
MÁRIO EMÍLIO TEIXEIRA DOURADO JÚNIOR · ii Catalogação da Publicação na Fonte Universidade...
147
i MÁRIO EMÍLIO TEIXEIRA DOURADO JÚNIOR SÍNDROME DE GUILLAIN BARRÉ: EPIDEMIOLOGIA, PROGNÓSTICO E FATORES DE RISCO Tese de doutorado apresentada ao Programa de Pós-Graduação em Ciências da Saúde da Universidade Federal do Rio Grande do Norte, como requisito para obtenção do título de Doutor em Ciências da Saúde. Orientadora: Selma Maria Bezerra Jerônimo NATAL/RN 2015
Transcript of MÁRIO EMÍLIO TEIXEIRA DOURADO JÚNIOR · ii Catalogação da Publicação na Fonte Universidade...

i
MÁRIO EMÍLIO TEIXEIRA DOURADO JÚNIOR
SÍNDROME DE GUILLAIN BARRÉ: EPIDEMIOLOGIA, PROGNÓSTICO E
FATORES DE RISCO
Tese de doutorado apresentada ao Programa de Pós-Graduação em Ciências da Saúde da Universidade Federal do Rio Grande do Norte, como requisito para obtenção do título de Doutor em Ciências da Saúde. Orientadora: Selma Maria Bezerra Jerônimo
NATAL/RN
2015

ii
Catalogação da Publicação na Fonte Universidade Federal do Rio Grande do Norte – UFRN
Dourado Júnior, Mário Emílio Teixeira. Síndrome de Guillain-Barré: epidemiologia, prognóstico e fatores de risco / Mário Emílio Teixeira Dourado Júnior. - Natal, 2015. 119f: il. Orientadora: Prof.ª Dr.ª Selma Maria Bezerra Jerônimo. Tese (Doutorado) - Programa de Pós-Graduação em Ciências da Saúde. Centro de Ciências da Saúde. Universidade Federal do Rio Grande do Norte. 1. Neuroimunologia - Tese. 2. Síndrome de Guillain-Barré - Tese. 3. Transcriptoma - Tese. I. Jerônimo, Selma Maria Bezerra. II. Título. RN/UF/BSA01 CDU 616.833-002

iii
MINISTÉRIO DA EDUCAÇÃO E CULTURA
UNIVERSIDADE FEDERAL DO RIO GRANDE DO NORTE
CENTRO DE CIÊNCIAS DA SAÚDE
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS DA SAÚDE
Coordenador do Programa de Pós-Graduação em Ciências da Saúde
Prof. Dr. Eryvaldo Socrates Tabosa do Egito

iv
MÁRIO EMÍLIO TEIXEIRA DOURADO JÚNIOR
SÍNDROME DE GUILLAIN BARRÉ: EPIDEMIOLOGIA, PROGNÓSTICO E
FATORES DE RISCO
Aprovada em _____/_____/_____
BANCA EXAMINADORA
Presidente da Banca – Profª Dra Selma Maria Bezerra Jerônimo
Membros:
Prof. Dr. Acary Souza Bulle de Oliveira
Prof. Dr. Osvaldo Jose Moreira Nascimento
Prof. Dr. João Paulo Matos Santos Lima
Prof. Dr. Marcos Romualdo Costa

v
DEDICATÓRIA
Dedico este trabalho a minha família.
Aos meus pais, Mário Dourado e Marluce, pelo amor que nos passa
A minha esposa, Cecília Carvalheira, minha amada e companheira
Aos meus filhos, Júlia, Laura e Pedro, a razão da minha vida
Aos meus irmãos, Neto, Lucimar, Rosiane, Carlyle, Maria Lúcia, Rodrigo e Marcos
Lima, presentes em todos os momentos da minha vida
Aos meus avós, representada pela professora Maria Dourado

vi
AGRADECIMENTOS
A professora Selma Jerônimo pela orientação e pela oportunidade dada à
minha formação. Que os meus olhos marejados, meu aperto de mão e um abraço
apertado e sincero sejam capazes de transmitir a admiração, respeito e gratidão
que tenho pela professora. Obrigado.
Quero agradecer de uma forma muito especial aos voluntários,
especialmente, aos que sofreram da Síndrome de Guillain-Barré. Obrigado pela
participação nesse trabalho.
Aos pesquisadores do Departamento de Bioquímica e do Instituto de
Medicina Tropical da UFRN, especialmente Núbia, Cláudio, Paulo, Freire e
Leonardo pela ajuda, ensinamentos e auxílio na realização desse trabalho.
Ao professor João Paulo Matos e ao pesquisador Raulzito Fernandes, pela
ajuda nas análises dos dados do transcriptoma.
Agradeço ao Laboratório de Imunogenética, Departamento de Bioquímica,
UFRN, alunos, funcionários, pesquisadores, professores que foram de essencial
importância para a realização do trabalho
Aos colegas médicos, enfermeiros, técnicos de enfermagem, fisioterapeuta e
nutricionistas de diferentes Hospitais do nosso Estado, que, com competência,
atenderam aos pacientes com a Síndrome de Guillain-Barré.
Aos meus amigos de profissão, especialmente Marcos Lima, Lúcio Flávio,
Ênio, Sydnei, Marcão, Aldair, Fábio Melo, que me proporcionaram muitos
momentos de alegria e compartilharam sonhos pessoais e profissionais.
E a todos que direta ou indiretamente colaboraram para a concretização
desse trabalho.

vii
RESUMO
Indrodução. A Síndrome de Guillain-Barré (SGB) é uma polineuropatia imuno-
mediada, sendo, atualmente, a mais frequente causa de paralisia aguda
neuromuscular. As principais variantes dessa síndrome são: a polineuropatia
desmielinizante inflamatória aguda (PDIA), a neuropatia axonal motora aguda
(NAMA), a neuropatia axonal motora e sensitiva aguda (NAMSA), e a síndrome de
Miller-Fisher. Há também diferenças na distribuição geográfica destas variantes. A
resposta imune aberrante, pós infecção, parece ser resultante de um mimetismo
molecular, devido a formação de autoanticorpos e ativação do sistema
complemento e de citocinas. São encontrados polimorfismos bialélicos nos genes
codificadores dos receptores das frações Fc das imunoglobulinas (FcRIIa, FcRIIIa
e FcRIIIb) que afetam a afinidade e eficiência na resposta imune celular, sugerindo
a existência de susceptibilidade individual no risco de desenvolver a SGB. No
Brasil, há poucos estudos epidemiológicos sobre a SGB e nenhum relato sobre a
frequência das variantes e suas manifestações clínicas. Os objetivos deste estudo
foram: (1) caracterizar a SGB e suas manifestações clínicas em uma coorte de
pacientes com SGB oriundos do Estado do Rio Grande do Norte (RN); (2)
determinar se polimorfismos em receptores FcR estão envolvidos com o risco de
doença, e (3) avaliar a expressão gênica global buscando identificar possíveis vias
que poderiam ser moduladas na fase inicial da doença e, consequentemente,
diminuir o tempo de doença.
Metodologia. Foram recrutados 149 casos de SGB diagnosticados entre 1994-
2013 no RN, tendo sido avaliados os dados clínicos e laboratoriais visando a
determinar a evolução. DNA e RNA foram extraídos do sangue periférico e
anticorpos antigangliosídeos foram determinados em amostras de soro. Foram
genotipados polimorfismos nos genes FCGR2A e FCGR3A, em pessoas com SGB
(n=141) e controles saudáveis (n=364), sendo ainda analisadas as expressões
gênicas globais de 12 pacientes com SGB, por RNAseq. As amostras de sangue
para os estudos de expressão gênica foram coletadas ao diagnóstico e pós-
recuperação.
Resultados. A incidência de SGB foi de 0,3/100 mil pessoas no RN, sem presença
de sazonalidade, com os casos ocorrendo em uma idade mais jovem. A SGB foi

viii
precedida por infecções em 63,7%, sendo a diarreia associada a variante axonal
(p=0,025). A PDIA foi a variante mais frequente (81,8%), seguida de NAMA
(14,7%) e de NAMSA (3,3%). A distribuição da fraqueza muscular correlacionou
com as variantes, sendo a proximal mais frequente na PDIA, enquanto a distal
predominou na variante axonal. O nadir < 10 dias ocorreu em 84,6% dos indivíduos
na variante axonal e 42,4% dos casos com PDIA (P<0,0001). A forma
desmielinizante apresentou uma recuperação na deambulação mais rápida do que
a variante axonal (P<0,0001). A mortalidade de SGB foi de 5,3%. O pior
prognóstico aos 12 meses estava associado com a variante axonal (OR 17,063; P
= 0,03) e no tempo de melhora um ponto na escala funcional de Hughes (OR
1,028; P = 0.03). As distribuições dos genótipos e alelos em FCGR2A (p=0,367) e
em FCGR3A (p=0,2430) não foram diferentes entre os pacientes com SGB e
controles. A análise da expressão gênica global mostrou variação na expressão
dos mRNAs de isoformas de proteínas associadas à fase sintomática da doença.
Conclusões. Não há sazonalidade na ocorrência da SGB no RN, havendo um
predomínio da variante desmielinizante e 50% dos casos tinham idade inferior a 20
anos. A variante axonal está associada ao mau prognóstico. O diagnóstico precoce
e a identificação da variante, acompanhada de intervenções adequadas, levam a
diminuição da morbidade a longo prazo. Variações polimórficas nos genes de
FCGR parecem não influenciar a susceptibilidade ou o curso da SGB nessa
população. Variações na expressão gênica apontam para vias de desregulação e
alterações em interações transcricionais, que podem ser utilizadas como potenciais
alvos de modulação.
Palavras Chaves: Síndrome de Guillain-Barré, Neuroepidemiologia,
Neuroimunologia, FcyR, polimorfismo, transcriptoma.

ix
ABSTRACT
Introduction. Guillain-Barré syndrome (GBS) is an immune-mediated
polyneuropathy and the principal cause of acute neuromuscular paralysis. The most
prominent GBS subtypes are: acute inflammatory demyelinating polyneuropathy
(AIDP), acute motor axonal neuropathy (AMAN), acute motor-sensory axonal
neuropathy (AMSAN) and Fisher syndrome (FS). Differences in geographical
distribution of variants have been reported. In Brazil, there are few studies
describing the characteristics of GBS, but none on the frequency of GBS variants
and their clinical manifestations. Infection-induced aberrant immune response
resulting from molecular mimicry and formation of cross-reacting antibodies,
contribute to complement activation. Functional biallelic polymorphism in
immunoglobulin receptors that influence the affinity of IgG subclasses and the type
of immune response have been described, suggesting genetic susceptibility to
developing disease. It remains unclear whether individuals carrying different FCGR
alleles have differential risk for GBS and⁄or disease severity. The goals of this study
were: (1) To characterize GBS and describe the clinical findings in a cohort of
patients with GBS from the state of Rio Grande do Norte, Brazil; (2) to determine
whether polymorphism in FCGR were associated with development of GBS, and (3)
to tease out whether the global gene expression studies could be a tool to identify
pathways and transcriptional networks which could be regulated and decrease the
time of disease.
Methods. Clinical and laboratory data for 149 cases of GBS diagnosed from 1994
to 2013 were analyzed. Genomic DNA and total RNA were extracted from whole
blood. Antigangliosides antibodies were determined in the sera. In addition, we
also assessed whether FCGR polymorphism are present in GBS (n=141) and blood
donors (n=364), and global gene expressions were determined for 12 participants
with GBS. Blood samples were collected at the diagnosis and post-recovery.
Results. AIDP was the most frequent variant (81.8%) of GBS, followed by AMAN
(14.7%) and AMSAN (3.3%). The incidence of GBS was 0.3 ⁄ 100,000 people for
the state of Rio Grande do Norte and cases occurred at a younger age. GBS was
preceded by infections, with the axonal variant associated with episodes of diarrhea
(P = 0.025). Proximal weakness was more frequent in AIDP, and distal weakness

x
predominant in the axonal variant. Compared to 42.4% of cases with AIDP
(P<0.0001), 84.6% of cases with the axonal variant had nadir in <10 days.
Individuals with the axonal variant took longer to recover deambulation (P<0.0001).
The mortality of GBS was 5.3%. A worse outcome was related to an axonal variant
(OR17.063; P=0.03) and time required to improve one point in the Hughes
functional scale (OR 1.028; P=0.03). The FCGR genotypes and allele frequencies
did not differ significantly between the patients with GBS and the controls (FCGR2A
p=0.367 and FCGR3A p=0.2430). Global gene expression using RNAseq showed
variation in transcript coding for protein isoforms during acute phase of disease.
Conclusions. The annual incidence of GBS was 0.3 per 100,00 and there was no
seasonal pattern. A predominance of the AIDP variant was seen, and the incidence
of the disease decreased with age. The distribution of weakness is a function of the
clinical variants, and individuals with the axonal variant had a poorer prognosis.
Early diagnosis and variant identification leads to proper intervention decreasing in
long-term morbidity. FCGR polymorphisms do not seem to influence susceptibility to
GBS in this population. This study found deregulated genes and signs of
transcriptional network alterations during the acute and recovery phases in GBS.
Identification of pathways altered during disease might be target for immune
regulation and with potential to ameliorate symptoms.
Key words Guillain-Barré Syndrome, Neuroepidemiology, Neuroimmunology,
FcyR; Polymorphism, transcriptome.

xi
LISTA DE SIGLAS E ABREVIATURAS
C.jejuni Campylobacter jejuni
CMV Citomegalovirus
EGR Eearly growth response 3 gene
FcR Receptor da porção Fc da imunoglobulina
FCGR Gene do receptor da porção Fc da imunoglobulina
FCGR2A Gene da classe 2 A do receptor da imunoglobulina
FCGR3A Gene da classe 3 A do receptor da imunoglobulina
FCGR3B Gene da classe 3 B do receptor da imunoglobulina
FcRIIa Receptor da porção Fc da imunoglobulina da classe IIa
FcRIIIa Receptor da porção Fc da imunoglobulina da classe IIIa
FcRIIIb Receptor da porção Fc da imunoglobulina da classe IIIb
Gg Gangliosídeos
GD1a Gangliosídeo disialo GD1a
GM1 Gangliosídeo monosialo GM1
IgEV Imunoglobulina endovenosa
IgG Imunoglobulinas
NAMA Neuropatia axonal motora aguda
NAMSA Neuropatia axonal motora e sensitiva aguda
PCR Polymerase chain reaction
PDIA Polineuropatia desmielinizante inflamatória aguda
RNAseq RNA sequencing
SGB Síndrome de Guillain Barré
SMF Síndrome de Miller Fisher
SOCS Supressors Of Cytokine Signaling gene

xii
LISTA DE FIGURAS
Figura 1 Variantes clínicas da SGB .................................................................... 17
Figura 2 Glicolipídeos associados a neuropatias inflamatórias .......................... 36
Figura 3 Os diversos tipos de FcR presentes nas células da resposta
imune ................................................................................................... 43
Figura 4 Esquema da resposta imunológica na SGB ......................................... 57
Figura 5 Incidência das variantes da SGB por idade entre 1994 e 2007............ 68
Figura 6 Incidência anual da SGB por 1,000,000 de pessoas no período
de 1994-2007, por variante clínica ....................................................... 68
Figura 7 Sazonalidade das variantes da Síndrome de Guillain Barré no
período de 1994-2007. ......................................................................... 69
Figura 8 Curva de sobrevivência apresentando a proporção de pacientes
que não recuperaram a deambulação entre as variantes
desmielinizante e axonal ....................................................................... 72
Figura 9 Gráfico de Dispersão gênica, baseado em número de genes, de
cada transcriptoma analisado (X1 a X24). Avaliação da
qualidade dos dados ............................................................................ 78
Figura 10 Análise de agregação hierárquica (Hierarchical clustering
analysis - HCA), provenientes do escalonamento
multidimensional dos transcriptomas analisados, durante a fase
sintomática e pós recuperação ............................................................. 78
Figura 11 Heatmap apresentando a variabilidade de expressão de genes
selecionados nos indivíduos na fase de recuperação
comparando com a fase aguda ............................................................ 83

xiii
LISTA DE TABELAS
Tabela 1 Receptores FCRIIa e FCRIIIa e variantes alélicas ............................... 46
Tabela 2 Características demográficas da população estudada ......................... 67
Tabela 3 Quadro clínico observado nos pacientes com SGB ............................. 71
Tabela 4 Análise, por regressão múltipla logístico, das variáveis
associadas ao mau prognóstico. .......................................................... 73
Tabela 5 Características dos casos com Síndrome de Guillain
genotipados para o receptor de Fc ....................................................... 74
Tabela 6 Distribuição da frequência dos genótipos e alelos nos controles e
nos casos ............................................................................................. 75
Tabela 7 Risco estimado do efeito genético sobre a doença através de
modelo de regressão logistica. ............................................................. 75
Tabela 8 Distribuição dos genótipos quanto a aspectos clínicos e
laboratoriais .......................................................................................... 76
Tabela 9 Distribuição da combinação de genótipos entre controles e
pacientes .............................................................................................. 76
Tabela 10 Características dos pacientes submetidos a análise do
transcriptoma ........................................................................................ 77
Tabela 11 Lista dos genes com aumento ou diminuição significativo da
expressão num caso da variante desmielinizante (caso 1) .................. 79
Tabela 12 Lista dos genes com aumento ou diminuição significativo da
expressão num caso da variante desmielinizante (caso 2) .................. 80
Tabela 13 Lista dos genes com aumento ou diminuição significativo da
expressão num caso da variante axonal (caso 3) ................................ 81
Tabela 14 Lista dos genes com aumento ou diminuição significativo da
expressão num caso da Síndrome de Miller Fisher (caso 7) ............... 82

xiv
SUMÁRIO
RESUMO ........................................................................................................... vii
ABSTRAT ........................................................................................................... ix
LISTA DE SIGLAS E ABREVIATURAS .............................................................. xi
LISTA DE FIGURAS .......................................................................................... xii
LISTA DE TABELAS .......................................................................................... xiii
1 INTRODUÇÃO ................................................................................................... 17
2 OBJETIVOS ....................................................................................................... 21
2.1 Objetivo geral ............................................................................................... 21
2.2 Objetivos específicos ................................................................................... 21
3 REVISÃO BIBLIOGRÁFICA .............................................................................. 22
3.1 Histórico ....................................................................................................... 22
3.2 Epidemiologia .............................................................................................. 25
3.3 Diagnóstico .................................................................................................. 26
3.4 História natural e prognóstico ...................................................................... 28
3.5 Classificação da SGB .................................................................................. 28
3.6 Patogênese .................................................................................................. 32
3.7 Gangliosídeos .............................................................................................. 34
3.8 Fatores de risco para a patogênese da SGB ............................................... 38
3.8.1 Infecção e mimetismo molecular .............................................................. 38
3.8.2 Vacinas ..................................................................................................... 40
3.8.3 História familiar ......................................................................................... 42
3.8.4 Susceptibilidade genética ......................................................................... 42
3.8.5 Polimorfismo dos genes para o receptor do domínio Fc de
imunoglobulina IgG .................................................................................. 42
3.8.5.1 Receptor Fc ........................................................................................... 42
3.8.5.2 Estrutura do FcR .................................................................................... 43
3.8.5.3 Função do FcR ...................................................................................... 45
3.8.5.4 Polimorfismo do FCGR .......................................................................... 46
3.8.5.5 Polimorfismo do FCGR em doenças infecciosas ................................... 47
3.8.5.6 Polimorfismo do FCGR em doenças autoimunes .................................. 50
3.8.5.7 Polimorfismo do FCGR na SGB ............................................................ 51
3.9 Tratamento .................................................................................................. 53

xv
3.10 Expressão global de genes na SGB. ......................................................... 54
4 PACIENTES E METODOLOGIA ....................................................................... 58
4.1 Desenho do estudo ...................................................................................... 58
4.2 Local do estudo ........................................................................................... 58
4.3 População estudada .................................................................................... 59
4.4 Métodos de coleta de dados ........................................................................ 59
4.5 Estratégia metodológica e análise dos dados .............................................. 61
4.5.1 Metodologia para realização do Objetivo 1 .............................................. 61
4.5.2 Metodologia para realização do Objetivo 2 ............................................... 61
4.5.3 Metodologia para realização do Objetivo 3 ............................................... 62
4.5.3.1 Extração do DNA ................................................................................... 62
4.5.3.2 Genotipagem do FCGR ......................................................................... 63
4.5.3.3 Análise estatística .................................................................................. 64
4.5.4 Metodologia para realização do Objetivo 4 ............................................... 64
4.5.4.1 Montagem do transcritos obtidos por RNA-Seq utilizando a
plataforma Illumina Hi-Seq2000 ........................................................... 64
4.5.4.2 Mapeamento e Expressão diferença de genes ...................................... 65
4.5. Considerações éticas ................................................................................. 65
5 RESULTADOS .................................................................................................. 67
5.1 Clínica .......................................................................................................... 67
5.2 Avaliação de polimorfismos nos genes codificadores dos receptores
FcRIIa e FcRIIIa........................................................................................... 73
5.3 Resultados preliminares da expressão gênica global em células
monucleares de sangue periférico durante a fase sintomática e pós
recuperação da SGB ................................................................................... 76
6 DISCUSSÃO ...................................................................................................... 84
6.1 Aspectos clínicos da SGB............................................................................ 84
6.2 Polimorfismo do FCGR ................................................................................ 92
6.3 Expressão gênica global em células monucleares de sangue
periférico durante na SGB .......................................................................... 94
7 CONCLUSÕES .................................................................................................. 96
8 REFERÊNCIAS ................................................................................................. 97
9 ANEXOS .......................................................................................................... 120
1. Carta do Prof Luis Barraquer ....................................................................... 120

xvi
2. Critérios diagnósticos da SGB ..................................................................... 122
3. Critérios eletrofisiológicos da SGB .............................................................. 123
4. Termo de consentimento livre e esclarecido................................................ 124
5. Questionário ................................................................................................ 126
6. Artigo publicado: Clinical characteristics of Guillain-Barré syndrome in
a tropical country: a Brazilian experience .................................................... 128
7. Artigo concluído: No association of FCGR2A and FCGR3A
polymorphisms in a Guillain Barré Syndrome in a Brazilian population ...... 135

17
1. INTRODUÇÃO
A Síndrome de Guillain-Barré (SGB) é a principal causa de paralisia flácida
aguda, após a erradicação da poliomielite.(1, 2) É uma polirradiculoneuropatia
inflamatória, geralmente pós-infecciosa e mediada pelo sistema imune. A patogenia
da doença não é bem conhecida, entretanto há evidências sólidas de que se trata
de uma resposta imune aberrante contra determinantes antigênicos presentes no
nervo periférico, especialmente glicolipídeos.(3)
Nos últimos anos, tem-se tornado evidente a heterogeneidade da SGB.
Estudos clínicos, eletrofisiológicos e imunopatológicos caracterizaram, além da
polineuropatia desmielinizante inflamatória aguda (PDIA), também chamada de
variante clássica, outras duas novas variantes, a neuropatia axonal motora aguda
(NAMA) e a neuropatia axonal motora e sensitiva aguda (NAMSA). A síndrome de
Miller-Fisher (SMF), também, é considerada uma variante da SGB. Essa
variabilidade clínica pode ser resultado de diferentes mecanismos patogênicos.(4-
6) A figura 1 mostra as principais variantes clínicas da SGB.
Degenaração axonal
secundária
PDIA
NAMSA
NAMA S. DE MILLER-FISHER
SGB
Figura 1. Principais variantes clínicas da Síndrome de Guillain Barré.
A síndrome está presente em todo o mundo e sua incidência anual é de 0.16
a 4 casos por 100.000 e com mínima variação sazonal. A SGB pode ocorrer em
qualquer idade, no entanto, nos EUA e Europa a incidência aumenta com a idade e
na China a doença predomina em criança, notadamente a variante axonal da
SGB.(2, 7) A distribuição geográfica das variantes é específica, com a PDIA
predominando na Europa e EUA,(8) enquanto na China,(4) Japão,(9, 10) e em
alguns países da América Latina, como México,(11) Argentina(12) e Ilha do
Coração,(13) predominam a NAMA. Esses estudos sugerem que a NAMA pode

18
contribuir com o maior número de casos da SGB no resto da América Latina.
Entretanto, em estudo anterior realizado no Rio Grande do Norte, Brasil,
observamos um predomínio da variante desmielinizante.(14)
No Brasil, há poucos estudos sobre as características epidemiológicas da
SGB e não existem estudos sobre a frequência das variantes e de suas
manifestações clínicas. Estima-se, na população menor de 15 anos, uma
incidência da SGB de 0.36 a 0.6 casos/100.000.(15)
A SGB é considerada uma afecção autoimune com participação sinérgica
entre a imunidade celular e a humoral.(3) O mimetismo molecular é o provável
mecanismo que desencadeia o processo inflamatório efetor na SGB.(16) A
resposta imune contra o agente infeccioso resulta na produção de anticorpos que
reconhecem epítopos semelhantes aos do microorganismo no nervo periférico,
desencadeando o ataque imune a este último. Entre os microorganismos que
compartilham epítopos com o nervo periférico estão o Campylobacter jejuni,
Haemophilus influenzae e Citamegalovírus (CMV).(17-19) Além dessas infecções,
o vírus da dengue é um potencial agente desencadeante da SGB.(20, 21)
O fato de apenas alguns indivíduos, de uma população de infectados, virem
a desenvolver a SGB levou à sugestão de susceptibilidade individual ao
desenvolvimento da doença.(22) Recentemente, foi demonstrado que polimorfismo
nos genes de receptores da porção Fc de imunoglobulinas IgG (FcγRs), presentes
nas membranas de células do sistema imune, poderia resultar em níveis diferentes
de susceptibilidade às doenças, incluindo a SGB.(23, 24) Entretanto, não há um
consenso na SGB; em outro estudo, a distribuição dos genótipos FCGR não era
diferente entre os indivíduos com a SGB e controles.(25)
Três subclasses de FcR (FcRIIa, FcRIIIa e FcRIIIb) apresentam
polimorfismos bialélicos que afetam a afinidade para as subclasses de IgG e a
eficiência da resposta imune celular. No FCGR2A o polimorfismo é atribuído a
substituição da histidina na posição 131 por uma arginina. O alótipo FCGR2A-
H131 possui alta afinidade para IgG2. A presença de valina ou fenilalanina na
posição 158 do FCGR3A (158V versus 158F) determina afinidade para receptor de
IgG1, IgG3 e IgG4; o alelo com valina exibe alta afinidade para estas subclasses
de imunoglobulina. Ambos FCGR2A e FCGR3A são expressos em macrófagos. O
FCGR3B é expresso somente em neutrófilos, e há polimorfismos descrito com os

19
alótipos NA1 e NA2. O alótipo FCGR3B-NA1 liga mais eficazmente
imunocomplexos contendo IgG1 e IgG3 do que FCGR3B-NA2. (26)
Vários estudos têm estabelecido a importância da interação entre as
imunoglobulinas e os FcRs na susceptibilidade a doenças infecciosas. Macrófagos
expressam na sua membrana FcyR. O polimorfismo do FCGR pode influenciar o
vigor da resposta inflamatória e pode contribuir para a diferença interindividual de
susceptibilidade a doenças inflamatórias e infecciosas, a diversidade alélica destes
receptores poderia determinar respostas inflamatórias distintas e,
consequentemente, diferentes formas clínicas.(27)
Entre as doenças infecciosas, a leishmaniose visceral, cujo agente etiológico
no Brasil é a Leishmania infantum, é resultado da disfunção da resposta imune
celular, com supressão celular Leishmania específica, e parece estar envolvida no
desenvolvimento de sintomas. O mesmo ocorre na hanseníase onde os
macrófagos infectados por Mycobacterium leprae exibem alteração na sua
propriedade de superfície, tal como sua capacidade de expressar receptores Fc.
Em ambas doenças, há indícios de que os anticorpos podem estar envolvidos na
patogênese dessas doenças.(28, 29)
Diante do exposto, podemos resumir que a SGB é autoimune, causada por
resposta imune aberrante contra o nervo periférico induzida por uma infecção, além
de determinados fatores individuais de susceptibilidade. Entretanto, a SGB é
heterogênea, com distintas variantes clínicas e patológicas refletindo diferentes
mecanismo da lesão nervosa. As alterações patológicas principais são: inflamação,
degeneração axonal e desmielinização. O mecanismo imunopatogênico, avaliado
por fluidos biológicos, não está totalmente compreendido, portanto novas
abordagens devem ser utilizadas para o entendimento completo dessa síndrome.
Os avanços da neurociência, genética e biologia molecular permitiram o
desenvolvimento de novas ferramentas que avaliam a interface genética e
ambiente. Células fenotipicamente idênticas podem variar dramaticamente em
relação ao seu comportamento durante o seu tempo de vida e essa variação se
reflete em sua composição molecular, devido a alterações transcricionais. A partir
da análise desses transcritos de RNA de uma célula (transcriptoma) é possível ter-
se uma visão sem precedentes sobre a genômica funcional, com identificação das
redes de sinais associadas com a patogenia. É possível melhor examinar as

20
diversas vias de interação entre os processos intrínsecos celular e estímulos
extrínsecos. A utilização de sequenciamento de transcriptos de next generation é
uma ferramenta poderosa e de grande impacto para compreendermos diversos
processos biológicos com amplas implicações para a pesquisa básica e clínica.(30)
Neste estudo caracterizamos a frequência das variantes da SGB no Rio
Grande do Norte, e como elas se manifestam. Nós consideramos um período
compreendido entre as duas últimas décadas, que corresponde a uma fase
transicional sociodemográfica no Brasil, especialmente para a região nordeste,
onde ocorreu intensa migração populacional para as áreas urbanas, o que resultou
em mudanças no padrão de distribuição de doenças, incluindo doenças
infecciosas. Examinamos se as variantes da SGB, desmielinizante e axonal,
apresentam manifestações clínicas diferentes e sua evolução após um ano do
início da afecção.
Ainda, nós tivemos como hipótese que heterogeneidade genética no FCGR
influencia o risco da SGB, considerando que os autoanticorpos participam na
patogenia dessa síndrome. Nós examinamos se os FcRs de alta afinidade são
mais frequentes na SGB do que na população normal e se estes estavam
associados à forma mais grave da SGB. Por último, através da abordagem para
transcriptoma usando tecnologia de sequenciamento “next generation” (RNA seq),
nós determinamos as diferenças de expressões gênicas nos indivíduos com a
SGB, comparando a fase aguda com a fase de recuperação.
O objetivo geral desse estudo foi compreender a evolução das respostas
celulares desencadeadas pela infecção e pelo sistema imunológico e determinar o
modelo molecular para a SGB. Para testar essas hipóteses, nós realizamos um
estudo de coorte no qual foram examinados aspectos epidemiológicos e clínicos, e
outro estudo caso controle para avaliação do polimorfismo do FCGR. Foram
incluídos neste estudo, controles saudáveis de banco de sangue da região. Para o
estudo do transcriptoma foram selecionados 12 indivíduos com a SGB.

21
2. OBJETIVOS
2.1 Geral:
Caracterizar as manifestações clínicas das variantes da Síndrome de
Guillain- Barré e determinar os fatores de riscos ambientais e genéticos envolvidos
com a doença no Estado do Rio Grande do Norte.
2.2 Objetivos específicos:
1. Determinar a incidência/prevalência da Síndrome de Guillain-Barré e dos
seus subtipos no Estado do Rio Grande do Norte.
2. Determinar as diferenças clínicas entre as variantes desmielinizante e
axonal e a evolução clínica.
3. Determinar se polimorfismos no receptor Fc estão associados com risco
mais elevedo de desenvolver a Síndrome de Guillain-Barré.
4. Identificar o padrão de expressão gênica na Síndrome de Guillain-Barré
sintomática e pós-recuperação.

22
3. REVISÃO BIBLIOGRÁFICA
A SGB constitui atualmente a causa mais frequente de paralisia flácida
aguda no mundo. Caracteriza-se, clinicamente, por paralisia ascendente,
anormalidades sensitivas e autonômicas de variados graus decorrentes de lesão
de raízes e nervos periféricos. Trata-se de uma afecção mediada pelo sistema
imune onde autoanticorpos e/ou células inflamatórias reagem contra epítopos das
raízes e nervos periféricos, promovendo lesão da mielina ou axônio ou ambos. A
etiologia e a fisiopatologia ainda não estão completamente entendidas. A
exposição a agentes infecciosos, provocando uma resposta imunológica por
mimetismo molecular, associada a fatores genéticos são considerados
determinantes na origem da doença.
3.1 Histórico
Apesar dos grandes avanços da neurofisiologia e da imunogenética, a
doença ainda é determinada por critérios clínicos já descritos, em 1916, por
Guillain, Barré e Strohl. Como muitas outras síndromes neurológicas, a
compreensão da SGB ocorreu numa sequência histórica de descrição de casos e
com os avanços da medicina contemporânea.
A primeira descrição da SGB ocorreu, em 1859, por Jean Baptiste Octave
Landry de Thézillat, neurologista francês. Ele publicou diversos casos e denominou
de paralisia aguda ascendente, apesar de um deles ter apresentado evolução
crônica. Outros autores publicaram casos e estes usavam como referência a
publicação de Landry. Em 1876, Westphal usou pela primeira vez o termo “paralisia
ascendente de Landry”.
Em 1916, três neurologistas franceses Georges Guillain, Jean-Alexandre
Barré, e André Strohl descreveram dois soldados com paralisia flácida aguda que,
subsequentemente, se recuperaram. Estudos do líquor cefalorraquidiano desses
pacientes, técnica descrita 25 anos antes por Quincke, mostrou aumento das
proteínas com contagem de células normal. Esse dado laboratorial, com

23
dissociação albumino-citológica, caracterizou essa síndrome diferenciando-a da
poliomielite e de outras doenças.
O termo Síndrome de Guillain-Barré foi introduzido por Draganescu e
Claudian, num congresso da Sociedade de Neurologia de Paris, em 1927, onde
Barré era o presidente. Não se conhece as razões da omissão do nome de Strohl,
mas certamente foi injusta. Strohl estudou medicina, matemática, química e física.
Obteve o título de doutor em medicina e física. Nos casos originais contribuiu para
o estudo eletrofisiológico. Na ocasião, ele era um jovem, com 29 anos de idade,
recém-graduado em medicina. Seus achados demonstrando retardos dos reflexos
anunciava presença de desmielinização como a principal alteração neurofisiológica
da variante clássica, polineuropatia desmielinizante inflamatória aguda, da SGB.
Ele publicou inúmeros trabalhos sobre reflexos e condução nervosa.(31)
Haymarker e Kernohan, em 1949, publicam uma série de 50 casos fatais da
SGB. O ponto forte dessa publicação foi a descrição dos achados patológicos da
SGB, que incluíram edema da porção proximal do nervo periférico, desintegração
da mielina, acompanhado posteriormente de infiltrado linfocitário e fagocitose.
Alterações retrógadas nas células da ponta anterior da medula, cromatólise,
também foram relatadas.
Por volta de 1955, um modelo experimental de Neurite Autoimune foi
desenvolvido por Raymond D Adams e Byron Waksman. A injeção de homegenato
de nervo em coelhos produzia manifestações clínicas e patológicas similares a
SGB. Neste modelo, o achado marcante foi a presença precoce de infiltrado
linfocitário no nervo periférico. A partir desse modelo, a SGB é tida como uma
doença inflamatória.
Miller Fisher, em 1956, descreveu três casos de oftalmoplegia aguda, ataxia
e arreflexia, após infecão de vias respiratórias. Num dos casos havia dissociação
albumino-citológica. O autor considerou esta entidade, agora conhecida com
Síndrome de Miller Fisher (SMF), como uma variante da SGB. Os pacientes com
SFM podem apresentar envolvimento dos nervos facial e bulbares, assim como
desenvolver paralisia flácida.(32)
Uma nova e clássica contribuição para a compreensão da SGB ocorreu em
1969. Asbury, Arnason e Adams publicaram, na revista Medicine, achados

24
patológicos de 19 pacientes com SGB. A presença de infiltrado mononuclear
perivascular adjacente às regiões de desmielinização segmentar era a
característica patológica principal. A inflamação não era restrita às raízes nervosas,
outras porções do nervo também estavam comprometidas.
Em 1985, um estudo multicêntrico nos Estados Unidos demonstrou benefício
da plasmaferese no tratamento da SGB, apoiando a hipótese de anticorpos anti-
neural no desenvolvimento dessa doença.(33) Em 1992, van der Meché e
colaboradores, publicaram o primeiro estudo randomizado mostrando efeito
positivo da imunoglobulina endovenosa (IgEV) em altas doses na SGB. Desde
então, o uso da IgEV, na dose de 0,4g/Kg por dia, por cinco dias consecutivos,
tornou-se o tratamento de eleição na maioria dos centros hospitalares, devido a
sua grande praticidade.(34)
Até então, a SGB era considerada originalmente uma doença
desmielinizante do nervo periférico. Entretanto, a partir da descrição clínica e
eletrofisiológica, por McKhann e colaboradores, em 1991, de casos de SGB no
norte da China, que ocorriam de forma episódica, foi identificada a variante
puramente motora a axonal da SGB. Essa condição é denominada de Neuropatia
Axonal Motora Aguda (NAMA) e ocorre em toda parte do mundo, mas em
diferentes proporções. A NAMA tem sido associada à infecção prévia por
Campylobacter jejuni e por ataque mediado por anticorpos contra o axolema do
nodo de Ranvier.(4, 35, 36) Recentemente, estudos em humanos e modelos
animais demonstraram que a NAMA pode ocorrer, em alguns casos, com bloqueio
da condução reversível sem degeneração axonal.(37, 38) Em 1988, anticorpos
contra gangliosídeos foram encontrados, pela primeira vez, na SGB.(39)
Gangliosídeos são glicoesfingolipídeos distribuídos na membrana celular, com sua
porção glicídica ancorada para o lado de fora da célula.
A paralisia ascendente tornou-se a descrição clássica da SGB. Entretanto, a
utilização de novas técnicas eletrofisiológicas permitiu subclassificar a SGB e
diferenciá-la quanto ao processo patológico envolvendo o nervo, desmielinizante
ou axonal, selecionando subpopulações de axônios comprometidos nestas
variantes, sensitivo e/ou motor. As principais variantes descritas são: polineuropatia
desmielinizante inflamatória aguda, neuropatia axonal motora aguda, neuropatia
axonal motora e sensitiva aguda, síndrome de Miller-Fisher. Mais recentemente,

25
novas variantes, notadamente com envolvimento focal, foram relatadas: SMF
sobrepondo com SGB, oftalmoparesia aguda sem ataxia, SGB com reflexos
conservados, variante regional faringo-cérvico-braquial, SGB atáxica, SGB atáxia
sensitiva, SGB sensitiva pura, encefalite de Bickerstaff, encefalite de Bickerstaff
sobrepondo com SGB.(3)
O Professor Lluis Barraquer-Bordas (1923-2010), uma das personalidades
mais marcantes da neurologia catalana, conheceu, quando jovem, o Dr. Guillain,
em um congresso internacional de neurologia realizado em Paris, em 1949. Prof
Barraquer afirmava que o Dr. Guillain mantinha uma ilusão de descobrir um
microorganismo causador da síndrome por ele descrita. Nessa época, foi
identificado o vírus responsável pela poliomielite. Essa hipótese de Guillain, que
infecção poderia estar envolvida na patogênese da SGB, é indicada em carta que
recebi do Prof Barraquer, que foi um dos meus tutores durante a minha residência
em neurologia, no Hospital de La Santa Creu I Sant Pau (1988-1993), (Anexo 1).
Quadro décadas depois do encontro de Barraquer com Guillain, estudos
epidemiológicos revelaram a associacão do Campilobacter jejuni com a SGB.(36)
Estudos contemporâneos demonstraram que o envoltório lipossacarídeo dessa
bactéria compartilha carboidratos que são similares aos dos gangliosídeos do
nervo periférico. Portanto, a doença se desenvolve por mimetismo molecular.(40,
41)
3.2 Epidemiologia
Após a erradicação da poliomielite, a SGB tornou-se a principal causa de
paralisia flácida no mundo. A afecção está presente em todo o mundo e sua
incidência anual encontra-se em 0,16 a 4 casos por 100.000 e com mínima
variação sazonal. Na Europa e nos Estados Unidos da América, a incidência
aumenta 20% a cada incremento de 10 anos de idade e os homens são mais
acometidos do que as mulheres. Antecedentes infecciosos precedem 70% dos
casos.(1, 2, 7) Como esses dados foram na maioria coletados dos EUA e Europa,
possíveis variações regionais podem ocorrer, especialmente na América Latina,
Ásia e África, que apresentam tipos distintos de patógenos.

