
Fúlvia de Souza Vieira Solange Macedo Aily · aplicado um questionário visando coletar dados da...
54
Fúlvia de Souza Vieira Solange Macedo Aily ANÁLISE FUNCIONAL DE PACIENTES COM SPOAN: Paraplegia Espástica com Atrofia Óptica e Neuropatia Tese apresentada à Universidade Federal de São Paulo – Escola Paulista de Medicina, para obtenção do Título de Especialista em Intervenção Fisioterapêutica em Doenças Neuromusculares. São Paulo 2007
Transcript of Fúlvia de Souza Vieira Solange Macedo Aily · aplicado um questionário visando coletar dados da...

Fúlvia de Souza VieiraSolange Macedo Aily
ANÁLISE FUNCIONAL DE PACIENTES COM SPOAN:Paraplegia Espástica com Atrofia Óptica e Neuropatia
Tese apresentada à Universidade Federal de São Paulo – Escola Paulista de Medicina, para obtenção do Título de Especialista em Intervenção Fisioterapêutica em Doenças Neuromusculares.
São Paulo2007

2
Fúlvia de Souza VieiraSolange Macedo Aily
ANÁLISE FUNCIONAL DE PACIENTES COM SPOAN:Paraplegia Espástica com Atrofia Óptica e Neuropatia
Tese apresentada à Universidade Federal de São Paulo – Escola Paulista de Medicina, para obtenção do Título de Especialista em Intervenção Fisioterapêutica em Doenças Neuromusculares.
Orientadora: Márcia Cristina Bauer CunhaCo – Orientadora: Zodja Graciani
São Paulo2007

3
Aos nossos pais, Aureo e Josete Aily, e João Carlos e Maria de Lourdes Vieira, pelo carinho, apoio, confiança e incentivo incondicional em
toda nossa trajetória de vida.
v

4
Aily, Solange Macedo; Vieira, Fúlvia de Souza Análise funcional de pacientes com SPOAN: Paraplegia Espástica com
Atrofia Óptica e Neuropatia. /Fúlvia de Souza Vieira; Solange Macedo Aily. -- São Paulo, 2007.
x, 65f.
Tese (Livre Docência) – Universidade Federal de São Paulo. Escola Paulista de Medicina. Programa de Pós-graduação em Intervenção Fisioterapêutica em Doenças Neuromusculares.
Título em inglês: Functional analysis of patients with SPOAN: Spastic Paraplegia, Optic Atrophy, and Neuropathy.
1. SPOAN. 2. Paraplegia Espástica Hereditária (PEH). 3. Qualidade de Vida. 4. Depressão. 5. Funcionalidade. 6. Marcha.

5
UNIVERSIDADE FEDERAL DE SÃO PAULO
ESCOLA PAULISTA DE MEDICINA
DEPARTAMENTO DE NEUROLOGIA E NEUROCIRURGIA
Chefe do Departamento: Prof. Dr. Acary Souza Bulle de Oliveira.
Coordenador(a) do Curso de Pós-graduação: Ft. Francis Meire Fávero.
iii

6
Fúlvia de Souza Vieira
Solange Macedo Aily
ANÁLISE FUNCIONAL DE PACIENTES COM SPOAN:
Paraplegia Espástica com Atrofia Óptica e Neuropatia
Presidente da banca:
____________________________________________
Prof. Dr. Acary Souza Bulle de Oliveira
BANCA EXAMINADORA
____________________________________________
Prof. Dra. Sissy Veloso Fontes
____________________________________________
Prof. Dra. Márcia Cristina Bauer Cunha
iv

7
LISTA DE GRÁFICOS
Gráfico 1. Avaliação das pacientes em nível de depressão com
BDI..................................................................................................................20
Gráfico 2. Diferença entre as pacientes de SPOAN em relação ao
auto-cuidado...................................................................................................21
Gráfico 3. Demonstra o controle respiratório e esfincteriano das
pacientes com SPOAN .................................................................................22
Gráfico 4. Diferenças na mobilidade no quarto e no banheiro das
pacientes com SPOAN..................................................................................23
Gráfico 5. Mobilidade das pacientes dentro e fora de casa............................................24
Gráfico 6. Escala de classificação da análise do andar..................................................24
viii

8
LISTA DE TABELAS
Tabela 1. PEH’s de herança autossômica recessiva......................................................4
Tabela 2. Comparação das pacientes com SPOAN na escala de
qualidade de vida...........................................................................................19
Tabela 3. Pontuação das pacientes com SPOAN na BDI ............................................20
Tabela 4. Apresenta o item de auto-cuidado da escala SCIM das
02 pacientes de SPOAN................................................................................20
Tabela 5. Apresenta o item de controle respiratório e esfincteriano
da escala SCIM das 02 pacientes de SPOAN .............................................21
Tabela 6. Apresenta a mobilidade no deslocamento das pacientes
com SPOAN .................................................................................................22
Tabela 7. Apresenta a mobilidade dentro e fora de casa avaliadas
pela escala SCIM ..............……....................................................................23
ix

9
SUMÁRIO
Dedicatória .........................................................................................................................v
Agradecimentos .................................................................................................................vi
Lista de Gráficos ..............................................................................................................viii
Lista de Tabelas ................................................................................................................ix
Resumo ..............................................................................................................................x
1 INTRODUÇÃO ..............................................................................................................1
1.1 Paraplegia Espástica Hereditária (PEH) ......................................................................1
1.2 Síndrome de SPOAN (Paraplegia Espástica com Atrofia Óptica e Neuropatia) ..........6
2 OBJETIVOS ................................................................................................................11
3 MÉTODOS ..................................................................................................................12
4 RESULTADOS ............................................................................................................15
5 DISCUSSÃO ...............................................................................................................26
6 CONCLUSÕES ...........................................................................................................34
7 ANEXOS ....................................................................................................................35
8 REFERÊNCIAS ..........................................................................................................63
Abstract
vii

10
AGRADECIMENTOS
À nossa orientadora Márcia Cristina Bauer Cunha, pelo grande auxílio,
paciência, orientação e cooperação para nossa formação profissional.
À nossa co–orientadora Zodja Graciani, pela amizade, carinho e
aconselhamento dado para a realização dessa pesquisa.
À Dra. Sissy Veloso Fontes e à Ms. Francis Meire Fávero, pelo auxílio nas
horas difíceis, onde, demonstrando paciência e simpatia, nos deram apoio
proporcionando calma.
Aos pacientes e cuidadores, pela confiança, dedicação e satisfação oferecida
durante toda a realização do trabalho.
Agradecemos o apoio oferecido pela instituição Universidade de Santo
Amaro, por nos ceder a oportunidade de realizar a pesquisa.
À todos os amigos e colegas, pela amizade e eventuais auxílios, e,
principalmente, por participarem intensamente das aflições e dos prazeres que este
trabalho proporcionou.
À Deus, por sempre nos iluminar e nos ajudar em todas as etapas de nossas
vidas.
À nossa coragem, perseverança e dedicação, que sempre nos acompanha,
induzindo, assim, uma ascendente trajetória profissional.
Vi

11
RESUMO
INTRODUÇÃO: O termo SPOAN (Paraplegia Espástica com Atrofia Óptica e Neuropatia) refere-se a uma síndrome neurodegenerativa, progressiva, de herança autossômica recessiva, recém descoberta por pesquisadores do Centro de Estudos do Genoma Humano e do Hospital das Clínicas da Universidade de São Paulo (USP). SPOAN caracteriza-se por atrofia congênita do nervo óptico, paraparesia espástica progressiva, sinais de neuropatia motora e sensitiva, entre outros. OBJETIVOS: Comparar as características clínicas e história da doença entre dois casos de pacientes com síndrome de SPOAN e o relatado na literatura, além de analisar a qualidade de vida, identificar o grau de depressão, verificar a funcionalidade, e comparar a marcha de uma delas com a marcha de pacientes com paraplegia espástica hereditária (PEH). MÉTODOS: Foram incluídas na amostra duas pacientes do sexo feminino, com idade entre 12 e 28 anos, com diagnóstico comprovado de síndrome de SPOAN, sendo aplicado um questionário visando coletar dados da história da doença, uma ficha de avaliação neurológica, uma escala de qualidade de vida, escala de depressão de Beck, escala de funcionalidade SCIM e escala de classificação da análise do andar. RESULTADOS: Ambas as pacientes A e Z apresentaram nistagmo como principal sinal de atrofia do nervo óptico, paraparesia espástica progressiva com início na infância, alterações de sensibilidade, entre outros, sendo que a paciente Z apresentou todos os sintomas em um grau de severidade mais elevado. A paciente Z mostrou-se mais sociável e comunicativa, mesmo passando a maior parte do tempo recolhida em sua casa. Analisando a escala de Beck, observou-se que a paciente A encontra-se emum nível de depressão de leve a moderada, e a paciente Z de moderada a grave. A paciente Z apresentou um acometimento de maior nível em relação à funcionalidade quando comparada com a paciente mais jovem. Analisando a marcha da paciente A, notou-se que esta apresentou alteração em todos os aspectos avaliados. CONCLUSÕES: As pacientes A e Z apresentam características clínicas e história da doença similar a dos casos dos pacientes com síndrome de SPOAN avaliados em Serrinha dos Pintos; a funcionalidade mostrou-se compatível com o perfil das pacientes, levando em consideração a idade e o tempo de doença, demonstrando que a qualidade de vida encontra-se adequada. As duas pacientes demonstraram um grau de insatisfação diante das impossibilidades pessoais, levando a baixa estima e falta de perspectiva diante da vida. A paciente A apresentou um grau de acometimento maior com relação à marcha, quando comparada aos pacientes com paraplegia espástica hereditária descritos na literatura.
PALAVRAS CHAVES: SPOAN, Paraplegia Espástica Hereditária (PEH), Qualidade de Vida, Depressão, Funcionalidade, Marcha.
x

12
1 INTRODUÇÃO
1.1 Paraplegia Espástica Hereditária (PEH):
O termo “Paraplegia Espástica Hereditária” ou “Paraparesia Espástica
Hereditária” (PEH) foi descrito primeiramente por Strumpell, em 1880, sendo utilizado
para descrever um grupo de doenças neurodegenerativas, hereditárias, clinicamente e
geneticamente heterogêneas, caracterizadas por espasticidade e fraqueza bilateral,
simétrica, de progressão lenta, afetando, primariamente, os membros inferiores (Vazza
et al., 2000; Hughes et al., 2001; Crosby, Proukakis, 2002; Fink, 2002; Hodgkinson et
al., 2002; Burgunder, Hunziker, 2003; Fink, 2003; Lesca et al., 2003; McDermott et al.,
2003; Fink, 2004; Klebe et al., 2004; Meijer et al., 2004; Orlacchio et al., 2004; Bouslam
et al., 2005; Santos et al., 2005). Espasticidade e fraqueza progressiva nas
extremidades inferiores também são características de muitas outras doenças
geneticamente herdadas, incluindo leucodistrofias, ataxia de Friedreich, doença de
Machado-Joseph, esclerose múltipla e doença de Alzheimer familiar. Alguns sinais
neurológicos, tais como neuropatia periférica, ataxia, demência e acometimento
pseudobulbar, diferem estas condições das PEH’s, cuja característica neurológica mais
incapacitante é a marcha espástica (Fink, 2003).
Clinicamente, as síndromes da PEH são classificadas como puras (também
chamadas de incomplexas ou assindrômicas) quando os sintomas se limitam ao
acometimento de neurônios motores superiores, incluindo espasticidade e fraqueza
progressiva nos membros inferiores, hiperreflexia nessas extremidades, e sinal de
Babinski, freqüentemente acompanhados por distúrbios urinários e déficit sensitivo
profundo nas extremidades distais. As PEH’s são classificadas como complexas ou
sindrômicas quando os sinais citados na forma pura ocorrem associados com outros
déficits neurológicos ou não-neurológicos, incluindo neuropatia motora e periférica,
atrofia óptica, atraso mental, demência, epilepsia, surdez, catarata, disartria,
retinopatia, amiotrofia, disfagia, sintomas extrapiramidais, entre outros (Vazza et al.,
2000; Crosby, Proukakis, 2002; Fink, 2002; Burgunder, Hunziker, 2003; Fink, 2003;
Klebe et al., 2004; Meijer et al., 2004; Bouslam et al., 2005; Santos et al., 2005). Nas
PEH’s puras, os sintomas se limitam aos membros inferiores, a força muscular, o tônus

