UNIVERSIDADE DO VALE DO ITAJAÍ CENTRO DE CIÊNCIAS ...siaibib01.univali.br/pdf/Fernanda...
112
UNIVERSIDADE DO VALE DO ITAJAÍ CENTRO DE CIÊNCIAS TECNOLÓGICAS DA TERRA E DO MAR CURSO DE CIÊNCIA DA COMPUTAÇÃO SISTEMA WEB PARA PROJETO E ANÁLISE DE PRIMERS Sistemas de Informação Fernanda Scariot Itajaí (SC), junho de 2004.
Transcript of UNIVERSIDADE DO VALE DO ITAJAÍ CENTRO DE CIÊNCIAS ...siaibib01.univali.br/pdf/Fernanda...

UNIVERSIDADE DO VALE DO ITAJAÍ
CENTRO DE CIÊNCIAS TECNOLÓGICAS DA TERRA E DO
MAR
CURSO DE CIÊNCIA DA COMPUTAÇÃO
SISTEMA WEB PARA PROJETO E ANÁLISE DE PRIMERS
Sistemas de Informação
Fernanda Scariot
Itajaí (SC), junho de 2004.

UNIVERSIDADE DO VALE DO ITAJAÍ
CENTRO DE CIÊNCIAS TECNOLÓGICAS DA TERRA E DO
MAR
CURSO DE CIÊNCIA DA COMPUTAÇÃO
RELATÓRIO DO TRABALHO DE CONCLUSÃO DE CURSO
SISTEMA WEB PARA PROJETO E ANÁLISE DE PRIMERS
Sistemas de Informação
Fernanda Scariot
Relatório apresentado à banca
examinadora do trabalho de conclusão
do curso de ciência da computação para
análise e aprovação.
Itajaí (SC), junho de 2004.

i
EQUIPE TÉCNICA
Acadêmica
Fernanda Scariot
Professor Orientador
Rafael Luiz Cancian, M.Sc.
Professor Co-orientador
André Oliveira de Souza Lima, Dr.
Coordenador dos Trabalhos de Conclusão de Curso
Anita Maria Fernandes da Rocha, Dra.
César Albenes Zeferino, Dr.
Coordenador do Curso
Luís Carlos Martins, Esp.

ii
DEDICATÓRIA
Dedico este trabalho aos meus pais que sempre
me incentivaram a seguir em frente e sempre
acreditaram no meu potencial.
Agradeço pelo amor infinito e pelas palavras de
alegria e força que recebi, mesmo estando longe.

iii
AGRADECIMENTOS
Agradeço a Deus pela vida, pela saúde e pela inteligência que me foi concedida, e por ter
enviado verdadeiros anjos para me acompanhar e me ajudar durante toda a vida.
Agradeço a minha mãe que sempre acreditou em mim e não deixou que eu desistisse em
nenhum momento e a meu pai que sempre teve gestos de amor incondicional. A meus irmãos que
sempre estiveram do meu lado e mostraram-se grandes amigos.
A meus magníficos amigos que sempre me auxiliaram, me fizeram sorrir, e nunca me
abandonaram. Em especial a Ariel, grandiosíssimo amigo, Carlos, meu "pai" e amigo de plantão, e
às minhas amigas Fernanda e Ana Paula, sempre grandes companheiras.
Agradeço a todos àqueles que se mostraram verdadeiros amigos em toda a minha vida
acadêmica. Descobri inúmeros novos amigos nessa caminhada...
Agradeço a meus mestres que me ensinaram e me auxiliaram em tudo que podiam, em
especial a Anita, Elisangela, Rudimar, Adhemar, e César.
A meu co-orientador professor André Lima e a pesquisadora Sheila, que contribuíram para
que esse trabalho fosse realizado.
Muito obrigada!

iv
SUMÁRIO
LISTA DE ABREVIATURAS E SIGLAS ............................................vii
LISTA DE FIGURAS ..............................................................................ix
LISTA DE TABELAS..............................................................................xi
RESUMO .................................................................................................xii
ABSTRACT ............................................................................................xiii
I – INTRODUÇÃO....................................................................................1
1. APRESENTAÇÃO.................................................................................................. 1
2. JUSTIFICATIVA.................................................................................................... 4
3. IMPORTÂNCIA ..................................................................................................... 5
4. OBJETIVOS ............................................................................................................ 6
4.1. Geral ....................................................................................................................... 6
4.2. Específicos.............................................................................................................. 6
5. METODOLOGIA ................................................................................................... 7
II – REVISÃO BIBLIOGRÁFICA........................................................10
1. MATERIAL GENÉTICO .................................................................................... 10
1.1. DNA e RNA ......................................................................................................... 11
1.2. Gene...................................................................................................................... 13
1.3. Proteínas ............................................................................................................... 14
1.4. Replicação ............................................................................................................ 16
1.5. Transcrição ........................................................................................................... 17
1.6. Tradução............................................................................................................... 18
1.7. Desnaturação e Renaturação do DNA.................................................................. 19
2. TÉCNICAS DE BIOLOGIA MOLECULAR .................................................... 20
2.1. Seqüenciamento e Clonagem ............................................................................... 20
2.2. PCR ...................................................................................................................... 21

v
2.2.1. Primers............................................................................................................... 24
2.3. Eletroforese .......................................................................................................... 26
3 – BIOINFORMÁTICA.......................................................................................... 27
3.1. Bancos de Dados Biológicos Públicos................................................................. 27
3.2. Alinhamento de Seqüências ................................................................................. 29
3.3. Árvores Filogenéticas........................................................................................... 32
3.4. Simuladores de Eletroforese................................................................................. 34
4 – SISTEMAS PARA WEB .................................................................................... 35
4.1. Perl........................................................................................................................ 36
4.1.1. Principais Módulos Perl .................................................................................... 37
4.1.2. BioPerl............................................................................................................... 38
4.2. HTML................................................................................................................... 39
4.2. PHP....................................................................................................................... 40
4.3. Banco de Dados.................................................................................................... 41
4.3.1. SQL ................................................................................................................... 42
4.3.2. MySQL.............................................................................................................. 42
4.3.3. PostgreSQL ....................................................................................................... 43
4.3.4. Comparação de Bancos de Dados ..................................................................... 44
III. DESENVOLVIMENTO...................................................................45
1. MODELAGEM DO SISTEMA ........................................................................... 45
1.1. Análise do Sistema ............................................................................................... 45
1.1.1. Diagrama de Contexto....................................................................................... 45
1.1.2. Diagramas de Fluxos de Dados......................................................................... 46
1.1.3. Dicionário de Dados Utilizados na Modelagem do Sistema ............................ 50
1.1.4. Diagrama de Casos de Uso................................................................................ 54
1.1.5. Diagrama de Classes ......................................................................................... 55
1.1.6. Diagramas de Seqüência ................................................................................... 57
1.1.7. Diagrama de Navegação do Sistema................................................................. 60

vi
1.1.8. Diagrama Entidade-Relacionamento ................................................................ 61
1.1.9. Dicionário de Dados das Tabelas ...................................................................... 63
1.2. Projeto do Sistema................................................................................................ 68
1.2.1. Especificação de Processos ............................................................................... 68
1.2.2. Modelo Físico do Banco de Dados ................................................................... 72
2. DESCRIÇÃO DO SISTEMA PROPOSTO........................................................ 74
2.1. Busca de Seqüências ............................................................................................ 75
2.2. Preparação de Seqüências .................................................................................... 80
2.3. Projeto de Primers ................................................................................................ 83
IV – CONCLUSÕES ...............................................................................89
BIBLIOGRAFIA .....................................................................................91
GLOSSÁRIO ...........................................................................................97

vii
LISTA DE ABREVIATURAS E SIGLAS
A Adenina
AIDS Síndrome da Imunodeficiência Adquirida
ASP Active Server Pages
BD Banco de Dados
BLOSUM Blocks Substitution Matrix
C Citosina
CGI Common Gateway Interface
DBI Database Interface
DNA Ácido Desoxirribonucléico
EMBL European Molecular Biology Laboratory
EST Expressed Sequence Tags
G Guanina
HIV Human Immunodeficiency Virus
HTML HyperText Markup Language
LWP.pm Library for Web Programming
NCBI National Center for Biotechnology Information
NIH National Institutes of Health
PAM Point Accepted Mutation
pb Pares de bases
PCR Polimerase Chain Reaction
PDB Protein Data Base
PDL Perl Data Language
Perl Practical Extraction and Report Language
RNA Ácido Ribonucléico
RNAm RNA mensageiro
RNAr RNA ribossômico
RNAt RNA transportador
RPM RPM Package Manager
SGBD Sistema Gerenciador de Banco de Dados
SQL Structured Query Language
T Timina

viii
Ta Temperatura de Anelamento
Tm Temperature Melting
U Uracila
UML Unified Modeling Language

ix
LISTA DE FIGURAS
Figura 1. Nucleotídeo Timina ............................................................................................................11
Figura 2. A Dupla hélice do DNA......................................................................................................12
Figura 3. Bases nitrogenadas que compõe o DNA.............................................................................13
Figura 4. Tabela de Tradução de Aminoácidos..................................................................................15
Figura 5. Replicação de cadeia de DNA ............................................................................................17
Figura 6. Reação em cadeia da polimerase – PCR.............................................................................22
Figura 7. Exemplo de gel de eletroforese...........................................................................................27
Figura 8. Alinhamento par-a-par entre seqüências realizado pelo Blast............................................30
Figura 9. Alinhamento múltiplo entre seqüências feito pelo ClustalW .............................................31
Figura 10. Árvore Filogenética feita pelo software ClustalW ...........................................................33
Figura 11. Menu de opções do pacote de programas Phylip..............................................................33
Figura 12. Tela do software Gel Electrophoresis Simulator ..............................................................34
Figura 13. Tela do software Simulation of Electrophoresis & Sequence Building of DNA .............35
Figura 14. Diagrama de Contexto. .....................................................................................................46
Figura 15. Diagrama de Fluxo de Dados, nível1................................................................................47
Figura 16. Diagrama de Fluxo de Dados decomposto do processo 1.4. ............................................48
Figura 17. Diagrama de Fluxo de Dados decomposto do processo 1.5. ............................................49
Figura 18. Diagrama de Fluxo de Dados decomposto do processo 1.6. ............................................50
Figura 19. Use Cases..........................................................................................................................54
Figura 20. Diagrama de Classes do Sistema ......................................................................................56
Figura 21. Diagrama de Seqüência com as ações iniciais do sistema................................................57
Figura 22. Diagrama de Seqüência do método DesignPrimers() .......................................................58
Figura 23. Diagrama de Seqüência do método _DesignIndividualPrimers().....................................58
Figura 24. Diagrama de Seqüência do método _DesignPairedPrimers()...........................................59
Figura 25. Diagrama de Seqüência do método _AnalysePrimers() ...................................................60
Figura 26. Diagrama de Navegação do Sistema ................................................................................61
Figura 27. Diagrama Entidade-Relacionamento, modelo Lógico......................................................62
Figura 28. Diagrama Entidade-Relacionamento, modelo Físico. ......................................................73
Figura 29. Tela de opções principais do sistema................................................................................74
Figura 30. Tela de parâmetros de busca de seqüências......................................................................76

x
Figura 31. Tela com o sumário de algumas seqüências encontradas. ................................................78
Figura 32. Tela de Digitação de Seqüências ......................................................................................79
Figura 33. Tela com o resultado do alinhamento feito pelo software ClustalW................................81
Figura 34. Figura com alinhamento feito pelo software ClustalW ....................................................83
Figura 35. Tela com os parâmetros de entrada para o projeto de primers. ........................................84
Figura 36. Tela com os parâmetros de entrada internos para o projeto de primers. ..........................85
Figura 37. Fitas sense e anti-sense .....................................................................................................86
Figura 38. Tela com os resultados do projeto de primers. .................................................................88

xi
LISTA DE TABELAS
Tabela 1. Tabela de Degenerações de Bases Nucléicas .....................................................................26
Tabela 2. Dicionário de Dados da Modelagem do Sistema. ..............................................................50
Tabela 3. Dicionário de Dados dos Fluxos de Dados. .......................................................................52
Tabela 4. Tabela Degeneracao ...........................................................................................................63
Tabela 5. Tabela Tipo_PCR ...............................................................................................................63
Tabela 6. Tabela Projeto_PCR ...........................................................................................................63
Tabela 7. Tabela Primer_Rejeitado....................................................................................................63
Tabela 8. Tabela Produto_Amplificado .............................................................................................64
Tabela 9. Tabela Corte .......................................................................................................................64
Tabela 10. Tabela Usuario .................................................................................................................64
Tabela 11. Tabela Enzima_Usada......................................................................................................65
Tabela 12. Tabela Enzima..................................................................................................................65
Tabela 13. Tabela Resultado_Alinhamento .......................................................................................65
Tabela 14. Tabela Organismo ............................................................................................................65
Tabela 15. Tabela Região_Similar .....................................................................................................66
Tabela 16. Tabela Similaridade..........................................................................................................66
Tabela 17. Tabela Posição_Anelamento ............................................................................................66
Tabela 18. Tabela Tipo_Est_Secundaria............................................................................................66
Tabela 19. Tabela Est_Secundaria .....................................................................................................66
Tabela 20. Tabela Primer ...................................................................................................................67
Tabela 21. Tabela Par_Primer ............................................................................................................67
Tabela 22. Tabela Codon ...................................................................................................................67
Tabela 23. Tabela Conjuntos_Organismos ........................................................................................67

xii
RESUMO
O presente trabalho descreve o desenvolvimento de um software para web que auxilia o
trabalho dos pesquisadores da área de biologia molecular, automatizando algumas das etapas
necessárias para o projeto e análise de primers, usados em reações de PCR (Polymerase Chain
Reaction). O desenvolvimento do software utilizou a linguagem de programação Perl e o módulo
BioPerl, específico para bioinformática, em conjunto com o banco de dados MySQL, e outros
softwares, como o ClustalW, executando em ambiente Linux. Por ser baseado em web, este
software pode ser acessao a partir de qualquer computador conectado à Internet, e em qualquer
plataforma. O software faz a busca de seqüências genômicas em bancos de dados públicos, a
preparação dessas sequências e o projeto de primers com base nas restrições impostas pelo
pesquiador, apresentando as características dos primers gerados e/ou analisados. Cada etapa é
automatizada, mas o software permite ainda a intervenção do pesquisador em cada passo, que pode
sempre usar sua experiência para corrigir problemas na automatização desse processo. O
mecanismo usado para o projeto de primers permite que se gerem primers confiáveis e específicos.
Os resultados apontam para um grande benefício aos pesquisadores dessa área, embora várias
características ainda possam ser incorporadas ao software.

xiii
ABSTRACT
This work describes the development of a web system that will help the work of molecular
biologists, automatizating some steps necessary to the Primer design and analisys, used in PCR
(Polymerase Chain Reaction). The development of software used the programming language Perl
and the BioPerl module, specific for bioinformatics, in set with the data base MySQL, and others
softwares, as the ClustalW, executing in Linux environment. For being based on web, this software
can be accessed from any computer conected to the internet, and in any platform. Software makes
the search of genomic sequences in public data bases, the preparation of these sequences and the
design of primers on the basis of the restrictions imposed for the research, presenting the generated
and/or analyzed characteristics of primers. Each stage is automatized, but software still allows the
intervention of the researcher in each step, that can always use his experience to correct problems
in the automatization of this process. The used mechanism for the design of primers allows that they
generate primers trustworthy and specific. The results point with respect to a great benefit the
researchers of this area, even so some characteristics still can be incorporated software.

I – INTRODUÇÃO
1. APRESENTAÇÃO
A bioinformática é uma nova ferramenta científica que une a bioquímica com a informática.
Criada nas últimas décadas para tratar a imensa massa de dados que vem sendo gerada pelos vários
projetos de genoma ao redor do mundo, a bioinformática permite explorar informações nunca antes
disponíveis, e pode gerar resultados nunca antes imaginados.
O termo bioinformática tem sido utilizado para descrever os processos computacionais
usados para dar apoio às pesquisas relacionadas com material genético (DNA, RNA e proteínas).
No mesmo sentido vem sendo utilizado o termo “Biologia Computacional” e, em geral, podem ser
considerados como sinônimos. Já o termo “Informática Aplicada à Biologia” é mais geral, e inclui,
além da bioinformática, qualquer sistema (software ou hardware) usado por pesquisadores da área
de ciências biológicas.
Em relação às pesquisas com material genético, alvo da bioinformática, o trabalho do
biólogo e pesquisador de área afim começa geralmente com o seqüenciamento de um trecho de
DNA (Ácido Desoxirribonucléico). O seqüenciamento pode ser feito de vários modos, mas, em
geral, o processo básico é o seguinte: uma amostra de DNA é retirada do organismo de interesse.
Esse DNA é fragmentado utilizando-se enzimas de restrição. Enzimas de restrição são proteínas
capazes de “cortar” a dupla fita de DNA em trechos específicos. Assim, obtêm-se vários pequenos
pedaços de DNA, capazes de serem seqüenciados com o equipamento adequado.
Como, em geral, a quantidade de DNA é pequena, é necessário “amplificar” (clonar) esse
DNA. Isso pode ser feito através de plasmídeos (técnica in vivo) ou PCR (técnica in vitro). PCR, ou
Polimerase Chain Reaction (Reação em Cadeia da Polimerase), permite aumentar milhões ou
bilhões de vezes a quantidade inicial de DNA. Para isso, são necessários apenas uma pequena
amostra de DNA, nucleotídeos livres, uma enzima chamada Taq DNA Polimerase e inicializadores
de polimeração, chamados primers.

