
PRODUÇÃO DE ÓXIDO DE TÓRIO NUCLEARMENTE PURO · A purificação de tório por extração com...
32
PRODUÇÃO DE ÓXIDO DE TÓRIO NUCLEARMENTE PURO K. J. BRIL e P. KRUMHOLZ PUBLICAÇÃO lEA N.' Dezembro — 1965 INSTITUTO DE ENERGIA ATÔMICA Caixa Postal 11049 (Pinheiros) lADE UNIVERSITÁRIA "ARMANDO DE SALLES OLIVEIRA" SÃO PAULO — BRASIL
Transcript of PRODUÇÃO DE ÓXIDO DE TÓRIO NUCLEARMENTE PURO · A purificação de tório por extração com...

P R O D U Ç Ã O D E ÓXIDO D E TÓRIO N U C L E A R M E N T E P U R O
K. J. BRIL e P. KRUMHOLZ
PUBLICAÇÃO lEA N.'
Dezembro — 1965
INSTITUTO DE ENERGIA ATÔMICA Caixa Postal 11049 (Pinheiros)
lADE UNIVERSITÁRIA "ARMANDO DE SALLES OLIVEIRA"
SÃO PAULO — BRASIL

PRODUÇÃO DE ÓXIDO DE TORIO NUCLEARMENTE PURO
K. Je Bril " ^ e P. Ki^Mholz'^^
Divisão de Engenharia Química
Instituto de Energia Atómica
Sao Paalo - Brasil
Publicação lEA m 1 1 5
Dezembro - 1955
(1) K.. J. Bril - Chefe da Divisão de Engenharia Química -
Instituto de Energis Atómica
(2) P, IQrumfaolz - Diretor, Laboratório de Pesquisas da
Orquima S«A<.

Comissão Kacíonai de Energia .Suelear
Presidente? Prof. Luiz Cliitra do Prado
Universidade de Sao Bs-olo
Reitor? Prof o Luiz Antonio da ümcia. e Silva
Instituto de Energia Atbmioa
Diretors Prof. Romulo Ribeiro Plerori
Conselíio Teenico-Científ ico do IFl/l
Prof. Helio Ijourenço ne Oliveira ) ) pela USP
Pro.f. Walter Bcraani )
Prof. Fiui Ribeiro Frax,iG<' )
) pela CNEH Prcf. ülieodoreto fLI, áe A,r:ruda Souto )

PRODUÇÃO DE ÓXIDO DE TORIO MtJCIfiARMEHTE PURO
K. J. Brll è P. Krumholz
RESUMO
Os progressos recentes no tratamento de minérios de tó
rio e nos processos de obtenção de óxido de torio nuclearmente
puro, são discutidos com referencia especial à experiencia indiis
títal e em escala piloto realizada no Brasil.
O ataque alcalino da monazlta, prfttlcado em Sao Paulo
desde 19^8 era escala industrial, apresenta do ponto de vista da
recuperação de torio, algumas vantagens sobre os outros métodos
de ataque. Os rendimentos são praticamente quantitativos e os
fatores de separação entre tório e terras raras, respectivamente
tório e fosfato são da ordem de 100. O concoitrado de tório re
sultante constitui um exoelente ponto de partida para a purifica
ção de tório por extração com solventes.
A purificação de tório por extração com tributilfosfa
to é discutida com «ifase especial sobre os problemas de:
a) extração de soluções não filtradas (suspensões);
b) separação de tório das terras raras;
c) separação de tório do uranio.
A formação de oaulsÕes durante a extração de suspen
sões pode ser evitada pelo uso de carvão ativo cc»no agente eoitl-
-emulslfloante.
A separação de tório das terras raras apresenta alguns

. 2 .
aspectos imprevistos quando se usa colunas pulsadas para as opera
ções de lavagem do extrato orgânico. Apesar de que os fatores de
separação aumentam rápidamente com a concentração crescente de tó
rio no solvente, as vantagens deste fenômeno estão mais que anula
das pelo aumento da altura de um estágio teórico numa coluna pul
sada operada em regime de concentrações elevadas de tório na fase
orgânica.
Separações muito eficientes entre torio e uranio podem
ser realizadas por meio de precipitação extrativa de sulfato de
torio. Fatores de separação da ordem de 20.000 podem ser obtidos
num único estagio. O processo assegura uma recuperação simultanea
de ácido nítrico.
A precipitação de sulfato do tório é seguida de uma
transformação de sulfato em hidroxicarbonato^ a partir da qual,
por càlcinaçao a 900 - 1000°C, obtém-se um óxido de tório de pure
za elevada.
ABSTRACT
Recent developments in the processing of thorium
bearing ores and in the production of nuclear grade thorium oxide
are reviewed with special reference to industrial and pilot plant
experience in Brazil.
The alkaline process for breaking up monazite, as
practiced in Brazil since I9V8 on an industrial scale, presents
from the point of view of thorium recovery, considerable advantage
over other methods. In particular, yields are practically
quantitative, separation factors of thorium from rare earths and
phosphate are of the order of 100, and thorium is obtained in a
form very suitable for solvent extraction purification.
Solvent extraction purification of thorium is discussed
with special emphasis on: a) slurry extraction, b) thorium

5 .
ucocontamination from rare earths and c) separation of thorium
and uranium.
Eriiuliilon formation in the extraction of unfiltered
solutions i s prt-:vented by the use of active carbon as. de-
emulsifying agent.
Satisfactory descontamination of thorium from the rare
earths, presen' is some peculiar problems if the scrubbing opera
tion is performed in pulsed extraction columns. Whereas increas
ing thorium concentrations in the tributylphosphate solvent
improves the thorium-rare earths single-stage separation factors,
it severely reduces the rate of transfer of the minor
constituents into the aqueous phase, resulting in high H.E.T.S.
values.
Highly efficient thorium-uranium separation is obtain
ed by using an extractive-precipitation of thorium sulfate.
Ihorium-uraniuOT separation factors of up to 20.000 are obtained
in a single-stage operation. A further advantage of the extractive
precipitation procedure is the easy recovery of nitric acid.
After the transforfnation of thorium sulfate into a basic
carbonate, a iiigh purity thorium oxide is obtained by calcination.
Les proipr'ès récentes dans le traitement des minerais de
thorium et dans la production d'oxyde de thorium nucleaireraent pur
sont discutés, en relation avec l'expérience à l'échelle industrie
le aussi bien que pilote, acquise au Brésil.
Du point de vue de la production de thorium, le procédé
alcalin du traitment de la monazite, utilisé au Brésil à l'échel
le industrielle déjà depuis 19^l8, offre un avantage considerable

en comparaison avec les autres techniques. En particulier, le ren
dement est pratiquement qua quantitatif, les facteurs de sépara -
tion du thorium des terres rares et du phosphate sont de l'ordre
de 100, et le thorium est obtenu dans une forme bien appropriée
pour une purification finale par extraction avec des solvents.
La purification du thorium par extraction avec des sol-
vents est discutée tout particulièrement en relation avec les pro
blêmes suivants: a) extraction des solutions non filtrées, b) dé
contamination du thorium des terres rares, c) séparation du tho
rium de l'uranium.
La formation des emulsions pendant l'extraction des so
lutions non filtrées, est éliminée par l'emploi du carbon actif
comme agent desémulsifiant.
Iftie decontamination satisfaisante du thorium des terres
rares présente un aspect particulier lorsque l'opération de lava
ge est faite dans des colonnes puisées à plaques perforées. L'aig
mentation de la concentration du thorium dans le tributyl phospha
te favorise les coefficients de séparation entre le thoriuum et
les terres rares. Cependant cette augmentation de la concentration
est accompagnée par une diminution de la vitesse de transport du
constituant mineur, avec augmentation résultante de la hautetar du
plateau théorique.
On obtient des séparations très nettes du thorium de l'
uranium par l'extraction et précipitation simultanée du sulfate
de thorium. On peut attendre des coefficients de séparation al
lant Jusque'à 20.000 en une seule opération. Le procédé permet
une récupération facile de l'acide nitrique.
Après transformation du sulfate de thorium en carbonate
basique on obtient un oxyde de thorium, de très haute pureté, par
calcination.

