Barreiras Rotacionais em Sistemas Amídicos. Estudo ... Rodrigo Pontes.pdf · RMND, por não se...
205
Universidade Estadual de Maringá Centro de Ciências Exatas Departamento de Química Tese de Doutorado Barreiras Rotacionais em Sistemas Amídicos. Estudo Experimental e Teórico de Congêneros de Carbamatos e Uréias Orientado: Rodrigo Meneghetti Pontes Orientador: Ernani Abicht Basso Maringá – 2006
Transcript of Barreiras Rotacionais em Sistemas Amídicos. Estudo ... Rodrigo Pontes.pdf · RMND, por não se...

Universidade Estadual de Maringá Centro de Ciências Exatas Departamento de Química
Tese de Doutorado
Barreiras Rotacionais em Sistemas Amídicos. Estudo Experimental e Teórico de Congêneros de
Carbamatos e Uréias
Orientado: Rodrigo Meneghetti Pontes
Orientador: Ernani Abicht Basso
Maringá – 2006

“Ainda que eu fale as línguas dos homens e dos anjos, se não tiver amor, serei como o bronze que soa ou como o címbalo que retine. Ainda que eu tenha o dom de profetizar e conheça todos os mistérios e toda a ciência; ainda que eu tenha tamanha fé, a ponto de transportar montes, se não tiver amor, nada serei. E ainda que eu distribua todos os meus bens entre os pobres e ainda que entregue o meu próprio corpo para ser queimado, se não tiver amor, nada disso me aproveitará. O amor é paciente, é benigno; o amor não arde em ciúmes, não se ufana, não se ensoberbece, não se conduz inconvenientemente, não procura seus interesses, não se exaspera, não se ressente do mal; não se alegra com a injustiça, mas regozija-se com a verdade; tudo sofre, tudo crê, tudo espera, tudo suporta. O amor jamais acaba; mas, havendo profecias, desaparecerão; havendo línguas, cessarão; havendo ciência, passará; porque, em parte, conhecemos e, em parte, profetizamos. Quando, porém, vier o que é perfeito, então, o que é em parte será aniquilado. Quando eu era menino, falava como menino, sentia como menino, pensava como menino; quando cheguei a ser homem, desisti das coisas próprias de menino. Porque, agora, vemos como em espelho, obscuramente; então veremos face a face. Agora, conheço em parte; então conhecerei como também sou conhecido. Agora, pois, permanecem a fé, a esperança e o amor, estes três; porém o maior destes é o amor.” (I Coríntios 13:1-13)

À minha mãe Cleuza,
Por sua coragem e sabedoria,
Pela dedicação e pelas lições de vida
Dedico esta tese

AGRADECIMENTOS
Ao Prof. Dr. Ernani Basso, pela orientação desde os primeiros dias de
laboratório, por sua ajuda em me fazer crescer como cientista, e pelo apoio
nos momentos difíceis.
À minha família, Ondina, Daniella, Mariana, Valdomiro, Cleonice, Cléia,
Jefferson, Clizeide, Voni, Raimundo, Renata, Luissy, Volney, Tiago, Renan,
Otávio e Vítor. In memorian ao meu avô Otávio e à Ana Maria.
À Rosangela Basso, pela amizade e incentivo.
Ao Cleverson, à Gisele, à Barbara, à Franciele, à Ana, ao Jaime e ao
Francisco. Cada um ao seu modo me ajudou a crescer um pouco.
Ao Dr. Paulo R. Oliveira, pelas primeiras lições de cálculo e por sua
prestatividade.
Aos pessoal da IC pelo convívio agradável.
Aos professores do DQI, particularmente ao Prof. Dr. Noboru Hioka pelo
incentivo no início da graduação.
Ao Agrinaldo, pela amizade e por suas mensagens de sabedoria.
Ao Prof. Dr. Gentil José Vidotti por assumir a co-orientação durante o
período de pós-doutorado do Prof. Ernani.
Ao Frimmel, por sua solicitude constante, principalmente durante a
realização dos experimentos de RMND.
À Dra Ivânia Schuquel pela paciência e empenho durante os experimentos de
RMND, por não se importar de trabalhar à noite e jantar salgadinhos da
cantina.
Ao Prof. Dr. Roberto Rittner pelo incentivo ao grupo ECO.
Ao Cláudio Tormena, pelo apoio, principalmente nos primeiros tempos.
À Cristina e ao Claudemir, por seu bom atendimento sempre que necessário.
À turma do bloco 31.

Ao pessoal da república, Rene, Luiz, Leonardo e Rogério.
Ao Augustinho, pela amizade e pelo bom exercício intelectual.
À CAPES, pelas bolsas de mestrado e doutorado.
Ao CENAPAD-SP, pelo uso do ambiente computacional.

RESUMO
Barreiras Rotacionais em Sistemas Amídicos. Estudo Experimental e
Teórico de Congêneros de Carbamatos e Uréias
Orientado: Rodrigo Meneghetti Pontes
Orientador: Ernani Abicht Basso
Nossa investigação tratou das barreiras rotacionais para ligações C-N conjugadas
em uma série de congêneros do N,N-dimetilcarbamato de O-metila [CH3―X―(C=Z)
―N(CH3)2, X=O,S e Z=O,S] e da N,N,N’-trimetiluréia [CH3―N(X)―(C=Z)―N(CH3)2,
X=H,CH3 e Z=O,S]. A Ressonância magnética nuclear dinâmica (RMND) foi
empregada para a determinação experimental das barreiras rotacionais em diversos
solventes. Foram realizados cálculos ab initio e com a teoria do funcional de
densidade (TFD-B3LYP) para o estudo teórico das barreiras rotacionais em fase
gasosa e sua resposta à polaridade do meio. Empregamos, ainda, simulações com
dinâmica molecular buscando avaliar o efeito das ligações de hidrogênio. Para
estes trabalhos, fez-se necessária a parametrização de campos de forças para os
solutos. Os resultados mostraram que é fundamental a inclusão dos efeitos de
solvatação para o cômputo correto das barreiras rotacionais, sendo que a combi-
nação do modelo de solvatação IPCM com cálculos de dinâmica molecular mostrou-
se bastante adequada para este propósito. Observamos aumento das barreiras
rotacionais devido à ligações de hidrogênio para dois compostos, enquanto nos
demais o efeito foi negativo, i.e. diminuição da barreira. A origem das barreiras foi
investigada através de cálculos com a teoria dos orbitais naturais de ligação (NBO).
Verificamos que a deslocalização eletrônica possui um papel fundamental, como se
supunha, mas precisa ser suplementada pelas modificações energéticas na
estrutura de Lewis, decorrentes, principalmente, das variações nos parâmetros
estruturais (comprimentos e ângulos de ligação).

ABSTRACT
Rotational Barriers in Amidelike Systems. An Experimental and
Theoretical Study of Carbamate and Urea Congeners
Author: Rodrigo Meneghetti Pontes
Advisor: Ernani Abicht Basso
Our investigation dealt with the rotational barriers for conjugated C-N bonds in
congeners of O-methyl N,N-dimethylcarbamate [CH3―X―(C=Z)―N(CH3)2, X= O,S and
Z=O,S] and N,N,N’-trimethylurea [CH3―N(X)―(C=Z)―N(CH3)2, X=H,CH3 and Z=O,S].
Dynamic nuclear magnetic resonance (DNMR) was employed for the experimental
determination of the rotational barriers in several solvents. Gas-phase rotational
barriers, as well as their response to the medium polarity, were studied through ab
initio and density functional theory (DFT-B3LYP). In addition, we performed
molecular dynamic simulations to evaluate the effect of hydrogen bonding. For
these latter, it was necessary to conduct a force field parameterization for each
solute. The results showed that it is indispensable to include solvation effects in
order to correctly calculate rotational barriers, and that combining the IPCM
solvation model with molecular dynamics calculations provide us with a suitable
way to approach the problem. Hydrogen bonding causes increase on the rotational
barrier of two compounds, but has a negative effect on the remaining, i.e.
decrease their barriers. The rotational barrier origin was studied through natural
bond orbital theory (NBO). Our findings showed that electron delocalization indeed
plays an important role, as it was supposed to do, but needs to be supplemented by
energetic modifications taking place in the Lewis structure, which originates mainly
from structural parameters variations (bond lengths and angles).

ÍNDICE
Abreviaturas e Símbolos
Definição das Estruturas
1. Introdução e Objetivos.....................................................................1
2. Fundamentação Teórica ...................................................................6
2.1. Métodos de Estrutura Eletrônica ....................................................7
2.1.1. O Método de Hartree-Fock......................................................8
2.1.1. Conjunto de Funções de Bases ............................................... 13
2.1.3. Correlação Eletrônica.......................................................... 16
2.1.4. Teoria do Funcional de Densidade........................................... 17
2.1.5. Otimização de Geometrias.................................................... 19
2.1.6. Propriedades Termodinâmicas ............................................... 21
2.1.7. Modelos de Solvatação......................................................... 23
2.1.8. Modelos de Solvatação Contínuos ............................................ 24
2.1.9. Teoria dos Orbitais Naturais de Ligação .................................... 26
2.2. Dinâmica Molecular ................................................................. 28
2.2.1. Visão Geral ...................................................................... 28
2.2.2. Campo de Forças ............................................................... 30
2.2.3. Função de Distribuição Radial de Pares..................................... 31
2.3. Ressonância Magnética Nuclear Dinâmica ....................................... 33
2.4. Barreiras Rotacionais em Sistemas Amídicos .................................... 37
3. Parte Experimental ....................................................................... 43
3.1. Ressonância Magnética Nuclear ................................................... 44
3.2. Cálculos Teóricos .................................................................... 46
3.2.1. Métodos de Estrutura Eletrônica ............................................. 46
3.2.2. Dinâmica Molecular ............................................................ 48
3.3. Preparação dos Compostos......................................................... 49
4. Resultados e Discussão ................................................................... 56
4.1. Obtenção dos Compostos e RMND ................................................. 57
4.1.1. Síntese dos Compostos ........................................................ 57
4.1.2. Determinação das Barreiras Rotacionais por RMND ....................... 60
4.2. Estudo Teórico ....................................................................... 67
4.2.1. Estruturas Estáveis em Fase Gasosa ......................................... 67

4.2.1.1. Conformações para o Grupo 1........................................... 67
4.2.1.2. Conformações para o Grupo 2........................................... 70
4.2.2. Barreiras Rotacionais no Vácuo e em Solução por Métodos Quânticos . 81
4.2.2.1. Grupo 1 ..................................................................... 81
4.2.2.2. Grupo 2 ..................................................................... 88
4.2.3. Inclusão do Efeito das Ligações de Hidrogênio por Dinâmica Molecular
Associadas a Cálculos Quânticos ................................................... 92
4.2.3.1. Grupo 1 ..................................................................... 92
4.2.3.1. Grupo 2 ....................................................................108
4.2.4. Origem das Barreiras Rotacionais ...........................................120
4.2.4.1. Grupo 1 ....................................................................120
4.2.4.2. Grupo 2 ....................................................................134
5. Conclusões ................................................................................142
Referências Bibliográficas..................................................................145
Apêndice I ....................................................................................152
Apêndice II ...................................................................................155
Apêndice III...................................................................................163

ABREVIATURAS, SÍMBOLOS E DEFINIÇÕES
SCF Self-Consistent Field (Campo Auto-Consistente)
HF Hartree-Fock
B3LYP Método de Três Parâmetros de Becke com Funcional de Correlação de Lee, Young e Par
TFD Teoria do Funcional de Densidade
IPCM Isodensisty Polarizable Continuum Model (Modelo do Contínuo Polarizável com uso de Superfície de Isodensidade)
DM Dinâmica Molecular
RMND Ressonância Magnética Nuclear Dinâmica
NBO Natural Bond Orbital (Orbital Natural de Ligação)
ef Estado fundamental
et Estado de transição
fdr Função de distribuição radial
g(r) Função de distribuição radial
Tc Temperatura de coalescência
Dn Separação das freqüências de absorção de dois núcleos que participam de um processo de troca
APB Análise do Perfil das Bandas
SEP Superfície de Energia Potencial
ε, s Parâmetros de Lennard-Jones
q Carga atômica
e Unidade de carga atômica (equivalente à carga de um elétron)
e Constante dielétrica
ei Energia do NBO i; também para energia, de forma geral, na função de partição
εi Energia do orbital SCF i
hartree Unidade de energia, 1 hartree = 627,51 kcal/mol
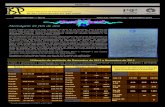
DEFINIÇÃO DE ESTRUTURAS Grupo 1
X
H3C
Z
N
CH3
CH3ef designação genérica da
estrutura planar
Z
NXH
HH
H
H
H
H
H
H
Z
NXH
HH
H
HH
H
H
H
et1 et2
estruturas geradas pela rotação
sobre a ligação C-N
X
Z
NH
HH
H
HH
H
H
H
X
Z
NH
HH
H
H
H
HH
H
X
Z
NH
HH
HH
H
H
HH
X
Z
NH
HH
HH
H
H
HH
ef1 ef2
ef3 ef4
estrutura planar com as conformações das
metilas definidas
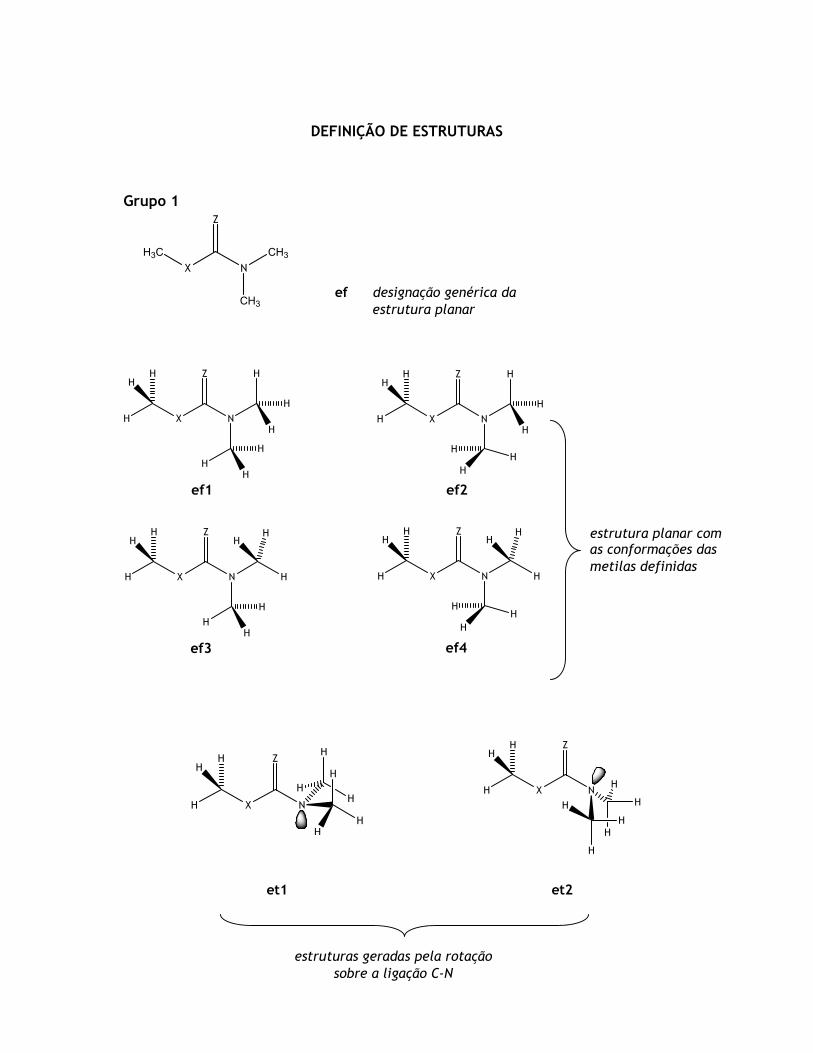
Grupo 2
N
H
Z
NH
HH
H
HH
H
H
H
N
H
Z
NH
HH
H
H
H
HH
H
N
H
Z
NH
HH
HH
H
H
HH
N
H
Z
NH
HH
HH
H
H
HH
N
H
Z
NH
HH
H
H
HH
H
H
N
H
Z
NH
H
H
HH
H
H
H
H
N
H
Z
N
HH
H
H
HH
H
H
H
N
H
Z
N
HH
H
H
HHH
H
H
ef1 ef2
ef3 ef4
ef5 ef6
ef7 ef8
N
H3C
H
Z
N
CH3
CH3ef designação genérica da
estrutura planar
estrutura planar com as conformações das
metilas definidas
z
NNH
HH
H
H
H
H
H
H
z
NNH
HH
H
HH
H
H
H
H
H
z
NN
H
H
H
H
H
H
H
z
NNH
HH
H
H
H
H
H
H
H
H
H
H
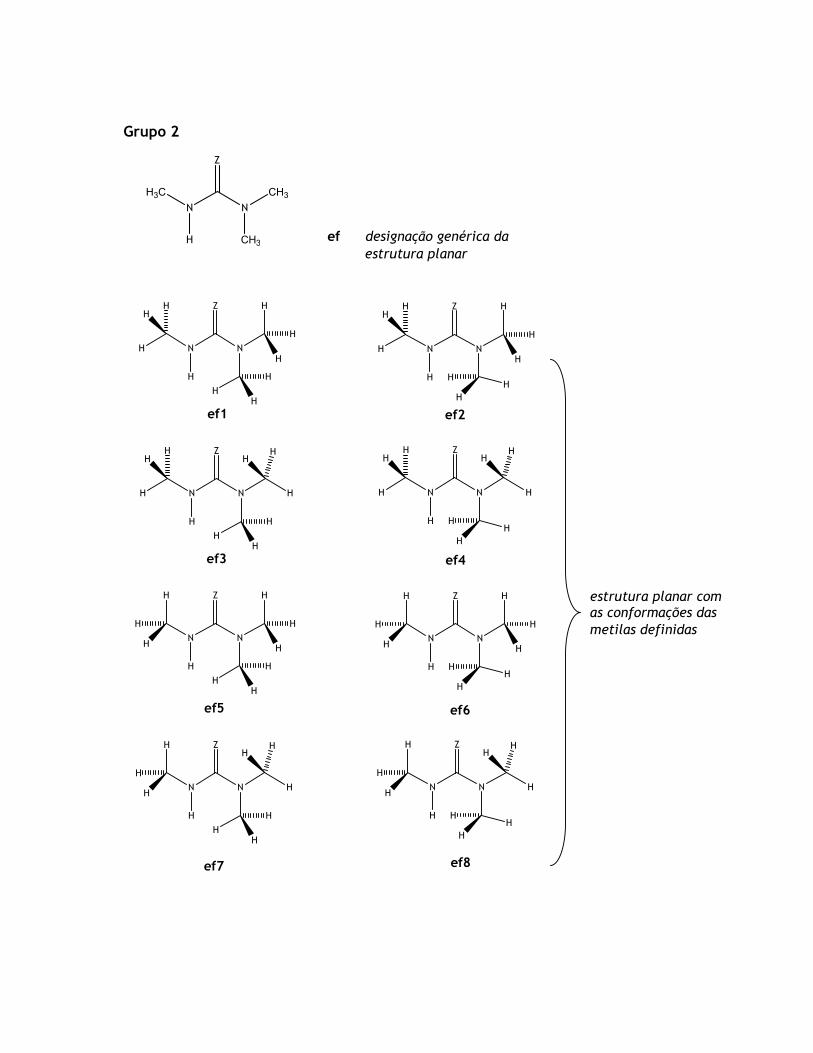
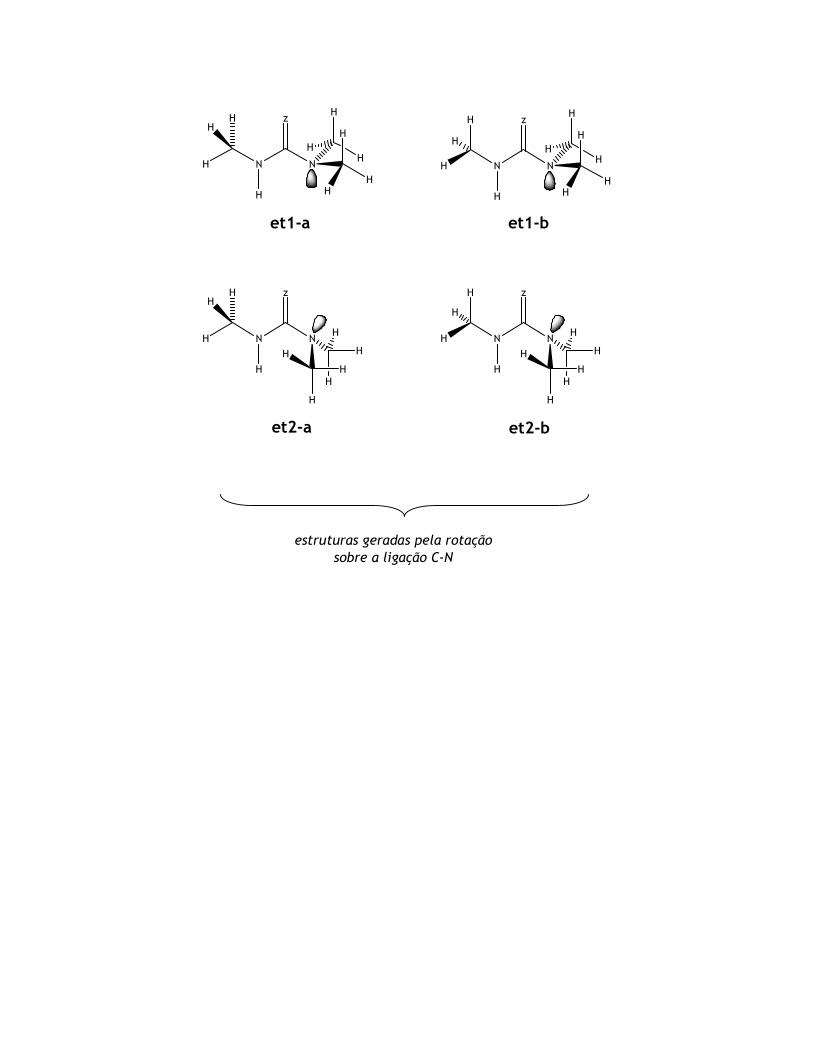
et1-a et1-b
et2-a et2-b
estruturas geradas pela rotação
sobre a ligação C-N

1
Capítulo 1:
Introdução e Objetivos

Introdução e Objetivos 2
Capítulo 1: Introdução e Objetivos
Os carbamatos (Fig. 1.1) são alguns dos mais importantes compostos com atividade
farmacológica.1-6 Doenças como a miastenia gravis e o mal de Alzheimer são
tratadas com drogas contendo esse grupo funcional, que também ocorre na
estrutura de certos pesticidas. Em vista disso, têm-se buscado estabelecer a
correlação estrutura-atividade com o intuito de desenvolver drogas mais potentes e
de menor toxidez, mas essas investigações esbarram na falta de conhecimento a
respeito da estrutura química dos sítios ativos das colinesterases.1-3 Mesmo assim, é
possível afirmar que a distância entre os grupos funcionais é um dos fatores
determinantes.1
Figura 1.1: Estrutura geral de amidas e carbamatos.
Recentemente, Furukawa et al.2 mostraram que a (S)-metacolina e a
(2S,4R,5S)-muscarina mudam sua conformação quando se ligam a um receptor de
acetilcolina. A principal implicação de tal descoberta é o fato de ser necessário
considerar, além da distância entre os grupos funcionais, a flexibilidade molecular,
pois esta pode ser responsável por um acomplamento enzima-substrato mais
efetivo. Obviamente, esta flexibilidade depende da facilidade com que se pode
rotacionar os grupos funcionais sobre as ligações químicas, ou em outras palavras,
das barreiras rotacionais. Portanto, a compreensão destas últimas é crucial para o
entendimento da atividade biológica.
Na última década, a descoberta de que as barreiras rotacionais em
carbamatos são insensíveis ao solvente fez com que esses compostos se tornassem
modelos bastante interessantes também do ponto de vista da química
fundamental.7, 8 No entanto, a atenção dedicada aos carbamatos ainda é pequena
se os compararmos às amidas (Fig. 1.1), que desempenham um papel importante
na química biológica, particularmente no que diz respeito à estrutura das proteínas
X
C
O
NAmidas: X = alquila ou arilaCarbamatos: X = OR(R = alquila ou arila)

Introdução e Objetivos 3
e enzimas.9-21 A grande atenção dedicada às amidas decorre do fato delas serem os
“blocos fundamentais” constituintes das proteínas, de modo que sua análise pode
ajudar na compreensão do comportamento desses polímeros biológicos.
O aspecto mais notável acerca das amidas é a elevada barreira de energia
para a rotação sobre a ligação (C=O)-N. Atribui-se essa barreira à deslocalização do
par de elétrons do nitrogênio sobre o orbital π antiligante da carbonila, nN → π*C=O,
ou ainda, na liguagem da teoria de ligação de valência, à possibilidade das
estruturas de ressonância apresentadas na Fig. 1.2.16 Contudo, nos últimos anos
alguns autores têm contestado a validade desse modelo, argumentando que o
mesmo não é capaz de suprir explicações para certas características tais como a
distribuição de carga elétrica.22-24
Figura 1.2. Estruturas de ressonância para amidas e carbamatos.
Situação similar foi encontrada para o etano, que embora difira dos
compostos de nosso interesse, ilustra bem a idéia que tentamos expressar.25-27
Recentemente, a comunidade química foi surpreendida com os artigos de Weinhold
e Goodman-Pophristic nos quais os autores contrariam um dos mais conhecidos
fundamentos da estereoquímica.25, 27 Nos referidos estudos, os autores demonstram
que a barreira rotacional no etano possui origem predominantemente
hiperconjugativa, e não estérica, como é ensinado em livros de química
orgânica.28-30 Essa conclusão é o resultado da aplicação de métodos modernos de
estrutura eletrônica a um problema que foi tratado de forma essencialmente
qualitativa quando de sua proposição. Muito se tem feito a esse respeito para as
amidas,15, 17-19, 23, 31-35 mas não conhecemos estudo similar para os carbamatos e
seus congêneros.
Além da origem das barreiras rotacionais, um segundo problema intriga os
pesquisadores desta área, trata-se da influência do ambiente químico, ou em
outras palavras, do solvente. Sabe-se que a barreira rotacional nas amidas aumenta
X
C
O
N X
C
O
N

Introdução e Objetivos 4
com o aumento da polaridade do meio.10, 19 Ademais, verificou-se que a formação
de ligações de hidrogênio soluto-solvente causa uma posterior elevação, como é o
caso da N,N-dimetilacetamida (DMA) que tem sua barreira acrescida por
~ 4 kcal/mol ao passar da fase gasosa para uma solução aquosa.19 Porém, quando
os estudos foram estendidos para carbamatos, surpreendentemente não se
encontrou qualquer variação, fosse com a polaridade do meio ou com a capacidade
de formar ligações de hidrogênio.7 As explicações que haviam sido elaboradas para
as amidas precisaram então ser revistas, uma vez que amidas e carbamatos
partilham do mesmo fragmento (C=O)-N e deveriam, em princípio, seguir
comportamentos similares.8 Este contraste entre as duas classes de compostos
revelou a necessidade da exploração de novos sistemas.
Tendo em vista os trabalhos que já haviam sido desenvolvidos pelo grupo de
estereoquímica de compostos orgânicos (ECO) desta Universidade,36-38 julgamos
que as moléculas mais apropriadas para uma investigação seriam os congêneros de
carbamatos. Consideramos também que a inclusão de outros modelos como as
uréias e seus congêneros poderia ser de grande valia, uma vez que sua estrutura
possui uma particularidade não presente nos outros compostos, viz. dois pares de
elétrons dos nitrogênios interagindo com a (tio)carbonila. Isto deu origem à série 1
a 8 apresentada na Fig. 1.3.
O presente trabalho foi proposto com o intuito de adquirir dados
experimentais para 1-8 e compreender estas observações com a ajuda de cálculos
teóricos. Para esses propósitos, prevíamos a realização de experimentos de RMN
dinâmica em diferentes solventes. Quanto ao estudo teórico, a natureza do
problema exigia o emprego de cálculos de estrutura eletrônica, mas também de
métodos que fossem capazes de tratar adequadamente de interações como as
ligações de hidrogênio. Sendo assim, o estudo teórico foi composto por métodos ab
initio e da teoria do funcional de densidade (TFD), além de dinâmica molecular.
De forma resumida, estes foram nossos objetivos:
� Obter os compostos 1-8 e determinar suas barreiras rotacionais em diversos
solventes por RMN dinâmica;

Introdução e Objetivos 5
� Explicar o efeito da polaridade do solvente e das ligações de hidrogênio por
meio de cálculos teóricos, tanto de estrutura eletrônica como clássicos;
� Avaliar a origem das barreiras rotacionais através de cálculos teóricos.
Figura 1.3. Compostos estudados.
C
O
NOCH3
CH3
H3C
C
O
NSCH3
CH3
H3C
C
S
NOCH3
CH3
H3C
C
S
NSCH3
CH3
H3C
C
O
NNCH3
CH3
H3C
H
C
S
NNCH3
CH3
H3C
H
C
O
NNCH3
CH3
H3C
CH3
C
S
NNCH3
CH3
H3C
CH3
N,N-Dimetilcarbamato de O-Metila
N,N-Dimetiltiocarbamato de S-Metila
(1)
(2)
(3)
N,N-Dimetiltiocarbamato de O-Metila
N,N-Dimetilditiocarbamato de S-Metila(4)
(5)
(6)
(7)
(8)
N,N,N'-Trimetiluréia
N,N,N'-Trimetiltiouréia
N,N,N',N'-Tetrametiluréia
N,N,N',N'-Tetrametiltiouréia

Introdução e Objetivos 6
Capítulo 2:
Fundamentação Teórica

Fundamentação Teórica 7
Capítulo 2: Fundamentação Teórica
No desenvolvimento de nossos trabalhos, fizemos uso de ressonância magnética
nuclear dinâmica (RMND) para o estudo experimental das barreiras rotacionais,
bem como de métodos teóricos para o entendimento das observações. No que
segue, abordaremos os conceitos necessários à compreensão do trabalho. A
linguagem do texto foi direcionada para o leitor com pouca ou nenhuma
experiência com métodos de estrutura eletrônica e dinâmica molecular, de forma
que as exposições, em certos casos, podem ser prolixas para o pesquisador com
experiência na área. Pedimos a compreensão do mesmo, que pode, à sua escolha,
seguir diretamente para o próximo capítulo. O mesmo se aplica à RMN dinâmica.
2.1 Métodos de Estrutura Eletrônica
A expressão “estrutura eletrônica” é utilizada para uma extensa classe de
métodos que tratam explicitamente do comportamento de elétrons e núcleos nas
espécies químicas. É utilizada para a diferenciação de um segundo conjunto de
métodos, baseados na física clássica, que descreve o comportamento dos átomos
através de um grupo de parâmetros ajustados para reproduzir dados experimentais,
sem contudo considerar a estrutura subatômica.
Os próprios métodos de estrutura eletrônica subdividem-se em três outras
categorias: ab initio, semi-empíricos e métodos híbridos. O método de Hartree-
Fock é o alicerce para todos os três e dele trataremos agora.

Fundamentação Teórica 8
2.1.1 O Método de Hartree-Fock39-45
“Método para a determinação dos orbitais espaciais ψi da função de onda
determinantal de um elétron, baseado na redução de equações diferenciais
não-lineares acopladas para as formas ótimas dos orbitais moleculares pelo uso
do método variacional. O operador Hamiltoniano de Hartree-Fock é definido
em termos desses orbitais através dos operadores de repulsão de Coulomb e de
troca. O procedimento geral para resolver as equações de Hartree-Fock
consiste em tornar os orbitais auto-consistentes com respeito ao campo que
eles produzem. Isto é alcançado através de um processo computacional de
tentativa e erro, e por esta razão o processo todo é chamado de método do
campo auto consistente.” (Glossary of Therms Used in Theoretical Organic
Chemistry, IUPAC Recomendations 1999).46
A descrição dos sistemas microscópicos segue os princípios da física quântica, cujo
desenvolvimento deu-se no primeiro terço do século passado com contribuições de
diversos cientistas, dentre eles, Planck, Einstein, Dirac, Heisenberg, Schrödinger,
Bohr e Born. Nessa teoria, um sistema físico é descrito pela seguinte equação de
movimento, a equação de Schrödinger*:
(2.1)
na qual H é o operador Hamiltoniano, Ψ é a função de onda, dependente das
coordenadas eletrônicas (r) e nucleares (R), e E é a energia do sistema. Resolver
essa equação significa encontrar as funções de onda Ψ que a satisfazem e os seus
autovalores E de energia.
Tendo em vista que as velocidades com que os elétrons se movimentam são
bastante superiores às dos núcleos, podemos tratar estes como fixos e resolver a
Eq. (2.1) apenas para os elétrons. Esta é a aproximação de Born-Oppenheimer. No
átomo de hidrogênio, os únicos termos que compõem o operador Hamiltoniano são
a energia cinética do elétron (assumindo o núcleo fixo) e a energia potencial de
* Em sua forma mais geral, a equação de Schrödinger é dependente do tempo. Como nosso interesse principal são os sistemas estacionários, trabalharemos com a forma independente do tempo, que provém de uma separação de variáveis a partir da Eq. (2.1).
( ) ( )rRrR ,, Ψ=Ψ EH

Fundamentação Teórica 9
interação elétron-núcleo. Para este sistema, é possível empregar o procedimento
conhecido como “separação de variáveis”, no qual a Eq. de Schrödinger é dividida
em três equações mais simples que podem ser resolvidas de forma exata. Quando
há vários elétrons, faz-se necessário considerar a interação repulsiva entre os
mesmos. É justamente esse termo de interação elétron-elétron que impede a
aplicação do processo de separação de variáveis e preclude a resolução exata da
equação de Schrödinger para sistemas multi-eletrônicos.
O recurso possível para esse problema é a busca por soluções numéricas ou
aproximadas. O primeiro passo nessa busca consiste em escrever a função de onda
Ψ(r1, r2,⋅⋅⋅,rn) – que depende das coordenadas dos elétrons 1, 2,...,n – em termos de
funções mais simples ϕi(ri), cada uma das quais dependendo das coordenadas
apenas do elétron i. As funções ϕi(ri) são chamadas de orbitais moleculares ou
atômicos, dependendo do sistema em estudo. Douglas Hartree trabalhou com
produtos das funções ϕi(ri) da seguinte forma:
Ψ(r1, r2,⋅⋅⋅,rn) = ϕ1(r1) ϕ2(r2)⋅⋅⋅ ϕn(rn)
A partir dessa função, Hartree conseguiu encontrar soluções numéricas
bastante satisfatórias. Mas o produto (2.2) não é condizente com o princípio da
exclusão de Pauli, cuja importância na descrição das partículas elementares
começava a ficar evidente na mesma época dos trabalhos de Hartree. Em termos
matemáticos, o princípio de Pauli requer que o sinal da função de onda seja
invertido quando se troca as coordenadas de dois elétrons. Isso não ocorre na
Eq. (2.2), pois ϕ1(r1)ϕ2(r2)⋅⋅⋅ ϕn(rn) é o mesmo que ϕ2(r2)ϕ1(r1)⋅⋅⋅ϕn(rn).
Vladimir Fock então utilizou uma função com seguinte forma:
Sendo !n1 uma constante de normalização. Ou seja, a função Ψ(r1, r2,⋅⋅⋅,rn) é
definida pelo determinante dos orbitais moleculares ϕi(ri); a barra sobre as funções
(2.2)
( )
( ) ( ) ( )( ) ( ) ( )
( )( )
( )( )
( )( )n
n
n
n
nn
r
r
r
r
r
r
rrr
rr
rrr
k
k
2k
2k
1k
1k
12111
12111
21
r
!
1,,,
ϕϕ
ϕϕ
ϕϕ
ϕϕϕϕϕϕ
L
LMOMM
L
L
L =Ψ (2.3)

Fundamentação Teórica 10
serve para identificar o spin eletrônico. Essa função foi proposta por Slater e por
isso é conhecida como “determinante de Slater”.
Como exemplo, vamos assumir que cada um dos dois elétrons do átomo de
hélio possa ser representado por um orbital atômico do tipo 1s (ϕi(ri)=1s). Neste
caso, o determinante de Slater teria a seguinte forma:
que após ser desenvolvido fica:
A função (2.5) tem seu sinal invertido quando trocamos as coordenadas dos
elétrons 1 e 2, e portanto satisfaz o requerimento de antissimetria exigido pelo
princípio de Pauli.
A partir do determinante de Slater, Fock deduziu a equação para a energia
molecular:
na qual εi é a energia do orbital i, ijJ é “integral de Coulomb” e diz respeito à
repulsão elétron-elétron; ijK é a “integral de troca”, que surge do princípio de
Pauli e não possui análogo clássico; NNV é a energia potencial de interação núcleo-
núcleo. O procedimento utilizado para se obter a energia molecular ou atômica por
meio da Eq. (2.6) é chamado “método de Hartre-Fock”, abreviadamente, HF.
A energia fornecida pela Eq. (2.6) não é exata, uma vez que a função de onda
(2.3) é uma aproximação para a função “verdadeira”. Deste modo, a Eq. (2.6) está
sujeita ao “princípio variacional”, segundo o qual a energia obtida por uma função
aproximada, de forma variacional, é sempre maior do que a energia exata.
Apesar das simplificações de Hartree-Fock em relação à função de onda
Ψ(r1,r2,⋅⋅⋅,rn), ainda é necessário conhecer a forma dos orbitais moleculares ϕi(ri) e
isso constitui um outro problema. Roothaan propôs uma simplificação adicional,
( )( ) ( )( ) ( )21
2121 s1s1
s1s1
2
1,
rr
rrrr =Ψ
( ) ( ) ( ) ( )[ ]122121 s1s1s1s12
1),( rrrrrr −=Ψ
(2.4)
(2.5)
( )∑∑∑= ==
+−−=2/
1
2/
1
2/
1HF 22
n
i
n
jNNijij
n
ii VKJE ε (2.6)

Fundamentação Teórica 11
que consite em representar os orbitais moleculares como combinações lineares de
orbitais atômicos:
sendo ikc os coeficientes e kχ os orbitais atômicos. As funções kχ utilizadas para a
construção do orbital molecular constituem um “conjunto de funções de base”.
Tendo em vista uma conveniência matemática, as funções mais comumente
utilizadas como bases atômicas são as Gaussianas, i.e. funções do tipo 2xe α− .
O problema passa então a ser a determinação do melhor conjunto de valores
para os coeficientes ikc . A substituição da Eq. (2.7) na Eq. (2.6), seguida de
rearranjos, fornece:
na qual,
,
e Frs é o “operador de Fock”.
O que está representado em (2.8) é um conjunto de n equações lineares
simultâneas. Nesse sistema, rsirs SF ε− são os coeficientes, ao passo que os
coeficientes dos orbitais atômicos, ikc , que desejamos encontrar, assumem o papel
de variáveis. Resolver o sistema (2.8), portanto, significa encontrar os valores dos
coeficientes dos orbitais atômicos.
As energias εi são os autovalores do operador de Fock:
e para conhecê-las, é preciso antes conhecer os orbitais moleculares iϕ , que por
sua vez dependem dos coeficientes ikc (Eq. 2.7). Mas para encontrar esses
coeficientes, precisamos dos valores de iε , ou seja, voltamos ao início. Um
problema desse tipo não pode ser resolvido de forma exata, e sim por um
( ) ∑ +++==k
niniikikii cccc χχχχϕ L2211r (2.7)
( )∑=
=−n
srsirssi SFc
1
0ε (2.8)
∫= τχχ dFF srrs* ∫= τχχ dS srrs
*
iiiF ϕεϕ = (2.9)

Fundamentação Teórica 12
procedimento de “iterações”. Esse processo consiste em sugerir um conjunto inicial
de coeficientes para obter os orbitais moleculares iϕ e seus autovalores iε iniciais.
Os coeficientes rsirs SF ε− em (2.8) são então utilizados para encontrar um conjunto
de valores melhorados para os ikc . A partir desse conjunto melhorado, obtém-se
um novo sistema de equações lineares, que por sua vez gera um conjunto
melhorado de coeficientes dos orbitais e assim por diante. Esse ciclo de cálculos é
repetido até que a diferença entre uma iteração e outra fique dentro de um limite
pré-estabelecido. Neste ponto, a densidade eletrônica da molécula atingiu sua
convergência. Lembremos que a densidade eletrônica determina a distribuição de
carga elétrica da molécula, ou de um orbital, e essa distribuição de carga gera um
campo elétrico, que não era conhecido no início dos cálculos. Se o campo elétrico
final (ou a densidade que o gera) for usado como ponto de partida para um novo
ciclo de cálculos, o resultado será o próprio campo. Diz-se então que o campo é
“auto-consistente”, ou seja, gera a si mesmo, e por isso a teoria de Hartree-Fock é
referida muitas vezes como teoria do “campo auto-consistente” (self-consistent
field, SCF).
Cálculos deste tipo podem até ser realizados manualmente para alguns
átomos, como Hartree fazia na década de 20 do século passado, mas na medida em
que aumentam a complexidade do sistema e a acurância almejada, torna-se
necessário recorrer a computadores. Na verdade, uma das primeiras aplicações dos
computadores pioneiros foi a resolução de cálculos científicos como os de Hartree,
com sua participação.42 Obviamente, o tempo que um computador gasta fazendo
um cálculo SCF depende da complexidade do sistema – do número e do tipo de
átomos – e da quantidade de funções utilizadas na expansão dos orbitais
moleculares (2.7). Num único ciclo de cálculos é necessário que o computador
resolva milhares de integrais. Sendo assim, o ponto de partida para a realização de
um cálculo HF é a escolha do conjunto de funções de bases, do qual trataremos
agora com mais detalhes.

Fundamentação Teórica 13
2.1.2 Conjunto de Funções de Bases39-45, 47
“Em química quântica, um conjunto de funções empregadas para a
representação de orbitais moleculares. Pode-se distinguir entre o conjunto de
bases mínimo (inclui uma função de base para cada orbital SCF atômico ocupado
com membros quânticos principal e angular distintos); conjunto de bases com
valência dividida (inclui duas ou mais funções de base para cada orbital de
valência); conjunto de bases de duplo zeta (DZ) (um conjunto de bases com
valência dividida que inclui exatamente o dobro de funções do conjunto de
bases mínimo); conjunto de bases extendido (o conjunto maior do que o
conjunto de bases de duplo zeta); conjunto de bases polarizado (incorpora
funções de base com número quântico de momento angular superior ao
necessário para o átomo em seu estado eletrônico fundamental; permite que os
orbitais se ajustem não somente em tamanho, mas também em sua forma);
conjunto de bases com funções difusas e outros.” (Glossary of Therms Used in
Theoretical Organic Chemistry, IUPAC Recomendations 1999).46
Como dissemos no ítem anterior, é a partir das funções atômicas que são
construídos os orbitais moleculares e, sendo assim, a escolha correta do conjunto
de funções de base é essencial para o resultado final, seja em termos de acurância
ou de rapidez.
A escolha mais obvia nos primeiros tempos da química computacional foi a
adoção do chamado conjunto mínimo, composto apenas dos orbitais atômicos do
átomo livre. Assim, o conjunto mínimo para o hidrogênio é um único orbital 1s, ao
passo qua para o carbono temos 1s, 2s, 2px, 2py e 2pz. Os orbitais para ao átomo de
hidrogênio, soluções exatas da equação de Schrödinger, são “orbitais do tipo
Slater” (Slater Typer Orbitals, STO) e possuem uma dependência radial exp(-ζr). O
problema com essas funções é a sua forma matemática, que acaba por dificultar os
cálculos. Por outro lado, funções Gaussianas, exp(-ζr2), possuem propriedades que
facilitam sua manipulação e se mostram atrativas para a substituição dos STO.
Porém, uma única Gaussiana não é capaz de reproduzir um STO, principalmente
próximo à origem. Faz-se necessário então combinar várias Gaussianas para
aproximar o comportamento de um STO, mas ainda assim o uso das Gaussianas traz
simplificações.

Fundamentação Teórica 14
Um dos primeiros conjuntos construídos com este intuito foi o STO-3G, no
qual três funções gaussianas são combinadas para aproximar um orbital do tipo
Slater; daí vem a notação “3G”. As funções Gaussianas utilizadas nessa construção
são denominadas “gaussianas primitivas”. Diz-se então que três Gaussianas com
simetria 1s são “contraídas” para formar um STO 1s.
A base STO-3G, apesar de ter sido amplamente utilizada nos primeiros tempos
da química computacional, possui pouca flexibilidade do ponto de vista químico.
Por exemplo, um orbital 2s de um carbono próximo a um átomo mais
eletronegativo deve ser mais compacto do que próximo a um átomo de maior
eletropositividade. Mas no STO-3G ambos os ambientes são representados pela
mesma base 2s. Visando resolver este problema, foram desenvolvidos os conjunto
de bases com “valência dividida”. Por exemplo, ao invés de combinar três
Gaussianas para descrever um orbital de valência numa única contração, pode-se
dividir essas três funções em dois grupos, um com duas e um outro com uma única
Gaussiana. Esses grupos podem então ser otimizados de forma separada e produzir
orbitais que se adaptam melhor às particularidades do ambiente químico:
+ =
1º grupo 2º grupo combinação
A base 3-21G representa cada orbital interno por uma combinação de três
Gaussianas primitivas, e cada orbital de valência é representado por dois grupos de
funções, um formado por duas Gaussianas primitivas e o outro por apenas uma. É
possivel ainda melhorar a base aumentando-se o número de Gaussianas. Esse é o
caso da base 6-31G, que utiliza 6 Gaussianas primitivas para representar cada
orbital interno e, à semelhença da 3-21G, divide cada orbital de valência em dois
grupos, sendo agora de 3 e de 1 Gaussianas primitivas. Os resultados das bases com
valência dividida são bastante superiores aos dos conjuntos mínimos, mas em
compensação o tempo de cálculo aumenta substancialmente.
A divisão da camada de valência melhora a flexibilidade das bases,
permitindo que os orbitais atômicos se expandam ou se contraim, mas não permite

Fundamentação Teórica 15
nenhuma polarização. Considere, por exemplo, como ficaria o orbital 1s do
hidrogênio em uma hidroxila. Experaríamos um deslocamento para o lado do
oxigênio. Esse tipo de efeito pode ser tratado acrescentando-se “funções de
polarização” ao conjunto de bases. No exemplo anterior, poderíamos combinar o
orbital 1s com um orbital p:
+ =
Os próprios orbitais p podem ser polarizados pela adição de orbitais d ou mesmo f.
As funções de polarização são fundamentais para a correta descrição dos
ângulos de ligações. Segundo Jasien et al.11, essas funções são também necessárias
para uma descrição adequada das barreiras rotacionais em amidas e,
conseqüentemente, nos tipos de sistemas objetos do presente estudo. Denota-se as
funções de polarização por uma letra entre parêntesis, ou por asteriscos:
6-31G(d,p) indica o uso da base 6-31G, como a definimos acima, com a inclusão de
funções do tipo p no hidrogênio e funções do tipo d nos átomos pesados; uma
notação alternativa é 6-31G**.
Há sistemas químicos cuja descrição em regiões distantes do núcleo merece
ser considerada com mais cautela, como é o caso dos ânions ou dos átomos com
pares isolados. Para esses sistemas, adiciona-se mais uma Gaussiana ao conjunto de
bases, possuindo esta Gaussiana um coeficiente (ζ ) pequeno. Isto faz com que a
função adicionada decaia “lentamente” com o raio atômico e possua valores
significativos nas regiões afastadas do núcleo. Por essa razão, são chamadas
“funções difusas” e são identificadas pelo símbolo “+”. Assim, a base 6-31+G(d,p)
possui todos os atributos anteriores e ainda uma função difusa em cada átomo
pesado. Pode-se ainda adicionar funções difusas ao hidrogênio e aí teríamos
6-31++G(d,p).