2
Para o seqüenciamento, nucleotídeos especialmente marcados com algum indicador
fluorescente também são adicionados à amostra. Esses compostos são colocados num equipamento
chamado termociclador que, por simples mudanças de temperatura (aquece a 95oC e resfria a cerca
de 60oC, várias vezes), faz com que as moléculas de DNA se repliquem, aumentando a quantidade
da amostra. Após ter DNA suficiente, é necessário separar essa amostra por peso molecular, e assim
obter sua seqüência. Para isso utiliza-se o seqüenciador. A amostra de DNA é colocada num gel de
eletroforese, que consiste numa placa de gel ligada a uma fonte que gera corrente elétrica. As
amostras tendem, por efeito do campo eletromagnético, a atravessar o gel, e as moléculas menores o
fazem mais rapidamente. Na parte de baixo do gel, um feixe de laser do seqüenciador mede a
freqüência da luminescência do indicador fluorescente colocado em alguns nucleotídeos. Com isso,
cada nucleotídeo terminal é identificado e a seqüência é, posteriormente, determinada.
Embora a construção do seqüenciador já necessite do auxílio da informática, é apenas após
essa etapa que a bioinformática realmente começa. Inicialmente foram desenvolvidos softwares
para avaliar a qualidade das seqüências obtidas, uma vez que, por problemas tecnológicos, as
seqüências obtidas pelo seqüenciador não são perfeitas. Também por restrições tecnológicas, esse
equipamento é capaz de seqüenciar apenas trechos pequenos de DNA (por isso ele é inicialmente
fragmentado). Então, programas que sobrepõe as inúmeras pequenas seqüências (shotguns) obtidas
para gerar uma seqüência completa final (contig) foram desenvolvidos.
Com a obtenção das seqüências de DNA dos organismos de interesse, o trabalho do
pesquisador de biologia está recém começando. Em geral, para saber como o DNA (genótipo) se
relaciona com alguma característica física (fenótipo), várias outras etapas que usam bioinformática
são necessárias. Inicialmente, é necessário descobrir quais trechos desse DNA correspondem aos
genes, pois são os genes que serão transcritos em RNA (Ácido Ribonucléico) e esses então vão para
o citoplasma onde são traduzidos em proteínas, e são as proteínas, em primeira instância, as
responsáveis pela atividade biológica de um organismo e suas características físicas.
Inúmeros algoritmos foram desenvolvidos para a predição de genes, e hoje vários deles
podem ser usados através da web. Sabe-se que qualquer gene possui um códon (seqüência de 3
pares de bases) de inicialização de tradução e um códon de terminação. Além disso, eles possuem
uma região promotora, anterior ao códon de inicialização, onde a RNA polimerase se ligará para dar
início à transcrição do RNA. Como esses códons e região promotora podem variar para cada

3
organismo, e sua existência não necessariamente indica um gene real, tais algoritmos apenas são
uma indicação que tal trecho pode ser um gene, e é sempre necessária a comprovação em
laboratório “molhado” (biológico).
A seqüência de um gene determina a seqüência de aminoácidos da proteína que será gerada,
sendo que cada 3 (três) pares de base serão traduzidos para um aminoácido. Programas que fazem
essa tradução foram então desenvolvidos. Entretanto, a seqüência de aminoácidos de uma proteína
(sua estrutura primária), não é suficiente para caracterizá-la ou obter informações sobre seu
funcionamento no organismo. A proteína dobra-se sobre si, criando estruturas secundárias, e essas
também possuem uma conformação tridimensional (estrutura terciária). Em geral, é a conformação
tridimensional da proteína que determina sua função. Em muitas proteínas, entretanto, várias
estruturas terciárias precisam agrupar-se (estrutura quaternária) para que possam exercer sua função
(PANDOLFI, 2003). Vários algoritmos e softwares, cada vez mais complexos, foram desenvolvidos
para predizer as estruturas de uma proteína. Sempre, após a análise in silico (em computador), é
necessário uma comprovação in vitro.
As proteínas, após terem sido produzidas, passam por outras transformações, reagindo com
outras proteínas e moléculas no ambiente celular. Essas transformações geram as vias metabólicas
desse organismo. Estudos e análises do metabolismo estão sendo agora incluídos em programas de
bioinformática.
Outras técnicas recentes de bioinformática incluem a análise de transcriptoma, que são as
moléculas de RNA presentes no citoplasma. Com isso, tem-se certeza de que genes estão ativos e
pode-se, através de programas especializados, medir a taxa de expressão desses genes,
relacionando-os de diversas formas, principalmente em condições ambientais diferentes. Pode-se,
por exemplo, medir a taxa de expressão de centenas de genes numa célula normal e numa célula
cancerígena, e verificar quais genes estão relacionados com essa doença. Para isso utilizam-se
técnicas de hibridização automatizadas, chamadas micro-arrays.
Cada novo software de bioinformática desenvolvido abre caminho para mais pesquisas e
novas descobertas. Um dos maiores objetivos a ser alcançado pela bioinformática é a produção de
fármacos com base na estrutura das proteínas. Este resultado trará drogas mais eficazes e com
menor tempo para a entrada no mercado (PROSDOCIMI, 2003). Para que este e outros objetivos

4
sejam alcançados, iniciou-se o estudo de seqüências de DNA e genes (genômica), e atualmente seu
foco está na análise de transcritos (ESTs – Expressed Sequence Tags) e proteínas (proteômica). E
será focado nas vias metabólicas (metabolômica) em pouco tempo. Prevê-se que, num futuro não
muito distante, estará focada na fisiologia do organismo (fisiolômica ?).
O desenvolvimento de algoritmos e softwares para tratar com essas informações é um
desafio complexo e constante, mas um desafio não menos importante e às vezes desconhecido do
público em geral (inclusive biólogos) é a estruturação desses dados. A criação de bancos de dados,
sua estrutura, integridade, tratamento de dados replicados, etc, também faz parte da bioinformática,
e é apenas com a representação eficaz e eficiente desses dados, que os algoritmos para seu
tratamento podem ser usados.
Dada a importância e uso do experimento de PCR em diversas técnicas de biologia
molecular e da relativa dificuldade em determinar seus componentes, este trabalho apresenta o
desenvolvimento de um sistema de bioinformática para estruturação de dados e automatização de
processos biológicos, no que se refere ao projeto de experimentos de PCR, da busca de seqüências
genômicas ao projeto e análise dos primers usados no experimento. Com isso espera-se contribuir
com pesquisadores da área biológica, médica e farmacêutica, e também contribuir com o
aperfeiçoamento da ciência e pesquisa em bioinformática,
2. JUSTIFICATIVA
A bioinformática é uma ferramenta recente e em pleno desenvolvimento. Imagina-se que
será a nova ciência do século XXI. Embora sirva como ferramenta ao trabalho de biólogos (entre
outros profissionais, como médicos, farmacêuticos, veterinários e agrônomos), seu desenvolvimento
depende principalmente da atuação de profissionais de Ciência da Computação, capazes de
especificar os algoritmos para execução em computador. A participação desse tipo de profissional,
incluindo estudantes de graduação, é que permitirá a criação de novos algoritmos e ferramentas.
Sob aspectos computacionais, a bioinformática é um grande desafio. Em geral, profissionais
de computação são capazes de especificar algoritmos para problemas que tem solução/teoria
conhecida. Ou seja, é necessário saber resolver o problema “à mão” para que se possa ensinar o
computador a resolvê-lo. Entretanto, no caso da bioinformática, em geral, não se conhece a teoria

5
associada ao problema. Não é conhecido como o DNA, genes e proteínas realmente funcionam, e
ainda há variações entre organismos, entre tecidos, entre células e até mesmo numa única célula, ao
longo do tempo. Portanto, um trabalho nessa área é tema relevante de pesquisa, até mesmo em nível
de graduação.
Por não se conhecer exatamente o funcionamento de sistemas biológicos (organismos), os
programas de bioinformática geralmente utilizam técnicas de mineração de dados e inteligência
artificial. Uma boa modelagem é indispensável, principalmente no que se refere à organização de
bancos de dados. Devido à grande quantidade de dados, e à necessidade de muita análise sobre
esses dados, a bioinformática é uma aplicação potencial para algoritmos heurísticos e
processamento paralelo. Assim, também do ponto de vista computacional, trata-se de uma área
realmente interessante.
3. IMPORTÂNCIA
A importância de desenvolvimento em bioinformática tem sido amplamente divulgada, e é
quase consenso mundial que ela pode ser capaz de resolver inúmeros problemas altamente
relevantes, dentre os quais apenas alguns serão mencionados.
Em relação à agricultura, a bioinformática e a engenharia genética estão melhorando a
qualidade dos alimentos, estão tornando-os mais resistentes a pragas e mais produtivos. Isso tem
aumentado, num nível nunca antes visto, a quantidade de alimento produzido no mundo. Como
aumento populacional dos últimos séculos, apenas o melhoramento genético é capaz de produzir
alimento para todos. O mesmo tem ocorrido à pecuária, onde o melhoramento de animais tem
produzido rebanhos mais resistentes e mais adequados ao consumo. Novas proteínas têm sido
introduzidas no leite bovino através de modificações genéticas nos animais (BACALTCHUK,
2003).
Em relação à espécie humana, a importância da bioinformática é ainda maior. A
identificação de problemas genéticos já é uma realidade, e genes responsáveis por inúmeras doenças
já foram identificados, como os genes GPRA e AAA1, presentes no cromossomo 7 e responsáveis
pela asma (AGENCIA EFE, 2004). Com o avanço da bioinformática, da genômica para a

6
proteômica e metabolômica, além da identificação de tais genes, será possível o projeto racional de
fármacos, atuando diretamente em nível genético. Com isso, não só a expectativa de vida será
maior, mas a própria qualidade de vida do ser humano será melhor.
Especificamente em relação a este trabalho, ele é de grande importância para inúmeros
pesquisadores que trabalham com biologia molecular, virologia e bioquímica. A idéia de sua
realização surgiu da necessidade de dois pesquisadores, um do Laboratório de Biologia Molecular
da UNIVALI e outro do Laboratório de Virologia da UEL (Universidade Estadual de Londrina). O
trabalho desses profissionais será significativamente facilitado, sendo concluído num intervalo de
tempo muito menor e com qualidade muito superior.
Este trabalho permitirá que os pesquisadores façam, num único ambiente web, a busca de
seqüências genômicas, o alinhamento dessas seqüências e o projeto dos primers que serão usados
em experimentos de PCR. Um avanço importante proposto neste trabalho é a análise da
especificidade dos primers, garantindo que eles anelarão em certas seqüências e não em outras.
Com isso, torna-se significativamemte mais fácil o projeto de primers que amplifiquem apenas
certas estirpes ou mutações de organismos de interesse. Como o trabalho está disponível na Web
para todos os pesquisadores do mundo, este sistema agilizará o trabalho de muitos biólogos e
pesquisadores de áreas afins.
4. OBJETIVOS 4.1. Geral
O objetivo geral deste trabalho é desenvolver um sistema baseado em web para busca de
seqüências genômicas, projeto e análise de primers para experimentos de PCR.
4.2. Específicos
• Armazenar e manipular, em banco de dados no servidor, de forma hierárquica, seqüências
de DNA, RNA, primers, enzimas de restrição e anotações genômicas.

7
• Acessar automaticamente, via internet, bancos de dados genômicos públicos, e diversas
ferramentas disponíveis (ClustalW, Blastn, Blastp). Acessar várias ferramentas já
disponíveis em forma de pipeline ou “linha de produção”. Comparar as seqüências locais
de DNA e RNA com seqüências em bancos de dados públicos: Blastn, par-a-par e
alinhamento múltiplo.
• Analisar um primer, apresentando: tamanho, peso molecular, temperatura de hibridização,
complementaridade, estruturas secundárias internas, entropia, entalpia, etc. Avaliar a
qualidade de primers segundo parâmetros pré-estabelecidos (restrições do usuário).
Definir o trecho da seqüência que o primer deve alinhar e tamanho de amplicon desejado.
Permitir a inclusão de sítios de restrição e bases degeneradas nos primers.
5. METODOLOGIA
Foram necessárias cinco etapas a fim de executar o projeto: estudo, modelagem,
desenvolvimento, validação e divulgação dos resultados. As etapas de estudo e modelagem foram
efetivadas no TCC1, sendo que as etapas de desenvolvimento, validação e divulgação científica
foram efetivadas no TCC2.
Essas são as etapas gerais de trabalhos científicos que usam o método hipotético-dedutivo. A
etapa de estudo compreende a definição clara do problema, o escopo do sistema e aquisição do
conhecimento prévio sobre o tema e soluções existentes. A etapa de modelagem consiste em
definição de nova hipótese ou proposta de nova solução ao problema estudado. A etapa de
desenvolvimento trata da experimentação da hipótese ou desenvolvimento da nova solução. A
validação visa corroborar a hipótese desenvolvida, além de dar base científica à solução
desenvolvida. Por fim, todo trabalho científico deve ser descrito de tal forma que possa ser
reproduzido e testado por outros pesquisadores.
Etapa 1 – Estudo – Definição do problema e revisão de soluções/técnicas existentes
Nesta etapa buscou-se compreender exatamente os conceitos envolvidos no trabalho, como
PCR, primers, eletroforese, alinhamento par-a-par e alinhamento múltiplo, homologia, genes

8
ortólogos e parólogos, etc. Esse conhecimento prévio foi necessário para entender o processo de
trabalho dos pesquisadores em biologia molecular. Posteriormente, foi necessário estudar as
ferramentas de bioinformática já desenvolvidas e que foram necessárias neste trabalho, como o
ClustalW e o GeneRunner. Além disso, foi necessário saber utilizar os bancos de dados biológicos
públicos, como o GenBank, disponível no site do NCBI (National Center for Biotechnology
Information) e o Expasy.
Para o desenvolvimento, foi necessário estudar também a criação e manutenção de bancos
de dados com MySQL, a linguagem de programação Perl (Practical Extraction and Report
Language), amplamente utilizada em bioinformática, e o ambiente Linux, no qual executam as
ferramentas de bioinformática que foram usadas neste trabalho. Por fim, também foi necessário ter
bom conhecimento do funcionamento do alinhamento múltiplo, bem como de todas as condições
para o projeto de primers diversos.
Etapa 2 – Modelagem – Levantamento de hipóteses e Proposta de nova solução
A etapa de modelagem compreende a especificação tanto dos bancos de dados quanto da
aplicação em si. A estrutura do banco de dados considerou os organismos, as seqüências de DNA,
enzimas de restrição, primers e seqüência consenso. A modelagem do sistema deve especificar
completamente o funcionamento do mesmo, e inclui tanto a Análise quanto o Projeto do Sistema.
Para tanto foi utilizada a Análise Estruturada para as etapas de busca de seqüências e preparação das
seqüências para projeto de primers, e análise orientada a objeto, e sua descrição com a linguagem
UML (Unified Modeling Language) para a etapa de projeto de primers.
Etapa 3 – Desenvolvimento – Experimentação – Implementação da solução proposta
O desenvolvimento compreende a implementação do sistema web, seguindo rigorosamente a
modelagem da etapa anterior. A implementação foi realizada em forma de componentes (módulos)
independentes, de forma a facilitar a depuração, testes, manutenção e reuso dos mesmos. Na
implementação do sistema foram utilizadas as classes de objetos da biblioteca BioPerl, já
desenvolvida por pesquisadores e disponível para bioinformatas. Todo o sistema foi executado a
partir de browsers, o que permite seu uso a qualquer pesquisador em qualquer plataforma de
hardware.

9
São acessados bancos de dados públicos, como o NCBI (NATIONAL CENTER FOR
BIOTECHNOLOGY INFORMATION, 2003), através da interface do sistema. Quando é acessada
a página do NCBI, tem-se como resposta uma página em HTML (HyperText Markup Language)
com os dados da pesquisa. A partir desta página são tiradas as informações que o usuário deseja.
Esta tarefa é simplificada quando se utiliza a linguagem Perl, que é excelente para o tratamento de
strings (seqüências de caracteres).
O objetivo de implementar a etapa de pesquisa em banco de dados públicos é de facilitar
tarefas que exigem que dados obtidos de um sistema, como o NCBI sejam enviados para outras
ferramentas, como o ClustalW. Assim, em uma única interface, teremos acesso a várias ferramentas
e vários processos serão interligados. Quando o usuário acessa a página do sistema, ele fornece
dados para que sejam processados pelo servidor. Os dados são então processados no servidor e
enviados apenas os resultados para o browser do usuário. Os dados obtidos através de pesquisa em
banco de dados públicos e outras ferramentas são armazenados nos bancos do sistema (no servidor)
para futuras consultas e para gerar relatórios com estatísticas pertinentes à pesquisa, como por
exemplo, o número de determinada base nucléica em algum trecho de DNA.
Etapa 4 – Validação – Teste das hipóteses levantadas – Validação da solução implementada
A etapa de validação e testes compreendeu a comparação dos resultados obtidos com o
sistema com resultados obtidos através de outros softwares existentes no mercado, como o
GeneRunner. E quando houver a possibilidade de realizar a experiência em laboratório serão
comparados os resultados gerados pelo software aos resultados obtidos na prática. O laboratório de
Bioquímica e Biologia Molecular usado será um laboratório específico da área de ciências
biológicas da UNIVALI, que tenha o material necessário para a validação.
Etapa 5 – Divulgação – Corroboração da nova solução desenvolvida
A divulgação dos resultados está sendo feita via página web, em servidor Apache. O
material sobre o projeto está disponível com o sistema.

II – REVISÃO BIBLIOGRÁFICA
1. MATERIAL GENÉTICO A evolução do planeta Terra produziu uma imensa diversidade de formas vivas. São
aproximadamente quatro milhões de espécies de animais, vegetais, protozoários e bactérias, cujos
comportamentos, morfologia e funções diferem um dos outros. As biologias celular e molecular
estudam a organização que é comum entre os seres vivos. Elas analisam as moléculas e os
componentes celulares com que são construídas todas as formas de vida (DE ROBERTIS e HIB,
2001).
A célula é considerada, por muitos autores, a menor unidade de vida, contendo as
características morfológicas e fisiológicas de todos os organismos vivos. Atualmente, aceita-se que
o gene seja a menor unidade de vida, desde que devidamente definido (DAWKINS, 1979). As
propriedades de um organismo vivo dependem de suas células individuais, cuja continuidade ocorre
através de seu material genético. As células são divididas em dois grandes grupos: as procarióticas e
as eucarióticas. As células procarióticas são estruturalmente mais simplificadas que as células
eucarióticas (ZAHA, 1996, p.13) sendo que a principal diferença entre ambos os tipos celulares
consiste no fato que os procarióticos não possuem membrana nuclear (DE ROBERTIS e HIB, 2001,
p.3).
Os componentes químicos das células são classificados em orgânicos (ácidos nucléicos,
hidratos de carbono, lipídeos e proteínas) e inorgânicos (água e sais minerais). A maior parte das
estruturas celulares contém lipídeos e moléculas muito grandes, denominadas polímeros, integrados
por monômeros, que se prendem entre si por ligações covalentes (ibidem).
Nos organismos existem três importantes polímeros: polissacarídeos, cujos monômeros são
os açúcares, os ácidos nucléicos, formados pelos nucleotídeos (monômeros), e as proteínas,
formadas pelos aminoácidos (ZAHA, 1996, p.18). Os polissacarídeos (hidratos de carbono),
compostos de carbono, hidrogênio e oxigênio, são a principal fonte de energia da célula (DE

11
ROBERTIS e HIB, 2001, p.25). Os ácidos nucléicos serão estudados mais profundamente no
decorrer do texto.
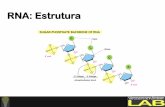
1.1. DNA e RNA
Ácidos nucléicos são polímeros lineares de nucleotídeos, unidos por ligação fosfodiéster.
Existem dois tipos de ácidos nucléicos: ácido desoxirribonucléico (DNA) e ácido ribonucléico
(RNA). Cada nucleotídeo é formado de um grupo fosfato, um açúcar (pentose) e uma base
nitrogenada (púrica e pirimídica). No DNA existem duas bases purinas (formadas por dois anéis de
carbono), adenina (A) e guanina (G), e duas bases pirimidínicas (formadas por um simples anel de
carbono), timina (T) e citosina (C). Entre DNA e RNA há duas principais diferenças: no RNA tem-
se a presença da uracila (U) em vez da timina; e a pentose no RNA é a ribose e no DNA, é a
desoxirribose (ZAHA, 1996, p 32). A Figura 1 mostra um exemplo de uma base nitrogenada, a
timina.
Figura 1. Nucleotídeo Timina
Fonte: Kaiser (2003)
Uma seqüência de nucleotídeos possui uma orientação química de extrema importância. Em
uma fita de DNA ou RNA, numa das extremidades há um grupo fosfato ligado ao 5C (carbono 5’)
do açúcar (extremidade 5’) e na outra há uma hidroxila ligada ao 3C (carbono 3’) do açúcar
(extremidade 3’). Assim, na extremidade 5’ da cadeia, um grupo fosfato está presente e, na
extremidade 3’, um grupo OH. Convencionou-se escrever e ler a seqüência nucleotídica da

12
esquerda para a direita, no sentido 5’ 3’ (ibidem). A representação de uma cadeia
polinucleotídica é feita através das letras das bases nitrogenadas: 5’ AAAGGCTTCGA 3’.
O DNA é composto de duas cadeias polinucleotídeas helicoidais que formam uma dupla
hélice em torno de um eixo central (ZAHA, 1996) Uma fita se une à outra por meio de pontes de
hidrogênio. As pontes de hidrogênio são formadas entre pares de nucleotídeos. A ilustração da
dupla hélice do DNA pode ser vista na Figura 2. Os pares de bases são: adenina que se une à timina,
e a citosina que se une à guanina. Entre A e T há a formação de duas pontes de hidrogênio e entre C
e G há três pontes de hidrogênio, o que faz com que sua ligação seja mais forte. As bases nucléicas
que compõem o DNA são exibidas na Figura 3. Geralmente, o comprimento de uma seqüência de
DNA é descrito em pares de bases (pb), quilobases (1000 pb), megabases (1 milhão pb) etc (GIBAS
e JAMBECK, 2001).
Figura 2. A Dupla hélice do DNA
Fonte: Access (2003)

13
Figura 3. Bases nitrogenadas que compõe o DNA.
Fonte: Kaiser (2003)
O RNA é uma molécula de ácido nucléico formada por uma só cadeia polimérica. Ele é
sintetizado a partir do molde de DNA e é utilizado na expressão da informação genética. A
seqüência de bases (estrutura primária) é similar à do DNA, exceto pela substituição da
desoxirribose por ribose e da timina por uracila (ZAHA, 1996). O RNA se divide em 3 tipos: RNA
ribossômico (RNAr), RNA transportador (RNAt) e RNA mensageiro (RNAm).
1.2. Gene
Cada molécula de DNA contém vários genes dispostos ao longo do seu comprimento.
Segundo De Robertis e Hib (2001), o gene é “a seqüência de DNA que contém a informação
necessária para sintetizar uma molécula de RNA e, se esta corresponde a um RNA mensageiro, a
partir dele construir uma proteína”. Segundo Zaha (1996), “o gene corresponde a uma seqüência
particular de DNA, codificadora de uma informação (proteína ou RNA)”.
Os genes são segmentos individualizados de uma molécula de DNA separados uns dos
outros por DNA intergênico (ou interveniente) (BROWN, 1999). Os genes constituem entidades
biológicas por meio das quais as características físicas dos pais são transmitidas aos filhos. As

14
mutações que ocorrem nos genes podem ser benéficas ou não para a adaptação da espécie (DE
ROBERTIS e HIB, 2001).
Uma definição interessante e abrangente é dada por Dawkins (1979): “um gene é definido
como qualquer porção do material cromossômico que dura potencialmente por um número
suficiente de gerações para servir como unidade da seleção natural” e ainda “um gene é um
replicador com alta fidelidade de cópia”.
Há três tipos de genes: os codificadores de proteína (que são modelos para gerar as
moléculas de proteína), os especificadores de RNA (que são modelos para as máquinas químicas), e
os genes não transcritos (que são regiões do DNA que possuem algum propósito funcional, mas não
alcançam esse propósito, sendo transcritos para criar outra molécula) (GIBAS e JAMBECK, 2001).
A informação biológica que um gene transporta é o conjunto de instruções para a síntese de
uma molécula de RNA que poderá, conseqüentemente, ser traduzida em proteína ou ainda orientar a
síntese de uma enzima. Em organismos complexos as seqüências dos genes codificantes
interrompidos são chamados de exons, e as seqüências intergênicas são chamadas de introns
(ZAHA,1996).
A expressão gênica é o processo pelo qual a informação biológica contida no gene se torna
disponível à célula. Esse processo é relativamente constante. A expressão gênica é composta de
várias fases: transcrição, tradução e a síntese da molécula de RNA ou de um polipeptídio (BROWN,
1999).
1.3. Proteínas
As proteínas constituem mais da metade do peso seco de uma célula. São polímeros que
desempenham inúmeras funções biológicas e também determinam a forma e a estrutura da célula.
Os monômeros que compõem as proteínas são os aminoácidos. Os aminoácidos são ácidos
orgânicos que possuem um grupo amina e um grupo carboxila. A união entre os vários aminoácidos
é feita através de ligações peptídicas (ZAHA, 1996).

