Universidade de São Paulo Instituto de Química · 2020. 3. 10. · O compósito contendo 75% de...
78
Universidade de São Paulo Instituto de Química Programa de Pós-Graduação em Química Andrei Martins Surkov Desenvolvimento de eletrodos compósitos de carbono e poliestireno para aplicações eletroanalíticas Versão corrigida da dissertação São Paulo Data de Depósito na SPG 11/10/2019
Transcript of Universidade de São Paulo Instituto de Química · 2020. 3. 10. · O compósito contendo 75% de...

Universidade de São Paulo
Instituto de Química
Programa de Pós-Graduação em Química
Andrei Martins Surkov
Desenvolvimento de eletrodos compósitos de carbono e
poliestireno para aplicações eletroanalíticas
Versão corrigida da dissertação
São Paulo
Data de Depósito na SPG
11/10/2019

Andrei Martins Surkov
Desenvolvimento de eletrodos compósitos de carbono e
poliestireno para aplicações eletroanalíticas
Dissertação apresentada para o Instituto
de Química da Universidade de São
Paulo para obtenção do título de Mestre
em Química.
Área de concentração: Química
analítica
Orientador: Prof. Dr. Lúcio Angnes
São Paulo
2019

Autorizo a reprodução e divulgação total ou parcial deste trabalho, por qualquer meioconvencional ou eletronico, para fins de estudo e pesquisa, desde que citada a fonte.
Ficha Catalográfica elaborada eletronicamente pelo autor, utilizando oprograma desenvolvido pela Seção Técnica de Informática do ICMC/USP e
adaptado para a Divisão de Biblioteca e Documentação do Conjunto das Químicas da USP
Bibliotecária responsável pela orientação de catalogação da publicação:Marlene Aparecida Vieira - CRB - 8/5562
S961dSurkov, Andrei Desenvolvimento de eletrodos compósitos decarbono e poliestireno para aplicaçõeseletroanalíticas / Andrei Surkov. - São Paulo, 2019. 69 p.
Dissertação (mestrado) - Instituto de Química daUniversidade de São Paulo. Departamento de QuímicaFundamental. Orientador: Angnes, Lúcio
1. Eletrodos Compósitos. 2. Eletroquímica . 3.Eletroanalítica. I. T. II. Angnes, Lúcio, orientador.


Agradecimentos
Ao Lúcio, pelo incentivo, orientação e toda a paciência ao longo do trabalho.
Aos meus pais, Vadim e Maria do Rosário, por toda educação e incentivo que me deram.
À minha irmã, Lara, por todo apoio que me deu.
À Cinthia, que me aguentou nos tempos mais difíceis e todo suporte do mundo.
Aos meus amigos: Alejandro, América, Caio, Calil, Calila, Carina, Fofal, Fofala, Gabs,
Guilherme, Japa, Jesus, João, Rafa, Snape...
Aos meus amigos André, Carlos e Lucas um agradecimento pelas risadas de sempre,
ânimo para o desenvolvimento do trabalho e amizade.
Ao Alexandre (Peixe) por toda ajuda no desenvolvimento do trabalho e amizade.
Aos membros do LAIA e pessoas que contribuíram diretamente ou indiretamente no
trabalho
Ao Instituto de Química, pela disponibilização do espaço para realização da pesquisa.
Ao CNPq pela bolsa concedida (processo 133279/2018-5).

“Nunca se afaste de seus sonhos, pois se eles se forem,
você continuará vivendo, mas deixado de existir.”
Mark Twain

Resumo
Um novo eletrodo compósito (G-PS) foi preparado a partir de grafite e poliestireno
(isopor). As partículas de grafite foram suspensas em clorofórmio e o poliestireno foi
adicionado à mistura para sua completa dissolução. O clorofórmio foi volatilizado a 80ºC,
e acetona foi adicionada para o amolecimento do material resultante, conferindo alto grau
de plasticidade ao mesmo, favorecendo assim a construção dos eletrodos compósitos. A
proporção de grafite foi otimizada com base nos sinais voltamétricos obtidos com o par
reversível Fe(CN)63–/Fe(CN)6
4–. O compósito contendo 75% de grafite (m/m) mostrou o
melhor desempenho, considerando sinal, estabilidade mecânica e alta reprodutibilidade
de resposta eletroquímica. Estudos de homogeneidade do compósito indicaram boa
uniformidade ao longo de um mesmo eletrodo e também entre eletrodos produzidos em
diferentes ensaios. A caracterização eletroquímica destes eletrodos demonstrou que os
processos são governados por transporte difusional de massa para ferricianeto de potássio.
A resistência de transferência de carga e a resposta eletroquímica do eletrodo compósito
foi diretamente comparada com eletrodo de carbono vítreo (GCE) e eletrodo de pasta de
carbono (CPE). Aplicações do eletrodo G-PS como sensor eletroquímico foram feitas
para paracetamol e dipirona. Análises destes fármacos foram realizadas associando o
eletrodo G-PS e um sistema de análise por injeção em fluxo. Os resultados obtidos foram
validados frente aos métodos padrões descritos nas farmacopeias americana e brasileira e
também comparados com método por UV-Vis. Os limites de detecção calculados foram
0,084 e 0,67 μmol L-1 para paracetamol e dipirona, respectivamente. A faixa linear de
trabalho para o paracetamol foi 0,28 – 204 μmol L-1, enquanto para dipirona foi 2,7 – 86
μmol L-1.
Palavras-chave: Eletrodo compósito, eletrodo de baixo custo, amperometria, grafite-
poliestireno.

Abstract
A new composite electrode (G-PS) was prepared from graphite and polystyrene
(Styrofoam). The graphite particles were suspended in chloroform and the polystyrene
was added to the mixture for complete dissolution. Chloroform was volatilized at 80ºC
and acetone was added to soften the resulting material, giving it a high degree of
plasticity, thus favoring the construction of the composite electrodes. The graphite ratio
was optimized based on the voltammetric signals obtained with the reversible pair
Fe(CN)63–/Fe(CN6
4–. The composite containing 75% graphite (w/w) showed the best
performance considering signal, mechanical stability and high reproducibility of
electrochemical response. Composite homogeneity studies indicated good uniformity
across the same electrode and also between electrodes produced in different assays. The
electrochemical characterization of these electrodes demonstrated that the processes are
governed by diffusional mass transport for potassium ferricyanide. The resistance of
charge transfer and the electrochemical response of the composite electrode were directly
compared with glassy carbon electrode (GCE) and carbon paste electrode (CPE).
Applications of the G-PS electrode as electrolytic sensor were made for acetaminophen
and dipyrone quantification. Analyses of these drugs were performed by associating the
G-PS electrode and a flow injection system. The results obtained were validated against
the standard methods described in the American and Brazilian pharmacopoeias and also
compared with the UV-Vis method. The detection limits calculated were 0.084 and 0.67
μmol L-1 for acetaminophen and dipyrone respectively. The linear working range for
acetaminophen was 0.28 - 204 μmol L-1, while for dipyrone it was 2.7 - 86 μmol L-1.
Keywords: Composite electrode, low cost electrode, amperometry, graphite-polystyrene.

Lista de Abreviaturas
FIA – Análise por injeção em fluxo (Flow Injection Analysis)
G-PS – Eletrodo compósito de grafite e poliestireno
GCE – Eletrodo de carbono vítreo
CPE – Eletrodo de pasta de carbono
IpA – Corrente de pico anódico
IpC – Corrente de pico catódico
EpA – Potencial de pico anódico
EpC – Potencial de pico catódico
ΔEp – Distância de pico anódico e catódico
RCT – Resistência de transferência de carga
CV – Ciclo voltamograma

Lista de Figuras
Figura 1 Aplicação de potencial em função do tempo em um estudo de voltametria
cíclica 21
Figura 2 Comportamento de decaimento das correntes capacitiva e faradaica em
função do tempo. 22
Figura 3
Esquema de um sistema para análise por injeção em fluxo, utilizando
gravidade como propulsão da solução carreadora, para uma cela
eletroquímica controlada por um potenciostato interfaceado com um
computador.
24
Figura 4 Circuito de Randles para um sistema eletroquímico simples envolvendo
transferência de carga. 25
Figura 5 Espectro de impedância para um sistema redox simples em que envolva
transferência de carga 36 27
Figura 6
(a) Diagrama de uma célula convencional de 3 eletrodos composta por fio
de platina como eletrodo auxiliar (AE), eletrodo compósito como eletrodo
de trabalho (WE), eletrodo de Ag/AgCl (KCl (sat)) como eletrodo de
referência (RE), imersa em 20 mL de eletrólito. (b) Diagrama da célula
utilizada para a análise em fluxo, composta pelo eletrodo de referência de
Ag/AgCl (KCl (sat) (RE), uma agulha de aço inox, posicionada na saída da
célula, como eletrodo auxiliar (AE) e eletrodo compósito de grafite e
poliestireno como eletrodo de trabalho (WE), posicionado na parte inferior
da célula.
30
Figura 7
Diagrama de produção do eletrodo G-PS: (a) repulverização do grafite; (b)
adição do isopor e clorofórmio com posterior homogeneização; (c)
aquecimento do material a aproximadamente 80 ºC por duas horas em
capela; (d) adição de acetona e geração de uma pasta maleável; (e) remoção
do excesso de acetona; (f) remoção do material do almofariz; (g) inserção do
material compósito no corpo do eletrodo; (h) Aquecimento de eletrodo
compósito a 40 ºC por 2 horas e (i) renovação da superfície
consecutivamente com lixas de diferentes granulometrias e papel sulfite.
32
Figura 8
Resposta eletroquímica do eletrodo compósito de grafite e poliestireno (65%
m/m em grafite), confeccionado somente acetona na etapa de dissolução do
poliestireno, em K3Fe(CN)6 1,0 mmol L-1 utilizando KCl 0,10 mol L-1 como
eletrólito suporte. Velocidade de varredura: 50 mV s-1.
36
Figura 9
Otimização da porcentagem de grafite (m/m) no eletrodo G-PS pelo
desenvolvimento das ondas voltamétricas do par reversível de Fe(CN)6-3
/Fe(CN)6-4, 1,0 mmol L-1, em KCl 0,10 mol L-1. Velocidade de varredura 10
mV s-1.
37
Figura 10 Resposta eletroquímica do eletrodo G-PS em K3Fe(CN)6 1,0 mmol L-1
utilizando KCl 0,10 mol L-1 como eletrólito suporte, considerando 6 39

superfícies do eletrodo em decorrência do desbaste. Velocidade de
varredura: 10 mV s-1.
Figura 11
Influência da velocidade de varredura (ν) nas correntes de pico anódico e
catódico das ondas voltamétricas (IpC e IpA) de K3Fe(CN)6 1 mmol L-1 em
KCl 0,10 mol L-1. (a) Registros dos voltamogramas com aumento da
velocidade de varredura: 5, 10, 20, 30, 40, 50, 75, 100, 150, 200 mV s-1. (b)
Relação entre o pico anódico e catódico em função da raiz da velocidade de
varredura (dados obtidos de (a)). (c) Relação linear entre o log de IpA (e log
IpC) e o log de ν (dados obtidos de (a)).
41
Figura 12
Imagem da superfície do eletrodo de grafite e poliestireno. (a) detalhamento
dos riscos provenientes do polimento na superfície do eletrodo. (b)
Detalhamento da superfície do eletrodo com aumento da ampliação. Regiões
“A” e “B” são regiões com menor e maior condução respectivamente.
42
Figura 13
Comparação entre os eletrodos G-PS, GCE e CPE. (a) Voltamogramas
cíclicos obtidos para K3Fe(CN)6 1,0 mmol L-1 em KCl 0,10 mol L-1 e (b)
somente em KCl 0,10 mol L-1. Velocidade de varredura: 10 mV s-1. (c)
Cronoamperometria obtida para K3Fe(CN)6 1 mmol L-1 em KCl 0,10 mol L-1
e fit linear comparando para cada um dos eletrodos. (d) Diagrama de Nyquist
de impedância eletroquímica utilizando K3Fe(CN)6 1,0 mmol L-1e
K4Fe(CN)6 1,0 mmol L-1 em KCl 0,1 mol L-1. Os valores de quí-quadrado
para G-PS, GC e CPE foram respectivamente (0,03 ± 0,02), (0,04 ± 0,02) e
(0,13 ± 0,05).
45
Figura 14 Faixa de trabalho para os eletrodos GCE, G-PS e CPE em (a) HNO3 0,10
mol L-1, (b) KNO3 0,10 mol L-1 e (c) KOH 0,10 mol L-1. Velocidade de
varredura: 50 mV s-1. 47
Figura 15
Voltamogramas cíclicos usados para estimar potencial de limite positivo e
negativo para as faixas de trabalho do eletrodo G-PS em soluções com e sem
prévia desaeração para (a) HNO3 0,10 mol L-1, (b) KNO3 0,10 mol L-1 e (c)
KOH 0,10 mol L-1. (d) Voltamogramas cíclicos com scans progressivos para
avaliação da dependência das reações redox em KOH 0,10 mol L-1.
Velocidade de varredura: 50 mV s-1.
49
Figura 16
Análise exploratória de cisteína, ácido ascórbico, ácido gálico, peróxido de
hidrogênio, riboflavina, nitrito de sódio, paracetamol e dipirona utilizando
G-PS. Branco: Tampão acetato 0,10 mol L-1 (pH 4,7). Velocidade de
varredura: 50 mV s-1.
50
Figura 17
Estudo de faixa linear de (a) paracetamol e (b) dipirona utilizando FIA-
Amperométrico com potenciais fixos em 0,70 V e 0,40 V respectivamente.
Detalhamento das concentrações mais baixas de (c) paracetamol (0,28 e 0,84
μmol L-1) e (d) dipirona (0,67 e 1,34 μmol L-1). Detalhamento de um único
pico para verificar razão sinal ruído para 0,28 μmol L-1 de paracetamol (e) e
0,67 μmol L-1 de dipirona (f). Velocidade de fluxo de 2,65 min L-1.
52
Figura 18 Sinais de corrente obtidas por FIA em uma ampla faixa de concentração de
paracetamol (a) e dipirona (c) e apresentação de suas respectivas regiões
lineares (b e d). 54

Figura 19
(a)Fiagrama para injeções consecutivas a-e de padrão e de amostra (S) para
paracetamol. (b) Regressão linear para os dados obtidos do Fiagrama.
Velocidade de fluxo de 2,65 min L-1. (c) Espectro de absorção molecular
para paracetamol com concentrações crescentes a-e de padrão e amostra (S).
(d) Regressão linear para os dados obtidos do espectro de absorção
molecular. Comprimento de onda avaliado: 243 nm.
57
Figura 20
(a)Fiagrama para injeções consecutivas a-e de padrão e de amostra (S) para
dipirona. (b) Regressão linear para os dados obtidos do Fiagrama.
Velocidade de fluxo de 2,65 min L-1. (c) Espectro de absorção molecular
para dipirona com concentrações crescentes a-e de padrão e amostra (S). (d)
Regressão linear para os dados obtidos do espectro de absorção molecular.
Comprimento de onda avaliado: 258 nm.
58

Lista de Tabelas
Tabela 1 Relação entre a superfície do eletrodo e a distância de pico em solução
contendo ferricianeto 1,0 mmol L-1e KCl 0,10 mol L-1. 36
Tabela 2 Relação entre a porcentagem de grafite nos eletrodos G-PS e a distância de
pico em solução contendo ferricianeto 1,0 mmol L-1 e KCl 0,10 mol L-1. 37
Tabela 3 Relação entre a altura do eletrodo frente ao contato elétrico e a distância de
pico em solução contendo ferricianeto 1 mmol L-1 e KCl 0,1 mol L-1. 39
Tabela 4
Comparação entre eletrodos de carbono vítreo, pasta de carbono e compósito
de grafite e poliestireno com relação a área efetiva, rugosidade, diferença entre
picos para uma solução de K3Fe(CN)6 1,0 mmol L-1 em KCl 0,1 mol L-1 e
resistência de transferência de carga (RCT).
45
Tabela 5 Limites positivos e negativos referentes à janela de potencial de trabalho para
os eletrodos GCE, CPE e G-PS, sendo o último realizado também com
soluções desareadas. 47
Tabela 6 Comparação com valores de faixa linear e limites de quantificação e detecção
para eletrodos de carbono associados a sistemas de injeção em fluxo para
paracetamol e dipirona encontrados na literatura. 55
Tabela 7 Resultados para análise de paracetamol e dipirona utilizando o método padrão,
Fia-amperométrico e UV-Vis. 59

Sumário
Introdução ............................................................................................................... 13
1.1 Eletrodos compósitos ....................................................................................... 14
1.1.1 Definição ....................................................................................................... 14
1.1.2 Produção ........................................................................................................ 14
1.1.3 Características ................................................................................................ 14
1.1.4 Eletroanalítica com compósitos ..................................................................... 16
1.1.5 Biossensores .................................................................................................. 19
1.1.6 Eletrodos compósitos de poliestireno ............................................................ 20
1.2 Técnicas eletroquímicas ................................................................................... 20
1.2.1 Voltametria ............................................................................................... 20
1.2.2 Amperometria ........................................................................................... 21
1.3 Técnicas em fluxo ............................................................................................ 23
1.4 Impedância eletroquímica ................................................................................ 24
Objetivos ................................................................................................................. 28
Materiais e métodos ................................................................................................ 29
3.1 Reagentes ......................................................................................................... 29
3.2 Instrumentação eletroquímica .......................................................................... 29
3.3 Preparação do eletrodo compósito (G-PS) ....................................................... 31
3.4 Caracterização do eletrodo de Grafite-Poliestireno (G-PS) ............................. 32
3.4.1 Voltametria cíclica .................................................................................... 32
3.4.2 Microscopia eletrônica de varredura ........................................................ 32
3.4.3 Impedância eletroquímica......................................................................... 33
3.4.4 Preparação dos eletrodos de pasta de carbono (CPE)............................... 33
3.5 Determinação de paracetamol e dipirona em fármacos ................................... 33
3.6 Validação ......................................................................................................... 34
Resultados e discussão ........................................................................................... 35
4.1 Eletrodo de grafite e poliestireno (G-PS) ........................................................ 35
4.2 Caracterização eletroquímica do eletrodo de Grafite-Poliestireno (G-PS) .......... 39
4.3 Comparação entre eletrodos de carbono .......................................................... 44
4.4 Aplicações ........................................................................................................ 49
4.5 Limite de detecção e quantificação .................................................................. 50
4.6 Quantificação de paracetamol e dipirona ......................................................... 56
Conclusões .............................................................................................................. 60
Perspectivas ............................................................................................................ 61

