
Hipinóticos e Sedativos Rodrigo Borges de Oliveira (Msc.) · Mudanças na arquitetura do sono,...
160
Rodrigo Borges de Oliveira (Msc.) Hipinóticos e Sedativos
Transcript of Hipinóticos e Sedativos Rodrigo Borges de Oliveira (Msc.) · Mudanças na arquitetura do sono,...

Rodrigo Borges de Oliveira (Msc.)
Hipinóticos e Sedativos

• A necessidade de sono e a latência para iniciar o sono sofrem grandes flutuações individuais;
• Idade é um fator importante:
• Tempo total de sono e períodos REM e profundo são maiores em crianças e adultos jovens do que em indivíduos idosos;
• Idosos: sono entrecortado por vários períodos despertos.
Sono normal

Hipóteses
• Restauração de energia gasta durante a vigília
• Menor consumo de energia durante o sono: a atividade motora émais baixa, assim como o fluxo sangüíneo, a respiração, a temperatura corporal e, conseqüentemente, a taxa metabólica global.
• Redução da temperatura cerebral (sono não REM), que aumenta gradativamente durante a vigília
• Recuperação da sensibilidade a neurotransmissores, como a noradrenalina, que diminui gradativamente durante a vigília
Funções do Sono

• Consiste na permanente ativação das vias tálamo-corticaispelas sinapses excitatórias glutamatérgicas das fibras aferentes (ex. transmissão de informação sensorial)
• Neurônios tálamo-corticais são mantidos ligeiramente despolarizados, com o potencial de membrana próximo ao potencial de disparo;
• Ativação massiva dos dendritos das células corticais;
• EEG dessincronizado
Vigília

• Vias ativadoras originárias dos neurônios histaminérgicos do hipotálamo posterior;
• Projetam amplamente para o córtex cerebral;
• Lesão desta região: coma e sincronização do EEG;
• Anti-histamínicos: sonolência
Vigília
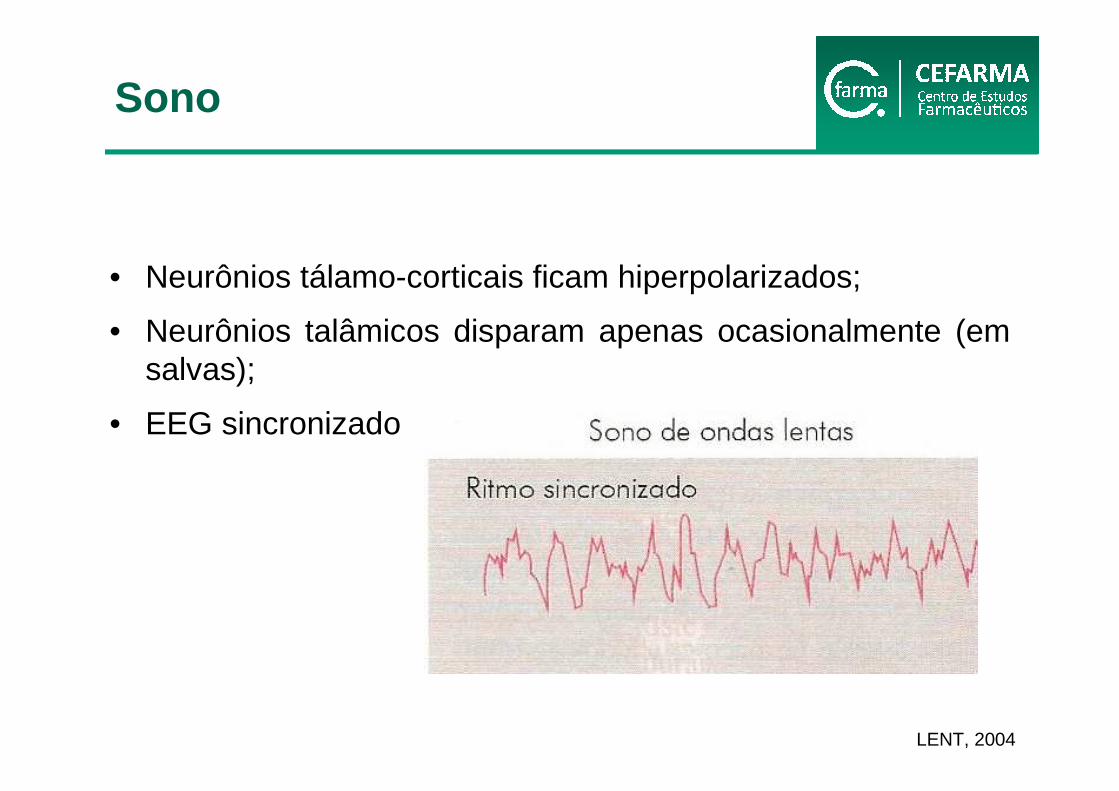
• Neurônios tálamo-corticais ficam hiperpolarizados;
• Neurônios talâmicos disparam apenas ocasionalmente (em salvas);
• EEG sincronizado
LENT, 2004
Sono

• Sistemas moduladores histaminérgicos: recebem inervação inibitória de neurônios gabaérgicos do hipotálamo anterior;
• Neurônios gabaérgicos do hipotálamo anterior: parecem entrar em ação no início do sono de ondas lentas, silenciando os neurônios histaminérgicos que mantêm a vigília;
• Neurônios colinérgicos do hipotálamo anterior: ativos durante o sono de ondas lentas;
• Mecanismo que dispara o processo: não esclarecido
Indução do Sono

• Sinais para acordar: ambiente externo e sistemas sensoriais ou sistema temporizador circadiano (mecanismo ainda desconhecido);
• Neurônios noradrenérgicos do locus ceruleus: aumentam a atividade na transição sono paradoxal e vigília.
Vigília

• Uma das queixas mais comuns na prática médica em geral;
• Hipnótico perfeito:
� Deveria permitir a ocorrência de um sono com arquitetura normal, não causaria efeitos no dia seguinte e não interagiria com outros medicamentos;
� Seu uso crônico não provocaria dependência ou insônia rebote após a interrupção;
• Exercício moderado regular: satisfaz todos estes critérios e pode ser eficaz para muitos casos.
Insônia

Crença, por parte do paciente, de que não está dormindo suficientemente;
Mudanças na arquitetura do sono, consideradas normais no processo de envelhecimento;
Aparecimento de problemas clínicos ou psiquiátricos que afetam o dormir;
Hipnóticos parecem ser utilizados em maior quantidade por mulheres e idosos.
GUIMARÂES, FS In: FUCHS et al., 2004
Insônia

Insônia Transitória•Dura menos de 3 dias;
•Habitualmente causada por um breve estressor ambiental e situacional (ex. após viagens aéreas intercontinentais);
Insônia a curto prazo•Dura de 3 dias à 3 semanas;
•Habitualmente causada por um estressor pessoal, como doença, sofrimentos ou problemas de trabalho.
Classificação da Insônia

Insônia a longo prazo
•Já durou mais de 3 semanas;
•Nenhum estressor específico pode ser identificado;
•Necessidade de uma avaliação médica mais completa, mas a maior parte não necessita que o sono seja estudado por toda a noite.
Classificação da Insônia

Insônia secundária a transtorno psiquiátrico
•Ansiedade, depressão, esquizofrenia, crise maníaca e outros;
•Tratamento deve objetivar a correção da causa primária do distúrbio do sono;
•Insônia comum em pacientes depressivos que usam ISRS – pacientes acabam melhorando por conta da melhora da depressão
•Associação de trazodona para pacientes cuja insônia persistente é efeito colateral dos ISRS
Classificação da insônia

Insônia secundária a transtorno psiquiátrico
•Insônia secundária a quadros esquizofrênicos – tratar com antagonistas dopaminérgicos
•Estes antipsicóticos são sedativos
•Associação com benzodiazepínicos também pode ser uma alternativa viável
Classificação da Insônia

Insônia condicionada (apreendida)•Não há doença psiquiátrica ou outra condição clínica importante;
•Esses pacientes associaram a atividade de dormir a atividades consistentes com a vigília e não com o sono
•Tratamento: realizar atividades da vigília fora do quarto de dormir. A cama deve ser utilizada apenas para dormir e para sexo.
Má percepção do estado de sono•Queixa de sono ruim, sem alterações mensuráveis
•Tratamento difícil
Insônia em pacientes idosos
Classificação da Insônia

• Insônia transitória constitui-se na única indicação bem comprovada de hipnóticos;
• Eficácia de benzodiazepínicos em diminuir a latência do sono e prolongar sua duração;
• Nos casos de insônia secundária: tratamento deve objetivar a correção da causa primária do distúrbio do sono;
• Faltam dados que justifiquem o uso dos hipnóticos mais comuns (benzodiazepínicos) em insônia crônica. Efeito tende a diminuir com o tratamento; alterações na arquitetura do sono e efeitos colaterais.
Tratamento da Insônia

• Benzodiazepínicos – mais utilizados;
• Zolpidem, zopiclona e zaleplona (utilização está aumentando)
• Outros depressores do sistema nervoso central - foram abandonados para esta indicação:
� Barbitúricos;
� Hidrato de cloral;
� Etclorvinol;
� Glutetimida;
� Meprobamato;
� Metiprilona;
� Paraldeído
Tratamento da Insônia

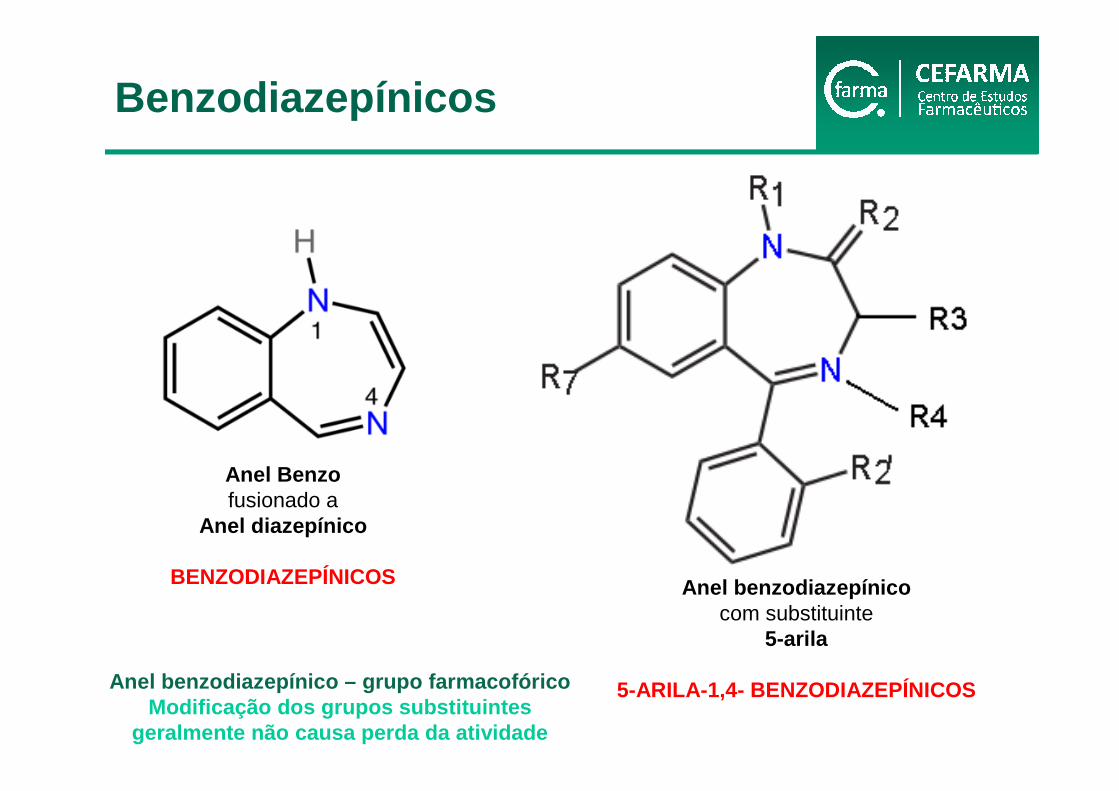
Anel Benzofusionado a
Anel diazepínico
BENZODIAZEPÍNICOS Anel benzodiazepínicocom substituinte
5-arila
5-ARILA-1,4- BENZODIAZEPÍNICOSAnel benzodiazepínico – grupo farmacofóricoModificação dos grupos substituintes
geralmente não causa perda da atividade
Benzodiazepínicos

A B
C
Benzodiazepínicos

diazepamclordiazepóxido bromazepamclonazepam flunitrazepam
midazolam triazolamlorazepam
clorazepato
estazolamoxazepam alprazolam
Clobazam(1,5-benzodiazepina)
Brotizolam(modificação no anel A)
Flumazenil (anel C -> cetona /Metila na posição 4 – ANTAGONISTA)
Benzodiazepínicos

• Hipnóticos mais utilizados;
• Maior índice terapêutico e menor potencial para dependência física;
• Não provocam indução enzimática hepática;
• Produzem um sono mais fisiológico
• Depressores inespecíficos
• Não são depressores generalizados (não causam analgesia e anestesia)
Benzodiazepínicos
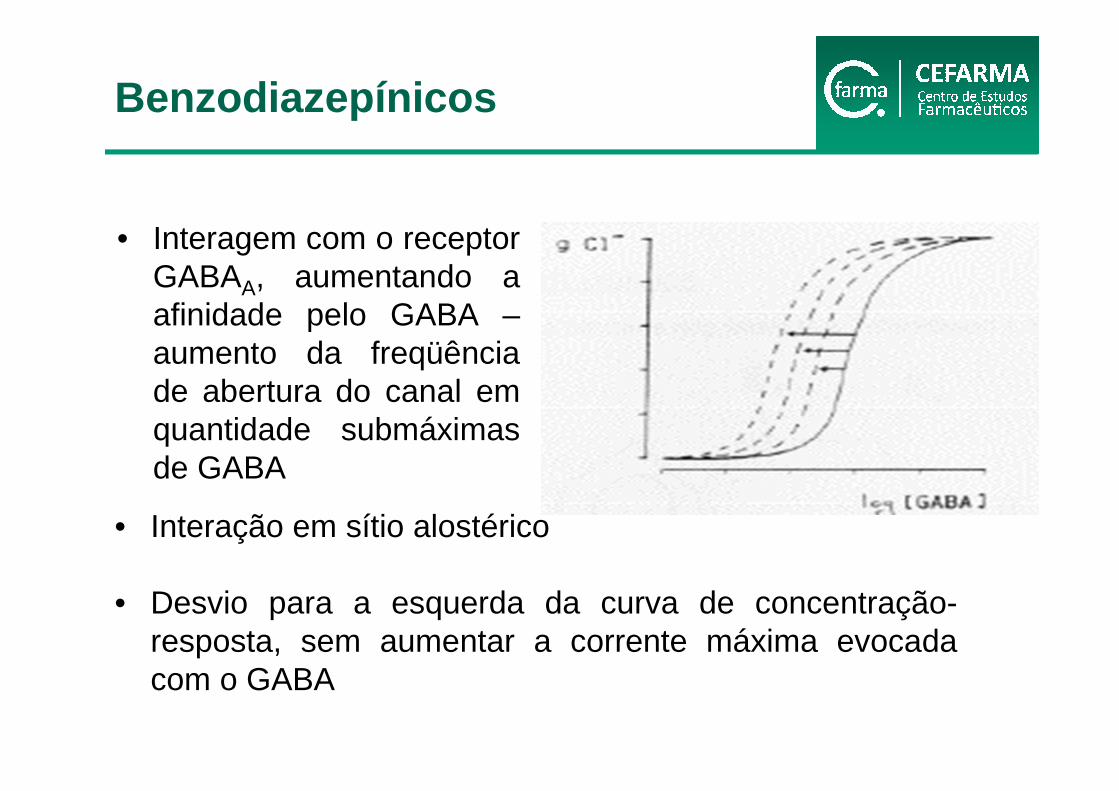
• Interagem com o receptor GABAA, aumentando a afinidade pelo GABA –aumento da freqüênciade abertura do canal em quantidade submáximas de GABA
• Interação em sítio alostérico
• Desvio para a esquerda da curva de concentração-resposta, sem aumentar a corrente máxima evocada com o GABA
Benzodiazepínicos

SONO:
•É um estado fisiológico cíclico;
•Caracterizado no ser humano por 5 estágios fundamentais, que se diferenciam de acordo com:
• O padrão do eletrencefalograma (EEG);
• A presença ou ausência de movimentos oculares rápidos (rapid eye movements: REM),
• Mudanças em diversas outras variáveis fisiológicas, como o tono muscular e o padrão cardiorespiratório.
Fisiologia do Sono

• Em sono, os indivíduos apresentam-se imóveis, ou com um repertório limitado de movimentos, os quais são de natureza involuntária, automática, sem propósitos definidos.
• A reatividade a estímulos auditivos, visuais, tácteis e dolorosos éreduzida ou abolida em relação à vigília, particularmente em fases de sono profundo, sendo necessário o aumento da intensidade do estímulo para trazer o indivíduo de volta à vigília, o que nem sempre é observado, mesmo sob estimulação intensa
• Durante o sono, o indivíduo mantém-se de olhos fechados ou entreabertos e não mostra interação produtiva com o ambiente.
Fisiologia do Sono

• O sono pode ser visto como um estado similar ao coma, especialmente nos casos de coma de menor profundidade, em que não há comprometimento das funções cardio-respiratórias.
• O grande diferencial entre tais estados, à simples observação do ser que dorme, é a característica de reversão espontânea e mais ou menos programada ao longo do tempo do estado de sono para a vigília, o que não é o caso do coma.
Fisiologia do Sono
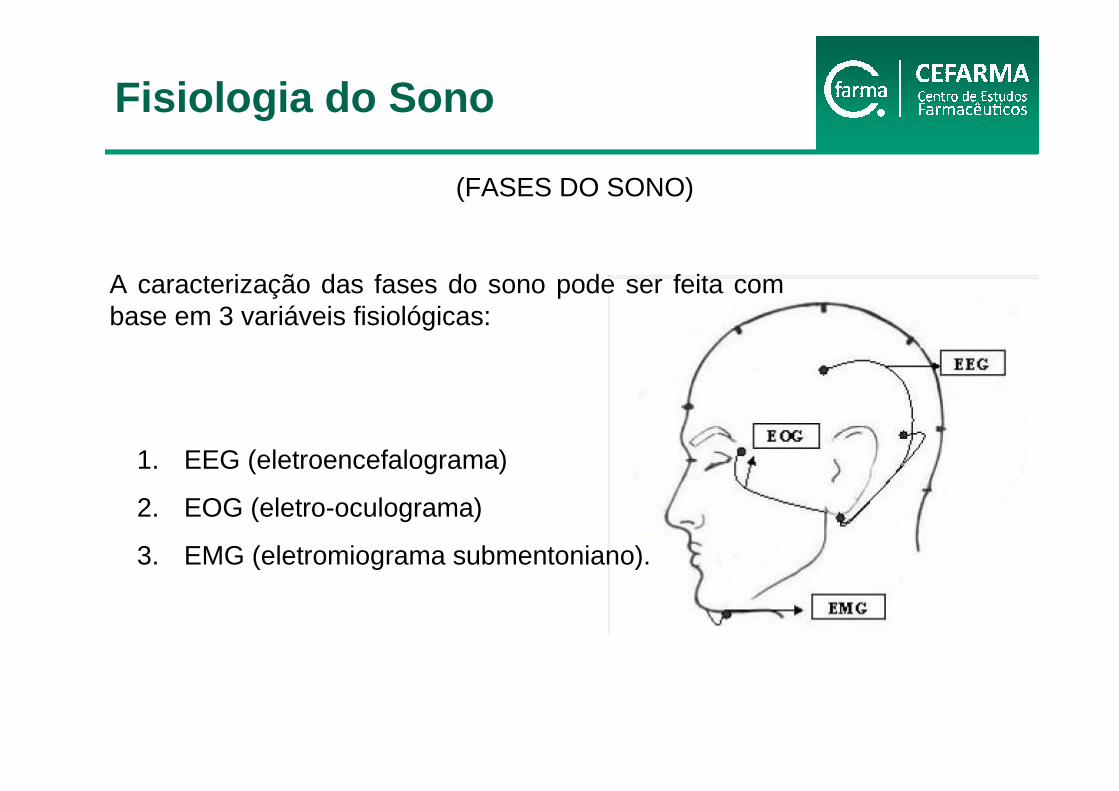
(FASES DO SONO)
A caracterização das fases do sono pode ser feita com base em 3 variáveis fisiológicas:
1. EEG (eletroencefalograma)
2. EOG (eletro-oculograma)
3. EMG (eletromiograma submentoniano).
Fisiologia do Sono

