
INSTITUTO OSWALDO CRUZ INSTITUTO DE TECNOLOGIA … · Tabela 28 - Incerteza padrão do Método tipo...
136
INSTITUTO OSWALDO CRUZ INSTITUTO DE TECNOLOGIA EM IMUNOBIOLÓGICOS Pós-Graduação em Biologia Celular e Molecular Mestrado Profissional em Tecnologia de Imunobiológicos Ana Lucia Martins de Luna Palmigiani Avaliação das Incertezas de Medições Analíticas em Implementação de um Modelo de Controle Rio de Janeiro 2005 Dissertação apresentada ao Instituto Oswaldo Cruz com parte dos requisitos para obtenção do título de Mestre em Tecnologia de Imunobiológicos.
Transcript of INSTITUTO OSWALDO CRUZ INSTITUTO DE TECNOLOGIA … · Tabela 28 - Incerteza padrão do Método tipo...

INSTITUTO OSWALDO CRUZ INSTITUTO DE TECNOLOGIA EM IMUNOBIOLÓGICOS
Pós-Graduação em Biologia Celular e Molecular Mestrado Profissional em Tecnologia de Imunobiológi cos
Ana Lucia Martins de Luna Palmigiani
Avaliação das Incertezas de Medições Analíticas em Implementação de um Modelo de Controle
Rio de Janeiro
2005
Dissertação apresentada ao Instituto Oswaldo Cruz com parte dos requisitos para obtenção do título de Mestre em Tecnologia de Imunobiológicos.

ii
Trabalho Realizado no Instituto de Tecnologia em
Imunobiológicos, no Departamento de Qualidade,
sob a orientação da Dra. Silvana do Couto Jacob.

iii
INSTITUTO OSWALDO CRUZ
INSTITUTO DE TECNOLOGIA EM IMUNOBIOLÓGICOS Pós-Graduação em Biologia Celular e Molecular
Mestrado Profissional em Tecnologia de Imunobiológi cos
Ana Lucia Martins de Luna Palmigiani
Avaliação das Incertezas de Medições Analíticas em Implementação de um Modelo de Controle
Orientadora: Doutora Silvana do Couto Jacob Aprovada em 29/06/2005 Examinadores: Dra. Akie Kawakami Ávila - Presidente Dra. Verônica Maria de Araújo Calado - Titular Dr. Laerte da Cunha Azeredo - T itular Dr. Josino Costa Moreira - Suplente Dra. Queenie Siu Hang Chui - S uplente
Rio de Janeiro, 29 de junho de 2005.

iv
Dedicatória
Ao meu filho amado, Lucas.

v
Agradecimentos
Aos meus pais pelo amor, dedicação e perseverança que conduziram a mim e aos meus irmãos. Mostrando-nos a importância de lutarmos por aquilo que
acreditamos. Ao meu marido, pela compreensão e entusiasmo nas dificuldades e nos momentos que achava que iria desistir. Sempre com paciência e palavras de carinho e amor. Ao meu filho querido, por entender que a dedicação a esse trabalho o roubou momentos de prazer e que acima de tudo acreditou que poderia tornar-me uma melhor mãe. Com todo o seu amor ! Ao meu irmão Sylvio, pelos acertos nos piores momentos com a “distância que separa o computador do encosto de seu operador”. Aos funcionários e amigos do LAFIQ, que deram muita força e energia para poder continuar. Esse trabalho é para vocês! Que ele sirva de ferramenta e propicie tranqüilidade e segurança nas tomadas de decisões. A Izabel Cristina Crespo pela paciência que dispensou a análises, correções e opiniões pertinentes. Foram bem positivas. A Cláudia Amorim pela força na organização com a impressão. Ao João Crispim, pela dedicação nas análises de águas. A Márcia Arissawa por ter dividido comigo aquele presente de Deus, o ás da informática. Ao Filipe Cavalcante pela ajuda preciosa nos programas de Excel. A Cláudia Maria Dias pela importante ajuda “Statistica”. A professora Verônica Maria Araújo Calado pelo carinho, atenção e esclarecimentos oportunos com diferentes pontos de vista.

vi
A Aline Xavier pela contribuição nas estimativas de incertezas com importantes opiniões. Ao Sr. Paulo Couto, INMETRO, pela ajuda no esclarecimento da norma ISO GUM. A Sra. Yukie Yrata pela valiosa e incontestável contribuição no tratamento dos dados e estimativas de incerteza, que demonstraram inovações. Foi muito importante! Obrigada. A Darcy Akemi Hokama, por acreditar que não iríamos deixar a “corda ruir”. A Zaíra Antunes e Sheila Farage pela paciência e zelo por todos nós e nossos trabalhos. Fizemos tudo que pensávamos não conseguir...Obrigada! Ao Diretor, Dr. Akira Homma, pela oportunidade, credibilidade, inovação, entusiasmo e acima de tudo por mostrar-nos tudo isso. Muito obrigada!
A todos aqueles que torceram e que de alguma forma tornaram isso tudo possível.

vii
Ao meu Deus

viii
Índice
Índice.............................................................................................................viii
Índice de Ilustrações ....................................................................................... xi
Índice de Tabelas........................................................................................... xii
Lista de Abreviações ..................................................................................... xiv
Resumo ........................................................................................................ xvi
Abstract ........................................................................................................xvii
1 - Introdução...................................................................................................1
1.1- Histórico ...................................................................................................1
1.2 - Regulamentação ......................................................................................4
1.3- Ferramentas de Qualidade ........................................................................9
1.4 - Validação............................................................................................... 14
1.5 - A Incerteza ........................................................................................... 24
1.6- A Condutividade...................................................................................... 32
1.7 - A cromatografia de íons ......................................................................... 39
2- Objetivo Geral............................................................................................ 41
2.1- Objetivos Específicos: .................................................................................... 41
3 - Materiais e Métodos .................................................................................. 42
3.1- Descrição do Método tipo I ...................................................................... 44
3.1.2- Materiais e condições de análise .......................................................... 44
3.1.3- Reagentes e Soluções ......................................................................... 45
3.1.3- Preparo da Curva de Calibração........................................................... 46
3.1.4- Preparo das Amostras.......................................................................... 46
3.2- Descrição do Método tipo III .................................................................... 47
3.2.1- Materiais do Método tipo III .................................................................. 47
3.2.2- Condições de Análise........................................................................... 48
4 - Resultados e Discussão: ........................................................................... 48
4.1 - Resultados para o Método tipo I ............................................................. 50
4.1.1- Primeira curva de calibração ................................................................ 50
4.1.1.1- Resultados obtidos pelo analista 1 .................................................... 53

ix
4.1.1.2- Cartas de Controle dos Resultados obtidos pelo Analista 1................ 54
4.1.1.4 - Resultados obtidos pelo Analista 2 ................................................... 56
4.1.1.5 - Cartas de Controle dos Resultados do Analista 2 ............................. 57
4.1.2 - Segunda Curva de Calibração ............................................................ 58
4.1.2.1 - Resultados obtidos pelo Analista 3 ................................................... 60
4.1.2.2 - Cartas de Controle dos Resultados do Analista 3 ............................. 61
4.1.3 - Estudo e tratamento dos dados obtidos pelos três Analistas ................ 62
4.1.3.1 - Teste F(Snedecor) ..................................................................... 62
4.1.3.2 - Verificação de Tendência ................................................................. 63
4.1.3.3 - Teste t, comparação da média experimental ..................................... 66
4.1.3.4 - Recuperação dos analistas 1 e 3 ...................................................... 68
4.1.4 - Validação do Método tipo I .................................................................. 69
4.1.4.1- Seletividade ...................................................................................... 69
4.1.4.2 - Faixa de trabalho ............................................................................. 70
4.1.4.3 - Linearidade ...................................................................................... 70
4.1.4.4 - Precisão........................................................................................... 70
4.1.4.5 – Exatidão .......................................................................................... 74
4.1.4.6 – Robustez ......................................................................................... 77
4.1.5 - Diagrama de Causa e Efeito................................................................ 77
4.1.6 - Relatando as fontes de incerteza estimadas do Método tipo I .............. 78
4.1.6.1 - Estudo das variáveis e suas contribuições........................................ 78
4.1.7 - Diagrama de Ishikawa “refinado” do método tipo I ............................... 86
4.1.8 - Contribuições para a Incerteza Padrão Combinada do Método tipo I.... 87
4.2- Método tipo III ......................................................................................... 88
4.2.1- Análise de estabilidade da amostra e carta de controle......................... 88
4.2.2- Comportamento da Amostra e carta de controle ................................... 90
4.2.3- Comportamento dos resultados gerados por três analistas do Método tipo III.
...................................................................................................................... 93
4.2.4- Parâmetros de Validação ..................................................................... 98
4.2.4.1 - Especificidade .................................................................................. 98
4.2.4.2 - Precisão........................................................................................... 98
4.2.4.3 - Robustez........................................................................................ 101
4.2.5 - Diagrama de Causa e Efeito do Método tipo III .................................. 101

x
4.2.6 - Estimativas de Incerteza ................................................................... 102
4.2.6.1 - Cálculo da Incerteza Padrão Combinada do Método tipo III ............ 103
4.2.6.2 - Estimativa de Incerteza Expandida do método tipo III ..................... 104
4.7- Diagrama de Causa e Efeito “refinado” do método tipo III ...................... 104
4.8 - Contribuições para a Incerteza Padrão Combinada do Método tipo III... 105
5 - Conclusão............................................................................................... 106
6- Anexos .................................................................................................... 108
ANEXO A ........................................................................................................ 108
ANEXO B ........................................................................................................ 109
ANEXO C......................................................................................................... 110
ANEXO D ........................................................................................................ 111
7 - Bibliografia.............................................................................................. 112

xi
Índice de Ilustrações Ilustração 1 - Estrutura Organizacional do Laboratório de Controle Físico-Químico........4
Ilustração 2: Demonstração gráfica das possíveis causas de erros nos resultados
analíticos. ................................................................................................................11
Ilustração 3: Método normalizado, segundo USP 25. ....................................................16
Ilustração 4: Avaliação gráfica da incerteza padrão de medição....................................25
Ilustração 5 - Primeira curva de calibração
[ ] pico) do áreay ( x ão)concentraç ( ==χ utilizada pelos Analistas 1 e 2 .....................50
Ilustração 6 - Carta de Controle dos dados individuais do Analista 1...........................54
Ilustração 7 -Carta de Controle dos dados médios do Analista 1...................................54
Ilustração 8 - Carta de Controle dos dados individuais do Analista 2.............................57
Ilustração 9 - Carta de Controle dos dados médios do Analista 2..................................57
Ilustração 10 - Segunda Curva de calibração [ ] pico) do áreay ( x ão)concentraç ( ==χ
utilizada pelo Analista 3...........................................................................................58
Ilustração 11 - Carta de Controle dos dados individuais do Analista 3...........................61
Ilustração 12 - Carta de Controle dos dados médios do Analista 3................................61
Ilustração 13 - Gráfico repetitividade x Reprodutibilidade ..............................................72
Ilustração 14 - Gráfico Box Whiskers dos três analistas ................................................73
Ilustração 15 - Comportamento dos dados dos três analistas relacionado as medições
................................................................................................................................74
Ilustração 16 - Gráfico de dispersão dos resíduos em relação ao valor ideal. ...............76
Ilustração 17: Gráfico de Ishikawa do método I..............................................................78
Ilustração 18 - Preparo das soluções de trabalho ..........................................................84
Ilustração 19- Diagrama de Ishikawa refinado do método tipo I....................................86
Ilustração 20 - Relação Fontes de Incerteza versus Percentual de contribuição ...........87
Ilustração 21: Carta de controle com as medidas de estabilidade .................................89
Ilustração 22 : Carta de controle do comportamento dos dados da água WFI ao longo
de um mês, com duas amostragens diárias............................................................92
Ilustração 23 - Medidas de condutividade do analista 1.................................................95
Ilustração 24 - Medidas de condutividade do Analista 2 ................................................95
Ilustração 25 - Medidas de condutividade do Analista 3 ................................................96
Ilustração 26 - Gráfico de Reprodutibilidade ................................................................100

xii
Ilustração 27 - Gráfico Box Whiskers dos três analistas ..............................................100
Ilustração 28: Diagrama de Ishikawa do método III......................................................101
Ilustração 29 - Diagrama de Causa e Efeito refinado do Método tipo II .......................104
Ilustração 30 - Relação de variáveis que contribuíram para a Estimativa de Incerteza
Padrão Combinada do Método tipo III...................................................................105
Índice de Tabelas Tabela 1 - Classificação de métodos e parâmetros analíticos recomendados à
validação. ................................................................................................................15
Tabela 2 - Percentuais de recuperação de amostras.....................................................20
Tabela 3 - Atualização da Curva de Horwitz ( AOAC , 2000 ). .......................................21
Tabela 4 - Relação de medidas de condutividade do estágio 1. ....................................36
Tabela 5 - Estágio 3 de condutividade. ..........................................................................37
Tabela 6 - Requerimentos de Qualidade Química conforme Portaria 1469 de
29/12/2000 – Ministério da Saúde, para água potável, USP 26 água purificada e
destilada e pop 106040-617....................................................................................38
Tabela 7 - Preparo da curva padrão...............................................................................46
Tabela 8 - Área dos picos obtidos para diferentes soluções padrão usadas para
confecção da curva de calibração 1. .......................................................................51
Tabela 9 - Parâmetros de regressão linear da primeira curva de calibração, obtidos
através do programa Excel. ....................................................................................51
Tabela 10 - Dados obtidos pelo Analista 1. ....................................................................53
Tabela 11 - Dados obtidos pelo Analista 2. ....................................................................56
Tabela 12 - Área dos picos obtidas para diferentes soluções padrão usadas para
confecção da curva de calibração 2. .......................................................................58
Tabela 13 - Regressão Linear da segunda curva de calibração. ...................................59
Tabela 14 - Resultados obtidos pelo Analista 3. ............................................................60
Tabela 15 - Relação de Variâncias dos três analistas....................................................62
Tabela 16 - Taxas de Recuperação dos dados obtidos pelos Analistas 1 e 3. ..............68
Tabela 17 - Tabela comparativa de valores corrigidos..................................................71

xiii
Tabela 18 -Relação de coeficientes variação e analistas...............................................71
Tabela 19 - Relação de resíduos entre os dados gerados por cada analista estudado. 75
Tabela 20 - Contribuição da variável Massa na estimativa de Incerteza do método tipo I.
................................................................................................................................78
Tabela 21 - Contribuição da variável Volume na estimativa de Incerteza do método tipo
I. ..............................................................................................................................79
Tabela 22 - Relação de desvios padrão antes da correção. ..........................................81
Tabela 23 - Resultados de condutividade para o estudo da estabilidade da amostra....88
Tabela 24 - Condutividade da WFI DE amostras coletadas por trinta dias consecutivos.
................................................................................................................................91
Tabela 25 - Dados estatísticos dos valores da condutividade ( comportamento da
amostra em 30 dias)................................................................................................92
Tabela 26 - Comportamento dos Analistas com a análise de condutividade em WFI....94
Tabela 27 - Relação desvio padrão x analista - Método III. ...........................................99
Tabela 28 - Incerteza padrão do Método tipo III...........................................................102

xiv
Lista de Abreviações
ABNT Associação Brasileira de Normas Técnicas
AMC Analytical Methods Committee
ANVISA Agencia Nacional de Vigilância Sanitária
AOAC Association of Official Analytical Chemist
ASTM American Society for Testing and Materials Standards
BPF Boas práticas de fabricação
BPL Boas Práticas de Laboratório
B+S Baush Strobel
CEP Controle estatístico de processo
CCQ Círculos de Controle de Qualidade
DTP Vacina contra a Difteria, o tétano e a pertussis
FAO Food and Agriculture Organization of the United Nations
FDA Food and Drug Administration
FIOCRUZ Fundação Oswaldo Cruz
GSK GlaxoSmithKline
Hib Haemophilus influenzae tipo b
HPIC High performance ion chromatography
HPLC High Performance Liquid Chromatography
INMETRO Instituto Nacional de Metrologia, Normalização e Qualidade Industrial
ICH International Conference on Harmonization
ISO International Standardization Organization
IUPAC International Union of Pure and Applied Chemistry
LALIO Laboratório de Liofilização.
LAFAM Laboratório de Febre Amarela
LAFIQ Laboratório Físico-Químico
Log Book Caderno de registro de operação/ utilização
LSL Lower Control Limit
NBR Norma Brasileira Registrada
NMKLC Nordic Comittee on Food Analyses

xv
OPAS Organização Pan Americana de Saúde
PNI Programa Nacional de Imunização
POP Procedimento operacional padronizado
PRP Polissacarídeo capsular purificado
PRR’P Poliribosilribitol fosfato
PW Purified Water
RDC Resolução da Diretoria Colegiada
SI Sistema Internacional
TOC Total Organic Carbon
USL Up Control Limit
USP United States Pharmacopeia
VIM Vocabulário internacional de metrologia
WFI Water for injection
WHO World Health Organization

xvi
Resumo O objetivo deste trabalho foi aprimorar a qualidade analítica do laboratório de
Controle Físico-Químico, LAFIQ, de Bio - Manguinhos através da implantação de
ferramentas que permitam avaliar o desempenho dos resultados do laboratório e criar
um modelo de controle. Foram estudados dois métodos de análise: determinação do
conteúdo de polissacarídeo na vacina contra Haemophillus influenzae tipo B – Hib que
corresponde a categoria de método tipo I, conforme Farmacopéia Americana e da
condutividade em água para injetáveis correspondente a categoria de método tipo III.
Os parâmetros de validação e adequação dos dois métodos foram estudados e cartas
controle foram construídas utilizando os dados das análises de amostras referência
realizadas por diversos analistas em dias diferentes. Com parâmetros calculados a
partir destas cartas foi possível avaliar quão correto é um resultado comparado ao valor
desejado, representado pelo valor da grandeza que está sendo medido. O parâmetro
desvio-padrão, e o correspondente intervalo de confiança foram utilizados para medir a
dispersão dos resultados através do cálculo das incertezas das medições. Para o
método tipo I as estimativas das incertezas padrão combinada e expandida foram u =
0,50 µg / mL e U = 1,00 µg / mL, respectivamente. Foram considerados os parâmetros
Reprodutibilidade, a concentração da solução de trabalho, do volume, da pureza do
reagente, e da massa, como os que mais contribuíram para a incerteza desta categoria
de método. Para o método tipo III foram encontradas as incertezas u = 0,03 µS / cm e U
= 0,06 µS / cm, como incertezas padrão combinada e padrão expandida. Os fatores que
mais contribuíram na incerteza deste método foram os relacionados a qualificação /
calibração do equipamento, a estabilidade da amostra e a precisão do procedimento
dos analistas. O uso destes recursos estatísticos desenvolvidos e estudados na
presente dissertação levarão ao aprimoramento do controle da qualidade dos
imunobiológicos garantindo a qualidade do trabalho dos analistas e a maior
confiabilidade dos resultados fornecidos pelo LAFIQ.

xvii
Abstract The objective of this work was to improve the quality of the Bio-Manguinhos’
Laboratory of Physico-Chemical Quality Control using tools that allow to evaluate the
analytical performance. The work was carried out studying two analytical methodologies:
determination of the content of polysaccharide in Haemophillus influenzae type b
vaccine, that is type I according to American Pharmacopeia’s and conductivity
measurement in injectable waters, type III. In this study it was suggested a model that
allows to guarantee the quality of results emitted by the Control Laboratory. The
validation of the chosen methods and the construction of chart controls were made by
evaluating the validation parameters, data for reference sample and estimating the
uncertainty of the measurements. This last one is considered the quantified attribute that
allows estimating if the result is correct compared to the certified value. The uncertainty
corresponds to the range of the established confidence that means the dispersion of the
results. All these statistics tools will improve the implementation of a Quality System. For
the method type I the uncertainty standard and expanded were u = 0,50 µg / mL and U =
1,00 µg / mL, respectively. The calculations of the measurement uncertainty were
performed considering the follow sources: Reproducibility, concentration of the standard
solutions, volume, purity of reagent, and mass. For the method type III, the values were
u = 0,02 µS/cm and U = 0,06 µS/cm. The parameters that contributed to the uncertainty
of this method were: the behavior of the sample, the qualification and performance of
equipment used and the stability of water samples. For both studied analytical methods
the main contribution for the analytical results uncertainties was the precision of the
analytical procedures.
The use of these statistic tools will improve the confidence on the analytical
results from the LAFIQ.

1
1 - Introdução
1.1- Histórico As vacinas foram criadas no século passado, a partir do estudo de Pasteur,
que criou as primeiras vacinas atenuadas virais e as bacterianas. A primeira vacina
foi descoberta em 1796 por Edward Jenner, que desenvolveu a vacina contra a
varíola, conforme Moulin, 1996 e Fernandes, 2000 . Com os avanços da biologia e
da genética, aliados à constatação de que as vacinas são o instrumento de saúde
pública que gera o maior benefício ao menor custo, levaram o governo a traçar, em
1991, a meta de auto-suficiência em imunobiológicos de maior impacto para o
quadro epidemiológico nacional. O mérito é do trabalho realizado pelo Instituto de
Tecnologia em Imunobiológicos de Manguinhos (Bio-Manguinhos) da Fundação
Oswaldo Cruz - FIOCRUZ, que é o responsável pela produção de vacinas e reativos
(kits de diagnóstico de doenças infecto-parasitárias). A história de Bio-Manguinhos
se confunde com a da própria FIOCRUZ, já que ela nasceu como Instituto
Soroterápico, destinado a produzir soros e vacinas. A partir de 1976, Bio-
Manguinhos começou a ganhar aspecto industrial que tem hoje, quando começou a
produzir mais de 100 milhões de doses anuais de vacinas e reativos, dados
fornecidos do IV Congresso Interno da FIOCRUZ, Termos de referência, 2002.
A Unidade Bio-Manguinhos está no contexto da FIOCRUZ, como produtora
de imunobiológicos. Ela atende a demanda do país, através do Programa Nacional
de Imunizações (PNI) e também à demanda externa através da exportação da
vacina contra a febre amarela. O Centro que integra o Complexo Tecnológico de
Vacinas é o maior produtor de imunizantes da América Latina e um dos maiores do
mundo, com capacidade de preparação de cerca de 200 milhões de doses anuais.
Em 2004/2005, a FIOCRUZ, em parceria com o Instituto Butantã, começou a
produzir a vacina Penta Brasil, uma associação das vacinas difteria, tétano e
caxumba - DTP, Haemophilus influenzae tipo b - Hib e da hepatite tipo B - Jornal de
Bio-Manguinhos ( BIONotícias), 2004.
Maior produtor brasileiro de vacinas, respondendo por 60% da produção
nacional, Bio-Manguinhos fabricou no ano de 2003 cerca de 80 milhões de doses.