. 5 *
Io i™omJSAO
A£ origeíis da :i.ndústria de tório ascendem ao início do
século XX.O A ffiatéi/ia prima :foi co.ustituida, pela monazita e o pro
duto final foi o nitrato de tório q.ue serviu pai-a confecção das
camisas para ilwíinaçao a gás= Entre as rj.iia,s guerras mundiais ,
este mercado de torio desapareceu progressivainentec A lenta renas_
cença do ...¡ercado dssts metal &oiía-se atusimente Intrinsicamente li
gada com o futiaro desenvolvlñiento dos programas de produção de
energia atômica^ já que as aplicações nao-nucleares de torio con-
tinuam secido de pouo:a iiiiportancJ,a, A estíerança de utilizar o to
rio em reatores de potencia jast Lf j.ca o interesse crescente nos
trabalhoíí fòobre aperf-aiçoa.Ticrit.0 di?, tecnologia deste metal. O obje
tivo do presente treV. iho í -Xç resumir os progressos realizados
no tratamento de miaÓT-los de torÃO e ua produção de óxido de tó
rio nuclearmente-; uuro, insistindo em primeiro lugar sobre a expe
riencia industrial e piloto que ja c?.:Jde \rário3 anos esta sendo
acumulada do Brasilo
necent.eiTiente. as reservas do rnimdo ocidental' em tório
de baixo custo íp - 1.0 Uo$/ib de lS-iO„) fortoíi estiííiadas ao redor
de 5 X 10"' tonela.ia; de aiiO^ aperiss ( 1 ) , rfeve-se portanto que a
fabricação futura da ooníbustJvel nuolear vai depender essencial -
mente das disponibilidades dos minarí j s de bai,xo teor de tório ^ ) .
Iftna bibliografia recente ccntán nvisritrosas referencias sobre a mi-
neralogia, prc;--p«cçào, explora-jao e reservr; lie torio (5)= O nume
ro dos inlnériop que o catem tório liltratassa 100. Entre eles mais
1 - Kaplan e c o i a b o r a d o r e K dlscutesn o s roc\;rños m i n e r a i s de
tório na íÃ';;.iã'i Sov-lstxcíi (sogurido J Iuc lc Sei, Abstr. l6 (1962\ n? 20^ 852..

„"U menus 60 contém ao reãor de 1% ds IhO,. . Apessr- o.XBSQ¡ alèm Xa
racRsaita que é a fonte principal de tóri-:<f t rianlta e torlta sao
os únicos comercialmente e.xplc>r£,d'.s Tío g[iie ce refei-'e ó. nionssita
O), a capacidade ds mlnere,çao atmlmonta instalada, ultrap.assal££
gamente o ritmo de cmsxmOf e a tondéncia ganer-ali ada nos países
produtores é de diminuir mais ain<Ja a producá :> (6)0
III. GBnmçÃo m oaminmuDOü t ^ r i o
Bn gerai, a materia prima para a fabricação de tório d©
pureza nuclear é cí>n.stituida per iro, concentrado, mñl.s oi?. menos x'l'
CO em sais de torio. D processo frsanees partindo de yjn ur&jio-to-
riari.ita particularmente? rico en; tc-:rio constituí ama notável e:xc3-
•jão (ver tabela l). Ape.sar d.loêo,, o dasenvolvimanto rápido da te£
nologia de extração com sclvaiba-s,, nos permite entrever pax'.a um
futuro não muito distante, a, obtenção de t ório muito pwo^ p9..rtx3
do até de minérios muito pobres (T).
A tabela I da um raciTmo do,-r. diferentes métodos da trata
msnto de minérios de tório, incluindo as tentatlvs.s muito recen
tes de recuperação de torio "caro" (até 500 üi flb - 'Bi «- ü) par ~
tindo de rainérioG de muito baixo teoí"- de torio e uranio.
Numerosos itiétodoiv de tratariientc- de nionazita foimm recen
temente discutidos ( 5 ) . A3 vantagens relativas destes métodos
continuam sendo d.iscutíysl,i. Ife práticia j.ndu;-triíi.l U3,ar;\-*a méto
dos baseados sobre o ataque seje com ácido í>vilfúr;ico scja com so
da cáustica. À análise do rusto de produção (7-9) dá ama pequena
vantagem aos processos que en^olvgm a-eí>.'3.Ui spJi'úrioo. Esta compa
ração entretanto nao dé. o devido valor às terras i'aras s ao tri
fosfato de sódio» O .ataque aloalíno perinite g. r-ealizaçat- de urna
separação partic-alar-it.ente nítida entre fosfato, torio e terras ra
ras. As terras raras, em p€,r-tií;'alar_, poáe.ni. ser obtidas pràtieaiten
te livres de ambos s torio e focí'atc. O :ítaqrs í¡,loñ.llno msrerc por

. 7 .
tanto a nossa preferencia desde q.ue se considere a possibilidade
de purificação futura de tório e/ou de separação do grupo das ter
ras raras em seus elementos individuais.
O processo brasileiro
O tratamento químico da monazita foi iniciado no Bra
sil em 19-'3. A capacidade de produção é da ordem de 5.000 tonela
das de monazita por ano, bem acima da produção atual. O processo
baseia-se sobre ataque com soda cáustica. Vários anos de experiai
cia industrial permitiram aperfeiçoar sensivelmente o processo
oial (10) .
A monazita é purificada até alcançar um concentrado de
• 9&.^'jl>> O minério é constituido por 6ks^ de óxidos de terras ra
ras (ingO^), 5-5 - 6.5^ de TliO e 0.I5 - 0.35^ de U^Og. A grande
pureza da areia facilita muito o tratamento químico ( 1 1 - 1 2 ) .
A monazita purificada é moída até conter 99^ minus 30O
raesh. O ataque com soda cáustica (-55^ p/p) é conduzido sob pre£
são de várias atmosferas a 170°G« A soda está sendo utilizada com
excesso de ~100^ sobre a quantidade teórica. Os hidróxidos de tó
rio e de terras raras são retomadas com água e separados do tri -
fosfato de sódio e do excesso de hidróxido de sódio por filtraçao.
Depois da cristalização de trifosfato de sódio, as águas maes
são concentradasj -50^ da soda cáustica, assim recuperada, pode
ser recirculada no ataque principal.
A adição de agentes redutores (p, ex. glucose) durante
o ataque cora soda cáustica permite evitar a oxidaçao de cério. O
cério oxidado pode trazer algumas dificuldades, que foram encon -
tradas também por outros autores (h-S-^k), durante a dissolução de
terras raras.
O processo que está sendo seguido para a separação de

