
CASO CLÍNICO - Departamento de Fisiologia e...
34
CASO CLÍNICO Clara Mota Randal Pompeu Faculdade de Medicina – UFC PET Medicina UFC
Transcript of CASO CLÍNICO - Departamento de Fisiologia e...

CASO CLÍNICO Clara Mota Randal Pompeu
Faculdade de Medicina – UFC
PET Medicina UFC

História Clínica
Identificação: AGM, 18 meses, sexo feminino, parda, natural de Orós (Ceará).
QP: caroços no corpo

HDA: Paciente apresentada ao serviço com adenomegalia generalizada. Apresentava cabelos cinza-prateados, fotofobia, hipocromia de pele (em face, tronco e membros). Foi hospitalizada com o diagnóstico de abcesso em região cervical posterior, otite média aguda e pneumonia lobar direita que evoluiu com derrame pleural sem identificação do agente etiológico.

Antecedentes:
Filha única de pais não consangüíneos, nasceu de parto normal a termo com peso de 2750g e estatura de 48cm.
Durante o primeiro mês de vida apresentou impetigo e aos três meses foi hospitalizada por gastroenterocolite aguda.

Antecedentes:
Nesta mesma ocasião, os cabelos e a pele, em região dorsal e membros, clarearam. Foi avaliada em serviço médico tendo sido cogitada a hipótese diagnóstica de vitiligo.
O seu desenvolvimento neuropsicomotor foi adequado e o esquema de vacinação completo para a idade.

Recebeu tratamento e, após a alta, continuou acompanhamento em regime de ambulatório. Achados:
Estado geral regular, levemente descorada, com déficit pôndero-estatural (peso e estatura abaixo do percentil 2,5).
Albinismo parcial óculo-cutâneo, fotofobia, cabelos cinza-prateados, amígdalas hipertrofiadas e adenomegalia submandibular e retroauricular bilaterais.

Realizada TC de tórax, pelve e abdome: discreta hepatoesplenomegalia.
Iniciou tratamento com vitamina C e ampicilina .
Durante quatro meses, não apresentou intercorrências.