2
Foram 30 milhões de doses da vacina contra febre amarela; 34 milhões da vacina
contra paralisia infantil (poliomielite); e 16 milhões da vacina tetravalente,
associação da vacina DTP - contra difteria, tétano e coqueluche - e da vacina contra
a bactéria Haemophilus influenzae tipo b, Hib, que pode causar pneumonia e um tipo
de meningite. No ano de 2004 a produção ficou na casa dos 120 milhões de doses,
dos quais 26 milhões foram produzidas com destino à exportação. Para atender ao
PNI fabricaram-se 20 milhões de doses de vacina tríplice viral, que corresponde à
combinação dos imunizantes sarampo, rubéola e caxumba, 16 milhões de doses de
vacina contra a febre amarela, 20 mil da Hib, 17 milhões da tetravalente e 40
milhões de doses da vacina contra a poliomielite, conforme dados levantados
oficialmente da área comercial.
Em 1999, Bio-Manguinhos passou a produzir o imunizante contra
Haemophilus influenzae tipo b - Hib, bactéria que provoca meningite, otite e
pneumonia sendo uma das principais causas de doenças e mortes de crianças em
todo o mundo. Sua produção no país é resultado de um acordo de transferência de
tecnologia, no qual Bio-Manguinhos importa da Bélgica o antígeno concentrado,
realizando a formulação, envasamento, liofilização e processamento final da vacina.
No ano de 2004, Bio-Manguinhos começou a desenvolver as fases de produção,
através de fermentações bacterianas e obtenção do polissacarídeo Haemophillus para
conjugação com a proteína monomérica tetânica. Essa vacina sendo totalmente
nacional possibilitará a imunização de cinco doenças, com somente uma dose aplicada
em crianças. Isto será possível através da seguinte combinação de vacinas: Hib, DTP e
hepatite B. Sua forma de apresentação é de cinco doses por frasco. O Brasil torna-se
auto-suficiente na imunização do Haemophilus influenzae tipo b.
A preparação da vacina conjugada contra Hib, é baseada na tecnologia de
Schneerson, na qual o polissacarídeo é preparado a partir da cepa virulenta 20752 e
após ativação com brometo de cianogênio e extração com um separador de hidrazida
adípica, é combinado à proteína monomérica tetânica, através da condensação com
carbodiimida. Após purificação o conjugado é liofilizado em presença de lactose, que é
o estabilizador - Schneerson et al, 1987.
Cada dose de vacina reconstituída (0,5mL) contém 10 µg de polissacarídeo
capsular purificado (PRP) de Haemophilus influenzae tipo b, ligado covalentemente a
cerca de 30µg de toxóide tetânico (na forma de proteína monomérica tetânica) , com

3
excipiente lactose. A reconstituição é feita em diluente salina fenolada ou em vacina
líquida DTP. Com a última maneira de reconstituição ela imuniza até quatro doenças e
ambas as formas são utilizadas de forma intramuscular, como informação de sua bula.
Atualmente esta última é a forma mais utilizada pelo programa nacional de imunização
(PNI).
Inicialmente a vacina desenvolvida contra a doença, era constituída somente
pelo polissacarídeo capsular tipo b. Após estudos clínicos, foi verificado que a vacina
conferia uma baixa imunogenicidade de polissacarídeo poliribosil ribitol (PRRP) em
crianças menores de 18 meses, portanto era ineficiente para o objetivo a que se
dispunha. Essa deficiência gerou estudos e o desenvolvimento da forma carreadora da
proteína monomérica tetânica, que é metodologia de Schneerson, 1980. Com essa
formulação, a vacina contra Hib ligada à proteína monomérica tetânica, confere
imunidade a crianças na faixa etária de 2 a 6 meses, sendo necessárias três doses, e
um reforço aos dois anos de idade, já que na fase infantil a doença torna-se mais
severa. Sendo necessários 12,5 µg de polissacarídeo Hib / dose.
Após implantação das boas práticas de fabricação, BPF, em 1999, o
controle interno da produção de imunobiológicos passou por uma série de
reformulações, que visaram atender as expectativas da Unidade Bio-Manguinhos,
conforme as recomendações do Ministério da Saúde e da Agência Nacional de
Vigilância Sanitária, ANVISA, também das normalizações de caráter internacional.
A área de Garantia da Qualidade, atuante nas BPF da Unidade Bio-
Manguinhos existe desde 20/03/1990. Isso contribuiu para o aprimoramento e
implantação das BPF, nas áreas de Produção e Controle de Qualidade.
O Controle de Qualidade existe quase que simultaneamente ao nascimento
da Unidade, e vem desenvolvendo-se a cada ano buscando novas metodologias
analíticas de controle dos produtos e sua missão é garantir a qualidade dos
imunobiológicos de Bio-Manguinhos.
O Laboratório Físico-químico - LAFIQ, está inserido nesse contexto e faz
parte do Departamento de Qualidade. Ele emite laudos diários das análises de todos
os materiais que entram no Almoxarifado da Unidade, desde as matérias-primas e
insumos, até os materiais de embalagem em geral. Além disso, análises rotineiras
que são realizadas em diversos tipos de águas produzidas, soluções de processo,
produtos intermediários, produtos em desenvolvimento, soluções reagentes para kit

4
e diagnóstico e produtos imunobiológicos acabados. Os laudos emitidos são os
registros dos ensaios físico-químicos, padronizados de acordo com o tipo de
material e produto. O organograma do Departamento de Qualidade e Laboratório de
Controle Físico-Químico é mostrado na ilustração 1.
Ilustração 1 - Estrutura Organizacional do Laborató rio de Controle Físico-Químico
Na ilustração 1 pode-se observar as atribuições do Departamento de
Qualidade, do Laboratório de Controle Físico-Químico de Qualidade e a interação
com outros Departamentos, serviços e setores de Bio-Manguinhos que o LAFIQ
atende como seus clientes.
1.2 - Regulamentação A ANVISA, sob orientação e respaldos técnicos da Organização Pan-
americana de Saúde, OPAS, e Organização Mundial de Saúde, OMS, tem como
Lab. Cont.Microbiológico
Lab. de Cont. Biológico
Lab. de Meio de Cultura
Lab. de Exp.Animal
Lab. de Neurovirulência
Setor deAmostragem
Setor dematérias-primas
Setor de Produto Final
Setor de Apoio e Pesquisa
Produção Desenvolvimento Manutenção Almoxarifado Reativos Administração

5
interesse maior o estabelecimento de critérios objetivando garantir a qualidade e
segurança dos produtos imunobiológicos. Essas ações incluem as inspeções
realizadas e a efetivação de uma consciência de Qualidade Total, a começar pela
compra de insumos, produção, controle de processos, controle de qualidade e
produto acabado. Para isso metodologias eficazes de análise em parceria com o
acompanhamento de métodos e produtos, contribuem para atingir a meta da
Qualidade total assegurada - Resolução da Diretoria Colegiada (RDC) 210, 2003 e
304 de dezembro de 2004.
Entende-se por Qualidade como um modo de pensar na organização, ou
empresa. A implantação do controle estatístico de processo (CEP) deve ser baseada
num ambiente em que a qualidade seja parte integrante dos valores e da rotina da
organização, isto é, um ambiente em que comportamento organizacional seja
realmente condicionado pelo pensamento em qualidade. Um diagnóstico preliminar,
realizado para verificar o comportamento organizacional frente ao aspecto qualidade
pode evitar desperdícios futuros.
De acordo com a ANVISA, Qualidade e Produtividade, são fatores
essenciais para a competitividade, que devem ser preocupação constante dos
setores produtivos. A qualidade observou diferentes abordagens ao longo do tempo,
sendo até hoje fator chave de sucesso .
Ao longo do tempo, desde a fase de produção artesanal até os dias de
hoje, pelo menos, quatro diferentes abordagens vem sendo observadas, como relata
Alfredo Lobo, 2003 - INMETRO.
o A fase da produção artesanal que caracterizou-se pela total
aproximação entre o produtor e o consumidor. A interação plena
entre ambos propiciava que este passasse diretamente para o
produtor suas expectativas.
o A fase da revolução industrial que provocou grandes mudanças em
termos de abordagem da qualidade. O aumento da escala de
produção introduziu o chamado controle da qualidade. Inicialmente,
com foco na inspeção do produto final, o controle da qualidade
passou por uma série de aperfeiçoamentos. A inspeção em

6
diferentes etapas do processo produtivo, o controle estatístico da
qualidade, as cartas de controle, dentre outros, se destacaram. De
qualquer forma, o controle da qualidade tinha ênfase na detecção de
defeitos. O distanciamento entre quem produzia e quem consumia e
a segmentação do controle da qualidade, como conseqüência da
produção seriada, diluíram a responsabilidade pela qualidade e
problemas com qualidade dos produtos surgiram com maior
intensidade.
o A exploração espacial, os programas nucleares e mais recentemente
a exploração de petróleo em águas profundas, cujas instalações
demandam maior confiabilidade, provocaram uma nova e importante
mudança na abordagem da questão da qualidade nas empresas.
Estudos demonstraram que a maior parte dos problemas de
qualidade tinha origem em falhas gerenciais e não técnicas. Essa
constatação deu origem aos chamados sistemas de gestão de
qualidade, que associam ações de controle que, como anteriormente
mencionado, têm ênfase na detecção de defeitos, com ações de
administração da qualidade, que tem ênfase na prevenção de
defeitos.
o Pré-qualificar os fornecedores, analisar criticamente os projetos,
elaborar e qualificar os procedimentos de execução e de inspeção,
treinar e qualificar pessoal, calibrar os instrumentos de medição,
identificar expectativas e avaliar o grau de satisfação dos clientes,
dentre outras, são ações típicas de prevenção de defeitos, ou de
administração da qualidade. A base normativa hoje mais utilizada
para a implantação de sistemas de gestão da qualidade é a norma
ISO 9001, 2000. Diante da necessidade de implantar sistemas de
gestão da qualidade, os países, em especial os desenvolvidos,
começaram a estabelecer normas nacionais de gestão da qualidade.
Tal fato causou transtornos para as empresas exportadoras, que
tinham que implantar sistemas de gestão da qualidade com base em

7
diferentes padrões normativos, para atender diversos países. O
mérito da ISO 9000 foi exatamente unir as diferentes bases
normativas em uma única, hoje universalmente aceita.
No Brasil, a partir do início da década de 90, vem sendo observado um
grande movimento em prol da melhoria da qualidade de produtos e serviços. A
criação pelo Governo Federal do Programa Brasileiro da Qualidade e Produtividade,
as aberturas econômicas, que expôs as empresas brasileiras a um ambiente de
grande competição, a evolução do cidadão brasileiro enquanto consumidor, que
passou a exercer mais plenamente seus direitos e deveres, e a estabilização da
moeda foram fatores indutores e decisivos para esse movimento. Nesse período,
qualidade deixou de ser preocupação exclusiva dos técnicos para ser de todos, mais
em particular do gerente. O conceito atual é de que qualidade é adequação ao uso,
cujos requisitos devem estar previamente estabelecidos.
O mercado globalizado vem demandando novas abordagens em termos da
questão da qualidade. Uma adequada gestão pela qualidade, que tem decisiva
contribuição e que alavanca a competitividade, passou a ser decisiva para a
sobrevivência das empresas, no ambiente de grande competição hoje observado.
Surgem as chamadas barreiras não tarifárias, ou barreiras técnicas,
estabelecidas através da promulgação de normas, regulamentos ou procedimentos
de avaliação da conformidade. O fato é que o espaço para dificultar o acesso a
mercados através do estabelecimento de tarifas acabou para a grande maioria dos
países, passando estes a fazê-lo através das barreiras técnicas.
Por avaliação da conformidade entende-se a implementação de uma
sistemática, com regras pré-estabelecidas e devidamente acompanhadas e
avaliadas, que propicie adequado grau de confiança de que um produto, processo
ou serviço atende aos requisitos de uma norma ou regulamento técnico. O
mecanismo de avaliação da conformidade mais comumente utilizado e conhecido é
a certificação.
A certificação de produtos caracteriza-se pela existência de parceria
independente entre o produtor e o consumidor, quando o objetivo maior é a
qualidade desses produtos.

8
O grande desafio da avaliação da conformidade é sua utilização como
regulador de mercados. A adoção de programas de avaliação da conformidade,
obedecendo práticas internacionais, propiciará o reconhecimento mútuo entre
programas de diferentes países, permitindo um natural fluxo de produtos, sem o
ônus da repetição dos ensaios e avaliações nos países compradores.
Hoje, no Brasil, existem 45 programas de avaliação da conformidade de
produtos de caráter compulsório e 82 de caráter voluntário. As estruturas de
credenciamento de organismos de avaliação da conformidade e de laboratórios de
calibração e de ensaios, coordenados pelo Instituto Nacional de Metrologia,
Normalização e Qualidade Industrial - INMETRO, são as únicas da América Latina
reconhecidas internacionalmente, o que representa uma vantagem competitiva para
as empresas brasileiras - fonte INMETRO, por Alfredo Lobo, 2003.
O setor de produção de vacinas no Brasil, é marcado por desafios
peculiares. Ele requer uma base científica e tecnológica intensa. Tratam-se de
produções que implicam em alto custo e têm ciclos longos de processos. A
organização da produção é contínua e está sempre submetida a exigências
regulatórias. A demanda na área de produção nacional é caracterizada pelo setor
público, onde o principal usuário é o mercado interno, conforme Reinaldo
Guimarães, 2002 .
O acompanhamento de produtos bem como os estudos pós marketing
caracterizam dados históricos de produtos, relacionados a qualidade desses
produtos, ao comportamento populacional e suas doenças. Os estudos clínicos
demonstram como de fato um produto novo, ou uma nova formulação é relacionada
à imunidade de doenças com o objetivo de atingir a erradicação da mesma no país.
A adoção de práticas adequadas de gestão da qualidade, a normalização, a
metrologia e avaliação da conformidade, tudo isso representa um diferencial na
economia globalizada e, portanto fundamental importância para a exportação
brasileira.
Como ferramenta da qualidade, prevista nas normas de Boas Práticas de
Fabricação, BPF, da ANVISA, estão as técnicas e os critérios de validação de
processos, métodos e produtos. Como exigência da legislação vigente, RDC 304 -
ANVISA, a validação tem com objetivo assegurar a conformidade dos produtos
farmacêuticos em relação as especificações estabelecidas, que devem atender

9
padrões adequados de exatidão e reprodutibilidade. Para essa finalidade o FDA
publicou em 01/03/1998, em conjunto com o ICH - “Internacional Conference on
Harmonization” da “Technical Requirements for Registration of Pharmaceuticals for
Human Use” um guia contendo diretrizes sobre a validação de procedimentos
analíticos. De acordo com a seção 501 deste guia, os ensaios e especificações
existentes nas monografias da United States Pharmacopeia (USP) e do “National
Formulary” constituem padrões legais. Portanto, as metodologias de análise devem
ser escolhidas de modo a atender padrões farmacopeicos vigentes
As Boas Práticas de Laboratório, BPL, fazem parte desse sistema de
qualidade assegurada e é o quesito que diz respeito à organização e às condições
sob as quais os estudos em laboratório e de campo são planejados, realizados,
monitorados, registrados, relatados e arquivados, conforme definição do INMETRO,
Norma NIT DICLA 028 revisão 00.
Laboratórios de ensaios são normalizados conforme a International
Standardization Organization, ISO 17025. Segundo esta norma a validação de uma
metodologia analítica pode ser definida como a “confirmação por exame e
fornecimento de evidência objetiva de que os requisitos específicos são atendidos”
(NBR ISO/ IEC 17025: 2001) . A referência, segundo parâmetros de performance
analítica é a farmacopéia americana, USP 27- capítulo <1225> 2004; ASTM
internacional, 2004; RDC 304, 2004; FDA, 2000; INMETRO, 2003; RDC 210, 2003;
ISO 17025, 2001; IUPAC, 2002.
Os procedimentos operacionais padronizados (POP), de equipamentos e os
registros de dados correspondentes abrangem, entre outros, a operação, manutenção
rotineira, manutenção não rotineira, calibração e utilização do mesmo. O equipamento
usado nos estudos não deve interferir com o sistema-teste.
1.3- Ferramentas de Qualidade
Conhecido como diagrama de Ishikawa, pois foi introduzido por Kaoru Ishikawa
e como diagrama Espinha de Peixe ou diagrama de causa e efeito, recebeu essa
denominação devido à sua aparência. Ishikawa nasceu em 1915, com formação em

10
química aplicada pela Universidade de Tóquio, foi o primeiro a utilizar o termo
“Controle de Qualidade” e desenvolveu as ferramentas de qualidade. O diagrama de
causa e efeito, é uma representação gráfica que ajuda a identificar, explorar e
mostrar as possíveis causas de uma situação ou problema específico. Cada
diagrama tem uma grande seta apontada para o nome de um problema. Os ramos
que saem dessa seta representam as categorias de causas, tais como: mão-de-obra,
materiais, máquinas, meio ambiente, medidas, métodos e etc. As setas menores
representam itens dentro de cada categoria e possibilita a discussão, detalhada,
sobre o funcionamento de um processo ou sobre um problema. As Ferramentas de
Ishikawa são, de acordo com Schissatti, 1998:
o Diagramas de causa e efeito (espinha de peixe ou diagrama de Ishikawa)
o Histogramas
o Folhas de verificação
o Gráficos de dispersão
o Fluxogramas
o Cartas de controle.
Ishikawa observou que nem todos os problemas poderiam ser resolvidos
por essas ferramentas, ele percebeu que ao menos 95% poderiam ser, e que
qualquer trabalhador fabril poderia efetivamente utilizá-las. Embora algumas dessas
ferramentas já fossem conhecidas há algum tempo, Ishikawa as organizou
especificamente para aperfeiçoar o Controle de Qualidade computacional.
Conforme a ilustração 2 o gráfico de Ishikawa, diagrama de causa e efeito, é
possível visualizar as possíveis fontes de erros e conseqüentemente variabilidade
em resultados analíticos.

11
Ilustração 2: Demonstração gráfica das possíveis ca usas de erros nos resultados analíticos.
Para fins de estudo, é importante simplificar essas fontes de erros
verificando quais são verdadeiramente importantes e que acarretam variabilidade em
resultados analíticos. As fontes de erros levam à maior incerteza, que comprometem o
resultado final. No trabalho abordaremos as possíveis causas de erros e suas fontes de
incerteza nos dois métodos.
A implantação de qualidade nas empresas deve ser considerada como um
processo de mudança organizacional, onde o controle estatístico de processo, CEP,
é muito mais do que a introdução de novas ferramentas de melhoria de processos. A
rotina de atividades das funções, as atitudes e crenças da organização são objetos
de mudança. Desta forma, a implantação deve ser planejada como um processo de
mudança organizacional e cultural.
RESULTADO
FINAL
Causas pessoais
Causas materiais
Calibração
Sensibilidade
Linearidade
Repetitividade
Instabilidade
Amostragem
Treinamento
Experiência Qualidade
Condições
Validação

12
O sistema de controle a ser desenvolvido para garantir que essas ações
sejam implementadas e efetivadas a contento, geralmente, acarretam mudanças na
postura e atribuições dos que terão a responsabilidade pela melhoria dos processos.
Uma segunda observação refere-se à função das cartas de controle. Essas são
ferramentas que indicam que é necessário intervir no processo para mantê-lo no
estado desejado. É necessário acima de tudo conhecimento, tanto para se
interpretar a carta de controle e perceber o aviso, quanto para identificar o que deve
ser ajustado no processo. Em outras palavras, o fator gerencial é predominante na
ingerência de atitudes e correções necessárias que são sinalizadas por essas
ferramentas. Quando o conhecimento de problemas é a ponte da partida para a
correção e resolução dos mesmos, de acordo com Chambers, 1992.
O controle de processo é uma ferramenta imprescindível para a
credibilidade de uma produção e conhecimento dos desvios que ocasionalmente
possam ocorrer. Com ele é permitido checar sua performance, a capacidade e
facilita intervenções e conseqüentemente correções necessárias. Em se tratando
especificamente do sistema de ações corretivas vale ressaltar que enquanto a
manutenção da variabilidade do processo em um determinado patamar depende
basicamente de ações operacionais, isto é, das ações executadas por aqueles que
utilizam diretamente as cartas de controle, a redução da variabilidade depende do
esforço conjunto daqueles que têm autoridade técnica sobre o processo, de acordo
com Controle de Contaminação - julho, 2004.
Algumas ferramentas que dão subsídio ao andamento adequado de uma
fábrica são:
• Sistemas de Controle e Gerenciamento de Produção (execução,
planejamento, relatórios);
• Controle Estatístico de Processo (CEP);
• Rastreabilidade de processo;
• Sistema de Controle de Qualidade e Gerenciamento de Paradas e perdas do
processo;
• Sistemas de Autocontrole e Gerenciamento de Processos (expedição e
recebimento de materiais para a produção);
• Sistemas de Manutenção Preditiva e Preventiva;
• Integração com Sistemas de Gestão Empresarial.

13
Esse controle faz parte da avaliação técnica da qualidade. Segundo Kume,
1993, este sistema foi proposto inicialmente por Walter Shewhart. Os problemas
decorrentes das causas especiais ou aleatórias são inevitáveis em qualquer
processo. Porém, é possível que exista uma causa assinalável dos problemas
decorrentes de causas comuns e que existam fatores relevantes a serem
investigados. Shewhart interpretou a variabilidade como sendo passível de duas
possibilidades. Aquela que ocorre dentro dos limites definidos pelo acaso, e aquela
que está fora destes limites. Estando fora ele acreditava que as causas pudessem
ser identificadas, ou seja, elas seriam assinaláveis de acordo com Deming, 1997. A
variação controlada é caracterizada por um estável e consistente padrão de variação
sobre o tempo. Shewhart atribuiu tal variação a causas aleatórias onde a variação
não controlada é caracterizada por um padrão de variação que muda ao longo do
tempo. Ele atribuiu as mudanças no padrão de variação às causas assinaláveis.
As cartas de controle 3б são muito utilizadas para separar causas comuns
de causas especiais. A partir de uma média de dados de determinado processo, são
estabelecidos dois limites, um superior e um inferior, distantes 3б desta mesma
média. Considerando-se a normalidade do processo, desde que o mesmo esteja
estabilizado, existe uma probabilidade de 99,73% de que qualquer valor extraído do
processo esteja entre eles. As variações aleatórias são produzidas pelas interações
entre mão-de-obra, máquinas, materiais e métodos. O resultado líquido da variação
é relativamente consistente ao longo do tempo. Por outro lado, juntando às causas
aleatórias, há ocasionalmente fatores especiais que têm um grande impacto nas
características do produto, e estas causas segundo Shewhart poderiam ser
identificadas, Deming,1990 e Deming, 1997, Introdution to Statistical Quality Control.
4th edition. USA, 2001. Daí a importância de resultados corretos e conhecimento de
variáveis que possam contribuir para variabilidades de qualidade.
A partir de um pioneirismo japonês, muitos têm adotado, como ênfase, o
melhoramento contínuo de processos, e só assim, obtendo conformidade total às
especificações.
Para efeito de estudo, no trabalho foram abordados os diagramas de causa
e efeito (diagrama de Ishikawa), as cartas de controle para resultados analíticos,
dispersão dos dados, gráfico de Pareto e as estimativas de incerteza, provenientes

14
de estudos de validação e adequação analítica de controle de qualidade. Essas
ferramentas de controle foram aplicados e realizados nas seguintes metodologias:
cromatografia, por HPLC de troca iônica, categoria tipo I – denominado Método I e
na medição de condutividade, que é o método de categoria III - Método III, conforme
parâmetros farmacopeicos, USP 27, 2004 e NBR ISSO/ IEC 17025, 2001. Eles
foram selecionados entre tantos outros por motivo de seus resultados sinalizarem
possíveis correções de processo e serem métodos quimicamente críticos para o
Laboratório. Para esses métodos serão utilizadas amostras de polissacarídeo Hib e
água para injetáveis - Water for Injection, WFI, respectivamente, com o objetivo de
utilizar essas ferramentas para melhorar a qualidade dos processos produtivos
através de resultados analíticos, de Bio-Manguinhos e do Laboratório de Controle
Físico Químico de Qualidade.
1.4 - Validação
O primeiro passo para a obtenção de resultados analíticos confiáveis está na
validação do método analítico escolhido. A validação pode ser definida como a
“confirmação por exame e fornecimento de evidência objetiva de que os requisitos
específicos são atendidos”, segundo a (NBR ISO/ IEC 17025: 2001) . Um método
analítico pode ser validado intralaboratorialmente ou interlaboratorialmente. No próprio
laboratório, através de testes de recuperação e um ou dois dos enfoques seguintes, o
analista pode validar um método analítico em comparação com um método
independente ou através do emprego de material de referência certificado.
Interlaboratorialmente, a validação pode ser obtida através de um estudo colaborativo
envolvendo vários laboratórios. Se o método escolhido já foi objeto de um estudo
colaborativo, ainda assim o analista é obrigado a validá-lo intralaboratorialmente para
demonstrar que pode ser usado em seu laboratório, ou pelo menos fazer sua
qualificação. Os requisitos de validação estão claramente demonstrados e associados a
clientes e métodos.

15
A seguir a recomendação de parâmetros e classificação dos métodos a
serem validados (USP 27, 2004): o Categoria I: Quantificação de macro componentes em substâncias ativas ou
ingredientes ativos em produtos farmacêuticos acabados.
o Categoria II: Determinação de impurezas em substâncias ativas ou
componentes de degradação em produtos farmacêuticos acabados.
o Categoria III: Determinação de características físico-químicas em substâncias
ativas ou em produtos acabados (ex. dissolução, tamanho de partículas,
liberação da droga).
o Categoria IV : Testes de identificação
De acordo com a classificação acima os métodos devem seguir a seguinte
normalização de validação, como descritos na tabela 1:
Tabela 1 - Classificação de métodos e parâmetros an alíticos recomendados à validação.
Nos casos de métodos normalizados não é necessário efetuar o processo
completo de validação desde que não ocorram alterações significativas dos mesmos e
quando o produto em questão é o indicado em monografia oficial, como a USP por
exemplo, in Bassani, 2004 . Conforme recomendação do FDA’s Guidance for Industry,
Parâmetros investigados e classificação dos métodos
* Pode ser exigido dependendo da natureza do ensaio e specífico
Parâmetro Categoria I Categoria II Categoria
analítico Quantitativo Qualitativo III I V
Exatidão sim sim * * não
Precisão sim sim não sim não
Seletividade sim sim sim * sim
Lim. Detec. não não sim * não
Lim. Quantif. não sim não * não
Linearidade sim sim não * não
Faixa sim sim * * não
Robustez sim sim sim sim não

16
2000, é necessário apenas verificar: a especificidade, a estabilidade da solução da
amostra e a precisão intermediária. Alguns conceitos que respaldam essas
recomendações:
o Abordagem para Métodos Normalizados
21 CFR 211 194 (a) (2) - Verificação
Que é a evidência documentada que um método previamente validado desempenha
como pretendido, no ambiente em que está sendo aplicado.
o INMETRO DOQ-CGCRE-008 :2002
Demonstrar a competência do laboratório se atinge o pretendido e que opera bem sob
as condições normais de uso. Na ilustração 3 é possível compreender o escopo da
aplicação de uma validação, USP, fonte Bassani, 2004. Se o método atende ou não a
sua finalidade.
Ilustração 3: Método normalizado, segundo USP 25.
Como o método de determinação de condutividade (Método III ), utilizado para
o cálculo de incerteza, é um método normalizado, somente os seguintes parâmetros
precisam ser verificados: a competência do Laboratório e o registro dos resultados.
Porém para efeito de estudo será demonstrada a validação e harmonização entre as
metodologias em estudo para esse trabalho. Foram verificadas a precisão intermediária
e a robustez, através de dados de estabilidade, já que ele responde a categoria III.
Definir escopo da aplicação
O método atende ?N
Validação completa
Verificar competência do laboratório
S
Documentar resultados

17
Entendemos como precisão de um procedimento como o grau de
concordância entre os resultados de uma medição, conforme VIM (Vocabulário
Internacional de Metrologia, 2003). É medida através da dispersão dos resultados
individuais do meio e geralmente é expressa como sendo o desvio padrão ou o
coeficiente de variação (desvio padrão relativo), quando o procedimento completo for
aplicado repetidamente para separar amostras idênticas do mesmo lote homogêneo do
material. Ela pode ser expressa pela repetitividade, pela reprodutibilidade e precisão
intermediária. A repetitividade é o grau de concordância entre os resultados de
medições sucessivas de um mesmo mensurando, efetuadas sob as mesmas condições
de medição - VIM, 2003. Já a Reprodutibilidade é entendida como o grau de
concordância entre os resultados das medições de um mesmo mensurando, efetuadas
sob condições modificadas de medição. Alguns autores a especificam como a
dispersão encontrada de um mesmo ensaio, realizado em diferentes laboratórios -
ASTM, 2004. Para efeito de estudo foi demonstrado a codificação “R” para representar
a reprodutibilidade e “r” a repetitividade, conforme recomendação de Horwitz, 1998.
A precisão intermediária é a forma de expressar as variações dentro de um
mesmo laboratório, com dias de análises diferentes, ou analistas diferentes, ou até
métodos diferentes (se for o caso), todos com um mesmo equipamento. Todas as
variáveis sob a mesma condição de amostra ou padrão, com definição prévia de quais
itens variarão de cada vez, INMETRO – DOQ – CGCRE – 008, 2003.
A precisão conforme a USP 27, Validation Compendia Methods <1225>
o Precisão intermediária e Reprodutibilidade – Precisão inter Laboratório com o
mínimo de 6 determinações de amostra homogênea com concentração de 100%
do analito em questão.
o Calculada pelo desvio padrão ou desvio padrão relativo (RSD).
A robustez do método é a medida da sensibilidade que este apresenta face a
pequenas variações. Um método é robusto quando não apresenta variação a pequenas
variações. Quanto maior for a robustez maior será a confiança de sua precisão,
INMETRO – DOQ – CGCRE – 008, 2003.

