

Síndromes: Causas, Conseqüências e Prescrição de Exercícios · • Linguagem: disfagia:...
108
Síndromes: Causas, Conseqüências e Prescrição de Exercícios Fabiana Guedes Universidade de São Paulo Universidade Gama Filho
Transcript of Síndromes: Causas, Conseqüências e Prescrição de Exercícios · • Linguagem: disfagia:...

Síndromes: Causas, Conseqüências e Prescrição de Exercícios
Fabiana Guedes
Universidade de São Paulo
Universidade Gama Filho

SÍNDROMES
• Síndrome de Down • Síndrome de Edwards • Síndrome do X Frágil • Síndrome Rett • Síndrome Klinefelter • Síndrome Turner • Síndrome Williams • Síndrome de Asperger
(Autismo) • Síndrome Alcóolica
Fetal

Síndrome de Down
• Fisionomia e personalidade: Face achatada, língua protusa e grande, fissura palpebral oblíqua. Geralmente sociáveis e carinhosos
• Coração: defeito do septo ventricular e comunicação interatrial
• Gastrointestinal: estenose duodenal e estreitamento anal
• Músculo esquelético: hiperflexibilidade, instabilidade cervical e ATM, displasia coxofemoral
• Visuais e Auditivos: miopia, catarata, rebaixamento auditivo
• Linguagem: disfagia: defict de deglutição • RN: baixo peso, hiporreflexia, hérnia
umbilical, hipotonia, atraso no DNPM • Neurológicos: retardo mental, microcefalia,
5% convulsão • Trissomia do cromossomo 21

Síndrome de Down
• Pode ser definida como uma deficiência múltipla, caracterizada por alterações físicas, orgânicas e intelectuais, provocadas por um distúrbio no cromossomo 21.

Síndrome de Down
• A SD é uma condição genética reconhecida há mais de um século por John Langdon Down, sendo uma das causas mais freqüentes de deficiência mental.
• 85% Trissomia 21 ( relação com o comando de enzimas que podem estar ligadas a alterações neurológicas).
• Não disjunção meiótica
• 1 a 2 – 1000 nascimentos
(SANVITO, 2008)

Síndrome de Down
Causas: Idade materna > 35 anos( óvulos
mais velhos correm maior risco de sofrer alterações genéticas)
Hereditariedade Casamento consangüíneo
Causas oriundas de exposição a
agentes químicos
(SANVITO, 2008)

Síndrome de Down
Características
Língua protrusa, dentes pequenos, pele seca;
Nuca reta, pescoço curto;
Mãos grossas e curtas; prega única na palma das mãos;
Cabelo falho e fino e nariz achatado;
Prega epicantal (no canto dos olhos);
Genitais pouco desenvolvidos;
Baixa estatura.

Síndrome de Down
• Visuais e Auditivos: miopia, catarata, rebaixamento auditivo
• Linguagem: disfagia: defict de deglutição
• RN: baixo peso, hiporreflexia, hérnia umbilical, hipotonia, atraso no DNPM
• Neurológicos: retardo mental, microcefalia, 5% convulsão

Síndrome de Edwards
• Fisionomia : Face triangular e fenda labial
• Coração: Défict Interventricular • Músculo esquelético:
Mielomeningocele (não fechamento do tubo neural), face triangular, fenda labial, escolioses, ossificação incompleta da clavícula
• RN: Baixo peso, prematuridade, hiperreflexia, hérnia umbilical, hipertonia espástica, atraso no Desenvolvimento Neuro Psico Motor
• Neurológicos: Retardo mental, microcefalia ou macrocefalia, atrofia cerebral- alterações na morfologia cerebelar (déficit de equilíbrio e ajuste postural)
• DNA: Trissomia do cromossomo 18

Síndrome de Edwards

Síndrome de Edwards
• 1960: Jonh H. Edwards
• Trissomia do 18 • Idade materna e gestação > 42 • 1:8000 nascimentos • 4 meninas:1menino • Não disjunção celular meiótica (Sanvito, 2008)

Síndrome de Edwards
Manifestações Clínicas:
• Baixo peso - 2000g
• 1/3 pré maturo
• Déficit mental
• Atrofia cerebral
• Alterações na morfologia cerebelar
• Microcefalia ou Macro
• Mielomeningocele
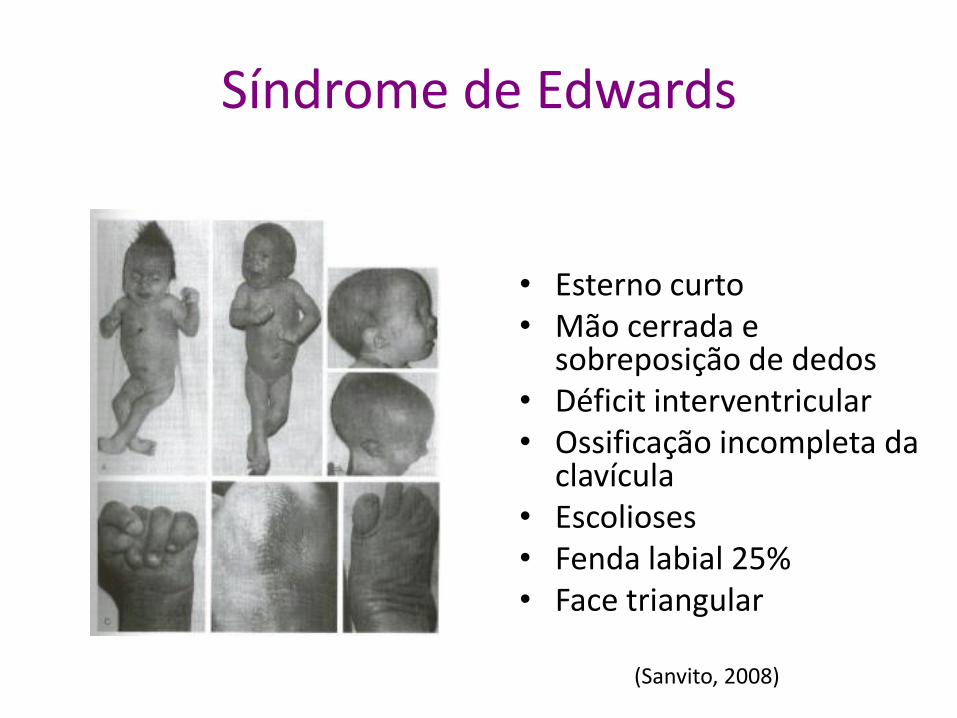
Síndrome de Edwards
• Esterno curto • Mão cerrada e
sobreposição de dedos • Déficit interventricular • Ossificação incompleta da
clavícula • Escolioses • Fenda labial 25% • Face triangular
(Sanvito, 2008)

