
Síntese de compostos triazólicos com potencial ação ... · ... de carbonato de potássio e ......
15
Departamento de Química Síntese de compostos triazólicos com potencial ação antitumoral para linhagens celulares de glioblastoma Aluno: Eduardo Ferrari Ometto Colombo Orientador: Camilla Djenne Buarque 1. Introdução O glioblastoma (gbm) é um tumor caracterizado pelo seu alto grau de proliferação, invasão tecidual e necrose sendo classificado como o mais maligno e incurável. As células tronco tumorais (CTT) são subpopulações de células tumorais que levam ao seu crescimento e recorrência. Os Gbms e CTT são resistentes aos tratamentos anti-câncer adotados atualmente, tais como quimio- e radioterapias (Biochim Biophys Acta, 2012). Apesar de avanços recentes, estudos adicionais dos mecanismos moleculares que levam a esta malignização são necessários para a melhor compreensão dos alvos terapêuticos e melhor planejamento dos fármacos. Dentre todos os casos de tumores que podem se desenvolver no cérebro, o gbm representa 15,4%. Para adultos com glioblastoma mais agressivo, mesmo em tratamento, a sobrevida média é de cerca de 14,6 meses. A sobrevivência de dois anos é de 30% e quase 10% dos pacientes com glioblastoma podem viver cinco anos ou mais. Os triazóis tem uma grande importância na área farmacológica. Estes são substâncias hetero-aromáticas de origem sintética que apresentam seis elétrons π, classificados de acordo com sua forma isomérica, como vicinais 1,2,3-triazóis ou simétricos 1,2,4-triazóis (MELO, et al 2006). A seletividade dessa reação depende da metodologia sintética adotada. Figura 1: Estruturas isoméricas do triazol. Um dos métodos mais versáteis para a obtenção de triazois é a cicloadição de Huisgen entre azida e alcinos terminais. Entretanto, esse método apresenta alguns inconvenientes, tais como a necessidade de altas temperaturas e a falta regiosseletividade (JIANG, et al 2014).
Transcript of Síntese de compostos triazólicos com potencial ação ... · ... de carbonato de potássio e ......

Departamento de Química
Síntese de compostos triazólicos com potencial ação antitumoral para
linhagens celulares de glioblastoma
Aluno: Eduardo Ferrari Ometto Colombo
Orientador: Camilla Djenne Buarque
1. Introdução
O glioblastoma (gbm) é um tumor caracterizado pelo seu alto grau de proliferação,
invasão tecidual e necrose sendo classificado como o mais maligno e incurável. As células
tronco tumorais (CTT) são subpopulações de células tumorais que levam ao seu crescimento e
recorrência. Os Gbms e CTT são resistentes aos tratamentos anti-câncer adotados atualmente,
tais como quimio- e radioterapias (Biochim Biophys Acta, 2012). Apesar de avanços recentes,
estudos adicionais dos mecanismos moleculares que levam a esta malignização são
necessários para a melhor compreensão dos alvos terapêuticos e melhor planejamento dos
fármacos.
Dentre todos os casos de tumores que podem se desenvolver no cérebro, o gbm
representa 15,4%. Para adultos com glioblastoma mais agressivo, mesmo em tratamento, a
sobrevida média é de cerca de 14,6 meses. A sobrevivência de dois anos é de 30% e quase
10% dos pacientes com glioblastoma podem viver cinco anos ou mais.
Os triazóis tem uma grande importância na área farmacológica. Estes são substâncias
hetero-aromáticas de origem sintética que apresentam seis elétrons π, classificados de acordo
com sua forma isomérica, como vicinais 1,2,3-triazóis ou simétricos 1,2,4-triazóis (MELO, et
al 2006). A seletividade dessa reação depende da metodologia sintética adotada.
Figura 1: Estruturas isoméricas do triazol.
Um dos métodos mais versáteis para a obtenção de triazois é a cicloadição de Huisgen
entre azida e alcinos terminais. Entretanto, esse método apresenta alguns inconvenientes, tais
como a necessidade de altas temperaturas e a falta regiosseletividade (JIANG, et al 2014).