18
A determinação do conteúdo de polissacarídeo (Método tipo I ) em estudo, fora
desenvolvido pela matriz do produto vacina contra Hib - na GlaxoSmithKline - Bélgica, e
fora validado previamente. Neste método foram checados todos os parâmetros
recomendados para validação da categoria tipo I, o que permitiu a adaptação e
conseqüente validação em Bio-Manguinhos, com a aceitação de seus resultados. Na
categoria I os critérios exatidão, precisão, seletividade, linearidade, faixa de trabalho e
robustez, já foram previamente verificados. Para efeito de estudo alguns critérios foram
revistos.
A exatidão é o grau de concordância entre o resultado de uma medição e um
valor verdadeiro - VIM, 2003, poderá ser determinada mediante a aplicação do
procedimento a amostras do material a ser examinado, que tenham sido preparadas
com precisão quantitativa. Deverão também ser preparadas amostras em que o
analisado seja incorporado em quantidades cerca de 10% superiores e em quantidade
abaixo da variação de valores esperada. A exatidão poderá também ser determinada
através da comparação dos resultados obtidos mediante a utilização de um
procedimento alternativo que tenha sido anteriormente validado.
A seletividade ao contrário da especificidade é a capacidade do método em
produzir resposta para vários analitos, porém as distingue de um analito para outro.
Tanto a seletividade quanto a especificidade são relacionadas a detecção. Ambas as
formas tentam abordar o mesmo problema o que medimos e o que pensamos que
medimos. Já a especificidade pode ser entendida como a medida do grau de
interferência, ou ausência da mesma, nas análises de matrizes complexas, sendo uma
condição de exatidão. Por exemplo impurezas presentes ou condições de instabilidade
de resultados, VIM, 2003.
A linearidade do procedimento analítico é a sua capacidade de produzir
resultados que sejam diretamente proporcionais à concentração do analisado na
amostra, em uma faixa de concentração. Ela é obtida por padronização interna ou
externa e formulada como expressão matemática usada no cálculo da concentração do
analito, expressa pela equação da reta ,
bxay += ∴

19
y é a resposta medida, x a concentração ( valor conhecido), a o coeficiente linear ou
intercepto da linha de regressão e b o coeficiente angular ou inclinação da linha de
regressão (slope). A tangente da reta, b, é um dos requisitos que torna possível medir a
performance do método.
Conforme recomendação vigente, o coeficiente de correlação de uma curva de
calibração, r, deve seguir os seguintes critérios:
O Critério de aceitação Ideal é: r > 0,999
e aceitável r > 0,90 ,
de acordo com INMETRO DOQ -CGCRE-008:2003.
Através da comparação de diferentes coeficientes angulares, que na verdade
pode ser entendido como a tangente da função calibração, é possível avaliar junto com
o desvio padrão residual, a sensibilidade, o desvio padrão e o coeficiente de variação
(desvio padrão relativo) do procedimento, as características de performance do método
analítico, conforme ISO 8466-2, 1993.
A faixa de trabalho é estabelecida como a faixa utilizada ideal para a
quantificação de interesse, de uma curva de calibração. Pode ser definida como de um
décimo até o dobro do valor do limite de restrição ou de 50 a 150% do valor real da
concentração do analito, de acordo com Thompson, 1999 e Inman, 1987, de 60 a
120%, conforme recomenda o FDA, Horwitz, 1998 e de 20 a 200%, como relata a ISO
8466, 1990 .
A precisão e robustez já foram definidas na descrição de validação do método
III.
Quando não é possível preparar matriz limpa da amostra (branco), são
recomendados ensaios de recuperação, para eliminar o efeito matriz. Para esses
testes é feita a adição do componente de interesse à matriz seguida da execução do
método que está sendo avaliado. Entende-se por recuperação como os testes que
são realizados cuja finalidade é testar o analito e verificar se há interferências no
método. O teor medido do componente adicionado é dividido pelo valor efetivamente
adicionado e multiplicado por 100, obtendo-se assim a percentagem de recuperação.
Este enfoque sofre com o fato que o analito (composto de interesse sendo

20
analisado) não sendo parte integrante da amostra, sua extração pode ser mais fácil
que de uma amostra que o contenha naturalmente. Porém, os resultados deste tipo
de teste quando associados a outros enfoques podem fornecer um panorama do
comportamento do método com relação ao composto de interesse no tipo de matriz
utilizada. Amostras formuladas sinteticamente e ainda, o método de adição, podem
também ser empregadas no teste de recuperação, conforme recomendação da
IUPAC, 2002, através da FAO/ WHO.
A tabela 2 apresenta os valores aceitáveis de percentuais de recuperação
de amostras nos testes de recuperação, de acordo com a concentração inicial
estimada.
Tabela 2 - Percentuais de recuperação de amostras.
Poderemos sempre perguntar: Quando os resultados podem ser considerados
bons? Esta é uma pergunta que qualquer analista se faz. A resposta está na
equação desenvolvida por Horwitz para precisão.
Na década de 80, William Horwitz publicou um estudo dos métodos
colaborativamente estudados e aceitos pela “Association of Official Analytical
Chemists”(AOAC). Ele demonstrou que o coeficiente de variação ou RSD inter
laboratorial de qualquer método não depende do instrumento utilizado, da matriz, ou
técnica empregada e sim do teor em que o componente analisado se encontra no
composto e que pode ser descrito pela equação denominada como função de
Horwitz, AMC Technical Brief No 17, 2004:
60 – 115 0,000 000 01 10 ppb
80 – 110 0,000 001 1 ppm
90 – 107 0,000 1 0,01 %
95 – 105 0,001 0,1 %
97 – 103 0,01 1 %
98 – 102 0,1 10 %
98 – 102 1,0 100 %
Recuperação média %
Concentração Fracional
Concentração

21
CV (%) = 2 (1-0,5 log C)
A concentração C é expressa como potência de 10. Por exemplo: 1
mg/Kg (ppm) = 10-6 g/kg, o que nos dá um CV % = 16%. Para o analista é útil
conhecer ainda algumas outras propriedades do método analítico que pretende
empregar. O conceito de limite de detecção tem sido objeto de diferentes definições
conforme a área de trabalho. É bom saber que o próprio analista pode criar sua
própria definição de limite de detecção desde que expresse claramente qual é o seu
conceito desta medida. De uma maneira geral pode-se definir como limite de
detecção a menor concentração do analito da qual podemos estar confiantes na sua
medição. Uma boa definição é proposta por Caulcutt e Boddy os quais nos diz ser o
limite de detecção a menor concentração do analito da qual temos 95% de confiança
que será detectada pelo método. Uma outra definição é ainda fornecida pelo Codex
Alimentarius, 2002, que declara ser o limite de detecção convencionalmente aceito
como 3 vezes o desvio padrão do branco da amostra. Limite de quantificação tem
sido usualmente definido como 5 vezes o limite de detecção, como declarado na
tabela 3, com os limites de quantificação.
Tabela 3 - Atualização da Curva de Horwitz ( AOAC , 2000 ).
Concentração Concentração Fracional
RSD r
%
RSD R
%
Exemplo
100 % 1,0 1,0 2,0 Compostos puros 10 % 0,1 1,5 2,8 Macro nutrientes 1 % 0,01 2,0 4,0 Prod. Farmacêuticos.
0,1 % 0,001 3,0 5,7 Micro nutrientes
0,01 % 0,000 1 5,0 8,0 Vitamina C de alimentos.
1 ppm 0,000 001 10 16 Resíduo de Pesticidas.
10 ppb 0,000 000 01 20 32 Aflatoxinas
Onde RSDr = desvio padrão relativo intra laboratorial (repetitividade) e RSDR
corresponde a precisão inter laboratorial (Reprodutibilidade ou precisão
intermediária).
Ao empregar material de referência certificado, deve-se escolher aquele
cujas matrizes (sejam os materiais que contêm tudo menos o composto de
interesse) mais se aproximem do tipo de amostra para o qual o método que está

22
sendo testado será usado. A ISO Guia 30, define material de referência como um
material ou substância na qual uma ou mais propriedades estão suficientemente
estabelecidas para serem usadas para a calibração de um instrumento, avaliação de
um método analítico ou para designar valores a materiais. Neste sentido, cada
laboratório pode criar seus próprios materiais de referência para o controle de
qualidade rotineiro. Para a validação de métodos, no entanto, emprega-se material
de referência certificado, onde teores de uma ou mais propriedades estão
certificados por procedimentos tecnicamente válidos, acompanhados por um
certificado proveniente de um organismo certificador acreditado. Alguns exemplos de
organizações que fornecem materiais de referência certificados: Community Bureau
of Reference (União Européia), Ministry of Agriculture, Fisheries and Food (Reino
Unido), National Institute for Standards and Technology (antigo National Bureau of
Standards, EUA), National Research Council (Canadá), National Institute for
Environmental Studies (Japão), entre outros.
Avaliação interlaboratorial de um método é realizada através de um estudo
colaborativo. O protocolo para execução destes estudos foi harmonizado pela ISO,
IUPAC e AOAC e é reconhecido pelo Codex Alimentarius - dentro da União
Européia. Neste programa, uma ou várias amostras homogêneas são distribuídas a
laboratórios, onde a análise é executada por analistas experientes. Os resultados
vão indicar a exatidão do método, sua precisão interlaboratorial (R,
reprodutibilidade ) e intralaboratorial (r, repetitividade ). Métodos estudados
colaborativamente são publicados por entidades governamentais ou privadas que se
dedicam a este tipo de estudo.
Segundo os protocolos normalizadores citados, as condições para a
realização de um estudo interlaboratorial são: participação de oito ou mais
laboratórios, utilização de amostras homogêneas e representativas do produto e um
mínimo de cinco materiais diferentes distribuídos entre os laboratórios participantes,
com um mínimo de um par de duplicatas “cegas” e analistas competentes para
executar a análise. A média de todos os resultados é considerada a melhor
estimativa da quantidade do composto a ser determinado.
O LAFIQ participou de Estudos Colaborativos com a FEEMA no ano de
2003 e 2005, em análises físico-químicas de água.

23
É oportuno lembrar que estudos interlaboratoriais podem servir para várias
finalidades, tais como: prover estimativas da repetitividade e da reprodutibilidade de
um método analítico, prover uma estimativa objetiva do desempenho do Laboratório,
encorajar a autocrítica e a percepção dos erros cometidos durante análises e
sobretudo ajudar na identificação das necessidades de treinamento de pessoal.
Em alguns setores da Química Analítica é agora uma exigência formal
(legislativa), que os laboratórios introduzam medidas de garantia da qualidade para
assegurar que são capazes e estão aptos a fornecer informação com qualidade
confiável. Tais medidas incluem: o uso de procedimentos com controle interno de
qualidade bem definido, participação em testes de competência – com crédito
baseado na NBR ISO/IEC 17025, 2001 e estabelecimento da rastreabilidade dos
resultados das medições através de relatórios e suas validações.
Até agora foi abordado a importância de validar uma metodologia analítica,
porém qual é a validade desse relatório gerado? Existem algumas questões que
precisam ser verificadas habitualmente no Laboratório. Foi sempre pensado que a
cada intervenção de manutenção, por exemplo, seria necessária uma revalidação de
um determinado método. Sabemos agora que não é bem assim. Se o escopo da
validação prévia foi alterada é importante verificar esses parâmetros modificados e
se ainda atendem ao método e aplicação propostos. De acordo com Furman, W,
1998, existem fatores que ocasionam modificações significantes em resultados de
um método analítico, tais como: análise de um novo produto ou nova formulação,
troca de condições principais de operação, por exemplo, novo analista, uso de
colunas de configurações e tamanhos diferentes, temperatura de análise para
métodos cromatográficos e sobretudo a troca de módulos de equipamento e
softwares de cálculos que possam influir predominantemente no resultado final. Se
houverem intervenções num equipamento, validado, porém se estas mantiverem as
mesmas condições previamente definidas - aplicabilidade e operacionalização do
sistema fidedigna, o método continua mantendo suas características de validação,
Inman E, 1987; NBR ISO/ IEC 17025, 2001; FDA, 2002.

24
1.5 - A Incerteza
Atualmente, diferentes setores da indústria, da saúde, da área de meio
ambiente, entre outros, utilizam os resultados de análises gerados em laboratórios
químicos para a tomada de decisões. Com base nestes resultados, aceitam-se ou
rejeitam-se matérias primas, diferenciam-se desempenho de fornecedores,
processos produtivos são modificados, atua-se sobre a saúde das pessoas e dos
animais.
Deste modo, os resultados das análises devem ser “bons”, ou melhor,
devem apresentar “qualidade aceitável” em função dos objetivos requeridos. Mas
como podemos definir melhor estas medições de boa qualidade? Caso os resultados
analíticos tivessem sido expressos apenas como médias, eles não poderiam ter sido
comparados com o valor de referência. Apenas quando as médias estão
acompanhadas de suas incertezas, é possível tomar as decisões adequadas.
Resultados médios sem as respectivas incertezas carecem de significado porque
não dão a informação completa sobre a medição.
Entende-se por incerteza, “o parâmetro, por exemplo o desvio-padrão (ou
um múltiplo dele), ou a metade de um intervalo correspondente a um nível aceitável
da confiança estabelecido, que caracteriza a dispersão dos valores que podem ser
razoavelmente atribuídos ao mensurando” – conforme o “Vocabulário Internacional
de Termos Fundamentais e Gerais de Metrologia – VIM”.
A atual preocupação da ciência da medição química é representada pela
questão da estimativa da incerteza associada aos resultados obtidos
experimentalmente. Esta preocupação originou-se nos laboratórios analíticos
prestadores de serviços, visto que a estimativa das incertezas é exigida por critérios
para o reconhecimento formal da competência técnica de laboratórios. Esta questão
é assunto ainda bastante controvertido no meio científico. Até mesmo o significado
do termo “incerteza” ainda não é perfeitamente entendido por muitos químicos, que
costumam confundi-lo com a repetitividade - Horwitz, W, 1998 FDA.
Quando é estabelecido um grau de confiança, pode-se determinar a
incerteza combinada expandida, através do critério do intervalo de confiança,
25
utilizando um fator de abrangência, k. Na maioria dos casos, usa-se o valor de k = 2
correspondentes ao estabelecimento do nível de confiança de aproximadamente
95%.
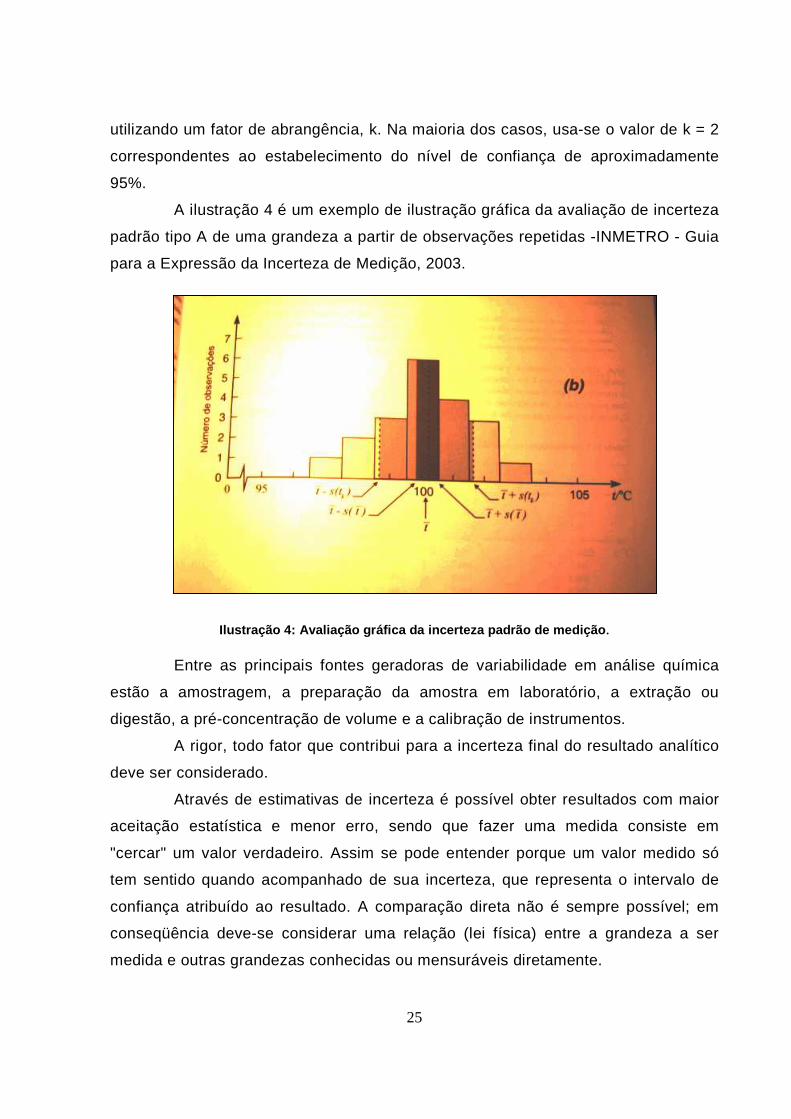
A ilustração 4 é um exemplo de ilustração gráfica da avaliação de incerteza
padrão tipo A de uma grandeza a partir de observações repetidas -INMETRO - Guia
para a Expressão da Incerteza de Medição, 2003.
Ilustração 4: Avaliação gráfica da incerteza padrão de medição . Entre as principais fontes geradoras de variabilidade em análise química
estão a amostragem, a preparação da amostra em laboratório, a extração ou
digestão, a pré-concentração de volume e a calibração de instrumentos.
A rigor, todo fator que contribui para a incerteza final do resultado analítico
deve ser considerado.
Através de estimativas de incerteza é possível obter resultados com maior
aceitação estatística e menor erro, sendo que fazer uma medida consiste em
"cercar" um valor verdadeiro. Assim se pode entender porque um valor medido só
tem sentido quando acompanhado de sua incerteza, que representa o intervalo de
confiança atribuído ao resultado. A comparação direta não é sempre possível; em
conseqüência deve-se considerar uma relação (lei física) entre a grandeza a ser
medida e outras grandezas conhecidas ou mensuráveis diretamente.

26
Com diferentes abordagens porém de forma válida e igualmente aceitas, as
estimativas de incerteza podem ser vistas de duas maneiras distintas, a visão dos
químicos e a dos metrologistas. Existe uma tendência dos químicos analíticos, em
aproveitar dados obtidos no estudo de validação dos métodos. Por outro lado, os
profissionais da área de metrologia utilizam a estimativa através da transferência de
abordagem usando métodos físicos, para processos metrológicos em química, ou
seja, utilizando resultado por resultado de cada componente. Contudo , de ambas as
formas, deve ser igual em todos os processos de medição, seja quimicamente ou
metrologicamente considerada.
Como relata Horwitz, 1998, a incerteza está sempre correlacionada a tipos
de erros, sejam eles sistemáticos - bias, ou aleatórios - randomizados. Há algum
tempo na química analítica os erros sistemáticos vem sendo minimizados com o uso
contínuo de instrumental calibrado e certificado. Como exemplo disso temos a linha
de base na cromatografia e o “drift” - correção de leitura de display em um
condutivímetro. Com essas correções prévias estudos comprovam que os erros que
se somam aos resultados analíticos são basicamente aleatórios e permitem eximir a
expressão de erros sistemáticos. Contudo os erros aleatórios podem ser
provenientes de analistas, preparo e injeção de amostras, curvas de calibração e
software de cálculos não validados.
Na literatura pertinente, diferentes abordagens de estimativas de incerteza são
encontradas:
o O guia preparado pelo NMKL, “Nordic Comittee on Food Analyses”,
baseia-se na determinação da estimativa de incerteza pelo intervalo
de confiança através da medida de precisão intermediária e a média
obtida nas repetições. Rejeita a hipótese de usar erros sistemáticos
para o cálculo da incerteza e recomenda que a estimativa de
incerteza seja feita por dados de um único laboratório, de acordo
com Horwitz, 1998.
o O guia ISO/TS 21748 in AOAC, 1998, baseia-se na utilização de
dados de estudos colaborativos, através da reprodutibilidade. Porém
é necessário verificar a presença de erros sistemáticos com regras
previamente estabelecidas.

27
o Conforme o guia IUPAC/ISO/AOAC, 1999, é sugerido o uso do
desvio-padrão calculado a partir da reprodutibilidade, expresso como
incerteza combinada padrão.
o Já a NBR ISO/IEC 17025, 2001, considera outras abordagens para
a estimativa da incerteza de medição em ensaios, diferentes da
anterior que estima componente por componente. Nesta Norma,
tanto os dados de precisão intermediária, da repetitividade quanto os
da reprodutibilidade, através de estudos colaborativos, podem ser
utilizados.
o O guia EURACHEM/ CITAC, 2001, recomenda o uso de dados de
repetitividade, precisão intermediária, reprodutibilidade como fontes
de dados de estudos e inter laboratoriais, através do desvio-padrão,
para as estimativas de cálculo da incerteza padrão, padrão
combinada e padrão combinada expandida.
o FAO/ WHO, 2002, recomenda o uso do intervalo de confiança para a
expressar a incerteza de medição. Com dados de desvio-padrão
provenientes de dados de precisão obtidos após estudo de
validação, correlacionada a erros aleatórios.
o O INMETRO, 2003, agrupa os componentes da incerteza em duas
categorias tipo “A” e tipo “B”. Essas duas são baseadas no método
de avaliação e se aplicam à incerteza e não são substitutas para os
termos aleatórios e sistemáticos. A incerteza de correção de um
efeito sistemático conhecido pode em alguns casos ser obtida por
uma avaliação do tipo A, enquanto que em outros casos, por uma
avaliação do tipo B, podendo-se assim obter do mesmo modo a
incerteza que caracteriza um efeito aleatório. A primeira, tipo A, é
obtida a partir de uma função densidade de probabilidade e só existe
se houver um controle estatístico, através de cartas de controle. A
segunda, tipo B, é obtida de uma suposta função densidade de
probabilidade, baseada no grau de credibilidade de que um evento
vá ocorrer, sendo portanto chamada de probabilidade subjetiva.

28
o A AMC, 2004, de acordo com Horwitz prevê o cálculo de incerteza
para métodos analíticos e questiona o custo da metodologia em
estudo.
Laboratórios normalmente constroem a curva de calibração com 4 a 5
pontos ao longo da faixa de trabalho desejada, com algumas repetições em cada
ponto. A média aritmética é o valor aceito e o desvio-padrão da média é calculado
para cada ponto, sendo que a incerteza pontual é dada pelo intervalo de confiança
da média, estabelecido o grau de confiança desejado. Calculadas desta maneira, as
incertezas não são representativas para a curva de calibração como um todo; trata-
se de incertezas pontuais, e nada se pode afirmar quanto aos pontos intermediários
da curva. O uso da análise pela variância do resíduo está sendo amplamente
difundido para calcular a incerteza ao longo da curva de calibração. A dispersão das
medições deve ser independente da concentração das soluções de referência, que é
a propriedade conhecida como homoscedasticidade em y, onde o cálculo de
concentração é proveniente da reta de linearidade. Deve ser válido o pressuposto de
que a variância é constante nos erros aleatórios, ou randomizados, já que os erros
sistemáticos tendem a ser mínimos nos métodos químicos que utilizam instrumental
calibrado.
Se por acaso a variável independente x estiver sujeita a erros, estes devem
ser desprezíveis quando comparados aos erros em y. Para verificar se a equação de
regressão é estatisticamente significativa efetuam-se os testes para verificação de
ajuste do modelo linear, validade da regressão, sua eficiência e sua eficiência
máxima, de acordo com dados da “Analytical Methods Committee” (AMC), 2000 e
INMETRO, 2003. A análise de variância uni variável, tabela de ANOVA, é uma
ferramenta importante para este estudo. Antes de utilizarmos a função calibração
estabelecida pela teoria dos mínimos quadrados, é preciso testar a linearidade,
procedendo-se à comparação de variâncias. O teste é feito através do cálculo da
razão das médias quadráticas.
Na prática a estimativa de incerteza é baseada na teoria dos erros, onde
ela é função das variáveis. Embora elas não signifiquem a mesma coisa, mas
correspondem a variação aceita em torno de um resultado correto e podem ser
expressas pelo menos por três formas.