8 .
tório das terras raras depende da forma na qual estas ultimas po
dem ser vendidas.
Si o produto final desejado são os cloretos de terras
raras, os hidróxidos mistos de tório e terras raras sao retomados
em água em quantidade necessária para assegurar uma concentração
final de JOO g/l de óxidos totaisj em seguida os cloretos de ter
ras raras sao dissolvidos seletivamente, a 70° com ácido clorí -
dricü (pH final ~ ^ . 5 ) . TÓrio é recuperado quantitativamente e o
uranio quase com.pletamente. Os fatores de descontaminaçao (D) de
tório das terras raras e dos fosfatos são da ordem de 100 (11-15)«
Uma separação satisfatória de tório das terras raras nao
pode ser obtida pela lixiviaçao seletiva com E^SO^„ Portanto ,
si o nosso desejo é obter as terras raras em forma de sulfates ou
sulfates duplos, os hidróxidos mistos sao retomados com água edi£
solvidos completamente em ácido sulfúrico a ^5° C, até alcançar
uma concentração final de 80 g/l. Ein seguida o tório é precipita
do com amoníaco até alcançar o pH final de 6, As terras raras oo-
-precipitadas são solubilizadas finalmente por meio de ácido clo
rídrico diluido.
Uma terceira alteniativa foi elaborada para produzir teta
ras raras livres de cério. Neste caso os hidróxidos mistos estão
sendo secados e cério oxidado por meio de ar ( 1 ^ ) , ou então reto
mados com água e cério oxidado com cloro ( 1 5)' Eiitão as terras ra
mSf livres de tório, sao solubilizadas seletivamente por meio de
ácido clorídrico. Bn seguida o tório é separado do cério por dis
solução em presença de um agente redutor (metanol, glucose, SO^ ,
etc). FinalüPente precipita-se o tório por meio de ajnoníaco.
O hidróxido de torio bruto obtido por um dos tres meto-
dos acima expostos, é em seguida tratado com uma solução de carbo
nato e bicarbonato de sódio. 70-80^ de uranio e ao mesmo tempo
uma grande jsrte de cloretos, são assim solubilizados. O urânio é
finalmente recuperado do filtrado na forma de diuranato de sódio.

. 9 .
O hidróxido de tório bruto assim obtido, tem a composi
ção indicada na tabela II. Durante um certo tempo foi produzido,
a partir deste hidróxido bruto, um sulfato de torio de pureza in
dustrial. Os detalhes deste processo foram publicados numa das
conferencias de Genebra por Krumholz e Gottdenker ( l6) .
IV. PREPARAÇÃO DE ÓXIDO DE TÓRIO DE PUREZA NUCLEAR
Até a presente data, no caso de tório, especificações
de pureza nuclear publicadas sao muito escassas. Bn geral, está
sendo exigido manter abaixo de 0.05 PPra as terras raras que pos
suem altas secçÕes de choque com os neutrons térmicos: samário ,
gadolinio, europio e disprósio. Considerando a produção de
o conteúdo de deveria ser também muito pequeno e de preferen
cia menor de 1 ppm ( 1 7 ) . Partindo de um produto de composição in
dicada no quadro II, chegamos à conclusão de que do processo de
purificação vamos ter que exigir um fator de descontaminaçao da 5 k ^
ordem de ICr para as terras rai?as e da ordem de 10 para o uranio.
Os métodos atuais de produção de tório de pureza nuclear
em escala industrial sao todos eles baseados sobre extração com
solventes, apesar de estar sendo também estudados, processos fa
zendo uso de resinas troca-íons (7-66-76-78). Tributilfosfato é o
solvente mais popular, Apesar disso, um esforço importante está
sendo dedicado à procura de solventes ainda melhores (80-89) so
bretudo para a eventualidade do retratajnento dos combustíveis ir
radiados (79-90-106-108). O estudo dos efeitos sinergéticos (95-
-98) e dos efeitos específicos dos diluentes (99) deve também cca
tribuir para melhorar os futuros processos de extração.
O uso industrial do tributilfosfato foi sistematicamen
te estudado na França {6^-6k), na índia {hl-9l), na Inglaterra
(20-2^-56-66), no Brasil (92-95), etc. O livro de CUTHBERT (7)
trás um excelente retrospecto dos processos usados nos Estados

. 10 .
Unidos. Vamos limitar a áiscussao aos métodos de preparação de
óxido de tório nuclearmente puro, partindo de concentrados qj.íe pro
vem do tratamento da monazita.
Segundo a descrição de JAMRA.CK (66) o processo qué esta
sendo seguido na Inglaterra inicia-se partindo de soluções nítrl--
cas com a extração de uranio por meio de tributilfosfato diluido
a 5fo em xilerio. 0 tório é separado em seguida das terras raras e
das outras impurezas, por extração com trj.butilfosfato a tO . Ca
da ciclo é composto de 5 estagies de extração, 5-6 estágios de la
vagem e 5 estágios de re-extraçao, O tório é finalmente re-extrai
do por meio de ácido nítrico 0.02 N. O produto final, dependendo
das características requeridas, é obtidc por desnitração ou por
precipitação de oxalato ou de hidróxido (por meio de amoníaco).l6n
processo muito semelhante foi estudado na índia ( 91 ) .
Seguindo o processo elaborado pf lo Instituto Battelle
nos Estados Unidos (kS-hg-^'^l), a separação de uranio está sendo
efetuada após a extração de tório por meio de tributilfosfato.I^
rio é re-extraido seletivajnente e o uranio porventura co-extraído
é mais uma vez extraído com solventa fresco. A fim de completara
descontaminaçao de uranio, o torio e precipitado em forma de cxa-
lato. Seguindo a alternativa estudada no Instituto de Ames (8-9-
-19 ) o concentrado de tório de partida é obtido via precipitação
de oxalatos de maneira que o «eu conteúdo em uranio, ao iniciar-
-se o processo de piarificaçao com. solventes, já é bastante reduzi_
do.
O processo brasileiro
O processo estudado no Brasil para a obtenção de oxido
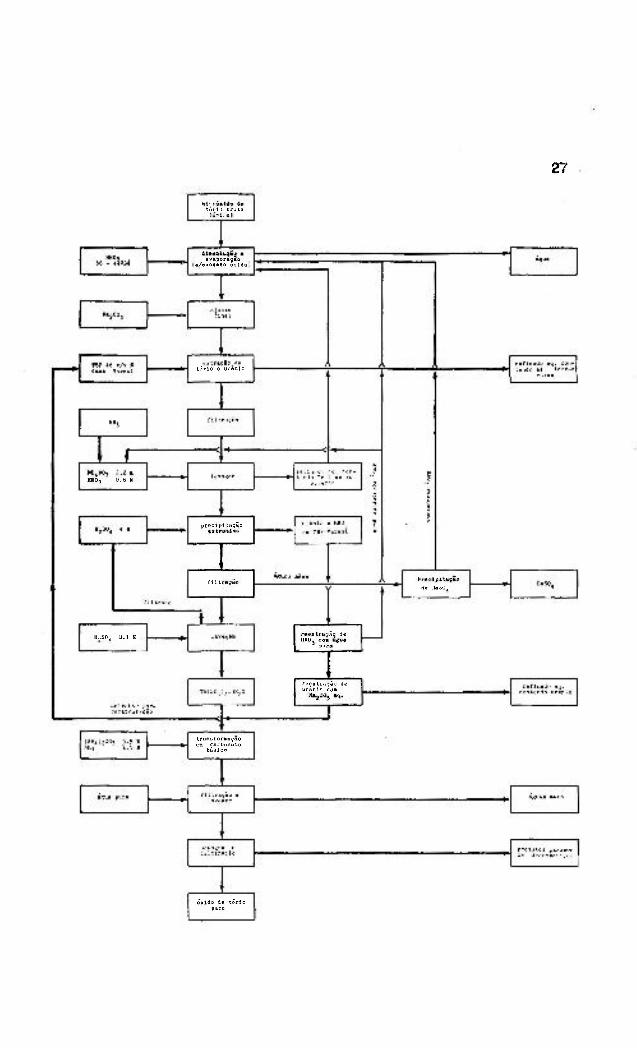
de tório de pureza nuclear (ver fig. I) difere em vários pontos
da prática corrente;
a) o tório o extraído a partir de uma solução ds teiíra