Departamento de Química
Diante de tais problemas, Sharpless e colaboradores desenvolveram uma metodologia seletiva
para a síntese de triazóis utilizando sais de cobre (I) como catalisador. A metodologia
desenvolvida facilitou de forma regioespecífica a cicloadição entre alcinos e azidas
produzindo apenas 1,2,3-triazol triazóis 1,4- dissubstituídos. Devido aos elevados
rendimentos, baixos tempos de reação e o amplo escopo, essas reações passaram a ser
conhecidas como reações click (KOLB e SHARPLESS, 2001). O campo emergente da reação
click chemistry oferece uma abordagem única para a síntese de moléculas contendo 1,2,3-
triazóis. Esta reação deve-se, em parte, à facilidade com a qual as azidas e alcinos podem ser
introduzidos numa molécula e a sua relativa estabilidade sob uma variedade de condições
(AGALAVE et al, 2011).
O grande interesse pelos compostos 1,2,3 triazóis na área farmacológica deve-se a sua
capacidade de atuação como grupo farmacofórico, sendo estes estáveis à degradação
metabólica e capazes de realizar ligações de hidrogênio e dipolo-dipolo, o que favorece a
ligação com alvos biomoleculares, além da melhora sua solubilidade (Manetsch et al., 2004;
Agalave et al., 2011).
Além disso, a relação bioisostérica do anel 1,2,3 triazólico com as amidas vem sendo
bastante investigada na literatura, devido a sua relativa planaridade e forte momento dipolo
serem semelhantes as propriedades físico-químicas das amidas, mas diferentemente destas, o
anel triazólico não sofre hidrólise química, sendo também resistente a oxidação e degradação
enzimática (Holub e Kirshenbaum, 2010; Freitas et al., 2011).
Existem inúmeros fármacos ou candidatos à fármacos contendo o grupo 1,2,3-triazóis
(Agalave, 2011). Dentre os compostos que atuam no sistema nervoso central para o
tratamento de câncer cerebral, destaca-se o triazol PIT-1 que atua em linhagens de
glioblastoma humano U87MG (Figura 1; Kommagalla, 2014).
Figura 2: Triazol PIT-1.

Departamento de Química
Justificativa
Devido à baixa eficiência no tratamento utilizado no combate aos glioblastomas, e o
alto índice de mortalidade relacionados a esse agressivo tumor cerebral, faz-se necessário a
busca por novos fármacos, capazes de inibir o desenvolvimento desse tipo de tumor. Dessa
forma, com base na importância biológica dos compostos contendo grupos 1,2,3-triazóis e
diante dos resultados preliminares obtidos para os compostos LSO-02 e LSO-03 (Esquema 1),
que apresentaram ação significativa em linhagens celulares de glioblastomas (GMB 02 e
GMB 72), o presente trabalho busca realizar modificações no composto LSO-02, composto
sintetizado em projetos anteriores, com a finalidade de potencializar sua ação biológica diante
das linhagens celulares gbms.
Esquema 1: Compostos que apresentaram atividade para linhagens de glioblastomas.
2. Objetivo
O presente trabalho visa realizar modificações estruturais no composto LSO-02,
utilizando grupos funcionais já conhecidos na literatura por apresentarem atividade
anticarcinogênica, tais como sufonilhidrazonas, benzoquinonas entre outros.
3. Metodologia
Os esquemas 2 e 3 representam as retrossínteses realizadas neste trabalho.