Fundamentação Teórica 16
2.1.3 Correlação Eletrônica39-43, 48
“O ajuste do movimento eletrônico para as posições instantâneas (diferente das
médias temporais) de todos os elétrons em um sistema molecular, i.e. a
tendência dos elétrons em correlacionar seu movimento de forma a manter-se o
mais distante possível por causa das restrições impostas pelo princípio da
exclusão de Pauli (repulsão de troca) e por causa das repulsões eletrostáticas
(repulsão Coulômbica).” (Glossary of Therms Used in Theoretical Organic
Chemistry, IUPAC Recomendations 1999).46
No método de Hartree-Fock cada elétron é representado por uma função de onda
que possui dependência apenas nas coordenadas daquele elétron (aproximação dos
orbitais). A probabilidade de se encontrar um elétron em um dado ponto nas
vizinhanças de um núcleo é determinada apenas pela posição em relação ao
núcleo, mas não em relação aos outros elétrons. Um certo elétron interage apenas
com o campo médio dos outros elétrons, mas não há dependência mútua nas
equações SCF e não se considera as interações instantâneas entre esses elétrons.
Em outras palavras, os movimentos não estão correlacionados.
A aproximação introduzida por Hartree-Fock, i.e. tratar os elétrons com
funções de onda independentes, possui conseqüências energéticas. A energia
negligenciada no cálculo HF é denominada “energia de correlação eletrônica”.
Formalmente, a energia de correlação é definida por:
Sendo Eexata a energia exata não-relativística do sistema.
Diversos métodos foram desenvolvidos para o cálculo de energias de
correlação, sendo que a maioria deles parte do resultado HF e sobre ele realiza
correções. Entre eles podemos citar Interação de Configurações, Teoria de
Perturbação de Møller-Plesset e Teoria do Funcional de Densidade (TFD).
A idéia da interação de configurações é a construção de uma função de onda
que leva em consideração não somente o estado fundamental, o
Ψ , do sistema, mas
também alguns de seus estados excitados,
HFexatacorr EEE −= (2.10)

Fundamentação Teórica 17
na qual os ci são os pesos de cada “configuração eletrônica”. Os resultados de um
cálculo com interação de configurações pode ser bastante acurado, mas o custo
computacional é elevado.
Na teoria de perturbação de Møller-Plesset, a correção para a correlação é
introduzida na forma, como o próprio nome sugere, de uma perturbação sobre a
função de onda. A função de onda é escrita como:
onde )(onΨ indica a correção de n-ésima ordem para a função de onda do estado
fundamental e λ é um parâmetro que varia de 0 a 1. Quando se utiliza apenas n=1
para truncar a série acima, tem-se uma perturbação de primeira ordem, MP1
(Møller-Plesset até 1ª. ordem), para n=2, MP2, e assim por diante. A acurância do
cálculo melhora com o aumento da ordem escolhida. O mais comum é a utilização
de MP2, pelo equilíbrio entre qualidade de resultados e tempo de cálculo, mas em
trabalhos de maior rigor também se emprega MP4.
A teoria do funcional de densidade, que utilizamos ao longo de nossos
trabalhos juntamento com HF, será considerada à parte.
2.1.4 Teoria do Funcional de Densidade39, 40, 42, 43, 49, 50
“Uma teoria que trata da descrição mecânica quântica de sistemas atômicos e
moleculares em termos da densidade eletrônica. Todas as propriedades são
funcionais dessa densidade, incluindo a energia cinética T[ρ] e a energia de
repulsão elétron-elétron Vee[ρ]. A energia eletrônica total de um sistema com N
elétrons é expressa como um funcional de suas densidades de uma partícula ρ(r)
E[ρ] = T[ρ] + Iν(r) ρ(r)dr + Vee[ρ]
onde ν(r) é o potencial devido aos núcleos; a energia acima é um mínimo
quando ρ é a densidade do estado fundamental correta. Na TFD, o termo de
energia de troca exata para um único determinante é substituído por uma
(2.11) L3311oo Ψ+Ψ+Ψ=Ψ ccc
(2.12) L+Ψ+Ψ+Ψ+Ψ+Ψ=Ψ (4)o
4(3)o
3(2)o
2(1)o
(o)o λλλλ

Fundamentação Teórica 18
expressão mais geral, o funcional de troca-correlação, que inclui termos tanto
para a energia de repulsão de troca como para a energia de correlação, omitida
no método de Hartree-Fock. A TFD fornece a base conceitual para um grande
número de conceitos químicos importantes como eletronegatividade, dureza e
moleza absolutas, a teoria dos orbitais de fronteira, etc.” (Glossary of Therms
Used in Theoretical Organic Chemistry, IUPAC Recomendations 1999).46
A Teoria do Funcional de Densidade (TFD) tem suas origens ainda nos primeiros
anos da física quântica moderna, na década de 1920, com trabalhos de Thomas,
Fermi e Dirac. A idéia básica desses autores consistia em expressar a energia de um
sistema como uma função de sua densidade eletrônica, ρ(r), ou seja, E=E[ρ(r)]. A
densidade eletrônica é, por sua vez, uma função das coordenadas espaciais, r. Por
esta razão, diz-se que a energia é um “funcional” da densidade eletrônica, pois o
papel de variável é desempenhado por uma outra função, ρ(r). Porém, as
considerações que precisavam ser feitas naquela época para se chegar a resultados
numéricos eram demasiadamente simplistas e não encontrariam uso na química
quântica moderna.
Passos importantes foram dados por Slater na década de 1950, mas pode-se
dizer que a estrutura moderna da TFD deve-se aos trabalhos de Hohenberg e Kohn,
em 1964,51 e Kohn e Sham, em 1965.52 Hohenberg-Kohn demonstraram, pela
primeira vez, que a densidade eletrônica determina de forma unívoca todas as
propriedades do estado fundamental de um sistema, colocando assim a teoria de
Thomas-Fermi-Dirac num patamar exato. Porém, faltou aos autores estabelecer
como calcular o funcional E[ρ(r)]. Para fazê-lo, Kohn-Sham desenvolveram uma
abordagem semelhante à de Hartree-Fock, que leva à seguinte expressão para a
energia molecular:
E = ET + EV +EJ + EXC
na qual ET é a energia cinética, EV inclui a contribuição da atração elétron-núcleo e
da repulsão núcleo-núcleo, EJ é a repulsão elétron-elétron e EXC é o termo de troca
e correlação. Este último deve-se essencialmente a efeitos quânticos e não possui
análogos clássicos como os três primeiros. O termo de EXC leva em consideração a
(2.13)

Fundamentação Teórica 19
energia de troca, que surge da antisimetria das funções de onda para elétrons
(Seção 2.1.1), e a energia de correlação, que resulta da influência que um elétron
exerce sobre o movimento dos demais (também chamada de correlação dinâmica).
O funcional de troca-correlação pode ainda ser dividido em duas partes:
EXC[ρ(r)] = EX[ρ(r)] + EC[ρ(r)]
sendo EX[ρ(r)] e EC[ρ(r)] os funcionais de troca e de correlação, respectivamante.
Recentemente, Becke49 percebeu que haveria vantagens em mesclar os
métodos de Hartree-Fock e TFD, o que deu origem aos métodos híbridos. O mais
popular deles nos dias de hoje é conhecido como B3LYP, sigla que identifica o uso
do funcional de troca-correlação de Becke no qual está incluído o funcional de
correlação desenvolvido por Lee, Yang e Parr.53 O número três vem do uso de três
parâmetros empíricos utilizados para compor o funcional EX. Por utilizar esses
parâmetros empíricos, é comum não classificar o método B3LYP como ab initio.
Convém mencionar que nem todos os métodos TFD são híbridos ou utilizam
parâmetros e, assim, poderiam ser chamados ab initio. Todavia, os parâmetros
empíricos estão presentes nos mais utilizados e é comum considerar a TFD
propriamente como uma classe à parte para diferenciá-la de métodos como HF e
MP2. Uma vantagem importante da TFD sobre os demais métodos de correlação
eletrônica é sua rapidez, o que fez com que essa teoria se tornasse popular nos
últimos anos entre a comunidade química.
2.1.5 Otimização de Geometrias40-44
Nas seções anteriores descrevemos os métodos para obter a energia
molecular, sem porém mencionar qual estrutura deve ser utilizada em tais
cálculos. Certamente a energia da molécula de H2 será diferente se os átomos
estiverem separados por 1 Å ou por 1,5 Å, mas como decidir qual o valor? Uma
opção é recorrer a dados experimentais como os de difração de raios-X ou
microondas, mas para a maioria dos casos estes dados não são disponíveis. No caso
do H2, poderíamos calcular a energia para diversas distâncias H—H e assim
(2.14)

Fundamentação Teórica 20
determinar qual o valor mínimo para o sistema (Fig. 2.1). Esse processo é chamado
de “otimização de geometria”.
Figura 2.1. Superfície de energia potencial hipotética para o H2.
Todavia, à medida em que a complexidade do sistema aumenta, também
aumenta o número de parâmetros a serem otimizados. Para a água, teríamos dois
comprimentos de ligação (ou um, se aplicarmos restrições de simetria) e um ângulo
de ligação. Para o peróxido de hidrogênio teríamos três comprimentos de ligação,
dois ângulos de ligação e um ângulo diedro, e assim por diante. Dessa forma,
adotar o procedimento de otimização que mencionamos acima para a molécula de
H2 não seria uma boa opção, muitas vezes até inviável. Imagine, por exemplo,
construir uma curva como a apresentada acima para cada parâmetro do cicloexano.
E ainda que o fizéssemos, teríamos os mínimos referentes a cada parâmetro, mas
dificilmente encontraríamos o mínimo global.
É possivel resolver esse problema se as derivadas da energia com respeito a
cada parâmetro forem determinadas, ou em outras paravras, pelo conhecimento do
gradiente da energia molecular com respeito aos parâmetros estruturais:
grad E = 3 ME / Mqi
Sabemos, do cálculo diferencial, que os extremos de uma função ocorrem nos
pontos onde sua derivada é zero. Pontos de mínimo e máximo são determinados
inequivocamente pela segunda derivada, que é positiva nos mínimos e negativa nos
máximos. Porém, o cômputo das segundas derivadas introduz complicações
(2.15)
Distância H-H
Ener
gia
Estrutura ótima

Fundamentação Teórica 21
adicionais e aumenta consideravelmente o tempo de cálculo, de forma que os
algoritmos utilizados atualmente trabalham em busca da condição:
grad E = 0
e são contruídos de modo a procurar por mínimos de energia e não máximos. As
derivadas da energia, tanto primeira como segunda, possuem expressões analíticas
conhecidas para boa parte dos métodos teóricos.
De forma simplificada, o processo de otimização de geometria funciona como
segue. Parte-se de uma estrutura inicial, com parâmetros estruturais aproximados
(ex. comprimentos de ligação C-C de 1,4 Å e C-H de 0,9 Å), calcula-se a energia da
molécula e determina-se o gradiente da energia em relação aos parâmetros. Se o
gradiente estiver abaixo de um valor preestabalecido (por exemplo 0,001) a
geometria é considerada otimizada. Caso contrário, varia-se os parâmetros,
seguindo-se para isso um algoritmo específico, e calcula-se novamente a energia
molecular e seu gradiente. O processo é repetido até que se obtenha a
convergência da estrutura para um ponto de mínimo, ou seja, grad E ≈ 0.
2.1.6 Propriedades Termodinâmicas39, 40, 43, 54
Até agora, apresentamos as ferramentas para determinar a geometria
molecular ótima e sua energia. Essa energia é normalmente chamada de energia
total, incluindo as contribuições eletrônica e de repulsão nuclear. Contudo, a
descrição completa dos sistemas químicos requer o conhecimento da energia livre
de Gibbs, G, e das quantidades relacionadas, entalpia H e entropia S. Para obtê-
las, utlizamo-nos da estrutura teórica da termodinâmica estatística, ramo da
ciência que cuida da determinação das propriedades termodinâmicas de um
sistema em termos de seu comportamento microscópico.54 Segundo a
termodinâmica estatística, o elo de ligação entre o macroscópico e o microscópico
é a “função de partição”, Q, cuja forma geral é:
Q = 3 exp(-ei / kBT)
(2.16)
(2.17)

Fundamentação Teórica 22
Sendo ei os níveis de energia do sistema, kB a constante de Boltzmaan e T a
temperatura absoluta. A função de partição corresponde ao número de estados
microscópicos acessíveis ao sistema em uma dada temperatura.
Tendo em vista que os níveis de energia de uma molécula dividem-se em
eletrônico, rotacional, vibracional e translacional, pode-se escrever:
Q = Qele Qvib Qrot Qtrans
na qual Qele representa a contribuição eletrônica, Qvib representa a contribuição
das vibrações moleculares, Qrot representa a contribuição da rotação molecular e
Qtrans diz repeito à translação da molécula. (À rigor, teríamos de incluir também a
contribuição da energia relativistica, E=mc2, mas as variações nesta energia são
desprezíveis na escala dos processos químicos, de modo que resultados acurados
podem ser obtidos sem o seu cômputo).
Para o cálculo da contribuição eletrônica, assume-se que a população dos
estados excitados é desprezível e trabalha-se apenas com o estado fundamental.
Uma vez que a função de partição é o número de estados acessíveis ao sistema, no
estado fundamental ela pode ser determinada pela multplicidade de spin da
molécula.
A contribuição vibracional é obtida tratando-se cada modo de vibração como
um oscilador harmônico. Essa simplificação é adequada na maioria dos casos, mas
pode causar erros para alguns modos que apresentam alto caráter anarmônico,
como a inversão do nitrogênio ou a torção de metilas.19
A parte translacional é obtida a partir dos níveis de energia da partícula na
caixa, ao passo que a contribuição rotacional é calculada pelos níveis de energia de
um rotor rígido. Na prática, a contribuição desses dois movimentos para as
variações nas propriedades termodinâmica são pouco importante para rotacões
sobre ligações, e o mesmo vale para a parte eletrônica. A maior contribuição
provém, portanto, das vibrações moleculares, pois as freqüências vibracionais são
mais sensíveis às variações que ocorrem durante a isomerização rotacional.
(2.18)

Fundamentação Teórica 23
2.1.7 Modelos de Solvatação39-41, 43, 45, 55-61
Apresentamos os resursos necessários para obter a energia molecular e as
propriedades termodinâmicas de um composto. No entanto, os procedimento
anteriores trabalham com a molécula no vácuo (ou em fase gasosa, se assumirmos o
comportamento de um gás ideal). Uma vez que a presente tese propõe, entre seus
objetivos, o estudo do efeito do solvente sobre as barreiras rotacionais, é preciso
descrever também as formas de se obter as energias moleculares em fase
condensada.
Há dois modos de se estudar o efeito do solvente sobre as propriedades
moleculares: os métodos contínuos e os métodos explícitos. No primeiro caso, o
solvente é tratato como um contínuo representado por alguns parâmetros, como a
constante dielétrica. Nesses modelos não se faz referência à estrutura molecular
do solvente, e sendo assim, interações fortes como as ligações de hidrogênio não
são incluídas. Por outro lado, nos modelos explícitos trabalha-se diretamente com
uma porção de moléculas de solvente, sendo essa porção grande o suficiente para
reproduzir as propriedades do liquido. Portanto, nos modelos explícitos as
interações específicas soluto-solvente (complexação ou ligações de hidrogênio) são
consideradas. Ademais, leva-se em conta a estrutura estatística do líquido, uma
vez que as proprieades são obtidas como uma média sobre um grande número de
configuração das posições moleculares.
A grande desvantagem dos modelos explícitos é a dimensão do sistema em
estudo. Tipicamente, é preciso cerca de 200 moléculas para se reproduzir
apropriadamente as propriedades de um líquido. Se o solvente for a água, teríamos
um sistema com 600 átomos, que é um número proibitivo para trabalhos ab initio
ou TFD. A dificuldade não surge da impossibilidade de calcular a energia de tal
sistema, isso é possivel, mas sim do tratamento das posições moleculares. Numa
caixa com 200 moléculas, teríamos um número inestimável de mínimos de energia
e todos contribuiriam de forma expressiva para as propriedades macroscópicas do
líquido. Aqui surge o primeiro problema, para se obter um único desses mínimos
seria necessário uma otimização da geometria de todo o sistema, e isso aumenta
drasticamente o custo computacional. Ainda que se realizasse essa otimização,
teríamos apenas um dos tantos mínimos e a cobertura da totalidade tornaria os

Fundamentação Teórica 24
trabalhos inviáveis. Por essa razão, os modelos explícitos utilizam simplificações
que apelam à mecânica clássica na busca da redução do tempo de cálculo. A seguir
descreveremos os modelos contínuos. Dos modelos explícitos, por constituirem uma
família de cálculos completamente diferente dos ab initio e TFD, trataremos após
descrever todos os fundamentos de estrutura eletrônica necessários aos nossos
trabalhos.
2.1.8 Modelos de Solvatação Contínuos40, 55, 57-61
O mais simples dos modelos de solvatação é o de Onsager,56, 60, 61 que baseia-
se na interação do dipolo do soluto com o campo elétrico do solvente. O dipolo
molecular induz um campo elétrico no solvente, que, por sua vez, interage com o
soluto e causa estabilização. Por essa razão, diz-se que este é um modelo de
campo de reação. Em sua forma atual, a energia obtida pelo modelo de Onsager é
calculada de forma auto-consistente, ou seja, o efeito do campo elétrico sobre a
energia molecular é incluído no Hamiltoniano eletrônico e participa do proceso SCF
(Seção 2.1). Esse procedimento também é conhecido como método SCRF (self-
consistent reaction field).
A maior limitação do método de Onsager é a forma da cavidade molecular. O
soluto é colocado em uma cavidade esférica, fora da qual localiza-se o contínuo
caracterizado pela constante dielétrica e. Uma cavidade esférica é uma
aproximação razoável apenas para moléculas pequenas. Em isomerização
rotacional, a forma molecular, particularmente o volume, não se modifica muito e
pode-se contar nestes casos com um cancelamento de erros. Uma outra limitação é
a consideração apenas do momento de dipolo, mas esta é mais branda do que a
forma da cavidade.
O modelo do contínuo polarizável (PCM, porarizable continuum model)57-59
melhora bastante as duas limitações que citamos para Onsager. A cavidade do
soluto é definida pela junção de um conjunto de esferas de tamanhos diferentes.
Além disso, nas fronteiras da cavidade situa-se um elevado número de cargas
pontuais que se adequam, durante os cálculos, de forma a melhor reproduzir o

Fundamentação Teórica 25
campo de resposta do solvente induzido pelo soluto. O tratamento desse modelo é
equivalente a considerar todos os momentos elétricos da molécula.
Um terceiro modelo melhora ainda mais a descrição da cavidade do soluto,
trata-se do modelo do contínuo polarizável com uso de superfície de isodensidade
(IPCM, isodensity polarizable continuum model).55 A abordagem é parecida com a
do PCM, mas a cavidade é definida por meio de uma superfície construída a partir
da densidade eletrônica do soluto (Fig. 2.2). O IPCM trabalha com um valor fixo de
isodensidade ao longo dos cálculos (tipicamente 0,0004 e/bohr2). Para melhorar
ainda mais a descrição do efeito do solvente, pode-se variar esse valor e otimizá-lo
de forma a fornecer um efeito mais realístico. Isso é feito no modelo do contínuo
polarizável com uso de superfície de isodensidade auto-consistente (SCF-IPCM, self-
consistente field isodensity polarizable continuum model). Embora melhor do que
o IPCM, o SCF-IPCM costuma apresentar problemas de convergência além de exigir
um tempo de cálculo muito maior. Na prática, o custo computacional dos modelos
contínuos segue a ordem SCF-IPCM > IPCM > PCM > Onsager.
Figura 2.2. Definição da cavidade molecular do modelo IPCM para a DMF.
Existe ainda um grande número de modelos de solvatação contínuos que não
descreveremos aqui.59 Ao longo de nossos trabalhos, utilizamos o IPCM, de modo
que nossa intenção com o que expusemos acima era apenas apresentar uma
hierarquia dos modelos e localizar o método que escolhemos.

Fundamentação Teórica 26
2.1.9 Teoria dos Orbitais Naturais de Ligação62-68
“Orbital natural de ligação (NBO) – o orbital que é formado a partir de orbitais
natutais híbridos. Para uma ligação s localizada entre os átomos A e B, o NBO é:
sAB = cAhA + cBhB
onde hA e hB são híbridos naturais centrados nos átomos A e B. Os NBOs
correspondem, de forma muito próxima, à representação de ligações localizadas
e pares isolados como as unidades básicas da estrutura molecular, de forma que
é possível interpretar convenientemente as funções de onda ab initio em termos
dos conceitos clássicos de estrutura de Lewis pela transformação destas funções
para a forma NBO.” (Glossary of Therms Used in Theoretical Organic Chemistry,
IUPAC Recomendations 1999).46
Os cálculos de orbital molecular (OM) podem fornecer resultados bastante
satisfatórios com respeito às energias relativas, e mesmo para as energias totais se
for incluída correlação eletrônica com um conjunto de bases grande o suficiente. A
partir das energias relativas podemos saber que a espécie X é mais estável do que a
espécie Y. A pergunta que segue a esta informação é sobre o porquê desse
comportamento, e neste ponto a teoria do orbital molecular já não se mostra tão
atrativa. Issso porque conceitos como pares isolados, ligações simples e duplas não
existem dentro do contexto OM, o que torna o “pensar químico” um tanto
desconfortável. Para esse propósito, é conveniente utilizar métodos que buscam
incorporar as idéias das da ligação de valência. O mais popular dentre eles é o dos
orbitais naturais de ligação (natural bond orbitals, NBO) que passaremos a
descrever.
Considere a seguinte expressão, denominada de matriz de densidade de um
elétron:
na qual aij são os coeficientes e ϕi são os orbitais que determinam a matriz. O que
a Eq. (2.19) descreve é simplesmente a densidade eletrônica molecular em termos
das densidades eletrônicas associadas a cada orbital ϕi. Em sua forma diagonal,
somente os elementos com i = j são diferentes de zero, i.e.
∑∑=i j
jiija φφρ *
(2.19)

Fundamentação Teórica 27
sendo nk o número de ocupação do k-ésimo orbital. Os orbitais χ que satisfazem à
equação acima são chamados de “orbitais naturais” e foram propostos por Löwdin69
numa tentativa de fornecer ferramentas para interpretar as funções de onda da
mecânica quântica.
Foster e Weinhold64 perceberam a aplicação do trabalho de Löwdin nos
conceitos de hibridização e ligação química. Deste modo, propuseram um
procedimento para construir orbitais naturais atômicos (semelhantes às soluções da
Eq. de Schrödinger para o hidrogênio) e, a partir destes, orbitais naturais híbridos
(sp, sp2, sp3 ...). Os orbitais híbridos podem ser combinados para formar “orbitais
naturais de ligação” (NBO). Os NBOs assim construídos permitem descrever a
estrutura química de uma molécula a partir de ligações entre dois átomos e pares
isolados, de forma muito similar às estrutulas de Lewis. A molécula de metano, por
exemplo, seria formada por quatro ligações sigma C-H, HCσ − , e pelos elétrons
internos do carbono.
No processo de construção dos NBOs, formam-se também orbitais
antiligantes. No caso do metano, teríamos os *H-Cσ e os orbitais de Rydberg, que são
orbitais antiligantes centrados nos átomos.65 A teoria NBO fornece ainda
ferramentas para analisar transferências de carga de orbitais ligantes para
antiligantes, bem como as implicações energéticas associadas. Isso é feito a partir
de um procedimento de perturbação de segunda ordem que fornece a seguinte
expressão para a energia de estabilização:
onde qi é a ocupação do orbital doador, ei a energia de cada NBO e Fij é o elemento
fora da diagonal da matriz de Fock.68
Badenhoop e Weinhold62 demonstraram que é possível avaliar também
interações não-ligantes por meio da teoria NBO. As interações de que essa teoria
pode tratar são de natureza quântica e derivam do princípio da exclusão de Pauli,
∑=k
kkkn χχ *ρ (2.20)
ij
ij
i
FqE
ee −=
2)2(
(2.21)

Fundamentação Teórica 28
sendo por isso referidas como “interações de Pauli” ou “repulsões de Pauli”, ou
ainda “energia de troca”.
Quando dois orbitais são ortogonais, sua integral de sobreposição é nula, ao
contrário de orbitais não-ortogonais. Badenhoop e Weinhold62 propuseram então
que o custo energético da ortogonalização de orbitais naturais poderia ser utilizado
para determinar a energia de troca do sistema. Essa energia de troca, ao contrário
daquela mencionada na teoria de Hartree-Fock, é de caráter desestabilizador.
Fisicamente, a energia de troca assim obtida é vista como uma espécie de “pressão
quântica” que tende a manter os elétrons separados.25 Portanto, pode-se calcular a
energia de troca para duas moléculas, digamos, dois confôrmeros, e comparará-las,
de forma que aquele que possuir o maior valor será desestabilizado por esse tipo de
interação.
2.2 Dinâmica Molecular
2.2.1 Visão Geral39, 41, 43, 70-73
“Um método de modelagem computacional das propriedades estáticas (no
equilíbrio) e dinâmicas (cinéticas) de sistemas de várias partículas baseado na
resolução da equação Newtoniana clássica, assumindo um potencial de forças e
um protocolo para a preparação do sistema em uma configuração de partida
racional predeterminados, visando monitorar o movimento das moléculas no
líquido ou em solução, definir as trajetórias e então a média sobre o tempo, e
obter a magnitude das funções examinadas.” (Glossary of Therms Used in
Theoretical Organic Chemistry, IUPAC Recomendations 1999).46
Nas seções anteriores descrevemos os métodos de cálculo que tratam
detalhadamente da estrutura eletrônica dos sistemas químicos. Esses métodos
tornam possível a predição de um grande número de propriedades moleculares,
cuja exatidão depende do nível de teoria que se usa para a realização dos cálculos.
Os métodos de estrutura eletrônica são bem sucedidos quando tratamos de
propriedades da fase gasosa ou quando as interações intermoleculares de uma fase

Fundamentação Teórica 29
condensada são pouco importantes, mas, à medida que interações como as ligações
de hidrogênio tornam-se importantes, a divergência entre experimento e teoria
fica mais acentuada. Como explicado na Seção 2.1.7, existem métodos de cálculo
que tentam reproduzir o efeito das vizinhanças sobre uma molécula através da
representação dessas vizinhanças por um campo de forças médio (o campo de
reação). Porém, esse procedimento ainda não é suficiente para tratar da influência
das interações de curto alcance.
Para formarmos uma idéia melhor sobre a importância de tratar
adequadamente a estrutura estatística dos líquidos, lembremos que o tempo de
aquisição dos espectros de RMN de 1H é da ordem de segundos. No entanto, as
modificações das configurações das moléculas de solvente ao redor do soluto
ocorrem em tempos da ordem de picosegundos (10-12 s).41 Em outras palavras, o
resultado obtido no espectro é uma média temporal dessas configurações, cujo
número, em nosso exemplo, é da ordem de 1012 (!). daí surge a necessidade de
métodos como Monte Carlo e dinâmica molecular;39, 41, 43 nos ocuparemos apenas
com este último.
De forma resumida, o que se faz em dinâmica molecular é preparar o
sistema em um estado inicial e, a partir das leis clássicas, reproduzir sua evolução
temporal. Durante esse processo, produz-se um grande número de configurações
espaciais que são acumuladas em certos intervalos de tempo. Ao final, pode-se
avaliar uma propriedade física de interesse tomando-se uma média temporal de
todas as configurações.
O primeiro passo numa simulação é o estabelecimento das posições e
velocidades iniciais, sendo essas últimas determinadas a partir de uma distribuição
de Maxwell. A seguir calcula-se as forças atuantes nas partículas em um tempo t
através da relação,
na qual V é o potencial do sistema e ri as posições das partículas. Por meio de uma
solução numérica da equação de movimento de Newton, chega-se às posições e
velocidades de cada partícula em um tempo t+∆t.
i
ir
VF
∂∂
−= (2.22)

Fundamentação Teórica 30
Existem certas condições utilizadas durante a simulação que ajudam a tornar
o modelo verossímil, trata-se das “condições periódicas de contorno”. Numa caixa
cúbica contendo moléculas de solvente, os movimentos moleculares estão
orientados em direções aleatórias, de forma que uma molécula pode migrar tanto
para o interior da caixa como para fora. Neste último caso, teríamos um problema
com a conservação de matéria. Esse problema pode ser corrigido cercando-se a
caixa de interesse por outras 21 imagens suas, o que também diminui a passagem
abrupta do solvente para o vácuo. Assim, quando uma molécula deixa a caixa por
uma extremidade, uma outra, com as mesmas características, adentra a caixa pela
face oposta. Além disso, volume, temperatura e pressão podem ser controlados,
estes dois últimos por meio de recursos que equivalem ao acoplamento do sistema
a “reservatórios de calor e pressão”. Mantendo-se o volume (V), a temperaruta (T)
e o número de partículas (N) constantes, teríamos um “ensemble”* NVT. Se
permitíssemos que o volume variasse e mantivéssemos a pressão constante,
teríamos um “ensemble” NPT, dentre outros.
2.2.2 Campo de Forças12, 13, 74-77
Para o cálculo do movimento das moléculas, é preciso conhecer o potencial
de interação entre as mesmas. O cômputo das energias potenciais em dinâmica
molecular precisa ser mais rápido do que em métodos ab initio e TFD, e por isso
recorre-se a formulações clássicas. Uma simplificação normalmente utilizada é a
negligência dos movimentos intramoleculares, o que torna desnecessário o cálculo
das interações entre os átomos de uma mesma molécula. As interações
intermoleculares são compostas por forças de Coulomb e de van der Waals. A
expressão matemática para um potencial Coulombiano é bem conhecida, e para a
representação das forças de van der Waals adota-se o potencial de Lennard-Jones.
Desta forma, a energia de interação entre duas moléculas é dada pela seguinte
equação:
* A palavra “conjunto” ou “coleção” aplicaria-se bem ao presente caso, mas seguiremos a tendência dos tradutores em manter o termo “ensemble”.
(2.23) ( ) ( )[ ]{ }∑∑ −+=∆i j
ijijijijijijji rrreqqE 6122int //4/ σσε

Fundamentação Teórica 31
sendo
,
na qual qi são as cargas atômicas, enquanto σ e εi são os parâmetros de Lennard-
Jones que determinam as interações de origem não-eletrostática. O conjunto de
parâmetros e a Eq. (2.23) determinam um “campo de forças”.
Quando bem parametrizado, um campo de forças pode fornecer resultados
comparáveis, e por vezes superiores, aos ab initio e TFD. Contudo, os cálculos de
estrutura eletrônica demoram horas ou dias, ao passo que o uso de campos de
forças reduz os tempos para a ordem de segundos. Em contrapartida, as
parametrizações possuem muita especificidade, isto é, um conjunto de parâmetros
ajustados para uma classe de compostos não se aplica a uma segunda classe, e
mesmo numa única classe pode haver resultados divergentes. Conseqüentemente,
em muitos casos é preciso, antes de realizar a simulação, parametrizar o campo de
forças para o sistema que se deseja estudar. Esse procedimento será descrito com
mais detelhes nas discussões.
2.2.3 Função de Distribuição Radial de Pares39, 41, 70
Como mencionado anteriormente, nas simulações de líquidos constrói-se modelos
que buscam reproduzir, de forma explícita, as interações de curto alcance entre as
moléculas. A partir dessas simulações pode-se fazer inferências a respeito do
arranjo relativo das moléculas, ou seja, a respeito da estrutura do líquido. Essa
informação pode ser obtida pela “função de distribuição radial de pares” (fdr),
definida por:
onde ( )rgij é a fdr entre os átomos i e j, ( )rrrNij ∆+, é o número de átomos j
encontrados na camada esférica r e rr ∆+ cujo centro é o átomo i, rr ∆24π é o
jiij εεε = jiij σσσ =
( )( )
j
ij
ijrr
rrrNrg
ρπ ∆
∆+= 24
,(2.24)

Fundamentação Teórica 32
volume da camada esférica e jρ é a densidade numérica média de átomos j na
porção de líquido utilizada na simulação.
Para ver de que forma a fdr descreve a organização das moléculas do
líquido, observemos primeiramente que a razão ( ) rrrrrNij ∆∆+ 24, π é a densidade
numérica de átomos j no interior da camada esférica de volume rr ∆24π , que
chamaremos de ( )rrrj ∆+,ρ , de modo que a Eq. (2.24) pode ser escrita como:
Consideremos agora um líquido no qual a densidade de átomos j é uniforme
ao londo de todo o volume. Nesse caso, a densidade local, ( )rrrj ∆+,ρ , seria igual
à densidade média, jρ , em todas as regiões do líquido e teríamos ( ) 1=rgij .
Suponhamos agora que em uma região próxima de r a densidade local ( )rrrj ∆+,ρ
fosse maior do que a densidade média jρ , teríamos então ( ) 1>rgij . Caso
contrário, isto é, ( )rrrj ∆+,ρ menor do que jρ , teríamos ( ) 1<rgij . Interpreta-se
( )rgij da seguinte forma: as regiões do líquido para as quais ( ) 1>rgij são regiões
para as quais a probabilidade de se encontrar átomos j é aumentada em relação às
regiões em que ( ) 1=rgij . O mesmo aplica-se a ( ) 1<rgij . Na Fig. 2.3 apresentamos
uma fdr típica.
Figura 2.3. Função de distribuição radial.
(2.25) ( )( )
j
j
ij
rrrrg
ρ
ρ ∆+=
,
0 2 4 6 8 10
0,0
0,3
0,6
0,9
1,2
1,5
g(r)
r (Å)
Rc

Fundamentação Teórica 33
As informações extraídas das fdrs podem ser analisadas de forma
quantitativa a partir de um processo de integração. Por exemplo, na Fig. 2.3 o
primeiro máximo corresponde à primeira camada de solvatação de átomos j ao
redor do átomo i, de forma que pela integração da fdr de 0 até o primeiro vale,
que identificaremos por um raio de corte Rc, teremos o número de átomos j
solvatando o átomo i dentro de Rc. Este número, ou número de coordenação
( )cij RC , é calculado por:
e para o caso de intervalos finitos,
Para a análise das frds é preciso considerar também o formato das bandas, e
não somente a existência de máximos e mínimos, como veremos nas discussões.
2.3 Ressonância Magnética Nuclear Dinâmica78-81
A determinação de barreiras rotacionais em sistemas conjugados é realizada
essencialmente através da ressonância magnética nuclear. A espectroscopia de
microondas, empregada para moléculas pequenas como o acetaldeído, não é
aplicada à modelos similares às amidas por causa da necessidade da resolução de
equações demasiadamente complexas para cada molécula.82 Por outro lado, a RMN
fundamenta-se numa base simples que pode ser facilmente aplicada sem a
necessidade de uma formulação teórica específica para cada sistema.83 Recebe a
denominação “RMN dinâmica” (RMND) a parte da RMN que se ocupa com os
processos de permuta química, tanto no âmbito intramolecular como
intermolecular. A RMND explora o fato de que o perfil dos sinais, sua intensidade e
largura, dependem da rapidez com que os processos de troca ocorrem – para
( ) ( ) drrrgRCcR
ijjcij2
0
4πρ ∫=
( ) ( ) rrrgRCcR
kkkijjcij ∆= ∑
=0
24πρ
(2.26)
(2.27)

Fundamentação Teórica 34
velocidades entre 10-1 e 103 Hz as bandas de 1H são afetadas, enquanto as bandas
de 13C sofrem modificações entre 10 e 5×103 Hz.83
Para melhor entender a técnica, consideremos como exemplo um dos
compostos estudados na presente tese, o N,N-dimetilditiocarbamato de S-metila
(4). A rotação sobre a ligação C-N faz com que as metilas amídicas invertam sua
posição:
Essa rotação ocorre um certo número de vezes por segundo e, assumindo-se
uma cinética de primeira ordem, pode ser caracterizada por uma constante de
velocidade k em Hz (ou s-1). Uma vez que as vizinhanças químicas de (A) e (X) são
diferentes, seus deslocamentos químicos devem ser distintos e poderiam ser
observados separadamente no espectro. Porém, o formato dos sinais depende da
velocidade da rotação sobre a ligação C-N, o que por sua vez depende da
temperatura em que é realizado o experimento. Em temperaturas baixas o
suficiente, observa-se sinais distintos, mas à medida em que a temperatura é
elevada, os sinais passam a se alargar e a se aproximar e, por fim, coalescem numa
única banda (Fig. 2.4).
Figura 2.4. Experimento de RMND de 1H para o composto 4 em CDCl3: a) espetros experimentais em diversas temperaturas; b) espectros simulados e suas respectivas constantes de velocidade (k).
C N
CH3
CH3
S
H3CS
C N
CH3
CH3
S
H3CS
(A)
(X)
(X)
(A)

Fundamentação Teórica 35
Informações sobre a cinética do processo podem ser obtidas modificando-se
as equações de Bloch – equações fundamentais da RMN – para processos com
permuta. Chega-se à seguinte expressão para o perfil do sinal em função da
freqüência n:
onde νA e νX são as frequências de ressonância dos núcleos A e X, M0 é a
magnetização e k é a constante de velocidade.81, 83
A derivada de I(ν) em relação a ν fornece dois máximos com uma separação
(∆ν’) que satisfaz a equação:
Na temperatura de coalescência (Tc) , ∆ν '= 0, o que nos leva a:
Vale salientar que a separação dos sinais (∆ν) refere-se à uma condição na
ausência de troca, na qual os sinais não estejam alargados. (Geralmente, é preciso
ir à temperaturas muito baixas para atingir esta condição, o que, por vezes,
inviabiliza a determinação de ∆ν por causa do congelamento do solvente).
A teoria do estado de transição de Eyring80 relaciona a constante de
velocidade com os parâmetros de ativação ∆G‡, ∆H‡ e ∆S‡ por meio da equação:
(2.28) ( ) ( )( )[ ] ( ) ( )2X
2A
22XA
2
2XA0
42 ννννννννν
ν−−+−+
−=
πk
kMI
(2.29) ( ) ( )[ ] 2122
XA '2
νννπ
∆−−=k
(2.30) 22
)( XAc
νπννπ ∆=
−=k
∆−=
RT
G
h
Tkk
‡B expκ
(2.31a)
∆
∆−=
R
S
RT
H
h
Tkk
‡‡B expexpκ (2.31b)

Fundamentação Teórica 36
na qual kB é a constante de boltzmann, h é a constante de Planck e κ é o
coeficiente de transmissão (usualmente tomado como 1). Isolando ∆G‡ da
Eq. (2.31a) e substituindo o valor das constantes, chegamos a:
que fornece a energia de Gibbs de ativação em cal/mol. Portanto, podemos utilizar
a constante de velocidade na coalescência dada pela Eq. (2.30) e conhecer assim o
∆G‡ para o processo. Na Fig. 2.4, o primeiro espectro inferior serviria para
determinar Dν, ao passo que do último superior se extrairia Tc.
Tal procedimento, contudo, não fornece a entalpia e a entropia de ativação.
Essas duas últimas precisam ser determinadas por um protocolo diferente. Se
linearizarmos a Eq. (2.31b), obtemos:
que nada mais é do que uma equação com a forma y=a-b(1/T). Assim, um gráfico
de ln(k/T) versus 1/T fornece os valores de ∆H‡ e ∆S‡ pelos coeficientes angular e
linear, respectivamente, e, a partir destes, o de ∆G‡ por meio de:
∆G‡ = ∆H‡ - T∆S‡
O problema deste último procedimento é a necessidade de se conhecer a
constante de velocidade em diversas temperaturas, ao passo que a Eq. (2.30) é
válida somente na temperatura de coalescência. Para este propósito, faz-se uso do
método de “análise do perfil das bandas” (APB).83 A Eq. (2.28) expressa o perfil das
bandas de RMN de dois núcleos em permuta como uma função da freqüência e de
alguns parâmetros, dentre eles, a constante de velocidade k. Podemos tomar um
espectro experimental e variar os parâmetros da Eq. (2.28) até que os sinais
experimentais e calculados coicidam. Nesta condição, teríamos a constante de
velocidade para a temperatura de realização daquele experimento. Este processo
pode ser repetido para várias temperaturas, donde se obtém as constantes de
velocidade necessárias à construção do gráfico. Atualmente, diversos programas
+=∆
k
TTG log32,1058,4‡
(2.32)
RT
H
R
S
h
k
T
k ‡‡Blnln
∆−
∆+
=
(2.33)
(2.34)

Fundamentação Teórica 37
computacionais implementam rotinas que facilitam o trabalho de ajuste das
bandas, como é o caso do WINDNMR,84 que adotamos na presente tese. Na Fig. 2.4
apresentamos uma comparação de espectros calculados e simulados.
Portanto, as energias de Gibbs de ativação podem ser obtidas tanto pelo
método da temperatura de coalescência como por análise do perfil das bandas,
sendo este último procedimento um pouco mais trabalhoso, porém também mais
informativo.
2.4 Barreiras Rotacionais em Sistemas Amídicos
A rotação sobre a ligação C-N pode ocorrer via dois estados de transição, nos quais
o par de elétrons do nitrogênio posiciona-se anti (et1) ou syn (et2) à ligação dupla
(Fig. 2.5).15, 17, 18, 22, 23, 31-33, 85-87 Nos estados de transição não há orientação
adequada para a conjugação *OCN =→ πn , de modo que a forma mais estável do
nitrogênio é a piramidal.
Figura 2.5. Definição das estruturas moleculares para as espécies existentes durante a
rotação sobre a ligação C-N.
Os estados de transição possuem propriedades químicas diferentes,
particularmente no que diz respeito a seus momentos de dipolo. A forma como

Fundamentação Teórica 38
esses estados de transição são afetados pelo meio serve de base para explicar o
comportamento de sistemas distantes.19, 20
Embora o estudo dos sistemas amídicos não seja algo novo,16, 88 investigações
sistemáticas acerca do efeito do solvente surgiram apenas na última década.7, 10, 19,
89, 90 Wiberg et al.19 determinaram, tanto experimentalmente como teoricamente,
as barreiras rotacionais para a N,N-dimetilacetamida (DMA) e para a N,N-
dimetilformamida (DMF). Seus estudos mostraram que a barreira da DMA aumenta
por 4 kcal/mol ao se passar da fase gasosa para água e a DMF sofre um aumento
mais modesto, de 2 kcal/mol. O comportamento distinto dessas amidas foi
explicados com base em cálculos IPCM no nível MP2/6-31+G(d,p), como segue.
Primeiramente, o estado de transição 1 (et1, Fig. 2.5) possui um momento de
dipolo menor do que o estado de transição 2 (et2), de acordo com os cálculos
teóricos, e ambos os et possuem momento de dipolo menor do que o estado
fundamental (ef). Na DMA, o estado de transição preferido é o et1, enquanto que
para a DMF a rotação ocorre preferencialmente por meio do et2. Sendo assim, a
rotação para a DMA é governada por uma diferença de momento de dipolo maior do
que aquela na DMF. À medida em que a polaridade do solvente aumenta, a barreira
rotacional sofre também um aumento proporcional à essa diferença de momentos
de dipolo, e isso faz com que a barreira na DMA seja mais afetada.19
Posteriomente, Wiberg e Rush20 estenderam os estudos para a N,N-
dimetiltioacetamida (DMTA) e para a N,N-dimetiltioformamida (DMTF). Os
resultados foram similares, porém as tioamidas mostraram-se ainda mais sensíveis
ao solvente. Rablen et al.89, 90 investigaram acrilonitrilas, extremamente similares
às amidas, e também constataram o aumento de suas barreiras rotacionais pelo
solvente.
À mesma época desses últimos estudos de Rablen, uma comunicação por Cox
e Lectka7 chamara a atenção para o fato de que um outro grupo de compostos, que
também continha um esqueleto amídico, possuia barreiras rotacionais insensíveis
ao solvente – tratava-se dos carbamatos. A situação tornou-se ainda mais intrigante
ao se calcular os momentos de dipolo dos carbamatos, pois as diferenças entre o
estado fundamental e os estados de transição eram similares às das amidas.8 Sendo
assim, seria esperado um efeito do solvente comparável àquele observado para as
amidas, e essa divergência impôs a necessidade de rever o modelo proposto por

Fundamentação Teórica 39
Wiberg et al.19 baseado nas diferenças de momentos de dipolo. Na Tabela 2.1
listamos alguns dos resultados de Cox e Lectka. 7
Tabela 2.1. Efeito do solvente sobre as barreiras rotacionais de amidas e carbamatos a,b,c
N
OMe
O
PhO
N
OMe
O
OBnO
Solvente ∆G‡ ∆G‡
CCl4 17,4 17,1
CDCl3 18,7 17,2
CD3OD 19,1 17,3
25% D2O/CD3OD 19,6 17,4
50% D2O/CD3OD 19,9 17,4
75% D2O/CD3OD 20,2 17,3 a Medidas por RMN de 1H com transferência de saturação. b Isomerisação cis-trans. c ± 0,2 kcal/mol.
Rablen8 baseou sua explicação nos momentos de dipolo dos estados
fundamentais. Em seus estudos, verificou que, apesar das diferenças de dipolo
entre ef e et serem similares para amidas e carbamatos, o momento de dipolo do
ef dos carbamatos era aproximadamente a metade daquele para as amidas.
Levando em consideração o modelo de solvatação de Onsager,61 que prevê uma
dependência quadrática com o momento de dipolo, Rablen propôs que o efeito do
solvente sobre a barreira rotacional seria proporcional ao momento de dipolo do
ef.8 Tendo as amidas momentos de dipolo próximos a 4 D e os carbamatos cerca de
2 D, esperar-se-ia uma razão de efeitos do solvente da ordem de 16/4.
Obviamente, se o efeito do solvente para os carbamatos fosse menor do que a
precisão experimental, não se verificaria modificações representativas. Portanto,
os momentos de dipolo dos ef, e não as diferenças de dipolo entre ef e et,
deveriam ser analisadas para racionalizar o efeito do solvente.
O modelo de Rablen descreve bem a relação entre amidas e carbamatos, mas,
de forma intrigante, os cálculos de Wiberg et al.19 mostraram que o momento de
dipolo do ef da DMF é levemente superior ao da DMA, ou seja, na ordem oposta à

Fundamentação Teórica 40
sensibilidade das barreiras. Para o caso das amidas, então, o modelo de Wiberg et
al. é mais adequado. Parecia, portanto, que cada modelo tinha sua validade para
uma circunstância, mas restava estabelecer o que determinava cada
comportamento. Esta era a situação no início dos trabalhos da presente tese.
Além do efeito do meio sobre as barreiras rotacionais, muito se tem feito com
o objetivo de compreender a origem destas barreiras.15, 17, 18, 22, 23, 31-33, 85-87, 91 O
modelo mais popular fundamenta-se na teoria de ressonância. Há, contudo,
aqueles que contestam a validade de tal modelo, como Lauvergnat e Hilberty.23
Esses autores utilizram cálculos ab initio baseados na teoria de ligação de valência
para estimar a contribuição da deslocalização do par de elétrons do nitrogênio na
formamida e na tioformamida. Concluiram que este efeito é responsável por
aproximadamente a metade da barreira de energia para a formamida e dois terços
para a tioformamida. De onde, então, viria o restante da energia para a rotação?
Laidig e Cameron22 utilizaram um método alternativo, que descreve a
estrutura molecular com base na soma de propriedades atômicas (átomos em
moléculas),92 e propuseram que a preferência pela hibridização sp2 no ef decorria
da estabilidade que o par de elétrons do nitrogênio conferiria ao sistema evitando
interações estéricas com a carbonila ou com a ligação C-H da carbonila (na
formamida e na tioformamida). Wiberg e Laidig24 sugeriram que as modilicações
nas cargas atômicas durante a rotação não são condizentes com o modelo de
ressonância. Observa-se uma grande variação no nitrogênio, mas a variação do
oxigênio é relativamente pequena. Por outro lado, há um aumento significativo na
carga do carbono carbonílico. Essas observações levaram os referidos autores24 a
concluir que a interação responsável pela barreira deve ocorrer entre o nitrogênio
e o carbono carbonílico, e que o oxigênio é um mero “expectador” durante a
rotação.
É possível, no entanto, reconciliar o modelo de ressonância com as
observações de Wiberg e Laidig. Lembremos que a interação responsável pela
barreira rotacional ocorre entre o par de elétrons do nitrogênio e o orbital
antiligante π da carbonila, *OCN =→ πn . O orbital antiligante tem seu maior lóbulo
na região do carbono, de forma que, numa transferência de carga para este orbital,
a maior parte se concentraria nessa região. Como o lóbulo na região do oxigênio é
menor, é razoável que a variação de carga sobre o mesmo seja menor que no

Fundamentação Teórica 41
carbono. Ainda assim, Wiberg e Rush20 atestaram que a barreira possui um
componente π e um σ, implicando que a explicação acima estaria incompleta.
Diversos estudos trataram da barreira rotacional dos carbamatos e seus
congêneros.5, 7, 8, 79, 93-109 Alguns fazem uso de cálculos teóricos, mas em nenhum
caso encontramos investigações a respeito da origem das barreiras.5, 7, 89, 103, 109 Por
serem sistemas parecidos, esperaríamos que os resultados encontrados para as
amidas servissem também para os carbamatos, mas assim fazendo, poderíamos
incorrer no mesmo erro que mencionamos a respeito do efeito do solvente. O único
estudo experimental existente sobre o efeito do solvente nas barreiras dos
carbamatos, além do que foi realizado nesta tese, é o de Cox e Lectka.7 Da mesma
forma, somente o trabalho de Rablen8 aborda o problema do ponto de vista
teórico, e ainda assim, com o uso apenas de cálculos ab initio e TFD. Quanto à
magnitude, Kleinpeter et al.110, 111 investigaram o efeito dos substituintes nas
barreiras de carbamatos e tiocarbamatos e seus resultados mostram que a variação
é discreta para substituintes alifáticos, mas aumenta significativamente para
aromáticos. Estudos de natureza similar foram conduzidos por Deetz et al.,103
segundo os quais a adição de um grupo pirimidil no nitrogênio faz a barreira
decrescer ao ponto de não ser possível sequer medí-la por RMN. Os grupos arila
ligados ao nitrogênio também diminuem a barreira, porém com menor intensidade.
Por outro lado, a substituição no oxigênio com ligações simples (-O-) faz com que a
barreira aumente.101
Os estudos experimentais acerca das barreiras rotacionais das uréias e seus
congêneros reportam valores abaixo daqueles encontrados para amidas e
carbamatos,112-117 como é o caso da uréia em dimetilformamida/dimetilsulfóxido
que apresenta uma barreira de 11,2 kcal/mol.117 A metilação deste composto
aparentemente diminui a barreira rotacional, indicando grande contribuição da
auto-associação.115 Não há, contudo, uma avaliação sistemática do efeito do
solvente, o que se deve principalmente às pequenas barreiras e à solubilidade das
uréias em temperaturas baixas.
Quanto ao aspecto teórico, existem estudos que tratam tanto da estrutura
como da origem das barreiras nas uréias e seus congêneros.118-121 Destaque-se o
trabalho de Kim et al.121, que utilizaram cálculos NBO e concluiram que a não-
planaridade da uréia em seu estado fundamental surge da competição dos pares de

Fundamentação Teórica 42
elétrons dos nitrogênios distintos pela interação com o orbital π antiligante da
carbonila. Há alguns estudos envolvendo simulações de líquidos, porém a maior
parte trata apenas das propriedades dos estados fundamentais.122-127

Parte Experimental 43
Capítulo 3:
Parte Experimental

Parte Experimental 44
Capítulo 3: Parte Experimental
3.1 Ressonância Magnética Nuclear
Os espectros de RMN foram adquiridos em um espectrômetro Varian Gemini 2000
BB operando a 300 MHz para 1H e, posteriormente, num Varian Mercury Plus BB a
300 MHz. Utilizamos as seguintes condições experimentais:
Parâmetro Gemini Mercury
Freqüência (MHz) 300,065 300,059
Largura espectral (Hz) ~ 2500 ~ 2500
Duração do pulso (ms) 6,7 7,2
Tempo de espera de reciclagem (s) 1,000 1,359
Número de transientes 16 16
Número de pontos de dados (K) 32 32
Alargamento da linha (Hz) 0,0 0,0
As amostras para os experimentos com variação de temperatura foram
preparadas pela adição de 15µL do composto líquido ou 20 mg do composto sólido
em 0,7 mL do solvente. As amostras que exigiram o uso de um tubo de inserção
foram preparadas com uma menor quantidade de solvente (0,5 mL). Nos casos em
que o dissulfeto de carbono foi usado como solvente, exigindo assim um tubo de
inserção, a acetona deuterada foi utilizada para ajustar a homogeneidade do
campo magnético. Para os experimentos realizados com água deuterada, a
referência empregada foi o 3-(trimetilsilil)propionato-2,2,3,3-d4 de sódio. Para as
demais amostras a referência foi o tetrametilsilano (TMS).
Nos experimentos à baixas temperaturas foi necessário o acoplamento, ao
aparelho de RMN, de um sistema de fornecimento de nitrogênio gasoso para a
injeção, ejeção e rotação da amostra na sonda. Esse procedimento foi necessário
tendo em vista que nessas temperaturas a umidade do ar se condensaria na sonda,

Parte Experimental 45
comprometendo o experimento. O sistema utilizado consistia de um tanque de
nitrogênio líquido com trocador de calor acoplado à saída (Fig. 3.1). O trocador de
calor foi construído a partir de um radiador de automóvel e tubos de cobre pelo
técnico MSc. Antonio Frimmel.
Figura 3.1. Sistema utilizado na produção de N2 gasoso para os experimentos à baixas temperaturas. A ampliação mostra o tracador de calor.
Em todos os experimentos com variação de temperatura aguardou-se por
cerca de 6 minutos, no mínimo, até que a amostra atingisse o equilíbrio térmico
antes da aquisição. Para a correta determinação das constantes de velocidade, a
temperatura da sonda foi calibrada por meio de padrões de metanol e etileno glicol
selados sob vácuo. No Apêncide I descrevemos com mais detalhes o processo de
calibração.