15
A constituição da proteína sintetizada depende diretamente do código genético, revelado no
RNA pela seqüência de códons. O número de códons na fita de RNAm determina o tamanho da
proteína. Existem, ao todo, 64 códons, que são resultantes das quatro letras (A, U, G e C) dispostas
em trincas de nucleotídeos (43 = 64). Como existem mais códons (64) que aminoácidos (20), quase
todos os aminoácidos podem ser reconhecidos por mais de um códon, isto porque algumas trincas
de nucleotídeos atuam como sinônimos. Esta situação resulta na degeneração do código genético
(DE ROBERTIS e HIB, 2001). Pode-se ver a tabela de aminoácidos e as proteínas que são
codificadas por cada trinca na Figura 4.
Figura 4. Tabela de Tradução de Aminoácidos
Fonte: Hilbers (2003)
Existem dois códons especiais: um é códon iniciador (AUG) que é aquele que inicia a
síntese protéica e o outro é códon de terminação (UAA, UAG e UGA), que sinaliza onde o
ribossomo deve finalisar a síntese protéica e a liberação da nova cadeia de polipeptídios. A Figura 4
mostra a tabela de tradução para a maioria dos organismos. No entanto, para as mitocôndrias e
alguns organismos, a tabela é diferente. Por exemplo, no DNA da mitocôndria humana, o códon
UGA do RNAm em vez de ser um códon finalizador, codifica o triptofano.

16
Quatro níveis estruturais são encontrados nas moléculas de proteínas: a estrutura primária,
secundária, terciária e quaternária. A estrutura primária é a seqüência de aminoácidos (BROWN,
1999, p 83). É ela que determina os demais níveis de organização da molécula de proteína. A
estrutura secundária se refere à forma espacial da proteína (sendo constituída, principalmente, por
α-helices, β-folhas e laços). A estrutura terciária é a formação de novos dobramentos originados de
ligações entre trechos da própria molécula. A estrutura quaternária resulta da combinação de dois
ou mais polipeptídios (DE ROBERTIS e HIB, 2001).
1.4. Replicação
A replicação é o processo pelo qual uma molécula de DNA se duplica, dando origem a duas
moléculas idênticas à molécula inicial. Para que esse processo ocorra, há a necessidade de um
conjunto de proteínas específicas (SILVA, 2001a). A seguir é descrito o processo básico
simplificado de replicação, do DNA, uma vez que o processo completo é demasiado complexo para
ser abordado totalmente neste trabalho.
Para que ocorra a replicação de uma molécula de DNA faz-se necessário separar as duas
fitas de nucleotídeos da molécula, através da enzima helicase. Durante esse processo, apenas uma
região limitada de bases está não-pareada. A separação de bases ocorre em regiões chamadas de
origem de replicação, e progridem ao longo da molécula na direção 3’ (BROWN, 1999). Essa etapa
resulta uma nova fita contínua e vários trechos de DNA que formam uma segunda nova fita
simples, então chamados de fragmentos de Okazaki. A replicação de uma fita de DNA e os
fragmentos de Okazaki podem ser vistos na Figura 5.

17
Figura 5. Replicação de cadeia de DNA
Fonte: Classic (2003)
A síntese de novas fitas é feita pela enzima DNA-polimerase. Essa enzima tem a função de
unir nucleotídeos soltos aos fragmentos de Okazaki. Como a enzima DNA-polimerase não pode
sintetizar outra fita a partir de nucleotídeos livres, ela necessita de um molde de fita dupla para
iniciar a síntese completa de uma fita dupla (SILVA, 2001a). Este molde, chamado de primer
(iniciador) nada mais é do que uma pequena cadeia de RNA de fita simples, onde, a partir da
posição 3’, a enzima DNA polimerase iniciará a incorporação de nucleotídeos, formando a fita
dupla do DNA. Logo após a síntese da fita dupla de DNA, a enzima ligase une os vários fragmentos
de Okazaki. Dessa forma, as duas fitas de DNA já estão terminadas e naturalmente se enrolam
formando a dupla hélice (SILVA, 2001a).
1.5. Transcrição
A transcrição é a síntese de moléculas de RNA usando moléculas de DNA como molde. A
síntese ocorre pela união entre si dos nucleotídeos de RNA que seguem a ordem dos nucleotídeos
da molécula de DNA (DE ROBERTIS, 2001). A enzima responsável pela transcrição é a RNA
polimerase (SILVA, 2001a).
Através da transcrição são sintetizados todos os RNAs da célula. O RNAm será usado para
transferir a informação genética do DNA para a síntese protéica. Os demais RNAs têm funções
finais na célula, tanto estruturais como catalíticas. Durante a transcrição ocorre o controle da

18
expressão gênica, que estabelece quais os genes são transcritos e quantos deles são necessários
(ZAHA, 1996).
A ligação inicial do DNA à RNA polimerase deve ocorrer em uma posição específica, um
pouco antes do gene a ser transcrito. Isso é importante porque em geral uma grande parte do DNA
não é gene e não deverá ser transcrito. O ponto inicial onde deve se unir a RNA polimerase é
chamado de promotor. Um promotor é uma pequena seqüência de nucleotídeos reconhecida por
uma RNA polimerase como o ponto ao qual deve-se ligar ao DNA a fim de começar a transcrição.
Durante a etapa de alongamento da transcrição, a RNA polimerase migra ao longo da molécula de
DNA desenrolando a dupla fita à medida que progride, enquanto que, seqüencialmente, une
ribonucleotídeos à extremidade 3’ da molécula crescente de RNA. A fita de DNA que foi
desenrolada volta ao seu estado normal. A finalização da transcrição ocorre, como na inicialização,
em um ponto especifico da molécula de DNAm chamado de códon de terminação. (BROWN,
1999).
1.6. Tradução
A síntese, ou tradução protéica é um processo que requer um filamento de RNAm, RNAt e
subunidades ribossômicas. No ribossomo, o RNAm é traduzido em uma proteína, processo no qual
também é necessária a intervenção dos RNAt. Os RNAt captam os aminoácidos do citosol e
conduzem-os até o ribossomo na ordem estabelecida pelos nucleotídeos do RNAm, que são os
moldes do sistema (DE ROBERTIS e HIB, 2001).
Cada RNAt pode ser distinguido por sua especificidade por um dos 20 aminoácidos
existentes. Por exemplo, o RNAtgli é específico para glicina, o RNAtyr é específico para tirosina.
Uma molécula de RNAt forma uma ligação com seu aminoácido (e com nenhum outro) e pode
reconhecer e se ligar a um códon que especifica aquele aminoácido. Pode haver mais de um RNAt
para um único aminoácido, o que reflete o fato de que o código genético é redundante, e que a
maioria dos aminoácidos é codificada por mais de um códon (BROWN, 1999).

19
Geralmente os códons que representam um mesmo aminoácido são parecidos entre si, e é
freqüente que sejam diferentes no terceiro nucleotídeo. Devido à pouca especificidade deste
nucleotídeo, diz-se que existe uma degeneração na terceira base da maioria dos códons (DE
ROBERTIS e HIB, 2001).
1.7. Desnaturação e Renaturação do DNA
Os termos desnaturação e renaturação são sinônimos de separação das fitas e reanelamento
das fitas, respectivamente. A desnaturação ocorre quando as pontes de hidrogênio entre as cadeias
complementares do DNA são rompidas e as fitas se separam. O processo inverso ocorre na
renaturação. Esses processos ocorrem normalmente na natureza e podem ser desencadeados in vitro.
A temperatura na qual 50% das moléculas de DNA se encontra desnaturado é chamado de Tm
(Temperature Melting) (ZAHA, 1996).
Para desencadear as reações acima citadas, uma das técnicas usadas é variação da
temperatura. Para o rompimento de um par CG são necessárias temperaturas mais elevadas, porque
entre a ligação das bases C e G há três pontes de hidrogênio, enquanto entre A e T há apenas duas
pontes. É possível calcular o valor de Tm de um dado DNA a partir do conteúdo de GC, usando a
fórmula: Tm (°C) = 69,3 + 0,41 (%GC) – 500 / tamanho (ibidem).
A renaturação pode ocorrer mesmo quando as moléculas de DNA estão completamente
separadas. Se uma solução contendo DNA desnaturado for lentamente resfriada, as fitas
reassociam-se. Esse anelamento ocorre geralmente a uma temperatura 25°C abaixo da Tm. Se um
resfriamento abrupto ocorre, a renaturação pode não ocorrer (ibidem).
Essas características do DNA são importantes também para os experimentos de PCR, que
realizam a replicação de DNA in vitro e baseiam-se justamente na desnaturação e renaturação do
DNA. A PCR é explicada na seção 2.2.

20
2. TÉCNICAS DE BIOLOGIA MOLECULAR 2.1. Seqüenciamento e Clonagem
Provavelmente, a técnica mais importante disponível aos pesquisadores é o seqüenciamento
de DNA, processo através do qual se pode determinar a ordem exata de nucleotídeos de um trecho
de DNA. Os métodos de seqüenciamento de DNA mais rápidos e eficientes estão disponíveis desde
a década de 70. A primeira molécula de DNA a ser totalmente seqüenciada foi o genoma do
bacteriófago X 174 com 5.386 nucleotídeos que foi completado em 1975 (BROWN, 1999).
Um dos requisitos para o seqüenciamento do DNA é a habilidade para obter fragmentos
definidos de DNA. Qualquer método de seqüenciamento de DNA começa com uma população de
um fragmento definido de DNA. A partir dessa população, são polimerizados conjuntos de
moléculas que diferem em tamanho devido à incorporação na extremidade 3´ de uma base
modificada, a qual interrompe o processo de síntese da cadeia complementar (nucleotídeo de
terminação). Essas moléculas são separadas por eletroforese de acordo com seu tamanho e por meio
do reconhecimento de qual base modificada está na extremidade 3´ é possível determinar a
seqüência original do DNA.
Existem vários motivos que levam ao seqüenciamento de um genoma completo, entre eles: a
descoberta das funções dos genes; a utilização de genes já conhecidos na produção de fármacos ou
outros produtos de interesse biotecnológico; a identificação de genes que causam doenças,
possibilitando a criação de terapias e uso em seres que ainda não desenvolveram os sintomas; e a
descoberta da função (se houver) dos trechos de DNA que ainda são ditos como extragênicos (sem
função alguma) (BROWN, 1999).
A clonagem de genes pode ser feita através da técnica de DNA recombinante. Nesta técnica
é inserido um pequeno trecho de DNA em uma molécula replicante. Esta se replicará, e junto com
ela, o trecho inserido será multiplicado também (GRIFFITHS et al, 2002).

21
2.2. PCR
A Polimerase Chain Reaction (PCR) é uma técnica que possibilita a reprodução de
milhares, milhões ou mesmo bilhões de cópias de um determinado fragmento de DNA. Através
dessa técnica, uma seqüência particular de interesse pode ser amplificada (clonada), tornando-se
majoritária na amostra de DNA, e assim a seqüência amplificada pode ser utilizada para outros fins
(TRIUNFOL, 2003).
PCR é útil em diagnósticos para verificar a presença de um gene ou um estado mutacional
de um gene específico, ou simplesmente na amplificação de um segmento específico (GRIFFITHS
et al, 2002). Depois que uma seqüência é amplificada, ela pode ser separada em um experimento de
eletroforese, por exemplo.
O processo de PCR utiliza múltiplos ciclos de desnaturação, anelamento com primers e
alongamento da seqüência por ação da polimerase (LIFE TECHNOLOGY, CA. 2000). Esses
primers são pequenos fragmentos de DNA complementares a cada uma das extremidades da
seqüência de DNA de interesse. Cada ciclo se inicia com a desnaturação da dupla-hélice do DNA,
elevado a altas temperaturas (95 °C), por aproximadamente 1 minuto. Esta etapa é seguida da etapa
de anelamento dos primers ao DNA molde, a temperaturas que variam de 55 ºC por 1 a 2 minutos.
Posteriormente ocorre a elongação de cadeia por ação da polimerase, em geral entre temperaturas
de 72 °C durante 2 a 5 minutos. Então cada cadeia sofre a desnaturação novamente e se tornam
moldes para mais um ciclo.
O número de ciclos, a temperatura de anelamento, o tempo de cada ciclo e outros
componentes de PCR variam de acordo com o objetivo e condições utilizadas (SILVA, 2001a). A
Figura 6 mostra uma representação da técnica de PCR. Ao final do primeiro ciclo, há duas fitas da
molécula original de DNA, mais duas cópias da região de interesse. Esses ciclos são repetidos
várias vezes, tipicamente 30 vezes, num aparelho chamado termocirculador. Ao final desses ciclos
de amplificação existem, tipicamente, mais que 1 milhão de cópias do segmento de DNA de
interesse para cada molécula molde original da amostra inicial. (TRIUNFOL, 2003).

22
Figura 6. Reação em cadeia da polimerase – PCR
Fonte: Triunfol (2003)
Para a amplificação de um trecho de DNA através da técnica de PCR são necessários dois
pares de primers em filamentos opostos. Para o seqüenciamento de um trecho de DNA é necessário
apenas um primer que se anele no início do trecho. Após a replicação do trecho entre os dois
primers, as duas novas seqüências são desnaturados por aquecimento para gerar moldes
unifilamentares, e um segundo ciclo inicia (GRIFFITHS et al, 2002).
Os parâmetros que afetam a eficiência da reação de PCR são: temperatura de anelamento
(Ta) e tempo de anelamento, temperatura de desnaturação (Tm, muito importante) e tempo de
desnaturação, PCR buffer (tampão que mantém o pH e a quantidade de sais necessários para a
atuação da enzima), concentração de MgCl2, concentração e projeto de primer e cossolventes, entre
vários outros.
Há dois termos muito utilizados em reações de PCR que não podem ser confundidos, a Tm e
a Ta. A Tm (temperature melting – temperatura de ‘derretimento’) é a temperatura na qual 50% do
DNA se encontra desnaturado (ZAHA, 1996). E a Ta (annealing temperature - temperatura de
anelamento) é a temperatura na qual o primer se anela à seqüência alvo. Geralmente, a Ta é
calculada 5 °C abaixo da temperatura estimada de Tm (ALKAMI, 1999).
Três são as métricas utilizadas para medir a qualidade do trecho amplificado pela reação de
PCR: especificidade, eficiência e fidelidade.

23
A especificidade é a medida para saber se a reação amplifica apenas a região-alvo e não
outras regiões da molécula, o que ocasionaria “falsos-positivos”, com a amplificação de DNA, mas
não o trecho desejado. Ela é muito influenciada pelo primer utilizado na reação, principalmente seu
tamanho e degeneração. A concentração de primers também é uma variável importante, sendo que
baixas concentrações aumentam a especificidade, enquanto altas concentrações facilitam o
anelamento não específico (ALKAMI, 1999).
A eficiência, que trata da rentabilidade da reação de PCR. A eficiência pode ser alta se o
produto amplificado for pequeno e se os ciclos da PCR forem bem ajustados, permitindo um
anelamento e polimerazação rápidos. Um aumento na concentração de íons Mg++ também pode
aumentar a eficiência da reação, embora aumentos exagerados tendem a causar reações menos
específicas (ALKAMI, 1999).
A última métrica a ser analisada é a fidelidade (exatidão ou acurária), que verifica se o
produto amplificado é uma cópia idêntica à original. Pode-se aumentar a fidelidade diminuindo a
concentração de dNTP e íons Mg++ numa proporção equimolar, mas, principalmente, a fidelidade
está relacionada ao tipo da polimerase utilizada na PCR. (ALKAMI, 1999).
Há alguns tipos de reações de PCR com processos e finalidades diferentes. Entre eles,
destacam-se no contexto deste trabalho: Nested PCR, Multiplex PCR, Universal PCR e Degenerate
PCR (SINGH e KUMAR, 2003).
• Nested PCR: é utilizado quando a quantidade de seqüência-alvo é muito pequena (< 100
moléculas). Este processo consiste em amplificar, primeiramente, uma região da seqüência,
e depois disso realiza-se uma segunda PCR que amplificará um trecho menor do resultado
da primeira reação de PCR. Para este tipo de PCR são necessários quatro primers.
• Multiplex PCR: esta técnica envolve a amplificação de dois ou mais trechos de uma mesma
seqüência-alvo. Preferencialmente é utilizado um único primer, mas pode-se utilizar mais
primers. No projeto de multiplex primers, a característica mais importante a ser analisada é a
temperatura de anelamento que deve ser similar entre os primers projetados. Nesta técnica,
deve ser levado em consideração o tamanho do produto, para que eles possam ser separados
por eletroforese com eficiência. Multiplex PCR é normalmente utilizado para detecção de
mutações.