29
Incerteza - é expressa através do desvio padrão.
Ela pode ser dada por: µ ± б, onde µ é a média das medidas e б o desvio-padrão da
média da população, se este for igual ao desvio-padrão da média, S, então o desvio-
padrão da média pode ser assumido como б = S. Ela pode ser utilizada quando se
conhece a natureza da amostra, sua estabilidade e as medições do analito em
questão estar sob controle estatístico, então é entendida como incerteza padrão
tipo A . O desvio- padrão deve ser determinado após conhecer a repetitividade do
método e reprodutibilidade entre técnicos ou Laboratórios, a um grau de liberdade
finito, conforme manual de Incerteza do INMETRO, 2003 e guia IUPAC/ISO/AOAC,
2002. Dividindo o valor do desvio-padrão encontrado pela raiz quadrada do número
de determinações.
Incerteza padrão tipo B - é a combinação de várias fontes de incerteza de um
método analítico como a provável probabilidade de um resultado correspondente ao
grau de liberdade “∞”. Elas podem ser provenientes de: dados de medições prévias,
conhecimento prévio de comportamento de instrumentos, certificados de calibração
e qualificação de equipamentos, dados de fornecedores, incertezas de manuais
entre outras, que não seja conhecido o grau de confiança ou o tratamento estatístico
dos dados. Cada técnico deve saber identificar as fontes de incerteza de cada
método e que não devem ser negligenciadas. Os componentes de incerteza
possuem distribuição de probabilidade peculiar de cada grandeza. Todas as fontes
dessas incertezas devem estar com distribuição normal ou serem normalizadas.
Para transformar os valores de desvio-padrão da média em incertezas
padrão com probabilidade normal, deve-se dividir o valor encontrado, seja do tipo A
ou B pelo fator de probabilidade correspondente ao grau estimado. Por exemplo,
para 95% de confiança , o valor deve ser dividido por k= 1,96 (fator de abrangência).
Dados de desvio-padrão sem o grau de confiança declarado podem ter:

30
o distribuição de probabilidade triangular, ou seja, quando pode-se estimar as
fronteiras (limites superior e inferior) e a maioria dos resultados decai sobre
os extremos do gráfico. Portanto, para sua normalidade de distribuição, o
valor pode ser considerado dividindo o valor da incerteza padrão por 6 . Por
exemplo a declaração de um valor sem nível de confiança declarado.
o Se a distribuição de probabilidade for do tipo retangular, onde suas fronteiras
permitirem estimativa de valores de limite superior, inferior da dispersão dos
valores sem informações suficientes para estimar o valor da probabilidade,
sua normalização será dividir o valor da incerteza padrão por 3 .
o E a distribuição bimodal, quando somente é possível estimar as fronteiras do
valor de entrada de grandeza e sua probabilidade de encontrar o valor
verdadeiro que está na extremidade dos intervalos. Normalizando essa
distribuição divide-se a incerteza padrão por 2 .
Fonte EURACHEM, 2002.
A incerteza padrão combinada total de um método é expressa pela soma quadrática
das incertezas padrão de cada componente, como por exemplo as incertezas de
entrada e saída com a mesma unidade. Expressa por :
uc = 222 )(...)()( nubuau +++
Sendo a,b,..,n as fontes de incerteza e u(a),u(b),...,u(n) os valores das incertezas
padrão de cada componente.
Quando as grandezas de entrada não têm a mesma unidade das de saída, deve-se
utilizar a regra da normalização da distribuição retangular.
Incerteza padrão expandida - É a combinação de todos os níveis de incerteza,
porém a um nível aceitável de confiança de 95 a 99%.

31
Para se calcular é necessário multiplicar a incerteza combinada por um fator de
abrangência que permitirá a definição de um intervalo com nível de confiança maior.
Contudo será preciso estimar o número de graus de liberdade, que depende do
tamanho da amostra. Ao conhecermos o número do grau de liberdade, maior será a
probabilidade de que o valor verdadeiro esteja dentro de uma faixa de incerteza
estimada.
O número de graus de liberdade para incertezas tipo A é dado pela equação:
i = n -1ט
Sendo טi o número de graus de liberdade do conjunto i de medidas e n o número de
medidas do conjunto.
Para incertezas tipo B, como a probabilidade de graus de liberdade no interior da
distribuição deve ser máxima, ao contrário da parte externa desse intervalo, deve ser
mínima.
Então, para as incertezas tipo B , pode-se padronizar como sempre infinito:
→∞ iט ∞ =iט ∴
Observação: Se houver diferença entre os graus de liberdade do tipo A e o B, dos
dados; Se por exemplo a do tipo A for finito e a do tipo B infinito. Deve-se calcular
como:
= efט( )[ ]
( )[ ]∑
=
n
i i
c
yi
y
1
4
4
ϑµ
µ ∴
ef = Graus de Liberdadeט
µc ( y) = Incerteza combinada do método

32
µi = Incerteza padrão da componente i
ϑ i = Graus de liberdade da componente i
Fonte Manual do INMETRO, 2003.
Atualmente, o usual é a expressão da estimativa de incerteza expandida ser
dada pela multiplicação do valor da incerteza padrão combinada por k = 2. Assumindo
um valor de k médio.
U = ± u x k EURACHEM, 2002.
1.6- A Condutividade
A condutividade de uma solução eletrolítica é a expressão numérica
quantitativa da sua capacidade de transportar a corrente elétrica. Ela é definida
como sendo o inverso da resistência elétrica de 1cm3 do líquido a uma temperatura
de 25ºC. Entendendo como condutividade a propriedade de conduzir eletricidade,
através de seus íons em solução. Ela é definida com o inverso da resistividade e tem
como unidade de medida, µS/cm.
O mecanismo responsável é o transporte de cargas elétricas na solução
através dos íons. Todos os íons presentes na solução participam dessa condução e,
por essa razão, pode-se dizer que a condutividade fornece uma informação global,
por natureza não específica. A maioria dos ácidos, bases e sais inorgânicos são
bons condutores da corrente elétrica ao passo que substâncias orgânicas, que não
se dissociam em solução (benzina, gasolina, açúcares por exemplo), não são boas
condutoras.
A condutividade depende de vários fatores:
o Da concentração global em espécies ionizadas. Quanto maior a quantidade
de íons numa solução, maior será sua condutividade. Em geral, para soluções
aquosas de eletrólitos inorgânicos, observa-se um aumento quase linear da

33
condutividade com o aumento da concentração (até concentrações de 10 ou
20% em peso).
o Do tipo de íons.
Quanto menor o íon, maior será sua mobilidade e, portanto, maior sua
condutividade. Para diâmetro igual, íons bivalentes e trivalentes conduzem
mais que os íons monovalentes.
o Da natureza do solvente;
Solventes polares (água, amônia, álcool metílico, etc.) exaltam a ionização das
substâncias dissolvidas e, portanto, favorecem a condutividade.
o Das interações entre o solvente e os íons dissolvidos.
Um solvente muito polar, como a água, pode ser visualizado como um conjunto de
dipolos elétricos em permanente movimento caótico. A tendência dos dipolos
associarem-se com íons é tanto maior, quanto menor forem os íons. É o caso do íon
Cs+ (Césio), relativamente pequeno, que fixa facilmente as moléculas de água,
tornando-se hidratado e, conseqüentemente, pouco condutor.
o Da temperatura.
Ao contrário da condutividade metálica, a condutividade eletrolítica aumenta com a
temperatura. De modo geral esse efeito é devido ao fato de que a mobilidade
individual dos íons aumenta com a temperatura e que a viscosidade do solvente
diminui. Para soluções aquosas, o coeficiente de temperatura da condutância iônica
varia entre 0,5 e 5% por grau centígrado, dependendo do tipo e da concentração do
íon condutor. Erros muito significativos podem ser cometidos na medição da
condutividade se esse efeito não for compensado (manual ou automaticamente),
dependendo do equipamento, condutivímetro.
Suas aplicações podem ser vistas como inúmeras:
o Controle da pureza em água destilada e deionizada, condensados,
substâncias orgânicas;
o Determinação dos eletrólitos residuais em água potável, água
desmineralizada, água para alimentação de caldeiras, efluentes;
o Concentração de sais em banhos de salmoura, salinas, fertilizantes, fibras e
têxteis, banhos de anodização, galvanização e eletrodeposição, soluções
fisiológicas (diálise), alimentos e sucos de frutas;

34
o Força de ácidos e bases em processamento dos ácidos e bases inorgânicos
diluídos e concentrados e soluções alcalinas corrosivas;
o Contaminações de sais em trocadores de calor, circuitos de arrefecimento;
o Processamento químico e detecção do final de lavagem de precipitados,
determinação da solubilidade de sais pouco solúveis, titulações
condutométricas.
A célula é um dos elementos mais importantes de um sistema de medição
da condutividade. Basicamente, ela consiste em duas placas metálicas cujas áreas e
distância são precisamente fixadas, montadas rigidamente numa cavidade
construída em material isolante, vidro ou plástico. Essa cavidade serve para delinear
um pequeno volume constante do líquido a medir e, conseqüentemente, torna a
medição da condutividade independente do volume total da amostra e da
proximidade de superfícies tais como paredes de tanques ou tubulações. As placas
metálicas, chamadas eletrodos, são construídas em metais nobres, geralmente
platina, e são revestidas por um depósito eletrolítico de negro de platina. Esse
depósito poroso limita os efeitos da polarização, conforme manual do condutivímetro
Metrohm.
A determinação da condutividade elétrica de uma solução consiste então
basicamente na medição da sua resistência elétrica. Para obter precisão e
sensibilidade nessa medição, é necessário que o valor da resistência eletrolítica se
mantenha entre determinados limites. No caso de medição e controle da
condutividade em processos industriais, para os quais são toleráveis erros de mais
ou menos 1%, é desejável que a resistência medida situe-se entre os limites
aproximativos de 100 até 100.000 Ohms. O critério usado na seleção de uma célula
é que ela tenha uma constante tal que a resistência da solução em teste esteja
dentro desses limites. Na prática, a aplicação desse critério leva a escolher uma
célula com baixo valor da constante para medir soluções de baixa condutividade e
alto valor da constante para soluções de condutividade elevada.
As moléculas de água dissociadas em íons em função do pH e da
temperatura resultam na medida da condutividade. Ela pode ser afetada pela
contaminação gasosa, por exemplo o dióxido de carbono dissolvido em água, que
altera a medição. Os íons estranhos à amostra - WFI, representados por íons
cloretos e íons sódios, comumente encontrados em índices residuais em águas cuja

35
concentração teórica máxima permitida é de 0,47ppm (mg/L) e 0,3 ppm (mg/L) de
íon amônio em solução, em balanço com os intrínsecos, são responsáveis pela
eletroneutralidade e pureza química da água para uso farmacêutico. A combinação
dos íons existentes e os desconhecidos em função do pH, estão descritos no
terceiro estágio de medição da Farmacopéia Americana, USP 27. Os dois
preliminares estágios de medição, inclusos nessa monografia, são requerimentos
para esse teste. Se os dois primeiros são satisfatórios não haverá necessidade de
passar ao terceiro. Porém o último torna-se necessário caso tenha havido resultados
falhos, dentro do limite de condutividade especificado, conforme USP 27.
Através das medições diárias de condutividade no Laboratório, a água
purificada, WFI, que é um dos parâmetros a ser estudado, método III, é fator
primordial de qualidade. Ela é o princípio de tudo, sendo base e veículo de diversos
processos produtivos e de Controle de Qualidade. Para uso farmacêutico, ela exige
tratamento de alta pureza para assegurar que não haja interferência de
contaminantes que afetem a qualidade das drogas e imunobiológicos produzidos
para injeção, o que é crucial para sua eficácia e para a saúde.
Embora as diferentes farmacopéias descrevam vários tipos de águas para uso
farmacêutico, em linhas gerais pode-se dividi-las em dois grandes grupos: a Água
Purificada (PW: Purified Water) e a Água para Injeção (WFI: Water for Injection).
Conforme a Farmacopéia Americana a tabela a seguir demonstra o comportamento
aceitável, para o primeiro estágio de condutividade. Caso os valores medidos não
estejam claramente definidos a seguir, os resultados não atendam à primeira
verificação, outros dois estágios de medição são recomendados, conforme
procedimento de análise de WFI, pop 106040.617. Podem ser também aceitos
resultados que estejam no estágio 2 e 3, sendo que a aceitação no primeiro exclui a
utilização dos próximos subseqüentes. Estes parâmetros foram descritos
primeiramente na farmacopéia americana, USP 23, como revisão anexa, efetivados na
USP 24 e em suas subseqüentes versões de revisões, USP 25, 26 e 27, que é a última
vigente, conforme descrito na tabela 4.

36
Tabela 4 - Relação de medidas de condutividade do e stágio 1.
Para o estágio 2, é recomendada a agitação por 5 minutos da amostra, com a
finalidade de haver contaminação controlada, por gás carbônico, CO2 e medir
novamente a condutividade. Caso não atenda ao critério de medida, que
corresponde ao valor menor que 2,1 µS/cm, é necessário passar a amostra para o
estágio 3 de medição de condutividade. Este remete à comparação com valores de
pH, conforme a tabela 5.

37
Tabela 5 - Estágio 3 de condutividade.
Os dois tipos de água são definidos da mesma forma do ponto de vista
químico, porém diferem quanto aos requisitos de pureza microbiológica. A
introdução das normas contidas na USP 23, e reafirmadas na USP 24, significou
uma mudança importante nas exigências de controle para águas farmacêuticas. Na
USP 23 a recomendação para produção de WFI poderia ser através de osmose
reversa e destilação. Porém na USP 24 já é recomendada a produção de WFI
somente proveniente de destilação. Qualquer outro tipo de processo de purificação
caracterizaria a produção de água purificada e não para uso injetável. Até o
surgimento das normas contidas na USP 23, com os parâmetros confirmados na
USP 27, em 2004, os controles a que se devia submeter uma água para uso
farmacêutico eram os ensaios qualitativos de laboratório, nos quais o seu conteúdo
químico era analisado com base em análise relativamente simples, com resultado de
“aprovado” ou “não aprovado”. Conforme a tabela 6, os parâmetros de qualidade de

38
uma água potável que é submetida ao processo de retirada de íons com
deionização, seguida de destilações múltiplas.
Tabela 6 - Requerimentos de Qualidade Química confo rme Portaria 1469 de 29/12/2000 – Ministério da Saúde, para água potável, USP 26 água purificada e destilada e pop 106040-617.
Carac. tipos
Odor pH (25°C)
Cor Condut. (µS/cm)
Cloreto Dureza Total
Nitritos e Nitratos
Sólidos dissolvidos
Ferro Sulfato *TOC *
Potável Nenhum
6,5-8,5 <15uH _ <250ppm
<500ppm <1000ppm <1000ppm <0,3ppm <400ppm _
Deionizada nenhum 5,0-7,0 incolor
<1,3
<1,0ppm _ <0,2ppm _ _ _ <500 ppb
Destilada (WFI)
_ “ “ <1,3 _ _ _ _ _ _ <500 ppb
*TOC – Carbono Orgânico Total
Os equipamentos utilizados para esse estudo, fotômetro, analisador de
carbono orgânico, potenciômetro e condutivímetro são rigorosamente calibrados,
com padrões, de acordo com as BPL. Essa calibração deve possibilitar
rastreabilidade das medidas de grandezas físicas fundamentais com os padrões. Os
certificados de calibração devem referir-se a padrões de medida nacionais ou
internacionais da mesma forma. Se uma unidade operacional mantém tais padrões,
estes devem ser calibrados por órgãos competentes a intervalos periódicos.

39
1.7 - A cromatografia de íons
A denominação inicial de cromatografia era que um sistema poderia separar
um tipo de produto em frações, por diferença de cor, daí a origem do nome.
Ela consiste de um método físico-químico de separação de fases, sendo
uma estacionária e a outra chamada de móvel, esta última é eluída e transpassa
através da anterior quando submetida a determinada pressão. Daí sua
denominação, cromatografia líquida de alta performance, advinda da denominação
de cromatografia líquida de alta pressão - high performance liquid chromatography -
HPLC.
A técnica de cromatografia de íons, troca iônica, é baseada na interação
reversível entre a carga da amostra e a do meio, que são as resinas das colunas
cromatográficas, pela diferença de pH - chamada “ions exchange chromatography”.
O método é utilizado para separação e quantificação de íons com base em resinas
de troca iônica. As resinas utilizadas nas colunas de separação têm interação com a
amostra e afinidade, por competição de cargas com o eluente.
De acordo com o The United States Food and Drug Administration
(U.S.FDA), agência regulatória do Estados Unidos, é requerido que produtos
farmacêuticos devam ser testados para garantir sua composição e verificar sua
identidade, qualidade e pureza. Muitos desses ingredientes ativos não têm cor, não
são cromóforos, portanto não podem ser detectados por absorvância. É o caso dos
carboidratos, glicóis , açúcares, álcoois, grupamentos aminados e compostos
sulfurados. Eles devem ser oxidados e detectados por amperometria. O método de
detecção é específico para esses tipos de analitos que após oxidação resultam em
monômeros ou compostos transparentes. A detecção amperométrica é uma
poderosa técnica com limites muito baixos de detecção.
A aplicação da cromatografia com detecção por amperometria pulsada, é
baseada no diferencial amperométrico e é denominada cromatografia de troca iônica
e utiliza diferentes tipos de colunas para vários tipos de produtos. As colunas do tipo
carbopac PA 10, utilizadas para análises de carboidratos, são compostas pelo
polímero poliestireno divinilbenzeno sulfonado e características hidrofílicas
moderadas. Seu mecanismo de retenção possibilita a exclusão iônica, a exclusão
estérica e a adsorção. Seu empacotamento, processo de enchimento de recheio,

40
consiste de 460 nm de diâmetro de látex do substrato de poliestireno divinilbenzeno
tipo “crosslinked” - tipo enovelado, com ligação cruzada. Sua natureza é específica
para designar alta seletividade para monossacarídeos e sua capacidade de troca
iônica é de 100 µeq/ coluna.
O objetivo da utilização dessa metodologia é o estudo da quantificação do
teor da vacina, através da medida das subunidades do polissacarídeo
poliribosilribitol, oriundo de carboidratos como fonte de carbono, proveniente do
metabolismo de fermentações bacterianas, misturas de nutrientes e massas
celulares. Esse açúcar de alto peso molecular é o produto característico de interesse
da vacina Haemophilus influenzae tipo b, Hib.
Após despolimerização com hidróxido de sódio as subunidades geradas, a
partir da separação das frações pelo método cromatográfico descrito, são
quantificadas por detector de amperometria pulsada. Os resultados encontrados são
analisados pelo cálculo das áreas dos picos de seus cromatogramas. As condições
de análise e seus materiais estão descritas na metodologia em estudo.
A dosagem do polissacarídeo, poliribosilribitol fosfato é fator importante
para medir a concentração do soluto da vacina. Este confere a integridade e é
através dessa quantificação que é possível verificar se o produto está apto para uso.
Através desse valor, associado a seu peso molecular, é possível diagnosticar se a
vacina tem poder imunogênico. A técnica utilizada para a dosagem desse teor é a
cromatografia de íons.

41
2- Objetivo Geral O objetivo do trabalho proposto é implementar ferramentas que permitam
avaliar os resultados gerados rotineiramente no LAFIQ, de modo a aprimorar o
controle da qualidade analítica do mesmo.
2.1- Objetivos Específicos:
1. Estudar os Parâmetros de adequação / validação, conforme exigência da
norma NBR ISO/IEC 17025 e USP 27 de dois tipos de métodos de análise
para duas categorias distintas:
o Determinação do conteúdo de polissacarídeo na vacina conjugada
contra a Hib, ( Método categoria tipo I );
o Determinação da condutividade em água para injetáveis (water for
injection- WFI) ( Método categoria tipo III ).
2. Construir cartas de controle, para os dois métodos escolhidos e através da
variabilidade dos dados encontrar os intervalos aceitáveis para os resultados
obtidos.
3. Calcular as incertezas das medições analíticas para os métodos categoria tipo
I e III.
4. Avaliar os fatores que realmente contribuem para a incerteza das medições,
trazendo melhorias para a Unidade.

42
3 - Materiais e Métodos
O estudo foi realizado com dois métodos analíticos utilizados na rotina do
LAFIQ cuja escolha já foi justificada anteriormente:
- determinação do conteúdo de polissacarídeo em amostra referência de
poliribosilribitol fosfato de Haemophilus influenzae tipo b, através de separação
cromatográfica por troca iônica, com detecção amperométrica pulsada
(Método de categoria tipo I ) e,
- determinação da condutividade em amostras de água para injetáveis, WFI
(Método de categoria tipo III ).
Embora os dois métodos escolhidos para estudo sejam métodos oficiais
presentes em monografias nacionais e internacionais, para cada um deles os
parâmetros de validação foram estudados e os resultados apresentados e
discutidos no capítulo 4. Para que uma análise seja confiável é necessário que a
mesma seja executada através de um método validado, ou seja, o método tenha
sido submetido a estudos laboratoriais que demonstrem que ele atende as
exigências requeridas a que se destina.
Para o método tipo I estudaram-se os parâmetros:
1. Seletividade;
2. Faixa de trabalho;
3. Linearidade;
4. Precisão;
5. Exatidão;
6. Estimativas de incerteza padrão das diversas fontes, padrão combinada e
padrão expandida.

43
Os parâmetros seletividade, faixa de trabalho e exatidão já foram previamente
estudados, na primeira validação do método, realizada no LAFIQ.
Para o método tipo III, os seguintes parâmetros foram estudados:
1. Precisão;
2. Estimativas de Incerteza de medição;
Foram estabelecidos os seguintes passos para a organização do trabalho e
para o cálculo da incerteza de medição de cada método em estudo:
A descrição sucinta do método cujos procedimentos operacionais
padronizados estão apresentados no anexo A;
Estudo e avaliação dos parâmetros de validação e adequação;
Os cálculos e especificações que devem ser aceitos;
Identificação das fontes de incerteza, demonstradas no diagrama de
causa e efeito;
A expressão dos cálculos de estimativas das incertezas padrão, padrão
combinada e incerteza expandida;
Avaliação da contribuição de cada fonte de incerteza parcial e a
importância das mesmas para a incerteza total através da construção do
gráfico de Pareto;
Diagrama de causa e efeito refinado;
Recomendação de melhorias que visem a implementação de um modelo
de controle.

44
3.1- Descrição do Método tipo I
O princípio do Método tipo I , consiste na determinação do conteúdo de
polissacarídeo por HPLC que é baseada na separação de unidades simples (fosfato
de ribosil ribitol) do polissacarídeo hidrolisado, PRRP, em coluna de troca iônica sob
condições alcalinas extremas. A detecção das unidades separadas pela
cromatografia é feita através de detector amperométrico pulsado cuja medição
quantitativa é realizada pela corrente resultante da oxidação dos carboidratos da
amostra em questão. A interpretação do teste é baseada no tempo de eluição do
pico característico da unidade simples de poli ribosil ribitol fosfato, que elui em
aproximadamente 20 minutos . O pico é integrado através de programa
computacional, chamado peaknet, do próprio equipamento e o conteúdo de
polissacarídeo é então quantificado. A concentração da amostra referência, utilizada
para estudo foi calculada com o resultado da integração de seu pico, pelo valor do
tempo de retenção, plotado na reta de regressão linear da curva padrão utilizada
previamente.
3.1.2- Materiais e condições de análise
Sistema cromatográfico - HPLC marca Dionex, modelo DX 500 (composto de
bomba, amostrador automático e seletor)
Coluna Borate Trap
Coluna Amino Trap
Coluna Analítica Carbopac PA 10
Balões volumétricos calibrados de 25 mL, 50 mL e 100 mL
Pipetas calibradas de 2, 2,5 , 3, 4 e 5 mL
Pipeta automática de volume variável de 1000 a 5000 µL
Balança para determinação de umidade “moisture analyser” com prato de
alumínio para pesagem.

45
3.1.3- Reagentes e Soluções
Solução para a fase móvel: Solução aquosa contendo 30 milimoles de NaOH e
100 milimoles de acetato de sódio por litro de água ultrapura.
Material de referência de PRRP fornecido pela GlaxoSmithKline, com pureza de
100±5%
Solução estoque contendo 250µg/mL de PRRP (solução mãe) - Foram pesados
inicialmente 118mg do material, PRRP referência e dessecados em balança de
lâmpada halógena, a 70°C, até peso constante. A mas sa da amostra seca, após
peso constante, foi de 107mg e foi dissolvida em água purificada e avolumada a
50mL, em balão volumétrico, originando uma concentração de 2,14 mg/mL.
Dessa solução foi tomada uma alíquota de 5,84 mL, medida em pipeta
automática calibrada, e avolumada para 50 mL, em balão volumétrico.
Soluções amostra contendo 15 µg /mL de PRRP: preparada a partir da diluição
de 6,0 mL solução estoque contendo 250 µg/mL em balão volumétrico de 100
mL.
Solução de trabalho (padrão) contendo 25 µg/mL de PRRP: preparada a partir da
diluição de 2,5 mL da solução estoque contendo 250 µg/mL em balão
volumétrico de 25 mL.
Soluções de calibração contendo 10, 15, 20 e 25 µg/mL de PRRP: preparadas a
partir da solução de trabalho contendo 25 µg/mL de PRRP, conforme dados da
tabela 7, cuja matriz é semelhante da amostra em estudo, preparadas em
duplicata.

