
Riella - Seção 02 - Distúrbios Hidroeletrolíticos
177
Capítulo 8 Compartimentos Líquidos do Organismo Miguel Carlos Riella, Maria Aparecida Pachaly e Leonardo Vidal Riella UNIDADES DE MEDIDA DE ÁGUA E DE ELETRÓLITOS Peso atômico Peso molecular Equivalente eletroquímico Pressão osmótica, osmol e miliosmol Concentração molar ou molaridade (M) Concentração molal ou molalidade (m) DIFUSÃO E OSMOSE OSMOLALIDADE E TONICIDADE Soluções isotônicas, hipertônicas e hipotônicas Soluções isosmóticas, hiperosmóticas e hiposmóticas ÁGUA TOTAL DO ORGANISMO Determinação da água corporal total COMPARTIMENTOS LÍQUIDOS Determinação do volume extracelular (VEC) Determinação do volume dos subcompartimentos extracelulares Plasma Volume intersticial-linfático Volume dos líquidos transcelulares Determinação do volume intracelular (VIC) COMPOSIÇÃO ELETROLÍTICA DOS COMPARTIMENTOS LÍQUIDOS DISTRIBUIÇÃO DA ÁGUA ENTRE COMPARTIMENTOS Adição de água ou solução hipotônica Adição de solução hipertônica de NaCl Adição de solução isotônica de NaCl TROCAS LÍQUIDAS ENTRE PLASMA E INTERSTÍCIO EXERCÍCIOS REFERÊNCIAS BIBLIOGRÁFICAS ENDEREÇOS RELEVANTES NA INTERNET RESPOSTAS DOS EXERCÍCIOS A água é o principal constituinte do corpo humano e de todos os organismos vivos. O próprio organismo é uma solução aquosa na qual estão dissolvidos vários íons e moléculas. Em circunstâncias normais, mesmo havendo variações na dieta, o conteúdo de água e eletrólitos é man- tido estável au6évés de modificações na excreção uriná- ria. 1 A distribuição desta solução aquosa e de seus vários constituintes no organismo é objeto de discussão nas pá- ginas seguintes. UNIDADES DE MEDIDA DE ÁGUA E DE ELETRÓLITOS O corpo humano é formado por uma solução aquosa que representa 45 a 60% do peso corporal. 2 Nesta solução, o solvente é a água e o soluto está representado por subs- tâncias orgânicas e inorgânicas. Para melhor compreensão das unidades que expressam a concentração dos solutos, os seguintes conceitos são importantes:
-
Upload
ismael-muniz -
Category
Documents
-
view
828 -
download
1
Transcript of Riella - Seção 02 - Distúrbios Hidroeletrolíticos

Capítulo
8Compartimentos Líquidos do Organismo
Miguel Carlos Riella, Maria Aparecida Pachaly e Leonardo Vidal Riella
UNIDADES DE MEDIDA DE ÁGUA E DE ELETRÓLITOS
Peso atômico
Peso molecular
Equivalente eletroquímico
Pressão osmótica, osmol e miliosmol
Concentração molar ou molaridade (M)
Concentração molal ou molalidade (m)
DIFUSÃO E OSMOSE
OSMOLALIDADE E TONICIDADE
Soluções isotônicas, hipertônicas e hipotônicas
Soluções isosmóticas, hiperosmóticas e hiposmóticas
ÁGUA TOTAL DO ORGANISMO
Determinação da água corporal total
COMPARTIMENTOS LÍQUIDOS
Determinação do volume extracelular (VEC)
Determinação do volume dos subcompartimentos
extracelulares
Plasma
Volume intersticial-linfático
Volume dos líquidos transcelulares
Determinação do volume intracelular (VIC)
COMPOSIÇÃO ELETROLÍTICA DOS COMPARTIMENTOS
LÍQUIDOS
DISTRIBUIÇÃO DA ÁGUA ENTRE COMPARTIMENTOS
Adição de água ou solução hipotônica
Adição de solução hipertônica de NaCl
Adição de solução isotônica de NaCl
TROCAS LÍQUIDAS ENTRE PLASMA E INTERSTÍCIO
EXERCÍCIOS
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET
RESPOSTAS DOS EXERCÍCIOS
A água é o principal constituinte do corpo humano ede todos os organismos vivos. O próprio organismo é umasolução aquosa na qual estão dissolvidos vários íons emoléculas. Em circunstâncias normais, mesmo havendovariações na dieta, o conteúdo de água e eletrólitos é man-tido estável au6évés de modificações na excreção uriná-ria.1
A distribuição desta solução aquosa e de seus váriosconstituintes no organismo é objeto de discussão nas pá-ginas seguintes.
UNIDADES DE MEDIDA DE ÁGUAE DE ELETRÓLITOS
O corpo humano é formado por uma solução aquosaque representa 45 a 60% do peso corporal.2 Nesta solução,o solvente é a água e o soluto está representado por subs-tâncias orgânicas e inorgânicas. Para melhor compreensãodas unidades que expressam a concentração dos solutos,os seguintes conceitos são importantes:
André
Material de Enfermagem

capítulo 8 91
Peso Atômico
Peso atômico é o peso total de um átomo ou a média dasmassas dos isótopos naturais de um elemento químico. Opeso de 1 átomo de oxigênio é 16 e serve como referênciapara o peso atômico de todas as substâncias. Assim, o pesoatômico do potássio é 39, em relação ao peso atômico dooxigênio.1
Peso Molecular
É a soma dos pesos atômicos de todos os elementosencontrados na fórmula de uma substância. O peso mole-cular expresso em gramas é igual a mol (M) e, em miligra-mas, é igual a milimol (mM).1 Exemplo:
SUBSTÂNCIA FÓRMULA PESO MOLECULAR MOL (M) MILIMOL(mM)
Cloreto de KCl 39 � 35,5 � 74,5 74,5 g 74,5 mgPotássio
Equivalente Eletroquímico
Partículas com carga positiva são chamadas cátions (porexemplo, Na� e K�) e partículas com carga negativa sãochamadas ânions (Cl� e HCO3
�). Quando cátions e ânionsse combinam, eles o fazem de acordo com sua carga iônica(valência) e não de acordo com seu peso.1
Equivalência eletroquímica se refere ao poder de com-binação de um íon. Um equivalente é definido como o pesoem gramas de um elemento que se combina com ou subs-titui 1 g de íon hidrogênio (H�). Também se obtém o equi-valente de uma determinada substância dividindo-se opeso molecular por sua valência.1 Para íons monovalentes,1 mol é igual a 1 equivalente. Para íons divalentes, 1 mol éigual a 2 equivalentes.
Como 1 g de H� é igual a 1 mol de H� (contendo apro-ximadamente 6,02 � 1023 partículas), um mol de qualquerânion monovalente (carga –1) se combinará como H� e seráigual a um equivalente (eq).
1 mol H� (1 g) � 1 mol Cl� (35,5 g) �
1 mol HCl (36,5 g)
Da mesma forma, 1 mol de um cátion monovalente (car-ga �1) também é igual a 1 equivalente, pois pode substituiro H� e combinar-se com 1 equivalente de algum ânion.
1 mol Na�(23 g) � 1 mol Cl� (35,5 g) �
1 mol NaCl (58,5 g)
Já o cálcio ionizado (Ca��) é um cátion divalente (carga
�2). Por exemplo, no cloreto de cálcio 1 mol de Ca�� com-bina-se com 2 moles de Cl� e é igual a 2 equivalentes.1
1 mol Ca�� (40 g) � 2 mol Cl� (71g) �
1 mol CaCl2 (111 g)
Por sua pequena concentração no organismo, os eletró-litos são comumente expressos em miliequivalentes (mEq).Um miliequivalente é igual a 10�3 equivalentes.
Pressão Osmótica, Osmol e MiliosmolOutra maneira de expressar o número de partículas de
soluto presentes é através da pressão osmótica, que deter-mina a distribuição de água entre os compartimentos. Apressão osmótica é proporcional ao número de partículaspor unidade do solvente e não se relaciona à valência oupeso das partículas.1 As unidades utilizadas são o osmol(Osm) e o miliosmol (mOsm). Um osmol é o número de íonspor mol ou a quantidade de substância que se dissocia emsolução para formar um mol de partículas osmoticamenteativas. Por exemplo, 1 mol de NaCl tem 2 osmóis de solu-to, pois se dissocia em Na e Cl. Um mol de glicose contémapenas 1 osmol de soluto, pois a glicose não é ionizável.
A pressão osmótica determina a distribuição de águaentre os espaço intra- e extracelular, como será discutidoao se abordar tonicidade (v. a seguir).
Concentração Molar ou Molaridade (M)É o número de moles do soluto por litro de solução, a
uma dada temperatura.
Concentração Molal ou Molalidade (m)É o número de moles do soluto por 1.000 gramas do
solvente.
DIFUSÃO E OSMOSE
A difusão é dividida em dois subtipos: a difusão sim-ples e a difusão facilitada. Na difusão simples, a passagemde íons ou moléculas através de uma membrana ocorredevido ao movimento cinético aleatório destas partículas,sem a necessidade de ligação com proteínas de transpor-te. A taxa de difusão simples depende da quantidade desubstância disponível, velocidade de movimento cinéticoe número de aberturas na membrana celular através dasquais as moléculas ou íons podem se mover. Na difusãofacilitada, há necessidade de interação com uma proteínatransportadora, a qual se liga quimicamente às moléculase facilita sua passagem através da membrana.5
A osmose ocorre quando duas soluções de concentra-ções diferentes encontram-se separadas por uma membra-
peso molecularvalência iônica1 Eq �

92 Compartimentos Líquidos do Organismo
na semipermeável. Há então um movimento de água dasolução menos concentrada para a mais concentrada, a qualsofre uma diluição progressiva, até que as duas soluçõesatinjam um equilíbrio.
OSMOLALIDADE ETONICIDADE
É importante diferenciar os conceitos de osmolalidadee tonicidade. A osmolalidade é determinada pela concen-tração total de solutos numa determinada solução ou com-partimento. Tonicidade é a capacidade que os solutos têmde gerar uma força osmótica que provoca o movimento deágua de um compartimento para outro.3,4 Para que ocorraaumento da tonicidade no espaço extracelular, por exem-plo, é necessário que solutos permaneçam confinados nesteespaço sem atravessar livremente as membranas celularese sem migrar para os demais compartimentos. Isto provo-cará o movimento de água do compartimento intracelularpara o extracelular (osmose) para estabelecer um equilíbrioosmótico, gerando também diminuição do volume dascélulas. Alguns dos solutos capazes de produzir este mo-vimento de água (osmóis efetivos) são: sódio, glicose, ma-nitol e sorbitol. O sódio permanece no espaço extracelularsem movimentar-se para outros compartimentos devido àação da bomba sódio-potássio ATPase, que continuamen-te bombeia o sódio para fora das células.
A glicose é um osmol efetivo, mas é normalmente me-tabolizada no interior das células, e desta forma não con-tribui significativamente para a tonicidade sob circunstân-cias normais. No diabetes mellitus descontrolado, a concen-tração elevada de glicose no plasma pode levar a um au-mento significativo da osmolalidade e da tonicidade, cau-sando movimento de água para dentro do espaço extrace-lular. A uréia contribui para a osmolalidade, mas atraves-sa livremente as membranas e não influi no movimento deágua entre compartimentos.3,4
Soluções Isotônicas, Hipertônicas eHipotônicas
As soluções isotônicas apresentam a mesma tonicidadeque o plasma, e conseqüentemente não induzem movimen-to de água através das membranas celulares e não provo-cam variação do volume celular. Exemplo de solução iso-tônica: solução salina a 0,9%; solução glicosada a 5%.
Soluções hipertônicas geram o movimento de água emdireção ao espaço extracelular, provocando diminuição dovolume celular. Exemplo: solução salina em concentraçãosuperior a 0,9%.
As soluções hipotônicas provocam o movimento deágua em direção ao compartimento intracelular, provocan-do edema celular.5 Exemplo: solução salina em concentra-
ção inferior a 0,9%. A Fig. 8.1 exemplifica os efeitos des-critos.
Soluções Isosmóticas, Hiperosmóticas eHiposmóticas
A osmolalidade de uma solução é determinada pelaquantidade total de partículas dissolvidas, incluindo ossolutos que atravessam as membranas celulares. Os termosisosmótico, hiperosmótico e hiposmótico se referem a umacomparação com o fluido extracelular normal. Por exem-plo, a solução salina a 0,9% é ao mesmo tempo isotônica(não provoca movimento de água) e isosmótica (apresen-ta o mesmo número de partículas de soluto) em relação aoespaço extracelular.
Pontos-chave:
• A osmolalidade depende do número totalde solutos numa solução ou compartimento
• Tonicidade é a capacidade que os solutostêm de provocar movimento de água de umcompartimento para outro. Esta propriedadedefine o que são soluções isotônicas,hipotônicas e hipertônicas
ÁGUA TOTAL DO ORGANISMO
A água total do organismo varia entre 45 e 60% do pesocorporal, de acordo com a idade, o sexo e a composiçãocorporal do indivíduo.3,7 Esta proporção variável é devidoàs diferentes quantidades de gordura presentes no orga-nismo, pois em gordura neutra quase não existe água.Assim, indivíduos obesos, embora mais pesados, possuemmenos água no organismo. Da mesma forma, por possuí-rem maior quantidade de gordura no organismo, as mu-lheres têm menor proporção de água corporal (50%). Já osidosos, por apresentarem menor massa muscular, têm ummenor conteúdo de água.3 Nas crianças, a água corporaltotal equivale a cerca de 70%-80% do peso, pois apresen-tam menor conteúdo de tecido adiposo.
Fig. 8.1 Efeito do contato de diferentes soluções com hemácias:solução isotônica (A); solução hipertônica (B); e solução hipotô-nica (C).
B CA

capítulo 8 93
Para efeitos práticos de cálculo, consideraremos a águatotal como sendo 60% do peso corporal, independentemen-te das variações anteriormente mencionadas.
Determinação daÁgua Corporal Total
O método laboratorial que determina a água total doorganismo baseia-se na técnica de diluição,5,8 fundamenta-da no seguinte princípio: quando se adiciona uma quan-tidade conhecida de soluto a um volume desconhecido desolvente, e dosa-se a concentração final da substância, épossível calcular o volume do solvente. Por exemplo, adi-cionando 1 kg (1.000 mg) de uma substância a um volu-me de solvente, e obtendo-se uma concentração final de100 mg/litro, chega-se à conclusão de que o volume dosolvente é igual a 10 litros. Acompanhe com a fórmulaabaixo:
Ci/Vf � Cf e Vf � Ci/Cf
Onde:
Ci: concentração (quantidade) inicial da substânciaadicionada;
Cf: concentração final da substância adicionada;Vf: volume final da solução.
1.000 mg/Vf � 100 mg/litro
Vf � 1.000/100 � 10 litros
A determinação da quantidade de água do organismoin vivo só foi possível após o emprego de isótopos da água:estáveis (deutério) ou radioativos (trítio). Um destes com-postos é injetado na circulação e aguarda-se um determi-nado período para que haja equilíbrio no plasma. Natural-mente, a quantidade da substância que é metabolizada eexcretada durante este período de equilíbrio deve ser con-siderada. A antipirina foi também uma substância bastan-te utilizada na determinação da água total do organismo.
COMPARTIMENTOS LÍQUIDOS
A água do organismo se distribui em compartimentos,em parte devido a diferentes composições iônicas (Fig. 8.2).No entanto, estes compartimentos não são estanques, ha-vendo um constante intercâmbio hidroeletrolítico. Basica-mente, identificam-se dois grandes compartimentos: intra-celular e extracelular.
O compartimento intracelular é composto pela águaexistente no citoplasma de todas as células. Já o comparti-mento extracelular, como o próprio termo indica, refere-se a toda a água externa às células e possui subcomparti-mentos: plasma, líquido intersticial e linfa, água dos ossose líquidos transcelulares (Fig. 8.2).
Os líquidos transcelulares representam coleções de lí-quidos que não são simples transudatos, mas são líquidossecretados e incluem: secreções das glândulas salivares,pâncreas, fígado e árvore biliar, além dos líquidos nas ca-vidades pleurais, oculares, peritoneal, no lúmen do tratogastrintestinal e líquido cefalorraquidiano.4
Terceiro espaço é um termo proposto por Randall, em1952, para descrever a situação na qual o líquido extrace-lular é perdido ou seqüestrado numa área do corpo ondenão participa das trocas, e conseqüentemente não satisfazàs necessidades hídricas do paciente. Exemplos: líquidono intestino na presença de íleo, líquido peritoneal na pe-ritonite, líquido peripancreático na pancreatite aguda e oedema do queimado. Por exemplo, no paciente com obs-trução intestinal ou íleo intenso, vários litros de fluidosricos em eletrólitos podem estar confinados ao intestino,sem que o paciente possa utilizá-los, mesmo que esteja hi-povolêmico.
Determinação do Volume Extracelular(VEC)
O método utilizado também se baseia no princípio da téc-nica de diluição, preferindo-se uma substância que seja ex-cluída das células e permaneça no espaço extracelular. Vá-rias substâncias têm sido utilizadas: 36Cl, sulfato, tiossulfatoe tiocianato, além de certos sacarídeos (manitol, inulina esucrose).8 Nenhuma destas substâncias é considerada ideal.Elas variam na sua capacidade de penetração nas células eos resultados da determinação do VEC são, portanto, diver-sos, variando de 16 a 28%. Na prática, considera-se que ovolume extracelular corresponde a 20% do peso corporal.5
Determinação do Volume dosSubcompartimentos Extracelulares
PLASMAO volume plasmático é determinado empregando-se
substâncias que ficam confinadas ao leito vascular. A al-Fig. 8.2 Compartimentos líquidos do organismo (percentual dopeso corporal).

94 Compartimentos Líquidos do Organismo
bumina ou eritrócitos podem ser utilizados. A albuminamarcada com 131I é a mais empregada, e o volume de dis-tribuição determinado está em torno de 4,5% do peso cor-poral. Entretanto, alguma 131I-albumina escapa do leitovascular para o interstício. Quando se empregam eritró-citos, eles são previamente marcados com crômio-51(51Cr).
VOLUME INTERSTICIAL-LINFÁTICOÉ calculado indiretamente, subtraindo-se o volume plas-
mático do volume extracelular, e aproxima-se de 20% daágua total ou 12% do peso corporal.
VOLUME DOS LÍQUIDOSTRANSCELULARES
É calculado pela soma das várias secreções e aproxima-se de 1,5% do peso corporal ou 2,5% da água total (Qua-dro 8.1).
Determinação do Volume Intracelular(VIC)
O volume intracelular não pode ser determinado dire-tamente e é calculado subtraindo-se o volume extracelu-lar da água corporal total. Na prática, considerando-se aágua total do organismo como sendo 60% do peso corpo-ral e o volume extracelular 20%, conclui-se que o volumeintracelular é de 40% do peso total.5
Pontos-chave:
• Regra 60:40:20• Água corporal total � 60% do peso
corporal.• Compartimentos:
Intracelular � 40% do peso corporalExtracelular � 20% do peso corporal
COMPOSIÇÃO ELETROLÍTICADOS COMPARTIMENTOS
LÍQUIDOS
A composição eletrolítica do plasma e dos líquidos in-tersticial e intracelular pode ser apreciada no Quadro 8.2.
No líquido extracelular o cátion mais abundante é osódio, e o cloro é seu principal ânion. Em menor concen-tração no líquido extracelular, observamos K�, Ca�� e Mg��
e os ânions HPO ,4 � � H PO2 4
� e SO .4 � � Além disso, muitos
ácidos orgânicos (láctico, pirúvico, cítrico) existem no líqui-do extracelular como ânions e podem estar elevados emdiversas enfermidades.5 O sódio no líquido extracelularrepresenta a metade de sua osmolalidade.
No líquido intracelular o cátion mais abundante é opotássio, e os ânions prevalentes são compostos orgânicoscomo os fosfatos, sulfatos e proteínas. Observam-se aindaMg��, Ca�� e os ânions inorgânicos Cl� e HCO3
�. Note queo total de íons intracelulares excede o do plasma e, no en-tanto, a osmolalidade intra- e extracelular é a mesma. Acre-dita-se que alguns destes íons intracelulares sejam osmo-ticamente inativos, isto é, ligados a proteínas e a outrosconstituintes celulares. Metade da osmolalidade do líqui-do intracelular é dada pelo K�.
A determinação de eletrólitos no interior das células étecnicamente difícil, além de variar de acordo com a ori-gem do tecido estudado. Por exemplo, apesar da possibi-lidade de acesso às hemácias do sangue periférico, a dosa-gem dos eletrólitos nestas células, que não possuem núcle-os e mitocôndrias, pode não refletir o que ocorre no tecidomuscular.6
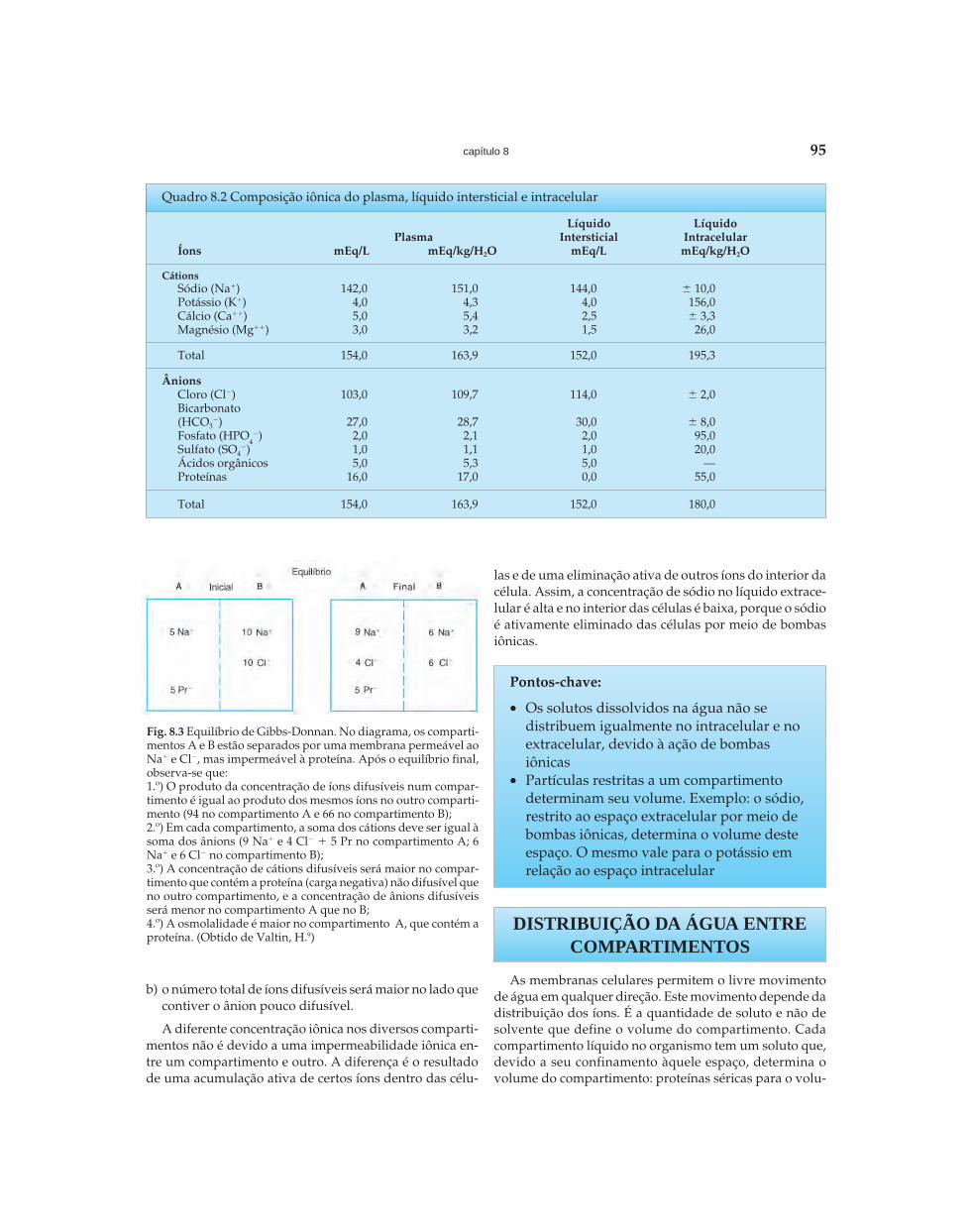
O líquido intersticial é um ultrafiltrado do plasma. Sen-do assim, não contém os elementos celulares (hemácias, leu-cócitos, plaquetas), e sim um líquido ultrafiltrado que pra-ticamente não contém proteínas. Note-se que a soma totalde íons no plasma é maior que a do líquido intersticial. Aexplicação está na distribuição de Gibbs-Donnan5,7,9 (Fig. 8.3):
a) quando há um ânion pouco difusível num dos lados damembrana (no caso, as proteínas no lado vascular), aconcentração de um íon positivo difusível será maiorneste lado, e a concentração de um ânion difusível serámenor;
Quadro 8.1 Distribuição da água total numadulto jovem*
% do Peso % da ÁguaCompartimento Corporal Total
Plasma 4,5 7,5
Líquido intersticial linfático 12,0 20,0
Tecido conjuntivo denso ecartilagem 4,5 7,5
Água do osso (inacessível) 4,5 7,5
Transcelular 1,5 2,5
Extracelular total 27,0 45,0
Extracelular funcional** 21,0 —
Água total 60,0 100,0
Água intracelular 33,0 55,0
*Modificado de Edelman, I. S. e Leibman, J.11
**O líquido extracelular funcional representa o extracelular total menosa água do osso e do líquido transcelular.
capítulo 8 95
b) o número total de íons difusíveis será maior no lado quecontiver o ânion pouco difusível.
A diferente concentração iônica nos diversos comparti-mentos não é devido a uma impermeabilidade iônica en-tre um compartimento e outro. A diferença é o resultadode uma acumulação ativa de certos íons dentro das célu-
las e de uma eliminação ativa de outros íons do interior dacélula. Assim, a concentração de sódio no líquido extrace-lular é alta e no interior das células é baixa, porque o sódioé ativamente eliminado das células por meio de bombasiônicas.
Pontos-chave:
• Os solutos dissolvidos na água não sedistribuem igualmente no intracelular e noextracelular, devido à ação de bombasiônicas
• Partículas restritas a um compartimentodeterminam seu volume. Exemplo: o sódio,restrito ao espaço extracelular por meio debombas iônicas, determina o volume desteespaço. O mesmo vale para o potássio emrelação ao espaço intracelular
DISTRIBUIÇÃO DA ÁGUA ENTRECOMPARTIMENTOS
As membranas celulares permitem o livre movimentode água em qualquer direção. Este movimento depende dadistribuição dos íons. É a quantidade de soluto e não desolvente que define o volume do compartimento. Cadacompartimento líquido no organismo tem um soluto que,devido a seu confinamento àquele espaço, determina ovolume do compartimento: proteínas séricas para o volu-
Quadro 8.2 Composição iônica do plasma, líquido intersticial e intracelular
Líquido LíquidoPlasma Intersticial Intracelular
Íons mEq/L mEq/kg/H2O mEq/L mEq/kg/H2O
CátionsSódio (Na�) 142,0 151,0 144,0 � 10,0Potássio (K�) 4,0 4,3 4,0 156,0Cálcio (Ca��) 5,0 5,4 2,5 � 3,3Magnésio (Mg��) 3,0 3,2 1,5 26,0
Total 154,0 163,9 152,0 195,3
ÂnionsCloro (Cl�) 103,0 109,7 114,0 � 2,0Bicarbonato(HCO3
�) 27,0 28,7 30,0 � 8,0Fosfato (HPO
4�) 2,0 2,1 2,0 95,0
Sulfato (SO4�) 1,0 1,1 1,0 20,0
Ácidos orgânicos 5,0 5,3 5,0 —Proteínas 16,0 17,0 0,0 55,0
Total 154,0 163,9 152,0 180,0
Fig. 8.3 Equilíbrio de Gibbs-Donnan. No diagrama, os comparti-mentos A e B estão separados por uma membrana permeável aoNa� e Cl�, mas impermeável à proteína. Após o equilíbrio final,observa-se que:1.º) O produto da concentração de íons difusíveis num compar-timento é igual ao produto dos mesmos íons no outro comparti-mento (94 no compartimento A e 66 no compartimento B);2.º) Em cada compartimento, a soma dos cátions deve ser igual àsoma dos ânions (9 Na� e 4 Cl� � 5 Pr no compartimento A; 6Na� e 6 Cl� no compartimento B);3.º) A concentração de cátions difusíveis será maior no compar-timento que contém a proteína (carga negativa) não difusível queno outro compartimento, e a concentração de ânions difusíveisserá menor no compartimento A que no B;4.º) A osmolalidade é maior no compartimento A, que contém aproteína. (Obtido de Valtin, H.9)

96 Compartimentos Líquidos do Organismo
me intravascular, sódio para o compartimento extracelu-lar e potássio para o intracelular. A rápida distribuiçãoproporcional de água entre os compartimentos assegurauma concentração osmolar intra- e extracelular essencial-mente idêntica.
A osmolalidade plasmática de um indivíduo normalestá em torno de 289 mOsm/kg H2O, atribuída principal-mente ao sódio e aos ânions uréia e glicose. A osmolalida-de plasmática é igual a duas vezes a concentração plasmá-tica do sódio, mais a osmolalidade da uréia, mais a osmo-lalidade da glicose. A osmolalidade plasmática poderá serdeduzida, considerando-se as seguintes concentraçõesnormais: sódio plasmático — 140 mEq/L; uréia plasmáti-ca — 30 mg/100 ml, e glicemia — 90 mg/100 ml.
Osmolalidade plasmática �
(Na � 2) � ( � 10) � ( � 10)
Na� � 2 � 140 mEq/L � 280 mOsm/kg H2O
Uréia: � 10 � 5 mOsm/kg H2O
Glicemia: � 10 � 5 mOsm/kg H2O
Então, a osmolalidade plasmática estimada com os da-dos acima é de 290 mOsm/kg H2O.
Para o cálculo da contribuição da uréia para a osmola-lidade, dividimos a concentração plasmática da uréia por60, que é seu peso molecular. Da mesma forma, dividimosa glicose por seu peso molecular, que é 180. Multiplicamosambos os cálculos por 10, a fim de convertermos mg/100ml em mg/L. Quando não se dispõe das concentrações deuréia e glicose, a osmolalidade do plasma pode ser estima-da multiplicando-se a concentração de sódio por dois.
Alguns líquidos transcelulares têm uma osmolalidademuito diferente dos outros compartimentos. Isto se deveao fato de estarem separados dos outros compartimentos
por uma camada de células e uma membrana pouco per-meável à água. Desta forma, secreções gastrintestinais e osuor são hiposmóticos.
Como a osmolalidade é a mesma dentro e fora das cé-lulas, a passagem de água do interior para fora das célu-las, ou vice-versa, só ocorre se houver mudança de osmo-lalidade e tonicidade. As seguintes circunstâncias, ilustra-das na Fig. 8.4 e baseadas na discussão de Robert Pitts, tra-duzem situações em que se alteram a osmolalidade e ovolume dos compartimentos extra- e intracelular.10
Adição de Água ou Solução HipotônicaSe administrarmos água ou solução hipotônica a um
indivíduo, seja por via oral ou endovenosa, e se conside-rarmos que não haverá diurese durante o período do es-tudo, a água distribui-se rápida e proporcionalmente en-tre os dois compartimentos. Observa-se uma redução uni-forme na osmolalidade e um aumento no volume dos doiscompartimentos (aumento maior no intracelular por sermaior que o extracelular)5,7 (Fig. 8.4).
Adição de SoluçãoHipertônica de NaCl
A infusão endovenosa de uma solução hipertônica deNaCl expande o compartimento extracelular e provocaum movimento passivo de água do compartimento in-tracelular (osmolalidade menor) para o extracelular (os-
Pontos-chave:
• Osmolalidade plasmática � (Na � 2) �
( � 10) �( � 10)
• Osmolalidade plasmática normal � 290mOsm/kg H2O
Uréia Glic60 180
Fig. 8.4 Alterações no volume e na osmolalidade dos compartimentos intra- e extracelulares, quando se adiciona: A) apenas água aoorganismo; B) uma solução salina hipertônica; C) uma solução salina isotônica. O estado inicial dos compartimentos intracelular (I)e extracelular (E) está representado pelas linhas contínuas e no final está representado por linhas interrompidas. A altura do com-partimento representa a osmolalidade, e a largura, o volume. (Modificado de Pitts, R.10)
Uréia60
Glic180
30 mg / 100 ml60
90 mg / 100 ml180

capítulo 8 97
molalidade maior devido à solução adicionada), até queambos os compartimentos se equilibrem e se tornem isos-móticos. A saída de água reduz o volume do comparti-mento intracelular e, conseqüentemente, aumenta a osmo-lalidade deste compartimento. No final, ambos os compar-timentos terão uma osmolalidade maior que a inicial5,7
(Fig. 8.4).
Adição de Solução Isotônica de NaCl
Como o sódio permanece principalmente no comparti-mento extracelular, há uma expansão do volume destecompartimento, mas não ocorre alteração na osmolalida-de intra- e extracelular e, tampouco, no volume intracelu-lar5,7 (Fig. 8.4).
Pontos-chave:
• Soluções de diferentes tonicidadesprovocam variações no volume doscompartimentos intra- e extracelular
• Soluções isotônicas de sódio aumentam oextracelular, pois o sódio se mantém nestecompartimento
• Soluções hipotônicas e água se distribuemno intra- e extracelular (maior proporção nointracelular)
• Soluções hipertônicas causam movimentode água do intra- para o extracelular,diminuindo o primeiro e aumentando osegundo
TROCAS LÍQUIDAS ENTREPLASMA E INTERSTÍCIO
A nutrição das células e a remoção dos produtos dometabolismo celular somente são possíveis devido à exis-tência de uma circulação capilar. Ela permite uma rápidatroca de nutrientes entre a circulação e as células atravésdo líquido intersticial. O transporte dos nutrientes e cata-bólitos pelo sangue depende da adequação da função cir-culatória e do volume líquido circulante. Portanto, man-ter o volume plasmático é essencial.
A pressão hidrostática determinada pela bomba cardí-aca num compartimento (vascular) altamente permeávelà água e aos solutos poderia determinar a passagem detodo o líquido intravascular rapidamente para o interstí-cio. Isto não ocorre porque a esta pressão hidrostática seopõe uma outra pressão — a pressão osmótica determina-da pelas proteínas, principalmente albumina, também co-nhecida como pressão coloidosmótica ou pressão oncóti-ca. A pressão oncótica está em torno de 25 mmHg. Já o lí-quido intersticial tem pouca proteína, tendo uma pressãooncótica em torno de 5 mmHg.2 A diferença, portanto, en-tre a pressão osmótica do plasma e a do interstício é de 20mmHg e esta força se opõe à pressão hidrostática.5,7
Foi Starling quem primeiro formulou o mecanismo dedistribuição de líquido entre os compartimentos vasculare intersticial (Fig. 8.5). Segundo ele, o sangue chega aoscapilares com uma certa força (pressão hidrostática), capazde determinar o retorno venoso ao coração. A pressão hi-drostática é determinada pela pressão mecânica geradapelo coração. A pressão média nas grandes artérias é de 95mmHg, mas, quando o sangue chega ao leito capilar, a
Fig. 8.5 Hipótese de Starling para troca de líquido entre plasma e interstício. Os fatores que determinam esta troca são denominadosforças de Starling. (Obtido de Valtin, H.9)

98 Compartimentos Líquidos do Organismo
pressão hidrostática cai para 40-45 mmHg. Esta pressãohidrostática de 40-45 mmHg determina a passagem de lí-quido intravascular para o interstício e a ela se opõem apressão oncótica das proteínas, em torno de 25-30 mmHg,e uma pressão do turgor intersticial de 2-5 mmHg. Destaforma, o balanço dessas forças resulta numa pressão defiltração positiva, em torno de 10-15 mmHg.5
Uma pequena quantidade de proteínas atravessa os ca-pilares, mas quase tudo retorna à circulação através do sis-tema linfático. No entanto, uma fração permanece no inters-tício e é responsável pela pressão oncótica intersticial de 3mmHg. Quando a coluna de sangue atinge o lado venosodo capilar, a pressão hidrostática está reduzida a 10-15mmHg e o balanço das forças é negativo, determinando areabsorção do líquido filtrado no lado venoso capilar.5
Acredita-se que o principal mecanismo que altera a pres-são hidrostática intracapilar não é a resistência ao longo docapilar e sim a atividade de esfíncteres pré-capilares (Fig.8.5). Quando há um relaxamento do esfíncter, a pressão hi-drostática intracapilar aumenta, favorecendo a filtração aolongo do capilar; quando o esfíncter se contrai, a pressãohidrostática cai, e talvez só ocorra reabsorção ao longo docapilar. Também é importante a área de superfície dos ca-pilares. Quando o esfíncter se contrai, muitos capilares sãodesviados da circulação arterial, reduzindo a área de super-fície capilar; quando o esfíncter se relaxa, ocorre o inverso.
Além do mais, o ritmo de fluxo líquido através do capi-lar endotelial não depende só das forças de Starling, mastambém do coeficiente de filtração, expresso pela seguin-te fórmula:9
q � Kf(Pc – Pt) – (pp – pt), onde:
q � ritmo de fluxo através do capilar;Kf � coeficiente de filtração;Pc � pressão hidrostática intracapilar;Pt � pressão do turgor tecidual;pp � pressão oncótica do plasma;pt � pressão oncótica intersticial.
Conclui-se que se a pressão hidrostática for excessiva,ou a pressão oncótica do plasma reduzida, haverá um ex-cesso de filtração de líquido para o interstício e, se for ul-
trapassada a capacidade de remoção pelos linfáticos, ha-verá edema.
EXERCÍCIOS
(Respostas no final do capítulo.)
1) Adulto jovem de 70 kg. Calcular a água corporal total, espaço ex-tracelular, volume plasmático e volume intracelular.
2) Em relação à proporção de água corporal total, que diferenças exis-tem em pacientes obesos, mulheres, crianças e idosos?
3) Qual a osmolalidade plasmática de um paciente que apresenta asseguintes dosagens plasmáticas: uréia � 240 mg/dl; glicose � 360mg/dl; sódio � 133 mEq/litro.
4) Frente à osmolalidade encontrada na questão anterior, o que ocorrecom os compartimentos intra- e extracelular?
5) O que ocorre com as forças de Starling em presença de hipoalbu-minemia?
6) Cite um exemplo de solução endovenosa que deve ser adminis-trada quando se deseja aumentar o volume do espaço extracelu-lar.
7) Cite um exemplo de solução endovenosa que se administra paraexpandir o espaço extracelular e contrair o espaço intracelular.
REFERÊNCIAS BIBLIOGRÁFICAS
1. ROSE, B.; POST, T.W. Units of solute measurement. Up to Date, vol.9, n. 1, Cap. 1B. 2000.
2. HAYS, R.M. Dynamics of body water and electrolytes, Cap. 1, pág.1. In: Clinical Disorders of Fluid and Eletrolyte Metabolism. Eds. Mor-ton H. Maxwell and C. R. Kleeman. McGraw-Hill Book Co., 1972.
3. PRESTON, R.A. Acid-Base, Fluids and Electrolytes Made RidiculouslySimple. Cap.1, pág. 3. MedMaster Inc., Miami, 1997.
4. OH, M.S. and CARROLL, H.J. Regulation of intracellular andextracellular volume. In: Fluid, Electrolyte and Acid-Base Disorders.Eds. Arieff, A.I. and DeFronzo, R.A. Cap. 1, pág. 1. Churchill Livin-gstone Inc. New York, 1995.
5. GUYTON, A.C. and HALL, J.E. The body fluid compartments:extracellular and intracellular fluids; interstitial fluid and edema. In:Textbook of Medical Physiology. Cap. 25, págs. 297-313. W.B. SaundersCo., 1996.
6. MAFFLY, R.H. The body fluids: volume, composition and physicalchemistry, Cap. 2, pág. 65. In: The Kidney. Eds. B. M. Brenner and F.C. Rector Jr. W. B. Saunders Co., 1976.
7. HALPERIN, M.L.; GOLDSTEIN, M.B. Sodium and water physio-logy. In: Fluid, Electrolyte and Acid-Base Physiology — A Problem-BasedApproach. Cap. 6, pág. 217. W.B. Saunders Co., 1994.
8. MALNIC, G. e MARCONDES, M. Fisiologia Renal. EPU, 1986.9. VALTIN, H. Renal Function: Mechanisms Preserving Fluid and Solute
Balance in Health. Cap. 2, pág. 20, Little, Brown and Co., Boston, 1995.10. PITTS, R.D. Physiology of the Kidney and Body Fluids. Cap. 2, pág. 11.
Year Book Medical Publishers Inc., 3rd edition, 1974.11. EDELMAN, I.S. and LEIBMAN, J. Am. J. Med., 27:256, 1959.
ENDEREÇOS RELEVANTES NA INTERNET
Química e soluçõeshttp://dbhs.wvusd.k12.ca.us
Forças de Starlingwww.liv.ac.uk/�petesmif/teaching/1bds - mb/notes/fluid/text.htm
Pontos-chave:
• A pressão hidrostática é a principal forçaque provoca o movimento de líquido parafora da luz do capilar
• A pressão coloidosmótica ou oncótica(determinada principalmente pelaalbumina) é a principal força que se opõe àhidrostática e provoca o movimento delíquido para dentro da luz do capilarsanguíneo

capítulo 8 99
Uréia60
Outroswww.physio.mcgill.ca/209A/Body - fluids/Body - fl3.htmwww.umds.ac.uk/physiology/rbm/bodyflu
RESPOSTAS DOS EXERCÍCIOS
1) Num adulto jovem de 70 kg:a. Água corporal total � 60% de 70 kg � 42 litrosb. Volume do espaço extracelular � 20% de 70 kg � 14 litrosc. Volume plasmático � 4,5% de 70 kg � 3,15 litrosd. Volume do espaço intracelular � 40% de 70 kg � 28 litros
2) A água corporal total encontra-se diminuída (menos que 60% dopeso corporal) em pacientes obesos e mulheres, devido ao maiorconteúdo de gordura que apresentam. Os idosos apresentam me-nor massa muscular, e conseqüentemente menor proporção deágua em relação ao peso. As crianças apresentam conteúdo degordura reduzido, e então a proporção de água corporal total émaior em relação ao peso.
3) Osmolalidade plasmática � (Na � 2) � ( � 10) � ( � 10),então:
Osmolalidade plasmática � (133 � 2) � (240/60 � 10) � (360/180� 10) � 326 mOsm/kg H
2O
4) No exemplo acima, com o aumento da osmolalidade e tonicidadedo plasma (a osmolalidade normal oscila entre 280 e 290 mOsm/kg H
2O), ocorre a passagem de água do espaço intracelular para o
extracelular até haver um equilíbrio osmótico entre os dois com-partimentos. Como resultado final, o volume do espaço intrace-lular sofre redução (pela perda de água) e o extracelular sofre oacréscimo de água, inclusive diluindo o sódio do intravascular.
5) Em presença de hipoalbuminemia, existe redução da pressão on-cótica, o que favorece a filtração de líquido para o interstício nolado venoso do capilar e dificulta a reabsorção de líquido intersti-cial no lado venoso do capilar; caso seja ultrapassada a capacida-de de absorção pelos linfáticos, isto resultará em edema.
6) Solução salina a 0,9% (chamada solução salina isotônica).
7) Solução salina hipertônica (concentração maior que 0,9%).
Glic180

Capítulo
9Metabolismo da Água
Miguel Carlos Riella e Maria Aparecida Pachaly
MECANISMO DA SEDE
VASOPRESSINA (HORMÔNIO ANTIDIURÉTICO)
Mecanismo de ação do hormônio antidiurético (HAD) —
aquaporinas
OUTROS HORMÔNIOS
Catecolaminas
Hormônio tireoidiano
Hormônios adrenocorticais
Sistema renina-angiotensina
MECANISMO RENAL DE REGULAÇÃO DA ÁGUA
Considerações anatômicas
Vascularização da medula renal
Concentração da urina — mecanismo de contracorrente
Fluxo sanguíneo medular
Papel da uréia no mecanismo de concentração
urinária
Recirculação medular da uréia
Diluição da urina
DISTÚRBIOS CLÍNICOS DO METABOLISMO DA ÁGUA
DÉFICIT DE ÁGUA — HIPERNATREMIA — ESTADO
HIPEROSMOLAR
Causas de hipernatremia e estado hiperosmolar
Hipernatremia com hipovolemia
Hipernatremia com hipervolemia
Hipernatremia com volemia aparentemente normal
Manifestações clínicas de hipernatremia
Manejo do paciente com hipernatremia
Linhas gerais
Cálculo do déficit de água
Tipo de fluido
Ritmo de correção
Evolução
EXCESSO DE ÁGUA — HIPONATREMIA — ESTADO
HIPOSMOLAR
Causas de hiponatremia
Pseudo-hiponatremia
Redistribuição de água
Intoxicação aguda pela água
Hiponatremia crônica
MANIFESTAÇÕES CLÍNICAS DE HIPONATREMIA
Diagnóstico
TRATAMENTO DA HIPONATREMIA
Linhas gerais
Cálculo do excesso de água
Tratamento da hiponatremia sintomática
Ritmo de correção
Complicações do tratamento
EXERCÍCIOS
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET
RESPOSTAS DOS EXERCÍCIOS
No dia-a-dia, a ingesta de líquidos deve igualar-se àsperdas através da respiração, suor, trato gastrintestinal ediurese.*1 Nos adultos, a água corresponde a 60% do peso
corporal, sendo a maior parte localizada no espaço intra-celular.
Para evitar que haja variações na osmolalidade plasmá-tica, a qual é determinada principalmente pela concentra-ção plasmática de sódio, devem ser feitos ajustes adequa-dos na ingesta e excreção de água. Estes ajustes são reali-zados de forma mais significativa sobre o controle da sede,
*O termo diurese refere-se a um fluxo de urina maior do que o normal,isto é, superior a 1 ml/min no adulto; antidiurese refere-se a um fluxourinário reduzido, geralmente inferior a 0,5 ml/min no adulto.

capítulo 9 101
secreção do hormônio antidiurético (HAD) e mecanismosrenais de conservação ou eliminação de água.1
Quando existe déficit de água no organismo, os rinsparticipam de um sistema de retroalimentação com osmor-receptores e hormônio antidiurético, minimizando a per-da de água. Já quando existe excesso de água no organis-mo, estes mecanismos se dirigem a uma maior excreção deágua pelos rins. 2
MECANISMO DA SEDE
Para equilibrar as perdas diárias de água, é necessáriohaver ingesta de líquido, que é regulada pelo mecanismoda sede. Sede é definida como o desejo consciente de inge-rir água.2
Acredita-se que os estímulos para a sede se originamtanto no compartimento intracelular como no extracelular.A sensação de sede origina-se no centro da sede, localizadonas porções anterior e ventromedial do hipotálamo. Naverdade, os neurônios que compõem o centro da sede sãoespecializados na percepção de variações de pressão osmó-tica do plasma, e por isso recebem a denominação de os-morreceptores. Um dos mais importantes estímulos para asede é o aumento da osmolaridade do líquido extracelu-lar, e o “limiar” para o surgimento da sede é em torno de290 mOsm/L. Nesta situação, os osmorreceptores sofremcerto grau de desidratação, gerando impulsos que são con-duzidos por neurônios especializados até centros corticaissuperiores, onde então a sede se torna consciente.2,3 Estemecanismo é ativado nas situações em que há aumento daosmolalidade do plasma, como no déficit de água e naadministração de soluções hipertônicas cujos solutos nãopenetram nas células.
Por sua vez, déficits no volume extracelular e na pres-são arterial também desencadeiam a sede, por vias inde-pendentes das estimuladas pelo aumento da osmolarida-de do plasma. Por exemplo, depleção do espaço extrace-lular (diarréia, vômitos) e a perda de sangue por hemorra-gia estimulam a sede mesmo sem haver modificação naosmolaridade do plasma. O mecanismo para que isto ocor-ra está relacionado ao estímulo de barorreceptores, que sãoreceptores de pressão existentes na circulação torácica.2 Umterceiro importante estímulo à sede é a angiotensina II.Fitzsimons acredita que a angiotensina e outras substân-cias vasoativas atuem em estruturas vasculares periventri-culares (seriam receptores mecânicos da sede no cérebro),reduzindo o volume vascular a esse nível e causando sede.4
Como a angiotensina II também é estimulada pela hipo-volemia e baixa pressão arterial, seu efeito sobre a sedeauxilia na restauração do volume sanguíneo e pressão ar-terial, juntamente com as ações renais da angiotensina II,reduzindo a excreção de fluidos.2
Alguns outros fatores influenciam a ingesta de água. Porexemplo, a falta de umidade da mucosa oral e do esôfago
desencadeia a sensação de sede. Nesta situação, a inges-tão de água pode provocar alívio imediato da sede, mes-mo antes de ter havido absorção da água no trato gastrin-testinal ou qualquer modificação na osmolaridade do plas-ma. Porém este alívio da sede é de curta duração, e o dese-jo de ingerir água só é efetivamente interrompido quandoa osmolaridade plasmática ou o volume extracelular retor-narem ao normal. De modo geral, a água é absorvida edistribuída no organismo cerca de 30-60 minutos após aingestão. O alívio imediato da sede, apesar de temporário,é um mecanismo que impede que a ingestão de água pros-siga indefinidamente, o que levaria ao excesso de água ediluição excessiva dos fluidos corporais. 2
Estudos experimentais demonstram que os animais nãoingerem quantidades de água superiores às necessáriaspara restaurar a osmolaridade plasmática e volemia ao nor-mal.2 Já em humanos, a quantidade de água ingerida va-ria de acordo com a dieta e a atividade do indivíduo, e emgeral é excessiva em relação às necessidades diárias. Estaingestão excessiva, que não é induzida por um déficit deágua e cujo mecanismo é desconhecido, é extremamenteimportante, pois assegura as necessidades futuras do in-divíduo.
Habitualmente, a sede e a ingesta líquida representamuma resposta normal a um déficit de água. Isto é o queocorre nos exemplos já mencionados, de vômitos, diarréia,diabetes insipidus, diabetes mellitus, hipocalemia, hipercalce-mia etc. No entanto, em algumas situações, o paciente temsede, mas não há um déficit de água. Este estado patológi-co pode ser devido à irritação contínua dos neurônios dasede por tumor, trauma ou inflamação, ingestão compul-siva de água, hiper-reninemia etc.
Hipodipsia (diminuição ou ausência de sede) é usual-mente causada por tumor (p.ex., craniofaringioma, glioma,pinealoma ectópico etc.) ou trauma. Além de afetarem ocentro da sede, estes exemplos podem também ocasionarlesão do sistema supra-óptico-hipofisário, causando diabe-tes insipidus, o que agrava o déficit de água e dificulta omanejo clínico.
VASOPRESSINA (HORMÔNIOANTIDIURÉTICO)
O hormônio antidiurético (HAD) interage com porçõesterminais do nefro, aumentando a permeabilidade destessegmentos à água, desta forma aumentando a conservaçãoda água e a concentração urinária.
Além do aumento da permeabilidade à água nos túbu-los coletores, o HAD tem uma importante participação narecirculação da uréia entre o ducto papilar e a porção finaascendente da alça de Henle, pois aumenta a permeabili-dade do ducto coletor à uréia, e este mecanismo auxilia namanutenção da hipertonicidade da medula renal.5
102 Metabolismo da Água
O HAD é um hormônio sintetizado no hipotálamo porgrupos de neurônios que formam os núcleos supra-ópticoe paraventricular, próximos ao centro da sede. Após a sín-tese, este decapeptídio (arginina-vasopressina em huma-nos) é armazenado em grânulos e transportado ao longodos axônios, em direção à neuro-hipófise (lobo posteriorda hipófise). No interior dos grânulos, o hormônio formaum complexo com uma proteína chamada neurofisina A ouneurofisina II. Parte destes grânulos pode ser liberada rapi-damente, através de exocitose, enquanto os demais servi-riam de estoque.3
A liberação deste hormônio está condicionada a estímu-los, que podem ser osmóticos ou não-osmóticos.
O estímulo osmótico refere-se a uma alteração da osmo-lalidade. Quando ocorre déficit de água no organismo, háum aumento na osmolalidade, reduzindo o volume dascélulas por desidratação celular* (inclusive das células dosnúcleos supra-óptico e paraventricular), estimulando as-sim a liberação do HAD. É necessário ressaltar que os os-morreceptores são estimulados apenas por variações reaisda tonicidade plasmática, isto é, por solutos que não atra-vessam as membranas. Solutos que atravessam as mem-branas celulares, como a uréia (e glicose nas células cere-brais), não aumentam a secreção de HAD.5,6
Por outro lado, quando há excesso de água no organis-mo, a hiposmolalidade que se estabelece inibe a liberaçãodo hormônio antidiurético. Tudo indica que a alteração dovolume celular altera a atividade elétrica dos neurônios dosnúcleos hipotalâmicos, afetando assim a liberação de va-sopressina.
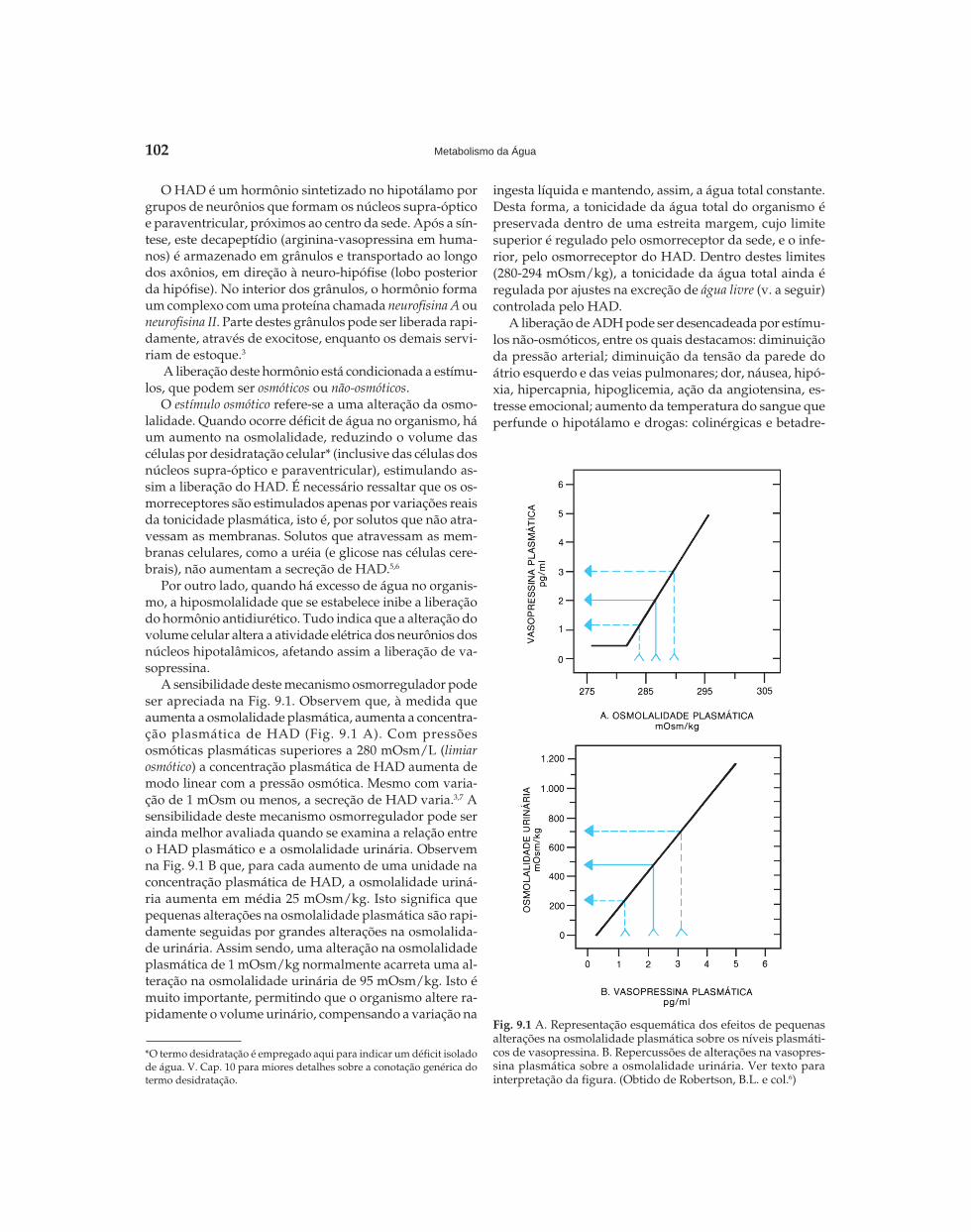
A sensibilidade deste mecanismo osmorregulador podeser apreciada na Fig. 9.1. Observem que, à medida queaumenta a osmolalidade plasmática, aumenta a concentra-ção plasmática de HAD (Fig. 9.1 A). Com pressõesosmóticas plasmáticas superiores a 280 mOsm/L (limiarosmótico) a concentração plasmática de HAD aumenta demodo linear com a pressão osmótica. Mesmo com varia-ção de 1 mOsm ou menos, a secreção de HAD varia.3,7 Asensibilidade deste mecanismo osmorregulador pode serainda melhor avaliada quando se examina a relação entreo HAD plasmático e a osmolalidade urinária. Observemna Fig. 9.1 B que, para cada aumento de uma unidade naconcentração plasmática de HAD, a osmolalidade uriná-ria aumenta em média 25 mOsm/kg. Isto significa quepequenas alterações na osmolalidade plasmática são rapi-damente seguidas por grandes alterações na osmolalida-de urinária. Assim sendo, uma alteração na osmolalidadeplasmática de 1 mOsm/kg normalmente acarreta uma al-teração na osmolalidade urinária de 95 mOsm/kg. Isto émuito importante, permitindo que o organismo altere ra-pidamente o volume urinário, compensando a variação na
ingesta líquida e mantendo, assim, a água total constante.Desta forma, a tonicidade da água total do organismo épreservada dentro de uma estreita margem, cujo limitesuperior é regulado pelo osmorreceptor da sede, e o infe-rior, pelo osmorreceptor do HAD. Dentro destes limites(280-294 mOsm/kg), a tonicidade da água total ainda éregulada por ajustes na excreção de água livre (v. a seguir)controlada pelo HAD.
A liberação de ADH pode ser desencadeada por estímu-los não-osmóticos, entre os quais destacamos: diminuiçãoda pressão arterial; diminuição da tensão da parede doátrio esquerdo e das veias pulmonares; dor, náusea, hipó-xia, hipercapnia, hipoglicemia, ação da angiotensina, es-tresse emocional; aumento da temperatura do sangue queperfunde o hipotálamo e drogas: colinérgicas e betadre-
*O termo desidratação é empregado aqui para indicar um déficit isoladode água. V. Cap. 10 para miores detalhes sobre a conotação genérica dotermo desidratação.
Fig. 9.1 A. Representação esquemática dos efeitos de pequenasalterações na osmolalidade plasmática sobre os níveis plasmáti-cos de vasopressina. B. Repercussões de alterações na vasopres-sina plasmática sobre a osmolalidade urinária. Ver texto parainterpretação da figura. (Obtido de Robertson, B.L. e col.6)

capítulo 9 103
nérgicas (acetilcolina e isoproterenol, respectivamente),morfina, nicotina, ciclofosfamida, barbitúricos etc.2,7 Entreos estímulos não-osmóticos para a liberação do HAD, es-tão os provenientes de áreas onde se encontram receptoresde pressão (barorreceptores): seio carotídeo, átrio esquerdoe veias pulmonares. Eles respondem a variações da pressãosobre a parede do órgão receptor, emitindo impulsos ner-vosos que modulam a liberação hipotalâmica de HAD.Quando há uma menor tensão na parede do órgão, há trans-missão de estímulos para a liberação central de HAD. Istopode ocorrer, por exemplo, na contração do volume extra-celular ou volume circulante efetivo e hipotensão arterial.8
Ao contrário, uma inibição não-osmótica da liberação deADH ocorre quando há: aumento da pressão arterial, au-mento da tensão da parede do átrio esquerdo e das veiaspulmonares, diminuição da temperatura do sangue queperfunde o hipotálamo e uso de algumas drogas (norepi-nefrina, clonidina, haloperidol, difenil-hidantoína, álcool).2
Mecanismo de Ação do HormônioAntidiurético (HAD) — Aquaporinas
O HAD modifica a membrana luminal das células prin-cipais dos túbulos distal final e coletor, causando aumentoda permeabilidade à água. O HAD interage com receptoresespecíficos da superfície (receptores V1 e V2), localizados namembrana basolateral. Esta interação produz efeitos sobreo cálcio e o AMPc intracelulares, que por sua vez modifi-cam a permeabilidade da membrana luminal à água. O re-ceptor V1 existe também no músculo liso vascular, sendoresponsável pelo efeito vasoconstritor do HAD, que por istotambém recebe o nome de vasopressina.5,7
Recentemente, foi evidenciada a existência de uma fa-mília de proteínas de membrana que exercem a função decanais de água em tecidos transportadores de fluidos (porexemplo, no cristalino, nos túbulos renais, etc).3,9 Estes ca-nais de água são hoje conhecidos como aquaporinas. Até omomento, já foram identificadas cinco aquaporinas que seexpressam nos rins (AQP 1, 2, 3, 4 e 6).10 Nas células prin-cipais dos túbulos distais e ductos coletores, está presentea aquaporina 2, que é um canal de água sensível ao HAD.Na presença de HAD, o receptor V2 é estimulado e ativa aadenil ciclase e o AMP cíclico. Com isto, vesículas especí-ficas no citoplasma se movem e se fundem com a membra-na apical (luminal). Estas vesículas contêm a aquaporina2, que, uma vez inserida na membrana luminal das célu-las principais dos túbulos distais e coletores, permite apassagem de água para dentro da célula.11 No bordo baso-lateral das células principais, estão presentes as aquapori-nas 3 e 4, que permitem o transporte de água de dentro dacélula para o interstício, porém neste ponto sem a partici-pação do HAD.5 As aquaporinas 1 e 6 estão relacionadas àabsorção de água, mas em outros segmentos tubulares,também sem dependência do HAD.10
O HAD é o principal hormônio atuante na regulação daexcreção de água. No entanto, outros hormônios afetam aexcreção de água, como veremos na seção seguinte.
Pontos-chave:
• A sede e a liberação de HAD sãodesencadeadas por um aumento daosmolalidade plasmática e têm por objetivomanter a osmolalidade estável
• No rim, o HAD ativa a fusão de canais deágua (aquaporina 2) com a membranaluminal dos túbulos coletores, permitindo areabsorção de água
OUTROS HORMÔNIOS
Catecolaminas
As catecolaminas afetam a excreção de água através deum mecanismo intra-renal e outro extra-renal. No meca-nismo intra-renal, os agentes adrenérgicos alteram a res-posta da membrana tubular renal ao HAD. Assim, os ago-nistas alfadrenérgicos tipo norepinefrina causam aumen-to do volume urinário, por diminuírem o efeito do HADsobre a permeabilidade da membrana tubular renal àágua. Já a estimulação betadrenérgica aumenta a perme-abilidade tubular à água, causando diminuição do volu-me urinário.12
No mecanismo extra-renal, a ação das catecolaminasse faz através de alterações na liberação de HAD, comojá mencionado. Várias outras substâncias vasoativas (an-giotensina II, prostaglandina E1, nicotina) têm efeitossobre os barorreceptores atriais, alterando a liberação deHAD.
Hormônio Tireoidiano
Sabe-se que pacientes hipotireóideos têm comprometi-da a sua capacidade de excretar uma carga de água. Poroutro lado, são desconhecidos os mecanismos pelos quaiso hormônio tireoidiano facilita a excreção de água. Umadas hipóteses é a de que o hormônio tireoidiano altera asensibilidade do túbulo renal ao HAD. Há evidência de quea maioria dos pacientes com hipotireoidismo e hiponatre-mia têm elevada concentração plasmática de HAD. Comoo hipotireoidismo cursa com débito cardíaco habitualmen-te diminuído,13 nestes casos a liberação de HAD pode es-tar sendo estimulada pela redução associada do volume ar-terial efetivo. Também se encontrou queda da taxa de fil-tração glomerular nestes pacientes, o que é revertido coma terapia hormonal apropriada.14

104 Metabolismo da Água
Hormônios AdrenocorticaisNa insuficiência adrenal, pode ser observado um com-
prometimento na excreção de água, cuja causa não estáesclarecida. Alguns autores acreditam que a deficiência deglicocorticóides seja responsável pela deficiente excreçãode água. Segundo eles, a deficiência de glicocorticóidesproduziria alguns efeitos hemodinâmicos sistêmicos (ta-quicardia, diminuição do volume sistólico), e estas altera-ções estimulariam o mecanismo barorreceptor de estímu-lo ao HAD, causando retenção de água.
Também tem sido investigada a participação da deficiên-cia dos mineralocorticóides na diminuição da excreção de águaexistente na insuficiência adrenal. Acredita-se que os minera-locorticóides influenciam a secreção de HAD indiretamente,pois ao manter o volume extracelular evitam a liberação não-osmótica de HAD observada na depleção de volume.
Sistema Renina-AngiotensinaO sistema renina-angiotensina também participa no
controle da secreção de HAD, principalmente quando aosmolalidade plasmática está aumentada. A angiotensinaestimula a liberação de HAD e aumenta a sensibilidade dosistema de osmorregulação.8
MECANISMO RENAL DEREGULAÇÃO DA ÁGUA
O tremendo progresso nesse campo deve-se basicamen-te à aplicação de técnicas de micropuntura in vivo no rim demamíferos, principalmente o rato, e mais recentemente peloavanço da biologia molecular.
Para que seja mantida a homeostase do organismo, énecessário que o rim apresente a capacidade de variar ovolume urinário de modo a reter ou eliminar água, ou seja,concentrar ou diluir a urina.
Diariamente o organismo humano necessita eliminar pro-dutos tóxicos resultantes do metabolismo (p.ex., uréia, ácidosorgânicos) e solutos em excesso (sódio, potássio, cálcio, mag-nésio). A média diária a ser eliminada é de cerca de 750mOsm/dia. Com a ingestão usual de água (2-2,5 L/dia), aosmolaridade urinária encontra-se entre 400 e 450 mOsm/L,o que requer um volume urinário de 1,5 litro/dia. Caso a in-gestão de água seja deficiente, a osmolaridade da urina podesubir até 1.300 mOsm/L, e então o volume urinário vai variarcorrespondentemente, da seguinte forma: 750 mOsm a seremeliminados � osmolaridade de 1.300 � volume urinário de 0,6litro.3 Esta variação decorre do efeito do HAD, conforme jádiscutido, causando a reabsorção de água no ducto coletor.
Da mesma forma, a capacidade de diluir a urina é im-portante para que o organismo elimine excessos de água.Isto é obtido através da redução da osmolaridade da uri-na até valores como 50 mOsm/L.3
Para melhor compreensão dos mecanismos de concen-tração e diluição da urina, vale a pena relembrar algunsconceitos anatômicos.
Considerações Anatômicas
Como sabemos, cada nefro (unidade funcional básicado rim) é constituído pelo glomérulo e por uma forma-ção tubular longa, onde os sucessivos segmentos apresen-tam diferentes características quanto a estrutura e função.Em sua maior parte, os nefros são superficiais, contendoalças de Henle curtas e sem ramo ascendente delgado. Osnefros restantes são justamedulares, e seus glomérulosestão situados próximo à junção corticomedular, possu-indo longas alças de Henle com ramo ascendente delga-do (Fig. 9.2).
Os trabalhos experimentais mostraram que o transpor-te de água e solutos no nefro distal ocorre em pelo menoscinco segmentos morfologicamente distintos: a) Ramo as-cendente espesso da alça de Henle; b) Mácula densa; c)Túbulo contornado distal; d) Ductos coletores corticais ee) Ductos coletores papilares.
O ramo ascendente espesso da alça de Henle estende-se da medula externa até a mácula densa. Este segmentoreabsorve NaCl através de uma membrana impermeávelà água, elaborando, portanto, um líquido hipotônico.
A mácula densa é um segmento mais curto, cujas célu-las parecem agir como sensoras no mecanismo reguladordo feedback túbulo-glomerular (v. Cap. 10).
Na mácula densa, inicia-se o túbulo contornado distal.O túbulo distal clássico sempre foi considerado como o seg-mento que se estende da mácula densa até a junção com
Fig. 9.2 Relação dos vários segmentos do nefro com o córtex e amedula renal.

capítulo 9 105
outro túbulo distal. Recentemente, foi mostrado que estesegmento, na verdade, está formado por dois segmentosdistintos: segmento proximal, cujo epitélio é similar ao doramo ascendente espesso, e segmento distal (também deno-minado túbulo coletor), cujo epitélio é similar ao do ductocoletor cortical15 (v. também Cap. 1).
O segmento distal (túbulo coletor) do túbulo contorna-do distal só responde à ação do hormônio antidiurético emalgumas espécies de animais. Já o segmento cortical do ductocoletor tem uma permeabilidade alta à água na presença deHAD e uma permeabilidade baixa na ausência deste.
A permeabilidade à uréia do segmento cortical do duc-to coletor é baixa, mesmo na presença de HAD. O segmentomedular interno-papilar do ducto coletor tem uma perme-abilidade à uréia mais alta que a do segmento cortical e,na presença de HAD, ela aumenta mais. A permeabilida-de deste segmento medular interno-papilar à água é altana presença de HAD e baixa na ausência deste.
Vascularização da Medula RenalA medula renal pode ser dividida em: a) Medula exter-
na, com uma faixa externa e outra interna (a faixa externaé também conhecida como zona subcortical), e b) Medulainterna (v. Fig. 9.2).
O sangue chega à medula renal através das arteríolaseferentes de glomérulos justamedulares. Estes vasos divi-dem-se na zona subcortical para formarem os vasa rectaarteriais, que atravessam a medula em feixes em forma decone e, às vezes, deixam estes feixes para suprirem umplexo capilar adjacente. Os plexos capilares são drenadospor vasa recta venosos que entram num destes feixes e as-cendem até a base do cone, na zona subcortical (Fig. 9.3).
No rato, uma secção transversal da medula externamostra três zonas concêntricas: a) área central, contendovasa recta arterial e venoso; b) anel periférico, contendo vasarecta venosos e a maioria dos ramos descendentes das al-ças de Henle, e c) por fora do anel, o ramo ascendente daalça de Henle, ducto coletor e plexo capilar.16
Acredita-se que os vasa recta têm a função de remover olíquido absorvido dos ductos coletores e segmento descen-dente da alça de Henle. O fluxo de plasma na parte terminaldos vasa recta ascendentes é maior que o fluxo de plasma naentrada dos vasa recta descendentes, e esta diferença é igualao ritmo de absorção de líquido do segmento descendente daalça de Henle e do ducto coletor. Isto é necessário, pois nãose conhece nenhuma outra via pela qual a água reabsorvidapossa chegar da medula à circulação sistêmica.
Concentração da Urina — Mecanismode Contracorrente
Recorde-se que são 180 litros de líquido filtrados pelos rinsdiariamente e que apenas 1,5 litro é excretado na urina. Isto
significa que, num adulto, aproximadamente 100 ml de fil-trado glomerular chegam aos túbulos proximais a cada mi-nuto. A maior parte da água filtrada (60 a 70%) é reabsorvidano túbulo contornado proximal, acompanhando a reabsor-ção de NaCl. Portanto, neste segmento a absorção de água épassiva. Cerca de 10% são reabsorvidos na pars recta do tú-bulo proximal pelo mesmo mecanismo. No ramo descendentedelgado da alça de Henle, ocorre a reabsorção (10 a 15%) deágua livre (sem soluto), devido ao gradiente osmótico exis-tente entre o túbulo e o interstício medular. Este gradienteosmótico se estabelece graças a um sistema de contracorren-te multiplicador (v. a seguir). O restante é reabsorvido nosductos coletores, sob a influência do hormônio antidiurético.O líquido que atinge o túbulo contornado distal é semprehipotônico e a eliminação de urina concentrada ou diluídadepende da reabsorção de água nos ductos coletores.
Foi observado inicialmente, em vários mamíferos, queo grau de concentração urinária por eles alcançado estavarelacionado com o comprimento do segmento delgado dasalças de Henle. Posteriormente, comprovou-se que apenasmamíferos e alguns pássaros podiam elevar a concentra-ção de urina acima da do plasma e que estes animais pos-suíam alças de Henle medulares (portanto, longas). Estefato sugeriu que a concentração de urina deveria ocorrerno interior das alças de Henle.
Fig. 9.3 Esquema da estrutura da medula renal no rato (zona in-terna e zona externa). VRA = vasa recta arteriais; VRV � vasa rectavenosos; RD � ramo descendente da alça de Henle; RA � ramoascendente da alça de Henle; DC � ducto coletor. (Modificadode Kriz, W. e Lever, A.F.16)

106 Metabolismo da Água
A hipótese do sistema de contracorrente multiplicador paraexplicar a concentração de urina ao longo dos túbulos foisugerida em 1942 por Werner Kuhn, baseada na configu-ração em U da alça de Henle. Ele observou que, devido aesta configuração, o líquido tubular fluiria em ramos ad-jacentes, mas em direções opostas. Sendo um físico-quími-co familiarizado com termodinâmica, ele sabia que um flu-xo contracorrente poderia estabelecer grandes gradientesde temperatura ao longo do eixo longitudinal de canaisadjacentes, enquanto são pequenos os gradientes de tem-peratura entre canais transversais (v. Fig. 9.5).17 Transpor-tando estes princípios para a pressão osmótica, ele imagi-nou que pequenas diferenças na concentração de solutosentre os dois ramos da alça de Henle poderiam resultar emgrandes diferenças de concentração ao longo dos túbulos.Além do mais, ele achou que estas grandes diferenças deconcentração poderiam ser transmitidas ao interstício quecerca os túbulos, criando assim um aumento progressivona concentração de soluto, paralelo aos túbulos.
Haveria necessidade, no entanto, de três fatores básicospara que o sistema de contracorrente multiplicador funci-onasse: a) fluxo contracorrente (proporcionado pela alça deHenle); b) diferenças de permeabilidade entre os túbulos(o ramo ascendente é praticamente impermeável à água),e c) uma fonte de energia (atualmente atribuída ao trans-porte ativo de cloro no ramo ascendente espesso).
Na presença destes elementos, o líquido tubular seriaconcentrado da seguinte maneira (Fig. 9.4):
1. No segmento espesso ascendente da alça de Henle, háuma reabsorção ativa de cloro. Esta reabsorção ativa criauma diferença transtubular de potencial elétrico, que éresponsável pela remoção passiva de sódio.
2. O segmento ascendente espesso tem uma baixa perme-abilidade à água, o que permite que o fluido tubularneste segmento se torne hiposmótico em relação ao dointerstício. No entanto, a uréia permanece no interior dotúbulo, pois este segmento tem uma permeabilidadebaixa à uréia.
3. No ducto coletor cortical já existe ação do HAD, e, napresença deste, a água é reabsorvida, tornando o líqui-do tubular isosmótico com o sangue. A permeabilida-de deste segmento à uréia é baixa, e, com a perda deágua, a concentração intraluminal de uréia aumentaainda mais.
4. Na medula externa, o interstício hiperosmolar (osmo-lalidade determinada em parte pela reabsorção de NaClno segmento ascendente espesso) retira mais água dolíquido tubular, aumentando ainda mais a concentraçãode uréia.
5. Na medula interna, tanto a água como a uréia são reab-sorvidas do ducto coletor na presença do HAD. Este
Fig. 9.4 Sistema de contracorrente multiplicador.* O diagrama mostra os ramos descendente e ascendente da alça de Henle, o túbulo distale o ducto coletor. O contorno mais espesso do ramo ascendente da alça de Henle indica que este ramo é impermeável à água. 1. Reabsorçãoativa de cloro e passiva de sódio, mecanismo que dilui o líquido tubular e torna o interstício medular hiperosmótico. 2. No segmento distal(túbulo coletor) do túbulo distal (em algumas espécies de animais) e � no ducto coletor, ocorre reabsorção de água através de um gradi-ente osmótico. A presença de HAD (v. texto) facilita este transporte passivo. Com a reabsorção de água, ocorre concentração intratubularda uréia. Na medula interna, a água e a uréia são reabsorvidas. 3. O acúmulo da uréia no interstício medular cria o gradiente osmótico paraa reabsorção passiva de água no ramo descendente da alça de Henle � e, assim, concentra o NaCl no ramo descendente da alça de Henle.O tamanho das letras dos solutos indica-lhes a concentração relativa.*Baseado na hipótese de Stephenson19 e Kokko e Rector.20

capítulo 9 107
segmento (medular interno do ducto coletor) tem umapermeabilidade mais alta à uréia do que o segmentocortical do ducto coletor; esta permeabilidade aumentamais na presença de HAD. Este segmento apresentauma permeabilidade alta à água na presença de HAD ebaixa na sua ausência.
6. O cloreto de sódio e a uréia no interstício exercem umaforça osmótica para retirar água do segmento delgadodescendente da alça de Henle. Este segmento é relati-vamente impermeável a uréia e NaCl. Esta perda deágua faz aumentar a concentração de NaCl no ramodescendente delgado, de tal forma que, na curva da alça,a concentração de NaCl será maior no interior do túbu-lo do que no interstício. No entanto, o líquido tubular aesse nível é isosmótico com o interstício papilar, cujaconcentração total de soluto está na maior parte consti-tuída pela uréia.
7. Quando líquido tubular atingir o ramo ascendente del-gado da alça de Henle (segmento impermeável e per-meável ao NaCl), o NaCl passará passivamente para ointerstício (devido ao gradiente de concentração). Comoa permeabilidade deste segmento é mais alta para oNaCl do que para a uréia, o NaCl sai do túbulo para ointerstício mais rapidamente que a uréia quando estapassa do interstício para o interior do túbulo. Com oaumento da concentração de NaCl no interstício, have-rá maior absorção de água na porção fina descendenteda alça, com conseqüente maior hipertonicidade do flui-do tubular, o que gera um maior fluxo de Na� e Cl� noramo fino ascendente da alça de Henle, constituindoassim um sistema de contracorrente multiplicador, apa-rentemente passivo na medula interna, que foi iniciadoe mantido pelo transporte de Na� e Cl� na porção es-pessa da alça na região medular externa.
8. O ramo espesso ascendente recebe, portanto, um flui-do diluído, que se tornará ainda mais diluído em virtu-de da reabsorção de NaCl neste segmento.
A urina final pode alcançar uma concentração próxima,mas não exceder a concentração do interstício medular. Nohomem, em condições de antidiurese, a concentração uri-nária máxima alcançada é de aproximadamente 1.200-1.300mOsm/kg, ou seja, quatro vezes a osmolalidade do plasma.
Apesar do progresso alcançado nos últimos anos emrelação aos mecanismos de concentração da urina, muitosaspectos ainda permanecem sem solução. Atualmente,aceita-se que a alça de Henle é o elemento multiplicadorno sistema de contracorrente e que o segmento delgado daalça é o multiplicador na medula interna.18 Pouca dúvidaresta também de que o segmento delgado ascendente daalça é a fonte de NaCl responsável pelo aumento na con-centração de NaCl desde a base da medula interna até apapila.18 A incerteza permanece em relação ao mecanismode reabsorção do NaCl no segmento delgado ascendente:se ativo ou passivo. Nos últimos anos, vários modelos ex-perimentais tentaram solucionar o problema, como os de
Stephenson,19 e ainda de Kokko e Rector.20,21 A descriçãoutilizada acima para o mecanismo de concentração do lí-quido tubular baseou-se no modelo de Kokko e Rector, queparte do pressuposto que não há um transporte ativo namedula interna (segmento delgado ascendente), no que dizrespeito ao mecanismo de concentração.
FLUXO SANGUÍNEO MEDULARComo já mencionamos, acredita-se que os vasa recta têm
a função de remover o líquido absorvido nos ductos cole-tores e segmento descendente da alça de Henle. Natural-mente, o fluxo sanguíneo medular deve ser de tal ordemque os solutos do interstício não sejam excessivamenteremovidos, o que eliminaria o gradiente osmótico medu-lar, tão importante na concentração urinária. Sabe-se, pois,que a concentração osmolar na ponta da papila é inversa-mente proporcional ao fluxo sanguíneo para esta área.
A manutenção deste interstício hiperosmolar deve-se:a) a um baixo fluxo sanguíneo medular (apenas 5% do flu-xo plasmático renal passam pela área medular e papilar);b) à presença dos vasa recta, responsáveis por um sistemade contracorrente trocador. A disposição anatômica da cir-culação capilar na medula tem todas as características deum sistema de contracorrente trocador.
O princípio deste sistema, conhecido em termodinâmi-ca, tem sido aplicado a sistemas biológicos e está ilustradona Fig. 9.5. Suponhamos um tubo ao qual fornecemos águaa 30°C e a um fluxo de 10 ml/min (Fig. 9.5 A). Esta águapassa por uma fonte de calor e recebe 100 calorias por mi-nuto. Logo, a água que sai do tubo está a uma temperatu-ra de 40°C. A seguir, dobramos o tubo, introduzindo, por-tanto, um fluxo contracorrente no sistema e mantendo afonte de calor no mesmo local (Fig. 9.5 B). O sistema émontado de tal maneira que o fluxo de saída passa próxi-mo do fluxo de entrada, propiciando a troca de calor entreos dois fluxos (entrada e saída). Desta forma, a água aque-cida (que está saindo) encontra a água fria (que está entran-do) e perde calor para ela. Portanto, a temperatura da águaque entra se eleva antes de atingir a fonte de calor. O pro-cesso continua até que se atinja um estado de equilíbrio. Atemperatura máxima alcançada no sistema de contracor-rente é maior que no fluxo retilíneo.
As mesmas considerações são válidas para a adição desoluto em vez de calor (Fig. 9.5 C). O soluto (NaCl) é adi-cionado ao interstício e o equilíbrio entre os capilares se fazatravés do interstício. A finalidade deste sistema é facili-tar ao máximo a transferência de uma molécula permeá-vel entre canais adjacentes, evitando o movimento dasmoléculas ao longo desses canais.
A arquitetura vascular da medula renal facilita a trocade água e solutos entre os vasa recta ascendentes e descen-dentes, minimizando a entrada de água e saída de solutoda medula renal da seguinte maneira22 (v. Fig. 9.6).
1. O sangue circula pelos vasa recta através do interstíciomedular, progressivamente mais hiperosmolar em dire-

108 Metabolismo da Água
ção à papila. A pressão hidrostástica transcapilar favore-ce a saída de líquido do capilar, e a pressão oncótica trans-capilar favorece a entrada de líquido para o capilar. Comoo sangue circula rapidamente, não há tempo para umequilíbrio osmótico entre o capilar e o interstício.
2. Como a concentração dos solutos no interstício é mai-or, a pressão osmótica transcapilar favorece a saída deágua do capilar descendente, aumentando a concentra-ção das proteínas plasmáticas.
3. Como os capilares são permeáveis a NaCl e uréia, e aconcentração destes no interstício é maior que no capi-lar, eles entram no capilar descendente.
4. Quando o sangue atinge o capilar ascendente, a concen-tração de solutos no plasma excede a do interstício (quese torna progressivamente menos hiperosmolar em di-reção ao córtex), e os solutos, então, deixam o capilar.
5. Da mesma forma, a pressão oncótica (determinada pe-las proteínas plasmáticas) está elevada quando o san-gue atinge o capilar ascendente. A soma da pressãooncótica e da pressão osmótica (determinada pelos so-lutos não-protéicos) determina a entrada de líquido nocapilar.
6. A quantidade de líquido que entra no capilar ascendenteé maior que a quantidade de líquido removida do capi-lar descendente, e a diferença é igual ao volume de lí-quido reabsorvido no ramo descendente da alça deHenle e nos ductos coletores.
7. Em resumo, os vasa recta preservam os solutos e remo-vem a água, mantendo a hiperosmolalidade da medularenal.
PAPEL DA URÉIA NO MECANISMO DECONCENTRAÇÃO URINÁRIA
A uréia é o produto final do metabolismo protéico nosmamíferos, sendo excretada quase unicamente pelos rins.Além da água e dos gases sanguíneos, a uréia é a substân-cia mais difusível no organismo.
Investigações passadas já haviam demonstrado que apresença de uréia era essencial para a obtenção de uma
Fig. 9.5 Princípios do sistema de contracorrente trocador. Observem que a temperatura máxima obtida no sistema de contracorrente(B) é maior que a obtida no sistema de fluxo linear (A). Em (C), representamos uma alça capilar em contato com o líquido intersticial.Notem que, no início (flechas), os sais de sódio penetram no capilar e, no final, retornam para o interstício (v. texto para uma expli-cação mais detalhada). (Modificado de Berliner R.W. e col.17)
Fig. 9.6 Sistema de contracorrente trocador pelos vasa recta. Pr �proteína plasmática. O tamanho das letras dos solutos indica aconcentração relativa de cada soluto com relação à sua localiza-ção na medula (v. texto para detalhes de funcionamento do sis-tema). Obtido de Jamison, R.L. e Maffly, R.H.22

capítulo 9 109
osmolalidade urinária máxima. Se um animal deficiente emproteínas recebia uréia, a capacidade de concentração uri-nária aumentava.
RECIRCULAÇÃO MEDULAR DA URÉIA
1. Uma quantidade mais ou menos constante de uréia éreabsorvida no túbulo proximal, independentemente dobalanço de água.
2. No ducto coletor cortical (e, em algumas espécies, notúbulo coletor), sob a influência do hormônio antidiuré-tico, a água é reabsorvida, o que determina um aumen-to da concentração intraluminal de uréia (Fig. 9.4).
3. No segmento medular interno-papilar do ducto coletor,a permeabilidade à uréia aumenta mesmo na ausênciado HAD, o qual, quando presente, parece aumentar ain-da mais esta permeabilidade. Desta forma, devido àdiferença transtubular da concentração de uréia, esta sedifunde para o interstício medular.
4. A uréia, então, torna a entrar no túbulo renal na pars rectado túbulo proximal ou ramo descendente de nefros su-perficiais e justamedulares. Como a alça delgada justa-medular está numa região contendo uma alta concen-tração de uréia no interstício, mais uréia entra no nefrojustamedular do que no superficial. Portanto, o fluxo deuréia que deixa o túbulo distal justamedular é maior doque o que deixa o nefro superficial.
Pontos-chave:
• Quando existe déficit de água, os rinsreabsorvem mais água pelo mecanismo deconcentração urinária, estimulado peloHAD
• A concentração urinária depende damanutenção de uma medula renalhipertônica pelo mecanismo decontracorrente e recirculação de uréia
Diluição da Urina
Não importa se a urina final será hiper- ou hipotônica:o líquido tubular que chega ao túbulo contornado distalserá sempre hipotônico. Os ductos coletores (segmento cor-tical e medular interno-papilar) e o segmento distal dotúbulo contornado distal são segmentos sensíveis à açãodo HAD. Quando há uma redução ou cessação na libera-ção de HAD, estes segmentos tornam-se relativamenteimpermeáveis à água. Em conseqüência, no sistema cole-tor o líquido hipotônico permanece hiposmótico em rela-ção ao plasma. No segmento medular interno-papilar doducto coletor, ocorre reabsorção de água, pois o segmentoainda é permeável à água (embora menos) na ausência deHAD.
Devido à ausência de HAD, a permeabilidade à uréiado segmento medular interno-papilar do ducto coletordiminui; logo, a reabsorção de uréia também diminui.Além disso, como há redução geral na reabsorção de água,o gradiente transtubular de uréia também diminui (recor-de-se que é a reabsorção de água dos segmentos poucopermeáveis à uréia que determina o aumento de sua con-centração intratubular), e logo se reduz a recirculaçãomedular do sistema coletor para a alça de Henle. E, comojá foi exposto, a uréia exerce um papel fundamental no sis-tema de contracorrente.
A capacidade de um indivíduo ingerir grande quantida-de de água, sem desenvolver um excesso de água, traduz acapacidade renal de excretar grande quantidade de urinadiluída. A osmolalidade mínima que pode ser alcançadapelo rim humano é de aproximadamente 50 a 60 mOsm/kg, permitindo volumes de urina de 15 a 20 litros por dia.
É necessário frisar alguns pontos importantes no meca-nismo de diluição da urina e expor os conceitos de clearan-ce osmolar e clearance de água livre.
Baseando-se no que já foi exposto nas páginas preceden-tes, conclui-se que a formação e a excreção de uma urinadiluída dependem de três fatores básicos: a) oferta adequa-da de líquido tubular ao segmento diluidor do nefro; b) re-absorção adequada de soluto no segmento diluidor do nefro;c) impermeabilidade do segmento diluidor do nefro à água.
Se analisarmos a urina, veremos que ela está constituí-da por uma fase aquosa na qual vários solutos estão dis-solvidos. Os solutos são ânions e cátions não-voláteis e osprodutos do metabolismo nitrogenado. Se relacionarmosa concentração destes solutos na urina (ou seja, a osmola-lidade urinária) com a osmolalidade plasmática, podere-mos ter três tipos de tonicidade urinária: urina isotônica,hipotônica e hipertônica em relação ao plasma (v. Fig. 9.7).Foi Homer Smith quem originalmente considerou a urinacomo contendo dois volumes virtuais: um volume conten-do uma quantidade de soluto excretado numa concentra-ção igual à do plasma (isotônica) e um outro volume con-tendo água sem soluto.23
Quando se considera o fluxo urinário (ml de urina porminuto), o volume de urina que contém os solutos numaconcentração igual à do plasma é denominado de clearan-ce osmolar e o volume de urina sem solutos refere-se ao cle-arance de água livre. O termo clearance de água livre é errô-neo, pois, na verdade, não indica a depuração de uma subs-tância e não é calculado pela fórmula clássica U � V/P, esim pela fórmula:
CH2O � V � Cosm
Onde:
CH2O � clearance de água livreV � volume de urina (fluxo urinário em ml/min)Cosm � clearance osmolar

110 Metabolismo da Água
Considerando de outra maneira, podemos dizer que o cle-arance de água livre refere-se à quantidade de água livre (águasem solutos) que precisa ser adicionada ou retirada da urinapara que a urina se torne isosmótica com o plasma.
Observem na Fig. 9.7 B que, quando a urina é isotônica,isto é, tem a mesma concentração osmolar que o plasma, oclearance de água livre é zero. Já na urina hipotônica, o clea-rance de água livre é positivo e, na hipertônica, negativo.Costuma-se empregar a expressão TCH2O quando o clearancede água livre for negativo. A letra C indica que a reabsorçãoocorre nos ductos coletores. Portanto, TCH2O � �CH2O.
O clearance osmolar, que se refere ao volume de urinanecessário para excretar todos os solutos urinários numaproporção isosmótica, é calculado através da fórmula clás-sica do clearance:
Cosm � Uosm � V
Posm
Onde:
Cosm � osmolalidade urinária (mOsm/L)V � fluxo urinário (ml/min)Posm � osmolalidade plasmática (mOsm/L)
Vejamos, nos dois exemplos seguintes, o cálculo do cle-arance osmolar e do clearance de água livre.1. Calcular o Cosm de um paciente que apresenta osmolali-
dade plasmática de 300 mOsm/L, osmolalidade uriná-ria de 100 mOsm/L e fluxo urinário de 5 ml/min:
Cosm � 100 � 5
� 1,66 ml/min300
2. Calcular o clearance de água livre de um paciente cujaurina apresenta osmolalidade de 600 mOsm/L, osmo-lalidade plasmática de 300 mOsm/L e fluxo urinário de1 ml/min:
CH2O � 1 � 600 � 1
� � 1300
Interpretação do clearance osmolar e do clearance deágua livre
É óbvio que variações na ingesta e na excreção osmolarnão causarão alterações na osmolalidade plasmática (poisa fração osmolar é sempre isosmótica). No entanto, paraque a osmolalidade seja mantida, a fração de água livreingerida deverá ser igual ao clearance de água livre. Se aingestão de água livre exceder o clearance de água livre,haverá uma diminuição da osmolalidade plasmática. Ficaclaro, portanto, a importância do mecanismo renal de di-luição da urina (excreção de água livre) na preservação daosmolalidade plasmática.
Pontos-chave:
• A diluição urinária é resultado daimpermeabilidade dos túbulos coletores àágua na ausência de HAD
• A excreção dos excessos de água é realizadaatravés da elaboração de urina finaldiluída
Fig. 9.7 Relação do clearance de água livre com a tonicidade da urina (v. texto). (Modificado de Hays, R.M. e Levine, S.D.33)
(significa urina hipertônica)

capítulo 9 111
DISTÚRBIOS CLÍNICOS DOMETABOLISMO DA ÁGUA
A integração do sistema sede-HAD-rim permite que mes-mo com grandes variações na ingesta líquida a osmolalidadeno organismo seja mantida mais ou menos constante. Quan-do há déficit de água, ocorre aumento da osmolalidade noorganismo, a qual estimula a sede e a liberação de HAD; estaaltera a permeabilidade do epitélio do ducto coletor, permi-tindo maior conservação de água. Na presença de excesso deágua, ocorre o inverso: hiposmolalidade, ausência de sede emenor liberação de HAD e conseqüente menor permeabilida-de à água no ducto coletor, causando, portanto, maior diure-se. Daí se deduz que alterações no mecanismo de concentra-ção e diluição da urina provocam distúrbios no metabolismoda água, que são a hipernatremia e a hiponatremia.
É importante também relembrar que os distúrbios dometabolismo da água estão relacionados a alterações naosmolalidade plasmática e são evidenciados pela dosagemdo sódio plasmático, o qual estará concentrado ou diluídono plasma, de acordo com a água corporal total do indiví-duo. Já os distúrbios do metabolismo do sódio são verifi-cados pela avaliação do estado do espaço extracelular, atra-vés do exame físico (v. Caps. 8 e 10).24
O termo desidratação refere-se à perda de água que levaa uma elevação do sódio plasmático e a um déficit de águaintracelular devido ao movimento de água das células parao líquido extracelular. Já o termo depleção de volume serefere à diminuição do espaço extracelular devido à perdade sódio e água, como ocorre, por exemplo, nas diarréias.24,25
DÉFICIT DE ÁGUA —HIPERNATREMIA —
ESTADO HIPEROSMOLAR
Hipernatremia ocorre quando a concentração plasmá-tica de sódio encontra-se acima de 145 mEq/L. A hiperna-tremia é um dos distúrbios eletrolíticos mais comuns empacientes hospitalizados. Chega a ser preocupante que,nesta população, uma importante causa de hipernatremiaé a iatrogenia, por reposição inadequada das perdas empacientes com acesso restrito à água.26
Um déficit de água no organismo é acompanhado porum aumento na concentração plasmática de sódio. Comojá foi abordado no Cap. 8, o sódio é o principal íon deter-minante da osmolalidade no compartimento extracelular,de forma que a hipernatremia tem grande importância clí-nica, por sua associação com hiperosmolaridade e conse-qüentes efeitos sobre o conteúdo celular de água. A hiper-natremia é a principal causa de hiperosmolaridade.
Uma série de adaptações ocorre em todo o organismopara minimizar o efeito da hiperosmolaridade sobre a es-
trutura e a função da célula, especialmente no cérebro. Ossintomas de hiperosmolaridade aparecem quando estesmecanismos de adaptação são ultrapassados.27
A membrana celular é de modo geral altamente perme-ável à água, o que torna o volume intracelular muito sus-cetível às variações da osmolaridade do extracelular. Ahiperosmolalidade induz um movimento de água do in-tracelular para o extracelular, reduzindo o volume celular.Esta alteração no volume celular leva a mudanças no vo-lume e função celulares.
Por razões anatômicas, o cérebro é especialmente vulne-rável às alterações no volume celular. Reduções agudas novolume cerebral podem levar a uma separação entre o cére-bro, as meninges e o crânio, com ruptura de vasos sanguíne-os e hemorragia. Porém, no cérebro, os astrócitos são capa-zes de restaurar o volume cerebral ao normal após transtor-nos osmóticos. No caso da hipernatremia, após algum tem-po estas células respondem com um aumento na concentra-ção intracelular de vários solutos osmoticamente ativos, in-cluindo o sódio, o potássio, o cloro. Além destes, progressi-vamente há acúmulo também dos chamados osmóis idiogêni-cos, que incluem aminoácidos (glutamato, glutamina, tauri-na, ácido gama-aminobutírico), creatina, fosfocreatina,mioinositol e glicerofosforilcolina. Na hipernatremia aguda,por não ter havido tempo suficiente para o acúmulo destassubstâncias, que manteriam o volume celular, é mais prová-vel ocorrer variação do volume celular cerebral, com mani-festações clínicas importantes. Na hipernatremia crônica, es-tes osmóis acumulados no interior das células levam à ma-nutenção do volume celular, com menor sintomatologia.27
Os outros mecanismos de adaptação à hipernatremiasão a liberação de HAD e a ativação do mecanismo dasede.27 Normalmente, o centro da sede é muito sensívelmesmo a pequenos aumentos da osmolalidade, da ordemde 1 a 2%. Porém, mesmo que o mecanismo da sede sejaativado, muitos pacientes podem não expressar a sedeadequadamente ou não ter acesso à água. Isto é observa-do em crianças pequenas e adultos com alterações do ní-vel de consciência, principalmente idosos. Além disso, acapacidade de concentração urinária e conservação de águadiminuem com a idade, e, nos idosos, a osmolalidade uri-nária máxima pode ser de apenas 500-700 mOsm/kg.28-30
Então, vários fatores tornam estes indivíduos mais propen-sos ao desenvolvimento de hipernatremia significativa.
Pontos-chave:
• Hipernatremia é diagnosticada comconcentração plasmática de sódio maior que145 mEq/L
• Hipernatremia produz hiperosmolalidade,uma vez que o sódio é o principaldeterminante da osmolalidade plasmática

112 Metabolismo da Água
Causas de Hipernatremia eEstado Hiperosmolar
No Quadro 9.1 podem ser observadas as principais cau-sas de hipernatremia. Uma abordagem também bastantedidática se baseia na determinação do estado do espaçoextracelular nos pacientes com hipernatremia, agrupandoas causas mais prováveis do distúrbio de acordo com avolemia do paciente e o sódio urinário31 (v. Quadro 9.10).
A hipernatremia é uma das causas de estado hiperos-molar, o qual pode também ser ocasionado por uréia, gli-cose e etanol.
HIPERNATREMIA COM HIPOVOLEMIAHipernatremia com depleção do espaço extracelular e
hipovolemia pode ser decorrente de perdas extra-renais ou
renais de fluidos hipotônicos.31 Há uma perda concomitan-te de água e sódio, embora haja proporcionalmente umamaior perda de água. Clinicamente, observam-se sinais decontração de volume: veias jugulares invisíveis, hipoten-são ortostática, taquicardia, pobre turgor da pele e muco-sas secas. Devido à hemoconcentração, o hematócrito e asproteínas plasmáticas estão elevados.
Perdas extra-renais podem ser decorrentes de sudoreseexcessiva ou diarréia, particularmente em crianças. Emalguns tipos de diarréia, principalmente nas osmóticas,ocorre perda de fluido hipotônico em relação ao plasma,provocando aumento na concentração plasmática de sódio.Isto pode ser observado também em crianças em que o flui-do de reposição é hipertônico. Como resposta às perdas,os rins são estimulados a conservar água e sódio, a urinamostra-se hipertônica e a concentração urinária de sódio ébaixa, menor que 20 mEq/L.31
Por sua vez, perda de fluidos hipotônicos pelos rinspode ser observada durante a diurese osmótica, como ocor-re na administração de manitol e no paciente diabéticodescompensado, com glicosúria. A glicosúria é a principalcausa de diurese osmótica em pacientes ambulatoriais.Não se evidencia conservação renal de água e sódio, poisa urina é justamente a fonte de perda. A urina pode ser iso-ou hipotônica e o sódio urinário é maior que 20 mEq/L.Em pacientes hospitalizados, outras causas de diurese os-mótica são encontradas: alimentação hiperprotéica (a uréiaage como agente osmótico); expansão do volume por so-lução salina e liberação de obstrução urinária bilateral. Aosmolalidade urinária nestas situações está geralmenteacima de 300 mOsm/kg, ao contrário da urina diluída dadiurese aquosa. Além do mais, a excreção de solutos (pro-duto da urina de 24 h � volume � osmolalidade) é nor-mal na diurese aquosa (600-900 mOsm/kg/dia) e aumen-tada na diurese osmótica.
HIPERNATREMIA COM HIPERVOLEMIAEsta categoria de hipernatremia é pouco freqüente. Ge-
ralmente ocorre em pacientes que receberam grandesquantidades de cloreto ou bicarbonato de sódio hipertô-nico. Ao exame físico há sinais do excesso de extracelular,
Fig. 9.8 Relação entre a osmolalidade plasmática e a ingesta e excreta osmolar e de água livre. Como a fração osmolar é sempre umafração isotônica, não há alterações na osmolalidade plasmática quando se modifica a ingesta ou excreta da fração osmolar. No en-tanto, variações na ingesta ou excreta de água livre modificam a osmolalidade plasmática. (Baseado no diagrama de Hays, R.M. eLevine, S.D.33)
Quadro 9.1 Causas de hipernatremia
Perda de água• Perdas insensíveis (respiração e sudorese)• Hipodipsia• Diabetes insipidus central• Diabetes insipidus nefrogênico
Perda de fluido hipotônico• Perdas renais
• Diurese osmótica• Diuréticos de alça• Fase poliúrica de NTA• Diurese pós-obstrutiva
• Perdas gastrintestinais• Vômitos, sondagem nasogástrica• Diarréia• Catárticos osmóticos
• Perdas cutâneas• Queimaduras
Sobrecarga de sódio• Administração de soluções hipertônicas de sódio• Enemas ricos em sódio• Hiperaldosteronismo primário• S. de Cushing

capítulo 9 113
como congestão pulmonar e ingurgitamento dos vasos dopescoço.31
HIPERNATREMIA COM VOLEMIAAPARENTEMENTE NORMAL
Este é o tipo mais freqüente de hipernatremia, e se devea perdas de água sem eletrólitos. Ao exame, o espaço ex-tracelular pode ser considerado normal. Devido à per-meabilidade das membranas celulares à água, um terçoda água perdida provém do extracelular, e dois terços, dointracelular. É por isso que a principal conseqüência daperda de água é a hipernatremia, e não a depleção do ex-tracelular.31
Hipernatremia com volemia normal pode ser decorrentede perdas insensíveis pelo suor e respiração, que, se nãoforem apropriadamente repostas, elevam a concentraçãoplasmática de sódio. Estas perdas em geral somam 0,6 ml/kg/hora, mas aumentam muito nas queimaduras, febre,taquipnéia e exercícios intensos.32
É causada principalmente por distúrbios que prejudi-cam os mecanismos normais de conservação renal de água,por baixa concentração plasmática de hormônio antidiuré-tico (diabetes insipidus pituitário ou central) ou por compro-metimento da resposta renal a níveis máximos de HAD(diabetes insipidus nefrogênico).
Se a perda líquida for através da pele e do trato respira-tório, a urina será hipertônica. A quantidade de sódio uri-nário é variável e reflete a ingesta diária. Se a perda líqui-da for de origem renal (diabetes insipidus central ou nefro-gênico), a urina será hipotônica, e a quantidade de sódiourinário, também variável.
Ponto-chave:
• Hipernatremia pode cursar com espaçoextracelular normal, diminuído ouaumentado
Diabetes insipidus (DI) pituitário ou centralCaracteriza-se por uma alteração central na síntese ou
secreção de HAD, limitando a capacidade renal de concen-trar a urina e causando graus variados de poliúria e poli-dipsia. A falta de HAD pode ser induzida por distúrbiosem um ou mais locais de secreção do HAD: osmorrecep-tores hipotalâmicos, núcleos supra-óticos ou paraventricu-lares; ou a porção superior do trato supra-ótico hipofisá-rio. Por outro lado, lesão do trato abaixo da eminênciamédia ou da parte posterior da hipófise produz apenasuma poliúria transitória. Nestes casos, o HAD produzidono hipotálamo ainda pode ser secretado na circulação sis-têmica através dos capilares portais da eminência média.
CAUSAS. As cirurgias de hipófise, tumores supra-se-lares e traumatismo craniano são causas de DI central33 (v.Quadro 9.2). As neoplasias primárias ou secundárias do
cérebro, que envolvam a região pituitário-hipotalâmica,podem cursar com DI central. Isto ocorre mais freqüente-mente com metástases de câncer de pulmão, leucemia oulinfoma. A incidência de DI varia com a extensão da lesão:10-20% na remoção transesfenoidal de adenoma hipofisá-rio restrito à cela e até 60-80% nos casos de grandes tumo-res que requerem hipofisectomia total. Alguns pacientesapresentam um padrão trifásico de polidipsia-poliúria nopós-operatório: na primeira fase, imediata à cirurgia, os pa-cientes apresentam polidipsia-poliúria; segue-se a segun-da fase, caracterizada por quatro a cinco dias de antidiu-rese; e, após vários dias, uma terceira fase, na qual a poli-úria reaparece. Acredita-se que, na primeira fase, ocorrauma lesão aguda dos núcleos hipotalâmicos e que, portan-to, não haja síntese e liberação de vasopressina. Já a segun-da fase ocorreria devido à liberação de vasopressina pelotecido neuro-hipofisário necrosado. Nesta fase, entre osdias 6 e 11, ingestão excessiva de água pode causar hipona-tremia. Pacientes com lesões menos graves podem ter umDI central transitório que começa 24-48 horas depois da ci-rurgia e melhora em uma semana. Além disto, nem todosos pacientes passam pelas três fases. É importante frisar quea maioria dos casos de poliúria após neurocirurgia não sãodecorrentes de DI central, mas devidos a um excesso de lí-quidos durante a cirurgia e diurese osmótica pelo uso demanitol e corticosteróides para minimizar o edema cerebral(que podem causar hiperglicemia e glicosúria). A diferenci-ação pode ser feita pela osmolalidade urinária, resposta àrestrição de água e administração exógena de HAD.
Aproximadamente 30% dos casos de DI central são denatureza idiopática, por um processo auto-imune com in-flamação linfocítica da haste hipofisária e da parte poste-rior. Uma causa mais rara é o diabetes insipidus central fa-miliar, habitualmente transmitido como um traço autossô-mico dominante. O DI central familiar parece estar associ-ado a uma mutação do gene que controla a síntese de HAD:preprovasopressina-neurofisina II. O precursor não é cli-vado em HAD, acumulando-se localmente e causando amorte de células produtoras de HAD.
Quadro 9.2 Causas de diabetes insipidus pituitário
Pós-hipofisectomiaIdiopáticoPós-traumáticoTumores supra- e intra-selares — metastático (pp. mama)
craniofaringiomapinealoma
CistosHistiocitoseGranulomas — tuberculose, sarcoidoseVasculares — aneurismas, trombose, síndrome de
SheehanInfecciosas e imunológicas — meningite, encefalite
síndrome de Guillain-Barré
114 Metabolismo da Água
A encefalopatia hipóxica (ou isquemia grave, como ocor-re na parada cardiocirculatória ou choque) causa uma di-minuição da liberação de HAD. A gravidade do defeitopode ser variável, desde uma discreta e assintomática po-liúria até uma forma mais evidente. Exemplo: síndrome deSheehan, onde a secreção de HAD é subnormal, mas amanifestação clínica é discreta.
Após um quadro de taquicardia supraventricular podeocorrer poliúria transitória devido à liberação aumentadado fator atrial natriurético e secreção diminuída de HAD.As alterações hormonais parecem ocorrer devido à ativa-ção de receptores locais de volume devido ao aumento dapressão no átrio esquerdo e da pressão sistêmica.
Na anorexia nervosa a liberação de HAD é subnormalou errática, talvez devido à disfunção cerebral. É um de-feito geralmente discreto, e quando ocorre poliúria, esta édecorrente do aumento na sede.
Diabetes insipidus nefrogênicoRefere-se à diminuição da capacidade de concentração
urinária que resulta da resistência à ação do HAD. Istopode refletir uma resistência no local de ação do HAD nosductos coletores ou interferência com o mecanismo contra-corrente devido à lesão medular ou diminuição na reab-sorção de NaCl no segmento medular espesso ascendenteda alça de Henle.
CAUSAS. As principais causas de DI nefrogênico estãoagrupadas no Quadro 9.4.
O diabetes insipidus nefrogênico hereditário é um distúr-bio infreqüente que resulta em graus variados de resistên-cia ao HAD. Há dois receptores diferentes para o HAD: osreceptores V1 e V2. Ativação dos receptores V1 induz va-
soconstrição e aumento da liberação de prostaglandinas,enquanto receptores V2 se relacionam a resposta antidiuré-tica, vasodilatação periférica e liberação do fator VIII e fa-tor de von Willebrand das células endoteliais. A transmis-são é ligada ao sexo (X-linked). Como a mutação é no re-ceptor V2, estão comprometidas as respostas antidiuréti-cas, vasodilatadoras e do fator de coagulação, enquanto osefeitos vasoconstritores e nas prostaglandinas estão intac-tos. A herança ligada ao sexo significa que os homens têmmarcada poliúria e as mulheres variam de um estado por-tador a uma importante poliúria. Recentemente uma for-ma autossômica recessiva foi descrita na qual o receptorV2 está intacto, assim como as respostas sobre a vasodila-tação e a coagulação; o defeito está nos “canais de água”coletores (chamados aquaporina-2). Estes canais normal-mente armazenados no citosol, sob influência do HAD,movem-se e se fundem com a membrana luminal, permi-tindo a reabsorção de água.
O diabetes insipidus nefrogênico adquirido é mais comumque o congênito e também menos grave, porque a capaci-dade renal de concentrar a urina até a osmolalidade doplasma está preservada. Assim, a polidipsia e a poliúria sãomoderadas: 3-5 litros por dia. As principais causas de DInefrogênico são abordadas a seguir.
As nefropatias crônicas podem causar DI nefrogênico,com comprometimento da capacidade renal de concentra-ção máxima da urina (geralmente quando a TFG for me-nor que 60 ml/min). Embora se possa encontrar hiposte-núria (osmolalidade urinária menor que a plasmática) emnefropatias crônicas avançadas, uma poliúria sintomáticaé rara. No entanto, a evidência mais precoce e mais gravedeste comprometimento na concentração urinária ocorreem enfermidades que afetam a região medular e papilardo rim, tais como: doença policística, doença cística medu-lar, amiloidose, pielonefrite, uropatia obstrutiva, anemiade células falciformes, etc. As causas deste defeito na con-
Quadro 9.3 Diferenciação de distúrbios poliúricospor desidratação e administração exógena devasopressina
Uosm Uosm
antes* depois**
Normal 1,067 ± 68,7 987,0 ± 79,4(N = 9)Diabetes insipidus 168 ± 13,0 445,0 ± 52,0(N = 18)Diabetes insipidus 437 ± 33,6 548,0 ± 28,2
incompleto(N = 12)Polidipsia 738 ± 52,9 779,8 ± 73,1
primária(N = 7)
Modificado de Berl, T. e cols.29 após adaptação do trabalho de Miller, M.e cols.38
N indica o número de casos estudados em cada grupo.Uosm � osmolalidade urinária.*antes — ao término do período de privação líquida e antes de recebervasopressina.**depois — após a administração de vasopressina.
Quadro 9.4 Causas de diabetes insipidus nefrogênico
Congênito
AdquiridoNefropatia crônicaDoença policísticaDoença cística medularAmiloidosePielonefriteUropatia obstrutivaAnemia de células falciformesDistúrbios eletrolíticos (hipercalcemia, hipocalemia)Alterações na dieta — redução na ingesta de
proteína e sódio— ingestão crônica excessiva
de águaAgentes farmacológicos: lítio, metoxiflurano,demeclociclina etc.