(FASES DO SONO)
O sono divide-se em dois tipos fisiologicamente distintos:
1. NREM
(Non Rapid Eye Movement )
� Fase 1
� Fase 2
� Fase 3
� Fase 4
2. REM
(Rapid Eye Movement )
• O sono NREM ocupa cerca de 75% do tempo do sono• O sono REM ocupa os 25 % restantes
Fisiologia do Sono

(FASES DO SONO)FASE NREM
VIGÍLIA - Fase (A); Vigilia descontraída. Predomina o ritmo alfa.ESTÁGIO 1 - Fase (B): adormecimento: diminuição do ritmo alfa. Aparecimento de baixas ondas teta, conhecida como estágio 1.ESTÁGIO 2 - Fase (C): sono leve. Continuação da diminuição da freqüência até as ondas delta. Fusos de sono de 12-15 Hz agrupados de permeio, conhecida como estágio 2.ESTÁGIO 3 E 4 - sono Profundo. Quase que exclusivamente grandes e lentas ondas delta, conhecida como estágios 3 e 4.
Fisiologia do Sono
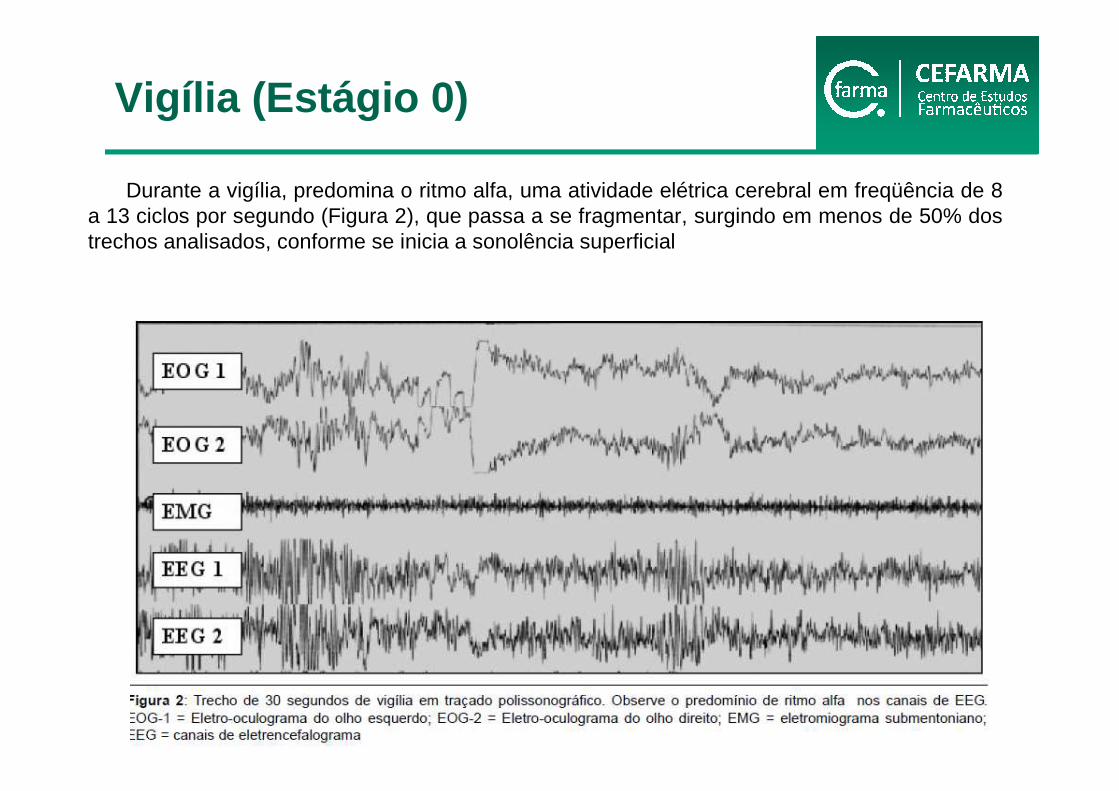
Durante a vigília, predomina o ritmo alfa, uma atividade elétrica cerebral em freqüência de 8 a 13 ciclos por segundo (Figura 2), que passa a se fragmentar, surgindo em menos de 50% dos trechos analisados, conforme se inicia a sonolência superficial
Vigília (Estágio 0)
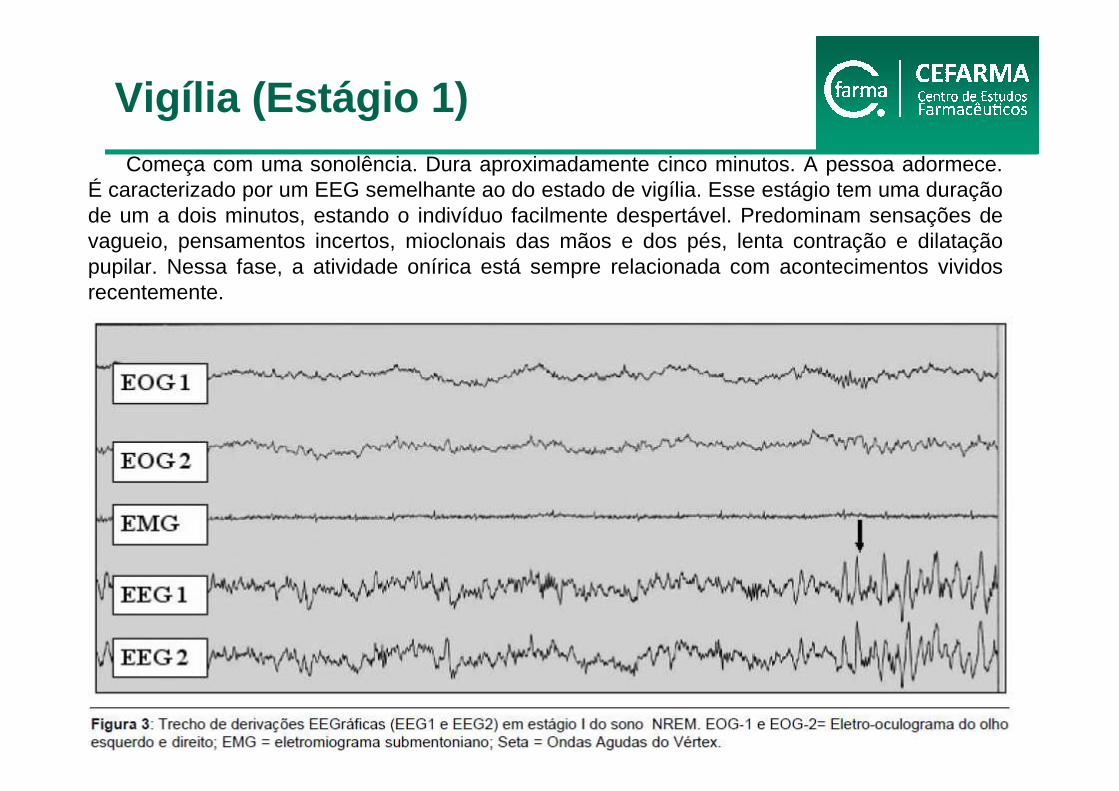
Começa com uma sonolência. Dura aproximadamente cinco minutos. A pessoa adormece. É caracterizado por um EEG semelhante ao do estado de vigília. Esse estágio tem uma duração de um a dois minutos, estando o indivíduo facilmente despertável. Predominam sensações de vagueio, pensamentos incertos, mioclonais das mãos e dos pés, lenta contração e dilatação pupilar. Nessa fase, a atividade onírica está sempre relacionada com acontecimentos vividos recentemente.
Vigília (Estágio 1)
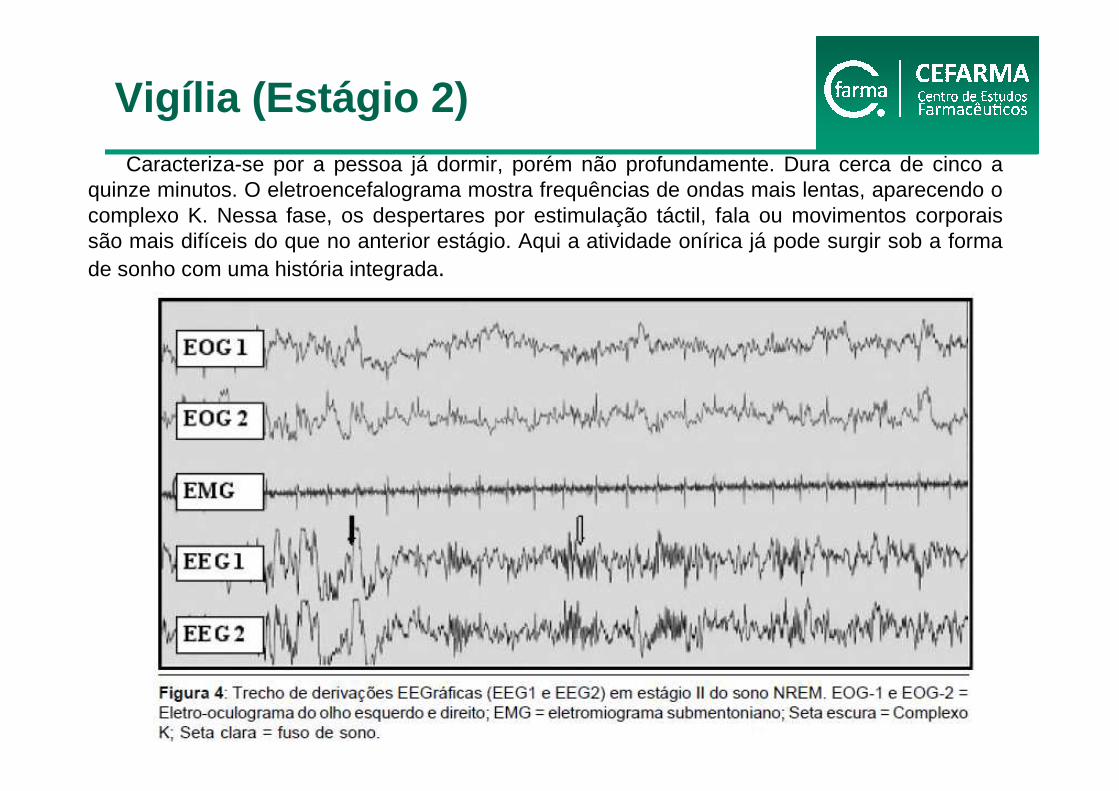
Caracteriza-se por a pessoa já dormir, porém não profundamente. Dura cerca de cinco a quinze minutos. O eletroencefalograma mostra frequências de ondas mais lentas, aparecendo o complexo K. Nessa fase, os despertares por estimulação táctil, fala ou movimentos corporais são mais difíceis do que no anterior estágio. Aqui a atividade onírica já pode surgir sob a forma de sonho com uma história integrada.
Vigília (Estágio 2)
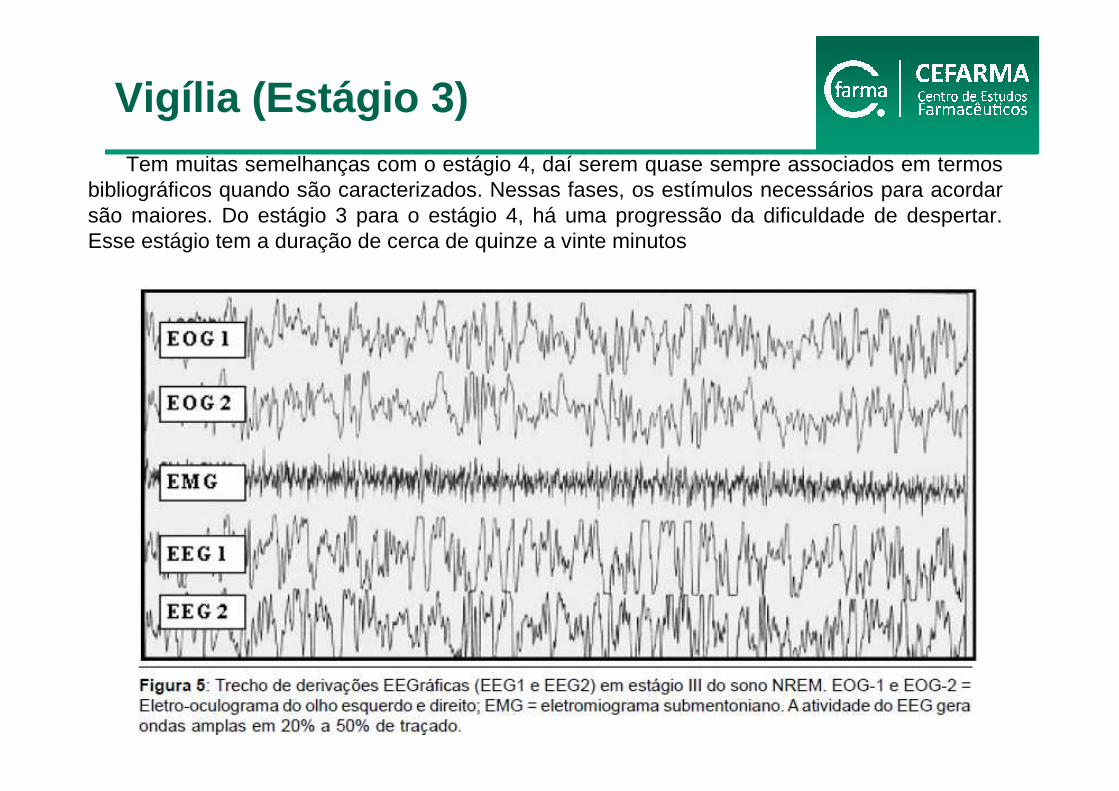
Tem muitas semelhanças com o estágio 4, daí serem quase sempre associados em termos bibliográficos quando são caracterizados. Nessas fases, os estímulos necessários para acordar são maiores. Do estágio 3 para o estágio 4, há uma progressão da dificuldade de despertar. Esse estágio tem a duração de cerca de quinze a vinte minutos
Vigília (Estágio 3)

São quarenta minutos de sono profundo. É muito difícil acordar alguém nessa fase de sono. Depois, a pessoa retorna ao terceiro estágio (por cinco minutos) e ao segundo estágio (por mais quinze minutos). Entra, então, no sono REM.
Este estágio NREM do sono caracteriza-se pela secreção do hormônio do crescimento em grandes quantidades, promovendo a síntese proteica, o crescimento e reparação tissular, inibindo, assim, catabolismo. O sono NREM tem, pois, um papel anabólico, sendo essencialmente um período de conservação e recuperação de energia física.
Vigília (Estágio 4)

• O sono REM caracteriza-se por uma intensa atividade registrada no EEG seguida por flacidez e paralisia funcional dos músculos esqueléticos. Nesta fase, a atividade cerebral é semelhante à do estado de vigília. Deste modo, o sono REM é também denominado por vários autores como sono paradoxal, podendo mesmo falar-se em estado dissociativo.
• Nesta fase do sono, a atividade onírica é intensa, sendo sobretudo sonhos envolvendo situações emocionalmente muito fortes.
• É durante essa fase que é feita iscugula da atividade cotidiana, isto é, a separação do comum do importante. Estudos também demonstram que é durante o REM que sonhos ocorrem. A fase representa 20 a 25% do tempo total de sono e surge em intervalos de sessenta a noventa minutos. É essencial para o bem-estar físico e psicológico do indivíduo.
(FASES DO SONO)FASE NREM
Fisiologia do Sono
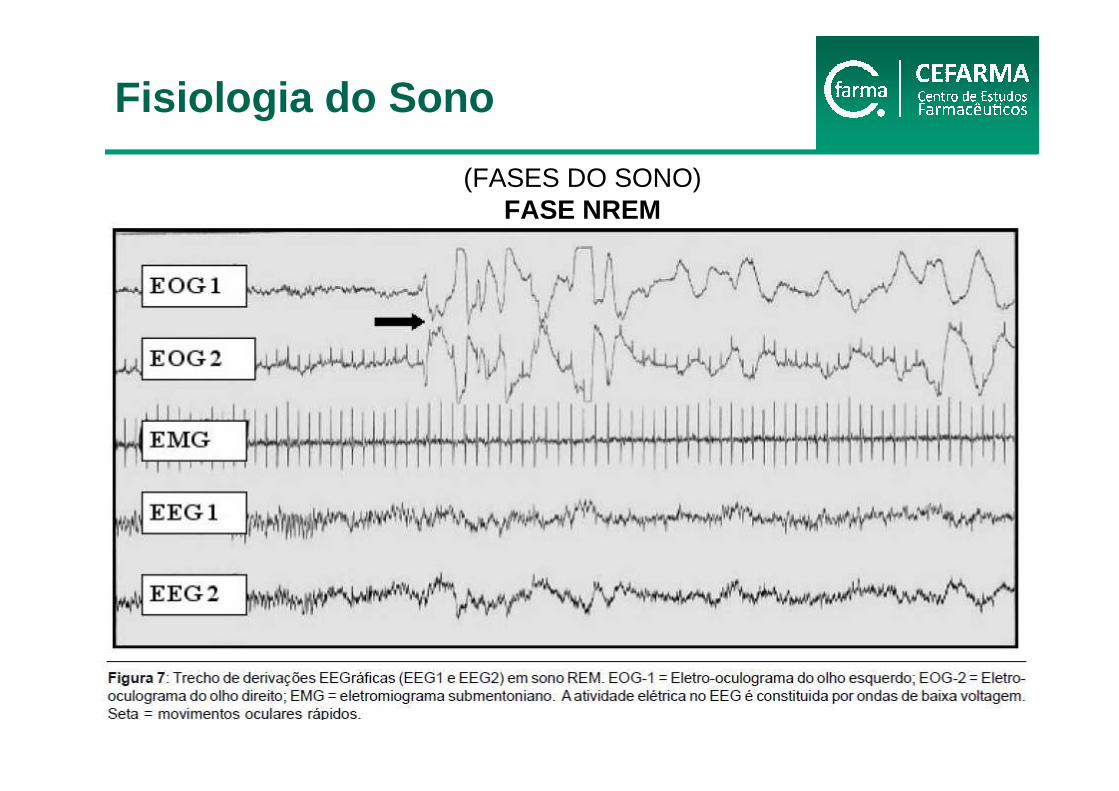
(FASES DO SONO)FASE NREM
Fisiologia do Sono
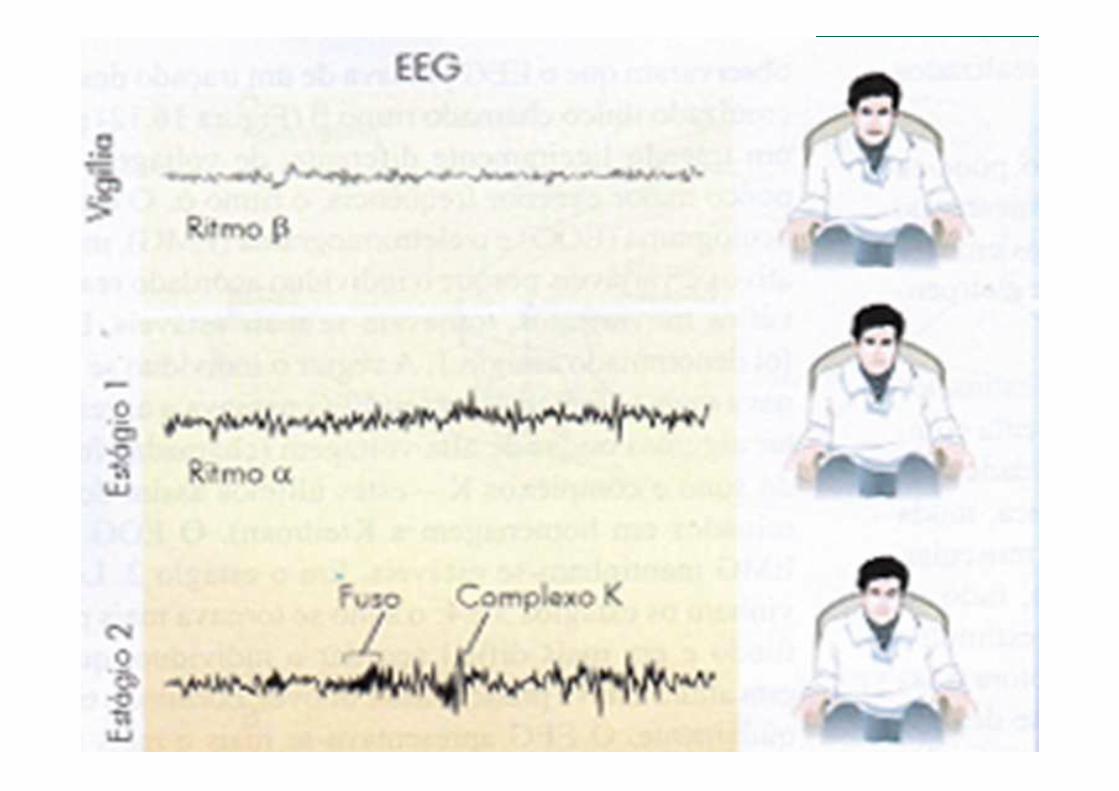
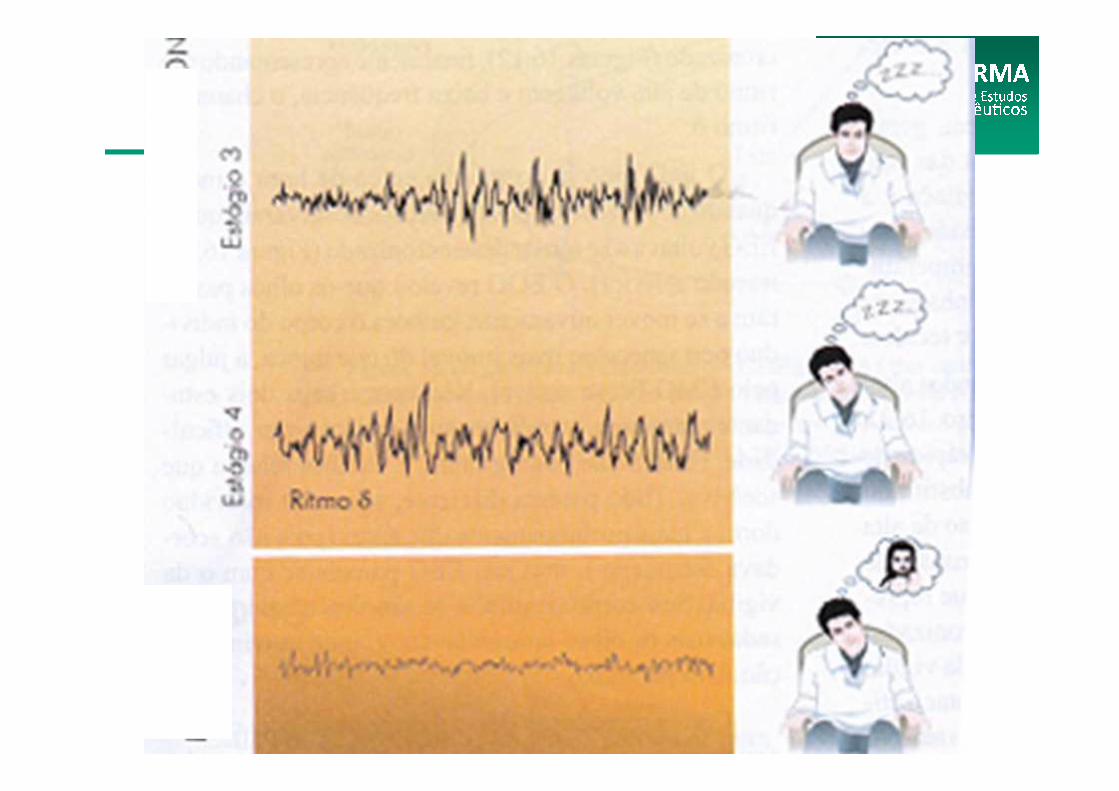

