidade histológica da doença que dificulta a reprodutibilidade; (ii) impacto das
variações técnicas, e finalmente, não podemos descartar (iii) diferenças na composição das
células H-RS e do microambiente entre adultos e crianças, previamente descritas (BARROS
et al., 2012).
Há também uma limitação relacionada ao número de casos neste estudo (78 casos, 18
eventos), cujo poder estatístico para descobrimento é baixo. Entretanto, esta limitação é
menor se entendemos o presente como um grupo de validação independente, para marcadores
já descritos na mesma patologia e utilizando a mesma abordagem metodológica.
Apontando para a complexidade biológica como responsável pela falta de
reprodutibilidade, existem discrepâncias inclusive entre o trabalho que originalmente
identificou os 11 genes associados a prognóstico por TLDA (SANCHEZ-ESPIRIDION et al.,
2009) e o trabalho de desenvolvimento do escore (SANCHEZ-ESPIRIDION et al., 2010) no
que diz respeito à super-expressão do perfil de marcadores de macrófagos (STAT1 e LYZ), que
no primeiro estudo está associado a prognóstico desfavorável, enquanto que no segundo, a um
prognóstico favorável. Recentemente, outro escore clínico para predizer a SG foi
desenvolvido baseado na expressão gênica por PCR digital a partir de RNA de parafina
(SCOTT et al., 2012). Este escore estava composto por 23 genes, incluindo também genes
associados a macrófagos como ALDH1, CD68, STAT1 e LYZ, cuja super-expressão foi
associada a sobrevida global mais curta em um grupo de pacientes adultos. Nesse trabalho, a
expressão de CASP3 não foi associada a prognóstico. Estes resultados, em conjunto com os
obtidos no presente trabalho indicam que, mesmo usando metodologias quantitativas
altamente reprodutíveis, como RT-qPCR, os perfis de expressão gênica não são uma

97
ferramenta que possa ser facilmente transposta para a clínica com resultados multicêntricos
aceitáveis.
Quanto à variabilidade técnica, controles de qualidade para avaliação da cinética das
reações de qPCR, assim como critérios de identificação de falta de amplificação de uma
amostra foram estritamente seguidos neste estudo. Alterações na cinética da curva,
especificamente na pendente são indicadores da presença de inibidores, que poderia dar como
resultado um falso negativo (BAR et al., 2003; TICHOPAD et al., 2010). SANCHEZ-
ESPIRIDION et al. (2010), utilizam como critérios de exclusão uma pureza do RNA de
λ260/230 <1,7; ou amplificação dos GDR em Cq>35 ciclos, ou falta de amplificação em 30%
dos genes. Nosso grupo decidiu aceitar as amostras de RNA com λ260/230 < 1,7; casos com
amplificação nos GDR Cq>35 ciclos foram excluídos, aqueles que mostrarem alterações na
curva sigmoide ou falta de amplificação em pelo menos 5 genes dos 11 do grupo total
avaliados eram encaminhados para uma nova extração de RNA e posteriormente excluídos na
presença do mesmo padrão.
É possível pensar que se a expressão gênica estivesse afetada extremamente pela
variabilidade biológica interindividual e pela variabilidade técnica, os níveis de expressão
gênica seriam randômicos e não seria possível descrever padrões e perfis no grupo total, o que
não foi o caso.
A análise de cluster hierárquico não supervisionado identificou um grupo com baixa
expressão dos genes da via do ciclo celular, majoritariamente EBV- e EN grau II. Este subtipo
da EN caracteriza-se por apresentar um maior número de células tumorais (BENNETT; TU;
HUDSON, 1981) e inicialmente foi associada a uma resposta desfavorável às terapias
(MACLENNAN et al., 1992). Em termos gerais, o ciclo celular na célula H-RS se encontra
desregulado, com alta expressão das ciclinas e de CDKs, assim como alterações no checkpoint
mitótico levando a uma proliferação e divisão aberrante causando uma multinucleação e
poliploidia (KUPPERS, 2001; GARCIA et al., 2003; BAI et al., 2005; SANCHEZ-
AGUILERA; GARCIA; et al., 2006; ALVARO et al., 2008; BARROS et al., 2009). A
diminuição da expressão dos genes do ciclo celular neste grupo poderia ser reflexo da
existência de alguma subpopulação de células H-RS com parada na divisão celular, o que
poderia estar associado potencialmente com resposta terapêutica desfavorável. Já foi descrita
uma relação entre maior fração proliferativa e eficácia maior das drogas quimioterápicas
(RADULOVIC; JELIC, 1997), inclusive no LHc de crianças por nosso grupo e por outros

98
autores (BARROS; ZALCBERG; HASSAN, 2008; DINAND et al., 2008; MORAIS et al.,
2009). Alternativamente, uma vez que não houve uma diminuição de Ki67 nas células
tumorais nesse grupo, é possível que o perfil de expressão reflita a composição particular do
MAT, caracterizado por um baixo número de células citotóxicas GRZB+ e macrófagos
CD163+ e baixa fração proliferativa linfocitária. Esta constelação de características
particulares nunca antes foi descrita e sua potencial associação com prognóstico desfavorável
(não mostrado, por falta de poder estatístico) estimula a ampliar o número de casos para
melhor descrever este possível grupo prognóstico.
A análise de componentes principais identificou três componentes que explicavam
~70% da variabilidade na expressão gênica no grupo de pacientes com LHc pediátricos. Isto
era esperado, visto a escolha dos alvos baseado em vias celulares, reforçando a ideia de que a
marcada variabilidade interindividual não impede a identificação de padrões com sentido
biológico no LHc. Este trabalho é o primeiro a analisar a expressão gênica exclusivamente em
crianças com LHc, mostrando a reprodutibilidade destes perfis nesse grupo etário. Chamou a
atenção o segundo PCA composto por dois genes do microambiente (STAT1 e LYZ) e dois
genes da via da apoptose (CASP3 e BCL2). Como o PCA captura grupos de genes em vetores
ortólogos, este PCA fornece uma informação que pode estimular a pesquisa da interação dos
macrófagos com a célula tumoral e outras células do microambiente, vista sua importância na
resposta terapêutica. Quais são as células expressando CASP3 e BCL2 moduladas pela
população de macrófagos, somente poderá ser respondido por ensaios funcionais. Neste
trabalho é mostrado que as células do microambiente expressam CASP3 ativada em
proporções variáveis, e sabe-se que BCL2 é uma proteína altamente expressa pelos linfócitos
B para sua sobrevivência dentro dos folículos dos nódulos linfáticos, o que justifica a
agrupação de genes do microambiente com os representantes da via da apoptose.
A análise supervisionada (comparação entre grupos selecionados) mostrou que o
subtipo histológico EN apresentou menor expressão dos genes CASP3 e CDK1 quando
comparados com a CM. Este perfil de expressão gênica pode ser, ao menos em parte, adstrito
ao MAT, uma vez que a EN tem baixo número de células citotóxicas, cuja associação com a
expressão de CASP3 ativada no microambiente foi mostrada neste estudo. Além disso, a
baixa expressão de CDK1 pode estar associada a um menor número de linfócitos proliferantes
no MAT, visto a correlação entre a expressão deste gene e o número de linfócitos Ki67+.

99
Um estudo de expressão gênica a partir da massa tumoral total utilizando a
metodologia de microarray (Affymetrix), foi realizado por BIRGERSDOTTER et al. (2009)
visando diferenciar os subtipos EN e CM. Este estudo mostrou que existem perfis
diferenciais, a EN apresenta uma alta expressão de genes relacionados à matriz extracelular e
seu remodelamento e inflamação, enquanto que a CM se caracteriza pela presença de
moléculas inflamatórias e genes associados à resposta citotóxica. A análise combinada da
expressão gênica e composição do microambiente tumoral por IHQ em nossos casos apoia a
caracterização gênica descrita por BIRGERSDOTTER et al. (2009).
Como mencionado anteriormente, no presente trabalho, uma alta expressão de CASP3
foi associada a uma pior SLP. Como este resultado é contra-intuitivo em relação ao papel da
CASP3 como efetora da apoptose, decidimos investigar a expressão da proteína CASP3
ativada por IHQ. Infelizmente, a imunomarcação de CASP3 nas células tumorais não pode ser
registrada com confiança, devido a problemas de índole técnica; a identificação das células H-
RS expressando CASP3 ativada e sua distinção dos macrófagos contendo corpos apoptóticos
no citoplasma viu-se dificultada pelo insucesso das duplas marcações com CD30, muito
provavelmente porque esta proteína desaparece da membrana celular durante as fases
precoces do apoptose (DUKERS et al., 2002). Duplas marcações com CD68 e CASP3
poderão ajudar a resolver esta questão. Isto é relevante, pois há controvérsias na literatura
sobre o valor prognóstico de CASP3 nas células H-RS. Um maior número de células tumorais
CASP3+ foram associadas a um melhor prognóstico (DUKERS et al., 2002), enquanto que
não tiveram associação significativa no estudo de (BAI et al., 2007). Esta discrepância pode
dever-se a diferenças nos critérios de registro de positividade nos dois estudos.
Assim, decidimos focar a análise no número de células CASP3+ no MAT, análise que
não tinha sido feita anteriormente na literatura, e compará-la com a expressão gênica e com as
diferentes subpopulações neste microambiente. Em nosso grupo de pacientes, a expressão de
CASP3 foi correlacionada com o número de células citotóxicas (CD8+, GRZB+, TIA1+) no
microambiente, assim como foi encontrada uma associação positiva entre o número de
linfócitos CASP3+ e o número de linfócitos citotóxicos CD8+. Não houve associação entre
expressão de CASP3 e número de células expressando CASP3 ativada, evidenciando a
preeminência da regulação pós-traducional destas (DENAULT; SALVESEN, 2002;
BOATRIGHT; SALVESEN, 2003).

100
A associação entre células citotóxicas no microambiente do LHc e a expressão de
proteínas pró e anti-apoptóticas nas células H-RS, inclusive CASP3, foi mostrada
anteriormente (DUKERS et al., 2002; ALVARO et al., 2008). A interpretação possível é que
os estímulos pró-apoptóticos mediados pelas células citotóxicas tenham como resultado a
indução da apoptose na células H-RS, ou uma resposta de escape, via por exemplo a super-
expressão de BCL2L1.
Os resultados da expressão de CASP3 nas células do microambiente devem ser
interpretadas desde outro ângulo. Em primeiro lugar, junto com a documentação da
proliferação dos linfócitos pela primeira vez na literatura sobre o LHc, mostra que há uma
“vida linfocitária” no linfonodo doente, ou seja, as células não somente são recrutadas, mas
também proliferam e morrem, certamente em relação a estímulos obtidos in situ. Desta
maneira, o modelo de partida segundo o qual o MAT no LHc reflete primariamente o que é
recrutável na periferia (POPPEMA, 2005) - exemplificado na constituição do MAT nos LHc
associados ao HIV (THOMPSON et al., 2004; BIGGAR et al., 2006; HARTMANN et al.,
2013) ou proposto por nossa equipe ao relacionar a histogênese do LHc de criança com a
condição imune no momento da transformação maligna (BARROS et al., 2011)- é uma
simplificação ou subestimação das interações celulares que ocorrem nos linfonodos tumorais.
Em segundo lugar, é preciso de perguntar sobre a natureza e estado fisiológico dos
parceiros dessas interações, e o grau em que os mecanismos de homeostase linfocitária no
linfonodo doente são propriamente saudáveis. Os resultados apresentados aqui são claramente
inadequados para responder estas perguntas, e a sua resposta cabal deverá esperar até o
desenvolvimento de modelos experimentais para esta doença. No entremeio, a linha de
interpretação baseada nos estudos de associação podem ajudar na formação de hipóteses.
Previamente em trabalhos desenvolvidos por nossa equipe no mesmo grupo de
pacientes, uma pior SLP foi associada a um maior número de células citotóxicas (GRZB+)
(BARROS et al., 2012), em concordância com a maioria dos estudos prévios que
investigaram prognóstico associado com o microambiente no LHc de adultos (ALVARO-
NARANJO et al., 2005; ALVARO et al., 2005; KELLEY et al., 2007; TZANKOV et al.,
2008).
A principal associação das células expressando CASP3+ no MAT é com as células T
CD8+ e com as células B. Isto permite oferecer uma primeira explicação para a observação do

101
pior prognóstico relacionado à alta expressão do gene CASP3: as células expressando este
gene não se limitam às H-RS (na qual se esperaria um efeito favorável na resposta
terapêutica), senão também, e em maior medida, CASP3 é expressa as células do MAT. Como
há número proporcional de células CD8+ e CASP3+, a causa do prognóstico desfavorável
poderia ter a mesma origem biológica
O resultado de um alto número de células GRZB+ associado a falha terapêutica é
oposto ao modelo de imunovigilância, em que as CTLs estariam controlando o crescimento
tumoral. Esta característica não tem ainda uma explicação científica, mas uma das
interpretações possíveis é que as CTLs estivessem controlando outra/s subpopulação/ções
com função anti-tumoral. A análise conjunta de CASP3 e Ki67 identificou duas populações
com “comportamentos” opostos no tumor. O número de células CD8+, assim como as células
diferenciadas para citotoxicidade aumenta concomitantemente ao marcador de proliferação,
independentemente a CASP3+, refletindo provavelmente sua natureza de célula ativada. A
população CD20+ aumenta na presença de altos números de células CASP3+ independente do
marcador de proliferação Ki67+. Isto poderia ser interpretado como que o número de
linfócitos B não dependesse tanto da proliferação, quanto da diminuição (apoptose) de uma
população que controla o seu número?
Por outro lado, a ativação de CASP3 pode também estar envolvida na diferenciação de
diferentes tipos celulares (ALAM et al., 1999; SCHWERK; SCHULZE-OSTHOFF, 2003).
Esta condição poderia explicar a expressão de CASP3+ em células em proliferação (Ki67+).
Novas estratégias de identificação celular devem ser implementadas com o intuito de
caracterizar as subpopulações expressando estes dois marcadores, juntos e separados. Estudos
com marcação de TUNEL devem ser realizados para identificar se as células expressando
CASP3+ estão de fato sendo encaminhadas para a apoptose ou a proteína está cumprindo
papeis diversos nos diferentes tipos celulares que compõem a massa tumoral. Por exemplo,
SORDET et al. (2002) mostraram que a ativação da CASP3 nem sempre leva à morte celular
em monócitos, podendo contribuir à diferenciação de macrófagos.
A presença e expressão de macrófagos no MAT é um motivo constante na literatura
recente sobre o LHc (SANCHEZ-ESPIRIDION et al., 2009; SANCHEZ-ESPIRIDION et al.,
2010; STEIDL et al., 2010; TZANKOV et al., 2010; BARROS; HASSAN; NIEDOBITEK,
2012; HARRIS et al.,2012; SCOTT et al., 2012; TAN et al., 2012; DEAU et al., 2013),
devido às evidências crescentes de seu papel desfavorável na resposta terapêutica (STEIDL;

102
FARINHA; GASCOYNE, 2011; BARROS; HASSAN; NIEDOBITEK, 2012; TAN et al.,
2012). Neste trabalho, foi encontrado que o número de macrófagos CD68+ e CD163+ esteve
altamente correlacionado com a expressão de STAT1, LYZ e IRF4. Isto reforça a assinatura
gênica dos macrófagos, tendo em conta que IRF4 é um importante fator de transcrição na
polarização de macrófagos M2 (SANCHEZ-ESPIRIDION et al., 2009; SATOH et al., 2010).
Entretanto, em nosso grupo de pacientes, esta assinatura não teve valor prognóstico, em
concordância com a própria variabilidade observada no estudo original (SANCHEZ-
ESPIRIDION et al., 2009; SANCHEZ-ESPIRIDION et al., 2010), e na possibilidade de que
a expressão não reflita a subpopulação de macrófagos realmente associada com o prognóstico.
Neste sentido, a perspectiva é que a IHQ quantitativa deva ser refinada para fornecer
biomarcadores de macrófagos com valor prognóstico reprodutível nas diversas situações
clínicas em que os pacientes são tratados. A heterogeneidade e plasticidade dos macrófagos
pode ser um empecilho para este objetivo.
Os macrófagos têm sido classificado em 3 categorias: M1 caracterizados pelo
marcador de membrana CD68 e a expressão de STAT1, M2 identificados pela marcação de
CD163 e a expressão de STAT6, e os macrófagos associados ao tumor (TAM) sendo
denominados como macrófagos M2 like que apresentam o marcador de membrana CD163
(MANTOVANI et al., 2002; ÁLVARO et al., 2006; BISWAS; MANTOVANI, 2010). Esta
visão está sendo confrontada por alguns trabalhos que mostram heterogeneidade na
diferenciação dentro de um mesmo tumor (PETTERSEN et al., 2011), inclusive por dados da
nossa equipe, em relação à polarização e a CD163 como marcador de macrófago M2
(BARROS et al., 2013, em preparação).
A presença do EBV nas células H-RS foi associada a um aumento da expressão gênica
de STAT1 e LYZ. Esta associação decorre do enriquecimento do MAT dos casos EBV+ com
células da linhagem monocítica/macrófago (CD14+, CD68+ e CD163+), observada por nós
nas crianças e por outros autores no LHc de adulto (KAMPER et al., 2011; STEIDL et al.,
2011; BARROS et al., 2012) . Os mecanismos de recrutamento dos macrófagos pelo vírus são
desconhecidos, assim como o estado de ativação destas células nos casos EBV+ e EBV-.
Entretanto, nas crianças com LHc, diferente do observado em adultos, o EBV parece modular
a resposta desfavorável associada aos macrófagos CD163+ (BARROS et al., 2012). De fato,
esta observação também foi feita no presente trabalho em relação a CASP3 e CDK1, em que a
presença do vírus amenizava o impacto prognóstico deste marcador.

103
O papel causal do EBV na patogênese da doença é bem estabelecido (HSI, 2008;
KUPPERS, 2009a; ALDINUCCI et al., 2010; KUPPERS et al., 2012), enquanto que seus
papeis na progressão da mesma e na resposta terapêutica ainda precisam ser melhor
entendidos. Nas crianças com LHc parece haver uma resposta imune antitumoral/antiviral
mais efetiva nos casos EBV+ que pode modular o efeito das quimioterapias (BARROS et al.,
2012). Isto seria responsáveis pela melhor sobrevida e resposta clínica nos casos EBV+ da
infância (CLAVIEZ et al., 2005). Os nossos resultados prévios, em conjunto com os deste
trabalho mostram que a presença do EBV é um importante modulador dos potenciais fatores
prognósticos investigados (BARROS, HASSAN, NIEDOBITEK, 2012; BARROS et al.,
2012). Estas características diferenciais podem ser também responsáveis pela falta de impacto
prognóstico de marcadores biológicos de reconhecida importância nos adultos.
Este conjunto de características que contribuem para a complexidade deste doença
precisam ser melhor entendidas, sobre tudo nesta época de grandes desenvolvimentos
terapêuticos alvo-específicos. O LHc de criança é uma doença pouco estudada e com este
trabalho esperamos ter feito uma contribuição em relação às condições de aplicação das
metodologias de estudo, assim como em atrair atenção para uma situação biológica, clínica e
terapêutica diferenciada dentro do LHc, que requer maiores esforços de pesquisa visto a curta
idade dos pacientes e a necessidade de tratamentos cada vez mais precisos.

104
6 CONCLUSÕES
Durante este estudo, uma metodologia para extração de RNA a partir de material
FFPE foi desenvolvida e aperfeiçoada, introduzindo modificações que aumentaram o
rendimento e a qualidade do RNA extraído. Esta metodologia foi utilizada com sucesso para
estudos de perfis de expressão gênica em pacientes com LHc. O método otimizado mostrou
aplicabilidade similar em amostras de arquivo independentemente do tempo de
armazenamento.
Um desenho experimental ANOVA tipo nested (hierárquico) foi realizado para avaliar
as fontes de variabilidade introduzidas em cada etapa da metodologia aplicada. Os genes
avaliados (LYZ, STAT1, IRF4, BCL2 e CASP3 como alvos e GUSB e HMBS como referência)
mostraram variabilidades similares às publicadas para tecidos a fresco, indicando que a
utilização de RNA de parafina não acarreta em uma variabilidade adicional que impeça ter
resultados com sentido biológico. Todos os genes mostraram alto impacto da variabilidade
biológica. Os genes de baixa expressão sofreram mais os efeitos da variabilidade técnica. O
uso de valores normalizados (∆Cq) resultou em uma diminuição da variabilidade. A etapa de
pré-Amp do cDNA é uma fonte sistemática de variabilidade técnica, devendo ser otimizada
previamente aos estudos com material primário.
Um método de quantificação das reações de PCR em tempo real baseado na análise da
cinética da curva (Cy0) foi aplicado, pondo em evidência o efeito potencial de inibidores nos
genes com mais variabilidade biológica e técnica (CASP3 e LYZ). Visto que as reações a
partir de RNA FFPE são sistematicamente afetadas pela presença de inibidores, os novos
métodos de quantificação mediante a cinética da curva podem ser uma alternativa mais
eficiente, entretanto, comparações que mostrem superioridade significativa com o método Cq
são ainda necessárias.
Este trabalho é o primeiro a analisar a expressão de genes relacionados às vias do ciclo
celular (CENPF, CDK1, CCNA2, CCNE2 e HMMR), apoptose (CASP3, BCL2, BCL2L1),
ativação de monócitos/macrófagos (STAT1, LYZ) e fator regulador de interferon 4 (IRF4) em
um grupo exclusivamente de crianças com LHc (N=84), mostrando a reprodutibilidade destes
perfis nesse grupo etário. A análise de componentes principais identificou três componentes
que explicavam ~70% da variabilidade na expressão gênica. O subtipo EN apresentou baixos
níveis dos genes CASP3 e CDK1 quando comparado com a CM.