o 1 1 .
acidez j
b) o tório é extraído sera filtraçao prévia, a partir dé
suspensões que podem conter até 150 g/l de sólidos ,
usando carvão ativo para prevenir a formação even
tual de eraulsoes ( 1 0 1 ) |
c) o tório é re-extraído por precipitação extrativa de
sulfato de torio ( 1 0 1 ) |
d) uma separação adicional de viranio, devido a alta se~
letividade da precipitação extrativa, toma-se desn£
cessáriaí
e) mais ou menos 60^ do ácido nítrico recuperado pode
ser recirculado na dissolução.
As dificuldades de preparação de suspensões cOm boa fil
trabilidade, por dissolução do hidróxido bruto de torio em ácido
nítrico, sao bem conhecidas, o que levou vários pesquisadores a
procurar conseguir a extração direta das dissoluções de hidróxido
de tório bruto. Os resultados foram pouco satisfatórios (10^-105).
Procedemos a um estudo sistemático das condições de dissolução e
introduzindo o uso do carvao ativo para evitar a formação de emul
soes, conseguimos vencer as dificuldades encontradas na extração
direta das sujspensoes.
A seletividade da separação entre tório e uranio por
precipitação de sulfato de tório aumenta extraordinariamente, se
a precipitação do sulfato de tório é feita diretamente partindo
de solução orgánica. A concentração resultante muito elevada em
nitrato e a presença de tributilfosfato, permitem manter sempre a
concentração de uranio na fase aquosa a um nivel extremamente bai.
xo e desta maneira diminuir a co-precipitaçao por um fator da or-
(Jem de 100 em comparação com o gráu de co-precipitaçao partindo
de uma solução aquosa pura ( 1 0 1 - 1 0 2 ) .

. 12 .
IV. 1 . Preparação de soluções para extração
Introduz-se mais ou menos l/3 da carga úmida do hidróxi^
do bruto de tório dentro do líquido que provém da etapa de lava
gem ("scrubbing"). Introduz-se em seguida, alternativamente, o
ácido nítrico recuperado e o hidróxido bruto. Mantém-se uma aci
dez livre deficiente, evaporando ao longo de todo o processo a
suspensão até alcançar 550 g/l de óxido de tório . Introduz-se
em seguida ácido fresco, Be°, até uma acidez livre de
2 - 2 .5 N. A suspensão é em seguida mantida dixrante 6 horas a
uma temperatura próxima de ebulição. A solução é finalmente neu
tralizada até uma acidez livre de 0.8 K com carbonato de sódio.
A suspensão assim preparada contém; 280 g/l de óxido de tório ,
2 . 2 M líHi NO * KaKO^ e 100-150 g/l de sólidos.
IV. 2 . Purificação por extração
Por razoes discutidas num outro trabalho (95) o solvai
te usado contém 1+6 v/v^ de tributilfosfato e v/vf de Varsol
(nome comercial de uma fração alifática de petróleo). Este diluai
te é previamente tratado com ácido sulfúrico para eliminar os hi
drocarbonetos nao saturados e aromáticos.
É bem conhecido que os coeficientes de distribuição de
muitos elementos diminuem rapidamente quando a concentração de t£
rio no tributilfosfato aproxima-se do limite de saturação (7 ) . A
fim de aproveitar no máximo este efeito, decidimos trabalhara con
centraçoes muito elevadas de tório em tributilfosfato. Todavia, o
solvente usado separa-se em duas fases quando a 50° C, a concen-
2 - Evaporando esta suspensão a 220 l/lOO kg de óxido de tório e introduzindo-a em 105 1« de ácido nítrico 56 Be°, a 70-90° , diluindo-a -kOO 1 . e digerindo-a durante 5 horas a 90° C, ob-tem-se suspensões que filtram com relativa facilidade.

1 3
tração de torio alcança mais ou menos I30 g/l de óxido de torx«.>o
Partindo de xmsa. suspensão que contem 28O g/l de oxido
de tório e ms.iitendo a concentração de óxido de tório na solução or
ganica de saída entre 1 2 5 - 1 2 6 g/l, o rendimento de extração foi
de 98 a 99i^ ^ 5 respectivamente em 5 estágios. Para uma concen
tração de saída de 1 2 0 g/l, o rendimento aumenta ate 99 e 9 9 « 5 ^
respectivamente. Usando a mesma concentração dos nitratos, o r ^
dimento extração de tório deveria ser superior a 99»8^ to=
dos os casos pre-citados. A diminuição do rendimento que observa
mos com sistema real, resulta de ação coraplexante dos fluoretos e
de fosfato presentes em pequenas quantidades no hidróxido de tório
bmrto,
A eficiência da descontaminaçao de tório das terras ra
ras foi seguida usando europio como indicador. O europio pode ser
facilmente analisado polarogràfioamente, até em presença de um
grande excesso de tório. Além disso, o fator de descontaminaçao
D_ , dao uma idéia muito realista sobre a eficiencia da separação •liU ^
de tório de todos os elementos deste grupo, já que o fator de se
paraçao, 1 ^ / A e entre os mais desfavoráveis dentro do grupo
das terras raras ( 9 2 ) . O conhecimento da variação experimental de
com a concentração de tório no tributilfosfato ( 9 3 ) * permite
predizer corretamente o comportamento do sistema. Itontendo ao re~
dor de 1 2 5 g/l a concentração de óxido de tório no tributilfosfa
to de saída, temos encontrado na secção de extração, em bom acor
do com os valores previstos, um coeficiente de descontaminaçao de
tório do europio ~ 1 2 0 e uma descontaminaçao concomitante melhor
ainda para as outras terras raras.
A maior parte das experiências de extração foi realiza=
da usando uma bateria de misturadores-decantadores em vicu^ (volu
me útil de 1 por estágio) e num misturador-decantador de aço
inoxidável de volume útil 1 0 1 .
Temos operado de preferencia com emulsoes do tipo agua-