Referências bibliográficas ...................................................................................... 62
Anexos .................................................................................................................... 69
Anexo 1 ...................................................................................................................... 69
Anexo 2 ...................................................................................................................... 70
Anexo 3 ...................................................................................................................... 71
Anexo 4 ...................................................................................................................... 73
Súmula curricular ................................................................................................... 75

13
Introdução
O desenvolvimento das técnicas voltamétricas teve início ainda no século XIX,
mas foi com Heyrowsky que recebeu impulso definitivo, quando desenvolveu o primeiro
polarógrafo. Este equipamento incomum, apresentado pela primeira vez em 1922 era
dotado de uma célula com dois eletrodos, sendo um dos eletrodos – denominado eletrodo
de trabalho - constituído por um eletrodo gotejante de mercúrio. O segundo eletrodo – o
eletrodo de referência – era constituído de um poço de mercúrio, recoberto por
calomelano e de uma solução de cloreto de potássio [1]. Este novo instrumento tornou
possível obter informações qualitativas e quantitativas para baixas concentrações de
espécies metálicas, bem como para compostos orgânicos.
Nas décadas seguintes foram testados diferentes materiais condutores (metais,
várias formas de carbono) e mais adiante (em 1958) Adams [2] demonstrou ser possível
realizar medidas eletroquímicas/eletroanalíticas utilizando um compósito, que viria a
denominar de eletrodos de pasta de carbono. Neste estudo, 1 g de pó de grafite foi
misturado a 7 mL de tribromometano, produzindo uma pasta relativamente compacta, que
foi utilizada para oxidar iodeto em meio ácido.
O estudo com o eletrodo de carbono proporcionou novos estudos principalmente
por possuir uma janela de potencial maior na região anódica se comparado ao eletrodo de
mercúrio, cujo metal é relativamente oxidado a óxido de mercúrio em potenciais
catódicos. Este estudo de Adams foi o precursor para o grande número de trabalhos com
eletrodos compósitos que existem na literatura (tanto de pasta de carbono como os que
envolvem outros materiais). Uma pesquisa na base webofknowledge indica a existência
de 11852 eletrodos de pasta de carbono e 34086 eletrodos compósitos (em setembro de
2019).

14
1.1 Eletrodos compósitos
1.1.1 Definição
Os compósitos constituem uma classe de material feito por dois ou mais
componentes, mantendo as propriedades de originais [3, 4] ou gerando um novo material
com desempenho diferenciado [4]. Os eletrodos compósitos geralmente são produzidos a
partir de uma fase isolante, responsável pela aglutinação do material, e uma fase
condutora [4-8] que pode ser constituída por exemplo de uma fase alotrópica de carbono
como grafite, negro de fumo, fibras de carbono, etc [3].
1.1.2 Produção
Há duas maneiras principais de produção destes eletrodos: (I) através da
dispersão da fase condutora no material que vai ser polimerizado, como visto em
eletrodos compósitos de adesivo epóxi [7-9]; e (II) através da dissolução de polímeros em
solvente, seguido da adição da fase condutora na solução gerada, formando uma “tinta
condutora”, se aproximando de eletrodos “screen-printed” [4, 9-11].
1.1.3 Características
Eletrodos compósitos contendo carbono possuem como características o custo
relativamente baixo e facilidade de produção (processo muitas vezes semelhante ao
eletrodo de pasta de carbono, porém com acréscimo de um tempo para cura), além de
baixa corrente de fundo, característica da maioria dos materiais carbonáceos.
A otimização da concentração das fases isolantes e condutoras neste tipo de
eletrodo é bastante importante. Navarro-Laboulais et. al. estudaram o efeito da
composição de grafite em um eletrodo compósito de epóxi. Os autores mostraram um
aumento no desempenho eletroquímico ao acréscimo da composição de grafite, devido

15
ao mesmo aumentar o contato entre partículas, até certo limite. Ultrapassando este limite,
a estrutura passa a ser menos estável mecanicamente. De acordo com os autores, o
aumento de grafite na composição do compósito aumenta a contribuição da capacitância
da dupla camada elétrica do eletrodo, desfavorecendo os processos de transferência de
carga [12].
Não somente o aumento da concentração da fase condutora, como também o uso
de fases condutoras mais “refinadas” como grafeno e nanotubos de carbono, oferecem
uma melhora significativa no sinal [13], porém com o inconveniente de um aumento
considerável de custo.
Diversas fases isolantes já foram utilizadas como “binder” para estes eletrodos.
Muitos dos estudos envolvem resina epóxi na produção dos eletrodos [5, 6, 9, 12-27],
mas outros materiais como poliuretano [24, 28-38], policloreto de vinila [39], óleo de
rícino [40], borracha de silicone [24, 27, 41, 42], poliéster [43, 44], parafina [45, 46],
polietileno [47], teflon [16, 48-54] e poliestireno [6, 7, 55, 56], entre outros, já foram
utilizados para manufatura destes compósitos. Um dos principais efeitos verificados
quando ocorre a troca de material polimérico é a alteração da faixa de potencial de
trabalho dos eletrodos [24]. Calixto et. al. compararam compósitos de grafite com
poliuretano, epóxi e borracha de silicone, sem apresentar diferenças significativas com
relação à resposta eletroquímica dos 3 materiais, porém demonstrando diferentes
comportamentos principalmente com relação aos potenciais limítrofes da região catódica
para diversos eletrólitos [24].
Uma das principais características dos eletrodos compósitos é a facilidade de
renovação da superfície. Para materiais preparados de forma homogênea, a renovação da
superfície gera resultados que não apresentam diferenças significativas a cada renovação.
Isto foi observado tanto para eletrodos mais simples com uma fase condutora e uma

16
isolante, quanto para eletrodos modificados. Wang et. al. demonstraram a resposta
eletroquímica de um eletrodo compósito de epóxi e grafite com boa homogeneidade
mesmo com renovação da superfície [25], assim como a homogeneidade de resposta em
eletrodos modificados com enzimas [19, 20].
1.1.4 Eletroanalítica com compósitos
Eletrodos compósitos preparados com epóxi e grafite foram explorados para
quantificação de metais pesados por stripping voltamétrico, como uma forma alternativa
aos eletrodos de mercúrio [5, 26]. A mesma técnica também foi explorada para a
quantificação de Au3+ em eletrodo preparado com epóxi e tubos de grafite impregnados
2-mercaptobenzotiazol [14]. Para quantificar iodeto, um eletrodo rotatório foi construído.
Para tal, foi preparado um compósito constituído por fibras de grafite randomicamente
orientadas e epóxi, tendo este eletrodo apresentado desempenho superior a um eletrodo
de carbono vítreo [22]. A quantificação de tioureia utilizando um compósito de epóxi e
grafite expandido foi demonstrada com o uso de cronoamperometria e voltametria cíclica
[6].
Eletrodos compósitos de grafite e poliuretano foram explorados na literatura para
quantificação de furosemida por voltametria de pulso diferencial e onda quadrada [29] e
imipramina [30] utilizando voltametria de onda quadrada. Amperometria, associada a um
sistema em fluxo, proporcionou-se a quantificação de atenolol [28] e paracetamol [31],
apresentando elevada frequência analítica. A comparação direta dos sinais gerados com
eletrodos compósitos frente à resposta proporcionada por um eletrodo de carbono vítreo
para detecção de atenolol, demonstrou um limite de detecção 8 vezes inferior [28] ao
passo que para paracetamol [31], o sinal foi 6 vezes menor. A quantificação de triptofano
foi realizada para este tipo de compósito utilizando nanopartículas de ouro como

17
modificador a partir da técnica de voltametria de pulso diferencial [32]. Com a adição de
grafeno em um compósito grafite-epóxi, a determinação de escitalopram foi realizada
com uma resposta superior à eletrodos não modificados e/ou modificados com nanotubos
de carbono [33]. Eletrodos compósitos impressos de grafite e poliuretano foram
explorados para quantificação de paracetamol [34], paracetamol e cafeína
simultaneamente [35] e epinefrina [36]. Os mesmos sensores também foram explorados
para a quantificação simultânea de Zn (II), Pb (II), Cu (II) e Hg (II) [37]. Em outro estudo,
dopamina pôde ser quantificada com a modificação destes eletrodos com uma base de
Schiff [38].
Eletrodos compósitos de PVC-grafite foram explorados para quantificar metais
(Cu2+, Hg2+ e Pb2+) por stripping voltamétrico. No mesmo estudo foi demonstrada a
potencialidade da utilização destes sensores associados a um sistema de injeção em fluxo
para a quantificação de catecol [39].
A construção de um compósito utilizando óleo de rícino (ou castor oil) e grafite
foi apontada como uma nova possibilidade de aglutinante. Este sensor foi utilizado para
análises por injeção em fluxo, resultando em sensibilidade mais elevada que a obtida com
eletrodos de carbono vítreo, frente à ferricineto de potássio, catecol e hidroquinona [40].
A resposta eletroquímica de compósitos de borracha de silicone foi comparada
com a de eletrodos compósitos de epóxi [24, 27] e poliuretano [24]. Estes eletrodos foram
explorados para a quantificação de rutina [41] e hidroquinona por pulso diferencial [42].
Eletrodos compósitos de grafite e poliéster modificados com óxido de zircônio
dopado com vanádio tetragonal e monoclínico foram utilizados para a quantificação de
peróxido de hidrogênio, utilizando voltametria linear e de pulso diferencial [43].

18
Eletrodos compósitos de grafite e parafina foram explorados para quantificar
citrato de sildenafil e furosemida por voltametria de onda quadrada [45]. Em outro estudo
foi descrito um sistema um pouco mais complexo, onde uma resina de cobre (II) é
associada ao grafite e parafina, possibilitando a quantificação de rutina utilizando a
técnica de voltametria linear [46].
Um eletrodo compósito constituído por polietileno, grafite e óxido de cobre
(Cu2O) foi associado à um sistema em fluxo, para a quantificação de manitol em meio
alcalino. Foi verificado melhora do desempenho do eletrodo em função do aumento
concentração de hidróxido [47].
O teflon utilizado como aglutinante teve bastante destaque na área de
biossensores [48-53]. A quantificação dos herbicidas dissulfiram e dissulfeto de
tetrametiltiuram foi realizada com compósito de teflon e grafite a partir da técnica de
stripping voltamétrico [54].
Compósitos à base de carbono foram menos explorados como sensores
potenciométricos em comparação com as aplicações voltamétricas e amperométricas.
Entretanto a literatura apresenta importantes aplicações, desde a determinação de ânions
como Cl-, NO3-, SO4
2-, H2PO4-, F- e HCO3
- [57] e cátions como Ag+ [58], Tl+ [59] e Hg2+
[60] com limites de detecção na ordem de 10-9 mol L-1. Em alguns estudos, diferentemente
do descrito para os sensores amperométricos e voltamétricos, são utilizados líquidos
iônicos como aglutinantes, além ionóforos na fabricação de sensores potenciométricos
[58-60], promovendo uma melhora na resposta eletroquímica do eletrodo e
proporcionando respostas Nernstianas, com estabilidade e melhora no tempo de resposta
[61].

19
Um eletrodo compósito, a base de carbono e polianilina, (construído na forma
de “werable electrode”), foi utilizado para monitoramento de pH em feridas infeccionadas
e não infeccionadas, demonstrando a versatilidade no uso de eletrodos compósitos. Este
sensor apresentou boa estabilidade mesmo quando aplicada uma tensão mecânica sobre
o eletrodo [62].
1.1.5 Biossensores
A imobilização dos modificadores biológicos na superfície de um eletrodo sólido
nem sempre é algo simples de ser realizado [19]. O uso de enzimas em associação com
os eletrodos compósitos pode ser mais simples em comparação com a imobilização
superficial. A incorporação do elemento biológico na etapa de produção dos eletrodos
permite a renovação da superfície do sensor, mesmo havendo enzimas imobilizadas no
interior do compósito. A enzima tirosinase foi bastante explorada como modificador em
diversos eletrodos como grafite-epóxi [16, 19, 21, 49], grafite-teflon [16, 49], carbono
vítreo reticulado e epóxi [16]. Diferentes fases poliméricas promovem interações distintas
entre a superfície do eletrodo e o ciclo catalítico da tirosinase, causando alterações nas
velocidades de reação para compostos fenólicos. Além do tempo de reação, materiais
diferentes oferecem tempos de vida útil distintos, respostas diferentes quanto a
homogeneidade e intensidade de corrente [16].
Muitos compósitos utilizam misturas de enzimas em sua composição com
destaque a misturas contendo a peroxidase. Esta enzima foi utilizada em misturas com
glicose oxidase para quantificação de glicose em vinhos [48], associada com lactato
oxidase e para determinação de L-lactato em amostras alimentícias [50], utilizando
eletrodos compósitos de grafite e teflon em ambos os casos. Colesterol oxidase e
peroxidase foram exploradas em outro estudo para determinação de colesterol em

20
alimentos [51]. Eletrodos compósitos de epóxi foram utilizados para incorporação de
enzimas em um estudo para quantificação de bilirrubina, com a modificação do compósito
a partir de bilirrubina oxidase e peroxidase [20].
1.1.6 Eletrodos compósitos de poliestireno
Eletrodos compósitos de carbono e poliestireno foram anteriormente relatados
na literatura [6, 7, 55, 56]. Corb et. al. utilizaram os eletrodos de grafite e poliestireno
expandido para determinação de Tioureia e compararam seu desempenho com o de
eletrodos compósitos de epóxi como fase isolante [6]. Rassei et. al. utilizaram eletrodos
de nanofibras de carbono e poliestireno como sensor voltamétrico para determinação de
Pb(II) por voltametria de redissolução anódica [7]. Manea et. al. avaliaram o
comportamento do eletrodo de grafite esfoliado e poliestireno para a determinação de
ácido oxálico, e compararam o seu desempenho com eletrodos de fases isolantes
diferentes (poliuretano e adesivo epóxi) [55]. Xu et. al. utilizaram um compósito de
nanotubos de carbono e poliestireno como detector eletroquímico para separação
eletroforética de rutina e quercetina por eletroforese capilar em microchip, em tampão
borato 50 mmol L-1 (pH 9,2) como eletrólito para separação [56].
1.2 Técnicas eletroquímicas
1.2.1 Voltametria
As técnicas voltamétricas são utilizadas para estudos dos processos ocorridos no
eletrodo, seja da transferência de carga da reação, processos adsortivos, processos
termodinâmicos, reversibilidade da reação, mecanismos de reação, cinética, etc [3, 63].
A voltametria cíclica é geralmente o primeiro teste realizado em estudos eletroquímicos

21
por fornecer de forma rápida os potenciais de redução e oxidação da espécie envolvida,
bem como o número de elétrons envolvidos na reação [3].
A ciclovoltametria se baseia na variação controlada de potencial em função do
tempo, de forma linear ou cíclica (Fig. 1), originando uma corrente referente ao processo
que ocorre no eletrodo, seja de natureza capacitiva (carga da dupla camada elétrica) ou
faradaica (referente ao processo de oxidação ou redução) [3, 63].
Figura 1 - Aplicação de potencial em função do tempo em um estudo de voltametria cíclica.
Fonte: Brett, 1993 [63].
1.2.2 Amperometria
A amperometria é a técnica baseada na medição da corrente em um potencial
fixo ao longo do tempo. A sua principal vantagem, frente as técnicas voltamétricas, é
apresentar um aumento da relação sinal ruído. Isso devido à medição da corrente ser
proveniente majoritariamente de natureza faradaica, e não capacitiva, possibilitando o uso
para quantificação de analitos eletroativos [3, 63].
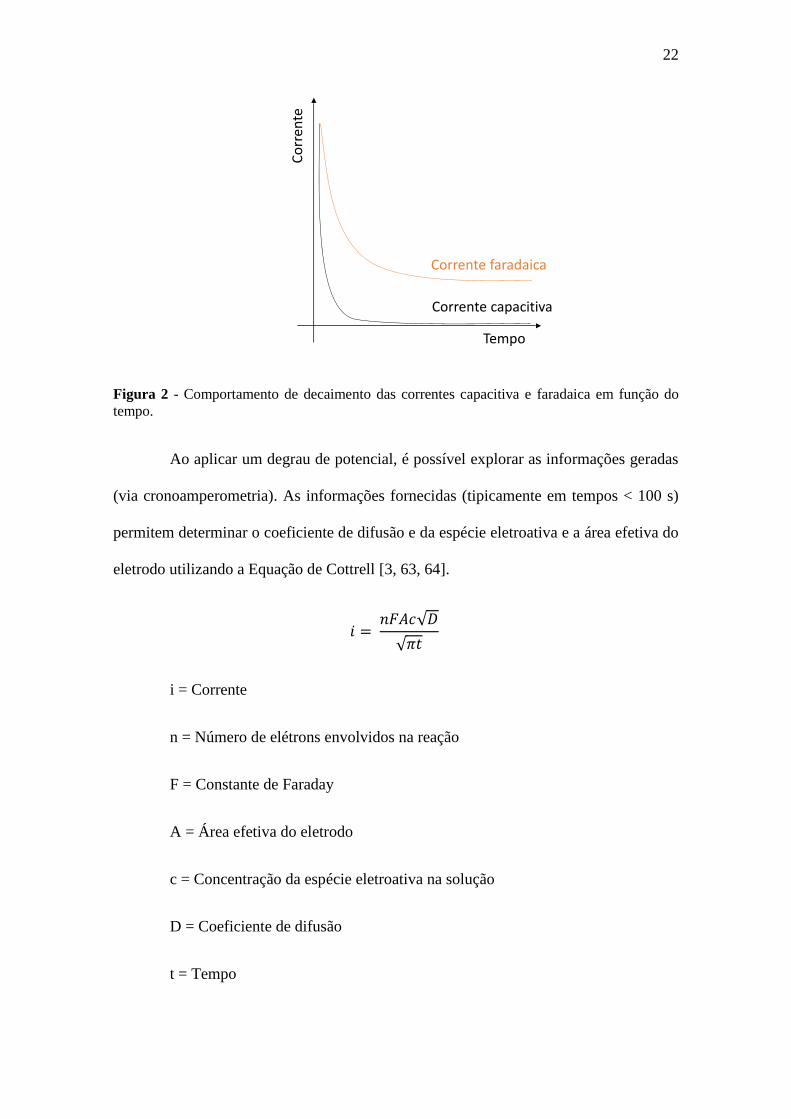
A Figura 2 apresenta o comportamento de decaimento das correntes (capacitiva
e faradaica) ao decorrer do tempo ao ser aplicado um degrau de potencial, demonstrando
uma queda mais significativa em curto espaço de tempo para a corrente capacitiva em
comparação com a corrente faradaica.
22
Figura 2 - Comportamento de decaimento das correntes capacitiva e faradaica em função do
tempo.
Ao aplicar um degrau de potencial, é possível explorar as informações geradas
(via cronoamperometria). As informações fornecidas (tipicamente em tempos < 100 s)
permitem determinar o coeficiente de difusão e da espécie eletroativa e a área efetiva do
eletrodo utilizando a Equação de Cottrell [3, 63, 64].
𝑖 = 𝑛𝐹𝐴𝑐√𝐷
√𝜋𝑡
i = Corrente
n = Número de elétrons envolvidos na reação
F = Constante de Faraday
A = Área efetiva do eletrodo
c = Concentração da espécie eletroativa na solução
D = Coeficiente de difusão
t = Tempo
Co
rren
te
Tempo
Corrente faradaica
Corrente capacitiva