Parte Experimental 46
Os espectros foram salvos em formato MS-DOS e editados no programa
NUTS.128 Os FIDs editados foram então submetidos à análise dos perfis de suas
bandas, na região das metilas amídicas, com o programa WINDNMR.84 Este programa
permite a simulação dos sinais a partir de certos dados definidos pelo usuário, a
saber: freqüência dos sinais, largura dos sinais à meia-altura na ausência de troca e
constante de velocidade, que é o valor que mais nos interessa.
Ao todo, foram adquiridos cerca de 330 espetros de RMN de 1H (6 compostos,
8 ou 9 solventes estudados para cada composto e uma média de 7 pontos para cada
solvente).
3.2 Cálculos Teóricos
3.2.1 Métodos de Estrutura Eletrônica
Para a realização dos cálculos, foram utilizados computadores com diversas
configurações, com processadores AMD Athlon de 1,0 a 3,2 GHz e memória RAM de
256 Kb a 1,0 Gb, rodando em ambiente Windows ou LINUX (Kurumin 4.0). Também
foram empregadas estações de trabalho do Centro Nacional de Processamento de
Alto Desempenho de São Paulo (CENAPAD-SP), com processadores trabalhando
tanto em série como em paralelo em ambiente UNIX. (O CENAPAD-SP faz parte do
programa SINAPAD, implementado pelo Ministério da Ciência de Tecnologia através
da FINEP, e disponibiliza para usuários de todo o país um ambiente computacional
equipado com softwares específicos, oferecendo ainda um serviço de suporte
técnico para auxílio aos usuários).
As otimizações de geometria foram realizadas com os métodos HF e B3LYP,
utilizando o conjunto de funções de base 6-311+G(2d,p). Nesta base, as camadas
de valência são representadas por três conjuntos de funções, um deles constituído
por três gaussianas primitivas e os outros dois por apenas uma. Além disso, cada
átomo pesado recebe dois conjuntos de orbitais d para polarização, adicionando-se
um orbital p aos hidrogênios. Ao longo dos trabalhos, utilizamos também as bases
6-31+G(d,p) e a 6-31G(d) – já descritas na Seção 2.1.2 – e a PTZV de Ahlrichs et

Parte Experimental 47
al..129 Esta última, à semelhança da 6-311+G(2d,p), divide a camada de valência
em três grupos de gaussianas e adiciona um conjunto de funções d nos átomos
pesados, além de p no hidrogênio.
As geometrias otimizadas foram submetidas a cálculos de freqüências para
caracterizá-las como pontos estacionários ou não, e para a obtenção das
propriedades termodinâmicas. As estruturas foram então utilizadas em cálculos de
ponto único para a obtenção das energias de solvatação pelo método IPCM. Em
alguns casos, foram construídas superfícies de energia potencial em uma ou duas
dimensões. No caso das superfícies 1D, variou-se o ângulo diedro C-N-C=Z em
incrementos de 10o, num total de 36 passos (B3LYP/6-31+G(d,p) ou B3LYP/
6-31G(d,p)). As superfícies 2D foram construídas com incrementos de 30o, cobrindo
um intervalo de 180o sobre os ângulos X-C-N-C de cada uma das metilas amídicas
variadas (B3LYP/6-31G(d)). Todos esses cálculos foram realizados com os
programas GAUSSIAN 98130 e GAUSSIAN 03131. Para manipulação de estruturas,
construção de matrizes e vizualização dos resultados, utilizamos os programas
gráficos MOLDEN132, MOLEKEL133 e GAUSSVIEW.
Os cálculos para análizes energéticas foram realizados através do módulo
NBO 5.0134 compilado no programa GAMESS135. Para a determinação das energias de
deslocalização, utilizamos a opção NOSTAR no grupo “DEL”. As energias de troca
foram obtidas por meio da opção STERIC no grupo “NBO”, enquanto que os pesos
dos híbridos de ressonância foram avaliados pela opção NRTSTR no mesmo grupo.
Neste último caso, é necessário especificar a natureza de cada uma das ligações da
estrutura, simples, duplas e pares isolados. Esses cálculos foram realizados no nível
HF/PTZV//HF/6-311+G(2d,p), i.e. cálculo de ponto único em HF/PTZV sobre a
estrutura otimizada em HF/6-311+G(2d,p).
Para o cômputo das energias de repulsão eletrostática, empregamos o
esquema de partição de energia de Mayer e Mamza136, 137 implementado no
programa APOST138. Neste esquema, a energia total molecular é repartida em
contribuições atômicas, podendo-se analisar a partir disso as energias de interação
de dois átomos que não estejam diretamente ligados. Essa energia de interação é
ainda particionada em componentes, um dos quais é o termo eletrostático que nos
interessa. O APOST toma como entrada um arquivo “.FChk” do GAUSSIAN, que é
obtido por meio da opção “FormCheck”. Esses cálculos foram realizados no nível

Parte Experimental 48
HF/PTZV//HF/6-311+G(2d,p). Para a análise do arquivo de saída do APOST,
escrevemos um programa simples em linguagem FORTRAN.
3.2.2 Dinâmica Molecular
Os cálculos com dinâmica molecular foram realizados utilizando o pacote de
programas TINKER139 (ANALIZE, XYZEDIT, MINRIGID, DYNAMICS, RADIAL,...). Dois solventes
foram empregados, sendo eles a água, representada pelo modelo de quatro pontos
TIP4P,12 e o metanol, representado por um modelo de três pontos do campo
OPLS.74 Como não havia parâmetros para os compostos em estudos, tivemos que
ajustar um campo de forças antes das simulações de líquidos. Para isso, calculamos
as energias de interação de uma série de complexos 1:1 soluto-solvente no nível
B3LYP/6-31+G(d,p). Essas energias de interação foram corrigidas para o “erro de
sobreposição de conjunto de funções de bases” (basis set superposition error,
BSSE),39 que nada mais é do que uma diminuição não-realística da energia do
complexo em relação às moléculas isoladas devida ao maior número de funções de
bases no primeiro. Para fazê-lo, calculamos a energia do soluto mantendo todas as
bases do complexo, porém sem os átomos de solvente, e fizemos o mesmo para as
moléculas de solvente, i.e. mantivemos as bases do complexo e retiramos os
átomos do soluto. No programa GAUSSIAN 98 esse procedimento pode ser realizado
através da opção “Massage”. De posse das energias de complexação, variamos
manualmente os parâmetros da Eq. (2.23), por meio do programa ANALIZE139, até
que a mesma fornecesse resultados próximos aos TFD.
Para as simulações, construímos caixas periódicas com 300 moléculas do
solvente, água ou metanol. Isso foi feito através do programa XYZEDIT139 e de uma
molécula do solvente em formato Cartesiano. A caixa de água tinha dimensões
20,80 × 20,80 × 20,80 Å3, sendo essas dimensões determinadas a partir da
densidade do líquido. A caixa de metanol tinha dimensões 27,275 × 27,275 ×
27,275 Å3. Submetemos as caixas a uma minimização inicial utilizando o programa
MINRIGID139, que efetua a otimização de um sistema de moléculas rígidas
(gradiente=1,0). Em seguida, a caixa foi equilibrada a 298 K no ensemble NVT por
20 ps (20.000 etapas) através do programa DYNAMICS139. Neste último caso,
empregamos um parâmetro de acoplamento de temperatura (τ) de 0,01 ps, e

Parte Experimental 49
0,001 ps para o tempo de cada etapa. Uma vez que a caixa encontrava-se
equilibrada, removemos quatro moléculas próximas ao seu centro por meio do
programa PCMODEL140 e, empregando novamente o XYZEDIT139, adicionamos a
molécula de interesse na lacuna das moléculas removidas. No caso da caixa de
metanol, utilizamos uma versão mais atualizado do código XYZEDIT139, que
corretamente remove as moléculas necessárias e adiciona o soluto sem a
necessidade de utilizar outro programa. Para este último caso, apenas uma
molécula de metanol foi removida. O sistema foi equilibrado por 30 ps nas mesmas
condições anteriores, após o que, τ foi aumentado para 0,3 ps e procedeu-se à
acumulação por mais 300 ps. As coordenadas foram salvas a cada 100 etapas.
A partir dos arquivos acumulados, construímos a função de distribuição radial
para cada par de átomos soluto-solvente por meio do programa RADIAL139. As fdrs
foram integradas com o auxílio do programa Origin.
3.3 Preparação dos Compostos • Solventes de Uso Geral
Para a preparação dos compostos foram utilizados solventes de qualidade técnica
ou P.A., cuja purificação foi realizada de acordo com procedimentos usuais.141 Os
solventes deuterados e o CCl4, empregados na aquisição dos espetros de RMN,
foram obtidos comercialmente e utilizados sem purificação adicional. O CS2,
empregado nos espectros de RMN, foi purificado por destilação em coluna de
Vigreux (p.e. 45 oC /~760 Torr) e armazenado com peneira molecular de 4 Å.
• Cloreto de N,N-dimetilcarbamoíla
Obtido comercialmente e destilado em coluna de Vigreux com pressão reduzida,
fornecendo uma fração média de p.e. 45 oC/4 Torr (lit.141 34 oC/1 Torr). O
destilado foi estocado sob atmosfera de N2 com peneira molecular de 4 Å.

Parte Experimental 50
• Cloreto de N,N-Dimetiltiocarbamoíla
Obtido comercialmente e seco à vácuo na presença de pentóxido de fósforo, sendo
utilizado sem purificação posterior. O produto foi estocado sob atmosfera de N2 em
um dissecador.
• N,N-Dimetilcarbamato de O-Metila (1) 36
Em um balão de fundo redondo de 125 mL com duas bocas, equipado com agitador
magnético e condensador de refluxo, e preenchido com nitrogênio gasoso,
adicionou-se 70 mL de tetraidrofurano (THF) seco, 2,9 mL (0,072 mol) de metanol,
previamente tratado com iodo e magnésio,141 e 1,7g (0,074 mol) de sódio metálico.
Manteve-se a mistura em refluxo por 30 min. Adicionou-se, com uma seringa e
durante 20 min, 6,4 mL de cloreto de N,N-dimetilcarbamoíla. Manteve-se a mistura
em refluxo e sob agitação por uma noite. Permitiu-se que a mistura atingisse a
temperatura ambiente e verteu-se a mesma sobre 100 mL de água destilada.
Transferiu-se a mistura para um funil de decantação, separou-se as fases e extraiu-
se o composto da fase aquosa com éter etílico (4 × 30mL). Juntou-se as fases
orgânicas e secou-se com sulfato de sódio anidro. Filtrou-se e removeu-se o
solvente em evaporador rotatório. O produto foi destilado em coluna de Vigreux,
obtendo-se 3,5 g (49 %) de um composto incolor de p.e. 128 oC/~760Torr (lit.107
130-132 oC/~760 Torr). RMN de 1H δ(300 MHz, CDCl3): 3,69 (3H, s); 2,91 (6H, s).
• N,N-Dimetiltiocarbamato de S-Metila (2) 107, 142
Em um balão de duas bocas de 250 mL, equipado com agitador magnético e
condensador de refluxo (em expiral), foram adicionados 29 g (0,402 mol) de
tiouréia, 15 mL (0,370 mol) de metanol e 60 mL (0,530 mol) de uma solução de HBr
48% m/m. Manteve-se a mistura em refluxo durante uma noite. Após este período,
conectou-se, com uma mangueira de silicone, o topo do condensador a um balão de

Parte Experimental 51
duas bocas de 125 mL – previamente flambado e preenchido com nitrogênio gasoso
– equipado com agitador magnético, contendo 60 mL de THF seco e 1,3 g (0,566
mol) de sódio metálico. Essa conexão foi feita por meio de um borbulhador cuja
extremidade foi ajustada de forma a fazer com que o gás que provinha do balão de
250 mL entrasse em contato com o THF a 1 cm do fundo do balão de 125 mL. A
segunda boca do balão continha um registro ligado à linha de nitrogênio gasoso.
Resfriou-se o balão de 125 mL a –40oC, com um banho de nitrogênio líquido e
etanol, e adicionou-se, com uma pipeta de Pasteur e em pequenas porções, 80 mL
(0,200 mol) de uma solução aquosa de NaOH 10% m/m ao balão de 250 mL. Neste
estágio, o fluxo de nitrogênio gasoso foi cessado e o registro conectado ao balão de
125 mL foi utilizado para regular a pressão no interior do balão, de forma a
permitir a passagem do gás pelo solvente. Ao término da adição de hidróxido de
sódio, retirou-se as conexões do balão de 125 mL, tampando-o com septos de
borracha. Aqueceu-se, lentamente, a 60 oC durante 40 min, com eventuais
inserções de uma agulha de metal para reduzir levemente a pressão interna. Após
este período, adaptou-se um condensador de refluxo (em expiral) e adicionou-se,
lentamente, 10 mL de cloreto de N,N-dimetilcarbamoíla (0,109 mol) ao balão de
125 mL. Aqueceu-se a 65oC com banho de glicerina e manteve-se em agitação por
uma noite. Permitiu-se, então, que a mistura atingisse a temperatura ambiente,
vertendo-a sobre 150 mL de uma solução gelada de bicarbonato de sódio a 3%.
Extraíu-se com éter etílico (5 vezes de 30mL), secou-se a fase orgânica com sulfato
de sódio anidro, filtrou-se e evaporou-se o solvente em evaporador rotatório,
obtendo-se 5,4 g (42 %) de um líquido levemente amarelado de p.e. 54 oC/>6 Torr
(lit.107 184 oC/~760 Torr). RMN de 1H δ(300 MHz, CDCl3): 2,96 (6H, s); 2,29 (3H, s).
• N,N-Dimetiltiocarbamato de O-Metila (3) 107
Em um balão de duas bocas de 125 mL, equipado com agitador magnético,
condensador de refluxo e preenchido com nitrogênio gasoso, adicionou-se 60 mL de
THF seco, 2,0 mL (0,049 mol) de metanol e 0,9 g (0,039 mol) de sódio metálico.
Permitiu-se que a mistura reagisse por 40 min. Resfriou-se o reator a 5 oC com

Parte Experimental 52
banho de gelo e adicionou-se 4,0 g (0,032 mol) de cloreto de N,N-
dimetiltiocarbamoíla. Retirou-se o banho de gelo e, lentamente, aqueceu-se a
mistura à 70oC, mantendo-se a mesma nesta temperatura por uma noite. Retirou-se
o aquecimento e permitiu-se que a mistura atingisse a temperatura ambiente,
vertendo-a em seguida sobre 150 mL de uma solução gelada de bicarbonato de
sódio a 3%. Extraiu-se o composto da fase aquosa com éter etílico (5 vezes de
30 mL), secou-se com sulfato de sódio anidro e removeu-se o solvente em
evaporador rotatório. Destilou-se o produto a vácuo obtendo-se 1,9 g (50 %) de um
composto incolor com p.e. 76 oC/>7 Torr (lit.107 87-92 oC/12 Torr). RMN de 1H δ(300
MHz, CDCl3): 4,02 (3H, s); 3,74 (3H, s); 3,12 (3H, s).
• N,N-Dimetilditiocarbamato de S-Metila (4)
1a Tentativa
Na primeira tentativa procedeu-se de forma análoga à preparação do composto 2,
substituindo-se o cloreto de N,N-dimetilcarbamoíla pelo cloreto de N,N-
dimetiltiocarbamoíla. No entanto, a quantidade de produto obtida foi
extremamente baixa e com muitas impurezas de difícil remoção.
2a Tentativa 107
Em um balão de três bocas de 100 mL, equipado com agitador magnético e
condensador de refluxo, adicionou-se 18 mL (0,260 mol) de solução aquosa de
N,N-dimetilamina a 40% m/m. Resfriou-se com banho de gelo e adicionou-se,
lentamente, 5,5 mL (0,088 mol) de dissulfeto de carbono. Retirou-se o banho,
permitindo-se que o reator atingisse a temperatura ambiente. Manteve-se a
mistura nessa condição por 20 min. Aqueceu-se, com banho de glicerina, até 45 oC
e manteve-se esta temperatura por mais 30 min com as bocas do balão abertas,
sem o condensador. Resfriou-se a mistura com banho de gelo e adicionou-se, de
forma lenta, 5,6 mL (0,088 mol) de iodeto de metila. Retirou-se o banho de gelo e
aqueceu-se, lentamente, à 50 oC por 5 h. Decorrido este período, retirou-se o

Parte Experimental 53
aquecimento e permitiu-se que a mistura atingisse a temperatura ambiente. Os
cristais formados foram removidos da fase aquosa e dissolvidos, sob aquecimento,
em uma mistura de etanol e água (1:1; 300 mL). Elevou-se a temperatura até que o
volume fosse reduzido pela metade. Imergiu-se a mistura em banho de gelo e
filtrou-se os cristais formados em funil de Büchner. Essa última etapa foi repetida
mais uma vez. Em seguida, dissolveu-se os cristais em éter etílico e secou-se com
sulfato de magnésio anidro. Filtrou-se e removeu-se o solvente em evaporador
rotatório, obtendo-se 2,0 g (17 %) de cristais incolores com p.f. 43-44 oC (lit.107 45-
47 oC). RMN de 1H δ(300 MHz, CDCl3): 3,57 (3H, s); 3,38 (3H, s); 2,65 (3H, s).
• N,N,N’-Trimetiluréia (5) 115
Em um balão de 125 mL de duas bocas, previamente flambado e equipado com
agitador magnético, adicionou-se 0,9 g de cloreto de N,N-dimetilcarbamoila
(0,0084 mol), 0,5 g (0,0074 mol) de cloreto de metilamônio e 60 mL de piridina
seca. Adaptou-se um condensador ao balão e manteve-se a mistura em agitação a
70 oC por 2 dias. Passado este período, retirou-se o aquecimento e aguardou-se até
que a mistura atingisse a temperatura ambiente. Verteu-se a mistura sobre 200 mL
de água destilada gelada. Adicionou-se cloreto de sódio até a saturação e extraiu-
se o produto com éter etílico (5 x de 30 mL). Secou-se a fase orgânica com sulfato
de magnésio anidro, filtrou-se e removeu-se o solvente em evaporador rotatório.
Adicionou-se diclorometano, secou-se, filtrou-se e removeu-se o solvente em
evaporador rotatório. Esta última etapa foi repetida até que toda a piridina tivesse
sido removida. Obteve-se 0,6 g (70 %) de um sólido branco com p.f. 75-76oC. RMN
de 1H δ(300 MHz, CDCl3): 4,47 (1H, bl); 2,90 (6H, s); 2,81/2,80(3H, d).
• Isotiocianato de Metila 143
Em um balão de 100 mL de três bocas, equipado com agitador magnético,
condensador de refluxo e funil de adição, adicionou-se 3 mL (0,050 mol) de
dissulfeto de carbono e 3,6 g (0,053mol) de cloreto de metilamônio dissolvidos em

Parte Experimental 54
6 mL de água. Resfriou-se a mistura a 10–15 oC com banho de gelo e adicionou-se,
com agitação e por um período de 20 min, 4,2 g (0,105 mol) de hidróxido de sódio
dissolvidos em 10 mL de água. Manteve-se a agitação e aqueceu-se a mistura com
banho de glicerina por duas horas (neste período, a temperatura gradualmente foi
elevada da ambiente para 80 oC). Retirou-se o aquecimento e esperou-se que a
mistura atingisse 38 oC. Adicionou-se, lentamente (30 min), 5 mL (0,088 mol) de
clorocarbonato de etila. Manteve-se a agitação por mais uma hora. A mistura foi
transferida para um funil de separação, recolhendo-se a fase que se formou na
parte inferior. Secou-se com sulfato de sódio anidro e destilou-se com coluna
simples obtendo-se 1,7 g (47 %) de um líquido incolor com p.e. 110 oC/~760 Torr
(lit.144 119 oC/760 Torr). Espectro de massas: m/e = 73.
• N,N,N’-Trimetiltiouréia (6) 145
1a Tentativa
Em um balão de 125 mL, previamente flambado, equipado com agitador magnético
e preenchido com nitrogênio gasoso, adicionou-se 70 mL de tetraidrofurano seco,
3,5 g (0,0283 mol) de cloreto de N,N-dimetiltiocarbamoíla e 2 mL de piridina
(0,0248 mol) . Em um segundo balão, de 250 mL, munido de um condensador de
refluxo em expiral e agitador magnético, adicionou-se 40 g (0,592 mol) de cloreto
de metilamônio. Adaptou-se ao topo do condensador uma mangueira de silicone
munida de um borbulhador em uma de suas extremidades. Este borbulhador foi
ajustado ao balão de 125 mL de modo a fazer com que o gás produzido entrasse em
contato com o líquido 1 cm acima do fundo. Resfriou-se o balão de 125 mL com
banho de gelo e adicionou-se lentamente, com pipeta de Pasteur, uma solução de
24 g (0,600 mol) de NaOH em 60 mL de água ao balão de 250 mL. Desligou-se o
fluxo de nitrogênio do balão de 125 mL, permitindo-se o fluxo da metilamina
gasosa produzida. Manteve-se a mistura em agitação e em refluxo por uma noite.
Passado este período, retirou-se o banho de aquecimento e aguardou-se até que o
sistema atingisse a temperatura ambiente. Transferiu-se a mistura para um funil de
separação, lavando-se a mesma com água destilada (2 x de 30 mL). Secou-se a fase

Parte Experimental 55
orgânica com sulfato de sódio anidro e removeu-se o solvente em evaporador
rotatório. Obteve-se um sólido de coloração escura contendo diversos subprodutos
(RMN), não sendo possível o isolamento do composto de interesse.
2a Tentativa
Em um balão de 125 mL de duas bocas, previamente flambado e equipado com
agitador magnético, adicionou-se 2 g (0,0162 mol) de cloreto de N,N-
dimetiltiocarbamoila, 2 g (0,0300) de cloreto de metilamônio e 60 mL de piridina
seca. Adaptou-se um condensador ao balão e manteve-se a mistura em agitação a
70 oC por 4 dias. Passado este período, retirou-se o aquecimento e aguardou-se até
que a mistura atingisse a temperatura ambiente, vertendo-se a mesma sobre
200 mL de água destilada gelada. Adicionou-se cloreto de sódio até a saturação e
extraiu-se o produto com éter etílico (4 x de 30 mL). Secou-se a fase orgânica com
sulfato de magnésio anidro, filtrou-se e removeu-se o solvente em evaporador
rotatório. Obteve-se um sólido laranja escuro contendo diversos subprodutos
(RMN), não sendo possível o isolamento do composto de interesse.
3a Tentativa
Em um balão de duas bocas de 100 mL, equipado com agitador magnético, e
condensador de refluxo, foram colocados 5,3 mL (2,1 g; 0,046 mol) de uma solução
aquosa de N,N-dimetilamina (44% m/m). Adicionou-se, com uma seringa e de forma
lenta, 1,7 g (0,023 mol) de isotiocianato de metila. Após a adição, manteve-se em
agitação por 20 min. Passado este período, aqueceu-se a mistura em banho de óleo
por 30 min para remover o excesso de dimetilamina. A solução foi aquecida com
carvão ativo, filtrada e imergida em uma bacia com gelo. O sobrenadante foi
separado dos cristais formados com uma pipeta de Pasteur. Os cristais foram
primeiramente secos em dissecador (uma noite) e depois secos à vácuo na presença
de pentóxido de fósforo. Obteve-se 0,65 g (24 %) de cristais brancos com p.f. 87-
88 oC (lit.146 87 oC). RMN de 1H δ(300 MHz, CDCl3): 3,27 (6H, s); 3,16/3,15 (3H, d)

Resultados e Discussão 56
Capítulo 4:
Resultados e Discussão

Resultados e Discussão 57
Capítulo 4: Resultados e Discussão
4.1 Obtenção dos Compostos e RMND
4.1.1 Síntese dos Compostos
Dentre os oito compostos apresentados na Introdução, 7 e 8 foram adquiridos
comercialmente e os seis primeiros foram sintetizados de acordo com os
procedimentos descritos no Cap. 3. A maioria das reações é conhecida e de fácil
execução, salvo aquelas em que se faz necessária a produção de reagentes gasosos.
O composto 1 foi preparado pela adição do metóxido à carbonila do cloreto
de N,N-dimetilcarbamoíla:
O produto é facilmente purificado por destilação em coluna de Vigreux e é
relativamente estável. Uma reação similar foi utilizada para obter o composto 3,
mas neste caso o reagente foi o cloreto de N,N-dimetiltiocarbamoíla. Ao contrário
de 1, o composto 3, após isolado, é extremamente sensível à umidade, de forma
que observávamos a evolução de uma fumaça branca quando o mantínhamos
exposto ao ar por apenas alguns segundos. Este comportamento foi limitante no
caso dos experimentos com RMND, como veremos a seguir, porque em razão disto
não foi possível adquirir espectros em solventes próticos para 3.
A idéia para a preparação de 2 segue a mesma reação de 1, mas o nucleófilo
é o tiometóxido. Este último é obtido a partir da tiouréia, tendo como
intermediário um sal de isotiurônio:
CH3O +C
O
NCl
CH3
CH3
C
O
NO
CH3
CH3
H3C
(1)

Resultados e Discussão 58
A preparação do tiometóxido representa um acréscimo significativo, pois,
após a adição de hidróxido, o metanotiol se desprende do reator na forma de um
gás e precisa ser transportado para um segundo balão contendo sódio metálico em
THF. O borbulhamento de metanotiol no THF ocorre pela diferença de pressão
entre o balão de produção e o que contém o THF. Essa diferença é determinada
pela temperatura do banho do balão com THF e pela taxa de adição do hidróxido,
mas o primeiro fator é mais significativo. No entanto, o banho que utilizamos para
manter o balão resfriado, etanol/nitrogênio líquido, é bastante sensível, de modo
que se não for controlado adequadamente, há o risco da solução contendo sódio
metálico ser projetada para o balão onde é produzido o metanotiol, causando uma
explosão. Por isso, é recomendável que não se utilize uma mangueira muito curta e
que se mantenha o balão com THF no nível mais baixo possível em relação ao balão
do metanotiol.
Tentamos preparar o composto 4 da mesma forma que 2, substituindo apenas
o cloreto de N,N-dimetilcarbamoíla pelo cloreto de N,N-dimetiltiocarbamoíla.
Contudo, os rendimentos foram extremamente baixos e a quantidade de impurezas
que se agregava ao composto era de difícil separação. Sendo assim, optamos por
uma reação alternativa:
C
H2N
S
NH2
+ HBr + CH3OHH3C
S
C
NH2
NH2 Br
CH3SH
CH3SH + Cl
C
O
N
CH3
CH3
S
C
O
N
CH3
CH3
H3CNao
1)
2)
CS2 + NH(CH3)2H
S
C
S
N
CH3
CH3
CH3I H3C
S
C
N
CH3
CH3
S
(2)
(4)
NaOH i)
ii)

Resultados e Discussão 59
Essa reação foi efetuada sem maiores problemas e forneceu o composto com
pureza adequada e em quantidade suficiente para a realização dos experimentos.
O composto 5 foi preparado de forma similar a 1 e 3, mas utilizando CH3NH2
como nucleófilo. Todavia, não foi possível aplicar esse procedimento para 6, uma
vez que os rendimento foram extremamente baixos e o composto encontrava-se
misturado com impuzeras de difícil remoção. Seguimos então um procedimento
para a preparação de tiouréias a partir de isotiocianatos. No nosso caso, foi
necessário preparar o isotiocianato de metila:
De posse do isotiocianato de metila, a N,N,N’-trimetiltiouréia (6) pôde ser
obtida pela reação com N,N-dimetilamina:
Na Tabela 4.1 estão apresentados os rendimentos e as propriedades físicas
dos compostos que sintetizamos, além de 7 e 8.
CH3NH2 + CS2 + NaOH + H2O
+
C
S
S
N
H3C
H
Na
C
S
S
N
H3C
H
Na C
O
Cl
O
C2H5
N
C
S
S
H3C C
O
O
C2H5
H
N
C
S
S
H3C C
O
O
C2H5
H
CH3-N=C=S + COS + C2H2OH
1)
2)
3)
CH3-N=C=S + (CH3)2NH
S
CN N
H3C
H
CH3
CH3(6)

Resultados e Discussão 60
Tabela 4.1. Propriedades físicas e rendimentos dos compostos estudados
Estado na T Amb. p.e./P (oC/Torr) p.f.(oC) Rendimento (%)
1 l 128 /760 - 49
2 l 54/6 - 45
3 l 76/7 - 50
4 s - 43-44 17
5 s - 75-76 70
6 s - 87-88 24
7 l 62-63/12 - - a
8 s - 75-77 - a
a De origem comercial (Aldrich).
4.1.2 Determinação das Barreiras Rotacionais por RMND
Há duas formas pelas quais se pode obter o valor das barreiras rotacionais por
RMND, por meio da temperatura de coalescência (Tc) e a separação limite dos
sinais (Dn), e por meio de análise do perfil das bandas (APB). O método da
temperatura de coalescência trabalha na região de temperaturas na qual o erro na
determinação da constante de velocidade deve ser mínimo, i.e. próximo à
coalescência.78 Contudo, este método requer a determinação da separação entre
os sinais na ausência de troca, algo muitas vezes limitado pela temperatura de
congelamento do solvente. No método APB, Tc e Dn não precisam ser conhecidas,
mas é necessária a obtenção do maior número possível de espectros em
temperaturas variadas, dando-se preferência para as temperaturas próximas à
coalescência.
Na Fig. 4.1, apresentamos uma seqüência de espectros simulados e
experimentais utilizados na análise do perfil das bandas. As Tabelas 4.2 e 4.3
apresentam os parâmetros de ativação determinados a partir dos gráficos de Eyring
(Fig. 4.2), como explicado no Cap. 2.

Resultados e Discussão 61
Figura 4.1. Experimentos de RMND de 1H para o composto 1 em CD3OD: à esquerda, espectros experimentais em diversas temperaturas e, à direita, os respectivos espectros simulados e suas constantes de velocidade (k).
Figura 4.2. Gráfico de Eyring para o composto 1 obtido em CD3OD.
3,50 3,55 3,60 3,65 3,70 3,75-5,0
-4,5
-4,0
-3,5
-3,0
-2,5Y = 22,1635 - 7159,5807*XCoeficiente de Correlação = -0,997
ln(k
/Tem
p)
1/Temp (103/K)

Resultados e Discussão 62
Tabela 4.2. Barreiras rotacionais para os compostos 1-4 determinadas por RMND de 1H (∆H‡ e ∆G‡ em kcal/mol e ∆S‡ em cal/K@mol)a
1 2
∆H‡ ∆S‡ ∆G‡ ∆H‡ ∆S‡ ∆G‡
CCl4 – – – – – –
Bz-d6 – – – – – –
CS2 10,8±0,3 -15,0±1,2 15,3±0,5 12,5±0,7 -5,4±2,8 14,0±1,1
CD2Cl2 10,4±0,6 -16,6±2,1 15,3±0,9 14,7±1,0 1,7±4,1 14,2±1,6
CD3OD 14,2±0,6 -3,1±2,1 15,2±0,9 12,6±1,1 -6,7±4,2 14,6±1,6
CD3CN 12,9±1,4 -7,0±5,4 15,0±2,1 13,5±0,7 -2,8±2,7 14,3±1,1
20% D2O/CD3OD 10,1±0,8 -17,1±2,9 15,2±1,2 9,7±0,3 -17,7±1,2 14,9±0,5
DMSO-d6 – – – – – –
D2O 11,6±1,0 -10,6±3,4 14,8±1,4 11,6±0,5 -10,8±1,7 14,8±0,7
3 4
∆H‡ ∆S‡ ∆G‡ ∆H‡ ∆S‡ ∆G‡
CCl4 17,4±0,3 -0,2±0,9 17,5±0,4 13,8±0,4 -2,7±1,2 14,6±0,5
Bz-d6 19,9±0,8 8,0±2,5 17,6±1,1 15,1±0,2 0,3±0,5 15,0±0,2
CS2 – – – – – –
CD2Cl2 – – – – – –
CD3OD – – – 14,2±0,1 -3,1±0,4 15,2±0,2
CD3CN 17,4±0,3 -0,2±0,9 17,5±0,4 15,7±0,1 0,4±0,3 15,6±0,1
20% D2O/CD3OD – – – 14,8±0,2 -2,5±0,6 15,5±0,3
DMSO-d6 18,7±0,3 2,7±0,7 17,9±0,3 16,0±0,2 0,0±0,5 16,0±0,2
D2O – – – 12,7±0,2 -10,8±0,6 15,9±0,3
a Erros estimados com um intervalo de confiança de 95 % para a distribuição de Student.
Os resultados das Tabelas 4.2 e 4.3 trazem erros experimentais que vão de
0,1 kcal/mol até 2,1 kcal/mol para ∆G‡, e em certos casos a magnitude desses
erros compromete a avaliação de variações representativas. Porém, para os
solventes em que se pôde obter dados adequados de Tc e Dn, espera-se erros de
aproximadamente 0,2 kcal/mol†, o que pode ser de grande valia em combinação
com os dados das Tabelas 4.2 e 4.3. A Fig. 4.3 descreve a variação das freqüências
† Este é na verdade um valor extremo, obtido a partir da Eq. (2.32) assumindo-se um erro de 3 K na determinação da temperatura de coalescência para um sistema parecido aos estudados, Dn = 15 Hz e Tc = 300 K.

Resultados e Discussão 63
de absorção do composto 1 com a temperatura em CD3OD. Neste caso, foi possível
obter convergência para Dn, de forma que a partir de -20 oC a separação dos sinais
permanece aproximadamente constante. Todavia, essa condição não foi possível
para todos os solventes, muitas vezes por limitações impostas pelo ponto de
congelamento dos mesmos. Para esses casos, a separação entre os sinais das
metilas pode vir a ser menor do que a separação limite, causando um aumento
irreal na barreira rotacional assim determinada. Nas Tabelas 4.4 e 4.5
apresentamos as barreiras rotacionais calculadas pelo método da temperatura de
coalescência.
Tabela 4.3. Barreiras Rotacionais para os Compostos 5-6 determinadas por RMND de 1H (∆H‡ e ∆G‡ em kcal/mol e ∆S‡ em cal/K@mol)a
5 6
∆H‡ ∆S‡ ∆G‡ ∆H‡ ∆S‡ ∆G‡
CCl4 – – – – – –
Bz-d6 – – – – – –
CS2 – – – – – –
CD2Cl2 – – – 10,3±0,1 1,5±0,7 9,8±0,3
CD3OD 6,5±0,3 -16,0±1,5 11,3±0,6 8,4±0,2 -6,7±0,8 10,5±0,3
CD3CN – – – – – –
10% D2O/CD3OD 5,0±0,1 -25,0±0,6 12,4±0,2 10,1±0,2 -1,3±0,8 10,5±0,3
DMSO-d6 – – – – – –
D2O – – – – – –
a Erros estimados com um intervalo de confiança de 95 % para a distribuição de Student.
Passemos agora à análise das magnitudes relativas das barreiras e de sua
resposta ao solvente. Tanto os dados de Tc como de APB mostram que o composto 1
não experimenta variação significativa com o solvente. Como dissemos na
Introdução, esse comportamento já era conhecido para os carbamatos, mas dados
para o N,N-dimetilcarbamato de metila (1) ainda não haviam sido obtidos. Para o
composto 2, os dados de APB mostram um aumento sistemático com a polaridade
do solvente e podería-se atribuir isso a um efeito real do solvente não fosse pelos
erros experimentais. Os dados de Tc para este composto nos fornecem informações
valiosas, salvo apenas o fato de que para a água não foi possível atingir uma boa
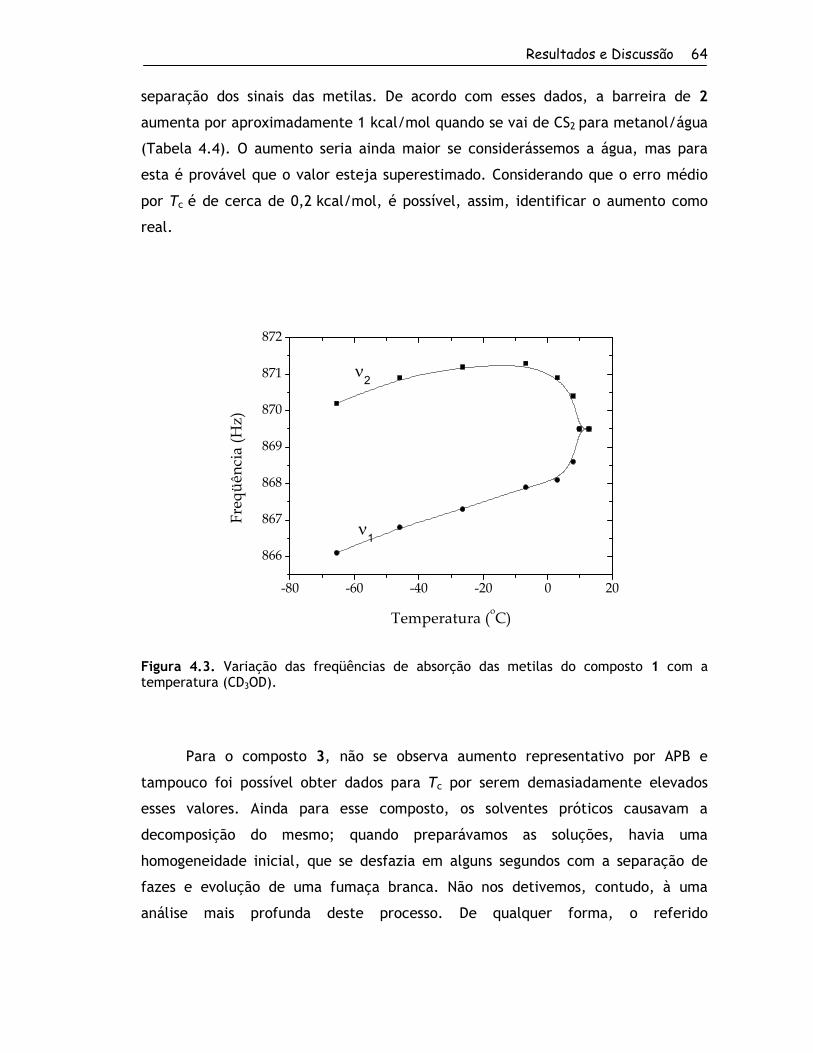
Resultados e Discussão 64
separação dos sinais das metilas. De acordo com esses dados, a barreira de 2
aumenta por aproximadamente 1 kcal/mol quando se vai de CS2 para metanol/água
(Tabela 4.4). O aumento seria ainda maior se considerássemos a água, mas para
esta é provável que o valor esteja superestimado. Considerando que o erro médio
por Tc é de cerca de 0,2 kcal/mol, é possível, assim, identificar o aumento como
real.
Figura 4.3. Variação das freqüências de absorção das metilas do composto 1 com a temperatura (CD3OD).
Para o composto 3, não se observa aumento representativo por APB e
tampouco foi possível obter dados para Tc por serem demasiadamente elevados
esses valores. Ainda para esse composto, os solventes próticos causavam a
decomposição do mesmo; quando preparávamos as soluções, havia uma
homogeneidade inicial, que se desfazia em alguns segundos com a separação de
fazes e evolução de uma fumaça branca. Não nos detivemos, contudo, à uma
análise mais profunda deste processo. De qualquer forma, o referido
-80 -60 -40 -20 0 20
866
867
868
869
870
871
872
ν2
ν1
Freq
üênc
ia (H
z)
Temperatura (oC)

Resultados e Discussão 65
comportamento nos dá evidências de um efeito drástico das ligações de hidrogênio
sobre a estrutura do composto 3.
Tabela 4.4. Barreiras rotacionais para os compostos 1-4 determinadas por RMND de 1H pelo método da temperatura de coalescência (Tc em oC, ∆ν em Hz e ∆G‡ em kcal/mol)
1 2 3 4
Tc ∆ν ∆G‡ Tc ∆ν ∆G‡ Tc ∆ν ∆G‡ Tc ∆ν ∆G‡
CCl4 − − − − − − − − − 31,5 35,10 15,2a
Bz-d6 − − − − − − − − − 54,7 149,0 15,5
CS2 21,6 10,9 15,4 -5,8 6,6 14,2 − − − − − −
CD2Cl2 5,9 3,6 15,2 -14,6 2,1 14,3 − − − − − −
CD3OD 11,6 4,1 15,4 -1,0 4,0 14,7 − − − 43,5 42,4 15,7
CD3CN -1,0 1,8 15,1 -7,8 2,3 14,6 − − − 43,6 43,5 15,7
D2O/Metb 11,8 6,0 15,2a 13,7 8,2 15,1 − − − 49,1 40,5 16,0
DMSO-d6 − − − − − − − − − 54,7 34,1 16,5
D2O 10,8 3,0 15,5a 17,7 9,3 15,3a − − − 61,1 38,3 16,7
a Não foi possível atingir uma boa separação para os sinais das metilas. b 20 % D2O/CD3OD.
Tabela 4.5. Barreiras rotacionais para os compostos 5 e 6 determinadas por RMND de 1H pelo método da temperatura de coalescência (Tc em oC, ∆ν em Hz e ∆G‡ em kcal/mol)
5 6
Tc ∆ν ∆G‡ Tc ∆ν ∆G‡
CCl4 − − − − − −
Bz-d6 − − − − − −
CS2 − − − − − −
CD2Cl2 − − − -53,9 102,6 10,3
CD3OD -75,3 18,3 10,0 -56,8 99,9 10,2
CD3CN − − − − − −
10% D2O/CD3OD -66,5 13,8 10,6 -45,2 90,2 10,2
DMSO-d6 − − − − − −
D2O − − − − − −
Tanto Tc como APB mostram que a barreira rotacional de 4 é afetada de
forma representativa pelo solvente, porém, os comportamentos são levemente

Resultados e Discussão 66
diferentes em cada método. Note que os erros experimentais para 4 segundo APB
são os menores entre os dados da Tabela 4.2. Segundo os dados desta Tabela, há
um aumento da barreira quando se vai de CCl4 para DMSO. Este aumento não
persiste ao se passar para a água, mas seria esperado, tendo em vista que a
barreira rotacional neste composto mostra-se sensível à polaridade. O fato de não
se observar um aumento posterior na água sugere que as ligações de hidrogênio
possuem um efeito negativo sobre a barreira rotacional de 4. Este comportamento
é menos óbvio nos dados de Tc, nos quais observa-se um aumento de 0,2 kcal/mol
passando do DMSO para a água. No entanto, 0,2 kcal/mol é o erro estimado para
este experimento, ou seja, não se pode tratar esta variação como significativa.
Como veremos mais adiante, os cálculos teóricos suportam um efeito negativo por
parte das ligações de hidrogênio.
Devido à baixíssima temperatura de coalescência para as uréias e tiouréias
(5-8), não foi possível fazer um estudo extensivo acerca do efeito do solvente para
estes compostos; obtivemos dados para um número modesto de solventes. Para a
N,N,N’,N’-tetrametiluréia (7) e a N,N,N’,N’-tetrametiltiouréia (8), não observamos
a descoalescência dos sinais em nenhuma das temperaturas experimentais, de
forma que não pudemos determinar as barreiras rotacionais destes compostos por
RMND. No caso dos compostos 5 e 6, os dados de APB são de longe mais confiáveis
do que os de Tc, pois a separação dos sinais das metilas era relativamente pobre.
Com os dados de APB, foi possível observar uma variação representativa nos
solventes para os quais os espectros eram adequados para análise. Tanto 5 como 6
têm suas barreiras aumentadas pelo solvente, mas aqui não é possível fazer uma
clara distinção do efeito da polaridade do solvente e das ligações de hidrogênio,
tendo em vista o limitado número de solventes passíveis de medidas de barreiras
(Tabela 4.3).
A barreira da N,N,N’-trimetiluréia (5) aumenta por ~1 kcal/mol ao passar do
metanol para metanol/água, o que constrasta com a N,N,N’-trimetiltiouréia (6)
para a qual a barreira permanece insensível à mesma mudança. Há, por outro lado,
uma pequena diferença entre a barreira em diclorometano e em metanol para 6. A
interpretação destes resultados não pode ser conduzida apenas com os dados que
apresentamos até agora, precisaremos, outrossim, do auxílio dos cálculos teóricos
que passaremos a descrever.