capítulo 9 115
centração urinária são múltiplas: destruição na medularenal das inter-relações anatômicas entre a alça de Henle,vasa recta e ducto coletor; talvez a presença de toxinas urê-micas na circulação, que antagonizam a ação da vasopres-sina, e a diurese osmótica a que são submetidos os nefrosremanescentes.
Alterações na dieta podem causar diabetes insipidus ne-frogênico. Em reduções crônicas na ingesta protéica, a con-centração máxima da urina está comprometida, e isto pa-rece estar relacionado com a menor formação de uréia, querepresenta mais ou menos 50% da tonicidade do interstí-cio medular. Da mesma forma, a restrição de sódio com-promete o mecanismo de concentração, pois o primeiropasso no mecanismo de contracorrente multiplicador é areabsorção ativa de cloro (e passiva de sódio) no segmen-to espesso ascendente da alça de Henle. A restrição de clo-reto de sódio resulta num aumento da reabsorção proxi-mal destes íons, e, portanto, a quantidade que chega à alçade Henle é menor. Por fim, a ingestão crônica de excessosde água, como ocorre nos bebedores compulsivos de água(polidipsia primária), reduz a tonicidade do interstíciomedular e compromete a capacidade de concentraçãomáxima da urina 34 (v. Quadro 9.4).
Alguns distúrbios eletrolíticos também são causa de di-abetes insipidus nefrogênico. Entre eles, a hipercalcemia ea hipocalemia. O mecanismo pelo qual a hipercalcemiacompromete a concentração urinária ainda não está escla-recido. A deposição de cálcio na medula renal e a contra-ção de volume que geralmente acompanha a hipercalce-mia são fatores a considerar. Uma ação direta a nível celu-lar alterando o equilíbrio osmótico também tem sido con-siderada. O defeito na concentração torna-se clinicamenteaparente quando a concentração plasmática de cálcio estápersistentemente acima de 11 mg/dl. Com concentraçãoplasmática de potássio persistentemente abaixo de 3 mEq/L,há indícios de que ocorre redução da reabsorção de NaClno segmento ascendente espesso da alça de Henle e umamenor resposta do túbulo coletor ao HAD. Tanto na hiper-calcemia como na hipocalemia, o defeito no mecanismo deconcentração é discreto, e, para explicarem a ingesta líqui-da superior às vezes a 3-5 litros, alguns autores sugeremum efeito destes eletrólitos no mecanismo da sede.
Uma outra causa de DI nefrogênico é a anemia de célu-las falciformes, em que há uma tendência das hemácias emadquirir a forma de foice no ambiente hipertônico e debaixa tensão de oxigênio na medula renal. Esta alteraçãona forma das hemácias compromete a circulação dos vasarecta e causa edema e infartos da papila renal, ocasionan-do a incapacidade de concentrar adequadamente a urina.
Existem drogas que interferem com a ação renal doHAD, prejudicando a reabsorção de água. Entre estas dro-gas, destacamos o lítio, a dimetilclortetraciclina, ometoxifluorano e as sulfoniluréias. O lítio é uma drogamuito usada em psiquiatria no manejo de psicose manía-co-depressiva. Aparentemente esta droga inibe a ação da
vasopressina na formação de adenosina-monofosfato cícli-co (cAMP) e induz poliúria reversível.35 Pacientes com acnetratados com doses altas de dimetilclortetraciclina (deme-clociclina) podem apresentar poliúria e polidipsia.36 Estadroga inibe a ação da vasopressina, possivelmente atravésde uma interferência na geração e ação de cAMP. Ela tam-bém se liga a uma proteína específica da célula epitelial,que é importante na ação do HAD. O metoxifluorano é umagente anestésico que pode causar diabetes insipidus nefro-gênico por induzir redução da permeabilidade do ductocoletor ou diminuição da tonicidade do interstício medu-lar.37
MANIFESTAÇÕES CLÍNICAS DO DI CENTRAL ENEFROGÊNICO. Além da poliúria, noctúria e da polidip-sia que pode chegar a 15 litros ao dia, a maior parte dospacientes portadores de DI central apresenta níveis de só-dio plasmático normal ou pouco aumentado, uma vez queo mecanismo da sede está intacto, repondo pelo menosparcialmente a perda de água. Porém, pode ocorrer hiper-natremia no DI central em que o paciente não tenha aces-so à água ou que tenha seu mecanismo da sede alterado.Com o tempo, pode ocorrer grande dilatação vesical e dosureteres, a ponto de não haver mais noctúria. Além disso,outras manifestações decorrem da doença de base.
Pontos-chave:
• Diabetes insipidus central é causado poralteração da produção e/ou liberação doHAD
• Diabetes insipidus nefrogênico decorre dainsensibilidade renal ao HAD
DIAGNÓSTICO DO DI CENTRAL, NEFROGÊNICOE OUTRAS FORMAS DE POLIÚRIA. Além da poliúria,polidipsia e hipernatremia com volemia normal, no diabe-tes insipidus central a densidade da urina é bastante baixa(1,001-1,005), embora formas parciais de DI, na vigência dedesidratação intensa, possam formar urina hipertônica. Háalguns testes para o diagnóstico de DI, como a restrição deágua, administração de solução salina hipertônica e admi-nistração exógena de hormônio antidiurético, como vere-mos a seguir.
A restrição simples de água é o teste mais utilizado edetermina a capacidade de o paciente elaborar HAD emresposta à hipertonicidade do plasma. O paciente é pesa-do e, a seguir, restringe-se a água por 12-16 horas ou atéque ele perca 3-5% do peso corporal. Cada amostra de uri-na é coletada para determinação do volume e densidadeurinária e/ou osmolalidade. Um indivíduo normal reduzo volume urinário para menos de 0,5 ml/min e aumenta aosmolalidade urinária (superior a 800 mOsm/kg). O paci-ente com DI mantém um alto volume urinário e uma os-molalidade urinária em torno de 200 mOsm/kg. Alguns

116 Metabolismo da Água
autores preferem um teste mais curto (6-8 horas) e compa-ram a osmolalidade sérica e urinária inicial com a final. Umlongo período de restrição líquida deve ser evitado devi-do ao risco de depleção de volume e hipernatremia, e al-guns autores sugerem períodos de restrição de água deapenas 2-3 horas. O volume e a osmolalidade urinária sãodeterminados a cada hora, e o sódio plasmático, a cada 2horas.
Com a administração de solução salina hipertônica (300ml de NaCl a 5%), ocorre aumento da osmolalidade plas-mática e, nos indivíduos normais, há uma liberação deHAD e conseqüente redução do volume urinário. Este testenão tem sido utilizado de rotina.
O aumento da osmolalidade plasmática em indivíduosnormais conduz a uma elevação progressiva da liberaçãodo HAD e, portanto, da osmolalidade urinária. Quando aosmolalidade plasmática atinge 295-300 mOsm/kg (nor-mal 275-290 mOsm/kg), a ação endógena do HAD no rimé máxima. Neste ponto, administrar HAD não eleva a os-molalidade urinária, a menos que haja um problema cen-tral na liberação de HAD, ou seja, DI central. O teste derestrição da água continua até que a osmolalidade uriná-ria atinja um nível normal (acima de 600 mOsm/kg), indi-cando liberação e ação intactas do HAD, a osmolalidadeurinária fique estável em duas medidas consecutivas, ape-sar de um aumento na osmolalidade plasmática, ou se aosmolalidade plasmática exceder 295-300 mOsm/kg. Nes-tas duas últimas situações, administra-se HAD exógeno (10mg de DDAVP por spray nasal). Monitora-se o volume e aosmolalidade urinária. Os padrões de resposta à restriçãode água e à administração de DDAVP são distintos, depen-dendo da causa do DI.29,38
No DI central, que é geralmente parcial, a liberação deHAD e a osmolalidade urinária podem aumentar com oaumento da osmolalidade plasmática. Porém, como a libe-ração de HAD é inadequada, a concentração urinária ob-tida não é máxima, e neste caso o HAD exógeno leva a umaumento da osmolalidade urinária e queda no débito uri-nário.
No DI nefrogênico a restrição de água causa elevaçãosubmáxima na osmolalidade urinária. O aumento da os-molalidade plasmática estimula a liberação de HAD, mascomo os pacientes com DI nefrogênico de modo geral sãoparcialmente resistentes ao HAD, pode haver um aumen-to pequeno na osmolalidade urinária. A administração deHAD exógeno também pode aumentar a osmolalidadeurinária.
Na polidipsia primária, a restrição de água aumenta aosmolalidade urinária. Como a liberação de HAD estánormal, não há resposta ao HAD exógeno. A capacidadede concentração urinária está diminuída, pois a poliúria ea polidipsia crônicas retiram solutos da medula renal, di-minuindo o gradiente intersticial medular.39
Talvez no futuro os resultados do teste de restrição àágua e administração de HAD possam ser confirmados
pela medida da excreção urinária de aquaporina-2, que éo “canal de água” do túbulo coletor. A excreção deaquaporina-2 aumenta muito após a administração deHAD em indivíduos normais e naqueles com DI central,podendo ser usada como um índice da ação deste hormô-nio no rim.39,40
Ponto-chave:
• Ο diagnóstico diferencial entre diabetesinsipidus central, nefrogênico e outrasformas de poliúria é realizado através dahistória clínica e dos testes de restrição deágua, infusão de salina hipertônica eadministração de HAD
TRATAMENTO DO DI CENTRAL. O tratamento doDI central visa a diminuição do débito urinário, através doaumento na atividade do HAD e reposição adequada dasperdas líquidas. O DI central é tratado com a administra-ção do hormônio antidiurético (HAD) ou com o uso deoutros medicamentos não-hormonais.41
Atualmente, está disponível a desmopressina (DDA-VP), que tem efeito antidiurético potente, sem efeitovasopressor. A desmopressina é apresentada na forma lí-quida e pode ser utilizada pela via intranasal, aplicada atra-vés de um pequeno tubo plástico ou na forma de spray.Inicia-se com dose de 5 µg à noite; dependendo dos efei-tos sobre a noctúria, a dose pode ser aumentada em 5 µg edepois acrescentadas doses diurnas. Nos EUA está dispo-nível uma apresentação oral de DDAVP, mas que tempotência de apenas 10-20% da forma nasal.41 O risco daadministração do DDAVP é a retenção de água e hipona-tremia, já que, sob o efeito desta droga, o paciente é inca-paz de excretar normalmente a água ingerida.
Para os pacientes que têm resposta incompleta à desmo-pressina, pode ser necessário acrescentar drogas que au-mentem a liberação de ADH, aumentem o efeito do ADHno rim (em DI central parcial) ou diminuam o débito uri-nário de maneira independente do HAD. Entre estas dro-gas, podem ser utilizadas a clorpropamida, clofibrato, ace-taminofen e tegretol, diuréticos tiazídicos e antiinflamató-rios não-hormonais.
A clorpropamida é uma droga utilizada no manejo dediabetes mellitus, mas também é eficaz no tratamento do DIcentral. Esta droga é capaz de reduzir o volume urinário eelevar a osmolalidade urinária em pacientes portadores deDI central. Acredita-se que potencialize os efeitos do HADcirculante, talvez sensibilizando o túbulo renal à ação daHAD. Ainda não está esclarecido se a clorpropamida temuma ação central (estimulando a liberação de HAD). Apóso diagnóstico, administram-se 250 mg de clorpropamidauma ou duas vezes ao dia, e o efeito será observado entreo terceiro e o sétimo dia após a administração. Ela não é

capítulo 9 117
efetiva na forma nefrogênica do DI e é menos efetiva quan-to mais grave for o DI. O maior problema é a hipoglicemiaque causa, sobretudo em crianças.
O clofibrato (droga usada no tratamento de dislipide-mias) parece aumentar a secreção pituitária de vasopres-sina e não possuir nenhuma ação sensibilizante ao nível detúbulo renal. Por não ter efeitos colaterais (como a hipo-glicemia da clorpropamida), pode ser utilizado no manejodo DI parcial. A dose de 500 mg cada 6 horas pode reduzira poliúria em DI central. A carbamazepina (usada no tra-tamento da epilepsia) parece aumentar a resposta tubularao HAD. A carbamazepina é utilizada numa dose de 100 a300 mg duas vezes ao dia. A clorpropamida, clofibrato ecarbamazepina podem reduzir o débito urinário no DIcentral em até 50%.41
A indução de discreta depleção de volume com umadieta baixa em sódio e diuréticos tiazídicos (hidroclorotia-zida, 25 mg uma ou duas vezes ao dia) são medidas efica-zes no tratamento do DI, reduzindo o débito urinário emcerca de 50%. A hipovolemia induzida aumenta a reabsor-ção proximal de água e sódio, reduzindo assim a oferta deágua aos locais HAD-sensíveis dos ductos coletores.41
Os antiinflamatórios não-hormonais (principalmente oibuprofeno) causam inibição da síntese de prostaglandinasrenais, e isto aumenta a capacidade de concentração uri-nária, já que as prostaglandinas normalmente antagonizama ação do HAD. Podem reduzir o débito urinário em 25-50%.41
TRATAMENTO DO DI NEFROGÊNICO. O tratamen-to se dirige à correção da doença de base e à diminuição dapoliúria. Os pacientes com DI nefrogênico não se benefici-am da administração de HAD ou drogas que aumentem suasecreção ou resposta renal, pois o defeito é justamente umaresistência renal (parcial ou completa) ao HAD. Ao invésdisso, apresentam efeitos favoráveis no tratamento do DInefrogênico: diuréticos tiazídicos, antiinflamatórios não-hormonais e dieta hipossódica e baixa em proteínas.
Como já mencionado, os diuréticos tiazídicos induzemuma depleção do extracelular, aumentando a reabsorçãoproximal de sódio e água, com isso diminuindo a oferta deágua aos locais sensíveis ao HAD nos túbulos coletores.Esta resposta é potencializada com o uso concomitante deamiloride ou outro diurético poupador de potássio. Osdiuréticos de alça induzem uma resistência relativa aoADH e não devem ser usados.42
Os antiinflamatórios não-hormonais apresentam no DInefrogênico os mesmos efeitos já discutidos com relaçãoao tratamento do DI central.
O débito urinário no DI nefrogênico pode ainda ser re-duzido com a utilização de uma dieta com pouco sal epouca proteína, que induz uma diminuição na excreção desolutos (sal e uréia) e no volume de água necessário paraexcretá-los.
Para os pacientes com DI nefrogênico parcial, talvez autilização de níveis suprafisiológicos de HAD possa au-
mentar a resposta renal a este hormônio. Dessa forma, adesmopressina pode ser utilizada em pacientes com poli-úria persistente após a utilização das outras medidas.
Pontos-chave:
• Ο princípio do tratamento do diabetesinsipidus central é a utilização de análogosdo HAD (DDAVP). Também são úteis:clorpropamida, clofibrato, acetaminofen,carbamazepina, tiazídicos eantiinflamatórios não-hormonais
• No diabetes insipidus nefrogênico,recomenda-se dieta com baixo teor de sal eproteínas, e o uso de tiazídicos eantiinflamatórios não-hormonais
Manifestações Clínicas de HipernatremiaAs manifestações clínicas de um estado hiperosmolar
dependem da existência ou não de alterações no volumedos compartimentos líquidos. Isto, por outro lado, depen-de de a substância que determina o estado hiperosmolarter livre acesso à água intracelular. O estado hiperosmolarpode ser classificado em dois grupos: devido à substânciacom fácil acesso à água intracelular (uréia, etanol) e devi-do ao acúmulo de solutos habitualmente excluídos do com-partimento intracelular (glicose, sódio).43 Como já menci-onamos, a hipernatremia é uma das causas mais importan-tes de estado hiperosmolar.
Como a uréia é altamente difusível, alterações na con-centração plasmática de uréia não são acompanhadas demudanças no volume dos compartimentos líquidos. Ape-nas quando é administrada rapidamente e em grandesdoses, a uréia pode causar um gradiente osmótico trans-celular e produzir mudanças nos compartimentos líquidos.A ingestão de etanol é uma causa comum de hiperosmo-lalidade, mas, da mesma forma que a uréia, tem fácil aces-so à água intracelular e, portanto, não causa mudanças novolume dos compartimentos líquidos. Apenas o álcool etí-lico pode causar um aumento da osmolalidade de signifi-cação clínica, pois cada 100 mg/100 ml elevam a osmolali-dade em 22 mOsm/L.
A glicose, por sua vez, é uma substância osmoticamenteativa, pois atravessa as membranas celulares muito lenta-mente. Diabetes mellitus e diálise peritoneal com glicosehipertônica são situações clínicas comuns de hiperosmo-lalidade plasmática. Durante a fase inicial de descompen-sação do diabetes mellitus, ocorre hiperglicemia sem glicosú-ria, enquanto o limiar renal de excreção da glicose não foiexcedido. Esta hiperglicemia inicial causa um aumento daosmolalidade plasmática, e o desvio da água do comparti-mento intracelular para o extracelular torna os dois com-

118 Metabolismo da Água
partimentos isosmóticos. O resultado final é um aumentoda osmolalidade nos dois compartimentos, aumento dovolume do compartimento extracelular e hiponatremiadevido à diluição do sódio no extracelular pela água pro-veniente do compartimento intracelular. Na segunda fase dedescompensação do diabetes mellitus, a hiperglicemia exce-de o limiar de excreção renal e aparece a glicosúria. Nestafase ocorre uma diurese osmótica, com grandes perdasurinárias de água e cloreto de sódio e conseqüente contra-ção do volume plasmático. No coma diabético hiperglicê-mico não-cetótico, a depleção de água pode ser tão grandeque, apesar da hiperglicemia (1.000 mg/100 ml), o sódioplasmático está normal ou elevado. O organismo reage àcontração do volume plasmático, desviando líquido dointerstício e, mais importante, desviando líquido das célu-las para expandir o compartimento extracelular. A águaintracelular sai, acompanhada de eletrólitos (K�, Cl�,HPO4
�), para que a isosmolalidade transcelular seja man-tida. O manejo desses pacientes requer, além da adminis-tração de insulina, a administração de líquidos e eletróli-tos. Se a osmolalidade inicial não for muito elevada, admi-nistra-se solução salina isotônica, a fim de restaurar o vo-lume plasmático. Particular atenção deve ser dada à repo-sição de potássio, pois, mesmo na presença de hipercale-mia, a administração de insulina e líquido é seguida derápida queda na concentração plasmática de potássio.Quando a osmolalidade plasmática inicial for muito ele-vada, recomenda-se a administração de uma solução sali-na hipotônica (NaCl a 0,45%).
O sódio tem um acesso limitado ao compartimento in-tracelular, e o estado hiperosmolar que acompanha a hi-pernatremia reflete um déficit de água total, sobretudo daágua intracelular. Este déficit de água pode ser acompa-nhado de um déficit de sódio, mas sempre em menor quan-tidade que a perda de água29 (v. Quadros 9.6 e 9.10). Alémda associação com hipovolemia, também é possível encon-trar hipernatremia com volemia normal ou aumentada. Énecessário avaliar o espaço extracelular através de um cui-dadoso exame físico, conforme será abordado no Cap. 10.
Entre as manifestações clínicas da própria hipernatre-mia, predominam aquelas que refletem disfunção do sis-tema nervoso central, principalmente se o aumento na con-centração do sódio se fez de forma rápida, ao longo de al-gumas horas. A maior parte dos pacientes não internadosque apresentam hipernatremia é muito jovem ou idosa.Estes grupos etários apresentam alterações do mecanismoda sede, redução da capacidade de concentração máximada urina e falha na resposta normal ao ADH.44
Em crianças, são comuns a hiperpnéia, fraqueza mus-cular, inquietude, choro, insônia, letargia e até mesmocoma. As crianças geralmente não apresentam sintomas atéque a concentração plasmática de sódio exceda 160 mEq/L.Se o paciente está consciente, a sede pode ser intensa. Onível de consciência se correlaciona com a gravidade da hi-pernatremia. Convulsões não ocorrem, a menos que o pa-
ciente receba sobrecarga de sódio ou reidratação muito in-tensa.
Entre os pacientes hospitalizados, as manifestações po-dem não ser tão nítidas, pois muitos deles apresentamdoença neurológica preexistente. Na maioria das vezes, háalterações sensoriais, como confusão mental, estupor e,eventualmente, coma. Pode haver hipotensão, taquicardiae até hipertermia. O volume urinário é pequeno, a menosque haja uma diurese osmótica ou uma síndrome poliúri-ca. A concentração plasmática das proteínas está elevadae, se houver um déficit de sódio associado, verifica-se umaelevação da hemoglobina e do hematócrito. O líquido ce-falorraquidiano pode ser xantocrômico ou sanguinolento,graças a um aumento da permeabilidade ou mesmo rup-tura dos capilares cerebrais devido à redução de volumedo cérebro.
Pontos-chave:
• As principais manifestações dahipernatremia se relacionam ao sistemanervoso central e dependem da idade dopaciente e da rapidez de instalação
• Os sintomas são mais intensos nahipernatremia aguda que na crônica, pois omecanismo de compensação (ganhointracelular de osmóis) não está ativado
Manejo do Paciente com Hipernatremia
LINHAS GERAISO tratamento da hipernatremia depende de dois fato-
res importantes: volume do compartimento extracelular eritmo de aparecimento da hipernatremia.
Na hipernatremia associada à depleção do volume ex-tracelular, o primeiro objetivo é restaurar a volemia com
Quadro 9.5 Mecanismos renais necessários para oclearance de água
A. Produção de um gradiente osmótico1. Número suficiente de nefros funcionantes2. Oferta suficiente de NaCl aos segmentos medulares3. Transporte suficiente de NaCl nos segmentos
medulares4. Conservação suficiente de uréia na medula renal
B. Utilização do gradiente osmótico1. Fluxo sanguíneo renal apropriado2. Ação apropriada da vasopressina nos ductos
coletores3. Resposta apropriada da vasopressina pelos ductos
coletores4. Fluxo urinário apropriado
capítulo 9 119
soro fisiológico. Se houver sinais de colapso circulatóriopela contração de volume, a solução salina isotônica deveser administrada até que a instabilidade hemodinâmicaseja corrigida. Posteriormente, podem ser utilizados o soroglicosado a 5% ou uma solução hipotônica (0,45%) de clo-reto de sódio. Se não houver instabilidade hemodinâmicainicial, inicia-se a administração simultânea de soro glico-sado a 5% e solução salina isotônica. Quando se dispuserde uma solução salina hipotônica (NaCl 0,45%), esta serápreferida.
O manejo dos pacientes com hipernatremia associada aum excesso de volume extracelular baseia-se na reposiçãode água por via oral ou parenteral e na remoção do sódiocom diuréticos de alça. Na presença de insuficiência renal,hipernatremia e excesso de volume são manejados atravésde diálise.
Finalmente, naqueles pacientes com hipernatremia evolemia normal o manejo baseia-se na interrupção da per-da continuada de líquido e na administração de água soba forma de soro glicosado a 5%. A administração de líqui-do pode ser feita por via oral, via sonda nasogástrica ouvia parenteral.46
CÁLCULO DO DÉFICIT DE ÁGUAConsidere um paciente com peso usual de 70 kg, apre-
sentando sódio plasmático atual de 155 mEq/L e sódionormal de 140 mEq/L:
1.º passo: Calcular a água total normal deste paciente:70 kg x 60% � 42 litros (alguns autores consideram a águatotal do homem como 60% do peso corporal, e 50% nasmulheres, por possuírem mais tecido adiposo e, logo, me-nos água. Além disso, consideram a água total atual como
sendo menor em pacientes hipernatrêmicos e que estãocom déficit de água; logo usam, em vez de 60% e 50%,valores de 50% e 40% para homens e mulheres, respecti-vamente).
2.° passo: Calcular a quantidade de água total que estepaciente possui com o sódio em 155 mEq/L.
Água atual � Água normal � Sódio normal
� 42 � 140
� 38 litrosSódio atual 155
3.º passo: Calcular o déficit de água: Água atual � águanormal � 38 � 42 � 4 litros de déficit de água. Esta é aquantidade de fluido hipotônico que o paciente necessi-ta receber para que seu sódio plasmático retorne a 140mEq/L.
TIPO DE FLUIDOA escolha do fluido a ser infundido para a correção da
hipernatremia depende da via de administração e da ne-cessidade de corrigir outro distúrbio hidroeletrolítico coe-xistente. Para uso enteral, podem ser utilizadas a águadestilada ou soluções eletrolíticas hipotônicas.27
Para reposição endovenosa, o fluido ideal é aquele quenão contém osmóis efetivos e ao mesmo tempo não ocasi-one o risco de hemólise por exposição dos eritrócitos a umfluido excessivamente hipotônico. Alguns autores sugeremque a correção com solução contendo glicose está associa-da a acidose láctica intracelular cerebral, devendo por istoser evitada.27
Em alguns casos, a solução salina a 0,9%, contendo 154mEq de sódio por litro, pode ser útil. Isto é verdadeiroquando coexiste depleção do espaço extracelular com a
Quadro 9.6 Interpretação e manejo da hipernatremia*
Distúrbio Sódio total Causas clínicas Osmolalidade Tratamentobásico do organismo urinária e NaU**
Perda de Sódio total Perdas renais: Urina iso - ou hipo- Solução salinaágua e sódio reduzido (diurese osmótica) tônica; NaU isotônica
� 20 mEq/LPerdas extra-renais: Urina hipertônica
sudorese NaU � 10 mEq/L
Perda de Sódio total Perdas renais: Urina iso-, hipo- ou Água ou soroágua normal diabetes insipidus, hipertônica glicosado a 5%
central ou nefrogênico NaÜ variávelPerdas extra-renais: Urina hipertônica
pele e trato respiratório NaÜ variável
Adição Excesso de Hiperaldosteronismo Urina iso - ou hiper- Água ou sorode sódio sódio total primário; síndrome de tônica NaU glicosado a 5%
Cushing; diálise � 20 mEq/L � diuréticoshipertônica; bicarbonatode sódio hipertônico
*Modificado de Berl, T. e cols.8
**NaÜ indica a concentração urinária de sódio.

120 Metabolismo da Água
hipernatremia. Esta solução (154 mEq/L) terá ainda umcerto efeito diluidor sobre o plasma em condições de hi-pernatremia muito intensa. Na maioria das vezes, entre-tanto, a correção de hipernatremia somente com soluçãosalina isotônica é um procedimento inadequado. É prefe-rível repor uma solução salina a 0,45%, o que pode serobtido pela infusão simultânea de volumes iguais de SG5% (ou água destilada) e solução salina isotônica (a 0,9%).27
Há autores que recomendam que a solução glicosada a5% seja utilizada nas situações em que existe a possibili-dade de sobrecarga de volume com a infusão de fluidoscontendo sódio, como na insuficiência cardíaca.27
RITMO DE CORREÇÃOUma correção rápida da hipernatremia é perigosa. Com
a hipernatremia ocorre saída de líquido das células cere-brais. Dentro de 1-3 dias o volume cerebral é restauradopor líquido cefalorraquidiano (aumentando o volume in-tersticial) e pela entrada de solutos nas células (atraindoágua para o interior das células e logo restaurando o volu-me). Em casos de hipernatremia aguda, que se desenvol-ve em algumas horas, a correção rápida é relativamentesegura e eficaz.
Porém, nas hipernatremias que se instalam ao longo devárias horas ou dias, é necessária uma abordagem maiscautelosa. Nesta situação crônica, uma correção rápidacausa movimento osmótico de água para dentro do cére-bro, aumentando o seu volume.27 Este edema cerebral podecausar convulsões, lesão neurológica irreversível e morte.Há evidência de que existe segurança com um ritmo decorreção entre 0,5-0,7 mEq/L por hora, acima do qual rea-ções adversas ocorrem.47 Nenhuma reação adversa ocorrequando o ritmo de correção não excede 0,5 mEq/L porhora. Assim, se o sódio plasmático for de 168 mEq/L, oexcesso de 28 mEq/L (168-140) deve ser corrigido em 56horas (28 divididos por 0,5 mEq).27
Algumas vezes, a taxa de correção não se iguala àquelaque foi calculada. Isto provavelmente se deve a perdascontinuadas de fluidos hipotônicos. Nestas circunstânci-as, o tratamento da doença de base deve ser revisado etodas as perdas fluidas devem ser reavaliadas e acrescen-tadas à reposição já calculada. Idealmente, deve ser feitauma monitorização laboratorial a cada 4-6 horas para ava-liar a eficácia do tratamento.27
A piora do quadro neurológico durante a reposição defluido hipotônico pode significar o desenvolvimento deedema cerebral e requer reavaliação imediata e interrup-ção temporária da reposição.44
EVOLUÇÃOAparentemente, a morbidade e a mortalidade pela hi-
pernatremia se relacionam principalmente com a rapidezde instalação do distúrbio, e não com sua intensidade.Mesmo com o tratamento, a mortalidade em adultos ultra-passa 40%, o que em parte pode ser conseqüência da do-
ença de base. Muitos dos pacientes que sobrevivem desen-volvem algum grau de dano cerebral permanente.27
Além disso, alguns autores relatam a possibilidade dea hipernatremia crônica acionar um processo catabólicosistêmico. A hipótese é que a diminuição do volume dascélulas hepáticas e musculares pela hipernatremia desen-cadearia um processo de catabolismo protéico, caquexia edegradação tecidual.27
Pontos-chave:
• Ο tratamento da hipernatremia é feito comsoluções hipotônicas
• Para evitar edema cerebral, a correção dosníveis plasmáticos de sódio não deveexceder 0,5 mEq/L por hora
EXCESSO DE ÁGUA —HIPONATREMIA — ESTADO
HIPOSMOLAR
Em condições normais, a concentração plasmática de só-dio é mantida dentro de limites estreitos, 135 a 145 mEq/L,devido à regulação da sede e adequada secreção e ação doHAD. A capacidade de o rim excretar água sem solutos(controlada pelo HAD) é um ponto fundamental no con-trole da tonicidade do organismo.45 A osmolalidade efeti-va ou tonicidade se refere à contribuição de solutos que nãopodem atravessar livremente todas as membranas celula-res (como o sódio e a glicose), induzindo assim desviostranscelulares de água (v. Cap. 8).48
A dificuldade na excreção de água livre é uma das cau-sas mais comuns de hiponatremia ou estado hiposmolarencontrado no paciente hospitalizado, correspondendo a1-2% dos pacientes admitidos por doença aguda ou crôni-ca.45 Os idosos apresentam diminuição da capacidade deeliminação de uma carga de água, o que pode explicar emparte a suscetibilidade deste grupo ao desenvolvimento dehiponatremia.44
As principais situações clínicas associadas à hiponatre-mia estão agrupadas no Quadro 9.7. A hiponatremia poderesultar de liberação excessiva de HAD, anormalidades nadiluição urinária e/ou desordens do mecanismo da sede.45
Enquanto a hipernatremia sempre implica hipertonici-dade e hiperosmolalidade, a hiponatremia pode cursarcom tonicidade baixa, normal ou aumentada.48
A hiponatremia dilucional ou hipotônica (também chama-da de hiponatremia real), que é a forma mais comum dehiponatremia, é causada por retenção de água e cursa comosmolalidade plasmática menor que 275 mOsm/kg. Se aingesta ou aporte de água é superior à capacidade de ex-creção renal, ocorrerá diluição dos solutos do organismo,

capítulo 9 121
resultando em hiposmolalidade e hipotonicidade. São cau-sas deste tipo de hiponatremia: insuficiência cardíaca, se-creção inapropriada de HAD e depleção do espaço extra-celular.48-50 A hiponatremia hiperosmolar ou hipertônica ocor-re na hiperglicemia e infusão de manitol e cursa com os-molalidade plasmática habitualmente superior a 290mOsm/kg.48,50 Por fim, a hiponatremia isosmolar ou isotô-nica é a causada por hiperproteinemia ou hiperlipidemiagraves (pseudo-hiponatremia) e cursa com osmolalidadeplasmática normal, de 275-290 mOsm/kg.49
A hiponatremia também pode ser classificada de acor-do com sua duração, sendo chamada de aguda, quandodura menos que 48 horas, e crônica, quando ultrapassa esteperíodo.51
Causas de Hiponatremia
PSEUDO-HIPONATREMIATanto a hiperproteinemia (por exemplo, no mieloma múl-
tiplo) como a hiperlipidemia podem resultar em dosagensaparentemente baixas de sódio, devido ao espaço que estassubstâncias ocupam na fase aquosa de uma amostra de san-gue.45,52 Se grandes quantidades de macromoléculas ou li-pídios estão presentes, a quantidade de água por unidadede volume de plasma está diminuída. Os laboratórios apre-sentam os resultados da dosagem de sódio por unidade devolume de plasma. Entretanto, a concentração real de sódioé a quantidade (mEq) em uma unidade de volume (1 litro)de plasma dividida pela percentagem de água no plasma(cerca de 93%). Os 7% restantes do plasma correspondemàs proteínas e lipídios. Uma vez que os íons sódio estão dis-solvidos somente na fase aquosa do plasma, uma concen-tração de sódio de 143 mEq/L no plasma total equivale auma concentração de 154 mEq/L na água do plasma (143� 0,93). Para evitar avaliações errôneas, o plasma pode sercentrifugado para separar e remover as proteínas e os lipí-dios, ou a dosagem pode ser feita diretamente com eletro-dos sensíveis a íons, que somente reconhecem a quantida-de de sódio dissolvido na água do plasma.45
A redução na dosagem de sódio causada por hipertri-gliceridemia pode ser calculada multiplicando-se a concen-tração plasmática dos triglicérides (mg/dl) por 0,002. Porexemplo, para uma concentração de triglicérides de 5.000mg/dl, a concentração de sódio diminuiria de 144 para 134mEq/L.45 Para pacientes com hiperproteinemia, calcula-sea repercussão sobre a dosagem plasmática de sódio multi-plicando-se a quantidade de elevação da proteína totalacima de 8 g/dl por 0,25. Por exemplo, para uma concen-tração plasmática de proteína de 17 g/dl, a concentraçãode sódio diminui apenas 2,25 mEq/L. A pseudo-hipona-tremia é tratada com a correção da doença que ocasiona odistúrbio.45
Em todo caso, para uma conclusão correta sobre umabaixa concentração de sódio, é prudente verificar que mé-todo está sendo utilizado pelo laboratório para a dosagemdeste íon.
REDISTRIBUIÇÃO DE ÁGUAOutra causa de hiponatremia em que a diminuição na
concentração de sódio não está associada com uma dimi-nuição na osmolalidade plasmática também merece umcomentário especial. Quando está presente no plasma gran-de quantidade de um soluto (que não o sódio) que não sedifunde livremente através das membranas celulares, cria-se um gradiente osmótico que favorece o movimento deágua do intracelular para o extracelular, resultando emhiponatremia com hipertonicidade.
A causa mais comum deste tipo de hiponatremia é ahiperglicemia, mas também tem sido relatada durante te-rapia com manitol hipertônico. Ao contrário do que ocor-re com a hiperlipidemia e hiperproteinemia, a baixa con-centração de sódio nestas circunstâncias é um reflexo realda concentração de sódio no espaço extracelular. O queocorre é a passagem de água do intracelular para o extra-celular, diluindo o sódio do plasma. O tratamento destetipo de hiponatremia deve ser dirigido à correção das con-centrações elevadas de glicose ou manitol, o que resultaráno movimento de água para o intracelular, com restaura-ção da concentração do sódio plasmático ao normal.45
Outra causa é a irrigação durante cirurgia de próstata,com grandes volumes de manitol, sorbitol, glicina ou águadestilada, que acabam sendo absorvidos através do leitocirúrgico cruento. Inicialmente, o soluto absorvido ficaconfinado ao espaço extracelular, trazendo água do intra-celular, a qual dilui o sódio plasmático, resultando numestado de hiponatremia isotônica. O manitol é imediata-mente excretado na urina, mas o sorbitol e a glicina sãometabolizados, causando severa hipotonicidade e desviode água para o intracelular. Sintomas neurológicos gravespodem ocorrer, especialmente com a glicina, devido à neu-rotoxicidade direta do aminoácido e níveis elevados deamônio gerados durante seu metabolismo.45
Para calcular a contribuição da glicose ou do manitolpara a osmolalidade plasmática, basta dividir a concentra-
Quadro 9.7 Situações clínicas associadas comhiponatremia*
1. Pseudo-hiponatremia2. Insuficiência cardíaca congestiva3. Cirrose hepática avançada4. Síndrome nefrótica5. Insuficiência renal crônica6. Contração de volume intravascular ou extravascular7. Estresse emocional e físico8. Distúrbios endócrinos9. Agentes farmacológicos
10. Síndrome de secreção inapropriada de vasopressina
*Obtido de Berl, T. e col.8

122 Metabolismo da Água
ção plasmática (mg/100 ml) pelo peso molecular da subs-tância (glicose e manitol têm peso molecular de 180). Mul-tiplica-se a concentração plasmática da substância por 10para transformar mg/100 ml em mg/L. Exemplo: se a con-centração plasmática da glicose for 180 mg/100 ml, a con-tribuição para a osmolalidade será: 180 � 10 � 180 � 10mOsm/L.
Pode-se também considerar que para cada 100 mg/dlde elevação na glicemia acima de 200 mg/dl, há uma re-dução de 1,6 mEq/L no sódio plasmático. Exemplo: a gli-cemia passou de 200 a 1.200 mg/dl. A concentração desódio plasmático deve cair de 140 para 124 mEq/L semalteração no conteúdo total de água ou de eletrólitos, masapenas com desvio de água do intracelular para o extrace-lular (1,6 mEq/L � 10 � 16 mEq).
INTOXICAÇÃO AGUDA PELA ÁGUAHiponatremia pode desenvolver-se agudamente em
pacientes que ingerem grandes quantidades de fluido hi-potônico. Isto ocorre em três situações: pacientes com taxade filtração glomerular (TFG) normal que ingerem gran-des quantidades de água (polidipsia psicogênica); pacien-tes com TFG muito reduzida que ingerem quantidadesmoderadas de água; e pacientes bebedores de cerveja.45
A polidipsia psicogênica ou ingestão compulsiva deágua é relatada em pacientes psiquiátricos, sendo que partedeles desenvolve hiponatremia sintomática. A ingesta agu-da de líquidos pode exceder 15-20 litros ao dia, superandoa capacidade máxima do rim em eliminar a sobrecarga deágua. De modo geral, a interrupção da ingesta excessiva euma diurese volumosa são suficientes para a correção dahiponatremia; estes pacientes raramente desenvolvem sin-tomas. Porém, um grupo de pacientes psiquiátricos desen-volve hiponatremia sintomática. Nestes, estudos demons-traram sensibilidade aumentada ao HAD, defeito na dilui-ção urinária independente do HAD ou mesmo níveis ele-vados de HAD. Alguns fatores, tais como a própria psico-se, náuseas, nicotina e várias drogas psicotrópicas, estimu-lam a secreção de HAD.45
Hiponatremia é bem descrita em indivíduos que inge-rem grandes quantidades de cerveja, sem aporte nutricio-nal adequado. Nesta situação, há redução da quantidadede urina diluída que pode ser formada, pois há poucossolutos na urina.
Na insuficiência renal, a diluição urinária não está com-prometida, mas a quantidade total de urina que pode serexcretada está muito reduzida devido ao comprometimen-to da TFG. Por exemplo, num paciente com TFG de 5 li-tros ao dia, apenas 30% do filtrado glomerular alcançamos segmentos diluidores do nefro, resultando em 1,5 litrode urina ao dia. Mesmo que os níveis de HAD estivessemcompletamente suprimidos, e que os 5 litros de filtradoalcançassem o segmento diluidor, o volume urinário nãopoderia exceder 5 litros. Então, no paciente com insufici-ência renal severa, a ingestão excessiva de água pode fa-
cilmente exceder a capacidade do rim de excretar uma car-ga de água, mesmo que o mecanismo de diluição estejaintacto.45
HIPONATREMIA CRÔNICAA abordagem racional ao paciente com hiponatremia
envolve uma avaliação correta do sódio corporal total eespaço extracelular (através do exame físico),31 osmolali-dade urinária e sódio urinário (v. Quadros 9.11 e 9.12). Aavaliação e a classificação do paciente hiponatrêmico combase na volemia têm sido utilizadas desde a década de1960.
Hiponatremia com Sódio Corporal Total AumentadoHiponatremia com um aumento no sódio corporal é
observada em três situações: cirrose, síndrome nefrótica einsuficiência cardíaca congestiva. O exame físico destespacientes demonstra sinais de sobrecarga e excesso doextracelular (v. Cap. 10). O denominador comum entreestas condições é um volume circulante efetivo diminuí-do, ao qual o rim responde como se estivesse sendohipoperfundido, com menor TFG e retendo sódio proxi-malmente. Esta diminuição do volume circulante efetivoativa a liberação não-osmótica de HAD, o sistema renina-angiotensina-aldosterona e o sistema simpático. A concen-tração urinária encontra-se aumentada, como resultado dasecreção excessiva de HAD e pelo menor fluxo urinário,que tem maior tempo de contato com o epitélio do ductocoletor, permitindo maior retrodifusão passiva de águapara o interstício. Com aumento da gravidade da cirrose,síndrome nefrótica ou insuficiência cardíaca congestiva,perde-se a capacidade de concentrar a urina, e uma urinaisotônica com o plasma, e com alto teor de sódio, é elabo-rada. Deve-se tomar cuidado ao avaliar a dosagem de só-dio urinário nos pacientes que recebem diuréticos, parti-cularmente os diuréticos de alça, pois também produzemurina hipotônica e com sódio alto.45
Hiponatremia com Sódio Corporal Total DiminuídoHiponatremia associada com diminuição do espaço
extracelular pode ocorrer por perdas renais ou não-renais.A semiologia evidencia sinais de contração do espaço ex-tracelular (v. Cap. 10).
As perdas não-renais incluem as perdas gastrintestinais(diarréia e vômitos), perdas cutâneas excessivas (queima-duras, raramente sudorese) ou acúmulo de terceiro espa-ço (pancreatite, peritonite, queimaduras, esmagamentomuscular). Em todas estas situações, a redução do espaçoextracelular resulta em hipoperfusão renal e diminuição daTFG. Isto provoca aumento da reabsorção de sódio no tú-bulo proximal, com menos sódio disponível para os seg-mentos diluidores distais. Também existe um estímulo aoHAD, com maior reabsorção de água. Recentemente temsido descrita a síndrome de hiponatremia dos maratonis-

capítulo 9 123
tas, em que os atletas perdem grandes quantidades de só-dio pelo suor e de modo geral ingerem fluidos de reposi-ção que contêm água, glicose e pouco sódio.45,53,54
Perdas renais de sódio são observadas com o uso dediuréticos, doença renal intersticial crônica e deficiênciade aldosterona. Todos os diuréticos, independentemen-te de seu local de ação, induzem um balanço negativo desódio. Esta depleção de sódio, por sua vez, desencadeiaa liberação não-osmótica de HAD. Na nefrite intersticialcrônica, há lesão direta das células tubulares nos segmen-tos diluidores distais e alteração da arquitetura renal nor-mal. Disso resultam uma perda renal de sódio e diminui-ção do clearance de água livre. Por fim, na deficiência dealdosterona, o defeito na diluição urinária está relaciona-do ao balanço negativo de sódio, que resulta em diminui-ção do sódio que chega aos segmentos diluidores distais,e à liberação não-osmótica de HAD induzida pela deple-ção do EEC.45
Hiponatremia com Sódio CorporalAparentemente Normal
Hiponatremia em um paciente com o espaço extracelu-lar aparentemente normal pode resultar de secreção ina-propriada de HAD (SIHAD) ou de um reajuste deosmostato.45
A SIHAD foi inicialmente descrita em 1957.55 É assimchamada, pois a secreção de HAD não se deve a um estí-mulo osmótico ou não-osmótico. Tem como característicasa hiponatremia, hipotonicidade, urina inapropriadamen-te concentrada, sódio urinário elevado e, freqüentemente,ácido úrico plasmático em níveis baixos.56 As causas destasíndrome podem ser observadas no Quadro 9.8. O meca-nismo básico da SIHAD é atividade HAD ou HAD-símileexcessiva, causando aumento da reabsorção de água noducto coletor, resultando em expansão do espaço extrace-lular. Como apenas um terço da água retida é distribuídano espaço extracelular, sinais de hipervolemia, como ede-ma ou ingurgitamento das veias do pescoço, não estãopresentes. Porém, uma discreta expansão do intravascularresulta em aumento do fluxo plasmático renal e TFG e di-minuição da reabsorção proximal de sódio. Como a secre-ção de aldosterona é normal ou tende a ser suprimida pelaexpansão crônica de volume, uma quantidade significati-va de sódio deixa de ser reabsorvida na alça de Henle etúbulo distal. Conseqüentemente, quantidades aumenta-das de sódio chegam ao túbulo coletor, que possui capaci-dade limitada de absorver sódio, e a excreção de sódio estáaumentada.45 A hipouricemia encontrada na SIHAD sedeve a uma menor reabsorção proximal de ácido úrico.57
Cabe aqui um comentário a respeito da hiponatremia empacientes com SIDA (síndrome da imunodeficiência adqui-rida). A hiponatremia é encontrada em 35-55% dos paci-entes aidéticos internados e é geralmente causada porSIHAD relacionada a pneumonia, neoplasia ou infecção dosistema nervoso central. Eventualmente perdas por diar-
réia podem causar depleção de volume circulante efetivo,ativando a liberação de HAD pelos mecanismos já descri-tos. Uma causa menos comum de hiponatremia em aidéti-cos é a insuficiência de adrenais, relacionada com infecçãopor citomegalovírus, micobactérias, pelo próprio HIV ouainda por infiltração e hemorragia por sarcoma de Kaposi.58
Os pacientes com um quadro compatível com reajustedo osmostato possuem um limiar de osmorregulação emtorno de uma hiposmolalidade plasmática. Estes pacien-tes conseguem suprimir o HAD adequadamente quandoa osmolalidade plasmática está baixa e a diluição urináriaé adequada. Em situação de hipertonicidade, há aumentoapropriado na secreção de HAD e concentração urinária.O reajuste de osmostato pode ser encontrado em qualqueruma das causas de SIHAD, estados hipovolêmicos, qua-driplegia, psicose, desnutrição e tuberculose.45,59 A hipona-tremia não é progressiva e melhora espontaneamente coma resolução da doença básica.45
Quadro 9.8 Situações clínicas associadas comSIHAD*
1. Produção excessiva de HAD por tumor• Pulmão, gastrintestinal, timo, próstata, linfoma
2. Aumento da liberação hipotálamo-hipofisária deHADa) Doença pulmonar
• Tuberculose, pneumonia, abcessob) Doenças do sistema nervoso central
• Trauma, convulsões, meningite, encefalite,abcesso
• Tumor• Hemorragia subdural, subaracnóide, aneurisma• Acidente vascular encefálico
c) Doenças endócrinas• Deficiência de glicocorticóides• Mixedema
d) Drogas• Opiáceos e barbitúricos• Ecstasy• Sulfoniluréias (clorpropamida, tolbutamida)• Nicotina• Clofibrato• Antidepressivos tricíclicos• Inibidores seletivos da recaptação de serotonina
(fluoxetina, sertralina)• Carbamazepina• Drogas antineoplásicas (vincristina, vinblastina)• Tiazídicos
e) AIDS
3. Administração exógena de HAD
4. Drogas que potencializam o efeito do HAD ou têmefeito HAD-símile• Clorpropamida• Ciclofosfamida64
• Ocitocina
*SIHAD = Síndrome da secreção inapropriada de HAD.

124 Metabolismo da Água
Pontos-chave:
• Ο diagnóstico de hiponatremia é feito comconcentrações plasmáticas de sódio � 135mEq/L
• Hiponatremia pode cursar com volemianormal, aumentada ou diminuída
MANIFESTAÇÕES CLÍNICAS DEHIPONATREMIA
O nível de hiponatremia que pode causar sinais e sinto-mas varia com o ritmo de queda do sódio plasmático e coma idade do paciente. Em geral, um paciente mais jovemtolera melhor um determinado nível de hiponatremia queum mais idoso. Entretanto, hiponatremia aguda pode de-terminar importantes sinais e sintomas do sistema nervo-so central: depressão do nível de consciência, convulsõese morte, mesmo com níveis de sódio plasmático entre 125e 130 mEq/L. Estas manifestações são atribuídas principal-mente a um edema cerebral, causado pela rápida reduçãona concentração plasmática de sódio.60 Isto ocorre porquenão há tempo para as células cerebrais eliminarem partí-culas osmoticamente ativas do seu interior, reduzindo as-sim o edema celular. Por outro lado, este mecanismo prote-tor contra o edema cerebral é muito efetivo na hiponatre-mia crônica, de forma que um paciente pode estar assinto-mático com um sódio plasmático inferior a 110 mEq/L.
Os sinais e sintomas se correlacionam com o grau deedema cerebral. Náuseas e mal-estar são sintomas preco-ces e podem ser observados quando a concentração plas-mática de sódio cai para 125-130 mEq/L. Na seqüênciaocorrem cefaléia, letargia, obnubilação e eventualmenteconvulsões, coma e parada respiratória, caso o sódio caiapara 115-120 mEq/L.60 Outros sinais e sintomas incluemcâimbras e anorexia, diminuição dos reflexos tendinososprofundos, reflexos patológicos, hipotermia e paralisiapseudobulbar. São particularmente suscetíveis ao edemacerebral mulheres jovens em pós-operatório, mulheres ido-sas usando diuréticos tiazídicos, crianças e pacientes hipo-xêmicos.51
Estão presentes também os sinais e sintomas relaciona-dos à doença de base que ocasionou a hiponatremia.45
Diagnóstico
Na avaliação de um paciente hiponatrêmico, a históriaclínica é de grande importância, assim como a verificaçãodo balanço hídrico, perdas e aporte de fluidos nos diasprecedentes.50
Além da dosagem do sódio plasmático e do sódio uri-nário, a osmolalidade plasmática, osmolalidade urinária,
potássio plasmático e gasometria são de utilidade no diag-nóstico diferencial das hiponatremias.
A osmolalidade plasmática encontra-se diminuída namaior parte dos pacientes hiponatrêmicos, uma vez que ébasicamente determinada pela concentração plasmática desódio. Mas, em alguns casos, a osmolalidade (e não a tonici-dade) do plasma está normal (como na hiperlipidemia e nahiperproteinemia) ou elevada (hiperglicemia, administraçãode manitol). Quando há osmolalidade plasmática elevada,ocorre movimento osmótico de água para fora das células, ea concentração de sódio no plasma diminui por diluição.57
A resposta renal apropriada em presença de um exces-so de água é excretar urina maximamente diluída. Quan-do isto não ocorre, deve-se suspeitar de que exista ação doADH ou anormalidade renal.61 Na urina, a osmolalidadeauxilia a diferenciar entre uma alteração na capacidade deexcretar urina diluída (presente na maior parte dos casos)e a polidipsia primária, na qual a excreção de água é nor-mal, mas a ingesta é tão volumosa que ultrapassa a capa-cidade de excreção. Na polidipsia primária, a resposta àhiponatremia é a supressão do HAD, resultando numaurina com osmolalidade abaixo de 100 mOsm/kg e densi-dade menor que 1,003. No restante dos casos, a secreçãode HAD continua apesar da hiponatremia, prejudicandoa diluição urinária e mantendo a osmolalidade urináriamaior ou igual a 300 mOsm/kg.57
Concentrações urinárias de sódio menores que 25 mEq/Lsugerem a participação de perdas não-renais de sódio nagênese da hiponatremia, enquanto concentrações superi-ores a 40 mEq/L sugerem secreção inapropriada de HAD.57
A dosagem do potássio e a verificação do estado ácido-básico podem auxiliar a diferenciar algumas situações: porexemplo, alcalose metabólica e hipocalemia indicam usode diuréticos ou vômitos; acidose metabólica e hipocale-mia sugerem diarréia ou uso de laxantes, e acidose meta-bólica e hipercalemia sugerem insuficiência adrenal.57
TRATAMENTO DAHIPONATREMIA
Linhas Gerais
Com exceção da pseudo-hiponatremia e da hiperglice-mia, a hiponatremia implica um desvio de água para den-tro das células e edema das células. Este desvio é particu-larmente importante no sistema nervoso central, uma vezque o cérebro está alojado no espaço inextensível da caixacraniana e o edema cerebral causa sintomas graves.61
A idade do paciente, rapidez de instalação da hipona-tremia, avaliação do volume do compartimento extracelu-lar e a concentração do sódio urinário são muito importan-tes no planejamento terapêutico dos pacientes com hipo-natremia (Quadros 9.9 e 9.11).45 A doença básica deve ser

capítulo 9 125
avaliada e tratada adequadamente. Deve ser interrompi-do o uso de qualquer agente farmacológico que interfiracom o manejo renal da água.45
A maior parte dos pacientes hiponatrêmicos são assin-tomáticos e apresentam concentração plasmática de sódiomaior que 120 mEq/L. Nestes, a correção da hiponatremiapode ser feita de modo mais lento e gradual, através darestrição de água livre,62 e o tratamento com solução sali-na hipertônica não é indicado.45 Com a restrição de águalivre para menos de 1 litro ao dia, ocorre balanço negativode água, e o sódio plasmático é corrigido lentamente. Empacientes que se alimentam normalmente por via oral, ataxa de correção do sódio com a restrição de água raramen-te excede 1,5 mEq/dia. Já nos que não estão recebendonutrição via oral, e são mantidos apenas com fluidos in-travenosos, o balanço entre as perdas insensíveis e a repo-sição pode estar próximo de zero, e será ainda mais difícilobter um balanço negativo de água.45
Em um paciente hiponatrêmico com depleção do extra-celular concomitante, a solução salina isotônica (154 mEqde sódio por litro) é a solução escolhida. A solução salinacausa repleção do extracelular, interrompendo o estímulo
para a liberação de HAD, permitindo que a água em ex-cesso seja eliminada. Além disso, a solução salina tambémauxilia na correção da hiponatremia por possuir uma con-centração de sódio mais elevada (154 mEq/L) que o plas-ma hiponatrêmico.62
Se o paciente apresenta excesso do extracelular conco-mitantemente, ou se o paciente estiver perdendo o sódioinfundido através da urina, pode ser administrado diuré-tico de alça juntamente com a salina hipertônica. Nestasituação, é necessário avaliar a dosagem do sódio na uri-na após início do tratamento, para que este sódio seja re-posto, ao menos parcialmente. Se a correção do sódio plas-mático for menor que a esperada, a infusão deve ser rea-justada. 45
Na hiponatremia que ocorre no diabetes, a correçãoda hiperglicemia fará a água retornar para o interior dascélulas, normalizando a concentração plasmática de só-dio.
A hiponatremia associada a um excesso de sódio totalno organismo ocorre na insuficiência cardíaca, insuficiên-cia renal, cirrose ou síndrome nefrótica. O manejo destespacientes com excesso de água e sal baseia-se na restrição
Quadro 9.9 Interpretação e manejo da hiponatremia*
ConcentraçãoDistúrbio Compartimento urinária debásico extracelular Causas clínicas sódio (NaU)** Tratamento
Déficit de água Depleção do Perdas renais: NaÜ > 20 mEq/L Solução salinatotal e déficit volume extra- excesso de isotônicamaior de sódio celular diuréticos;total Deficiência de
mineralocorticóide;Nefrite perdedora
de sal;Acidose tubular
renal combicarbonatúria
Perdas extra-renais: NaÜ � 10 mEq/Lvômitos, diarréias,terceiro espaço;queimaduras,pancreatite
Excesso de Discreto excesso de Defic. de glicocorticóide; NaÜ � 20 mEq/L Restrição de águaágua total volume extrace- Hipotireoidismo;
lular (sem edema) Dor, emoção, drogas;Síndrome de secreção
inapropriada de HAD
Excesso de Excesso do Síndrome nefrótica; NaÜ � 10 mEq/L Restrição de águasódio total volume extra- Insuf. cardíaca;e maior celular (edema) Cirrose hepáticaexcesso deágua total Insuf. renal aguda NaÜ � 20 mEq/L
e crônica
*Modificado de Berl, T. e cols.8
**NaÜ indica a concentração urinária de sódio.
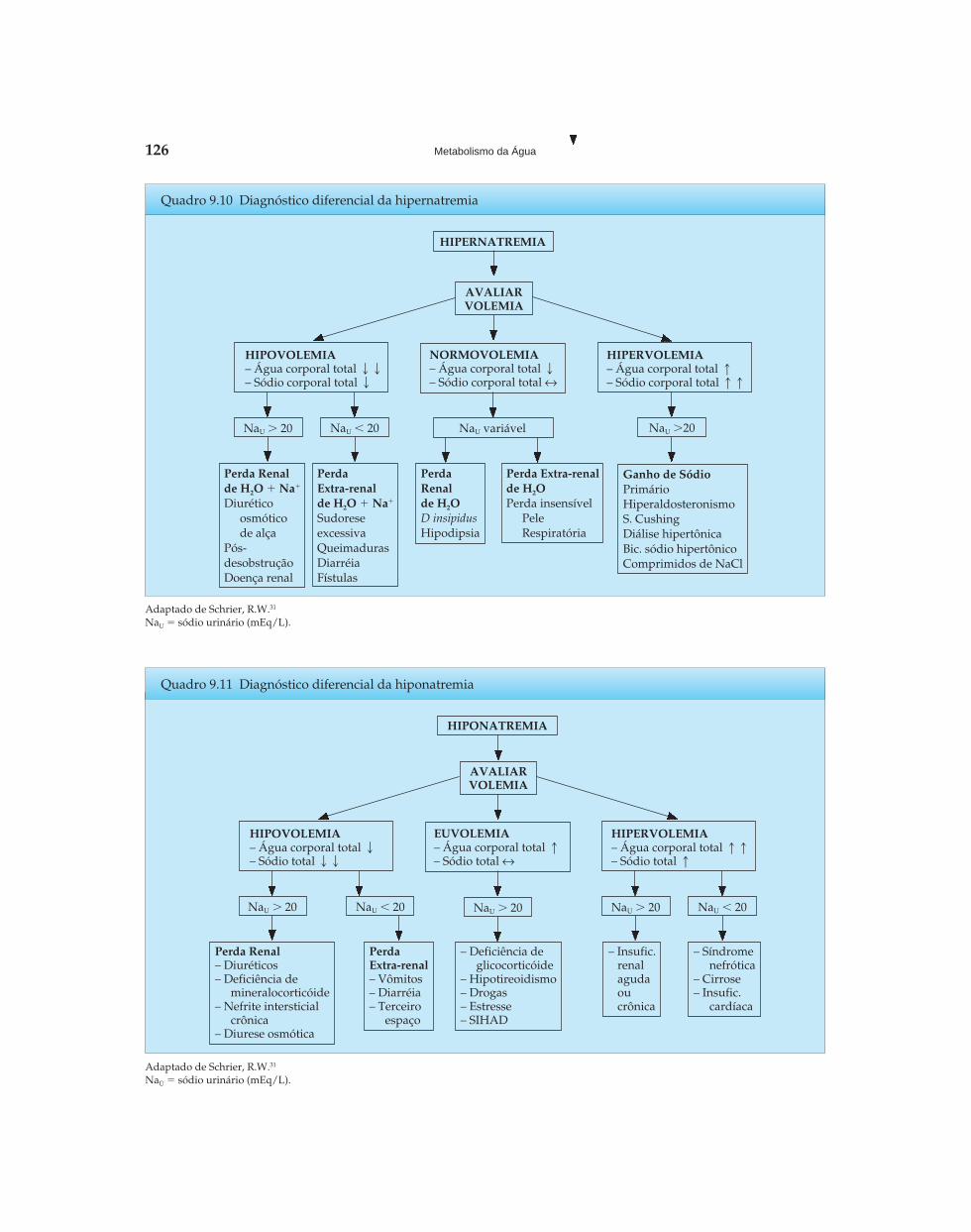
126 Metabolismo da Água
Quadro 9.10 Diagnóstico diferencial da hipernatremia
HIPERNATREMIA
AVALIARVOLEMIA
NORMOVOLEMIA– Água corporal total �– Sódio corporal total ↔
HIPOVOLEMIA– Água corporal total ��– Sódio corporal total �
HIPERVOLEMIA– Água corporal total �– Sódio corporal total ��
NaU variável NaU �20NaU � 20 NaU � 20
Ganho de SódioPrimárioHiperaldosteronismoS. CushingDiálise hipertônicaBic. sódio hipertônicoComprimidos de NaCl
Perda Extra-renalde H2OPerda insensível
PeleRespiratória
PerdaRenalde H2OD insipidusHipodipsia
PerdaExtra-renalde H2O � Na�
SudoreseexcessivaQueimadurasDiarréiaFístulas
Perda Renalde H2O � Na�
Diuréticoosmóticode alça
Pós-desobstruçãoDoença renal
Adaptado de Schrier, R.W.31
NaU � sódio urinário (mEq/L).
Quadro 9.11 Diagnóstico diferencial da hiponatremia
Adaptado de Schrier, R.W.31
NaÜ � sódio urinário (mEq/L).
HIPONATREMIA
AVALIARVOLEMIA
EUVOLEMIA– Água corporal total �– Sódio total ↔
HIPOVOLEMIA– Água corporal total �– Sódio total ��
HIPERVOLEMIA– Água corporal total ��– Sódio total �
NaU � 20 NaU � 20 NaU � 20 NaU � 20NaU � 20
PerdaExtra-renal– Vômitos– Diarréia– Terceiro
espaço
Perda Renal– Diuréticos– Deficiência de
mineralocorticóide– Nefrite intersticial
crônica– Diurese osmótica
– Síndromenefrótica
– Cirrose– Insufic.
cardíaca
– Insufic.renalagudaoucrônica
– Deficiência deglicocorticóide
– Hipotireoidismo– Drogas– Estresse– SIHAD

capítulo 9 127
de água e sal e no uso apropriado de diuréticos. Conside-rar hemodiálise nos casos de concomitante insuficiênciacardíaca congestiva ou síndrome nefrótica.
O manejo dos pacientes com hiponatremia e depleçãodo volume extracelular baseia-se na expansão do volumecirculante com solução salina isotônica. Os diuréticos, seem uso, deverão ser suspensos, e potássio deverá ser ad-ministrado, se houver hipocalemia. No caso da insufici-ência de adrenal, deve ser feita a adequada reposição hor-monal.
Nos pacientes com hiponatremia e sem sinais de altera-ção do sódio total do organismo, como ocorre na SIHAD ereajuste do osmostato, o manejo básico é a restrição líqui-da, que geralmente normaliza a concentração plasmáticado sódio. Apenas quando há sintomas de intoxicação aquo-sa, há necessidade de uma correção mais rápida (estupor,coma, convulsões). Em caso de necessidade de uso de so-lução contendo sódio, considerar que o manejo renal dosódio na SIHAD está intacto, ao contrário da depleção doextracelular, em que o sódio é retido. Isto significa que osódio administrado será eliminado na urina, e para issonecessitará de um volume de água. Por exemplo, ao seadministrar 1 litro de solução salina isotônica (300 mOsm),o sódio será eliminado juntamente com cerca de 500 ml deágua. Os 500 ml restantes terminarão por diluir ainda maiso plasma hiponatrêmico. Se for administrada uma soluçãohipertônica a 3% (1.026 mOsm/L), o sódio será eliminadopela urina, mas para isso necessitando de um volume maiorde água, o que produz um balanço negativo de água, cola-borando para a correção da hiponatremia. Concluindo, nahiponatremia sintomática da SIHAD a osmolalidade dofluido administrado deve exceder a osmolalidade da uri-na (que nesta síndrome geralmente é superior a 300mOsm/L). Portanto, a solução salina é de pouca utilidadenesta situação. Pode haver benefício também na adminis-tração de diurético de alça, o qual inibe a reabsorção decloro no ramo ascendente espesso da alça de Henle, o queinterfere com o mecanismo de contracorrente e induz umestado de resistência ao ADH. A demeclociclina e o lítiodiminuem a responsividade do túbulo coletor ao HAD eaumentam a excreção de água.62
Para os pacientes hiponatrêmicos com insuficiência car-díaca, cirrose ou SIHAD, uma perspectiva para o futuro éa utilização de um antagonista seletivo dos receptores V2(antidiuréticos) do HAD, atualmente em fase de testes. Esteagente produziria um balanço negativo de água sem pro-duzir mudanças na excreção de sódio e potássio.62,63
Cálculo do Excesso de ÁguaCalcular qual o excesso de água em um paciente de 70
kg, com sódio plasmático de 120 mEq/L.
1.° passo: Calcular qual seria a água total normal deste pa-ciente: 70 kg � 60% � 42 litros.
2.° passo: Calcular a quantidade de água total que este pa-ciente possui com o sódio em 120 mEq/L.
Água atual � Água normal � Sódio normal
� 42 � 140
� 49 litrosSódio atual 120
3.º passo: Excesso de água: Água atual � água normal� 49 � 42 � 7 litros de excesso de água.
Tratamento da Hiponatremia SintomáticaA hiponatremia sintomática é uma emergência médica,
e muitas vezes os pacientes necessitam de suporte avan-çado de vida, dada a intensidade do edema cerebral. Ossinais neurológicos e sintomas já foram descritos. Esta sín-drome pode ocorrer em qualquer estado hiposmolar, in-dependente do volume extracelular do paciente. Mesmopacientes com hiponatremia e grave depleção de volumepodem desenvolver edema cerebral.
Nestas circunstâncias, é necessária correção mais ágil dodistúrbio (v. Quadro 9.12). Por isso, a restrição de água nãoé considerada terapia adequada para a hiponatremia sin-tomática, uma vez que promove correção lenta do sódioplasmático.45 Nos indivíduos com hiponatremia sintomá-tica, o tratamento de escolha é a administração de soluçãosalina hipertônica (a 3%).
O cálculo da quantidade de sódio necessária para ele-var a concentração plasmática a um determinado valor éfeito com a fórmula a seguir:
Na necessário (mEq) � Água corporal normal � (Nadesejado � Na atual)
Por exemplo, quantos mEq de sódio são necessários paraelevar o sódio plasmático de 110 para 120 mEq/L numpaciente de 70 kg? Na necessário (mEq) � 42 L � (120 �110) � 420 mEq
Então, são necessários 420 mEq de sódio.Uma vez que a solução salina a 3% contém aproxima-
damente 514 mEq de sódio por litro, serão necessários cercade 800 ml desta solução para atingir o objetivo, o que podecausar sobrecarga de volume, principalmente nos pacien-tes com baixa reserva cardíaca. Quando a solução salina a3% não estiver disponível, pode ser preparada a partir dasolução salina isotônica a 0,9%, acrescentando 10 ml de clo-reto de sódio a 20% para cada 100 ml de salina isotônica.Observe que, no exemplo acima, a correção de 10 mEq es-taria dentro do limite de segurança para as 24 horas, mas,na presença de sintomas, a correção inicial pode chegar a1,5-2 mEq nas primeiras 3-4 horas, até a melhora dos mes-mos (v. Quadro 9.12).
Este modo de correção não deve ser usado para restaurar osódio plasmático a níveis normais! A utilização da salina hiper-tônica visa a melhora dos sintomas neurológicos mais graves.
Durante o intervalo em que a correção da hiponatremiasintomática estiver sendo feita, devem ser monitorados os

128 Metabolismo da Água
Quadro 9.12 Tratamento da hiponatremia, com base na duração e nos sintomas
Baseado em Berl, T.51
HIPONATREMIA
AGUDA CRÔNICA AGUDA CRÔNICA
Solução salinahipertônica
1-2 ml/kg/h�
Furosemide
A correção nãodeve ultrapassar 2mEq/L por hora
Não énecessáriacorreçãoimediata
Restriçãode
água livre
Solução salinahipertônica
1-2 ml/kg/h�
Furosemide
A correção nãodeve ultrapassar10-12 mEq/dia
SINTOMÁTICA ASSINTOMÁTICA
eletrólitos plasmáticos, até que o paciente esteja neurolo-gicamente estável.45 Além disso, há necessidade de semonitorar a volemia, se possível com medida da pressãocentral venosa (considerando suas limitações potenciais)ou pressão em capilar pulmonar com o cateter de Swan-Ganz.
Em 1973, Hantman e colaboradores propuseram o em-prego de furosemida no manejo da hiponatremia.64 Isto seaplica sobretudo aos pacientes que não podem tolerar umaexpansão do compartimento extracelular. A administraçãoendovenosa de furosemida induz um balanço negativo deágua, quando ao mesmo tempo se repõem as perdas ele-trolíticas (sódio e potássio) através de uma solução maisconcentrada. Os autores propõem a administração inicialde 1 mg/kg de furosemida. A concentração urinária desódio e potássio é determinada a cada hora, e a quantida-de excretada é reposta através de uma solução salina hi-pertônica (3%) com a quantidade apropriada de potássio.Nesta circunstância, a infusão de salina hipertônica deveser igual às perdas de sódio, potássio e cloro. O balançonegativo de água assim obtido é a diferença entre o fluxourinário e a quantidade de solução hipertônica adminis-trada. Doses subseqüentes de furosemida são administra-das para manter o balanço líquido negativo.
No caso de uma correção muito rápida ocorrer e serprontamente reconhecida, deve-se suspender temporari-amente a correção da hiponatremia e administrar DDAVPpara os pacientes com osmolalidade urinária baixa, pois oADH é suprimido pela hiponatremia. No caso da SIHAD,suspender a salina hipertônica. Os dados obtidos experi-mentalmente sugerem que o benefício deste tipo de abor-
dagem ocorre se o tratamento for iniciado antes do apare-cimento de sintomas neurológicos, ou seja, nas primeiras24 horas. Não há benefício se a desmielinização já se insta-lou.62
Ritmo de CorreçãoNão se sabe ao certo com que rapidez se deve corrigir
uma hiponatremia grave. Em pacientes assintomáticos,considera-se adequado corrigir cerca de 10-12 mEq/dia (0,5mEq/hora).
Já os pacientes sintomáticos necessitam de uma corre-ção mais rápida, com outra estratégia, mas mantendo oslimites de segurança. Nos pacientes sintomáticos, com con-vulsões ou outros sintomas graves, recomenda-se umacorreção inicial mais rápida, cerca de 1,5-2 mEq/hora, nasprimeiras 3-4 horas, ou até melhora dos sintomas neuro-lógicos. A correção no primeiro dia também não deve ul-trapassar 12 mEq.
Complicações do TratamentoA adaptação que preserva o volume cerebral na hipo-
natremia crônica protege contra o aparecimento de edemacerebral, mas cria problemas no momento do tratamento,pois um aumento rápido na concentração de sódio no plas-ma durante a correção pode levar à mielinólise pontinacentral (ou desmielinização osmótica).
O termo mielinólise pontina central pode não ser o maisadequado, uma vez que a desmielinização é geralmentemais difusa e muitas vezes não envolve a ponte. Estas al-

capítulo 9 129
terações podem ocasionar graves repercussões neurológi-cas que permanecem transitória ou definitivamente apóso tratamento.
Na hiponatremia crônica (desenvolve-se em mais de48 horas) há perda de osmóis intracelulares como prote-ção contra o edema cerebral. Porém, estes osmóis nãopodem ser rapidamente repostos quando o cérebro dimi-nui de volume durante a elevação do nível de sódio nosangue. Como resultado, o volume do cérebro diminuidurante a correção rápida da hiponatremia. É nas áreasonde o reacúmulo de osmóis é mais lento que as lesõesde mielinólise são mais intensas. Um mecanismo possí-vel é que a diminuição de volume dos axônios induzidapela variação osmótica produza a desmielinização pelaruptura de conexões dos axônios com sua bainha de mi-elina.60
De maneira geral, as manifestações clínicas de desmi-elinização osmótica ocorrem 2-6 dias após a correção dosníveis de sódio. Os sintomas incluem disartria, disfagia,letargia, paraparesia ou quadriparesia e até coma. Estessintomas podem não ser reversíveis.62 Evidências de-monstram que é a rapidez de correção nas primeiras 24horas que determina a ocorrência de lesões desmielini-zantes. Estas lesões são mais freqüentes quando a corre-ção ultrapassa 20 mEq/dia ou quando o sódio se elevapara mais de 140 mEq/L, e mais raras com correçõesabaixo de 0,5 mEq/hora ou 10-12 mEq/dia. Lesões des-mielinizantes não são vistas quando a correção é maislenta.62
A tomografia computadorizada e a ressonância magné-tica detectam as lesões de desmielinização, sendo este úl-timo método o preferido.65 Às vezes são necessárias atéquatro semanas para as lesões serem detectadas.62
Encontram-se em maior risco para o desenvolvimentoda desmielinização osmótica: mulheres na fase pré-meno-pausa usando tiazídicos, etilistas, desnutridos, queimados,pacientes depletados em potássio e crianças pré-púberese pacientes em insuficiência respiratória.51,66 Os pacientespsiquiátricos que desenvolvem polidipsia com hiponatre-mia de modo geral corrigem rapidamente a hiponatremia,sem seqüelas.60,62
Pontos-chave:
• O tratamento da hiponatremia depende dagravidade dos sintomas e rapidez deinstalação. Os sintomas mais gravesdecorrem de edema cerebral
• A hiponatremia sintomática é corrigida coma administração de solução salinahipertônica a 3%
• A correção da hiponatremia sintomática nãodeve ultrapassar 0,5 mEq/L/hora
EXERCÍCIOS
1) Um paciente de 35 anos sofreu trauma cranioencefálico grave e foi in-ternado em coma, escala de Glasgow 5, evoluindo para Glasgow 3. Seudébito urinário nos primeiros dois dias foi de aproximadamente 7 li-tros/dia. Além de receber 2 litros de solução salina isotônica e 1 litrode solução glicosada a 5% a cada dia, manitol era administrado na dosede 70 ml a cada 8 horas. Seus exames atuais demonstraram: Na� � 165mEq/litro. Responda:a) Existe distúrbio hidroeletrolítico? Qual?b) Qual a causa mais provável para o mesmo?c) Como você corrigiria este distúrbio?
2) Para um sódio plasmático de 150 mEq/litro, num paciente de 70 anosde idade, com 60 kg e assintomático, calcule:a) Qual a água normal?b) Qual a água atual?c) Como corrigir este distúrbio?
3) Mulher de 55 anos, usuária de fluoxetina, internada por broncopneu-monia. Na admissão, espaço extracelular aparentemente normal,contactuando adequadamente. Na� � 128 mEq/litro. Durante a in-ternação atual, tornou-se confusa e progressivamente sonolenta. Na�
� 117 mEq/litro. Peso = 55 kg.a) Existe distúrbio hidroeletrolítico? Qual?b) Qual a causa mais provável?c) Como tratar?
4) Homem portador de síndrome nefrótica, em anasarca, internado portromboflebite em membro inferior. Sem outros sintomas. Peso = 72kg. Na� � 125 mEq/L.a) Qual a água normal?b) Qual a água atual?c) Qual o tratamento?
REFERÊNCIAS BIBLIOGRÁFICAS
1. ROSE, B.D.; POST, T.W. Cap. 9A: Water balance and regulation ofplasma osmolality. Up To Date, v.9, n.3, 2001.
2. GUYTON, A.C.; HALL, J.E. Regulation of extracellular fluidosmolarity and sodium concentration. In: Textbook of Medical Physi-ology, pp. 349-365, Saunders, 1996.
3. SEGURO, A.C.; ZATZ, R. Distúrbios da tonicidade do meio inter-no: regulação do balanço de água. In: ZATZ, R. (ed.) FisiopatologiaRenal, pp. 189-208, Atheneu, 2000.
4. FITZSIMONS, J.T. The physiological basis of thirst. Kidney Int.,10(1):3, 1976.
5. MAGALDI, A.J.B. Mecanismos de concentração e diluição urinári-as. In: ZATZ, R. (ed.). Fisiopatologia Renal, pp. 57-69, Atheneu, 2000.
6. ROBERTSON, G.L. e col. The osmoregulation of vasopressin. Kid-ney Int., 10(1):25, 1976.
7. ROSE, B.D.; POST, T.W. Cap. 6B: Antidiuretic hormone and waterbalance. Up To Date, v.9, n.3, 2001.
8. BERL, T.; ROBERTSON, G.L. Pathophysiology of water metabolism.In: BRENNER, B.; RECTOR, F. (eds) The Kidney, pp. 866-924, Saun-ders, 2000.
9. KNEPPER, M.A.; VERBALIS, J.G.; NIELSEN, S. Role of aquaporinsin water balance disorders. Nephrology and hypertension – Currentopinion, jul. 1997.
10. KWON, T.H.; HAGER, H.; NEJSUM, L.N.; ANDERSEN, M.L.E.;FROKIAER, J.; NIELSEN, S. Physiology and pathophysiology ofrenal aquaporins. Seminars in Nephrology, 21(3):231-238, 2001.
11. ZEIDEL, M.L. Recent advances in water transport. Seminars in Ne-phrology, 18(2):167-177, 1998.
12. MCDONALD, K.M. e col. Hormonal control of renal water excreti-on. Kidney Int., 10(1):38, 1976.

130 Metabolismo da Água
13. ROSE, B.D. Hyponatremia in hypothyroidism. Up To Date, v.9, n.3,2001.
14. CAPASSO, G.; De TOMMASO, G.; ANASTASIO, P. Glomerularhemodynamics and renal sodium handling in hypothyroid andhyperthyroid patients. J. Am. Soc. Nephrol., 9:68A, 1998.
15. WOODHALL, P.B. e TISHER, C.C. Response of the distal tubule andcortical collecting duct to vasopressin in the rat. J. Clin. Invest.,52:3095, 1975.
16. KRIZ, W. e LEVER, A.F. Renal countercurrent mechanisms: struc-ture and function. Am. Heart J., 78.
17. BERLINER, R.W. e col. Dilution and concentration of the urine andthe action of antidiuretic hormone. Am. J. Med., 24:730, 1958.
18. BERLINER, R.W. The concentrating mechanism in the renalmedulla. Kidney Int., 9(2):214, 1976.
19. STEPHENSON, J.L. Concentration of urine in a central core of therenal counterflow system. Kidney Int., 2:85, 1972.
20. KOKKO, J.P. e RECTOR, Jr., F.C. Countercurrent multiplicationsystem without active transport in inner medulla. Kidney Int., 2:214,1972.
21. KOKKO, J.P. e TISHER, C.C. Water movement across nephronsegments involved with the countercurrent multiplication system.Kidney Int., 10(1):64, 1976.
22. JAMISON, R.L. e MAFFLY, R.H. The urinary concentrating mecha-nism. N. Engl. J. Med., 295:1059, 1976.
23. AYUS, J.C. Hypo and hypernatremia – Pathogenesis and diagnosis.Part 1 and 2. American Society of Nephrology Board Review Course,sep. 1998 (slide and audio symposium – www.hdcn.com).
24. ROSE, B.D.; POST, T.W. Cap. 8D: Volume regulation versus osmo-regulation. Up To Date, v.9, n.3, 2001.
25. MANGE, K.; MATSUURA, D.; CIZMAN, B.; SOTO, H.; ZIYADEH,F.N.; GOLDFARB, S.; NEILSON, E.G. Language guiding therapy:the case of dehydration versus depletion. Ann. Intern. Med.,127(9):848-853, 1997.
26. PALEVSKY, P.M.; BHAGRATH, R.; GREENBERG, A. Hypernatre-mia in hospitalized patients. Ann. Intern. Med., 124:197-203, jan., 1996.
27. AYUS, J.C.; BRENNAN, S. Hipernatremia. In: De FRONZO, R.;ARIEFF, A.I. (eds) Fluid, Electrolyte, and Acid-Base Disorders, pp.304-317, Churchill-Livingstone, 1995.
28. ROSE, B.D.; POST, T.W. Cap. 9B: Renal water excretion and reab-sorption. Up To Date, v.9, n.3, 2001.
29. BERL, T. e col. Clinical disorders of water metabolism. Kidney Int.,10(1):117, 1976.
30. ROSE, B.D. Causes of hypernatremia. Up To Date, v.9, n.3, 2001.31. SCHRIER, R. The patient with hyponatremia or hypernatremia. In:
Manual of Nephrology, pp. 20-36, Little, Brown, 1994.32. FRIED, L.F.; PALEVSKY, P.M. Hyponatremia and hypernatremia.
Med. Clin. N. Am., 81(3):585-609, 1997.33. HAYS, R.M. e LEVINE, S.D. Pathophysiology of water metabolism.
Cap. 15, p. 553 in The Kidney. Eds. B.M. Brenner e F.C. Rector Jr. W.B.Saunders Co., 1976.
34. SINGER, I. e FORREST Jr., J.N. Drug-induced states of nephrogenicdiabetes insipidus. Kidney Int., 10(1): 82, 1976.
35. COX, M. e SINGER, I. Lithium and water metabolism. Am. J. Med.,59:153, 1975.
36. MILLER, M. e MOSES, A.M. Drug-induced states of impaired waterexcretion. Kidney Int., 10(1): 96, 1976.
37. MAZZE, R.I. e col. Renal dysfunction associated with methoxyflu-rane anesthesia: a randomized, prospective clinical evaluation.JAMA, 216:278, 1971.
38. MILLER, M. e col. Recognition of partial defects in antidiuretic hor-mone secretion. Ann. Inter. Med., 73:721, 1970.
39. ROSE, B.D. Diagnosis of polyuria and diabetes insipidus. Up To Date,v.9, n.3, 2001.
40. KANNO, K.; SASAKI, S.; HIRATA, Y. e cols. Urinary excretion ofaquaporin-2 in patients with diabetes insipidus. N. Engl. J. Med.,332:1540-5, jun. 1995.
41. ROSE, B.D. Treatment of central diabetes insipidus. Up To Date, v.9,n.3, 2001.
42. ROSE, B.D. Treatment of nephrogenic diabetes insipidus. Up To Date,v.9, n.3, 2001.
43. LOEB, J.N. The hyperosmolar state. The New Engl. J. Med., 290:1184, 1974.44. KUGLER, J.P.; HUSTEAD, T. Hyponatremia and hypernatremia in
the elderly. Am. Fam. Phys., 61:3623-30, 2000.45. De FRONZO, R.; ARIEFF, A.I. Disorders of sodium metabolism –
Hyponatremia. In: Fluid, Electrolyte, and Acid-Base Disorders, pp.255-303, Churchill-Livingstone, 1995.
46. ADROGUÉ, H.J.; MADIAS, N.E. Hypernatremia. N. Engl. J. Med.,342(20):1493-1499, may, 2000.
47. ROSE, B.D. Treatment of hypernatremia. Up To Date, v.9, n.3, 2001.48. ADROGUÉ, H.J.; MADIAS, N.E. Hyponatremia. N. Engl. J. Med.,
342(21):1581-1589, may, 2000.49. FALL, P.J. Hyponatremia and hypernatremia. A systematic
approach to causes and their correction. Postgrad. Med., 107(5):75-82, may 2000.
50. PRESTON, R.A. Hyponatremia. In: Acid-Base, Fluids and ElectrolytesMade Ridiculously Simple, pp. 39-64, MedMaster, Inc., 1997.
51. BERL, T. Therapy of hypo and hypernatremia. Parts 1, 2, 3. Ameri-can Society of Nephrology Board Review Course, sep. 1998 (slideand audio symposium – www.hdcn.com).
52. ROSE, B.D. Causes of hyponatremia. Up To Date, v.9, n.3, 2001.53. DAVIS, D.P.; VIDEEN, J.S.; VILKE, G.M. e cols. Exercise-associated
hyponatremia in marathon runners: a two-year experience. J. Emerg.Med, 21(1):47-57, jul. 2001.
54. SPEEDY, D.B.; NOAKES, T.D.; SCHNEIDER, C. Exercise-associatedhyponatremia: a review. Emerg. Med., 13(1):5-6, mar., 2001.
55. SCWARTZ, W.B. e col. Syndrome of renal sodium loss and hipona-tremia probably resulting from inappropriate secretion ofantidiuretic hormone. Am. J. Med., 23:529, 1957.
56. ROSE, B.D. Causes of the SIADH. Up To Date, v.9, n.3, 2001.57. ROSE, B.D. Diagnosis of hyponatremia. Up To Date, v.9, n.3, 2001.58. ROSE, B.D. Electrolyte disturbances with HIV infection. Up To Date,
v.9, n.3, 2001.59. ROSE, B.D. Treatment of hyponatremia: SIADH and reset osmostat.
Up To Date, v.9, n.3, 2001.60. ROSE, B.D. Symptoms of hyponatremia and hypernatremia. Up To
Date, v.9, n.3, 2001.61. HALPERIN, M.L.; GOLDSTEIN, M.B. Sodium and water physiolo-
gy. In: Fluid, Electrolyte and Acid-Base Physiology. A problem-basedapproach, pp.217-320. W.B. Saunders, 1994.
62. ROSE, B.D. Treatment of hyponatremia. Up To Date, v.9, n.3, 2001.63. GROSS, P. Correction of hyponatremia. Seminars in Nephrology,
21(3):269-272, may 2001.64. HANTMAN, D. e col. Rapid correction of hyponatremia in the syndro-
me of inappropriate secretion of Antidiuretic Hormone. An alternati-ve treatment to hypertonic saline. Ann. Intern. Med., 78:870, 1973.
65. PIRZADA, N.A. Central pontine myelinolysis. Mayo Clin. Proc.,76(5):559-62, may, 2001.
66. ARIEFF, A.L.; AYUS, J.C. Outcome in hyponatremic encephalopathyis unrelated to rate of correction. J. Am. Soc. Nephrol., 10:119A, 1999.
ENDEREÇOS RELEVANTES NA INTERNET
http://www.kidneyatlas.org/book1/adk1_01.pdf —Excelente capítulo do Atlas on-line de Doenças Renais deRobert Schrier. Ótimas figuras.http://www.postgradmed.com/issues/2000/05_00/fall.htm — Artigo interessante sobre hipo- e hipernatremia.http://www.aafp.org/afp/20000615/3623.html — Artigosobre hipo- e hipernatremia em idosos.http://www.emedicine.com/emerg/topic263.htm — Boarevisão sobre hipernatremia.http://www.emedicine.com/emerg/topic275.htm — Boarevisão sobre hiponatremia.http://www.learndoctor.com/chapterpages/chapter19.htm— Auto-avaliação em metabolismo da água.http://www.swmed.edu/stars/resources/toto.pdf —Grupo de slides muito bons sobre hipernatremia.

capítulo 9 131
http://www.curriculum.som.vcu.edu/m2/renal/ppt/Homeostasis/ — Grupo de slides sobre distúrbios dometabolismo do sódio e da água.http://www.ndif.org/Translation/jtran-160.html —Resumo de um artigo da Medical Clinics of NorthAmerica de maio de 1997, pela Nephrogenic DiabetesInsipidus Foundation.
RESPOSTAS DOS EXERCÍCIOS
OBS.: Nestes exercícios utilizaremos 60% como a percentagem de águaem relação ao peso corporal, para homens e mulheres.
1) 35 anos, trauma cranioencefálico, sódio � 165 mEq/litro.a) Existe distúrbio hidroeletrolítico? Sim. Qual? Hipernatremia.b) Qual a causa mais provável? Este paciente apresenta pelo me-
nos três causas em potencial para o desenvolvimento de hiper-natremia. A primeira é o trauma cranioencefálico, que podecausar dano à secreção ou liberação de HAD, tornando o paci-ente incapaz de concentrar a urina, o que explicaria a poliúriaapresentada. Em segundo lugar, a administração de manitolinduz à produção de urina hipotônica. E por último, as perdasde água livre através da respiração e pela urina não estão sen-do adequadamente repostas.
c) Para corrigir esta hipernatremia, deveria ser reposta uma solu-ção hipotônica. O déficit de água que o paciente apresenta é de:
Sódio atual � água atual � sódio normal � água normalÁgua atual � 140 � (70 � 0,6)/165 � 35,6 litrosDéficit de água � água atual � água normal � 35,6 � 42 � 6,36 litros
Portanto, para que o sódio retorne ao normal (140 mEq/litro), é ne-cessário administrar 6,36 litros de solução salina hipotônica ou SG 5%. Acorreção não deve ultrapassar 0,5 mEq/litro/hora, em pelo menos 50horas (a dosagem de sódio está 25 mEq/litro acima do normal; 25 dividi-dos pela taxa de 0,5 � 50 horas).
2) 70 anos de idade, 60 kg, sódio � 150 mEq/litro.a) Água normal � 60% do peso � 60 � 0,6 � 36 litrosb) Sódio atual � água atual � sódio normal � água normal
Água atual � 140 � 36/150 � 33,6 litrosDéficit de água � 33,6 � 36 � 2,4 litros
c) Deve ser administrada solução salina hipotônica (2,4 litros) em20 horas (a dosagem de sódio está 10 mEq/litro acima do nor-mal; 10 divididos pela taxa de 0,5 � 20 horas).
3) 55 anos, broncopneumonia. Sódio � 117 mEq/litro.a) Trata-se de hiponatremia.b) Existem algumas possibilidades: a primeira é que a paciente
tenha uma SIHAD pela broncopneumonia, daí a impossibili-dade de eliminar urina diluída. Em segundo lugar, está em usode fluoxetina, que pode induzir aumento na liberação de HAD.Neste caso, deveria ser cuidadosamente verificado o balançode fluidos dos dias antecedentes, para excluir a participaçãode uma reposição excessiva de soro glicosado a 5%.
c) Como a paciente tornou-se agudamente sintomática, deve re-ceber solução salina hipertônica (3%). A quantidade de sódionecessária para elevar o sódio plasmático para 125 mEq é:
Sódio necessário � água corporal normal � (sódio desejado � atual)Sódio necessário � (55 � 60%) � (125 � 117) � 33 � 8 � 264 mEq
Sabendo que a solução salina hipertônica tem 514 mEq/litro, serãonecessários aproximadamente 500 ml desta solução. Nas primeiras 3-4horas, o ritmo de correção pode ser mais rápido (1,5-2 mEq/hora), e de-pois manter 0,5 mEq/hora.
Observe que em 264 ml desta solução há tanto sódio como em 1.700ml de salina isotônica. Além de corrigir a hiponatremia sintomática, estesódio também estará provocando expansão do extracelular, com o riscode congestão circulatória.
4) Paciente com síndrome nefrótica, em anasarca. Sódio � 125 mEq/litro.a) Água normal � (72 � 0,6) � 43 litros.b) Água atual � 43 � 140/125 � 48 litros.c) Este paciente apresenta excesso de 5 litros de água e está as-
sintomático. Deve ser restrita a ingestão de água e administra-do diurético, pois apresenta extracelular aumentado.

Capítulo
10Metabolismo do Sódio e Fisiopatologia do Edema
Miguel Carlos Riella, Maria Aparecida Pachaly e Leonardo Vidal Riella
INTRODUÇÃO
Balanço do sódio
RESPOSTA DO RIM ÀS ALTERAÇÕES NA INGESTA DE
SÓDIO
QUEM PERCEBE E REGULA AS ALTERAÇÕES DO
VOLUME EXTRACELULAR?
REGULAÇÃO INTRA-RENAL DA EXCREÇÃO DE SÓDIO
Auto-regulação renal
Filtração glomerular — balanço glomérulo-tubular
Reabsorção e propriedades físicas no capilar peritubular
Pressão oncótica peritubular
Pressão hidrostática no capilar peritubular
Balanço glomérulo-tubular e fatores humorais intra-renais
Reabsorção dependente da velocidade do fluxo de
líquido tubular
Reabsorção dependente do volume do túbulo proximal
TIPOS DE TRANSPORTE DE SÓDIO
REABSORÇÃO NOS DIFERENTES SEGMENTOS DO
NEFRO
Túbulo contornado proximal (TCP)
Segmentos delgados da alça de Henle
Segmento ascendente espesso da alça de Henle
(segmento diluidor)
Túbulo contornado distal (TCD)
Ducto coletor
OUTROS FATORES QUE REGULAM A EXCREÇÃO DE
SÓDIO
Redistribuição do filtrado glomerular
Angiotensina II
Aldosterona
Fatores físicos e volume do espaço extracelular
Hormônio natriurético
Fator natriurético atrial (FNA)
Fatores derivados do endotélio
Prostaglandinas
Sistema nervoso simpático
Diurese pressórica
DISTÚRBIOS CLÍNICOS DO METABOLISMO DO SÓDIO
Depleção de sódio ou do volume extracelular
Dados laboratoriais
Conseqüências da depleção do volume extracelular
Tratamento da depleção
Tipo de solução
Velocidade de administração
Volume a ser infundido (grau de depleção)
Monitorização do tratamento
EXCESSO DE VOLUME EXTRACELULAR—EDEMA
Fisiopatologia do edema
Edema localizado
Edema generalizado
Fisiopatologia do edema em situações clínicas específicas
Insuficiência cardíaca congestiva (ICC)
Cirrose hepática
Síndrome nefrótica
Glomerulonefrite aguda
Edema observado em mulheres
Causas diversas de edema
Princípios gerais no tratamento do edema
Tratamento da doença básica
Adequação da ingesta de sal e água
Mobilização do edema
Indução de balanço negativo de sódio
EXERCÍCIOS
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET
RESPOSTAS DOS EXERCÍCIOS

capítulo 10 133
INTRODUÇÃO
O sódio é o íon mais abundante do compartimento ex-tracelular, e a quantidade de sódio neste compartimento éque determina o seu volume. O sódio e seus dois princi-pais ânions, o cloro e o bicarbonato, constituem 90% oumais da quantidade de soluto no líquido extracelular. Poroutro lado, a quantidade de sódio no líquido intracelularé pequena, devido a mecanismos que ativamente eliminamo sódio das células.
A concentração de solutos é a mesma nos compartimen-tos intra e extracelular devido à livre movimentação daágua pelas membranas celulares, em resposta a um gradi-ente osmótico. Portanto, se há retenção de sódio no líqui-do extracelular, a pressão osmótica deste compartimentoaumenta e a água intracelular move-se para o comparti-mento extracelular até que haja equilíbrio osmótico. A hi-perosmolalidade do líquido extracelular também podeestimular a sede e a liberação do hormônio antidiurético,ambos determinando um balanço positivo de água. Então,o resultado final de um aumento de sódio no líquido ex-tracelular é um aumento do volume extracelular. Da mes-ma forma, uma diminuição da quantidade de sódio no lí-quido extracelular determina uma redução do volume ex-tracelular. Tudo indica, portanto, que o sistema que con-trola o balanço de sódio faz parte integrante do sistema quecontrola o volume extracelular.
Tendo em vista que a maior parte do volume líquidoextracelular corresponde à água, seria legítimo supor quea regulação daquele volume fosse realizada por intermé-dio dos mecanismos que controlam o balanço de água.1 Noentanto, as alterações na liberação de HAD e na excreçãode água são mediadas principalmente pela tonicidade doslíquidos no organismo, a qual é controlada pelo sistemaosmorregulador e não pelo sistema de controle do volu-me extracelular. Desde que o balanço de sódio é preserva-do, o controle da tonicidade serve para manter o volumede líquido extracelular constante.
Contudo, em algumas situações, a excreção de água éregulada primariamente pelo volume e não pela tonicida-de. Isto ocorre, por exemplo, quando há uma intensa con-tração do volume extracelular. Neste caso, a água é conti-nuamente reabsorvida (apesar da hipotonicidade que seestabelece), na tentativa de restaurar o volume extracelu-lar. Nesta situação, a regulação do volume tem preferên-cia sobre a osmorregulação.
Num indivíduo normal, o volume de líquido extracelu-lar e o balanço de sódio variam dentro de limites estreitos,mesmo em face de grandes variações na ingesta e excre-ção renal de água e sal. E é o rim que mantém o volumeextracelular constante, modulando a excreção de sódio.Assim, qualquer distúrbio que reduza o volume do com-partimento extracelular é acompanhado por uma reduçãoda excreção de sódio, enquanto um aumento de volume
do compartimento extracelular determina aumento naexcreção de sódio.
Se determinarmos a osmolalidade plasmática ou sérica,teremos a relação da soma dos solutos osmoticamente ati-vos (intra e extracelulares) com o volume de água nestescompartimentos. Como o sódio é o principal soluto no lí-quido extracelular, a concentração do sódio no plasma ousoro indica a relação existente entre a quantidade total desoluto e água no organismo.
Normalmente, a excreção de sódio na urina não depen-de da concentração plasmática de sódio, e vários experi-mentos demonstram isto. Por exemplo, quando se expan-de o volume extracelular com solução salina isotônica, aexcreção urinária de sódio aumenta. Da mesma forma, aingestão de água, combinada à administração de vasopres-sina, causa retenção de água que, eventualmente, acarretaexpansão do volume extracelular. Com o volume extrace-lular expandido, há aumento na excreção urinária de só-dio, apesar da hiponatremia causada pela administraçãosimultânea de água e vasopressina. Um outro exemplo é asituação em que o organismo só perde água, o que causadiminuição do volume extracelular e, conseqüentemente,diminuição da excreção urinária de sódio, apesar da hiper-natremia.
Balanço do SódioA ingestão média de cloreto de sódio em um adulto
normal é de 7 g ou 150 mEq por dia.1 Para manter o equi-líbrio, a mesma quantidade deve ser excretada.2 Ao con-trário da água, cuja ingestão é controlada pela sede, nãoexiste no ser humano um apetite específico para sódio.
Uma vez absorvido, o íon sódio distribui-se no orga-nismo da seguinte maneira: 45% para o líquido extrace-lular, 7% para o líquido intracelular e 48% para o esque-leto. O sódio do esqueleto se apresenta sob duas formas:permutável (50%) e não-permutável (50%). Esta divisãoé baseada na maior ou menor facilidade com que o sódiose liberta do osso para a circulação. O sódio não-permu-tável integra áreas firmemente mineralizadas, sendomenos acessível à circulação e, portanto, dificilmente seliberta do esqueleto. O sódio permutável pode libertar-se do osso em condições especiais como a acidose meta-bólica, onde o carbonato de sódio dos cristais deposita-dos na matriz óssea neutraliza o íon H�, trocando-o pelosódio.1
A concentração plasmática de sódio está entre 135 e 145mEq/L, sendo a concentração intracelular em torno de 10%da concentração plasmática. O sódio é eliminado do orga-nismo na urina, fezes e suor. Para efeito de balanço, o queimporta é a excreção urinária de sódio. A eliminação pelosuor adquire importância somente em casos de sudoreseprofusa, pois a concentração de sódio no suor é baixa. Damesma forma, diarréias graves podem determinar perdasconsideráveis de sódio nas fezes.