Departamento de Química
Esquema 2: Análise retrossintética para a síntese dos triazóis
Para obtenção do composto (7) realizou-se a reação de adição à carbonila presente no LSO-
02. Esta reação consiste na fluoração para obtenção do composto (7). A obtenção do
composto (9) ocorreu através da redução da carbonila presente no LSO-02. Para obtenção do
composto (11) realizou-se a reação de adição à carbonila presente no LSO-02. A síntese do
composto (13) foi realizada por meio da reação de Baeyer-Villiger. A sintese do LSO-02 foi
feita via click chemistry, na presença de sulfato de cobre (II) como catalisador, de acordo com
a metodologia proposta por Sharpless et al (2001) 4. Os precursores do LSO-02 foram feitos
pela azidação do 2- nitrobenzaldeido (1) via substituição nucleofílica aromática para a
obtenção do 2- azidobenzaldeido (2) e a reação do propargil-fenol (5) foi realizada via
substituição nucleofílica.
Esquema 3: Análise retrossintética para a síntese do triazol 15
A obtenção do composto (15) ocorreu via click chemistry, entre o 2-azido benzaldeído e o
fenil acetileno (14), na presença de sulfato de cobre (II) como catalisador.
4. Parte experimental
4.1 Síntese dos precursores do LSO-02
a. 2-Azidobenzaldeido

Departamento de Química
Esquema 4: Síntese da 2-azidobenzaldeido a partir do 2-nitrobenzaldeido.
Pesou-se 6,62 mmol do 2-nitrobenzaldeído (1) e 26,48 mmol (4 equivalentes) da azida de
sódio (2) e estes foram dissolvidos em 20 mL do DMF em um balão de 50 mL. A mistura
ficou sob agitação a 55°C durante 3 dias (96 horas). Ao meio reacional foi adicionado 30 ml
de éter etílico e então este foi lavado 3 vezes com 20 mL de água. A fase orgânica foi seca
com NaSO4 e o solvente foi evaporado no rota-evaporador. Por fim o produto foi isolado por
meio da coluna cromatográfica usando como eluente Acetato/Hexano (3%).
b. Propargil-fenol
Esquema 5: Síntese da 2-azidobenzaldeido a partir do 2-nitrobenzaldeido.
Pesou-se 10,59 mmol de fenol (3) e 21,19 mmol (2 equivalentes) de carbonato de potássio e
estes foram dissolvidos em 20 mL de acetonitrila em um balão de 50 mL, em seguida
adicionou-se lentamente com auxílio de uma seringa graduada 12,71 mmol (1,2 equivalentes)
de brometo de propargila (4). A mistura ficou sob agitação em refluxo, com atmosfera
modificada (N2), por 4 horas. Em seguida, foi adicionando à mistura 20 mL de água e em
seguida 20 mL de diclometano para realizar a separação liquido-liquido. A fase orgânica foi
lavada mais 2 vezes com 15 mL de água e então foi saca com NaSO4. O solvente foi
evaporado no rota-evaporador.
4.2 Síntese do LSO-02
Esquema 6: Síntese do LSO-02.

Departamento de Química
Pesou-se 1 mmol da azida (2) e 1 mmol do alcino (5) e estes foram dissolvidos em 6 mL de
uma solução aquosa de álcool terc-butílico (1:1). Em seguida adicionou-se 0,1 mmol de
ascorbato de sódio e 0,01 mmol de sulfato de cobre penta-hidratado. A mistura heterogênea
foi mantida sob agitação constante à temperatura ambiente. A mistura reacional foi extraída
com 20 mL de diclorometano e água (2 x 15mL), a fase orgânica foi seca com NaSO4 anidro e
então o solvente foi evaporado no rota-evaporador.
4.3 Modificações no composto LSO-02
a. Fluoração do LSO-02 via DAST
Esquema 7: Fluoração via DAST.
Em um balão de 5 mL seco e com atmosfera modificada, com N2, adicionou-se 0,21 mmol do
LSO-02 e 2 mL de diclorometano seco. Em seguida, 0,525 mmol (2,5 equivalentes) de DAST
(6) foi adicionado gota-a-gota. A mistura foi mantida sob agitação por 24 horas à temperatura
ambiente. Em seguida, adicionou-se 4 mL de uma solução saturada de bicarbonato de sódio
gelada e em seguida, 10 mL de diclorometano. Foi feita uma separação liquido-liquido e a
fase orgânica foi lavada 2 vezes com 10 mL de brine. Para retirar os resíduos da reação, foi
feita uma coluna baixa cuja fase móvel foi de 20% acetato/hexano.
b. Redução da carbonila presente no LSO-02
Esquema 8: Redução da carbonila para obtenção do álcool benzílico 9.
Dissolveu-se 0,21 mmol de LSO-02 em 3 mL de metanol em um balão de 10 mL. Em
seguida, a mistura foi colocada em um banho de gelo e mantida sob agitação e então
adicionou-se 0,546 mmol (2,6 equivalentes) de boro-hidreto de sódio (8). Após 30 minutos,
observou-se que a mistura fica transparente, indicando o fim da reação. Esta foi extraída com
15 mL de diclorometano, que foi lavado 2 vezes com 10 mL de água. A fase orgânica foi seca
com sulfato de sódio anidro e então o solvente foi evaporado no rota-evaporador.