Resultados e Discussão 67
4.2 Estudo Teórico
4.2.1 Estruturas Estáveis em Fase Gasosa
Como dissemos anteriormente, o propósito desta parte do estudo é o entendimento
das observações experimentais, uma vez que os métodos teóricos podem nos
fornecer informações detalhadas a respeito dos processos ocorrendo em fase
condensada e sobre a origem das barreiras rotacionais. Para assim o fazermos,
precisamos antes conhecer as barreiras rotacionais dos compostos em fase gasosa,
isoladas de qualquer interação com o solvente. As estruturas obtidas nesta etapa
serão também utilizadas nos estudos com o método NBO, que permite a
investigação de efeitos de deslocalização eletrônica. O primeiro passo nesse
sentido é o estabelecimento das conformações dos compostos, que a seguir
descrevemos. Afim de melhor sistematizar este estudo, dividimos os compostos em
dois grupos: os congêneros de carbamatos (Grupo 1) e os congêneros de uréias
(Grupo 2).
4.2.1.1 Conformações para o Grupo 1
Os estudos de Goodman et al.147-149 sugerem que em grupos metila vizinhos a um
oxigênio sp3 a conformação adotada é aquela apresentada na Fig. 4.4, na qual um
dos ângulos H-C-O-C é 180 o.
Figura 4.4. Conformação do éter dimetílico.
OH H
H HH H

Resultados e Discussão 68
Uma conformação semelhante foi obtida para todos os compostos do Grupo 1
e não realizamos nenhuma avaliação posterior para essa porção das moléculas. O
fragmento amídico é um caso mais complicado. Sabe-se que em grupos metila
vizinhos a insaturações, a conformação mais estável é aquela em que um dos
hidrogênios permanece eclipsado com a ligação dupla (Fig. 4.5).150 Uma das
estruturas de ressonância do grupo amida (Fig. 1.2) possui uma ligação dupla C=N.
Por analogia com os sistemas vinílicos, esperaríamos uma conformação para as
metilas como a da Fig. 4.5. Contudo, não temos aqui uma ligação dupla formal,
mas sim uma ligação com caráter de dupla, o que faz com que o efeito observado
nos sistemas vinílicos possua, nas amidas, uma intensidade menor.
Figura 4.5. Conformação de grupos metila em alcenos e amidas.
Os relatos de geometrias moleculares de amidas encontrados na literatura
apresentam estruturas como a da Fig. 4.5.19, 33 Esse foi também o nosso ponto de
partida para as otimizações de geometria das moléculas que estudamos, mas no
nosso caso obtínhamos freqüências imaginárias. Sendo assim, foi necessário realizar
um estudo sobre a conformação das metilas amídicas. Com esse fim, consideramos
as possibilidades apresentadas na Fig. 4.6. Cada uma dessas estruturas foi
submetida a uma otimização seguida por cálculos de freqüências com os métodos
HF e B3LYP. Os resultados estão apresentados na Tabela 4.6.
Os compostos 1 e 2 seguiram o comportamento das amidas, ou seja, a única
conformação estável que obtivemos foi a ef1, com os dois níveis de teoria. Por
outro lado, obtivemos duas conformações estáveis para os compostos 3 e 4, e
nesses casos os resultados dos métodos HF e B3LYP são diferentes.
H
HH
HH
H
N
O
H
HH
HH
H

Resultados e Discussão 69
Figura 4.6. Possíveis confôrmeros para os compostos 1-4.
Tabela 4.6. Resultados da pesquisa conformacional para os compostos 1-4 a,b,c,d
ef1 ef2 ef3 ef4
B3LYP -363,175947 (2i) -363,175144 (1i) -363,174575 (1i) -363,173821 (2i) (1)
HF -361,011323 (2i) -361,010695 (1i) -361,010219 (1i) -361,009507 (2i)
B3LYP -686,146239 (2i) → ef1 → ef1 -686,146239 (2i) (2)
HF -683,676750 (2i) → ef1 → ef1 -683,652169 (2i)
B3LYP -686,123893 (2i) -686,124065 (1i) -686,123935 (1i) -686,123838 (1i) (3)
HF -683,633783 (2i) -683,634383 (1i) -683,634350 (1i) -683,634290 (1i)
B3LYP → ef4 -1009,096894 (1i) → ef4 -1009,097170 (1i)
(4) HF -1006,279158 (2i) → ef4 -1006,280370 (1i) -1006,280687 (1i)
a 6-311+G(2d,p). b Energias em hartrees. c As freqüências imaginarias estão representadas entre parênteses. d “→”: converge para.
No Caso da conformação ef3 de 3, o hidrogênio da metila anti eclipsado com
a ligação (C=O)-N está a 2,197 Å do oxigênio (-O-) no método HF, sendo esta
distância um pouco menor com B3LYP, 2,188 Å. Some-se a isso o fato de que a
X
Z
NH
HH
H
HH
H
H
H
X
Z
NH
HH
H
H
H
HH
H
X
Z
NH
HH
HH
H
H
HH
X
Z
NH
HH
HH
H
H
HH
ef1 ef2
ef3 ef4

Resultados e Discussão 70
densidade eletrônica em B3LYP tende a concentrar-se nas periferias das moléculas
mais do que em HF,151 e teríamos uma repulsão estérica maior para o método
B3LYP que poderia explicar a instabilidade de ef3 neste método. Já a instabilidade
do ef2 de 4 em HF pode ser entendida com base na tendência do método HF em
fornecer comprimentos de ligações mais curtos do que os métodos de correlação
eletrônica.151 A distância entre o carbono da metila syn e o enxofre da dupla é
2,973 Å em HF, contra 2,994 Å em B3LYP. Portanto, numa conformação como ef2 o
hidrogênio da metila syn eclipsado com a ligação (C=S)-N sofre maior repulsão
estérica em HF, o que faz como que a metila rotacione e assuma a configuração de
ef4.
No caso dos estados de transição, utilizamos as estruturas da Fig. 4.7. Não há
relatos de estruturas diferentes dessas para os estados de transição, e a simetria
dos mesmos nos leva à conclusão de que ambas as metilas amídicas devem adotar a
mesma conformação. A concordância com os dados experimentais justifica nossa
escolha.
et1 et2
Figura 4.7. Estados de transição para o cálculo das barreiras rotacionais de 1-4.
4.2.1.2 Conformações para o Grupo 2
Tratemos primeiramente da N,N,N’-trimetiluréia (5) e da N,N,N’-trimetiltiouréia
(6). As possibilidades conformacionais para estes compostos são mais complexas do
que nos casos anteriores. Assumindo que cada metila possa ser arranjada de duas
Z
NXH
HH
H
H
H
H
H
H
Z
NXH
HH
H
H
H
H
H
H
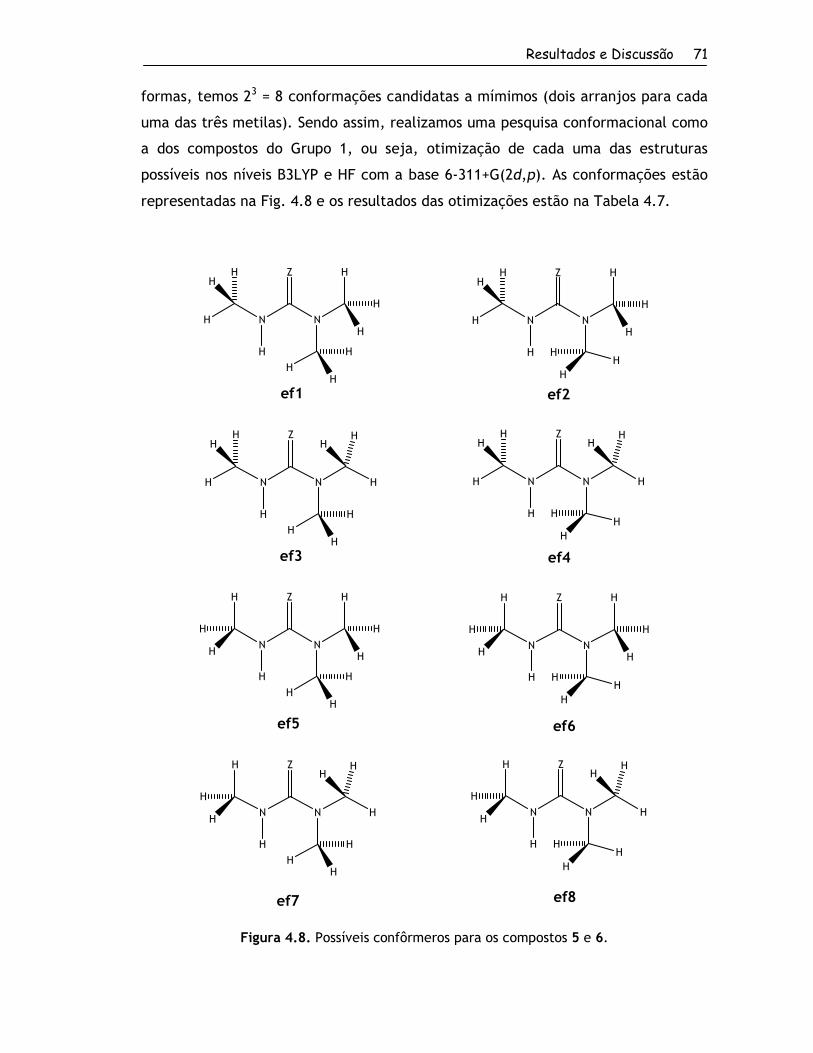
Resultados e Discussão 71
formas, temos 23 = 8 conformações candidatas a mímimos (dois arranjos para cada
uma das três metilas). Sendo assim, realizamos uma pesquisa conformacional como
a dos compostos do Grupo 1, ou seja, otimização de cada uma das estruturas
possíveis nos níveis B3LYP e HF com a base 6-311+G(2d,p). As conformações estão
representadas na Fig. 4.8 e os resultados das otimizações estão na Tabela 4.7.
Figura 4.8. Possíveis confôrmeros para os compostos 5 e 6.
N
H
Z
NH
HH
H
HH
H
H
H
N
H
Z
NH
HH
H
H
H
HH
H
N
H
Z
NH
HH
HH
H
H
HH
N
H
Z
NH
HH
HH
H
H
HH
N
H
Z
NH
HH
H
H
HH
H
H
N
H
Z
NH
H
H
HH
H
H
H
H
N
H
Z
N
HH
H
H
HH
H
H
H
N
H
Z
N
HH
H
H
HHH
H
H
ef1 ef2
ef3 ef4
ef5 ef6
ef7 ef8

Resultados e Discussão 72
A primeira estrutura estável que obtivemos foi o ef2. Afim de economizar
tempo, decidimos somente realizar cálculos de freqüências para as outras
estruturas caso fossem diferentes do ef2. Como pode ser observado na Tabela 4.7,
a única estrutura estável para o ef, tanto para 5 como para 6, é de fato o ef2.
Tabela 4.7. Resultados da pesquisa conformacional para a 5 e 6 a,b,c
(5) (6)
B3LYP HF B3LYP HF
ef1 → ef2 → ef2 → ef2 → ef2
ef2 -343,300404 -341,170901 -666,253401 -663,799793
ef3 → ef2 → ef2 → ef2 → ef2
ef4 → ef2 → ef2 → ef2 → ef2
ef5 → ef2 → ef2 → ef2 → ef2
ef6 → ef2 → ef2 → ef2 → ef2
ef7 → ef2 → ef2 → ef2 → ef2
ef8 → ef2 → ef2 → ef2 → ef2
a 6-311+G(2d,p). b Energias em hartrees. c “→”: converge para.
Ainda visando estabelecer corretamente as conformações, adotamos um
procedimento alternativo. Construímos uma superfície de energia potencial (SEP)
variando as posições das duas metilas ligadas ao mesmo nitrogênio simultanea-
mente. Esses cálculos foram realizados no nível B3LYP/6-31G* com incremento de
30 o para um intervalo de 180 o. Empregamos este último método como uma forma
de validação do procedimento de variar a posição da metilas, do modo que fizemos
para o Grupo 1, uma vez que para os próximos compostos o número de
conformações geradas pelo primeiro procedimento é consideravelmente maior e a
utilização de SEP mostra-se mais econômica. Os gráficos para a 5 e 6 estão
apresentados nas Figs. 4.9 e 4.10, respectivamente.
A SEP para a 5 apresenta três mínimos distintos (Fig. 4.9a 4.9b). Cada um
deles (Fig. 4.9c) assemelha-se ao ef2, como obtido pelo primeiro procedimento.
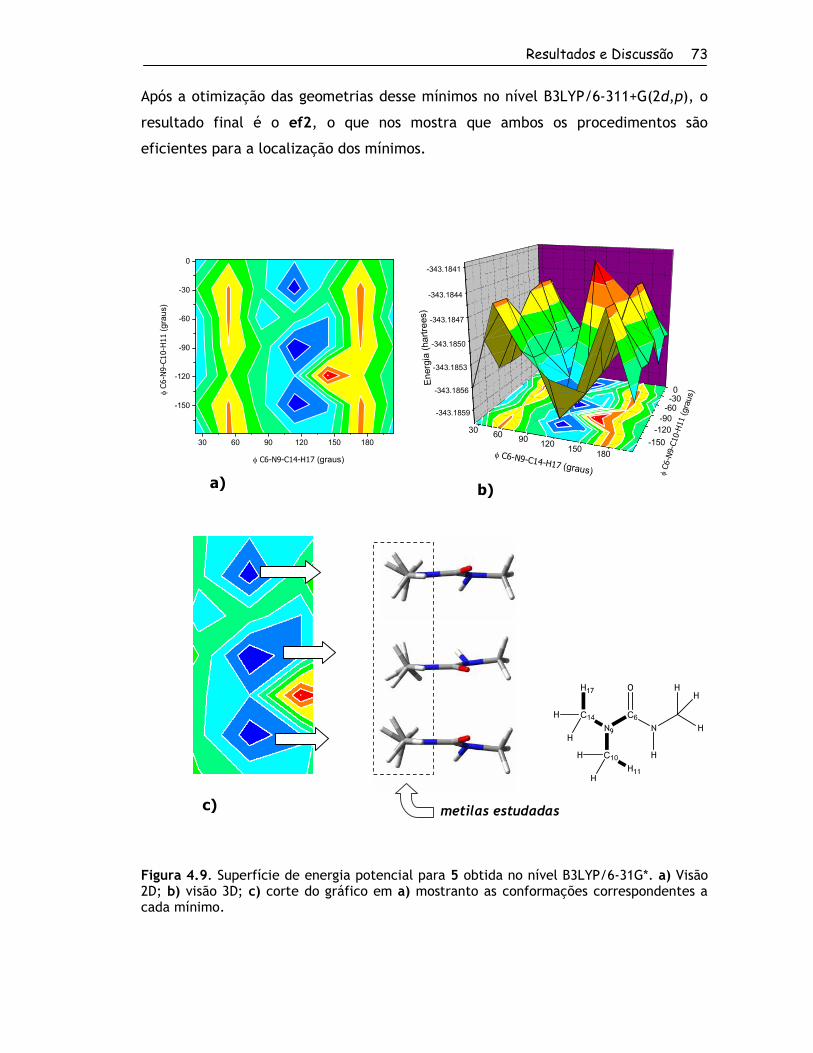
Resultados e Discussão 73
Após a otimização das geometrias desse mínimos no nível B3LYP/6-311+G(2d,p), o
resultado final é o ef2, o que nos mostra que ambos os procedimentos são
eficientes para a localização dos mínimos.
Figura 4.9. Superfície de energia potencial para 5 obtida no nível B3LYP/6-31G*. a) Visão 2D; b) visão 3D; c) corte do gráfico em a) mostranto as conformações correspondentes a cada mínimo.
c)
30 60 90 120 150 180
-150
-120
-90
-60
-30
0
φ C6-N9-C14-H17 (graus)
φ C6-N9-C10-H11 (
gra
us)
3060
90120
150180
-343.1859
-343.1856
-343.1853
-343.1850
-343.1847
-343.1844
-343.1841
-150
-120
-90
-60-300
En
erg
ia (
ha
rtre
es)
φ C6-N9-C10-H11 (
gra
us)
φ C6-N9-C14-H17 (graus)
a) b)
metilas estudadas
N
C6
H
O
N9
C14
C10
H
HH
H
HH11
H17
H
H

Resultados e Discussão 74
Figura 4.10. Superfície de energia potencial para a 6 obtida no nível B3LYP/6-31G*. a) Visão 2D; b) visão 3D; c) corte do gráfico em a) mostranto as conformações correspondentes a cada mínimo.
A SEP para 6 (Fig. 4.10) apresenta, semelhantemente, três mínimos. Há
outros mínimos visíveis na periferia da SEP, mas trata-se de repetições dos três
centrais. Assim como no caso de 6, os mínimos assemelham-se ao ef2 e convergem
para este quando otimizados no nível B3LYP/6-311+G(2d,p).
4060
80100
120140
160180
200
-666.1395
-666.1390
-666.1385
-666.1380
-666.1375
-666.1370
-160
-140
-120
-100
-80
-60
-40
-20
0
Energ
ia (h
artre
es)
φ C6-N9-C10-H11
(gra
us)
φ C6-N9-C14-H17 (graus)
30 60 90 120 150 180
-150
-120
-90
-60
-30
0
φ C6-N9-C14-H17 (graus)
φ C6-N9-C10-H11 (
gra
us)
a) b)
c) metilas estudadas
N
C6
H
S
N9
C14
C10
H
HH
H
HH11
H17
H
H

Resultados e Discussão 75
Passemos aos estados de transição. Para os compostos do Grupo 1,
trabalhamos com as possibilidades representadas na Fig. 4.7. Não nos deparamos
com nenhum problema quanto à otimização dessas estruturas, de forma que tanto
et1 como et2 apareceram como estados de transição de primeira ordem (uma
freqüência imaginária apenas) para os quatro compostos. Por outro lado, quando
fizemos as otimizações para o Grupo 2, verificamos que a metila do nitrogênio
secundário adota uma conformação oposta à dos compostos do Grupo 1. Para este
caso, não conhecemos relatos da literatura, e deste modo foi necessário estudar as
conformações apresentadas na Fig. 4.11. Os resultados estão compilados na Tabela
4.8.
Figura 4.11. Possíveis confôrmeros para os estados de transição dos compostos 5 e 6.
Os resultados dos métodos HF e B3LYP são diferentes para o composto 5,
mas se equivalem para 6. Por HF, o et1-a é estável para 5, ao contrário do
resultado B3LYP, que prediz apenas o et1-b. O et2-a de 5 é o único estável em HF,
mas em B3LYP ambos et1-a e et2-a são estáveis. Apesar das diferenças na posição
das metilas, os dois métodos concordam no fato de que o et1 é significativamente
Y
NNH
HH
H
H
H
H
H
H
Y
NNH
HH
H
HH
H
H
H
H
H
Y
NN
H
H
H
H
H
H
H
Y
NNH
HH
H
H
H
H
H
H
H
H
H
H
et1-a et1-b
et2-a et2-b

Resultados e Discussão 76
mais estável do que o et2, seja nas forma a ou b (5 a 6 kcal/mol). Esta diferença
deve-se principalmente à interação repulsiva entre o par de elétrons do nitrogênio
e a ligação dupla.
Tabela 4.8. Pesquisa conformacional para os estados de transição da N,N,N’-trimetiluréia (5) e da N,N,N’-trimetiltiouréia (6)a,b
N,N,N’-Trimetiluréia (5)
HF B3LYP
Energia #i Energia #i
et1-a -341,158723 1 -343,280692 1 c
et1-b -341,161232 2 -343,290087 1
et2-a -341,151284 1 -343,280692 1
et2-b → et2-a -343,280083 1
N,N,N’-Trimetiltiouréia (6)
HF B3LYP
Energia #i Energia #i
et1-a -663,790329 1 -666,243511 1
et1-b → et1-a → et1-a
et2-a -663,778956 1 -666,232775 1
et2-b → et2-a → et2-a
a 6-311+G(2d,p). b Energias em hartree. c A freqüência imaginária corresponde à inversão do nitrogênio secundário, não à rotacão C-N.
Os resultados distintos em HF e B3LYP para o et1 de 5 podem ser entendidos
com base no efeito da correlação eletrônica sobre os comprimentos de ligação.
Sabe-se que o método de Hartree-Fock geralmente fornece comprimentos de
ligação um pouco mais curtos do que os métodos de correlação.151 A análise dos
parâmetros otimizados nos mostra que as ligações (C=O)-NH e H3C-NH são
realmente mais curtas na geometria HF, viz. r((C=O)-NH)=1,339 Å e r(H3C-
NH)=1,444 Å, enquanto que os parâmetros B3LYP são r((C=O)-NH)=1,354 Å e r(H3C-
NH)=1,450 Å. Deste modo, se a geometria do et1 em HF fosse similar à B3LYP, o
hidrogênio eclipsado com a ligação (C=O)-N experimentaria maior interação
estérica com a região da carbonila. Existe também interação estérica com o
hidrogênio do grupo NH na geometria estável HF, mas, ao que parece, a interação
com a carbonila é mais intensa.

Resultados e Discussão 77
No caso da N,N,N’-trimetiltiouréia (6), apenas os et2-a foram obtidos. Isto
reforça a interpretação acima, pois a conjugação nN → π*C=S deve ser mais forte do
que a nN → π*C=O, fazendo com que o comprimento da ligação (C=S)-NH seja mais
curto na tiouréia. Sendo assim, o mesmo raciocínio aplicado ao caso HF no
parágrafo anterior vale aqui para o método B3LYP. Os comprimentos de ligação
suportam este argumento: r((C=S)-NH)=1,316 Å e r(H3C-NH)=1,450 Å em HF, e
r((C=S)-NH)=1,338 Å e r(H3C-NH)=1,451 Å em B3LYP. Apesar do aumento de
r(H3C-NH) em HF na N,N,N’-trimetiltiouréia (6), a nuvem eletrônica da tiocarbonila
é mais ampla do que a da carbonila e compensa a variação geométrica.
Tendo em vista que não foi possível realizar determinações experimentais
das barreiras rotacionais de 7 e 8, o interesse teórico nesses compostos ficou em
segundo plano e a pesquisa conformacional para os mesmos foi mais simples.
Basicamente, construímos as SEPs para 7 e 8 e otimizamos os mínimos resultantes.
A partir desses mínimos, rodamos uma varredura sobre a ligação C-N para localizar
os estados de transição, que foram então otimizados sem dar maior atenção à
posição das metilas como fizemos nos casos anteriores. As SEPs e os mínimos
resultantes estão apresentados nas Fig. 4.12 e 4.13.
A SEP para o composto 7 apresenta, à primeira vista, dois mínimos distintos
que se repetem periodicamente. Quando analisados, descobrimos que, embora as
periferias desses mínimos sejam diferentes, sua estrutura e energia são
essencialmente iguais, ou seja, há somente um mínimo de energia para 7. A
situação é similar para 8, mas para este a presença de um único mínimo é mais
evidente. A não ocorrência de outros mínimos nas SEPs destes compostos deve-se,
provavelmente, à grande interação estérica entre as metilas de nitrogênios
diferentes, que acaba por reduzir as opções estáveis de arranjos espaciais
O mesmo tipo de interação mencionado no parágrafo anterior faz com que as
metilas anti à ligação C=Z fiquem fora do plano N-(C=O)-N. Antes mesmo de levar
adiante os estudos com os compostos 7 e 8 podemos deduzir o porquê da
impossibilidade de detectar as barreiras por RMND, bastando notar que as
interações mencionadas colocam os pares de elétrons dos nitrogênios em uma
posição que desfavorece a deslocalização nN → π*C=X e diminui a barreira rotacional
(Figs. 4.12 e 4.13).
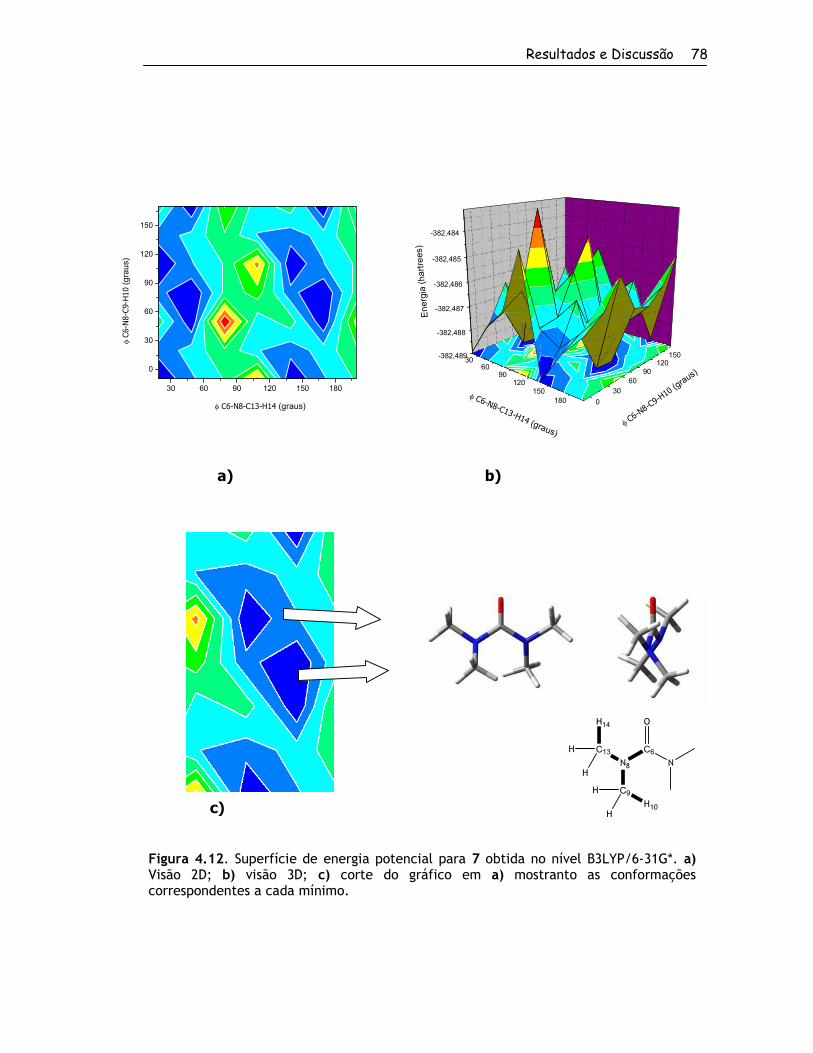
Resultados e Discussão 78
Figura 4.12. Superfície de energia potencial para 7 obtida no nível B3LYP/6-31G*. a) Visão 2D; b) visão 3D; c) corte do gráfico em a) mostranto as conformações correspondentes a cada mínimo.
a) b)
c)
N
C6
O
N8
C13
C9
H
HH10
H14
H
H
3060
90120
150
180
-382,489
-382,488
-382,487
-382,486
-382,485
-382,484
0
30
60
90120
150
En
erg
ia (
ha
rtre
es)
φ C6-N8-C9-H10 (
graus)
φ C6-N8-C13-H14 (graus)
30 60 90 120 150 180
0
30
60
90
120
150
φ C6-N8-C13-H14 (graus)
φ C6-N8-C9-H10 (
gra
us)
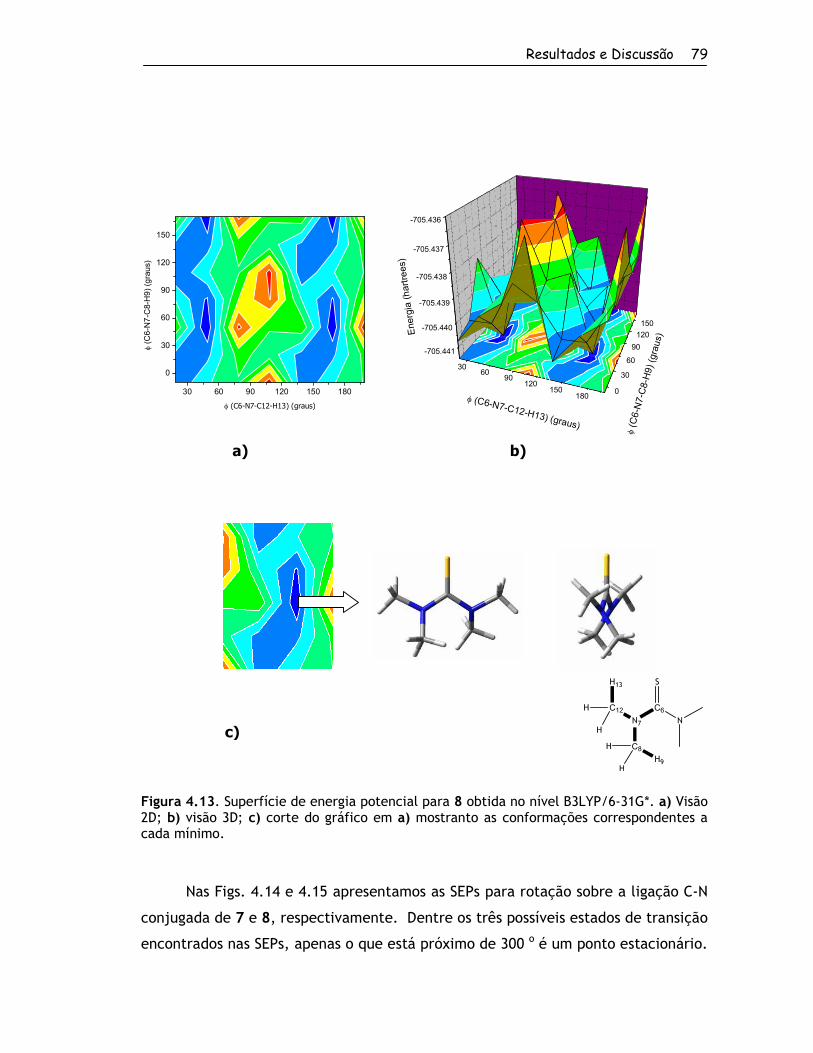
Resultados e Discussão 79
Figura 4.13. Superfície de energia potencial para 8 obtida no nível B3LYP/6-31G*. a) Visão 2D; b) visão 3D; c) corte do gráfico em a) mostranto as conformações correspondentes a cada mínimo.
Nas Figs. 4.14 e 4.15 apresentamos as SEPs para rotação sobre a ligação C-N
conjugada de 7 e 8, respectivamente. Dentre os três possíveis estados de transição
encontrados nas SEPs, apenas o que está próximo de 300 o é um ponto estacionário.
3060
90120
150180
-705.441
-705.440
-705.439
-705.438
-705.437
-705.436
0
30
60
90
120
150
Energ
ia (
hart
rees)
φ (C
6-N
7-C
8-H
9)
(gra
us)
φ (C6-N7-C12-H13) (graus)
30 60 90 120 150 180
0
30
60
90
120
150
φ (C6-N7-C12-H13) (graus)
φ (C
6-N
7-C
8-H
9)
(gra
us)
a) b)
c) N
C6
S
N7
C12
C8
H
HH9
H13
H
H

Resultados e Discussão 80
Este ponto corresponde ao et1 dos compostos anteriores e a Fig. 4.16 ilustra sua
forma para 7 e 8. A provável causa para não haver um análogo ao et2 é a presença
da metila adicional em 7 e 8, que promoveria alta interação estérica caso estes
compostos assumissem um arranjo similar aos et2 dos compostos anteriores.
Chegando a este ponto, temos em mãos as estruturas otimizadas em fase
gasosa que servirão de base para toda a seqüência dos trabalhos. Passemos então
ao estudo das barreiras rotacionais no vácuo e em solução pelo modelo IPCM.
Figura 4.14. Superfície de energia potencial 2D para 7 obtida no nível B3LYP/6-31+G**.
Figura 4.15. Superfície de energia potencial 2D para a 8 obtida no nível B3LYP/6-31G**.
0 50 100 150 200 250 300 350
0
2
4
6
8
10
12
14
∆E
(kca
l/m
ol)
τ (O7-C6-N8-C13) (graus)
0 40 80 120 160 200 240 280 320 360
0
2
4
6
8
10
12
14
τ (S20-C6-N7-C12) (graus)
∆E (kcal/mol)
N
C6
S20
N7
C12
C8
H
HH9
H13
H
H
N
C6
O7
N8
C13
C9
H
HH10
H14
H
H

Resultados e Discussão 81
Figura 4.16. Estados de transição obtidos a partir das superfícies de energia potencial 2D para os compostos 7 e 8.
4.2.2 Barreiras Rotacionais no Vácuo e em Solução por Métodos
Quânticos
4.2.2.1 Grupo 1
Uma vez determinadas as conformações, passamos ao cálculo das barreiras
rotacionais no vácuo e em solução utilizando o modelo de solvatação IPCM. Na
Tabela 4.9 apresentamos os parâmetros de ativação no vácuo e na Tabela 4.10
listamos as barreiras rotacionais com a inclusão do efeito da polaridade do meio.
Para estes cálculos utilizamos apenas as conformações de menor energia livre de
Gibbs. Além disso, as energias de solvatação determinadas pelo método IPCM
tratam apenas da parte eletrônica, não incluem as contribuições dos movimentos
nucleares (rotação, vibração e translação) e nem os efeitos das interações de curto
alcance – complexações ou ligações de hidrogênio.
As barreiras no vácuo (Tabela 4.9) evidenciam a necessidade de se incluir os
efeitos de solvatação. Tomemos por exemplo o composto 2, para o qual o valor
obtido com HF (11,66 kcal/mol) fica 1,4 kcal/mol abaixo do valor experimental

Resultados e Discussão 82
para o solvente de menor polaridade (CS2). O valor obtido com o método B3LYP é
um pouco melhor, mas ainda assim insatisfatório. No caso do composto 1, a
barreira com HF fica próxima dos valores experimentais, mas o valor fornecido pelo
método B3LYP é ruim. Tanto para 3 como para 4 obtém-se uma aparente
concordância com os experimento em ambos os níveis de teoria.
A inclusão da polaridade do solvente (Tabela 4.10) resulta em valores
concordantes com os experimentais para o composto 1 em HF. Para ser mais
exatos, os valores calculados são, em média, um pouco superiores ao obtidos por
APB, mas em pleno acordo com os obtidos por Tc. As barreiras em B3LYP são
aproximadamente 2 kcal/mol maiores do que as experimentais.
Tabela 4.9. Parâmetros de ativação no vácuo para os compostos 1-4 calculados com os nível de teoria HF/6-311+G(2d,p) e B3LYP/6-311+G(2d,p) (∆G ‡ e ∆H ‡ em kcal/mol, ∆S ‡ em cal/K⋅mol, T=298,15 K)
HF B3LYP
∆H ‡ ∆S ‡ ∆G ‡ ∆H ‡ ∆S ‡ ∆G ‡
(1)
et1 12,86 -7,08 14,97 13,86 -8,28 16,33
et2 13,89 -6,71 15,89 14,77 -7,90 17,12
efet. a 14,86 16,19
(2)
et1 9,71 -6,53 11,66 11,66 -5,52 13,40
et2 13,51 -5,90 15,29 14,82 -5,80 16,55
efet. a 11,66 13,40 (3) a
et1 15,16 -9,00 17,84 14,52 -9,21 17,27
et2 16,14 -8,46 18,66 15,97 -8,86 18,61
efet. a 17,71 17,21 (4) b
et1 11,12 -11,56 14,56 11,45 -7,78 13,77
et2 15,38 -10,59 18,53 15,54 -6,90 17,60
efet. a 14,56 13,77
a Barreira efetiva, incorporando a contribuição dos dois estados de transição. b Conformação ef2. c Conformação ef4.

Resultados e Discussão 83
Tabela 4.10. Barreiras rotacionais (∆G‡, em kcal/mol) para os compostos 1-4 calculadas nos níveis HF/6-311+G(2d,p) e B3LYP/6-311+G(2d,p) com os efeitos de solvatação incluídos através do modelo IPCM
et1 et2 efet, a
HF B3LYP HF B3LYP HF B3LYP (1)
e = 1,00 14,97 16,33 15,89 17,12 14,9 16,2 2,23 15,41 16,81 16,03 17,41 15,2 16,6 2,27 15,42 16,82 16,03 17,41 15,2 16,6 2,61 15,47 16,88 16,04 14,44 15,3 16,7 8,82 15,75 17,19 16,06 17,57 15,5 16,9 32,61 15,84 17,29 16,03 17,59 15,5 17,0 35,69 15,84 17,30 16,03 17,59 15,5 17,0 41,76 15,85 17,30 16,03 17,59 15,2 17,0 46,83 15,85 17,30 16,03 17,59 15,5 17,0 78,36 15,86 17,32 16,02 17,60 15,5 17,0 (2)
e = 1,00 11,66 13,40 15,29 16,55 11,7 13,4 2,23 12,56 14,60 15,45 17,84 12,6 14,6 2,27 12,57 16,62 15,45 17,84 12,6 14,6 2,61 12,70 14,75 15,47 17,88 12,7 14,8 8,82 12,84 15,46 15,52 18,04 12,8 15,5 32,61 13,70 15,72 15,51 18,08 13,7 15,7 35,69 13,70 15,73 15,51 18,08 13,7 15,7 41,76 13,72 15,74 15,51 18,09 13,7 15,7 46,83 13,73 15,75 15,51 18,09 13,7 15,7 78,36 13,76 15,78 15,51 18,09 13,7 15,8 (3) b
e = 1,00 17,84 17,27 18,66 18,61 17,7 17,2 2,23 19,17 18,03 19,06 18,70 18,7 17,9 2,27 19,19 18,06 19,07 18,71 18,7 17,9 2,61 19,37 18,17 19,14 18,73 18,8 18,0 8,82 20,39 18,80 19,48 18,86 19,4 18,4 32,61 20,76 19,04 19,59 18,90 19,5 18,6 35,69 20,77 19,04 19,59 18,90 19,5 18,6 41,76 20,79 19,06 19,60 18,91 19,5 18,6 46,83 20,80 19,06 19,60 19,91 19,5 18,6 78,36 20,84 19,09 19,61 18,91 19,5 18,6 (4) c
e = 1,00 14,56 13,77 18,53 17,60 14,6 13,8 2,23 15,41 14,31 19,65 18,38 15,4 14,3 2,27 15,42 14,32 19,66 18,39 15,4 14,3 2,61 15,54 14,40 19,80 18,50 15,5 14,4 8,82 16,26 14,87 20,50 18,97 16,6 14,9 32,61 16,55 15,06 2073 19,11 16,6 15,1 35,69 16,56 15,06 20,74 19,11 16,6 15,1 41,76 16,57 15,07 20,75 19,12 16,6 15,1 46,83 16,58 15,08 20,76 19,13 16,6 15,1 78,36 16,62 15,10 20,78 19,14 16,6 15,1
a Barreira efetiva, incorporando a contribuição dos dois estados de transição. b Conformação ef2. c Conformação ef4. Constantes dielétricas corrigidas para 25 oC 152; a ordem dos solventes, depois do vácuo (e = 1,00), é a mesma que aparece na Tabela 4.2 (pg. 62)

Resultados e Discussão 84
As barreiras em solução calculadas para o composto 2 divergem dos
experimentos tanto em HF como em B3LYP. Para este último, a concordância é um
pouco melhor, mas assim como para 1, os valores B3LYP ficam acima dos
experimentais. O composto 3 serve como um bom exemplo de advertência para a
prática comum de se trabalhar com barreiras rotacionais sem a inclusão do efeito
do solvente, pois as barreiras no vácuo, que eram próximas às experimentais, ficam
demasiadamente elevadas. Para os solventes menos polares, as barreiras em B3LYP
ainda estão em uma faixa aceitável, mas com o aumento da polaridade a
divergência se acentua. O mesmo se pode dizer acerca de 4 – há uma inicial
concordância entre os valores em B3LYP e os experimentos, mas, como antes, essa
concordância diminui com o aumento da polaridade. As barreiras em HF concordam
com os valores obtidos por Tc, que são um pouco maiores do que os APB. Porém,
como já dissemos, para 4 esperamos informações mais exatas por APB.
Neste ponto do trabalho, não há como julgar a performance dos métodos HF
e B3LYP, uma vez que percorremos apenas metade do caminho para a reprodução
das barreiras experimentais. Contudo, o que parece confuso neste estágio tornar-
se-á claro com a inclusão da contribuição das ligações de hidrogênio.
Analisemos agora as variações nas barreiras rotacionais previstas
teoricamente pelo método IPCM, i.e. o efeito da polaridade do solvente. A barreira
para 1 sofre em ambos os métodos um leve aumento, 0,2 kcal/mol em HF e 0,3
kcal/mol em B3LYP, quando se passa do CS2 para H2O. Ou seja, embora os métodos
HF e B3LYP divirjam na magnitude da barreira, ambos predizem uma variação
pequena, condizente com as observações experimentais (Tabelas 4.2 e 4.4). Já
para o composto 2, os cálculos apontam uma variação de 1 kcal/mol para a mesma
mudança de solventes. Isto está de acordo com os experimentos, mas as
magnitudes das barreiras ainda não são satisfatórias.
Passando-se de CCl4 para DMSO, há uma variação para 3 de 0,8 e 0,9
kcal/mol em HF e B3LYP, respectivamente. Esta variação não é observada
experimentalmente. Para 4 as variações, de CCl4 para H2O, são 1,2 e 0,8 kcal/mol
em HF e B3LYP. Estes valores, particularmente o HF, são coerentes com a mudança
experimental de 1,3 kcal/mol.

Resultados e Discussão 85
Resumindo, variações mensuráveis são previstas para 2, 3 e 4, mas somente
para 2 e 4 essas variações foram observadas experimentalmente. Vejamos então o
que causa essas respostas à polaridade do meio.
Como dissemos no Cap. 2, a resposta das barreiras rotacionais ao solvente
tem sido explicada com base na diferença de momento de dipolo entre ef e et ou
com base no momento de dipolo do ef. É possível, contudo, desenvolver uma
formulação que nos permita agregar a contribuição de ambos e assim realizar uma
análise mais completa. Para fazê-lo, consideremos a energia de solvatação tal
como expressa no modelo de Onsager:61
na qual e é a constante dielétrica do solvente, ao é o raio de uma cavidade que
contém o soluto e m é o momento de dipolo do soluto. O momento de dipolo do et
pode ser expresso como met = mef + δm, sendo δm a diferença entre os momentos de
dipolo do ef e do et. Com isso, chegamos ao seguinte resultado para a energia de
solvatação do et:
onde
O efeito do solvente resultante da polaridade do meio pode ser calculado
como a diferença entre as energias de solvatação do et e do ef,
Uma vez que o primeiro termo da Eq. (4.2) é simplesmente a energia de
solvatação do ef, chegamos a:
( )( ) 3
o
2
solv 11
aG
µ+−
=∆e
e
2
( ) ( ) ( ) 2oefo
2efo
etsolv ,,2, δµξδµµξµξ aaaG eee −−−=∆
( ) ( )( ) 3
oo
1121
,a
a+−
=e
eeξ
efsolv
etsolvSolventedoEfeito GG ∆−∆=
( ) ( ) 2oefo ,,2SolventedoEfeito δµξδµµξ aa ee −−=
(4.1)
(4.2)
(4.3)
(4.4)
(4.5)
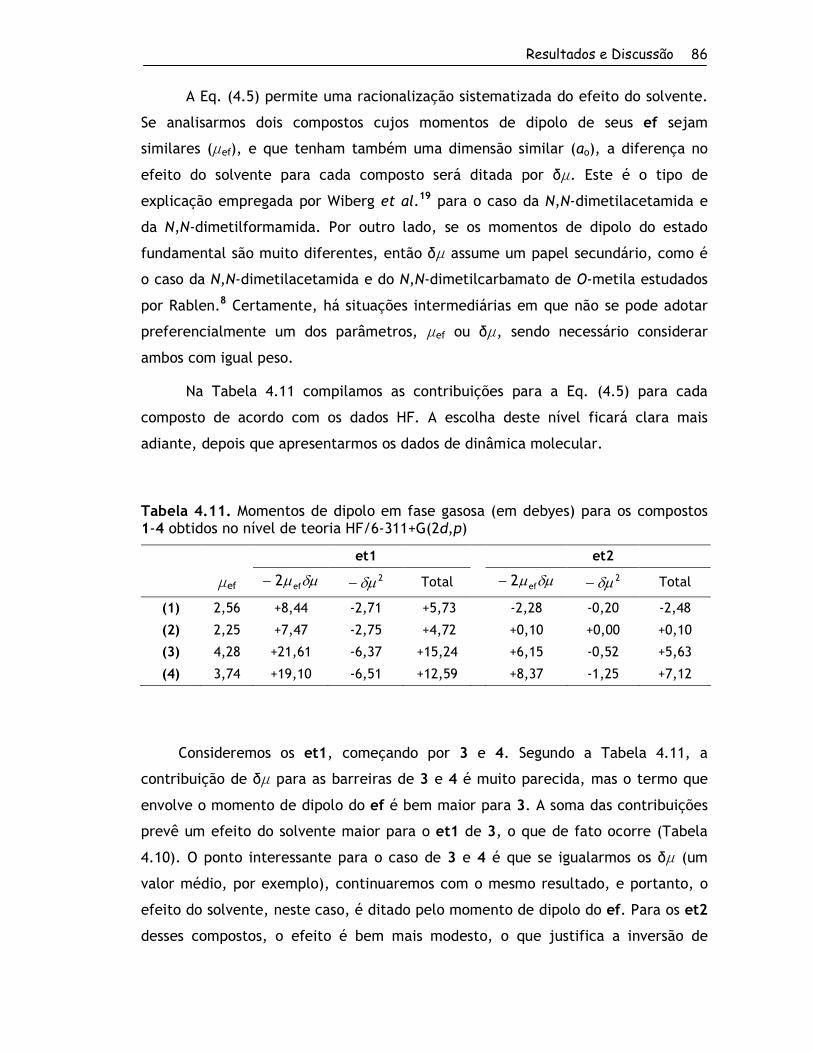
Resultados e Discussão 86
A Eq. (4.5) permite uma racionalização sistematizada do efeito do solvente.
Se analisarmos dois compostos cujos momentos de dipolo de seus ef sejam
similares (mef), e que tenham também uma dimensão similar (ao), a diferença no
efeito do solvente para cada composto será ditada por δm. Este é o tipo de
explicação empregada por Wiberg et al.19 para o caso da N,N-dimetilacetamida e
da N,N-dimetilformamida. Por outro lado, se os momentos de dipolo do estado
fundamental são muito diferentes, então δm assume um papel secundário, como é
o caso da N,N-dimetilacetamida e do N,N-dimetilcarbamato de O-metila estudados
por Rablen.8 Certamente, há situações intermediárias em que não se pode adotar
preferencialmente um dos parâmetros, mef ou δm, sendo necessário considerar
ambos com igual peso.
Na Tabela 4.11 compilamos as contribuições para a Eq. (4.5) para cada
composto de acordo com os dados HF. A escolha deste nível ficará clara mais
adiante, depois que apresentarmos os dados de dinâmica molecular.
Tabela 4.11. Momentos de dipolo em fase gasosa (em debyes) para os compostos 1-4 obtidos no nível de teoria HF/6-311+G(2d,p)
et1 et2
mef δµµef2− 2δµ− Total δµµef2− 2δµ− Total
(1) 2,56 +8,44 -2,71 +5,73 -2,28 -0,20 -2,48
(2) 2,25 +7,47 -2,75 +4,72 +0,10 +0,00 +0,10
(3) 4,28 +21,61 -6,37 +15,24 +6,15 -0,52 +5,63
(4) 3,74 +19,10 -6,51 +12,59 +8,37 -1,25 +7,12
Consideremos os et1, começando por 3 e 4. Segundo a Tabela 4.11, a
contribuição de δm para as barreiras de 3 e 4 é muito parecida, mas o termo que
envolve o momento de dipolo do ef é bem maior para 3. A soma das contribuições
prevê um efeito do solvente maior para o et1 de 3, o que de fato ocorre (Tabela
4.10). O ponto interessante para o caso de 3 e 4 é que se igualarmos os δm (um
valor médio, por exemplo), continuaremos com o mesmo resultado, e portanto, o
efeito do solvente, neste caso, é ditado pelo momento de dipolo do ef. Para os et2
desses compostos, o efeito é bem mais modesto, o que justifica a inversão de

Resultados e Discussão 87
estabilidade observada para os et de 3, i.e. embora a barreira aumente para ambos
os et de 3, o aumento é mais expressivo para o et1. Isso ocorre porque o momento
de dipolo dos et2 é aproximadamente o dobro dos momentos de dipolo dos et1.
O caso de 1 e 2 é um pouco mais complexo. De acordo com a Tabela 4.10, a
barreira através de et1 para 2 aumenta por cerca de 2 kcal/mol, enquanto a
barreira para 1 sofre uma elevação de menos de 1 kcal/mol. No entanto, A Tabela
4.11 aponta um efeito do solvente maior para 1, revelando uma falha em nosso
modelo baseado nos momentos de dipolo em fase gasosa. Temos que considerar,
contudo, que o composto 2 possui um átomo de enxofre e isso acaba por aumentar
a polarizabilidade do composto. Isso é confirmado ao compararmos as componentes
do tensor de hiperpolarizabilidade: (69,375, -1,292, 59,611, -0,281, 0,062, 46,282)
para 1 e (94,555, -1,590, 74,280, -0,199, 0,094, 56,407) para 2. Sendo assim,
espera-se que a densidade eletrônica de 2 varie mais na presença do campo
elétrico do solvente. Na Tabela 4.12 listamos os resultados para os cálculos em
água. A diferença anteriomente observada agora se inverteu, ainda que por uma
pequena quantidade, em favor do composto 2. Vale lembrar que estamos
utilizando um modelo relativamente simples para guiar nossas interpretações, de
forma que temos que considerar a possibilidade de não encontrarmos valores
totalmente condizentes com os experimentos e com os cálculos IPCM. Mesmo
assim, o modelo de Onsager pode ser empregado na interpretação qualitativa dos
resultados.
Tabela 4.12. Momentos de dipolo em água (em debyes) para os compostos 1-4 obtidos no nível de teoria HF/6-311+G(2d,p)
et1 et2
mef δµµef2− 2δµ− Total δµµef2− 2δµ− Total
(1) +3,61 +15,95 -4,88 +11,07 -3,19 -0,19 -3,38
(2) +3,63 +16,56 -5,21 +11,35 +1,17 -0,03 +1,15
(3) +6,04 +46,28 -14,67 +31,61 +15,58 -1,66 +13,92
(4) +5,39 +34,10 -10,19 +23,91 +18,90 -3,13 +15,77
Do que expusemos acima, decorre que não apenas os momentos de dipolo
devem ser considerados na análise da resposta ao solvente das barreiras

Resultados e Discussão 88
rotacionais, é preciso ter em mente também a importância da polarizabilidade
molecular, ou seja, a forma como a densidade eletrônica muda na presença do
campo elétrico do solvente.
4.2.2.2 Grupo 2
As barreiras rotacionais no vácuo para os compostos 5-8 estão apresentadas na
Tabela 4.13, seguidas das barreiras em solução na Tabela 4.14.
Tabela 4.13. Parâmetros de ativação no vácuo para os compostos 5-8 calculados com os nível de teoria HF/6-311+G(2d,p) e B3LYP/6-311+G(2d,p) (∆G ‡ e ∆H ‡ em kcal/mol, ∆S ‡ em cal/K⋅mol, T=298,15 K)
HF B3LYP ∆H ‡ ∆S ‡ ∆G ‡ ∆H ‡ ∆S ‡ ∆G ‡ (5)
et1-a 5,02 -3,78 6,14 et1-b 5,71 -5,62 7,39 et2-a 11,18 -0,39 11,30 11,23 -3,94 12,40 et2-b 11,29 -1,60 11,77 efet. a 6,14 7,39
(6)
et1-a 5,15 -8,37 7,64 5,37 -8,41 7,88 et2-a 11,93 -7,50 14,17 11,72 -7,77 14,03 et2-b efet. a 7,64 7,88
(7)
et1 3,90 -1,07 4,22 3,72 -3,71 4,82
(8) et1 3,12 -2,81 3,95 3,60 -3,31 4,59
a Barreira efetiva, incorporando a contribuição de todos os estados de transição.
Assim como no Grupo 1, as barreiras no vácuo em B3LYP são maiores do que
as HF, porém as diferenças aqui são menos acentuadas. Segundo ambos os
métodos, a barreira para a N,N,N’-trimetiltiouréia (6) é maior do que para a
N,N,N’-trimetiluréia (5), enquanto que a situação se inverte para a N,N,N’,N’-
tetrametiluréia (7) e a N,N,N’,N’-tetrametiltiouréia (8).

Resultados e Discussão 89
Tabela 4.14. Barreiras rotacionais (∆G‡, em kcal/mol) para os compostos 5-8 calculadas nos níveis HF/6-311+G(2d,p) e B3LYP/6-311+G(2d,p) com os efeitos de solvatação incluídos através do modelo IPCM
N,N,N’-Trimetiluréia (5) HF B3LYP et1-a et2-a efet.a et1-b et2-a et2-b efet.a
e b = 1,00 6,14 11,30 6,1 7,39 12,40 11,77 7,4 2,61 7,00 10,88 7,0 8,33 12,39 11,66 8,2 8,82 7,61 10,69 7,6 8,99 12,36 11,50 9,0 20,49 7,79 10,61 7,8 9,19 12,34 11,42 9,2 32,61 7,85 10,59 7,8 9,25 12,33 11,40 9,2 37,18 7,86 10,58 7,9 9,26 12,33 11,39 9,2 78,36 7,90 10,56 7,9 9,31 12,32 11,37 9,3
N,N,N’-Trimetiltiouréia (6) HF B3LYP et1-a et2-a efet.a et1-b et2-a efet.a
e = 1,00 7,64 14,17 7,6 7,88 14,03 7,9 2,61 9,13 13,33 9,1 9,24 13,60 9,2 8,82 10,29 12,70 10,3 10,40 13,33 10,4 20,49 10,65 12,48 10,6 10,77 13,23 10,8 32,61 10,75 12,41 10,7 10,88 13,20 10,9 37,18 10,78 12,40 10,7 10,91 13,20 10,9 78,36 10,86 12,34 10,8 11,00 13,17 11,0 N,N,N’,N’-Tetrametiluréia (7) HF B3LYP et1 et1 e = 1,00 4,22 4,82
2,61 4,31 5,21 8,82 4,56 5,65 20,49 4,67 5,82 32,61 4,71 5,87 37,18 4,72 5,88 78,36 4,75 5,92 N,N,N’,N’-Tetrametiltiouréia (8) HF B3LYP et1 et1 e = 1,00 3,95 3,59
2,61 4,25 3,99 8,82 4,38 4,20 20,49 4,38 4,24 32,61 4,38 4,25 37,18 4,38 4,26 78,36 4,38 4,26
a Barreira efetiva, incorporando a contribuição de todos os estados de transição. b Constantes dielétricas corrigidas para 25 oC.152 Solventes, pela ordem: vácuo, CS2, CH2Cl2, (CH3)2CO, CH3OH, CD3CN, H2O.