26
Sazonalidade foi relatada no norte da China, onde durante os meses de
verão de 1991 e 1992, na zona rural, houve um aumento da variante puramente
motora da SGB.(4, 6) Já nos Estados Unidos da América, a incidência é maior no
inverno.(42) Os estudos epidemiológicos no Brasil são escassos. Dias-Tosta e
Kückelhaus, analisando o banco de dados do programa da vigilância
epidemiológica das paralisias agudas e flácidas (PAF) da Fundação Nacional de
Saúde, entre 1990 e 1996, estimaram a incidência annual de 0,39 a 0,63
casos/100.000 para casos de SGB na população menor de quinze anos.(15) Outro
estudo, realizado em São Paulo, analisando retrospectivamente os prontuários de
um único centro, no período de 1995 a 2002, observou menor incidência da doença
em pacientes com idade abaixo de 15 anos (18,9%) e acima de 60 anos (16,9%). A
frequência maior foi observada no subgrupo com idade entre 15 e 60 anos (66,2%).
A incidência anual foi de 0,6 casos/100.000 habitantes.(43)
3.3 Diagnóstico
A SGB tem início agudo ou subagudo, com evolução monofásica,
manifestando com parestesias, formigamentos, fraqueza, dor ou a combinação de
alguns desses sintomas. O quadro típico é uma paralisia simétrica e ascendente
que piora num período de 12 horas a 28 dias (nadir, tempo para alcançar o déficit
máximo) seguido de um período de platô. Os pacientes apresentam hipo ou
arreflexia. Aproximadamente 25% dos pacientes apresentam insuficiência
respiratória, necessitando ventilação mecânica, por comprometimento dos
músculos diafragma e intercostal. Envolvimento de nervos cranianos é frequente,
incluindo o VII par craniano resultando em paralisia facial, os oculomotores e
bulbares. Disautonomia, com alterações cardíacas (hipotensão postural,
bradiarritmias, assistolia), gastrointestinais (íleo, constipação) e hiponatremia
podem ocorrer. O estudo do líquor mostra a dissociação clássica albumino-
citológica, com aumento da proteína sem aumento da celularidade. História de
infecção respiratória ou diarreia três dias a seis semanas antes do inicio do quadro
é frequente.(1, 44)
A distribuição da fraqueza muscular correlaciona com as variantes da SGB.

27
É proximal na PDIA, distal na NAMA e difusa na NAMSA.(1) A presença de dor é
frequente, ocorrendo em 55 a 89% dos pacientes, e, muitas vezes,
negligenciada.(45-47) Ela ocorre na fase aguda da doença e pode persistir durante
a reabilitação por meses, habitualmente grave. A presença de dor na fase aguda
correlaciona com a ocorrência de dor tardialmente.(48, 49). Os mecanismos
reponsáveis pela dor ainda não estão esclarecidos.
Além do quadro clínico e do estudo do líquor, o diagnóstico da SGB é
auxiliado pela eletroneuromiografia. O estudo da condução nervosa e a
eletromiografia por agulha apresentam evidências de envolvimento dos nervos
periféricos, assim como diferencia os subtipos desmielinizante e axonal. Vários
critérios eletrofisiológicos para o diagnóstico da SGB foram publicados.(6, 50-52)
Estudos realizados na primeira semana da doença podem ser normais. Nesta fase,
alterações sugestivas de desmielinização podem ser na verdade disfunção
reversível do axolema motor ao nível do nodo de Ranvier. Recomenda-se
realização de pelo menos dois estudos eletrofisiológicos.(53, 54)
Apesar da importância da eletroneuromiografia e do estudo do líquor, esses
exames não são obrigatórios para o diagnóstico da SGB. Recentemente, um grupo
de estudo (Brighton Collaboration GBS Working Group), formado por
pesquisadores de diferentes nacionalidades, elaborou um guia para definir a SGB e
SMF. Na impossibilidade de realizar a eletroneuromiografia e a punção lombar, a
SGB é definida como: paralisia flácida e bilateral das extremidades, mais hipo ou
arreflexia generalizada, mais padrão evolutivo monofásico, com o déficit máximo da
fraqueza entre 12 horas e 28 dias seguido de uma fase de platô, mais ausência de
diagnóstico alternativo para a fraqueza muscular.(55)
A SMF é diagnosticada pela presença de oftalmoplegia, ataxia e arreflexia,
mais ausência de fraqueza nas extremidades, mais padrão evolutivo monofásico,
com o déficit máximo da fraqueza entre 12 horas e 28 dias seguido de uma fase de
platô, mais ausência de alteração da consciência ou sinais de lesão da via córtico-
espinhal, mais dissociação albumino-citológica no líquor, mais eletroneuromiografia
normal ou alteração da condução sensitiva, mais ausência de diagnóstico
alternativo.(55)
Existem várias doenças, do sistema nervoso central ou periférico, que
apresentam quadro de paralisia flácida similar a SGB. O diagnóstico diferencial de

28
neuropatia aguda, tais como vasculite, deficiência vitamínica, neuropatia tóxica,
porfiria, devem ser consideradas e excluídas, tendo como norteadores a história
clínica e os exames complementares. Encefalite do tronco cerebral, mielopatia
aguda, doença da junção neuromuscular ou miopatia podem confundir com a SGB
e devem ser consideradas no diagnóstico diferencial.
3.4 História natural e prognóstico
O dado clínico principal da SGB é a paralisia ascendente rapidamente
progressiva. O paciente, geralmente, alcança o déficit máximo até quatro semanas
após o início dos sintomas, no entanto, a maioria dos casos ocorre após poucas
horas ou dentro de duas semanas. Segue-se uma fase de platô, que varia de dias,
semanas a meses. Posteriormente, ocorre recuperação, lenta, na dependência da
área neural afetada pelo processo inflamatório.(44, 56) A evolução é favorável, na
maioria dos casos, entretanto, 20% destes morrem ou ficam sequelados, apesar da
disponibilidade de cuidados intensivos modernos e de tratamento específico.(57) A
extensão da lesão nervosa na fase aguda e a capacidade de recuperação, na
convalescência, são determinantes da evolução. Idade avançada, infecção prévia,
progressão rápida da doença, necessidade de ventilação mecânica e degeneração
axonal na eletroneuromiografia estão associados a pior prognóstico.(58-61)
3.5 Classificação da Síndrome de Guillain-Barré
Similar a tantas outras áreas da investigação científica, o caminho para
entender a SGB inicia com o estudo da fenomenologia. Um grupo de fenótipos,
alguns com aparência similar e outros bastante díspares, são chamados de SGB.
O avanço das técnicas eletrofisiológicas contribuíram na classificação desta
doença, permitindo identificar o envolvimento seletivo da mielina e/ou do axônio e
subclassificando-a em diferentes variantes. Primariamente, a SGB é classificada
em dois subtipos: desmielinizante ou axonal. Entretanto, além dessas duas
variantes, outras são reconhecidas: neuropatia axonal motora e sensitiva aguda

29
(NAMSA), síndrome de Miller-Fisher (SMF), SMF sobrepondo com SGB,
oftalmoparesia aguda sem ataxia, SGB com reflexos conservados, variante
regional faringo-cérvico-braquial, SGB atáxica, SGB atáxia sensitiva, SGB sensitiva
pura, encefalite de Bickerstaff, encefalite de Bickerstaff sobrepondo com SGB. No
entanto, a questão a saber é se elas representam uma única doença, com
manifestações clínicas diversas, ou se são entidades distintas.(62)
A classificação, desmielinizante e axonal, tem distribuição geográfica
variável. A variante desmielinizante (PDIA) é mais prevalente na Europa e Estados
Unidos.(8) Já a variante axonal predomina na China (4, 6), Japão (10), Bangladesh
(63) e alguns países da Americana Latina, tais como México (11), Argentina (12) e
Ilha Coração (13). Em Israel, a variante axonal é mais frequente do que nos países
Europeus e na América do Norte. (64) Não sabemos a frequência das variantes da
SGB no Brasil. Os escassos estudos publicados não classificaram a SGB entre as
duas divisões principais, desmielinizante e axonal, utilizando a
eletroneuromiografia.(15, 43, 65)
A eletroneuromiografia é uma importante ferramenta clínica para diferenciar
as variantes desmielinizante e axonal. Entretanto a sua interpretação na fase
aguda da SGB não é fácil. O bloqueio da condução que é considerado nos critérios
diagnósticos, até então aceitos, como indicadores de desmielinização pode
acontecer na variante axonal. O valor da velocidade de condução motora a partir
do qual é indicativo de desmielinizante não é uniforme entre diferentes estudos.(66)
Vários critérios foram criados para dignosticá-la.(5, 6, 50, 51). Para
considerar desmielinização (PDIA) é necessário demonstrar alterações
consequentes ao processo inflamatório e lesão multifocal da mielina: bloqueio da
condução motora e/ou dispersão temporal do potencial de ação composto
muscular, prolongamento da onda F, aumento da latência distal motora, diminuição
da velocidade de condução em pelo menos dois a três nervos motores segundo o
critério utilizado. Na PDIA, a inflamação segmentar na mielina é responsável pelo
déficit de condução do impulso elétrico no nervo periférico, resultando no bloqueio
da condução traduzido clinicamente por paralisia flácida e arrefléxica. O déficit
motor é de predomínio proximal. Envolvimento de nervos cranianos, notadamente
o VII, com ou sem envolvimento respiratório e disautonomia, são frequentes.
A NAMA é uma variante da SGB com comprometimento exclusivo do axônio

30
motor. Os achados eletrofisiológicos característicos são: diminuição da amplitude
dos potenciais compostos de ação muscular ou ausência do potencial motor, com
pouca ou discreta alteração da velocidade de condução motora, mais preservação
dos potenciais de ação sensitivos. Clinicamente, manifesta-se por tetraparesia
ascendente, flácida e arrefléxica, sem envolvimento sensitivo. A fraqueza é de
predomínio distal. Pode ocorrer insuficiência respiratória. Sinais de irritação
meníngea e de disautonomia estão presentes em alguns pacientes. A recuperação
habitualmente é lenta, mas em alguns casos ocorre rapidamente. Durante a fase
de recuperação, alguns pacientes podem desenvolver hiperreflexia.(4, 67)
A classificação da SGB através da eletroneuromiografia é sujeita a
equívocos, notadamente na fase precoce da doença. Alguns pacientes com a
variante NAMA associados a anticorpos anti-gangliosídeos são classificados
erroneamente como PDIA. Um estudo italiano mostrou que a proporção de
pacientes com PDIA diminuía de 67% para 58% e a proporção de pacientes com
NAMA aumentava de 18% para 38% após realização de um segundo estudo
eletrofisiológico.(53) Um estudo de colaboração entre Japão e Itália demonstrou
que a NAMA era subdiagnosticada, já que muitos pacientes diagnosticados com
PDIA, num estudo inicial, tinham na verdade NAMA.(68) Esses indivíduos
apresentam uma falha reversível da condução motora, provavelmente por bloqueio
dos canais de sódios por anti-Gg no nódulo de Ranvier, caracterizados na
eletroneuromiografia por bloqueio da condução e aumento da latência distal
motora. Num segundo estudo eletrofisiológico, os indivíduos restauram o bloqueio
da condução sem desenvolver a dispersão temporal do potencial de ação
composto muscular que é o característico do processo de remielinização. Portanto,
um único estudo eletroneuromiográfico não é capaz de classificar corretamente as
variantes, sendo necessário realizar uma série de exames por cada paciente. Esse
novo mecanismo, bloqueio de condução reversível, não estava contemplado nos
critérios de diagnóstico eletrofisiológico. Por consequência, a prevalência da NAMA
deve ser maior do que as referidas nas séries publicadas.(54)
Em 2003, Capasso e colaboradores descreveram dois pacientes que
apresentaram fraqueza muscular aguda sem sinais ou sintomas sensitivos. Esses
indivíduos tinham anticorpos IgG contra gangliosídeos (GM1, GD1a e GD1b). O
estudo de condução revelava bloqueio da condução motora com potenciais

31
sensitivos normais. Houve melhora clínica e do bloqueio da condução em duas a
cinco semanas, sem desenvolver dispersão temporal ou sinais de desnervação na
eletromiografia por agulha. Mais um nova variante foi considerada e denominada
de neuropatia motora aguda com bloqueio da condução.(69)
A caracterização da NAMSA é dada pela ausência de alterações
desmielinizantes, diminuição da amplitude distal do potencial de ação composto
muscular e redução de < 50% da amplitude do potencial sensitivo. Manifesta-se
clinicamente por tetraplegia de início fulminante e necessidade de ventilação
mecânica, entre dois a cinco dias, após o início dos sintomas. A recuperação
desses pacientes é lenta e incompleta, pois depende da regeneração axonal, que,
geralmente, ocorre num ritmo de 1 a 3mm/dia.(70)
A Síndrome de Miller Fisher é uma variante rara, ocorrendo em 5% dos
casos da SGB.(2) O diagnóstico é clinico, caracterizado pela tríade: oftalmoplegia,
ataxia e arreflexia. A história prévia de infecção dentro das quatro semanas antes
do início do quadro, a dissociação albumino-citológica e a presença de anticorpo
anti-GQ1b auxiliam no diagnóstico. Envolvimento de outros pares cranianos
(paralisia facial, disfagia) e parestesias distais nas extremidades não são
infrequentes. O estudo da condução motora e sensitiva é variável. A condução
motora pode ser normal ou apresentar discretas alterações indicativas de
desmielinização, tais como aumento da latência distal e diminuição da velocidade
de condução.(71) O envolvimento do nervo facial pode ser demonstrado por
eletrofisiologia, em especial em casos que o comprometimento do facial é
subclínico, através da observação da redução da amplitude do potencial de ação
composto muscular.(72)
A condução sensitiva na SMF se apresenta com diminuição da amplitude do
potencial de ação sensitivo desproporcional ao prolongamento das latências distais
ou da redução das velocidades de condução sensitivas.(71, 72) Isto demonstra
inequívoca disfunção sensitiva na SMF, mas existem controvérsias se a neuropatia
sensitiva está relacionada com os sinais clínicos de arreflexia e ataxia. Em muitos
pacientes a ataxia é mais acentuada do que a neuropatia sensitiva e em outros há
ataxia com potenciais sensitivos normais.(73)
A ataxia é observada na SGB nas variantes de SMF, na variante atáxica da
SGB e na variante sensitiva da SGB. Alguns pacientes podem apresentar ataxia

32
aguda, com ausência do sinal de Romberg, sem oftalmoplegia, hipo ou arreflexia,
parestesias distais e dissociação albumino-citológica no líquor. Esses casos são
denominados de variante atáxica da SGB.(74) Na neuropatia sensitiva atáxica
aguda, sem oftalmoplegia, com presença de sinal de Romberg e arreflexia, há
envolvimento seletivo de fibras aferentes do grupo Ia do fuso muscular sendo
responsável pela ataxia.(75) Ito e colaboradores estudaram pacientes com ataxia
aguda sem oftalmoplegia e classificaram segundo a presença ou ausência do sinal
de Romberg. Não encontraram diferenças clínicas e nem laboratoriais (estudo do
líquor, anti-gangliosídeos) entre esses grupos. Eles denominaram essas variantes
de neuropatia atáxica aguda, com finalidade de evitar confusões nosológicas, e
consideram formas frustas da SMF.(76)
3.6 Patogênese
As alterações patológicas da SGB são heterogêneas refletindo as variantes
clínicas. Na variante clássica da SGB (PDIA) as alterações patológicas
encontradas são infiltrados inflamatórios, presença de células T e macrófagos
elevada, e áreas de desmielinização focal, frequentemente associadas com sinais
de degeneração axonal secundária. Esses achados podem ser detectados nas
raízes nervosas, bem como em nervos motor e sensitivo, e são indicativos de
mecanismo celular. Estudos de autopsia realizados numa fase precoce da PDIA
mostraram ocorrência de ativação do complemento e depósito de componentes
ativos de complementos na mielina, antes da invasão de macrófagos. A formação
do complexo membrana-ataque inicia a degeneração vesicular.(77, 78) A ligação
de anticorpos contra epítopos da célula de Schwann e lesão da mielina mediada
por complemento, mais a presença de anticorpos anti-gangliosídeos em supostos
pacientes com PDIA e a evolução temporal da SGB, sugerem predomínio do
mecanismo humoral sobre o celular.(79)
A neurite experimental autoimune, induzida por imunização com proteínas
de nervo periférico ou transferida para os animais por células T sensibilizadas com
essas proteínas, é o modelo animal de PDIA. Esses animais desenvolvem
características clínicas e patológicas semelhantes a PDIA. O antígeno é

33
apresentado pelo macrófago à célula, que penetra no endotélio, reconhece um
antígeno cruzado, resultando na liberação de citocinas e ativação de macrófagos
endoneural. Ocorre liberação de enzimas e óxido nítrico que invadem a mielina
compacta. Em paralelo, as células T ativadas liberam citocinas, ativam células B
para produzirem anticorpos. Esses anticorpos cruzam a barreira hemato-nervosa,
se ligam à célula de Schwann, ativam complemento e promovem a
desmielinização.
Na NAMA, anticorpos da classe IgG e componentes do sistema
complemento ativados ligam-se ao axolema da fibra motora no nodos de Ranvier,
seguido de formação do complexo membrana-ataque. Gangliosídeos GM1 e GD1a
são expressados em grande quantidade nesta região, onde estão localizados os
canais de sódio voltage-dependente. Evidências apontam para os Gg como
epítopos da resposta imune. Autoanticorpos contra GM1 ou GD1a ligam-se ao
axolema nodal, provocando desaparecimento dos canais de Na e o descolamento
da mielina paranodal; como consequência, ocorre falha da condução nervosa e
fraqueza muscular. O resultado desse processo é uma degeneração axonal sem
infiltrado linfocitário e nem desmielinização.(35, 66)
Em modelos animais de NAMA há três tipos de alterações: degeneração
axonal, bloqueio de condução transitório e a neuropatia motora aguda com
bloqueio da condução. Coelhos sensibilizados com GM1 desenvolvem paralisia
flácida e anticorpos anti-GM1. Esses anticorpos e complemento ativado se ligam
ao axolema da fibra motora nos nodos de Ranvier. Nessa região estão localizados
os canais de sódio voltage-dependente e os gangliosídeos GM1 e GD1a estão
expressados em grande quantidade. O processo autoimune provoca
desaparecimento dos canais de Na, o descolamento da mielina paranodal e
eventualmente degeneração axonal.(37, 80)
A sequência de alterações imunopatológicas e eletrofisiológicas observada
nos modelos animais sugere que na fase aguda ocorre redução de fatores de
segurança para condução nervosa: desequilíbrio iônico na região nodal devido à
formação de poros na membrana por ativação do complemento, a redução do
número de canais de sódio, o alogamento do nodo e as fugas de corrente na
região paranodal. Essas alterações podem ser reversíveis em um período
relativamente curto, como na NAMA com bloqueio da condução reversível ou na

34
neuropatia motora axonal com bloqueio da condução. Se a reação inflamatória
continuar, proteases são ativadas e promovem degradação do citoesqueleto
seguida de degeneração axonal. Em conclusão, o processo fisiopatológico da
NAMA varia de leve disfunção axonal reversível à degeneração axonal.(38, 81)
Já na forma clássica da SMF poucos estudos patológicos foram publicados,
refletindo o curso clínico benigno e a recuperação completa na maioria dos casos.
Um caso publicado demonstrou alterações de desmielinização similar a PDIA, sem
envolvimento do sistema nervoso central.(82) Um outro estudo revelou
desmielinização segmentar e edema axonal de nervos periféricos e oculomotores,
associada à leve cromatólise no tronco cerebral numa mulher com 64 anos. (83)
Evidências de lesão do sistema nervoso central têm se limitado a estudos de
neuroimagem e eletrofisiológicos.(3, 84)
Não é bem compreendido por que acontece o envolvimento seletivo de
axônio, motor e/ou sensitivo. Várias são as hipotéses estudadas, entre elas,
diferenças entre propriedades dos canais iônicos, maior concentração de GM1 na
fibra motora, diferença na ceramida modificando a configuração tridimensional do
açúcar do gangliosídeo, maior ligação de anticorpo anti-GD1a na fibra motora,
diferença nas propriedades biofísicas tornando a fibra motora mais vulnerável a
bloqueio de condução.(66)
A fisiopatologia da dor não é bem compreendida na SGB. Certamente, por
se tratar de uma doença mediada pelo sistema imune, o processo inflamatório
deve ser o principal responsável pela dor neuropática e a intensidade do processo
inflamatório e a expressão de citocinas estão associadas ao grau de dor
neuropática.(85) Outros mecanismos implicados na dor: irritação do nervi nervorum
que inerva troncos nervosos e ramos dorsais, desmielinização de fibras grossas
sensitivas, disfunção de fibras finas, compressão nervosa, dor musculo-
esquelética.(49, 86, 87)
3.7 Gangliosídeos
Os gangliosídeos (Gg) são glicoesfingolipídeos de membrana plasmática
encontrados, principalmente, nas membranas das células nervosas. São

35
compostos de lipídeo, ceramida, unidos a um ou mais açúcares (hexoses) e ácido
siálico. Eles recobrem parte da superfície celular, onde a ceramida adere à
membrana e o oligossacarídeo fica projetado para o meio extracelular.
Dependendo do número de ácido siálico eles são classificados em mono (GM1), di
(GD1a, GD1b), tri (GT1b, GT1a) e quatro (GQ1b). As letras e os números refletem
a diferentes posições do ácido siálico.(1) A figura 2 mostra os principais tipos de
gangliosídeos com as respectivas variantes clínicas da SGB associadas a
presença de anticorpo anti-gangliosídeos.
A presença de anticorpos contra diferentes gangliosídeos varia de 2 a 40%.
Há vários motivos para explicar esta variabilidade, tais como a frequência de
antecedentes infecciosos, o tempo de doença, a definição de valores normais, a
localização geográfica, diferentes sistemas, individual e comercial, de análise dos
anti-gangliosídeos.(88) Exceto a presença do anticorpo anti-GQ1b, que é o
marcador para SMF, há controvérsias se os demais anticorpos contra determinado
Gg são capazes de diferenciar as demais variantes.
Para alguns autores, os anticorpos anti GM1 e GD1a da classe IgG estão
associados com NAMA e não a PDIA,(68, 69) hipótese apoiada por modelos
utilizando animais experimentais, como coelhos sensibilizados com GM1, onde os
resultados são similares aos pacientes com SGB, ou seja, degeneração axonal
sem desmielinização.(89) Em 2003, publicamos nossos resultados da frequência
de anticorpos anti-GM1 em pacientes com SGB no Estado do Rio Grande do Norte.
Todos os pacientes com NAMA tinham anticorpos anti-GM1 (100%), contra 44% de
positividade dos pacientes com PDIA (p=0.01).(14) No entanto, para outros
autores, a presença de anticorpos anti-GM1 não é exclusiva da variante puramente
motora, possivelmente devido ao fato de tanto o nervo motor como o sensitivo
expressarem quantidades similares de GM1 e GD1a.(5, 90, 91)

36
Figura 2: Glicolipídeos associados a neuropatias inflamatórias. Gal = galactose; GalNAc = N-acetilgalactosamine; Glc = glucose; GlcNAc = N-acetilglucosamine; NeuNAc = ácido N-acetilneuraminico; GlcUA = ácido glucuronico; Cer = ceramide; LM1 = SPG, sialosilparaglobosido; Hex-LM1 = SLPG, sialosilactosaminilparaglobosido; SGPG = glucuronil paraglobosido sulfatado; SGLPG = glucuronil lactosaminil paraglobosido sulfatado. Anticorpos contra GM1 e GD1a estão associados a variante axonal, anticorpos anti-GD1b na variante sensitiva e anticorpos anti-GQ1b na síndrome de Miller-Fisher.
Também há controvérsias se a presença de anti-GM1 está associada à
gravidade no nadir da doença e à recuperação incompleta.(91-94) Um estudo
recente encontrou dois tipos de anticorpos anti-GM1 nos pacientes com SGB: um
que tem reação cruzada com anti-GD1b e outro grupo que não tem. A forma grave
da SGB está associada com o grupo que não tem reação cruzada com anti-GD1b.
Essa diferença de afinidade dos anticorpos, demonstrada por experimento onde o
antígeno foi estudado num contexto celular, denominado Cell-ELISA, indica que
diferentes regiões do GM1 estão envolvidas na ligação do anticorpo e influencia na

37
gravidade da doença. Nas células, GM1 interage com outros componentes da
membrana de tal forma que somente algumas áreas do oligossacarídeo estão
expostas ao ambiente extracelular e capazes de interagir com anticorpos.(95)
A patogenicidade da lesão nervosa por anticorpos anti-Gg é complexa. Por
um lado fatores neurobiológicos, tais como local e extensão da lesão associada à
capacidade regenerativa, estão associados ao prognóstico. Por outro lado, fatores
imunológicos relacionados com as características antígeno-anticorpo, tais como,
afinidade e especificidade fina, densidade de antígeno, isótopo do anticorpo
influenciando a meia vida deste, também participam na patogenia. Estudos
sorológicos para anticorpos anti-gangliosídeos em pacientes com SGB
provavelmente são pouco preditores já que os ensaios realizados em meios sólidos
(placas) não são capazes de informar sobre esses fatores imunológicos
relacionados com a patogenia.(96)
Anticorpo anti-GQ1b, reagindo cruzadamente com GT1a, está associado
com a SMF e suas variantes incompletas, oftalmoparesia sem ataxia ou ataxia sem
oftalmoparesia. (97-99) Também está associado a uma variante com envolvimento
do sistema nervoso central, encefalite de Bikcerstaff, caracterizada por
oftalmoparesia aguda, ataxia e alteração da consciência após síndrome
infecciosa.(100) Pacientes com fraqueza da musculatura faríngea-cervical-braquial,
variante focal da SGB, estão associados com anticorpos anti-GT1a.(101)
Esses padrões clínicos com envolvimento ocular, ataxia e bulbar são
justificados pela forte expressão dos gangliosídeos em determinados nervos. O
GQ1b é fortemente expressado nos nervos oculomotor, troclear e abducento,
assim como os fusos musculares.(76, 97, 102, 103) Já o GT1a é fortemente
expressado nos nervos vago e glossofaríngeo.(103)
Recentemente, demonstrou-se que um complexo de gangliosídeos
constituído por dois diferentes gangliosídeos pode ser o epítopo para a SGB e para
SMF.(104) O anticorpo não reage para cada gangliosídeo isoladamente, mas para
um novo epítopo formado por um complexo de ambos os gangliosídeos. A
presença de anticorpos contra o complexo GD1a/GD1b foi associado com
insuficiência ventilatória e déficit de nervos cranianos na SGB.(104) Anticorpos
anti-complexos GM1/GalNAC-GD1a foram associados à forma motora pura da
SGB.(105, 106) A formação de complexos de gangliosídeos proporciona uma nova

38
configuração na membrana plasmática, deixando-os mais acessíveis aos
autoanticorpos.
Os anticorpos anti-gangliosídeos na SGB são altamente variáveis com
relação à especificidade fina. O método ideal para determinar esta especificidade é
a cromatografia de capa fina, pouco usada na rotina clínica. Já o teste de ELISA,
comercialmente disponível, não é custo-efetivo porque, para determinar o perfil de
anticorpos, deve-se testar cada um dos anticorpos com um ensaio separado.(107)
3.8 Fatores de risco para a patogênese da Síndrome de Guillain-Barré
3.8.1 Infecção e mimetismo molecular
Aproximadamente 2/3 dos casos da SGB são precedidos de quadro
infeccioso. Entre os agentes infecciosos mais frequentes estão o Campilobacter
jejuni, CMV, vírus do Epstein-Barre, Mycoplasma pneumonie e influenza.(88, 108-
110)
Entre 12 a 60% dos casos de SGB apresentam sorologia e/ou cultura
positiva para C. jejuni. Essa bactéria é gram negativa, e é uma das principais
causadoras de diarreia aguda. A SGB, precedida por infecção por C. jejuni, está
associada à presença de anti-GM1, degeneração axonal, recuperação lenta e
incompleta.(22, 91, 111, 112) Não se detectaram anticorpos anti-GM1 em
pacientes com enterite por C. jejuni que não desenvolviam SGB.(113).
Há controvérsias se a infecção por C. jejuni pode provocar a variante
desmielinizante da SGB.(114) Alguns estudos demonstram que a infecção por C.
jejuni, apesar de estar fortemente associada à variante axonal, também precede a
variante desmielinizante.(14, 88, 115). Num outro estudo, pacientes com infecção
por C. jejuni e que desenvolveram a SGB, a eletroneuromiografia seriada revelou
que a SGB era exclusivamente axonal.(116) Alguns desses pacientes
apresentaram alterações eletrofisiológicas iniciais que simulavam desmielinização,
mas, na verdade, eram bloqueio da condução reversível detectados na fase
precoce da NAMA associada a anticorpos anti-GM1.(92, 117)

39
Anticorpos contra epítopos da membrana celular de certas bactérias têm
sido demonstrado, em estudos histopatológicos (118, 119) e em modelos animais
(120, 121), reagindo contra gangliosídeos do sistema nervoso periférico,
proporcionando evidência de que o mimetismo molecular é o responsável pela
patogênese da SGB.(79) Coelhos sensibilizados com capa lipossacarídica de C.
jejuni desenvolvem NAMA, similar aos coelhos sensibilizados com GM1. As
alterações patológicas do nervo periférico nesses animais são similares aos
indivíduos com SGB.(122)
A elucidação da participação do C. jejuni na patogênese da SGB é
corroborada com os detalhes do conhecimento da biossíntese e estrutura da
membrana celular deste agente infeccioso, isolado de pacientes com a neuropatia.
A variação na estrutura da membrana celular dessa bactéria é geneticamente
determinada.(123) Vários genes estão envolvidos na síntese de oligossacarídeos
da capa lipossacarídica do C jejuni agrupando-o em diferentes classes.(124) Uma
classe específica, entretanto, o gene sialiltranferase (cst-II), é essencial para a
síntese de gangliosídeos-like da capa lipossacarídica e está associada com o
desenvolvimento da SGB. A cst-II codifica uma enzima que transfere o ácido siálico
para a capa lipossacarídica. A cst-II sialiltransferese consiste em 291 aminoácidos,
e o 51 determina a atividade enzimática. A cst-II (Thr51) tem somente uma
atividade 2,3-sialiltransferase atividade (monofuncional) e produz GM1-like e
GD1a-like. Em contrapartida, a cst-II (Asn51) tem duas atividades 2-3 e a 2,8-
sialiltransferase (bifuncional), e elabora GT1a-like e GD1c-like mimetizando o
GQ1b. Em outras palavras, dependendo do gene que regula a capa lipossacarídica
da C. jejuni, o indivíduo infectado não tem a SGB, tem a variante NAMA ou a
SMF.(125)
O CMV é a segunda infecção mais freqüente na SGB, ocorrendo entre 11 a
15%.(109, 126, 127) Os pacientes com SGB associadas à CMV são jovens, com
evolução inicial grave, insuficiência respiratória, comprometimento de nervos
cranianos, déficit sensitivo intenso, variante desmielinizante e presença de
anticorpo anti-GM2.(88, 126) A presença de anti-GM2 está associada à infecção
por CMV sem ou com SGB.(19) Epítopos GQ1b e GM1-like da membrana celular
do Hemophilus influenza foram observados em pessoas com SFM e variante
axonal da SGB.(16, 128)

40
A infecção por M. pneumonie precede a SGB em 3 a 6% dos caso.(88, 109)
Os pacientes com SGB associada a M. pneumonie são jovens, mas não há outros
dados que diferenciem de outros casos da SGB. A presença de estruturas
similares ao GM1 no M. pneumonie aponta para o mimetismo molecular como
responsável pela patogênese.(129)
A presença de celularidade aumentada no líquor e a alta incidência de
insuficiência respiratória são dados comuns na SGB associada à infecção pelo
vírus de Epstein-Barr. Esse agente infeccioso provoca a SGB em 1% dos
casos.(88) A variante desmielinizante é a mais comum após essa infecção.(108) O
vírus da dengue pode determinar SGB, além de várias outras manifestações
neurológicas.(130) A dengue é causada por um arbovírus da família dos
Flaviviridae e é transmitido pelo mosquito Aedes egypti. Houve ressurgimento
desse vírus em vários países da América Latina, incluindo o Brasil, décadas após
ter sido eliminado.
3.8.2 Vacinas
Várias publicações têm documentado a ocorrêcia da SGB após vacinações,
mas a relação específica ainda é discutida.(131-137) O debate surgiu
principalmente após a observação de um aumento na incidência de SGB após
vacinas contra a gripe suína, em 1976, nos EUA. Na ocasião, após um surto de
gripe, em Nova Jersey, EUA, o governo promoveu uma campanha de imunização
que vacinou 40 milhões de americanos, temendo potenciais efeitos, semelhantes à
pandemia de gripe de 1918, quando morreram mais de 30 milhões de pessoas no
mundo. Porém, a campanha foi interrompida três meses após surgirem relatos da
SGB, até 6 semanas após a vacinação. O risco foi de 8,8 casos por milhão entre
as pessoas que foram vacinadas.(138, 139)
Em 2009, uma nova pandemia do vírus da gripe (H1N1), parcialmente de
origem suína, propagou-se pelo mundo e em resposta foi desenvolvida uma vacina
que vem sido utilizada internacionalmente. Mais uma vez o debate sobre o risco de
desenvolver doenças após a vacinação tornou-se pública.