Síndrome de Edwards
• Espasticidade (distúrbio motor caracterizado pelo aumento
do tônus muscular, dependente da velocidade, associado à exacerbação do reflexo miotático.
Baixa expectativa de vida: 5 - 10% vivem até 1 ano Sobrevida relacionada a gravidade das má formações
Foram relatados pacientes com 13,15 e 18 anos (Schinzel, 1984)

Síndrome do X Frágil
Fisionomia e personalidade: Aparentemente normal, orelhas grandes, face alongada, ansiedade e hiperatividade
Músculo esquelético: Déficit coordenação e de movimentos finos, hipermobilidade articular, escolioses, pectus escavatum
Visuais e Auditivos: Estrabismo e miopia
Linguagem:Dificuldade de comunicação, fala rápida e confusa
RN: Baixo peso, hiporreflexia, hipotonia, atraso no Desenvolvimento Neuro Psico Motor
Neurológicos: Retardo mental, macrocefalia, convulções
DNA:Alteração no GENE FRM 1 - Fragile mental retardation do cromossomo X

Síndrome do X Frágil

Síndrome do X Frágil
• 1:4.000 H – 1: 6.000 M
• Segunda causa mais freqüente de comprometimento mental
• 80-90% das famílias portadoras ainda não foram diagnosticadas
• Mudança no gene do cromossomo X
• Fragilidade do braço longo FMR-1

Síndrome do X Frágil
• 1969: Herbert Lubs – Descoberta da fragilidade
• 1970: Grant Stherland – Denominou X frágil
• 1991: FMR1 foi descoberta com
base em estudos moleculares do
DNA

Síndrome do X Frágil
FMR-1
• Fragile mental retardation
• Apresenta cópias dos trinucleoídeos CGG/DNA
• Número normal: 6-50
• X frágil: 200 +
• Síntese da proteína FMRP
(Intelecto- capacidade mental)

Síndrome do X Frágil
Manifestações Clínicas:
• Fisionomia aparentemente normal
• Reconhecimento associado a deficiência mental
• RN- macrocefalia e hipotonia
• Orelhas grandes e face alongada

Síndrome do X Frágil
• Atraso no DNPM
• Déficit Coordenação
• Movimentos finos
• Convulções
• Hipermobilidade articular
• Escolioses
• Pectus escavatum
(Sanvito, 2008)

Síndrome do X Frágil
Hiperatividade
Ansiedade
Humor instável
Estrabismo e miopia
Dificuldade de comunicação
Fala rápida e confusa
Alguns têm prejuízos muito pequenos, com desempenho praticamente normal.
Outros têm comprometimentos moderados, mas com atendimentos especializados chegam a bons resultados sociais e funcionais

Síndrome de Rett
Fisionomia e personalidade: Aparentemente normal por um período, afastamento do convívio social
Gastrointestinal: órgãos aumentados Músculo esquelético: Déficit na deambulação
e movimentos finos das mãos, retardo no crescimento, pés e mãos pequenos e finos, hipertonia
Visuais e Auditivos: Retinopatia, atrofia óptica e catarata
Linguagem: Perda de palavras aprendidas, prejuízos na compreensão, raciocínio e comunicação.
RN: Desenvolvimento pré e perinatal aparentemente normal- DNPM sem alterações até os 6 anos de idade
Neurológicos: Perda progressiva de funções neurológicas, movimentos involuntários e repetitivos de mãos, atrofia do perímetro cefálico após os 6 anos
DNA: Mutação do gene MECP2 (importante no funcionamento de outros genes) do cromossomo X

Síndrome de Rett

Síndrome de Rett
• 1954: Andreas Rett
• 1 em cada 10.000 a 15.000 meninas nascidas vivas
Clinicamente:
• Perda progressiva das funções neurológicas e motoras após um período de
desenvolvimento aparentemente normal, que vai de 6 a 18 meses de idade.
• Após esta idade, as habilidades adquiridas (como fala, capacidade de andar e uso intencional das mãos) são perdidas gradativamente
• Surgem as estereotipias manuais (movimentos repetitivos e involuntários das mãos), que é característica marcante da doença.

Síndrome de Rett
• 1999: Zoghbi - mutações (alterações) em um gene localizado no cromossomo X.
• Gene MECP2 (do inglês methyl-CpG-binding protein 2) "mec-pê-dois"
• Produz uma proteína (MeCP2) que controla a expressão de vários genes importantes para o desenvolvimento dos neurônios
• Na Rett, a MeCP2 não é capaz de funcionar corretamente e todo o desenvolvimento dos neurônios do embrião fica comprometido
É importante lembrar que mutações no DNA ocorrem em todas as nossa células, ao longo de nossas vidas, todos os dias, sem que possamos saber
ou controlar.

Síndrome de Rett
Diagnóstico:
Critérios necessários (presentes em todas as pacientes):
◦ Desenvolvimento pré-natal (antes do nascimento) e perinatal (pouco
tempo depois de nascer) aparentemente normal
◦ DNPM normal até os 6 meses de idade
◦ Perímetro cefálico normal ao nascimento
◦ Desaceleração do perímetro cefálico após 6 meses de idade
◦ Perda do uso propositado das mãos
◦ Movimentos manuais estereotipados (torcer, aperta, agitar, esfregar, bater palmas, "lavar as mãos" ou levá-las à boca)
◦ Afastamento do convívio social, perda de palavras aprendidas
◦ Prejuízos na compreensão, raciocínio e comunicação.