13
e a destreza em membros superiores, a fala e a deglutição permanecem sem
alterações (Fink, 2003).
A prevalência da PEH é de 2.0 – 9.6 / 100.000 habitantes (Klebe et al.,
2004).
O aparecimento das primeiras manifestações clínicas da PEH varia desde a
infância até após a sexta década de vida. O início dos sintomas, assim como sua
gravidade, o índice de progressão da doença e o grau de incapacidade variam entre as
diferentes formas genéticas de PEH, entre famílias diferentes com a mesma forma
genética, e entre indivíduos afetados da mesma família que contém exatamente a
mesma mutação genética. Geralmente, quando os sintomas surgem na infância, a
perda funcional é relativamente pequena com o passar dos anos. Por outro lado,
quando os primeiros sinais aparecem durante ou após a adolescência, o distúrbio na
marcha progride insidiosamente. A paraparesia espástica é o sinal clínico mais grave e
incapacitante da PEH. Conforme os sintomas progridem, e a fraqueza e a
espasticidade nos membros inferiores se acentuam, os indivíduos afetados passam a
necessitar de bengala, andador e cadeira de rodas. Aqueles com início dos sintomas
precoce, PEH aparentemente não-progressiva, se diferem daqueles com diplegia
espástica apenas pela história familiar (Fink, 2002; Hodgkinson et al., 2002; Fink, 2003;
Fink, 2004; Klebe et al., 2004).
O diagnóstico para PEH é confirmado quando há presença de: (1) sintomas
típicos de distúrbios na marcha (cujo início varia desde a infância, diplegia espástica
não-progressiva, até a fase adulta, na qual a fraqueza e a espasticidade nos membros
inferiores tem progressão insidiosa), freqüentemente associados a distúrbios urinários;
(2) achados neurológicos de acometimento do trato corticoespinal (espasticidade,
fraqueza, hiperreflexia, sinal de Babinski) sendo limitados às extremidades inferiores
(os membros superiores podem apresentar reflexos ativos, porém não patológicos, e
tônus muscular normal); (3) história familiar com distúrbio similar que se caracteriza a
doença de herança autossômica dominante, recessiva ou ligada ao X, e (4) exclusão
de outras doenças, incluindo esclerose múltipla, leucodistrofia, anormalidades
estruturais envolvendo o córtex ou a medula espinal, e distonia (Fink, 2003).
Esta doença é caracterizada pela heterogeneidade genética: pode ser de
herança autossômica dominante, autossômica recessiva ou autossômica recessiva
ligada ao X, havendo relatos de casos para ambas as formas puras e complexas, para
cada um dos tipos citados (Fink, 2003). PEH de herança autossômica dominante é a

14
forma mais comum, sendo responsável por aproximadamente 70 – 80% dos casos
(Vazza et al., 2000; Klebe et al., 2004). A forma pura ocorre mais comumente que a
complexa (Bouslam et al., 2005; Santos et al., 2005).
Atualmente, uma grande variedade de tipos de PEH tem sido definida por
meio de análise genética. O termo PEH é denominado SPG (“Paraplegia Espástica”)
para diferenciar suas diversas formas (Fink, 2003; Klebe et al., 2004). Apesar desse
número estar crescendo a cada ano, estudos recentes se contradizem quanto ao
número correto de SPG’s existentes até o momento. O estudo mais recente utilizado
para a realização dessa pesquisa mostra 27 SPG’s identificados, variando de 1 SPG à
27 SPG, de acordo com a ordem de suas descobertas, e mais uma nova forma, SPG
28, recém descoberta em uma família marroquina (Bouslam et al., 2005). A maior parte
dos estudos cita 10 SPG’s encontrados na forma dominante e 3 SPG’s na forma ligada
ao X (Crosby, Proukakis, 2002; Fink, 2002; Fink, 2003; Fink, 2004; Meijer et al., 2004;
Orlacchio et al., 2004). Bouslam et al. (2005) descrevem 11 SPG’s na forma recessiva,
e um número maior para a forma dominante. Atualmente, são dez mutações genéticas
identificadas para PEH, mostrando as diversas alterações patofisiológicas envolvidas
nesta doença (Fink, 2004; Meijer et al., 2004). A forma autossômica recessiva, sendo
menos comum que a forma dominante, apresenta um número menor de PEH’s e genes
identificados. Até o momento, totalizam-se três genes descobertos para a forma
recessiva: paraplegina (SPG 7), espartina (SPG 20) e maspardina (SPG 21) (Fink,
2004; Meijer et al., 2004; Bouslam et al., 2005; Santos et al., 2005). Entre os diversos
tipos de PEH de herança recessiva conhecida atualmente, podemos citar 11 SPG’s
sendo 3 SPG’s na forma pura (Bouslam et al., 2005). A localização no cromossomo e
as características associadas nas formas complexas podem ser observadas na tabela
1 (Crosby, Proukakis, 2002; Fink, 2002; Hodgkinson et al., 2002; Fink, 2003; Meijer et
al., 2004; Bouslam et al., 2005; Santos et al., 2005).

15
Tabela 1. Paraplegias Espásticas Hereditárias (PEH’s) de herança autossômica
recessiva.
Tipo Cromossomo/Localização
Gene/ proteína
Formas/ Síndromes Características associadas
SPG 5 8q desconhecido Pura
SPG 7 16q24.3 paraplegina complexa ou puraAtrofia óptica e cerebelar ou cortical, disartria, disfagia, neuropatia periférica, lesões vasculares, alterações mitocondriais, pé cavo
SPG 11 15q13-14 desconhecido complexa ou puraAtraso mental, corpo caloso estreito, neuropatia periférica, disartria, fraqueza distal, nistagmo
SPG 14 3q27-28 desconhecido Complexa Atraso mental, neuropatia periférica
SPG 15 14q22-24 desconhecido complexa (Sd. de Kjellin)
Atraso mental, demência, neuropatia periférica, amiotrofia distal, disartria, maculopatia pigmentada
SPG 20 13q12.3 espartina complexa (Sd. de Troyer)
Disartria, fraqueza distal, atraso no desenvolvimento e baixa estatura
SPG 21 15q22.31 maspardina complexa (Sd. de Mast)
Demência, sinais cerebelares e extrapiramidais, disartria, amiotrofia
SPG 23 1q24-32 desconhecido complexa (Sd. de Lison)
Alterações na pigmentação cutânea e capilar, neuropatia periférica
SPG 25 6q23-24 desconhecido Complexa Prolapso do disco intervertebral
SPG 27 10q22-24 desconhecido PuraSPG 28 14q21-22 desconhecido Pura
Descobertas recentes de muitos genes da PEH têm gerado, rapidamente,
novos conceitos de mecanismos patofisiológicos para a doença (Fink, 2003). Estudos
neuropatológicos mostram que a maioria das PEH’s, incluindo a maior parte na forma
pura, envolve neurodegeneração axonal progressiva das porções distais do trato
corticoespinal, do fascículo grácil, e do trato espinocerebelar, que juntas constituem as
fibras motoras e sensitivas mais longas do sistema nervoso central (Crosby, Proukakis,
2002; Hodgkinson et al., 2002; Burgunder, Hunziker, 2003; Fink, 2003; McDermott et
al., 2003). O trato corticoespinal é o mais gravemente afetado, seguido pelas fibras do
corno posterior da medula (McDermott et al., 2003). Degeneração progressiva de
axônios longos da medula não é causa única da PEH, também ocorre em indivíduos
normais com o passar dos anos, assim como em várias outras doenças como
esclerose lateral amiotrófica, esclerose lateral primária e alterações na medula espinal
(Fink, 2004). Um padrão específico de degeneração é visto na PEH, no qual os corpos

16
celulares permanecem intactos enquanto a degeneração se limita, principalmente, aos
axônios, podendo ocorrer o que é chamado de axonopatia de “morte retrógrada”,
acometendo inicialmente porções distais, progredindo para regiões mais proximais até
atingir o corpo celular (Crosby, Proukakis, 2002; Burgunder, Hunziker, 2003;
McDermott et al., 2003).
Muitos estudos moleculares têm sido realizados visando descobrir os
mecanismos envolvidos nessa degeneração, que leva à axonopatia nas PEH’s, mas a
causa ainda é desconhecida, e algumas hipóteses são discutidas (Crosby, Proukakis,
2002; Burgunder, Hunziker, 2003; Fink, 2003; McDermott et al., 2003). A preferência da
degeneração por axônios longos pode ser explicada pela falta de produção de ATP
nessas fibras, por deficiência de mitocôndrias e outras organelas. Mutações nos genes
envolvidos nesses processos resultam no bloqueio do transporte dessas substâncias
ao longo dos microtúbulos. Esse transporte é um processo vital, no qual a célula
transporta uma grande quantidade de componentes de uma região para outra, e
axônios longos são particularmente mais suscetíveis a essa interrupção. Esse bloqueio
evita que cargas adequadas, vindas de determinadas substâncias, cheguem ao seu
destino correto na célula, fazendo com que a demanda de energia destas células
aumente. Isso poderia explicar porque mutações nestes genes levam à degeneração
de tipos celulares tão específicos. Outra hipótese para esta degeneração seria
anormalidade na mielinização do trato corticoespinal, talvez por efeitos tóxicos nos
oligodendrócitos, induzidos por um tráfego intracelular anormal (Crosby, Proukakis,
2002; Burgunder, Hunziker, 2003; Fink, 2003). A identificação dessas mutações
genéticas está ajudando os pesquisadores a descobrir a verdadeira causa da doença a
nível molecular (Crosby, Proukakis, 2002; Burgunder, Hunziker, 2003; McDermott et al.,
2003).
Não existe tratamento para minimizar a progressão da doença. O tratamento
para PEH se limita a medicamentos anti-espásticos (Baclofen, Tizanidine, Tetrazepam,
Dantrolene e toxina botulínica) e fisioterapia, principalmente para prevenir
complicações futuras como contraturas musculares, e para preservar as capacidades
funcionais existentes (Fink, 2003; Klebe et al., 2004).

17
1.2 Síndrome de SPOAN (Paraplegia Espástica com Atrofia Óptica e Neuropatia):
O termo SPOAN (em inglês spastic paraplegia, optic atrophy, and
neuropathy) refere-se a uma síndrome neurodegenerativa, progressiva, de herança
autossômica recessiva, recém descoberta por pesquisadores do Centro de Estudos do
Genoma Humano e do Hospital das Clínicas da Universidade de São Paulo (USP), e
descrita em maio de 2005 pelos mesmos pesquisadores, em um artigo publicado na
revista norte-americana Annals of Neurology.
Santos et al. (2005) descrevem o caso de uma família com um número
enorme de uniões consangüíneas, com 25 indivíduos afetados, dos quais 22 foram
avaliados clinicamente, portando uma doença neurológica previamente desconhecida.
A família é natural de Serrinha dos Pintos, cidade de 4.300 habitantes,
situada 370 quilômetros a oeste de Natal - Rio Grande do Norte, cuja população é
totalmente carente de serviços de saúde, onde fisioterapia não existe. Até agora os
cientistas identificaram 26 pessoas vivas com SPOAN, das quais 17 são mulheres e 9
homens. Todos os indivíduos afetados são brancos, caucasianos, provavelmente de
ascendência portuguesa, ou talvez holandesa, e originários dessa cidade serrana. A
maioria ainda mora em sua terra natal e todos descendem de casais aparentados, de
19 uniões consangüíneas. Análises adicionais da história familiar revelam muitos outros
parentes afetados de antepassados europeus, que se instalaram em Serrinha dos
Pintos há mais de 150 anos, com SPOAN estando presente em mais de oito gerações.
Casamentos entre primos sempre foi, e continua sendo, um hábito local entre os
moradores da cidade, sendo que estes casamentos consangüíneos apresentam maior
risco de gerar crianças com alguma doença hereditária. A síndrome foi reconhecida
nessa região no final do século XIX, e mais seis casos novos de indivíduos afetados
foram relatados nos últimos anos, sabendo-se também que quatro indivíduos com a
mesma síndrome morreram nos últimos 20 anos. O conceito de que essa condição é
geneticamente pré-determinada, curiosamente, não está presente entre os habitantes
da comunidade, e muitas outras causas foram postuladas para explicar esse
fenômeno, a maioria delas relacionadas à sífilis (Pivetta, 2005; Santos et al., 2005).
Os pesquisadores classificam SPOAN, após a realização de diversos
estudos genéticos, como uma doença de herança autossômica recessiva. Homens e
mulheres têm a mesma probabilidade de apresentar a doença, a despeito de o número