Departamento de Química
b. Adição de sufonilhidrazina à carbonila
Para obtenção do composto (11) realizou-se a reação de adição à carbonila presente no LSO-
02. Foi utilizada a sufonilhidrazina (10) como nucleófilo, descrita no esquema 7.
Esquema 9: Obtenção da sulfonilhidrazona 11.
Adicionou-se 0,40 mmol de LSO-02 e 0,48 mmol (1,2 equivalentes) de sufonilhidrazina (10),
dissolvidos em 3 mL de metanol, em um balão de 10 mL. A mistura foi mantida em refluxo
sob agitação por 2 horas a 50°C. Em seguida, foi evaporado aproximadamente metade do
solvente presente no balão no rota-evaporador e, em seguida, o balão foi mantido na geladeira
por duas horas para que o produto precipite. O precipitado (de coloração branca) foi filtrado, e
lavado com etanol gelado.
c. Reação de Baeyer-Villiger
A síntese do composto (13) foi realizada por meio da reação de Baeyer-Villiger, utilizando
AMCPB (12), descrita no esquema 8.
Esquema 10: Reação de Baeyer Villiger.
Para a primeira etapa desta síntese, em um balão de 5 mL seco e com atmosfera modificada,
com N2, adicionou-se 0,287 mmol do LSO-02 e 0,430 mmol (1,5 equivalentes) de AMCPB
(12) e em seguida, estes foram dissolvido em 2 mL de diclorometano seco. A mistura foi
mantida sob agitação por 6 horas à temperatura ambiente sem contato com luz. Em seguida

Departamento de Química
foi feita uma separação liquido-liquido com 7 mL de acetato de etila, lavado 3 vezes com 3
mL de uma solução saturada de carbonato de sódio e mais 3 vezes com brine. O solvente foi
seco no rota-evaporador.
Para a segunda etapa, foi adicionado 2 mL de metanol e 310 µL de uma solução de hidróxido
de sódio 6 molar. Esta mistura foi mantida sob agitação por 30 minutos à temperatura
ambiente. Em seguida foi feita uma separação liquido-liquido com 10 mL de acetato de etila
lavado 3 vezes com 5 mL de brine. A fase orgânica foi seca com sulfato de sódio anidro e
então o solvente foi evaporado no rota-evaporador.
4.4 Modificação do composto LSO-02 a partir do fenilacetileno
A obtenção do composto (15) ocorreu via click chemistry, entre o 2-azido benzaldeído e o
fenil acetileno (14), na presença de sulfato de cobre (II) como catalisador, descrita no
esquema 9.
Esquema 11: Modificação do composto LSO-02 via click chemistry com o fenilacetileno
Pesou-se 1,25 mmol da azida (2) e 1,5 mmol do alcino (14) e estes foram dissolvidos em 5
mL de uma solução aquosa de álcool terc-butílico (1:1). Em seguida adicionou-se 0,125 mmol
de ascorbato de sódio e 0,0125 mmol de sulfato de cobre penta-hidratado. A mistura
heterogênea foi mantida sob agitação constante à temperatura ambiente por 8 horas. Ao fazer
uma TLC, observou-se que ainda havia azida (2) porém não havia mais alcino (14) no meio
reacional, então foi adicionado mais 1,5 mmol do mesmo e a reação foi mantida sob agitação
por mais 8 horas. A mistura reacional foi extraída com 20 mL de diclorometano e água (2 x
15mL), a fase orgânica foi seca com NaSO4 anidro e então o solvente foi evaporado no rota-
evaporador.
5. RESULTADOS E DISCUSSÕES
5.1 Síntese do LSO-02 e seus precursores
Os precursores utilizados para a síntese do LSO-02, o 2-azido benzaldeído (2) e propargil
fenol (5), foram obtidos com rendimentos de 60% e 70% respectivamente. Para a síntese do 2-
azido benzaldeído realizou-se a azidação do 2- nitrobenzaldeido (1) via substituição