Síndrome de Rett
• Critérios de suporte (presentes em algumas pacientes): – Distúrbios respiratórios: hiperventilação, apneia, dificuldade
expulsão forçada de ar, broquiectasia e saliva excessiva. – Bruxismo – Distúrbios do sono – Tônus muscular anormal: espasticidade – Distúrbios vasomotores periféricos (pés e mãos frios ou cianóticos) – Cifose/escoliose progressiva – Retardo no crescimento – Pés e mãos pequenos e finos

Síndrome de Rett
• Classificação:
• Clássica
• Atípica

Síndrome de Rett
Clássica: evolui em quatro estágios definidos:
Estágio 1 - de 6 a 18 meses de idade: ◦ Desaceleração do perímetro cefálico ◦ Alteração do tônus muscular (Hipotonia Hipertonia) ◦ A criança interage pouco, perde o interesse por brinquedos.
Neste estágio, os primeiros sintomas da doença estão surgindo, mas
muitas vezes nem são percebidos pelos pais (especialmente se são "marinheiros de primeira viagem") ou pelos médicos (muitos deles desconhecem a síndrome de Rett).

Síndrome de Rett
• Estágio 2 - de 2 a 4 anos de idade:
• Ocorre regressão do desenvolvimento
• Inicia-se a perda da fala e uso intencional das mãos, que é substituído pelas esteretipias manuais
• Distúrbios respiratórios, distúrbios do sono (acordam à noite com ataques de risos ou gritos)
• Manifestações de comportamento autístico

Síndrome de Rett
• Estágio 3 - de 4 a 10 anos de idade:
• A regressão é severa dos problemas motores
• Crises convulsivas
• Escoliose marcantes
• Melhora da interação social e comunicação
• Tornam-se mais tranquilas e as características autísticas diminuem

Síndrome de Rett
Estágio 4 - a partir dos 10 anos de idade:
Perdem completamente a capacidade de andar, embora algumas nunca tenham adquirido esta habilidade
Escoliose, rigidez muscular e distúrbios vasomotores periféricos mantidos
Os movimentos manuais involuntários diminuem em frequência e intensidade
DI em todos os estágios desde o nascimento !
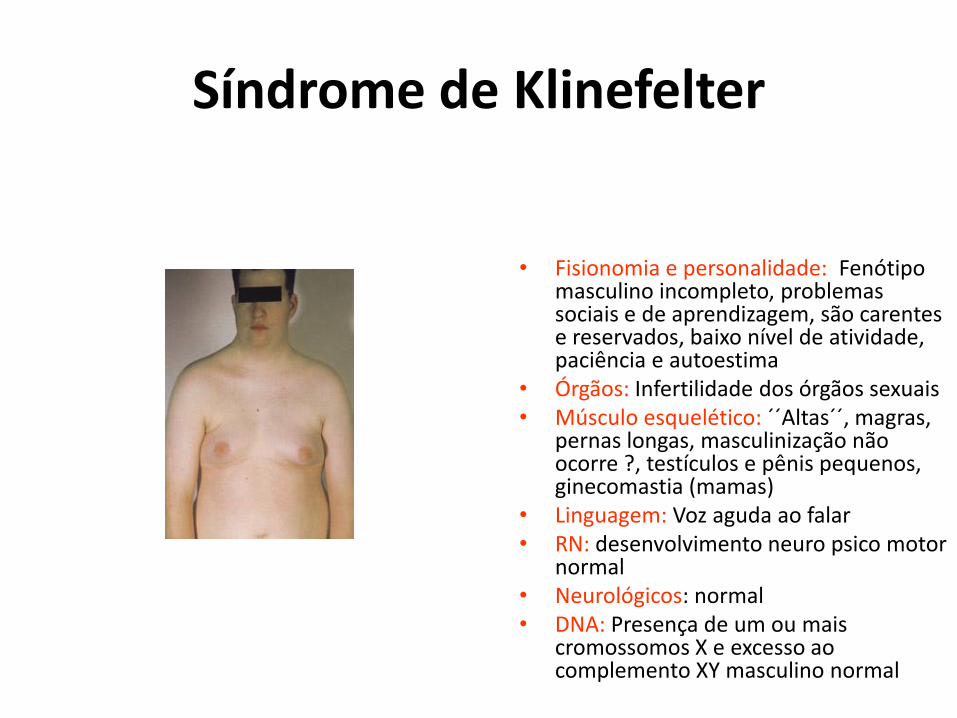
Síndrome de Klinefelter
• Fisionomia e personalidade: Fenótipo masculino incompleto, problemas sociais e de aprendizagem, são carentes e reservados, baixo nível de atividade, paciência e autoestima
• Órgãos: Infertilidade dos órgãos sexuais • Músculo esquelético: ´´Altas´´, magras,
pernas longas, masculinização não ocorre ?, testículos e pênis pequenos, ginecomastia (mamas)
• Linguagem: Voz aguda ao falar • RN: desenvolvimento neuro psico motor
normal • Neurológicos: normal • DNA: Presença de um ou mais
cromossomos X e excesso ao complemento XY masculino normal