23
A associação da amperometria com sistemas de análise por injeção em fluxo
favorece a obtenção de sinais virtualmente livres da contribuição capacitiva, com elevada
sensibilidade, uma vez que o transporte de massa para a superfície do eletrodo é
extremamente mais eficiente que em processos difusionais.
1.3 Técnicas em fluxo
As técnicas em fluxo tiveram início com Skeggs [65] em 1957 e se tornaram
ferramentas importantes, especialmente para processos que envolvam um grande número
de amostras e análises, com aplicações nas mais diversas áreas, dentre as quais a
industrial, ambiental e na área clínica [66].
A técnica de análise por injeção em fluxo (FIA) surgiu com os estudos realizados
por Ruzicka e Hansen [67], e possui pelo menos 3 etapas importantes: propulsão da
solução carreadora, injeção da amostra, e detecção [68]. Em muitos processos (em
especial envolvendo a espectrofotometria, onde uma reação ocorre em fluxo) um quarto
processo é necessário: a reação durante o percurso da amostra até o detector. A propulsão
pode ser realizada de diferentes formas, sendo as mais comuns as baseadas em bombas
peristálticas, seringas motorizadas, sistemas pressurizados ou mesmo por gravidade,
promovendo um fluxo “constante”. O injetor é responsável por transferir o analito para o
fluxo com a solução carreadora, seguindo para a reação (quando se aplica) e detecção do
sinal [68]. Para estudos espectrofotométricos, geralmente a reação da amostra ocorre
previamente à detecção, com o uso de bobinas para mistura de reagentes. Com relação a
medições eletroquímicas, há necessidade de apenas três etapas, visto que geralmente não
há necessidade de uma reação prévia e consequentemente de bobina de reação, que não
contribuiria em nada e ainda favoreceria a diluição da amostra ao longo do caminho (Fig.
3). Diferentes técnicas analíticas foram exploradas em associação com a injeção em fluxo

24
como amperometria [69-71], quimiluminescência [72, 73], absorção atômica [74, 75],
espectrofotometria [76, 77], entre outras.
Figura 3 - Esquema de um sistema para análise por injeção em fluxo, utilizando gravidade como
propulsão da solução carreadora, para uma cela eletroquímica controlada por um potenciostato
interfaceado com um computador.
1.4 Impedância eletroquímica
A impedância eletroquímica oferece a possibilidade de caracterização de
diversos parâmetros de materiais condutores e suas relações interfaciais [78]. A técnica
de impedância mais comumente utilizada se baseia na perturbação da corrente aplicada
ou potencial aplicado em um sistema com frequência única, sendo possível avaliar a
amplitude e a fase do sinal correspondente comparando-a com o sinal enviado. Este sinal
pode ser convertido para o domínio de tempo utilizando uma transformada de Fourier,
sendo assim possível medir os valores de impedância real e imaginária para um
determinado sistema à uma única frequência. Ao se estudar a impedância em uma faixa
de frequências é possível caracterizar os processos da célula eletroquímica [63, 78].
AE
RE
SE
WE
Graphite/PS composite layer
PTFE
Graphite/PS composite
REAE
WE
I RE
Acrylic bodyWE
AERubber
Separator
Inlet
(a)(b)
(c)
SE
AE
RE
SE
WE
Graphite/PS composite layer
PTFE
Graphite/PS composite
REAE
WE
I RE
Acrylic bodyWE
AERubber
Separator
Inlet
(a)(b)
(c)
SE
AE
RE
SE
WE
Graphite/PS composite layer
PTFE
Graphite/PS composite
REAE
WE
I RE
Acrylic bodyWE
AERubber
Separator
Inlet
(a)(b)
(c)
SE
Descarte
Eletrodo de referência
Eletrodo de trabalho
Eletrodo auxiliar
Injetor
Sistema de propulsão por
gravidade
Sistema de propulsão por gravidade
Injetor
Eletrodo de referência
Eletrodo auxiliarDescarte
Eletrodo de trabalho
SeringaAmostra

25
Sistemas eletroquímicos podem ser representados por circuitos elétricos
correspondentes, utilizando uma combinação de resistores e capacitores para explicação
do modelo [63]. O modelo de Randles é um dos modelos mais conhecidos e trabalhados
para sistemas eletroquímicos (Fig. 4).
Figura 4 - Circuito de Randles para um sistema eletroquímico simples envolvendo transferência
de carga. Adaptado de Brett, 1993 [63]
A partir da Figura 4, pode-se correlacionar o elemento de fase “Cd” com a dupla
camada elétrica do sistema eletroquímico; o resistor “RΩ” com a resistência não
compensada da solução; o resistor “RCT” com a resistência de transferência de carga
correlacionada com as características condutivas do material e a impedância de Warburg
“W”, correlacionado com o processo faradaico [63].
As equações que regem as impedâncias real e imaginária para um sistema com
modelo de circuito equivalente de Randles [63] são:
𝑍′ = 𝑅𝛺 + 𝑅𝑐𝑡 + 𝜎𝜔−0,5
(𝜎𝜔0,5𝐶𝑑 + 1)2 + 𝜔2𝐶𝑑2(𝑅𝑐𝑡 + 𝜎𝜔−0,5)2
W
R2R1
C
RΩRCT
W
Cd

26
−𝑍′′ = 𝜔𝐶𝑑(𝑅𝑐𝑡 + 𝜎𝜔−0,5)2 + 𝜎2𝐶𝑑 + 𝜎𝜔−0,5
(𝜎𝜔0,5𝐶𝑑 + 1)2 + 𝜔2𝐶𝑑2(𝑅𝑐𝑡 + 𝜎𝜔−0,5)2
Z’ = Impedância real
-Z’’ = Impedância imaginária
σ = Densidade de carga superficial
ω = Frequência angular
RΩ = Resistência da solução
RCT = Resistência de transferência de carga
Cd = Capacitância
Em frequências elevadas, impedância imaginária torna-se muito baixa, tornando
a contribuição maior do sistema a parte real de sua impedância. A medida que se diminui
a frequência, -Z’’ aumenta, e a corrente passa no circuito equivalente tanto pelo elemento
de fase, quanto pelo resistor. Após a região do centro do semicírculo, a reatância
capacitiva do capacitor começa a se elevar, diminuindo a contribuição da impedância
imaginária, favorecendo a passagem de corrente somente pelo capacitor à medida que se
decresce mais a frequência [63].
Há duas regiões que regem o espectro de impedância (Fig. 5), região em que há
o controle cinético (semicírculo) e controle difusional (transporte de massa). O controle
cinético se dá quando a relação entre a resistência de transferência de carga é muito maior
que o valor da impedância de Warburg [63].

27
Figura 5 – Espectro de impedância para um sistema redox simples em que envolva transferência
de carga (Adaptado de BRETT,1993) [63].
Sistemas eletroquímicos mais complexos em que há a presença de membranas
ou de reações acopladas, por exemplo, é necessário o estudo do circuito equivalente que
melhor corresponde ao sistema, levando em consideração que não há somente um circuito
que represente o sistema [63, 78].
Controle cinético
Controle por
transporte
de massa

28
Objetivos
Objetivo geral
- Desenvolver eletrodos compósitos de grafite e poliestireno provindo de isopor
com baixo custo de produção, alta homogeneidade e boa resposta eletroquímica.
Objetivos específicos
- Determinar e comparar a resposta eletroquímica do eletrodo compósito
desenvolvido com outros eletrodos de carbono.
- Determinar paracetamol e dipirona utilizando o eletrodo desenvolvido por
amperometria associado à técnica de injeção em fluxo e comparar com métodos padrão
da literatura.
- Abrir a possibilidade de incorporação de outras espécies catalíticas a este
material, gerando sensores de construção simples e de baixo custo.

29
Materiais e métodos
3.1 Reagentes
Todas as soluções foram preparadas com água deionizada a partir de um sistema
de purificação Nanopure (ρ> 18,2 MΩ cm) e reagentes de grau analítico. O clorofórmio
(triclorometano) e a acetona (n-propanona) usados como veículo e solventes de
modelagem, respectivamente, foram ambos de grau espectroscópico. O pó de grafite foi
de grau analítico, adquirido da LabSynth (Brasil). A fonte de poliestireno foi o
poliestireno extrudado, que está disponível mundialmente nas mais diversas formas e
pode ser encontrado em lojas convencionais de papelaria com o nome comercial de
isopor. Alternativamente, é possível reaproveitar embalagens (de isopor) descartadas.
Para confecção dos eletrodos de pasta de carbono (CPEs) o aglutinante usado
foi o óleo mineral CAS 8020-83-5 (0,838 g mL-1) da Aldrich. Para preparar soluções de
Fe(CN)63–, foi utilizado o sal de ferricianeto de potássio (K3[Fe(CN)6]) do LabSynth
(Brasil) diretamente dissolvido em KCl 0,1 mol L-1. O íon Fe(CN)64- foi proveniente do
sal de ferrocianeto de potássio trihidratado (K4[Fe(CN)6] . 3H2O) do Carlos Erba
Reagents.
Paracetamol (4-Acetamido-Fenol) foi adquirido da Sigma (CAS 103-90-2),
assim como dipirona (metamizol) (CAS 68-89-3).
3.2 Instrumentação eletroquímica
Os experimentos voltamétricos foram realizados em temperatura ambiente
(~25ºC) com um potenciostato PGSTAT20 da Metrohm controlado pelo software NOVA.
Uma célula convencional de 3 eletrodos (Fig. 6a) foi utilizada para as medições
voltamétricas, com o eletrodo compósito de grafite e poliestireno (G-PS) como eletrodo

30
de trabalho, um fio de platina como eletrodo auxiliar e um eletrodo de Ag/AgCl(KCl sat.)
que foi produzido no próprio laboratório (utiliza uma ponteira de pipeta de Eppendorf e
separador de baterias como membrana porosa) [79]. Um eletrodo de carbono vítreo
comercial da Metrohm foi utilizado para fazer medições com as mesmas soluções, com a
finalidade de comparar os resultados gerados com o novo eletrodo. Para a determinação
em fluxo do acetaminofeno e metamizol, foi confeccionada uma célula eletroquímica no
próprio laboratório, como mostrada na Figura 6b. Para tal foi feito uso de um sistema de
injeção em fluxo em linha única. Para esta célula, foi utilizada uma agulha de seringa de
aço inox como eletrodo auxiliar posicionado na saída da solução, um eletrodo de
referência posicionado exatamente acima (em frente ao centro do eletrodo de trabalho -
G-PS) que foi posicionado na parte de baixo da célula. Um espaçador de borracha foi
utilizado para delimitar volume da célula (~50 μL).
Figura 6 – (a) Diagrama de uma célula convencional de 3 eletrodos composta por fio de platina
como eletrodo auxiliar (EA), eletrodo compósito como eletrodo de trabalho (ET), eletrodo de
Ag/AgCl (KCl (sat)) como eletrodo de referência (ER), imersa em 20 mL de eletrólito (KCl 1 mmol
L-1 por exemplo). (b) Diagrama da célula utilizada para a análise em fluxo, composta pelo eletrodo
de referência de Ag/AgCl (KCl (sat) (ER), uma agulha de aço inox, posicionada na saída da célula,
como eletrodo auxiliar (EA) e eletrodo compósito de grafite e poliestireno como eletrodo de
trabalho (ET), posicionado na parte inferior da célula.
EA
ER
SE
ET
Grafite/PsCamadado eletrodo
Corpo de Teflon
Grafite/PsCompósito
ER
Corpo de acrílicoET
EASeparadorde borracha
Entrada
(a) (b)

31
3.3 Preparação do eletrodo compósito (G-PS)
Pesa-se 600 mg de pó de grafite que posteriormente é repulverizado com auxílio
de um pistilo e almofariz (Fig. 7a). Adiciona-se 10 mL de clorofórmio (resultando em
uma suspensão de pó de grafite) e adiciona-se 200 mg de isopor (Fig. 7b). Homogeneíza-
se muito bem esta suspensão (10 min), que a seguir é deixada para secar em estufa por 2
horas à 80 ºC (Fig. 7c). Retira-se o almofariz da estufa, espera-se o esfriamento até a
temperatura ambiente e adiciona-se 5 mL de acetona, mistura-se o material, que irá gerar
uma massa maleável (Fig. 7d). O excesso de acetona é removido (Fig. 7e), e então o
compósito é manipulado para ser inserido em um tubo de teflon (Ø = 3,6 mm) (Fig. 7 f-
g). Para estabelecer facilmente o contato elétrico, um parafuso foi rosqueado no interior
do tubo de teflon. Os tubos foram preenchidos com profundidade controlada de 6 mm
(para os testes de homogeneidade ao longo do eletrodo) e 3 mm para os demais estudos.
Após o preenchimento do eletrodo, leva-se à estufa com temperatura de 40ºC por 2 horas
(Fig. 7h). Após aquecimento o compósito fica bastante rígido, sendo necessário para
nenovar a superfície do eletrodo o uso de lixas de diferentes granulometrias, (200 e 1200).
A etapa final consistiu em polimento diretamente sobre uma folha de papel (Fig. 7i).
Verificou-se que esta última etapa produzia uma superfície mais homogênea em
comparação com as superfícies apenas lixadas.

32
Figura 7 – Diagrama de produção do eletrodo G-PS: (a) repulverização do grafite; (b) adição do
isopor e clorofórmio com posterior homogeneização; (c) aquecimento do material a
aproximadamente 80 ºC por duas horas em capela; (d) adição de acetona e geração de uma pasta
maleável; (e) remoção do excesso de acetona; (f) remoção do material do almofariz; (g) inserção
do material compósito no corpo do eletrodo; (h) Aquecimento de eletrodo compósito a 40 ºC por
2 horas e (i) renovação da superfície consecutivamente com lixas de diferentes granulometrias e
papel sulfite.
3.4 Caracterização do eletrodo de Grafite-Poliestireno (G-PS)
3.4.1 Voltametria cíclica
A caracterização do eletrodo compósito foi realizada pela execução de
voltamogramas cíclicos a taxas crescentes de velocidade de varredura (de 5 a 200 mV s-1)
para K3Fe(CN)6 1,0 mmol L-1 em KCl 0,10 mol L-1. A comparação entre os eletrodos de
carbono (G-PS, GC e CPE), assim como o teste de homogeneidade ao longo do eletrodo
(desbaste) foram realizados utilizando K3Fe(CN)6 1,0 mmol L-1 em meio de KCl 0,10 mol
L-1 como “padrão” para as medições comparativas.
3.4.2 Microscopia eletrônica de varredura
As imagens da superfície do eletrodo G-PS foram obtidas por microscopia
eletrônica de varredura com emissão de campo, obtidas no microscópio modelo JSM–
7401–F da Jeol, com tensão de aceleração de 3kV.
(a) (b) (c) (d) (e)
(f)(g)(h)(i)
P36
1200
Sulfite

33
3.4.3 Impedância eletroquímica
Para os estudos de impedância eletroquímica foi utilizando o potenciostato
PGSTAT 302N com módulo FRA 32M da Metrohm controlado pelo software NOVA.
Os registros foram realizados utilizando uma concentração equimolar de K3Fe(CN)6 e
K4Fe(CN)6 de (1.0 mmol L-1 de cada) em KCl 0,10 mol L-1 com frequências variando de
100 kHz a 0,1 Hz. A resistência de transferência de carga foi determinada utilizando como
modelo de fitting o circuito modificado de Randles, com um componente de fase
capacitivo e a impedância de Warburg como componente para o caráter difusional em
baixas frequências.
3.4.4 Preparação dos eletrodos de pasta de carbono (CPE)
Os eletrodos de pasta de carbono utilizados para comparação foram
confeccionados utilizando a proporção de 570 mg de grafite para 430 mg de óleo Nujol,
formando uma pasta com 57 % m/m de grafite, também inseridos em tubo de teflon com
o mesmo diâmetro dos utilizados para o eletrodo G-PS.
3.5 Determinação de paracetamol e dipirona em fármacos
Para a determinação do paracetamol e dipirona utilizou-se o eletrodo compósito
de grafite-poliestireno como eletrodo de trabalho em uma célula de fluxo já descrita
anteriormente (Item 3.2, Fig. 6b). Utilizou-se tampão acetato 0,10 mol L-1 (pH 4,7) como
eletrólito suporte para ambas as determinações.
Para a quantificação dos analitos em amostras reais, adicionou-se um
comprimido em 50 mL de água deionizada e sua solubilização foi efetivada em banho de
ultrassom (42 kHz e 160W) por 20 minutos. Filtrou-se a solução para remoção dos

34
excipientes e transferiu-se uma alíquota de 50 µL para um balão de 25 mL, posteriormente
completado com tampão acetato. A solução resultante foi utilizada como amostra. O sinal
amperométrico foi monitorado em 700 mV para o paracetamol e 400 mV para dipirona
(vs. Ag/AgCl (KCl sat.)). Para a comparação dos resultados obtidos, foram utilizadas análises
por espectroscopia de UV/Vis e HPLC para paracetamol e UV/Vis e titulação de oxi-
redução para a dipirona.
3.6 Validação
Para validação do método eletroquímico utilizado para quantificar paracetamol,
a mesma amostra foi analisada por HPLC, técnica recomendada pela farmacopeia
americana volume 32–NF 27 [80]. A análise por HPLC foi realizada com um
equipamento da Shimadzu Modelo Prominence LC - 2030, coluna Shimadzu (Shim-
pack VP-ODS 4,6 x 250 mm) com monitoramento a 243 nm. As análises
espectrofotométricas de paracetamol e dipirona foram realizadas com o espectrômetro
Agilent 8453 e os sinais foram monitorados em 243 nm e 258 nm para paracetamol e
dipirona respectivamente. De acordo com a 5ª edição da farmacopeia brasileira [81], o
método oficial para quantificar dipirona envolve a análise iodométrica. Os resultados
obtidos por HPLC e por titulação iodométrica foram muito similares aos obtidos por via
espectrofotométrica e todos os resultados obtidos para cada analito (dipirona e
paracetamol) foram bastante convergentes.