41
Wise e colaboradores do CDC, EUA, observaram que a vigilância ativa entre
45 milhões americanos durante a epidemia de gripe 2009-2010, a incidência da
SGB durante os 42 dias após a vacina contra H1N1 era 57% mais frequente que
fora desta janela de tempo. A estimativa de excesso de casos da SGB associada à
vacina foi de menos de 1 (0,74 casos) por milhão de pessoas vacinadas.(140) Igual
a outras vacinações prévias (141) e dez vezes menos do que a campanha de
1979.
Numerosos estudos têm demonstrado a efetividade da vacina contra H1N1
em prevenir a infecção por influenza A (H1N1). Considerando que o excesso da
SGB após a vacinação é de menos de 1, este risco é pequeno comparando com a
redução da morbidade e mortalidade promovida pela vacinação.(140)
Não se sabe porque as novas vacinas contra influenza têm menos
associação com a SGB. Nachamki e colaboradores levantaram a hipótese que a
vacina de influenza utilizada em 1976, produzida em ovos de galinha, poderia ter
contido proteínas contaminadas de C. jejuni que mimetizam os gangliosídeos
humanos, ou que os componentes da vacina poderiam ter provocado a produção
de anticorpos antigangliosídeos em alguns indivíduos vacinados. Quando injetado
em camundongos, esta vacina, utilizada em 1976, não induziu a produção de
anticorpos anti-Campylobacter, mas promoveu a produção de anticorpos anti-GM1.
Entretanto, as vacinas contra influenza de 1991-1992 até 2004-2005 também o
fizeram.(142) Uma explicação possível da redução do risco persistente da SGB
após as novas vacinas contra influenza é o incremento das fases de filtragem e
purificação requeridas para a preparação dessas vacinas, com a finalidade de
minimizar as reações alérgicas do ovo ou de hipersensibilidade.(137) Outras
vacinas, por exemplo, contra hepatite, poliomielite e tétano, também estão
associadas a SGB.(135)
No Brasil, um estudo analisou o banco de dados do programa da vigilância
epidemiológica das paralisias agudas e flácidas (PAF) da Fundação Nacional de
Saúde, entre 1990 e 1996. De 3619 notificações de PAF, encontrou-se 1678 com
SGB, com incidência anual de 0,39-0,63 casos/100.000. Não se comprovou
variação sazonal nem relação com a vacina.(15)

42
3.8.3 História familiar
A SGB é considerada uma doença causada por fatores ambientais e
genéticos, mais do que uma doença por uma única herança mendeliana.
Entretanto, casos familiares têm sido relatados apoiando a participação genética na
patogenia da doença.(143-145) Também há relatos de casos familiares na
SMF.(146)
3.8.4 Imunossusceptibilidade genética
Considerando que o mimetismo molecular não é demonstrado em
aproximadamente 40% dos pacientes com SGB,(147) e nem todas cepas de C.
jejuni isoladas contêm epítopos similar ao GM1 e induzem anticorpos anti-
gangliosídeos,(22) além do que somente 0,01% dos pacientes com enterite por C.
jejuni apresentam a SGB(148), suportam a hipótese de fatores individuais
influenciadores da SGB após a infecção. Apesar disso, poucos estudos foram
conduzidos para identificar um potencial fator de risco do hospedeiro associado à
susceptibilidade de desenvolver a SGB. Estudos avaliando polimorfismos em HLA
classe II, receptores CD14 e TLR4 não identificaram associação com a SGB.(149,
150) Associação da gravidade da doença foi demonstrada com genes que
codificam para manose-lectina, FcR, metaloproteinases de matriz 9, e fator de
necrose tumoral α.(23, 151, 152) Esses estudos requerem confirmação num maior
número de pessoas afetadas, e precisa ser demonstrado um efeito funcional
destas associações genéticas.
3.8.5 Polimorfismo dos genes para o receptor do domínio Fc de
imunoglobulina IgG
3.8.5.1 Receptor do domínio Fc de IgG (FcR)
Os FcRs humanos correspondem a um grupo de glicoproteínas de superfície

43
que pertencem à superfamília das imunoglobulinas (IgG). São os receptores para
os domínios Fc de IgG, presentes nas membranas celulares de células do sistema
imune, mas também encontrados como moléculas solúveis. Os FcRs são definidos
pela sua especificidade para os isotipos de imunoglobulina: FcR ligam IgG, FcαR
ligam IgA, FcεR ligam IgE, FcμR ligam IgM, e FcδR ligam IgD.(153)
Os FcR têm importante função de modular a resposta imune. São
considerados cruciais na ligação entre a imunidade celular e humoral. A interação
com FcR confere aos anticorpos função efetora potente. Inúmeras evidências
sugerem relação entre o polimorfismo dos genes para o FcR (FCGR) e
susceptibilidade a doenças.(26, 154, 155)
3.8.5.2 Estrutura dos FcR
A complexidade da família dos FcR é espelhada pela presença de quatro
diferentes subclasses de IgG no ser humano (IgG1 a IgG4), que se ligam com
grande afinidade e especificidade para diferentes FcRs. Em humanos, IgG1 e IgG3
são mais pró-inflamatórias. Os FcRs são classificados pela sua afinidade a
diferentes subclasses de IgG e pela sua ação ativadora ou inibitória.(156) Os
diferentes tipos de receptores FcR são mostrados na Figura 3.
Figura 3. Os diversos tipos de FcR presentes nas células da resposta imune.
Adaptado de Horgarth M P et al. (157)
Small chemical entities
Chemical compounds that are
less than 500 Da in size.
Immunoreceptor
tyrosine-based activation
motif
(ITAM). An amino acid
sequence (12–14 amino
acids long) containing two
precisely spaced tyrosine
residues that, upon
phosphorylation
by the SRC family of tyrosine
kinases and following receptor
aggregation, initiate an
intracellular signalling cascade
that leads to cell activation.
This motif is typically found
in small, often dimeric
membrane proteins that are
non-covalently associated with
major immunoreceptors such
as Fc receptors and antigen
receptors.
Immunoreceptor tyrosine-
based inhibition motif
(ITIM). An amino acid
sequence, distinct from
an immunoreceptor
tyrosine-based activation
motif, that is present within
the cytoplasmic tail of some
membrane receptors and
functions solely in the
transduction of inhibitory
signals. ITIMs are normally
composed of a six-amino-acid
sequence containing a tyrosine
residue, a hydrophobic residue
two amino acids upstream of
the tyrosine residue and a
leucine residue three amino
acids downstream of the
tyrosine residue. The inhibitory
function of the ITIM is activated
by phosphorylation of the
tyrosine residue, which
is mediated by the SRC family
of kinases. This leads to the
recruitment and activation
of tyrosine and/or lipid
phosphatases including SHP
(SH2 domain-containing
protein tyrosine phosphatase)
and SHIP (SH2 domain-
containing inositol-5-
phosphatase), respectively.
The ITIM of the Fc receptor
Fc RIIb is the archetypal ITIM.
small chemical entities
(FIG. 1)
REFS 8,9
The human leukocyte FcRs
(FIG. 1; TABLES 1,2)
FCGR1A
FCGR2A
FCGR2B
FCGR2C
FCGR3A
FCGR3B
FCGR2C
FCGR2A FCGR2B
FCGR2B (TABLES 1,2)
(FIG. 2)
immunoreceptor tyrosine-based activation motif
immunoreceptor tyrosine-based inhibition motif
Nature Reviews | Drug Discovery
FcγRI FcγRI FcγRIFcγRIIa FcγRIIb FcγRIIIa FcγRIIIbFcγRIIc
ITAM ITIMCommon FcRγ-chain
γ subunit
Figure 1 | Diagrammatic representation of the human leukocyte Fc receptors. The Fc receptors (FcRs) consist of an immunoglobulin-binding polypeptide chain containing two immunoglobulin-like domains that form the extracellular region. Fc RI is an exception as it has three such domains, the first two of which are related to those of the other Fc Rs and Fc RI, the third domain is unique. Fc R, which also has two extracellular domains, is more closely related to the leukocyte immunoglobulin-like receptors. Signal transduction by the activating FcRs occurs via immunoreceptor tyrosine-based activation motif (ITAM)-dependent mechanisms, usually via a non-covalent association of the immunoglobulin-binding chain with a dimeric, ITAM-containing subunit, typically the ‘common’ FcR -chain. In mast cells, Fc RI also associates with a multimembrane-spanning and signal-amplifying -subunit (the same
applies to Fc RIIIa; not shown). Fc RIIa, Fc RIIb and Fc RIIc are unusual as their signalling motifs are contained in the cytoplasmic tail of the ligand-binding chain. This comprises an ITAM for the activating Fc RIIa or Fc RIIc, and an immunoreceptor tyrosine-based inhibition motif (ITIM) for the inhibitory Fc RIIb.
REVIEWS
312 www.nature.com/reviews/drugdisc
© 2012 Macmillan Publishers Limited. All rights reserved
Neutrófilo
Macrófago
Monócito
NK
Monócito
Macrófago
NK
Monócito
Macrófago

44
A família dos FcR é formada por 3 classes principais: FcRI (CD64), FcRII
(CD32), FcRIII (CD16), além do receptor neonatal para IgG (FcRn). Os FcRs
ativadores incluem o receptor de alta afinidade FcRI e a família de receptores de
baixa afinidade, FcRIIa, FcRIIc, FcRIIIa e FcRIIIb em humanos. O FcRIIb é o único
FcR que tem função inibitória. Todos eles caracterizam-se por terem dois ou três
domínios extracelulares apresentando uma homologia aos das imunoglobulinas,
domínios transmembranas e domínios citoplasmáticos. Os FcRs são expressos
em células do sistema hematopoético. Células do sistema imune inato, tais como
monócitos, macrófagos, células dendríticas, basófilos e mastócitos expressam
FcRs ativadores e inibidores. Células matadoras naturais expressam somente
receptor ativador FcRIII e as células B somente expressam o receptor inibitório
FcRIIB. (155)
O FcRIIb é o único FcR expresso em células B, além de outras células,
incluindo células dendríticas, macrófagos, neutrófilos, células da mast e basófilos.
Quando expresso nessas células, o FcRIIb inibe a produção de anticorpos, a
fagocitose e a liberação de citocinas.(158, 159)
Esses receptores são codificados por oito genes distintos que estão
localizados no braço longo do cromossoma 1. O FcRI é uma glicoproteína de 70-
Kda que se liga à IgG monomérica com alta afinidade (108 - 109M-1). Ele possui
uma distribuição mais restrita do que os outros receptores, presente quase
exclusivamente nos fagócitos mononucleares. Os monócitos têm 15 a 40 mil
receptores por célula, e os macrófagos têm 50 mil por célula. Essas células, após
serem incubadas com IFN-γ, sofrem um incremento de 5 a 10 vezes no número de
receptores. Três genes são responsáveis por quatro transcritos: FcRIa1, FcRIb1,
FcRIb2, FcRIc. Transcritos FcRIb1 e FcRIc contêm códon de parada em suas
regiões extracelular e podem codificar receptores solúveis (s FcγR).
O receptor FcRII, glicoproteína de 40-Kda, encontra-se amplamente
distribuído em várias células (polimorfonucleares, plaquetas, linfócitos granular,
natural kiler, linfócitos B, monócitos e macrófagos) com baixa afinidade (<107 M-1)
às subclasses de imunoglobulinas. O número de receptores é similar em todas as
células, e sua expressão não se modifica com citocinas. Não se liga a
imunoglobulina monomérica, mas reage com complexos imune de IgG e opsoniza

45
partícula. Três genes (2A, B e C) geram seis transcritos (FcγRIIa1, 2; IIb1, 2 e 3;
IIc), onde o FcγRIIa2 falta informação para a região transmembrana.
O domínio citoplasmático dos FcRI e FcRII contém uma sequência estrutural
denominada de motivo de ativação de imuno-receptor baseada em tirosina (ITAM:
imunorecpetor tyrosine-based activation motifs) que é responsável por transmitir
para o interior da célula o sinal da ligação da IgG ao seu receptor.(160)
O FcRIII é extremamente glicosilado e manifesta-se como uma molécula
com um peso molecular que varia de 50-70 KDa. Esses receptores ligam IgG na
forma de imunocomplexos. Dois genes distintos e altamente homólogos codificam
o FcγRIII (IIIa e b) e dois transcritos são descritos (FcγRIIIa e b). O FcRIIIa
expressa-se em macrófagos, polimorfonucleares, células matadoras naturais, e em
algumas células T, além de interagir com IgG complexada e monomérica. O FcRIII
GPI-ligado expressa-se seletivamente em neutrófilos e possui uma baixa afinidade
pela IgG (<107 M-1). As sequências de ITAM, transdutoras de sinal intracelular,
estão presentes apenas no FcRIIIa e não no FcRIIIb. (155)
3.8.5.3 Função do FcR
O equilíbrio entre vias de sinalização ativadora e inibitória gera uma boa
resposta imunológica. Como efeito, uma expressão aberrante ou a presença de
variantes alélicas de FCGR com funcionalidade alterada foram observadas em
doenças autoimunes, como lúpus eritematoso sistêmico e artrite reumatoide, além
de doenças infecciosas e neoplasias.(155)
A IgG tem importante função de defesa contra patógenos. Indivíduos com
hipogamaglobulinemia apresentam aumento da susceptibilidade à infecção. A
interação entre o patógeno ligado a IgG e a ativação direta do FcR promove a
citotoxicidade celular dependente de anticorpo, degranulação de células
citotóxicas, fagocitose, liberação de citocinas e outros mediadores
inflamatórios.(155)
A ativação dos FcRs por imuno complexos resulta em fosforilação de
sinalizadores, ITAM, presentes no domínio citoplasmático dos receptores FcRIIa e

46
FcRIIc ou na cadeia γ dos receptores FcRI e FcRIIIa. A fosforilação desses ITAM
resulta em ativação de moleculas SYK e iniciação de sinais em cascata.(154)
É no domínio IgG-like proximal à membrana presentes nos receptores onde
existem segmentos de ligação com IgG. Já o domínio distal está relacionado com
optimização da interação entre ligante e receptor. A presença de um terceiro
domínio provavelmente altera a alta afinidade para FcRIa. O FcRII contém ao
menos três “spots” relacionados com a ligação com IgG: domínio aminoácidos
nas posições 109-116, 129-135 e 154-161. Mutações em cada uma dessas áreas
compromete a afinidade, variando até a falta total de ligação (154-161). Neste
último caso pode ser atribuído a vários resíduos, entre os quais está o aminoácido
131. O aminoácido desta posição pode ser arginina (R) ou histidina (H), devido ao
polimorfismo geneticamente determinado.
3.8.5.4 Polimorfismo do FCGR em doenças infecciosas e autoimunes
A capacidade para o FcR iniciar a resposta específica biológica é
heterogênea. FcRI, por exemplo, é altamente eficiente para promover
apresentação de antígeno in vitro e in vivo. Já o FcRII é mais eficiente em iniciar
fagocitose. FcRI, IIa e IIIa são todos capazes de iniciar citotoxicidade, porém o
FcRIIIb pode iniciar fagocitose de pequenas partículas, mas não citotoxicidade.
Três subclasses de FCRG (FCGR2A, FCGR3A e FCGR3B) exibem polimorfismos
bialélicos que afeta a afinidade para as subclasses de IgG e a eficiência da
resposta imune celular. A tabela 1 mostra os receptores FCRIIa e FCRIIIa e suas
variantes alélicas.
Tabela 1. Receptores FCRIIa e FCRIIIa e variantes alélicas.
Gene Produto Expresso Polimorfismo Variantes
alélicas
Afinidade
IgG
Ação
FCGR2A FcRIIa (CD32) Neutrofilos,
monócitos,
macrófagos,
células dentritas,
plaquetas
rs1801274:
histidina (H) por
argninina (R),
posição 131
FGCR2A-H-
131
FCGR2A-R-
131
IgG2 e
IgG1 -
FGCR2A-H-
131
Fagocitose;
citotoxicidade
célula; produção
citocinas
FCGR3A FcRIIIa (CD16) células natural
killer e
macrófagos
rs396991:valina
(V) por fenilalanina
(F), posição 158
FCGR3A-V-
158
FCGR3A-P-
158
IgG2 e
IgG1
FGCR2A-V-
131
Fagocitose;
citotoxicidade
célula; produção
citocinas

47
No FCGR2A o polimorfismo é atribuído a uma única diferença de
nucleotídeo na base 494 (A ou G) que resulta na expressão de dois alelos com
diferentes aminoácidos na posição 131. No FCGR2A R131 (alelo raro), com o
códon CGT, codifica o aminoácido arginina (R); o FCGR2A H 131 (alelo normal,
selvagem) expressa o aminoácido histidina (H), codificado pelo códon CAT.(161) O
FCGR H131 se liga com maior afinidade a IgG3, sendo o único que se liga
eficientemente a IgG2.(162)
A presença de valina ou fenilalanina na posição 158 do FCGR3A (158V
versus 158F) determina afinidade para receptor de IgG1, IgG3 e IgG4; a primeira
exibe alta afinidade para estas subclasses e é o alelo normal. A atividade das
células NK induzidas por IgG é aumentada entre as pessoas com FCGR3A-
V/V158. Ambos FCGR2A e FCGR3A são expressados em macrófagos.
FCGR3B unicamente é expresso em neutrófilos, e apresenta polimorfismo
NA1-NA2. FCGR3B-NA1 (alelo normal, selvagem) é mais eficiente em ligar
complexos imunes do tipo IgG1 e IgG3 do que FCGR3B-NA2. Quanto maior a
afinidade do receptor pelo ligante, mais eficiente é a função efetora dos leucócitos,
incluindo a fagocitose, a citotoxicidade celular e a produção de citocinas. O
polimorfismo do FCGR pode influenciar o vigor da resposta inflamatória e pode
contribuir para a diferença interindividual de susceptibilidade a doenças
inflamatórias e infecciosas.
3.8.5.5 Doenças infecciosas
A ineficiência da fagocitose do complexo IgG2-bactéria opsonizada
incrementa o risco de infecções por bactérias encapsuladas. Pacientes com
deficiência do componente tardio do complemento não são capazes de lisar
bactérias gram negativa através da formação do complexo ataque de membrana
(MAC), e o paciente só depende da fagocitose para a defesa. Foram relatados
aumento da incidência de doença meningocócica em pacientes com deficiência do
componente tardio do complemento e com genótipo FCGR2AR/R131- FCGR3B-
NA2/2. (163-165) Indivíduos com genótipo FCGR2AH/H131 têm menor frequência
de infecção por S.pneumoniae.(166)

48
A periodontite do adulto, uma das principais causas de perda de dente no
adulto, é uma doença infecciosa resultante do efeito direto da bactéria e do
processo inflamatório. A persistência ou a recidiva da doença após o tratamento
está relacionada, além da infecção, a fatores de risco tais como susceptibilidade
individual. Vários estudos encontraram associação entre polimorfismo do FCGR2A
e FCGR3B.(167-170) Os indivíduos com alelo FCGR2A H131 ou com genótipo
FCGR2A-H/H131 apresentam a periodontite com maior frequência ou
agressividade comparado com controles.(168, 169, 171)
Os leucócitos polimorfonucleares com o genótipo H/H131 ligam-se de forma
eficiente com IgG2 e exibem maior capacidade fagocítica do que os leucócitos
expressando o genótipo R/R131. Na verdade, tem sido proposto que a alta
afinidade da FCGR2A -H131 com IgG pode contribuir para liberação de citocina
pró-inflamatória por monócitos / macrófagos, possivelmente levando a um risco
aumentado para a periodontite crônica marginal.(169) O alelo FCGR3B-NA2
também tem sido associado com periodontite.(170) Os leucócitos que expressam
FCGR3B-NA2 são menos eficientes para fagocitar complexos imunes de IgG1 e
IgG3 com Porphyromonas gingivalis, sugerindo redução do clearance de bactérias
patogênicas.(167) Os FCGR2A e FCGR3B atuam sinergicamente na estimulação
de neutrófilos. Siqueira e colaboradores, na cidade do Rio de Janeiro, observaram
que a combinação do genótipos FCGR2A-H131 e FCGR3B-NA2 estava associada
com periodontite apical pós-tratamento,(171), mas não detectaram associação com
o polimorfismo do gene FCGR3A.(172)
Os FcR são também importantes em infecções virais. A dengue
hemorrágica, forma grave da dengue, ocorre comumente numa infecção
secundária por um subtipo de vírus diferente do que causou a infecção primária.
Os anticorpos preexistentes, obtidos quando da infecção prévia por outro tipo viral,
não neutralizam o segundo vírus infectante de tipo diferente e amplificam a
infecção, facilitando ao novo tipo infectante a penetração em macrófagos. Os vírus
utilizam a porção Fc dos anticorpos ligados ao envelope para a ligação com os
receptores de membrana FcR, presentes na membrana celular macrofágica(18).
Trata-se do fenômeno de facilitação por anticorpos da penetração viral em
macrófagos (antibody dependent enhancement – ADE).(173) O genótipo FCGR2A-
R/R131 está associado a menor risco de dengue hemorrágica em crianças

49
vietinamitas(174), já o genótipo FCGR2A-H/H131 está relacionado com a dengue
hemorrágica em Cuba.(175)
A associação de infecção parasitária com FCGR tem sido descrita. O FcRIIa
participa ativamente na elicitação da resposta efetora de monócitos e macrófagos.
Este receptor tem baixa afinidade para se ligar a IgG, mas se liga eficientemente
com complexos imunes.(176) Os primeiros estudos mostravam que pacientes
infectados pelo Plasmodium falcipurum apresentavam a forma mais grave da
malária na presença do genótipo FCGR2A H131/H131, e que o alelo FCGR2A
R131 era um fator protetor da doença.(177-179) Entretanto, estudos recentes
demontram o contrário, o genótipo FCGR2A R131/131 está associado com formas
severas da malária. (180, 181) Indivíduos da etnia Fulani que vivem em Burkina
Faso e Mali são menos acometidos de Malária do que outras etnias. Esta proteção
está associada à alta prevalência do genótipo FCGR2A H131/H131 e do alelo
FCGR2A H131.(181) O FcRIIa H131 se liga com maior afinidade a IgG3 e o único
que se liga eficientemente a IgG2. Já o FcRIIaR131 tem baixa afinidade. A IgG2 é
pouca ativadora do padrão clássico do complemento, ja o FcRIIa-H131 é essencial
para a manipulaçao dos complexos imunes. Esta discrepância de resultados pode
ser explicado por diferenças genéticas entre populações, diferenças entre as
interações genética-ambiental e por diferentes padrões de transmissão da malária
em distintas áreas estudadas.(180)
O FcR também participa no processo de internalização de outros parasitas,
como a Leishmania sp.(182). As Leishmanioses são um complexo de espécies que
podem causar doenças com formas clínicas que variam de leishmaniose
tegumentar a leishmaniose visceral. As leishmanioses são doenças complexas e
fatores genéticos e ambientais determinam se um paciente desenvolverá formas
leves ou graves desta doença. Altos níveis de anticorpos são observados na
leishmaniose visceral (LV), mas os anticorpos não são protetores e há indicação
que podem estar envolvidos na patogênese, facilitando a internalização da
Leishmania via receptores de Fc.(183) Ainda, outros estudos em animais sugerem
que IgG em associação com FcR participam na patogênese da doença e que o
fenômeno de facilitação por anticorpos da penetração parasitária em macrófagos
(antibody dependent enhancement – ADE) influencia a produção de citocinas,

50
promovendo a produção de IL-10 e inibindo a resposta celular, desta maneira
evitando a erradicação do patógeno intracelular.(28)
Contrariamente, Woelbing e colaboradores descreveram, em modelos
animais, que esses receptores são importantes como protetor contra a
leishmaniose tegumentar americana. Nesse estudo, a IgG opsonizada pela
leishmania ligada ao FcR das células dentríticas induz à produção de IL-12.
Consequentemente o animal infectado devenvolve pequenas lesões e há redução
da carga parasitária.(182)
Um estudo recente, realizado por Oliveira e colaboradores, em Góias, Brasil,
não identificou diferenças entre o genótipo e a distribuição alélica do FCGR2A
entre pacientes com leishmaniose tegumentar americana comparando com
controles.(29) Entretanto, os autores comentam que os seus resultados não podem
ser generalizados para outras populações, uma vez que existe diversidade
genética entre diferentes grupos étnicos relacionados com o gene que codifica o
FCGR2A e diferentes genes, tais como o gene do receptor de quimiocina CXCRI e
genes de citocinas, que foram associados à progressão da leishmaniose.(184, 185)
3.8.5.6 Doenças autoimunes
O Lupus Eritematoso Sistêmico (LES) é caracterizado por produção de
anticorpos contra constituintes nucleares e desenvolvimento de inflamação em
múltiplos órgãos. Nessa doença, a função imperfeita do FcR afeta o clearance de
complexos imunes que se depositam em vários tecidos com consequente lesão
tissular. Os alelos FCGR2A-R131, FCGR2B 232 treonina/treonina e FCGR3A-F158
foram associados à susceptibilidade ao LES.(186-188)
Autoanticorpos diretamente contra o domínio Fc da IgG ocorre na artrite
reumatoide (fator reumatoide) promovendo a destruição do osso e da cartilagem. O
fenótipo FCGRIIIA-F158 é considerado um fator de risco para o desenvolvimento
da artrite reumatoide, possivelmente pela baixa afinidade deste receptor com a IgG
e ineficiência do clearence de complexos imunes.(189)

51
A púrpura trombocítica idiopática (PTI) é uma doença autoimune causada
por destruição das plaquetas por autoanticorpos. As plaquetas se ligam a IgG e
são fagocitadas por macrófagos no fígado e baço, mediadas pela interação com
FcR. Os pacientes com PTI mais frequentemente apresentam os fenótipos
FCGR3B-NA1 homozigotos, FCGR3A-V158/F158 heterozigotos e FCGR2A-H131.
A maior interação do FCGR3B-NA1 com os complexos imunes IgG1 e IgG3 pode
contribuir para o aumento do clearence das plaquetas e redução do seu número
nesses indivíduos.(190)
A patogênese da trombocitopenia induzida por heparina está relacionada
com anticorpos que facilitam o clearence das plaquetas através do FcR dos
macrófagos. A trombose pode ocorrer na trombocitopenia induzida por heparina
quando o clearence das plaquetas é ineficiente e isto tem sido relatado na
presenca do alelo FCGR2A-R131, que tem baixa afinidade por IgG.(191)
Os genótipos FCGR2A-R131/R131, FCGR3A-F158/F158 e FCGR3B-
NA1/NA1 são fatores de risco para granulomatose de Wegner. Trata-se de uma
vasculite sistêmica que é causada por anticorpos anticitoplasma de neutrófilos. A
ocupação do FcR por autoanticorpo induz a liberação de IL-8, fator quimiotático
para neutrófilos.(190)
Em estudo numa população holandesa foi observado que o genótipo
FCGR2A-R/R131 é um fator de risco para miastenia gravis.(192) Esse genótipo
resulta em proteína com uma interação relativamente ineficiente com complexos
imunes IgG2/IgG3.(193)
Na Esclerose Múltipla, uma doença desmielinizante e inflamatória do
sistema nervoso central, os FcR não correlacionavam com a susceptibilidade de
desenvolver a doença. Entretanto, o genótipo FCGR3B-NA1/NA1 estava associado
com uma forma leve da doença. Esse receptor tem alta afinidade por IgG1 e IgG3,
sendo importante no clearance de autoanticorpos e de complexos imunes
circulantes.(194)
3.8.5.7 Síndrome de Guillain-Barré
Várias são as evidências do papel dos FcRs na patogênese da SGB. Os

52
macrófagos são ativados após a ocupação dos seus FcR por anticorpos anti-
gangliosídeos, consequentemente ocorre destruição da mielina e/ou axônio. Outra
evidência é a melhora clínica com o tratamento da doença com imunoglobulina
humana endovenosa. Esse medicamento age através do bloqueio do FcR.(34, 195)
O FcR tem sido demonstrado em células de Schwann, células perineurais, células
endoteliais e macrófagos endoneurais, também em nervos fetais de
aproximadamente 10 semanas de gestação, indicando que esses receptores são
componentes inatos do sistema nervoso periférico.(196)
Embora os anticorpos participem na patogênese da SGB, estudos com
diferentes FcR são escassos nessa doença. Além disso, não está claro se
indivíduos portadores de diferentes alelos FcR têm maior risco para desenvolverem
a SGB ou tê-la de forma mais grave. Na Holanda, 31 pacientes com SGB
apresentaram aumento da frequência de alelos FCGR2A-H131 comparado com
187 doadores. O homozigoto FCGR2A-H131 apresentou maior risco de
desenvolver a forma mais severa da doença.(197) Em outro estudo, realizado na
Noruega, foram avaliados os mesmos polimorfismos, FCGR2A, FCGR3A e
FCGR3B, em 62 pacientes com SGB e 89 controles sãos. Não foi encontrada
diferença significativa entre a genotipagem e alelos dos pacientes e controles.
Entretanto, estratificando a doença em severa e leve, os pacientes homozigotos
para o alelo FCGR3B-NA1 apresentavam significativamente a doença numa forma
leve.(25)
van Sorge e colaboradores, avaliando coorte de pacientes da Holanda e
Inglaterra, também observaram associação da gravidade com o FCGR3B: quanto
mais frequente a presença do alelo FCGR3B-NA2 mais grave era a doença. Foi
levantada a hipótese que o FCGR3B-NA2 diminui o clearence de complexos
imunes, aumentando assim as chances de respostas inflamatórias localizadas.(23)
Sinha et al, demonstraram que o genótipo FCGR2A-H/H131 estava
associado com um risco 10x maior de desenvolver a SGB na Índia. Também,
nesse estudo, os autores identificaram maior freqüência da SGB em indivíduos
com o genótipo FCGR3A-V/V158.(24) Ambos receptores, FCGR2A-H/H131 e
FCGR3A-V/V158, são expressos em macrófagos, que por sua vez participam
ativamente na patogênese da SGB.(198, 199) A alta afinidade de ligação desses
receptores influencia no vigor da resposta inflamatória ao aumentar a eficiência das

53
funções efetoras dos leucócitos tais como fagocitose, citotoxicidade celular e
produção de citocinas. Provavelmente, a acentuada resposta imune gerada pelos
macrófagos em indivíduos homozigotos para receptores de alta afinidade
predispõe a doenças autoimunes como a SGB. Neste trabalho da Índia, não se
demonstrou associação do FCGR3B N/NA2 com a gravidade da doença. Para os
autores, a relevância desta associação é questionável uma vez que o FCGR3B só
é expressado em neutrófilos e a participação dos neutrófilos na patogênese da
SGB está para ser demonstrada.(24)
Assim, é notório que são necessários estudos para compreender a
repercussão do FCGR na patogênese da SGB em grandes amostras, de diferentes
populações e etnias.
3.9 Tratamento
Apesar dos avanços da medicina, 5% dos pacientes com SGB falecem por
complicações, tais como septicemia, embolia pulmonar, assistolia relacionada a
disautonomia.(3) É necessário cuidado multidisciplinar para prevenir e tratar as
complicações potencialmente fatais. Insuficiência respiratória ocorre em 25% dos
casos. Monitorização regular da função pulmonar, incluindo medidas da
capacidade vital forçada, e transferência precoce para uma unidade intensiva são
fundamentais. A entubação deve ser realizada antes que ocorra a fadiga ou uma
parada respiratória, ou seja, deve ser preventiva. Pacientes com fraqueza bulbar
grave necessitam de entubação, mesmo que a capacidade vital e as trocas
gasosas estejam relativamente normais.(1)
A deglutição deve ser avaliada e a passagem de tubo nasogástrico deve ser
colocada nos pacientes com risco de broncoaspiração. Todos os pacientes devem
ser monitorados para possíveis arritmias cardíacas, hipertensão ou hipotensão
arterial. O uso de heparina subcutânea e meia de compressão graduada estão
indicadas em pacientes não ambulantes para evitar trombose venosa profunda.
Outras complicações, tais como, dor, úlcera de córnea por paralisia facial, úlcera
de decúbito, retenção de urina, disfunção gastrointestinal, também devem ser
prevenidas e manejadas adequadamente. O reconhecimento e tratamento da dor,

54
fisioterapia e suporte psicológico são essenciais.(1, 8) A dor, em forma de
parestesia, radiculopatia, síndrome meningeia, musculoesquelética, artralgia, pode
anteceder a fraqueza muscular e pode persistir em um terço dos pacientes um ano
após o início da doença.(49)
A plasmaférese foi o primeiro tratamento efetivo para auxiliar a recuperação
do paciente com SGB, notadamente quando iniciado nas duas primeiras semanas.
Ela remove autoanticorpos, citocina e complemento. O regime de tratamento
recomendado são 5 trocas de plasma num período de 2 semanas.(33, 200)
O tratamento com IgEV é mais efetivo do que a plasmaférese.(34, 201)
Acredita-se que a IgEV atue através de bloqueio dos FcyR, neutralização de
anticorpos patogênicos e da ativação do complemento e regulação das células
T.(202) Ela tornou-se tratamento de escolha na maioria dos centros médicos
devido à sua maior facilidade para administração. A dose recomendada é de 2g por
Kg de peso, divididos em cinco dias consecutivos.(34)
O uso de corticoide, oral ou intravenoso, não é benéfico para os pacientes
com a SGB.(203). A combinação de imunoglobulina e metilprednisonola
intravenosa não é mais efetivo do que a imunoglobulina isoladamente.(204) Na
SGB, a lesão neuronal ocorre por disfunção dos canais de sódio, nos nodos de
Ranvier, por anticorpos anti-gangliosídeos e depósito de complemento, sem
infiltração de macrófagos. Essas células são observadas na fase de
recuperação.(80) Portanto, a ação do corticoide de inibir a proliferação celular não
terá benefício na SGB, uma vez que a lesão já ocorreu.
3.10 Expressão global de genes na SGB
As evidências atuais são convicentes que a SGB é autoimune, causada por
resposta imune aberrante contra o nervo periférico induzida por uma infecção, além
de determinados fatores individuais de susceptibilidade. Entretanto, a SGB é
heterogênea, com distintas variantes clínicas e patológicas refletindo diferentes
mecanismos da lesão nervosa. As alterações patológicas principais são:
inflamação, degeneração axonal e desmielinização. O mecanismo
imunopatogênico não está totalmente compreendido.(3)

55
Em resumo ao que apresentamos anteriormente: a variante axonal é a
melhor estudada. Através de mimetismo molecular entre o envoltório
lipossacarídeo do C. jejuni e os gangliosídeos do nervo periférico, há produção de
anticorpos e depósito de complemento no nodo de Ranvier. O alvo do ataque são
os canais de sódio voltagem dependente, altamente expressos nessa região. Os
indivíduos desenvolvem uma falha da condução motora, caracterizado
eletrofisiologicamente por bloqueio da condução, seguido de desaparecimento
desses canais, formação de poros na membrana, deslocamento da mielina
paranodal e degeneração axonal. A ativação do complemento aparece
precocemente e a invasão de macrófagos só é observada dentro da primeira
semana após lesão neuronal mediada pelo complemento. Eventualmente a falha
de condução motora é transitória, sem degeneração axonal. Nessa variante,
predomina o mecanismo humoral (Figura 4). (205)
Na variante clássica, são encontrados infiltrados inflamatórios, células T e
macrófagos. O alvo do sistema imune é a mielina resultando em desmielinização
focal. Em paralelo, ocorre produção de citocinas e de auto-anticorpos. Há também
ativação precoce do sistema do complemento. Portanto, tanto o mecanismo celular
quanto humoral estão envolvidos nessa variante. Outra evidência da participação
do sistema humoral é a melhora com tratamento de plasmaférese ou
imunoglobulina humana endovenosa.(66)
Considerando que apenas 0,01% dos pacientes com enterite por C. jejuni
desenvolve a SGB, é provável que fatores individuais influenciem a
susceptibilidade de desenvolver a síndrome ou modifiquem o curso da doença.
Entretanto, várias tentativas de identificar fatores de susceptibilidade
imunogenética na SGB foram mal sucedidas.(1) Diante do exposto, o mecanismo
imunopatogênico, avaliado por fluidos biológicos, não está totalmente
compreendido, portanto, para tal, novas abordagens devem ser utilizadas.
O transcriptoma é o conjunto completo de transcritos (RNAs mensageiros,
RNAs ribossômicos, RNAs transportadores e os microsRNAs) em uma célula. O
perfil do transcriptoma é dado em tipo e em quantidade e varia segundo o estado
fisiológico, diferentes estímulos ou doença do indivíduo. Os avanços recentes na
tecnologia de sequenciamento tornam possíveis obter informações sobre os
transcriptomas, que podem ser instrutivas no que diz respeito à forma como as

56
células repondem aos sinais e outros estímulos ambientais ou quando ela adquire
fenótipo aberrante. Todas as células de um organismo individual têm um genótipo
praticamente idêntico, mas os transcriptomas individuais refletem a expressão de
um subconjunto de genes, que é determinado pelo seu estado epigenético. A
análise deve ser realizada em um única célula, já que cada célula tem seus
transcriptomas exclusivos.(30, 206)
Várias tecnologias foram desenvolvidas para caracterizar o transcriptoma de
uma população de células, incluindo métodos de análise em grande escala como
SAGE (serial analysis of gene expression) e os microarrays ou microarranjos
(microarranjos de cDNA, os microarranjos de oligos e os DNA-chips).
Recentemente, uma nova ferramenta foi desenvolvida, chamada de RNA-seq, para
sequenciamento de cDNA por NGS (sequenciadores de próxima geração, next
generation sequence). Essa técnica permite a quantificação dos níveis de
expressão gênica, mesmo em transcritos que possuem níveis mais baixos de
expressão devido a sua alta sensibilidade. Também permite identicar exons e
introns, mapear seus limites e identificar porções 5’e 3’ dos genes. É útil para
descobrir novas transcrições, identificar mutações, splicing alternativos.(207)
São escassos os trabalhos que determinam o modelo molecular para SGB.
Um estudo, em Taiwan, analisou a expressão genética, através de microarranjos,
de sete pacientes com SGB e sete controles. Duzentos e cinquenta e seis genes e
18 redes genéticas estavam associadas com a SGB. Entre esses genes, quatro
eram os mais expressos (FOS, PTGS2, HMGB2 e MMP9). Os genes das funções
biológicas mais alteradas significativamente estavam envolvidas em resposta
inflamatória, doença infecciosa e doença respiratória. Já os mecanismos
moleculares implicados na SGB foram morte celular, desenvolvimento celular e
movimento celular. A análise do padrão canônico apresentou que GnRH, hormônio
liberador da gonadotrofina, e o sinal ERK/MAPK eram os padrões mais
significantes e aumentados na SGB. (208)
Nós decidimos estudar os possíveis eventos moleculares responsáveis pela
evolução da SGB, tendo como pressuposto que seria possível trazer mais
conhecimento sobre a interação entre os processos intrínsecos celular e estímulos
extrínsecos e compreender a patogênese dessa doença.