Síndrome de Klinefelter
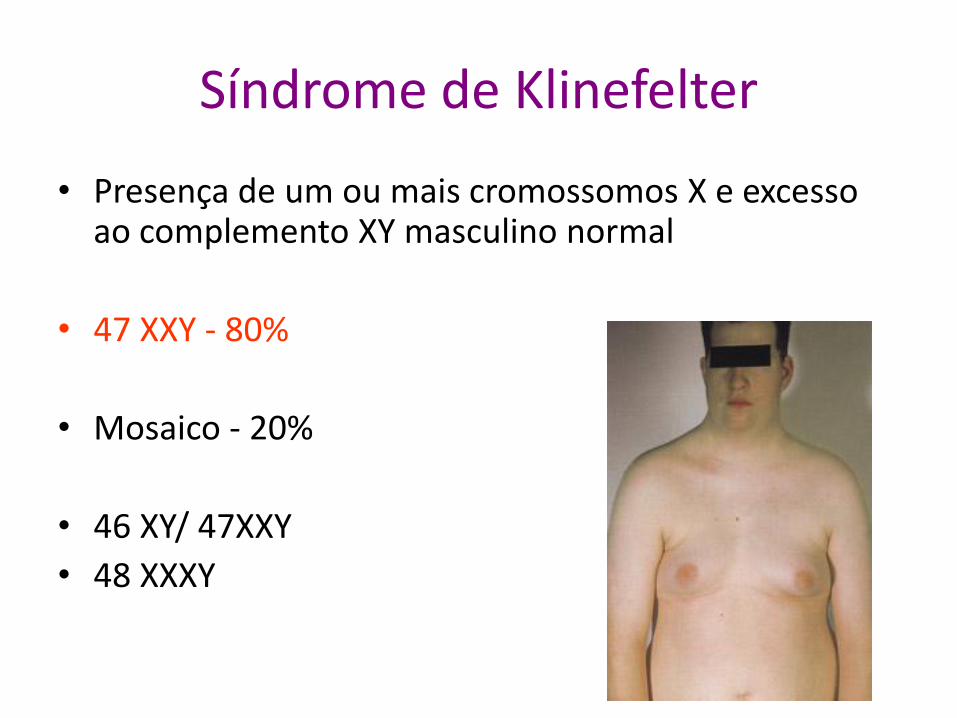
Síndrome de Klinefelter
• Presença de um ou mais cromossomos X e excesso ao complemento XY masculino normal
• 47 XXY - 80%
• Mosaico - 20%
• 46 XY/ 47XXY
• 48 XXXY


Síndrome de Klinefelter
• 1: 1000
• Não disjunção meiótica
• Garante um fenótipo masculino
• Metade são perdidos em abortos espontâneos

Síndrome de Klinefelter
Características da síndrome de klinefelter: - Esterilidade - Problemas sociais e/ou de aprendizagem - Alterações posturais Outras características são relatadas por seus familiares: - Preferência por jogos calmos - São freqüentemente carentes e reservados - Tremores na mãos - Frustração causa explosão de temperamento - Dificuldade de concentração - Baixo grau de paciência - Dificuldade de despertar pela manhã - Baixa auto-estima

Síndrome de Klinefelter
Manifestações Clínicas: ´´Altas´´, magras Pernas longas Masculinização não ocorre ? Testículos e pênis pequenos Voz aguda Osteoporose? Ginecomastia Infertilidade: diminuição execessiva da testoterona

Osteoporose Juvenil
Na infância, a formação excede a reabsorção, e a remodelação óssea é intensa
Picos de crescimento:
11 e 14 anos nas meninas
13 e 17 anos nos meninos
Fatores intrínsecos : raça, sexo e fatores hormonais hormônio de crescimento, estrógeno e testosterona (HIPOGONADISMO)
Fatores extrínsecos: aspectos nutricionais, hábitos, presença de doenças crônicas e uso de medicamentos
(Pereira, 2003)
Fratura

Alterações Posturais na Infância e Adolescência
Hipercifose Hiperlordose Escoliose
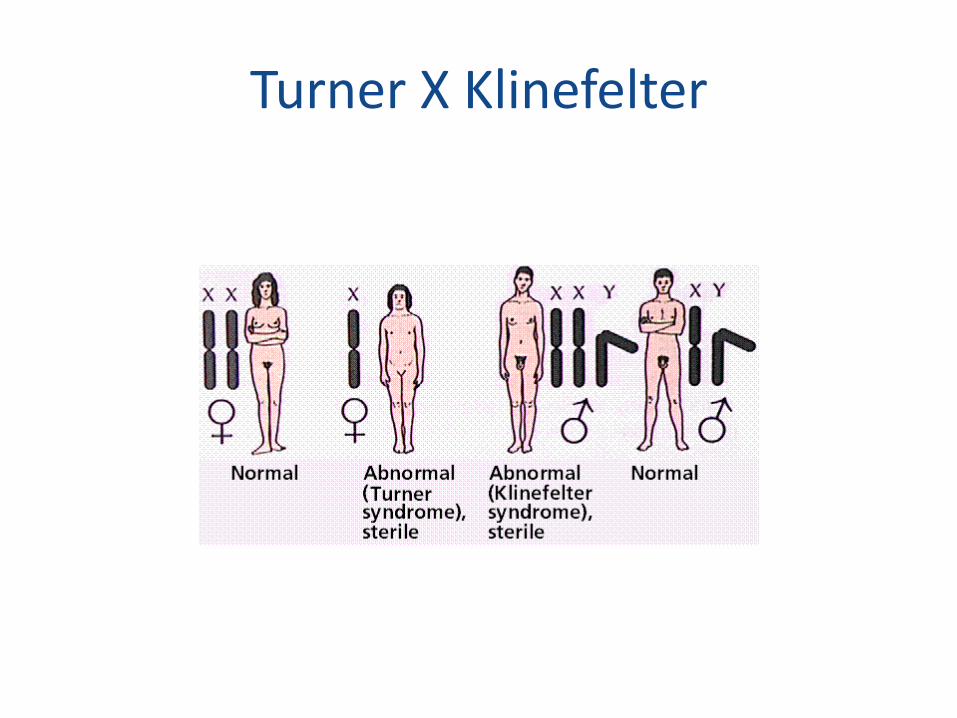
Turner X Klinefelter

