
Estudo de dois membros da família das proteínas da...
58
Universidade de Lisboa Faculdade de Ciências Departamento de Biologia Vegetal Estudo de dois membros da família das proteínas da membrana externa de Helicobacter pylori, hopM e hopN Andrea Sofia Rebelo Santos Mestrado em Biologia Molecular e Genética 2011
-
Upload
trinhthuan -
Category
Documents
-
view
213 -
download
0
Transcript of Estudo de dois membros da família das proteínas da...

Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Estudo de dois membros da família das
proteínas da membrana externa de
Helicobacter pylori, hopM e hopN
Andrea Sofia Rebelo Santos
Mestrado em Biologia Molecular e Genética
2011


Universidade de Lisboa
Faculdade de Ciências
Departamento de Biologia Vegetal
Estudo de dois membros da família das
proteínas da membrana externa de
Helicobacter pylori, hopM e hopN
Andrea Sofia Rebelo Santos
Mestrado em Biologia Molecular e Genética
Dissertação orientada pela Doutora Mónica Oleastro do Instituto
Nacional de Saúde Dr. Ricardo Jorge e pelo Professor Doutor Pedro
Silva da Faculdade de Ciências da Universidade de Lisboa.
2011

A presente dissertação não foi redigida segundo o novo Acordo
Ortográfico da Língua Portuguesa por opção do autor.


Índice
ÍNDICE:
Agradecimentos……………………………………………………………………………………....i
Lista de Abreviaturas…………………………………………………………………………….…ii
Lista de Figuras………………………………………………………………..……………………iii
Lista de Tabelas……………………………………………………………………………………..iv
Resumo…………………………………………………………………………..……………………v
Abstract……………………………………………………………………………………………….vi
Capítulo 1: Introdução……………………………………………………………………………1
1. Helicobacter pylori- considerações gerais………..…………….…………………….…..1
2. Características da infecção por Helicobacter pylori…..…………………….……….….1
3. Diversidade Genética de Helicobacter pylori…………………………………….……..…2
4. Diversidade Geográfica de Helicobacter pylori…………………………………….…..…3
5. Factores de virulência de Helicobacter pylori……………………………………..……...4
5.1. Citotoxina vacuolizanteVacA…………………………………………………………….4
5.2. Ilhéu de patogenicidade cag……………………………………………………………..5
5.3. Proteínas da membrana externa………………………………………………………..5
5.3.1 Mecanismos de regulação da expressão das proteínas de membrana externa…7
Capítulo 2: Objectivos……………………………………………………………………………9
Capítulo3: Materiais e Métodos………………………………………………………………10
1. Estirpes de Helicobacter pylori e material genómico……………………………..…...10

Índice
2. Avaliação da diversidade genómica dos genes hopM e hopN…………………….....10
3. Avaliação da variabilidade genética, análise filogenética e parâmetros
evolutivos dos genes hopM e hopN…………………………………….…………………12
4. Análise Estatística…………………………………………………………………………….12
Capítulo 4: Resultados………………………………………………………………………….13
1. Avaliação da diversidade genómica dos genes hopM e hopN………………...……..13
1.1. Distribuição dos genes hopM e hopN por região geográfica…………………………..….14
1.2. Distribuição dos genes hopM e hopN por patologia………………………………………..14
1.3. Associação de hopM e hopN com o genótipo de virulência……………………………… 16
2. Análise filogenétiva, avaliação da variabilidade e parâmetros evolutivos dos
genes hopM e hopN……………………………………………………………………………....16
3. Variação Alélica………………………………………………………………………………….20
3.1 Distribuição dos alelos por gene e origem geográfica…………………………………22
3.2 Distribuição dos alelos dos genes hopM e hopN de acordo com a patologia
e perfil de virulência………………………………………………………………………………23
3.2.1 Distribuição dos alelos por patologia…………………………………………………..23
3.2.2 Distribuição dos alelos por perfil de virulência………………………………………..24
Capítulo 5: Discussão e Conclusão……………………………………………….…..26
Bibliografia…………………………………………………………………...……………….30
Anexos…………………………………………………………………………………….…….37

Agradecimentos
Andrea Santos i
Em primeiro lugar, não poderia deixar de agrader à Mónica Oleastro pela oportunidade
dada para a realização deste trabalho, pela orientação desta tese e pelo apoio científico
proporcionado ao longo destes 7 anos de trabalho.
Ao Professor Pedro Silva pela sua disponibilidade e ajuda nos momentos difíceis. A sua
orientação e a luz que trouxe à minha mente foram sem dúvida essenciais para a
elaboração desta tese.
Ao meu amigo e colega de trabalho João Benoliel, pela amizade, ajuda e apoio
oferecidos ao longo destes 7 anos de convivência. Sem ti, todo este trabalho seria bem mais
difícil e certamente ainda não teria terminado. Obrigada pela paciência para ouvires os meus
desabafos, pelas inúmeras palavras de incentivo e por assegurares o laboratório por mim,
libertando-me para a dedicação “quase” exclusiva à minha tese.
À Rita Cordeiro pela sua amizade, dedicação, orientação e pela imensa paciência
demonstradas durante a elaboração deste trabalho. Obrigada pela tua disponibilidade e
conhecimentos transmitidos durante todo o processo de desenvolvimento desta tese. Sem ti
nada disto seria possível, MUITO OBRIGADA!!
À Alexandra Nunes pela sua disponibilidade e ajuda na interpretação de alguns
resultados.
À Ana Pelerito, pela amizade, pelos sorrisos e momentos de descontracção
proporcionados nos momentos de trabalho.
A todos os meus amigos pela compreensão, apoio e amizade. Obrigada por fazerem
parte da minha vida!
Por fim, mas seguramente com maior importância…
Ao Nuno, por todo o amor, carinho e compreensão mas principalmente pelo seu grande
apoio e força nos momentos mais difíceis. És o meu porto de abrigo, quem me apazigua e
acalma…Obrigada meu querido!
À minha querida maninha, pelo seu carinho e incentivo ao longo de toda a nossa vida.
Aos meus queridos Pais, que ao longo de toda a minha caminhada têm estado ao meu
lado, incentivando e apoiando cada passo que tenho dado. Sem vocês nada disto seria
possível e agradeço todos os sacrifícos que fizeram para que chegasse até aqui…. Vocês
são o exemplo que pretendo seguir na minha vida… Este trabalho é dedicado a vocês!

Lista de Abreviaturas
Andrea Santos ii
Lista de Abreviaturas:
ADN Ácido Desoxirribonucleico
Ba bA Blood group antigen binding adhesin
cagA cytotoxin associated gene A
cag PAI ilhéu de patogenicidade cag
CG Cancro gástrico
CT Citosina/Timina
DDI Departamento de doenças infecciosas
EUA Estados Unidos da América
G Gastrite
G+C Guanina+Citosina
H.pylori Helicobacter pylori
IL-8 Interleucina-8
INSA Instituto Nacional de Saúde Dr. Ricardo Jorge
Ka Substituições não sinónimas
Kb kilobases
kDA Kilodaltons
Ks Substituições sinónimas
Leb Lewis B Blood Group
MALT Mucosa associated lymphoid tissue
MLST Multilocus sequence typing
min. Minuto
N.D. Não definido
OipA Outer inflammatory protein A
OMPs Outer membrane protein
ORF Open reading frame
pb Pares de bases
PCR Polimerase chain reaction
s Segundo
SabA Sialic acid binding adhesin
SST4 Sistema de secreção do tipo IV
UP Úlcera péptica
vacA Vacuolating cytotoxin gene A

Lista de Figuras
Andrea Santos iii
LISTA DE FIGURAS
Figura 1: Análise filogenética das cinco famílias de parólogos da estirpe de referência da
estirpe de referência J99.-------------------------------------------------------------------------------------6
Figura 2: Distribuição do número de cópias e localização genómica de hopM e hopN, por
região geográfica.------------------------------------------------------------------------------------------------14
Figura 3: Frequência dos genes hopM e hopN nas estirpes de Helicobacter pylori de origem
ocidental e asiática de acordo com a patologia.---------------------------------------------------------15
Figura 4: Análise filogenética de 379 sequências de Helicobacter pylori, 147 do gene hopM
e 232 do gene hopN.------------------------------------------------------------------------------------------17
Figura 5: Representação gráfica da similaridade entre as 379 sequências de nucleótidos
dos genes hopM e hopN de estirpes de Helicobacter pylori.-----------------------------------------18
Figura 6: Representação gráfica da similaridade entre 147 sequências de nucleótidos do
gene hopM, identificando os 4 alelos.--------------------------------------------------------------------21
Figura 7: Alelos hopM e hopN. (A) Alinhamento representativo das variações ao nível das
sequências de aminoácidos do segmento 4 de hopM e hopN. (B) Esquema representativo
dos 4 alelos demonstrando que as variantes AIII e AIV são putativos recombinantes das
variantes AI e AII. -----------------------------------------------------------------------------------------------21
Figura 8: Gráficos de similaridade entre sequências de nucleótidos do gene hopM por
variante alélica. --------------------------------------------------------------------------------------------------21
Figura 9: Distribuição dos alelos por gene, na população geral. -----------------------------------22
Figura 10: Distribuição dos alelos de cada gene de acordo com a origem geográfica das
estirpes de Helicobacter pylori em estudo. ---------------------------------------------------------------22

Lista de Figuras
Andrea Santos iii
Figura 11: Frequência das quatro variantes alélicas de acordo com a patologia, na
população geral (A) e por origem geografia (B e C). ------------------------------------------------23
Figura 12: Frequência dos alelos hopM+hopN de acordo com genótipo de virulência da
estirpe na população geral. -----------------------------------------------------------------------------------24
Figura 13: Distribuição das quatro variantes alélicas na população ocidental, considerando
os genes em conjunto (A) e separados (B e C) de acordo com o genótipo de virulência das
estirpes de Helicobacter pylori. ------------------------------------------------------------------------------25

Lista de Tabelas
Andrea Santos iV
LISTA DE TABELAS:
Tabela 1: Primers utilizados na amplificação e sequenciação dos loci hopM e hopN. -------11
Tabela 2: Análise das distâncias moleculares, substituições sinónimas e não sinónimas para
as sequências correspondentes ao gene completo e segmentos 1,2,3 e 4 dos genes hopM e
hopN. ---------------------------------------------------------------------------------------------------------------19
Tabela 3: Análise das percentagens de identidade obtidas entre as sequências do segmento
4 de cada um das variantes alélicas de hopM e hopN. -----------------------------------------------20

Resumo
Andrea Santos V
RESUMO
Neste trabalho pretendeu-se estudar a diversidade genética e evolução dos genes hopM
e hopN, que codificam proteínas de membrana externa de H. pylori, bem como contribuir
para o esclarecimento do seu papel na virulência desta bactéria, através da identificação de
possíveis associações a patologias específicas e a marcadores moleculares de virulência
(cagA e vacAs1).
Foi utilizado um painel de 234 estirpes clínicas e sequências de 23 estirpes de referência,
isoladas de doentes de origem geográfica diferente e com diversas patologias gástricas.
A reconstrução filogenética destes dois genes sugere evolução divergente entre eles. Em
cada um dos genes, foram identificados dois clusters predominantes, sendo a segregação
entre eles motivada pela origem geográfica das estirpes, havendo uma diferenciação clara
entre as estirpes de origem ocidental e as de origem asiática.
Foi observada diversidade relativamente ao número de cópias de cada gene e à sua
localização genómica, sendo esta diferença notória entre grupos geográficos diferentes. Na
população ocidental ambos os genótipos, de cópia única e dupla cópia (mesmo gene ou
genes diferentes), foram observados. Contrariamente, na população asiática apenas foram
observados os genótipos de cópia única, enquanto no grupo africano apenas os genótipos
de dupla cópia foram apresentados.
Apesar de nenhum destes genótipos ser marcador da doença gastroduodenal, observou-
se uma associação estatisticamente significativa, na população ocidental, entre a dupla
cópia hopN e o genótipo mais virulento, e entre a dupla cópia hopM e o genótipo menos
virulento. No grupo de origem oriental, não se observou nenhuma associação
estatisticamente significativa com o perfil de virulência da estirpe.
A análise das sequências dos dois genes sugere a sua regulação por variação de fase e
ainda a possibilidade da ocorrência de fenómenos de recombinação intragenómica ou
intergenómica, podendo os mecanismos de duplicação ou conversão génica estar
implicados na regulação da sua expressão.
Os genes hopM e hopN revelaram um elevado grau de similaridade entre si, com uma
distância molecular de 0,146±0,006 ao nível da sequência de nucleótidos e de 0,161±0,011
ao nível da sequência de aminoácidos. Dentro de cada gene, verificou-se um grau de
conservação ainda superior, com distâncias moleculares e taxas de substituições de
nucleótidos muito similares, quando comparados os valores obtidos para cada gene.
Verificou-se ainda uma taxa superior de substituições sinónimas (Ks) do que não sinónimas
(Ka), sendo a razão Ka/Ks inferior a um, o que aponta para a existência de uma pressão

Resumo
Andrea Santos V
evolutiva para a conservação destes genes, através da acção da selecção negativa ou
purificadora.
Foram identificadas quatro variantes alélicas, comuns a ambos os genes, designadas por
AI, AII, AIII e AIV. No geral, foram observados dois alelos predominantes, os alelos AI
(39,2%) e o AII (42%,), enquanto os alelos AIII e AIV, putativos recombinantes dos alelos AI
e AII, foram observados com menor frequência nas estirpes estudadas (12,9% e 5,8%,
respectivamente). Na população ocidental e africana todos os alelos foram observados,
sendo o mais prevalente o alelo AI (49,8% e 69,6%, respectivamente). Nestas populações
não se verificou nenhuma associação entre as variantes alélicas e a patologia. No entanto,
na população ocidental, os alelos AII e AIII foram significativamente associados com o
genótipo de virulência da estirpe, mais concretamente o alelo AII com o genótipo menos
virulento e o AIII com o genótipo mais virulento. Na população asiática, o alelo AII foi o mais
prevalente (85,2%) e o alelo AIV não foi observado. Nesta população, não se verificou
nenhuma associação com suporte estatístico, entre as variantes alélicas e a patologia
gástrica ou o grau de virulência da estirpe.
Globalmente estes resultados sugerem que hopM e hopN fazem parte do grupo de genes
que codificam OMPs de H. pylori implicadas tanto na interacção da bactéria com o seu
hospedeiro humano, como na variação antigénica, contribuindo para a persistência e
sucesso da infecção. Mais, as variantes alélicas podem constituir biomarcadores de
virulência da estirpe em determinadas populações.
Palavras-chave: Helicobacter pylori; OMPs; hopM; hopN.

Abstract
Andrea Santos vi
ABSTRACT
This work aimed to study the genetic diversity and evolution of hopM and hopN genes,
which encode outer membrane proteins of H. pylori, as well as contribute to the clarification
of its role in the virulence of this bacterium, by identifying possible associations to specific
diseases and virulence markers (cagA and vacAs1).
We used a panel of 234 clinical strains and of 23 reference strains sequences isolated
from patients from different geographical origins and presenting different gastroduodenal
pathologies. The phylogenetic reconstruction of these two genes suggests a divergent
evolution between them. It also revealed a geographic segregation in each gene, with two
dominant clusters, with a clear differentiation among strains of Western and East Asian
origin.
Diversity was observed regarding the number of copies and their genomic localization,
with East Asian and Western strains presenting different genotypes. In the Western strains
both, the single and double copy genotypes were observed. In contrast, in the East Asian
group, only the single copy was presented, while in the African group only the double copy
genotype was observed.
While no association was found between these genotypes and gastroduodenal disease,
statistically significant associations between the hopN double copy genotype and the more
virulent profile, and between the hopM double copy genotype and the less virulent profile,
were observed in the Western group. In the East Asian group, there was no association
between hopM/hopN genotypes and the virulence profile of the strain.
The sequence analysis of these genes suggests regulation by phase variation. It also
indicates possible recombination events, leading to gene duplication or hopM/hopN
conversion which may be implicated in the regulation of these genes as well.
The hopM and hopN genes revealed a high degree of similarity between them, with a
moelcular distance of 0,146±0,006 at nucleotide level and of 0,161±0,011 at the aminoacid
level. Within each gene, there was an even higher degree of conservation, with very close
molecular distances and rates of nucleotide substitutions, when comparing the values
obtained for each gene. It was observed a higher level of synonymous substitutions (Ks) than
non-synonymous (Ka), and the ratio Ka/Ks<1, suggests an evolutionary pressure to maintain
the two genes by negative or purifying selection.
Four allelic variantes were identified in both genes and in all strains studied although with
different frequencies. In general, two dominant alleles were observed, alleles AI (39,3%) and
AII (42%), and two less predominant, putative recombinants of the dominant variants, alleles

Abstract
Andrea Santos vi
AIII (12,9%) e AIV (5,8%). In Western and in African strains all the alleles were observed,
althoug the AI allele was the most frequently found (49,8% and 69,6%, respectively). In these
populations, there were no correlation between any allele and disease. However in the
Western group, AII and AIII alleles were statiscally associated with the virulence genotype of
the strain, more specifically, the AII allele with the less virulent genotype and AIII with the
most virulent genotype. In East Asian strains, the allele AII was the most prevalent (85,2%)
and the allele AIV was not observed. In this group, despite the apparent associations
between some allelic variants and certain diseases, no statistical association was observed,
nor between hopM/hopN alleles and the virulence genotype of the strain.
Overall, the results of this study suggest that hopM and hopN are good candidates to be
part of the pool of H. pylori OMPs implicated in host-bacteria interface and also contributing
to the generation of antigenic variability, and thus involved in persistence and success of H.
pylori infection. Moreover, some allelic variants may constitute biomarkers of the virulence of
the strain in some populations.
Keywords: Helicobacter pylori; OMPs; hopM; hopN.

Introdução
Andrea Santos
1
Capítulo1: Introdução
1. Helicobacter pylori- Considerações gerais
Helicobacter pylori (H. pylori) é uma bactéria Gram negativa espiralada que coloniza o
estômago humano [1,2]. Possui cinco a sete flagelos unipolares com bainha e extremidades
bulbares [3,4], essenciais para a motilidade e sobrevivência no estômago.
A primeira referência a esta bactéria, remonta a 1893 por Bizzozero, o qual descreve a
presença de “espiroquetas” no estômago de gatos e cães [5,6]. Desde essa data e até aos
anos oitenta, vários relatos foram feitos relativamente à presença deste microrganismo em
tecido gástrico, nomeadamente de doentes com úlcera péptica e cancro gástrico ou de
amostras colhidas post-mortem [4,5,7]. Só em 1983, o gastrenterologista Barry Marshall e o
patologista Robert Warren conseguem a primeira cultura bacteriana a partir de uma amostra
gástrica. Em 1989, é então criado o género Helicobacter [3,8].
Este género é actualmente composto por mais de 27 espécies [9,10] todas microaerófilas
[9]. A maioria das espécies são catalase e oxidase positivas e algumas, como H. pylori, são
também urease positiva [9]. A colonização do estômago e do tracto intestinal pelo género
Helicobacter é ubiquitária no reino animal, sendo as espécies intestinais incapazes de
colonizar o estômago e vice-versa. Há assim uma divisão em dois grupos dentro do género,
definidos de acordo com o seu tropismo para determinados órgãos [11].
H.pylori tem como nicho o epitélio gástrico humano e de outros primatas [11]. Apesar de
conseguir sobreviver em condições de pH bastante ácidas (pH<5), normalmente coloniza o
antro gástrico (parte inferior) onde a produção de ácido é menor. A sobrevivência deste
microrganismo nesta gama de pH deve-se à presença da enzima urease, que hidrolisa a
ureia em amoníaco e dióxido de carbono, levando à tamponização do microambiente que
rodeia a bactéria [2]. Mais concretamente, H.pylori localiza-se abaixo da camada de muco, à
superfície das células epiteliais, podendo encontrar-se aderente ou móvel e utiliza o pH do
muco como orientação quimiotática [2,12]. Dados mais recentes sugerem que pode também
invadir as células da mucosa gástrica, alternando entre o espaço intra e extracelular, aptidão
que pode estar implicada na persistência e patogenicidade de H.pylori [13].
2.Características da Infecção por Helicobacter pylori
O conhecimento actual sugere que a aquisição de H. pylori ocorre predominantemente na
infância, sendo o contágio intrafamiliar a forma mais comum de transmissão [14-16]. Uma
vez estabelecida no estômago, a infecção irá persistir durante toda a vida do hospedeiro se
não for tratada, levando ao aparecimento de doença gástrica severa, com as manifestações
clínicas a sobrevir sobretudo na idade adulta [17].