Resultados e Discussão 90
A inclusão do efeito do solvente causa aumentos expressivos nas barreiras
rotacionais de 5 e 6, mas tem um efeito mais modesto sobre as barreiras de 7 e 8
(Tabela 4.14). A barreira de 6 é mais sensível ao solvente do que a de 5, uma
situação similar à observada entre amidas e tioamidas.20
A magnitude relativa fornecida pelos cálculos IPCM é inversa à experimental,
uma vez que os experimentos mostram que a barreira de 5 é maior do que a de 6.
Lembremos aqui que só foi possível obter dados de RMND para 5 em solventes
próticos, que incorporam efeitos não reproduzidos no modelo IPCM. Mas as
barreiras calculadas para 6, tanto por HF como por B3LYP, são coerentes com as
experimentais, o que nos dá indícios de que 6 é pouco afetado por ligações de
hidrogênio.
Mesmo com a inclusão do efeito do solvente, o maior valor que se obtém
dentre aqueles calculados para 7 e 8 é 5,9 kcal/mol. Isso é quase a metade do
valor calculado para 5, sendo esta a causa de não ter sido possível determinar as
barreiras para 7 e 8.
Da mesma forma como procedemos para o Grupo 1, compilamos na
Tabela 4.15 os momentos de dipolo e diferenças de momentos de dipolo para os
compostos do Grupo 2. Uma vez que os dois et2 obtidos para 5 em B3LYP são
próximos em energia, e visto que nenhum deles faz contribuição significativa à
barreira, tomamos apenas o et2-b para essas análises.
Tabela 4.15. Momentos de dipolo em fase gasosa (em debyes) para os compostos 5-8 obtidos no nível de teoria HF/6-311+G(2d,p)
et1 et2
mef δµµef2− 2δµ− Total δµµef2− 2δµ− Total
(5) 4,00 +8,11 -1,03 +7,08 -5,37 +0,45 -4,92
(6) 5,98 +21,15 -3,13 +18,03 +3,86 +0,10 +3,96
(7) 3,40 +2,86 -0,18 +2,68 − − −
(8) 5,29 +6,59 -0,39 +6,20 − − −
O maior efeito da polaridade do solvente observado para 6 em relação a 5
deve-se tanto ao momento de dipolo do ef como à maior δm. É interessante notar
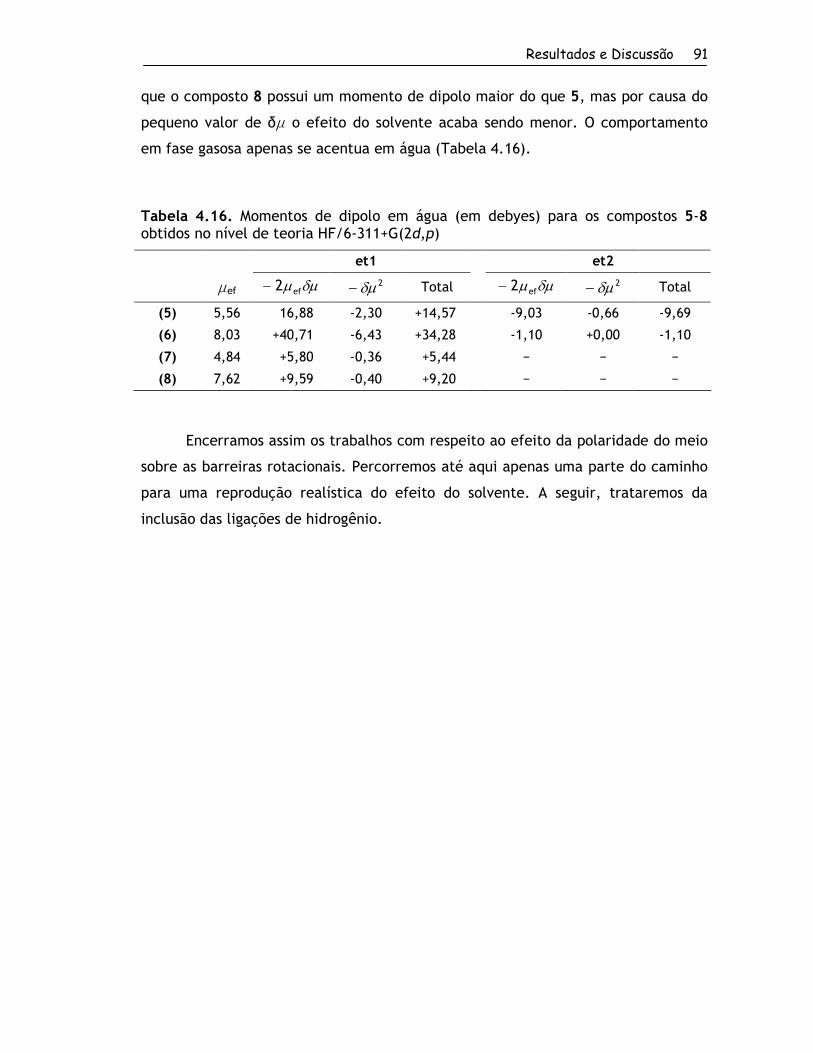
Resultados e Discussão 91
que o composto 8 possui um momento de dipolo maior do que 5, mas por causa do
pequeno valor de δm o efeito do solvente acaba sendo menor. O comportamento
em fase gasosa apenas se acentua em água (Tabela 4.16).
Tabela 4.16. Momentos de dipolo em água (em debyes) para os compostos 5-8 obtidos no nível de teoria HF/6-311+G(2d,p)
et1 et2
mef δµµef2− 2δµ− Total δµµef2− 2δµ− Total
(5) 5,56 16,88 -2,30 +14,57 -9,03 -0,66 -9,69
(6) 8,03 +40,71 -6,43 +34,28 -1,10 +0,00 -1,10
(7) 4,84 +5,80 -0,36 +5,44 − − −
(8) 7,62 +9,59 -0,40 +9,20 − − −
Encerramos assim os trabalhos com respeito ao efeito da polaridade do meio
sobre as barreiras rotacionais. Percorremos até aqui apenas uma parte do caminho
para uma reprodução realística do efeito do solvente. A seguir, trataremos da
inclusão das ligações de hidrogênio.

Resultados e Discussão 92
4.2.3 Inclusão do Efeito das Ligações de Hidrogênio por Dinâmica
Molecular Associada a Cálculos Quânticos
4.2.3.1 Grupo 1
Mencionamos no Cap. 2 a importância da incorporação das interações de
curto alcance no cálculo das energias de solvatação. Existe na literatura alguns
trabalhos em que essa contribuição é obtida pela adição de uma molécula do
solvente a uma região receptora de prótons.8, 18 Esse procedimento constitui uma
aproximação relativamente pobre, pois não considera adequadamente a estrutura
estatística do líquido.
Como dissemos ainda no Cap. 2, a realização de cálculos com dinâmica
molecular requer a especificação de um conjunto de parâmetros que descreve as
interações entre o soluto e o solvente (campo de forças). Infelizmente, não existe
parametrização para o Grupo 1, e sendo assim, nosso primeiro passo foi obter os
parâmetros de Lennard-Jones e de Coulomb para cada um dos compostos desse
grupo, tanto no estado fundamental quanto nos estados de transição.
O procedimento que adotamos é semelhante ao descrito por Jorgensen et
al.10 (Parte Experimental). Na Fig. 4.17 apresentamos os complexos utilizados na
parametrização. Ao longo desses cálculos, mantivemos a geometria do soluto fixada
nos valores obtidos com o nível B3LYP/6-311+G(2d,p) e a geometria da molécula de
água fixada nos valores do modelo TIP4P.12 Apenas a distância entre a molécula de
água e o soluto foi otimizada. A energia de complexação ( compE∆ ) foi então
calculada por:
sendo compE a energia total do complexo solutoּּּágua. Os valores para águaE e
solutoE foram obtidos de forma a corrigir o erro de sobreposição de conjunto de
funções de bases.39
( )solutoáguacompcomp EEEE +−=∆ (4.6)

Resultados e Discussão 93
ef et1 et2
c1
c2
c3
c4
c5
c6
Figura 4.17. Complexos utilizados na parametrização dos campos de forças para os compostos 1-4.

Resultados e Discussão 94
Uma vez obtidas as energias de complexação, foi necessário ajustar os
parâmetros q, ε e σ para cada átomo ou grupo de átomos. No modelo que
adotamos, os hidrogênios dos grupos metila não são tratados explicitamente, ao
invés disso, os três hidrogênios e o carbono constituem um único agregado descrito
por um conjunto de valores q, ε e σ. Isso simplifica consideravelmente os cálculos
durante as simulações, tornando-as mais rápidas e sem perda significativa da
qualidade dos resultados. Portanto, temos um modelo de sete pontos para os
solutos do Grupo 1.
A otimização simultânea de todos os parâmetros da Eq. (2.23) é uma tarefa
extremamente laboriosa. Apenas para o soluto teríamos 18 parâmetros a ser
variados simultaneamente. Por isso, adotamos uma simplificação adicional em que
os parâmetros de Lennad-Jones são mantidos fixos enquanto variamos apenas as
cargas atômicas.10 Iniciamos os trabalhos utilizando parâmetros otimizados para
amidas10, 77 e alcoóis74 e empregamos o programa ANALIZE139 para calcular as
energias de complexação a partir do campo de forças que definimos. Os valores
finais para o conjunto de parâmetros estão apresentados na Tabela 4.17 e as
energias de complexação calculadas pela Eq. (2.23) estão listadas na Tabela 4.18.
A concordância entre os valores calculados por B3LYP e pelo campo de forças
mostra que o procedimento adotado para a otimização dos parâmetros é
justificável, de forma que temos um campo de forças que descreve apropriada-
mente as interações entre o soluto e a água.
Com respeito às cargas pontuais, é interessante observar a forma como os
valores da Tabela 4.17 são coerentes com o modelo de ressonância. Por exemplo, a
Fig. 1.2 sugere que a ressonância desloca carga elétrica do nitrogênio para o
oxigênio carbonílico, efeito esse que só ocorre no ef. Assim, com o desfavo-
recimento da conjugação nos et, a carga do oxigênio carbonílico deveria diminuir,
ao passo que a carga do nitrogênio deveria aumentar. Como pode ser visto na
Tabela 4.17, é o que de fato ocorre.
Uma vez determinados os parâmetros de Coulomb e Lennard-Jones,
procedemos às simulações conforme descrito na Parte Experimental. Na Fig. 4.18
apresentamos um “instantâneo” da caixa periódica utilizada.
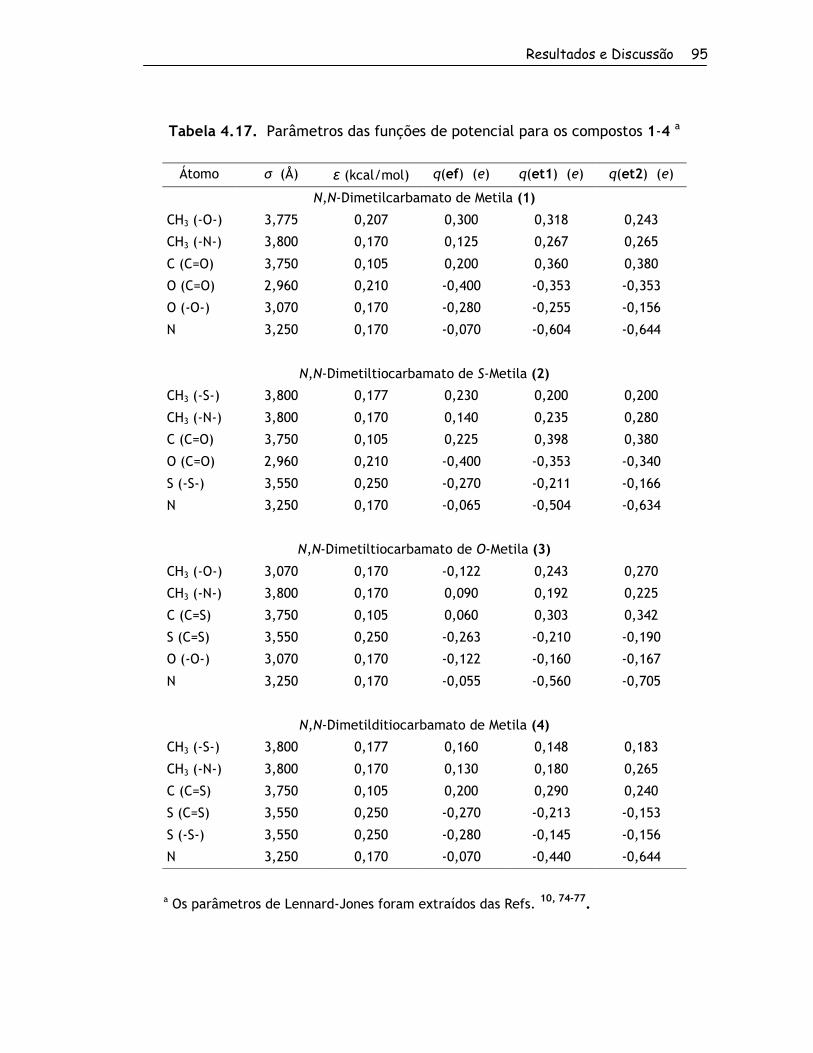
Resultados e Discussão 95
Tabela 4.17. Parâmetros das funções de potencial para os compostos 1-4 a
a Os parâmetros de Lennard-Jones foram extraídos das Refs. 10, 74-77.
Átomo σ (Å) ε (kcal/mol) q(ef) (e) q(et1) (e) q(et2) (e)
N,N-Dimetilcarbamato de Metila (1)
CH3 (-O-) 3,775 0,207 0,300 0,318 0,243
CH3 (-N-) 3,800 0,170 0,125 0,267 0,265
C (C=O) 3,750 0,105 0,200 0,360 0,380
O (C=O) 2,960 0,210 -0,400 -0,353 -0,353
O (-O-) 3,070 0,170 -0,280 -0,255 -0,156
N 3,250 0,170 -0,070 -0,604 -0,644
N,N-Dimetiltiocarbamato de S-Metila (2)
CH3 (-S-) 3,800 0,177 0,230 0,200 0,200
CH3 (-N-) 3,800 0,170 0,140 0,235 0,280
C (C=O) 3,750 0,105 0,225 0,398 0,380
O (C=O) 2,960 0,210 -0,400 -0,353 -0,340
S (-S-) 3,550 0,250 -0,270 -0,211 -0,166
N 3,250 0,170 -0,065 -0,504 -0,634
N,N-Dimetiltiocarbamato de O-Metila (3)
CH3 (-O-) 3,070 0,170 -0,122 0,243 0,270
CH3 (-N-) 3,800 0,170 0,090 0,192 0,225
C (C=S) 3,750 0,105 0,060 0,303 0,342
S (C=S) 3,550 0,250 -0,263 -0,210 -0,190
O (-O-) 3,070 0,170 -0,122 -0,160 -0,167
N 3,250 0,170 -0,055 -0,560 -0,705
N,N-Dimetilditiocarbamato de Metila (4)
CH3 (-S-) 3,800 0,177 0,160 0,148 0,183
CH3 (-N-) 3,800 0,170 0,130 0,180 0,265
C (C=S) 3,750 0,105 0,200 0,290 0,240
S (C=S) 3,550 0,250 -0,270 -0,213 -0,153
S (-S-) 3,550 0,250 -0,280 -0,145 -0,156
N 3,250 0,170 -0,070 -0,440 -0,644

Resultados e Discussão 96
Tabela 4.18. Energias de complexação (kcal/mol) soluto-água calculadas no nível B3LYP/6-31+G(d,p) e através do campo de forças clássico a
a As espécies c1, c2,... são definidas na Fig. 4.17.
c1 c2 c3 c4 c5 c6
N,N-Dimetilcarbamato de O-Metila (1)
TFD -6,21 -5,91 -1,20 -2,24 -0,79 -0,83 ef C. Forças -6,32 -5,98 -1,32 -2,37 -0,65 -0,80
TFD -5,38 -4,96 -5,91 -1,62 -1,32 -0,54
et1 C. Forças -5,28 -4,90 -5,93 -1,56 -1,54 -0,98
TFD -5,36 -4,44 -5,88 -1,04 -1,51 -0,55 et2 C. Forças -5,53 -4,16 -5,55 -0,93 -1,24 -1,11
N,N-Dimetiltiocarbamato de S-Metila (2)
TFD -4,87 -5,51 -1,07 -2,01 -0,24 -0,97 ef C. Forças -4,43 -5,89 -0,80 -1,74 -0,84 -1,22
TFD -4,64 -4,49 -4,47 -1,35 -0,57 -0,30
et1 C. Forças -4,39 -4,66 -4,61 -1,30 -1,05 -1,01
TFD -4,46 -3,92 -5,45 -1,73 -0,74 -0,56 et2 C. Forças -4,34 -4,05 -5,32 -1,28 -1,16 -1,30
N,N-Dimetiltiocarbamato de O-Metila (3)
TFD -2,79 -2,46 -1,23 -0,51 -0,85 -1,02 ef C. Forças -2,93 -2,60 -1,42 -1,22 -0,57 -0,63
TFD -2,26 -1,58 -5,13 -1,05 -1,21 -0,36
et1 C. Forças -2,16 -1,67 -5,10 -0,50 -1,22 -0,50
TFD -2,32 -1,28 -5,68 -0,42 -1,52 -0,39 et2 C. Forças -2,32 -1,29 -5,66 -0,46 -1,66 -0,54
N,N-Dimetilditiocarbamato de S-Metila (4)
TFD -2,77 -2,42 -0,68 -2,30 -0,28 -1,15 ef C. Forças -2,68 -2,57 -0,56 -2,30 -0,42 -1,11
TFD -2,20 -1,54 -3,99 -1,03 -0,72 -0,21
et1 C. Forças -2,11 -1,77 -3,95 -0,91 -0,86 -0,73
TFD -2,29 -1,25 -5,49 -1,36 -0,69 -0,48 et2 C. Forças -2,17 -1,32 -5,07 -1,26 -1,14 -1,02

Resultados e Discussão 97
Figura 4.18. Instantâneo da caixa periódica utilizada nas simulações com água. A caixa possui dimensões 20,8 Å ×20,8 Å ×20,8Å e contém 296 moléculas de água.
As funções de distribuição radial (g(r)) para o compostos do Grupo 1 estão
apresentadas nas Figs. 4.19-4.22. Como dissemos na Fundamentação Teórica, esses
gráficos refletem a probabilidade de se encontrar determinados tipos de átomos
próximos um ao outro. Uma fdr, contudo, não fornece diretamente probabilidades
no sentido quantitativo, outrossim, é construída de forma que gxy(r)→ 1 à medida
que ∞→r (Cap. 2). Nas regiões em que os átomos x e y estão próximos, a fdr pode
diferir da unidade, e caso isso ocorra, teremos um indicador de estruturação no
líquido: máximos indicam regiões favoráveis para se encontrar átomos do tipo y, ao
passo que mínimos indicam diminuição da quantidade dos mesmos naquela região.
Seguiremos os passos seguintes para incorporar o efeito das interações
específicas nas barreiras rotacionais:
1) Análise dos aspectos qualitativos das fdrs para identificar as camadas de
solvatação, quando existentes;
2) Integração das fdrs para obter o número de moléculas de água nas camadas
de solvatação;

Resultados e Discussão 98
3) Cálculo da energia total de interação das moléculas de água da camada de
solvatação do soluto;
4) Cálculo das barreiras rotacionais incorporando as contribuições determi-
nadas no ítem anterior.
Vejamos, separadamente, os aspectos mais importantes das fdrs para os
compostos 1-4.
N,N-Dimetilcarbamato de O-Metila (1) (Fig. 4.19). As fdrs das metilas, tanto do
fragmento amídico como do estérico, apresentam bandas largas e com máximos
tipicamente em torno de 4 Å. Essa observação indica que as moléculas de água se
arranjam ao redor das metilas, mas não se prendem a elas por interações fortes – a
estruturação das moléculas de água ocasionada por esse tipo de interação é pouco
significante.10, 77 Por outro lado, vê-se claramente que as fdrs para o oxigênio
carbonílico, (C=)OAAAH(água), apresentam máximos agudos para o ef, o que indica
interações fortes e grande restrição geométrica. A distância entre os máximos das
fdrs com o oxigênio e o hidrogênio da água é 0,95 Å, aproximadamente o valor do
comprimento da ligação O-H utilizada no modelo TIP4P (0,9572 Å),12 o que mostra
que existe uma complexação eficiente entre o oxigênio carbonílico e as moléculas
de água. Observa-se ainda máximos pouco resolvidos nas fdrs envolvendo o carbono
carbonílico, mas trata-se de máximos em regiões relativamente distantes do
referido carbono. Ao compararmos, por exemplo, o máximo da fdr (O=)CAAAH(água)
com máximo da fdr (C=)OAAAH(água), vemos que a distância entre eles é de
aproximadamente 1,1 Å, um valor muito próximo ao do comprimento da ligação
C=O. Portanto, os máximos que aparecem na fdr (O=)CAAAH(água) resultam da
presença das moléculas complexadas com o oxigênio carbonílico. A ausência de
máximos nas fdrs do nitrogênio, juntamente com as observações acima, nos dão
bases para concluir que o oxigênio carbonílico é o único sítio de solvatação do ef do
composto 1.
O comportamento é um pouco diferente para os et, visto que ambos
apresentam máximos tanto para as fdrs do oxigênio carbonílico como para as do
nitrogênio. Como no caso do ef, há máximos nas fdrs do carbono carbonílico e a
explicação que expusemos anteriormente se aplica aqui. O fato de agora existirem

Resultados e Discussão 99
máximos nas fdrs no nitrogênio é uma simples conseqüência da mudança de sua
hibridização, de sp2 para sp3, pois o par de elétrons não se encontra mais envolvido
na conjugação com a carbonila e fica disponínel para a formação de ligações de
hidrogênio.
N,N-Dimetiltiocarbamato de S-Metila (2) (Fig. 4.20). As fdrs para 2 são, de forma
geral, semelhantes às de 1. A diferença mais acentuada é a intensidade dos
máximos que aparecem nos et em relação à intensidade dos máximos dos ef. Em 2,
o máximo do ef é mais pronunciado do que o do et, e de uma forma mais intensa
do que em 1, ou seja, devemos observar um efeito do solvente mais pronunciado
em 2.
N,N-Dimetiltiocarbamato de O-Metila (3) (Fig. 4.21). Neste composto, a diferença
essencial para os anteriores é a ausência de picos nas fdrs do ef, indicando que
nessa espécie não ocorre ligação de hidrogênio. De fato, as energias de
complexação para a água com a tiocarbonila (Tabela 4.18) são significativamente
menores do que para a carbonila. Quanto aos nitrogênios dos et, as fdrs indicam
ligações de hidrogênio em ambos. Dessa forma, 3 deve seguir um comportamento
oposto a 2, ou seja, como os et são estabilizados pelas ligações de hidrogênio, a
barreira deve diminuir em solventes próticos.
N,N-Dimetilditiocarbamato de Metila (4) (Fig. 4.22). O composto 4 segue o mesmo
comportamento de 3, com a diferença de que o pico para as fdrs do N são pouco
evidentes no et1. Também para esse composto devemos esperar uma diminuição
da barreira rotacional em solventes próticos.
Vemos, portanto, que em seus aspectos qualitativos as fdrs são coerentes com
as observações experimentais, excetuando-se o composto 3, para o qual não foi
possível realizar medidas em solventes próticos.

Resultados e Discussão 100
Figura 4.19. Funções de distribuição radial para o composto 1: a) ef; b) et1; c) et2).
a)
b)
c)

Resultados e Discussão 101
Figura 4.20. Funções de distribuição radial para o composto 2: a) ef; b) et1; c) et2).
a)
b)
c)

Resultados e Discussão 102
Figura 4.21. Funções de distribuição radial para o composto 3: a) ef; b) et1; c) et2).
a)
b)
c)

Resultados e Discussão 103
Figura 4.22. Funções de distribuição radial para o composto 4: a) ef; b) et1; c) et2).
a)
b)
c)

Resultados e Discussão 104
Podemos, agora, quantificar as ligações de hidrogênio. Como dissemos no
Cap. 2, a integração de um pico na fdr fornece o número médio de átomos, e por
conseguinte, de moléculas que se encontram complexados ao soluto. Com base nas
observações anteriores, realizamos a integração das fdrs para os compostos do
Grupo 1; os resultados estão apresentados na Tabela 4.19.
Tabela 4.19. Análise das funções de distribuição radial para os compostos 1-4
C=Z N Total ∆∆E e
Espécie N.C.a ∆E b N.C.a ∆E b N.C.c ∆E d
N,N-Dimetilcarbamato de Metila (1)
ef 1,69 (2,53) -10,21 − f − f 1,69 -10,21
et1 1,08 (2,43) -5,60 0,89 (2,63) -5,28 1,97 -10,88 -0,67
et2 1,12 (2,38) -5,50 0,92 (2,63) -5,41 2,04 -10,91 -0,70
N,N-Dimetiltiocarbamato de S-Metila (2)
ef 1,56 (2,48) -8,09 − f − f 1,56 -8,09
et1 0,93 (2,38) -4,24 0,44 (2,68) -1,97 1,37 -6,21 +1,88
et2 0,71 (2,18) -2,95 0,84 (2,68) -4,58 1,55 -7,53 +0,56
N,N-Dimetiltiocarbamato de O-Metila (3)
ef − f − f − f − f
et1 − f − f 0,92 (2,73) -4,72 0,92 -4,72 -4,72
et2 − f − f 1,01 (2,68) -5,71 1,01 -5,71 -5,71
N,N-Dimetilditiocarbamato de Metila (4)
ef − f − f − f − f
et1 − f − f 0,36 (2,73) -1,44 0,36 -1,44 -1,44
et2 − f − f 0,93 (2,68) -5,10 0,93 -5,10 -5,10
a Número de coordenação obtido pela integração das fdrs (C=)Z⋅⋅⋅H(água) ou N⋅⋅⋅H(água); os raios de corte são dados em parêntesis e em Å. b Energias de complexação soluto⋅⋅⋅água (kcal/mol) calculadas com base nos valores da Tabela 4.18 e nos números de coordenação. c Número de coordenação total obtido pela soma das contribuições dos dois sítios (C=)Z e N. d Energia total de interação soluto⋅⋅⋅água (kcal/mol) obtida pela soma das contribuições dos dois sítios (C=)Z e N. e Contribuição total das ligações de hidrogênio para as barreiras rotacionais (kcal/mol). f Nenhuma camada de solvatação identificada nas fdrs.
Para a obtenção das energias de complexação listadas na Tabela 4.19,
utilizamos os valores da Tabela 4.18. Assim, a complexação de uma molécula de
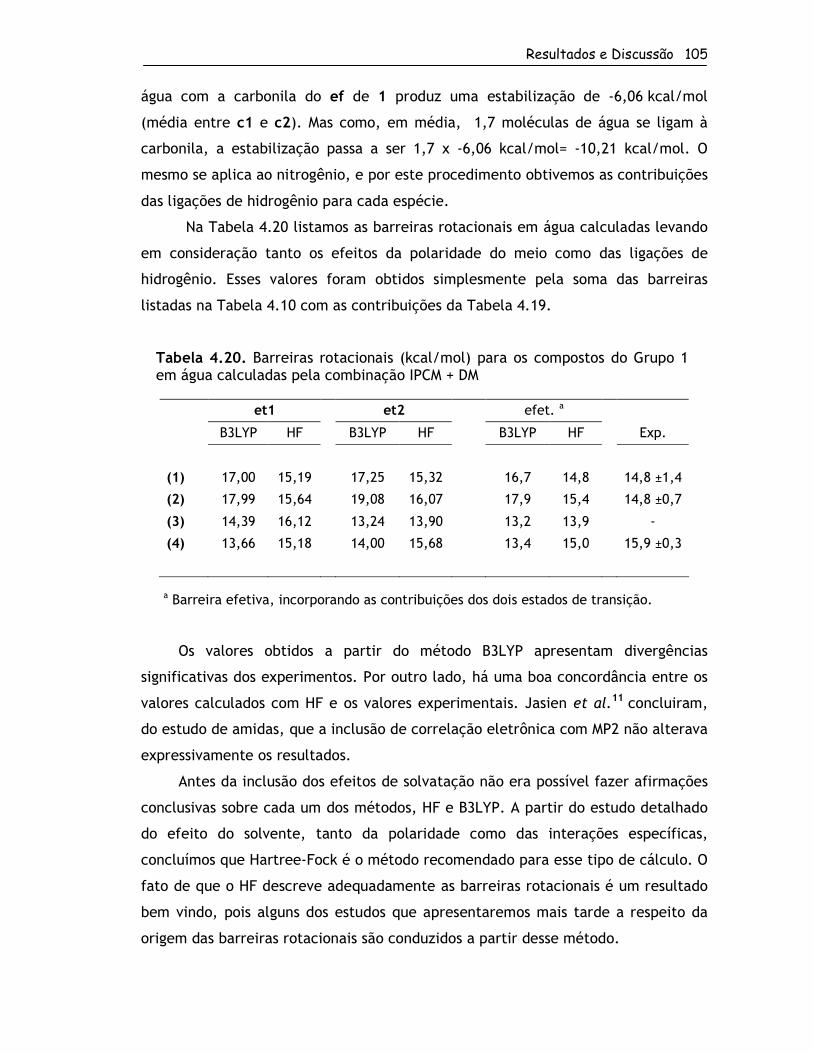
Resultados e Discussão 105
água com a carbonila do ef de 1 produz uma estabilização de -6,06 kcal/mol
(média entre c1 e c2). Mas como, em média, 1,7 moléculas de água se ligam à
carbonila, a estabilização passa a ser 1,7 x -6,06 kcal/mol= -10,21 kcal/mol. O
mesmo se aplica ao nitrogênio, e por este procedimento obtivemos as contribuições
das ligações de hidrogênio para cada espécie.
Na Tabela 4.20 listamos as barreiras rotacionais em água calculadas levando
em consideração tanto os efeitos da polaridade do meio como das ligações de
hidrogênio. Esses valores foram obtidos simplesmente pela soma das barreiras
listadas na Tabela 4.10 com as contribuições da Tabela 4.19.
Tabela 4.20. Barreiras rotacionais (kcal/mol) para os compostos do Grupo 1 em água calculadas pela combinação IPCM + DM
et1 et2 efet. a
B3LYP HF B3LYP HF B3LYP HF Exp.
(1) 17,00 15,19 17,25 15,32 16,7 14,8 14,8 ±1,4
(2) 17,99 15,64 19,08 16,07 17,9 15,4 14,8 ±0,7
(3) 14,39 16,12 13,24 13,90 13,2 13,9 -
(4) 13,66 15,18 14,00 15,68 13,4 15,0 15,9 ±0,3
a Barreira efetiva, incorporando as contribuições dos dois estados de transição.
Os valores obtidos a partir do método B3LYP apresentam divergências
significativas dos experimentos. Por outro lado, há uma boa concordância entre os
valores calculados com HF e os valores experimentais. Jasien et al.11 concluiram,
do estudo de amidas, que a inclusão de correlação eletrônica com MP2 não alterava
expressivamente os resultados.
Antes da inclusão dos efeitos de solvatação não era possível fazer afirmações
conclusivas sobre cada um dos métodos, HF e B3LYP. A partir do estudo detalhado
do efeito do solvente, tanto da polaridade como das interações específicas,
concluímos que Hartree-Fock é o método recomendado para esse tipo de cálculo. O
fato de que o HF descreve adequadamente as barreiras rotacionais é um resultado
bem vindo, pois alguns dos estudos que apresentaremos mais tarde a respeito da
origem das barreiras rotacionais são conduzidos a partir desse método.

Resultados e Discussão 106
Concentremos agora a nossa atenção ao comportamento de cada composto.
Exceto para 2, as barreiras rotacionais decrescem como conseqüência das ligações
de hidrogênio, tanto para o et1 como para o et2 (Tabela 4.20). Vale lembrar que
os casos relatados na literatura, até o período da redação desta tese, sempre
descreviam um comportamento oposto, i.e. aumento da barreira rotacional em
solventes próticos10, 19, 20, 89, 90. O efeito mais discreto ocorre em 1. Como
mencionado na análise das fdrs desse composto, os estados de transição são
protonados tanto no nitrogênio como no oxigênio carbonílico, ao passo que
somente o ef forma ligações de hidrogênio neste último sítio. Mesmo sendo a
interação C=O⋅⋅⋅H-O-H mais forte no ef, a soma das interações C=O⋅⋅⋅H-O-H e
N⋅⋅⋅H-O-H acaba estabilizando mais os et como mostram os dados da Tabela 4.19.
Notemos ainda que esse comportamento é coerente com o modelo de ressonância
para os carbamatos, ou seja, a ressonância no ef aumenta a carga negativa no
oxigênio carbonílico, tornando-o um doador de carga melhor, mas ao mesmo tempo
diminui a carga negativa no nitrogênio e desfavorece a protonação neste ponto.
Nos et, o efeito de ressonância desaparece e diminui a quantidade de carga no
oxigênio, não obstante, agora o nitrogênio também é um sítio favorável para
formação de ligações de hidrogênio:
ef et
No composto 2 os efeitos são qualitativamente parecidos a 1, exceto que o
nitrogênio não é um doador de carga tão bom. Para mostrá-lo, observemos
primeiramente as cargas de Mulliken no nitrogênio. No et1 de 1 há -0.326 e em
O
N
CH3
CH3
O
H3C
H
O
H
O
NO
H3C
H3CCH3
H
O
H
O
H
H

Resultados e Discussão 107
excesso contra apenas -0.058 e em 2 [HF/6-311+G(2d,p)]. Para o et2, os valores
são -0.325 e no caso de 1 e -0.159 e para 2. As cargas ajustadas para DM na Tabela
4.17 indicam a mesma tendência, embora com valores diferentes. Some-se a isso
um outro fator, no et1 de 2 o átomo de enxofre, em razão de seu volume,
atrapalha a aproximação da molécula de água como conseqüência de uma maior
interação estérica. Portanto, no et1 do composto 2 há dois efeitos – carga menor e
repulsão provocada pelo enxofre – ao passo que no et2 tem-se apenas a diminuição
de carga em relação ao composto 1. Considerando esses fatores, o et1 é
desfavorecido em relação ao et2 na formação de ligações de hidrogênio, o que fica
evidente na Tabela 4.19, e ambos são desfavorecidos em relação aos et de 1.
Consideremos agora os compostos 3 e 4. Para ambos a barreira rotacional
diminui, mas diminui de forma mais acentuada em 3. Nesses compostos, não há
formação de ligação de hidrogênio na região da ligação dupla, fato que pode ser
racionalizado pelo modelo das durezas de ácidos e bases.153 O hidrogênio é um
ácido de Lewis duro e, portanto, irá interagir de forma efetiva com uma base de
Lewis dura, como o oxigênio. O enxofre, por sua vez, é uma base mole, e, sendo
assim, interage de forma fraca com o hidrogênio. Desta forma, somente os et serão
estabilizados por ligações de hidrogênio e isso causa a diminuição das barreiras. O
segundo ponto de que devemos considerar é o porquê da diferença entre 3 e 4.
Essencialmente, em 3 os dois et são estabilizados de forma comparável um
ao outro (Tabela 4.19), enquanto que em 4 a estabilização do et1 é
expressivamente mais fraca do que a do et2. Os motivos dessa diferença podem ser
entendidos por uma explicação similar à utilizada para 2, baseada em interações
estéricas ocasionadas pelo átomo de enxofre. Como no caso anterior, o volumoso
átomo de enxofre atrapalha a aproximação da molécula de água no et1 de 4, mas
não de 3. Todavia, em 4 há dois átomos de enxofre e seria esperado, pelo menos
em princípio, que também o et2 experimentasse um efeito parecido ao do et1. A
ausência deste efeito pode ser entendida com base em alguns aspectos estruturais
dos dois estados de transição. Para tanto, consideremos uma ligação O-H na qual o
átomo de hidrogênio situa-se à 2 Å do nitrogênio e a ligação O-H orienta-se de
forma a contituir um tetraedro, na direção do par de elétrons do nitrogênio
(Fig. 4.23). Como o ângulo ∠S-C-N no et1 é menor do que o ângulo ∠S=C-N no et2

Resultados e Discussão 108
(104,2 o contra 124,4 o), o oxigênio da água estará mais próximo do enxofre no et1,
o que faz com que a repulsão estérica seja maior neste.
Figura 4.23. Diferenças estruturais para complexos de água com et1 e et2 do composto 4.
Resumindo, embora os efeitos das ligações de hidrogênio incluídos através de
DM possam diferir levemente dos efeitos experimentais, o método que aplicamos
prevê a tendência correta das barreiras rotacionais em água e nos permite
entender, com detalhes, quais aspectos da solvatação de cada espécie determina
este comportamento.
4.2.3.2. Grupo 2
N,N,N-Trimetiluréia e N,N,N’-Trimetiltiouréia
Nas simulações de líquidos realizadas com esses compostos, tendo em vista os
resultados experimentais, utilizamos o metanol como solvente. A determinação das
funções de potencial de interação entre moléculas de 5/6 e metanol foi conduzida
de forma similar à dos compostos do Grupo 1, exceto que agora não realizamos
otimização das distâncias entre as moléculas de soluto e solvente. Procedemos
assim fundamentados no princípio de que a informação essencial necessária ao
S
NS
H
OS
NS
H
O
104,4o
124,4o
3,213 Å
2,930 Å
et1 et2

Resultados e Discussão 109
processo de parametrização é a energia de complexo em função das coordenadas
espaciais dos átomos, não havendo necessidade de se trabalhar exatamente num
mínimo de energia. De fato, durante a simulação de dinâmica molecular não é
realizado qualquer processo que envolva a otimização de geometrias – calcula-se as
forças atuantes entre as moléculas e determina-se assim o seu movimento. O ponto
negativo deste processo é o fato de que, se a estrutura do complexo estiver muito
distante de um mínimo, a utilização do mesmo para estimar a energia de
complexação na etapa seguinte pode prejudicar os resultados. Porém, notamos que
para a maioria dos casos anteriores a distância típica de uma ligação de hidrogênio
é de 2 Å. Assim, adotamos esse valor como padrão.
Os complexos utilizados para a parametrização estão apresentados na
Fig. 4.24. Note que foram empregados sete complexos, e não seis como antes, em
razão do hidrogênio adicional dos compostos 5 e 6.
Existem diversos estudos na literatura utilizando dinâmica molecular para
uréia e para a tetrametiluréia, mas todos eles tratam apenas do estado
fundamental dessas moléculas. Como vimos anteriormente, há diferenças
significativas entre os parâmetros para os estados fundamentais e os estados de
transição. Mesmo assim, poderíamos, em princípio, utilizar os parâmetros da
literatura apenas para os estados fundamentais.123-127 Procedemos dessa forma,
mas não obtivemos resultados satisfatórios para as energias de complexação entre
os solutos e o metanol. Por conta disso, fomos novamente compelidos ao processo
de ajuste utilizado no Grupo 1, ou seja, utilizamos os parâmetros de van der Waals
da literatura e variamos as cargas atômicas. O modelo que adotamos para 5 e 6
consiste de 8 pontos e os parâmetros ajustados estão apresentados na Tabela 4.21,
enquanto as energias de complexação estão compiladas na Tabela 4.22.
De forma geral, o campo de forças reproduz satisfatoriamente os resultados
TFD, com ligeiras exceções aos complexos com as metilas de 5. Nesses casos, o
campo de forças sempre superestima os valores B3LYP. Contudo, como pode ser
visto nas fdrs para os compostos do Grupo 1, quando se tem energias de
complexação menores do que c.a. 4 kcal/mol, observa-se apenas interações do tipo
dipolar. As regiões de maior interesse da molécula, isto é, aquelas onde
potencialmente poderíamos observar ligações de hidrogênio, são bem descritas.

Resultados e Discussão 110
ef et1 et2
c1
c2
c3
c4
c5
c6
c7
Figura 4.24. Complexos utilizados na parametrização dos campos de forças para os compostos 5 e 6.

Resultados e Discussão 111
Tabela 4.21. Parâmetros das funções de potencial para os compostos 5 e 6 a
aParâmetros de Lennard-Jones extraídos das Refs. 123-127.
Nas Figs. 4.25 e 4.26 apresentamos as fdrs para 5 e 6, respectivamente.
Analisemos primeiramente o composto 5. Os picos indicativos de ligações de
hidrogênio aparecem de forma clara nas fdrs (C=)O⋅⋅⋅H(água) e (C=)O⋅⋅⋅O(água)
tanto para o estado fundamental como para os estados de transição. Vê-se,
contudo, que o pico da fdr é acentuadamente mais intenso no ef, o que sugere que
este último é estabilizado por ligações de hidrogênio e que estas causam um
aumento na barreira rotacional. Essa é a conclusão mais importante a respeito das
fdrs da Fig. 4.25, mas alguns outros aspectos chamam a atenção. Primeiramente,
Átomo σ (Å) ε (kcal/mol) q(ef2) (e) q(et1-a) (e) q(et2-a) (e)
N,N,N’-Trimetiluréia (5)
N1 3,250 0,1699 -0,485 -0,030 0,060
C2 3,7538 0,1280 0,143 0,143 0,123
C3 3,750 0,1050 0,853 0,283 0,303
H4 0,000 0,000 0,136 0,092 0,012
O5 2,960 0,210 -0,555 -0,376 -0,376
N6 3,250 0,1270 -0,296 -0,412 -0,442
C78 3,7538 0,170 0,102 0,150 0,160
N,N,N’-Trimetiltiouréia (6)
N1 3,250 0,1699 -0,100 0,255 0,249
C2 3,7538 0,1280 0,000 0,010 0,000
C3 3,750 0,1050 -0,024 0,041 0,016
H4 0,000 0,000 0,120 -0,022 0,090
S5 3,550 0,250 -0,060 0,041 0,085
N6 3,250 0,1270 0,084 -0,209 -0,780
C78 3,7538 0,128 -0,010 -0,058 0,170
Z5
C3
N1 N6
C78
C78
C2
H4

Resultados e Discussão 112
os nitrogênios de 5 não são bons receptores de prótons; exceto pelo pequeno
“ombro” que aparece na fdr [(CH3)2]N⋅⋅⋅ H(água) do et2-a, não há indícios de
ligações de hidrogênio tanto no ef como no et1. Além disso, os máximos dos picos
de ligações de hidrogênio normalmente situam-se em 2 Å, mas no caso da referida
fdr do et2-a, a saliência inicial é pouquíssimo intensa nessa região e ganha
intensidade na medida em que r aumenta. O nitrogênio secundário deveria, de
fato, comportar-se desta maneira, uma vez que é quase planar tanto no ef como
nos et. Mas o nitrogênio terciário, que muda bastante sua hibridização durante a
rotação, deveria, pelo menos em princípio, ser uma boa base de Lewis.
Tabela 4.22. Energias de complexação (kcal/mol) soluto-água calculadas no nível B3LYP/6-31+G(d,p) e através do campo de forças clássico para 5 e 6
c1 c2 c3 c4 c5 c6 c7
N,N,N’-Trimetiluréia (5)
TFD −6,31 −5,93 −1,63 −2,96 −0,59 +0,08 −2,21
ef2 C. Forças −6,23 −6,27 −1,47 −2,69 −0,87 −0,71 −2,58
TFD −5,67 −5,48 −3,91 +2,33 −0,34 −0,19 −2,11
et1-a C. Forças −5,83 −5,45 −3,37 +2,05 −0,88 −0,55 −2,50
TFD −5,74 −5,24 −4,62 +1,44 −0,66 −0,22 −1,67
et2-a C. Forças −5,94 −5,06 −4,21 +1,15 −1,47 −0,72 −1,91
N,N,N’-Trimetiltiouréia (6)
TFD +0,51 +0,54 −1,01 −1,66 −0,50 −0,30 +0,58 ef2
C. Forças +0,22 +0,22 −1,45 −2,20 −0,28 −0,27 +1,52
TFD +1,54 +1,86 −3,11 +2,91 −0,91 −0,49 +1,10 et1-b
C. Forças +1,44 +2,30 −3,07 +3,02 −1,32 −0,23 +0,46
TFD +1,54 +2,47 −4,43 +4,51 −1,09 −0,12 −2,33 et2-b
C. Forcas +1,35 +2,45 −4,37 +3,86 −0,99 +0,02 −2,13
Alguns aspectos estruturais explicam a baixa tendência em formar ligações de
hidrogênio na região do nitrogênio terciário de 5. Tomemos para comparação o
composto 1. No et2 desse composto, a média dos ângulos ∠C-N-C é 112,8 o, ao
passo que no et2 de 5 esse valor é 114,0 o. Tais valores demonstram que o

Resultados e Discussão 113
nitrogênio terciário em 5 é piramidalizado de forma menos intensa. Segundo
cálculos NBO (próxima seção), o par de elétrons do nitrogênio no et2 de 1 possui
86,96% de caráter p, enquanto que em 5 o valor é 89,01%. Em outras palavras, o
nitrogênio em 1 aproxima-se mais de uma hibridização sp3 do que em 5, e
portanto, há uma maior densidade de probabilidade na região externa do topo da
pirâmide >N- em 1. No composto 5 a densidade de probabilidade se distribui mais
entre as duas faces do N e deixa a face voltada para o metanol com menor
densidade.
Outro aspecto intrigante das fdrs de 5 é a diferença de intensidade das fdrs
envolvendo o hidrogênio e o oxigênio do metanol. A intensidade do pico da fdr
(C=)O⋅⋅⋅H(MeOH) é aproximadamente duas vezes a do pico da frd (C=)O⋅⋅⋅O(MeOH).
Lembremos, porém, que para obter o número de coordenação não integramos a fdr
diretamente, mas sim a função gij(r)ρj4pr2dr, na qual ρj é a densidade numérica de
átomos j (Cap. 2). Quando propriamente integradas, as duas fdrs acima fornecem o
mesmo número de coordenação. Por exemplo, para o ef o valor é 0,83 no caso da
frd (C=)O⋅⋅⋅H(MeOH) e 0,85 para a fdr (C=)O⋅⋅⋅O(MeOH). Por fim, observa-se picos de
solvatação nas fdrs do carbono carbonílico. Esse efeito, como explicado para o caso
dos compostos do Grupo 1, resulta presença de moléculas de metanol em outras
regiões de solvatação do composto. No caso do ef, é a solvatação do oxigênio
carbonílico que causa o aparecimento do sinal na fdr do carbono.
Nas fdrs de 6 (Fig. 2.26), não há picos de solvatação para o enxofre, efeito
similar ao dos compostos 3 e 4. A solvatação é evidenciada somente nas fdrs do
nitrogênio terciário do et2-b. Lembremos, contudo, que os cálculos de estrutura
eletrônica mostram que a barreira para o et2-b é expressivamente maior do que a
do et1-b (10 kcal/mol contra 15 kcal/mol, Tabela 4.14). Desta forma, a maior
contribuição para a barreira rotacional de 6 provém do et1-b, que não é afetado
pelas ligações de hidrogênio assim como o ef2. A ligações de hidrogênio no et2-b,
que o estabilizam, farão com que sua contribuição para o processo de rotação
aumente e, sendo assim, a constante de velocidade para o et2-b passará a ter
importância perto da constante para o et1-b. Como consequência, a barreira
rotacional deve sofrer uma pequena diminuição.

Resultados e Discussão 114
Figura 4.25. Funções de distribuição radial para a N,N,N’-Trimetiluréia (5).

Resultados e Discussão 115
Figura 4.26. Funções de distribuição radial para a N,N,N’-Trimetiltiouréia (6).

Resultados e Discussão 116
Com base nas observações acima, realizamos a integração dos picos das fdr
para o oxigênio de 5 e os nitrogênios terciários de 5 e 6, Tabela 4.23. Na Tabela
4.24 apresentamos as barreiras rotacionais calculadas levando-se em consideração
o efeito das ligações de hidrogênio. Os valores calculados para 5, em ambos os
níveis utilizados, são coerentes com a medida experimental, embora difiram por
1 kcal/mol entre si. Por outro lado, a barreira para 6 fica signicativamente abaixo
do valor experimental nos dois níveis de teoria e chega a diferir por 2 kcal/mol no
HF.
Os valores IPCM predizem uma barreira rotacional maior para 6, ao contrário
dos experimentos. Sendo assim, esperaríamos realmente que a inclusão das
ligações de hidrogênio contribuisse para a diminuição da barreira de 6, como de
fato se observou. Esse efeito parece ter sido excessivamente acentuado em nossos
cálculos, mas o resultado qualitativo é coerente com as observações experimen-
tais. Ao compararmos 6 com os dois casos do Grupo 1 para os quais também
observamos diminuição das barreiras, 3 e 4, parece-nos sensata a conclusão de que
compostos amídicos que possuam um átomo de enxofre na tiocarbonila terão suas
barreiras rotacionais diminuídas frente a solventes próticos, algo até o presente
não apontado na literatura.* A causa principal é a baixa afinidade do enxofre por
prótons. Como mencionado na discussão do Grupo 1, no estado fundamental o
nitrogênio terciário adota uma configuração aproximada-mente planar e não forma
ligações de hidrogênio, de forma que somente o calcogênio da ligação dupla
poderia servir como sítio de complexação. Isso ocorre na carbonila, mas não na
tiocarbonila e assim, neste último caso, não se formam ligações de hidrogênio no
estado fundamental. Nos estados de transição, o nitrogênio assume uma
configuração piramidalizada e passa a ser um bom receptor de prótons. Portanto,
as ligações de hidrogênio nos et acabam por estabilizá-los em relação ao ef
fazendo com que a barreira rotacional diminua.
Como pode ser visto, também para o caso de 5 e 6 a combinação IPCM + DM
mostra-se efetiva para o estudo das barreiras rotacionais. Refinamentos do
método, como um maior número de complexos para a parametrização e
otimizações de geometria, poderiam corrigir as divergência observadas para 6.
* Os dados experimentais de Wiberg e Rush 20. Wiberg, K. B.; Rush, D. J., J. Am. Chem. Soc. 2001, 123, 2038-2046.) para a DMTF e a DMTA sugerem a ausência de complexação.