Síndrome de Turner
Fisionomia e personalidade: Face triangular, problemas sociais e
de aprendizagem, são carentes e reservados, baixo nível de atividade
Órgãos: Envelhecimento do ovário e não desenvolvimento do mesmo , déficit de hormônio do crescimento e estrógeno
Músculo esquelético: Linfedema de membros, displasia coxofemoral, Baixa estatura 150,cm, pescoço curto, pouca mama, tórax largo, hipercifose e escoliose
Visuais e Auditivos: Déficit de visão- Estrabismo e audição
RN: Desenvolvimento neuro psico motor comprometido
Neurológicos: 1/5 DM DNA: Monossomia do cromossomo
X (um a menos)
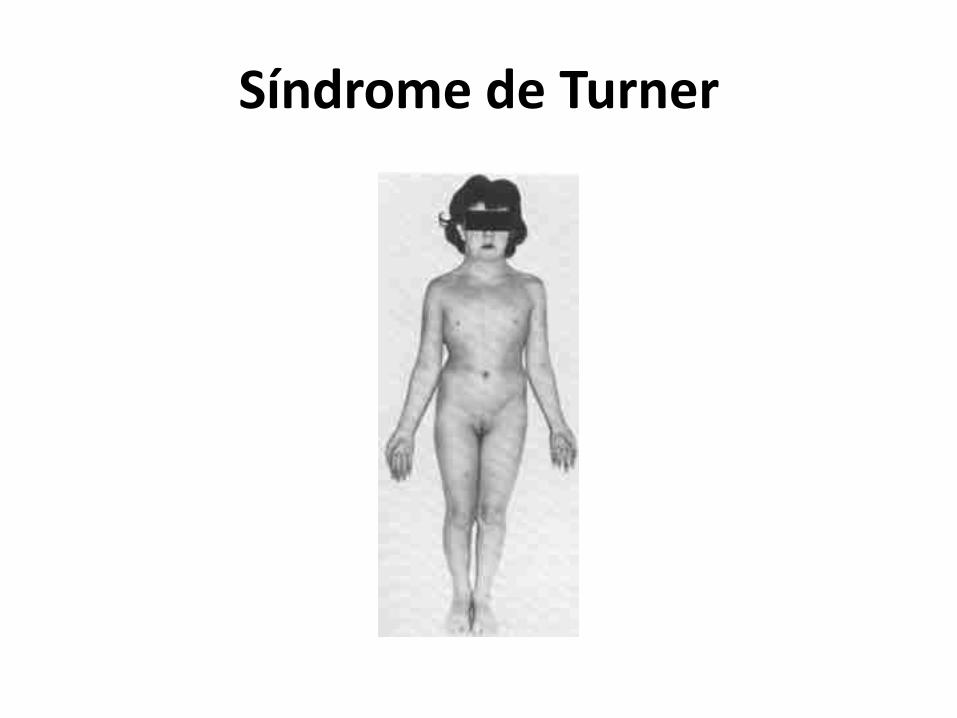
Síndrome de Turner

Síndrome de Turner
• 1:5000
• Monossomia do cromossomo X
• Quase todos são abortados
• 45 X – 75%
• Mosaico – 15% - 45 X/ 46 XX
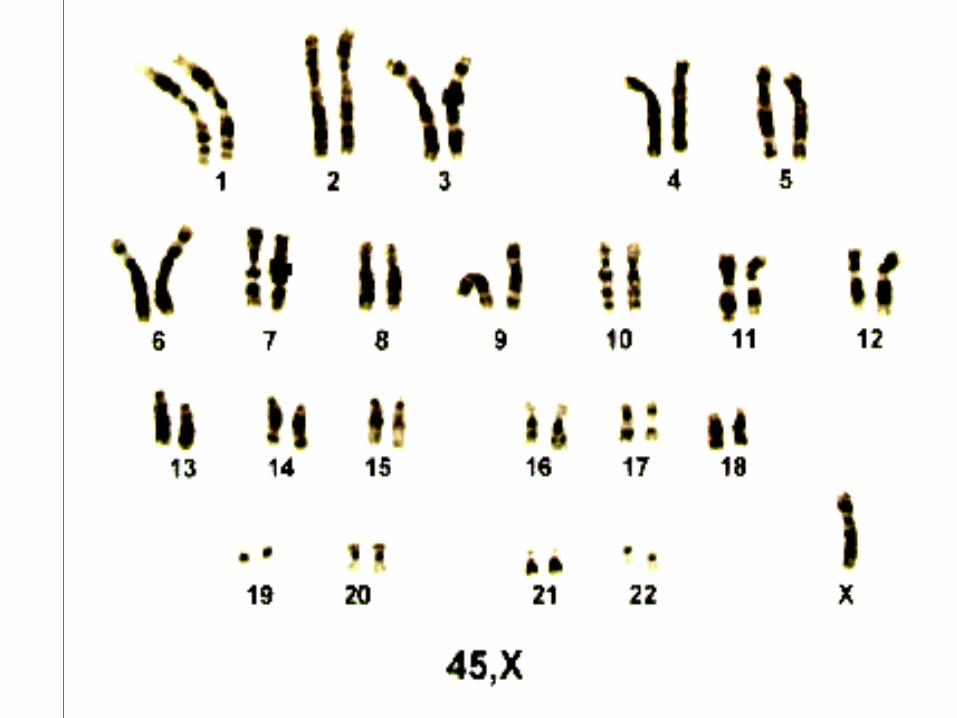

Síndrome de Turner
Manifestações Clínicas: • Baixa estatura 150,cm • Pescoço curto • Pouca mama • Tórax largo • Hipercifose e escoliose • Déficit de audição e visão • 1/5 DM

Síndrome de Turner
Face triangular e estrabismo
Envelhecimento do ovário e não desenvolvimento do mesmo
Déficit de hormônio do crescimento e estrógeno
Linfedema de membros
Displasia coxofemoral

Síndrome de Williams
• Fisionomia e personalidade: face expressiva, face de duende, nariz pequeno empinado, sorriso freqüente, bochechas proeminentes. Pessoa incompreendente, irritação e reclusa, não sociáveis e comunicáveis
• Coração: alterações cardiovasculares freqüentes, Ex: Estenose aórtica supravalvular, prolapso da valva mitral
• Órgãos: anomalias gênito urinárias • Músculo esquelético: baixa estatura, déficit de
coordenação e equilíbrio, escolioses e contraturas musculares
• Visuais e Auditivos: normal • Linguagem: Dificuldade para ler e escrever, voz
anasalada, dificuldade de expressão da linguagem
• RN: Desenvolvimento Neuro Psico Motor insuficiente, sua maioria apresenta dificuldade de alimentar-se no primeiro ano de vida
• Neurológicos: microcefalia e retardo mental • DNA: mutação genética esporádica, perda do
gene 7q11 do cromossomo 7

Síndrome de Williams

Síndrome de Williams
• 1960: médico Dr. J.C.P. Williams, Nova Zelândia
• Retardo mental • Dificuldade para ler e escrever • Dificuldade em realizar operações matemáticas • Face expressiva • Alterações cardiovasculares • Talento musical incompreendente

Síndrome de Williams
• Rara desordem genética freqüentemente não diagnosticada
• Não tem causa ambiental, médica
ou influencia psicossocial
• É uma mutação que
ocorre esporádicamente