46
3.1.3- Preparo da Curva de Calibração O preparo das soluções padrão foi realizado a partir de uma solução de
trabalho contendo 25µg/mL. A curva-padrão foi preparada da seguinte forma, como
detalhado na tabela 7 e cada solução padrão foi injetada em duplicata.
Tabela 7 - Preparo da curva padrão.
Padrões
Concentração (µg/mL)
Volume solução trabalho (mL)
Volume de água Milli-Q (mL)
P1 10 2 3 P2 15 3 2 P3 20 4 1 P4 25 5 ---
3.1.4- Preparo das Amostras Os três analistas tomaram como amostra uma alíquota de 6,0 mL da solução
estoque, medida em pipeta automática calibrada, (250 µg/mL), que foi transferida para
balão volumétrico de 100 mL e avolumado até o traço de aferição com água tipo Milli-Q,
resultando numa concentração de 15 µg/mL. Foram tomadas alíquotas de 5,0 mL
dessa solução e transferidas para 10 diferentes frascos (em duplicata) do amostrador
automático, totalizando um quantitativo de 20 replicatas por analista. Foram
adicionados 500 µL de NaOH 1 M em cada frasco e a mistura foi homogeneizada, para
proceder a hidrólise do polissacarídeo. As soluções amostra mais hidróxido de sódio,
foram deixadas à temperatura ambiente por 16 horas para a hidrólise completa,

47
conforme estudo prévio realizado pela GSK, garantindo que não haja produtos de
degradação que possam interferir, antes de proceder a análise.
Cada analista efetuou suas análises em dias diferentes, respeitando-se as
condições operacionais do sistema e do Laboratório, este número de injeções diárias foi
o limite permitido. Horwitz W, 1997, recomenda o uso de no mínimo 6 replicas para a
concentração em estudo .
3.2- Descrição do Método tipo III
O Método tipo III , determinação da condutividade em água para injetáveis,
WFI, consiste na medição numérica da capacidade elétrica de transportar e conduzir
a corrente em solução. O inverso da resistência elétrica de 1cm3 do líquido na
temperatura de 25°C. Ela é medida diariamente no La boratório em amostras de
diversos tipos de águas purificadas e potáveis.
3.2.1- Materiais do Método tipo III
O material utilizado e as condições de análise têm as seguintes
características:
Condutivímetro 712 METROHM com célula de medição 0,77 cm-1;
Eletrodo AG 9101 Herisau;
Termômetro acoplado;
Padrão de condutividade com certificado fornecido pela METROHM,
concentração de 1408 µS/cm.

48
3.2.2- Condições de Análise
Para conhecimento do comportamento da água WFI estudada , foram
coletadas amostras diárias num período de trinta dias consecutivos por um único
analista e demonstradas as variabilidades dos resultados através de sua carta de
controle, pois existe somente padrão de calibração do equipamento e não padrão de
amostra com mesma matriz.
As condutividades foram obtidas com diferentes tempos a fim de descobrir
até que ponto sua estabilidade seria mantida. Foram medidos pontos com 0, 30, 60
e 90 minutos após a coleta e sua temperatura foi diminuindo gradativamente com o
passar do tempo. A temperatura inicial da amostra foi de cerca de 90°C e após 90
minutos estava à temperatura ambiente.
Para esse estudo foram sempre analisadas amostras coletadas de um
mesmo sistema de produção de WFI.
Por fim foram verificadas as condições de análise de precisão intra
Laboratório, através da repetitividade, dos dados obtidos pelo mesmo analista -
precisão intra dia - e com três analistas diferentes em dias diferentes, todos com
mesmo equipamento - caracterizando a Reprodutibilidade. As variabilidades foram
verificadas através dos desvios padrão e coeficientes de variação, desvios padrão
relativos. Conforme recomendado pela NBR ISO/ IEC 17095 e Farmacopéia
americana, de acordo com os parâmetros de validação analítica para esse método o
importante seria somente testar sua adequação, já que é um método padronizado.
O estudo foi realizado seguindo a mesma organização feita para o método tipo I.
4 - Resultados e Discussão :
Os resultados das estimativas de incerteza de medição dos dois métodos em
estudo estão apresentados de acordo com a ordem descrita no capítulo 3. Os dados
obtidos foram utilizados para a confecção de cartas de controle, como recomendado

49
pela ASTM, 2004, utilizando o programa STATiSTICA versão 6.0. Não foi feita a
exclusão de pontos fora de controle com o objetivo de visualizar o comportamento dos
analistas e das amostras rotineiramente. As causas prováveis para alguns pontos
estarem fora de controle podem estar associadas a erros sistemáticos, visto que 95%
dos dados estão dentro dos limites de controle, caracterizando a presença de erros
aleatórios conforme Ryan, 2000. Foram tomadas medidas de correção através do
teste t e recuperação.
Para cada analista, os primeiros histogramas são referentes aos dados
tratados de maneira individual, com o objetivo de visualizar graficamente o
comportamento de cada conjunto de resultados obtidos por cada analista. Na
distribuição normal, o cálculo dos limites superior e inferior, foi considerado pelo
programa Statistica utilizado como ± 3σ, que corresponde a 6σ, com desvio padrão da
distribuição, dado por:
∴=dR
2
σ
σ = Desvio padrão da distribuição
R = Amplitude média
d2= parâmetro de distribuição ou amplitude relativa, tabelado em função de “n”
medições
(Desenvolvido por Shewhart, de acordo com Montgomery, 2001.)
Para efeito de cálculo e parâmetros de validação foram utilizados dados
provenientes das cartas de controle construídas com a média das médias dos
resultados de modo a obter a precisão intra-ensaio e intra laboratorial. Esses gráficos
estão apresentados após as cartas de controle das médias de valores individuais
obtidos por cada um dos três analistas.

50
4.1 - Resultados para o Método tipo I
4.1.1- Primeira curva de calibração
Foi construída uma curva de calibração padrão (concentração x área) e
através do cálculo das áreas dos picos de cada um desses pontos foram
calculados os valores de concentração real para cada ponto. A concentração das
soluções amostras foi obtida pela interpolação dos dados na curva de calibração,
pela reta de regressão linear, usando o programa Excel como ferramenta de
cálculo. Estes resultados foram verificados em máquina de calcular científica. Para
a integração das áreas dos picos referentes as soluções amostra e padrão
utilizou-se o programa software Peaknet do próprio equipamento.
Os analistas 1 e 2, utilizaram a primeira curva de calibração, enquanto
que o analista 3 utilizou os dados da segunda curva. O uso de curvas diferentes
foi necessário devido ao término da primeira solução eluente e conseqüente
preparação de uma outra identicamente preparada.
Ilustração 5 - Primeira curva de calibração [ ] pico) do áreay ( x ão)concentraç ( ==χ utilizada pelos Analistas 1 e 2
y = 0,5892x + 0,1315r = 1
0
2
4
6
8
10
12
14
16
0 5 10 15 20 25 30

51
Tabela 8 - Área dos picos obtidos para diferentes s oluções padrão usadas para confecção da curva de calibração 1.
Concentração Nominal (µg/mL)
Área (nCmin)
Concentração Real
(µg/mL) x y x
10 6,0236 9,9995 15 8,9844 15,0243 15 8,9646 14,9907 20 11,907 19,9850 20 11,908 19,9865 25 14,869 25,0110 25 14,864 25,0030
O estudo estatístico dos dados obtidos pela primeira curva de calibração, estão
apresentados na tabela 9:
Tabela 9 - Parâmetros de regressão linear da pri meira curva de calibração, obtidos através do programa Excel.
Estatística de regressão
R múltiplo 1
R-Quadrado 1
R-quadrado ajustado 1
Erro padrão 1,12347E-14Observações 7
ANOVAgl SQ MQ F F de significação
Regressão 1 64,48002913 64,48002913 5,10863E+29 1,01752E-73Resíduo 5 6,31089E-28 1,26218E-28Total 6 64,48002913
Coeficientes Erro padrão Stat t valor-P 95% inferiores 95% superioresInterseção 0,13155 1,58882E-14 8,27975E+12 4,87774E-64 0,13155 0,13155Variável X 1 0,589235 8,24396E-16 7,14747E+14 1,01752E-73 0,589235 0,589235

52
Observa-se que o valor de r, coeficiente de correlação encontrado foi
adequado, pois quanto mais próximo de 1 resulta num ajuste perfeito entre os pontos,
demonstrando o grau de correlação entre x e y. Através da tabela 9 de ANOVA foi
possível comprovar a análise de variância uni variável, ou seja, a variação em torno
da média, que é a soma quadrática total, originada a partir do somatório das somas
quadráticas da regressão e dos resíduos, relacionados a erros, sendo possível
conhecer os erros relacionados para “a e b”, coeficientes linear e angular,
respectivamente. Quanto maior for o termo da soma quadrática da regressão, melhor
será o ajuste, pois implica que a relação com os resíduos é pequena. O valor - p,
correspondente à probabilidade cumulativa, com valor menor que o nível de
significância de 0,05, relacionado a 95% de confiança, que caracterizou valores de
interseção e coeficiente angular estatisticamente significantes.
Segundo manual do INMETRO, 2003, a maneira de “aninhar” os valores de
diferentes grandezas dependentes, consistindo de arranjos que ajudam a identificar e
quantificar efeitos aleatórios, é chamada de análise de variância e isso é possível pelo
estudo da ANOVA.

53
4.1.1.1- Resultados obtidos pelo analista 1
Tabela 10 - Dados obtidos pelo Analista 1.
Amostras Área (nCmin)
Valores individuais de Concentração (µg/mL)
Valores Médios de Concentração (µg/mL)
1 8,8629 14,8191 14,8495 1 8,8987 14,8798 2 8,8635 14,8201 14,8578 2 8,9080 14,8956 3 8,8663 14,8248 14,8170 3 8,8572 14,8094 4 8,7164 14,5704 14,6466 4 8,8063 14,7230 5 8,7324 14,5976 14,6728 5 8,8211 14,7481 6 8,6769 14,5034 14,4024 6 8,5580 14,3016 7 8,4842 14,1763 14,3634 7 8,7047 14,5506 8 8,6269 14,4185 14,3067 8 8,4951 14,1948 9 8,5363 14,2648 14,1950 9 8,4541 14,1253 10 8,6831 14,5139 14,5416 10 8,7158 14,5694
Média 14,5653
14,5653
Desvio-Padrão
0,2507
0,2403
O desvio padrão da média utilizado no cálculo é igual a ( )
∴−
−=∑
=
11
n
x
S
n
iIx
x i= medida
x= média das medidas
n= número de medidas

54
4.1.1.2- Cartas de Controle dos Resultados obtidos pelo Analista 1
Ilustração 6 - Carta de Controle dos dados individ uais do Analista 1
Ilustração 7 -Carta de Controle dos dados médios do Analista 1

55
De acordo com as cartas de controle, ilustrações 6 e 7, foi possível visualizar o
decréscimo da concentração ao longo das análises, aproximadamente após a metade
dos dados, e uma tendência dos pontos em estar nos limites externos da curva de
Gauss, conforme o histograma de observações. Com a ajuda do software Statistica
construiu-se dois tipos de gráficos que mostram o comportamento dos dados obtidos
por cada analista. No histograma de observações é possível conhecer a dispersão dos
dados diante da curva de Gauss, com distribuição normal. Nas cartas de controle, das
médias dos dados individuais e das médias dos dados médios observa-se a
variabilidade dos resultados dentro dos limites de controle, 6б (desvio padrão da
distribuição), em torno de suas médias que delimitam regiões com probabilidade de
99,7% - Montgomery, 2001.
As observações aqui apresentadas serviram também para julgamento das
próximas cartas de controle, construídas com dados obtidos pelos analistas 2 e 3.
É importante ressaltar que antes da injeção das amostras e padrões, o
sistema cromatográfico foi estabilizado, quanto a linha de base, conforme
recomendado pelo fabricante, visando a eliminação de possíveis erros sistemáticos
provenientes de ruídos do equipamento. Porém, esse procedimento não garantiu a
presença somente de erros aleatórios nas análises originados pelos analistas. Pelo
comportamento dos dados observados nas cartas de controle e pelo motivo das
injeções terem sido realizadas automaticamente, nota-se que há sinais de possíveis
erros aleatórios somados aos sistemáticos. Com a finalidade de eliminar estes últimos
procedeu-se a correção dos dados, conforme demonstrada no item 4.1.3.4.

56
4.1.1.4 - Resultados obtidos pelo Analista 2
Tabela 11 - Dados obtidos pelo Analista 2.
Amostras Área (nCmin)
Valores individuais de Concentração (µg/mL)
Valores Médios de Concentração (µg/mL)
1 8,7481 14,6242 14,7074 1 8,8461 14,7906 2 8,9243 14,9233 14,9185 2 8,9188 14,9140 3 8,9029 14,8870 14,8435 3 8,8518 14,8002 4 - - 14,9011 4 8,9112 14,9011 5 8,8958 14,8749 14,8641 5 8,8830 14,8532 6 8,9248 14,9241 15,2880 6 9,3536 15,6519 7 9,1147 15,2464 15,3291 7 9,2122 15,4119 8 9,1185 15,2529 15,2773 8 9,1474 15,3019 9 9,0232 15,0911 15,1619 9 9,1067 15,2329 10 8,8809 14,8496 14,8496 10 8,7481 -
Média 15,0293 15,0161
Desvio- Padrão
0,2636
0,2423

57
4.1.1.5 - Cartas de Controle dos Resultados do Anal ista 2
Ilustração 8 - Carta de Controle dos dados individu ais do Analista 2
Ilustração 9 - Carta de Controle dos dados médios d o Analista 2
Esse analista obteve dados com comportamento de tendência, porém inverso
aos do analista 1, uma vez que os resultados foram elevados, demonstrando um
súbito crescimento, observado a partir do décimo ponto da carta de controle.

58
4.1.2 - Segunda Curva de Calibração
Ilustração 10 - Segunda Curva de calibração [ ] pico) do áreay ( x ão)concentraç ( ==χ utilizada pelo Analista 3
Tabela 12 - Área dos picos obtidas para diferentes soluções padrão usadas para confecção da curva de calibração 2.
Concentração Nominal (µg/mL)
Área (nCmin)
Concentração Real
(µg/mL) x y x
10 6,4876 10,0973 10 6,3497 9,8797 15 9,5685 14,9587 15 9,6748 15,1265 20 12,7998 20,0574
20 12,6626 19,8410 25 15,8979 24,9460 25 15,9913 25,0934
O estudo estatístico dos dados obtidos pela segunda curva de calibração estão
apresentados na tabela 13:
y = 0,6337x + 0,0884r = 0,99985
0
2
4
6
8
10
12
14
16
18
0 5 10 15 20 25 30

59
Tabela 13 - Regressão Linear da segunda curva de ca libração.
Estatística de regressão
R múltiplo 1
R-Quadrado 1
R-quadrado ajustado 1
Erro padrão 2,53818E-15Observações 8
ANOVAgl SQ MQ F F de significação
Regressão 1 100,4420104 100,442 1,55909E+31 1,78112E-92Resíduo 6 3,86542E-29 6,44E-30Total 7 100,4420104
Coeficientes Erro padrão Stat t valor-P 95% inferiores 95% superioresInterseção 0,088435 2,94866E-15 3E+13 9,27489E-80 0,088435 0,088435Variável X 1 0,633748 1,60502E-16 3,95E+15 1,78112E-92 0,633748 0,633748
Observa-se que os valores de r, da soma quadrática, erro padrão e p-valor
estão coerentes com aqueles da tabela 9, relativa aos dados da primeira curva de
calibração.
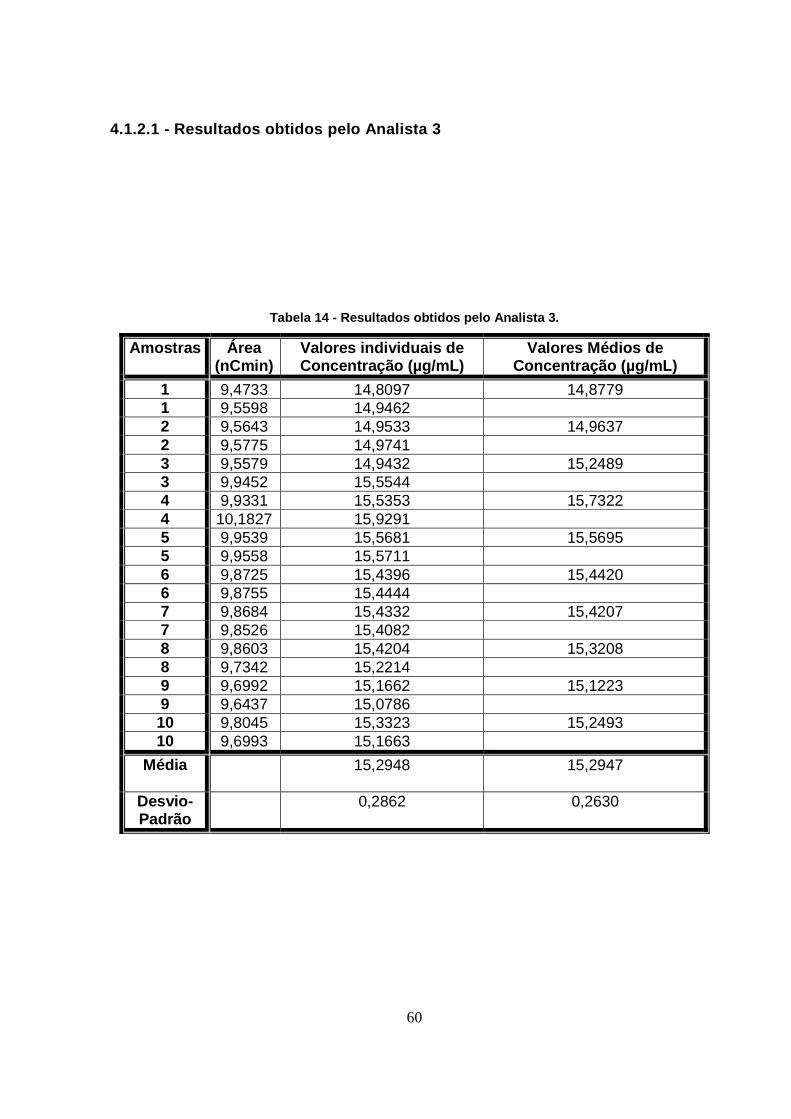
60
4.1.2.1 - Resultados obtidos pelo Analista 3
Tabela 14 - Resultados obtidos pelo Analista 3.
Amostras Área (nCmin)
Valores individuais de Concentração (µg/mL)
Valores Médios de Concentração (µg/mL)
1 9,4733 14,8097 14,8779 1 9,5598 14,9462 2 9,5643 14,9533 14,9637 2 9,5775 14,9741 3 9,5579 14,9432 15,2489 3 9,9452 15,5544 4 9,9331 15,5353 15,7322 4 10,1827 15,9291 5 9,9539 15,5681 15,5695 5 9,9558 15,5711 6 9,8725 15,4396 15,4420 6 9,8755 15,4444 7 9,8684 15,4332 15,4207 7 9,8526 15,4082 8 9,8603 15,4204 15,3208 8 9,7342 15,2214 9 9,6992 15,1662 15,1223 9 9,6437 15,0786 10 9,8045 15,3323 15,2493 10 9,6993 15,1663
Média 15,2948
15,2947
Desvio- Padrão
0,2862
0,2630

61
4.1.2.2 - Cartas de Controle dos Resultados do Anal ista 3
Ilustração 11 - Carta de Controle dos dados individ uais do Analista 3
Ilustração 12 - Carta de Controle dos dados médios do Analista 3
De acordo com as cartas de controle construídas com os dados gerados pelo
terceiro analista é notório que estes apresentaram-se comportamento diferente dos
demais. Porém aproxima-se do tipo de comportamento daqueles dados obtidos pelo
analista 2.

62
4.1.3 - Estudo e tratamento dos dados obtidos pelos três Analistas
4.1.3.1 - Teste F(Snedecor)
Este teste foi realizado com o objetivo de comparar a semelhança do
comportamento entre os três analistas através de suas variâncias obtidas no
tratamento dos dados individuais de cada um deles. Na tabela 15 estão expressas as
variâncias dos três analistas relacionadas aos valores individuais de concentração e o
desvio padrão da média.
Tabela 15 - Relação de Variâncias dos três analista s.
Analistas Desvio- padrão (S) Variância (S 2)
1 0,2507 0,0628
2 0,2636 0,0695
3 0,2862 0,0819
Comparação de variâncias entre o Analista 2 e o Analista 1:
F calculado = SS
2
1
2
2 = 0628,0
0695,0 = 1,10668 ≡ 1,11
F crítico ou tabelado (para n-1 graus de liberdade = 19 e 95% de confiança)
F(19,19) e α = 0,05 = 2,16
Como F calculado < F crítico , não há diferença significativa entre as variâncias obtidas
através dos dados destes dois analistas, então podemos considerar que elas são
estatisticamente homogêneas.

63
Comparação entre o Analista 3 e o Analista 2:
F calculado = SS
2
2
2
3 = 0695,0
0819,0 = 1,18
F crítico ou tabelado (para n-1 graus de liberdade) = F(19,19) e α = 0,05 = 2,16
Como F calculado < F crítico , não há diferença significativa entre as variâncias dos dados
obtidos por estes analistas, então as variâncias e as médias são estatisticamente
homogêneas.
Comparação entre o Analista 3 e o Analista 1:
F calculado = SS
2
1
2
3 = 0628,0
0819,0 = 1,30
F crítico ou tabelado (para n-1 graus de liberdade) = F(19,19) e α = 0,05 = 2,16
Como F calculado < F crítico , não há diferença significativa entre as variâncias dos dados
dos analistas, então elas são estatisticamente homogêneas.
4.1.3.2 - Verificação de Tendência ( teste t para v erificar a média de duas séries
de dados independentes, obtidos por dois analistas diferentes usando o
mesmo método e mesmo equipamento.)
Conforme VIM, 2003, “a tendência de um instrumento de medição é
normalmente estimada pela média dos erros de indicação de um número apropriado
de medições repetidas”. Para esse teste é recomendado que tenha sido realizado o
teste F, para saber qual tipo de teste t utilizar. Como não houve diferenças

64
significantes entre a comparação das variâncias dos analistas, foi utilizado o teste t
para a verificação da média de duas séries de dados e sua tendência.
Pela forma de comportamento da distribuição dos dados, através de suas
cartas de controle, foi possível suspeitar que houvesse tendência do comportamento
dos dados obtidos pelos diferentes analistas. As distribuições tiveram caráter de
distribuição normal não padronizadas tipo bi caudais. Foram estudados os dados
individuais para a correção dos mesmos.
De acordo com sua definição, a tendência é independente do número de
medidas, sendo constante ou variável de forma previsível. Podendo ser denominada
de erro sistemático ou erro de tendência, conforme VIM, 2003. Visando a sua
eliminação estas foram expressas como recuperação analítica, que é o quociente
entre o valor observado e o valor esperado. Foi demonstrado se a mesma é
desprezível ou compensada, como recomenda o guia EURACHEM, 2002.
Para verificar a tendência dos resultados gerados pelos três analistas primeiramente
calculou-se o desvio padrão ponderado utilizando a equação abaixo:
Sc = ( ) ( )
2
11
21
2
2
2
1
−+−×+−×
nnSS nn
∴
Sc = Desvio padrão ponderado
S21 = Variância encontrada pelo analista 1
S22= Variância encontrada pelo analista 2
n1= Número de determinações obtidas pelo analista 1
n2= Número de determinações obtidas pelo analista 2
Em seguida aplicou-se o teste t para a comparação de duas séries de dados
independentes (utilizando o mesmo método, mesmo equipamento). Foram
considerados os valores médios de cada analista, com suas respectivas médias das
médias :

65
t =
nnS
XX
C21
2,1
21
11 +
− ∴
X1 = Média obtida pelo analista 1
X 2 = Média obtida pelo analista 2
2,1SC = Desvio padrão ponderado dos analistas para os dois analistas
n1 e n2 = Número de determinações obtidas pelos dois analistas
considerando t crítico = 1,734 para 18 graus de Liberdade correspondente a n=10
para cada analista ≡ (10+10)-2 .
Comparação entre o analista 1 e 2:
SC 1 e 2 = ( ) ( )[ ] ( ) ( )[ ] =
−+−+−
22020
1202636,012025068,0 22 xx
38
320214,1193969,1 += 0,2572
t calculado =
+
−
20
1
20
12572,0
0293,155653,14 =
0813,0
464,0= 5,70

66
Comparação entre o analista 1 e 3:
SC 1 e 3 = ( ) ( )[ ] ( ) ( )[ ] =
−+−+−×
22020
120286201,0,012025068,0 22 x =+38
5563,11939,10,2690
t c =
+
−
20
1
20
12690,0
2948,155653,14=
0851,0
7295,0 = 8,57
Comparação entre o analista 2 e 3:
SC 2 e 3 = ( ) ( )[ ] ( ) ( )[ ] =
−+−+−
22020
120286201,01202636,0 22 xx 27,0
38
5563,13202,1 =+
t c =
+
−
20
1
20
127,0
2948,150293,15=
0854,0
2655,0 = 3,11
Os dados obtidos pelos analistas, que serviram de parâmetro comparativo das
análises dos três analistas e o valor tabelado de t tab ou crítico = 1,734, obtiveram-se
calculadost com valores maiores que o t tab. Então mesmo com valores de desvio-
padrão e coeficientes de variação próximos e a precisão do método, existe diferença
significativa entre os analistas. A menor diferença encontrada foi entre os analistas 2
e 3.
4.1.3.3 - Teste t, comparação da média experimental e um valor conhecido
(valor referência)
O teste t foi utilizado como ferramenta estatística para comparar as médias
dos resultados obtidos pelos analistas com o valor médio nominal desejado para as