(FASES DO SONO)
Fisiologia do Sono

• O sono noturno se inicial pelo estágio 1do sono NREM, após um tempo de latência aproximada de 10 minutos.
• Após uns poucos minutos em sono I, háo aprofundamento para o estágio 2 , em que se torna mais difícil o despertar do indivíduo.
• Após 30 a 60 minutos, instala-se o sono de ondas lentas, respectivamente, os estágios 3 e 4 (aproximadamente 90 min), com interpenetrações de ambos no decorrer desta etapa mais profunda do sono NREM.
• Após a fase 4, o indivíduo retorna ao estágio 3, estágio 2 e entra no sono REM (5 a 10 minutos), completando- se o primeiro ciclo NREM-REM do sono noturno.
• O primeiro sono REM é mais curto que os seguintes.
• Desta forma, cumprem-se cerca de 5 a 6 ciclos de sono NREM-REM, durante uma noite de 8 horas de sono, com diminuição graduação do tempo de sono profundo (estágios 3 ou 4) e aumento do sono REM

Receptores GABA: 2 subtipos principais
GABAA: ionotrópico(5 subnunidades: 2 α, 2 β e 1 γ)
GABAB: metabotrópico(acoplado à proteína G)
Cuidado!!!GABAC: ionotrópico (variação de GABAA )(todas as subunidades são ρ)
Receptor GABAérgico
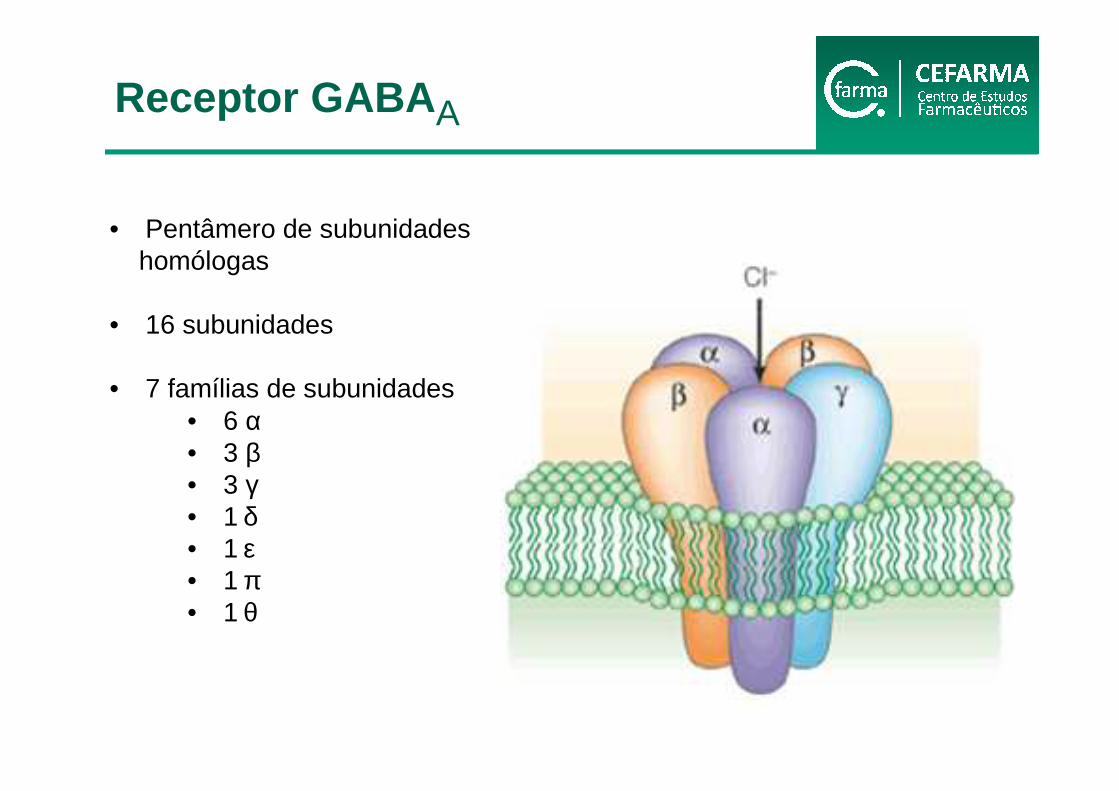
• Pentâmero de subunidadeshomólogas
• 16 subunidades
• 7 famílias de subunidades• 6 α• 3 β• 3 γ• 1 δ• 1 ε• 1 π• 1 θ
Receptor GABA A

• Complexidade adicional por conta de variantes de remontagem do RNA de aglumas dessas subnidades (ex. g2 e a6)
• α2, β2 e γ -> confere alta sensibilidade aos BZD (α1-6, β1-3, e γ1-3)
• Três combinações predominam em adultos: α1β2γ2, α2β3γ2, α3β3γ2
• Receptores formados só por subunidades α e β – são funcionais (barbitúricos também se ligam nesses receptores – barbitúricos se ligam bem à subunidade β)
Receptor GABA A

• GABA se liga na interface entre as subnidades α1 e β2
• BZD se ligam na interface entre as subunidades α e γ.
Receptor GABA A

Verkman e Gallieta, 2009
Mecanismo de ação dos BZD

• Se liga ao receptor BZD (interface entre as subnidades α1 e β2), alterando a conformação do receptor GABAérgico, tornando o mesmo mais ávido ao GABA.
• A conformação alterada do receptor GABAérgico aumenta a frequência de abertura do canal de cloreto promovida pelo GABA, mesmo em quantidades submáximas do mesmo.
• O GABA, ao ligar ao receptor GABAérgico, promove a abertura do canal de cloreto.
• O cloreto entra na célula e hiperpolariza a membrana.• A hiperpolarização impede a geração de um potencial de ação e o efeito
final é inibitório.
• Os BZDs não possuem capacidade de abrir diretamente o canal de Cl-, eles apenas aumentam a afinidade do receptor pelo GABA. O GABA sim, é capaz de promover a abertura dos canais de Cl-.
• Os barbitúricos, contrariamente aos BZDs, são capazes de, diretamente, abrir os canais de Cl-.
Mecanismo de ação dos BZD
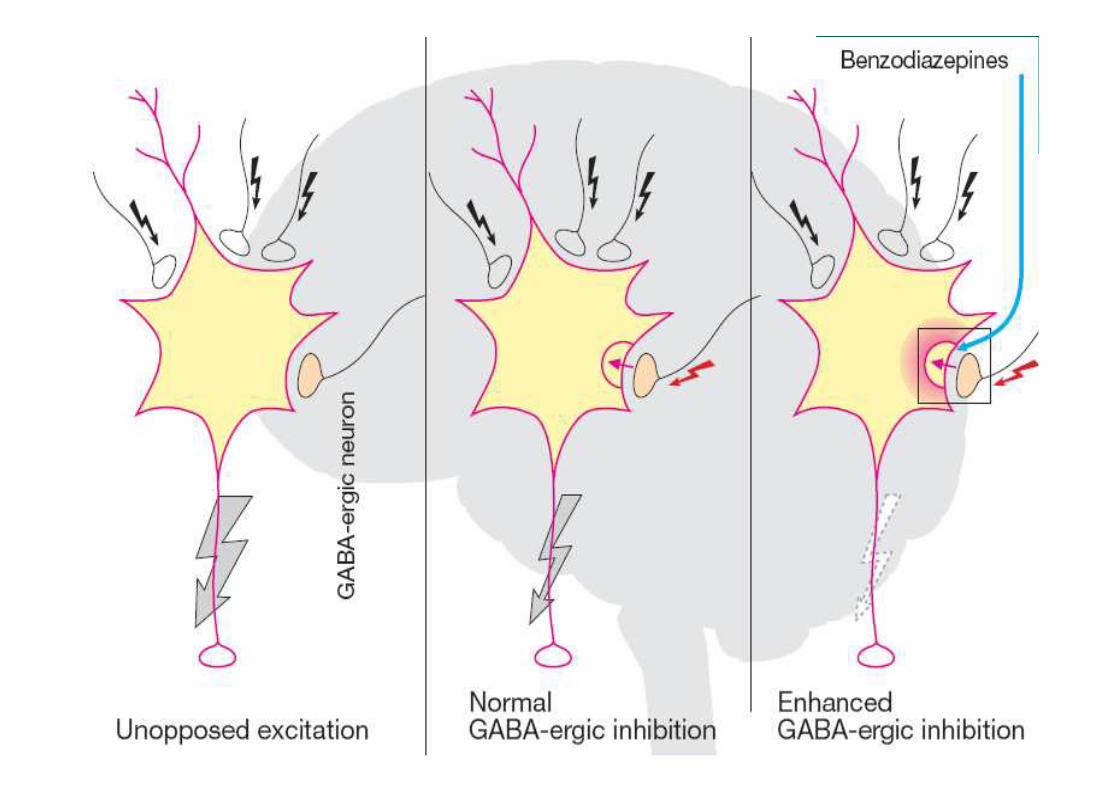

• Multiplicidade de subunidades gera variedade de receptores GABAA –diversidade dos efeitos farmacológicos dos BZD
• Teoricamente, podem ser gerados cerca de 1 milhão de receptores GABAA – esse número é menor por conta de restrições na montagem dos receptores
• Diversidade de receptores com distribuição variável em diferentes regiões do cérebro, controlando diferentes circuitos neuronais.
Receptor GABA A

• O conhecimento de quais subunidades do receptor GABAA são responsáveis por cada efeito particular dos BZD estáemergindo.
• A ligação dos BZD com a subunidade α requer um resíduo de histidina
• α1, α2, α3 e α5 contém resíduo de histidina
• α4 e α6 não contém resíduo de histidina (mas de arginina) -> BZD não tem afinidade para α4 e α6
Receptor GABA A

• Em termos de mecanismo de ação dos BZD, suas similaridades são muito grandes para separá-los em categorias individuais (ex. ansiolíticos ou hipnóticos)
• Um BZD comercializado como hipnótico, mesmo que administrado em baixas doses produzirá também efeitos ansiolíticos.
• Um BZD comercializado como ansiolítico induzirá o paciente ao sono quando administradas doses altas.
• Ligação dos BZD às subunidades:• α1 – mais associados a efeitos hipnóticos e anticonvulsivantes• α2 e/ou α3 – mais associados a efeitos ansiolíticos
Receptor GABA A

• O conhecimento de quais subunidades do receptor GABAA são responsáveis por cada efeito particular dos BZD estáemergindo.
• A ligação dos BZD com a subunidade a requer um resíduo de histidina
• α1, α2, α3 e α5 contém resíduo de histidina
• α4 e α6 não contém resíduo de histidina (mas de arginina) -> BZD não tem afinidade para α4 e α6
Receptor GABA A
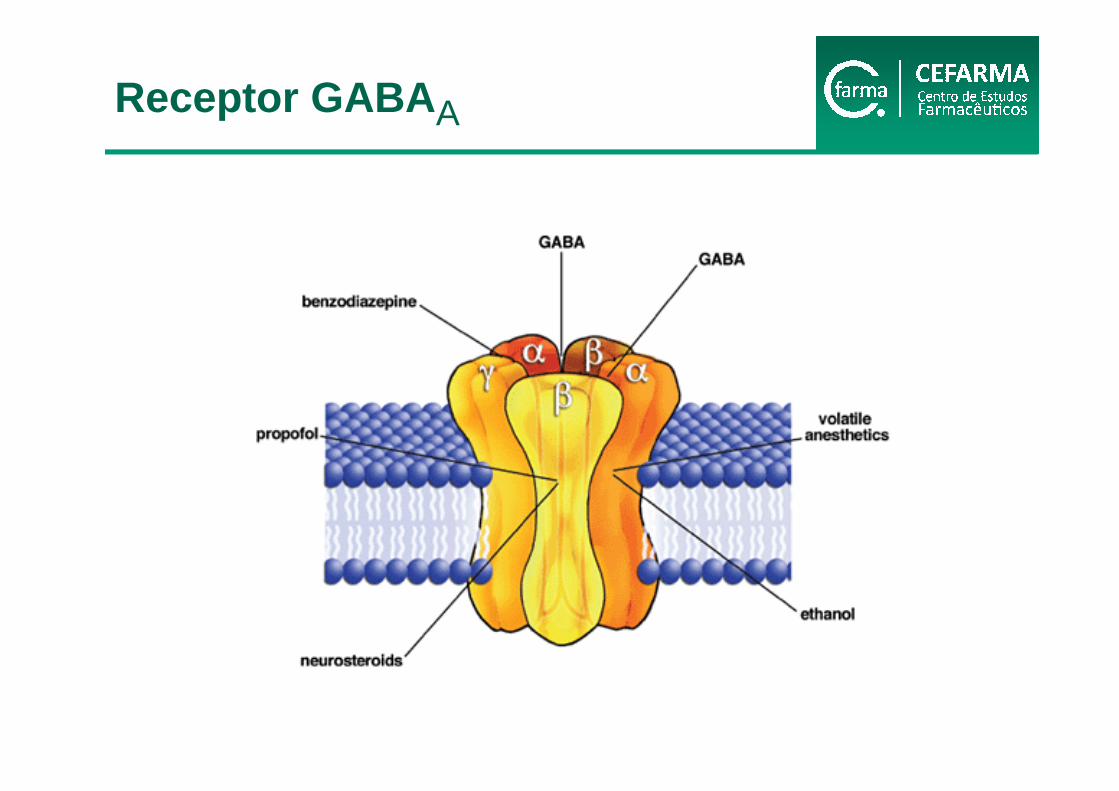
Verkman e Gallieta, 2009
Receptor GABA A
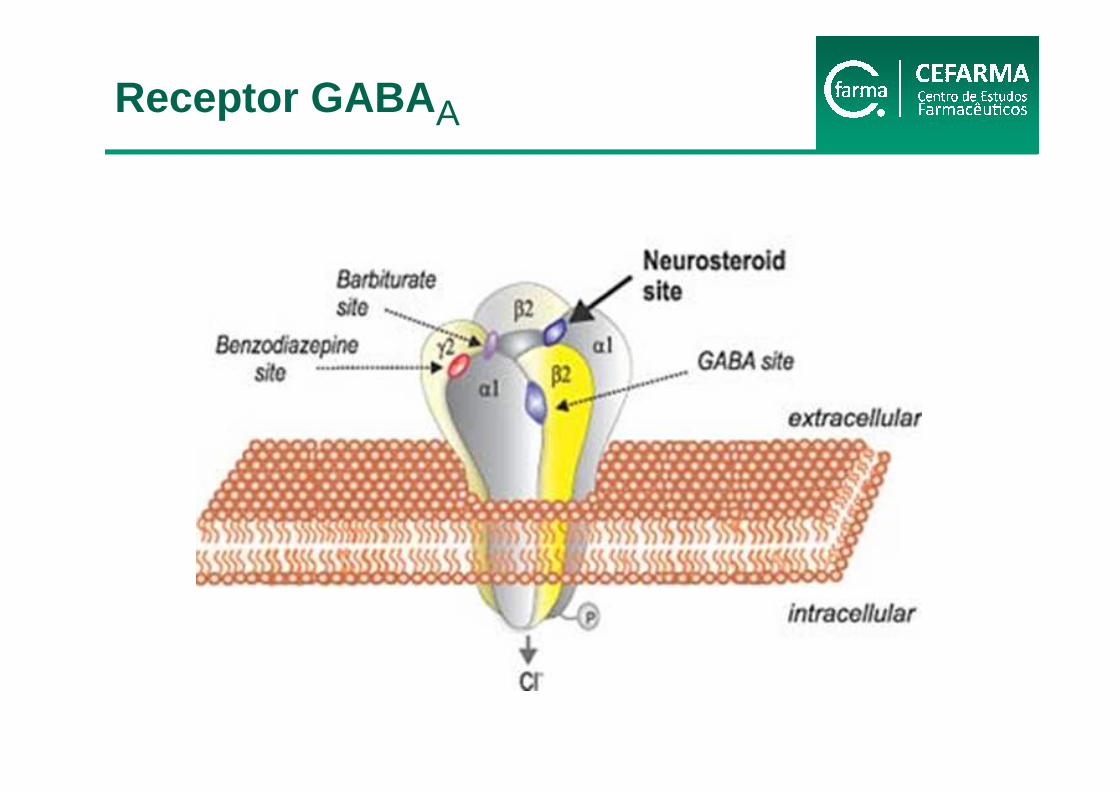
Verkman e Gallieta, 2009
Receptor GABA A
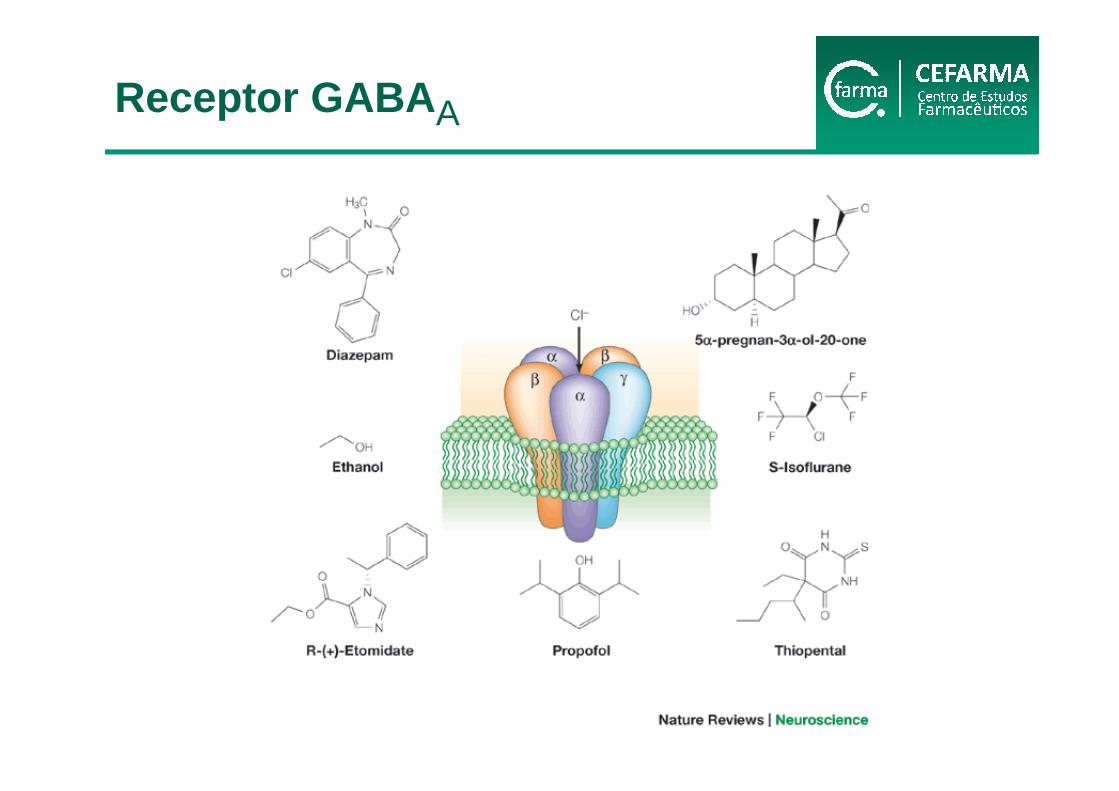
Verkman e Gallieta, 2009
Receptor GABA A