35
Resultados e discussão
4.1 Eletrodo de grafite e poliestireno (G-PS)
Inicialmente, diversos solventes foram testados para a dissolução do
poliestireno, incluindo acetona, clorofórmio, acetato de etila, n-hexano, n-butanol,
tetracloreto de carbono e éter de petróleo. Dos solventes listados, o n-butanol, n-hexano
e éter de petróleo não foram capazes de dissolver o isopor. A acetona utilizada como
solvente dissolveu parcialmente o isopor e originou um eletrodo com baixa
homogeneidade e resposta eletroquímica insatisfatória (Fig. 8). Os solventes restantes
foram capazes de dissolver completamente o poliestireno, porém apresentam toxicidade
elevada (clorofórmio e tetracloreto de carbono) ou inflamabilidade (acetato de etila). O
solvente escolhido foi o clorofórmio, apesar da conhecida toxicidade, o que nos levou a
utilizar EPIs adequados e capela.
Para os primeiros testes de construção dos eletrodos compósitos, optou-se por
adotar o percentual de 65% de grafite para 35% de poliestireno e como solvente apenas
acetona para dissolver o polímero. Como pode ser visto nos resultados abaixo, o eletrodo
compósito resultante (Fig. 8) não apresentou uma boa homogeneidade ao longo do seu
corpo. Em diferentes seções, foram observadas respostas do eletrodo com diferenças
significativas entre o pico catódico e anódico (ΔEp) apresentados na Tabela 1.
Foram testadas diferentes estratégias para uniformizar o compósito de grafite e
isopor. A melhor condição foi realizar a dissolução do poliestireno com clorofórmio, que
é evaporado, a seguir a acetona é adicionada para tornar o material altamente maleável,
favorecendo assim obter um material bem mais uniforme.

36
Figura 8 - Resposta eletroquímica do eletrodo compósito de grafite e poliestireno (65% m/m em
grafite), confeccionado somente acetona na etapa de dissolução do poliestireno, em K3Fe(CN)6
1,0 mmol L-1 utilizando KCl 0,10 mol L-1 como eletrólito suporte. Velocidade de varredura: 50
mV s-1.
Tabela 1 – Relação entre a superfície do eletrodo e a distância de pico em solução contendo
ferricianeto 1,0 mmol L-1e KCl 0,10 mol L-1.
Superfície ΔEp (mV)
A 158
B 211
C 174
D 143
E 208
F 105
G 122
H 85
I 143
J 105
K 100
Para otimizar a composição destes eletrodos, as composições contendo 55, 60,
65, 70 e 75% de grafite foram testadas. Os testes envolveram a resposta frente ao processo
redox de Fe(CN)6-3/Fe(CN)6
-4 estudada por voltametria cíclica (Fig. 9). Foi observada
uma tendência de diminuição da distância entre o pico anódico e catódico com o aumento
-0,2 0,0 0,2 0,4 0,6 0,8-8
-6
-4
-2
0
2
4
6
F
Co
rren
te (
)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
3,1 mm
3,4 mm
3,8 mm
4,4 mm
4,8 mm
5,0 mm
HJK GI
-0,2 0,0 0,2 0,4 0,6 0,8-8
-6
-4
-2
0
2
4
6
Corr
ente
(
)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
Superfície
0,8 mm
1,6 mm
A
CB
-0,2 0,0 0,2 0,4 0,6 0,8-8
-6
-4
-2
0
2
4
6
D
Co
rren
te (
)
Potencial aplicado (V vs. Ag/AgCl(sat.)
)
1,8 mm
2,1 mm
E
B
Superfície do
eletrodo (A)0,0 mm
0,5 mm
1,0 mm
1,5 mm
2,0 mm
2,5 mm
3,0 mm
3,5 mm
4,0 mm
4,5 mm
5,0 mm
5,5 mm
6,0 mm
CDE
FG
H
I
JK
Contato
elétrico

37
da porcentagem de grafite na mistura até alcançar 75 %. Em porcentagens maiores, há
uma desagregação do material na superfície do eletrodo, atribuída à falta de material
polimérico para sua agregação. Os valores de diferença entre os picos catódico e anódico
são apresentados na Tabela 2.
Figura 9 – Otimização da porcentagem de grafite (m/m) no eletrodo G-PS pelo desenvolvimento
das ondas voltamétricas do par reversível de Fe(CN)6-3/Fe(CN)6
-4, 1,0 mmol L-1, em KCl 0,10 mol L-1.
Velocidade de varredura 10 mV s-1.
Tabela 2 – Relação entre a porcentagem de grafite nos eletrodos G-PS e a distância de pico em
solução contendo ferricianeto 1,0 mmol L-1e KCl 0,10 mol L-1.
% Grafite ΔEp (mV) Média
(mV)
Desvio padrão
(mV) Eletrodo 1 Eletrodo 2 Eletrodo 3
55% 159 129 132 140 17
60% 140 112 144 132 17
65% 85 114 105 101 15
70% 78 95 73 82 11
75% 71 75 80 75 5
-0.2 0.0 0.2 0.4 0.6 0.8-10
-8
-6
-4
-2
0
2
4
6
Co
rren
te (
A)
Potencial aplicado (V vs. Ag/AgCl(KCl Sat))
55%
60%
65%
70%
75%

38
Com pode ser visto na Tabela 2, a média dos valores de ΔEp apresentam uma
tendência de diminuição da distância com o aumento da porcentagem de grafite no
compósito. Considerando a dispersão de cada distribuição, de 65 % em diante, não há
diferença estatística relevante quanto a diminuição na distância dentre os potenciais de
pico catódico e anódico (α = 0,05) (Anexo 1), e, portanto, não há um melhora
considerável com a diminuição de ΔEp. Entretanto, o valor do desvio padrão do
compósito cuja composição foi de 75 % de grafite, se apresentou consideravelmente mais
baixo do que as outras composições (menor que a metade dos outros valores), o que ficou
comprovado ser verdade no decorrer deste trabalho. Portanto, a composição de 75 % de
grafite foi escolhida para as etapas posteriores devido à boa estabilidade mecânica e
resposta eletroquímica reprodutível. Ainda, com o acréscimo do grafite no compósito, o
desenvolvimento de uma onda de redução no potencial de -0,20 V torna-se evidente, e é
melhor explicado mais adiante (Fig. 15). Resumidamente, esta onda é referente ao
processo de redução do oxigênio dissolvido na solução, gerando peróxido de hidrogênio
(O2/H2O2). Este significativo aumento da corrente de redução ainda não está claro, uma
vez que a área ativa do eletrodo permanece praticamente a mesma.
Para avaliar a resposta eletroquímica do eletrodo ao longo de sua extensão, foram
preparados eletrodos com extensão de 6 mm e medidas com a superfície inicial foram
comparadas com a constituição interna do eletrodo, após desbastes de 1 em 1 mm. Na
Figura 10 são apresentados os voltamogramas cíclicos registrados para cada secção de
um eletrodo. Foi utilizada uma solução de Fe(CN)63- para avaliar as diferentes superfícies
ao longo do desbaste do eletrodo. Os valores de diferença de pico catódico e anódico em
função do desbaste da superfície de três eletrodos distintos foram reunidos na Tabela 3.
Não houve diferença significativa entre as médias dos resultados obtidos para as

39
diferentes profundidades do material (α = 0,05) (Anexo 2), o que atesta uma
homogeneidade aceitável da dispersão do grafite em toda a extensão do compósito.
Figura 10 - Resposta eletroquímica do eletrodo G-PS em K3Fe(CN)6 1,0 mmol L-1 utilizando
KCl 0,10 mol L-1 como eletrólito suporte, considerando 6 superfícies do eletrodo em
decorrência do desbaste. Velocidade de varredura: 10 mV s-1.
Tabela 3 – Relação entre a altura do eletrodo frente ao contato elétrico e a distância de pico em
solução contendo ferricianeto 1 mmol L-1e KCl 0,1 mol L-1.
Altura
do
eletrodo
ΔEp (mV) Média
(mV)
Desvio Padrão
(mV) Eletrodo 1 Eletrodo 2 Eletrodo 3
6 mm 78 95 83 85 9
5 mm 76 78 85 80 5
4 mm 73 68 81 74 7
3 mm 71 73 88 77 9
2 mm 76 71 81 76 5
1 mm 71 71 83 75 7
4.2 Caracterização eletroquímica do eletrodo de Grafite-Poliestireno (G-PS)
Para avaliar as características do novo material, experimentos utilizando uma
sonda reversível (como Fe(CN)63-) foram realizados. Os resultados mostraram valores
A
B
C
D
E
F
-0.2 0.0 0.2 0.4 0.6 0.8-12
-9
-6
-3
0
3
6
Co
rren
te (
A)
Potencial aplicado (V vs. Ag/AgCl(KCl Sat)
)
Superfície do
eletrodo (A)6,0 mm
5,0 mm
4,0 mm
3,0 mm
2,0 mm
1,0 mm
Contato
elétrico
B
C
D
E
F

40
que seguem exatamente as condições previstas pela equação de Nernst, (transporte
difusional na ausência de convecção) como pode ser visto na Figura 11. Experimentos
por voltametria cíclica correlacionando as correntes de pico com o aumento da velocidade
de varredura (Fig. 11a) mostram um comportamento linear quando é relacionada a
corrente com a raiz quadrada da velocidade de varredura. A distância entre o pico anódico
e catódico variou de 71 mV para 90 mV com o aumento da velocidade de varredura de 5
a 200 mV s-1. Os valores de corrente de pico anódico (IpA) e catódico (IpC) apresentaram
uma boa correlação linear com a raiz quadrada da velocidade de varredura (ν1/2), com
coeficiente (R2) de 0,995 e 0,983 respectivamente (Fig. 11b). A regressão linear entre o
log Ip vs. log ν mostrou uma boa concordância com os demais dados (Fig. 11c), com
resultados de coeficiente angular próximos a 0,5 (0,482 ± 0,007 e 0,51 ± 0,01),
concordando com uma cinética regida por transporte segundo a equação de Randles-
Sevcik [64]. Apesar do valor teórico de distância entre pico catódico e anódico para um
sistema reversível ser de 59,16 mV/e– [64], o valor encontrado para o eletrodo é bastante
satisfatório. Considerando os valores de corrente de pico anódico e catódico (IpA e IpC)
e ν1/2 (Fig. 11b), calculou-se a área efetiva do eletrodo para transferência de elétrons com
coeficiente de difusão de 7,20 x 10-6 cm2 s–1 [82] e 1,0 x 10-3 mmol L-1 de Fe(CN)63–,
resultando em uma área efetiva de 0,079 cm2 utilizando a equação de Randles-Sevcik
[64]. Considerando que a área geométrica do eletrodo é de 0,10 cm2, se o eletrodo fosse
completamente plano, haveria 79% da área condutora. Este valor está próximo a 75%,
teor de grafite utilizado na construção dos eletrodos.

41
Figura 11 – Influência da velocidade de varredura (ν) nas correntes de pico anódico e catódico
das ondas voltamétricas (IpC e IpA) de K3Fe(CN)6 0,10 mmol L-1 em KCl 0,10 mol L-1. (a)
Registros dos voltamogramas com aumento da velocidade de varredura: 5, 10, 20, 30, 40, 50, 75,
100, 150, 200 mV s-1. (b) Relação entre o pico anódico e catódico em função da raiz da velocidade
de varredura (dados obtidos de (a)). (c) Relação linear entre o log de IpA (e log IpC) e o log de ν
(dados obtidos de (a)).
A imagem da superfície do eletrodo pode ser visualizada com o uso da
microscopia eletrônica de varredura (Fig. 12). Observa-se que apesar de utilizar lixas de
diferentes granulometrias (partindo da maior para a menor), o polimento sobre papel não
é capaz de eliminar os riscos causados pelos grânulos da lixa, os quais não prejudicam a
resposta eletroquímica do eletrodo. As imagens obtidas por microscopia eletrônica (Figs.
12a e 12b) mostram regiões claras (não condutoras) e escuras (condutoras) indicam que
há uma certa heterogeneidade no compósito. Na Figura 12b há duas regiões distintas
demarcadas: A região “A” compreende uma região mais isolante em que os elétrons
-0.2 0.0 0.2 0.4 0.6 0.8
-30
-25
-20
-15
-10
-5
0
5
10
15
20
25
30
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl Sat))
Log IpA
Log IpC
-2,6 -2,4 -2,2 -2,0 -1,8 -1,6 -1,4 -1,2 -1,0 -0,8 -0,6
-5,4
-5,2
-5,0
-4,8
-4,6
y = -5,775 + 0,506x
R2 = 0,994
y = -5,702 + 0,482x
R2 = 0,998
Lo
g C
orr
ente
(lo
g A
)
Log (log V s-1)
0,1 0,2 0,3 0,4 0,5-30
-25
-20
-15
-10
-5
0
5
10
15
20
25
30
y = 3,629E-7 - 5,624E-5x
R2 = 0,983
y = 3,064E-7 + 5,685E-5x
R2 = 0,995
IpA
IpC
Co
rren
te (
A)
1/2
(V1/2
s-1/2
)
(a) (b)
(c)

42
emitidos pelo SEM se acumulam por não estar eficientemente aterrada. A região “B”
apresenta o contraste escuro, por conter uma concentração de grafite na superfície maior,
tornando-a facilmente aterrável por ser um material condutor.
Figura 12 – Imagem da superfície do eletrodo de grafite e poliestireno. (a) detalhamento dos
riscos provenientes do polimento na superfície do eletrodo. (b) Detalhamento da superfície do
eletrodo com aumento da ampliação. Regiões “A” e “B” são regiões com menor e maior condução
respectivamente.
Apesar da maioria dos dados apontarem positivamente para um comportamento
reversível da sonda Fe(CN)63–, a distância entre picos é ainda suscetível ao aumento da
velocidade de varredura e apresenta valores acima do teórico de 59,16 mV/e– a 25ºC.
Sabe-se que a velocidade de transferência de carga dos eletrodos de carbono é mais lenta
que a de eletrodos de metais nobres [83, 84], levando à voltamogramas mal definidos e
com menor reversibilidade, assim como para o processo Fe(CN)63–/Fe(CN)6
4– aqui
estudado. Até mesmo para eletrodos de ouro policristalino, a melhora da resposta
eletroquímica é bastante dependente do pré-tratamento químico da superfície do eletrodo
[85]. Para melhorar a resposta de eletrodos compósitos de fibra de carbono ou grafite-
epóxi, altas densidade de corrente (2 A cm–2) se mostraram eficientes, enquanto o
polimento da superfície para eletrodos de carbono vítreo ou outro material de carbono
com α-alumina aumenta a taxa de redução na superfície do ferricianeto [83]. O
200 μm 20 μm
(a) (b)
B
A

43
aquecimento dos substratos do carbono vítreo a 500ºC em pressões reduzidas também
melhora suas propriedades eletroquímicas. Então, a especiação dos grupos da superfície
nos eletrodos de carbono promove efeitos altamente pronunciados sobre o desempenho
observado na voltametria. Em outro estudo, foi observado o aumento da resposta
eletroquímica para polimentos em alta velocidade com partículas sucessivamente
menores de carbeto de silício, pasta de diamante e α-alumina, seguida de limpeza com
ultrassom. Tal desempenho foi atribuído a uma maior concentração de oxigênio na
superfície do eletrodo em comparação com a concentração no bulk ou mesmo em pontos
não ativos na superfície do eletrodo, o que foi especulado como sendo uma associação
benéfica de grupos fenólicos [86].
Após o começo das investigações do grafeno e do óxido de grafeno, a maioria
das relações entre a superfície dos grupos de carbono e a reatividade eletroquímica
ficaram mais claras [87]. Uma grande parte das características diferenciadas do óxido de
grafeno reduzido quimicamente e utilizado como sensor, provém de sua elevada
condutividade, associada a uma grande abundância de defeitos em sua estrutura bem
como em grupos funcionais presentes em sua estrutura [88-91]. Para o processo
Fe(CN)63–/Fe(CN)6
4– envolvendo eletrodos de carbono vítreo modificados com óxido de
grafeno reduzido quimicamente, a distância entre os picos variou de 61,5 para 73 mV,
utilizando a velocidade de 10 mV s–1 para diferentes eletrodos [88, 92-95], enquanto para
o eletrodo de grafite e poliestireno estudado com a mesma velocidade de varredura e
eletrólito suporte, a distância entre picos foi de 71 a 80 mV. É importante mencionar que
o par Fe(CN)63–/Fe(CN)6
4– é considerado uma sonda sensível à superfície, mas insensível
a óxidos [88, 96]. Isso sugere de que o eletrodo G-PS oferece um coeficiente de
transferência de carga para o processo de redução do ferricianeto a ferrocianeto
consistente com os eletrodos de oxido de grafeno quimicamente reduzidos, apesar da sua