57
Figura 4: esquema da resposta imunológica, fase efetora, na SGB. Mimetismo molecular entre
gangliosídeos compartilhados entre o C jejuni e o nervo periférico. Os anticorpos atuam através da
citotoxicidade celular dependente do ac, por ativação do complemento (formação de poros), por
bloqueio da condução nervosa.
Músculo
Lesão da mielina - Clássica
Lesão do axônio - AMAN ou AMANS
Complemento e macrófago
B
C.jejuni - Penner 19
Ac. Anti-
GM1 IgG Epítopo
GM1
T
Complemento
Mácrofago
C5b-9 TNF-alfa
IFN-gama
IL-4
IL-6

58
4. PACIENTES E METODOLOGIA
4.1. Desenho de estudo
Trata-se de um estudo onde descrevemos as características demográficas,
clínicas e evolutivas de 149 indivíduos consecutivos diagnosticados da SGB em
Natal, Brasil, entre os meses de junho de 1994 a dezembro de 2007, que foram
acompanhados prospectivamente. Esses casos foram classificados, por critérios
eletroneuromiográficos, em duas variantes, desmielinizante e axonal, comparadas
entre si em busca de associações.
Avaliamos também fatores de risco para desenvolvimento de SGB, através
de um estudo tipo caso-controle sobre polimorfismo dos genes FCGR2A e
FCGR3A, para determinar se variabilidades dessa proteína influencia no
desenvolvimento de SGB ou das distintas formas clínicas. Para esse estudo, foram
estudadas as genotipagens de 141 indivíduos com SGB recrutados de 1994 a
2014.
A expressão gênica durante a fase sintomática e após a recuperação foi
avaliada num subgrupo dos participantes da coorte, recrutados em 2013,
objetivando identificar mediadores envolvidos na patogênese da Síndrome de
Guillain Barré.
4.2. Local do estudo:
A maioria dos indivíduos com SGB que participaram do estudo foram
recrutados no Hospital Monsenhor Walfredo Gurgel, em Natal, Rio Grande do
Norte. Durante o período do estudo, 1994 a 2007, este era o único hospital público
de referência para hospitalização relacionada a doenças neurológicas agudas,
tanto pediátrica quanto adulta, em todo o Estado do Rio Grande do Norte.
Inicialmente, realizamos reuniões com a equipe médica e enfermagem da urgência,
além das unidades de terapia intensiva, para capacitação no diagnóstico e

59
tratamento da SGB. Toda suspeita de paralisia flácida aguda naquele período era
comunicada ao neurologista, ou seja, a mim. Após avaliação clínica, o paciente era
submetido à punção lombar e solicitada eletroneuromiografia. Em alguns pacientes
tive que utilizar recursos próprios (por exemplo, transporte) para realizar a
eletroneuromiografia. Os dados demográficos e clínicos foram coletados e
registrados consecutivamente em planilha do Excel. Alguns indivíduos com a SGB,
em menor proporção, foram acompanhados em hospitais da rede privada.
Os estudos imunogenéticos foram realizados no Departamento de
Bioquímica, Laboratório de Imunogenética da Universidade Federal do Rio Grande
do Norte e no Institudo de Medicina Tropical da UFRN.
4.3. População estudada
Indivíduos com paralisia flácida aguda definida clinicamente pelos critérios
de Asbury e Cornblath (anexo 2).(44) Os pacientes foram classificados nas
variantes desmielinizante (PDIA) e axonal, NAMA (sem envolvimento sensitivo) e
NAMSA (com envolvimento sensitivo) segundo critérios eletrofisiológicos (anexo
3).(6, 209) A Síndrome de Miller-Fisher era caracterizada pela tríade de ataxia,
arreflexia e oftalmoparesia sem fraqueza nas extremidades.(32)
Para o estudo de caso-controle, utilizamos o sangue de doadores de sangue
saudáveis, com idade acima dos 35 anos, de banco de sangue da mesma área dos
pacientes com a SGB,.
4.4 Métodos de coleta de dados
Após a seleção dos participantes que preencheram os critérios de inclusão,
foi lido o termo de consentimento livre e esclarecido (anexo 4). Para os indivíduos
selecionados para o estudo e que assinaram o termo de consentimento, foram
aplicados os questionários individuais padronizados (anexo 5).

60
Esse questionário continha as variáveis: idade, sexo, procedência,
antecedente de infecção entre uma a quatro semanas antes do início do quadro,
data de início, sintomas motores ou sensitivos, distribuição da fraqueza (proximal X
distal), déficit de pares cranianos, presença de dor ou disautonomia, tempo para
alcançar o déficit máximo (nadir), grau de incapacidade funcional no pico da
doença, dias de ventilação mecânica, dias para recuperar a deambulação, dias
para ganhar um ponto na escala funcional e presença de sequelas e/ou dor
neuropática após, no mínimo, seis meses do início do quadro.
Todos os pacientes foram examinados e submetidos ao exame
eletroneuromiográfico por um único pesquisador (METD). O estudo da condução
foi realizada em pelo menos quatro nervos motores e quatro sensitivos. Assim,
todos foram re-avaliados por pelo menos duas vezes após um ano da doença.
Mensurou-se a força muscular a partir da soma de escores de três músculos
em cada extremidade, através do Medical Research Council scores. Neste caso, a
soma de escores variou de 60 (normal) até 0 (tetraplegia).
Utilizou-se uma escala funcional para determinar a incapacidade (Escala de
Hughes): 6 = morte; 5 = requer suporte ventilatório; 4 = restrito ao leito ou cadeira;
3 = capaz de andar 10 metros com ajuda; 2 = capaz de andar sem ajuda; 1 =
mínimos sinais e sintomas, capaz de correr; 0 = normal.(210) O grau da
incapacidade era definido no pico da doença. Todos pacientes com ≥ 4 da Escala
de Hughes e nível de IgA normal, receberam tratamento com imunoglobulina
humana (2g/Kg).
Durante a fase aguda da doença e antes de receber tratamento com
imunoglobulina humana, foram coletados 15ml de sangue em veia periférica e
estocados em tubos de Vacutair® secos e com EDTA para avaliação laboratorial de
anticorpos anti-gangliosídeos e para estudo genético. Posteriormente, os soros
foram armazenados em tubos Eppenddorfs® e congelados a -200 até a realização
do teste.
Para o estudo da expressão gênica global foram coletados 9ml, na fase
aguda e na fase de recuperação, estocados em tubo com heparina para extração
do RNA total. Posteriormente, o sangue foi armazenado em tubos Eppenddorfs® e
congelados a -800C até a realização do teste.

61
Todos os dados coletados foram comparados entre as variantes
desmielinzante e axonal.
4.5 Estratégia metodológica e análise dos dados
4.5.1. Metodologia para realização do Objetivo 1
A incidência anual de casos por 100.000 foi calculada considerando a
estimativa da população de acordo com o DATASUS (www.datsus.gov.br), bem
como a incidência média no período de 1994-2007 e a incidência por idade. A
relação entre incidência e idade foi avaliada utilizando-se um modelo de regressão
logística. A incidência de SGB por idade foi calculada usando como denominador a
idade estimada para a população.
Os indivíduos com SGB foram divididos nas duas principais variantes,
desmielinizante e axonal, através de critérios eletroneuromiográficos. As diferentes
percentagens destas variantes foram comparadas utilizando o teste de Fisher ou
qui-quadrado, e as médias ou medianas com o teste de Wilcoxon. Para variáveis
de análise do tipo qualitativa como Sexo, Procedência etc, forão feitos cruzamentos
por tabelas de contingência com as variáveis de diagnóstico e aplicado o teste do
qui-quadrado de máxima verossimilhança para testar hipótese de associação. A
hipótese de "não associação" é rejeitada quando o valor de p (M-L Chi-square) é
menor que 0.05.
4.5.2 Metodologia para realização do Objetivo 2
Utilizou-se uma escala funcional, Escala de Hughes,(210) para determinar a
gravidade no pico da doença. Os casos foram classificados em severos ou leves
(Escala de Hughes ≥ 4 e ≤ 3, respectivamente). Pacientes com evolução
desfavorável foram definidos tendo EH ≥ 3 após 1 ano de início de doença e/ou a
presença de dor neuropática.

62
Durante o seguimento foram monitorados os dias de ventilação mecânica,
os dias para ganhar um ponto na escala funcional de Hughes, os dias para início
da deambulação, os dias de internamento hospitalar, além de presença de
sequelas.
As diferentes percentagens dessas variantes foram analisadas utilizando o
teste de Fisher ou qui-quadrado, e as médias com o teste de Wilcoxon. O tempo
para deambular e para ganhar um ponto na escala de Hughes foi analisado pelo
método de Kaplan e Meier.
Utilizou-se um modelo de regressão múltipla logístico tendo como resposta o
logt da variável evolução e as predictoras selecionadas pelo método forward–LR
sobre os sequintes conjuntos de variáveis: diagnóstico (axonal X desmielinizante),
idade, sexo, antecedentes de gripe, antecedentes de diarreia, fraqueza proximal,
fraqueza distal, paralisia facial, paralisia bulbar, disautonomia, nadir, dias para
melhorar um ponto na escala de Hughes e dias para andar.
4.5.3 Metodologia para realização do Objetivo 3
Para alcançar o objetivo três foi realizada extração de DNA do sangue
periférico de pacientes com SGB e pessoas saudáveis com idade de 35 anos,
oriundos do Estado do Rio Grande do Norte. Em seguida, utilizando PCR em
tempo real pelo TaqMan SPN Genotyping reagentes, nós analisamos a frequência
dos genótipos e a distribuição dos alelos para os genes FCGR2A e FCGR3A nos
indivíduos com a SGB comparando com controles normais. Utilizou-se a Escala
funcional de Hughes para determinar a severidade no pico da doença. Os casos
foram classificados em graves ou leves (Escala de Hughes ≥ 4 e ≤ 3,
respectivamente).
4.5.3.1 Extração do DNA
A extração de DNA foi realizada segundo a técnica descrita por Grimberg e
col.,1989.(211) O DNA foi transferido para tubos de microcentrífuga (Tubos

63
prolypropileno-v.1.7ml) (Labcon North American) e lavado com etanol 70% (C2H6O
– etanol absoluto) e secado à temperatura ambiente por 30 minutos, na capela de
fluxo laminar (Baker, B2 classe II, TIPO alb3, Sanford, Maine, EUA). O DNA foi
solubilizado em 500mL de água estéril, determinada a concentração através da
absorção em 260 nm (Espectrofotômetro – Hitachi, Japão), sendo aliquotado em
tubos de microcentrífuga e estocado a -20°C, para posterior análise.
4.5.3.2. Genotipagem do FCGR
Os genótipos de rs1801274 (A>G; H131R) em FCGR2A e de rs396991
(T>G; P158V) para FCGR3A foram determinados por PCR em tempo real pelo
TaqMan SPN Genotyping reagentes (C_9077561_20 and C_25815666_10,
respectivamente, fornecido pela Applied Biosystems, CA, USA), seguindo o
protocolo do fabricante.(212)
Em resumo, a mistura de reagentes para realização da reação em cadeia
da polimerase (PCR) continha 5 ul de 2X TaqMan Universal PCR Master Mix (Life
Technologies) para rs1801274 e para rs396991. Em Ambas as reações usamos ~
80 ng de ADN genômico e 1ul de 10X ensaio TaqMan genotipagem de SNP, com o
volume final para 10 ul.
As condições de ciclagem para ambos os genótipos foram: pré-
aquecimento a 60°C durante 1 min, seguido de um ciclo de 2 minutos a 50°C para
ativação da uracil-DNA glicosilase e e outro de 92°C por 10 minutos para ativação
da AmpliTaq Gold, uma desnaturação de 95°C por 15 segundos e
anelamento/extensão a 60°C por um minuto, repetidos durante 40 vezes e seguido
por uma post-read a 60°C por 1 minuto.
Após a amplificação dos moldes de ADN utilizando o sistema de PCR ABI
PRISM® 7500 Real-Time (Life Technologies), as determinações foram realizadas
com a intensidade de fluorescência dos alelos, usando o software 7500 System
(Life Technologies).

64
4.5.3.3 – Análise estatística
A distribuição dos genótipos e alelos foi comparada com o teste de qui-
quadrado. Com o objetivo de estimar o efeito genético, os genótipos foram
codificados assumindo um modelo log-aditivo, e incluídos em um modelo de
regressão logística como variável explicativa. Idade e sexo foram adicionados ao
modelo como co-variáveis. A análise foi realizada em Stata 11.2 e os valores de
p,0.05 foram considerados significativos.
4.5.4 Metodologia para realização do Objetivo 4
Doze pacientes consecutivos, que tiveram a SGB no ano de 2013, foram
selecionados para a análise da expressão gênica global usando RNAseq. O
sangue foi coletado de veia periférica, entre a primeira e segunda semana após o
início da doença. Uma segunda amostra foi coletada na fase de recuperação (100
dias média, SD 54.77). O RNA foi extraído de células mononucleares obtidas do
sangue periférico. As células foram lisadas por Tryzol e congeladas até a
purificação de RNA. RNA foi isolado e purificado por colunas, seguindo o protocolo
do fabricante (Quiagen, EUA). Após purificação, as amostras foram digeridas com
DNASE I e a qualidade do RNA foi avaliada pelo nanodrop e subsequente
bioanalisador (Agilent INC, EUA). Amostras com RIN acima de 8 foram utilizadas
para o RNA seq.
4.5.4.1 Montagem do transcritos obtidos por RNA-Seq utilizando a plataforma
Illumina Hi-Seq2000
Após a extração de RNA, a montagem das bibliotecas do tipo Paired End foi
realizada por facility (DNA core Facility – Universidade de Iowa) para os 12
pacientes humanos, ao diagnóstico da Síndrome Guillain-Barré e pós-recuperação,
das várias formas clínicas. O RNA extraído foi utilizado como substrato para
construção das bibliotecas de cDNA em quadruplicatas. Cada amostra extraída de

65
RNA foi considerada uma unidade experimental. O sequenciamento foi feito pela
plataforma Illumina HiSeq. O desenho experimental foi duas condições (Durante e
Após a Síndrome) x quatro repetições da unidade experimental.
4.5.4.2. Mapeamento e Expressão Diferencial de Genes
O processamento, a montagem e análise dos dados de transcriptoma foram
realizados em um servidor computacional Linux/UNIX contendo capacidade total de
72 núcleos de processamento, 128 Gb de memória RAM, alocado no Instituto de
Medicina Tropical do Rio Grande do Norte (IMT-RN). Os dados brutos no formato
fastq (contendo informações de sequência e qualidade) foram avaliados com base
em valores de Phred pelo programa FastQC.(213) O genoma de referência Homo
sapiens (GCF_000001405.26) foi retirado do Genbank, National Center for
Biotechnology Information (NCBI) e indexado e alinhado usando o programa
Bowtie 1.1.1 (214) O mapeamento foi realizado com a ferramenta TopHat 2.0.13.
Foram descartadas da análise as leituras com bases de Phred >= 33. (215, 216) O
pacote de programas Cufflinks 2.2.1 foi utilizado para a montagem do
transcriptoma, comparações entre amostras e obtenção da expressão diferencial
global dos transcritos e genes. (217) Os arquivos de saída da ferramenta Cuffdiff,
do Cufflinks, foram analisados e expressos de forma gráfica por meio da utilização
do pacote CummeRbund 2.7.2, do ambiente de linguagem estatística
computacional, R (http://www.r-project.org).
4.5 Considerações éticas
O protocolo foi avaliado e aprovado pelo Comitê de Ética em Pesquisa da
Universidade Federal do Rio Grande do Norte (CEP-UFRN 046/03 e CEP-UFRN
198/09) e Comissão Nacional de Ética em Pesquisa (CONEP: 9061). O Certificado
de aprovação é 0215.0.051.000-09 (http://portal2.saude.gov.br/sisnep/)
Todos os detalhes do protocolo foram discutidos com todos os sujeitos que
foram convidados a participar do estudo. Os pacientes foram questionados se

66
consentiam em participar do estudo e o termo de consentimento livre e esclarecido
foi utilizado (ANEXO). Os indivíduos (ou familiares ou guardiões legais dos sujeitos
menores de 18 anos de idade) foram convidados a ler ou, nos casos de
analfabetos, a ouvirem a leitura do termo de consentimento e assinar o
consentimento.
Todas as informações dos pacientes foram mantidas sob sigilo e são
acessíveis apenas à equipe do estudo. Esses dados foram armazenados em fichas
e em bancos de dados mantidos no Laboratório de Imunogenética do
Departamento de Bioquímica da UFRN.
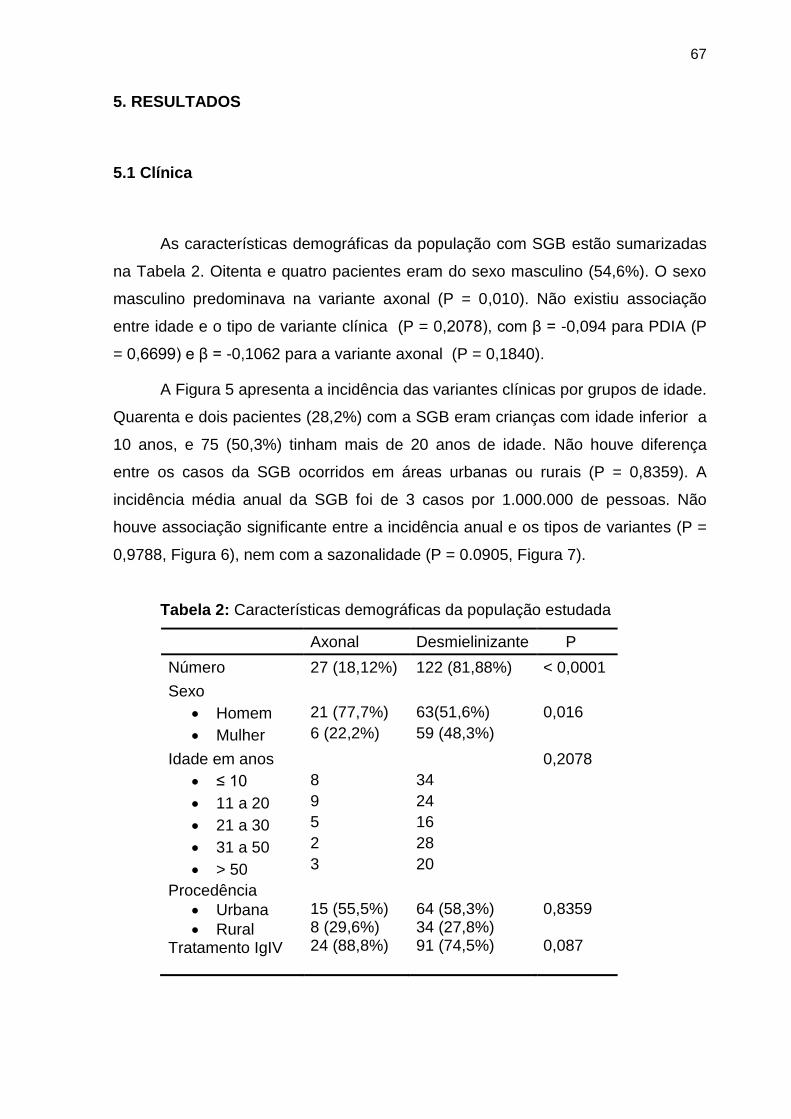
67
5. RESULTADOS
5.1 Clínica
As características demográficas da população com SGB estão sumarizadas
na Tabela 2. Oitenta e quatro pacientes eram do sexo masculino (54,6%). O sexo
masculino predominava na variante axonal (P = 0,010). Não existiu associação
entre idade e o tipo de variante clínica (P = 0,2078), com β = -0,094 para PDIA (P
= 0,6699) e β = -0,1062 para a variante axonal (P = 0,1840).
A Figura 5 apresenta a incidência das variantes clínicas por grupos de idade.
Quarenta e dois pacientes (28,2%) com a SGB eram crianças com idade inferior a
10 anos, e 75 (50,3%) tinham mais de 20 anos de idade. Não houve diferença
entre os casos da SGB ocorridos em áreas urbanas ou rurais (P = 0,8359). A
incidência média anual da SGB foi de 3 casos por 1.000.000 de pessoas. Não
houve associação significante entre a incidência anual e os tipos de variantes (P =
0,9788, Figura 6), nem com a sazonalidade (P = 0.0905, Figura 7).
Tabela 2: Características demográficas da população estudada
Axonal Desmielinizante P
Número 27 (18,12%) 122 (81,88%) < 0,0001
Sexo
Homem
Mulher
21 (77,7%)
6 (22,2%)
63(51,6%)
59 (48,3%)
0,016
Idade em anos
≤ 10
11 a 20
21 a 30
31 a 50
> 50
8
9
5
2
3
34
24
16
28
20
0,2078
Procedência
Urbana
Rural Tratamento IgIV
15 (55,5%) 8 (29,6%) 24 (88,8%)
64 (58,3%) 34 (27,8%) 91 (74,5%)
0,8359 0,087

68
Figura 5. Incidência das variantes da SGB por idade entre 1994 e 2007, considerando o número de
casos por faixa etária usando como denominador a estimativa da população nas respectivas faixas
para o ano de 1999.
Figura 6. Incidência anual da SGB por 1.000.000 de pessoas no período de 1994-2007, por
variante clínica. Não foi observada associação entre as variantes clínicas e o ano.
0
10
20
30
40
50
60
70
≤ 9 10 - 19 20 - 39 40 - 59 ≥ 60
Inci
dê
nci
a p
or
1.0
00
.00
0
Idade em anos
Total PDIA Axonal
0,0
1,0
2,0
3,0
4,0
5,0
6,0
7,0
Inci
dên
cia
po
r 1
.00
0.0
00
Ano
Axonal PDIA Total

69
Figura 7. Distribuição dos casos de Síndrome de Guillain Barré no período de 1994-2007.
Observou-se um pico em novembro, mas não foi significante (P = 0.0905)
Dos 149 casos da SGB, 122 (81,9%) foram classificados como PDIA e 27
(18,1%) como da variante axonal (P < 0,0001). Dentro da variante axonal, 22
(14,7%) apresentavam comprometimento exclusivamente motor (NAMA) e 5 (3,3%)
o envolvimento era motor e sensitivo (NAMSA). Não foram observados casos com
a tríade clássica da SMF, mas cinco casos (3,3%) tinham, além da tetraparesia,
oftalmoparesia (um com III nervo craniano bilateral, três com VI nervo craniano
bilateral e um com oftalmoplegia completa). Destes cinco casos, um apresentava
eletroneuromiografia compatível com a variante axonal e os outros quatro com a
variante desmielinizante. Dois desses pacientes necessitaram de ventilação
mecânica. As manifestações clínicas estão detalhadas na Tabela 3.
História de infecção de via respiratória alta foi relatada, duas semanas antes
do início dos sintomas, por 83 pacientes (55,7%), e diarreia por 12 (8%). A diarreia
era mais frequente na variante axonal (P=0,0254). O nadir foi alcançado dentro dos
10 primeiros dias de doença em 50,0 % dos pacientes. Na variante axonal, 84,6%
alcançaram o déficit máximo dentro de 10 dias, comparado com 42,4% da variante
desemielinizante (P < 0,0001). Muitos pacientes desenvolveram a fraqueza
muscular de predomínio proximal (65,1%), notadamente na variante
desmielinizante (P<0,0001). Em pacientes com a variante axonal, predominava a
fraqueza distal (P<0,0001) e a fraqueza era global predominantemente na NAMSA
(P = 0,040). Alterações sensitivas ocorreram em 73,2% dos pacientes,
0,0%
5,0%
10,0%
15,0%
20,0%
25,0%
Jan Feb Mar Apr Mai Jun Jul Ago Set Out Nov Dec
Meses
Axonal PIDA Total

70
significativamente mais frequente na variante desmielinizante (P<0,0001). A
paralisia facial periférica era mais frequente na variante desmielinizante, com
prevalência de 66,9% (P=0,0026). Na variante axonal, a paralisia facial periférica
eram menos frequente na NAMA (22%) comparando com NAMSA (80%).
Dor foi um sintoma frequente, ocorrendo em aproximadamente 60% dos
casos, mas sem diferença entre as variantes. A presença de sinais de irritação
meníngea ocorreu em 22,2% a 17,2%, nas variantes axonal e desmielinizante
respectivamente. Noventa e cinco pacientes (63,8%) apresentaram disfagia e/ou
disfonia, e muitos necessitaram de alimentação por sonda. Disautonomia foi
diagnosticada em 45,6% dos pacientes e ventilação mecânica foi instituída em
33,6% dos casos, sem diferença significativa entre as variantes. O tempo médio de
ventilação mecânica foi de 17,87 dias, variando de 2 a 150 dias; variante axonal
43,63 (5 a 150) e na desmielinizante 13,77 (2 a 40). Traqueostomia foi realizada
em 13,4% dos pacientes, sendo mais frequente na variante NAMSA (60%; P =
0,040).
Dos 115 pacientes (77,1%) que foram tratados com IgEV, 91 (74,5%) tinham
a variante desmielinizante e 24 (88,8%) a variante axonal. A maioria (85,2%)
estava acamada ou recebia ventilação mecânica (Escala de Hughes ≥ 4). Evolução
desfavorável ocorreu em 13,4% dos casos, com maior frequência nos pacientes
com a variante axonal (P< 0,0001), ver Tabela 3. Os indivíduos com a variante
axonal tinham mais tempo em ventilação mecânica (P = 0,0052), passavam mais
tempo para ganhar um ponto na escala de Hughes (P<0,0001) e necessitavam de
mais tempo para recuperar a deambulação (P< 0,0001), Figura 8. Oito pacientes
faleceram (5,3%). Os pacientes com a variante axonal tinham longa permanência
hospitalar, 31,04 (± 23,1), quando comparados com a variante desmielinizante,
22,54 (± 13.26 dias), P = 0,0234.
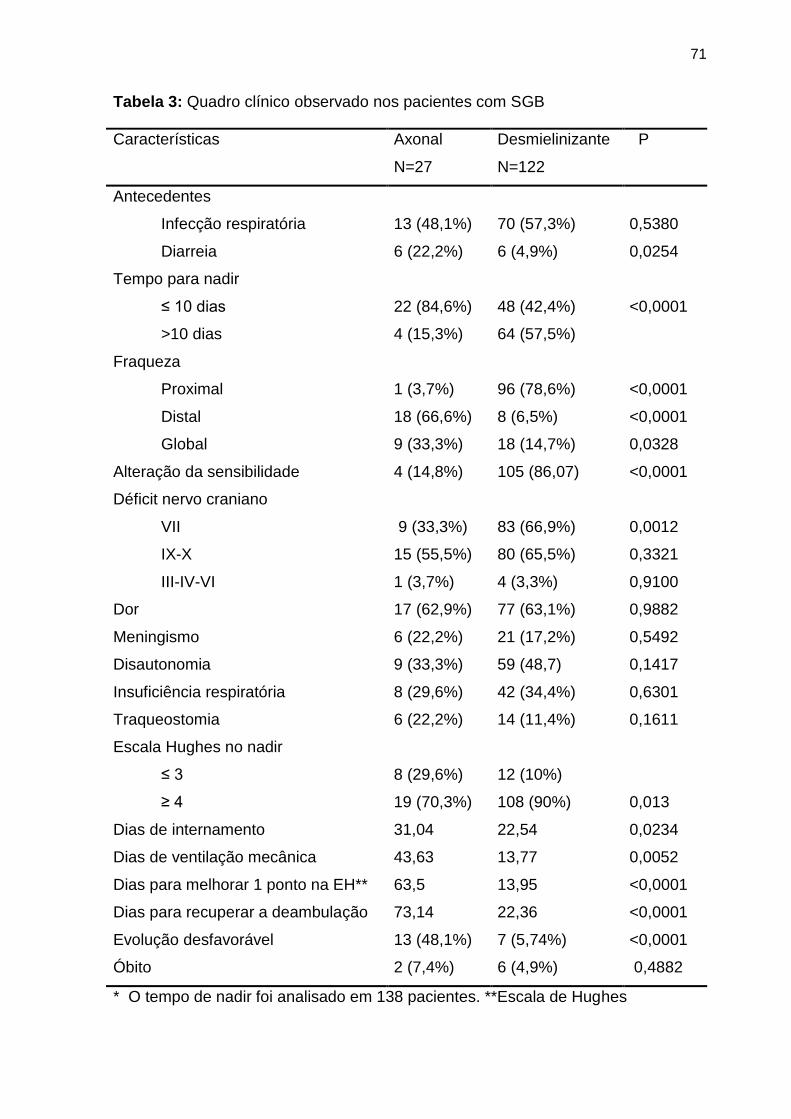
71
Tabela 3: Quadro clínico observado nos pacientes com SGB
Características Axonal
N=27
Desmielinizante
N=122
P
Antecedentes
Infecção respiratória
Diarreia
13 (48,1%)
6 (22,2%)
70 (57,3%)
6 (4,9%)
0,5380
0,0254
Tempo para nadir
≤ 10 dias
>10 dias
22 (84,6%)
4 (15,3%)
48 (42,4%)
64 (57,5%)
<0,0001
Fraqueza
Proximal
Distal
Global
1 (3,7%)
18 (66,6%)
9 (33,3%)
96 (78,6%)
8 (6,5%)
18 (14,7%)
<0,0001
<0,0001
0,0328
Alteração da sensibilidade 4 (14,8%) 105 (86,07) <0,0001
Déficit nervo craniano
VII
IX-X
III-IV-VI
9 (33,3%)
15 (55,5%)
1 (3,7%)
83 (66,9%)
80 (65,5%)
4 (3,3%)
0,0012
0,3321
0,9100
Dor 17 (62,9%) 77 (63,1%) 0,9882
Meningismo 6 (22,2%) 21 (17,2%) 0,5492
Disautonomia 9 (33,3%) 59 (48,7) 0,1417
Insuficiência respiratória 8 (29,6%) 42 (34,4%) 0,6301
Traqueostomia 6 (22,2%) 14 (11,4%) 0,1611
Escala Hughes no nadir
≤ 3
≥ 4
8 (29,6%)
19 (70,3%)
12 (10%)
108 (90%)
0,013
Dias de internamento 31,04 22,54 0,0234
Dias de ventilação mecânica 43,63 13,77 0,0052
Dias para melhorar 1 ponto na EH** 63,5 13,95 <0,0001
Dias para recuperar a deambulação 73,14 22,36 <0,0001
Evolução desfavorável 13 (48,1%) 7 (5,74%) <0,0001
Óbito 2 (7,4%) 6 (4,9%) 0,4882
* O tempo de nadir foi analisado em 138 pacientes. **Escala de Hughes
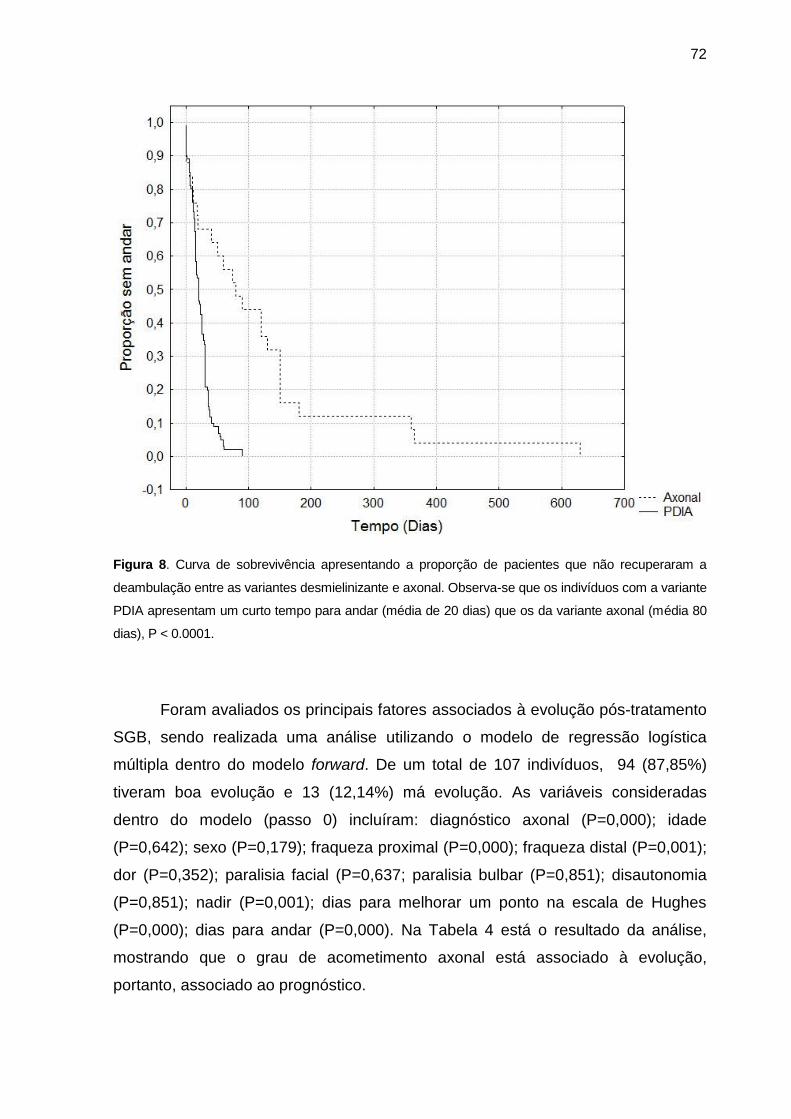
72
Figura 8. Curva de sobrevivência apresentando a proporção de pacientes que não recuperaram a
deambulação entre as variantes desmielinizante e axonal. Observa-se que os indivíduos com a variante
PDIA apresentam um curto tempo para andar (média de 20 dias) que os da variante axonal (média 80
dias), P < 0.0001.
Foram avaliados os principais fatores associados à evolução pós-tratamento
SGB, sendo realizada uma análise utilizando o modelo de regressão logística
múltipla dentro do modelo forward. De um total de 107 indivíduos, 94 (87,85%)
tiveram boa evolução e 13 (12,14%) má evolução. As variáveis consideradas
dentro do modelo (passo 0) incluíram: diagnóstico axonal (P=0,000); idade
(P=0,642); sexo (P=0,179); fraqueza proximal (P=0,000); fraqueza distal (P=0,001);
dor (P=0,352); paralisia facial (P=0,637; paralisia bulbar (P=0,851); disautonomia
(P=0,851); nadir (P=0,001); dias para melhorar um ponto na escala de Hughes
(P=0,000); dias para andar (P=0,000). Na Tabela 4 está o resultado da análise,
mostrando que o grau de acometimento axonal está associado à evolução,
portanto, associado ao prognóstico.

73
Tabela 4: variáveis associadas ao mau prognóstico (Escala de Hughes ≥ 3)
OR P Intervalo confiança
95%
Diagnóstico axonal 17,063
0,003
2,566-113,483
Dias para melhorar um ponto na escala de Hughes
1,028 0,003 1,010-1,047
5.2 Avaliação de polimorfismos nos genes codificadores dos receptores
FcRIIa e FcRIIIa
As características dos 141 casos de SGB genotipados para polimorfismos
nos FCGR2A e FCGR3A estão apresentados na Tabela 5. A média de idade foi
29,75. A presença de infecção precedendo a SGB foi observada em 91 casos
(64,53%) e 100 (70,92%) foram classificados com PDIA. Foram selecionados como
controles 364 pessoas saudáveis, recrutadas após passarem por critérios para
serem doadores de sangue (310 homens; média de idade 35,8 anos). Os homens
correspondem, normalmente, a 75% dos doadores de sangue na nossa
comunidade. As frequências dos genótipos no grupo controle estavam em
equilíbrio de Hardy-Weinberg (dado não apresentado). Nas tabelas 6 e 7 estão a
distribuição da genotipagem e dos alelos. Para o genótipo FCGR2A a frequência
nos casos e controles foi: genótipo H ⁄ H 26 (18,57%) e 74 (20,44%), genótipo R ⁄
R 40 (28,57%) e 106 (29,28%), genótipo H ⁄ R 74 (52,86%) e 182 (50,28%),
respectivamente. Para a distribuição do genótipo FCGR3A nos casos da SGB 66
(47,14%) tinham o genótipo P / P, 14 (10%) tinham o genótipo V ⁄ V e 60 (42,86%)
eram heterozigotos com o genótipo P / V. Nos controles, 180 (49,59%) tinham o
genótipo P ⁄ P, 35 (9,64%) apresentavam o genótipo V ⁄ V e 148 (40,77%) eram
heterozigotos com o genótipo P / V. A frequência dos genótipos e dos alelos não
diferiu significantemente entre casos e controles.
Analisamos a distribuição dos genótipos de acordo com a presença de
antecedentes infecciosos, com as variantes da SGB (axonal X desmielinizante) e
com a positividade dos anticorpos anti-gangliosídeos e não foi observada
correlação (tabela 8). A distribuição das diferentes combinações de genótipos
também não era diferente entre controles e os indivíduos com SGB (tabela 9).