• Os BZD são agonistas totais
• Foram identificados agonistasinversos, que reduzem a eficácia do GABA e, portanto, são ansiogênicos, mas não são usados clinicamente.
• Acredita-se que há ligantes endógenos para os receptores de BZD, mas sua função fisiológica é desconhecida
• Os BZD não afetam os receptores de outros aminoácidos, como glicina e glutamato
Receptor GABA A


• Sedação
• Hipnose
• Redução da ansiedade e da agressão
• Relaxamento muscular
• Amnésia anterógrada
• Atividade anticonvulsivante• Efeitos são principalmente devido à ação do SNC
• Apenas 2 dos efeitos resultam da ação periférica
� Vasodilatação coronária (observada após administrarção i.v. de alguns BZD em doses terapêuticas)
� Bloqueio neuromuscular (observado após doses muito altas)
Efeitos Farmacológicos

• Modificam atividade em todos os neuroeixos, embora algumas estruturas podem ser preferencialmente afetadas.
• Não produzem o mesmo grau de depressão que os barbitúricos e anestésicos voláteis (analgesia e anestesia).
• Todos os BZD têm perfis farmacológicos similares, mas diferem em seletividade e, por isso, a utilidade clínica pode variar.
• À medida que se aumenta a dose, a sedação progride para a hipnose e daí para o estupor.
• Não produzem anestesia geral verdadeira, pois a consciência geralmente persiste. Além disso, não se observa relaxamento suficiente para produzir cirurgia.
• Ainda é difícil separar ansiedade e sedação
Propriedades Farmacológicas no SNC

(sedação e indução do sono)
• Diminuem a latência do sono, ou seja, o tempo para começar a dormir (diminuição da atividade alfa na vigília).
• Aumentam a duração total do sono (em pacientes que dormem menos de 6 horas).
• Diminuem o número de despertares.
• Tempo no estágio 1 diminui e há uma proeminente redução no tempo gasto no sono de ondas lentas (estágios 3 e 4).
• Grande prolongamento do estágio 2 (que é a principal fração do sono NREM).
Propriedades Farmacológicas no SNC

• Redução do sono REM.
• Todos os hipnóticos reduzem o sono REM (sono REM possui função benéfica), embora os BZD reduzam menos que os outros hipnóticos.
• O zolpidem e a zaleptona reduzem o sono REM ainda menos que os BZD (superioridade deles como hipnóticos em relação aos BZD).
• Interrupção artificial do sono REM causa irritabilidade e ansiedade.
• Este efeito é compensado pelo aumento no número de ciclos de sono REM.
• Ocorre redução do número de deslocamentos para os estágios de sono mais leve (0 e 1) e dos movimentos corporais.
• Os picos noturnos de secreção de hormônio do crescimento, prolactina e hormônio luteinizante não são afetados.
(sedação e indução do sono)
Propriedades Farmacológicas no SNC

• Com o uso crônico de BZD, os efeitos sobre os vários estágios de sono habitualmente declinam em poucas noites – TOLERÂNCIA.
• Quando o uso de BZD é interrompido, pode ocorrer “efeito rebote”, podendo haver aumento na quantidade e profundidade de sono REM.
• Se a dose não for excessiva, os pacientes vão notar apenas um encurtamento do sono, ao invés de exacerbação da insônia.
• Embora o uso prolongado dos BZD como hipnóticos seja desagradável (tolerância e dependência), o uso ocasional é eficaz (ex. viagens de avião).
• Encurtamento do tempo de sono ou insônia
• Os efeitos dos diferentes BZDs sobre o sono estão mais relacionados às suas propriedades farmacocinéticas do que farmacodinâmicas
(sedação e indução do sono)
Propriedades Farmacológicas no SNC

• São capazes de reduzir a ansiedade e agressão
• Utilizados no tratamento da síndrome do pânico
• Não têm efeitos antidepressivos, com a possível exceção do alprazolam
• Pode ocorrer agressividade e irritabilidade paradoxal – mais comum com os de ação ultra curta (triazolam – retirado da Inglaterra). É uma manifestação provável de síndrome de abstinência
• Tolerância em doses utilizadas para tratar ansiedade é motivo de debate
(ansiedade e agressão)
Propriedades Farmacológicas no SNC

• Tratam principalmente as reações de ansiedade aguda, embora estejam perdendo campo para os antidepressivos para esta indicação.
• São eficazes na ansiedade grave com atividade autossômica (transtorno do pânico) – principalmente os de alta potência:
• Alprazolam
• Clonazepam
• Lorazepam
Obs.: vários antidepressivos também são eficazes para este tipo de ansiedade (com frequência os BZD são associados para tratar pânico ou utilizados como SOS durante a crise).
• São eficazes para no tratamento da ansiedade generalizada ou inespecífica (a escolha de qual BZD parece fazer pouca diferença)
(redução da ansiedade e agressão)
Propriedades Farmacológicas no SNC

• Pacientes com ansiedade associada com depressão, usam BZD + antidepressivos, mas os BZD não tratam a depressão (possível exceção talvez seja o alprazolam)
• Tratamento prolongado de pacientes que apresentam sintomas persistentes ou recidivantes de ansiedade – uso de BZD de alta potência (tratamento controverso)
Uso prolongado causa tolerância
• Idosos e pacientes com disfunção hepática
• Oxazepam em doses fracionadas é preferível (ação curta e éconjugado diretamente e eliminado)
• Lorazepam também compartilha tal propriedade, mas não o
(redução da ansiedade e agressão)
Propriedades Farmacológicas no SNC

• BZD reduzem comportamento agressivo
• Podem aumentar a irritabilidade e agressão de forma paradoxal.
• Mais associado com os BZD de ação ultracurta
• Afeito associado com síndrome de abstinência
• Triazolam (retirado do Reino Unido por este motivo)
• Estudos posteriores mostraram que em doses baixas não diferem de outros BZD acerca destes efeitos.
(redução da ansiedade e agressão)
Propriedades Farmacológicas no SNC

• BZD possuem ação anticonvulsivante
• Frequentemente utilizados (ex. diazepam) em situações de emergência onde o paciente apresenta convulsão
• O clonazepam, nitrazepam e nodazepam têm atividade anticonvulsivante mais seletiva que a maioria dos outros benzodiazepínicos.
• O desenvolvimento de tolerância aos efeitos anticonvulsivantes limitou a sua utilidade no tratamento das desordens convulsivas em seres humanos
(efeitos antivonvulsivantes)
Propriedades Farmacológicas no SNC

• Possuem propriedades relaxantes musculares
• O tônus muscular aumentado é comum em estados de ansiedade, contribuindo para dor e cefaléia, que incomodam estes pacientes
• A ação miorrelaxante dos BZD contribui para a melhora deste quadro
• Os BZD aumentam a incoordenação motora, mas é possível obter efeitos miorrelaxanntes sem perda apreciável da coordenação
• Embora os BZD afetem a coordenação, a cognição é muito menos afetada.
• O clonazepam em doses não-sedativas causa relaxamento muscular, ao passo que, em tais doses, o dazepam e vários outros BZD não.
(redução do tônus muscular e da coordenação)
Propriedades Farmacológicas no SNC

• Os BZD bloqueiam a memória de eventos experimentados enquanto sob sua influência (tal efeito não é geralmente obtido com outros depressores do SNC)
• Pequenos procedimentos cirúrgicos e procedimentos de diagnósticos, como endoscopia, frequentemente utilizam BZD.
• Em casos de procedimentos endoscópicos, os de ação rápida, como midazolam, são preferenciais (para que o paciente não demore a se recuperar)
(amnésia retrógrada)
Propriedades Farmacológicas no SNC

• Doses hipnóticas de BZD não têm efeitos sobre a respiração em indivíduos normais
• Atenção para crianças e pacientes com comprometimento hepático
• Em doses mais altas (como as usadas na pré-anestesia ou para procedimentos endoscópicos) os BZD deprimem levemente a ventilação alveolar e causam acidose respiratória (por estimulação do estímulo hipóxico).
• A depressão alveolar e acidose respiratória se tornam exarcebadosem pacientes com DPOC (doença pulmonar obstrutiva crônica) resultando em hipóxia alveolar e/ou narcose por CO2.
• Este último efeito pode causar hipoventilação e hipóxia em alguns pacientes com DPOC grave
Propriedades Farmacológicas sobre a Respiração

• Pacientes com AOS (apneia obstrutiva do sono), doses hipnóticas dos BZD podem diminuir o tônus muscular das vias respiratórias superiores e exagerar o impacto dos episódios apneicos sobre a hipóxia alveolar, a hipertensão pulmonar e a carga ventricular cardíaca.
• A presença de DPOC e AOS é considerada por alguns especialistas como contraindicação para o uso de BZDs (além de álcool
• Apesar da grande segurança dos BZD em indivíduos normais, a presença de DPOC e AOS aumenta os riscos de depressão respiratória grave, podendo haver morte de tais indivíduos.
Propriedades Farmacológicas sobre a Respiração

• Os efeitos cardiovasculares dos BZD não são importantes em indivíduos normais, exceto na intoxicação grave
• Em doses pré-anestésicas, todos os BZD diminuem a pressão sanguínea e aumentam a frequência cardíaca
• O diazepam aumenta o fluxo coronário possivelmente por provocar um aumento das concnetrações intersticiais de adenosina (que causa relaxamento do músculo liso endotelial)
• Em grandes doses, o midazolam diminui consideravelmente o fluxo sanguíneo e a assimilação de oxigênio cerebrais
• Não há descrição de qualquer impacto destes fármacos sobre dano letal em pacientes com doença cardíaca
Propriedades Farmacológicas sobre a o trato gastrintestinal
• Melhoram desordens do TGI relacionadas com ansiedade
• Há pouca evidência de ação direta no TGI
Propriedades Farmacológicas sobre o Sistema Cardiovascular


• Bem absorvidos por via oral
• Têm elevado coeficiente de partição óleo e água na sua forma molecular (lipofílicos)
• Grau de lipofilicidade pode variar até 50 vezes entre os diferentes BZD devido a grupamentos polares presentes
• Oxazepam e lorazepam são absorvidos mais lentamente
(Absorção)
Farmacocinética dos BZD
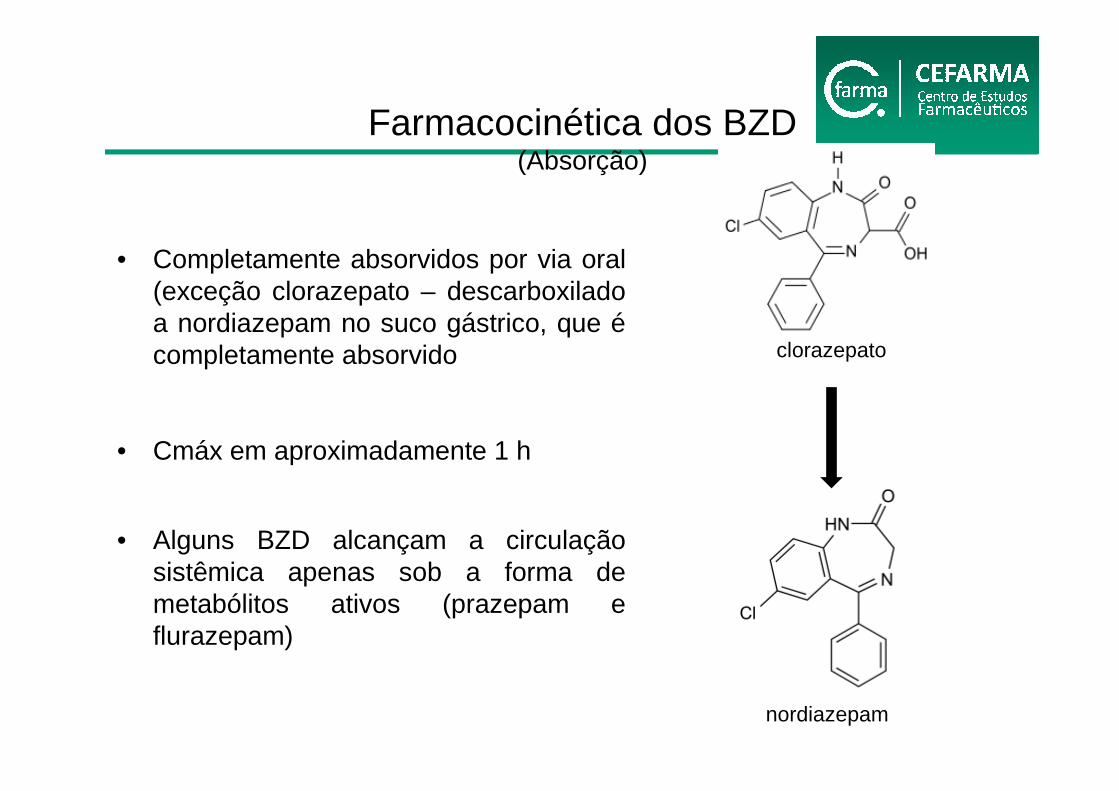
• Completamente absorvidos por via oral (exceção clorazepato – descarboxiladoa nordiazepam no suco gástrico, que écompletamente absorvido
• Cmáx em aproximadamente 1 h
• Alguns BZD alcançam a circulação sistêmica apenas sob a forma de metabólitos ativos (prazepam e flurazepam)
Farmacocinética dos BZD(Absorção)
clorazepato
nordiazepam

• Se ligam às proteínas plasmáticas
• A extensão da ligação varia com a solubilidade -> quanto mais lipossolúvel, maior a taxa de ligação:
• Diazepam – 99 %
• Alprazolam – 70 %
• Embora possa haver competição com outros fármacos pelo sítio das PTNs plasmáticas, não há qualquer consequência clínica importante
• A concentração no líquido cérebro-espinhal éaproximadamente a mesma do fármaco livre no plasma.
Farmacocinética dos BZD(distribuição)

• Modelo bicomartimental
• Rápida captação dos BZD pelo cérebro e outros órgãos muito perfundidos, seguida de redistribuição para tecidos menos perfundidos, como músculos e gorduras (após administração intravenosa ou oral com rápida absorção)
• A redistribuição é mais rápida com os BZD mais lipossolúveis.
• Extremamente lipofílicos podem apresentar modelo tricompartimental(acúmulo em lipídeos com liberação mais demorada, alterando a cinética de eliminação)
(Distribuição)
Farmacocinética dos BZD

• Cinéticas de redistribuição de fármacos lipofílicos (ex. diazepam) tornam-se ainda mais complexas pela circulação êntero-hepática
• Grandes volumes de distribuição e em muitos casos maiores nos pacientes idosos
• Nos regimes usados para sedação noturna, a taxa de redistribuição pode ter às vezes uma influência maior que a biotransformação sobre a duração dos efeitos no SNC
• Atravessam a barreira placentária e são secretados também no leite materno
(Distribuição)
Farmacocinética dos BZD

• São biotransformados por enzimas do citocromoP450, particularmente CYP3A4 e CYP2C19 (reações de fase I)
• Os metabólitos gerados pelas reações de fase I geralmente são ativos
• Fármacos inibidores dessas enzimas podem aumentar o tempo de meia vida dos BZD
• Interações farmacocinéticas importantes: eritromicina, claritromicina, ritonavir, intraconazol, cetoconazol, nefazodona e suco de pomelo são inibidores da CYP3A4 (acúmulo de BZD)
• Cimetidina e anticonceptivos orais inibem a N-desalquilação e 3-hidroxilação(reações de fase I) dos BZD (acúmulo de BZD)
• Após sofrerem biotransformação por reações de fase I, os BZD sofrem conjugação, principalmente com ácido glicurônico
(Biotransformação)
Farmacocinética dos BZD