134 Metabolismo do Sódio e Fisiopatologia do Edema
RESPOSTA DO RIM ÀSALTERAÇÕES NA INGESTA DE
SÓDIO
Quando se altera a ingesta de sódio, a adaptação naexcreção renal de sódio é lenta, podendo levar muitos diaspara que se iguale à ingesta.3 Observem na Fig. 10.1 que,quando a ingestão de NaCl aumenta, apenas uma partedeste incremento é eliminada no primeiro dia. O restanteé retido, juntamente com água, resultando numa expansãodo volume extracelular. A expansão do volume extracelu-lar estimula progressivamente um aumento na excreção desódio, até que a quantidade excretada se iguale à ingerida.Por outro lado, se a ingesta de sódio for reduzida abrupta-mente, levará muitos dias para que a excreção de sódio sejareduzida a uma quantidade igual à ingesta.
O mecanismo pelo qual alterações no volume extrace-lular modificam a excreção de sódio não está totalmenteesclarecido e será abordado a seguir. Normalmente, aquantidade de sódio excretado na urina está em torno de0,5% da quantidade filtrada pelo rim. Na Fig. 10.2, umúnico nefro representa a função total de ambos os rins.Considerando uma filtração glomerular de 125 ml/min eum sódio plasmático de 140 mEq/L, o sódio total filtradopor dia será de 25.200 mEq. Aproximadamente 67% dosódio filtrado são reabsorvidos no túbulo contornado pro-ximal e 10% na parte reta do túbulo proximal. Isto signifi-ca que a reabsorção proximal de sódio está em torno de 80%da carga filtrada, enquanto 20% do sódio filtrado são reab-sorvidos em segmentos distais ao túbulo proximal.
Considerando-se um fluxo urinário normal de 1 ml porminuto (1.440 minutos em 24 horas), o volume urinário es-tará em torno de 1.500 ml. Se a concentração urinária desódio for de 100 mEq/L, a excreção urinária diária de sódioserá em torno de 150 mEq ou 0,6% do sódio total filtrado.
Fig. 10.1 Balanço de sódio no homem. Observe que, quando a ingesta de sódio é subitamente elevada, apenas cerca da metade doincremento aparece na urina no primeiro dia. O restante do incremento fica retido no organismo e aumenta o volume de líquidoextracelular, que se traduz por um aumento do peso. Nos dias subseqüentes, uma fração menor de sódio é retida, e a excreção desódio aumenta progressivamente, até que em três a cinco dias a excreção se iguala à ingestão. O estímulo para o aumento na excre-ção de sódio se deve à expansão do volume extracelular. Observe também que, quando se reduz abruptamente a ingesta, a diminui-ção na excreção de sódio é também gradual e os mesmos mecanismos operam, só que de maneira inversa. (Obtido de Earley, L.E.3)
Fig. 10.2 Filtração e excreção diária de sódio num adulto normal.No diagrama, o nefro representa toda a população de nefros deambos os rins. Observe que cerca de 80% do sódio filtrado são re-absorvidos no nefro proximal e que no final apenas 0,6% da cargafiltrada aparece na urina. Observe, também, que a quantidade ex-cretada é mais ou menos igual à quantidade ingerida, o que indicaque há um balanço. (Baseado na concepção de Valtin, H. 53)
RFG � 125 ml/min = 180 L/DIAPNa
� � 140 mEq/L
UNa� � 100 mEq/L

capítulo 10 135
Pelo exposto, poderíamos deduzir que uma alteração dafiltração glomerular ou da reabsorção tubular de sódiopode comprometer o balanço de sódio e, conseqüentemen-te, o volume dos compartimentos líquidos do organismo.
Pontos-chave:
• A concentração plasmática de sódio é de135-145 mEq/L
• A adaptação renal às variações na ingestade sódio é lenta
• A excreção urinária diária de sódio deveequilibrar-se com a ingesta
• Apenas 0,6% de todo o sódio filtrado éeliminado na urina
QUEM PERCEBE E REGULA ASALTERAÇÕES DO
VOLUME EXTRACELULAR?
A homeostase dos fluidos é essencial para a manuten-ção da estabilidade circulatória. Pequenas modificações novolume extracelular devem ser prontamente identificadase corrigidas, para que o equilíbrio seja mantido.4 Existemestruturas no organismo que agem como receptores devolume, e, através de mecanismos nervosos, humorais ehormonais, provocam adaptações funcionais em váriosórgãos e fornecem aos rins os elementos para correção dosdesvios no volume extracelular1 (Quadro 10.1). Por exem-plo, a expansão de volume ativa uma seqüência de sinaisprovenientes de vários destes receptores, aumentando aexcreção de sódio. Ao contrário, a resposta à depleção devolume é a conservação renal de sal e água.4
A redistribuição interna do volume intravascular, mes-mo sem mudança no volume circulante, provoca alteraçãona excreção de sódio. Por exemplo, quando um indivíduose deita, a excreção de sódio aumenta, e, quando fica de
pé, a excreção de sódio diminui.3 Isto significa que a pos-tura influi sobre a excreção de sódio. Epstein e cols. verifi-caram que, quando se comprimia externamente uma fís-tula arteriovenosa grande, a excreção de sódio na urinaaumentava.5 No caso da fístula arteriovenosa, a compres-são externa impede a passagem do sangue arterial para osistema venoso, causando aumento do volume arterial efe-tivo. Isto sugere que o volume arterial efetivo exerce con-trole sobre o volume extracelular.
Há receptores de volume no leito vascular venoso epulmonar (intratorácicos),6 capazes de perceber reduçõesno retorno venoso e ativar uma diminuição na excreçãourinária de sal. Isto ocorre, por exemplo, quando o indiví-duo fica muito tempo em pé, quando se aplicam tornique-tes nas pernas ou em indivíduos em ventilação com pres-são positiva. De modo inverso, o aumento do retorno ve-noso torácico aumenta a excreção urinária de sódio, comose observa em indivíduos em decúbito dorsal.
O tônus simpático e a secreção de adrenalina e noradre-nalina são ativados quando existe queda no débito cardía-co ou queda de pressão arterial. Esta redução na pressãoativa os receptores cardíacos e arteriais, aumentando asdescargas em tronco cerebral que aumentam o tônus sim-pático, iniciando eventos que levam à normalização daperfusão, entre eles um aumento da reabsorção tubular desódio.7
Talvez a demonstração mais convincente da influênciada volemia intratorácica e receptores cardiopulmonares nanatriurese derive de estudos com indivíduos normais imer-sos em água até o pescoço. A pressão hidrostática do líqui-do de imersão ocasiona a redistribuição do fluido intravas-cular e do interstício dos membros inferiores para o tórax.O conseqüente aumento no volume circulante central pro-voca natriurese e aumento da diurese. Resposta similar éobtida em pacientes cirróticos, que excretam pouco sódioem condições basais.7
Foram identificados receptores de volume localizados nosátrios, seio carotídeo e arco aórtico. Quando existe queda napressão arterial ou débito cardíaco, o tônus simpático e asecreção de adrenalina e noradrenalina são ativados porestes receptores, iniciando eventos que levam à normaliza-ção da perfusão, entre eles aumento da reabsorção tubularde sódio.7 Além disso, estes receptores estão associados aocontrole da liberação de HAD (v. Cap. 9).
A liberação de HAD e a sede, mecanismos de restaura-ção do déficit de água, podem também ser estimulados poraumento da osmolalidade plasmática e pela contração isos-mótica do volume extracelular (através do sistema renina-angiotensina).
O rim percebe alterações no volume e na pressão intra-vascular através de um sistema barorreceptor localizadono aparelho justaglomerular da arteríola aferente e célu-las da mácula densa no túbulo distal (v. Cap. 7). Estes re-ceptores influenciam a atividade do sistema renina-angi-otensina-aldosterona, endotelina e óxido nítrico.7 Uma re-
Quadro 10.1 Receptores mecânicos sensíveis aalterações regionais da volemia
Receptores de volume intratorácicosAurículasVentrículo direitoCapilares pulmonares
Receptores de volume no sistema arterialArtérias carótidasArco aórtico
Receptores de volume no rimReceptores de volume no sistema nervoso centralReceptores de volume no fígado

136 Metabolismo do Sódio e Fisiopatologia do Edema
dução na pressão de perfusão renal promove liberação derenina do aparelho justaglomerular, com formação de an-giotensina II, liberação de aldosterona e retenção de sódio.
A administração de soluções distintas causa diferentestaxas de excreção de sódio. Uma expansão do comparti-mento intravascular com a administração de plasma ousangue, por exemplo, causa natriurese menos significati-va do que a obtida com quantidades equivalentes de solu-ção salina isotônica. Todavia, a administração de uma so-lução hipertônica de albumina expande o intravascular econtrai o compartimento intersticial, podendo não modi-ficar a excreção de sódio. Isto indica que outros estímulos,além da expansão absoluta do volume extracelular, sãoimportantes na excreção de sódio.3
Há sugestões de que o fígado também possua recepto-res especiais e participe da regulação da excreção de águae sal. Estudos demonstraram que a infusão de solução sa-lina isotônica ou hipertônica no sistema porta causa umanatriurese mais significativa do que se a mesma soluçãofosse infundida numa veia sistêmica.8
Pontos-chave:
• O sódio é o principal cátion do extracelular• A quantidade de sódio no organismo
determina o volume do espaço extracelular• Para manter a estabilidade circulatória, o
volume extracelular deve seradequadamente controlado
• Os sensores de volume e pressãodesencadeiam mecanismos de regulação doextracelular, aumentando ou diminuindo aexcreção de sódio
REGULAÇÃO INTRA-RENAL DAEXCREÇÃO DE SÓDIO
Num indivíduo sadio a quantidade reabsorvida de só-dio é superior a 99% da quantidade filtrada. Como a quan-tidade filtrada excede em muito a excretada, torna-se cla-ro que o rim deve possuir um sistema de conservação desódio altamente desenvolvido.
Auto-regulação RenalVários mecanismos mantêm a quantidade de sódio fil-
trada relativamente constante. Os rins são capazes de man-ter a taxa de filtração glomerular constante, mesmo quehaja amplas variações da pressão de perfusão renal. Estefenômeno é chamado auto-regulação renal. Respostas namusculatura lisa das arteríolas aferentes ocorrem em di-reta proporção com mudanças na pressão de perfusão re-
nal, mantendo estáveis o fluxo sanguíneo renal, TFG e só-dio filtrado.9
Porém, somente modificações na TFG não são suficien-tes para explicar os ajustes na excreção de sódio.4
Filtração Glomerular — BalançoGlomérulo-tubular
Observou-se que uma diminuição da filtração glomeru-lar, causada por hemorragia ou constrição da artéria renal,diminuía a excreção de sódio. Já um aumento na filtraçãoglomerular causado pela administração de solução salinaera acompanhada por aumento na excreção de sódio. Por-tanto, estes estudos demonstravam um paralelo entre fil-tração glomerular e excreção de sódio.
Entretanto, De Wardener10 e outros investigadores de-monstraram que o aumento na excreção de sódio queocorre com a expansão do volume extracelular permane-ce mesmo quando se reduz a filtração glomerular e con-seqüentemente a quantidade de sódio filtrada. Por outrolado, ao se produzir um aumento na filtração glomeru-lar, mas sem expandir o volume extracelular, a excreçãode sódio permanece inalterada ou aumenta muito pou-co. Isto tudo indica que as alterações na filtração glome-rular não são essenciais para o rim regular o volume ex-tracelular.6 O ponto principal na regulação do equilíbriode sódio é o controle de sua reabsorção,2 como veremos aseguir.
Numerosas investigações demonstraram que alteraçõesna filtração glomerular são acompanhadas por alteraçõesproporcionais na reabsorção de líquido no túbulo proxi-mal, de modo que a fração do volume filtrado que é reab-sorvida pelo túbulo proximal permanece mais ou menosconstante.1 Normalmente, 80% do filtrado glomerular sãoreabsorvidos pelo túbulo proximal.
O fenômeno pelo qual alterações na taxa de filtraçãoglomerular se acompanham de modificações correspon-dentes na reabsorção tubular de sódio é chamado de ba-lanço glomérulo-tubular (v. Quadro 10.2).1,2 Este balançoevita alterações excessivas na excreção de sódio quando afiltração é abruptamente aumentada ou diminuída. Osprincipais mecanismos responsáveis pelo balanço glomé-rulo-tubular são: pressão oncótica e hidrostática peritubu-
Quadro 10.2 Balanço glomérulo-tubular
Filtração Reabsorção Fração de Volume nãoGlomerular Proximal Reabsorção Reabsorvido(ml/min) (ml/min) (%) (ml/min)
150 120 80 30100 80 80 2050 40 80 10
Obtido de Malnic, G. e Marcondes, M.1

capítulo 10 137
lares, fatores humorais intra-renais, velocidade do fluxotubular e volume do túbulo proximal.11 Estes mecanismossão descritos a seguir.
Reabsorção e Propriedades Físicas noCapilar Peritubular
PRESSÃO ONCÓTICA PERITUBULARAlterações na concentração de albumina e pressão on-
cótica nos capilares peritubulares afetam o movimentotranstubular de sódio. A concentração de albumina no ca-pilar peritubular é determinada pela concentração plasmá-tica de albumina na arteríola eferente e pela fração de fil-tração (porção do fluxo plasmático renal que é filtrada).Portanto, um aumento no ritmo de filtração glomerularaumenta a fração de filtração, formando o ultrafiltrado(plasma sem proteínas), retirando água e eletrólitos docapilar glomerular e aumentando a concentração relativade albumina no capilar peritubular. Este aumento da pres-são oncótica favorece a reabsorção de sal e água. A dimi-nuição da filtração glomerular tem efeito oposto.
Brenner e cols. demonstraram que a diminuição da reab-sorção de sódio no túbulo proximal, que ocorre durante aexpansão do volume extracelular com solução salina iso-tônica, é decorrente da diminuição da pressão oncótica docapilar peritubular. Quando os autores perfundiam o ca-pilar peritubular com uma solução de albumina, normali-zando a pressão oncótica, a inibição da reabsorção de só-dio era corrigida.12,13
PRESSÃO HIDROSTÁTICA NOCAPILAR PERITUBULAR
Earley e cols. sugeriram que alterações na pressão hidros-tática do capilar peritubular seriam responsáveis por mo-dificações na reabsorção de sal e água.14 Um aumento dapressão capilar peritubular causaria natriurese, e a diminui-ção da pressão capilar teria um efeito oposto. O mesmo gru-po de investigadores demonstrou que a natriurese induzi-da por aumento na pressão hidrostática do capilar peritu-bular poderia ser inibida por um aumento da pressão oncó-tica do plasma. Estas observações levaram o grupo a postu-lar que o ritmo de reabsorção de sódio pode ser influencia-do pelo balanço das forças de Starling (v. Cap. 8).
Existem importantes diferenças no movimento transca-pilar de líquido entre os capilares periféricos, glomerula-res e peritubulares. As forças de Starling que norteiam atroca de líquido no capilar periférico já foram abordadasno Cap. 8, enquanto as forças que governam a filtraçãoglomerular foram abordadas no Cap. 3.
No capilar peritubular são muito distintas as forças res-ponsáveis pela troca de líquido. A arteríola eferente, fun-cionando como um vaso de resistência, contribui para aredução da pressão hidrostática entre o glomérulo e o ca-pilar peritubular. Além do mais, como o capilar peritubu-
lar recebe sangue do glomérulo, a pressão oncótica plas-mática é alta no início do capilar devido ao ultrafiltradoglomerular (líquido sem proteína). Logo, quanto maior foro ritmo de filtração glomerular em relação ao fluxo plas-mático (fração de filtração), maior será a concentração pro-téica na arteríola eferente. Assim sendo, ao contrário docapilar periférico e glomerular, o capilar peritubular é ca-racterizado por valores elevados de pressão oncótica queem muito excedem a pressão hidrostática, resultando emabsorção de líquido. Apesar de a pressão oncótica no ca-pilar peritubular diminuir ao longo do capilar, à medidaque o líquido é reabsorvido, esta pressão permanece mai-or que a pressão hidráulica.
BALANÇO GLOMÉRULO-TUBULAR EFATORES HUMORAIS INTRA-RENAIS
A participação de um fator luminal na reabsorção desódio foi sugerida por Leyssac.15 Segundo este autor, umaumento na reabsorção tubular proximal reduz a pressãointraluminal e, conseqüentemente, aumenta as forças quepromovem a filtração glomerular. Um maior ritmo de fil-tração glomerular aumenta a quantidade de líquidoofertado ao túbulo proximal, restaurando o balanço glo-mérulo-tubular. Uma diminuição na reabsorção tubularaumentaria a pressão intraluminal, a qual diminuiria a fil-tração glomerular.
Thuray e Schnermann, por sua vez, propuseram ummecanismo diferente para explicar a relação entre a filtra-ção glomerular e a reabsorção tubular de sódio.16 Segundoestes autores, a quantidade de sódio que atinge a máculadensa do nefro pode, por um mecanismo de feedback (con-trole retrógrado), controlar a filtração glomerular destenefro, através da liberação local de renina e geração deangiotensina II, que é um potente constritor de músculoliso.
Um aumento na filtração glomerular aumenta a quan-tidade de sal e água que chega à mácula densa. Isto pro-move a liberação de renina e formação de angiotensina II.A angiotensina II causa constrição da arteríola aferente,diminuindo a filtração glomerular e restaurando, assim, obalanço glomérulo-tubular. Uma redução da filtração glo-merular resulta em diminuição da quantidade de sal e águaque atinge a mácula densa, havendo então redução na li-beração de renina. Com isso, menos angiotensina II é for-mada, resultando em vasodilatação da arteríola aferente,o que causa aumento na filtração glomerular. Posterior-mente, os mesmos autores concluíram que não era a con-centração de sódio intraluminal na mácula densa que da-ria o sinal para liberação de renina, e sim a quantidade desódio transportada pelas células da mácula densa e queentraria em operação somente quando houvesse aumentono transporte de sódio a esse nível. No entanto, até o mo-mento esta teoria é conflitante e talvez não tenha partici-pação na regulação da filtração glomerular em condiçõesfisiológicas.

138 Metabolismo do Sódio e Fisiopatologia do Edema
REABSORÇÃO DEPENDENTE DA VELOCIDADEDO FLUXO DE LÍQUIDO TUBULAR
Alguns estudos mostram que a reabsorção de líquido émaior no segmento inicial do túbulo contornado proximaldo que nos segmentos mais distais. Postulou-se, então, queo acúmulo de um soluto pouco reabsorvível nos segmen-tos iniciais do túbulo contornado proximal (acúmulo de-vido à reabsorção de água, que progressivamente concen-tra este soluto) inibiria a reabsorção de sal nos segmentosmais distais. Entretanto, túbulos isolados e perfundidos invitro não exibiram esta característica de reabsorção aumen-tada no segmento inicial do túbulo contornado proximal(TCP). Mas, quando o líquido perfundido utilizado foi umultrafiltrado do plasma, esta relação entre fluxo e reabsor-ção de sódio foi novamente detectada.17 A conclusão é deque esta relação fluxo/reabsorção ainda padece de de-monstração mais convincente.
REABSORÇÃO DEPENDENTE DO VOLUMEDO TÚBULO PROXIMAL
Esta teoria propõe que o ritmo de absorção de líquidodo túbulo proximal é diretamente proporcional ao volu-me tubular. Segundo os proponentes desta teoria, a varia-ção do volume tubular é importante, pois expõe o filtradoglomerular a uma maior ou menor área de reabsorção epermite um maior tempo de contato do líquido intratubu-lar com as paredes do túbulo proximal.18 Assim sendo, umaumento na filtração glomerular proporciona um volumemaior de filtrado e, conseqüentemente, maior volume tu-bular, que se acompanha de aumento na sua capacidadede reabsorção. Uma redução da filtração glomerular reduzo volume de filtrado, e, portanto, o volume tubular, redu-zindo a capacidade reabsortiva. Em face de outras investi-gações, que concluíram que o volume tubular não é fatorimportante no balanço glomérulo-tubular, a hipótese ori-ginal não é por todos aceita.
Em resumo, pode-se afirmar que alterações na filtraçãoglomerular podem ou não ser acompanhadas de alteraçõesna excreção de sódio. Tudo depende de como se alterou a
filtração glomerular. Se o volume extracelular não é alte-rado, um aumento na filtração glomerular acompanha-sede pouco ou nenhum aumento na excreção de sódio. Poroutro lado, uma expansão do volume extracelular semprecausa aumento na excreção de sódio, mesmo que não sereduza a filtração glomerular.
Atualmente, vários investigadores têm tentado esclare-cer o papel destes fatores na reabsorção distal de sódio.Alguns estudos sugerem que as alterações nas forças deStarling são também capazes de alterar a reabsorção desódio no nefro distal.
TIPOS DE TRANSPORTEDE SÓDIO
O transporte ativo de Na� através de tecidos epiteliaisé o processo fisiológico primário responsável pela manu-tenção do balanço de sal em vertebrados.
O conhecimento que se tem sobre o transporte tubularde sódio deve-se ao estudo de segmentos isolados do nefroatravés da técnica de micropunção em animais como o rato(Quadro 10.3). Nesta técnica, obtêm-se amostras do líqui-do tubular através de micropipetas. Além disto, os segmen-tos do nefro podem ser isolados e perfundidos in vitro,observando-se sua função. Mais recentemente, a evoluçãodas técnicas de micropunção (patch-clamp) e a biologiamolecular trouxeram grandes progressos no entendimen-to do transporte de íons e solutos através de membranasbiológicas.
Pela técnica patch-clamp uma pipeta cheia de líquido écolocada contra a superfície da célula e leve sucção é apli-cada, permitindo o estudo do movimento de íons peloscanais existentes nesta área. É possível até mesmo obterdados de um único canal e saber quanto tempo permane-ce aberto ou fechado (gating).
Os mecanismos de entrada de sódio nas células tubula-res são:
a) Via canais de sódio: Esta entrada é característica dotúbulo distal (contornado) e ducto coletor, e ocorre pelamembrana apical. Estes canais são especificamente blo-queados pelo diurético amiloride.
b) Acoplada ao movimento de outros íons ou solutos:Estes sistemas de co-transporte são encontrados em todoo nefro e são as vias predominantes de transporte api-cal de Na� no túbulo proximal e ramo espesso ascenden-te da alça de Henle. Os sistemas de co-transporte sãoclassificados em symporters ou antiporters. Os symportersoperam o movimento de Na� e o íon ou soluto acopla-do na mesma direção. Por exemplo, o transportador deNa�/glicose, em que ambos são transportados paradentro da célula. Já os antiporters trocam o Na� por ou-tro íon ou soluto. Um exemplo de sistema antiporter é oco-transporte de Na�/H�.
Pontos-chave:
• O ponto principal na regulação do balançodo sódio é o controle de sua reabsorção
• Balanço glomérulo-tubular: é ummecanismo de ajuste na reabsorção de sódiopelos túbulos, de acordo com a filtraçãoglomerular
• Variações nas pressões oncótica ehidrostática peritubulares, pressão e volumetubulares e fatores hormonais afetam aexcreção de sódio
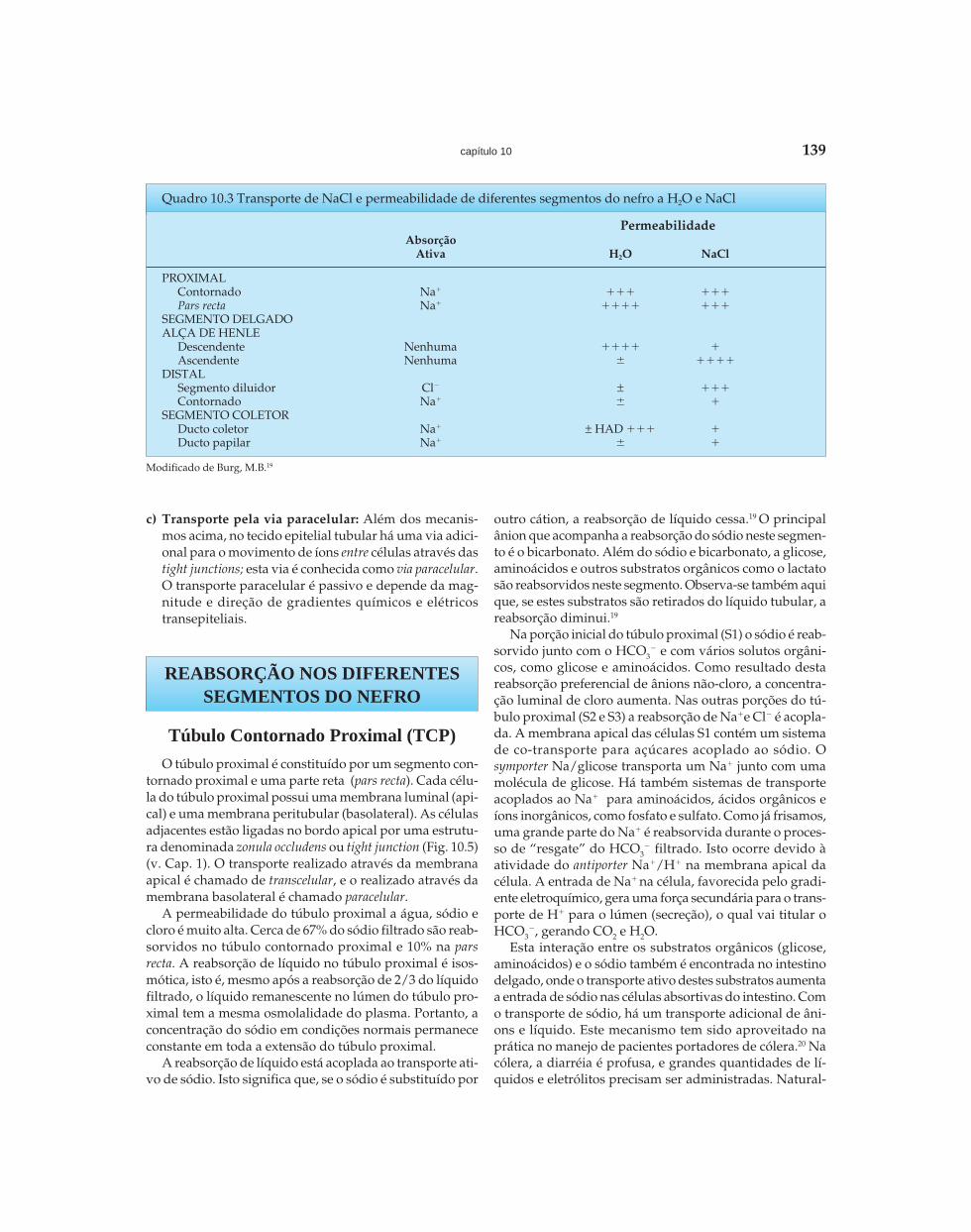
capítulo 10 139
c) Transporte pela via paracelular: Além dos mecanis-mos acima, no tecido epitelial tubular há uma via adici-onal para o movimento de íons entre células através dastight junctions; esta via é conhecida como via paracelular.O transporte paracelular é passivo e depende da mag-nitude e direção de gradientes químicos e elétricostransepiteliais.
REABSORÇÃO NOS DIFERENTESSEGMENTOS DO NEFRO
Túbulo Contornado Proximal (TCP)
O túbulo proximal é constituído por um segmento con-tornado proximal e uma parte reta (pars recta). Cada célu-la do túbulo proximal possui uma membrana luminal (api-cal) e uma membrana peritubular (basolateral). As célulasadjacentes estão ligadas no bordo apical por uma estrutu-ra denominada zonula occludens ou tight junction (Fig. 10.5)(v. Cap. 1). O transporte realizado através da membranaapical é chamado de transcelular, e o realizado através damembrana basolateral é chamado paracelular.
A permeabilidade do túbulo proximal a água, sódio ecloro é muito alta. Cerca de 67% do sódio filtrado são reab-sorvidos no túbulo contornado proximal e 10% na parsrecta. A reabsorção de líquido no túbulo proximal é isos-mótica, isto é, mesmo após a reabsorção de 2/3 do líquidofiltrado, o líquido remanescente no lúmen do túbulo pro-ximal tem a mesma osmolalidade do plasma. Portanto, aconcentração do sódio em condições normais permanececonstante em toda a extensão do túbulo proximal.
A reabsorção de líquido está acoplada ao transporte ati-vo de sódio. Isto significa que, se o sódio é substituído por
outro cátion, a reabsorção de líquido cessa.19 O principalânion que acompanha a reabsorção do sódio neste segmen-to é o bicarbonato. Além do sódio e bicarbonato, a glicose,aminoácidos e outros substratos orgânicos como o lactatosão reabsorvidos neste segmento. Observa-se também aquique, se estes substratos são retirados do líquido tubular, areabsorção diminui.19
Na porção inicial do túbulo proximal (S1) o sódio é reab-sorvido junto com o HCO3
� e com vários solutos orgâni-cos, como glicose e aminoácidos. Como resultado destareabsorção preferencial de ânions não-cloro, a concentra-ção luminal de cloro aumenta. Nas outras porções do tú-bulo proximal (S2 e S3) a reabsorção de Na�e Cl� é acopla-da. A membrana apical das células S1 contém um sistemade co-transporte para açúcares acoplado ao sódio. Osymporter Na/glicose transporta um Na� junto com umamolécula de glicose. Há também sistemas de transporteacoplados ao Na� para aminoácidos, ácidos orgânicos eíons inorgânicos, como fosfato e sulfato. Como já frisamos,uma grande parte do Na� é reabsorvida durante o proces-so de “resgate” do HCO3
� filtrado. Isto ocorre devido àatividade do antiporter Na�/H� na membrana apical dacélula. A entrada de Na� na célula, favorecida pelo gradi-ente eletroquímico, gera uma força secundária para o trans-porte de H� para o lúmen (secreção), o qual vai titular oHCO3
�, gerando CO2 e H2O.Esta interação entre os substratos orgânicos (glicose,
aminoácidos) e o sódio também é encontrada no intestinodelgado, onde o transporte ativo destes substratos aumentaa entrada de sódio nas células absortivas do intestino. Como transporte de sódio, há um transporte adicional de âni-ons e líquido. Este mecanismo tem sido aproveitado naprática no manejo de pacientes portadores de cólera.20 Nacólera, a diarréia é profusa, e grandes quantidades de lí-quidos e eletrólitos precisam ser administradas. Natural-
Quadro 10.3 Transporte de NaCl e permeabilidade de diferentes segmentos do nefro a H2O e NaCl
PermeabilidadeAbsorção
Ativa H2O NaCl
PROXIMALContornado Na� ��� ���Pars recta Na� ���� ���
SEGMENTO DELGADOALÇA DE HENLE
Descendente Nenhuma ���� �Ascendente Nenhuma � ����
DISTALSegmento diluidor Cl� ± ���Contornado Na� � �
SEGMENTO COLETORDucto coletor Na� ± HAD ��� �Ducto papilar Na� � �
Modificado de Burg, M.B.19

140 Metabolismo do Sódio e Fisiopatologia do Edema
mente a via oral é mais prática e mais econômica. No en-tanto, a administração de uma solução de água e eletróli-tos acompanha-se de uma reabsorção intestinal pequena,insuficiente para corrigir as perdas. No entanto, se a solu-ção eletrolítica contiver glicose, ocorre aumento na reab-sorção intestinal de sódio e, conseqüentemente, de outrosânions e líquido.
Do total de NaCl reabsorvido, estima-se que 2/3 mo-vem-se pela via transcelular e 1/3 pela via paracelular. Comoa concentração intracelular de sódio é baixa, a entrada desódio do lúmen para a célula depende de um gradienteeletroquímico. Já a principal via de saída do Na� da célulaé pela membrana basolateral, através da Na,K-ATPase.Além disto, o Na� sai através do symporter 1 Na�/3HCO3
�.O transporte de sódio para fora da célula é ativo (Fig. 10.4).
O transporte paracelular de NaCl é passivo. É movidopor gradientes químicos e elétricos transepiteliais (trans-porte difuso) ou por fluxo de líquido através do epitélio(transporte convectivo ou solvent drag effect — efeito arras-tão). A via paracelular tem uma alta permeabilidade a NaCle água. Já mencionamos também que a composição do lí-quido tubular é diferente nas porções iniciais e finais dotúbulo proximal. Assim, no segmento inicial do TP há umaqueda dramática na concentração de HCO3
�, glicose eaminoácidos, e um aumento concomitante no cloreto. Naparte final do TP este cloreto se difunde para o interstíciopassivamente e a geração de voltagem proporciona a for-ça para a reabsorção difusa de Na�.
A reabsorção de água pelo TP proporciona um meca-nismo adicional para o transporte paracelular de NaCl.Com a reabsorção de solutos, o líquido luminal fica umpouco hipotônico em relação ao interstício. Este pequenogradiente osmótico é suficiente para causar a reabsorçãode grande quantidade de água e junto levar o NaCl peloefeito de arrasto.
O sódio parece entrar na célula passivamente, atravésda membrana apical, e é transportado para o espaço in-tercelular. Isto causa aumento na concentração (osmola-lidade) no espaço intercelular, o que atrai água passiva-mente devido ao gradiente osmótico. Com a chegada deágua, a pressão hidrostática aumenta no espaço interce-lular e o líquido é forçado a sair através da membranabasal (Fig. 10.5). Portanto, a pressão hidrostática elevadado espaço intercelular cria um gradiente de pressão en-tre este espaço e o interstício, fazendo com que este líqui-do passe para o interstício. Daí para o capilar, há um ou-tro gradiente de pressão determinado pela pressão hi-drostática intracapilar (que favorece a saída de líquido) epela pressão oncótica do plasma (que se opõe à filtraçãodo líquido). Os solutos orgânicos transportados para oespaço intercelular aumentam a osmolalidade, explican-do em parte por que eles, quando presentes no líquidotubular, aumentam a reabsorção de líquido. Naturalmen-te, o líquido tubular contém vários íons e o movimentode sódio altera o ritmo de absorção destes íons. Quando
o ambiente hiperosmolar do espaço intercelular criadopela reabsorção ativa de sódio atrai água, também atraioutros solutos. (efeito arrastão). Isto explica por que, quan-do se expande o volume extracelular e se reduz a reab-sorção proximal de sal e água, também se percebe dimi-nuição na reabsorção de potássio, cloro, bicarbonato, cál-cio e fosfato.
O balanço dos gradientes de pressão oncótica e hidros-tática é que determina a força que move o líquido do in-terstício para o capilar peritubular. Se a pressão hidrostá-tica aumentar, ou a pressão oncótica diminuir, menos lí-quido passará do interstício para o capilar. A presença demais líquido no interstício aumenta a pressão hidrostáticano local. Haverá, então, inversão do gradiente de pressãono espaço intercelular e fluxo retrógrado de sal e água parao lúmen tubular. Além disto, poderá haver redução notransporte ativo de sódio para o espaço intercelular devi-
Fig. 10.3 Repercussões sobre a excreção urinária de sódio quan-do se aumenta o ritmo de filtração glomerular, com ou sem ex-pansão simultânea do volume extracelular, através de soluçãosalina isotônica e hormônio da paratireóide (PTH), respectiva-mente. Observe que, quando se administra PTH, a carga filtradade sódio (CFNa) aumenta aproximadamente 6.000 mEq/min, en-quanto a excreção de sódio (UNaV) aumenta somente 100 mEq/min. Durante a expansão do volume, a CFNa aumentou 1.200mEq/min com uma natriurese significativa (1.600 mEq/min).(Obtido de Slatopolski, E. e col.54)
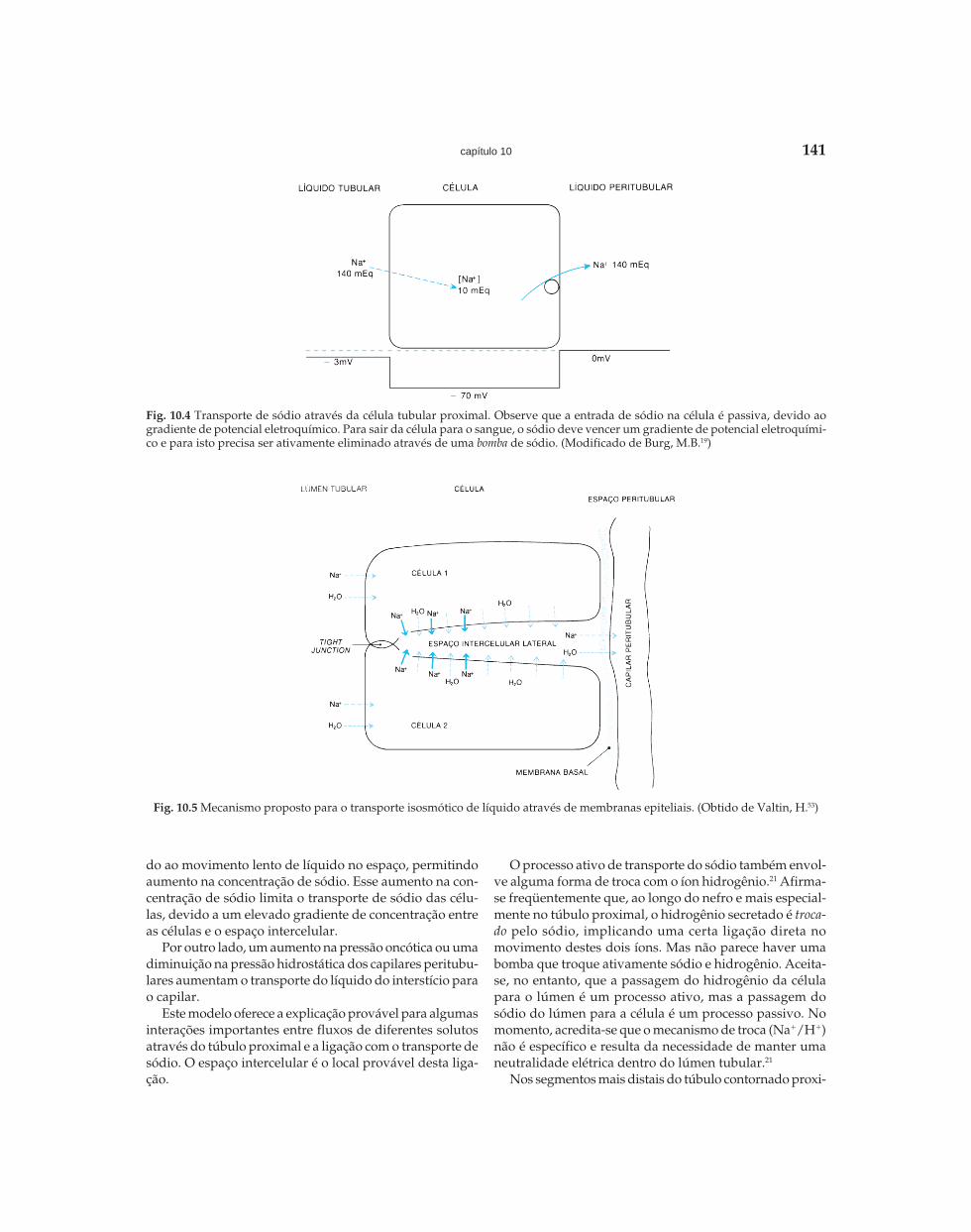
capítulo 10 141
do ao movimento lento de líquido no espaço, permitindoaumento na concentração de sódio. Esse aumento na con-centração de sódio limita o transporte de sódio das célu-las, devido a um elevado gradiente de concentração entreas células e o espaço intercelular.
Por outro lado, um aumento na pressão oncótica ou umadiminuição na pressão hidrostática dos capilares peritubu-lares aumentam o transporte do líquido do interstício parao capilar.
Este modelo oferece a explicação provável para algumasinterações importantes entre fluxos de diferentes solutosatravés do túbulo proximal e a ligação com o transporte desódio. O espaço intercelular é o local provável desta liga-ção.
O processo ativo de transporte do sódio também envol-ve alguma forma de troca com o íon hidrogênio.21 Afirma-se freqüentemente que, ao longo do nefro e mais especial-mente no túbulo proximal, o hidrogênio secretado é troca-do pelo sódio, implicando uma certa ligação direta nomovimento destes dois íons. Mas não parece haver umabomba que troque ativamente sódio e hidrogênio. Aceita-se, no entanto, que a passagem do hidrogênio da célulapara o lúmen é um processo ativo, mas a passagem dosódio do lúmen para a célula é um processo passivo. Nomomento, acredita-se que o mecanismo de troca (Na�/H�)não é específico e resulta da necessidade de manter umaneutralidade elétrica dentro do lúmen tubular.21
Nos segmentos mais distais do túbulo contornado proxi-
Fig. 10.4 Transporte de sódio através da célula tubular proximal. Observe que a entrada de sódio na célula é passiva, devido aogradiente de potencial eletroquímico. Para sair da célula para o sangue, o sódio deve vencer um gradiente de potencial eletroquími-co e para isto precisa ser ativamente eliminado através de uma bomba de sódio. (Modificado de Burg, M.B.19)
Fig. 10.5 Mecanismo proposto para o transporte isosmótico de líquido através de membranas epiteliais. (Obtido de Valtin, H.53)

142 Metabolismo do Sódio e Fisiopatologia do Edema
mal, o transporte ativo de sódio ainda é o processo básicoresponsável pela absorção de líquido. Como no segmentoproximal do TCP a reabsorção de bicarbonato foi mais rá-pida que a de cloro (devido ao processo de acidificação),neste segmento distal a concentração de bicarbonato nolíquido tubular é menor e a do cloro maior, e é possível queo transporte de cloro neste segmento seja passivo, devidoao gradiente de concentração entre o lúmen e o sangue.Alguns acreditam que a difusão do cloro, através destegradiente químico, possa ser a força primária na reabsor-ção de água e sal nestes segmentos mais distais do TCP.Devido à extensa reabsorção no segmento inicial do TCP,a concentração de glicose, aminoácidos e outros substra-tos orgânicos diminui no segmento distal, e, conseqüente-mente, o ritmo de absorção de líquido também diminui. Apars recta é relativamente inacessível à micropuntura, ra-zão pela qual tem sido estudada em preparações in vitro.O transporte de sódio é ativo, e o de cloro, provavelmentepassivo.
Segmentos Delgados da Alça de HenleAs características de permeabilidade dos segmentos
delgados à água e solutos são bastante importantes para acompreensão do transporte destes elementos.19
No segmento delgado descendente a permeabilidade àágua é alta, enquanto no segmento delgado ascendente ébaixa. A permeabilidade ao sódio e à uréia é maior no seg-mento delgado ascendente do que no descendente. Nosegmento ascendente a permeabilidade ao sódio excede ada uréia.
A evidência atual é de que não há transporte ativo deNaCl nos segmentos delgados da alça de Henle, e as ca-racterísticas de permeabilidade anteriormente descritasexplicam o transporte passivo de NaCl e uréia nos segmen-tos delgados da alça de Henle.
No segmento descendente ocorre concentração de so-luto devido à saída passiva de água, determinada pelogradiente osmótico. Alguns autores sugeriram que o au-mento na concentração de soluto também se dá devido àentrada de soluto do interstício para o lúmen tubular (de-vido ao gradiente osmótico), embora em menor proporçãoque a saída de água. Na curva da alça, o líquido é hiperos-molar e tem a mesma osmolalidade que o interstício, masa concentração de NaCl é superior à do interstício. Aisosmolalidade é dada pela uréia, cuja concentração nointerstício é maior que a do lúmen tubular. Devido a estascaracterísticas de concentração e de permeabilidade dosegmento ascendente delgado, o NaCl difunde-se do lú-men para o interstício. A uréia não se difunde tão rapida-mente do interstício para o lúmen, porque o segmento émais permeável ao sódio do que à uréia.
Desta forma, ocorre a reabsorção de NaCl e diluição dolíquido tubular no segmento ascendente delgado da alçade Henle (v. Cap. 4).
Segmento Ascendente Espesso da Alçade Henle (Segmento Diluidor)
Este segmento estende-se do ramo ascendente delgadoà mácula densa. A permeabilidade à água é baixa e a reab-sorção de sal em excesso (em relação à água) gera um flui-do tubular diluído. No segmento espesso ascendente, areabsorção ativa de cloro gera uma diferença de potencialcapaz de reabsorver passivamente o sódio.
O ritmo de reabsorção de NaCl no segmento diluidordepende da quantidade absoluta de NaCl que chega. Poroutro lado, o ritmo de transporte de NaCl no segmentodiluidor depende da concentração de NaCl no lúmen. Seaumenta a quantidade absoluta do NaCl que chega ao seg-mento diluidor, aumenta a concentração de NaCl no seg-mento e, portanto, aumenta a reabsorção de NaCl. Se areabsorção de NaCl no túbulo proximal diminui, aumen-ta a quantidade de NaCl que chega ao segmento diluidor,e logo aumenta a reabsorção de NaCl, minimizando as al-terações na quantidade de NaCl ofertada ao túbulo con-tornado distal.
Este segmento normalmente absorve 20% da carga fil-trada de NaCl. A entrada de Na� e Cl� ocorre através damembrana apical por um symporter eletroneutro: 1 Na�:1K�:2 Cl�. Os diuréticos de alça são inibidores específicosdeste transportador. O gradiente de Na� do lúmen para acélula gera um grande componente da força propulsorapara reabsorção destes íons. O gradiente de Na� é manti-do pela Na,K-ATPase na membrana basolateral, que ati-vamente elimina o Na� do interior da célula. Além da viatranscelular, o Na� é reabsorvido pela via paracelular. Comodurante o transporte transcelular se gera uma voltagemtransepitelial, a absorção de Na� se faz pela via paracelu-lar (aproximadamente 50% da reabsorção de Na�).
Túbulo Contornado Distal (TCD)
Aproximadamente 7% da carga filtrada de NaCl é aquireabsorvida. Estende-se da mácula densa até a junção comoutro túbulo contornado, formando, a partir de então, oducto coletor cortical.
A reabsorção de sal continua neste segmento e a reab-sorção de água depende da resposta deste segmento aoHAD. O líquido tubular que chega ao TCD é hiposmóticodevido à reabsorção de NaCl no segmento diluidor. Emalgumas espécies de animais, como o cão e o macaco, olíquido permanece hiposmótico porque a parte distal doTCD (túbulo coletor) não responde à ação do HAD. Emoutras espécies animais, a osmolalidade do líquido aumen-ta, e isto porque o segmento distal do TCD responde à açãodo HAD.
Acredita-se que Na� e Cl� entram na célula por um sis-tema de transporte eletroneutro e a força propulsora é ogradiente de Na� do lúmen para a célula. O gradiente é

capítulo 10 143
mantido pela atividade da Na,K-ATPase na membranabasolateral. A reabsorção de cloro ocorre de modo ativo epassivo.
Ducto ColetorNormalmente este segmento reabsorve 3% da carga fil-
trada de sódio. Entretanto, é nesta porção que existem osmaiores gradientes de concentração entre sangue e urinae onde são feitos os ajustes finais para a excreção de íons.
Os ductos coletores vão desde o córtex externo até aponta da papila. São divididos em três segmentos. O pri-meiro segmento (ducto coletor cortical) se estende do cór-tex externo até a junção corticomedular. Contém dois ti-pos de células: célula principal e célula intercalada. A cé-lula principal é local de reabsorção de Na� e K�, e a célulaintercalada está envolvida na acidificação da urina. A reab-sorção ativa de Na� se faz pela atividade da Na,K-ATPaselocalizada na membrana basolateral. Com esta atividade,estabelece-se um grande gradiente eletroquímico para aentrada do Na� na célula através de um canal seletivo deNa�, sensível ao amiloride. O segundo segmento (ductocoletor medular externo) vai da junção corticomedular atéa junção da medula interna e externa. O transporte de Na�
parece ser o mesmo do ducto coletor cortical. O terceirosegmento (ducto coletor medular interno) é um segmentomuito ramificado com um único tipo de célula. Pouco sesabe sobre o transporte de íons neste segmento.
Pontos-chave:
• O túbulo proximal (parte contornada eparte reta) é o principal local de reabsorçãodo sódio filtrado — cerca de 77% do sódiofiltrado são aí reabsorvidos
• O restante do sódio é reabsorvido nossegmentos distais ao túbulo proximal
OUTROS FATORES QUEREGULAM A EXCREÇÃO DE
SÓDIO
A regulação da excreção de sódio depende em últimaanálise do controle da diferença entre a quantidade de só-dio filtrada e a quantidade reabsorvida. Teoricamente, aexcreção de sódio pode ser regulada por alterações na fil-tração glomerular ou reabsorção tubular. Mas, como já foimencionado, a filtração glomerular não é peça crítica naexcreção de sódio, e, portanto, alterações na excreção sãoresultado de alterações da reabsorção tubular. Os fatoresque parecem ter um papel importante na regulação daexcreção de sódio são apresentados a seguir.11
Redistribuição do Filtrado Glomerular
O rim do mamífero é formado por uma população he-terogênea de nefros. Aproximadamente 85% dos nefros sãosuperficiais, localizados próximo ao córtex (nefros corti-cais), e possuem alças de Henle curtas. Os nefros restan-tes, mais ou menos 15%, estão localizados na junção docórtex com a medula (nefros justamedulares) e possuemalças de Henle longas.
A excreção renal de sódio pode ser influenciada poruma redistribuição de filtrado glomerular entre os nefroscorticais e justamedulares. Os nefros corticais (alça curta)teriam mais chances de deixar o sódio escapar do que osjustamedulares (alça longa). Por outro lado, uma redistri-buição do filtrado dos nefros corticais para os justamedu-lares facilitaria a retenção de sódio. Embora seja uma hi-pótese atraente, ainda faltam dados mais convincentespara aceitá-la.
Angiotensina II
A angiotensina II é produzida quando a renina é libera-da pelo aparelho justaglomerular. A angiotensina integrao sistema renina-angiotensina-aldosterona (v. Cap. 7). Umadiminuição do volume circulante efetivo é estímulo à pro-dução de renina, que gera angiotensina; esta estimula asecreção de aldosterona, que, por sua vez, aumenta a reab-sorção tubular de sódio, tentando restaurar o volume cir-culante.
O principal efeito renal da angiotensina II é estimular areabsorção de NaHCO3
� no túbulo contornado proximal.Como o fluido deve permanecer isosmótico neste local, aágua é reabsorvida, e o cloro intraluminal aumenta. Esteaumento cria uma diferença de concentração que leva àreabsorção passiva de cloro (arrastando sódio pela eletro-neutralidade e água pela isosmolalidade). A angiotensi-na II é também potente vasoconstritora seletiva de arterí-olas eferentes. Com isso, ocorre aumento na fração de fil-tração, alterando a reabsorção proximal devido a fatoresfísicos.2
Aldosterona
É um hormônio secretado pela zona glomerulosa dasglândulas adrenais. É capaz de estimular o transporte deeletrólitos por células epiteliais de glândulas salivares, tratogastrintestinal e túbulos renais. A aldosterona tem umpapel importante na manutenção da homeostase do Na�,e chega a ser responsável por 5% da reabsorção total desódio.
A secreção de aldosterona é estimulada pela angioten-sina, concentração de potássio plasmático e hormônio adre-nocorticotrófico (ACTH). Aparentemente a aldosteronaentra na célula por difusão, migra até o núcleo e induz a

144 Metabolismo do Sódio e Fisiopatologia do Edema
síntese de proteínas que aumentam a entrada de sódio domeio externo para o interior da célula.
No epitélio tubular, a aldosterona induz aumento dapermeabilidade da membrana apical ao sódio e, ao mes-mo tempo, excreção de potássio. Após ser absorvido, osódio é então removido para o capilar peritubular pelabomba de sódio. O transporte ao nível da bomba de sódio tam-bém está vinculado ao de potássio. À medida que o sódioé expulso da célula, aumenta a concentração intracelularde potássio, o qual, devido ao gradiente químico que seestabelece entre o meio intracelular e o meio extracelular,sai passivamente da célula1 (v. também Cap. 12).
Fatores Físicos e Volume doEspaço Extracelular
Como já abordamos, há evidência de que fatores físicosinfluenciam o ritmo de absorção de líquido do túbulo con-tornado proximal. Os principais fatores são: hematócrito,concentração plasmática de proteínas e as pressões hidros-táticas na artéria renal, veia renal e ureter.22
O papel das pressões oncótica e hidrostática do capilarperitubular já foi comentado. Com relação à pressão venosarenal, demonstrou-se que um aumento desta pressão di-minui a reabsorção de sódio no nefro proximal, desde quenão haja redução da filtração glomerular. Quando o volu-me do espaço extracelular está reduzido, a urina elimina-da contém quantidades muito pequenas de sódio. O inver-so ocorre quando o espaço extracelular encontra-se expan-dido. Nos indivíduos euvolêmicos, o rim excreta a cargadiária de NaCl. Então, não se costumam definir valores“normais” de sódio na urina, pois os mesmos devem seravaliados de acordo com o estado fisiológico e a ingestapelo paciente.2
Quanto ao hematócrito, uma redução deste causa au-mento na excreção de sódio e redução da fração de filtra-ção e da resistência vascular renal. Estes efeitos podem sermediados pela alteração da viscosidade do sangue na cir-culação pós-glomerular, a qual, alterando a fração de fil-tração e a resistência vascular renal, altera as pressõesperitubulares oncótica e hidrostática, respectivamente.
Hormônio Natriurético
Observações experimentais conduziram ao conceito daexistência de um regulador da bomba Na,K-ATPase hámais de 30 anos.10 Foram as experiências de De Wardenere cols. que demonstraram que a natriurese que ocorria coma infusão de solução salina não dependia dos dois fatoresaté então considerados importantes no controle da excre-ção de sódio, isto é, ritmo de filtração glomerular e aldos-terona.10 Os experimentos iniciais foram feitos com circu-lação cruzada entre animais, um dos quais tinha o volumeextracelular expandido.10 Os efeitos natriuréticos da expan-
são do espaço extracelular em um animal também ocorri-am no segundo animal. A expansão do intravascular comsolução salina provocava diurese ativa, sem modificaçõesna pressão de perfusão renal, taxa de filtração glomerular,ou atividade mineralocorticóide. Presumiu-se que a natriu-rese era devida a uma substância circulante que exerciaseus efeitos diretamente nos processos de reabsorção tu-bular de sódio.
Experimentos posteriores confirmaram que extratos doplasma, urina e certos tecidos eram natriuréticos in vivo eapresentavam um efeito direto no transporte transepiteli-al do sódio. Entre os vários fatores natriuréticos isolados,o fator isolado por Bricker e cols. parece apresentar a me-lhor correlação com a manipulação renal de sódio. Estefator foi encontrado também no sangue e na urina de pa-cientes urêmicos.23,24 Estas substâncias possuem caracterís-ticas semelhantes aos digitálicos. A descoberta destas subs-tâncias nos tecidos dos mamíferos e a existência de isofor-mas de Na,K-ATPase com diferentes afinidades pelos gli-cosídeos cardíacos sugerem que a bomba Na,K-ATPase éendogenamente regulada por este composto. Porém, ain-da não foi esclarecido se o hormônio natriurético e o inibi-dor digital-like da bomba Na,K-ATPase são a mesma mo-lécula. Possivelmente o local de origem do hormônio na-triurético é o hipotálamo. Cogita-se que esta substância seorigina nas adrenais.25,26
O hormônio natriurético induz:
a) natriurese in vivo;b) inibição do transporte ativo de sódio in vitro;c) inibição da Na,K-ATPase;d) inotropismo positivo ee) reatividade vascular aumentada (pode estar envolvido
na gênese da hipertensão essencial).
Recentemente a estrutura química do inibidor endóge-no da Na,K-ATPase foi caracterizada como um isômero doglicosídeo cardíaco ouabaína. É possível que mais de umcomposto digital-like esteja presente em humanos.25
Outros hormônios conhecidos afetam a excreção de só-dio. A ocitocina pode aumentar a excreção de sódio, masnão há evidência de que normalmente participe da regu-lação da excreção de sódio. A vasopressina, quando admi-nistrada por muito tempo, pode aumentar a excreção desódio, parecendo isto ocorrer por expansão do volumeextracelular, devido à retenção de água.
A angiotensina, quando administrada em doses capa-zes de elevar a pressão arterial, pode aumentar a excreçãode sódio, na ausência de uma elevação da filtração glome-rular. O efeito parece ser devido a um aumento na pressãohidrostática do capilar peritubular.
Fator Natriurético Atrial (FNA)Na década de 60, estudos demonstraram a presença de
grânulos nos miócitos atriais. Em 1981 confirmou-se que

capítulo 10 145
estes grânulos produzem substâncias que possuem impor-tante participação na regulação do volume extracelular. Ainvestigação inicial demonstrou que a administração en-dovenosa de um extrato atrial causava uma abrupta diu-rese, natriurese, caliurese e uma diminuição da pressãoarterial. Mais recentemente verificou-se que este fator atrialnatriurético é um peptídeo, cuja seqüência de aminoácidosjá foi identificada e sintetizada. Em seres humanos estepeptídeo provoca redução da pressão arterial média, ele-vação do ritmo de filtração glomerular, do fluxo urinárioe aumento da excreção de sódio e potássio. A elevação doritmo de filtração produzida se acompanhou de fluxo plas-mático renal inalterado ou diminuído.26,27
O mecanismo pelo qual o fator atrial eleva a filtraçãoglomerular não está elucidado. É possível que exerça efei-to vasoconstritor aferente e eferente,26 elevando a pressãocapilar glomerular e, portanto, o ritmo de filtração. Outrashipóteses seriam: redistribuição da filtração glomerularpara nefros mais profundos e elevação do coeficiente defiltração. O FNA também diminui a reabsorção de sódiono túbulo proximal, através da liberação local de dopami-na e inibição da liberação de renina pelo rim, inibição daliberação de aldosterona pelas adrenais e inibição da rea-bsorção proximal mediada pela angiotensina II.26,27 A re-dução da secreção de renina pode ser devida em parte aum aumento na carga de sódio para a mácula densa gera-da pela elevação do ritmo de filtração glomerular. No mús-culo liso de grandes artérias isoladas e pré-constritas, lei-tos vasculares periféricos e músculo liso intestinal, o FNAproduz relaxamento.
Aparentemente, o estiramento das paredes dos átrioscardíacos é o principal estímulo à síntese do fator natriuré-tico atrial, como ocorre na sobrecarga de volume.25 Porém,as células ventriculares podem ser recrutadas para a suaprodução.24 Em pacientes com doença cardíaca ou pulmo-nar, o FNA pode ser utilizado como marcador de prognós-tico, pois existe correlação entre os níveis de FNA circu-lantes e as pressões de átrio direito e esquerdo.25
A principal forma circulante de FNA é um peptídeo de28 aminoácidos, consistindo nos aminoácidos 99 a 126 daextremidade C da pró-FNA. Além desta forma, já foramisolados e descritos outros tipos de agentes natriuréticos,que podem ter importância similar ou superior ao FNA emtermos de natriurese.26 Estas substâncias diferem do FNApela seqüência de aminoácidos envolvida: além de pelomenos quatro subtipos de FNA, existem ainda o peptídeonatriurético cerebral (BNF) e o peptídeo atrial natriuréticotipo C (CNF). O local de produção varia de um tipo paraoutro, mas estas substâncias mantêm funções similares aoFNA.26,27
Estes agentes natriuréticos e diuréticos, com certo efei-to vasodilatador renal seletivo, têm potencial terapêuticoem situações clínicas tais como: insuficiência renal aguda,síndrome hepatorrenal e insuficiência cardíaca congesti-va. Além disso, podem ser úteis no manejo da retenção
de sódio e sobrecarga de volume da insuficiência renal crô-nica.26,27
Fatores Derivados do EndotélioO endotélio é importante fonte de substâncias capazes
de regular o tônus vascular, tais como a endotelina, o óxi-do nítrico (antes conhecido como fator de relaxamentoderivado do endotélio — FRDE) e a prostaciclina. Estassubstâncias estão envolvidas no equilíbrio do sódio e água,pois têm propriedades vasodilatadoras e vasoconstritorasque regulam a pressão de perfusão dos rins, coração e vas-culatura.4
A endotelina tem efeitos vasoconstritores, com reduçãodo fluxo sanguíneo renal e TFG e retenção de sódio e água.O óxido nítrico pode ser produzido na mácula densa e temefeito vasodilatador aferente,28 com aumento da natriure-se por inibição da Na,K-ATPase e aumento da diurese.4
ProstaglandinasAs prostaglandinas têm efeitos sobre o fluxo sanguíneo
renal e sobre o manejo tubular de água e sal. Aparentemen-te, os resultados finais da estimulação da síntese de pros-taglandinas pelo rim são: vasodilatação, aumento da per-fusão renal, natriurese e facilitação da excreção de água.Quando se bloqueia a ciclo-oxigenase com antiinflamató-rios não-hormonais, existe diminuição da excreção de só-dio, aumento da resposta vasoconstritora renal à angioten-sina II e queda da TFG.4
Sistema Nervoso Simpático
O tônus simpático aumenta a reabsorção de sódio pe-los túbulos por um efeito direto e pela secreção de angio-tensina II e aldosterona.7
Diurese Pressórica
Em indivíduos normais, mesmo pequenas elevações dapressão arterial são acompanhadas de um aumento naexcreção renal de sódio e água, por diminuição da reab-sorção no túbulo proximal e alça de Henle. Possivelmenteo aumento da pressão arterial sistêmica seja transmitido aointerstício, desencadeando estas alterações. As prostaglan-dinas e o óxido nítrico podem estar envolvidos.29
Ponto-chave:
• O aumento ou diminuição da excreção renalde sódio resulta de uma ampla rede deeventos, em que participam fatores físicos,hemodinâmicos, humorais e hormonais

146 Metabolismo do Sódio e Fisiopatologia do Edema
DISTÚRBIOS CLÍNICOS DOMETABOLISMO DO SÓDIO
Distúrbios do equilíbrio do sódio são diagnosticados atra-vés de uma avaliação do volume extracelular. Um déficit desódio total no organismo causa depleção do volume extracelu-lar, e as manifestações clínicas dependem da magnitudedesta depleção. Um excesso de sódio total no organismoexpande o volume extracelular e, se a expansão for consi-derável, poderá manifestar-se clinicamente por edema.
O termo desidratação, freqüentemente empregado,pode causar confusão. Partilhamos da opinião de outros,segundo os quais as expressões excesso ou depleção do volu-me extracelular refletem melhor a idéia de que distúrbios dosódio são distúrbios de volume e envolvem déficit ou ex-cesso de uma solução isotônica de sódio, o que tem tam-bém implicações terapêuticas.30 Os pacientes com depleçãodo extracelular perderam sal e água, e a concentração plas-mática de sódio é de modo geral normal.
Ao contrário, os distúrbios do balanço de água são dis-túrbios da osmolalidade plasmática, traduzida por altera-ções na concentração de sódio plasmático e indicados pelaterminologia déficit ou excesso de água. Talvez o termo de-sidratação seja melhor empregado em situações em queexiste déficit de água, como nas hipernatremias.31 É preci-so salientar que os distúrbios do balanço de água depen-dem somente da quantidade relativa de água (em relação àquantidade de soluto), e não da quantidade absoluta deágua. Assim, um paciente com edema pode ter aumentona água total do corpo, mas desde que o sódio e a águaretidos no extracelular sejam isotônicos, não haverá alte-ração na água intracelular e, portanto, não haverá distúr-bio do balanço de água.
Pontos-chave:
• A avaliação e o diagnóstico dos distúrbiosclínicos do metabolismo do sódio e doespaço extracelular são feitos através dahistória clínica e do exame físico,detectando-se a depleção ou o excesso(edema)
• O diagnóstico de distúrbios do metabolismoda água é feito através da dosagem do sódioplasmático
Depleção de Sódio ou doVolume Extracelular
As causas de depleção do espaço extracelular encon-tram-se listadas no Quadro 10.4, sendo divididas basica-mente em causas renais e não-renais.
Habitualmente grande parte do volume secretado na luzdo trato gastrintestinal é reabsorvida, resultando numvolume fecal de cerca de 100-200 ml ao dia. Porém, em si-tuações em que a reabsorção se encontra diminuída, comonas diarréias e sondagem gástrica, perdas significativas defluido extracelular podem ocorrer, resultando em deple-ção.32
Os rins possuem um sistema de ajuste para equilibrar aexcreção com a ingesta. Mas se este sistema falha e a ex-creção é excessiva, a depleção pode instalar-se. São exem-plos disso situações como o uso de diuréticos, nefropatiasperdedoras de sal e o hipoaldosteronismo.32
Não existe nenhum método laboratorial prático para sedeterminar o volume extracelular. O diagnóstico se baseiana história clínica, exame físico e alguns exames laborato-riais. O dado mais importante no diagnóstico é a históriade perda de líquido que contém sódio.
Na história clínica, o paciente relata vômitos e/ou diar-réia, sudorese profusa, poliúria etc. O diagnóstico de de-pleção do volume extracelular, na ausência de história deperda de líquido que contém sódio, obriga-nos a questio-nar e rever o diagnóstico. Isto porque, se a ingesta de só-dio cessa, o mecanismo renal de conservação do sódio é tãoeficiente que um déficit de sódio não se estabelecerá.
O paciente pode inicialmente apresentar fraqueza, ano-rexia, náuseas e, a seguir, tonturas, síncope e, finalmente,um estado de colapso circulatório.
Os sintomas resultam de inadequado volume circulantee dependem de quatro fatores principais: a) magnitude daperda de volume; b) velocidade na perda de volume; c)natureza do fluido perdido, se somente água, água comsódio, ou sangue; e d) resposta vascular à redução de vo-lume.4
Por exemplo, a perda aguda de 1 litro de sangue porhemorragia gastrintestinal resulta em oligúria e manuten-
Quadro 10.4 Causas de depleção de sódio
1. Perdas renaisA. Ausência de doença renal
a. Diurese osmótica (glicosúria, manitol etc.)b. Diuréticos (tiazídicos, furosemida etc.)c. Insuficiência adrenal (primária)d. Secreção inapropriada de HAD
(primária)B. Enfermidades renais
a. Nefropatia crônica (particularmente doençamedular cística e nefrite intersticial)
b. Fase diurética da necrose tubular agudac. Uropatia pós-obstrução
2. Perdas extra-renaisA. Gastrintestinal: vômitos, diarréia, fístulas etc.B. Pele: sudorese, queimadurasC. Iatrogênicas: paracentese, toracocenteseD. Terceiro espaço: pancreatite aguda, fraturas,
esmagamentos, íleo
Modificado de Chapman, W.H. e col.30

capítulo 10 147
ção do hematócrito, com pouca contribuição do fluido in-tersticial em expandir o intravascular. A perda mais lentada mesma quantidade de sangue permite que haja trans-ferência de fluido do intersticial para o intravascular, comqueda conseqüente do hematócrito. Com a parcial restau-ração do volume sanguíneo, o volume de urina e a respos-ta hemodinâmica à contração de volume podem estar pou-co afetados.4
Os achados clínicos também dependem do tipo de flui-do perdido. A perda de 1 litro de água sem eletrólitos numpaciente de 70 kg reduz o volume sanguíneo em 2,5%, e ahemodinâmica renal e sistêmica são pouco afetadas. Aperda de 1 litro de fluido extracelular reduz o volume desangue em 6,6%, e instalam-se oligúria e taquicardia dis-cretas com o paciente deitado. A perda de 1 litro de san-gue reduz o volume em 20%, resultando em oligúria gra-ve e choque.4
Entre os sinais mais sensíveis no diagnóstico de um ina-dequado volume circulante, destacamos as alterações ortos-táticas de pressão arterial e a determinação simultânea dopulso periférico. Portanto, determinam-se a pressão arteri-al e o pulso com o paciente deitado, sentado no leito, comos pés para fora da cama e de pé, quando possível. Fazer opaciente sentar-se no leito, sem que os pés fiquem penden-tes para fora da cama, pode não ser suficiente para produ-zir uma queda ortostática da pressão arterial. Normalmen-te, quando o paciente muda da posição deitada para a sen-tada ou de pé, a sua pressão sistólica quase não se altera, e apressão diastólica aumenta 5 ou 10 mm Hg. Se há um ina-dequado volume circulante, as pressões sistólica e diastóli-ca caem 10 mm Hg ou mais, e nota-se aumento da freqüên-cia cardíaca ou pulso periférico. Uma queda ortostática dapressão arterial também pode ocorrer independente do vo-lume circulante e estar relacionada com comprometimentodo sistema nervoso autônomo periférico, tal como ocorre nodiabetes mellitus, insuficiência renal crônica ou com o uso dedrogas, especialmente bloqueadores adrenérgicos. É neces-sário salientar que pressão arterial aparentemente normalpode ser encontrada em indivíduos previamente hiperten-sos que estejam depletados.32
Os sinais chamados clássicos de depleção do volumeextracelular, como diminuição do turgor da pele, diminui-ção do volume da língua ou diminuição do tônus ocular,têm pouco valor clínico. Quando estes sinais são detectá-veis, o grau de depleção do volume extracelular é de talordem que o paciente está quase em choque. Por outrolado, pessoas obesas, jovens ou com depleções leves podemapresentar turgor de pele normal.32
Um outro sinal clínico bastante útil é a avaliação do en-chimento venoso no pescoço. Quando um paciente está emdecúbito dorsal, as veias jugulares são visíveis até quase oângulo da mandíbula. Se as veias jugulares não forem vi-síveis ou mostrarem pobre enchimento, suspeita-se dedepleção do volume extracelular. É necessário, no entan-to, salientar que, em algumas pessoas normais, as veias
jugulares são invisíveis e, em outras, se apresentam cheiaspor possuírem válvulas ou alterações da elasticidade, semrefletirem o volume circulante. Desta forma, em algunscasos, necessitamos da determinação direta da pressão ve-nosa central.
Quando a depleção de volume é intensa, o débito car-díaco cai, o mesmo ocorrendo com a pressão venosa sistê-mica intratorácica. Portanto, a determinação da pressãovenosa central (PVC) poderia ser um indicador sensível deredução no retorno venoso e débito cardíaco. Entretanto,como os limites de normalidade são muito amplos em in-divíduos diferentes, é impossível definir hipovolemianuma única determinação. Por outro lado, uma única de-terminação do volume sanguíneo não dá idéia do grau dedeficiência e de como o coração vai tolerar a restauraçãodo volume. Quando se correlacionaram o volume sanguí-neo e a PVC em pacientes em choque, observou-se que acorrelação era pobre33 (v. Fig. 10.6). Talvez o melhor guiada adequação do volume sanguíneo circulante não sejauma única determinação da PVC ou do volume sanguíneo,e, sim, a observação da resposta cardiovascular à expan-são do volume (v. próxima seção). Para uma boa interpre-tação da PVC, os seguintes princípios são importantes:33
1. Uma PVC reduzida não permite uma conclusão eviden-te de que o volume sanguíneo está reduzido.
2. Num paciente com insuficiência circulatória (choque),uma PVC baixa indica que uma expansão do volumeserá benéfica. No entanto, uma PVC alta não contra-in-dica uma expansão do volume sanguíneo, mas devepermanecer a mesma ou cair à medida que o volumesanguíneo aumenta. Por outro lado, se a PVC inicial éelevada e continua a elevar-se à medida que a expan-são de volume prossegue, a infusão deve ser suspensa.
3. Uma elevação da PVC acima do normal, durante a ex-pansão, indica que a expansão está sendo excessiva.
É preciso lembrar que o controle da PVC fornece-nosuma idéia mais ou menos precisa da pressão de enchimentodo ventrículo direito, mas não nos esclarece nada sobre afunção do ventrículo esquerdo. Num indivíduo normal, aexpansão de volume eleva simetricamente as pressões deátrio direito e esquerdo, o que não ocorre em indivíduoscom insuficiência ventricular esquerda. A pressão venosaintratorácica, normalmente, não deve exceder 8 cm deágua, podendo ser determinada através de um cateter emveia cava superior e tomando-se o zero do manômetro naaltura da linha axilar média.
DADOS LABORATORIAISEntre os exames de laboratório, a elevação do hemató-
crito e da concentração plasmática das proteínas acompa-nha a depleção do volume extracelular, pois ambos estãoconfinados ao espaço intravascular. Uréia e creatinina po-dem estar elevadas, dependendo do grau de redução dataxa de filtração glomerular.32
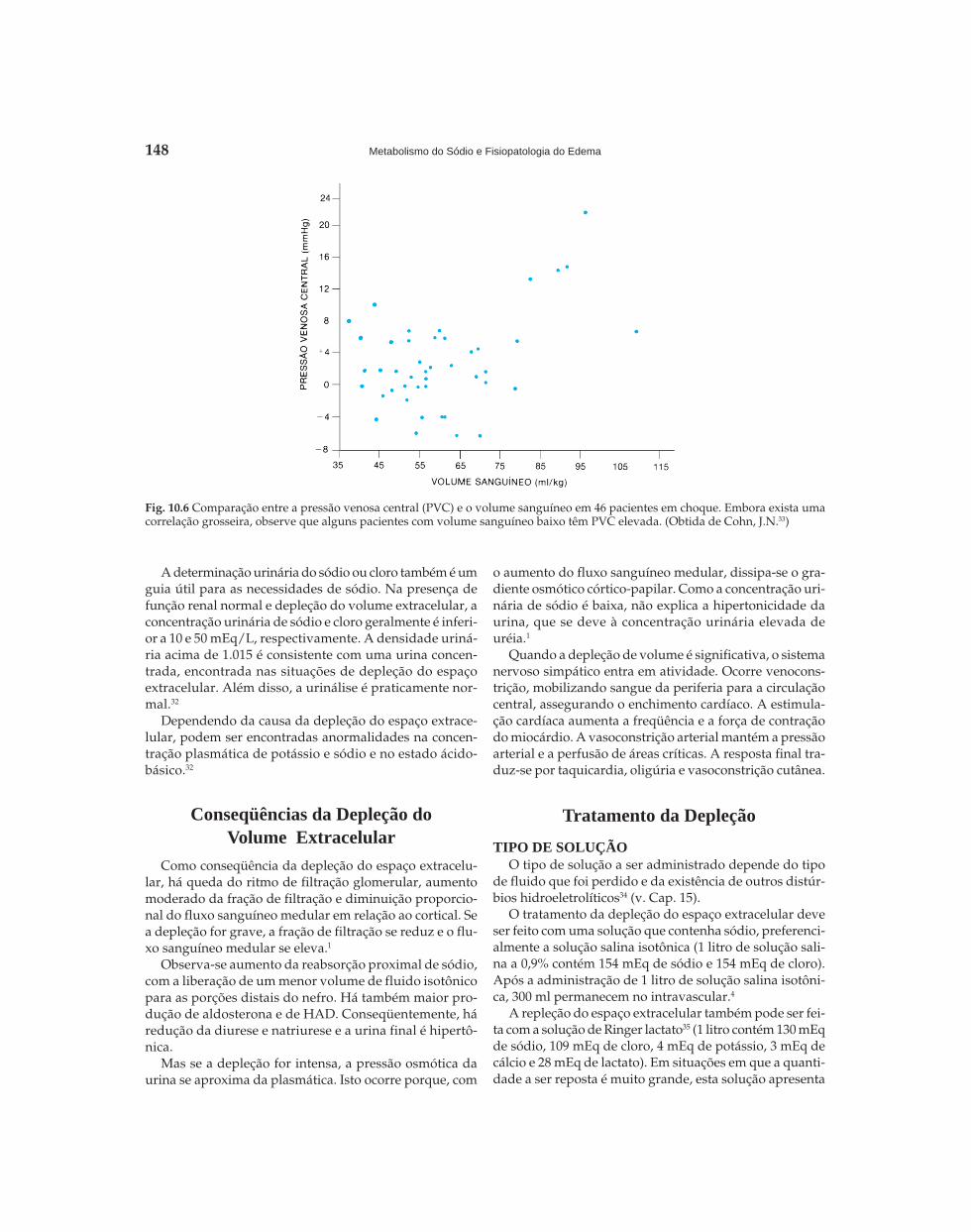
148 Metabolismo do Sódio e Fisiopatologia do Edema
A determinação urinária do sódio ou cloro também é umguia útil para as necessidades de sódio. Na presença defunção renal normal e depleção do volume extracelular, aconcentração urinária de sódio e cloro geralmente é inferi-or a 10 e 50 mEq/L, respectivamente. A densidade uriná-ria acima de 1.015 é consistente com uma urina concen-trada, encontrada nas situações de depleção do espaçoextracelular. Além disso, a urinálise é praticamente nor-mal.32
Dependendo da causa da depleção do espaço extrace-lular, podem ser encontradas anormalidades na concen-tração plasmática de potássio e sódio e no estado ácido-básico.32
Conseqüências da Depleção doVolume Extracelular
Como conseqüência da depleção do espaço extracelu-lar, há queda do ritmo de filtração glomerular, aumentomoderado da fração de filtração e diminuição proporcio-nal do fluxo sanguíneo medular em relação ao cortical. Sea depleção for grave, a fração de filtração se reduz e o flu-xo sanguíneo medular se eleva.1
Observa-se aumento da reabsorção proximal de sódio,com a liberação de um menor volume de fluido isotônicopara as porções distais do nefro. Há também maior pro-dução de aldosterona e de HAD. Conseqüentemente, háredução da diurese e natriurese e a urina final é hipertô-nica.
Mas se a depleção for intensa, a pressão osmótica daurina se aproxima da plasmática. Isto ocorre porque, com
o aumento do fluxo sanguíneo medular, dissipa-se o gra-diente osmótico córtico-papilar. Como a concentração uri-nária de sódio é baixa, não explica a hipertonicidade daurina, que se deve à concentração urinária elevada deuréia.1
Quando a depleção de volume é significativa, o sistemanervoso simpático entra em atividade. Ocorre venocons-trição, mobilizando sangue da periferia para a circulaçãocentral, assegurando o enchimento cardíaco. A estimula-ção cardíaca aumenta a freqüência e a força de contraçãodo miocárdio. A vasoconstrição arterial mantém a pressãoarterial e a perfusão de áreas críticas. A resposta final tra-duz-se por taquicardia, oligúria e vasoconstrição cutânea.
Tratamento da Depleção
TIPO DE SOLUÇÃOO tipo de solução a ser administrado depende do tipo
de fluido que foi perdido e da existência de outros distúr-bios hidroeletrolíticos34 (v. Cap. 15).
O tratamento da depleção do espaço extracelular deveser feito com uma solução que contenha sódio, preferenci-almente a solução salina isotônica (1 litro de solução sali-na a 0,9% contém 154 mEq de sódio e 154 mEq de cloro).Após a administração de 1 litro de solução salina isotôni-ca, 300 ml permanecem no intravascular.4
A repleção do espaço extracelular também pode ser fei-ta com a solução de Ringer lactato35 (1 litro contém 130 mEqde sódio, 109 mEq de cloro, 4 mEq de potássio, 3 mEq decálcio e 28 mEq de lactato). Em situações em que a quanti-dade a ser reposta é muito grande, esta solução apresenta
Fig. 10.6 Comparação entre a pressão venosa central (PVC) e o volume sanguíneo em 46 pacientes em choque. Embora exista umacorrelação grosseira, observe que alguns pacientes com volume sanguíneo baixo têm PVC elevada. (Obtida de Cohn, J.N.33)

capítulo 10 149
benefícios, pois o lactato é convertido a bicarbonato no fí-gado e ameniza ou evita uma acidose dilucional. Não deveser utilizada em pacientes hipercalêmicos e com funçãorenal comprometida.
As soluções colóides (plasma, albumina) expandemprincipalmente o intravascular, pois suas grandes molécu-las não ultrapassam o endotélio capilar. Este tipo de flui-do deve ser reservado para situações graves, em que a ex-pansão do intravascular necessita ser rápida e efetiva, co-mo, por exemplo, em queimaduras extensas e choque. Nãose justifica a administração destas soluções em outras si-tuações. Devem também ser levados em conta fatores comoo alto custo e a meia-vida curta destas soluções.4 Mais re-centemente, tem sido utilizado o amido hidroxietílico(hetastarch), cujas moléculas têm cerca de 200.000 daltonse que permanece por até 24-36 horas no compartimentointravascular. No Brasil, estão disponíveis as apresentaçõesa 6% e 10% (Haes-steril®), que em 1 litro contém 60-100 gdo amido e 154 mEq de sódio.
Ao se administrar sangue, este permanece inteiramen-te no intravascular. Deve ser administrado quando hemor-ragia tiver sido a causa da depleção e das alterações hemo-dinâmicas já mencionadas.4 O hematócrito não deve serelevado acima de 35%.36
A administração de solução glicosada a 5% não é ade-quada no tratamento da depleção do extracelular, poisequivale à administração de água sem sódio, que se distri-bui uniformemente na água corporal total e não permane-ce em volume suficiente no intravascular. Por exemplo,após a administração de 1 litro de solução glicosada a 5%,permanecem no intravascular apenas 75-100 ml.
VELOCIDADE DE ADMINISTRAÇÃOA velocidade de administração da solução salina depen-
de da magnitude da insuficiência circulatória. Desde quenão haja cardiopatia, pode-se administrar um litro de so-lução salina por hora ou até em menor intervalo, em casosgraves. Não há necessidade de que todo o déficit de volu-me seja corrigido em poucas horas. O importante é que ossinais de hipovolemia grave desapareçam. A partir de en-tão, a reposição de volume pode ser mais lenta.
Um dos elementos muito importantes no manejo clíni-co é o controle dos fatores precipitantes: sangramento,vômitos, diarréia etc. Não havendo mais perdas, umamaior parcela do líquido administrado permanecerá noespaço extracelular, restaurando o seu volume.
VOLUME A SER INFUNDIDO(GRAU DE DEPLEÇÃO)
O grau de depleção do volume extracelular pode serestabelecido pela história clínica e achados de exame físi-co, sendo o cálculo aproximado. Por exemplo: um indiví-duo de 70 kg tem 14 litros, aproximadamente, de volumeextracelular (20% do peso corporal).
Uma depleção leve (10-15% de redução no EEC) nãocursa com sinais clínicos muito significativos, mas há his-tória de perda. Uma depleção moderada está entre 20 e 30%de redução no volume extracelular.37 O paciente pode apre-sentar, em decúbito dorsal, pressão arterial normal, mas aomesmo tempo ter taquicardia, pobre perfusão capilar ediminuição da temperatura da pele (devido à vasoconstri-ção). Uma determinação dos sinais vitais, na posição sen-tada ou em pé, aumenta os sinais de insuficiência circula-tória. Considerando o paciente acima, o déficit seria de 2,8a 4,2 litros de solução salina isotônica (v. Cap. 15 paramaiores detalhes sobre reposição hidroeletrolítica).
Uma depleção intensa representa 40 a 50% de reduçãodo volume extracelular.37 Clinicamente, o paciente apresen-ta hipotensão arterial mesmo em decúbito dorsal, ou já estáem choque. O déficit de volume extracelular será, portan-to, de 5,6 a 7 litros. Além disso, os pacientes em choquehipovolêmico apresentam intensa ativação adrenérgica,caracterizada por taquicardia, extremidades frias com en-chimento capilar lento, cianose de extremidades, oligúriae agitação e confusão mental, que se devem à diminuiçãodo fluxo sanguíneo cerebral.32
MONITORIZAÇÃO DO TRATAMENTOEm pacientes com reserva cardíaca normal, o efeito de
um desafio líquido pode ser monitorizado pela avaliaçãodo pulso, pressão arterial e fluxo urinário. Em pacientescom função cardíaca comprometida, a determinação seri-ada da PVC ou preferencialmente da pressão em capilarpulmonar (PCap) e débito cardíaco através de um cateterde Swan-Ganz possibilitam o diagnóstico precoce de so-brecarga de volume secundária ao desafio hídrico. Estasmedidas devem ser seriadas e sua avaliação dinâmica, ouseja, à medida que se vai expandindo o volume circulante.Administra-se rapidamente um volume de 100 ml e obser-vam-se as mudanças na PVC e PCap. Durante a expansãode volume, a PVC ou a pressão em capilar pulmonar po-dem inicialmente subir para depois cair. Esta elevação ini-cial se deve à infusão de fluidos num leito vascular vaso-
Pontos-chave:
• São sinais sensíveis para o diagnóstico dedepleção do espaço extracelular: alteraçõesortostáticas da pressão arterial e pulso,enchimento das jugulares e débito urinário
• A depleção pode ser classificada como leve,moderada e intensa, dependendo dasalterações encontradas no exame físico
• O tratamento geral da depleção doextracelular consiste na administração desolução isotônica contendo sódio

150 Metabolismo do Sódio e Fisiopatologia do Edema
constrito.4 Enquanto persistirem o choque, a hipotensão, ouse a PVC não se elevar, a expansão do volume é conside-rada inadequada.
Um outro dado útil na avaliação da adequação do volu-me sanguíneo é o volume urinário horário. Se, durante a repo-sição do volume, o volume urinário aumentar de 0-10 ml/hpara 50 ml/h ou mais, isto indica um adequado plano de re-posição. Por outro lado, a queda do volume urinário indicaque a reposição não está sendo suficientemente rápida.
EXCESSO DE VOLUMEEXTRACELULAR — EDEMA
Um excesso de sódio total no organismo acompanha-sede expansão do volume extracelular, que, se considerável,se manifestará por edema. Edema é o acúmulo anormal defluido em qualquer parte do organismo. Geralmente istoocorre em pacientes com cardiopatia, nefropatia, hepato-patia ou hipoproteinemia.
Fisiopatologia do EdemaEdema significa um acúmulo excessivo de líquido no
compartimento intersticial, ou seja, na parte não-vasculardo compartimento líquido extracelular. A passagem parao interstício de fluido ultrafiltrado do plasma (sem prote-ínas), decorrente da alteração das forças de Starling, é de-nominada transudação.38 São exemplos deste mecanismo osedemas decorrentes de obstrução venosa, insuficiênciacardíaca e edema pulmonar cardiogênico.
Outro tipo de edema ocorre por aumento da permeabi-lidade dos capilares a determinados solutos, tais como asproteínas, num mecanismo de exsudação. 38 Este mecanis-mo de formação de edema é observado em queimaduras,trauma, abcessos.
O edema pode ser bem localizado, como numa peque-na inflamação, ou generalizado, como na insuficiência car-díaca.
EDEMA LOCALIZADOO edema localizado resulta de fatores inflamatórios ou
físicos que aumentam a formação ou diminuem a remo-ção de líquido intersticial em uma região do corpo.9 Omecanismo de formação do edema localizado pode seradequadamente explicado com base numa alteração dasforças de Starling que controlam a troca de líquido entre oplasma e o interstício. Estas forças estão relacionadas naseguinte expressão: 39
q. � Kf [(Pc � Pt) – (�p � �t)
onde: q.� ritmo do fluxo de líquido através da parede ca-
pilar
Kf � coeficiente de filtração (proporcional à permeabi-lidade capilar e à área do leito capilar)
Pc � pressão hidrostática intracapilarPt � pressão do turgor tecidual�p � pressão oncótica do plasma�t � pressão oncótica intersticial
O edema localizado ocorre quando as alterações nas for-ças de Starling estão restritas a um órgão ou a um determi-nado território vascular. Normalmente, o balanço de forçasde Starling na porção arteriolar do capilar é de tal ordem queocorre filtração de líquido para o interstício. Com isto ocor-re diminuição da pressão hidráulica capilar e aumento dapressão coloidosmótica do plasma (v. também Cap. 8). Deacordo com a visão clássica de distribuição de líquido trans-capilar, a reversão do balanço das forças de Starling ocorriana porção terminal venosa do capilar, havendo então reab-sorção do líquido filtrado. Assim sendo, havendo equilíbrioentre o líquido filtrado e reabsorvido, apenas uma pequenaquantidade deveria retornar ao sistema vascular via linfáti-cos. No entanto, recentemente demonstrou-se que a pres-são hidráulica transcapilar excede a pressão coloidosmóti-ca do plasma em toda a extensão do capilar, de sorte que afiltração ocorre ao longo de todo o capilar.40 O líquido fil-trado retorna à circulação via linfáticos. Desta forma a cir-culação linfática passa a ter um papel importante no con-trole da formação do edema.
Também existe vasodilatação que aumenta a saída delíquido do capilar principalmente através de aumento dapressão hidrostática intracapilar e do coeficiente de filtra-ção. O aumento do Kf ocorre devido à abertura de novoscapilares, dilatação dos capilares e aumento da permeabi-lidade. Uma diminuição da �p e um aumento da �t tam-bém contribuem para a saída de líquido do capilar (Qua-dro 10.5).
Edema Generalizado
É a principal manifestação clínica da expansão do vo-lume líquido do compartimento extracelular e está invari-avelmente associado a uma retenção renal de sódio. É umamanifestação comum em situações clínicas tais como: in-suficiência cardíaca, cirrose hepática e síndrome nefrótica,onde a retenção renal de sódio é apenas uma resposta re-nal a um distúrbio hemodinâmico determinado pela enfer-midade básica (Quadro 10.6).
A distribuição do edema generalizado é afetada por fa-tores locais e gravitacionais. Assim sendo, o líquido inters-ticial em excesso pode acumular-se nos membros inferio-res de pacientes ambulatoriais e na região pré-sacra depacientes acamados. A baixa pressão do turgor tecidual nasregiões periorbital e escrotal pode acentuar o edema nes-tas áreas.9
O edema classifica-se em dois tipos: edema duro e ede-ma mole.42 O edema mole revela o sinal do cacifo quando a

capítulo 10 151
pressão digital deixa uma depressão transitória na pele,como ocorre por exemplo na insuficiência cardíaca. O ede-ma duro não revela o sinal do cacifo, pois a pressão digitalnão consegue mobilizar o líquido intersticial devido a obs-trução linfática (linfedema), fibrose do tecido subcutâneo,como pode ocorrer na obstrução venosa crônica, ou aumen-to da matriz intersticial, como no mixedema.43
É importante salientar que pode haver um acúmulo de4 a 5 litros de líquido no compartimento extracelular antesque o paciente ou o médico percebam o edema com sinaldo cacifo presente. Há, no entanto, sinais e sintomas suges-tivos do excesso de líquido no organismo: ganho de peso,flutuações diárias no peso (mais pesado à noite), reduçãoda diurese, noctúria, tosse ou dispnéia ao deitar-se e disp-néia aos esforços.
A intensidade do edema é graduada em cruzes (�, ��,��� ou ����/4�), dependendo da profundidade dadepressão criada com a compressão digital e também deacordo com a extensão do edema. Por exemplo, um paci-ente com síndrome nefrótica com moderado edema demembros inferiores até os joelhos tem um edema de ��/4�. Já um paciente com edema até a raiz das coxas, edemade parede abdominal e sinais de ascite tem um edema de����/4� e anasarca.42
A fisiopatogenia do edema em situações clínicas diver-sas será abordada na próxima seção.
Quadro 10.5 Fatores que contribuem para aformação do edema*
1. Dilatação arteriolarA. InflamaçãoB. CalorC. ToxinasD. Excesso ou déficit neuro-humoral
2. Redução da pressão osmóticaA. Hipoproteinemia
a. Desnutriçãob. Cirrose hepáticac. Síndrome nefróticad. Gastroenteropatia perdedora de
proteínaB. Aumento da permeabilidade capilar
a. Inflamaçãob. Queimadurasc. Traumad. Reação alérgica ou imunológica
C. Obstrução linfática3. Aumento da pressão venosa
A. Insuficiência cardíaca congestivaB. TromboflebiteC. Cirrose hepática
4. Retenção de sódioA. Ingesta excessiva de salB. Elevada reabsorção tubular renal de sódio
a. Redução da perfusão renalb. Aumento da secreção de renina-
angiotensina-aldosterona
*Baseado em Leaf, A. e Cotran, R.S.41
Quadro 10.6 Causas de edema generalizado
1. Enfermidades renaisA. Glomerulonefrite agudaB. Síndrome nefróticaC. Insuficiência renal agudaD. Insuficiência renal crônica
2. Insuficiência cardíacaA. Baixo débitoB. Alto débito (anemia, beribéri, tireotoxicose,
sepse etc.)3. Enfermidades hepáticas
A. CirroseB. Obstrução da drenagem hepática venosa
4. Enfermidades confinadas a mulheresA. GravidezB. Toxemia gravídicaC. Síndrome da tensão pré-menstrualD. Edema cíclico idiopático
5. Enfermidades vascularesA. Fístulas arteriovenosasB. Obstrução das veias do tórax
a. Veia cava inferiorb. Veia cava superior
6. Distúrbios endócrinosA. HipotireoidismoB. Excesso de mineralocorticóidesC. Diabetes mellitus
7. DrogasA. Estrogênios, anticoncepcionais oraisB. Agentes anti-hipertensivos
8. MiscelâneaA. Hipocalemia crônicaB. Anemia crônicaC. Edema nutricionalD. Síndrome da permeabilidade capilar elevada
Pontos-chave:
• Um dos principais sinais de excesso desódio no organismo é o edema
• O edema pode ser localizado ougeneralizado, e forma-se por transudação ouexsudação
Fisiopatologia do Edema em SituaçõesClínicas Específicas
INSUFICIÊNCIA CARDÍACA CONGESTIVA (ICC)A ICC ocorre quando o coração falha na sua função de
bomba e está habitualmente associada a uma retenção re-nal de sal e água e com edema pulmonar ou periférico. Hámuito se discutem os fatores que estariam envolvidos naretenção renal de sódio na insuficiência cardíaca. A teoriade “insuficiência retrógrada” propõe que à medida que ocoração falha, as pressões venosas periféricas e centraisaumentam, elevando a pressão hidráulica transcapilar e

152 Metabolismo do Sódio e Fisiopatologia do Edema
conseqüentemente promovendo a transudação de líquidono espaço intersticial, edema e contração do volume cir-culante. A teoria da “insuficiência anterógrada” diz que,com o comprometimento da função cardíaca e do ventrí-culo esquerdo, a periferia, incluindo o rim, passa a ser malperfundida, o que estimula mecanismos renais e intra-re-nais para a retenção renal de sódio. É provável que hajauma interdependência entre as duas teorias, e que o acon-tecimento básico seria uma retenção renal de sódio, e a tran-sudação transcapilar seria um evento secundário.
Na insuficiência cardíaca os rins estão funcionando ade-quadamente e retêm sódio numa tentativa de restaurar ovolume circulante efetivo. Este mecanismo, denominado“subpreenchimento” (underfilling) é também observado nacirrose hepática e síndrome nefrótica.38
Volume Sanguíneo Arterial EfetivoNa insuficiência cardíaca congestiva há um distúrbio na
relação normal do volume intravascular (volume efetivo) ea capacidade do leito vascular. Há sugestões de que o au-mento da reabsorção tubular renal de sódio seja decorrentede alterações circulatórias percebidas por sensores de volu-me nos átrios cardíacos e grandes vasos torácicos. Como jámencionado anteriormente, talvez os efeitos na excreçãorenal de sódio sejam oriundos da estimulação mecânica dosátrios cardíacos, através da liberação de um peptídio atrialnatriurético e por reflexos neurais bem estabelecidos.
A importância do fluxo sanguíneo no circuito arterial paracontrole da volemia foi demonstrada pela resposta renal àabertura e fechamento de uma fístula arteriovenosa.44 O fe-chamento da fístula acarretava uma rápida natriurese semalteração no ritmo de filtração glomerular, enquanto a aber-tura da fístula novamente reduzia a excreção de sódio. Nes-tas circunstâncias, as pressões hidráulicas nos átrios e cir-culação pulmonar diminuíam com o fechamento da fístulae aumentavam com a abertura da fístula.
A percepção arterial ocorre em vários locais do leitovascular arterial. Existem os barorreceptores carotídeos eos barorreceptores intra-renais no aparelho justaglomeru-lar. Uma redução da pressão de perfusão renal estimula aliberação de renina do aparelho justaglomerular resultan-do na formação de angiotensina II, aldosterona e retençãode sódio (Fig. 10.7). Esta retenção de sódio é na verdadeum mecanismo protetor para preservar a adequação dovolume circulante.
Papel do Rim na Retenção de SódioNa ICC há aumento do tônus simpático e das cate-
colaminas circulantes, responsáveis por aumento da re-sistência vascular periférica. No rim também ocorre au-mento da resistência vascular e freqüentemente reduçãodo ritmo de filtração glomerular (RFG). Mas não é a re-dução do RFG a responsável pela retenção de sódio, poisesta ocorre mesmo na ausência de qualquer alteração no
Fig. 10.7 Esquema dos mecanismos envolvidos na retenção de sódio e edema da insuficiência cardíaca. (Baseado em Schrier, R.W.9)

capítulo 10 153
RFG. Na ICC os nefros apresentam elevada fração de fil-tração, decorrente de aumento da resistência arteriolareferente. Com a elevação da fração de filtração há aumen-to da pressão oncótica pericapilar tubular, alterando asforças peritubulares de Starling e acarretando aumentoda reabsorção de sódio a nível de túbulo proximal (Fig.10.8).
Outras alterações hemodinâmicas intra-renais podemestar envolvidas: talvez o aumento do tônus simpático anível renal cause uma redistribuição do fluxo sanguíneopara nefros justamedulares (alças de Henle longas) quepodem reabsorver sódio mais avidamente que nefros cor-ticais.
Sistema Renina-angiotensina-aldosterona (SRAA)Como já frisamos, a diminuição da perfusão renal esti-
mula a liberação de renina com formação de angiotensinaI e II e aldosterona. A manutenção da pressão arterial emface de uma redução do volume sanguíneo arterial é ex-plicada pela elevação da angiotensina II. A retenção renalde sódio decorre da ação hemodinâmica da angiotensinaII (vasoconstrição da arteríola glomerular eferente e au-mento da fração de filtração), da sua ação direta no túbuloproximal e do hiperaldosteronismo.
ProstaglandinasMesmo que haja variação no volume plasmático, a in-
teração entre angiotensina II e prostaglandinas mantém ofluxo sanguíneo renal quase constante. A inibição da sín-tese da prostaglandina em animais normovolêmicos nãocompromete a filtração glomerular, mas quando há deple-ção de volume e níveis elevados de angiotensina II, o blo-
queio da síntese de prostaglandina reduz o fluxo sanguí-neo renal e a filtração glomerular. Da mesma maneira, ainibição da síntese de prostaglandina só reduz a excreçãode sódio se houver concomitante depleção de volume oucomprometimento intrínseco da função renal.
Em resumo, os níveis elevados de substâncias vasocons-tritoras, especialmente angiotensina II e catecolaminas, têmum importante papel na preservação de um adequado flu-xo sanguíneo renal na ICC.
Fator NatriuréticoA infusão contínua deste fator atrial causa uma redu-
ção da pressão arterial média, com elevação do ritmo defiltração glomerular, do fluxo urinário e da excreção desódio e potássio. A influência do FNA na pressão arterialrelaciona-se à sua capacidade de suprimir níveis plasmá-ticos de renina e em relaxar diretamente os vasos sanguí-neos. Como o FNA pode aumentar a filtração glomerularem doses que diminuem a pressão arterial e o fluxo san-guíneo renal, pode vir a ser útil no tratamento agudo docoração insuficiente.45
Ao estudarem as anormalidades na excreção de sódio eágua na ICC, Mettaurer et al. verificaram que os principaisfatores determinantes na excreção de sódio eram a ativa-ção do sistema renina-angiotensina e a função ventricular.46
Com relação à excreção de água, os fatores mais importan-tes foram os níveis plasmáticos de vasopressina e norepi-nefrina, a função renal e o grau de comprometimento dafunção ventricular esquerda. Um dos principais mecanis-mos de que o organismo lança mão para compensar a que-da do débito cardíaco é a ativação de sistemas neuro-hu-morais. Na ICC, a secreção de vasopressina e a ativação
Fig. 10.8 Controle peritubular da reabsorção de líquido do túbulo proximal.�p � pressão hidráulica transcapilar�� � pressão oncótica transcapilarA elevação da resistência vascular renal na ICC reduz a �� . O aumento da fração de filtração na ICC aumenta a ��. As alteraçõesem ambas as pressões aumentam a reabsorção proximal de sódio.

154 Metabolismo do Sódio e Fisiopatologia do Edema
dos sistemas simpático e renina-angiotensina servem paraotimizar a pré-carga e aumentar a contratilidade do mio-cárdio.
CIRROSE HEPÁTICAAs alterações hepáticas estruturais terminam por cau-
sar obstrução à drenagem venosa hepática, hipertensãoportal e shunt sanguíneo porta-sistêmico. Além destas al-terações hemodinâmicas, a função hepatocelular está com-prometida, causando redução na síntese de albumina efatores de coagulação. Há comprometimento na excreçãode sal e água e redução do ritmo de filtração glomerular(RFG). De modo semelhante à insuficiência cardíaca, a re-tenção renal de sódio e água não se deve a uma anor-malidade intrínseca dos rins, mas a mecanismos extra-re-nais que regulam a excreção renal destes elementos.
Alguns autores, como Levy, Wexler e Allotey propõemque um mecanismo de overflow esteja presente ao menosnas fases iniciais da cirrose. De acordo com este conceito,uma retenção de sódio pelo rim, não dependente de volu-me, é o distúrbio primário na homeostase do sódio empacientes com cirrose. Nesta teoria, a retenção de sódio eexpansão plasmática resultam da ausência do “escape” demineralocorticóides e antecedem o “subpreenchimento”.A predileção pelo acúmulo de líquido no peritônio, sob
forma de ascite, deve-se às alterações localizadas das for-ças de Starling, pela hipertensão portal. Aqueles autoresdemonstraram aumento no volume efetivo de sangue nasfases iniciais da cirrose. A retenção de sódio ocorreu inde-pendentemente de débito cardíaco, pressão arterialmédia, fluxo sanguíneo esplâncnico e hepático, TFG, flu-xo sanguíneo renal, níveis de aldosterona, estrógenos eprogesterona ou atividade simpática.4
Há várias outras influências independentes do volu-me sistêmico que sustentam a hipótese de overflow. A per-cepção de uma obstrução da drenagem venosa hepáticae elevada pressão hepática intra-sinusoidal através deuma via neural reflexa podem ser importantes mecanis-mos na retenção renal de sal e água, efetivada através deaumento na atividade simpática renal e cardiopulmonar(Fig. 10.9).47
Mesmo que o volume plasmático total esteja elevado nacirrose, o enchimento relativo do leito vascular arterialestará reduzido devido à redução da resistência vascularperiférica, inclusive com comprometimento dos reflexosvasomotores autônomos e diminuição da resposta pressó-rica a angiotensina II e catecolaminas.48 Isto resulta numleito vascular dilatado, hiporreativo a alterações de vole-mia e comprometido na sua capacidade de regular o tônuse a capacidade. Assim sendo, pacientes cirróticos se tornam
Fig. 10.9 Esquema dos mecanismos envolvidos na retenção de sódio e edema na cirrose hepática. (Baseado em Schrier, R.W.9 e Seifter,J.L. et al.47)

capítulo 10 155
muito vulneráveis e sujeitos a um colapso hemodinâmicoquando sofrem uma perda de volume aguda, como numahemorragia ou diurese agressiva.47 A percepção por sen-sores intratorácicos e arteriais da redução do volume san-guíneo arterial efetivo promove a retenção de sódio. A re-dução da resistência vascular periférica observada em cir-rose hepática avançada está relacionada, pelo menos emparte, a shunts arteriovenosos, mas talvez um vasodilata-dor (produzido, ou não inativado pelo fígado) tenha algu-ma participação. A seqüestração venosa esplâncnica queocorre secundária à hipertensão portal também contribuipara a redução da volemia.
Com a obstrução da drenagem hepática venosa, os si-nusóides hepáticos (altamente permeáveis a proteínas)permitem a passagem para o interstício de um elevado flu-xo de filtrado rico em proteínas, resultando num aumentoda formação de linfa hepática, principal responsável pelaascite em cirróticos. Quando o ritmo de formação da linfahepática excede o ritmo de retorno do líquido extracelularà circulação via ducto torácico, ocorre diminuição do vo-lume intravascular.47 O sucesso de procedimentos taiscomo o shunt peritoniovenoso, nos cirróticos com ascite,parece estar relacionado a uma rápida elevação do volu-me intravascular. Além disso, a hipoalbuminemia freqüen-temente presente nos cirróticos e a resultante redução dapressão coloidosmótica do plasma contribuem para a tran-sudação de líquido no compartimento intersticial e cavi-dade abdominal.
Em conjunto, estes fatores levariam a um “subpreenchi-mento” da árvore arterial levando à ativação do sistemarenina-angiotensina-aldosterona e do eixo simpático e li-beração de vasopressina. Estes eventos causariam a reten-ção de sódio e água pelo rim em fases mais avançadas dacirrose hepática.4
Função Renal na Cirrose HepáticaOs distúrbios característicos de função renal na cirrose
são a retenção de sódio e o comprometimento no clearancede água livre.47 A retenção renal de sódio pode ocorrer nacirrose na vigência de um RFG normal. Com a redução dovolume intravascular efetivo, há um aumento na reabsor-ção tubular proximal de sódio e uma redução da oferta delíquido aos túbulos distais, sendo esta última a causa daredução do clearance de água livre.
Renina-angiotensina-aldosteronaEmbora as causas de diminuição do volume sanguíneo
arterial efetivo sejam distintas na cirrose e na insuficiênciacardíaca, são similares os eventos subseqüentes que cau-sam retenção renal de sódio e água. A resistência vascularrenal está elevada nos cirróticos com ascite. A angiotensi-na II determina aumento da resistência da arteríola glome-rular eferente, causando aumento da fração de filtração,aumento da pressão oncótica pericapilar tubular e conse-
qüentemente aumento da reabsorção de sódio ao nível dotúbulo proximal. O aldosteronismo secundário ocorre de-vido à elevação de angiotensina II, esta última procuran-do preservar a pressão arterial. Portanto, face a uma redu-ção do volume intravascular, a ativação do eixo renina-angiotensina-aldosterona serve para preservar a pressãoarterial numa situação em que a capacidade vascular estámuito aumentada. Além da estimulação da angiotensinaII sobre a produção de aldosterona, a redução do fluxosanguíneo hepático compromete a degradação da aldos-terona e contribui ainda mais para a elevada atividade daaldosterona na cirrose. Entretanto, como na insuficiênciacardíaca, antagonistas da aldosterona não são efetivos emaumentar a excreção de sódio no tratamento do edema eda ascite do cirrótico.9
Na síndrome hepatorrenal existe caracteristicamenteuma pronunciada redução do fluxo sanguíneo renal comisquemia cortical e elevada resistência vascular renal,provavelmente devido à ação de substâncias vasoconstri-toras como a angiotensina II e norepinefrina.
ProstaglandinasA função das prostaglandinas na cirrose descompensa-
da é provavelmente a mesma de outros estados hipovolê-micos: manutenção do fluxo sanguíneo renal e ritmo defiltração glomerular através do antagonismo aos efeitospressóricos da angiotensina II e outros vasoconstritores namicrovasculatura renal.
SÍNDROME NEFRÓTICAPacientes com síndrome nefrótica apresentam proteinú-
ria maciça, hipoalbuminemia, edema periférico ou gene-ralizado (anasarca) e hipercolesterolemia.49 O fenômenoprimário na síndrome nefrótica é a perda maciça de prote-ínas pelo rim.
Estudos iniciais revelam uma correlação entre a concen-tração sérica de albumina e o grau de edema em pacientesnefróticos. Em face destas observações, achava-se que ahipoalbuminemia, através da redução da pressão oncóti-ca do plasma, era responsável pela saída de líquido docompartimento intravascular para o intersticial. Entretan-to, investigações experimentais não corroboraram esta hi-pótese: diminuições da concentração plasmática de prote-ína no homem e em animais eram acompanhadas de vo-lume plasmático constante ou elevado. Logo, ponderou-se que ajustes nos mecanismos de troca transcapilar peri-férico deveriam ocorrer: queda da pressão oncótica do lí-quido intersticial, aumento na pressão hidráulica do líqui-do intersticial e aumento do fluxo linfático e proliferaçãolinfática.50
Outros estudos recentes demonstraram que a perme-abilidade do capilar periférico à albumina varia direta-mente com as alterações na concentração sérica de albu-mina e inversamente com as alterações do volume plas-

156 Metabolismo do Sódio e Fisiopatologia do Edema
mático.40,51 Portanto, há certos mecanismos protetores con-tra a formação de edema em estados hipoproteinêmicos(Quadro 10.7).
Parece, então, que o grau de edema não está tão relaci-onado com o grau de hipoalbuminemia per se, mas comalterações de mecanismos renais de controle do volumeextracelular. Na síndrome nefrótica por lesões mínimas nacriança, a hipoalbuminemia tem um papel importantíssi-mo na formação do edema. Nestes casos a redução do vo-lume intravascular ativa a retenção renal de sódio (meca-nismo de underfilling). A seqüência de eventos que deter-minam aumento na reabsorção renal de sódio pode serapreciada na Fig. 10.10 e é semelhante à que ocorre na in-suficiência cardíaca e cirrose.
Entretanto, convém salientar que muitos pacientes comsíndrome nefrótica podem ter volume plasmático elevado.O perfil renina-angiotensina-aldosterona também tem va-
riado de acordo com o volume plasmático. Ativação doeixo renina-angiotensina-aldosterona é encontrada noscasos de volume plasmático reduzido e supressão do eixonos casos de volume plasmático elevado. Logo, parece nãohaver um único mecanismo para explicar a retenção renalde sal na síndrome nefrótica.
Como na insuficiência cardíaca e cirrose hepática, a ati-vidade simpática e o nível de catecolaminas circulantesestão elevados, refletindo-se num aumento de resistênciavascular renal. Entretanto, o fluxo sanguíneo renal e o RFGnão estão uniformemente diminuídos na síndrome nefró-tica e, em algumas circunstâncias, o RFG está elevado. Estafiltração elevada é devida à hipoalbuminemia, que dimi-nui a pressão oncótica do capilar glomerular e, portanto,tende a aumentar a pressão de filtração glomerular. Poroutro lado, em situações de importante hipoalbuminemia,a vasoconstrição da arteríola aferente do glomérulo podediminuir a pressão hidrostática do capilar glomerular ereduzir o aumento do RFG. Portanto, na síndrome nefró-tica, o RFG pode estar normal, elevado ou reduzido, de-pendendo do balanço entre o efeito da redução da pressãooncótica do plasma, a resistência vascular renal e a pres-são de filtração glomerular.
Outro aspecto do edema nefrótico quando comparadocom o cirrótico ou cardíaco é o seguinte: há uma maiordiminuição na reabsorção tubular proximal de sódio e águadevido à redução da pressão oncótica peritubular causa-da pela hipoalbuminemia. Além disso, quando se bloqueiaa reabsorção distal de sódio com diuréticos, os nefróticosexcretam uma fração maior da carga filtrada de sódio.Logo, nefróticos podem responder melhor do que cardía-cos e cirróticos a diuréticos que agem no nefro distal. Es-tes achados sugerem que o principal local de retenção desódio na síndrome nefrótica está no nefro distal. Não sesabe se a elevada atividade da aldosterona explica esteachado.
Em certos casos de síndrome nefrótica causada porglomerulonefrites do tipo membranosa e membranopro-liferativa, pode existir lesão renal que afete a capacidadeintrínseca do rim em excretar sódio, resultando na reten-ção líquida e edema pelo mecanismo de overflow.38
GLOMERULONEFRITE AGUDAGlomerulonefrite proliferativa difusa aguda e outras
formas de lesão glomerular aguda podem causar retençãode sódio e água e formação de edema sem muitas altera-ções na concentração plasmática de albumina. Este balan-ço positivo de sódio e água aumenta o volume sanguíneoe a pressão arterial. Se houver elevação também da pres-são hidráulica capilar, há desequilíbrio nas forças deStarling, com passagem de fluido intravascular para o in-terstício. Se as defesas do interstício forem vencidas (au-mento do fluxo linfático, características físicas do interstí-cio), ocorre edema. Este mecanismo de retenção de líqui-do devido a uma incapacidade renal de excretar sódio e
Quadro 10.7 Mecanismos protetores contra aformação do edema periférico em estadoshipoalbuminêmicos*
Elevada drenagem linfáticaVasoconstrição pré-capilarDiluição da proteína do líquido intersticialBaixa complacência do tecido intersticialAjustes da permeabilidade da parede capilar à albumina
*Obtido de Skorecki, K.L. et al.50
Fig. 10.10 Esquema dos mecanismos atuantes na retenção de só-dio e edema da síndrome nefrótica. (Baseado em Schrier, R.W.9)

capítulo 10 157
água é conhecido como “transbordamento”(overflow) e étambém observado na insuficiência renal crônica.38 Osmecanismos envolvidos na retenção de sódio na glomeru-lonefrite aguda (Fig. 10.11) são discutidos a seguir.
Comprometimento do Coeficiente de UltrafiltraçãoA lesão glomerular compromete o coeficiente de ultrafil-
tração (Kf) causando redução do ritmo de filtração glome-rular (RFG), o qual causa redução na excreção de sódio.Havendo manutenção da ingestão normal de sódio, have-rá balanço positivo de sódio com expansão do volume ex-tracelular. Em condições normais esta expansão do volu-me extracelular acarretaria uma série de reações que alte-rariam a reabsorção tubular de sódio, aumentando a ex-creção fracional de sódio e restaurando o balanço. Por ra-zões desconhecidas, na glomerulonefrite aguda estas adap-tações na reabsorção de sódio não ocorrem.
Alterações na Função Tubular RenalNão é surpresa que lesões obstrutivas e inflamatórias
dos capilares glomerulares resultem em alterações signifi-cativas das forças de Starling do capilar peritubular, mo-dificando o ritmo de absorção tubular.
Um achado característico na glomerulonefrite aguda éuma queda da fração de filtração (FF), que se acompanhade diminuição da pressão oncótica capilar, a qual, transmi-
tida ao capilar peritubular, resulta numa redução de reab-sorção de líquido no túbulo proximal. Há, no entanto, pou-ca evidência de que as alterações na reabsorção proximal desódio sejam o principal mecanismo na retenção de sódio daglomerulonefrite aguda. Existem evidências de que o nefrodistal participe ativamente na reabsorção de sódio da nefri-te aguda. Com a redução do coeficiente de ultrafiltração edo RFG, diminui a oferta distal de sódio e conseqüentementecai a excreção absoluta e fracional de sódio.
A atividade plasmática da renina está reduzida face àexpansão do volume extracelular e a secreção de aldoste-rona habitualmente não está elevada.
Insuficiência CardíacaA insuficiência cardíaca que pode ocorrer na glomeru-
lonefrite aguda, tanto pela elevação da pré-carga (volume)como da pós-carga (hipertensão arterial), acaba sendo maisum mecanismo que determina retenção de sódio.
O edema na glomerulonefrite aguda resulta de uma ex-pansão do volume extracelular e elevação da pressão intra-capilar sistêmica, alterando as forças de Starling nos capila-res periféricos. Com isto há saída de sal e água para o in-terstício, e, dependendo do grau de volume e pressão dolíquido intersticial, haverá evidência clínica de edema.
EDEMA OBSERVADO EM MULHERES
Edema da GravidezNuma gravidez normal há aumento na retenção renal
de sal, expansão do volume plasmático e ganho de peso.Há também aumento significativo do RFG, fluxo plasmá-tico renal e do débito cardíaco. Esta retenção de sódio nagravidez é considerada fisiológica para satisfazer as neces-sidades do feto, o aumento da capacidade vascular mater-na e a seqüestração de líquido na cavidade amniótica. Al-guns dos fatores importantes na retenção de sódio da gra-videz estão enumerados no Quadro 10.8.29 Alterações defatores físicos atuantes no túbulo renal parecem ser impor-tantes na retenção de sódio. O RFG está mais elevado doque o fluxo plasmático renal, resultando num aumento dafração de filtração.42,43 Edema localizado nas extremidadesinferiores ocorre em 75% das mulheres grávidas. Este ede-ma ocorre por várias razões:
• Efeito mecânico do útero aumentando a pressão veno-sa nos membros inferiores;
• Perfusão elevada nas pernas devido a um aumento nodébito cardíaco e diminuição da resistência vascularperiférica;
• Aumento do volume plasmático e redução da pressãooncótica do plasma;
• Outros fatores enumerados no Quadro 10.8.
Edema generalizado pode ocorrer em até 20% das mu-lheres grávidas e na ausência de toxemia é considerado atéfisiológico.
Fig. 10.11 Fisiopatologia do edema nefrítico. (Baseado em Glas-sock, R.J. et al.56)