67
soluções amostra, do método, pressupondo a distribuição normal dos valores da
população (valores nominais), servindo de orientação para verificação dos dados
experimentais.
t calculado =
nS
X µ−, onde
X = média das amostras
µ = média da população (valor nominal)
S = desvio padrão das amostras
n = número de determinações
Para n -1 graus de liberdade.
Analista 1 - t calculado =
2025068,0
155653,14 −= 7,75
Analista 2 - t calculado =
202636,0
150293,15 −= 0,50
Analista 3 - t calculado =
20286201,0
152948,15 −= 4,61
t crítico para 19 graus de liberdade = 2,14
Conclusão: somente os dados provenientes da análise do Analista 2 não tiveram
comportamento de tendência significativa podendo ser desprezível, pois seu
calculadost < t crítico e seus resultados são estatisticamente compatíveis com o valor
verdadeiro. Os dados dos Analistas 1 e 3 deverão ser corrigidos a fim de eliminar o
erro sistemático. Seus resultados encontrados foram significativamente diferentes do

68
valor verdadeiro, 15 µg/mL, para nível de confiança de 95%, verificado pelo teste t
tipo 1 (estudo comparativo da média da amostra com o valor verdadeiro, nominal).
4.1.3.4 - Recuperação dos analistas 1 e 3
Resultado do Analista 1 = édiacuperaçãom
staMédiaAnali
Re
1
Resultado do Analista 1 = Resultado corrigido para o erro sistemático
Média do Analista 1 = Média obtida sem correção
Recuperação Média = Média dos valores de recuperação obtidos pelo Analista 1
(Recuperação = valor obtido / valor verdadeiro)
Tabela 16 - Taxas de Recuperação dos dados obtidos pelos Analistas 1 e 3.
Amostra Analista 1 Analista 3 1 0,98794 0,9873 1 0,99199 0,9964 2 0,98801 0,9969 2 0,99304 0,9983 3 0,98832 0,9962 3 0,98729 1,0370 4 0,97136 1,0357 4 0,98153 1,0619 5 0,97317 1,0379 5 0,98321 1,0381 6 0,96689 1,0293 6 0,95344 1,0296 7 0,94509 1,0289 7 0,97004 1,0272 8 0,96124 1,0280 8 0,94632 1,0148 9 0,95098 1,0111 9 0,94168 1,0052
10 0,96759 1,0222 10 0,97129 1,0111
Desvio-Padrão 0,01671 0,0191 Média 0,97102 1,0197

69
Resultado médio do Analista 1 corrigido = 155653,14
5653,14 =
97102,0
5653,14 = 15
A correção dos dados obtidos pelo analista se dá dividindo-se a unidade pelo fator de
correção: Fcorreção = 1 / 0,97102 = 1,0298. Portanto, é possível obter os valores
corrigidos do analista 1, multiplica-se os valores encontrados pelo seu fator de
correção.
Resultado médio do Analista 3 corrigido = 152948,15
2948,15 = 14,9993 ≡ 15
Fcorreção = 1/ 1,0197 = 0,98068
4.1.4 - Validação do Método tipo I
Os parâmetros relacionados ao método, categoria tipo I de classificação de métodos, já
são conhecidos pois, o mesmo já fora validado anteriormente e foram também
verificadas as condições de análise de sua origem, GlaxoSmithKline, no LAFIQ (“In
house Validation”). Para efeito de estudo os parâmetros seletividade, faixa de
trabalho, linearidade, precisão e exatidão foram reavaliados.
4.1.4.1- Seletividade O método é seletivo e estável, baseado em estudo de validação prévio, conforme anexo
C1. Foram realizados anteriormente estudos de interferência do branco sem a matriz,
para verificar a seletividade do método e a estabilidade foi verificada em diferentes
temperaturas com as amostras, conforme relatório prévio de validação exposto no
anexo C. Os coeficientes angulares, b, das curvas de calibração demonstram que o
método tem a mesma sensibilidade para as duas curvas. Com o coeficiente de
correlação r > 0,9999, conforme Consideration of Harmonized IUPAC Guidelines for the
In-house Validation of Methods of Analyses, WHO, 2002; ISO 8466-1, 1990.

70
4.1.4.2 - Faixa de trabalho A faixa de trabalho utilizada foi de 67 a 167% da concentração do polissacarídeo de 10
a 25µg/mL, para o valor ideal de 15µg/mL, seguindo a mesma relação de 50 a 150%,
conforme recomendado pela USP 24, Thompson, 1999 e Inman, 1987; de 60 a 120%,
conforme recomenda o FDA, Horwitz, 1998 e de 20 a 200%, como relata a ISO 8466,
1990 .
4.1.4.3 - Linearidade O método é linear, conforme comprovado pelos parâmetros de linearidade da reta de
regressão discutidos e apresentados nas tabelas 9 e 13.
4.1.4.4 - Precisão
Reprodutibilidade
Pelo resultado dos três analistas, pode-se concluir que o método tem precisão, pois
apresentou desvios padrão com valores muito próximos e CV% ( RSD) ≤ 2% para o
sistema, Instrumento e analista, que de acordo com Horwitz, 2004, que caracteriza a
Reprodutibilidade, para o caso de detecção de macrocomponentes.
Repetitividade
Para um método ter repetitividade o critério é CV%< 1,0%. O método não pode ser
julgado repetitivo pelos dados médios avaliados, como já discutido anteriormente pelo
estudo da tendência dos dados e correção dos erros.
Considerando-se as correções dos dados individuais dos analistas 1 e 3 com os dados
individuais, foi possível obter os fatores de correção para os analistas 1 e 3, que
originaram a correção dos valores médios dos dois analistas, como é possível conferir
na tabela 17:

71
F c (analista 1) = 1,0298
F c (analista 3) = 0,9807
Tabela 17 - Tabela comparativa de valores corrigid os.
Analistas Médias
sem correção
(µg/mL)
Médias corrigidas
(µg/mL)
Desvios-padrão
sem correção
1 14,5653 14,9993 0,25068
2 15,0293 - 0,26360
3 15,2948 14,9993 0,28620
Conforme o cálculo do coeficiente da variação, desvio padrão relativo, na
aplicação dos dados corrigidos dos analistas 1 e 3, os dados dos analistas geraram
valores em conformidade com a recomendação de Horwitz, para a precisão do sistema,
analista versus equipamento, de acordo com a tabela 18.
CV% = ΧS
× 100 ∴
S = Desvio-padrão
Χ = Média
Tabela 18 -Relação de coeficientes variação e anali stas.
CV% sem correção
CV% após correção
Analista 1 1,72 1,65
Analista 2 1,61 -
Analista 3 1,87 1,72

72
A ilustração 13 refere-se ao diagrama qualitativo representativo da
repetitividade com a Reprodutibilidade do método tipo I. Ele mostra os valores dos
desvios das duas réplicas medidas por cada analista em relação ao valor médio das
vinte determinações e a dispersão entre as réplicas. Como forma de reforçar o estudo,
foi possível demonstrar a variabilidade dos dados obtidos por cada analista e entre eles.
O analista 2 obteve menor variabilidade dos dados entre as réplicas que os
demais. E sua média está melhor relacionada a média da análise, representada pelo
valor médio mais próximo do desvio zero em relação à média global. Portanto, como
verificado anteriormente e provado graficamente, este apresentou melhores resultados,
sem ter havido necessidade de correções.
Ilustração 13 - Gráfico repetitividade x Reprodutib ilidade
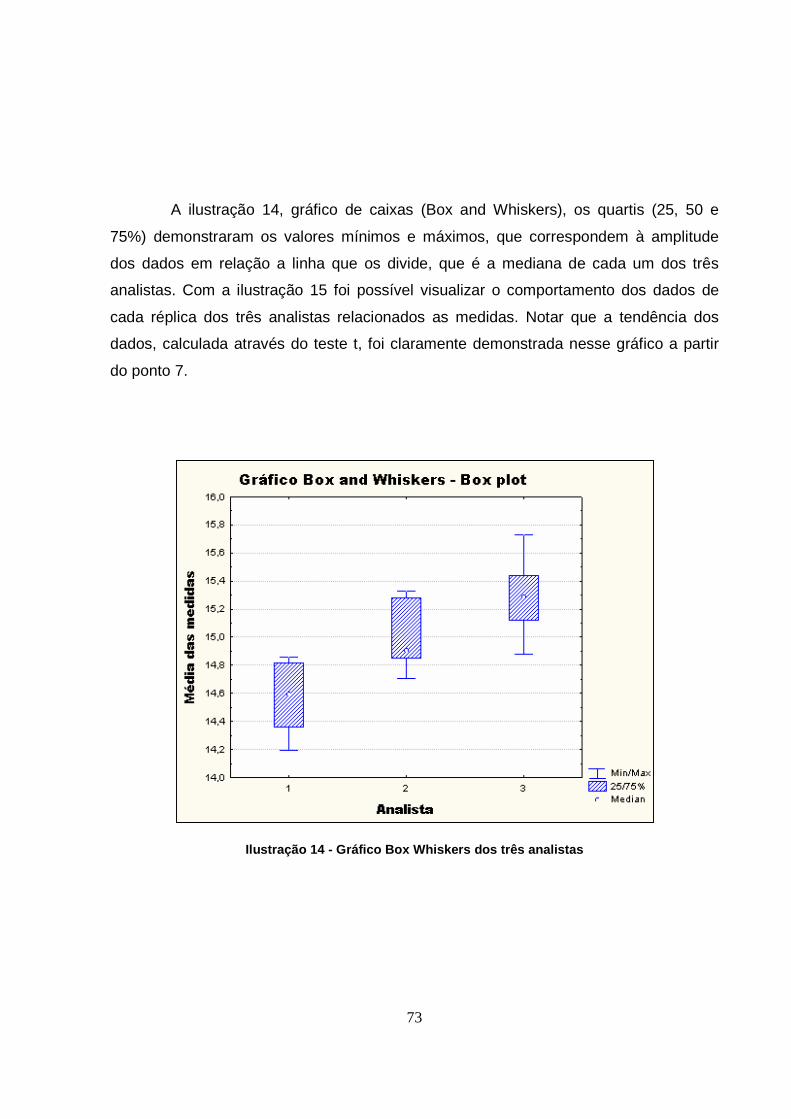
73
A ilustração 14, gráfico de caixas (Box and Whiskers), os quartis (25, 50 e
75%) demonstraram os valores mínimos e máximos, que correspondem à amplitude
dos dados em relação a linha que os divide, que é a mediana de cada um dos três
analistas. Com a ilustração 15 foi possível visualizar o comportamento dos dados de
cada réplica dos três analistas relacionados as medidas. Notar que a tendência dos
dados, calculada através do teste t, foi claramente demonstrada nesse gráfico a partir
do ponto 7.
Ilustração 14 - Gráfico Box Whiskers dos três anali stas
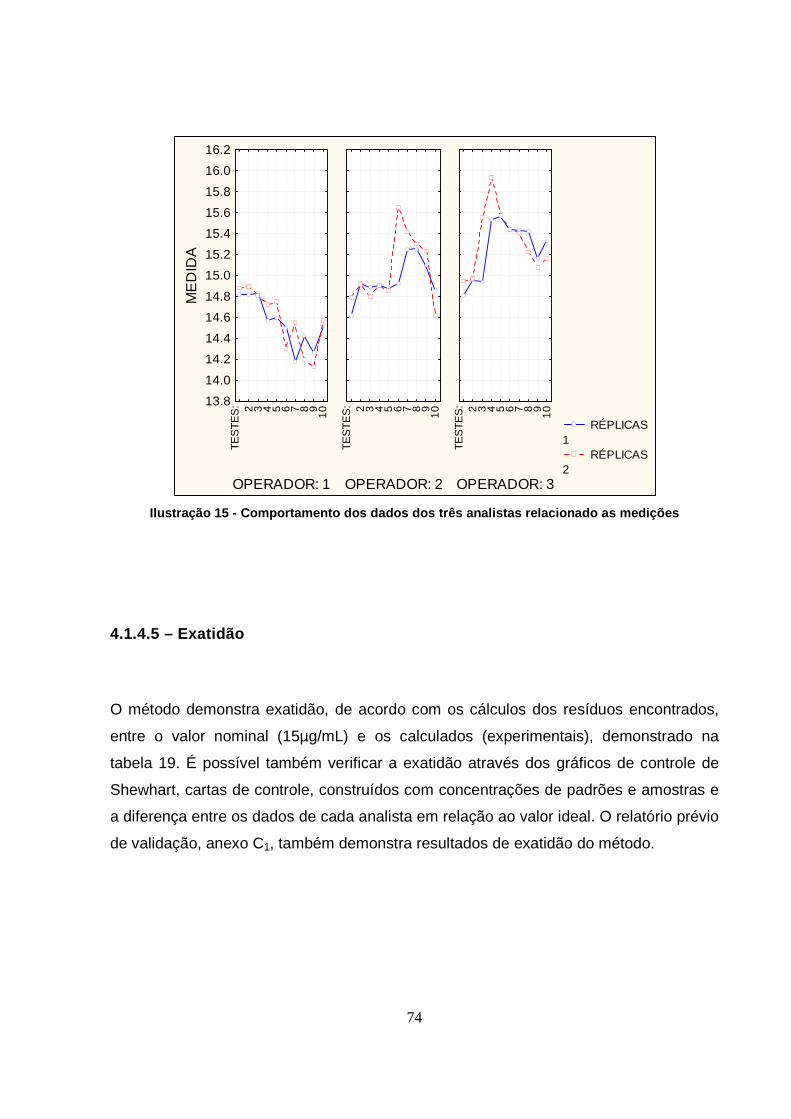
74
Ilustração 15 - Comportamento dos dados dos três an alistas relacionado as medições
4.1.4.5 – Exatidão O método demonstra exatidão, de acordo com os cálculos dos resíduos encontrados,
entre o valor nominal (15µg/mL) e os calculados (experimentais), demonstrado na
tabela 19. É possível também verificar a exatidão através dos gráficos de controle de
Shewhart, cartas de controle, construídos com concentrações de padrões e amostras e
a diferença entre os dados de cada analista em relação ao valor ideal. O relatório prévio
de validação, anexo C1, também demonstra resultados de exatidão do método.
RÉPLICAS1
RÉPLICAS2
OPERADOR: 1
TES
TE
S: 2 3 4 5 6 7 8 9
10
13.8
14.0
14.2
14.4
14.6
14.8
15.0
15.2
15.4
15.6
15.8
16.0
16.2
ME
DID
A
OPERADOR: 2
TES
TE
S: 2 3 4 5 6 7 8 9
10
OPERADOR: 3
TES
TE
S: 2 3 4 5 6 7 8 9
10

75
Tabela 19 - Relação de resíduos entre os dados gera dos por cada analista estudado.
Analista1 Analista2 Analista3
0,1809 0,3758 0,1903 0,1202 0,2094 0,0538 0,1799 0,0767 0,0467 0,1044 0,086 0,0259 0,1752 0,113 0,0568 0,1906 0,1998 0,5544 0,4296 - 0,5353 0,277 0,0989 0,9291
0,4024 0,1251 0,5681 0,2519 0,1468 0,5711 0,4966 0,0759 0,4396 0,6984 0,6519 0,4444 0,8237 0,2464 0,4332 0,4494 0,4119 0,4082 0,5815 0,2529 0,4204 0,8052 0,3019 0,2214 0,7352 0,0911 0,1662 0,8747 0,2329 0,0786 0,4861 0,1504 0,3323 0,4306 - 0,1663
O cálculo dos valores residuais entre cada concentração real, que corresponde ao valor
de concentração encontrada e a nominal, que é correlacionada ao valor teórico, é feito
subtraindo-se o valor encontrado do valor teórico esperado, dado por:
Resíduo = EncontradoTeorico XX −
Onde
XTeórico é o valor nominal da concentração de interesse igual a 15µg/mL;
XEncontrado é o valor real resultante da análise.
A fórmula é estimada em módulo conforme os dados na tabela 19 e na ilustração 16,
estão expressos com seus sinais reais, com a finalidade de demonstrar graficamente o
comportamento dos dados.
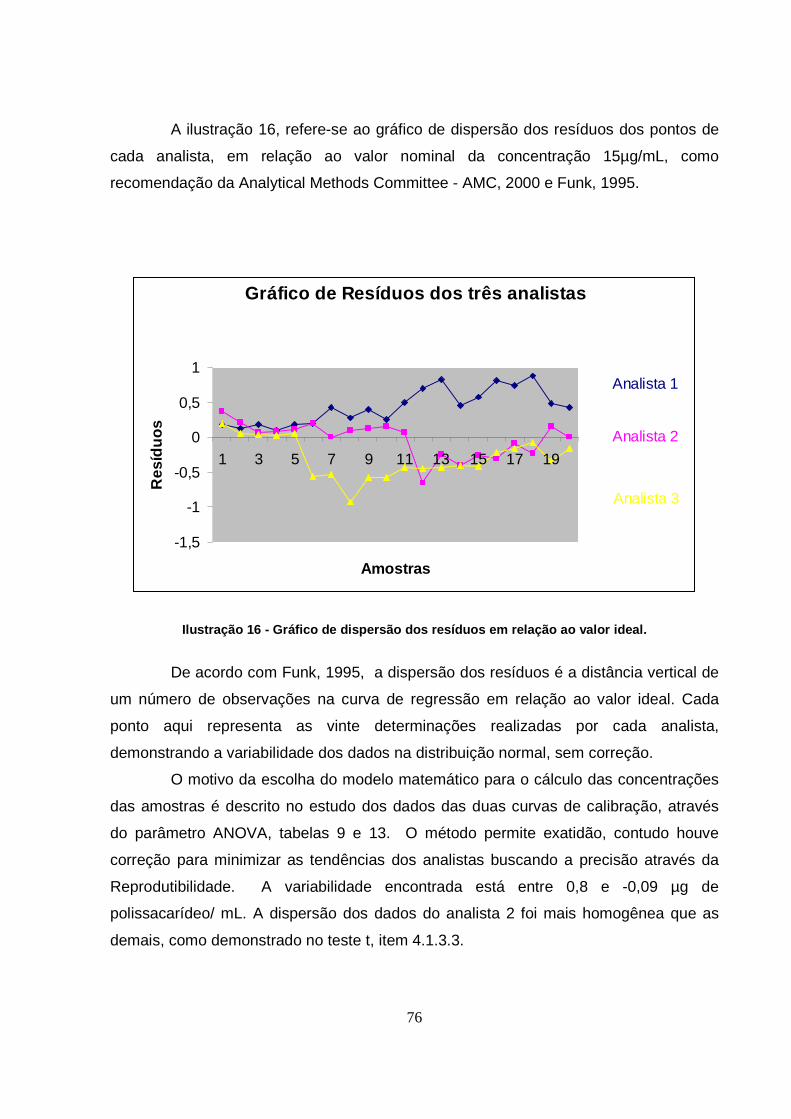
76
A ilustração 16, refere-se ao gráfico de dispersão dos resíduos dos pontos de
cada analista, em relação ao valor nominal da concentração 15µg/mL, como
recomendação da Analytical Methods Committee - AMC, 2000 e Funk, 1995.
Ilustração 16 - Gráfico de dispersão dos resíduos e m relação ao valor ideal.
De acordo com Funk, 1995, a dispersão dos resíduos é a distância vertical de
um número de observações na curva de regressão em relação ao valor ideal. Cada
ponto aqui representa as vinte determinações realizadas por cada analista,
demonstrando a variabilidade dos dados na distribuição normal, sem correção.
O motivo da escolha do modelo matemático para o cálculo das concentrações
das amostras é descrito no estudo dos dados das duas curvas de calibração, através
do parâmetro ANOVA, tabelas 9 e 13. O método permite exatidão, contudo houve
correção para minimizar as tendências dos analistas buscando a precisão através da
Reprodutibilidade. A variabilidade encontrada está entre 0,8 e -0,09 µg de
polissacarídeo/ mL. A dispersão dos dados do analista 2 foi mais homogênea que as
demais, como demonstrado no teste t, item 4.1.3.3.
Gráfico de Resíduos dos três analistas
-1,5
-1
-0,5
0
0,5
1
1 3 5 7 9 11 13 15 17 19
Amostras
Res
íduo
s
Analista 1
Analista 2
Analista 3

77
4.1.4.6 – Robustez
Através de estudo de validação e adequação prévias, conforme anexo C1, o método é
robusto para sua utilização. Foi verificada previamente o desempenho do método e sua
variação quanto aos parâmetros importantes descritos, garantindo que o mesmo atende
a sua finalidade.
4.1.5 - Diagrama de Causa e Efeito
Como abordado anteriormente, o diagrama de causa e efeito é uma importante
ferramenta para ajudar na visualização das possíveis causas de erros e conseqüentes
fontes de incerteza de um método. A seta central é representada pelo resultado
analítico desejado, mensurando, que no método tipo I corresponde a concentração de
polissacarídeo. As setas laterais representam as variáveis da equação e as setas
secundárias, que estão representadas pelos fatores que afetam a equação. Todas as
variáveis e os fatores identificados são representativos do método.
Para o método tipo I , A variável massa, por exemplo, teve contribuição na
estimativa da incerteza de medição com a calibração da balança, pois quando ela foi
calibrada pelo laboratório de Metrologia de Bio-Manguinhos, seus parâmetros de
repetitividade foram considerados para o cálculo do desvio-padrão e assim considerada
como grandeza de entrada para a estimativa de incerteza. Estes se somarão a outras
variáveis, que são: volume explicitado pela calibração das pipetas automáticas e balões
volumétricos e a variável qualificação do cromatógrafo, que originou em dados também
de repetitividade. Todas as variáveis e seus fatores que contribuíram para a incerteza
foram considerados da mesma forma, respeitando-se os dados provenientes de
calibrações, através da repetitividade dos dados cujos certificados estão no anexo C,
visando o resultado final do mensurando – conteúdo de polissacarídeo.

78
Ilustração 17: Gráfico de Ishikawa do método I
4.1.6 - Relatando as fontes de incerteza estimadas do Método tipo I
4.1.6.1 - Estudo das variáveis e suas contribuições
1. Contribuição da Massa
Tabela 20 - Contribuição da variável Massa na estim ativa de Incerteza do método tipo I.
Fonte de
Incerteza
Unidade Tipo Distribuição Valor Divisor Incerteza
(grandeza)
Balança mg A Normal 0,001 K=2 0,0005g ≡ 0,5 mg
Erro Relativo A Retangular 0,001 3 0,0006 ≡ 0,6
Incerteza Padrão Relativa 1,1
Calibração de pipetas e balões
Reprodutibilidade

79
A balança de lâmpada halógena, moisture analyser, foi calibrada pelo
Laboratório de Metrologia de Bio-Manguinhos, sendo portanto um dado de
entrada que contém carta de controle e conseqüentemente seu valor
proveniente do certificado, anexo C; pode ser considerado de uma fonte
de incerteza do tipo A, como recomenda manual EURACHEM.
Foi calculado o erro relativo, no mesmo certificado sendo este valor
considerado para o cálculo da incerteza padrão relativa da massa.
O valor da incerteza da massa foi aquele obtido no ponto de pesagem do
certificado de calibração, 0,1g, pois é o que mais se aproxima da massa
pesada no método em estudo.
2. Contribuição do Volume
Tabela 21 - Contribuição da variável Volume na esti mativa de Incerteza do método tipo I.
Fonte de
Incerteza
Unidade Tipo Distribui-
ção
Valor Divisor Incerteza
(grandeza)
Balão de 50mL mL A Normal 0,01 K=2,16 0,0046 mL
Erro Relativo A Retangular -0,0521 3 -0,030
Incerteza Padrão Relativa 0,0254 *
Fonte de
Incerteza
Unidade Tipo Distribui-
ção
Valor Divisor Incerteza
(grandeza)
Balão de 100mL mL A Normal 0,03 K=2,11 0,0142 mL
Erro Relativo A Retangular 0,0063 3 0,0036
Incerteza Padrão Relativa 0,0178

80
Fonte de
Incerteza
Unidade Tipo Distribui-
ção
Valor Divisor Incerteza
(grandeza)
Pipeta de
5000µL
mL A Normal 4 K=2,32 1,7241 µL ≡
0,0017 mL
Erro Relativo A Retangular - 0,5 3 0,29 ≡
0,00029
Incerteza Padrão Relativa 0,00199
Fonte de
Incerteza
Unidade Tipo Distribui-
ção
Valor Divisor Incerteza
(grandeza)
Pipeta de 1000
µL
mL A Normal 2 K=2,32 0,862 µL ≡
0,0009 mL
Erro Relativo A Retangular 1,3 3 0,75 µL ≡
0,0007
Incerteza Padrão Relativa 0,0016
* A incerteza padrão relativa do balão de 50 mL, tem valor numérico
negativo porém, conforme recomendação do INMETRO, não é possível
estimar esse tipo de resultado com sinal negativo.
Para que não fossem consideradas mais de uma vez, foi escolhido o pior
caso, que corresponde à maior incerteza relativa, do balão de 100mL ,
u =0,0178.