Departamento de Química
nucleofílica aromática. Para obtenção do propargil-fenol (5) foi realizada uma reação de
substituição nucleofílica do brometo de propargila (4) pelo fenol (3). Ambos os processos
estão descritos no Esquema 12.
Esquema 12: Síntese dos precursores do LSO-02.
Após a obtenção dos precursores, sintetizou-se a LSO-02 via click chemistry, na presença de
sulfato de cobre (II) como catalisador. Obtendo-se um rendimento de 60% como descrito no
esquema 13.
Esquema 13: Síntese do LSO-02.
Todos os compostos sintetizados, foram caracterizados por espectros de RMN de hidrogênio e
carbono, entretanto nesse trabalho só serão discutidos os espectros de hidrogênio.
5.2 Fluoração do LSO-02 via DAST
A obtenção do composto (7) se deu por meio da reação de fluoração nucleofílica, utilizando-
se o DAST (6) como reagente de fluoração, como representado no esquema 14.
Esquema 14: Fluoração do aldeído LDO-02 via DAST.

Departamento de Química
O composto sintetizado no esquema 14 foi obtido com rendimento de 72%, abaixo encontra-
se o espectro de RMN de hidrogênio, onde os sinais obtidos estão de acordo com a estrutura
proposta.
Figura 3: Espectro de RMN de hidrogênio do composto (7).
Pode-se observar no espectro acima, o sinal característico do hidrogênio presente no anel
triazóico, na forma de um singleto em torno de 7,96 ppm, este encontra-se identificados na
figura 3 como a. Observa-se também os sinais de hidrogênio do novo grupo fluorometileno
(CHF2) agora presente na molécula, em 6,75 ppm na forma de um tripleto relativo ao
acoplamento do H com os dois átomos de flúor, este está identificado na figura 3 como g.
Todos os outros sinais observados no espectro estão de acordo com o composto obtido.
5.3 Redução da carbonila presente no LSO-02
Para sintetizar o composto (9) foi feita uma redução da carbonila presente no LSO-02 com
borohidreto de sódio (8), descrita no esquema 15.
a g

Departamento de Química
Esquema 15: Redução da carbonila presente no LSO-02.
Foi possível sintetizar, com um rendimento de 65%, o composto (9). Abaixo, na figura 4,
encontra-se o espectro de RMN de hidrogênio. Este foi utilizado para a certificação de que o
composto desejado foi sintetizado com êxito.
Figura 4: Espectro de RMN de hidrogênio do composto (9).
O sinal característico do hidrogênio no triazol se encontra, na forma de um singleto, em 8,03
ppm, este está identificados na figura 4 como a. Também pode-se observar o sinal de
hidrogênio no novo grupo, agora presente na molécula, em 4,51 ppm, na forma de um dupleto
cuja integração é 2, e o hidrogênio da hidroxila em 3,41. Ambos os sinais se encontram
destacados na figura 4 e identificados como h e l, respectivamente. Todos os outros sinais
observados no espectro estão de acordo com o composto obtido.
5.4 Adição de sufonilhidrazina à carbonila
a h l