Introdução
Andrea Santos
2
Os principais factores de risco relacionados com a aquisição da infecção incluem práticas
sanitárias inadequadas, classe social mais baixa, elevada densidade populacional, entre
outros [14,17-19]. A prevalência mundial da infecção tem por isso uma variação bastante
acentuada entre países desenvolvidos e em desenvolvimento. Nos países em
desenvolvimento a taxa de infecção ronda os 50% nas crianças e mais de 90% dos adultos,
enquanto nos países desenvolvidos tem vindo a diminuir, atingindo cerca de 20-50% dos
adultos e 10% das crianças [20,21]. Em Portugal, a prevalência desta infecção atinge
valores muito próximos dos observados nos países em desenvolvimento, atingindo cerca de
80% da população adulta e variando entre aproximadamente 20% nas crianças até aos 5
anos e 50% nas crianças com idades compreendida entre 10 e 15 anos [22-24].
Vários estudos sugerem que a transmissão directa, pessoa a pessoa, é a via mais
provável de aquisição da infecção, quer seja por via oral-oral, gástrica-oral ou fecal-oral,
tendo sido detectado ADN de H. pylori no vómito, saliva, placa dentária, suco gástrico e
fezes [9,14,15,17,21,25,26]. Foram também sugeridas outras vias de transmissão, como
através da água, de animais e de endoscópios esterilizados incorrectamente [9,14,17,21,27-
31].
Clinicamente esta infecção é caracterizada pela presença de dores abdominais
persistentes, dores epigástricas pós-prandiais, vómitos e indigestão. A análise histológica da
biopsia gástrica destes doentes revela sempre a presença de gastrite crónica, que pode
evoluir para doença severa (15-20% dos indivíduos infectados), como úlcera duodenal ou
úlcera gástrica [17,32,33]. Menos frequentemente (cerca de 0,1- 4% dos casos) [17], a
infecção por H.pylori pode também progredir para carcinoma gástrico ou para o linfoma de
MALT (Mucosa Associated Lymphoid Tissue). No desenvolvimento destas patologias, para
além da infecção por H. pylori, os factores genéticos do hospedeiro e os factores
ambientais, desempenham um papel preponderante [17,34,35].
3. Diversidade Genética de Helicobacter pylori
Durante a longa convivência com os seres humanos, H. pylori tem desenvolvido
estratégias complexas de adaptação, no sentido de manter a inflamação do epitélio gástrico
num limiar que impeça a sua própria eliminação [17,32]. Esta adaptação constante a um
hospedeiro específico, traduz-se na grande diversidade encontrada nas estirpes de H. pylori
e são o reflexo das inúmeras alterações genómicas sofridas pela bactéria, essencialmente
por mecanismos de recombinação e mutação, durante o longo período de infecção [36,37].
Vários métodos moleculares permitiram demonstrar que é quase impossível encontrar dois
isolados independentes de H. pylori com perfis genómicos idênticos, o que sugere uma das
maiores variabilidades genómicas conhecidas entre as bactérias [36,37,38].

Introdução
Andrea Santos
3
Actualmente existem 31 genomas de H.pylori totalmente sequenciados e anotados,
obtidos de isolados clínicos independentes. A sua comparação permitiu verificar que cerca
de 10% dos genes presentes no genoma de H.pylori são específicos de estirpe [39-48] e
estão localizados em duas regiões genómicas denominadas de zonas de plasticidade 1 e 2
[44,49]. Estas regiões têm um conteúdo em G+C diferente do restante genoma (35%G+C,
em vez de 39%-percentagem média de G+C do restante genoma), o que sugere que a sua
aquisição possa ter resultado de transferência horizontal. As zonas de plasticidade contêm
várias sequências de inserção, bem como genes responsáveis pela transcrição de proteínas
com elevada homologia com algumas recombinases, integrases e topoisomerases. A
presença destas sequências sugere que estas regiões genómicas estariam implicadas em
eventos de recombinação, incluindo a transposição, levando a rearranjos genómicos entre
as estirpes [39,44,47,49].
A recombinação genética parece ser o mecanismo mais envolvido na adaptação desta
bactéria ao seu hospedeiro, resultando da troca de genes entre estirpes em colonização
mista [50-53]. Este mecanismo é possível porque H. pylori é naturalmente competente,
permitindo a entrada de ADN exógeno, cromossomal ou plasmídico, de outras estirpes de H.
pylori, normalmente sob a forma de pequenos fragmentos (417 pares de bases(pb)), que
são integrados no cromossoma [52,53]. A ocorrência de recombinação intragenómica,
também já foi descrita em H. pylori, especialmente em genes da família das proteínas de
membrana externa (OMPs, Outer membrane protein), levando a variações genómicas na
ausência de colonização mista [54,55].
As mutações pontuais são outro mecanismo que promove a elevada diversidade genética
de H. pylori, estando envolvido, por exemplo, no desenvolvimento de resistências aos
antibióticos [51,56]. Muitos genes identificados neste microrganismo contêm sequências
repetidas, homopoliméricas ou dinucleótidicas, que podem constituir locais preferenciais
para a ocorrência de mutações (hotspots), geradas pelo mecanismo de slipped strand
misparing, durante a replicação do ADN [39-48,57,58]. Este evento constitui um mecanismo
de regulação da expressão genética, alterando a funcionalidade dos genes ou afectando a
actividade do promotor, através da alteração da grelha de leitura desses genes [59,60].
Alguns autores sugerem que a elevada taxa de mutação desta bactéria, poderá dever-se
também à aparente ausência dos sistemas de reparação do ADN [36,61].
4. Diversidade geográfica de Helicobacter pylori
A comparação de sequências de várias categorias de genes de H. pylori, de diferentes
regiões geográficas, tem vindo a permitir identificar alelos distintos em determinados genes,
específicos ou mais expressos numa determinada região geográfica [36,38,53,62,63].
Assumindo que a infecção por H. pylori ocorre no seio familiar e que cada estirpe se adapta

Introdução
Andrea Santos
4
a um hospedeiro específico, é provável que a evolução desta bactéria tenha vindo a ocorrer
de forma particular em diferentes regiões geográficas [36,38,53]. Alguns autores sugerem
que os diferentes isolados analisados, reflectem tanto, as características iniciais da
população bacteriana que foi introduzida numa determinada região, como as pressões
antigénicas ou variações funcionais a que esteve sujeita, para persistir numa dada
população hospedeira [45,53,64]. Vários autores têm por isso utilizado as subdivisões
geográficas das estirpes de H. pylori, para traçar as migrações modernas e ancestrais do
Homem [53,65]. De uma forma geral, apesar da elevada taxa de recombinação observada,
as espécies de H. pylori podem ser divididas em seis populações e várias subpopulações
com distribuição geográfica distinta, baseadas nas diferenças de perfis de MLST (Multilocus
sequence typing) considerando sete genes housekeeping. Estas populações são
designadas por: hpEuropa (Europa e Países colonizados por Europeus); hpAfrica1 composta
pelas subpopulações hspWAfrica (África Oriental, África do Sul e América) e hspSAfrica
(África do Sul); hpEastAsian que é constituída pela subpopulação hspMaori (Polinésia,
Malásia e Tailândia), hspAmerind (Índios Americanos) e hspEAsia (Ásia Oriental);
hpNEAfrica (África do Norte); hpAfrica2 (população muito específica da África do Sul);
hpAsia2 (Ásia do Sul e Central) [53,65,66]. Recentemente foi identificada mais uma
população, a hpSahul que é constituída por estirpes isoladas de Aborígenes da Austrália e
de algumas tribos da Nova Zelândia [67,68].
Estas populações modernas são o resultado de dezenas de milhares de anos de
evolução conjunta com o Homem, reflectindo tanto as migrações, como o isolamento
geográfico de algumas populações humanas e as recombinações inter-estirpes [2,53,66,69].
5. Factores de virulência de Helicobacter pylori
São vários os factores de virulência já identificados em H. pylori, estando implicados tanto
na colonização e lesão do epitélio gástrico como na adaptação e sobrevivência do
microrganismo no estômago humano [9]. De entre eles destacam-se os flagelos, a enzima
urease e algumas adesinas (BabA, SabA, etc), assim como a presença da forma activa da
citotoxina vacuolizante VacA e do ilhéu de patogenicidade cag [9].
5.1 Citotoxina Vacuolizante VacA
A proteína VacA (vacuolating cytotoxin A) é uma toxina secretada por algumas estirpes
de H.pylori tendo sido inicialmente identificada devido à sua actividade vacuolizante das
células epiteliais gástricas [17,70]. A citotoxina é constituída por dois domínios, p33 e p55
que são clivados após secreção da toxina pela bactéria. O domínio p55 medeia a ligação às
células do hospedeiro, enquanto o p33, em conjunto com o N-terminal do domínio p55, é
responsável pela vacuolização celular [69,71,72]. Dependendo do pH do meio, a toxina pode

Introdução
Andrea Santos
5
apresentar-se sob a forma de oligómeros ou monómeros que são introduzidos na camada
lipídica da membrana das células epiteliais, formando canais iónicos [73,74]. Vários efeitos
celulares têm sido descritos para a proteína VacA, quer nas células epiteliais, interferindo
com a sua permeabilidade ou levando à formação de vacúolos ou à apoptose, quer em
células do sistema imunitário, interferindo com a proliferação das células T ou com a
apresentação do antigénio pelas células B [69,74-76]. Embora o gene vacA esteja presente
em 100% dos isolados de H. pylori, exibe um polimorfismo significativo que lhe confere
diferentes níveis de expressão e toxicidade [17,69,77]. Apresenta uma estrutura em
mosaico, resultante da diversidade alélica observada em três regiões distintas do gene: a
região 5’(s) que codifica o péptido sinal e para a região N-terminal da proteína madura
(alelos s1 ou s2), a região média (m) que codifica parte do domínio p55 (alelos m1 ou m2) e
a região intermédia (i) responsável pelo domínio p33 (alelos i1 ou i2) [12,69,78]. Todas as
combinações entre os alelos são possíveis, no entanto a combinação mais tóxica é a
s1m1i1 que corresponde a uma forma completamente activa da toxina, encontrada
frequentemente em estirpes isoladas de doentes com úlcera péptica e carcinoma gástrico
[17,69], sendo a combinação s2m2i2 a forma inactiva devido à presença de uma extensão
hidrofílica no N-terminal da proteína, que bloqueia as actividades de vacuolização e
formação de poros [69,79].
5.2 Ilhéu de patogenicidade cag
O ilhéu de patogenicidade cag (cag PAI) é uma região de aproximadamente 40 kb que
contém cerca de 31 genes implicados na virulência das estirpes de H.pylori [5]. Este ilhéu
pode estar ausente, presente ou não funcional, devido à interrupção do cag PAI pela
inserção de uma sequência IS605 ou por um grande fragmento de ADN cromossomal
[5,69,73].
Um cluster específico destes genes codifica componentes do sistema de secreção do tipo
IV (SST4), está envolvido no desencadear da resposta inflamatória e é responsável pela
injecção de moléculas bacterianas no interior das células do hospedeiro, como a proteína
CagA e o peptidoglicano [17,60,80-82]. O gene cagA, presente em cerca de 50-70% das
estirpes isoladas em países Ocidentais e em 90% das estirpes da Ásia Oriental, tem uma
associação epidemiológica forte com a úlcera duodenal e o carcinoma gástrico [83-87]. Ao
ser translocada para o interior das células epiteliais gástricas, a proteína CagA é fosforilada
em determinadas sequências repetidas (motivos EPIYA), por cinases do hospedeiro [88,89],
interferindo nas vias de sinalização das células epiteliais gástricas e alterando os seus
padrões morfológicos e de proliferação [90-92]. A interacção entre o SST4 e as células
hospedeiras resulta na indução de citocinas pró-inflamatórias in vivo [94,95], entre as quais
a interleucina-8 (IL-8) que é o principal mediador da inflamação na mucosa gástrica [69,96].

Introdução
Andrea Santos
6
5.3 Proteínas da membrana externa
As proteínas da membrana externa bacterianas (OMPs) estão envolvidas na interacção
entre a bactéria e o meio envolvente, desempenhando um papel essencial quer na sua
sobrevivência, quer na sua patogenicidade. Dependendo das espécies, estas proteínas
podem estar envolvidas no transporte de moléculas, funcionando como porinas, na
aderência e colonização do hospedeiro ou na evasão à resposta imunitária [97]. A análise
das sequências do genoma das estirpes de referência J99 e 26695 revelou a presença de
uma grande família de genes que codificam proteínas da membrana externa em H. pylori,
correspondendo a cerca de 4% do total de regiões codificantes de cada estirpe [39,58].
Foram identificadas cinco famílias de genes parálogos, contendo entre 3 e 33 membros que
variam em tamanho entre 27 e 135 kDA [97] (Figura 1).
ho
rD
0.05
ho
rD
0.05
fecA
hom
frpB
hof
hef
hop e hor
Figura 1: Análise filogenética das cinco famílias de parólogos da estirpe de referência J99 [Adaptado
de 98]. O tracejado identifica os 5 pares de genes duplicados.
A família 1 é constituída pelas proteínas Hop que contêm as regiões N e C terminais
altamente conservados e pelas proteínas Hor que não têm o motivo Hop no N-terminal [97].
Nesta família, as regiões conservadas podem ter estruturas e funções semelhantes, tais
como a integração da proteína na membrana externa ou a interacção proteína-proteína com

Introdução
Andrea Santos
7
outros membros da família, enquanto as regiões variáveis parecem estar envolvidas em
funções específicas, como por exemplo, a capacidade de agir como adesina ou porina [97].
Sabe-se actualmente que as proteínas HopA e HopE funcionam como porinas [99],
enquanto as proteínas HopB (AlpB), HopC (AlpA), HopS (BabA), HopP (SabA), HopZ e
HopH (OipA), funcionam como adesinas [96,100,101,102].
A família 2 é constituída por oito membros sem função conhecida e que exibem um
elevado nível de similaridade ao nível das proteínas (>95%) [97].
A terceira família é constituída por 4 membros (HomA, HomB, HomC e HomD) que
apresentam os terminais C e N conservados e domínio central variável, estando envolvidos
tanto na adaptação a hospedeiros específicos como na virulência de H. pylori. Até à data, os
membros melhor caracterizados desta família, são as proteínas HomA e HomB. O gene
homB é 90% homólogo com o homA [63,96,97]. Estudos recentes sugerem que o gene
homB constitui um novo factor de virulência dado a sua associação com a doença ulcerosa
péptica, enquanto o gene homA, foi correlacionado com a dispepsia não ulcerosa
[96,102,103]. O genótipo homB parece ainda constituir um marcador molecular na distinção
entre o cancro gástrico e a úlcera péptica [104]. Foi também demonstrado que a proteína
HomB é antigénica e é capaz de activar a secreção da IL-8 in vitro, através do seu papel
como adesina [96,103].
A família 4 (FecA e FrpB) tem homologia com OMPs de outras bactérias, envolvidas na
regulação do ferro, enquanto a família Hef (família 5) é constituída por três membros (HefA,
HefD e HefG) altamente conservados, homólogos a genes que codificam sistemas de efluxo
[97].
Dada a importância destas proteínas na relação entre bactéria e hospedeiro, H. pylori
dispõe de vários mecanismos implicados na regulação e diversidade ao nível da expressão
das OMPs, que fazem variar a composição proteica da membrana externa, bem como a
antigenicidade dos seus constituintes. De entre esses mecanismos destacam-se a
duplicação, delecção ou conversão génica, a regulação por variação de fase, a diversidade
alélica e a presença de genes específicos de estirpe.
5.3.1 Mecanismos de regulação da expressão das proteínas da membrana externa
A análise das sequências de diversas OMPs revelou que alguns genes hop apresentam
duas cópias no mesmo genoma. Ambas as estirpes de referências 26695 e J99 contêm 5
pares de genes duplicados: hopJ/hopK, hopM/hopN, hopS(babA)/hopT(babB),
hopP(sabA)/hopO(sabB) e homA/homB que também estão presentes na maioria das
estirpes clínicas [96,97] (Figura 1). A presença destes genes duplicados pode dever-se a
processos evolutivos em curso onde os genes duplicam, por recombinação intragenómica,
acumulando posteriormente mutações para divergirem gradualmente em genes que

Introdução
Andrea Santos
8
codificam novas proteínas, funcionalmente distintas. Este processo é provavelmente similar
ao que gerou a família Hop inteira [97]. Foi também sugerido que a duplicação destes genes
representaria uma forma de regulação da expressão de proteínas para a adaptação às
mudanças na inflamação e na resposta imunitária do hospedeiro [55,97], bem como na
evolução da doença gástrica. Por exemplo, o genótipo de dupla cópia homB foi associado à
úlcera péptica, o que sugere que a duplicação não é um fenómeno aleatório e pode
constituir uma vantagem biológica facilitando a adaptação a um hospedeiro específico [96].
Para além da duplicação, alguns genes hop contêm repetições de dinucleótidos CT na
região 5’ e são regulados por variação de fase [97,101,102,105]. Durante a replicação do
ADN, na zona de repetições pode ocorrer um desemparelhamento entre as bases
complementares (misparing), o que leva ao “deslize” da polimerase na grelha de leitura,
resultando na adição ou delecção de unidades CT. Estas alterações no número de
repetições podem originar a adição ou remoção de codões STOP, alterando a expressão da
proteína devido à mudança de fase on/off [60]. Os genes já identificados regulados por este
mecanismo em H. pylori são: babB, sabA e sabB, hopZ, oipA e homB. A variação de fase
como mecanismo de diversidade é utilizado por várias espécies bacterianas e tem mostrado
contribuir para a variação antigénica e virulência durante a infecção [58,96].
A variação alélica é também um mecanismo de diversidade em vários genes de H. pylori,
como babA, hopQ, hopZ, homA e homB [63,96,102,106,107]. Em todos estes genes é
observado um perfil conservado de segmentação, com a região variável localizada na região
média do gene e com a existência de pelo menos duas variantes alélicas conservadas.
Estas variantes podem associar-se em alguns casos, a determinadas apresentações
clínicas da infecção e podem também contribuir para a diversidade antigénica [106]. Por
exemplo no gene hopQ, a família alélica do tipo I é mais frequente em estirpes isoladas de
doentes com úlcera, estando associada ao genótipo virulento, cagPAI+/vacAs1 [106]. No
entanto, nos genes babA e babB, foram descritas 3 e 5 variantes alélicas respectivamente,
não estando nenhuma delas relacionadas com o genótipo de virulência ou com a
capacidade de adesão da estirpe [107]. O mesmo acontece com os genes homA e homB
[63]. Assim, nestes casos a variação alélica parece ser um mecanismo utilizado por H. pylori
para originar estirpes com diferentes perfis antigénicos que vão permitir evadir-se à resposta
imunitária [63,107].
Para além de todos estes mecanismos, algumas estirpes contêm ainda genes específicos
que codificam OMPs, tais como, hopU e homB, fenómeno que pode representar uma
vantagem adaptativa a hospedeiros específicos [97].