81
3. Contribuição dos analistas e do método.
Precisão do Método (Dados de Reprodutibilidade corr igidos)
Tabela 22 - Relação de desvios padrão antes da corr eção.
Analista Desvio Padrão (µg/mL)
1 0,25068
2 0,26360
3 0,28620
Para os dados do analista 2 não foi necessária a correção, conforme teste
t, demonstrado no item 4.1.3.3. Sua contribuição para a incerteza, como
grandeza de entrada referente a incerteza padrão relativa, do método foi
descrita anteriormente e o valor utilizado do desvio padrão sem correção
foi de, u = 0,26360 µg/mL .
Para os Analistas 1 e 3, foi feita a correção para minimizar a tendência.
Foram utilizados os dados calculados no item 4.1.3.4, que geraram a contribuição para
a estimativa de incerteza padrão relativa.
Correção para o Analista 1:
u (Analista 1 corrigido)=
= Resultado do Analista corrigido x
2
1
2
1
1
Re
+
diacuperçãoMe
uXu oanalistarecuperaçã
analista
analista
u (Analista 1 corrigido)= 15 x
22
97102,0
01671,0
5653,14
25068,0
+
= 0,52 µg/mL

82
Correção para o Analista 3:
Utilizando os dados calculados no item 4.1.3.4.
u (Analista 3 corrigido)= 15 x
22
0197,1
0191,0
2948,15
2636,0
+
= 0,54 µg/mL
Cálculo do Desvio-Padrão Ponderado para os três Ana listas:
Sc=( ) ( ) ( )
3
1311
321
22
2
2
1
−++
−×+−×+−×
nnnSSS nnn
Sc = ( ) ( ) ( )
3101010
1101936,01100676,01102916,0
−++−×+−×+−×
= 0,43 µg/mL
A incerteza padrão da Reprodutibilidade para o método tipo I foi de u = 0,43 µg/mL.
4. Contribuição da Concentração da Solução de Traba lho
Concentração = lãoVolumedoBa
FatormédioPurezarãomassadoPad diluição××
Onde,
Massa pesada = 107 mg
A incerteza padrão relativa da balança é de u =1,1 mg .

83
Pureza = 100% ± 5%
Considerada a 100% da massa de PRRP, correlacionada a 38% D- Ribose, de acordo
com o certificado do fabricante , que é o monômero correspondente após hidrólise
alcalina, com fator de conversão de 2,5, correlacionado ao valor de PRRP.
Significando a correlação de 100% de PRRP --------------- 2,5 x 38% (= 95%)
e 100% corresponde ------ 95% de monômeros após a
hidrólise. Então a variação considerada foi de 95 a 105%, assumindo-se 5% de
incerteza relacionada a pureza. Foi estimada a contribuição da incerteza, como tendo
uma distribuição retangular, tendo sido necessária a divisão pelo valor da distribuição
retangular, 3 para obter a incerteza padrão referente à pureza, como recomendado
pelo guia EURACHEM 3
05,0 = u = 0,029.
Volume do Balão = 50 mL
Com contribuição de incerteza, considerando o pior caso, u = 0,0178.
Fator de Diluição médio = 0,1085
Considerando-se a média dos dois fatores de diluição: 2
1,0117,0 += 0,1085, dividindo-se
por 6 , estimada como distribuição triangular a incerteza relativa foi u = 0,044, de
acordo com o preparo das soluções de trabalho, ilustração 18.

84
107 mg 50 mL 2,14 mg/mL
5,84 mL
50 mL
SoluçãoMãe 250 µg/mL
Fator de diluição = 0,117
2,5 mL
25 mL
Fator de diluição = 0,1
25µg/mL
Ilustração 18 - Preparo das soluções de trabalho
Pela fórmula da concentração do método foi possível calcular a concentração
da solução de trabalho:
Concentração = 10050
10001085,01,0107
××××
= 0,232 µg/ mL
A contribuição da incerteza da concentração é dada pela fórmula resumida de Kragten,
segundo Guia EURACHEM, 2002:

85
u (concentração) = Concentração x 222
+
+
volume
u
pureza
u
massa
u volumepurezamassa
Então,
u (concentração) = 0,232 x 222
50
044,00178,0
1,0
029,0
107
1,1
++
+
=
u (concentração) = 0,232 x ( )2001236,029,001028,0 ++ u (concentração) = 0,0699 ≡ 0,07 µg/mL
5 . Cálculo da Incerteza padrão combinada do método tipo I
u (método tipo I) = ( ) ( )[ ]2Re
2idadeprodutobilãoconcentraç uu +
u (método tipo I) = 0,07 + 0,43 = 0,49926 ≡ 0,50 µg/mL 6. Cálculo da Incerteza padrão expandida do método tipo I U = u x k Onde k= fator de abrangência a 95% de confiança = 2 U = 0,50 x 2 = 1,00 µg/mL → 15 ± 1,00 µg/mL.

86
4.1.7 - Diagrama de Ishikawa “refinado” do método t ipo I
Um novo diagrama de Ishikawa, ilustração 19, foi construído considerando-
se os fatores que realmente contribuíram para as estimativas de incerteza do
método. No diagrama anterior, ilustração 17, considerou-se uma variável
proveniente da qualificação do equipamento e a linearidade de injeção. Porém com
a consideração das fontes de incerteza que contribuíram para a concentração das
soluções de trabalho e da Reprodutibilidade do método, esse fator anterior ficou
implícito. Com o desvio-padrão da Reprodutibilidade, calculado a partir dos dados
obtidos nas diversas repetições, foi possível envolver todas as variabilidades do
processo de medidas, para os cálculos das curvas de calibração e das amostras. O
uso da fórmula de Kragten do manual EURACHEM, 2002, possibilitou o cálculo
simplificado da incerteza do método cromatográfico.
Ilustração 19- Diagrama de Ishikawa refinado do mé todo tipo I
Calibração de pipetas e balões
Reprodutibilidade
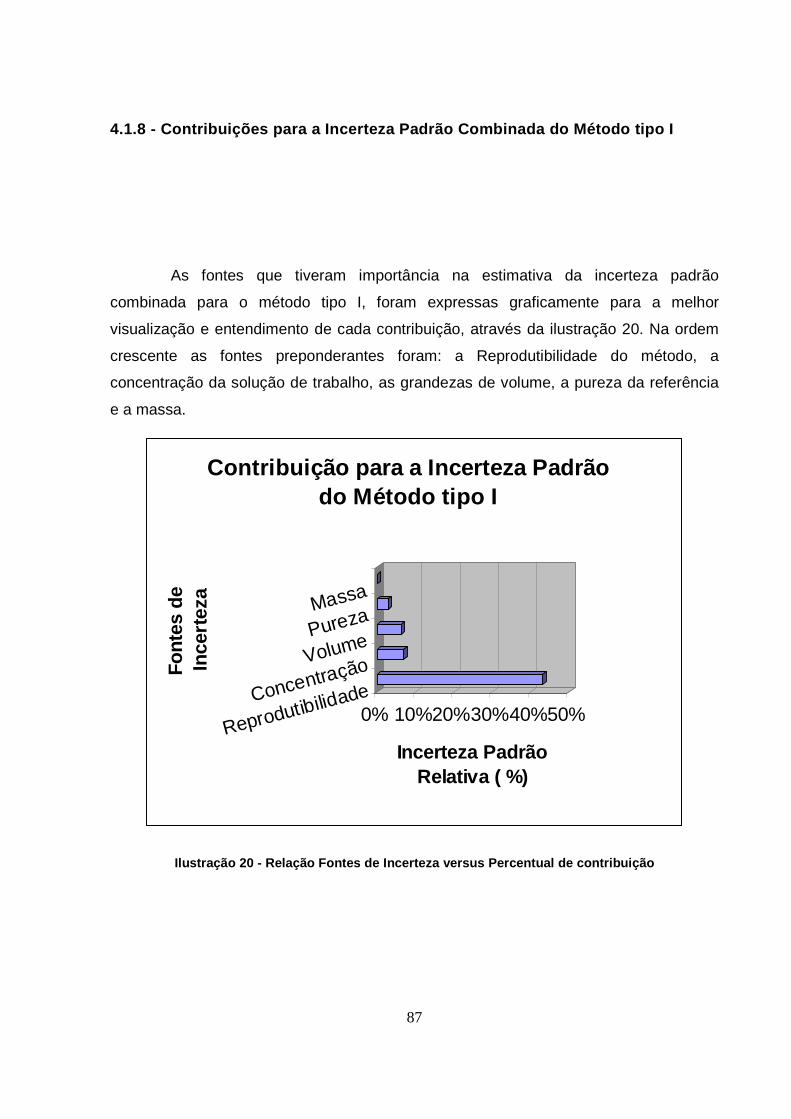
87
4.1.8 - Contribuições para a Incerteza Padrão Combi nada do Método tipo I
As fontes que tiveram importância na estimativa da incerteza padrão
combinada para o método tipo I, foram expressas graficamente para a melhor
visualização e entendimento de cada contribuição, através da ilustração 20. Na ordem
crescente as fontes preponderantes foram: a Reprodutibilidade do método, a
concentração da solução de trabalho, as grandezas de volume, a pureza da referência
e a massa.
Ilustração 20 - Relação Fontes de Incerteza versus Percentual de contribuição
0% 10%20%30%40%50%
Incerteza Padrão Relativa ( %)
ReprodutibilidadeConcentraçãoVolumePurezaMassa
Font
es d
e In
cert
eza
Contribuição para a Incerteza Padrão do Método tipo I
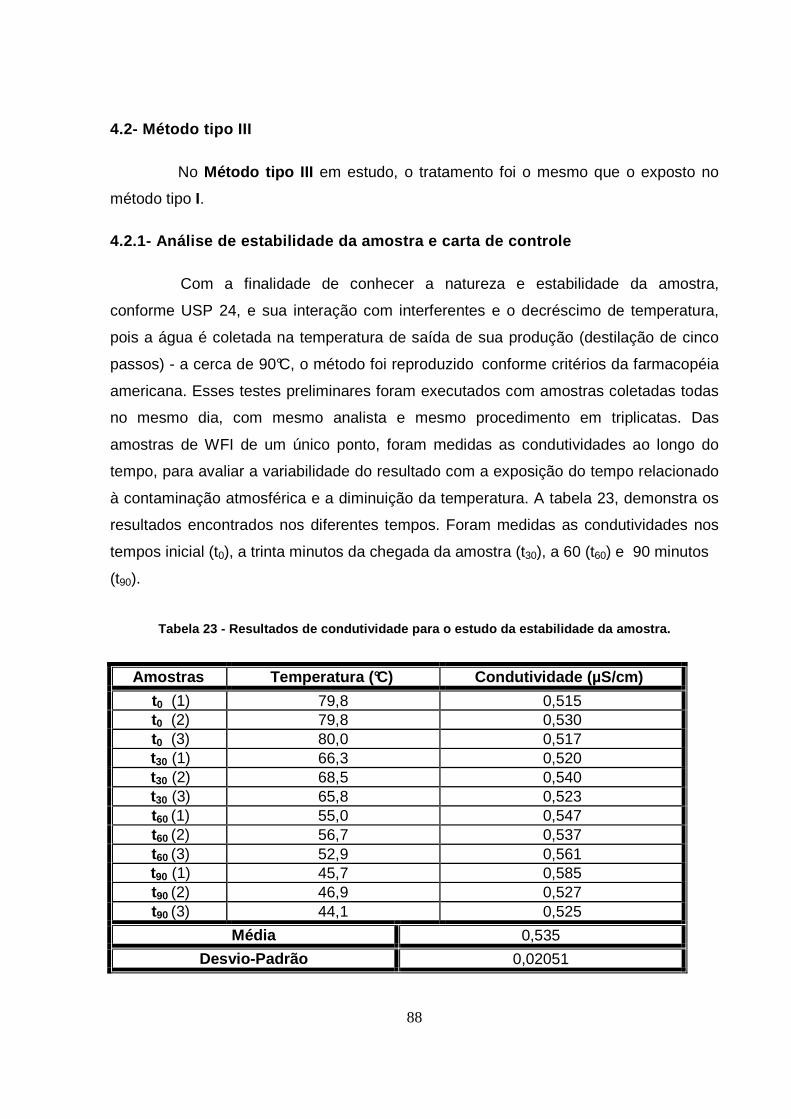
88
4.2- Método tipo III
No Método tipo III em estudo, o tratamento foi o mesmo que o exposto no
método tipo I.
4.2.1- Análise de estabilidade da amostra e carta d e controle Com a finalidade de conhecer a natureza e estabilidade da amostra,
conforme USP 24, e sua interação com interferentes e o decréscimo de temperatura,
pois a água é coletada na temperatura de saída de sua produção (destilação de cinco
passos) - a cerca de 90°C, o método foi reproduzido conforme critérios da farmacopéia
americana. Esses testes preliminares foram executados com amostras coletadas todas
no mesmo dia, com mesmo analista e mesmo procedimento em triplicatas. Das
amostras de WFI de um único ponto, foram medidas as condutividades ao longo do
tempo, para avaliar a variabilidade do resultado com a exposição do tempo relacionado
à contaminação atmosférica e a diminuição da temperatura. A tabela 23, demonstra os
resultados encontrados nos diferentes tempos. Foram medidas as condutividades nos
tempos inicial (t0), a trinta minutos da chegada da amostra (t30), a 60 (t60) e 90 minutos
(t90).
Tabela 23 - Resultados de condutividade para o estu do da estabilidade da amostra.
Amostras Temperatura (°C) Condutividade (µS/cm)
t0 (1) 79,8 0,515 t0 (2) 79,8 0,530 t0 (3) 80,0 0,517 t30 (1) 66,3 0,520 t30 (2) 68,5 0,540 t30 (3) 65,8 0,523 t60 (1) 55,0 0,547 t60 (2) 56,7 0,537 t60 (3) 52,9 0,561 t90 (1) 45,7 0,585 t90 (2) 46,9 0,527 t90 (3) 44,1 0,525
Média 0,535
Desvio-Padrão 0,02051
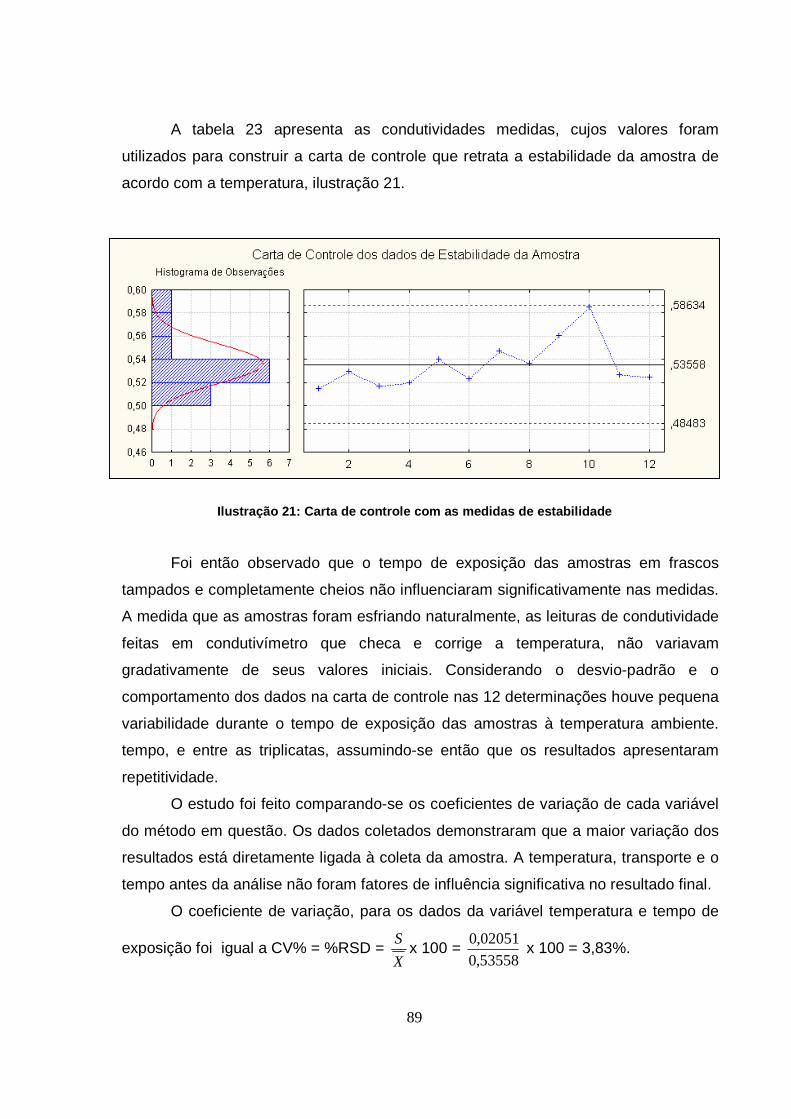
89
A tabela 23 apresenta as condutividades medidas, cujos valores foram
utilizados para construir a carta de controle que retrata a estabilidade da amostra de
acordo com a temperatura, ilustração 21.
Ilustração 21: Carta de controle com as medidas de estabilidade
Foi então observado que o tempo de exposição das amostras em frascos
tampados e completamente cheios não influenciaram significativamente nas medidas.
A medida que as amostras foram esfriando naturalmente, as leituras de condutividade
feitas em condutivímetro que checa e corrige a temperatura, não variavam
gradativamente de seus valores iniciais. Considerando o desvio-padrão e o
comportamento dos dados na carta de controle nas 12 determinações houve pequena
variabilidade durante o tempo de exposição das amostras à temperatura ambiente.
tempo, e entre as triplicatas, assumindo-se então que os resultados apresentaram
repetitividade.
O estudo foi feito comparando-se os coeficientes de variação de cada variável
do método em questão. Os dados coletados demonstraram que a maior variação dos
resultados está diretamente ligada à coleta da amostra. A temperatura, transporte e o
tempo antes da análise não foram fatores de influência significativa no resultado final.
O coeficiente de variação, para os dados da variável temperatura e tempo de
exposição foi igual a CV% = %RSD = X
Sx 100 =
53558,0
02051,0 x 100 = 3,83%.

90
De acordo com Horwitz, o desvio padrão relativo para um sistema ter precisão
adequada, em ensaios de características físico-químicas, e que garanta a
Reprodutibilidade do mesmo deve ser menor ou igual a 10%. Para a repetitividade é
recomendada uma variação inferior a 5%, que é o caso do método tipo III. Portanto,
ambos os parâmetros foram atendidos, o que sugere que o estudo da temperatura
serviu de base para conhecimento da amostra.
Foi verificado o comportamento da água para injetáveis por um período de
trinta dias consecutivos. A carta de controle, ilustração 22, representa o comportamento
dos dados e fornece subsídios para a compreensão do processo de produção da WFI.
Os dados de condutividade utilizados para a confecção da carta de controle foram
apresentados na tabela 24.
4.2.2- Comportamento da Amostra e carta de controle A tabela 24 apresenta os dados de condutividade da WFI obtidos por um mesmo
analista ao longo de trinta dias consecutivos. A tabela 25 apresenta os dados
estatísticos obtidos com os valores da condutividade .

91
Tabela 24 - Condutividade da WFI DE amostras coleta das por trinta dias consecutivos.
pH (25 ºC) CONDUTIVIDADE (µS/cm)
DIAS AMOSTRA 1 AMOSTRA 2 AMOSTRA 1 AMOSTRA 2
1 5,9 5,9 0,44 0,47
2 6,0 5,9 0,41 0,41
3 5,9 5,9 0,50 0,49
4 5,9 5,9 0,45 0,44
5 5,8 5,8 0,44 0,44
6 5,9 5,9 0,60 0,59
7 6,0 6,0 0,51 0,50
8 5,9 5,9 0,49 0,49
9 5,9 5,9 0,50 0,49
10 6,0 5,9 0,56 0,56
11 5,9 5,9 0,46 0,48
12 5,9 5,9 0,50 0,50
13 5,8 5,8 0,46 0,49
14 5,9 5,9 0,43 0,44
15 5,8 5,8 0,45 0,46
16 5,9 5,9 0,46 0,47
17 5,9 5,9 0,45 0,44
18 5,9 5,9 0,44 0,45
19 5,8 5,8 0,49 0,48
20 5,9 5,9 0,50 0,49
21 5,8 5,8 0,52 0,52
22 5,7 5,8 0,65 0,60
23 5,9 5,8 0,48 0,43
24 5,8 5,8 0,44 0,44
25 5,7 5,8 0,56 0,64
26 5,9 5,9 0,54 0,52
27 5,7 5,7 0,46 0,46
28 5,9 5,9 0,57 0,51
29 5,9 5,9 0,48 0,47
30 5,9 5,9 0,50 0,49

92
Tabela 25 - Dados estatísticos dos valores da condu tividade ( comportamento da amostra em 30 dias).
Desvio-padrão (µS/cm)
0,0519
Média (µS/cm)
0,490
Coeficiente de variação
10,59
A carta de controle, ilustração 22, foi construída originando uma carta de
controle, com os resultados das medidas de condutividade das amostras de água
coletadas num período de trinta dias consecutivos de um único sistema de produção
de água, coletadas por um único analista. A tabela 25 mostra os valores de desvio-
padrão, média e coeficiente de variação, atendendo aos critérios de precisão com a
Reprodutibilidade do sistema, segundo os critérios de Horwitz.
Ilustração 22 : Carta de controle do comportamento dos dados da água WFI ao longo de um mês, com duas amostragens diárias.
Ela teve como objetivo demonstrar a variabilidade do processo de produção da
água para injetáveis desse período juntamente com a análise, visto que para esse

93
método em estudo uma das possíveis fontes de erros é a proveniente da coleta da
amostra.
4.2.3- Comportamento dos resultados gerados por trê s analistas do Método tipo III.
Mesmo não tendo sido recomendado pela USP avaliar os parâmetros de
precisão, para o método tipo III pois é oficial, padronizado e já é conhecida sua
performance em Laboratórios de Controle, foi importante conhecer a variação dos
resultados de acordo com a técnica e os dados gerados pelos três analistas, visto
que para o cálculo da estimativa da incerteza é importante conhecer o
comportamento de ambos e também o desempenho do Laboratório em estudo.
Com a finalidade de conhecer o comportamento dos dados e a adequação
do método em relação à amostra, como sugerido pela USP 24, estas foram
coletadas, em dias diferentes, no mesmo ponto e analisadas por três diferentes
analistas. O comportamento da amostra já fora conhecido e estudado anteriormente,
conforme os dados das cartas de controle, ilustrações 21 e 22. Com os dados da
tabela 26, foi possível demonstrar a precisão, através da Reprodutibilidade,
realizada pela análise de três diferentes analistas, no mesmo sistema de produção,
mesmo ponto de coleta, mesmo equipamento de análise e em dias diferentes.
Na rotina diária do Laboratório é importante conhecer a variabilidade dos
resultados obtidos por analistas que podem executar análises em geral, para
avaliação dos resultados e melhor alocação de pessoas a diferentes tipos de
atividades.

94
Tabela 26 - Comportamento dos Analistas com a análi se de condutividade em WFI.
Analista 1 Analista 2 Analista 3
0,596 0,591 0,550 0,578 0,607 0,549 0,587 0,609 0,548 0,590 0,560 0,550 0,568 0,564 0,551 0,588 0,561 0,546
Média 0,584 0,582 0,549 Desvio-padrão 0,00995 0,02317 0,00179
Variância 0,000099 0,000540 0,000003
As cartas de controle referentes aos dados da tabela 26 facilitaram a
visualização dos dados, a variabilidade e a diferença de resultados entre cada
analista. Foi importante levar em consideração que os resultados variaram conforme
o esperado, dentro dos limites de controle e muito abaixo da especificação
permitida, que é menor que 1,3µS/cm. Os valores de desvio-padrão e variância
encontraram-se dentro de parâmetros adequados. As ilustrações 23, 24 e 25,
demonstraram o comportamento dos dados de acordo com a ordem da coleta das
amostras.

95
Ilustração 23 - Medidas de condutividade do analist a 1
Com o comportamento dos dados obtidos pelo analista 1 foi possível
visualizar os valores de condutividade, em seis determinações, e sua variabilidade
em torno da média, correspondendo a faixa de normalidade ideal, conforme o
histograma de observações que demonstra medidas sob controle com tendência
central. O número de determinações diárias foi determinante de acordo com o tempo
de coleta e o andamento da análise, que coincidentemente é o número mínimo
recomendado de determinações para ensaios de precisão.
Ilustração 24 - Medidas de condutividade do Analist a 2

96
O segundo analista obteve variabilidade em torno da média, porém com
comportamento diferente do anterior.
Ilustração 25 - Medidas de condutividade do Analist a 3
Os resultados provenientes da análise do terceiro analista tiveram
comportamento semelhante ao do primeiro, com distribuição normal centralizada.
Para efeito de comparação entre os três analistas foi feito o teste F de
Snedecor, que mede a precisão entre analistas através da comparação das
variâncias. A verificação foi feita em duplas. Para n-1 graus de liberdade e 95% de
confiança.
F = 22
21
S
S ∴
F = Teste de Snedecor

97
=21S Variância do analista 1 = 0,000099
=22S Variância do analista 2 = 0,000537
=23S Variância do analista 3 = 0,000003 = menor variabilidade entre os dados
Comparando o analista 1 com o 2 : F = 0,000537 ÷ 0,000099 = 5,42
Analista 1 com o 3 : F = 0,000099 ÷ 0,000003 = 33
Analista 2 com o 3 : F = 0,000537 ÷ 0,000003 = 189
O valor de Ftabelado , para n= 6, n-1 graus de liberdade e 95% de confiança é igual a
5,05.
Então pôde-se concluir que por comparação de variâncias dos dados dos três
analistas estes apresentaram diferença estatística de variância, pois Ftabelado <
Fcalculado.
O analista 3 obteve resultados mais precisos, provenientes de melhor treinamento e
experiência na coleta de amostras. A comparação foi adequada para medir o
comportamento de cada um, independente da análise da precisão do método em
estudo. Todos apresentaram bons resultados e vale aqui a ressalva do treinamento
e tempo de experiência. Esses fatores foram determinantes e comprovam o tempo
da experiência adquirida pelo terceiro analista, que obteve melhores dados de
dispersão. Houve diferença , expressa em µS/cm, de uma ordem de grandeza.
Os fatores que se fizeram importantes, no decorrer do estudo para o
método tipo III, foram:
o A coleta da amostra, com a amostragem da água, diagnosticado pelo o
correto procedimento do analista;
o O comportamento do sistema de produção de WFI, mostrado ao longo de
trinta dias consecutivos, que pode ser variável a cada dia.