-dentro-do-solvente,
Usando um solvente que contém 0 . 2 - 0 . 5 g/l de carvão
ativo conseguimos evitar completamente a formagao de emulsoes es
táveis. As experiencias preliminares de uma operação contínua fo
ram perfeitamente satisfatórias.
A fim de eliminar as impurezas coloidais e coalescer tra
ços da fase aquosa arrastada, o solvente carregado de tório é fil
trado na saída do sistema de extração, através de um filtro que
contém terra diatomácea.
A solução filtrada é submetida a lavagem ("scrubbing")
com nitrato de amonio 2 . 2 M • ácido nítrico 0 . 8 N. Em vista da
descontaminaçao realizada durante a operação de extração, o sist£
ma de lavagem deve assegurar para as terras raras, um fator de
descontaminaçao de apenas 1 0 ^ . Baseeindo-se nas curvas de equilí
brio ( 9 5 ) podemos concluir que, partindo de uma solução que con
tém 1 2 0 - 1 2 5 g/l de ThD^, este fator de descontaminaçao pode ser
alcançado em 3 estágios usando uma relação entre os volumes das
soluções aquosa e orgânica de 0 . 1 . Esta conclusão foi plenamen
te confirmada experimentalmente usando uma bateria de misturado
res-decantadores descontínuos.
O funcionamento de collonas pulsadas durante a operação
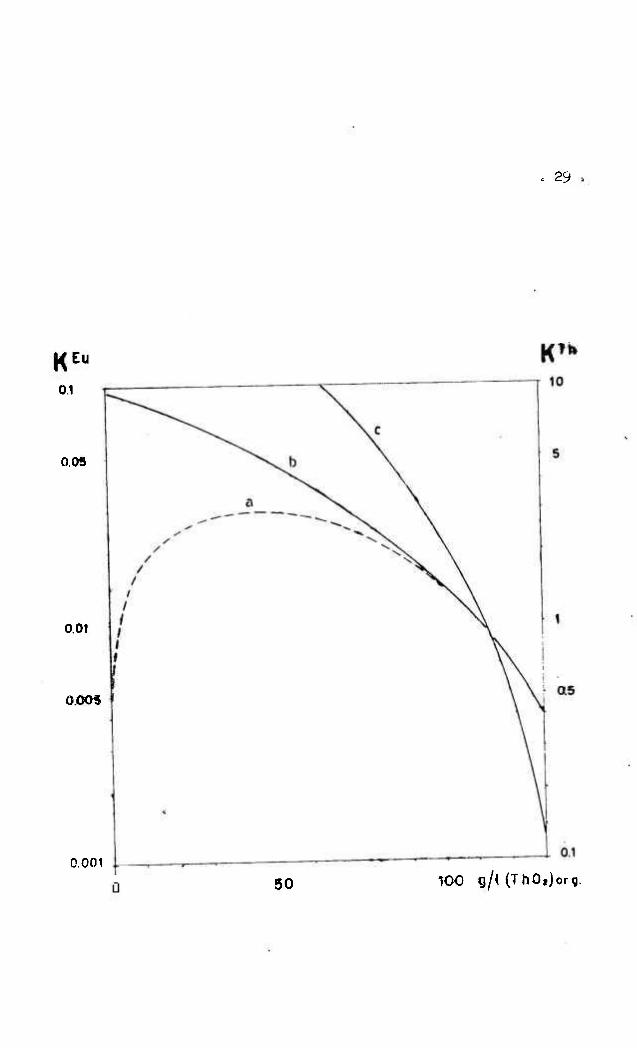
de lavagem é bastante surpreendente. A variação de D?^ com a con bu —
centraçao de tório no tributilfosfato nao obedece às previsões teo
ricas. Quando a concentração de tório ultrapassa 1 1 0 g/l, o coefi
ciente de descontaminaçao diminui, em lugar de melhorar, com a
concentração crescente de ThO^ no solvente. Para uma concentra
çao de 1 1 0 g/l de óxido de tório por litro, observamos uma altura
do estágio teórico da ordem de 5 0 cm. já em 1 2 5 s/l de óxido de
tório, a altura de um estágio aumentou até 2 m. Deste fato resu¿
ta uma diminuição do coeficiente de descontaminaçao efetivo por
um fator de 5 . Um efeito semelhante foi encontrado usando colu
nas pulsadas durante a separação de uranio do tório ( I O 3 ) .

. 1 5 .
No que se refere aos elementos mencionados na tabela 2 ,
apenas arsénico, boro, molibdenio e vanadio possuem coeficientes
de extração apreciáveis (95)° Ito sistema de lavagem de 5 estágios
assegura todavia, até para estes elementos, urna descontaminaçao
satisfatória. Devido à decomposição do tributilfosfato, o compor
tamento de fósforo é difícil de ser previsto.
IV. 5 . Precipitação extrativa de sulfato de tório
O solvente carregado é agora introduzido dentro de um
pequeno excesso de ácido sulfúrico k lü. A precipitação de sulfa
to de tório é rápida. Após filtraçao, as águas maes contem entre
3 - 1 0 g/l de óxido de tório (dependendo da temperatura de preci
pitação). Após eliminação de sulfates, por precipitação de sulfa
to de bário, o ácido nítrico resultante (~3N) é recirculado no
processo de dissolução conforme mostra a figura 1 . Os cristais de
sulfato de tório são lavados e o filtrado usado para preparar áci
do sulfúrico h N para um novo ciclo de precipitação.
Após o primeiro contato com HgSOj h N, o solvente con
tém ainda 2 - 5 g/l de óxido de tório. Ele é contactado com mais
uma porção de ácido sulfúrico 4 K. O tório remanescente é desta
maneira re-extraido e o solvente decantado. Introduz-se em segul
da ania nova porção do solvente carregado o que inicia o ciclo se
guinte da precipitação extrativa.
A precipitação extrativa de sulfato de tório, permite ~ Th h
alcançar coeficientes de descontaminaçao, da ordem de 2 x 1 0
( 1 0 1 ) . No intervalo de concentrações de 10 - 1 0 0 ppm, os fato
res de descontaminaçao para sódio, potássio, cálcio, níquel, co-
balto, cobre, zinco, cadmio, cromo, ferro, molibdenio e titanio
são da ordem de 1 0 0 . O fator de descontaminaçao de boro é ao re
dor de 1 0 ^ . As terras raras e o chumbo são descontaminados por um
fator de 1 0 e o fósforo por um fator de 5 ( 1 0 2 ) . Por pequeno que