Objectivos
Andrea Santos
9
Capítulo 2: Objectivos
As OMPs estão envolvidas na relação entre H. pylori e a mucosa gástrica humana,
contribuindo para a colonização e persistência da infecção [108].
Aproximadamente 4% do genoma de H. pylori contém informação para a codificação de
pelo menos 32 OMPs [97,109], sendo que as principais proteínas associadas com a
patogenicidade e adesão de H. pylori são: BabA, SabA, OipA, AlpA, AlpB e HomB
[59,96,103,109,110]. A adesina BabA interage com as células epiteliais gástricas, através da
ligação ao antigénio do grupo sanguíneo Lewis b (Leb), que também é produzido ao nível
destas células [108,110]. A interacção BabA-Leb levou a que esta OMP tenha sido
considerada, até à data, a principal adesina de H. pylori [108-110]. O gene babA tem um
gene parólogo, babB, cuja função é ainda desconhecida [110]. Foi demonstrado no entanto
que a recombinação entre os dois loci contribui para a dinâmica de adesão da bactéria às
células gástricas, aumentando a sua fitness [113].
Um estudo do “complexoma” de H. pylori mostrou que BabA e BabB formam complexos
com outras OMPs, nomeadamente HopM e HopN [109]. Estudos recentes sugerem que
nenhuma das proteínas BabA ou BabB é capaz de induzir resposta imunitária em macacos,
nem são os antigénios imunodominantes em humanos [109,111,112]. Uma hipótese que
poderia explicar este facto seria a de que as proteínas de membrana que interagem com
BabA ou BabB, por exemplo, HopM e HopN, poderiam mascarar os epítopos daquelas
proteínas e consequentemente serem elas próprias expostas e assim antigénicas [109].
Esta hipótese é ainda corroborada pelo facto de HopN/HopM terem demonstrado
reactividade contra soros de pacientes positivos para H. pylori [109,113]. Estes dados
sugerem que estas proteínas poderão desempenhar um papel importante na relação entre a
bactéria/hospedeiro, nomeadamente, constituindo prováveis antigénios e consequentemente
importantes alvos vacinais; por outro lado, o facto de formarem complexos com BabA
poderá indiciar o seu papel na adesão de H. pylori à célula epitelial gástrica [109,110].
Este trabalho tem como objectivo o estudo dos genes hopM e hopN, nomeadamente a
sua diversidade genética, a análise filogenética e evolutiva. Pretende-se também contribuir
para o esclarecimento do seu papel na virulência de H. pylori, através da identificação de
possíveis associações destes genes a patologias específicas e a marcadores moleculares
de virulência. Para tal, foi estudada uma amostra de estirpes clínicas de H. pylori, isoladas
de biopsias gástricas de doentes de diferentes regiões geográficas, apresentando diversas
patologias gástricas, nomeadamente, úlcera péptica, gastrite e cancro gástrico. Foram
também utilizadas as sequências nucleotídicas de 23 estirpes de referência de H. pylori,
cujos genomas já se encontram sequenciados e anotados.

Materiais e Métodos
Andrea Santos
10
Capítulo 3: Materiais e Métodos
1.Estirpes de Helicobacter pylori e material genómico
Foram estudadas 234 estirpes clínicas de H. pylori isoladas de biopsias gástricas de
doentes de diversas origens geográficas, apresentando diferentes patologias
gastroduodenais, nomeadamente dispepsia não ulcerosa com gastrite (G), doença ulcerosa
péptica (UP) gástrica ou duodenal, e cancro gástrico (CG) (Anexo I). As estirpes de H. pylori
estudadas pertencem à colecção de estirpes bacterianas da Unidade de
Campylobacter/Helicobacter do Departamento de Doenças Infecciosas (DDI) do Instituto
Nacional de Saúde Dr. Ricardo Jorge (INSA); o material genómico de algumas estirpes de
H. pylori não pertencentes à colecção do INSA, foi gentilmente cedido pelo Doutor Yoshio
Yamaoka, Department of Medicine, Michael E. DeBakey Veterans Affairs Medical Center
and Baylor College of Medicine, Houston, Texas, EUA.
Foram ainda utilizadas três estirpes de referência de H. pylori, as estirpes 26695 (ATCC
700392), J99 (ATCC 700824) e HPAG1, cujos genomas já se encontram sequenciados,
como controlos das reacções de amplificação e de sequenciação [39,46,58].
Cultura das estirpes de Helicobacter pylori
As estirpes de H. pylori conservadas a -80ºC em meio TSB com glicerol, foram cultivadas
em meio HP selectivo e não selectivo (Biogerm, Maia, Portugal), e incubadas a 37ºC numa
atmosfera de microaerófilia (5% de O2; 10% de CO2; 85% de N2), durante 48 horas. Uma
placa de cultura foi utilizada para extracção de ADN e a outra para congelar a estirpe de
modo a repor a colecção de bactérias (Anexo II).
Extracção de ADN genómico
A extracção de ADN genómico foi realizada utilizando o kit comercial QIAamp® DNA Mini
(Qiagen GmbH, Hilden, Alemanha), de acordo com as instruções do fabricante [114].
2. Avaliação da diversidade genómica dos genes hopM e hopN
Para o estudo da diversidade genómica dos genes hopM e hopN, na amostra de estirpes
em estudo, foram identificados os loci de cada gene alvo, por análise dos genomas de
estirpes sequenciadas (Anexo III), e desenhados oligonucleótidos iniciadores (doravante
designados por primers) nas respectivas regiões genómicas flaqueantes e no interior dos
genes (Tabela 1). Todos os primers descritos neste trabalho foram desenhados com recurso
ao software Web Primer3 (http://www.broad.mit.edu/cgi-bin/primer/primer3_www.cgi).
Procedeu-se depois à amplificação e sequenciação de cada locus, pelas técnicas de PCR e
sequenciação.
A reacção de amplificação foi realizada num volume final de 25μl contendo: 1μl de ADN,
Tampão 1X (Bioline Ltd, Londres, Reino Unido), 2,8mM de MgCl2, 200μM de cada dNTP

Materiais e Métodos
Andrea Santos
11
(Roche Diagnostics GmbH, Mannheim, Alemanha), 0,5μM de cada primer, 1U de Bio-X-
ActTM Long DNA Polimerase e água destilada estéril. Em cada ensaio usou-se um controlo
negativo constituído por água destilada estéril, e como controlos positivos o ADN das três
estirpes de referência. A reacção de amplificação consistiu num ciclo inicial de 5min a 95ºC
e 1min a 57ºC, seguido de 35 ciclos constituídos por extensão de 3min a 68ºC,
desnaturação a 95ºC durante 30s e hibridação dos primers a 57ºC durante 30s, com um
ciclo de extensão final de 10min a 68ºC, realizada num Termociclador GeneAmp® PCR
System 9700 (Applied Biosystems Inc., Foster City, EUA).
Tabela 1: Primers utilizados na amplificação e sequenciação dos loci hopM e hopN. Os primers internos utilizados são os mesmos nas duas regiões genómicas estudadas.
Região Genómica Primer Sequência (5’ 3´) TM (ºC)
hopM (primers externos) hopMF2 GCCATTCTTTTTGTATCTTTT 50,1 hopMR2EXT CTGTTGATTCTTCCATAAAAAACTC 50,6
hopN (primers externos) hopNF2 TCCCTATCGCTTATAGCTTGA 55,9 hopNR1 TATGCGGGTATTGGTTTTGC 45
hopMR4 CGCTAAAGCCACAGCTTGATA 47 hopMR3 GCGTTTTCATTAGCCTTAAG 43
hopM/hopN hopMR5 CCTTAACCCGCACCTTCT 56
(primers internos) hopMR6 AGGCCCTTGAATGCTGTG 56
hopMR7 TAGGGCCAGGGCCACAGC 52
hopMR8 GGTTTAAAGCGGTTTGGAT 42
Os produtos de amplificação foram detectados por electroforese em gel de agarose 1% e
corado com GelRedTM (1:10000) (Biotium Inc., Hayward, EUA), em tampão de corrida TBE
0,5X (Anexo IV). Foi utilizado como marcador de peso molecular o 1Kb DNA Ladder com
fragmentos de ADN de 500 pb a 12 Kb (Invitrogen S.A, Barcelona, Espanha).
Os produtos de amplificação foram purificados com o kit QIAquick® PCR Purification Spin
(Qiagen GmbH, Hilden, Alemanha), segundo instruções do fabricante [115] e seguidamente
sujeitos a sequenciação enzimática utilizando o Big Dye® Terminator (Applied Biosystems
Inc.) e o Sequenciador automático Genetic Analyser Abi-Prism 3130 xl (Applied Biosystems
Inc.). A reacção de sequenciação foi realizada num volume final de 10μl contendo: 2μl de
Big Dye® V1.1 (Applied Biosystems Inc.), 0,5μM de primer, 0,5 a 7,5μl de ADN purificado e
água destilada estéril. O programa de sequenciação consistiu num ciclo inicial de
desnaturação de 30s a 96ºC, seguido de 25 ciclos constituídos por uma desnaturação de
10s a 96ºC, uma hibridação de 5s a 45ºC e uma extensão de 4min a 60ºC. Estas reacções
foram realizadas num Termociclador GeneAmp® PCR System 9700 (Applied Biosystems
Inc.). Após a reacção cíclica os produtos foram entregues na Unidade de Tecnologia e
Inovação do INSA para sequenciar. As sequências de nucleótidos obtidas foram
visualizadas no programa Chromas LITE (Version 2.01) e comparadas recorrendo ao
programa MultAlin (http://multalin.toulouse.inra.fr/multalin/). Em seguida, foram traduzidas

Materiais e Métodos
Andrea Santos
12
em sequências de aminoácidos correspondentes recorrendo ao programa EMBOSS transeq
(www.ebi.ac.uk/emboss/transeq/).
3. Avaliação da variabilidade genética, análise filogenética e dos parâmetros
evolutivos dos genes hopM e hopN
De modo a avaliar a variabilidade genética e os parâmetros filogenéticos e evolutivos, foi
realizada uma análise bioinformática das sequências nucleotídicas e respectivas sequências
de aminoácidos previstas. Foram também utilizadas as sequências nucleotídicas das 23
estirpes de referência de H. pylori (Anexo III).
A variabilidade genética foi determinada a partir de gráficos de similaridade, utilizando
alinhamentos gerados pelo programa BioEdit Sequence Alignment Editor 7.0.9
(http://www.mbio.ncsu.edu/BioEdit/bioedit.html), com recurso à aplicação do programa
SimPlot 3.5.1 (http://sray.med.som.jhmi.edu/SCRoftware/simplot/).
A análise da filogenia foi realizada através de árvores filogenéticas criadas pelo programa
MEGASoftware 4.0.2 (Molecular Evolutionary Genetics Analysis)
(http://www.megasoftware.net/), a partir de alinhamentos múltiplos das sequências de
nucleótidos e respectivas sequências de aminoácidos previstas, obtidos no BioEdit
Sequence Alignment Editor 7.0.9 pela aplicação ClustalW. Foi utilizado o teste de filogenia
Bootstrap Neighbor Joining Tree com 1000 réplicas e o modelo de Kimura 2-parameter
[63,116,117].
Os parâmetros evolutivos foram determinados recorrendo ao programa MEGASoftware
4.0.2, utilizando alinhamentos também gerados no BioEdit Sequence Alignment Editor 7.0.9,
calculando-se: distância molecular pelo método p-distance, frequência de substituições
sinónimas (Ks), frequência de substituições não-sinónimas (Ka) e razão entre substituições
não-sinónimas e substituições sinónimas (Ka/Ks), pelo método de Nei Gojobori [63,116].
4. Análise Estatística
Para testar associações entre duas variáveis qualitativas, foram utilizadas tabelas de
contingência de 2x2, recorrendo-se ao teste Exacto de Fisher com recurso à aplicação do
programa EpiInfoTM 3.5.3. (http://www.cdc.gov/epiinfo). Todos os testes utilizados foram
bilaterais. Foi estabelecido em 5% o nível de significância dos testes, tendo-se rejeitado a
hipótese nula quando a probabilidade de significância do teste (p value) foi inferior a este
valor. Os valores p apresentados referem-se à comparação entre ter ou não o gene ou um
determinado alelo por patologia ou por genótipo de virulência (genótipo de virulência ou
genótipo não virulento).

Resultados
Andrea Santos
13
Capítulo 4: Resultados
1.Avaliação da diversidade genómica dos genes hopM e hopN
Considerando a classificação da estirpe de referência HPAG1, os genes hopM e hopN,
estão localizados no locus HPAG1_0230 (locusM) e no locus HPAG1_1289 (locusN),
respectivamente. Nas estirpes clínicas foram obtidos produtos de amplificação que variaram
entre 2604-2616 pb para o locusM e 2559-2589 pb para o locusN. Neste último, verificou-se
ainda para algumas estirpes (24,6%), a amplificação de produtos de menor peso molecular,
variando entre 200 e 1500 pb. Todos estes produtos de amplificação foram posteriormente
sequenciados e as respectivas sequências de nucleótidos foram analisadas recorrendo ao
programa MultAlin, sendo posteriormente traduzidas para as sequências de aminoácidos
correspondentes, recorrendo ao programa EMBOSS transeq.
Verificou-se em todas as estirpes estudadas (clínicas e de referência), que os genes
hopM e hopN ocupavam ambos os loci (M/N) indiferentemente. Os produtos de menor
tamanho localizados no locusN correspondiam a uma região intergénica (estirpes asiáticas),
a uma ORF (Open Reading Frame) mais curta com homologia com a região 3’ dos genes
hopM e hopN (estirpes ocidentais) ou ainda a uma ORF que correspondia à fusão entre a
região codificante do péptido sinal de HopM e HopN e a região 3’ de uma DNA-
metiltransferase de H. pylori (dnpA) (estirpes asiáticas).
Foram obtidas 468 sequências a partir de 234 estirpes clínicas, 132 correspondendo ao
gene hopM, 206 ao gene hopN, 115 Loci “vazios” e 15 sequências que não pertenciam a
nenhuma destas três categorias e que serão descritas seguidamente. Os dois genes foram
identificados com recurso à aplicação BLAST, Basic Local Alignment Search Tool
(http://blast.ncbi.nml.nih.gov/Blast.cgi). Apesar do elevado grau de similaridade entre hopM e
hopN (94% similaridade, calculado para as sequências da estirpe de referência HPAG1), os
genes diferenciam-se ao nível da extremidade 5’ na região compreendidade entre os 396 e
900 pb (Anexo V e Figura 5).Foram identificadas três estirpes ocidentais que apresentavam
uma sequência que correspondia a uma fusão entre os dois genes (quimeras hopM/N)
(Anexo V). Nestas três estirpes, duas apresentavam a quimera apenas no locusM, enquanto
a outra apresentava este putativo gene recombinante em ambos os loci.
Ambos os genes possuem na extremidade 5’ repetições de dinucleótidos CT. Verificou-se
em cinco estirpes clínicas e numa estirpe de referência (6/234, 2,6%) a existência de
proteínas putativas truncadas em ambos os loci, devido a alterações no número de
repetições.
As quimeras e as ORF’s truncadas foram eliminadas da restante análise de resultados.

Resultados
Andrea Santos
14
1.1 Distribuição dos genes hopM e hopN por região geográfica
Foram incluídas neste trabalho as sequências hopM/hopN de 226 estirpes clínicas e 23
estirpes de referência (n=249 estirpes) (Anexos I e III). Pelo menos uma cópia de hopM ou
hopN foi detectada em todas as estirpes analisadas. Foi observada diversidade
relativamente ao número de cópias de cada gene e à sua localização genómica (Figura 2). A
distribuição dos genes hopM e hopN relativamente ao número de cópias e loci que ocupam,
por origem geográfica, encontram-se representados na figura 2. Nas estirpes ocidentais os
genótipos de cópia única e dupla cópia (mesmo gene e genes diferentes) foram observados,
sendo o genótipo de dupla cópia mais prevalente. Contrariamente, nas estirpes de origem
asiática, apenas o genótipo de cópia única foi observado, enquanto na população africana
todas as estirpes estudadas possuíam o genótipo de dupla cópia. Verificou-se ainda que no
caso de cópia única, o gene ocupava sempre o locusM enquanto no caso de dupla cópia os
dois genes ocupavam indiferentemente os dois loci.
A) ESTIRPES OCIDENTAIS (n=167)
Genótipo cópia única
LocusM LocusN
Genótipo dupla cópia
LocusM Locus N
hopM
hopNhopN
hopM
17%
11,3%
hopM
hopM hopN
hopN
46,5%
hopN
hopM
x
x
18,9%
6,3%
x
x
B) ESTIRPES ASIÁTICAS (n=71)
LocusM Locus N
hopN
hopM 31,2%
68,8%
C) ESTIRPES AFRICANAS (n=11)
LocusM Locus N
hopM
hopNhopN
hopM
50%
16,7%
hopM hopN
hopN hopM
33,3%
Genótipo cópia única
Genótipo dupla cópia
Figura 2: Distribuição do número de cópias e localização genómica de hopM e hopN, por região geográfica. As
percentagens indicam a frequência de cada genótipo nas populações Ocidental, Asiática e Africana. O X representa ausência do gene hopM ou hopN completo.
1.2 Distribuição dos genes hopM e hopN por patologia
Para realizar esta análise foram incluídas apenas as estirpes para as quais havia
informação sobre a situação clínica do doente de onde tinha sido isolada (n= 224), com a
seguinte distribuição: 127 (56,7%) casos de úlcera péptica (UP), 82 (36,6%) de gastrite (G) e
15 (6,7%) de cancro gástrico (CG). Todas as estirpes com uma cópia de cada gene no seu
genoma (71/224, 31,7%) foram numa primeira abordagem eliminadas.
Verificou-se que o gene hopN foi o mais prevalente em todas as patologias, estando
presente em 73,3% das estirpes de UP (63/86), em 62,5% das G (35/56) e em 90,9% das
estirpes de CG (10/11).

Resultados
Andrea Santos
15
Com o objectivo de perceber se, para além da presença dos genes, a forma como eles se
distribuem no genoma (número de cópias e loci que ocupam), estaria associada com
patologias específicas, foi efectuada a análise da distribuição dos diferentes genótipos por
patologia. Considerando todas as estirpes, verificou-se que o genótipo mais frequentemente
encontrado na UP foi a cópia única hopN (35,4%), seguido da dupla cópia MN (31,5%). Na
gastrite, os genótipos mais frequentes foram a dupla cópia MN (31,7%) e a cópia única
hopN (26,8%) e no CG, o genótipo mais frequentemente encontrado foi o de cópia única
hopN (76,9%).
Posteriormente efectuou-se o mesmo tipo de análise por origem geográfica (Figura 3). Na
população ocidental o gene hopM foi mais frequentemente encontrado na G do que na UP,
enquanto o hopN foi mais frequente na UP. Na população asiática, o gene hopM foi mais
frequentemente identificado na G enquanto o gene hopN foi mais prevalente na UP e CG.
Na população africana todas as estirpes foram isoladas de UP sendo o gene hopN o mais
frequente (n=10, 85,7% hopN). Apesar de existir uma associação aparente entre a presença
do gene e a patologia, nenhuma associação é suportada estatisticamente, uma vez que o
hopN é o gene mais prevalente em todas as populações.
População Ocidental (n=83)A)
0
10
20
30
40
50
60
70
80
hopM hopN
26,1%
73,9%
37,8%
62,2%
UP G
% d
e e
sti
rpes p
osit
ivas
(n=46) (n=37)
0
10
20
30
40
50
60
70
80
hopM hopN
29,7%
70,3%
35%
65%
23,1%
76,9%
UP G CG
População Asiática (n=70)B)
(n=37) (n=20) (n=13)
% d
e e
sti
rpes p
osit
ivas
Figura 3: Frequência dos genes hopM e hopN nas estirpes de H. pylori de origem ocidental (A) e asiática (B) de
acordo com a patologia (úlcera péptica (UP), gastrite (G) e cancro gástrico (CG)). As percentagens foram calculadas para cada patologia.
Relativamente ao genótipo hopM/hopN, na população ocidental o mais prevalente foi a
dupla cópia MN tanto nas estirpes isoladas de UP (47,5%, 38/80) como nas de G (42,6%,
26/61). Na população asiática, o genótipo mais prevalente nas estirpes isoladas de UP foi a
cópia única hopN (70,3%, 26/37 UP), assim como em 65% (17/20) das G e em 76,9%
(10/13) dos CG. Na população africana, as estirpes foram todas isoladas de doentes com
UP, sendo o genótipo mais frequente a dupla cópia hopN (6/10, 60%). Assim, apesar de se
verificarem diferenças relativamente à prevalência dos diferentes genótipos por origem
geográfica, esse genótipo é também o mais encontrado em todas as patologias nessa
população.