Síndrome de Williams
• 1: 10.000 (Grimm e Wesselhoeft, 1980)
• Afeta tanto homens como mulheres
• Sua maioria apresenta dificuldade de alimentar-se no primeiro ano de vida
• Irritação e Reclusa

Síndrome de Williams
Etiologia:
• Microdeleção hemizigótica
• Perda de uma das cópias dos
genes, localizado na região
7q11 do cromossomo 7

Síndrome de Williams
Quadro Clínico:
• Face de duende
• Nariz pequeno empinado
• Lábios cheios
• Sorriso frequente
• Bochechas proeminentes
• Voz anasalada
• Alteração da Tiróide
• Produção de vit D deficitária

Síndrome de Williams
• Estenose aórtica supravalvular
• Hipoplasia da aorta
• Prolapso da valva mitral
• 65% microcefalia
• 95% retardo mental com QI de 56
• Impulsividade, autismo?
• Alterações de linguagem (expressão)
• Estatura Baixa

Síndrome de Williams
• Anomalias gênito-urinárias
• Micropênis
• Estenose da uretra
• Calculo renal
• Déficit coordenação e equilíbrio
• Atraso no desenvolvimento motor
• A maioria não são sociáveis e
comunicáveis (gestos e expressões)

Síndrome de Williams
Hipertensão arterial
Otite crônica
Função renal
Pesquisar a presença de escoliose e contratura das articulações.
O comportamento e aproveitamento escolar, quando problemáticos carecem de medidas de apoio.

Autismo
• O autismo é uma alteração cerebral, caracterizada como uma desordem que compromete o desenvolvimento psiconeurológico e afeta a capacidade da pessoa se comunicar, compreender e falar e afeta seu convívio social.
• Impedimento que prejudica a entrada sensorial e a maneira como as informações são processadas.
• O cérebro com autismo dá prioridade igual a todos os estímulos provocando uma enxurrada opressiva de informações sensoriais para ser manipulada.

Autismo
• O autismo infantil é um transtorno do desenvolvimento que manifesta-se antes dos 3 anos de idade, e é mais comum em meninos que em meninas e não é necessariamente acompanhado de retardo mental, pois existem casos de crianças que apresentam inteligência e fala intactas.

Autismo
• Fisionomia e personalidade: não apresenta capacidade de estabelecer relação com outras pessoas e responder adequadamente ao ambiente, isolamento, medo
• Músculo esquelético: atividades repetitivas, movimentos esteriotipados, tônus normal
• Visuais e Auditivos: normal, porém age como se
fosse surdo • Linguagem: impossibilidade de comunicação por
dificuldade de relacionamento interpessoal e atraso na fala
• RN: distúrbio do desenvolvimento, severa e
incapacitante, acometimento do sistema límbico - emocional
• Neurológicos: Normal, QI elevado, 70% QI reduzido com retardo mental associado
• DNA: normal, x frágil?alterações por infecção viral?
Causa ainda desconhecida

Síndrome de Asperger

Síndrome de Asperger
Fisionomia e personalidade: não consegue se sintonizar socialmente, formar família e dificuldade de arrumar emprego, desajeitado e solitários em sua maioria
Músculo esquelético: movimentos esteriotipados,
tônus normal Visuais e Auditivos: normal Linguagem: A fala se desenvolve na idade normal,
repetidas frases e palavras, só conversam sobre aquilo que lhes interessa, comunicação deficitária, voz robotizada
RN: transtorno invasivo do desenvolvimento de
causa desconhecida Neurológicos: capazes ou mais inteligentes que
outras pessoas, autismo de auto desempenho DNA: normal

Síndrome de Asperger
• 1944: médico austríaco Hans Asperger
• Grego: ´´autos´´= Próprio (Kanner, 1943)
• Título: Psicopatia Autista
• Desordem de Asperger

Síndrome de Asperger
• Asperger acreditava que os pacientes possuíam inteligência normal, e que não
existia atraso no desenvolvimento cognitivo e na fala
• Este estudo foi publicado na segunda guerra mundial em alemão e recebeu pouca atenção durante quase 40 anos

Síndrome de Asperger
• A síndrome não era reconhecida antes do 3 anos de idade
• A fala se desenvolve na idade normal
• Repetidas frases e palavras
• Brando Autismo
Ao conjunto de determinadas variações, chamamos de Espectro do Autismo, pois somam-se as
características autísticas, outras específicas de cada grupo de outros sintomas.

Síndrome de Asperger
Não possui a capacidade inata de ler as emoções expressas no comportamento das pessoas.
Não consegue se sintonizar socialmente
Só conversam sobre aquilo que lhes interessa
Os Aspies (como são conhecidos) ou ASP, normalmente não conseguem arrumar emprego, formar famíllia, mesmo que sejam tão capazes ou mais inteligentes que outras pessoas.

Síndrome de Asperger
• Apresentam elevadas habilidades cognitivas
• QI normal as vezes indo até faixas mais altas.

Síndrome de Asperger Tipos: Autismo Infantil (AI)- 70%
Transtorno esquizóide de personalidade e esquizofrenia infantil
Autismo de Alto Funcionamento ou Alto Desempenho
(AAF) ou (HFA)- 30% (Bosa, 2002) Super dotados

Síndrome de Asperger
• Andar desajeitado
• Pouco olhar para o intelocutor
• Voz sem modulação: robotizada
• Afetividade superficial
• Dificuldade de compreensão de piadas e significados superficiais (amigo x colega)

Asperger x Autismo Infantil
As diferenças fundamentais são: AI X SA
Gravidade do caso + Retardo Mental 70% + Alterações cognitivas + Atraso significativo da fala + Usa a 3ª pessoa pronominal (ele, ou seu nome) no
lugar da 1ª (eu) + QI executivo mais alto + Diagnóstico possível antes dos 3 anos +

Asperger x Autismo Infantil
AI x AS Diagnóstico de certeza só após 6 anos e idade + Inteligência verbal + Pedantismo (detalhista) + Busca ativa de interação social + Dá a impressão de possuir um estilo antigo, excêntrico + Pode dar a impressão de super -dotado + Pais com quadro similar +

Autismo
Relação com outras pessoas comprometida
1: 1.000 pessoas
Medo
Perda do contato com a realidade, o que acarretava uma grande
dificuldade ou impossibilidade de comunicação
É um distúrbio do desenvolvimento- Sistema Límbico- Emoções !!! (Gadia et al, 2004)

Autismo
• ASA - Autism Society of América, (1999) :
• Comunicação verbal e não verbal deficitária
• Geralmente se evidencia antes dos 3 anos
• Prejudica o desempenho educacional da criança !!!
Resistência a mudança no ambiente ou na rotina diária
Respostas incomuns a experiências sensoriais.