Departamento de Química
O composto (11) foi sintetizado fazendo uma adição à carbonila presente no LSO-02. Foi
utilizada a sufonilhidrazina (10) como nucleófilo, descrita no esquema 16.
Esquema 16: Adição de sufonilhidrazina à carbonila.
Com rendimento de 70%, o composto (11) foi sintetizado. Na figura 5 abaixo encontra-se o
espectro de RMN de hidrogênio, usado para caracterizar o mesmo.
Figura 5: Espectro de RMN de hidrogênio do composto (11).
Como esperado, existe no espectro um sinal na forma de um singleto em 8,59 ppm. Este é o
sinal do hidrogênio presente no anel triazóico, e está identificado na figura 5 como a.
Observa-se também o sinal do hidrogênio da sufonilhidrazona em 8,11 ppm que está
identificado na figura 5 como b. Todos os outros sinais observados no espectro estão de
acordo com o composto obtido.
b a

Departamento de Química
5.5 Reação de Baeyer-Villiger
Não foi possível a obtenção do Composto (13) por meio da reação de Baeyer-Villiger,
utilizando AMCPB (12), descrita no esquema 17. Era observado que o reagente (LSO-02) não
era consumido ao decorrer da reação, mesmo aumentando a equivalência de MCPBA (12).
Dessa forma, outras metodologias serão investigadas para a obtenção do composto (13).
Esquema 17: Reação de Baeyer-Villiger
5.6 Modificação do composto LSO-02 a partir do fenilacetileno
A obtenção do composto (15) ocorreu via click chemistry, entre o 2-azido benzaldeído e o
fenil acetileno (14), descrita no esquema 18.
Esquema 18: Modificação do composto a partir do fenilacetileno.
Ao analisar o espectro (figura 6), pode-se afirmar que de fato foi sintetizado o composto (15).
Porém, este foi obtido com um rendimento de 30%, como descrito no esquema (13). Então,
uma nova metodologia será estudada para se chegar no composto (15) com maiores
rendimentos.
Departamento de Química
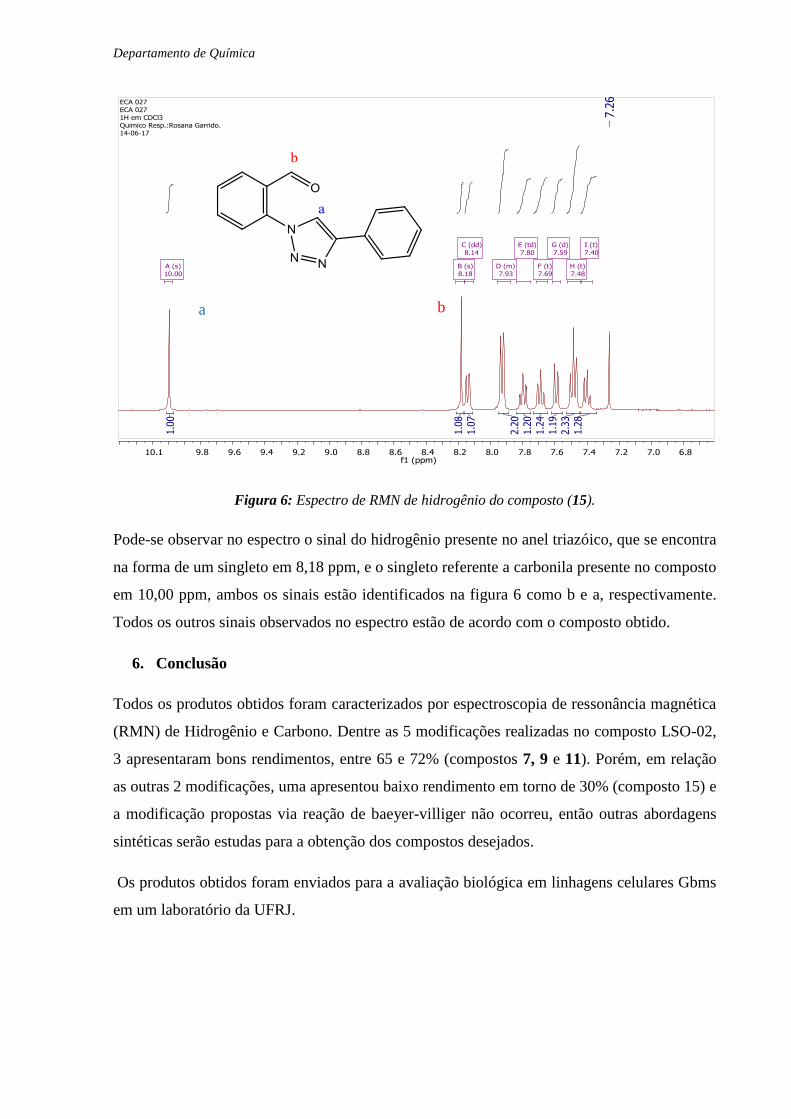
Figura 6: Espectro de RMN de hidrogênio do composto (15).
Pode-se observar no espectro o sinal do hidrogênio presente no anel triazóico, que se encontra
na forma de um singleto em 8,18 ppm, e o singleto referente a carbonila presente no composto
em 10,00 ppm, ambos os sinais estão identificados na figura 6 como b e a, respectivamente.
Todos os outros sinais observados no espectro estão de acordo com o composto obtido.
6. Conclusão
Todos os produtos obtidos foram caracterizados por espectroscopia de ressonância magnética
(RMN) de Hidrogênio e Carbono. Dentre as 5 modificações realizadas no composto LSO-02,
3 apresentaram bons rendimentos, entre 65 e 72% (compostos 7, 9 e 11). Porém, em relação
as outras 2 modificações, uma apresentou baixo rendimento em torno de 30% (composto 15) e
a modificação propostas via reação de baeyer-villiger não ocorreu, então outras abordagens
sintéticas serão estudas para a obtenção dos compostos desejados.
Os produtos obtidos foram enviados para a avaliação biológica em linhagens celulares Gbms
em um laboratório da UFRJ.
a b