Resultados
Andrea Santos
16
1.3 Associação dos genes hopM e hopN com o genótipo de virulência
Foram estudadas 222 estirpes com o objectivo de verificar a associação entre a
presença de hopM e hopN com outros genes de H.pylori associados à virulência,
nomeadamente a presença do gene cagA, marcador da presença do ilhéu de
patogenicidade cag e do alelo s do gene vacA. Definiram-se como estirpes virulentas as
estirpes positivas para o gene cagA e para o alelo citotóxico vacAs1, e estirpes não
virulentas as estirpes negativas para o gene cagA e com o alelo vacAs2 (alelo não
citotóxico) [79,83]. De uma forma geral, verificou-se a associação entre a presença de
dupla cópia M e da dupla cópia MN e o genótipo não virulento (57,9% MM vs 26,6%;
47,2% MN vs 20,7% outros genótipos, respectivamente) e a associação entre a cópia
única N e genótipo virulento (88,7% cópia única hopN vs 62,3%), sendo estas associações
suportadas estatisticamente (p=0,01, p<0,001 e p<0,001, respectivamente). Por origem
geográfica, na população ocidental observou-se que 80,8% das estirpes com dupla cópia N
eram virulentas, contra 56,5% de outros genótipos (p=0,03) e que 64,7% das estirpes com
dupla cópia M eram não virulentas, contra 37,5% de outros genótipos (p=0,05). Na
população asiática, não foi possível observar qualquer tipo de associação com o genótipo
de virulência, uma vez que todas as estirpes são cagA e vacAs1 positivas. Na população
africana, 60% das estirpes virulentas apresentam a dupla cópia N no seu genoma, não
tendo no entanto esta associação suporte estatístico, provavelmente devido ao pequeno
número de estirpes estudadas.
2.Análise filogenética, avaliação da variabilidade genética e parâmetros evolutivos
dos genes hopM e hopN
Para todas as estirpes em estudo (n=249), foi obtida uma sequência codificante em pelo
menos um dos loci analisados. Todas as sequências obtidas foram identificadas por
homologia com o respectivo gene, hopM ou hopN, recorrendo à aplicação BLAST. As
sequências correspondentes ao gene hopM variaram entre os 2082 e 2097 pb, enquanto
para o gene hopN variaram entre 2075 e 2100 pb.
A reconstrução filogenética das 379 sequências obtidas em ambos os loci (147 hopM e
232 hopN) mostra dois ramos independentes para cada um dos genes o que sugere
evolução divergente (Figura 4). Dois clusters predominantes foram identificados em ambos
os genes, sendo a segregação entre eles motivada pela origem geográfica das estirpes
(bootstrap=86). O cluster I foi identificado como grupo Ocidental e é composto por estirpes
isoladas de Portugal, França, Alemanha, Suécia, Brasil, Colômbia e EUA, enquanto o cluster
II foi identificado como grupo Oriental/Ameríndio e é composto por estirpes originárias da
Ásia Oriental (Japão, Singapura e Coreia do Sul) e da América do Sul (Peru e Venezuela)
(Figura 4). As estirpes africanas (Burquina-Faso, Gâmbia e África do Sul) encontram-se

Resultados
Andrea Santos
17
mais próximas das estirpes ocidentais e o facto de não se observar a formação de um
cluster específico, deve-se provavelmente ao baixo número de estirpes analisadas (n=11).
Esta hipótese é corroborada pela análise filogenética por gene, onde se observa a formação
de um pequeno cluster de estirpes africanas, embora não seja sustentado por valores de
bootstrap (resultado não apresentado).
Cluster I
Cluster I
JPJ2
3ho
pN1P
U
JPJ1
87h
opN
1P
U
JPJ2
22ho
pN1G
CJP
J42h
opN
1GC
JPJ6
3hop
N1G
C
JPJ2
15ho
pN1P
U
JPJ1
43ho
pN1G
C
JPJ4
4hop
N1G
C
JPJ1
41A
hopN
1PU
JPJ2
13ho
pN1P
U
JPJ6
4hop
N1G
C
JPJP
T42C
hopN
1G
SG
1200
2hop
N1
JP51
Bho
pN1G
JPF30
hopN
1PU
F30hop
N1
KRDual61
hopN
1PU
JPJ1
19AhopN1PU
KRNUD26hopN1G
KRNUD21hopN1G
SG8038hopN1
JPF32hopN2GC
F32hopN2
JPJ206hopN1PU
KRTriple90hopN1PU
KRDual2hopN1PU
KRDual9hopN1PU
JPJP101ADhopN1PU
JPJPT395ChopN1G
JPJ272hopN1PU
JPJ258hopN1PU
JPJ214hopN1PUKRNUD49hopN1GJPJ147hopN1GC
KRDual18hopN1PUJPF16hopN2GF16hopN2
KRNUD40hopN1GJPJ90hopN1PUKRDual58hopN1PU
JPJ80hopN1GC
SG12001hopN1
KRDual74hopN1PUSG7003hopN1
KRCA108hopN1GCKRNUD23hopN1G
KRDual24hopN1PU
NO35AhopN1
35AhopN1PUKRDual88hopN1PU
FRAnt170hopN1PU
SE24hopN1PU
DE948hopN1G
SE38hopN1PUSE59hopN1G
DE959hopN2G
DE959hopN1G
PT23hopN2G
PT23hopN1G
BF3hopN2PU
BF3hopN1PU
US2721ChopN2G
US2721ChopN1GPT70hopN1PU
US1904DhopN1PU
PTB26hopN2G
CO967DhopN1PU
PTB50hopN2
PTB50hopN1PTB13hopN2G
PTB47hopN1
PT559/02hopN1PUPT565/99hopN1G
PT219/99hopN1PU
PT285/99hopN1PU
PT228/99hopN1G
PTB53hopN1G
CO152ChopN1G
SE27hopN1G
NON29hopN2PU
SE47hopN1G
SES570hopN1PU
FR901hopN1G
CO216DhopN1G
COCol83hopN1G
PT28hopN1PU
SJM180hopN2G
PT38hopN1G
PTB59hopN2
PTB19hopN1
KRNUD6hopN
1GPTB
44hopN2G
PT1776/05hopN
1
PT255/99hopN
2PU
CO
Col635hopN
2PU
CO
817DhopN
1PU
CO
221ChopN
2GC
O928D
hopN2P
U
BF18A
BFhopN
1PU
BF2A
BFhopN
1PU
BF2A
BFhopN
2PU
BF18A
BFhopN
2PU
Gam
bia94/24hopN1
BR
198.96hopN1G
C
CO
817hopN2P
U
US
2766ChopN
2G
US
2791ChopN
1G
BF
16AB
FhopN
2PU
BF
16AB
FhopN
1PU
CO
Col635D
hopN1P
U
BR
198.95hopN1G
CB
R198.96hopN
2GC
BR
313
.03h
opN
1G
PT
1198
/04ho
pN2P
UP
T11
98/04
hopN
1P
U
US
166
5Dh
opN
1P
U
BF
5hop
N2
PU
US
157
8Dh
opN
1P
UU
S1
578D
hop
N2
PU
BF
25A
BF
hop
N2
PU
BF
25
AB
Fh
op
N1
PU
BR
131
1.95ho
pN1G
C
US
277
7Ch
opN
1G
US
277
9hop
N1G
US
278
7Ch
opN
2G
US
278
7Ch
opN
1G
BR
112
6.95ho
pN1P
UB
F1A
BF
hopN
1PU
BF
19A
BF
hop
N2P
U
BF
19AB
FhopN
1PU
PT
2hop
N1
PU
PT
B27
hop1
N
PT
B16
hopN
1P
UIn
dia
7hop
N1
PT
1089
/03ho
pN2P
U
PT
1089
/03h
opN
1PU
PT
372/
99ho
pN2P
U
PT
372/
99ho
pN1P
U
PT
B21
hopN
1G
ITG
27ho
pN1
G27
hopN
2
PTB
15ho
pN1P
U
PTB
51ho
pN1P
U
PT12
4/99
hopN
1PU
PT14
5/99
hopN
2PU
B8h
opN
1PU
PTB
8hop
N1
PTB
10ho
pN1G
PT7
71/9
9hop
N1P
U
US16
75Dho
pN1P
U
PT4
1hop
N2G
PT41
hopN
1G
PTB
68ho
pN1P
U
PTB17ho
pN1
PT255/
99hop
N1P
U
PT133/
99hop
N1PU
PTB9hop
N1PU
PTB18hopN1
PT36/00hopN2G
PT107hopN2PU
BR55.01hopN2G
BR55.01hopN1G
KRDual67hopN1PU
KRDual33hopN1PU
KRNUD9hopN1G
DE940hopN1PUSouthAfrica7hopN1
SG8033hopN1
JPJ103hopN1GC
52hopN2PU
DE951hopN1PU
GBN83hopN1PU SE30hopN1PU
SE3hopN2G
SE3hopN1G
PT95hopN2PU
SE1hopN1PU
SE10hopN1G
DE938hopN1PUPT194/99hopN2PU
PT194/99hopN1PU
SEEN35hopN1PU
PT137/99hopN2PU
US2752ChopN2GUS2752ChopN1G
FR2432hopN1G
DE946hopN1PU
DE937hopN1PU
DE944hopN1PUGBEN37hopN1PU
26695hopN1G
GB26695hopN1G
26695hopN2GGB26695hopN2GNON28hopN1PU
SE6hopN1PUSE7hopN1PUFRS583hopN1PU
FRS583hopN2PUSEEN24hopN1PU
GBEN36hopN2PUHPAG1hopN2GDE956hopN1G
DE943hopN1PUDE953hopN1G
DE963hopN1G
PTB3hopN1PU
COCol83hopN2GPT1hopN2G
PT1hopN1G
DE952hopN1G
DE942hopN1PUPTB59hopN1
PTB31hopN1GPT351/99hopN1PU
PT197/99hopN2PU
PT197/99hopN1PU
PTB14hopN1PT499/02hopN2PU
PT1152/04hopN1PU
PTB71hopN1
FRF93hopN1PU
PT1786/05hopN2G
PT1846/05hopN2PU
PT1786/05hopN1G
PT1846/05hopN1PU
PTB35hopN1G
PTB26hopN1G
PTB6hopN1
PTB22hopN1G
PTB34hopN1G
BR1361.95hopN1PU
PTB60hopN2PU
PTB60hopN1PU
PT181/99hopN1PU
PT417/02hopM2PT417/02hopM1PUCO152ChopM2GKRDU852hopM1PUPTB27hopM2FRHorhopM2PU
FRHorhopM1PUFRB38hopM1ML
FRB38hopM2MLPTB57hopM1G
PT124/99hopM2PU
PTB57hopM2G
PT95hopM1PU
US2781ChopM2G
US2781ChopM1GFRAnt170hopM2PU
PT655/99hopM1G
PT655/99hopM2G
PTB34hopM2G
PT224/99hopM1PUPT351/99hopM2
KRDual30hopM1PU
SEHPAG1hopM1G
DE952hopM2G
SE58hopM1PU
CO216ChopM2G
SES580hopM1PU
PT137/99hopM1PU
DE950hopM1G
PT41hopM2G
BR1361.95hopM2PU
PTB44hopM1G
PTB3hopM2PU
US3058ChopM1G
US3058C
hopM2G
BR313.03hopM
2G
GM
Gam
bia94/24hopM2
BF5hopM
1PUBF1A
BFhopM
2PU
US2777C
hopM2G
BR
198.95hopM2P
U
PTB
22hopM2G P
ESJM
180hopM1G
PE
PeC
an4hopM1G
C
PT28hopM
2PU
NO
N29hopM
1PU
SE
S569hopM
1PU
DE
P12hopM
1PU
DE
P12hopM
2PU
PT181/99hopM
2PU
PTB
65hopM1G
PTB
36hopM1
DE
955hopM1GS
E56hopM
1GG
BE
N24hopM
2PU
NO
N28hopM
2PU
PTB
71hopM2
PTB
69hopM2
PT
69hopM1
PT
B65hopM
2G
US
2766ChopM
1G
SE
S569hopM
2PU
GB
EN
35hopM2P
U
JPJP
30AD
hopM1P
U
JPJ167hopM
1PU
KR
NU
D36hopM
1G
JPJ188hopM
1PU
KR
NU
D34hopM
1G
JPF
57hopM1G
C
KR
NU
D47hopM
1GK
RD
ual23
hopM
1P
U
KR
NU
D14
hopM
1GK
RD
ual17
hopM
1P
UJP
JPT
1160
Cho
pM1G
JPJ83
hopM
1GC
VE
v225d
hopM
1G
PE
Cuz2
0hop
M1
PE
Sh
i470
hopM
1G
PE
Sa
t464h
opM
1
JPJ1h
opM
1P
U
KR
dua
l5hop
M1
PU
US
305
2Ch
opM
1G
KR
51h
opM
1PU
KR
dua
l15ho
pM1P
U
SE
5hop
M1P
U GB
EN
30ho
pM1
PU
GB
EN
36ho
pM1
PU
NO
N3
0hop
M1
PU
SE
10ho
pM2
PU
GB
EN
37ho
pM2
PU
ZA
Sou
thA
frica7ho
pM2
DE
942
hopM
2P
U
PT
B36
hopM
2
PT
285/9
9hop
M2P
UP
T17
76/05
hopM
2P
TB
68ho
pM2P
UP
TB
13ho
pM1G
PT
B25
hopM
2G
PT
B25hopM
1G
BR
358.96hopM2P
U
BR
358.96hopM1P
UFR
2432hopM2G
PTB
31hopM2G
PT38hopM
2G
US
2718ChopM
2G
US
2718ChopM
1G
PTB
15hopM2P
U
PTB
6hopM2
PTB
2hopM1P
U
DE948hopM
2G
PTB
19hopM2P
U
PT224/99hopM
2PU
PTB
21hopM2G
PTB47hopM2
PT228/99hopM
2G
PT207/99hopM2G
PT207/99hopM1G
US1904hopM2PU
SE57hopM1G
USJ99hopM1PU
USJ99hopM2PU
PT173/00hopM2G
PT173/00hopM1G
SE18hopM1PU
PT70hopM2PU
PTB2hopM2PUUS2779ChopM2G
BR1126.95hopM2PU
BR1311.95hopM2GC
BF11ABFhopM2PU
BF11ABFhopM1PU
PTB51hopM2PU
PT107hopM1G
US1675DhopM2PU
PTB17hopM2
PT145/99hopM1PU
FRF93hopM2PU
PT2hopM2PU
USB8hopM2PU
PT771/99hopM2
SG8033hopM2PTB16hopM2PU
PTB10hopM2GPT219/99hopM2PU
PTB8hopM2 CO967DhopM2PU PTB18hopM2 PTB9hopM2PUPTB14hopM2
0,02
100
hopN
hopM
Cluster II
Cluster II
86
86
Figura 4: Análise filogenética de 379 sequências, (147 hopM e 232 hopN) de H. pylori. O país de origem (PT,
Portugal; FR, França; SE, Suécia; DE, Alemanha; GB, Inglaterra; NO, Noruega; US, EUA; CO, Colômbia; JP, Japão; KR, Coreia; SP, Singapura; BF, Burquina-Faso) e a patologia (PU, úlcera péptica; G, gastrite; GC, cancro gástrico) estão representados no início e no fim da designação de cada estirpe clínica, respectivamente. O valor do índice de comprimento dos ramos está representado na árvore. Os números junto dos ramos principais indicam os valores de bootstrap>75 depois de 1000 interacções. Os círculos tracejados assinalam os dois clusters originados por segregação geográfica das estirpes. Cluster I, a azul corresponde ao grupo ocidental; cluster II, a vermelho corresponde ao grupo asiático.
Seguidamente foi avaliado o grau de similaridade entre as sequências de nucleótidos
de hopM e hopN. A observação do gráfico de similaridade sugere a existência de 4 regiões
distintas nos dois genes, à semelhança do que foi descrito recentemente num trabalho
analisando apenas estirpes de referência [118], designadas por segmento 1 (hopM: 1-
360pb; hopN: 1-360pb), segmento 2 (hopM: 360-873pb; hopN: 360-867pb), segmento 3
(hopM: 873-1824pb; hopN: 867-1818pb) e segmento 4 (hopM: 1824-2085pb; hopN: 1818-
2079pb) (Figura 5).