• Metabólitos ativos são biotransformados que os compostos originais – duração da ação de muitos BZD tem pouca relação com o composto originalmente administrado
• Ex.: metabólito ativo biotransformado lentamente
• Flurazepam (t ½ = 2-3 h)
• N-desalquilflurazepam (t ½ = 50 h ou mais)
• De forma oposta, a taxa de biotransformação de agentes inativados pela reação incial éimportante na determinação da duração da ação
• Oxazepam, lorazepam
(Biotransformação)
Farmacocinética dos BZD
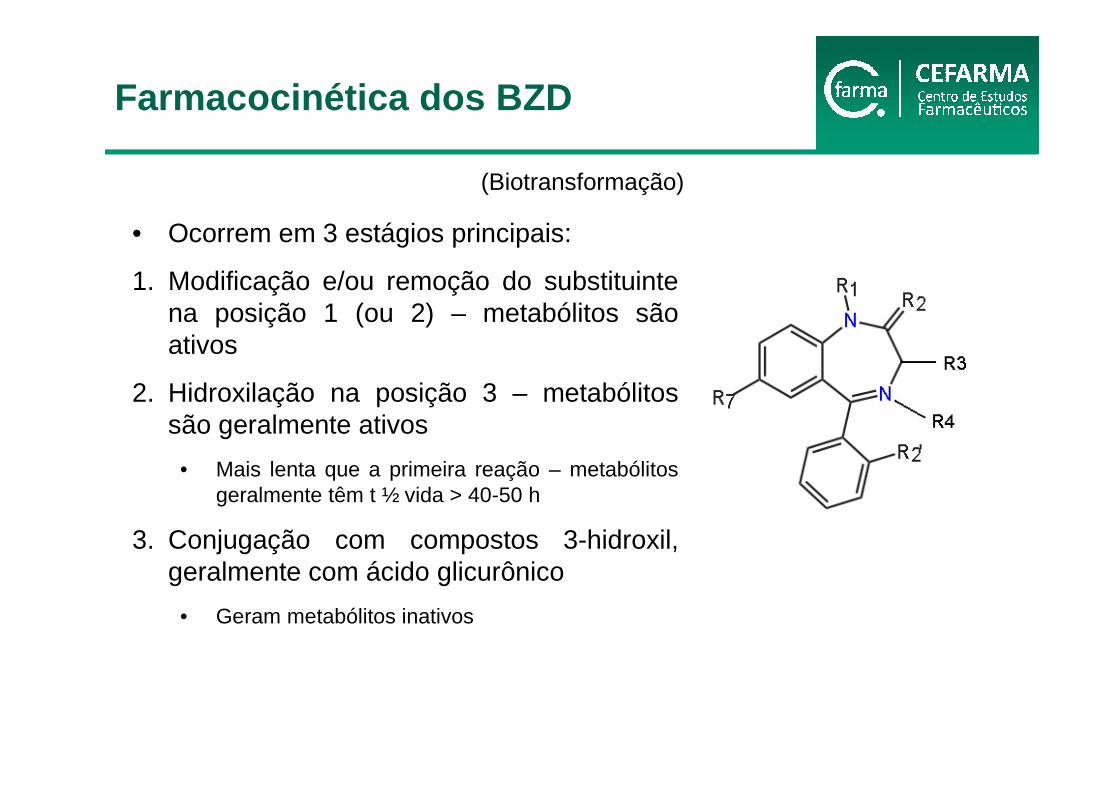
• Ocorrem em 3 estágios principais:
1. Modificação e/ou remoção do substituinte na posição 1 (ou 2) – metabólitos são ativos
2. Hidroxilação na posição 3 – metabólitos são geralmente ativos
• Mais lenta que a primeira reação – metabólitos geralmente têm t ½ vida > 40-50 h
3. Conjugação com compostos 3-hidroxil, geralmente com ácido glicurônico
• Geram metabólitos inativos
(Biotransformação)
Farmacocinética dos BZD
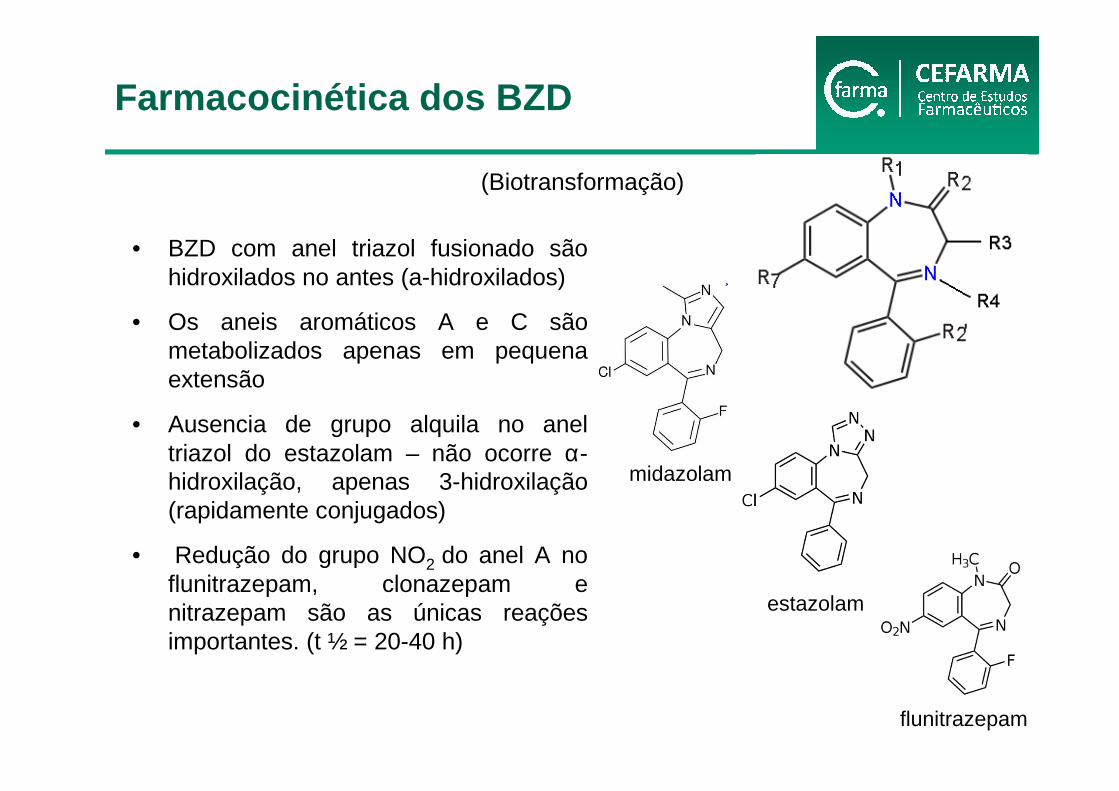
• BZD com anel triazol fusionado são hidroxilados no antes (a-hidroxilados)
• Os aneis aromáticos A e C são metabolizados apenas em pequena extensão
• Ausencia de grupo alquila no anel triazol do estazolam – não ocorre α-hidroxilação, apenas 3-hidroxilação(rapidamente conjugados)
• Redução do grupo NO2 do anel A no flunitrazepam, clonazepam e nitrazepam são as únicas reações importantes. (t ½ = 20-40 h)
flunitrazepam
midazolam
estazolam
(Biotransformação)
Farmacocinética dos BZD
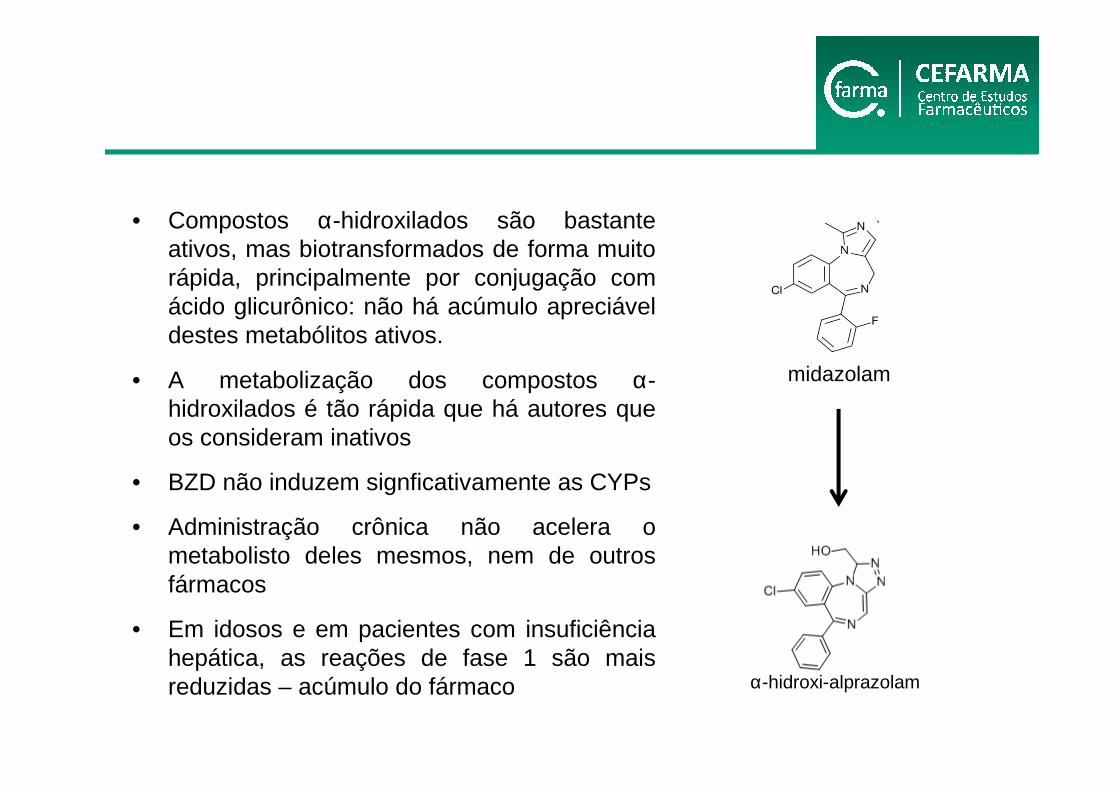
• Compostos α-hidroxilados são bastante ativos, mas biotransformados de forma muito rápida, principalmente por conjugação com ácido glicurônico: não há acúmulo apreciável destes metabólitos ativos.
• A metabolização dos compostos α-hidroxilados é tão rápida que há autores que os consideram inativos
• BZD não induzem signficativamente as CYPs
• Administração crônica não acelera o metabolisto deles mesmos, nem de outros fármacos
• Em idosos e em pacientes com insuficiência hepática, as reações de fase 1 são mais reduzidas – acúmulo do fármaco
midazolam
α-hidroxi-alprazolam
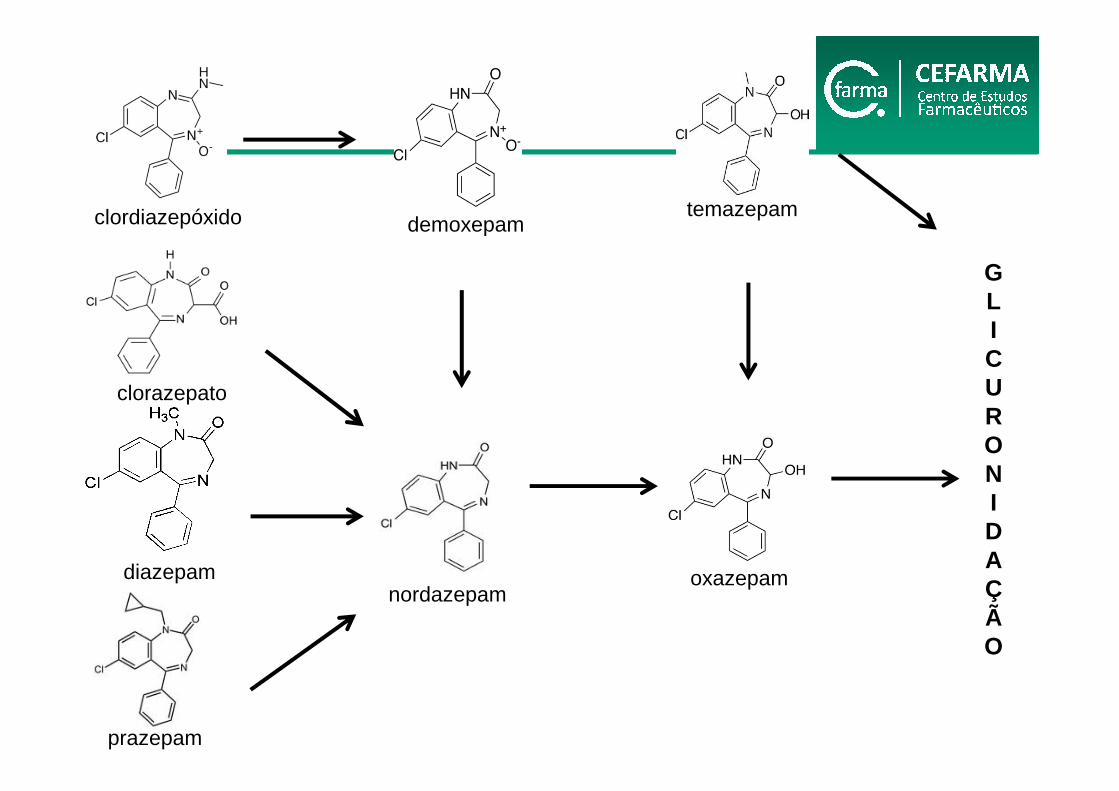
diazepam
clordiazepóxido
clorazepato
nordazepamoxazepam
demoxepamtemazepam
GLICURONIDAÇÃO
prazepam
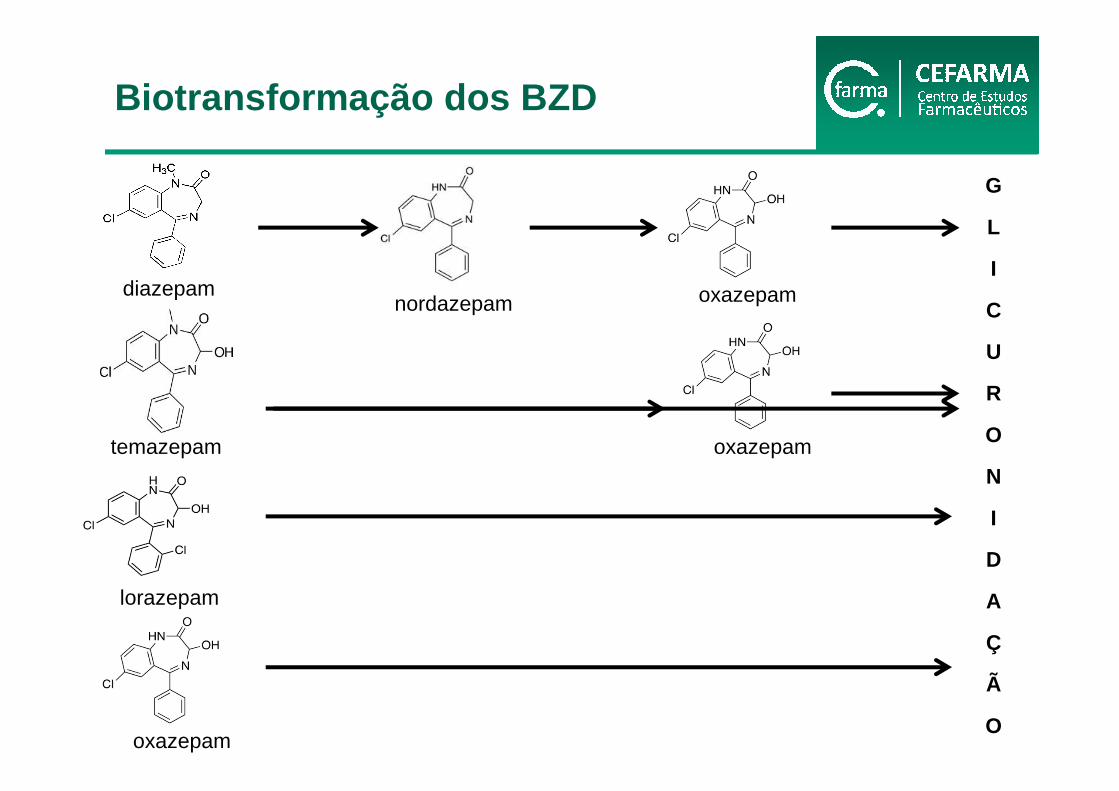
diazepamnordazepam oxazepam
G
L
I
C
U
R
O
N
I
D
A
Ç
Ã
O
lorazepam
oxazepam
temazepam oxazepam
Biotransformação dos BZD
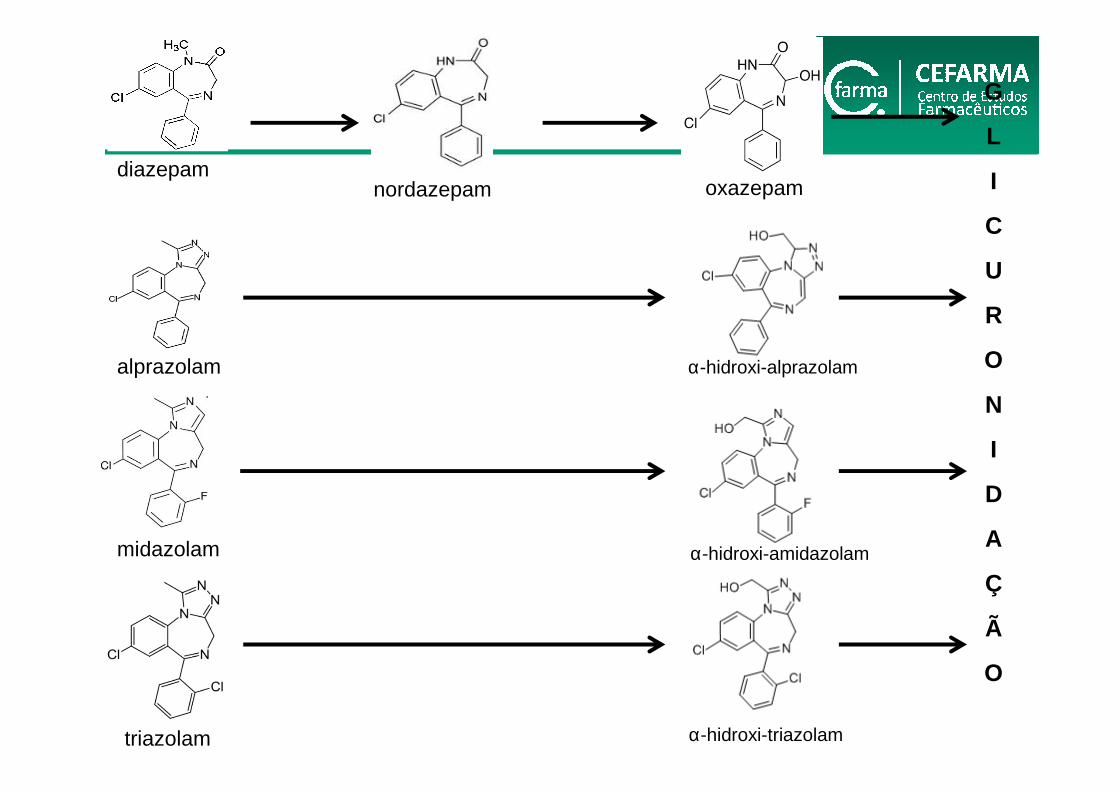
diazepamnordazepam oxazepam
G
L
I
C
U
R
O
N
I
D
A
Ç
Ã
O
midazolam
triazolam
alprazolam α-hidroxi-alprazolam
α-hidroxi-amidazolam
α-hidroxi-triazolam

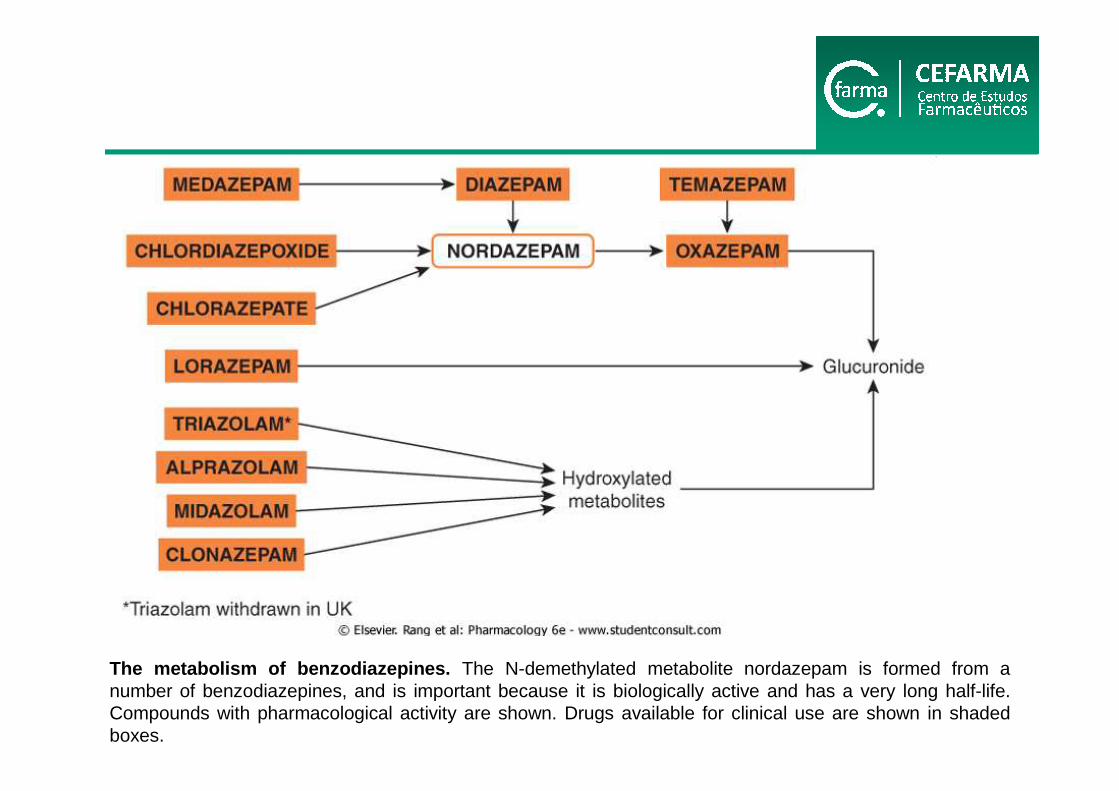
The metabolism of benzodiazepines. The N-demethylated metabolite nordazepam is formed from a number of benzodiazepines, and is important because it is biologically active and has a very long half-life. Compounds with pharmacological activity are shown. Drugs available for clinical use are shown in shadedboxes.