18
de casos conhecidos em Serrinha dos Pintos ser maior entre o sexo feminino. Uma
pessoa só vai desenvolver SPOAN se as duas cópias do ainda desconhecido gene
associado à síndrome, uma vinda do pai e outra da mãe, carregarem a mutação que
leva ao problema. Portanto, os pais de um doente com SPOAN são necessariamente
heterozigotos, portam a mutação, mas não desenvolvem a síndrome. Cada filho de
pais heterozigotos tem 25% de chance de manifestarem a doença, 50% de somente
portarem a mutação, e 25% de não serem portadores de nenhuma alteração, sendo
saudáveis e incapazes de passar adiante a mutação. Os números para essa condição
em Serrinha dos Pintos é assustador, sendo estimado que um em cada 250 habitantes
dessa cidade é afetado pela síndrome SPOAN, e que um entre nove indivíduos dessa
comunidade é heterozigoto, carregando a mutação genética responsável pelo
desenvolvimento da doença. De acordo com o censo brasileiro de 2000, de 5.507
municípios brasileiros, Serrinha dos Pintos está posicionada em 38° entre as 50
comunidades com maior porcentagem de portadores de deficiências físicas, com quase
6% de seus habitantes apresentando algum tipo de deficiência. A síndrome de SPOAN,
sem dúvida, foi a que mais contribuiu para essa posição (Pivetta, 2005; Santos et al.,
2005).
Ainda, não se sabe em qual gene está a mutação que causa a doença.
Foram analisadas amostras de DNA de 74 moradores da cidade, entre doentes e
sadios, e os resultados dos estudos indicam que o gene causador da SPOAN se
encontra em alguma região lateral do cromossomo 11q13, ligados aos marcadores
D11S1908 e D11S1889. Até hoje, não foi identificada nenhuma doença neurológica
associada a genes nesse intervalo dessa região do cromossomo 11 (Pivetta, 2005;
Santos et al., 2005). Apesar disso, uma forma de PEH complexa, de herança
autossômica dominante, caracterizada por espasticidade nos membros inferiores
associada à fraqueza de mãos e pés, e amiotrofia distal, a síndrome de Silver, já foi
identificada no cromossomo 11q12-14, porém com uma localização diferente da região
pressuposta da síndrome de SPOAN (Santos et al., 2005). Os cientistas ainda
procuram a mutação que provoca a SPOAN e, assim que identificada, os mesmos
criarão um teste pré-natal capaz de detectar a alteração genética em bebês, ainda em
gestação, e identificar os indivíduos heterozigotos para essa anomalia (Pivetta, 2005).
Vinte e dois pacientes com idade entre 9 e 63 anos foram avaliados
clinicamente por Santos et al. (2005) no artigo publicado, apresentando combinações
de características nunca relatadas anteriormente. Essa condição apresenta sintomas

19
que acrescentam às formas complexas de outras doenças neurodegenerativas como,
neuropatia motora e sensitiva hereditária (NMSH), paraplegia espástica hereditária
(PEH), e atrofia óptica hereditária.
As principais características clínicas dos indivíduos afetados pela SPOAN
incluem sintomas relacionados à atrofia congênita do nervo óptico, notados nos
primeiros meses de vida, e aparentemente não-progressiva. O nistagmo fixo pode ser
observado ainda no bebê recém-nascido acometido pela síndrome, perceptível apenas
por quem tem um histórico de doentes na família. Esses movimentos anormais e
involuntários do globo ocular reduzem o campo de visão dos afetados, fazendo com
que eles consigam enxergar os dedos de uma mão a uma distância máxima de 2
metros (Pivetta, 2005; Santos et al., 2005).
Os primeiros sinais de déficit motor aparecem nos primeiros anos de vida,
sendo a maioria antes de um ano. Atraso do desenvolvimento motor e paraparesia
espástica progressiva nos membros inferiores levando à uma marcha realizada na
ponta dos pés são as características mais incapacitantes da doença. Na maioria dos
casos, até mesmo os indivíduos mais precocemente fragilizados conseguem se
locomover, sozinhos ou amparados, com quedas e tropeções freqüentes, até certa
idade, mas geralmente, esta marcha é perdida por volta dos 10 anos de idade. Todos
os tipos de auxílio para ajudar na marcha, tornando-a o mais independente possível,
incluindo andador e bengalas, geralmente são perdidos antes dos 20 anos, por não
serem mais úteis, fazendo com que o uso da cadeira de rodas seja imprescindível.
Sinais de liberação piramidal como clônus e Babinski, não são comuns. Resposta de
tríplice-flexão (reflexo de retirada) é vista em alguns pacientes, mesmo não tendo
movimento voluntário nos membros inferiores. O acometimento dos membros
inferiores, além de ser mais intenso, sempre precede ao dos membros superiores, que
comumente começa por volta dos 20 anos. A hipertonia espástica (espasticidade) pode
ser observada em todos os pacientes com a síndrome (Pivetta, 2005; Santos et al.,
2005).
Como sinal de neuropatia motora e sensitiva, comumente com início no final
da adolescência, está presente a amiotrofia distal, ocorrendo geralmente em pacientes
com mais de 20 anos. Hiperhidrose, alteração na sensibilidade tátil, e falta de
sensibilidade profunda (vibratória e cinestésica) distal, são sinais muito comuns entre
os pacientes com SPOAN. A percepção à dor e temperatura não é afetada, até mesmo
em fases mais avançadas da doença, não havendo relato de dor espontânea. Perda

20
severa da musculatura distal de extremidades superiores e inferiores é característica
de todos os pacientes com mais de 20 anos. Não há sinal de fasciculação. Devido a
uma combinação de sinais piramidais e periféricos, os reflexos proximais são obtidos
mais facilmente que os distais; os reflexos bicipital, adutor e patelar, geralmente estão
aumentados; e os reflexos estilo-radial e calcâneo estão ausentes (Santos et al., 2005).
Disartria associada com baixo tom de voz (disfonia) também é uma
característica muito freqüente entre esses pacientes, acometendo indivíduos com mais
de 20 anos. Em casos mais severos, a disartria e disfonia são tão intensas que
dificilmente consegue-se entender a fala do paciente. Alguns sinais extrapiramidais
como, sinal de roda denteada, distonia no pescoço e mioclonia no polegar são raros de
ocorrer, havendo relato de quatro casos para a roda denteada, e um para as demais
(Santos et al., 2005).
Outra característica própria destes pacientes, podendo ser observada
precocemente ainda na infância, é uma resposta motora abrupta ao ouvir sons
inesperados. Curiosamente, até mesmo pacientes com ausência total de movimentos
voluntários nos membros inferiores, apresentam contrações musculares involuntárias
diante desses sons. Esta resposta está presente em todos os indivíduos com a
síndrome, observada até mesmo depois da fase adulta, sendo facilmente provocada,
incluindo casos mais severos e avançados da doença (Santos et al., 2005).
Deformidades na coluna e articulares, assim como limitação na mobilidade,
principalmente das articulações do tornozelo, joelho, punho e cotovelo, estão presentes
em todos os pacientes, com graus variados de limitação. Aumento da cifose cervico-
toracica e escoliose são sinais muito comuns entre esses pacientes. Em casos mais
severos, estas deformidades na coluna são tão intensas, impossibilitando o indivíduo
de sentar de forma independente. Não há evidências clínicas de doença do tecido
conjuntivo (Santos et al., 2005).
A intensidade dos sintomas pode variar entre os pacientes, porém a doença
é completamente perspicaz, e seus efeitos sobre a qualidade de vida dos afetados pela
síndrome são devastadores. Pode-se mencionar que, alteração cognitiva, atraso
mental, ataxia e surdez não estão presentes nestes pacientes, não fazendo parte,
portanto, dos sintomas clínicos da síndrome (Pivetta, 2005; Santos et al., 2005).
Conforme os sintomas progridem, com o passar dos anos, os indivíduos
acometidos se fecham e vivem apartados do dia-a-dia, recolhidos ao interior de suas
casas, totalmente dependentes dos cuidados de familiares. Sem o devido

21
acompanhamento médico, ninguém toma medicamentos para reduzir a rigidez e o
enfraquecimento dos membros (Pivetta, 2005).
Para a gente hospitaleira e humilde de Serrinha dos Pintos, ainda é muito
difícil entender sobre uma doença tão complexa quanto SPOAN, e eles a definem
como um “problema de família” (Pivetta, 2005).

22
2 OBJETIVOS
Primário:
Comparar as características clínicas e história da doença entre dois casos
de pacientes com síndrome de SPOAN e o relatado na literatura.
Secundários:
Analisar a qualidade de vida de duas pacientes com síndrome de SPOAN,
através do Teste de Qualidade de Vida;
Identificar o grau de depressão destas pacientes através da escala Beck
Depression Inventory (BDI);
Verificar, quantitativamente, a funcionalidade das pacientes através da
escala Spinal Cord Independence Measure (SCIM);
Comparar a marcha de paciente com síndrome de SPOAN e pacientes com
paraplegia espástica hereditária (PEH).

23
3 MÉTODOS
O presente estudo foi realizado no setor de neurologia do serviço
ambulatorial de fisioterapia da Universidade de Santo Amaro. Foram incluídas duas
pacientes com diagnóstico de SPOAN, que estão em acompanhamento clínico no
ambulatório, sendo que estas pacientes ou seus responsáveis concordaram e
assinaram o Termo de Consentimento Livre e Esclarecido (Anexo I).
Ambas as pacientes são do sexo feminino, com idade entre 12 e 28 anos,
diagnosticadas SPOAN. O diagnóstico destas pacientes foi baseado em critérios
clínicos, e exames eletroneuromiográficos, fundo de olho, eletrorretinograma, e DNA. A
confirmação do diagnóstico foi realizada no Centro de Estudos do Genoma Humano e
do Hospital das Clínicas da Universidade de São Paulo (USP).
Critérios de inclusão:
Pacientes com diagnóstico de SPOAN confirmado por meio de exames clínicos e
laboratoriais;
Estar matriculado no setor de neurologia do serviço ambulatorial da Universidade de
Santo Amaro;
Estar de acordo com a pesquisa e assinar o Termo de Consentimento Livre e
Esclarecido.
Critério de exclusão:
Possuir outra doença associada.
Foi aplicado um questionário (Anexo II), elaborado pelas próprias autoras da
pesquisa, visando coletar dados pessoais, queixa principal, histórico da doença,
evolução da mesma, casos de antecedente familiar com a doença, casamento
consangüíneo na família, presença de familiares em São Paulo com a doença,
familiares em Serrinha dos Pintos, história dos sinais e sintomas, entre outros. Também
foi aplicada uma ficha de avaliação (Anexo III), desenvolvida por especialistas na área