105
Dos 11 genes com valor prognóstico relatado em adultos, apenas a alta expressão de
CASP3 foi associada com pior SLP. Quando estratificados pelo status do EBV, os casos EBV
negativos mostraram uma sobrevida ainda pior na presença de altos níveis de CASP3. A
expressão da CASP3 não reflete somente as células H-RS, uma vez que a presença da proteína
CASP3 ativada foi detectada no MAT. Foi detectada uma associação consistente e com
provável valor clínico entre expressão gênica de CASP3, populações citotóxicas no MAT e
expressão de CASP3 ativada nestas mesmas células.
Escores clínicos proposto em pacientes com LHc em adultos não são reprodutíveis em
pacientes pediátricos.
Os casos associados ao EBV mostraram uma super-expressão de STAT1 e LYZ, em
concordância com um maior número de monócitos e macrófagos no MAT. O número de
células citotóxicas (CD8+, GRZB+, TIA1+), mostrou uma correlação positiva com a
expressão de CASP3, CDK1 e BCL2, STAT1 e/ou LYZ. Houve uma correlação inversa entre a
presença de alto número de eosinófilos com a expressão dos genes das vias do ciclo celular
(CENPF e CDK1), apoptose (CASP3 e BCL2), e relacionados a macrófagos (STAT1 e LYZ).
A abordagem integrada de expressão gênica e IHQ quantitativa pode fornecer pistas
sobre processos de interação patogênica entre as células H-RS e as células do MAT, que
possam estar envolvidos na resposta clínica do LHc pediátrico.

106
7 REFERÊNCIAS BIBLIOGRÁFICAS
ALAM, A. et al. Early activation of caspases during T lymphocyte stimulation results in
selective substrate cleavage in nonapoptotic cells. J Exp Med, v. 190, n. 12, p. 1879-90, Dec
20 1999.
ALDINUCCI, D. et al. The classical Hodgkin's lymphoma microenvironment and its role in
promoting tumour growth and immune escape. J Pathol, v. 221, n. 3, p. 248-63, Jul 2010.
ALLAVENA, P. et al. The inflammatory micro-environment in tumor progression: the role of
tumor-associated macrophages. Crit Rev Oncol Hematol, v. 66, n. 1, p. 1-9, Apr 2008.
ALVARO-NARANJO, T. et al. Tumor-infiltrating cells as a prognostic factor in Hodgkin's
lymphoma: a quantitative tissue microarray study in a large retrospective cohort of 267
patients. Leuk Lymphoma, v. 46, n. 11, p. 1581-91, Nov 2005.
ÁLVARO, T. et al. The presence of STAT1-positive tumor-associated macrophages and their
relation to outcome in patients with follicular lymphoma. Haematologica v. 91, p. 1605-
1612, 2006.
ALVARO, T. et al. Tumor-infiltrated immune response correlates with alterations in the
apoptotic and cell cycle pathways in Hodgkin and Reed-Sternberg cells. Clin Cancer Res, v.
14, n. 3, p. 685-91, Feb 1 2008.
ALVARO, T. et al. Outcome in Hodgkin's lymphoma can be predicted from the presence of
accompanying cytotoxic and regulatory T cells. Clin Cancer Res, v. 11, n. 4, p. 1467-73, Feb
15 2005.
ANDERSEN, C. L.; JENSEN, J. L.; ORNTOFT, T. F. Normalization of real-time quantitative
reverse transcription-PCR data: a model-based variance estimation approach to identify genes
suited for normalization, applied to bladder and colon cancer data sets. Cancer Res, v. 64, n.
15, p. 5245-50, Aug 1 2004.
ANIKEEVA, N.; SYKULEV, Y. Mechanisms controlling granule-mediated cytolytic activity
of cytotoxic T lymphocytes. Immunol Res, v. 51, n. 2-3, p. 183-94, Dec 2011.

107
ANTICA, M. et al. Gene expression in formalin-fixed paraffin-embedded lymph nodes. J
Immunol Methods, v. 359, n. 1-2, p. 42-6, Jul 31 2010.
APPLIED. TaqMan® PreAmp Master Mix Kit Protocol. USA: 22 p. 2007.
ARMSTRONG, A. A. et al. Association of Epstein-Barr virus with pediatric Hodgkin's
disease. Am J Pathol, v. 142, n. 6, p. 1683-8, Jun 1993.
ARZT, L. et al. Evaluation of formalin-free tissue fixation for RNA and microRNA studies.
Exp Mol Pathol, v. 91, n. 2, p. 490-5, Oct 2011.
AUDIC, S.; CLAVERIE, J. M. The significance of digital gene expression profiles. Genome
Res, v. 7, n. 10, p. 986-95, Oct 1997.
AZAMBUJA, D. et al. Lack of association of tumor-associated macrophages with clinical
outcome in patients with classical Hodgkin's lymphoma. Ann Oncol, v. 23, n. 3, p. 736-42,
Mar 2012.
BAI, M. et al. B-cell differentiation immunophenotypes in classical Hodgkin lymphomas.
Leuk Lymphoma, v. 47, n. 3, p. 495-501, Mar 2006.
BAI, M. et al. Expression of bcl2 family proteins and active caspase 3 in classical Hodgkin's
lymphomas. Hum Pathol, v. 38, n. 1, p. 103-13, Jan 2007.
BAI, M. et al. Cell cycle and apoptosis deregulation in classical Hodgkin lymphomas. In
Vivo, v. 19, n. 2, p. 439-53, Mar-Apr 2005.
BAR, T.; KUBISTA, M.; TICHOPAD, A. Validation of kinetics similarity in qPCR. Nucleic
Acids Res, v. 40, n. 4, p. 1395-406, Feb 2012.
BAR, T. et al. Kinetic Outlier Detection (KOD) in real-time PCR. Nucleic Acids Res, v. 31,
n. 17, p. e105, Sep 1 2003.
BARROS, M. H. Fatores genéticos e ambientais na patogênese, constituição do
microambiente tumoral e prognóstico clínico do linfoma de Hodgkin pediátrico. 2011.

108
192 f. Tese (Doutor) - Pós-Graduação em Oncologia, Instituto Nacional de Câncer, Rio de
Janeiro, Brasil.
BARROS, M. H.; HASSAN, R.; NIEDOBITEK, G. Disease patterns in pediatric classical
Hodgkin lymphoma: a report from a developing area in Brazil. Hematol Oncol, v. 29, n. 4, p.
190-5, Dec 2011.
______. Tumor-associated macrophages in pediatric classical hodgkin lymphoma: association
with epstein-barr virus, lymphocyte subsets, and prognostic impact. Clin Cancer Res, v. 18,
n. 14, p. 3762-71, Jul 15 2012.
BARROS, M. H. et al. Cell cycle characteristics and Epstein-Barr virus are differentially
associated with aggressive and non-aggressive subsets of Hodgkin lymphoma in pediatric
patients. Leuk Lymphoma, v. 51, n. 8, p. 1513-22, Aug 2010.
BARROS, M. H. et al. Tumor microenvironment composition in pediatric classical Hodgkin
lymphoma is modulated by age and Epstein-Barr virus infection. Int J Cancer, v. 131, n. 5,
p. 1142-52, Sep 1 2012.
BARROS, M. H.; ZALCBERG, I.; HASSAN, R. Syncytial neoplastic cells in paediatric
Hodgkin lymphoma. Eur J Haematol, v. 82, n. 1, p. 81-2, Jan 2009.
BARROS, M. H.; ZALCBERG, I. R.; HASSAN, R. Prognostic impact of CD15 expression
and proliferative index in the outcome of children with classical Hodgkin lymphoma. Pediatr
Blood Cancer, v. 50, n. 2, p. 428-9; author reply 430, Feb 2008.
BAUER, K. et al. Comparison of chemotherapy including escalated BEACOPP versus
chemotherapy including ABVD for patients with early unfavourable or advanced stage
Hodgkin lymphoma. Cochrane Database Syst Rev, n. 8, p. CD007941, 2011.
BAUMFORTH, K. R. et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus
nuclear antigen 1 in Hodgkin's lymphoma cells mediates Up-regulation of CCL20 and the
migration of regulatory T cells. Am J Pathol, v. 173, n. 1, p. 195-204, Jul 2008.

109
BECHTEL, D. et al. Transformation of BCR-deficient germinal-center B cells by EBV
supports a major role of the virus in the pathogenesis of Hodgkin and posttransplantation
lymphomas. Blood, v. 106, n. 13, p. 4345-50, Dec 15 2005.
BENNETT, M. H. et al. The prognostic significance of cellular subtypes in nodular sclerosing
Hodgkin's disease: an analysis of 271 non-laparotomised cases (BNLI report no. 22). Clin
Radiol, v. 34, n. 5, p. 497-501, Sep 1983.
BENNETT, M. H.; TU, A.; HUDSON, G. V. Analysis of grade 1 Hodgkin's disease (Report
no 6). Clin Radiol, v. 32, n. 5, p. 491-8, Sep 1981.
BIGGAR, R. J. et al. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS.
Blood, v. 108, n. 12, p. 3786-91, Dec 1 2006.
BIRGERSDOTTER, A. et al. Inflammation and tissue repair markers distinguish the nodular
sclerosis and mixed cellularity subtypes of classical Hodgkin's lymphoma. Br J Cancer, v.
101, n. 8, p. 1393-401, Oct 20 2009.
BISWAS, S. K.; MANTOVANI, A. Macrophage plasticity and interaction with lymphocyte
subsets: cancer as a paradigm. Nat Immunol, v. 11, n. 10, p. 889-96, Oct 2010.
BOATRIGHT, K. M.; SALVESEN, G. S. Mechanisms of caspase activation. Curr Opin Cell
Biol, v. 15, n. 6, p. 725-31, Dec 2003.
BOGGY, G. J.; WOOLF, P. J. A mechanistic model of PCR for accurate quantification of
quantitative PCR data. PLoS One, v. 5, n. 8, p. e12355, 2010.
BOHMANN, K. et al. RNA extraction from archival formalin-fixed paraffin-embedded
tissue: a comparison of manual, semiautomated, and fully automated purification methods.
Clin Chem, v. 55, n. 9, p. 1719-27, Sep 2009.
BRAUNINGER, A. et al. Typing the histogenetic origin of the tumor cells of lymphocyte-
rich classical Hodgkin's lymphoma in relation to tumor cells of classical and lymphocyte-
predominance Hodgkin's lymphoma. Cancer Res, v. 63, n. 7, p. 1644-51, Apr 1 2003.

110
BRINK, A. A. et al. Low p53 and high bcl-2 expression in Reed-Sternberg cells predicts poor
clinical outcome for Hodgkin's disease: involvement of apoptosis resistance? Mod Pathol, v.
11, n. 4, p. 376-83, Apr 1998.
BUSTIN, S. A. Absolute quantification of mRNA using real-time reverse transcription
polymerase chain reaction assays. J Mol Endocrinol, v. 25, n. 2, p. 169-93, Oct 2000.
BUSTIN, S. A. et al. MIQE precis: Practical implementation of minimum standard guidelines
for fluorescence-based quantitative real-time PCR experiments. BMC Mol Biol, v. 11, p. 74,
2010.
BUSTIN, S. A. et al. The MIQE guidelines: minimum information for publication of
quantitative real-time PCR experiments. Clin Chem, v. 55, n. 4, p. 611-22, Apr 2009.
BUTLER, J. J.; PUGH, W. C. Review of Hodgkin's disease. Hematol Pathol, v. 7, n. 2, p.
59-77, 1993.
CAO, S. et al. The protooncogene c-Maf is an essential transcription factor for IL-10 gene
expression in macrophages. J Immunol, v. 174, n. 6, p. 3484-92, Mar 15 2005.
CARBONE, A. et al. The Germinal centre-derived lymphomas seen through their cellular
microenvironment. Br J Haematol, v. 145, n. 4, p. 468-80, May 2009.
CARBONE, A. et al. Classical Hodgkin's lymphoma arising in different host's conditions:
pathobiology parameters, therapeutic options, and outcome. Am J Hematol, v. 86, n. 2, p.
170-9, Feb 2011.
CARBONE, P. P. et al. Report of the Committee on Hodgkin's Disease Staging
Classification. Cancer Res, v. 31, n. 11, p. 1860-1, Nov 1971.
CHABAY, P. A. et al. Pediatric Hodgkin lymphoma in 2 South American series: a distinctive
epidemiologic pattern and lack of association of Epstein-Barr virus with clinical outcome. J
Pediatr Hematol Oncol, v. 30, n. 4, p. 285-91, Apr 2008.

111
CHEN, J.; BYRNE, G. E., JR.; LOSSOS, I. S. Optimization of RNA extraction from
formalin-fixed, paraffin-embedded lymphoid tissues. Diagn Mol Pathol, v. 16, n. 2, p. 61-72,
Jun 2007.
CHETAILLE, B. et al. Molecular profiling of classical Hodgkin lymphoma tissues uncovers
variations in the tumor microenvironment and correlations with EBV infection and outcome.
Blood, v. 113, n. 12, p. 2765-3775, Mar 19 2009.
CLAVIEZ, A. et al. Impact of latent Epstein-Barr virus infection on outcome in children and
adolescents with Hodgkin's lymphoma. J Clin Oncol, v. 23, n. 18, p. 4048-56, Jun 20 2005.
CRAWFORD, D. H. Biology and disease associations of Epstein-Barr virus. Philos Trans R
Soc Lond B Biol Sci, v. 356, n. 1408, p. 461-73, Apr 29 2001.
CRONIN, M. et al. Measurement of gene expression in archival paraffin-embedded tissues:
development and performance of a 92-gene reverse transcriptase-polymerase chain reaction
assay. Am J Pathol, v. 164, n. 1, p. 35-42, Jan 2004.
D'AMELIO, M.; CAVALLUCCI, V.; CECCONI, F. Neuronal caspase-3 signaling: not only
cell death. Cell Death Differ, v. 17, n. 7, p. 1104-14, Jul 2010.
DEAU, B. et al. Macrophage, mast cell and T lymphocyte infiltrations are independent
predictive biomarkers of primary refractoriness or early relapse in classical Hodgkin
lymphoma. Leuk Lymphoma, v. 54, n. 1, p. 41-5, Jan 2013.
DELEEUW, R. J. et al. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in
cancer: a critical review of the literature. Clin Cancer Res, v. 18, n. 11, p. 3022-9, Jun 1
2012.
DENARDO, D. G.; ANDREU, P.; COUSSENS, L. M. Interactions between lymphocytes and
myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev, v. 29, n. 2,
p. 309-16, Jun 2010.
DENAULT, J. B.; SALVESEN, G. S. Caspases: keys in the ignition of cell death. Chem Rev,
v. 102, n. 12, p. 4489-500, Dec 2002.

112
DENNING, K. M. et al. A molecular expression signature distinguishing follicular lesions in
thyroid carcinoma using preamplification RT-PCR in archival samples. Mod Pathol, v. 20, n.
10, p. 1095-102, Oct 2007.
DEVILARD, E. et al. Gene expression profiling defines molecular subtypes of classical
Hodgkin's disease. Oncogene, v. 21, n. 19, p. 3095-102, May 2 2002.
DIEHL, V.; THOMAS, R. K.; RE, D. Part II: Hodgkin's lymphoma--diagnosis and treatment.
Lancet Oncol, v. 5, n. 1, p. 19-26, Jan 2004.
DINAND, V. et al. Proliferative index and CD15 expression in pediatric classical Hodgkin
lymphoma. Pediatr Blood Cancer, v. 50, n. 2, p. 280-3, Feb 2008.
DOLESHAL, M. et al. Evaluation and validation of total RNA extraction methods for
microRNA expression analyses in formalin-fixed, paraffin-embedded tissues. J Mol Diagn, v.
10, n. 3, p. 203-11, May 2008.
DUKERS, D. F. et al. High numbers of active caspase 3-positive Reed-Sternberg cells in
pretreatment biopsy specimens of patients with Hodgkin disease predict favorable clinical
outcome. Blood, v. 100, n. 1, p. 36-42, Jul 1 2002.
EICH, H. T. et al. Intensified chemotherapy and dose-reduced involved-field radiotherapy in
patients with early unfavorable Hodgkin's lymphoma: final analysis of the German Hodgkin
Study Group HD11 trial. J Clin Oncol, v. 28, n. 27, p. 4199-206, Sep 20 2010.
ELENITOBA-JOHNSON, K. S. et al. P53 expression in Reed-Sternberg cells does not
correlate with gene mutations in Hodgkin's disease. Am J Clin Pathol, v. 106, n. 6, p. 728-
38, Dec 1996.
ELLSWORTH, R. E. et al. Differential gene expression in primary breast tumors associated
with lymph node metastasis. Int J Breast Cancer, v. 2011, p. 142763, 2011.
FARRELL, K.; JARRETT, R. F. The molecular pathogenesis of Hodgkin lymphoma.
Histopathology, v. 58, n. 1, p. 15-25, Jan 2011.

113
FERREIRA, J. M. et al. Lymphoma subtype incidence rates in children and adolescents: first
report from Brazil. Cancer Epidemiol, v. 36, n. 4, p. e221-6, Aug 2012.
FIELDS, B. et al. Fields virology. 4th ed. Philadelphia: 2001.
FISHER, R. A. The arrangement of field experiments. J Min Agric G Br, v. 33, p. 503-13,
1926.
FLEIGE, S.; PFAFFL, M. W. RNA integrity and the effect on the real-time qRT-PCR
performance. Mol Aspects Med, v. 27, n. 2-3, p. 126-39, Apr-Jun 2006.
GANDHI, M. K.; TELLAM, J. T.; KHANNA, R. Epstein-Barr virus-associated Hodgkin's
lymphoma. Br J Haematol, v. 125, n. 3, p. 267-81, May 2004.
GARCIA, J. F. et al. Hodgkin and Reed-Sternberg cells harbor alterations in the major tumor
suppressor pathways and cell-cycle checkpoints: analyses using tissue microarrays. Blood, v.
101, n. 2, p. 681-9, Jan 15 2003.
GARCIA, J. F. et al. Nucleolar p14(ARF) overexpression in Reed-Sternberg cells in
Hodgkin's lymphoma: absence of p14(ARF)/Hdm2 complexes is associated with expression
of alternatively spliced Hdm2 transcripts. Am J Pathol, v. 160, n. 2, p. 569-78, Feb 2002.
GATAULT, S. et al. Involvement of eosinophils in the anti-tumor response. Cancer
Immunol Immunother, v. 61, n. 9, p. 1527-34, Sep 2012.
GODFREY, T. E. et al. Quantitative mRNA expression analysis from formalin-fixed,
paraffin-embedded tissues using 5' nuclease quantitative reverse transcription-polymerase
chain reaction. J Mol Diagn, v. 2, n. 2, p. 84-91, May 2000.
GUESCINI, M. et al. A new real-time PCR method to overcome significant quantitative
inaccuracy due to slight amplification inhibition. BMC Bioinformatics, v. 9, p. 326, 2008.
GUIDUCCI, C. et al. Redirecting in vivo elicited tumor infiltrating macrophages and
dendritic cells towards tumor rejection. Cancer Res, v. 65, n. 8, p. 3437-46, Apr 15 2005.

114
HANAHAN, D.; WEINBERG, R. A. The hallmarks of cancer. Cell, v. 100, n. 1, p. 57-70,
Jan 7 2000.
______. Hallmarks of cancer: the next generation. Cell, v. 144, n. 5, p. 646-74, Mar 4 2011.
HARRIS, J. A. et al. CD163 versus CD68 in tumor associated macrophages of classical
Hodgkin lymphoma. Diagn Pathol, v. 7, p. 12, 2012.
HARTMANN, S. et al. Spindle-shaped CD163+ rosetting macrophages replace CD4+ T-cells
in HIV-related classical Hodgkin lymphoma. Mod Pathol, Jan 11 2013.
HASENCLEVER, D.; DIEHL, V. A prognostic score for advanced Hodgkin's disease.
International Prognostic Factors Project on Advanced Hodgkin's Disease. N Engl J Med, v.
339, n. 21, p. 1506-14, Nov 19 1998.
HASSAN, R. et al. Epstein-Barr virus (EBV) detection and typing by PCR: a contribution to
diagnostic screening of EBV-positive Burkitt's lymphoma. Diagn Pathol, v. 1, p. 17, 2006.
HERTEL, C. B. et al. Loss of B cell identity correlates with loss of B cell-specific
transcription factors in Hodgkin/Reed-Sternberg cells of classical Hodgkin lymphoma.
Oncogene, v. 21, n. 32, p. 4908-20, Jul 25 2002.
HIGUCHI, R. et al. Simultaneous amplification and detection of specific DNA sequences.
Biotechnology (N Y), v. 10, n. 4, p. 413-7, Apr 1992.
HINZ, M. et al. Nuclear factor kappaB-dependent gene expression profiling of Hodgkin's
disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and
activator of transcription 5a activity. J Exp Med, v. 196, n. 5, p. 605-17, Sep 2 2002.
HJALGRIM, H. et al. Risk of Hodgkin's disease and other cancers after infectious
mononucleosis. J Natl Cancer Inst, v. 92, n. 18, p. 1522-8, Sep 20 2000.
HODGKIN. On some Morbid Appearances of the Absorbent Glands and Spleen. Med Chir
Trans, v. 17, p. 68-114, 1832.