Foi atendida novamente com otite média à direita, sinusite e adenite cervical à esquerda.
Realizado tratamento com antimicrobianos.
Apesar do tratamento, desenvolveu pneumonia em lobos inferiores, adenomegalia dolorosa e generalizada, acompanhada por hepatoesplenomegalia.
Foi hospitalizada.
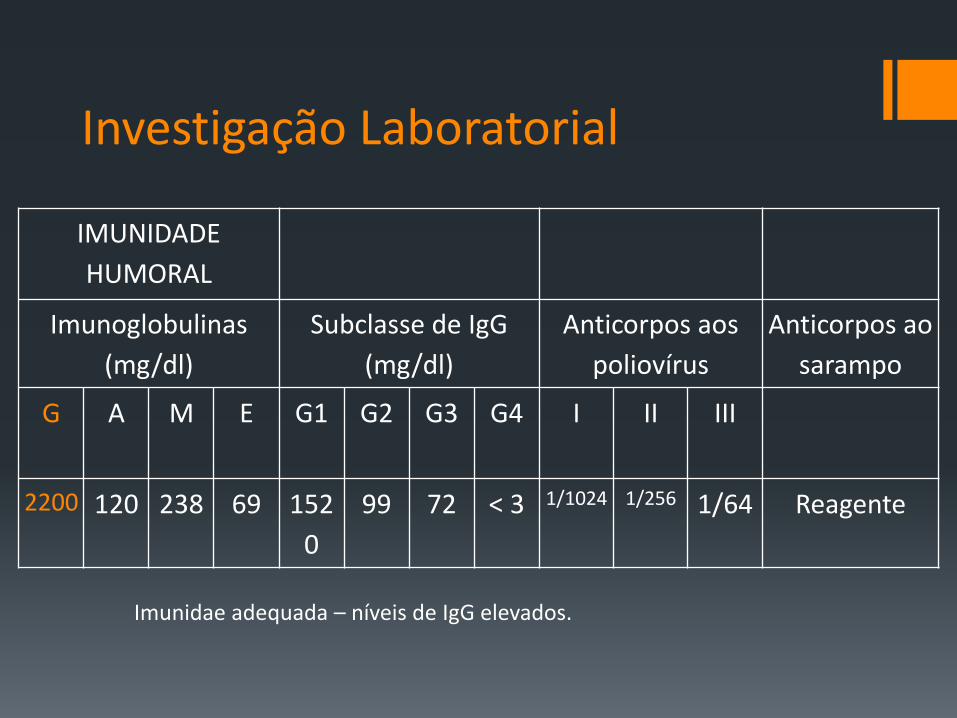
Investigação Laboratorial
IMUNIDADE
HUMORAL
Imunoglobulinas
(mg/dl)
Subclasse de IgG
(mg/dl)
Anticorpos aos
poliovírus
Anticorpos ao
sarampo
G A M E G1 G2 G3 G4 I II III
2200 120 238 69 152
0
99 72 < 3 1/1024 1/256 1/64 Reagente
Imunidae adequada – níveis de IgG elevados.
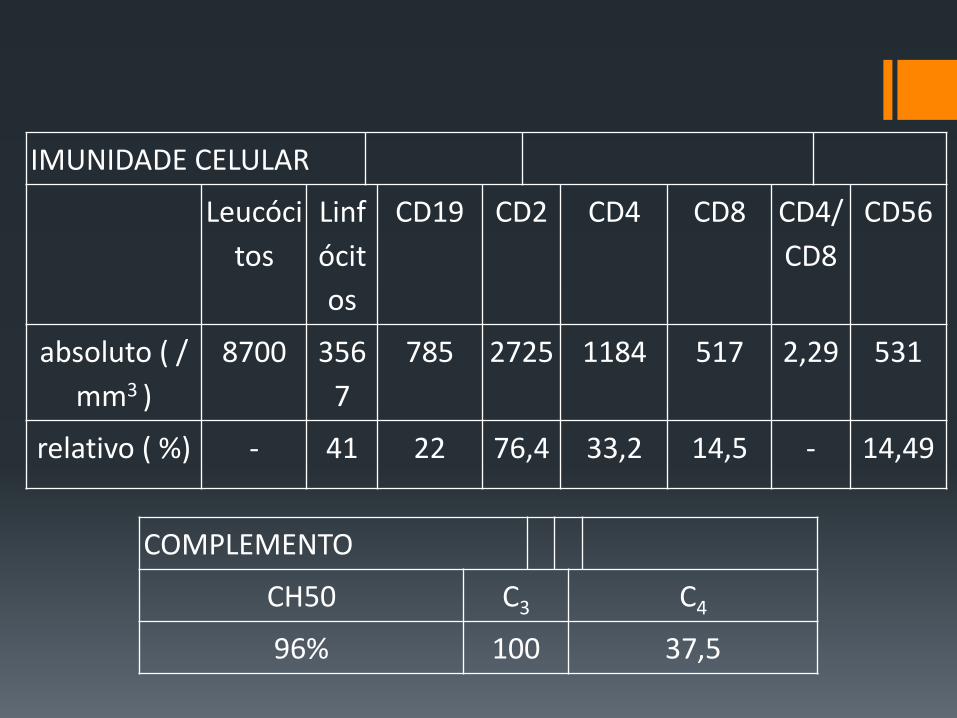
IMUNIDADE CELULAR
Leucóci
tos
Linf
ócit
os
CD19 CD2 CD4 CD8 CD4/
CD8
CD56
absoluto ( /
mm3 )
8700 356
7
785 2725 1184 517 2,29 531
relativo ( %) - 41 22 76,4 33,2 14,5 - 14,49
COMPLEMENTO
CH50 C3 C4
96% 100 37,5

AVALIAÇÃO DE
FAGÓCITOS
Quimiotaxia Migração
espontânea
Fagocitose % Bactérias
mortas
NBT
PNM MMN PMN MMN
P C P C P C P C P C P C
68,5 101,75 21,5 39,5 13 13,
4
6,4 8,9 36% 44% 38
%
51
%
Norm
al
P = Paciente C = Controle PMN = Polimorfonuclear MMN - Monomorfonuclear NBT = Nitroblue Tetrazolium Test