158 Metabolismo do Sódio e Fisiopatologia do Edema
Toxemia GravídicaOs fatores responsáveis pela elevada retenção de sódio
na toxemia são desconhecidos. Os níveis de renina-angio-tensina-aldosterona diminuem com o aparecimento datoxemia assim como diminuem o RFG e o fluxo sanguíneorenal. Postula-se que a retenção de sódio pode ser devidaa um comprometimento do balanço glomérulo-tubularresultante de uma hiper-reabsorção do filtrado, a exemplodo que ocorre numa glomerulonefrite proliferativa aguda,pois na toxemia há importante lesão endotelial com depo-sição de material fibrinóide.
Edema Cíclico IdiopáticoEsta é uma síndrome observada predominantemente em
mulheres obesas, adultas, que ainda não entraram na me-nopausa. A síndrome é caracterizada por períodos de ede-ma, cefaléia, irritabilidade e distensão abdominal. A in-vestigação não revela alterações cardíacas, renais ou he-páticas.
Como a maioria destas pacientes apresenta boa diuresee natriurese quando em repouso no leito, questiona-se sea elevada reabsorção de sódio não estaria associada à po-sição ortostática. Além do componente ortostático de re-tenção de líquido, há considerável evidência de que estaspacientes têm diminuição do volume plasmático.
Entre outros fatores aventados para explicar o edemadestacam-se: defeito na permeabilidade capilar e elevadosníveis de prolactina. Muitas pacientes usam ou usaramdiuréticos. Como os diuréticos causam contração do volu-me circulante, há um estímulo à retenção de sódio comelevação dos níveis de renina-angiotensina-aldosterona eparticipação de outros mecanismos. O edema parece ocor-rer principalmente após a cessação do uso dos diuréticos.A magnitude do ganho de peso está aumentada com umadieta alta em sal e em carboidratos.
Edema Pré-menstrualO edema geralmente faz parte da síndrome pré-mens-
trual caracterizada por nervosismo, irritabilidade e cefaléia.A causa da retenção de sódio não é conhecida mas é pro-vavelmente devida a um distúrbio endócrino como umaalteração na relação estrógeno/progesterona ou, comosugerido mais recentemente, uma elevação dos níveis plas-máticos de prolactina.52
CAUSAS DIVERSAS DE EDEMA
Síndrome da Permeabilidade Capilar ElevadaHá relatos de alguns pacientes que apresentaram angi-
oedema generalizado recorrente. Desconhece-se a causa daelevada permeabilidade capilar, sendo a única anormali-dade detectada a presença de uma paraproteína monoclo-nal IgG.53
Hipocalemia CrônicaAlguns pacientes com depleção crônica de potássio
podem apresentar edema periférico. Não se conhece a cau-sa da elevada reabsorção tubular de sódio.
MedicamentosVárias substâncias administradas e pacientes podem
determinar um aumento na reabsorção de sódio: estrogê-nios (anticoncepcionais); diazóxido; hidralazina; minoxi-dil e outras drogas simpatolíticas como metildopa, guane-tidina e clonidina. Mais recentemente antiinflamatóriosnão-esteróides foram incluídos neste grupo de drogas.
O mecanismo da retenção de sódio dos estrógenos nãoé conhecido, mas provavelmente relaciona-se a uma açãoa nível tubular.
Os vasodilatadores utilizados na hipertensão arterialreduzem a resistência vascular periférica, alterando a re-lação volume plasmático/capacitância vascular.
Microangiopatia Capilar do Diabetes MellitusHá relatos de alguns diabéticos com função renal nor-
mal que apresentam edema idiopático. Para estes casos temsido sugerido que, na posição ereta, pode haver uma pas-sagem excessiva de líquido para o interstício devido a umamicroangiopatia capilar, com conseqüente retenção desódio e edema.
Pontos-chave:
• A fisiopatogênese do edema na insuficiênciacardíaca, cirrose, síndrome nefrótica esíndrome nefrítica tem a participação dosmecanismos de subpreenchimento e/outransbordamento
• O tratamento medicamentoso do edema éfeito com diuréticos
Quadro 10.8 Possíveis fatores importantes naretenção renal de sódio da gravidez normal*
1. Obstrução ureteral devida ao útero grávido2. Efeitos da postura no RFG e na perfusão renal3. Efeitos da postura na seqüestração venosa nos
membros inferiores4. Possível aumento no apetite por sal5. Mecanismos responsáveis pela retenção tubular
renal de sódioa. Níveis elevados de aldosterona e outros
mineralocorticóidesb. Níveis elevados de estrogêniosc. Presença de fatores humorais retentores
de sódio ?d. Diminuição da resistência vascular
periféricae. Aumento anatômico da capacidade vascular
*Obtido de Levy & Seely.57

capítulo 10 159
Princípios Gerais no Tratamentodo Edema
TRATAMENTO DA DOENÇA BÁSICAComo a redução do volume sanguíneo arterial efetivo
é um denominador comum na retenção de sódio da insu-ficiência cardíaca, cirrose hepática e síndrome nefrótica, omanejo clínico deve ser dirigido para a correção deste dis-túrbio básico. Assim sendo, na insuficiência cardíaca me-lhorar o débito cardíaco restaura o volume circulante efe-tivo. Na síndrome nefrótica por lesões mínimas o uso decorticosteróides reduz a proteinúria e conseqüentementea hipoalbuminemia.
ADEQUAÇÃO DA INGESTA DE SAL E ÁGUAEmbora a restrição de sódio seja efetiva na prevenção
do aumento do edema, ela não causa um balanço negati-vo de sódio. A diurese de pacientes cardíacos hospitaliza-dos e colocados em dietas hipossódicas está mais relacio-nada ao efeito benéfico do repouso no débito cardíaco doque resultante da dieta hipossódica.
Pacientes que estão formando edema retêm uma fraçãoda ingesta diária de sal a fim de restaurar o volume san-guíneo arterial efetivo. A excreção urinária diária de sódiodestes pacientes reflete a capacidade de excreção renal.Conhecendo-se a oferta de sódio na dieta, a determinaçãoda excreção de sódio nas 24 horas permite saber se o ba-lanço de sódio é positivo ou negativo. Concentrações uri-nárias de sódio da ordem de 10 a 15 mEq/L geralmenteindicam um balanço positivo, ou seja, maior quantidadede sódio está sendo reabsorvida nos túbulos renais.
A maior parte dos pacientes edemaciados tem umcomprometimento na excreção renal de água. A ingestadiária de líquido deve ser ajustada para as perdas insensí-veis (500 a 700 ml) por dia mais as perdas urinárias.
MOBILIZAÇÃO DO EDEMAO repouso no leito é capaz de induzir diurese devido à
redução da seqüestração venosa na periferia, aumentan-do assim o volume sanguíneo arterial efetivo. Efeito simi-lar possuem as meias elásticas.
INDUÇÃO DE BALANÇO NEGATIVO DE SÓDIOÉ possível induzir balanço negativo de sódio com a uti-
lização de diuréticos (v. Cap. 43). Com a eliminação desódio provocada por estas drogas, há redução do volumecirculante, diminuição da pressão capilar e conseqüentemovimentação de fluido do interstício para o intravascu-lar, devido à modificação das forças de Starling. O fluidoassim trazido ao intravascular torna-se disponível para afiltração glomerular.38
Deve ser salientado, porém, que a redução no volumeintravascular obtida com os diuréticos pode provocar hi-povolemia e insuficiência renal. Recomenda-se que nos
pacientes em uso de diuréticos seja feita cuidadosa moni-torização diária do peso, volume urinário e pressão arteri-al com o paciente deitado, sentado e em pé.38 Além disso,é essencial o conhecimento da potência, local de ação ecomplicações do uso de diuréticos (ver Cap. 43).
A presença de edema per se não é uma indicação de usode diuréticos. Em geral, o uso dos diuréticos deve ficarrestrito a situações tais como: comprometimento da fun-ção cardíaca e/ou respiratória; desconforto físico devidoao acúmulo excessivo de líquido e permitir liberalizaçãodo sal na alimentação de pacientes que toleram pouco di-etas hipossódicas (Quadro 10.9).
EXERCÍCIOS
(Respostas no final do capítulo.)
1) Num indivíduo de 70 kg, qual o volume do espaço extracelular?
Nos exercícios 2 e 3, responda às seguintes perguntas:
a. Qual o distúrbio do extracelular que este paciente apresenta?b. Qual a intensidade deste distúrbio (em percentagem aproxi-
mada)?c. Que tipo de solução administrar?d. Qual a quantidade de solução a infundir?e. Em quantas horas deve ser administrada esta solução?
2) Tome como exemplo o mesmo indivíduo acima, com história dedois dias de evolução com vômitos e diarréia profusa. Ao examefísico apresenta queda de 15 mmHg na pressão sistólica e diastó-lica quando fica em pé. A mucosa oral está seca e as jugulares têmenchimento lento.
3) Considere uma paciente de 60 kg, que permaneceu internada portrês dias em outra cidade, com quadro de encefalite, com drena-gem por sonda nasogástrica de aproximadamente 2 litros de esta-se ao dia, utilizando manitol, e recebendo solução glicosada 2.000ml/dia. Esta paciente é admitida, no hospital onde você é planto-nista, com PA 60 � 30 mmHg, FC � 132 bpm, extremidades friase perfusão periférica comprometida, enchimento capilar lento,jugulares colabando com a inspiração e anúria. Além disso, encon-
Quadro 10.9 Princípios gerais no tratamento doedema*
1. Avaliação da adequação do tratamento da doençabásica responsável pelo edema
2. Avaliação do grau de ingesta de água e sal3. Mobilização do edema4. Avaliação da indicação do uso de diuréticos
A. Comprometimento da função respiratóriaa. Edema pulmonarb. Ascite com elevação dos diafragmas e
associada a atelectasiasB. Comprometimento da função cardiovascular
secundária a sobrecarga de volumeC. Excesso de líquido comprometendo a atividade
física e causando desconfortoD. Permitir maior liberalização do sal na dieta,
aumentando o paladar dos alimentosE. Indicação cosmética
*Obtido de Schrier, R.W.9

160 Metabolismo do Sódio e Fisiopatologia do Edema
tra-se confusa e sonolenta. Assim que a paciente chega, você pun-ciona uma veia jugular e encontra uma PVC de 3 cm H
2O.
REFERÊNCIAS BIBLIOGRÁFICAS
1. MALNIC, G. e MARCONDES, M. Fisiologia Renal, pág. 91, 3.ª parte— Regulação do volume extracelular. EDART — São Paulo Livra-ria Ltda. 1972.
2. HALPERIN, M.L.; GOLDSTEIN, M.B. Sodium and water physio-logy. In: Fluid, Electrolyte and Acid-Base Physiology — A Problem BasedApproach, Eds. Halperin, M.L.; Goldstein, M.B., pp. 217-251. W.B.Saunders Co, 1994.
3. EARLEY, L.E. Sodium metabolism. Cap. 3, pp. 95-119. In: ClinicalDisorders of Fluid and Electrolyte Metabolism. Eds. Maxwell, M.H. andKleeman, C.R. McGraw-Hill Book Co., 1972.
4. WINAVER, J.; ABASSI, Z.; GREEN, J.; SKORECKI, K.L. Control ofextracellular fluid volume and the pathophysiology of edema for-mation. Cap. 19, pp. 795-866. In: The Kidney, Eds. Brenner, B.M.; Rec-tor Jr, F.C. W.B. Saunders Co., 2000.
5. EPSTEIN, F.H. e cols. Effects of an arteriovenous fistula on renalhemodynamics and electrolyte excretion. J. Clin. Invest., 32:233, 1955.
6. DIRKS, J.H. e cols. Control of extracellular fluid volume and thepathophysiology of edema formation. Cap. 4, pp. 495-552. In: TheKidney, Eds. Brenner, B.M. and Rector Jr, F.C. W.B. Saunders Co.,1976.
7. ROSE, B.D.; POST, T.W. Up to Date v. 9., n. 3. Cap. 8B: Regulation ofthe effective circulating volume, 2001.
8. HABERICH, F.J. Osmoreception in the portal circulation. Fed. Proc.,27:1.137, 1968.
9. BICHET, D.G.; ANDERSON, R.J.; SCHRIER, R.W. Renal sodiumexcretion, edematous disorders, and diuretic use. In: Renal andElectrolyte Disorders, Ed. Schrier, R.W. Cap. 2, pp. 89-159, Little, Bro-wn and Co, 1992.
10. DE WARDENER, H.E. e cols. Studies on the efferent mechanism ofthe sodium diuresis, which follows the administration of intrave-nous saline in the dog. Clin. Sci., 21:249, 1961.
11. KLAHR, S. e SLATOPOLSKY, E. Renal regulation of sodium excre-tion. Function in health and in edema-forming states. Arch. Intern.Med., 131:780, 1973.
12. BRENNER, B.M. e cols. Postglomerular vascular protein concentra-tion: evidence for a causal role in governing fluid reabsorption andglomerulotubular balance by the renal proximal tubule, J. Clin.Invest., 50:336, 1971.
13. BRENNER, B.M. e col. Quantitative importance of changes inpostglomerular colloid osmotic pressure in mediating glomerulo-tubular balance in the rat. J. Clin. Invest., 52:190, 1973.
14. EARLEY, L.E. e FIEDLER, R.N. The effect of combined renalvasodilatation and pressor agents on renal hemodynamics and thetubular reabsorption of sodium. J. Clin. Invest., 45:542, 1966.
15. LEYSSAC, P.P. Dependence of GFR on proximal tubular reabsorp-tion of salt. Acta Physiol. Scand., 58:236, 1963.
16. THURAY, K. e SCHNERMANN, J. Die Natriumkonzentration an denMacula Densa-Zellen als regulierender faktor für das glomerulumfil-trat (Mikropunktions-versuche). Klin. Wochenschr., 43:410, 1965.
17. BARTOLI, E. e EARLEY, L.E. Evidence for the intraluminal actionof plasma factors on proximal sodium reabsorption. Clin. Res., 20:586,1972.
18. BRUNNER, F.P. e col. Mechanism of glomerulotubular balance: II.Regulation of proximal tubular reabsorption by tubular volume, asstudied by stopped-flow microperfusion. J. Clin. Invest., 54:603, 1966.
19. BURG, M.B. The renal handling of sodium chloride. Cap. 7, pp. 272-298. In: The Kidney. Eds. Brenner, B.M. and Rector Jr, F.C. W.B. Saun-ders Co., 1976.
20. PEIRCE, N.F. e col. Replacement of water and electrolyte losses incholera by an oral glucose-electrolyte solution. Ann. Inter. Med.,70:1.173, 1969.
21. GIEBISCH, G. Coupled ion and fluid transport in the kidney. TheNew Engl. J. Med., 287:913, 1972.
22. SCHRIER, R.W. e DE WARDENER, H.E. Tubular reabsortion ofsodium ion: influence of factors other than aldosterone and glome-rular filtration. N. Engl. J. Med., 285:1231, 1971 e 285:1292, 1971.
23. BOUGOIGNIE, J.J. e col. The presence of a natriuretic factor in uri-ne of patients with chronic uremia. J. Clin. Invest., 53:1559, 1974.
24. BRICKER, N.S.; ZEA, L.; SHAPIRO, M., et al. Biologic and physicalcharacteristics of the non-peptidic, non-digitalis-like natriuretic hor-mone. Kidney Int., 44:937, 1993.
25. HAUPERT, G.T. Natriuretic hormones. In: Cecil Textbook of Medici-ne, W.B. Saunders Co, Cap. 234, pp. 1194-1198, 2000.
26. ROSE, B.D. Natriuretic hormones: atrial peptides and ouabain-likehormone. Up to Date v. 9, n. 3, 2001.
27. LEVIN, E.R.; GARDNER, D.G.; SAMSON, W.K. Natriuretic Pepti-des. The New Engl. J. Med., 339(5):321-328, 1998.
28. HIGA, E.M.S. Óxido nítrico e o rim. In: Atualidades em Nefrologia, Eds:Cruz, J.; Barros, R.T., vol. 4, pp. 357-365, Sarvier, 1996.
29. ROSE, B.D.; POST, T.W. Regulation of renal Na� excretion. Cap. 8C,Up to Date v. 9, n. 3, 2001.
30. CHAPMAN, W.H. e col. The Urinary System. An Integrated Approach.W.B. Saunders Co. 1973.
31. POST, T.W.; ROSE, B.D. Dehydration is not synonymous with hypo-volemia. Up to Date v. 9, n. 3, 2001.
32. POST, T.W.; ROSE, B.D. Clinical manifestations and diagnosis ofvolume depletion, Up to Date v. 9, n. 3, 2001.
33. COHN, J.N. Central venous pressure as a guide to volume expansi-on. Ann. Intern. Med., 66:1283, 1967.
34. ROSE, B.D. Fluid replacement in volume depletion. Up to Date v. 9,n. 3, 2001.
35. PRESTON, R.A. IV solutions and IV orders. In: Acid-Base, Fluids andElectrolytes Made Ridiculously Simple. Ed. Preston, R.A., Cap. 2, pp.31-38, MedMaster, Inc., 1997.
36. ROSE, B.D.; MANDEL, J. Treatment of severe hypovolemia or hypo-volemic shock. Up to Date v. 9, n. 3, 2001.
37. SCRIBNER, B.H. Apostila para o curso de Equilíbrio Hidroeletrolí-tico (Syllabus), 1953.
38. SEGURO, A.C.; HELOU, C.M.B.; ZATZ, R. Fisiopatologia do edema.Cap. 2, pp. 151-172, In: Fisiopatologia Renal. Ed. Zatz, R., Atheneu, 2000.
39. VALTIN, H. Renal Dysfunction: Mechanisms Involved in Fluid andSolute Imbalance. Cap. 3, p. 65 Little, Brown and Co. 1979.
40. INTAGLIETTA, M.E.; ZWEIFACH, B.W. Microcirculatory basis offluid exchange. Adv. Biol. Med. Phys., 15:11, 1974.
41. LEAF, A., COTRAN , R.S. Renal Pathophysiology. Cap. 6, p. 136.Oxford University Press, 1976.
42. PORTO, C.C. Exame físico geral. In: Porto, C.C. Semiologia Médica,Cap. 7, pp. 65-114. Guanabara Koogan, 1997.
43. BAKRIS, G.L.; STEIN, J.H. Sodium metabolism and maintenanceof extracellular fluid volume. In: Fluid, Electrolyte and Acid-Base Di-sorders, Eds. Arieff, A.I.; DeFronzo, R.A. Cap. 2, pp. 29-49. Chur-chill-Livingstone, 1995.
44. EPSTEIN, F.H.; POST, R.S.; McDOWELL, M. Effects of an arterio-venous fistula on renal hemodynamics and electrolyte excretion. J.Clin., Invest., 32:233, 1953.
45. NEEDLEMAN, P.; GREENWALD, J.E. Atriopeptin: a cardiac hor-mone intimately involved in fluid, electrolyte and blood pressurehomeostasis. New Engl. J. Med., 314:828, 1986.
46. METTAURER, B.; ROULEAU, J.L.; BICHET, D. et al. Sodium andwater excretion abnormalities in CHF. Ann Intern. Med., 105:161,1986.
47. SEIFTER, J.L.; SKORECKI, K.L.; STIVELMAN, J.C.; HAUPER, G.;BRENNER, B.M. Control of extracellular fluid volume and patho-physiology of edema formation. Cap. 10, p. 343. In: The Kidney. Eds.Brenner B.M., Rector Jr., F.C. W.B. Saunders Co. 1986.
48. LUNZER, M.R.; NEWMAN, S.P.; BERNARD, A.G. et al. Impairedcardiovascular responsiveness in liver disease. Lancet, 2:382, 1975.
49. ROSE, B.D. Approach to the patient with edema. Up to Date, v. 9, n.3, 2001.

capítulo 10 161
50. SKORECKI, K.L.; NADLER, S.P.; BADR, K.F.; BRENNER, B.M. Re-nal and systemic manifestations of glomerular diseases. Cap. 21. p.891. In: The Kidney, Eds. Brenner, B.M., Rector Jr, F.C. W.B. SaundersCo., 1986.
51. WRIGHT, E.P. Capillary permeability of protein as a factor in thecontrol of plasma volume. J. Physiol., 237:39, 1974.
52. HALBREICH, U.; ASSAEL, M.; BEN-DAVID, M.; BORNSTEIN, R.Serum prolactin in women with premenstrual syndrome. Lancet,2:654, 1976.
53. ATKINSON, J.P.; WALDMANN, T.A.; STEIN, S.F. et al. Systemiccapillary leak syndrome and monoclonal IgG gammopathy. Medi-cine, 56:225, 1977.
54. VALTIN, H. Renal Function: Mechanisms Preserving Fluid and SoluteBalance in Health, p. 114. Little, Brown and Co., 1973.
55. SLATOPOLSKI, E. e cols. Studies on the characteristics of the con-trol system governing sodium excretion in uremic man. J. Clin.Invest., 47:521, 1968.
56. GLASSOCK, R.J.; COHEN, A.H.; BENNET, C.M.; MARTINEZ-MALDONADO, M. Primary glomerular diseases. Cap. 26. p. 1351.In: The Kidney. Eds. Brenner, B.M.; Rector, Jr, F.C. W.B. Saunders Co.,1981.
57. LEVY, M.; SEELY, J.F. Pathophysiology of edema formation. TheKidney, Eds. Brenner, B.M.; Rector, Jr. F.C. W.B. Saunders Co., p. 723,1981.
ENDEREÇOS RELEVANTES NA INTERNET
http://www.kidneyatlas.org/book1/adk1-02.pdf — Ex-celente capítulo do atlas on-line editado pelo Dr Schrier.http://umed.med.utah.edu/ms2/renal/Word%20files/c)%20Disorders%20of%20Volume-Ed.htm — página queaborda a fisiopatogenia e tratamento do edema.http://www.medonline.com.br/med-ed/med10/orimicc.htm — artigo que aborda as alterações renais en-contradas na insuficiência cardíaca.
http://www.geocities.com/HotSprings/4234/cirrose.html— página que entre outros itens descreve a fisiopatogeniadas alterações renais encontradas na cirrose hepática.http://www.learndoctor.com/chapterpages/chapter22.htm— site com questões de auto-avaliação.
RESPOSTAS DOS EXERCÍCIOS
1) Espaço extracelular � 20% do peso. Paciente de 70 kg � 14 litros.2) Paciente de 70 kg com diarréia e queda de PA e aumento da FC
ortostáticas.a. Depleção do espaço extracelular.b. 20-30% de depleção.c. Solução salina isotônica.d. 70 kg � 14 litros de EEC; 20-30% de DEEC � 14 � 0,2-0,3 �
2,8-4,2 litros de solução a infundir, pois este é o déficit apre-sentado.
e. Na primeira hora infundir volume suficiente para que os sinaishemodinâmicos encontrados sejam melhorados; o restante dovolume infundir nas próximas horas.
3) Paciente de 60 kg com história de perda por sonda gástrica e usode diurético osmótico.a. Esta paciente apresenta um grau avançado de depleção do es-
paço extracelular, com sinais de choque hipovolêmico.b. Depleção de 40-50% do espaço extracelular.c. Solução salina isotônica.d. 60 kg � 12 litros de EEC; 40-50% de DEEC � 12 � 0,4-0,5 � 4,8-
6 litros de solução a infundir, pois este é o déficit apresentado.e. Na primeira hora é importante infundir volume suficiente para
desaparecerem os sinais de comprometimento hemodinâmico.A monitorização da diurese auxilia a verificar a adequação dareposição; continuar monitorizando a PVC, avaliando este pa-râmetro sem esquecer de suas limitações.

Capítulo
11Metabolismo Ácido-Básico
Miguel Carlos Riella e Maria Aparecida Pachaly
INTRODUÇÃO
CONCEITOS E PRINCÍPIOS QUÍMICOS
Ácido
Base
Sistema tampão
pH
Lei de ação das massas
Equação de Henderson-Hasselbalch
Eletroneutralidade
METABOLISMO ÁCIDO-BÁSICO
SISTEMAS TAMPÃO
Sistema tampão ácido carbônico-bicarbonato
Proteínas plasmáticas
Hemoglobina
Tamponamento nos ossos
CONTROLE RESPIRATÓRIO DA PCO2
CONTROLE RENAL DO EQUILÍBRIO ÁCIDO-BÁSICO
Reabsorção tubular do bicarbonato filtrado
Secreção tubular de H�
Fatores que influenciam na reabsorção do bicarbonato
filtrado
Excreção de acidez titulável (AT)
Excreção de amônio (NH4�)
Produção proximal e secreção de NH4�
Gradiente intersticial corticopapilar para NH4�/NH3
Secreção de amônia nos ductos coletores (NH3)
Difusão não-iônica
DISTÚRBIOS CLÍNICOS DO METABOLISMO
ÁCIDO-BÁSICO
Acidose metabólica
Causas
Manifestações clínicas e efeitos sistêmicos
Achados laboratoriais
Tratamento
Alcalose metabólica
Causas de alcalose metabólica
Geração da alcalose metabólica
Manutenção da alcalose metabólica
Mecanismos de defesa do pH na alcalose
metabólica
Manifestações clínicas
Dados laboratoriais
Tratamento
Acidose respiratória
Causas
Conseqüências clínicas
Conseqüências fisiológicas
Tratamento
Alcalose respiratória
Causas
Conseqüências clínicas
Conseqüências fisiológicas
Tratamento
Distúrbios ácido-básicos mistos
Diagnóstico dos distúrbios ácido-básicos
Roteiro para interpretação dos distúrbios
ácido-básicos
Alguns exemplos
EXERCÍCIOS
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET
RESPOSTAS DOS EXERCÍCIOS

capítulo 11 163
INTRODUÇÃO
Para que seja mantida a estabilidade do meio interno,deve haver equilíbrio entre a produção e a remoção de íonshidrogênio (H�) em nosso organismo. Os rins são funda-mentais na eliminação do H�, mas o controle da concen-tração deste íon envolve ainda outros mecanismos, comoo tamponamento realizado pelo sangue, células e pul-mões.1
A quantidade de íon hidrogênio é mantida dentro delimites estreitos, num processo extremamente sensível,uma vez que a quantidade de hidrogênio no extracelular(40 nanoequivalentes/litro � 0,00004 mEq/litro) é cercade 1 milionésimo das concentrações do sódio, potássio oucloro.2
A manutenção desta baixa concentração hidrogeniôni-ca é essencial para a função celular normal. Os íons hidro-gênio são altamente reativos, particularmente com porçõesde moléculas protéicas com carga negativa.2 Assim, varia-ções na concentração de hidrogênio produzem grandeimpacto sobre as funções celulares, pois quase todos ossistemas enzimáticos de nosso organismo e proteínas en-volvidas na coagulação e contração muscular são influen-ciados pela concentração de íons hidrogênio.2,3
CONCEITOS E PRINCÍPIOSQUÍMICOS
Ácido
Substância capaz de doar íons H� (prótons). Exemplos:H2CO3, NH4
�, HCl. Um ácido forte como o HCl se dissociarapidamente e libera grandes quantidades de H�. Os áci-dos fracos têm uma menor tendência à dissociação, libe-rando H� com menor intensidade. O acúmulo excessivo deíons H� é chamado de acidose.1,4
Base
Substância (íon ou molécula) capaz de receber íons H�.Exemplos: HCO3
�, NH3, HPO4�. Uma base forte (p.ex., o
OH�) reage de maneira rápida e intensa com o H�, remo-vendo-o de uma solução. Uma base fraca reage de manei-ra pouco intensa. O termo base é usado como sinônimo deálcali. Álcali é uma molécula formada pela combinação deum metal alcalino (p. ex., sódio, potássio) com um íon for-temente básico, como o íon hidroxila (OH�). Os íons hidro-xila reagem rapidamente com os íons hidrogênio, portan-to são bases típicas. A remoção excessiva de íons H� doslíquidos corporais é chamada de alcalose. No equilíbrioácido-básico normal, a maior parte dos ácidos e bases exis-tentes no espaço extracelular é fraca.1
Sistema TampãoÉ o sistema formado por um ácido e uma base a ele con-
jugada, cuja finalidade é a de minimizar alterações na con-centração hidrogeniônica [H�] de uma solução. Em outraspalavras, uma base fraca se liga aos H� dissociados de umácido forte para formar um ácido fraco pouco dissociável,tamponando e, portanto, minimizando as alterações naconcentração de H�. Além disso, um sistema tampão tam-bém pode doar H�.5
pHComo a concentração hidrogeniônica [H�] é muito
baixa, torna-se mais simples expressar esta concentraçãoem escala logarítmica, utilizando as unidades de pH. OpH é inversamente proporcional à concentração hidro-geniônica. Um baixo pH corresponde a uma alta concen-tração de íons hidrogênio, enquanto um pH alto corres-ponde a uma concentração hidrogeniônica baixa. Portan-to, a atividade dos íons H� em uma solução determinaa sua acidez.1,6
pH � log 1/H� � � log [H�]
Para a [H�] normal de 40 mEq/litro, o pH é:
pH � � log [0,00000004] � 7,4
Nos líquidos corporais e diferentes tecidos existe umaampla variação de pH. O pH arterial normal é 7,40, sen-do um pouco menor no sangue venoso e interstício(7,35), devido à quantidade de CO2 que se difunde dostecidos. O pH urinário pode variar de 4,5 a 8,0, depen-dendo do estado ácido-básico do fluido extracelular. Noestômago, a produção de HCl pode reduzir o pH para0,8.1
Considera-se o pH como normal se estiver entre 7,35 e7,45. Os limites de pH sanguíneo compatível com a vidasão 6,8 e 8,0.1
Lei de Ação das MassasA lei de ação das massas estabelece que a velocidade de
uma determinada reação química é proporcional à concen-tração dos reagentes. Por exemplo, na reação abaixo, a ve-locidade com que a reação ocorre para a direita ou para aesquerda é uma constante que depende da concentraçãodos substratos.
HPO4�� � H� ↔ H2PO4
�
Em equilíbrio, são iguais as constantes para cada ladoda equação. Porém, se houver maior quantidade de subs-trato em um lado, a reação se dirige para o lado oposto. Alei de ação das massas é útil para descrever a dissociaçãode todos os ácidos e bases do organismo. Por exemplo, paraa dissociação de um ácido HA em H� � A�: 7

164 Metabolismo Ácido-Básico
Ka � [H�] � [A�]
[HA]
Onde:Ka � constante de dissociação para este ácido (há um va-lor para cada ácido).
Equação de Henderson-Hasselbalch
A equação que acabamos de ver pode ser reorganiza-da, originando a equação de Henderson-Hasselbalch, quequando aplicada ao sistema tampão ácido carbônico-bicar-bonato, um dos mais importantes de nosso organismo,define a relação entre pH, PCO2 e HCO3
�. Neste caso, pK éa constante de dissociação do ácido carbônico. Fica assimdemonstrado que o pH do sangue é determinado pela con-centração de bicarbonato e tensão de CO2.
6,7
pH � pK � log [HCO3
�]log [H2CO3]
Eletroneutralidade
É o princípio segundo o qual não pode haver acúmulode quantidades significativas de cargas elétricas em siste-mas biológicos, pois isto geraria diferenças muito altas depotencial elétrico nos tecidos. Então, ao ser absorvido umcátion, é necessário que seja reabsorvido um ânion, ou eli-minado outro cátion, de forma que resulte o mesmo nú-mero de cargas positivas e negativas.8
METABOLISMO ÁCIDO-BÁSICO
O metabolismo de gorduras e carboidratos origina CO2
e H2O. Aproximadamente 20.000 mEq de CO2 são produzi-dos diariamente. Ao observar a reação abaixo, percebe-seque se o CO2 não fosse eliminado, a reação se dirigiria nosentido de produção do H2CO3, que se dissociaria e aumen-taria a quantidade de hidrogênio no organismo, resultandoem acidose. A eliminação do CO2 é realizada pelos pulmões;por este motivo o CO2 é chamado de ácido volátil.2
CO2 � H2O ↔ H2CO3 ↔ H� � HCO3�
Além da produção de ácido volátil, são produzidosoutros ácidos em nosso metabolismo. A dieta ocidentalcontém aminoácidos e outras substâncias ácidas. Por exem-plo, o cloreto de lisina é metabolizado em ácido clorídricoe uréia; a hidrólise de proteínas e ácidos nucléicos formaácido fosfórico, e a oxidação de aminoácidos que contêmenxofre gera ácido sulfúrico. Desta forma, produz-se umacarga ácida diária da ordem de 1 mEq/kg/dia. Além dis-so, a oxidação incompleta da glicose pode originar 20-30mEq de ácidos orgânicos por dia.9
A produção endógena de ácidos é um processo normal,mas pode estar aumentada na presença de certas influên-cias hormonais, substratos exógenos ou interrupção dasvias de controle. Alguns estados patológicos se caracteri-zam por um aumento significativo na produção de ácidosorgânicos, como os cetoácidos formados no diabetes meli-to descompensado, alcoolismo ou jejum prolongado. Dro-gas e toxinas podem acelerar a produção de ácidos orgâ-nicos, como o ácido fórmico a partir do metanol; ácidooxálico a partir do etilenoglicol, e ácido salicílico a partirda aspirina. Outro mecanismo para acúmulo de ácido ocor-re quando seu metabolismo e excreção estiverem compro-metidos. Exemplo disso é o acúmulo de ácido láctico, casosua conversão para glicose (ciclo de Cori) seja interrompi-da por algum motivo; como o tecido muscular produzimensas quantidades deste ácido todos os dias, ele rapida-mente se acumularia.9 Ao contrário do CO2, que pode sereliminado pelos pulmões, os demais ácidos são denomi-nados ácidos não-voláteis ou fixos e devem ser eliminadospelo rim.
Além do ganho diário de ácidos voláteis e não-voláteis,nosso organismo também deve compensar as perdas fisi-ológicas de substâncias alcalinas, de cerca de 20-30 mEq debicarbonato por dia. Em algumas doenças diarréicas, estaperda pode aumentar dez vezes.1
Frente a todos estes dados, percebemos que existe emnosso organismo uma predominância de mecanismos quelevam a um excesso de ácidos. A manutenção de um pHnormal nos fluidos corporais frente a uma carga ácida re-quer a integração de mecanismos fisiológicos que impedemque haja variações muito intensas na concentração de hi-drogênio.
A primeira linha de defesa que atua na manutenção deum pH fisiológico frente à adição de ácidos são os tampões(bicarbonato e outros tampões extracelulares), que ageminstantaneamente. Já a segunda linha de defesa envolve osistema respiratório e consiste na variação da PCO2 de acor-do com a [H�] em minutos a horas. Por último, há a tercei-ra linha de defesa, que envolve o sistema renal através docontrole da concentração de bicarbonato. A eficácia máxi-ma deste último sistema é atingida 24 a 48 horas após oinício do desequilíbrio.2,10
Desta maneira, e voltando à equação de Henderson-Hasselbalch, podemos compreender que o organismo atuana normalização do pH atuando nas variáveis que deter-minam o pH: PCO2 e HCO3
�.O desvio do pH arterial abaixo de 7,35 ou acima de 7,45
é referido como acidemia e alcalemia, respectivamente. Osprocessos que tendem a reduzir ou elevar o pH são cha-mados acidose e alcalose. Desta maneira, poderemos terquatro alterações primárias do estado ácido-básico:
1. acidose metabólica: quando o HCO3� diminuir, ou quan-
do a concentração de H� aumentar;2. alcalose metabólica: quando o HCO3
� estiver elevado ouquando ocorrer uma perda de H�;

capítulo 11 165
3. acidose respiratória: quando ocorrer um aumento naPCO2;
4. alcalose respiratória: quando a PCO2 for reduzida.
Porém, há situações em que duas ou mais anormalida-des estão presentes, caracterizando os distúrbios ácido-básicos mistos.2
Pontos-chave:
• Os ácidos voláteis e não-voláteis,produzidos diariamente, são eliminadospelos pulmões e rins, respectivamente
• pH normal � 7,35-7,45. Para preservar asfunções celulares, variações de pH devemser corrigidas, através das seguintes linhasde defesa:1.ª (instantânea): Sistemas tampão2.ª (minutos): Componente respiratório3.ª (horas a dias): Componente renal (lento)
SISTEMAS TAMPÃO
A manutenção de um pH relativamente constante noorganismo se deve à integração renal-respiratória, já men-cionada, e à atuação de sistemas tampão (componentequímico), que minimizam as variações de pH conseqüen-tes a uma carga ácida ou alcalina.
Os sistemas tampão são de modo geral formados por áci-dos fracos (e o sal correspondente ou base), que não se disso-ciam completamente e, portanto, têm a capacidade de rece-ber ou doar H� quando a concentração de H� se altera. Porexemplo, quando um ácido forte é introduzido no sangue, elese dissocia completamente e aumenta a concentração de H�. Aoentrar em contato com o sistema tampão, o hidrogêniodissociado do ácido forte liga-se ao sal do sistema tampão,reduzindo a atividade de H�. Assim, o ácido forte é substitu-ído por um ácido fraco, de dissociação menos intensa.1,11
Ácido forte � base fraca ↔ sal neutro � ácido fracoExemplo: HCl � Na2HPO4 ↔ NaCl � NaH2PO4
Ao acrescentar uma base forte a um sistema tampão, elaé substituída por seu sal de base e um ácido fraco.1,11
Base forte � ácido fraco ↔ base fraca � águaExemplo: NaOH � NaH2PO4 ↔ Na2HPO4 � H2O
A capacidade do sistema tampão em resistir às altera-ções do pH é dependente da concentração e do pK do sis-tema tampão (Fig. 11.1). Quanto mais próximo do pK dosangue estiver o pK do tampão, maior será a sua capaci-dade de tamponamento.
Quando se adiciona ácido (H�) ao organismo, parte deleé tamponada quimicamente no líquido extracelular, e parte
difunde-se para dentro das células (Fig. 11.2). Aproxima-damente 60% são tamponados nas células e nos ossos, numprocesso que envolve troca de H� por Na� ou K�. Os 40%restantes são tamponados no líquido extracelular pelostampões existentes. Quando se adiciona uma substânciaalcalina, aproximadamente 70% são tamponados em líqui-do extracelular e o restante nas células.12 O movimento deH�, OH� ou HCO3
� através da membrana celular é impor-tante para o tamponamento de variações de pH que ocor-rem no extracelular ou intracelular.10
No organismo, os seguintes sistemas tampão são impor-tantes: bicarbonato, proteínas plasmáticas (extracelulares)e hemoglobina, fosfato, complexos organofosfatados, amô-nio, proteínas intracelulares e cristais de apatita do osso.De acordo com o princípio iso-hídrico, todos os tampões emuma solução estão em equilíbrio com a mesma concentra-ção de hidrogênio. Estes vários sistemas tampão não agemisoladamente; eles atuam ao mesmo tempo, cada qual comseu pK e concentração. Quando ocorre uma variação naconcentração de hidrogênio, ocorrem modificações emtodos os sistemas tampão. Qualquer condição que modifi-que o equilíbrio de um sistema tampão altera o equilíbriode todos os outros.1,8
Sistema Tampão Ácido Carbônico-Bicarbonato
É o principal sistema tampão do organismo. Observe queas reações químicas deste sistema tampão obedecem à quan-tidade existente de substrato e acontecem ao mesmo tempono sangue e nos túbulos renais. Quando íons H� são adici-
Fig. 11.1 Alteração no pH de uma solução tampão, à medida queum ácido é adicionado à solução. Observem que, quando o tam-pão estiver 50% livre e 50% combinado com H� (pK do tampão),haverá pouca alteração do pH. Portanto, o tampão será mais efi-ciente em soluções com um pH nesta faixa. (Obtido de Makoff,D.L.49)

166 Metabolismo Ácido-Básico
onados ao organismo, combinam-se com o HCO3� do plas-
ma, formando H2CO3, que se dissocia em água e CO2, o qualpode ser removido pelos pulmões. Neste sistema, o pH dolíquido extracelular é controlado pela eliminação ou recupe-ração de HCO3
� pelos rins e remoção de CO2 pelos pulmões.
H� � HCO3� ↔ H2CO3 ↔ CO2 � H2O
Devido à sua importância no equilíbrio ácido-básico, osistema tampão ácido carbônico-bicarbonato será aborda-do em mais detalhe ao longo deste capítulo.
Proteínas PlasmáticasAs proteínas e aminoácidos do sangue e intracelulares
são tampões importantes, pois possuem grupos químicoscapazes de receber ou liberar H�, comportando-se comoácidos ou bases. As proteínas possuem numerosos grupos
carboxila (�COOH), que podem perder um próton e for-mar �COO�. Também apresentam grupos amino (�NH2),que podem receber um próton e formar NH3.
10 A açãotamponante de uma proteína pode ser vista na Fig. 11.3.
A carga elétrica das proteínas varia com o pH do extra-celular. Para uma determinada proteína, a carga é deter-minada pelo equilíbrio entre seus grupos de carga negati-va e positiva. Uma proteína pode ser caracterizada pelo seuponto isoelétrico, isto é, o pH em que não apresenta car-gas negativas. Para as proteínas plasmáticas, o ponto isoe-létrico está em torno de 5,1-5,7, ou seja, bem abaixo do pHnormal de nosso organismo. Por isso, de modo geral asproteínas plasmáticas se comportam como poliânions.10
A albumina realiza uma parte significativa da açãotamponante do plasma que não é executada pelo bicarbo-nato, pois há vários grupos imidazol em sua molécula. Suacapacidade tamponante é superior à da globulina.10
Fig. 11.2 Mecanismos de defesa frente a um excesso de ácido. Quando ocorre alcalose, as reações se processam em sentido inverso.(Obtido de Makoff, D.L.49)
Fig. 11.3 Representação esquemática da ação tamponante de uma proteína.

capítulo 11 167
As proteínas localizadas no espaço intracelular tambémcontribuem para o tamponamento do H�. Por exemplo, asproteínas intracelulares do músculo esquelético colaboramcom 60% do tamponamento não realizado por bicarbona-to, sendo os 40% restantes realizados por fosfatos orgâni-cos e inorgânicos.10
HemoglobinaA hemoglobina é responsável pela maior parte do tam-
ponamento plasmático não realizado pelo bicarbonato,devido à sua alta concentração nas hemácias e sua grandecapacidade de tamponamento, por possuir vários gruposácidos ou básicos em sua molécula: carboxila (�COOH),amino (�NH2), amônia (�NH3).
O CO2 proveniente do metabolismo tissular difunde-separa dentro das hemácias. A hemoglobina reduzida, pre-sente ao nível tecidual, tem máxima afinidade por radicaisácidos, favorecendo a captação e o transporte de CO2. Den-tro das hemácias, apenas uma pequena parte do CO2 per-manece dissolvida. A maior parte do CO2 que adentra acélula sofre hidratação, por ação da anidrase carbônica(presente em grandes quantidades nas hemácias), forman-do H2CO3, que se dissocia em H� e HCO3
�. O hidrogênioassim liberado é tamponado por grupos amino da hemo-globina, a qual se transforma em H-Hb. 10
CO2 � H2O ↔ H2CO3 ↔ H� � HCO3�
�anidrase carbônica (AC)
Com o aumento da concentração intra-eritrocitária debicarbonato, este se difunde para o plasma devido ao gra-diente de concentração. Portanto, é nas hemácias que seforma parte do bicarbonato plasmático. Com a saída deHCO3
�, o Cl� adentra a célula, a fim de manter a eletroneu-tralidade.10
No sangue que transita pelos pulmões, a reação quími-ca anterior sofre uma inversão, e o CO2 é eliminado.10
Tamponamento nos OssosOs ossos contêm cerca de 60% do CO2 do organismo,
sendo a maior parte sob a forma de carbonato, formandocomplexos com cálcio, sódio e outros cátions. O restanteexiste sob a forma de bicarbonato, associado à hidroxiapa-tita. Existem evidências demonstrando que na acidose crô-nica (como na insuficiência renal crônica) a necessidade detamponamento leva à dissolução óssea, com liberação detampões fosfato e carbonato, num mecanismo possivel-mente mediado pelo paratormônio.10
CONTROLE RESPIRATÓRIODA PCO2
A segunda linha de proteção contra distúrbios ácido-básicos é o controle da concentração de CO2 pelos pulmões.A equação de Henderson-Hasselbalch demonstra que avariação da PCO2 através da respiração é uma importantemaneira de normalizar o pH. Assim, quando há aumentoda concentração de H�, este se combina com o bicarbona-to, formando ácido carbônico (H2CO3), que se dissocia emH2O e CO2. O CO2 continuamente produzido pelo meta-bolismo e resultante das reações dos sistemas tampão érapidamente eliminado pelos pulmões.
H� � HCO3� ↔ H2CO3 ↔ H2O � CO2 � respiração
�metabolismo
Além disso, a ventilação alveolar é estimulada ou inibi-da por variações na [H�]. Quando a concentração hidro-geniônica está elevada, o centro respiratório é estimulado,aumentando a amplitude dos movimentos respiratórios(hiperventilação alveolar), eliminando mais CO2. Uma ini-bição do centro respiratório (hipoventilação alveolar) ocor-re se a concentração de hidrogênio está baixa, por ummecanismo de feedback.1
CONTROLE RENAL DOEQUILÍBRIO ÁCIDO-BÁSICO
Apesar da eficiência dos sistemas tampão e do controlerespiratório, estes mecanismos proporcionam proteçãotemporária, minimizando alterações do pH quando ácidosfortes ou bases são adicionados ao organismo, ou quandoa concentração de CO2 se altera.
Um mecanismo mais duradouro é realizado pelos rins,através da reabsorção de quase todo o bicarbonato filtra-do e recuperação do HCO3
� que foi consumido no proces-
Pontos-chave:
• Tampões são substâncias capazes de doarou receber íons hidrogênio, atenuandovariações de pH
• Os principais tampões existentes em nossoorganismo são:BicarbonatoProteínas plasmáticas e intracelularesHemoglobinaOssos
• Cerca de 95% dos ácidos voláteis sãotamponados no intracelular. Dos ácidosfixos, 50% são tamponados no intracelular e50% no extracelular
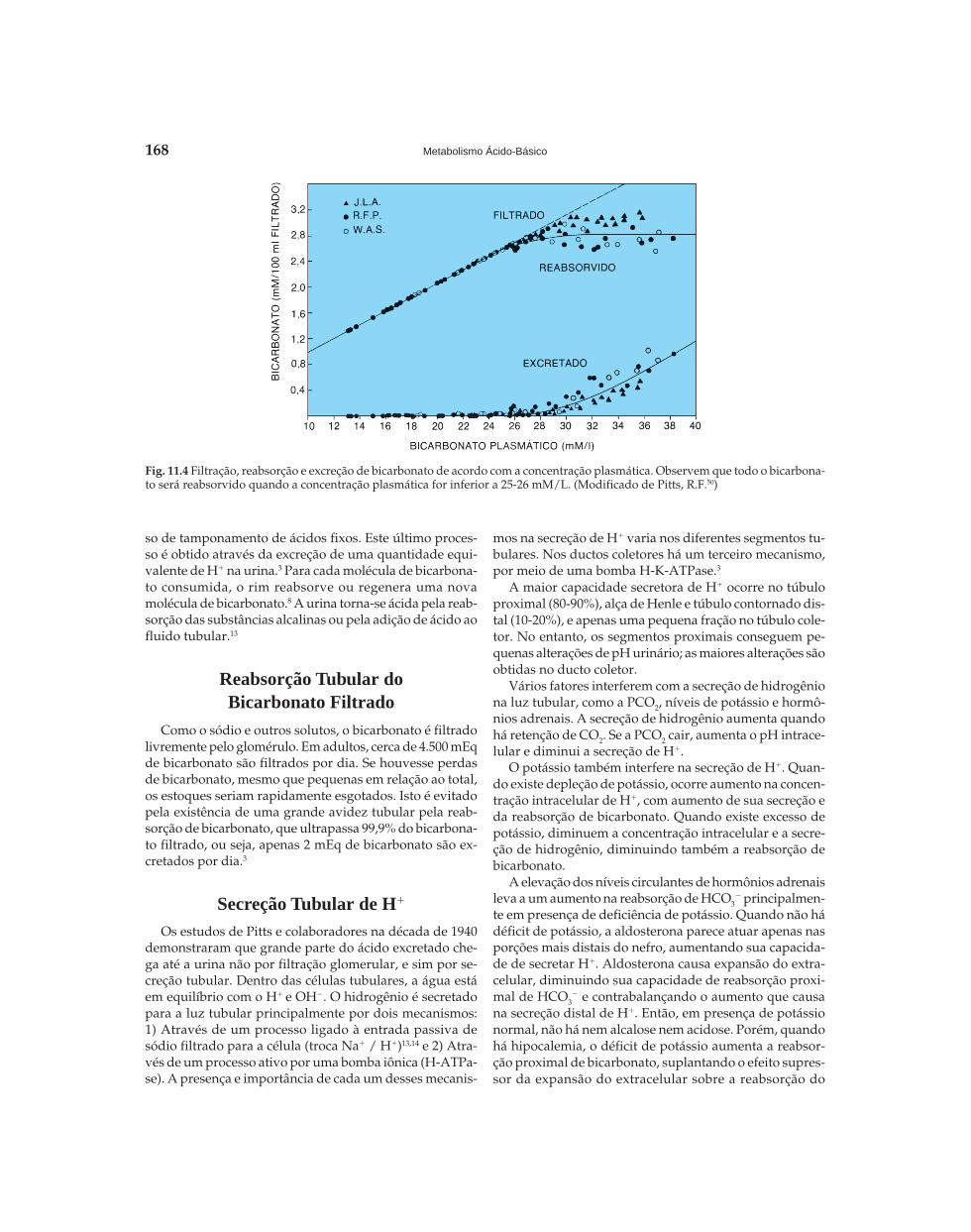
168 Metabolismo Ácido-Básico
so de tamponamento de ácidos fixos. Este último proces-so é obtido através da excreção de uma quantidade equi-valente de H� na urina.3 Para cada molécula de bicarbona-to consumida, o rim reabsorve ou regenera uma novamolécula de bicarbonato.8 A urina torna-se ácida pela reab-sorção das substâncias alcalinas ou pela adição de ácido aofluido tubular.13
Reabsorção Tubular doBicarbonato Filtrado
Como o sódio e outros solutos, o bicarbonato é filtradolivremente pelo glomérulo. Em adultos, cerca de 4.500 mEqde bicarbonato são filtrados por dia. Se houvesse perdasde bicarbonato, mesmo que pequenas em relação ao total,os estoques seriam rapidamente esgotados. Isto é evitadopela existência de uma grande avidez tubular pela reab-sorção de bicarbonato, que ultrapassa 99,9% do bicarbona-to filtrado, ou seja, apenas 2 mEq de bicarbonato são ex-cretados por dia.3
Secreção Tubular de H�
Os estudos de Pitts e colaboradores na década de 1940demonstraram que grande parte do ácido excretado che-ga até a urina não por filtração glomerular, e sim por se-creção tubular. Dentro das células tubulares, a água estáem equilíbrio com o H� e OH�. O hidrogênio é secretadopara a luz tubular principalmente por dois mecanismos:1) Através de um processo ligado à entrada passiva desódio filtrado para a célula (troca Na� / H�)13,14 e 2) Atra-vés de um processo ativo por uma bomba iônica (H-ATPa-se). A presença e importância de cada um desses mecanis-
mos na secreção de H� varia nos diferentes segmentos tu-bulares. Nos ductos coletores há um terceiro mecanismo,por meio de uma bomba H-K-ATPase.3
A maior capacidade secretora de H� ocorre no túbuloproximal (80-90%), alça de Henle e túbulo contornado dis-tal (10-20%), e apenas uma pequena fração no túbulo cole-tor. No entanto, os segmentos proximais conseguem pe-quenas alterações de pH urinário; as maiores alterações sãoobtidas no ducto coletor.
Vários fatores interferem com a secreção de hidrogêniona luz tubular, como a PCO2, níveis de potássio e hormô-nios adrenais. A secreção de hidrogênio aumenta quandohá retenção de CO2. Se a PCO2 cair, aumenta o pH intrace-lular e diminui a secreção de H�.
O potássio também interfere na secreção de H�. Quan-do existe depleção de potássio, ocorre aumento na concen-tração intracelular de H�, com aumento de sua secreção eda reabsorção de bicarbonato. Quando existe excesso depotássio, diminuem a concentração intracelular e a secre-ção de hidrogênio, diminuindo também a reabsorção debicarbonato.
A elevação dos níveis circulantes de hormônios adrenaisleva a um aumento na reabsorção de HCO3
� principalmen-te em presença de deficiência de potássio. Quando não hádéficit de potássio, a aldosterona parece atuar apenas nasporções mais distais do nefro, aumentando sua capacida-de de secretar H�. Aldosterona causa expansão do extra-celular, diminuindo sua capacidade de reabsorção proxi-mal de HCO3
� e contrabalançando o aumento que causana secreção distal de H�. Então, em presença de potássionormal, não há nem alcalose nem acidose. Porém, quandohá hipocalemia, o déficit de potássio aumenta a reabsor-ção proximal de bicarbonato, suplantando o efeito supres-sor da expansão do extracelular sobre a reabsorção do
Fig. 11.4 Filtração, reabsorção e excreção de bicarbonato de acordo com a concentração plasmática. Observem que todo o bicarbona-to será reabsorvido quando a concentração plasmática for inferior a 25-26 mM/L. (Modificado de Pitts, R.F.50)

capítulo 11 169
mesmo, e ainda secretando mais hidrogênio. Como resul-tado, estabelece-se uma alcalose metabólica.
Outro fator que interfere com a secreção do H� é a pre-sença de ânions não-reabsorvíveis em alta concentração notúbulo distal, como carbenicilina e penicilina. Isto aumen-ta o fluxo e a eletronegatividade intraluminal, favorecen-do a secreção de hidrogênio e potássio, resultando em al-calose metabólica.15,16
Uma vez na luz tubular, o hidrogênio secretado se com-bina com HCO3
� filtrado, formando H2CO3, que é conver-tido em CO2 e H2O. No túbulo proximal e ramo ascenden-te espesso da alça de Henle (mas não em segmentos maisdistais), esta reação ocorre em milissegundos, sob influên-cia da anidrase carbônica, que é uma enzima presente namembrana luminal das células e que não existe no fluidotubular. A anidrase carbônica é encontrada na porção con-tornada do túbulo proximal, porção ascendente espessa daalça de Henle e túbulo contornado distal. A inibição destaenzima (p.ex., pela acetazolamida) bloqueia a reabsorçãode bicarbonato e acidificação urinária.
O CO2 assim formado dentro do lúmen se difunde paradentro da célula, onde se combina com o OH� que resultada dissociação da água, e novamente, sob ação da anidra-se carbônica, forma-se HCO3
�. O HCO3� então se difunde
passivamente para o fluido peritubular e sangue. Em mui-tos segmentos do nefro o HCO3
� atravessa a membranabasolateral por difusão facilitada, acompanhando o Na�
(por um co-transportador), ou em troca por Cl�. Apesar deque algum Na� que acompanha o HCO3
� então atravessea célula passivamente, a maior parte é transportada ativa-mente para o fluido peritubular e sangue, pela bomba Na-K-ATPase. Assim, para cada H� secretado um HCO3
� re-torna ao fluido peritubular e sangue, e praticamente todoo bicarbonato filtrado é recuperado. Note que este não éum mecanismo puro de secreção de hidrogênio, pois o CO2
formado dentro dos túbulos pelo H� secretado retorna àcélula, formando mais H� por hidroxilação. Até aqui, nãohouve secreção verdadeira de hidrogênio.3
Como se observa na Fig. 11.5, a maior parte da reabsor-ção de bicarbonato (70-85%) ocorre nos segmentos iniciaisdo túbulo proximal e proporções variáveis na alça de Hen-le, túbulo distal e ducto coletor. 3
Fatores que Influenciam na Reabsorçãodo Bicarbonato Filtrado
A proporção de bicarbonato que retorna ao sangue éafetada por fatores que interagem entre si, como: a) quan-tidade de bicarbonato apresentada aos túbulos; b) estadodo espaço extracelular; e c) PCO2 arterial. É possível queestes fatores alterem a reabsorção de bicarbonato princi-palmente através de modificações na ativação ou no nú-mero de trocadores Na/K e H-ATPases. Alguns hormôni-os e substâncias vasoativas (paratormônio, hormôniosadrenais, angiotensina II, catecolaminas e dopamina) afe-tam a reabsorção de bicarbonato, através de mecanismosainda não muito compreendidos. Outros fatores, como adeficiência de potássio e cloro, exercem influência impor-tante apenas em presença de doença.3
1) A quantidade de bicarbonato filtrado e apresentadoaos túbulos varia de acordo com a concentração plas-mática de bicarbonato e a taxa de filtração glomeru-lar. Se as outras variáveis estiverem constantes (p.ex.,o volume do extracelular), a quantidade de bicarbo-nato reabsorvido é quase igual à quantidade filtrada.O mecanismo deste efeito ainda não está esclarecido,mas a taxa de reabsorção parece estar ligada à reab-sorção de sódio, principalmente no túbulo proximal.Isto pode ser em parte decorrente da necessidade deconservar sódio e manter o espaço extracelular.3,13
2) Efeito do volume do extracelular: quando o volumeestá bastante expandido, ocorre diminuição da reab-sorção de bicarbonato filtrado; o oposto ocorre quan-do o extracelular está contraído. Novamente, o me-canismo parece estar ligado a modificações na reab-sorção de sódio impostas pelas variações no volumeextracelular.3
3) Influência de modificações prolongadas na PCO2:quando ocorre diminuição da PCO2 (como, por exem-plo, por hiperventilação crônica), a reabsorção dobicarbonato diminui; quando há elevação da PCO2,aumenta a reabsorção de bicarbonato. Dois mecanis-mos parecem estar envolvidos nesta variação de re-absorção: a) mudança na quantidade de bicarbonatofiltrado e apresentado aos túbulos (isto só ocorre emdistúrbios crônicos, pois, nos agudos, a concentraçãoplasmática de bicarbonato muda muito pouco); e b)efeito direto da PCO2 sobre a atividade da H-ATPa-se e H-K-ATPase.3
Fig. 11.5 Mecanismo de reabsorção do bicarbonato filtrado. Vero texto. (Adaptado de Valtin, H.; Schafer, J.A.3)

170 Metabolismo Ácido-Básico
Como já foi mencionado, a dieta ocidental rica em pro-teínas produz vários ácidos não-voláteis (fixos), como oácido sulfúrico, fosfórico e ácidos orgânicos. Estes ácidossão tamponados nos seguintes tipos de reação:
2 H� � SO4�� � 2 Na� � 2 HCO3
�
↔ 2 Na� � SO4�� � 2 H2O � 2 CO2
2 H� � HPO4�� � 2 Na� � 2 HCO3
�
↔ 2 Na� � HPO4�� � 2 H2O � 2 CO2
Nestes exemplos, o CO2 assim produzido é eliminadopelos pulmões, e os dois sais neutros, Na2SO4 e Na2PO4, sãofiltrados pelo glomérulo. Se estes sais fossem excretadospela urina, o organismo ficaria em déficit de bicarbonatode sódio (NaHCO3), o principal tampão extracelular utili-zado na neutralização dos ácidos fixos. Os rins evitam estedéficit de bicarbonato de sódio através da excreção de NH4
�
e de acidez titulável. Em ambas as operações, o bicarbona-to recém-formado nas células tubulares renais é absorvi-do para o sangue peritubular, juntamente com o sódio quefoi filtrado.3
Excreção de Acidez Titulável (AT)
Se considerarmos uma urina com pH de 5,2, podemosadicionar a ela uma substância alcalina até que seu pH seiguale ao pH do sangue, ou seja, 7,4. A quantidade desubstância alcalina (em ml) necessária para titular a uri-na até se igualar ao pH do sangue é equivalente à quan-tidade de H� ligada aos tampões filtrados. Esta quanti-dade de ácido assim excretada é calculada e denomina-da acidez titulável.
Com a reabsorção de bicarbonato, a urina nos túbulosrenais se torna ácida. O hidrogênio secretado para a luztubular se combina com outros tampões que foram filtra-dos. Como parte deste último processo, o sal neutroNa2HPO4 é convertido no sal ácido NaH2PO4
�, principalmaneira de excreção de acidez titulável. Outros tampõesfiltrados, como ânions orgânicos, citrato, acetato e 3-hidro-xibutirato, são também titulados, mas de modo geral con-tribuem pouco para a AT, devido à sua baixa concentra-ção e baixo pK.3
O esquema de formação da AT urinária é mostrado naFig. 11.6 (note as semelhanças com a Fig. 11.5). A principalreação que gera o hidrogênio secretado parece ser a disso-ciação da água; o OH� que é simultaneamente liberado secombina com o CO2 intracelular, sob ação da anidrase car-bônica. Forma-se HCO3
�, que é adicionado ao fluido peri-tubular e sangue. No lúmen tubular, o H� secretado secombina com Na� e HPO4
��, formando NaH2PO4�, que é
excretado como ácido titulável na urina. Estas reações ocor-rem no túbulo proximal, túbulo distal e ductos coletores.O efeito aqui obtido é reabastecer o sangue com um bicar-bonato para cada bicarbonato consumido no processo detamponamento de um ácido fixo.3
Excreção de Amônio (NH4�)
Se a formação de acidez titulável fosse o único mecanis-mo para excretar H�, a quantidade de hidrogênio elimina-do na urina seria muito limitada pela quantidade de fos-fato e outros tampões que são filtrados. A observação deque na acidose existe um aumento não só da AT mas tam-bém do NH4
� na urina gerou a hipótese de que o NH4�
pudesse constituir um mecanismo adicional. Note que oamônio aparece na urina sob forma de sais neutros (p.ex.,cloreto de amônio — NH4Cl), o que serve para excretar H�
sem uma maior diminuição no pH urinário.3
O provável mecanismo para a excreção de NH4� é de-
monstrado nas Figs. 11.7 e 11.8. Este processo consta de trêsetapas: 1) produção e secreção de NH4
� nos túbulos proxi-mais; 2) mecanismo de contracorrente multiplicador deNH4
� nas alças de Henle, resultando no desenvolvimentode um gradiente corticopapilar para NH4
�/ NH3 dentro dointerstício medular; e c) difusão não-iônica de NH3 paradentro dos ductos coletores.3
PRODUÇÃO PROXIMAL E SECREÇÃO DE NH4�
Esta primeira etapa ocorre predominantemente nas cé-lulas tubulares proximais, onde a deaminação da glutami-na produz dois íons NH4
� e um íon de alfa-cetoglutarato.O metabolismo do último para glicose, ou para CO2 e água,produz dois novos íons HCO3
�. Assim como na excreçãode AT, esta reação adiciona um HCO3
� para cada H� queé excretado — neste caso, como parte do NH4
�. O sódio queacompanha o HCO3
� pode adentrar o fluido peritubularatravés da Na-K-ATPase ou via co-transportador HCO3
�.Em muitas circunstâncias, o NH4
� produzido no túbuloproximal é responsável por quase todo o NH4
� excretadona urina.3 É importante lembrar que nos quadros de aci-
Fig. 11.6 Mecanismo de formação de acidez titulável. Ver o tex-to. (Adaptado de Valtin, H.; Schafer, J.A.3)

capítulo 11 171
dose metabólica há um aumento significativo na produ-ção de NH3 a partir da glutamina, tornando-se a molécu-la de NH4
� o principal meio de excreção dos íons H� naurina. Além disso, a hipocalemia aumenta a produção deNH4
�, levando a uma maior secreção de H� para o lúmentubular.
GRADIENTE INTERSTICIAL CORTICOPAPILARPARA NH4
�/NH3
Nas alças de Henle, há um mecanismo contracorrentemultiplicador de NH4
� que produz um gradiente paraNH4
�/NH3 no interstício medular. Nos segmentos ascen-dentes espessos, o NH4
� é reabsorvido principalmente portransporte ativo secundário, substituindo o K� no co-trans-portador Na:K:2Cl que se localiza na membrana apical.
Nos segmentos ascendentes delgados a reabsorção deNH4
� pode ser passiva. A secreção de NH4� nos ramos
descendentes pode ocorrer mais por secreção paralela deH� e NH3 do que por secreção de NH4
�. O efeito final é omesmo, e a conseqüência importante é que a concentraçãointersticial de amônia total (isto é, NH4
� e NH3) se elevacom a proximidade da papila.3
SECREÇÃO DE AMÔNIA NOSDUCTOS COLETORES (NH3)
O segmento distal dos túbulos coletores e o ducto cole-tor são constituídos por pelo menos dois tipos principais decélulas, uma das quais, a célula intercalada alfa, secreta H�
mas não reabsorve Na�. Nesta célula, o H� que é derivadoda dissociação da água é secretado na luz tubular por doisco-transportadores, H�-ATPase e H�-K�-ATPase. O H� se-cretado se combina com o NH3 para formar NH4
�, que éentão excretado sob a forma de sais neutros, como o NH4Clou (NH4)2SO4. O NH3 pode difundir-se passivamente dointerstício onde é gerado pelo mecanismo de contracorren-te multiplicador, através da célula, para a luz tubular.3
O HCO3� formado pela dissociação da água cruza a mem-
brana basolateral para o fluido peritubular por difusão fa-cilitada, através de um trocador HCO3
�/Cl�. Então, comona excreção de AT e com o mecanismo do NH4
� dos túbu-los proximais, o resultado da reação nos ductos coletores éa recuperação de um HCO3
� para cada H� que é excretado,ou seja, exatamente o que é preciso após um HCO3
� ter sidoconsumido no tamponamento de um H� adicionado. O só-dio filtrado é reabsorvido pelas células principais.3
DIFUSÃO NÃO-IÔNICAA amônia (NH3) é um gás que atravessa a membrana
celular com grande facilidade, por ser lipossolúvel, e pode
Fig. 11.7 Produção de amônio (NH4�) nos túbulos proximais, a
partir da glutamina. (Adaptado de Valtin, H.; Schafer, J.A.3)
Fig. 11.8 Produção de amônio nas células intercaladas alfa dos ductos coletores. (Adaptado de Valtin, H.; Schafer, J.A.3)

172 Metabolismo Ácido-Básico
difundir-se do interstício para o lúmen tubular. Pratica-mente todo o NH3 que se difunde é transformado em NH4
�,pois o fluido tubular é ácido. Quanto mais ácida for a uri-na, maior é esta transformação. Devido à impermeabili-dade do segmento, o NH4
� formado não pode difundir-se novamente através do epitélio, e então tem que ser ex-cretado. Mais de 98% da amônia total (NH3 � NH4
�) es-tão sob a forma de NH4
�, pois o pH urinário está na faixade 4,4-7,4.3
A excreção ácida total corresponde à soma da acideztitulável e amônio urinário, menos o bicarbonato restantena urina (AT � NH4
� � HCO3� urinário).17
Ponto-chave:
• O controle renal do equilíbrio ácido-básico érealizado através dos seguintesmecanismos:Reabsorção do HCO3
� filtradoRegeneração de HCO3
� através da excreçãode H� ligado a tampões (AT) e na forma deamônio (NH4
�)
DISTÚRBIOS CLÍNICOS DOMETABOLISMO ÁCIDO-BÁSICO
O estado ácido-básico é avaliado através da gasometria,e não há diferenças significativas entre uma amostra arte-rial ou venosa com relação ao pH, bicarbonato e PCO2. Nosangue arterial, porém, é possível avaliar também as vari-áveis de oxigenação, como a PO2 e a saturação arterial deoxigênio, que permitem considerações sobre a ventilaçãodo paciente. Tomar o cuidado de não utilizar garrote eheparinizar a seringa adequadamente. Após a coleta dosangue, homogeneizar o conteúdo, eliminar as bolhas dear e vedar a seringa, encaminhando a amostra imediata-mente para laboratório ou mantendo-a refrigerada até omomento da análise. A demora em processar a amostrapromove o consumo de oxigênio e a produção de CO2,modificando os resultados.18,19
Como mencionamos há pouco, a observação da equaçãode Henderson-Hasselbalch indica que quatro distúrbiosprimários do metabolismo ácido-básico podem ocorrer: aci-dose metabólica, acidose respiratória, alcalose metabólica ealcalose respiratória. Em princípio, pode parecer que o diag-nóstico de anormalidade metabólica ou respiratória pode serfeito apenas conhecendo-se o bicarbonato plasmático e aPCO2, respectivamente. Em realidade, isto não é possível,pois cada distúrbio ácido-básico primário produz uma rea-ção compensatória secundária. Além das reações compen-satórias normais, podem surgir distúrbios ácido-básicosmistos, como veremos nas próximas seções.
Acidose MetabólicaA acidose metabólica é um distúrbio em que há eleva-
ção na concentração de hidrogênio, gerando pH baixo nofluido extracelular. O bicarbonato encontra-se diminuído,por estar sendo consumido no tamponamento do excessode ácido (H�). O hidrogênio em excesso estimula o centrorespiratório, provocando hiperventilação como mecanis-mo compensatório, eliminando mais CO2.
19
CAUSASA acidose metabólica pode ser resultado de um aumento
na produção ou diminuição na excreção renal de ácido, ouainda, perda de bicarbonato (v. Quadro 11.1).
Produção Aumentada de ÁcidoQuando existe aumento na produção de ácidos, pode ocor-
rer acidose grave, causando significativa diminuição no bi-carbonato plasmático. São exemplos disso a acidose láctica, acetoacidose diabética ou alcoólica e a intoxicação por algumasdrogas (como, por exemplo, o ácido acetilsalicílico).19
ACIDOSE LÁCTICA. O ácido láctico é normalmenteproduzido em nosso organismo, sendo quase todo conver-tido em glicose ou piruvato, no fígado e nos rins. O lactatoacumula-se quando sua produção está aumentada ou suautilização diminuída.19
Quadro 11.1 Causas de acidose metabólica
Produção ácida aumentadaa) Acidose láctica
• Hipoperfusão tecidual• Metformin• Etilismo• Doenças malignas• Infecção por HIV• Acidose D-láctica
b) Cetoacidose• Diabetes melito• Etilismo
c) Toxinas ingeridas• Aspirina• Etilenoglicol• Metanol
Perda de bicarbonato pela urina ou fezesa) Diarréiab) Fístulas pancreáticas, biliaresc) Acidose tubular renal proximal (tipo 2)
Redução na excreção renal de ácidoa) Insuficiência renalb) Acidose tubular renal tipo 1c) Acidose tubular renal tipo 4
(hipoaldosteronismo)
Outras• Dilucional
Adaptado de Rose, B.D.19

capítulo 11 173
A produção deste ácido aumenta em situações em quea oferta de oxigênio para os tecidos é inferior às necessi-dades, como, por exemplo, na hipoperfusão presente nochoque hipovolêmico, cardiogênico ou séptico. Nestas cir-cunstâncias, além de o piruvato ser preferencialmente con-vertido a lactato, sua utilização está diminuída, devido àsalterações na perfusão do fígado e rins.19 Menos freqüen-temente, a produção de ácido láctico pode aumentar ou seumetabolismo diminuir, por doenças hepáticas ou deficiên-cias enzimáticas hereditárias.20
O uso de metformin no diabetes melito pode produziracidose láctica, principalmente em presença de disfunçãorenal, hepática, ou etilismo. Eventualmente, pacientes eti-listas apresentam acidose láctica, causada por hipoperfu-são ou diminuição da utilização hepática de lactato.21
Nas doenças malignas, o metabolismo anaeróbio queocorre dentro de massas celulares mal vascularizadas podeocasionar acidose láctica. Em pacientes com SIDA, a aci-dose láctica está relacionada à doença hepática ou miopa-tia induzidas pela zidovudina, ou à presença de deficiên-cia de riboflavina.21
A acidose D-láctica ocorre em pacientes submetidos abypass jejuno-ileal, ressecção de intestino delgado ou ou-tras causas de síndrome do intestino curto. Nestas situa-ções, na presença de crescimento exagerado de bactériasanaeróbicas, o cólon converte glicose e amido em ácido D-láctico, que é absorvido pela circulação. A desidrogenaseL-láctica, que metaboliza o L-lactato fisiológico em piru-vato, não atua sobre o ácido D-láctico. Os pacientes apre-sentam anormalidades neurológicas após sobrecarga decarboidratos.22
CETOACIDOSE. A cetoacidose diabética é uma desor-dem em que a deficiência de insulina e o excesso de gluca-gon produzem aumento da síntese hepática de cetoácidos,principalmente ácido beta-hidroxibutírico e ácido acetoa-cético.19
O jejum prolongado também pode produzir cetoacido-se, mas de modo geral os ácidos gerados não consomemmais do que 3-4 mEq de bicarbonato/litro. Em etilistas, aassociação de um aporte deficiente de carboidratos com osefeitos do álcool inibindo a gliconeogênese e estimulandoa lipólise também pode produzir cetoacidose. A presençade diabetes agrava esta condição.23
INGESTÃO DE TOXINAS. Em nosso organismo, oácido acetilsalicílico é convertido em ácido salicílico. Aintoxicação por altas doses deste ácido produz acidosemetabólica devido à interferência com o metabolismo oxi-dativo, levando ao acúmulo de ácidos orgânicos, como olactato e cetoácidos. Em doses menores, o ácido acetilsali-cílico pode induzir alcalose respiratória, por estimulaçãodireta do centro respiratório.19,24
A intoxicação pelo metanol produz um quadro caracte-rístico de sintomatologia do sistema nervoso central, ocu-lar e abdominal. Agudamente os pacientes apresentam sin-tomas de embriaguez, confusão mental, dor abdominal e
vômitos, podendo evoluir com pancreatite. As alteraçõesoculares, como hiperemia conjuntival, diplopia e amauro-se, acompanham-se de alteração da fundoscopia, que de-monstra neurite óptica. O metabolismo do metanol produzácido fórmico, responsável pela acidose.24,25
O etilenoglicol está presente em produtos anticongelantese fluido de radiador, e é também utilizado em algumas eta-pas na indústria de bebidas. O etilenoglicol ingerido é meta-bolizado em compostos tóxicos, como o ácido oxálico, pelaação da desidrogenase alcoólica. Estes compostos tóxicosprovocam disfunção neurológica aguda, com ataxia, confu-são, convulsões e coma. Nos rins, determinam a deposiçãode cristais de oxalato de cálcio e insuficiência renal aguda.25
Perda de BicarbonatoPara cada molécula de base que é perdida, um próton deixa
de ser tamponado, resultando em acúmulo de ácido fixo.20 Aperda de secreções alcalinas do pâncreas e árvore biliar e asdiarréias induzidas ou não por laxantes podem causar aci-dose metabólica.19 Na acidose tubular renal proximal ocor-re perda de grandes quantidades de bicarbonato.
Redução na Excreção Renal de ÁcidoPara que o equilíbrio ácido-básico seja mantido na in-
suficiência renal, é necessário que ocorram adaptações nosnefros restantes. Inicialmente, há aumento da excreção deamônio (NH4
�) por nefro. Porém, quando a taxa de filtra-ção glomerular cai para menos de 30-40% do normal, co-meça a haver retenção da carga ácida diária; acidose ocor-re quando a massa renal remanescente estiver em torno de20%. A diminuição da excreção ácida na falência renal écausada principalmente pela pequena quantidade denefros funcionantes. Aumento de PTH, expansão volêmi-ca e diurese de solutos, observados na insuficiência renal,inibem a reabsorção de bicarbonato. Também ocorre dimi-nuição da produção de amônia (NH3). Como o bicarbona-to está sendo consumido, outros tampões acabam sendoacumulados (sulfato e fosfato).24 Os tampões plasmáticossão utilizados para neutralizar parte do ácido retido, masa principal forma de tamponamento nesta situação é feitadentro das células e nos ossos.19
As acidoses tubulares do tipo 1 (distal) e 4 (hipoaldos-teronismo) são raras. Na ATR tipo 1, o acúmulo de ácidoresulta de uma incapacidade de diminuir o pH urináriopara menos que 5,5-6. O pH urinário alcalino que resultaimpede os mecanismos de produção de acidez titulável eaprisionamento da amônia no lúmen tubular sob forma deamônio.19 Na acidose distal tipo 4, a deficiência de aldos-terona impede a secreção distal de hidrogênio e potássio,resultando em acidose metabólica e hipercalemia.18
OutrasCabe aqui um comentário sobre a acidose dilucional.
Esta acidose, de modo geral discreta, resulta da diluição

174 Metabolismo Ácido-Básico
do bicarbonato plasmático pela infusão rápida de grandesquantidades de fluido que não contém bicarbonato ou seusprecursores (p.ex., o lactato). Habitualmente a queda nobicarbonato não ultrapassa 10% e é rapidamente corrigi-da pelos rins.18,25
MANIFESTAÇÕES CLÍNICAS EEFEITOS SISTÊMICOS
As manifestações clínicas da acidose metabólica depen-dem da doença primária que está produzindo a acidose eda velocidade de instalação do distúrbio. Porém, em cir-cunstâncias graves, pode haver sintomas decorrentes daprópria acidose metabólica.
Como já foi mencionado, a acidose metabólica produzuma hiperventilação, com movimentos respiratórios pro-fundos (respiração de Kussmaul), observada ao exame fí-sico, principalmente quando o pH é menor que 7,20.
Observam-se vômitos, dores pelo corpo e fadiga. Com oaumento da gravidade da acidose, geralmente com bicar-bonato inferior a 10 mEq/litro, observa-se diminuição dacontratilidade miocárdica, dilatação arteriolar, venoconstri-ção periférica e arteriolar pulmonar. Conseqüentemente hádiminuição do débito cardíaco, hipotensão arterial, diminui-ção do fluxo sanguíneo para os rins e fígado, maior sensibi-lidade a arritmias cardíacas e diminuição da responsivida-de cardiovascular às catecolaminas. A associação destasmanifestações gera um ambiente propício para o desenvol-vimento de insuficiência cardíaca congestiva.
Há também manifestações neurológicas, com progres-siva diminuição do nível de consciência e até coma. Obser-va-se também maior degradação protéica e redução dadensidade óssea, principalmente nas acidoses crônicas.26
ACHADOS LABORATORIAISA acidose metabólica caracteristicamente causa uma
diminuição do pH, diminuição do bicarbonato e diminui-ção da PCO2. A compensação respiratória se inicia na primeirahora e se completa em até 24 horas. Esta compensação cau-sa a queda de 1,2 mmHg na PCO2 para cada redução de 1mEq/litro na concentração de bicarbonato.
� [HCO3�] � 1,2 � � [CO2]
(podem ser aceitas diferenças de 2 mEq/litro)27
Por exemplo, para um bicarbonato de 18 (redução de 6em relação ao normal), a hiperventilação deverá trazer aPCO2 para cerca de 32,8 (PCO2 normal de 40 � 7,2). Se aPCO2 estiver maior ou menor que este valor, o paciente temum distúrbio misto: além da acidose metabólica, acidoseou alcalose respiratória, respectivamente.25,27
Pode haver hipercalemia, causada pelo desvio iônico con-seqüente à necessidade de tamponamento do excesso dehidrogênio dentro das células.26 Um íon hidrogênio entrana célula, mas ao mesmo tempo, para manter a eletroneu-tralidade, deve sair da célula um outro íon de carga posi-tiva — o potássio, principal cátion do intracelular. Talvez
esta saída do potássio da célula se deva a uma inibição dabomba Na-K-ATPase celular pela acidose. Ao se corrigir aacidose, o potássio retorna para dentro das células, pois nãoexiste mais necessidade de tamponamento intracelular.
Além dos dados da história clínica, uma medida queauxilia no diagnóstico causal da acidose metabólica é ocálculo do anion gap (hiato-iônico).28 A necessidade demanter a eletroneutralidade faz com que o número de cá-tions no plasma seja igual ao número de ânions. Os cáti-ons são representados principalmente pelo sódio (o potás-sio não é habitualmente incluído no cálculo, pois sua in-terferência é pequena), e os ânions, pelo cloro e bicarbona-to. Porém, há outros ânions, que não são dosados habitu-almente, mas que contribuem para a fração aniônica doplasma: proteínas, lactato fosfato e sulfato. Esta fração deânions é identificada ao se verificar que a soma dos ânionsmedidos não é igual à dosagem do sódio.24
Anion gap � Na� � (Cl� � HCO3�)
Utilizando as concentrações normais dos eletrólitos nafórmula acima (Na � 140, HCO3
� � 24, e Cl� � 105), veri-ficamos que entre cátions e ânions existe uma diferença de8-16 mEq/litro, e que corresponde aos ânions que não fo-ram medidos (ânions “não mensuráveis”), mas que estãopresentes no plasma e contribuem para contrabalançar ascargas catiônicas.20,24 Possivelmente os ânions que consti-tuem o hiato iônico sejam os tampões aniônicos do espaçoextracelular. 20
Observe a fórmula do hiato iônico. Se a concentração decloro se mantém constante na acidose metabólica, mesmohavendo queda no bicarbonato (usado no tamponamentodo hidrogênio dissociado), a manutenção da eletroneutra-lidade se faz à custa do aumento de algum ânion que nãoo cloreto.20 Os fosfatos e as proteínas não sofrem variaçõesrápidas, de forma que existe pequena possibilidade de quesejam os responsáveis pelo aumento. Então, a eletroneu-tralidade deve estar sendo mantida pelo aumento de algumânion que em condições normais não está presente no plas-ma. Exemplos disso são: a) lactato, que se acumula na aci-dose láctica; b) beta-hidroxibutirato na cetoacidose; c) au-mento dos ânions sulfato, fosfato e ácidos orgânicos, nainsuficiência renal crônica; d) ácido fórmico na intoxicaçãopelo metanol; oxalato e glicolato na intoxicação por etile-noglicol, e lactato e cetonas na intoxicação pelo ácido ace-tilsalicílico.27 Esse tipo de acidose metabólica, em que ocloro permanece normal, é chamada de acidose normoclorê-mica, ou com anion gap (hiato iônico) aumentado.20,27
Ao contrário, nas acidoses causadas por perda de bicar-bonato, como as diarréias, não há retenção de ânions anô-malos, e o hiato iônico praticamente não se altera, já que àmedida que diminui o bicarbonato, pela perda intestinal,aumenta a reabsorção de cloro, para manter a eletroneu-tralidade. Este tipo de acidose, em que há perda de bicar-bonato, com aumento do cloro, é chamada de acidose hiper-clorêmica, ou com anion gap normal (v. Fig. 11.10).20
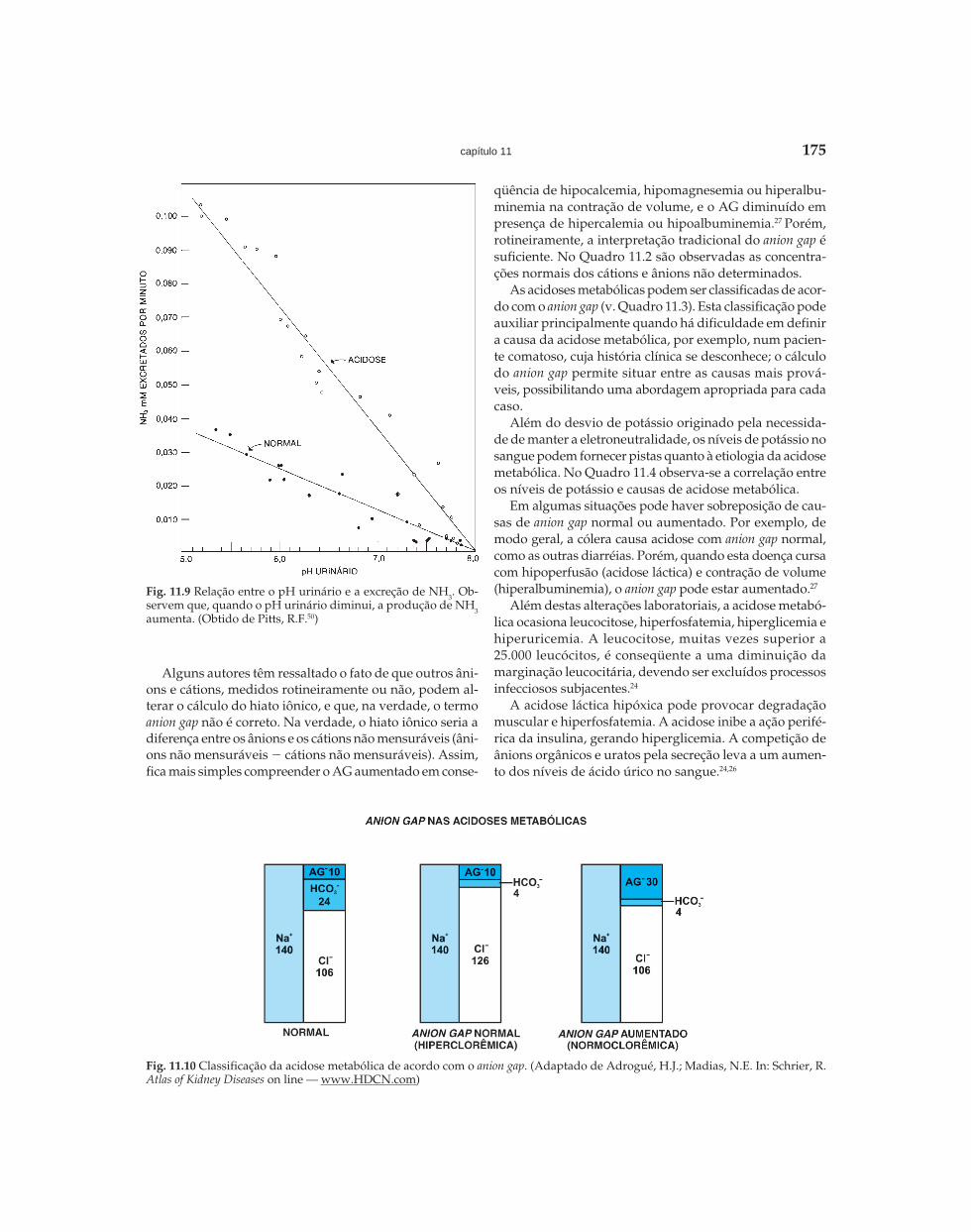
capítulo 11 175
Alguns autores têm ressaltado o fato de que outros âni-ons e cátions, medidos rotineiramente ou não, podem al-terar o cálculo do hiato iônico, e que, na verdade, o termoanion gap não é correto. Na verdade, o hiato iônico seria adiferença entre os ânions e os cátions não mensuráveis (âni-ons não mensuráveis � cátions não mensuráveis). Assim,fica mais simples compreender o AG aumentado em conse-
qüência de hipocalcemia, hipomagnesemia ou hiperalbu-minemia na contração de volume, e o AG diminuído empresença de hipercalemia ou hipoalbuminemia.27 Porém,rotineiramente, a interpretação tradicional do anion gap ésuficiente. No Quadro 11.2 são observadas as concentra-ções normais dos cátions e ânions não determinados.
As acidoses metabólicas podem ser classificadas de acor-do com o anion gap (v. Quadro 11.3). Esta classificação podeauxiliar principalmente quando há dificuldade em definira causa da acidose metabólica, por exemplo, num pacien-te comatoso, cuja história clínica se desconhece; o cálculodo anion gap permite situar entre as causas mais prová-veis, possibilitando uma abordagem apropriada para cadacaso.
Além do desvio de potássio originado pela necessida-de de manter a eletroneutralidade, os níveis de potássio nosangue podem fornecer pistas quanto à etiologia da acidosemetabólica. No Quadro 11.4 observa-se a correlação entreos níveis de potássio e causas de acidose metabólica.
Em algumas situações pode haver sobreposição de cau-sas de anion gap normal ou aumentado. Por exemplo, demodo geral, a cólera causa acidose com anion gap normal,como as outras diarréias. Porém, quando esta doença cursacom hipoperfusão (acidose láctica) e contração de volume(hiperalbuminemia), o anion gap pode estar aumentado.27
Além destas alterações laboratoriais, a acidose metabó-lica ocasiona leucocitose, hiperfosfatemia, hiperglicemia ehiperuricemia. A leucocitose, muitas vezes superior a25.000 leucócitos, é conseqüente a uma diminuição damarginação leucocitária, devendo ser excluídos processosinfecciosos subjacentes.24
A acidose láctica hipóxica pode provocar degradaçãomuscular e hiperfosfatemia. A acidose inibe a ação perifé-rica da insulina, gerando hiperglicemia. A competição deânions orgânicos e uratos pela secreção leva a um aumen-to dos níveis de ácido úrico no sangue.24,26
Fig. 11.9 Relação entre o pH urinário e a excreção de NH3. Ob-
servem que, quando o pH urinário diminui, a produção de NH3
aumenta. (Obtido de Pitts, R.F.50)
Fig. 11.10 Classificação da acidose metabólica de acordo com o anion gap. (Adaptado de Adrogué, H.J.; Madias, N.E. In: Schrier, R.Atlas of Kidney Diseases on line — www.HDCN.com)
176 Metabolismo Ácido-Básico
retidos resulta em rápida regeneração do bicarbonato, comresolução parcial ou completa da acidemia. O álcali podeaté mesmo retardar a recuperação, por aumentar a cetogê-nese hepática. Em pacientes com cetoacidose diabética e pHinferior a 7,10, pequenas doses de bicarbonato podem seradministradas com o objetivo de minimizar a depressãomiocárdica e hipoperfusão tecidual.29
A cetoacidose alcoólica é corrigida com a apropriadareposição de nutrientes e interrupção da ingestão de eta-nol. A infusão de glicose estimula a secreção de insulinamas inibe a secreção de glucagon, promovendo a regene-ração dos estoques de bicarbonato a partir do metabolis-mo dos cetoácidos retidos.29
Nos casos de acidose láctica causada por oxigenaçãotecidual inadequada, o ponto essencial no tratamento é acorreção da mesma, com repleção do volume circulanteefetivo, suporte ventilatório, agentes inotrópicos e trata-mento da septicemia. Na acidose láctica resultante de in-toxicação por metanol ou etilenoglicol, está indicada a di-álise para remoção das toxinas, além da administração degrandes quantidades de álcali. Etanol é o antagonista dometanol. 29
Tratamento da Acidose MetabólicaPara pacientes com acidemia leve ou moderada (pH
7,20), ou quando o processo subjacente possa ser rapidamentecontrolado, muitas vezes a administração de álcali não é ne-cessária. Porém, em pacientes com acidose grave (pH menorque 7,20; bicarbonato inferior a 8), já existem depressão mio-cárdica e disfunções enzimáticas significativas, e a adminis-
Quadro 11.3 Causas de acidose metabólica deacordo com o hiato iônico
Hiato iônico normal (hiperclorêmica)Perdas de bicarbonato
a) Gastrointestinal• Diarréia• Fístulas pancreáticas, biliares
b) Renal• Inibidores da anidrase carbônica• Acidose tubular renal
Outras• Acidose dilucional• Nutrição parenteral
Hiato iônico aumentado (normoclorêmica)Produção ácida aumentada
• Cetoacidose diabética ou alcoólica• Acidose láctica• Erros inatos do metabolismo
Ingestão de substâncias tóxicas• Intoxicação por salicilato• Ingestão de metanol• Ingestão de etilenoglicol
Falha na excreção ácida• Insuficiência renal aguda ou crônica
Adaptado de Shapiro, J.I.18
TRATAMENTOO tratamento é dirigido à doença básica e, em algumas
situações, à própria acidose metabólica, como veremos aseguir.
Tratamento da Doença de BaseA acidose metabólica é manifestação de uma doença
primária, e o tratamento deve ser dirigido à correção des-ta doença.
Na cetoacidose diabética, o ponto fundamental no tra-tamento é a administração de insulina e a correção dosdistúrbios da água, sódio e potássio. Não se deve adminis-trar álcali de rotina, pois o metabolismo dos cetoácidos
Quadro 11.2 Concentrações normais dos cátions eânions não mensurados rotineiramente
Cátions não Ânions nãodeterminados mEq/L determinados mEq/L
K� 4,5 Proteína 15
Ca�� 5 PO4�� 2
Mg�� 1,5 SO4�� 1
Ácidos orgânicos 5
Total 11 23
Quadro 11.4 Correlação entre os níveis depotássio, anion gap e causas de acidose metabólica
Anion gap normal Anion gap aumentado
Potássio sérico Potássio sérico normalreduzido ou elevado
Diarréia Cetoacidosediabética
Inibição da anidrase Cetoacidosecarbônica alcoólica
Acidose tubular Acidose lácticarenal
Intoxicação porsalicilato
Potássio sérico Metanolelevado
Administração de Ingestão deNH4Cl paraldeído
Pielonefrite crônica Etilenoglicol
Uropatia obstrutiva Insuficiência renal

capítulo 11 177
tração de bicarbonato de sódio pode ser benéfica. A acidosedeve ser tratada se estiver causando disfunções orgânicasgraves.18 Para calcular a quantidade necessária de bicarbona-to a ser administrada, utilizamos a fórmula a seguir:
Bic necessário � (Bicdesejado � Bicatual) � espaço do Bic18
Onde:Bicnecessário � quantidade de bicarbonato de sódio a admi-nistrar (em mEq)Bicdesejado � nível desejado de bicarbonatoBicatual � bicarbonato dosado no sangueEspaço do Bic � 50% do peso corporal
O espaço de bicarbonato é uma estimativa da capacida-de total de tamponamento do organismo, que inclui o bi-carbonato do extracelular, proteínas intracelulares e carbo-nato do osso. Com bicarbonato normal ou pouco reduzi-do, o excesso de hidrogênio é tamponado proporcional-mente na água corporal total, e o espaço aparente de bi-carbonato é de 50% do peso magro do indivíduo.18,30 Esteespaço aumenta na acidose metabólica grave, pois as cé-lulas e o osso passam a contribuir cada vez mais para otamponamento, podendo chegar a 70% do peso corporalquando a concentração de bicarbonato cai abaixo de 10mEq/litro; com bicarbonato menor que 5 mEq/litro, o es-paço pode ser de 100%.29-31
Por exemplo, um paciente de 70 kg tem um bicarbona-to de 9 mEq/litro, que se deseja elevar para 15 mEq/litro.O espaço de bicarbonato é de 70% e 50% para estas con-centrações, respectivamente. Considere então como espa-ço de bicarbonato a média entre 70% e 50%, ou seja, 60%.
Bicnecessário � (Bicdesejado � Bicatual) � espaço do Bic
Bicnecessário � (15 � 9) � (0,7 � 70 kg) � 6 � 49 � 294 mEq
Então, de acordo com este cálculo, cerca de 290 mEq deálcali (geralmente bicarbonato de sódio intravenoso) po-dem ser administrados nas primeiras 4-6 horas. Algunsautores sugerem que sempre se utilize o valor de 50% parao espaço de bicarbonato, independente do valor do bicar-bonato plasmático.29 Deve ser assinalado que esta estima-tiva não é exata, e são necessárias avaliações do pH extra-celular pelo menos 30 minutos após o término da infusão.Com o pH em nível mais seguro, não é mais necessáriareposição intravenosa, pois os rins serão capazes de rege-nerar o bicarbonato necessário.30
O tratamento da acidose metabólica é controverso, emfunção dos potenciais efeitos deletérios do bicarbonatoadministrado.18 A infusão de grandes quantidades de bi-carbonato de sódio a 8,4% (1 mEq/ml) pode ocasionar hi-pernatremia, hiperosmolalidade, diminuição da fraçãoionizada do cálcio, hipocalemia e aumento da produção deácidos orgânicos.26 Outra complicação que ocorre principal-mente em pacientes cardiopatas ou nefropatas é a sobre-carga de volume ocasionada pelo sódio da solução, quepode ser evitada ou tratada com o uso de diuréticos de alça,
e, se necessário, diálise. Outro aspecto desfavorável é apossibilidade de alcalose muito abrupta, quando a corre-ção da acidose for muito agressiva.29
O tamponamento de prótons pelo bicarbonato liberaCO2 (HCO3
� � H� ↔ H2CO3 ↔ H2O � CO2), elevando a
PCO2 nos líquidos corporais. Este efeito pode ser prejudi-cial em pacientes com reserva ventilatória limitada, falên-cia circulatória ou que estão sendo submetidos a ressusci-tação cardiopulmonar. Nestas circunstâncias, paradoxal-mente pode ocorrer piora da acidose intracelular e extra-celular, se a PCO2 exceder a fração de HCO3
�. No sistemanervoso central isto traz conseqüências graves, pois o CO2
em maior quantidade atravessa rapidamente a barreira li-quórica, elevando a PCO2 do líquor e piorando a acidosedo sistema nervoso central.29,32
De acordo com os consensos mais recentes da SociedadeAmericana de Cardiologia sobre parada cardiorrespiratória,o uso de bicarbonato de sódio na parada cardiorrespirató-ria é considerado Classe 3 (tratamento inadequado, semevidência científica de validade, e que pode ser prejudici-al). Porém, em situações especiais, e sob monitorização ade-quada, o bicarbonato de sódio pode vir a ser utilizado: a)Quando houver acidose e hipercalemia comprovada (Clas-se 1 — considerado tratamento útil e efetivo); b) No trata-mento de acidose metabólica responsiva a bicarbonato (Clas-se 2a — existência de evidências favoráveis ao seu uso); e c)Para controle de acidose pós-circulação espontânea em pa-rada cardiorrespiratória de longa duração e como coadju-vante na parada cardiorrespiratória desencadeada por an-tidepressivos tricíclicos (Classe 2b — tratamento não vali-dado em estudos clínicos, podendo ser útil em alguns do-entes e provavelmente sem reações adversas).33
Nas acidoses metabólicas crônicas, o bicarbonato desódio pode ser administrado por via oral.18 No Brasil estádisponível o bicarbonato de sódio em pó, contendo 12 mEqde bicarbonato e 12 mEq de bicarbonato por grama.
Pontos-chave:
• A acidose metabólica é classificada deacordo com o hiato iônico, que indica qual acausa mais provável: hiato iônico � Na� �(HCO3
� � Cl�)Hiato iônico aumentado: acréscimo de ácidoHiato iônico normal: perda de bicarbonato
• O mecanismo esperado de compensação é aeliminação de CO2, através dehiperventilação
• A administração de bicarbonato temindicações precisas, e a quantidade écalculada pela fórmula:Bicnecessário � (Bicdesejado � Bicatual) � espaço doBic

178 Metabolismo Ácido-Básico
Como alternativa à administração de bicarbonato, quetem como inconveniente a produção de CO2, poderia serutilizada uma mistura de bicarbonato de sódio com car-bonato de sódio (Carbicarb® — ainda não disponível parauso clínico), que gera mais bicarbonato do que CO2; alémdisso, o carbonato de sódio reage com o ácido carbônico,consumindo o CO2. Esta solução não evita hipervolemia ehipertonicidade.29
Alcalose MetabólicaÉ a situação clínica em que há pH elevado (alcalino),
baixa concentração hidrogeniônica, aumento na concentra-ção de bicarbonato e PCO2 elevada.
A alcalose é um distúrbio ácido-básico relativamentecomum, e sua importância pode ser melhor avaliada quan-do se correlacionam mortalidade e grau de alcalose. Em umgrupo de 177 pacientes cirúrgicos intensamente alcalóticos,verificou-se que, num pH de 7,54 a 7,56, a mortalidade foide 40%, e num pH de 7,65 a 7,7, ela atingiu 80%.34
CAUSAS DE ALCALOSE METABÓLICAAo se avaliar um paciente com alcalose metabólica, é
necessário esclarecer dois pontos fundamentais: o motivoque levou ao aumento do bicarbonato (fase de geração daalcalose metabólica) e os fatores que evitaram a excreçãode bicarbonato pelos rins, permitindo a persistência daalcalose (fase de manutenção)2,35 (v. Quadros 11.4 e 11.5).
GERAÇÃO DA ALCALOSE METABÓLICA
Perda de HidrogênioO íon H� pode ser perdido do líquido extracelular atra-
vés do trato gastrintestinal, dos rins ou por um desvio parao interior das células. Se a perda for maior que o ganho deácido proveniente da dieta e catabolismo, ocorrerá umaumento da concentração plasmática de bicarbonato. OQuadro 11.4 mostra as diversas situações clínicas em queesta perda de H� pode ocorrer.
PERDA GASTROINTESTINAL DE H�. Em indivídu-os normais, a secreção de ácido pelo estômago não leva aalcalose metabólica, pois esta perda de hidrogênio equili-bra-se com uma perda de bicarbonato nas secreções pan-creáticas. Porém, quando o suco gástrico é eliminado atra-vés de vômitos ou drenagem gástrica por sondas, há ten-dência para alcalose metabólica por dois motivos: perdapura do hidrogênio e ausência de estímulo para a secre-ção de bicarbonato. Quando se perde hidrogênio, a reaçãodo sistema ácido carbônico-bicarbonato gera HCO3
�, deforma que para cada mEq de hidrogênio perdido é gerado1 mEq de bicarbonato.2
CO2 � H2O ↔ H2CO3 ↔ H� � HCO3�
PERDA RENAL DE H�. É possível haver perda renalde hidrogênio quando a secreção distal deste íon estiver
aumentada. Isto ocorre em situações em que existe aporteadequado de sódio e água aos sítios tubulares distais eaumento dos níveis de aldosterona. Além de estimular abomba H-ATPase, a aldosterona estimula a reabsorção desódio, tornando a luz tubular mais eletronegativa e mini-mizando a retrodifusão dos íons hidrogênio para fora daluz tubular. A secreção distal de potássio também estáaumentada, resultando em hipocalemia.35
O excesso primário de mineralocorticóides cursa comalcalose metabólica e freqüentemente com hipertensãoarterial. Porém, os pacientes com hiperaldosteronismo se-cundário (p.ex., na cirrose ou insuficiência cardíaca) demodo geral não apresentam alcalose metabólica ou hipo-calemia, pois o efeito estimulatório da aldosterona é con-trabalançado pelo menor aporte distal de sódio e menorvolume urinário. Estes fatores reduzem a quantidade dehidrogênio e potássio na urina final. Se um ânion nãoreabsorvível (p.ex., penicilina) for administrado na vigên-cia de depleção de volume, a excreção deste ânion obrigaa perda de H� ou K� para manter a eletroneutralidade,levando então a hipocalemia e alcalose metabólica.35
O uso de diuréticos de alça ou tiazídicos produz aumen-to do aporte distal de sódio e água, possibilitando a indu-ção de excreção aumentada de hidrogênio. Uma diuresevolumosa pode produzir algum grau de depleção, contri-buindo para o desenvolvimento de alcalose metabólica.35
A acidose respiratória crônica leva a um aumento nasecreção de hidrogênio, ao mesmo tempo em que o bicar-bonato do plasma aumenta, para normalizar o pH (meca-nismo de compensação). Mas quando se reduz abrupta-mente a PCO2 (p.ex., em ventilação mecânica), desenvol-ve-se alcalose metabólica, por não ter havido tempo paraos rins eliminarem o excesso de bicarbonato. Nesta situa-ção, podem desenvolver-se graves anormalidades neuro-lógicas, pois o pH no cérebro aumenta rapidamente com adiminuição da PCO2. Estas complicações justificam a ne-cessidade de redução gradual da PCO2 em pacientes comacidose respiratória crônica.35
DESVIO DO HIDROGÊNIO PARA O INTRACELU-LAR. O desvio do íon hidrogênio para o espaço intracelu-lar pode ocorrer na hipocalemia (v. Cap. 12). Com o obje-tivo de repor o potássio do espaço extracelular, a hipoca-lemia induz a saída do potássio do intracelular; para man-ter a eletroneutralidade, o hidrogênio entra nas células,diminuindo os níveis plasmáticos e aumentando o pH.
Adição de Bicarbonato ao Líquido ExtracelularA administração de bicarbonato ou seus precursores,
tais como lactato, citrato ou acetato, num ritmo maior quea produção diária de ácido elevará os níveis plasmáticosde bicarbonato. Se a função renal for normal, uma cargade bicarbonato é quase toda excretada, causando pequenavariação no pH (v. Quadro 11.5). Porém, se a capacidadede excreção renal for ultrapassada, a alcalose metabólicase estabelece.

capítulo 11 179
Outro fato a ser considerado é que o lactato (na acidoseláctica) e o beta-hidroxibutirato (na cetoacidose diabética)regeneram bicarbonato quando são metabolizados. Nestasduas circunstâncias, a administração de bicarbonato exó-geno representaria um excesso de álcali, resultando emalcalose metabólica.
O citrato utilizado em anticoagulação para hemodiáli-se em pacientes com risco de sangramento, ou na anticoa-gulação de hemoderivados, pode também ser convertidoa bicarbonato. A administração de mais de oito unidadesde sangue estocado ou plasma fresco congelado produzeste efeito.35
Perda de Líquido Contendo GrandesQuantidades de Cloro
Quando se perde sódio, cloro e pouco bicarbonato, comoocorre na administração de diurético de alça, há contraçãodo extracelular com aumento relativo na concentração dobicarbonato.
Em certas situações, porém, há perda de fluidos mui-to ricos em cloro. São exemplos disso a perda de secreçõesgástricas em pacientes com acloridria, a diarréia no ade-noma viloso do cólon e cloridorréia congênita (esta últimaum defeito raro na reabsorção intestinal de cloro e secre-ção de bicarbonato, com diarréia crônica). Note que gran-de parte dos adenomas vilosos do cólon, que constituem5% dos pólipos intestinais e que têm potencial de malig-nidade, produzem acidose metabólica hiperclorêmica,pela perda de grandes volumes de fluido contendo po-tássio e bicarbonato. Cerca de 10-20% destes tumores têmum padrão secretor diverso, com secreção preferencial decloro.36
A síndrome de Bartter é uma desordem rara, diagnosti-cada principalmente em crianças, e que causa hipocalemiae alcalose metabólica resistente ao cloreto (v. próximasseções). Os pacientes apresentam cloro urinário elevado,alcalose metabólica, hiperplasia do aparelho justaglome-rular (inespecífica), gradiente transtubular de potássio ina-propriadamente alto e hiperaldosteronismo hiper-reninêmico, sem hipertensão arterial. É causada por umaalteração na função do co-transportador potássio/cloreto.37
A síndrome de Gitelman tem características semelhan-tes à síndrome de Bartter, porém com hipomagnesemia ehipocalciúria. É causada por alteração na função do co-transportador sódio/cloreto no túbulo contornado distal.37
MANUTENÇÃO DA ALCALOSE METABÓLICAComo já foi mencionado, normalmente os rins são ca-
pazes de excretar os excessos de bicarbonato. Portanto,para que uma alcalose metabólica persista, é necessária apresença de dois grupos de anormalidades: 1) Perda con-tinuada de hidrogênio, desvio transcelular de hidrogênio,administração de bicarbonato ou alcalose de contração; e2) Aumento na reabsorção renal de bicarbonato ou dimi-nuição na secreção distal de bicarbonato.35
Em presença de função renal normal, o aumento oumanutenção da reabsorção de bicarbonato pelos rins sedeve a pelo menos um dos seguintes fatores: a) Depleçãodo volume circulante efetivo; b) Depleção de cloro; c) Hi-pocalemia, e d) Hipoventilação e hipercapnia.35
Estes fatores acima mencionados são responsáveis pelamanutenção da alcalose metabólica, pois impedem a atu-ação dos mecanismos renais fisiológicos de eliminação demaiores quantidades de bicarbonato que levariam à nor-malização do bicarbonato no plasma. O esclarecimento dequal o fator envolvido auxilia na classificação das alcalosesmetabólicas e no planejamento terapêutico posterior.
Volume ExtracelularA depleção de volume aumenta a reabsorção de sódio
e o resgate de bicarbonato no túbulo proximal. No túbulodistal, também ocorre um aumento na reabsorção de só-dio (mediada por mineralocorticóide) em troca da secre-ção de H� ou K�. Com um aumento da secreção de H�,ocorre regeneração de bicarbonato.
Um aumento na reabsorção distal de sódio tambémpode ocorrer na ausência de depleção de volume extrace-lular, devido a um excesso de mineralocorticóide, como nohiperaldosteronismo primário. A elevada reabsorção dis-tal de sódio pode gerar e manter uma concentração eleva-da de bicarbonato se os hormônios mineralocorticóidesestimularem a secreção de H�.18
Deficiência de CloroPara que seja mantida a eletroneutralidade, quando a
concentração plasmática de bicarbonato se eleva, a con-
Quadro 11.5 Etiologia e classificação da alcalosemetabólica
Responsiva ao cloreto (cloro urinário menor que10 mEq/L)
a) Distúrbios gastrointestinais• Vômitos• Drenagem gástrica• Adenoma viloso do cólon• Cloridorréia congênita
b) Uso de diuréticosc) Correção de hipercapnia crônicad) Fibrose cística
Resistente ao cloreto (cloro urinário maior que20 mEq/L)
a) Excesso de mineralocorticóide• Hiperaldosteronismo• Síndrome de Cushing• Síndrome de Bartter• Alcaçuz
b) Hipocalemia
Adaptado de Shapiro, J.I.18