Autismo
Não:
Socializa
Espontaneidade
Compartilhar prazer interesses pessoais
Reciprocidade sócio emocional
Compreende de brincadeiras de faz de conta
Reciprocidade nas conversas
Expressão da linguagem (pouca)


Asperger x Autismo Infantil
O AUTISTA ESTÁ ISOLADO EM SEU PRÓPRIO MUNDO.
O ASPERGER ESTÁ EM NOSSO MUNDO PORÉM VIVENDO SEU ESTILO PRÓPRIO DE FORMA
ISOLADA.

Treatment and Education of Autistic and Communication Handicapped
Children (TEACCH)
• 1972: Departamento de Psiquiatria
• Universidade da Carolina do Norte
• Pensam em figuras, não em palavras
• Pensamentos funcionam como um “vídeo-tape”
• Rever algumas imagens sequenciais
• Dificuldades com sequências longas de informações ?
• Dificuldades em permanecer com uma informação no pensamento?

Treatment and Education of Autistic and Communication Handicapped
Children (TEACCH)
APRENDIZADO FUNCIONAL NATURAL
• Funcional - habilidade a ser ensinada, e que deve respeitar a idéia de “o mais funcional possível” ao sujeito que se dirige
• Natural - ambiente e situações de ensino que serão criados para que se dê o aprendizado, que deverão se aproximar ao máximo das situações vivenciadas no dia-a-dia.
• A aplicação de tal metodologia tem resultado na redução e/ou extinção dos comportamentos inapropriados


Autismo e Atividade Física
• De modo geral, os alunos com autismo mostram pouca habilidade motora (Morin & Reid apud Winnick 2004, p. 172). Conseqüentemente, os programas motores devem enfatizar habilidades e padrões motores fundamentais, jogos e esportes individuais e atividades de desenvolvimento que aumentem a proficiência física.
• Os programas de Educação Física e exercícios não devem se concentrar no ensino de movimentos como fins em si, mas na utilidade de seu aprendizado, destacando as possibilidades de avanços em adaptação, usos sociais das atividades promovidas, e aumento na qualidade de vida. (OLIVEIRA et al, 2009).

Síndrome de Asperger e Autismo
• Se faz necessário um acompanhamento neurológico, psiquiátrico e neuro-psicológico
• Desenvolvimento de programas pedagógicos
• Orientação a família e escola Método
Sonrise

Síndrome Alcoólica Fetal
Fisionomia e personalidade: face plana, nariz curto, labios finos, comunicativo e inteligente, irritabilidade
Órgãos: sopro cardíaco, estenose pulmonar e aórtico
Músculo esquelético: baixa estatura, hipertonia espástica, hiperreflexia, encurtamentos musculares e rigidez, reflexos primitivos presentes, má formações óssea, schuerman, escolioses e sinostoses radio ulnar
Visuais e Auditivos: normal em sua maioria porém podem apresentar alterações visuais- estrabismo e miopia
Linguagem: deficitária em acometimentos mais graves, pela espasticidade e normal em acometimentos leves
RN: prematuridade e desnutrição, baixo peso, atraso no Desenvolvimento Neuro Psico Motor, retardo no crescimento e divisão celular
Neurológicos: microcefalia, hipóxia (diminuição de aporte sanguíneo), acometimento neurológico, didtúrbio de comportamento e desenvolvimento, retardo mental em acometimentos extensos
DNA: normal

Síndrome Alcoólica Fetal

Síndrome Alcoólica Fetal
• Ingestão de bebida alcoólica pela mãe durante a gestação
• Efeitos irreversíveis • Leve à grave • Duração e quantidade • EUA: 3:1000 ...... Brasil ?

Síndrome Alcoólica Fetal
Fisiopatologia:
• Etanol atravessa a barreira placentária
• Alteração das membrana celulares
• Altera o transporte de aminoácidos no sangue – reduz a nutrição - prematuridade
• Retardo do crescimento e divisão celular

Síndrome Alcoólica Fetal
• Interferência na maturação neuronal
• Colapso momentâneo na vasculatura umbilical pela presença do álcool –
hipóxia
• Consumo alto de O2 pelo fígado, 99,9%
• Anormalidades neurológicas – convulsões, mielomeningocele, PC
• ´´Atraso intelectuais´´ e no DNPM • Distúrbios de comportamento e desenvolvimento

Síndrome Alcoólica Fetal
Alguns sintomas:
Irritabilidade, excitação e até disfunções neurológicas e
visuais, durante a amamentação mães que continuaram fumando, crianças com 7 anos em
média apresentaram atraso de aprendizado de cerca de 6 meses comparadas a crianças
de mães não fumantes.