Exame Realizado Resultado
Biópsia de pele da região
dorsal
Melanossomas gigantes na
camada basal da epiderme
Fundo de olho Rarefação do epitélio pigmentar
da retina de ambos os olhos
Análise de fio de cabelo Grânulos gigantes e grosseiros de
melanina
Esfregaço de sangue
periférico (ponta de dedo)
Inclusões grosseiras no interior
de linfócitos e de granulações
azurófilas em segmentados

Mielograma (punção aspirativa
de crista ilíaca)
Granulações grosseiras no interior
de células da linhagem
granulocítica, por vezes com
formações gigantes de coloração
eosinofílica. O mesmo foi observado
em células de linhagem monocítica
Biópsia de linfonodo inguinal
esquerdo
Hiperplasia linfóide reacional
Estudo imunohistoquímico
(método strepto-avidina-
biotina-peroxidase)
Compatível com o diagnóstico de
hiperplasia linfóide reacional

Evoluiu com derrame pleural à direita e insuficiência respiratória, aumento rápido de volume dos gânglios, principalmente os cervicais, edema palpebral e hepatoesplenomegalia (fígado a 8cm do RCD e baço a 10 cm do RCE).
Evoluiu com septicemia, sangramentos, aparecimento de massa de provável origem linfoproliferativa.
Faleceu em 48 horas.


Síndrome de Chediak-Higashi!

Síndrome de Chediak-Higashi
Raro distúrbio autossômico recessivo
Mutação no gene CHS1/LYST do cromossomo 1q42.1-2
Proteína de transporte organelar defeituosa – leva a defeitos na fusão e transporte de lisossomos
Menos de 500 casos reportados nos últimos 20 anos.
Comum em pais consanguíneos
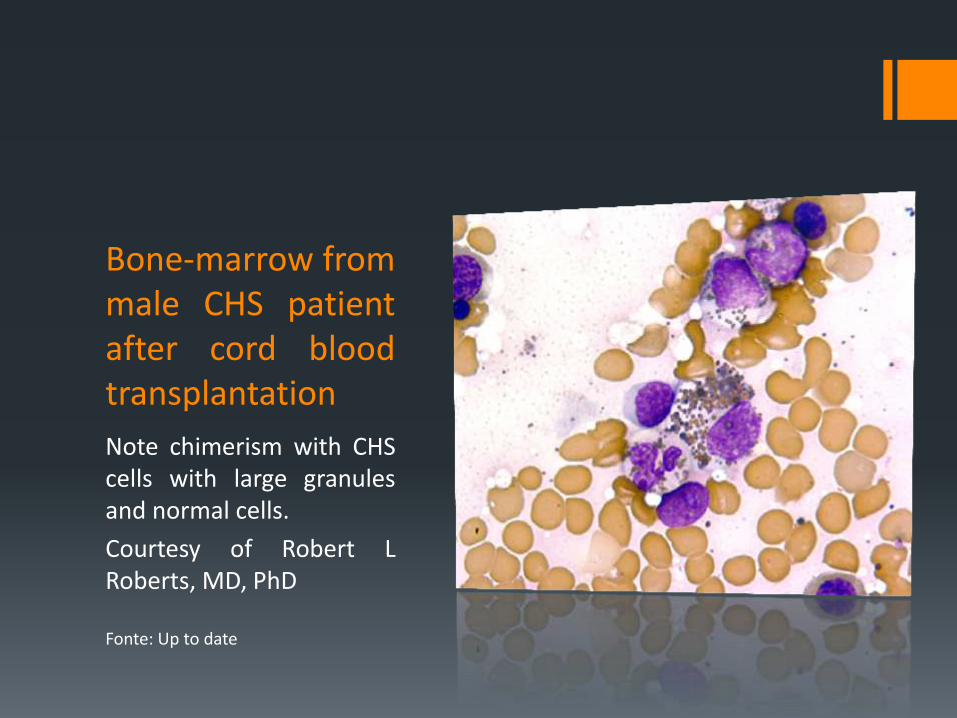
Bone-marrow from male CHS patient after cord blood transplantation
Note chimerism with CHS cells with large granules and normal cells.
Courtesy of Robert L Roberts, MD, PhD
Fonte: Up to date

Alterações Melanócitos: apresentam grânulos pigmentados
aumentados, que não são transferidos propriamente para queratinócitos e células epiteliais = albinismo óculo-cutâneo parcial
Neutrófilos: grânulo azurófilos gigantes, que não liberam seus conteúdos propriamente no caso de infecções virais e bacterianas, diminuindo a capacidade bactericida.
Neutrófilos: quimiotaxia reduzida.
Deficiência no pool de plaquetas = diástase sanguíneas
Reparo de membrana prejudicado.