. 1 6
3„ja o fator de descontaminaçao de fosforo, é particularmente pre
ciosoo
o tório é recuperado com rendimento de 96 - 97/ • Os re¿
taates 5 - sao reclrculados na dissolução.
As experiencias foram executadas num recipiente de 1 5 1»
de aço inoxidável. O sulfato de tório foi filtrado com urna altu
ra do bolo de 20 - 30 cm, e os cristais lavados com um pouco de
álcool para eliminar quaisquer traços de tributilfosfato aderente»
A precipitação extrativa de sulfato de tório apresenta
alguns problemas cuja solução satisfatória nao foi ainda encontra
da. O problema em questão relaciona-se com a formação de crestas
do sulfato de torio sobre as paredes do recipiente de precipita -
çao. Esperamos poder vencer esta dificuldade reclrculando diiran-
te a precipitação, uma quantidade importante dos cristais a fim
de oferecer uma grande superfície de cristalização.
Experiencias preliminares de precipitação extrativa con
tínua foram realizadas com sucesso em colunas agitadas.
IV. ho Recuperação de uranio e de ácido nítrico
O solvente livre de tório contem ainda mais ou menos 30^
do ácido inicial, assim como a totalidade de uranio. O ácido ní
trico pode ser extraído seletivamente com água e recirculado na
dissolução de hidróxido de tório bruto (ver figura 1 ) . Tomando em
consideração o ácido nítrico recuperado das águas mães de sulfato
de tório, a quantidade de ácido nítrico novo^ necessário para a
dissolução do hidróxido de tório bruto representa apenas ko^ da
quantidade total estequiométrica. Após a re-extração do ácido ní
trico o loranio é extraído por sua vez com carbonato de sódio e re
cuperado em forma de diuranato de sódio. O tratamento com carbona
to de sódio serve ao mesmo tempo para regenerar o solvente antes
de sua recirculação no processo de purificação de tório.

. 17 .
IV. 5 ' Transformação de sulfato de tório em carbonato básico
A transformação de sulfato de tório em carbonato básico
é feita a 70° C, introduzindo dentro de uma suspensão aquosa de
sulfato de torio, uma solução de 5"5 N de carbonato de amonio e
1.5 N de amoníaco. Após uma lavagem exaustiva consegue-se desta
maneira preparar um carbonato básico contendo 50 - 55/ de tório e
100 - 150 ppm de sulfato. A caleinação a 900°, 1100° e 1250° C
permite abaixar o teor em sulfato a 75> 25 e 10 ppm respectivamen
te.
A pureza de uma amostra típica de óxido de tório assim
preparada, é ilustrada no quadro III.
A produção de óxido de tório por intermedio de um carbo
nato básico deveria permitir alcançar um produto facilmente slnte
rizável (77).
V. REFEREHCIAS
(1) H.H. ADLER, USAEC Rept. TID-7650, (I962), 19/28.
(2) J.A. LAHE, ibid. 758/761.
(5) Geology of üranitim and Ihorium, Bibliographical Ser. n?
Int. At. Energy Agency, Vienna, (1962), 892 references.
(4) G.E. KAPLAN, T.A. USPENSKAYA, Yu. I. ZARMBO e I.V. CHIRKOV,
Thorium Mineral Resources, Chemistry and Technology,
Moskow, Atomizdat, (I96O).
(5) K.J. BRIL, Mass Extraction and Separation, in Progress in
the Chemistry and Technology of the Rare Earths (R.C.
Vickery ed) Pergamon Press (196^).

. 1 8 .
(6) J.G. PAFUffiR, Ihoriura, in Mineral Yearbook (Cli.W. MERRIL ed) US Bin?eau of Mines, Washington, (I962).
(7) F.L. CUTHBERT, T5ioriuin Production Technology, Adison-Wesley Publ. Comp. Inc., Reading ( 1 9 5 8 ) .
(8) J.J. BARGHUSEN, Univ. Microf. An Arbor, Michigan, L.C. Card, Mie 5 8 - 1 0 2 7 , ( 1 9 5 8 ) ; and M. SMUTZ, USAEC Rept. ISC-947
( 1 9 5 7 ) ; idem Ind. Eng. Chem. 5 0 , ( 1 9 5 8 ) , 175V5.
(9) K.G. SHAW, M. SMUTZ e G.L. BRIDGER, USAEC Rept. 1*07 ( 1 9 5 ^ ) ; e M.E. WHATLEY, Chem. Qig, Progr. Syrap, Ser. I5 ^0 ( 1 9 5 ^ ) , 1 6 7 / 7 0 .
(10) Ch. de RODDEN e M. PELTIER, US Pat. 2 . 785 . I25 , Feb. 2 6 ,
( 1 9 5 7 )
( 1 1 ) P. KRUMHOLZ, Symposium on Rare Metals, Indian Inst, of Metals, Dec. ( 1 9 5 7 ) , 7 8 ,
( 1 2 ) V. DEMAHT, F. GOTTDENKER e P, KRUMHOLZ, Brazil. Pat. pending.
( 1 5 ) ORQUIMA S.A., Brazil. Pat, pending,
{Ih) F. GOTTDENKER e P. KRUMHOLZ, US Pat. 5 , 1 1 1 , 5 7 5 , Nov. I9 , ( 1 9 6 3 ) , French Pat. 1 , 2 1 0 . 1 0 5 , Sept. 28 ( 1 9 5 8 ) , etc.
( 1 5 ) P. KRUMHOLZ, US Pat, 5 . 1 1 2 , 9 9 0 , Dec. 3, ( I 9 6 5 ) , French Pat. 1 . 2 3 2 . 9 3 8 , Sept, 1 , ( 1 9 5 8 ) , etc.
(16 ) P. KRUMHOLZ e F. GOITDENKER, Proc, UN Conf. PUAE, 8 ,
( 1 9 5 6 ) , 1 2 6 .
( 1 7 ) A.D. ARNOLD e R.P. WISHOW, USAEC Rept. ORNL-2056, ( 1 9 5 6 ) .
(18) G.L. BRIDGER et al., US Pat. 2 . 8 1 5 . 2 6 2 , Dec. 5 , ( 1 9 5 7 ) .

, 1 9 .
( 1 9 ) MoAc WEI.T e Mo SMUTZ, US Pat. 2o8i+9.286, Aug. 2 6 , ( 1 9 5 8 ) -
(20) L. GRAUGER, Uranium and Tb,orium, G. Newnes ed., London ( 1 9 5 8 ) .
( 2 1 ) E.G. PITZER, US Pat. 2 . 7 1 5 « 5 5 4 ( 1 9 5 5 ) .
(22) ANOTOIOUS, Chera, Eng. 66 , no 1 5 , ( 1 9 5 9 ) , 62
(25) Sh. laSHIMURA, Mem, Col. Sci, Uaiv, Kyoto, Ser. B. 2^^
( 1 9 5 9 ) , 2631 idem ibid. 2 6 , ( 1 9 5 9 ) , 1 7 3 / 9 1 -
i2h) A, AUDSI£Y et al,, in Extraction and Refining of the Rare Metals (Inst- of Mining and Metall,, ed) London ( 1 9 5 7 ) , 3 5 1 / 3 8 0 ,
(25) A. AUDSLEY et al,, Proc. ;:ad UN Int. Conf. PUAE, ^ ( 1 9 5 8 ) , 2 1 6 ,
(26) a. CARTETi et al,, J. Appl. Chem. (London), 1 0 , (1960)^
1U9; idem US Pat, 5 , 0 8 7 . 9 ^ , April 5 0 , (I963)»
(27) C.V, KLim e W.R. EEMETT, US Pat. 3 .01+7 .359 , July 5 1 , ( 1 9 6 2 ) ,
(28) 0 ,M, HILAL et al,, Proc. 2nd UN Int. Conf. PUAE, , ( 1 9 5 8 ) , 5 7 5 .
(29) E.S. PILKtNGTON e A.W. WYIZE, J. Soc. Chem, Ind. London, 66, ( 1 9 ^ 7 ) , 3 8 7 j J» Appl. Chem, London 2 , ( I 9 6 2 ) , 265^
ibid, hj ( 1 9 5 ^ ) , 5 6 8 ,
(30) A, AUDSLEY e RoW, BLUNDELL, Brit. Pat. 8 0 1 . 5 7 3 , Sept, 1 7 ,
( 1 9 5 8 ) ,
( 3 1 ) R.W. BLUNDELL, Brit, Pat. 7 8 3 . 6 2 8 , Sept. 2 5 , ( 1 9 5 7 ) .