24
• Universal PCR: são PCRs que utilizam primers projetados a partir da seqüência de
aminoácidos de uma proteína de interesse. É utilizado quando não se conhece a seqüência-
alvo (genômica), apenas a proteína possivelmente sintetizada por um gene dessa seqüência-
alvo.
• Degenerate PCR: pesquisadores aspiram a clonagem de genes novos, e nem sempre a
seqüência é totalmente conhecida. Então, pode ser usada uma união de primers degenerados
com universal primers para descobrir a seqüência de interesse. Para esta técnica utilizam-se
primers degenerados (explicado na seção 2.2.1). Primers degenerados baseadas em
seqüências de aminoácidos de regiões conservadas podem ser usados para a procura de
membros de uma família de genes, genes homólogos de diferentes espécies e vírus
relacionados.
2.2.1. Primers
Primer, ou iniciador, é uma pequena cadeia de DNA de fita simples, onde, a partir da
posição 3’, a enzima DNA polimerase iniciará a incorporação de nucleotídeos, formando a fita
dupla do DNA. Os primers são produzidos in vivo ou in vitro. Na forma natural, os primers são
produzidos por uma RNA polimerase especial chamada Primase (SILVA, 2001a).
O projeto (design) de primer é uma das mais importantes etapas para que se tenha uma
reação de PCR bem sucedida (ALKAMI, 1999), uma vez que são eles os responsáveis por prover
um pequeno trecho de dupla fita no qual a taq-Polimerase possa iniciar a replicação. Existem muitos
pontos que devem ser levados em consideração para se projetar um primer de boa qualidade.
Segundo Life Technology CA (2000), Afseth (1997), Innis (1990), os principais pontos de análise
são:
• Tamanho da seqüência do primer. Normalmente os primers têm entre 18 e 24 nucleotídeos.
O primer deve ser comprido o suficiente para ser único na seqüência e reduzir a
probabilidade da seqüência ser encontrada em locais não alvos na seqüência. Entretanto,
primers muito grandes são muito caros para fazer; não conferem grande sensibilidade; e
hibridizam mais lentamente, fazendo com que o rendimento diminua.
• Temperatura de anelamento: deve ser suficiente baixa para garantir a eficiência do
anelamento do primer com a seqüência alvo, e deve ser suficiente alta para minimizar o
anelamento com seqüências não alvo e formação de estruturas secundárias. Quando há dois

25
primers na mesma solução, é muito importante ter uma pequena diferença de temperatura de
hibridização entre os primers. Normalmente a diferença de Ta entre dois primers é de 5 °C.
• Quantidade de nucleotídeos G e C no primer: geralmente é preferível que a porcentagem de
GC no primer esteja entre 50% a 60%, ou na mesma proporção da seqüência original.
• Existe uma relação entre a temperatura de anelamento com a quantidade de GC do primer.
Como existe uma tripla ligação entre as bases G e C e uma dupla ligação entre A e T, a
temperatura de anelamento sofre alteração devido à quantidade de bases GC e AT. Quanto
maior for a quantidade de GC em um primer, maior será a temperatura de anelamento.
Existem duas fórmulas para calcular a Ta de um primer:
primers com 20 ou menos pares de bases: Ta = [4(G + C) + 2(A + T)] - 5°C;
primers com mais de 20 pares de bases: Ta = 62.3°C + 0.41°C (%GC) - 500/tamanho - 5°C
• Nucleotídeos na extremidade 3’: deve-se evitar trechos complementares na extremidade 3’,
a fim de diminuir a ocorrência de dímeros de primers, que ocorrem quando os primers
anelam-se um ao outro, e não se anelam à seqüência alvo.
• Deve-se evitar primers com potencial para formar estruturas secundárias internas, isto é, o
primer anelar-se em si mesmo e não anelar à seqüência alvo.
• Deve-se evitar uma extremidade 3’ rica em GC. Um valor bom é ter primers com três
nucleotídeos A ou T dentre os cinco últimos nucleotídeos.
• Os últimos três nucleotídeos precisam anelar-se perfeitamente à seqüência alvo para a
enzima polimerase fazer o alongamento da seqüência.
• Deve-se evitar um trecho com nucleotídeos contínuos iguais, por que estes trechos são
facilmente encontrados em diversas regiões do genoma, geralmente no início e no fim, e
assim não se consegue uma boa especificidade.
• Um método utilizado é a inserção de bases degeneradas nos primers. As bases degeneradas
podem ser um de dois ou mais nucleotídeos, conforme a Tabela 1. Primers degenerados
possuem pelo menos uma posição em sua seqüência que pode conter doi ou mais
nucleotídeos diferentes. O número de bases degeneradas é inversamente proporcional à
especificidade do primer. Não se deve incluir bases degeneradas na extremidade 3’ do
primer, para que não inicie a polimerização em trecho errado da seqüência. É recomendado
usar altas concentrações de primers degenerados em PCR porque muitos dos primers em
uma solução degenerada não são específicos.

26
Tabela 1. Tabela de Degenerações de Bases Nucléicas
IUPAC Base IUPAC Base A Adenina W A or T C Citosina K G or T G Guanina M A or C T / U Timina/Uracila B C or G or T R A or G D A or G or T Y C or T H A or C or T S G or C V A or C or G W A or T N qualquer base
Fonte: Triunfol (2003)
Existem várias ferramentas computacionais que projetam primers. Um exemplo de
ferramenta disponível na internet é Web Primer (WEB PRIMER, 2003), e outra ferramenta,
também livre, é Gene Runner. A ferramenta Web Primer aceita dois tipos de entrada de dados: uma
maneira é através do identificador do GenBank da seqüência a ser amplificada, e a outra maneira é a
entrada da seqüência de aminoácidos. Esta ferramenta provê primers para duas finalidades distintas,
seqüenciamento ou PCR. A ferramenta Gene Runner para Windows está disponível para download
gratuitamente em Gene Runner (2003). Esta ferramenta provê vários serviços, como projeto de
primers, alinhamento se seqüências, tradução para proteínas, e outros. Para o projeto de primers
esta ferramenta não é muito eficiente, pois ela não restringe os primers de acordo com os
parâmetros de entrada.
2.3. Eletroforese
Eletroforese é a técnica pela qual fragmentos de DNA de diferentes tamanhos são separados
por peso molecular. As seqüências de DNA são colocadas em um equipamento que contém canais
preenchidos de gel, e elas percorrem esse gel, movidos por uma diferença de potencial elétrico. Se o
peso molecular de uma seqüência é maior que o de outra, o primeiro percorre o gel mais lentamente
que o segundo (TRIUNFOL, 2003). A Figura 7 mostra um gel de eletroforese. Nas colunas de 1 a
14 há poços contendo diferentes seqüências de DNA. Pode-se ver, no lado esquerdo dessa Figura, a
indicação dos tamanhos dos fragmentos separados no gel.

27
Figura 7. Exemplo de gel de eletroforese
Fonte: Threlfall et al (2003)
Trechos de DNA, depois de amplificados por PCR, e possivelmente cortados por enzimas de
restrição, podem ser separados pela eletroforese e comparados a outras seqüências, para verificar se
duas seqüências possuem o mesmo gene, por exemplo.
3 – BIOINFORMÁTICA 3.1. Bancos de Dados Biológicos Públicos
Os bancos de dados em biologia molecular são importantes principalmente para
proporcionar à comunidade científica um grande repositório de dados de fácil acesso

28
(PROSDOCIMI, 2003a). Esses bancos de dados possuem seqüências de DNA, RNA, proteínas,
genes, ESTs, revistas e artigos de ciências biológicas e médicas, entre outras informações.
Os bancos de dados estão se expandindo rapidamente. De abril de 1988 até o final de 1999,
os projetos genoma cobriram 83 espécies com 21 projetos completados (predominantemente
microorganismos) e no ano de 2000 existiam 62 projetos em andamento. Esses projetos são
realizados através das técnicas de PCR e pelos sistemas de seqüenciamento automático (QUEIROZ,
2002). Segundo National Center For Biotechnology Information (2003), o número de seqüências
depositadas no GenBank em 2002 era de mais de 22 milhões de seqüências. Com o advento do
seqüenciamento do DNA e, principalmente, a partir da década de 1990, do seqüenciamento em
larga escala, foi necessária a construção de bancos de dados mais robustos para abrigar o número de
seqüências obtidas pelos pesquisadores.
O NCBI, por exemplo, foi criado pelo NIH (National Institutes of Health) em 1988 para
abrigar esse tipo de informação (PROSDOCIMI, 2003a). Para criar um índice para suas entradas,
foi criado um identificador, o GenInfo Identifier (gi), que é um número inteiro. Este número é único
para cada seqüência (TRIUNFOL, 2003).
O NCBI, em cooperação com o EMBL (European Molecular Biology Laboratory) e outras
organizações internacionais, fornece a seqüência mais completa de dados de seqüências de DNA
disponível no mundo, assim como o PubMed, um banco de dados taxionômico (classificação), e um
ponto de acesso alternativo para dados de estruturas e seqüências de proteínas. Esse banco de dados,
conhecido como GenBank, pode ser acessado a partir do site do NCBI (GIBAS e JAMBECK,
2001). O NCBI mantém dados de seqüências de todos os organismos, todas as fontes e todos os
tipos de moléculas de DNA, RNA, estruturas 3D de proteínas,...
O PubMed é um serviço do National Library of Medicine, e está disponível no site do
NCBI. Ele inclui mais de 11 milhões de artigos biomédicos. O PubMed inclui muitos artigos
completos e outras fontes relacionadas, como revistas científicas.

29
3.2. Alinhamento de Seqüências A busca de similaridades entre duas ou mais seqüências pode ser feita através das técnicas
de alinhamento. Alinhar duas seqüências significa estabelecer uma correspondência entre as bases
nucléicas ou aminoácidos de ambas seqüências, procurando dar prioridade a monômeros iguais. O
alinhamento de seqüências é uma das ferramentas de bioinformática mais utilizada.
O alinhamento pode ser feito para vários propósitos. Entre eles podem ser citados dois
principais: busca de similaridade e busca de homologia. Os termos similaridade e homologia são
muitas vezes usados de forma errônea. O termo similaridade refere-se à presença de locais similares
e idênticos em duas seqüências. O termo homologia indica que duas seqüências possuem uma
probabilidade alta de compartilharem o mesmo ancestral (GIBAS e JAMBECK, 2001).
Em um alinhamento, quando há monômeros iguais em duas ou mais seqüências, diz-se que
há match. Quando um monômero não pode ser alinhado a outro igual na outra seqüência, há a
substituição por um sinal (por exemplo, um “–“), ou a inserção de um espaço em branco (gap). Na
Figura 8, há um exemplo de alinhamento entre duas bases de seqüências. Para cada match,
substituição, remoção ou inserção há uma pontuação (score) específica. Uma pontuação maior
representa um melhor alinhamento, ou seja, mais similares são as seqüências. O alinhamento pode
ser feito entre duas ou mais seqüências. Se o alinhamento é entre duas seqüências, é chamando de
alinhamento par-a-par; se forem entre três ou mais seqüências, é chamado de alinhamento múltiplo
(GUIMARÃES e MELO, 2003).
Há dois tipos de algoritmos para alinhamento par-a-par: alinhamento global e alinhamento
local. O primeiro faz o alinhamento de duas seqüências por todo o seu comprimento. O Align é uma
ferramenta que se baseia no alinhamento global. O segundo tipo de alinhamento é o local. O
alinhamento local é muito utilizado quando não se conhecem as seqüências inteiras a serem
alinhadas. Muitas vezes é necessário procurar em outras seqüências disponíveis apenas trechos que
sejam iguais (GIBAS e JAMBECK, 2001). Por exemplo, pode-se utilizar o alinhamento local para
descobrir a quais seqüências um primer se anela. A principal ferramenta disponível que utiliza
alinhamento local é o Blast (Basic Local Alignment Search Tool). Esse tipo de alinhamento será
utilizado neste trabalho para verificar a especificidade do primer.

30
Através de interfaces web pode-se alinhar uma determinada seqüência a todas as outras
disponíveis em banco de dados biológicos públicos na internet (alinhamento par-a-par). Com a
ferramenta Blast, disponível no site do NCBI, pode-se fazer o alinhamento par-a-par da seqüência
desejada a todas as outras disponíveis nos repositórios do próprio site. E além disso, o alinhamento
é utilizado para outras ferramentas, como análise filogenética. As seqüências de DNA podem ser
comparadas (alinhadas) para localizar semelhanças que sugerem uma relação das moléculas pela
forma ou função ou ancestralidade comum (GIBAS e JAMBECK, 2001). Na Figura 8 há a
ilustração de um alinhamento feito pelo software Blast. Na parte de cima da Figura o software
mostra linhas coloridas significando cada umas das seqüências do banco que alinham-se a
seqüência procurada. Quando o mouse passa sobre as linhas, vê-se a pontuação do alinhamento
dessa seqüência em relação à seqüência procurada. Ao clicar com o mouse sobre as linhas pode-se
ver o alinhamento. Na parte de baixo da Figura 8 pode-se ver um alinhamento, destacando um
alinhamento perfeito entre duas bases (1), um gap (2) e um não-alinhamento (3)
Figura 8. Alinhamento par-a-par entre seqüências realizado pelo Blast
Fonte: Blast (2003)

31
Um alinhamento múltiplo é utilizado para estudar grupos de genes ou proteínas
relacionados, para verificar relações evolutivas entre os genes e para descobrir padrões que sejam
compartilhados entre grupos de seqüências funcional ou estruturalmente relacionadas (ibidem). O
programa mais comumente utilizado para alinhamentos múltiplos é o ClustalW. Esse software está
disponível em Clustalw (2003). Além de ser acessível por sua interface web disponível
gratuitamente, ele também pode ser instalado no computador do usuário, incluindo versão para
Linux. Na Figura 9 há a ilustração de um alinhamento feito pelo software ClustalW. Do lado
esquerdo da Figura há o identificador das seqüências no GenBank que foram utilizadas no
alinhamento. Do lado direito da Figura há o alinhamento, onde destacamos o alinhamento perfeito
entre duas bases (1), um gap (2) e um não-alinhamento (3). Pode-se notar que no início do
alinhamento há uma extensa faixa com gaps, para melhorar o alinhamento.
Figura 9. Alinhamento múltiplo entre seqüências feito pelo ClustalW
Fonte: ClustalW (2003)
Vários são os algoritmos de pontuação para o alinhamento de seqüências par-a-par.
Alinhamento de seqüências de aminoácidos relativamente semelhantes recebem pontuação positiva
e não semelhantes recebem pontuação negativa (lembrando que várias seqüências diferentes podem
gerar o mesmo aminoácido). Um dos métodos mais utilizado nos últimos anos foi PAM (Point

32
Accepted Mutation). Esse método analisa seqüências que sofreram divergência evolucionária. A
pontuação reflete a probabilidade de as seqüências terem sofrido mutações.
Outro método utilizado é o BLOSUM (Blocks Substitution Matrix). Ele tem se mostrado
superior à matriz PAM para detectar os parentescos biológicos entre seqüências moderadamente
divergentes (BONATO, BONATO e PEREIRA, 2003). Para relações mais próximas entre as
seqüências é utilizada a matriz BLOSUM62, e para relações mais distantes é utilizada a matriz
BLOSUM45 (GIBAS e JAMBECK, 2001). Um algoritmo utilizado para alinhamentos múltiplos é
conhecido como SP (Sum-of-Pairs). Cada linha de uma matriz recebe uma das seqüências a serem
alinhadas. Este método consiste em atribuir uma pontuação para cada coluna e tomar a soma desses
valores como sendo o score do alinhamento. O score de cada coluna é a soma das pontuações de
cada par de alinhamentos (GUIMARÃES e MELO, 2003).
3.3. Árvores Filogenéticas
Segundo Gibas e Jambeck (2001), “a inferência filogenética é o processo de desenvolver
hipóteses sobre a relação evolutiva sobre organismos com base nas suas características
observáveis”. Antigamente, a teoria da evolução se baseava em características visíveis dos seres.
Lineu classificou os seres em reinos, filos, classes, ... pela simples observância da anatomia de cada
um deles e desenhou uma árvore filogenética, que mostra a descendência entre os seres.
As árvores filogenéticas mostram a relação de descendência entre os organismos e o tempo
que se passou entre o ancestral comum de duas espécies (PROSDOCIMI, 2003). As árvores
filogenéticas servem, por exemplo, para, procurar entender a origem do homem ou reconstituir a
história epidemiológica da AIDS a partir de dados do genoma do vírus HIV (PROSDOCIMI et al.,
2003).
Quando se trata de evolução das espécies há dois termos que não podem ser esquecidos:
ortólogos e parólogos. Os ortólogos são genes que têm a mesma função e parólogos não possuem o
mesmo papel, mas possuem um ancestral em comum. Ortólogos ocorrem em espécies diferentes, e
parólogos existem no mesmo genoma mas têm funções diferentes (GIBAS e JAMBECK, 2001).
Árvores geralmente são utilizadas para distinguir ortólogos de parólogos (HIGGINS e TAYLOR,
2000).

33
A Figura 10 mostra a árvore filogenética de quatro seqüências de nucleotídeos. Essas são as
mesmas seqüências usadas no exemplo de alinhamento múltiplo, da Figura 9. Nota-se que entre a
primeira seqüência e a segunda (1) há uma descendência próxima, porque a distância entre elas na
arvore é pequena. Mas entre as duas primeiras seqüências e duas últimas, há distância entre elas é
maior (2). Logo, o ancestral comum entre a primeira e a segunda seqüência é mais próximo.
Figura 10. Árvore Filogenética feita pelo software ClustalW
Fonte: ClustalW (2003)
Há softwares que geram a árvore filogenética baseada em alinhamentos. O mais utilizado
atualmente é o pacote Phylip (PHYLIP, 2003). Ele é um conjunto de 30 programas, executados
separadamente, através da linha de comandos. Este pacote de softwares permite a análise de vários
tipos de dados, como DNA, proteínas e outros, e gera árvores por vários métodos. Este pacote tem
na linha de comandos um menu de opções que facilitam a interação com o pesquisador. O menu do
programa DNAPARS, que faz parte do programa Phylip, pode ser visualizado na Figura 11.
Figura 11. Menu de opções do pacote de programas Phylip
Fonte: Phylip (2003)

34
3.4. Simuladores de Eletroforese
Existem softwares disponíveis na web que simulam a eletroforese. Um exemplo, é o Gel
Electrophoresis Simulator (COWAN, 2003), que pode ser visto na Figura 12. Ele tem a
possibilidade de ter no máximo oito fragmentos de DNA e o gel utilizado é agarose. O maior
problema desse simulador é que ele não mostra o tempo de corrida dos fragmentos no gel. Mas
podem ser alterados o potencial de eletricidade e a concentração do gel de agarose.
Figura 12. Tela do software Gel Electrophoresis Simulator
Fonte: Cowan (2003)
Outra ferramenta disponível na web é Simulation of Electrophoresis & Sequence Building of
DNA (CRAIG, 2003). A tela desse software pode ser vista na Figura 13.

35
Figura 13. Tela do software Simulation of Electrophoresis & Sequence Building of DNA
Fonte: Craig (2003)
4 – SISTEMAS PARA WEB
Com o desenvolvimento e popularização da Internet, intensificaram-se as tecnologias e
ferramentas que auxiliam na criação de sistemas que executam através da Internet, principalmente
através de aplicações para web (world wid web). Esse tipo de sistema possui, como vantagens
principais, (i) a independência de plataforma de hardware, pois o código correspondente, ou é
interpretado, ou executa apenas no servidor; (ii) não há necessidade de instalar o sistema
localmente, uma vez que o mesmo é acessado diretamente por um navegador padrão (brownser);
(iii) a disponibilidade mundial do sistema; e (iv) a possibilidade de monitorar cada execução do
sistema.

36
4.1. Perl
Em bioinformática há uma grande variedade de ferramentas e um montante cada vez maior
de dados disponíveis em bancos de dados públicos. Uma linguagem muito utilizada para analisar e
extrair apenas a informação necessária de grandes quantidades é a Perl (Practical Extraction and
Report Language – Linguagem Prática para Extração e Relatórios) (GIBAS e JAMBECK, 2001).
Essa linguagem contém inúmeras facilidades para comparação de strings. Ela faz uso intensivo de
expressões regulares que são muito úteis em análises de seqüências da DNAs e proteínas, por
exemplo.
Muitos dos dados biológicos são armazenados em bancos de dados e grandes arquivos de
texto. A Perl é mais eficiente para extrair esses dados que outras linguagens, como C ou Java, pelas
funcionalidades incluídas em sua sintaxe. Com sua capacidade altamente desenvolvida para detectar
padrões em dados, e especialmente seqüências de caracteres em texto, tornou-se a opção mais
utilizada para esta tarefa (GIBAS e JAMBECK, 2001).
A riqueza de códigos Perl existentes para a bioinformática, a integração sem problemas dos
códigos com sistemas baseados em Unix, a portabilidade para várias plataformas e uma comunidade
de usuários incrivelmente entusiástica tornam a Perl a linguagem de scripts preferida pelos
bioinformatas (ibidem). Outro motivo que leva o uso da Perl é que está disponível gratuitamente
para vários sistemas operacionais, incluindo Linux. A Perl está disponível em Christiansen (2003).
Em várias versões do Linux, o compilador Perl está presente, sem que haja a necessidade de instalá-
lo (SILVA, 2001b).
Outra grande vantagem pertinente à Perl é a possibilidade de se utilizar alguns módulos Perl,
como o CGI (Common Gateway Interface), por exemplo, para tarefas específicas. Os módulos são
bibliotecas com funções relacionadas entre si. Por exemplo, o módulo DBI (Database Interface) é
usado para escrever programas que interagem com bancos de dados relacionais (GIBAS e
JAMBECK, 2001). Os módulos Perl estão disponíveis em CPAN (2003). Os módulos são gratuitos
e estão divididos em quatro pastas: por autores, por categoria, por módulo e mais recentes. Para a
instalação dos módulos estão disponíveis arquivos readme ou install juntamente com os módulos
(SIEVER, SPAINHOUR e PATWARDHAN, 1999).