Departamento de Química
7. Referências
1- AGALAVE, S.G.; MAUJAN, S. R.; PORE, V. S. Click Chemistry: 1,2,3-Triazoles as
Pharmacophores. Chem. Asian J. 6, 2696 – 2718, 2011.
2- KOLB, H. C.; FINN, M. G.; SHARPLESS, K. B. Click chemistry: Diverse chemical
function from a few good reactions. Angewandte Chemie-International Edition, v. 40,
n. 11, p. 2004-+, 2001.
3- KOLB, H.C.; SHARPLESS, K. B. Drug Discovery Today, 8, 1128 – 1137, 2003
4- Melo,J. O. F.; Donnici, C. L.; Augusti R.; Ferreira V.F.; de Souza M. C. B. V.;
Ferreira M. L. G.; Cunha, A. C. Heterociclos 1,2,3-triazólicos: histórico, métodos de
preparação, aplicações e atividades farmacológicas. Química Nova, v.29, São Paulo,
2006.
5- STOKES, J. B.; LIU, S.; DRIVER, T.G. Rh2(II)-Catalyzed Nitro Migration
Reactions: Selective Synthesis of 3-Nitroindoles from β-Nitro Styryl Azides. J. Am.
Chem. Soc., p. 4702–4705, 2011.
6- KOMMAGALLA, Y.; CORNEA, S.; RIEHLE, R.; TORCHILIN, V.; DEGTEREV,
A.; RAMANA, C.V. Med. Chem. Comm. v. 5, p. 1359-1363, 2014
7- http://www.abta.org/brain-tumor-information/types-of-tumors/glioblastoma.html?
referrer=https://www.google.com.br/. Acessado em: 29 julho 2017.
8- Alejandro F. Barrero,* Enrique J. Alvarez-Manzaneda and Rachid Chahboun,
Synthesis of Wiedendiol-A and Wiedendiol-B from Labdane Diterpenes, Received 5
February 1998; revised 10 March 1998; accepted 12 March 1998


