Resultados
Andrea Santos
18
Segmento 1 Segmento 2 Segmento 3 Segmento 4
Sim
ilari
dade
(%
)
Posição (pb)
hopM
hopN
Região que diferencia os dois genes
Figura 5: Representação gráfica da similaridade entre as 379 sequências de nucleótidos dos genes hopM e hopN de estirpes de Helicobacter pylori. Os gráficos foram gerados com o modelo Kimura 2-parameter, uma janela de 200pb, um passo de 20pb, sem Gap Strip e como referência a sequência hopM da estirpe HPAG1.
O segmento 1 corresponde à região 5’ e é uma zona relativamente conservada entre os
dois genes, apresentando uma similaridade superior a 84% na região mais extensa deste
segmento. Observa-se no entanto que nos primeiros 100 pb em ambos os genes, esta
similaridade diminui para aproximadamente 64%, o que pode reflectir a variação no número
de repetições CT observada nesta zona. O segmento 2 é a região que apresenta maior
variabilidade (similaridade mínima≈34%) e que corresponde à região de diferenciação dos
dois genes, enquanto o segmento 3 é uma zona bastante conservada, tanto entre genes
como intragene (similaridade≈80%). No segmento 4, observa-se um grau de polimorfismo
considerável (similaridade mínima≈68%), mas ao contrário dos segmentos anteriores, a
diversidade é observada quer entre sequências do mesmo gene, quer entre sequências dos
dois genes. Esta observação aponta para a existência de variantes alélicas, com zonas de
cruzamento entre as sequências das diferentes estirpes, sugerindo fenómenos de
recombinação.
Relativamente aos parâmetros evolutivos, calcularam-se para os dois genes as distâncias
moleculares das sequências de nucleótidos e de aminoácidos, e foi determinado o número
de substituições sinónimas (Ks) e não sinónimas (Ka), assim como a razão Ka/Ks. Dentro de
cada gene, as distâncias moleculares obtidas para as sequências nucleotídicas foram para o
gene hopM de 0,084±0,003 e para hopN de 0,079±0,003 (Tabela 2). Observou-se ainda que
as duas sequências da dupla cópia do mesmo gene (parólogos) eram mais próximas entre
si (0,025±0,004 para a dupla cópia hopM e 0,026±0,003 para a dupla cópia hopN), do que o
mesmo gene em estirpes diferentes (ortólogos) (Tabela 2). Relativamente à sequência de
aminoácidos, as distâncias moleculares obtidas foram bastante similares (Tabela 2). As
frequências Ks e Ka foram semelhantes para os dois genes, bem como o valor da razão
Ka/Ks (Tabela 2), verificando-se uma maior proporção de substituições sinónimas. Assim,

Resultados
Andrea Santos
19
tendo em conta que as substituições sinónimas são mais frequentes do que as não
sinónimas (Ks>Ka) e que o resultado da razão Ka/Ks é inferior a um, podemos especular
que o tipo de selecção que está a actuar sobre estes genes é a selecção negativa ou
purificadora [117]. Esta hipótese foi testada pelo método Codon-Based Z-test [119], sendo
suportada estatisticamente para os dois genes (pZ-test<0,001).
Foram também comparados os dois genes entre si e obteve-se para a sequência de
nucleótidos a distância molecular de 0,146±0,006 e para a proteína a distância de
0,161±0,011, o que corresponde a uma percentagem de identidade de 85,4% de 83,9%,
respectivamente.
Tabela 2: Análise das distâncias moleculares, substituições sinónimas e não sinónimas para as sequências correspondentes ao gene completo e segmentos 1, 2, 3 e 4 dos genes hopM e hopN.
hopM (n=147) Distância Molecular Ks
& Ka
& Ka/Ks
#
(nt) (a.a) Gene inteiro 0,084±0,003* 0,073±0,005 0,287±0,016 0,041±0,003 0,143±0,013 Segmento 1 0,073±0,007 0,041±0,009 0,280±0,040 0,025±0,006 0,089±0,025 Segmento 2 0,062±0,005 0,056±0,009 0,187±0,023 0,031±0,005 0,166±0,034 Segmento 3 0,088±0,005 0,076±0,008 0,327±0,026 0,041±0,005 0,110±0,015 Segmento 4 0,128±0,012 0,142±0,023 0,427±0,077 0,084±0,016 0,197±0,052
hopN (n=232) Distância Molecular Ks
& Ka
& Ka/Ks
#
(nt) (a.a) Gene inteiro 0,079±0,003 0,071±0,005 0,266±0,016 0,038±0,003 0,143±0,014 Segmento 1 0,076±0,008 0,044±0,010 0,291±0,039 0,026±0,006 0,089±0,024 Segmento 2 0,055±0,005 0,063±0,010 0,147±0,020 0,032±0,005 0,218±0,045 Segmento 3 0,079±0,005 0,066±0,008 0,300±0,025 0,035±0,005 0,117±0,019 Segmento 4 0,126±0,012 0,143±0,023 0,427±0,076 0,084±0,015 0,197±0,050
n, número de sequências analisadas. nt, nucleótidos; aa, aminoácidos &Ks, substituições sinónimas; Ka, substituições não-sinónimas.
* valor±erro. # pZ-test<0,001 para hipótese purificadora (Ka/Ks<1).
De uma maneira geral, dentro de cada gene, observa-se que o segmento 2 é o mais
conservado, enquanto o segmento 4 é o que apresenta maior distância molecular, quer ao
nível da sequência de nucleótidos quer ao nível da sequência de aminoácidos, corroborando
a análise do gráfico de similaridade (Figura 5). Curiosamente o segmento 2 do gene hopN,
apresenta uma distância molecular maior nas sequências de aminoácidos, do que nas
nucleotídicas, o que está de acordo com a maior razão Ka/Ks observada neste segmento,
comparativamente com os restantes segmentos. Relativamente às frequências Ks e Ka é de
realçar a maior taxa de substituição no segmento 4, com particular ênfase nas substituições
não sinónimas (aproximadamente quatro vezes superior). No global, os parâmetros
evolutivos mostram que este segmento é o mais variável dentro de cada gene, apoiando a
hipótese de constituir uma região de variação alélica (Tabela 2). À semelhança do que foi
observado para o gene completo, o valor da razão Ka/Ks obtido para cada um dos

Resultados
Andrea Santos
20
segmentos, foi inferior a um, sugerindo a existência de uma pressão evolutiva para a
manutenção das sequências de cada segmento. Esta hipótese foi testada pelo método
Codon-Based Z-test [119], sendo suportada estatisticamente (pZ-test<0,001) nos quatro
segmentos de ambos os genes.
3. Variação alélica
O segmento 4 de ambos os genes revelou-se bastante polimórfico, apresentando
diferenças nucleótidicas acentuadas, tanto entre genes (Figura 5) como dentro de cada
gene (Figura 6). Este polimorfismo é consistente com a elevada taxa de substituição de
nucleótidos observada neste segmento (Tabela 2). Para além disso, observa-se uma maior
distância molecular das sequências deste segmento, comparativamente com os outros três,
sendo esta distância ainda maior se compararmos as sequências de aminoácidos, o que
corrobora a ideia da existência de variantes alélicas.
Assim, a partir de um alinhamento gerado com sequências de aminóacidos
correspondentes ao segmento 4 dos genes hopN e hopM (Figura 7A), observou-se a
existência de 4 variantes alélicas distintas, designadas por AI, AII, AIII e AIV, comuns a
ambos os genes e com grau de similaridade entre genes (Tabela 3) idêntico ao grau de
similaridade dentro do mesmo gene (resultado não mostrado). Estas variantes foram
identificadas em todas as estirpes estudadas, embora com frequências diferentes. Assim
foram observados dois alelos predominantes, os alelos AI (39,2%, 149/379) e o AII (42%,
159/379), sendo as frequências dos alelos AIII e AIV de 12,9% (49/379) e de 5,8% (22/379),
respectivamente.
Tabela 3: Análise das percentagens de identidade obtidas entre as sequências do segmento 4 de cada uma das
variantes alélicas de hopM e hopN.
Alelo hopM vs alelo hopN (n=379) % Similaridada AI (n=149) AII (n=159) AIII (n=49) AIV (n=22)
nt 94,5 93,3 93,7 94,2 aa 95 94,8 94,6 96,4
n, número de sequências analisadas. nt, sequência de nucleótidos; aa, sequência de aminoácidos
A análise dos gráficos de similaridade por variante alélica (Figura 8), em conjunto com a
análise do gráfico de similaridade considerando todos os alelos (Figura 6), suporta a
definição do segmento 4 como a região alélica, já que dentro de cada alelo esta região é das
mais conservadas (Figura 8) e a mais diversa quando se consideram todos os alelos do
mesmo gene (Figura 6). A análise dos gráficos de similaridade considerando todos os alelos
(Figura 5 e 6), bem como a análise das sequências de aminoácidos do segmento 4 (Figura
7A) sugere que os alelos AIII e AIV resultam de fenómenos de recombinação entre as
variantes AI e AII. Esta hipótese é ainda suportada pela análise da similaridade dos alelos
dois a dois, verificando-se que os pares mais afastados são o AI/AII e o AIII/AIV
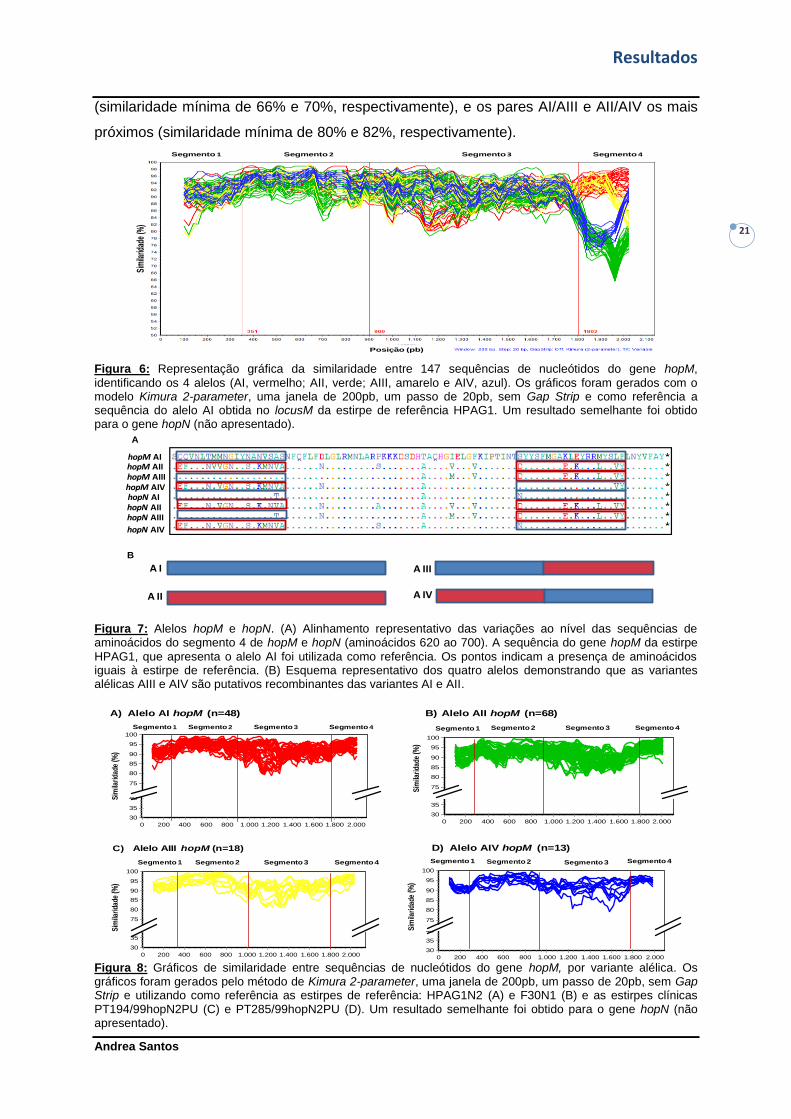
Resultados
Andrea Santos
21
(similaridade mínima de 66% e 70%, respectivamente), e os pares AI/AIII e AII/AIV os mais
próximos (similaridade mínima de 80% e 82%, respectivamente). Si
mila
ridad
e (%
)
Posição (pb)
Segmento 4Segmento3Segmento2Segmento 1
Figura 6: Representação gráfica da similaridade entre 147 sequências de nucleótidos do gene hopM,
identificando os 4 alelos (AI, vermelho; AII, verde; AIII, amarelo e AIV, azul). Os gráficos foram gerados com o modelo Kimura 2-parameter, uma janela de 200pb, um passo de 20pb, sem Gap Strip e como referência a sequência do alelo AI obtida no locusM da estirpe de referência HPAG1. Um resultado semelhante foi obtido para o gene hopN (não apresentado).
A I A III
A II A IV
hopM AIhopM AII
hopN AIII
hopN AIV
hopN AIIhopN AI
hopM AIIIhopM AIV
A
B
Figura 7: Alelos hopM e hopN. (A) Alinhamento representativo das variações ao nível das sequências de aminoácidos do segmento 4 de hopM e hopN (aminoácidos 620 ao 700). A sequência do gene hopM da estirpe
HPAG1, que apresenta o alelo AI foi utilizada como referência. Os pontos indicam a presença de aminoácidos iguais à estirpe de referência. (B) Esquema representativo dos quatro alelos demonstrando que as variantes alélicas AIII e AIV são putativos recombinantes das variantes AI e AII.
PT285/99hopM1PU
PTB25hopM2G
PTB36hopM2
BF11ABFhopM1PU
PTB13hopM2G
DE942hopM2PU
US2779ChopM2G
FR2432hopM2G
PTB31hopM1G
PTB68hopM1PU
PT38hopM1G
Window : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y S
core
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
PT285/99hopM1PU
PTB25hopM2G
PTB36hopM2
BF11ABFhopM1PU
PTB13hopM2G
DE942hopM2PU
US2779ChopM2G
FR2432hopM2G
PTB31hopM1G
PTB68hopM1PU
PT38hopM1G
Window : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y S
core
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
USJ99hopM2PU
PESJM180hopM1G
PTB47hopM2
PTB19hopM2PU
PTB21hopM2G
PTB15hopM2PU
PT28hopM2PU
DE948hopM2G
DE952hopM2G
CO216ChopM2G
BR313.03hopM2G
NON29hopM1PU
PTB2hopM1PU
PT224/99hopM2PU
Window : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y S
core
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
USJ99hopM2PU
PESJM180hopM1G
PTB47hopM2
PTB19hopM2PU
PTB21hopM2G
PTB15hopM2PU
PT28hopM2PU
DE948hopM2G
DE952hopM2G
CO216ChopM2G
BR313.03hopM2G
NON29hopM1PU
PTB2hopM1PU
PT224/99hopM2PU
Window : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y S
core
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
PTB51hopM1PU
BR1126.95hopM2PU
NON30hopM1PU
US3052ChopM1G
JPJ83hopM1GC
JPJP30ADhopM1PU
JPJPT160ChopM1G
JPJ1hopM1PU
JPJ167hopM1PU
KRDual17hopM1PU
KRNUD14hopM1G
KRNUD47hopM1G
KRNUD34hopM1G
KRNUD36hopM1G
KRDual15hopM1PU
KRDual5hopM1PU
PEShi470hopM1G
KR51hopM1PU
JPF57hopM1GC
PECuz20hopM1
PESat464hopM1Window : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y Sc
ore
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
PT655/99hopM2G
US2718ChopM2G
SE58hopM1PU
DE950hopM1G
DE955hopM1G
KRDU852hopM1PU
KRDual30hopM1PU
PEPeCan4hopM1GC
PTB25hopM1G
PTB57hopM2G
PTB57hopM1G
PT224/99hopM1PU
PT417/02hopM2
PT417/02hopM1PU
BR358.96hopM2PU
BR358.96hopM1PU
SES569hopM1PU
FRHorhopM2PU
FRHorhopM1PU
US2781ChopM2G
US2781ChopM1GWindow : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y Sc
ore
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
PTB51hopM1PU
BR1126.95hopM2PU
NON30hopM1PU
US3052ChopM1G
JPJ83hopM1GC
JPJP30ADhopM1PU
JPJPT160ChopM1G
JPJ1hopM1PU
JPJ167hopM1PU
KRDual17hopM1PU
KRNUD14hopM1G
KRNUD47hopM1G
KRNUD34hopM1G
KRNUD36hopM1G
KRDual15hopM1PU
KRDual5hopM1PU
PEShi470hopM1G
KR51hopM1PU
JPF57hopM1GC
PECuz20hopM1
PESat464hopM1Window : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y Sc
ore
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
PT655/99hopM2G
US2718ChopM2G
SE58hopM1PU
DE950hopM1G
DE955hopM1G
KRDU852hopM1PU
KRDual30hopM1PU
PEPeCan4hopM1GC
PTB25hopM1G
PTB57hopM2G
PTB57hopM1G
PT224/99hopM1PU
PT417/02hopM2
PT417/02hopM1PU
BR358.96hopM2PU
BR358.96hopM1PU
SES569hopM1PU
FRHorhopM2PU
FRHorhopM1PU
US2781ChopM2G
US2781ChopM1GWindow : 200 bp, Step: 20 bp, GapStrip: Off, Kimura (2-parameter), T/t: Variable
Position
2.0001.8001.6001.4001.2001.0008006004002000
Sim
ilarit
y S
core
100
95
90
85
80
75
70
65
60
55
50
45
40
35
30
Alelo AI hopM (n=48)
Sim
ilari
dade
(%)
A)
Segmento 1 Segmento2
Alelo AII hopM (n=68)
Alelo AIII hopM (n=18) Alelo AIV hopM (n=13)
Sim
ilari
dade
(%)
Sim
ilari
dade
(%)
Sim
ilari
dade
(%)
C)
B)
D)
Segmento 1 Segmento 2 Segmento 3 Segmento 4 Segmento 2 Segmento 3 Segmento 4Segmento 1
Segmento 1 Segmento 2 Segmento 3 Segmento 4Segmento 3 Segmento4
Figura 8: Gráficos de similaridade entre sequências de nucleótidos do gene hopM, por variante alélica. Os gráficos foram gerados pelo método de Kimura 2-parameter, uma janela de 200pb, um passo de 20pb, sem Gap Strip e utilizando como referência as estirpes de referência: HPAG1N2 (A) e F30N1 (B) e as estirpes clínicas PT194/99hopN2PU (C) e PT285/99hopN2PU (D). Um resultado semelhante foi obtido para o gene hopN (não apresentado).

Resultados
Andrea Santos
22
3.1 Distribuição dos alelos por gene e origem geográfica
No geral, foram observados dois alelos predominantes em ambos os genes, os alelos AI
e o AII. No gene hopM o alelo AI estava presente em 32,7% das estirpes (48/147) e o alelo
AII em 46,3% (68/147). No gene hopN o alelo mais frequente foi o AI em 43,5% das estirpes
(101/232), seguido do AII em 39,2% das estirpes (91/232). Os alelos AIII e AIV foram
encontrados menos frequentemente em ambos os genes (Figura 9).
AI32,7%
AII46,3%
AIII12,2%
AIV8,8%
AI43,5%
AII39,2%
AIII13,4%
AIV3,9%
Alelos hopM (n=147) Alelos hopN (n=232)
Figura 9: Distribuição dos alelos por gene, na população geral.
Posteriormente, foi estimada a frequência de cada alelo por grupo geográfico (Figura 10).
AI35,7%
AII37,5%
AIII15,2%
AIV11,6%
Grupo Ocidental (n=275)
Alelos hopM (n=112)
AI59,5%
AII14,1%
AIII17,8%
AIV8,6%
Alelos hopN (n=163)
AI11,1%
AII81,5%
AIII7,4%
Grupo Asiático (n=81)
Alelos hopM (n=27)
AI9,3%
AII87,0%
AIII3,7%
Alelos hopN (n=54)
AI37,5%
AII25%
AIII25%
AIV12,5%
Alelos hopM (n=8)
Grupo Africano (n=23)
AI86,7%
AII13,3%
Alelos hopN (n=15)
Figura 10: Distribuição dos alelos de cada gene de acordo com a origem geográfica das estirpes de H. pylori.
Na população ocidental observou-se a presença das quatro variantes alélicas em cada
gene, sendo os alelos mais frequentes os alelos AI no gene hopN e o alelo AII no gene
hopM. No grupo de estirpes de origem asiática, o alelo AII foi o mais frequentemente
observado em ambos os genes e o alelo AIV não foi detectado. Na população africana, no
gene hopM foram identificados as quatro variantes alélicas, sendo a mais frequente a
variante AI. No gene hopN o alelo AI foi igualmente o mais prevalente e curiosamente não
foram detectadas as variantes recombinantes (AIII e AIV) (Figura 10).

Resultados
Andrea Santos
23
Globalmente, verificou-se que na população ocidental e africana o alelo AI foi o mais
prevalente, com frequências de 49,8% (137/275) e de 69,6% (16/23) respectivamente,
enquanto o AII foi o alelo mais prevalente na população de origem asiática (85,2%, 69/81).
3.2 Distribuição dos alelos dos genes hopM e hopN de acordo com a patologia e
perfil de virulência
Com o intuito de verificar uma possível associação entre os alelos hopM/hopN e a
patologia ou o perfil de virulência das estirpes, foi avaliada a distribuição dos alelos dos dois
genes em conjunto e separadamente, tendo em conta estes dois parâmetros. Para esta
análise foram excluídas as estirpes isoladas de doentes sem informação da patologia
associada ou para as quais não tinha sido previamente determinado o genótipo de virulência
(cagA e vacAs1 positivos). Os valores de p apresentados referem-se à comparação entre ter
ou não o alelo por patologia (G ou UP) ou o alelo por genótipo de virulência (virulento/não
virulento).
3.2.1 Distribuição dos alelos hopM/hopN de acordo com a patologia
No geral, verifica-se uma frequência muito semelhante de cada um dos alelos tanto na
UP como na G, à expeção do alelo AIV que apresenta uma frequência superior na G
relativamente à UP. O alelo AII foi o mais prevalente nas estirpes de CG (Figura 11).
% d
e e
sti
rp
es p
osit
ivas
(n=191)
(n=127)
(n=15)
População Geral (n=333)
0
10
20
30
40
50
60
70
80
90
I II III IV
45,5
34,0
15,2
5,2
43,3
33,9
12,6 10,213,3
86,7
UP
G
CG
0
10
20
30
40
50
60
I II III IV
52,7
22,9
16,8
7,6
52,9
25,5
10,8 10,8
25,0 25,0
50,0
UP G CG
Grupo Ocidental (n=237)
(n=131) (n=102) (n=4)
% d
e e
sti
rp
es p
osit
ivas
0
20
40
60
80
100
I II
13,5
86,5
4,5
77,3
5,9
70,6
UP G CG
% d
e e
sti
rp
es p
osit
ivas
Grupo Asiático (n=76)
(n=37) (n=22) (n=17)
A
B C
Figura 11: Frequência das 4 variantes alélicas de acordo com a patologia, na população geral (A) e por origem
geográfica (B e C).