115
HSI, E. D. Biologic features of Hodgkin lymphoma and the development of biologic
prognostic factors in Hodgkin lymphoma: tumor and microenvironment. Leuk Lymphoma,
v. 49, n. 9, p. 1668-80, Sep 2008.
IARC WORKING GROUP ON THE EVALUATION OF CARCINOGENIC RISKS TO
HUMANS, I. A. F. R. O. C. Epstein-Barr virus and Kaposi's sarcoma herpesvirus/human
herpesvirus 8. Lyon, France: Distributed by IARC Press and by the World Health
Organization, 1997.
INSTITUTO NACIONAL DE CÂNCER JOSÉ ALENCAR GOMES DA SILVA, C. G. D.
A. E., COORDENAÇÃO DE PREVENÇÃO E VIGILÂNCIA Estimativa 2012: Incidencia
de Câncer no Brasil. Rio de Janeiro, Brasil: 2011. 118
JARRETT, R. F. et al. Impact of tumor Epstein-Barr virus status on presenting features and
outcome in age-defined subgroups of patients with classic Hodgkin lymphoma: a population-
based study. Blood, v. 106, n. 7, p. 2444-51, Oct 1 2005.
KAINZ, P. The PCR plateau phase - towards an understanding of its limitations. Biochim
Biophys Acta, v. 1494, n. 1-2, p. 23-7, Nov 15 2000.
KAMPER, P. et al. Tumor-infiltrating macrophages correlate with adverse prognosis and
Epstein-Barr virus status in classical Hodgkin's lymphoma. Haematologica, v. 96, n. 2, p.
269-76, Feb 2011.
KANZLER, H. et al. Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the
outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp
Med, v. 184, n. 4, p. 1495-505, Oct 1 1996.
KAPATAI, G.; MURRAY, P. Contribution of the Epstein Barr virus to the molecular
pathogenesis of Hodgkin lymphoma. J Clin Pathol, v. 60, n. 12, p. 1342-9, Dec 2007.
KEEGAN, T. H. et al. Epstein-Barr virus as a marker of survival after Hodgkin's lymphoma:
a population-based study. J Clin Oncol, v. 23, n. 30, p. 7604-13, Oct 20 2005.

116
KELLEY, T. W. et al. The ratio of FOXP3+ regulatory T cells to granzyme B+ cytotoxic
T/NK cells predicts prognosis in classical Hodgkin lymphoma and is independent of bcl-2 and
MAL expression. Am J Clin Pathol, v. 128, n. 6, p. 958-65, Dec 2007.
KIS, L. L. et al. IL-21 imposes a type II EBV gene expression on type III and type I B cells
by the repression of C- and activation of LMP-1-promoter. Proc Natl Acad Sci U S A, v.
107, n. 2, p. 872-7, Jan 12 2010.
KITCHEN, R. R.; KUBISTA, M.; TICHOPAD, A. Statistical aspects of quantitative real-time
PCR experiment design. Methods, v. 50, n. 4, p. 231-6, Apr 2010.
KLUMB, C. E. et al. Long-term outcome of children and adolescents treated with the COPP-
ABV hybrid protocol for Hodgkin's disease: balance of risks. Clin Oncol (R Coll Radiol), v.
19, n. 8, p. 628-9, Oct 2007.
KOCH, I. et al. Real-time quantitative RT-PCR shows variable, assay-dependent sensitivity
to formalin fixation: implications for direct comparison of transcript levels in paraffin-
embedded tissues. Diagn Mol Pathol, v. 15, n. 3, p. 149-56, Sep 2006.
KUBISTA, M. et al. The real-time polymerase chain reaction. Mol Aspects Med, v. 27, n. 2-
3, p. 95-125, Apr-Jun 2006.
KUPPERS, R. Evidence that Hodgkin and Reed-Sternberg cells in Hodgkin disease do not
represent cell fusions. Blood, v. 97, n. 3, p. 818-821, 2001.
______. The biology of Hodgkin's lymphoma. Nat Rev Cancer, v. 9, n. 1, p. 15-27, Jan
2009a.
______. Molecular biology of Hodgkin lymphoma. Hematology Am Soc Hematol Educ
Program, p. 491-6, 2009b.
KUPPERS, R.; ENGERT, A.; HANSMANN, M. L. Hodgkin lymphoma. J Clin Invest, v.
122, n. 10, p. 3439-47, Oct 1 2012.

117
KUPPERS, R. et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from
histological sections show clonal immunoglobulin gene rearrangements and appear to be
derived from B cells at various stages of development. Proc Natl Acad Sci U S A, v. 91, n.
23, p. 10962-6, Nov 8 1994.
LENEHAN, P. F. et al. Generation and external validation of a tumor-derived 5-gene
prognostic signature for recurrence of lymph node-negative, invasive colorectal carcinoma.
Cancer, v. 118, n. 21, p. 5234-44, Nov 1 2012.
LI, J. et al. Improved RNA quality and TaqMan Pre-amplification method (PreAmp) to
enhance expression analysis from formalin fixed paraffin embedded (FFPE) materials. BMC
Biotechnol, v. 8, p. 10, 2008.
LIVAK, K. J.; SCHMITTGEN, T. D. Analysis of relative gene expression data using real-
time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, v. 25, n. 4, p. 402-8,
Dec 2001.
LUKES, R. J.; BUTLER, J. J. The pathology and nomenclature of Hodgkin's disease. Cancer
Res, v. 26, n. 6, p. 1063-83, Jun 1966.
LUU-THE, V. et al. Improved real-time RT-PCR method for high-throughput measurements
using second derivative calculation and double correction. Biotechniques, v. 38, n. 2, p. 287-
93, Feb 2005.
MACLENNAN, K. A. et al. Diagnosis and grading of nodular sclerosing Hodgkin's disease: a
study of 2190 patients. Int Rev Exp Pathol, v. 33, p. 27-51, 1992.
MAGGIO, E. et al. Chemokines, cytokines and their receptors in Hodgkin's lymphoma cell
lines and tissues. Ann Oncol, v. 13 Suppl 1, p. 52-6, 2002.
MALUMBRES, R. et al. Paraffin-based 6-gene model predicts outcome in diffuse large B-
cell lymphoma patients treated with R-CHOP. Blood, v. 111, n. 12, p. 5509-14, Jun 15 2008.
MANCAO, C. et al. Rescue of "crippled" germinal center B cells from apoptosis by Epstein-
Barr virus. Blood, v. 106, n. 13, p. 4339-44, Dec 15 2005.

118
MANTOVANI, A. et al. Macrophage plasticity and polarization in tissue repair and
remodelling. J Pathol, v. 229, n. 2, p. 176-85, Jan 2013.
MANTOVANI, A.; LOCATI, M. Orchestration of macrophage polarization. Blood, v. 114, n.
15, p. 3135-6, Oct 8 2009.
MANTOVANI, A. et al. Macrophage polarization: tumor-associated macrophages as a
paradigm for polarized M2 mononuclear phagocytes. Trends Immunol, v. 23, n. 11, p. 549-
55, Nov 2002.
MARAFIOTI, T. et al. Hodgkin and reed-sternberg cells represent an expansion of a single
clone originating from a germinal center B-cell with functional immunoglobulin gene
rearrangements but defective immunoglobulin transcription. Blood, v. 95, n. 4, p. 1443-50,
Feb 15 2000.
MASSINI, G.; SIEMER, D.; HOHAUS, S. EBV in Hodgkin Lymphoma. Mediterr J
Hematol Infect Dis, v. 1, n. 2, p. e2009013, 2009.
MASUDA, N. et al. Analysis of chemical modification of RNA from formalin-fixed samples
and optimization of molecular biology applications for such samples. Nucleic Acids Res, v.
27, n. 22, p. 4436-43, Nov 15 1999.
MCDONALD, J. H. Handbook of Biological Statistics. Baltimore, Maryland: Sparky
House Publishing, 2009.
MEYER, R. M. et al. ABVD alone versus radiation-based therapy in limited-stage Hodgkin's
lymphoma. N Engl J Med, v. 366, n. 5, p. 399-408, Feb 2 2012.
MONTALBAN, C. et al. Influence of biologic markers on the outcome of Hodgkin's
lymphoma: a study by the Spanish Hodgkin's Lymphoma Study Group. J Clin Oncol, v. 22,
n. 9, p. 1664-73, May 1 2004.
MONTESINOS-RONGEN, M. et al. Mutation of the p53 gene is not a typical feature of
Hodgkin and Reed-Sternberg cells in Hodgkin's disease. Blood, v. 94, n. 5, p. 1755-60, Sep 1
1999.

119
MORAIS, A. et al. Number of involved anatomic areas as a risk predictor in pediatric
Hodgkin's lymphoma: a retrospective study. J Pediatr (Rio J), v. 85, n. 3, p. 236-42, May-
Jun 2009.
MORENTE, M. M. et al. Adverse clinical outcome in Hodgkin's disease is associated with
loss of retinoblastoma protein expression, high Ki67 proliferation index, and absence of
Epstein-Barr virus-latent membrane protein 1 expression. Blood, v. 90, n. 6, p. 2429-36, Sep
15 1997.
MURDOCH, C. et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat
Rev Cancer, v. 8, n. 8, p. 618-31, Aug 2008.
NOUTSIAS, M. et al. Preamplification techniques for real-time RT-PCR analyses of
endomyocardial biopsies. BMC Mol Biol, v. 9, p. 3, 2008.
NUTT, S. L.; TARLINTON, D. M. Germinal center B and follicular helper T cells: siblings,
cousins or just good friends? Nat Immunol, v. 12, n. 6, p. 472-7, Jun 2011.
OBERG, H. H. et al. Regulation of T cell activation by TLR ligands. Eur J Cell Biol, v. 90,
n. 6-7, p. 582-92, Jun-Jul 2011.
OKELLO, J. B. et al. Comparison of methods in the recovery of nucleic acids from archival
formalin-fixed paraffin-embedded autopsy tissues. Anal Biochem, v. 400, n. 1, p. 110-7, May
1 2010.
PARK, I. S. et al. Syncytial variant of nodular sclerosis Hodgkin's lymphoma assessed by fine
needle aspiration cytology. Cytopathology, v. 19, n. 6, p. 394-7, Dec 2008.
PETTERSEN, J. S. et al. Tumor-associated macrophages in the cutaneous SCC
microenvironment are heterogeneously activated. J Invest Dermatol, v. 131, n. 6, p. 1322-30,
Jun 2011.
PFAFFL, M. W. Livestock Transcriptomics: Quantitative mRNA Analytics in Molecular
Endocrinology and Physiology. 2003. 41 f. Habilitation - Faculty Center of Life and Food
Sciences, Technische Universität München, Freising, Germany

120
PFAFFL, M. W. Quantification strategies in real time PCR In: BUSTIN, S. A. (Ed.). A-Z of
quantitative PCR. La Jolla, CA: International University Line, 2004.
PFAFFL, M. W.; HAGELEIT, M. Validities of mRNA quantification using recombinant
RNA and recombinant DNA external calibration curves in real-time RT-PCR. Biotechnology
Letters, v. 23, p. 275-282, 2001.
PILERI, S. A. et al. Hodgkin's lymphoma: the pathologist's viewpoint. J Clin Pathol, v. 55,
n. 3, p. 162-76, Mar 2002.
POPPEMA, S. Immunobiology and pathophysiology of Hodgkin lymphomas. Hematology
Am Soc Hematol Educ Program, p. 231-8, 2005.
PORTER, A. G. Flipping the safety catch of procaspase-3. Nat Chem Biol, v. 2, n. 10, p.
509-10, Oct 2006.
RADULOVIC, S.; JELIC, S. General principles of chemotherapy. Acta Chir Iugosl, v. 44-
45, n. 1-1, p. 7-10, 1997.
REED, D. On the pathological changes in Hodgkin’s disease with special reference to its
relation to tuberculosis. John Hopkins Hosp. Rep. , v. 10, p. 133-193, 1902.
RENAULT, I. Z. et al. The significance of major and stable molecular responses in chronic
myeloid leukemia in the tyrosine kinase inhibitor era. Rev Bras Hematol Hemoter, v. 33, n.
6, p. 455-60, 2011.
ROBERTS, L. et al. Identification of methods for use of formalin-fixed, paraffin-embedded
tissue samples in RNA expression profiling. Genomics, v. 94, n. 5, p. 341-8, Nov 2009.
RUIJTER, J. M. et al. Evaluation of qPCR curve analysis methods for reliable biomarker
discovery: Bias, resolution, precision, and implications. Methods, v. 59, n. 1, p. 32-46, Jan
2013.
RUIJTER, J. M. et al. Amplification efficiency: linking baseline and bias in the analysis of
quantitative PCR data. Nucleic Acids Res, v. 37, n. 6, p. e45, Apr 2009.

121
SAHA, B. et al. Gene modulation and immunoregulatory roles of interferon gamma.
Cytokine, v. 50, n. 1, p. 1-14, Apr 2010.
SANCHEZ-AGUILERA, A. et al. Hodgkin's lymphoma cells express alternatively spliced
forms of HDM2 with multiple effects on cell cycle control. Oncogene, v. 25, n. 18, p. 2565-
74, Apr 27 2006.
SANCHEZ-AGUILERA, A. et al. Tumor microenvironment and mitotic checkpoint are key
factors in the outcome of classic Hodgkin lymphoma. Blood, v. 108, n. 2, p. 662-8, Jul 15
2006.
SANCHEZ-ESPIRIDION, B. et al. A molecular risk score based on 4 functional pathways for
advanced classical Hodgkin lymphoma. Blood, v. 116, n. 8, p. e12-7, Aug 26 2010.
SANCHEZ-ESPIRIDION, B. et al. A TaqMan low-density array to predict outcome in
advanced Hodgkin's lymphoma using paraffin-embedded samples. Clin Cancer Res, v. 15, n.
4, p. 1367-75, Feb 15 2009.
SANT, A. J.; MCMICHAEL, A. Revealing the role of CD4(+) T cells in viral immunity. J
Exp Med, v. 209, n. 8, p. 1391-5, Jul 30 2012.
SATOH, T. et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host
responses against helminth infection. Nat Immunol, v. 11, n. 10, p. 936-44, Oct 2010.
SCHMITTGEN, T. D.; LIVAK, K. J. Analyzing real-time PCR data by the comparative C(T)
method. Nat Protoc, v. 3, n. 6, p. 1101-8, 2008.
SCHMITZ, R. et al. Pathogenesis of classical and lymphocyte-predominant Hodgkin
lymphoma. Annu Rev Pathol, v. 4, p. 151-74, 2009.
SCHNELL, S.; MENDOZA, C. Theoretical description of the polymerase chain reaction. J
Theor Biol, v. 188, n. 3, p. 313-8, Oct 7 1997.
SCHWARTZ, C. L. Prognostic factors in pediatric Hodgkin disease. Curr Oncol Rep, v. 5,
n. 6, p. 498-504, Nov 2003.

122
SCHWERING, I. et al. Loss of the B-lineage-specific gene expression program in Hodgkin
and Reed-Sternberg cells of Hodgkin lymphoma. Blood, v. 101, n. 4, p. 1505-12, Feb 15
2003.
SCHWERK, C.; SCHULZE-OSTHOFF, K. Non-apoptotic functions of caspases in cellular
proliferation and differentiation. Biochem Pharmacol, v. 66, n. 8, p. 1453-1458, 2003.
SCOTT, D. W. et al. Gene Expression-Based Model Using Formalin-Fixed Paraffin-
Embedded Biopsies Predicts Overall Survival in Advanced-Stage Classical Hodgkin
Lymphoma. J Clin Oncol, Nov 26 2012.
SEGGES, P. Investigação de proteínas nucleolares potencialmente envolvidas na
desregulação do ciclo celular no Linfoma de Hodgkin Clássico. 2012. 81 f. (Qualificação
de doutorado) - Laboratorio de Oncovirologia-CEMO, Pós-Graduação em Oncologia, INCA
SHANKAR, A.; DAW, S. Nodular lymphocyte predominant Hodgkin lymphoma in children
and adolescents--a comprehensive review of biology, clinical course and treatment options.
Br J Haematol, v. 159, n. 3, p. 288-98, Nov 2012.
SMITH, M. R.; JIN, F.; JOSHI, I. Bortezomib sensitizes non-Hodgkin's lymphoma cells to
apoptosis induced by antibodies to tumor necrosis factor related apoptosis-inducing ligand
(TRAIL) receptors TRAIL-R1 and TRAIL-R2. Clin Cancer Res, v. 13, n. 18 Pt 2, p. 5528s-
5534s, Sep 15 2007.
SMOLEWSKI, P. et al. Expression of proliferating cell nuclear antigen (PCNA) and p53, bcl-
2 or C-erb B-2 proteins on Reed-Sternberg cells: prognostic significance in Hodgkin's disease.
Neoplasma, v. 45, n. 3, p. 140-7, 1998.
SMOLEWSKI, P. et al. Prognostic factors in Hodgkin's disease: multivariate analysis of 327
patients from a single institution. Clin Cancer Res, v. 6, n. 3, p. 1150-60, Mar 2000.
SORDET, O. et al. Specific involvement of caspases in the differentiation of monocytes into
macrophages. Blood, v. 100, n. 13, p. 4446-53, Dec 15 2002.

123
SPECHT, L. et al. Radiotherapy versus combined modality in early stages. Ann Oncol, v. 3
Suppl 4, p. 77-81, Sep 1992.
SPECTOR, N. et al. p53 Expression as a prognostic indicator in Hodgkin's lymphoma. J Clin
Oncol, v. 23, n. 13, p. 3158-9; author reply 3159-60, May 1 2005.
SRINIVASAN, M.; SEDMAK, D.; JEWELL, S. Effect of fixatives and tissue processing on
the content and integrity of nucleic acids. Am J Pathol, v. 161, n. 6, p. 1961-71, Dec 2002.
STAHLBERG, A. et al. Properties of the reverse transcription reaction in mRNA
quantification. Clin Chem, v. 50, n. 3, p. 509-15, Mar 2004.
STEFANOFF, C. G. et al. Laboratory strategies for efficient handling of paraffin-embedded
tissues for molecular detection of clonality in non-hodgkin lymphomas. Diagn Mol Pathol, v.
12, n. 2, p. 79-87, Jun 2003.
STEIDL, C.; CONNORS, J. M.; GASCOYNE, R. D. Molecular pathogenesis of Hodgkin's
lymphoma: increasing evidence of the importance of the microenvironment. J Clin Oncol, v.
29, n. 14, p. 1812-26, May 10 2011.
STEIDL, C. et al. Gene expression profiling of microdissected Hodgkin Reed-Sternberg cells
correlates with treatment outcome in classical Hodgkin lymphoma. Blood, v. 120, n. 17, p.
3530-40, Oct 25 2012.
STEIDL, C.; FARINHA, P.; GASCOYNE, R. D. Macrophages predict treatment outcome in
Hodgkin's lymphoma. Haematologica, v. 96, n. 2, p. 186-9, Feb 2011.
STEIDL, C. et al. Tumor-associated macrophages and survival in classic Hodgkin's
lymphoma. N Engl J Med, v. 362, n. 10, p. 875-85, Mar 11 2010.
STERNBERG, C. Übe r eine eigenartige unter dem Bilde der Pseudoleukämie verlaufende
Tuberkolose des lymphatischen Apparates. Z. Heilkunde, v. 19, p. 21-90, 1898.
SUN, J. C.; LANIER, L. L. NK cell development, homeostasis and function: parallels with
CD8(+) T cells. Nat Rev Immunol, v. 11, n. 10, p. 645-57, Oct 2011.

124
SUP, S. J. et al. Expression of bcl-2 in classical Hodgkin's lymphoma: an independent
predictor of poor outcome. J Clin Oncol, v. 23, n. 16, p. 3773-9, Jun 1 2005.
SUSLOV, O.; STEINDLER, D. A. PCR inhibition by reverse transcriptase leads to an
overestimation of amplification efficiency. Nucleic Acids Res, v. 33, n. 20, p. e181, 2005.
SWERDLOW, S. H. et al. WHO classification of tumours of haematopoietic and
lymphoid tissues. 4th edn. Lyon, France: International Agency for Research on Cancer,
2008.
TAN, K. L. et al. Tumor-associated macrophages predict inferior outcomes in classic
Hodgkin lymphoma: a correlative study from the E2496 Intergroup trial. Blood, v. 120, n. 16,
p. 3280-7, Oct 18 2012.
TANG, W. et al. Quality assurance of RNA expression profiling in clinical laboratories. J
Mol Diagn, v. 14, n. 1, p. 1-11, Jan 2012.
THOMPSON, L. D. et al. HIV-associated Hodgkin lymphoma: a clinicopathologic and
immunophenotypic study of 45 cases. Am J Clin Pathol, v. 121, n. 5, p. 727-38, May 2004.
THORLEY-LAWSON, D. A. Epstein-Barr virus: exploiting the immune system. Nat Rev
Immunol, v. 1, n. 1, p. 75-82, Oct 2001.
TIACCI, E. et al. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the
molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood, v. 120, n. 23, p.
4609-20, Nov 29 2012.
TICHOPAD, A. Quantitative real-time RT-PCR based transcriptomics: Improvement of
evaluation methods. 2004. 49 f. Thesis (Doctorate) - Lehrstuhl für Physiologie, Fakultät
Wissenschaftszentrum Weihenstephan, Technische Universität München, Munich, German.
TICHOPAD, A. et al. Quality control for quantitative PCR based on amplification
compatibility test. Methods, v. 50, n. 4, p. 308-12, Apr 2010.