Resultados e Discussão 117
Tabela 4.23. Análise das funções de distribuição radial para a 5 e 6
C=Y N Total ∆∆E e
Espécie N.C.a ∆E b N.C.a ∆E b N.C.c ∆E d
N,N,N’-Trimetiluréia (5)
ef2 0,86 (2,73) -5,26 − f − f 0,86 -5,26
et1-a 0,35 (2,78) -1,95 − f − f 0,35 -1,95 +3,31
et2-a 0,74 (2,88) -4,06 0,09 (2,53) -0,42 0,83 -4,48 +0,78
N,N,N’-Trimetiltiouréia (6)
ef2 − f − f − f − f 0,00 0,00
et1-b − f − f − f − f 0,00 0,00 0,00
et2-b − f − f 0,88 (2,83) -3,90 0,88 -3,90 -3,90
a Número de coordenação obtido pela integração das fdrs (C=)Z⋅⋅⋅H(MeOH) or N⋅⋅⋅H(MeOH); os raios de corte são dados em parêntesis e em Å. b Energias de interação soluto⋅⋅⋅MeOH (kcal/mol) calculadas com base nos valores da Tab. 5 e nos números de coordenação. c Número de coordenação total obtido pela soma das contribuições dos dois sítios (C=)Z e N. d Energia total de interação soluto⋅⋅⋅MeOH (kcal/mol) obtida pela soma das contribuições dos dois sítios (C=)Z e N. e Contribuição total das ligações de hidrogênio para as barreiras rotacionais (kcal/mol). f Nenhuma camada de solvatação identificada nas fdrs. Tabela 4.24. Barreiras rotacionais (kcal/mol) para 5 e 6 em metanol, calculadas pela combinação IPCM + DM
et1 et2 efet. a
B3LYP HF B3LYP HF B3LYP HF Exp.
(5) 12,56 11,16 12,18 11,37 11,9 10,9 11,3 ±0,6
(6) 10,91 10,75 9,34 8,51 9,30 8,5 10,5 ±0,3
a Barreira efetiva, incorporando a contribuição dos dois estados de transição.

Resultados e Discussão 118
N,N,N’,N’-Tetrametiluréia e N,N,N’,N’-Tetrametiltiouréia
Para estes dois compostos não realizamos parametrizações como nos casos
anteriores. Assumindo um comportamento similar, utilizamos os resultados de 5 e
6. Sendo assim, as contribuições para as barreiras rotacionais de 7 e 8 devida às
ligações de hidrogênio foram obtidas a partir da Tabela 4.23. Tendo em vista que
apenas o et1 participa da rotação desses últimos compostos, a barreira de 7 deve
ser acrescida por aproximadamente 3,31 kcal/mol, enquanto que a barreira de 8
não deve sofrer variação alguma por conta de ligações de hidrogênio. Na
Tabela 4.25 listamos os resultados assim obtidos.
Tabela 4.25. Barreiras rotacionais (kcal/mol) para 7 e 8 em metanol calculadas pela combinação IPCM + DM
et1 B3LYP HF
(7) 9,13 8,02 (8) 4,38 4,26
A barreira para 8 está definitivamente fora do alcance da RMND para
solventes cujo ponto de fusão seja próximo de -100 oC. Por outro lado, o menor
valor que determinamos experimentalmente para os compostos do Grupo 1 foi a
barreira em CD2Cl2 de 6, viz. 9,8 kcal/mol, o que sugere que a barreira de 7
poderia também ser vefificada nas mesmas condições. Precisamos, contudo, levar
em conta que a obtenção de espectros com separação de sinais depende não
apenas da magnitude da barreira, mas também do quão quimicamente
inequivalentes são as metilas amídicas. Lembremos que as metilas de 7 estão
“torcidas” em relação às de 5 e 6 (ver Fig. 4.12), e isso impõe uma condição
diferente. Se as freqüências de absorção das metilas fossem muito próximas, não
ocorreria o fenômeno de descoalescência, i.e. a observação de dois sinais distintos,
nas temperaturas acessíveis aos nossos experimentos.

Resultados e Discussão 119
Com os comentários da página anterior, finalizamos o estudo dos efeitos de
solvatação sobre as barreiras rotacionais. De forma geral, verificamos que todos os
compostos têm suas barreiras modificadas pelo solvente, em menor ou em maior
grau. Em alguns casos, porém, as variações são pequenas demais para serem
observadas experimentalmente, como ocorre com os carbamatos. As ligações de
hidrogênio podem tanto aumentar como diminuir as barreiras, e destaquemos aqui
que diminuições das barreiras pelas ligações de hidrogênio é algo até então não
relatado na literatura.
Na seção seguinte, buscaremos entender as causas das barreiras rotacionais
para cada composto em fase gasosa.

Resultados e Discussão 120
4.2.4 Origem das Barreiras Rotacionais
4.2.4.1 Grupo 1
Contribuições Energéticas para as Barreiras Rotacionais
Segundo a teoria de ligação de valência, os compostos do Grupo 1 podem ser
representados pelas seguintes estruturas de ressonância em seu estado
fundamental:
X=O,S Z=O,S
Figura 4.27. Estruturas de ressonância para os compostos do Grupo 1.
A rigor, o número de estruturas de ressonância para sistemas da
complexidade dos estudados aqui é substancialmente maior. Nos restringiremos,
contudo, às quatro acima, de vez que estão mais diretamente ligadas à barreira
rotacional e devem possuir os maiores pesos. A rotação sobre a ligação C-N, que
produz os estados de transição, elimina a estrutura c e causa o aumento da energia
molecular. Essa é a explicação mais popular para a origem das barreiras
rotacionais, como dissemos na Fundamentação Teórica. Entretanto, esse modelo
diz respeito somente ao efeito de deslocalização eletrônica, e assim assume que a
contribuição das repulsões não-ligantes e as modificações intra-ligações são
desprezíveis. Como veremos a seguir, a deslocalização eletrônica realmente tem
um papel fundamental na formação das barreiras, mas as energias de desloca-
lização, puramente, não são suficientes para explicar as magnitude relativas dessas
barreiras entre os compostos.
O principal método que adotaremos para esta parte do estudo é o dos orbitais
naturais de ligação (NBO), descrito no Capítulo 2. Além dele, faremos uso do
método desenvolvido por Mayer137 que permite a partição da energia molecular em
C
Z
X N
C
Z
X N
C
Z
X N
C
Z
X N
a b c d

Resultados e Discussão 121
componentes de um e dois centros e nos dá uma informação da qual o NBO não
trata, a saber, a energia de interação eletrostática entre átomos que não estão
ligados diretamente. Antes, porém, uma questão de validação. Por limitações
físicas do computador no qual rodamos os cálculos desta seção, tivemos que
ulitizar um conjunto de bases menor do que o empregado para as otimizações de
geometria. Adotamos, assim, o conjunto PTZV, um conjunto com a valência
dividida em três grupos, à semelhança do 6-311, e com funções de polarização d
para os átomos pesados e p para o hidrogênio. Para nos certificar de que os
resultados fornecidos pelo método HF/PTZV são apropriados, rodamos cálculos de
ponto único com as estruturas otimizadas em HF/6-311+G(2d,p), ou seja,
HF/PTZV//HF/6-311+G(2d,p)); os resultados estão apresentados na Tabela 4.26. Ao
longo desses cálculos, trabalhamos com ef2 para o composto 3. É importante
mencionar que esses valores não incluem as correções térmicas para os movimentos
moleculares; correspondem apenas às contribuições da energia total (eletrônica +
repulsão nuclear). Procedemos deste modo porque os cálculos NBO tratam apenas
dessa energia.
Tabela 4.26. Barreiras rotacionais para 1-4 calculadas em diferentes níveis a
HF/6-311+G(2d,p) HF/PTZV//HF/6-311+G(2d,p)
et1 et2 efet. b et1 et2 efet. b
(1) 13,75 14,86 13,67 14,42 15,21 14,28
(2) 10,69 14,57 10,69 11,07 14,60 11,07
(3) 16,25 17,35 16,16 17,10 18,12 17,00
(4) 12,29 16,68 12,29 13,12 17,36 13,12
a kcal/mol. b Barreira efetiva, incorporando a contribuição dos dois estados de transição.
Apesar das barreiras rotacionais fornecidas pelo método HF/PTZV//HF/6-
311+G(2d,p) ficarem, em média, 0,6 kcal/mol acima daquelas calculadas com
HF/6-311+G(2d,p), os valores são satisfatórios, pois as magnitudes relativas são
mantidas.
Seguindo o exemplo de Weinhold,66 poderíamos dividir a barreira rotacional,
∆E, da seguinte forma:
DE = DELewis + DEdesl (4.7)

Resultados e Discussão 122
na qual ELewis é a energia de uma estrutura de Lewis idealizada, isto é, sem
deslocalização. Na Tabela 4.27 listamos esses valores.
Tabela 4.27. Decomposição de energia para as barreiras rotacionais de 1-4 a,b
(1) (2) (3) (4)
et1 et2 et1 et2 et1 et2 et1 et2
∆ELewis -17,09 -20,56 -26,02 -25,42 -25,80 -32,68 -35,59 -38,26
∆Edesl 31,50 35,77 37,09 40,02 42,89 50,80 48,70 55,61
∆E 14,42 15,21 11,07 14,60 17,10 18,12 13,12 17,36
a HF/PTZV//HF/6-311+G(2d,p). b Em kcal/mol.
Vemos que, de fato, a deslocalização é o principal componente das barreiras,
e além disso, que a estrutura de Lewis dos et é mais estável do que a dos ef (o
valor de ∆ELewis é negativo). Entretanto, os valores relativos das energias de desloca-
lização não são condizentes com as barreiras calculadas, isto é, não se observa uma
correlação linear entre ambos. Por exemplo, tanto 4 como 2 possuem maior DEdesl
do que 1, mas a barreira em fase gasosa deste último é a maior. É preciso,
portanto, entender o que acontece nas estruturas de Lewis com respeito às
interações não-ligantes e às contribuições intramoleculares. Comecemos pelas
interações não-ligantes, compiladas na Tabela 4.28.
À exceção do et1 de 4, para todas as demais espécies as interações não-
ligantes desestabilizam o ef em relação aos et. Isso faz sentido, pois nos ef as
ligações N-CH3 ficam eclipsadas com a dupla e com a C-X (Fig. 4.6 e 4.7), ao passo
que nos et o arranjo é alternado. O fato da contribuição eletrostática dos et de 4
não seguir essa tendência deve-se provavelmente ao maior volume da nuvem
eletrônica dessa molécula. Assim, no ef tem-se uma metila interagindo com cada
ligação, C-S ou C=S, enquanto que em cada et tem-se duas metilas interagindo com
uma ligação e ainda um par de elétrons interagindo com a ligação oposta. Este
efeito também está presente nos outros compostos, mas, por serem suas nuvens
eletrônicas mais compactas, as interações entre as ligações eclipsadas nos ef
acabam sendo mais importantes.
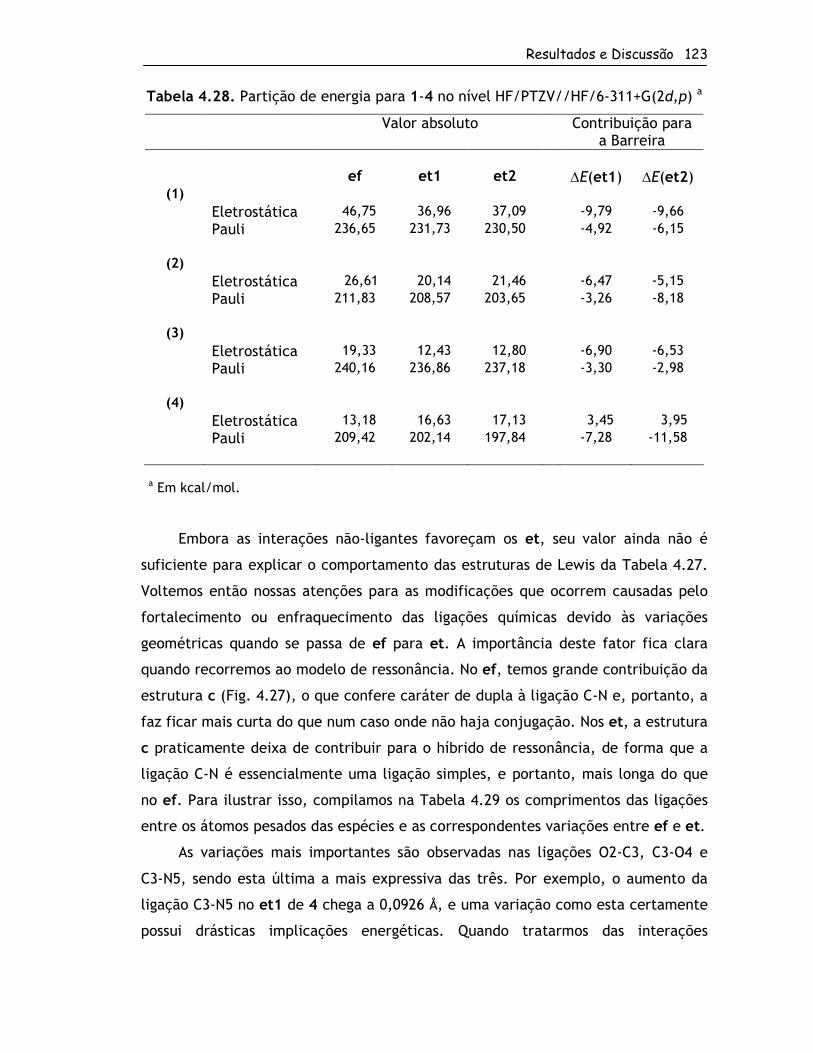
Resultados e Discussão 123
Tabela 4.28. Partição de energia para 1-4 no nível HF/PTZV//HF/6-311+G(2d,p) a
Valor absoluto Contribuição para a Barreira
ef et1 et2 ∆E(et1) ∆E(et2) (1) Eletrostática 46,75 36,96 37,09 -9,79 -9,66 Pauli 236,65 231,73 230,50 -4,92 -6,15
(2) Eletrostática 26,61 20,14 21,46 -6,47 -5,15 Pauli 211,83 208,57 203,65 -3,26 -8,18
(3) Eletrostática 19,33 12,43 12,80 -6,90 -6,53 Pauli 240,16 236,86 237,18 -3,30 -2,98
(4) Eletrostática 13,18 16,63 17,13 3,45 3,95 Pauli 209,42 202,14 197,84 -7,28 -11,58
a Em kcal/mol.
Embora as interações não-ligantes favoreçam os et, seu valor ainda não é
suficiente para explicar o comportamento das estruturas de Lewis da Tabela 4.27.
Voltemos então nossas atenções para as modificações que ocorrem causadas pelo
fortalecimento ou enfraquecimento das ligações químicas devido às variações
geométricas quando se passa de ef para et. A importância deste fator fica clara
quando recorremos ao modelo de ressonância. No ef, temos grande contribuição da
estrutura c (Fig. 4.27), o que confere caráter de dupla à ligação C-N e, portanto, a
faz ficar mais curta do que num caso onde não haja conjugação. Nos et, a estrutura
c praticamente deixa de contribuir para o híbrido de ressonância, de forma que a
ligação C-N é essencialmente uma ligação simples, e portanto, mais longa do que
no ef. Para ilustrar isso, compilamos na Tabela 4.29 os comprimentos das ligações
entre os átomos pesados das espécies e as correspondentes variações entre ef e et.
As variações mais importantes são observadas nas ligações O2-C3, C3-O4 e
C3-N5, sendo esta última a mais expressiva das três. Por exemplo, o aumento da
ligação C3-N5 no et1 de 4 chega a 0,0926 Å, e uma variação como esta certamente
possui drásticas implicações energéticas. Quando tratarmos das interações
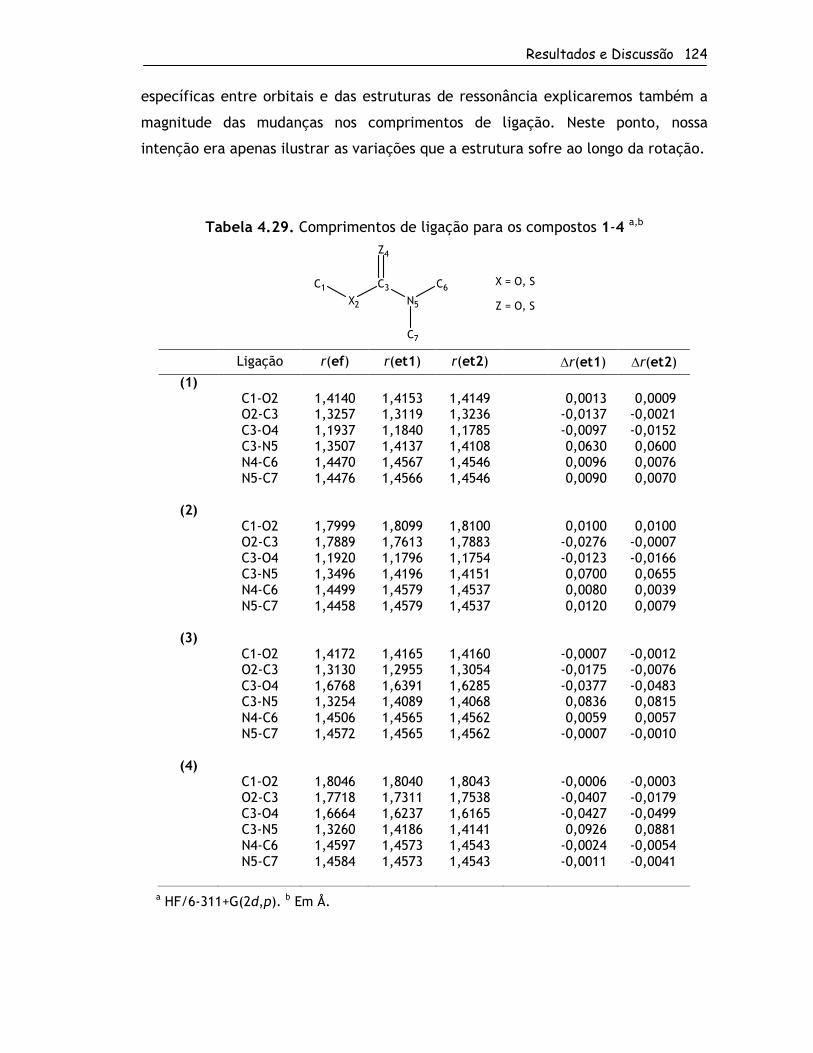
Resultados e Discussão 124
específicas entre orbitais e das estruturas de ressonância explicaremos também a
magnitude das mudanças nos comprimentos de ligação. Neste ponto, nossa
intenção era apenas ilustrar as variações que a estrutura sofre ao longo da rotação.
Tabela 4.29. Comprimentos de ligação para os compostos 1-4 a,b
Ligação r(ef) r(et1) r(et2) ∆r(et1) ∆r(et2)
(1) C1-O2 1,4140 1,4153 1,4149 0,0013 0,0009 O2-C3 1,3257 1,3119 1,3236 -0,0137 -0,0021 C3-O4 1,1937 1,1840 1,1785 -0,0097 -0,0152 C3-N5 1,3507 1,4137 1,4108 0,0630 0,0600 N4-C6 1,4470 1,4567 1,4546 0,0096 0,0076 N5-C7 1,4476 1,4566 1,4546 0,0090 0,0070
(2) C1-O2 1,7999 1,8099 1,8100 0,0100 0,0100 O2-C3 1,7889 1,7613 1,7883 -0,0276 -0,0007 C3-O4 1,1920 1,1796 1,1754 -0,0123 -0,0166 C3-N5 1,3496 1,4196 1,4151 0,0700 0,0655 N4-C6 1,4499 1,4579 1,4537 0,0080 0,0039 N5-C7 1,4458 1,4579 1,4537 0,0120 0,0079
(3) C1-O2 1,4172 1,4165 1,4160 -0,0007 -0,0012 O2-C3 1,3130 1,2955 1,3054 -0,0175 -0,0076 C3-O4 1,6768 1,6391 1,6285 -0,0377 -0,0483 C3-N5 1,3254 1,4089 1,4068 0,0836 0,0815 N4-C6 1,4506 1,4565 1,4562 0,0059 0,0057 N5-C7 1,4572 1,4565 1,4562 -0,0007 -0,0010
(4) C1-O2 1,8046 1,8040 1,8043 -0,0006 -0,0003 O2-C3 1,7718 1,7311 1,7538 -0,0407 -0,0179 C3-O4 1,6664 1,6237 1,6165 -0,0427 -0,0499 C3-N5 1,3260 1,4186 1,4141 0,0926 0,0881 N4-C6 1,4597 1,4573 1,4543 -0,0024 -0,0054 N5-C7 1,4584 1,4573 1,4543 -0,0011 -0,0041
a HF/6-311+G(2d,p). b Em Å.
C1
X2
C3
Z4
N5
C6
C7
X = O, S
Z = O, S

Resultados e Discussão 125
O método NBO fornece uma maneira de avaliar a estabilidade de uma ligação
química através dos autovalores de energia dos orbitais de ligação. Na Tabela 4.30
apresentamos esses valores, tanto para os orbitais ligantes localizados entre dois
átomos como para os pares de elétrons isolados. As energias apresentadas nessa
tabela levam em conta a população eletrônica em cada orbital, ρi, ou seja, são o
produto do autovalor do orbital, εi, por essa população, E = ρiei, de modo que as
variações das energias dos orbitais ligantes são dadas por:
DE = ρe te e t < ρe fee f
A identificação “caroço” que aparece na Tabela 4.30 diz respeito aos elétrons
internos de cada átomo, que não participam diretamente nas ligações químicas mas
que também são afetados pelas variações estruturais.
A forma como as energias dos NBO refletem a estabilidade das ligações pode
ser ilustrada pela variação da energia do orbital N5C3σ − . A estrutura de ressonância
c implica um caráter de ligação dupla entre os átomos C3-N5 no ef, mas não nos
et, de maneira que, no curso da rotação, a ligação C3-N5 se torna mais longa
(Tabela 4.29) e perde seu caráter de dupla, ficando assim menos estável. Como
pode ser visto na Tabela 4.30, o orbital N5C3σ − sofre uma desestabilização próxima a
120 kcal/mol em 1. No caso da ligação C3-Z4, os componentes ρ e π seguem em
direções opostas, mas o balanço líquido é negativo, ou seja, a ligação C3-Z4 é mais
estável no et do que no ef. Como no caso anterior, esse comportamento é previsto
pelo modelo de ressonância, tendo em vista que, no ef, a ligação C3-Z4 possui
forte caráter de dupla, e esse caráter, devido à estrutura c (Fig. 4.27), é eliminado
nos et. A estrutura d ainda permanece nos et, e de forma mais intensa que no ef,
mas sua contribuição não é tão importante para o et como a de c é para o ef.
Outra variação importante é a do par isolado do nitrogênio. No ef, esse par
de elétrons assume uma configuração essencialmente p, ao passo que nos et, com a
piramidalização do nitrogênio, o mesmo orbital passa a ser um híbrido aproxima-
damente sp3. Como pode ser visto na Tabela 4.30, isso leva a uma expressiva
diminuição da energia do orbital. Além disso, a diminuição da energia é acentuada
pelo aumento da população eletrônica. Por exemplo, no ef de 1 a população é de
1,76177 elétrons contra 1,90598 elétrons no et1, um reflexo claro da transferência
(4.8)

Resultados e Discussão 126
de carga como resultado da estrutura de ressonância c (Fig. 4.27). Para se ter uma
idéia do que esses números significam, no orbital N5C3σ − , que também sofre uma
intensa variação de energia, os valores são 1,98711 e 1,98261 no ef e no et1,
respectivamente, ou seja, a diferença de carga é bem menor do que no par de
elétrons isolado.
Tabela 4.30. Variação das energias dos NBO entre ef e et dos compostos 1-4 a,b,c
(1) (2) (3) (4)
et1 et2 et1 et2 et1 et2 et1 et2
X2C1σ − 0,11 -8,87 8,07 1,62 -0,62 -7,07 0,10 -6,59
C3X2σ − -37,49 -19,18 -40,43 -16,77 -55,24 -41,41 -62,43 -43,61
Z4C3σ − -158,14 -165,44 -168,63 -169,95 -739,98 -747,35 -787,09 -782,80
Z4C3π − 61,30 64,75 55,04 59,16 617,74 618,39 636,98 639,25
N5C3σ − 125,37 119,68 131,48 126,56 166,02 160,65 169,86 164,96
C6N5σ − 39,77 32,04 39,60 24,96 60,39 59,96 51,50 42,95
C7N5σ − 50,52 42,82 61,73 47,09 53,37 53,00 66,53 58,02
Xn 0,28 -8,22 -1,87 -4,53 2,25 -2,18 1,80 2,49
'
Xn 16,28 1,56 15,62 2,68 28,80 11,56 33,51 17,27
Zn -15,02 -9,22 -9,77 -9,72 -30,50 -25,25 -28,42 -25,63
'
Zn -32,72 -27,40 -35,99 -33,23 -24,98 -18,24 -26,34 -23,33
Nn -129,54 -116,89 -137,17 -110,95 -143,03 -139,74 -153,66 -131,11
Caroço -39,26 -48,38 -57,10 -78,95 -110,24 -105,30 -121,89 -133,22
Soma -118,53 -142,75 -139,43 -162,03 -176,02 -182,99 -219,55 -221,36
a HF/PTZV//HF/6-311+G(2d,p). b Em kcal/mol. c O apóstrofo serve para diferenciar os dois pares isolados dos calcogênios.
C1
X2
C3
Z4
N5
C6
C7
X = O, S
Z = O, S

Resultados e Discussão 127
Por fim, como a estrutura c desaparece nos et, a estrutura d passa a ter
maior peso, uma vez que agora o calcogênio Z recebe carga apenas da ligação
X2-C3. Sendo assim, o caráter de dupla da ligação X2-C3 aumenta nos et e, por
conseqüência, também a sua estabilidade, como de fato se observa na Tabela 4.30.
Vejamos agora o balanço total das energias dos orbitais. Em cada caso, os et
são favorecidos pelas energias dos orbitais de ligação ao somarmos todos os
valores, como pode ser visto ao final da Tabela 4.30. Comparemos 1 com 2. Os
valores para esse último são mais negativos, o que siginifica que, no total, os et de
2 são mais estabilizados pelas variações nas ligações do que os de 1. Assim, a maior
DEdesl (Tabela 4.27) de 2 é compensada pelas variações intra-ligações. Explicação
similar aplica-se a 4. Na Tabela 4.27 vemos que esse composto possui as maiores
DEdesl, mas em compensação, as variações intra-moleculares também são significa-
tivamente maiores em 4 do que nos outros compostos e o resultado é a queda de
sua barreira rotacional. O composto 3 possui DEdesl menores do que 4, mas também
são menores as variações nas energias dos orbitais (Tabela 4.30), de modo que o
balanço acaba fazendo com que a barreira de 3 seja maior. As variações nas
energias dos orbitais de 3 são maiores do que em 1 e 2, mas esse fato parece ser
compensado pela maior DEdesl em 3.
Em resumo, a maior contribuição para as barreiras rotacionais vem da
deslocalização eletrônica e os demais fatores atuam como uma espécie de “ajuste
fino” das magnitudes das barreiras. Essas magnitudes são, portanto, o resultado de
um delicado balanço entre os efeitos acima mencionados, particularmente,
energias de deslocalização e interações intra-ligações.
Híbridos de Ressonância
Concentremos agora nossa atenção apenas nos efeitos de deslocalização
eletrônica. Os valores da Tabela 4.27 correspondem à deslocalização total das
moléculas, ou seja, a todo tipo de interações entre os orbitais ligantes e anti-
ligantes. Desejamos, no entanto, conhecer os tipos mais importantes de interações,
aquelas que possuem maior influência na composição das barreiras rotacionais. É
nossa intenção estabelecer também a validade do modelo de ressonância como
representado pelas estruturas a-d (Fig. 4.27). Comecemos por esse último ponto.

Resultados e Discussão 128
Na teoria de ressonância,154 cada uma das estruturas a-d (Fig. 4.27) é
representada por uma função de onda, iψ , à qual está associada um peso iw . O
híbrido de ressonância, representado pela função de onda Ψ é o resultado da
combinação de todas as estruturas de ressonância possíveis. Como dissemos
anteriormente, há muitas outras estruturas além de a-d para os compostos em
estudo, mas a-d devem possuir os maiores pesos e fornecer uma boa descrição do
sistema e uma boa base para entender as modificações eletrônicas. Portanto,
nossos híbridos de ressonância serão dados por:
A pergunta que nos fazemos agora é: qual o peso iw de cada uma das
estruturas a-d ? Para responder a isso, utilizaremos a “teoria de ressonância
natural”, derivada do método NBO.155-157 Os pesos na Eq. (4.9) são determinados
procurando-se pela melhor combinação de valores que represente a densidade
molecular em termos das densidades eletrônicas das estruturas de ressonância.
Esse tipo de cálculo foi realizado com o módulo NBO 5.0 do programa GAMESS (Parte
Experimental) e os resultados estão apresentados na Tabela 4.31.
Alguns aspectos gerais chamam a atenção. Primeiro, a estrutura b, usualmen-
te incluída para justificar a polarização da carbonila, praticamente não participa
do híbrido. Segundo, como esperado, a estrutura a possui o maior peso, tanto nos
ef como nos et. Nos ef a estrutura c, responsável pelo caráter de dupla da ligação
C-N, possui o segundo maior peso, seguida pela estrutura d. Nos et, esta ordem se
inverte, ou seja, a estrutura d passa a ser a segunda de maior peso enquanto c
diminui para para aproximadamente 1%. A diminuição do peso de c é uma
conseqüência imediata da quebra de conjugação que ocorre nos et, eliminando o
caráter de dupla da ligação C-N.
A importância da deslocalização pode ser avaliada pelo peso da estrutura a,
pois quanto menor for a contribuição de qualquer uma das outras três estruturas,
maior será 1w . Comparemos os compostos 1 e 2. Ambos possuen aproximadamente
o mesmo peso 1w nos ef e nos et, mas 2 possui uma contribuição maior da
estrutura c para o ef e isso faz com que sua DEdesl seja maior. Essa observação é
acompanhada pela menor contribuição de d ao ef de 2, o que indica que a
44332211 ψψψψ wwww +++=Ψ (4.9)
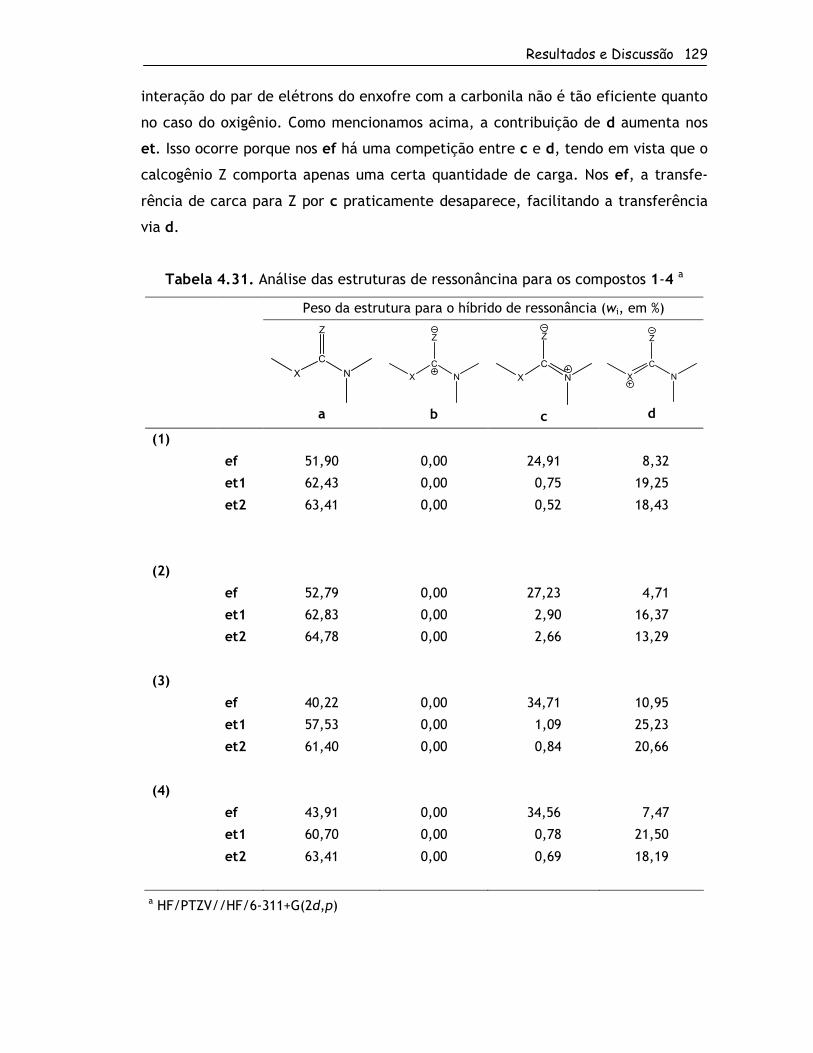
Resultados e Discussão 129
interação do par de elétrons do enxofre com a carbonila não é tão eficiente quanto
no caso do oxigênio. Como mencionamos acima, a contribuição de d aumenta nos
et. Isso ocorre porque nos ef há uma competição entre c e d, tendo em vista que o
calcogênio Z comporta apenas uma certa quantidade de carga. Nos ef, a transfe-
rência de carca para Z por c praticamente desaparece, facilitando a transferência
via d.
Tabela 4.31. Análise das estruturas de ressonâncina para os compostos 1-4 a
Peso da estrutura para o híbrido de ressonância (wi, em %)
C
Z
X N
a
C
Z
X N
b
C
Z
X N
c
C
Z
X N
d
(1)
ef 51,90 0,00 24,91 8,32
et1 62,43 0,00 0,75 19,25
et2 63,41 0,00 0,52 18,43
(2)
ef 52,79 0,00 27,23 4,71
et1 62,83 0,00 2,90 16,37
et2 64,78 0,00 2,66 13,29
(3)
ef 40,22 0,00 34,71 10,95
et1 57,53 0,00 1,09 25,23
et2 61,40 0,00 0,84 20,66
(4)
ef 43,91 0,00 34,56 7,47
et1 60,70 0,00 0,78 21,50
et2 63,41 0,00 0,69 18,19
a HF/PTZV//HF/6-311+G(2d,p)

Resultados e Discussão 130
A diferença nas contribuições das estruturas de ressonância para os
compostos 3 e 4 é muito sutil. A maior diferença está na contribuição de d, que é
menor para 4. Tendo em vista que esta estrutura estabiliza os et e diminui a
barreira, pode-se justificar a maior energia de deslocalização para 4 na
Tabela 4.27 justamente pela menor contribuição de d.
Como conclusão geral acerca dos híbridos de ressonância, pode-se dizer que a
estrutura responsável pelo caráter de ligação dupla nos ef, estrutura c, possui de
fato um peso considerável. Todavia, tentar entender as magnitudes relativas das
barreiras rotacionais recorrendo aos modelos das Fig. 1.2 ou 4.27, que representam
apenas os ef, não é uma prática recomendável. Por exemplo, os valores da
Tabela 4.31 para 1 e 2 claramente indicam maior energia de deslocalização para 2,
o que é contrário às barreiras rotacionais, pois já vimos que a barreira de 1 é
maior.
Buscaremos agora estudar os efeitos de ressonância com base nas interações
entre orbitais ligantes e anti-ligantes
Interações Entre Orbitais
Na Tabela 4.32 apresentamos energias de deslocalização associadas às
principais interações, a saber:
• *ZCN =→ πn , interação entre o par isolado do nitrogênio, Nn , e o orbital
pi antiligante da (tio)carbonila, *ZC=π . Associada à estrutura de
ressonância c;
• *ZCN σ =→n , interação entre o par isolado do nitrogênio e o orbital
sigma antiligante da (tio)carbonila, *ZCσ = . Associada à estrutura de
ressonância c;
• *ZCX =→ πn , interação entre o par isolado sp3, Xn , do calcogênio X e o
orbital pi antiligante da carbonila. Associada à estrutura de ressonância
d;

Resultados e Discussão 131
• *ZCX σ =→n , interação entre o par isolado sp3 do calcogênio X com o
orbital sigma antiligante da (tio)carbonila. Associada à estrutura de
ressonância d;
• *ZC
'X =→ πn , interação entre o par isolado s do calcogênio X, '
Xn , e o
orbital pi antiligante da (tio)carbonila. Associada à estrutura de
ressonância d; e
• *ZC
'X σ =→n , interação entre o par isolado s do calcogênio X e o orbital
sigma antiligante da (tio)carbonila. Associada à estrutura de
ressonância d.
Em primeiro lugar, a Tabela 4.32 nos mostra que a contribuição da
deslocalização para a barreira é dominada pela interação *ZCN =→ πn , o que é
coerente com a observação de que a estrutura c possui o segundo maior peso no
híbrido de ressonância. Porém, a magnitude relativa das barreiras dos quatro
compostos não se correlaciona bem com a energia da referida interação. Em 2, a
interação *ZCN =→ πn é maior do que em 1, ao contrário das barreiras rotacionais.
Em compensação, as interações que diminuem a barreira são maiores em 2 e isso
poderia contrabalancear o efeito *ZCN =→ πn , mas este não é o caso, pois a soma de
tais interações ainda deixa o resultado líquido superior para 2. Esse é o mesmo
resultado a que chegamos considerando as deslocalizações totais comentadas
anteriormente (Tabela 4.27).
Os compostos 3 e 4 apresentam um comportamento parecido, suas energias
de deslocalização são de magnitudes próximas, mas as interações que diminuem a
barreira rotacional são maiores em 4. Neste caso, os resultados da Tabela 4.32 não
seguem fielmente as energias de deslocalização totais sugerindo que o conjunto de
interações apresentados na tabela deve ser complementado por outras de menor
importância, da ordem de 1 kcal/mol. Contudo, a deslocalização *ZCN =→ πn corre-
laciona-se muito bem com as deslocalizações totais, justificando a abordagem
comum de se explicar as barreiras rotacionais através da mesma e da estrutura de
ressonância c. Contudo, vale mais uma vez reforçar que as energias de desloca-
lização por si só não explicam completamente as barreiras rotacionais e precisam
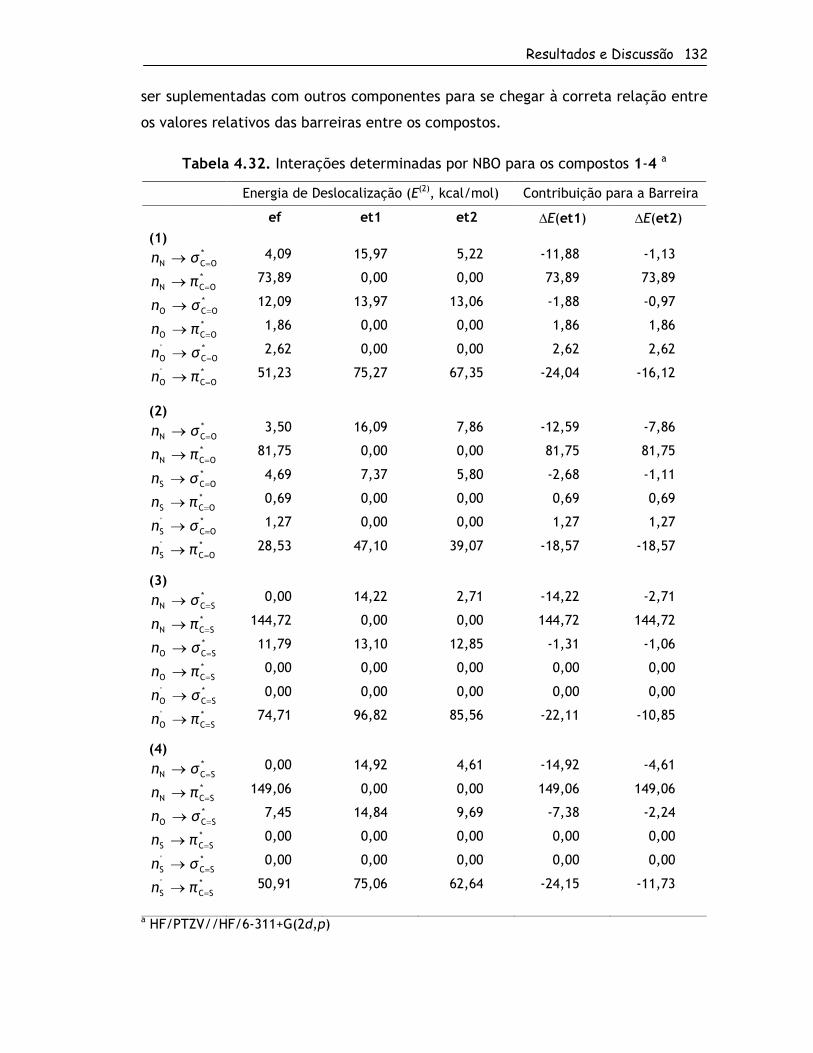
Resultados e Discussão 132
ser suplementadas com outros componentes para se chegar à correta relação entre
os valores relativos das barreiras entre os compostos.
Tabela 4.32. Interações determinadas por NBO para os compostos 1-4 a
Energia de Deslocalização (E(2), kcal/mol) Contribuição para a Barreira
ef et1 et2 ∆E(et1) ∆E(et2) (1)
*OCN =→ σn 4,09 15,97 5,22 -11,88 -1,13
*OCN =→ πn 73,89 0,00 0,00 73,89 73,89
*OCO =→ σn 12,09 13,97 13,06 -1,88 -0,97
*OCO =→ πn 1,86 0,00 0,00 1,86 1,86
*OC
'O =→ σn 2,62 0,00 0,00 2,62 2,62
*OC
'O =→ πn 51,23 75,27 67,35 -24,04 -16,12
(2)
*OCN =→ σn 3,50 16,09 7,86 -12,59 -7,86
*OCN =→ πn 81,75 0,00 0,00 81,75 81,75
*OCS =→ σn 4,69 7,37 5,80 -2,68 -1,11
*OCS =→ πn 0,69 0,00 0,00 0,69 0,69
*OC
'S =→ σn 1,27 0,00 0,00 1,27 1,27
*OC
'S =→ πn 28,53 47,10 39,07 -18,57 -18,57
(3)
*SCN =→ σn 0,00 14,22 2,71 -14,22 -2,71
*SCN =→ πn 144,72 0,00 0,00 144,72 144,72
*SCO =→ σn 11,79 13,10 12,85 -1,31 -1,06
*SCO =→ πn 0,00 0,00 0,00 0,00 0,00
*SC
'O =→ σn 0,00 0,00 0,00 0,00 0,00
*SC
'O =→ πn 74,71 96,82 85,56 -22,11 -10,85
(4)
*SCN =→ σn 0,00 14,92 4,61 -14,92 -4,61
*SCN =→ πn 149,06 0,00 0,00 149,06 149,06
*SCO =→ σn 7,45 14,84 9,69 -7,38 -2,24
*SCS =→ πn 0,00 0,00 0,00 0,00 0,00
*SC
'S =→ σn 0,00 0,00 0,00 0,00 0,00
*SC
'S =→ πn 50,91 75,06 62,64 -24,15 -11,73
a HF/PTZV//HF/6-311+G(2d,p)

Resultados e Discussão 133
Na Fig. 4.28 ilustramos a interação *ZCN =→ πn para os compostos 1 e 3
(carbonila e tiocarbonila). Vemos claramente o alinhamento dos orbitais nos ef
favorecendo a interação. Nos et, o par de elétrons isolado do nitrogênio fica
perpendicular ao orbital anti-ligante da carbonila e, desta forma, a interação decai
significativamente. Notemos ainda que, no ef, na região onde ocorre o contato
entre as superfícies mostradas dos dois orbitais, os lóbulos em 3 são maiores do
que os de 1, favorecendo a sobreposição dos orbitais. Este é um dos fatores
responsáveis pela maior energia de deslocalização para a interação *ZCN =→ πn no
composto 4 (Tabela 4.32).
ef et1 et2
Figura 4.28. Interação *ZCN =→ πn para os compostos 1 (a) e 3 (b).
nN
πC=Z*

Resultados e Discussão 134
4.2.4.2 Grupo 2
Contribuições Energéticas para as Barreiras Rotacionais
Na Tabela 4.33 compilamos as contribuições para a barreira rotacional da
deslocalização eletrônica e da estrutura de Lewis. Por limitação de recursos
computacionais, tivemos que realizar os cálculos para 7 e 8 sem funções de
polarização nos hidrogênios, e com isso esperamos uma diferença em relação a 5 e
6 com origem puramente nas funções de base. Não obstante, as diferenças
calculadas são tão expressivas que seguramente podemos projetá-las para fora do
problema das bases, além do fato das análises NBO não variarem muito com o
conjunto de funções utilizadas.158
Tabela 4.33. Decomposição de energia para as barreiras rotacionais de 5-8 a
(5) b (6) b (7) c (8) c
et1 et2 et1 et2 et1 et1
∆ELewis -6,15 -3,06 -18,22 -19,39 8,438 3,112
∆Edesl 12,05 15,30 24,40 32,91 -3,703 0,811
∆E 5,90 12,24 6,18 13,52 4,735 3,923
a kcal/mol. b HF/PTZV//HF/6-311+G(2d,p). c HF/PTZV//HF/6-311+G(2d,p) sem funções d nos hidrogênios.
O primeiro ponto que notamos é a coerência entre as energias de
deslocalização de 5 e 6 com as magnitudes relativas de suas barreiras, ou seja, a
barreira em fase gasosa de 6 é maior do que a de 5, tendência refletida pela maior
energia de deslocalização de 6. No entanto, a energia de deslocalização de 5 é a
metade da de 6, enquanto que suas barreiras rotacionais diferem por apenas
0,28 kcal/mol. Ocorre aqui uma compensação por parte das estruturas de Lewis,
que são mais estáveis nos et do que no ef para 5 e 6, porém de forma mais intensa
neste último. O comportamento se inverte completamente no caso de 7 e 8.
Surpreendentemente, a energia de deslocalização de 7 é maior no et e faz a
barreira rotacional diminuir. De forma similar a energia de deslocalização de 8 é,
ainda que positiva, de apenas 0,8 kcal/mol. Em ambos os casos, a barreira
rotacional é dominada essencialmente pelas variações energéticas na estrutura de

Resultados e Discussão 135
Lewis. Embora seja este um resultado conspícuo, sua interpretação é simples,
bastando observar as estruturas do ef e do et (Figs. 4.29). A repulsão entre as
metilas anti à ligação dupla no ef faz com que as mesmas se direcionem para fora
do plano molecular, colocando os pares isolados dos nitrogênios em orientação
desfavorável para a interação com o orbital antiligante p da dupla. Quando uma
das ligações C-N é rotacionada para gerar o et1, a interação entre as metilas
ocorre de forma simétrica e faz com que as metilas que estão na parte não-
rotacionada da molécula posicionem-se num mesmo plano, colocando o par de
elétrons do nitrogênio num arranjo ótimo para a interação com o orbital p
antiligante da carbonila ou tiocarbonila *ZCN =→ πn . Isso explica o comportamento
das energias de deslocalização de 7 e 8.
Figura 4.29. Influência da repulsão entre as metilas de 7 sobre a posição do par de elé-
trons do nitrogênio.
Precisamos agora examinar o comportamento das estruturas de Lewis com
mais detalhes. Comecemos pelas interações não-ligantes, eletrostática e de Pauli,
compiladas na Tabela 4.34.
Em todos os casos as interações repulsivas não-ligantes favorecem os et. Isto
era esperado para 5 e 6 por sua semelhança com os compostos do Grupo 1. i.e. a
barreira formada pela deslocalização é diminuída pela estrutura de Lewis.
O
H3C
N
CH3
H3C
repulsão afastanto as metilas
alinhamento ótimo para interação
H3C CH3
N
CH3
CH3
alinhamento ótimo para interação
repulsões balanceadas deixama metila no centro
ef
et

Resultados e Discussão 136
Entretanto, 7 e 8 não têm suas barreiras formadas pela deslocalização, como
dissemos acima, e sim pelas estruturas de Lewis. Mas as interações não-ligantes
nessas estruturas favorecem os et à semelhança de 5 e 6, de forma que a única
fonte que resta para a composição da barreira são as variações intra-ligações.
Consideremos então a Tabela 4.35.
Tabela 4.34. Partição de energia para 5-8 no nível HF/PTZV//HF/6-311+G(2d,p) a,b
Valor absoluto Contribribuição para a Barreira
ef et1 et2 ∆E(et1) ∆E(et2)
(5)
Eletrostática +59,36 +52,46 +53,65 -6,90 -5,71
Pauli +234,46 +232,94 +230,15 -1,52 -4,31
Total -8,42 -10,02
(6)
Eletrostática +28,61 +12,93 +19,52 -15,68 -9,09
Pauli +235,25 +234,55 +231,44 -0,70 -3,81
Total -16,38 -12,90
ef et1 ∆E(et1)
(7)
Eletrostática +62,249 +60,555 -1,694
Pauli +282,81 +280,47 -2,340
Total -7,737
(8)
Eletrostática +51,142 +45,746 -5,396
Pauli +289,13 +287,40 -1,73
Total -6,315
a Sem funções p nos hidrogênios para 7 e 8. b kcal/mol.