Resultados
Andrea Santos
24
Por origem geográfica, na população ocidental (Figura 11B) verifica-se a mesma
tendência observada na população geral, não se observando nenhuma associação
estatisticamente significativa entre alelo e patologia. Os dados mostram no entanto, uma
associação entre o alelo AIII e o CG, embora sem significado estatístico dado o pequeno
número de estirpes estudadas. Na população asiática (Figura 11C), o alelo AII foi mais
frequentemente encontrado em todas as patologias, devido provavelmente à sua elevada
prevalência nesta população. No grupo africano, as estirpes foram todas isoladas de
doentes com UP, sendo a distribuição dos alelos AI, AII, AIII e AIV respectivamente de 65%
(13,20), 15% (3,20), 15% (3,20) e 5% (1/20).
3.2.2 Distribuição dos alelos do gene hopM e hopN por perfil de virulência
Foi também avaliada a associação entre os alelos hopM/hopN e os genótipos de
virulência de H. pylori. No geral, verifica-se uma maior prevalência dos alelos AII e AIII nas
estirpes com perfil mais virulento e dos alelos AI e AIV com o genótipo menos virulento,
apesar das diferenças observadas não serem suportadas estatisticamente (Figura 12).
0
5
10
15
20
25
30
35
40
45
50
I II III IV
42,7
36,2
16,1
5,0
48,3
34,8
7,9 9,0
virulento
não virulento
% d
e es
tirp
es p
osi
tiva
s
População Geral (n=288)
(n=199)
(n=89)
Figura 12: Frequência dos alelos hopM+hopN de acordo com genótipo de virulência da estirpe, na população
geral.
Considerando a distribuição dos alelos por genótipo de virulência em cada grupo
geográfico, verifica-se na população ocidental (Figura 13), que os alelos AI e AIII estão
associados ao genótipo de virulência, sendo a associação do AIII ao perfil mais virulento,
estatisticamente significativa. Os alelos AII e AIV são mais prevalentes no grupo de estirpes
com genótipo menos virulento, tendo a associação do Alelo AII ao genótipo menos virulento
suporte estatístico. Estas mesmas associações foram observadas considerando apenas os
alelos hopN (Figura 13C), não se verificando na distribuição dos alelos hopM (Figura 13B).
Na população asiática, as estirpes são todas do genótipo virulento, independemente do
alelo que apresentam, de acordo com o que foi descrito anteriormente [84,103]. No entanto,
o alelo AII foi o mais frequentemente detectado, com uma frequência de 85,2% (69/81) o
que parece indicar uma forte relação deste alelo com esta população. Na população

Resultados
Andrea Santos
25
africana, o alelo AIV não foi encontrado associado ao genótipo de virulência e os alelos AII e
AIII foram identificados apenas em estirpes com perfil virulento, apresentando ambos uma
frequência de 18,2% (2/11) respectivamente. Relativamente ao alelo AI, foi observado tanto
em estirpes virulentas como em não virulentas, sendo a sua frequência respectivamente de
63,6% (7/11) no grupo de estirpes com perfil virulento e de 100% (n=3) no grupo menos
virulento.
0
10
20
30
40
50
60
I II III IV
57,8
13,3
21,1
7,8
45,9
35,3
8,210,6
Virulento
não virulento
Alelos hopM + hopN (n=213)A
% d
e e
sti
rp
es p
osit
ivas
(n=128)
(n=85)
0
10
20
30
40
50
I II III IV
37,9
27,6
20,7
13,8
35,1
48,6
8,1 8,1
Virulento não virulento
Alelos hopMB
% d
e e
sti
rp
es p
osit
ivas
(n=29) (n=37)
0
10
20
30
40
50
60
70
I II III IV
63,6
9,1
21,2
6,1
54,2
25,0
8,312,5
virulento não virulento
% d
e e
sti
rp
es p
osit
ivas
Alelos hopNC
(n=99) (n=48)
p=0,003
p=0,02
p=0,02p=0,05
Figura 13: Distribuição das quatro variantes alélicas na população ocidental, considerando os genes em conjunto (A) e separadamente (B e C), de acordo com o genótipo de virulência das estirpes de H.pylori.

Discussão e Conclusão
Andrea Santos
26
Capítulo 5: Discussão e Conclusão
O presente trabalho teve como objectivo o estudo de dois membros das OMPs de H.
pylori, hopM e hopN, mais concretamente o estudo da sua diversidade genética, evolução e
a avaliação da sua associação com a patologia gástrica e com marcadores moleculares de
virulência de H. pylori, nomeadamente com os genes cagA e vacA.
Foram estudadas 234 estirpes clínicas e 23 estirpes de referência isoladas de doentes de
origem geográfica diferente e apresentando diversas gastropatologias. Em todas as estirpes
foi identificada pelo menos uma cópia de um destes genes no seu genoma, o que sugere a
existência de pressão selectiva para serem mantidos na bactéria, como já foi descrito para
outros genes codificantes de OMPs, tais como homA/homB, babA/babB, sabA e oipA
[99,104,120].
A análise de ambos os genes revelou diversidade tanto relativamente ao número de
cópias de cada gene como à sua localização genómica. Esta diversidade não foi associada
à patologia da qual a estirpe foi isolada, mas mostrou especificidade geográfica, havendo
uma diferença clara entre as estirpes de origem oriental e as de origem ocidental, à
semelhança do que foi descrito para outros factores de virulência de H. pylori [63,64,88,121-
123]. Na população ocidental todos os genótipos possíveis de hopM/hopN foram
identificados (cópia única ou dupla cópia, do mesmo gene ou genes diferentes), enquanto
na população asiática apenas os genótipos de cópia única foram observados.
Curiosamente, na população africana apenas foram identificados os genótipos de dupla
cópia, embora dado o número reduzido de estirpes analisadas, não se possa concluir se
este resultado reflecte uma tendência desta população, ou não. Vários estudos apontam
para que a variação no número de cópias de genes que codificam OMPs, esteja implicada
na adaptação da bactéria a hospedeiros específicos, sendo um factor crucial para o
desenvolvimento de uma infecção crónica e persistente [56,100,120]. Assim, sugere-se que
a especificidade geográfica no número de cópias hopM/hopN reflicta igualmente o papel
destes genes neste processo adaptativo.
O facto de estes genes terem demonstrado uma elevada similaridade tanto ao nível da
sequência de nucleótidos (Tabela 2) como na sequência de aminoácidos, especialmente na
região 5’ e 3’ (Tabela 2, segmentos 1 e 3), sugere a possibilidade de ocorrência de
fenómenos de recombinação intra e intergenómica, que podem levar a duplicação, delecção
ou conversão génica entre hopM e hopN, como resposta a variações ambientais. A
presença de uma ORF mais pequena no locusN de todas as estirpes asiáticas e de algumas
estirpes ocidentais, sugere que nestes casos o gene foi perdido, deixando um remanescente
que pode possibilitar a sua integração novamente no genoma, tal como foi sugerido para

Discussão e Conclusão
Andrea Santos
27
outros genes codificantes de OMPs de H.pylori (homA/homB, babA/babB) [64].
Curiosamente, estas proteínas putativas mais pequenas correspondem nas estirpes
ocidentais à região alélica (segmento 4) e no caso das estirpes asiáticas à fusão do péptido
sinal de hopM/hopN com a região 3’ de uma ADN-metiltransferase, constituindo assim locais
onde a elevada homologia com os genes, facilitam a recombinação e a integração do gene
no locus vazio. Um estudo recente sugere que estes fenómenos de perda e ganho de hopM
e hopN estão associados a um mecanismo complexo de recombinação, no qual ocorre uma
duplicação seguida de inversão de uma extensa região cromossómica [118,124].
A análise das sequências dos genes hopM e hopN mostrou, na maioria das estirpes
estudadas, a presença de uma ORF completa, havendo no entanto cerca de 2,6% das
estirpes com sequências truncadas. Em todas estas estirpes o desfasamento da grelha de
leitura deveu-se ao desemparelhamento entre as bases complementares (misparing),
resultando na adição ou delecção de unidades CT na região 5’ de ambos os genes. Esta
alteração do número de repetições pode originar a adição ou remoção de codões STOP,
alterando a expressão da proteína devido à mudança de fase on/off [60]. Assim, em ambos
os genes pode ocorrer variação de fase e regulação da expressão génica pelo mecanismo
de slipped-strand misparing, à semelhança do que já foi descrito para outros genes de
OMPs, tais como babB, sabA e sabB, hopZ, oipA e homB [64,104,105,108,125]. A variação
de fase como mecanismo de diversidade é utilizada por várias espécies bacterianas e tem
sido sugerido como marcador consistente de genes envolvidos da adaptação a um nicho
específico ou na evasão à resposta imunitária, contribuindo para a variação antigénica e/ou
virulência durante a infecção [60,127,128]. Um exemplo deste mecanismo foi demonstrado
para os genes babA e babB, no qual a mudança de fase de leitura possibilita à bactéria a
constituição de uma população heterogénea, com dinâmicas de adesão diferentes
permitindo-lhe adaptar-se a diferentes condições ambientais [99]. Assim, tendo em conta a
possibilidade de ambos os genes hopM e hopN serem regulados por variação de fase e por
eventos de recombinação intra e intergenómica, podemos mais uma vez especular que
estes genes estão envolvidos na adaptação de H. pylori ao seu hospedeiro humano.
Um estudo de complexos de proteínas de H. pylori mostrou que BabA e BabB formam
complexos com HopM e HopN [109]. Por outro lado, estudos recentes sugerem que
nenhuma das proteínas BabA ou BabB é capaz de induzir resposta imunitária em macacos
[109,111], nem são os antigénios imunodominantes em humanos [109,112]. Uma hipótese
que poderia explicar este facto seria a de que as proteínas de membrana que interagem
com BabA ou BabB, por exemplo, HopM e HopN, poderiam mascarar os epítopos daquelas
proteínas e consequentemente serem elas próprias expostas e assim antigénicas [109].
Esta hipótese é ainda corroborada pelo facto de as proteínas HopN e HopM terem
demonstrado reactividade contra soros de pacientes positivos para H. pylori [109,113].

Discussão e Conclusão
Andrea Santos
28
Todos estes dados sugerem que as proteínas HopM e HopN poderão desempenhar um
papel importante na relação entre a bactéria e o seu hospedeiro, nomeadamente,
constituindo prováveis antigénios e consequentemente importantes alvos vacinais [109,110],
ou por outro lado, poderão desempenhar um papel na adesão de H. pylori à célula epitelial
gástrica, visto que formam complexos com uma das principais adesinas, BabA [109,110].
Com o objectivo de tentar clarificar o papel destes genes na virulência das estirpes de
H.pylori, foram avaliadas as associações entre a presença dos genes hopM/hopN e a
patologia e o perfil de virulência da estirpe. No geral, foi verificada uma associação
estatisticamente significativa entre a cópia única hopN e o genótipo mais virulento (cagA e
vacAs1 positivas), e entre a dupla cópia hopM e a dupla cópia MN e o genótipo menos
virulento (cagA negativo, vacAs2). Verificou-se ainda, na população de origem ocidental, a
associação estatisticamente significativa entre a dupla cópia hopN e o genótipo mais
virulento e da dupla cópia hopM e o perfil menos virulento. Mais uma vez, estes dados
sugerem um papel determinante destes genes na relação entre a bactéria e o seu
hospedeiro. A clarificação das suas funções é por isso crucial e será posteriormente
avaliada através de estudos funcionais das respectivas proteínas, nomeadamente a
avaliação das propriedades antigénicas e da contribuição de HopM e HopN para as
características pró-inflamatórias de H. pylori, bem como o esclarecimento do envolvimento
destas proteínas na adesão à mucosa gástrica.
A reconstrução filogenética dos genes hopM e hopN sugere uma segregação baseada na
origem geográfica da estirpe, com as estirpes de origem ocidental e asiática a apresentarem
uma maior divergência entre si. Esta divergência foi também observada noutros genes de H.
pylori, tais como, genes que codificam outras OMPs (homA/homB e babA/babB), genes de
virulência (cagA e vacA) e genes housekeeping [38,63,107,129].
Os dois genes revelaram um elevado grau de similaridade entre si, com uma distância
moleular de 0,146±0,006 ao nível da sequência de ADN e de 0,161±0,011 na sequência de
aminoácidos. Estes resultados são bastante similares aos observados para outros genes
duplicados de OMPs [54,97] o que sugere a existência de fenómenos de duplicação ou
conversão génica e de recombinação intra e intergenómica entre eles.
Globalmente, os genes hopM e hopN apresentaram taxas de substituição de nucleótidos
muito semelhantes, com predomínio das substituições sinónimas, sugerindo que os dois
genes estão sujeitos aos mesmos constrangimentos funcionais, sendo estes mantidos por
acção da selecção negativa ou purificadora.
De uma maneira geral, dentro de cada gene, observa-se uma maior conservação do
segmento 2 (Figura 5 e 6 e Tabela 2), enquanto o segmento 4 apresenta maior distância
molecular, quer ao nível da sequência de nucleótidos quer ao nível da sequência de
aminoácidos. Mais concretamente, o segmento 4 de ambos os genes revelou um elevado

Discussão e Conclusão
Andrea Santos
29
polimorfismo, apresentando diferenças acentuadas, tanto entre genes (Figura 5) como
dentro de cada gene (Figura 6). Este polimorfismo foi consistente com a elevada taxa de
substituição de nucleótidos observada neste segmento (Tabela 2) e com a maior distância
molecular das sequências deste segmento quando comparado com os outros três,
permitindo identificá-lo como região alélica. Assim, no segmento 4, foi possível identificar
quatro variantes alélicas, comuns aos dois genes, à semelhança do que foi previamente
descrito num trabalho utilizando apenas estirpes de referência [118]. A existência de
variação alélica entre genes que codificam OMPs, é um mecanismo de diversidade
amplamente identificado, tendo sido já várias vezes descrito em H.pylori
[63,64,105,109,110]. Para hopM e hopN, observaram-se duas variantes alélicas globalmente
mais prevalentes (alelos AI e AII) e toda a análise realizada ao nível das sequências sugere
que as outras duas variantes (alelos AIII e AIV), menos prevalentes a nível mundial,
resultaram de fenómenos de recombinação intra ou intergenómicas, entre as variantes mais
frequentes.
No global, nenhum alelo foi significativamente associado com a patologia ou a origem
geográfica da estirpe, provavelmente devido à maior frequência dos tipos AI e AII, tanto nas
diferentes patologias estudadas como nas diferentes origens geográficas. No entanto, são
de sublinhar duas diferenças marcadas, o facto de o alelo AII ser o mais prevalente nas
estirpes asiáticas e não o AI como nas estirpes ocidentais e africanas, e o alelo AIV não ter
sido detectado em nenhuma estirpe asiática.
A distribuição das variantes alélicas pelo genótipo de virulência, na totalidade da
população em estudo, não mostrou nenhuma associação. No entanto, considerando cada
grupo geográfico individualmente, verificou-se na população ocidental a associação
estatisticamente significativa entre o alelo AII e o genótipo menos virulento e do alelo AIII
com o genótipo mais virulento, para os dois genes em conjunto e também para estes alelos
do gene hopN. Estas diferenças verificadas em populações de origem geográfica diferente
podem reflectir a adaptação da bactéria a pressões selectivas distintas, encontradas em
hospedeiros específicos, afectando a sua virulência [63,64]. Assim, a variação alélica
observada nos genes hopM e hopN, para além de contribuir para a variação antigénica,
pode ter um papel importante na virulência da estirpe, numa determinada população
humana, reflectindo o complexo processo evolutivo que ocorre durante uma infecção
persistente, resultado das pressões antigénicas ou variações funcionais a que esteve
sujeita, para persistir numa população hospedeira específica.
Em suma, os resultados obtidos no presente trabalho, sugerem que os genes hopM e
hopN contribuem para a adaptação e logo para a persistência da infecção por H. pylori.
Mais, as variantes alélicas podem constituir biomarcadores da virulência da estirpe em
determinadas populações.

Bibliografia
Andrea Santos
30
Bibliografia:
1. Eaton KA. Motility as a factor in the colonization of gnotobiotic piglets by Helicobacter pylori. Journal of Medical Microbiology 1992; 37: 123-127.
2. Montecucco C, Rappuoli R. Living dangerously: How Helicobacter pylori survive in the human stomach. Nature Reviews Molecular Cell Biology 2001; 2: 457-466.
3. Goodwin SC, Worsley BW. Microbiology of Helicobacter pylori. Gastroenterology Clinics of North America 1993; 22: 5-19.
4. Warren JR. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983; 1: 1273.
5. McNulty CA, Dent JC, Curry A, UFF JS, Ford GA, Gear MWL, Wilkinson SP. New spiral bacterium in gastric mucosa. Journal of Clinical Pathology 1989; 42: 585-591.
6. Doenges JL. Spirochaetes in gastric glands of macacus rhesus and humans without definite history of related disease. Proceedings of the Society for Experimental Biology and Medicine 1938; 38: 536-538.
7. Freeberg AS, Barron LE. The presence of spirochetes in human gastric mucosa. American Journal of Digestive Diseases 1940; 7: 443-445.
8. Goodwin CS, Armstrong JA, Chilvers T, Peters M, Collins MD, Sly L, McConnell W, Harper WES. Transfer of Campylobacter pylori and Campylobacter mustelae to Helicobacter gen. nov. as Helicobacter pylori comb. nov. and Helicobacter mustelae comb. nov., respectively. International Journal of Systematic Bacteriology 1989; 39: 397-405.
9. Andersen LP, Wadstrom T. Basic Bacteriology and Culture. In Mobley HLT, Mendz GL, Hazell SL, Ed. Helicobacter pylori: Physiology and Genetics. Washington DC: ASM Press, 2001; 27-38.
10. Fox JG. The non-H. pylori Helicobacters: their expanding role in gastrointestinal and systemic diseases. Gut 2002; 50: 273-283.
11. Solnick JV, Schauer DB. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clinical Microbiology Reviews 2001; 14: 59-97.
12. Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling HO, Josenhans C, Suerbaum S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proceedings of the National Academy of Sciences of the United States of America 2004; 101: 5024-5029.
13. Dubois A, Boren T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cellular Microbiology 2007; 9: 1108-1116.
14. Brown LM. Helicobacter pylori: Epidemiology and routes of transmission. Epidemiologic Reviews 2000; 22: 283-297.
15. Rowland M, Daly L, Vaughan M, Higgins A, Bourke B, Drumm B. Age-specific incidence of Helicobacter pylori. Gastroenterology 2006; 130: 65-72.
16. Fialho A, Braga A, Neto M, Carneiro J, Rocha A, Queiroz D, Braga L. Younger siblings play a major role in Helicobacter pylori transmission among children from a low-income community in the northeast of Brazil. Helicobacter 2010; 15: 491-496.
17. Portal-Celhay C, Perez-Perez GI. Immune responses to Helicobacter pylori colonization: mechanisms and clinical outcomes. Clinical Science 2006; 110: 305-314.
18. Dattoli V, Veiga R, Cunha S, Pontes-de-Carvalh L, Barreto M, Alcântara-Neves N. Seroprevalence and potential risk factors for Helicobacter pylori infection in Brazilian children. Helicobacter 2010; 15: 213-278.
19. Shi R, Xu S, Zhang H, Ding Y, Huang X, Chen X, Li X, Yan Z, Zhang G. Prevalence and risk factors for Helicobacter pylori infection in Chinese populations. Helicobacter 2008; 13: 157-165.
20. Bardhan PK. Epidemiological features of Helicobacter pylori infection in developing countries. Clinical Infectious Diseases 1997; 25: 973-978.