74
Tabela 5: Características dos casos com Síndrome de Guillain-Barré genotipados
para o receptor de Fc
Características Síndrome de Guillain-Barré Controles*
Numero 141 364
Sexo (masculino/feminino) 82/59 54/310
Idade (anos,DP) 29,8 (±20,3) 35,87 (10,43)
Infecção prévia NA
Diarréia 21
Infecção respiratória 69
Diarréia + infecção
respiratória
1
Vacinas 3
Ausente 47
Escore de severidade, n (%) NA
Leve (1-3) 27/135 (20)
Severo (4-5) 108/135 (80)
Anticorpos antigangliosídeos, n(%) NA
Anti-GM1 24/116 (20,6)
Anti-GD1a 8/92 (8,7)
Anti-GM1 e/ou anti-GD1a 32/116 (27,6)
Classificação eletrofisiológica, n (%) NA
Desmielinizante 100 (70,92)
Axonal 36 (25,55)
Não classificada 3 (7,31)
Miller-Fisher 2 (4,87)
* *Os controles foram oriundos de banco de sangue

75
Tabela 6: Distribuição da frequência dos genótipos e alelos nos controles e nos
casos
Gene
(variante)
Genótipos
e alelos
Controle,
n (%)
Casos SGB, n (%)
Leve Grave Total p-valora p-valorb
FCGR2A
(R131H)
R/R 106 (29,3) 31 (29,0) 8 (29,6) 40 (28.6)
0,955 0,849 R/H 182 (50,3) 56 (52,3) 15 (55,6) 74 (52.9)
H/H 74 (20,4) 20 (18,7) 4 (14,8) 26 (18.6)
R 394 (0,54) 118 (0,55) 31 (0,57) 154 (0.55) 0,905 0,869
H 330 (0,46) 96 (0,45) 23 (0,43) 126 (0.45)
FCGR3A
(P158V)
F/F 180 (49,6) 48 (44,4) 13 (50,0) 66 (47.1)
0,606 0,886 F/V 148 (40,8) 51 (47,2) 9 (34,6) 60 (42.9)
V/V 35 (9,6) 9 (8,3) 4 (15,4) 14 (10.0)
F 508 (0,70) 147 (0,68) 35 (0,67) 192 (0.69) 0,817 0,665
V 218 (0,30) 69 (0,32) 17 (0,33) 88 (0.31)
Tabela 7. Risco estimado do efeito genético sobre a doença através de modelo de
regressão logística.a
Alelo risco SGB total SGB leve SGB grave
OR (95% CI) p OR (95% CI) p OR (95% CI) p
FCGR2A
(H131) 0,92 (0,67 – 1,27) 0,624 0,87 (0,62 – 1,24) 0,453
0,86 (0,47 –
1,57) 0,630
FCGR3A
(V158) 1,00 (0,71 – 1,40) 0,982 0,99 (0,68 - 1,44) 0,961
1,12 (0,60 –
2,10) 0,713
a Modelo de regressão usando o grupo controle como referência e idade e sexo como co-
variáveis.

76
Tabela 8: Distribuição dos genótipos quanto a aspectos clínicos e laboratoriais
Gene
(variante)
Genótipos
e alelos
Variante
desmielizante*
n = 135
P-valor Presença
de infecção
n = 134
p-valor Ac anti-Gg
positivo
n = 115
p-valor
FCGR2A
(R131H)
R/R 29 (29,00)
0,411
22 (24,44)
0,230
6 (25,00)
0,907 R/H 50 (50,00) 52 (57,78) 14 (58,33)
H/H 21 (21,00) 16 (17,78) 4 (16,67)
FCGR3A
(P158V)
F/F 51 (51,52)
0,113
41 (45,56)
0,835
9 (37,50)
0,583 F/V 38 (38,38) 41 (45,56) 13 (54,17)
V/V 10 (10,10) 8 (8,89) 2 (8,33)
Tabela 9: distribuição da combinação de genótipos entre controles e pacientes
Combinação genótipos Controles
n=364 (%)
SGB
n=141 (%)
p-valor
H/H + V/V 13 (3,57) 6 (4,26) 0,925
Outros 283 (77,75) 108 ( 76,60)
R/R + PP 68 (18,68) 27 (19,15)
5.3. Resultados preliminares da expressão gênica global em células
monucleares de sangue periférico durante a fase sintomática e pós-
recuperação da Síndrome de Guillain-Barré
As características clínicas dos pacientes com SGB que foram avaliados na
análise da expressão gênica global estão mostradas na Tabela 10. Doze pacientes,
sendo 6 com forma desmielienizante (50%), quatro com forma axonal (33,3%) e
dois com forma Miller-Fisher (16,7%) foram avaliados, sendo 58% dos casos do
sexo masculino e a média de idade de foi 41,7 anos (±18,9). A segunda amostra de
sangue foi coletada dentro de 102 (±57,7) dias pós-diagnóstico.

77
Tabela 10: Características dos pacientes submetidos a análise do transcriptoma
Caso Idade Sexo Variante Na
dir
EH
*
Dias para
melhorar**
Anti-Gg Dias para 2
amostra
1 15 M Desmielnizante 10 3 5 - 60
2 27 M Desmielinizante 8 4 60 - 120
3 27 F Axonal 5 4 45 - 90
4 35 M Miller-Fisher 7 3 7 GQ1b 120
5 39 M Desmielinizante 10 4 22 - 60
6 42 F Axonal 7 3 40 GM1 60
7 51 F Miller-Fisher 10 4 10 GQ1b 60
8 52 F Axonal 2 4 546 GM1,
GD1a,
GD1b
120
9 55 F Desmielinizante 10 4 25 - 240
10 61 M Desmieliniante 25 3 15 - 60
11 61 M Axonal 3 4 100 GM1 60
12 71 M Desmielinizante 8 5 10 - 180
* Escala Hughes no nadir da doença. **Dias para melhorar um ponto na escala de Hughes.
***Intervalo de dias para coleta da segunda amostra de RNA
A Figura 9 mostra a dispersão gênica de cada amostra sequenciada,
considerando os escores das leituras obtidas em relação ao genoma humano,
enquanto a figura 10 mostra a análise de agregação hierárquica, indicando a
qualidade dos dados obtidos. As tabelas 11-14 apresentam exemplos de formas
clínicas de SGB quanto a expressão diferencial gênica na fase sintomática e pós-
recuperação. A figura 11 ilustra a expressão dos mRNAs na fase de recuperação
dos indivíduos com SGB em relação com a fase aguda, chamando atenção para
modulação positiva dos transcritos FOS, PTGS2, IL8, THBS1.

78
Figura 9. Gráfico de Dispersão gênica, baseado em número de genes, de cada transcriptoma
analisado (X1 a X24). Avaliação da qualidade dos dados oriundos do sequenciamento com base na
distribuição dos escores em FPKM (Fragments Per Kilobase Of Exon Per Million Fragments
Mapped) entre as amostras.
Figura 10. Análise de agregação hierárquica (Hierarchical clustering analysis - HCA), provenientes do Escalonamento Multidimensional dos transcriptomas analisados, considerando a expressão gênica individual durante a fase sintomática e pós- recuperação, incluindo os dados de todos os genes.

79
Tabela 11. Expresssão gênica na forma desmielinizante (caso 1), tendo como base a fase sintomática e após a recuperação.
Gene Número de vezes
aumentado
P valor Gene Número de vezes
diminuído
P valor
OR7E7P 2.00706 0.00285 SOCS3 -4.46362 0,00005
HBB 2.02611 0,00005 CXCL9 -3.47409 0,00005
RP11-277P12.20 2.0314 0,00005 IL1B -2.60224 0,00005
LOC102724811 2.25884 0,00005 ADM -2.54109 0,00005
RP11-219B17.1 2.02507 0,00005 THBD -2.49162 0,00005
HBA2 2.31921 0,00005 BRE-AS1 -2.45422 0.02175
TJP1P 2.63469 16 LRG1 -2.41885 0,00005
LOC102723543 2.01652 0.0078 RSG1 -2.34808 0.00025
ZNF441 2.02537 0,00005 HCAR2 -2.30974 0.0003
VN1R83P 2.23444 0.01065 IGKV1-8 -2.3076 0.0005
PSMD10P1 2.14852 0.02915 EGR1 -2.24752 0,00005
HNRNPU-AS1 2.03305 0,00005 MAFB -2.20889 0,00005
ZNF638-IT1 2.09552 0.00365 SAC3D1 -2.19186 0,00005
IGKV1-17 2.42133 0,00005 C19orf59 -2.15811 0,00005
ERMN 2.16883 0,00005 LIM2 -2.13901 0.02745
LOC200772 2.11955 0,00005 HOMEZ -2.11603 0,00005
ASB14 2.04256 0,00005 MMP9 -2.1028 0,00005
HLX -2.09785 0,00005

80
Tabela 12. Expresssão gênica na forma desmielinizante (caso 2), tendo como base a fase sintomática e após a recuperação.
Gene Número vezes aumentado
P valor Gene Número vezes diminuido
P valor
CD180 2135 0,00005 EGR3 -7.05214 0,00005
HEMGN 2.31029 0,00005 EGR2 -5.49446 0,00005
HBB 3.53071 0,00005 EGR1 -5.28461 0,00005
HBD 3.44243 0.0196 IL8 -4.75186 0,00005
MEG3 2.1408 0,00005 BRE-AS1 -4.33809 0.0091
HBM inf 0,00005 EREG -4.12878 0,00005
HBA2 3.3941 0,00005 PTGS2 -4.05352 0,00005
HBA1 3.86009 0,00005 NR4A2 -4.03351 0,00005
C17orf97 2.24826 0.0015 CXCL2 -3.68913 0.0174
NOG 3.12911 0,00005 G0S2 -3.67112 0,00005
VN2R21P inf 0.00755 THBS1 -3.53325 0,00005
SEPT5-GP1BB 2.49496 0.0153 RGS1 -3.4347 0,00005
VPREB3 2.71009 0.00455 HBEGF -3.36044 0,00005
PDGFB 2.41627 0,00005 FOSB -3.34282 0,00005
RPS18 2.07393 0.00545 NAMPT -3.11667 0,00005
RP5-1057J7.6 2.30318 0,00005 CD83 -2.99274 0,00005
ID3 2.73469 0.0006 ENC1 -2.97353 0,00005
CISH 2.16965 0,00005 DUSP2 -2.86138 0,00005
TM4SF19 2.88836 0.0176 NAMPTL -2.80878 0,00005
IL1B -2.65368 0,00005
SGK1 -2.59757 0,00005
IGLV3-27 -2.56395 0.0179
LOC100506036 -2.47348 0.00055
B3GNT7 -2.45452 0,00005
MAFF -2.41303 0,00005
SOCS3 -2.40304 0,00005
PDE4D -2.31975 0,00005
PER1 -2.31707 0,00005
LOC102724428 -2.29859 0,00005
PLAUR -2.24437 0,00005
AVPI1 -2.1941 0.0001
CXCR4 -2.15451 0,00005
LINC00264 -2.15022 0.00225
TNFAIP3 -2.08004 0,00005
DDIT4 -2.06868 0,00005
LINC00936 -2.06583 0,00005
SIK1 -2.05117 0,00005
TRIB1 -2.02711 0,00005
CCNB2 -2.02009 0,00005
NFIL3 -2.01148 0,00005
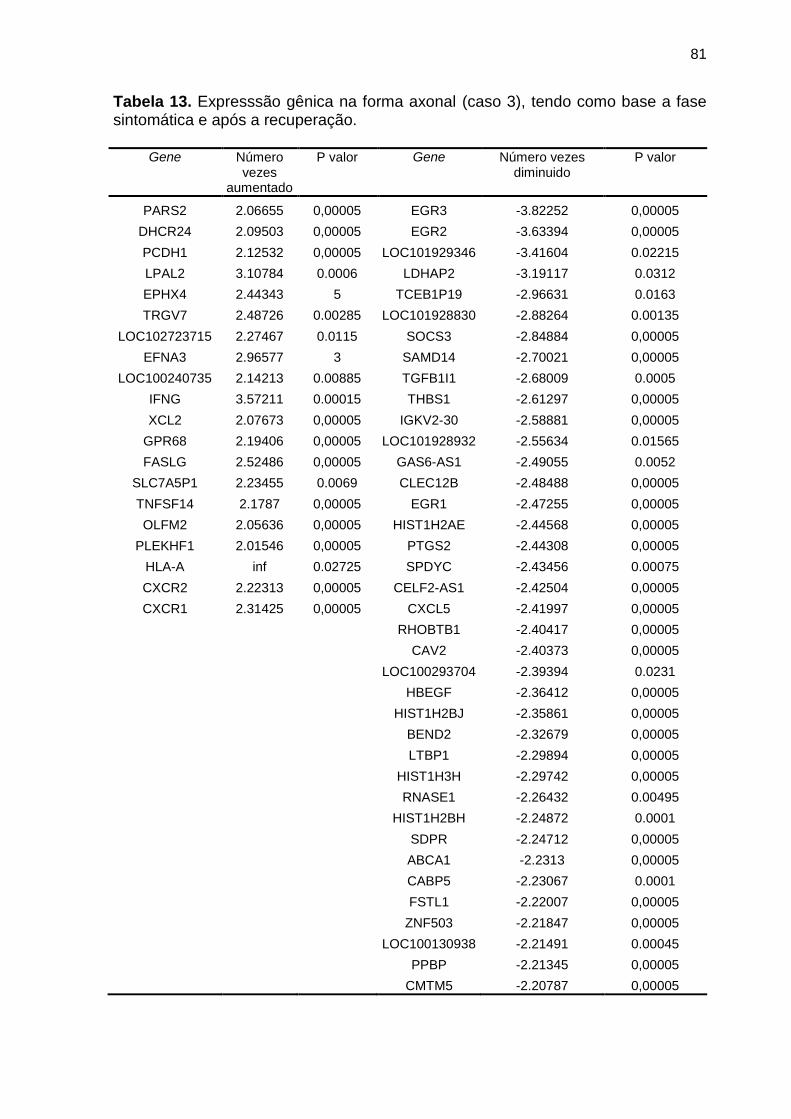
81
Tabela 13. Expresssão gênica na forma axonal (caso 3), tendo como base a fase sintomática e após a recuperação.
Gene Número
vezes aumentado
P valor Gene Número vezes diminuido
P valor
PARS2 2.06655 0,00005 EGR3 -3.82252 0,00005
DHCR24 2.09503 0,00005 EGR2 -3.63394 0,00005
PCDH1 2.12532 0,00005 LOC101929346 -3.41604 0.02215
LPAL2 3.10784 0.0006 LDHAP2 -3.19117 0.0312
EPHX4 2.44343 5 TCEB1P19 -2.96631 0.0163
TRGV7 2.48726 0.00285 LOC101928830 -2.88264 0.00135
LOC102723715 2.27467 0.0115 SOCS3 -2.84884 0,00005
EFNA3 2.96577 3 SAMD14 -2.70021 0,00005
LOC100240735 2.14213 0.00885 TGFB1I1 -2.68009 0.0005
IFNG 3.57211 0.00015 THBS1 -2.61297 0,00005
XCL2 2.07673 0,00005 IGKV2-30 -2.58881 0,00005
GPR68 2.19406 0,00005 LOC101928932 -2.55634 0.01565
FASLG 2.52486 0,00005 GAS6-AS1 -2.49055 0.0052
SLC7A5P1 2.23455 0.0069 CLEC12B -2.48488 0,00005
TNFSF14 2.1787 0,00005 EGR1 -2.47255 0,00005
OLFM2 2.05636 0,00005 HIST1H2AE -2.44568 0,00005
PLEKHF1 2.01546 0,00005 PTGS2 -2.44308 0,00005
HLA-A inf 0.02725 SPDYC -2.43456 0.00075
CXCR2 2.22313 0,00005 CELF2-AS1 -2.42504 0,00005
CXCR1 2.31425 0,00005 CXCL5 -2.41997 0,00005
RHOBTB1 -2.40417 0,00005
CAV2 -2.40373 0,00005
LOC100293704 -2.39394 0.0231
HBEGF -2.36412 0,00005
HIST1H2BJ -2.35861 0,00005
BEND2 -2.32679 0,00005
LTBP1 -2.29894 0,00005
HIST1H3H -2.29742 0,00005
RNASE1 -2.26432 0.00495
HIST1H2BH -2.24872 0.0001
SDPR -2.24712 0,00005
ABCA1 -2.2313 0,00005
CABP5 -2.23067 0.0001
FSTL1 -2.22007 0,00005
ZNF503 -2.21847 0,00005
LOC100130938 -2.21491 0.00045
PPBP -2.21345 0,00005
CMTM5 -2.20787 0,00005
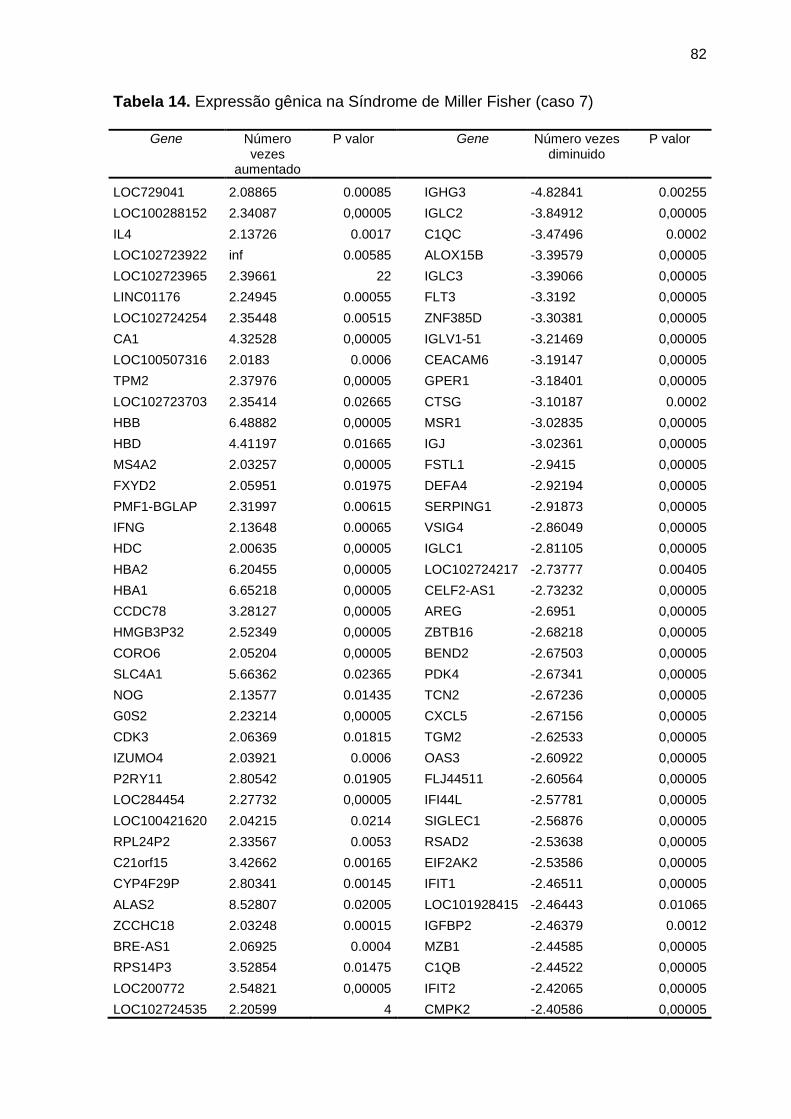
82
Tabela 14. Expressão gênica na Síndrome de Miller Fisher (caso 7)
Gene Número vezes
aumentado
P valor Gene Número vezes diminuido
P valor
LOC729041 2.08865 0.00085 IGHG3 -4.82841 0.00255
LOC100288152 2.34087 0,00005 IGLC2 -3.84912 0,00005
IL4 2.13726 0.0017 C1QC -3.47496 0.0002
LOC102723922 inf 0.00585 ALOX15B -3.39579 0,00005
LOC102723965 2.39661 22 IGLC3 -3.39066 0,00005
LINC01176 2.24945 0.00055 FLT3 -3.3192 0,00005
LOC102724254 2.35448 0.00515 ZNF385D -3.30381 0,00005
CA1 4.32528 0,00005 IGLV1-51 -3.21469 0,00005
LOC100507316 2.0183 0.0006 CEACAM6 -3.19147 0,00005
TPM2 2.37976 0,00005 GPER1 -3.18401 0,00005
LOC102723703 2.35414 0.02665 CTSG -3.10187 0.0002
HBB 6.48882 0,00005 MSR1 -3.02835 0,00005
HBD 4.41197 0.01665 IGJ -3.02361 0,00005
MS4A2 2.03257 0,00005 FSTL1 -2.9415 0,00005
FXYD2 2.05951 0.01975 DEFA4 -2.92194 0,00005
PMF1-BGLAP 2.31997 0.00615 SERPING1 -2.91873 0,00005
IFNG 2.13648 0.00065 VSIG4 -2.86049 0,00005
HDC 2.00635 0,00005 IGLC1 -2.81105 0,00005
HBA2 6.20455 0,00005 LOC102724217 -2.73777 0.00405
HBA1 6.65218 0,00005 CELF2-AS1 -2.73232 0,00005
CCDC78 3.28127 0,00005 AREG -2.6951 0,00005
HMGB3P32 2.52349 0,00005 ZBTB16 -2.68218 0,00005
CORO6 2.05204 0,00005 BEND2 -2.67503 0,00005
SLC4A1 5.66362 0.02365 PDK4 -2.67341 0,00005
NOG 2.13577 0.01435 TCN2 -2.67236 0,00005
G0S2 2.23214 0,00005 CXCL5 -2.67156 0,00005
CDK3 2.06369 0.01815 TGM2 -2.62533 0,00005
IZUMO4 2.03921 0.0006 OAS3 -2.60922 0,00005
P2RY11 2.80542 0.01905 FLJ44511 -2.60564 0,00005
LOC284454 2.27732 0,00005 IFI44L -2.57781 0,00005
LOC100421620 2.04215 0.0214 SIGLEC1 -2.56876 0,00005
RPL24P2 2.33567 0.0053 RSAD2 -2.53638 0,00005
C21orf15 3.42662 0.00165 EIF2AK2 -2.53586 0,00005
CYP4F29P 2.80341 0.00145 IFIT1 -2.46511 0,00005
ALAS2 8.52807 0.02005 LOC101928415 -2.46443 0.01065
ZCCHC18 2.03248 0.00015 IGFBP2 -2.46379 0.0012
BRE-AS1 2.06925 0.0004 MZB1 -2.44585 0,00005
RPS14P3 3.52854 0.01475 C1QB -2.44522 0,00005
LOC200772 2.54821 0,00005 IFIT2 -2.42065 0,00005
LOC102724535 2.20599 4 CMPK2 -2.40586 0,00005

83
Figura 11: Heatmap apresentando a variabilidade de expressão de genes selecionados nos
indivíduos na fase de recuperação comparando com a fase aguda. Vermelho e verde representam
upregulation e downregulation, respectivamente.

84
6. DISCUSSÃO
6.1 Aspectos Clínicos da Síndrome de Guillain Barré
Após o controle e erradicação da poliomielite na maior parte dos países, a
síndrome de Guillain Barré é a neuropatia aguda mais frequente. No entanto, são
escassos os dados sobre essa doença nos países em desenvolvimento. Em nosso
estudo, a incidência média anual da SGB foi de 0,3/100.000, sem presença de
sazonalidade. Houve um predomínio da variante desmielinizante e muitos dos
casos eram crianças. A variante axonal estava associada ao mal prognóstico. A
incidência anual da SGB em outras localidades varia de 0,16 a 4/100.000, onde
também se observa um aumento linear com a idade.(2, 7)
No Estado do Rio Grande do Norte, onde este estudo foi conduzido, a
incidência anual não tem aumentado, 50,3% dos indivíduos tinham menos de 20
anos de idade e a incidência diminuía com a idade. A distribuição encontrada neste
estudo é similar ao observado em outros estados brasileiros, em outros países da
America Latina, em Bangladesh e no norte da China.(4, 15, 218, 219) O
predomínio de jovens pode ser explicado por a alta exposição a fatores
desencadeantes, infecção ou após vacinas.
A nossa coorte não representa um estudo populacional, apesar de
provavelmente termos recrutado a maioria dos casos de SGB ocorrido no Estado
do Rio Grande do Norte nos últimos 14 anos. A incidência observada
(0.3/100.000/ano) foi próxima do estudo populacional brasileiro em crianças com
menos de 15 anos de idade (0,4/100.000/ano).(15) Além disso, não podemos
descartar o viés de seleção em nosso estudo devido a potencial falta de
diagnóstico dos casos leves em idosos ou a maior facilidade de diagnóstico em
crianças dado o eficiente sistema de vigilância de paralisia flácida aguda no Brasil
para monitorização da poliomielite. Há um temor que vacinação em excesso pode
levar a complicações neurológicas. No entanto, Dias-Tosta e Kuckelhaus não
confirmaram a correlação entre a vacinação da pólio oral e a SGB.(15)
Neste estudo, observamos um predomínio na variante desmielinizante,
similar aos estudos Europeu e da América do Norte, mas diferente de outros

85
países da América Latina.(11-13) A frequência da variante axonal era também
similar a outros estudos.(91, 220) Nas Américas Central e do Sul, Japão, Israel e
China a frequência da variante axonal varia de 30% a 47%.(4, 10-13, 64) A razão
para a diferença da frequência da variante axonal entre os países não está
esclarecida, mas pode ser por causa da suscetibilidade genética, bem como
fatores ambientais que podem desencadear uma variante particular. É importante
chamar atenção que há poucos estudos sobre a frequência e distribuição da SGB
no Brasil, e dentre estes nenhum utilizou a eletroneuromiografia para classificar as
variantes.(15, 43, 65)
A SMF ocorre habitualmente em 5% dos casos da SGB. Na nossa
população estudada não observamos nenhum caso com a tríade clássica da SFM.
Entretanto, encontramos cinco pacientes que apresentaram tetraparesia com
oftalmoparesia, sendo que dois evoluíram para insuficiência respiratória. A
oftalmoparesia tem sido relatada na SGB. Essa condição é definida como SMF
sobreposição com SGB (SMF/SGB).(221) Os pacientes com SMF/SGB
frequentemente necessitam de ventilação mecânica.(222)
A SGB representa o protótipo de doença pós-infecciosa, com 2/3 dos casos
precedidos de infecção. Infecções respiratórias e enterites são os antecedentes
mais frequentes na SGB. Em nosso estudo, 83 pacientes relataram sintomas
infecciosos antes do início da doença, sendo infecção respiratória a mais frequente
(55.7%), seguido de diarreia (8%). A diarreia foi observada mais frequentemente na
variante axonal (P = 0.01). No estudo de Rocha e colaboradores, realizado em
São Paulo, a infecção respiratória era a mais frequente, seguida da
gastroenterite.(43) A melhora recente da saúde pública no Brasil, incluindo a
qualidade da água e a melhora do nível socioeconômico pode ter influenciado na
redução na diarreia infantil. Num estudo anterior, detectamos a presença de
anticorpos anti-C. jejuni em oito de 21 pacientes estudados (32%) e associados
com a presença de anticorpos anti-GM1.(14)
Num estudo realizado no Brasil, 30% das manifestações neurológicas
durante a infecção por dengue era da SGB.(130). Em regiões endêmicas, países
tropicais e subtropicais, a infecção pode ser oligossintomática dificultando o
diagnóstico neurológico associado à infecção por dengue.(20) Durante a dengue,
há resposta imune aberrante incluindo produção de citocinas e quimiocinas,

86
ativação do complemento e ativação celular. Por consequência, resposta
autoimune pode ocorrer, notadamente na forma hemorrágica da doença. Indivíduos
com dengue produzem anticorpos. Esses mecanismos, provavelmente, estão
associados ao desenvolvimento das manifestações neurológicas. Encontramos três
casos de SGB com uma história recente de dengue (dados não mostrados).
Não observamos variação sazonal significante nos nossos casos. Nas
regiões equatoriais onde há pouca variação da temperatura, diarreia ocorre durante
os meses quentes e chuvosos. Estudo no norte do Brasil demonstrou que
infecções respiratórias ocorrem durante todo o ano.(223) Não está claro na
literatura se há sazonalidade. Num estudo de revisão sistemática, Mcgrogran e
colaboradores também não observaram um padrão de sazonalidade.(2).
Entretanto, num estudo nos Estados Unidos da América, observou-se um aumento
de 15% dos casos durante os meses de frio, consistente com o aumento de
infecções respiratórias superiores nesses meses.(7) Já no norte da China,
epidemias da variante axonal da SGB ocorreram durante o verão, por águas
contaminadas, causadas por infecção por Campylobacter jejuni (4). Na América
Central e México os pesquisadores identificaram aumento da frequência da SGB
nos meses chuvosos.(11, 224) No Brasil, Dias-Tosta e Kickelhaus(15) não
encontraram maior incidência em determinado período do ano, mas o estudo de
Rocha e colaboradores (43), realizado em São Paulo, encontrou uma alta
incidência nos meses de primavera e verão.
A média de tempo para alcançar o nadir foi ao redor de duas semanas.(1)
Observamos que pacientes com a variante axonal tinham uma evolução rápida da
doença, nadir < 10 dias, quando comparado com a variante desmielinizante (P =
0.00005). A heterogeneidade clínica tornou-se evidente na SGB. A distribuição da
fraqueza pode ser variável; alguns apresentam fraqueza difusa, outros têm
predomínio proximal ou distal.(1) Todos os nossos pacientes apresentavam
fraqueza muscular e arreflexia, com padrão evolutivo monofásico e déficit máximo
em menos de 30 dias. Na ausência de outro diagnóstico, isso é suficiente para
diagnosticar a SGB segundo os novos critérios estabelecidos por pesquisadores de
diferentes nacionalidades (Brighton Collaboration GBS Working Group).(55) Porém,
sem o estudo eletroneuromiográfico não é possível, só com o exame clínico,
distinguir entre as variantes axonal e desmielinizante. Em nosso estudo, a

87
distribuição da fraqueza correlacionava com a variante; a fraqueza distal era
indicativa da NAMA, variante axonal puramente motora; a fraqueza proximal estava
associada à variante desmielinizante e a fraqueza difusa indicava a NAMSA,
variante axonal motora e sensitiva. O envolvimento do nervo facial e as alterações
sensitivas predominaram na variante desmielinizante. O predomínio da fraqueza
distal é característico da NAMA, além do pouco envolvimento do nervo facial e da
ausência de déficit sensitivo.(220) Alterações da sensibilidade eram frequentes nos
nossos pacientes, notadamente na variante desmielinizante. Na variante axonal,
estava ausente no subtipo puramente motor, mas era grave no subtipo NAMSA.
A presença de dor, incluindo sinais de irritação meníngea, ocorreu em 81,8%
dos nossos pacientes, tendo sido relatada na variante axonal puramente motora,
além na variante desmielinizante. Sinais de irritação meníngea estavam presentes
nos pacientes com NAMA do norte da China.(4) A frequência da dor está descrita
em 55 a 89% dos pacientes com a SGB.(45-47) Sua presença pode anteceder a
fraqueza muscular confundindo com outras doenças e retardando o diagnóstico. É
importante reconhecê-la e tratá-la, especialmente nos pacientes que são incapazes
de falar devido à entubação. Dois tipos de dores são distinguidas. O primeiro inicia
antes do início da fraqueza permanecendo até a alta hospitalar; tem localização
nas extremidades e tem componente radicular, parestesias e dor muscular. O outro
tipo ocorre após a alta e persiste durante a reabilitação, localizando-se nas
extremidades e se comporta como parestesias, dor muscular e artralgia. A dor é
intensa durante o curso da doença, porém mais acentuada na fase aguda. A
presença de dor está associada a distúrbios da sensibilidade, a maior déficit motor
e maior incapacidade. A presença de dor na fase aguda correlaciona com a
ocorrência de dor tardiamente.(48, 49)
A fisiopatologia da dor na SGB não é bem conhecida. O envolvimento das
raízes nervosas pode explicar a dor lombar ou a dor lombar com irradiação para as
extremidades. Fatores inflamatórios podem gerar dor via nervi nervorum. A dor
muscular ou articular possivelmente é decorrente da imobilização. Dor neuropática
provocada por atividade espontânea ou anormal das vias sensitivas mielinizadas
podem explicar as disestesias e parestesias nas extremidades.(49) No entanto,
estudos recentes demonstraram o envolvimento das fibras finas, amielinizadas, na
SGB, como causa da dor e da disfunção autonômica.(86, 225)

88
Curiosamente, a dor tem sido relatada na variante axonal puramente motora.
Ruts L e colaboradores observaram que 49% dos pacientes com NAMA tinham
dor.(49) O mesmo grupo, num estudo posterior, observou redução difusa da
densidade de fibra nervosa intradérmica, tanto amielínica como mielínica, da SGB
e suas variantes, incluindo a puramente motora, correlacionando com a presença
de dor na fase aguda e com incapacidade aos seis meses.(87) Esse resultado leva
ao questionamento quanto a NAMA ter um componente puramente motor e se os
critérios ou ferramentas utilizadas seriam suficientes para determinar a existência
de um componente motor puro.
Observamos alta prevalência de envolvimento da musculatura bulbar (63%),
coincidindo com um grande número de casos graves (85.2%) no nosso coorte. A
paralisia dos músculos bulbares é observada tipicamente nos pacientes com a
SGB grave, em pacientes ventilados, na variante SMF/SGB e na variante faringeo-
cervico-braquial.(222, 226)
Disautonomia ocorreu em 45,9% dos nossos pacientes, tanto na variante
axonal como na desmielinizante. Essa frequência está próxima da descrita nas
grandes séries; 2/3 dos casos da SGB apresentaram disautonomia.(227-229) A
extensa distribuição dos nervos autonômicos resulta numa constelação de sinais e
sintomas por falha ou hiperatividade do simpático e parassimpático. Os sintomas
incluem arritmias cardíacas, flutuações da pressão arterial, resposta hemodinâmica
anormal a medicamentos, alteração do suor, anormalidades pupilares, disfunção
vesical e intestinal, hiponatremia. O seu reconhecimento é importante para o
tratamento. Uma das principais causas de morte súbita na SGB é a disautonomia
cardíaca. Em alguns casos é necessário a colocação de marcapasso cardíaco
transcutâneo.(230)
Aproximadamente 20 a 30% dos pacientes com SGB necessitam de
ventilação mecânica.(226, 231). Em nosso estudo, 50 pacientes (33.5%)
apresentaram insuficiência respiratória e foram entubados. A insuficiência
respiratória ocorreu tanto na variante axonal como na desmielinizante; entretanto,
quando a variante axonal foi subdividida, esta insuficiência predominava na
NAMSA (60%) do que na NAMA (22%). A alta frequência de insuficiência nos
nossos pacientes reflete a gravidade dos casos selecionados.
Não há consenso entre os estudos sobre os indicadores da insuficiência

89
ventilatória. Um estudo com 722 pacientes, dos quais 313 necessitaram de
ventilação mecânica, identificou tempo do início da doença e do internamento
menor de sete dias; incapacidade para tossir, incapacidade para ficar de pé,
incapacidade de flexionar o pescoço e aumento das enzimas hepáticas como
variáveis determinantes de evolução.(232) Para Durand e colaboradores, os
indicadores de insuficiência respiratória foram a presença de bloqueio da condução
do nervo fibular e a redução da capacidade vital; seus pacientes em ventilação
mecânica tinham mais frequentemente a variante desmielinizante.(226) Outro
estudo associou a ocorrência de insuficiência respiratória na SGB com escore de
Hughes elevado no internamento, na rápida progressão dos sintomas, na paralisia
bulbar e na paralisia facial periférica bilateral.(233)
Assim, a insuficiência respiratória na SGB é comum e com risco de morte. A
falha respiratória deve ser detectada precocemente por observação clínica ou
testes da função pulmonar. Pacientes que necessitam de ventilação mecânica são
de alto risco para sofrerem complicações, incluindo pneumonia, sepsi,
sangramento gastrointestinal, embolia pulmonar, entre outras.
O tempo médio de duração de ventilação mecânica nos estudos variavam
de 15 a 43 dias, sugerindo que uma proporção de pacientes pode ser poupada da
traqueostomia.(200, 234-236) Na nossa série, a média de dias em ventilação
mecânica foi de 17,87. A traqueostomia foi realizada em 20 pacientes (13,4%), a
maioria do subtipo NAMSA (60%, p = 0,04). Entre os pacientes que necessitaram
ventilação mecânica, Lawn e Wijdicks observaram que a duração da ventilação e a
traqueostomia estava aumentada em pacientes idosos e na presença de doença
pulmonar prévia.(237)
A melhoria dos cuidados intensivos e a utilização de tratamento específico,
por exemplo imunoglobulina humana, tem levado a um melhor prognóstico na
SGB. Entretanto, ainda de 2 a 10% dos pacientes falecem, em torno de 20%
permanecem sem andar após seis meses do início do quadro, outros apresentam
dor crônica e/ou fadiga que impactam nas atividades da vida diária.(238-241)
A evolução desfavorável, no nosso estudo, ocorreu em 48.1% dos casos da
variante axonal contra 5,74% da variante desmielinizante (P<0,0001) e a
mortalidade foi de 5,3%. Os nossos pacientes com variante axonal tinham longa
permanência hospitalar, tardavam mais tempo para melhorar um ponto na escala

90
de Hughes e para recuperar a deambulação. Analisando a evolução dos nossos
pacientes, utilizando regressão múltipla logística com modelo de seleção forward-
LR, identificamos que as variáveis diagnóstico axonal e o tempo para melhorar um
grau na escala de Hughes estão associadas ao mau prognóstico. O curso clínico
da SGB é determinado pela extensão da lesão neural, notadamente do axônio
motor, que ocorre durante a fase aguda.
Na verdade, as variações na taxa e extensão da recuperação dessa
síndrome torna difícil determinar o prognóstico. Além da degeneração axonal
motora, que pode ser determinada pela eletroneuromiografia, outros fatores estão
associados ao mau prognóstico, a saber: idosos, infecção prévia, progressão
rápida da doença, longa duração da fase de platô, amplitude do potencial de ação
composto muscular, necessidade de ventilação mecânica.(59-61, 242)
Rinske van Koningsveld e colaboradores elaboraram, na Holanda, um
escore para determinar o prognóstico após seis meses da doença.(243) O escore,
de 1 a 7, foi desenvolvido através de análise da idade (3 categorias), presença ou
ausência de diarreia (2 categorias) e a escala de incapacidade de Hughes (5
categorias). Pacientes com escore elevado (7) têm 80% de chance de estar sem
caminhar independentemente aos seis meses do início da doença. Esse escore
deve ser validado em outros países, inclusive no Brasil, haja vista a discrepância
entre a faixa etária e, possivelmente, diferentes agentes infecciosos. Já referimos,
anteriormente, que os nossos pacientes são predominantemente jovens, com
menos de 20 anos, diferente da Europa onde a incidência aumenta com a idade.
Alem disso, não observamos que a idade e os antecedentes de diarreia
correlacionavam ou associavam com o mau prognóstico (p=0,3978 e p=0,3765,
respectivamente). Assim, é necessário determinar marcadores de prognóstico, que
provavelmente não são os mesmos entre diferentes regiões, para esta grave
afecção que demanda o melhor tratamento.
A classificação da SGB através da eletroneuromiografia é sujeita a
equívocos, notadamente na fase precoce da doença. Os critérios eletrofisiológicos
para o diagnóstico da variante NAMA se baseia na demonstração de degeneração
axonal motora e ausência de desmielinização.(6) Entretanto, sabemos que muitos
pacientes com NAMA apresentam bloqueio da condução reversível que são
classificados erroneamente de PDIA, subestimando assim a frequência da