Resultados e Discussão 137
Os resultados para 5 e 6 mostram que as estruturas de Lewis de seus ef são
desestabilizadas em relação aos et. Além disso, esta desestabilização é maior no
composto 6, compensando assim sua maior energia de deslocalização e
aproximando sua barreira de 5 (Tabela 4.33). Nos compostos 7 e 8 ocorre o
contrário, i.e. suas estruturas de Lewis são mais estáveis nos ef e isto nos dá mais
uma evidência de que as barreiras rotacionais destes compostos possui uma
natureza diferente das dos anteriores.
Tabela 4.35. Variação das energias dos NBO entre ef e et dos compostos 5-8 a
(5) b (6) b (7) c (8) c
et1 et2 et1 et2 et1 et1
C1N2−σ -20,87 -22,04 -17,97 -17,13 -16,30 -9,88
C3N2−σ -51,84 -42,35 -52,39 -43,81 -68,43 -71,40
X8N1−σ -17,08 -28,81 2,92 -7,48 -24,63 -20,45
Z4C3−σ -43,68 -48,77 -73,01 -77,48 -7,67 -17,49
Z4C3−π -13,10 -10,55 -25,82 -25,97 -4,19 -8,94
N5C3−σ 115,42 103,57 156,31 144,20 100,57 135,49
C7N5−σ 53,93 28,85 66,25 46,14 27,58 42,10
C6N5−σ 31,71 7,21 47,06 26,96 34,71 40,78
N2n 60,64 45,10 40,07 22,32 73,68 59,97
N5n -114,54 -73,15 -144,03 -109,00 -92,15 -118,03
Z4n -1,52 0,47 -18,12 -14,55 4,21 -4,44 'Z4n -14,24 -11,21 -13,64 -8,62 0,94 -3,23
Caroço 0,26 -35,97 -41,07 -70,81 -9,76 -40,53
Soma -14,90 -87,64 -73,44 -135,22 +18,56 +30,42
a kcal/mol. b HF/PTZV//HF/6-311+G(2d,p). c HF/PTZV//HF/6-311+G(2d,p) sem funções p nos hidrogênios.
C1
N2
C3
Z4
N5
C6
C7
X = H, CH3
Z = O, S
X8

Resultados e Discussão 138
Lembremos agora da explicação anterior para a baixa contribuição da energia
de deslocalização dos compostos 7 e 8, ou seja, nos et há um arranjo mais
favorável para transferência de carga do par de elétrons isolado para o orbital p
antiligante da dupla. Com base nisso, e nas discussões anteriores para os compostos
do Grupo 1, podemos apontar uma tendência comum a todos os compostos
estudados: as estruturas de Lewis são mais estáveis para as conformações onde a
conjugação é mínima. Mas enfatizamos que esta conclusão deve ser estendida
apenas a sistemas com o fragmento amídico. Há dois compo-nentes principais para
este comportamento, sendo eles as energias dos orbitais da ligação C=Z e dos pares
isolados dos nitrogênios. Primeiramente, quando a conjugação é possível, a ligação
C=Z adquire caráter de ligação simples, ou seja, mais fraca. Segundo, quando a
conjugação é possível, o par de elétrons do nitrogênio assume hibridização sp2,
sendo que o arranjo natural de um par de elé-trons em um nitrogênio é ~ sp3. Se
combinarmos ambos os efeitos, vemos que a estrutura do estado fundamental dos
compostos 1-6 deve ser desestabilizada em relação ao et, o que é confirmado pelas
variações de energias dos orbitais mencio-nados listadas nas Tabelas 4.30 e 4.35.
Em 7 e 8, o et é tão ou mais estabilizado por conjugação quanto o ef, de forma
que para estes compostos ocorre o oposto, isto é, as estruturas de Lewis dos et é
que são desestabilizadas em relação às dos ef.
Híbridos de Ressonância
Na Tabela 4.36 apresentamos os pesos das estruturas de ressonância para 5-8.
Assim como para o Grupo 1, a estrutura b não participa do híbrido de ressonância,
que é portanto descrito de forma satisfatória por a, c e d. No estado fundamental
de 5 a estrutura a contribui com 52,28 % para o híbrido, enquanto que no composto
6 este número cai para 36,10 %. Isto demonstra que a conjugação é mais efetiva
em 6, de acordo com as energias de deslocalização calculadas (Tabela 4.33).
Observa-se também que cada uma das estruturas c e d possui um peso menor do
que a estrutura c dos compostos do Grupo 1. Conseqüentemente, o caráter de
dupla da ligação C-N no Grupo 2 é menor, o que é coerente com as menores
barreiras rotacionais. Isso ocorre porque há dois pares de elétrons interagindo com
a (tio)carbonila, mas esta suporta apenas uma certa quantidade de carga e por isso

Resultados e Discussão 139
cada uma das estruturas c e d possui um peso menor do que o esperado se hou-
vesse apenas um nitrogênio.
Tabela 4.36. Análise das estruturas de ressonâncina para os compostos 5-8 a
Peso da estrutura para o híbrido de ressonância (%)
C
Z
N N
a
C
Z
N N
b
C
Z
N N
c
C
Z
N N
d
(5)
ef 52,28 0,00 16,07 17,13
et1 55,84 0,00 0,53 27,38
et2 57,56 0,00 0,61 24,60
(6)
ef 36,10 0,00 25,59 24,91
et1 47,15 0,00 0,55 35,74
et2 51,41 0,00 0,53 30,97
(7)
ef 54,74 0,00 13,31 13,75
et1 52,88 0,00 0,68 22,97
(8)
ef 41,43 0,00 20,05 20,95
et1 43,86 0,00 0,56 36,79
a HF/PTZV//HF/6-311+G(2d,p). c HF/PTZV//HF/6-311+G(2d,p) sem funções p nos hidrogênios.
No caso de 7 e 8, os pesos dos estados fundamentais são maiores, refletindo
uma menor conjugação. Assim como nos dois compostos anteriores, a
deslocalização é maior no derivado com enxofre. Em todos os casos, a contribuição
de d aumenta ao longo da rotação, pois somente um dos nitrogênios interage
efetivamente com a ligação C-Z.
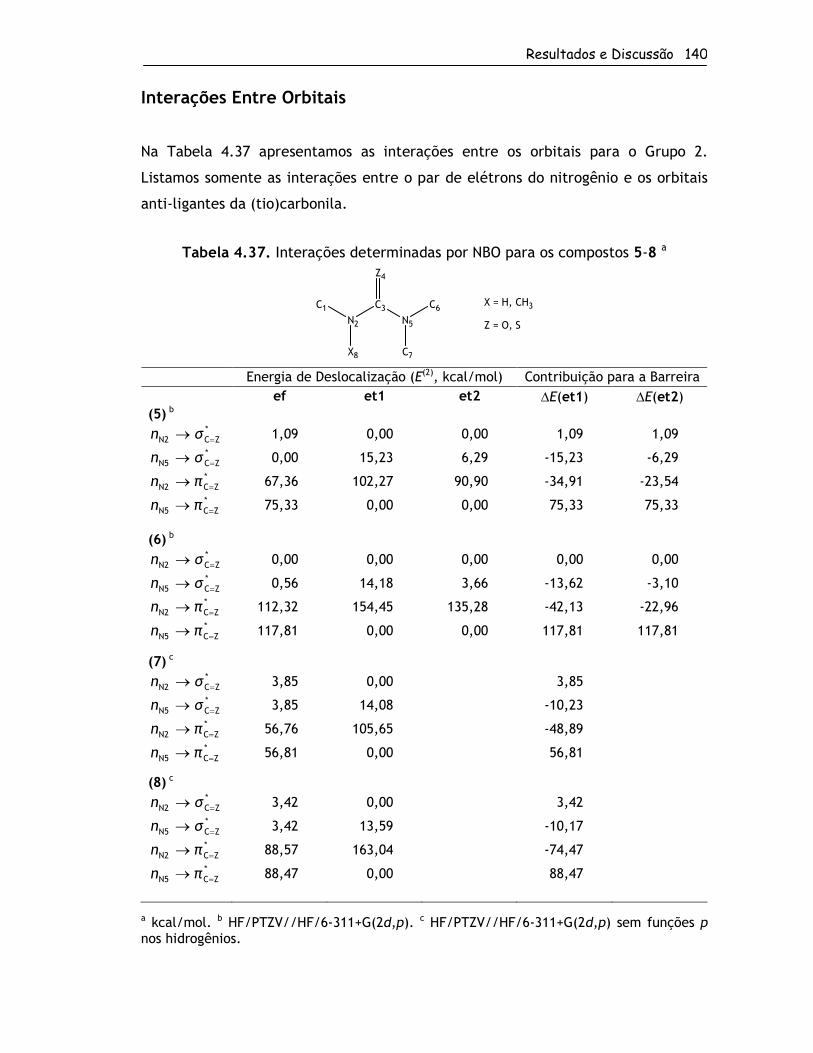
Resultados e Discussão 140
Interações Entre Orbitais Na Tabela 4.37 apresentamos as interações entre os orbitais para o Grupo 2.
Listamos somente as interações entre o par de elétrons do nitrogênio e os orbitais
anti-ligantes da (tio)carbonila.
Tabela 4.37. Interações determinadas por NBO para os compostos 5-8 a
Energia de Deslocalização (E(2), kcal/mol) Contribuição para a Barreira ef et1 et2 ∆E(et1) ∆E(et2) (5) b
*ZCN2 =→ σn 1,09 0,00 0,00 1,09 1,09
*ZCN5 =→ σn 0,00 15,23 6,29 -15,23 -6,29
*ZCN2 =→ πn 67,36 102,27 90,90 -34,91 -23,54
*ZCN5 =→ πn 75,33 0,00 0,00 75,33 75,33
(6) b
*ZCN2 =→ σn 0,00 0,00 0,00 0,00 0,00
*ZCN5 =→ σn 0,56 14,18 3,66 -13,62 -3,10
*ZCN2 =→ πn 112,32 154,45 135,28 -42,13 -22,96
*ZCN5 =→ πn 117,81 0,00 0,00 117,81 117,81
(7) c *
ZCN2 =→ σn 3,85 0,00 3,85 *
ZCN5 =→ σn 3,85 14,08 -10,23 *
ZCN2 =→ πn 56,76 105,65 -48,89 *
ZCN5 =→ πn 56,81 0,00 56,81
(8) c *
ZCN2 =→ σn 3,42 0,00 3,42 *
ZCN5 =→ σn 3,42 13,59 -10,17 *
ZCN2 =→ πn 88,57 163,04 -74,47 *
ZCN5 =→ πn 88,47 0,00 88,47
a kcal/mol. b HF/PTZV//HF/6-311+G(2d,p). c HF/PTZV//HF/6-311+G(2d,p) sem funções p nos hidrogênios.
C1
N2
C3
Z4
N5
C6
C7
X = H, CH3
Z = O, S
X8

Resultados e Discussão 141
Em ambos os derivados com enxofre, 6 e 8, as energias de deslocalização são
maiores, corroborando com as observações acerca das estruturas de ressonância
comentadas acima. Notamos ainda que em 5 e 6 as delocalizações formadoras
superam expressivamente aquelas que diminuem a barreira. Em 7 e 8 as interações
que aumentam e diminuem a barreira são de magnitude próxima, concordando com
a conclusão de que nestes compostos a barreira não é oriunda de deslocalização.
Finalizamos assim a parte do estudo que trata das origem das barreiras
rotacionais. A conclusão de maior importância desta parte dos estudos diz respeito
à forma como as barreiras rotacionais são afetadas por outros efeitos além da
deslocalização eletrônica.

Conclusões 142
Capítulo 5:
Conclusões

Conclusões 143
Capítulo 5: Conclusões
No que diz respeito aos experimentos, foi possível obter dados para seis dos oito
compostos estudados, ficando os compostos 7 e 8 fora do alcance da RMND.
Observou-se variação da barreira rotacional com o solvente para 2, 4, 5 e 6. O
composto 1 mostrou-se insensível tanto à polaridade como às interações espe-
cíficas. Os dados de RMND para 3 sugerem insensibilidade à polaridade e forte
interação com os solventes próticos, visto que nestes não foi possível sequer
adquirir espectros por causa da decomposição da amostra.
Os cálculos teóricos foram de muita valia para a interpretação dos resultados
e possibilitaram o entendimento dos dois fatores estudados, polaridade e ligações
de hidrogênio, para o efeito do solvente. Verificamos que é imprescindível a
inclusão dos efeitos de solvatação para o cálculo correto das barreiras rotacionais.
O maior acréscimo às barreiras rotacionais pelas ligações de hidrogênio foi
observado para 2. Por outro lado, encontramos casos em que as ligações de
hidrogênio diminuem as barreiras rotacionais, uma situação diferente das repor-
tadas na literatura. Este comportamento ocorre nos compostos com enxofre.
O método que foi aplicado, combinando dinâmica molecular e cálculos quân-
ticos, mostrou-se satisfatório. Há, em alguns casos, um certo exagero nos efeitos,
como ocorre para o composto 6. Entretanto, julgamos que isso pode ser corrigido
por refinamentos do método, incluindo um número maior de complexos na parame-
trização, realizando otimizações de geometria ou diminuindo as restrições durante
as otimizações, simulações no ensemble NPT e simulações com modelos flexíveis
tanto para soluto como para o solvente. Evidentemente, a inclusão de tais melho-
ramentos faz crescer exponencialmente os recursos computacionais e o trabalho
de pesquisa, mas na medida em que for sendo acumulado um conjunto de dados a
respeito dos compostos que estudamos, estes melhoramentos podem vir a ser
utilizados em ferramentas de rotina.
Quanto à origem das barreiras rotacionais, nos fica claro que as
especificidades de cada sistema precludem a completa racionalização em termos
de um único efeito, como é prática comum neste assunto. A deslocalização eletrô-
nica desempenha, sem dúvida, o papel mais importante, mas somente com este

Conclusões 144
conceito não é possível explicar as magnitudes relativas das barreiras rotacionais.
Verificamos que há um delicado balanço entre um conjunto de efeitos, sendo
necessário considerar as modificações nas interações não-ligantes, bem como nas
forças que mantém próximos os átomos diretamente ligados. A deslocalização
eletrônica não parece ser responsável pela barreira rotacional dos compostos 7 e 8.
Nestes, a principal contribuição vem das forças intra-ligações, e isto ocorre devido
à forma como os grupos metilas se arranjam no ef fazendo com que o nitrogênio
fique numa posição desfavorável à conjugação. O modelo de ressonância é válido
para explicar as energias de deslocalização, mas não a barreira rotacional por
completo.
De forma geral, a combinação dos dados experimentais e teóricos nos
permitiu avançar no conhecimento acerca das barreiras rotacionais, sobre sua
resposta ao solvente e sobre suas causas. Desejamos que o material aqui
apresentado possa ser útil para o planejamento de modelos com aplicações
práticas, ou como ponto de partida para novas investigações a respeito de
fundamentos químicos.

Referências Bibliográficas 145
Referências Bibliográficas
1. Beers, W. H.; Reich, E., Nature 1970, 228, 917-922.
2. Furukawa, H.; Hamada, T.; Hayashi, M. K.; Haga, T.; Muto, Y.; Hirota, H.; Yokoyama, S.; Nagasawa, K.; Ishiguro, M., Mol. Pharmacol. 2002, 62, 778-787.
3. Oliveira, P. R. O.; Wiectzycoski, F.; Basso, E. A.; Gonçalves, R. A. C.; Pontes, R. M., J. Mol. Struct. 2003, 657, 191-198.
4. Song, J.; Gordon, M. S.; Deakyne, C. A.; Zheng, W., J. Phys. Chem. A 2004, 108, 11419-11432.
5. Souza, W. F.; Kambe, N.; Sonoda, N., J. Phys. Org. Chem. 1996, 9, 179-186.
6. Tezer, N., J. Mol. Struct. (Theochem) 2005, 714, 133-136.
7. Cox, C.; Lectka, T., J. Org. Chem. 1998, 63, 2426-2427.
8. Rablen, P. R., J. Org. Chem. 2000, 65, 7930-7937.
9. Cox, C.; Lectka, T., Acc. Chem. Res. 2000, 33, 849-858.
10. Duffy, E. M.; Severance, D. L.; Jorgensen, W. L., J. Am. Chem. Soc. 1992, 114, 7535-7542.
11. Jasien, P. G.; Stevens, W. J.; Krauss, M., J. Mol. Struct. (Theochem) 1986, 139, 197-206.
12. Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D., J. Chem. Phys. 1983, 79, 926-935.
13. Jorgensen, W. L.; Gao, J., J. Am. Chem. Soc. 1988, 110, 4212-4216.
14. LeMaster, C. B.; True, N. S., J. Phys. Chem. 1989, 93, 1307-1311.
15. Lim, K.-T.; Francl, M. M., J. Phys. Chem. 1987, 91, 2716-2721.
16. Stewart, W. E.; Siddall III, T. H., Chem. Rev. 1970, 70, 517-551.
17. Wiberg, K. B.; Breneman, C. M., J. Am. Chem. Soc. 1992, 114, 831-840.
18. Wiberg, K. B.; Rablen, P. R., J. Am. Chem. Soc. 1995, 117, 2201-2209.
19. Wiberg, K. B.; Rablen, P. R.; Rush, D. J.; Keith, T. A., J. Am. Chem. Soc. 1995, 117, 4261-4270.
20. Wiberg, K. B.; Rush, D. J., J. Am. Chem. Soc. 2001, 123, 2038-2046.
21. Wiberg, K. B.; Rush, D. J., J. Org. Chem. 2002, 67, 826-830.
22. Laidig, K. E.; Cameron, L. M., J. Am. Chem. Soc. 1996, 118, 1737-1742.
23. Lauvergnat, D.; Hilberty, P. C., J. Am. Chem. Soc. 1997, 119, 9478-9482.
24. Wiberg, K. B.; Laidig, K. E., J. Am. Chem. Soc. 1987, 109, 5935-5943.
25. Pophristic, V.; Goodman, L., Nature 2001, 411, 565-568.
26. Schreiner, P. R., Angw. Chem. Int. Ed. 2002, 41, 3579-3581.

Referências Bibliográficas 146
27. Weinhold, F., Nature 2001, 411, 539-541.
28. Allinger, N.; Cava, M. P.; Jongh, D. C.; Lebel, N. A.; Stevens, C. L., Química Orgânica. 2 ed.; LTC: Rio de Janeiro, 1976.
29. Carey, C.; Sundberg, R. J., Advanced Organic Chemistry. 2 ed.; Wiley: New York, 1986; Vol. 1.
30. Solomons, G.; Fryhle, C., Química Orgânica. 7 ed.; LCT: Rio de Janeiro, 2001; Vol. 1.
31. Fogarasi, G.; Szalay, G., J. Phys. Chem. A 1997, 101, 1400-1408.
32. Mo, Y.; Schleyer, P. R.; Wu, W.; Lin, M.; Zhang, Q.; Gao, J., J. Phys. Chem. A 2003, 107, 10011-10018.
33. Vassilev, N. G.; Dimitrov, V., J. Mol. Struct. 1999, 484, 39-47.
34. Wiberg, K. B., Acc. Chem. Res. 1999, 32, 922-929.
35. Wiberg, K. B.; Hadad, C. M.; Rablen, P. R.; Cioslowski, J., J. Am. Chem. Soc. 1992, 114, 8644-8654.
36. Basso, E. A.; Oliveira, P. R. O.; Caetano, J.; Schuquel, I. T. A., J. Braz. Chem. Soc. 2001, 12, 215-222.
37. Oliveira, P. R. O. Dissertação de Mestrado. Universidade Estadual de Maringá, Maringá, 2000.
38. Silva, R. A. C. Dissertação de Mestrado. Universidade Estadual de Maringá, Maringá, 1995.
39. Cramer, J. C., Essentials of Computational Chemistry. John Wiley & Sons: Cornwall, 2003.
40. Foresman, J. B.; Frisch, Æ., Exploring Chemistry with Electronic Structure Methods. 2 ed.; Gaussian, Inc.: Pittsburgh, 1996.
41. Grant, G. H.; Richards, W. G., Computational Chemistry. Oxford University Press: Oxford, 1995.
42. Levine, I. N., Quantum Chemistry. 4 ed.; Prentice-Hall International, Inc.: New York, 1991.
43. Young, D., Computational Chemistry. John Wiley & Sons: New York, 2001.
44. Custódio, R.; Politi, J. R. S.; Segala, M.; Haiduke, R. L. A.; Cyrillo, M., Quím. Nova 2002, 25, 159-170.
45. Freitas, L. C. G., Quím. Nova 1999, 22, 293-298.
46. Minkin, V. I.; (Ed.), Pure. Appl. Chem. 1999, 71, 1919-1981.
47. Shavitt, I., Isr. J. Chem. 1993, 33, 357-367.
48. Raghavachari, K.; Anderson, J. B., J. Phys. Chem. 1996, 100, 12960-12973.
49. Becke, A. D., J. Chem. Phys. 1993, 98, 1372-1377.
50. Kohn, W.; Becke, A. D.; Parr, R. G., J. Phys. Chem. 1996, 100, 12974-12980.
51. Hohenberg, P.; Kohn, W., Phys. Rev. B 1964, 136, 864-871.
52. Kohn, W.; Sham, L., Phys. Rev. A 1965, 140, 1133-1138.

Referências Bibliográficas 147
53. Lee, C.; Yang, W.; Parr, R. G., Phys. Rev. B 1988, 37, 785-789.
54. Atkins, P. W., Physical Chemistry. 6 ed.; Oxford University Press: Oxford, 1998.
55. Foresman, J. B.; Keith, T. A.; Wiberg, K. B.; Snoonian, J.; Frish, M. J., J. Phys. Chem. 1996, 100, 16098-16104.
56. Kirkwood, J. G., Chem. Phys. 1934, 7, 351-361.
57. Miertuš, S.; Srocco, E.; Tomasi, J., Chem. Phys. 1981, 55, 117-129.
58. Miertuš, S.; Tomasi, J., Chem. Phys. 1982, 65, 239-245.
59. Tomasi, J.; Persico, M., Chem. Rev. 1994, 94, 2017-2094.
60. Wong, M. W.; Wiberg, K. B.; Frish, M. J., J. Chem. Phys. 1991, 95, 8991-8998.
61. Onsager, L., J. Am. Chem. Soc. 1936, 58, 1486-1493.
62. Badenhoop, J. K.; Weinhold, F., J. Chem. Phys. 1997, 107, 5406-5421.
63. Carpenter, J. E.; Weinhold, F., J. Mol. Struct. (Theochem) 1988, 169, 41-62.
64. Foster, J. P.; Weinhold, F., J. Am. Chem. Soc. 1980, 102, 7211-7218.
65. Reed, A. E.; Curtiss, L. A.; Weinhold, F., Chem. Rev. 1988, 88, 899-926.
66. Reed, A. E.; Weinhold, F., Isr. J. Chem. 1991, 31, 227-285.
67. Reed, A. E.; Weinhold, F., J. Chem. Phys. 1983, 78, 4066-4073.
68. Weinhold, F., J. Chem. Educ. 1999, 76, 1141-1146.
69. Löwdin, P., Phys. Rev. 1955, 97, 1474-1489.
70. Barlette, V. E.; Freitas, L. C. G., Quím. Nova 1999, 22, 254-262.
71. Berendsen, H. J. C.; Postma, J. P. M.; van Gunsteren, W. F.; DiNota, A.; Haak, J. R., J. Chem. Phys. 81, 81, 3684-3690.
72. van Gunsteren, W. F.; Mark, A. E., J. Chem. Phys. 1998, 108, 6109-6116.
73. Jorgensen, W. L.; Buckner, J. K., J. Phys. Chem. 1987, 91, 6083-6085.
74. Jorgensen, W. L., J. Phys. Chem. 1986, 90, 1276-1284.
75. Jorgensen, W. L., J. Phys. Chem. 1986, 90, 6379-6388.
76. Jorgensen, W. L.; Madura, J. D.; Swenson, C. J., J. Am. Chem. Soc. 1984, 106, 6638-6646.
77. Jorgensen, W. L.; Swenson, C. J., J. Am. Chem. Soc. 1985, 107, 569-578.
78. Allerhand, A.; Gutwsky, H. S.; Jonas, J.; Meinzer, R. A., J. Am. Chem. Soc. 1966, 88, 3185-3194.
79. Jackman, L. M.; Cotton, F. A., Dynamic Nuclear Magnetic Resonance Spectroscopy. Academic Press: Nova York, 1975.
80. Kincaid, J. F.; Eyring, H.; Stearn, A. E., Chem. Rev. 1941, 28, 301-635.
81. McConnell, H. M., J. Chem. Phys. 1958, 28, 430-431.
82. Lin, C. C.; Swalwn, J. D., Rev. Mod. Phys. 1959, 31, 841-892.

Referências Bibliográficas 148
83. Gil, V. M. S.; Geraldes, C. F. G. C., Ressonância Magnética Nuclear: Funadmentos, Métodos e Aplicações. Fundação Calouste Gulbenkian: Lisboa, 1987.
84. Reich, H. J., J. Chem. Educ. Software 3D2.
85. Basch, H.; Hoz, S., Chem. Phys. Lett. 1998, 294, 117-125.
86. Bhraratam, P. V.; Moudgil, R.; Kaur, D., J. Phys. Chem. A 2003, 107, 1627-1634.
87. Wiberg, K. B.; Laidig, K. E., J. Am. Chem. Soc. 1987, 109, 5935-5943.
88. Fraenkel, G.; Franconi, C., J. Am. Chem. Soc. 1960, 82, 4478-4483.
89. Rablen, P. R.; Miller, D. A.; Bullock, V. R.; Hutchinson, P. H.; Gorman, J. A., J. Am. Chem. Soc. 1999, 121, 218-226.
90. Rablen, P. R.; Pearlman, S. A.; Miller, D. A., J. Am. Chem. Soc. 1999, 121, 227-237.
91. Wiberg, K. B.; Rablen, P. R., J. Am. Chem. Soc. 1993, 115, 9234-9242.
92. Bader, R. F. W., Atoms in Molecules: A Quantum Theory. Claredon Press: Oxford, 1990.
93. Gayathri Devi, K. R.; Sathyanarayana, D. N.; Manogaran, S., Spectrochim. Acta 1981, 37A, 31-36.
94. Julià, S.; Ginebreda, A.; Sala, P.; Sancho, M.; Annunziata, R. C., F., Org. Magn. Reson. 1983, 21, 573-575.
95. Kornberg, N.; Kost, D., J. Chem. Soc. Perkin Trans. 2 1979, 1661-1664.
96. Kost, D.; Kornberg, N., Tetrahedron Lett. 1978, 35, 3275-3276.
97. Lemire, A. E.; Thompson, J. C., Can. J. Chem. 1970, 48, 824-829.
98. Marcovici-Mizrahi, D.; Gottlieb, H. E.; Marks, V.; Nudelman, A., J. Org. Chem. 1996, 61, 8402-8406.
99. Moraczewski, A. L.; Banaszynski, L. A.; From, A. M.; White, C. E.; Smith, B. D., J. Org. Chem. 1998, 63, 7258-7262.
100. Schlottmann, B. U., Tetrahedron Lett. 1971, 1221-1224.
101. Yamagami, C.; Takao, N.; Takeuchi, Y., Aust. J. Chem. 1986, 39, 457-463.
102. Zhengyan, L.; Mattes, R.; Schnockel, H.; Thunemann, M.; Hunting, E.; Hohnke, U.; Mendel, C., J. Mol. Struct. 1984, 117, 117-127.
103. Deetz, M. J.; Forbes, C. C.; Jonas, M.; Malerich, J. P.; Smith, B. D.; Wiest, O., J. Org. Chem. 2002, 67, 3949-3952.
104. Martin, M. L.; Marbon, F.; Trierweller, M., J. Phys. Chem. 1981, 85, 76-78.
105. Smith, B. D.; Goodenough-Lashua, D. M.; D'Souza, C. J. E.; Norton, K. J.; Schmidt, L. M.; Tung, J. C., Tetrahedron Lett. 2004, 45, 2747-2749.
106. Valega, T. M., J. Org. Chem. 1966, 31, 1150-1153.
107. Yoder, H. C.; Komoriya, A.; Kochanowski, J. E.; Suydam, F. H., J. Am. Chem. Soc. 1971, 93, 6515-6518.

Referências Bibliográficas 149
108. Lustig, E.; Benson, W. R.; Duy, N., J. Org. Chem. 1967, 32, 851-852.
109. Sun, H., Macromolecules 1993, 26, 5924-5936.
110. Kleinpeter, E.; Kretschemer, M.; Borsdorf, R.; Widera, R.; Mühlstädt, M., J. prakt. Chem. 1980, 322, 793-797.
111. Kleinpeter, E.; Widera, R.; Mühlstädt, M., J. prakt. Chem. 1977, 319, 133-139.
112. Hartman, J. S.; Schrobilgen, J., Can. J. Chem. 1973, 51, 99-103.
113. Haushalter, K. A.; Lau, J.; Roberts, J. D., J. Am. Chem. Soc. 1996, 118, 8891-8896.
114. Isaksson, G.; Sandström, J., Acta Chem. Scand. 1970, 24, 2565-2582.
115. Martin, M. L.; Filleux-Blanchard, M. L.; Martin, G. J., Org. Magn. Reson. 1980, 13, 396-402.
116. Tompa, A. S.; Barefoot, R. D.; Price, E., J. Phys. Chem. 1969, 73, 435-438.
117. Zhao, Y.; Raymond, M. K.; Tsai, H.; Roberts, J. D., J. Phys. Chem. 1993, 97, 2910-2913.
118. Bryantsev, V. S.; Firman, T. K.; Hay, B. P., J. Phys. Chem. A 2005, 109, 832-842.
119. Feigel, M.; Strassner, T., J. Mol. Struct. (Theochem) 1993, 283, 33-48.
120. Godfrey, P. D.; Brown, R. D.; Hunter, A., J. Mol. Struct. 1997, 413-414, 405-414.
121. Kim, W.; Lee, H.-J.; Choi, Y. S.; Choi, J.-H.; Yoon, C.-J., J. Chem. Soc., Faraday Trans. 1998, 94, 2663-2668.
122. Belletato, P.; Freitas, L. C. G.; Arêas, E. P. G.; Santos, P. S., Phys. Chem. Chem. Phys. 1999, 1, 4769-4776.
123. Bertran, C. A.; Cirino, J. J. V.; Freitas, L. C. G., J. Braz. Chem. Soc. 2002, 13, 238-244.
124. Cirino, J. J. V.; Bertran, C. A., Quím. Nova 2002, 25, 358-363.
125. Mezei, M.; Jancsó, G., Chem. Phys. Lett. 1995, 239, 237-240.
126. Tovchigrechko, A.; Rodnikova, M.; Barthel, J., J. Mol. Liq. 1999, 79, 187-201.
127. Smith, L. J.; Berendsen, H. J. C.; Gunsteren, W. F., J. PHys. Chem. B 2004, 108, 1065-1071.
128. Acorn NMR, "NUTS - NMR Utility Transform Software". www.acornnmr.com (2002)
129. Schäfer, A.; Huber, C.; Alhrichs, R., J. Chem. Phys. 1994, 100, 5829-5835.
130. Frish, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery Jr., J. A.; Stramann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Rega, N.; Salvador, P.; Dannenberg, J. J.; Malick,

Referências Bibliográficas 150
D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Lianshenko, A.; Piskorzs, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A., Gaussian 98, Revision A.11.2. Gaussian, Inc.: Pittsburgh PA, 2001.
131. Frish, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery Jr., J. A.; Stramann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Rega, N.; Salvador, P.; Dannenberg, J. J.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Lianshenko, A.; Piskorzs, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A., Gaussian 03, Revision B.04. Gaussian, Inc.: Pittsburgh PA, 2003.
132. Schaftenaar, G.; Noordik, J. H., Molden: a pre- and post-processing program for molecular and eletronic structures. J. Comp.-Aided Mol. Design 2000, 14, 123-134.
133. Flükiger, P.; Lüthi, H. P.; Portmann, S.; Weber, J., MOLEKEL: An interactive molecular graphics tool. Chemia 2000, 54, 766-770.
134. Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Bohmann, J. A.; Morales, C. M.; Weinhold, F., NBO 5.0. Theoretical Chemistry Institute, University of Wiscosion: Madison, 2001.
135. Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S. J.; Windus, T. L.; Dupuis, M.; Montgomery, J. A., GAMESS (20 Jun. 2002). J. Comp. Chem. 1993, 14, 1347-1363.
136. Hamza, A.; Mayer, I., Theor. Chem. Acc. 2003, 109, 91-98.
137. Mayer, I., Chem. Phys. Lett. 2000, 332, 381-388.
138. Mayer, I.; Hamza, A., Program "APOST", Version 1.05. Chemical Research Center, Hungarian Academy of Sciences: Budapest, 2003.
139. Ponder, J. W., TINKER: Software tools for molecular design, Version 4.1. Washington University School of Medicine: St. Louis, 2003.
140. PCMODEL for Windows, Version 8.00.1. Serena Software: 2001.
141. Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R., Purification of Laboratory Chemicals. Pergamond Press: Oxford, 1983.
142. Frank, R. L.; Smith, P. V., J. Am. Chem. Soc. 1946, 68, 2103-2104.
143. Moore, M. L.; Crossley, F. S., Organic Synthesis. John Wiley & Sons: Nova York, 1955; Vol. 3, p. 599-600.

Referências Bibliográficas 151
144. Occupational Safety & Health Administration U. S. Departement of Labor. http://www.osha.gov/dts/chemicalsampling/data/CH_254225.html (19/10/2005),
145. Moore, M. L.; Crossley, F. S., Organic Synthesis. John Wiley & Sons: Nova York, 1955; Vol. 3, p 617-618.
146. Shawn, W. H. R.; Walker, D. G., J. Am. Chem. Soc. 1957, 79, 4329-4331.
147. Goodman, L.; Pophristic, V., Chem. Phys. Lett. 1996, 259, 287-295.
148. Pophristic, V.; Goodman, L., J. Phys. Chem. A 2000, 104, 3231-3238.
149. Pophristic, V.; Goodman, L.; Guchhait, N., J. Phys. Chem. A 1997, 101, 4290-4297.
150. Hehre, W. J.; Pople, J. A.; Devaquet, A. J. P., J. Am. Chem. Soc. 1976, 98, 664-668.
151. Hadad, C. M.; Rablen, P. R.; Wiberg, K. B., J. Org. Chem. 1998, 63, 8668-8681.
152. Lide, D. R. (Ed.), CRC Handbook of Chemistry and Physics. 77 ed.; CRC Press: Boca Raton, 1996.
153. Pearson, R. G., J. Chem. Educ. 1987, 64, 561-567.
154. Pauling, L., The Nature of the Chemical Bond. 3 ed.; Cornell University Press: Ithaca, 1960.
155. Glendening, E. D.; Badenhoop, J. K.; Weinhold, F., J. Comp. Chem. 1998, 19, 628-646.
156. Glendening, E. D.; Weinhold, F., J. Comp. Chem. 1998, 19, 593-609.
157. Glendening, E. D.; Weinhold, F., J. Comp. Chem. 1998, 19, 610-627.
158. Badenhoop, J. K.; Weinhold, F., Int. J. Quant. Chem 1999, 72, 269-280.

Apêndice I 152
Apêndice I
Calibração do Termômetro
da Sonda de RMN

Apêndice I 153
Apêndice I: Calibração do Termômetro da Sonda de RMN
A calibração da sonda fundamenta-se no fato de que o deslocamento químico de-
pende da temperatura. Desta forma, utiliza-se uma substância padrão, para a qual
a dependência do deslocamento químico com a temperatura é conhecida, e compa-
ra-se o valor dos deslocamentos químicos obtidos no aparelho com aqueles relata-
dos para essa substância de referência. O procedimento foi realizado de acordo
com as instruções do manual de operação do aparelho, do qual também se extraiu
os valores para as referências. Na Tabela A1.1 apresentamos o conjunto de dados
utilizados na calibração.
Tabela A1.1 Dados para a calibração do sistema de controle de temperatura da sonda de RMN para os aparelhos Gemini e Mercury
Gemini a Mercury b
temp. observada temp. corrigida temp. observada temp. corrigida Metanol -40,0 -36,1 -70,0 -66,7 -30,0 -26,7 -60,0 -56,8 -20,0 -16,4 -50,0 -47,1 -10,0 -6,6 -35,0 -32,4 0,0 3,2 -25,0 -22,6 10,0 12,3 -15,0 -13,5 15,0 17,8 -5,0 -3,2 5,0 6,2 15,0 15,9 Etilenoglicol 20,0 22,1 23,4 23,8 30,0 31,0 25,0 25,2 40,0 40,5 30,0 29,6 50,0 50,0 40,0 38,4 60,0 59,0 50,0 48,0 70,0 68,2 60,0 57,5 70,0 67,0 80,0 75,9
a ± 0,4 oC. b ± 0,1 oC.
Os padrões empregados foram o metanol (para temperaturas baixas) e o
etilenoglicol (para temperaturas altas). Obtivemos espectros em diversas tempe-
raturas nas regiões de interesse, sendo que em cada temperatura aguardamos por

Apêndice I 154
cerca de 6 min, após o ajuste, para a estabilização térmica do sistema. Na Tabe-
la A2.1 apresentamos as equações das retas ajustadas aos dados da Tabela A1.1.
Tabela A1.2. Equações ajustadas aos dados da Tabela A1.1
Gemini Mercury
Metanol obscorr
97822,098088,2 tt ⋅+= obscorr
97079,042110,1 tt ⋅+=
Etilenoglicol obscorr
92571,047619,3 tt ⋅+= obscorr
92605,087291,1 tt ⋅+=
a obs: refere-se à temperatura observada; corr: refere-se à temperatura corrigida. As equações fornecem valores em oC.
É importante ressaltar que cada equação é válida apenas na faixa de
temperaturas na qual foi estudada. Por exemplo, a equação para a correção das
temperaturas no espectrômetro Gemini obtida com metanol é válida na faixa
aproximada de –40 a 15 oC (temperatura observada).

Apêndice II 155
Apêndice II
Espectros de RMN

Apêndice II 156
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.507.007.50
1.0
0
1.9
92.8502.8752.9002.9252.950
CO
O
NCH3
CH3
H3C
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.507.007.50
1.0
0
2.0
0
2.8502.8752.9002.925
Figura A1.1. Espectro de RMN de 1H para o N,N-dimetilcarbamato de O-metila (1) em CD3OD a 300 MHz: superior a 25 oC, inferior a -70 oC.

Apêndice II 157
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
2.0
0
2.9503.0003.050
CS
O
NCH3
CH3
H3C
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
2.0
9
2.9503.0003.050
Figura A1.2. Espectro de RMN de 1H para o N,N-dimetiltiocarbamato de S-metila (2) em CD3OD a 300 MHz: superior a 10 oC, inferior a -70 oC.

Apêndice II 158
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.501
.00
1.9
7
2.903.003.103.203.303.403.50
CO
S
NCH3
CH3
H3C
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
1.9
7
2.903.003.103.203.303.40
Figura A1.3. Espectro de RMN de 1H para o N,N-dimetiltiocarbamato de O-metila (3) em CD3CN a 300 MHz: superior a 75 oC, inferior a 23 oC.

Apêndice II 159
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
2.0
3
3.203.303.403.503.603.703.80
CS
S
NCH3
CH3
H3C
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
1.9
9
3.303.403.503.603.70
Figura A1.4. Espectro de RMN de 1H para o N,N-dimetilditiocarbamato de S-metila (4) em CDCl3 a 300 MHz: superior a 53 oC, inferior a 22 oC.

Apêndice II 160
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
2.0
3
2.8002.8502.9002.950
CN
O
NCH 3
CH 3
H 3C
H
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
2.0
8
2.7502.8002.8502.9002.950
Figura A1.5. Espectro de RMN de 1H para a N,N,N’-trimetiluréia (5) em CD3OD a 300 MHz: superior a 25 oC, inferior a -95 oC.

Apêndice II 161
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
0
1.9
0
3.1003.1503.2003.2503.300
CN
S
NCH3
CH3
H 3C
H
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
1.0
00
.89
1.1
3
2.903.003.103.203.303.40
Figura A1.6. Espectro de RMN de 1H para a N,N,N’-trimetiltiouréia (6) em CD3OD a 300 MHz: superior a -30 oC, inferior a -80 oC.

Apêndice II 162
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
CN
O
NCH3
CH3
H3C
CH3
ppm
0.000.501.001.502.002.503.003.504.004.505.005.506.006.50
CN
S
NCH 3
CH3
H 3C
CH3
Figura A1.7. Espectro de RMN 1H para a N,N,N’,N’-tetrametiluréia (7) (superior, -40 oC) e para a N,N,N’,N’-Tetrametiltiouréia (8) (inferior, -30 oC) em CD3OD a 300 MHz.

Apêndice III 163
Apêndice III
Matrizes das Estruturas
Otimizadas

164
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.41398667
r3=1.32566882
a3=116.37200204
r4=1.3507289
a4=112.61288012
d4=-177.78685488
r5=1.4476427
a5=122.71188196
d5=-10.96772162
r6=1.19372954
a6=122.66210236
d6=1.32145108
r7=1.44704498
a7=118.2953979
d7=-171.78332434
r8=1.07995929
a8=110.62721011
d8=60.87372287
r9=1.07977533
a9=110.64791757
d9=-60.38731682
r10=1.07905628
a10=105.62810119
d10=-179.78175674
r11=1.08770894
a11=111.32508573
d11=96.94823916
r12=1.07616445
a12=109.73103611
d12=-23.60122238
r13=1.08348594
a13=109.2726163
d13=-143.32451833
r14=1.07613518
a14=110.84890705
d14=33.9370355
r15=1.08773718
a15=111.71828113
d15=-87.64628481
r16=1.08286719
a16=108.56986166
d16=153.07801765
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.43461022
r3=1.36118763
a3=114.66597218
r4=1.36215754
a4=112.06684473
d4=-178.79391
r5=1.45354115
a5=124.30973892
d5=-3.2938553
r6=1.2160609
a6=122.88023959
d6=0.83780338
r7=1.45258372
a7=118.89612304
d7=-176.85152912
r8=1.08970153
a8=110.6782902
d8=61.05324943
r9=1.08951789
a9=110.67234132
d9=-60.01823254
r10=1.08773228
a10=105.31005819
d10=-179.50515335
r11=1.09554437
a11=110.7173896
d11=113.26135478
r12=1.08587491
a12=109.12493947
d12=-6.90840866
r13=1.0940558
a13=110.11977964
d13=-126.98690395
r14=1.08435191
a14=110.4563299
d14=12.32697354
r15=1.09604042
a15=110.88596166
d15=-108.46664482
r16=1.09397323
a16=109.59403364
d16=132.2776719
N,N-Dimetilcarbamato de O-Metila (1) ef 6-311+G(2d,p)
B
3
L
Y
P
H
F

165
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.41398667
r3=1.32566882
a3=116.37200204
r4=1.3507289
a4=112.61288012
d4=-177.78685488
r5=1.4476427
a5=122.71188196
d5=-10.96772162
r6=1.19372954
a6=122.66210236
d6=1.32145108
r7=1.44704498
a7=118.2953979
d7=-171.78332434
r8=1.07995929
a8=110.62721011
d8=60.87372287
r9=1.07977533
a9=110.64791757
d9=-60.38731682
r10=1.07905628
a10=105.62810119
d10=-179.78175674
r11=1.08770894
a11=111.32508573
d11=96.94823916
r12=1.07616445
a12=109.73103611
d12=-23.60122238
r13=1.08348594
a13=109.2726163
d13=-143.32451833
r14=1.07613518
a14=110.84890705
d14=33.9370355
r15=1.08773718
a15=111.71828113
d15=-87.64628481
r16=1.08286719
a16=108.56986166
d16=153.07801765
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.43988951
r3=1.33971149
a3=115.35766027
r4=1.42942386
a4=110.30025217
d4=179.9999274
r5=1.46781576
a5=110.70425803
d5=-117.69452096
r6=1.20652266
a6=123.75385326
d6=-0.00030136
r7=1.46781612
a7=110.70406366
d7=117.69280871
r8=1.08964069
a8=110.35356833
d8=60.38063613
r9=1.08964012
a9=110.35369768
d9=-60.37293936
r10=1.08686523
a10=105.4100609
d10=-179.99631553
r11=1.09857565
a11=112.81853903
d11=64.52406744
r12=1.09096917
a12=109.66377548
d12=-57.04858555
r13=1.0908869
a13=108.32570974
d13=-174.73416423
r14=1.09096921
a14=109.66367205
d14=57.04828507
r15=1.09857531
a15=112.81878589
d15=-64.52458934
r16=1.09088711
a16=108.32555901
d16=174.73342962
N,N-Dimetilcarbamato de O-Metila (1) et1 6-311+G(2d,p)
B
3
L
Y
P
H
F

166
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.41487755
r3=1.32358989
a3=117.02902384
r4=1.4107451
a4=112.72702033
d4=179.99883032
r5=1.45460261
a5=112.58966954
d5=64.50297568
r6=1.17848914
a6=123.10895027
d6=0.0021583
r7=1.45460341
a7=112.58797526
d7=-64.53977327
r8=1.08034304
a8=110.4819771
d8=60.51282428
r9=1.08034226
a9=110.48246092
d9=-60.50978724
r10=1.07875526
a10=105.88912862
d10=-179.99872211
r11=1.08858219
a11=113.03497415
d11=70.32217219
r12=1.08286807
a12=109.68401273
d12=-51.41402955
r13=1.08238375
a13=108.40695942
d13=-168.97029726
r14=1.08286842
a14=109.68440379
d14=51.3882378
r15=1.08857994
a15=113.03589443
d15=-70.34771465
r16=1.08238421
a16=108.40609824
d16=168.94395882
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.43879174
r3=1.35274366
a3=115.79088986
r4=1.42489946
a4=112.12881643
d4=179.99942753
r5=1.46523147
a5=112.76979047
d5=64.53489911
r6=1.20091428
a6=123.2462744
d6=0.
r7=1.4652304
a7=112.76944025
d7=-64.53820809
r8=1.08965446
a8=110.33154344
d8=60.34553164
r9=1.08965473
a9=110.33129554
d9=-60.34907153
r10=1.08729442
a10=105.59732292
d10=179.99836389
r11=1.09899164
a11=113.22859845
d11=70.42135417
r12=1.09086551
a12=109.59684637
d12=-51.44190387
r13=1.09139058
a13=108.31243015
d13=-168.91505772
r14=1.09086499
a14=109.59684448
d14=51.43639705
r15=1.09899204
a15=113.22880547
d15=-70.4270311
r16=1.09139112
a16=108.31238201
d16=168.90957892
N,N-Dimetilcarbamato de O-Metila (1) et2 6-311+G(2d,p)
B
3
L
Y
P
H
F

167
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.80858155
r3=1.79512795
a3=99.26221766
r4=1.34965289
a4=115.92943814
d4=-178.04935735
r5=1.44705957
a5=123.62197255
d5=-9.68416134
r6=1.19253876
a6=120.07337439
d6=1.39488593
r7=1.4509602
a7=118.21989386
d7=-172.35869182
r8=1.07777981
a8=110.43923796
d8=61.4272504
r9=1.07767304
a9=110.40109744
d9=-60.57538636
r10=1.08185545
a10=105.61584744
d10=-179.58791669
r11=1.08712129
a11=111.06340498
d11=98.76729671
r12=1.07578683
a12=109.68414763
d12=-21.68821689
r13=1.08321772
a13=109.22707512
d13=-141.48012349
r14=1.07877645
a14=111.37952272
d14=38.45291631
r15=1.08663235
a15=112.00691504
d15=-83.81149951
r16=1.0818768
a16=108.43848081
d16=156.54164141
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.82019156
r3=1.82135846
a3=98.05207001
r4=1.36287262
a4=115.06575397
d4=-177.13888427
r5=1.45253087
a5=124.11610651
d5=-3.98829002
r6=1.21545047
a6=120.74774894
d6=2.81197372
r7=1.45717732
a7=118.74978512
d7=-173.76517717
r8=1.08698903
a8=110.50059764
d8=63.34621709
r9=1.08668581
a9=110.19637963
d9=-58.40312614
r10=1.0896867
a10=105.62672712
d10=-177.52822006
r11=1.09532372
a11=110.72628408
d11=109.68339067
r12=1.08584069
a12=109.0090486
d12=-10.41529239
r13=1.09307435
a13=109.87057439
d13=-130.42600542
r14=1.08810874
a14=110.92371034
d14=28.64844375
r15=1.09564896
a15=111.87699725
d15=-93.10276707
r16=1.09192492
a16=108.89049789
d16=147.02423655
N,N-Dimetiltiocarbamato de S-Metila (2) ef 6-311+G(2d,p)
B
3
L
Y
P
H
F

168
C
S,1,r2
C,2,r3,1,a3
O,3,r4,2,a4,1,d4,0
N,3,r5,2,a5,1,d5,0
C,5,r6,3,a6,2,d6,0
C,5,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,6,r11,5,a11,3,d11,0
H,6,r12,5,a12,3,d12,0
H,6,r13,5,a13,3,d13,0
H,7,r14,5,a14,3,d14,0
H,7,r15,5,a15,3,d15,0
H,7,r16,5,a16,3,d16,0
Variables:
r2=1.80986985
r3=1.76130094
a3=100.93279907
r4=1.17961984
a4=123.99154304
d4=0.00351015
r5=1.41959128
a5=110.78831674
d5=-179.99732911
r6=1.45787893
a6=111.12499976
d6=116.93125261
r7=1.45788033
a7=111.1253157
d7=-116.94883069
r8=1.07887148
a8=110.38030176
d8=60.89827541
r9=1.07887365
a9=110.3799796
d9=-60.88705016
r10=1.0815625
a10=106.38044786
d10=-179.99444371
r11=1.08758639
a11=112.56461669
d11=66.20787177
r12=1.08275892
a12=109.66269815
d12=-55.09817723
r13=1.08217509
a13=108.45541743
d13=-172.9493931
r14=1.08275821
a14=109.66243469
d14=55.0976558
r15=1.08758573
a15=112.56545439
d15=-66.20836599
r16=1.08217522
a16=108.45511493
d16=172.94846858
C
S,1,r2
C,2,r3,1,a3
O,3,r4,2,a4,1,d4,0
N,3,r5,2,a5,1,d5,0
C,5,r6,3,a6,2,d6,0
C,5,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,6,r11,5,a11,3,d11,0
H,6,r12,5,a12,3,d12,0
H,6,r13,5,a13,3,d13,0
H,7,r14,5,a14,3,d14,0
H,7,r15,5,a15,3,d15,0
H,7,r16,5,a16,3,d16,0
r2=1.82401
r3=1.76906
a3=100.40762
r4=1.20434
a4=124.83944
d4=0.06837
r5=1.43764
a5=109.34529
d5=-180.01268
r6=1.46881
a6=111.09605
d6=117.13607
r7=1.46889
a7=111.10694
d7=-117.20927
r8=1.08776
a8=110.12302
d8=60.81738
r9=1.08775
a9=110.11377
d9=-60.47187
r10=1.0893
a10=106.58081
d10=-179.82627
r11=1.09795
a11=112.75023
d11=66.03797
r12=1.0908
a12=109.53397
d12=-55.36044
r13=1.09101
a13=108.38925
d13=-173.20153
r14=1.0908
a14=109.53164
d14=55.38546
r15=1.09796
a15=112.74928
d15=-66.00829
r16=1.09102
a16=108.40515
d16=173.22655
N,N-Dimetiltiocarbamato de S-Metila (2) et1 6-311+G(2d,p)
B
3
L
Y
P
H
F