Manifestações Clínicas
Albinismo óculo-cutâneo parcial
Infecções piogênicas recorrentes
Mecha prateada no cabelo

Manifestações Clínicas
Pigmentação ocular reduzida Fotofobia Acuidade visual diminuída Nistagmo Estrabismo Infecções: pele, trato respiratório e mucosas
(principalmente Staphylococcus aureus. Streptococcus pyogenes e Pneumococcus).
Gengivite e úlceras orais Enterocolite Função renal diminuída (reportada em animais).

Manifestações Clínicas
Defeitos de coagulação – sangramentos tornam-se mais importantes durante a fase acelerada
Hepatoesplenomegalia
Linfadenomegalia
Neurológicas: fraqueza e déficit sensorial, por conta de neuropatia periférica; ataxia; tremores; paralisia de nervos cranianos; declínio intelectual progressivo; convulsões; degeneração espino-cerebelar, com distúrbios de movimentação e demência.

Fase Acelerada
Infiltração linfocítica
Piora da imunodeficiência
Febre
Aumento da hepatoesplenomegalia
Adenomegalias
Pancitopenia
Sangramentos
Ocorre em 80% dos pacientes e costuma ser letal!

Exames
Neutropenia
Atividade quimiotática reduzida
Atividade bactericida reduzida
Hipergamaglobulinemia
Atrofia cerebral e medular
EEG: convulsões
Grânulos gigantes nas células de Schwann e musculares

Diagnóstico
Esfregaço de sangue periférico: grânulos azurófilos gigantes em neutrófilos, eosinófilos e outros granulócitos. Também encontrados em células da medula óssea, melanócitos, sistema nervoso, epitélio tubular renal, fibroblastos e mucosa gástrica.
Melanócitos: melanossomos
Pesquisa de mutação
Diagnóstico pré-natal: vilosidades coriônicas, sangue fetal e amostra de cabelo.

Diagnóstico Diferencial
Síndrome de Griscelli: imunodeficiência, albinismo parcial óculo-cutâneo, ausência de grânulos azurófilos.
Síndrome de Hermansky-Pudlak: albinismo óculo-cutâneo parcial, distúrbios de coagulação

Tratamento Medidas de suporte:
antitérmicos;
higiene oral e cuidados dentários;
higiene do ambiente físico e orientação nutricional;
fisioterapia respiratória e motora;
transfusão de glóbulos vermelhos e plaquetas se necessário; transfusão de glóbulos brancos não irradiados: tem aparentemente alterado o curso clínico dos pacientes;
vacinas contra pneumococo e H. influenzae b

Tratamento
Tratamento com antibióticos Antibioticoterapia profilática A associação
Sulfametoxazol-Trimetoprim é usada em alguns pacientes
Antifúngicos: usado quando não houver resposta às drogas antibacterianas ou quando se comprova a etiologia fúngica
Antivirais: Aciclovir: quando a fase acelerada se relaciona com infecção viral
Ácido ascórbico: na dose de 1g/dia, com baixa toxicidade.

Tratamento
Fase acelerada
colchicina
prednisona, vincristina e ciclofosfamida
Interferon
Gamaglobulina endovenosa
Interleucina 2 (IL-2)
Esplenectomia

Transplante de Medula Óssea
Transplante de medula óssea (TMO): Tto de escolha, sobretudo na fase precoce Única opção de cura Dificuldades concomitantes com drogas
imunossupressoras; infecção e complicações graves a longo prazo como reação enxerto versus hospedeiro fazem desta uma opção para poucos pacientes
O TMO alogênico tem sido proposto como tratamento curativo da SCH e capaz de prevenir a fase acelerada40.

Bibliografia
Site Up To Date: Chediak-Higashi syndrome
Site Up To Date: Primary disorders of phagocytic function: An overview


