91
NAMA.(53, 68). Estudos em humanos e modelos animais de NAMA mostraram que
o mecanismo principal das neuropatias axonais aguda associadas a anticorpos
anti-gangliosídeos era a disfunção nodal mediada por complemento.(35, 37) Essas
alterações são reversíveis, o que explica a resolução do bloqueio da condução, a
ausência de degeneração axonal e a rápida melhora clínica.(37, 81)
Por consequência, a prevalência da NAMA deve ser maior do que as
referidas nas séries publicadas. Outros fatores, tais como o número de nervos
estudados, a distribuição desigual da lesão, e a consciência do fato de que as
anormalidades podem mudar ao longo do curso da doença, também devem ser
considerados. Isso implica numa reclassificação da variante após a repetição do
teste. Portanto, um único estudo eletroneuromiográfico não é capaz de classificar
corretamente as variantes, sendo necessário realizar uma série de exames por
cada paciente e estabelecer novos critérios eletrofisiológicos para a NAMA.(53, 54,
244)
Nos últimos anos, a SGB tem sido descrita com espectro de apresentações,
algumas muito similares e outras bastante diferentes. A questão é saber se elas
representam um processo patológico comum com diversas manifestações clínicas,
ou se são doenças distintas, desta forma à espera de uma resposta definitiva.(62)
Todos os nossos casos foram examinados pelo mesmo neurologista (MEDJ), que
também realizou o estudo eletroneuromiográfico usando o mesmo protocolo. A
eletroneuromiografia foi repetida, pelo menos uma vez, em todos os pacientes com
a variante axonal e em muitos com a variante desmielinizante. Realmente, na fase
aguda da SGB a distinção entre as variantes axonal e desmielinizante pode ser
difícil ou impossível em determinados pacientes. O bloqueio da condução devido à
falha da condução na região nodal pode ocorrer na fase inicial da NAMA e da
neuropatia motora aguda com bloqueio da condução, seguida ou não de
degeneração axonal.(38, 117) Isso explica porque alguns pacientes com NAMA,
variante axonal puramente motora, não têm má evolução. Nossos pacientes com a
variante axonal tinham evolução desfavorável, mas cinco pacientes com NAMA
iniciaram a deambulação com menos de 10 dias do início do quadro. Certamente
esses novos conhecimentos, determinando diagnósticos refinados, obriga elaborar
uma nova classificação para a SGB. Por fim, o reconhecimento dos diferentes

92
subtipos da SGB é importante porque, provavelmente, eles têm diferentes
mecanismos imunopatogênicos e tratamentos.
6.2 Polimorfismo do FCGR
No presente estudo, a frequência dos genótipos e dos alelos dos FCGR2A e
FCGR3A não eram diferentes entre os pacientes e os controles. Também não
encontramos associação com a gravidade da doença. Os alelos mais prevalentes
nesse estudo foram o FCGR2A-R131 e FCGR3A-F158. Isso está de acordo com
outras populações brasileiras e é similar aos reportados nos indivíduos brancos norte-
americanos e europeus.(171, 172, 245)
A tentativa de identificar fatores de risco imunogenéticos nos pacientes com a
SGB não tem sido uniforme. A avaliação do polimorfismo dos genes FCGR foi
realizada em diferentes populações, holandesa, norueguesa, inglesa e indiana. Num
estudo de meta-análise, que incluía população inglesa e holandesa, (23) e no estudo
da Noruega (25) não havia associação entre o polimorfismo do FCGR com a
ocorrência da SGB. Num outro estudo holandês (197) e outro indiano, (24) o genótipo
FCGR2A H/H131 predominava nos pacientes com SGB. O genótipo FCGR3A-V/V158
também estava associada a SGB na população indiana. (24)
Os resultados também foram diferentes quando avaliada a frequência dos
genes e alelos com a SGB estratificada por severidade. O estudo holandês detectou
associação do genótipo FCGR2A H/H com a SGB severa. (197) Na população
norueguesa, o alelo FCGR3B NA1 estava associado com a forma leve da doença.
(25) O mesmo foi observado no estudo de meta-análise, onde o alelo FCGR3B Na2
correlacionava com a forma grave da SGB. (23) Nesse mesmo estudo, o genótipo
FCGR3A-F/F158 estava associado com formas leves da doença.
O polimorfismo genético dos FCGR, introduzindo variações entre os indivíduos,
é um dos responsáveis pela variedade de respostas biológicas. Os genótipos
FCGR2A H/H, FCGR3A V/V e FCGR3B NA1/NA1 são mais eficientes em ligar a IgG e
desempenhar suas funções, influenciando o vigor da resposta inflamatória. Ambos,
FCGR2A H/H e FCGR3A-V/V158, são expressados em macrófagos. (26)

93
Possivelmente, uma resposta imune mais acentuada, gerada por macrófagos, nos
indivíduos homozigotos para os receptores da alta afinidade pode predispor a SGB.
A importância da associação do FCGR3B (não estudado no nosso trabalho) é
questionável já que ele é expressado apenas nos neutrófilos e a participação dessas
células na patogênese da SGB ainda não está esclarecida. Estudos anteriores
mostram que os alelos FCGR3B NA1 e FCGR3B NA2 predispõem a desenvolver
forma mais leve e grave da doença, respectivamente. Interessante, o FCGR3B
NA1/NA1 aumenta a atividade do FCGR2A H/H. (246, 247) De fato, esses efeitos
autocrinos e paracrinos podem proporcionar uma base para os alelos dos receptores
FcR influenciarem outros componentes do sistema imune. Portanto, diferente dos
resultados citados, esperávamos que os indivíduos homozigotos FCGR3B fossem
mais susceptíveis a desenvolver a SGB e/ou ter a forma mais grave da doença.
Essas disparidades podem ser reflexo das diferenças étnicas, da real
heterogeneidade genética, ou por amostras pequenas e de baixo poder estatístico, o
que torna as comparações entre os estudos muito difícil. Os nossos resultados
contribuem com a disparidade, principalmente porque são diferentes dos encontrados
nos estudos anteriores. Não observamos qualquer efeito significante. Isso parece
indicar a robustez de nossos resultados negativos nesta grande amostra de pacientes
e controles.
Possivelmente, na SGB, o modelo receptor-uma função seja inadequado.
Outros mecanismos devem ser explorados na SGB, tais como, interações dos
neutrófilos com outros componentes do sistema imune, modulação das proteínas
tirosino quinases, fatores de transcrição. Vários genes, mapeados na proximidade do
locus do FCGR, que influenciam a resposta inflamatória, podem ser fonte de
explicações alternativas. (23) Além disso, recentemente, estudos demonstraram que
variação no número de cópias (VNC) de genes pode estar envolvida na
susceptibilidade de doenças, incluindo doenças autoimunes. É descrito que há VNC
no locus (1q23) onde os genes FCGR2A e FCGR3A estão localizados. A deleção de
um alelo em FCGR3A correlaciona com redução da expressão do FcRIIIa nas células
NK e com redução da função citotóxica. Até o momento, não foi relatado VNC no
FCGR2A. (26, 212) Não avaliamos a VNC nos nossos casos e controles.

94
Em resumo, este estudo sugere que genótipos FCGR2A e FCGR3A não estão
associados com a susceptibilidade para a SGB ou com o curso da doença. Nós
concluímos que os polimorfismos dos FCGR2A e FCGR3A não são fatores de risco
para SGB. Estudos são necessários para analisar a região do FCGR, usando
sequenciamento de novas gerações, e para entender como mecanismos relacionados
com FCGR podem interferir no desenvolvimento da SGB.
6.3 Expressão gênica global em células mononucleares de sangue periférico
durante a Síndrome de Guillain Barré
A análise preliminar mostra diferenças na expressão de fatores
transcricionais (EGR, FOSB, SOCS3,HLA) e quimiocinas e citocinas (IFNG,
CXCR2, CXCR1), além de outras proteínas inflamatórias e proteolíticas (PTGS2,
MMM-9, THBS1).
De relevância é a expressão de EGR (early growth response 3), gene que
regula a expressão de mais de 330 genes, a maioria dos quais está relacionada
com a resposta imune e processos inflamatórios. O EGR induz a expressão de
citocinas, fatores de crescimento e fatores de modelação de matriz. (248)
O gene FOS codifica um fator de transcrição que regula a proliferação,
diferenciação e transformação celular. Em cooperação com outros fatores, o FOS
regula expressão de várias interleucinas, TNF-alfa, IFN gama, CD40L, CD5 e
CD25. (249)
O gene IFNGR1 codifica a cadeia alfa do Interferon gama. Essa citocina
produzida pelas celulas T é o principal marcador de resposta Th1. As funções do
interferon-gama são ativar macrofagos, célula importante na patogenia da SGB,
induzir expressão de MHCI e II, mudança de classe de células B, apoptose de
células T, aumentar a produção de outras citocinas, tais como TFN-alfa e IL-1b.
(250, 251)
O gene Supressor Of Cytokine Signaling (SOCS) produz proteína
intracellular que inibe os sinais das citocinas em várias células. O grande número
de infecções virais está associada com o aumento da expressão desse gene. A

95
SOCS 3 é expressada na célula de Schwann e é um fator negativo de sinais de
citocinas em macrófagos. Modelos animais de lesão de nervo periférico mostram a
importância da SOCS na degeneração Waleriana. (252)
A proteína codificada pelo gene PTGS2 é uma enzima, da família ciclo-
oxigenase (COX-2), que converte o ácido aracdônico em prostaglandinas. O
PTGS2 está envolvido com eventos biológicos de lesão, inflamação ou
proliferação. Na SGB e em outras neuropatias imunomediadas foi descrito aumento
na expressão do PTGS2 no nervo sural. (253) Em modelos animais da SGB, a
administração de inibidores da COX-2 tem efeito neuroprotetor. (254)
As metaloproteinases da matriz extracelular, MMP-9 e THBS-1 por exemplo,
são responsáveis pela degradação dos elementos constituintes da membrana
basal e da matriz extracelular. A MMP-9 está aumentada no soro dos pacientes
com SGB e correlaciona com a gravidade, assim como o seu nível aumenta na
neuropatia alérgica experimental. Inibidores da MMP-9 diminui a gravidade da
neuropatia alérgica experimental. (255, 256) THBS-1 é uma glicoproteína da matriz
extracelular secretada por plaquetas, músculo liso, astrócitos, células endoteliais e
várias células tumorais. O aumento da expressão do gene THBS-1 está associado
com proteólise em glioblastoma. (257)
Esse achados preliminaries validam estudo anterior (208) e apontam novas
vias metabólicas potencialmente envolvidas na fisiopatologia da Síndrome de
Guillain-Barré, passíveis de serem reguladas e possibilitando novas estratégias
terapêuticas.

96
7. CONCLUSÕES
1. A incidência da Síndrome de Guillain-Barré no Rio Grande do Norte é
semelhante àquela observada para outras regiões do Brasil.
2. A Síndrome de Guillain-Barré predomina em jovens.
3. A variante desmielinizante é a mais frequente.
4. A variante axonal está associada à diarreia, nadir curto e déficit motor
distal, enquanto a forma desmielinizante apresenta déficit motor proximal,
alterações sensitivas e envolvimento do nervo facial.
5. Fase de platô prolongada está associada à variante axonal
6. As variáveis diagnóstico axonal e o tempo para melhorar um ponto na
escala de Hughes estão associadas ao mau prognóstico.
7. Os genótipos e alelos FCGR2A e FCGR3A não estão associados à
síndrome de Guillain-Barré ou a sua gravidade.
8. Há diferenças significativas na expressão global da Sindrome de
Guillain-Barré, com aumento de genes relacionados à resposta
inflamatória, fatores de crescimento e fatores transcricionais.

97
8 REFERÊNCIAS
1. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet neurology. 2008 Oct;7(10):939-50. PubMed PMID: 18848313. 2. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology. 2009;32(2):150-63. PubMed PMID: 19088488. 3. Yuki N, Hartung HP. Guillain-Barre syndrome. The New England journal of medicine. 2012 Jun 14;366(24):2294-304. PubMed PMID: 22694000. 4. McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Annals of neurology. 1993 Apr;33(4):333-42. PubMed PMID: 8489203. 5. Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, et al. Electrophysiological classification of Guillain-Barre syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Annals of neurology. 1998 Nov;44(5):780-8. PubMed PMID: 9818934. 6. Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, et al. Guillain-Barre syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain : a journal of neurology. 1995 Jun;118 ( Pt 3):597-605. PubMed PMID: 7600081. 7. Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36(2):123-33. PubMed PMID: 21422765. 8. Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005 Nov 5;366(9497):1653-66. PubMed PMID: 16271648. 9. Hiraga A, Mori M, Ogawara K, Hattori T, Kuwabara S. Differences in patterns of progression in demyelinating and axonal Guillain-Barre syndromes. Neurology. 2003 Aug 26;61(4):471-4. PubMed PMID: 12939419. 10. Nagasawa K, Kuwabara S, Misawa S, Fujii K, Tanabe Y, Yuki N, et al. Electrophysiological subtypes and prognosis of childhood Guillain-Barre syndrome in Japan. Muscle & nerve. 2006 Jun;33(6):766-70. PubMed PMID: 16506153. 11. Nachamkin I, Arzarte Barbosa P, Ung H, Lobato C, Gonzalez Rivera A, Rodriguez P, et al. Patterns of Guillain-Barre syndrome in children: results from a Mexican population. Neurology. 2007 Oct 23;69(17):1665-71. PubMed PMID: 17898327. 12. Paradiso G, Tripoli J, Galicchio S, Fejerman N. Epidemiological, clinical, and electrodiagnostic findings in childhood Guillain-Barre syndrome: a reappraisal.

98
Annals of neurology. 1999 Nov;46(5):701-7. PubMed PMID: 10553986. 13. van Koningsveld R, Rico R, Gerstenbluth I, Schmitz PI, Ang CW, Merkies IS, et al. Gastroenteritis-associated Guillain-Barre syndrome on the Caribbean island Curacao. Neurology. 2001 Jun 12;56(11):1467-72. PubMed PMID: 11402102. 14. Dourado ME, Duarte RC, Ferreira LC, Queiroz JW, Illa I, Perez-Perez G, et al. Anti-ganglioside antibodies and clinical outcome of patients with Guillain-Barre Syndrome in northeast Brazil. Acta neurologica Scandinavica. 2003 Aug;108(2):102-8. PubMed PMID: 12859286. 15. Dias-Tosta E, Kuckelhaus CS. Guillain Barre syndrome in a population less than 15 years old in Brazil. Arquivos de neuro-psiquiatria. 2002 Jun;60(2-B):367-73. PubMed PMID: 12131933. 16. Yuki N, Taki T, Inagaki F, Kasama T, Takahashi M, Saito K, et al. A bacterium lipopolysaccharide that elicits Guillain-Barre syndrome has a GM1 ganglioside-like structure. The Journal of experimental medicine. 1993 Nov 1;178(5):1771-5. PubMed PMID: 8228822. Pubmed Central PMCID: 2191246. 17. Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barre and Fisher syndromes. The Lancet infectious diseases. 2001 Aug;1(1):29-37. PubMed PMID: 11871407. 18. Houliston RS, Koga M, Li J, Jarrell HC, Richards JC, Vitiazeva V, et al. A Haemophilus influenzae strain associated with Fisher syndrome expresses a novel disialylated ganglioside mimic. Biochemistry. 2007 Jul 10;46(27):8164-71. PubMed PMID: 17567050. 19. Yuki N, Tagawa Y. Acute cytomegalovirus infection and IgM anti-GM2 antibody. Journal of the neurological sciences. 1998 Jan 21;154(1):14-7. PubMed PMID: 9543317. 20. Soares CN, Cabral-Castro M, Oliveira C, Faria LC, Peralta JM, Freitas MR, et al. Oligosymptomatic dengue infection: a potential cause of Guillain Barre syndrome. Arquivos de neuro-psiquiatria. 2008 Jun;66(2A):234-7. PubMed PMID: 18545789. 21. Santos NQ, Azoubel AC, Lopes AA, Costa G, Bacellar A. Guillain-Barre syndrome in the course of dengue: case report. Arquivos de neuro-psiquiatria. 2004 Mar;62(1):144-6. PubMed PMID: 15122449. 22. Sheikh KA, Nachamkin I, Ho TW, Willison HJ, Veitch J, Ung H, et al. Campylobacter jejuni lipopolysaccharides in Guillain-Barre syndrome: molecular mimicry and host susceptibility. Neurology. 1998 Aug;51(2):371-8. PubMed PMID: 9710005. 23. van Sorge NM, van der Pol WL, Jansen MD, Geleijns KP, Kalmijn S, Hughes RA, et al. Severity of Guillain-Barre syndrome is associated with Fc gamma Receptor III polymorphisms. Journal of neuroimmunology. 2005 May;162(1-2):157-

99
64. PubMed PMID: 15833371. 24. Sinha S, Prasad KN, Jain D, Nyati KK, Pradhan S, Agrawal S. Immunoglobulin IgG Fc-receptor polymorphisms and HLA class II molecules in Guillain-Barre syndrome. Acta neurologica Scandinavica. 2010 Jul;122(1):21-6. PubMed PMID: 20105138. 25. Vedeler CA, Raknes G, Myhr KM, Nyland H. IgG Fc-receptor polymorphisms in Guillain-Barre syndrome. Neurology. 2000 Sep 12;55(5):705-7. PubMed PMID: 10980740. 26. Gillis C, Gouel-Cheron A, Jonsson F, Bruhns P. Contribution of Human FcgammaRs to Disease with Evidence from Human Polymorphisms and Transgenic Animal Studies. Frontiers in immunology. 2014;5:254. PubMed PMID: 24910634. Pubmed Central PMCID: 4038777. 27. Nimmerjahn F. Activating and inhibitory FcgammaRs in autoimmune disorders. Springer seminars in immunopathology. 2006 Dec;28(4):305-19. PubMed PMID: 17115158. 28. Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. The Journal of experimental medicine. 2005 Mar 7;201(5):747-54. PubMed PMID: 15753208. Pubmed Central PMCID: 1351290. 29. Oliveira CR, Pereira LI, Pereira AJ, Ferreira AA, Crespo AM, Silveira LA. Allelic polymorphism of human FcgammaRIIA-H/R131 receptor in American tegumentary leishmaniasis. International journal of immunogenetics. 2011 Jun;38(3):225-31. PubMed PMID: 21324097. 30. Saliba AE, Westermann AJ, Gorski SA, Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic acids research. 2014 Aug;42(14):8845-60. PubMed PMID: 25053837. Pubmed Central PMCID: 4132710. 31. Pritchard J, Hughes RA. Guillain-Barre syndrome. Lancet. 2004 Jun 26;363(9427):2186-8. PubMed PMID: 15220044. 32. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). The New England journal of medicine. 1956 Jul 12;255(2):57-65. PubMed PMID: 13334797. 33. Plasmapheresis and acute Guillain-Barre syndrome. The Guillain-Barre syndrome Study Group. Neurology. 1985 Aug;35(8):1096-104. PubMed PMID: 4022342. 34. van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. The New England journal of medicine. 1992 Apr 23;326(17):1123-9. PubMed PMID: 1552913.

100
35. Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Annals of neurology. 1996 Oct;40(4):635-44. PubMed PMID: 8871584. 36. Kaldor J, Speed BR. Guillain-Barre syndrome and Campylobacter jejuni: a serological study. British medical journal. 1984 Jun 23;288(6434):1867-70. PubMed PMID: 6428580. Pubmed Central PMCID: 1441765. 37. Susuki K, Yuki N, Schafer DP, Hirata K, Zhang G, Funakoshi K, et al. Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Experimental neurology. 2012 Jan;233(1):534-42. PubMed PMID: 22178332. Pubmed Central PMCID: 3268874. 38. Kokubun N, Nishibayashi M, Uncini A, Odaka M, Hirata K, Yuki N. Conduction block in acute motor axonal neuropathy. Brain : a journal of neurology. 2010 Oct;133(10):2897-908. PubMed PMID: 20855419. 39. Ilyas AA, Willison HJ, Quarles RH, Jungalwala FB, Cornblath DR, Trapp BD, et al. Serum antibodies to gangliosides in Guillain-Barre syndrome. Annals of neurology. 1988 May;23(5):440-7. PubMed PMID: 3133978. 40. Gilbert M, Brisson JR, Karwaski MF, Michniewicz J, Cunningham AM, Wu Y, et al. Biosynthesis of ganglioside mimics in Campylobacter jejuni OH4384. Identification of the glycosyltransferase genes, enzymatic synthesis of model compounds, and characterization of nanomole amounts by 600-mhz (1)h and (13)c NMR analysis. The Journal of biological chemistry. 2000 Feb 11;275(6):3896-906. PubMed PMID: 10660542. 41. Godschalk PC, Kuijf ML, Li J, St Michael F, Ang CW, Jacobs BC, et al. Structural characterization of Campylobacter jejuni lipooligosaccharide outer cores associated with Guillain-Barre and Miller Fisher syndromes. Infection and immunity. 2007 Mar;75(3):1245-54. PubMed PMID: 17261613. Pubmed Central PMCID: 1828588. 42. Shui IM, Rett MD, Weintraub E, Marcy M, Amato AA, Sheikh SI, et al. Guillain-Barre syndrome incidence in a large United States cohort (2000-2009). Neuroepidemiology. 2012;39(2):109-15. PubMed PMID: 22846726. 43. Rocha MS, Brucki SM, Carvalho AA, Lima UW. Epidemiologic features of Guillain-Barre syndrome in Sao Paulo, Brazil. Arquivos de neuro-psiquiatria. 2004 Mar;62(1):33-7. PubMed PMID: 15122430. 44. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barre syndrome. Annals of neurology. 1990;27 Suppl:S21-4. PubMed PMID: 2194422. 45. Forsberg A, Press R, Einarsson U, de Pedro-Cuesta J, Widen Holmqvist L, Swedish Epidemiological Study G. Impairment in Guillain-Barre syndrome during the first 2 years after onset: a prospective study. Journal of the neurological sciences. 2004 Dec 15;227(1):131-8. PubMed PMID: 15546603.

101
46. Moulin DE, Hagen N, Feasby TE, Amireh R, Hahn A. Pain in Guillain-Barre syndrome. Neurology. 1997 Feb;48(2):328-31. PubMed PMID: 9040715. 47. Ropper AH, Shahani BT. Pain in Guillain-Barre syndrome. Archives of neurology. 1984 May;41(5):511-4. PubMed PMID: 6721720. 48. Pentland B, Donald SM. Pain in the Guillain-Barre syndrome: a clinical review. Pain. 1994 Nov;59(2):159-64. PubMed PMID: 7892013. 49. Ruts L, Drenthen J, Jongen JL, Hop WC, Visser GH, Jacobs BC, et al. Pain in Guillain-Barre syndrome: a long-term follow-up study. Neurology. 2010 Oct 19;75(16):1439-47. PubMed PMID: 20861454. 50. Cornblath DR. Electrophysiology in Guillain-Barre syndrome. Annals of neurology. 1990;27 Suppl:S17-20. PubMed PMID: 2194420. 51. Meulstee J, van der Meche FG. Electrodiagnostic criteria for polyneuropathy and demyelination: application in 135 patients with Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. Journal of neurology, neurosurgery, and psychiatry. 1995 Nov;59(5):482-6. PubMed PMID: 8530930. Pubmed Central PMCID: 1073708. 52. Kalita J, Misra UK, Das M. Neurophysiological criteria in the diagnosis of different clinical types of Guillain-Barre syndrome. Journal of neurology, neurosurgery, and psychiatry. 2008 Mar;79(3):289-93. PubMed PMID: 17615164. 53. Uncini A, Manzoli C, Notturno F, Capasso M. Pitfalls in electrodiagnosis of Guillain-Barre syndrome subtypes. Journal of neurology, neurosurgery, and psychiatry. 2010 Oct;81(10):1157-63. PubMed PMID: 20870864. 54. Uncini A, Kuwabara S. Electrodiagnostic criteria for Guillain-Barre syndrome: a critical revision and the need for an update. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2012 Aug;123(8):1487-95. PubMed PMID: 22480600. 55. Sejvar JJ, Kohl KS, Gidudu J, Amato A, Bakshi N, Baxter R, et al. Guillain-Barre syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2011 Jan 10;29(3):599-612. PubMed PMID: 20600491. 56. Van der Meche FG, Van Doorn PA, Meulstee J, Jennekens FG, Centre GB-cgotDNRS. Diagnostic and classification criteria for the Guillain-Barre syndrome. European neurology. 2001;45(3):133-9. PubMed PMID: 11306855. 57. Hahn AF. Guillain-Barre syndrome. Lancet. 1998 Aug 22;352(9128):635-41. PubMed PMID: 9746040. 58. Hughes RA, Hadden RD, Rees JH, Swan AV. The Italian Guillain-Barre Study Group. The prognosis and main prognostic indicators of Guillain-Barre

102
syndrome: a multicentre prospective study of 297 patients. Brain : a journal of neurology. 1998 Apr;121 ( Pt 4):767-9. PubMed PMID: 9577400. 59. A prospective study on the incidence and prognosis of Guillain-Barre syndrome in Emilia-Romagna region, Italy (1992-1993). Emilia-Romagna Study Group on Clinical and Epidemiological Problems in Neurology. Neurology. 1997 Jan;48(1):214-21. PubMed PMID: 9008520. 60. Visser LH, Schmitz PI, Meulstee J, van Doorn PA, van der Meche FG. Prognostic factors of Guillain-Barre syndrome after intravenous immunoglobulin or plasma exchange. Dutch Guillain-Barre Study Group. Neurology. 1999 Aug 11;53(3):598-604. PubMed PMID: 10449126. 61. Chio A, Cocito D, Leone M, Giordana MT, Mora G, Mutani R, et al. Guillain-Barre syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003 Apr 8;60(7):1146-50. PubMed PMID: 12682322. 62. Sladky JT. Guillain-Barre syndrome: blind men describe an elephant? Neurology. 2007 Oct 23;69(17):1647-9. PubMed PMID: 17954779. 63. Islam Z, Jacobs BC, van Belkum A, Mohammad QD, Islam MB, Herbrink P, et al. Axonal variant of Guillain-Barre syndrome associated with Campylobacter infection in Bangladesh. Neurology. 2010 Feb 16;74(7):581-7. PubMed PMID: 20157160. 64. Kushnir M, Klein C, Pollak L, Rabey JM. Evolving pattern of Guillain-Barre syndrome in a community hospital in Israel. Acta neurologica Scandinavica. 2008 May;117(5):347-50. PubMed PMID: 17995988. 65. Franca MC, Jr., Deus-Silva L, de Castro R, Garibaldi SG, Pfeilsticker BH, Nucci A, et al. Guillain-Barre syndrome in the elderly: clinical, electrophysiological, therapeutic and outcome features. Arquivos de neuro-psiquiatria. 2005 Sep;63(3B):772-5. PubMed PMID: 16258654. 66. Franssen H, Straver DC. Pathophysiology of immune-mediated demyelinating neuropathies-Part II: Neurology. Muscle & nerve. 2014 Jan;49(1):4-20. PubMed PMID: 24037667. 67. McKhann GM, Cornblath DR, Ho T, Li CY, Bai AY, Wu HS, et al. Clinical and electrophysiological aspects of acute paralytic disease of children and young adults in northern China. Lancet. 1991 Sep 7;338(8767):593-7. PubMed PMID: 1679153. 68. Sekiguchi Y, Uncini A, Yuki N, Misawa S, Notturno F, Nasu S, et al. Antiganglioside antibodies are associated with axonal Guillain-Barre syndrome: a Japanese-Italian collaborative study. Journal of neurology, neurosurgery, and psychiatry. 2012 Jan;83(1):23-8. PubMed PMID: 22010183. 69. Capasso M, Caporale CM, Pomilio F, Gandolfi P, Lugaresi A, Uncini A. Acute motor conduction block neuropathy Another Guillain-Barre syndrome variant. Neurology. 2003 Sep 9;61(5):617-22. PubMed PMID: 12963751.

103
70. Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WF, et al. An acute axonal form of Guillain-Barre polyneuropathy. Brain : a journal of neurology. 1986 Dec;109 ( Pt 6):1115-26. PubMed PMID: 3790970. 71. Sauron B, Bouche P, Cathala HP, Chain F, Castaigne P. Miller Fisher syndrome: clinical and electrophysiologic evidence of peripheral origin in 10 cases. Neurology. 1984 Jul;34(7):953-6. PubMed PMID: 6539872. 72. Fross RD, Daube JR. Neuropathy in the Miller Fisher syndrome: clinical and electrophysiologic findings. Neurology. 1987 Sep;37(9):1493-8. PubMed PMID: 2819783. 73. Aranyi Z, Kovacs T, Sipos I, Bereczki D. Miller Fisher syndrome: brief overview and update with a focus on electrophysiological findings. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2012 Jan;19(1):15-20, e1-3. PubMed PMID: 21631649. 74. Richter RB. The ataxic form of polyradiculoneuritis (Landry-Guillain-Barre syndrome). Clinical and pathologic observations. Journal of neuropathology and experimental neurology. 1962 Apr;21:171-84. PubMed PMID: 14492053. 75. Kuwabara S, Asahina M, Nakajima M, Mori M, Fukutake T, Hattori T, et al. Special sensory ataxia in Miller Fisher syndrome detected by postural body sway analysis. Annals of neurology. 1999 Apr;45(4):533-6. PubMed PMID: 10211482. 76. Ito M, Matsuno K, Sakumoto Y, Hirata K, Yuki N. Ataxic Guillain-Barre syndrome and acute sensory ataxic neuropathy form a continuous spectrum. Journal of neurology, neurosurgery, and psychiatry. 2011 Mar;82(3):294-9. PubMed PMID: 21252265. 77. Hafer-Macko CE, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Annals of neurology. 1996 May;39(5):625-35. PubMed PMID: 8619548. 78. Koski CL, Sanders ME, Swoveland PT, Lawley TJ, Shin ML, Frank MM, et al. Activation of terminal components of complement in patients with Guillain-Barre syndrome and other demyelinating neuropathies. The Journal of clinical investigation. 1987 Nov;80(5):1492-7. PubMed PMID: 3680509. Pubmed Central PMCID: 442409. 79. Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain : a journal of neurology. 2002 Dec;125(Pt 12):2591-625. PubMed PMID: 12429589. 80. Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, et al. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007 Apr 11;27(15):3956-67. PubMed PMID: 17428969.

104
81. Uncini A. A common mechanism and a new categorization for anti-ganglioside antibody-mediated neuropathies. Experimental neurology. 2012 Jun;235(2):513-6. PubMed PMID: 22507308. 82. Pulitano S, Viola L, Genovese O, Chiaretti A, Piastra M, Mariotti P, et al. Miller-Fisher syndrome mimicking intracranial hypertension following head trauma. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2005 Jun;21(6):473-6. PubMed PMID: 15830202. 83. Barontini F, Di Lollo S, Maurri S, Lambruschini P. Localization of the pathological process in Miller Fisher syndrome. Italian journal of neurological sciences. 1992 Apr;13(3):221-5. PubMed PMID: 1624278. 84. Lo YL. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle & nerve. 2007 Nov;36(5):615-27. PubMed PMID: 17657801. 85. Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet neurology. 2012 Jul;11(7):629-42. PubMed PMID: 22710756. 86. Martinez V, Fletcher D, Martin F, Orlikowski D, Sharshar T, Chauvin M, et al. Small fibre impairment predicts neuropathic pain in Guillain-Barre syndrome. Pain. 2010 Oct;151(1):53-60. PubMed PMID: 20696121. 87. Ruts L, van Doorn PA, Lombardi R, Haasdijk ED, Penza P, Tulen JH, et al. Unmyelinated and myelinated skin nerve damage in Guillain-Barre syndrome: correlation with pain and recovery. Pain. 2012 Feb;153(2):399-409. PubMed PMID: 22154920. 88. Caudie C, Quittard Pinon A, Taravel D, Sivadon-Tardy V, Orlikowski D, Rozenberg F, et al. Preceding infections and anti-ganglioside antibody profiles assessed by a dot immunoassay in 306 French Guillain-Barre syndrome patients. Journal of neurology. 2011 Nov;258(11):1958-64. PubMed PMID: 21516465. 89. Susuki K, Nishimoto Y, Yamada M, Baba M, Ueda S, Hirata K, et al. Acute motor axonal neuropathy rabbit model: immune attack on nerve root axons. Annals of neurology. 2003 Sep;54(3):383-8. PubMed PMID: 12953272. 90. Vriesendorp FJ, Mishu B, Blaser MJ, Koski CL. Serum antibodies to GM1, GD1b, peripheral nerve myelin, and Campylobacter jejuni in patients with Guillain-Barre syndrome and controls: correlation and prognosis. Annals of neurology. 1993 Aug;34(2):130-5. PubMed PMID: 8338337. 91. Rees JH, Gregson NA, Hughes RA. Anti-ganglioside GM1 antibodies in Guillain-Barre syndrome and their relationship to Campylobacter jejuni infection. Annals of neurology. 1995 Nov;38(5):809-16. PubMed PMID: 7486873. 92. Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, et al. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barre syndrome. Annals of neurology. 1998 Aug;44(2):202-

105
8. PubMed PMID: 9708542. 93. Jacobs BC, Schmitz PI, van der Meche FG. Campylobactery jejuni Infection and treatment for Guillain-Barre Syndrome. The New England journal of medicine. 1996 Jul 18;335(3):208-9. PubMed PMID: 8657230. 94. Vriesendorp FJ, Triggs WJ, Mayer RF, Koski CL. Electrophysiological studies in Guillain-Barre syndrome: correlation with antibodies to GM1, GD1B and Campylobacter jejuni. Journal of neurology. 1995 Jul;242(7):460-5. PubMed PMID: 7595678. 95. Lardone RD, Yuki N, Odaka M, Daniotti JL, Irazoqui FJ, Nores GA. Anti-GM1 IgG antibodies in Guillain-Barre syndrome: fine specificity is associated with disease severity. Journal of neurology, neurosurgery, and psychiatry. 2010 Jun;81(6):629-33. PubMed PMID: 19965859. 96. Lopez PH, Zhang G, Zhang J, Lehmann HC, Griffin JW, Schnaar RL, et al. Passive transfer of IgG anti-GM1 antibodies impairs peripheral nerve repair. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010 Jul 14;30(28):9533-41. PubMed PMID: 20631181. Pubmed Central PMCID: 3038609. 97. Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barre syndrome: clinical and immunohistochemical studies. Neurology. 1993 Oct;43(10):1911-7. PubMed PMID: 8413947. 98. vd Kruijk RA, Lampe AS, Endtz HP. Bilateral abducens paresis following Campylobacter jejuni enteritis. The Journal of infection. 1992 Mar;24(2):215-6. PubMed PMID: 1569314. 99. Kusunoki S, Chiba A, Kanazawa I. Anti-GQ1b IgG antibody is associated with ataxia as well as ophthalmoplegia. Muscle & nerve. 1999 Aug;22(8):1071-4. PubMed PMID: 10417789. 100. Ito M, Kuwabara S, Odaka M, Misawa S, Koga M, Hirata K, et al. Bickerstaff's brainstem encephalitis and Fisher syndrome form a continuous spectrum: clinical analysis of 581 cases. Journal of neurology. 2008 May;255(5):674-82. PubMed PMID: 18274803. 101. Nagashima T, Koga M, Odaka M, Hirata K, Yuki N. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. Archives of neurology. 2007 Oct;64(10):1519-23. PubMed PMID: 17923636. 102. Liu JX, Willison HJ, Pedrosa-Domellof F. Immunolocalization of GQ1b and related gangliosides in human extraocular neuromuscular junctions and muscle spindles. Investigative ophthalmology & visual science. 2009 Jul;50(7):3226-32. PubMed PMID: 19255160. 103. Koga M, Yoshino H, Morimatsu M, Yuki N. Anti-GT1a IgG in Guillain-Barre

106
syndrome. Journal of neurology, neurosurgery, and psychiatry. 2002 Jun;72(6):767-71. PubMed PMID: 12023422. Pubmed Central PMCID: 1737908. 104. Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M, et al. Ganglioside complexes as new target antigens in Guillain-Barre syndrome. Annals of neurology. 2004 Oct;56(4):567-71. PubMed PMID: 15389898. 105. Kaida K, Sonoo M, Ogawa G, Kamakura K, Ueda-Sada M, Arita M, et al. GM1/GalNAc-GD1a complex: a target for pure motor Guillain-Barre syndrome. Neurology. 2008 Nov 18;71(21):1683-90. PubMed PMID: 19015484. 106. Ogawa G, Kaida K, Kuwahara M, Kimura F, Kamakura K, Kusunoki S. An antibody to the GM1/GalNAc-GD1a complex correlates with development of pure motor Guillain-Barre syndrome with reversible conduction failure. Journal of neuroimmunology. 2013 Jan 15;254(1-2):141-5. PubMed PMID: 23000056. 107. Alaedini A, Briani C, Wirguin I, Siciliano G, D'Avino C, Latov N. Detection of anti-ganglioside antibodies in Guillain-Barre syndrome and its variants by the agglutination assay. Journal of the neurological sciences. 2002 Apr 15;196(1-2):41-4. PubMed PMID: 11959155. 108. Hadden RD, Karch H, Hartung HP, Zielasek J, Weissbrich B, Schubert J, et al. Preceding infections, immune factors, and outcome in Guillain-Barre syndrome. Neurology. 2001 Mar 27;56(6):758-65. PubMed PMID: 11274311. 109. Jacobs BC, Rothbarth PH, van der Meche FG, Herbrink P, Schmitz PI, de Klerk MA, et al. The spectrum of antecedent infections in Guillain-Barre syndrome: a case-control study. Neurology. 1998 Oct;51(4):1110-5. PubMed PMID: 9781538. 110. Tam CC, O'Brien SJ, Petersen I, Islam A, Hayward A, Rodrigues LC. Guillain-Barre syndrome and preceding infection with campylobacter, influenza and Epstein-Barr virus in the general practice research database. PloS one. 2007;2(4):e344. PubMed PMID: 17406668. Pubmed Central PMCID: 1828628. 111. Rees JH, Soudain SE, Gregson NA, Hughes RA. Campylobacter jejuni infection and Guillain-Barre syndrome. The New England journal of medicine. 1995 Nov 23;333(21):1374-9. PubMed PMID: 7477117. 112. Koga M, Yuki N, Ariga T, Morimatsu M, Hirata K. Is IgG anti-GT1a antibody associated with pharyngeal-cervical-brachial weakness or oropharyngeal palsy in Guillain-Barre syndrome? Journal of neuroimmunology. 1998 Jun 1;86(1):74-9. PubMed PMID: 9655474. 113. Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990 Dec;40(12):1900-2. PubMed PMID: 2247243. 114. Kuwabara S. Does Campylobacter jejuni infection elicit axonal or demyelinating Guillain-Barre syndrome, or both? Journal of neurology, neurosurgery, and psychiatry. 2011 Mar;82(3):238. PubMed PMID: 21325023.