24
de fisioterapia neurológica, da Universidade de Santo Amaro, visando colher dados
como diagnóstico fisioterapêutico, tônus muscular, sensibilidade, atividades reflexas,
presença de sinais piramidais de liberação, limitações de amplitude de movimento,
equilíbrio, coordenação, cognição, entre outros. Também foram aplicadas quatro
escalas:
Escala de qualidade de vida (Anexo IV), sendo um questionário que se
divide em quatro áreas: social, afetiva, profissional e saúde. Para responder ao
questionário as pacientes tiveram que utilizar sim ou não para cada pergunta (Lippi,
Rocha, 1996);
Escala de BDI (Beck Depression Inventory) consiste em um auto-relato que
mensura o grau de depressão, constituída por 21 itens, cada um com quatro
alternativas (Anexo V). A pontuação varia entre 0 e 3 pontos, subentendendo graus
crescentes da gravidade da depressão. A depressão foi considerada relevante com
uma pontuação igual ou acima de 20 pontos (Gorenstein, Andrade, 1998);
Escala SCIM (mensuração de independência em lesão medular), que foi
desenvolvida por Catz et al. em 1997, para verificar as diferenças funcionais e níveis de
independência entre lesados medulares (Anexo VI). Esta cobre as três principais áreas
da função: auto-cuidado, controle respiratório e de esfíncteres e mobilidade. O primeiro
inclui os seguintes itens: alimentação, banho, vestir-se e higiene pessoal; os pontos
desta área variam de 0 a 20. Controle respiratório e de esfíncteres: respiração, controle
da bexiga, controle do intestino, e uso do banheiro; nessa área os pontos variam de 0 a
40. Mobilidade é dividida entre: itens de atuação no quarto e banheiro, e itens de
atuação em todos os cômodos, dentro e fora de casa. Na primeira temos: mobilidade
na cama e ação para prevenir pontos de pressão, transferência da cama - cadeira de
rodas, e cadeira de rodas - vaso sanitário. A segunda inclui mobilidade de pequena,
média e longa distância, controle em escadas e transferência da cadeira de rodas -
carro. Nesta, os pontos variam de 0 a 40. Apesar de inicialmente ter sido criada para
portadores de lesão medular, a escala mostra-se sensível para observar a
funcionalidade, por isso esta foi incluída em nosso estudo (Catz, Itzkovich, 1997);
A escala de classificação da análise do andar (Anexo VII) foi aplicada na
paciente A, com o intuito de quantificar a alteração da marcha ainda existente nessa
paciente. Nessa escala, podemos observar 3 categorias com perguntas que possuem
escores de 0 a 3. O primeiro item, sendo categoria geral contém 5 perguntas, a
pontuação geral se baseia na soma das 5 perguntas, sendo que 0 indica dados de um

25
individuo normal e 15 do indivíduo alterado; a categoria da extremidade inferior consta
4 perguntas, sendo 0 indivíduo normal e 12 indivíduo alterado; e a categoria do tronco,
da cabeça e da extremidade superior encontra-se 7 perguntas, com o total geral de 0
ponto para indivíduo normal e 21 para indivíduo alterado. Na análise visual da escala,
observa-se cadência e os passos indicando três opções de escolha; o percurso, sendo
observado pela distância, duração e velocidade; as fases de apoio e balanço,
mostrando a seqüência feita por um indivíduo normal, assim descrevendo a seqüência
feita pela paciente da amostra; e a duração das fases de apoio, de balanço e o ciclo
para cada membro. Para isso, foi feito um vídeo da marcha da paciente, pois este é o
critério incluído no método de avaliação da escala, realizado na sala do ambulatório de
neurologia da Faculdade de Fisioterapia da Universidade de Santo Amaro, onde foi
solicitado para a paciente andar uma distância de 6 metros, em solo plano, descalça,
apoiando os membros superiores na coxa da terapeuta, que permaneceu sentada
sobre um banco com rodinhas, acompanhando os movimentos da paciente. Deve-se
destacar que não foi solicitada nenhuma correção de postura e movimentos durante a
marcha, para que a paciente pudesse andar da maneira mais confortável possível.
Essa escala foi desenvolvida para avaliar a marcha de pacientes que andam sem
apoio, porém a paciente incluída na amostra não é capaz de andar sem o devido
auxílio. Apenas os itens do grupo de perguntas da categoria do tronco, da cabeça e da
extremidade superior, tiveram que ser adaptados de forma que as avaliadoras
analisassem a marcha considerando o apoio de membros superiores sobre a coxa da
terapeuta (Wolfsan, Whipple, 1990).
Além da escala de classificação da análise do andar, foi utilizada também, a
ficha de avaliação neurológica (Anexo III), no item avaliação da marcha, onde
descrevemos, de forma simples, a dissociação entre cinturas, fase de oscilação, fase
de apoio plantar, transferência de peso, tamanho do passo, velocidade, ritmo,
movimentos associados, marcha em linha reta e marcha sob comando; para que se
pudesse obter informações adicionais para comparação com a marcha de pacientes
com paraplegia espástica hereditária (PEH), descrita por Klebe et al. em 2004.
As escalas e dados coletados foram aplicados uma única vez, e preenchidos
pelas pesquisadoras, que leram as questões e/ou alternativas, e anotaram as
respostas, sem interferir nas mesmas, pois as pacientes não possuíam habilidade para
escrever.

26
4 RESULTADOS
Casuística:
Foram avaliadas e incluídas no estudo duas pacientes do sexo feminino,
entre 12 e 28 anos. O tempo de diagnóstico variou entre 3 e 7 anos. Uma das
pacientes é ex-moradora de Serrinha dos Pintos, já a outra não apresenta co-ligação
nenhuma com a cidade do Rio Grande do Norte, porém, esta última é filha de pais que
tiveram casamento consangüíneo. As duas referem que a dificuldade às perdas foi
segmentada, ocorrendo a longo prazo. O questionário expôs diferença entre elas como
sendo a paciente A, estudante que se encontra cursando o ensino fundamental, e a
paciente Z que encerrou os estudos na pré- escola. Podemos observar com quantas
pessoas estas pacientes moram, e qual o salário familiar existente; encontramos na
paciente A quatro moradores, sendo a renda familiar obtida por duas pessoas; na
paciente Z temos cinco moradores e três pessoas que trabalham e ajudam nos gastos.
Citaremos a seguir a história de cada uma das pacientes:
Paciente A (Figura 1): 12 anos, natural da cidade de São Paulo, filha de pais
saudáveis primos de primeiro grau, possui um irmão também saudável, relata não ter
antecedentes familiares com a doença. A paciente apresenta tetraparesia espástica
leve com predomínio crural. Relata ter nascido de parto cesáreo, sem traumas ou
doenças na infância, referindo demora ao andar (1 ano e 6 meses), perda de equilíbrio
com início aos 3 anos, e uma alteração na marcha notada na mesma época (andava
na ponta dos pés e puxava a perna para andar). A mãe refere ter ido ao médico
diversas vezes, porém estes entravam em conflito quanto ao diagnóstico correto,
relatando, portanto, que a paciente era portadora de paralisia cerebral. A paciente
nunca andou normalmente, sendo capaz de andar a uma distância maior que 200
metros e correr com dificuldade até os 7 anos. Foi relatado que aos 4/5 anos de idade
necessitou de ajuda para andar, havendo piora do quadro desde os 8 anos com a
paciente tendo quedas mais freqüentes e cansaço com facilidade, utilizando, em alguns
momentos, a cadeira de rodas por fraqueza nos membros inferiores, porém faz uso
freqüente desta há 6 meses, mesmo conseguindo andar, ainda com dificuldade, por

27
pouco tempo, com apoio. O nistagmo foi diagnosticado, através de exames, aos 5
anos, sendo visível aos 7, não havendo progressão nesse aspecto, porém há uma
redução do campo visual, com a paciente apresentando dificuldade de enxergar os
dedos de uma mão a uma distância maior que 2 metros. Desde os 3 anos de idade, foi
notado um aumento da espasticiade, porém foi há uns 3 anos que houve piora desse
quadro. Há 2 anos, percebeu fraqueza na musculatura distal dos membros inferiores,
com perda da sensibilidade tátil e profunda notada aos 7 anos. A paciente não
apresenta distúrbios urinários ou fecais, e desde 1 ano se assusta com facilidade
diante de sons inesperados. O tom de voz sempre foi baixo, e não apresenta disfagia.
Não há alteração cognitiva. Foi realizada avaliação clínica, onde a paciente refere
fraqueza nos membros inferiores como sendo sua queixa principal. Há presença de
hipotrofismo, principalmente na musculatura de tríceps sural bilateral. A força muscular
nos membros superiores foi avaliada como sendo grau 4, e nos membros inferiores
grau 2. Há ausência de clônus e do reflexo aquileu. Os reflexos bicipital e tricipital estão
diminuídos, já o patelar apresenta-se levemente aumentado. A articulação do quadril
encontra-se limitada para extensão passiva, as articulações dos membros superiores
apresentam-se livres de restrições. A amplitude de movimento ativo dos membros
inferiores não foi possível avaliar devido à fraqueza muscular. O equilíbrio na posição
ajoelhada e em pé apresenta-se alterado, de forma que a paciente não consegue
permanecer na posição por muito tempo. As provas de coordenação foram realizadas
com facilidade, exceto calcanhar-joelho, na qual a paciente encontrou dificuldade para
elevar o membro. Há presença de sinal de Romberg. As manobras de Barre e
Mingazzini também estão alteradas. A paciente deambula com auxílio, apresentando
hiperlordose lombar e escoliose discreta côncava à esquerda. Não há sinais de
acometimento cerebelar e distonia. A sensibilidade superficial e profunda encontram-se
diminuídas nos membros inferiores, principalmente nos pés. A sensibilidade térmica
encontra-se sem alteração.
Paciente Z (Figura 1): 28 anos, cadeirante, natural de Serrinha dos Pintos,
mora em São Paulo desde os 3 anos, relata sua história com dificuldade, pois sua voz
apresenta-se muito baixa, além de um sinal muito nítido, a disartria. Refere ter
antecedentes familiares com SPOAN em Serrinha (25 pessoas), sendo que seus pais
são primos de 2º grau, e seus familiares residentes de São Paulo não apresentam a
doença. É a única filha acometida entre três irmãos. Em relação à avaliação clínica, a
paciente apresenta tetraparesia espástica grave com predomínio crural. Relata como

28
sendo sua queixa atual, uma forte dor nas costas que iniciou esse ano. Nasceu de
parto normal, sem história de trauma na infância, porém apresentou algumas doenças
na idade entre 5 e 7 anos, como pneumonia, sarampo, catapora e hepatite, e diarréia,
febre e gripe eram freqüentes. Refere ter iniciado a marcha com 1 ano e 3 meses,
sendo que esta apresentava-se na ponta dos pés, em uma velocidade rápida, gerando
quedas frequentes para trás desde o início. A mãe relata ter procurado o médico
algumas vezes, mas assim como na paciente A, estes não sabiam o diagnóstico
correto, referindo ter uma síndrome desconhecida. A paciente foi submetida à primeira
cirurgia para aplicação de toxina botulínica nos membros inferiores aos 5 anos,
inicialmente na musculatura adutora, e mais tarde, nos flexores plantares. A fraqueza
nos membros inferiores iniciou-se aos 7 anos, evoluindo da região distal para proximal,
com a paciente andando se apoiando nas paredes. A marcha foi sendo perdida aos
poucos, com o passar dos anos, sendo que a paciente era capaz de andar a uma
distância maior que 200 metros e correr com dificuldade até os 8 anos. Com 12 anos,
relata ter sofrido uma queda no banheiro de casa, ficando traumatizada e com medo de
acontecer novamente. A partir dessa época, a paciente parou de andar, perdendo a
marcha por completo, após ganhar uma cadeira de rodas. O nistagmo apareceu na
mesma época em que começou a fraqueza nos membros inferiores, quando a paciente
tinha 7 anos, este não progrediu, porém o campo de visão apresenta-se reduzido, com
a paciente conseguindo enxergar os dedos de uma mão a uma distância de 1 metro.
Relata que a dificuldade com as mãos e a fraqueza nos membros superiores iniciou na
mesma época que foi para a cadeira de rodas. A diminuição da sensibilidade tátil nos
membros inferiores foi notada por volta dos 4 anos, e o aumento da espasticidade,
quando parou de andar. Não apresenta distúrbios urinários ou fecais. Desde a infância
se assusta com facilidade diante de sons inesperados, porém esse quadro piorou aos
12 anos. Assim que foi para a cadeira de rodas, iniciaram-se os problemas na coluna.
O tom de voz sempre foi baixo. Atualmente, a paciente apresenta disfagia leve, sem
alteração cognitiva, nem sinais de acometimento cerebelar. A musculatura de tríceps
sural apresenta-se hipotrófica bilateralmente. A força muscular nos membros inferiores
foi avaliada como sendo grau 0 e nos membros superiores grau 3, principalmente na
musculatura distal, e na musculatura do tronco. Há ausência de clônus e do reflexo
aquileu, os reflexos bicipital e tricipital estão presentes, já o patelar e o de retirada
estão aumentados. Apresenta limitação nas articulações do tornozelo, joelho, ombro e
punho bilateral, com encurtamento, principalmente, dos músculos isquiotibiais,

29
iliopsoas e tríceps sural. Devido ao fato de a paciente não ficar nas posições sentada,
ajoelhada, quatro apoios e em pé sem apoio, não foi possível avaliar o equilíbrio. As
provas de coordenação foram realizadas com dificuldade, lentamente, exceto
calcanhar-joelho, mostrando-se incapaz de realizar devido à ausência de movimento
voluntário nessas extremidades. O sinal de Romberg encontra-se alterado, pois a
paciente não permanece na posição ortostática. As manobras de Barre e Mingazzini
são positivas e a dos membros superiores estendidos e Stewart Homes são negativas.
Apresenta aumento da cifose cervico-toracica e escoliose em “S” com concavidade
torácica à esquerda, muito severa, impossibilitando a paciente de estender a cabeça.
Ela permanece flexionada o tempo todo, impossibilitando-a de sentar sem apoio. A
sensibilidade superficial e profunda encontram-se diminuídas nos membros inferiores,
principalmente nos pés, e nas mãos em menor intensidade. A sensibilidade térmica
encontra-se normal.
Figura 1. Paciente A: 12 anos, deambula com auxílio da barra paralela com adução
dos joelhos, característica da marcha espástica. A paciente Z: 28 anos, restrita a
cadeira de rodas, apresenta aumento da cifose cervico-toracica e escoliose, assim
como padrão flexor dos membros superiores.