37
Para bioinformatas o módulo mais usado é o BioPerl, do Projeto Bioperl (juntamente com
Biopython, Biojava e Bioxml). Este projeto dedica-se à criação de uma biblioteca de código aberto
de módulos de pesquisa em bioinformática (GIBAS e JAMBECK, 2001).
4.1.1. Principais Módulos Perl
Os principais módulos Perl utilizados em bioinformática são: CGI.pm, LWP, DBI, GD e
PDL e BioPerl. No texto a seguir há uma breve descrição dos módulos (com exceção do módulo
BioPerl, descrito em 3.1.3), segundo Gibas e Jambeck (2001):
• CGI.pm (Simple Common Gateway Interface Class) é um módulo para programação de
páginas web interativas. As funções que fornece estão voltadas para a formatação de páginas
web, e para a criação e processamento de formulários nos quais os usuários podem digitar
informações. Esta biblioteca define objetos CGI, entidades que contêm o valor da string de
busca (por exemplo, o endereço URL - Uniform Resource Locator) e outras variáveis de
estado. Usando métodos de um objeto CGI, pode-se examinar palavras-chave e parâmetros
passados para seu script, e criar formulários que iniciam com valores que vieram dos
parâmetros (BIOPERL, 2003)
• LWP.pm (Library for Web Programming) Utilizando LWP, os programas Perl podem
enviar dados para formulários, recuperar páginas da web e eliminar grande parte do tédio de
interagir manualmente com os serviços da web utilizando um navegador. Para recuperar
uma página web, pode-se utilizar o módulo LWP::Simple e o comando print get
(“http://www.univali.br”);. Este comando retornará o código fonte da página
solicitada.
• DBI: O módulo DBI (Database Interface) é um módulo para escrever programas que
interagem com banco de dados relacionais. Permite escrever programas que colocam dados
em bancos de dados, consultam os bancos de dados, e extraem registros de banco de dados,
sem prestar atenção ao banco de dados específico que está sendo utilizado. Um script de
banco de dados para Oracle, por exemplo, pode ser utilizado em um banco de dados
MySQL, sendo necessárias apenas alterações secundárias. Esse módulo define um conjunto
de métodos, variáveis e convenções que fornecem uma interface consistente para banco de
dados (BIOPERL, 2003).

38
• GD: O módulo GD permite gerar gráficos utilizando programas Perl. É muito utilizado para
criar esquemas simples e personalizados em páginas da web, como estatísticas de utilização
de servidor da web (GIBAS e JAMBECK, 2001). O GD define três classes: GD::Image,
GD::Font e GD::Polygon (BIOPERL, 2003) O Paint-Blast.pm, um módulo que gera
representações gráficas de alinhamentos de seqüências a partir de saídas Blast, é um
exemplo de aplicativo GD. Está disponível no ScriptCentral do Bioperl.
• PDL: O módulo PDL (Perl Data Language) é uma biblioteca para fazer cálculos com
matrizes. Em biologia computacional, é muito utilizada para cálculos com microarrays e
imagens. Por exemplo, géis bidimensionais que medem a interação entre proteínas são
geralmente armazenados como imagens, que nada mais são que matrizes, e métodos de
processamento de imagens podem localizar e comparar características dos géis. Sem o
módulo PDL, para processar uma matriz de n dimensões, é necessário ter n loops de
controle. Para cada dimensão se faz necessário ter um loop para percorrer toda a matriz,
indexando linhas, colunas, etc. Se for usado o módulo PDL, o programa Perl interpreta cada
matriz como se fosse uma variável escalar, sem a necessidade de identificar a posição da
matriz. Por exemplo, para somar duas matrizes e colocar o resultado em uma terceira o
comando seria o seguinte: $a = $b + $c.
4.1.2. BioPerl
Bioperl é uma coleção de módulos Perl que facilitam o desenvolvimento de programas Perl
para aplicações em bioinformática. Ele fornece classes Perl reutilizáveis que facilitam a escrita de
scripts. O BioPerl não fornece programas completos, e sim métodos para tarefas comuns em
bioinformática, como manipulação de seqüências, acesso à banco de dados usando uma gama de
formatos disponíveis dos vários programas de biologia molecular incluindo Blast e ClustalW. O
bioinformata é responsável por escrever programas que unam estes módulos (BIOPERL, 2003).
O BioPerl está disponível em BioPerl (2003). Juntamente com a instalação do módulo
BioPerl está disponível o arquivo de ajuda e manual de instalação para os maiores sistemas
operacionais, incluindo Linux e Windows (TISDALL, 2001).

39
O módulo Bio::Perl fornece algumas funções simples, como por exemplo, este script
abaixo recupera uma seqüência do banco de dados do SwissProt (SWISS-PROT, 2003) e escreve-a
em formato Fasta. use Bio::Perl;
# Este script executa apenas em computador conectado internet.
$seq_object = get_sequence('swissprot',"ROA1_HUMAN");
write_sequence(">roa1.fasta",'fasta',$seq_object);
O Bio::Perl tem inúmeras outras funções para facilitar algumas tarefas, incluindo:
• get_sequence – recupera uma seqüência de um banco de dados.
• read_sequence – lê uma seqüência de um arquivo.
• read_all_sequences – lê todas as seqüências de um arquivo
• new_sequence – produz uma seqüência Bioperl de uma string apenas
• write_sequence – escreve uma seqüência ou um vetor de seqüências em um arquivo
• translate – fornece a tradução de uma seqüência de DNA em proteína
• translate_as_string – fornece a tradução de uma seqüência, devolvendo apenas uma string
• blast_sequence – faz um alinhamento BLAST (par-a-par) com todas as seqüências do NCBI.
• write_blast – escreve um relatório Blast em um arquivo.
4.2. HTML
A HTML (HyperText Markup Language) é uma linguagem de marcação: um conjunto de
indicadores (chamados de elementos) que definem o texto e os objetos do documento. A HTML
marca o documento com notas (conhecidas como tags) que têm o objetivo de informar ao software
que está lendo o documento, por exemplo, onde deverá ser iniciado um novo parágrafo ou se
determinado texto corresponde ao título de uma seção ou deve ser enfatizado (SAVOLA,
WESTENBROEK e HECK, 1997).
Não é necessário ter uma plataforma de computador específica para visualizar documentos
feitos em HTML, pois ela é uma linguagem portável, que pode ser transportada para qualquer
ambiente computacional (ibidem). Essa é uma das maiores facilidades encontradas na HTML.

40
A HTML é utilizada para fazer páginas da web. Ela produz desde textos simples até
formulários. Mas com a HTML não é possível fazer páginas web dinâmicas ou fazer páginas que
acessem algum banco de dados. Então, para solucionar este problema, geralmente é usada outra
linguagem para o acesso a banco de dados, como PHP e ASP (Active Server Pages), por exemplo.
Uma vantagem da HTML é que ela pode ser utilizada em conjunto com outras linguagens, como
por exemplo, com PHP e Java. Isso quer dizer que o código fonte de uma página web pode ter
códigos em PHP, HTML e Java.
4.2. PHP
PHP é uma linguagem em forma de script (parte de código HTML que é interpretado pelo
Browser ou pelo servidor web) que interage ao lado do servidor para a criação de páginas dinâmicas
na web (ANSELMO, 2000). O código PHP é escrito em um documento HTML para conferir-lhe a
capacidade de gerar instruções específicas (CASTAGNETTO et al, 2001).
O PHP pode ser obtido no site PHP oficial em PHP (2003), gratuitamente (ANSELMO,
2000). O pacote PHP básico é distribuído através de código-fonte e binários pré-compilados. Estes
são disponíveis como distribuições RPM (RPM Package Manager – utilitário usado para instalar,
desinstalar e atualizar pacotes de software, disponível em Herrold (2003)) para instalações Linux
que suportem o formato RPM (CASTAGNETTO et al, 2001).
As páginas que contêm código PHP são visualizadas no browser do usuário em formato
HTML apenas, preservando o código do programador. O servidor web fica encarregado de executar
o código PHP no servidor. Normalmente a saída de um código PHP é HTML, que é apresentada ao
usuário. Por exemplo, se uma página web mostra dados de uma tabela do banco de dados, o código
para essa tarefa pode ser feito em PHP. Mas o código que é mostrado na página do usuário tem
apenas tags HTML com os dados da tabela.
Os servidores web mais comuns são: Apache (principalmente para Linux) e IIS (Microsoft
Internet Information Server para Windows). No sistema descrito por este trabalho, será usado o
servidor Apache juntamente com o sistema operacional Linux.

41
A sintaxe do PHP é muito semelhante à do C e Perl, e de fácil aprendizado. Além disso,
outras vantagens que fazem do PHP uma linguagem muito utilizada são: com PHP pode-se criar e
alterar cookies (arquivos que determinadas páginas guardam no disco rígido do usuário para obter
referência do próprio usuário), autenticações, redirecionar usuários, e principalmente conectividade
com os mais variados bancos de dados (ANSELMO, 2000).
4.3. Banco de Dados
Em conseqüência da grande quantidade de informações de seqüências de nucleotídeos e de
aminoácidos que são produzidas atualmente, principalmente em projetos genoma, transcriptoma e
proteoma, o uso dos bancos de dados (BD) vem assumindo uma importância crescente na
bioinformática (PROSDOCIMI, 2003b).
Um banco de dados é um conjunto de informações manipuláveis de mesma natureza
inseridas em um mesmo local, obedecendo a um padrão de armazenamento. Ele é basicamente um
sistema de manutenção de informações por computador, capaz de manter as informações
organizadas e torná-las disponíveis quando solicitadas, com rapidez e confiabilidade. Existem
várias ferramentas para gerenciamento de banco de dados relacionais como MySQL, Oracle e
PostGreSQL (SILVA, 2001b).
Os modelos de banco de dados podem ser divididos em: modelos lógicos com base em
objetos, modelos lógicos com base em registros e modelos físicos. Cada um deles se divide em mais
categorias. O modelo lógico com base em registros se divide em: modelo relacional, modelo de rede
e modelo hierárquico. Este projeto usará o modelo relacional que é caracterizado pela utilização de
um conjunto de tabelas. Elas são usadas para representar tanto os dados como a relação entre eles.
Cada tabela possui múltiplas colunas (campos) e cada uma possui um nome único. Cada linha da
tabela é chamada de registro e contém os dados da tabela. O SGBD (Sistema Gerenciador de Banco
de Dados) é um sistema que tem como objetivo proporcionar um ambiente para a recuperação e
armazenamento das informações do banco de dados (SILBERSCHATZ, KORTH e SUDARSHAN,
1999).

42
4.3.1. SQL
Para trabalhar com um SGBD, é necessário utilizar uma linguagem de acesso ao banco. A
SQL (Structured Query Language) é certamente o padrão para a linguagem de banco de dados
relacionais. Ela é usada para consulta em banco de dados, para a definição da estrutura de dados,
para a modificação de dados no banco e para especificação de restrições de segurança no banco
(SILBERSCHATZ, KORTH e SUDARSHAN, 1999).
Os comandos SQL podem ser passados para o SGBD por outro programa (por exemplo, um
script que coleta dados em um formulário da web) ou digitados manualmente. É óbvio que a
primeira opção é ideal, especialmente para informar grandes quantidades de registros em um banco
de dados.
Dentro de um código SQL podem ser mencionadas variáveis do Perl. A aplicação acontece
de forma bem simples, sem necessidades de concatenação ou combinação de apóstrofo e aspas. Por
exemplo, pode ser digitado o seguinte comando no código fonte de programa em Perl: (“SELECT *
FROM Ofertas WHERE codi_ofe = $codigo”), onde $codigo é uma variável do Perl (SILVA,
2001b).
4.3.2. MySQL
O MySQL é um SGBD relacional simples, bastante fácil de instalar e executar. Ele está
disponível no site oficial do MySQL (MySQL, 2003), juntamente com toda a documentação
(SILVA, 2001b). Em Anexos I há a instalação do BD MySQL.
O MySQL é um SGBD de código aberto. Seu uso é gratuito, exceto na versão Windows ou
no uso profissional (lucrativo). Esse gerenciador de banco de dados possui arquitetura cliente-
servidor (SILVA, 2001b).
O MySQL é relativamente fácil de configurar e usar, e está disponível para várias
plataformas, entre elas: Linux, Windows, FreeBSD. Sua base de dados pode ser acessada por C,

43
C++, Eiffel, Java, Perl, PHP e Python, sendo o PHP a linguagem mais comum para acessar esse
gerenciador de banco de dados (ibidem).
O MySQL possui um conjunto pequeno de operações sobre os dados, em comparação com
outros bancos encontrados no mercado, como Oracle. É mais indicado para aplicações que
gerenciem BD pequenos ou médios, e não para aplicações que fazem uso intensivo de BD ou
possuem grandes quantidades de informação. Como o MySQL é um SGBD estritamente relacional,
ele é utilizado para armazenar tipos de dados usuais. Para a maioria das aplicações-padrão de banco
de dados, inclusive bioinformática, o MySQL é um excelente ponto de partida (GIBAS e
JAMBECK, 2001).
4.3.3. PostgreSQL
O PostgreSQL é um sofisticado sistema de gerenciamento de banco de dados relacional e
orientado a objetos, com mais de 15 anos de desenvolvimento. Ele é considerado objeto-relacional
por implementar, além das características de um SGBD relacional, algumas características de
orientação a objetos, como herança e tipos personalizados. Suporta quase todas as construções SQL,
incluindo subseleções, transações, tipos definidos pelo usuário e funções. É extremamente robusto e
confiável, além de ser extremamente flexível e rico em recursos. O PostgreSQL utiliza o modelo
cliente-servidor (PROSDOCIMI, 2003b).
Pela riqueza de recursos e conformidade com os padrões, ele é um SGBD muito adequado
para o estudo universitário do modelo relacional, além de ser uma ótima opção para empresas
implementarem soluções de alta confiabilidade sem altos custos de licenciamento.
É um programa que possui o seu código fonte disponível e o seu uso livre para aplicações
comerciais ou não. Atualmente está disponível seu código fonte, além de binários pré-compilados
em diversas plataformas (entre elas Linux) (POSTGRESQL, 2003). A versão atual é PostgreSQL
7.3, lançada em novembro de 2002 (PROSDOCIMI, 2003b).

44
O PostgreSQL é um pouco limitado em sua capacidade de administrar grande quantidade de
operações, em comparação com o Oracle e outros SGBD comerciais, mas para banco de dados
médio é um produto excelente (GIBAS e JAMBECK, 2001).
4.3.4. Comparação de Bancos de Dados Ambos os SGBDs citados anteriormente são muito utilizados nos sistemas web disponíveis.
Para bioinformatas, o BD mais comumente usado é o MySQL. Muitas das ferramentas de
bioinformática que utilizam bancos de dados têm sua base instalada em um servidor MySQL.
O MySQL tem uma principal vantagem sobre o PostGreSQL, que é a documentação. Há
muitos livros e páginas na internet que explicam como integrar MySQL com o servidor Apache e a
linguagem PHP. Além disso, existe o software PHPTriad, que instala e configura conjuntamente o
BD MySQL, servidor Apache e a linguagem PHP. Como este sistema está disponível na web, é
muito importante que seja usado um banco de dados que seja fácil de integrar com linguagens como
PHP e Perl, sendo que já existem módulos Perl que fazem a interação com MySQL. A instalação do
MySQL é relativamente fácil. Mas o principal ponto que deve ser considerado na hora de escolher
entre esses dois SGBDs é que o postgreSQL não é muito utilizado pela dificuldade no seu
gerenciamento (PROSDOCIMI, 2003b).

III. DESENVOLVIMENTO
1. MODELAGEM DO SISTEMA
A funcionalidade do sistema pode ser divida em 3 etapas: a busca de seqüências, a
preparação das seqüências e o projeto de primers. Para as duas primeiras etapas foi utilizada a
Análise Estruturada, e para a terceira etapa foi utilizada a modelagem Orientada a Objeto, com a
linguagem UML.
Para a Análise Estruturada são apresentados o diagrama de Contexto, os diagramas de
Fluxo de Dados e o Dicionário de Dados. Para a Análise Orientada a Objeto são apresentados o
Diagrama de Caso de Uso, o Diagrama de Classes e os Diagramas de Seqüência dos processos mais
complexos. Além desses, é apresentado o Diagrama Entidade-Relacionamento, e a descrição das
informações das tabelas do banco de dados do diagrama ER. No projeto do sistema é apresentada a
especificação dos processos da análise estruturada.
As ferramentas utilizadas na modelagem foram: Rational Rose 2000, para a modelagem
em UML, PowerDesigner ProcessAnalyst 6.1.0 para a modelagem Estruturada e AllFusion ERwin
Data Modeler 4.0 para o diagrama ER.
1.1. Análise do Sistema
1.1.1. Diagrama de Contexto
O Diagrama de Contexto é um diagrama de fluxo de dados composto por um único
processo que representa todo o sistema. Sua função é apresentar, graficamente, a comunicação do
sistema com o ambiente externo. A Figura 14 exibe o diagrama de contexto do sistema descrito
nesse trabalho.

46
Esse diagrama mostra os dados que são enviados ao sistema e os dados que o sistema
retorna. O usuário insere alguns dados no sistema, esse processa e retorna os resultados para o
usuário. O sistema, além de enviar os resultados para o usuário, envia dados para o GenBank e para
a classe Primer_Design (classe que projeta os primers). Estas duas entidades externas (simbolizadas
com um quadrado na Figura 14) retornam dados para o sistema, e este os apresenta ao usuário.
Figura 14. Diagrama de Contexto.
1.1.2. Diagramas de Fluxos de Dados
O Diagrama de Fluxo de Dados (DFD) é um elemento de modelagem usada para descrever
a transformação dos dados ao longo dos processos realizados pelo sistema. Na Figura 15 pode-se
ver o DFD de nível 1 (mais abstrato), com todos os processos e os fluxos associados. Os processos
(simbolizados como círculos) transformam as entradas recebidas das entidades externas e de outros
procesos e os depósitos de dados (simbolizados como retângulos), e produz novos dados que são
enviados a outros processos ou às entidades externas.

47
Figura 15. Diagrama de Fluxo de Dados, nível1.

48
Os processos 1.4, 1.5 e 1.6 são relativamente complexos e foram decompostos em sub-
processos. Nas Figuras 16, 17, 18 são exibidos os DFD’s oriundos da decomposição do DFD de
nível 1.
A Figura 16 mostra os sub-processos oriundos da decomposição do processo 1.4
(Exibir_Resultado_Busca_Sequencia). Este diagrama mostra os dados vindos do processo
Exibir_Param_Busca_ou_Primers e enviados para o processo 1.4.1. Este último faz a busca de
seqüências no GenBank. Então é executado o processo 1.4.2 que exibe as seqüências do banco e as
trazidas do GenBank para o usuário. Cada uma das seqüências contém um checkbox para o usuário
marcar se a seqüência deve ser considerada e se a seqüência deve anelar no primer.
Figura 16. Diagrama de Fluxo de Dados decomposto do processo 1.4.
A Figura 17 mostra os sub-processos oriundos da decomposição do processo 1.5
(Incluir_Seqüências_BD). Este diagrama mostra os dados que saem do processo
Exibir_Resultado_Busca_Seqüências e entram no processo 1.5.1. Nesse processo o sistema atualiza
o banco de dados com as seqüências selecionadas ou não na tela que exibe as seqüências. Para as
seqüências que não estão no banco e foram selecionadas na análise, o sistema acessa o GenBank e
traz as informações pertinentes para o processo 1.5.1.

49
Figura 17. Diagrama de Fluxo de Dados decomposto do processo 1.5.
A Figura 18 mostra a etapa de preparação das seqüências para o projeto de primers. O
processo 1.6.1 acessa o banco de dados, lê as seqüências que devem ser consideradas e coloca-as no
arquivo, em formato FASTA. Através do BioPerl, o processo 1.6.2 lê as seqüências do arquivo e
gera a fita consenso degenerada e a fita match. O processo 1.6.3 mostra no browser as fitas
consenso e match. As fitas consenso são as regiões nas quais os primers devem anelar.