20 .
(32) R.V. NAIR e S.S. MOOSSATH, Buli. Centr. Res. Inst. Univ. Travancore, Ser. A , k, ( 1 9 6 5 ) , 6 3 / 8 6 9 / 7 3 .
( 3 3 ) R.K. DUTTA, J. Sci. Ind. Research 1 ^ ( 1 9 5 5 ) , ^95, 88.
(54) K.B. BROWN et al., Proc. 2nd UN Int. Conf. PUAE, , ( 1 9 5 8 ) , 1+72.
(55) D.J. CROUSE e K.B. BROWN, USAEC Rept. ORNL-2720, ( 1 9 5 9 ) -
(36) S.R. BORROWMAN e J.B. ROSEMBAUM, US Bureau of Mines Rept. BM-RI-5917, ( 1 9 6 1 ) .
(57) K. KAWAMÜRA e T. TAKEUCHI, Nippon Genshiryouku Gakkalshi, k, ( 1 9 6 2 ) , Tlh.
(58) W. RYAN, UKAEA Rept. DSIR C R L / A E 1 5 5 -
(39) I-C. KRAITZER, Can. Pat. 6 5 5 . 1 7 9 , Nov. 2 7 , (I962).
(ho) R . A . WELLS et al., Brit. Pat. 900.U52, July h, ( 1 9 6 2 ) ;
idem ibid. 899-284, June 20 , ( I 9 6 2 ) .
(hi) A . J . HEAD et al., UKAEA Rept. DSIR C R L / A E I 6 6 ( 1 9 5 8 ) ;
ibid. J. Appl. Chem. 2 , ( 1 9 5 9 ) , 599-
(h2) G.D. CALKINS, US Pat. 2 . 8 5 8 . 5 7 0 , June 1 0 , ( 1 9 5 8 ) .
(45) R . A . NAGLE e T.K. MURPHY, The Analyst 84, ( 1 9 5 9 ) , 5 7 -
(44) R.W. BANE, US Pat. 2 . 9 0 2 . 5 3 8 , Sept. 9, ( 1 9 5 9 ) .
(45) Sh.Tu e C . L . CHOW. Chemistry (Formosa), ( 1 9 5 8 ) , 1 7 7 , 1 8 1 .
(46) J . H . GROSS, US Pat. 2 - 9 1 5 . 5 6 3 , Dec. 1 , ( 1 9 5 9 ) .
(47) H.N. SETHNA e S. FAREEDUDDIN in Symposium on Rare Metals,

. 21 .
Indian Inst, of Metals, Dec. (1957), 68.
(U8) A,E, BEARSE et al., CSiem. Eng. Progress (195^),
255/9.
(U9) G.D. CALKINS e E . G . B0HLÍ4ANN, US Pat. 2.815.264, Dec. J, (1957).
(50) ANONYMOUS, Chem. Eng. 65, (1959), 62, 64, 104.
(51) G.D. CALKINS, US Pat. 2.8ll.4ll, Oct. 29, (1957).
(52) G o E . KAPLAN e T.E. USPENSKAYA, Proc. 2nd UN Int. Conf. PUAE, 2, (1958), 578.
(55) G.A. MEERSON et al., Soviet J. Atomic Eiiergy, , (1957), 1054.
(54) A.E. EBERLE, USAEC Rept. NyD-204l, (1955).
(55) J.R. RUHOFF et al., US Pat. 5.029.131, April 10, (I962).
(56) J.M. FLETCHER e G.J. ASHWORTH, Brit. Pat. 783.195, Sept. 18, (1957).
(57) R.H. POIKEER et al., Ind. Ehg. Chem., 0, (1958), 6I3.
(58) K.N. KURUP e S.S. MOOSATH, Bull. Res. Inst. Uhiv. Kerela Trivandrum Ser. A ^ n9 1, (1957), 15/21j ibid. 6, n9 1 (1959), 1.
(59) K.N. KARTHA, Bull. Centr. Res. Inst. Uhiv. Travancore, Trivandrum Ser. A 4, (1955), 55«
(60) T. ISHINO e H. TAMURA, Technol. Repts. Osaka IMiv. 8,
(1958), 427.

22 .
(61) B. SARMA e J. GUPTA, J. Sci. Ind. Research (India), ikB, (1955) , 82; Y.W. GOKHALLE et al., ibid. 1 ^ (I960), 422 /5 .
(62) O.M. HILAL e F.A. EL GOHARY, Ind. Chem. Eng., 2 , ( 196 l ) ,
997.
(65) M. BRODSKY et al., in Progress in Nuclear Chemistry, Series III, (1958), ed. Pergamon Press, vol. 2 , 68.
(64) c. BRAUN et al., Proc. 2nd UN Int. Conf. PUAE, 4, (1958) , 202.
(65) Ch. LORRAIN, private communication.
(66) W.D. JAMRACK, Rare Earth Metal Extraction, Pergamon Press, (1965) .
(67) ANONYMOUS, Chem. Eng., April 50, (I962), 65.
(68) W. RYAN, UKÂEA Rept. SDIR CRL-AE l62 .
(69) T.V. ARDEN et al., J. Appl. Chem., (1959), ^ 6 .
(70) K.B. BROWN, USAEC Rept. CF-60-7-108, (I96O).
(71) K.B. BROWN et al., USAEC Rept. IID-765O, (I962), I9 /28 .
(72) H. BROWN e L.T. SILVER, Proc. UN Int. Conf. PUAE, 8,
(1956), 129 .
(75) K.B. BROWN et al.. In USAEC Repts.s a) ORNL-331^ (1962), b) ORNL-TM-449 (1962), c) ORHL-IM-107 (I96I) , d) CF-6O-1 1 - 1 2 6 (i960), e) ORHL-3155 (1961) , f) CF-61-3-141 (I96I) .
(74) F.J. FRANCIS, US Bureau of Mines Inf. Circ. nff 8124, (1962) .