30
As tabelas e gráficos serão exibidos com a inicial do primeiro nome, de
acordo com o critério citado no termo de consentimento livre e esclarecido.
Na tabela 2, temos o teste de qualidade de vida onde consideramos os itens:
social, afetivo, profissional e da saúde; os números referem-se às perguntas citadas no
teste. No quadrante social, podemos verificar que não houve diferença significante; no
item profissional houve diferença significante, pois o avanço da doença trouxe
incapacidade e incompatibilidade de dar continuidade à vida profissional (na paciente
de menor idade a vida profissional é considerada como estudante); já no item referente
à saúde, a paciente A demonstra-se melhor.
Tabela 2. Comparação das pacientes com SPOAN na escala de qualidade de vida.
QUALIDADE DE VIDA
SOCIAL 1 2 3 4 5 6 7 8 9 10
A N N S N N S S S N S
Z N N S N S S N N N S
AFETIVO 1 2 3 4 5 6 7 8 9 10
A N S S S S S S S S S
Z N S S S S S S S S S
PROFISSIONAL/ ESCOLAR 1 2 3 4 5 6 7 8 9 10
A S S S S S N S N S S
Z N N N N N N N N N N
SAÚDE 1 2 3 4 5 6 7 8 9 10
A N S S N N S S S N S
Z N N N N N N N N N N
SAÚDE 11 12 13 14 15
A N S S S S
Z N N N N N
A tabela 3 apresenta o escore da BDI, que variou de 13 a 24; a paciente com
13 pontos encontra-se em depressão de leve a moderada, e a de 24 pontos de
moderada a grave.
O questionário apresenta as perguntas de forma numérica (01 a 21), mas,
para facilitar a visualização dos resultados, alteramos a forma numérica para letras (A
até U).

31
Tabela 3. Pontuação das pacientes com SPOAN na BDI.
ESCALA DE BECK A B C D E F G H I J K L M N O P Q R S T U SOMA
A 0 0 0 0 0 2 0 2 0 1 0 0 2 2 1 0 2 0 0 1 0 13Z 0 3 0 1 0 0 2 3 0 1 1 0 2 1 3 1 0 0 0 3 3 24
O gráfico 1 demonstra a diferença entre as pacientes com SPOAN; a
denominada como Z apresenta o nível de depressão elevado em relação à outra.
Gráfico 1. Avaliação das pacientes em nível de depressão com BDI.
A tabela 4 é referente ao escore da escala SCIM, onde os pontos variam de
0 a 5.
No item de auto-cuidado, observa-se que a paciente A tem maior
independência; a média do item alimentação é a mais elevada, e as duas pacientes
declaram comer a comida cortada, não necessitando de adaptações, somente de
assistência.
Tabela 4. Apresenta o item de auto-cuidado da escala SCIM, das 02 pacientes com
SPOAN.
Auto-CuidadoAlimentação Banho Vestuário Higiene
A 4 4 2 2
ESCALA DE BECK
0
1
2
3
A B C D E F G H I J K L M N O P Q R S T U
A Z

32
Z 4 3 0 2Média 4 3,5 1 2
Observando o gráfico 2 visualizamos a discrepância no item vestuário, a
paciente Z necessita de assistência total para se vestir, sendo que no estágio mais
avançado da doença é indispensável à ajuda.
Gráfico 2. Diferença entre as pacientes de SPOAN em relação ao auto-cuidado.
O controle respiratório e esfincteriano na tabela 5 refere-se ao escore da
escala SCIM, onde os pontos variam de 0 a 10; no item controle da bexiga os pontos
variam de 0 a 15. Para a respiração, controle da bexiga e uso do vaso, os escores
foram máximos, mas para o controle intestinal houve alteração, devido à evolução da
doença.
Tabela 5. Apresenta o item de controle respiratório e esfincteriano da escala SCIM, das
02 pacientes de SPOAN.
Controle Respiratório e Esfincteriano
Respiração Controle Bexiga Controle Intestino Uso do vasoA 10 15 10 4Z 10 15 5 4
Média 10 15 7,5 4
O gráfico 3 apresenta diferença significante em relação ao item controle de
AUTO-CUIDADO
0
1
2
3
4
5
Alimentação Banho Vestuário Higiene
A Z

33
intestino da paciente A sobre a Z; nos outros itens não houve alteração.
Gráfico 3. Demonstra o controle respiratório e esfincteriano das pacientes com
SPOAN.
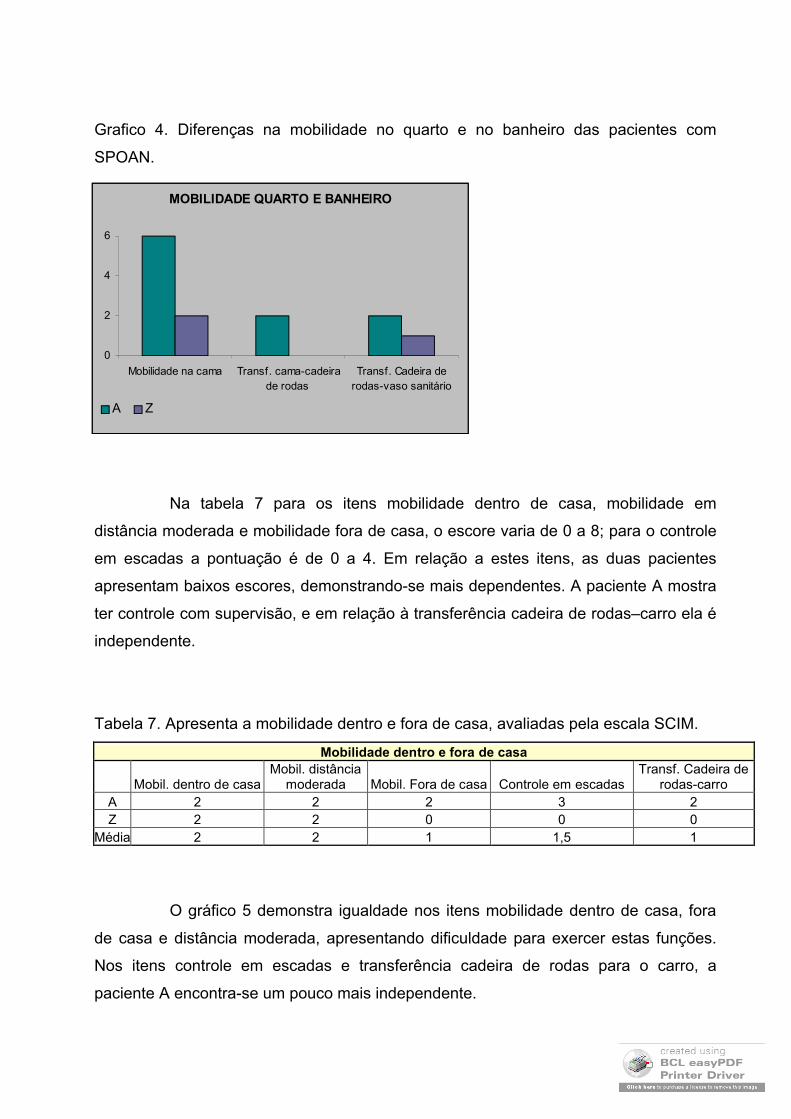
A tabela 6 mostra a mobilidade no quarto e no banheiro das pacientes
incluídas no trabalho. No item mobilidade na cama, a pontuação varia de 0 a 6, as
transferências tanto da cama para a cadeira de rodas como da cadeira de rodas para o
vaso sanitário a pontuação varia de 0 a 2. A paciente A apresenta melhor pontuação na
tabela referente a mobilidade no quarto e banheiro.
Tabela 6. Apresenta a mobilidade no deslocamento das pacientes com SPOAN.
Mobilidade Quarto e Banheiro
Mobilidade na camaTransf. cama-cadeira de
rodas Transf. Cadeira de
rodas-vaso sanitário
A 6 2 2Z 2 0 1
Média 4 1 1,5
A mobilidade reduzida, apresentada pela paciente Z, indica um nivel de
dependência pessoal. A transferência cadeira de rodas – vaso sanitário é feita pelas
duas, porém, há quedas frequentes citadas pela paciente Z, principalmente quando
esta se dispõe a realizar essa atividade sozinha.
CONTROLE RESPIRATÓRIO E ESFINCTERIANO
0
3
6
9
12
15
Respiração Controle Bexiga Controle Intestino Uso do vaso
A Z
34
Grafico 4. Diferenças na mobilidade no quarto e no banheiro das pacientes com
SPOAN.
Na tabela 7 para os itens mobilidade dentro de casa, mobilidade em
distância moderada e mobilidade fora de casa, o escore varia de 0 a 8; para o controle
em escadas a pontuação é de 0 a 4. Em relação a estes itens, as duas pacientes
apresentam baixos escores, demonstrando-se mais dependentes. A paciente A mostra
ter controle com supervisão, e em relação à transferência cadeira de rodas–carro ela é
independente.
Tabela 7. Apresenta a mobilidade dentro e fora de casa, avaliadas pela escala SCIM.
Mobilidade dentro e fora de casa
Mobil. dentro de casa Mobil. distância
moderada Mobil. Fora de casa Controle em escadasTransf. Cadeira de
rodas-carroA 2 2 2 3 2Z 2 2 0 0 0
Média 2 2 1 1,5 1
O gráfico 5 demonstra igualdade nos itens mobilidade dentro de casa, fora
de casa e distância moderada, apresentando dificuldade para exercer estas funções.
Nos itens controle em escadas e transferência cadeira de rodas para o carro, a
paciente A encontra-se um pouco mais independente.
MOBILIDADE QUARTO E BANHEIRO
0
2
4
6
Mobilidade na cama Transf. cama-cadeirade rodas
Transf. Cadeira derodas-vaso sanitário
A Z

35
Gráfico 5. Mobilidade das pacientes dentro e fora de casa.
Importante observar que foi aplicada a escala de classificação da análise do
andar somente na paciente A, pois obtém a marcha com auxilio, sendo assim uma das
terapeutas realizou o auxilio para a aplicação.
No gráfico 6 podemos observar o primeiro item de categoria geral,
mostrando um total máximo de pontos, estando 100% alterada com relação ao normal
(0%). Na categoria da extremidade inferior podemos observar que o total da paciente A
é 11 e a pontuação máxima é 12, encontrando-se 91,5% alterada. Na categoria do
tronco, cabeça e extremidade superior o total foi de 15 pontos, estando 71,5% alterada.
Assim consideremos o individuo em todas as categorias alteradas.
Gráfico 6. Escala de classificação da análise do andar.
MOBILIDADE DENTRO E FORA DE CASA
0
2
4
6
8
Mobil. dentrode casa
Mobil.distancia
moderada
Mobil. Forade casa
Controle emescadas
Transf.Cadeira derodas-carro
A Z
100%
91%
71%
0%
100%
categoria geral
categoria de extremidadeinferior
categoria do tronco,cabeça e da extremidadesuperior