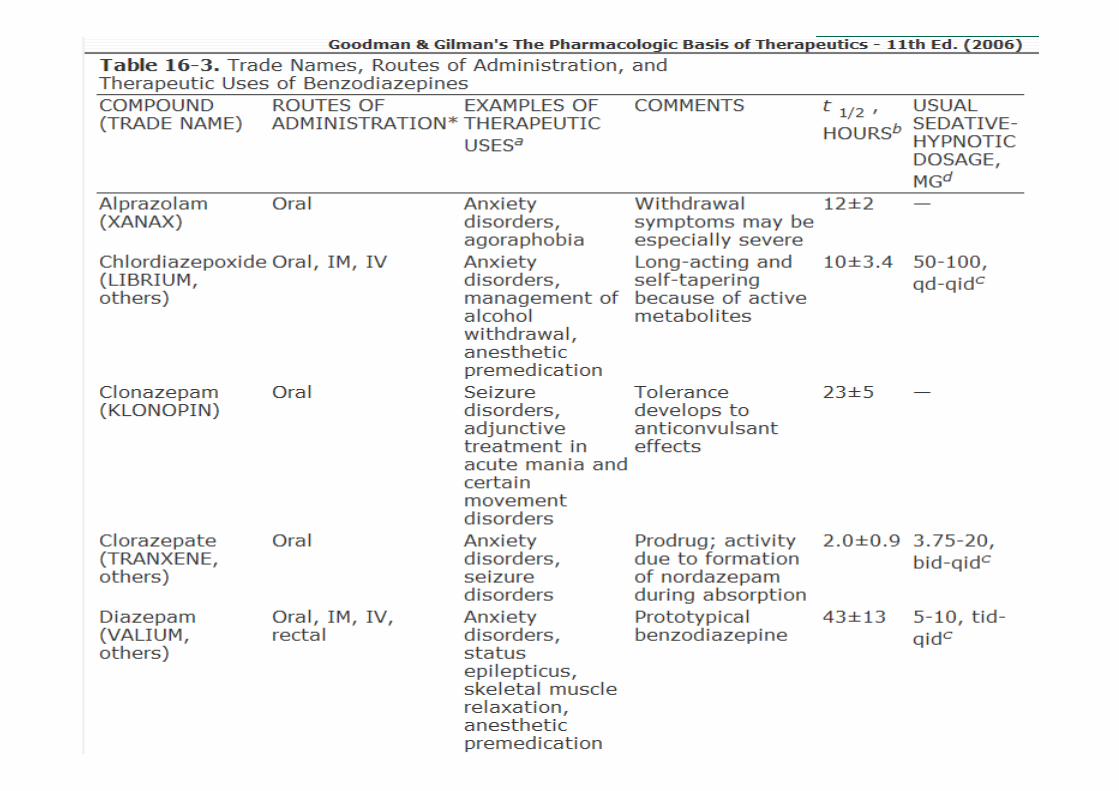
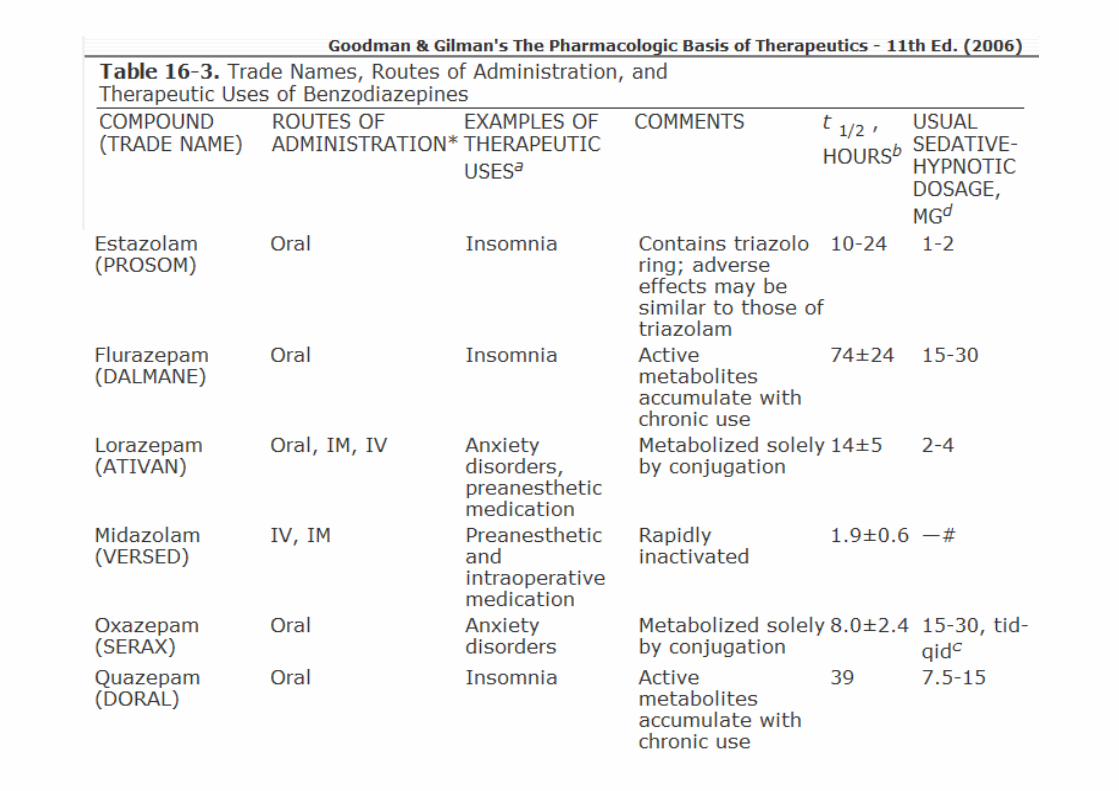
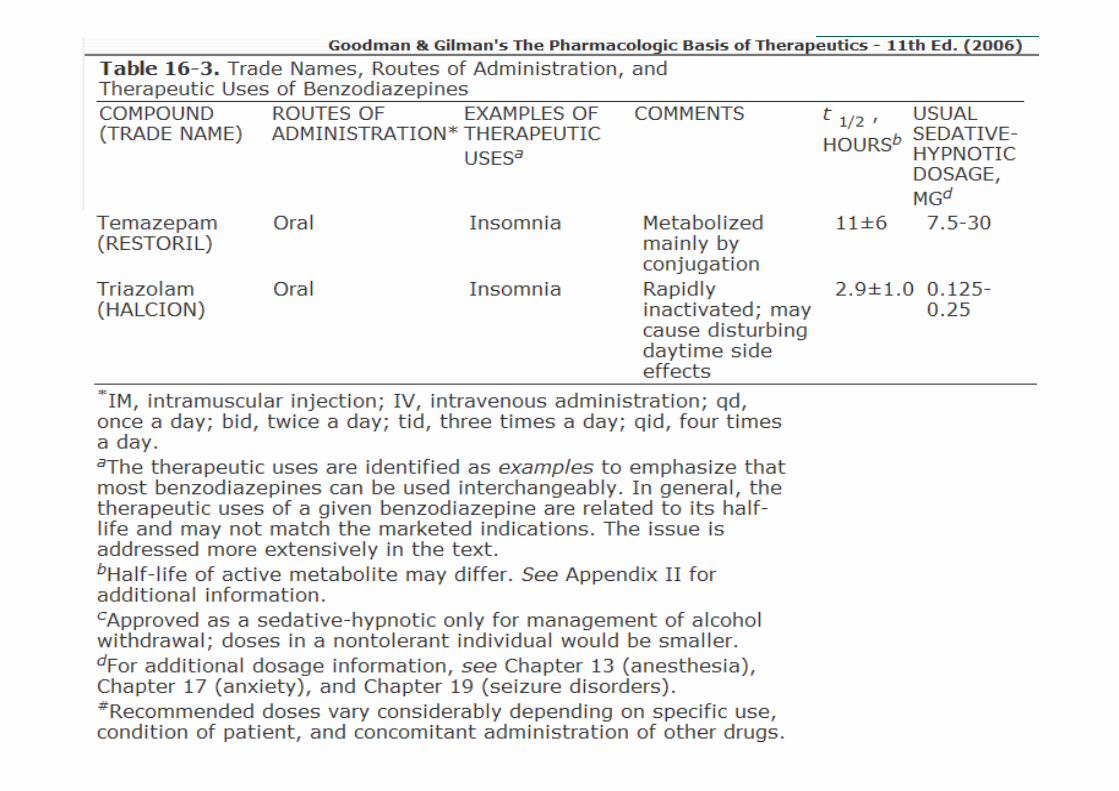
• Ação ultra-rápida
• Ação curta, com meias-vidas de menos de 6 h: triazolam;
• Ação intermediária, com meias-vidas de 6 a 24 h: estazolame temazepam;
• Longa ação: meias-vidas maiores que 24 h: flurazepam, diazepam
Classificação com base no tempo de ação

• Os metabólitos dos BZD são excretados predominantente por via renal, havendo também excreção fecal (bile)
• Pode haver excreção no leite
• Neonatos de mães que utilizaram BZD com frequência durante a gravidez podem ter síndrome de abstinência
(excreção)
Farmacocinética dos BZD


Dependem da meia-vida;• Hipnóticos:
� Meia-vida de eliminação curta, embora isso traga a desvantagem de maior propensão a uso abusivo e de maior gravidade da abstinência após a interrupção do fármaco;
� Meia-vida de eliminação longa nos casos de pacientes que também necessitam de efeitos ansiolíticos durante o dia e toleram a sedação diurna
• Ansiedade: meia-vida longa
• Anticonvulsivantes (estado epiléptico): meia-vida longa e entrada rápida no cérebro;
• Dependência de etanol: diazepam
Uso Terapêuticos

• Risco de dependência e uso abusivo;
• Sintomas de abstinência: insônia, ansiedade, disforia, irritabilidade, suores, sonhos desagradáveis, tremores, anorexia, síncopes e tonteiras –especialmente se ocorre subitamente;
• Necessária redução gradual de dose quando o tratamento está para ser interrompido.
• Tolerância : ocorre com todos benzodiazepínicos
� Menos acentuada do que com os barbitúricos (estes também induzem tolerância farmacocinética)
• Síndrome de abstinência : Ansiedade e insônia, tremores, disforia, irritabilidade, sudorese, tonturas, sonhos desagradáveis, tonteiras. -> mais abrupta para os BZD de curta duração
• Dependência e uso abusivo : uso crônico. Não ocorre com a mesma extensão observada com os sedativos mais antigos e outros fármacos de reconhecido potencial para uso indiscriminado.
Tolerância e Dependência

• Atenção especial ao FLUNITRAZEPAM (Rohypnol®) – extremamento potente; produz sedação intensa após 20 min de injestão que pode durar horas e causa significativa amnésia retrógrada
1. Uso abusivo
2. Uso concomitante com
etanol e outras drogas
3. BZD + etanol ->
potencialmente perigoso
4. Acidentes
5. Estupros
6. Envenenamentos
7. Roubos
8. Sequestros
date rape drug
Benzodiazepínicos –grande importância forense

• Quando Cmáx é alcançado em doses hipnóticas:
� delírio
� lassidão
� aumento dos tempos de reação
� incoordenação motora
� comprometimento das funções menais e motoras
� confusão, amnésia anterógrada
• Os efeitos acima podem comprometer a capacidade de dirigir veículos e outras habilidades psicomotoras, especialmente em combinação com etanol
• Persistência desses efeitos ao acordar são adversos
• Sonolência diurna residual
Efeitos adversos dos BZD

• Intensidade e icidência de toxicidade no SNC aumenta com a idade: fatores farmacodinâmicos e farmacocinéticos
• Outros efeitos: fraqueza, dor de cabeça, visão borrada, vertigem, náuseas e vômitos, desconforto epigástrico e diarréia
• Pode haver aumento da convulsão em pacientes com epilepsia (efeito adverso paradoxal)
• Flurazepam aumenta a incidência de pesadelos ocasionalmente com efeitos paradozais (tagarelice, ansiedade, irritabilidde, alucinações e comportamento maníaco)
• Jà foram descritos efeitos paradoxal para vários BZD: amnésia, euforia, inquietação, alucinações e comportamentos hipomaníacos.
• Reações de desinibição e descontrole podem ocorrer (comportamento bizarro, desinibido, hostilidade e raiva)
• Ocasionalmente pode haver paranoia, depressão, ideação suicida
Efeitos adversos dos BZD

• Os efeitos adversos devido a doses excessivas agudas são menos perigosos do que a maioria dos outros fármacos ansiolíticos/hipnóticos, pois os BZD possuem elevado índice terapêutico
• Em doses elevadas há sono prolongado, sem depressão séria da respiração ou da função cardiovascular
• Exceção a danos sérios do sistema respiratório pode ocorrer em pacientes com AOS
• O uso de BZD com outros depressores do SNC (álcool e barbitúricos), carbamazepina e antidepressivos tricíclicos pode provocar depressão respiratória severa e levar à morte
Intoxicação Aguda

• Intoxicação por BZD: varia de sedação leve, sem depressão respiratória (na maioria absoluta dos casos) até coma profundo, com insuficiência respiratória (com associação a outros depressores do SNC). Ocorre frquentemente lassidão, diminuição dos reflexos, ataxia, confusão mental e amnésia. A depressão respiratória com BZD isolado éraríssima.
• Efeitos cardiovasculares em doses terapêuticas são pequenos. Nasoverdose, pode ocorrer diminuição da frequência cardíaca e da pressão arterial.
• Intoxicações por flunitrazepam e midazolam: mdiminuição do débidocardíaco e do fluxo sanguíneo cerebral
• Diazepam: aumenta o fluxo sanguíneo coronariano
• Os BZD diminuem a secreção de ácido no estômago, mas não retardam o esvaziamento gástrico.
Intoxicação Aguda

• Descontaminação gástrica (até 2 horas após ingestão) -> lavagem gástrica (não induzir vômito)
• Administração de carvão ativado após a lavagem gástrica
• Intubação endotraquela em pacientes em coma para proteção das vias aéreas e garantia de ventilação/oxigenação
• Diurese forçada não funciona (intensa metabolização hepática)
Tratamento do paciente intoxicado

• Antídoto específico: antatonista competitivo de receptor benzodiazepínico – FLUMAZENIL
• Administração de uma série de injeções é preferível àde uma injeção em bolus;
• A ausência de resposta a 5 mg de flumazenil sugere que a sedação não está sendo causada por um benzodiazepínico;
• Contraindicações para uso do flumazenil (intoxicação associada com antidepressivos tricíclicos, o que provoca convulsões, e arritmias cardíacas)
• Além de tratar a intoxicação, o flumazenil também éusado como ferramenta de diagnóstico
• Reversão dos efeitos sedativos produzidos por benzodiazepínicos administrados durante a anestesia geral ou durante procedimentos diagnósticos/terapêuticos
Flumazenil
Tratamento do paciente intoxicado

• Não é eficaz na superdosagem de barbitúricos ou de antidepressivos tricíclicos usados isoladamente;
• Neste contexto, a administração pode desencadear convulsões, especialmente em pacientes envenenados por antidepressivos tricíclicos;
• Convulsões ou outros sintomas de abstinência também podem ser precipitados em pacientes que estiveram tomando benzodiazepínicos por períodos prolongados e nos quais pode ter se
desenvolvido tolerância e/ou dependência.
Flumazenil
Tratamento do paciente intoxicado



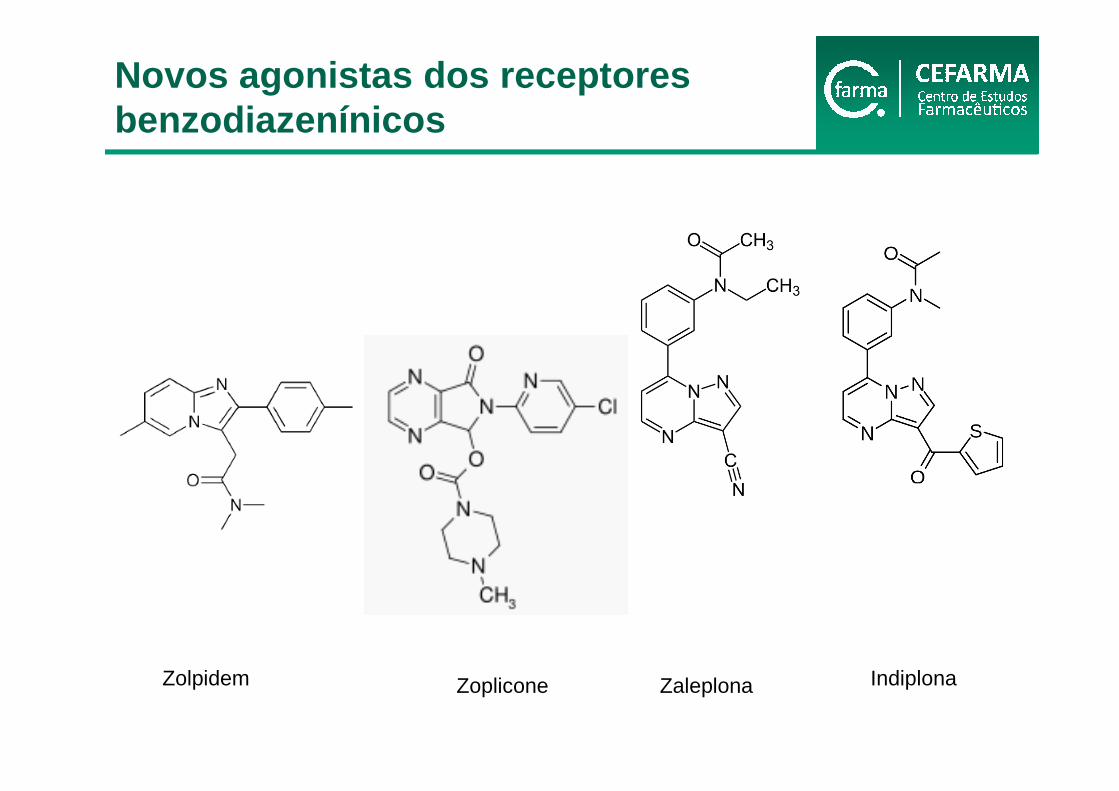
Zolpidem ZaleplonaZoplicone Indiplona
Novos agonistas dos receptores benzodiazenínicos