180 Metabolismo Ácido-Básico
centração de cloro deve reduzir-se. Porém, com a perda desódio, e conseqüente contração do volume extracelular, oestímulo para restaurar o volume extracelular supera oestímulo para aumentar a excreção de bicarbonato. O pa-pel do cloro é crucial nesta situação, pois é o único outroânion, além do bicarbonato, que pode acompanhar a rea-bsorção de sódio. Portanto, para se elevar ou manter a reab-sorção de sódio enquanto simultaneamente se eleva a ex-creção de bicarbonato, um ânion reabsorvível (cloro) pre-cisa estar presente para acompanhar a reabsorção de só-dio. Se há deficiência de cloro, os rins reabsorvem outroânion, o bicarbonato, perpetuando a alcalose metabólica.18
Depleção de PotássioÉ um fator importante na origem e manutenção da al-
calose metabólica. Com a saída de potássio das células,aumenta a concentração de H� intracelular, inclusive nascélulas tubulares renais. Havendo mais H� para secreção,maior será o resgate de bicarbonato. Além disso, em pre-sença de hipocalemia, as bombas H-K-ATPase (que pro-movem reabsorção de potássio e secreção de hidrogênio)e a síntese de NH3 são estimuladas, resultando em elimi-nação de maiores quantidades de H�, na forma de NH4
�.18,35
Hipoventilação e HipercapniaDa mesma forma que a depleção de potássio, a hipercap-
nia aumenta a concentração intracelular de H� disponívelpara secreção e, portanto, para resgate de bicarbonato.
MECANISMOS DE DEFESA DO pH NAALCALOSE METABÓLICA
Com a elevação do bicarbonato plasmático por um dostrês mecanismos básicos já mencionados, os mecanismosde defesa do organismo entram em ação, na tentativa denormalizar o pH.
Sistema TampãoA fase de tamponamento é controlada pelo imediato
tamponamento químico. Aproximadamente 1/3 do exces-so de bicarbonato é tamponado pelo H� intracelular, quesai das células para o líquido extracelular. Exemplo distoé a saída de lactato das células musculares, para tamponaro espaço extracelular.
Compensação RespiratóriaA segunda fase do mecanismo de defesa do pH é con-
trolada pelo sistema respiratório. Para que o pH retorne aonormal, em face de uma elevação na concentração de bi-carbonato, a PCO2 deve ser elevada. Isto ocorre através dahipoventilação alveolar, com retenção de CO2 e elevaçãoda PCO2. O grau de compensação é limitado pelas neces-sidades de O2, já que a pO2 será reduzida com a hipoventi-lação. O limite superior de elevação compensatória daPCO2 é geralmente aceito como 55 mmHg, mas há relatos
de elevação até 60-75 mmHg em indivíduos normais. De-vido a estes fatores, a compensação respiratória na alcalo-se metabólica é menos intensa que na acidose metabólica.
Correção RenalO rim é responsável pela terceira fase do mecanismo de
defesa do pH. O rim tem a capacidade de eliminar o ex-cesso de bicarbonato, a não ser que outros fatores compro-metam esta capacidade renal (v. a seguir). Esta eliminaçãode base é bem mais rápida que a capacidade renal de ex-cretar H�.
MANIFESTAÇÕES CLÍNICASNa maioria das vezes, os sinais e sintomas da enfermi-
dade básica dominam o quadro clínico e dificilmente po-derão ser separados. Não há sintomas ou sinais patogno-mônicos. A avaliação do espaço extracelular fornece dadosmuito importantes. Num paciente depletado, com defici-ência de potássio, a causa provável da alcalose metabólicaé a perda renal (diuréticos) ou gastrintestinal (vômitos).Além destes sintomas, há os referentes à hipocalemia, comofraqueza ou paralisia muscular, distensão abdominal, íleoe arritmias cardíacas, poliúria e aumento da produção deamônia (que aumenta o risco de encefalopatia em hepa-topatas).38 Um extracelular expandido, com hipertensãoarterial e hipocalemia, leva à suspeita de hiperaldostero-nismo.37
O elevado risco de intoxicação digitálica, intervalo QTprolongado e ondas U são complicações conhecidas daalcalose. A resistência vascular cerebral é sensível à PCO2
e a hipocapnia é uma potente força vasoconstritora cere-bral. Um fluxo sanguíneo cerebral reduzido pode justifi-car muitos sinais e sintomas neurológicos observados,como cefaléia, convulsões, letargia, delirium e estupor.38
DADOS LABORATORIAISO padrão diagnóstico no sangue arterial é elevação do
pH, da concentração de bicarbonato e PCO2. O padrão ele-trolítico é de hipocloremia e hipocalemia. A hipocalemia ébasicamente conseqüente à perda urinária de potássio quese deve a uma elevada secreção distal.
Como o mecanismo de compensação da alcalose é a re-tenção de CO2 através de hipoventilação, em alguns casosobserva-se hipóxia, dependendo da função pulmonar pré-via do paciente.
A concentração urinária de cloro é muito útil na avalia-ção inicial da alcalose metabólica. Concentração de cloronuma amostra de urina inferior a 10 mEq/litro indica queo rim está reabsorvendo sódio avidamente, compatívelcom situações associadas à depleção de volume e que res-pondem à infusão de cloreto de sódio (“sensíveis” ao clo-reto de sódio) (v. a seguir).
Concentração urinária de cloro superior a 20 mEq/litrodemonstra que não há depleção de volume e que o cloro

capítulo 11 181
não é um elemento crucial na manutenção da alcalose; esteperfil geralmente corresponde às alcaloses resistentes aocloreto de sódio. O sódio urinário não é útil nestas circuns-tâncias porque pode estar elevado durante períodos debicarbonatúria.
Como a alcalemia estimula a glicólise anaeróbica e au-menta a produção de ácido láctico e cetoácidos, pode ha-ver moderada elevação no anion gap.
A alcalemia aguda reduz a liberação de oxigênio paraos tecidos, por aumentar a afinidade entre oxigênio e he-moglobina. A alcalemia crônica anula este efeito, aumen-tando a concentração de ácido 2,3 difosfoglicérico nas he-mácias.38
TRATAMENTOPelo exposto, fica evidente a necessidade de serem cor-
rigidos os mecanismos que impedem os rins de excretaremquantidades maiores de bicarbonato. Abordaremos o tra-tamento da alcalose metabólica de acordo com sua classi-ficação.
Alcalose Metabólica Responsiva ao CloretoApesar de a correção do déficit de cloreto ser essencial,
a seleção do cátion que o acompanha em solução (sódio,potássio ou próton) depende do estado do espaço extrace-lular, da presença e do grau de depleção de potássio asso-ciada, e do grau e reversibilidade de qualquer diminuiçãoda taxa de filtração glomerular. Quando a função renal énormal, ao se repor cloreto o excesso de bicarbonato seráeliminado pelos rins.36
Se existe depleção de cloreto e do extracelular concomi-tantemente (que é a situação mais comum), a administra-ção de solução salina isotônica (NaCl 0,9%) é adequada ecorrige os dois déficits. Em presença de sinais de depleçãodo extracelular, a quantidade a ser administrada está emtorno de 3-5 litros de solução salina isotônica. Porém, se nãohá sinais de depleção do extracelular, o déficit de cloropode ser calculado pela fórmula: 0,2 � peso (kg) � aumen-to desejado no cloreto plasmático (mEq/litro).
As perdas continuadas de cloro e potássio devem sercalculadas e acrescentadas à reposição. Como se instaladiurese alcalina com a correção do cloreto, recomenda-seacrescentar 10-20 mEq de potássio por litro de soluçãoadministrada, para evitar que se some uma hipocalemia.36
Na presença de sobrecarga de volume, está contra-in-dicada a reposição de grandes quantidades de volumecontendo sódio; então repor cloreto sob forma de cloretode potássio, em doses de 10-20 mEq.
O HCl é indicado se o NaCl ou KCl não puderem serusados, ou se houver necessidade de correção imediata, porexemplo, se o pH for maior que 7,55, ou na presença deencefalopatia hepática, arritmia cardíaca, intoxicação digi-tálica ou alteração do estado mental. A quantidade neces-sária de HCl, administrado como solução 0,1 ou 0,2 M, écalculada pela fórmula: 0,5 � peso (kg) � redução deseja-
da no bicarbonato plasmático (mEq/L). O objetivo do tra-tamento com HCl é reverter uma alcalose grave, e inicial-mente deve-se calcular uma correção parcial do bicarbo-nato, e não total. Pode-se preparar uma solução isotônicade HCl adicionando-se 150 ml de ácido clorídrico 1 N em1 litro de água destilada. A infusão de 1 a 2 litros destasolução, em 24 horas, corrige a alcalose na maioria doscasos.36 (Obs: solução 0,1-0,2 N é a solução contendo 100-200 mEq de hidrogênio por litro.)38
O HCl deve ser administrado em ambiente de terapiaintensiva, por cateter em veia cava ou outra veia central degrande calibre, sendo a posição do cateter necessariamen-te confirmada por RX, já que a administração de HCl forado vaso provocaria repercussões dramáticas.36 A velocida-de de infusão pode chegar a 25 ml/hora. RecentementeKnutsen mostrou a possibilidade de se administrar, atra-vés de uma veia periférica, ácido clorídrico 0,15 N em umasolução de aminoácidos e emulsão lipídica.39
Alternativas ao HCl são: o cloreto de amônio (NH4Cl) ea arginina mono-hidrocloreto. O cloreto de amônio (374mEq de hidrogênio por litro) pode ser administrado porveia periférica, em quantidade não superior a 300 mEq nas24 horas; é contra-indicado na insuficiência renal ou hepá-tica.36 A arginina mono-hidrocloreto (475 mEq de H� porlitro) pode causar hipercalemia grave em pacientes cominsuficiência renal, principalmente se houver doença he-pática concomitante.38
Se a taxa de filtração glomerular for adequada, o uso deacetazolamida, que é um diurético inibidor da anidrasecarbônica, na dose de 250-500 mg via oral ao dia aumentasignificativamente a excreção renal de bicarbonato e potás-sio. É benéfico para pacientes que tenham sobrecarga devolume e particularmente útil para os pacientes em que senecessita manter eliminação de sódio ou quando o potássioestiver elevado. Se não houver hipocalemia, é aconselhávela reposição de potássio, pela alta probabilidade de se desen-volver hipocalemia na vigência de diurese alcalina.18,36
Caso não haja resposta renal após a repleção de cloro oufor necessária diálise para o controle da insuficiência re-nal, a diálise corrigirá a alcalose metabólica. Porém, se sóestiverem disponíveis os líquidos de diálise com altas con-centrações de bicarbonato ou seus precursores, pode serrealizada diálise peritoneal de emergência com soluçãosalina isotônica, sendo a manutenção de potássio, cálcio emagnésio feita pela via intravenosa.36
No caso de a alcalose ser conseqüência de perdas conti-nuadas de suco gástrico, são úteis os antieméticos. Na al-calose da gastrocistoplastia, a administração de um inibi-dor da bomba de prótons, como o omeprazol, bloqueará asecreção gástrica na neobexiga.
Alcalose Metabólica Resistente ao CloretoQuando a hipocalemia estiver associada com uma alca-
lose discreta a moderada, a administração de 40-60 mEqde KCl quatro vezes ao dia é de modo geral suficiente. No

182 Metabolismo Ácido-Básico
entanto, se estiver presente arritmia cardíaca ou situaçãode ameaça à vida, o KCl pode ser administrado na propor-ção de 40 mEq/hora, em concentrações não superiores a60 mEq/litro, sob monitorização eletrocardiográfica. Aglicose deve ser inicialmente omitida da solução de repo-sição, pois a secreção de insulina pode diminuir ainda maisa concentração de potássio. Uma vez iniciada a reposiçãode potássio, a presença de glicose na solução auxilia narepleção celular de potássio.36
Quando a causa for um excesso de mineralocorticóide,o tratamento é dirigido à remoção cirúrgica da fonte oubloqueio da mesma. Os efeitos do mineralocorticóide so-bre o sódio, o potássio e o bicarbonato podem ser reverti-dos com a espironolactona, diurético poupador de potás-sio. Além disso, podem ser úteis a restrição de sódio e oacréscimo de potássio na dieta.36
Nas síndromes de Bartter e Gitelman, o principal obje-tivo do tratamento é diminuir a perda urinária de potás-sio. Na síndrome de Bartter, os inibidores da enzima con-versora reduzem a produção de angiotensina II e diminu-em a secreção de aldosterona. Como a síntese de prosta-glandinas está elevada nesta síndrome, e pode contribuirpara as perdas de sódio, cloro e potássio, inibidores daprostaglandina sintetase podem melhorar a alcalose me-tabólica. Na síndrome de Gitelman, os diuréticos poupa-dores de potássio e a suplementação dietética de potássiosão necessários.36
Pontos-chave:
• A alcalose metabólica apresenta as fases degeração e manutenção. Na fase demanutenção a eliminação de bicarbonatopelos rins está prejudicada
• Classificação: responsiva ou resistente aocloreto
• O tratamento se baseia na correção de:Espaço extracelularDeficiência de potássioDeficiência de cloro
• Em casos graves, pode ser necessária aadministração de ácido clorídrico
Acidose Respiratória
Ocorre quando há uma retenção de CO2 (hipercapnia)no organismo e traduz-se por uma elevação da PCO2 nosangue. Isto ocorre quando a produção de CO2 nos tecidosexcede a capacidade de remoção pelos pulmões.
CAUSASMais comumente são distúrbios neuromusculares (le-
sões do sistema nervoso central, da parede torácica e mio-
patias) ou enfermidades pulmonares (asma, enfisema etc.).O denominador comum é uma hipoventilação alveolar,que pode ser causada por uma simples obstrução das viasaéreas superiores. V. Quadro 11.6.
CONSEQÜÊNCIAS CLÍNICASClinicamente, há uma diferença entre o estabelecimen-
to rápido e o gradual da retenção de CO2. Os pacientes seadaptam melhor quando a elevação é gradual.
A retenção de CO2 pode causar confusão mental, tremordo tipo flapping e coma. O único sinal clínico fidedigno dehipercapnia é a demonstração de PCO2 elevada no sangue.A PCO2 venosa é geralmente 6 mmHg mais elevada que aarterial.
CONSEQÜÊNCIAS FISIOLÓGICASObservando-se a equação de Henderson-Hasselbalch,
fica claro que, para o organismo manter o pH sanguíneo,a concentração plasmática de bicarbonato deve variar.
Os tampões celulares desempenham o papel principalna resposta a alterações agudas da concentração de CO2.Quando a PCO2 aumenta, aumenta também a concentra-ção de H2CO3, e, portanto, a concentração de H�. O H�
Quadro 11.6 Causas de acidose respiratória(aguda e crônica)
Acidose respiratória agudaa) Anormalidades neuromusculares
• Lesão neurológica (tronco, medula alta)• Síndrome de Guillain-Barré, miastenia gravis• Drogas
b) Obstrução de vias aéreas• Corpo estranho• Edema ou espasmo de laringe• Broncoespasmo grave
c) Desordens tóraco-pulmonares• Tórax instável• Pneumotórax• Pneumonia grave• Inalação de fumaça• Edema pulmonar
d) Doença vascular pulmonar• Embolia pulmonar maciça
e) Ventilação mecânica controlada• Parâmetros inadequados (freqüência, volume
corrente)• Espaço morto aumentado
Acidose respiratória crônicaa) Anormalidades neuromusculares
• Paralisia diafragmática• Síndrome de Pickwick
b) Desordens tóraco-pulmonares• Doença pulmonar obstrutiva crônica• Cifoescoliose• Doença pulmonar intersticial terminal
Baseado em Kaehny W.D.43

capítulo 11 183
entra na célula em troca por Na� e K� e é tamponado pelasproteínas celulares, deixando o bicarbonato no líquidoextracelular. Este tamponamento celular é responsável poraproximadamente 50% do aumento agudo na concentra-ção plasmática de bicarbonato.40
Ao mesmo tempo, parte do CO2 entra na hemácia, for-mando H2CO3, o qual, dissociando-se, libera H� e HCO3
�.O íon H� é tamponado pela hemoglobina, e o bicarbonatoentra no líquido extracelular em troca de cloro. Este meca-nismo é responsável por aproximadamente 30% do aumen-to agudo na concentração plasmática de bicarbonato. Nohomem, a magnitude do aumento na concentração de bi-carbonato plasmático é pequena, sendo inferior a 5 mEqquando a PCO2 aumenta gradualmente de 40 para 80 mmHg.40,41
Quando a hipercapnia continua, a capacidade de tam-ponamento se esgota rapidamente. A necessidade de com-pensação leva a um aumento na excreção de H� e na reab-sorção e produção de bicarbonato.
Schwartz e cols. mostraram, em cães expostos a umaatmosfera de CO2, que o rápido aumento que ocorria nasprimeiras 24 horas no bicarbonato plasmático não se acom-panhava de um aumento na excreção urinária de H�. Mas,entre três e seis dias, o bicarbonato plasmático continuavaaumentando, até atingir um platô. O autor, então, demons-trou que este último aumento no bicarbonato estava asso-ciado a um aumento na excreção urinária de H�, sob a for-ma de NH4
�, e, durante esta fase, o rim restaurou os tam-pões celulares e extracelulares consumidos durante a faseaguda, gerando novo bicarbonato (v. Fig. 11.9).42 Portanto,na retenção crônica de CO2, o limiar da reabsorção de bi-carbonato está elevado, assim como há uma excreção ele-vada de cloro. É preciso mencionar que, no homem comretenção crônica de CO2, não há uma compensação com-pleta.
TRATAMENTOÉ dirigido à causa da hipoventilação alveolar. Exemplo:
desobstrução das vias aéreas superiores, alívio do bronco-espasmo do asmático, etc.
Alcalose RespiratóriaOcorre quando há uma redução de CO2 no organismo e
traduz-se por uma diminuição da PCO2 no sangue. Estasituação é conhecida como hipocapnia e é o resultado deuma hiperventilação alveolar.
CAUSASQualquer condição que estimule a ventilação pulmonar
poderá ocasionar uma redução da PCO2. Exemplos: dor,ansiedade, salicilatos, tumores cerebrais ou acidentes vas-culares encefálicos, estados de hipóxia (cardiopatias cianó-ticas, altitudes, insuficiência cardíaca congestiva, anemia
etc.), estados infecciosos (septicemias), estados hipermeta-bólicos (febre, delirium tremens), insuficiência hepática, es-tados conversivos, etc.43
CONSEQÜÊNCIAS CLÍNICASClinicamente, a hiperventilação pulmonar, além das
manifestações clínicas da enfermidade básica, pode seracompanhada de outros sintomas e sinais, possivelmenterelacionados com o pH do sangue, circulação cerebral enível de cálcio iônico: parestesias nas extremidades e re-gião perioral, alteração na consciência e espasmoscarpopedais.
CONSEQÜÊNCIAS FISIOLÓGICASQuando há redução da PCO2 (hipocapnia), ocorrem re-
ações em sentido inverso ao daquelas que mencionamosdurante retenção de CO2. Os tampões intracelulares libe-ram H� e trocam cloro e bicarbonato na direção oposta.40
Estes processos causam redução do bicarbonato plasmáti-co. Geralmente, esta redução é da ordem de 7-8 mEq/Lquando a PCO2 é reduzida de 40 para 15 mmHg. Há tam-bém redução do limiar de reabsorção renal de bicarbona-to e retenção de cloro pelo rim.
TRATAMENTOÉ dirigido ao distúrbio que originou a hiperventilação
alveolar. No entanto, a PCO2 pode ser rapidamente eleva-da, fazendo-se o paciente respirar uma mistura de gás car-bônico a 5%, ou aumentando o espaço morto e diminuin-do o volume-minuto quando em uso de ventilador.
Distúrbios Ácido-básicos MistosChamamos distúrbio ácido-básico misto à ocorrência de
dois ou mais distúrbios ácido-básicos simultaneamente nomesmo paciente. Assim, as desordens combinadas podemmascarar umas às outras, resultando em pH relativamen-te normal. Distúrbios ácido-básicos graves podem passardespercebidos, a menos que uma abordagem passo a pas-so seja utilizada na avaliação das gasometrias.44
DIAGNÓSTICO DOS DISTÚRBIOSÁCIDO-BÁSICOS
História clínica e exame físico completos devem ser rea-lizados, verificando antecedentes de perdas fluidas, uso demedicamentos e estado do espaço extracelular. Verifiqueos valores encontrados na gasometria (arterial ou venosa)e compare com os valores normais (Quadro 11.7).
Alguns autores sugerem que, antes de iniciar a avalia-ção dos resultados da gasometria, seja verificada a valida-de interna dos dados obtidos, através da fórmula deHenderson: [H�] � 24 � PCO2/[HCO3
�]. A concentraçãohidrogeniônica (em mEq/litro) para cada pH é encontra-da no Quadro 11.8. Os valores intermediários podem ser
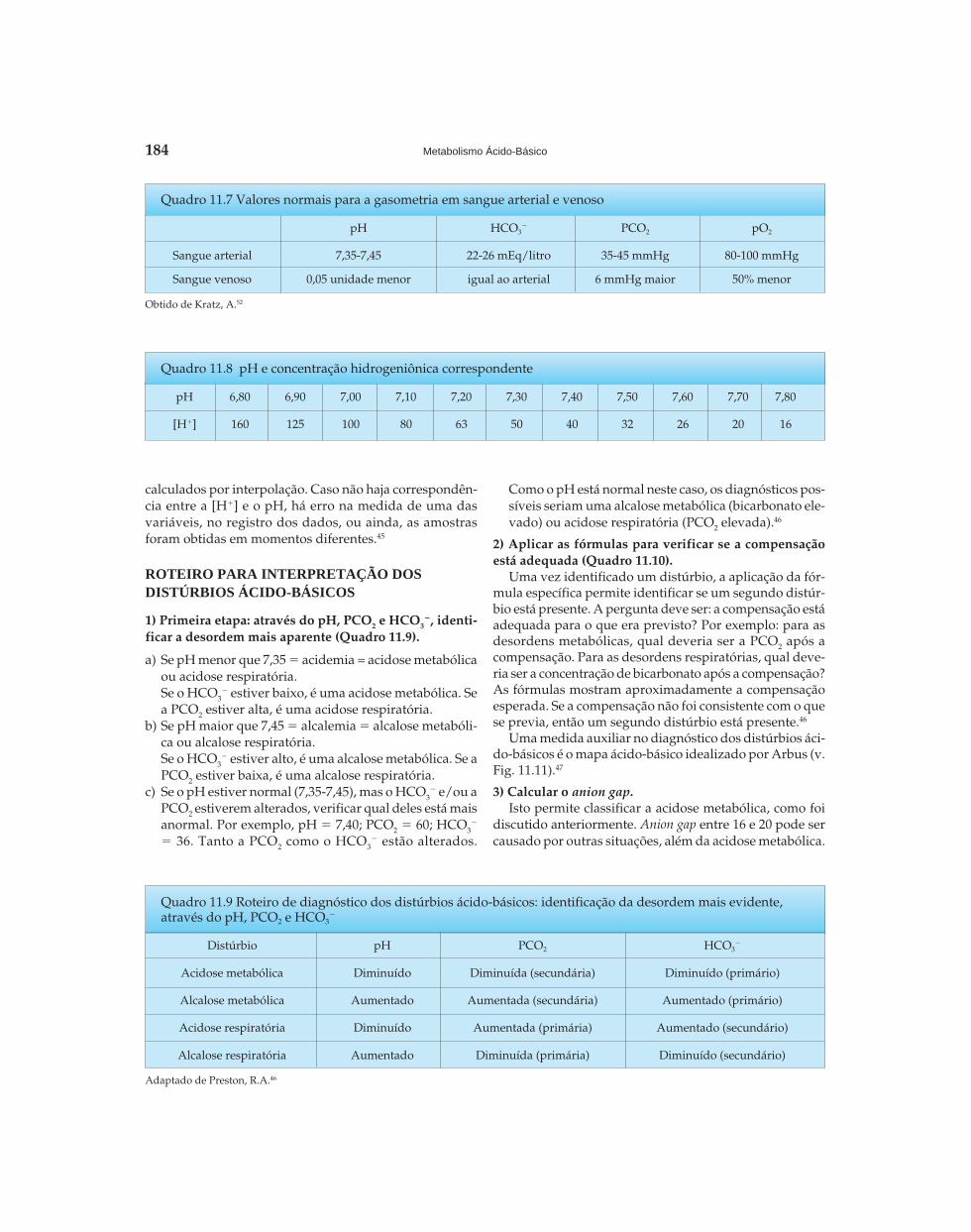
184 Metabolismo Ácido-Básico
calculados por interpolação. Caso não haja correspondên-cia entre a [H�] e o pH, há erro na medida de uma dasvariáveis, no registro dos dados, ou ainda, as amostrasforam obtidas em momentos diferentes.45
ROTEIRO PARA INTERPRETAÇÃO DOSDISTÚRBIOS ÁCIDO-BÁSICOS
1) Primeira etapa: através do pH, PCO2 e HCO3�, identi-
ficar a desordem mais aparente (Quadro 11.9).
a) Se pH menor que 7,35 � acidemia = acidose metabólicaou acidose respiratória.Se o HCO3
� estiver baixo, é uma acidose metabólica. Sea PCO2 estiver alta, é uma acidose respiratória.
b) Se pH maior que 7,45 � alcalemia � alcalose metabóli-ca ou alcalose respiratória.Se o HCO3
� estiver alto, é uma alcalose metabólica. Se aPCO2 estiver baixa, é uma alcalose respiratória.
c) Se o pH estiver normal (7,35-7,45), mas o HCO3� e/ou a
PCO2 estiverem alterados, verificar qual deles está maisanormal. Por exemplo, pH � 7,40; PCO2 � 60; HCO3
�
� 36. Tanto a PCO2 como o HCO3� estão alterados.
Como o pH está normal neste caso, os diagnósticos pos-síveis seriam uma alcalose metabólica (bicarbonato ele-vado) ou acidose respiratória (PCO2 elevada).46
2) Aplicar as fórmulas para verificar se a compensaçãoestá adequada (Quadro 11.10).
Uma vez identificado um distúrbio, a aplicação da fór-mula específica permite identificar se um segundo distúr-bio está presente. A pergunta deve ser: a compensação estáadequada para o que era previsto? Por exemplo: para asdesordens metabólicas, qual deveria ser a PCO2 após acompensação. Para as desordens respiratórias, qual deve-ria ser a concentração de bicarbonato após a compensação?As fórmulas mostram aproximadamente a compensaçãoesperada. Se a compensação não foi consistente com o quese previa, então um segundo distúrbio está presente.46
Uma medida auxiliar no diagnóstico dos distúrbios áci-do-básicos é o mapa ácido-básico idealizado por Arbus (v.Fig. 11.11).47
3) Calcular o anion gap.Isto permite classificar a acidose metabólica, como foi
discutido anteriormente. Anion gap entre 16 e 20 pode sercausado por outras situações, além da acidose metabólica.
Quadro 11.7 Valores normais para a gasometria em sangue arterial e venoso
pH HCO3� PCO2 pO2
Sangue arterial 7,35-7,45 22-26 mEq/litro 35-45 mmHg 80-100 mmHg
Sangue venoso 0,05 unidade menor igual ao arterial 6 mmHg maior 50% menor
Obtido de Kratz, A.52
Quadro 11.9 Roteiro de diagnóstico dos distúrbios ácido-básicos: identificação da desordem mais evidente,através do pH, PCO2 e HCO3
�
Distúrbio pH PCO2 HCO3�
Acidose metabólica Diminuído Diminuída (secundária) Diminuído (primário)
Alcalose metabólica Aumentado Aumentada (secundária) Aumentado (primário)
Acidose respiratória Diminuído Aumentada (primária) Aumentado (secundário)
Alcalose respiratória Aumentado Diminuída (primária) Diminuído (secundário)
Adaptado de Preston, R.A.46
Quadro 11.8 pH e concentração hidrogeniônica correspondente
pH 6,80 6,90 7,00 7,10 7,20 7,30 7,40 7,50 7,60 7,70 7,80
[H�] 160 125 100 80 63 50 40 32 26 20 16

capítulo 11 185
Valores acima de 30 sempre significam acidose metabóli-ca com anion gap aumentado. Para valores acima de 20,existe alta probabilidade de ser acidose metabólica comanion gap aumentado.46
Observação: Os elementos BE (base excess) e BD (base defi-cit) da gasometria refletem o excesso de álcalis na alcalosee a falta de bases na acidose metabólica. Valores normais:BE � �2 mEq/L; BD � �2mEq/L. Na alcalose metabóli-
Fig. 11.11 Mapa ácido-básico. A área central (N) representa a área de normalidade. Conhecendo-se pelo menos duas das variáveis(PCO2, pH e HCO3
�), traça-se uma linha pelos respectivos valores, e o ponto de encontro de duas linhas indica o distúrbio ácido-básico e a variação normal de compensação que pode ocorrer. Se o ponto de encontro das linhas cair fora das áreas sombreadas, aschances são de que o paciente tenha um distúrbio ácido-básico misto. (Obtido de Arbus, G.S.47)
PCO2 (mmHg)
Quadro 11.10 Roteiro de diagnóstico dos distúrbios ácido-básicos: aplicar as fórmulas para verificar se acompensação está adequada
Acidose metabólica PCO2 � 1,5 � [HCO3�] � 8 ou
� [HCO3�] � 1,2 � � [CO2]
Variação aceita nos distúrbios simples: 2 mEq/litro
Alcalose metabólica PCO2 � 40 � 0,7 � [HCO3� atual � HCO3
� normal]Variação aceita nos distúrbios simples: 5 mEq/litro
Acidose respiratória Aguda: [HCO3�] aumenta 1 mEq para cada 10 mmHg de aumento na PCO2
Crônica: [HCO3�] aumenta 3,5 mEq para cada 10 mmHg de aumento na PCO2
Alcalose respiratória Aguda: [HCO3�] diminui 2 mEq para cada 10 mmHg de queda na PCO2
Crônica: [HCO3�] diminui 5 mEq para cada 10 mmHg de queda na PCO2
Adaptado de Preston, R.A.46
BICARBONATOmEq/l
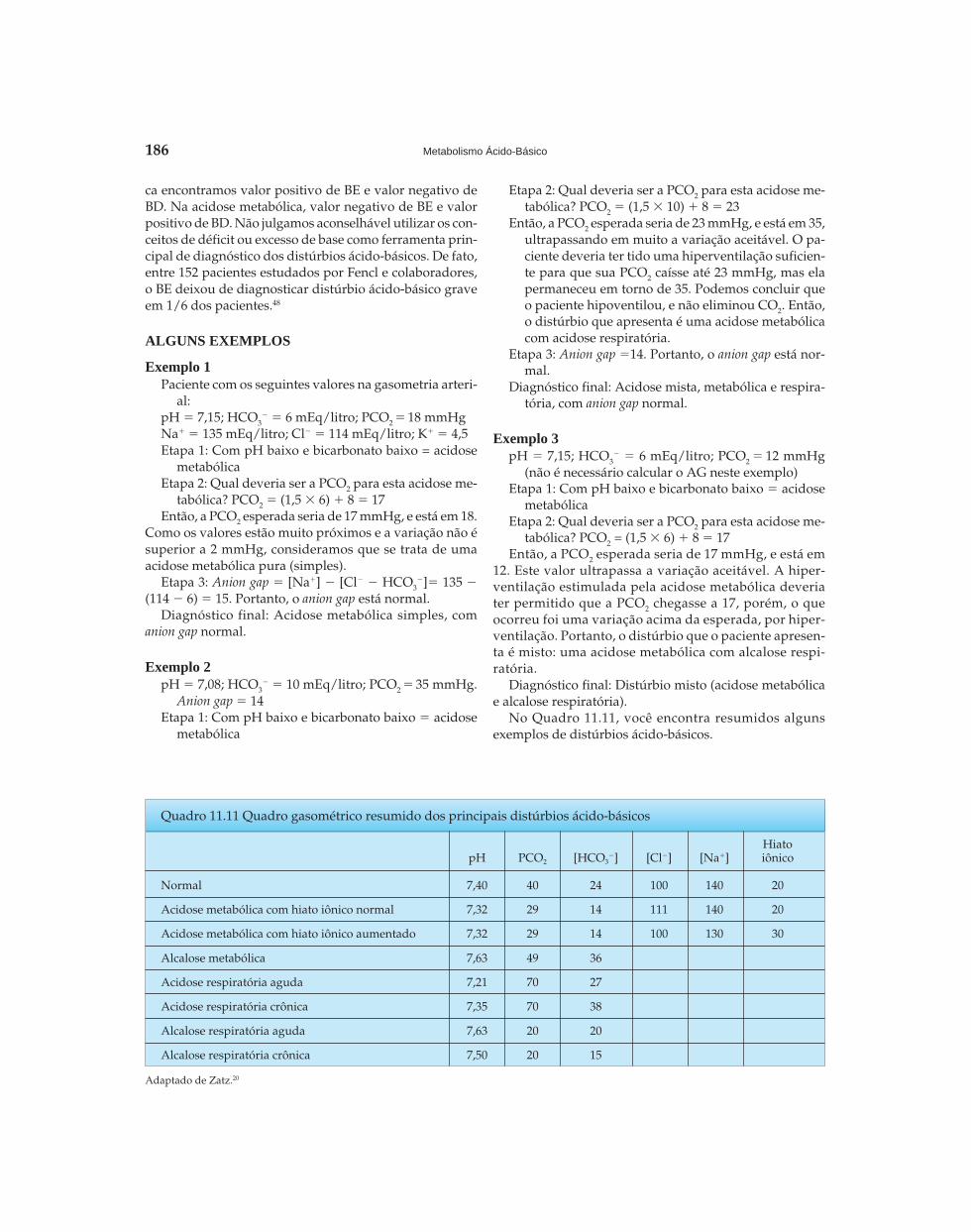
186 Metabolismo Ácido-Básico
ca encontramos valor positivo de BE e valor negativo deBD. Na acidose metabólica, valor negativo de BE e valorpositivo de BD. Não julgamos aconselhável utilizar os con-ceitos de déficit ou excesso de base como ferramenta prin-cipal de diagnóstico dos distúrbios ácido-básicos. De fato,entre 152 pacientes estudados por Fencl e colaboradores,o BE deixou de diagnosticar distúrbio ácido-básico graveem 1/6 dos pacientes.48
ALGUNS EXEMPLOS
Exemplo 1Paciente com os seguintes valores na gasometria arteri-
al:pH � 7,15; HCO3
� � 6 mEq/litro; PCO2 � 18 mmHgNa� � 135 mEq/litro; Cl� � 114 mEq/litro; K� � 4,5Etapa 1: Com pH baixo e bicarbonato baixo = acidose
metabólicaEtapa 2: Qual deveria ser a PCO2 para esta acidose me-
tabólica? PCO2 � (1,5 � 6) � 8 � 17Então, a PCO2 esperada seria de 17 mmHg, e está em 18.
Como os valores estão muito próximos e a variação não ésuperior a 2 mmHg, consideramos que se trata de umaacidose metabólica pura (simples).
Etapa 3: Anion gap � [Na�] � [Cl� � HCO3�]� 135 �
(114 � 6) � 15. Portanto, o anion gap está normal.Diagnóstico final: Acidose metabólica simples, com
anion gap normal.
Exemplo 2pH � 7,08; HCO3
� � 10 mEq/litro; PCO2 � 35 mmHg.Anion gap � 14
Etapa 1: Com pH baixo e bicarbonato baixo � acidosemetabólica
Etapa 2: Qual deveria ser a PCO2 para esta acidose me-tabólica? PCO2 � (1,5 � 10) � 8 � 23
Então, a PCO2 esperada seria de 23 mmHg, e está em 35,ultrapassando em muito a variação aceitável. O pa-ciente deveria ter tido uma hiperventilação suficien-te para que sua PCO2 caísse até 23 mmHg, mas elapermaneceu em torno de 35. Podemos concluir queo paciente hipoventilou, e não eliminou CO2. Então,o distúrbio que apresenta é uma acidose metabólicacom acidose respiratória.
Etapa 3: Anion gap �14. Portanto, o anion gap está nor-mal.
Diagnóstico final: Acidose mista, metabólica e respira-tória, com anion gap normal.
Exemplo 3pH � 7,15; HCO3
� � 6 mEq/litro; PCO2 � 12 mmHg(não é necessário calcular o AG neste exemplo)
Etapa 1: Com pH baixo e bicarbonato baixo � acidosemetabólica
Etapa 2: Qual deveria ser a PCO2 para esta acidose me-tabólica? PCO2 = (1,5 � 6) � 8 � 17
Então, a PCO2 esperada seria de 17 mmHg, e está em12. Este valor ultrapassa a variação aceitável. A hiper-ventilação estimulada pela acidose metabólica deveriater permitido que a PCO2 chegasse a 17, porém, o queocorreu foi uma variação acima da esperada, por hiper-ventilação. Portanto, o distúrbio que o paciente apresen-ta é misto: uma acidose metabólica com alcalose respi-ratória.
Diagnóstico final: Distúrbio misto (acidose metabólicae alcalose respiratória).
No Quadro 11.11, você encontra resumidos algunsexemplos de distúrbios ácido-básicos.
Quadro 11.11 Quadro gasométrico resumido dos principais distúrbios ácido-básicos
HiatopH PCO2 [HCO3
�] [Cl�] [Na�] iônico
Normal 7,40 40 24 100 140 20
Acidose metabólica com hiato iônico normal 7,32 29 14 111 140 20
Acidose metabólica com hiato iônico aumentado 7,32 29 14 100 130 30
Alcalose metabólica 7,63 49 36
Acidose respiratória aguda 7,21 70 27
Acidose respiratória crônica 7,35 70 38
Alcalose respiratória aguda 7,63 20 20
Alcalose respiratória crônica 7,50 20 15
Adaptado de Zatz.20

capítulo 11 187
EXERCÍCIOS
Nos exercícios a seguir, avalie os dados clínicos e laboratoriais, e uti-lizando o roteiro sugerido, responda: a) Qual o distúrbio ácido-básico?b) Qual a compensação esperada? c) Qual o hiato iônico?
1) pH � 7,54; PCO2 � 53; HCO3� � 42; Na� � 141; K� � 3,1; Cl� � 88.
2) pH � 7,27; PCO2 � 26; HCO3� � 12; Na� � 142; K� � 3,6; Cl� � 100.
3) pH � 7,10; PCO2 � 20; HCO3� � 11; Na� � 140; K� � 3,8; Cl� � 110.
4) pH � 7,54; PCO2 � 32; HCO3� � 16; Na� � 141; K� � 3,1; Cl� �
88. Paciente ingeriu 6 g de ácido acetilsalicílico há 12 horas. Fre-qüência respiratória: 32 mrm.
5) pH � 7,18; PCO2 � 65; HCO3� � 48; Na� � 137; K� � 4,3; Cl� �
95. Paciente enfisematoso, internado com extensa broncopneumo-nia. Creatinina � 4,5 mg/dl.
REFERÊNCIAS BIBLIOGRÁFICAS
1. GUYTON, A.C.; HALL, J.E. Regulation of acid-base balance. In:Guyton, A.C.; Hall, J.E. (eds) Textbook of Medical Physiology, pp. 385-403, W.B. Saunders, 1996.
2. ROSE, B.D.; RENNKE, H.G. Acid-base physiology and metabolicalkalosis. In: Renal Pathophysiology — The Essentials, pp. 123-151,Williams & Wilkins.
3. VALTIN, H.; SCHAFER, J.A. H� balance. In: Valtin, H.; Schafer, J.A.Renal Function, pp.183-133.
4. RECTOR Jr, F.C. Renal acidification and ammonia production;chemistry of weak acids and bases; buffer mechanisms. Cap. 9, p.318. In: The Kidney. Eds. B.M. Brenner e F.C. Rector Jr. W.B. Saun-ders Co. 1976.
5. SCRIBNER, B.H. Teaching Syllabus for the Course on Fluid and EletrolyteBalance. University of Washington, 7th revision, 1969.
6. CHAPMAN, W.H. e col. The Urinary System. An integrated approa-ch. W.B. Saunders Co. 1973.
7. ROSE, B.D.; POST, T.W. Acids and bases. Up To Date, v.9, n.3, 2001.8. SEGURO, A.C.; MAGALDI, A.J.B.; HELOU, C.M.B.; MALNIC, G.;
Zatz, R. Processamento de água e eletrólitos pelos túbulos renais.In: Zatz, R. Fisiopatologia Renal, pp. 71-96, Atheneu, 2000.
9. BRENNER, B.; COE, F.L.; RECTOR, F.C. Acid base homeostasis. In:Brenner, B.; Coe, F.L.; Rector, F.C. Renal Physiology in Health andDisease, pp. 112-131. W.B. Saunders Co, 1987.
10. BIDANI, A.; DuBOSE, T.D, Jr. Cellular and whole-body acid-baseregulation. In: Arieff, A.I.; DeFronzo, R.A. (eds) Fluid, Electrolyte, andAcid-base Disorders, pp. 69-103. Churchill, Livingstone, 1995.
11. MALNIC, G. e MARCONDES, M. Fisiologia Renal. Pág. 125, Edart-São Paulo, 1972.
12. SWAN, R.C. e PITTS, R.F. Neutralization of infused acid by nephrec-tomized dogs. J. Clin. Invest., 34:205, 1955.
13. RECTOR Jr, F.C. Acidification of the urine. Cap. 14, p. 431. In: Han-dbook of Physiology. Ed. Geiger, S.R. Waverly Press, Inc., Baltimore,1973.
14. PITTS, R.F. e ALEXANDER, R.S. The nature of the renal tubular me-chanism for acidifying the urine. Am. J. Physiol., 144:239, 1945.
15. CLAPP, J.R. e col. Effects of unreabsorbed anions on proximal anddistal transtubular potentials in rats. Am. J. Physiol., 202:781, 1962.
16. SCHWARTZ, W.B. e col. Effect of chronic hypercapnia on eletrolyteand acid-base equilibrium. II. Recovery with special reference to theinfluence of chloride intake. J. Clin. Invest., 40:1238, 1961.
17. ROSE, B.D.; POST, T.W. Renal hydrogen excretion. Up To Date, v.9,n.3, 2001.
18. SHAPIRO, J.I.; KAEHNY, W.D. Pathogenesis and management ofmetabolic acidosis and alkalosis. In: Schrier, R.W. (ed). Renal andElectrolyte Disorders, pp. 161-210. Little, Brown, 1992.
19. ROSE, B.D.; RENNKE, H.G. Metabolic acidosis. In: Renal Pathophy-siology — The Essentials, pp. 152-168, Williams & Wilkins.
20. ZATZ, R.; MALNIC, G. Distúrbios do equilíbrio ácido-base. In: Zatz,R. Fisiopatologia Renal, pp. 209-244, Atheneu, 2000.
21. ROSE, B.D.; POST, T.W.; NARINS, R.G. Causes of lactic acidosis. UpTo Date, v.9, n.3, 2001.
22. ROSE, B.D. D-Lactic acidosis. Up To Date, v.9, n.3, 2001.23. ROSE, B.D. Alcoholic and fasting ketoacidosis. Up To Date, v.9, n.3, 2001.24. NARINS, R.G. Introduction to metabolic acidosis, Part 1, 2 and 3.
American Society of Nephrology Board Review Course, sep. 1998(www.hdcn.com).
25. PRESTON, R.A. Metabolic Acidosis. In: Acid-base, Fluids, andElectrolytes Made Ridiculously Simple, pp. 97-115, MedMaster Inc., 1997.
26. ADROGUÉ, H.J.; MADIAS, N.J. Disorders of acid-base balance. In: Schrier,R.W. Atlas of Kidney Diseases (on line - http://www.kidneyatlas.org/),Blackwell Science, 1999.
27. POST, T.W.; ROSE, B.D. Approach to the patient with metabolicacidosis. Up To Date, v.9, n.3, 2001.
28. OH, M.S. e CARROL, H.J. Current concepts: the anion gap. New Engl.J. Med., 297:814, 1977.
29. ADROGUÉ, H.J.; MADIAS, N.E. Management of life-threateningacid-base disorders. Part I. N. Engl. J. Med., 38(1):26-34, 1998.
30. ROSE, B.D. Treatment of metabolic acidosis. Up To Date, v.9, n.3, 2001.31. GARELLA, S. e col. Severity of metabolic acidosis as a determinant
of bicarbonate requirements. New Engl. J. Med., 289:121, 1973.32. POSNER, J.B. e PLUM, F. Spinal fluid pH and neurologic symptoms
in systemic acidosis. New Engl. J. Med., 277:605, 1967.33. MATTAR, J.A. Bicarbonato de sódio na parada cardiorrespiratória.
Rev. Soc. Cardiol. Est. São Paulo, 1997;1.34. WILSON, R.F. e col. Severe alkalosis in critically ill surgical patients.
Arch Surg., 105:197, 1972.35. BLACK, R.M.; ALFRED, H.J.; FAN, P.Y.; STOFF, J.S. Metabolic alka-
losis. In: Rose & Black’s Problems in Nephrology, pp. 64-73, Little, Bro-wn and Co., 1996.
36. GALLA, J.H. Metabolic alkalosis. J. Am. Soc. Nephrol., 11:369-375,2000.
37. DuBOSE, T., Jr. Metabolic alkalosis. American Society of NephrologyBoard Review Course, sep. 1998 (www.hdcn.com).
38. ADROGUÉ, H.J.; MADIAS, N.E.; Management of life-threateningacid-base disorders. Part 2. N. Engl. J. Med., 38(2):107-111, 1998.
39. KNUTSEN, O.H. New method for administration of hydrochloricacid in metabolic alkalosis. Lancet, 2:953, 1983.
40. RASTEGAR, A. e THIERS, S.O. Physiologic consequence and bodilyadaptations to hyper- and hypocapnia. Chest., 62:283, 1972.
41. BRACKETT, N.C. e col. Carbon dioxide titration in man. New Engl.J. Med., 272:6, 1965.
42. SCHWARTZ, W.B. e col. The response of extracellular hydrogen ionconcentration to grade degree of chronic hypercapnia: the physio-logic limit of the defense of pH. J. Clin. Invest., 44:291, 1965.
43. KAEHNY, W.D. Pathogenesis and management of respiratory andmixed acid-base disorders. In: Schrier, R.W. (ed) Renal and Electro-lyte Disorders, pp. 211-230. Little, Brown, 1992.
44. MCCURDY, D.K. Mixed metabolic and respiratory acid-basedisturbance: diagnosis and treatment. Chest, 62:35S, 1972.
45. FALL, P.A stepwise approach to acid-base disorders. Postgrad. Med.,107(3):249-263, 2000.
46. PRESTON, R.A. Mixed acid-base disorders. In: Acid-base, Fluids, andElectrolytes Made Ridiculously Simple, pp. 125-143, MedMaster Inc.,1997.
47. ARBUS, G.S. An in vivo acid-base nomogram for clinical use. Can.Med. Assoc. J., 109:291, 1973.
48. FENCL, V.; JABOR, A.; KAZDA, A.; FIGGE, J. Diagnosis of metabolicacid-base disturbances in critically ill patients. Am. J. Respir. Crit. CareMed., 162(6):2246-51, 2000.
49. MAKOFF, D. L. Acid-base metabolism. Cap. 8, p. 297. In: Clinical Di-sorders of Fluid and Electrolyte Metabolism. Eds. M.H. Maxwell e C.R.Kleeman. McGraw-Hill Book Co., 1972.
50. PITTS, R.F. Physiology of the Kidney and Body Fluids, 3rd Edition. YearBook Medical Publishers Inc., 1974.
51. SELDIN, D.W e RECTOR Jr, F.C. The generation and maintenanceof metabolic alkalosis. Kidney Int., 1:306, 1972.

188 Metabolismo Ácido-Básico
ENDEREÇOS RELEVANTES NA INTERNET
http://www.kidneyatlas.org/book1/adk1 – 06.pdf —Excelente capítulo do Atlas de Doenças Renais on line deRobert Schrier.http://www.biology.arizona.edu/biochemistry/problem – sets/medph/01q.html — Tutorial muito in-teressante com perguntas e respostas comentadas.http://perfline.com/cursos/cursos/acbas/acbas.htm —Revisão geral do equilíbrio ácido-básico e testes.
RESPOSTAS DOS EXERCÍCIOS
1) pH � 7,54; PCO2 � 53; HCO3� � 42.
a) Distúrbio ácido-básico: pH alto, bicarbonato alto, PCO2 alta � al-calose metabólica.
b) Compensação esperada para a alcalose metabólica é a hipoventi-lação alveolar, com aumento na PCO2, como se observa nesta ga-sometria. Aplicando a fórmula para verificar se a compensação daalcalose metabólica é adequada:(PCO2 � 40 � 0,7 � [HCO3
� atual � HCO3� normal]) � 53 � 40 �
0,7 � (42 � 24) � 53 � 52,6. Portanto, a compensação está dentrodo que era esperado, e se trata de um distúrbio simples.
c) Anion gap � Na� � (HCO3� � Cl�) � AG � 11.
2) pH � 7,27; PCO2 � 26; HCO3� � 12; Na� � 142; K� � 3,6; Cl� � 100.
a) Distúrbio ácido-básico: pH baixo, bicarbonato baixo, PCO2 baixa� acidose metabólica.
b) A compensação esperada para a acidose metabólica é a hiperventi-lação alveolar, com diminuição na PCO2, como se observa nestagasometria. Aplicando a fórmula para verificar se a compensaçãoda acidose metabólica é adequada: PCO2 � 1,5 � [HCO3
�] � 8 �26 � (1,5 � 12) � 8 � 26 � 26. Portanto, a compensação está ade-quada: a acidose estimulou a hiperventilação, reduzindo a PCO2 aonível que era esperado.
c) Anion gap � Na� � (HCO3� � Cl�) � AG � 142 – (12 � 100) �
AG � 30. O anion gap está aumentado. Verificar quais as causasprováveis.
3) pH � 7,10; PCO2 � 32; HCO3� � 11; Na� � 140; K� � 3,8; Cl� � 110.
a) Distúrbio ácido-básico: pH baixo, bicarbonato baixo, PCO2 baixa� acidose metabólica.
b) Compensação esperada para a acidose metabólica é a hiperventi-lação alveolar, com diminuição na PCO2, como se observa nestagasometria. Aplicando a fórmula para verificar se a compensaçãoda alcalose metabólica é adequada: PCO2 � 1,5 � [HCO3
�] � 8 �32 � (1,5 � 11) � 8 � 24,5 � 17. O mecanismo de compensaçãofoi insuficiente e não reduziu a PCO2 aos níveis esperados. Por-tanto, trata-se de uma acidose mista (acidose metabólica � acido-se respiratória).
c) Anion gap � Na� � (HCO3� � Cl�) � AG � 140 – (11 � 110) �
AG � 19. O anion gap está normal. Verifique as causas prováveis.
4) pH � 7,54; PCO2 � 32; HCO3� � 16; Na� � 141; K� � 3,1; Cl� � 88.
Paciente ingeriu 6 g de ácido acetilsalicílico há 12 horas. Freqüênciarespiratória: 32 mrm.a) Distúrbio ácido-básico: pH alto, bicarbonato baixo, PCO2 baixa �
alcalose respiratória.b) Compensação esperada para a alcalose respiratória é a eliminação
de bicarbonato e retenção de ácido pelo rim. Aplicando a fórmulade alcalose respiratória (aguda) para verificar se a compensação éadequada: [HCO3
�] deveria diminuir 2 mEq para cada 10 mmHgde queda na PCO2. Como a PCO2 caiu 8 mmHg, a concentraçãode bicarbonato deveria cair para cerca de 22,4 mEq/L. Porém, aqueda no bicarbonato foi superior, chegando a 16 mEq/L. O me-canismo de compensação foi inadequado, e conclui-se que estepaciente apresenta um distúrbio ácido-básico misto: alcalose res-piratória e acidose metabólica.
c) AG � Na� � (HCO3� � Cl�) � AG � 37.
5) pH � 7,18; PCO2 � 65; HCO3� � 28; Na� � 137; K� � 4,3; Cl� � 95.
Paciente enfisematoso, internado com extensa broncopneumonia.Creatinina � 4,5 mg/dl.a) Distúrbio ácido-básico: pH baixo, bicarbonato alto, PCO2 alta �
acidose respiratória.b) Compensação esperada para a acidose respiratória é a retenção de
bicarbonato pelo rim. Aplicando a fórmula de acidose respirató-ria (crônica) para verificar se a compensação é adequada: [HCO3
�]Deve aumentar 3,5 mEq para cada 10 mmHg de aumento na PCO2.Como a PCO2 aumentou 25 mmHg, o bicarbonato deveria estarem torno de 32,75. Observe que o bicarbonato elevou-se pouco,frente ao que era esperado, talvez devido ao comprometimento defunção renal que este paciente apresenta. Então, o distúrbio apre-sentado por ele é uma acidose mista (metabólica � respiratória).
c) AG � Na� � (HCO3� � Cl�) � AG � 14.

Capítulo
12Metabolismo do Potássio
Miguel Carlos Riella e Maria Aparecida Pachaly
INTRODUÇÃO
DISTRIBUIÇÃO DO POTÁSSIO NO ORGANISMO
MÉTODOS DE AVALIAÇÃO DA QUANTIDADE DE
POTÁSSIO NO ORGANISMO
Concentração plasmática do potássio
Determinação do potássio total com 40K
Determinação do potássio trocável
Outros métodos
INTERPRETAÇÃO DO POTÁSSIO PLASMÁTICO
FATORES QUE AFETAM A DISTRIBUIÇÃO
TRANSCELULAR DE POTÁSSIO
BALANÇO DO POTÁSSIO
Ingesta e excreta
Excreção renal de potássio
Transporte tubular renal de potássio
Canais de potássio
Túbulo proximal
Ramo descendente da alça de Henle (RDAH)
Ramo ascendente da alça de Henle (RAAH)
Túbulo distal (TD)
Reciclagem medular de potássio
Fatores que influenciam a secreção de potássio nos
túbulos distal e coletor
SISTEMAS HORMONAIS ATUANTES NA HOMEOSTASIA
DO POTÁSSIO
Insulina
Glucagon
Catecolaminas
Hormônios adrenocorticais
Como age a aldosterona?
ADAPTAÇÃO A NÍVEIS ELEVADOS DE POTÁSSIO
Adaptação renal ao potássio
Adaptação extra-renal ao potássio
PAPEL DO BALANÇO ÁCIDO-BÁSICO
HOMEOSTASIA DO POTÁSSIO NA INSUFICIÊNCIA RENAL
Papel do sistema renina-angiotensina-aldosterona
Excreção gastrintestinal de potássio
Tolerância celular ao potássio
AÇÃO DOS DIURÉTICOS
DISTÚRBIOS CLÍNICOS DO METABOLISMO DO POTÁSSIO
Depleção de potássio (hipocalemia)
Causas de hipocalemia
Manifestações clínicas
Diagnóstico diferencial
Tratamento da hipocalemia
Cálculo do déficit de potássio
Reposição de potássio em algumas situações especiais
Excesso de potássio (hipercalemia)
Causas de hipercalemia
Diagnóstico diferencial
Manifestações clínicas
Tratamento da hipercalemia
EXERCÍCIOS
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET
RESPOSTAS DOS EXERCÍCIOS

190 Metabolismo do Potássio
INTRODUÇÃO
O potássio é o cátion intracelular mais abundante e suainfluência se faz sentir em vários processos metabólicos dacélula. A função neuromuscular e os potenciais de mem-brana dependem de maneira crítica da relação entre a con-centração de potássio intracelular e extracelular.
Em vista disso, os mecanismos que regulam a concen-tração de potássio devem ser bastante precisos. Embora aconcentração de potássio no líquido extracelular seja redu-zida, quando comparada com a concentração intracelular,a variação é pequena (3,5 a 5,0 mEq/L). As repercussõesclínicas de pequenas variações nesta concentração extra-celular de potássio são, no entanto, dramáticas. Cabe ao rimgrande parte da responsabilidade pelo controle da concen-tração de potássio.
DISTRIBUIÇÃO DO POTÁSSIO NOORGANISMO
O potássio total do corpo está em torno de 55 mEq/kg,e portanto, num indivíduo de 70 kg, há aproximadamente3.500 mEq de potássio, sendo pelo menos 90% intracelula-res1,2 e 10% extracelulares (Fig. 12.1). Porém, apenas 2% dopotássio extracelular se encontram no plasma e fluido in-tersticial (50-70 mEq); o restante encontra-se no tecido ós-seo, de onde pode ser mobilizado lentamente.3
A maior parte do potássio intracelular (em torno de3.000 mEq) está no interior das células musculares, o quenão implica um acúmulo relativo de potássio no músculo,mas apenas reflete a preponderância da massa muscularem relação à massa corporal.
A acentuada diferença de concentração entre os espa-ços intracelular e extracelular é mantida pela bomba iôni-ca sódio-potássio-ATPase (Na-K-ATPase), que ativamen-
te transporta o potássio para dentro e o sódio para fora dascélulas.4
O papel do potássio intracelular com relação à água éanálogo ao papel do sódio no líquido extracelular, isto é,cada um é o principal determinante da osmolalidade doseu compartimento e a quantidade absoluta de cada umestá relacionada com o volume do compartimento intra-ou extracelular.5
A facilidade com que se pode determinar a concentra-ção de sódio no líquido extracelular contrasta com as difi-culdades existentes na determinação direta do potássiointracelular.
MÉTODOS DE AVALIAÇÃO DAQUANTIDADE DE POTÁSSIO NO
ORGANISMO
Concentração Plasmática do PotássioDemonstrou-se que há uma correlação entre a quantidade
de potássio no plasma e a quantidade total de potássio noorganismo de um indivíduo normal.6 Embora alguns es-tudos não tenham mostrado uma correlação entre a con-centração plasmática de potássio e o potássio total do orga-nismo, há muita evidência na literatura que demonstra quea concentração plasmática de potássio reflete a quantida-de total de potássio no organismo.5
Determinação do Potássio Total com 40KA administração de potássio radioativo (40K) permite a
detecção externa de toda a radiação emitida pelo 40K pro-veniente do corpo.7 Por este método, chegou-se à conclu-são de que o potássio total do homem está em torno de 55mEq/kg, e o da mulher, em torno de 49 mEq/kg.1 A dife-
Fig. 12.1 Distribuição do potássio num adulto pesando 70 kg. Observe que a maior parte do potássio está contida nas células mus-culares. (Obtido de Black, D.A.K.1)

capítulo 12 191
rença deve-se ao fato de as mulheres possuírem maiorquantidade de tecido adiposo e menor massa muscular.
Determinação do Potássio Trocável*
O potássio trocável representa 92 a 99% do potássio to-tal e refere-se ao potássio que se mobiliza com mais facili-dade. O método baseia-se na administração de uma quan-tidade conhecida de 42K, e, após um período de equilíbrio,a concentração de 42K, multiplicada pela dose administra-da, fornece o potássio trocável.
Outros Métodos
A determinação do potássio total ou trocável não nospermite saber a concentração intracelular de potássio. Paraisto haveria necessidade de determinar a água do organis-mo e o volume do compartimento extracelular.5 Estas de-terminações são difíceis e não muito precisas. Felizmente,existem outras maneiras de expressar os dados de potás-sio: o potássio do organismo pode ser relacionado com opeso do indivíduo (v. Quadro 12.2), com a sua massa cor-poral sem gordura e com a altura e excreção de creatinina.
Além disso, há métodos de análise tissular. A biópsiade músculo é útil, pois o músculo contém aproximadamen-te 60% do potássio do organismo, e uma estimativa dopotássio muscular total dá uma idéia grosseira do potás-sio total do organismo.5
A determinação do potássio intracelular em eritrócitose leucócitos também tem sido utilizada para a estimativado potássio total.
Os vários métodos existentes refletem as dificuldadesencontradas pelos investigadores.
INTERPRETAÇÃO DO POTÁSSIOPLASMÁTICO
Scribner e Burnell desenvolveram a idéia de que deple-ção e excesso de potássio devem ser definidos em face dasalterações do potássio total do organismo, tomando-se umponto de referência.8 Os autores acreditavam que um ponto
de referência era essencial, pois que alterações no potássiototal, per se, não tinham significado. Exemplificavam como paciente em jejum, que perde potássio mas não se tornadeficiente em potássio porque, ao mesmo tempo, destróimassa protéica (devido ao jejum). O ponto de referênciaescolhido foi denominado capacidade total do potássio (totalpotassium capacity)§ e refere-se à soma de todos os ânions eoutros grupos químicos fora do líquido extracelular e ca-pazes de reter íons K� ou ligarem-se a estes. A capacidadedo potássio teria vários componentes (v. Fig. 12.2). As cé-lulas musculares contribuiriam com a maior parcela, alémdo fígado, glicogênio, hemácias e ossos.
Desta maneira, define-se depleção de potássio como umadiminuição do potássio total em relação à capacidade dopotássio. Exemplo: depleção de potássio devido a perdasgastrintestinais ou renais, sem ingesta adequada (v. Fig.12.2).
*A determinação da massa de eletrólitos no corpo está intimamente rela-cionada com a determinação do volume dos líquidos no corpo. Quandose administra sódio ou potássio radioativo, eles são diluídos pelos isóto-pos, que ocorrem normalmente no corpo. Alguns eletrólitos do corpoestão em solução e se equilibram rapidamente com os eletrólitos marca-dos por substâncias radioativas. Outros eletrólitos estão incorporados emfáscias, tendões, ossos etc. e se equilibram mais lentamente com os ele-trólitos marcados. Isto dificulta o cálculo da massa total de determinadoeletrólito. A massa de eletrólito que se equilibra ou se troca rapidamentecom o eletrólito marcado é denominada massa trocável ou permutável. Daías expressões sódio ou potássio trocável, de troca ou permutável. É óbvio quea massa trocável será sempre inferior à massa total do organismo.7
Quadro 12.1 Alterações no potássio sérico
Distribuição transcelular alterada1. Ácido-básico
a. Acidose: para cada 0,1 unidade de pH que cai, opotássio se eleva em 0,6 mEq/L
b. Alcalose: para cada 0,1 unidade de pH que sobe, opotássio diminui em 0,1 mEq/L
2. Insulina3. Aldosterona4. Agentes �-adrenérgicos (epinefrina)
Alteração das reservas de potássio1. Depleção — 1 mEq/L de redução para um déficit de
200-300 mEq2. Retenção — 1 mEq/L de aumento reflete um excesso
de 200 mEq
Modificado de Tannen, R.L. Manual of Nephrology. Edit. Robert Schrier.Little, Brown and Co., 1981.
Quadro 12.2 Depleção de potássio: algumas causasgastrintestinais
DiarréiaFezes líquidas: cólera, síndrome de Zollinger-EllisonFezes formadas: esteatorréia, pós-gastrectomia
Secreção de tumores: adenoma vilosoExsudato inflamatório: colite ulcerativaVômito e diarréia: gastroenteriteVômito: estenose pilóricaAspiração gástrica contínuaFístulas: biliar, pancreática, gastrocólicaOutras: abuso de purgativos, enemas
Modificado de Black, D.A.K.1
§O termo capacidade total do potássio talvez não traduza com fidelidade osignificado do termo total potassium capacity.
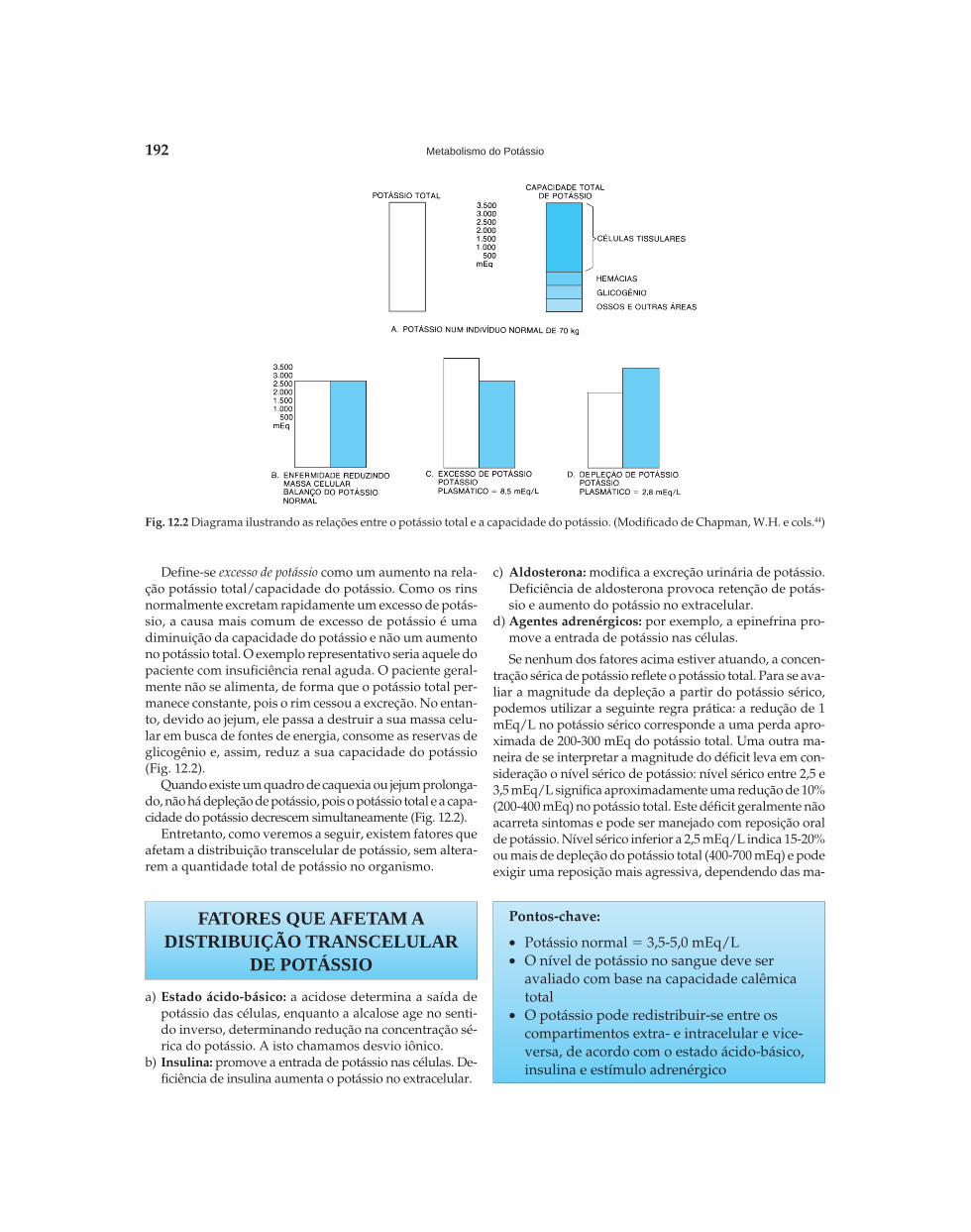
192 Metabolismo do Potássio
Define-se excesso de potássio como um aumento na rela-ção potássio total/capacidade do potássio. Como os rinsnormalmente excretam rapidamente um excesso de potás-sio, a causa mais comum de excesso de potássio é umadiminuição da capacidade do potássio e não um aumentono potássio total. O exemplo representativo seria aquele dopaciente com insuficiência renal aguda. O paciente geral-mente não se alimenta, de forma que o potássio total per-manece constante, pois o rim cessou a excreção. No entan-to, devido ao jejum, ele passa a destruir a sua massa celu-lar em busca de fontes de energia, consome as reservas deglicogênio e, assim, reduz a sua capacidade do potássio(Fig. 12.2).
Quando existe um quadro de caquexia ou jejum prolonga-do, não há depleção de potássio, pois o potássio total e a capa-cidade do potássio decrescem simultaneamente (Fig. 12.2).
Entretanto, como veremos a seguir, existem fatores queafetam a distribuição transcelular de potássio, sem altera-rem a quantidade total de potássio no organismo.
FATORES QUE AFETAM ADISTRIBUIÇÃO TRANSCELULAR
DE POTÁSSIO
a) Estado ácido-básico: a acidose determina a saída depotássio das células, enquanto a alcalose age no senti-do inverso, determinando redução na concentração sé-rica do potássio. A isto chamamos desvio iônico.
b) Insulina: promove a entrada de potássio nas células. De-ficiência de insulina aumenta o potássio no extracelular.
c) Aldosterona: modifica a excreção urinária de potássio.Deficiência de aldosterona provoca retenção de potás-sio e aumento do potássio no extracelular.
d) Agentes adrenérgicos: por exemplo, a epinefrina pro-move a entrada de potássio nas células.
Se nenhum dos fatores acima estiver atuando, a concen-tração sérica de potássio reflete o potássio total. Para se ava-liar a magnitude da depleção a partir do potássio sérico,podemos utilizar a seguinte regra prática: a redução de 1mEq/L no potássio sérico corresponde a uma perda apro-ximada de 200-300 mEq do potássio total. Uma outra ma-neira de se interpretar a magnitude do déficit leva em con-sideração o nível sérico de potássio: nível sérico entre 2,5 e3,5 mEq/L significa aproximadamente uma redução de 10%(200-400 mEq) no potássio total. Este déficit geralmente nãoacarreta sintomas e pode ser manejado com reposição oralde potássio. Nível sérico inferior a 2,5 mEq/L indica 15-20%ou mais de depleção do potássio total (400-700 mEq) e podeexigir uma reposição mais agressiva, dependendo das ma-
Fig. 12.2 Diagrama ilustrando as relações entre o potássio total e a capacidade do potássio. (Modificado de Chapman, W.H. e cols.44)
Pontos-chave:
• Potássio normal � 3,5-5,0 mEq/L• O nível de potássio no sangue deve ser
avaliado com base na capacidade calêmicatotal
• O potássio pode redistribuir-se entre oscompartimentos extra- e intracelular e vice-versa, de acordo com o estado ácido-básico,insulina e estímulo adrenérgico

capítulo 12 193
nifestações clínicas. É difícil imaginar-se o déficit quando onível sérico é inferior a 1,8-2,0 mEq/L. Em caso de hiperca-lemia, um aumento de 1 mEq/L no potássio sérico refletepelo menos 200 mEq de excesso de potássio total.
BALANÇO DO POTÁSSIO
Ingesta e ExcretaNormalmente, a quantidade diária de potássio ingeri-
da varia entre 50 e 150 mEq. A quantidade de potássioexcretada pela pele através do suor é pequena, cerca de 16a 18 mEq/L. A excreção de potássio nas fezes é da ordemde 5 a 10 mEq por dia, mas perdas consideráveis ocorremnas diarréias, esteatorréias e com uso de laxantes.1
Em vista da pequena excreção cutânea e intestinal depotássio, é óbvio que a maior responsabilidade pela excre-ção do potássio cabe ao rim.2
Excreção Renal de PotássioA excreção renal de potássio depende de três processos:a) taxa de filtração glomerular do potássio (que é igual
à taxa de filtração glomerular � concentração plasmáticade potássio); b) taxa de transporte de potássio do lúmentubular para o sangue (reabsorção), e c) taxa de transportedo potássio do sangue para o lúmen tubular (secreção). Emcondições habituais, a taxa de filtração do potássio é man-tida constante, e a maior parte do potássio excretado nãoresulta do processo de filtração glomerular, e sim do pro-cesso de secreção tubular. Em circunstâncias em que a taxade filtração glomerular está reduzida, como a insuficiên-cia renal, pode haver acúmulo de potássio com graves re-percussões clínicas.2
De maneira geral, as porções iniciais do nefro reabsor-vem potássio e as mais distais o secretam. No entanto, al-
guma excreção também ocorre nos segmentos proximais,enquanto alguma reabsorção ocorre no ducto coletor. Cercade 65% do potássio filtrado são reabsorvidos no túbuloproximal, e 25-30% na alça de Henle, principalmente noramo ascendente espesso. Como estes segmentos tubula-res mais proximais executam principalmente processos dereabsorção de potássio, a maior parte da variação em suaexcreção é causada por ajustes na secreção nos segmentostubulares mais distais (como os túbulos distais e túbuloscoletores).2
Transporte TubularRenal de Potássio
CANAIS DE POTÁSSIOSabe-se atualmente que o movimento passivo de íons e
água através de membranas biológicas é facilitado por umgrupo de proteínas conhecidas como canais. Canal de íoné definido como uma proteína transmembrana com umorifício ou poro através do qual os íons podem passar poreletrodifusão.
Canais de potássio (K�) constituem um grupo de prote-ínas de membrana que facilitam o movimento passivo (gui-ado pelo gradiente eletroquímico para K�) de K� atravésde membranas celulares. Um ou mais tipos de canais deK� podem ser detectados em virtualmente todas as célu-las de mamíferos. Os canais de K� que se abrem e fechamem resposta a alterações na voltagem da membrana sãochamados de canais voltagem-dependentes (Kv). Umasubclasse de canais Kv necessita de cálcio para ativação esão conhecidos como maxicanais K�. Recentemente veri-ficou-se que canais Kv têm um papel crucial na regulaçãoda contração vascular da musculatura lisa e portanto naresistência vascular periférica e pressão arterial.
Os íons K� atravessam as membranas fundamentalmen-te por dois mecanismos: via canais ou carregadores. A forçapropulsora do movimento de potássio através do canal éa diferença de potencial eletroquímico. O transporte depotássio mediado por carregador envolve a ligação com umaproteína específica carregadora, e a alteração na conforma-ção desta proteína é necessária para atravessar a barreiracelular.
Embora a importância fisiológica de canais Kv não possaser imediatamente óbvia no epitélio renal, está claro quevários destes genes se expressam no rim e que os Kv po-dem ter um papel na secreção de potássio no ducto cole-tor cortical e na reciclagem de K na medula interna.9
TÚBULO PROXIMALApós a filtração, 60-65% do potássio no líquido tubular
são reabsorvidos no túbulo contornado proximal. O túbu-lo proximal funciona como um epitélio de baixa resistên-cia, onde ocorre uma extensa reabsorção de água, sódio,potássio e outros íons. Duas forças passivas promovem
Fig. 12.3 Desvio iônico do potássio em presença de acidose e al-calose. Na acidose, para cada 0,1 de queda no pH, há uma eleva-ção de 0,6 mEq/L no potássio sérico. Na alcalose, para cada 0,1de aumento no pH, o nível do potássio sérico cai 0,1 mEq/L.
ACIDOSE�0,1 pH ��K 0,6 mEq/L
ALCALOSE�0,1 pH ��K 0,1 mEq/L
H�H� H�
H�H�
H�H�
H�H�
K�
K�K�
K�
K�
K�
K� K� K�K�
K�K�

194 Metabolismo do Potássio
reabsorção transepitelial de potássio: a) o movimento delíquido através de junções intercelulares provoca um ar-rasto de potássio no mesmo sentido (solvent drag effect); b)uma força eletroquímica, determinada por uma diferençade potencial transepitelial que varia de valores positivosno túbulo proximal, favorecendo a reabsorção, a valoresnegativos nos segmentos distais (túbulo coletor), favore-cendo a secreção de potássio. Desta forma, ocorre uma re-absorção passiva por eletrodifusão.4
Além destas forças passivas, há evidência de uma viatranscelular ativa para reabsorção de potássio. Esta infor-mação deriva de experimentos em que a reabsorção de lí-quido e sódio é marcadamente reduzida e a reabsorção depotássio continua.
A saída de potássio da célula para o líquido peritubu-lar e capilar peritubular é exclusivamente passiva. Istoocorre pelo gradiente eletroquímico e pela alta permeabi-lidade da membrana celular baso-lateral.
RAMO DESCENDENTE DA ALÇADE HENLE (RDAH)
Atualmente, acredita-se que o potássio seja secretado nolíquido tubular neste segmento do nefro. Jamison e cols.mostraram que, no final deste segmento, a quantidade depotássio excede a filtrada e concluíram que este potássiosecretado provém do potássio absorvido no ramo ascen-dente da alça de Henle (v. a seguir) e que o ritmo de secre-ção depende do gradiente existente entre o interstício me-dular e o lúmen tubular. Portanto, o mecanismo de trans-porte parece ser passivo.10
RAMO ASCENDENTE DA ALÇADE HENLE (RAAH)
Está bem estabelecido que a reabsorção de potássio atra-vés da membrana luminal se faz contra um gradiente ele-troquímico e através de um mecanismo de co-transporte,de tal forma que um Na�, um K� e dois Cl� são translocadossimultaneamente. Este processo eletricamente neutro cons-titui o transporte ativo secundário de potássio. A forçapromotora origina-se da extrusão ativa de sódio através damembrana baso-lateral da célula. A saída de potássio dacélula se faz pela membrana baso-lateral e pode ser pordifusão através de canais de potássio ou acoplado a íonscloro via um co-transportador KCl.
TÚBULO DISTAL (TD)A porção do túbulo distal responsável pela secreção de
potássio parece estar restrita à parte final do segmentoentre a mácula densa e a confluência de dois túbulos dis-tais: a parte mais distal do TD e o túbulo coletor cortical. Aparte convoluta do TD (parte inicial) não participa funcio-nalmente do transporte de potássio.
Há dois tipos de células no túbulo distal que participamdo transporte de potássio: as células principais (claras), maisnumerosas e responsáveis pela reabsorção e secreção depotássio, e as células intercaladas (escuras), que regulam areabsorção de potássio e a secreção de íons H�.4
A célula principal transporta o K� através da membra-na baso-lateral pela atividade Na-K-ATPase. O movimen-to preferencial do K� se faz para o lúmen, e isto ocorre pelaeletrodifusão de sódio do lúmen para a célula pela mem-brana apical. A secreção de potássio pode ser poderosa-mente influenciada por qualquer coisa que altere a entra-da de sódio (íons) na célula através da membrana apical.A aldosterona aumenta a condução de sódio pela membra-na apical, aumentando secundariamente a secreção e asaída de potássio.
Um segundo tipo de reabsorção de potássio está nosductos coletores medulares. É possível que o transporte depotássio e hidrogênio esteja ligado neste local. A estimu-lação da secreção de H� aumenta o potencial positivo dolúmen, aumentando a reabsorção passiva de potássio, evice-versa.
RECICLAGEM MEDULAR DE POTÁSSIOHá evidência recente de que é diferente o transporte de
potássio entre os nefros superficiais (corticais) e os profun-dos (justamedulares). A base da alça de Henle contém maisK� do que está presente no filtrado glomerular. Há evidên-cia de que este K� adicionado à alça de Henle provém doducto coletor medular. Desta forma, o K sofre uma recicla-gem na medula renal, similar ao que ocorre com a uréia. Aalta concentração medular de K origina um gradiente quefavorece a secreção passiva de potássio na pars recta e ramofino descendente da alça de Henle. A reciclagem de K
Fig 12.4 Reabsorção tubular de potássio nos diferentes segmen-tos do nefro. Adaptado de DeFronzo, R.A.; Smith, J.D.47
CARGA FILTRADA600-700 MEQ/DIA
REABSORÇÃO DE K�
60-70% SECREÇÃO DE K�
INICIAL MÉDIA FINAL
SECREÇÃODE K�
Túbulo proximalREABSORÇÃO
20-30%
K�
Túbulocoletor
EXCREÇÃOURINÁRIA90 mEq/dia
Túbulo distal

capítulo 12 195
proporciona ótimas condições para o nefro distal excretarK. Quando ocorre uma alta ingesta de K, a urina deve ex-cretar o excesso. Assim, a alta concentração de K no ductocoletor não se dissipa para o interstício devido à alta con-centração de K na medula.
FATORES QUE INFLUENCIAM A SECREÇÃODE POTÁSSIO NOS TÚBULOS DISTAL ECOLETOR
a) Ingesta de potássio: a secreção de potássio aumentaquando o potássio dietético é elevado e diminui quan-do este é reduzido. O efeito do aporte de potássio sobrea secreção é mediado por alterações na concentraçãoplasmática de potássio, aumentando ou diminuindo aatividade da enzima sódio-potássio-ATPase da mem-brana baso-lateral. Além disso, a elevação dos níveis depotássio estimula a secreção de aldosterona, que aumen-ta a secreção de potássio.2,4
b) Fluxo de líquido tubular distal e concentração intra-celular: se o fluxo é maior, aumenta a secreção de po-tássio.3 Porém, a secreção depende também da concen-tração intracelular de potássio: mesmo que haja umaumento de fluxo tubular, se a concentração intracelu-lar de potássio for baixa, não há aumento em sua secre-ção.4,11
c) Aporte de sódio aos segmentos distais: como já men-cionamos, a concentração de sódio intraluminal a essenível pode potencialmente modificar o ritmo de secre-ção de potássio. A entrada de sódio pela membrana lu-minal das células principais diminui a negatividade in-tracelular, favorecendo a secreção de potássio. Com oaumento da concentração intracelular de sódio, aumentatambém a atividade da sódio-potássio-ATPase baso-la-teral, o que aumenta o potássio intracelular e aumentasua secreção. Então, quando a concentração de sódio doTCD aumenta, a secreção de potássio também aumen-ta.12 Isto explica por que situações em que existe aumen-to da oferta de sódio às porções finais do túbulo distal(por exemplo, uso de diuréticos) podem levar a um dé-ficit de potássio.3 Quando se remove o sódio do lúmen,a secreção de potássio diminui.12
d) Aldosterona: é um hormônio produzido pelas glându-las adrenais; influencia diretamente alguns dos princi-pais determinantes da secreção de potássio, tais comoconcentração de potássio intracelular, permeabilidadeda membrana luminal ao potássio e diferença de poten-cial transepitelial4 (v. adiante).
e) Ânions não absorvíveis na luz tubular: o gradientetransepitelial distal é lúmen-negativo devido à contínuareabsorção ativa de sódio; a presença de ânions comobicarbonato, sulfato e fosfato ajuda a manter negativa adiferença de potencial elétrico entre luz e interstício,favorecendo a secreção de potássio. Quanto mais nega-tivo o gradiente, maior é a secreção de potássio.3,4
f) Modificações agudas no estado ácido-básico: a alcalo-se aguda aumenta e a acidose aguda diminui a secre-ção de potássio. É possível que com elevações na con-centração de íons H� (acidose) haja diminuição da ati-vidade da Na-K-ATPase das células, gerando acúmulode potássio no extracelular. O pH ácido pode tambémaumentar a permeabilidade celular à saída de potássio.Nas células principais, isto ocasiona redução na secre-ção, sendo o resultado final uma retenção de potássio.Nas alcaloses, o movimento de potássio é do extracelu-lar para o intracelular, levando à hipocalemia.3,4
Pontos-chave:
• A principal forma de excreção do potássio éatravés de secreção nos segmentos maisdistais do nefro
• A excreção renal de potássio sofre ainfluência dos níveis plasmáticos do íon,aldosterona, fluxo tubular e estado ácido-básico
SISTEMAS HORMONAISATUANTES NA HOMEOSTASIA
DO POTÁSSIO
A regulação da concentração do potássio extra- e intra-celular e da sua excreção pelo rim parece estar sob a influ-ência de vários sistemas hormonais. E eles se inter-relaci-onam de maneira a garantir a existência de um mecanis-mo de segurança contra falhas. Se ocorrer elevação dosníveis de potássio, todo o sistema é acionado, procurandoreduzir sua concentração.
Insulina
A insulina provoca a entrada de potássio para den-tro das células, de modo independente de sua ação so-bre o metabolismo da glicose.3 Este efeito se deve à ca-pacidade da insulina de ativar a Na-K-ATPase, aumen-tando a concentração intracelular de potássio e diminu-indo a de sódio. A interação insulina-receptor tambémativa um contratransportador Na�-H�, que resulta ementrada de sódio na célula e que estimula ainda mais a Na-K-ATPase, com os efeitos já descritos. Além disso, a hiper-calemia aguda estimula a liberação de insulina pelo pân-creas.3,13
Há muito tempo já se reconhecia que a administraçãode glicose reduzia a concentração de potássio no plasma ena urina. Hoje, sabe-se que a insulina liberada pela hiper-glicemia promove a transferência de potássio para muitos

196 Metabolismo do Potássio
tecidos, sobretudo fígado e músculo esquelético. Esta ca-pacidade da insulina em transferir potássio para dentro dascélulas pode ser clinicamente observada durante o trata-mento da cetoacidose diabética e tem uma extraordináriaimportância prática na terapêutica da hipercalemia.13,14
Uma discreta hipercalemia num indivíduo normal éacompanhada de uma liberação de insulina. Isto faz pres-supor que um indivíduo com deficiência de insulina seriamais propenso a desenvolver hipercalemia. Porém, osmecanismos de defesa contra uma hipercalemia não de-pendem só da insulina, mas também de aldosterona, a qualtem uma ação mais retardada. A implicação prática des-ta inter-relação é a propensão de pacientes diabéticos adesenvolverem hipercalemia quando recebem uma dro-ga que interfere com a ação da aldosterona, tipo triamte-rene.14,15
Assim como a alteração no metabolismo dos carboidra-tos provoca mudanças no metabolismo do potássio, o in-verso é também verdadeiro. Há evidências na literatura deque uma deficiência de potássio compromete o metabolis-mo dos carboidratos. Demonstrou-se que o uso de diuré-ticos tiazídicos, em pacientes com curva anormal de tole-rância à glicose, era capaz de causar diabetes mellitus sinto-mático.14,16 Esta intolerância à glicose que se desenvolve empacientes que recebem tiazídicos pode ser corrigida comsuplementação de potássio. A implicação prática é de queuma intolerância aos carboidratos clinicamente importan-te associada a diuréticos ocorre mais provavelmente empacientes diabéticos ou com diabetes mellitus latente. Tal-vez pela deficiência de insulina, pode não haver hipocale-mia, o que pode levar o médico a não suspeitar de um dé-ficit de potássio.
GlucagonA administração de doses farmacológicas de glucagon
pode causar hiperglicemia e hipercalemia agudas. O glu-cagon tem efeito glicogenolítico potente, responsável pelahiperglicemia. A hipercalemia é proveniente da liberaçãode potássio pelo fígado.17
CatecolaminasOs efeitos das catecolaminas na concentração de potás-
sio do espaço extracelular são complexos e dependem dotipo de receptor estimulado.
Os estímulos aos receptores �2-adrenérgicos estimulamo movimento de potássio para dentro das células, prova-velmente via Na-K-ATPase, podendo causar hipocale-mia.3,13 Este mecanismo pode envolver um aumento noAMP cíclico e, como resultado, fosforilação e ativação dasódio-potássio-ATPase. As catecolaminas também podematuar de modo indireto, estimulando a glicogenólise, queleva a hiperglicemia e liberação de insulina pelas células β
do pâncreas. A insulina, por sua vez, causa a entrada depotássio nas células.
Com a estimulação �-adrenérgica há passagem de po-tássio para dentro das células do músculo esquelético. Asimplicações são as seguintes:14
1.º) Alguns agentes que possuem atividade estimuladorade receptor �-adrenérgico podem ser úteis no trata-mento da hipercalemia aguda;
2.º) Agentes �-bloqueadores como o propranolol, que evi-tam a entrada de potássio no músculo esquelético,podem ser úteis em estados hipocalêmicos nos quaisa entrada de potássio no músculo está acelerada. Exem-plo: paralisia periódica.
3.º) Pacientes que recebem �-bloqueadores podem desen-volver hipercalemia, pelo menos em cinco situações:deficiência de insulina, insuficiência renal, exercício,administração de KCl e quando ingerem simultanea-mente drogas que interferem com a ação da aldoste-rona, tipo espironolactona.
A infusão endovenosa de epinefrina ou nor-epinefrinapode causar uma hipercalemia aguda transitória que pa-rece ocorrer por liberação de potássio do fígado.18 A epi-nefrina aumenta a produção de glucagon pelas células alfado pâncreas e estimula a produção de glicose pelo fígado.Ambos os mecanismos podem estimular a liberação deinsulina, a qual, como já mencionamos, é capaz de reduziro potássio plasmático.
A estimulação α-adrenérgica causa efeitos opostos, po-dendo originar hipercalemia pela saída de potássio dascélulas e inibição da liberação de insulina pelo pâncreas.12
Hormônios AdrenocorticaisA aldosterona é um dos mais potentes mineralocorticói-
des naturais e tem uma participação importantíssima naregulação da quantidade de sódio e potássio no organis-mo. Este hormônio, atuando nos túbulos renais, aumentaa reabsorção de sódio e a secreção de potássio. Embora asações sejam opostas, o balanço de sódio permanece está-vel, mesmo quando a ingesta de potássio varia muito, evice-versa.
Um aumento de 0,3 mEq/L na concentração de potás-sio é suficiente para produzir um aumento significativo nasecreção de aldosterona.19,20 A administração de potássioaumenta a secreção de aldosterona, ao passo que a deple-ção a diminui. Além dos níveis de potássio, outro fator deestímulo à síntese de aldosterona pelas adrenais são osníveis de angiotensina II.
A depleção de volume ou de sódio ativa a secreção derenina pelas células dos aparelhos justaglomerulares dosrins. A renina age sobre um substrato plasmático chama-do angiotensinogênio, convertendo-o em angiotensina I, oqual, sob o efeito da enzima conversora no pulmão, con-verte-se em angiotensina II. Esta estimula a secreção de

capítulo 12 197
aldosterona, que causa secreção tubular de potássio e reab-sorção de sódio, restaurando a volemia, a qual inibe o es-tímulo inicial para produção de renina. Como se podeobservar, estes fatores não atuam isoladamente, e o con-junto recebe o nome de sistema renina-angiotensina-aldoste-rona (SRAA).2,13
Uma concentração elevada de potássio estimula a secre-ção de aldosterona, a qual, atuando nos túbulos renais,aumenta a excreção de potássio, normalizando o potássioplasmático. Quando a concentração de potássio plasmáti-co cai, desaparece o estímulo para secreção de aldostero-na, completando-se um sistema fechado de controle retró-grado. Simultaneamente, o potássio plasmático elevadoinibe diretamente a secreção de renina e vice-versa.
COMO AGE A ALDOSTERONA?Estudos mostram que a aldosterona e os mineralocorti-
cóides atuam no túbulo coletor cortical e não no túbulo con-tornado distal, como se pensava anteriormente. Acredita-seque a aldosterona entra na célula pelo lado sanguíneo e seliga a um receptor de proteína no citoplasma, o qual se unecom o núcleo para promover síntese protéica. As proteínasassim sintetizadas poderiam aumentar a permeabilidade damembrana plasmática apical ao sódio, aumentando o apor-te de sódio para o lado sanguíneo da célula (local do trans-porte ativo). A bomba de sódio na face peritubular, estimu-lada pela maior síntese protéica, aumenta a extrusão de só-dio da célula para o espaço extracelular. Este maior trans-porte de sódio determina um maior gradiente elétrico trans-tubular, criando condições para maior secreção de potássio.6
A entrada de potássio pela membrana peritubular em trocapelo sódio é mediada pela Na-K-ATPase. Cargas de potás-sio aumentam a atividade de Na-K-ATPase, independenteda secreção de aldosterona.
Pontos-chave:
• A insulina e os estímulos �2-adrenérgicosestimulam a captação do potássio pelascélulas
• A aldosterona atua no túbulo coletorcortical, aumentando a reabsorção de sódioe a secreção de potássio
ADAPTAÇÃO A NÍVEISELEVADOS DE POTÁSSIO
Atualmente, aceita-se a existência de um mecanismo deadaptação que explica a tolerância de animais a doses ele-vadas de potássio. Por exemplo, quando se administrampor via endovenosa doses elevadas de potássio a animaissubmetidos a uma ingestão alta de potássio, há uma rápi-
da secreção urinária deste íon. Da mesma forma, na insu-ficiência renal crônica, os nefros remanescentes aumentama sua capacidade de excretar potássio.21
Adaptação Renal ao PotássioEm vista do que mencionamos acima, concluímos que
o rim tem uma capacidade intrínseca de responder a umacarga de potássio, excretando mais potássio na urina. Omecanismo responsável por esta secreção elevada de po-tássio reside na atividade das células do nefro distal, jáabordada anteriormente.
São um pouco contraditórios os dados experimentaiscom relação ao local no nefro responsável pela adaptaçãoao potássio. Parece não haver dúvida de que o túbulo dis-tal tem um papel crítico na secreção de potássio, mas aparticipação do sistema coletor não está definida. Wrighte cols., por exemplo, mostraram que, em ratos submetidosà ingestão crônica de potássio, só o túbulo distal era res-ponsável pela excreção elevada de potássio. No entanto,se os animais não recebiam sódio, o sistema coletor contri-buía significativamente para a excreção de potássio. Estu-dos mostraram que o epitélio do sistema coletor é poten-cialmente capaz de secretar potássio.22
Adaptação Extra-renal ao PotássioEm situações de excesso de potássio, outros órgãos po-
dem contribuir para a homeostase do potássio. Há váriasevidências de que a aldosterona age em outros tecidos demodo semelhante ao observado nos túbulos renais.3 Porexemplo, o cólon pode aumentar a excreção de potássio,num mecanismo mediado pela aldosterona. No tecidomuscular, a aldosterona parece deslocar o potássio para ointracelular.3 Experimentalmente, a entrada de potássio nascélulas é maior em animais submetidos à ingestão eleva-da crônica de potássio (e presumivelmente com níveis ele-vados de aldosterona), do que em animais submetidos auma ingesta normal de potássio.23
As inter-relações potássio-insulina-glucagon e catecola-minas já foram analisadas nas páginas precedentes.
PAPEL DO BALANÇOÁCIDO-BÁSICO
Existe evidência de que a produção de amônia está inti-mamente relacionada com a homeostase do potássio.24,25
Assim, durante uma depleção de potássio, há um aumentona excreção de amônio (NH4
�), possivelmente devido a umaumento na produção renal de amônia (NH3). Simultane-amente, observa-se um aumento no pH urinário, o quelevou alguns autores a postular a possível coexistência deum defeito no gradiente de hidrogênio.

198 Metabolismo do Potássio
Existe um pouco de controvérsia quanto ao distúrbioácido-básico que uma depleção de potássio produz. Algunsinvestigadores demonstraram que, no cão, a depleção depotássio causa acidose sistêmica, e esta seria responsávelpela produção aumentada de amônia.26 Já no rato, ocorrealcalose metabólica e no homem não há alteração ou ocor-re discreta alcalose metabólica. Em vista desta discrepân-cia, acredita-se, no momento, que não é o estado ácido-básico sistêmico que influi sobre a produção de amônia epH urinário.24
Em face de um excesso de potássio, ocorre uma dimi-nuição na excreção de amônio.
O metabolismo do sódio parece estar intimamente re-lacionado com a homeostase potássio/ácido-básico. A in-ter-relação, embora ainda controvertida, seria da seguintemaneira:19
A depleção de potássio aumenta a atividade da renina plas-mática e diminui a secreção de aldosterona. Parece tambémresultar num aumento da reabsorção de sódio no nefroproximal e numa diminuição da reabsorção do nefro dis-tal.27 É provável que a diminuição da reabsorção de sódiono nefro distal seja mediada pela diminuição na secreçãode aldosterona.
Um excesso de potássio diminui a atividade da renina eestimula a secreção de aldosterona. Além disto, diminui areabsorção proximal de sódio e estimula a sua reabsorçãodistal. O aumento da secreção de aldosterona contribuipara a reabsorção distal elevada de sódio.
Estes ajustes na reabsorção de sódio servem para man-ter a homeostase do sódio e do potássio quando a ingestade potássio é modificada. Assim, na presença de um défi-cit de potássio, como há um aumento na reabsorção proxi-mal de sódio, menos sódio chega ao nefro distal, ondenormalmente ocorre a troca Na�-K�, e como a secreção dealdosterona também está diminuída, a reabsorção distal desódio também é reduzida. Assim, o balanço de sódio émantido, enquanto a excreção de potássio é diminuída.Quando há um excesso de potássio, ocorre o inverso.
Várias observações indicam que a reabsorção de sódiotambém influencia a excreção de hidrogênio no nefro dis-tal.28 Acredita-se que a produção de amônia possa minimi-zar as alterações ácido-básicas quando a reabsorção desódio é modificada.
Se existe menos amônia para tamponar o H� no lúmen,o pH urinário cai muito, elevando o gradiente transtubu-lar para a secreção de H� e, portanto, diminuindo a excre-ção de ácido.24
Na presença de uma depleção de potássio, há uma dimi-nuição na reabsorção distal de sódio e um aumento naprodução de amônia. A amônia tampona o H� no lúmen,transformando-se em amônio (NH4
�). Com isto, o pH nolúmen não cai muito e, por conseguinte, o gradiente trans-tubular para a secreção de H� também não é muito gran-de, e logo a excreção de ácido não é reduzida. Portanto, opapel da amônia é manter a excreção de ácido na vigência
de uma diminuição na reabsorção distal de sódio, a qual,como mencionamos anteriormente, se acompanha de umadiminuição na excreção de ácido.24
Uma das implicações práticas do aumento na produçãode amônio foi dada em 1963. É clássico o conceito de quehipocalemia pode precipitar coma hepático. Como empacientes cirróticos muitas vezes se administram diuréti-cos, estes podem causar hipocalemia, a qual aumenta aprodução de amônia, e o paciente com disfunção hepáticapode ser incapaz de metabolizar a amônia, predispondo-se à instalação de coma hepático.29
A secreção de K� e H� depende muito da concentraçãointracelular destes íons. Por exemplo, numa alcalose agu-da (respiratória ou metabólica), o potássio passa do líqui-do extracelular para o interior das células, e, numa acido-se (respiratória ou metabólica), o potássio sai das células.
O mecanismo deste movimento transcelular não estábem esclarecido. Portanto, na alcalose, a concentração in-tracelular de potássio aumenta (inclusive na célula tubu-lar renal), e mais potássio está disponível para excreção.Na acidose, ocorre o contrário.
Uma alcalose sistêmica aumenta a perda urinária depotássio, enquanto uma acidose sistêmica diminui a excre-ção renal de potássio. Mas, na verdade, o potássio e o hi-drogênio não competem pela secreção, e os dados experi-mentais mostram que, enquanto a secreção de hidrogênioaumenta, a de potássio também aumenta, e vice-versa.12
HOMEOSTASIA DO POTÁSSIO NAINSUFICIÊNCIA RENAL
A manutenção do balanço de potássio, durante a insta-lação de insuficiência renal crônica, reflete a participaçãoprogressiva de mecanismos de adaptação.30
A concentração plasmática de potássio aumenta apenasna fase terminal da insuficiência renal crônica. Isto impli-ca que, à medida que cai o ritmo de filtração glomerular, afração do potássio filtrado também aumenta.
Bank e cols. demonstraram que, em ratos com insufici-ência renal causada por nefrectomia subtotal, não havia al-teração na fração de reabsorção de potássio ao longo do tú-bulo distal (quando comparados com o grupo-controle), masaumentava muito a secreção de potássio no ducto coletor.31
Tanto na insuficiência renal como na ingestão crônicade potássio, a adaptação renal resulta de um aumento deatividade da Na-K-ATPase.
Papel do SistemaRenina-Angiotensina-Aldosterona
A aldosterona é um estimulador potente da secreçãotubular de potássio. A evidência baseada em dados expe-rimentais é de que uma produção elevada de aldosterona

capítulo 12 199
não é indispensável para a manutenção do equilíbrio depotássio na uremia.
Vários autores mostraram que a concentração plasmá-tica de aldosterona na insuficiência renal terminal é nor-mal, desde que a renina e o potássio plasmático estejamdentro do normal. Quando aumenta a concentração plas-mática de potássio e/ou renina, aumenta a concentraçãode aldosterona.32
A conclusão é de que há necessidade, pelo menos, deníveis normais de aldosterona, pois se uma insuficiênciarenal se complica com hipoaldosteronismo, ocorre hiper-calemia.33
Excreção Gastrintestinal de Potássio
Normalmente, a quantidade de potássio excretada nasfezes representa uma quantidade pequena da ingesta diá-ria. No entanto, o intestino é potencialmente uma fonte deperda de potássio, como ocorre nas diarréias. Estudos emindivíduos normais e urêmicos, numa dieta normal depotássio, mostraram que, enquanto nos indivíduos nor-mais a excreção fecal era de 12% da ingesta, em urêmicosera de 34%.34
Tem sido sugerido que o mecanismo da excreção intesti-nal aumentada de potássio seja mediado pela aldosterona.
Tolerância Celular ao PotássioQuando se administra potássio a urêmicos, o potássio
sérico aumenta muito mais do que em pacientes normais.Isto indica que a tolerância celular ao potássio diminui nainsuficiência renal. Conclui-se, portanto, que um mecanis-mo de adaptação renal existe em indivíduos normais eurêmicos, mas um mecanismo de adaptação extra-renal sóexiste em normais.30
Ponto-chave:
• Na insuficiência renal, existe umaadaptação aos níveis elevados de potássio,com aumento da excreção renal e intestinalfrente a cargas de potássio, pela ação daaldosterona
AÇÃO DOS DIURÉTICOS
Como já mencionamos, um dos fatores determinantesdo ritmo de secreção distal de potássio é o fluxo de urinapelo segmento do nefro. Portanto, quanto maior o fluxo deurina pelo túbulo distal cortical, maior é a excreção depotássio. E os diuréticos são agentes que aumentam o flu-xo de urina.12
Como alguns diuréticos inibem a reabsorção proximalde sódio, uma maior quantidade de sódio chega ao nefrodistal, e postulou-se inicialmente que a caliurese que ocor-ria com estes diuréticos era resultado da maior concentra-ção intraluminal de sódio no túbulo distal cortical.
Atualmente, não se acredita que esta concentração in-traluminal de sódio limite a secreção de potássio (apenaspotencialmente, como já foi frisado). Mas há evidência deque, no sistema coletor (cortical e medular), a concentra-ção intraluminal de sódio limita a secreção de potássio.Assim, um aumento da oferta de sódio ao sistema coletoraumenta a secreção de potássio (v. também Cap. 43).
DISTÚRBIOS CLÍNICOS DOMETABOLISMO DO POTÁSSIO
Depleção de Potássio (Hipocalemia)Refere-se a uma diminuição do potássio total em rela-
ção à capacidade do potássio ou resultado de uma distri-buição transcelular e traduz-se habitualmente por umaredução na sua concentração plasmática (hipocalemia � 3,5mEq/L). A alcalose é a causa mais comum de alteração nadistribuição transcelular. Um déficit real de potássio resultageralmente de perdas gastrintestinais ou renais.
CAUSAS DE HIPOCALEMIAA depleção a que nos referimos é a que se deve à perda
do íon K� e não pela redução da massa celular (capacida-de do potássio). Isto pode ocorrer durante um período deingesta reduzida de potássio, não compensada por umaredução na excreção de potássio. Isto não é freqüente, poisquando a ingesta diminui por letargia, anorexia, coma etc.,a excreção também diminui. Portanto, depleção de potás-sio por falta de ingesta só ocorre se os rins forem impedi-dos de conservar potássio.
A causa mais comum de depleção de potássio é umaperda elevada de potássio do corpo. Como a perda depotássio pela pele é desprezível (a não ser em sudoreseprofusa), restam o rim e o trato gastrintestinal como viasimportantes na perda de potássio.
Desvio Transcelular ou RedistribuiçãoApenas uma pequena fração do potássio corporal total
está localizada no espaço extracelular, e pequenos desviospara o intracelular produzem grandes variações na concen-tração plasmática de potássio.35 Estes desvios podem sercausados por:
a) Alterações do estado ácido-básico: na alcalose metabó-lica ou respiratória, íons hidrogênio saem das célulaspara minimizar as mudanças no pH do extracelular. Anecessidade de manter a eletroneutralidade entre os