50
Figura 18. Diagrama de Fluxo de Dados decomposto do processo 1.6.
1.1.3. Dicionário de Dados Utilizados na Modelagem do Sistema
O dicionário de dados consiste numa lista de todos os dados e fluxos utilizados nos
diagramas de Fluxo de Dados e a sua respectiva descrição, para uma melhor compreensão do
sistema.
Tabela 2. Dicionário de Dados da Modelagem do Sistema.
Nome Tipo Descrição
Accession Escalar *valor do atributo accession na página do GenBank*
Acesso Escalar *atributo do banco de dados*
Anelar Escalar *atributo do banco de dados*
Ano_Max Escalar {digito} *ano máximo da seqüência *
Ano_Min Escalar {digito} *ano mínimo da seqüência *
Autor Escalar *autor da seqüência*
Buffer Escalar *variável passada entre as telas*
Cabecalho Escalar *definição da seqüência no formato fasta*
Cbx_Alinhar_GI Escalar *nome do checkbox*

51
Cbx_Alinhar_Num_Seq Escalar *nome do checkbox*
Cbx_Considerar_GI Escalar *nome do checkbox*
Cbx_Considerar_Num_Seq Escalar *nome do checkbox*
Cbx_Padrão_GI Escalar *nome do checkbox*
Cbx_Padrão_Num_Seq Escalar *nome do checkbox*
Código_HTML Escalar *código HTML que retorna da pagina do GenBank*
Com_Palavra_chave Escalar *restriçao para query de busca de seqüências.Keyword*
Consenso Escalar *fita consenso gerada pelo alinhador*
Count Escalar {digito} *Count contém o numero de sequencias trazidas
do GenBank*
Definição Escalar *atributo do banco de dados*
Definição_BD Escalar *valor da definição que está no BD*
Definition Escalar *valor do atributo definition na página do GenBank*
Digitada Escalar *atributo do banco de dados*
Fonte Escalar *atributo do banco de dados*
Gene Escalar *gene da seqüência *
GI_atual Escalar *GI da seqüência atual*
GI_BD Escalar *Gi que está no BD*
GI_Organismo Escalar *GI da sequencia*
Ids Array *array que contem os numeros de gi das sequencias
trazidas do GenBank*
Keyword Escalar *palavra-chave de busca de seqüências*
Match Escalar *fita match gerada pelo alinhamento*
MeuEndereco Escalar *variável que contem a URL para acessar*
Nova_Definicao Escalar *Valor a ser inserido no banco*
Nova_Fonte Escalar *Valor a ser inserido no banco*
Nova_GI Escalar *Valor a ser inserido no banco*
Nova_ID Escalar *Valor a ser inserido no banco*
Nova_Seq Escalar *Valor a ser inserido no banco*
Num_Sequencia Escalar *Chave primaria do banco de dados*
Num_Seqüências_Exibidas Escalar {digito}*Número de seqüências para mostrar na tela de
seqüências trazidas do NCBI*

52
Opção Escalar [projeto | analise]
Organism Escalar *valor do atributo organism na página do GenBank*
Organismo Escalar *organismo da seqüência
Origin Escalar *valor do atributo origin na página do GenBank*
Parâmetros_primer Escalar *string com os parametros de primer*
Parametros_primers_avanca
dos
Escalar *string com os parametros internos de primer*
Primer_pronto Escalar *primer pronto*
Resultados_primer Escalar *resultados do primer para ser mostrado na tela*
Selecionada Escalar *atributo do banco de dados*
Sem_palavra_chave Escalar *restrição para query de busca de seqüências*
Sequence Escalar *sequence para arquivo fasta*
Sequencia Escalar *seqüência de DNA*
Sequencias Escalar *seqüências de DNA*
Source Escalar *valor do atributo source na página do GenBank*
Sumario Escalar *sumario das seqüências trazidas do GenBank*
Tam_Max Escalar {digito}*tamanho máximo da seqüência *
Tam_Min Escalar {digito}*tamanho mínimo da seqüência *
Accession Escalar *valor do atributo accession na página do GenBank*
Acesso Escalar *atributo do banco de dados* Anelar Escalar *atributo do banco de dados*
Ano_Max Escalar {digito} *ano máximo da seqüência *
Ano_Min Escalar {digito} *ano mínimo da seqüência *
Tabela 3. Dicionário de Dados dos Fluxos de Dados.
Nome Descrição
Buffer_cbx_Selecionados Buffer
Buffer_Consenso Buffer
Buffer_Opção_Yes_No Buffer
Buffer_Par_Avançados Parâmetros_primer_avancados
Buffer_Query_Busca Buffer

53
Buffer_Vazio Buffer
Código_HTML Código_HTML
Consenso_Match Consenso + match
Dados_Buscados_Seqüência Definition + accession + source + organism + origin
Dados_Para_Análise Seqüências + primer_pronto
Dados_Seq_Selecionadas Anelar + Selecionada + Digitada + Num_Sequencia
Dados_Seqüência_Digitada Acesso + Definição + Digitada + fonte + GI_Organismo +
Selecionada + Sequencia
Dados_Seqüência_Selecionadas Acesso + Anelar + Definição + Digitada + fonte +
GI_Organismo + fonte + Num_Seqüência + Selecionada +
Sequencia
GI_Seqüência_Atual GI_Atual
Ids_Count Ids + count
Linha_Consenso Consenso + match
Meu_Endereço_URL MeuEndereço
Opção_Principal_Escolhida buffer
Opção_Principal_Sistema Opção
Parâmetros_Primer Parâmetros_Primers + Parâmetros_primers_avancados
Restrições_Busca_Usuário ano_Max + ano_min + Autor + com_palavra_chave + Gene +
Num_sequencias_exibidas + Organismo + Keyword +
sem_palavra_chave + Tam_Max + Tam_Min
Resultado_Final resultados_primer
Resultados_Primer resultados_primer
Seq_Fasta_Para_Arq Sequence + cabecalho
Seqüência_Digitada nova_seq + nova_ID + nova_Definição + nova_GI + nova_Fonte
Seqüência_e_Cabeçalho Sequence + cabecalho
Sequencia_Primer_Analise Seqüências + primer_pronto
Seqüências_Digitadas Definição + GI_Organismo
Seqüências_Encontradas Count + ids + sumario + definicao_BD + GI_BD
Seqüências_Já_Cadastradas Acesso + Definição + GI_Organismo + Sequencia
Seqüências_para_Alinhamento Acesso + Definição + GI_Organismo + Sequencia
Seqüências_Selecionadas cbx_Padrao_GI + cbx_Alinhar_GI + cbx_Considerar_GI +

54
cbx_Padrao_Num_Seq + cbx_Alinhar_Num_Seq +
cbx_Considerar_Num_Seq
Sumário Sumário
Ultima_Seqüência_Arquivo sequence
URL_GenBank MeuEndereco
Usuário_Parâmetros_Primer parametros_primers
Após as etapas de busca e preparação das seqüências, é realizado o projeto/análise de
primers. A modelagem desta etapa é feita através da orientação a objetos, e inicia com o diagrama
de casos de uso.
1.1.4. Diagrama de Casos de Uso
Os Casos de Uso ou Use Cases descrevem as ações permitidas ao usuário do sistema. Na
Figura 19 são exibidos os use cases Analisa_Primer e Projeta_Primer.
Figura 19. Use Cases
Abaixo são apresentadas as características dos casos de uso exibidos na Figura 19. No
caso da análise, os primers já estão prontos, e deve-se apenas analisar suas características. No caso
de projeto, o sistema deve projetar primers cujas características satisfaçam as restrições impostas
pelo usuário.

55
Use Case: Analisa_Primer
Classe que dispara: Usuário
Funcionamento Básico
Usuário insere parâmetros para Análise de Primers Sistema exibe resultados de Análise de Primers
Use Case: Projeta_Primer
Classe que dispara: Usuário
Funcionamento Básico
Usuário insere parâmetros para Projeto de Primers
Sistema exibe resultados de Projeto de Primers
1.1.5. Diagrama de Classes
O diagrama de classes mostra as classes do sistema, atributos e métodos pertinentes a cada
classe e a relação entre as classes. Na Figura 20 é exibido o diagrama de classes do sistema descrito
nesse trabalho. Há quatro classes principais: TPrimerDesign, TPrimer, TPrimerParameters e
TBiblioteca. As classes Usuário e Interface são utilizadas na modelagem dos diagramas de
seqüência.
A classe TPrimerDesign é utilizada para guardar as informações do projeto de primers.
Essa classe armazena o a seqüência de DNA, a seqüência de primer, a enzima de restrição, o tipo de
primer (amplificação ou seqüenciamento), número de primers rejeitados por cada um dos
parâmetros, e os primers aprovados na fita sense e anti-sense. Os métodos dessa classe projetam ou
analisam o primer. Os métodos InternalParameters e DesignParameters apenas chamam os métodos
de mesmo nome implementados na classe TPrimerParameters.

56
Figura 20. Diagrama de Classes do Sistema
A classe TPrimerDesign cria um objeto da classe TPrimer, para que essa armazene as
informações do primer que está sendo analisado no método _DesignIndividualPrimers().O único
método dessa classe é o método construtor Create. A classe TBiblioteca possui funções para
manipulação de string e valores numéricos. Esta classe é utilizada como apoio às outras classes. A
classe TPrimerParameters possui os parâmetros de entrada inseridos pelo usuário.

57
1.1.6. Diagramas de Seqüência
O diagrama de seqüência mostra a troca de mensagens entre as classes em ordem
temporal. Na parte superior do diagrama estão as classes que fazem parte do processo e nas linhas
verticais visualiza-se a dimensão tempo. A importância desse tipo de diagrama é a visualização da
interação entre as classes e métodos necessários.
Na Figura 21 é apresentado o diagrama de seqüência das ações executadas no momento
em que o usuário aperta no botão para Projetar Primers da tela de parâmetros de primers. O sistema
atribui os parâmetros de projeto à classe TPrimerDesign. Se o usuário escolhe a opção Análise de
Primers, é executado o método DesignPrimers(); se o usuário escolhe a opção Projeto de Primers, é
executado o método AnalysePrimers().
Figura 21. Diagrama de Seqüência com as ações iniciais do sistema.
Na Figura 22 é apresentado o diagrama de seqüência das ações executadas no método
DesignPrimers(). Este método executa três outros métodos da própria classe. O método
DesignPrimers() é o método público principal da classe.
A Figura 23 mostra o diagrama de seqüência do método _DesignIndividualPrimers(). Este
método percorre a fita sense e a fita anti-sense (no caso de amplificação), e forma os primers de
acordo com as restrições do usuário. No final do método, é criada uma lista com todos os primers
aprovados na fita sense e outra lista para os primers aprovados na anti-sense.

58
Figura 22. Diagrama de Seqüência do método DesignPrimers()
Figura 23. Diagrama de Seqüência do método _DesignIndividualPrimers()
Após o método _DesignIndividualPrimers() formar os primers, o método
_DesignPairedPrimers() forma pares entre os primers já selecionados para a fita sense e anti-sense.

59
Os pares são formados analisando a diferença de temperatura de anelamento entre os primers, o
tamanho do amplicon, o número de dimmer entre os primers. Se foi formado um par de primers,
este é armazenado no atributo aPairs, da classe TPrimer. A Figura 24 mostra o diagrama de
seqüência do método _DesignPairedPrimers().
Figura 24. Diagrama de Seqüência do método _DesignPairedPrimers()
Se o usuário escolheu a opção inicial “Analisar Primers” ao invés de “Projetar Primers”, o
sistema executa o processo _AnalysePrimers(). Este processo mostra para o usuário as
características do primer que o usuário inseriu. A Figura 25 exibe o diagrama de seqüência do
método _AnalysePrimers().

60
Figura 25. Diagrama de Seqüência do método _AnalysePrimers()
1.1.7. Diagrama de Navegação do Sistema
O usuário inicia sempre a navegação do sistema na tela “Exibir Opções Principais”, e
finaliza o sistema na tela “Mostrar Resultados Primer”. Na primeira tela o usuário deve selecionar
umas das opções: Análise de Primers ou Projeto de Primers. Dependendo de qual opção o usuário
selecionar, a tela apresentada para o usuário será “Mostrar Parâmetros Análise” ou “Mostrar
Parâmetros Busca”. A Figura 26 mostra a navegação entre as telas do sistema.

61
Figura 26. Diagrama de Navegação do Sistema
1.1.8. Diagrama Entidade-Relacionamento
Para modelar o banco de dados do sistema utilizou-se o Diagrama Entidade-
Relacionamento (ER), dividido em modelo lógico (conceitual) e modelo físico. A Figura 27 mostra
o modelo lógico do banco de dados. O modelo Físico é apresentado na seção 1.2.2.
Várias tabelas que estão presentes na modelagem não foram utilizadas. Elas são
necessárias para o cumprimento dos objetivos secundários deste trabalho, não realizados, e
permanecem no banco visando a expansão do sistema atual.

62
Figura 27. Diagrama Entidade-Relacionamento, modelo Lógico.

63
1.1.9. Dicionário de Dados das Tabelas
As tabelas a seguir mostram o dicionário de dados do diagrama de entidade-
relacionamento utilizado no sistema. Em cada tabela está o nome da tabela do banco de dados, o
nome de cada campo, o tipo e a descrição do campo.
Tabela 4. Tabela Degeneracao
Nome: Degeneração
Nome do campo Tipo do campo Descrição do campo
Id_degeneracao UniqueID Chave primária
Letra Varchar (1) Letra que identifica a degeneração
Nucleotideo Varchar (6) Nucleotídeos que são identificados por esse degeneração
Tabela 5. Tabela Tipo_PCR
Nome: Tipo_PCR
Nome do campo Tipo do campo Descrição do campo
Id_tipo_pcr UniqueID Chave primária
Descricao_pcr Varchar (20) Tipo da pcr (seqüenciamento ou amplificacao)
Tabela 6. Tabela Projeto_PCR
Nome: Projeto_PCR
Nome do campo Tipo do campo Descrição do campo
Id_projeto_PCR UniqueID Chave primária
Nome_projeto Varchar (20) Nome do projeto dado pelo usuário
Tabela 7. Tabela Primer_Rejeitado
Nome: Primer_Rejeitado
Nome do campo Tipo do campo Descrição do campo
Id_primer_rejeitado UniqueID Chave primária
Reject_Ta Integer Número de primers rejeitados por temp. anelamento

64
Reject_GC Integer Número de primers rejeitados por percentual de GC
Reject_CG3L Integer Número de primers rejeitados por percentual CG 3’
Reject_3L Integer Número de primers rejeitados por alinhamento perfeito 3’
Reject_DiffTa Integer Número de primers rejeitados por dif. temp anelam.
Reject_Palindr Integer Número de primers rejeitados por palindromes
Reject_2ndHairpin Integer Número de primers rejeitados por num. de hairpins
Reject_2ndBudges Integer Número de primers rejeitados por num. de budges
Reject_2ndDimmer Integer Número de primers rejeitados por num. de dimmers
Reject_Degener Integer Número de primers rejeitados por num. degenerações
Reject_Degener3L Integer Número de primers rejeitados por num. degenerações 3’
Reject_Amplicon Integer Número de primers rejeitados por tam. amplicon
Tabela 8. Tabela Produto_Amplificado
Nome: Produto_Amplificado
Nome do campo Tipo do campo Descrição do campo
Id_produto_amplificado UniqueID Chave primária
Tamanho Integer Tamanho do produto amplificado
Tabela 9. Tabela Corte
Nome: Corte
Nome do campo Tipo do campo Descrição do campo
Id_corte UniqueID Chave primária
Pos_corte Integer Posição do corte da enzima usada no produto
amplificado
Tabela 10. Tabela Usuario
Nome: Usuário
Nome do campo Tipo do campo Descrição do campo
Id_usuario UniqueID Chave primária
Nome_usuário Varchar (20) Nome do usuário
Login_usuario Varchar (20) Login do usuário
Senha_usuário Varchar (20) Senha do usuário

65
Num_logins_usuário Integer Número de logins efetuados por esse usuário
Tabela 11. Tabela Enzima_Usada
Nome: Enzima_Usada
Nome do campo Tipo do campo Descrição do campo
Id_enzima_usada UniqueID Chave primária
Tabela 12. Tabela Enzima
Nome: Enzima
Nome do campo Tipo do campo Descrição do campo
Id_enzima UniqueID Chave primária
Nome_enzima Varchar (20) Nome da enzima
Sequencia_enzima Varchar (20) Seqüência de bases nucléicas da enzima
Pos_corte_enzima Integer Número da posição de corte dessa enzima
Tabela 13. Tabela Resultado_Alinhamento
Nome: Resultado_Alinhamento
Nome do campo Tipo do campo Descrição do campo
Id_resultado_alinhamento UniqueID Chave primária
Seqüência_alinhada Varchar (20) Letras que identificam a seq. Alinhada (por
exemplo: agtc---s) onde “s” é uma degeneração e
“–“ é um gap
Tabela 14. Tabela Organismo
Nome: Organismo
Nome do campo Tipo do campo Descrição do campo
Num_sequencia UniqueID Chave primária
Seqüência MediumText Seqüência a ser feita o primer
GI_Organismo Varchar (20) Número de gi de identificação no GenBank
Anelar Boolean O primer deve ou não anelar a essa seqüência
Selecionada Boolean Considerar ou não essa seqüência para fazer o primer
Digitada Boolean Seqüência digitada ou buscada no GenBank
Acesso Varchar (20) ID acesso do organismo (dado do GenBank)

66
Definição Varchar (20) Definição do organismo (dado do GenBank)
Fonte Varchar (20) Fonte do organismo (dado do GenBank)
Tabela 15. Tabela Região_Similar
Nome: Regiao_Similar
Nome do campo Tipo do campo Descrição do campo
Id_regiao_similar UniqueID Chave primária
Pos_inicio_regiao_similar Integer Posição inicial da região similar
Tamanho_região_similar Integer Tamanho da região similar
Tabela 16. Tabela Similaridade
Nome: Similaridade
Nome do campo Tipo do campo Descrição do campo
Id_reg1 UniqueID Identificador da primeira região similar
Id_reg1 UniqueID Identificador da segunda região similar
Tabela 17. Tabela Posição_Anelamento
Nome: Posicao_Anelamento
Nome do campo Tipo do campo Descrição do campo
Id_posicao_anelamento UniqueID Chave primária
Sense Boolean Sense ou anti-sense
Posicao_anelamento Integer Posição que o primer se anela à seqüência
Tabela 18. Tabela Tipo_Est_Secundaria
Nome: Tipo_Est_Secundaria
Nome do campo Tipo do campo Descrição do campo
Id_tipo_est_secundaria UniqueID Chave primária
Descricao_est_secundaria Varchar (20) Nome da estrutura secundaria (loop, hairping...)
Tabela 19. Tabela Est_Secundaria
Nome: Est_Secundaria
Nome do campo Tipo do campo Descrição do campo
Id_est_secundaria UniqueID Chave primária

67
Seq_est_secundaria Varchar (20) Seqüência da estrutura secundária
Tabela 20. Tabela Primer
Nome: Primer
Nome do campo Tipo do campo Descrição do campo
Id_primer UniqueID Chave primária
Seqüência_primer Varchar (20) Seqüência do primer
Tm Float Temperatura de desnaturação do primer
Ta Float Temperatura de anelamento do primer
Tabela 21. Tabela Par_Primer
Nome: Par_Primer
Nome do campo Tipo do campo Descrição do campo
Id_par_primer UniqueID Chave primária
Par_primer_selecionado Boolean Primer foi selecionado ou não
Tabela 22. Tabela Codon
Nome: Códon
Nome do campo Tipo do campo Descrição do campo
Id_codon UniqueID Chave primária
Codon Varchar (3) Três letras que identificam o codon (exemplo: atc)
Amino Varchar (20) Nome do aminoácido traduzido pelo codon
Tabela 23. Tabela Conjuntos_Organismos
Nome: Conjuntos_organismos
Nome do campo Tipo do campo Descrição do campo
Id_conj UniqueID Chave primária
Descricao Varchar (20) Nome do conjunto de organismos que contém o codon. O
mesmo codon pode traduzir diferentes aminoácidos em
diferentes organismos.