• Estrutura química diferentes dos BZD
• São agonistas dos receptores GABAA (mesmos locais que os BZD)
• Atuam seletivamente em receptores benzodiazepínicos ômega 1, envolvidos na sedação, porém, não nos receptores ômega 2, concentrados em áreas do cérebro que regulam a cognição, a memória e as funções motoras;
• Possuem eficácia hipinótica prolongada, sem ocorrência de insônia de rebote na interrupção súbita
• Têm graus similares de eficácia
• Possuem efeitos anticonvulsivantes e relaxntes musculares modestos mesmo em doses elevadas e não são aprovados para tais usos
• Seus efeitos são revertidos com flumazenil
Novos agonistas dos receptores benzodiazenínicos
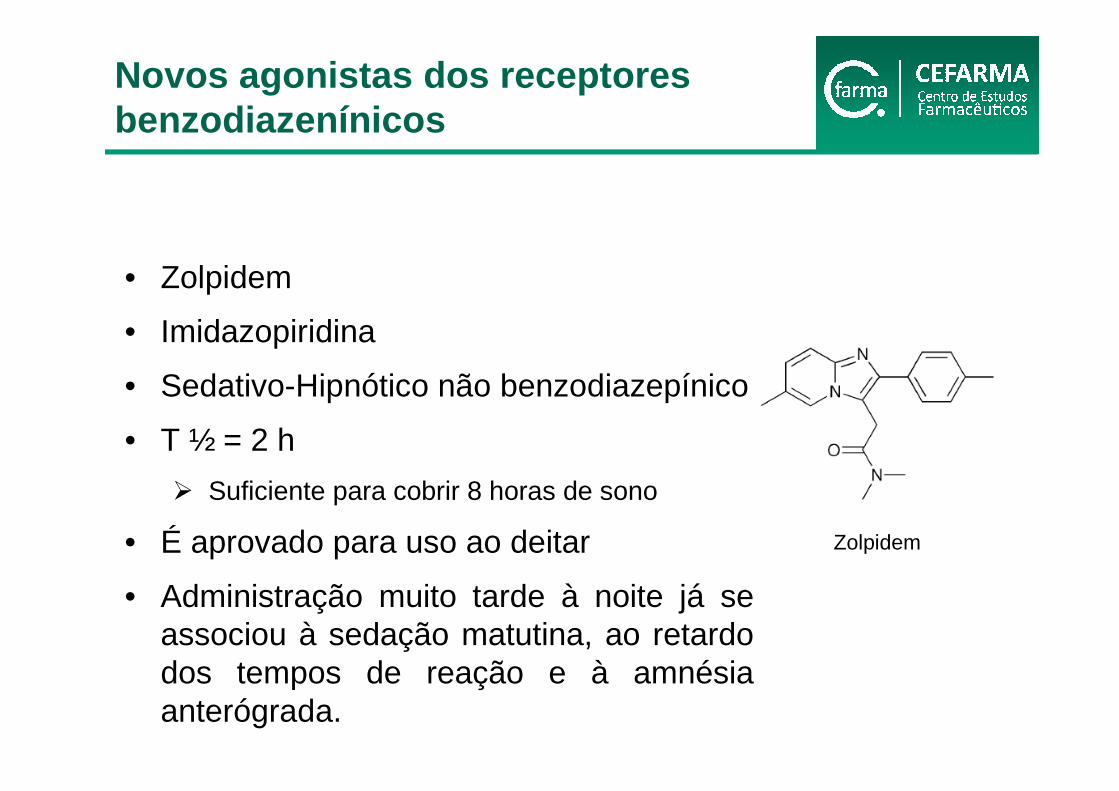
• Zolpidem
• Imidazopiridina
• Sedativo-Hipnótico não benzodiazepínico
• T ½ = 2 h
� Suficiente para cobrir 8 horas de sono
• É aprovado para uso ao deitar
• Administração muito tarde à noite já se associou à sedação matutina, ao retardo dos tempos de reação e à amnésia anterógrada.
Zolpidem
Novos agonistas dos receptores benzodiazenínicos

• Produz apenas efeitos anticonvusivantesfracos em animais
• Ações sedativas relativamente fortes parecem mascarar efeitos ansiolíticos em modelos animais de ansiedade
• Tem poucos efeitos nos estágios do sono
• Tão eficaz quanto os BZD para diminuir a latência do sono e em prolongar o tempo de sono total
• Após sua interrupção, os efeitos benéficos do sono persistem por até uma semana (embora já se descreveu insônia rebote na primeira noite)
• Tolerância e dependência física são raras
Novos agonistas dos receptores benzodiazenínicos

• Melhora do tempo de sono induzido por zolpidemmanteve-se por 6 meses d tratamento sem sinais de abstinência ou rebote após sua interrupção
• Em doses terapêuticas, a sedação diurna e aminésia não são frequentes.
• Superdosagens não causam depressão respiratória, a menos que co-administrado com outros agentes (etanol)
• Doses hipnóticas aumentam a hipóxia e a hipercarbia dos pacientes com AOS, tal como os BZD
Novos agonistas dos receptores benzodiazenínicos

• Zolpidem
• Bem absorvido no TGI
• Metabilismo hepático de primeira passagem resulta em biodisponibilidade de 70 % (valor menor ainda se ingerido com alimentos, que retardam a sua absorção e aumenta o fluxo sanguíneo hepático)
• A eliminação se dá quase que inteiramente por conversão a produtos inativos no fígado, em grande parte pro oxidação dos grupos metila sobre os aneis fenila e imidazopiridina aos correspondentes ácidos carboxílicos
• T ½ pode aumentar 2 vezes ou mais em indivíduos com cirrose e idosos (ajuste da dose)
Novos agonistas dos receptores benzodiazenínicos
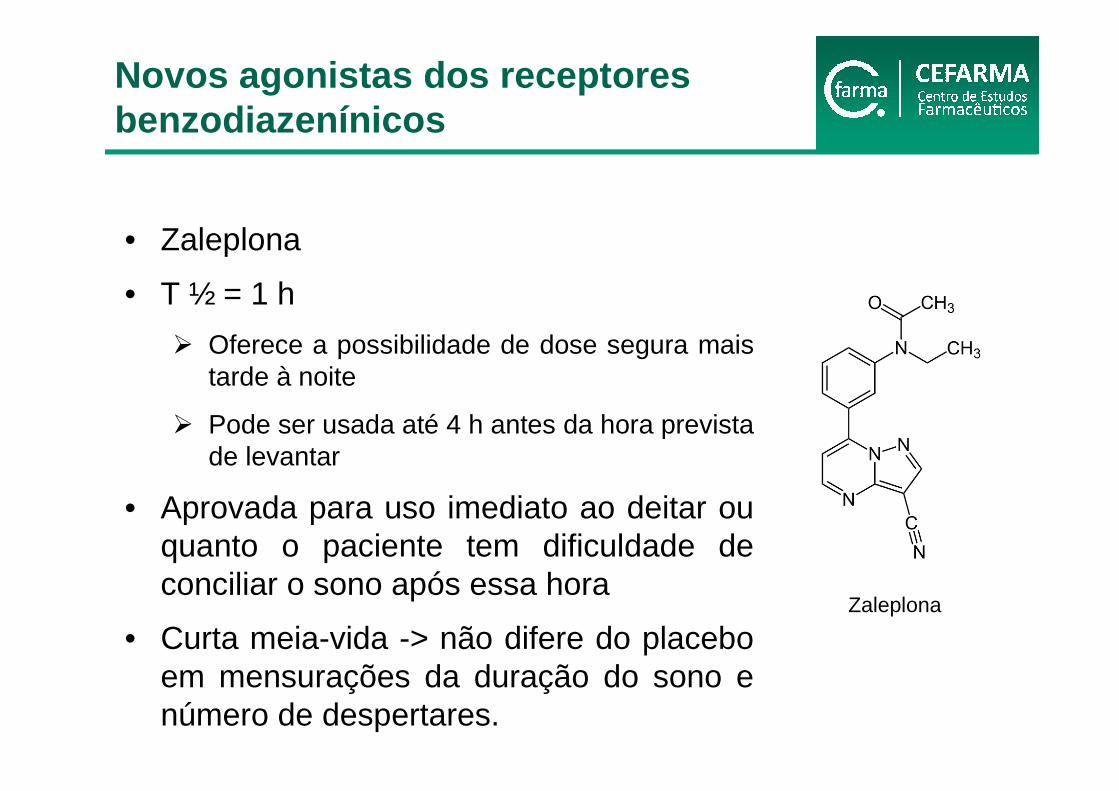
• Zaleplona
• T ½ = 1 h
� Oferece a possibilidade de dose segura mais tarde à noite
� Pode ser usada até 4 h antes da hora prevista de levantar
• Aprovada para uso imediato ao deitar ou quanto o paciente tem dificuldade de conciliar o sono após essa hora
• Curta meia-vida -> não difere do placebo em mensurações da duração do sono e número de despertares.
Zaleplona
Novos agonistas dos receptores benzodiazenínicos
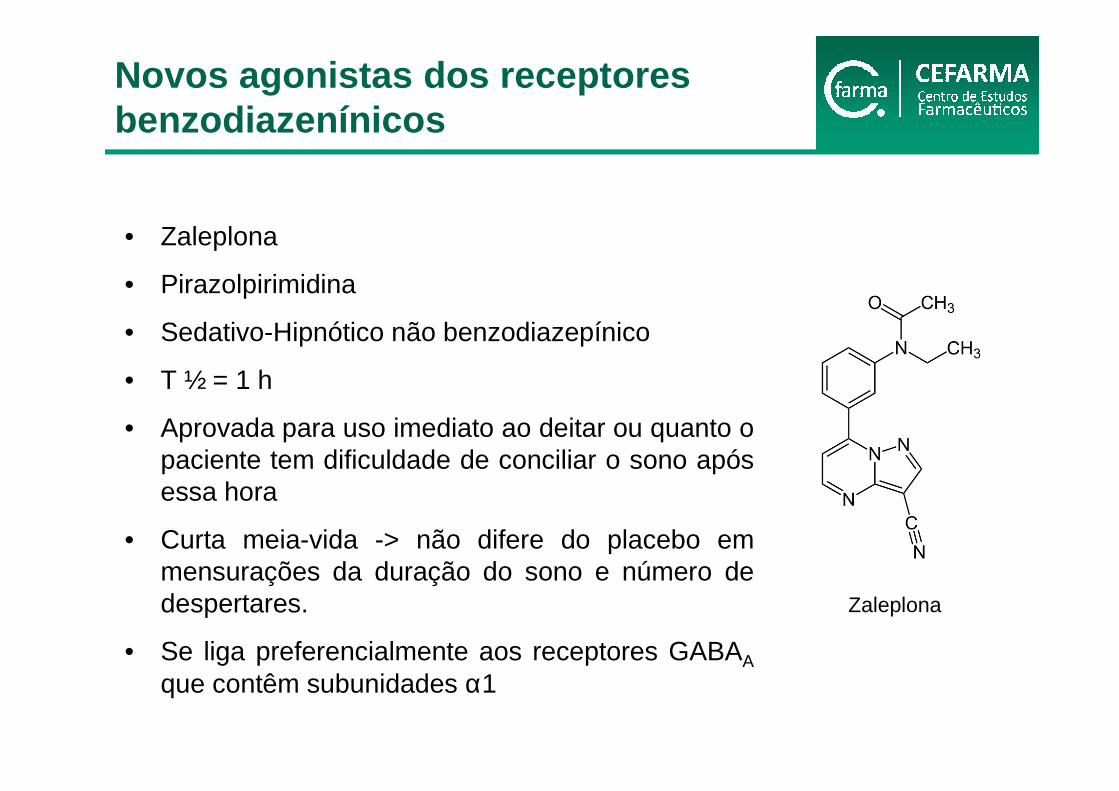
• Zaleplona
• Pirazolpirimidina
• Sedativo-Hipnótico não benzodiazepínico
• T ½ = 1 h
• Aprovada para uso imediato ao deitar ou quanto o paciente tem dificuldade de conciliar o sono após essa hora
• Curta meia-vida -> não difere do placebo em mensurações da duração do sono e número de despertares.
• Se liga preferencialmente aos receptores GABAA
que contêm subunidades α1
Zaleplona
Novos agonistas dos receptores benzodiazenínicos
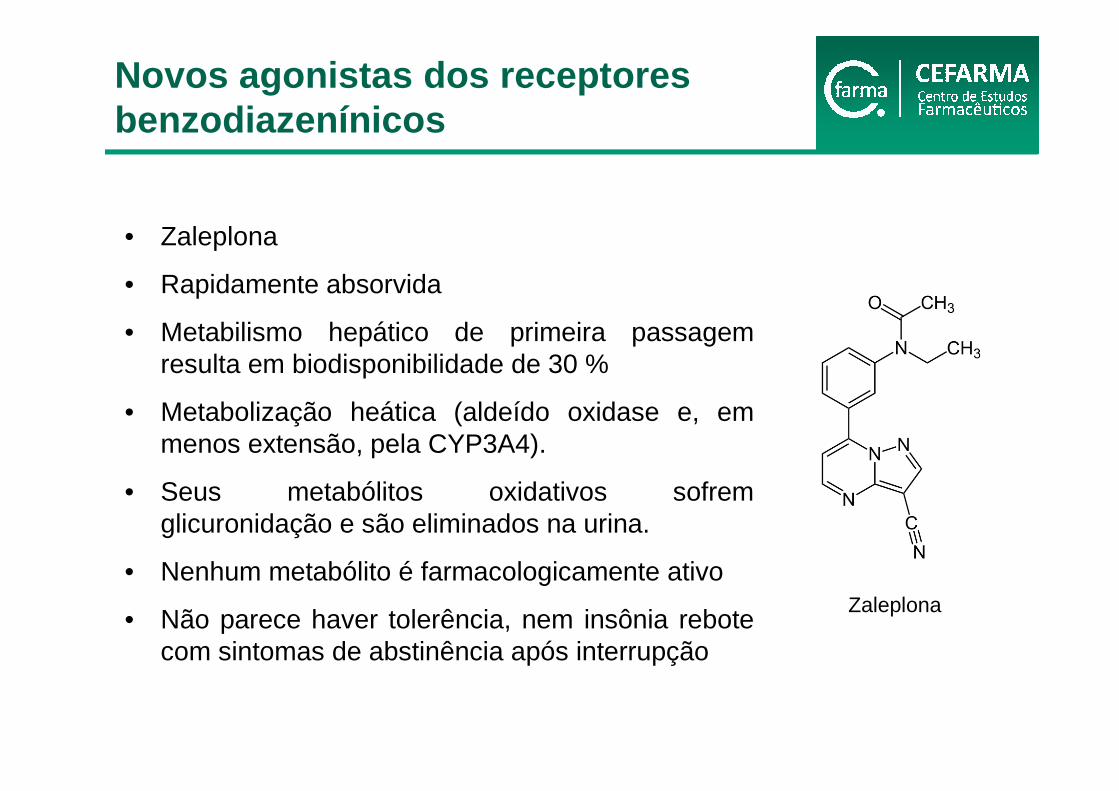
• Zaleplona
• Rapidamente absorvida
• Metabilismo hepático de primeira passagem resulta em biodisponibilidade de 30 %
• Metabolização heática (aldeído oxidase e, em menos extensão, pela CYP3A4).
• Seus metabólitos oxidativos sofrem glicuronidação e são eliminados na urina.
• Nenhum metabólito é farmacologicamente ativo
• Não parece haver tolerência, nem insônia rebote com sintomas de abstinência após interrupção
Zaleplona
Novos agonistas dos receptores benzodiazenínicos
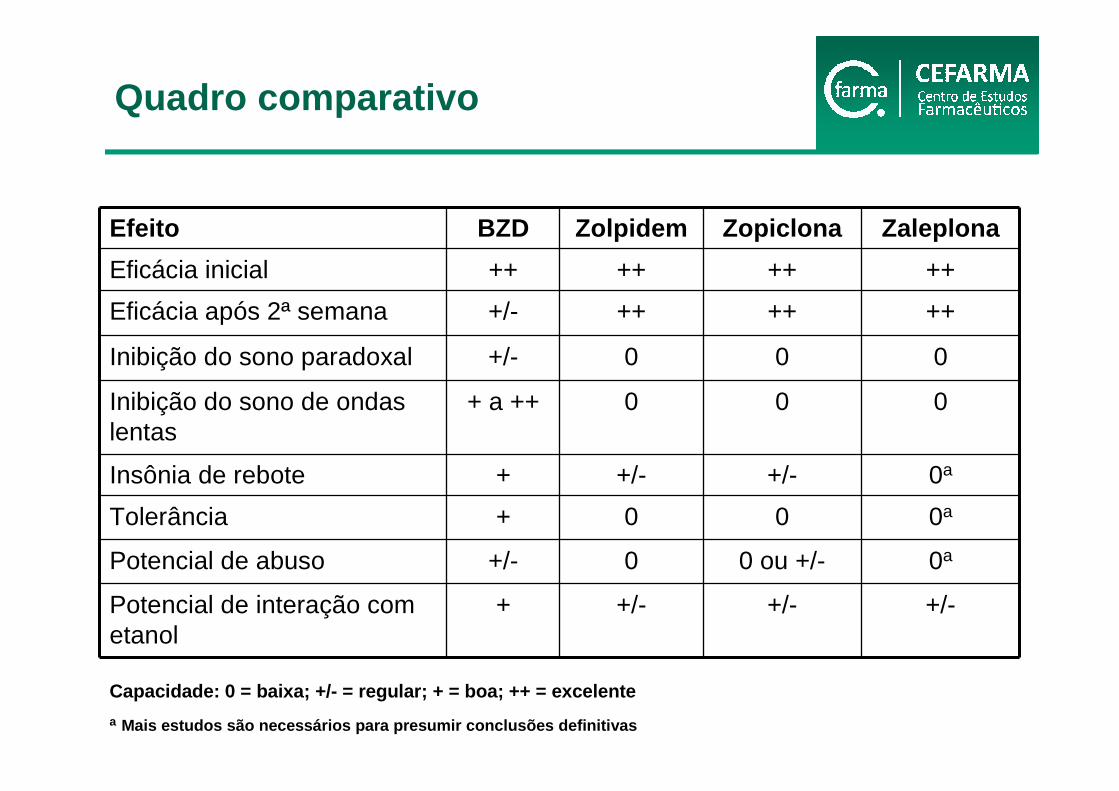
Efeito BZD Zolpidem Zopiclona Zaleplona
Eficácia inicial ++ ++ ++ ++
Eficácia após 2ª semana +/- ++ ++ ++
Inibição do sono paradoxal +/- 0 0 0
Inibição do sono de ondas lentas
+ a ++ 0 0 0
Insônia de rebote + +/- +/- 0a
Tolerância + 0 0 0a
Potencial de abuso +/- 0 0 ou +/- 0a
Potencial de interação com etanol
+ +/- +/- +/-
Capacidade: 0 = baixa; +/- = regular; + = boa; ++ = excelente
a Mais estudos são necessários para presumir conclusõ es definitivas
Quadro comparativo

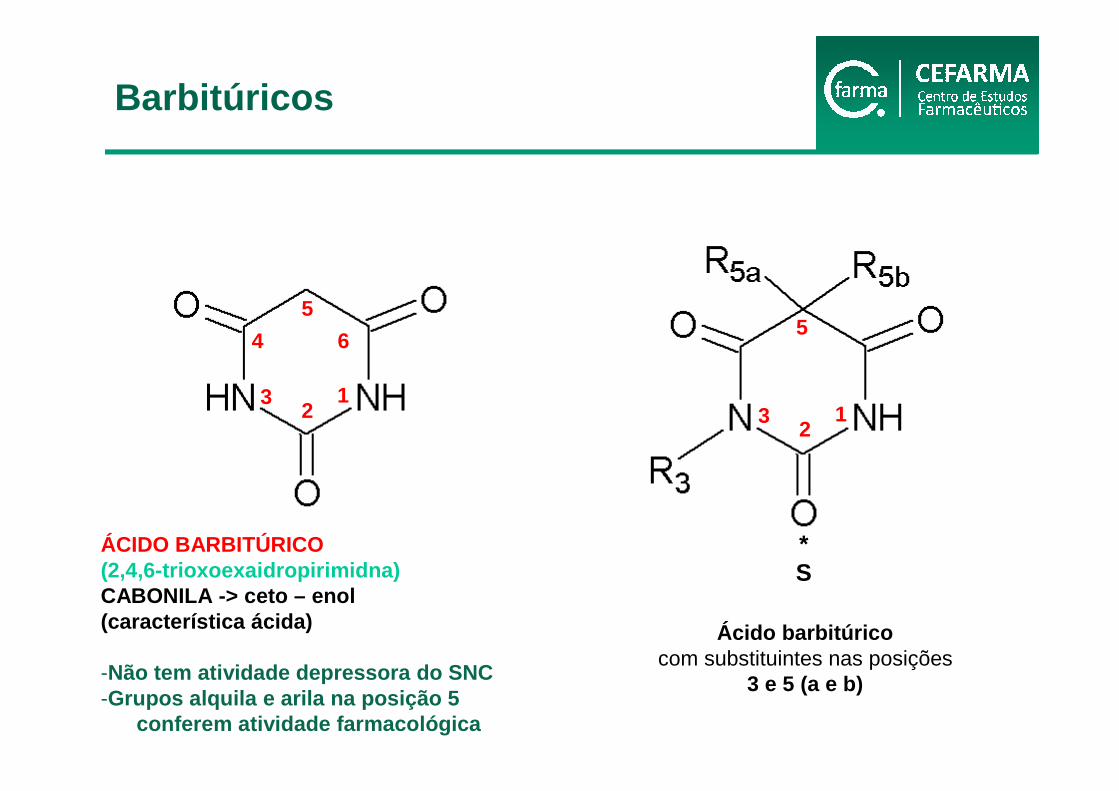
ÁCIDO BARBITÚRICO(2,4,6-trioxoexaidropirimidna)CABONILA -> ceto – enol(característica ácida)
-Não tem atividade depressora do SNC-Grupos alquila e arila na posição 5
conferem atividade farmacológica
Ácido barbitúricocom substituintes nas posições
3 e 5 (a e b)
12
3
5
*S
12
3
5
64
Barbitúricos