200 Metabolismo do Potássio
compartimentos leva à entrada de potássio nas células.Este efeito produz um aumento de 0,6 mEq/L no potás-sio do extracelular para cada 0,1 unidade de pH que cai,no caso da alcalose metabólica, e 0,1 mEq/L no caso dealcalose respiratória.36
b) Ação da insulina: como já comentado anteriormente, ainsulina promove a entrada de potássio nas célulasmusculares e hepáticas, reduzindo os níveis plasmáti-cos. Este efeito pode ser observado após a administra-ção de insulina na hiperglicemia grave ou na cetoacidosediabética.36
c) Infusão de glicose: a concentração plasmática de potás-sio diminui com a administração de glicose, por meca-nismo similar à insulina.36
d) Atividade �-adrenérgica: a estimulação de receptores �2-adrenérgicos promove a entrada de potássio nas células.Então, hipocalemia transitória pode ser observada emsituações em que há liberação de epinefrina, como, porexemplo, intoxicação por teofilina e isquemia coronaria-na. A infusão de aminas vasoativas também pode pro-vocar este efeito, que pode ser utilizado terapeuticamen-te na hipercalemia: a administração de um agonista �-adrenérgico (como a terbutalina e o albuterol) reduz osníveis de potássio em cerca de 0,5-1 mEq/L.36
e) Paralisia periódica hipocalêmica: um raro distúrbio ca-racterizado por ataques recorrentes de paralisia flácidadesde a infância, acompanhados de hipocalemia devi-do a uma redistribuição do potássio para o interior dascélulas.36,37
f) Envenenamento pelo bário (carbonato de bário): podeproduzir paralisia flácida e hipocalemia devido a umbloqueio dos canais de potássio na membrana, que nor-malmente permitem a passagem de potássio para o ex-tracelular. O sulfato de bário utilizado em exames radi-ográficos não acarreta risco para os pacientes.36
g) Tratamento de anemias graves: resulta em rápida assi-milação do potássio para dentro das hemácias que es-tão sendo produzidas, levando a hipocalemia. Este efeitohabitualmente é observado dois dias após o início dotratamento da anemia.35
h) Outras causas: hipotermia, intoxicação por teofilina,cloroquina.35,36
Perdas GastrintestinaisAs principais causas gastrintestinais de hipocalemia
estão enumeradas no Quadro 12.2.
a) Aporte dietético insuficiente: pode ocorrer em pacien-tes idosos e etilistas, em que a ingesta de potássio é ina-dequada, e em pacientes em fase de rápida síntese ce-lular, como os submetidos a hiperalimentação.
b) Diarréias: normalmente, a excreção de potássio para umvolume fecal habitual de 200 ml não excede 10 mEq/dia,mas pode elevar-se muito em certas situações, como nasdiarréias agudas ou crônicas e abuso de laxativos. As
hipocalemias causadas pelas diarréias podem cursartambém com acidose metabólica pela perda de bicarbo-nato. A acidose provoca um desvio iônico que mesmoem vigência de hipocalemia provoca a saída de potás-sio de dentro das células, mascarando os níveis plasmá-ticos de potássio.
Normalmente, a resposta à perda de potássio pelointestino é a conservação renal de potássio, através dadiminuição de sua secreção tubular. Porém, esta respos-ta sofre um efeito antagônico: como a diarréia provocadepleção de sódio e hipovolemia, e estas ocasionammaior produção de aldosterona, a secreção de potássiopode estar elevada.3
c) Ureterossigmoidostomia: resulta em absorção anormalde cloreto de sódio em associação com secreção de po-tássio e bicarbonato para a luz da alça intestinal. Causatambém acidose metabólica do tipo hiperclorêmica.37
d) Vômitos: o teor de potássio no suco gástrico não é ele-vado, mas os vômitos ou a drenagem nasogástrica po-dem ocasionar hipocalemia. Isto se deve mais à perdade ácido clorídrico do que à perda de potássio.3,38 A per-da de ácido leva à alcalose metabólica, a qual produz umdesvio iônico de potássio para dentro das células e se-creção de potássio pelas células tubulares distais. Tam-bém está ativo o sistema renina-angiotensina-aldostero-na, pela perda de água e sódio, o que acelera a perda depotássio pelos rins.3
Perdas RenaisJá apresentamos, nas páginas precedentes, muita evi-
dência da importância do rim como via final de controleda homeostase do potássio. Muitas vezes, a resposta renalé apropriada pela interferência dos mecanismos de controledo balanço de potássio. Outras vezes, a resposta renal in-dica uma nefropatia ou um distúrbio na ação dos mecanis-mos de controle, como ocorre, por exemplo, com o uso dediuréticos.
a) Diuréticos: o uso de diuréticos é, talvez, a causa maisfreqüente de hipocalemia na prática clínica. Todos osdiuréticos provocam excreção de potássio, exceto oschamados poupadores de potássio (v. Cap. 43 para maio-res informações).
Os tiazídicos causam maior perda de potássio porqueaumentam o fluxo de urina pelos segmentos corticais donefro distal, além de, em parte, serem inibidores da ani-drase carbônica.12
O furosemide e o ácido etacrínico inibem a reabsorçãoativa de cloro no ramo ascendente da alça de Henle,responsável provável pela reabsorção passiva de potás-sio neste segmento. Ademais, além de produzirem ummaior fluxo de urina, estes agentes parecem inibir a re-absorção proximal de potássio, promovendo caliurese.12
Os inibidores da anidrase carbônica, tipo acetazolami-da, não afetam o transporte proximal de potássio mas

capítulo 12 201
aumentam a secreção de potássio no nefro distal. O me-canismo parece ser duplo: a inibição da secreção de H�
no nefro distal causa hiperpolarização transtubular, queé uma força para o movimento passivo do potássio dacélula para a urina. Além disto, como estes agentes ini-bem a reabsorção proximal de bicarbonato, mais bicar-bonato chega ao nefro distal e, sendo ele pouco reabsor-vível, induz um aumento do fluxo de urina, como fazemoutros agentes.12 Algumas drogas utilizadas na práticaclínica, como a anfotericina e a carbenicilina, tambémaumentam a perda de potássio.
Os diuréticos osmóticos, tipo manitol, também acelerama excreção de potássio por elevarem o fluxo de líquidotubular no nefro distal.
b) Hiperaldosteronismo: a produção excessiva de aldos-terona por um tumor ou hiperplasia adrenais (hiperal-dosteronismo primário) ou por hipovolemia e hipoper-fusão renal (hiperaldosteronismo secundário) determi-na um aumento na excreção de potássio pelos mecanis-mos já abordados anteriormente, com conseqüente hi-pocalemia. O mesmo ocorre com a estenose de artériarenal.38
O alcaçuz (Glycyrrhiza glabra, elemento utilizado nafabricação de laxantes, indústria de doces, tabaco e cer-vejarias) contém um esteróide, o ácido glicirrízico, o qualinibe uma enzima que converte o cortisol em cortisona.Desta forma o cortisol em níveis elevados induz umaumento na atividade mineralocorticóide.38
c) Alterações tubulares: como as estruturas tubulares donefro distal excretam a maior parte do potássio ingeri-do, é fácil compreender que alterações tubulares podemlevar a uma excreção excessiva de potássio. Exemplos:acidose tubular renal, síndrome de Fanconi, pielonefri-te, fase poliúrica da necrose tubular aguda, etc.
d) Alterações genéticas: a síndrome de Bartter é uma desor-dem rara que se manifesta na infância e cursa com hi-pocalemia, alcalose metabólica, hiper-reninemia, hipe-raldosteronismo, hiperplasia do aparelho justaglomeru-lar e, algumas vezes, hipomagnesemia. São comunspoliúria, polidipsia, hipercalciúria. Mais rara é a ocor-rência de hipomagnesemia. Também existe aumento naliberação renal de prostaglandinas vasodilatadoras, oque pode explicar a pressão arterial normal. Resulta deanormalidades na função tubular, primariamente notransporte de cloreto de sódio na porção espessa da alçade Henle. Com isso, ocorre uma discreta depleção devolume, que ativa o sistema renina-angiotensina-aldos-terona. A combinação de hiperaldosteronismo e aumen-to do fluxo distal (pelo defeito reabsortivo) aumenta asecreção de potássio e hidrogênio nos túbulos coletores,levando a hipocalemia e alcalose metabólica.36
A síndrome de Gitelman cursa com os mesmos acha-dos da síndrome de Bartter, porém o defeito é no co-transportador sódio-potássio do segmento inicial dotúbulo distal.3 Nesta síndrome, perda de magnésio é
mais comum, e podem ocorrer tetania e fadiga. Geral-mente é diagnosticada em crianças maiores ou adultosjovens.36
e) Ânions não reabsorvíveis: normalmente o gradienteelétrico negativo no túbulo coletor, gerado pela reabsor-ção de sódio, é equilibrado pela reabsorção de cloreto.Em algumas situações, o sódio chega ao nefro distalacompanhado de um ânion não reabsorvível (por exem-plo, bicarbonato, penicilina). Nestes casos, parte do só-dio será reabsorvida em troca com o potássio, aumen-tando sua excreção.36
f) Hipomagnesemia: uma grande parte dos pacientes comhipocalemia apresentam hipomagnesemia (por uso dediuréticos, diarréia). A hipomagnesemia induz à perdarenal de potássio por mecanismos complexos. É comumencontrar hipomagnesemia em pacientes em que existedificuldade para correção da hipocalemia; nestes casos,só se conseguirá corrigir o potássio após a reposição demagnésio.35,36
g) Anfotericina B: este medicamento modifica a permea-bilidade celular através da interação com esteróis damembrana, promovendo secreção de potássio.36
h) Outras causas: gentamicina e cisplatina têm efeito tóxi-co direto sobre as células tubulares, induzindo à perdarenal de potássio.35
Ponto-chave:
• A hipocalemia (potássio � 3,5 mEq/L) podeser causada por redistribuição, perdasgastrintestinais e renais
MANIFESTAÇÕES CLÍNICAS
MetabólicasA hipocalemia pode afetar o metabolismo protéico e
gerar dificuldade em obter balanço nitrogenado positivodurante nutrição parenteral. Testes de tolerância à glicosepodem estar alterados, possivelmente devido a uma me-nor resposta das células beta do pâncreas à glicose. Alémdisso, encontram-se comprometidas, também, a liberaçãode aldosterona e hormônio de crescimento.37
CardiovascularesOcorrem irregularidades do ritmo cardíaco, caracteriza-
das por batimentos ectópicos e alterações eletrocardiográ-ficas: alargamento do QRS, depressão do segmento ST,diminuição de ondas T e, eventualmente, o aparecimentode ondas U após as ondas T (Fig. 12.7). Estas alteraçõesrefletem o impacto da hipocalemia sobre o potencial demembrana. A depleção de potássio também aumenta orisco de arritmias em pacientes recebendo digital. Estespacientes costumam receber diuréticos e uma dieta pobre
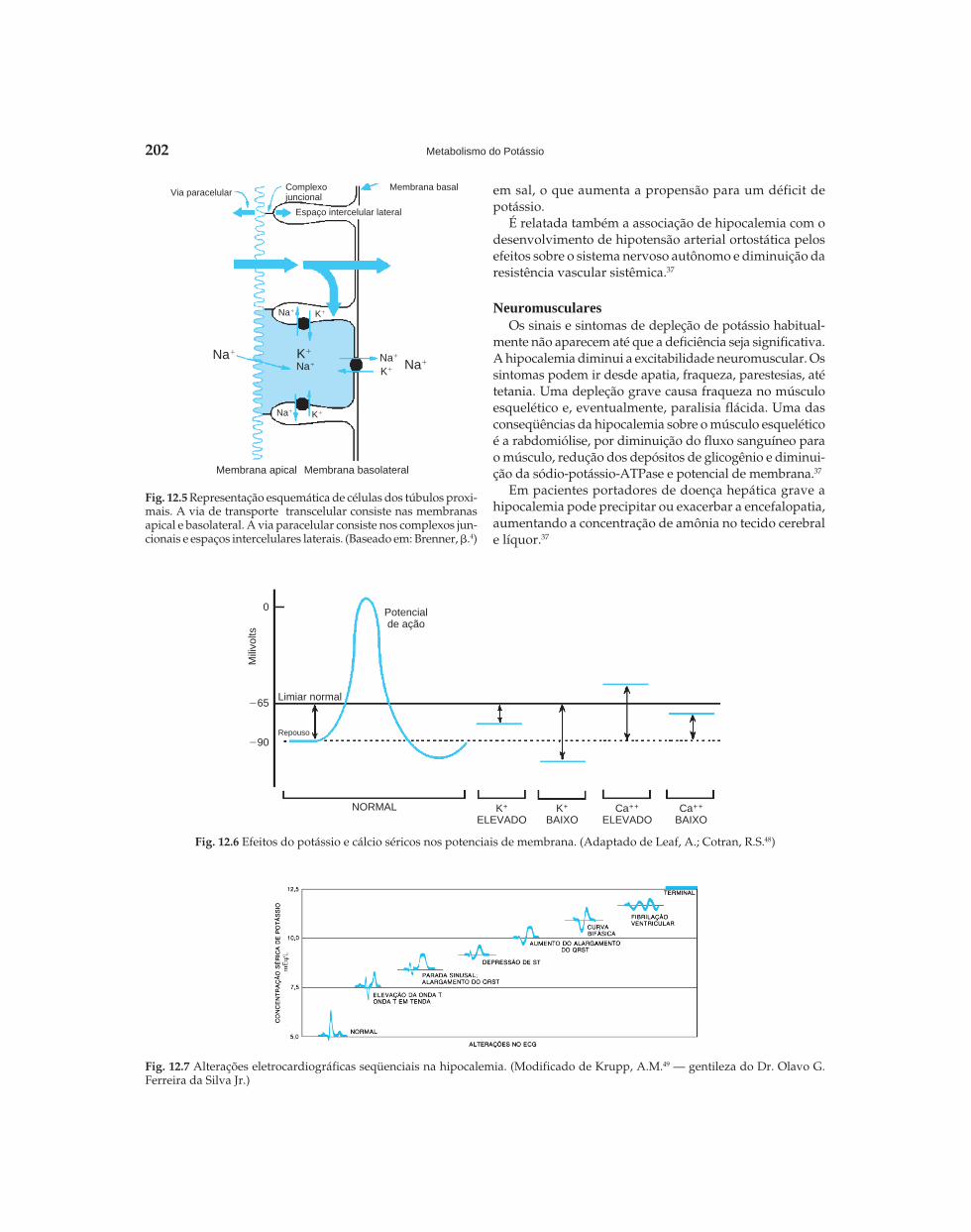
202 Metabolismo do Potássio
em sal, o que aumenta a propensão para um déficit depotássio.
É relatada também a associação de hipocalemia com odesenvolvimento de hipotensão arterial ortostática pelosefeitos sobre o sistema nervoso autônomo e diminuição daresistência vascular sistêmica.37
NeuromuscularesOs sinais e sintomas de depleção de potássio habitual-
mente não aparecem até que a deficiência seja significativa.A hipocalemia diminui a excitabilidade neuromuscular. Ossintomas podem ir desde apatia, fraqueza, parestesias, atétetania. Uma depleção grave causa fraqueza no músculoesquelético e, eventualmente, paralisia flácida. Uma dasconseqüências da hipocalemia sobre o músculo esqueléticoé a rabdomiólise, por diminuição do fluxo sanguíneo parao músculo, redução dos depósitos de glicogênio e diminui-ção da sódio-potássio-ATPase e potencial de membrana.37
Em pacientes portadores de doença hepática grave ahipocalemia pode precipitar ou exacerbar a encefalopatia,aumentando a concentração de amônia no tecido cerebrale líquor.37
Fig. 12.5 Representação esquemática de células dos túbulos proxi-mais. A via de transporte transcelular consiste nas membranasapical e basolateral. A via paracelular consiste nos complexos jun-cionais e espaços intercelulares laterais. (Baseado em: Brenner, �.4)
Fig. 12.6 Efeitos do potássio e cálcio séricos nos potenciais de membrana. (Adaptado de Leaf, A.; Cotran, R.S.48)
Fig. 12.7 Alterações eletrocardiográficas seqüenciais na hipocalemia. (Modificado de Krupp, A.M.49 — gentileza do Dr. Olavo G.Ferreira da Silva Jr.)
Via paracelularComplexojuncional
Membrana basal
Espaço intercelular lateral
Na� K�
K�
Na�Na�
Na�
K� Na�
Na� K�
Membrana apical Membrana basolateral
Repouso
Limiar normal
Mili
volts
0
�65
�90
NORMAL K�
ELEVADOK�
BAIXOCa��
ELEVADOCa��
BAIXO
Potencialde ação

capítulo 12 203
DigestivasPodem ocorrer sintomas digestivos, como náuseas e
distensão abdominal e de alças intestinais (íleo paralítico).
RenaisComo conseqüência da hipocalemia, os mecanismos de
conservação de potássio encontram-se ativados, e a con-centração urinária de potássio está diminuída.
Além disso, vários estudos, no ser humano e em animais,demonstraram que a depleção de potássio está associada auma vacuolização das células epiteliais tubulares, mais pro-nunciada no túbulo proximal, todavia também vista no tú-bulo contornado distal. Tudo indica que as lesões são rever-síveis, pelo menos nas fases iniciais da depleção.39 Há umasugestão na literatura, baseada em observações clínicas eexperimentais, de que a depleção de potássio torna os indi-víduos (e animais) suscetíveis à pielonefrite.40
Podem ocorrer ainda polidipsia por estímulo da sede epoliúria pela incapacidade de concentrar maximamente aurina, como um diabetes insípido nefrogênico. Aparente-mente, a hipocalemia causa uma dificuldade de o ADHformar o segundo mensageiro, o AMP cíclico.37,38
DIAGNÓSTICO DIFERENCIALNaturalmente para se determinar a causa da hipocale-
mia devemos verificar se a mesma resulta de uma redistri-buição do potássio ou representa realmente um déficit. Ascausas de alteração na distribuição (alcalose, insulina, al-dosterona e drogas �-adrenérgicas) já foram abordadas. Sea causa da hipocalemia não estiver na redistribuição dopotássio, estaremos frente a um déficit real de potássio, edevemos determinar se a perda de potássio é renal ou ex-tra-renal (Quadros 12.3 e 12.7).
Pela própria história clínica podemos ter idéia da causa
Quadro 12.3 Diagnóstico diferencial de hipocalemia
I - Perda extra-renal (K urinário � 20 mEq/dia)A. Ácido-básico normal B. Acidose metabólica
1. Ingesta inadequada 1. Perdas gastrintestinaisa. anorexia nervosa a. diarréiab. dieta de chá c/torradas b. fístula
2. Pele c. adenoma vilosoa. suor d. abuso de laxativos
II - Perda renal (K urinário � 20 mEq/dia)A. Acidose metabólica B. Ácido-básico variável
1. Acidose tubular renal 1. Síndrome de Fanconia. distal (tipo I) 2. Fase diurética (NTA, pós-obstrução)b. proximal (tipo II) 3. Nefrite intersticial
2. Diamox 4. Leucemia3. Cetoacidose diabética 5. Antibióticos (penicilina, carbenicilina)4. Enterostomia ureteral 6. Depleção de magnésio
a. ureterossigmoidostomia a. adquiridab. ureteroileostomia b. perda renal hereditária
C. Alcalose metabólicaCloro urinário baixo 1. Vômitos ou perda gástrica(cloro urinário < 10 mEq/dia) 2. Diuréticos
3. Pós-hipercapnia4. Diarréia perdedora de Cl (congênita)
Cloro urinário elevado(cloro urinário � 10 mEq/dia)Excesso de mineralocorticóide
(hipertensão arterial)� Aldosterona � Renina 1. Hiperaldosteronismo primário
a. adenomab. hiperplasia
� Renina 1. Hipertensão renovascular2. Hipertensão maligna3. Tumor secretor de renina
Aldosterona N ou ↓ 1. Excesso de corticosterona ou DOC2. Alcaçuz3. Síndrome de Liddle4. Síndrome de Cushing5. ACTH ectópico
Outros 1. Diuréticos, síndrome de Bartter, depleção grave de K
Modificado de Narins, R.G.; Heilig, C.W.; Kupin, W.L.41

204 Metabolismo do Potássio
do distúrbio, porém alguns dados laboratoriais além dadosagem do potássio plasmático podem fornecer significati-vas informações. Por exemplo, a dosagem do potássio em urinade 24 horas pode auxiliar a determinar se a causa da hipo-calemia é uma perda urinária ou não. Caso o potássio uri-nário esteja acima de 20 mEq/litro, suspeita-se de perdarenal. Se menor que 20 mEq/litro, demonstra que a con-servação renal de potássio está ocorrendo, e a causa dahipocalemia é extra-renal. A dosagem de potássio emamostra aleatória de urina pode ser usada, mas é menosprecisa.36
Também a gasometria venosa, além de demonstrar a pos-sibilidade de desvio iônico, pode evidenciar uma causa pro-vável para o distúrbio: por exemplo, vômitos e síndrome deBartter cursam com alcalose; alguns distúrbios tubularesrenais e cetoacidose diabética cursam com acidose.
Ponto-chave:
• Além da dosagem plasmática de potássio,auxiliam no diagnóstico de hipocalemia:
Dosagem de potássio na urinaGasometria venosa
TRATAMENTO DA HIPOCALEMIAEstá indicada a reposição de potássio para os pacientes
que apresentem hipocalemia cuja causa não seja a redis-tribuição entre compartimentos.38
A hipocalemia é raramente uma emergência, e, sempreque possível, a via oral deverá ser empregada para reposi-ção de soluções de potássio, preferencialmente sob a for-ma de cloreto.35 No Brasil, estão disponíveis as seguintesapresentações de cloreto de potássio: drágeas de 500 mg,drágeas de liberação lenta contendo 600 mg e xarope con-tendo 900 mg em 15 ml. Na prática, a correção de hipoca-lemia somente pela ingestão de alimentos com alto teor depotássio não é adequada.
A via endovenosa só será utilizada se houver necessi-dade de uma administração mais rápida ou se o pacientenão puder ingerir. A urgência na administração do potás-sio depende basicamente das repercussões cardíacas e neu-romusculares. Pacientes com envolvimento muscular sig-nificativo ou alterações eletrocardiográficas deverão rece-ber quantidades maiores e em menor tempo.
A maior parte da literatura indica que não mais de 40mEq de potássio devam ser colocados em cada litro desolução para uso endovenoso e que a administração nãodeve ser inferior a 60 minutos. Hamill sugere que a infu-são de até 0,5 mEq/kg em uma hora é segura para pacien-tes gravemente doentes.42 Outros sugerem 0,75 mEq/kg ou30 mEq/m2 em pessoas obesas durante 1 a 2 horas. Asquantidades de potássio a serem administradas serão tan-to maiores quanto maior a depleção, pois primeiramente
o potássio adentra as células e refaz os estoques intracelu-lares, para em seguida iniciar a normalização dos níveis noextracelular.
É importante lembrar que a administração de potássioem solução que contenha glicose pode reduzir ainda maisos níveis de potássio; se for possível, a reposição inicialdeve ser feita em solução salina isotônica.36
Numa hipocalemia grave (� 2,0 mEq/L) e associada aarritmias cardíacas, até 80-100 mEq deverão ser adminis-trados em 1 hora para suprimir a irritabilidade cardíaca.O fator limitante nestas altas doses é a dor no trajeto veno-so durante a infusão. Uma solução para este problema se-ria a administração através de dois acessos periféricos, cadainfusão contendo 40-50 mEq/L. Se houver problema deexcesso de volume, podemos concentrar a solução, mas aídevemos utilizar uma veia de alto fluxo, como por exem-plo uma veia femoral. A infusão de grandes quantidadesatravés das veias subclávia, jugular ou através de cateteratrial não é recomendada, pois as altas concentrações in-tracardíacas de potássio podem causar arritmias. Sempreque for urgente a reposição de potássio, esta deverá serefetuada sob controle eletrocardiográfico.
No Brasil, a apresentação de cloreto de potássio maisutilizada para uso endovenoso é na concentração de 19,1%,onde cada ml tem 2,5 mEq de potássio e 2,5 mEq de cloro.
Os riscos da utilização de potássio dependem da via deadministração, idade e presença de co-morbidades, comopor exemplo a insuficiência renal. Mesmo administradopor via oral, o potássio pode ocasionar parada cardíaca porhipercalemia, sendo este fato mais observado em pacien-tes idosos, pacientes com insuficiência renal, pacientes querecebem simultaneamente potássio por via oral e endove-nosa e naqueles que recebem potássio e diuréticos poupa-dores de potássio.43
As drágeas de potássio para liberação entérica eventu-almente provocam ulceração do intestino delgado. Já aspreparações líquidas de potássio não têm bom paladar,mas raramente causam ulcerações intestinais.
CÁLCULO DO DÉFICIT DE POTÁSSIONa ausência de um distúrbio ácido-básico, a magnitu-
de do déficit pode ser calculada considerando-se a capaci-dade para potássio (massa muscular) do paciente44 (Qua-dro 12.4) ou utilizando-se as regras práticas já enumera-das. Portanto, se o potássio total pode ser estimado (consi-derando-se o peso e a massa muscular do paciente), pode-se calcular o déficit de potássio em mEq (v. exercícios adi-ante).
Se desejarmos usar o potássio plasmático como guia daterapêutica, há necessidade de uma estimativa grosseira dainfluência do distúrbio ácido-básico na relação entre opotássio plasmático e o intracelular. Esta relação é expos-ta na Fig. 12.9, a qual indica a influência do pH sanguíneona concentração do potássio plasmático sem que haja alte-ração no potássio total. Pode-se verificar que, para cada

capítulo 12 205
alteração no pH de 0,1 unidade, ocorre uma alteração nopotássio plasmático de 0,6 mEq/L. Portanto, tendo-se o pH,pode-se deduzir o potássio plasmático, como se não hou-vesse distúrbio ácido-básico (v. exercícios adiante).
REPOSIÇÃO DE POTÁSSIO EM ALGUMASSITUAÇÕES ESPECIAIS
Em pacientes não edemaciados e que desenvolvem hi-pocalemia durante a administração de diuréticos tiazídi-cos, pode-se normalizar o potássio plasmático administran-do-se 60 mEq de cloreto de potássio por dia.43 Apenas al-guns permanecem hipocalêmicos mesmo que se adminis-trem 100 mEq por dia.45 A administração de diuréticos quepoupam potássio normaliza o potássio plasmático duran-te a terapia com diuréticos tiazídicos ou de alça, mas a ex-periência clínica mostra que a administração de cloreto depotássio em quantidades suficientes tem o mesmo efeito.O bom senso atual indica que, em pacientes não edemaci-ados recebendo diuréticos de modo crônico, não há neces-sidade de administrar potássio profilaticamente. Nestespacientes, recomenda-se um controle laboratorial a cadaum ou dois meses e, se a concentração plasmática do po-tássio chegar a menos de 3 mEq/L, administra-se umasolução de potássio a 10% por via oral, proporcionando-se 50-60 mEq por dia.43
A administração de sais de potássio ou diuréticos pou-padores de potássio a pacientes edemaciados está particu-larmente indicada naqueles que recebem digital ou que sãosuscetíveis ao desenvolvimento de coma hepático. A ad-ministração diária de 40-80 mEq de uma solução de potás-sio é em geral suficiente. Se a administração de sais depotássio por via oral não corrige o déficit, podem-se em-pregar agentes bloqueadores da secreção de potássio nonefro distal. A espironolactona é eficiente, mas o custo éelevado e a terapia prolongada pode causar ginecomastia.O custo do triamterene é menor, mas ele já é menos efici-ente.
Em pacientes com alcalose metabólica e hipocalemia, aadministração de sais de potássio, sob a forma de acetato,gluconato ou lactato, não corrige o déficit de potássio, a nãoser que o déficit de cloro seja corrigido através da admi-nistração de cloreto de potássio ou através da administra-ção simultânea de um destes sais de potássio e uma outrafonte de cloro (v. Cap. 11).
Excesso de Potássio(Hipercalemia)
O excesso de potássio é definido como um aumento narelação potássio total/capacidade de potássio ou devido auma redistribuição transcelular e é geralmente identifica-do por um aumento da concentração plasmática acima dosvalores normais (hipercalemia > 5 mEq/L).
CAUSAS DE HIPERCALEMIAAs situações que mais comumente resultam em hiper-
calemia são aquelas em que o rim não mais consegue ex-cretar o potássio ingerido ou proveniente de uma libera-ção endógena. A capacidade de excreção renal do potás-sio é muito grande, e, em indivíduos normais, a ingestãoexcessiva de potássio não produz um excesso de potássio.
Pseudo-hipercalemiaRefere-se à elevação da concentração sérica ou plasmá-
tica de potássio por movimento deste íon para fora dascélulas durante ou após a coleta de sangue. Geralmente istose relaciona a trauma durante a coleta, quando o garrote émantido por muito tempo antes da punção venosa, ouquando há demora no processamento da amostra, resul-tando em liberação de potássio das hemácias por hemóli-se.36,46 Leucócitos acima de 100.000/mm3 ou plaquetas aci-ma de 400.000/mm3 podem resultar em pseudo-hiperca-lemia, pois estas são células ricas em potássio, que podeser liberado durante o processo de coagulação.36
O ECG pode ser útil na diferenciação entre a hipercale-mia verdadeira e a factícia, pois alterações só ocorrem nahipercalemia verdadeira.
RedistribuiçãoA entrada de íons hidrogênio em excesso pelas células,
como ocorre nas acidoses, leva a um movimento de potás-sio para fora das células com o objetivo de manter a ele-troneutralidade. Para cada 0,1 unidade de pH que cai, opotássio extracelular sobe 0,6 mEq/L.
Uma liberação rápida de potássio pode ocorrer tambémem destruição celular maciça após cirurgia, trauma comesmagamento e lesão muscular (rabdomiólise), infecçõesextensas ou hemólise maciça.38 Estes quadros geralmentese acompanham de um comprometimento da função re-nal e conseqüente redução na excreção de potássio.
Outras causas de hipercalemia por redistribuição seri-am: uso de �-bloqueadores, intoxicação digitálica, parali-sia periódica familiar hipercalêmica, exercícios extenuan-tes e administração de succinilcolina.38
Insuficiência Renal AgudaNa insuficiência renal aguda, há uma redução impor-
tante na excreção do potássio, pois se estabelece um qua-dro de oligúria ou anúria, geralmente com destruição ce-
Quadro 12.4 Estimativa da capacidade do potássio
Potássio Total (mEq/kg)Massa Muscular Homens Mulheres
Normal 45 35Perda moderada 32 25Perda acentuada 23 20
Modificado de Chapman, W.H. e col.44

206 Metabolismo do Potássio
lular num paciente hipercatabólico, diminuindo a capaci-dade do potássio e lançando na circulação o potássio libe-rado das células. Hipercalemia em insuficiência renal crô-nica não é comum, por razões já abordadas nas páginasprecedentes. Cumpre apenas salientar que vários estudosmostram que a secreção de potássio na insuficiência renalcrônica está aumentada, talvez pelo maior aporte de sódioao nefro distal. De modo geral, pacientes renais crônicossem aporte excessivo de potássio podem manter-se semhipercalemia enquanto o clearance de creatinina estiveracima de 5-10 ml/min.35
Insuficiência AdrenalOs principais estímulos fisiológicos para a liberação de
aldosterona são a angiotensina II (gerada pela liberação derenina pelos rins) e a elevação do potássio plasmático. Des-te modo, a hipercalemia por diminuição do efeito da aldos-terona se deve geralmente a doença renal (prejudicando asecreção de renina), disfunção adrenal (alterando a libera-ção de aldosterona) ou resistência tubular à ação da aldos-terona.
Na insuficiência adrenal com hipoaldosteronismo, se opaciente ingere uma dieta adequada em sal, não ocorrehipercalemia, talvez porque, havendo uma oferta adequa-da de sódio ao nefro distal, haverá secreção de potássio,apesar do hipoaldosteronismo. A hipercalemia é mais fre-qüentemente observada na crise addisoniana, que depen-de de uma depleção de sódio.1
Existe uma situação chamada hipoaldosteronismo hi-porreninêmico, que acomete principalmente idosos diabé-ticos com algum grau de insuficiência renal. Neles, a hi-percalemia seria causada por uma baixa produção de re-nina devido à lesão de células justaglomerulares. Esta se-ria também uma explicação para o fato de que os pacien-tes diabéticos são mais suscetíveis a desenvolverem hi-percalemia quando utilizam diuréticos poupadores de po-tássio.
A heparina e inibidores da enzima conversora tambémpodem suprimir a produção de aldosterona.
Diuréticos Poupadores (Retentores) de PotássioA utilização de espironolactona, amiloride e triamtere-
ne pode causar hipercalemia, sobretudo se empregados empacientes com insuficiência renal. Como já mencionamosnas páginas precedentes, a administração de diuréticospoupadores de potássio a pacientes diabéticos os predis-põe à hipercalemia.
UreterojejunostomiaO jejuno absorve o potássio existente na urina, provo-
cando elevação dos níveis sanguíneos deste íon.
Outras CausasTrimetoprim, antiinflamatórios não-esteróides.
Pontos-chave:
• A hipercalemia (potássio � 5,0 mEq/L)pode ocorrer por problemas durante acoleta ou por redistribuição, insuficiênciaadrenal e insuficiência renal
• É raro ocorrer hipercalemia sem disfunçãorenal
DIAGNÓSTICO DIFERENCIALAo se identificar uma hipercalemia, devemos diferen-
ciar entre uma falsa determinação laboratorial (pseudo-hipercalemia), fenômeno de redistribuição e um aumentoreal do potássio total (Quadro 12.5). Mais uma vez, a his-
Quadro 12.5 Diagnóstico diferencial dehipercalemia
I - Pseudo-hipercalemia1. Hemólise2. Trombocitose3. Leucocitose
II - Redistribuição1. Acidose2. Insulina3. Bloqueio �-adrenérgico4. Infusão de arginina5. Succinilcolina6. Intoxicação digitálica (superdose)7. Paralisia periódica
III - Retenção de potássio
RFG � 5 ml/min — 1. Oligoanúria2. Carga de potássio
a. exógenab. endógena — necrose
tissularhemólisehipercata-bolismo
RFG � 20 ml/min
� Aldosterona 1. Doença de Addison2. Hipoaldosteronismo
hiporreninêmico3. Inibição de prostaglandina
sintetaseAldosterona 1. Tubulopatias primárias
normal a. Adquiridas— transplante renal— lúpus eritematoso— amilóide— anemia de células
falciformesb. Hereditárias
2. Drogasa. espironolactonab. amiloridec. triamterene
Modificado de Narins, R.G.; Heilig, C.W.; Kupin, W.L.41
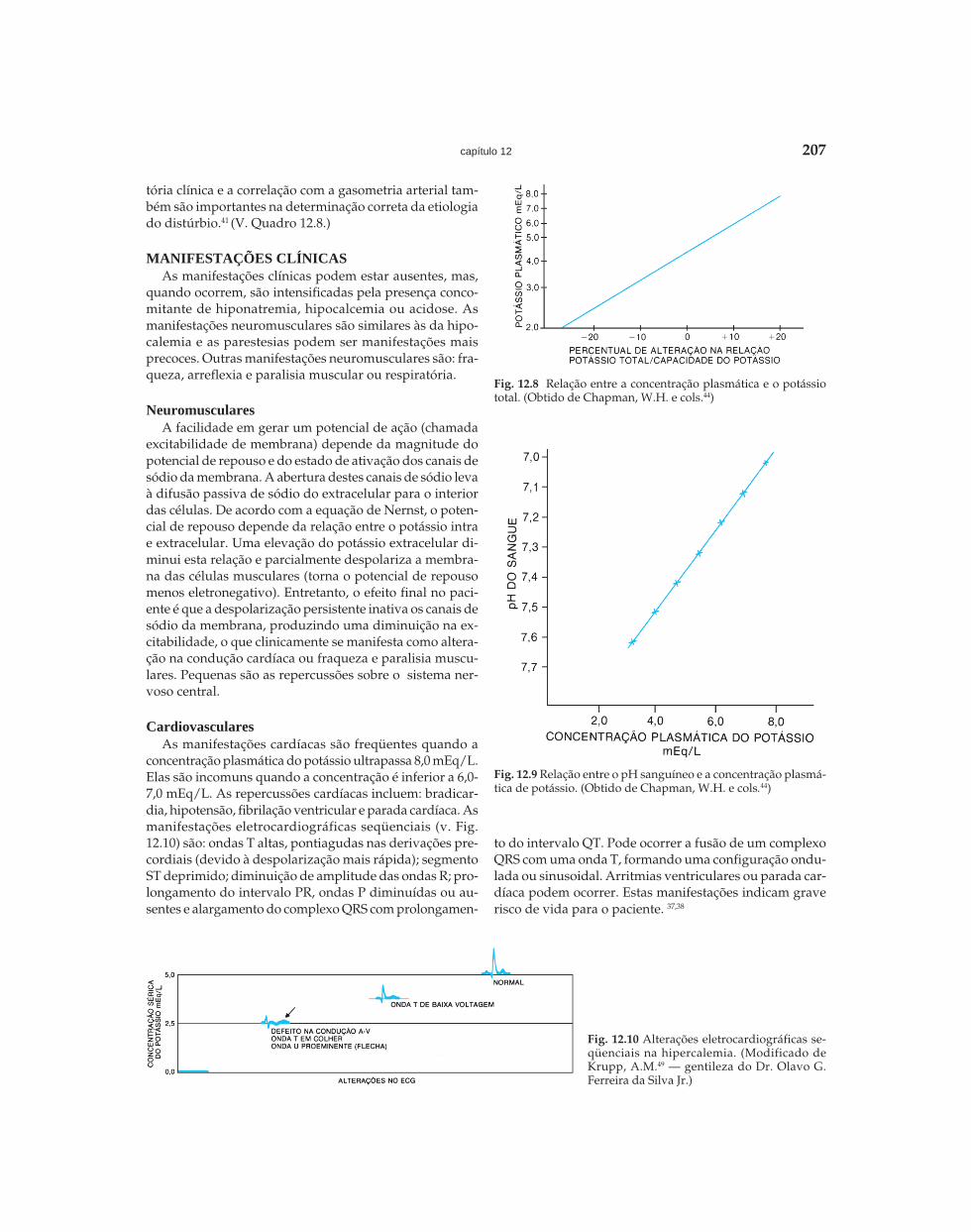
capítulo 12 207
tória clínica e a correlação com a gasometria arterial tam-bém são importantes na determinação correta da etiologiado distúrbio.41 (V. Quadro 12.8.)
MANIFESTAÇÕES CLÍNICASAs manifestações clínicas podem estar ausentes, mas,
quando ocorrem, são intensificadas pela presença conco-mitante de hiponatremia, hipocalcemia ou acidose. Asmanifestações neuromusculares são similares às da hipo-calemia e as parestesias podem ser manifestações maisprecoces. Outras manifestações neuromusculares são: fra-queza, arreflexia e paralisia muscular ou respiratória.
NeuromuscularesA facilidade em gerar um potencial de ação (chamada
excitabilidade de membrana) depende da magnitude dopotencial de repouso e do estado de ativação dos canais desódio da membrana. A abertura destes canais de sódio levaà difusão passiva de sódio do extracelular para o interiordas células. De acordo com a equação de Nernst, o poten-cial de repouso depende da relação entre o potássio intrae extracelular. Uma elevação do potássio extracelular di-minui esta relação e parcialmente despolariza a membra-na das células musculares (torna o potencial de repousomenos eletronegativo). Entretanto, o efeito final no paci-ente é que a despolarização persistente inativa os canais desódio da membrana, produzindo uma diminuição na ex-citabilidade, o que clinicamente se manifesta como altera-ção na condução cardíaca ou fraqueza e paralisia muscu-lares. Pequenas são as repercussões sobre o sistema ner-voso central.
CardiovascularesAs manifestações cardíacas são freqüentes quando a
concentração plasmática do potássio ultrapassa 8,0 mEq/L.Elas são incomuns quando a concentração é inferior a 6,0-7,0 mEq/L. As repercussões cardíacas incluem: bradicar-dia, hipotensão, fibrilação ventricular e parada cardíaca. Asmanifestações eletrocardiográficas seqüenciais (v. Fig.12.10) são: ondas T altas, pontiagudas nas derivações pre-cordiais (devido à despolarização mais rápida); segmentoST deprimido; diminuição de amplitude das ondas R; pro-longamento do intervalo PR, ondas P diminuídas ou au-sentes e alargamento do complexo QRS com prolongamen-
to do intervalo QT. Pode ocorrer a fusão de um complexoQRS com uma onda T, formando uma configuração ondu-lada ou sinusoidal. Arritmias ventriculares ou parada car-díaca podem ocorrer. Estas manifestações indicam graverisco de vida para o paciente. 37,38
Fig. 12.8 Relação entre a concentração plasmática e o potássiototal. (Obtido de Chapman, W.H. e cols.44)
Fig. 12.10 Alterações eletrocardiográficas se-qüenciais na hipercalemia. (Modificado deKrupp, A.M.49 — gentileza do Dr. Olavo G.Ferreira da Silva Jr.)
Fig. 12.9 Relação entre o pH sanguíneo e a concentração plasmá-tica de potássio. (Obtido de Chapman, W.H. e cols.44)
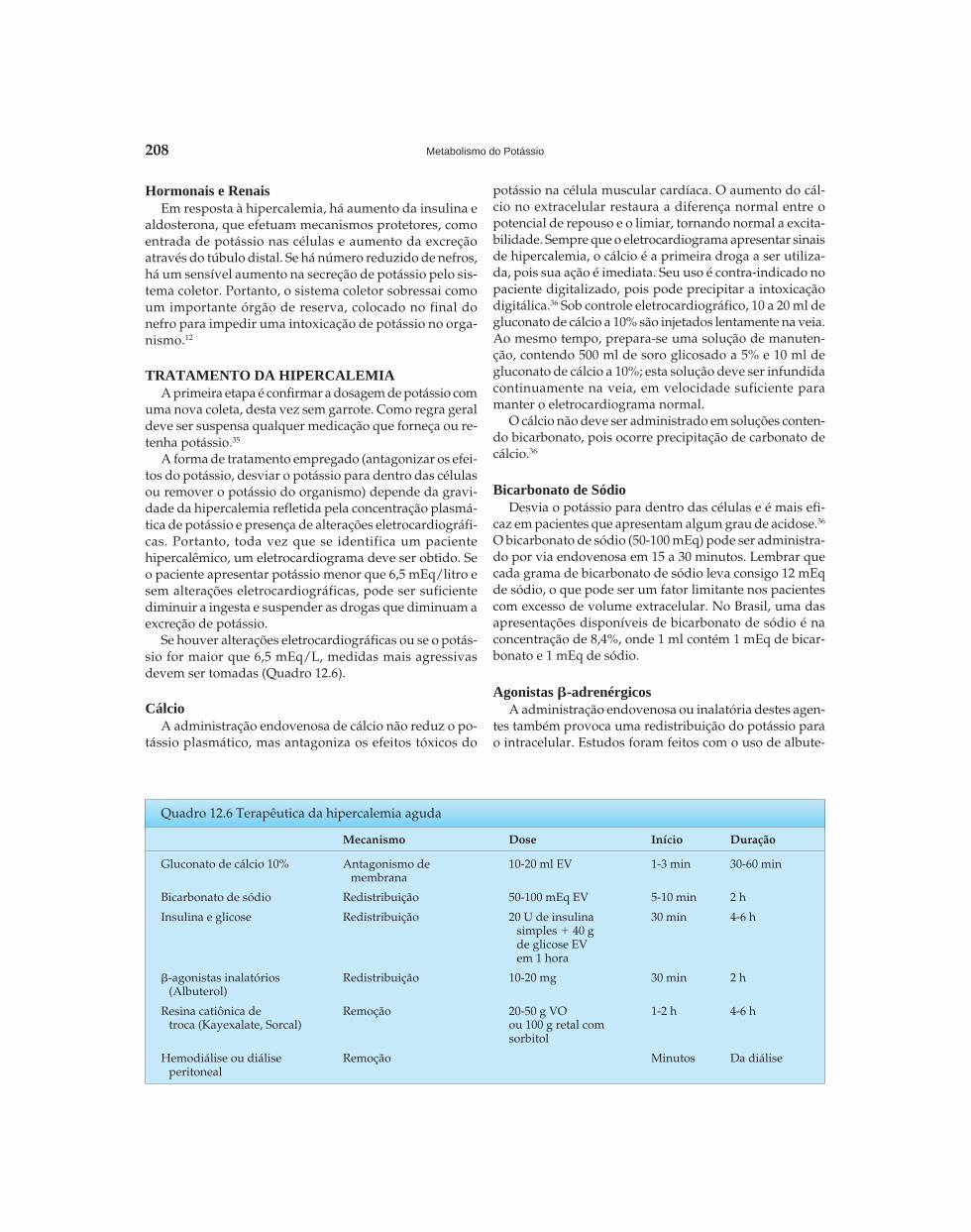
208 Metabolismo do Potássio
Hormonais e RenaisEm resposta à hipercalemia, há aumento da insulina e
aldosterona, que efetuam mecanismos protetores, comoentrada de potássio nas células e aumento da excreçãoatravés do túbulo distal. Se há número reduzido de nefros,há um sensível aumento na secreção de potássio pelo sis-tema coletor. Portanto, o sistema coletor sobressai comoum importante órgão de reserva, colocado no final donefro para impedir uma intoxicação de potássio no orga-nismo.12
TRATAMENTO DA HIPERCALEMIAA primeira etapa é confirmar a dosagem de potássio com
uma nova coleta, desta vez sem garrote. Como regra geraldeve ser suspensa qualquer medicação que forneça ou re-tenha potássio.35
A forma de tratamento empregado (antagonizar os efei-tos do potássio, desviar o potássio para dentro das célulasou remover o potássio do organismo) depende da gravi-dade da hipercalemia refletida pela concentração plasmá-tica de potássio e presença de alterações eletrocardiográfi-cas. Portanto, toda vez que se identifica um pacientehipercalêmico, um eletrocardiograma deve ser obtido. Seo paciente apresentar potássio menor que 6,5 mEq/litro esem alterações eletrocardiográficas, pode ser suficientediminuir a ingesta e suspender as drogas que diminuam aexcreção de potássio.
Se houver alterações eletrocardiográficas ou se o potás-sio for maior que 6,5 mEq/L, medidas mais agressivasdevem ser tomadas (Quadro 12.6).
CálcioA administração endovenosa de cálcio não reduz o po-
tássio plasmático, mas antagoniza os efeitos tóxicos do
potássio na célula muscular cardíaca. O aumento do cál-cio no extracelular restaura a diferença normal entre opotencial de repouso e o limiar, tornando normal a excita-bilidade. Sempre que o eletrocardiograma apresentar sinaisde hipercalemia, o cálcio é a primeira droga a ser utiliza-da, pois sua ação é imediata. Seu uso é contra-indicado nopaciente digitalizado, pois pode precipitar a intoxicaçãodigitálica.36 Sob controle eletrocardiográfico, 10 a 20 ml degluconato de cálcio a 10% são injetados lentamente na veia.Ao mesmo tempo, prepara-se uma solução de manuten-ção, contendo 500 ml de soro glicosado a 5% e 10 ml degluconato de cálcio a 10%; esta solução deve ser infundidacontinuamente na veia, em velocidade suficiente paramanter o eletrocardiograma normal.
O cálcio não deve ser administrado em soluções conten-do bicarbonato, pois ocorre precipitação de carbonato decálcio.36
Bicarbonato de SódioDesvia o potássio para dentro das células e é mais efi-
caz em pacientes que apresentam algum grau de acidose.36
O bicarbonato de sódio (50-100 mEq) pode ser administra-do por via endovenosa em 15 a 30 minutos. Lembrar quecada grama de bicarbonato de sódio leva consigo 12 mEqde sódio, o que pode ser um fator limitante nos pacientescom excesso de volume extracelular. No Brasil, uma dasapresentações disponíveis de bicarbonato de sódio é naconcentração de 8,4%, onde 1 ml contém 1 mEq de bicar-bonato e 1 mEq de sódio.
Agonistas �-adrenérgicosA administração endovenosa ou inalatória destes agen-
tes também provoca uma redistribuição do potássio parao intracelular. Estudos foram feitos com o uso de albute-
Quadro 12.6 Terapêutica da hipercalemia aguda
Mecanismo Dose Início Duração
Gluconato de cálcio 10% Antagonismo de 10-20 ml EV 1-3 min 30-60 minmembrana
Bicarbonato de sódio Redistribuição 50-100 mEq EV 5-10 min 2 h
Insulina e glicose Redistribuição 20 U de insulina 30 min 4-6 hsimples � 40 gde glicose EVem 1 hora
�-agonistas inalatórios Redistribuição 10-20 mg 30 min 2 h(Albuterol)
Resina catiônica de Remoção 20-50 g VO 1-2 h 4-6 htroca (Kayexalate, Sorcal) ou 100 g retal com
sorbitol
Hemodiálise ou diálise Remoção Minutos Da diáliseperitoneal

capítulo 12 209
rol, 10-20 mg por via inalatória em 4 ml de solução salina,ou 0,5 mg via endovenosa (no Brasil, o albuterol não é dis-ponível). Também pode ser utilizada a epinefrina via en-dovenosa (0,05 �g/kg/minuto). Deve ser lembrado que aabsorção via inalatória é errática e a administração endo-venosa é potencialmente arritmogênica. Outros efeitos in-cluem: taquicardia e angina de peito em indivíduos susce-tíveis. Então, estes agentes devem ser evitados em pacien-tes com doença coronariana. Em pacientes renais crônicos,que muitas vezes têm doença coronariana subclínica, deveser feita monitorização cuidadosa.36
Infusão de Glicose-InsulinaDesvia o potássio para dentro das células, causando
rápida redução do potássio plasmático. Pode-se utilizar 1unidade de insulina para cada 2 g de glicose. Se o pacientenão estiver alimentando-se e para evitar hipoglicemia, re-comenda-se administrar 4 g de glicose para cada unida-
de de insulina. Costuma-se gotejar na veia 200 ml de soroglicosado a 20% com 20 unidades de insulina, durante 60minutos. É necessária cuidadosa observação para sinais dehipoglicemia, como sonolência, sudorese, taquicardia.
Resinas de TrocaAs resinas de troca removem o potássio do organismo,
mas atuam mais lentamente. As resinas são substânciasque, administradas por via oral ou retal, promovem a tro-ca de sódio ou cálcio (dependendo da resina empregada)pelo potássio plasmático. Elas são capazes de remover 1mEq de potássio por grama de resina. É importante lem-brar que as resinas que trocam sódio por potássio (1,7 a 2,5mEq de Na�/mEq de K�) podem acarretar um excesso desódio no organismo e, conseqüentemente, determinar so-brecarga cardiovascular. No Brasil, a resina disponível é àbase de poliestirenossulfonato de cálcio (Sorcal), apresen-tada em envelopes de 30 gramas.
Quadro 12.7 Diagnóstico da hipocalemia
RedistribuiçãoPseudo-hipocalemia
Adaptado de Narins, R.G.; Heilig, C.W.; Kupin, W.L.41
Depleção real de potássio
Perda extra-renal(potássio urinário � 20 mEq/L)
Perda renal(potássio urinário � 20 mEq/L)
Bicarbonato Pressão Arterial
Baixo Normal Alto Elevada Normal
Renina Plasmática Bicarbonato
Alta AltoBaixa Baixo
Aldosterona Acidose TubularRenal
Cloreto Urinário
� 10 mEq/dia� 10 mEq/diaAlta Baixa
VômitosHiperaldos-teronismoprimário
DiuréticosVômitosFístulagástrica
Sudoreseprofusa
DiarréiasFístulasintestinaisbaixas
Hipertensão:MalignaRenovascular
Túbulo secretorrenina
S. de CushingMineralocorticóideHiperpasia
congênitade adrenais
DiuréticosS. de BartterHipomagnesemiaHiperaldos-teronismocom pressãonormal
Depleção extremade potássio
HIPOCALEMIA

210 Metabolismo do Potássio
DiáliseQuando os métodos conservadores falham, o tratamen-
to dialítico remove o potássio do organismo (v. Cap. 48).
MineralocorticóidesA fludrocortisona é usada no tratamento dos pacien-
tes com hipoaldosteronismo, porém com monitorização
apropriada para sintomas de sobrecarga de volume, umavez que retém sódio ao mesmo tempo em que eliminapotássio.
EXERCÍCIOS
1) Um homem de 70 kg e sem perda aparente de massa muscular chegaao hospital após um quadro de gastroenterite, e a investigação labo-ratorial mostra um potássio plasmático de 2,8 mEq/L. Calcular opotássio total e a percentagem de déficit.
2) Um paciente chega ao hospital após três dias de vômitos e a investi-gação mostra um pH de 7,6 e um potássio plasmático de 3,0 mEq/L.Qual seria a concentração de potássio com pH de 7,4?
3) Um paciente etilista, com quadro de vômitos há três dias, vem aopronto-socorro. Seu espaço extracelular está reduzido em 20%. Potás-sio = 2,3 mEq/L; pH de 7,52 e bicarbonato de 40 mEq/L. Que distúr-bio de potássio apresenta e qual a causa?
4) Ao ser chamado(a) para avaliar uma paciente diabética, renal crôni-ca, com potássio de 6,8, qual sua conduta?
5) Paciente de 27 anos, admitido na UTI em mal epiléptico após overdosede cocaína. pH � 6,9; bicarbonato � 12 mEq/L; potássio � 8,5 mEq/L.
Quadro 12.8 Diagnóstico da hipercalemia
HIPERCALEMIA
PSEUDO-HIPERCALEMIA REDISTRIBUIÇÃOGarrote AcidoseHemólise HiperglicemiaLeucocitose Beta-bloqueadoresTrombocitose Succinilcolina
Intoxicação digitálicaParalisia periódica
EXCESSO REAL DE POTÁSSIO
TFG � 10 ml/min TFG � 20 ml/minOligúria de qualquer causaAporte de potássio
ExógenoEndógeno
HemóliseNecrose de tecidoHipercatabolismo
Aldosterona baixa
Renina plasmática baixaHipoaldosteronismo
hiporreninêmicoInibição da PG sintetaseCiclosporina
Renina plasmáticanormal ou alta
Doença de AddisonDefeitos hereditários na síntese de aldosteronaHeparinaInibidores da enzima conversora
Adaptado de Narins, R.G.; Heilig, C.W.; Kupin, W.L.41
Aldosterona normal ou altaDesordens tubulares primárias
Transplante renalLúpus eritematosoAmiloidoseAnemia falciformeUropatia obstrutiva
DrogasEspironolactonaTriamtereneAmilorideTrimetoprim
Pontos-chave:
• A hipercalemia é um distúrbio grave,principalmente por suas repercussões sobrea condução cardíaca
• Eletrocardiograma sempre deve sersolicitado na hipercalemia
• Os achados no ECG determinam a rapidezcom que deve ser tratada a hipercalemia

capítulo 12 211
Urina acastanhada, positiva para hemoglobina. Enzimas musculareselevadas. Explique os motivos pelos quais este paciente apresentahipocalemia e qual é o potássio real para um pH de 7,4.
REFERÊNCIAS BIBLIOGRÁFICAS
1. BLACK, D.A.K. Potassium metabolism. Cap. 4, p. 121. In: ClinicalDisorders of Fluid and Electrolyte Metabolism. Eds. Maxwell, M.H. eKleeman, C.R. McGraw-Hill Book Co., 1972.
2. GUYTON, A.C. and HALL, J.E. Integration of renal mechanisms forcontrol of blood volume and extracelular fluid volume; and renalregulation of potassium, calcium, phosphate and magnesium. In:Textbook of Medical Physiology. Cap. 29, p. 367-383. W.B. Saunders Co.,1996.
3. SEGURO, A.C.; MALNIC, G.; ZATZ, R. Distúrbios do metabolismodo potássio. In: Fisiopatologia Renal, Ed. Zatz, R. Cap. 8, p. 123-150.Atheneu, 2000.
4. BRENNER, B.; COE, F.L.; RECTOR, F.C. Potassium Homeostasis. In:Renal Physiology in Health and Disease. Eds: Brenner, B.; Coe, F.L.;Rector, F.C. W.B. Saunders Co., 1987.
5. PATRICK, J. Assessment of body potassium stores. Kidney Int.,11(6):476, 1977.
6. BODDY, K. e cols. The relation between potassium in body fluidsand total body potassium in healthy and diabetic subjects. Clin. Sci.Mol. Med., 49:385, 1975.
7. BLAHD, W.H. Radioisotope techniques, Cap. 15, p. 613. In: ClinicalDisorders of Fluid and Electrolyte Metabolism. Eds. Maxwell, M.H. eKleeman, C.R. McGraw-Hill Book Co., 1972.
8. SCRIBNER, B.H. e BURNELL, J.M. Interpretation of the serum po-tassium concentration. Metabolism, 5:468, 1956.
9. Symposium on Ion Channels. Kidney International, vol. 48, out. 1995.10. JAMISON, R.L. e cols. Potassium secretion by the descending limb
of pars recta of the juxtamedullary nephron in vivo. Kidney Int., 9:323,1976.
11. KHURI, R.N. e col. Effects of flow rate and potassium intake on dis-tal tubule potassium transfer. Am. J. Physiol., 228:1.249, 1975.
12. GRANTHAM, J.J. Renal transport and excretion of potassium. Cap.8, p. 299. In: The Kidney. Eds. Brenner, B.M. e Rector Jr., F.C. W.B.Saunders Co., 1976.
13. HALPERIN, M.L.; GOLDSTEIN, M.B. Potassium Physiology. In:Fluid, Electrolyte and Acid-Base Physiology – A Problem-Based Appro-ach. Cap. 9, p. 321-424. W.B. Saunders Co., 1994.
14. KNOCHEL, J.P. Role of glucoregulatory hormones in potassiumhomeostasis. Kidney Int., 11(6):443, 1977.
15. WALKER, B.R. e col. Hyperkalemia after triamterene in diabeticpatients. Clin. Pharmacol. Ther., 13:643, 1972.
16. SHAPIRO, A.P. e cols. Effect of thiazides on carbohydrate meta-bolism in patients with hypertension. N. Engl. J. Med., 265:1028,1961.
17. ELLIS, S. e BECKETTS, S.B. Mechanism of the potassium mobilizingaction of epinephrine and glucagon. J. Pharmacol. Exp. Ther., 142:318,1963.
18. CRAIG, A.B.J.R. e col. Blockade of hyperkalemia and hyperglycemiainduced by epinephrine in frog liver and in cats. Am. J. Physiol.,197:52, 1959.
19. SEALEY, J.E. e LARAGH, J.H.A. Proposed cybernetic system forsodium and potassium homeostasis: Coordination of aldosteroneand intrarenal physical factors. Kidney Int., 6:281, 1974.
20. BODY, J.E. e MULROW, P.J. Further studies of the influence of po-tassium upon aldosterone production in the rat. Endocrinology,90:299, 1972.
21. SILVA, P. e cols. Adaptation to potassium. Kidney Int., 11(6):466,1977.
22. WRIGHT, F.S. Relation of electrical potencial difference to potassiumsecretion by the distal renal tubule. Int. Congr. Physiol. Sci., 11:115,1974.
23. ALEXANDER, E.A. e LEVINSKY, N.G. An extrarenal mechanismof potassium adaptation. J. Clin. Invest., 47:740, 1968.
24. TANNER, R.L. Relationship of renal ammonia production and po-tassium homeostasis. Kidney Int., 11(6):453, 1977.
25. PITTS, R.F. Control of renal production of ammonia. Kidney Int.,1:297, 1972.
26. BURNELL, J.M. e col. Metabolic acidosis accompanying potassiumdeprivation. Am. J. Physiol., 227:329, 1974.
27. BRANDIS, M. e cols. Potassium induced inhibition of proximal tu-bular fluid reabsorption in rats. Am. J. Physiol., 222:421, 1972.
28. SCHWARTZ, W.B. e cols. Acidification of urine and increasedammonium excretion without change in acid-base equilibrium:sodium reabsorption as a stimulus to acidifying process. J. Clin.Invest., 34:673, 1955.
29. BAERTL, J.M. e cols. Relation of acute potassium depletion to renalammonium metabolism in patients with cirrhosis. J. Clin. Invest.,42:696, 1963.
30. van YPERSELE de STRIHOU, C. Potassium homeostasis in renalfailure. Kidney Int., 11(6):491, 1977.
31. BANK, N. e AYNEDJIAN, H.S. A micropuncture study of potas-sium excretion by the remnant kidney. J. Clin. Invest., 52:1.480, 1973.
32. WEIDMAN, P. e cols. Plasma aldosterone in terminal renal failure.Ann. Intern. Med., 78:13, 1973.
33. GERSTEIN, A.R. e cols. Aldosterone deficiency in chronic renal fai-lure. Nephron, 5:90, 1968.
34. HAYES, C.P. e cols. An extrarenal mechanism of the maintenanceof potassium balance in severe chronic renal failure. Trans. Assoc. Am.Physicians, 80:207, 1967.
35. PRESTON, R.A. Hypokalemia and Hyperkalemia. In: Acid-Base,Fluids and Electrolytes Made Ridiculously Simple. Caps. 5 e 6, p. 77-96.Medmaster Inc. Miami, 1997.
36. ROSE, B. Potassium. In:. Up to Date, vol. 9, n.1, 2000.37. GABOW, P.A.; PETERSON, L.N. Disorders of potassium metabo-
lism. In: Renal and Electrolyte Disorders. Cap. 5, p. 231-285, Ed: Schrier,R.W. Little, Brown, 4th edition, 1992.
38. ROSE, B.D.; RENNKE, H.G. Disorders of potassium balance. In: Re-nal Pathophysiology — the Essentials. Cap. 7, p. 169-190. Eds: Rose, B.D.;Rennke, H.G. Williams & Wilkins.
39. SUKI, W.N. e EKNOYAN, G. Tubulo-interstitial disease. Cap. 25,p. 1.126. In: The Kidney. Eds. Brenner, B. M. and Rector Jr, F.C. W.B.Saunders Co., 1976.
40. MUEHRCKE, R.C. e McMILLAN, J.C. The relationship of chronicpyelonephritis to chronic potassium deficiency. Ann. Intern. Med.,59:427, 1963.
41. NARINS, R.G.; HEILIG, C.W.; KUPIN, W.L. The patient with hypo-kalemia or hyperkalemia. In: Manual of Nephrology, Cap. 3, p. 37-54.Ed.: Schrier, R.W. Little, Brown, 4th edition, 1995.
42. HAMILL, R.J. Efficacy and safety of potassium infusion therapy inhypokalemia critically ill patients. Critical Care Medicine, 19(6):694,1991.
43. KASSIRER, J.P. e HARRINGTON, J.T. Diuretics and potassium me-tabolism: a reassessment of the need, effectiveness and safety ofpotassium therapy. Kidney Int., 11(6):505, 1977.
44. CHAPMAN, W.H. e cols. The Urinary System. Cap. 4, p. 114. W.B.Saunders Co., 1973.
45. SCHWARTZ, A.B. e SWARTZ, C.D. Dosage of potassium chlorideelixir to correct thiazide-induced hypokalemia. JAMA, 230:702, 1974.
46. BOEDEKER, E.C. e DAUBER, J.H. Manual of Medical Therapeutics.Cap. 2, p. 44, 21st edition. Little, Brown and Co., Boston, 1974.
47. DeFRONZO, R.A.; SMITH, J.D. Disorders of potassium metabolism.In: Fluid, Electrolyte and Acid-Base Disorders, Eds: Arieff, A.I.; DeFron-zo, R.A., Churchill Livingstone, 1995.
48. LEAF, A.; COTRAN, R.S. Renal Pathophysiology, 2nd edition, NewYork, Oxford University Press, 1980.
49. KRUPP, A.M. Fluid and electrolyte disorders. Ap. 2, pp. 13-30. In:Current Diagnosis and Treatment. Eds: Krupp, M.A. e Chatton, M.J.Lange Medical Publications, 1973. (Gentileza do Dr. Olavo G. Fer-reira da Silva Jr.)

212 Metabolismo do Potássio
ENDEREÇOS RELEVANTES NA INTERNET
http://www.emedicine.com/emerg/topic273.htm — comexemplos de ECG.http://www.learndoctor.com/chapterpages/chapter22.htm — questões para self-assessment com respostas dis-cutidas – Chris O’Callaghan e Barry Brenner.http://www.seaox.com/lz/lz20-b.html — exemplos deECG em hipo- e hipercalemia.http://www.barttersite.com/hyper&hypoK.htm — pági-na muito boa, com um artigo de revisão da Medical Cli-nics of North America de 1997.http://www.medinfo.ufl.edu/year2/clinmed/Nephro-logy/Potassium.PDF — artigo com bom resumo da clíni-ca de hipo- e hipercalemia, inclusive com as alterações deECG e potencial de ação.
RESPOSTAS DOS EXERCÍCIOS
1) Com a ajuda do Quadro 12.4, obtém-se: 45 mEq � 70 kg � 3.150 mEq.Como não há distúrbio ácido-básico, verificamos, na Fig. 12.8, que umpotássio plasmático de 2,8 corresponde a um déficit de aproximada-mente 13% do potássio total, ou seja, em torno de 400 mEq.
2) Na Fig. 12.9, verificamos que, se não houver alteração no potássio total,a concentração normal de potássio para um pH de 7,6 seria 3,0 mEq/L.
Isto significa que, se o pH fosse corrigido para 7,4, o potássio plasmá-tico seria de 4,5 mEq/L.
3) Este paciente apresenta hipocalemia (potássio menor que 3,5 mEq/L),que provavelmente se deve à perda renal de potássio, uma vez que adepleção do espaço extracelular ativa o sistema renina-angiotensina-aldosterona, aumentando a excreção renal de potássio. Além disso, obicarbonato age como um ânion pouco reabsorvível, carregando só-dio para o túbulo coletor, o que também aumenta a secreção de po-tássio na luz tubular. A alcalose metabólica que este paciente apre-senta pode ter ocasionado um desvio iônico de cerca de 0,6 mEq/Lde potássio para o intracelular; seu potássio real deve ser em torno de2,3 � 0,6 � 2,9 mEq/L.
4) Interromper qualquer administração de potássio. Obter um eletrocar-diograma. A presença de ondas T apiculadas confirma a hipercale-mia verdadeira. Neste caso, é necessária intervenção imediata paraantagonizar os efeitos tóxicos do potássio sobre a fibra cardíaca (ad-ministrar cálcio EV). Prosseguir com as outras etapas de tratamentoda hipercalemia: bicarbonato, glicose-insulina, agentes �2-adrenérgi-cos, resinas de troca e diálise. Afastar a possibilidade de redistribui-ção. Afastar a possibilidade de pseudo-hipercalemia.
5) Este paciente apresenta dados compatíveis com rabdomiólise, possi-velmente decorrente das convulsões prolongadas. Além disso, apre-senta acidose metabólica, que pode ter sido causada pelo metabolis-mo anaeróbio induzido pela hipoxemia e convulsões. O potássio do-sado é de 8,5 para um pH de 6,9. O potássio real deste paciente paraum pH de 7,4 é de 5,5. As causas da hipercalemia neste caso poderi-am ser: redistribuição, pela acidose metabólica, e destruição de célu-las musculares, principal reservatório de potássio no organismo. De-vemos realizar um ECG imediatamente e tratar a hipercalemia deacordo com a seqüência já mencionada.

Capítulo
13Metabolismo do Cálcio, Fósforo e Magnésio
Marcelo Mazza do Nascimento, Miguel Carlos Riella e Marcos Alexandre Vieira
CÁLCIO
Introdução
Homeostase do cálcio
Distribuição do cálcio
Absorção, excreção, balanço interno
Fatores que regulam a homeostase do cálcio
PTH e vitamina D
Funções no organismo
Hipocalcemia
Definição
Causas de hipocalcemia
Diagnóstico
Quadro clínico
Tratamento
Hipercalcemia
Definição
Resposta adaptativa
Causas de hipercalcemia
Quadro clínico
Diagnóstico
Tratamento
FÓSFORO
Introdução
Homeostase do fósforo
Distribuição
Absorção, excreção e balanço interno
Mecanismos de transporte
Fatores que regulam a excreção de fósforo
Funções do fósforo no organismo
Hipofosfatemia
Introdução
Causas
Quadro clínico
Diagnóstico
Formas de apresentação
Hiperfosfatemia
Introdução
Causas
Pseudo-hiperfosfatemia
Quadro clínico
Tratamento
MAGNÉSIO
Homeostase do magnésio
Distribuição
Unidades de medida
Absorção, excreção e balanço interno
Fatores que influenciam a excreção de magnésio
Funções do magnésio no organismo
Hipomagnesemia
Causas
Quadro clínico
Diagnóstico
Tratamento
Hipermagnesemia
Definição
Causas de hipermagnesemia
Quadro clínico
Tratamento
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET

214 Metabolismo do Cálcio, Fósforo e Magnésio
absorvido no duodeno (400 mg). O suco digestivo acrescecerca de 200 mg de cálcio nas 24 horas, perfazendo no to-tal uma absorção diária de 600 mg.
Os mecanismos de transporte do cálcio são realizadostanto de forma ativa quanto passiva. O transporte ativo sedá principalmente pela presença de sódio na luz intestinal,baixa concentração de cálcio e ação do calcitriol. O meca-nismo não dependente de energia ocorre quando a concen-tração de cálcio no lúmen intestinal é alta (13 mg/dl).1,2,3
RinsA filtração renal do cálcio se dá pela sua porção difusí-
vel (complexos na forma de vários sais e fração ionizável),isto é, 60% do cálcio total. A reabsorção tubular do cálcioacontece principalmente no túbulo contornado proximal,ramo ascendente espesso da alça de Henle e no túbulocontornado distal.
Ponto-chave:
• Cálculo da concentração plasmática decálcio na presença de hipoalbuminemia[Ca] corrigido � [Ca] medido � 0,8 � (4,5 �[albumina]
[Ca] medido em mg/dlAlbumina medida em g/dlExemplo: [Ca] medido 7,6 mg/dlAlbumina 2,5 g/dl
[Ca] corrigido � 7,6 � 0,8 � 2 � 9,2 mg/dl
No túbulo contornado proximal, o cálcio é reabsorvidoconjuntamente com o sódio, e em estados de depleção devolume extracelular a sua reabsorção é aumentada. Emsituações de expansão do espaço extracelular, porém, ocor-re o inverso. Cerca de 60% do cálcio filtrado é reabsorvidono túbulo contornado proximal.
No ramo espesso ascendente da alça de Henle, outros20 a 25% do cálcio filtrado são reabsorvidos, e drogas queatuam neste segmento específico do néfron, como o furo-semide, aumentam a excreção de cálcio, como se verá pos-teriormente.
CÁLCIO
Introdução
A manutenção da homeostase do cálcio é de fundamen-tal importância, do ponto de vista fisiológico, metabólicoe estrutural, em nosso organismo. Sua participação na cas-cata da coagulação, reações enzimáticas e na transmissãoneuromuscular dá a dimensão de sua importância para quese mantenham níveis plasmáticos normais.
Os mecanismos fisiológicos necessários à manutençãode níveis séricos normais de cálcio, bem como as alteraçõesdeste equilíbrio (hipocalcemia, hipercalcemia), serão dis-cutidos a seguir.
Homeostase do Cálcio
DISTRIBUIÇÃO DO CÁLCIOCerca de 99% do cálcio do nosso organismo encontra-
se no esqueleto. Um indivíduo normal de 70 kg contémaproximadamente 1,2 kg de cálcio. Deste total, 5,3 g estãono fluido intracelular, 1,3 g no fluido extracelular (exclu-indo-se ossos) e mais de 1 kg encontra-se nos ossos sob aforma de cristais de hidroxiapatita.
A distribuição sanguínea do cálcio se dá da seguintemaneira: cerca de 50% na forma difusível (cálcio ionizávele na forma de complexos) e o restante, não-difusível, liga-do às proteínas plasmáticas.
Como a albumina é a proteína mais abundante no plas-ma, 90% do cálcio ligado às proteínas encontra-se ligado aela. Sendo assim, a diminuição dos níveis séricos de albu-mina determina alterações na concentração de cálcio séri-co total. Por exemplo, a diminuição em 1,0 g/dl da concen-tração sérica de albumina diminui a concentração de cál-cio total em 0,8 mg/dl. As alterações da concentração séri-ca de globulinas determinam menores variações na con-centração de cálcio sérico (1,0 g/dl de globulina para 0,12mg/dl de cálcio total).
A porção do cálcio difusível se divide em fração ionizá-vel, 90% do total (ultrafiltrável), e o restante formandocomplexos com bicarbonato, citrato, fosfato, lactato e sul-fato. A fração ionizável de cálcio varia com o pH sanguí-neo, sendo que a alcalose diminui a concentração de cál-cio ionizável, ao contrário da acidose. Alteração em 0,1unidade no pH sérico modifica a ligação proteína-cálcio em0,12 mg/dl.1,2,3
ABSORÇÃO, EXCREÇÃO, BALANÇO INTERNO
Absorção Intestinal Um indivíduo normal ingere aproximadamente 1.000
mg de cálcio elementar ao dia (15 mg/kg/dia). Dependen-do da concentração de 1,25-diidroxivitamina D3 (calcitri-ol) e do conteúdo de cálcio na dieta, 20 a 40% deste total é
Ponto-chave:
• No túbulo contornado proximal o cálcio éreabsorvido conjuntamente com o sódio ena presença de depleção extracelular a suareabsorção aumenta. Na presença deexpansão extracelular, ocorre o inverso, eisto pode ser usado no tratamento dahipercalcemia

capítulo 13 215
A regulação da reabsorção do cálcio ocorre no túbulocontornado distal (10% do total), pela ação do PTH e docalcitriol. Estas substâncias aumentam a reabsorção localatravés de mecanismos ativos da bomba de cálcio e trocasde sódio por cálcio.
Fatores que Regulam aHomeostase do Cálcio
PTH E VITAMINA DA vitamina D3 é formada a partir da dieta e da clivagem
fotolítica na pele do 7-desidrocolesterol. A vitamina D2, pro-veniente de fonte dietética (ergosterol), juntamente com avitamina D3, são as formas ativas da vitamina D no sangue.
No fígado a vitamina D sofre a ação da 25-hidroxivita-mina D3 (calcidiol), que nos rins, pela ação da 1�-hidroxi-lase diidroxivitamina D3 (1�-hidroxilase), transforma-seem calcitriol.
Em situações de hipocalcemia ou em estados de deman-da de cálcio, é feita a conversão de calcidiol em calcitriol,porém em estados de normocalcemia o calcitriol não é for-mado em grande quantidade.1,2,3
O calcitriol aumenta o transporte de cálcio no intestino,age no néfron distal aumentando a reabsorção de cálcio enos ossos aumenta a mobilização de cálcio.
O PTH em situações de hipocalcemia tem sua sínteseaumentada e agirá sobre os ossos aumentando a ativida-de das células reabsortivas (osteoclastos). Nos rins, no tú-bulo distal aumenta a reabsorção de cálcio e estimula aatividade da enzima 1�-hidroxilase, com conseqüentemaior síntese de calcitriol. A normocalcemia e o aumentodo calcitriol agem como inibidores da secreção de PTH.
Já em situações de hipercalcemia, dar-se-á uma inibição daprodução de PTH e um estímulo à liberação de calcitonina
pela tireóide, fazendo com que aconteça uma diminuição naprodução de calcitriol, normalizando os níveis de cálcio.
Os principais reguladores da atividade da 1�-hidroxi-lase são o PTH (estímulo) e o fósforo inorgânico, sendo quea hiperfosfatemia possui uma ação inibidora da atividadeenzimática, ao contrário da hipofosfatemia.
Funções no OrganismoA função do cálcio no organismo humano a nível celu-
lar se dá principalmente pela estabilização das membra-nas celulares e pelo transporte de sódio e potássio. Seupapel em processos como a endocitose e a exocitose estábem estabelecido.1,2,3,7
No osso exerce função estrutural. A transmissão neuro-muscular e a excitação nervosa são dependentes do cálcio, jáque este regula a entrada de sódio e potássio no interior dacélula, necessária para a propagação do potencial de ação.7
No músculo o cálcio se liga à superfície da célula deter-minando o nível de despolarização necessária para que seinicie a contração. Também a intensidade de contraçãodepende da concentração de cálcio ionizado a nível intra-celular. Daí se percebe que as alterações para cima ou parabaixo dos níveis de cálcio causam sinais e sintomas princi-palmente a nível neuromuscular.
Hipocalcemia
DEFINIÇÃOA queda do cálcio sérico total abaixo de 8,8 mg/dl é
indicativo de hipocalcemia, porém isto não define umadiminuição da concentração da fração ionizável, já queexiste uma ligação do cálcio à albumina. Para cada quedade albumina em 1,0 g/dl abaixo de 4,0 g/dl, adicionamos0,8 mg/dl à concentração total de cálcio plasmático (v. fór-mula mais precisa no ponto-chave, anteriormente).
Nas situações de hipocalcemia, a resposta do organismose dá pela atuação das paratireóides na liberação de PTH.
Quando a normocalcemia é atingida, diminui a secre-ção de PTH, e este efeito de retroalimentação negativo éexercido e estimulado pelo aumento do calcitriol e norma-lização dos níveis de cálcio.7,8
Fig. 13.1 Demonstração das áreas e da proporção de reabsorçãode cálcio.
HIPOCALCEMIA
PTH
Fração excretora de cálcio MobilizaçãoósseaAtividade da 1�-hidroxilase
25(OH)D3
Calcitriol
NORMOCALCEMIA

216 Metabolismo do Cálcio, Fósforo e Magnésio
CAUSAS DE HIPOCALCEMIASão inúmeras as causas de hipocalcemia. As principais
que se apresentam na prática clínica serão comentadas aseguir (v. Quadro 13.1).
HipoparatireoidismoIdiopático. A forma idiopática se encontra ligada a de-
feitos na embriogênese ou sendo parte de síndromespoliglandulares. Na primeira forma, há uma ausência con-gênita das quatro glândulas que, quando associada à au-sência de timo, é conhecida como síndrome de DiGeorge.1,10
Quando associado a síndromes poliglandulares, incluiinsuficiência supra-renal, hipogonadismo primário, diabe-tes mellitus, hepatite crônica ativa, má absorção, anemiaperniciosa. Anticorpos antiparatireóide são encontradosem até 40% dos casos.10
Pós-cirúrgico. A forma mais comum de hipoparatireoi-dismo é a cirurgia na região cervical (doença de Graves,das paratireóides, câncer de tireóide). Estes pacientes po-dem desenvolver hipocalcemia grave com 24 horas de pós-operatório. A hipofosfatemia e a hipomagnesemia acom-panham o quadro clínico.10
Pseudo-hipoparatireoidismoNesta síndrome há uma resistência periférica (rins e
esqueleto) à ação do PTH. A osteodistrofia hereditária deAlbright (pseudo-hipoparatireoidismo do tipo 1), com suasalterações somáticas características (face arredondada,pescoço grosso, retardo mental, encurtamento de falange,tórax em barril), é o exemplo clássico deste quadro, que temcomo característica uma não-formação de AMP cíclico(AMPc) em resposta ao PTH.1,2,10
Os pacientes apresentam sinais de hipocalcemia crôni-ca (catarata, achados neuromusculares, dentição anormal,
alterações cardiovasculares). O diagnóstico é feito por umnão-aumento do AMP cíclico urinário à infusão de PTH.
No pseudo-hipoparatireoidismo tipo 2, há a formaçãode AMPc urinário, porém com uma resposta fosfatúricaprejudicada (diminuída). Os níveis de PTH no sangue seapresentam normais ou elevados.
Hipomagnesemia (v. Hipomagnesemia)A hipocalcemia vista nos pacientes com deficiência em
magnésio acontece principalmente nos etilistas, que con-comitantemente apresentam má absorção intestinal e dé-ficit de vitamina D. Níveis séricos menores que 0,8 mEq/Lde magnésio atuam sobre as paratireóides diminuindo aliberação e a ação do PTH; nos ossos, reduzindo a mobili-zação de cálcio e inibindo sua ação diretamente no túbulorenal. A hipocalcemia nesta situação só será corrigida comreposição de magnésio.11
Hiperfosfatemia (v. Hiperfosfatemia)A hiperfosfatemia causa diminuição na produção de
calcitriol, pela inibição da atividade da 1�-hidroxilase, comconseqüente menor formação de calcitriol diminuindo aabsorção intestinal e óssea de cálcio. A infusão de fósforopode fazer com que haja precipitação de cálcio quando oproduto cálcio � fósforo atinge 70.2,3,13,14
Drogas AnticonvulsivantesCerca de 20% dos pacientes epilépticos recebendo dro-
gas anticonvulsivantes apresentam hipocalcemia e osteo-malácia.
Níveis subnormais de calcitriol, por inibição da 1�-hi-droxilase ou maior degradação enzimática do calcidiol nohepatócito, são hipóteses que tentam explicar este achado.
Relacionadas à Vitamina DMá Absorção. Encontrada em etilistas, idosos e pacien-
tes com esteatorréia (lipossolubilidade da vitamina D), queapresentam absorção diminuída de vitamina D.
Drogas. Rifampicina, isoniazida e cetoconazol podemdiminuir a síntese de calcitriol e calcidiol. A gentamicinapode causar hipocalcemia por mecanismo indireto devi-do à perda de magnésio pela urina.
Pontos-chave:
• Hipocalcemia resistente ao tratamento podeser secundária a hipomagnesemia e sómelhora com a correção dos níveis séricosde magnésio
• Hiperfosfatemia inibe a atividade da 1�-hidroxilase, diminuindo a produção decalcitriol e logo diminuindo a reabsorçãointestinal de cálcio
Quadro 13.1 Causas de hipocalcemia
HIPOPARATIREOIDISMO PRIMÁRIOIdiopáticoPós-cirúrgico
PSEUDO-HIPOPARATIREOIDISMOHIPOCALCEMIA ASSOCIADA A DOENÇA MALIGNAHIPOMAGNESEMIASÍNDROME DO CHOQUE TÓXICO NEONATALPANCREATITE AGUDAINSUFICIÊNCIA RENALHIPERFOSFATEMIA RELACIONADA À VITAMINA D
Dietético (baixa ingesta)Má absorçãoTerapia anticonvulsivanteDoença hepáticaRaquitismo dependente de vitamina D
DOENÇAS TUBULARES RENAISDROGAS (mitramicina, colchicina, furosemide, citrato
endovenoso, drogas anticonvulsivantes)

capítulo 13 217
Doença Hepática Crônica. A hipocalcemia pode ocor-rer nesta situação pela deficiência na 25-hidroxilação davitamina D e deficiência na formação de bile (diminuiçãona absorção intestinal da vitamina D).
Raquitismo Dependente de Vitamina D. Há dois tiposfundamentais:
Tipo I – Deficiência enzimática da 1�-hidroxilase.Tipo II – Resistência periférica à ação ao calcitriol.
Estes pacientes desenvolvem hipofosfatemia grave, pordiminuição da absorção intestinal de fósforo, e fosfatúriadevido ao hiperparatireoidismo secundário, ocasionandodeformidades ósseas significativas.8,10,12
Causas RenaisSíndrome Nefrótica. A diminuição dos níveis de
calcidiol tem sido relatada, proporcionalmente à intensi-dade da proteinúria e da hipoalbuminemia. Suspeita-seque a perda da proteína ligante de vitamina D seja elimi-nada na urina.
Disfunções Tubulares. As disfunções tubulares distaise proximais podem causar hipocalcemia e raquitismo. Ainterferência da acidose na produção de calcitriol tem sidodescrita.
Insuficiência Renal Crônica. A hipocalcemia aparecedevido à retenção de fósforo e diminuição da produção decalcitriol, com conseqüente hiperparatireoidismo secundá-rio.1,2,3,6,13 A concentração sérica elevada ou normal decalcidiol, com calcitriol diminuído, sugere a presença deinsuficiência renal crônica ou raquitismo dependente devitamina D.
Outras CausasDoenças Malignas. Câncer de próstata, mama, ou leu-
cemia aguda podem causar hipocalcemia (não devido àhipoalbuminemia) em decorrência de lesões osteoblásticasno esqueleto que captam cálcio.
Pancreatite Aguda. Várias causas concorrem para a hi-pocalcemia nesta situação: insuficiência renal aguda, ní-veis elevados de calcitonina, necrose gordurosa, que nes-ta situação formam sais insolúveis de gordura com o cál-cio.
Hipocalcemia Pré-natal. Níveis baixos de PTH e níveiselevados de calcitonina nos três primeiros dias de nasci-mento podem ser responsáveis por esta síndrome. Ocorremais em filhos de mães diabéticas, prematuros e com an-gústia respiratória.
Síndrome do Choque Tóxico. Ocorre em mulheres jo-vens que utilizam tampões durante a menstruação e é pro-duzida por algumas cepas de estafilococo que provocamo choque endotóxico.
Alcalose Respiratória. Aumenta a ligação do cálcio io-nizável à albumina.
DIAGNÓSTICOO diagnóstico de hipocalcemia deve levar em conta a sua
fração ionizável, que se detecta por medidas diretas (nor-mal de 4,75 a 5,2 mg/dl) ou do cálcio sérico total, conside-rando as correções quanto à medida de albumina sérica epH (v. discussão anterior).
O comportamento dos níveis séricos de fósforo podeauxiliar na descoberta da etiologia da hipocalcemia. A hi-perfosfatemia sugere hipoparatireoidismo, pseudo-hipo-paratireoidismo e insuficiência renal, enquanto a hipofos-fatemia é comumente observada nos casos de hiperparati-reoidismo secundário (diminuição na produção renal decalcitriol) e em outros distúrbios da vitamina D.
Medidas séricas do PTH podem distinguir os pacientescom hipoparatireoidismo primário de pacientes com pseu-do-hipoparatireoidismo.
A medida na urina de fósforo, cálcio e AMP cíclico apósinfusão de PTH (teste de Ellsworth-Howard) auxilia nodiagnóstico diferencial de hipoparatireoidismo primário,que apresenta aumento dos níveis de AMPc e fósforo comdiminuição da excreção de cálcio, não havendo nenhumamudança destes parâmetros quando da suspeita de pseu-do-hipoparatireoidismo.
A concentração de calcidiol se encontra diminuída nospacientes com má absorção intestinal e déficit de vitaminaD. A suspeita de hipomagnesemia, como causa de hipo-calcemia, dá-se quando os níveis plasmáticos de magné-sio se encontram abaixo de 1,2 mg/dl.1,2,3,7,8,12,13,14 Nos paci-entes com concentração diminuída de calcidiol, a presen-ça de hipocalcemia e hipofosfatemia são indicadores debaixa absorção e ingestão de alimentos. As concentraçõesde PTH devem ser dosadas conjuntamente com cálcio sé-rico e variam conforme a causa de hipocalcemia. Pacien-tes com hipomagnesemia podem ter PTH elevado, normalou baixo. Sua concentração geralmente é reduzida nospacientes com hipoparatireoidismo. Anormalidades comopseudo-hipoparatireoidismo ou distúrbios no metabolis-mo da vitamina D apresentam concentrações de PTH ele-vadas.
QUADRO CLÍNICOAs principais manifestações clínicas encontradas na hi-
pocalcemia são principalmente de caráter neuromuscular(v. Quadro 13.2).
Neuromuscular. Tetania e convulsões são as manifes-tações mais graves. A tetania latente pode ser demonstra-da pelo sinal de Chvostek (encontrado em 10% da popu-lação normal) percutindo-se o nervo facial após sua saídado canal auditivo, sendo positivo quando se observa umacontração da musculatura da hemiface.1,2,3
Outro sinal para se detectar tetania incipiente é o deTrousseau, que não se encontra em pessoas normais econsiste em se insuflar o manguito do aparelho de pres-são arterial 3 mmHg acima da pressão arterial sistólica por

218 Metabolismo do Cálcio, Fósforo e Magnésio
3 min, observando-se então a pressão de contração espas-módica dos músculos da região. O sinal é negativo em 34%dos pacientes com hipocalcemia latente.
Convulsões e distúrbios emocionais, como irritabilida-de, labilidade emocional, alucinações e depressão, tambémsão observados.1,2,3
Cardiovascular. Hipotensão arterial e arritmias (cujoeletrocardiograma aponta prolongamento do intervalo QTe alterações de onda T) têm sido descritas.
Lesões Dermatológicas. Anormalidades da pele, unhas,dentes e oculares são vistas na hipocalcemia crônica.
Gastroenterológicas. Constipação e dor abdominalpodem fazer parte do quadro. Diarréia com deficiência deabsorção de vitamina B6 e gorduras ocasionalmente podemaparecer.
TRATAMENTODeve-se tratar a hipocalcemia quando o valor corrigido
de cálcio sérico total é inferior a 7 mg/dl e naqueles paci-entes cujos sintomas neuromusculares (tetania, parestesi-as, convulsões) estão presentes.
A terapêutica também se divide quanto à apresentaçãona forma aguda e crônica.
Forma Aguda. A situação clínica mais evidente nestaforma de apresentação é pós-paratireoidectomia. A abor-dagem nesta situação deve ser encarada como urgente. Ossintomas geralmente estão presentes quando os valores decálcio total são menores que 7,0 mg/dl.
A administração endovenosa de gluconato de cálcioa 10% (1 a 2 g de gluconato de cálcio-cálcio intravenoso,100 a 200 mg de cálcio elementar), infundindo num tem-po não inferior a 10 minutos, é a abordagem inicial. Ou-tras formas de apresentação incluem o cloreto e o citratode cálcio. O gluconato de cálcio é a apresentação esco-lhida geralmente pela menor propensão a necrose teci-
dual quando ocorre infusão rápida ou no extravasamentotecidual.
A administração de cálcio deve ser feita até o desapare-cimento dos sintomas, repetindo a infusão de gluconato a10% lentamente na dose de 0,5 a 1,5 mg/kg/hora (90 mgde cálcio elementar em 10 ml da ampola), com monitora-ção dos níveis séricos, até atingir uma concentração decálcio total de 8,0 mg/dl.
Alguns cuidados devem ser tomados quanto à infusãode cálcio.
• Pacientes tomando digital. A infusão de cálcio aumen-ta a sensibilidade miocárdica à intoxicação por digital,devendo-se fazer a monitoração cardíaca durante a in-fusão.
• Hipopotassemia e hipomagnesemia devem ser corrigi-das.
• Irritação endovenosa pode acontecer, se a solução formuito concentrada.
• A solução não deve ter bicarbonato ou fosfato, pois po-dem formar complexos insolúveis com o cálcio.
Forma Crônica. Administração oral de cálcio e vitami-na D nas situações de deficiência vitamínica e diminuiçãoda função das paratireóides.
Cálcio na forma oral deve ser dado para que se atinjauma concentração de 1 g de cálcio elementar ao dia. Asformas de apresentação incluem o carbonato de cálcio emcomprimidos de 500 mg com 400 mg de cálcio elementar,e acetato de cálcio, comprimidos de 350 mg com 87,5 mgde cálcio elementar.
Quadro 13.2 Manifestações clínicas da hipocalcemia
NEUROMUSCULARESTetaniaConvulsõesPapiledemaAnsiedade, depressão, psicose
ECTODÉRMICASPele secaPerda de cabeloCatarataEczema
CARDIOVASCULARESHipotensãoArritmiasInsuficiência cardíaca
GASTRINTESTINALEsteatorréia
Pontos-chave:
• Hipocalcemia: Ca�� � 8,8 mg/dl• Relação cálcio-albumina: Redução de 1,0 g/
dl no valor da albumina abaixo de 4,0 eleva0,8 no valor do cálcio total
• Diagnóstico: Nível de cálcio total corrigidoou cálcio ionizável. Correlacionar comníveis de fósforo e PTH e calcidiol parafacilitar o diagnóstico. Pesquisar sinais deTrousseau e Chvostek
• Quadro clínico: Caracteriza-se pormanifestações neuromusculares
• Tratamento: Forma aguda — Gluconato decálcio 10% lentamente. Correção domagnésio e potássio concomitante senecessário
• Tratamento: Forma crônica — Cálcio oral,vitamina D ou tiazídicos conforme aetiologia

capítulo 13 219
Vitamina D. A vitamina D, como já foi visto, aumentaos níveis séricos de cálcio, conseqüentemente provocandohipercalciúria. Devido a este fato, nefrocalcinose e calcifi-cação de tecidos moles podem ser observadas.
A monitoração dos níveis de cálcio no sangue e na uri-na deve ser feita periodicamente. As formas de vitaminaD mais utilizadas são o calcitriol (biologicamente maispotente — de 0,5 a 1,0 �g) e a outra menos ativa, o ergo-calciferol (vitamina D3 — 1 a 10 �g/dia).
A hiperfosfatemia pode acontecer com a correção dahipocalcemia pela vitamina D e aumentar os riscos de ne-frocalcinose e calcificação de partes moles. Manutenção dosníveis de fósforo através da dieta e drogas como acetozo-lamide pode ser de utilidade clínica.
Tiazídicos. Limitam a excreção urinária de cálcio, dimi-nuindo as necessidades de vitamina D e cálcio (v. Hiper-calcemia).1,2,3,13
Hipercalcemia
DEFINIÇÃOA hipercalcemia é definida quando os níveis séricos de
cálcio total são superiores a 10,5 mg/dl. Em aproximada-mente 80% dos casos, as causas mais comuns são hiperpa-ratireoidismo e tumores malignos.
RESPOSTA ADAPTATIVAOs eventos metabólicos principais que ocorrem em res-
posta à hipercalcemia são apresentados abaixo:A hipercalcemia provoca a diminuição na liberação de
PTH e aumento na produção de calcitonina, provocandodiminuição na atividade da 1�-hidroxilase e conseqüen-
CAUSAS DE HIPERCALCEMIAAs duas principais causas de hipercalcemia são hi-
perparatireoidismo e malignidade. As etiologias prin-cipais deste distúrbio serão descritas a seguir (v. Qua-dro 13.3).
Hiperparatireoidismo PrimárioIncidência. O hiperparatireoidismo primário apresen-
ta uma média anual de incidência na população de 22 ca-sos por 100.000, havendo um aumento progressivo com aidade, sendo duas vezes mais comum nas mulheres do quenos homens.
Causas. Cerca de 85% dos pacientes com hiperparati-reoidismo primário têm como causa principal o adenomasimples de uma das quatro glândulas da paratireóide. Orestante se deve à hiperplasia e carcinoma, este responsá-vel por menos de 1% dos casos.
As causas podem ser de origem genética ou devido àirradiação da região cervical. A causa genética mais conhe-cida é a neoplasia endócrina do tipo I. Prévia irradiação dacabeça e pescoço pode dar origem a adenomas, numa in-cidência que pode atingir 4 a 11%. A hipercalcemia se de-senvolve pelo aumento da produção de PTH, com conse-qüente aumento na reabsorção tubular de cálcio e diminui-ção de sua excreção. O conseqüente estímulo da atividadeosteoclástica e aumento do turnover ósseo é demonstradopelo aumento dos níveis de fosfatase alcalina, osteocalci-na e hidroxiprolina urinária.
O aumento dos níveis de calcitriol determina incrementoda absorção intestinal de cálcio. O PTH também eleva a
Atividade 1�-hidroxilase25(OH)D3
Fração excretora de cálcio Mobilizaçãoóssea
Calcitriol
NORMOCALCEMIA
Liberação de PTHe
Calcitonina
HIPERCALCEMIA
Quadro 13.3 Causas de hipercalcemia
HIPERPARATIREOIDISMO• Primário• Terciário: Má absorção/Insuficiência renal crônica
(IRC)
ASSOCIADA A DOENÇAS MALIGNASASSOCIADA A DOENÇAS ENDÓCRINAS• Hipertireoidismo• Feocromocitoma• Insuficiência adrenal
SARCOIDOSE E OUTRAS DOENÇASGRANULOMATOSAS
INTOXICAÇÃO POR VITAMINA DINTOXICAÇÃO POR VITAMINA ASÍNDROME ÁLCALI-LEITEIMOBILIZAÇÃO PROLONGADAASSOCIADA A DROGAS• Diuréticos tiazídicos• Carbonato de lítio• Estrógenos
DOENÇA DE PAGETHIPERCALCEMIA IDIOPÁTICA DA INFÂNCIA
te redução do calcitriol. Por conseguinte, isto se traduz naredução da absorção e reabsorção do cálcio na luz intes-tinal e no túbulo distal, respectivamente, com aumentoda fração excretora de cálcio e diminuição da mobiliza-ção do cálcio ao nível ósseo, levando à normocalce-mia.1,2,5,17

220 Metabolismo do Cálcio, Fósforo e Magnésio
excreção de fósforo e bicarbonato urinário, devido à dimi-nuição da reabsorção no túbulo proximal destes íons eocasionando uma acidose metabólica hiperclorêmica, alémde elevação do AMPc urinário.
Diagnóstico. Além da hipercalcemia, a elevação dosníveis de PTH (tanto fração terminal como região médiada molécula) é característica da doença. Os pacientes seapresentam habitualmente com cálculos renais recorren-tes e ocasionalmente com alterações ósseas características(aumento da reabsorção óssea pelo aumento da atividadeosteoclástica), levando à osteíte fibrosa e à osteopenia. Éimportante notar que pacientes apresentam carcinoma deparatireóide em 10% dos casos.
Tratamento. A cirurgia com remoção do tecido anormalda paratireóide é o tratamento de escolha. Naqueles paci-entes com quadro discreto, assintomáticos, o tratamentoclínico pode estar indicado, devendo o paciente ser acom-panhado freqüentemente (v. tratamento da hipercalcemia).Em mãos experimentadas, a cura pela cirurgia pode che-gar a 95% dos casos.1,2,5,6,12,17
MalignidadeIncidência. É a causa mais comum de hipercalcemia
encontrada em pacientes internados, sendo a segunda cau-sa mais freqüente, depois do hiperparatireoidismo. Estima-se a incidência de 135 casos de câncer por ano que desen-volvem hipercalcemia. Os principais tumores envolvidosestão descritos no Quadro 13.4.
Causas. Os mecanismos principais envolvidos no desen-volvimento de hipercalcemia incluem:
Produção de PTHrP (Peptídio Relacionado ao Parator-mônio). Esta substância produzida pelo tumor, com estru-tura de aminoácidos semelhante ao PTH, liga-se aos seusreceptores, aumentando a reabsorção tubular de cálcio etambém ao nível ósseo.
Os tumores mais envolvidos na produção de PTHrP são:tumor de células escamosas do pulmão, pescoço e carci-noma de células renais. A abordagem da hipercalcemiainduzida por câncer poderá ocorrer com a redução da li-beração de proteína relacionada ao PTH. O uso de análo-
gos do calcitriol, como 22-oxacalcitriol, pode ser uma al-ternativa no futuro.54
Produção de Fatores que Estimulam Osteoclastos eAnálogos da Vitamina D. Tumores hematológicos comoo mieloma causam hipercalcemia devido à liberação decitocinas produzidas pelas células malignas, que são osfatores de ativação do osteoclasto nas superfícies ósseas tra-beculares. Linfomas de células T podem produzir calcitri-ol. Os tumores de mama, além de poderem aumentar aabsorção óssea diretamente através de suas células malig-nas, podem produzir prostaglandinas que estimulam aatividade osteoclástica.2,5,17,18
Tireotoxicose É uma causa relativamente freqüente de hipercalcemia,
com incidência chegando a 10 a 20% dos pacientes porta-dores deste distúrbio. O hormônio tireoidiano age direta-mente no osso, acelerando o turnover ósseo. O tratamentodo hipertireoidismo é eficaz na diminuição dos níveis decálcio.2,5
Doenças GranulomatosasDentre as doenças granulomatosas, como tuberculose,
histoplasmose, candidíase e coccidioidomicose, destaca-sea sarcoidose como a principal causa de hipercalcemia.
A hipercalcemia na sarcoidose se deve ao fato dos ma-crófagos (localizados no pulmão destes pacientes) conver-terem o calcidiol em calcitriol. Isto provoca um aumentoda reabsorção intestinal de cálcio, com conseqüente su-pressão da produção de PTH, resultando em hipercalci-úria, formação de cálculos de oxalato de cálcio e nefro-calcinose.
O acometimento renal pela sarcoidose associado a ne-frocalcinose leva à insuficiência renal observada neste dis-túrbio. A medida da concentração sérica do PTH é de fun-damental importância no diagnóstico diferencial, pois o hi-perparatireoidismo pode ocorrer conjuntamente com a sar-coidose.1,2,5
ImobilizaçãoA imobilização prolongada é uma causa conhecida de
hipercalcemia e hipercalciúria. A perda de massa óssea éacompanhada de paralisia muscular de qualquer etiologia— a chamada osteoporose de desuso.
A hipercalcemia suprime a produção de PTH e forma-ção de calcitriol, promovendo a hipercalciúria e conseqüen-te nefrolitíase. A causa principal de imobilização é o trau-ma raquimedular, porém outras situações como poliomi-elite, síndrome de Guillain-Barré e queimaduras extensassão outras causas descritas.1,2,5
Intoxicação por Vitamina DA maior parte dos casos desenvolve-se durante o trata-
mento com vitamina D em casos de hipoparatireoidismo,
Quadro 13.4 Hipercalcemia e malignidade
INCIDÊNCIA(em porcentagem)
Pulmão 35Mama 25Hematológico 14
(mieloma, linfoma)Cabeça/Pescoço 6Renal 3Próstata 3Origem desconhecida 7Outros 8
Adaptado de Mundy, G.R. e Martin, T.J. Metabolism, 31:1247-77, 1982.

capítulo 13 221
doenças ósseas ou tentativas de minorar os efeitos do cor-ticóide sobre o esqueleto a longo prazo.
Também doses excessivas de suplementos vitamínicospodem causar intoxicação. A hipercalcemia é devida tan-to a um aumento na absorção óssea, como a um aumentona absorção intestinal. A presença de hiperfosfatemia, di-minuição da função renal (nefrocalcinose) e deposição te-cidual de cálcio nos tecidos são outros achados. Altas con-centrações de vitamina D aumentam a 25�-hidroxilaçãohepática, elevando os níveis de calcidiol. Por outro lado, a1�-hidroxilação no rim é inibida por altas concentraçõesde calcitriol, determinando nesta situação níveis altos decalcidiol e normais de calcitriol.1,2,5,8,16,17
DrogasDiuréticos Tiazídicos. A maior parte de seus efeitos no
metabolismo de cálcio pode ser explicada pela contraçãodo volume plasmático, associada à dieta hipossódica ge-ralmente prescrita aos pacientes hipertensos (aumentan-do a reabsorção proximal de cálcio conjuntamente com osódio, devido à depleção de volume extracelular). Especu-la-se um efeito potencializador do PTH nos rins.
Geralmente não se observam elevações significativasdos níveis séricos de cálcio (acima de 11 mg/dl), ocorren-do na maioria das vezes a reversão da hipercalciúria e hi-percalcemia. Elevações significativas do cálcio sérico comuso crônico de tiazídico devem levar à suspeita de outrasdoenças subjacentes, em especial o hiperparatireoidis-mo.1,2,5,17
Diuréticos de Alça. Agem diminuindo a reabsorção decálcio na alça de Henle, porém este efeito pode ser masca-rado pela contração de volume. Nestes casos, geralmenteassociados a dieta hipossódica, maior quantidade de cál-cio é reabsorvida no túbulo proximal e menos cálcio che-ga até a alça de Henle, podendo piorar estados prévios dehipercalcemia.1,2,5,17,39
Carbonato de Lítio. Têm sido descritos casos (em tor-no de 5%) de pacientes em uso desta mediação nos quais asuspensão da droga faz com que haja retorno dos níveisde cálcio sérico aos valores normais.
Pontos-chave:
• Hiperparatireoidismo e doença maligna sãoas principais causas de hipercalcemia
• Neoplasias de pulmão e mama são as maisfreqüentemente associadas à hipercalcemia
• Hipercalcemia e hipofosfatemia sugeremhiperparatireoidismo ou malignidade
• Mais recentemente o uso indiscriminado demultivitamínicos contendo vitamina D temsido associado à hipercalcemia
Aminofilina. A toxicidade por aminofilina tem sidorelatada como causa de hipercalcemia em até 20% dos ca-sos. A causa do distúrbio é desconhecida.1,2,5
Aspirina. Níveis tóxicos de ácido acetilsalicílico podemcausar hipercalcemia, sem aumento nos níveis de albumi-na.
Estrógenos e Antiestrógenos. Podem causar hipercal-cemia no tratamento do câncer de mama metastático.
Outras CausasSíndrome Álcali-leite. Esta doença está associada à in-
gestão de carbonato de sódio (forma de antiácido) maisleite. A fisiopatologia deste distúrbio envolve a hiperab-sorção intestinal de cálcio e álcali, como também a excre-ção urinária inadequada de cálcio, diminuição da funçãorenal e alcalose metabólica. Hoje em dia esta síndrome émenos comum, restringindo-se aos casos de uso de cálciono tratamento da osteoporose.2,5,17
Intoxicação por Vitamina A. A vitamina A é um fatorde estímulo à atividade do osteoclasto e, quando ingeridanuma quantidade superior a 50.000 UI/dia, pode causarosteopenia por diminuição da função do osteoblasto. Oachado radiológico característico é a calcificação laminarperiosteal que pode ser vista na radiografia das mãos.5,17
Hipercalcemia Hipocalciúrica Familiar. É uma patolo-gia que se caracteriza pela presença de hipercalcemia ediminuição na fração excretora de cálcio (menor que 100mg/g de creatinina), transmitindo-se como um traço au-tossômico dominante. Difere do hiperparatireoidismo porapresentar uma diminuição na fração excretora de cálcio eníveis normais de PTH. A presença de familiares com estedistúrbio auxilia no diagnóstico. A maioria dos pacientesnão requer tratamento.1,2,5,17
Doença de Addison. Os mecanismos envolvidos nestapatologia devem-se principalmente à contração de volu-me aumentando a reabsorção tubular de cálcio e deficiên-cia na ação do glicocorticóide, que normalmente possuiuma ação antivitamina D.17
Insuficiência Renal Aguda. A hipercalciúria pode, porsi só, causar insuficiência renal (mecanismo de vasocons-trição renal), principalmente na sarcoidose, mieloma, in-toxicação por vitamina D, ou ser sua conseqüência, comoocorre na fase de recuperação da insuficiência renal agu-da e na rabdomiólise.1,2,5,17
Insuficiência Renal Crônica. É observada principal-mente nos pacientes em hemodiálise em que a água usadano tratamento contém alta concentração de alumínio. Istoocorre pelo fato de que o alumínio, ao ser depositado noosso, retarda a formação óssea e inibe a atividade osteo-blástica. A osteomalácia resultante não responde à vitami-na D, e o osso não atua mais como depósito de cálcio, re-sultando em hipercalcemia.
A hipocalcemia crônica da IRC pode levar ao desenvol-vimento de hiperparatireoidismo secundário, por estímu-lo contínuo na glândula, tornando-se esta autônoma. O uso

222 Metabolismo do Cálcio, Fósforo e Magnésio
continuado de quelantes de fósforo, que contenham cálcio(carbonato de cálcio e acetato de cálcio), e de calcitriol podelevar à hipercalcemia.1,2,5,17
Pseudo-hipercalcemia. Elevações na concentração deproteínas plasmáticas no sangue podem levar ao aumen-to do cálcio sérico total porém sem aumento da fração li-vre. Isto pode ocorrer após infusão de grande quantidadede plasma (tratamento da púrpura trombocitopênicatrombótica) e no mieloma múltiplo, quando a proteína domieloma se liga ao cálcio, aumentando sua concentraçãosérica.1,5,17
QUADRO CLÍNICOSintomas Gerais. A hipercalcemia na sua forma leve
pode não apresentar sintomas, porém nos quadros maisgraves sintomas como anorexia, náuseas, vômitos, obnu-bilação, cefaléia, poliúria e nictúria podem estar presentes(v. Quadro 13.5).
Sistemas AfetadosNervoso. Embora os mecanismos não estejam comple-
tamente estabelecidos, o aumento do cálcio livre no siste-ma nervoso central pode acarretar diminuição da condu-ção nervosa nos terminais nervosos, traduzindo-se em le-targia em casos mais graves, confusão mental, coma.
Cardiovascular. Pacientes portadores de hipercalcemiadesenvolvem hipertensão arterial provavelmente por me-canismos de vasoconstrição.
No coração, o cálcio provoca um aumento da contratili-dade cardíaca. As alterações eletrocardiográficas mais co-muns são encurtamento do espaço PR e do QT, bloqueioAV de primeiro grau e alterações da onda T.
Gastrintestinal. A ação do cálcio na musculatura lisa econdução nervosa, além de seu efeito sobre a produção degastrina, aponta para as principais manifestações clínicas,
que são: constipação, anorexia, náuseas, vômitos e úlceraduodenal.
Renal. A litíase renal pode ser observada nos quadrosde hiperparatireoidismo. A nefrocalcinose (calcificaçãoparenquimatosa principalmente a nível da medula renal)não é necessariamente associada à litíase, sendo que nospacientes com hiperparatireoidismo as duas condiçõespodem ocorrer separadamente.
A insuficiência renal é multifatorial e decorre de:
• Obstrução tubular• Depósito parenquimatoso (nefrite intersticial)• Vasoconstrição renal, depleção do volume extracelular.
A correção da hipercalcemia pode reverter e melhorarsignificativamente o ritmo de filtração glomerular.
Outra anormalidade causada pela hipercalcemia é aresistência à ação do ADH nos túbulos coletores, cujo me-canismo exato não está estabelecido. Alcalose metabólica,devido ao aumento da capacidade de tamponamento ós-seo, pelo acometimento do esqueleto nos tumores malig-nos, pode ser responsável por este distúrbio ácido-básico.
Acidose tubular renal pode ser ocasionada nos pacien-tes com hiperparatireoidismo pela ação do PTH no túbulocontornado proximal, resultando em perda de bicarbona-to e conseqüente acidose metabólica hiperclorêmica. Per-das renais de sódio, magnésio e potássio também são des-critas na hipercalcemia.1,2,5,15,16,18
Pontos-chave:
• Hipercalcemia pode acarretar depleção dovolume extracelular e contribuir paraaumentar a reabsorção proximal de cálcio
• Na hipercalcemia há uma resistência à açãodo hormônio antidiurético nos túbuloscoletores, contribuindo para a poliúriaobservada na hipercalcemia
DIAGNÓSTICOCerca de 80 a 90% dos casos de hipercalcemia são cau-
sados por hiperparatireoidismo ou tumores malignos. Es-tes mais encontrados em pacientes hospitalizados e aque-le, em pacientes assintomáticos. A história clínica, o exa-me físico e a dosagem de PTH sérico oferecem uma preci-são diagnóstica em 99% dos casos.
Além disto, é importante destacar que no hiperparati-reoidismo alguns detalhes clínicos são de fundamentalimportância no auxílio diagnóstico, destacando-se:
• Hipercalcemia assintomática• História familiar ou evidência de neoplasia endócri-
na• Irradiação prévia na região cervical• Mulheres na menopausa.
Quadro 13.5 Sinais e sintomas de hipercalcemia
NEUROLÓGICOS: Confusão mental, estupor,irritabilidade, coma.
CARDIOVASCULARES: Aumento da contratilidademiocárdica, alterações no ECG (aumento do QTc,bloqueio AV de primeiro grau, etc.), hipertensãoarterial sistemática.
GASTRINTESTINAIS: Constipação, náusea, vômito,úlcera duodenal.
RENAIS: Nefrocalcinose, litíase renal, insuficiência renal,diabetes insipidus nefrogênico, distúrbios ácido-básicos(acidose e alcalose metabólica), perdas renais defosfato, magnésio, potássio, glicose e aminoácidos.
OCULAR: Calcificação da conjuntiva e da córnea.
HEMATOLÓGICO: Fibrose de medula óssea nos casosde hiperparatireoidismo secundário.

capítulo 13 223
Fósforo. A hipofosfatemia só acontece nas situações deelevação do PTH sérico, como no hiperparatireoidismo, ouna presença do PTHrP nos tumores malignos, em conse-qüência destes aumentarem a excreção de fósforo pelosrins.
A hiperfosfatemia estará presente nas outras situaçõesonde não acontece uma maior excreção de fósforo uriná-rio, como se presencia nas doenças granulomatosas, into-xicação por vitamina D, síndrome leite-álcali e tireotoxico-se, entre outras.
Cálcio Urinário. A dosagem do cálcio urinário é umimportante auxílio diagnóstico principalmente na síndro-me de hipercalcemia hipocalciúrica familiar, quando aevidência de uma dosagem de cálcio na urina menor que100 mg/g de creatinina faz o diagnóstico. Outras duas si-tuações em que se presencia a hipocalciúria são a síndro-me álcali-leite e o uso de tiazídicos.
Cloro. Devido à redução de bicarbonato (bicarbonatú-ria), evidenciada no hiperparatireoidismo, a dosagem decloro pode ser de ajuda diagnóstica, já que concentraçõesacima de 103 mEq/L podem ser encontradas. Na síndro-me leite-álcali se verá a situação inversa, ou seja, a presen-ça de alcalose metabólica com dosagem de cloro inferior a100 mEq/L.
RX. A presença de alterações radiológicas característi-cas da osteíte fibrosa — reabsorção subperiosteal falangi-ana, lesões císticas na clavícula e imagens de “pimenta esal” no crânio — é observada em 5% dos casos de hiperpa-ratireoidismo.
PTH. A dosagem da fração intacta do PTH, pelo méto-do imunorradiométrico, é o exame de escolha no diagnós-tico do hiperparatireoidismo primário. A presença de ní-veis de PTH normais deve ser vista com cuidado, pois ahipercalcemia persistente suprime a sua produção. A pre-sença de elevação do PTHrP acontece nos tumores malig-nos.17,18
Vitamina D. Quando as dosagens de PTH e PTHrP es-tão normais, e não for encontrada nenhuma evidência deneoplasia maligna, deve-se proceder à dosagem de calci-triol e calcidiol. O aumento do calcidiol sugere intoxicaçãopor vitamina D. O calcitriol se elevará nas seguintes con-dições clínicas: doenças granulomatosas, linfomas, produ-ção renal aumentada causada por hiperparatireoidis-mo.1,2,5,16,17,18
Deve-se iniciar o tratamento da hipercalcemia naquelespacientes sintomáticos ou que apresentam cálcio séricoacima de 15 mg/dl. Conforme o mecanismo fisiopatológi-co causador da hipercalcemia, modalidades terapêuticasdiferenciadas são instituídas (v. Quadro 13.6).
TRATAMENTOAborda-se este distúrbio tentando, conforme a causa
subjacente, agir sobre o mecanismo desencadeador da hi-percalcemia, promovendo:
• Diminuição da absorção intestinal de cálcio• Aumento na excreção urinária• Diminuição na reabsorção óssea• Quelação do cálcio ionizado.
Os pacientes assintomáticos que apresentam cálcio sé-rico com valores menores ou iguais a 13 mg/dl tambémdevem ser tratados, pelos efeitos deletérios da hipercalce-mia crônica. As principais drogas utilizadas no tratamen-to da hipercalcemia serão discutidas a seguir.1,2,5,16,17,18,19
Corticóides. São utilizados em pacientes nos quais acausa da hipercalcemia é uma maior absorção de cálciointestinal. Agem diretamente no epitélio intestinal, inibin-do a absorção de cálcio. Na sarcoidose e em outras doen-ças granulomatosas, têm seu efeito direto sobre a ativida-de da doença.
São eficazes nas doenças malignas em 30% dos casos, eespecialmente naqueles com doença hematológica. Têmseu efeito em torno de 7 a 10 dias do início de seu uso, uti-lizando-se prednisona na dose de 1 mg/kg/dia.1,2,5,17,19
Solução Salina e Furosemide. O cálcio é principalmen-te reabsorvido no túbulo contornado proximal e na alçade Henle, devido ao gradiente elétrico criado pela reab-sorção concomitante de sódio e cloro, neste segmento donéfron.
Desta forma, a inibição da reabsorção de sódio no tú-bulo contornado proximal inibe o transporte passivo decálcio, promovendo a calciúria. A expansão de volumeplasmático ocasionada pela infusão salina leva à natriure-se e conseqüentemente à excreção concomitante de cálciopela urina. Como há uma maior oferta de sódio, cálcio eágua na alça de Henle, a adição de furosemide inibe o trans-porte destes íons, incrementando o efeito calciúrico da in-fusão salina.
Deve-se ter cuidado antes da infusão de furosemide, jáque a maioria destes pacientes podem estar depletados
Quadro 13.6 Tratamento da hipercalcemia
Diminuição da absorção intestinal• Corticóide• Fosfato oral
Aumento da excreção urinária• Solução salina + furosemide
Diminuição na reabsorção óssea• Calcitonina• Mitramicina• Difosfonatos• Nitrato de gálioDiálise
Quelação do cálcio ionizado• EDTA• Fosfato endovenoso ou oral