44
simplicidade e baixíssimo custo de produção. Entendemos que se trata de um compósito
promissor.
4.3 Comparação entre eletrodos de carbono
A comparação do comportamento eletroquímico do eletrodo G-PS, eletrodo de
carbono vítreo (GCE) e eletrodo de pasta de carbono (CPE) em presença de um sistema
redox reversível de FeCN63- é apresentado na Figura 13a. Os valores de diferença de pico
de corrente foram 77 ± 7 mV para eletrodo compósito (n = 19), 85 ± 3 mV para eletrodo
de carbono vítreo (n = 3) e 121 ± 21 mV para eletrodo de pasta de carbono (n = 3). Todos
estes valores são significativamente diferentes do valor esperado de 59,16 mV para um
sistema redox reversível que envolve um elétron a 25 ° C, sendo que os eletrodos G-PS e
GCE apresentaram valores mais próximos do teórico. Os valores do CPE para a diferença
de pico podem ser explicados devido à sua maior resistência e menor velocidade na
transferência de carga [97-100] em comparação com os eletrodos GCE e G-PS. A relação
entre o pico de corrente anódica e catódica foi de (1,07 ± 0,03), (1,04 ± 0,01) e (1,16 ±
0,03) para G-PS, GC e CPE, respectivamente.
A comparação dos três eletrodos mostra uma baixa corrente de fundo para o
GCE, em comparação com G-PS e CPE (Fig. 13b), mesmo considerando sua área efetiva
calculada utilizando como base a cronoamperometria do ferricianeto (Fig. 13c) e a
equação de Cottrell [64]. O valor da área efetiva para G-PS, GCE e CPE, calculada com
a equação de Cottrell é respectivamente de (7,3 ± 0,3), (7,8 ± 0,4) e (10 ± 1) mm2.
Para avaliar a resistência de transferência de carga (RCT), um ensaio de
impedância foi realizado (Fig. 13d) e demonstrou valores de RCT bastante próximos para
GCE e G-PS, (0,77 ± 0,13) e (0,89 ± 0,03) kΩ, respectivamente. Um valor mais elevado

45
de RCT foi observado para o eletrodo de pasta de carbono (2,8 ± 0,7) kΩ devido à sua
maior resistência.
Figura 13 – Comparação entre os eletrodos G-PS, GC e CPE. (a) Voltamogramas cíclicos obtidos
para K3Fe(CN)6 1,0 mmol L-1 em KCl 0,10 mol L-1 e (b) somente em KCl 0,1 mol L-1. Velocidade
de varredura: 10 mV s-1. (c) Cronoamperometria obtida para K3Fe(CN)6 1,0 mmol L-1 em KCl
0,10 mol L-1 e fit linear comparando para cada um dos eletrodos. (d) Diagrama de Nyquist de
impedância eletroquímica utilizando K3Fe(CN)6 1,0 mmol L-1e K4Fe(CN)6 1,0 mmol L-1 em KCl
0,10 mol L-1. Os valores de quí-quadrado para G-PS, GC e CPE foram respectivamente (0,03 ±
0,02), (0,04 ± 0,02) e (0,13 ± 0,05).
Tabela 4 – Comparação entre eletrodos de carbono vítreo, pasta de carbono e compósito de grafite
e poliestireno com relação a área efetiva, rugosidade, diferença entre picos para uma solução de
K3Fe(CN)6 1,0 mmol L-1 em KCl 0,10 mol L-1 e resistência de transferência de carga (RCT).
Eletrodo
Área
geométrica
(mm2)
Área real (mm2)
(Cottrell) Rugosidade ΔEp (mV) RCT (kΩ)
Carbono
vítreo 7,1
7,8 ± 0,4 1,09
85 ± 3 0,77 ± 0,13
(n = 3) (n = 3) (n = 3)
G-PS 10,2 7,3 ± 0,3
0,72 77 ± 7 0,89 ± 0,03
(n = 3) (n = 3) (n = 19)
CPE 10,2 10 ± 1
0,98 121 ± 21 2,8 ± 0,7
(n = 3) (n = 3) (n = 3)
GCE
G-PS
CPE
Fit linear de GC
Fit linear de G-PS
Fit linear de CPE
0 2 4 6 8 10-60
-50
-40
-30
-20
-10
0
Corr
ente
(
A)
t-1/2
(s-1/2
)
(c)
GCE
G-PS
CPE
0 2 4 60,0
0,5
1,0
1,5
2,0
2,5
-Z
''(k
)
Z' (k)
(d)W
-0,2 0,0 0,2 0,4 0,6 0,8
-12
-8
-4
0
4
8
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl Sat.)
)
GCE
G-PS
CPE
(a) (b)
-0,2 0,0 0,2 0,4 0,6 0,8
-2,5
-2,0
-1,5
-1,0
-0,5
0,0
0,5
1,0
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl Sat.)
)
GCE
G-PS
CPE

46
Para verificar a janela de potencial do G-PS, foram realizados ensaios com 3
eletrólitos: HNO3, KNO3 e KOH (Fig. 14). Observa-se para as três soluções uma faixa de
trabalho menor para o G-PS e CPE em relação ao GCE. A reação que limita a região
anódica da faixa de trabalho dos eletrodos pode ser associada com a oxidação de H2O
para O2 e, para a região catódica, a redução de H2O para H2 [101].
• Região anódica:
2H2O O2 + 2H+ + 2e-
• Região catódica
2H2O + 2e- H2 + 2OH-
Os valores para os potenciais limite para os eletrodos G-PS, GCE e CPE são
apresentados na Tabela 5. Os valores de potencial limite da região anódica para o CPE e
G-PS são significantemente menores que os valores encontrados para o GCE, sendo este
processo atribuído a redução de oxigênio dissolvido a peróxido, juntamente com a
redução do peróxido a água.
O2 + 2H+ + 2e- H2O2
H2O2 + 2H+ + 2 e- 2H2O
Uma possível explicação para os resultados do G-PS e CPE, seria que eletrodos
com fases isolantes de natureza hidrofóbica (casos do poliestireno e do óleo mineral), têm
maior afinidade entre o oxigênio molecular e a superfície do eletrodo, levando à um efeito
eletrocatalítico através da reação de redução de oxigênio comparando com eletrodo de
carbono vítreo antecipando a onda de redução observada. Este aspecto precisa ser
elucidado, pois poderá levar à diversas aplicações interessantes [102].

47
Tabela 5 – Limites positivos e negativos referentes à janela de potencial de trabalho para os
eletrodos GCE, CPE e G-PS, sendo o último realizado também com soluções desareadas.
Limite negativo
Solução GCE (V vs.
Ag/AgCl)
CPE (V vs.
Ag/AgCl)
G-PS (V vs.
Ag/AgCl)
G-PS Desareado
(V vs. Ag/AgCl)
0,10 mol L-1
HNO3 –0,8 –0,15 –0,15 –0,60
0,10 mol L-1
KNO3 –1,4 –0,30 –0,20 –0,70
0,10 mol L-1
KOH –1,5 –0,30 –0,25 –0,25
Limite positivo
Solução GCE (V vs.
Ag/AgCl)
CPE (V vs.
Ag/AgCl)
G-PS (V vs.
Ag/AgCl)
G-PS Desareado
(V vs. Ag/AgCl)
0,10 mol L-1
HNO3 1,2 1,2 1,2 1,2
0,10 mol L-1
KNO3 1,1 1,1 1,1 1,1
0,10 mol L-1
KOH 0,6 0,6 0,6 0,5
Figura 14 - Faixa de trabalho para os eletrodos GCE, G-PS e CPE em (a) HNO3 0,10 mol L-1, (b)
KNO3 0,10 mol L-1 e (c) KOH 0,10 mol L-1. Velocidade de varredura: 50 mV s-1.
-1,0 -0,5 0,0 0,5 1,0 1,5 2,0
-60
-40
-20
0
20
40
60
Co
rren
te (
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
GC
G-PS
CPE
-2,0 -1,5 -1,0 -0,5 0,0 0,5 1,0 1,5 2,0-80
-60
-40
-20
0
20
40
60
80
100
120
Co
rren
te (
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
GC
G-PS
CPE
-2,0 -1,5 -1,0 -0,5 0,0 0,5 1,0-40
-20
0
20
40
60
80
Co
rren
te (
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
GC
G-PS
CPE
(a) (b)
(c)

48
Para verificar a influência do oxigênio em solução, foram realizados
experimentos sem e com prévia desaeração do sistema (Fig. 15) para HNO3, KNO3 e
KOH. Foi possível aumentar a janela de trabalho para o G-PS para as soluções de HNO3
(Fig. 15a) e KNO3 (Fig. 15b) em aproximadamente 0,5 V na região catódica (Tabela 5),
demonstrando a dependência da reação envolvida com o oxigênio dissolvido. O limite da
região catódica para a solução de KOH 0,10 mol L-1 não apresentou mudanças com a
desaeração do sistema (Fig. 15c), devido a um processo redox distinto. A Figura 15d
demonstra a resposta eletroquímica do G-PS em solução de KOH 0,10 mol L-1 em 2
varreduras consecutivas. Observa-se para a primeira varredura a ausência de picos de
oxidação até o limite da região anódica para este eletrodo, enquanto para a volta do ciclo,
apresenta uma onda de redução em aproximadamente 0,45 V. A partir do segundo
voltamograma, um pico de oxidação próximo a zero é observado, demonstrando uma
dependência da redução ocorrida na varredura anterior. Estes resultados sugerem uma
menor estabilidade do compósito em meio alcalino. Especula-se que alguns grupos
superficiais do polímero estejam na forma oxidada. Quando a varredura atinge a região
catódica (~0,4 V) estes grupos são reduzidos. No ciclo seguinte, ao alcançar a região
anódica (~0,2 V) tais grupos voltam a ser oxidados na superfície. Repetições do ciclo
mostra a repetição dos sinais da segunda varredura.

49
Figura 15 – Voltamogramas cíclicos usados para estimar potencial de limite positivo e negativo
para as faixas de trabalho do eletrodo G-PS em soluções com e sem prévia desaeração para (a)
HNO3 0,10 mol L-1, (b) KNO3 0,10 mol L-1 e (c) KOH 0,10 mol L-1. (d) Voltamogramas cíclicos
com scans progressivos para avaliação da dependência das reações redox em KOH 0,10 mol L-1.
Velocidade de varredura: 50 mV s-1.
4.4 Aplicações
A literatura descreve a utilização de eletrodos à base de carbono para analises de
um grande número de compostos químicos [103-113]. Os eletrodos compósitos
desenvolvidos neste estudo foram utilizados para a avaliação da resposta voltamétrica de
diversos compostos, dentre os quais: cisteína, ácido ascórbico, ácido gálico, peróxido de
hidrogênio, nitrito, riboflavina, dipirona e paracetamol, cujos CVs estão apresentados na
Figura 16.
Dentre estes compostos, dipirona e paracetamol foram escolhidos para estudos
seguintes mais detalhados e também em associação com um sistema de injeção em fluxo.
-1,0 -0,5 0,0 0,5 1,0
-120
-80
-40
0
40
80
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
G-PS
G-PS desareado
-1,0 -0,5 0,0 0,5 1,0 1,5-250
-200
-150
-100
-50
0
50
100
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
G-PS
G-PS desareado
-1,0 -0,5 0,0 0,5 1,0 1,5-40
-20
0
20
40
60
80
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
G-PS
G-PS desareado
(a) (b)
(c)
-0,8 -0,4 0,0 0,4 0,8
-50
0
50
100
150
200
Corr
ente
(
A)
Potencial aplicado (V vs. Ag/AgCl(KCl sat)
)
Scan 1
Scan 2
(d)

50
Figura 16 - Análise exploratória de cisteína, ácido ascórbico, ácido gálico, peróxido de
hidrogênio, riboflavina, nitrito de sódio, paracetamol e dipirona utilizando G-PS. Branco: Tampão
acetato 0,10 mol L-1 (pH 4,7). Velocidade de varredura: 50 mV s-1.
4.5 Limite de detecção e quantificação
O limite de detecção foi calculado a partir da relação conhecida: LD = 3Sb/m,
em que “Sb” é o desvio padrão de 10 injeções de branco e “m” é a inclinação da curva de
calibração. No caso da análise por injeção em fluxo, a medição do branco pode ser obtida
a partir da variação de sinal durante a passagem de eletrólito. Para avaliar o limite de
detecção, foram utilizadas soluções cujas concentrações se encontravam próximas do
valor de LD calculado a partir da somatória dos resíduos das curvas de calibração. Os
valores de LD obtidos para o paracetamol e a dipirona foram 5,08 e 1,32 µmol L–1 para o
FIA-amperometrico. Foram preparadas soluções com 1,3 vezes a concentração do LD
calculado, e o desvio padrão proveniente do sinal desenvolvido em 12 amostras foi
utilizado como “Sb”. Nesta aproximação, é considerado que tanto o branco, como LD e
1,3LD compreendem variáveis homocedásticas aleatórias.
Analito
Branco
Analito
Branco
Analito
Branco
-0,4 0,0 0,4 0,8 1,2
Analito
Branco
Potencial aplicado (V vs. Ag/AgCl(KCl Sat))
Analito
Branco
Analito
Branco
Ácido gálico
Ácido ascórbico
Cisteína
-0,4 0,0 0,4 0,8 1,2
Analito
Branco
Potencial aplicado (V vs. Ag/AgCl(KCl Sat))
Nitrito de sódio
Riboflavina
Peroxido de hidrogênio
-0,4 0,0 0,4 0,8 1,2
Analito
Branco
Potencial aplicado (V vs. Ag/AgCl(KCl Sat))
Dipirona
Paracetamol

51
As concentrações das soluções usadas foram de 6,88 μmol L-1 e 1,72 μmol L-1
para paracetamol e dipirona respectivamente.
Para o limite de quantificação (LQ), foi utilizado a mesma aproximação do
“Sb”, porém agora com a fórmula: LQ = 10Sb/m.
O estudo da faixa linear de resposta dos medicamentos foi realizado a partir de
injeções de soluções com concentração crescente a partir dos valores próximos ao limite
de quantificação calculados de 0,28 μmol L-1 para paracetamol e 0,67 μmol L-1 para
dipirona (Fig. 17a e 17b). Os potenciais utilizados para oxidação do paracetamol e
dipirona foram +0,7 V e +0,4 V, respectivamente. A escolha dos potenciais foi feita com
base nos sinais obtidos por voltametria cíclica (Fig. 16).

52
Figura 17 – Estudo de faixa linear de (a) paracetamol e (b) dipirona utilizando FIA-
Amperométrico com potenciais fixos em 0,70 V e 0,40 V respectivamente. Detalhamento das
concentrações mais baixas de (c) paracetamol (0,28 e 0,84 μmol L-1) e (d) dipirona (0,67 e 1,34
μmol L-1). Detalhamento de um único pico para verificar razão sinal ruído para 0,28 μmol L-1 de
paracetamol (e) e 0,67 μmol L-1 de dipirona (f). Velocidade de fluxo de 2,65 min L-1.
Foram preparadas soluções de com concentrações de 0,28; 0,84; 2,52; 7,56;
22,68; 68,0; 204 ;612 ;1840 ;2760 e 5510 μmol L-1 (a-k) para paracetamol e 0,67; 1,34;
2,68; 5,36; 10,7; 21,4; 42,8; 85,7; 171; 343; 686; 1370; 2740; 5490 μmol L-1 (a-n) para
dipirona.
608 616 624
15
18
21
Corr
ente
(n
)
Tempo (s)
192 198 204
108
111
114
Corr
ente
(n
)
Tempo (s)
400 600 800
0,01
0,02
0,03
0,04
b
Corr
ente
(
)
Tempo (s)
a
0 500 1000 1500 2000 2500 30000
2
4
6
8
10
12
14
e
k
j
i
h
f
g
dcba
Corr
ente
(
)
Tempo (s)
0 500 1000 1500 2000 2500 3000 3500 40000
1
2
3
4
a b c d e f gh
i
j
k
l
m
Corr
ente
(
)
Tempo (s)
n
200 400 600
0,08
0,10
0,12
b
Corr
ente
(
)
Tempo (s)
a
(a) (b)
(c) (d)
(e) (f)

53
As Figuras 17c e 17d demonstram o perfil do fiagrama para as soluções de
menor concentração utilizadas na faixa linear para dipirona e paracetamol (Fig. 17c e
17d), apresentando ruídos de grande intensidade em relação ao sinal, provenientes do
manuseio da válvula injetora. No caso do paracetamol, a concentração mais baixa
estudada (0,28 µmol L–1) apresenta valores próximo de 10 vezes o tamanho do sinal em
relação ao ruído da linha base para a concentração mais baixa (Fig.17e), o que caracteriza
esta concentração como uma boa aproximação do limite de quantificação, enquanto que
para a dipirona (0,67 µmol L–1), a relação do sinal com o ruído da linha base aparenta ser
em torno de 3 vezes, caracterizando esta condição como uma aproximação mais
consistente com o valor de limite de detecção (Fig.17f).
A Figura 18 apresenta a faixa linear para o paracetamol e a dipirona (Fig 18a e
18c) e a regressão linear com a curva de maior sensibilidade (Fig 18b e 18d). O
paracetamol e a dipirona possuem uma faixa linear para o eletrodo G-PS utilizando FIA-
amperométrico de 0,28 a 204 μmol L-1 e 2,7 a 86 μmol L-1. Para concentrações mais
elevadas, observa-se o desvio da linearidade, como seria de esperar.