Bibliografia
Andrea Santos
31
21. Mitchell H, Megraud F. Epidemiology and diagnosis of Helicobacter pylori infection. Helicobacter 2002; 1: 8-16.
22. Monteiro L, Oleastro M, Pelerito A, Falcão M, Rabiais S, Baptista I, Barros I, Cordeiro M, Cabral J, Lopes AI, Ramalho P. Study of prevalence of Helicobacter pylori infection in a paediatric population in Lisbon. Helicobacter 2003; 8: A06.13.
23. Quina MG, G.E.H.P. Helicobacter pylori: the Portuguese scene. Grupo de Estudo Português do Helicobacter pylori (GEPHP). European Journal of Cancer Prevention 1994; 3 (Suppl.2): 65-67.
24. Oleastro M, Pelerito A, Nogueira P, Benoliel J, Santos A, Cabral J, Lopes AI, Ramalho P, Monteiro M. Prevalence and incidence of Helicobacter pylori infection in a healthy pediatric population in the Lisbon area. Helicobacter 2011; 16(5): 363-72.
25. Weyermann M, Rothenbacher D, Brenner H. Acquisition of Helicobacter Pylori in early childhood: independent contributions of infected mothers, fathers and siblings. American Journal of Gastroenterology 2009; 104(1): 182-189.
26. Konno M, Fujii N, Yokota S, Sato K, Takahashi M, Mino E, Sugiyama T. Five year follow-up study of mother-to-child transmission of Helicobacter pylori infection detected by a random amplified polymorphic DNA fingerprinting method. Journal of Clinical Microbiology 2005; 43: 2246-2250.
27. Dore MP, Sepulveda AR, El Zimaity H, Yamaoka Y, Osato MS, Mototsugu K, Nieddu AM, Realdi G, Graham DY. Isolation of Helicobacter pylori from sheep - Implications for transmission to humans. American Journal of Gastroenterology 2001; 96: 1396-1401.
28. Handt L, Fox J, Dewhirst F, Fraser G, Paster B, Yan L, Rozmiarek H, Rufo R, Stalis I. Helicobacter pylori isolated from the domestic cat: public health implications. Infection and Immunity 1994; 62: 2367-2374.
29. Akamatsu T, Tabata K, Hironga M, Kawahami H, Uyeda M. Transmission of Helicobacter pylori via flexible feberoptic endoscopy. American Journal of Infection Control 1996; 24: 396-401.
30. Fantry GT, Zheng QX, James SP. Conventional cleaning and disinfection techniques eliminate the risk of endoscopic transmission of Helicobacter pylori. American Journal of gastroenterology 1995; 90: 227-232.
31. Tytgat GN. Endoscopic transmission of Helicobacter pylori. Alimentary Pharmacology and Therapeutics 1995; 9: 105-110.
32. Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. British Medical Bulletin 1998; 54: 139-150.
33. Marshall BJ, Armstrong JA, McGechie DB, Glancy RJ. Attemps to fulfil Koch's postulates for pyloric Campylobacter. Medical Journal of Australia 1985; 142: 436-439.
34. Akhter Y, Ahmed I, Devi SM, Ahmed N. The co-evolved Helicobacter pylori and gastric cancer: trinity of bacterial virulence, host susceptibility and lifestyle. Infectious Agents and Cancer 2007; 2: 2.
35. Polk D, Peek R. Helicobacter pylori: gastric cancer and beyond. Nature Reviews Cancer 2010; 10: 403-414.
36. Falush D, Kraft C, Taylor NS, Correa P, Fox JG, Achtman M, Suerbaum S. Recombination and mutation during long term gastric colonization by Helicobacter-pylori: Estimates of clock rates, recombination size, and minimal age. Proceedings of the National Academy of Sciences of the United States of America 2001; 98: 15056-15061.
37. Suerbaum S, Achtman M. Evolution of Helicobacter pylori: the role of recombination. Trends in Microbiology 1999; 7: 182-184.
38. Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Molecular Microbiology 1999; 32: 459-470.
39. Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. Genomic-sequence comparison

Bibliografia
Andrea Santos
32
of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1999; 397:176-180.
40. Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, Stein M, Salama NR, Guillemin K. The complete genome sequence of Helicobacter pylori strain G27. Journal of Bacteriology 2009; 191:447-448.
41. Devi S, Taylor T, Avasthi T, Kondo S, Suzuki Y, Megraud F, Ahmed N. Genome of Helicobacter pylori Strain 908. Journal of Bacteriology 2010; 192: 6488-6489.
42. Farnbacher M, Jahns T, Willrodt D, Daniel R, Haas R, Goesmann A, Kurtz S, Rieder G. Sequencing, annotation, and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics 2010; 11:335.
43. Fischer W, Windhager L, Rohrer S, Zeiller M, Karnholz A, Hoffmann R, Zimmer R, Haas R. Strain specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Research 2010; 38: 6089-6101.
44. Gressmann H, Linz B, Ghai R, Pleissner KP, Schlapbach R, Yamaoka Y, Kraft C, Suerbaum S, Meyer TF, Achtman M: Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genetics 2005, 1: e43
45. Kersulyte D, Kalia A, Gilman RH, Mendez M, Herrera P, Cabrera L, Velapatiño B, Balqui J, Paredes Puente de la Vega F, Rodriguez Ulloa CA, Cok J, Hooper CC, Dailide G, Tamma S, Berg DE. Helicobacter pylori from Peruvian amerindians: traces of human migrations in strains from remote Amazon, and genome sequence of an Amerind strain. PLoS One 2010; 5: e15076.
46. Oh JD, Kling-Backhed H, Giannakis M, Xu J, Fulton RS, Fulton LA, Cordum HS, Wang C, Elliott G, Edwards J, Mardis ER, Engstrand LG, Gordon JI. The complete genome sequence of a chronic atrophic gastritis Helicobacter pylori strain: evolution during disease progression. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103: 9999-10004.
47. Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proceedings of the National Academy of Sciences of the United States of America 2000; 97: 14668-14673.
48. Thiberge JM, Boursaux-Eude C, Lehours P, Dillies MA, Creno S, Coppée JY, Rouy Z, Lajus A, Ma L, Burucoa C, Ruskoné-Foumestraux A, Courillon-Mallet A, De Reuse H, Boneca I, Lamarque D, Megraud F, Delchier JC, Médigue C, Bouchier C, Labigne A, Raymond J. From array-based hybridization of Helicobacter pylori isolates to the complete genome sequence of an isolate associated with MALT lymphoma. BMC Genomics 2010; 11:368.
49. Occhialini A, Marais A, Alm R, Garcia F, Sierra R, Megraud F. 2000. Distribution of open reading frames of plasticity region of strain J99 in Helicobacter pylori strains isolated from gastric carcinoma and gastritis patients in Costa Rica. Infection and Immunity. 68:6240-6249.
50. Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proceedings of the National Academy of Sciences of the United States of America 2001; 98: 14625-14630.
51. Kersulyte D, Chalkauskas H, Berg DE. Emergence of recombinant strains of Helicobacter pylori during human infection. Molecular Microbiology 1999; 31: 31-43.
52. Kraft C, Stack A, Josenhans C, Niehus E, Dietrich G, Correa P, Fox JG, Falush D, Suerbaum S. Genomic changes during chronic Helicobacter pylori infection. Journal of Bacteriology 2006; 188: 249-254.
53. Suerbaum S, Achtman M. Helicobacter pylori: recombination, population structure and human migrations. International Journal of Medical Microbiology 2004; 294: 133-139.
54. Pride DT, Blaser MJ. Concerted evolution between duplicated genetic elements in Helicobacter pylori. Journal of Molecular Biology 2002; 316: 629-642.
55. Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proceedings of the National Academy of Sciences of the United States of America 2004; 101: 2106-2111.

Bibliografia
Andrea Santos
33
56. Megraud F. Helicobacter pylori resistance to antibiotics: prevalence, mechanism, detection. What's new? Canadian Journal of Gastroenterology 2003; 17: 49B-52B.
57. Mane S, Dominguez-Bello M, Blaser M, Sobral B, Hontecillas R, Skoneczka J, Mohapatra S, Crasta O, Evans C, Modise T, Shallom S, Shukla M, Varon C, Megraud F, Maldonado-Contreras A, Williams K, Bassaganya-Riera J. Host-interactive genes in Amerindian Helicobacter pylori diverge from their old world homologs and mediate inflammatory responses. Journal of Bacteriology 2010; 192: 3078-3092.
58. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997; 388: 539-47.
59. Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity H, Reddy R, Arnqvist A, Graham D. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut 2006; 55: 775-778.
60. Henderson RI, Owen P, Nataro JP. Molecular switches-the ON and OFF of bacterial phase variarion. Molecular Microbiology 1999; 33: 919-932.
61. Dong QJ, Wang Q, Xin YN, Li N, Xuan SY. Comparative genomics of Helicobacter pylori. World Journal of Gatroenterology 2009; 32: 3984-3991.
62. Oleastro M, Cordeiro R, Menard A, Gomes JP. Allelic diversity among Helicobacter pylori outer membrane protein genes homB and homA generated by recombination. Journal of Bacteriology 2010; 192: 3961-3968.
63. Oleastro M, Cordeiro R, Menard A, Yamaoka Y, Queiroz D, Megraud F, Monteiro L. Allelic diversity and phylogeny of homB, a novel co-virulence marker of Helicobacter pylori. BMC Microbiology 2009; 9: 248.
64. Disotell T. Discovering human history from stomach bacteria. Genome Biology 2003; 4: 213.
65. Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez Perez GI, Yamaoka Y, Megraud F, Otto K, Reichard U, Katzowitsch E, Wang XY, Achtman M, Suerbaum S. Traces of human migrations in Helicobacter pylori populations. Science 2003; 299: 1582-1585.
66. Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, Yamaoka Y, Graham DY, Perez-Trallero E, Wadstrom T, Suerbaum S, Achtman M. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007; 445: 915-918.
67. Moodley Y, Linz B, Yamaoka Y, Windsor H, Breurec S, Wu JY, Maady A, Bernhöft S, Thiberge JM, Phuanukoonnon S, Jobb G, Siba P, Graham D, Marshall B, Achtman M. The peopling of the Pacific from a bacterial perspective. Science 2009; 323: 527-530.
68. Yamaoka Y. Helicobacter pylori typing as a tool for tracking human migration. Clinical Microbiology and Infection 2009; 15: 829-34.
69. Atherton J, Blaser M. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. Journal of Clinical Investigation 2009; 119: 2475-2487.
70. Cover T, Blaser M. Purification and characterization of the vacuolating toxin from Helicobacter pylori. Journal of Biological and Chemistry 1991; 267: 10570-10575.
71. Telford JL, Ghiara P, Dell’Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce M, Censini S, Covacci A, Xiang Z, Papini E, Montecucco C, Parente L, Rappuoli R. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. Journal of Experimental Medicine 1994; 179: 1653-1658.
72. de Bernard M, Burroni D, Papini E, Rappuoli R, Telford J, Montecucco C. Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infection and Immunity 1998; 66: 6014-6016.

Bibliografia
Andrea Santos
34
73. Cover TL, Hanson PI, Heuser JE. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. Journal of Cell Biology 1997; 138: 759-769.
74. Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B, Telford JL, Rappuoli R, Montecucco C, Papini E, Zoratti M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO Journal 1999; 18: 5517-5527.
75. Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amadei A, D’Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T Cell activation by two independent mechanisms. The Journal of Experimental Medecine 2003; 198: 1887-1897.
76. Molinari B, Salio M, Galli C, Norais N, Rappuoli R, Lanzavecchia A, Montecucco C. Selective inhibition of Li-dependent Antigen presentation by Helicobacter pylori toxin VacA. The Journal of Experimental Medecine 1998; 187: 135-140.
77. Cover TL, Cao P, Lind DL, Tham KT, Blaser MJ. Correlation between vacuolating cytotoxin production by Helicobacter pylori isolates in vitro and in vivo. Infection and Immunity 1993; 61: 5008-5012.
78. Atherton JC, Cao P, Peek RM, Tummuru MKR, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori: Association of specific vacA types with cytotoxin production and peptic ulceration. Journal of Biological Chemistry 1995; 270: 17771-17777.
79. Letley DP, Rhead JL, Twells RJ, Dove B, Atherton JC. Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. Journal of Biological Chemistry 2003; 278: 26734-26741.
80. Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, Suzuki T, Sasakawa C. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. Journal of Experimental Medicine 2000; 191: 593-602.
81. Christie PJ, Vogel JP. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends in Microbiology 2000; 8: 354-360.
82. Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proceedings of the National Academy of Sciences of the United States of America 1993; 90: 5791–5795.
83. Gunn MC, Stephens JC, Stewart JAD, Rathbone BJ, West KP. The significance of cagA and vacA subtypes of Helicobacter pylori in the pathogenesis of inflammation and peptic ulceration. Journal of Clinical Pathology 1998; 51: 761-764.
84. Maeda S, Ogura K, Yoshida H, Kanai F, Ikenoue T, Kato N, Shiratori Y, Omata M. Major virulence factors, VacA and CagA, are commonly positive in Helicobacter pylori isolates in Japan. Gut 1998; 42: 338-343.
85. Nomura AMY, Perez Perez GI, Lee J, Stemmermann G, Blaser MJ. Relation between Helicobacter pylori cagA status and risk of peptic ulcer disease. American Journal of Epidemiology 2002; 155: 1054-1059.
86. Oliveira AG, Santos A, Guerra JB, Rocha GG, Rocha AMC, Oliveira CA, Cabral M, Nogueira A, Queiroz DMM. babA2 and cagA-positive Helicobacter pylori strains are associated with duodenal ulcer and gastric carcinoma in Brazil. Journal of Clinical Microbiology 2003; 41: 3964-3966.
87. Palli D, Masala G, Del Giudice G, Plebani M, Basso D, Berti D, M EN, Ceroti M, Peeters PH, de Mesquita HB, Buchner FL, Clavel-Chapelon F, Boutron-Ruault MC, Krogh V, Saieva C, Vineis P, Panico S, Tumino R, Nyren O, Siman H, Berglund G, Hallmans G, Sanchez MJ, Larranaga N, Barricarte A, Navarro C, Quiros JR, Key T, Allen N, Bingham S, Khaw KT, Boeing H, Weikert C, Linseisen J, Nagel G, Overvad K, Thomsen RW, Tjonneland A, Olsen A, Trichoupoulou A, Trichopoulos D, Arvaniti A, Pera G, Kaaks R, Jenab M, Ferrari P, Nesi G, Carneiro F, Riboli E, Gonzalez CA. CagA+ Helicobacter pylori infection and gastric cancer risk in the EPIC-EURGAST study. International Journal of Cancer 2007; 120: 859-867.

Bibliografia
Andrea Santos
35
88. Rohde M, Puls J, Buhrdorf R, Fischer W, Haas R. A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Molecular Microbiology 2003; 49: 219-234.
89. Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proceedings of the National Academy of Sciences of the United States of America 2000; 97: 1263-1268.
90. Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proceedings of the National Academy of Sciences of the United States of America 2002; 99:14428-14433.
91. Yamazaki S, Yamakawa A, Ito Y, Ohtani M, Higashi H, Hatakeyama M, Azuma T. The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to SHP-2 in human gastric mucosa. Journal of Infectious Diseases 2003; 187: 334-337.
92. Moese S, Selbach M, Kwok T, Brinkmann V, Konig W, Meyer TF, Backert S. Helicobacter pylori induces AGS cell motility and elongation via independent signaling pathways. Infection and Immunity 2004; 72: 3646–3649.
94. Naumann M, Wessler S,Bartsch C, Wieland B, Covacci A, Haas R, Meyer TF. Activaqtion of activator 1 and stress reponse kinases in the epithelial cells colonized by Helicobacter pylori enconding the cag pathogenicity island. Jounal of Biological Chemistry 1999; 274:14559-14564.
95. Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proceedings of the National Academy of Sciences of the United States of America 1999; 96: 14559-14564.
96. Oleastro M, Cordeiro R, Ferrand J, Nunes B, Lehours P, Carvalho-Oliveira I, Mendes AI, Penque D, Monteiro L, Megraud F, Menard A. Evaluation of the clinical significance of homB, a novel candidate marker of Helicobacter pylori strains associated with peptic ulcer disease. Journal of Infectious Diseases 2008; 198: 1379-87.
97. Alm RA, Bina J, Andrews BM, Doig P, Hancock REW, Trust TJ. Comparative genomics of Helicobacter pylori: Analysis of the outer membrane protein families. Infection and Immunity 2000; 68: 4155-4168.
98. Oleastro, M. (2008). Peptic ulcer in children: search for new virulence factors in Helicobacter Pylori. Dissertação de Doutoramento não publicada. Faculdade de Ciências, Universidade de Lisboa, Lisboa, Portugal
99. Exner MM, Doig P, Trust TJ, Hancock RE. Isolation and characterization of a family of porin proteins from Helicobacter pylori. Infection and Immunity 1995; 63: 1567-1572.
100. Dossumbekova A, Prinz C, Mages J, Lang R, Kusters JG, Van Vliet AH, Reindl W, Backert S, Saur D, Schmid RM, Rad R. Helicobacter pylori HopH (OipA) and bacterial pathogenicity: genetic and functional genomics analysis of hopH gene polymorphisms. Journal of Infectious Diseases 2006; 194: 1346-1355.
101. Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraia S, Wadström T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002; 297: 573-578.
102. Peck B, Ortkamp M, Diehl KD, Hundt E, Knapp B. Conservation, localization and expression of HopZ, a protein involved in adhesion of Helicobacter pylori. Nucleic Acids Research 1999; 27: 3325-3333.
103. Oleastro M, Cordeiro R, Yamaoka Y, Queiroz D, Megraud F, Monteiro L, Menard A. Disease association with two Helicobacter pylori duplicate outer membrane protein genes, homB and homA. Gut Pathogens 2009; I: 12.
104. Jung SW, Sugimoto M, Graham DY, Yamaoka Y. homB status of Helicobacter pylori as a novel marker to distinguish gastric cancer from duodenal ulcer. Journal of Clinical Microbiology 2009; 47: 3241-3245.

Bibliografia
Andrea Santos
36
105. Backstrom A, Lundberg C, Kersulyte D, Berg DE, Boren T, Arnqvist A. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proceedings of the National Academy of Sciences of the United States of America 2004; 101: 16923-16928.
106. Cao P, Cover TL. Two different families of hopQ alleles in Helicobacter pylori. Journal of Clinical Microbiology 2002; 40: 4504-451.
107. Pride DT, Meinersmann RJ, Blaser MJ. Allelic variation within Helicobacter pylori babA and babB. Infection and Immunity 2001; 69: 1160-1171.
108. Odenbreit S, Swoboda K, Barwig I, Ruhl S, Borén T, Koletzko S, Haas R. Outer membrane protein expression profile in Helicobacter pylori clinical isolates. Infection and Immunity 2009; 77: 3782-3790.
109. Bernarde C, Lehours P, Lasserre JP, Castroviejo M, Bonneu M, Mégraud F, Ménard A. Complexomics study of two Helicobacter pylori pathological origins. Molecular & Cellular Proteomics 9.12 2010; 2796-2826.
110. Yamaoka Y. Roles of Helicobacter pylori BabA in gastroduodenal pathogenesis. World Journal of Gastroenterology 2008; 27: 4265-4272.
111. Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proceedings of the National Academy of Sciences of the United States of America 2002; 101: 2106-2111.
112. Haas G, Karaali G, Ebermayer G, Metzger K, Lamer S, Zimny-Arndt U, Diescher S, Goebel UB, Vogt K, Roznowski AB, Wiedenmann BJ, Meyer TF, Aebischer T, Jungblut PR. Immunoproteomics of Helicobacter pylori infection and relation to gastric disease. Proteomics 2002; 2: 313-324.
113. Meinke A, Storm M, Henics T, Gelbmann D, Prustomersky S, Kovács Z, Minh DB, Noiges B, Stierschneider U, Berger M, vonGabain A, Engstrand L, Nagy E. Composition of the ANTIGENome of Helicobacter pylori defined by human serum antibodies. Vaccine 2009; 27: 3251-3250.
114. Qiagen® Sample and Assay Technologies (2010). DNA Purification from Tissue.
http://www.qiagen.com/selectlocation.aspx?redirect=%2fliterature%2frender.aspx%3fid%3d200373.
115. Qiagen® Sample and Assay Technologies (2010). PCR purification kit. http://www.qiagen.com/literature/default.aspx?Term=PCR+purification+kit&Language=EN&LiteratureType=1+2+3+4+53&ProductCategory=0.
116. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 1980; 16: 111-120.
117. Graur D, Li WH (2000). Fundamentals of Molecular Evolution: Sinauer Associates, INC.
118. Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N, Takahashi N, Yoshida M, Azuma T, Hattori M, Uchiyama I, Kobayashi I. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiology, 2011; 11:1-27.
119. Nei M, Kumar S. Synonymous substitutions and non synonymous nucleotide substitutions. In Molecular Evolution and Phylogenetics Volume 1. Ed. Nei M. New York: Oxford University Press; 2000:52-61.
120. Ilver D, Arnqvist A, Ögren J, Frick I-M, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998; 279: 373-377.
121. Cao P, Lee KJ, Blaser MJ, Cover TL. Analysis of hopQ alleles in East Asian and Western strains of Helicobacter pylori. FEMS Microbiology letters 2005; 252: 3-43.
122. vanDoorn LJ, Figueiredo C, Megraud F, Pena S, Midolo P, Queiroz DM, Carneiro F, Vanderborght B, Pegado MD, Sanna R e tal. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology 1999; 116:823-830.