125
TICHOPAD, A.; DIDIER, A.; PFAFFL, M. W. Inhibition of real-time RT-PCR quantification
due to tissue-specific contaminants. Mol Cell Probes, v. 18, n. 1, p. 45-50, Feb 2004.
TICHOPAD, A. et al. Standardized determination of real-time PCR efficiency from a single
reaction set-up. Nucleic Acids Res, v. 31, n. 20, p. e122, Oct 15 2003.
TICHOPAD, A. et al. Design and optimization of reverse-transcription quantitative PCR
experiments. Clin Chem, v. 55, n. 10, p. 1816-23, Oct 2009.
TZANKOV, A. et al. Rare expression of T-cell markers in classical Hodgkin's lymphoma.
Mod Pathol, v. 18, n. 12, p. 1542-9, Dec 2005.
TZANKOV, A.; MATTER, M. S.; DIRNHOFER, S. Refined prognostic role of CD68-
positive tumor macrophages in the context of the cellular micromilieu of classical Hodgkin
lymphoma. Pathobiology, v. 77, n. 6, p. 301-8, 2010.
TZANKOV, A. et al. Correlation of high numbers of intratumoral FOXP3+ regulatory T cells
with improved survival in germinal center-like diffuse large B-cell lymphoma, follicular
lymphoma and classical Hodgkin's lymphoma. Haematologica, v. 93, n. 2, p. 193-200, Feb
2008.
VAN 'T VEER, L. J. et al. Gene expression profiling predicts clinical outcome of breast
cancer. Nature, v. 415, n. 6871, p. 530-6, Jan 31 2002.
VAN DE VIJVER, M. J. et al. A gene-expression signature as a predictor of survival in breast
cancer. N Engl J Med, v. 347, n. 25, p. 1999-2009, Dec 19 2002.
VAN SPRONSEN, D. J. et al. Disappearance of prognostic significance of histopathological
grading of nodular sclerosing Hodgkin's disease for unselected patients, 1972-92. Br J
Haematol, v. 96, n. 2, p. 322-7, Feb 1997.
VANDESOMPELE, J. et al. Accurate normalization of real-time quantitative RT-PCR data
by geometric averaging of multiple internal control genes. Genome Biol, v. 3, n. 7, p.
RESEARCH0034, Jun 18 2002.

126
VANDESOMPELE, J.; KUBISTA, M.; PFAFFL, M. W. Rerence gene validation Software
for improve Normalization. In: LOGAN, J., et al (Ed.). Real Time PCR: current technology
and applications Caister Academic Press, 2009.
VASSILAKOPOULOS, T. P. et al. Development and validation of a clinical prediction rule
for bone marrow involvement in patients with Hodgkin lymphoma. Blood, v. 105, n. 5, p.
1875-80, Mar 1 2005.
VESELY, M. D. et al. Natural innate and adaptive immunity to cancer. Annu Rev Immunol,
v. 29, p. 235-71, 2011.
VIKSTROM, I.; TARLINTON, D. M. B cell memory and the role of apoptosis in its
formation. Mol Immunol, v. 48, n. 11, p. 1301-6, Jun 2011.
WHITE, J. A. et al. Guidelines for human gene nomenclature (1997). HUGO Nomenclature
Committee. Genomics, v. 45, n. 2, p. 468-71, Oct 15 1997.
XIE, Y. et al. Robust gene expression signature from formalin-fixed paraffin-embedded
samples predicts prognosis of non-small-cell lung cancer patients. Clin Cancer Res, v. 17, n.
17, p. 5705-14, Sep 1 2011.
YOUNG, L. S.; RICKINSON, A. B. Epstein-Barr virus: 40 years on. Nat Rev Cancer, v. 4,
n. 10, p. 757-68, Oct 2004.
ZHAO, S.; FERNALD, R. D. Comprehensive algorithm for quantitative real-time polymerase
chain reaction. J Comput Biol, v. 12, n. 8, p. 1047-64, Oct 2005.
ZHU, J.; PAUL, W. E. CD4 T cells: fates, functions, and faults. Blood, v. 112, n. 5, p. 1557-
69, Sep 1 2008.
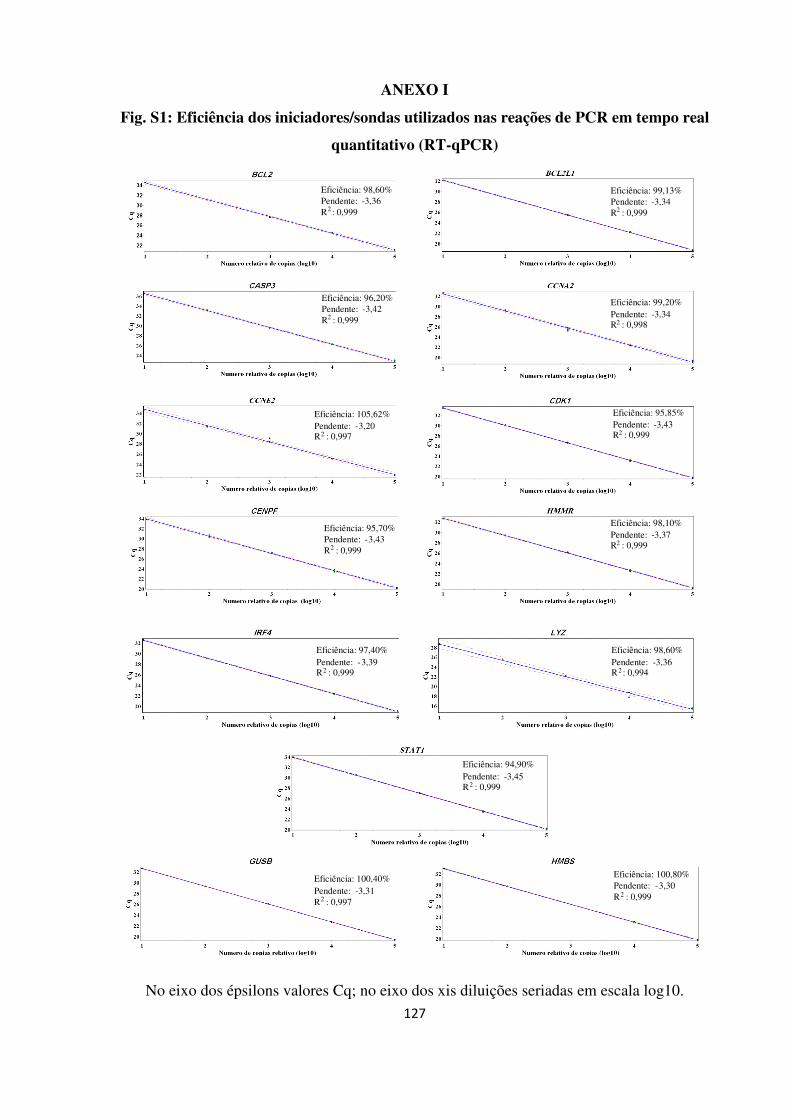
127
ANEXO I
Fig. S1: Eficiência dos iniciadores/sondas utilizados nas reações de PCR em tempo real
quantitativo (RT-qPCR)
No eixo dos épsilons valores Cq; no eixo dos xis diluições seriadas em escala log10.
Eficiência: 100,40%Pendente: - 3,31R2 : 0,997
Eficiência: 100,80% Pendente: - 3,30R 2 : 0,999
Eficiência: 94,90%Pendente: - 3,45 R2 : 0,999
Eficiência: 98,60%Pendente: - 3,36R2 : 0,994
Eficiência: 97,40%Pendente: - 3,39 R2 : 0,999
Eficiência: 98,60%Pendente: - 3,36R2 : 0,999
Eficiência: 96,20% Pendente: -3,42R 2 : 0,999
Eficiência: 99,20%Pendente: -3,34 R2 : 0,998
Eficiência: 99,13%Pendente: -3,34 R2 : 0,999
Eficiência: 105,62%Pendente: -3,20 R2 : 0,997
Eficiência: 95,70%Pendente: -3,43R2 : 0,999
Eficiência: 95,85%Pendente: - 3,43 R2 : 0,999
Eficiência: 98,10%Pendente: -3,37 R2 : 0,999

128
ANEXO II
INFORMAÇÃO MÍNIMA PARA PUBLICAÇÃO EM EXPERIMENTOS DE
QUANTIFICAÇÃO EM PCR EM TEMPO REAL (MIQE), SUGERIDAS POR
BUSTIN et al. (2009).
1 Desenho experimental
• Grupo experimental: linfonodos de crianças e adolescentes com linfoma de Hodgkin
clássico (LHc) e tonsilas com Hiperplasia Reativa
• Número de pacientes: 93 LHc e 6 LHR
2 Amostras
• Descrição: linfonodos fixados e impregnados em parafina (FFPE)
• Quantidade de amostra processada: 5 cortes de parafina de 3 µm cada um
• Procedimento do processamento de fixação: formol 10% por 24 horas.
• Tempo e condições de armazenamento das amostras: 6-12 anos a 20-22°C em
ambiente fechado
3 Extração do RNA
• Kits utilizados: 1) Máster PureTM RNA purification kit (MP) (Epicentre, Madison,
WI) com 480 µL de solução de lise celular e de tecido e 60 µL de proteinase K 60
mg/dL a 65°C por 20 horas. 2) RecoverAllTM total nucleic acid isolation kit (RA)
(AmbionTM por Life TechnologiesTM, Carlsbad, CA) com 200 µL de tampão de
digestão e 60 µL de proteinase K (60mg/dL), incubado a 50°C por 15 minutos e
depois 20 horas a 65°C
• Reagentes adicionais usados: proteinase K (Invitrogen por Life TechnologiesTM,
Carlsbad, CA) a 60 mg/dL.
• Tratamento com DNase: 1) Método MP: 200 µL da solução DNase I (5 µL de DNase I
e 200 µL de 1X de tampão de DNase) supridos pelo kit a 37°C por 30 minutos. 2)
Método RA: 60 µL de mix DNase (6 µL tampão de DNAse 10X, 4 µL DNase e 50 µL
água livre de nucleases) supridos pelo kit a temperatura ambiente por 30 minutos.
• Avaliação de contaminação por DNA: não se aplica
• Quantificação do RNA:

129
� Método e aparelho: espectrofotometria no Nanodrop (ND-1000
Spectrophotometer, Wilmington, USA).
� Pureza: λ260/280: 1,80 + 0,14
λ260/230: 1,39 + 0,66
• Integridade do RNA:
� Método e aparelho: análise de RNA pela metodologia microfluídica na plataforma
2100 bioanalyzer (Agilent Technologies)
� RIN:1,2 – 3,6
� Eletroforese: gel de agarose 1,2% em tampão fosfato (Na2HPO4 0,01M) com
coloração de brometo de etídio (0,5 µg/mL).
• Teste de inibição: não realizado. Problema de inibição abordado na análise de qPCR.
• Armazenamento do RNA: -70°C
4 Transcrição reversa
• Condições da reação: 2 µL tampão RT 10X, 0,8 µL dNTPs 25X, 2 µL iniciadores
randômicos 10X, 1 µL de MultiScribe RT 50U/µL, 4,2 µL água livre de nucleases.
• Quantidade de RNA e volume da reação: 0,5 µg de RNA diluído em 10µL de água
para um volume final da reação de 20µL.
• Estratégia de iniciadores: Uso de iniciadores hexaméricos randômicos
• Transcritase reversa e concentração: MultiScribe™ Reverse Transcriptase em 50
U/µL.
• Temperatura e tempo: 25°C por 10 minutos, 37°C por 120 minutos e 85°C por 5
minutos.
• Fabricante e número de catálogo: Applied por Life TechnologiesTM, Carlsbad, CA
com o número 4368814.
• Estocagem do cDNA: -70°C

130
5 Alvos de PCR
Símbolo Gene*
Eficiência (%)
# Acesso sequência
Localiz amplicon
Iniciador exon
ID do ensaio Tam amp
(pb) BCL2
BCL2L1
CCNA2
CCNE2
CDK1
CENPF
CASP3
HMMR
IRF4
LYZ
STAT1
GUSB
HMBS
98,60
99,13
99,20
105,62
95,85
95,70
96,20
98,10
97,40
98,6
94,90
100,4
100,8
NM_000633.2
NM_138578.1
NM_001237.3
NM_057749.2
NM_001170407.1
NM_001786.4
NM_033379.4
NM_016343.3
NM_004346.3
NM_001142556.1
NM_001142557.1
NM_012484.2
NM_012485.2
NM_001195286.1
NM_002460.3
NM_000239.2
NM_007315.3
NM_139266.2
NM_000181.3
NM_000190.3
1081
930
520
705
121
121
121
8155
250
1448
1221
1445
1400
1339
1342
358
1171
1171
1925
186
2-3
2-3
1-2
7-8
1-2
1-2
1-2
14-15
2-3
11-12
8-9
11-12
10-11
8-9
8-9
2-3
9-10
9-10
11-12
1-2
Hs00608023_m1
Hs00236329_m1
Hs00153138_m1
Hs00180319_m1
Hs00176469_m1
Hs00193201_m1
Hs00234385_m1
Hs00234864_m1
Hs00180031_m1
Hs00426231_m1
Hs00234829_m1
Hs99999908_m1
Hs00609297_m1
81
65
110
92
101
99
66
98
88
64
79
81
64
* De acordo com HUGO (WHITE et al., 1997). Localiz amplicon: localização do amplicon segundo o RefSeq; Tam amp: tamanho do amplicon
• Screening de especificidade in silico (BLAST): não se aplica
• Variantes de splicing nos alvos: não se aplica
6 Oligonucleotideos da qPCR
• Sequência do iniciador: Descrito de acordo com a localização na RefSeq
• Sequência da sonda: Descrito de acordo com a localização na RefSeq
• Localização e identidade de alguma modificação: não se aplica
• Fabricante dos oligonucleotideos: Applied por Life TechnologiesTM, Carlsbad, CA
7 Protocolo de qPCR
• Condições completa da reação: volume final da reação de 15 µL: 7,5 µL TaqMan
universal PCR Máster Mix 2X, 0,75 µL Iniciador/Sonda 20X, 2,75 µL água livre de
nucleases e 4 µL cDNA pré-amplificado.

131
• Volume da reação e quantidade de cDNA: volume final de 15 µL com 4 µL cDNA
específico pré-amplificado previamente diluído 1:20 em água livre de nucleases.
• Concentração de iniciadores, sonda, Mg++ e dNTP: padrão no reagente TaqMan
universal PCR Máster Mix (Applied, Life TechnologiesTM)
• Identificação e concentração da polimerase: padrão no reagente TaqMan universal PCR
Máster Mix (Applied, Life TechnologiesTM)
• Aditivos: não foram usados
• Fabricante das placas e número de catálogo: Applied Biosystems, número de catálogo
N8010560
• Parâmetros completos da termociclagem: 50°C por 2 minutos, 95°C por 10 minutos. 40
ciclos de 95°C por 15 segundos e 60°C por 1 minuto.
• Fabricante do instrumento de qPCR: ABI 7000, Applied Biosystems.
8 Validação de qPCR
• Especificidade: gel de agarose 3%
• NTC: Cq ˃40
• Curva standard com slope e intersecção em y: vide Anexo I
• Eficiência da PCR (slope): (-3,30) – (-3,43)
• R2 da curva standard: 0,994 - 0,999
• Rango dinâmico linear: 4-5 log10
• Variação do Cq no limite baixo: DP ≤ 0,5
• Evidencia para o limite de detecção: não determinado
• Multiplex, eficiência e LOD para cada ensaio: não determinado
9 Data analise
• Programa de análise de qPCR (fonte, versão): Genex enterprise (MultiD analyses)
• Método de determinação Cq: 2-∆Cq
• Identificação e disposição dos outliers: teste Grubbs e KOD
• Resultados dos NTCs: Cq˃40
• Justificação do número de genes de referência selecionados: foram selecionados os dois
genes mais estável descritos para LHc reportado por Sanchez-Espiridion et al, Clin
Cancer Res 2009;1367:15(4).
• Descrição do método de normalização: não se aplica

132
• Número e estado (qPCR) de replicatas técnicas: 2 qPCR
• Reprodutibilidade (variação inter-ensaio): BCL2 (1,51%), BCL2L1 (1,96%), CASP3
(0,70%), CCNA2 (1,71%), CCNE2 (1,67%), CDK1 (2,07%), CENPF (2,69%), HMMR
(1,55%), IRF4 (1,43%), LYZ (1,18%), STAT1 (2,15%), GUSB (1,35%) e HMBS
(1,55%).
• Métodos estatísticos para a significância dos resultados: teste Mann-Whitney para 2
amostras independentes e teste Spearman para correlações bivariadas.
• Software (fonte e versão): SPSS 20.0

Carta de aprovação do estudo pelo comitê de ética em pesquisa
133
ANEXO III
Carta de aprovação do estudo pelo comitê de ética em pesquisa
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Carta de aprovação do estudo pelo comitê de ética em pesquisa