68
1.2. Projeto do Sistema
1.2.1. Especificação de Processos
A Especificação de Processos determina os passos a serem seguidos para a sua
implementação, ou seja, as etapas necessárias para a execução de uma atividade. A seguir são
apresentadas as Especificações dos Processos do sistema, descritas em pseudocódigo:
Especificação o Processo 1.1, Exibir_Opçoes_Principais. O usuário escolhe uma das opçoes. O usuário clica em "Next". Chama "Exibir_Param_Busca_ou_Primers"
Especificação o Processo 1.2, Exibir_Param_Busca_ou_Primers. /*recebe buffer que contém o valor da opção que o usuário escolheu na tela anterior/* SE opção = projeto ENTAO ENCONTRA Seqüência, Acesso, GI_Organismo, Definição EM Organismo ENQUANTO existir registro FACA mostrar tabela com registros FIM ENQUANTO mostra botão “Type Sequences” SE usuario clicar em botão “Type Sequences” ENTAO chama Digitar_Sequencia mostra parâmetros para busca de seqüências mostra botões SE usuário clicar em botao “Next” ENTAO chama Exibir_Resultado_Busca_Sequencias SENAO SE usuário clicar em botao “Back” ENTAO chama Exibir_Opçoes_Principais SENAO mostra parâmetros para analise de primers. mostra botões SE usuário clicar em botão “Next” ENTAO chama Imprimir_Resultados_Primers SENAO SE usuário clicar em botão “Back” ENTAO chama chama Exibir_Opçoes_Principais FIM SE FIM SE
Especificação o Processo 1.3, Digitar_Sequencia. dados_sequencias = buffer SE usuário digitou seqüência ENTAO INSERIR Acesso, Definição, GI_Organismo, Fonte, Seqüência, Anelar, Selecionada, Digitada EM Organismo mostra formulário com campos para inserção de seqüências SE usuário clicar em botão Include ENTAO

69
chama este mesmo procedimento FIM SE ENCONTRA Seqüência, Acesso, GI_Organismo, Definição EM Organismo ENQUANTO existir registros FACA mostra registro FIM ENQUANTO SE usuário clicar em botão “Next” ENTAO chama Exibir_Param_Busca_ou_Primers FIM SE
Especificação o Processo 1.7, Imprimir_Parametros_Primer. mostra (cabecalho_HTML) mostra (formulario_com_campos_parametros_projeto_primers) */campo para usuário inserir seqüência recebe a linha consenso ou a seqüência de DNA /* SE (usuário clicar em "Next") ENTAO Envia Dados para Classe_Primer chama Imprimir_Resultados_Primers SENAO SE (usuário clicar em "BACK") ENTAO chama Fazer_Mult_Align SENAO SE (usuário clicar em "Clear") ENTAO Limpa os campos da tela FIM SE mostra (formulario_com_botao_parametros_avancados_projeto_primers) SE (usuário clicar em "Others Parameters") ENTAO chama Imprimir_Parameters_Avancados_Primers FIM SE
Especificação o Processo 1.8, Imprimir_Parametros_Avancados_Primers. mostra (cabecalho_HTML) mostra (formulario_com_campos_parametros_avancados_projeto_primers) SE (usuário clicar em "BACK") ENTAO Imprimir_Parametros_Primers FIM SE
Especificação o Processo 1.9, Imprimir_Resultados_Primers. recebe dados da classe Primer_Design mostra Resultados_Primers mostra botão finalizar SE (usuário clicar em botão “conclude”) ENTAO Apaga as seqüências que estão no banco FIM SE
Especificação o Processo 1.4.1, Executar_Busca_GenBank. mostra (cabecalho HTML da Tela) variaveis_de_restricoes = buffer query_string = query_string + variaveis_de_restricoes chama Imprimir_sequencias_encontradas

70
Especificação o Processo 1.4.2, Imprimir_sequencias_encontradas. ESCREVE count /*sequencias digitadas que estão no banco/* ENCONTRAR Num_Sequencia, GI_Organismo, Definicao EM Organismo COM Digitada = 1 LE registro ENQUANTO existir registro FACA mostra registro FIM ENQUANTO */seq que estão no banco, que foram buscadas no GenBank/* ENCONTRAR GI_Organismo EM Organismo COM Digitada = 0 LE registro ENQUANTO existir registro FACA meuendereco = URL base GenBank + registro sumario = HTML da seqüência ids[i] sumario = texto do HTML entre as tags de HTML "<dd>" e "<br></dd>" mostra tabela com registro encontrado e sumario FIM ENQUANTO PARA i DE 0 ATE count FACA ENCONTRAR Num_Sequencia EM Organismo COM GI_Organismo =ids[i] AND Digitada = 0 LE registro SE não existe registro ENTAO meuendereco = URL base GenBank + ids[i] sumario = HTML da seqüência ids[i] sumario = texto do HTML entre as tags "<dd>" e "<br></dd>" mostra tabela com ids[i] e sumario FIM SE FIM PARA mostra (formulario com botões de Back, Next, e Clear) SE (usuário clicar em "Next") ENTAO chama Incluir_Sequencias_BD SENAO SE (usuário clicar em "BACK") ENTAO chama Exibir_Param_Busca_ou_Primers FIM SE
Especificação o Processo 1.5.1, Atualizar_BD. mostra (cabecalho HTML da Tela) */buffer é a variavel que vem da tela anterior/* */em buffer há apenas os checkboxes selecionados e outros componentes/* PARA CADA checkbox selecinado na tela anterior ENTAO vetor = nome do checkbox FIM SE PARA i ATE tamanho_vetor FACA SE padrao ESTÁ CONTIDO EM vetor[i] ENTAO SE Digitado ESTÁ CONTIDO EM vetor[i] ENTAO Num_Sequencia_Atual = (numero que é obtido atraves de expressões regulares, a partir do nome do checkbox. O nome do checkbox tem o numero do índice da seqüência no banco) SE considerar ESTÁ CONTIDO EM vetor[i + 1] ENTAO alinhar = (se alinhar ESTÁ CONTIDO EM vetor [i + 2]) ALTERAR (Anelar, Selecionada, Digitada) EM Organismo PARA (0,0,1) COM Num_Sequencia = Num_Sequencia_atual; SENAO

71
ALTERAR (Anelar, Selecionada, Digitada) EM Organismo PARA (Alinhar, 1, 1) COM Num_Sequencia = Num_Sequencia_atual; FIM SE SENAO GI_atual = (numero que é obtido através de expressões regulares, a partir do nome do checkbox. O nome do checkbox tem o numero do GI. SE considerar ESTÁ CONTIDO EM VETOR[I + 1] ENTAO alinhar = (se alinhar ESTÁ CONTIDO EM vetor [i + 2]) ENCONTRA Num_Sequencia EM Organismo COM Digitada = 1 and GI_Organismo = GI_Atual; SE existe registro ENTAO ALTERA (Anelar) EM Organismo PARA (alinhar) COM Num_Seqüência = registro SENAO chama Recuperar_Sequencia INSERIR (Acesso, Definicao, GI_Organismo, Fonte, Sequencia, Anelar, Selecionada, Digitada) EM Organismo VALORES (accession, definition,GI_atual,source,origin,alinhar, 1, 0) FIM SE SENAO ENCONTRA Num_Sequencia EM Organismo COM Digitada = 0 AND GI_Organismo = GI_atual SE (existe registro) ENTAO REMOVER EM Organismo COM Num_Sequencia = registro FIM SE FIM SE FIM SE FIM SE FIM PARA SE (Btn_Yes_No = Yes) ENTAO chama Fazer_Mult_Align SENAO chama Exibir_Resultado_Busca_Sequencias FIM SE
Especificação o Processo 1.5.2, Recuperar_Sequencia. meuendereco = URL base do GenBank + gi_atual codigo = HTML da seqüência de numero gi_atual codigo = texto do HTML entre as tags "<pre>" e "</pre>" definition = texto da variavel codigo entre as palavras "DEFINITION" e "ACCESSION" accession = texto da variavel codigo entre as palavras "ACCESSION" e "VERSION" source = texto da variavel codigo entre as palavras "SOURCE" e "ORGANISM" organism = texto da variavel codigo entre as palavras "ORGANISM" e "REFERENCE" origin = texto dda variavel codigo entre as palavras "ORIGIN" e "//"
Especificação o Processo 1.6.1, Colocar_Seq_em_Arq. ENCONTRAR GI_Organismo, Acesso, Definicao, Sequencia EM Organismo COM Selecionada = 1 AND Anelar = 1 LE registro ABRE arquivo ENQUANTO existir registro FACA cabecalho = registro[0] + gb + registro[1] + | + registro[2] sequence = registro[3] ESCREVE em arquivo cabecalho e sequence

72
FIM ENQUANTO FECHA arquivo
Especificação o Processo 1.6.2, Fazer_Consenso. ENCONTRAR COUNT (num_sequencia) EM Organismo COM Selecionada = 1 AND Anelar = 1 LE count /*numero de registros do banco*/ SE count > 1 ENTAO LE sequencias do arquivo mostra sequencias atraves do BioPerl faz o alinhamento com as sequencias do arquivo atraves do BioPerl gera a fita consenso atraves do BioPerl gera a fita match SENAO SE count > 0 ENTAO consenso = sequence SENAO consenso = ' '; FIM SE
Especificação o Processo 1.6.3, Fazer_Match. ESCREVE consenso ESCREVE match SE usuário clica em “Next” ENTAO chama Imprimir_Parametros_Primer SENAO SE usuário clica em “Back” ENTAO chama Exibir_Resultado_Busca_Sequencias FIM SE
1.2.2. Modelo Físico do Banco de Dados
A Figura 28 mostra o modelo físico do banco de dados.

73
Figura 28. Diagrama Entidade-Relacionamento, modelo Físico.

74
2. DESCRIÇÃO DO SISTEMA PROPOSTO
O sistema descrito nesse trabalho encontra-se disponível no site do grupo de
bioinformática da Univali, em http://bioinformatica.cttmar.univali.br/express/TCC_opcoes.html.
O usuário do sistema pode optar entre duas opções iniciais. A primeira opção é Projeto de
PCR Completo e a segunda opção é Análise de Primers. A primeira opção inclui três etapas. As
etapas são Busca e/ou Inclusão de Seqüências, Alinhamento Múltiplo das seqüências a serem
analisadas e o Projeto de Primers. Na segunda opção, apenas há a Análise de Primers, que tem
como finalidade exibir as características do(s) primer(s) inseridos na análise. A Figura 29 exibe a
primeira tela do sistema.
Figura 29. Tela de opções principais do sistema.

75
2.1. Busca de Seqüências
A busca de seqüências tem como objetivo definir as seqüências que serão submetidas ao
experimento de PCR. Pode-se especificar que primers amplifiquem ou não trechos dessas
seqüências. Por enquanto nada é realizado com as seqüências com as quais os primers não devem
anelar. O usuário pode digitar diretamente as seqüências ou pode simplesmente apresentar palavras-
chaves e outras restrições, e o sistema irá, automaticamente, acessar bancos de dados públicos e
recuperar as seqüências relacionadas a essas palavras.
A busca de seqüências é realizada no banco de dados genômicos GenBank. Através de
palavras-chaves e outras restrições que o usuário insere, o sistema acessa remotamente o banco de
dados GenBank disponível em NCBI (NATIONAL CENTER FOR BIOTECHNOLOGY
INFORMATION, 2003). As restrições de busca são: Organismo, Autor, Com as palavras-chaves,
Sem as palavras-chaves, Tamanho da seqüência, Nome do Gene, Ano de Publicação da seqüência.
A Figura 30 mostra a tela de parâmetros de busca de seqüências. Essas restrições são muito
importantes para o usuário, pois minimiza o número de seqüências que não interessam. Se o usuário
não deseja fazer busca de seqüências no GenBank, ele pode simplesmente limpar os campos de
busca com o botão Limpar e avançar para a próxima tela.

76
Figura 30. Tela de parâmetros de busca de seqüências

77
Após o acesso ao GenBank, o sistema exibe o número de seqüências encontradas e o
sumário de cada uma. O sumário contém uma pequena descrição das seqüências e é um link para a
descrição completa. Se o resultado não for satisfatório, o usuário pode voltar à busca e alterar seus
parâmetros. Para cada uma das seqüências exibidas, existe um campo onde o usuário define se a
seqüência deve fazer parte do projeto de primer, e outro campo onde o usuário define se os primers
projetados devem anelar na seqüência ou não.
Depois que o usuário selecionou as seqüências que devem fazer parte da análise, o sistema
acessa novamente o GenBank e traz as seqüências e outras características para serem armazenadas
no banco de dados no servidor. As características das seqüências e as próprias seqüências são
encontradas na página HTML da seqüência no GenBank e analisadas através de expressões
regulares. As características das seqüências armazenadas no banco de dados são: definição, acesso,
fonte, organismo e a seqüência. A tabela utilizada nessa etapa do projeto preenche a tabela
Organismo.
A Figura 31 mostra a relação das seqüências encontradas, de acordo com os parâmetros
exibidos na Figura 30. Segundo os parâmetros foram encontradas 68 seqüências no GenBank, mas
na Figura 31 está sendo mostradas apenas 3 das seqüências encontradas. A primeira seqüência é
uma seqüência que já está cadastrada no banco de dados no servidor. A diferença entre as
seqüências digitadas e as trazidas do GenBank é que as seqüências buscadas no GenBank contêm
um link para a página do NCBI, e as outras não contêm link nenhum.

78
Figura 31. Tela com o sumário de algumas seqüências encontradas.
O usuário do sistema pode digitar as seqüências e informações a respeito das mesmas. Esta
característica do sistema permite ao usuário inserir as seqüências com as quais ele costuma
trabalhar. As informações que o usuário pode inserir a respeito das seqüências digitadas são: ID, GI,
definição e fonte (organismo) da seqüência, como mostra a Figura 32. Para as seqüências digitadas

79
também há a possibilidade do usuário definir a quais delas o primer deve se anelar e quais
seqüências devem fazer parte do projeto de PCR. Se as seqüências digitadas não são consideradas
no projeto, elas permanecem no banco de dados, para que o usuário possa voltar e considerar a
seqüência sem ter que inserí-la no banco novamente.
Figura 32. Tela de Digitação de Seqüências

80
Esta característica do sistema de inserir as seqüências possibilita ao usuário definir as
seqüências e fazer o projeto de primers, sem a necessidade de utilizar mais que um sistema para o
projeto de PCR. Não há a necessidade do usuário procurar as seqüências em uma página web e fazer
o projeto de PCR em outro sistema.
O acesso ao site do NCBI para a busca de seqüências no banco de dados GenBank é feita
através da biblioteca BioPerl. O módulo utilizado é Bio::DB::Query::GenBank. O código abaixo
executa a busca de seqüências através das palavras-chave($query_string) e retorna no atributo
count o número de seqüências encontradas e no atributo ids os números de gi das seqüências
encontradas. my $query = Bio::DB::Query::GenBank->new (-db => 'nucleotide', -query=> $query_string, -mindate=> $ano_min, -maxdate=> $ano_max); my $count = $query->count; #número de seqüências encontradas my @ids = $query->ids; #os números de GI das seqüências encontradas
2.2. Preparação de Seqüências
Esta etapa é necessária para o caso de um único primer dever anelar em duas ou mais
seqüências (multiplex) ou para o caso do projeto de primers degenerados. Para que um primer
amplifique mais de uma seqüência é necessário que o primer projetado seja complementar a todas
as seqüências. Nesta etapa é feito um alinhamento múltiplo com todas as seqüências que o primer
deve anelar, e o primer será gerado de acordo com a fita consenso do alinhamento. Se houver
apenas uma seqüência para a análise, esta etapa não será executada.

81
Figura 33. Tela com o resultado do alinhamento feito pelo software ClustalW.
Uma das vantagens desse sistema é permitir projetos de PCR nos quais os primers se
anelam a mais de uma seqüência. Isso pode ser útil nos casos de detecção de genes patógenos em

82
qualquer estirpe de organismo. Para que isso ocorra, é necessário encontrar regiões comuns a todas
as seqüências as quais o primer deve anelar (seqüências positivas). Além disso, essas regiões não
podem ser encontradas nas seqüências em que o primer não deve anelar (seqüências negativas).
Para tanto, é realizado o alinhamento múltiplo com as seqüências positivas. Regiões de grande
conservação (pouca degeneração) estão identificadas na fita consenso resultante. A fita consenso
resultante então é repassada à etapa de projeto de primers e armazenada na tabela
Resultado_Alinhamento. Neste projeto não está sendo feito um alinhamento par-a-par com as
seqüências nas quais os primers não devem anelar.
Por exemplo, o usuário pode requisitar um primer que se anele a todos as seqüências de
Papillomavirus (ex: Human papillomavirus type 81, 89, 90) para descobrir se a seqüência contém
um determinado gene. Nesse caso é necessário encontrar uma região conservada entre as seqüências
para fazer um único primer, se possível. A região mais conservada é aquela que contém um número
menor de bases degeneradas.
O alinhamento das seqüências para o projeto de primers é feito no software ClustalW para
Linux. Este software é acessado através da biblioteca BioPerl. Os módulos utilizados são:
Bio::Tools::Run::Alignment::Clustalw, Bio::SimpleAlign, Bio::PrimarySeq e Bio::SeqIO. Os
principais parâmetros de entrada utilizados para o ClustalW são: o arquivo contendo as seqüências e
a definição da cada uma, a matriz utilizada (BLOSUM) e o arquivo de saída (extensão .aln). O
arquivo com extensão “.aln”, contém o alinhamento múltiplo. Depois do arquivo gerado, é utilizado
o método Bio::SimpleAlign::consensus_iupac para a geração da fita consenso degenerada. Para a
geração de uma segunda fita consenso, chamada de fita “match”, é utilizado o método
Bio::SimpleAlign::match_line(). Este método gera uma fita com o símbolo * nas posições onde o
alinhamento é perfeito. O pedaço do código utilizado para o alinhamento é exibido abaixo. $str = Bio::SeqIO->new(-file => 'TCC_Sequences.fasta', '-format' => 'Fasta' ); @seq_array = (); while ( my $seq = $str->next_seq() ) { push (@seq_array, $seq) ;} $seq_array = \@seq_array; $factory = Bio::Tools::Run::Alignment::Clustalw->new(@params); @params = ('matrix' =>'BLOSUM', 'OUTFILE' => 'TCC_Sequences.aln' ); $aln = $factory->align($seq_array); $consenso = $aln->consensus_iupac(); $match = $aln->match_line();

83
A Figura 34 mostra um pedaço do arquivo gerado pelo alinhamento feito pelo ClustalW,
com extensão .aln. PileUp MSF: 714 Type: N Check: 2251 .. Name: gi|48374930 oo Len: 714 Check: 4033 Weight: 50.0 Name: gi|48374929 oo Len: 714 Check: 8218 Weight: 50.0 // gi|48374930 .......... ........TG TATGTGCTTG TAAATATTAA GTTGTATGTG gi|48374929 TATATGTTTG TATGTGCTTG TATGTGCTTG TAAATATTAA GTTGTATGTG gi|48374930 TGTTTGTATG TATGGTATAA TAAACACGTG TGTATGTGTT TTTAAATGCT gi|48374929 TGTTTGTATG TATGGTATAA TAAACACGTG TGTATGTGTT TTTAAATGCT gi|48374930 TGTGTAACTA TTGTGTCATG CAACATAAAT AAACTTATTG TTTCAACACC gi|48374929 TGTGTAACTA TTGTGTCATG CAACATAAAT AAACTTATTG TTTCAACACC gi|48374930 TACTAATTGT GTTGTGGTTA TTCATTGTAT ATAAACTATA TTTGCTACAT gi|48374929 TACTAATTGT GTTGTGGTTA TTCATTGTAT ATAAACTATA TTTGCTACAT gi|48374930 CCTGTTTTTG TTTTATATAT ACTATATTTT GTAGCGCCAG TGGCCATTTT gi|48374929 CCTGTTTTTG TTTTATATAT ACTATATTTT GTAGCGCCAG CGGCCATTTT gi|48374930 GTAGCTTCAA CCGAATTCGG TTGCATGCTT TTTGGCACAA AATGTGTTTT gi|48374929 GTAGCTTCAA CCGAATTCGG TTGCATGCTT TTTGGCACAA AATGTGTTTT Figura 34. Figura com alinhamento feito pelo software ClustalW
2.3. Projeto de Primers
Esta etapa visa projetar um ou mais primers de acordo com a(s) seqüência(s) que o usuário
informa ou analisar um ou vários primers prontos fornecidos pelo usuário. Através de uma ou várias
seqüências fornecidas pelo usuário e outros parâmetros de entrada o sistema apresenta os primers
encontrados que satisfaz as restrições. Se o usuário selecionar apenas uma seqüência, o sistema
projetará um primer para a seqüência. Mas se o usuário inserir mais de uma seqüência, o sistema
projetará um primer baseado na fita consenso gerada na etapa de preparação das seqüências.

84
As Figuras 35 e 36 mostram as telas de parâmetros de primers. A primeira tela (Figura 35)
contém os campos para os parâmetros mais simples, enquanto que a segunda tela (Figura 36),
contém os campos para o usuário inserir os parâmetros internos de primers.
Figura 35. Tela com os parâmetros de entrada para o projeto de primers.