169
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.80995024
r3=1.78824471
a3=100.61502066
r4=1.41512083
a4=115.44064502
d4=179.97086166
r5=1.45374286
a5=114.21205663
d5=66.66967681
r6=1.17540228
a6=121.43460763
d6=-0.02691269
r7=1.45373881
a7=114.21066888
r8=1.07836001
a8=110.2624685
d8=60.93586616
r9=1.07834608
a9=110.25728845
d9=-60.79055296
r10=1.08139855
a10=106.2146199
d10=-179.92624919
r11=1.0890602
a11=113.39871134
d11=74.95585904
r12=1.08201581
a12=109.75955128
d12=-46.85130158
r13=1.08258584
a13=108.29063865
d13=-164.53643532
r14=1.08201608
a14=109.76008782
d14=46.8624081
r15=1.08906212
a15=113.40012563
d15=-74.94524823
r16=1.0825855
a16=108.2907891
d16=164.54730514
d7=-66.68462903
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
O,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.82185145
r3=1.8050922
a3=99.97632493
r4=1.42962155
a4=114.8021973
d4=179.99994105
r5=1.46381111
a5=114.11627738
d5=66.34457402
r6=1.19975575
a6=121.76406337
d6=-0.00007572
r7=1.46381121
a7=114.11627742
r8=1.08734994
a8=110.05550208
d8=60.62557614
r9=1.08734966
a9=110.05550143
d9=-60.62570516
r10=1.08925904
a10=106.41881977
d10=179.99994023
r11=1.0995436
a11=113.55281789
d11=74.22500716
r12=1.09008656
a12=109.66727724
d12=-47.64145952
r13=1.09172007
a13=108.25869454
d13=-165.29029259
r14=1.09008652
a14=109.66728802
d14=47.64142097
r15=1.09954346
a15=113.55279566
d15=-74.22504968
r16=1.09171994
a16=108.25870251
d16=165.2902838
d7=-66.34440116
N,N-Dimetiltiocarbamato de S-Metila (2) et2 6-311+G(2d,p)
B
3
L
Y
P
H
F

170
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.41723341
r3=1.3129619
a3=121.3785883
r4=1.32536504
a4=110.7824084
d4=-179.99893011
r5=1.45719881
a5=120.58356731
d5=-0.00482682
r6=1.67679915
a6=123.10936082
d6=-0.00354304
r7=1.45058344
a7=123.04533312
d7=179.98950641
r8=1.07843123
a8=110.54028155
d8=60.78688586
r9=1.07843135
a9=110.54017386
d9=-60.78601082
r10=1.079094
a10=104.80439279
d10=-179.99955447
r11=1.08437082
a11=109.66935996
d11=120.27828921
r12=1.07360233
a12=110.07061681
d12=-0.00999132
r13=1.08437025
a13=109.66814254
d13=-120.29774415
r14=1.08148627
a14=111.2922408
d14=60.72075994
r15=1.08147596
a15=111.29174005
d15=-60.6764692
r16=1.07956836
a16=107.95670075
d16=-179.97758811
C
O,1,B1
C,2,B2,1,A1
N,3,B3,2,A2,1,D1,0
C,4,B4,3,A3,2,D2,0
S,3,B5,2,A4,1,D3,0
C,4,B6,3,A5,2,D4,0
H,1,B7,2,A6,3,D5,0
H,1,B8,2,A7,3,D6,0
H,1,B9,2,A8,3,D7,0
H,7,B10,4,A9,3,D8,0
H,7,B11,4,A10,3,D9,0
H,7,B12,4,A11,3,D10,0
H,5,B13,4,A12,3,D11,0
H,5,B14,4,A13,3,D12,0
H,5,B15,4,A14,3,D13,0
Variables:
B1=1.41693039
B2=1.3130116
B3=1.32509336
B4=1.45483052
B5=1.67497507
B6=1.45472475
B7=1.07852307
B8=1.07852214
B9=1.07916572
B10=1.08066081
B11=1.08061982
B12=1.08065934
B13=1.08424792
B14=1.08424829
B15=1.07340387
A1=121.11054422
A2=112.65658017
A3=123.64746254
A4=123.40195849
A5=120.31789131
A6=110.57135544
A7=110.57043174
A8=104.79542669
A9=111.02568063
A10=107.90168524
A11=111.02441824
A12=109.65671283
A13=109.65728083
A14=110.98513136
D1=-179.97503888
D2=-0.02117992
D3=-0.02013689
D4=179.97463949
D5=60.81085991
D6=-60.8115856
D7=179.99974406
D8=-60.35693008
D9=-179.99926311
D10=60.35875486
D11=-120.43912903
D12=120.43772191
D13=-0.00094422
N,N-Dimetiltiocarbamato de O-Metila (3) HF/6-311+G(2d,p)
ef3 ef2

171
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.43407912
r3=1.34842907
a3=119.10763021
r4=1.35022301
a4=110.10278602
d4=-177.95348874
r5=1.46248303
a5=121.34598134
d5=-2.85051529
r6=1.67233748
a6=123.67315485
d6=1.95115131
r7=1.45525184
a7=121.83537637
d7=-174.38585992
r8=1.08910816
a8=110.61656637
d8=60.53322257
r9=1.08899129
a9=110.54475159
d9=-60.58474605
r10=1.08819417
a10=104.66301888
d10=179.97381407
r11=1.09497027
a11=110.48756089
d11=108.65246516
r12=1.08523101
a12=109.29199213
d12=-11.42522954
r13=1.09247158
a13=109.56062716
d13=-131.56273165
r14=1.08724759
a14=110.8703952
d14=44.67150826
r15=1.09445125
a15=111.54509806
d15=-76.38509104
r16=1.08899832
a16=108.19056823
d16=164.1307192
N,N-Dimetiltiocarbamato de O-Metila (3) B3LYP/6-311+G(2d,p)
ef2

172
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.41650606
r3=1.29547756
a3=121.1177767
r4=1.40893818
a4=109.88024118
d4=-179.99490331
r5=1.4564693
a5=112.88043333
d5=-115.49803032
r6=1.63908081
a6=125.10379028
d6=0.00021287
r7=1.45648093
a7=112.87514963
d7=115.43194322
r8=1.07964216
a8=110.35154602
d8=-60.58920878
r9=1.07821554
a9=105.10463979
d9=-179.99751775
r10=1.07964396
a10=110.35331101
d10=60.59355332
r11=1.08713271
a11=112.62713442
d11=69.60353036
r12=1.08220606
a12=109.8273211
d12=-51.89856101
r13=1.08241382
a13=108.08660727
d13=-169.61596856
r14=1.0822042
a14=109.82891629
d14=51.88552086
r15=1.08713187
a15=112.62943438
d15=-69.61708159
r16=1.08241338
a16=108.08451055
d16=169.60402794
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.43705206
r3=1.32929611
a3=119.13331339
r4=1.41977459
a4=109.22250733
d4=-179.99642592
r5=1.46655418
a5=112.65184335
d5=-115.9285187
r6=1.64777567
a6=125.05639695
d6=0.00687914
r7=1.4665519
a7=112.65196802
d7=115.98000136
r8=1.08979309
a8=110.22072221
d8=-60.29675105
r9=1.08718756
a9=104.96205086
d9=179.9682574
r10=1.08977886
a10=110.22209031
d10=60.22726275
r11=1.09841132
a11=112.56089376
d11=68.0010378
r12=1.09023312
a12=109.72064536
d12=-53.41468521
r13=1.09114185
a13=108.19595731
d13=-171.26399017
r14=1.09023132
a14=109.72056255
d14=53.41369299
r15=1.09841748
a15=112.5606645
d15=-68.00151056
r16=1.09114578
a16=108.19980019
d16=171.26296377
N,N-Dimetiltiocarbamato de O-Metila (3) et1 6-311+G(2d,p)
B3LYP HF

173
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.41602832
r3=1.30538593
a3=121.34225653
r4=1.40684079
a4=111.9452138
d4=179.99981866
r5=1.45623398
a5=112.64035765
d5=64.70291712
r6=1.62848585
a6=125.2825361
d6=-0.00020597
r7=1.45623114
a7=112.64284743
d7=-64.67879333
r8=1.07959983
a8=110.30314905
d8=60.54302547
r9=1.07960044
a9=110.30318434
d9=-60.54317063
r10=1.07863384
a10=105.28014996
d10=180.
r11=1.08848143
a11=113.08154765
d11=69.59116571
r12=1.0823573
a12=109.50974827
d12=-52.20073874
r13=1.08215912
a13=108.2084426
d13=-169.75314375
r14=1.08235516
a14=109.51011337
d14=52.14527068
r15=1.08848493
a15=113.08016289
d15=-69.64468287
r16=1.08215935
a16=108.2092768
d16=169.69854767
C
O,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.43600252
r3=1.33904304
a3=119.68068498
r4=1.41637507
a4=111.4583668
d4=179.98829748
r5=1.46577845
a5=113.15179794
d5=65.10764467
r6=1.63888171
a6=124.99611001
d6=-0.01257148
r7=1.46576989
a7=113.15109599
d7=-65.0962294
r8=1.08981053
a8=110.17295091
d8=60.24746955
r9=1.08980481
a9=110.1743346
d9=-60.17341927
r10=1.08769934
a10=105.19060792
d10=-179.9649841
r11=1.09909157
a11=113.28219515
d11=69.99438027
r12=1.0906379
a12=109.36807997
d12=-51.79226061
r13=1.09126034
a13=108.25195372
d13=-169.36062551
r14=1.09063509
a14=109.36709827
d14=51.72268335
r15=1.09909913
a15=113.28219365
d15=-70.05895941
r16=1.09126158
a16=108.2543316
d16=169.29233907
N,N-Dimetiltiocarbamato de O-Metila (3) et2 6-311+G(2d,p)
B3LYP HF

174
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.80459497
r3=1.7717509
a3=104.48523998
r4=1.32598572
a4=114.11952219
d4=179.94733922
r5=1.4583948
a5=122.23691471
d5=0.25334067
r6=1.66636821
a6=122.06622126
d6=-0.14247745
r7=1.45965466
a7=119.45910183
d7=179.22037177
r8=1.07711771
a8=110.78194044
d8=61.40501919
r9=1.07712243
a9=110.78329508
d9=-61.4096197
r10=1.08289326
a10=104.20981313
d10=-179.99985474
r11=1.08063042
a11=110.64403157
d11=60.21573289
r12=1.0807951
a12=110.67608418
d12=-59.68049403
r13=1.07868481
a13=108.50386532
d13=-179.72632439
r14=1.08301234
a14=111.26620613
d14=63.1100067
r15=1.08218494
a15=111.1430939
d15=-58.384308
r16=1.07716177
a16=108.74474843
d16=-177.48519318
N,N-Dimetilditiocarbamato de S-Metila (4) ef HF/6-311+G(2d,p)
ef4

175
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
Variables:
r2=1.81298714
r3=1.79435744
a3=102.94175316
r4=1.35181215
a4=112.58382342
d4=-178.6053091
r5=1.46160514
a5=122.40621796
d5=-2.74616592
r6=1.66820125
a6=122.12989868
d6=1.60321642
r7=1.46032445
a7=121.64949958
d7=-175.88393775
r8=1.08690892
a8=110.65385668
d8=61.50781391
r9=1.08691651
a9=110.5536815
d9=-60.63122407
r10=1.09105922
a10=104.52329482
d10=-179.56778124
r11=1.09465907
a11=110.31592117
d11=107.84142354
r12=1.08491418
a12=109.32182257
d12=-12.18255323
r13=1.09203648
a13=109.32747775
d13=-132.42984797
r14=1.09092449
a14=111.02516217
d14=51.82520487
r15=1.0940429
a15=111.60343134
d15=-69.69104519
r16=1.08799373
a16=108.44612407
d16=170.67563652
C
S,1,B1
C,2,B2,1,A1
N,3,B3,2,A2,1,D1,0
C,4,B4,3,A3,2,D2,0
S,3,B5,2,A4,1,D3,0
C,4,B6,3,A5,2,D4,0
H,1,B7,2,A6,3,D5,0
H,1,B8,2,A7,3,D6,0
H,1,B9,2,A8,3,D7,0
H,7,B10,4,A9,3,D8,0
H,7,B11,4,A10,3,D9,0
H,7,B12,4,A11,3,D10,0
H,5,B13,4,A12,3,D11,0
H,5,B14,4,A13,3,D12,0
H,5,B15,4,A14,3,D13,0
Variables:
B1=1.81315092
B2=1.79349592
B3=1.34992783
B4=1.46245697
B5=1.66655147
B6=1.46326645
B7=1.08700626
B8=1.08701133
B9=1.09092323
B10=1.0919403
B11=1.08791326
B12=1.09043817
B13=1.08684915
B14=1.09408555
B15=1.0914268
A1=102.66259456
A2=113.20633888
A3=122.1675183
A4=122.96834698
A5=119.19439759
A6=110.53659137
A7=110.61405981
A8=104.62917679
A9=110.85013154
A10=108.58531072
A11=110.48022542
A12=108.8633787
A13=111.58744277
A14=110.81856645
D1=179.08724443
D2=1.57557952
D3=-1.28966288
D4=177.19111009
D5=60.69839802
D6=-61.36041559
D7=179.67424938
D8=-61.09979239
D9=178.47031782
D10=58.10560709
D11=-170.67794149
D12=69.31388276
D13=-51.77994341
N,N-Dimetilditiocarbamato de S-Metila (4) ef B3LYP/6-311+G(2d,p)
ef4 ef2

176
C
S,1,r2
C,2,r3,1,a3
S,3,r4,2,a4,1,d4,0
N,3,r5,2,a5,1,d5,0
C,5,r6,3,a6,2,d6,0
C,5,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,6,r11,5,a11,3,d11,0
H,6,r12,5,a12,3,d12,0
H,6,r13,5,a13,3,d13,0
H,7,r14,5,a14,3,d14,0
H,7,r15,5,a15,3,d15,0
H,7,r16,5,a16,3,d16,0
r2=1.80401675
r3=1.73108639
a3=105.03598243
r4=1.62370723
a4=126.38470778
d4=0.00762008
r5=1.41859447
a5=108.01905669
d5=-179.99519879
r6=1.45729912
a6=112.87214624
d6=115.37860539
r7=1.45729182
a7=112.87242423
r8=1.07899361
a8=110.63170521
d8=61.04569863
r9=1.07899592
a9=110.63207187
d9=-61.07290093
r10=1.08221604
a10=105.53548181
d10=179.98641963
r11=1.08699885
a11=112.54069145
d11=69.0586485
r12=1.08199083
a12=109.68448839
d12=-52.32404185
r13=1.08241818
a13=108.14283521
d13=-170.18340718
r14=1.08199109
a14=109.68499757
d14=52.31440142
r15=1.08700177
a15=112.54077281
d15=-69.0669537
r16=1.08241905
a16=108.14372045
d16=170.17448343
d7=-115.37779728
C
S,1,r2
C,2,r3,1,a3
S,3,r4,2,a4,1,d4,0
N,3,r5,2,a5,1,d5,0
C,5,r6,3,a6,2,d6,0
C,5,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,6,r11,5,a11,3,d11,0
H,6,r12,5,a12,3,d12,0
H,6,r13,5,a13,3,d13,0
H,7,r14,5,a14,3,d14,0
H,7,r15,5,a15,3,d15,0
H,7,r16,5,a16,3,d16,0
r2=1.81656
r3=1.74034
a3=104.10567
r4=1.64227
a4=126.72234
d4=-0.00642
r5=1.43098
a5=107.18373
d5=179.99231
r6=1.46721
a6=112.72407
d6=115.74271
r7=1.46722
a7=112.72017
r8=1.08848
a8=110.20351
d8=60.49508
r9=1.08848
a9=110.20746
d9=-60.59074
r10=1.09003
a10=106.03636
d10=179.95236
r11=1.09785
a11=112.51683
d11=67.69644
r12=1.09024
a12=109.54314
d12=-53.51458
r13=1.09118
a13=108.2948
d13=-171.51623
r14=1.09024
a14=109.54367
d14=53.49276
r15=1.09785
a15=112.51461
d15=-67.71655
r16=1.09118
a16=108.29472
d16=171.49614
d7=-115.69505
N,N-Dimetilditiocarbamato de S-Metila (4) et1 6-311+G(2d,p)
B3LYP HF

177
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.80427004
r3=1.75383487
a3=105.23503451
r4=1.41409831
a4=113.44892155
d4=-179.99809534
r5=1.45426821
a5=114.78813003
d5=67.53256687
r6=1.6164598
a6=124.71665469
d6=0.00178773
r7=1.45426837
a7=114.7885472
r8=1.07842322
a8=110.5259788
d8=61.06227056
r9=1.07842294
a9=110.52608552
d9=-61.06353486
r10=1.08213292
a10=105.30447918
d10=179.99933778
r11=1.08901628
a11=113.49189731
d11=74.80664446
r12=1.08161514
a12=109.57244248
d12=-47.01217196
r13=1.08258962
a13=108.13011617
d13=-164.6679758
r14=1.08161528
a14=109.57242917
d14=47.01267054
r15=1.0890161
a15=113.49182973
d15=-74.80617241
r16=1.08258959
a16=108.13014148
d16=164.66839128
d7=-67.52735011
C
S,1,r2
C,2,r3,1,a3
N,3,r4,2,a4,1,d4,0
C,4,r5,3,a5,2,d5,0
S,3,r6,2,a6,1,d6,0
C,4,r7,3,a7,2,d7,0
H,1,r8,2,a8,3,d8,0
H,1,r9,2,a9,3,d9,0
H,1,r10,2,a10,3,d10,0
H,7,r11,4,a11,3,d11,0
H,7,r12,4,a12,3,d12,0
H,7,r13,4,a13,3,d13,0
H,5,r14,4,a14,3,d14,0
H,5,r15,4,a15,3,d15,0
H,5,r16,4,a16,3,d16,0
r2=1.81464422
r3=1.76804896
a3=104.46857446
r4=1.42207261
a4=112.93399471
d4=-179.98930573
r5=1.46280304
a5=115.14608268
d5=67.93983546
r6=1.63615105
a6=124.56485718
d6=0.01185224
r7=1.4628049
a7=115.14381215
r8=1.08804073
a8=110.11801456
d8=-60.56029966
r9=1.09023872
a9=105.90412042
d9=179.9541057
r10=1.08803935
a10=110.11538574
d10=60.46853339
r11=1.09989449
a11=113.65608042
d11=74.83453495
r12=1.0900158
a12=109.41715967
d12=-46.89952064
r13=1.09179093
a13=108.2953875
d13=-164.59516161
r14=1.09001738
a14=109.41722127
d14=46.91608254
r15=1.09989147
a15=113.65829428
d15=-74.82031749
r16=1.09178997
a16=108.29314433
d16=164.61051286
d7=-67.95295114
N,N-Dimetilditiocarbamato de S-Metila (4) et2 6-311+G(2d,p)
B3LYP HF

178
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,4,D3,0
H,1,B6,6,A5,2,D4,0
O,6,B7,1,A6,2,D5,0
N,6,B8,1,A7,2,D6,0
C,9,B9,6,A8,1,D7,0
H,10,B10,9,A9,6,D8,0
H,10,B11,9,A10,6,D9,0
H,10,B12,9,A11,6,D10,0
C,9,B13,6,A12,1,D11,0
H,14,B14,9,A13,6,D12,0
H,14,B15,9,A14,6,D13,0
H,14,B16,9,A15,6,D14,0
Variables:
B1=1.44857731
B2=1.08477317
B3=1.07887023
B4=1.0823563
B5=1.36603626
B6=0.99049873
B7=1.20035888
B8=1.37044185
B9=1.44838169
B10=1.08033296
B11=1.08343993
B12=1.08697994
B13=1.45090087
B14=1.08264622
B15=1.08814099
B16=1.07599897
A1=112.46438933
A2=109.78299515
A3=108.27614066
A4=118.62012421
A5=117.30517736
A6=121.25655594
A7=116.2704536
A8=121.17065509
A9=108.41653742
A10=111.93910874
A11=112.67533004
A12=116.5427371
A13=108.93334978
A14=111.95793599
A15=109.6001213
D1=120.62152833
D2=118.6573598
D3=57.26326316
D4=-147.16109665
D5=-10.93754949
D6=169.41962631
D7=19.49570408
D8=178.68536818
D9=-63.96786107
D10=58.85895505
D11=170.28834784
D12=155.13644471
D13=-84.60667759
D14=35.77651714
N
C,1,r2
H,2,r3,1,a3
H,2,r4,1,a4,3,d4,0
H,2,r5,1,a5,3,d5,0
C,1,r6,2,a6,3,d6,0
H,1,r7,2,a7,3,d7,0
O,6,r8,1,a8,2,d8,0
N,6,r9,1,a9,2,d9,0
C,9,r10,6,a10,1,d10,0
H,10,r11,9,a11,6,d11,0
H,10,r12,9,a12,6,d12,0
H,10,r13,9,a13,6,d13,0
C,9,r14,6,a14,1,d14,0
H,14,r15,9,a15,6,d15,0
H,14,r16,9,a16,6,d16,0
H,14,r17,9,a17,6,d17,0
Variables:
r2=1.45536937
r3=1.09483753
a3=112.66639422
r4=1.08777293
a4=109.25893867
d4=120.03760518
r5=1.09033054
a5=108.66740757
d5=-121.12593907
r6=1.38086134
a6=118.92266798
d6=-69.9297639
r7=1.00612345
a7=116.30565229
d7=81.01496569
r8=1.22495154
a8=121.42458861
d8=-11.3912975
r9=1.38350352
a9=115.88310266
d9=169.38324147
r10=1.45356679
a10=122.12197297
d10=11.61285581
r11=1.089133
a11=108.58763412
d11=-178.45439499
r12=1.09405127
a12=111.82290464
d12=-61.12454893
r13=1.09674161
a13=112.76749864
d13=61.69421574
r14=1.45622885
a14=117.5489496
d14=169.99079431
r15=1.09223031
a15=109.65662248
d15=143.55149808
r16=1.09712437
a16=111.80674309
d16=-95.88109964
r17=1.08586728
a17=108.73434195
d17=23.90311039
N,N,N’-Trimetiluréia (5) ef2 6-311+G(2d,p)
B3LYP HF

179
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
H,1,B6,6,A5,2,D4,0
O,6,B7,1,A6,2,D5,0
N,6,B8,1,A7,2,D6,0
C,9,B9,6,A8,1,D7,0
H,10,B10,9,A9,6,D8,0
H,10,B11,9,A10,6,D9,0
H,10,B12,9,A11,6,D10,0
C,9,B13,6,A12,1,D11,0
H,14,B14,9,A13,6,D12,0
H,14,B15,9,A14,6,D13,0
H,14,B16,9,A15,6,D14,0
Variables:
B1=1.44406587
B2=1.08151151
B3=1.08246693
B4=1.08244095
B5=1.33880426
B6=0.99004839
B7=1.19435809
B8=1.42824605
B9=1.45710346
B10=1.08725569
B11=1.08328129
B12=1.08276292
B13=1.45709724
B14=1.0827624
B15=1.08327955
B16=1.08725709
A1=108.56647237
A2=111.04470009
A3=111.03325816
A4=122.08331334
A5=117.18870048
A6=123.37088523
A7=112.68740082
A8=110.64358442
A9=112.48628015
A10=109.77808963
A11=108.67950145
A12=110.64549966
A13=108.67980764
A14=109.77729965
A15=112.48633488
D1=119.74245889
D2=-119.73530099
D3=179.93015107
D4=-179.93997812
D5=-0.02740134
D6=179.97459983
D7=-117.8765031
D8=-64.88292804
D9=56.17754146
D10=174.20803736
D11=117.89415966
D12=-174.19737247
D13=-56.16600865
D14=64.89315804
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
H,1,B6,6,A5,2,D4,0
O,6,B7,1,A6,2,D5,0
N,6,B8,1,A7,2,D6,0
C,9,B9,6,A8,1,D7,0
H,10,B10,9,A9,6,D8,0
H,10,B11,9,A10,6,D9,0
H,10,B12,9,A11,6,D10,0
C,9,B13,6,A12,1,D11,0
H,14,B14,9,A13,6,D12,0
H,14,B15,9,A14,6,D13,0
H,14,B16,9,A15,6,D14,0
Variables:
B1=1.45037695
B2=1.08788329
B3=1.09276439
B4=1.09276487
B5=1.35394917
B6=1.00720982
B7=1.21697502
B8=1.45197348
B9=1.46855065
B10=1.09787215
B11=1.09128718
B12=1.09138461
B13=1.46855109
B14=1.09138467
B15=1.09128695
B16=1.0978716
A1=107.87442351
A2=111.06914866
A3=111.06907862
A4=123.46964338
A5=115.90602712
A6=124.57636239
A7=111.26763221
A8=110.31232189
A9=112.60075724
A10=109.69486981
A11=108.61773347
A12=110.31222855
A13=108.61774664
A14=109.69487324
A15=112.60073574
D1=119.36665694
D2=-119.36667254
D3=0.
D4=179.99992734
D5=0.00007586
D6=180.
D7=-118.45033518
D8=-63.72503595
D9=57.42257822
D10=175.47524205
D11=118.45035024
D12=-175.4749474
D13=-57.42230509
D14=63.72531681
N,N,N’-Trimetiluréia (5) et1 6-311+G(2d,p)
HF – et1-a B3LYP – et1-b

180
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,4,D3,0
H,1,B6,6,A5,2,D4,0
O,6,B7,1,A6,2,D5,0
N,6,B8,1,A7,2,D6,0
C,9,B9,6,A8,1,D7,0
H,10,B10,9,A9,6,D8,0
H,10,B11,9,A10,6,D9,0
H,10,B12,9,A11,6,D10,0
C,9,B13,6,A12,1,D11,0
H,14,B14,9,A13,6,D12,0
H,14,B15,9,A14,6,D13,0
H,14,B16,9,A15,6,D14,0
Variables:
B1=1.44543059
B2=1.08257577
B3=1.08185804
B4=1.08192822
B5=1.34957558
B6=0.98959625
B7=1.18911103
B8=1.42513813
B9=1.44831821
B10=1.08298101
B11=1.09311906
B12=1.08229361
B13=1.44850132
B14=1.09293869
B15=1.08300765
B16=1.08224421
A1=111.16494662
A2=108.58259579
A3=110.86971846
A4=121.68528755
A5=119.13469345
A6=122.08868866
A7=115.86262362
A8=114.2382769
A9=108.85597332
A10=113.773194
A11=109.78229286
A12=114.21884869
A13=113.74645511
A14=108.84663778
A15=109.76572335
D1=119.86278307
D2=119.64855257
D3=-178.92030693
D4=177.6412225
D5=0.92609868
D6=-179.22088359
D7=66.89927036
D8=164.93089552
D9=-74.81302755
D10=46.95009519
D11=-66.8232232
D12=74.73621244
D13=-164.96807202
D14=-46.98806742
N
C,1,r2
H,2,r3,1,a3
H,2,r4,1,a4,3,d4,0
H,2,r5,1,a5,3,d5,0
C,1,r6,2,a6,3,d6,0
H,1,r7,2,a7,3,d7,0
O,6,r8,1,a8,2,d8,0
N,6,r9,1,a9,2,d9,0
C,9,r10,6,a10,1,d10,0
H,10,r11,9,a11,6,d11,0
H,10,r12,9,a12,6,d12,0
H,10,r13,9,a13,6,d13,0
C,9,r14,6,a14,1,d14,0
H,14,r15,9,a15,6,d15,0
H,14,r16,9,a16,6,d16,0
H,14,r17,9,a17,6,d17,0
Variables:
r2=1.45373
r3=1.08966
a3=108.88579
r4=1.09035
a4=110.25527
d4=119.73804
r5=1.09267
a5=111.42851
d5=-120.33756
r6=1.36094
a6=121.92792
d6=-172.23194
r7=1.00645
a7=118.96434
d7=11.29842
r8=1.21278
a8=122.40409
d8=1.4105
r9=1.44495
a9=115.34322
d9=-178.9828
r10=1.45717
a10=114.19885
d10=66.05704
r11=1.09221
a11=108.93498
d11=165.49424
r12=1.10491
a12=113.96921
d12=-74.41283
r13=1.0905
a13=109.7624
d13=47.46675
r14=1.45766
a14=114.06574
d14=-66.72073
r15=1.10466
a15=113.8715
d15=74.204
r16=1.09221
a16=108.94534
d16=-165.61103
r17=1.09044
a17=109.74063
d17=-47.56272
N,N,N’-Trimetiluréia (5) et2-a 6-311+G(2d,p)
B3LYP HF

181
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
H,1,B6,6,A5,2,D4,0
O,6,B7,1,A6,2,D5,0
N,6,B8,1,A7,2,D6,0
C,9,B9,6,A8,1,D7,0
H,10,B10,9,A9,6,D8,0
H,10,B11,9,A10,6,D9,0
H,10,B12,9,A11,6,D10,0
C,9,B13,6,A12,1,D11,0
H,14,B14,9,A13,6,D12,0
H,14,B15,9,A14,6,D13,0
H,14,B16,9,A15,6,D14,0
Variables:
B1=1.45247658
B2=1.09274017
B3=1.08736429
B4=1.09314904
B5=1.36359046
B6=1.00735253
B7=1.21222291
B8=1.44566576
B9=1.45702738
B10=1.0922561
B11=1.10477668
B12=1.09046509
B13=1.45715854
B14=1.10473289
B15=1.09223112
B16=1.09046097
A1=110.96644303
A2=107.77169453
A3=111.20244282
A4=123.36645063
A5=118.17111097
A6=123.3358492
A7=114.77429864
A8=114.25641682
A9=108.92684046
A10=113.98288533
A11=109.7389873
A12=114.20086305
A13=113.95426158
A14=108.93938193
A15=109.74024679
D1=119.32222897
D2=-121.36096257
D3=-122.01336961
D4=179.2440865
D5=0.41912032
D6=-179.68538268
D7=66.39240095
D8=165.14427014
D9=-74.74087406
D10=47.1362716
D11=-66.74640317
D12=74.63698799
D13=-165.22423109
D14=-47.19562474
N,N,N’-Trimetiluréia (5) et2-b B3LYP/6-311+G(2d,p)

182
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,4,D3,0
H,1,B6,6,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
S,6,B16,1,A15,2,D14,0
Variables:
B1=1.44923032
B2=1.08150311
B3=1.07926063
B4=1.08255268
B5=1.34029381
B6=0.98945143
B7=1.34377866
B8=1.45120084
B9=1.07808794
B10=1.08673261
B11=1.08470134
B12=1.4544381
B13=1.08162867
B14=1.08521942
B15=1.07497651
B16=1.68860895
A1=111.64250124
A2=110.75310548
A3=107.39351632
A4=124.35285808
A5=118.54462083
A6=116.02194054
A7=121.29316156
A8=108.81455572
A9=112.05619723
A10=111.60674134
A11=121.20107393
A12=108.37573199
A13=111.45497786
A14=110.185418
A15=120.71695934
D1=121.02513165
D2=119.10468797
D3=59.91214338
D4=-167.53276418
D5=177.04576788
D6=10.30048443
D7=169.08768675
D8=-72.22084678
D9=50.41300297
D10=179.933538
D11=154.49909669
D12=-85.36158811
D13=35.18895008
D14=-3.58436896
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,4,D3,0
H,1,B6,6,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
S,6,B16,1,A15,2,D14,0
Variables:
B1=1.45411031
B2=1.09219416
B3=1.08927416
B4=1.09080201
B5=1.36166805
B6=1.00597338
B7=1.36544817
B8=1.45554269
B9=1.08783668
B10=1.09768385
B11=1.09374023
B12=1.4563478
B13=1.09420542
B14=1.09418072
B15=1.0854166
B16=1.6877006
A1=111.76522405
A2=110.51267783
A3=107.77257393
A4=123.48732832
A5=118.6655178
A6=115.18005192
A7=121.76249064
A8=108.72371064
A9=112.47627544
A10=111.43606201
A11=122.08506025
A12=110.11853233
A13=110.3851591
A14=108.95720227
A15=120.71067922
D1=120.23234175
D2=119.32193039
D3=59.28576401
D4=-164.17295431
D5=177.66979464
D6=3.21316081
D7=170.29680538
D8=-70.83686924
D9=51.86884516
D10=-175.82086456
D11=121.61901221
D12=-118.21417157
D13=1.55997496
D14=-3.28989401
N,N,N’-Trimetiltiouréia (6) ef2 6-311+G(2d,p)
B3LYP HF

183
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,5,D3,0
H,1,B6,6,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
S,6,B16,1,A15,2,D14,0
Variables:
B1=1.4459852
B2=1.08141947
B3=1.08128124
B4=1.08142326
B5=1.31573892
B6=0.9937625
B7=1.42378117
B8=1.45804012
B9=1.08603647
B10=1.08244646
B11=1.08280623
B12=1.45803955
B13=1.08280629
B14=1.08244622
B15=1.08603618
B16=1.65980632
A1=110.72690022
A2=108.02097951
A3=110.72977068
A4=125.353414
A5=115.34799861
A6=111.31466493
A7=112.41094487
A8=112.48126702
A9=109.83833159
A10=108.17871123
A11=112.41115021
A12=108.17863436
A13=109.83826296
A14=112.48131275
A15=124.05251525
D1=119.87375782
D2=-120.24845999
D3=-60.14769079
D4=-179.99359683
D5=179.99740319
D6=-116.3768129
D7=-68.64774163
D8=52.53760945
D9=170.52051339
D10=116.3834011
D11=-170.52079415
D12=-52.53796472
D13=68.64745804
D14=-0.00067896
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
H,1,B6,6,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
S,6,B16,1,A15,2,D14,0
Variables:
B1=1.45057641
B2=1.09172081
B3=1.09173026
B4=1.08928527
B5=1.3376194
B6=1.01027572
B7=1.4411358
B8=1.46851031
B9=1.09730809
B10=1.09054802
B11=1.09140164
B12=1.4685133
B13=1.09140135
B14=1.09054751
B15=1.09730973
B16=1.66364403
A1=110.60327215
A2=110.60998311
A3=108.48621195
A4=124.82624869
A5=115.18937805
A6=110.77474872
A7=112.02401361
A8=112.39231091
A9=109.73309957
A10=108.3186486
A11=112.02201867
A12=108.31915311
A13=109.73296396
A14=112.39174623
A15=124.01393215
D1=119.32203305
D2=-120.33805424
D3=-59.59121278
D4=179.99680252
D5=-179.99895379
D6=-117.07236149
D7=-66.57638754
D8=54.47404926
D9=172.63038766
D10=117.06715964
D11=-172.62545698
D12=-54.46869309
D13=66.58114509
D14=0.00178721
N,N,N’-Trimetiltiouréia (6) et1-a 6-311+G(2d,p)
B3LYP HF

184
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,5,D3,0
H,1,B6,6,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
S,6,B16,1,A15,2,D14,0
Variables:
B1=1.44724103
B2=1.08129036
B3=1.08177053
B4=1.08131261
B5=1.32529408
B6=0.99238494
B7=1.42032352
B8=1.44961831
B9=1.0824942
B10=1.09316637
B11=1.08176221
B12=1.44961466
B13=1.09316984
B14=1.08249376
B15=1.08176286
B16=1.64902513
A1=110.72879405
A2=108.04569426
A3=110.73438314
A4=125.10733266
A5=117.40062837
A6=115.1371477
A7=114.29088964
A8=108.60418858
A9=113.87675343
A10=109.56226832
A11=114.29219299
A12=113.877546
A13=108.60428231
A14=109.56254202
A15=123.59517676
D1=119.88138302
D2=-120.23193528
D3=-60.15122525
D4=-179.96368721
D5=179.99125867
D6=67.29850954
D7=165.98121912
D8=-73.79321137
D9=48.03777146
D10=-67.29215421
D11=73.79615303
D12=-165.97923893
D13=-48.03575381
D14=-0.00778151
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
H,1,B6,6,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
S,6,B16,1,A15,2,D14,0
Variables:
B1=1.45228259
B2=1.09160659
B3=1.09155695
B4=1.0900389
B5=1.34586426
B6=1.00906734
B7=1.4346746
B8=1.45795164
B9=1.09178499
B10=1.10487933
B11=1.09028961
B12=1.45795526
B13=1.10487678
B14=1.09178524
B15=1.09028891
B16=1.65564227
A1=110.60416839
A2=110.61339355
A3=108.50347108
A4=124.72057568
A5=117.64609673
A6=114.77969918
A7=114.3721499
A8=108.77391318
A9=114.08458767
A10=109.46621204
A11=114.37049447
A12=114.08351908
A13=108.77434435
A14=109.46603442
A15=123.32200899
D1=119.29738498
D2=-120.33083195
D3=-59.68506429
D4=179.97004646
D5=-179.98144674
D6=67.02938774
D7=166.45790132
D8=-73.38314308
D9=48.41992103
D10=-67.0458401
D11=73.38313753
D12=-166.4574905
D13=-48.41892955
D14=0.01353618
N,N,N’-Trimetiltiouréia (6) et2-b 6-311+G(2d,p)
B3LYP HF

185 N,N,N’,N’-Tetrametiluréia (7) ef HF/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
O,6,B6,1,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
C,1,B16,6,A15,7,D14,0
H,17,B17,1,A16,6,D15,0
H,17,B18,1,A17,6,D16,0
H,17,B19,1,A18,6,D17,0
Variables:
B1=1.44976178
B2=1.07797122
B3=1.08340936
B4=1.08715289
B5=1.37803933
B6=1.19778675
B7=1.37808417
B8=1.45217315
B9=1.07654473
B10=1.08841106
B11=1.08421764
B12=1.44976875
B13=1.07796971
B14=1.08340918
B15=1.08715504
B16=1.45216726
B17=1.08422506
B18=1.07654557
B19=1.08840306
A1=110.17234052
A2=108.38336865
A3=112.14157784
A4=115.24789691
A5=121.82541274
A6=116.34863452
A7=120.21519474
A8=111.28072175
A9=110.71583275
A10=109.91266022
A11=115.24348312
A12=110.17180312
A13=108.3835561
A14=112.14214726
A15=120.22564692
A16=109.91832138
A17=111.27828948
A18=110.71169797
D1=119.16024959
D2=-120.7061872
D3=45.97307289
D4=5.79124529
D5=-174.20655419
D6=44.49123818
D7=-28.01701699
D8=92.04012387
D9=-148.54781146
D10=-174.20422953
D11=45.97568962
D12=165.13616292
D13=-74.72988981
D14=-135.54521485
D15=-148.45934648
D16=-27.92369146
D17=92.12826792

186 N,N,N’,N’-Tetrametiluréia (7) ef B3LYP/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,3,D3,0
O,6,B6,1,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
C,1,B16,6,A15,7,D14,0
H,17,B17,1,A16,6,D15,0
H,17,B18,1,A17,6,D16,0
H,17,B19,1,A18,6,D17,0
Variables:
B1=1.4564407
B2=1.08664894
B3=1.09237637
B4=1.09704219
B5=1.38972984
B6=1.22431912
B7=1.38972336
B8=1.45836057
B9=1.08610999
B10=1.09729086
B11=1.09431458
B12=1.45643748
B13=1.08664812
B14=1.09237745
B15=1.09704249
B16=1.45836136
B17=1.0943123
B18=1.08610664
B19=1.09729411
A1=109.7499815
A2=108.81553612
A3=112.02806918
A4=116.04946364
A5=121.98054906
A6=116.03868093
A7=121.68105104
A8=110.72837255
A9=110.27219453
A10=110.99723336
A11=116.05129235
A12=109.7495716
A13=108.81599438
A14=112.02798567
A15=121.68021592
A16=110.99539676
A17=110.72947885
A18=110.27307076
D1=119.7012965
D2=120.18134778
D3=37.57485287
D4=7.57488038
D5=-172.41286687
D6=40.85995707
D7=-16.96723925
D8=102.37437907
D9=-137.99648886
D10=-172.42621862
D11=37.56312692
D12=157.26465976
D13=-82.5541639
D14=-139.13017392
D15=-138.02071905
D16=-16.99217476
D17=102.35038243

187 N,N,N’,N’-Tetrametiluréia (7) et1 HF/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
O,6,B6,1,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
C,1,B16,6,A15,7,D14,0
H,17,B17,1,A16,6,D15,0
H,17,B18,1,A17,6,D16,0
H,17,B19,1,A18,6,D17,0
Variables:
B1=1.44642802
B2=1.07400121
B3=1.08625854
B4=1.08626359
B5=1.3470198
B6=1.19757016
B7=1.42802405
B8=1.45646297
B9=1.08363032
B10=1.08299684
B11=1.08702915
B12=1.45646217
B13=1.08363076
B14=1.08702967
B15=1.08299681
B16=1.4473792
B17=1.08592628
B18=1.07537742
B19=1.08593328
A1=110.56703938
A2=109.97153137
A3=109.9734067
A4=125.43920005
A5=123.02440836
A6=115.23798245
A7=110.44195193
A8=109.81467839
A9=108.68280118
A10=112.512021
A11=110.44189944
A12=109.81486233
A13=112.51215085
A14=108.68273865
A15=119.47702802
A16=110.11934057
A17=109.84821553
A18=110.12325647
D1=120.65676645
D2=-120.65795061
D3=0.02492896
D4=179.98368055
D5=-0.01816625
D6=-117.95989155
D7=56.85373172
D8=174.85042411
D9=-64.16023947
D10=117.96482323
D11=-56.85395504
D12=64.16000873
D13=-174.85061913
D14=0.00546734
D15=-120.42713295
D16=-0.04185883
D17=120.34432592

188 N,N,N’,N’-Tetrametiluréia (7) et1 B3LYP/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,3,D3,0
O,6,B6,1,A5,2,D4,0
N,6,B7,1,A6,2,D5,0
C,8,B8,6,A7,1,D6,0
H,9,B9,8,A8,6,D7,0
H,9,B10,8,A9,6,D8,0
H,9,B11,8,A10,6,D9,0
C,8,B12,6,A11,1,D10,0
H,13,B13,8,A12,6,D11,0
H,13,B14,8,A13,6,D12,0
H,13,B15,8,A14,6,D13,0
C,1,B16,6,A15,7,D14,0
H,17,B17,1,A16,6,D15,0
H,17,B18,1,A17,6,D16,0
H,17,B19,1,A18,6,D17,0
Variables:
B1=1.45444664
B2=1.08437988
B3=1.09512955
B4=1.09512875
B5=1.36240562
B6=1.22095846
B7=1.44912704
B8=1.46760638
B9=1.09159307
B10=1.09158857
B11=1.09785719
B12=1.46761054
B13=1.09159573
B14=1.09786142
B15=1.09158834
B16=1.45504033
B17=1.09447687
B18=1.08559496
B19=1.09448626
A1=109.54930187
A2=110.15728294
A3=110.15532205
A4=125.13919249
A5=123.13064624
A6=114.53125648
A7=110.1609946
A8=109.73058147
A9=108.66551689
A10=112.58138117
A11=110.16095166
A12=109.73092726
A13=112.58115219
A14=108.66646676
A15=119.18210982
A16=110.23553596
A17=109.13284128
A18=110.2414099
D1=120.5164016
D2=-120.51777786
D3=-0.01046913
D4=179.99017169
D5=-0.0124959
D6=-118.4727748
D7=58.19770703
D8=176.25451445
D9=-62.84364108
D10=118.40900666
D11=-58.18432988
D12=62.85667839
D13=-176.24171476
D14=0.00215564
D15=-120.33436312
D16=-0.05030872
D17=120.23346775

189 N,N,N’,N’-Tetrametiltiouréia (8) ef HF/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,3,D3,0
N,6,B6,1,A5,2,D4,0
C,7,B7,6,A6,1,D5,0
H,8,B8,7,A7,6,D6,0
H,8,B9,7,A8,6,D7,0
H,8,B10,7,A9,6,D8,0
C,7,B11,6,A10,1,D9,0
H,12,B12,7,A11,6,D10,0
H,12,B13,7,A12,6,D11,0
H,12,B14,7,A13,6,D12,0
C,1,B15,6,A14,7,D13,0
H,16,B16,1,A15,6,D14,0
H,16,B17,1,A16,6,D15,0
H,16,B18,1,A17,6,D16,0
S,6,B19,1,A18,2,D17,0
Variables:
B1=1.45117834
B2=1.07665289
B3=1.08350165
B4=1.08510286
B5=1.35407147
B6=1.35413988
B7=1.45556508
B8=1.07650511
B9=1.08521565
B10=1.08477418
B11=1.45117464
B12=1.08350416
B13=1.08510466
B14=1.07665485
B15=1.45555362
B16=1.08477631
B17=1.07650635
B18=1.08520871
B19=1.6780359
A1=110.5407119
A2=107.75905837
A3=111.81728728
A4=120.34413998
A5=115.83415565
A6=121.30377849
A7=111.09456412
A8=109.07666281
A9=111.28667151
A10=120.33906384
A11=107.75820473
A12=111.81899909
A13=110.5403157
A14=121.31346565
A15=111.2895757
A16=111.0926707
A17=109.07408932
A18=122.08352002
D1=119.10246508
D2=119.8541681
D3=43.24564367
D4=-168.4270555
D5=38.60534836
D6=-4.80272671
D7=114.10836406
D8=-126.56439181
D9=-168.41040852
D10=162.35787356
D11=-77.78768418
D12=43.25680509
D13=38.5391482
D14=-126.50638253
D15=-4.74286279
D16=114.16719615
D17=11.57854989

190 N,N,N’,N’-Tetrametiltiouréia (8) ef B3LYP/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,3,D3,0
N,6,B6,1,A5,2,D4,0
C,7,B7,6,A6,1,D5,0
H,8,B8,7,A7,6,D6,0
H,8,B9,7,A8,6,D7,0
H,8,B10,7,A9,6,D8,0
C,7,B11,6,A10,1,D9,0
H,12,B12,7,A11,6,D10,0
H,12,B13,7,A12,6,D11,0
H,12,B14,7,A13,6,D12,0
C,1,B15,6,A14,7,D13,0
H,16,B16,1,A15,6,D14,0
H,16,B17,1,A16,6,D15,0
H,16,B18,1,A17,6,D16,0
S,6,B19,1,A18,2,D17,0
Variables:
B1=1.45657373
B2=1.08647127
B3=1.09231304
B4=1.09552661
B5=1.37420368
B6=1.37450968
B7=1.46258254
B8=1.08606232
B9=1.09540821
B10=1.09404322
B11=1.45661688
B12=1.09230461
B13=1.09555553
B14=1.08646689
B15=1.462452
B16=1.09406679
B17=1.08606773
B18=1.09537566
B19=1.68014408
A1=110.11549232
A2=108.19216555
A3=111.82971239
A4=119.90027765
A5=115.03444843
A6=121.24127343
A7=110.53159271
A8=109.40649566
A9=111.51571557
A10=119.86866691
A11=108.18288474
A12=111.83906038
A13=110.11654568
A14=121.29827085
A15=111.52948904
A16=110.52175941
A17=109.3944166
A18=122.48214611
D1=119.68125573
D2=120.10810239
D3=40.19635605
D4=-167.63274126
D5=39.7035837
D6=-7.0542264
D7=111.93675022
D8=-128.48102025
D9=-167.60363007
D10=160.04224004
D11=-79.85438438
D12=40.36490864
D13=39.38650236
D14=-128.16477315
D15=-6.73124683
D16=112.2576688
D17=12.37357402

191 N,N,N’,N’-Tetrametiltiouréia (8) et1 HF/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,4,D2,0
C,1,B5,2,A4,5,D3,0
N,6,B6,1,A5,2,D4,0
C,7,B7,6,A6,1,D5,0
H,8,B8,7,A7,6,D6,0
H,8,B9,7,A8,6,D7,0
H,8,B10,7,A9,6,D8,0
C,7,B11,6,A10,1,D9,0
H,12,B12,7,A11,6,D10,0
H,12,B13,7,A12,6,D11,0
H,12,B14,7,A13,6,D12,0
C,1,B15,6,A14,7,D13,0
H,16,B16,1,A15,6,D14,0
H,16,B17,1,A16,6,D15,0
H,16,B18,1,A17,6,D16,0
S,6,B19,1,A18,16,D17,0
Variables:
B1=1.45647258
B2=1.08439204
B3=1.08438878
B4=1.0724045
B5=1.31979681
B6=1.42132928
B7=1.45666826
B8=1.08581962
B9=1.08280991
B10=1.0832692
B11=1.4566698
B12=1.08280931
B13=1.08582002
B14=1.08326921
B15=1.45599507
B16=1.08084951
B17=1.08084106
B18=1.08028454
B19=1.67166936
A1=109.43928428
A2=109.43794299
A3=110.4813663
A4=124.72668153
A5=114.88397253
A6=112.62309418
A7=112.54739737
A8=109.89382762
A9=108.21452588
A10=112.6225893
A11=109.8937734
A12=112.54740963
A13=108.21437625
A14=120.17620774
A15=110.6955899
A16=110.69415564
A17=108.29916511
A18=123.16098375
D1=118.62486505
D2=120.68722576
D3=-0.01636707
D4=0.01336718
D5=-115.99006223
D6=-68.708745
D7=52.40643511
D8=170.36367111
D9=115.98329002
D10=-52.40532195
D11=68.70996448
D12=-170.36262616
D13=179.99907736
D14=-59.97690744
D15=59.96252146
D16=179.99256119
D17=-0.00218996

192 N,N,N’,N’-Tetrametiltiouréia (8) et2 B3LYP/6-311+G(2d,p)
N
C,1,B1
H,2,B2,1,A1
H,2,B3,1,A2,3,D1,0
H,2,B4,1,A3,3,D2,0
C,1,B5,2,A4,5,D3,0
N,6,B6,1,A5,2,D4,0
C,7,B7,6,A6,1,D5,0
H,8,B8,7,A7,6,D6,0
H,8,B9,7,A8,6,D7,0
H,8,B10,7,A9,6,D8,0
C,7,B11,6,A10,1,D9,0
H,12,B12,7,A11,6,D10,0
H,12,B13,7,A12,6,D11,0
H,12,B14,7,A13,6,D12,0
S,6,B15,1,A14,2,D13,0
C,1,B16,6,A15,7,D14,0
H,17,B17,1,A16,6,D15,0
H,17,B18,1,A17,6,D16,0
H,17,B19,1,A18,6,D17,0
Variables:
B1=1.46021463
B2=1.09124391
B3=1.09127759
B4=1.08874153
B5=1.34559003
B6=1.43686773
B7=1.46677034
B8=1.09734698
B9=1.09089984
B10=1.09180149
B11=1.46676812
B12=1.09180144
B13=1.09090189
B14=1.09734799
B15=1.67396952
B16=1.4624633
B17=1.09355739
B18=1.09357067
B19=1.08366041
A1=110.54698383
A2=110.55357195
A3=108.59750441
A4=119.69548578
A5=114.35348106
A6=112.2815271
A7=112.40625728
A8=109.8000728
A9=108.39043297
A10=112.28230134
A11=108.39102708
A12=109.80055345
A13=112.40591654
A14=122.94069286
A15=124.34217209
A16=109.81797822
A17=109.82006607
A18=109.34470502
D1=118.91265381
D2=-120.54437855
D3=-179.9200113
D4=179.99729554
D5=-116.61629151
D6=-66.27418323
D7=54.64598252
D8=172.80195922
D9=116.61329734
D10=-172.80115247
D11=-54.64470523
D12=66.27471503
D13=-0.00044653
D14=-0.0493473
D15=120.5231955
D16=-120.41055552
D17=0.05650285