Síndrome Alcoólica Fetal
Manifestações Clínicas:
• Microcefalia
• Face plana
• Nariz curto
• Fissura palpebral pequena
• Lábio superior fino
• Estrabismo
• Miopia
• Sopro cardíaco, estenose pulmonar e aórtico

Síndrome Alcoólica Fetal
• Déficit ou presença de reflexos primitivos
• Hiperexcitabilidade
• Irritabilidade
• Baixa estatura
• Retardo mental
• Déficit concentração
• Má formações vertebrais, scheuerman, escolioses, sinostose radio ulnar
•

Síndrome Alcoólica Fetal
Conhecido como: ósteo condromatose (Exostose)
Beniga disfunção do desenvolvimento da formação e direção da cartilagem endocondral de crescimento
Ossificação progressiva Femuro - tibial

Síndrome Alcoólica Fetal
• Apenas cerca de 17% conseguem acompanhar o desenvolvimento normal
• Problema de saúde pública
• Conscientizar e orientar a sociedade
• Equipe TRANSDICIPLINAR
A metade tem que freqüentar uma escola para crianças com dificuldades de aprendizagem, 1/5 vão a uma
escola para deficientes
Será que isso realmente é eficaz?
GABY

O professor deve adequar a metodologia a ser adotada:
• Qual grupo de educandos apresenta maior facilidade de aprendizado?
• Por quanto tempo o aluno pode permanecer atento as atividades solicitadas?
• Qual interesse do aluno? • Adaptar o programa e planejamento de aula, com
criatividade para alcançar os objetivos (Educação física e inclusão: considerações para a
prática pedagógica na escola Freitas, P. Cidade, R. 2009)

Deficiência Intelectual Plano de Aula
(Marteniuk, 1976)
Deficientes Mentais leves Aprendizagem lenta Capacidade de dominar habilidades acadêmicas básicas Capacidade de adaptação social e pessoa Capacidade de freqüentar escola comum em classes especial ou regular Deficientes Mentais Moderados Atraso significativo na aprendizagem Distúrbios psicomotores visíveis Adaptação a programas sistematizados Capacidade de formar hábitos higiênicos de rotina Capacidade de ajustamento satisfatório em relação à família, à escola e à comunidade. Deficientes Mentais Severos Acentuado prejuízo na comunicação; Acentuado prejuízo na mobilidade; Alcance de resultados no trabalho condicionado e repetitivo, com supervisão e ajuda
constantes. Deficientes Mentais Profundos Dependência completa; Limitações extremamente acentuadas na aprendizagem.

Deficiência Intelectual Plano de Aula
(Marteniuk, 1976)
Psicomotoras Lentidão na marcha Gestos e posturas inadequados Deficiência na coordenação de movimentos globais e finos Distúrbios de equilíbrio Distúrbios na relação espaço – temporal Deficiência ventilatória – funcional Afetivos-Sociais Baixa resposta frente a eventos sociais Desconhecimento das limitações e possibilidades Desajuste em atividades em grandes grupos Demonstração de reações emocionais ao ser solicitado individuais ou em grupo Cognitivas Atenção dirigida limitada Limitações de aprendizagem espontânea Memória falha nas questões espaciais e temporais Lentidão para aquisição de fala Distúrbios na aquisição de imagem corporal

Deficiência Intelectual Plano de Aula
(Marteniuk, 1976)
Normalização • Implica garantir vida a mais normal possível para a pessoa portadora de
deficiência, proporcionando-lhe oportunidades humanas, que não se distanciem de sua realidade, possibilitando-lhe condições de estar mais pronto para o exercício da vida diária. Implica tratamento normal, que não significa desconhecimento de suas limitações e impedimentos, proporcionados pela deficiência.
Integração • Significa igualdade de oportunidade, de poder tomar partes nas mesmas
oportunidades oferecidas por sua comunidade e que venham a oferecer seu desenvolvimento e aprendizado.
Individualização • Significa o exercício do conhecimento particular de cada indivíduo,
garantindo-lhe melhores condições para adaptar-se ao grupos, que se constituiem da sua escola, da sua comunidade, etc. Cada pessoa necessita de uma solução individual, embora isso não represente atendimento individual, mas sim garantir-lhe condições de aprendizado compatíveis com sua individualidade.

Deficiência Intelectual Plano de Aula
(Marteniuk, 1976)
Perfil do Professor O professor deve ser: Seguro; Estimulador; Versátil; Persiste, Comunicativo e Paciente. O professor deve ter: Rapidez de raciocínio, bom senso, atenção, auto-domínio, resistência à rotina
e capacidade de observação
Diretrizes para Exercícios Passar a informação do exercício de modo que possa ser assimilada com facilidade
Incluir estratégias para evitar distrações e promover motivação Promover aprendizado de tarefas simples e progredir para complexas
Realizar repetição do ato constantemente - Princípio da aprendizagem motora Ao mudar a tarefa sempre parta de algo de base já aprendido
Utilizar estratégias necessárias para adequar o movimento ou tarefa motora Busque sempre conhecimentos a cerca da patologia, conheça seu aluno, oriente
os pais ou cuidadores, realize seu trabalho de forma consciente e com amor.

E AGORA
Tratamento Atividades Específicas

Tratamento e Atividades Específicas
• Conhecer a patologia
• Avaliação adequada
• Adequar tônus
• Minimizar dificuldade e
estimular o aprendizado
• Respeitar grau de deficiência mental

Tratamento e Atividades Específicas
Atividade física:
• Promover independência nas AVD`S
• Prevenir contraturas e deformidades
• Evitar complicações
• Força e resistência muscular
• Controle de tronco
• Estabilidade

Tratamento e Atividades Específicas
Estimular o DNPM Rolar... Controle de cabeça Padrão flexor Estímulos visuais e
auditivos
Flacidez ? Espasticidade? DOWN- CUIDADO: IAA

Tratamento e Atividades Específicas
Com o crescimento:
• Atividades ativas e rápidas no caso do down
• Com resistência
• Equilíbrio
• Diferentes estímulos sensoriais

Possíveis Soluções dos Problemas
Déficit de atenção e apatia:
Mudança do tom de voz - liderança
Realização de brincadeiras durante uma orientação
Apresentação da novidade e desafio
Materiais coloridos que imitem sons
(Gorgartti, 2008)

Possíveis Soluções dos Problemas
Déficit de linguagem e comunicação:
Estimular outras formas de comunicação não verbal
Estimular atividades rítmicas e expressivas
(Gorgartti, 2008)

Possíveis Soluções dos Problemas
• Déficit de compreensão:
• Clareza na apresentação de informações
• Uso de diferentes canais sensoriais para a transmissão da mesma informação
(Gorgartti, 2008)

Possíveis Soluções dos Problemas
Agressividade:
Rigidez no início Refletir sobre a estrutura da atividade Requisitar auxilio de monitor Afastar o aluno da turma por um período Utilizar o aluno como ajudante na manutenção da disciplina Deixar claro que a punição é relacionada ao comportamento
(Gorgartti, 2008)


