36
A escala de classificação da análise do andar apresenta um item como
análise visual, podendo-se observar cadência/passos da paciente A que foi de 22 por
minuto, entrando, assim na colocação do item de menor que 70 por minuto.
O percurso foi cronometrado em uma distância de 3 metros, onde a duração
foi de 36 segundos e a velocidade de 0,083m/s, sendo que a unidade colocada para a
velocidade é de 1,46m/s para o indivíduo normal.
PARÃMETRO VALOR UNIDADE
Distância 3 metros
Duração 36 segundos
Velocidade 0,083 (1,46) m/s
A fase de apoio inicia com o toque do calcanhar, apoio médio, impulso e
ausente, sendo que a paciente A teve como resultado uma troca nessa seqüência,
iniciando com apoio médio, depois toque do calcanhar, completando as duas últimas
fases com êxito. Na fase de balanço, a paciente A fez a seqüência correta que inicia
com aceleração, balanço médio, desaceleração e ausente.
A duração condiz com a fase de apoio, balanço e do ciclo do membro inferior
direito (MID) e membro inferior esquerdo (MIE), podendo-se obter resultados como:
FASE MID MIE UNIDADE
Apoio 1,5 2,06 segundoBalanço 1,36 2,75 segundo
Ciclo 5,2 5,29 segundo

37
5 DISCUSSÃO
A combinação de paraplegia espástica hereditária, neuropatia axonal,
disartria, resposta motora abrupta diante de sons inesperados, e atrofia congênita do
nervo óptico, observados nos pacientes com síndrome de SPOAN, nunca foi descrita
anteriormente (Santos et al., 2005).
A síndrome de neuropatia motora e sensitiva hereditária (NMSH) associada
à atrofia do nervo óptico consiste no tipo VI no qual esse grupo de doenças é dividido.
As NMSH’s são classificadas, de acordo com Dyck, em sete tipos, apresentando em
comum, fraqueza de musculatura distal associado à atrofia e graus variados de
distúrbios sensitivos. Dois grupos principais podem se diferenciar: tipo I, primariamente
afeta a bainha de mielina, e tipo II, mostrando uma degeneração axonal. Dyck
introduziu três tipos (V, VI, VII), com o objetivo de reunir os sintomas frequentemente
associados, como paraplegia espástica, atrofia óptica, e retinopatia. Já foram descritos
casos em que os pacientes portadores de NMSH apresentavam características
incompatíveis com essa classificação. O primeiro caso relatado de NMSH de herança
autossômica recessiva tipo VI, foi descrito em 1937, com dois irmãos portando
neuropatia e atrofia do nervo óptico (Chalmers et al., 1997; Dillmann et al., 1997;
Santos et al., 2005).
Em 1997, Dillmann et al. descreveram o caso de dois irmãos, filhos de pais
com união consangüínea, portando neuropatia motora e sensitiva hereditária com
atrofia óptica e paraplegia espástica. A história da doença e dos sintomas é parecida
com a descrita nesse estudo pela síndrome de SPOAN, porém, o tempo em que os
pacientes permanecem com a marcha mostram-se diferentes, cuja média descrita por
Dillmann et al. (1997), foi de 35 anos, enquanto Santos et al. (2005) cita uma média de
15 anos para os pacientes com SPOAN perderem completamente a marcha. A época
em que estes pacientes notaram algum distúrbio de visão foi na segunda década de
vida, inicialmente com uma progressão lenta, depois mais rápida; já os pacientes
descritos nesta pesquisa apresentaram distúrbios similares antes dos 10 anos,
mostrando uma atrofia óptica não-progressiva. As alterações de sensibilidade nas
extremidades superiores e inferiores mostraram-se parecidas quanto ao início do
surgimento. Alguns sinais como disartria e resposta motora abrupta diante de sons
inesperados, não foram citados por Dillmann et al. (1997). As duas doenças, apesar de

38
apresentarem história e quadro clínico similares, estas se diferenciam, principalmente,
pelo tempo de progressão dos sintomas.
De acordo com o artigo publicado em 2005, foi descrito que a maioria dos
pacientes com SPOAN iniciou os primeiros sinais de déficit motor antes de 1 ano de
idade, tendo um atraso no desenvolvimento neuro-psico-motor. Nos primeiros meses
de vida, é possível notar que a maioria desses pacientes apresenta nistagmo. O caso
das pacientes A e Z descritos nesse estudo se contradiz quanto à idade de início desse
sinal, quando comparado à maioria da população com a mesma doença, onde estas
manifestaram esse tremor involuntário do globo ocular aos 7 anos. Assim como a
maioria dos pacientes, estas também apresentaram uma redução do campo visual não-
progressiva.
Aos 12 anos, a paciente A ainda apresenta marcha, embora com dificuldade,
se apoiando, não a mantendo por muito tempo devido à fraqueza nos membros
inferiores, tendo que se aliar à cadeira de rodas. Já a paciente Z perdeu a marcha,
mesmo com auxílio, aos 12 anos. O artigo publicado por Santos et al. (2005) cita perda
total da marcha independente entre 6 e 10 anos, e a marcha com apoio, entre 10 e 15
anos para a maioria dos pacientes.
Um atraso ao iniciar a marcha também é característico destes pacientes,
assim como um padrão típico de marcha com passos rápidos e na ponta dos pés. A
paciente A iniciou a marcha com 1 ano e 6 meses, e a paciente Z com 1 ano e 3
meses, apresentando o mesmo padrão característico, associado com quedas
freqüentes.
Foram observados alguns sinais clínicos na avaliação neurológica das
pacientes A e Z: hipertonia espástica com predomínio crural, presença de sinal de
canivete à movimentação passiva, disfonia, resposta motora abrupta diante de sons
inesperados, com a paciente Z não apresentando contração voluntária em musculatura
de membros inferiores, disartria, deformidades articulares e da coluna que levassem à
limitação de movimento, fraqueza mais intensa nas extremidades distais, reflexos
proximais aumentados e distais diminuídos, alteração na sensibilidade tátil, vibratória e
cinestésica, principalmente nas extremidades distais dos membros inferiores.
As pacientes A e Z possuem os mesmos sinais clínicos que foram
observados na maioria dos pacientes avaliados por Santos et al. (2005).
Uma diminuição na sensibilidade dolorosa foi observada nas duas pacientes,
o que não corrobora com o artigo, onde a maioria não apresenta alteração nesse

39
aspecto, mesmo na fase mais avançada da doença. Este artigo também relata sobre
aumento do reflexo bicipital apresentado pelos pacientes de Serrinha dos Pintos, não
ocorrendo o mesmo com as pacientes avaliadas na amostra. Estas não apresentaram
sinal de Babinski, fasciculação, ataxia, atraso mental, alteração cognitiva, e sinais
extrapiramidais tais como distonia e mioclonia (Santos et al., 2005).
Pôde-se observar que a intensidade dos sintomas variou entre as pacientes
A e Z, como era esperado. Devido ao fato de a paciente Z ser mais velha que a
paciente A, esta apresentou todos os sintomas, sem exceção, em um grau de
severidade mais elevado, tornando-a mais dependente que a paciente mais jovem, em
todos os aspectos. A idade de início e progressão dos sintomas coincidiu entre as
pacientes, destacando que a paciente A, por ter apenas 12 anos, não apresenta alguns
sinais como disartria e amiotrofia distal nos membros superiores.
Ao analisar a funcionalidade destas pacientes com síndrome de SPOAN,
observamos acometimento de maior nível na paciente Z, quando comparada com a
paciente A, por se encontrar em um estágio mais avançado da doença. Embora, alguns
itens como alimentação e controle de bexiga coincidam em pontuação máxima entre as
pacientes, mostrando independência total nesse aspecto, os itens relacionados à
higiene, controle respiratório, uso do vaso, mobilidade dentro de casa e mobilidade à
distância moderada, também se mostram no mesmo nível de pontuação, porém, com
menor grau de independência. Podemos observar que em vestuário, transferência
cama-cadeira de rodas, mobilidade fora de casa, controle em escadas e transferência
cadeira de rodas-carro, apenas a paciente A realiza estas atividades, apresentando
uma pontuação menor em relação à independência, quando comparados aos itens
citados anteriormente. Isso se deve ao fato de que esta paciente apresenta algum grau
de força muscular em membros inferiores, enquanto que a paciente Z não apresenta
nenhum movimento voluntário nessas extremidades, além de que a fraqueza nos
membros superiores desta apresenta–se em um estágio mais avançado, dificultando a
realização de determinadas atividades.
A escala de funcionalidade SCIM é um instrumento de avaliação que
mensura as alterações funcionais mais significativas em pacientes que sofreram lesão
medular, analisando a capacidade destes indivíduos ao executar as tarefas diárias. Um
estudo feito por Catz et al. em 2001 confirmou o que outros estudos concluíram, que a
escala SCIM possui maior nível de confiabilidade e sensibilidade em relação à
alterações funcionais de pacientes com lesão medular quando comparada à outras

40
escalas. No estudo feito, comparou-se a sensibilidade das escalas SCIM e FIM
(Functional Independence Measure) no que diz respeito à funcionalidade. Outro estudo
feito pelos mesmos autores, em 2002, que teve como objetivo validar a versão II da
escala SCIM aplicando-a em uma população de 202 pacientes com lesão medular,
mostrou que o item alimentação foi a tarefa mais facilmente executada dentro da área
de auto-cuidado, e os itens mobilidade na cama e dentro de casa os mais fáceis dentro
da mobilidade, coincidindo com os resultados obtidos pelas pacientes A e Z. As tarefas
mais difíceis foram vestuário, transferência cadeira de rodas – vaso sanitário e controle
em escadas. As pacientes A e Z apresentaram maior dificuldade em vestuário,
transferência cama – cadeira de rodas e mobilidade fora de casa. No artigo, o que diz
respeito à respiração e controle esfincteriano, o item respiração se mostrou mais fácil
do que os demais itens dessa área. O controle de intestino mostrou-se um pouco
melhor do que o controle de bexiga, e ambos foram mais fáceis quando comparados ao
uso do vaso. As pacientes da amostra apresentaram mesmo grau de dificuldade em
relação ao controle de intestino e uso do vaso sanitário (Itzkovich et al., 2002). Devido
ao fato da escala SCIM ter sido desenvolvida para pacientes com lesão medular, não
há estudos na literatura que mostram o uso dessa escala em doenças
neuromusculares.
Observando a escala de qualidade de vida, nota-se que no quadrante social,
curiosamente, a paciente Z mostrou-se mais sociável e comunicativa, mesmo
passando a maior parte do tempo recolhida em sua casa, enquanto a paciente A
mostrou-se o oposto, apresentando uma vida social mais ativa que confere a de
pessoas da mesma idade. A escala mostra introspectiva em relação à paciente Z.
Nota-se, nas duas pacientes, o mesmo grau de sensibilidade com relação ao
item afetivo da escala acima. Relatam afeto aos que as rodeiam e concordam com uma
pergunta do teste, referindo gostar e admirar a si mesmas, porém, no momento da
resposta a expressão facial das duas pacientes mostrou-se confusa, não concordando
completamente com a resposta dada. A vaidade é algo indispensável à vida destas
pacientes, tendo unhas feitas e cabelos sempre bem cuidados. A paciente A,
discretamente, relata ter interesse em um garoto da escola e, curiosamente, não
demonstra expectativa alguma quanto a algum relacionamento, assim como a paciente
Z. A paciente A refere, no quadrante de saúde, estar com o peso dentro da média,
porém, relata que necessita emagrecer usando, algumas vezes, uma cinta abdominal,
enquanto a paciente Z diz estar acima da média do seu peso, enquanto observamos ter