107
115. Drenthen J, Yuki N, Meulstee J, Maathuis EM, van Doorn PA, Visser GH, et al. Guillain-Barre syndrome subtypes related to Campylobacter infection. Journal of neurology, neurosurgery, and psychiatry. 2011 Mar;82(3):300-5. PubMed PMID: 21270063. 116. Kokubun N, Shahrizaila N, Koga M, Hirata K, Yuki N. The demyelination neurophysiological criteria can be misleading in Campylobacter jejuni-related Guillain-Barre syndrome. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2013 Aug;124(8):1671-9. PubMed PMID: 23514735. 117. Kokubun N, Shahrizaila N, Hirata K, Yuki N. Reversible conduction failure is distinct from neurophysiological patterns of recovery in mild demyelinating Guillain-Barre syndrome. Journal of the neurological sciences. 2013 Mar 15;326(1-2):111-4. PubMed PMID: 23394975. 118. Griffin JW, Li CY, Ho TW, Tian M, Gao CY, Xue P, et al. Pathology of the motor-sensory axonal Guillain-Barre syndrome. Annals of neurology. 1996 Jan;39(1):17-28. PubMed PMID: 8572662. 119. Kaida K, Kusunoki S, Kamakura K, Motoyoshi K, Kanazawa I. Guillain-Barre syndrome with antibody to a ganglioside, N-acetylgalactosaminyl GD1a. Brain : a journal of neurology. 2000 Jan;123 ( Pt 1):116-24. PubMed PMID: 10611126. 120. Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y, et al. Animal model of axonal Guillain-Barre syndrome induced by sensitization with GM1 ganglioside. Annals of neurology. 2001 Jun;49(6):712-20. PubMed PMID: 11409422. 121. Usuki S, Taguchi K, Cawthraw SA, Shibata K, Ariga T, Newell DG, et al. Human and chicken antibodies to gangliosides following infection by Campylobacter jejuni. Experimental neurology. 2006 Jul;200(1):50-5. PubMed PMID: 16500643. 122. Komagamine T, Yuki N. Ganglioside mimicry as a cause of Guillain-Barre syndrome. CNS & neurological disorders drug targets. 2006 Aug;5(4):391-400. PubMed PMID: 16918391. 123. Gilbert M, Karwaski MF, Bernatchez S, Young NM, Taboada E, Michniewicz J, et al. The genetic bases for the variation in the lipo-oligosaccharide of the mucosal pathogen, Campylobacter jejuni. Biosynthesis of sialylated ganglioside mimics in the core oligosaccharide. The Journal of biological chemistry. 2002 Jan 4;277(1):327-37. PubMed PMID: 11689567. 124. Fry BN, Feng S, Chen YY, Newell DG, Coloe PJ, Korolik V. The galE gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and virulence. Infection and immunity. 2000 May;68(5):2594-601. PubMed PMID: 10768949. Pubmed Central PMCID: 97464. 125. Yuki N, Koga M. Bacterial infections in Guillain-Barre and Fisher syndromes. Current opinion in neurology. 2006 Oct;19(5):451-7. PubMed PMID: 16969154.

108
126. Visser LH, van der Meche FG, Meulstee J, Rothbarth PP, Jacobs BC, Schmitz PI, et al. Cytomegalovirus infection and Guillain-Barre syndrome: the clinical, electrophysiologic, and prognostic features. Dutch Guillain-Barre Study Group. Neurology. 1996 Sep;47(3):668-73. PubMed PMID: 8797462. 127. Winer JB, Hughes RA, Anderson MJ, Jones DM, Kangro H, Watkins RP. A prospective study of acute idiopathic neuropathy. II. Antecedent events. Journal of neurology, neurosurgery, and psychiatry. 1988 May;51(5):613-8. PubMed PMID: 3404161. Pubmed Central PMCID: 1033063. 128. Mori M, Kuwabara S, Miyake M, Dezawa M, Adachi-Usami E, Kuroki H, et al. Haemophilus influenzae has a GM1 ganglioside-like structure and elicits Guillain-Barre syndrome. Neurology. 1999 Apr 12;52(6):1282-4. PubMed PMID: 10214761. 129. Susuki K, Odaka M, Mori M, Hirata K, Yuki N. Acute motor axonal neuropathy after Mycoplasma infection: Evidence of molecular mimicry. Neurology. 2004 Mar 23;62(6):949-56. PubMed PMID: 15037698. 130. Soares CN, Faria LC, Peralta JM, de Freitas MR, Puccioni-Sohler M. Dengue infection: neurological manifestations and cerebrospinal fluid (CSF) analysis. Journal of the neurological sciences. 2006 Nov 1;249(1):19-24. PubMed PMID: 16870213. 131. Haber P, DeStefano F, Angulo FJ, Iskander J, Shadomy SV, Weintraub E, et al. Guillain-Barre syndrome following influenza vaccination. JAMA : the journal of the American Medical Association. 2004 Nov 24;292(20):2478-81. PubMed PMID: 15562126. 132. Hughes R, Rees J, Smeeton N, Winer J. Vaccines and Guillain-Barre syndrome. Bmj. 1996 Jun 8;312(7044):1475-6. PubMed PMID: 8664637. Pubmed Central PMCID: 2351205. 133. Lasky T, Terracciano GJ, Magder L, Koski CL, Ballesteros M, Nash D, et al. The Guillain-Barre syndrome and the 1992-1993 and 1993-1994 influenza vaccines. The New England journal of medicine. 1998 Dec 17;339(25):1797-802. PubMed PMID: 9854114. 134. Nakayama T, Onoda K. Vaccine adverse events reported in post-marketing study of the Kitasato Institute from 1994 to 2004. Vaccine. 2007 Jan 5;25(3):570-6. PubMed PMID: 16945455. 135. Souayah N, Nasar A, Suri MF, Qureshi AI. Guillain-Barre syndrome after vaccination in United States: data from the Centers for Disease Control and Prevention/Food and Drug Administration Vaccine Adverse Event Reporting System (1990-2005). Journal of clinical neuromuscular disease. 2009 Sep;11(1):1-6. PubMed PMID: 19730016. 136. Tuttle J, Chen RT, Rantala H, Cherry JD, Rhodes PH, Hadler S. The risk of Guillain-Barre syndrome after tetanus-toxoid-containing vaccines in adults and children in the United States. American journal of public health. 1997 Dec;87(12):2045-8. PubMed PMID: 9431302. Pubmed Central PMCID: 1381255.

109
137. Nelson KE. Invited commentary: Influenza vaccine and Guillain-Barre syndrome--is there a risk? American journal of epidemiology. 2012 Jun 1;175(11):1129-32. PubMed PMID: 22582208. 138. Schonberger LB, Bregman DJ, Sullivan-Bolyai JZ, Keenlyside RA, Ziegler DW, Retailliau HF, et al. Guillain-Barre syndrome following vaccination in the National Influenza Immunization Program, United States, 1976--1977. American journal of epidemiology. 1979 Aug;110(2):105-23. PubMed PMID: 463869. 139. Sencer DJ, Millar JD. Reflections on the 1976 swine flu vaccination program. Emerging infectious diseases. 2006 Jan;12(1):29-33. PubMed PMID: 16494713. Pubmed Central PMCID: 3291400. 140. Wise ME, Viray M, Sejvar JJ, Lewis P, Baughman AL, Connor W, et al. Guillain-Barre syndrome during the 2009-2010 H1N1 influenza vaccination campaign: population-based surveillance among 45 million Americans. American journal of epidemiology. 2012 Jun 1;175(11):1110-9. PubMed PMID: 22582209. 141. Juurlink DN, Stukel TA, Kwong J, Kopp A, McGeer A, Upshur RE, et al. Guillain-Barre syndrome after influenza vaccination in adults: a population-based study. Archives of internal medicine. 2006 Nov 13;166(20):2217-21. PubMed PMID: 17101939. 142. Nachamkin I, Shadomy SV, Moran AP, Cox N, Fitzgerald C, Ung H, et al. Anti-ganglioside antibody induction by swine (A/NJ/1976/H1N1) and other influenza vaccines: insights into vaccine-associated Guillain-Barre syndrome. The Journal of infectious diseases. 2008 Jul 15;198(2):226-33. PubMed PMID: 18522505. 143. Davidson DL, O'Sullivan AF, Morley KD. HLA antigens in familial Guillain-Barre syndrome. Journal of neurology, neurosurgery, and psychiatry. 1992 Jun;55(6):508-9. PubMed PMID: 1619424. Pubmed Central PMCID: 1014913. 144. Geleijns K, Brouwer BA, Jacobs BC, Houwing-Duistermaat JJ, van Duijn CM, van Doorn PA. The occurrence of Guillain-Barre syndrome within families. Neurology. 2004 Nov 9;63(9):1747-50. PubMed PMID: 15534275. 145. Yuki N, Tsujino Y. Familial Guillain-Barre syndrome subsequent to Campylobacter jejuni enteritis. The Journal of pediatrics. 1995 Jan;126(1):162. PubMed PMID: 7815216. 146. Peeples E. Familial Miller Fisher syndrome. Journal of child neurology. 2011 May;26(5):645-8. PubMed PMID: 21325125. 147. Godschalk PC, Bergman MP, Gorkink RF, Simons G, van den Braak N, Lastovica AJ, et al. Identification of DNA sequence variation in Campylobacter jejuni strains associated with the Guillain-Barre syndrome by high-throughput AFLP analysis. BMC microbiology. 2006;6:32. PubMed PMID: 16594990. Pubmed Central PMCID: 1513382. 148. McCarthy N, Giesecke J. Incidence of Guillain-Barre syndrome following

110
infection with Campylobacter jejuni. American journal of epidemiology. 2001 Mar 15;153(6):610-4. PubMed PMID: 11257070. 149. Geleijns K, Schreuder GM, Jacobs BC, Sintnicolaas K, van Koningsveld R, Meulstee J, et al. HLA class II alleles are not a general susceptibility factor in Guillain-Barre syndrome. Neurology. 2005 Jan 11;64(1):44-9. PubMed PMID: 15642902. 150. Geleijns K, Jacobs BC, Van Rijs W, Tio-Gillen AP, Laman JD, van Doorn PA. Functional polymorphisms in LPS receptors CD14 and TLR4 are not associated with disease susceptibility or Campylobacter jejuni infection in Guillain-Barre patients. Journal of neuroimmunology. 2004 May;150(1-2):132-8. PubMed PMID: 15081257. 151. Geleijns K, Roos A, Houwing-Duistermaat JJ, van Rijs W, Tio-Gillen AP, Laman JD, et al. Mannose-binding lectin contributes to the severity of Guillain-Barre syndrome. Journal of immunology. 2006 Sep 15;177(6):4211-7. PubMed PMID: 16951387. 152. Geleijns K, Emonts M, Laman JD, van Rijs W, van Doorn PA, Hermans PW, et al. Genetic polymorphisms of macrophage-mediators in Guillain-Barre syndrome. Journal of neuroimmunology. 2007 Oct;190(1-2):127-30. PubMed PMID: 17761309. 153. Daeron M. Fc receptor biology. Annual review of immunology. 1997;15:203-34. PubMed PMID: 9143687. 154. Takai T. Roles of Fc receptors in autoimmunity. Nature reviews Immunology. 2002 Aug;2(8):580-92. PubMed PMID: 12154377. 155. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nature reviews Immunology. 2008 Jan;8(1):34-47. PubMed PMID: 18064051. 156. Dijstelbloem HM, van de Winkel JG, Kallenberg CG. Inflammation in autoimmunity: receptors for IgG revisited. Trends in immunology. 2001 Sep;22(9):510-6. PubMed PMID: 11525942. 157. Hogarth PM, Pietersz GA. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nature reviews Drug discovery. 2012 Apr;11(4):311-31. PubMed PMID: 22460124. 158. Amigorena S, Bonnerot C, Choquet D, Fridman WH, Teillaud JL. Fc gamma RII expression in resting and activated B lymphocytes. European journal of immunology. 1989 Aug;19(8):1379-85. PubMed PMID: 2550246. 159. Ravetch JV, Kinet JP. Fc receptors. Annual review of immunology. 1991;9:457-92. PubMed PMID: 1910686. 160. Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Annals of hematology. 1998 Jun;76(6):231-48. PubMed PMID: 9692811.

111
161. Norris CF, Pricop L, Millard SS, Taylor SM, Surrey S, Schwartz E, et al. A naturally occurring mutation in Fc gamma RIIA: a Q to K127 change confers unique IgG binding properties to the R131 allelic form of the receptor. Blood. 1998 Jan 15;91(2):656-62. PubMed PMID: 9427722. 162. Clark MR, Stuart SG, Kimberly RP, Ory PA, Goldstein IM. A single amino acid distinguishes the high-responder from the low-responder form of Fc receptor II on human monocytes. European journal of immunology. 1991 Aug;21(8):1911-6. PubMed PMID: 1831131. 163. Fijen CA, Bredius RG, Kuijper EJ. Polymorphism of IgG Fc receptors in meningococcal disease. Annals of internal medicine. 1993 Oct 1;119(7 Pt 1):636. PubMed PMID: 8363182. 164. Platonov AE, Shipulin GA, Vershinina IV, Dankert J, van de Winkel JG, Kuijper EJ. Association of human Fc gamma RIIa (CD32) polymorphism with susceptibility to and severity of meningococcal disease. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 1998 Oct;27(4):746-50. PubMed PMID: 9798027. 165. Bredius RG, Derkx BH, Fijen CA, de Wit TP, de Haas M, Weening RS, et al. Fc gamma receptor IIa (CD32) polymorphism in fulminant meningococcal septic shock in children. The Journal of infectious diseases. 1994 Oct;170(4):848-53. PubMed PMID: 7930726. 166. Sanders LA, van de Winkel JG, Rijkers GT, Voorhorst-Ogink MM, de Haas M, Capel PJ, et al. Fc gamma receptor IIa (CD32) heterogeneity in patients with recurrent bacterial respiratory tract infections. The Journal of infectious diseases. 1994 Oct;170(4):854-61. PubMed PMID: 7930727. 167. Kobayashi T, Yamamoto K, Sugita N, van der Pol WL, Yasuda K, Kaneko S, et al. The Fc gamma receptor genotype as a severity factor for chronic periodontitis in Japanese patients. Journal of periodontology. 2001 Oct;72(10):1324-31. PubMed PMID: 11699473. 168. Loos BG, Leppers-Van de Straat FG, Van de Winkel JG, Van der Velden U. Fcgamma receptor polymorphisms in relation to periodontitis. Journal of clinical periodontology. 2003 Jul;30(7):595-602. PubMed PMID: 12834496. 169. Yamamoto K, Kobayashi T, Grossi S, Ho AW, Genco RJ, Yoshie H, et al. Association of Fcgamma receptor IIa genotype with chronic periodontitis in Caucasians. Journal of periodontology. 2004 Apr;75(4):517-22. PubMed PMID: 15152814. 170. de Souza RC, Colombo AP. Distribution of FcgammaRIIa and FcgammaRIIIb genotypes in patients with generalized aggressive periodontitis. Journal of periodontology. 2006 Jul;77(7):1120-8. PubMed PMID: 16805673. 171. Siqueira JF, Jr., Rocas IN, Provenzano JC, Daibert FK, Silva MG, Lima KC. Relationship between Fcgamma receptor and interleukin-1 gene polymorphisms

112
and post-treatment apical periodontitis. Journal of endodontics. 2009 Sep;35(9):1186-92. PubMed PMID: 19720214. 172. Siqueira JF, Jr., Rocas IN, Provenzano JC, Guilherme BP. Polymorphism of the FcgammaRIIIa gene and post-treatment apical periodontitis. Journal of endodontics. 2011 Oct;37(10):1345-8. PubMed PMID: 21924179. 173. Halstead SB, O'Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. The Journal of experimental medicine. 1977 Jul 1;146(1):201-17. PubMed PMID: 406347. Pubmed Central PMCID: 2180729. 174. Loke H, Bethell D, Phuong CX, Day N, White N, Farrar J, et al. Susceptibility to dengue hemorrhagic fever in vietnam: evidence of an association with variation in the vitamin d receptor and Fc gamma receptor IIa genes. The American journal of tropical medicine and hygiene. 2002 Jul;67(1):102-6. PubMed PMID: 12363051. 175. Garcia G, Sierra B, Perez AB, Aguirre E, Rosado I, Gonzalez N, et al. Asymptomatic dengue infection in a Cuban population confirms the protective role of the RR variant of the FcgammaRIIa polymorphism. The American journal of tropical medicine and hygiene. 2010 Jun;82(6):1153-6. PubMed PMID: 20519616. Pubmed Central PMCID: 2877427. 176. Braga EM, Scopel KK, Komatsu NT, da Silva-Nunes M, Ferreira MU. Polymorphism of the Fcgamma receptor IIA and malaria morbidity. Journal of molecular and genetic medicine : an international journal of biomedical research. 2005;1(1):5-10. PubMed PMID: 19565007. Pubmed Central PMCID: 2702062. 177. Cooke GS, Aucan C, Walley AJ, Segal S, Greenwood BM, Kwiatkowski DP, et al. Association of Fcgamma receptor IIa (CD32) polymorphism with severe malaria in West Africa. The American journal of tropical medicine and hygiene. 2003 Dec;69(6):565-8. PubMed PMID: 14740869. 178. Omi K, Ohashi J, Patarapotikul J, Hananantachai H, Naka I, Looareesuwan S, et al. Fcgamma receptor IIA and IIIB polymorphisms are associated with susceptibility to cerebral malaria. Parasitology international. 2002 Dec;51(4):361-6. PubMed PMID: 12421634. 179. Shi YP, Nahlen BL, Kariuki S, Urdahl KB, McElroy PD, Roberts JM, et al. Fcgamma receptor IIa (CD32) polymorphism is associated with protection of infants against high-density Plasmodium falciparum infection. VII. Asembo Bay Cohort Project. The Journal of infectious diseases. 2001 Jul 1;184(1):107-11. PubMed PMID: 11398118. 180. Nasr A, Iriemenam NC, Troye-Blomberg M, Giha HA, Balogun HA, Osman OF, et al. Fc gamma receptor IIa (CD32) polymorphism and antibody responses to asexual blood-stage antigens of Plasmodium falciparum malaria in Sudanese patients. Scandinavian journal of immunology. 2007 Jul;66(1):87-96. PubMed PMID: 17587350.

113
181. Nasr A, Iriemenam NC, Giha HA, Balogun HA, Anders RF, Troye-Blomberg M, et al. FcgammaRIIa (CD32) polymorphism and anti-malarial IgG subclass pattern among Fulani and sympatric ethnic groups living in eastern Sudan. Malaria journal. 2009;8:43. PubMed PMID: 19284648. Pubmed Central PMCID: 2660360. 182. Woelbing F, Kostka SL, Moelle K, Belkaid Y, Sunderkoetter C, Verbeek S, et al. Uptake of Leishmania major by dendritic cells is mediated by Fcgamma receptors and facilitates acquisition of protective immunity. The Journal of experimental medicine. 2006 Jan 23;203(1):177-88. PubMed PMID: 16418399. Pubmed Central PMCID: 2118064. 183. Buxbaum LU. A detrimental role for IgG and FcgammaR in Leishmania mexicana infection. Immunologic research. 2008;42(1-3):197-209. PubMed PMID: 19002608. 184. Castellucci L, Jamieson SE, Miller EN, Menezes E, Oliveira J, Magalhaes A, et al. CXCR1 and SLC11A1 polymorphisms affect susceptibility to cutaneous leishmaniasis in Brazil: a case-control and family-based study. BMC medical genetics. 2010;11:10. PubMed PMID: 20089160. Pubmed Central PMCID: 2823618. 185. Jeronimo SM, Duggal P, Ettinger NA, Nascimento ET, Monteiro GR, Cabral AP, et al. Genetic predisposition to self-curing infection with the protozoan Leishmania chagasi: a genomewide scan. The Journal of infectious diseases. 2007 Oct 15;196(8):1261-9. PubMed PMID: 17955446. Pubmed Central PMCID: 2330094. 186. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000 Aug;13(2):277-85. PubMed PMID: 10981970. 187. Koene HR, Kleijer M, Swaak AJ, Sullivan KE, Bijl M, Petri MA, et al. The Fc gammaRIIIA-158F allele is a risk factor for systemic lupus erythematosus. Arthritis and rheumatism. 1998 Oct;41(10):1813-8. PubMed PMID: 9778222. 188. Karassa FB, Trikalinos TA, Ioannidis JP, Fcgamma R-SLEM-AI. Role of the Fcgamma receptor IIa polymorphism in susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Arthritis and rheumatism. 2002 Jun;46(6):1563-71. PubMed PMID: 12115187. 189. Nieto A, Caliz R, Pascual M, Mataran L, Garcia S, Martin J. Involvement of Fcgamma receptor IIIA genotypes in susceptibility to rheumatoid arthritis. Arthritis and rheumatism. 2000 Apr;43(4):735-9. PubMed PMID: 10765917. 190. Ivan E, Colovai AI. Human Fc receptors: critical targets in the treatment of autoimmune diseases and transplant rejections. Human immunology. 2006 Jul;67(7):479-91. PubMed PMID: 16829303. 191. Arepally G, McKenzie SE, Jiang XM, Poncz M, Cines DB. Fc gamma RIIA H/R 131 polymorphism, subclass-specific IgG anti-heparin/platelet factor 4

114
antibodies and clinical course in patients with heparin-induced thrombocytopenia and thrombosis. Blood. 1997 Jan 15;89(2):370-5. PubMed PMID: 9002937. 192. van der Pol WL, Jansen MD, Kuks JB, de Baets M, Leppers-van de Straat FG, Wokke JH, et al. Association of the Fc gamma receptor IIA-R/R131 genotype with myasthenia gravis in Dutch patients. Journal of neuroimmunology. 2003 Nov;144(1-2):143-7. PubMed PMID: 14597109. 193. Dijstelbloem HM, Bijl M, Fijnheer R, Scheepers RH, Oost WW, Jansen MD, et al. Fcgamma receptor polymorphisms in systemic lupus erythematosus: association with disease and in vivo clearance of immune complexes. Arthritis and rheumatism. 2000 Dec;43(12):2793-800. PubMed PMID: 11145038. 194. Vedeler CA, Myhr KM, Nyland H. Fc receptors for immunoglobulin G--a role in the pathogenesis of Guillain-Barre syndrome and multiple sclerosis. Journal of neuroimmunology. 2001 Aug 30;118(2):187-93. PubMed PMID: 11498253. 195. Dalakas MC. Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology. 1998 Dec;51(6 Suppl 5):S2-8. PubMed PMID: 9851723. 196. Vedeler CA, Conti G, Bannerman P, Pleasure D. Expression of genes encoding receptors for IgG (FcRIII) and for C3b/C4b (Crry) in rat sciatic nerve during development and Wallerian degeneration. Journal of neuroscience research. 1992 Apr;31(4):654-61. PubMed PMID: 1533683. 197. van der Pol WL, van den Berg LH, Scheepers RH, van der Bom JG, van Doorn PA, van Koningsveld R, et al. IgG receptor IIa alleles determine susceptibility and severity of Guillain-Barre syndrome. Neurology. 2000 Apr 25;54(8):1661-5. PubMed PMID: 10762510. 198. Hartung HP, Pollard JD, Harvey GK, Toyka KV. Immunopathogenesis and treatment of the Guillain-Barre syndrome--Part I. Muscle & nerve. 1995 Feb;18(2):137-53. PubMed PMID: 7823972. 199. Griffin JW, George R, Ho T. Macrophage systems in peripheral nerves. A review. Journal of neuropathology and experimental neurology. 1993 Nov;52(6):553-60. PubMed PMID: 7693878. 200. Efficiency of plasma exchange in Guillain-Barre syndrome: role of replacement fluids. French Cooperative Group on Plasma Exchange in Guillain-Barre syndrome. Ann Neurol. 1987 Dec;22(6):753-61. PubMed PMID: 2893583. 201. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barre syndrome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Lancet. 1997 Jan 25;349(9047):225-30. PubMed PMID: 9014908. 202. Dalakas MC. Intravenous immunoglobulin in autoimmune neuromuscular diseases. JAMA : the journal of the American Medical Association. 2004 May

115
19;291(19):2367-75. PubMed PMID: 15150209. 203. Hughes RA, Swan AV, van Doorn PA. Corticosteroids for Guillain-Barre syndrome. Cochrane database of systematic reviews. 2010 (2):CD001446. PubMed PMID: 20166061. 204. van Koningsveld R, Schmitz PI, Meche FG, Visser LH, Meulstee J, van Doorn PA, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barre syndrome: randomised trial. Lancet. 2004 Jan 17;363(9404):192-6. PubMed PMID: 14738791. 205. Kuwabara S, Yuki N. Axonal Guillain-Barre syndrome: concepts and controversies. Lancet neurology. 2013 Dec;12(12):1180-8. PubMed PMID: 24229616. 206. Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nature methods. 2011 Apr;8(4 Suppl):S6-11. PubMed PMID: 21451510. Pubmed Central PMCID: 3408593. 207. Mantione KJ, Kream RM, Kuzelova H, Ptacek R, Raboch J, Samuel JM, et al. Comparing bioinformatic gene expression profiling methods: microarray and RNA-Seq. Medical science monitor basic research. 2014;20:138-42. PubMed PMID: 25149683. Pubmed Central PMCID: 4152252. 208. Chang KH, Chuang TJ, Lyu RK, Ro LS, Wu YR, Chang HS, et al. Identification of gene networks and pathways associated with Guillain-Barre syndrome. PloS one. 2012;7(1):e29506. PubMed PMID: 22253732. Pubmed Central PMCID: 3254618. 209. Feasby TE, Hahn AF, Brown WF, Bolton CF, Gilbert JJ, Koopman WJ. Severe axonal degeneration in acute Guillain-Barre syndrome: evidence of two different mechanisms? Journal of the neurological sciences. 1993 Jun;116(2):185-92. PubMed PMID: 8336165. 210. Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet. 1978 Oct 7;2(8093):750-3. PubMed PMID: 80682. 211. Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic acids research. 1989 Oct 25;17(20):8390. PubMed PMID: 2813076. Pubmed Central PMCID: 334995. 212. Moriya H, Saito K, Helsby N, Hayashi N, Sugino S, Yamakage M, et al. Single-nucleotide polymorphisms and copy number variations of the and genes in healthy Japanese subjects. Biomedical reports. 2014 Mar;2(2):265-9. PubMed PMID: 24649108. Pubmed Central PMCID: 3917743. 213. Kroll KW, Mokaram NE, Pelletier AR, Frankhouser DE, Westphal MS, Stump PA, et al. Quality Control for RNA-Seq (QuaCRS): An Integrated Quality Control

116
Pipeline. Cancer informatics. 2014;13(Suppl 3):7-14. PubMed PMID: 25368506. Pubmed Central PMCID: 4214596. 214. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10(3):R25. PubMed PMID: 19261174. Pubmed Central PMCID: 2690996. 215. Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome research. 1998 Mar;8(3):186-94. PubMed PMID: 9521922. 216. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009 May 1;25(9):1105-11. PubMed PMID: 19289445. Pubmed Central PMCID: 2672628. 217. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012 Mar;7(3):562-78. PubMed PMID: 22383036. Pubmed Central PMCID: 3334321. 218. Landaverde JM, Danovaro-Holliday MC, Trumbo SP, Pacis-Tirso CL, Ruiz-Matus C. Guillain-Barre syndrome in children aged <15 years in Latin America and the Caribbean: baseline rates in the context of the influenza A (H1N1) pandemic. The Journal of infectious diseases. 2010 Mar;201(5):746-50. PubMed PMID: 20102270. 219. Islam Z, Jacobs BC, Islam MB, Mohammad QD, Diorditsa S, Endtz HP. High incidence of Guillain-Barre syndrome in children, Bangladesh. Emerging infectious diseases. 2011 Jul;17(7):1317-8. PubMed PMID: 21762603. Pubmed Central PMCID: 3381380. 220. Visser LH, Van der Meche FG, Van Doorn PA, Meulstee J, Jacobs BC, Oomes PG, et al. Guillain-Barre syndrome without sensory loss (acute motor neuropathy). A subgroup with specific clinical, electrodiagnostic and laboratory features. Dutch Guillain-Barre Study Group. Brain : a journal of neurology. 1995 Aug;118 ( Pt 4):841-7. PubMed PMID: 7655882. 221. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology. 2001 Apr 24;56(8):1104-6. PubMed PMID: 11320188. 222. Funakoshi K, Kuwabara S, Odaka M, Hirata K, Yuki N. Clinical predictors of mechanical ventilation in Fisher/Guillain-Barre overlap syndrome. Journal of neurology, neurosurgery, and psychiatry. 2009 Jan;80(1):60-4. PubMed PMID: 18948360. 223. de Arruda E, Hayden FG, McAuliffe JF, de Sousa MA, Mota SB, McAuliffe MI, et al. Acute respiratory viral infections in ambulatory children of urban northeast Brazil. The Journal of infectious diseases. 1991 Aug;164(2):252-8. PubMed PMID: 1649871.

117
224. Ramirez-Zamora M, Burgos-Ganuza CR, Alas-Valle DA, Vergara-Galan PE, Ortez-Gonzalez CI. [Guillain-Barre syndrome in the paediatric age: epidemiological, clinical and therapeutic profile in a hospital in El Salvador]. Revista de neurologia. 2009 Mar 16-31;48(6):292-6. PubMed PMID: 19291652. Sindrome de Guillain-Barre en edad pediatrica. Perfil epidemiologico, clinico y terapeutico en un hospital de El Salvador. 225. Pan CL, Tseng TJ, Lin YH, Chiang MC, Lin WM, Hsieh ST. Cutaneous innervation in Guillain-Barre syndrome: pathology and clinical correlations. Brain : a journal of neurology. 2003 Feb;126(Pt 2):386-97. PubMed PMID: 12538405. 226. Durand MC, Porcher R, Orlikowski D, Aboab J, Devaux C, Clair B, et al. Clinical and electrophysiological predictors of respiratory failure in Guillain-Barre syndrome: a prospective study. Lancet neurology. 2006 Dec;5(12):1021-8. PubMed PMID: 17110282. 227. Singh NK, Jaiswal AK, Misra S, Srivastava PK. Assessment of autonomic dysfunction in Guillain-Barre syndrome and its prognostic implications. Acta neurologica Scandinavica. 1987 Feb;75(2):101-5. PubMed PMID: 3577674. 228. Winer JB, Hughes RA. Identification of patients at risk of arrhythmia in the Guillain-Barre syndrome. The Quarterly journal of medicine. 1988 Sep;68(257):735-9. PubMed PMID: 3256902. 229. Zochodne DW. Autonomic involvement in Guillain-Barre syndrome: a review. Muscle & nerve. 1994 Oct;17(10):1145-55. PubMed PMID: 7935521. 230. Hughes RA, Wijdicks EF, Benson E, Cornblath DR, Hahn AF, Meythaler JM, et al. Supportive care for patients with Guillain-Barre syndrome. Archives of neurology. 2005 Aug;62(8):1194-8. PubMed PMID: 16087757. 231. Moore P, James O. Guillain-Barre Syndrome: incidence, management and outcome of major complications. Critical care medicine. 1981 Jul;9(7):549-55. PubMed PMID: 7238063. 232. Sharshar T, Chevret S, Bourdain F, Raphael JC, French Cooperative Group on Plasma Exchange in Guillain-Barre S. Early predictors of mechanical ventilation in Guillain-Barre syndrome. Critical care medicine. 2003 Jan;31(1):278-83. PubMed PMID: 12545029. 233. Walgaard C, Lingsma HF, Ruts L, Drenthen J, van Koningsveld R, Garssen MJ, et al. Prediction of respiratory insufficiency in Guillain-Barre syndrome. Annals of neurology. 2010 Jun;67(6):781-7. PubMed PMID: 20517939. 234. McKhann GM, Griffin JW, Cornblath DR, Mellits ED, Fisher RS, Quaskey SA. Plasmapheresis and Guillain-Barre syndrome: analysis of prognostic factors and the effect of plasmapheresis. Annals of neurology. 1988 Apr;23(4):347-53. PubMed PMID: 3382169. 235. Double-blind trial of intravenous methylprednisolone in Guillain-Barre

118
syndrome. Guillain-Barre Syndrome Steroid Trial Group. Lancet. 1993 Mar 6;341(8845):586-90. PubMed PMID: 8094828. 236. Appropriate number of plasma exchanges in Guillain-Barre syndrome. The French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Annals of neurology. 1997 Mar;41(3):298-306. PubMed PMID: 9066350. 237. Lawn ND, Wijdicks EF. Tracheostomy in Guillain-Barre syndrome. Muscle & nerve. 1999 Aug;22(8):1058-62. PubMed PMID: 10417787. 238. Merkies IS, Schmitz PI, Samijn JP, van der Meche FG, van Doorn PA. Fatigue in immune-mediated polyneuropathies. European Inflammatory Neuropathy Cause and Treatment (INCAT) Group. Neurology. 1999 Nov 10;53(8):1648-54. PubMed PMID: 10563607. 239. Bernsen RA, de Jager AE, Schmitz PI, van der Meche FG. Residual physical outcome and daily living 3 to 6 years after Guillain-Barre syndrome. Neurology. 1999 Jul 22;53(2):409-10. PubMed PMID: 10430437. 240. Bernsen RA, de Jager AE, Schmitz PI, van der Meche FG. Long-term impact on work and private life after Guillain-Barre syndrome. Journal of the neurological sciences. 2002 Sep 15;201(1-2):13-7. PubMed PMID: 12163188. 241. Dornonville de la Cour C, Jakobsen J. Residual neuropathy in long-term population-based follow-up of Guillain-Barre syndrome. Neurology. 2005 Jan 25;64(2):246-53. PubMed PMID: 15668421. 242. The prognosis and main prognostic indicators of Guillain-Barre syndrome. A multicentre prospective study of 297 patients. The Italian Guillain-Barre Study Group. Brain : a journal of neurology. 1996 Dec;119 ( Pt 6):2053-61. PubMed PMID: 9010009. 243. van Koningsveld R, Steyerberg EW, Hughes RA, Swan AV, van Doorn PA, Jacobs BC. A clinical prognostic scoring system for Guillain-Barre syndrome. Lancet neurology. 2007 Jul;6(7):589-94. PubMed PMID: 17537676. 244. Hiraga A, Kuwabara S, Ogawara K, Misawa S, Kanesaka T, Koga M, et al. Patterns and serial changes in electrodiagnostic abnormalities of axonal Guillain-Barre syndrome. Neurology. 2005 Mar 8;64(5):856-60. PubMed PMID: 15753422. 245. Kuwano ST, Bordin JO, Chiba AK, Mello AB, Figueiredo MS, Vieira-Filho JP, et al. Allelic polymorphisms of human fcgamma receptor IIa and Fcgamma receptor IIIb among distinct groups in Brazil. Transfusion. 2000 Nov;40(11):1388-92. PubMed PMID: 11099670. 246. Salmon JE, Edberg JC, Kimberly RP. Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. The Journal of clinical investigation. 1990 Apr;85(4):1287-95. PubMed PMID: 1690757. Pubmed Central PMCID: 296565.