98
Outros fatores tais como, a temperatura, a contaminação atmosférica e a calibração
diária do equipamento não foram determinantes para esse método como se
suspeitava. A temperatura, embora influencie diretamente na condutividade, é
corrigida automaticamente pelo equipamento no ato de sua leitura. Não havendo
opção de medida de condutividade sem a correção da mesma, portanto não foi fator
que contribuísse para a estimativa de incerteza do método. A contaminação
atmosférica da amostra não teve relevância para essa medição, pois para a
amostragem correta a contaminação não é significativa, conforme provado
experimentalmente no teste de estabilidade de WFI. Quanto à calibração do
equipamento, ela é realizada diariamente permitindo a correção do mesmo através
de artifício mecânico do sistema de medida, antes da leitura das amostras e na
leitura de conferência do padrão de condutividade como recomendado pelo
fabricante, através do drift de calibração.
4.2.4- Parâmetros de Validação
4.2.4.1 - Especificidade Foram avaliadas as possíveis interferências na análise, tais como a exposição das
amostras ao longo de um período determinado e queda de temperatura, conferindo
assim sua estabilidade da água em questão, já que não existe padrão de
condutividade com mesma matriz e faixa de especificação.
4.2.4.2 - Precisão
1. Reprodutibilidade
Esta á a comparação da variabilidade em condições diferentes, analistas e dias
diferenciados, considerando a variabilidade do sistema de produção de WFI. Pela
análise dos desvios padrão relativos dos três analistas em dias diferentes e pelos

99
dados do comportamento da água estudados, o método demonstra precisão entre
analistas, através da Reprodutibilidade. Para fins de estudo e de cálculos de
incerteza, será utilizado o desvio padrão ponderado.
Tabela 27 - Relação desvio padrão x analista - Méto do III.
Analista Desvio padrão relativo (%)
1 1,70 2 3,98 3 0,32
Ao verificar, conforme Horwitz, que a precisão decresce quando a
concentração decresce, ou seja, quanto menor a concentração pior é a precisão, é
importante que a coleta da amostra seja adequada e homogênea. Mesmo com
diferenças significativas entre os analistas, segundo critérios já expostos, de acordo
com a tabela 27, existe precisão entre eles caracterizada pela Reprodutibilidade.
Com a ilustração 26 é possível visualizar a variabilidade dos dados dos
diferentes analistas, com os desvios médios de cada um nas determinações.
Observou-se que o analista 3 obteve valor médio de análise inferior ao valor médio
global entre os dados dos três analistas. Os analistas 1 e 2 obtiveram
comportamento semelhantes.
Na ilustração 27, gráfico de caixas, estão apresentados além da
variabilidade dos três analistas, a amplitude dos dados gerados por cada um deles e
a mediana. Foi notória a pequena variabilidade dos dados obtidos pelo analista 3.

100
Ilustração 26 - Gráfico de Reprodutibilidade
Ilustração 27 - Gráfico Box Whiskers dos três anali stas

101
4.2.4.3 - Robustez Com a avaliação dos resultados de condutividade e estabilidade das amostras e da
análise, foi possível concluir que o método é robusto para essa finalidade, conforme
USP 27.
4.2.5 - Diagrama de Causa e Efeito do Método tipo I II
No diagrama de causa e efeito do método categoria tipo III, ilustração 28, foram
consideradas as fontes de incerteza provenientes de resultados de repetitividade dos
dados e a qualificação do condutivímetro, cujo certificado de calibração do fornecedor
encontra-se no anexo C7. Todos estes parâmetros contribuíram com a variabilidade
rotineira do Laboratório. Estas variáveis aceitas como dados de entrada foram
avaliadas ao longo do trabalho e discutidas de acordo com seus resultados,
especificações e referencias bibliográficas.
Ilustração 28: Diagrama de Ishikawa do método III

102
4.2.6 - Estimativas de Incerteza
A tabela 28, apresenta as grandezas de entrada e valores necessários para os
cálculos da estimativa de incerteza padrão para o Método tipo III, seguida de
observações referentes a cada uma das grandezas.
Tabela 28 - Incerteza padrão do Método tipo III.
Fontes de Incerteza
Tipo de Incerteza
Unidade Valor Distribuição de
Probabilidade
Incerteza Padrão Relativa
1) Comportamento
da Amostra
A
µS/cm
0,0519
Normal
30
0,0095
2) Estabilidade da Amostra com a
temperatura
A
µS/cm
0,0205
Normal
12
0,0059
3) Reprodutibilidade
A
µS/cm
-
-
0,013
4) Calibração do
Condutivímetro
B
µS/cm
0,01
Normal
K=2
0,0050
5) Resolução do
Display
B
µS/cm
0,008
Retangular
3
0,0046
Com a finalidade de entendimento dos dados da tabela 28, as seguintes considerações
fizeram-se necessárias:
1. O comportamento da amostra ao longo de trinta dias, demonstrou a estabilidade
do sistema de produção de WFI, considerado-se o valor de desvio-padrão como
contribuição da incerteza tipo A.
2. A estabilidade da amostra com a temperatura, através da carta de controle
verificou-se o comportamento das réplicas com o decréscimo de temperatura.
3. Através dos dados de Reprodutibilidade, obtidos pelo cálculo do desvio-padrão
ponderado, estimou-se a contribuição para a incerteza padrão.

103
Sc=( ) ( ) ( )
3
1311
321
22
2
2
1
−++
−×+−×+−×
nnnSSS nnn
=
=( ) ( ) ( )
3666
16000003,01600054,0160,00001
−++−×+−×+−×
= 0,013 µS/cm
4. Os dados obtidos da qualificação do equipamento – foi utilizado o certificado do
padrão para a calibração, expresso em µS/cm .
5. A qualificação da performance do condutivímetro - utilizou-se os dados
fornecidos pelo representante. Os dados de entrada são provenientes de testes
de resposta elétrica do display do equipamento (cover closed - cover open), cuja
resistência foi expressa em ± Ω transformada para µS/cm, através da fórmula S
=Ω1
. O dado do somatório do relatório de qualificação é igual a ± 0,08 µS/cm .
4.2.6.1 - Cálculo da Incerteza Padrão Combinada do Método tipo III Para efeito de cálculo não foram utilizados os dados de estabilidade da
amostra, item 2, pois assumiu-se que a contribuição tivesse sido considerada através
dos dados do comportamento da amostra ao longo de trinta dias, pois essa contribuição
apresentou o pior desvio padrão (maior valor de dispersão). Com os dados da tabela
28, foi possível fazer o somatório pela fórmula de Kragten.
u(y)= ( ) ( ) ( ) ( )2222 0046,00050,0013,00095,0 +++ = 0,032 ≡ 0,03 µS/cm

104
4.2.6.2 - Estimativa de Incerteza Expandida do méto do tipo III Incerteza padrão combinada x k (para 95% de confiança) U = 0,03 x 2 = 0,06 µS/cm . Calculando um valor médio de condutividade, já que não dispomos de padrão de
condutividade como um valor nominal, de todas as etapas igual a 0,548 µS/cm,
correspondente à incerteza de ± 0,06 µS/cm. A especificação aceita de condutividade
para água do tipo WFI é menor que 1,3 µS/cm.
4.7- Diagrama de Causa e Efeito “refinado” do métod o tipo III De acordo com os dados acima, cálculos das estimativas de incerteza padrão
combinada e expandida, foi possível concluir que o gráfico de causa e efeito deveria ser
alterado para as reais condições comprovadas experimentalmente, como demonstrada
na ilustração 29.
Ilustração 29 - Diagrama de Causa e Efeito refinado do Método tipo II
Estabilidadedo Sistema
( Comportamento da Amostra)
Qualificação de padrão

105
Todas as variáveis representadas no Diagrama de Ishikawa refinado, foram as
que realmente contribuíram no processo de estimativas de incerteza do método tipo III.
4.8 - Contribuições para a Incerteza Padrão Combina da do Método tipo III
Ilustração 30 - Relação de variáveis que contribuír am para a Estimativa de Incerteza Padrão
Combinada do Método tipo III
Conforme a ilustração 30, gráfico de Pareto, foi possível concluir que as fontes
que mais contribuíram para a Incerteza Padrão Combinada do Método tipo III, e que
mais influenciaram na estimativa da incerteza, em ordem decrescente, foram: a
Reprodutibilidade do Método, o comportamento da amostra, a calibração do
condutivímetro e a resolução do display do equipamento.
0% 5% 10% 15%
Incerteza Padrão Relativa (%)
Repro
dutib
ilidad
e
Compo
rtamen
to da
Amo..
.
Calibr
ação
do C
ondu
tiví...
Resolu
ção d
o Disp
lay
Fon
tes
de
Ince
rteza
Contribuição para a Incerteza do Método tipo III

106
5 - Conclusão Com os resultados obtidos ao longo deste trabalho foi possível mostrar que as
metodologias utilizadas no LAFIQ para as determinações do teor de PRRP em vacinas
e a condutividade em amostras de água produzidas para injetáveis são adequadas ao
propósito conforme mostrado pelos parâmetros de validação estudados - precisão,
repetitividade e reprodutibilidade obtida em diversos dias por analistas diferentes.
Como ferramentas de controle visando o melhoramento contínuo dos
resultados gerados no laboratório de controle de qualidade têm-se o uso contínuo do
diagrama de Ishikawa para outros métodos que propiciarão o melhor entendimento das
metodologias analíticas utilizadas; a implementação de cartas de controle para avaliar a
performance dos analistas e dos métodos; aplicação de testes estatísticos que
permitam comparar os resultados obtidos com aqueles esperados e por fim as
estimativas de incertezas de medição, que mostram os parâmetros que podem
influenciar no resultado analítico.
Através das cartas de controle construídas com os dados gerados por diversos
analistas em dias diferentes vem sendo possível avaliar rotineiramente se o processo (a
análise) está fora de controle e identificar as possíveis causas desta situação.
Através do cálculo das incertezas de medição dos dois métodos estudados
pode-se identificar os parâmetros que mais contribuem para a estimativa de incerteza
dos resultados finais. Para o método tipo I, determinação do conteúdo de
polissacarídeo em vacina contra a Hib, usando cromatografia de troca iônica com
detecção eletroquímica, demonstrando que os parâmetros que mais contribuíram para
o valor da incerteza foram: a reprodutibilidade do método, seguida da concentração das
soluções de trabalho, a contribuição do volume, da pureza do padrão, e por último as
incertezas provenientes de massa. O resultado encontrado para a incerteza padrão
combinada foi de 15 ± 0,50 µg / mL e a incerteza padrão expandida 15 ± 1,00 µg / mL.
Para o método tipo III, medida da condutividade em água para injetáveis, as variáveis
que influenciaram para a estimativa da incerteza do método foram àquelas provenientes

107
da reprodutibilidade do método, o comportamento da amostra, a qualificação e
resolução do equipamento. Os valores para as incertezas padrão combinada e
expandida foram considerados em relação à média das medições de condutividade em
estudo igual a 0,548 µS / cm.Então podemos considerar os valores das incertezas
padrão combinada encontrada igual a 0,548 ± 0,03 µS / cm e 0,548 ± 0,06 µS / cm,
para a incerteza padrão expandida, para o nível de confiança de 95%.
Como avaliação crítica, muitos autores discutem a verdadeira necessidade de
expressar as incertezas das medições, considerando o tempo e custo gastos em
estudos para calcular este valor. Entretanto as normas de qualidade sejam da área da
química analítica ou das normalizações técnicas, fazem recomendações dessa
estimativa devido aos benefícios que ela pode proporcionar. No entanto, é necessário o
consenso para o cálculo e a expressão da incerteza, a fim de facilitar a interpretação
apropriada dos resultados em análises químicas. A estimativa deste valor para um
método analítico é fundamental para expressar adequadamente os resultados finais
pois o valor da incerteza que “cerca” o resultado é importante para a aceitação de um
produto e a garantia da execução da análise química com qualidade.
Em trabalhos futuros pretende-se implementar as ferramentas aqui
desenvolvidas e estudadas como recursos para a melhoria na expressão de todos os
resultados analíticos do laboratório de Controle Físico-químico garantindo assim a
qualidade do trabalho dos analistas e a confiabilidade do Laboratório.
Como disse Chambers e Wheeler, 1992 “ se o gerenciamento sinaliza que
atingir as especificações é satisfatório, o resultado invariavelmente será a falha” .

108
6- Anexos
ANEXO A
Pop relacionados aos métodos tipo I e tipo III. A1 – Determinação do conteúdo de polissacarídeo na vacina contra a Hib, por HPLC. A2 – Determinação colorimétrica do conteúdo de polissacarídeo na vacina Hib, PSTT e
conteúdo de ribose no PRRP.
A3 – Monitoramento dos sistemas de purificação de água. A4 - Análise físico-química de águas purificadas.

109
ANEXO B Cromatogramas obtidos pelos três analistas do método tipo I. B1 - Cromatograma dos pontos da primeira curva de calibração. B2 - Cromatograma de amostras obtidos pelo analista 1. B3 - Cromatogramas de amostras obtidos pelo analista 2. B4 – Cromatogramas dos pontos da segunda curva de calibração. B5 – Cromatogramas de amostras obtidos pelo analista 3.

110
ANEXO C
Certificados de calibração e validação. C1 – Primeiro relatório de validação do método tipo I. C2 – Certificado de análise de amostra referência utilizada no método tipo I. C3 – Documentação de qualificação do equipamento, HPLC, utilizado no método tipo I. C4 – Certificados de calibração de pipetas automáticas utilizadas no método tipo I. C5 – Certificados de calibração de balões volumétricos utilizados no método tipo I. C6 – Certificado de calibração da balança utilizada no método tipo I. C7 – Documentação de qualificação do equipamento, condutivímetro, utilizado no
método tipo III.

111
ANEXO D Tabelas Estatísticas.
D1 – t “ Student”. D2 – F “ Snedecor”. D3 – Fator de abrangência. D4 – Número de graus de liberdade.
D5 - Fatores para construção das vaiáveis das Cartas de Controle.

112
7 - Bibliografia
1) ABNT - INMETRO. Guia para a Expressão da Incerteza de Medição. Terceira
Edição Brasileira. Edição revisada de agosto de 2003: Guide to the Expression of
Uncertainty in Measurement; NMKL, 1997 – Estimation and expression of
measurement uncertainty in chemical analysis. Procedure n°5. NBR ISO/IEC 17025,
2001. Requisitos Gerais para a Competência de Laboratórios de Calibração e de
ensaios. ABNT/RJ, Brasil.
2) Agência Nacional de Vigilância Sanitária, ANVISA. RDC 210; 2003.
3) AMC Background paper. Analytical Methods Committee Background n°1; June,
2004: What is Uncertainty from sampling, and why is it important? The Royal Society
of Chemistry.
4) AMC, Royal Society Chemistry. The estimation and use of recovery factors. Analytical
Methods Committee; London, 2000.
5) ANALION - Fabricante de condutivímetros: A análise da Condutividade.
[http://www.analion.com.br/condutividade.htm ] Capturado em 16 de novembro de
2004, Rio de Janeiro.
6) ANVISA. RDC 304; 2004. Diário Oficial da União de 07/12/2004.
7) AOAC. Horwitz, W. Official Methods of Analysis of the Association of Official
Analytical Chemists. 17th ed. Vol. 2, chapter 50 (MA-CQ. 039). Gaithersburg; Maryland,
2000.

113
8) Associação Brasileira de Normas Técnicas: ABNT: Requisitos Gerais para
competência de Laboratórios de Ensaio e Calibração. NBR ISO/ IEC 17025; Brasil,
Rio de Janeiro, 2001.
9) Barros, CB, Hirata, YS. Validação de Métodos Analíticos. Revisão 05. São
Paulo;Outubro de 1999.[Apostila do curso de Validação de Metodologia Analítica] -
EXPOLABOR.
10) Bassani, C. Validação de metodologias analíticas. [Apresentação ao Seminário
de Validação Analítica]: Consultoria a Bio-Manguinhos/ FIOCRUZ. Brasil; Rio de
Janeiro;2004.
11) Bruce, P, Riekkola, ML. Pratical Method Validation: Validation Sufficient for an
Analysis Method. Finland; 1998.
12) Bujard E, Finot PA. Mesure de la disponibilité et du blocage de la lysine dans les
laits industriels. Ann.Nutr.Alim.:1978, 32, 291-305.
13) Procedimentos operacionais padronizados em anexo
o Determinação de condutividade em águas para injetáveis (WFI):pop
106040.617.
o Determinação do conteúdo de polissacarídeo em vacinas: pop 106040.588.
14) Dhillon, BS. Quality Control Reliability, and Engineering Design; 1985; USA.
15) Douglas C Montgomery, George C Runger, John Willey & Sons, Inc. Applied
Statistics and Probability for Engineers; Third Edition; 2003.
16) Environmental Protection Agency. Science and Ecosystem Support. Manual EPA
– Division: Region 4; December, 1997.
17) EURACHEM / EA Guide 04/10 – rev 02. Acreditation for Microbiological
Laboratories; July 2002 .

114
18) FDA. Horwitz, W; Evaluation of Analytical Methods Used for Regulation of Foods
and Drugs; USA; January, 1982.
19) FDA: Guidance for Industry - Analytical Procedures and Methods Validation,
Chemistry, Manufacturing and Controls Documentation. USA: August; 2000.
20) Finot, PA et al. The extend of the maillard reaction during the processing of milk.
Prog. Ed. Nutr.Sci. Vol 5,pp.345-355. Britain, 1981.
21) Flaig, JJ. Estimating the Process Sigma. Applied Technology; 2004;USA.
22) Funk W, Dammann V ; Donnevert G; Quality Assurance in Analytical Chemistry.
Review of the Germain edition; 1995.
23) Furman, W B, Dorsey J G, Testes de Sensibilidade do Sistema para Métodos
Oficiais de Cromatografia Líquida e Gasosa. Pharmaceutical Technology; agosto
1998.
24) Gimenéz, EC. The Uncertainty in the calculation of the blocked and reactive lysine in
milk products, as determined by furosine method - Practitioner’s Report. Switzerland.
July, 2004.
25) Guia EURACHEM/ CITAC: Determinando a Incerteza na Medição Analítica.
Segunda Edição- Editores: SRL Elisson ( LGC, UK), M Rosslein ( EMPA- Switzerland),
A Williams ( UK); 2002, Versão Brasileira.
26) Guimarães R. Pesquisa no Brasil: A Reforma Tardia. Universidade Federal do
Rio de Janeiro, UFRJ; 2002.
27) Hirata, Y S. Gráficos de Controle para Laboratórios de Ensaios; 2002; São
Paulo, Brasil.

115
28) INCQS. ALINORM 03/23. Proposta de diretrizes para avaliar métodos de ensaio.
Apêndice VII; 11/04/2003.
29) Inman, E L, Frischmann, J K et al. General Method Validation Guidelines for
Pharmaceutical Samples . Journal of Chromatographic Science, vol 25; june 1987.
30) INMETRO. NIT-DICLA-028; Revisão 00 . BPL – Boas Práticas de Laboratório.
31) INMETRO:Orientações Sobre Validação de Métodos de Ensaios Químicos,
(Brasil). DOQ-CGCRE-008; revisão 01:março de 2003.
32) Instituto Oswaldo Cruz. Relatório do IV Congresso Interno da FIOCRUZ. Termo
de referência; 2002.
33) International Standards Organization: ISO 8466-1: Water Quality. Calibration and
Evaluation of Analytical Methods and Estimations of Performance Characteristics.
First edition; 1990.
34) IUPAC Guidelines for The In-House Validation of Methods of Analysis. Joint
FAO/WHO Food Standards Programme. Codex Committee on Methods of Analysis
and Sampling, twenty-fourth session. Single Laboratory Validation. Consideration of
Harmonized. Budapest, Hungary: 18-22; November 2002.
35) IUPAC Technical Report. Harmonized Guidelines for Single-Laboratory
Validation of Methods of Analysis: Thompson, M, Ellison Stephen LR, and Wood, R.
UK; 1999.
36) Joint FAO/ WHO Food Standards Programme. Codex Committee on Methods of
Analysis and Sampling. Twenty-fourth Session: Single-Laboratory Validation. Budapest,
Hungary; November, 2002.
37) Journal of AOAC International. Horwitz, W. Uncertainty - a chemist's view; 1998.

116
38) Lobo A. “Qualidade e Produtividade”in:[www.inmetro.gov.br]; Capturado em
novembro de 2003, Rio de Janeiro.
39) Manual do condutivímetro - 712 Conductometer - METRHON AG CH-9101
Herisau - Schweiz.
40) Manual do HPLC de troca iônica – DIONEX DX 500 - USA.
o Monitoramento dos sistemas de purificação de água: 106040.008.
41) Montgomery C D. Introdution to Statistical Quality Control. 4th edition. USA; 2001.
Moulin, 1996; Fernandes A, 2000. “A Vacinologia é hoje uma Ciência em vias de
autonomia”.
42) Oliveira A M S, Oliveira R & Hamerski A. Estudo de indicadores de qualidade em
obras repetitivas.
43) Pharmaceutical Technology. Brittain H G. Validação de Métodos Analíticos Não
Cromatográficos. NJ,USA; Junho de 1998.
44) Quantification of Carbohydrates and Glycols in Pharmaceuticals [Applications
note 117. DIONEX Reference Library; 2004; USA].
45) Royal Society of Chemistry. Thompson, M. AMC Technical Brief n° 17: The
Amazing Horwitz Function; July 2004.
46) Ryan, TP. Statistical Methods for Quality Improvement. Second Edition; 2000;
USA.
47) Schissatti, M Luiz. Dissertação submetida à Universidade Federal de Santa
Catarina para obtenção do grau de mestre em engenharia. Uma metodologia de
implantação de cartas de shewhart para o controle de processos; Capítulo IV ;
Dezembro de 1998; Santa Catarina; Brasil.

117
48) Schneerson R et al. Vaccines composed of polyssaccharide protein conjugates:
current status, unanswered questions, and prospects for the future. In: Bell R,
Torrigiani G, eds. Towards better carbohydrate vaccines. Chichester, Wiley, 1987:
307 - 327 ( published on behdef of the World Health Organization).
49) Schneerson R, Barrera O. Sutton A & Robbins JB. Utilização da proteína
monomérica tetânica na vacina Haemophillus influenzae tipo b :(1980); J. Exp. Med.
152 (361-376).
50) Small, H, Bowman, B. Ion Chromatography: a historical perspective. American
Laboratory. Ca, USA; 1998.
51) Statistics For Analytical Chemistry, Second Edition, Miller, J C; Miller, J N; New
York, 1988.
52) StatSoft 6: Software STATISTICA. Versão 6.0; Tulsa USA.
53) The Determination of Carbohydrates, Alcohols and Glycols in Fermentation
Broths. [ Application note 122. DIONEX Reference Library; 2004; USA].
54) Thomas P. Ryan. Statistical Methods for Quality Improvement; Second Edition;
2000.
55) Triola, F M. Introdução à Estatística. Sétima edição;1999.
56) United States Pharmacopeia. USP 27. NF 22. The Official Compendia of
Standards; 2004.
57) VALE L M. Como Obter Resultados Confiáveis em Cromatografia. Boletim
Técnico No. 19: UNICAMP; São Paulo, Brasil.
58) Valente, L S. Como obter resultados confiáveis em cromatografia; 2001.
59) VIM - 3ª Edição. Rio de Janeiro, 2003.

118
60) Web´s
Página da FIOCRUZ
http://www.fiocruz.br/ccs/novidades/out03
Guia de Navegatión Especializada em Salud
http://www.medicentro.com.co
Ciência Tecnologia e Meio Ambiente – ABR – RADIOBRAS
http://www.radiobras.gov.br
ANVISA – Legislação - Resoluções
http://www.anvisa.gov.br/legis/resol/309-99.htm
Instituto brasileiro de Pesos e Medidas
www.inmetro.gov.br
Instituto baiano de Metrologia e Qualidade
www.ibametro.gov.br
PDF created with Fine Print PDF Factory trial version
http://www.fineprint.com
Portal CAPES
www.periodicos.capes.gov.br
Instituto Nacional de Propriedade Industrial
o Transferência de Tecnologia
o Informações Tecnológicas
o Patentes
www.inpi.gov.br

119
Trabalhos Científicos sobre pesquisa de Incerteza de Medições Analíticas
www.scielo.org.br
Trabalhos Científicos sobre pesquisa de Incerteza de Medições Analíticas
www.labsgoogle.com/ gviewer.html
61) Wheeler, DJ, Chambers, DS. Understating Statistical Process Control; 1992.


