134
ANEXO IV
TABELAS SUPLEMENTARES
• Tabela S1: Descrição dos anticorpos utilizados para imunohistoquímica no estudo das
células do microambiente tumoral no linfoma de Hodgkin clássico.
• Tabela S2: Distribuição da variabilidade introduzida em porcentagem em cada etapa
da metodologia utilizada na avaliação de amostras FFPE.
• Tabela S3: Correlações entre a expressão gênica do microambiente do linfoma de
Hodgkin clássico pediátrico, avaliadas por PCR em tempo real quantitativo (qPCR).
• Tabela S4: Caracterização e quantificação das células do microambiente tumoral em
pacientes com linfoma de Hodgkin clássico pediátricos.
• Tabela S5: Correlações entre células infiltrantes no microambiente do linfoma de
Hodgkin clássico pediátrico, avaliadas por marcações de imunohistoquimica
quantitativa.
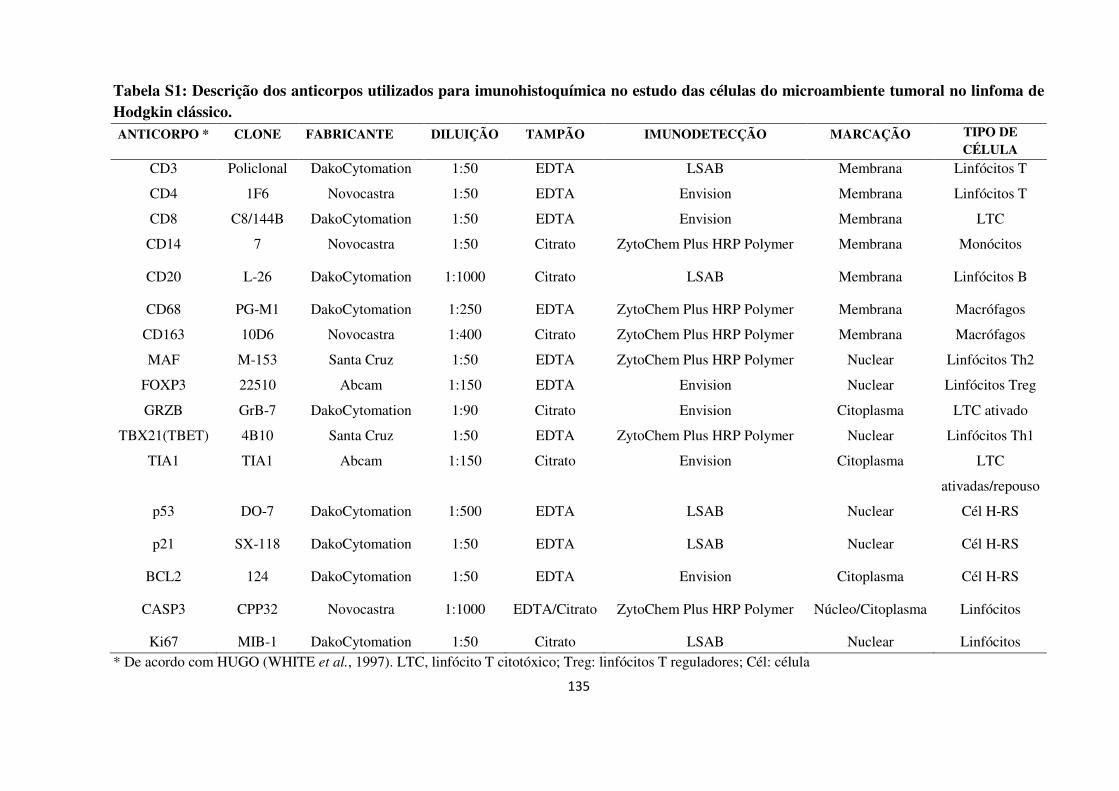
135
Tabela S1: Descrição dos anticorpos utilizados para imunohistoquímica no estudo das células do microambiente tumoral no linfoma de Hodgkin clássico. ANTICORPO * CLONE FABRICANTE DILUIÇÃO TAMPÃO IMUNODETECÇÃO MARCAÇÃO TIPO DE
CÉLULA
CD3 Policlonal DakoCytomation 1:50 EDTA LSAB Membrana Linfócitos T
CD4 1F6 Novocastra 1:50 EDTA Envision Membrana Linfócitos T
CD8 C8/144B DakoCytomation 1:50 EDTA Envision Membrana LTC
CD14 7 Novocastra 1:50 Citrato ZytoChem Plus HRP Polymer Membrana Monócitos
CD20 L-26 DakoCytomation 1:1000 Citrato LSAB Membrana Linfócitos B
CD68 PG-M1 DakoCytomation 1:250 EDTA ZytoChem Plus HRP Polymer Membrana Macrófagos
CD163 10D6 Novocastra 1:400 Citrato ZytoChem Plus HRP Polymer Membrana Macrófagos
MAF M-153 Santa Cruz 1:50 EDTA ZytoChem Plus HRP Polymer Nuclear Linfócitos Th2
FOXP3 22510 Abcam 1:150 EDTA Envision Nuclear Linfócitos Treg
GRZB GrB-7 DakoCytomation 1:90 Citrato Envision Citoplasma LTC ativado
TBX21(TBET) 4B10 Santa Cruz 1:50 EDTA ZytoChem Plus HRP Polymer Nuclear Linfócitos Th1
TIA1 TIA1 Abcam 1:150 Citrato Envision Citoplasma LTC
ativadas/repouso
p53 DO-7 DakoCytomation 1:500 EDTA LSAB Nuclear Cél H-RS
p21 SX-118 DakoCytomation 1:50 EDTA LSAB Nuclear Cél H-RS
BCL2 124 DakoCytomation 1:50 EDTA Envision Citoplasma Cél H-RS
CASP3 CPP32 Novocastra 1:1000 EDTA/Citrato ZytoChem Plus HRP Polymer Núcleo/Citoplasma Linfócitos
Ki67 MIB-1 DakoCytomation 1:50 Citrato LSAB Nuclear Linfócitos * De acordo com HUGO (WHITE et al., 1997). LTC, linfócito T citotóxico; Treg: linfócitos T reguladores; Cél: célula
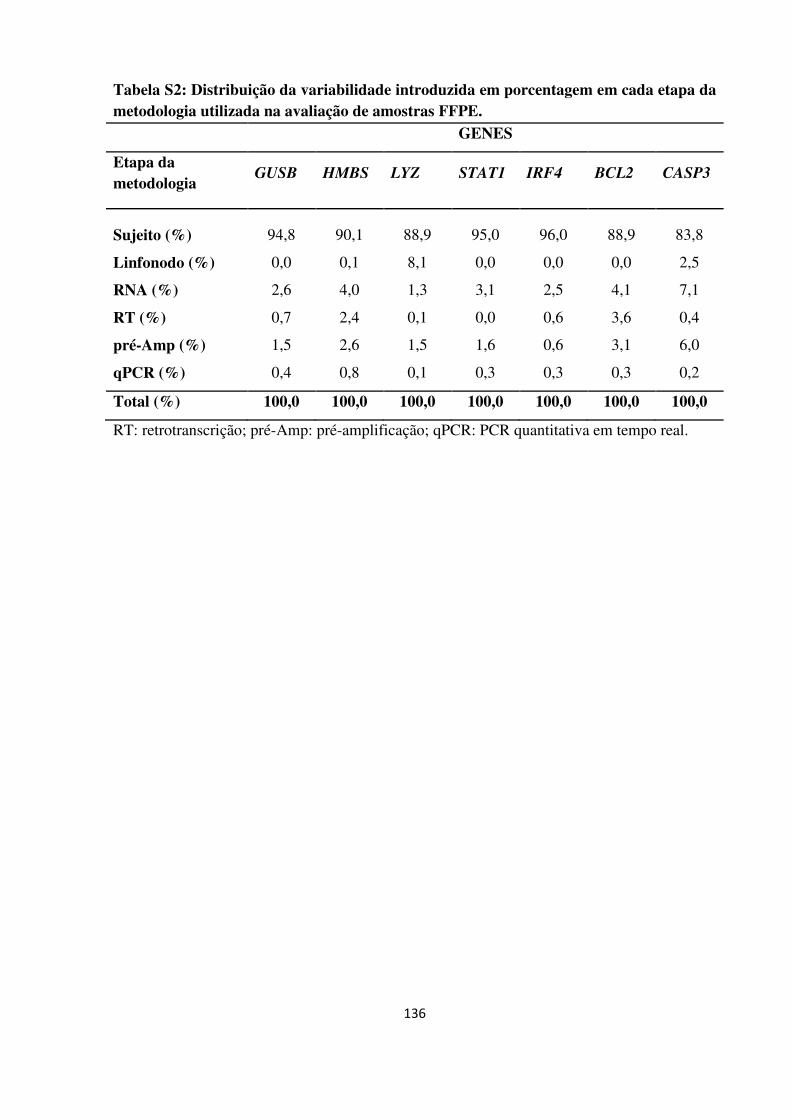
136
Tabela S2: Distribuição da variabilidade introduzida em porcentagem em cada etapa da metodologia utilizada na avaliação de amostras FFPE. GENES
Etapa da metodologia
GUSB HMBS LYZ STAT1 IRF4 BCL2 CASP3
Sujeito (%) 94,8 90,1 88,9 95,0 96,0 88,9 83,8
Linfonodo (%) 0,0 0,1 8,1 0,0 0,0 0,0 2,5
RNA (%) 2,6 4,0 1,3 3,1 2,5 4,1 7,1
RT (%) 0,7 2,4 0,1 0,0 0,6 3,6 0,4
pré-Amp (%) 1,5 2,6 1,5 1,6 0,6 3,1 6,0
qPCR (%) 0,4 0,8 0,1 0,3 0,3 0,3 0,2
Total (%) 100,0 100,0 100,0 100,0 100,0 100,0 100,0
RT: retrotranscrição; pré-Amp: pré-amplificação; qPCR: PCR quantitativa em tempo real.
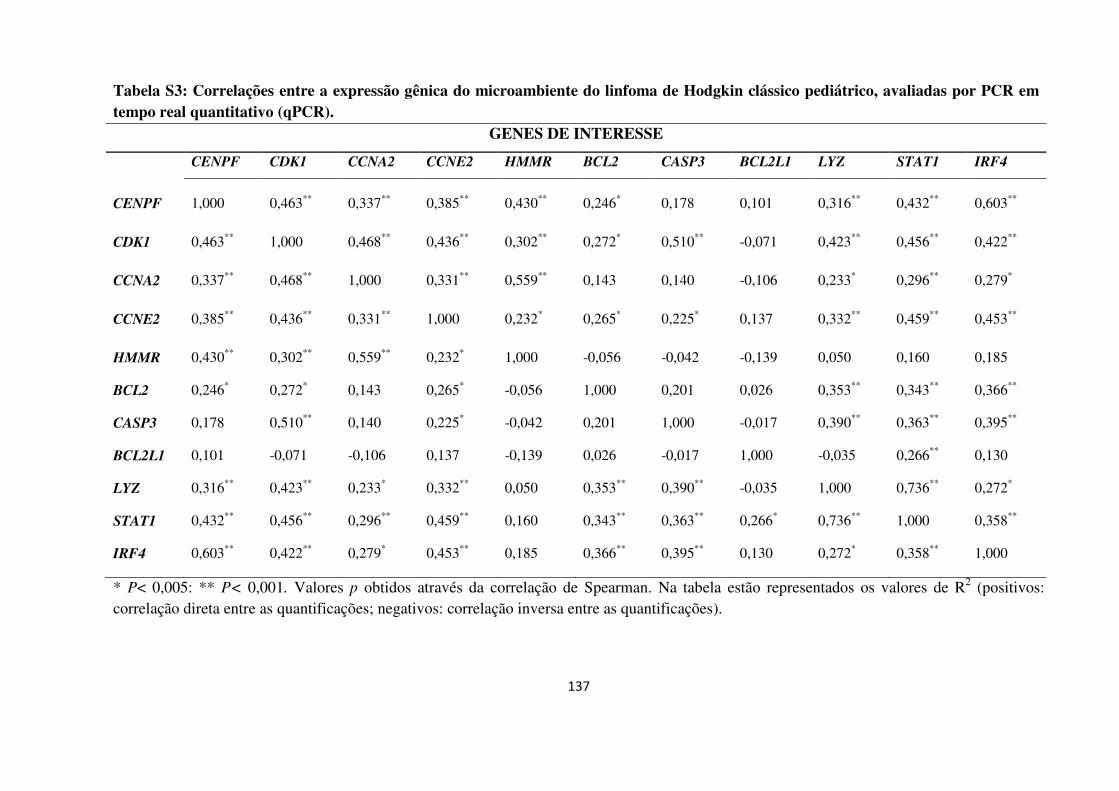
137
Tabela S3: Correlações entre a expressão gênica do microambiente do linfoma de Hodgkin clássico pediátrico, avaliadas por PCR em tempo real quantitativo (qPCR).
GENES DE INTERESSE
CENPF CDK1 CCNA2 CCNE2 HMMR BCL2 CASP3 BCL2L1 LYZ STAT1 IRF4
CENPF 1,000 0,463** 0,337** 0,385** 0,430** 0,246* 0,178 0,101 0,316** 0,432** 0,603**
CDK1 0,463** 1,000 0,468** 0,436** 0,302** 0,272* 0,510** -0,071 0,423** 0,456** 0,422**
CCNA2 0,337** 0,468** 1,000 0,331** 0,559** 0,143 0,140 -0,106 0,233* 0,296** 0,279*
CCNE2 0,385** 0,436** 0,331** 1,000 0,232* 0,265* 0,225* 0,137 0,332** 0,459** 0,453**
HMMR 0,430** 0,302** 0,559** 0,232* 1,000 -0,056 -0,042 -0,139 0,050 0,160 0,185
BCL2 0,246* 0,272* 0,143 0,265* -0,056 1,000 0,201 0,026 0,353** 0,343** 0,366**
CASP3 0,178 0,510** 0,140 0,225* -0,042 0,201 1,000 -0,017 0,390** 0,363** 0,395**
BCL2L1 0,101 -0,071 -0,106 0,137 -0,139 0,026 -0,017 1,000 -0,035 0,266** 0,130
LYZ 0,316** 0,423** 0,233* 0,332** 0,050 0,353** 0,390** -0,035 1,000 0,736** 0,272*
STAT1 0,432** 0,456** 0,296** 0,459** 0,160 0,343** 0,363** 0,266* 0,736** 1,000 0,358**
IRF4 0,603** 0,422** 0,279* 0,453** 0,185 0,366** 0,395** 0,130 0,272* 0,358** 1,000
* P< 0,005: ** P< 0,001. Valores p obtidos através da correlação de Spearman. Na tabela estão representados os valores de R2 (positivos: correlação direta entre as quantificações; negativos: correlação inversa entre as quantificações).
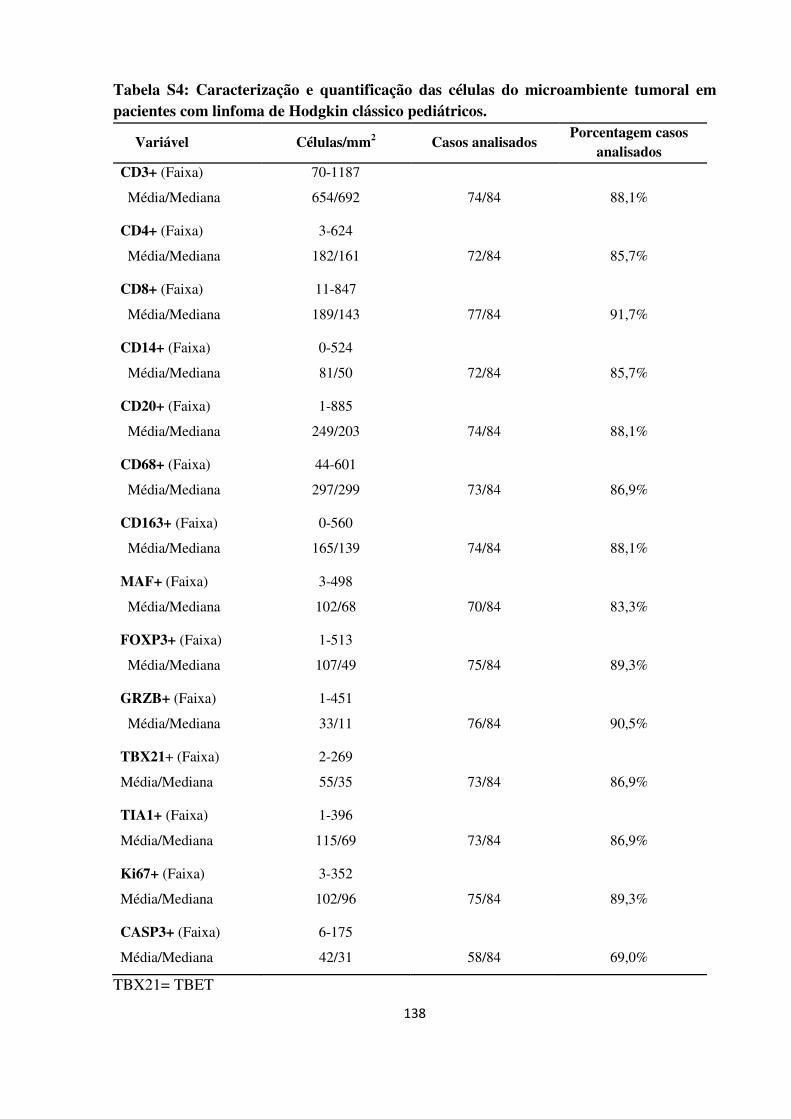
138
Tabela S4: Caracterização e quantificação das células do microambiente tumoral em pacientes com linfoma de Hodgkin clássico pediátricos.
Variável Células/mm2 Casos analisados Porcentagem casos
analisados CD3+ (Faixa) 70-1187
Média/Mediana 654/692 74/84 88,1%
CD4+ (Faixa) 3-624
Média/Mediana 182/161 72/84 85,7%
CD8+ (Faixa) 11-847
Média/Mediana 189/143 77/84 91,7%
CD14+ (Faixa) 0-524
Média/Mediana 81/50 72/84 85,7%
CD20+ (Faixa) 1-885
Média/Mediana 249/203 74/84 88,1%
CD68+ (Faixa) 44-601
Média/Mediana 297/299 73/84 86,9%
CD163+ (Faixa) 0-560
Média/Mediana 165/139 74/84 88,1%
MAF+ (Faixa) 3-498
Média/Mediana 102/68 70/84 83,3%
FOXP3+ (Faixa) 1-513
Média/Mediana 107/49 75/84 89,3%
GRZB+ (Faixa) 1-451
Média/Mediana 33/11 76/84 90,5%
TBX21+ (Faixa) 2-269
Média/Mediana 55/35 73/84 86,9%
TIA1+ (Faixa) 1-396
Média/Mediana 115/69 73/84 86,9%
Ki67+ (Faixa) 3-352
Média/Mediana 102/96 75/84 89,3%
CASP3+ (Faixa) 6-175
Média/Mediana 42/31 58/84 69,0%
TBX21= TBET
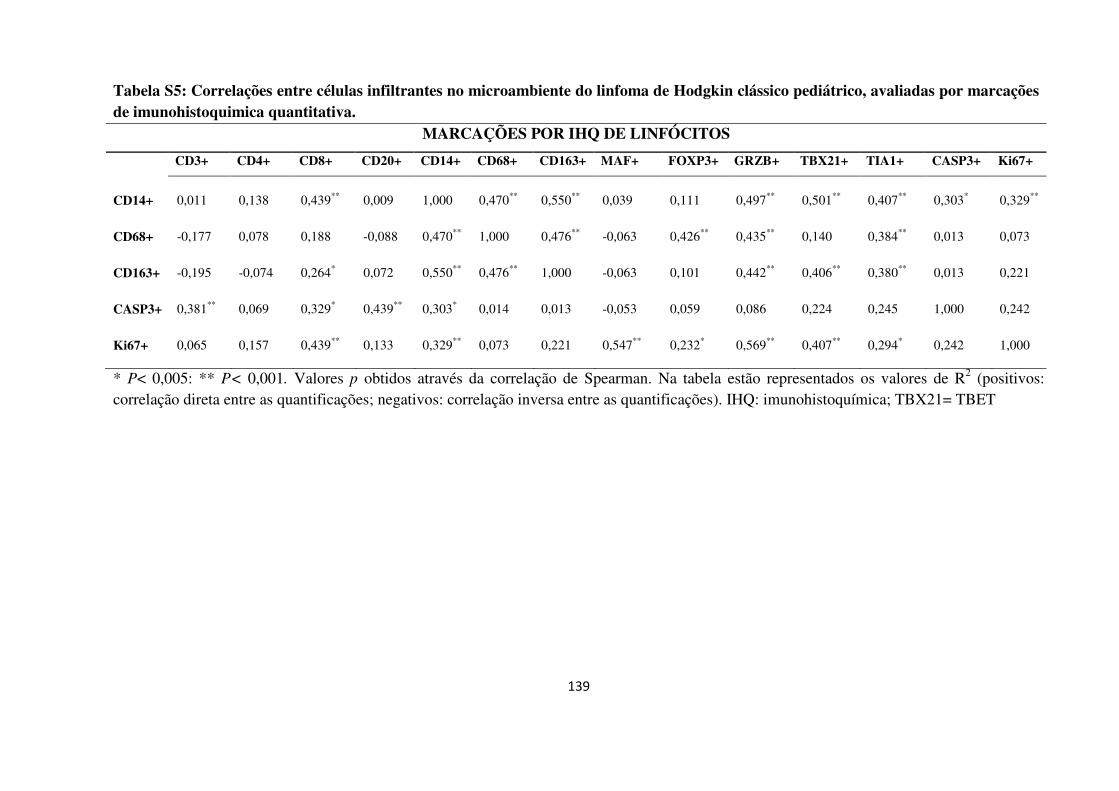
139
Tabela S5: Correlações entre células infiltrantes no microambiente do linfoma de Hodgkin clássico pediátrico, avaliadas por marcações de imunohistoquimica quantitativa.
MARCAÇÕES POR IHQ DE LINFÓCITOS
CD3+ CD4+ CD8+ CD20+ CD14+ CD68+ CD163+ MAF+ FOXP3+ GRZB+ TBX21+ TIA1+ CASP3+ Ki67+
CD14+ 0,011 0,138 0,439** 0,009 1,000 0,470** 0,550** 0,039 0,111 0,497** 0,501** 0,407** 0,303* 0,329**
CD68+ -0,177 0,078 0,188 -0,088 0,470** 1,000 0,476** -0,063 0,426** 0,435** 0,140 0,384** 0,013 0,073
CD163+ -0,195 -0,074 0,264* 0,072 0,550** 0,476** 1,000 -0,063 0,101 0,442** 0,406** 0,380** 0,013 0,221
CASP3+ 0,381** 0,069 0,329* 0,439** 0,303* 0,014 0,013 -0,053 0,059 0,086 0,224 0,245 1,000 0,242
Ki67+ 0,065 0,157 0,439** 0,133 0,329** 0,073 0,221 0,547** 0,232* 0,569** 0,407** 0,294* 0,242 1,000
* P< 0,005: ** P< 0,001. Valores p obtidos através da correlação de Spearman. Na tabela estão representados os valores de R2 (positivos: correlação direta entre as quantificações; negativos: correlação inversa entre as quantificações). IHQ: imunohistoquímica; TBX21= TBET

140
ANEXO V
TRABALHO PUBLICADO E APRESENTAÇÕES EM CONGRESSO
• VERA-LOZADA MG, BARROS MH, STEFANOFF CG, HASSAN R. Expressão de
marcadores de ativação e diferenciação de macrófagos no linfoma de Hodgkin clássico
como marcadores prognóstico: a propósito das condições técnicas de aplicação. In:
Congresso Brasileiro de Hematologia, Hemoterapia e Terapia Celular – HEMO 2012,
Rio de Janeiro.
• VERA-LOZADA MG, BARROS MH, SCHOLL V, CAPPELLETTI P,
STEFANOFF CG, HASSAN R. Avaliação de perfis de expressão gênica com valor
prognóstico no linfoma de Hodgkin clássico da infância e adolescência. In: Congresso
Brasileiro de Hematologia, Hemoterapia e Terapia Celular – HEMO 2012, Rio de
Janeiro.
• BARROS, M.H., VERA-LOZADA, G., SOARES, F.A., NIEDOBITEK, G. e
HASSAN R. Tumor microenvironment composition in pediatric classical Hodgkin
lymphoma is modulated by age and Epstein-Barr virus infection. Int J Cancer, v.
131, n. 5, p. 1142-52, Sep 1 2012.