85
Figura 36. Tela com os parâmetros de entrada internos para o projeto de primers.
O usuário pode deixar parâmetros de entrada das características dos primers em branco. O
sistema utiliza valores padrão, ou desconsidera o parâmetro, ou em alguns casos, calcula o valor

86
baseado em outros parâmetros. Por exemplo, o sistema calcula o tamanho do primer baseado na
percentagem de CG da seqüência e na Tm desejada.
O projeto inicia-se definindo os tamanhos mínimo e máximo para o primer, para que o
mesmo tenha uma Ta (temperatura de anelamento) compatível com os valores indicados de Ta,
%GC e bases na ponta 3’(tail bases). A seguir, a seqüência é percorrida, e para cada base
percorrida, de LenMin (tamanho mínimo do primer) até LenMax (tamanho máximo) bases
seguintes são analisadas.
O princípio é rejeitar o primer assim que alguma restrição não for satisfeita. Inicia-se
avaliando as restrições que possam permitir percorrer a seqüência mais rapidamente, economizando
tempo de processamento. Exemplificando: o número máximo permitido de seqüências contínuas
iguais é maxCont. Se o sistema está buscando primers da posição 1 até a posição 20, e encontra ‘X’
bases contínuas iguais a partir da base 12, então o sistema só começa a procura de novos primers a
partir da posição (12 + X – maxCont + 1). Isso elimina a procura de primers em muitas bases.
O primer é formado a partir do trecho da seqüência sob análise. No caso da fita sense, o
primer é seu complemento e sua ponta 3’ está no início da seqüência. No caso da fita anti-sense, o
primer é a própria seqüência sense, e sua ponta 3’ está no final de sua seqüência. A Figura 37
mostra o esquema de uma fita sense e uma fita anti-sense, os primers anelados e o sentido de
amplificação das fitas.
Figura 37. Fitas sense e anti-sense
Uma vez com a seqüência do possível primer determinada, iniciam-se as verificações de
cumprimento das demais restrições. Cada restrição pode ser testada ou não, dependendo se o
usuário estabelecer limites ou não. As restrições estabelecidas podem ser:
• Degenerações: procuram-se bases degeneradas a partir do final do primer. Se o máximo de
degenerações for encontrada na posição K do primer, avança-se a busca na seqüência para a

87
posição (posição_atual + K). Considera-se uma degeneração uma base alternativa, assim, S
(C ou G) é considerada uma degeneração, B (T ou C ou G) é considerada 2 degenerações e
N (A ou T ou C ou G) é considerada 3 degenerações.
• Degenerações 3’: esta restrição segue a mesma regra da descrita anteriormente, mas verifica
apenas as degenerações na extremidade 3’.
As restrições acima permitem avançar a busca de primers na seqüência em várias bases.
As próximas restrições apenas rejeitam primers, mas a busca continua a partir da base atual.
Exceção para harpin, dimmer e budge que consomem tempo de processamento e são deixadas para
o final dos testes de restrição. As próximas restrições são:
• %GC 3’: Nessa verificação conta-se o número de bases G e bases C e divide-se pelo
tamanho da ponta 3’ (geralmente 7 bases). Bases degeneradas são consideradas da seguinte
maneira: C, G ou S (C ou G) = 1; B (C ou G ou T) = 2/3; H (A ou C ou T) = 1/3, etc.
• %GC: idem à restrição anterior, mas considera todo o primer. Só verifica-se todo o primer
se a restrição %GC 3’ foi satisfeita.
• Ta: a temperatura de anelamento do primer é calculada a partir da fórmula:
[62,3oC + 0,41 (C + G) – 500/length] – 5oC
onde:
C + G = número de bases C + número de bases G, incluindo suas degenerações.
Length = tamanho da seqüência do primer
• Alinhamento 3’: verifica-se se há alinhamento perfeito nas últimas ‘x’ bases.
• Hairpins: o procedimento para achar hairpins e budges é o mesmo. O que os diferencia é
que os hairpins são buldges nos quais a estrutura secundária inclui as 3 últimas bases da
ponta 3’.
A Figura 38 apresenta a tela com o resultado do projeto de primers. O resultado é
apresentado da mesma forma para a análise e projeto de primers. Para a análise são apresentadas as
características dos primers dados pelo usuário. Para o projeto são apresentados os primers
aprovados na fita sense e anti-sense, e os pares formados. Para cada primer da fita sense são
apresentados os primers compatíveis na fita anti-sense.

88
Figura 38. Tela com os resultados do projeto de primers.
Os primers gerados pelo sistema foram avaliados manualmente e verificou-se que todos
atendem às restrições importas pelo pesquisador, sendo, em princípio, adequados ao uso
estabelecido pelo mesmo.

IV – CONCLUSÕES
O Sistema Web para Projeto e Análise de Primers é um sistema disponibilizado na Web
para auxílio à preparação de experimentos de PCR. Através desse sistema o pesquisador pode fazer
a análise de primers já projetados ou projetar novos primers a partir de uma série de restrições
(parâmetros de projetos) e seqüências genônicas inseridas pelo pesquisador ou trazidas de bancos de
dados públicos automaticamente pelo sistema
Esse sistema deve ser de grande utilidade para os pesquisadores da área de biologia
molecular, pois diminui o tempo para o projeto de primers, e tende a melhorar a qualidade dos
primers projetados em relação aos softwares existentes. Normalmente o pesquisador acessa o banco
de dados público (GenBank), recupera as seqüências genômicas de interesse, e se for necessário,
executa o alinhamento múltiplo em outra ferramenta, e por fim, faz o projeto de primers numa
terceira ferramenta. Uma vantagem adicional desse sistema é possibilitar ao pesquisador utilizar
todas essas ferramentas em único ambiente, e algumas vezes de forma transparente. A etapa de
projeto de primers provê primers para seqüenciamento ou amplificação.
Os primers projetados podem ainda ser de vários tipos: degenerados, primers para
anelarem em mais de uma seqüência, primers com inserção de trechos específicos na ponta 3’ (que
podem ser enzimas de restrição ou trechos para mutação sítio-dirigida). Uma análise manual dos
primers projetados verificou que todos eles satisfazem todas as restrições impostas pelo
pesquisador, sendo primers consistentes. Entretanto, validação final dos primers depende de sua
confecção por uma indústria farmacêutica e de seu uso em experimentos de PCR reais, realizado em
laboratório de biologia molecular com os equipamentoe e reagentes necessários, o que envolve
aspectos que extrapolam o âmbito deste trabalho. Porém, os objetivos principais propostos neste
trabalho, no âmbito computacional, foram atingidos.
O desenvolvimento desse sistema apresentou uma série de dificuldades, entre elas: (i) Pouco
conhecimento inicial na área de biologia molecular, não pertencente à ciência da computação; (ii)
Problemas inesperados na configuração e administração de ambiente Linux e mesmo na instalação
de módulos e programas necessários, como Perl, MySQL, BioPerl, ClustalW e Blast. O Sistema
Operacional Linux exigiu tempo e esforço adicionais não dimensionados inicialmente; e (iii) A

90
linguagem de programação Perl, como outras para desenvolvimento de sistemas para Web, não
possui recursos para depuração do código, o que demonstrou ser um grande limitador.
Destacam-se ainda alguns avanços e melhorias necessários ao sistema para seu uso eficaz:
(i) A inclusão de verificação de similaridade entre a fita concenso das seqüências selecionadas para
anelamento dos primers com as seqüências selecionadas para o não anelamento dos primers, com o
objetivo de eliminar da fita concenso regiões não específicas, onde os primers não deveriam anelar-
se; (ii) Após o projeto dos primers, fazer um alinhamento par-a-par dos mesmos com as seqüências
existentes no GenBank. Essas melhorias devem prover uma análise de especificidade muito ampla;
(iii) Na análise das características dos primers, alterar as fórmulas de cálculo para que representem
a estabilidade química das moléculas através da entropia e entalpia das mesmas; (iv) Incluir
mecanismos (ou acesso a ferramentas já existentes) para tradução de proteína para DNA, de forma a
permitir também o projeto de primers concenso; e (v) Aumentar a abrangência desse sistema,
realizando também a análise do produto amplificado pela PCR, cortes por enzimas de restrição e a
simulação do gel de eletroforese, apresentando os resultados finais da PCR. Deste modo, o sistema
poderia ser usado em praticamente todas as etapas da PCR.
Além dessas melhorias, sugere-se que futuros trabalhos nessa área considerem também os
seguintes pontos cruciais: (i) Sistemas para bioinformática envolvem, normalmente, uma
quantidade de dados realmente muito grande, então sugere-se (a) o uso de estruturas de dados
criadas especificamente para facilitar o acesso a esses dados e (b) o uso intensivo de processamento
paralelo; (ii) Atividades em bioinformática requerem, normalmente, a execução de inúmeras sub-
atividades para as quais normalmente também já existem sistemas desenvolvidos, então sugere-se:
(c) o conhecimento dos sistemas existentes e de sua inter-relação, o que facilitaria o
desenvolvimento de atividades de mais alto nível; e (d) o conhecimento do funcionamento interno
(algoritmo) desses sistemas e os pressupostos biológicos envolvidos, para que não lhes seja dado
um uso inapropriado.

91
BIBLIOGRAFIA
ACCESS Excellence. DNA - A More Detailed Description. Disponível em:
<http://www.accessexcellence.org/AB/GG/dna2.html>. Acesso em: 14 out. 2003.
AFSETH, Guy. PCR Primer: Strategies to Improve Results. Given at Northwestern University, 9
jun. 1997. Disponível em: < http://www.biotechlab.nwu.edu/pe/>. Acesso em: 18 nov. 2003.
AGENCIA EFE. Cientistas identificam dois genes vinculados à asma. Quinta, 8 de abril de 2004,
17h29. Disponível em: < http://noticias.terra.com.br/ciencia/interna/0,,OI291089-EI296,00.html >.
Acesso em 08 abr. 2004.
ALKAMI Biosystems. Alkami Quick Guide for PCR. 1. ed. United States of America: Guanabara
Koogan, 1999.
ANSELMO, Fernando. PHP e MySQL para Windows. Florianópolis: Visual Books, 2000. 144p.
BACALTCHUK, Benami. A Percepção Social Nacional e Internacional Frente à Biotecnologia
no Contexto Agropecuário. Disponível em:
<http://www.cnpt.embrapa.br/livros/biotecnologia/bio1.htm>. Acesso em: 03 set. 2003.
BIOMANIA. Disponível em: <http://www.biomania.com.br/biotecnologia/enzima_rest.php>.
Acesso em: 15 set. 2003.
BIOPERL. Disponível em: < http://www.bioperl.org/>. Acesso em: 11 set. 2003.
BLAST. Disponível em: <http://www.ncbi.nlm.nih.gov/BLAST/Blast.cgi>. Acesso em: 22 nov.
2003.
BONATO, Chris; BONATO, Cezar e PEREIRA, Maria do Socorro Vieira. Biologia na Web.
Disponível em: <http://www.biologianaweb.com/genomica/multi.html>. Acesso em: 16 nov. 2003.

92
BROWN, T. A.. Genética: Um Enfoque Molecular. 3. ed. Rio de Janeiro: Guanabara Koogan,
1999. 336 p. Título Original: A Molecular Approach.
CASTAGNETTO, Jesus et al. Professional PHP Programando. São Paulo: Makron Books do
Brasil, 2001. 770 p.
CHRISTIANSEN, Tom. Perl.com. Disponível em: <http://www.perl.com/>. Acesso em: 15 set.
2003.
CLASSIC Experiments in DNA Replication. Disponível em:
<http://www.blc.arizona.edu/marty/411/Modules/classic.html>. Acesso em: 18 nov. 2003.
CLUSTALW. Disponível em: <http://www.ebi.ac.uk/clustalw/>. Acesso em: 22 nov. 2003.
COWAN, John. Gel Electrophoresis Simulator. Disponível em:
<http://webphysics.davidson.edu/applets/biogel/biogel.html>. Acesso em: 23 nov. 2003.
CPAN: Comprehensive Perl Archive Network. Disponível em: <http://www.perl.com/cpan/>.
Acesso em: 18 set. 2003.
CRAIG, Paul. Simulation of Electrophoresis & Sequence Building of DNA. Disponível em:
<http://www.rit.edu/~pac8612/electro/dna/DNA.html#>. Acesso em: 23 nov. 2003.
DAWKINS, R. O gene egoísta. Belo Horizonte: Ed. Itatiaia, 1979. 230 p.
DE ROBERTIS, E. M. F., HIB, Jose. Bases da Biologia Molecular e Celular. 3. ed. Rio de
Janeiro: Guanabara Koogan, 2001. 418 p.
GENE RUNNER for Windows. Disponível em: <http://www.generunner.com>. Acesso em: 20 set.
2003.
GIBAS, Cynthia, JAMBECK, Per. Desenvolvendo Bioinformática: ferramentas de software
para aplicações em biologia. Rio de Janeiro: Campus, 2001. 440 p.

93
GRIFFITHS, A. et al. Introdução a Genética. 7. ed. Rio de Janeiro: Guanabara Koogan, 2002.
GUIMARÃES, Kátia Silva; MELO, Jeane Cecília Bezerra de. Uma Introdução à Análise de
Seqüências e Estruturas Biológicas. In: JAI’03 – XXII Jornadas de Atualização em Informática,
Capítulo 1, ago. 2003.
HIGGINS, Des e TAYLOR, Willie. Bioinformatics: Sequence, Structure and Databanks. New
York: Oxford University, 2000. 249p.
HILBERS, Peter. Bioinformatics. Disponível em:
<http://wwwpa.win.tue.nl/hilbers/d.ogo2.1inf11sept2001/node19.html#SECTION00019000000000
0000000>. Acesso em: 05 out. 2003.
INNIS, Michael A. et al. PCR Protocols: A Guide to Methods and Applications. United States:
Academic Press, 1990. 482 p.
INTRODUÇÃO às Expressões Regulares. Disponível em:
<http://www.helpscribble.com.br/webhelp/navbar/hs30000.htm>. Acesso em: 29 out. 2003.
JONGENEEL, C. Victor. Transcription - types d'ARN. Disponível em: <http://ludwig-
sun1.unil.ch/~vjongene/molbio/chapt_7.htm>. Acesso em: 24 out. 2003.
KAISER, Gary E. Tutorial – DNA, DNA Replication, and RNA. Atualizado em: 07 jan. 2003.
Disponível em: <http://www.cat.cc.md.us/~gkaiser/goshp.html>. Acesso em: 14 out. 2003.
LIFE TECHNOLOGY. PCR/RT-PCR Applications Guide. Induslab. [S.I. : s.n.], [ca. 2000]
29 p.
MySQL. Disponível em: <www.mysql.com>. Acesso em: 18 set. 2003.
NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION. Disponível em:
<http://www.ncbi.nlm.nih.gov>. Acesso em: 22 set. 2003.

94
PANDOLFI, Valesca. Estruturas protéicas envolvidas nos processos biológicos. Disponível em:
<http://www.cnpt.embrapa.br/livros/biotecnologia/bio18.htm>. Acesso em: 03 set. 2003.
PHP. Disponível em: <http://www.php.net>. Acesso em: 16 set. 2003.
PHYLIP. Disponível em: <http://evolution.genetics.washington.edu/phylip.html>. Acesso em: 14
nov. 2003.
POSTGRESQL. Disponível em: <http://www.postgresql.org/>. Acesso em: 18 set. 2003.
PROJETO POSTGRESQL BRASIL. Disponível em: <http://pgsqlbr.querencialivre.rs.gov.br/>.
Acesso em: 28 set. 2003.
PROSDOCIMI, Francisco. Chico On Line. Disponível em:
<http://www.icb.ufmg.br/~franc/cool/index.htm>. Acesso em: nov. 2003.
PROSDOCIMI, Francisco et al. Bioinformática: Manual do Usuário. Disponível em:
<http://www.biotecnologia.com.br/bio29/bioinfo.html>. Acesso em: 14 set. 2003.
Atualizado em: 15/06/03.
QUEIROZ, Alexandre. Apostila de Introdução à Bioinformática. Disciplina de Bioinformática.
Universidade Federal do Rio Grande do Norte. Fevereiro de 2002.
SAVOLA, Tom; WESTENBROEK, Alan; HECK, Joseph. Usando HTML. Rio de Janeiro: Editora
Campus LTDA, 1997. 681p.
SCHWARTZ, Randal L.. Learning Perl. United States of America: O´Reilly & Associates, Inc,
1994. 246 p.
SIEVER, Ellen; SPAINHOUR, Stephen; PATWARDHAN, Nathan. Perl – Guia completo. Rio de
Janeiro: Ciência Moderna, 1999. 698p.

95
SILBERSCHATZ, Abraham; KORTH, Henry F. e SUDARSHAN, S. Sistema de Banco de Dados.
3 ed. São Paulo: Makron Books, 1999.
SILVA, Flávio Henrique (Resp.). Módulo: Biologia Molecular. I Escola Brasileira de Inteligência
Artificial e Bioinformática – InBio - São Carlos: [s.n.], 2001a.
SILVA, Luciano Carlos da. Banco de Dados para Web do Planejamento à Implementação. São
Paulo: Érica, 2001b. 240 p.
SINGH, Vinay K. e KUMAR, Anil. PCR Primer Design. Bioinformatics Sub-centre, School of
Biotechnology, Devi Ahilya University, Índia. Disponível em:
<http://faculty.fullerton.edu/rmc89/bioinformatics/PCR_PrimerDesign.htm>. Acesso em: 19 nov.
2003.
SWISS-PROT Database Access. Disponível em: <http://www.ebi.ac.uk/swissprot/access.html>.
Acesso em: 16 set. 2003.
TASSO, E.B. et al. Genética. Universidade Federal De São Paulo. Escola Paulista de Medicina.
Disponível em: <http://www.virtual.epm.br/cursos/genetica/htm/base.htm>. Acesso em: 15 nov.
2003.
THOMPSON, M., MCINNES, R., WILLARD, H. Genética Médica. Rio de Janeiro:
GUANABARA KOOGAN, 1998. p. 23-25.
THRELFALL, E. John et al. Application of Pulsed-Field Gel Electrophoresis to an
international Outbreak of Salmonella agona. Laboratory of Enteric Pathogens, Central Public
Health Laboratory, London, United Kingdom. Abr-jun 1996. Disponível em:
<http://www.cdc.gov/ncidod/eid/vol2no2/threlfal.htm>. Acesso em: 16 dez. 2003.
TISDALL, James D.. Beginning Perl for Bioinformatics. United States of America: O´Reilly &
Associates, Inc, 2001. 368 p.
TRIUNFOL, Márcia L. O DNA vai à escola. Disponível em: <http://www.odnavaiaescola.com/>.
Acesso em: 24 out 2003.

96
VALTAS, Marco. Genética Molecular e Tecnologia do DNA Recombinante. Disponível em:
<http://morpheus.fmrp.usp.br/td/>. Acesso em: 14 ago. 2003.
WEB PRIMER: DNA and Purpose Entry. Disponível em: <http://seq.yeastgenome.org/cgi-
bin/SGD/web-primer>. Acesso em: 14 nov. 2003.
ZAHA, Arnaldo (Coord). Biologia Molecular Básica. 1. ed. Porto Alegre: Mercado Aberto, 1996.
336p. (Série Ciência XXI).

GLOSSÁRIO
Árvore filogenética mostra a relação de descendência entre os organismos e o tempo que se
passou entre o ancestral comum de duas espécies
Códon uma trinca de nucleotídeos que codifica um aminoácido
Contig um conjunto contíguo de seqüencias superpostas de DNA
DNA Ácido desoxirribonucléico, o material genético
Eletroforese separação de moléculas com base em sua carga elétrica total
Exons uma das regiões codificantes de um gene descontínuo
Gene o gene corresponde a uma seqüência particular de DNA, codificadora de uma
informação
Homologia indica que duas seqüências possuem uma probabilidade alta de
compartilharem o mesmo ancestral
Introns em um gene descontínuo, um dos segmentos que não contém informação
biológica
Ortólogos genes que têm a mesma função
Parólogos genes que não possuem o mesmo papel, mas possuem um ancestral em
comum
PCR técnica que possibilita a reprodução de milhares de cópias de um determinado
fragmento de DNA
Primer pequena cadeia de DNA de fita simples, onde, a partir da posição 3’, a enzima
DNA polimerase iniciará a incorporação de nucleotídeos, formando a fita
dupla do DNA.
Score pontuação
Shotguns pequenas seqüências de DNA para formar uma única seqüência
Similaridade refere-se à presença de locais similares e idênticos em duas seqüências
trechos conservados