54
Figura 18 – Sinais de corrente obtidas por FIA em uma ampla faixa de concentração de
paracetamol (a) e dipirona (c) e apresentação de suas respectivas regiões lineares (b e d).
Com o objetivo de se comparar a resposta eletroanalítica do eletrodo G-PS com
outros na literatura, a Tabela 6 reúne diversos parâmetros analíticos de outros eletrodos
de carbono [31, 114-119], utilizados em associação com análise em fluxo de paracetamol
e de dipirona.
0 1 2 3 4 5 60
2
4
6
8
10
12
14
Corr
ente
(
A)
Paracetamol (mmol L-1)
0 50 100 150 2000,0
0,5
1,0
1,5
2,0
Corr
ente
(
Paracetamol (mol L-1)
0 1 2 3 4 5 60
1
2
3
4
Co
rren
te (
A)
Dipirona (mmol L-1)
(a) (b)
(d)(c)
R2 = 0,999
R2 = 0,995
0 20 40 60 80 1000,0
0,1
0,2
0,3
0,4
Co
rren
te (
A)
Dipirona (mol L-1)
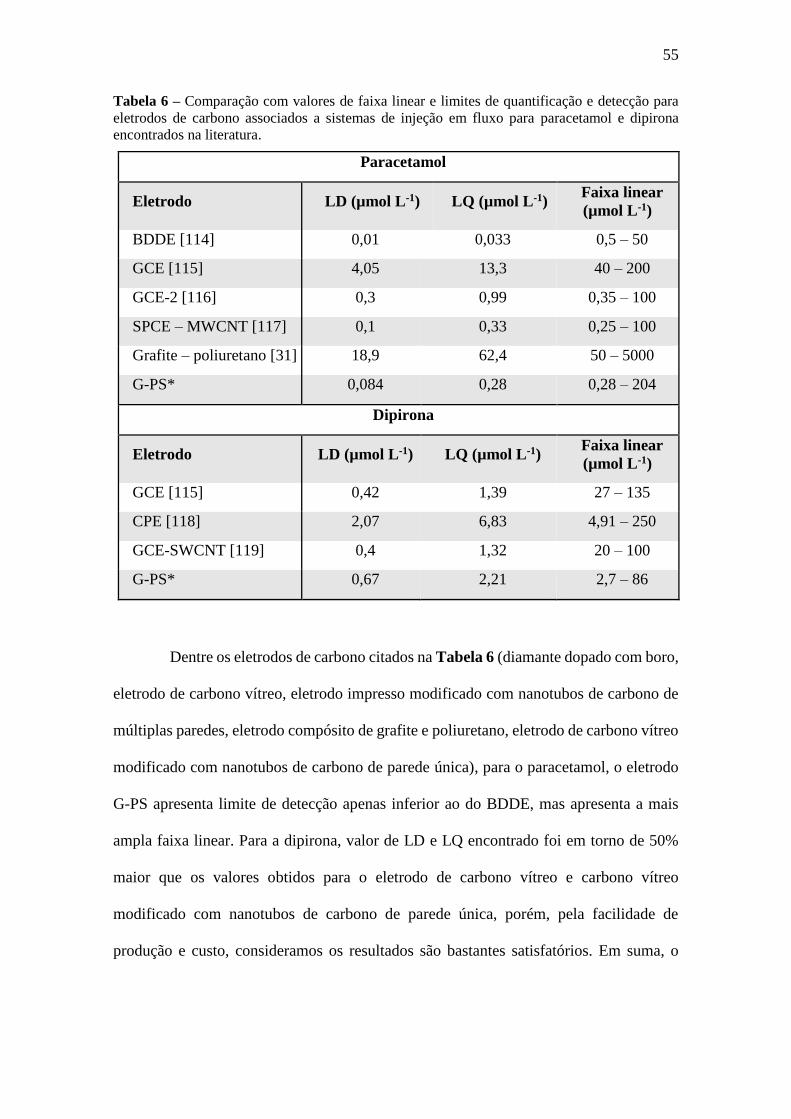
55
Tabela 6 – Comparação com valores de faixa linear e limites de quantificação e detecção para
eletrodos de carbono associados a sistemas de injeção em fluxo para paracetamol e dipirona
encontrados na literatura.
Paracetamol
Eletrodo LD (µmol L-1) LQ (µmol L-1) Faixa linear
(µmol L-1)
BDDE [114] 0,01 0,033 0,5 – 50
GCE [115] 4,05 13,3 40 – 200
GCE-2 [116] 0,3 0,99 0,35 – 100
SPCE – MWCNT [117] 0,1 0,33 0,25 – 100
Grafite – poliuretano [31] 18,9 62,4 50 – 5000
G-PS* 0,084 0,28 0,28 – 204
Dipirona
Eletrodo LD (µmol L-1) LQ (µmol L-1) Faixa linear
(µmol L-1)
GCE [115] 0,42 1,39 27 – 135
CPE [118] 2,07 6,83 4,91 – 250
GCE-SWCNT [119] 0,4 1,32 20 – 100
G-PS* 0,67 2,21 2,7 – 86
Dentre os eletrodos de carbono citados na Tabela 6 (diamante dopado com boro,
eletrodo de carbono vítreo, eletrodo impresso modificado com nanotubos de carbono de
múltiplas paredes, eletrodo compósito de grafite e poliuretano, eletrodo de carbono vítreo
modificado com nanotubos de carbono de parede única), para o paracetamol, o eletrodo
G-PS apresenta limite de detecção apenas inferior ao do BDDE, mas apresenta a mais
ampla faixa linear. Para a dipirona, valor de LD e LQ encontrado foi em torno de 50%
maior que os valores obtidos para o eletrodo de carbono vítreo e carbono vítreo
modificado com nanotubos de carbono de parede única, porém, pela facilidade de
produção e custo, consideramos os resultados são bastantes satisfatórios. Em suma, o

56
eletrodo G-PS pode reunir sensibilidade de materiais de carbono diferenciados com uma
ampla faixa linear presente nos compósitos convencionais.
4.6 Quantificação de paracetamol e dipirona
Os resultados obtidos a partir da amperometria, associados ao método de injeção
em fluxo para dipirona e paracetamol com a utilização do eletrodo G-PS foram
comparados com os obtidos com espectroscopia UV-Vis e os métodos padrão da
farmacopeia americana (USP) para o paracetamol [80] e farmacopeia brasileira para a
dipirona [81]. A metodologia desenvolvida foi aplicada para a análise de amostras reais.
Comprimidos contendo 750 mg de paracetamol e comprimidos com 500 mg de dipirona
foram analisados e os resultados comparados com os obtidos por outros métodos. O sinal
eletroquímico foi monitorado em +0,7 V vs. Ag/AgCl(sat.) para o paracetamol (Fig. 19a)
e +0,4 V vs. Ag/AgCl(sat.) para dipirona (Fig. 20a). Os resultados obtidos por FIA foram
(0,74 ± 0,02) g e (0,50 ± 0,02) g para paracetamol e dipirona, ao passo que os resultados
obtidos para os métodos oficiais (HPLC) foi de (0,754 ± 0,002) g para paracetamol e
(0,491 ± 0,009) g para dipirona (Titulação redox). Ainda, a análise dos dois compostos
foi feita por espectrofotometria, tendo sido obtidos UV-Vis (0,727 ± 0,003) g e (0,500 ±
0,004) g respectivamente. As Figuras 19 e 20 apresentam os fiagramas e os espectros
UV-Vis, bem como todas as curvas de calibração destas medições.
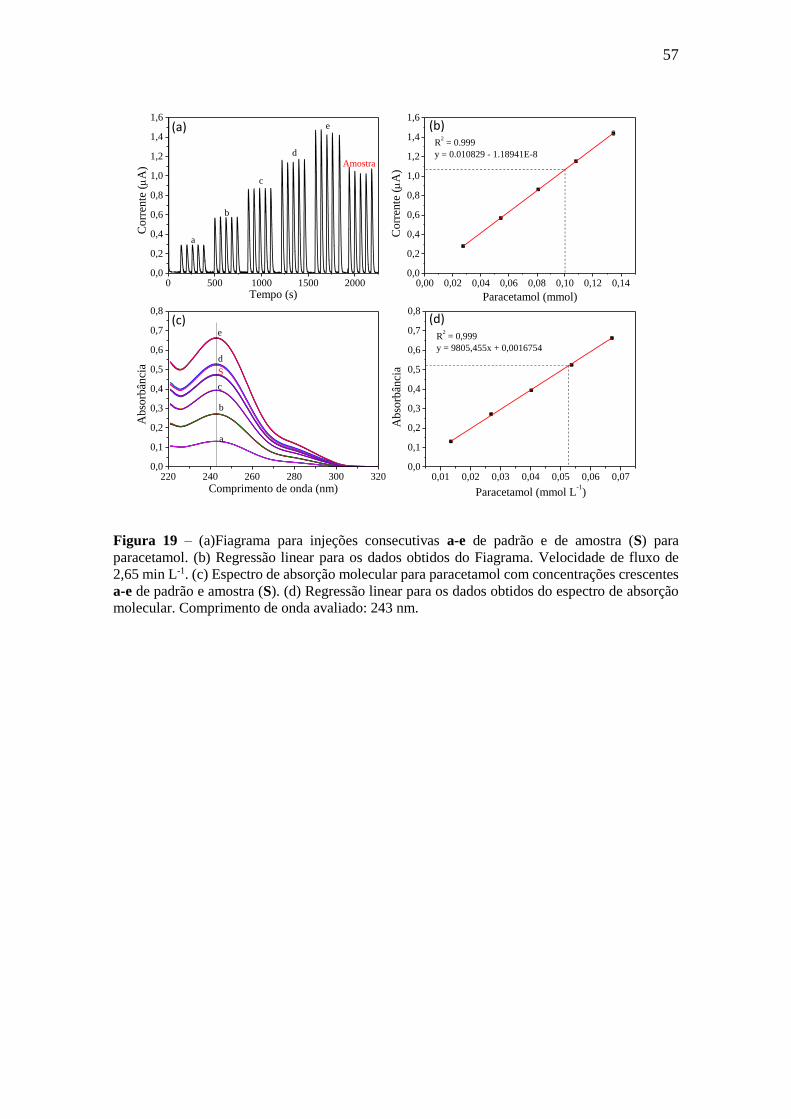
57
Figura 19 – (a)Fiagrama para injeções consecutivas a-e de padrão e de amostra (S) para
paracetamol. (b) Regressão linear para os dados obtidos do Fiagrama. Velocidade de fluxo de
2,65 min L-1. (c) Espectro de absorção molecular para paracetamol com concentrações crescentes
a-e de padrão e amostra (S). (d) Regressão linear para os dados obtidos do espectro de absorção
molecular. Comprimento de onda avaliado: 243 nm.
0 500 1000 1500 20000,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Amostra
e
d
c
b
Co
rren
te (
A)
Tempo (s)
a
0,00 0,02 0,04 0,06 0,08 0,10 0,12 0,140,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Co
rren
te (
A)
Paracetamol (mmol)
R2 = 0.999
y = 0.010829 - 1.18941E-8
0,01 0,02 0,03 0,04 0,05 0,06 0,070,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Ab
sorb
ânci
a
Paracetamol (mmol L-1)
R2 = 0,999
y = 9805,455x + 0,0016754
220 240 260 280 300 3200,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
S
c
d
b
e
Ab
sorb
ânci
a
Comprimento de onda (nm)
a
(a) (b)
(c) (d)

58
Figura 20 – (a)Fiagrama para injeções consecutivas a-e de padrão e de amostra (S) para dipirona.
(b) Regressão linear para os dados obtidos do Fiagrama. Velocidade de fluxo de 2,65 min L-1. (c)
Espectro de absorção molecular para dipirona com concentrações crescentes a-e de padrão e
amostra (S). (d) Regressão linear para os dados obtidos do espectro de absorção molecular.
Comprimento de onda avaliado: 258 nm.
Todas as curvas obtidas para o método eletroanalítico e UV-Vis demonstraram
uma boa correlação linear (R2 = 0,989 no mínimo). Os resultados provenientes do método
padrão e UV-Vis foram comparados com os do método eletroanalítico utilizando os testes
estatísticos (F e T), apresentando diferença entre as variâncias (teste F) para o paracetamol
mas não apresentando diferença entre os resultados das técnicas com 95% de confiança e
4 graus de liberdade (teste T) para o paracetamol e a dipirona (Anexo 3 e 4). A Tabela 7
resume os resultados obtidos para os diferentes métodos utilizados.
0 500 1000 1500 20000,0
0,1
0,2
0,3
0,4
0,5
0,6
Amostra
e
d
c
b
a
Co
rren
te (
A)
Time (s)
0,00 0,01 0,02 0,03 0,04 0,05 0,06 0,070,0
0,1
0,2
0,3
0,4
0,5
Corr
ente
(
A)
Dipirona (mmol L-1)
R2 = 0,989
y = 0,006373x + 3,17513E-8
220 230 240 250 260 270 2800,0
0,1
0,2
0,3
0,4
0,5
0,6
a
b
S
c
d
Ab
sorb
ânci
a
Comprimento de onda (nm)
e
(a) (b)
0,00 0,01 0,02 0,03 0,04 0,05 0,060,0
0,1
0,2
0,3
0,4
0,5
0,6
Ab
sorb
ance
Dipirona (mmol L-1)
R2 = 0.999
y = 8889.066x - 0.0054766
(d)(c)

59
Tabela 7 – Resultados para análise de paracetamol e dipirona utilizando o método padrão, Fia-
amperométrico e UV-Vis.
FIA – amperometrico
Concentração (mg) Slope (A L mol-1)
Paracetamol 740 ± 20 0,01083
Dipirona 500 ± 20 0,00637
Método padrão
Concentração (mg) Slope (A L mol-1)
Paracetamol 754 ± 2 -
Dipirona 491 ± 9 -
UV-Vis
Concentração (mg) Slope (L mol-1)
Paracetamol 727 ± 3 9805
Dipirona 500 ± 4 8889

60
Conclusões
O eletrodo G-PS apresentou uma boa resposta eletroquímica considerando seu
baixo custo de produção e boa homogeneidade intra e entre eletrodos. A resistência de
transferência de carga pode ser comparada ao eletrodo de carbono vítreo, se bem que a
janela de potencial é mais restrita. Esta nova forma de construção de eletrodos apresenta
potencial de aplicação em diversos fármacos e de outras espécies de interesse industrial,
clínica e ambiental. Há possibilidade de sua exploração para a análise de metais (o que
vai requerer a eliminação do oxigênio dissolvido), uma vez que sua janela de potencial é
limitada na região catódica). Paracetamol e dipirona foram quantificados pelo método
eletroquímico utilizando o eletrodo G-PS e os dados foram validados frente aos métodos
padrão da farmacopeia americana e brasileira estatisticamente.

61
Perspectivas
- Estudo mais detalhado da reação que ocorre no eletrodo em meio alcalino;
- Produção de eletrodos compósitos descartáveis impressos utilizando como base fita
adesiva (scotch tape);
- Quantificação dos analitos testados por voltametria cíclica em amostras reais;
- Modificação do eletrodo com nanotubos de carbono, carbon black, nanoparticulas de
ouro, etc;
- Modificação com enzimas para produção de biossensores;
- Desenvolvimento de eletrodo compósito utilizando uma fase condutora metálica em
lugar (ou associada) de grafite;
- Desenvolvimento de eletrodos de trabalho compósitos para células eletroquímicas de
microfluídica.

62
Referências bibliográficas
1. Heyrovský, J., Elektrolýza se rtuťovou kapkovou kathodou. Chemické Listy, 1922. 16: p. 256-264.
2. Adams, R.N., Carbon Paste Electrodes. Analytical Chemistry, 1958. 30: p. 1576. 3. Wang, J., Analytical Electrochemistry. Second edition ed. 2001: John Wiley & Sons, Inc. 4. Céspedes, F., E. MartÍnez-Fàbregas, and S. Alegret, New materials for electrochemical
sensing I. Rigid conducting composites. Trends In Analytical Chemistry, 1996. 15(7): p. 296-304.
5. Carrégalo, S., A. Merkoçi, and S. Alegret, Application of Graphite-Epoxy Composite Electrodes in Differential Pulse Anodic Stripping Voltammetry of Heavy Metals. Microchimica Acta, 2004. 147(4).
6. Corb, I., et al., Carbon-based Composite Electrodes: Preparation, Characterization and Application in Electroanalysis. Sensors, 2007. 7: p. 2626-2635.
7. Rassaei, L., et al., Carbon Nanofiber–Polystyrene Composite Electrodes for Electroanalytical Processes. Electroanalysis, 2007. 19(14): p. 1461-1466.
8. Tallman, D.E. and S.L. Petersen, Composite Electrodes for Electroanalysis: Principles and Applications. Electroanalysis, 1990. 2: p. 499-510.
9. da Silva, R.A.B., et al., Desenvolvimento, caracterização e aplicação eletroanalítica de um compósito fluido de adesivo epóxi, grafite e ciclo-hexanona. Química Nova, 2010. 33(6): p. 1398-1402.
10. Thiyagarajan, N., et al., Disposable electrochemical sensors: A mini review. Electrochemistry Communications, 2014. 38: p. 86-90.
11. Nascimento, V.B. and L. Angnes, Eletrodos Fabricados por “Silk-Screen”. Química Nova, 1998. 21(5): p. 614-629.
12. Navarro-Laboulais, J., et al., Voltammetric determination of optimal conductive load proportion in graphite-epoxy composite electrodes. Journal of Electroanalytical Chemistry, 1994. 379: p. 159-163.
13. Pumera, M., A. Merkoçi, and S. Alegret, Carbon nanotube-epoxy composites for electrochemical sensing. Sensors and Actuators B: Chemical, 2006. 113(2): p. 617-622.
14. Ye, R. and S.B. Khoo, Cathodic stripping voltammetric determination of ultratrace gold(iii) at a bulk modified epoxy–graphite tube composite electrode in flow systems. The Analyst, 1998. 124: p. 353-360.
15. Lawrence, N.S., et al., Carbon–epoxy electrodes: unambiguous identification of authentic triple-phase (insulator/solution/electrode) processesElectronic supplementary information (ESI) available: AFM image of the carbon–epoxy electrode. . Chemical Communications, 2002(10): p. 1028-1029.
16. Serra, B., et al., Composite electrochemical biosensors: a comparison of three different electrode matrices for the construction of amperometric tyrosinase biosensors. Biosensors & Bioelectronics, 2002. 17: p. 217-226.
17. Kirgöz, Ü.A., et al., A biosensor based on graphite epoxy composite electrode for aspartame and ethanol detection. Analytica Chimica Acta, 2006. 570(2): p. 165-169.
18. Šebková, S., T. Navrátil, and M. Kopanica, Graphite Composite Electrode in Voltammetry. Analytical Letters, 2005. 38(11): p. 1747-1758.
19. Wang, J. and K. Varughese, Polishable and Robust Biological Electrode Surfaces. Analytical Chemistry, 1990. 62(3): p. 318-320.
20. Wang, J. and M. Ozsoz, A Polishable Amperometric Biosensor for Bilirubin. Electroanalysis, 1990. 2: p. 647-650.
21. Onnerfjord, P., et al., Tyrosinase graphite-epoxy based composite electrodes for detection of phenols. Biosensors & Bioelectronics 1995. 10: p. 607-619.