119
247. Salmon JE, Millard SS, Brogle NL, Kimberly RP. Fc gamma receptor IIIb enhances Fc gamma receptor IIa function in an oxidant-dependent and allele-sensitive manner. The Journal of clinical investigation. 1995 Jun;95(6):2877-85. PubMed PMID: 7769129. Pubmed Central PMCID: 295975. 248. Decker EL, Nehmann N, Kampen E, Eibel H, Zipfel PF, Skerka C. Early growth response proteins (EGR) and nuclear factors of activated T cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic acids research. 2003 Feb 1;31(3):911-21. PubMed PMID: 12560487. Pubmed Central PMCID: 149206. 249. Wagner EF, Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunological reviews. 2005 Dec;208:126-40. PubMed PMID: 16313345. 250. Schmidt B, Stoll G, van der Meide P, Jung S, Hartung HP. Transient cellular expression of gamma-interferon in myelin-induced and T-cell line-mediated experimental autoimmune neuritis. Brain : a journal of neurology. 1992 Dec;115 ( Pt 6):1633-46. PubMed PMID: 1486454. 251. Girkontaite I, Urbonaviciute V, Maseda D, Neubert K, Herrmann M, Voll RE. Apoptotic cells selectively suppress the Th1 cytokine interferon gamma in stimulated human peripheral blood mononuclear cells and shift the Th1/Th2 balance towards Th2. Autoimmunity. 2007 Jun;40(4):327-30. PubMed PMID: 17516220. 252. Girolami EI, Bouhy D, Haber M, Johnson H, David S. Differential expression and potential role of SOCS1 and SOCS3 in Wallerian degeneration in injured peripheral nerve. Experimental neurology. 2010 May;223(1):173-82. PubMed PMID: 19576891. Pubmed Central PMCID: 2849922. 253. Hu W, Mathey E, Hartung HP, Kieseier BC. Cyclo-oxygenases and prostaglandins in acute inflammatory demyelination of the peripheral nerve. Neurology. 2003 Dec 23;61(12):1774-9. PubMed PMID: 14694045. 254. Miyamoto K, Oka N, Kawasaki T, Miyake S, Yamamura T, Akiguchi I. New cyclooxygenase-2 inhibitors for treatment of experimental autoimmune neuritis. Muscle & nerve. 2002 Feb;25(2):280-2. PubMed PMID: 11870698. 255. Sharshar T, Durand MC, Lefaucheur JP, Lofaso F, Raphael JC, Gherardi RK, et al. MMP-9 correlates with electrophysiologic abnormalities in Guillain-Barre syndrome. Neurology. 2002 Nov 26;59(10):1649-51. PubMed PMID: 12451218.
256. Redford EJ, Smith KJ, Gregson NA, Davies M, Hughes P, Gearing AJ, et al. A combined inhibitor of matrix metalloproteinase activity and tumour necrosis factor-alpha processing attenuates experimental autoimmune neuritis. Brain : a journal of neurology. 1997 Oct;120 ( Pt 10):1895-905. PubMed PMID: 9365379.
257. Seliger C, Leukel P, Moeckel S, Jachnik B, Lottaz C, Kreutz M, et al. Lactate-modulated induction of THBS-1 activates transforming growth factor (TGF)-beta2 and migration of glioma cells in vitro. PloS one. 2013;8(11):e78935. PubMed PMID: 24223867. Pubmed Central PMCID: 3815307.

120
ANEXOS
Anexo 1

121

122
Anexo 2: Critérios diagnósticos da SGB
NECESSÁRIO PARA O DIAGNÓSTICO
Fraqueza muscular progressiva em > 1 extremidade
Arreflexia ou hiporreflexia
FORTEMENTE INDICATIVO DO DIAGNÓSTICO
Progressão dura entre 1-4 semanas
Sinais e sintomas relativamente simétricos
Sinais e sintomas sensitivos leves
Acometimento de nervos cranianos, especialmente VII bilateral
Disautonomia
Ausência de febre no início
Proteinorraquia, com poucas células (< 10)
Alterações eletrofisiológicas típicas
QUADRO QUE FAZ DUVIDAR O DIAGNÓSTICO
Nível sensitivo
Sintomas e sinais assimétricos persistentes
Retenção vesical marcada e persistente
Mais de 50 células no LCR (exceto na SGB associada ao HIV)
QUADRO QUE EXCLUE O DIAGNÓSTICO
Botulismo, poliomielites ou polineuropatia tóxica, porfiria, difteria,
síndrome sensitiva pura, sem fraqueza.

123
Anexo 3: Critérios eletroneuromiográficos para as variantes da SGB (6, 196)
Alteração em dois ou mais nervos durante as primeiras duas semanas da
doença
Desmielinizante
Velocidade de condução motora <90% do limite inferior (<85% se a
amplitude do potencial de ação composto muscular distal for < 50% do
limite inferior da normalidade.
Latência distal motora > 110% do limite superior da normalidade( >120%
se a amplitude do potencial de ação composta muscular distal for <
100% do limite inferior da normalidade)
Evidência inequívoca de dispersão temporal
Latência mínima da onda F > 120%
Axonal
Neuropatia axonal motora aguda
Ausência de evidência de desmielinização
Amplitude distal do potencial de ação composto muscular < 80% do
limite inferior da normalidade
Amplitude e velocidade de condução do potencial de ação sensitivo
normais
Neuropatia axonal motora e sensitiva aguda
Ausência de evidência de desmielinização
Amplitude distal do potencial de ação composto muscular < 80% do
limite inferior da normalidade
Amplitude do potencial de ação sensitivo <50% do limite inferior da
normalidade

124
Anexo 4: Termo de consentimento livre e esclarecido
Título do Projeto: Anticorpos Antigangliosídeos em neuropatias periféricas
Pesquisadores: Mário Emílio Dourado e Selma M.B. Jeronimo
Estamos propondo realizar uma pesquisa sobre um grupo de doenças denominadas de neuropatias. Vários fatores podem influenciar o desenvolvimento dessas doenças que podem impossibilitar diversas ações do indivíduo, como por exemplo, o andar. Algumas pessoas com essas doenças podem apresentar dano de tal sorte que podem ficar impossibilitadas de se locomoverem temporariamente ou permanentemente. Vários são os fatores que influenciam o desenvolvimento dessas doenças. Para podermos compreender melhor as neuropatias, estamos solicitando sua participação como voluntário. Caso você concorde em participar desta pesquisa, iremos solicitar que você assine este documento quando terminarmos esta explicação. Se você for o pai ou a mãe ou o guardião legal de um menor que esteja sendo convidado a participar deste estudo, o termo “você” se aplica a este menor. O responsável legal, por este menor, será solicitado a ler e dar permissão, por escrito, para que o menor participe deste estudo.
Este estudo tem como objetivo compreender por que as pessoas adoecem com a doença conhecida com o nome de Síndrome de Guillain-Barré ou outros tipos de neuropatias inflamatórias. Iremos determinar no sangue se há presença de um tipo de substância (anticorpo antigangliosídeo) que pode estar relacionado com o dano ou pode ser um marcador desta doença.
Esta pesquisa se propõe também a estudar informações contidas na molécula do DNA. DNA é a substância encontrada dentro das nossas células, parte do nosso corpo, que é herdada dos nossos pais e transmitida por nós aos nossos filhos.
Os procedimentos a serem realizados incluem uma consulta médica, onde obteremos dados sobre a história da sua doença; coleta de sangue, para realização de hemograma, isolamento das células, DNA, plasma e soro, e caso o seu médico solicite a realização de uma punção medular, solicitamos permissão para estudar parte do líquido obtido. Usaremos o líquor coletado para pesquisar uma substância produzida pelo sistema de defesa (citocinas). Você somente realizará esse exame caso seu médico solicite-o para esclarecer sua doença. Você não o fará somente com o objetivo de colher dados para esta pesquisa. Você também poderá ser submetido a um exame denominado eletroneuromiografia. Esse exame é padrão e importante para confirmar a presença da neuropatia e determinar qual alteração patológica está ocorrendo no seu nervo.
Os riscos associados à participação neste estudo são sangramentos ou manchas arroxeadas no local da coleta do sangue, infecção e desmaio. A eletroneuromiografia é um exame seguro e bem tolerado, não precisando de preparo especial para o estudo. Pode haver sangramento cutâneo no local da agulha de eletromiografia que é facilmente estancado. Está contra-indicado em indivíduos que usam marcapasso cardíaco.
Os benefícios em participar deste estudo são decorrentes de acompanharmos a sua evolução, após o término do seu tratamento, através de exames periódicos. Os procedimentos relativos ao tratamento relativos a sua doença serão custeados pelo SUS e/ou convênio, referentes ao custo de hospitalização e exames necessários para condução do seu tratamento. Os exames relativos à pesquisa, incluindo determinação de anticorpos anti-gangliosídeos, sorologia para CMV, EBV e C. jejuni, serão custeados por recursos por nós obtidos para a realização deste estudo. Os estudos de eletromiografia serão realizados pelo Dr. Mário Emílio Dourado. Os resultados dos estudos genéticos a serem realizados serão obtidos a longo prazo, não temos previsão de quando os resultados desta pesquisa poderão trazer diretamente um benefício para você.
Informações sigilosas sobre a sua saúde são aquelas que identificam você e estejam relacionadas com a sua saúde física e mental, seja passada, presente ou futura. Essas informações, geralmente, estão presentes no seu prontuário ou em anotações realizadas por profissionais de saúde. Ao assinar este termo de consentimento, você estará autorizando a liberação dos dados existentes, para que nós possamos estudá-los. Essas informações serão mantidas em sigilo e nós não liberaremos para terceiros.
Registro da sua participação neste estudo será mantido em sigilo. Nós guardaremos os registros de cada pessoa, em sala trancada, e somente os Doutores Mário Emílio Dourado e Selma Jeronimo ou pesquisadores trabalhando. Se houver algum dano decorrente deste estudo ou devido à negligência de algum membro da nossa equipe, tratamento médico será fornecido sem ônus e será providenciado pelo Dr. Mário Emílio Dourado. Se houver problemas médicos não relacionados aos procedimentos aqui descritos, o próprio indivíduo será responsável. Tentaremos resolver os problemas médicos mais simples e encaminharemos aqueles mais complexos a hospitais

125
especializados ou postos de saúde. Se houver algum dano decorrente da participação nesta pesquisa ou se for resultante de negligência de funcionários da Universidade Federal do Rio Grande do Norte compensação será disponibilizada. A UFRN não fornecerá compensação se o dano não for decorrente de procedimentos realizados devido a esta pesquisa.
Todas as informações obtidas serão sigilosas e seu nome não será identificado em nenhum momento. Os dados coletados serão armazenados de forma sigilosa, sendo os pesquisadores e o próprio indivíduo participante os únicos autorizados a conhecer os resultados individuais. Não serão entregues os resultados dos dados obtidos a terceiros. Se você tiver algum gasto que seja devido à sua participação na pesquisa, você será ressarcido, caso solicite. Em qualquer momento, se você sofrer algum dano comprovadamente decorrente desta pesquisa, você terá direito a indenização.
Como esta pesquisa envolve o estudo de DNA, assumimos o compromisso que o mesmo será usado apenas para o que foi esclarecido aqui. Pedimos sua permissão para guardar o DNA, o soro e o plasma coletados nessa pesquisa, no Laboratório de Imunogenética, Departamento de Bioquímica, na Universidade Federal do Rio Grande do Norte. Este material será guardado em tubos identificados por um código durante um período de 5 anos. Qualquer outro estudo que venhamos a desenvolver e que usará informação sobre sua doença ou material do seu sangue será primeiramente submetido ao Comitê de Ética em Pesquisa da UFRN, à Comissão Nacional de Ética em Pesquisa (CONEP), e solicitada sua autorização. Lembramos que você poderá remover sua permissão para o armazenamento do material em qualquer época, sem qualquer prejuízo para você.
Você ficará com uma cópia deste Termo e toda a dúvida que você tiver a respeito desta pesquisa, poderá perguntar diretamente para Selma Jerônimo ou pesquisadores envolvidos no projeto, no Departamento de Bioquímica do Centro de Biociências no campus central da Universidade Federal do Rio Grande do Norte, na Avenida Senador Salgado Filho, Lagoa Nova, Natal-RN, CEP = 59078-970, ou pelo telefone (84) 3215-3428.
Dúvidas a respeito da ética desta pesquisa poderão ser questionadas ao Comitê de Ética em Pesquisa da UFRN no endereço Praça do Campus Universitário, Lagoa Nova. Caixa Postal 1666, CEP 59072-970 Natal/RN, Telefone/Fax (84)215-3135.
Consentimento Livre e Esclarecido
Declaro que compreendi os objetivos desta pesquisa, como ela será realizada, os riscos e benefícios envolvidos e concordo em participar voluntariamente da pesquisa “Anticorpos Antigangliosídeos em neuropatias periféricas”.
Impressão digital para aqueles impossibilitados de escreverem seu nome.
Nome (letra de forma) Assinatura Data
Pais ou responsáveis:
Pesquisador Responsável Assinatura Data

126
Anexo 5: Questionário de avaliação
NEUROPATIAS INFLAMATÓRIAS Data do registro:
1. Nome: 2. Data nascimento: 3. Sexo: 4. Fone 5. Procedência: zona rural X zona urbana 6. Diagnóstico
a. SGB desmielinizante b. NAMA c. NAMSA d. SGB associada a SMF e. SMF f. Variantes faríngea SGB
7. LCR a. Hipercelularidade b. Dissociação proteína-citológica
8. Tratamento a. IgEV b. PF c. Corticoide d. Outros
9. Antecedentes a. Vacina b. Infecção respiratória vias altas c. Gastroenterite, diarreia d. Cirurgia
10. Presença de dor dentro dos 3 meses antes da SGB; tipo de dor e se usou analgésicos 11. Presenca de dor nas duas semanas antes do início da fraqueza 12. Dor – localização
a. Lombar b. Muscular c. Interescapular d. Pescoço e. Extremidades tronco
13. Dor – tipo a. Radicular b. Parestesias dolorosas c. Disestesias d. Dor articular e. Dor muscular f. Meningismo
14. Dor – intensidade* a. 0 (sem dor) a 10 (pior dor possível) = b. Classificação
i. Leve: 0 a 4 ii. Moderada: 5 a 7 iii. Severa: 8 a 10
15. Uso diário de analgésicos a. Não b. Paracetamol c. AINE d. Opioides e. Antidepressivos
16. Anticonvulsivantes 17. Fraqueza
a. Proximal b. Distal

127
c. Global 18. Escore da soma da força em 6 músculos (0 a 60) 19. Incapacidade p/ flexionar pescoço(≤2/5) 20. Déficit sensitivo 21. Oftalmoparesia (III, IV, VI) 22. Paralisia facial (uni ou bilateral) 23. Disfagia 24. Alimentação enteral 25. Disautonomia**
a. HAS b. Taquicardia c. Bradiarritmia d. Parada cardíaca e. Ileo-constipação f. Pupila atônica
26. Nadir – tempo para déficit máximo 27. INCAT sensitivo*** 28. Insuficiência respiratória 29. Dias de VM 30. Traqueostomia 31. Escala Hughes no nadir**** 32. Tempo para melhorar um ponto na escala Hughes 33. Tempo para recuperar a deambulação 34. Severidade
a. Sim b. Não
35. Evolução a. Boa b. Má
36. Óbito 37. Complicações
a. Infecção b. Escara c. Trombose venosa d. Alergia e. Estenose traqueia
38. Sorologias a. HIV b. HTLV c. Dengue d. CMV e. VEB f. H inflenza g. VHC h. HBSAG i. M pneumonia j. C jejuni
39. Imunologia a. FAN
Gangliosídeos b. GM1, GM3, GD1a, GD1b, GT1b, GQ1b, GD1a-GD1b, GD1b-GT1b

128
Anexo 6: Artigo Clinical characteristics of Guillain-Barré syndrome in a tropical
country: a Brazilian experience publicado na Acta Neurologica Scandinavica

129
130

131

132

133

134

135
Anexo 7: Artigo concluído No association of FCGR2A and FCGR3A polymorphisms
in a Guillain Barré Syndrome in a Brazilian population
No association between FCGR2A and FCGR3A polymorphisms in Guillain Barré Syndrome in a Brazilian population
Mario Emilio Dourado Junior1,2, Leonardo Capistrano Ferreira,3 Francisco Freire Neto, 3,4 Selma M.B. Jeronimo3,4,5 1Department of Integrative Medicine, 2Health Post-Graduate Program, Health Sciences Center; 3Department of Biochemistry, Bioscience Center; 4Institute of Tropical Medicine, Federal University of Rio Grande do Norte, Natal, RN, Brazil, and 5Institute of Science and Technology of Tropical Diseases, Salvador, BA, Brazil. Abstract
The pathogenesis of Guillain Barré Syndrome (GBS) is not entirely understood, but
includes infection-induced aberrant immune responses resulting from molecular
mimicry and cross-reacting antibodies in addition to complement activation and host
susceptibility. Human FCGR polymorphisms that modulate affinity for human IgG
subclasses have been associated with GBS. However, earlier FcγR association
studies were performed with small cohorts of GBS patients, yielding conflicting
results. We assessed whether polymorphisms in FCGR2A and FCGR3A were
associated with Guillain Barré syndrome in a Brazilian population. We genotyped
141 GBS patients and 364 healthy controls for FCGR2A and FCGR3A. The FCGR
genotypes and allele frequencies did not differ significantly between the patients
with GBS and the controls. None of the FCGR genotypes or alleles was associated
with either mild or severe disease among GBS patients. We conclude that FCGR
polymorphisms are not associated with susceptibility or clinical outcome in Guillain
Barré Syndrome in this Brazilian population.

136
Introduction
Guillain-Barré syndrome (GBS) is an immune-mediated polyneuropathy and
is currently the principal cause of acute neuromuscular paralysis. The three main
GBS subtypes are acute motor axonal neuropathy (AMAN), Miller-Fisher syndrome
(MFS), and acute inflammatory demyelinating polyneuropathy (AIDP).(1) There are
differences in the geographic distribution the clinical subtypes. In Brazil, Europe and
North America the demyelinating Guillain–Barré syndrome accounts for up 80 to
90% of cases (1-3), whereas in China, Japan, Bangladesh and Mexico the
frequency of the axonal Guillain–Barré syndrome is prevalent. The annual incidence
of GBS is reported to be between 0.6 and 4 cases per 100,000.(1, 2) In Brazil, the
mean annual incidence of GBS is 0.3 cases per 100,000 people and there seems to
be some regional variation in the incidence.(3)
Infection-induced aberrant immune responses resulting from molecular
mimicry and presence of cross-reacting antibodies, along with complement
activation, have been associated with the pathogenesis of GBS. (4) About two-
thirds of patients have a prior history of infections with pathogens such as
Campylobacter jejuni, cytomegalovirus and Epstein-Barr virus. The viruses were
associated with AIDP, whereas C. jejuni infection has been strongly associated with
AMAN.(1, 2) Pure motor symptoms and positive serum IgG antibodies to
gangliosides, such as GM1, GD1a or CalNAc-GD1a, are associated with AMAN.(5-
8) The association between antiglycolipid antibodies and AIDP is less well
established. It is hypothesized that the association between C. jejuni infection and
AMAN could be caused by cross-reactivity between epitopes in the lipo-
oligosaccharide present in the bacterial wall and gangliosides expressed on the
peripheral motor axons.(9) Serum samples from patients with AMAN after a
confirmed C. jejuni infection contained high titers of antibodies against GM1 or
GD1a. The terminal structures of lipo-oligosaccharides from C. jejuni were identical

137
to those of GM1 and GD1a present in human cells. This seems to be a mechanism
leading to production of antibodies aimed at bacterial lipo-oligosaccharide, but also
cross reacting with ganglioside present on the surface of peripheral motor axons. In
a rabbit model of GBS, immunization with a GM1-like lipo-polioligosaccharide
induced antiGM1 antibodies and manifested clinically as axonal neuropathy, similar
to what is found in patients with GBS from which the C. jejuni was isolated.(10)
However, not all C. jejuni contain epitopes similar to those recognized by GM1
gangliosides antibodies, (11)(12) and furthermore only 0.01% of patients with C.
jejuni enteritis present GBS.(13)
FCGR polymorphisms have been associated with human diseases, including
autoimmune disease. Three classes of human FCGR (FCGR2A, FCGR3A and
FCGR3B) contain allelic polymorphisms that influence their affinity for IgG
subclasses and consequently the efficacy of IgG-mediated effector functions.(14,
15) The role of FcγR in the development of GBS is supported by activation of
macrophages after interaction of complex ganglioside antibodies with the
immunoglobulin receptor, causing damage on the myelin or in the axon.(16)
Moreover, intravenous IgG, effective in GBS, acts by blocking FcγR and improves
recovery time.(17, 18)
Although there are numerous reports suggesting an association between
polymorphisms in genes for FCGR2A and FCGR3A and susceptibility to GBS, the
results of the distribution of these genotypes in GBS in different populations have
been inconsistent. Some studies have shown an association (19, 20), whereas
others showed no association (21, 22). In this study, we determined FCGR2A and
FCGR3A genotypes in 141 GBS patients and 364 healthy controls in the state of
Rio Grande do Norte, Brazil. We did not find an association in alleles or genotypes
of FCGR2A and FCGR3A in this population.

138
Materials and methods:
Subjects: A total of 141 individuals with GBS were recruited from the State of Rio
Grande do Norte, Brazil. GBS was defined by the Asbury and Cornblath criteria.
(23) Electrophysiological studies of the patients with GBS were used to classify the
cases as demyelinating, demyelinating and axonal, or axonal degeneration,
according to criteria as described previously.(6) The Miller-Fisher variant included
those subjects presenting with the triad of ataxia, areflexia, and ophthalmoplegia in
the absence of limb weakness.(24) The degree of severity was assessed at the
peak of illness using a functional disability scale described by Hughes. (25) Cases
were classified as severe (GBS disability scores of four or more) or mild (score of 3
or less). A total of 364 healthy blood donors from the same region were recruited as
controls.
Antiganglioside antibodies
Blood samples were collected from GBS patients during the acute phase of
disease. Serum samples were used to test for the presence of GM1 and GD1a
antibodies using ELISA, as described in a previous study. (8)
FcγR polymorphisms
Genomic DNA was extracted from leucocytes separated from whole blood using a
standard protocol. (26) FCGR2A and FCGR3A genotypes were determined by the
Real-Time PCR TaqMan® SNP Genotyping Assays (the assay IDs were
C_9077561_20 and C_25815666_10, respectively) using a 7500 Real-Time PCR
System (Applied Biosystems, CA, USA) in accordance with manufacturer’s
instruction.
Statistical analysis
Genotype and allele distributions were compared by the chi-squared test. To
estimate the genetic effect, we coded the genotypes assuming a log-additive model
and included it, as explanatory variable, in a logistic regression model adjusting for

139
age and sex. The analyses were performed in Stata 11.2 assuming a significance
level of α = 0.05.
Ethical considerations
The protocol was reviewed and approved by the Universidade Federal do Rio
Grande do Norte Ethical Committee (CEP-UFRN 046 ⁄ 03 and CEP- UFRN 198 ⁄ 09)
and by the Brazilian National Ethical Committee (CONEP: 9061). The certificate of
approval is 0215.0.051.000-09 (http://portal2. saude.gov.br/sisnep/). All subjects or
their legal guardian signed the informed consent.
(http://portal2.saude.gov.br/sisnep/)
Results
Patient characteristics
Of the 141 patients recruited, 82 (58.2%) were males and the median age was 29.4
years (±20.3). A group of 364 healthy blood donors from the same region were
recruited as controls (310 male, 54 female; median age=35.86 years). Controls
were purposely selected from an older population to allow for greater exposure to
the environmental risk factors, as infections, associated with GBS. The patient
characteristics are presented in Table 1. Evidence of recent infection for one or
more pathogen(s) was detected in 91 (64.5%) patients. Of those, 69 (49%)
presented a previous episode of respiratory tract infection. The majority of the cases
had a severe score at diagnosis. Of the 141 GBS cases, 100 (70.9%) were
classified as AIDP.
Genotype analysis
In comparing genotype and allele distributions between the GBS cases and
the controls, no difference was detected for either the R131H (FCGR2A) or P158V
(FCGR3A) polymorphism (Table 2). We also performed logistic regression analyses
adjusting for age and sex, but no genetic association was found (Table 3). Lastly,

140
we conducted two additional determinations in which the cases were classified as
either Axonal or Demyelinating and as either positive or negative antigangliosides
antibody status; no statistical difference was observed in either determination (data
not shown). The distribution of the FCGR genotype combinations were similar for
subjects in the GBS and control groups, and there was no association with mild or
severe forms of the disease (data not shown).
Table 1: Characteristics of the patients with Guillain-Barré syndrome
Characteristics Total
Number 141
Male/female 82/59
Age, y, mean (SD) 29.8 (±20.3)
Previous infection, n
Respiratory tract infection 69
Diarrhea 21
Diarrhea and Respiratory tract infection 1
vaccine 3
none 47
Severity score, n (%)
Mild (1-3) 27/135 (20)
Severe (4-5) 108/135 (80)
Antiganglioside antibodies, n (%)
Anti-GM1 24/116 (20.6)
Anti-GD1a 8/92 (8.7)
Anti-GM1 and/or Anti-GD1a 32/116 (27.6)
Electrophysiologic classification, n(%)
Demyelinating 100 (70.9)
AXONAL 36 (25.5)
Unclassified 3 (7.3)
Miller Fisher 2 (4.9)
SD, standard deviation; n, number.

141
Table 2. Genotypes and alleles distribution in the control and case groups.
Gene (variant)
Genotypes and alleles
Control, n (%)
GBS cases, n (%)
Mild Severe Total p-valuea p-valueb
FCGR2A (R131H)
R/R 106 (29.3) 31 (29.0) 8 (29.6) 40 (28.6) 0.955 0.849 R/H 182 (50.3) 56 (52.3) 15 (55.6) 74 (52.9)
H/H 74 (20.4) 20 (18.7) 4 (14.8) 26 (18.6)
R 394 (0.54) 118
(0.55) 31 (0.57)
154 (0.55)
0.905 0.869 H 330 (0.46) 96 (0.45) 23 (0.43)
126 (0.45)
FCGR3A (P158V)
F/F 180 (49.6) 48 (44.4) 13 (50.0) 66 (47.1) 0.606 0.886 F/V 148 (40.8) 51 (47.2) 9 (34.6) 60 (42.9)
V/V 35 (9.6) 9 (8.3) 4 (15.4) 14 (10.0)
F 508 (0.70) 147
(0.68) 35 (0.67)
192 (0.69) 0.817 0.665
V 218 (0.30) 69 (0.32) 17 (0.33) 88 (0.31) a Chi-squared test for comparison among Control, Mild and Severe groups. b Chi-squared test for comparison between Control and Total GBS groups. GBS, Guillain Barré Syndrome; n, number.
Table 3. Genetic effect on disease risk estimated through logistic regression models a
Risk allele
Total GBS Mild GBS Severe GBS
OR (95% CI) p OR (95% CI) p OR (95% CI) p
FCGR2A (H131)
0.92 (0.67 - 1.27)
0.624 0.87 (0.62 -
1.24) 0.45
3
0.86 (0.47 - 1.57)
0.630
FCGR3A (V158)
1.00 (0.71 - 1.40)
0.982 0.99 (0.68 -
1.44) 0.96
1
1.12 (0.60 - 2.10)
0.713
a Regression model using control as the reference group and age and sex as covariates. GBS, Guillain Barré Syndrome; OR, odds ratio; CI, confidence interval
Discussion
In this study, the FCGR genotypes and allele frequencies did not differ
significantly between the GBS patients and the controls. In addition, FCGR
genotypes did not correlate with disease outcome. The most frequent alleles
detected in this study were FCGR2A-R131 and FCGR3A-F158. This is in
agreement with the FCGR genotype distribution in healthy donors from other

142
Brazilian populations and similar to those reported for whites in the United States
and Europe. (27-30)
Attempts to identify immunogenetic risk factors in patients with GBS have not
been uniform. Evaluations of FCGR gene polymorphisms have been performed on
populations of Dutch, Norwegian, English and Indian. A meta-analysis, which
included English and Dutch populations (22) and a study on Norwegians, (21) did
not confirm an association between the polymorphism of FCGR and occurrence of
GBS. However, FCGR2A H/H131 genotype frequency was significant increased in
both Dutch (19) and Indian (20) patients with GBS. The FCGR3A-V/V158 genotype
also was associated with increased risk for developing GBS.(20) The results were
also different when GBS was evaluated with respect to severity and the frequency
of genes and alleles. The study of Dutch patients showed an association of
FCGR2A H/H131 genotype with severe disease (19).
Genetic polymorphisms of FCGR were associated with a variety of biological
responses. Genotypes FCGR2A H/H, FCGR3A V/V and FCGR3B NA1/NA1 are
more efficient in binding IgG. Both FCGR2A and FCGR3A are expressed on
macrophages and the rationale is that individuals homozygous for high affinity
receptor H-131 and V-158 alleles undergo phagocytosis and produce higher levels
of proinflammatory cytokines than individuals with R-131 and F-158 alleles.
Because FCGR3B is found only on neutrophils and the role of these cells in
the pathogenesis of GBS is unknown, the association with FCGR3B polymorphism
is questionable. In the Norwegian study, subjects with FCGR3B NA1/NAI had a
higher risk for mild disease, (21) whereas subjects with FCGR3B NA2/NA2 had a
greater risk for severe disease.(22) In the same study, the FCGR3A-F/F158
genotype was associated with mild forms of the disease.

143
Interestingly, the FCGR3B NA1/NA1, not genotyped in this work, increases
the activity of FCGR2A H/H. (31, 32) In fact, these autocrine and paracrine effects
may provide a basis for the FcγR receptors influencing the immune system.
Therefore, unlike previous studies (21,22), we expected that the homozygous
FCGR3B NA1/NA1 were more likely to develop GBS and / or have the most severe
form of the disease. The difference in the findings may reflect ethnic differences, the
real genetic heterogeneity, or small and low statistical power samples, which makes
comparisons between studies difficult. Our results contribute to this disparity. We
did not observe any significant effect at the 0.05 cut-off level. This seems to indicate
the robustness of our negative findings in this large sample of subjects with GBS
from a single ethnic background.
It is possible that the receptor model is inappropriate for GBS. Other
mechanisms may be exploited in GBS, such as neutrophil interactions with other
components of the immune system, the modulation of protein tyrosine kinases or
the involvement of transcription factors. Several genes, mapped in the vicinity of the
FCGR locus, that influence the vigor of the inflammatory response may provide an
alternative explanation.(22) More recently, studies have shown copy number
variation (CNV) could be involved in susceptibility to diseases, including
autoimmune diseases. There is CNV for the locus 1q23, where FCGR2A and
FCGR3A are located. Deletion in FCGR3A leads to reduction of FcRIIIa expression
in NK cells (?), resulting in a decrease in their cytotoxic function. There is no
description of CNV for FCGR2A.(15, 33) We did not assess CNV for FCGR2A and
FCGR3A in our population. In summary, our findings suggest that FCGR genotypes
are not associated with GBS susceptibility or disease course. We conclude that
FCGR polymorphisms are not risk markers for GBS. Further studies are needed to

144
better understand the possible FCGR relationship to molecular mechanisms
involved in development of GBS, including differences in copy number variation.
References
1. Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005 Nov 5;366(9497):1653-66. PubMed PMID: 16271648. 2. Yuki N, Hartung HP. Guillain-Barre syndrome. The New England journal of medicine. 2012 Jun 14;366(24):2294-304. PubMed PMID: 22694000. 3. Dourado ME, Felix RH, da Silva WK, Queiroz JW, Jeronimo SM. Clinical characteristics of Guillain-Barre syndrome in a tropical country: a Brazilian experience. Acta neurologica Scandinavica. 2012 Jan;125(1):47-53. PubMed PMID: 21428966. 4. van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet neurology. 2008 Oct;7(10):939-50. PubMed PMID: 18848313. 5. Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990 Dec;40(12):1900-2. PubMed PMID: 2247243. 6. Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, et al. Guillain-Barre syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain : a journal of neurology. 1995 Jun;118 ( Pt 3):597-605. PubMed PMID: 7600081. 7. Odaka M, Yuki N, Hirata K. Anti-GQ1b IgG antibody syndrome: clinical and immunological range. Journal of neurology, neurosurgery, and psychiatry. 2001 Jan;70(1):50-5. PubMed PMID: 11118247. Pubmed Central PMCID: 1763462. 8. Dourado ME, Duarte RC, Ferreira LC, Queiroz JW, Illa I, Perez-Perez G, et al. Anti-ganglioside antibodies and clinical outcome of patients with Guillain-Barre Syndrome in northeast Brazil. Acta neurologica Scandinavica. 2003 Aug;108(2):102-8. PubMed PMID: 12859286. 9. Yuki N, Susuki K, Koga M, Nishimoto Y, Odaka M, Hirata K, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2004 Aug 3;101(31):11404-9. PubMed PMID: 15277677. Pubmed Central PMCID: 509213. 10. Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, et al. Anti-GM1 antibodies cause complement-mediated disruption of sodium

145
channel clusters in peripheral motor nerve fibers. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007 Apr 11;27(15):3956-67. PubMed PMID: 17428969. 11. Godschalk PC, Bergman MP, Gorkink RF, Simons G, van den Braak N, Lastovica AJ, et al. Identification of DNA sequence variation in Campylobacter jejuni strains associated with the Guillain-Barre syndrome by high-throughput AFLP analysis. BMC microbiology. 2006;6:32. PubMed PMID: 16594990. Pubmed Central PMCID: 1513382. 12. Sheikh KA, Nachamkin I, Ho TW, Willison HJ, Veitch J, Ung H, et al. Campylobacter jejuni lipopolysaccharides in Guillain-Barre syndrome: molecular mimicry and host susceptibility. Neurology. 1998 Aug;51(2):371-8. PubMed PMID: 9710005. 13. McCarthy N, Giesecke J. Incidence of Guillain-Barre syndrome following infection with Campylobacter jejuni. American journal of epidemiology. 2001 Mar 15;153(6):610-4. PubMed PMID: 11257070. 14. Takai T. Roles of Fc receptors in autoimmunity. Nature reviews Immunology. 2002 Aug;2(8):580-92. PubMed PMID: 12154377. 15. Gillis C, Gouel-Cheron A, Jonsson F, Bruhns P. Contribution of Human FcgammaRs to Disease with Evidence from Human Polymorphisms and Transgenic Animal Studies. Frontiers in immunology. 2014;5:254. PubMed PMID: 24910634. Pubmed Central PMCID: 4038777. 16. van Sorge NM, van den Berg LH, Geleijns K, van Strijp JA, Jacobs BC, van Doorn PA, et al. Anti-GM1 IgG antibodies induce leukocyte effector functions via Fcgamma receptors. Annals of neurology. 2003 May;53(5):570-9. PubMed PMID: 12730990. 17. van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. The New England journal of medicine. 1992 Apr 23;326(17):1123-9. PubMed PMID: 1552913. 18. Dalakas MC. Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology. 1998 Dec;51(6 Suppl 5):S2-8. PubMed PMID: 9851723. 19. van der Pol WL, van den Berg LH, Scheepers RH, van der Bom JG, van Doorn PA, van Koningsveld R, et al. IgG receptor IIa alleles determine susceptibility and severity of Guillain-Barre syndrome. Neurology. 2000 Apr 25;54(8):1661-5. PubMed PMID: 10762510. 20. Sinha S, Prasad KN, Jain D, Nyati KK, Pradhan S, Agrawal S. Immunoglobulin IgG Fc-receptor polymorphisms and HLA class II molecules in Guillain-Barre syndrome. Acta neurologica Scandinavica. 2010 Jul;122(1):21-6. PubMed PMID: 20105138.

146
21. Vedeler CA, Raknes G, Myhr KM, Nyland H. IgG Fc-receptor polymorphisms in Guillain-Barre syndrome. Neurology. 2000 Sep 12;55(5):705-7. PubMed PMID: 10980740. 22. van Sorge NM, van der Pol WL, Jansen MD, Geleijns KP, Kalmijn S, Hughes RA, et al. Severity of Guillain-Barre syndrome is associated with Fc gamma Receptor III polymorphisms. Journal of neuroimmunology. 2005 May;162(1-2):157-64. PubMed PMID: 15833371. 23. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barre syndrome. Annals of neurology. 1990;27 Suppl:S21-4. PubMed PMID: 2194422. 24. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). The New England journal of medicine. 1956 Jul 12;255(2):57-65. PubMed PMID: 13334797. 25. Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet. 1978 Oct 7;2(8093):750-3. PubMed PMID: 80682. 26. Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic acids research. 1989 Oct 25;17(20):8390. PubMed PMID: 2813076. Pubmed Central PMCID: 334995. 27. Siqueira JF, Jr., Rocas IN, Provenzano JC, Daibert FK, Silva MG, Lima KC. Relationship between Fcgamma receptor and interleukin-1 gene polymorphisms and post-treatment apical periodontitis. Journal of endodontics. 2009 Sep;35(9):1186-92. PubMed PMID: 19720214. 28. Siqueira JF, Jr., Rocas IN, Provenzano JC, Guilherme BP. Polymorphism of the FcgammaRIIIa gene and post-treatment apical periodontitis. Journal of endodontics. 2011 Oct;37(10):1345-8. PubMed PMID: 21924179. 29. Kuwano ST, Bordin JO, Chiba AK, Mello AB, Figueiredo MS, Vieira-Filho JP, et al. Allelic polymorphisms of human fcgamma receptor IIa and Fcgamma receptor IIIb among distinct groups in Brazil. Transfusion. 2000 Nov;40(11):1388-92. PubMed PMID: 11099670. 30. Vigato-Ferreira IC, Toller-Kawahisa JE, Pancoto JA, Mendes-Junior CT, Martinez EZ, Donadi EA, et al. FcgammaRIIa and FcgammaRIIIb polymorphisms and associations with clinical manifestations in systemic lupus erythematosus patients. Autoimmunity. 2014 Nov;47(7):451-8. PubMed PMID: 24896836. 31. Salmon JE, Edberg JC, Kimberly RP. Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. The Journal of clinical investigation. 1990 Apr;85(4):1287-95. PubMed PMID: 1690757. Pubmed Central PMCID: 296565.

147
32. Salmon JE, Millard SS, Brogle NL, Kimberly RP. Fc gamma receptor IIIb enhances Fc gamma receptor IIa function in an oxidant-dependent and allele-sensitive manner. The Journal of clinical investigation. 1995 Jun;95(6):2877-85. PubMed PMID: 7769129. Pubmed Central PMCID: 295975. 33. Moriya H, Saito K, Helsby N, Hayashi N, Sugino S, Yamakage M, et al. Single-nucleotide polymorphisms and copy number variations of the and genes in healthy Japanese subjects. Biomedical reports. 2014 Mar;2(2):265-9. PubMed PMID: 24649108. Pubmed Central PMCID: 3917743.


