41
um corpo magro, com a possibilidade de estar abaixo da média. Ainda observando a
saúde das mesmas, estas demonstram preocupação ao cuidar do corpo, tendo alguns
pontos de discordância, enquanto a paciente A refere ter cefaléias freqüentes, porém,
costuma fazer exercício físico, a paciente Z não tem esta possibilidade, relatando ter
dificuldade para se desligar dos problemas e dormir. Estes dados condizem com a
escala de Beck, onde a paciente Z relata, em sua história, sentir dores nas costas por
ficar muito tempo na cadeira de rodas. Isso fez com que não houvesse estímulo para
seguir uma vida profissional, nem mesmo estudou, assim, passando por uma vida de
calmaria. Temos o oposto com a outra paciente, onde esta tem uma vida escolar ativa,
proporcionando um aumento do ritmo da atividade diária, apresentando, assim, um
nível de leve a moderado de depressão para escala de Beck, onde demonstra estar se
sentindo culpada, punida, chorosa, relatando mudanças e perda funcional, pelo
cansaço e dificuldade de iniciar um esforço. O nível de depressão de moderado a grave
apresentado pela paciente Z mostrou o desânimo em relação ao futuro, desprazer em
relação à vida pessoal, irritação, choro, impossibilidade de fazer algum trabalho, onde
relata se sentir preocupada vendo os anos passarem, estar ficando velha e sem
atrativo. Estes são itens ainda não observados pelas outras escalas aplicadas,
demonstrando que a doença gera incapacidades importantes na vida pessoal destas
pacientes.
O inventário de depressão de Beck (IDB) é um instrumento que tem o intuito
de avaliar a presença de sintomas depressivos. A escolha deste se justifica pelo fato de
haver dados recentes quanto à sua validação no Brasil. A incidência de transtornos
psicopatológicos, principalmente de depressão grave, é alta na esclerose múltipla, e
pode até mesmo ser mais elevada do que em outras doenças neurológicas crônicas e
incapacitantes. Estudos feitos mostram que a depressão acomete de 27 a 54% dos
pacientes, apresentando-se como moderada ou grave. Um estudo feito por Haase et al.
em 2004, que teve como objetivo avaliar o funcionamento psicossocial em 34 pacientes
com esclerose múltipla através das características psicométricas de quatro medidas de
auto-relato, entre elas o IDB, indicou presença de níveis elevados de sintomas de
sofrimento psicológico entre estes pacientes. Na amostra, a mediana da distribuição
situou-se sobre o ponto de corte 15 no IDB e 23,53% dos portadores de esclerose
múltipla obtiveram escores iguais ou maiores que 20 indicando nível de depressão de
moderada a grave, assim como a apresentada pela paciente Z. Outro estudo, feito por
Mendes et al. em 2003, teve como objetivo verificar a freqüência de depressão em 84

42
pacientes com esclerose múltipla e a sua correlação com a duração da doença e
incapacidade funcional. O estudo mostrou que a depressão esteve presente em 17,9%,
e que os maiores escores da escala de Beck correlacionaram-se com maior
incapacidade funcional, porém não estão associados ao tempo de doença. Não foi
encontrado nenhum artigo que cita o uso da escala de Beck em doenças
neuromusculares, assim como o teste de qualidade de vida.
No estudo feito por Klebe et al. (2004), onde foi feita uma análise da marcha
de pacientes com paraplegia espástica hereditária, verificou-se uma redução
significante da velocidade, quando comparada à marcha de indivíduos saudáveis.
Enquanto estes apresentaram uma velocidade de 1,15 m/s, os pacientes com PEH
apresentaram 0,71 m/s. Já a paciente da amostra apresentou uma velocidade, também
diminuída, com 0,083 m/s, de acordo com a escala de classificação da análise do
andar. Esta também indica que a paciente demora 36 segundos para andar uma
distância de 3 metros, sendo o valor de uma pessoa normal de 3,3 segundos para
percorrer a mesma distância (Asencio et al., 2001). Talvez isso se deva aos baixos
valores do comprimento do passo (stride length), onde os indivíduos normais
apresentaram 1,16 metro, os acometidos pela síndrome, 0,91 metro, e a paciente A
0,55 metro; e da cadência, com 120 passos/minuto para os indivíduos sem alteração, e
94 passos/minuto para os pacientes com PEH; a paciente incluída na pesquisa
apresentou 22 passos/minuto. O afastamento dos pés durante a marcha mostrou
resultados curiosos, sendo que a largura do passo nos indivíduos saudáveis foi de 11,5
cm, enquanto nos indivíduos com a síndrome foi de 15,8 cm, associado a uma base
alargada. Este resultado não confere à da paciente da amostra que apresentou 4 cm
de afastamento entre o pés. Talvez isso se deva ao fato de esta paciente apresentar
uma espasticidade maior em musculatura adutora, fazendo com que, em algumas
vezes, uma perna cruze a outra ao dar o passo, tornando visível o joelho valgo. Klebe
et al. (2004) citam uma diminuição do ângulo de flexão do joelho na fase de oscilação
para esses pacientes, assim como na paciente incluída na pesquisa. Isso se deve ao
fato de os pacientes com PEH apresentarem uma altura do passo diminuída em
relação aos indivíduos saudáveis, com estes apresentando 18,1 cm contra 11,4 cm nos
acometidos. A paciente A não realiza dorsiflexão do tornozelo durante a fase de
oscilação, fazendo com que os artelhos entrem em contato com o solo durante todo o
trajeto, mostrando fraqueza em musculatura dorsiflexora. A paciente realiza uma
pequena elevação do calcanhar, em torno de 10 cm, fazendo uma flexão plantar do

43
tornozelo, durante a fase de oscilação. Isso confere ao resultado obtido na escala,
onde a paciente realiza o apoio médio do pé antes mesmo do toque do calcanhar,
durante o início da fase de apoio. Devido ao fato de os pacientes com PEH
apresentarem um ângulo de flexão do joelho diminuída, uma diminuição do ângulo de
flexão do quadril também é observada nestes indivíduos durante a fase de oscilação.
Os pacientes com PEH descritos no artigo de Klebe et al. (2004), mostraram
redução dos parâmetros cinemáticos e temporal da marcha, como velocidade,
cadência, comprimento do passo, amplitude de movimento do joelho, devido à
paraparesia espástica nas extremidades inferiores, porém, estes pacientes
apresentaram resultados significantemente melhor quando comparados à paciente A
incluída na amostra, pois ela apresenta maior grau de acometimento.
A dissociação entre cinturas mostrou-se alterada na paciente incluída na
pesquisa, onde esta realiza uma depressão do ombro oposto à perna que faz o impulso
para dar o passo, também realiza uma anteriorização do ombro do mesmo lado da
perna que vai à frente. A fase de oscilação apresenta-se com um tempo aumentado
(1,36 segundo para o membro inferior direito, e 2,75 para o membro inferior esquerdo)
sendo o valor normal de 0,36 segundo para o indivíduo saudável (Asencio et al., 2001).
Ainda nesta fase, a paciente apresenta uma rotação interna do pé associado a uma
flexão do joelho da perna de apoio antes mesmo de dar o impulso com a mesma. A
fase de apoio plantar também apresenta um tempo aumentado (1,50 segundo para o
membro inferior direito, e 2,06 para o membro inferior esquerdo) sendo o valor normal
de 0,62 segundo para cada membro de um indivíduo saudável (Asencio et al., 2001).
Nesta fase a paciente realiza uma hiperextensão do joelho ao apoiar o pé no solo. A
descarga de peso dos membros superiores sobre a coxa da terapeuta apresenta-se
aumentada, e nos membros inferiores não transfere peso para o calcanhar, só para o
antepé, gerando uma anteriorização do centro de massa. A paciente manteve o mesmo
ritmo na maior parte do teste, tendo que parar algumas vezes, por alguns segundos,
para descansar. A marcha é compensada por uma hiperlordose lombar, levando a uma
flexão do quadril bilateral durante toda a marcha nas fases de oscilação e apoio,
aumentando esse grau na fase de oscilação; e movimentos escapulares, também
sendo observada uma inclinação e leve rotação do tronco para o lado oposto ao da
perna que vai a frente, sendo pior esta inclinação para o lado direito. Talvez isso, assim
como as demais compensações realizadas pela paciente, se deva à fraqueza de todos
os grupos musculares de membros inferiores. Ao fazer inclinação do tronco para um

44
lado, a paciente realiza flexão do cotovelo do mesmo lado e extensão do mesmo do
lado oposto. A cabeça mantém-se semi-flexionada, acompanhando os movimentos do
tronco, direciona o quadril para o mesmo lado da inclinação ao dar o impulso com a
perna.
Como foi observado nos dados obtidos na escala e na avaliação da marcha,
o hemicorpo esquerdo da paciente da amostra apresenta-se um pouco mais
comprometido que o direito. A paciente apresentou 100% de alteração para a categoria
geral da escala, 91,5% para a categoria da extremidade inferior, e 71,5% para a
categoria do tronco, da cabeça e da extremidade superior, quando comparada a
indivíduos normais. Essa alteração significante da marcha, observada na paciente A,
deve-se, tanto à fraqueza muscular em membros inferiores quanto à alteração de
equilíbrio da mesma.
Está comprovado que a neuropatia periférica é uma disfunção comum entre
as pessoas idosas, o que aumenta o risco de alterações na mobilidade e quedas. Isso
se deve ao fato destes pacientes se tornarem incapazes de desenvolver e executar
uma estratégia de proteção apropriada para evitar a queda diante de obstáculos
inesperados ou perturbações de grandes escalas durante a locomoção. Um estudo
feito por Richardson et al. em 2005 teve como objetivo, diante destas informações,
identificar alterações na marcha relacionadas à história de queda entre um grupo de
indivíduos idosos com neuropatia periférica em dois ambientes diferentes: padrão
(superfície plana com iluminação normal) e alterado (superfície irregular com
iluminação baixa). Os dados cinemáticos da marcha foram obtidos através de 42
pacientes com idade média de 64 anos, onde 52,4% relatam história de pelo menos
uma queda. Na superfície plana, os pacientes com neuropatia periférica com e sem
história de queda apresentaram resultados similares quanto aos parâmetros avaliados
na marcha. Porém, na superfície irregular, os indivíduos com história de queda
apresentaram um aumento do tempo e largura do passo, e uma diminuição significante
do comprimento e velocidade, além de se submeterem a ajustes posturais mais
severos, quando comparados aos indivíduos sem história de queda. Não foram
encontrados na literatura artigos científicos citando o uso da escala de classificação da
análise do andar em pacientes neurológicos.

45
6 CONCLUSÕES
Com base nos resultados obtidos neste trabalho, pode-se concluir:
. As pacientes A e Z apresentam características clínicas e história da doença
similar a dos casos dos pacientes com síndrome de SPOAN avaliados em Serrinha dos
Pintos.
. Analisando as escalas de Qualidade de vida e SCIM, observa-se que a
funcionalidade mostrou-se compatível com o perfil das pacientes, levando em
consideração a idade e o tempo de doença, demonstrando que a qualidade de vida
encontra-se adequada, com boa comunicação pessoal e uma aproximação social.
. Em relação à depressão, ambas as pacientes demonstram um grau de
insatisfação diante das impossibilidades pessoais, levando a baixa estima e falta de
perspectiva diante da vida.
. A paciente A apresenta um grau de acometimento maior com relação à
marcha, quando comparada aos pacientes com paraplegia espástica hereditária
descritos na literatura.
Sugerimos que estudos futuros sejam realizados para avaliar o equilíbrio
destas pacientes, assim como outros fatores, comparando-os com a funcionalidade e
qualidade de vida de pacientes com outras síndromes de paraplegia espástica
hereditária, além de verificar os efeitos da intervenção fisioterapêutica nestes
pacientes.

46
7. ANEXOS
Ficha de Dados Pessoais e Histórico da Doença
Data:______________
Nome:____________________________________________________________
Idade:___________________
Sexo ( ) feminino ( ) masculino
Data de nascimento:____-____-____
Naturalidade:____________________Nacionalidade:_______________________
Endereço:__________________________________________________________
Telefone:____________________________
Estado Civil:___________________Escolaridade:__________________________
Estuda:_____________série_________________
Hobby:_____________________
Salário Familiar:_____________________________________________________
Quantos moram na casa:_________________
Trabalha:__________________________________________________________
Data do Diagnóstico_____________________
Queixa Principal?
História da Doença?
Evolução da Doença?
Cronologia?
Antecedentes familiares apresentam a doença?
Casamento consangüíneo na família ou paternos?
Presença de familiares em São Paulo, estes apresentam a doença?
Apresenta família em Serrinha dos Pintos?
Sinais e Sintomas da DoençaAbertura ocular
Pré-natal
Tipo de parto
Trauma na infância
Doença na infância
Quando começou a andar?
Como foi o desenvolvimento psicomotor (rolar, sentar, engatinhar, andar)?