63
22. Nacamulli, L. and E. Gileadi, Electrochemical properties of graphite fibre/epoxy composite electrodes. Journal of Applied Electrochemistry, 1983. 12: p. 73-78.
23. O’Hare, D., J.V. Macpherson, and A. Willows, On the microelectrode behaviour of graphite–epoxy composite electrodes. Electrochemistry Communications, 2002. 4: p. 245-250.
24. Calixto, C.M.F., et al., Development of Graphite-Polymer Composites as Electrode Materials. Materials Research, 2007. 10(2): p. 109-114.
25. Wang, J., et al., Polishable and Robust Modified Graphite Epoxy Electrodes. Analytical Chemistry, 1989. 61: p. 508-512.
26. Moreno-Baron, L., A. Merkoçi, and S. Alegret, Graphite-epoxy composite as an alternative material to design mercury free working electrodes for stripping voltammetry. Electrochimica Acta, 2003. 48(18): p. 2599-2605.
27. Ramírez-García, S., et al., Carbon composite electrodes: surface and electrochemical properties. The Analyst, 2002. 127(11): p. 1512-1519.
28. Cervini, P. and É.T.G. Cavalheiro, Graphite-Polyurethane Composite Electrode as an Amperometric Flow Detector in the Determination of Atenolol. Analytical Letters, 2008. 41(10): p. 1867-1877.
29. Semaan, F.S., et al., A Graphite-Polyurethane Composite Electrode for the Analysis of Furosemide. Electroanalysis, 2008. 20(21): p. 2287-2293.
30. de Toledo, R.A., et al., Use of Graphite Polyurethane Composite Electrode for Imipramine Oxidation—Mechanism Proposal and Electroanalytical Determination. Analytical Letters, 2006. 39(3): p. 507-520.
31. Cervini, P. and É.T.G. Cavalheiro, Determination of Paracetamol at a Graphite-Polyurethane Composite Electrode as an Amperometric Flow Detector. Journal of the Brazilian Chemical Society, 2008. 19(5): p. 836-841.
32. Mattioli, I.A., et al., Electrochemical investigation of a graphite-polyurethane composite electrode modified with electrodeposited gold nanoparticles in the voltammetric determination of tryptophan. Journal of Electroanalytical Chemistry, 2019. 835: p. 212-219.
33. Baccarin, M., P. Cervini, and E.T.G. Cavalheiro, Comparative performances of a bare graphite-polyurethane composite electrode unmodified and modified with graphene and carbon nanotubes in the electrochemical determination of escitalopram. Talanta, 2018. 178: p. 1024-1032.
34. Saciloto, T.R., P. Cervini, and É.T. Gomes Cavalheiro, New Screen Printed Electrode Based on Graphite and Polyurethane Composite for the Determination of Acetaminophen. Analytical Letters, 2013. 46(2): p. 312-322.
35. Saciloto, T.R., P. Cervini, and É.T.G. Cavalheiro, Simultaneous Voltammetric Determination of Acetaminophen and Caffeine at a Graphite and Polyurethane Screen-Printed Composite Electrode. Journal of the Brazilian Chemical Society, 2013.
36. Iarb, D., et al., Determination of Epinephrine at a Screen-Printed Composite Electrode Based on Graphite and Polyurethane. Journal of Analytical & Bioanalytical Techniques, 2017. 08(01).
37. Saciloto, T.R., P. Cervini, and É.T.G. Cavalheiro, Simultaneous Voltammetric Determination of Zn(II), Pb(II), Cu(II), and Hg(II) in Ethanol Fuel Using an Organofunctionalized Modified Graphite-Polyurethane Composite Disposable Screen-Printed Device. Electroanalysis, 2014. 26(12): p. 2664-2676.
38. Santos, S.X.d. and É.T.G. Cavalheiro, Using of a Graphite-Polyurethane Composite Electrode Modified with a Schiff Base as a Bio-Inspired Sensor in the Dopamine Determination. Journal of the Brazilian Chemical Society, 2014.
39. Albertús, F., et al., A PVC-graphite composite electrode for electroanalytical use. Preparation and some applications. Analytica Chimica Acta, 1997. 355: p. 23-32.

64
40. Mendes, R.K., S. Claro-Neto, and É.T.G. Cavalheiro, Evaluation of a new rigid carbon–castor oil polyurethane composite as an electrode material. Talanta, 2002. 57: p. 909–917.
41. Santos, S.X.d., L.H. Mazo, and É.T.G. Cavalheiro, The Use of a Graphite-Silicone Rubber Composite Electrode in the Determination of Rutin in Pharmaceutical Formulation. Journal of the Brazilian Chemical Society, 2008. 19(8): p. 1600-1606.
42. de Oliveira, A.C., S.X. dos Santos, and É.T.G. Cavalheiro, Graphite–silicone rubber composite electrode: Preparation and possibilities of analytical application. Talanta, 2008. 74(4): p. 1043-1049.
43. Doménech, A. and J. Alarcón, Determination of hydrogen peroxide using glassy carbon and graphite/polyester composite electrodes modified by vanadium-doped zirconias. Analytica Chimica Acta, 2002. 452: p. 11-22.
44. Doménech, A. and J. Alarcón, Electrochemistry of vanadium-doped tetragonal and monoclinic ZrO2 attached to graphite/polyester composite electrodes. Journal of Solid State Electrochemistry, 2002. 6(7): p. 443-450.
45. Mendonça, R.X.d., et al., Voltammetric Determination of Sildenafil Citrate and Furosemide at Composite Electrodes of Graphite-Paraffin for Use in Samples of Pharmaceutical and Toxicological Interests. Revista Virtual de Química, 2015. 7(5).
46. Freitas, K.H.G., R.A. Medeiros, and O. Fatibello-Filho, Voltammetric Determination of Rutin Using a Carbon Composite Electrode Modified with Copper(II)-Resin. Analytical Letters, 2009. 42(6): p. 881-897.
47. Cataldi, T.R.I. and D. Centonze, Development of a carbon composite electrode made from polyethylene and graphite powder modified with copper(I) oxide. Analytica Chimica Acta, 1996. 326: p. 107-115.
48. Cerro, M.A.d., et al., Graphite-Teflon-Peroxidase Composite Electrodes. Application to the Direct Determination of Glucose in Musts and Wines. Electroanalysis, 1997. 9(14): p. 1113-1119.
49. Serra, B., et al., Graphite-teflon-tyrosinase composite electrodes for the monitoring of phenolic compounds in predominantly non aqueous media. Analusis, 1999. 27(7): p. 592-599.
50. Serra, B., et al., Graphite-Teflon composite bienzyme electrodes for the determination of L-lactate: Application to food samples. Biosensors & Bioelectronics, 1999. 14: p. 505-513.
51. Pena, N., et al., Graphite-Teflon Composite Bienzyme Electrodes for the Determination of Cholesterol in Reversed Micelles. Application to Food Samples. Analytical Chemistry, 2001. 73.
52. Wang, J. and M. Musameh, Carbon Nanotube/Teflon Composite Electrochemical Sensors and Biosensors. Analytical Chemistry, 2003. 75: p. 2075-2079.
53. Serafín, V., et al., Electrochemical biosensor for creatinine based on the immobilization of creatininase, creatinase and sarcosine oxidase onto a ferrocene/horseradish peroxidase/gold nanoparticles/multi-walled carbon nanotubes/Teflon composite electrode. Electrochimica Acta, 2013. 97: p. 175-183.
54. Fernández, C., A.J. Reviejo, and J.M. Pingarron, Development of graphite-poly( tetrafluoroethylene) composite electrodes Voltammetric determination of the herbicides thiram and disulfiram. Analytica Chimica Acta, 1995. 305: p. 192-199.
55. Manea, F., et al., Electrochemical Oxidation and Determination of Oxalic Acid at an Exfoliated Graphite-Polystyrene Composite Electrode. Sensors, 2007. 7.
56. Xu, J., H. Zhang, and G. Chen, Carbon nanotube/polystyrene composite electrode for microchip electrophoretic determination of rutin and quercetin in Flos Sophorae Immaturus. Talanta, 2007. 73(5): p. 932-937.

65
57. Colilla, M., et al., Amino-polysiloxane hybrid materials as carbon composite electrodes for potentiometric detection of anions. Journal of Materials Chemistry, 2005. 15(35-36): p. 3844.
58. Afkhami, A., et al., New nano-composite potentiometric sensor composed of graphene nanosheets/thionine/molecular wire for nanomolar detection of silver ion in various real samples. Talanta, 2015. 131: p. 548-555.
59. Bagheri, H., et al., A new nano-composite modified carbon paste electrode as a high performance potentiometric sensor for nanomolar Tl(I) determination. Journal of Molecular Liquids, 2014. 197: p. 52-57.
60. Shirzadmehr, A., A. Afkhami, and T. Madrakian, A new nano-composite potentiometric sensor containing an Hg2+-ion imprinted polymer for the trace determination of mercury ions in different matrices. Journal of Molecular Liquids, 2015. 204: p. 227-235.
61. Safavi, A., et al., Ionic Liquids Modify the Performance of Carbon Based Potentiometric Sensors. Electroanalysis, 2007. 19(5): p. 582-586.
62. Rahimi, R., et al., Highly Stretchable Potentiometric pH Sensor Fabricated via Laser Carbonization and Machining of Carbon−Polyaniline Composite. ACS Applied Materials & Interfaces, 2017. 9(10): p. 9015-9023.
63. Brett, C.M.A. and A.M.O. Brett, Electrochemistry: principles, methods, and applications. 1993: Oxford University Press.
64. Bard, A.J. and L.R. Faulkner, Electrochemical Methods: Fundamentals and Applications. Second edition ed. 2001: John Wiley & Sons, Inc.
65. Skeggs, L.T., An Automatic Method For Colorimetric Analysis. American Journal of Clinical Pathology, 1957. 28(3): p. 311-322.
66. Trojanowicz, M. and K. Kołacińska, Recent advances in flow injection analysis. The Analyst, 2016. 141(7): p. 2085-2139.
67. Růžička, J. and E.H. Hansen, Flow Injection Analysis: Principles, Applications And Trends. Analytica Chimica Acta, 1975. 78: p. 147-157.
68. Zagatto, E.A.G. and C.C. Oliveira, Classificação e definição dos métodos de análises em fluxo (recomendações - iupac 1994). Química Nova, 1999. 22(1): p. 143-146.
69. Felix, F.S. and L. Angnes, Fast and Accurate Analysis of Drugs Using Amperometry Associated With Flow Injection Analysis. Journal of Pharmaceutical Sciences, 2010. 99(12): p. 4784-4804.
70. Stefano, J.S., et al., Flow-Injection Analysis with Multiple-Pulse Amperometry for Simultaneous Determination of Paracetamol and Naproxen Using a Homemade Flow Cell for Screen-Printed Electrodes. Journal of the Brazilian Chemical Society, 2014.
71. Matos, R.C., et al., Flow injection analysis-amperometric determination of ascorbic and uric acids in urine using arrays of gold microelectrodes modified by electrodeposition of palladium. Analytica Chimica Acta, 2000. 404: p. 151-157.
72. Murillo Pulgarín, J.A., L.F. García Bermejo, and A. Carrasquero Durán, Determination of Antioxidant Activity of Hibiscus Flowers by Flow Injection Analysis with Chemiluminescence Detection. Analytical Letters, 2016. 50(1): p. 186-196.
73. Spilstead, K.B., et al., Evaluation of coloured materials in microfluidic flow-cells for chemiluminescence detection. Analytica Chimica Acta, 2017. 968: p. 66-73.
74. Petrovich, M.B., V.R.A. Filho, and J.A.G. Neto, Direct determination of Calcium in milk by atomic absorption spectrometry using flow-injection analysis. Eclética Química, 2007. 32(3): p. 25-30.
75. Gómez, A.V.L. and J.M. Calatayud, Determination of cyanide by a flow injection analysis-atomic absorption spectrometric method. The Analyst, 1998. 123: p. 2103-2107.
76. Meneses, S., N. Maniasso, and E. Zagatto, Spectrophotometric flow-injection determination of sulphate in soil solutions. Talanta, 2005. 65(5): p. 1313-1317.

66
77. Rufino, J.L., et al., Flow-Injection Spectrophotometric Determination Of Tetracycline And Doxycycline In Pharmaceutical Formulations Using Chloramine-T As Oxidizing Agent. Química Nova, 2009. 32(7): p. 1764-1769.
78. Barsoukov, E. and J.R. Macdonald, Impedance Spectroscopy Theory, Experiment, and Applications. 2005: John Wiley & Sons, Inc.
79. Pedrotti, J.J., L. Angnes, and I.G.R. Gutz, Miniaturized reference electrodes with microporous polymer junctions. Electroanalysis, 1996. 8(7): p. 673-675.
80. United States Pharmacopeia 32 - NF 27. Vol. 32 - NF 27. 2009. 2221. 81. Farmacopeia Brasileira. 5 ed. Vol. 2. 2010. 1448. 82. Konopka, S.J. and B. McDuffie, Diffusion coefficients of ferri- and ferrocyanide ions in
aqueous media, using twin-electrode thin-layer electrochemistry. Analytical Chemistry, 1970. 42(14): p. 1741-1746.
83. Wightman, R.M., Methods to Improve Electrochemical Reversibility at Carbon Electrodes. Journal of The Electrochemical Society, 1984. 131(7): p. 1578.
84. Stutts, K.J., et al., Enhanced Electrochemical Reversibility at Heat-Treated Glassy Carbon Electrodes. Analytical Chemistry, 1983. 55: p. 1632-1634.
85. Carvalhal, R.F., R. Sanches Freire, and L.T. Kubota, Polycrystalline Gold Electrodes: A Comparative Study of Pretreatment Procedures Used for Cleaning and Thiol Self-Assembly Monolayer Formation. Electroanalysis, 2005. 17(14): p. 1251-1259.
86. Kamau, G.N., W.S. Willis, and J.F. Rusling, Electrochemical and Electron Spectroscopic Studies of Highly Polished Glassy Carbon Electrodes. Analytical Chemistry, 1985. 57: p. 545-551.
87. Ambrosi, A., et al., Electrochemistry of Graphene and Related Materials. Chemical Reviews, 2014. 114(14): p. 7150-7188.
88. Shao, Y., et al., Graphene Based Electrochemical Sensors and Biosensors: A Review. Electroanalysis, 2010. 22(10): p. 1027-1036.
89. Park, S. and R.S. Ruoff, Chemical methods for the production of graphenes. Nature Nanotechnology, 2009. 4(4): p. 217-224.
90. Stankovich, S., et al., Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon, 2007. 45(7): p. 1558-1565.
91. Schniepp, H.C., et al., Functionalized Single Graphene Sheets Derived from Splitting Graphite Oxide. Journal of Physical Chemistry B, 2006. 110(17): p. 8535-8539.
92. Yang, S., et al., A facile method for preparation of graphene film electrodes with tailor-made dimensions with Vaseline as the insulating binder. Electrochemistry Communications, 2009. 11(10): p. 1912-1915.
93. Tang, L., et al., Preparation, Structure, and Electrochemical Properties of Reduced Graphene Sheet Films. Advanced Functional Materials, 2009. 19(17): p. 2782-2789.
94. Shang, N.G., et al., Catalyst-Free Efficient Growth, Orientation and Biosensing Properties of Multilayer Graphene Nanoflake Films with Sharp Edge Planes. Advanced Functional Materials, 2008. 18(21): p. 3506-3514.
95. Wang, J., et al., Comparative studies on electrochemical activity of graphene nanosheets and carbon nanotubes. Electrochemistry Communications, 2009. 11(10): p. 1892-1895.
96. McCreery, R.L., Advanced Carbon Electrode Materials for Molecular Electrochemistry. Chemical Reviews, 2008. 108: p. 2646–2687.
97. Nicholson, R.S., Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics. Analytical Chemistry, 1965. 37(11): p. 1351-1355.
98. Morrin, A., A.J. Killard, and M.R. Smyth, Electrochemical Characterization of Commercial and Home-Made Screen-Printed Carbon Electrodes. Analytical Letters, 2003. 36(9): p. 2021-2039.