224 Metabolismo do Cálcio, Fósforo e Magnésio
pelas condições clínicas subjacentes às quais estão sujeitose devido ao próprio efeito natriurético da hipercalcemia.A infusão prévia de solução salina, antes de administrarfurosemide, faz-se necessária.
O regime sugerido é a administração de solução salinaisotônica (3 a 5 L/dia), com a verificação e eventual repo-sição de potássio e magnésio sendo feita de acordo com asmedidas séricas. Utilizando-se este esquema, espera-senormalizar os níveis de cálcio em 12 a 24 horas.1,5,17
Regimes com maior infusão salina são descritos, inici-ando-se a infusão de 1 a 2 litros de solução salina isotônicaem período de 1 hora, acrescido de furosemide, 80 mg acada 2 horas, a fim de se manter um débito urinário nãoinferior a 250 ml/h. Este regime produz uma maior excre-ção urinária de sódio, fósforo, cálcio, cloro, magnésio eágua, sendo que se este tratamento tiver duração maior que100 horas, a infusão de magnésio se faz necessária, numritmo de 15 mg/h.
O volume urinário deve ser reposto a cada hora, comsolução contendo soro glicosado a 5%, 90 a 120 mEq/L desódio e 10 a 20 mEq/L de potássio. Haverá queda do cálciosérico, de 2 a 4 horas, havendo normalização em até 24 ho-ras. Deve-se lembrar da necessidade de reposição dos ou-tros eletrólitos (sódio, fósforo, cloro, magnésio), através desuas medidas na urina coletadas a intervalos de 4 horas.2
O regime deve ser cuidadosamente avaliado em paci-entes portadores de insuficiência cardíaca e renal onde asobrecarga de volume pode ser uma complicação. O diuré-tico só é utilizado quando se descarta a presença de deple-ção do espaço extracelular.1,2,5,17,18
Pontos-chave no manejo da hipercalcemia:
• Lembrar que a expansão do volumeextracelular inibe o transporte passivo decálcio, promovendo a calciúria
• A adição de um diurético de alça(furosemide) inibe a reabsorção de cálcio aeste nível mas não deve ser usado sem antescorrigir-se a depleção do volumeextracelular
Bifosfonatos. Este grupo de drogas se utiliza no trata-mento da hipercalcemia na malignidade, conjuntamentecom a infusão salina. Sua ação se dá pela inibição da ativi-dade osteoclástica, diminuindo os níveis de cálcio sérico ediminuindo a dor nos pacientes com lesões osteolíticas sig-nificativas.
O efeito máximo dos bifosfonatos se dá entre o quinto eo sétimo dia. Nos Estados Unidos, o etidronato e o pami-dronato são as drogas disponíveis, existindo uma terceiradroga, o clodronato, sendo maior a experiência clínica comas duas primeiras.
O etidronato (EDHP) deve ser iniciado na forma endo-venosa, 7,5 mg/kg/dia em 250 ml de solução salina em umperíodo não inferior a 4 horas, por três dias. O prolonga-mento do tratamento de três para cinco dias aumenta aresposta em 60 a 100%.
A duração da normocalcemia é de 1 a 7 semanas. A te-rapia deve ser mantida por via oral, na dose de 10 mg/kg/dia, e é ineficaz sem o uso intravenoso prévio. Os efeitoscolaterais mais importantes são a hiperfosfatemia, devidoa uma maior reabsorção de fosfato. A dosagem deve serreduzida em 50% na presença de insuficiência renal.
O pamidronato é mais potente que o etidronato e causamenor desmineralização óssea, sendo a droga de escolhaentre os bifosfonatos. É utilizada na forma intravenosa,numa dose inicial de 30 mg num período de infusão de 4horas em dose única, podendo ser repetida em setedias.2,5,17,18,19
Em caso de hipercalcemia grave, a dose pode ser de até90 mg. Várias preparações orais estão sendo propostas parasubstituir o pamidronato como agente de escolha na hiper-calcemia associada a malignidade.
Os bifosfonatos têm sido utilizados no tratamento dospacientes com hiperparatireoidismo primário, porém commenor eficácia do que nos pacientes portadores de neo-plasias.
Calcitonina. Utilizada na maioria das vezes em pacien-tes com hipercalcemia associada a malignidade e nos pa-cientes com função renal alterada. Em situações onde osbifosfonatos são contra-indicados, a calcitonina, na dose de4 UI/kg intramuscular ou subcutânea, age rapidamente,normalizando o cálcio em até 2 a 3 horas. A resposta ocor-re em 60 a 70% dos pacientes que a utilizam. Há contro-vérsia na literatura quanto ao uso simultâneo de corticói-de, no sentido de diminuir o aparecimento de resistênciaà calcitonina. Quando utilizado, administra-se hidrocorti-sona na dose de 100 mg a cada 6 horas.2,5,17,18
Mitramicina. É uma droga antineoplásica que inibe asíntese de DNA dependente de RNA e é altamente efetivano tratamento da hipercalcemia, principalmente associa-da às neoplasias. Utiliza-se na dose de 25 �g/kg em umperíodo de 6 horas, com pico máximo de ação em 12 ho-ras, durando seu efeito por alguns dias e repetindo-se adose a cada 3 a 7 dias.
Hepatotoxicidade, nefrotoxicidade e disfunção plaque-tária limitam o seu uso.2,5,17,18
Fósforo Oral. Tem utilidade no tratamento dos pacientesportadores de hiperparatireoidismo primário, com hipofos-fatemia (menor que 3,5 mg/dl), na dose de 1 a 3 g/dia.
Outros quelantes do cálcio ionizado, como o EDTA (áci-do etileno diaminotetracético), e o fósforo endovenosopodem determinar insuficiência renal, precipitação de cál-cio e fósforo em partes moles e arritmias fatais, limitandoa sua utilização.1,2,5,17,18
Estrógenos. Nos pacientes portadores de hiperparati-reoidismo primário, a cirurgia é a terapia de eleição, po-

capítulo 13 225
rém, quando a cirurgia é contra-indicada, em quadros dehipercalcemia sintomática, ou níveis superiores a 11 mg/dl nas mulheres em menopausa, os estrógenos podem serutilizados.
O seu modo de ação é desconhecido, podendo ser utili-zado como estilbestrol ou preparação equivalente de eti-nil estradiol.1,2,5,17
Cloroquina e Cetoconazol. Utilizados na sarcoidose.Devido a não existir grande experiência na utilização des-tes medicamentos, serão utilizados nos casos de ausênciade resposta ao corticóide e na contra-indicação destes.1,2,5,17
Nitrato de Gálio. É um inibidor da reabsorção óssea,com relatos de ser mais potente que o etidronato, sendoadministrado pela via endovenosa por até cinco dias, tor-nando seu uso de alto custo. Tem efeito nefrotóxico com-provado.1,2,5,17
Diálise. Está reservada àqueles pacientes que apresen-tam insuficiência cardíaca ou insuficiência renal em que ainfusão salina não pode ser suportada. A utilização de diá-lise peritoneal é uma alternativa à hemodiálise.1,2,5,15
Agentes calcimiméticos, como a norcalcina, ligam-se aosreceptores sensíveis ao cálcio e suprimem a liberação dePTH, podendo ser um tratamento futuro do hiperparati-reoidismo primário. O uso de anticorpos contra os hor-mônios responsáveis pela hipercalcemia pode ser feitofuturamente. Imunização em pacientes com hipercalcemiapor carcinoma de paratireóide também pode ter resulta-dos benéficos.59,60,61
FÓSFORO
Introdução
Embora o fósforo não seja o principal ânion em nossoorganismo (é o sexto mais abundante), seu papel é de fun-damental importância na manutenção do metabolismocelular, processo de mineralização óssea e manutenção doequilíbrio ácido-básico, entre outras funções.
A sua homeostase é dependente da interação entre ossistemas digestivo, ósseo e dos rins, cabendo ao parator-mônio (PTH) e à vitamina D a sua regulação.
Homeostase do Fósforo
DISTRIBUIÇÃOO fósforo representa 1% do peso corporal total. A sua
distribuição é a seguinte: 85% se encontram nos ossos, 14%nos tecidos moles e 1% no fluido extracelular.
O fósforo se apresenta no sangue principalmente naforma de fosfolipídio (fósforo orgânico), cerca de 70% dototal, sendo os 30% restantes na forma inorgânica. Nestaúltima forma, 15% estão ligados a proteínas e 75% são for-mas livres; destas, 50% formam fosfato monovalente e di-
valente, e outros 40% formam sais com sódio, magnésio ecálcio. Menos de 0,01% existe na forma de PO4. Os valoresnormais nos adultos vão de 2,5 a 4,5 mg/dl (0,81 a 1,45mmol).
ABSORÇÃO, EXCREÇÃO E BALANÇO INTERNOOs mecanismos envolvidos na regulação do fósforo
envolvem o trato gastrintestinal, rins e ossos. O PTH, cál-cio, vitamina D e a calcitonina desempenham papel fun-damental na homeostase deste ânion.
Absorção Intestinal. A ingestão diária varia de 800 a1.850 mg/dia; deste total, 65% são absorvidos principal-mente a nível de duodeno e jejuno.
No duodeno, sua absorção ocorre por meio de transpor-te ativo, intimamente relacionado e estimulado pela pre-sença de vitamina D (cujo mecanismo é independente daabsorção de cálcio), e pela concentração de sódio no lúmenintestinal.
Outro mecanismo envolvido é o transporte passivo doíon, que ocorre no jejuno e no íleo e é diretamente propor-cional à concentração de fósforo nestes segmentos.
O PTH age indiretamente na absorção de fosfato poraumentar os níveis de calcitriol e conseqüentemente au-mentando a absorção intestinal de fósforo.
O cálcio e o magnésio em altas concentrações no lúmendigestivo se ligam ao fósforo, diminuindo sua absorção. Oalumínio também forma complexos insolúveis no tratodigestivo com o fósforo, sendo esta uma terapia utilizadana hiperfosfatemia da insuficiência renal crônica, por tem-po limitado.1,2,21,23,25
Ossos. O fósforo participa de maneira direta e indiretado processo de mineralização óssea. Indiretamente, atra-vés da ação do PTH e da vitamina D. A hipofosfatemia levaa defeitos da mineralização óssea, além de aumento daatividade absortiva do osso por aumento dos níveis devitamina D. Diretamente, pela ação do fosfato na matura-
Quadro 13.7 Fatores que influenciam a excreçãode fósforo
HIPERCALCEMIA DIMINUI
PTH AUMENTA
ACIDOSE AUMENTA
ALCALOSE DIMINUI
VITAMINA D DIMINUI
INSULINA DIMINUI
GLUCAGON AUMENTA
DIURÉTICOS AUMENTA
EXPANSÃO DE VOLUME AUMENTA
CALCITONINA AUMENTA
CORTICÓIDE DIMINUI

226 Metabolismo do Cálcio, Fósforo e Magnésio
ção e mineralização da matriz óssea, participando da pro-dução de colágeno.20
Rins. Em um adulto normal, cerca de 5,25 g de fós-foro inorgânico (Pi) são filtrados diariamente. Deste total,80 a 97% são reabsorvidos nos túbulos renais. No túbulocontornado proximal (TCP) acontece 80% da reabsor-ção, com reabsorção quase nula a nível de alça de Hen-le. No túbulo contornado distal, 10% do total de Pi fil-trado é reabsorvido, sendo controversa a reabsorção nosductos coletores.
MECANISMOS DE TRANSPORTEO principal mecanismo de transporte, no túbulo proxi-
mal, é transcelular, dependente de energia, mantido pelogradiente de sódio gerado pela bomba Na-K-ATPase namembrana basolateral.
Este mecanismo é saturável, necessitando da presençade sódio no local de entrada na célula (bordo em escovada membrana), sugerindo assim um mecanismo carreador.
O transporte se dá em torno de uma molécula de fósfo-ro para duas moléculas de sódio, ou seja, sódio e fósforosão co-transportados.23,24
FATORES QUE REGULAM A EXCREÇÃO DEFÓSFORO
Gradiente de Sódio e pH. Estes fatores fazem a regula-ção renal a curto prazo (modificação alostérica). Quandoa concentração de sódio luminal está aumentada, ocorreuma maior absorção de fósforo pelos mecanismos já des-critos.
A atividade de co-transporte de sódio e fósforo é redu-zida pelo aumento da concentração luminal de íons hidro-gênio, sendo o efeito luminal da acidose maior quando aconcentração de sódio no lúmen tubular diminui; isto podeser explicado por um efeito inibitório da acidose no trans-porte de sódio.20,23,24,25
PTH. É o fator hormonal mais importante na reabsor-ção do fósforo nos rins, especialmente no túbulo contor-nado proximal. Sua ação a nível intracelular parece sermediada pelo AMPc e proteína cinase C, regulando o co-transporte de sódio e Pi e inibindo a reabsorção local defósforo.23,24
Vitamina D. A vitamina D, em especial o calcitriol, temação independente do PTH, pela maior absorção de fósfo-ro no trato digestivo, promovendo sua maior reabsorçãono túbulo contornado proximal.
Calcitonina. Tem efeito hiperfosfatúrico pela diminui-ção do cálcio ionizado plasmático, diminuindo assim areabsorção de fósforo no túbulo proximal.
Insulina, Glicose e Glucagon. A insulina tem um efei-to independente, aumentando a entrada de fósforo no in-terior da célula e levando à hipofosfatemia. A inibição daneoglicogênese diminui a concentração do fósforo citosó-lico e aumenta a reabsorção tubular de fósforo.
A administração de glicose provoca fosfatúria devido àdiurese osmótica por ela provocada.
Cálcio. A hipercalcemia provoca alterações na quanti-dade de Pi filtrado no túbulo, principalmente provocadapela saída de fósforo do interior da célula e conseqüenteformação de complexos com o cálcio [Ca(PO4)2]; este efei-to independe da ação do PTH. O efeito indireto se dá pelaação do PTH na hipercalcemia anteriormente descrita nometabolismo do cálcio.
Corticóides. O corticóide diminui a reabsorção tubularde fósforo agindo diretamente no túbulo proximal, comoacontece na síndrome de Cushing.
FUNÇÕES DO FÓSFORO NO ORGANISMONos ossos, o fósforo tem papel fundamental na minera-
lização óssea, pois ele é depositado na forma de cristais dehidroxiapatita, na matriz orgânica do osso. A sua defici-ência pode ocasionar osteomalácia e raquitismo.
Nos tecidos moles é componente das membranas celu-lares, material genético (DNA e RNA) e fator intermediá-rio no metabolismo celular. Os fosfolipídios são os consti-tuintes essenciais das membranas celulares e das organe-las intracelulares.
No eritrócito, o 2,3-difosfoglicerato (2,3-DPG) exerceinfluência direta na disponibilidade de oxigênio aos teci-dos. Na deficiência de fósforo há redução na síntese de 2,3-DPG, aumentando a afinidade da hemoglobina com o oxi-gênio e diminuindo sua disponibilidade aos tecidos.
O fósforo faz parte da formação da adenosina trifosfato(ATP), que fornece energia para vários processos metabó-licos fundamentais na vida da célula.1,2,20,25
Pontos-chave:
• Em torno de 80% do fósforo filtrado éreabsorvido no túbulo contornado proximal
• PTH é o fator mais importante nareabsorção de fósforo pelos rins
Hipofosfatemia
INTRODUÇÃOCerca de 1% do fósforo se distribui no espaço extrace-
lular, sendo que deste total 30% representam a sua fraçãoinorgânica, e é esta fração que é medida no plasma (Pi).Sendo assim, pode-se ter uma depleção do fósforo corpo-ral total com concentrações “normais” no sangue.
A hipofosfatemia é considerada leve quando os níveisde Pi estão em torno de 1 a 2,5 mg/dl, e grave quando estaconcentração se encontra abaixo de 1,5 mg/dl. Um grupoespecial de pacientes, os etilistas, desenvolvem mais fre-qüentemente este distúrbio, chegando à incidência de 10%em pacientes hospitalizados.25,26

capítulo 13 227
CAUSASSão três os mecanismos responsáveis pela diminuição
da concentração plasmática de Pi: redistribuição do fos-fato extracelular para dentro da célula, diminuição da ab-sorção intestinal e aumento das perdas urinárias. As prin-cipais causas de hipofosfatemia são listadas no Quadro13.8.
Redistribuição InternaNutrição. No processo de nutrição (enteral ou paren-
teral) em pacientes desnutridos, há um consumo maior defósforo intracelular. Se quantidades insuficientes de fós-foro são fornecidas na repleção nutricional destes pacien-tes, e concomitantemente grandes quantidades de carboi-drato (estímulo à liberação de insulina) forem fornecidas,a hipofosfatemia aguda pode desenvolver-se. Pacientessubmetidos a nutrição parenteral podem, por má absor-ção, jejum e aumento do metabolismo, desenvolver omesmo quadro se a repleção de fosfato não for adequa-da.2,21
Alcalose Respiratória. A queda da pressão parcial deCO2 provoca a saída de CO2 do interior da célula, não sen-do acompanhada na mesma proporção pelo bicarbonato.Isto desencadeia uma alcalose intracelular, com ativaçãoda glicólise (ativada pela fosfofrutoquinase) e deslocamen-to do fósforo do extra- para o intracelular.
É a causa mais comum de hipofosfatemia em pacienteshospitalizados. Situações clínicas como alcoolismo, síndro-me de abstinência, queimaduras, hiperalimentação, uso decorticóides, calcitonina e catecolaminas determinam estedistúrbio ácido-básico, levando à hipofosfatemia.31
Leucose. Contagens superiores a 100.000 leucócitos emleucemias ou crise blástica podem aumentar o consumo defósforo pela intensa proliferação celular.
Síndrome do Osso Faminto. Deposição de cálcio e fós-foro nos ossos após cirurgia na região cervical (paratireoi-dectomia) causa hipocalcemia e hipofosfatemia, pela inten-sa deposição de cálcio e fósforo ósseo.
Diminuição da Absorção IntestinalA causa mais comum nesta situação é o uso de antiáci-
dos, que formam complexos insolúveis com o fósforo e nãosão absorvidos. Doenças do trato gastrintestinal que cau-sam dificuldade de absorção de fósforo (doença de Crohn,síndrome do intestino curto, doença celíaca, entre outras)e má absorção de vitamina D também são responsáveispela diminuição na absorção.
A diminuição da ingesta de fósforo é raramente causaisolada de hipofosfatemia, mas freqüentemente esta situ-ação é associada a diarréia crônica, onde a deficiência devitamina D também desempenha o seu papel, causandohiperparatireoidismo secundário e aumentando a excreçãode fósforo.
O jejum prolongado por si só raramente causa deficiên-cia de fósforo, já que nesta situação há uma hipoinsuline-mia e aumento do catabolismo celular, liberando fósforoda célula. Quando esses pacientes são novamente alimen-tados, este processo se inverte sem reposição de fósforo, ea hipofosfatemia aparecerá.2,28,30
Aumento da Excreção UrináriaHiperparatireoidismo e Raquitismo. A hipersecreção
de PTH ou análogo (PTHrP) leva a um quadro de hipofos-fatemia por aumento na secreção de fosfato. A forma he-reditária de raquitismo (raquitismo resistente à vitaminaD) ou em associação a tumores na vida adulta (osteomalá-cia oncogênica) provoca defeitos seletivos tubulares nareabsorção de fósforo.
Raquitismo severo, osteomalácia e retardo do cresci-mento manifestam-se nas crianças. Os níveis de calcitriolsão reduzidos nas duas situações. Outra forma mais rarade raquitismo é aquele conhecido como hereditáriohipercalciúrico hiperfosfatêmico de transmissão autossô-mica recessiva, apresentando níveis elevados de calcitrioldevido à hipercalciúria2,21,23,26
Diabetes Mellitus. Pacientes que apresentam diabetesdescompensado com glicosúria, poliúria e acidose aumen-tam a excreção de fósforo na urina em grandes quantida-des.
No quadro de cetoacidose ocorre uma maior produçãode fósforo intracelular, havendo maior liberação para oplasma e aumentando ainda mais a sua excreção renal.
A correção da cetoacidose com insulina e repleção dovolume extracelular leva a queda rápida dos níveis de fós-foro, porém a níveis dificilmente inferiores a 1 mg/dl, tor-nando sua reposição na maioria das vezes desnecessá-ria.21,25,26
Síndrome de Fanconi. É uma disfunção tubular proxi-mal que acontece no adulto geralmente em decorrência demieloma múltiplo e na criança devido a cistinose ou do-ença de Wilson, que se traduz por glicosúria, aminoacidú-ria e hipouricemia com acidose tubular renal tipo 2, alémde hiperfosfatúria.
Quadro 13.8 Causas de hipofosfatemia
REDISTRIBUIÇÃO INTERNAAumento de insulina, durante nutriçãoAlcalose respiratória agudaSíndrome do osso faminto
DIMINUIÇÃO DA EXCREÇÃO URINÁRIAHiperparatireoidismo primário e secundárioRaquitismo resistente à vitamina D• ligado ao X (infância)• osteomalácia oncogênica (no adulto)Raquitismo hereditário hipofosfatêmico com
hipercalciúriaSíndrome de FanconiAcetazolamida

228 Metabolismo do Cálcio, Fósforo e Magnésio
QUADRO CLÍNICOA hipofosfatemia causa uma variedade de sinais e sin-
tomas. O quadro clínico é decorrente da diminuição dosníveis de 2,3-difosfoglicerato (2,3-DPG) e de compostosenergéticos fundamentais à base de fósforo que mantêm ometabolismo celular (adenosina trifosfato-ATP).
Os pacientes sintomáticos apresentam níveis de Pi abai-xo de 1,0 mg/dl. As condições clínicas mais associadas àsintomatologia são: alcoolismo crônico, hiperalimentaçãosem fosfato e ingestão crônica de antiácidos.
A cetoacidose diabética e a hiperventilação causam hi-pofosfatemia grave, porém em menor freqüência por nãocausar uma depleção crônica. Os principais sistemas atin-gidos com suas respectivas repercussões clínicas serãodescritos adiante.2,21,26,27,28
Disfunção HematológicaHemácias. A diminuição intracelular do ATP leva a uma
maior rigidez do eritrócito, promovendo a hemólise quan-do as concentrações de fósforo são inferiores a 0,5 mg/dl.
Leucócitos. Também a diminuição do ATP intracelularleva a defeitos na fagocitose e quimiotaxia.
Plaquetas. Trombocitopenia com defeitos na retração docoágulo é observada.
Sistema Nervoso CentralUm quadro de encefalopatia metabólica pode ocorrer,
levando a sintomas de irritabilidade, confusão mental, es-tupor e até mesmo coma, por provável mecanismo de hi-póxia.
Sistema Músculo-esqueléticoOs músculos necessitam de grande quantidade de ATP
para manter atividades de contração e manutenção depotencial de membrana, que são prejudicadas pela hipo-fosfatemia.
A hipofosfatemia crônica leva a um acúmulo de água,sódio e cloro no interior da célula. O quadro clínico podeapresentar-se como uma miopatia proximal ou disfagia(atingindo a musculatura lisa).
A rabdomiólise pode ocorrer quando a hipofosfatemiaaguda acontece em um paciente que já apresenta depleçãoprévia de fósforo. Tais situações são vistas no alcoolismocrônico e em pacientes recebendo hiperalimentação semsuplemento de fósforo. Elevação da creatinina fosfoquinaseaponta para necrose muscular.29
Sistema ÓsseoA hipofosfatemia leva a um aumento de calcitriol que
dá origem a uma maior reabsorção óssea, levando à hiper-calciúria pela liberação de cálcio do osso. Hipofosfatemiaprolongada leva ao raquitismo na infância e à osteomalá-cia no adulto, por defeitos na mineralização óssea.30
Sistema CardiopulmonarA depleção de ATP prejudica a contratilidade miocárdi-
ca, levando à insuficiência cardíaca de baixo débito, poden-do levar à falência miocárdica franca quando os níveis defósforo atingem limites inferiores a 1,0 mg/dl. O acometi-mento da musculatura diafragmática e insuficiência respi-ratória podem acontecer em casos graves de hipofosfatemia.21
DIAGNÓSTICONa maioria das vezes a causa da hipofosfatemia é apa-
rente, pelos dados de história e exame físico. Quando adepleção de fosfato se estabelece, a reabsorção renal émáxima: sendo assim, podem-se diferenciar as situaçõesclínicas calculando-se a fração excretora de fósforo oumedindo-se a sua concentração urinária nas 24 horas.
A fração excretora de fósforo é calculada da seguintemaneira:
Fep
fósforo urinário creatinina plasmáticafósforo plasmático creatinina urinária
��
�� 100
Uma fração excretora de fósforo abaixo de 5% ou umaconcentração urinária menor que 100 mg na urina de 24horas afasta o diagnóstico de perda renal de fosfato.
As principais causas envolvidas nesta situação se devemou a um desvio intracelular de fósforo ou a uma diminui-ção da absorção intestinal. O desvio intracelular aumenta-do acontece mais freqüentemente se o paciente recebeuinfusões de glicose ou insulina, como no tratamento dodiabetes mellitus descompensado ou realimentação. A alca-lose respiratória se encontra dentro das principais causas.Diarréia crônica, uso de antiácidos ou deficiência de vita-mina D são as principais causas de má absorção.
Quando a fração excretora de fósforo é maior que 20%ou a urina de 24 horas apresenta uma concentração maiorque 100 mg, a perda renal de fosfato está presente, deven-do-se investigar como causas principais hiperparatireoidis-mo primário e secundário (hipercalcemia, hipofosfatemia,perda urinária de fósforo), defeitos tubulares já anterior-mente descritos, como a síndrome de Fanconi, raquitismoresistente à vitamina D (na criança) e osteomalácia onco-gênica no adulto.1,2,21,25
Os sintomas de hipofosfatemia se iniciam quando osníveis séricos de fósforo atingem níveis inferiores a 1,5 mg/dl; a maioria dos pacientes hipofosfatêmicos são assinto-máticos, de maneira que o tratamento da causa subjacenteé o principal objetivo.
Por outro lado, nos pacientes sintomáticos, ou nos quetenham defeitos tubulares crônicos e que venham a desen-volver hipofosfatemia, a correção com suplementos à basede fósforo deve ser feita.
O fósforo dosado no plasma é a forma elementar (inor-gânico), e o fosfato se encontra nos sistemas biológicos,embora na prática não se faça esta distinção. A concentra-ção plasmática de fosfato (2,5 a 4,5 mg/dl) é medida em

capítulo 13 229
mg/dl ou mmol/L, sendo que a conversão de uma unida-de em outra obedece aos seguintes cálculos:2
1 mmol de fosfato � 31 mg de fósforo elementar
1 mmol de fosfato � 3,1 mg/dl de fósforo
1 mg de fósforo � 0,032 mmol de fosfato
1 mg/dl de fósforo � 0,32 mmol/L de fosfato
A reposição é preferível na forma oral, já que a reposi-ção endovenosa apresenta riscos de precipitação com cál-cio, insuficiência renal e arritmias cardíacas. A administra-ção oral se dá numa dose de 2,5 a 3,5 g (80 a 110 mmol)diários em doses divididas.
Valores séricos abaixo de 1,0 mg/dl podem causar da-nos importantes ao paciente, como rabdomiólise, sendo aadministração endovenosa necessária.58
No paciente sintomático a administração endovenosa nãodeve ultrapassar 2,5 mg (0,08 mmol/L)/kg a cada 6 horas,com monitorização dos níveis de fósforo, cálcio, potássio emagnésio a cada 6 horas, podendo a dose ser dobrada se asmanifestações clínicas são muito graves. A infusão deve sersuspensa quando os níveis de fósforo atingem 2,0 mg/dl eos de cálcio estão menores que 8,0 mg/dl.1,2,21,25
FORMAS DE APRESENTAÇÃO
IntravenosaFosfato de Potássio. Cada ml contém 3 mmol de fosfa-
to (93 mg de fósforo) e 4,4 mEq de potássio. Ampola de 5ml e 15 ml são disponíveis.
Fosfato de Sódio. Cada ml contém 3 mmol de fosfato(93 mg de fósforo) e 4 mEq de sódio. Ampolas de 15 e 30ml são disponíveis.
Via OralK/Phos Neutro. Cada tablete possui 250 mg de fósforo,
13 mEq de sódio e 1,1 mEq de potássio.
Neutra-Phos. 75 ml de solução contém 250 mg e fósfo-ro e 7,1 mEq de sódio e potássio.
Neutra-Phos-K. Cada cápsula ou 75 ml de solução con-tém 350 mg de fósforo e 14,2 mEq de potássio.
Prie demonstrou que o uso de dipiridamol na dose de75 mg quatro vezes ao dia pode ser útil para aumentar osníveis de fósforo em pacientes com aumento idiopático dafosfatúria.69
Hiperfosfatemia
INTRODUÇÃOA hiperfosfatemia, na maioria das vezes, é resultado da
incapacidade dos rins em excretar, de maneira eficiente, ofosfato do organismo. Em indivíduos normais, elevaçõesna ingesta de fósforo não acarretam elevações similares naconcentração plasmática. A hiperfosfatemia é diagnostica-da quando o nível plasmático de fósforo se encontra aci-ma de 4,5 mg/dl.1,2,22
CAUSASAs principais causas de hiperfosfatemia são conseqüên-
cias de:
• Aumento da ingesta• Diminuição de sua excreção• Desvios intracelulares de fósforo.
A seguir se discutirão as principais causas dos distúr-bios e suas conseqüências clínicas (v. Quadro 13.9).
Pontos-chave:
• Hipofosfatemia: Fósforo � 2,5 mg/dl• Freqüente em alcoólatras• Diagnóstico através do quadro clínico e
exame físico• Fração excretora de fósforo auxilia o
diagnóstico• Dieta geralmente é o suficiente para tratar o
déficit• Quando presentes sintomas graves, preferir
a reposição endovenosa• Dipiridamol parece elevar os níveis de
fósforo
Quadro 13.9 Causas de hiperfosfatemia
DIMINUIÇÃO NA EXCREÇÃO RENALInsuficiência renal• aguda• crônica
HipoparatireoidismoPseudo-hiperparatireoidismoAcromegaliaDifosfonatosCalcinose tumoral
DESVIOS TRANSCELULARESInfecçõesEstados hipercatabólicosLeucose/leucemiaAcidose metabólica e respiratóriaSíndrome de esmagamento (rabdomiólise)HipertermiaAnemia hemolítica
PSEUDO-HIPERFOSFATEMIAParaproteinemiasMieloma múltiploMacroglobulinemia de WaldenströmHiperlipidemiaRefrigeração prolongadaContaminação por heparina sódica

230 Metabolismo do Cálcio, Fósforo e Magnésio
Diminuição na ExcreçãoInsuficiência Renal. A hiperfosfatemia acontece quan-
do o ritmo de filtração glomerular cai em torno de 20 a 25ml/min, mantendo-se uma ingesta normal de fósforo. Nainsuficiência renal crônica, a carga de fósforo filtrada pornéfron aumenta, porém à custa da elevação dos níveis plas-máticos. Já na insuficiência renal aguda, quando há umaqueda repentina do ritmo de filtração glomerular, este fe-nômeno compensatório não acontece, observando-se mai-ores elevações nos níveis plasmáticos de fósforo.
Hipoparatireoidismo. As situações clínicas de deficiên-cia na produção ou resistência na ação do PTH (pseudo-hipoparatireoidismo) levam à hiperfosfatemia. A diferen-ciação entre estas duas situações clínicas se dá pela medi-da dos níveis de PTH (que se encontram elevados no pseu-do-hipoparatireoidismo) e pela medida do AMP cíclicourinário (diminuído no hipoparatireoidismo).
Acromegalia/Hipertireoidismo. Cerca de um terço dospacientes com hipertireoidismo podem apresentar hiper-fosfatemia leve devido a uma maior reabsorção tubular eóssea de fósforo. Na acromegalia por ação do hormônio decrescimento se dá uma maior reabsorção tubular e ósseade fósforo. Na acromegalia por ação do hormônio de cres-cimento se dá uma maior reabsorção tubular e óssea defósforo, porém na maioria das vezes de forma discreta, semrepercussão clínica.33
Drogas. Os bifosfonatos usados no tratamento da hiper-calcemia e doença de Paget diminuem a excreção, bemcomo provocam desvios intracelulares de fósforo, elevan-do seus níveis séricos. Outras medicações, como uso abu-sivo de enemas e laxativos à base de fósforo, bem como aadministração endovenosa de fosfato e derivados de vita-mina D (calcitriol) em portadores de insuficiência renal, sãooutras causas bem estabelecidas.
Calcinose Tumoral. É uma síndrome rara, encontradaem pacientes jovens da raça negra que apresentam calcifi-cações ao longo de suas articulações. Trata-se de uma anor-malidade genética causada por um aumento na reabsor-ção tubular de fósforo. Os níveis de cálcio e PTH são nor-mais, porém há elevação dos níveis de calcitriol. O fósforoforma complexos com o cálcio causando hipocalcemia. Osníveis de cálcio permanecem normais devido a uma mai-or reabsorção no néfron distal, não havendo evidência dehipoparatireoidismo ou pseudo-hipoparatireoidismo.
Desvios Intracelulares de FósforoSendo o fósforo o ânion predominante no espaço intra-
celular, o intenso catabolismo celular ou sua destruiçãopermite a passagem de fósforo do interior da célula para omeio extracelular. Situações clínicas que provocam necro-se celular, tais como hepatite fulminante, hipertermia ma-ligna e síndrome de esmagamento com rabdomiólise, cau-sam hiperfosfatemia.
A terapia citotóxica em doenças hematológicas, comoa leucemia linfoblástica aguda e linfomas, provoca a cha-
mada síndrome de lise tumoral, caracterizada por hiper-fosfatemia, hipocalcemia, hiperuricemia e hiperpotasse-mia. Quando existe precipitação de ácido úrico nos túbu-los renais, ocorre insuficiência renal, podendo agravar ahiperfosfatemia. Na cetoacidose diabética, apesar de ha-ver uma diminuição do fósforo corporal total devido àdiurese osmótica, há um desvio de fósforo (Pi) do intra-celular para o extracelular (v. hipofosfatemia), revertidocom o tratamento e que posteriormente evolui para hipo-fosfatemia.1,2,22,32
A acidose metabólica provoca um maior metabolismodo fósforo orgânico para inorgânico e conseqüente libera-ção para o extracelular.2
PSEUDO-HIPERFOSFATEMIASituações como a hemólise durante a coleta de sangue,
ou a presença de gamopatias monoclonais (provocandouma maior ligação do fósforo com as paraproteínas), po-dem causar elevações falsas dos níveis plasmáticos.35
QUADRO CLÍNICOAs manifestações clínicas da hiperfosfatemia se dão em
função de sua ação sobre os níveis de cálcio sérico, PTH,calcitriol e na ação inibitória sobre a atividade da 1α-hidro-xilase. A hiperfosfatemia grave leva à hipocalcemia, devi-do aos depósitos de cálcio e fósforo nos tecidos moles, alémdo efeito inibitório sobre o calcitriol.
Este processo de deposição é visto quando o produtocálcio e fósforo ultrapassa o valor de 70 (normal de 40), e acalcificação se dá nos vasos sanguíneos, pulmão, córnea,rins, pele e mucosas.2,22,32,33
A síndrome do olho vermelho, devido à calcificação dacórnea, e deposição periarticular atingindo articulações dosdedos, costelas e ombros são outros achados.
A hipocalcemia sintomática aparecerá, levando a con-vulsões em casos graves de hiperfosfatemia. Mesmo assim,elevações súbitas dos níveis de fósforo, que atinjam 6 mg/dl, podem causar sintomas.2
Pontos-chave:
• A insuficiência renal é a principal causa dehiperfosfatemia
• Sendo o fósforo o principal ânionintracelular, situações clínicas de destruiçãocelular (ex., rabdomiólise) se acompanhamde hiperfosfatemia
TRATAMENTOOs princípios do tratamento da hiperfosfatemia são
aqueles que procuram atingir a causa subjacente do distúr-bio, diminuindo a absorção e promovendo a maior excre-ção renal deste íon. Em pacientes que apresentam função

capítulo 13 231
renal normal, o aumento da ingesta de fósforo raramentecausa hiperfosfatemia.
Na síndrome de lise tumoral, quando da quimioterapia,a promoção de uma diurese vigorosa (como aquela pro-movida na hipercalcemia), com infusão de solução salinae uso de acetazolamida 15 mg/kg ou 500 mg a cada 6 ho-ras, diurético que alcaliniza a urina impedindo a precipi-tação de cristais de ácido úrico e promovendo a natriure-se, é eficaz na produção de uma maior excreção de fósfo-ro.2,22
Nos pacientes portadores de insuficiência renal crôni-ca, quando o ritmo de filtração glomerular atinge 20 a 25ml/min, a restrição da ingesta de fósforo em 600 a 900 mg/dia se faz necessária, porém a utilização de substâncias quese liguem ao fósforo na luz intestinal (quelantes), impedin-do a sua absorção, é de uso corriqueiro.34 O hidróxido dealumínio na dose de 500 a 1.800 mg 3 a 6 vezes por dia comas refeições ou 20 minutos após as refeições está indicadopara pacientes com hiperfosfatemia visando à reduçãomais rápida dos níveis séricos de fósforo.
As formas de apresentação podem ser cápsulas, líqui-do, suspensão e tabletes. As cápsulas se apresentam nasdoses de 400 mg ou 500 mg; líquido, 600 mg/5 ml; suspen-são oral, 320 mg/5 ml, 450 mg/5 ml ou 675 mg/5 ml; e ta-bletes de 300 mg, 500 mg ou 600 mg.51,63 No Brasil a formamais comum de apresentação parece ser a suspensão oral.
O carbonato de cálcio na dose de 8,5 g (variando de 2,5a 20 g/dia), com efeito máximo de 1 g junto às refeições,liga-se ao fosfato tanto exógeno como endógeno (secreta-do pelo pâncreas e parótidas) na luz intestinal e inibe demaneira eficaz a absorção do fósforo.1,2,22,34 A dose de car-bonato de cálcio é aumentada gradualmente até o fósforoplasmático atingir uma concentração entre 4,5 e 5,5 mg/dl. Hipercalcemia é uma complicação comum com o usode carbonato de cálcio, ocorrendo mais freqüentementequando usadas preparações de vitamina D (calcitriol).62,63
O uso crônico de hidróxido de alumínio em pacientes emhemodiálise pode levar à intoxicação por este metal, comquadro de encefalopatia, osteomalácia resistente à vitami-na D, anemia e miopatia, devendo-se fazer substituiçãopelo carbonato de cálcio. Quando há hipercalcemia, cálcioem torno de 11 mg/dl com hiperfosfatemia persistente emníveis elevados, o acréscimo de hidróxido de alumínio deveser feito por um período provisório até o melhor controlede cálcio e fósforo.22
Um novo agente não contendo cálcio, alumínio e mag-nésio, assim evitando os problemas das medicações con-tendo estes íons, está sendo usado com bons resultados. Osevelamar é um polímero catiônico que quela o fósforo portroca iônica. Trabalhos mostraram que o sevelamar foi tãoefetivo quanto os quelantes habitualmente usados, comocarbonato de cálcio ou acetato de cálcio, não alterando aconcentração plasmática de cálcio e controlando os níveisde fósforo. Também foi constatado um efeito na reduçãodos níveis de colesterol total. O uso de sevelamer (Rena-
gel®) fica reservado, geralmente, para pacientes com hi-percalcemia, devido aos seus custos.64,65,68
Em pacientes portadores de hipoparatireoidismo, a ad-ministração de PTH aumentaria a excreção de fosfato uri-nário, porém o uso a longo prazo determina a formação deauto-anticorpos, limitando sua ação terapêutica. A impor-tância de sua utilização se dá na diferenciação do hipopara-tireoidismo primário do pseudo-hipoparatireoidismo, quan-do se mede o fósforo urinário nestas duas situações, apóssua administração. Observar-se-á que, no pseudo-hiperpa-ratireoidismo, não há aumento na excreção urinária de fós-foro, ao contrário do que ocorre no hipoparatireoidismo.
Os genes responsáveis pelo transporte fosfato-sódiodependente foram recentemente isolados. Novas drogasestão sendo estudadas para tratar a hiperfosfatemia. Estasdrogas atuam nos transportadores fosfato-sódio, como oácido fosfomórfico (PFA), que inibe o transporte fosfato-sódio-dependente no túbulo renal. Em ratos esta drogaaumentou a fração de excreção de fósforo, resultando emmelhora dos valores séricos. O uso de PFA em humanos élimitado pela toxicidade renal.66,67
Pontos-chave:
• Hiperfosfatemia: Fósforo 4,5 mg/dl• Quadro clínico: Semelhante à hipocalcemia.
Predomínio de sintomas neurológicos.Depósitos nos tecidos moles quandoproduto cálcio � fósforo 70
• Tratamento da causa. Restrição dietética defósforo. Evitar prescrever hidróxido dealumínio devido a doença óssea relacionadaao alumínio. Uso restrito para reduçãorápida do fósforo
• Sevelamer: Uso na presença dehipercalcemia concomitante
MAGNÉSIO
Homeostase do Magnésio
DISTRIBUIÇÃOO magnésio (Mg��) é o quarto íon mais abundante do
organismo, sendo a nível intracelular o segundo mais pre-valente, após o potássio. Um adulto normal possui cercade 24 g de Mg, sendo a fração sérica muito pequena em re-lação ao magnésio corporal total, distribuindo-se da se-guinte maneira:
• 60% nos ossos• 39% no espaço intracelular• 1% no espaço extracelular.

232 Metabolismo do Cálcio, Fósforo e Magnésio
No plasma cerca de 60% do magnésio se encontram li-vres (fração iônica),30 35% ligados às proteínas e 5 a 10%formando complexos com bicarbonato, citrato e fosfato.
UNIDADES DE MEDIDAO magnésio é mensurado em três unidades; mmol/L,
mg/dl e mEq/L. 1 mEq/L corresponde a 0,5 mmol/L e 1,2mg/dl.
O valor definido como normal para a concentração sé-rica de magnésio é de 1,4 a 1,7 mEq/L.
ABSORÇÃO, EXCREÇÃO E BALANÇOINTERNO
Absorção IntestinalA dieta habitual de Mg�� é de aproximadamente 4 mg/
kg/dia. Deste total, 25 a 60% são absorvidos no intestinodelgado. Os mecanismos envolvidos neste processo são:difusão passiva e difusão facilitada. O movimento de águana luz intestinal tem papel relevante na absorção de Mg��.Os principais fatores que influenciam na absorção intesti-nal de magnésio são:
• proteínas, carboidratos, sódio, água e vitamina D ⇒estimulam a absorção
• fosfato ⇒ inibe a absorção.
A quantidade de magnésio na dieta é de fundamentalimportância, pois dietas com baixo teor de magnésio au-mentam a capacidade de absorção intestinal em até 90%do total ingerido. A excreção diária de magnésio é em tor-no de 30 a 40 mg/dia pelas fezes.1,2,36
RinsO Mg�� difere da absorção de outros ânions pelo fato
de o túbulo contornado proximal não ser o responsávelprincipal pela sua reabsorção e sim a alça de Henle (ramoascendente espesso). Do total de 3.400 mg/dia de Mg��
filtrado, 15 a 25% são reabsorvidos no TCP e 5 a 10% notúbulo distal, sendo o restante na alça de Henle.1,2,36
Os mecanismos responsáveis pela absorção de magné-sio na alça de Henle não estão completamente estabeleci-dos, porém o transporte paracelular por difusão devido aum gradiente elétrico favorável, gerado pela reabsorção decloreto de sódio, é a teoria mais aceita.36 A reabsorção pa-racelular parece ser facilitada por uma proteína chamadaparacelin 1 (PCLN-1). A perda de magnésio estaria relaci-onada a mutações no gene da proteína PCLN-1, que estãolocalizadas na alça espessa de Henle.52
A ação do PTH, aumentando a reabsorção local, é umfator relevante no transporte deste íon no ramo ascenden-te espesso da alça de Henle. Também o transporte passivode Mg�� se dá devido a um gradiente eletronegativo nointerior da célula, gerado por uma concentração intrace-lular de magnésio de 1,0 mEq/L, facilitando o transportedo lúmen tubular para o interior da célula.2,36
A célula tubular da alça de Henle na membrana basola-teral possui um processo ativo de transporte de Mg�� parafora da célula, através de bomba ativa ou troca de Na� porMg��.
No túbulo proximal, a absorção do magnésio é de 15 a25%, sendo o mecanismo unidirecional e dependente daquantidade de magnésio na luz tubular. Já no túbulo dis-tal, 10% do Mg filtrado é ofertado a este segmento, ondesomente uma pequena fração é reabsorvida, através decanais de magnésio da membrana luminal e com mecanis-mos na membrana basolateral semelhantes àqueles da alçade Henle.1,2,36
Ponto-chave:
• Reabsorção de magnésio: Principalmente noramo espesso da alça de Henle (70 a 75%),ao contrário de outros íons
FATORES QUE INFLUENCIAM A EXCREÇÃO DEMAGNÉSIO
Os principais fatores envolvidos na excreção do mag-nésio são:
Hipo- e Hipermagnesemia. A concentração de magné-sio plasmático é a principal responsável pela excreção uri-nária, principalmente no segmento cortical ascendente daalça de Henle. A hipermagnesemia diminui a reabsorção,ao contrário da hipomagnesemia. Há alguma evidência deque a concentração intracelular de Mg�� regula esta res-posta, modificando o número de canais de Mg�� na mem-brana luminal.36
Hipo- e Hipercalcemia. A hipercalcemia parece aumen-tar a excreção de magnésio devido ao fato de o cálcio com-
Fig. 13.2 Demonstração das áreas de reabsorção de magnésio nonéfron justamedular e néfron cortical superficial.
Néfron Justamedular Néfron Cortical Superficial

capítulo 13 233
petir com o transporte passivo de magnésio. Por outro lado,a hipocalcemia pode aumentar a reabsorção de Ca�� eMg��. Este fato se reflete nos pacientes portadores da sín-drome de hipercalcemia hipocalciúrica que apresentamhipermagnesemia devido à ausência do efeito inibitório dahipercalcemia na reabsorção de magnésio.
PTH. Como já foi visto, o PTH aumenta a reabsorção demagnésio, principalmente na alça de Henle.
Diuréticos. Tanto os diuréticos de alça como os tiazídi-cos e diuréticos osmóticos causam hipermagnesiúria, prin-cipalmente por diminuir o transporte de sódio, cloro e cál-cio.38
Expansão de Volume. A expansão de volume causauma diminuição na reabsorção de sódio, água e magnésio,por um aumento do fluxo tubular que chega à alça deHenle, gerando um menor gradiente elétrico transtubularcomprometendo a reabsorção.
Ossos. Aproximadamente 60% do magnésio total seencontra nos ossos, na superfície óssea na forma de cris-tais, pronto para a mobilização em estados de deficiência.A hipocalcemia que se dá em situações de hipomagnese-mia pode em parte ser explicada pela troca a nível da su-perfície óssea do cálcio pelo magnésio.1,2,36
FUNÇÕES DO MAGNÉSIO NO ORGANISMOO magnésio participa de múltiplas funções no organis-
mo. É importante para a ação de cerca de 300 enzimas, naglicogenólise e respiração celular, nas funções da membra-na e aderência celular, transporte transmembrana de só-dio, potássio e cálcio. Participa das funções de contração erelaxamento muscular, neurotransmissão e condução dopotencial de ação e influencia na função de proteínas emitocôndrias. Também auxilia na estrutura do ribossomoe na ligação do RNA mensageiro ao ribossomo.70,71
Hipomagnesemia
Define-se hipomagnesemia quando a concentração sé-rica de magnésio é menor que 1,4 mEq/L (0,7 mmol/L ou1,7 mg/dl). A incidência deste distúrbio chega a 12% dospacientes hospitalizados, chegando em unidades de trata-mento intensivo a 65%. Desnutrição, hipoalbuminemia euso de aminoglicosídeos contribuem para esta maior inci-dência, nas unidades de tratamento intensivo.37,44
CAUSASHá três mecanismos principais causando a hipomagne-
semia: redução na absorção intestinal, aumento da perdaurinária e desvio intracelular do íon. As causas principaisde hipomagnesemia se encontram no Quadro 13.10.
Perdas GastrintestinaisAs principais causas de perdas gastrintestinais se devem
a quadros de má absorção intestinal, como o espru tropi-
cal, ressecção intestinal, fístulas biliares, causadoras deperdas significativas de magnésio. Outras condições, comoa pancreatite aguda, causam deficiência de magnésio.
A esteatorréia, outra situação de perda gastrintestinal,forma “sabões” na luz intestinal, com perda de magnésio.
Aspiração nasogástrica contínua sem reposição conco-mitante de magnésio causa hipomagnesemia, já que o flui-do gástrico tem aproximadamente 1 mEq/L de magnésio.Outra situação mais rara é um erro inato do metabolismocaracterizado por deficiência seletiva na absorção de mag-nésio, sendo que esta desordem se apresenta no períodoneonatal, quando ocorre hipocalcemia, que se corrige comreposição de magnésio. O desenvolvimento de hipomag-nesemia por ingestão diminuída é uma causa rara do dis-túrbio. O abuso de laxativos e diarréia crônica são outrascausas.2,36,37,45
Perdas RenaisAs perdas renais de magnésio se dão ou por defeitos
tubulares específicos no transporte de magnésio, ou pordefeitos tubulares específicos no transporte de sódio, comconseqüente déficit na reabsorção de magnésio nos seg-mentos do néfron onde ocorre o transporte passivo de só-dio e magnésio.36,45
Diuréticos. Os diuréticos de alça e os tiazídicos inibem areabsorção de magnésio, enquanto os poupadores de potás-sio aumentam o transporte de magnésio do lúmen tubularpara o interior da célula no túbulo coletor. A diurese osmó-tica, provocada por estados de hiperglicemia, e a diuresepós-obstrutiva causam perdas de magnésio na urina.2,36,41
Nefrotoxinas. Os aminoglicosídeos causam hipomag-nesemia, hipocalcemia e hipopotassemia. A cisplatina é
Quadro 13.10 Causas principais dehipomagnesemia
PERDAS GASTRINTESTINAISDiarréia, pancreatite aguda, síndrome do intestino curto,
hipomagnesemia intestinal primária, esteatorréia
PERDAS RENAISDiuréticos (de alça e tiazídicos), cisplatina,
aminoglicosídeos, anfotericina B, pentamidina,ciclosporina
ÁlcoolExpansão de volumeHipercalcemiaTransplante renalDiurese pós-obstrutivaSíndrome de BartterPerda renal primária de magnésio
MISCELÂNEASíndrome do osso famintoFoscarnetPós-operatório

234 Metabolismo do Cálcio, Fósforo e Magnésio
outra droga que causa hipomagnesemia, chegando a 50%dos pacientes em algumas séries. Outras drogas são a an-fotericina B, que causa acidose tubular renal e hipomag-nesemia leve, e a ciclosporina, que ocasiona perda renal demagnésio após transplante renal e de medula óssea.1,2,36,43,45
Álcool. Vários mecanismos estão envolvidos no desen-volvimento de hipomagnesemia nos pacientes etilistas:diarréia, baixa ingestão e efeito direto do álcool no túbulorenal, causando perda urinária de magnésio.
Hipercalcemia. Cálcio e magnésio parecem competirpelo mesmo local de reabsorção no ramo espesso ascenden-te da alça de Henle, onde a hipercalciúria nesta situaçãoprovoca maior perda de magnésio na urina. Isto pode sercomprovado pela hipomagnesemia leve encontrada no hi-perparatireoidismo primário.2,36
Disfunção da Alça de Henle. Disfunções aí localizadas,como na fase de recuperação da necrose tubular aguda,diurese pós-obstrutiva e na síndrome de Bartter (defeitocongênito que promove a perda renal de potássio, alcalo-se metabólica, hipercalciúria e hipomagnesemia).45
Expansão de Volume. O maior exemplo é o que acon-tece nos estados de hiperaldosteronismo cuja expansão dovolume extracelular leva a um menor efeito de reabsorçãodo magnésio através de seus mecanismos passivos.
Miscelânea. Dentre outras causas, destacamos a cetoa-cidose diabética antes do tratamento, pelo quadro hiper-catabólico, e a perda urinária devida à diurese osmóticaintensa.
Na síndrome do osso faminto pós-tireoidectomia comressecção inadvertida da paratireóide ou paratireoidecto-mia pode ocorrer uma maior deposição de magnésio aonível ósseo.
Na insuficiência renal crônica, acidose tubular renal enefrite intersticial, pode-se observar uma perda maior demagnésio na urina.1,2,36,41,43,44,45
QUADRO CLÍNICOO quadro clínico da hipomagnesemia é acompanhado
na maioria das vezes por outros distúrbios metabólicos,como hipopotassemia, hipocalcemia e alcalose metabóli-ca, além de depender da velocidade de instalação do dis-túrbio.
Manifestações Neuromusculares. A tetania é um acha-do comum, quando associada a hipocalcemia, sendo rarana ausência deste distúrbio. O sinal de Chvostek é maiscomum que o de Trousseau na hipomagnesemia. Convul-sões, tremores e mioclonia também são outros achados.2 Ossinais neuromusculares são mais comuns em etilistas epacientes com má absorção intestinal.
Manifestações Cardiovasculares. As manifestações car-diovasculares mais importantes são as arritmias ventricu-lares, especialmente durante os fenômenos isquêmicos.Muitos estudos não controlados têm apontado uma maiorincidência de arritmias ventriculares em pacientes com hi-pomagnesemia do que com níveis normais de magnésio.42,45
Outros estudos demonstram que a administração demagnésio após eventos isquêmicos nas primeiras 24 horasdiminui a incidência de arritmias neste período. O risco deintoxicação digitálica pode ser observado na hipomagne-semia, pela perda intracelular de potássio. Isto decorre dofato de que a diminuição na concentração de magnésiointracelular provoca uma diminuição na atividade do ATP,responsável por inibir a secreção de potássio do interior dacélula, abrindo-se os canais permeáveis ao potássio comconseqüente secreção deste para o interior do lúmen tubu-lar. A hipocalemia se desenvolve e só é corrigida com areposição concomitante de magnésio.39
O papel que desempenha o magnésio na patogênese dahipertensão arterial é investigado, parecendo haver umacorrelação inversa entre a ingestão de magnésio e a inci-dência de hipertensão.2,36
Hipocalcemia. O sinal mais proeminente de hipomag-nesemia grave é a hipocalcemia, onde são encontradosníveis de PTH normais ou baixos. A hipomagnesemia su-prime a secreção de PTH, aumenta a resistência óssea aohormônio e diminui os níveis de AMPc em resposta à açãodo PTH.2,36,42,45
DIAGNÓSTICOA hipomagnesemia deve ser suspeitada na presença de:
diarréia crônica, uso de diuréticos, hipocalcemia, hipopo-tassemia refratária, arritmias ventriculares particularmenteapós eventos isquêmicos. Para diferenciar se a causa dahipomagnesemia é de origem renal ou gastrintestinal,deve-se medir a excreção de magnésio nas 24 horas. Amedida em 24 horas e não em uma amostra é importantedevido às variações diurnas na excreção do magnésio.53 Afração de excreção de magnésio se calcula através da se-guinte fórmula:
FE Mg � MgU � CrP / (0,7 � MgP) � CrP
Onde U e P são as amostras das concentrações urináriae plasmática de magnésio e creatinina, respectivamente. Aconcentração plasmática de magnésio é multiplicada por0,7 devido ao fato de 70% do magnésio se encontrar livreno plasma; este produto é então multiplicado pela concen-tração urinária de creatinina. A coleta da amostra paradeterminação do magnésio sérico pode alterar-se quandoocorre hemólise, elevando in vitro a sua concentração. Paracada 1 g/L de queda de hemoglobina por lise, há elevaçãode 0,05 mmol/L. Valores de FE Mg maiores que 2% oumagnésio medido nas 24 horas maior que 10 mg represen-tam perdas devido ao uso de drogas (aminoglicosídeos,cisplatina, diuréticos).1,2,44
TRATAMENTOA hipomagnesemia leve (níveis em torno de 1,4 mg/dl
a 1,7 mg/dl) não necessita de tratamento, mas de correçãoda causa subjacente. Alguns autores recomendam terapiaoral com tabletes de magnésio para pacientes assintomáti-

capítulo 13 235
cos. O uso de 2 a 4 tabletes com 5 a 7 mEq por tablete pare-ce ser suficiente. Nos casos mais severos deve-se aumen-tar para 6 a 8 tabletes.
Já em casos de emergência, pacientes apresentando con-vulsões ou tetania, as primeiras medidas são infusão de 200mg (8,2 mmol) de sulfato de magnésio a 50%, ou 4 ml deMgSO4 a 50% em 100 ml de solução salina isotônica, de-vendo ser administrados em 10 minutos e os níveis demagnésio novamente medidos em 30 minutos. Pode serrepetida a dose quando necessário.1,2,36
Em casos menos urgentes, uma infusão constante de 0,5mmol/kg nas 24 horas ou 2 ml de sulfato de magnésio a50% (4,1 mmol ou 100 mg) intramuscular a cada 3 ou 4horas podem ser administrados no primeiro dia, com pos-terior redução da dose.
Outra forma de reposição, quando há presença de ar-ritmia ou tetania, é realizar a infusão de 50 mEq de mag-nésio via endovenosa em 8 a 24 horas, com o intuito demanter a concentração de magnésio acima de 1,0 mg/dl.
Em pacientes sob nutrição parenteral, a adição de 4,1mmol (100 mg) previne o desenvolvimento de hipomag-nesemia. Adultos com perda intestinal podem receber te-rapia oral na dose de 240 a 720 mg/dia.1,36,45 Como o mag-nésio plasmático é o principal responsável pela reabsorçãorenal, elevações abruptas no plasma podem levar à elimi-nação de 50% do magnésio infundido, pela abolição doestímulo de conservação do magnésio.36
Pontos-chave:
• Hipomagnesemia geralmente éacompanhada de outros distúrbiosmetabólicos, como hipocalcemia ehipopotassemia. A hipocalcemia é umindicador de gravidade da hipomagnesemia
• Hipomagnesemia leve: sem tratamento• Hipomagnesemia com sintomas severos:
terapia endovenosa
Hipermagnesemia
DEFINIÇÃODefine-se hipermagnesemia quando os níveis de mag-
nésio são superiores a 2,1 mEq/L (1 mmol/L ou 2,6 mg/dl). Em indivíduos com ingestão normal, ao redor de 3%do magnésio ingerido é excretado na urina, em especial noramo ascendente espesso da alça de Henle.2,48,49
CAUSAS DE HIPERMAGNESEMIAAs causas principais de hipermagnesemia encontram-
se no Quadro 13.11. Como o aumento de magnésio nãopossui um sistema hormonal regulador e o rim é o princi-pal responsável pela sua excreção, a hipermagnesemia se
desenvolverá em casos de insuficiência renal ou devido aoabuso de magnésio administrado sob a forma oral (antiá-cidos e laxativos), enema ou endovenosa.1,2,48,49,50
Insuficiência Renal. Como se sabe, o rim tem grandecapacidade de excretar o excesso de magnésio do organis-mo. Desta maneira, a hipermagnesemia acontece nos pa-cientes portadores de insuficiência renal crônica e em pa-cientes que estão em hemodiálise. O uso, nesta população,de antiácidos, enemas e dialisados com alta concentraçãode magnésio são causas deste distúrbio.
Aumento da Ingestão. Em pacientes com função renalnormal, a administração excessiva de magnésio por viaoral, retal ou endovenosa pode ser responsável pelo au-mento dos níveis de magnésio no plasma.
O exemplo clássico é o tratamento da eclâmpsia, quan-do níveis de 6 a 8,5 mg/dl podem causar hipocalcemiamaterna (inibição da liberação de PTH) e hipopotassemianeonatal.
Quantidades substanciais de magnésio são absorvidaspelo intestino grosso na forma de enemas. Por exemplo, 400a 800 mmol/d de magnésio via retal aumentam a concen-tração plasmática de 7,2 até 19,2 mg/dl.
Outras Causas. A insuficiência supra-renal e o hiperpa-ratireoidismo têm sido relatados como causa de hipermag-nesemia, por provocarem contração de volume plasmáti-co no primeiro caso e pelo efeito direto do PTH no segun-do, aumentando a reabsorção tubular de magnésio. A hi-percalcemia diminui a reabsorção renal de magnésio, con-trabalançando o efeito do PTH e deixando os níveis demagnésio normais e até baixos. Nos pacientes com a sín-drome familiar da hipercalcemia hipocalciúrica, a ausên-cia do efeito inibitório do cálcio no túbulo renal provoca ahipermagnesemia.
O desvio de magnésio para o extracelular pode dar-se
Quadro 13.11 Causas principais dehipermagnesemia
AUMENTO DA INGESTA• Administração excessiva de magnésio: oral, retal; rara
em pacientes com função renal normal
DIMINUIÇÃO DA EXCREÇÃO RENAL• Aumento da ingesta de substâncias que contenham
magnésio, como antiácidos, laxativos, lítio, diuréticos,poupadores de potássio
• Conteúdo alto de magnésio no dialisado
OUTRAS CAUSAS• Hiperparatireoidismo primário• Hipercalcemia hipocalciúrica familiar• Cetoacidose diabética• Estados hipercatabólicos• Tratamento da intoxicação por teofilina• Síndrome álcali-leite• Insuficiência supra-renal

236 Metabolismo do Cálcio, Fósforo e Magnésio
em casos de acidose, feocromocitoma, estados hipercata-bólicos e síndrome de lise tumoral.1,2,48,49
QUADRO CLÍNICOA hipermagnesemia é uma situação rara na ausência de
insuficiência renal ou administração de substâncias quecontenham magnésio. A gravidade e a presença dos sinto-mas vão variar com a intensidade do distúrbio que, quan-do leve (menor que 3 mEq/L, 3,6 mg/dl ou 5 mmol/L),causa poucos sintomas.
Neuromuscular. O aumento dos níveis de magnésiodiminui o impulso nervoso através da junção neuromus-cular, provocando um efeito curarizante. Há diminuiçãodos reflexos profundos, notados quando os níveis atingem4 a 6 mEq/L (4,8 a 7,2 mg/dl ou 2 a 3 mmol/L); se houvermaior elevação dos níveis plasmáticos, poder-se-ão obser-var quadriplegia flácida e paralisia respiratória.47
Cardiovascular. No coração o magnésio tem efeito atra-vés do bloqueio dos canais de cálcio e de potássio, levan-do a efeito inotrópico negativo e arritmogênico, quando suaconcentração atinge 4 a 5 mEq/L (4,8 a 7,2 mg/dl ou 2,0 a2,5 mmol/L), com conseqüente hipotensão arterial e bra-dicardia. Em quadros mais graves, bloqueio atrioventricu-lar total poderá ocorrer.46
Hipocalcemia. Hipermagnesemia leve a moderadapode levar a inibição na secreção de PTH, levando a umaredução transitória na concentração de cálcio e na maioriadas vezes não associada a sintomas, além de exercer umefeito bloqueador sobre os canais de cálcio.2,46,48
Hiperpotassemia. A hipermagnesemia provoca o blo-queio dos canais de secreção de potássio.2,48
TRATAMENTOA maioria dos casos de hipermagnesemia pode ser evi-
tada, como nos renais crônicos, não utilizando produtosque contenham magnésio. Quando a função renal é nor-mal, a parada de infusão de magnésio determina a resolu-ção do distúrbio.
Nos pacientes com quadro de risco de vida a infusão degluconato de cálcio, 100 a 200 mg, infundido em 5 a 10minutos, agindo como antagonista do magnésio (o mag-nésio é um bloqueador dos canais de cálcio), deve ser feitaimediatamente. A associação de insulina e glicose aumen-ta a entrada de magnésio para o interior da célula.48,49
Nos pacientes em hemodiálise se fará o tratamento comum dialisado livre de magnésio.2,48,49
REFERÊNCIAS BIBLIOGRÁFICAS
1. SUTTON, R.A.L., DIRKS, J.H. Disturbance of calcium and magne-sium metabolism. In: Brenner, B.M., Rector Jr, F.C. The Kidney, Phi-ladelphia: W.B. Saunders, 841, 1991.
2. BLACK, R.M.; ALFRED, H.J.; FAN, P.Y.; STOFF, J.S. Disorders ofcalcium, phosphorus, and magnesium. In: Rose, D.; Black, R.M. Cli-
nical Problems in Nephrology. New York: Little, Brown and Company,1995, 96-120.
3. KUMAR, R. Calcium metabolism. In: Jacobson, H.R.; Stiker, G.E.; Klahr,S. The Principles and Practice of Nephrology. St Louis: Mosby, 964, 1995.
4. MUNDY, G.R.; REASNER, C.A. Hypocalcemia. In: Jacobson, H.R.;Stiker, G.E.; Klahr, S. The Principles and Practice of Nephrology. St Louis:Mosby, 971, 1995.
5. MUNDY, G.R.; REASNER, C.A. Hypocalcemia. In: Jacobson, H.R.;Stiker, G.E.; Klahr, S. The Principles and Practice of Nephrology. St Louis:Mosby, 977, 1995.
6. WASSERMANN, R.H. and FULMER, C.S. Calcium transportproteins, calcium absorption, and vitamin D. Ann. Rev. Physiol.,45:375, 1983.
7. KUROKAWA, K. Calcium regulating hormones and the kidney.Kidney Int., 32:760, 1987.
8. REICHEL, H.; KOEFFLER, H.; NORMANN, A. The role of vitaminD endocrine system in health and disease. N. Engl. J. Med., 320:980,1989.
9. DESAI, T., CALRSON, R., GEHEB, M. Prevalence and clinical im-plications of hypocalcemia in acutely ill pacients in a medical inten-sive care setting. Am. J. Med., 84:209, 1988.
10. BRESLAU, N.A. and PAK, C.Y. Hypoparathyroidism. Metabolism,28:1261, 1979.
11. GRABER, M.L. and SCHULMAN, G. Hypomagnesemic hypocalce-mia independent of parathyroid hormone. Ann. Int. Med., 104:804,1986.
12. PAPAPOULOS, S.E.; CLEMENS, T.L.; FRAHER, L.J. et al. Metabo-lites of vitamin D in human vitamin D deficiency: Effect of vitaminD, or 1,25-dihydroxycholecalciferol. Lancet, 2:612, 1980.
13. THOMES, J.F., BILEZKIAN, J.P. Hypocalcemic emergencies. Endo-crinol. Metab. Clin. North Am., 22:363, 1993.
14. ATTIE, M. Hypocalcemic symptoms following parathyroidectomy.JAMA, 265:2888, 1991.
15. MERIC, F.; YAP, P.; BIA, M. Etiology of hypercalcemia in hemodi-alysis pacients on calcium carbonate therapy. Am. J. Kidney Dis.,16:459, 1990.
16. ADAMS, J. Vitamin D metabolite related hypercalcemia. Endocrinol.Metab. Clin. North Am., 18:765, 1989.
17. MALLETE, L.E. The hypercalcemias. Semin. Nephrol., 12:159, 1992.18. ADAMI, S.; ROSSINI, M. Hypercalcemia of malignancy: pathophy-
siology and treatment. Bone, 13 Suppl 1:51, 1992.19. MORTON, A.R.; FRIEFELD, J.; HALPERIN, F. Done of biphospho-
nate of malignancy. Lancet, 335:1496, 1990.20. HRUSKA, K.A.; KOVACH, K.L. Phosphate balance and metabolism.
In: Jacobson, H.R.; Sticker, G.E.; Klahr, S. The Principles and Practiceof Nephrology. St Louis: Mosby, 986, 1995.
21. HRUSKA, K.A.; KOVACH, K.L. Hypophosphatemia. In: Jacobson,H.R.; Sticker, G.E.; Klahr, S. The Principles and Practice of Nephrology.St Louis: Mosby, 986, 1995.
22. HRUSKA, K.A.; KOVACH, K.L. Hypophosphatemia. In: Jacobson,H.R.; Sticker, G.E.; Klahr, S. The Principles and Practice of Nephrology.St Louis: Mosby, 1000, 1995.
23. GMAJ, P.; MURER, H. Cellular mecanisms of inorganic phosphatetransport in kidney. Physiol. Rev., 66:36, 1986.
24. HAMMERMAN, M.R. Phosphate transport across renal proximaltubular cell membranes. Am. J. Physiol., 251:385, 1986.
25. YU, C.G.; LEE, D.B.N. Clinical disorders of phosphorus metabolism.West. J. Med., 147:569, 1987.
26. CAMP, M.A.; ALLON, M. Severe hypophosphatemia in hospitali-zed pacients. Miner. Electrol. Metab., 16:365, 1990.
27. KNOCHEL, J.P. The clinical status of hypophosphatemia. N. EnglandJ. Med., 313:447, 1985.
28. KNOCHEL, J. The pathophysiology and clinical characteristics ofsevere hypophosphatemia. Arch. Intern. Med., 137:203, 1977.
29. KNOCHEL, J. Hypophosphatemia and rhabdomyolisis. Am. J. Med.,92:455, 1992.
30. GLORIEUX, F.H. Calcitriol treatment in vitamin D dependent andvitamin D resistant rickets. Metabolism, 39:10, 1990.

capítulo 13 237
31. KRAPF, R.; JAEGER, P.; HULTER, H.N. Chronic respiratory alka-losis induces renal PTH resistance, hyperphosphatemia and hypo-calcemia in humans. Kidney Int., 42:727, 1992.
32. DELMEZ, J.A.; SLATOPOLSKY, E. Hyperphosphatemia: its conse-quence and treatment in pacients with chronic renal disease. Am. J.Kidney Dis., 19:303, 1992.
33. ALFREY, A.C.; ZHU, J.M. The role of hyperphosphatemia. Am. J.Kidney Dis., 17:53, 1991.
34. SLATOPOLSKY, E.; RUTHEFORD, W.E.; ROSENBAUM, R. et al.Hyperphosphatemia. Clin. Nephrol., 7:138, 1977.
35. McCLOSKEY, E. et al. Pseudohyperphosphatemia in multiple mye-loma. Br. Med. J., 299:1381, 1989.
36. SUTTON, R.A.L.; SAKHAEE, K. Magnesium balance and metabo-lism. In: Jacobson, H.R.; Striker, G.E.; Klahr, S. The Principles andPractice of Nephrology, St Louis: Mosby, 1005, 1995.
37. SUTTON, R.A.L.; SAKHAEE, K. Hypomagnesemia. In: Jacobson,H.R.; Striker, G.E.; Klahr, S. The Principles and Practice of Nephrology,St Louis: Mosby, 1008, 1995.
38. AL-GHAMDI, S.M.G., CAMERON, E.C.; SUTTON, R.A.L. Magne-sium deficiency: Pathophysiologic and clinical overview. Am. J. Kid-ney Dis., 24:737, 1994.
39. RYAN, M. Diuretics and potassium/magnesium depletion: Direc-tions for treatment. Am. J. Med., 82 (3A):38, 1987.
40. SALEM, M., KASISKI, N., ANDREI, A.M.; BRUSSEL, T.; GOLD,M.R.; CONN, A.; CHERNOW, B. Hypomagnesemia is a frequentfinding in the emergency department in pacients with chest pain.Arch. Intern. Med., 151:2185, 1991.
41. WHANG, R.; RYDER, K. Frequency of hypomagnesemia and hyper-magnesemia. Requested vs routine. JAMA, 263:3063, 1990.
42. RASMUSSEN, H. et al. Magnesium infusion reduces the incidenceof arrythmias in acute miocardial infarction. Arch. Intern. Med.,147:753, 1987.
43. MARTINS, B.J.; BLACK, J.; McLELLAND, A.S. Hypomagnesemiain elderly hospital admissions: a study of clinical significance. Q. J.Med., 78:177, 1991.
44. REINHART, R.A.; DESBIENS, N.A. Hypomagnesemia in pacientsentering the ICU. Crit. Care Med., 13:506, 1985.
45. WHANG, R. Magnesium deficiency: pathogenesis, prevalence, andclinical implications. Am. J. Med., (suppl 3A):24, 1987.
46. ZWERLING, H. Hypermagnesemia induced hypotension and hypo-ventilation. JAMA, 266:2374, 1991.
47. KRENDELL, D. Hypermagnesemia and neuromuscular transmis-sion. Semin. Neurol., 10:42, 1990.
48. SUTTON, R.A.L.; SAKHAEE, K. Hypermagnesemia. In: Jacobson,H.R., Striker, G.E.; Klahr, S. The Principles and Practice of Nephrology,St Louis: Mosby, 1013, 1995.
49. ELLYN, R.J. Magnesium metabolism in health and disease. DiseaseA Month, 34:161, 1988.
50. JUPPNER, H.; SCHIPANI, E. Receptors for parathyroid hormoneand parathyroid hormone-related peptide. Curr. Opin. Nephrol. Hy-pertens., 5(4):300-6, 1996.
51. LACY, C. Drug Information Handbook, 1978.52. SIMON, D.B. et al. Science , jul 2;285 (5424):103-6, 1999.53. FLEMING, C.R.; GEORGE, L.; STONER, G.L. et al. The importance
of urinary magnesium values in patients with gut failure. Mayo Clin.Proc. 71:21, 1996.
54. YU, J.; RMI, J.; GOLTZMAN, D. et al. Vitamin D analogs as potenci-al for the treatment of cancer associated with hypercalcemia (CAH).Proc. Annu. Meet. Am. Assoc. Cancer Res., 34:A1526, 1993.
55. RYZEN, E.; NELSON, T.A.; RUDE, R.K. Low blood mononuclearcell magnesium content and hypocalcemia in normomagnesemic pa-tients. West J. Med., 147:549, 1987.
56. BIBER, J. Celular aspects of proximal phosphate reabsorption. Kid-ney Int, 36:360, 1989.
57. MURER, H. Celular mechanisms in proximal tubular Pi reabsorpti-on: Some answers and more questions. J. Am. Soc. Nephrol., 2:1649,1992.
58. LENTZ, R.D.; BROWN, D.M.; KJELLSTRAND, C.M. Treatment ofsevere hypophosphatemia. Intern. Med. Dec; 89 (6):941-4, 1978.
59. ANTONSEN, J.L.; SHERRARD, D.J.; ANDRÉS, D.L. A calcimimeticagent acutely suppresses parathyroid hormone levels in patientswith cronic renal failure. Rapid communication. Kidney Int., 53:223,1998.
60. COLLINS, M.; SKARUKIS, M.C.; BILEZIKIAN, J.P. et al. Treatmentof hypercalcemia secondary to parathyroid carcinoma with a novelcalcimimetic agent. J. Clin. Endocrinol. Metab.; 83:1083, 1998.
61. BRADWELL, A.R.; HARVEY, T.C. Control of hypercalcemia ofparathyroid carcinoma by immunization. Lancet, 353:370, 1999.
62. FOURNIER, A.; MONIERE, P.; BEN HAMIDA, F. et al. Use ofalkaline calcium salts as phosphate binder in uremic patients. Kid-ney Int. Suppl., 38:S50, 1992.
63. SLATOPOLSKY, E.; WEERTS, C.; LOPEZ-HIKER, S. et al. Calciumcarbonate as a phosphate binder in patients with chronic renal fai-lure undergoing dialysis. N. Engl. J. Med., 315:317, 1986.
64. CHERTOW, G.M.; BURKE, S.K.; LAZARUS, J.M.; et al. Polyallyla-mine hydrochloride (RenaGel): A noncalcemic phosphate binder forthe treatment of hyperphosphatemia in chronic renal failure. Am. J.Kidney Dis., 29:66, 1997.
65. BLEYER, A.J.; BURKE, S.K.; DILLON, M. et al. A comparison of thecalcium-free phosphate binder sevelamar hydrochloride with cal-cium acetate in the treatment of hyperphosphatemia in hemodialy-sis patients. Am. J. Kidney Dis., 33:694, 1999.
66. MURER, H.; BIBER, J. Molecular mechanisms in renal phosphatereabsorption. Nephrol. Dial. Transplant, 10:1501, 1995.
67. BROOKS, D.P.; ALI, S.M.; CONTINO, L.C. et al. Phosphate excretionand phosphate transporter messenger RNA in uremic rats treatedwith phosphonoformic acid. J. Pharmacol. Exp. Ther., 281:1440, 1997.
68. SLATOPOLSKY, E.A.; BURKE, S.K.; DILLON, M.A. and THE RE-NAGEL STUDY GROUP. Renagel®, a nonabsorbed calcium- andaluminium free phosphate binder, lowers serum phosphorus andparathyroid hormone. Kidney Int., 55;299:307, 1999.
69. PRIE, D.; BLANCHET, F.B.; ESSIG, M.; JOURDAIN, J.P., FRIE-DLANDER, G. Dipyridamole decreases phosphate leak andaugments serum phosphorus in patients with low renal phosphatethreshold. J. Am. Soc. Nephrol., jul; 9(7);1264-9, 1998.
70. SWAMINATHAN, R. Hypo-hypermagnesemia In: Oxford Textbookof Clinical Nephrology, 2nd ed., edited by Davison A.M.; Cameron, J.S.;Grunfeld, J.P.; Kerr, D.N.S.; Ritz, E.; Winearls, C.G. Oxford Univer-sity Press, 271-310, 1998.
71. FAUCI, A.S.; BRAUNVALD, E.; ISSELBACHER, K.J.; WILSON, J.D.;MARTIN, J.B.; KASPER, D.L.; HAUSER, S.L.; LONGO, D.L.; Labo-ratory values of clinical importance. In: Harrison´s Principles of Inter-nal Medicin, 14th international ed., McGraw-Hill Book Company: A1-A9, 1998.
ENDEREÇOS RELEVANTES NA INTERNET
www.hdcn.comwww.nejm.orgwww.immunologyed.comwww.acpjc.orgwww.ajkd.orgwww.annals.orgwww.nature.comwww.science.comwww.jcb.orgwww.jci.orgwww.ovid.comwww.utol.com

Capítulo
14Metabolismo do Ácido Úrico
Paulo Henrique Fraxino e Miguel Carlos Riella
INTRODUÇÃO
METABOLISMO DAS PURINAS E SÍNTESE DO ÁCIDO
ÚRICO
METABOLISMO DO ÁCIDO ÚRICO
Produção de ácido úrico
Produção exógena de ácido úrico
Produção endógena de ácido úrico
Excreção de ácido úrico
Aparelho gastrintestinal
Aparelho urinário
ESTADOS DE HIPERURICEMIA
Definição
Classificação
Hiperuricemia primária
Hiperuricemia secundária
Epidemiologia
Apresentação clínica
Gota
Nefropatia aguda pelo ácido úrico
Nefropatia crônica pelo ácido úrico
Nefropatia hiperuricêmica familiar
Nefrolitíase pelo ácido úrico
Hiperuricemia no transplante renal
Manejo clínico e farmacológico dos estados de
hiperuricemia
Hiperuricemia assintomática
Gota
Nefropatia aguda pelo ácido úrico
Nefropatia crônica pelo ácido úrico
Nefropatia hiperuricêmica familiar
Nefrolitíase pelo ácido úrico
Hiperuricemia no transplante renal
ESTADOS DE HIPOURICEMIA
Definição
Diminuição na produção de ácido úrico
Deficiência da xantina-oxidase
Excreção aumentada de ácido úrico
REFERÊNCIAS BIBLIOGRÁFICAS
ENDEREÇOS RELEVANTES NA INTERNET
litíase renal, patologias geralmente relacionadas a estadosde hiperuricemia. Ou ainda, situações clínicas observadasem associação a estados de hipouricemia, a saber: na defi-ciência de xantina-oxidase, em doenças hepáticas, na sín-drome de Fanconi, na síndrome da imunodeficiência ad-quirida, entre outras.
É objetivo deste capítulo revisar a síntese do ácido úrico,como ocorre sua produção e excreção, as patologias decorren-tes das alterações do seu metabolismo, as manifestações clíni-cas destas doenças e seus manejos clínicos e terapêuticos.
INTRODUÇÃO
O conhecimento das particularidades do metabolismodo ácido úrico torna-se imprescindível para a compreen-são da gênese de diversas patologias relacionadas, bemcomo dos seus tratamentos.
É sabido que alterações dos níveis séricos do ácido úri-co poderão implicar complicações sistêmicas importantes,como gota, nefropatia aguda e crônica pelo ácido úrico e
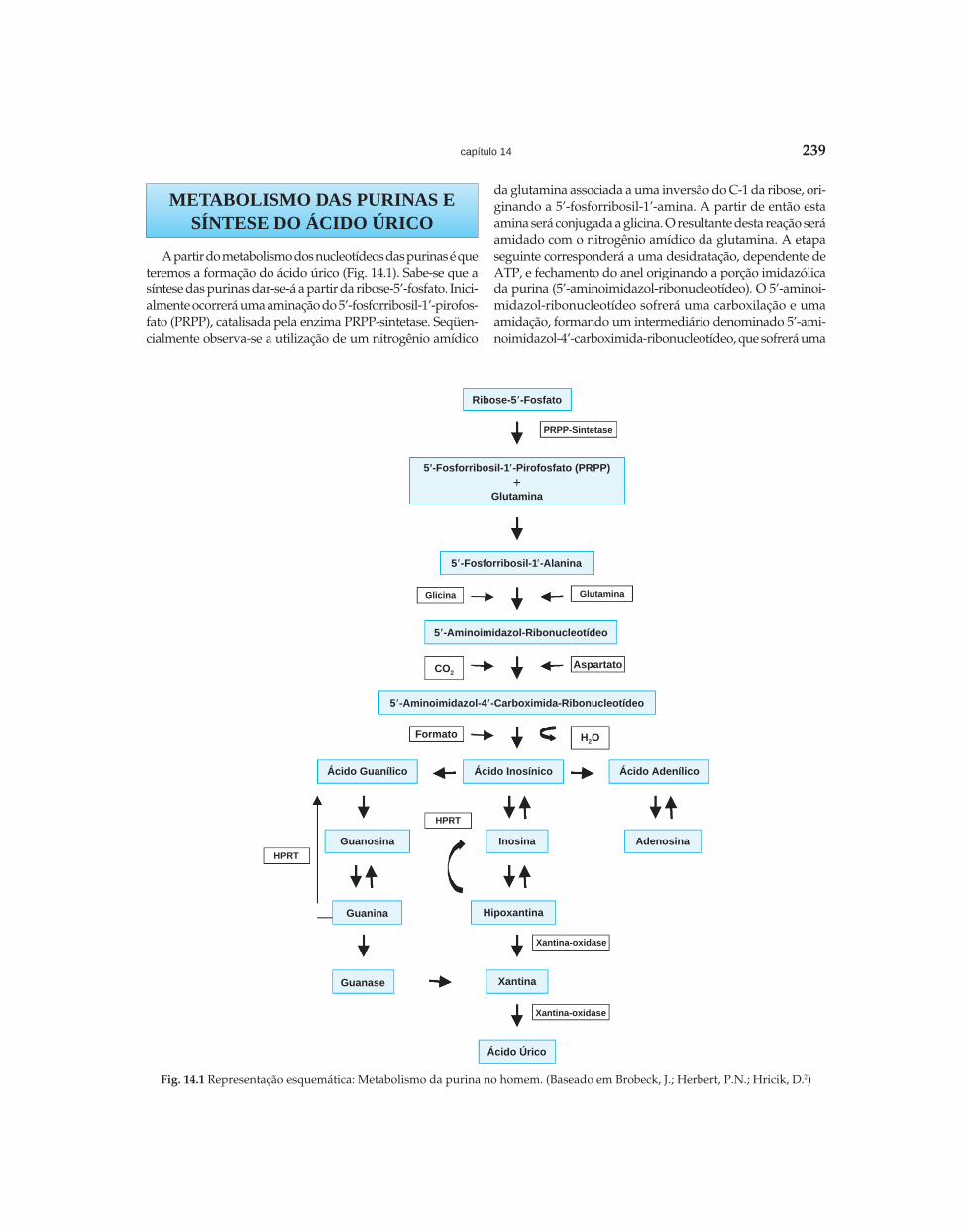
capítulo 14 239
METABOLISMO DAS PURINAS ESÍNTESE DO ÁCIDO ÚRICO
A partir do metabolismo dos nucleotídeos das purinas é queteremos a formação do ácido úrico (Fig. 14.1). Sabe-se que asíntese das purinas dar-se-á a partir da ribose-5’-fosfato. Inici-almente ocorrerá uma aminação do 5’-fosforribosil-1’-pirofos-fato (PRPP), catalisada pela enzima PRPP-sintetase. Seqüen-cialmente observa-se a utilização de um nitrogênio amídico
da glutamina associada a uma inversão do C-1 da ribose, ori-ginando a 5’-fosforribosil-1’-amina. A partir de então estaamina será conjugada a glicina. O resultante desta reação seráamidado com o nitrogênio amídico da glutamina. A etapaseguinte corresponderá a uma desidratação, dependente deATP, e fechamento do anel originando a porção imidazólicada purina (5’-aminoimidazol-ribonucleotídeo). O 5’-aminoi-midazol-ribonucleotídeo sofrerá uma carboxilação e umaamidação, formando um intermediário denominado 5’-ami-noimidazol-4’-carboximida-ribonucleotídeo, que sofrerá uma
Ribose-5�-Fosfato
PRPP-Sintetase
5’-Fosforribosil-1�-Pirofosfato (PRPP)�
Glutamina
5�-Fosforribosil-1�-Alanina
GlutaminaGlicina
5�-Aminoimidazol-Ribonucleotídeo
CO2Aspartato
5�-Aminoimidazol-4�-Carboximida-Ribonucleotídeo
Formato H2O
Ácido Guanílico Ácido Inosínico Ácido Adenílico
HPRT
Guanosina Inosina Adenosina
HPRT
Guanina Hipoxantina
Xantina-oxidase
Guanase Xantina
Xantina-oxidase
Ácido Úrico
Fig. 14.1 Representação esquemática: Metabolismo da purina no homem. (Baseado em Brobeck, J.; Herbert, P.N.; Hricik, D.2)

240 Metabolismo do Ácido Úrico
cursores de uratos é oriunda da dieta alimentar; sabe-se quefórmulas dietéticas livres de purinas chegam a reduzir a ex-creção urinária de ácido úrico em aproximadamente 40%.33
O processo de produção de uratos envolve a quebra dosnucleotídeos de purinas, o ácido guanílico (GMP), o ácidoinosínico (IMP) e o ácido adenílico (AMP). Finalmente, aguanina e a hipoxantina são então metabolizadas em xan-tina e esta, sob a ação irreversível da xantina-oxidase, emácido úrico.
PRODUÇÃO EXÓGENA DE ÁCIDO ÚRICOUma dieta alimentar sem restrição de purinas será sufi-
ciente para a manutenção da excreção urinária de ácidoúrico. Esta, por sua vez, dependerá da quantidade e do tipode purina existentes na dieta.34 Adultos jovens e hígidos,submetidos a uma dieta sem purina, reduzirão a concen-tração sérica de ácido úrico em cerca de 37% em 10 dias ea excreção urinária em torno de 33%, no mesmo período.4
PRODUÇÃO ENDÓGENA DE ÁCIDO ÚRICOA síntese continuada e o turnover endógenos das puri-
nas mantêm a excreção urinária do ácido úrico em tornode 300 a 400 mg/dia, fato este que independerá até mes-mo de uma restrição dietética de purinas.34
Excreção de Ácido ÚricoDe uma maneira geral, o organismo humano não é ca-
paz de metabolizar o urato, o que significa dizer que paraa manutenção da homeostase de seu metabolismo o uratodeverá ser eliminado através dos rins ou intestino.
Um indivíduo adulto do sexo masculino apresenta umpool de ácido úrico de aproximadamente 1.200 mg, sendoque, nas mulheres, esta quantidade se reduz a 600 mg. Dia-riamente, 50 a 60% das quantidades acima citadas serão re-novadas através dos metabolismos endógeno e exógeno. Aexcreção diária média através da urina será em torno de 450mg, e a intestinal, de aproximadamente 200 mg.4
APARELHO GASTRINTESTINALSabe-se que a entrada de urato através do intestino ocor-
re por um processo passivo variável de acordo com a con-centração sérica do ácido úrico. As bactérias do trato in-testinal são capazes de degradar o ácido úrico em dióxidode carbono e amônia, através da ação das uricases,alantoinases, alantoicases e ureases, num processo deno-minado “uricólise intestinal”.4,34 A quantidade de ácidoúrico encontrada nas fezes, apesar de pequena, poderáestar aumentada em alguns estados patológicos, como nassituações de insuficiência renal.
APARELHO URINÁRIOOs processos que envolvem a excreção renal de ácido
úrico têm sido definidos através de estudos de fisiologia e
formilação, recebendo um último átomo de carbono. Após asua desidratação completar-se-á a síntese do ribonucleotídeoda hipoxantina, o ácido inosínico (IMP).34
Como podemos observar na Fig. 14.1, que corresponde àrepresentação esquemática do metabolismo das purinas nosseres humanos, a partir da formação do ácido inosídico (IMP)teremos uma ramificação na via de biossíntese das purinas.Para um lado ocorrerá oxidação e aminação do IMP na de-pendência do ATP, originando o ácido guanílico (GMP), epara outro, dependendo agora do GTP, o IMP sofrerá ami-nação para originar o ácido adenílico (AMP). Seguindo-se avia de biossíntese, observamos que o ácido inosídico (IMP)passará a inosina e esta a hipoxantina, que por ação da enzi-ma xantino-oxidase originará a xantina e esta o ácido úricona dependência da ação da mesma enzima.34
Uma vez revisado o metabolismo das purinas, vale res-saltar que a velocidade de formação ou síntese do ácidoúrico dependerá da concentração intracelular da PRPP.Portanto, é correto afirmar que uma ação maior da enzi-ma PRPP-sintetase implicará concentração maior de PRPPe conseqüente biossíntese acelerada de purinas com mai-or formação de ácido úrico. Outra forma possível de au-mento da PRPP seria por uma deficiência ou menor ativi-dade da enzima hipoxantina-guanina-fosforribosil-trans-ferase (HPRT), enzima esta responsável pela conversão dahipoxantina em IMP e da guanina em GMP.34
Segundo a literatura, cerca de 10% dos pacientes com pro-dução aumentada de ácido úrico teriam como causa princi-pal uma deficiência parcial de HPRT.2 Tanto esta alteraçãoquanto a hiperatividade da PRPP-sintetase são defeitos fami-liares herdados como caráter ligado ao cromossomo X.3
Pontos-chave:
• A maioria das espécies mamíferas temníveis muito baixos de ácido úrico porque omesmo é convertido em alantoína, umproduto excretado e altamente solúvel
• Em humanos o ácido úrico é o produto finaldo metabolismo das purinas porque ohomólogo humano do gene uricase dosmamíferos foi modificado e não temexpressão (pseudogene)
METABOLISMO DO ÁCIDOÚRICO
Produção de Ácido Úrico
O ácido úrico é produzido no fígado a partir da degrada-ção de purinas sintetizadas de forma endógena ou ingeridasatravés da alimentação. Uma quantidade significativa de pre-

capítulo 14 241
Túbulo ProximalApós sofrer a filtração glomerular, o urato que chega ao
túbulo proximal passará por três processos distintos:4,35
• Reabsorção, no início do túbulo proximal, de cerca de90 a 100% de tudo que foi filtrado.
• No segmento S2 do túbulo proximal, haverá secreção deurato, proporcionando um retorno de 50% do que foifiltrado novamente ao lúmen tubular.
• Finalmente no segmento S3 ocorrerá a dita absorção pós-secretória.
Os mecanismos através dos quais estes processos ocor-rem não estão completamente definidos. Acredita-se, noentanto, que as trocas aniônicas desempenhem um impor-tante papel. A reabsorção de urato poderá ser mediada porum urato-OH (ou urato-HCO3) contratransportador namembrana luminal do túbulo proximal que opera em pa-ralelo a uma troca Na-H (Fig. 14.2).
A secreção de uratos no segmento S2 do túbulo proxi-mal envolve mecanismos mais complexos no que diz res-peito à troca de ânions. Essa secreção depende, na realida-de, de um transporte ativo secundário que envolve umprocesso de co-transporte de sódio, que permite a excre-ção renal não somente do ácido úrico mas também de fár-macos, como aspirina, antibióticos e diuréticos.43 Desta for-ma, identificam-se situações de hiperuricemia resultantesda redução da secreção habitual de uratos por ação de al-guns destes ácidos.35 Modernamente, tenta-se explicar estemecanismo com a teoria de carreadores de ânions na mem-brana basolateral e, talvez, na membrana luminal.7,8,9 Onúmero e a distribuição de cargas negativas parecem seros principais determinantes do grau de ligação. Os com-postos formados adentram a célula através de um co-trans-
Fig. 14.2 Representação esquemática. Reabsorção de ácido úrico no túbulo proximal.
farmacologia renal em animais de experimentação e emseres humanos. Cerca de 5% do ácido úrico circulante estáligado a proteínas, o que significa dizer que todo o restan-te poderá ser filtrado pelos glomérulos renais livremente.5,6
Além da filtração glomerular, outras três etapas do seumetabolismo poderão ser identificadas como de responsa-bilidade renal: a reabsorção pré-secretória, a secreção e areabsorção pós-secretória, todas ocorrendo no túbulo pro-ximal.5, 35
Pontos-chave:
• A uricólise intestinal é responsável poraproximadamente 1/3 do metabolismo totalde urato e é responsável pela eliminaçãoextra-renal de todo o urato
• A excreção urinária de urato é responsávelpelos 2/3 restantes do ácido úricoproduzido diariamente
• O clearance de ácido úrico, no entanto, estáem 7-12% de toda a carga filtrada
Filtração GlomerularComo já foi visto, apenas uma pequena porcentagem do
ácido úrico circulante está ligado à proteína. Conclui-se,desta forma, que uma grande quantidade deste será filtra-da pelos glomérulos diariamente. Apesar disto, o clearan-ce do ácido úrico em adultos normais encontra-se em tor-no de 7 a 12% de toda a carga filtrada, justificado pelo fatode que cerca de 90% do urato filtrado sofre reabsorção tu-bular.
Célula do Túbulo Proximal
Lúmen Capilar Peritubular
3 Na�
H�
Na�
OH� Ur�
2 K�
ATP ase
Ur�
Ur�
A�(?Cl�)

242 Metabolismo do Ácido Úrico
porte com o sódio e, também, podem ser produzidos den-tro destas células criando um gradiente favorável que ser-virá como um mediador nas trocas aniônicas7,8,34,43 (Fig.14.3). Além disso, um gradiente elétrico favorável poderápropiciar um transporte por difusão facilitada de um âni-on orgânico para o interior da luz tubular, permitindo,desta forma, a sua secreção.8
Após o exposto, conseguimos compreender a importân-cia dos mecanismos que permitem ao túbulo proximal se-cretar e, especialmente, reabsorver ácido úrico. A concen-tração urinária deste será reflexo direto daquilo que acon-tece principalmente nos segmentos S2 e S3 do túbulo pro-ximal. Em termos numéricos, sabe-se que apenas 12% doácido úrico filtrado aparecerão na urina.4
ESTADOS DE HIPERURICEMIA
DefiniçãoA melhor definição para o que é uma hiperuricemia ba-
seia-se no limite de solubilidade dos uratos nos fluidos hu-manos, ou seja, ocorrerá quando a concentração de uratosséricos corresponder a um estado de maior saturação nestecompartimento orgânico. Esta definição físico-química cor-responde à concentração de urato excedendo 7 mg/dl quan-do utilizarmos métodos enzimáticos (uricase) na sua men-suração. Um valor inferior em até 1 mg/dl poderá ser acei-to quando métodos calorimétricos sejam utilizados.
A persistência de níveis séricos elevados de ácido úrico(hiperuricemia) é uma alteração bioquímica relativamen-te comum em nosso meio. Tal situação ocorrerá, de maneira
geral, como resultado de uma excessiva produção de ura-tos, pela diminuição da sua excreção renal ou por umacombinação de ambos os fatores.36 Baseados neste conhe-cimento, poderemos classificar a hiperuricemia em duascategorias: primária e secundária.
Classificação
HIPERURICEMIA PRIMÁRIACorresponde às situações em que níveis séricos eleva-
dos de ácido úrico são identificados sem doenças coexis-tentes ou uso de drogas que possam diminuir a sua excre-ção ou aumentar a sua produção (Quadro 14.1).
HIPERURICEMIA SECUNDÁRIANesta categoria encontramos as situações resultantes de
uma excessiva produção de uratos (Quadro 14.2) ou quan-do se identifica uma diminuição de seu clearance renal(Quadro 14.3) por uso de drogas, toxinas, dieta ou outradoença associada.
Uma situação clínica que merece discussão especial é ahiperuricemia assintomática. Neste caso a situação dehiperuricemia não se encontra associada a nenhum acha-do clínico específico, como artrite, tofo ou litíase urinária.Embora a hiperuricemia esteja freqüentemente associadaa outras entidades clínicas, como hipertensão, obesidade,dislipidemias ou abuso no consumo de álcool, não há evi-dência clínica de que o ácido úrico seja um fator causal paraelas.39 Alguns indivíduos apresentam este estado hiperu-ricêmico durante toda a sua vida sem o desenvolvimentode qualquer outro tipo de complicação de maior gravida-de.10,37,38
Fig. 14.3 Representação esquemática. Secreção de ácido úrico no túbulo proximal.
Célula do Túbulo Proximal
Lúmen
3 Na�
Capilar Peritubular
2 K�
Ur�
Ur�
Ur�
A�
A�(?Cl�)
ATP ase

capítulo 14 243
Quadro 14.3 Causas de hiperuricemia secundáriapelo decréscimo do clearance renal
A. Alterações Clínicas:1. Insuficiência renal crônica de qualquer etiologia.2. Depleção de volume.3. Nefropatia por chumbo.4. Cetoacidose diabética.5. Acidose láctica.6. Pré-eclâmpsia.7. Obesidade.8. Hiperparatireoidismo.9. Hipotireoidismo.
10. Sarcoidose.11. Nefropatia hereditária associada a hiperuricemia ou
gota.
B. Induzida por Droga ou Dieta:1. Diuréticos tiazídicos e de alça.2. Ciclosporina.3. Salicilatos em baixas doses.4. Etambutol.5. Pirazinamida.6. Etanol.7. Levodopa.8. Abuso de laxantes (alcalose).9. Restrição de sal.
Modificado de Hellmann, D.B.11
Quadro 14.1 Origem das hiperuricemias
Hiperuricemias Primárias
A. Produção Aumentada de Purina:1. Idiopática2. Defeitos enzimáticos (s. de Lesch-Nyhan, doenças do
armazenamento de glicogênio)
Hiperuricemias Secundárias
A. Aumento do Catabolismo e do Turnover de Purina1. Distúrbios mieloproliferativos2. Distúrbios linfoproliferativos3. Sarcoma e carcinoma disseminados4. Anemias hemolíticas crônicas5. Drogas citotóxicas6. Psoríase
B. Diminuição do Clearance Renal do Ácido Úrico:1. Doença renal intrínseca2. Alteração da função de transporte tubular• Induzido por drogas (tiazídicos, probenecide)• Lactacidemia (acidose láctica, alcoolismo)• Cetoacidose (diabetes)• Diabetes insipidus• Síndrome de Bartter
B. Diminuição do Clearance Renal do Ácido Úrico:1. Idiopático
Quadro 14.2 Causas de hiperuricema secundária.Aumento da biossíntese de purinas e/ou daprodução de uratos
A. Defeitos Enzimáticos Genéticos:1. Deficiência de hipoxantina-guanina-fosforribosil-
transferase.2. Deficiência de fosfatase-6-glicose.3. Hiperatividade da fosforribosil-pirofosfato-sintetase.
B. Alterações Clínicas que Cursam com Aumento daProdução de Purinas ou Uratos:
1. Doenças mieloproliferativas.2. Doenças linfoproliferativas.3. Hemólise.4. Psoríase.5. Hipóxia tecidual.6. Síndrome de Down.7. Doenças malignas.
C. Aumento da Produção de Purinas Induzida porDrogas, Dieta e Toxinas:
1. Etanol.2. Dieta rica em purinas.3. Frutose.4. Vitamina B12 (pacientes com anemia perniciosa).5. Ácido nicotínico.6. Drogas citotóxicas.7. Warfarina.

244 Metabolismo do Ácido Úrico
EpidemiologiaA incidência de hiperuricemia difere entre os sexos mas-
culino e feminino, especialmente quando as mulheres en-contram-se em idade reprodutiva; este fato justifica-se de-vido a um maior clearance de uratos por ação estrogênica.41
De uma maneira geral, os homens hiperuricêmicos já apre-sentam início de elevações dos níveis de ácido úrico du-rante a puberdade, e as manifestações clínicas se fazem pre-sentes, em média, duas décadas após.
Apresentação ClínicaA hiperuricemia poderá apresentar-se clinicamente de
diversas formas; abordaremos a seguir as principais: gota,nefropatia aguda pelo ácido úrico, nefropatia crônica peloácido úrico, nefropatia hiperuricêmica familiar e nefrolití-ase pelo ácido úrico.
GOTAA gota é um transtorno metabólico de natureza hetero-
gênea e familiar, decorrente de alterações no metabolismodas purinas, que se caracteriza, principalmente, por hipe-ruricemia associada ao depósito de ácido úrico em diver-sas estruturas (preferencialmente articulações). Sendo as-sim, as crises agudas de artrite, geralmente monoarticula-res, são achados freqüentes. Com a evolução do quadroclínico a artrite torna-se crônica, associando-se a deformi-dades articulares e ao aparecimento de tofos, que são de-pósitos de monourato de sódio (Figs. 14.4 a 14.6).
A maior incidência de gota nos homens ocorre entre 30e 45 anos de idade, e nas mulheres, entre 55 e 70 anos (pós-menopausa). Cerca de 90% dos pacientes com gota primá-ria são do sexo masculino.11 Campion e cols, em 1982, apósum acompanhamento de 2.046 homens saudáveis por 15anos, evidenciaram uma incidência de gota em 4,9%, 0,5%e 0,1% em decorrência de um aumento dos níveis séricosde ácido úrico maiores que 9,0 mg/dl, entre 7,0 e 8,9 mg/dl e inferiores a 7,0 mg/dl, respectivamente.37 Já Langforde cols, em 1987, demonstraram que apenas 12% dos paci-entes com níveis de ácido úrico sangüíneo entre 7,0 e 7,9
mg/dl desenvolveram gota num período de estudo de 14anos.42
Alguns autores afirmam que 90% dos casos de gotapossam estar relacionados com uma excreção de ácidoúrico deficiente.2 Tal situação será identificada quando ti-vermos uma menor filtração glomerular de uratos, umaumento na reabsorção tubular, uma menor secreção tu-bular ou ainda uma combinação dos fatores citados.34 Nosquadros de insuficiência renal, aguda ou crônica, a redu-ção do clearance renal do ácido úrico poderá resultar emhiperuricemia; contudo, a gota raramente se manifesta nospacientes renais, talvez por uma diminuição da respostainflamatória aos cristais de ácido úrico proporcionada pelauremia.2
Berger e Yu afirmam que a gota por si só raramente le-vará a uma deterioração da função renal.12,13,14 Gota acom-panhada de insuficiência renal grave poderá ser vista emassociações com outras patologias subjacentes, como litía-se urinária, hipertensão arterial sistêmica, infecção uriná-ria e outras.34 A maioria dos investigadores acredita que anefropatia gotosa é uma manifestação dependente do graue da duração da hiperuricemia.34
Vários estudos correlacionam achados histopatológicosencontrados em biópsias renais com a ocorrência concomi-tante de hiperuricemia.12,15,16,17,18Fig. 14.4 GOTA: Deformidade articular e tofo (gonagra).
Fig. 14.5 GOTA: Deformidade articular e tofo.
Fig. 14.6 Monourato de sódio.

capítulo 14 245
Pontos-chave:
• As mulheres pré-menopausa têm umclearance maior de uratos devido à açãoestrogênica
• Cerca de 90% dos pacientes com gotaprimária são homens
• Gota per se raramente causa deterioração dafunção renal
Quadro ClínicoClinicamente, manifesta-se por um quadro de artrite
aguda, de aparecimento súbito, que ocorre na maioria dasvezes durante a noite, extremamente doloroso, que se se-gue a flutuações rápidas dos níveis de ácido úrico apósingestão excessiva de álcool ou certos tipos de alimentos,cirurgias, infecção, diuréticos ou drogas uricosúricas.34,40
Febre de até 39°C poderá estar presente.11 O quadro artrí-tico acomete preferencialmente a primeira articulaçãometatarsofalangiana (podagra), entretanto, outras articu-lações poderão estar comprometidas, como os joelhos(gonagra — Fig. 14.4) e, menos freqüentemente, os punhos(quiragra).40 Nas mãos a articulação mais afetada é a inter-falangiana do quinto pododáctilo. As apresentaçõespoliarticulares são infreqüentes, e quando presentes carac-terizam-se por serem assimétricas.
De uma maneira geral, após a primeira crise (monoar-trite aguda), crises poliarticulares poderão surgir. Com aevolução da doença o período intercrítico se reduz progres-sivamente, acabando por instalar-se uma artrite crônicaque sofre períodos de agudização. Nesse momento depó-sitos de monourato de sódio em tecidos moles começam aser reconhecidos, sendo denominados de tofos. Estes aco-metem preferencialmente as mãos, pés, olécrano, patela epavilhão auricular. A aspiração do material contido nostofos confirma a deposição dos cristais birrefringentes deurato de sódio que poderão aparecer livres ou no interiorde neutrófilos.
O comprometimento articular crônico, caracterizado porlesões de reabsorção osteocartilaginosa em “saca-bocado”e deformidades, aparecerá com a evolução da doença (Fig.14.5).
Diagnóstico Laboratorial
1. Níveis elevados de ácido úrico (�7,5 mg/dl), excetuan-do-se os casos em que drogas para sua redução tenhamsido empregadas.
2. VHS elevado nos surtos agudos.3. Elevação na contagem de células brancas poderá acom-
panhar também os quadros agudos.4. Cristais de urato de sódio observados na aspiração do
conteúdo dos tofos ou líquido sinovial confirmam odiagnóstico (Fig. 14.6).
Diagnóstico por Imagem
1. Ausência de achados radiológicos nos quadros iniciais(radiografia negativa pode não afastar a gota).
2. O aparecimento de cavidades ou erosões marginais nasextremidades ósseas poderá ser identificado nos qua-dros de mais longa duração.
3. Edema, do tipo granuloso, nos tecidos moles de paci-entes portadores de tofo gotoso.19
É importante a observação de que achados radiológicossemelhantes aos da gota poderão ser identificados na ar-trite reumatóide, sarcoidose, mieloma múltiplo, hiperpa-ratireoidismo e na doença de Hand-Schüller-Christian.11
Diagnóstico Diferencial
1. Celulite.2. Artrite piogênica aguda.3. Condrocalcinose aguda (pseudogota), onde se identi-
ficam depósitos de pirofosfato de cálcio no líquido si-novial, raio X positivo e nível sérico de ácido úrico nor-mal.
4. Artrite reumatóide, sarcoidose, mieloma múltiplo, hi-perparatireoidismo e doença de Hand-Schüller-Christian.
NEFROPATIA AGUDA PELO ÁCIDO ÚRICOA característica desta patologia é o aparecimento de um
quadro de insuficiência renal oligúrica ou anúrica decor-rente da precipitação intratubular de ácido úrico.20,21,34 Talsituação relaciona-se a uma produção ou excreção aumen-tada de ácido úrico em pacientes portadores de linfoma,leucemia, doenças mieloproliferativas (policitemia vera),particularmente naqueles submetidos à radioterapia ouquimioterapia, em decorrência de uma intensa lise celular.Outras causas, porém com menor freqüência, são: crisesconvulsivas que levam a um maior catabolismo celular,tratamento de tumores sólidos, síndrome de deficiência daenzima hipoxantina-guanina-fosforribosil-transferase(HPRT) ou na síndrome Fanconi-like por diminuição na re-absorção de uratos no túbulo proximal.20,21,22,34
Quadro ClínicoDeve-se suspeitar do diagnóstico em pacientes que desen-
volvam um quadro de insuficiência renal associada às situ-ações clínicas anteriormente mencionadas e que cursem comquadro de hiperuricemia. Os níveis de ácido úrico geralmen-te são superiores a 15 mg/dl, diferente de outras situaçõesde insuficiência renal aguda, onde estes valores geralmentesão inferiores a 12 mg/dl (faz-se exceção às de etiologia pré-renal).
Sintomas urinários não necessariamente se fazem pre-sentes, podendo-se observar dor lombar ou em flanco re-ferida por pacientes que apresentem litíase associada.