• Em meio alcalino o tautômero enol é favorecido pela sua localização entre os dois nitrogênios amido eletronegativos, o que resulta na formação de sais (ex.: barbituratos de sódio)
• Posição 2 é O -> oxobarbitúrico
• Posição 2 é S -> tiobartitúrico (mais lipossolúveis)
• Em geral, as diferentes alterações estruturais que aumentam a lipofilicidade:
� Diminuem a duração da ação
� Reduzem a latência do início da atividade
� Aceleram a degradação metabólica
� Aumentam a potência hipnótica
Barbitúricos

Barbitúricos
butalbitalamobarbital butabarbital fenobarbital
mefobarbital metoexital pentobarbital secobarbital tiopental
aprobarbital

• Já foram extensamente usados como sedativos-hipnóticos.
• Exceto por uns poucos usos especializados foram em grande parte substituídos pelos BZD (mais seguros)
• Deprimem reversivelmente a atividade de todos os tecidos excitáveis
• O SNC é extraordinariamente sensível, mas efeitos diretos sobre tecidos periféricos são muito fracos mesmo quando administrados em concentrações anestésicas
• Entretanto, déficits sérios na função cardiovascular e outras funções periféricas ocorrem na intoxicação por barbitúricos
Barbitúricos

• Se ligam ao recetor GABAA, entretanto em uma região distinta dos BZD (subunidades α e β são necessárias, mas a γ não)
• Intensificam a ação do GABA sobre os receptores GABAérgicos em baixas concentração
• Potencializam as correntes de Cl- induzidas por GABA por prolongar os períodos (de abertura) durante os quais ocorrem os surtos de abertura do canal (ao invés de aumentar a frequência de abertura, como fazem os BZD)
• Facilitam a ligação dos BZD (quando coadministrados)
Mecanismo de Ação
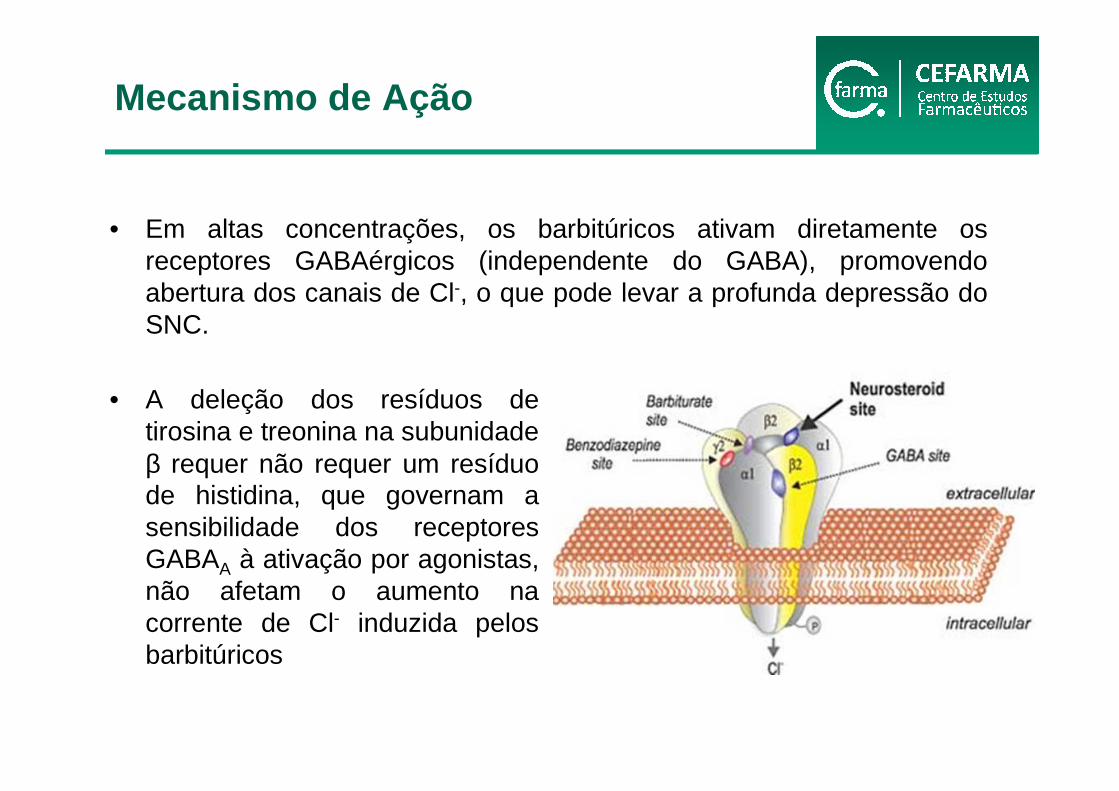
• Em altas concentrações, os barbitúricos ativam diretamente os receptores GABAérgicos (independente do GABA), promovendo abertura dos canais de Cl-, o que pode levar a profunda depressão do SNC.
• A deleção dos resíduos de tirosina e treonina na subunidade β requer não requer um resíduo de histidina, que governam a sensibilidade dos receptores GABAA à ativação por agonistas, não afetam o aumento na corrente de Cl- induzida pelos barbitúricos
Mecanismo de Ação

• Concentrações subanestésicas podem reduzir as despolarizações induzidas por glutamato
• Apenas os subtipos AMPA de receptores de glutamato sensíveis a cainato ou quisqualtaparecem ser afetados
• Em concentrações mais altas que produzem anestesia, o pentobarbital(ambos os isômeros) suprime o disparo neuronal repetitivo de alta frequência, aparentemente como resultado da inibição da função dos canais de Na+ sensíveis àtetrodoxina dependente de voltagem
Mecanismo de Ação

• Os barbitúricos exercem vários efeitos distintos sobre a transmissão sináptica excitatória e inibitória:
• O (-)-pentobarbital potencializa dos aumentos induz Cl- e, em concentrações abaixo de 10 mM, deprime as correntes de Ca+2
ativiadas por voltagem em neurônios de hipocampo isolados. Em concentrações acima de 100 mM, a condutância do Cl- aumenta mesmo na ausência do GABA.
• O fenobarbital é menos eficaz e menos potente na produção desses efeitos, ao passo que o (+)-pentobarbital tem apenas fraca atividade
• As propriedades anticonvulsivantes mais seletivas do fenobarbital e o seu índice terapêutico mais alto podem ser explicados por sua capacidade mais baixa de deprimir a função neuronal, quando comparado com os barbitúricos anestésicos
Mecanismo de Ação
• Exceto pelas propriedades anticonvulsivantes do fenobarbital e seus congêneres, os barbitúricos possuem baixo grau de seletividade e pequeno índice terapêutico
• Não é possível obter um efeito desejado sem depressão do SNC
• A percepção e reação à dor são relativamente poupados até quase o momento da inconsciência
• Em pequenas doses, os barbitúricos causam hiperalgesia e, por isso, não se pode confiar neles para produzir sedação ou sono na presença de dor, ainda que moderada.
fenobarbital
Propriedades Farmacológicas

• Doses hipnóticas aumentam o tempo de sono total e alteram os estágios de sono de modo dose-dependente
• Como os BZD, eles também diminuem a latência do sono, o número de despertares e as durações do sono REM e de ondas lentas
• Em poucos dias ocorre tolerância aos efeitos do sono, podendo o efeito sobre o tempo total reduzir em até 50 % em 2 semanas
• A interrupção ao tratamento contínuo leva a efeito rebote em todos os parâmetros para os quais se descreve uma redução por barbitúricos
Propriedades Farmacológicas

• Deprimem seletivamente a transmissão dos gânglios autônomos e reduzem a excitação nicotínica pelos ésteres de colina (lembre-sem: vasos sanguíneos sótêm invervação simpática)
• Este efeito é responsável, em parte, pela queda da pressão arterial produzida pelos oxibarbitúricos i.v. e intoxicações graves
• Nas funções neuromusculares esqueléticas, os efeitos bloqueadores da tubocurarina e do decametônio são igualmente intensificados durante a anestesia por barbitúricos, pois os barbitúricos inibem (em concentrações hipnóticas e anestésicas) a passagem de corrente através dos receptores colinérgicos nicotínicos
Propriedades Farmacológicas

• Deprimem o impulso respiratório e os mecanismos responsáveis pelo caráter rítmico da respiração
• Esse impulso neurogênio é reduzido em doses hipnóticas, mas não mais do que seria normalmente reduzido no sono normal
• Entretanto, esse impulso é eliminado em doses 3 vezes superiores às doses normalmente usadas para induzir o sono
• As doses elevadas também suprimem o impulso hipóxico
Propriedades Farmacológicas

• Entretanto, a margem de segurança entre os planos mais superficiais da anestesia cirúrgica e a depressão respiratória grave é suficiente grande para permitir o uso de barbitúricos de ação ultracurta como anestésicos
• Os barbitúricos deprimem apenas levemente os reflexos protetores, atéque o grau de intoxicação seja suficientemente grave para produzir depressão respiratória severa.
• Por isso, tosse, espirros, soluços e laringoespasmos podem ocorrem durante a anestesia por barbitúricos
Propriedades Farmacológicas
• Não produzem efeitos cardiovasculares significativas v.o. em doses sedativas e hipnóticas (ocorre apenas leve diminuição da pressão arterial e da frequência cardíaca, como ocorre no sono)
• Efeitos do tiopental (anestésico) sobre o coração são benignos comparativamente aos anestésicos voláteis
• Não há habitualmente alteração ou queda na pressão arterial média
Propriedades Farmacológicas

• Os reflexos cardiovasculares ficam reduzidos (pela inibição ganglionar parcial), mas só traz prejuízos a pacientes com insuficiência cardíaca congestiva ou choque hipovolêmico (cujos reflexos já operam no limite), podendo os barbitúricos causar queda elevada da pressão
• Quando tiopental e outros barbitúricos administrados i.v. são admnistradosapós medicação pré-anestésica convencional, é comum redução do fluxo sanguíneo renal e cerebral, com notável queda na pressão do LCR
• Arritimias cardíacas são raras, mas podem aumentar a incidência quando coadministrados com adrenalina ou halotano.
Propriedades Farmacológicas

• Concentrações anestésicas têm efeito direto no coração:
� Deprimem os canais de Na+
� Reduzem a função de dois tipos de canais de K+
• Entretanto, depressão direta da contratilidade cardíaca ocorre apenas quando são administradas doses várias vezes superiores às necessárias para se anestesiar.
• Isso contribui para a depressão cardiovascular que acompanha o envenenamento agudo por barbitúrico
• Hipotensão no envenenamento grave pode provocar oligúria grave ou anúria (toxicidade renal)
Propriedades Farmacológicas

• Os oxibarbitúricos tendem a diminuir o tônus da musculatura do TGI e a amplitude das contrações rítimicas.
• Efeito: parte periférico, parte central (depende da dose)
• Dose hipnótica não retarda significativamente o esvaziamento gástrico
• Alívio de sintomas GI por doses sedativas se deve à ação depressora central principalmente
Propriedades Farmacológicas
• Como sedativos-hipnóticos: são geralmente administrados por v.o.
• Absorção rápida e completa
• Sais de sódio mais rapidamente absorvidos que os ácidos livres correspondentes
• Pode-se usar i.m. (administração profunda para evitar necrose)
• Há supositários
• Via i.v. é reservada para tratar epilepsia (fenobarbital sódico) ou para indução e/ou manutenção de e anesteria geral (tiopental ou metoexital)
(absorção)
Farmacocinética

• Bem distribuídos
• Atravessam a barreira placentária
• Altamente lipossolúveis (principalmente aqueles usados para produzir anestesia)
• Baixa taxa de ligação a proteínas plasmáticas
• Sofrem redistribuição após injeção intravenosa
• Tiopental e metoexital -> pacientes despertam 5-15 min após injeção de doses anestésicas habituais
(distribuição)
Farmacocinética

• Eliminação é quase que completamente por biotransformação hepática (exceções são os menos lipossólúveis – aprobarbitale fenobarbital – cerca de 35 % destes são excretados inalterados pela urina)
• Sofrem conjugação hepática antes da excreção renal
• A oxidação em C5 é a mais importante biotransformação e cessa a atividade farmacológica
• Oxidação resulta na formação de álcoois, cetonas, fenóis e ácidos carboxílicos que podem ser excretados na urina como tais ou como conjugados do ácido glicurônico
(biotransformação)
12
3
5
fenobarbitalaprobarbital
Farmacocinética

• N-glicosilação é uma via metabólica importante para o fenobarbital
• Outras biotransformações incluem a N-hidroxilação, a dessulfuração de tiobarbitúricos a oxibarbitúricos, a abertura do anel do ácido barbitúrico e a N-desalquilação dos N-alquilbarbitúricos
IMPORTANTE!!!• A N-desalquilação dos N-
alquilbarbitúricos gera METABÓLITOS ATIVOS.
12
3
5
fenobarbital mefobarbital
(biotransformação)
Farmacocinética

• A eliminação metabólica é mais rápida em jovens do que em idosos e lactentes
• T1/2 vida aumenta durante a gestação
• Doença hepática crônica (cirrose) aumenta meia-vida plasmática
• Os barbitúricos administrados cronicamente alteram notavelmente o teor de proteínas e lipídeos do retículo endoplasmático liso hepático, bem como o aumento da atividade da glicuroniltrasnferase e das CYP 12A, 2C9, 2C19 e 3A4.
• Indução enzimática aumenta o metabolismo dos próprios barbitúricos e de outros fármacos (contribui para TOLERÂNCIA)
(biotransformação)
Farmacocinética

• A indução dessas enzimas também aumenta o metabolismo de esteróides, colesterol, sais biliares e vitaminas K e D
• Anestésicos, outros sedativo-hipnóticos e etanol, que são metabolizados pelas mesmas enzimas, têm seu metabolismo reduzido (tolerância cruzada – interação medicamentosa)
• O efeito indutor vai além das CYP: induz o aumento da sintetase do ácido δ-aminolevulínico (AAL) e aldeído desidrogenase
(biotransformação)
Farmacocinética

• Amplamente substituídos pelos BZD para sedação -> fenobarbital e butabarbitalainda estão disponíveis para este fim
• Fenobarbital e butabarbital também são usados para antagonizar efeitos de estimulantes do SNC, como efedrina, dextroanfetamina e teofilina (abordagem melhor é ajustar a dose dos estimulantes)
• Fenobarbital ainda é usado para abstinência de hipnossedativos
• Tratamento emergencial das convulsões (tétano, eclâmpsia, estado epilético, hemorragia cerebral e envenenamento por convulsivantes) -> BZD são superiores para este fim (diazepam)
Usos terapêuticos

• Fenobarbital sódico -> frequentemente usado como anticonvulsivante – entretanto, mesmo após administração i.v.leva-se 15 min para atingir concentrações de pico no cérebro
• Agentes de ação ultracurta (tiopenal e metoexital) são frequentemente empregados como anestésicos intravenosos
• Supositório de metoexital pode ser empregado para indução de anestesia ou sedação para procedimentos de imagem em crianças
• Doses anestésicas de barbitúricos atenuam o edema cerebral resultante de cirurgias, traumatismo cefálico ou isquemia cerebral e podem diminuir o tamanho do infarto e aumentar a sobrevida
Usos terapêuticos

Usos terapêuticos

• Sonolência
• Depressão do SNC residual no dia seguinte
• Alterações sutis no humor
• Comprometimento de julgamento e atividades motoras finas
• 200 mg de secobarbital compromete o desempenho para dirigir veículos ou pilotar aviões por 10-22 horas.
• Efeitos residuais podem tomar a forma de vertigens, náuseas, vômitos ou diarréia
• Excitação paradoxal pode ocorrer (inquietação, excitação, aspecto de embriaguez)
• Depressão respiratória grave
• Injeção i.v. rápida pode causar colapso cardiovascular
• Hipotensão arterial e choque
Efeitos adversos

• Administração com outros depressores do SNC, principalmente etanol (mas também BZD)– grave depressão respiratória, sendo frequentemente letal
IMPORTANTE!!!

• Tolerância é rapidamente desenvolvida em usuários crônicos, sendo necessário cerca de 5-10 vezes a dose habitual para provocar sedação
• Tolerância farmacodinâmica (mais importante) e farmacocinética
• Com o passar de algumas semanas a tolerância farmacocinética alcança seu pico, ao passo que a tolerância farmacodinâmica continua a se desenvolver
• A tolerância aos efeitos sobre o humor, a sedação e a hipnose ocorre mais prontamente e é maior que a tolerância aos efeitos anticonvulsivantes e letais (por isso, à medida que se aumenta a tolerância, diminui-se o índice terapêutico)
• A tolerância farmacodinâmica confere tolerância a todos os outros depressores do SNZ (incluindo o álcool) – tolerância cruzada
• Outros depressores e etanol potenciam a ação depressora, diminuindo drasticamente a dose tóxica
Tolerância e Dependência

• Principais sintomas são devidos aos efeitos no SNC e no sistema cardiovascular
• Sedação que pode progredir para coma e colapso respiratório por depressão do centro respiratório, nos casos graves.
• Efeito paradoxal em alguns pacientes: agitação psicomotora por depressão dos centros inibitórios (período de agitação é seguido de sonolência e até coma profundo em casos graves)
• A depressão do SNC é generalizada -> letargia, gala arrastada, ataxia. Pode haver diminuição e até ausência de reflexos.
• Sequelas podem ocorrer nos casos de hipóxia e hipoglicemia prolongadas
• Hipotermia
• Colapso cardiovascular com hipotensão grave (depressão miocárdica direta e vasodilatação periférica) sugere ingestão de altas doses
• Arritmias cardíacas são raras
Intoxicação Aguda

• Ventilação lenta e superficial podendo evoluir para insuficiência respiratória
• Hipóxia e acidose respiratória grave pode ocorrer
• Risco de aspiração deve ser considerado sempre, pois pode gerar pneumonia de aspiração e síndrome da angústia respiratória do adulto (SARA), responsáveis por maior morbidade e mortalidade nas intoxicações por barbitúricos
• Diminuição do tônus muscular e do peristaltismo
• Oligúria e anúria quando há hipotensão grave
Intoxicação Aguda

• Principais sintomas são devidos aos efeitos no SNC e no sistema cardiovascular
• Sedação que pode progredir para coma e colapso respiratório por depressão do centro respiratório, nos casos graves.
• Efeito paradoxal em alguns pacientes: agitação psicomotora por depressão dos centros inibitórios (período de agitação é seguido de sonolência e até coma profundo em casos graves)
• A depressão do SNC é generalizada -> letargia, gala arrastada, ataxia. Pode haver diminuição e até ausência de reflexos.
• Sequelas podem ocorrer nos casos de hipóxia e hipoglicemia prolongadas
Intoxicação Aguda

• Não há antídoto
• Tratamento de suporte
• Manter as vias aéreas
• Administrar oxigênio
• Avaliar necessidade de intubaçãoendotraqueal e assistência ventilatória
• Hipotensão arterial deve ser tratada agressivamente (canulação da veia periférica com agulha calibrosa e reposição volêmica)
Tratamento do paciente intoxicado

• Lavagem gástrica até 24 h após ingestão maciça, pois há retardo no esvaziamento gástrico
• Nunca induzir vômitos
• Usar carvão ativado (por até 72 h)
• Alcalinização da urina (aumenta a excreção em até 10 vezes) –barbitúricos mais hidrossolúveis (fenobarbital)
• Diurese forçada em pacientes com boa função renal e cardíaca
• Hemodiálise e hemoperfusão
Tratamento do paciente intoxicado





Não mais usados como sedativo-hipnóticos
Para-aldeído : polímero do acetaldeído. Reações de abstinência (especialmente delirium tremens em pacientes hospitalizados) e outros estados psiquiátricos caracterizados por excitação.
Hidrato de cloral : rapidamente reduzido a tricloroetanol, em grande parte, pela álcool desidrogenase hepática;
Tricloroetanol: in vitro, exerce efeitos semelhantes aos dos barbitúricos sobre o canal do receptor GABAA
Etclorvinol: ações farmacológicas semelhantes às dos barbitúricos; propriedades anticonvulsivantes e relaxantes musculares;
Meprobamato: propriedades farmacológicas lembram a dos benzodiazepínicos.

Propofol
•Utilidade na sedação durante os cuidados intensivos em adultos;
•Procedimentos de endoscopia gastrintestinal;
•Recuperação transvaginal de oócitos;
•Mecanismos não muito esclarecidos: ação sobre GABAA (?), outros receptores, acoplados à proteína G (?)


