47
Quando começou a perceber alteração de equilíbrio?
Nasceu com nistagmo? ( ) não ( ) sim
Quando apareceram os primeiros sinais de nistagmo? _______________________
Consegue enxergar coisas ao seu redor, contar os dedos em uma distância de 2m?
Dificuldades gerais a perda foi segmentada a longo prazo ou de imediato?
Dificuldade para subir e descer escada e segurar objetos?
Dificuldade de higiene pessoal?
Quando diminuiu a sensibilidade nos membros?
Quando percebeu o aumento da espasticidade em MMII?
Quando percebeu fraqueza nos MMII e MMSS?
Quando perdeu a capacidade de correr?
Quando parou de andar e usar a cadeira de rodas?
Tem quedas freqüentes?
O tom de voz era mais alto na infância, quando começou a ficar mais fraco?
Assusta com facilidade a estímulos auditivos exacerbados?
Ficha de Avaliação Neurológica da
Universidade de Santo Amaro
IDENTIFICAÇÃO:
Nome DN
Naturalidade
Sexo Cor
Profissão Grau de instrução
Endereço
Cidade
Fone
Diagnóstico Fisioterapêutico
Diagnóstico Médico
ANAMNESE:
EXAME FISIOTERAPÊUTICO:

48
Padrões Posturais
Tônus Muscular
Sensibilidade
Tátil
Térmica
Dolorosa
Cinestésica
Discriminativa
Atividades Reflexas
Reflexos tendinosos:
Patelar
Aquileu
Bicipital
Tricipital
Sinais de liberação reflexa:
Babinski
Clônus
Reações associadas
Impulso extensor
Extenção cruzada
Sincinesias
Limitação de ADM
Ativa
Passiva
Reações de equilíbrio, retificação e proteção
Sentado

49
Ajoelhado
Em pé
Manobras deficitárias
Braços estendidos
Romberg
Mingazzini
Barre
Stewart Homes
Provas de coordenação
Index-index
Index-nariz
Calcanhar-joelho
Movimentos alternados
Avaliação da Marcha
Dissociação entre cinturas
Fase de oscilação
Fase de apoio plantar
Transferência de peso
Tamanho do passo
Velocidade
Ritmo
Movimentos Associados
Marcha em linha reta
Marcha sob comando

50
Termo de Informação e Consentimento Livre e Esclarecido
As alunas Fúlvia de Souza Viera e Solange Macedo Aily, orientadas pela Profª Dr
Márcia Cristina Bauer Cunha desenvolverão um estudo denominado “Características
Funcionais de Pacientes com SPOAN: Paraplegia Espástica com Atrofia Óptica e
Neuropatia”.
As informações à seguir, serão oferecidas para sua participação voluntária nesse
estudo. Leia atentamente as informações aqui contidas, pedindo esclarecimento caso
ocorra dúvidas.
Após o seu consentimento, o Sr. (a) será avaliado por um fisioterapeuta que perguntará
sobre alguns dos seus dados pessoais antecedentes pessoais, familiares e
hereditários, e a história da doença atual.
Você deverá colaborar respondendo à (1) questionário contendo perguntas
relacionadas à qualidade de vida, (1) questionário relacionado a estado emocional
atual, (1) questionário classificando a funcionalidade, somente uma vez cada
questionário.
Os questionários serão lidos pelas pesquisadoras e as respostas anotadas pelas
mesmas.
Seus dados pessoais serão de caráter confidencial, ficando sob responsabilidade das
pesquisadoras, assim sendo inspecionado pelas pesquisadoras envolvidas no estudo e
pelas autoridades legais.
Os dados desse estudo se forem divulgados em forma de relatório ou for publicada,
isto será feito de forma codificada para que a confidencialidade seja mantida.
A participação não inclui nenhum ônus por parte do terapeuta e nem do participante.
Você poderá receber mais informações ou mesmo desistir da pesquisa em qualquer
momento.
Eu________________________________________, RG___________________,
declaro ter sido esclarecido(a) quanto ao objetivo desta pesquisa e concordo
participar livremente deste trabalho, cujos dados autorizo que sejam utilizados para fins
científicos e apresentações específicas.
________________________(assinatura do paciente)

51
________________________(assinatura do acompanhante no caso do paciente
possuir incapacidade física)
São Paulo, _________________________________
Declaro que expliquei pessoalmente o termo de Consentimento Livre e
Esclarecido ao paciente ou representante legal para a participação deste estudo.
_________________________data______________
(assinatura do responsável pelo estudo)

52
8 REFERÊNCIAS
1. Asencio G, Blanc Y. A Marcha Humana, a Corrida e o Salto. São Paulo: Manole; 2001. p.18-113, passim.
2. Bouslam N, Benomar A. Mapping of a New Form of Pure Autosomal Recessive Spastic Paraplegia (SPG28). Ann Neurol. 2005;57(4):567-71.
3. Burgunder J-M, Hunziker W. Hereditary Spastic Paraplegia: Clues from a Rare Disorder for a Common Problem? IUBMB Life. 2003;55(6):347-52.
4. Catz A, Itzkovich M. SCIM- Spinal Cord Independence Measure: a new disability scale for patients with spinal cord lesions. A new SCL disability scale. 1997;35:850-56.
5. Catz A, Itzkovich M. The spinal cord independence measure (SCIM): Sensitivity to functional changes in subgroups of spinal cord lesion patients. Spinal Cord.2001;39(2):97-100.
6. Chalmers RM, Riordan-Eva P. Autosomal recessive inheritance of hereditary motor and sensory neuropathy with optic atrophy. J Neurol Neurosurg Psychiatry. 1997;62:385-87.
7. Crosby AH, Proukakis C. Is the Transportation Highway the Right Road for Hereditary Spastic Paraplegia? Am J Hum Genet. 2002;71(5):1009-16.
8. Dillmann U, Heide G. Hereditary motor and sensory neuropathy with spastic paraplegia and optic atrophy: report on a family. J Neurol. 1997;244:562-65.
9. Fink JK. Hereditary Spastic Paraplegia: The Pace Quickens. Ann Neurol. 2002;51(6):669-72.
10. Fink JK. Advances in the hereditary spastic paraplegias. Exp Neurol. 2003;184:106-10.
11. Fink JK. The Hereditary Spastic Paraplegias: Nine Genes and Counting. Arch Neurol. 2003;60(8):1045-49.
12. Fink JK. Hereditary Spastic Paraplegia: Spastin Phenotype and Function. ArchNeurol. 2004;61(6):830-33.
13. Gorenstein C, Andrade L. Inventário de depressão de Beck: propriedades psicométricas da versão em português. Rev Psiq Clin. 1998;25(5):245-50.
14. Haase V, Lacerda S. Avaliação do funcionamento psicossocial na esclerose múltipla: características psicométricas de quatro medidas de auto-relato. Arq Neuro-Psiquiatr. 2004;62(2a):282-90.

53
15. Hodgkinson CA, Bohlega S. A novel form of autosomal recessive pure hereditary spastic paraplegia maps to chromosome 13q14. Neurology. 2002;59(12):1905-09.
16. Hughes CA, Byrne PC. SPG15, A new locus for autosomal recessive complicated HSP on chromosome 14q. Neurology. 2001;56:1230-33.
17. Itzkovich M, Tripolski M. Rasch analysis of the Catz-Itzkovich spinal cord independence measure. Spinal Cord. 2002;40(8):396-407.
18. Klebe S, Stolze H. Gait analysis of sporadic and hereditary spastic paraplegia. J Neurol. 2004;251(5):571-78.
19. Lesca G, Eymard-Pierre E. Infantile ascending hereditary spastic paralysis (IAHSP): Clinical features in 11 families. Neurology. 2003;60(4):674-82.
20. Lippi M, Rocha J. Stress, Hipertensão e Qualidade de Vida. Campinas: Papirus; 1996. p.13-18.
21. Many pathways lead to hereditary spastic paraplegia. Lancet Neurol. 2003;2(4):210.
22. McDermott CJ, Grierson AJ. Hereditary Spastic Paraparesis: Disrupted Intracellular Transport Associated with Spastin Mutation. Ann Neurol. 2003;54(6):748-59.
23. Meijer IA, Cossette P. A Novel Locus for Pure Recessive Hereditary Spastic Paraplegia Maps to 10q22.1 – 10q24.1. Ann Neurol. 2004;56(4):579-82.
24. Mendes M, Tilbery C. Depressão na esclerose múltipla forma remitente-recorrente. Arq Neuro-Psiquiatr. 2003;61(3a):591-95.
25. Orlacchio A, Kawarai T. Hereditary Spastic Paraplegia: Clinical Genetic Study of 15 Families. Arch Neurol. 2004;61:849-55.
26. Pivetta M. SPOAN: Uma Nova Doença. Pesquisa do Estado de São Paulo (FAPESP). 2005 Jul.
27. Richardson JK, Thies SB. Gait Analysis in a Challenging Environment Differentiates Between Fallers and Nonfallers Among Older Patients With Peripheral Neuropathy. Arch Phys Med Rehabil. 2005;86(8):1539-44.
28. Santos S, Kok F. Spastic Paraplegia, Optic Atrophy, and Neuropathy Is Linked to Chromosome 11q13. Ann Neurol. 2005;57:730-37.
29. Vazza G, Zortea M. A New Locus for Autosomal Recessive Spastic Paraplegia Associated with Mental Retardation and Distal Motor Neuropathy, SPG14, Maps to Chromosome 3q27 – q28. Am J Hum Genet. 2000;67:504-09.
30. Wolfsan L, Whipple R. Gait Assessment In The Elderly: A Gait Abnormality Rating Scale and Its Relation to Falls. J Gerontol. 1990;45(1):12-19.

54
ABSTRACT
BACKGROUND: SPOAN (Spastic Paraplegia, Optic Atrophy, and Neuropathy) termrefers to a neurodegenerative and progressive disease, from autossomal recessive inheritance, discovered by scientists from Genome Human Study Centre and Clinical Hospital from University of São Paulo (USP). SPOAN is mainly characterized by congenital optic atrophy, progressive spastic paraparesis, motor and sensory axonal neuropathy. OBJECTIVES: To compare the clinical features and disease history between two patients with SPOAN syndrome and the literature reported, beyond analyse the quality of life, identify the depression level, verify the functionality and to compare the gait from one of them with the gait from patients with Hereditary Spastic Paraplegia (HSP). METHODS: Two female patients were included in this study, at the age of 12 and 28 years, with confirmed diagnosis of SPOAN syndrome. It was applied a questionnaire, to get pieces of information from disease history, a neurological evaluation card, a quality of life test, the Beck Depression Inventory, the Spinal Cord Independence Measure (SCIM), and the Gait Abnormality Rating Scale. RESULTS: Both patients showed spontaneous fixation nystagmus as symptom related to congenital optic atrophy, progressive spastic paraparesis with onset in early childhoodwas present, beyond sensibility disorders, and other, considering that the patient Z had all symptoms more severe. The patient Z is more communicative and sociable than patient A, even wasting most of the time indoors. Beck Depression Inventory showed a light to moderate level of depression for patient A, and a moderate to more severe level for patient Z. This patient showed herself more deficient in relation to functionality when compared with the younger patient. When analysing the patient A gait, we observed significantly lower values in all parameters evaluated. CONCLUSIONS: The patients A and Z showed clinical features and disease history similar from patients with SPOAN syndrome evaluated in Serrinha dos Pintos; the functionality and the quality of lifeshowed suitable with the patients profile, considering the age and disease time. Both patients showed a high insatisfaction level for individual disabilities, leading to low esteem and non perspective for life. The patient A showed herself more deficient in relation to gait, when compared with patients with Hereditary Spastic Paraplegia (HSP) reported.
KEYWORDS: SPOAN, Hereditary Spastic Paraplegia (HSP), Quality of Life,Depression, Functionality, Gait.






![Aula 10 - Avaliação Neurológica (1) [Modo de Compatibilidade]](https://static.fdocumentos.com/doc/165x107/55cf94f1550346f57ba5808b/aula-10-avaliacao-neurologica-1-modo-de-compatibilidade.jpg)