141

142

Tumor microenvironment composition in pediatric classicalHodgkin lymphoma is modulated by age and Epstein-Barr virusinfectionMario Henrique M. Barros1,2, Gabriela Vera-Lozada1, Fernando A. Soares3, Gerald Niedobitek2 and Rocio Hassan1
1 Bone Marrow Transplantation Center, Instituto Nacional de Cancer (INCA), Rio de Janeiro, Brazil2 Institute for Pathology, Unfallkrankenhaus Berlin, Berlin, Germany3 Pathology Department, Hospital A.C. Camargo, Sao Paulo, Brazil
Classical Hodgkin lymphoma (cHL) is characterized by a small number of neoplastic cells in a background of reactive cells.
Children and adults differ in constitution and functionality of the immune system and it is possible that there may be age-
related differences in tumor microenvironment composition in cHL. One hundred children with pediatric cHL were studied.
Tumor-infiltrating lymphocytes were analyzed by immunohistochemistry (IHC) and image analysis. Epstein-Barr virus (EBV)
status was determined by EBER-specific in situ hybridization and IHC. Results were analyzed in the context of age-group,
histological characteristics and clinical follow-up. EBV-status was not associated with age-group. Children <10 years and
EBV1 cases were characterized by a more intense T cell infiltrate, exhibiting a cytotoxic/Th1 profile, characterized by higher
numbers of CD31, CD81, TIA11 and TBET1 lymphocytes. Extranodal disease (p 5 0.016) and high number of GranzymeB1
lymphocytes (p 5 0.04) were independently associated with reduced progression-free survival (PFS). Yet, in EBV1 cases,
improved outcome was observed in cases with low numbers of FOXP31 lymphocytes (p 5 0.046), FOXP3/CD8 ratio < 1 (p 5
0.021) and TBET/CMAF ratio < 1 (p 5 0.017). By contrast, in EBV2 cases, poor survival was observed in cases with
extranodal disease (p 5 0.028), MC subtype (p 5 0.009) and high numbers of TIA11 (p 5 0.044) and GranzymeB1 (p 5
0.04) lymphocytes. The results suggest that in EBV1 cHL an effective immune response directed against viral or tumor
antigens may be triggered in the tumor microenvironment and that physiological and age-related changes of the immune
system may also modulate the tumor microenvironment in pediatric cHL.
Classical Hodgkin lymphoma (cHL) is characterized by asmall number of neoplastic, Hodgkin and Reed-Sternberg(HRS) cells, in a background of inflammatory non-neoplasticcells, mainly B and T cells.1 The clinical and pathologicalcharacteristics of cHL are thought to result from an immunedysfunction, characterized by the deregulated expression ofseveral cytokines and chemokines,2 and an inadequate
immune response due to poor immunogenicity of HRS cells,the immunosuppressive effects exerted by the tumor cellsand/or the poor response of the host immune system.2
At a local level, the tumor microenvironment is main-tained by a crosstalk between malignant and reactive cells inthe microenvironment, mediated by cytokines and chemo-kines expressed by both, HRS and inflammatory cells.2
Increasing evidence of the importance of tumor microenvir-onment in the pathogenesis of and clinical response to cHLhas come from immunohistochemical and molecular stud-ies.2–4 Particularly, the number of several intratumoral cellpopulations, such as cytotoxic and regulatory (Treg) Tcells,2,5,6 or the ratio between these populations7–9 have beendescribed as being associated with therapeutic response.
Up to now, however, all these studies have been con-ducted in adult patients or a mixture of adolescents andadults. It is possible that children and adults differ in theirresponse to the tumor, not only with respect to the recruitedcell types, but also to the functional status of these cells. Inhealthy individuals, the numbers of T and B lymphocytes inthe peripheral blood differ between children and adults.10–13
Furthermore, in adults aging of the immune system charac-terized by the accumulation of CD8!CD28" cells may con-tribute to the shaping of the immune response to tumor orviral antigens.14–16
Key words: Hodgkin lymphoma, children, Epstein-Barr virus,lymphocyte, tumor microenvironment, prognosisAdditional Supporting Information may be found in the onlineversion of this article.Grant sponsor: INCT para Controle do Cancer; Grant number:
CNPq 573806/2008-0; Grant sponsor: FAPERJ; Grant number:
E26/170.026/2008 (Brazil); Grant sponsor: Swissbridge Foundation(Switzerland), Grant sponsor: Fundaçao de Amparo a Pesquisa doEstado do Rio de Janeiro (FAPERJ); Grant number: E26/111.432/2010; Grant sponsor: CAPES-DAAD program (Brazil-Germany)DOI: 10.1002/ijc.27314History: Received 26 Aug 2011; Accepted 26 Sep 2011; Online 25Oct 2011Correspondence to: Mario Henrique M. Barros, Institute forPathology, Unfallkrankenhaus Berlin, Warener Str. 7, 12683 Berlin,Germany, Tel: !49 30 5681 3750; Fax: !49 30 5681 3753,E-mail: [email protected]
Tum
orIm
mun
olog
y
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC
International Journal of Cancer
IJC

The presence of Epstein-Barr virus (EBV), which clonallyinfects HRS cells in a proportion of cases, is another variablecapable of modulating the tumor microenvironment byinducing the expression of cytokines involved in cell migra-tion and of viral immunogenic proteins.17–19 In fact, molecu-lar profiling studies have identified a distinct immune molec-ular signature for EBV-associated cHL.18,20
In this study, we have analyzed the composition of the tu-mor microenvironment and its prognostic impact in a largeseries of pediatric cHL comprising comparable numbers ofEBV! and EBV" cases.
Material and MethodsPatients
One hundred children and adolescents (up to 18 year old)diagnosed with cHL at the Instituto Nacional do Cancer(INCA, Brazil) between 1999 and 2006 were included in thisstudy, based on availability of formalin-fixed paraffin-embed-ded tissue and complete clinical data for retrospective analy-sis. All patients were negative for the human immunodefi-ciency virus (HIV) infection. Patients were classified in twoage groups (#10 years vs. >10 years) to investigate microen-vironment characteristics in younger vs. older children.10–13,21
Pretreatment factors evaluated were disease stage accordingto the Ann Arbor staging criteria,22 presence of B symptomsand mediastinal mass, number of involved anatomic areas(IAA) (nodal and extranodal), presence of anemia (children<12 years: Hb <12 g/dl; male 12–18 years: Hb <13.5 g/dl;female 12–18 years: 11.5 g/dl), leukopenia (<4,000 leukocytes/mm3) and lymphopenia (<800 lymphocytes /mm3). Evalua-tion of IAA was performed as previously described.20 Chil-dren with Stages I, IIA or IIIA were classified as favorableclinical presentation and children with Stages IIB, IIIB or IVwere classified as unfavorable clinical presentation, accordingpediatric oncology guidelines.23 All patients were treatedaccording pediatric protocols and received adriblastine-basedchemotherapy:24,25 53 children (53%) were treated accordingto the ABVD-protocol (adriamycin, bleomycin, vinblastineand dacarbazine) and 47 children (47%) according to theGerman pediatric protocol HD90, consisting of OPPA (vin-cristine, procarbazine, prednisone and doxorrubicine) forgirls, OEPA (vincristine, procarbazine, prednisone and doxor-rubicine) for boys and COPP (cyclophosphamide, vincristine,procarbazine and prednisone) for both when they were notpresenting with early stage disease (Stages I and IIA).25
Radiotherapy was administered only when was proposed bythe protocols.24,25 This study was approved by the INCAEthics Committee.
Histology
Diagnosis of cHL was established by morphologic criteriaaccording to the WHO classification.1 All cases were inde-pendently reviewed by two pathologists (MHMB and GN);discordant cases were discussed at a double-headedmicroscope. In cases no agreement was reached, cases were
designated ‘‘unclassifiable’’. The interfollicular pattern wasdefined as the presence of frequent residual follicles or exten-sive follicular hyperplasia with interfollicular tumor infil-trates.26 All histological parameters were evaluated in conven-tional H&E-stained slides.
Tissue microarray design
A Tissue arrayer device (Beecher Instrument, SilverSpring,MD) was used to assemble the tissue microarray (TMA)blocks. From each case, two 1-mm-diameter cores, selectedfrom two different representative tumor areas, were included.
Immunohistochemistry
TMA blocks were sectioned at a thickness of 3 lm. Sectionswere dried for 12 hr at 56$C. Dewaxed paraffin sections wererehydrated and submitted to endogenous peroxidase block-ade. Antigen retrieval was performed by heat treatment in apressure-cooker for 1 min. Buffers used for antigen retrievaland primary antibodies are listed in the Supporting Informa-tion Table S1. Briefly, the antibodies used were anti- CD3,CD4, FOXP3, CD8, TIA1, Granzyme B and CD20. Anti-TBET and anti-CMAF antibodies were used for characteriza-tion of Th1 and Th2 cells, respectively, as previouslyreported.27 Proliferative activity of reactive non-neoplasticlymphocytes was evaluated by Ki67 immunostaining.
After incubation with primary antibody, immunodetectionwas performed using labeled streptavidin biotin (LSAB) visu-alization system (Dako, Glostrup, Denmark), Envision kit(Dako) or ZytoChem Plus HRP polymer kit (Zytomed Sys-tems, Berlin, Germany) (Supporting Information Table S1),employing diaminobenzidine (DAB) chromogen as substrate.Sections were counterstained with hematoxylin. External andinternal controls included in the TMA were taken into con-sideration to interpret stainings.
EBV detection
Latent EBV infection was determined in all patients by insitu hybridization (ISH) with fluorescein-conjugated probesfor EBV-encoded RNAs (EBER-ISH) and by IHC againstlatent membrane protein 1 (LMP1) as described.20
Computer-assisted microscopical analysis
For lymphocyte subset evaluation, each core was photo-graphed using AxioCam MRc camera (Zeiss, Germany) at a200% magnification. The numbers of labeled lymphocyteswere determined per 1 mm2 using the image analysis soft-ware HISTO (Biomas, Elangen, Germany). Germinal centerswithout colonization by neoplastic cells, when present in thecore, were not considered in the cell count.
Threshold
For each lymphocyte subset, the 25th and 50th percentileswere used to categorize the intensity of the lymphocyte infil-tration. Cases displaying no labeled lymphocytes in bothcores were considered not evaluable for technical reasons.
Tum
orIm
mun
olog
y
Barros et al. 1143
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC

Statistical analysis
Pearson’s chi-square and Fisher’s exact test were used to testassociation between dichotomous variables. Mann-Whitneytest was used to test association between dichotomous andcontinuous variables, while Spearman’s correlation was usedto test association between continuous variables. Differenceswere considered significant at p < 0.05 in two-tailed tests.Primary treatment was defined as a failure if the lymphomahad progressed at any time after the initiation of therapy;treatment success was defined as the absence of progressionor relapse. Progression-free survival (PFS) was the interval(in months) from diagnosis to progression at any time,relapse from complete response, or initiation of new, previ-ously unplanned treatment or to the last follow-up in thepatients with treatment success. Overall survival (OS) refersto the interval (in months) from the diagnosis to death orlast follow-up. Survival distributions were estimated by theKaplan-Meier method and differences were compared usinglog-rank test. Multivariate Cox proportional hazard regres-sion method was used to determine the independent prog-nostic value of statistically significant variables in univariateanalyses. Data were analyzed using Statistical Package for theSocial Sciences 13.0 (SPSS).
ResultsClinico-pathological parameters and EBV status
The age at diagnosis ranged from 3 to 18 years (median 14years), with a male:female ratio of 1.7:1 (64 males and 36females). Similar numbers of patients were allocated to favor-able and unfavorable clinical presentation groups (52.6% vs.47.4%).The number of IAA ranged from 1 to 9 (median 3).
NS was the predominant subtype (69/100, 69%), followedby mixed cellularity (MC) (23/100, 23%). The presence ofgranulomas was observed in 37% of cases (37/100 cases), pre-dominantly in MC subtype (14/23 MC cases with granulomasvs. 21/69 NS cases with granulomas, p & 0.007, v2). Granulo-mas were found preferentially in the young age-group (15/27children # 10 years had granulomas, and 22/73 children >10 years had granulomas. p & 0.019, v2). There was no asso-ciation of this histological feature with EBV status or otherclinical-histological variables.
Overall, EBV was detected in 44.8% cases, with the MCsubtype (15/22, 68.1%) showing a higher association than theNS subtype (28/74, 37.8%) (p & 0.012); there was no associa-tion with age-groups (# vs. >10 years; p & 0.185). The mainclinical and histological characteristics are listed in Table 1.
Lymphocyte subsets in pediatric cHL
CD3! T cells as well as T-cell subsets showed no specificdistribution in the tumor microenvironment, being some-times observed in the vicinity of HRS cells in a rosette-likefashion, but without predominance of this feature (Fig. 1).The CD20! B cells were distributed uniformly in the tumormicroenvironment, without specific localization.
A detailed summary of the numbers of each lymphocytepopulation per mm2 is given in the Table 2. A higher numberof CD3! T cells in comparison to CD20! B lymphocytes wasobserved (median 645 cells/mm2 vs. 196 cells/mm2, respec-tively). However, CD20! B cells were observed at least as fre-quently as any T cell subset individually [CD4! (median 155cells/mm2), FOXP3! (median 49 cells/mm2), CMAF! (median68 cells/mm2), TBET! (median 32 cells/mm2), CD8! (median143 cells/mm2), TIA1! (median 69 cells/mm2) and GranzymeB! (median 11 cells/mm2)] (Fig. 2).
Ki67 immunostaining was evaluated in the small maturebenign lymphocytes. A large variation was observed regard-ing the number of non-neoplastic Ki67! lymphocytes (3–352cells/mm2, median: 91 cells/mm2). These lymphocytesshowed no specific distribution in the tumor microenviron-ment. No association with histological features or with EBV-status was found. A direct correlation (Spearman’s correla-tion) was observed between the number of benign Ki67!lymphocytes and the number of FOXP3! (p & 0.042),CD8! (p < 0.0005), TIA1! (p & 0.015), Granzyme B! (p< 0.0005), TBET! (p < 0.0005) and CMAF! (p < 0.0005)lymphocytes (Supporting Information Fig. S1).
Lymphocyte subsets are associated with clinical and
histological features
Age groups. The younger age group (#10 years) was charac-terized by a more intense T cell infiltrate, exhibiting a cyto-toxic/Th1 profile, characterized by higher numbers of CD3!(p & 0.025; Mann-Whitney test), CD8! (p & 0.016, Mann-Whitney test), TIA1! (p & 0.008, Mann-Whitney test) andTBET! (p & 0.045, Mann-Whitney test) lymphocytes,whereas in older children (>10 years) FOXP3!, CMAF!and CD4! cells were more prevalent (Table 3).
Clinical characteristics. In general, favorable clinical parame-ters were associated with a more intense T lymphocyte infil-trate. Stages I/II were associated with high numbers of CD3!(median 748 cells/mm2 vs. 510 cells/mm2 in III/IV stages,p & 0.004, Mann-Whitney test), CD4! (median 209 cells/mm2 vs. 99 cells/mm2 in III/IV stages, P & 0.005, Mann-Whitney test) and CMAF! (median 94 cells/mm2 vs. 29cells/mm2 in III/IV stages, p & 0.003, Mann-Whitney test)lymphocytes. Conversely, children with '4IAA exhibited lownumbers of CD3! (median 457 cells/mm2 vs. 752 cells/mm2
with <4IAA, p < 0.0005, Mann-Whitney test), CD4! (me-dian 91 cells/mm2 vs. 209 cells/mm2 with <4IAA, p & 0.027,Mann-Whitney test), CMAF! (median 44 cells/mm2 vs. 94cells/mm2 with <4IAA, p & 0.032, Mann-Whitney test) andCD8! (median 111 cells/mm2 vs. 183 cells/mm2 with<4IAA, p & 0.008, Mann-Whitney test) lymphocytes in thetumor microenvironment.
Histopathological characteristics. MC subtype was associatedwith high numbers of CD8! (p & 0.049, Mann-Whitneytest), TIA1! (p & 0.036, Mann-Whitney test) and GranzymeB! (p & 0.019, Mann-Whitney test) lymphocytes, typical ofa cytotoxic profile, when compared with all other types. This
Tum
orIm
mun
olog
y
1144 Tumor Microenvironment in Pediatric Classical Hodgkin Lymphoma
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC
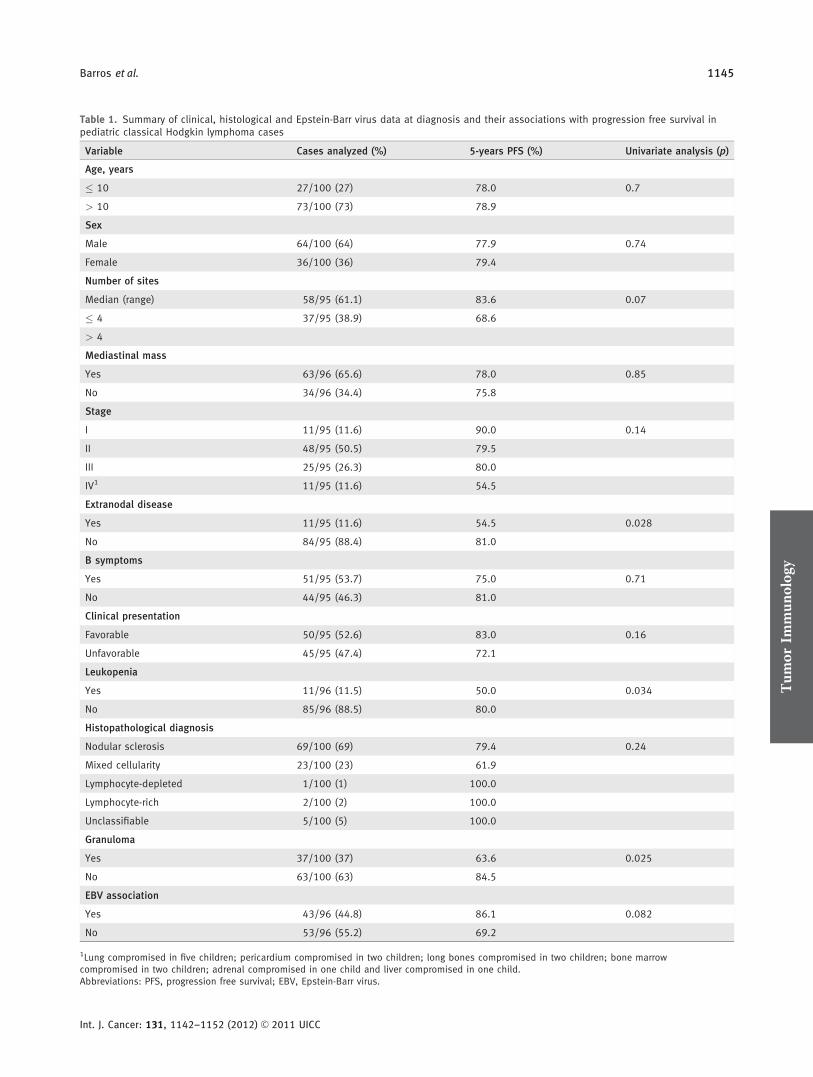
Table 1. Summary of clinical, histological and Epstein-Barr virus data at diagnosis and their associations with progression free survival inpediatric classical Hodgkin lymphoma cases
Variable Cases analyzed (%) 5-years PFS (%) Univariate analysis (p)
Age, years
# 10 27/100 (27) 78.0 0.7
> 10 73/100 (73) 78.9
Sex
Male 64/100 (64) 77.9 0.74
Female 36/100 (36) 79.4
Number of sites
Median (range) 58/95 (61.1) 83.6 0.07
# 4 37/95 (38.9) 68.6
> 4
Mediastinal mass
Yes 63/96 (65.6) 78.0 0.85
No 34/96 (34.4) 75.8
Stage
I 11/95 (11.6) 90.0 0.14
II 48/95 (50.5) 79.5
III 25/95 (26.3) 80.0
IV1 11/95 (11.6) 54.5
Extranodal disease
Yes 11/95 (11.6) 54.5 0.028
No 84/95 (88.4) 81.0
B symptoms
Yes 51/95 (53.7) 75.0 0.71
No 44/95 (46.3) 81.0
Clinical presentation
Favorable 50/95 (52.6) 83.0 0.16
Unfavorable 45/95 (47.4) 72.1
Leukopenia
Yes 11/96 (11.5) 50.0 0.034
No 85/96 (88.5) 80.0
Histopathological diagnosis
Nodular sclerosis 69/100 (69) 79.4 0.24
Mixed cellularity 23/100 (23) 61.9
Lymphocyte-depleted 1/100 (1) 100.0
Lymphocyte-rich 2/100 (2) 100.0
Unclassifiable 5/100 (5) 100.0
Granuloma
Yes 37/100 (37) 63.6 0.025
No 63/100 (63) 84.5
EBV association
Yes 43/96 (44.8) 86.1 0.082
No 53/96 (55.2) 69.2
1Lung compromised in five children; pericardium compromised in two children; long bones compromised in two children; bone marrowcompromised in two children; adrenal compromised in one child and liver compromised in one child.Abbreviations: PFS, progression free survival; EBV, Epstein-Barr virus.
Tum
orIm
mun
olog
y
Barros et al. 1145
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC
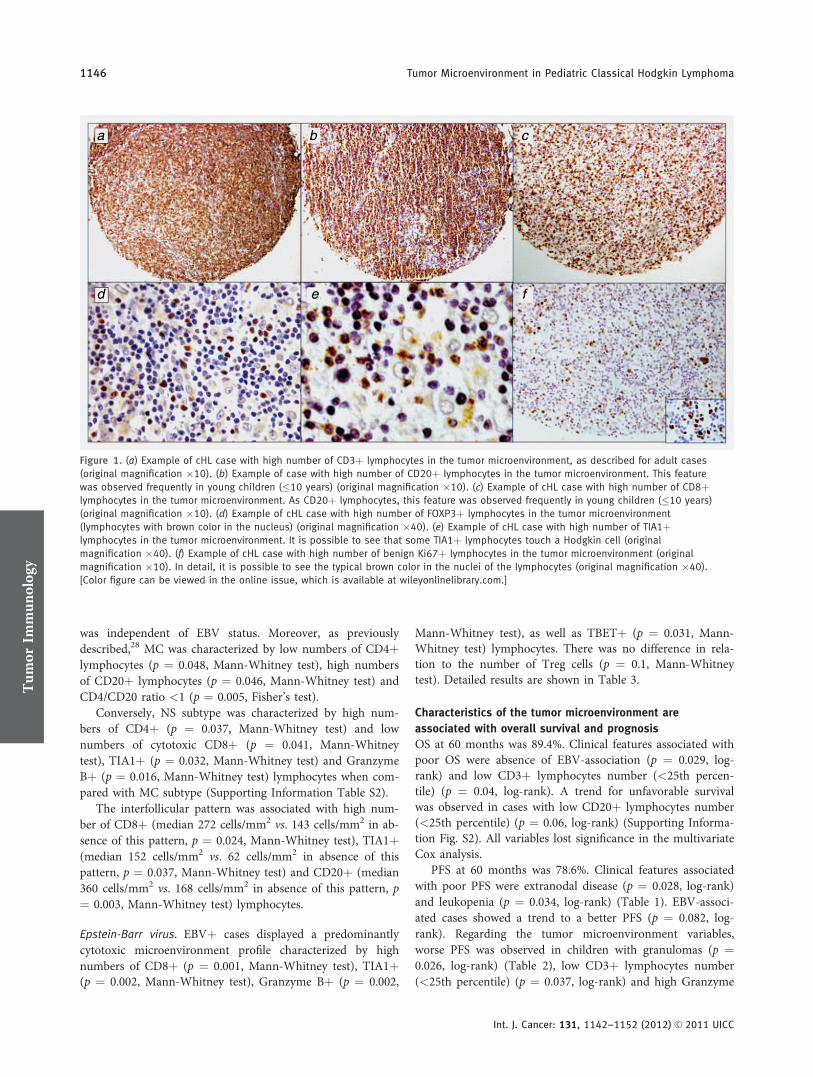
was independent of EBV status. Moreover, as previouslydescribed,28 MC was characterized by low numbers of CD4!lymphocytes (p & 0.048, Mann-Whitney test), high numbersof CD20! lymphocytes (p & 0.046, Mann-Whitney test) andCD4/CD20 ratio <1 (p & 0.005, Fisher’s test).
Conversely, NS subtype was characterized by high num-bers of CD4! (p & 0.037, Mann-Whitney test) and lownumbers of cytotoxic CD8! (p & 0.041, Mann-Whitneytest), TIA1! (p & 0.032, Mann-Whitney test) and GranzymeB! (p & 0.016, Mann-Whitney test) lymphocytes when com-pared with MC subtype (Supporting Information Table S2).
The interfollicular pattern was associated with high num-ber of CD8! (median 272 cells/mm2 vs. 143 cells/mm2 in ab-sence of this pattern, p & 0.024, Mann-Whitney test), TIA1!(median 152 cells/mm2 vs. 62 cells/mm2 in absence of thispattern, p & 0.037, Mann-Whitney test) and CD20! (median360 cells/mm2 vs. 168 cells/mm2 in absence of this pattern, p& 0.003, Mann-Whitney test) lymphocytes.
Epstein-Barr virus. EBV! cases displayed a predominantlycytotoxic microenvironment profile characterized by highnumbers of CD8! (p & 0.001, Mann-Whitney test), TIA1!(p & 0.002, Mann-Whitney test), Granzyme B! (p & 0.002,
Mann-Whitney test), as well as TBET! (p & 0.031, Mann-Whitney test) lymphocytes. There was no difference in rela-tion to the number of Treg cells (p & 0.1, Mann-Whitneytest). Detailed results are shown in Table 3.
Characteristics of the tumor microenvironment are
associated with overall survival and prognosisOS at 60 months was 89.4%. Clinical features associated withpoor OS were absence of EBV-association (p & 0.029, log-rank) and low CD3! lymphocytes number (<25th percen-tile) (p & 0.04, log-rank). A trend for unfavorable survivalwas observed in cases with low CD20! lymphocytes number(<25th percentile) (p & 0.06, log-rank) (Supporting Informa-tion Fig. S2). All variables lost significance in the multivariateCox analysis.
PFS at 60 months was 78.6%. Clinical features associatedwith poor PFS were extranodal disease (p & 0.028, log-rank)and leukopenia (p & 0.034, log-rank) (Table 1). EBV-associ-ated cases showed a trend to a better PFS (p & 0.082, log-rank). Regarding the tumor microenvironment variables,worse PFS was observed in children with granulomas (p &0.026, log-rank) (Table 2), low CD3! lymphocytes number(<25th percentile) (p & 0.037, log-rank) and high Granzyme
Figure 1. (a) Example of cHL case with high number of CD3! lymphocytes in the tumor microenvironment, as described for adult cases(original magnification %10). (b) Example of case with high number of CD20! lymphocytes in the tumor microenvironment. This featurewas observed frequently in young children (#10 years) (original magnification %10). (c) Example of cHL case with high number of CD8!lymphocytes in the tumor microenvironment. As CD20! lymphocytes, this feature was observed frequently in young children (#10 years)(original magnification %10). (d) Example of cHL case with high number of FOXP3! lymphocytes in the tumor microenvironment(lymphocytes with brown color in the nucleus) (original magnification %40). (e) Example of cHL case with high number of TIA1!lymphocytes in the tumor microenvironment. It is possible to see that some TIA1! lymphocytes touch a Hodgkin cell (originalmagnification %40). (f) Example of cHL case with high number of benign Ki67! lymphocytes in the tumor microenvironment (originalmagnification %10). In detail, it is possible to see the typical brown color in the nuclei of the lymphocytes (original magnification %40).[Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Tum
orIm
mun
olog
y
1146 Tumor Microenvironment in Pediatric Classical Hodgkin Lymphoma
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC
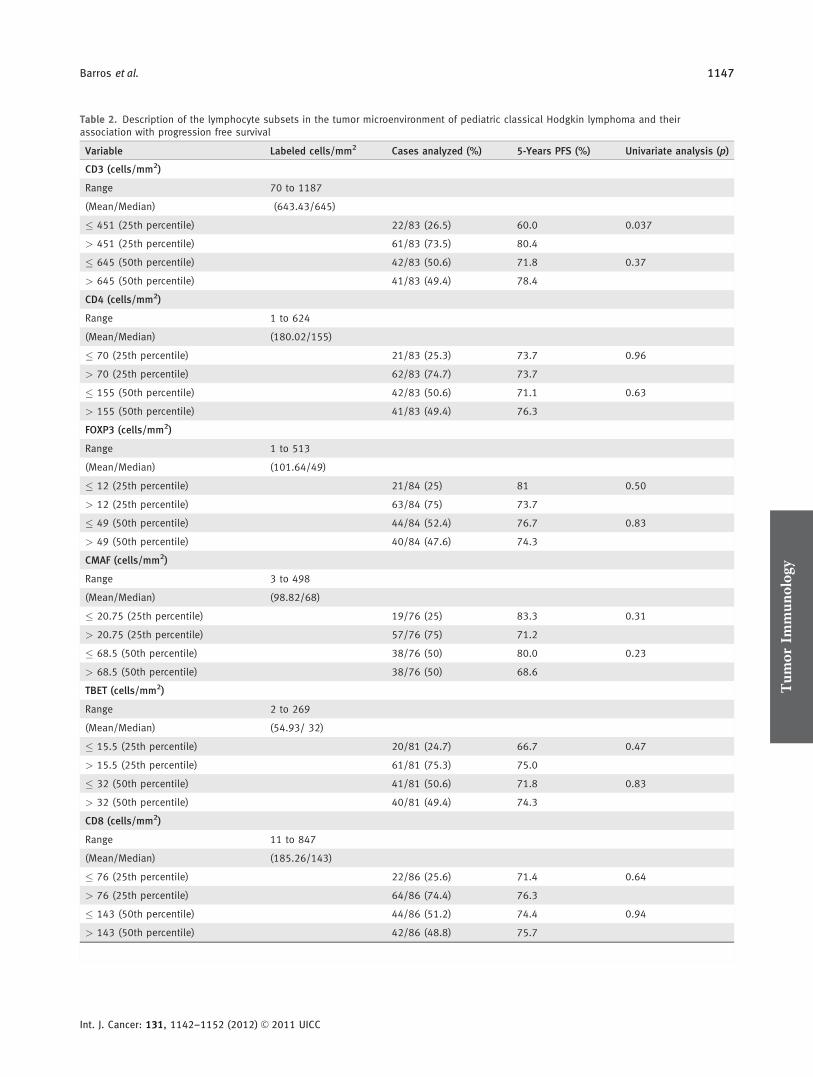
Table 2. Description of the lymphocyte subsets in the tumor microenvironment of pediatric classical Hodgkin lymphoma and theirassociation with progression free survival
Variable Labeled cells/mm2 Cases analyzed (%) 5-Years PFS (%) Univariate analysis (p)
CD3 (cells/mm2)
Range 70 to 1187
(Mean/Median) (643.43/645)
# 451 (25th percentile) 22/83 (26.5) 60.0 0.037
> 451 (25th percentile) 61/83 (73.5) 80.4
# 645 (50th percentile) 42/83 (50.6) 71.8 0.37
> 645 (50th percentile) 41/83 (49.4) 78.4
CD4 (cells/mm2)
Range 1 to 624
(Mean/Median) (180.02/155)
# 70 (25th percentile) 21/83 (25.3) 73.7 0.96
> 70 (25th percentile) 62/83 (74.7) 73.7
# 155 (50th percentile) 42/83 (50.6) 71.1 0.63
> 155 (50th percentile) 41/83 (49.4) 76.3
FOXP3 (cells/mm2)
Range 1 to 513
(Mean/Median) (101.64/49)
# 12 (25th percentile) 21/84 (25) 81 0.50
> 12 (25th percentile) 63/84 (75) 73.7
# 49 (50th percentile) 44/84 (52.4) 76.7 0.83
> 49 (50th percentile) 40/84 (47.6) 74.3
CMAF (cells/mm2)
Range 3 to 498
(Mean/Median) (98.82/68)
# 20.75 (25th percentile) 19/76 (25) 83.3 0.31
> 20.75 (25th percentile) 57/76 (75) 71.2
# 68.5 (50th percentile) 38/76 (50) 80.0 0.23
> 68.5 (50th percentile) 38/76 (50) 68.6
TBET (cells/mm2)
Range 2 to 269
(Mean/Median) (54.93/ 32)
# 15.5 (25th percentile) 20/81 (24.7) 66.7 0.47
> 15.5 (25th percentile) 61/81 (75.3) 75.0
# 32 (50th percentile) 41/81 (50.6) 71.8 0.83
> 32 (50th percentile) 40/81 (49.4) 74.3
CD8 (cells/mm2)
Range 11 to 847
(Mean/Median) (185.26/143)
# 76 (25th percentile) 22/86 (25.6) 71.4 0.64
> 76 (25th percentile) 64/86 (74.4) 76.3
# 143 (50th percentile) 44/86 (51.2) 74.4 0.94
> 143 (50th percentile) 42/86 (48.8) 75.7
Tum
orIm
mun
olog
y
Barros et al. 1147
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC
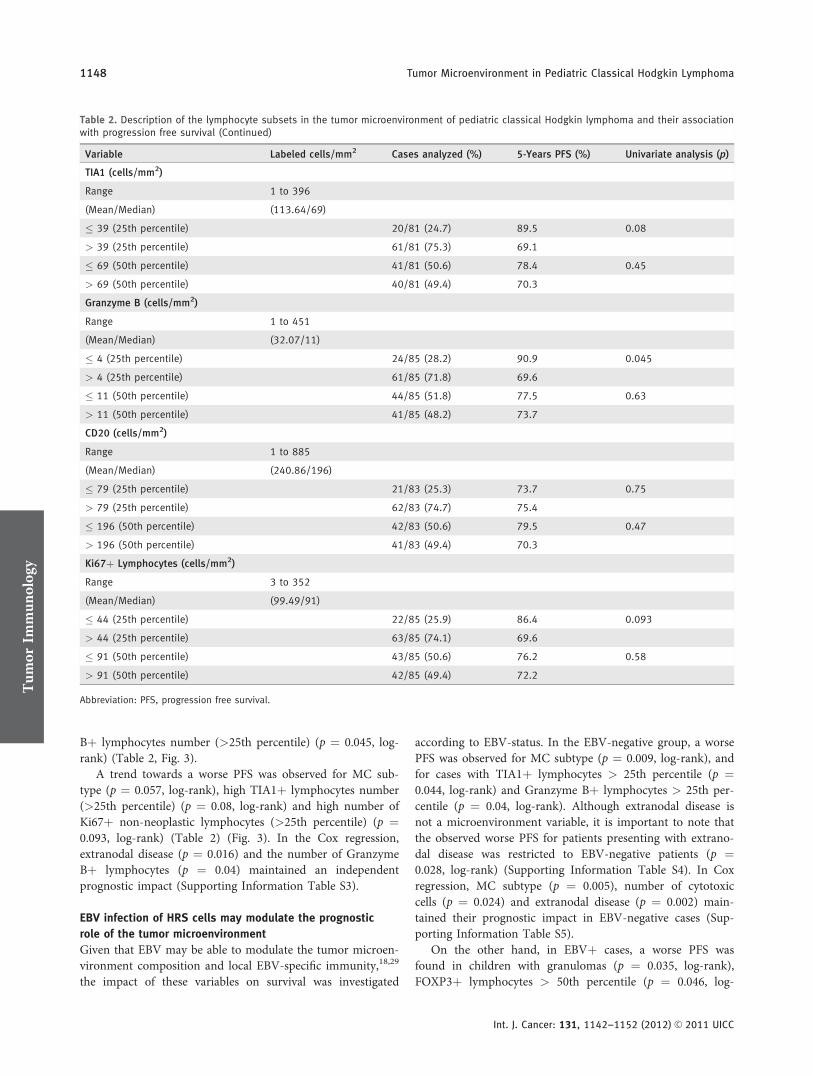
B! lymphocytes number (>25th percentile) (p & 0.045, log-rank) (Table 2, Fig. 3).
A trend towards a worse PFS was observed for MC sub-type (p & 0.057, log-rank), high TIA1! lymphocytes number(>25th percentile) (p & 0.08, log-rank) and high number ofKi67! non-neoplastic lymphocytes (>25th percentile) (p &0.093, log-rank) (Table 2) (Fig. 3). In the Cox regression,extranodal disease (p & 0.016) and the number of GranzymeB! lymphocytes (p & 0.04) maintained an independentprognostic impact (Supporting Information Table S3).
EBV infection of HRS cells may modulate the prognostic
role of the tumor microenvironment
Given that EBV may be able to modulate the tumor microen-vironment composition and local EBV-specific immunity,18,29
the impact of these variables on survival was investigated
according to EBV-status. In the EBV-negative group, a worsePFS was observed for MC subtype (p & 0.009, log-rank), andfor cases with TIA1! lymphocytes > 25th percentile (p &0.044, log-rank) and Granzyme B! lymphocytes > 25th per-centile (p & 0.04, log-rank). Although extranodal disease isnot a microenvironment variable, it is important to note thatthe observed worse PFS for patients presenting with extrano-dal disease was restricted to EBV-negative patients (p &0.028, log-rank) (Supporting Information Table S4). In Coxregression, MC subtype (p & 0.005), number of cytotoxiccells (p & 0.024) and extranodal disease (p & 0.002) main-tained their prognostic impact in EBV-negative cases (Sup-porting Information Table S5).
On the other hand, in EBV! cases, a worse PFS wasfound in children with granulomas (p & 0.035, log-rank),FOXP3! lymphocytes > 50th percentile (p & 0.046, log-
Table 2. Description of the lymphocyte subsets in the tumor microenvironment of pediatric classical Hodgkin lymphoma and their associationwith progression free survival (Continued)
Variable Labeled cells/mm2 Cases analyzed (%) 5-Years PFS (%) Univariate analysis (p)
TIA1 (cells/mm2)
Range 1 to 396
(Mean/Median) (113.64/69)
# 39 (25th percentile) 20/81 (24.7) 89.5 0.08
> 39 (25th percentile) 61/81 (75.3) 69.1
# 69 (50th percentile) 41/81 (50.6) 78.4 0.45
> 69 (50th percentile) 40/81 (49.4) 70.3
Granzyme B (cells/mm2)
Range 1 to 451
(Mean/Median) (32.07/11)
# 4 (25th percentile) 24/85 (28.2) 90.9 0.045
> 4 (25th percentile) 61/85 (71.8) 69.6
# 11 (50th percentile) 44/85 (51.8) 77.5 0.63
> 11 (50th percentile) 41/85 (48.2) 73.7
CD20 (cells/mm2)
Range 1 to 885
(Mean/Median) (240.86/196)
# 79 (25th percentile) 21/83 (25.3) 73.7 0.75
> 79 (25th percentile) 62/83 (74.7) 75.4
# 196 (50th percentile) 42/83 (50.6) 79.5 0.47
> 196 (50th percentile) 41/83 (49.4) 70.3
Ki67! Lymphocytes (cells/mm2)
Range 3 to 352
(Mean/Median) (99.49/91)
# 44 (25th percentile) 22/85 (25.9) 86.4 0.093
> 44 (25th percentile) 63/85 (74.1) 69.6
# 91 (50th percentile) 43/85 (50.6) 76.2 0.58
> 91 (50th percentile) 42/85 (49.4) 72.2
Abbreviation: PFS, progression free survival.
Tum
orIm
mun
olog
y
1148 Tumor Microenvironment in Pediatric Classical Hodgkin Lymphoma
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC

rank), FOXP3/CD8 ratio >1 (p & 0.021, log-rank), FOXP3/TIA1 ratio >1 (p & 0.06, log-rank) and TBET/CMAF ratio>1 (p & 0.017, log-rank) (Supporting Information Table S4).Because of the small number of children in this group, Coxanalysis was not performed.
DiscussionThe tumor microenvironment in cHL has been considered tobe a manifestation of host immune reactions to malignantcells.2 In this context, it is important to note that as yet all cHLtumor microenvironment studies were performed with adultsor a mix of adolescents and adults and that it is not clear if chil-
dren and adolescents have a particular composition of tumormicroenvironment. This is a pertinent point because immuno-logical differences exist between children and adults.10,11 It isconceivable that such differences could affect the immunologi-cal response to HRS cells or to viral antigens in EBV! cases.
In our pediatric group, we have observed higher numbersof CD3! over CD20! lymphocytes in the tumor microenvir-onment, in line with previous studies. However, an analysisby T cell populations showed that B lymphocytes were morefrequent than any of the T cell subsets investigated, includingCD4! cells. Thus, this first study of pediatric cHL suggeststhat the tumor microenvironment composition is indeed dif-ferent from that observed and adults with cHL, where CD4!cells are more prevalent than CD20! lymphocytes2,4,30 andthat the described predominance of infiltrating CD4! Tcells3,5,8 is not an invariable attribute of cHL.
This distinct feature of pediatric cHL may reflect thehigher number of CD20! in relation to CD4! lymphocytesobserved in peripheral blood of healthy children, when com-pared to adults.12,13,21 The quantitative contribution ofCD20! lymphocytes to the cHL microenvironment is start-ing to attract attention, since this lymphocyte subset has beenassociated with a favorable response to therapy.18,31 In ourstudy, the CD20! population showed a marginal impact onoverall survival. A more detailed characterization of intratu-moral B-cells, for example with reference to B-1 and B-2cells, will be required to fully appreciate any effects of B-cellson outcome in pediatric cHL.
It is possible that the shift from a cytotoxic/Th1 tumormicroenvironment to a more regulatory/Th2 profile, asobserved in older children in our study, may mirror thephysiological behavior of lymphocyte populations in the
Figure 2. Box-plot graph showing the numerical distribution ofeach lymphocyte population studied in the tumormicroenvironment of pediatric classical Hodgkin lymphoma.
Table 3. Description of the lymphocyte subsets by age-group and EBV-association in pediatric classical Hodgkin lymphoma
Variable
Age group EBV
#10 years1 (median) '10 years1 (median) p Positive1 (median) Negative1 (median) p
CD3! cells 161–1170 (803) 70–1187 (621) 0.025 234–1170 (700) 70–1187 (664) 0.4
CD4! cells 14–478 (155) 1–624 (167) 0.8 1–434 (187) 3–6249 (136) 0.9
FOXP3! cells 1–416 (41) 1–513 (79) 0.3 2–387 (79) 1–51310 (44) 0.1
TBET! cells 2–264 (58) 4–2692 (32) 0.045 2–269 (61) 4–26411 (29) 0.03
CMAF! cells 3–4983 (64) 3–387 (70) 0.9 3–387 (67) 3–498 (71) 0.9
CD8! cells 41–847 (272) 11–533 (170) 0.016 49–847 (264) 11–533 (132) 0.001
TIA1! cells 36–396 (143) 1–3964 (58) 0.008 22–396 (152) 1–35812 (61) 0.002
GRANZYME B! cells 1–1425 (12) 1–4516 (17) 0.4 1–451 (32) 1–88 (10) 0.002
CD20! cells 44–8857 (222) 1–8798 (181) 0.1 20–698 (258) 1–88513 (190) 0.1
The p value is from Mann-Whitney Test.1The numbers represent labeled cells/mm2 and the numbers in the bracket represent the median value. 2In this range, there are three outlier caseswith 167, 201 and 269 TBET! cells/mm2. 3In this range, there is one outlier case with 498 CMAF! cells/mm2. 4In this range, there are four outliercases with 325, 360, 376 and 396 TIA1! cells/mm2. 5In this range, there is one outlier case with 142 Granzyme B! cells/mm2. 6In this range,there are four outlier cases with 90, 123, 211 and 451 Granzyme B ! cells/mm2. 7In this range, there is one outlier case with 885 CD20! cells/mm2. 8In this range, there are two outlier cases with 783 and 879 CD20! cells/mm2. 9In this range, there is one outlier case with 624 CD4! cells/mm2. 10In this range, there are four outlier cases with 349, 416, 457 and 513 FOXP3! cells/mm2. 11In this range, there are three outlier cases with114, 184 and 264 TBET! cells/mm2. 12In this range, there are two outlier cases with 325 and 358 TIA1! cells/mm2. 13In this range, there are twooutlier cases with 783 and 885 CD20! cells/mm2.Abbreviation: EBV, Epstein-Barr virus.
Tum
orIm
mun
olog
y
Barros et al. 1149
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC

periphery. In line with this, Faria et al. showed that age-related changes in peripheral lymphocyte subsets in healthyBrazilian populations are not progressive and equally steadyfor all cell populations, but instead show an oscillatorybehavior which is characteristic for each cell population indifferent age groups.21 Previous studies have emphasized therole of cytokines and chemokines produced by the neoplasticcells in shaping the microenvironment of cHL. Our resultsnow raise the possibility that physiological and age-relatedchanges of the immune system may also modulate the tumormicroenvironment in pediatric cHL.
In our cases, an intense T cell infiltrate was associatedwith favorable disease presentation (low disease stages andtumor burden). In colorectal cancer it is well recognized thathigh numbers of intratumoral CD3! lymphocytes are associ-ated with a less aggressive disease course.32,33 In cHL such anassociation has not been confirmed and may reflect the pres-ence of a systemic mechanism of immunosurveillance.
In this pediatric series, we were not able to confirm thevalue of FOXP3! Treg population for prediction of favorableoutcome. As in adults,5–7 higher numbers of cytotoxic cellswere independently associated with worse PFS.
The positive correlation between the number of benignKi67! lymphocytes and different lymphocyte subsets, described
for the first time in this study, indicates that lymphocytes fromtumor microenvironment are able to proliferate in situ inresponse to antigens and/or cytokine stimuli. High numbers ofKi67! lymphocytes showed a borderline association with unfav-orable outcome in univariate analysis. This is in apparent con-trast to the positive prognostic effect of a high CD3! T-cellcount and suggests that proliferation may be restricted to T-cellsubsets with adverse prognostic impact. Additional studies arerequired to establish the phenotype and functional status of theKi67! benign lymphocytes.
This pediatric series was composed of comparable num-bers of EBV! and EBV" cases, allowing an analysis of therole of the EBV in modulating the immune response at themicroenvironment level. Although previous in vitro studiesshowed that EBV can induce the production of cytokinesinvolved in the Treg-CD4! cell migration,34 we did notobserve differences in the number of CD4!, FOXP3! andCMAF! cells between EBV! and EBV" cases. On the otherhand, EBV! cases were characterized by high numbers ofintratumoral CD8!, TIA1!, Granzyme B! and TBET! cells.This is in line with seminal research that showed that, com-pared with EBV" cases, EBV! cHL expresses significantlyhigher levels of MCH Class I, an intact antigen presentationmachinery and higher numbers of intratumoral activated
Figure 3. Kaplan-Meier curves of the variables associated with the progression-free survival in pediatric classical Hodgkin lymphoma.(a) Mixed cellularity; (b) Granuloma; (c) CD3! lymphocyte >451/mm2 (25th percentile); (d) Granzyme B! lymphocytes >39/mm2 (25thpercentile); (e) Ki67! lymphocytes >44/mm2 (25th percentile). [Color figure can be viewed in the online issue, which is available atwileyonlinelibrary.com.]
Tum
orIm
mun
olog
y
1150 Tumor Microenvironment in Pediatric Classical Hodgkin Lymphoma
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC

cytotoxic T lymphocytes.35–37 Recently, Chetaille et al.,18 usingsamples of adults and children, described a molecular signa-ture of EBV! cHL that is characterized by genes associatedwith Th1 and antiviral responses, which is validated in ourexclusively pediatric population. However, in adults, it is pos-sible that cytotoxic T cells (in parts responsible for this antivi-ral signature) are not effective in the immune response againstEBV in the HRS cells, as previously shown by Frisan et al.29
In addition, acquisition of genetic alterations by HRS cells canresult in resistance to apoptosis and therefore resistance to cy-totoxic T cell-mediated killing.38–40
In the EBV! group, characterized by a better therapeuticresponse, a worse PFS was observed in cases with high numberof FOXP3 lymphocytes, FOXP3/CD8 ratio >1 and FOXP3/TIA1 ratio >1 suggesting that: (1) the immune response againstEBV is mediated by CD8! cytotoxic lymphocytes and (2)FOXP3! lymphocytes are inhibiting the cytotoxic responsedirected against EBV, as demonstrated by several studies.19,41,42
EBV is able to modulate immune escape mechanisms2,17,35,43,44
and several studies point to an adverse prognosis for EBV-asso-ciated adult cHL.45–47 Nevertheless, it appears likely that in pedi-atric cHL, the interaction between EBV! HRS and the immunesystem follows a different scenario. In fact, a favorable outcomewas described for EBV! young cHL patients,48,49 including theseries reported here, suggesting that in children with EBV!
cHL, EBV may elicit an effective immune response, which isnegatively modulated by a suppressor microenvironment.42,50
Cytotoxic T-cells are an adverse factor in EBV" but not inEBV! cases. On the contrary, FOXP3! cells had a negativeprognostic effect in EBV! but not in EBV" cases. This becameparticularly evident when examining the ratio of FOXP3! Tregover cytotoxic effector cell populations. This reinforces our hy-pothesis that in pediatric patients, an EBV-specific immuneresponse may be mounted locally and that inhibition of thisimmune response by FOXP3! cells may negatively affect out-come. If confirmed, this would have significant implications forthe application of microenvironment targeted therapies in thepediatric settings.
We are, nevertheless, aware that the number of cases inthis study imposes a limitation in relation to the prognosticimpact of the described variables. However, we think our se-ries of sequential cases with similar distribution in relation toEBV-status is appropriate for the immunological evaluationperformed here.
In conclusion, this study suggests that the tumor microen-vironment of pediatric cHL has particular characteristics, notonly with respect to the number of lymphocytes, but also inrelation to the functional status of these cells. Validationstudies are necessary to confirm the prognostic impact of thevariables described in this study.
References
1. Swerdlow SH, International Agency for Researchon Cancer., World Health Organization. WHOclassification of tumours of haematopoietic andlymphoid tissues, 4th edn. Lyon, France:International Agency for Research on Cancer,2008.
2. Aldinucci D, Gloghini A, Pinto A, De Filippi R,Carbone A. The classical Hodgkin’s lymphomamicroenvironment and its role in promotingtumour growth and immune escape. J Pathol2010;221:248–63.
3. Alvaro T, Lejeune M, Garcia JF, Salvado MT,Lopez C, Bosch R, Jaen J, Escriva P, Pons LE.Tumor-infiltrated immune response correlateswith alterations in the apoptotic and cell cyclepathways in Hodgkin and Reed-Sternberg cells.Clin Cancer Res 2008;14:685–91.
4. Steidl C, Connors JM, Gascoyne RD. Molecularpathogenesis of hodgkin’s lymphoma: increasingevidence of the importance of themicroenvironment. J Clin Oncol 2011;29:1812–26.
5. ten Berge RL, Oudejans JJ, Dukers DF, MeijerJW, Ossenkoppele GJ, Meijer CJ. Percentage ofactivated cytotoxic T-lymphocytes in anaplasticlarge cell lymphoma and Hodgkin’s disease: anindependent biological prognostic marker.Leukemia 2001;15:458–64.
6. Alvaro-Naranjo T, Lejeune M, Salvado-UsachMT, Bosch-Princep R, Reverter-Branchat G, Jaen-Martinez J, Pons-Ferre LE. Tumor-infiltratingcells as a prognostic factor in Hodgkin’slymphoma: a quantitative tissue microarray studyin a large retrospective cohort of 267 patients.Leuk Lymphoma 2005;46:1581–91.
7. Alvaro T, Lejeune M, Salvado MT, Bosch R,Garcia JF, Jaen J, Banham AH, Roncador G,
Montalban C, Piris MA. Outcome in Hodgkin’slymphoma can be predicted from the presence ofaccompanying cytotoxic and regulatory T cells.Clin Cancer Res 2005;11:1467–73.
8. Kelley TW, Pohlman B, Elson P, Hsi ED. Theratio of FOXP3! regulatory T cells to granzymeB! cytotoxic T/NK cells predicts prognosis inclassical Hodgkin lymphoma and is independentof bcl-2 and MAL expression. Am J Clin Pathol2007;128:958–65.
9. Tzankov A, Meier C, Hirschmann P, Went P,Pileri SA, Dirnhofer S. Correlation of highnumbers of intratumoral FOXP3! regulatory Tcells with improved survival in germinal center-like diffuse large B-cell lymphoma, follicularlymphoma and classical Hodgkin’s lymphoma.Haematologica 2008;93:193–200.
10. Bartlett JA, Goldklang AR, Schleifer SJ, Keller SE.Immune function in healthy inner-city children.Clin Diagn Lab Immunol 2001;8:740–6.
11. Luebke RW, Chen DH, Dietert R, Yang Y,Luster MI. Immune system maturity andsensitivity to chemical exposure. J ToxicolEnvironmental Health A: Current Issues 2006;69:811–25.
12. Erkeller-Yuksel FM, Deneys V, Yuksel B, HannetI, Hulstaert F, Hamilton C, Mackinnon H, StokesLT, Munhyeshuli V, Vanlangendonck F, et al.Age-related changes in human bloodlymphocyte subpopulations. J Pediatr 1992;120:216–22.
13. Heldrup J, Kalm O, Prellner K. Blood T and Blymphocyte subpopulations in healthy infantsand children. Acta Paediatr 1992;81:125–32.
14. Sharma G, Hanania NA, Shim YM. The agingimmune system and its relationship to the
development of chronic obstructive pulmonarydisease. Proc Am Thorac Soc 2009;6:573–80.
15. Weng NP, Akbar AN, Goronzy J. CD28(-) Tcells: their role in the age-associated decline ofimmune function. Trends Immunol 2009;30:306–12.
16. Simone R, Zicca A, Saverino D. The frequency ofregulatory CD3!CD8!CD28- CD25! Tlymphocytes in human peripheral blood increaseswith age. J Leukoc Biol 2008;84:1454–61.
17. Baumforth KR, Birgersdotter A, Reynolds GM,Wei W, Kapatai G, Flavell JR, Kalk E, Piper K,Lee S, Machado L, Hadley K, Sundblad A, et al.Expression of the Epstein-Barr virus-encodedEpstein-Barr virus nuclear antigen 1 in Hodgkin’slymphoma cells mediates Up-regulation ofCCL20 and the migration of regulatory T cells.Am J Pathol 2008;173:195–204.
18. Chetaille B, Bertucci F, Finetti P, Esterni B,Stamatoullas A, Picquenot JM, Copin MC,Morschhauser F, Casasnovas O, Petrella T,Molina T, Vekhoff A, et al. Molecular profiling ofclassical Hodgkin lymphoma tissues uncoversvariations in the tumor microenvironment andcorrelations with EBV infection and outcome.Blood 2009;113:2765–3775.
19. Heller KN, Arrey F, Steinherz P, Portlock C,Chadburn A, Kelly K, Munz C. Patients withEpstein Barr virus-positive lymphomas havedecreased CD4(!) T-cell responses to the viralnuclear antigen 1. Int J Cancer 2008;123:2824–31.
20. Barros MH, Scheliga A, De Matteo E, MinnicelliC, Soares FA, Zalcberg IR, Hassan R. Cell cyclecharacteristics and Epstein-Barr virus aredifferentially associated with aggressive and non-aggressive subsets of Hodgkin lymphoma in
Tum
orIm
mun
olog
y
Barros et al. 1151
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC

pediatric patients. Leuk Lymphoma 2010;51:1513–22.
21. Faria AM, de Moraes SM, de Freitas LH, SpezialiE, Soares TF, Figueiredo-Neves SP, Vitelli-AvelarDM, Martins MA, Barbosa KV, Soares EB,Sathler-Avelar R, Peruhype-Magalhaes V, et al.Variation rhythms of lymphocyte subsets duringhealthy aging. Neuroimmunomodulation 2008;15:365–79.
22. Lister TA, Crowther D, Sutcliffe SB, Glatstein E,Canellos GP, Young RC, Rosenberg SA, ColtmanCA, Tubiana M. Report of a committee convenedto discuss the evaluation and staging of patientswith Hodgkin’s disease: Cotswolds meeting. J ClinOncol 1989;7:1630–6.
23. Pizzo PA, Poplack DG. Principles and practice ofpediatric oncology, 3rd edn. Philadelphia:Lippincott-Raven, 1997.
24. Fryer CJ, Hutchinson RJ, Krailo M, Collins RD,Constine LS, Hays DM, Heller RM, Davis PC,Nachman J, O’Brien RT, et al. Efficacy andtoxicity of 12 courses of ABVD chemotherapyfollowed by low-dose regional radiation inadvanced Hodgkin’s disease in children: a reportfrom the Children’s Cancer Study Group. J ClinOncol 1990;8:1971–80.
25. Schellong G, Potter R, Bramswig J, Wagner W,Prott FJ, Dorffel W, Korholz D, Mann G, Rath B,Reiter A, Weissbach G, Riepenhausen M, et al.High cure rates and reduced long-term toxicity inpediatric Hodgkin’s disease: the German-Austrian multicenter trial DAL-HD-90. TheGerman-Austrian Pediatric Hodgkin’s DiseaseStudy Group. J Clin Oncol 1999;17:3736–44.
26. Zhou XG, Sandvej K, Li PJ, Ji XL, Yan QH,Zhang XP, Da JP, Hamilton-Dutoit SJ. Epstein-Barr virus (EBV) in Chinese pediatric Hodgkindisease: Hodgkin disease in young children is anEBV-related lymphoma. Cancer 2001;92:1621–31.
27. Schreck S, Friebel D, Buettner M, Distel L,Grabenbauer G, Young LS, Niedobitek G.Prognostic impact of tumour-infiltrating Th2 andregulatory T cells in classical Hodgkinlymphoma. Hematol Oncol 2009;27:31–9.
28. Barros MH, Hassan R, Niedobitek G. Diseasepatterns in pediatric classical Hodgkinlymphoma: a report from a developing area inBrazil. Hematol Oncol 2011, in press.
29. Frisan T, Sjoberg J, Dolcetti R, Boiocchi M, DeRe V, Carbone A, Brautbar C, Battat S, BiberfeldP, Eckman M, et al. Local suppression of Epstein-Barr virus (EBV)-specific cytotoxicity in biopsiesof EBV-positive Hodgkin’s disease. Blood 1995;86:1493–501.
30. Hudnall SD, Betancourt E, Barnhart E, Patel J.Comparative flow immunophenotypic features ofthe inflammatory infiltrates of Hodgkinlymphoma and lymphoid hyperplasia. CytometryB Clin Cytom 2008;74:1–8.
31. Steidl C, Lee T, Shah SP, Farinha P, Han G,Nayar T, Delaney A, Jones SJ, Iqbal J,Weisenburger DD, Bast MA, Rosenwald A, et al.Tumor-associated macrophages and survival inclassic Hodgkin’s lymphoma. N Engl J Med 2010;362:875–85.
32. Lee WS, Park S, Lee WY, Yun SH, Chun HK.Clinical impact of tumor-infiltrating lymphocytesfor survival in stage II colon cancer. Cancer 2010;116:5188–99.
33. Deschoolmeester V, Baay M, Van Marck E,Weyler J, Vermeulen P, Lardon F, Vermorken JB.Tumor infiltrating lymphocytes: an intriguingplayer in the survival of colorectal cancerpatients. BMC Immunol 2010;11:19.
34. Baumforth KR, Young LS, Flavell KJ,Constandinou C, Murray PG. The Epstein-Barrvirus and its association with human cancers.Mol Pathol 1999;52:307–22.
35. Oudejans JJ, Jiwa NM, Kummer JA, Horstman A,Vos W, Baak JP, Kluin PM, van der Valk P,Walboomers JM, Meijer CJ. Analysis of majorhistocompatibility complex class I expression onReed-Sternberg cells in relation to the cytotoxicT-cell response in Epstein-Barr virus-positive and-negative Hodgkin’s disease. Blood 1996;87:3844–51.
36. Murray PG, Constandinou CM, Crocker J, YoungLS, Ambinder RF. Analysis of majorhistocompatibility complex class I, TAPexpression, and LMP2 epitope sequence inEpstein-Barr virus-positive Hodgkin’s disease.Blood 1998;92:2477–83.
37. Lee SP, Constandinou CM, Thomas WA, Croom-Carter D, Blake NW, Murray PG, Crocker J,Rickinson AB. Antigen presenting phenotype ofHodgkin Reed-Sternberg cells: analysis of the HLAclass I processing pathway and the effects ofinterleukin-10 on epstein-barr virus-specificcytotoxic T-cell recognition. Blood 1998;92:1020–30.
38. Izban KF, Ergin M, Martinez RL, Alkan S.Expression of the tumor necrosis factor receptor-associated factors (TRAFs) 1 and 2 is acharacteristic feature of Hodgkin and Reed-Sternberg cells. Mod Pathol 2000;13:1324–31.
39. Brink AA, Oudejans JJ, van den Brule AJ, KluinPM, Horstman A, Ossenkoppele GJ, van HeerdeP, Jiwa M, Meijer CJ. Low p53 and high bcl-2expression in Reed-Sternberg cells predicts poorclinical outcome for Hodgkin’s disease:involvement of apoptosis resistance? Mod Pathol1998;11:376–83.
40. Durkop H, Hirsch B, Hahn C, Stein H. cIAP2 ishighly expressed in Hodgkin-Reed-Sternberg cellsand inhibits apoptosis by interfering withconstitutively active caspase-3. J Mol Med (Berl)2006;84:132–41.
41. Juszczynski P, Ouyang J, Monti S, Rodig SJ,Takeyama K, Abramson J, Chen W, Kutok JL,
Rabinovich GA, Shipp MA. The AP1-dependentsecretion of galectin-1 by Reed Sternberg cellsfosters immune privilege in classical Hodgkinlymphoma. Proc Natl Acad Sci USA 2007;104:13134–9.
42. Gandhi MK, Lambley E, Duraiswamy J, Dua U,Smith C, Elliott S, Gill D, Marlton P, Seymour J,Khanna R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with thesuppression of latent membrane antigen-specificCD8! T-cell function in Hodgkin lymphomapatients. Blood 2006;108:2280–9.
43. Marshall NA, Culligan DJ, Tighe J, JohnstonPW, Barker RN, Vickers MA. Therelationships between Epstein-Barr virus latentmembrane protein 1 and regulatory T cells inHodgkin’s lymphoma. Exp Hematol 2007;35:596–604.
44. Chapman AL, Rickinson AB, Thomas WA,Jarrett RF, Crocker J, Lee SP. Epstein-Barr virus-specific cytotoxic T lymphocyte responses in theblood and tumor site of Hodgkin’s diseasepatients: implications for a T-cell-based therapy.Cancer Res 2001;61:6219–26.
45. Stark GL, Wood KM, Jack F, Angus B, ProctorSJ, Taylor PR. Hodgkin’s disease in the elderly: apopulation-based study. Br J Haematol 2002;119:432–40.
46. Jarrett RF, Stark GL, White J, Angus B,Alexander FE, Krajewski AS, Freeland J, TaylorGM, Taylor PR. Impact of tumor Epstein-Barrvirus status on presenting features and outcomein age-defined subgroups of patients with classicHodgkin lymphoma: a population-based study.Blood 2005;106:2444–51.
47. Diepstra A, van Imhoff GW, Schaapveld M,Karim-Kos H, van den Berg A, Vellenga E,Poppema S. Latent Epstein-Barr virus infection oftumor cells in classical Hodgkin’s lymphomapredicts adverse outcome in older adult patients.J Clin Oncol 2009;27:3815–21.
48. Keegan TH, Glaser SL, Clarke CA, Gulley ML,Craig FE, Digiuseppe JA, Dorfman RF, MannRB, Ambinder RF. Epstein-Barr virus as amarker of survival after Hodgkin’s lymphoma: apopulation-based study. J Clin Oncol 2005;23:7604–13.
49. Engel M, Essop MF, Close P, Hartley P, PallesenG, Sinclair-Smith C. Improved prognosis ofEpstein-Barr virus associated childhoodHodgkin’s lymphoma: study of 47 South Africancases. J Clin Pathol 2000;53:182–6.
50. Ouyang J, Juszczynski P, Rodig SJ, Green MR,O’Donnell E, Currie T, Armant M, TakeyamaK, Monti S, Rabinovich GA, Ritz J, Kutok JL,et al. Viral induction and targeted inhibitionof galectin-1 in EBV! posttransplantlymphoproliferative disorders. Blood 2011;117:4315–22.
Tum
orIm
mun
olog
y
1152 Tumor Microenvironment in Pediatric Classical Hodgkin Lymphoma
Int. J. Cancer: 131, 1142–1152 (2012) VC 2011 UICC


