2 5
( 7 5 ) S.R. BORRöWMÄI e J.B. ROSENMÜM, US Bureau of Mines
Rept. BÄ~Rl~59l6 ( i 9 6 0 ) .
( 7 6 ) M o l e V ñ M í E S í K O O I , US Pat. 5.067.004, A p p l . Oct. 5 , ( 1 9 5 9 ) .
r r r ) x. íIAMDk st al», J. Am, Ceram. Soc, 4^, ( 1 9 6 2 ) , 2 5 3/7.
( 7 8 ) J, KCRKÍSCH e G.E, JAHAUER, Talanta, ( I 9 6 2 ) , 9 5 7 / 8 5 .
( 7 9 ) A o L . JÖTTS et al,, USÄEG Rept, TID-765O, ( 1 9 6 2 ) , 35.
(80) I , l A G I , Kogyo I gaku /.asshl, (1962) , 27/9.
( 8 1 ) D.I. PEPPARD et al., üSAEC Rept, TTD- 1 5 3 1 5 , ( 1 9 6 2 ) .
ifiZ) DoW, MimmM e R.W. GAa?a!RALL, J, Inog. & Nuclear Chem.,
2 1 , (196I) , 55V8"
( 8 3 ) ? o G , M A M I N ö , Can, J, Chem,, hö, (I962), 1 6 8 4 / 9 .
(84) wmm TAim et a l o , Nippon Kagaku Zasshi, 8 I , ( 1 9 6 O ) ,
1 5 5 ^ i - / 8 o
( 8 5 ) H, SÄirCHO, Bull. Chem. Soc, Japan, ( I 9 6 I ) , 1 2 5 4/7-
(86) W.J, MC irmKÄ, e K.A, ALLEN, J. Phys. Chem., 6 ^ , ( 1 9 6 l ) ,
i3;5S/6"
{ 8 7 ) VU'bc BROWN e C P . COLEMAN, in Progress in Nuclear
E S õ r c / ç Series H I , (P.R. Bruce, J.M. Fletcher e HH.
i^pmis. e à b ) , Pergamon Press, (1^8).
( 8 8 ) M.E, WHITLEY et al,, USAEC Rept, ORNL-3N-410, ( I 9 6 3 ) .
( 8 9 ) n,G, PKHRCW et al., USAEC Rept. TID-6839, (I96O).
;iO) R = E. BIA*:;Ü et al,^ USAEC Rept, T I D - 7 6 5 O , ( 1 9 6 2 ) , 384;
iblà. a R H I / - 3 2 1 9 , ( 1 9 6 2 ) ; R.H. RAINEY e J.G. MOOIiE,

2h
us P a t o 5 o 0 ^ 9 o 4 0 0 , Augo l 4 , ( 1 9 6 2 ) ,
( 9 1 ) S o PAREEDÜDDIH e t a l o , Proc, 2nd Ü K I n t , Confo PUAE, 4,
( 1 9 5 8 ) , 208 .
(92) K o J a BRIL e P o KRUMHOLZ, in "inter -Am, Sympo Peaceful
Appl. N u c l c Energy^ 3rd^ Rio d e Janeiro, i 9 6 0 " , p» 3 7 ,
Pan American IMion, Washington, D , C o ( 1 9 6 1 ) 0
(93) Po KRUMHOLZ e K c J c DRIL, to be published.
(94) Co CHRISTENSEN e J o D . PLATER, Brit» Pat. 9 0 7 , 1 0 7 , O c t ,
3 , ( 1 9 6 2 ) ,
(95) Lo NEWr-yiPI e P o KLOTZ, USAEC Rept, B N L ^ 6 l 4 l , (I962);
ibid. Bi1L»6l88, ( 1 9 6 5 ) 0
(96) H, IRVING e D.No EDGINGTON, J , I n o r g o & Nuolear Chem.,
2 0 , ( 1 9 6 1 ) , 514/5O1 idem ibid, 3 2 l / 3 4 | idem ibid. 2 1 ,
1 6 9 / 8 0 o
(97) T ,Vo HEALY, i b i d o I9, (I96I), 3 1 4 / 2 7 ,
(98) Solvent E x t n o Symposium Gatlinburg, Tenn., O c t , 2 3 - 2 6 ,
( 1 9 6 2 ) 0
(99) S o SIEKIERSKL, J , Inorg, & Nuclear Chera,, 2 4 , ( I 9 6 2 ) ,
2 0 5 / 1
(100) J.B. MADJAR, K . J o BRIL e P, KRUMHOLZ, Brit, Pat, 8 8 7 . 3 9 5 ,
May 9 , ( 1 9 6 2 ) , Can, Pat, 6 3 8 , 7 5 9 , r^rch 2 7 , ( 1 9 6 2 ) , etc.
( 1 0 1 ) K.J, BRIL e P, KRWÍI-I0I2;, US Pat, 5 c l 0 4 , 9 4 0 , Sept, 24,
( 1 9 6 5 ) , Brito Fat. 8 8 0 . 0 4 6 , Feb, 7 , ( 1 9 6 2 ) , etc,
(102) K,J. BRIL e P . G o DE SABOIA ARAÚJO, Ind, Eng, Chem.
(Process Design and Development), ¿, ( 1 9 6 4 ) , 8/IO,

. 25 .
VI. LISTA DE FIGURAS
Fig. 1 - Esquema de produção de óxido de tório nuclearmente
puro.
Fig. 2 - Variação dos coeficientes de distribuição de tório
( K ^ ) e de europio ( K ^ ) com a concentração de tó
rio no tributilfosfato 46 v/v^ e Varsol 54 v/v^.
( 1 0 5 ) J. BEHTíOIRAS, K.J. BRIL e P. KRUMHOLZ, ibid. 1 , ( I 9 6 2 ) ,
64/68.
(104) R.J. Mc NAMEE e R.P. WISHOW, USAEC Rept. ORNL-I8T5.
(105 ) W.A. MEELEY et al., USAEC Rept. BMI-946, ( 1 9 5 4 ) .
(106) A.T. GRESKY et al., USAEC Rept. ORIJL-5574, ( 1 9 6 5 ) ;
ibid. ORUL-m-464, ( 1 9 6 5 ) .
(107) P.B. CARBON, US Pat. 2 . 7 7 6 . 8 7 7 , Jan. 8 , ( 1 9 5 7 ) ; K. TADA
e H. KATO, Japan Pat. 8 l 5 7 ( ' 5 4 ) , Dec. 1 1 , ( 1 9 5 4 ) .
(108) T.F. GANNOLLY, USAEC Rept. ORNL-2971, (I96O),
T.H. SIDDALL, ibid. HD-1 8 2 9 9 , (I965).

. 26 .
Fig. 1 - Esquema de produ fio de oxido de torio nuolearmentQ puroo
túfl'J bruto
*vtipor«;ao «/oicaiBo neldo
t>;rlo o uranio
27
HNO-. O.t) N
HySO, U.l S
precli 1tagSo
fll t
nriü eos pul
ño de
ruextrai.-áo de urúnlD eo»
-le naáO.
lí-üristix-iiiiiíao eiTi carlumutp Lúsico
¿sido i» tirio puro

28 .
Fig. 2 - Variação dos coeficientes de distribuição
de tório (k''*) e de europio (kÍ^) com a
ooncentração de tório em tributilfosfato
U6 v/v^ e Varsol 54 v/v^.
Ctirva a:
Curva b:
Curva 01
(fase aquosa •> solução ajustada para
extração).
(fase aquosa: HaNO^ 2.2 M • HNO^ 0.8 N).
(fase aquosa: NHi NO 2.2 M • HNO^ a8 N).
. 29
0.1
0.05
0.01
0.00S
0.001 50 100 g/( (ThOtjorg.


