Bibliografia
Andrea Santos
37
123. van Doorn L, Figueiredo C, Sanna R, Plaisier A, Schneeberger P, De Boer W, Quint W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology 1998; 115: 58-66.
124. Furuta Y, Kawai M, Yahara K, Takahshi N, Handa N, Tsuru T, Oshima K, Yoshida M et al. Birth and death of genes linked to chromosomal inversion. Proceedings of the National Academy of Science USA 2011; 108:1501-1505.
125. Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proceedings of the National Academy of Sciences of the United States of America 1999; 96: 12778-12783.
126. Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (OipA) of Helicobacter pylori. Proceedings of the National Academy of Sciences of the United States of America 2000; 97: 111-133.
127. Salum L, Snyder LA, Sauders NJ. Apatation by phase variation in pathogenic bacteria. Advances in Applied Microbiology 2003; 52:263-301.
128. van der Woude MW, Baumler AJ. Phase and antigenic variation in bacteria. Clinical Microbiology Reviews 2004; 17: 581-611.
129. Kersulyte D, Mukhopadhyay AK, Velapatino B, Su WW, Pan ZJ, Garcia C, Hernandez V, Valdez Y, Mistry RS, Gilman RH, Yuan Y, Gao H, Alarcon T, Lopez Brea M, Nair GB, Chowdhury A, Datta S, Shirai M, Nakazawa T, Ally R, Segal I, Wong BCY, Lam SK, Olfat FO, Boren T, Engstrand L, Torres O, Schneider R, Thomas JE, Czinn S, Berg DE. Differences in genotypes of Helicobacter pylori from different human populations. Journal of Bacteriology 2000; 182: 3210-3218.
130. Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Molecular Microbiology 2001; 42:1337–1348.
131. Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nature Reviews Microbiology 2007; 5: 441–452.

Anexos
Andrea Santos
Anexo I: DISTRIBUIÇÃO DAS ESTIRPES CLÍNICAS DAS Helicobacter pylori (n=234) em ESTUDO
DE ACORDO COM A REGIÃO GEOGRÁFICA, PATOLOGIA, SEXO E IDADE DOS DOENTES
Origem Patologia N.º de estirpes
(n=234)
Sexo masculino
(%)
Média idade ±
SD (anos)
Países Ocidentais
Portugal
UP 36
47,3 51,8 ± 15,4 G 26
ND 12
Total 74
Alemanha UP 8
50,0 58,6 ± 11,9 G 9
Total 17
Suécia UP 13
58,8 66,6 ± 11,2 G 8
Total 21
França UP 4
ND ND G 2
Total 6
Noruega UP 3 ND ND
Inglaterra UP 7 ND ND
EUA UP 4
ND ND G 13
Total 17
Brasil
UP 4
52,5 52,8 ± 16,4 G 2
CG 2
Total 8
UP 4 57,9 50,0 ±12,7 Colômbia G 3
Total 7
Países Ásia Oriental
Japão
UP 16
57,9 44,3 ± 12,7 G 5
CG 10
Total 31
UP 17
76,1 44,7 ± 9,9 Coreia do Sul G 11
CG 1
Total 29
Singapura ND 5 ND ND
Países África Ocidental Burquina-Faso UP 9 ND ND
Nº, número; SD, Desvio padrão; G, gastrite; UP, úlcera péptica; CG, cancro gástrico; ND, Não disponível

Anexos
Andrea Santos
Anexo II: ISOLAMENTO, IDENTIFICAÇÃO E CONSERVAÇÃO DE ESTIRPES DE Helicobacter pylori
e OBTENÇÃO DE MATERIAL GENÓMICO
A) Isolamento e conservação das estirpes
Reagentes e Material:
Meio HP selectivo (Agar Wilkins Chalgrin suplementado com Vancomicina (10mg/ml),
Trimetroprim (5mg/ml), Fungizona (1ml), Cefsulodina (2mg/ml) e com 10% de sangue de
cavalo) (Biogerm)
Meio HP não selectivo (Agar Wilkins Chalgrin suplementado com 10% de sangue de
cavalo) (Biogerm)
Meio TSB (Trypticase Soya Broth) com 15% de Glicerol
Jarras de anaerobiose Mart® Microbiology (Mart Microbiology B.V., Drachten,
Holanda)
Equipamento:
Sistema gerador de gás AnoxomatTM (Mart Microbiology B.V.)
Procedimento:
Isolamento:
Descongelou-se um tubo contendo a estirpe conservada a -80ºC em TBS com 15% de
glicerol, e semeou-se o homogeneizado em meio selectivo (HP selectivo) e em meio não
selectivo (HP não selectivo). De seguida, as placas foram incubadas a 37ºC, em atmosfera
de microaerofilia (5% de O2, 10% de CO2 e 85% de N2). A atmosfera foi obtida por um
sistema gerador de gás AnoxomatTM. Prolongou-se a incubação durante dois dias, com
renovação da atmosfera e observação diária das placas.
Identificação presuntiva:
Foi observada a morfologia típica das colónias de H.pylori (pequenas, brilhantes,
convexas e translúcidas). Foi realizada coloração de Gram de forma a confirmar a
morfologia espiralada e a percentagem de células cocóides.
Conservação:
Após 48h, recuperou-se a cultura bacteriana, tendo-se preparado uma suspensão
bacteriana em 0,5ml de meio TBS com 15% de glicerol, e distribuiu-se em dois tubos de
congelação, que foram guardados a -80ºC.
B) EXTRACÇÃO DE DNA GENÓMICO
A extracção de DNA genómico foi realizada pelo kit QIAamp® DNA Mini (Qiagen GmbH),
segundo as instruções do fabricante (114).

Anexos
Andrea Santos
Anexo III: CARACTERÍSTICAS GERAIS DAS ESTIRPES DE REFERÊNCIA DE Helicobacter pylori
TOTALMENTE SEQUENCIADAS E ANOTADAS (n=23) QUE FORAM INCLUÍDAS NO ESTUDO
Estirpe N.º de acesso
de GenBank # Patologia Origem População* Referência
26695 AE000511 G Reino Unido Ocidental (hpEurope) 58
J99 AE001439 UP EUA Africano (hpsWAfrica) 39
HPAG1 CP000241 G Suécia Ocidental (hpEurope) 46
Shi470 CP001072 G Peru Ameríndio(hpsAmerind) 45
G27 CP001173 ND Itália Ocidental (hpEurope) 40
P12 CP001217 UP Alemanha Ocidental (hpEurope) 130
B38 FM991728 MALT França Ocidental (hpEurope) 48
B8 FN598874 UP EUA Ocidental (hpEurope) 42
SJM180 CP002073 G Peru Ocidental (hpEurope) ND
51 CP000012 UP Coreia do Sul Oriental (hpsEAsia) ND
52 CP001680 UP Coreia do Sul Oriental (hpsEAsia) ND
35 A CP002096 UP Coreia do Sul Oriental (hpsEAsia) ND
F30 AP011941 UP Japão Oriental (hspEAsia) 44
F16 AP011940 G Japão Oriental (hpsEAsia) 44
F32 AP011943 CG Japão Oriental (hpsEAsia) 44
F57 AP011945 CG Japão Oriental (hpsEAsia) 44
V225d CP001582 G Venezuela Ameríndio (hpsAmerind) 57
Sat464 CP002071 ND Peru Ameríndio (hpsAmerind) ND
PeCan4 CP002074 CG Peru Ameríndio (hpsAmerind) ND
Cuz20 CP002076 ND Peru Ameríndio (hpsAmerind) ND
Gambia94/24 CP002332 ND Gâmbia Africano (hpsWAfrica) ND
SouthAfrica7 CP002336 ND África do Sul (hspSAfrica) ND
India7 CP002331 ND Índia (hspAsia2) ND
G, gastrite; UP, úlcera péptica; C, cancro gástrico; MALT, Mucosa Associated Lymphoid Tissue;
N.D., Não disponível;
# http://www.ncbi.nlm.nih.gov/genomes/lpoks.cgi;
* Classificação da população de H. pylori segundo a distribuição geográfica das estirpes adaptada
de 66,131
.

Anexos
Andrea Santos
Anexo IV: DETECÇÃO DE PRODUTOS DE AMPLIFICAÇÃO POR ELECTROFORESE EM GEL DE
AGAROSE
Reagentes:
Agarose UltraPureTM (Invitrogen S.A)
Tampão TBE 0,5X (0,045M Tris; 0,045M Ácido bórico; 0,01M EDTA)
GelRedTM (10,000x) (Biotium Inc)
Solução de aplicação (0,25% Azul bromofenol; 0,25% Xileno cianol; 15% Ficoll;
120mM EDTA)
Marcador de peso molecular 1Kb DNA Ladder (Invitrogen S.A)
Equipamento:
Tina de electroforese de mini géis horizontal i Mupid (Eurogentec S.A, Seraing,
Bélgica)
Transluminador Gel Doc 2000 (Bio Rad Laboratories Ltd.)
Aparelho fotográfico P93 (Mitsubishi Electric Europe B.V., Barcelona, Espanha)
Micropipetas (Eppendorf)
Procedimento:
A agarose foi pesada (1,2g para gel de 1%) e dissolvida, por meio de calor, em tampão
TBE 0,5X (120ml). Após o arrefecimento da agarose, adicionou-se o Gel RedTM (1,5μl) e
distribui-se o preparado pelo tabuleiro, colocando o pente apropriado para a aplicação das
amostras. Estes volumes permitem preparar dois géis de 25ml e quatro de 15ml.
Depois de o gel ter solidificado, colocou-se na tina de electroforese e encheu-se a tina
com tampão TBE 0,5X, de modo a que o gel fique completamente imerso. De seguida,
colocaram-se as amostras (5μl produto de amplificação + 2μl solução de aplicação) e o
marcador de peso molecular nos poços e procedeu-se à corrida com uma voltagem
constante de 100V, durante 30min. No fim, monitorizou-se o gel no transiluminador e tirou-
se uma fotografia.

Anexos
Andrea Santos
Anexo V: Alinhamento representativo de algumas sequências de nucleótidos dos genes hopM e hopN, de estirpes clínicas e de referência em estudo, da região onde é possível distinguir os dois genes (300 a 800 pb). 350 500
SE13MUP GTGGCAAGTCATAGCCCTTTTTATCGGCTGTGGCCCTGGCCCTA-------CC-----------GATAATCAAAGCTATCAATCGTTTGGTAACACACCAG---------------CCCTTAATAG---GACAACCACCACTTGCAATGG
PT499/02MU GTGGCAAGTCATAGCCCTTTTTATTGGCTGCGGCCCTGGCCCTA-------CC-----------AACAATCAAAGCTACCAATCGTTTGGTAACACACCAG---------------CCCTTAATGG---GACAACCACCACTTGCAATCA
USJ99MUP GTGGCAAGTCATAGCCTTTGGCATCAGCTGTGGCCCTGGCCCCAATCTTGGCCCAGAACATTTAGAAAATGGGGGCGTTCGATCGTTTGACAACACGCCAAACTACAGCTACAACACCGGTAGCGGAACGACCACCACCACTTGTAATGG
PTB66MG GTGGCAAGTCATAGCCTTTGGCATCAGCTGTGGCCCTGGCCCCAATCTTGGCCCAGAACATTTAGAAAATGGGGGCGTTCGATCGTTTGACAACACGCCAAACTACAGCTACAACACCGGTAGCGGAACGACCACCACCACTTGCAATCA
PTB66NG GTGGCAAGTCATAGCCTTTGGCATCAGCTGTGGCCCTGGCCCCAATCTTGGCCCAGAACATTTAGAAAATGGGGGCGTTCGATCGTTTGACAACACGCCAG---------------CCCTTAATGG---GACCACCACCACTTGCAATCA
SEHPAG1NG GTGGCAAGTCATAGCCCTTTTTATTGGCTGCGGCCCTGGTCCTA--------CCA----------ACAATCAAAGCTACCAATCGTTTGGTAACACACCAG---------------CCCTTAATGG---GACAACCACCACTTGCAATCA
GB26695hNG GTGGCAAGTCATAGCCCTTTTTATTGGCTGTGGCCCTGGCCCTA--------CCA----------ATAATCAAAGCTATCAATCGTTTGGTAACACACCAG---------------CCCTTAATGG---GACCACCACCACTTGCAATCA
Consensus GTGGCAAGTCATAGCCcTTtttATcgGCTGtGGCCCTGGcCCtA.......cCca.........gA.AATcaaaGCtatCaATCGTTTGgtAACACaCCAg...............CCctTAatgG...GACcACCACCACTTGcAATca
501 650
SE13MUP GGCTAGTAATGTGGGGCCCAATGGCATTCTATMTAGCAGSGAATACCAGGTTCTCAATACCGCTTATCAAACTATCCAAACCGCTTTAAACCAAAACCAAGGAGGCGGGATGCCTGCCTTGAATGGCTCTAAAAATAT---GGTAGTCAA
PT499/02MU AGCGTATGGGACAGGCCCTAATGGCATTCTATCCATTGATGAATACCAAAAACTCAACCAAGCTTATCAAATTATCCAAACCGCTTTAAACCAAAACCAAGGAGGCGGGATGCCTGCCTTGAACAGCTCCAAAAATAT---GGTAGTCAA
USJ99MUP AGCCAGTAATGTAGGGCCCAATGGTATCCTATCTAGCAGCGAATACCAGGTTCTCAATACCGCTTATCAAACTATCCAAACCGCTTTAAACCAAAACCAAGGAGGCGGGATGCCTGCCTTGAATAGCTCCAAAAATAT---GGTAGTCAA
PTB66MG AGCATACGGGACAGGCCCTAATGGCATTCTATCCATTGATGAATACCAAAAACTCAACCAAGCTTATCAAATTATCCAAACCGCTTTAAACCAAAACCAAGGAGGCGGGATGCCTGCCTTGAATGACACCACCAAAACAGGGGTAGTCAA
PTB66NG AGCATACGGGACAGGCCCTAATGGCATTCTATCCATTGATGAATACCAAAAACTCAACCAAGCTTATCAAATTATCCAAACCGCTTTAAACCAAAACCAAGGAGGCGGGATGCCTGCCTTGAATGACACCACCAAAACAGGGGTAGTCAA
SEHPAG1NG AGCGTATGGGACAGGCCCTAATGGCATTCTATCCATTGACGAATACCAAAAACTCAACCAAGCTTATCAGATCATCCAAACCGCTTTAAACCAAAATCAAGGGGGTGGGATGCCTGCCTTGAATGACACCACCAAAACAGGGGTAGTCAA
GB26695hNG AGCATATGGGACAGGCCCTAATGGCATCCTATCTATTGATGAATACCAAAAACTCAACCAAGCTTATCAGATCATCCAAACCGCTTTAAACCAAAATCAAGGGGGTGGGATGCCTGCCTTGAATGACACCACCAAAACAGGGGTAGTCAA
Consensus aGC.tatgggacaGGcCCtAATGGcATtCTATccAttgatGAATACCAaaaaCTCAAccaaGCTTATCAaAttATCCAAACCGCTTTAAACCAAAAcCAAGGaGGcGGGATGCCTGCCTTGAAtggCtCcAaaAAtAt...GGTAGTCAA
651 800
SE13MUP TATCAATCAAACTTT------------CACAAAAAACCCTACAACAGAATACACTTACCCTAA---TGGGAATGG-----CAAT-TATTATTCAGGCGGATCATCAATCCCAATCCAGCTAAAGATTAGTAGCGTCAATGACGCTGAAAA
PT499/02MU TATCAATCAAACTTT------------CACAAGAAACCCTACCACAGAATACACTTACTCTGA---TGGGAATGG-----CAAT-TATTATTCAGGCGGTTCATCAATCCCAATCCAGCTAAAGATTAGCAGCGTCAATGACGCTGAAAA
USJ99MUP TATCAATCAAACTTT------------CACAAAAAACCCTACCACAGAATACACTTACCCCGA---TGGGAATGG-----CAAT-TATTATTCAGGCGGTTCATCAATCCCAATCCAGCTAAAGATTAGTAGCGTCAATGACGCTGAAAA
PTB66MG CATACAACAAACCAATTATAAGACCACCACACAAAACAATATCATACAGCATTATTATGATGAGAATGGGAAAGAGATTCCAACCTATTATTCAGGCGGATCATCATTCTCGCCTACGATACAATTGAAATACAATAATAACGCTGAATA
PTB66NG CATACAACAAACCAATTATAAGACCACCACACAAAACAATATCATACAGCATTATTATGATGAGAATGGGAAAGAGATTCCAACCTATTATTCAGGCGGATCATCATTCTCGCCTACGATACAATTGAAATACAATAATAACGCTGAATA
SEHPAG1NG CATACAACAAACCAATTATAGAACCACTACACAAAACAATATCATAGAGCATTATTATACAGAGAATGGGAAAGAGATCCCAACCCATTATTCAGGCGGATCATCATTCTCGCCTACAATACAATTGACATACCACAATGACGCTGAATA
GB26695hNG CATACAACAAACCAATTATAGGACCACCACACAAAACAATATCATAGAGCATTATTATACAGAGAATGGGAAAGAGATCCCAGTCTCTTATTCAGGCGGATCATCATTCTCGCCTACAATACAATTGACATACCATAATAACGCTGAAAA
Consensus tATcaAtCAAACttt............cACAaaAAACccTAccAcAgAatAcacTTAc.ctgA...TGGGAAtGg.....CAat.taTTATTCAGGCGGaTCATCAaTCcCaatccagcTAaAgaTtAg.agCgtcAATgACGCTGAAaA
hopN
hopN
hopM
hopM
hopN
hopM
Nota: Foram incluídas as sequências correspondentes às quatro quimeras, identificadas da seguinte
forma: SE13MUP, PT499/02MU, PTB66MG e PTB66NG. As setas das cores respectivas ao lado de
cada gene (por exemplo:. hopN) indicam se a sequência é similar a hopM ou hopN, ao longo da
região localizada entre os 300 e os 800 pb destes genes. A posição da segunda seta de cada cor
indica a base a partir da qual uma determinada sequência alterou de um gene para outro. O
alinhamento foi efectuado com as estirpes de referência USJ99M (jhp0212, hopM), GB26695N
(HP1342, hopN) e HPAG1N (HPAG1_1289, hopN). O N/M identificam o locus de onde a sequência
foi obtida.


