246 Metabolismo do Ácido Úrico
Diagnóstico Laboratorial
1. Hiperuricemia.2. Função renal alterada.3. Urinálise evidenciando cristais de ácido úrico. Quando
normal não afasta o diagnóstico.4. A relação entre ácido úrico (mg)/creatinina (mg) em
uma amostra de urina será maior que 1,0. Nas demaiscausas de insuficiência renal aguda costuma variar en-tre 0,60 e 0,75.20
5. Hipercalemia, hiperfosfatemia e hipocalcemia poderãoser identificadas nos pacientes que apresentem síndro-me de lise tumoral.33,44
NEFROPATIA CRÔNICA PELO ÁCIDO ÚRICOEsta é uma forma de insuficiência renal crônica decor-
rente da deposição de cristais de urato de sódio no inters-tício medular, originando microtofos. Tal deposição deter-minará uma resposta inflamatória crônica que levará a umafibrose intersticial.44 A deposição de cristais de urato nointerior dos túbulos renais também poderá ocorrer, causan-do lesão epitelial e obstrução intratubular. Alguns autoressugerem que a deposição intersticial ocorra como conse-qüência dos depósitos intratubulares de ácido úrico, quepromoveriam uma ruptura da membrana basal com pos-terior retubulização.23
A hiperuricemia como causa primária de insuficiênciarenal crônica não é algo comum.34 A nefropatia por uratojá foi relacionada num passado à gota tofácea. Na atuali-dade a formação de tofos e, especialmente, o comprometi-mento da função renal são infreqüentes.16
Quadro ClínicoUma manifestação inicial comum da nefropatia crôni-
ca pelo ácido úrico é a albuminúria que, normalmente, éleve e de caráter intermitente.34 Com a progressão da do-ença renal, aparecem os sinais de uremia. Neste estágio, édifícil diferenciar se a doença renal é causa ou conseqüên-cia da hiperuricemia.34
Diagnóstico LaboratorialConsidera-se uma elevação dos níveis séricos de ácido
úrico desproporcional ao grau de insuficiência renal quandoeste excede 9 mg/dl frente a uma creatinina plasmática igualou inferior a 1,5 mg/dl, 10 mg/dl nas situações em que aconcentração de creatinina esteja entre 1,5 e 2,0 mg/dl, e 12mg/dl nas situações de insuficiência renal mais avançada.17
NEFROPATIA HIPERURICÊMICA FAMILIARA nefropatia hiperuricêmica familiar foi descrita no iní-
cio da década de 60 por Duncan e Dixon.24 Embora seja umapatologia rara, vários relatos sobre ela permeiam a litera-tura médica nas últimas décadas.25 A sua etiologia estárelacionada com uma irregularidade na mobilização tubu-lar renal de urato, que resultaria de uma incapacidade das
células tubulares renais em fazer a remoção de ácido úricodo interstício.3 Não há portanto, nestes pacientes, sínteseacelerada de purinas.25,26
A exemplo do que foi descrito na nefropatia crônica peloácido úrico, a presença de uratos no interstício renal levariainicialmente a uma reação inflamatória local que se seguiráde fibrose e comprometimento progressivo da função renal.
NEFROLITÍASE PELO ÁCIDO ÚRICOA incidência de nefrolitíase pelo ácido úrico pode ser
bastante variável, estando relacionada diretamente com apopulação analisada. As características nutricionais, gené-ticas e ambientais parecem ser bastante significativas no quediz respeito à sua epidemiologia. Nos Estados Unidos daAmérica e Europa a sua prevalência é de aproximadamen-te 5 a 10% do total de casos relatados de nefrolitíase.4 Empaíses em desenvolvimento esta prevalência poderá chegara 40%, especialmente naqueles de clima árido e quente, nosquais há uma maior tendência de se observar um volumeurinário menor e um pH urinário mais ácido, favorecendo-se assim a precipitação de cristais de ácido úrico. Um estu-do multicêntrico acerca da litíase renal no Brasil observouhiperuricosúria em aproximadamente 30% dos litiásicos.49
Sabe-se que este tipo de cálculo pode incidir também numapopulação sem história prévia de gota; contudo, cerca de20% dos portadores de gota acabam por desenvolvê-lo.4
Mais de 80% dos cálculos de urato, encontrados empacientes portadores de gota, são exclusivamente de áci-do úrico. Nos demais casos geralmente se observa oxalatode cálcio ou fosfato de cálcio circundando um núcleo cen-tral de urato. A prevalência de cálculos de oxalato de cál-cio entre pacientes com gota chega a ser 10 a 30 vezes mai-or que na população não-gotosa.
Na gota primária, a incidência na formação de cálculosvariará de acordo com a quantidade de ácido úrico excre-tada. Incidirá em 10 a 20% dos pacientes com excreçãourinária normal (800 mg/dia no homem e 750 mg/dia namulher), podendo variar entre 40 e 50% quando a excre-ção de ácido úrico atinja 1.000 mg/dia.28,52,53
A formação de cálculos de ácido úrico, decorrente daprecipitação urinária de seus cristais, está na dependênciadireta de dois fatores: a sua alta concentração urinária e opH urinário ácido. Observe a equação a seguir:
H� � Urato� ↔ Ácido Úrico
O desvio desta reação converterá sais relativamentesolúveis de urato em ácido úrico insolúvel.27 A solubilida-de total do ácido úrico na urina cai de 200 mg/dl num pHurinário de 7,0 para 15 mg/dl num pH de 5,0.27
Outras situações clínicas também poderão estar relaci-onadas com a formação de litíase por ácido úrico, como:aumento na produção do ácido úrico nas doenças mielo-proliferativas, uso de drogas uricosúricas (aspirina, probe-necide) diminuindo a sua reabsorção tubular, nas diarréi-as crônicas em virtude da diminuição do volume urinário

capítulo 14 247
associado a uma queda do pH urinário27,28 ou pelo aumentoda excreção de ácido úrico proporcionado pelo uso de al-guns hormônios, como estrógenos e corticosteróides.4
Houve a preocupação de alguns clínicos com a possibi-lidade de que aqueles pacientes que apresentavam hipe-ruricemia induzida pelo uso de tiazídicos pudessem desen-volver nefrolitíase no momento em que se associasseminibidores da enzima de conversão da angiotensina oumesmo antagonistas dos receptores de angiotensina II.54 Talfato parece infundado em virtude de drogas como o losar-tan, por exemplo, serem capazes de aumentar o pH uriná-rio pela redução da reabsorção de bicarbonato.
Para finalizar, poderíamos citar ainda casos idiopáticosde nefrolitíase pelo ácido úrico, acometendo pacientes queapresentam sua concentração plasmática e renal normaisporém com uma tendência à acidificação urinária semoutras anormalidades da função renal.4
Quadro ClínicoOs achados clínicos são comuns às demais situações de
litíase urinária com ou sem uropatia obstrutiva (ver capí-tulo específico) (Figs. 14.7 e 14.8).
Nestes pacientes o que nos chama a atenção é a presen-ça de quadro sugestivo de urolitíase subjacente a outraspatologias que cursem com hiperuricemia e/ou hiperse-creção urinária de ácido úrico.
Diagnóstico Laboratorial
1. Dosagem sérica de ácido úrico.2. pH urinário.3. Dosagem da concentração urinária de ácido úrico.4. Análise bioquímica do cálculo eliminado e/ou retirado.
Diagnóstico por Imagem
1. Os cálculos puros de ácido úrico não são radiopacos,desta forma o exame radiológico simples poderá sernegativo.
2. Ultra-som e/ou tomografia de rins e vias urinárias po-derão identificar a presença do cálculo.
3. A urografia excretora revelará uma lesão intraluminarradiotransparente, sendo imprescindível o diagnósticodiferencial com tumores e presença de coágulos.
HIPERURICEMIA NO TRANSPLANTE RENALLin e colaboradores, em 1989, demonstraram a incidên-
cia de hiperuricemia em 84% dos pacientes transplantadosem uso de ciclosporina comparada a 30% naqueles paci-entes que tinham a sua imunossupressão feita com azatio-prina e prednisona.29 A artrite gotosa tem sido relatada em7 a 24% dos pacientes tratados com ciclosporina,29,30 sendoo diagnóstico inicial mais freqüentemente feito entre osmeses 17 e 24 após o transplante renal.29
Além da ciclosporina, que pode promover hiperurice-mia por uma diminuição do fluxo plasmático renal, outrosfatores também poderão ser relacionados, como o uso dediuréticos e as situações de insuficiência renal decorrentede episódios de rejeição.29,30,31
Manejo Clínico e Farmacológico dosEstados de Hiperuricemia
HIPERURICEMIA ASSINTOMÁTICA
Considerações GeraisA hiperuricemia assintomática na grande maioria das
vezes (80 a 90%) ocorrerá por um excesso no consumo depurinas, por uma secreção diminuída de uratos ou umasoma destes dois fatores. Um grupo menor de pessoaspoderão apresentá-la devido a um aumento da sua produ-ção endógena.
As causas etiológicas de hiperuricemia secundária de-verão ser investigadas e tratadas individualmente (Qua-dro 14.2, Quadro 14.3). A coleta da urina de 24 horas, comdosagem de ácido úrico e da creatinina, em pessoas comfunção renal normal (recebendo uma dieta standard, comexclusão de álcool e drogas que alterem o metabolismo doácido úrico) geralmente poderá estabelecer se estamos fren-te a uma superprodução de ácido úrico (� 800 mg/dia ou12 mg/kg/dia) ou uma diminuição de seu clearance renal.Fig. 14.7 Nefrolitíase. Hidronefrose por uropatia obstrutiva.
Fig. 14.8 Nefrolitíase. Dilatação ureteral, pielocalicial e hidrone-frose por uropatia obstrutiva.

248 Metabolismo do Ácido Úrico
A relação entre o clearance de urato e creatinina na urinade 24 horas menor que 6% define um déficit de excreção.Pacientes que persistem com níveis urinários superiores a670 mg/dia, mesmo após uma dieta baixa em purinasdurante um período de cinco dias (Quadro 14.4), deverãoser considerados inicialmente como superprodutores.
Tratamento FarmacológicoQuando analisamos riscos e benefícios, o tratamento
com drogas hipouricemiantes nestes pacientes, na maio-ria das vezes, não se fará necessário. Entretanto, três situ-ações clínicas deverão merecer atenção especial, com con-seqüente instituição de tratamento famacológico, são elas:
• Paciente em radioterapia ou quimioterapia deve-rá receber alopurinol na profilaxia da nefropatiaaguda pelo ácido úrico.21
• Níveis séricos de ácido úrico persistentemente al-tos, 13 mg/dl no homem e 10 mg/dl na mulher.34
• Alopurinol deverá ser prescrito para paciente queapresente excreção urinária de ácido úrico maiorque 1.000 mg/dia, quando o controle dietético nãoestá sendo satisfatório.34 Deve-se objetivar uma ex-creção urinária de 800 mg/dia.
GOTA
Considerações GeraisUma abordagem equivocada relativamente freqüente,
nos portadores de gota, é o manejo simultâneo do quadro
artrítico agudo e da hiperuricemia.34,56 Sabe-se que redu-ções súbitas nos níveis séricos de ácido úrico poderão pre-cipitar episódios de artrite gotosa.34 Pelo exposto, o mane-jo do quadro de hiperuricemia deverá ser postergado atéa resolução do quadro artrítico agudo.
Tratamento Farmacológico do Quadro Agudo
• RepousoO paciente deverá ser mantido em repouso por pelo
menos 24 horas após melhora dos sintomas agudos. Istoporque a deambulação precoce poderá precipitar a recor-rência do quadro artrítico.56
• Antiinflamatórios não-hormonais (AINH)Os AINH têm sido as drogas de escolha no manejo do
quadro artrítico agudo. Dentre eles, tradicionalmente, pres-creve-se a indometacina, embora outros antiinflamatóriostenham bons resultados. A dose preconizada é de 25 a 50mg a cada 8 horas por 5 a 10 dias, período este em que ossintomas deverão estar resolvidos.10,11,34,56 Nos casos em quehá risco do desenvolvimento de sangramento digestivouma opção seria o uso dos inibidores da COX-2,56 nas do-ses recomendadas pela farmacopéia. É importante salien-tarmos, no entanto, que o risco do desenvolvimento denefropatia pelos antiinflamatórios ditos tradicionais ouaqueles inibidores da COX-2 é similar.57
• ColchicinaA colchicina é uma droga que poderá ser empregada tan-
to nos períodos intercrise como no manejo do quadro agudoda gota. Esta droga é capaz de inibir a fagocitose de cristaisde urato pelos neutrófilos, não interferindo no metabolismodos uratos.34 Sua excreção se dará através da bile, secreçõesintestinais e urina.34 Sua administração deverá ser iniciadapoucas horas após o início dos sintomas.56 O esquema poso-lógico preconizado é de 0,5 a 0,6 mg via oral a cada hora atéque sintomas gastrintestinais apareçam, como náuseas, vô-mitos ou dor abdominal.56 A dose total necessária geralmen-te variará entre 4 e 6 mg e não deverá jamais exceder 8 mg.56
O uso endovenoso poderá ser uma opção para que não te-nhamos sintomas gastrintestinais, contudo dor local, extra-vasamento com dano tecidual e supressão de medula ósseasão complicações possíveis. A dose endovenosa inicial seráde 1 a 2 mg diluídos em 20 a 50 ml de solução salina adminis-trados através de catéter intravascular.10,11,32,56 Duas doses adi-cionais de 2 mg poderão ser administradas em intervalos deseis horas.56 Não se deverá exceder um total de 4 mg, e a col-chicina não deverá ser administrada pela via oral por pelomenos três semanas.56 Pacientes portadores de insuficiênciarenal ou hepática e indivíduos idosos deverão ter a dose re-duzida em 50%. É importante salientar que o risco de toxici-dade estará aumentado para aqueles pacientes que fazem usosimultaneamente de drogas inibidoras da enzima P-450 (eri-tromicina, cimetidina, tolbutamina).32 Frente à associação
Quadro 14.4 Conteúdo de purina nos alimentos
A. Alimentos com Pouca Purina:1. Cereais refinados e seus produtos, flocos de milho,
arroz branco, massa, araruta, sagu, farinha de milho,bolos, pães, fubá, tapioca.
2. Leite e seus derivados, ovos.3. Açúcar, doces, gelatina.4. Manteiga, margarina poliinsaturada, outras
gorduras.5. Tomate, vegetais de folhas verdes (algumas exceções).6. Frutas, nozes, manteiga de amendoim.7. Sopas ou cremes feitos com vegetais permitidos e sem
carnes.8. Água, suco de frutas, bebidas carbonatadas, chá,
café.
B. Alimentos com Muita Purina:1. Todos os tipos de carnes.2. Extratos e molhos de carne.3. Fermento e derivados, cerveja, outras bebidas
alcoólicas.4. Feijão, ervilha, lentilha, grão-de-bico, espinafre,
aspargo, couve-flor, soja, cogumelos.5. Cereais integrais (arroz, trigo, centeio, aveia).6. Coco, castanha-do-pará, castanha de caju.

capítulo 14 249
entre doença hepática e renal, a via de administração endo-venosa deverá ser proscrita11
• CorticóidesOs corticóides estarão bem indicados para aqueles pa-
cientes que apresentarem contra-indicação para o uso deAINH.56 Uma possibilidade para o seu uso seria através deinjeções intra-articulares nos pacientes que apresentemcomprometimento monoarticular, desde que o diagnósti-co de artrite séptica já tenha sido afastado.34 A administra-ção intra-articular poderá ser feita com o uso de triancino-lona, 10 a 40 mg, na dependência do tamanho da articula-ção comprometida.56 Nos casos de gota com comprometi-mento poliarticular, a via endovenosa deverá ser prioriza-da, com a administração de metilprednisolona, 40 mg aodia, com redução da dose e retirada dentro de sete dias.56
O uso oral de corticóides também poderá ser uma opçãode tratamento; preconiza-se o uso de prednisona, 40 a 60mg ao dia, com retirada da droga em sete dias.56
• AnalgésicoPoderemos lançar mão dos opióides somente nos casos
de dor intensa.
Tratamento Farmacológico do Período Intercrise
• Orientações dietéticasAs purinas contidas na dieta usualmente não contribui-
rão com mais que 1 mg/dl na concentração sérica de uratos.32
Mesmo com uma pequena contribuição aparente, a orien-tação dietética deverá sempre ser feita, especialmente paraaqueles pacientes com alta ingesta de purinas (Quadro 14.4).A obesidade, o uso abusivo de álcool bem como períodosprolongados de jejum deverão ser desencorajados. Um dé-bito urinário superior a 2 litros ao dia deverá ser estimula-do através de uma ingesta hídrica adequada.
• ColchicinaA colchicina aparece como uma das melhores opções na
profilaxia dos quadros agudos. A dose preconizada é de 0,5a 0,6 mg duas vezes ao dia.10,11,32,56 Pacientes com disfunçãohepática ou renal deverão receber uma única dose ao dia,reduzindo-se assim o risco do desenvolvimento de neuropa-tia periférica e miosite.56 A interrupção da droga poderá serfeita quando não mais ocorrerem crises agudas num períodode 6 a 8 semanas.10,11,32 Além da prevenção dos quadros agu-dos, o uso da colchicina também estará indicado no momen-to em que iniciarmos a administração de drogas uricosúricasou alopurinol, evitando-se quadros agudos precipitados pormudanças abruptas nos níveis séricos de ácido úrico.11
• Antiinflamatórios não-hormonais (AINH)Doses diárias de indometacina ou seus equivalentes
poderão ser utilizadas nos casos em que a colchicina isola-da falha na prevenção de quadros agudos.32
• Evitar medicamentos hiperuricemiantesDiuréticos de alça e tiazídicos inibem a secreção renal
de ácido úrico e portanto devem ser evitados. O uso debaixas doses de aspirina (� 3 g/dia) também agrava a hi-peruricemia.56
Redução dos Níveis Séricos de Ácido Úrico
• Agentes uricosúricosEstas drogas diminuem o pool de uratos pelo bloqueio
de sua reabsorção tubular. Seu emprego é ineficaz em pa-cientes com creatinina maior que 2 mg/dl.11 Sua principalindicação seria nos casos em que há um aumento na fre-qüência ou gravidade dos ataques agudos, desde que aexcreção urinária diária de ácido úrico seja inferior a 800mg.11 O probenecide pode ser usado na dose de 500 mg pordia, chegando até 1 a 2 gramas ao dia.32,56 A sulfinpirazonaé utilizada em dose inicial de 50 a 100 mg duas vezes aodia, com aumentos graduais até 200 a 400 mg duas vezesao dia.32,56 Para minimizar o risco de precipitação de cris-tais de ácido úrico com conseqüente formação de cálculos,sempre que optarmos pelo uso destas drogas deveremosmanter o pH urinário em torno de 6,0 (citrato de potássio,30 a 80 mEq/dia) e um volume urinário superior a 2 litrosao dia.
• AlopurinolPacientes hiperuricêmicos, que apresentem uma excre-
ção urinária diária de ácido úrico superior a 800 mg, sebeneficiarão com o uso de alopurinol. Esta droga é umainibidora da xantina-oxidase e prontamente diminui osníveis plasmáticos e urinários de ácido úrico. A dose inici-al é de 100 mg ao dia por sete dias, com aumento da dosecaso os níveis séricos de ácido úrico permaneçam elevados.Os melhores resultados serão obtidos com doses entre 200a 300 mg de alopurinol ao dia.32,56
NEFROPATIA AGUDA PELO ÁCIDO ÚRICOA nefropatia aguda pelo ácido úrico é uma entidade
clínica que acontece como parte da síndrome de lise tumo-ral, com já foi descrito anteriormente. A sua prevençãoparece ser a melhor conduta terapêutica. Pacientes queserão submetidos a radioterapia ou quimioterapia paratratamento de neoplasias, que possuem um alto turnovercelular, deveriam receber profilaticamente alopurinol emdoses elevadas (600 a 900 mg/dia).21,45 O débito urináriodeverá ser mantido elevado, acima de 2,5 litros ao dia, quepoderá ser conseguido através da administração de solu-ção salina e até mesmo manitol.
A alcalinização da urina com o uso de acetazolamida oubicarbonato é controversa na literatura. Conger e colabo-radores, em 1976, num trabalho clássico demonstraram quea simples hidratação com solução salina seria tão efetivaquanto a alcalinização no sentido de diminuir a precipita-ção de cristais de ácido úrico.45 A alcalinização da urina

250 Metabolismo do Ácido Úrico
objetivaria transformar o ácido úrico em sais de urato, maissolúveis e portanto com menor risco de precipitação. Con-tudo, tal conduta poderia promover a precipitação de fos-fato de cálcio em pacientes com hiperfosfatemia.
Pacientes que evoluem com a instalação de um quadro deinsuficiência renal aguda devem ser manejados com a pres-crição de alopurinol, hidratação vigorosa e diuréticos de alça,estando contra-indicado o uso de bicarbonato de sódio. Otratamento dialítico (hemodiálise) deverá ser restrito aos ca-sos em que se necessita remover o excesso de ácido úrico cir-culante porém não se consegue induzir a diurese.
Outros agentes têm sido usados no manejo destes paci-entes, que são a uricase e o polietileno-glicol-uricase (PEG-uricase), ainda em fase experimental. Sabe-se que a uricaseou urato oxidase é uma enzima que catalisa a oxidação doácido úrico em compostos mais solúveis.58 O seu uso atual-mente tem sido limitado a pacientes com câncer que desen-volvem hiperuricemia induzida pela quimioterapia. Istoporque a sua administração associa-se com certa freqüên-cia a reações alérgicas com possibilidade de anafilaxia.59
NEFROPATIA CRÔNICA PELO ÁCIDO ÚRICOUma vez que se instale a insuficiência renal crônica, o
tratamento mais efetivo no que diz respeito à remoção deuratos é a hemodiálise. Conseguimos uma depuração de150 ml/min utilizando-se um fluxo de bomba de sangueem torno de 300 a 400 ml/min. Estes valores são muitosuperiores àqueles obtidos através da diálise peritoneal.4
Mejias e Maldonado referem a possibilidade de uma redu-ção superior a 50% da concentração plasmática inicial deácido úrico em um período de 6 horas de hemodiálise.4
Pacientes que já estejam em programa de tratamento di-alítico regular e que mesmo assim persistam com níveis sé-ricos de ácido úrico acima dos valores desejados devem sertratados com alopurinol, para que se previnam surtos de ar-trite recorrente. Sabendo-se que o alopurinol é uma drogaque depende da excreção renal para sua eliminação, o ajus-te de dose se faz necessário.47 Uma sugestão de prescriçãobaseada no clearance de creatinina seria a seguinte: paci-entes com Clcreatinina entre 20 e 50 ml/min deveriam rece-ber apenas 1/3 da dose habitual, enquanto pacientes comClcreatinina inferior a 20 ml/min, 1/6 da dose diária recomen-dada.48
NEFROPATIA HIPERURICÊMICA FAMILIARO tratamento desta entidade patológica deve fundamen-
tar-se no uso de agentes uricosúricos, do alopurinol, quetem demonstrado alguns bons resultados, como demons-traram Reitter e colaboradores em 1995,3 além do controlerigoroso dos níveis pressóricos.
NEFROLITÍASE PELO ÁCIDO ÚRICOExistem três pontos fundamentais que regem o trata-
mento dos pacientes portadores de litíase urinária peloácido úrico: 27,28
• Deve-se manter um débito urinário em torno de 2 litrosao dia no intuito de se diminuir a concentração uriná-ria de ácido úrico.
• Alcalinizar a urina, pois se sabe que, em torno de umpH de 6,5, cerca de 90% do ácido úrico urinário estarásob a forma de urato, minimizando-se assim o risco deprecipitação. Tal eficácia poderá ser comprovada pelaobservação da equação de Henderson-Hasselbalch, quedemonstra a relação entre urato e ácido úrico:
pH � 5,35 � log ([urato] � [ácido úrico])
• Uso de alopurinol, para que se reduza a produção deácido úrico e conseqüentemente a sua excreção.
A administração de bicarbonato ou citrato de potássio,na dose de 60 a 80 mEq/dia,28,37 pode ser eficaz na dissolu-ção dos cálculos já formados ou na prevenção da forma-ção de novos cálculos. A alcalinização utilizando-se sais desódio não produz o efeito desejado, pois a expansão devolume resultante de sua administração aumentará a ex-creção de sódio e secundariamente de cálcio.27,28 A hiper-calciúria resultante poderá trazer conseqüências indesejá-veis, pois o ácido úrico poderá atuar como um nicho paraa formação de cálculos de oxalato de cálcio.27,28
Habitualmente, os pacientes que mantêm uma excreçãode ácido úrico diária superior a 1.000 mg e que não respon-dem a alcalinização e hidratação requerem o uso continu-ado de alopurinol.27 Agentes uricosúricos são proscritos.
Procedimentos como litotripsia extracorpórea geral-mente não são necessários, visto que as recomendaçõesacima mencionadas podem levar à dissolução dos cálcu-los de ácido úrico.
HIPERURICEMIA NO TRANSPLANTE RENALSabendo-se que o tratamento da hiperuricemia nestes
pacientes não é isento de riscos, recomenda-se que paci-entes assintomáticos não deverão ser tratados. Dado ogrande número de interações medicamentosas, especial-mente no que diz respeito às drogas imunossupressoras,a hiperuricemia ou gota em pacientes transplantados re-nais só deverá ser conduzida por profissionais experimen-tados nesta área.
• ColchicinaÉ a droga de escolha para os casos de artrite gotosa agu-
da em pacientes transplantados. A dose recomendada va-riará de 0,15 a 0,6 mg ao dia, prescrito somente para paci-entes que não apresentem disfunção renal. Convém lem-brar que a administração simultânea de ciclosporina outacrolimus à colchicina diminui seu clearance.60,61
• Antiinflamatórios não-hormonais (AINH)O uso deste grupo farmacológico poderá aumentar os
riscos de nefrotoxicidade à ciclosporina em função da di-minuição da taxa de filtração glomerular possibilitada pela

capítulo 14 251
inibição da síntese renal de prostaglandinas. Riscos e be-nefícios deverão ser avaliados antes da prescrição dosAINH.
• CorticóidesAumento nas doses de prednisona para 20 ou 30 mg ao
dia poderá ser uma medida eficaz frente a quadros artríti-cos agudos.
• AlopurinolPacientes que fazem uso de azatioprina não deverão
receber alopurinol. O seu uso implicará o acúmulo de ummetabólito ativo da azatioprina denominado 6-mercapto-purina, que acarretará maior risco de toxicidade à medulaóssea.62,63
Em situações em que o uso do alopurinol seja impres-cindível, duas alternativas se apresentam: a primeira de-las seria a redução na dose diária de azatioprina em pelomenos 50%, com monitorização rigorosa da contagem decélulas brancas, ou até mesmo a descontinuação da droga;e a segunda seria a prescrição do micofenolato em lugarda azatioprina.
• Agentes uricosúricosO uso de drogas como probenecide ou sulfinpirazona
só poderá ser aventado para aqueles pacientes que apre-sentem função renal normal e não tenham história de cál-culos renais. Lembrando que a sulfinpirazona reduz osníveis de ciclosporina.
ESTADOS DE HIPOURICEMIA
Definição
A hipouricemia, por definição, corresponderia a todasas situações clínicas em que nos deparamos com um nívelsérico de ácido úrico igual ou inferior a 2 mg/dl.4 Esta si-tuação poderá ser identificada em até 2% dos pacienteshospitalizados e em menos de 0,5% na população em ge-ral.64 Os estados de hipouricemia de maneira geral resul-tarão de uma diminuição na produção de uratos ou doaumento de sua excreção.
Diminuição na Produção de Ácido Úrico
Muitos mecanismos poderão estar envolvidos nesteprocesso; em seguida descreveremos alguns deles.
DEFICIÊNCIA DA XANTINA-OXIDASE
• AlopurinolO alopurinol é uma droga que atua na redução dos ní-
veis séricos de ácido úrico através da inibição da ação da
enzima xantina-oxidase. O emprego desta droga talvez sejaa causa mais comum de hipouricemia, porém os níveisséricos dificilmente serão inferiores a 2,5 mg/dl.
• Doenças hepáticasComprometimentos hepatocelulares graves poderão
culminar com uma perda da ação enzimática da xantina-oxidase hepática, levando a uma situação de hipouricemia.
• Xantinúria hereditáriaA xantinúria é resultante de uma marcada redução da
atividade da enzima xantina-oxidase e está associada comos mais profundos graus de hipouricemia no homem. Estedefeito enzimático leva à síntese reduzida de ácido úricocom acúmulo de seus precursores, hipoxantina e xantina.A concentração sérica de ácido úrico na xantinúria é usu-almente inferior a 1 mg/dl.4,34
EXCREÇÃO AUMENTADA DE ÁCIDO ÚRICO
• Expansão de volume extracelularA hipouricemia nesta situação será induzida pela redu-
ção na reabsorção de sódio e ácido úrico no túbulo proxi-mal, decorrente da expansão do volume extracelular. Talsituação poderá ser identificada nos pacientes que estãorecebendo grandes quantidades de líquido endovenoso,nos portadores de síndrome da secreção inapropriada dohormônio antidiurético, ou ainda naqueles com polidipsiapsicogênica.52,65
• Síndrome de FanconiEsta síndrome mais freqüentemente é observada em
crianças portadoras de cistinose e em adultos com mielo-ma múltiplo. Observa-se uma redução na reabsorção deácido úrico nos túbulos proximais e também de glicose,fosfato, potássio, bicarbonato e aminoácidos.
• Hipouricemia renal familiarEsta é uma síndrome de herança autossômica caracte-
rizada por um defeito tubular no transporte de uratos.
• Síndrome da imunodeficiência adquiridaA hipouricemia tem sido identificada em alguns paci-
entes portadores de SIDA e relacionada a algum compro-metimento intracraniano, a doença disseminada relaciona-da e a um pobre prognóstico. Um outro fator associado quepoderá justificar a sua presença seria o uso de altas dosesde sulfametoxazol-trimetoprim no tratamento das infec-ções por Pneumocystis carinii.55
• DrogasTalvez uma das causas mais comuns de hipouricemia
fosse secundária ao uso de alguns fármacos. Alguns auto-res referem que este tipo de etiologia poderia representarcerca de 66% do total de casos de hipouricemia.4 Algumas

252 Metabolismo do Ácido Úrico
drogas poderiam induzir a uricosúria diminuindo a liga-ção de urato às proteínas plasmáticas, inibindo a reabsor-ção do urato filtrado ou dificultando a secreção de uratona porção média do túbulo proximal. Exemplos clássicossão os salicilatos em altas doses, certos tipos de contrastesradiológicos, o sulfametoxazol-trimetoprim e ainda algunsantagonistas dos receptores de angiotensina II, como porexemplo o losartan.
REFERÊNCIAS BIBLIOGRÁFICAS
1. BROBECK, J.R. Fisiologia das purinas e pirimidinas. In: ———. AsBases Fisiológicas da Prática Médica, 9.ª ed. Rio de Janeiro: GuanabaraKoogan, 1976, pp. 116-124.
2. HERBERT, P.N.; HRICIK, D. Hiperuricemia e gota. In: Andreoli, T.E.;Carpenter, C.C.J.; Plum, F.; Smith, L. Cecil — Medicina Interna Bási-ca, 2.ª ed. Rio de Janeiro: Guanabara Koogan, 1991, pp. 337-380.
3. REITTER, L.; BROWN, M.; EDMONDS, J. Familial hyperuricemicnephropathy. American Journal of Kidney Diseases, v. 25, n. 2, pp. 235-241, february, 1995.
4. MEJIAS, E.; MALDONADO, M.M. Disturbances of uric acid meta-bolism. In: Handbook of Renal Therapeutics (Edit. Maldonado, M.M.).Plenum Medical Book Co., 1983, Cap. 8, pp. 155-171.
5. KAHN, A.M. Effect of diuretics on the renal handling of urate.Seminars in Nephrology, 8:305, 1988.
6. GUGGINO, S.E.; MARTIN, G.J.; ARONSON, P.S. Specificity andmodes of the anion exchanger in dog renal microvillus membranes.American Journal of Physiology, 244:F612, 1983.
7. BURCKHARDT, G.; ULLRICH, K.J. Organic anion transport acrosscontraluminal membrane-dependence on sodium. Kidney Internati-onal, 36:370, 1989.
8. PRITCHARD, J.B.; MILLER, D.S. Comparative insights into themechanisms of renal organic anion and cation secretion. AmericanJournal of Physiology, 261:R1329, 1991.
9. ULLRICH, K.J.; RUMRICH, G. Contraluminal transport systems inthe proximal renal tubule involved in secretion of organic anion.American Journal of Physiology, 254:f453, 1988.
10. KAHL, L.E. Artrite e doenças reumatológicas. In: Woodley, M.;Alison, W. Manual de Terapêutica Clínica, 27.ª ed. Rio de Janeiro: Me-dsi — Editora Médica e Científica Ltda, 1994, pp. 637-643.
11. HELLMANN, D.B. Arthritis and musculoskeletal disorders. In: Ti-erney, L.M.; McPhee, S.J.; Papadakis, M.A. Current Medical Diagno-sis and Treatment, 3.ªed. East Norwalk, Connecticut: Appleton andLange, 1994, pp. 668-672.
12. YU, T.F.; BERGER, L. Impaired renal function in gout: its associati-on with hypertensive vascular disease and intrinsic renal disease.American Journal of Medicine, 72:95-100, 1982.
13. BERGER, L.; YU, T.F. Renal function in gout. IV. An analysis of 524gouty subjects including long-term follow up studies. American Jour-nal of Medicine, 59:605-613, 1975.
14. YU, T.F.; BERGER, L.; DORPH, D.J.; SMITH, H. Renal function ingout. V. Factors influencing the renal hemodynamics. American Jour-nal of Medicine, 67:766-771, 1979.
15. TARNG, D.C. et al. Renal function in gout patients. American Jour-nal of Nephrology, 15:31-37, 1995.
16. BECK, L.H. Requiem for gouty nephropathy. Kidney International,30:280-287, 1986.
17. MURRAY, T.; GOLDBERG, M. Chronic intersticial nephritis. Annalsof Internal Medicine, 82:453-459, 1975.
18. FESSEL, W.J. Renal outcomes of gout and hyperuricemia. Ameri-can Journal of Medicine, 67:74-82, 1979.
19. PAUL, L.W.; JUHL, J.H. Doenças das articulações. In: ———. Inter-pretação Radiológica. Rio de Janeiro: Guanabara Koogan, 1977, pp. 246-248.
20. ROSE, B.D. Pathophysiology of renal disease. In: ———. Pathophy-siology of Renal Disease, 2nd ed. New York, USA: McGraw-Hill, 1987,pp. 418-425.
21. KJELLSTRAND, C.M.; CAMPBELL, D.C. et al. Hyperuricemic acu-te renal failure. Archives of Internal Medicine, 133:349, 1974.
22. HRICIK, D.E.; GOLDSMITH, G.H. Uric acid nephrolithiasis and acu-te renal failure due to streptozocin nephrotoxicity. American Jour-nal of Medicine, 84:153, 1988.
23. EMMERSON, B.T. The kidney and gout. In: Hamburger, J.; Crosnier,J.; Grunfeld, J.P. (Eds) Nephrology. New York, USA, Wiley, 1979, pp.747-756.
24. DUNCAN, H.; DIXON, A. Gout, familial hyperuricemia, and renaldisease. Q.J. of Medicine, 113:127-135, 1960.
25. CAMERON, J.S. et al. Gout, uric acid and purine metabolism inpaediatric nephrology. Pediatric-Nephrology, 7:105-118, 1993.
26. MORO, F. et al. Familial juvenile gouty nephropathy with renal uratehyposecretion preciding renal disease. Clinical Nephrology, 35:263-269, 1991.
27. COE, F.L. Uric acid and calcium oxalate nephrolithiasis. Kidney Inter-national, 24:392, 1983.
28. RIESE, R.J. et al. Uric acid nephrolithiasis: patogenesis and treatment.Journal of Urology, 148:765, 1992.
29. LIN, H. et al. Cyclosporine-induced hyperuricemia and gout. TheNew England Journal of Medicine, 321:287, 1989.
30. NOORDZIJ, T.C. et al. Renal handling of urate and incidence of goutyarthritis during cyclosporine and diuretic use. Transplantation, 52:64,1991.
31. DERAY, G.; BEHMIDA, M.; LEHOANG, P. et al. Renal function andblood pressure in patients receiving long-term, low dose cyclospo-rine therapy for idiopathic uveitis. Annals of Internal Medicine,117:578, 1992.
32. EMMERSON, B.T. The management of gout. The New England Jour-nal of Medicine, February, v. 334, n. 7, p. 445-451, 1996.
33. GRIEBSCH, A.; ZOLLNER, N. Effects of ribonucleotides given orallyon uric acid production in man. Adv. Exp. Med. Biol., 4:4, 1974.
34. FRAXINO, P.H.; CUBAS, J.A.M.; RIELLA, M.C. Metabolismo do áci-do úrico. In: Riella, M.C. Princípios de Nefrologia e Distúrbios Hidroe-letrolíticos, 3.ª ed. Rio de Janeiro; Guanabara Koogan S.A., 1996, pp.161-170.
35. KAHN, A.M. Indirect coupling between sodium and urate transportin the proximal tubule. Kidney International, 36:378, 1989.
36. WYNGAARDEN, J.B.; KELLEY, W.N. Gout and hyperuricemia.Grune and Stratton, New York, 1976.
37. CAMPION, E.W.; GLYNN, R.J.; DeLABRY, L.O. Assintomatichyperuricemia. Risks and consequences in the normative aging stu-dy. American Journal of Medicine, 82:421, 1982.
38. HALL, A.P.; BARRY, P.E.; DAWBER, T.R.; McNAMARA, P.M. Epi-demiology of gout and hyperuricemia: A long term population stu-dy. American Journal of Medicine, 42:27, 1967.
39. JOHNSON, R.J.; KIVLIGHN, S.D.; KIM, Y.G. et al. Reappraisal of pa-thogenesis and consequences of hyperuricemia in hypertension,cardiovascular disease and renal disease. American Journal of Kid-ney Disease, 33:225, 1999.
40. PORTO, C.C. e colaboradores. Sinais e Sintomas. In: Porto, C.C. Exa-me Clínico, 3.ª ed. Rio de Janeiro: Guanabara Koogan S.A., 1996, pp.116.
41. ANTON, F.M.; PUIG, J.G.; RAMOS, T. et al. Sex differences in uricacid metabolism in adults. Evidence for a lack of influence of estra-diol 17 (E2) on the renal handling of urate. Metabolism, 35:343, 1988.
42. LANGFORD, H.G.; BLAUFOX, M.D.; BORHANI, N.O. et al. Isthiazide-produced uric acid elevation harmful? Analysis of datafrom the hypertension detection and follow-up program. Archivesof Internal Medicine, 147:645, 1987.
43. SEGURO, A.C.; MAGALDI, A.J.B.; HELOU, C.M.B.; MALNIC, G.;ZATZ, R. Processamento de água e eletrólitos pelos túbulos renais.In: Zatz, R. Série Fisiopatologia Clínica — Fisiopatologia Renal (vol. 2),1.ª ed. São Paulo: Editora Atheneu, 2000, pp. 71-96.
44. MONBALLYU, J.; ZACHEE, P.; VERBECKMOES, R.; BOOGAERTS,M.A. Transient acute renal failure due to tumor-lysis-induced severe

capítulo 14 253
phosphate load in a patient with Burkitt’s lymphoma. Clinical ofNephrology, 22:47, 1984.
45. RAZIS, E.; ARLIN, Z.A.; AHMED, T. et al. Incidence and treatmentof lysis syndrome in patients with acute leukemia. Acta Haematology,91:171, 1994.
46. CONGER, J.D.; FALK, S.A. et al. A micropuncture study of the ear-ly phase of acute urate nephropathy. Journal of Clinical Investigation,58:681, 1976.
47. KOZENY, G.A., HANO, J.E. Rheumatologic disease. In: Daugirdas,J.E., Ing, T.S. Handbook of Dialysis, 2.ª ed. USA: Little Brown, 1994,pp. 665-666.
48. GUZMAN, N.J. Use of drugs in renal failure. In: ———. Nephrology,3.ª ed. Baltimore, Maryland, USA: William and Wilkins, 1995, p. 289.
49. LARANJA, S.M.R.; HEILBERG, I.P.; COÊLHO, S.T.S.N. e colabora-dores. Estudo multicêntrico de litíase renal no Brasil (Multilit). In:Calculose Renal — Fisiopatologia, Diagnóstico e Tratamento. São Paulo:Editora Sarvier, 1995, pp. 295-298.
50. PAK, C.Y.; SAKHAEE, K.; FULLER, C. Successful treatment of uricacid nephrolithiasis with potassium citrate. Kidney International,30:442, 1986.
51. YU, T.F. Urolithiasis in hyperuricemia and gout. Journal of Urology,126:424, 1981.
52. BECK, L.H. Hypouricemia in the syndrome of inappropriate secre-tion of antidiuretic hormone. The New England Journal of Medicine,301:528, 1979.
53. YU, T.F.; GUTMAN, A.B. Uric acid nephrolithiasis in gout: Predis-posing factors. Annals of Internal Medicine, 67:1133, 1967.
54. SHAHINFAR, S.; SIMPSON, R.L.; CARIDES, A.D. et al. Safety of lo-sartan in hypertensive patients with thiazide-induced hyperurice-mia. Kidney International, 56:1879, 1999.
55. CHERTOW, G.M.; SEIFTER, J.L.; CHRISTIANSEN, C.L.; O’DONNEL,W.J. Trimethoprim-sulfamethoxazole and hypouricemia. ClinicalNephrology, 46:187, 1996.
56. HELLMAN, D.B.; STONE, J.H. Arthritis & musculoskeletal disor-ders. In: Tierney Jr, L.M.; McPhee, S.J.; Papadakis, M.A. Current —Medical Diagnosis & Treatment, 2001, pp. 817-821.
57. PERAZELLA, M.A.; TRAY, K. Selective cyclooxygenase-2 inhibitors:a pattern of nephrotoxicity similar to traditional nonsteroidal anti-inflammatory drugs. American Journal of Medicine, 111(1): 64-67, 2001.
58. LOUYOT, P.; MONTET, Y.; ROLAND, J. et al. L’urate oxydase dansle traitement de la goutte et de l’hyperuricemie. Ver. Rheum. Mal.Oteortic., 37:795, 1970.
59. MONTAGNAC, R.; SCHILLINGER, F. Accident anaphylactique liéà l’injection intraveineuse d’urate-oxydase chez une dialysée.Nephrologie, 11:59, 1990.
60. BURACK, D.A.; GRIFFITH, B.P.; THOMPSON, M.E. et al. Hyperu-ricemia and gout among heart transplant recipients receiving cyclos-porine. American Journal of Medicine, 92:141, 1992.
61. SIMKIN, P.A.; GARDNER, G.C. Colchicine use in cyclosporine tre-ated transplant recipients: how little is too much? [editorial]. Jour-nal of Rheumatology, 27:1334, 2000.
62. ELLION, G.B.; CALLAHAN, S.; NATHAN, H. Potentiation by inhi-bition of drug degradation: 6-substituted purine and xanthine oxi-dase. Biochem. Pharmacol., 12:85, 1963.
63. RAGAB, A.H.; GILKERSON, E.; MYERS, M. The effect of 6-mercaptopurine and allopurinol on granulopoiesis. Cancer Res.,34:2246, 1974.
64. OGINO, K.; SAITOH, M. et al. Clinical significance of hypouricemiain hospitalized patients. J. Med., 22:76, 1991.
65. PERETZ, A.; DECAUX, G.; FAMAEY, J.P. Hypouricemia and intra-venous infusions. Journal of Rheumatology, 10:66, 1983.
ENDEREÇOS RELEVANTES NA INTERNET
Nephrology, Dialysis and Transplantationhttp://ndt.oupjournals.org
American Society of Nephrologyhttp://www.asn-online.com/
International Society of Nephrologyhttp://www.isn-online.org
The New England Journal of Medicinehttp://www.nejm.org/content/index.asp
Cyber Nephrologyhttp://www.cybernephrology.org
American Society of Hypertensionhttp://www.ash-us.org
American College of Rheumatologyhttp://www.rheumatology.org
Annals of the Rheumatic Diseasehttp://www.anrheumdis.org
Arthritis Foundationhttp://www.arthritis.org
Johns Hopkins Arthritis Centerhttp://www.hopkins-arthritis.org

Capítulo
15Terapia Parenteral. Reposição Hidroeletrolítica
Miguel Carlos Riella e Maria Aparecida Pachaly
INTRODUÇÃO
COMO SE FORMULA O PLANO PARENTERAL DIÁRIO?
CÁLCULO DA NECESSIDADE BÁSICA
Perdas urinárias
Volume
Sódio
Potássio
Cloro
Sensível e insensível
Perdas gastrintestinais
Volume
Eletrólitos
CÁLCULO DAS CORREÇÕES
Correções para a água
Correções para o sódio
O terceiro espaço
Sangue e plasma
Ácido-básico
Potássio
PRINCÍPIOS GERAIS DO PLANO PARENTERAL
PLANO DE ADMINISTRAÇÃO
PRESCRIÇÃO MÉDICA
EXEMPLOS
APÊNDICE
Soluções cristalóides
Soluções colóides
Outras soluções e aditivos para uso parenteral
REFERÊNCIAS BIBLIOGRÁFICAS
sencorajando os investigadores. Apenas quando Floren-ce Seibert descobriu por que havia substâncias pirogênicasna água destilada, o progresso da terapia parenteral foimais rápido.
No entanto, a grande utilidade da terapia parenteral nopós-operatório foi restringida durante muitas décadas, peloconceito de que o paciente cirúrgico apresentava uma in-tolerância ao sal. Isto se baseava na observação de que, nopós-operatório, a excreção urinária de sódio diminuía mui-to, chegando a quase zero quando se administravam peque-nas quantidades de soluções salinas. Na época, acreditou-se que isto refletia uma incapacidade do rim, pós-cirurgia,de tolerar grandes quantidades de sal. Em vista disso, pa-cientes no pós-operatório receberam, por muitos anos,apenas uma solução de água e glicose. É evidente que,numa análise retrospectiva, muitas das complicações pós-operatórias, como o íleo prolongado, insuficiência renal,hipotensão, catabolismo excessivo, etc., podem ser atribu-
INTRODUÇÃO
O desenvolvimento da terapia parenteral iniciou-se porvolta de 1616, quando William Harvey descobriu a circu-lação do sangue. Mas foi só em 1818 que Blundell realizoua primeira transfusão humana. No início, as complicaçõesforam muitas. Os grupos sangüíneos não eram conhecidose as reações fatais eram freqüentes, a ponto de a troca desangue humano ter sido proibida por lei.
Atribuiu-se a Thomas Latta, da Escócia, em 1831, o mé-rito de ter sido o primeiro a empregar a terapia parenteralde maneira racional. Ele administrou uma solução salinaa pacientes com cólera e diarréia intensa.
Quando Karl Landsteiner descobriu os grupos sangüí-neos em 1901, reavivou-se o interesse na transfusão desangue e na terapia parenteral. Porém, os problemas comas infecções e as reações pirogênicas continuavam de-

capítulo 15 255
ídas a déficits de volume e sódio.1 Apenas quando se evi-denciou que a redução de sódio urinário no pós-operató-rio era uma resposta compensatória, é que passaram a seradministradas soluções mais balanceadas.
Nas últimas décadas, têm havido grandes progressosnesta área. Técnicas mais sofisticadas permitiram uma aná-lise da composição corporal, de seus vários compartimen-tos líquidos e de seus constituintes. Foram determinadas asnecessidades básicas diárias do organismo com relação àágua, a eletrólitos, minerais, vitaminas e, inclusive, necessi-dades energéticas (calorias) e suas fontes: lipídios, carboi-dratos e proteínas. Com isto, tornou-se possível modificar anecessidade básica, para corrigir déficits decorrentes deperdas anormais de água, solutos e fontes de energia.
O suporte nutricional e a hiperalimentação passaram ater um lugar de destaque na terapia parenteral, comple-mentando a terapia hidroeletrolítica. A escolha entre a re-posição hidroeletrolítica e a de agentes nutritivos (nutri-ção parenteral) passou a depender do período em que opaciente permanecerá em jejum. A reposição de água eeletrólitos não deverá prolongar-se por mais de sete dias(em média), sem um suporte nutricional. A partir de en-tão, a nutrição parenteral poderá atender às necessidadesbásicas de água, eletrólitos e substratos energéticos.
O capítulo atual integra os conhecimentos adquiridosnos capítulos anteriores sobre a fisiologia e distúrbios doscompartimentos líquidos, água, sódio, potássio e equilíbrioácido-básico, abordando os princípios da reposição hidro-eletrolítica. As indicações, técnica, complicações e resulta-dos da nutrição parenteral são os assuntos do capítulo se-guinte.
COMO SE FORMULA O PLANOPARENTERAL DIÁRIO?
A etapa inicial para a formulação do plano parenteral éa obtenção de todos os dados possíveis da história clínica,exame físico e dados laboratoriais.2
Na história, alguns sintomas podem sugerir distúrbioshidroeletrolíticos específicos. Por exemplo, se o pacienterelatar que está vomitando, é mais provável que apresen-te uma alcalose metabólica e um déficit de sódio e potás-sio. Se ele tiver sintomas de insuficiência cardíaca conges-tiva, poderá apresentar um excesso de sódio. Rápidasmudanças no peso geralmente traduzem ganho ou perdalíquida. As informações sobre ingesta e excreta são extre-mamente úteis.2
Há necessidade de uma anotação diária do volume delíquido administrado e da quantidade excretada sob a for-ma de urina, perdas gastrintestinais, drenagem etc. A deter-minação diária do peso, quando possível, pode servir comoguia para as necessidades diárias de sódio (v. a seguir).
As determinações das concentrações plasmáticas de
sódio, potássio, cloro, bicarbonato, glicose, uréia e creati-nina já são rotina na maioria dos hospitais e, como vere-mos, são de extrema valia no diagnóstico e correção dosdistúrbios hidroeletrolíticos.
O método delineado a seguir, para a reposição hidroe-letrolítica, foi idealizado e aperfeiçoado pelo Dr. BeldingH. Scribner, da Universidade de Washington, em Seattle,Estados Unidos.2 Ele acredita que o método é útil porquepermite a formulação de um plano parenteral diário paracada paciente. Portanto, o plano é individualizado, de acor-do com as necessidades do paciente naquele momento.Acreditamos, particularmente, que a sua grande utilidadetambém está em proporcionar um plano de trabalho parao diagnóstico e o tratamento de problemas complexos.
Uma vez obtida toda a informação possível do pacien-te, a formulação do plano obecede à seguinte ordem:
1) Cálculo da necessidade básica: refere-se à quantidadede líquidos e eletrólitos que se prevê como perdas parao paciente nas próximas 24 horas. Estas perdas incluem:perdas urinárias, digestivas e perdas sensíveis e insen-síveis (pele e pulmão).
2) Cálculo das correções hidroeletrolíticas em face dos dis-túrbios detectados através de uma avaliação clínica elaboratorial.
3) O balanço entre a necessidade básica e as correções in-dica o total de líquido e eletrólitos a ser administrado.
CÁLCULO DA NECESSIDADEBÁSICA
O plano parenteral básico tem por objetivo a reposiçãode perdas de fluidos e eletrólitos ocorridas em 24 horas,através da pele, pulmões, urina e outros fluidos corporais.
A necessidade básica de líquidos e eletrólitos correspon-de à somatória das perdas ocorridas nas últimas 24 horas.Os volumes e a quantidade de eletrólitos necessários encon-tram-se expostos no Quadro 15.1. As estimativas baseiam-se em valores médios de populações saudáveis. Porém,quando o paciente se encontra internado, e estiver sendomonitorizada a diurese ou a dosagem dos eletrólitos uriná-rios, estes valores são mais exatos e devem ser utilizados.
Recomendamos que seja utilizado o Quadro 15.3, paraorganizar a anotação dos volumes das perdas líquidas eeletrolíticas de cada paciente. Uma vez tabulados todos osdados de forma sistematizada, torna-se muito mais fácilcalcular os subtotais, assegurando que todas as perdas se-jam consideradas e repostas.
Perdas Urinárias
VOLUMEO volume urinário para um indivíduo normal varia
entre 500 ml (em condições de restrição hídrica intensa) e

256 Terapia Parenteral. Reposição Hidroeletrolítica
2.500 ml ao dia. O volume urinário de 1.500 ml, utilizadopara cálculo, representa um valor médio entre os volumesurinários mínimo e máximo excretados habitualmente.Desta forma, se o volume líquido administrado for exces-sivo em relação às necessidades do paciente, o rim excre-tará o excesso, e se porventura for insuficiente, ele conser-vará o máximo possível de líquido. É necessário lembrartambém que a urina contém dois componentes líquidos:um, correspondente à água sem eletrólitos, e outro, em quea água veicula eletrólitos. Por exemplo, num volume uri-nário de 1.500 ml, com sódio de 75 mEq/litro, concluímosque cerca de 500 ml são suficientes para a eliminação dosódio sob forma de uma solução isotônica, enquanto osrestantes 1.000 ml correspondem a água livre.
Quando o paciente apresenta um distúrbio da funçãorenal, os rins não são capazes de variar a excreção de águae eletrólitos de acordo com a ingesta. Por exemplo: a) se opaciente apresenta oligúria devido a um comprometimentoorgânico do rim, haverá uma incapacidade do rim em re-gular o balanço de água. A administração excessiva de lí-quido em relação ao volume excretado causará um exces-so de água no organismo. Nestes casos, o volume urinárioda necessidade básica deverá ser igual ao volume de uri-na excretada (v. também manejo da insuficiência renalaguda — Cap. 20); b) da mesma forma, a presença de ede-ma implica um excesso de volume extracelular e, portan-to, de sódio total. É preciso, então, reduzir a necessidadebásica de sódio a zero.
É necessário lembrar que o metabolismo de proteínas,gorduras e carboidratos produz a chamada água endóge-na, num volume de cerca de 400 ml ao dia. O metabolismode 1 g de lipídios gera 1 ml de água; de 1 g de glicose, 0,64ml de água, e de 1 g de proteína, 0,4 ml de água. Este volu-me de água pode, em algumas circunstâncias especiais,como a insuficiência renal anúrica, contribuir para o apa-recimento de hiponatremia dilucional.
SÓDIOA ingesta média diária de sódio é de 135 a 170 mEq (8 a
10 g de sal). Os rins são capazes de conservar ou excretar
mais sódio quando há modificações da dieta, num proces-so de adaptação que é efetivo após alguns dias (v. Cap. 10).Para atender às necessidades básicas, costumamos admi-nistrar 50-75 mEq diários de sódio, permitindo ao rim eli-minar uma maior ou menor quantidade, de acordo com asnecessidades.3
POTÁSSIOA perda diária habitual pela urina e fezes é de 40 mEq
(v. Cap. 12).3 Na necessidade básica, administramos estes40 mEq, observando que caberá ao rim modular a excre-ção deste íon, de acordo com as necessidades.
CLOROA necessidade básica de cloro é deduzida pela soma da
necessidade dos dois cátions: Na� e K�.
SENSÍVEL E INSENSÍVELHabitualmente consideramos, para a necessidade bá-
sica, uma perda líquida diária pela pele e pulmões da or-dem de 1.000 ml. A perda diária através da pele está emtorno de 400 ml, mas aumenta muito por sudorese profu-sa, febre, ambientes quentes e de pouca umidade. As per-das eletrolíticas na sudorese e respiração são desprezíveis(v. Quadro 15.1: zero nas colunas de sódio, potássio e clo-ro), e a reposição é feita apenas com água. Caso haja fe-bre, acrescentar mais 100 ml de água para cada grau aci-ma de 38°C. Em presença de taquipnéia, adicionar 100-200 ml para cada 4 movimentos respiratórios por minu-to acima de 20 no homem e 16 na mulher. Se a sudoresefor excessiva, haverá perdas eletrolíticas que deverão serrepostas.
Perdas Gastrintestinais
VOLUMENo plano parenteral básico são levadas em conta as
perdas ocorridas pela drenagem de fluidos corporais, atra-vés de sondas e fístulas. Procura-se fazer uma estimativa
Quadro 15.1 Necessidades básicas diárias
Perdas ÁGUA ELETRÓLITOS (mEq/dia)
(ml/dia) Sódio Potássio Cloro
Urina 1.500 75 40 115
Sensível e Insensível 1.000 0 0 0
Gastrintestinalb apH � 4 50 mEq/L 10 mEq/L 100 mEq/LpH � 4 100 mEq/L 10 mEq/L 100 mEq/L
a) indica-se o volume perdido no dia anterior.b) a secreção gástrica contém ainda 90 mEq de H� por litro.

capítulo 15 257
antecipada do volume a ser eliminado nas próximas 24horas, baseando-se nas perdas ocorridas em dias anterio-res. Isto é, se um paciente vem eliminando 1.000 ml de sucogástrico ao dia, é natural esperar que ele elimine a mesmaquantidade nas próximas 24 horas. No entanto, é impor-tante salientar que, se uma avaliação ao final das primei-ras oito horas revela um volume eliminado próximo doesperado para as 24 horas, há necessidade de revisar o pla-no terapêutico traçado.
ELETRÓLITOSSem dúvida, o melhor meio de avaliar as perdas eletro-
líticas em um determinado fluido do trato gastrintestinalé proceder à análise bioquímica do líquido. Como isto nãoé realizado rotineiramente, utilizamos algumas regras prá-ticas. No caso do suco gástrico, costuma-se utilizar o se-guinte raciocínio: suco gástrico de pH superior a 4 tem umaconcentração de sódio em torno de 100 mEq/L, ou 10% dovolume eliminado; se o pH for inferior a 4, a concentraçãode sódio será de 50 mEq/L, ou 5% do volume eliminado.De modo geral, consideramos que o suco gástrico elimina-do apresenta pH menor que 4. Exemplo: volume de sucogástrico eliminado � 1.500 ml, com pH � 6; quantidadeprovável de sódio eliminado: 10% de 1.500 � 150, ou seja,150 mEq de sódio.
A perda de potássio no suco gástrico é pequena e nãovaria com a acidez do líquido. O cálculo é geralmente feitona base de 10 mEq/L, ou 1% do volume eliminado. A con-centração habitual de cloro está em torno de 100 mEq/L(Quadro 15.2).
Para as demais secreções do trato gastrintestinal, o Qua-dro 15.2 demonstra as concentrações eletrolíticas médiasnos fluidos pancreáticos, biliares, intestinais, etc. Estasperdas também devem ser repostas no plano básico.
Quadro 15.2 Conteúdo eletrolítico dos fluidos corporais (mEq/L)
LÍQUIDO Na� K� Cl� HCO3� Volume (L/dia)
Saliva 30 20 35 15 1-1,5Suco gástrico — pH � 4 50 10 100 - 2,5Suco gástrico — pH � 4 100 10 100 - 2Bile 145 5 110 40 1,5Duodeno 140 5 80 50 -Pâncreas 140 5 75 90 0,7-1Íleo 130 10 110 30 3,5Ceco 80 20 50 20 -Cólon 60 30 40 20 -Suor 50 5 55 - 0-3Ileostomia — recente 130 20 110 30 0,5-2Ileostomia — adaptada 50 5 30 25 0,4Colostomia 50 10 40 20 0,3
Adaptado de Koch, S.M.7
CÁLCULO DAS CORREÇÕES
A segunda fase do plano parenteral tem por objetivo acorreção de distúrbios encontrados em cada uma das ca-tegorias enumeradas a seguir: 1) água; 2) sódio; 3) ácido-básico; 4) potássio, e 5) sangue e plasma. Deve ser rotinei-ramente verificada a presença de distúrbios em cada umdestes elementos. Isto será extremamente útil na aborda-gem dos distúrbios hidroeletrolíticos mais complexos.
Na folha de reposição hidroeletrolítica, há uma seçãoespecífica para correções (Quadro 15.3). Se não há distúr-bios a corrigir, deve-se colocar um zero na coluna apropri-ada. Um sinal de adição (�) ou subtração (�) indica se aquantidade deverá ser adicionada ou retirada do planoparenteral.
Correções para a Água
Naturalmente as considerações feitas no Cap. 9 são va-liosas para a análise e a compreensão dos distúrbios dometabolismo da água. Como foi frisado, a maneira maisprática de avaliar a necessidade de água é determinar osódio plasmático, que reflete a osmolalidade plasmática.O objetivo é administrar uma quantidade de água quemantenha o sódio plasmático entre 130 e 135 mEq/L.
Considerando que a água corporal total (ACT) equiva-le a cerca de 60% do peso corporal, o déficit ou excesso deágua podem ser calculados pela fórmula abaixo. Ao secomparar a água corporal normal com a atual, será possí-vel verificar a magnitude do excesso ou déficit.
Água atual
Água normal Sódio normalSódio atual
�
�
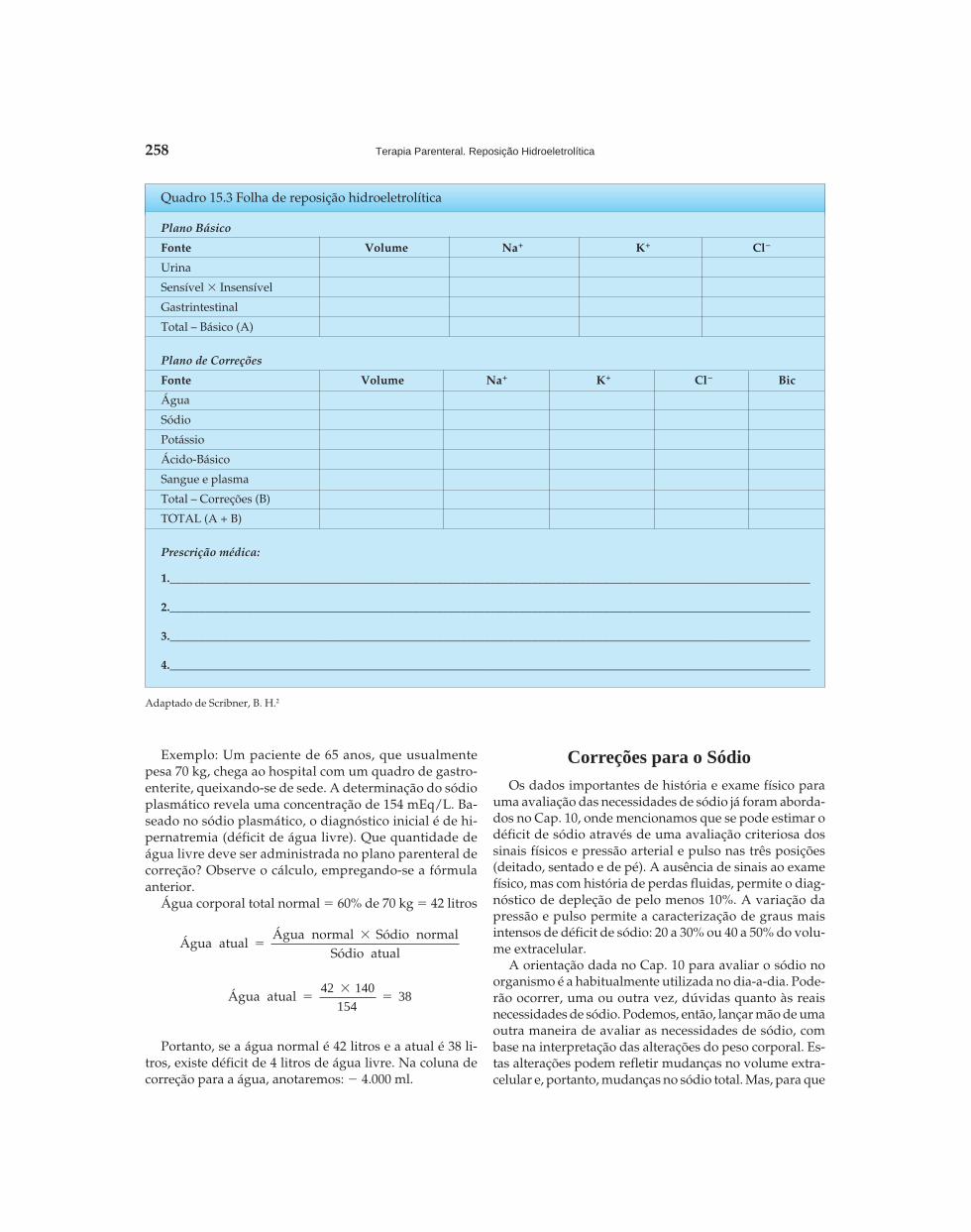
258 Terapia Parenteral. Reposição Hidroeletrolítica
Exemplo: Um paciente de 65 anos, que usualmentepesa 70 kg, chega ao hospital com um quadro de gastro-enterite, queixando-se de sede. A determinação do sódioplasmático revela uma concentração de 154 mEq/L. Ba-seado no sódio plasmático, o diagnóstico inicial é de hi-pernatremia (déficit de água livre). Que quantidade deágua livre deve ser administrada no plano parenteral decorreção? Observe o cálculo, empregando-se a fórmulaanterior.
Água corporal total normal � 60% de 70 kg � 42 litros
Água atual
Água normal Sódio normalSódio atual
�
�
Água atual
�
��
42 140
15438
Portanto, se a água normal é 42 litros e a atual é 38 li-tros, existe déficit de 4 litros de água livre. Na coluna decorreção para a água, anotaremos: � 4.000 ml.
Correções para o SódioOs dados importantes de história e exame físico para
uma avaliação das necessidades de sódio já foram aborda-dos no Cap. 10, onde mencionamos que se pode estimar odéficit de sódio através de uma avaliação criteriosa dossinais físicos e pressão arterial e pulso nas três posições(deitado, sentado e de pé). A ausência de sinais ao examefísico, mas com história de perdas fluidas, permite o diag-nóstico de depleção de pelo menos 10%. A variação dapressão e pulso permite a caracterização de graus maisintensos de déficit de sódio: 20 a 30% ou 40 a 50% do volu-me extracelular.
A orientação dada no Cap. 10 para avaliar o sódio noorganismo é a habitualmente utilizada no dia-a-dia. Pode-rão ocorrer, uma ou outra vez, dúvidas quanto às reaisnecessidades de sódio. Podemos, então, lançar mão de umaoutra maneira de avaliar as necessidades de sódio, combase na interpretação das alterações do peso corporal. Es-tas alterações podem refletir mudanças no volume extra-celular e, portanto, mudanças no sódio total. Mas, para que
Quadro 15.3 Folha de reposição hidroeletrolítica
Plano Básico
Fonte Volume Na� K� Cl�
Urina
Sensível � Insensível
Gastrintestinal
Total – Básico (A)
Plano de Correções
Fonte Volume Na� K� Cl� Bic
Água
Sódio
Potássio
Ácido-Básico
Sangue e plasma
Total – Correções (B)
TOTAL (A + B)
Prescrição médica:
1.____________________________________________________________________________________________________________
2.____________________________________________________________________________________________________________
3.____________________________________________________________________________________________________________
4.____________________________________________________________________________________________________________
Adaptado de Scribner, B. H.2

capítulo 15 259
o peso reflita o volume extracelular, duas correções sãonecessárias: uma para o catabolismo e outra para a águaintracelular.
Estas correções são necessárias, pois é óbvio que, se umindivíduo perdeu 2 kg nas últimas 48 horas, parte pode tersido devido a uma diminuição do volume extracelular,parte a um déficit de água, e o restante, ao catabolismo porjejum, infecção etc. Atribui-se ao catabolismo uma perdadiária de peso (massa protéica e gordurosa) entre 0,3 e 0,5kg, dependendo do grau de catabolismo. A seguinte equa-ção indica os fatores que causam alterações no peso:
� peso � � VEC � � LIC � Perda de massa protéica e gor-durosa, onde:
� peso � diferença entre o peso inicial e final;
� VEC � diferença entre o volume de líquido extracelularinicial e final;
� LIC � diferença entre a quantidade de líquido (água)intracelular inicial e final;
Perda de massa protéica e gordurosa � diferença na mas-sa celular devido ao catabolismo diário.
A água intracelular equivale a 40% do peso corporal, esupõe-se que alterações na água intracelular reflitam alte-rações na osmolalidade plasmática e, conseqüentemente,alterações no sódio plasmático. Desta forma, a diferença nolíquido intracelular será:
� LIC � LIC � PNa
PNa � diferença entre o sódio plasmático inicial (PNai) e osódio plasmático final (PNaf) em relação ao sódio plasmáti-co inicial.
Pode-se também usar a percentagem de alteração nosódio plasmático (� % � Na). Logo, � LIC � (0,4 � peso)� (PNai - PNaf)/ PNai
A equação final será:
� peso � � VEC � (0,4 � peso) � (PNai � PNaf)/PNai � (0,3� n.º dias)
ou, substituindo (PNai � PNaf)/PNai por % � Na:
� peso � � VEC � (0,4 � peso) � % � Na � (0,3 � n.o dias)
Exemplo: Um paciente de 60 kg é submetido a uma gas-trectomia total, recebendo apenas água e eletrólitos por viaparenteral. No 10.º dia de pós-operatório, seu peso é de 58kg. O sódio plasmático inicial e agora no 10.º dia é o mes-mo: 140 mEq/L. Qual foi a alteração no volume extracelu-lar?
Aplicando a equação anterior, teremos:
� 2 kg � � VEC � (24 litros � 0 � 3kg)
� 2 kg � � VEC � (0 � 3 kg)
� 2 kg � � VEC � 3 kg
� VEC � � 1 litro.
Comentário: A análise dos dados deste paciente permi-te deduzir que, no 10.º dia de pós-operatório, ele deveriater perdido 3 kg à custa do catabolismo. No entanto, eleperdeu só 2 kg, e, como não houve variação no sódio plas-mático, deduz-se que não houve variação na água intrace-lular. Portanto, o aumento de 1 kg foi à custa de um au-mento no volume extracelular.
Suponhamos agora que, no mesmo exemplo anterior, osódio plasmático esteja em 126 mEq/L no 10.º dia de pós-operatório. Vejamos qual a alteração no volume extrace-lular.
� peso � � VEC � (0,4 � peso) � (PNai � PNaf)/PNai � (0,3� n.º dias)
� 2 kg � � VEC � (0,4 � 60) � (140 � 126)/140 � 0,3 � 10
� 2 kg � � VEC � 24 � 10% � 3
� 2 kg � � VEC � 2,4 � 3
� 2 kg � � VEC � 0,6
� VEC � � 1,4 litro.
Comentário: Como houve uma redução do sódio plas-mático da ordem de 10% (140 � 126 � 14 ou 10% de 140),este paciente ganhou 10% do volume de água intracelular(24 litros), ou seja, 2,4 litros. Como no final de 10 dias eledeveria ter perdido 3 kg devido ao catabolismo e adquiri-do 2,4 kg pelo ganho de água, a redução de peso deveriaser de apenas 0,6 kg. Mas, como ele perdeu 2 kg, isto sig-nifica que o volume extracelular foi reduzido em 1,4 litro,como se deduziu acima.
A correção para sódio implica a administração de umasolução isotônica de água e sódio. Se chegarmos à conclu-são de que há um déficit de sódio da ordem de 1.000 ml,colocamos na coluna de volume o valor de 1.000 ml prece-dido do sinal �. Nas colunas do sódio e cloro, colocamoso valor 150 mEq, que se refere à quantidade de sódio e clo-reto existente por litro de solução salina isotônica.
Na presença de edema e, portanto, de excesso de sódiono organismo, nenhuma solução contendo sódio será ad-ministrada, e a coluna de Na� terá apenas zeros.
O TERCEIRO ESPAÇOEste termo foi criado para descrever um compartimen-
to físico ou fisiológico no qual líquidos do organismo, es-pecialmente o líquido extracelular, acumulam-se em decor-rência de uma lesão e não mais participam do volume cir-culante.1,4 Seria talvez mais preciso imaginar este líquidocomo um volume seqüestrado internamente e oriundo dolíquido extracelular. Desta forma, pode haver uma enor-me diminuição no volume extracelular, sem que haja alte-ração do peso. Como dissemos, este líquido localiza-semais comumente em tecidos lesados, como na pele, apósqueimaduras; na superfície peritoneal, após uma agressãoquímica ou bacteriana; na massa muscular esquelética,após trauma ou esmagamento; acúmulo intraluminal de

260 Terapia Parenteral. Reposição Hidroeletrolítica
secreções digestivas no caso de uma obstrução intestinal eo próprio líquido ascítico. Até que exista um restabeleci-mento da integridade celular dos tecidos lesados, este lí-quido acumulado não tem valor funcional. É importanterelembrar que, como este líquido se origina do extracelu-lar, inicialmente há uma redução do volume extracelular,e o organismo responde com retenção de água e sal, quese traduz por aumento do peso.
A redução da excreção de sódio urinário que ocorre nopós-operatório, que por muitos anos foi interpretada comouma intolerância do rim ao sódio (v. introdução do capí-tulo), nada mais é que uma resposta fisiológica face a umaredução do volume extracelular, decorrente de uma se-qüestração de líquido (terceiro espaço) na área de incisãocirúrgica, área de dissecção e nos espaços manipulados,como ocorre com o edema das alças intestinais pós-mani-pulação.
Sangue e PlasmaSe houver uma redução importante do volume globu-
lar ou evidência de sangramento ativo, a administração desangue pode estar indicada. Da mesma forma, nos proces-sos inflamatórios intraperitoneais (peritonites) ou no gran-de queimado, a perda de plasma é significativa, e a sua re-posição será importante na manutenção de um bom volu-me circulante.
É importante salientar que o volume plasmático e ovolume extracelular podem variar em direções opostas. Porexemplo, na presença de hipoproteinemia e edema, o vo-lume extracelular está aumentado e o volume plasmáticoreduzido, podendo haver sinais de hipovolemia.
Ácido-básicoO processo diagnóstico de um distúrbio ácido-básico já
foi abordado no Cap. 11. Ficou explícito que, se houver umaalcalose metabólica, a correção da depleção do volumeextracelular e do déficit de potássio, em geral, será sufici-ente. Raramente há necessidade da administração de áci-dos minerais.
Se o diagnóstico é de acidose metabólica, calculamos aquantidade de bicarbonato de sódio a ser administrada (jáabordada no Cap. 11) e anotamos na coluna do sódio. Lem-brar de anotar, na coluna de volume, a quantidade de lí-quido que será utilizada para administrar o bicarbonato.Também é necessário deduzir, da necessidade básica ou dacorreção para sódio, a quantidade de sódio administradacom o bicarbonato de sódio.
PotássioO potássio plasmático nos dá uma idéia do potássio total
do organismo. Uma vez determinado o déficit (método
exposto no Cap. 12), anotamos o valor na coluna do potás-sio e do cloro. Um outro modo de fazer um cálculo aproxi-mado do déficit de potássio é o seguinte:3
1. Se K� sérico � 3 mEq/L: para elevar o K� sérico em1 mEq/L, há necessidade de administrar de 100 a 200mEq de potássio.
2. Se K� sérico � 3 mEq/L: para elevar o K� sérico em1 mEq/L, há necessidade de administrar de 200 a 400mEq de potássio.
3. Para cada alteração no pH de 0,1 unidade, há uma alte-ração inversa de 0,6 mEq/L na concentração sérica de K�.Exemplo: pH � 7,3; K� � 4,6 mEq/L. Como houve umaredução de 0,1 no pH, o K� sérico se elevou em 0,6 mEq/L.Com a correção do pH para 7,4, o K� sérico voltará a 4,0mEq/L.
PRINCÍPIOS GERAIS DO PLANOPARENTERAL
1. É necessário que se faça apenas uma estimativa da mag-nitude do distúrbio, a qual servirá de guia para a repo-sição. Uma determinação exata não é possível e tampou-co necessária.
2. À medida que se faz a correção do distúrbio, o planoterapêutico seguinte deverá aproximar-se da necessida-de básica e permitir que o próprio rim faça os ajustesfinais.
3. Nunca há necessidade de corrigir o distúrbio completa-mente nas primeiras 24 horas.
4. Cálcio, magnésio e fósforo normalmente não são acres-centados às soluções hidrossalinas que se destinam a umareposição hidroeletrolítica de poucos dias de duração,porém são essenciais na nutrição parenteral. No Cap. 13se encontram as diretrizes para o diagnóstico e o trata-mento dos distúrbios relacionados a esses elementos.
PLANO DE ADMINISTRAÇÃO
Na folha de reposição hidroeletrolítica, determinam-seos totais combinados de volume e eletrólitos da necessida-de básica e correções.
Sódio. É administrado sob a forma de solução salinaisotônica, na qual cada 1.000 ml possui 150 mEq de sódio.Se a quantidade de sódio a ser determinada for de 300 mEq,são necessários 2.000 ml de solução salina isotônica (sorofisiológico). Este volume (2.000 ml) é deduzido do volumetotal do líquido previsto na reposição.
Água. É administrada sob a forma de uma solução deglicose a 5% (isotônica). Soluções de glicose mais concen-tradas (10, 20 ou 50%) poderão ser utilizadas, mas por veiacentral, já que em veia periférica soluções hipertônicascausam flebite.

capítulo 15 261
Potássio. É encontrado sob a forma de cloreto de potás-sio, acetato de potássio e fosfato de potássio. Na reposiçãohidroeletrolítica, geralmente utilizamos o cloreto de potás-sio. As outras formas de apresentação são reservadas paraa nutrição parenteral. O KCl a 19,1% (ampolas de 10 ml)contém 2,5 mEq de K� por ml. A quantidade de potássioprevista na reposição é distribuída preferencialmente pe-los frascos de soro glicosado a 5%. Evita-se a colocação depotássio em soro fisiológico porque, numa emergência (p.ex., choque), o líquido a ser administrado rapidamente é osoro fisiológico e nunca o soro glicosado. Se o soro fisioló-gico contiver K�, sua administração rápida poderá causarsérias arritmias cardíacas. Evitar uma concentração de K�
superior a 30 mEq/L, pois concentrações maiores causamirritação e dor ao longo da veia. Se o paciente se apresentaoligúrico ou com retenção nitrogenada, é preferível nãoadicionar potássio ao primeiro frasco de solução. Se hou-ver boa diurese em resposta à reposição líquida, adiciona-se potássio aos demais frascos.
PRESCRIÇÃO MÉDICA
A prescrição do plano parenteral:
a) especifica a solução básica a ser administrada: soro fisi-ológico, soro glicosado a 5% etc.;
b) especifica o volume de cada solução básica: 1.000 ml,3.000 ml etc.;
c) identifica os frascos de cada solução por um númeroconsecutivo: p. ex., soro fisiológico, 3.000 ml; frascos 1,2 e 3;
d) indica os aditivos a serem usados na solução: p. ex.,adicionar 10 ml de KCl 19,1% aos frascos 4, 5, 6 e 7 desoro glicosado a 5%;
e) indica a velocidade de infusão, ou gotejamento por mi-nuto. Aproximadamente, utilizando-se equipos comunsde infusão, a seguinte relação é válida:
gotas/min ml/h L/24 h
6 21 0,512 42 118 63 1,524 84 2
EXEMPLOS
Exemplo n.º 1: Uma jovem de 28 anos é submetida a umacolecistectomia e, 24 horas após, apresenta-se bem, apenascom sede. Dados vitais: PA 140/80 mm Hg, deitada; pul-so: 80 b.p.m.; T � 36,2°C; FR � 10 m.r.m.; peso 60 kg; diu-rese das 24 horas: 600 ml; sódio e potássio plasmáticos: 147mEq/L e 3,9 mEq/L, respectivamente; drenagem nasogás-trica: 2.500 ml (pH � 6,0). Formular o plano parenteral paraas próximas 24 horas. Acompanhe pelo Quadro 15.4.
1.ª etapa — cálculo do plano básico:
• Perda por diurese � 600 ml, com 30 mEq de Na�; 15 mEqde K� e 45 mEq de cloreto.
• Perda sensível e insensível � 1.000 ml (sem eletrólitos).• Perda gastrintestinal � 2.500 ml (é previsto um volume
de perda igual ao do dia anterior). Como o pH do sucogástrico é elevado, a perda de sódio equivale a 10% dovolume eliminado, ou seja, 250 mEq; a perda de potás-sio geralmente é de 1% do volume eliminado: 25 mEq.
2.ª etapa — cálculo do plano de correções:
• Água: A análise deste caso mostra que há um déficit deágua (traduzido por hipernatremia). No cálculo do dé-ficit, verificamos que a água corporal normal desta pa-ciente deveria ser 36 litros; porém, com sódio plasmáti-co de 147 mEq/L, a água corporal (atual) se encontra em34,2 litros. Existe, portanto, um déficit de 1.800 ml.
• Sódio: Não são evidenciados sinais de depleção ou ex-cesso do extracelular, apesar de uma certa redução nodébito urinário em relação ao esperado para um adultonormal. Observe que os dados de pressão arterial e pulsoestão normais. Não é necessária correção.
• Potássio: O potássio sérico está normal. Não é necessá-ria correção.
• Ácido-básico: Não há dados.• Sangue e plasma: Não há dados.
Exemplo n.º 2: Um homem de 35 anos é trazido para oServiço de Emergência do hospital após ter sido encontra-do por amigos num estado semi-estuporoso. Segundo osamigos, ele vinha bebendo muito nos últimos dias. A his-tória médica pregressa era irrelevante, a não ser por umtratamento ambulatorial de úlcera péptica. Ao exame físi-co, ele se apresentava obnubilado, com os seguintes dadosvitais:
PA (deitado): 100/60 mm HgPA (sentado): 40/? mm HgPulso (deitado): 100 b.p.m.Pulso (sentado): 140 b.p.m.Freq. Resp.: 18 m.r.m.Temp � 38°CPeso: 60 kg
As veias jugulares não eram visíveis em decúbito dorsal.O exame do abdome acusou dor epigástrica e ruídos hi-droaéreos hipoativos. Não havia edema. Os exames de la-boratório revelaram: hematócrito � 45%; 10.500 leucócitoscom 75% de polimorfonucleares; glicemia � 120 mg/100ml; sódio plasmático � 125 mEq/L; potássio plasmático �3,0 mEq/L; cloro plasmático � 75 mEq/L; bicarbonatoplasmático � 25 mEq/L; creatinina � 1,8 mg/100 ml; pHarterial � 7,41; pCO2 � 38 e pO2 � 60. Formular o planoparenteral para as próximas 24 horas (Quadro 15.5).
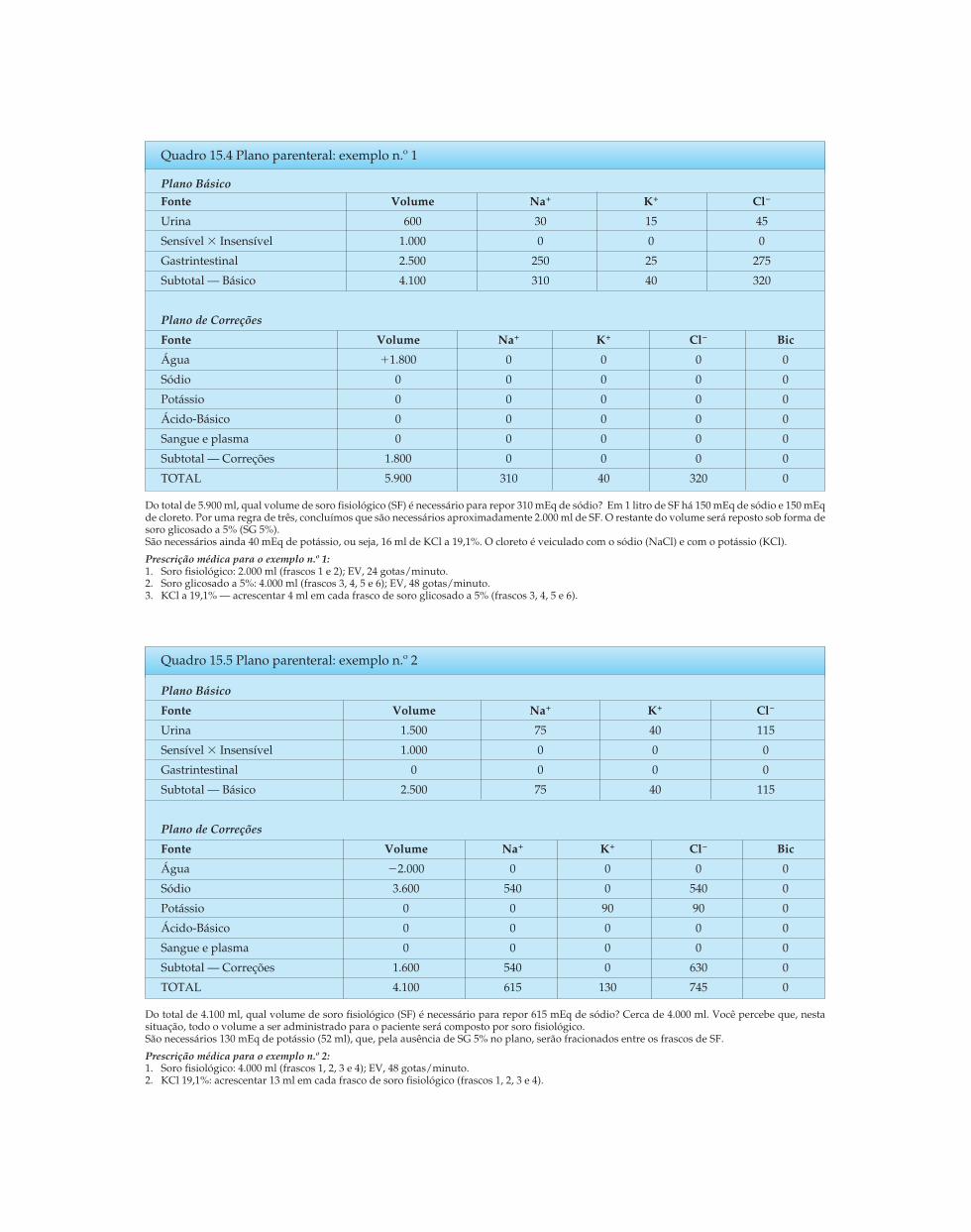
Quadro 15.4 Plano parenteral: exemplo n.º 1
Plano BásicoFonte Volume Na� K� Cl�
Urina 600 30 15 45
Sensível � Insensível 1.000 0 0 0
Gastrintestinal 2.500 250 25 275
Subtotal — Básico 4.100 310 40 320
Plano de Correções
Fonte Volume Na� K� Cl� Bic
Água �1.800 0 0 0 0
Sódio 0 0 0 0 0
Potássio 0 0 0 0 0
Ácido-Básico 0 0 0 0 0
Sangue e plasma 0 0 0 0 0
Subtotal — Correções 1.800 0 0 0 0
TOTAL 5.900 310 40 320 0
Do total de 5.900 ml, qual volume de soro fisiológico (SF) é necessário para repor 310 mEq de sódio? Em 1 litro de SF há 150 mEq de sódio e 150 mEqde cloreto. Por uma regra de três, concluímos que são necessários aproximadamente 2.000 ml de SF. O restante do volume será reposto sob forma desoro glicosado a 5% (SG 5%).São necessários ainda 40 mEq de potássio, ou seja, 16 ml de KCl a 19,1%. O cloreto é veiculado com o sódio (NaCl) e com o potássio (KCl).
Prescrição médica para o exemplo n.º 1:1. Soro fisiológico: 2.000 ml (frascos 1 e 2); EV, 24 gotas/minuto.2. Soro glicosado a 5%: 4.000 ml (frascos 3, 4, 5 e 6); EV, 48 gotas/minuto.3. KCl a 19,1% — acrescentar 4 ml em cada frasco de soro glicosado a 5% (frascos 3, 4, 5 e 6).
Quadro 15.5 Plano parenteral: exemplo n.º 2
Plano Básico
Fonte Volume Na� K� Cl�
Urina 1.500 75 40 115
Sensível � Insensível 1.000 0 0 0
Gastrintestinal 0 0 0 0
Subtotal — Básico 2.500 75 40 115
Plano de Correções
Fonte Volume Na� K� Cl� Bic
Água �2.000 0 0 0 0
Sódio 3.600 540 0 540 0
Potássio 0 0 90 90 0
Ácido-Básico 0 0 0 0 0
Sangue e plasma 0 0 0 0 0
Subtotal — Correções 1.600 540 0 630 0
TOTAL 4.100 615 130 745 0
Do total de 4.100 ml, qual volume de soro fisiológico (SF) é necessário para repor 615 mEq de sódio? Cerca de 4.000 ml. Você percebe que, nestasituação, todo o volume a ser administrado para o paciente será composto por soro fisiológico.São necessários 130 mEq de potássio (52 ml), que, pela ausência de SG 5% no plano, serão fracionados entre os frascos de SF.
Prescrição médica para o exemplo n.º 2:1. Soro fisiológico: 4.000 ml (frascos 1, 2, 3 e 4); EV, 48 gotas/minuto.2. KCl 19,1%: acrescentar 13 ml em cada frasco de soro fisiológico (frascos 1, 2, 3 e 4).
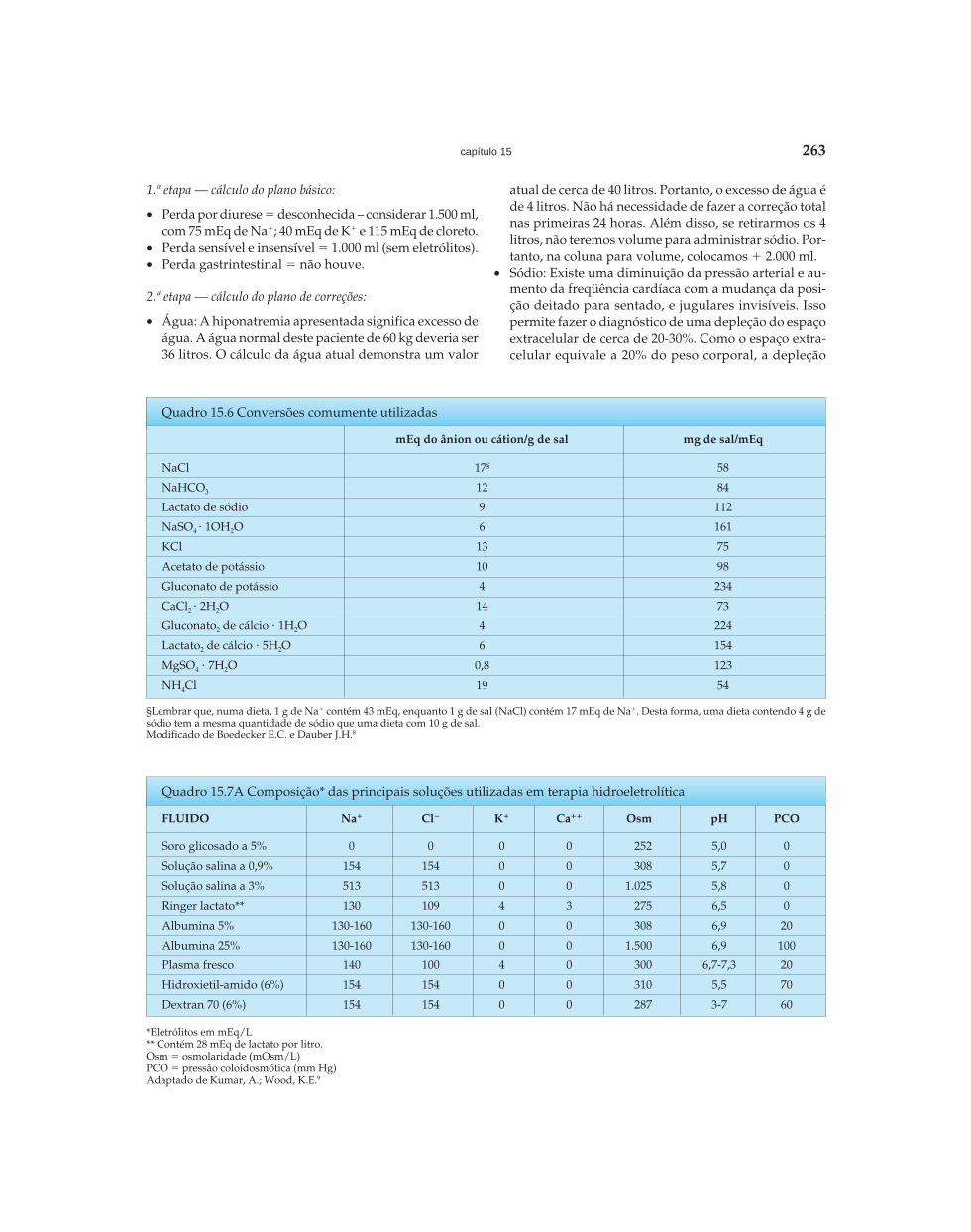
capítulo 15 263
1.ª etapa — cálculo do plano básico:
• Perda por diurese � desconhecida – considerar 1.500 ml,com 75 mEq de Na�; 40 mEq de K� e 115 mEq de cloreto.
• Perda sensível e insensível � 1.000 ml (sem eletrólitos).• Perda gastrintestinal � não houve.
2.ª etapa — cálculo do plano de correções:
• Água: A hiponatremia apresentada significa excesso deágua. A água normal deste paciente de 60 kg deveria ser36 litros. O cálculo da água atual demonstra um valor
atual de cerca de 40 litros. Portanto, o excesso de água éde 4 litros. Não há necessidade de fazer a correção totalnas primeiras 24 horas. Além disso, se retirarmos os 4litros, não teremos volume para administrar sódio. Por-tanto, na coluna para volume, colocamos � 2.000 ml.
• Sódio: Existe uma diminuição da pressão arterial e au-mento da freqüência cardíaca com a mudança da posi-ção deitado para sentado, e jugulares invisíveis. Issopermite fazer o diagnóstico de uma depleção do espaçoextracelular de cerca de 20-30%. Como o espaço extra-celular equivale a 20% do peso corporal, a depleção
Quadro 15.6 Conversões comumente utilizadas
mEq do ânion ou cátion/g de sal mg de sal/mEq
NaCl 17§ 58
NaHCO3 12 84
Lactato de sódio 9 112
NaSO4 1OH2O 6 161
KCl 13 75
Acetato de potássio 10 98
Gluconato de potássio 4 234
CaCl2 2H2O 14 73
Gluconato2 de cálcio 1H2O 4 224
Lactato2 de cálcio 5H2O 6 154
MgSO4 7H2O 0,8 123
NH4Cl 19 54
§Lembrar que, numa dieta, 1 g de Na� contém 43 mEq, enquanto 1 g de sal (NaCl) contém 17 mEq de Na�. Desta forma, uma dieta contendo 4 g desódio tem a mesma quantidade de sódio que uma dieta com 10 g de sal.Modificado de Boedecker E.C. e Dauber J.H.8
Quadro 15.7A Composição* das principais soluções utilizadas em terapia hidroeletrolítica
FLUIDO Na� Cl� K� Ca�� Osm pH PCO
Soro glicosado a 5% 0 0 0 0 252 5,0 0
Solução salina a 0,9% 154 154 0 0 308 5,7 0
Solução salina a 3% 513 513 0 0 1.025 5,8 0
Ringer lactato** 130 109 4 3 275 6,5 0
Albumina 5% 130-160 130-160 0 0 308 6,9 20
Albumina 25% 130-160 130-160 0 0 1.500 6,9 100
Plasma fresco 140 100 4 0 300 6,7-7,3 20
Hidroxietil-amido (6%) 154 154 0 0 310 5,5 70
Dextran 70 (6%) 154 154 0 0 287 3-7 60
*Eletrólitos em mEq/L** Contém 28 mEq de lactato por litro.Osm � osmolaridade (mOsm/L)PCO � pressão coloidosmótica (mm Hg)Adaptado de Kumar, A.; Wood, K.E.9

Quadro 15.7B Expansão inicial de volume (� 3 horas) com alguns fluidos intravenosos (ml)*
FLUIDO EIC EEC EIT PL
Soro glicosado a 5% 600 40 255 85
Solução salina a 0,9% � 100 1.100 825 275
Solução salina a 3% � 2.950 3.950 2.690 990
Ringer lactato 0 1.000 670 330
Albumina 5% 0 1.000 100 900
Albumina 25% 0 1.000 � 3.500 4.500
Papa de hemácias 0 1.000 130 870
Plasma fresco 0 1.000 0 1.000
Sangue total 0 1.000 0 1.000
Dextran 70 (6%) 0 1.000 � 1.000 2.000
HAES-steril 0 1.000 � 500 1.500
*Após infusão de 1 litro de solução.EIC � espaço intracelularEIT � espaço intersticial
Quadro 15.8 Principais aditivos utilizados
ADITIVOS ELETRÓLITOS – mEq/ml
Na� K� Cl� Ca�� Mg�� HCO3�§
NaCl 20% 3,4 - 3,4 - - -
KCl 19,1% - 2,5 2,5 - - -
Gluc. Cálcio 10% - - 4,8 - - 4,8
CaCl2 10% - - 13,6 13,6 - -
Sulfato de Mg 10% - - - - 8,1 -
NaHCO3 10% 1,2 - - - - 1,2
NH4Cl 20% - - 3,75 - - -
§Incluídos lactato, gluconato, acetato.Modificado de Faintuch, J.11
EEC � espaço extracelularPL � volume plasmáticoAdaptado de Carlson, R.W.; Rattan, S.; Haupt, M.10
Quadro 15.9 Perda estimada de líquido e sangue de acordo com os dados clínicos iniciais do paciente
Classe I Classe II Classe III Classe IV
Perda de sangue (ml) Até 750 750-1.500 1.500-2.000 � 2.000
Perda de sangue (% volume sanguíneo) Até 15% 15-30% 30-40% � 40%
Pulso (b.p.m.) � 100 � 100 � 120 � 140
Pressão de pulso (mm Hg) N ou � � � �
Freq. respiratória (m.r.m.) 14-20 20-30 30-40 � 35
Diurese (ml/h) � 30 20-30 5-15 Desprezível
Estado mental / SNC Ansiedade Ansiedade Ansiedade e Confusão e
leve moderada confusão letargiaReposição volêmica(regra 3:1) Cristalóide Cristalóide Cristalóide e Cristalóide e
sangue sangue
A regra 3:1 se baseia no fato de que a maior parte dos pacientes em choque hemorrágico necessita de 300 ml de solução eletrolítica para cada 100 mlde sangue perdido. A avaliação clínica contínua de cada paciente pode minimizar as dificuldades existentes para o cálculo exato da quantidade etipo de fluidos a administrar. Baseado em: Advanced Trauma Life Support.12

capítulo 15 265
apresentada neste caso corresponde a 2.400-3.600 ml.Neste caso, optamos por reposição de 3.600 ml, pois aPA e o pulso em decúbito dorsal poderiam ser conside-rados alterados.
• Potássio: O potássio sérico encontra-se diminuído (2,5mEq/litro). Como não há distúrbio ácido-básico nemdesvio iônico, a necessidade de potássio deste pacienteestá entre 200 e 400 mEq. Outra forma de calcular a ne-cessidade de potássio é através da Fig. 12.5, onde veri-ficamos que para um potássio de cerca do 3,0, corres-ponde uma deficiência de 10%. Calculando o potássiototal (45 mEq/kg � 45 � 60 � 2.700 mEq), concluímosque o déficit é de 270 mEq. Não há necessidade de cor-rigir este déficit nas primeiras 24 horas, e, além do mais,como estamos restringindo água livre, não temos volu-me para administrar o potássio, pois não desejamos ul-trapassar a concentração de 30 mEq/L. Em vista disso,optamos pela correção de apenas 1/3 do déficit total eanotamos 90 mEq na coluna do potássio e cloro.
• Ácido-básico: sem distúrbios.• Sangue e plasma: sem distúrbios.
APÊNDICE
Soluções CristalóidesSão soluções verdadeiras em que sólidos cristalinos es-
tão dissolvidos em água, sob a forma de íons ou molécu-las. Exemplo: solução salina isotônica, solução de Ringerlactato, solução glicosada 5%. Os cristalóides são infundi-dos no espaço intravascular, mas distribuem-se em todo oespaço extracelular e, eventualmente, para o intracelular.5
1) Soro glicosado a 5% (SG 5%): é uma solução hipotônica,que veicula água e pequena quantidade de glicose. Emcondições normais, a glicose é assimilada pelas células enão causa alterações na glicemia do paciente. Porém, nodiabetes melito, pode desenvolver-se hiperglicemia. Numpaciente não-diabético, ao administrarmos SG 5% junta-mente com SSI, a SSI permanecerá no espaço intravascu-lar; a glicose será metabolizada, e a água livre se distri-buirá no espaço extracelular e intracelular. É útil no tra-tamento da hipernatremia, como forma de administraçãode água livre, veículo para a administração de medica-mentos, manutenção de acessos venosos permeáveis.Soluções mais concentradas de glicose (10, 20 ou 50%)podem ser utilizadas, mas causam flebite quando infun-didas em veias periféricas. Como não contém sódio, nãoé adequada para repleção do extracelular.6
2) Solução salina a 0,9% — isotônica (SSI): esta solução édenominada isotônica por apresentar tonicidade seme-lhante à do plasma. É utilizada quando se necessita ex-pandir o espaço extracelular, pois o sódio é o principalcátion deste espaço, e determina seu volume. Uma so-
lução que contenha sódio tende a se distribuir no espa-ço de distribuição do sódio, ou seja, no extracelular.Soluções hipotônicas contêm um maior teor de água li-vre, que se distribuirá parte para o extracelular e partepara o intracelular. A solução salina isotônica é adequa-da para a correção de depleção do espaço extracelular,manejo líquido em pós-operatório (em que soluçõeshipotônicas causariam hiponatremia), correção inicialdo choque, hemorragias e queimaduras. Por ser isotô-nica, esta solução não provoca desvios de líquido entrecompartimentos. Em 1 litro desta solução há aproxima-damente 150 mEq de sódio.5,6
3) Ringer lactato: é uma solução levemente hipotônica, quecontém sódio e lactato. No fígado, o lactato é converti-do em bicarbonato. Sua utilização atenua a acidosemetabólica dilucional que poderia ocorrer em situaçõesem que é necessária a reposição de grandes volumes desolução salina isotônica.
4) Solução salina a 3%: é uma solução cristalóide hipertô-nica, que promove desvios de água do intracelular parao intravascular. É utilizada no tratamento da hiponatre-mia sintomática.
Soluções Colóides
São suspensões de partículas muito grandes, que nãoatravessam membranas semipermeáveis. Sua presença emum dos lados da membrana exerce uma força de atração(pressão oncótica) que é proporcional à sua concentração.Os colóides são utilizados para manter o volume plasmá-tico, produzindo uma expansão efetiva do volume circu-lante, com pouca ou nenhuma perda para o interstício. Apermanência destas soluções no intravascular (quando oendotélio está íntegro) aumenta a duração de sua ação. Seo endotélio estiver lesado, pode haver escape de soluçãocolóide para o interstício. Devido às características da dis-tribuição destas soluções, doses menores de colóide cau-sam maior expansão do intravascular que os cristalóides.De modo geral, na ausência de lesão endotelial significati-va, são necessários três volumes de solução cristalóide parapromover um efeito equivalente a 1 volume de soluçãocolóide em expansão do intravascular (“regra 3:1”). Estadistribuição modifica-se muito no choque séptico. Sãoexemplos de colóides: a albumina, o hidroxietil-amido, osdextrans e as gelatinas.5
As referências bibliográficas 13 a 23 demonstram a con-trovérsia atual existente em torno da escolha da soluçãomais adequada a ser administrada em situações especiais.
1) Albumina (Albumina Humana 20%): é a principal pro-teína do soro, contribuindo com 80% da pressão oncó-tica do plasma. É disponível em solução a 20%. Dosesacima de 20 ml/kg causam maior aumento no intravas-cular que o volume infundido, pois o incremento napressão oncótica provoca movimento de líquido para o

266 Terapia Parenteral. Reposição Hidroeletrolítica
intravascular. A meia-vida intravascular da albumina éde 16 horas. É um efetivo expansor de volume no trau-ma e choque. São argumentos contra seu uso a possibi-lidade de transmissão de doenças infecciosas (hepatitee SIDA) e a ocorrência de eventuais reações anafiláticas.5
2) Hidroxietil-amido (Haes-Steril): é um polímero rami-ficado da glicose, com peso molecular e clearance variá-veis. É um expansor efetivo de volume. Acima de 20 ml/kg, pode causar coagulopatia. Não possui o risco detransmitir infecções; a possibilidade de reações anafilá-ticas é pequena.5
3) Dextrans (Dextran 40): são misturas de polímeros daglicose, de vários tamanhos e pesos moleculares (dex-tran 40 e dextran 70). A expansão de volume causadapor estas soluções depende do peso molecular, quanti-dade, velocidade de administração e taxa de eliminação.A infusão de dextran 70 causa expansão mais prolon-gada e efetiva que o dextran 40. Estas soluções modifi-cam as propriedades reológicas do sangue na microcir-culação (diminuem a viscosidade), podendo melhoraro consumo de oxigênio em pacientes gravemente doen-tes. Como os outros colóides sintéticos, pode causar re-ações de hipersensibilidade e efeitos sobre a coagulação.5
4) Gelatinas (Haemacel e Hisocel a 3,5%): o nível e aduração de seu efeito sobre o volume plasmático depen-dem da taxa de infusão. De modo geral não alteram acoagulação e são eliminadas inalteradas pelos rins eintestino. Experimentalmente, demonstrou-se que estasolução pode extravasar para o compartimento inters-ticial com certa rapidez.5
Outras Soluções e Aditivos paraUso Parenteral
1) Cloreto de potássio a 19,1% (KCl 19,1%): é o aditivo uti-lizado para repor as perdas e deficiências de potássio,principalmente em pacientes intolerantes ao potássioadministrado por via oral. A dose prescrita deve sercuidadosamente observada. O potássio é um agente ir-ritante para as veias, dependendo de sua diluição (semaior que 30 mEq/litro). Mais importante, porém, é quepacientes com disfunção renal podem desenvolver hi-percalemia fatal.6 Neste caso é preferível não adicionarpotássio ao primeiro frasco de solução. Se houver boadiurese em resposta à reposição líquida, adiciona-sepotássio aos demais frascos. O potássio pode ser admi-nistrado com o soro glicosado ou com solução salinaisotônica. Como apresentado no Cap. 12, a infusão comsoro glicosado causa a entrada de potássio mais rapida-mente nas células, devido à liberação de insulina, o quedificultaria a correção do potássio no sangue. Por outrolado, após a correção de uma hipocalemia grave, evita-se colocar o potássio em soro fisiológico, pois, numaemergência (p.ex., o choque), o líquido a ser adminis-
trado rapidamente é o soro fisiológico e nunca o soroglicosado. Se o soro fisiológico contiver potássio, suaadministração poderá causar complicações cardíacas.Cada ml desta solução contém 25 mEq de potássio.
2) Bicarbonato de sódio: está disponível a solução de bi-carbonato de sódio a 8,4%, que contém 1 mEq de bicar-bonato e 1 mEq de sódio por ml. Frascos de 250 ml.
REFERÊNCIAS BIBLIOGRÁFICAS
1. DUKE, J.H. Jr. e BOWEN, J.C. Fluids and electrolytes: basic conceptsand recent developments. Contemporary Surgery, 7:19, 1975.
2. SCRIBNER, B.H. Teaching Syllabus for the Course on Fluid and Electro-lyte Balance, 1969, University of Washington, Seattle.
3. ARIEF, A.I. Principles of parenteral therapy and parenteral nutriti-on. Cap. 13, pág. 567. Clinical Disturbance of Fluid and Electrolyte Meta-bolism. Edits. M.M. Maxwell e C.R. Kleeman. McGraw-Hill Co., 1972.
4. CHAPMAN, W.H. et al. The Urinary System. An Integrated Approach.Cap. 4, pág. 89. W.B. Saunders Co., 1973.
5. McCUNN, M.; KARLIN, A. Nonblood fluid resuscitation. Anesth.Clin. North America, 17(1):107-123, 1999.
6. PRESTON, R.A. IV solutions and IV orders. In: Preston, R.A. Acid-Base, Fluids, and Electrolytes Made Ridiculously Simple, Cap. 2, pp. 31-38, MedMaster Inc., 1997.
7. KOCH, S.M. Appendix: Critical care catalog. In: Civetta, J.M.; Taylor,R.W.; Kirby, R.R. Critical Care, 1997.
8. BOEDECKER, E.C. e DAUBER, J.H. Manual of Medical Therapeutics,21 st. edition. Little, Brown and Co., 1974.
9. KUMAR, A.; WOOD, K.E. Hemorrhagic and hypovolemic shock. In:Parrillo, J.E. Current Therapy in Critical Care Medicine, 1997.
10. CARLSON, R.W.; RATTAN, S.; HAUPT, M. Anesth. Rev., 17 (sup-pl.3):14, 1990.
11. FAINTUCH, J. Hidratação no pós-operatório. Cap. 38, pág. 311.Manual de Pré- e Pós-Operatório. Edits. J. Faintuch, M.C.C. Machadoe A.A. Raia. Edit. Manole Ltda., 1978.
12. AMERICAN COLLEGE OF SURGEONS Advanced Trauma Life Su-pport, 1993.
13. BUNN, F.; ALDERSON, P.; HAWKINS, V. Colloid solutions for fluidresuscitation (Cochrane Review) — The Cochrane Library, Issue 1, 2002.
14. ALDERSON, P.; SCHIERHOUT, G.; ROBERTS, I.; BUNN, F.Colloids versus crystalloids for fluid resuscitation (Cochrane Revi-ew) — The Cochrane Library, Issue 1, 2002.
15. KWAN, I.; BUNN, F.; ROBERTS, I. (WHO Pre-Hospital Trauma CareSteering Committee). Timing and volume of fluid administration forpatients with bleeding following trauma. (Cochrane Review) — TheCochrane Library, Issue 1, 2002.
16. WHATLING, P.J. Intravenous fluids for abdominal aortic surgery(Cochrane Review) — The Cochrane Library, Issue 1, 2002.
17. ORLINSKY, M.; SHOEMAKER, W.; REIS, E.; KERSTEIN, M.D.Current controversies in shock and resuscitation. Surgical Clinics ofNorth America, 81(6), dec. 2001.
18. NGUYEN, T.T.; GILPIN, D.A.; MEYER, N.A.; HERNDON, D.N.Current treatment of severely burned patients. Annals of Surgery,223(1):15-26, 1996.
19. ASTIZ, M.E.; RACKOW, E.C. Crystalloid-colloid controversyrevisited. Critical Care Medicine, 27(1):34-35, 1999.
20. INTERNATIONAL TASK FORCE Practice parameters for hemody-namic support of sepsis in adult patients. Critical Care Medicine,27(3):639-660, 1999.
21. HENRY, S.; SCALEA, T.M. Resuscitation in the new millennium.Surgical Clinics of North America, 79(6):1259-1267, 1999.
22. WAXMAN, K. Are resuscitation fluids harmful? Critical Care Medi-cine, 28(1):264-265, 2000.
23. DROBIN, D.; HAHN, R.G. Volume kinetics of Ringer’s solution in hy-povolemic volunteers. Anesthesiology, 90(1):81-91, 1999.


