67
99. Urbaniczky, C. and K. Lundstrom, Voltammetric Studies on Carbon Paste Electrodes: The Influence of Paste Composition on Electrode Capacity and Kinetics. Journal of Electroanalytical Chemistry, 1984. 176: p. 169-182.
100. Chen, P., M.A. Fryling, and R.L. McCreery, Electron Transfer Kinetics at Modified Carbon Electrode Surfaces: The Role of Specific Surface Sites. Analytical Chemistry, 1995.
101. Dekanski, A., et al., Glassy carbon electrodes. Carbon, 2001. 39(8): p. 1195-1205. 102. Mounfield III, W.P., et al., Electrochemical Oxygen Reduction for the Production of
Hydrogen Peroxide. Chem 2018. 4(1): p. 18-19. 103. Enache, T.A. and A.M. Oliveira-Brett, Boron doped diamond and glassy carbon
electrodes comparative study of the oxidation behaviour of cysteine and methionine. Bioelectrochemistry, 2011. 81(1): p. 46-52.
104. Zhou, M., et al., Electrochemical Behavior of L-Cysteine and Its Detection at Ordered Mesoporous Carbon-Modified Glassy Carbon Electrode. Analytical Chemistry, 2007. 79: p. 5328-5335.
105. Hu, I.-F. and T. Kuwana, Oxidative Mechanism of Ascorbic Acid at Glassy Carbon Electrodes. Analytical Chemistry, 1986. 58(14): p. 3235-3239.
106. Ngai, K.S., et al., Voltammetry Detection of Ascorbic Acid at Glassy Carbon Electrode Modified by Single-Walled Carbon Nanotube/Zinc Oxide. International Journal of Electrochemical Science, 2013. 8: p. 10557 - 10567.
107. Abdel-Hamid, R. and E.F. Newair, Electrochemical behavior of antioxidants: I. Mechanistic study on electrochemical oxidation of gallic acid in aqueous solutions at glassy-carbon electrode. Journal of Electroanalytical Chemistry, 2011. 657(1-2): p. 107-112.
108. Aoki, K., M. Ishida, and K. Tokuda, Electrode kinetics of the oxidation of hydrogen peroxide at pretreated glassy carbon and carbon fiber electrodes. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1988. 251: p. 63-71.
109. Sophia, J. and G. Muralidharan, Amperometric sensing of hydrogen peroxide using glassy carbon electrode modified with copper nanoparticles. Materials Research Bulletin, 2015. 70: p. 315-320.
110. Kozub, B.R., N.V. Rees, and R.G. Compton, Electrochemical determination of nitrite at a bare glassy carbon electrode; why chemically modify electrodes? Sensors and Actuators B: Chemical, 2010. 143(2): p. 539-546.
111. Shiu, K.-K. and K. Shi, Selective Determination of Vitamin B2 at electrochemically Activated Glassy Carbon Electrode. Electroanalysis, 1999. 12(2): p. 134-139.
112. Bacil, R.P., et al., Mechanism of Electro-Oxidation of Metamizole using Cyclic Voltammetry at a Glassy Carbon Electrode. ECS Transactions, 2012. 43(1): p. 251-258.
113. Li, Y. and S.-M. Chen, The Electrochemical Properties of Acetaminophen on Bare Glassy Carbon Electrode. International Journal of Electrochemical Science, 2012. 7: p. 2171-2187.
114. Wangfuengkanagul, N. and O. Chailapakul, Electrochemical analysis of acetaminophen using a boron-doped diamond thin film electrode applied to flow injection system. Journal of Pharmaceutical and Biomedical Analysis, 2002. 28: p. 841–847.
115. dos Santos, W.T.P., et al., Simple Flow Injection Amperometric System for Simultaneous Determination of Dipyrone and Paracetamol in Pharmaceutical Formulations. Journal of the Brazilian Chemical Society, 2009. 20(7): p. 1249-1255.
116. Messina, G.A., I.E. De Vito, and J. Raba, On-line microfluidic sensor integrated with an enzyme-modified pre-cell for the monitoring of paracetamol in pharmaceutical samples. Analytica Chimica Acta, 2006. 559(2): p. 152-158.
117. Fanjul-Bolado, P., et al., Electrochemical study and flow injection analysis of paracetamol in pharmaceutical formulations based on screen-printed electrodes and carbon nanotubes. Anal Chim Acta, 2009. 638(2): p. 133-8.

68
118. Marcolino-Júnior, L.H., et al., Flow injection amperometric determination of dipyrone in pharmaceutical formulations using a carbon paste electrode. Il Farmaco, 2003. 58(10): p. 999-1004.
119. Rumin, M., et al., Voltammetry and Flow Injection Analysis with Amperometric Detection for Sensitive Sodium Metamizole Determination on Glassy Carbon Electrode Modified with SWCNTs/Nafion. ECS Journal of Solid State Science and Technology, 2016. 5(8): p. M3005-M3011.
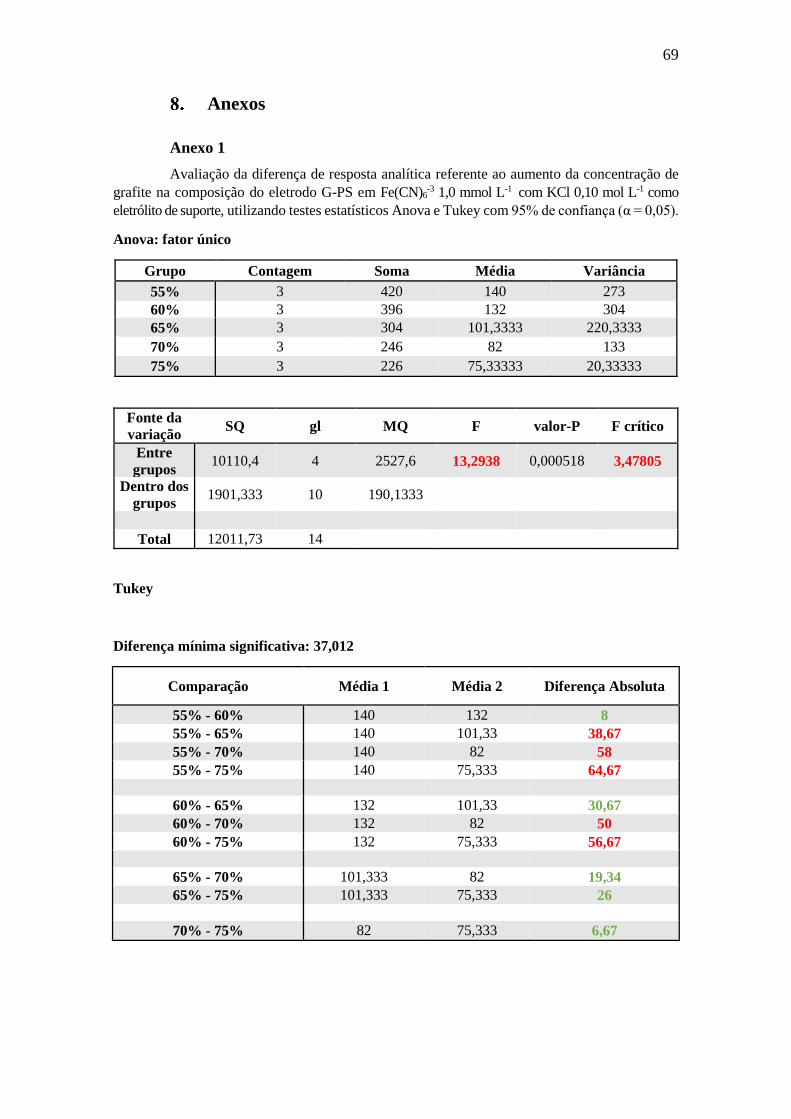
69
Anexos
Anexo 1
Avaliação da diferença de resposta analítica referente ao aumento da concentração de
grafite na composição do eletrodo G-PS em Fe(CN)6-3 1,0 mmol L-1 com KCl 0,10 mol L-1 como
eletrólito de suporte, utilizando testes estatísticos Anova e Tukey com 95% de confiança (α = 0,05).
Anova: fator único
Grupo Contagem Soma Média Variância
55% 3 420 140 273
60% 3 396 132 304
65% 3 304 101,3333 220,3333
70% 3 246 82 133
75% 3 226 75,33333 20,33333
Fonte da
variação SQ gl MQ F valor-P F crítico
Entre
grupos 10110,4 4 2527,6 13,2938 0,000518 3,47805
Dentro dos
grupos 1901,333 10 190,1333
Total 12011,73 14
Tukey
Diferença mínima significativa: 37,012
Comparação Média 1 Média 2 Diferença Absoluta
55% - 60% 140 132 8
55% - 65% 140 101,33 38,67
55% - 70% 140 82 58
55% - 75% 140 75,333 64,67
60% - 65% 132 101,33 30,67
60% - 70% 132 82 50
60% - 75% 132 75,333 56,67
65% - 70% 101,333 82 19,34
65% - 75% 101,333 75,333 26
70% - 75% 82 75,333 6,67
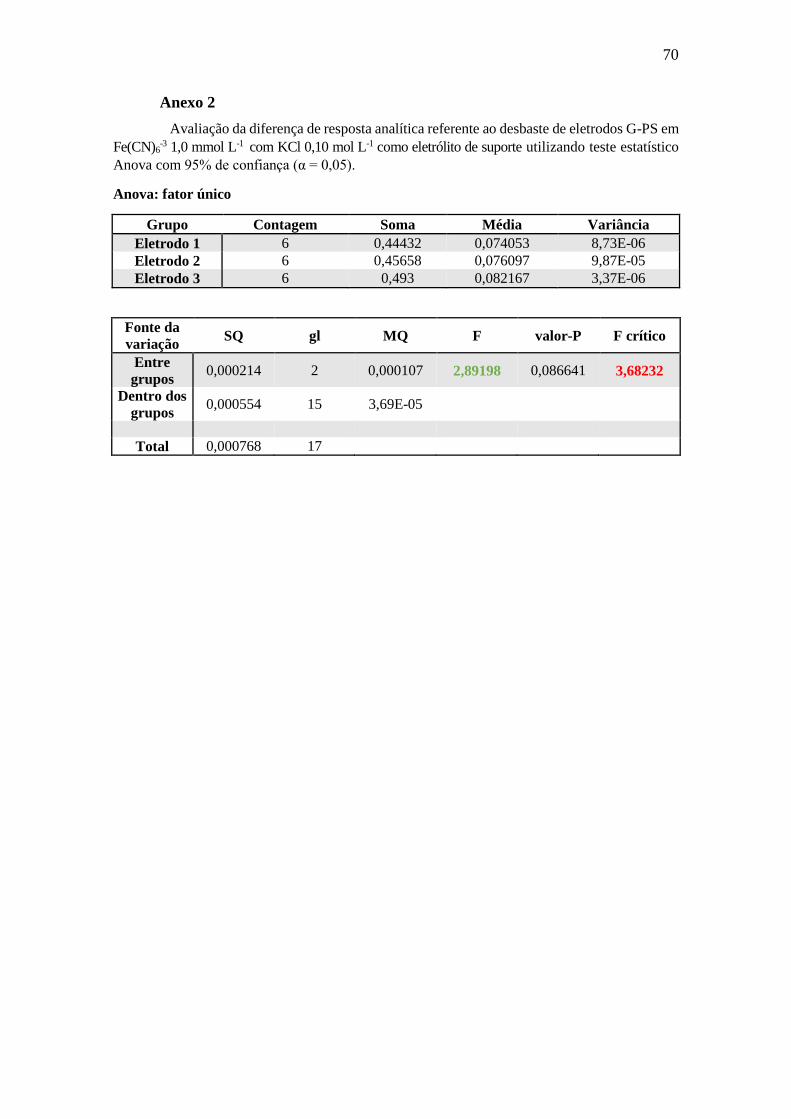
70
Anexo 2
Avaliação da diferença de resposta analítica referente ao desbaste de eletrodos G-PS em
Fe(CN)6-3 1,0 mmol L-1 com KCl 0,10 mol L-1 como eletrólito de suporte utilizando teste estatístico
Anova com 95% de confiança (α = 0,05).
Anova: fator único
Grupo Contagem Soma Média Variância
Eletrodo 1 6 0,44432 0,074053 8,73E-06
Eletrodo 2 6 0,45658 0,076097 9,87E-05
Eletrodo 3 6 0,493 0,082167 3,37E-06
Fonte da
variação SQ gl MQ F valor-P F crítico
Entre
grupos 0,000214 2 0,000107 2,89198 0,086641 3,68232
Dentro dos
grupos 0,000554 15 3,69E-05
Total 0,000768 17

71
Anexo 3
Resumo dos resultados da quantificação de paracetamol utilizando as 3 metodologias:
Uv-Vis, FIA amperométrico e HPLC.
UV-Vis (g) FIA (g) HPLC (g)
0,730 0,765 0,758
0,723 0,736 0,755
0,727 0,72 0,754
0,726 0,717 0,752
0,727 0,754 0,751
Avaliação das variâncias entre as técnicas (Teste F) e entre as médias dos resultados
(Teste T) para 95% de confiança (α = 0,05).
Teste – F: UV-Vis e FIA
UV-Vis FIA
Média 0,726653 0,738549
Variância 6,78E-06 0,000436
Observações 5 5
gl 4 4
F 0,01555
P(F<=f) uni-caudal 0,000696
F crítico uni-caudal 0,15654
Teste-F: Fia e HPLC
FIA HPLC
Média 0,738549 0,753699
Variância 0,000436 6,97E-06
Observações 5 5
gl 4 4
F 62,5495
P(F<=f) uni-caudal 0,000735
F crítico uni-caudal 6,38823

72
Teste-T: Presumindo variâncias equivalentes (UV-Vis e FIA)
UV-Vis FIA
Média 0,726653 0,738549
Variância 6,78E-06 0,000436
Observações 5 5
Variância agrupada 0,000221
Hipótese da diferença de média 0
gl 8
Stat t -1,264
P(T<=t) uni-caudal 0,120903
t crítico uni-caudal 1,859548
P(T<=t) bi-caudal 0,241806
t crítico bi-caudal 2,306
Teste-T: Presumindo variâncias diferentes (FIA e HPLC)
FIA HPLC
Média 0,738549 0,753699
Variância 0,000436 6,97E-06
Observações 5 5
Hipótese da diferença de média 0
gl 4
Stat t -1,6096
P(T<=t) uni-caudal 0,091389
t crítico uni-caudal 2,131847
P(T<=t) bi-caudal 0,182778
t crítico bi-caudal 2,77645

73
Anexo 4
Resumo dos resultados da quantificação de dipirona utilizando as 3 metodologias: Uv-
Vis, FIA amperométrico e HPLC.
UV-Vis (g) FIA (g) HPLC (g)
0,493 0,515 0,501
0,501 0,484 0,493
0,502 0,500 0,488
0,500 0,521 0,494
0,501 0,494 0,478
Avaliação das variâncias entre as técnicas (Teste F) e entre as médias dos resultados
(Teste T) para 95% de confiança (α = 0,05).
Teste-F: UV-Vis e FIA
UV-Vis FIA
Média 0,499665 0,502833
Variância 1,32E-05 0,000231
Observações 5 5
gl 4 4
F 0,05708
P(F<=f) uni-caudal 0,008433
F crítico uni-caudal 0,15654
Teste-F: FIA e HPLC
FIA HPLC
Média 0,502833 0,490888
Variância 0,000231 7,30E-05
Observações 5 5
gl 4 4
F 3,1571
P(F<=f) uni-caudal 0,145757
F crítico uni-caudal 6,38823
74
Teste-T: Presumindo variâncias equivalentes (UV-Vis e FIA)
UV-Vis FIA
Média 0,499665 0,502833
Variância 1,32E-05 0,000231
Observações 5 5
Variância agrupada 0,000122
Hipótese da diferença de média 0
gl 8
Stat t -0,4538
P(T<=t) uni-caudal 0,331014
t crítico uni-caudal 1,859548
P(T<=t) bi-caudal 0,662027
t crítico bi-caudal 2,306
Teste-T: Presumindo variâncias equivalentes (FIA e HPLC)
FIA HPLC
Média 0,502833 0,490888
Variância 0,000231 7,30E-05
Observações 5 5
Variância agrupada 0,000152
Hipótese da diferença de média 0
gl 8
Stat t 1,53304
P(T<=t) uni-caudal 0,081904
t crítico uni-caudal 1,859548
P(T<=t) bi-caudal 0,163807
t crítico bi-caudal 2,306

75
Súmula curricular
Andrei Martins Surkov
e-mail: [email protected]
Mestrando em Química Analítica pelo Instituto de Química de São Paulo (IQ/USP) –
desde - 02/2017
Graduado em Bacharelado em Química pela Faculdade Oswaldo Cruz – 2016
Técnico em Química pela Etec Irmã Agostina – 2012
Experiência profissional
Instituto de Química da Universidade de São Paulo – Monitor das disciplinas:
Fundamentos de Química experimental para graduação em química – 1º Semestre de
2017;
Química analítica para graduação em farmácia - 2º Semestre de 2017;
Química analítica para graduação em oceanoigrafia - 1º Semestre de 2018.
Artigo em elaboração
Very Low-Cost Graphite-Polystyrene Composites with Enhanced Electrochemical and
Electroanalytical Performances.
Congressos
Poster
Desenvolvimento de Eletrodos Compósitos de Carbono com Poliestireno para
Aplicações Eletroanalíticas no 19ºENQA/7ªCIAQA realizado de 16 a 19 de setembro
de 2018 em Caldas Novas/GO.
New low cost flexible screen-printed electrode as a sensor for electroanalytical
applications no 22ºSIBEE realizado 01-05 de setembro de 2019 em Ribeirão Preto/SP.
Apresentação oral
Very Low-Cost Graphite-Polystyrene Composite with Enhanced Electrochemical
and Electroanalytical Performances no 22ºSIBEE realizado 01-05 de setembro de
2019 em Ribeirão Preto/SP.